WO2012118189A1 - アミノ-カルボン酸により修飾されたヒアルロン酸誘導体 - Google Patents
アミノ-カルボン酸により修飾されたヒアルロン酸誘導体 Download PDFInfo
- Publication number
- WO2012118189A1 WO2012118189A1 PCT/JP2012/055421 JP2012055421W WO2012118189A1 WO 2012118189 A1 WO2012118189 A1 WO 2012118189A1 JP 2012055421 W JP2012055421 W JP 2012055421W WO 2012118189 A1 WO2012118189 A1 WO 2012118189A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hyaluronic acid
- diyl
- hydrogen atom
- acid derivative
- alkyl
- Prior art date
Links
- 0 CCC(CC)C(C(**)C1OC(C(*)C(*)C2O*(CC)CC=*)OC2C(*)=O)OC(C*)C1N=O Chemical compound CCC(CC)C(C(**)C1OC(C(*)C(*)C2O*(CC)CC=*)OC2C(*)=O)OC(C*)C1N=O 0.000 description 3
- XDTMQSROBMDMFD-UHFFFAOYSA-N C1CCCCC1 Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- LCIICDVIYIMVNX-UHFFFAOYSA-N CCC[N](C)(C)CC(C)C Chemical compound CCC[N](C)(C)CC(C)C LCIICDVIYIMVNX-UHFFFAOYSA-N 0.000 description 1
- GBMDYACPQYNUGA-UHFFFAOYSA-N CNC(CC1CCCCC1)C(O)=O Chemical compound CNC(CC1CCCCC1)C(O)=O GBMDYACPQYNUGA-UHFFFAOYSA-N 0.000 description 1
- NVXKJPGRZSDYPK-UHFFFAOYSA-N CNC(CCc1ccccc1)C(O)=O Chemical compound CNC(CCc1ccccc1)C(O)=O NVXKJPGRZSDYPK-UHFFFAOYSA-N 0.000 description 1
- XRBFRWVSTGFFCB-UHFFFAOYSA-N CNC(Cc1cc2ccccc2cc1)C(O)=O Chemical compound CNC(Cc1cc2ccccc2cc1)C(O)=O XRBFRWVSTGFFCB-UHFFFAOYSA-N 0.000 description 1
- FWVHRCVITNUYFV-UHFFFAOYSA-N CNCc(cc1)ccc1C(O)=O Chemical compound CNCc(cc1)ccc1C(O)=O FWVHRCVITNUYFV-UHFFFAOYSA-N 0.000 description 1
- BYTJBOHWWSBSNS-ZBOXLXRLSA-N C[C@@H](CCCNC)CC1(C)N(C)C1 Chemical compound C[C@@H](CCCNC)CC1(C)N(C)C1 BYTJBOHWWSBSNS-ZBOXLXRLSA-N 0.000 description 1
Images




























































































Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B37/00—Preparation of polysaccharides not provided for in groups C08B1/00 - C08B35/00; Derivatives thereof
- C08B37/006—Heteroglycans, i.e. polysaccharides having more than one sugar residue in the main chain in either alternating or less regular sequence; Gellans; Succinoglycans; Arabinogalactans; Tragacanth or gum tragacanth or traganth from Astragalus; Gum Karaya from Sterculia urens; Gum Ghatti from Anogeissus latifolia; Derivatives thereof
- C08B37/0063—Glycosaminoglycans or mucopolysaccharides, e.g. keratan sulfate; Derivatives thereof, e.g. fucoidan
- C08B37/0072—Hyaluronic acid, i.e. HA or hyaluronan; Derivatives thereof, e.g. crosslinked hyaluronic acid (hylan) or hyaluronates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/715—Polysaccharides, i.e. having more than five saccharide radicals attached to each other by glycosidic linkages; Derivatives thereof, e.g. ethers, esters
- A61K31/726—Glycosaminoglycans, i.e. mucopolysaccharides
- A61K31/728—Hyaluronic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/61—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule the organic macromolecular compound being a polysaccharide or a derivative thereof
Definitions
- the present invention relates to a compound having amino and carboxy such as alkylamine and amino acid (amino-carboxylic acid) or a hyaluronic acid derivative modified with an amide compound thereof (amino-carboxylic acid amide), and the hyaluronic acid derivative and the drug are bonded.
- a pharmaceutical composition comprising the hyaluronic acid derivative, particularly a complex of the hyaluronic acid derivative and a drug.
- PEG Polyethylene glycol
- Non-patent Documents 1 and 2 a phenomenon in which clearance at the second administration is abnormally fast (Accelerated Blood Clearance phenomenon, hereinafter also referred to as “ABC phenomenon”) has been reported (Non-patent Documents 1 and 2). Therefore, it is difficult to say that the safety and effectiveness of PEGylated drugs are well established.
- hyaluronic acid is an example of a polymer already used in clinical practice.
- Hyaluronic acid (hereinafter also referred to as “HA”) It is a polysaccharide isolated from the vitreous body of cattle eyes by Meyer and has long been known as the main component of the extracellular matrix.
- HA is a kind of glycosaminoglycan composed of a disaccharide unit in which D-glucuronic acid and N-acetylglucosamine are linked by a ⁇ (1 ⁇ 3) glycosidic bond as a repeating unit.
- Hyaluronic acid has no species difference in its chemical and physical structure, and there exists a metabolic system of hyaluronic acid in humans.
- hyaluronic acid is known as a very safe biomaterial in terms of immunity or toxicity.
- hyaluronic acid as a physiologically active substance related to the induction of cell adhesion, proliferation, and migration has attracted attention.
- high-molecular-weight hyaluronic acid can be mass-produced by microorganisms, and drug delivery systems using hyaluronic acid (hereinafter also referred to as “DDS”) have been actively studied.
- DDS drug delivery systems using hyaluronic acid
- a hyaluronic acid derivative has been developed and its usefulness has been demonstrated by highly modifying the carboxy of the glucuronic acid moiety of hyaluronic acid to realize prolonged retention in blood (Patent Document 7).
- Patent Document 7 the retention of the hyaluronic acid derivative in the blood is prolonged by increasing the carboxy modification rate of the glucuronic acid moiety.
- the two do not correlate linearly and change abruptly at a certain threshold.
- oligonucleotides such as antisense DNA / RNA and siRNA, which are being developed as nucleic acid pharmaceuticals, are susceptible to degradation by nucleolytic enzymes inside and outside the body, and are administered intravenously as a single nucleic acid (hereinafter also referred to as “iv”). Then, it is quickly decomposed.
- nucleic acid drugs In order to express the efficacy of nucleic acid drugs, it is essential to deliver the nucleic acid into the cytoplasm or nucleus.
- pharmaceuticals that have proteins and peptides as active ingredients have mainly been those that act on extracellular targets. In order to develop more innovative drugs, there is a need for means for allowing proteins and peptides to act on intracellular targets.
- a method for delivering a protein or peptide into the cytoplasm that is, a method for efficiently releasing a pharmaceutically active ingredient from the endosome into the cytoplasm after being taken into the cell by endocytosis is required.
- cationic synthetic polymers can electrostatically condense negatively charged genes and deliver genes into the cytoplasm, they have been effective as gene carriers.
- examples of such cationic synthetic polymers include poly-L-lysine (Patent Document 8, Non-Patent Document 3), polyethyleneimine (Non-Patent Document 4), synthetic polymer having imidazolyl (Non-Patent Document 5), polyamidoamine dendrimer ( Synthetic polymers such as Non-Patent Document 6) have been reported.
- a synthetic polymer having a secondary amine-containing polyethyleneimine or imidazolyl is taken into the cytoplasm with high introduction efficiency by exerting a proton sponge effect.
- Non-patent Document 7 chitosan which is a cationic polymer of polysaccharide
- Non-patent Document 8 a method of modifying with a cell-penetrating peptide (cpp) (Non-patent Document 8) and an attempt to introduce a protein or peptide complex into a cytoplasm by using a cationic liposome as a carrier have been reported. (Non-patent Document 9), the introduction efficiency is not necessarily high.
- Non-patent Document 10 polyamine PEGylation, or an approach in which HA is modified with polyamine (Non-patent Document 10), or a polyamine is modified with a functional group that desorbs in a pH-responsive manner (for example, at low pH). By doing so, an approach to reduce its toxicity is underway. However, since polyamine is still inherent, further contrivance and examination are necessary for its toxicity (Non-patent Document 11).
- Non-patent document 12 Poly (ethylacrylic acid) (Non-patent document 12) and poly (propylacrylic acid) (Non-patent document 13) are known as such anionic polymers, and a polycarboxylic acid polymer having a pKa of about 5 is a cell.
- Non-patent Document 14 succinate-modified polyglycidol
- Non-patent Document 15 pseudo-peptide in which the side chain of poly (L-lysine-isophthalamide) is modified with L-phenylalanine have pH responses.
- anionic polymer exhibiting sex it has been reported that it is effective for gene / drug release from endosomes, that is, for the incorporation of drugs into the cytoplasm via endocytosis.
- An example of modification of carboxy in hyaluronic acid with an amino acid is, for example, produced by using 2-chloro-4,6-dimethoxy-1,3,5-triazine in the presence of N-methylmorpholine.
- Modification of glycine with ethyl ester using 4,6-dimethoxy-1,3,5-triazine) -4-methylmorpholium (hereinafter also referred to as DMT-MM) as a condensing agent is possible. Is 20% at the maximum (Non-patent Document 16).
- Non-patent Document 18 hyaluronic acid introduced with alanine has been reported as an example of using a triazine-based compound as a condensing agent. Modifications have also been reported for amino acids (Non-patent Document 19, Patent Document 10).
- EDC 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride
- EDC 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride
- leucine methyl ester hydrochloride valine methyl ester hydrochloride
- Water-insoluble biocompatible modified with hydrochloride, proline methyl ester hydrochloride, phenylalanine methyl ester hydrochloride, arginine methyl ester hydrochloride and histidine methyl ester hydrochloride, gelled without deprotection
- the modification rate is unknown (Patent Document 9).
- the problem to be solved by the invention is to provide a hyaluronic acid derivative having both biodegradability and blood retention characteristics and / or a hyaluronic acid derivative capable of delivering a gene and a drug into the cytoplasm. It is. Another problem to be solved by the invention is to provide a conjugate in which the hyaluronic acid derivative and the drug are bound, and a pharmaceutical composition containing the hyaluronic acid derivative, particularly a complex of the hyaluronic acid derivative and the drug. It is.
- the present inventor has intensively studied to solve such problems. As a result, the carboxy of the glucuronic acid moiety of hyaluronic acid or a salt thereof is changed to a specific alkylamine, a specific amino-carboxylic acid or a specific amino-carboxylic acid.
- the present inventors have found that the hyaluronic acid derivative obtained by reacting with an amide to convert to an amide has both biodegradability and blood retention properties, and completed the present invention. Further, the present inventors have found that a hyaluronic acid derivative modified with a specific amino-carboxylic acid is effective in releasing a gene / drug from the endosome into the cytoplasm, thereby completing the present invention.
- the present invention relates to a hyaluronic acid derivative having both biodegradability and blood retention, and a hyaluronic acid that suppresses in vivo degradation of a compound having pharmacological activity, particularly a nucleic acid drug, and enables delivery into the cytoplasm. It relates to acid derivatives. Furthermore, the present invention relates to a method for producing the hyaluronic acid derivative, a drug and a pharmaceutical composition containing the hyaluronic acid derivative, and a method for producing the same.
- R 1 , R 2 , R 3 , and R 4 are each independently selected from a hydrogen atom, C 1-6 alkyl, formyl, and C 1-6 alkylcarbonyl; R 8 is a hydrogen atom, formyl or C 1-6 alkylcarbonyl;
- X is a group represented by —NR x —AB—Z;
- A is selected from —CR a R b —, and C 3-8 cycloalkylene, B is a direct bond, Z is —COOR y or —CONR ya R yb ; or —A—B—Z is , C 1-6 alkyl; or
- A is —CH 2 — or —CH 2 —CH 2 —, B is phenylene (wherein phenylene is substituted with one or more groups selected from hydroxy and halogen atoms) Selected from C 3-8 cycloalkylene and phenylmethane-1,1-diyl;
- A is C 3-8 cycloalkylene and B is a direct bond;
- A is —CH 2 — or —CH 2 —CH 2 —, and B is phenylene (wherein phenylene may be substituted with one or more groups selected from hydroxy and halogen atoms), C 3- Selected from 8 cycloalkylene and phenylmethane-1,1-diyl; or
- A is 2-cyclohexylethane-1,1-diyl, 2- (2-naphthyl) ethane-1,1-diyl, 3-phenylpropane
- the hyaluronic acid derivative according to the above (1) selected from -1,1-diyl, cyclohexylmethane-1,1-diyl and 4-hydroxyphenylmethane-1,1-diyl, wherein B is a direct bond.
- A is —CH 2 — and B is cyclohexane-1,1-diyl, benzene-1,4-diyl, benzene-1,3-diyl, 2-chlorobenzene-1,4-diyl and phenylmethane Selected from -1,1-diyl;
- A is —CH 2 CH 2 — and B is benzene-1,4-diyl; or Wherein A is selected from 2-cyclohexylethane-1,1-diyl, 2- (2-naphthyl) ethane-1,1-diyl and 3-phenylpropane-1,1-diyl;
- B is a direct bond,
- R 1 , R 2 , R 3 , and R 4 are each independently selected from a hydrogen atom, C 1-6 alkyl, formyl, and C 1-6 alkylcarbonyl;
- R 8 is a hydrogen atom, formyl or C 1-6 alkylcarbonyl;
- X is a group represented by —NHCH 3 , —NH (CH 2 ) 2 CH 3 , —NR x —AB—COOR y or —NR x —CHR c —CONR ya R yb ;
- R x and R y are each independently selected from a hydrogen atom and C 1-6 alkyl;
- A represents —CR a R b —, C 3-8 cycloalkylene, 2-cyclohexylethane-1,1-diyl, 2- (2-naphthyl) ethane-1,1-diyl, 3-phenylpropane-1, Selected from 1-diyl,
- B is selected from phenylene (wherein phenylene may be substituted with one or more groups selected from hydroxy and halogen atoms), C 3-8 cycloalkylene and phenylmethane-1,1-diyl Is;
- R a is selected from a hydrogen atom and C 1-6 alkyl;
- R b is selected from a hydrogen atom and C 1-6 alkyl (wherein C 1-6 alkyl may be substituted with one or more groups selected from hydroxy, carboxy and carbamoyl);
- R c represents C 1-6 alkyl optionally substituted with carbamoyl;
- R ya and R yb are each independently selected from a hydrogen atom, C 1-6 alkyl, formyl and C 1-6 alkylcarbonyl]
- X is —NHCH 3 , —NH (CH 2 ) 2 CH 3 or —NR x —CHR c —CONR ya R yb , or A is —CR a R b — or C 3-8 cyclo
- A is C 3-8 cycloalkylene and B is a direct bond; A is —CH 2 — or —CH 2 —CH 2 —, and B is phenylene (wherein phenylene may be substituted with one or more groups selected from hydroxy and halogen atoms), C 3- Selected from 8 cycloalkylene and phenylmethane-1,1-diyl; or A is 2-cyclohexylethane-1,1-diyl, 2- (2-naphthyl) ethane-1,1-diyl, 3-phenylpropane
- the hyaluronic acid derivative according to (6) above selected from -1,1-diyl, cyclohexylmethane-1,1-diyl and 4-hydroxyphenylmethane-1,1-diyl, wherein B is a direct bond.
- A is —CH 2 — and B is cyclohexane-1,1-diyl, benzene-1,4-diyl, benzene-1,3-diyl, 2-chlorobenzene-1,4-diyl and phenylmethane Selected from -1,1-diyl;
- A is —CH 2 CH 2 — and B is benzene-1,4-diyl; or Wherein A is selected from 2-cyclohexylethane-1,1-diyl, 2- (2-naphthyl) ethane-1,1-diyl and 3-phenylpropane-1,1-diyl;
- B is a direct bond, The hyaluronic acid derivative according to (6) or (8).
- R 1 , R 2 , R 3 , and R 4 are each independently selected from a hydrogen atom, C 1-6 alkyl, formyl, and C 1-6 alkylcarbonyl;
- R 8 is a hydrogen atom, formyl or C 1-6 alkylcarbonyl;
- X is a group represented by —NR x —AB—COOR y ;
- R x and R y are each independently selected from a hydrogen atom and C 1-6 alkyl;
- A represents —CR a R b —, C 3-8 cycloalkylene, 2-cyclohexylethane-1,1-diyl, 2- (2-naphthyl) ethane-1,1-diyl, and 3-phenylpropane-1 , 1-diyl and B is a direct bond; or
- A is —CH 2 — and B is selected from phenylene and C 3-8 cycloalkylene;
- R a is selected
- A is —CR a R b — or C 3-8 cycloalkylene.
- A is C 3-8 cycloalkylene and B is a direct bond;
- A is —CH 2 — and B is selected from phenylene and C 3-8 cycloalkylene; or
- A is 2-cyclohexylethane-1,1-diyl, 2- (2-naphthyl) ethane-1,1
- A is —CH 2 — and B is selected from cyclohexane-1,1-diyl and benzene-1,4-diyl; or Wherein A is selected from 2-cyclohexylethane-1,1-diyl, 2- (2-naphthyl)
- R 1a , R 2a , R 3a , and R 4a are each independently selected from a hydrogen atom, C 1-6 alkyl, formyl, and C 1-6 alkylcarbonyl;
- R 8a is a hydrogen atom, formyl or C 1-6 alkylcarbonyl;
- X a is selected from hydroxy and —O ⁇ Q + ; where Q + represents a counter cation]
- the hyaluronic acid derivative according to any one of (1) to (13), further comprising a disaccharide unit represented by: (15) Formula (III):
- R 1b , R 2b , R 3b , and R 4b are each independently selected from a hydrogen atom, C 1-6 alkyl, formyl and C 1-6 alkylcarbonyl;
- R 8b is a hydrogen atom, formyl or C 1-6 alkylcarbonyl;
- X b is a group represented by —NR e —Y b —R d ;
- R e is a hydrogen atom or C 1-6 alkyl;
- R d is a hydrogen atom, C 1-6 alkyl, —CO—C (R 7 ) ⁇ CH 2 , or —CO—G 4 —X c ;
- R 7 is a hydrogen atom or methyl;
- G 4 is selected from phenylene, C 3-8 cycloalkylene, or -G 5- (C 1-10 alkylene) -G 6- , wherein the C 1-10 alkylene moiety is 1-3 phenylene or C 3-8 cyclo
- a group represented by: Y b represents —CH 2 — (CHR 5 ) 1 -2 —CH 2 —NH—, —CH 2 — (CHR 6 ) p-2 —CH 2 —O—, — (CH 2 ) j —S— or -(CH 2 ) a- (Y 1- (CH 2 ) b ) c -G-; l, p, and j are each independently an integer selected from 2 to 10, and R 5 and R 6 are each independently a hydrogen atom or hydroxy; a is an integer selected from 2 to 10; b is an integer independently selected from 2 to 10; c is an integer selected from 1 to 200; Y 1 is an oxygen atom or —NR n —; G is an oxygen atom, a sulfur atom or —NH—; R n represents a hydrogen atom, C 1-6 alkyl, —CO— (CH 2 ) d —R o , — (CH 2 ) e —R
- the non-derivatized hyaluronic acid having a main chain structure corresponding to the hyaluronic acid derivative has a weight average molecular weight of 20 to 120 kilodaltons, and the non-derivatized hyaluronic acid has the formula (II) Wherein R 1a , R 2a , R 3a and R 4a are hydrogen atoms, R 8a is acetyl and Xa is hyaluronic acid consisting only of disaccharide units of —O ⁇ Na +.
- a pharmaceutical composition comprising the hyaluronic acid derivative according to any one of (1) to (17) as a carrier.
- a hyaluronic acid derivative-drug conjugate in which one or more drugs are bound to the hyaluronic acid derivative according to any one of (1) to (17) above.
- biodegradable drug carrier comprising the hyaluronic acid derivative according to any one of (1) to (17) above.
- a carrier for introducing a drug into the cytoplasm comprising the hyaluronic acid derivative according to any one of (1) to (17) above.
- the hyaluronic acid according to any one of (1), (2), (5) to (7), (10), (11), and (14) to (17) Biodegradable drug carriers comprising the derivatives are provided.
- the hyaluronic acid derivative according to any one of (1), (3) to (6), (8) to (10), and (12) to (17) is included.
- Carriers for the cytoplasmic introduction of drugs are provided.
- a method for administering a drug comprising administering to the subject a therapeutically effective amount of the drug together with the hyaluronic acid derivative according to any one of (1) to (17) above.
- a method for introducing a drug into the cytoplasm comprising administering a therapeutically effective amount of the drug together with the hyaluronic acid derivative according to any one of (1) to (17) above.
- the hyaluronic acid according to any one of (1), (2), (5) to (7), (10), (11), and (14) to (17)
- a method of administering a drug comprising administering to the subject a therapeutically effective amount of the drug along with the derivative.
- the therapeutic effect is effective together with the hyaluronic acid derivative according to any one of (1), (3) to (6), (8) to (10), and (12) to (17).
- a method of introducing a drug into the cytoplasm comprising administering an amount of the drug is provided.
- Example 2 is an example of 1 H-NMR spectrum of HA-TBA prepared in Example 1-2.
- 1 is an example of 1 H-NMR spectrum of HA-FL / TBA prepared in Example 1-3.
- 1 is an example of 1 H-NMR spectrum of HA-Ala / FL prepared in Example 1-4-1.
- 2 is an example of 1 H-NMR spectra of HA-Ser / FL and HA-Ser-OEt / FL prepared in Example 1-4-2.
- It is an example of 1 H-NMR spectrum of HA-Glu / FL prepared in Example 1-4-3.
- 2 is an example of 1 H-NMR spectra of HA-Gly / FL and HA-Gly-OEt / FL prepared in Example 1-4-4.
- 1 is an example of 1 H-NMR spectrum of HA-Val / FL prepared in Example 1-4-5.
- 1 is an example of 1 H-NMR spectrum of HA-Leu / FL prepared in Example 1-4-6.
- 1 is an example of 1 H-NMR spectrum of HA-Ile / FL prepared in Example 1-4-7.
- 1 is an example of 1 H-NMR spectrum of HA-Thr / FL prepared in Example 1-4-8.
- 1 is an example of 1 H-NMR spectrum of HA-Asp / FL prepared in Example 1-4-9.
- 1 is an example of 1 H-NMR spectrum of HA-cACHCA / FL prepared in Example 1-4-10.
- 1 is an example of 1 H-NMR spectrum of HA-tACHCA-OEt / FL prepared in Example 1-4-11.
- 1 is an example of 1 H-NMR spectrum of HA-Aib / FL prepared in Example 1-4-12.
- 1 is an example of 1 H-NMR spectrum of HA-ACBuCA-OEt / FL prepared in Example 1-4-13.
- 1 is an example of 1 H-NMR spectrum of HA-Asn / Rh prepared in Example 1-4-14.
- 1 is an example of 1 H-NMR spectrum of HA-Ala-NH 2 / Rh prepared in Example 1-4-15.
- 1 is an example of 1 H-NMR spectrum of HA-Val-NH 2 / Rh prepared in Example 1-4-16.
- 1 is an example of 1 H-NMR spectrum of HA-Asn-NH 2 / Rh prepared in Example 1-4-17.
- 1 is an example of 1 H-NMR spectra of HA-Me and HA-Me / FL prepared in Example 1-4-18.
- 1 is an example of 1 H-NMR spectra of HA-Pr and HA-Pr / FL prepared in Example 1-4-19.
- 1 is an example of 1 H-NMR spectrum of HA-AMCHCA prepared in Example 1-5-1.
- 1 is an example of 1 H-NMR spectrum of HA-pcACHCA prepared in Example 1-5-2.
- 1 is an example of 1 H-NMR spectrum of HA-Nal prepared in Example 1-5-3.
- 1 is an example of 1 H-NMR spectrum of HA-APBA prepared in Example 1-5-4.
- 1 is an example of 1 H-NMR spectrum of HA-Cha prepared in Example 1-5-5.
- 1 is an example of 1 H-NMR spectrum of HA-AMBA prepared in Example 1-5-6.
- 1 is an example of 1 H-NMR spectrum of HA-3AMBA prepared in Example 1-5-7.
- 1 is an example of 1 H-NMR spectrum of HA-APhPA prepared in Example 1-5-8.
- 1 is an example of 1 H-NMR spectrum of HA-AEBA prepared in Example 1-5-9.
- 1 is an example of 1 H-NMR spectrum of HA-AMClBA prepared in Example 1-5-10.
- 1 is an example of 1 H-NMR spectrum of HA-AMSA prepared in Example 1-5-11.
- 1 is an example of 1 H-NMR spectrum of HA-4AMCHCA prepared in Example 1-5-12.
- 1 is an example of 1 H-NMR spectrum of HA-Chg prepared in Example 1-5-13.
- 1 is an example of 1 H-NMR spectrum of HA-pHPhg prepared in Example 1-5-14.
- 1 is an example of 1 H-NMR spectrum of HA-FL prepared in Comparative Example 1-1-1 and HA-EDOBEA having a low modification rate.
- 1 is an example of 1 H-NMR spectra of HA-EDOBEA-Ac / FL prepared in Comparative Example 1-1-2 and HA-EDOBEA having a high modification rate.
- 2 is an example of 1 H-NMR spectrum of HA-Phe / FL prepared in Comparative Example 1-1-3.
- 2 is an example of 1 H-NMR spectrum of HA-Tyr / FL prepared in Comparative Example 1-1-4.
- 2 is an example of 1 H-NMR spectrum of HA-MePhe / FL prepared in Comparative Example 1-1-5.
- 2 is an example of 1 H-NMR spectrum of HA-Pro-OMe / FL prepared in Comparative Example 1-1-6.
- 7 is an example of 1 H-NMR spectrum of HA-Gly-NH 2 / Rh prepared in Comparative Example 1-1-7.
- 2 is an example of 1 H-NMR spectrum of HA-Ser-NH 2 / Rh prepared in Comparative Example 1-1-8.
- 2 is an example of 1 H-NMR spectrum of HA-Leu-NH 2 / Rh prepared in Comparative Example 1-1-9.
- 1 is an example of 1 H-NMR spectrum of HA-Ile-NH 2 / Rh prepared in Comparative Example 1-1-10.
- 1 is an example of 1 H-NMR spectrum of HA-Thr—NH 2 / Rh prepared in Comparative Example 1-1-11.
- 1 is an example of 1 H-NMR spectrum of HA-Gln-NH 2 / Rh prepared in Comparative Example 1-1-12.
- 2 is an example of 1 H-NMR spectrum of HA-Nle prepared in Comparative Example 1-2-1.
- 2 is an example of 1 H-NMR spectrum of HA-tLeu prepared in Comparative Example 1-2-2.
- 2 is an example of 1 H-NMR spectrum of HA-pFPhe prepared in Comparative Example 1-2-3.
- 2 is an example of 1 H-NMR spectrum of HA-Phg prepared in Comparative Example 1-2-4.
- 2 is an example of 1 H-NMR spectrum of PEG-FL prepared in Comparative Example 1-3. It shows the group mean value of the sample concentration transition in rat plasma calculated in Example 3-2-2. It is the graph which plotted the group average value of the sample concentration transition in the rat plasma computed in Example 3-2-2. It is the graph which plotted the group average value of the sample concentration transition in the rat plasma computed in Example 3-2-2.
- 2 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Ala / FL prepared in Example 1-4-1 to size exclusion chromatography.
- FIG. 2 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Ser / FL prepared in Example 1-4-2 to size exclusion chromatography.
- FIG. 3 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Glu / FL prepared in Example 1-4-3 to size exclusion chromatography.
- FIG. FIG. 2 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Gly / FL prepared in Example 1-4-4 to size exclusion chromatography.
- FIG. 6 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Val / FL prepared in Example 1-4-5 to size exclusion chromatography.
- FIG. 6 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Leu / FL prepared in Example 1-4-6 to size exclusion chromatography.
- FIG. 3 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Ile / FL prepared in Example 1-4-7 to size exclusion chromatography.
- FIG. 6 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Thr / FL prepared in Example 1-4-8 to size exclusion chromatography.
- FIG. FIG. 3 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Asp / FL prepared in Example 1-4-9 to size exclusion chromatography.
- FIG. 3 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-cACHCA / FL prepared in Example 1-4-10 to size exclusion chromatography.
- 2 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-tACHCA-OEt / FL prepared in Example 1-4-11 to size exclusion chromatography.
- 2 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Aib / FL prepared in Example 1-4-12 to size exclusion chromatography.
- FIG. 3 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-ACBuCA-OEt / FL prepared in Example 1-4-13 to size exclusion chromatography.
- FIG. 3 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Asn / Rh prepared in Example 1-4-14 to size exclusion chromatography.
- FIG. 6 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Ala-NH 2 / Rh prepared in Example 1-4-15 to size exclusion chromatography.
- 6 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Val-NH 2 / Rh prepared in Example 1-4-16 to size exclusion chromatography.
- FIG. 3 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Asn-NH 2 / Rh prepared in Example 1-4-17 to size exclusion chromatography.
- 2 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Me / FL prepared in Example 1-4-18 to size exclusion chromatography.
- 2 is a chromatogram obtained by subjecting a rat urine sample administered with HA-Pr / FL prepared in Example 1-4-19 to size exclusion chromatography.
- 2 is a chromatogram obtained by subjecting a rat urine sample administered with HA-FL prepared in Comparative Example 1-1-1 to size exclusion chromatography.
- 2 is a chromatogram obtained by subjecting a rat urine sample administered with HA-EDOBEA-Ac / FL prepared in Comparative Example 1-1-2 to size exclusion chromatography.
- FIG. 3 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Phe / FL prepared in Comparative Example 1-1-3 to size exclusion chromatography.
- 4 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Tyr / FL prepared in Comparative Example 1-1-4 to size exclusion chromatography.
- 6 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-MePhe / FL prepared in Comparative Example 1-1-5 to size exclusion chromatography.
- FIG. 6 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Pro-OMe / FL prepared in Comparative Example 1-1-6 to size exclusion chromatography.
- FIG. 7 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Gly-NH 2 / Rh prepared in Comparative Example 1-1-7 to size exclusion chromatography.
- 6 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Ser-NH 2 / Rh prepared in Comparative Example 1-1-8 to size exclusion chromatography.
- 10 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Leu-NH 2 / Rh prepared in Comparative Example 1-1-9 to size exclusion chromatography.
- 2 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Ile-NH 2 / Rh prepared in Comparative Example 1-1-10 to size exclusion chromatography.
- 2 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Thr-NH 2 / Rh prepared in Comparative Example 1-1-11 to size exclusion chromatography.
- 2 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Gln-NH 2 / Rh prepared in Comparative Example 1-1-12 to size exclusion chromatography. It is a table
- FIG. 6 is a chromatogram obtained by subjecting a urine sample of a rat administered with HA-Ala-PTH / Rh prepared in Example 5-5 to size exclusion chromatography.
- the hyaluronic acid derivative of the present invention is not particularly limited as long as it is a hyaluronic acid derivative containing one or more disaccharide units represented by the formula (I).
- the ratio of the disaccharide unit of the formula (I) to the disaccharide repeating unit present in the hyaluronic acid derivative of the present invention is, for example, 70% or more, preferably 75% or more, more preferably 90%. % Or more.
- the upper limit may be 100% or less.
- a hyaluronic acid derivative comprising one or more disaccharide units represented by the formula (I) and one or more disaccharide units represented by the formula (II) and / or (III): Provided.
- the hyaluronic acid derivative is substantially composed of disaccharide units of the formulas (I) and (II).
- the hyaluronic acid derivative is, for example, 80% or more, preferably 90% or more, more preferably 95% of the disaccharide repeating units composed of D-glucuronic acid and N-acetylglucosamine contained in the derivative.
- the above is the disaccharide unit of the formula (I) or (II). In one embodiment of the present invention, it is composed only of the disaccharide units represented by the above formulas (I) and (II).
- the hyaluronic acid derivative is substantially composed of disaccharide units of the formulas (I), (II) and (III).
- the hyaluronic acid derivative is, for example, 80% or more, preferably 90% or more, more preferably 95% of the disaccharide repeating units composed of D-glucuronic acid and N-acetylglucosamine contained in the derivative.
- the above is the disaccharide unit of the formula (I), (II) or (III). In one embodiment of the present invention, it is composed only of disaccharide units represented by the above formulas (I), (II) and (III).
- the ratio of the specific disaccharide unit to the repeating unit of the disaccharide present in the hyaluronic acid derivative of the present invention is preferably included in the fixed amount of the hyaluronic acid derivative of the present invention which is a polysaccharide having the disaccharide unit as a repeating unit.
- R 1 , R 2 , R 3 , and R 4 are all hydrogen atoms.
- R 8 is preferably a hydrogen atom or C 1-6 alkylcarbonyl, more preferably a hydrogen atom or acetyl, and even more preferably acetyl.
- R 1a , R 2a , R 3a and R 4a , and R 1b , R 2b , R 3b and R 4b are preferably all hydrogen atoms.
- R 8a and R 8b are preferably a hydrogen atom or C 1-6 alkylcarbonyl, more preferably a hydrogen atom or acetyl, and more preferably acetyl.
- R x is a hydrogen atom or C 1-6 alkyl such as methyl or ethyl, and is preferably a hydrogen atom.
- R y is a hydrogen atom or C 1-6 alkyl such as methyl or ethyl, and is preferably a hydrogen atom, methyl or ethyl.
- examples thereof include 1,1-cyclohexylene, 1,2-cyclohexylene, 1,3-cyclohexylene, 1,4-cyclohexylene.
- examples include silene, 1,1-cyclopentylene, 1,2-cyclopentylene, 1,3-cyclopentylene, 1,1-cyclobutylene, 1,2-cyclobutylene, 1,3-cyclobutylene, and the like.
- Preferred examples include 1,1-cyclohexylene, 1,2-cyclohexylene, 1,4-cyclohexylene and 1,1-cyclobutylene.
- C 3-8 cycloalkylene is preferably 1,1-cyclohexylene, 1,2-cyclohexylene, or 1,1-cyclobutylene. From the viewpoint of delivering the drug into the cytoplasm, C 3-8 cycloalkylene is preferably 1,4-cyclohexylene.
- B when B is phenylene, examples thereof include 1,2-phenylene, 1,3-phenylene, and 1,4-phenylene. Preferable examples include 1,4-phenylene and 1,3-phenylene.
- B when B is phenylene, 1,4-phenylene is a particularly preferable example.
- Phenylene may be substituted with one or more groups selected from hydroxy and halogen atoms.
- substituted phenylene include 2-hydroxy-1,4-phenylene, 3-hydroxy-1,4-phenylene, 2,3-dihydroxy-1,4-phenylene, and 3,5-dihydroxy-1 , 4-phenylene, 5-hydroxy-1,3-phenylene, 3-hydroxy-1,2-phenylene, 4-hydroxy-1,2-phenylene; 2-chloro-1,4-phenylene, 2-iodo-1 , 4-phenylene, 3-bromo-1,4-phenylene, 2,6-difluoro-1,4-phenylene, 3,5-dichloro-1,4-phenylene, 5-chloro-1,3-phenylene, 3, -Bromo-1,2-phenylene, 4-chloro-1,2-phenylene, 6-fluoro-2-hydroxy-1,4-phenylene, 5-chloro-3-iodo-1,4-pheny 2-bromo-3-hydroxy-1,4
- R c is C 1-6 alkyl optionally substituted with carbamoyl (—CONH 2 ), preferably methyl, isopropyl or carbamoylmethyl, more preferably isopropyl or carbamoylmethyl.
- R ya and R yb are each independently selected from a hydrogen atom, C 1-6 alkyl, formyl and C 1-6 alkylcarbonyl, preferably both are hydrogen atoms; one is a hydrogen atom and the other is C 1 -6 alkyl; one is a hydrogen atom and the other is a C 1-6 alkylcarbonyl, more preferably both are hydrogen atoms; one is a hydrogen atom and the other is methyl; one is a hydrogen atom and the other is ethyl, Preferably, both are hydrogen atoms.
- Examples of X in the formula (I) include the following groups.
- the exemplified groups are preferable groups from the viewpoint of having both biodegradability and blood retention properties.
- R y , R ya , R yb are as defined herein].
- a defined by formula (I) is -CR a R b- (R a is as defined above.
- R b is selected from a hydrogen atom and C 1-6 alkyl (C 1-6 alkyl may be substituted with a group selected from hydroxy and carboxy), and B is Preferably, it is a direct bond; or A is C 3-8 cycloalkylene and B is a direct bond.
- X defined by formula (I) is —NHCH 3 , —NH (CH 2 ) 2 CH 3 or NR x is —CHR c —CONR ya R yb ;
- A is —CR a R b — (R a is as defined above, R b is a hydrogen atom and a C 1-6 alkyl (C 1 -6 alkyl is optionally substituted with a group selected from hydroxy, carboxy and carbamoyl) and B is a direct bond; or A is C 3-8 cycloalkylene And B is preferably a direct bond.
- R a a hydrogen atom and methyl are more preferable, and a hydrogen atom is more preferable.
- R b is preferably a hydrogen atom and C 1-4 alkyl (C 1-4 alkyl may be substituted with a group selected from hydroxy and carboxy), and may be methyl optionally substituted with hydroxy Further preferred. Examples of preferred R b include a hydrogen atom and C 1-4 alkyl (C 1-4 alkyl may be substituted with a group selected from hydroxy, carboxy and carbamoyl).
- B is preferably cyclohexane-1,2-diyl and cyclobutane-1,1-diyl, more preferably cyclohexane-1,2-diyl.
- a steric configuration of —NR x — and —COOR y on the cyclohexane ring both cis configuration and trans configuration are preferable.
- the hyaluronic acid derivative of the present invention may contain only the cis configuration or the trans configuration as the configuration of —NR x — and —COOR y on the cyclohexane ring, or the cis configuration and the trans configuration exist in an arbitrary ratio. May be. Or the stereochemistry of each group may be single and may exist in arbitrary ratios.
- preferable X is, for example, a group represented by the following formula:
- X include the following groups:
- X include the following groups:
- particularly preferred X include the following groups:
- the proportion of the disaccharide unit of the formula (I) in the disaccharide repeating unit present in the hyaluronic acid derivative of the present invention is preferably 70% or more, More preferably, it is 75% or more, More preferably, it is 90% or more.
- a as defined in formula (I) is C 3-8 cycloalkylene and B is a direct bond;
- A is —CH 2- and B is selected from phenylene and C 3-8 cycloalkylene; or
- A is 2-cyclohexylethane-1,1-diyl, 2- (2-naphthyl) ethane-1,1-diyl and It is preferably selected from 3-phenylpropane-1,1-diyl and B is a direct bond.
- a as defined in formula (I) is —CH 2 — and B is selected from cyclohexane-1,1-diyl and benzene-1,4-diyl; or A is 2- Selected from cyclohexylethane-1,1-diyl, 2- (2-naphthyl) ethane-1,1-diyl and 3-phenylpropane-1,1-diyl.
- a as defined by formula (I) is C 3-8 cycloalkylene and B is a direct bond; CH 2 — or —CH 2 —CH 2 —, wherein B is phenylene (wherein phenylene may be substituted with one or more groups selected from hydroxy and halogen atoms), C 3-8 cycloalkylene And phenylmethane-1,1-diyl; or A is 2-cyclohexylethane-1,1-diyl, 2- (2-naphthyl) ethane-1,1-diyl, 3-phenylpropane-1 , 1-diyl, cyclohexylmethane-1,1-diyl and 4-hydroxyphenylmethane-1,1-diyl, preferably B is a direct bond.
- a defined by formula (I) is —CH 2 —, and B is cyclohexane-1,1-diyl, benzene-1,4-diyl, benzene-1,3-diyl, 2-chlorobenzene.
- A is —CH 2 CH 2 — and B is benzene-1,4-diyl; or
- A is 2 Selected from cyclohexylethane-1,1-diyl, 2- (2-naphthyl) ethane-1,1-diyl and 3-phenylpropane-1,1-diyl;
- X include the following groups:
- each asymmetric point includes only the R configuration, only the S configuration, and the R configuration and the S configuration in an arbitrary ratio. May be.
- Q + is not particularly limited as long as it is a counter cation that forms a salt with carboxy in water, and when it is divalent or more, it forms a plurality of salts with carboxy depending on the valence.
- counter cations include metal ions such as lithium ion, sodium ion, rubidium ion, cesium ion, magnesium ion, calcium ion; formula: N + R j R k R l R m (where R j , R k , R 1 and R m are each independently selected from a hydrogen atom and C 1-6 alkyl), preferably sodium ion, potassium ion, tetraalkylammonium ion ( For example, tetra n-butylammonium ion).
- R j , R k , R 1 and R m are preferably the same group selected from C 1-6 alkyl, and are preferably n-butyl.
- examples of Xb include the following groups: -HN- (CH 2 ) j -SH; —HN—CH 2 —CH 2 — (Y 1 —CH 2 —CH 2 ) c —SH; —HN— (CH 2 ) p —O—CO—C (R 7 ) ⁇ CH 2 ; —HN— (CH 2 ) 1 —NHCO—C (R 7 ) ⁇ CH 2 ; —HN—CH 2 —CH 2 — (Y 1 —CH 2 —CH 2 ) c —NHCO—C (R 7 ) ⁇ CH 2 ; or —HN—CH 2 —CH 2 — (Y 1 —CH 2 —CH 2 ) c— O—CO—C (R 7 ) ⁇ CH 2 [Wherein j, Y l , c, p, R 7 , and l are as previously defined herein].
- C 1-6 alkyl means linear or branched alkyl having 1 to 6 carbon atoms, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, “C 1-4 alkyl” such as s-butyl, i-butyl, t-butyl and the like, and n-pentyl, 3-methylbutyl, 2-methylbutyl, 1-methylbutyl, 1-ethylpropyl, n-hexyl 4-methylpentyl, 3-methylpentyl, 2-methylpentyl, 1-methylpentyl, 3-ethylbutyl, 2-ethylbutyl and the like.
- C 1-6 alkylcarbonyl means an alkylcarbonyl having a linear or branched alkyl having 1 to 6 carbon atoms, such as acetyl, propionyl, butyryl, isobutyryl, pivaloyl, valeryl. , Isovaleryl, hexanoyl and the like.
- aryl means an aromatic carbocyclic group, for example, an aromatic carbocyclic group having 6 to 14 carbon atoms.
- aryl include phenyl, naphthyl (1-naphthyl, 2-naphthyl and 3 -Naphthyl).
- C 3-8 cycloalkyl means a cyclic alkyl having 3 to 8 carbon atoms and includes, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl.
- C 3-8 cycloalkylene means a divalent cyclic alkyl having 3 to 8 carbon atoms, such as cyclopropylene, cyclobutylene, cyclopentylene, cyclohexylene, cycloheptylene, and cyclopropylene.
- Octylene is included. More specific examples include 1,1-cyclohexylene, 1,2-cyclohexylene, 1,3-cyclohexylene, 1,4-cyclohexylene, 1,1-cyclopentylene, 1,2-cyclopentylene. And the like include 1,3-cyclopentylene, 1,1-cyclobutylene, 1,2-cyclobutylene, 1,3-cyclobutylene, and the like.
- Preferred examples include 1,1-cyclohexylene, 1,2-cyclohexylene, 1,4-cyclohexylene and 1,1-cyclobutylene.
- phenylene means a divalent group in which two hydrogen atoms of benzene are substituted. Benzene-1,2-diyl, benzene-1,3-diyl, benzene-1,4- Jile is included.
- C 1-10 alkylene means a linear or branched alkylene group having 1 to 10 carbon atoms, such as methylene, ethane-1,2-diyl, propane-1.3- Diyl, butane-1,4-diyl, pentane-1,5-diyl, hexane-1,6-diyl, heptane-1,7-diyl, octane-1,8-diyl, nonane-1,9-diyl, Decane-1,10-diyl, propane-1,2-diyl, 2-methylpropane-1,3-diyl, butane-2,4-diyl, 3-methylbutane-1,4-diyl, 2-methylpentane- 1,5-diyl, 4-ethylhexane-1,6-diyl, 4-methylheptane-2,7-diyl, 5-ethyloctan
- C 1-10 alkylene having 1 to 3 C 3-8 cycloalkylene or phenylene inserted refers to 1 to 3, preferably 1 phenylene or C 3-8 cycloalkylene inserted. Or a C 1-10 alkylene group.
- G 4 include, for example, phenylene or a group represented by the following formula:
- the “halogen atom” means a fluorine atom, a chlorine atom, a bromine atom and an iodine atom.
- Xc is a halogen atom, a chlorine atom, a bromine atom and an iodine atom are preferable, and a bromine atom and an iodine atom are preferable from the viewpoint of reactivity with mercapto in a drug.
- the drug carrier is biodegradable
- the drug carrier detected in urine is reduced in molecular weight between 2 weeks after intravenous administration to rats and / or humans.
- intracellular means the inside of a cell membrane including endosomes and cell nuclei.
- intracytoplasm means the inside of the cell membrane excluding endosomes and cell nuclei from “intracellular”.
- endosome means a vesicle formed of a biological membrane formed in the process of taking up a substance by a cell and a vesicle formed by fusing them with a lysosome.
- “Making low molecular weight” can be determined by measuring the size of a drug carrier excreted in urine by subjecting it to size exclusion column chromatography (see Example 3-3 in this specification). reference). At this time, the peak top of the urine-derived drug carrier observed at any one time point during the period up to 2 weeks after administration or a period of 24 hours is derived from the urine sample collected at a previous time point or period. If the drug carrier has shifted to the low molecular weight side compared to the peak top of the drug carrier (that is, the retention time in the column chromatogram becomes longer), it is determined that the drug carrier is biodegradable.
- the hyaluronic acid derivative of the present invention containing one or more disaccharide units represented by the formula (I) is, for example, a hyaluronic acid or a derivative thereof substantially consisting of a disaccharide unit represented by the formula (II), more preferably It is synthesized using hyaluronic acid (including salt and the like) composed only of the disaccharide unit represented by the formula (II) as a raw material.
- the raw material hyaluronic acid has a weight average molecular weight of 20 to 120 kilodaltons, preferably 20 to 30 kilodaltons, and 50 to 120 kilodaltons.
- Preferable weight average molecular weights include, for example, 25 kilodalton and 99 kilodalton. From the viewpoint of improving blood retention, the lower limit of the weight average molecular weight may be 5 kilodaltons or more, preferably 50 kilodaltons or more, and more preferably 99 kilodaltons or more. The upper limit of the weight average molecular weight is preferably 250 kilodaltons or less.
- the preferred weight average molecular weight is 50 to 120 kilodaltons, for example, 99 kilodalton, and from the viewpoint of cytoplasmic transferability, the preferred weight average molecular weight is 20 to 120 kilodaltons, for example, 25 and 99 kilodaltons.
- the weight average molecular weight of hyaluronic acid (including salt) composed of only a disaccharide unit represented by the formula (II) used as a raw material of 20 to 120 kilodaltons is the hyaluronic acid derivative of the present invention.
- all of R 1a , R 2a , R 3a , and R 4a are hydrogen atoms
- R 8a is acetyl
- X a is —O ⁇ Na +. It means the weight average molecular weight when converted as.
- the weight average molecular weight is 400 kilodaltons.
- the weight average molecular weight calculated in accordance with the above is calculated to be 20 to 120 kilodaltons, it is included in one preferred embodiment of the present invention.
- the method for measuring the HA weight average molecular weight is, for example, a light scattering method, an osmotic pressure method, a viscosity method described in Seiichi Nakahama et al., “Essential Polymer Science” (published by Kodansha, ISBN4-06-153310-X).
- various known methods can be used.
- the weight average molecular weight measured by the light scattering method shown in the present specification is usually used in the technical field to which the present invention belongs, such as using a multi-angle light scattering detector (SEC-MALLS) connected to a size exclusion chromatography apparatus. It can be measured by the method used.
- SEC-MALLS multi-angle light scattering detector
- the hyaluronic acid derivative of the present invention is substantially composed of a disaccharide unit represented by the formula (II) having a weight average molecular weight of 500 kilodaltons, preferably 250 kilodaltons or less.
- the hyaluronic acid (including salts and the like) can be produced as a raw material.
- the hyaluronic acid derivative of the present invention is a hyaluronic acid derivative substantially composed of disaccharide units of formulas (I) and (II).
- the hyaluronic acid derivative of the present invention containing a disaccharide unit represented by the formula (I) can be produced by converting carboxy in the glucuronic acid moiety into an amide.
- a raw material hyaluronic acid (including a salt) preferably a hyaluronic acid composed only of a disaccharide unit represented by the formula (II) is converted into a tetraalkylammonium salt (for example, a tetrabutylammonium (TBA) salt.
- TSA tetrabutylammonium
- the hyaluronate and the formula: HNR x -AB-COOR z (wherein R z is an ester-forming group for protecting carboxy). And R x , A and B are as defined herein) or a compound of formula: HNR x -A-CONR ya R yb is reacted with a protecting group A method of performing deprotection is mentioned.
- the ester forming group is not particularly limited, and is not particularly limited as long as it is a group usually used for protecting carboxy. Examples of the ester forming group, C 1-6 alkyl, benzyl, C 1-6 alkoxy C 1-6 alkyl, and benzyloxy C 1-6 alkyl.
- the groups in the formula (I): —NR x —AB—COOR z and —NR x —A—CONR ya R yb may be the same or different in each of a plurality of disaccharide units.
- the above reaction may be performed using different types of compounds represented by formulas: HNR x -AB-COOR z and / or HNR x -A-CONR ya R yb .
- the condensing agent that can be used in the above reaction is not particularly limited.
- DMT-MM 4- (4,6-dimethoxy-1,3,5-triazine) -4-methylmorpholium
- CDI N , N′-carbonyldiimidazole
- DCC N, N′-dicyclohexylcarbodiimide
- EEDQ N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline
- PyBOP 4- (4,6-dimethoxy-1,3,5-triazine) -4-methylmorpholium
- CDI N , N′-carbonyldiimidazole
- DCC N, N′-dicyclohexylcarbodiimide
- DMT-MM is preferable in that the reaction proceeds with high efficiency even in a mixed solvent of water and an organic solvent.
- DMT-MM as a condensing agent, it is possible to highly selectively form an amide bond with amino and carboxy while suppressing the formation of an ester bond in a system in which many hydroxy groups coexist.
- this condensing agent for example, alcohol as a solvent reacts with carboxy of the hyaluronic acid moiety, or carboxy and hydroxy simultaneously present in the hyaluronic acid moiety are bonded within the molecule or between the molecules, which is undesirable. It is possible to prevent the formation of a crosslink.
- Solvents used in the above reaction include water, dimethyl sulfoxide (DMSO), dimethylformamide (DMF), dimethylacetamide (DMAc), 1,3-dimethyl-2-imidazolidinone (DMI), sulfolane (SF), N- Mention may be made of methylpyrrolidone (NMP), dioxane (for example, 1,4-dioxane), methanol, ethanol, propanol, butanol, acetonitrile, tetrahydrofuran, dichloromethane, chloroform, hexane, diethyl ether, ethyl acetate, and mixed solvents thereof. .
- DMSO dimethyl sulfoxide
- DMF dimethylformamide
- DMAc dimethylacetamide
- DI 1,3-dimethyl-2-imidazolidinone
- SF sulfolane
- NMP methylpyrrolidone
- dioxane
- DMSO dimethyl methacrylate
- water / DMSO mixed solvent it is preferable to use DMSO alone or a water / DMSO mixed solvent.
- amino-carboxylic acid it may be used in the reaction as a methanol solution or dioxane solution.
- HNR x -AB-COOR z examples include, for example, alanine ester, serine ester, glutamic acid diester, glycine ester, valine ester, leucine ester, isoleucine ester, threonine ester, aspartic acid diester, cis- 2-amino-1-cyclohexylcarboxylic acid ester, trans-2-amino-1-cyclohexylcarboxylic acid ester, 2-aminoisobutanoic acid ester, 1-amino-1-cyclobutanoic acid ester, 1-aminomethyl-1-cyclohexane acid Esters, cis-4-aminocyclohexane ester, L-2-naphthylalanine ester, 2-aminophenylbutanoate ester, cyclohexyl-L-alanine ester, 4-aminomethylbenzoate ester, etc.
- ester is, for example, C 1-6 alkyl ester, aryl ester, C 1-6 alkoxy C 1-6 alkyl ester, aryl C 1-6 alkyl ester, etc., preferably methyl ester, ethyl ester Benzyl ester and the like.
- HNR x -A-CONR ya R yb examples include alanine amide, asparagine amide, aspartic acid amide, glutamine amide, glutamic acid amide, glycinamide, isoleucine amide, leucine amide, phenylalanine amide, serine amide, Examples include threonine amide, tyrosine amide, and valine amide.
- a method for producing a derivative in which X is —NHCH 3 and —NH (CH 2 ) 2 CH 3 includes, for example, A method of reacting the tetrabutylammonium salt of hyaluronic acid with the compound represented by NH 2 CH 3 and NH 2 (CH 2 ) 2 CH 3 in a solvent in the presence of a suitable condensing agent. Is mentioned.
- the condensing agent and the solvent already described in the present specification can be used.
- the modification rate by each amine can be adjusted by controlling, for example, the equivalent number of the condensing agent and / or amine for the HA unit, the reaction temperature, the reaction time, and the like.
- the hyaluronic acid derivative of the present invention containing the disaccharide unit represented by the formula (III), for example, the above-mentioned tetrabutylammonium salt of hyaluronic acid and a suitable condensing agent in the presence of the solvent.
- R e , Y b , R 7 , G 4 , and X c are as previously defined herein
- a method of reacting with a compound to be removed and deprotecting when a protecting group is present In the above reaction, the condensing agent and the solvent already described in the present specification can be used.
- protecting group used in the above reaction include T.I. W. Greene, P.M. G. M.M. Wuts, Protective Groups in Organic Synthesis, Third Edition, John Wiley & Sons, Inc. , New York, 1999.
- hydroxy protecting groups include C 1-6 alkylcarbonyl, arylcarbonyl, heteroarylcarbonyl, C 1-6 alkoxycarbonyl, C 1-6 alkoxy C 1-6 alkyl, C 1-6 alkylaminocarbonyl, di (C 1-6 alkyl) aminocarbonyl, aryl C 1-6 alkyl, heteroaryl C 1-6 alkyl, aryl C 1-6 alkylaminocarbonyl, C 1-6 alkyl, C 1-6 alkylsulfonyl, ((amino C 1-6 alkyl) carbonyloxy) C 1-6 alkyl, unsaturated heterocyclic carbonyloxy C 1-6 alkyl, aryldi (C 1-6 alkyl) silyl, tri (C 1-6 alkyl) silyl and the like.
- Preferred hydroxy protecting groups include acetyl and the like.
- Examples of —NH— or amino protecting groups include C 1-6 alkylcarbonyl, aryl C 1-6 alkylcarbonyl, arylcarbonyl, heteroarylcarbonyl, C 1-6 alkoxycarbonyl, C 1-6 alkylaminocarbonyl, Examples include di (C 1-6 alkyl) aminocarbonyl, aryl C 1-6 alkyl, heteroaryl C 1-6 alkyl, (arylC 1-6 alkyl) aminocarbonyl, and the like.
- Preferred amino protecting groups include acetyl, t-butoxycarbonyl, 9-fluorenylmethoxycarbonyl and the like.
- amino may be protected to form a saturated or unsaturated heterocyclic group such as phthalic imide, succinimide, glutaric imide, and 1-pyrrolyl.
- Examples of mercapto protecting groups include C 1-6 alkylthio such as ethylthio and t-butylthio, substituted phenylthio such as 2-nitrophenylthio and 2-carboxyphenylthio, and heteroarylthio such as 2-pyridylthio. .
- a preferred example is 2-pyridylthio.
- Examples of groups represented by —NR e —Y b —R d in the above formula (III) include those represented by the formula: -NH-CH 2- (CHR 5 ) 1-2 -CH 2 -NH 2 ; —NH—CH 2 —CH 2 — (Y 1 —CH 2 —CH 2 ) c —NH 2 ; —NH—CH 2 — (CHR 6 ) p-2 —CH 2 —OH; —NH—CH 2 —CH 2 — (Y 1 —CH 2 —CH 2 ) c —OH; —NH— (CH 2 ) j —SH; —NH—CH 2 —CH 2 — (Y 1 —CH 2 —CH 2 ) c —SH; —NH— (CH 2 ) p —O—CO—C (R 7 ) ⁇ CH 2 ; —NH— (CH 2 ) 1 —NHCO—C (R 7 ) ⁇ CH 2 ; —NH—CH
- the number of CHR 5 and CHR 6 in which R 5 and R 6 are hydroxy contained in the molecule of the hyaluronic acid derivative is 0 to 8, preferably 0 to 3, more preferably 0 and 1.
- the solubility of the hyaluronic acid derivative of the present invention in water can be adjusted.
- l is preferably 2 to 6, and specific examples thereof include 2 and 6.
- one of R 5 is hydroxy a specific example of 1 is 3.
- Y 1 is an oxygen atom
- 2 is given as a specific example of c.
- specific examples of c include 1 to 3.
- Specific examples of l and p include 3.
- — (CH 2 ) a — (Y 1 — (CH 2 ) b ) c —G— include, for example, — (CH 2 ) 2 — (O—CH 2 —CH 2 ) c —O—, — (CH 2 ) 2 — (O—CH 2 —CH 2 ) c —NH—, — (CH 2 ) 3 — (O—CH 2 —CH 2 —CH 2 ) c —O—, — (CH 2 ) 3 — (O—CH 2 —CH 2 —CH 2 ) c —NH—, — (CH 2 ) 2 —NR n — (CH 2 ) 2 —O—, — (CH 2 ) 2 —NR n — (CH 2 ) 2 —NH—, — (CH 2 ) 3 —NR n — (CH 2 ) 4 —O—, — (CH 2 ) 3 —NR n — (CH 2 )
- R d bonded to these — (CH 2 ) a — (Y 1 — (CH 2 ) b ) c —G— include, for example, a hydrogen atom, —CO—CH ⁇ CH 2 , —CO—C (CH 3 ) ⁇ CH 2 , —CO—CH 2 —Cl, —CO—CH 2 —Br, —CO—CH 2 —I, —CO—CH 2 —SH, —CO—CH 2 —CH 2 —SH Etc.
- carboxy (—COOH) of the glucuronic acid moiety of hyaluronic acid is represented by the formula: H 2 N—CH 2 — (CHR 5) is reacted with diamine represented by l-2 -CH 2 -NH 2 wherein: -CONH-CH 2 - (CHR 5) was converted to an amide represented by l-2 -CH 2 -NH 2, Further, there is a method in which the terminal amino is modified and converted to an amide represented by the group: —CONH—CH 2 — (CHR 5 ) 1 -2 —CH 2 —NHR d .
- diamine examples include, for example, H 2 N— (CH 2 ) 2 —NH 2 , H 2 N— (CH 2 ) 3 —NH 2 , H 2 N— (CH 2 ) 4 —NH 2 , H 2 N— (CH 2 ) 5 —NH 2 , H 2 N— (CH 2 ) 6 —NH 2 , H 2 N— (CH 2 ) 7 —NH 2 , H 2 N— (CH 2 ) 8 —NH 2 H 2 N— (CH 2 ) 9 —NH 2 , H 2 N— (CH 2 ) 10 —NH 2 , H 2 N—CH 2 —CHOH—CH 2 —NH 2 , H 2 N—CH 2 —CHOH — (CH 2 ) 2 —NH 2 , H 2 N—CH 2 — (CHOH) 2 —CH 2 —NH 2 , H 2 N—CH 2 —CHOH— (CH 2 ) 3 —NH 2 , H 2 N— (CH 2) 2 -CHOH-CH-
- carboxy (—COOH) of the glucuronic acid moiety of hyaluronic acid is represented by the formula: H 2 N—CH 2 — (CHR 5) is reacted with hydroxylamine represented by p-2 -CH 2 -OH formula: -CONH-CH 2 - (CHR 5) was converted to an amide represented by p-2 -CH 2 -OH, further Examples thereof include a method in which a terminal amino is modified to convert it to an amide represented by a group: —CONH—CH 2 — (CHR 5 ) p-2 —CH 2 —OR d .
- hydroxyamine examples include, for example, H 2 N— (CH 2 ) 2 —OH, H 2 N— (CH 2 ) 3 —OH, H 2 N— (CH 2 ) 4 —OH, H 2 N — (CH 2 ) 5 —OH, H 2 N— (CH 2 ) 6 —OH, H 2 N— (CH 2 ) 7 —OH, H 2 N— (CH 2 ) 8 —OH, H 2 N— ( CH 2 ) 9 —OH, H 2 N— (CH 2 ) 10 —OH, H 2 N—CH 2 —CHOH—CH 2 —OH, H 2 N—CH 2 —CHOH— (CH 2 ) 2 —OH, H 2 N—CH 2 — (CHOH) 2 —CH 2 —OH, H 2 N—CH 2 —CHOH— (CH 2 ) 3 —OH, H 2 N— (CH 2 ) 2 —CHOH— (CH 2 ) 2- OH, H 2 N—CH 2 — (CHOH)
- the group: —NR e —Y b —R d in formula (III) may be the same or different in each of a plurality of disaccharide units.
- the above reaction may be performed using different types of compounds represented by the formula: HNR e —Y b —R d .
- X in formula (I) is —NR x —AB—COOR y
- X b is not only present at the position indicated in the disaccharide unit represented by formula (III), but also Part or all may be substituted with —OR y, and X may be —NR x —AB—CO—X b .
- the non-derivatized hyaluronic acid having a main chain structure corresponding to the hyaluronic acid derivative of the present invention has a weight average molecular weight of 20 to 120 kilodaltons.
- the non-derivatized hyaluronic acid (including a salt) is represented by the formula (II) wherein R 1a , R 2a , R 3a and R 4a are hydrogen atoms, R 8a is acetyl, and X a is ⁇ It is hyaluronic acid consisting only of disaccharide units that are O ⁇ Na + .
- the non-derivatized hyaluronic acid has a main chain structure corresponding to the hyaluronic acid derivative of the present invention
- the number of disaccharide units present in the non-derivatized hyaluronic acid is present in the hyaluronic acid derivative of the present invention. This is consistent with the number of disaccharide units
- the hyaluronic acid derivative in this aspect can be produced, for example, by using hyaluronic acid having a weight average molecular weight of 20 to 120 kilodalton or a sodium salt thereof as a raw material.
- the ratio of the disaccharide unit represented by the formula (II) to the disaccharide repeating unit present is preferably 50% or less, more preferably 30% or less, and 20% or less. Is more preferable. The lower limit of the ratio may be 0% or more.
- 50% or more of carboxy (—COOH) of the hyaluronic acid derivative is a group: —CONHCH 3 , —CONH (CH 2 ) 2 CH 3 , —CONR x —AB—COOR y , —CONR x —A -CONR ya R yb or -CONR e -Y b -R d .
- a hyaluronic acid derivative containing a carbon-carbon double bond reactive in the disaccharide unit represented by the formula (III) is a crosslinking agent having two or more mercaptos (for example, dithiothreitol: DTT, butanedithiol, polyethylene glycol) Dithiol) can be subjected to a crosslinking reaction.
- a hyaluronic acid derivative containing a mercapto in the disaccharide unit represented by the formula (III) is disulfide formed with a cross-linking agent having two or more mercaptos (for example, dithiothreitol: DTT, butanedithiol, polyethylene glycol dithiol).
- a crosslinking reaction using a crosslinking agent containing two or more reactive carbon-carbon double bonds for example, divinylsulfone.
- a hyaluronic acid derivative modified with amino and a crosslinking agent having succinimidyl ester or other imide ester at both ends of C 2-20 alkylene for example, bis [sulfosuccinimidyl] Crosslinking by condensation reaction with suberate (BS 3 ), ethylene glycol-bis [sulfosuccinimidyl] succinate (Sulfo-EGS), dimethyl adipimidate hydrochloride (DMA), etc .
- Hyaluronic acid derivative modified with amino And a crosslinking agent having formyl at both ends of C 2-20 alkylene for example, glutaraldehyde
- oxidation conditions of a hyaluronic acid derivative modified with mercapto for example, in the presence of sodium tetrathionate (STT))
- STT sodium tetrathionate
- the method for producing a hyaluronic acid derivative of the present invention includes a step of condensing a diamine with carboxy of a glucuronic acid moiety, and only a part of the diamine, for example, 10% remains unutilized by binding to a drug.
- the hyaluronic acid derivative contains amino, it is treated with, for example, dicarboxylic anhydrides such as succinic anhydride, maleic anhydride, glutaric anhydride and adipic anhydride, or dicarboxylic acids such as maleic acid, glutaric acid and adipic acid.
- the terminal functional group By reacting in the presence of a condensing agent, the terminal functional group can be returned to carboxy, and the cationic property of the remaining amino can be lost, or the total charge can be anionic.
- the terminal functional group is converted to an amide by treating with carboxylic acid anhydrides such as acetic anhydride and benzoic anhydride, or by reacting carboxylic acids such as acetic acid and benzoic acid in the presence of a condensing agent.
- carboxylic acid anhydrides such as acetic anhydride and benzoic anhydride
- carboxylic acids such as acetic acid and benzoic acid
- the chemical cross-linking structure of the gel of the hyaluronic acid derivative of the present invention may include a structure that decomposes in vivo.
- a group having an ester bond and methacryloyl may be used as a group for the crosslinking reaction.
- the crosslinking agent a compound having an ester bond, such as Sulfo-EGS or EDMA, or a compound having a peptide spacer that is degraded by an enzyme in a living body may be used.
- the gel crosslinked by the disulfide bond formed by the oxidation of mercapto is decomposed in a living body by a disulfide exchange reaction or a reduction reaction.
- the hyaluronic acid derivative of the present invention can be used as a carrier for a pharmaceutical composition.
- a hyaluronic acid derivative containing a disaccharide unit represented by formula (I) and formula (III) or formula (I), formula (II) and formula (III) is used as a crosslinking agent.
- low molecular weight compounds include, for example, anticancer agents (eg, alkylating agents, antimetabolites, alkaloids, etc.), immunosuppressive agents, anti-inflammatory agents (steroids, non-steroidal anti-inflammatory agents, etc.), antirheumatic agents. And antibacterial agents ( ⁇ -lactam antibiotics, aminoglycoside antibiotics, macrolide antibiotics, tetracycline antibiotics, new quinolone antibiotics, sulfa drugs, etc.).
- anticancer agents eg, alkylating agents, antimetabolites, alkaloids, etc.
- immunosuppressive agents e.g., anti-inflammatory agents, steroids, non-steroidal anti-inflammatory agents, etc.
- antirheumatic agents e.g., antirheumatic agents.
- antibacterial agents ⁇ -lactam antibiotics, aminoglycoside antibiotics, macrolide antibiotics, tetracycline antibiotics, new quinolone antibiotics, sulfa drugs
- proteins and peptides include, for example, an anemia treatment agent, an organ protective agent erythropoietin (EPO), a neutropenia treatment granulocyte colony stimulating factor (G-CSF), interferon- ⁇ , ⁇ , ⁇ , (INF- ⁇ , ⁇ , ⁇ ), thrombopoietin (TPO), serial neurotrophic factor (CNTF), tumour necrosis factor (TNF), tumour necrosis factor binding protein (TNFbp), interleukin-10 ( IL-10), FMS-like tyrosine kinase (Flt-3), growth hormone (GH), insulin, insulin-like growth factor-1 (IGF-1), platelet-derived growth factor (PDGF), interleukin-1 receptor antagonist (IL-1ra), blur -Derived neurotrophic factor (BDNF), keratinocyte growth factor (KGF), stem cell factor (SCF), megacaryosite growth differentiation factor (MGDF), osteoprotegerin (OPG), leptin
- nucleic acid examples include, for example, DNA, RNA, antisense, decoy, ribozyme, small interfering RNA, microRNA, RNA aptamer, and those chemically modified.
- proteins, peptides and nucleic acids are preferable, and by encapsulating these drugs, the composition of the present invention containing proteins, peptides and nucleic acids can be obtained.
- a hyaluronic acid derivative-drug conjugate in which one or more of the above drugs are bound to a hyaluronic acid derivative containing a disaccharide unit represented by the formula (I).
- the hyaluronic acid derivative comprising a disaccharide unit represented by formula (I) and formula (III) or formula (I), formula (II) and formula (III) is used as the hyaluronic acid derivative.
- a drug with a hydroxy, amino, mercapto or reactive carbon-carbon double bond (methacryloyl, acryloyl, etc.) contained in the group —NR e —Y b —R d of formula (III)
- the hyaluronic acid derivative-drug conjugate can be prepared.
- Part or all of the group —NR e —Y b —R d may be present in formula (I) as —NR x —AB—CO—NR e —Y b —R d. .
- R s , R t and R u are independently selected from a hydrogen atom and C 1-6 alkyl, i is an integer selected from 1 to 10 and q is selected from 2 to 10 An integer, k is an integer selected from 1 to 100, and r and m are independently an integer selected from 1 to 10) A spacer represented by may be inserted.
- Glucuronic acid 4-position and hydroxy of acetylglucosamine 1-position existing at the end of the main chain of the hyaluronic acid derivative of the present invention may be converted to other groups, for example, C 1-6 alkoxy, formyloxy and C It may be 1-6 alkylcarbonyloxy.
- the drug is encapsulated in the gel of the hyaluronic acid derivative of the present invention, or the hyaluronic acid derivative-drug conjugate of the present invention is one or more pharmaceutically acceptable diluents, wetting agents, emulsifiers.
- a pharmaceutical composition containing a dispersing agent, adjuvant, preservative, buffer, binder, stabilizer and the like can be administered in any suitable form depending on the intended route of administration.
- the route of administration may be a parenteral route or an oral route.
- a highly safe sustained-release preparation capable of long-term sustained release of drugs such as proteins, peptides, nucleic acids, and low molecular weight compounds, which cannot be obtained by conventional sustained-release preparations, and having appropriate biodegradability
- drugs such as proteins, peptides, nucleic acids, and low molecular weight compounds
- the hyaluronic acid derivative of the present invention has a function of promoting the release of the drug from the endosome into the cytoplasm in the pathway where the drug is taken into the cell via endocytosis, so that the drug is efficiently taken into the cytoplasm. It is also possible to provide pharmaceutical formulations and pharmaceutical compositions.
- the HA unit means a repeating unit (1 unit) of N-acetylglucosamine-glucuronic acid in hyaluronic acid.
- NMR measurement was performed using a nuclear magnetic resonance apparatus (JNM-ECA500, manufactured by JEOL Ltd.). NMR measurement conditions are shown below.
- Example 1 Synthesis of hyaluronic acid derivative (Example 1-1) Tetrabutylammonium (TBA) chloride cation exchange resin DOWEX (registered trademark) 50WX-8-400 (manufactured by Sigma-Aldrich) in ultrapure water The suspension was suspended, and the resin was washed about 3 times with ultrapure water by decantation. A 40 wt% tetrabutylammonium hydroxide aqueous solution (TBA-OH) (manufactured by Sigma-Aldrich) was added in an amount of about 1.5 times the molar equivalent of the cation exchange capacity of the resin and stirred for 30 minutes. After the excess TBA-OH solution was removed by decantation, washing with excess ultrapure water was repeated, and finally filtration was performed using a 0.45 ⁇ m filter to obtain a cation exchange resin that had been TBA-chlorinated.
- TBA-OH Tetrabutylammonium hydroxide aqueous solution
- Example 1-2 Preparation of HA TBA salt (HA-TBA) Hyaluronic acid sodium salt (HA-Na, manufactured by Shiseido Co., Ltd.) having a molecular weight of 25 kDa and 99 kDa was dissolved in ultrapure water at a concentration of 15 mg / mL. did.
- the TBA salified cation exchange resin obtained in Example 1-1 was added in an amount equivalent to 5 times the molar amount of the HA unit (unit molecular weight 401.3) in terms of the ion exchange capacity of the resin. After stirring for 15 minutes, filtration was performed using a 0.45 ⁇ m filter, and the filtrate was freeze-dried to obtain HA-TBA as a white solid.
- FIG. 1 shows a 1 H-NMR spectrum using 99 kDa HA—Na as a starting material and D 2 O as a solvent.
- the amount ratio of TBA to the HA unit was calculated from the integral value of 1.7 ppm; 16H), and the unit average molecular weight of HA-TBA was calculated from this ratio.
- the unit average molecular weight of HA-TBA starting from 99 kDa HA-Na was 752.6.
- Example 1-3 Synthesis of HA Fluorescein (FL) Labeled TBA Salt (HA-FL / TBA) HA-Na (99 kDa) synthesized in Example 1-2 as a starting material
- An anhydrous DMSO solution of TBA (10 mg / mL) was prepared. Thereafter, 5- (aminomethyl) fluorocein hydrochloride (manufactured by Invitrogen) was added in an amount of 0.05 molar equivalent to the HA unit.
- 4- (4,6-dimethoxy-1,3,5-triazin-2-yl) -4-methylmorpholinium chloride (DMT-MM, manufactured by Kokusan Chemical Co., Ltd.) was added to the HA unit.
- FIG. 2 shows a 1 H-NMR spectrum of a product using D 2 O as a measurement solvent.
- the integrated value of the acetyl-derived peak of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integration of four methyl ((N (CH 2 CH 2 CH 2 CH 3 ) 4 , 1.0 ppm; 12H) of TBA From this value, the amount ratio of TBA to the HA unit was calculated, and the unit average molecular weight of HA-TBA was calculated from this ratio.
- the integrated value of the acetyl-derived peak of glucosamine and 3- (trimethylsilyl) propion used as an internal standard The HA unit content per weight was quantified from the integrated value of the methyl-derived peak (—Si (CH 3 ) 3 , 0.0 ppm; 9H) of sodium acid-d 4 (TSP-d 4 ). Dissolve in 50 mM carbonate buffer (pH 9.0) at 0.05 mg / mL, and determine FL content per weight from its absorbance at 491 nm , Modification ratio by .FL calculating the modification rate by FL at HA units was 3-6% in each lot.
- Example 1-4 Synthesis of HA Derivative from HA-TBA or HA-FL / TBA
- Example 1-4-1 HA derivative modified with L-alanine (Ala) (HA-Ala / FL)
- An anhydrous DMSO solution (5 mg / mL) of HA-TBA synthesized in Example 1-2 using HA-Na (99 kDa) as a starting material was prepared. Thereafter, L-alanine ethyl ester hydrochloride (manufactured by Aldrich) was added in an amount equivalent to 3 times the molar amount of HA unit and 0.15 times the equivalent amount of 5- (aminomethyl) fluorocein hydrochloride.
- a 6-fold molar equivalent of DMT-MM was added to the HA unit and stirred overnight at room temperature.
- the reaction solution was dialyzed in the order of DMSO, 0.3 M NaCl aqueous solution, and ultrapure water (Spectrapore 4, fractional molecular weight (MWCO): 12 k-14 kDa).
- MWCO fractional molecular weight
- 2N NaOH was added to adjust the pH to 12.5 or higher, and the mixture was stirred for 1 hour to hydrolyze the ethyl ester, and carboxy was deprotected. Thereafter, the mixture was neutralized with 2N HCl, dialyzed, and lyophilized to obtain HA-Ala / FL as a yellow solid.
- FIG. 3-1 shows the 1 H-NMR spectrum of the product measured under the same conditions as described in Example 1-3. From the integrated value of the peak derived from the acetyl of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the peak derived from methyl in alanine (—CH 3 , 1.4 ppm; 3H), The modification rate by alanine in the HA unit was calculated (Table 1).
- Example 1-4-2 Synthesis of HA derivative (HA-Ser / FL) modified with L-serine (Ser) Except that L-serine ethyl ester hydrochloride (manufactured by Aldrich) was used instead of L-alanine ethyl ester hydrochloride, the same procedure as in Example 1-4-1 was carried out, and HA-Ser / FL was yellow. Obtained as a solid. A part of the dialysis solution before deprotection of carboxy was taken and freeze-dried to obtain a modification rate calculation sample (HA-Ser-OEt / FL).
- L-serine ethyl ester hydrochloride manufactured by Aldrich
- FIG. 3-2 shows the 1 H-NMR spectrum of the modification rate calculation sample measured under the same conditions as described in Example 1-3. From the integrated value of the acetyl-derived peak (—COCH 3 , 2.0 ppm; 3H) of glucosamine and the integrated value of the methyl-derived peak (—CH 3 , 1.3 ppm; 3H) in the ethyl ester of serine, Example In the same manner as in 1-4-1, the modification rate by serine in the HA unit was calculated (Table 1). Further, FIG. 3-2 shows a 1 H-NMR spectrum obtained by measuring the deprotected sample under the same conditions as described in Example 1-3.
- Example 1-4-3 Synthesis of HA derivative (HA-Glu / FL) modified with L-glutamic acid (Glu) L-glutamic acid diethyl ester hydrochloride (Tokyo Chemical Industry Co., Ltd.) instead of L-alanine ethyl ester hydrochloride HA-Glu / FL was obtained as a yellow solid in the same manner as in Example 1-4-1 except that Co., Ltd. was used.
- FIG. 3-3 shows the 1 H-NMR spectrum of the product measured under the same conditions as described in Example 1-3. From the integrated value of the peak derived from acetyl of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the peak derived from methylene of glutamic acid (—CH 2 CH 2 COOH, 2.3 ppm; 2H), Example 1 The modification rate of glutamic acid in the HA unit was calculated in the same manner as in 4-1 (Table 1).
- the peak derived from acetyl of glucosamine is overlapped with another methylene-derived peak of glutamic acid (—CH 2 CH 2 COOH, 2.1 ppm; 2H), so that the integration of 1.7 to 2.2 ppm peak is integrated.
- the value obtained by subtracting the integrated value of the peak at 2.3 ppm from the value was used as a peak derived from acetyl of glucosamine and used for calculation of the modification rate.
- Example 1-4-4 Synthesis of HA derivatives (HA-Gly and HA-Gly / FL) modified with glycine (Gly)
- An anhydrous DMSO solution of HA-TBA synthesized in Example 1-2 (5 mg / mL)
- HA-FL / TBA synthesized in Example 1-3 was used as an ultrapure water / DMSO mixed solution (1: 3) solution (about 4 mg / mL) as a starting material.
- Glycine ethyl ester hydrochloride manufactured by Wako Pure Chemical Industries, Ltd. was added at a 5-fold molar equivalent to the HA unit.
- FIG. 3-4 shows the 1 H-NMR spectrum of the sample for calculating the modification rate measured under the same conditions as described in Example 1-3. From the integrated value of the acetyl-derived peak (-COCH 3 , 2.0 ppm; 3H) of glucosamine and the integrated value of the methyl-derived peak (-CH 3 , 1.3 ppm; 3H) in the ethyl ester of glycine, The modification rate by glycine in the HA unit was calculated in the same manner as in 1-4-1 (Table 1). In addition, FIG. 3-4 shows a 1 H-NMR spectrum obtained by measuring the deprotected sample under the same conditions as described in Example 1-3.
- Example 1-4-5 Synthesis of HA derivatives (HA-Val and HA-Val / FL) modified with L-valine (Val) L-valine ethyl ester hydrochloride (Watanabe) instead of glycine ethyl ester hydrochloride Except for using Chemical Industries, Ltd.), the same method as in Example 1-4-4 was performed to obtain HA-Val as a white solid and HA-Val / FL as a yellow solid.
- FIG. 3-5 shows the 1 H-NMR spectrum of the product measured under the same conditions as described in Example 1-3. From the integrated value of the acetyl-derived peak of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the two methyl-derived peaks (—CH (CH 3 ) 2 , 0.9 ppm; 6H) in valine Then, the modification rate by valine in the HA unit was calculated in the same manner as in Example 1-4-1 (Table 1).
- acetyl peak of glucosamine is overlapped with the hydrogen peak at the 3rd position of valine (—CH (CH 3 ) 2 , 2.1 ppm; 1H), so the peak of 1.8 to 2.2 ppm A value obtained by subtracting a value obtained by multiplying the integral value of the 0.9 ppm peak by 1/6 from the integral value was used as a peak derived from acetyl of glucosamine to calculate the modification rate.
- Example 1-4-6 Synthesis of HA Derivatives Modified with L-Leucine (Leu) (HA-Leu and HA-Leu / FL) L-leucine ethyl ester hydrochloride (Tokyo) instead of glycine ethyl ester hydrochloride Except for using Kasei Kogyo Co., Ltd., the same method as in Example 1-4-4 was performed to obtain HA-Leu as a white solid and HA-Leu / FL as a yellow solid.
- FIG. 3-6 shows the 1 H-NMR spectrum of the product measured under the same conditions as described in Example 1-3. From the integrated value of the acetyl-derived peak of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the two methyl-derived peaks (—CH (CH 3 ) 2 , 0.9 ppm; 6H) in leucine In the same manner as in Example 1-4-1, the modification rate of leucine in the HA unit was calculated (Table 1).
- Example 1-4-7 Synthesis of HA derivatives modified with L-isoleucine (Ile) (HA-Ile and HA-Ile / FL)
- Anhydrous DMSO was used as a solvent, and instead of glycine ethyl ester hydrochloride Except that L-isoleucine methyl ester hydrochloride (manufactured by Watanabe Chemical Industry Co., Ltd.) was used in the same manner as in Example 1-4-4.
- HA-Ile was a white solid and HA-Ile / FL was Obtained as a yellow solid.
- FIG. 3-7 shows the 1 H-NMR spectrum of the product measured under the same conditions as described in Example 1-3.
- acetyl-derived peak of glucosamine is overlapped with the hydrogen peak at the 3-position of isoleucine (—CH (CH 3 ) CH 2 CH 3 , 1.9 ppm; 1H), so 1.8 to 2.2 ppm
- a value obtained by subtracting a value obtained by multiplying the integrated value of the peak at 0.9 ppm by 1/6 from the integrated value of the peak was used as the peak derived from acetyl of glucosamine, and used for calculation of the modification rate.
- Example 1-4-8 Synthesis of HA Derivatives Modified with L-Threonine (Thr) (HA-Thr and HA-Thr / FL) Hydrochloride of L-threonine methyl ester instead of glycine ethyl ester hydrochloride ( Except for using Bachem), the same procedure as in Example 1-4-4 was carried out to obtain HA-Thr as a white solid and HA-Thr / FL as a yellow solid.
- FIG. 3-8 shows the 1 H-NMR spectrum obtained by analyzing the product under the same conditions as described in Example 1-3. From the integrated value of the acetyl-derived peak (—COCH 3 , 2.0 ppm; 3H) of glucosamine and the integrated value of the methyl-derived peak (—CH 3 , 1.2 ppm; 3H) in threonine, Example 1-4 In the same manner as in -1, the modification rate of threonine in the HA unit was calculated (Table 1).
- Example 1-4-9 Synthesis of HA derivatives (HA-Asp and HA-Asp / FL) modified with L-aspartic acid (Asp) L-aspartic acid diethyl ester hydrochloride instead of glycine ethyl ester hydrochloride Except for using Watanabe Chemical Co., Ltd., the same method as in Example 1-4-4 was carried out to obtain HA-Asp as a white solid and HA-Asp / FL as a yellow solid.
- FIG. 3-9 shows the 1 H-NMR spectrum obtained by analyzing the product under the same conditions as described in Example 1-3. From the integrated value of the acetyl-derived peak (-COCH 3 , 2.0 ppm; 3H) of glucosamine and the integrated value of the methylene-derived peak (-CH 2 COOH, 2.7, 2.8 ppm; 2H) in aspartic acid In the same manner as in Example 1-4-1, the modification rate of the HA unit with aspartic acid was calculated (Table 1).
- Example 1-4-10 Synthesis of HA derivative (HA-cACHCA / FL) modified with cis-2-amino-1-cyclohexylcarboxylic acid (cACHCA) Hydrochloric acid of cACHCA ethyl ester instead of glycine ethyl ester hydrochloride
- cACHCA cis-2-amino-1-cyclohexylcarboxylic acid
- the HA-cACHCA / FL was converted to yellow using the same method as in Example 1-4-4, except that the salt (Acros) was used and carboxy deprotection was performed using 5N NaOH at pH 13.2. Obtained as a solid.
- FIG. 3-10 shows the 1 H-NMR spectrum of the product measured under the same conditions as described in Example 1-3. From the integrated value of the acetyl-derived peak of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the peak derived from the cyclohexane ring in cACHCA (—CHCOO—, 2.5 ppm; 1H), Example 1 The modification rate by cACHCA in the HA unit was calculated in the same manner as in 4-1 (Table 1).
- Example 1-4-11 Synthesis of HA derivative (HA-tACHCA-OEt / FL) modified with trans-2-amino-1-cyclohexylcarboxylic acid ethyl ester (tACHCA-OEt) HA-tACHCA was carried out in the same manner as in Example 1-4-4, except that tACHCA-OEt hydrochloride (manufactured by Acros) was used in place of glycine ethyl ester hydrochloride and carboxy was not deprotected. -OEt / FL was obtained as a yellow solid.
- FIG. 3-11 shows the 1 H-NMR spectrum obtained by analyzing the product under the same conditions as described in Example 1-3.
- the integrated value of the peak from 1.2 to 2.1 ppm is changed to the integrated value of the peak of 2.2 ppm.
- a value obtained by subtracting a value obtained by multiplying 11 was used as a peak derived from acetyl of glucosamine and used for calculation of the modification rate.
- Example 1-4-12 Synthesis of HA derivative (HA-Aib / FL) modified with 2-aminoisobutanoic acid (Aib) Hydrochloride of Aib ethyl ester instead of glycine ethyl ester hydrochloride (Watanabe Chemical Co., Ltd.) HA-Aib / FL was obtained as a yellow solid in the same manner as in Example 1-4-4, except that carboxy was deprotected using 5N NaOH at pH 13.2. It was.
- FIG. 3-12 shows the 1 H-NMR spectrum of the product measured under the same conditions as described in Example 1-3. From the integrated value of the acetyl-derived peak (—COCH 3 , 2.0 ppm; 3H) of glucosamine and the integrated value of the dimethyl-derived peak (—C (CH 3 ) 2 —, 1.5 ppm; 6H) in Aib, The modification rate by Aib in the HA unit was calculated in the same manner as in Example 1-4-1 (Table 1).
- Example 1-4-13 Synthesis of HA derivative (HA-ACBuCA-OEt / FL) modified with 1-amino-1-cyclobutanoic acid ethyl ester (ACBuCA-OEt) ACBuCA- in place of glycine ethyl ester hydrochloride Except that OEt hydrochloride (manufactured by Sigma Aldrich) was used and carboxy was not deprotected, the procedure was the same as Example 1-4-4, and HA-ACBuCA-OEt / FL was converted into a yellow solid. Obtained.
- FIG. 3-13 shows the 1 H-NMR spectrum of the product measured under the same conditions as described in Example 1-3. Based on the integrated value of the acetyl-derived peak (-COCH 3 , 2.0 ppm; 3H) of glucosamine and the integrated value of the peak (-CH 3 , 1.3 ppm; 3H) derived from ester-protected methyl in ACBuCA-OEt In the same manner as in Example 1-4-1, the modification rate by ACBuCA-OEt in the HA unit was calculated (Table 1).
- Example 1-4-14 Synthesis of HA Derivatives Modified with L-Asparagine (Asn) (HA-Asn and HA-Asn / Rh) HA-Asn was synthesized as follows.
- HA-TBA in anhydrous DMSO 5 mg / mL
- L-asparagine methyl ester hydrochloride manufactured by Bachem
- DMT-MM was added in a 3 to 6-fold molar equivalent with respect to the HA unit, and the mixture was stirred overnight at room temperature.
- the reaction solution was reprecipitated with diethyl ether, and the precipitate was dissolved in ultrapure water. After adding 5N NaOH solution to pH 12.5 or higher, the mixture was stirred for 1 hour to hydrolyze the ester and decarboxylate the carboxy. Went. Thereafter, neutralization was performed using a 5N HCl solution, followed by dialysis purification in the order of 0.3 M NaCl aqueous solution, distilled water and ultrapure water, and lyophilization to obtain HA-Asn as a white solid.
- HA-Asn / Rh was synthesized as follows.
- L-asparagine methyl ester hydrochloride and DMT-MM were sequentially added to the HA unit in 5-fold molar equivalents and 3-6-fold molar equivalents, respectively, and stirred overnight at room temperature.
- the reaction solution was purified by dialysis against DMSO. Piperidine (manufactured by Wako Pure Chemical Industries, Ltd.) was added to the resulting dialysate to a concentration of 20% and stirred for 2 hours to remove the Fmoc group. Then, it dialyzed and refined in order of DMSO, 0.3M NaCl aqueous solution, and distilled water.
- a 5N NaOH solution was added to the resulting dialysate to adjust the pH to 12.5 or higher, and the mixture was stirred for 1 hour to hydrolyze the ester and decarboxylate. Thereafter, neutralization was performed using a 5N HCl solution, followed by dialysis purification in the order of distilled water and ultrapure water, and lyophilization to obtain HA-Asn / EDOBEA.
- HA-Asn / EDOBEA 10 mg / mL was diluted 2-fold with 100 mM PB (pH 7.4), and NHS-rhodamine (5 / 6-carboxytetramethylrhodamine succinimidyl ester, manufactured by Thermo Fisher Scientific Inc.) was added in an amount of 0.05 times the molar equivalent of the HA unit and stirred overnight at room temperature. Subsequently, 40-fold molar equivalent of acetic anhydride (manufactured by Wako Pure Chemical Industries, Ltd.) was added to the HA unit, and the mixture was stirred for 1 hour to acetylate the excess terminal amino acid of EDOBEA.
- NHS-rhodamine 5 / 6-carboxytetramethylrhodamine succinimidyl ester, manufactured by Thermo Fisher Scientific Inc.
- 5N NaOH solution was added to the reaction solution to adjust the pH to 12.5 or higher, and the mixture was stirred for 1 hour, neutralized with 5N HCl solution, and purified by dialysis in the order of 0.3M NaCl aqueous solution, distilled water, and ultrapure water. Lyophilized to obtain HA-Asn / Rh as a red solid.
- Spectrapore 4 fractionated molecular weight (MWCO): 12 to 14 kDa was used for all dialysis membranes.
- FIG. 3-14 shows the 1 H-NMR spectrum obtained by analyzing the product under the same conditions as described in Example 1-3. From the integrated value of the peak derived from acetyl of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the peak derived from methylene of asparagine (—CH 2 CONH 2 , 2.8 ppm; 2H), Example 1 The modification rate by asparagine in the HA unit was calculated in the same manner as in 4-1 (Table 1).
- Example 1-4-15 Synthesis of HA derivatives (HA-Ala-NH 2 and HA-Ala-NH 2 / Rh) modified with L-alaninamide (Ala-NH 2 ) L-asparagine methyl ester hydrochloride Except that L-alaninamide hydrochloride (manufactured by Tokyo Kasei Kogyo Co., Ltd.) was used instead of and no operation related to reprecipitation and carboxy deprotection was performed. The method was performed to obtain HA-Ala-NH 2 as a white solid and HA-Ala-NH 2 / Rh as a red solid.
- L-alaninamide hydrochloride manufactured by Tokyo Kasei Kogyo Co., Ltd.
- FIG. 3-15 shows the 1 H-NMR spectrum of the product measured under the same conditions as described in Example 1-3. From the integrated value of the peak derived from acetyl of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the peak derived from methyl alanamide (—CH 3 , 1.5 ppm; 3H), Example 1-4 In the same manner as -1, the modification rate of alanine amide in the HA unit was calculated (Table 1).
- Example 1-4-16 Synthesis of HA derivatives (HA-Val-NH 2 and HA-Val-NH 2 / Rh) modified with L-valine amide (Val-NH 2 ) Instead of L-alaninamide hydrochloride Except that L-valine amide hydrochloride (manufactured by Tokyo Chemical Industry Co., Ltd.) was used in the same manner as in Example 1-4-15, HA-Val-NH 2 was a white solid, HA-Val-NH 2 / Rh was obtained as a red solid.
- L-valine amide hydrochloride manufactured by Tokyo Chemical Industry Co., Ltd.
- FIG. 3-16 shows the 1 H-NMR spectrum obtained by analyzing the product under the same conditions as described in Example 1-3. From the integrated value of the acetyl-derived peak of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the two methyl-derived peaks of valineamide (—CH (CH 3 ) 2 , 1.0 ppm; 6H), In the same manner as in Example 1-4-1, the modification rate of the HA unit with valine amide was calculated (Table 1).
- acetyl peak of glucosamine is overlapped with the hydrogen peak at the 3-position of valineamide (—CH (CH 3 ) 2 , 2.1 ppm; 1H), so that the peak of 1.8 to 2.2 ppm
- a value obtained by subtracting a value obtained by multiplying the integral value of the 1.0 ppm peak by 1/6 from the integrated value was used as a peak derived from acetyl of glucosamine, and used for calculation of the modification rate.
- Example 1-4-17 Synthesis of HA derivatives (HA-Asn-NH 2 and HA-Asn-NH 2 / Rh) modified with L-asparagine amide (Asn-NH 2 ) Instead of using L-asparaginamide hydrochloride (manufactured by Kokusan Kagaku Co., Ltd.), the same procedure as in Example 1-4-15 was carried out to prepare HA-Asn-NH 2 as a white solid, HA-Asn- the NH 2 / Rh was obtained as a red solid.
- FIG. 3-17 shows the 1 H-NMR spectrum obtained by analyzing the product under the same conditions as described in Example 1-3. From the integrated value of the acetyl-derived peak of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the methylene-derived peak of asparagine amide (—CH 2 CONH 2 , 2.8 ppm; 2H), Example 1 The modification rate by asparagine amide in the HA unit was calculated in the same manner as in 4-1 (Table 1).
- Example 1-4-18 Synthesis of HA Derivatives Modified with Methylamine (Me) (HA-Me and HA-Me / FL) HA-Me was synthesized as follows. An anhydrous DMSO solution (5 mg / mL) of HA-TBA synthesized in Example 1-2 using HA-Na (99 kDa) as a starting material was prepared.
- Methylamine Methylamine
- reaction solution was dialyzed in the order of 0.3 M NaCl aqueous solution and ultrapure water (Spectrapore 4, fractional molecular weight (MWCO): 12 k-14 kDa) and then lyophilized to obtain an intermediate as a white solid.
- MWCO fractional molecular weight
- the obtained intermediate was dissolved in ultrapure water so as to be 10 mg / mL, and then diluted 2-fold with 100 mM phosphate buffer solution (pH 7.4) to prepare a 5 mg / mL solution.
- NHS-fluorescein in an amount of 0.1-fold molar equivalent to the HA unit was added to this solution as a DMSO solution and stirred at room temperature for 1 hour, and then 40-fold molar equivalent of acetic anhydride (Wako Pure Chemical Industries) to the HA unit.
- the terminal amino of excess ethylenediamine was acetylated by adding and stirring for 1 hour.
- reaction solution was dialyzed in the order of 0.3 M NaCl aqueous solution and ultrapure water (Spectrapore 4, molecular weight cut off (MWCO): 12 k-14 kDa) in the order of light, then freeze-dried and HA-Me / FL was obtained as a yellow solid.
- MWCO molecular weight cut off
- FIG. 3-18 shows the 1 H-NMR spectrum of each product measured under the same conditions as described in Example 1-3. From the integrated value of the acetyl-derived peak (—COCH 3 , 2.0 ppm; 3H) of glucosamine and the integrated value of the methyl-derived peak (—CH 3 , 2.8 ppm; 3H), Example 1-4-1 Similarly, the modification rate by methyl in the HA unit was calculated (Table 1).
- Pr Propylamine
- reaction solution was dialyzed in the order of 0.3 M NaCl aqueous solution and ultrapure water (Spectrapore 4, fractional molecular weight (MWCO): 12 k-14 kDa) and then freeze-dried to obtain HA-Pr as a white solid.
- MWCO fractional molecular weight
- reaction solution was dialyzed in the order of 0.3 M NaCl aqueous solution and ultrapure water (Spectrapore 4, fractional molecular weight (MWCO): 12 k-14 kDa) and then lyophilized to obtain an intermediate as a white solid.
- MWCO fractional molecular weight
- the obtained intermediate was dissolved in ultrapure water so as to be 10 mg / mL, and then diluted 2-fold with 100 mM phosphate buffer solution (pH 7.4) to prepare a 5 mg / mL solution.
- 100 mM phosphate buffer solution pH 7.4
- 0.06 times the molar equivalent of NHS-fluorescein with respect to the HA unit was added as a DMSO solution and stirred at room temperature for 1 hour, and then 40 times the molar equivalent of acetic anhydride (Wako Pure Chemical Industries, Ltd.) with respect to the HA unit.
- the terminal amino of excess ethylenediamine was acetylated by adding and stirring for 1 hour.
- reaction solution was dialyzed in the order of 0.3 M NaCl aqueous solution and ultrapure water in the dark (Spectrapore 4, fractional molecular weight (MWCO): 12 k-14 kDa), freeze-dried, and HA-Pr / FL was obtained as a yellow solid.
- MWCO fractional molecular weight
- FIG. 3-19 shows the 1 H-NMR spectrum of each product measured under the same conditions as described in Example 1-3. From the integrated value of the acetyl-derived peak of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the methyl-derived peak (—CH 3 , 0.9 ppm; 3H) belonging to propyl, Example 1-4 The modification rate by methyl in the HA unit was calculated in the same manner as in -1 (Table 1).
- Example 1-5) Synthesis of HA derivative from HA-TBA (25 kDa or 99 kDa) (Example 1-5-1) HA derivative modified with 1-aminomethyl-1-cyclohexane acid (AMCHCA) (HA- Synthesis of AMCHCA)
- AMCHCA 1-aminomethyl-1-cyclohexane acid
- DMT-MM was added at a ratio shown in Table 2 with respect to the HA-TBA unit, and the mixture was stirred overnight at room temperature.
- the reaction solution was purified by dialysis against a large excess of 0.3 M NaCl aqueous solution (Spectrapore 4, fractional molecular weight (MWCO): 12 k-14 kDa), and the resulting dialysate was added with 5 N NaOH aqueous solution to adjust the pH to 13.
- the mixture was stirred at room temperature for 1 hour and further neutralized by adding 5N HCl.
- a 1 H-NMR spectrum using D 2 O as a solvent when the HA molecular weight is 99 kDa and the addition ratio of the reagent is HA-TBA unit / DMT-MM / AMCHCA-OMe ⁇ HCl 1/3/5. It is shown in Fig. 4-1. From the integrated value of the acetyl-derived peak of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the peak derived from cyclohexane belonging to AMCHCA (C 6 H 10 , 1.2 to 1.9 ppm; 10H), The modification rate by AMCHCA in the HA unit was calculated in the same manner as in Example 1-4-1 (Table 2).
- Example 1-5-2 Synthesis of HA derivative (HA-pcACHCA) modified with cis-4-aminocyclohexane acid (pcACHCA) cis-4-amino-1-cyclohexylcarboxylic acid instead of AMCHCA-OMe ⁇ HCl
- pcACHCA cis-4-amino-1-cyclohexylcarboxylic acid instead of AMCHCA-OMe ⁇ HCl
- methyl ester hydrochloride pcACHCA-OMe ⁇ HCl, manufactured by Iris Biotech
- the amount of reagent added is shown in Table 2.
- Example 1-5-3 Synthesis of HA derivative (HA-Nal) modified with L-2-naphthylalanine (Nal) L-2-naphthylalanine benzyl ester paratosylate salt instead of AMCHCA-OMe ⁇ HCl
- Preparation was carried out in the same manner as in Example 1-5-1 except that (Nal-Obzl ⁇ p-Ts, manufactured by Bachem) was used, and the target product (HA-Nal) was obtained as a white solid.
- the amount of reagent added is shown in Table 2.
- Example 1-5-4 Synthesis of HA derivative (HA-APBA) modified with 2-amino-4-phenylbutanoic acid (APBA) 2-amino-4-phenylbutanoic acid instead of AMCHCA-OMe ⁇ HCl
- APBA 2-amino-4-phenylbutanoic acid instead of AMCHCA-OMe ⁇ HCl
- Example 1-5-5-5-5-5-5-5-5-5-5-5-5-5-5-5-5-5 Synthesis of HA derivative (HA-Cha) modified with cyclohexyl-L-alanine (Cha) Cyclohexyl-L-alanine methyl ester hydrochloride (Cha-OMe) instead of AMCHCA-OMe ⁇ HCl Preparation was performed in the same manner as in Example 1-5-1 except that HCl (manufactured by Watanabe Chemical Co., Ltd.) was used to obtain the target product (HA-Cha) as a white solid. The amount of reagent added is shown in Table 2.
- Example 1-5-6 Synthesis of HA derivative (HA-AMBA) modified with 4-aminomethylbenzoic acid (AMBA) 4-aminomethylbenzoic acid methyl ester hydrochloride (AMBA) instead of AMCHCA-OMe ⁇ HCl
- AMBA 4-aminomethylbenzoic acid methyl ester hydrochloride
- the product (HA-AMBA) was obtained as a white solid by the same method as in Example 1-5-1 except that -OMe.HCl (manufactured by Sigma-Aldrich) was used.
- the amount of reagent added is shown in Table 2.
- the 1 H-NMR spectrum of this HA-AMBA is shown in FIG. 4-6.
- Example 1-5-7 Synthesis of HA derivative (HA-3AMBA) modified with 3-aminomethylbenzoic acid (3AMBA) 3-aminomethylbenzoic acid methyl ester hydrochloride (AMBA) instead of AMCHCA-OMe ⁇ HCl
- 3-aminomethylbenzoic acid methyl ester hydrochloride (AMBA) instead of AMCHCA-OMe ⁇ HCl
- the product (HA-3AMBA) was obtained as a white solid by the same method as in Example 1-5-1 except that -OMe.HCl (manufactured by Fluorochem) was used.
- the amount of reagent added is shown in Table 2.
- Example 1-5-8 Synthesis of HA derivative (HA-APhPA) modified with 3-amino-2-phenylpropionic acid (APhPA) 3-amino-2-phenylpropionic acid instead of AMCHCA-OMe ⁇ HCl
- Preparation was carried out in the same manner as in Example 1-5-1 except that ethyl ester hydrochloride (APhPA-OEt ⁇ HCl, manufactured by Tyger) was used, and the target product (HA-APhPA) was obtained as a white solid.
- the amount of reagent added is shown in Table 2.
- Example 1-5-9 Synthesis of HA derivative (HA-AEBA) modified with 4- (2-aminoethyl) benzoic acid (AEBA) 4- (2-aminoethyl) instead of AMCHCA-OMe ⁇ HCl
- AEBA benzoic acid methyl ester hydrochloride
- HA-AEBA target product
- Example 1-5-10 Synthesis of HA derivative (HA-AMClBA) modified with 4-aminomethyl-3-chlorobenzoic acid (AMClBA) 4-aminomethyl-3-chloro instead of AMCHCA-OMe ⁇ HCl
- Preparation was performed in the same manner as in Example 1-5-1 except that benzoic acid methyl ester hydrochloride (AMClBA-OMe ⁇ HCl, manufactured by Anichem) was used, and the target product (HA-AMClBA) was converted into a white solid. Obtained.
- the amount of reagent added is shown in Table 2.
- Example 1-5-11 Synthesis of HA derivative (HA-AMSA) modified with 5-aminomethylsalicylic acid (AMSA) 5-aminomethylsalicylic acid methyl ester hydrochloride (AMSA-OMe) instead of AMCHCA-OMe ⁇ HCl
- HCl manufactured by Oakwood
- the amount of reagent added is shown in Table 2.
- Example 1-5-12 Synthesis of HA derivative (HA-4AMCHCA) modified with trans-4-aminomethylcyclohexane acid (4AMCHCA) trans-4-aminomethylcyclohexane acid methyl ester instead of AMCHCA-OMe ⁇ HCl
- Preparation was carried out in the same manner as in Example 1-5-1 except that hydrochloride (4 AMCHCA-OMe.HCl, manufactured by AK scientific) was used, and the target product (HA-4AMCHCA) was obtained as a white solid.
- the amount of reagent added is shown in Table 2.
- Example 1-5-13 Synthesis of HA derivative (HA-Chg) modified with L-cyclohexylglycine (Chg) L-cyclohexylglycine methyl ester hydrochloride (Chg-OMe ⁇ HCl instead of AMCHCA-OMe ⁇ HCl)
- the product (HA-Chg) was obtained as a white solid by the same method as in Example 1-5-1 except that INDOFINE Chemical Company was used.
- the amount of reagent added is shown in Table 2.
- Example 1-5-14 Synthesis of HA derivative (HA-pHPhg) modified with (R) -amino- (4-hydroxyphenyl) acetic acid (pHPhg) (R) -amino instead of AMCHCA-OMe ⁇ HCl
- Preparation was carried out in the same manner as in Example 1-5-1 except that-(4-hydroxyphenyl) acetic acid methyl ester hydrochloride (pHPhg-OMe ⁇ HCl, manufactured by Sigma-Aldrich) was used.
- HA-pHPhg) was obtained as a white solid.
- the amount of reagent added is shown in Table 2.
- HA unit / BOP manufactured by Wako Pure Chemical Industries, Ltd.
- EDOBEA 2,2 ′-(ethylenedioxy) bis (ethylamine)
- EDOBEA 2,2 ′-(ethylenedioxy) bis (ethylamine)
- BOP 2,2 ′-(ethylenedioxy) bis (ethylamine)
- FIG. 5A shows the 1 H-NMR spectrum of HA-EDOBEA with a low modification rate measured under the same conditions as described in Example 1-3. From the integrated value of the peak derived from acetyl of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the peak derived from methylene at the end of EDOBEA (—CH 2 NH 2, 3.2 ppm; 2H), Example 1-3 and Similarly, the modification rate by EDOBEA in the HA unit was calculated (Table 3).
- the obtained low-modification HA-EDOBEA was dissolved in ultrapure water so as to be 10 mg / ml, and then diluted 2-fold with 100 mM phosphate buffer solution (pH 7.4) to give 5 mg / ml.
- a solution was prepared.
- 0.1-fold molar equivalent of NHS-fluorescein (5 / 6-carboxyfluorocein succinimidyl ester, NHS-FL, manufactured by PIERCE) as a HA unit was added as a DMSO solution for 1 hour at room temperature.
- FIG. 5-1 A 1 H-NMR spectrum of the product measured under the same conditions as described in Example 1-3 is shown in FIG. 5-1.
- the product was dissolved in 50 mM carbonate buffer (pH 9.0) at 0.05 mg / mL, the FL content per weight was determined from the absorbance at 491 nm, and the modification rate by FL in the HA unit was calculated (Table 3). .
- FIG. 5-2 shows the 1H-NMR spectrum of HA-EDOBEA with a high modification rate measured under the same conditions as described in Example 1-3.
- NaOD was added so that it might become 0.0046N in the case of a measurement solution, and it was made alkaline.
- the obtained highly modified HA-EDOBEA was dissolved in ultrapure water so as to be 10 mg / ml, and then diluted 2-fold with 100 mM phosphate buffer solution (pH 7.4) to obtain 5 mg / ml.
- a solution was prepared.
- 0.04 mole equivalent of NHS-fluorescein with respect to the HA unit was added as a DMSO solution and stirred at room temperature for 1 hour, and then 40 mole equivalent of acetic anhydride (Wako Pure Chemical Industries, Ltd.) with respect to the HA unit. Co., Ltd.) was added and the mixture was further stirred for 1 hour to acetylate the excess terminal amino acid of EDOBEA.
- reaction solution was dialyzed in the order of 0.3 M NaCl aqueous solution and ultrapure water in the dark (Spectrapore 4, fractional molecular weight (MWCO): 12 k-14 kDa), then freeze-dried and HA-EDOBEA-Ac. / FL was obtained as a yellow solid.
- MWCO fractional molecular weight
- FIG. 5-2 shows the 1 H-NMR spectrum of the product measured under the same conditions as described in Example 1-3.
- the modification rate by FL in the HA unit of the product was determined in the same manner as in Comparative Example 1-1-1 (Table 3).
- FIG. 5-3 shows the 1 H-NMR spectrum of the product measured under the same conditions as described in Example 1-3. From the integrated value of the acetyl-derived peak (—COCH 3 , 2.0 ppm; 3H) of glucosamine and the integrated value of the phenyl-derived peak (—C 6 H 5 , 7.2 to 7.4 ppm; 5H) in phenylalanine In the same manner as in Example 1-4-1, the modification rate of the HA unit with phenylalanine was calculated (Table 4).
- FIG. 5-4 shows the 1 H-NMR spectrum of the product measured under the same conditions as described in Example 1-3. Integration value of acetyl-derived peak of glucosamine (—COCH 3 , 2.0 ppm; 3H) and integration of peak derived from hydroxyphenyl in tyrosine (—C 6 H 4 OH, 6.8, 7.2 ppm; 4H) From the value, the modification rate by tyrosine in the HA unit was calculated in the same manner as in Example 1-4-1 (Table 4).
- FIG. 5-5 shows the 1 H-NMR spectrum obtained by analyzing the product under the same conditions as described in Example 1-3. From the integrated value of the acetyl-derived peak (—COCH 3 , 2.0 ppm; 3H) of glucosamine and the integrated value of the methyl-derived peak (—CH 3 , 1.5 ppm; 3H) in MePhe, Example 1-4 The modification rate by MePhe in the HA unit was calculated in the same manner as -1 (Table 4).
- FIG. 5-6 shows the 1 H-NMR spectrum obtained by analyzing the product under the same conditions as described in Example 1-3. From the integrated value of the peak derived from the acetyl of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the peak for 1H derived from the pyrrolidine ring in Pro-OMe (—CHCOO—, 2.4 ppm; 1H) In the same manner as in Example 1-4-1, the modification rate by Pro-OMe in the HA unit was calculated (Table 4).
- FIG. 5-7 shows the 1 H-NMR spectrum obtained by analyzing the product under the same conditions as described in Example 1-3. From the integrated value of the peak derived from acetyl of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the peak derived from methylene of glycinamide (—CH 2 —; 2H), Example 1-4-1 Similarly, the modification rate by glycinamide in the HA unit was calculated (Table 4).
- FIG. 5-8 shows the 1 H-NMR spectrum obtained by analyzing the product under the same conditions as described in Example 1-3. Similar to Example 1-4-1, from the integrated value of the acetyl-derived peak of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the methylene-derived peak of serine amide (—CH 2 —; 2H). Then, the modification rate with serine amide in the HA unit was calculated (Table 4).
- FIG. 5-9 shows the 1 H-NMR spectrum obtained by analyzing the product under the same conditions as described in Example 1-3. From the integrated value of the acetyl-derived peak of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the two methyl-derived peaks of leucine amide (—CH (CH 3 ) 2 , 0.9 ppm; 6H) In the same manner as in Example 1-4-1, the modification rate of leucine amide in the HA unit was calculated (Table 4).
- FIG. 5-10 shows the 1 H-NMR spectrum obtained by analyzing the product under the same conditions as described in Example 1-3.
- acetyl-derived peak of glucosamine is overlapped with the hydrogen peak at the 3-position of isoleucine amide (—CH (CH 3 ) CH 2 CH 3 , 1.9 ppm; 1H).
- a value obtained by subtracting a value obtained by multiplying the integral value of the 0.9 ppm peak by 1/6 from the integral value of the 2 ppm peak was used as a peak derived from acetyl of glucosamine, and used for calculation of the modification rate.
- FIG. 5-11 shows the 1 H-NMR spectrum obtained by analyzing the product under the same conditions as described in Example 1-3. From the integrated value of the peak derived from acetyl of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the peak derived from methyl of threonine amide (—CH 3 , 1.2 ppm; 3H), Example 1-4 In the same manner as in -1, the modification rate of threonine amide in the HA unit was calculated (Table 4).
- FIG. 5-12 shows the 1 H-NMR spectrum obtained by analyzing the product under the same conditions as described in Example 1-3. From the integrated value of the acetyl-derived peak of glucosamine (—COCH 3 , 2.0 ppm; 3H) and the integrated value of the methylene-derived peak of glutamine amide (—CH 2 CH 2 CONH 2 , 2.4 ppm; 2H) In the same manner as in Example 1-4-1, the modification rate of glutamic amide in the HA unit was calculated (Table 4).
- a 1 H-NMR spectrum using D 2 O as a solvent when the HA molecular weight is 99 kDa and the addition ratio of the reagent is HA-TBA unit / DMT-MM / Cha-OMe ⁇ HCl 1/3/5. It is shown in Fig. 6-1. From the integrated value of the acetyl-derived peak (—COCH 3 , 2.0 ppm; 3H) of glucosamine and the integrated value of the methyl-derived peak (—CH 3 , 0.9 ppm; 3H) in Nle, Example 1-4 The modification rate by Nle in the HA unit was calculated in the same manner as -1 (Table 5).
- a 1 H-NMR spectrum using D 2 O as a solvent when the HA molecular weight is 99 kDa and the addition ratio of the reagent is HA-TBA unit / DMT-MM / Cha-OMe ⁇ HCl 1/3/5. It is shown in Fig. 6-2. Acetyl-derived peak of glucosamine; the integral value of the (-COCH3,2.0ppm 3H), 3 one methyl from peaks in tLeu (-C (CH 3) 3 , 1.0ppm; 9H) from the integrated value of, The modification rate by tLeu in the HA unit was calculated in the same manner as in Example 1-4-1 (Table 5).
- a 1 H-NMR spectrum using D 2 O as a solvent when the HA molecular weight is 99 kDa and the addition ratio of the reagent is HA-TBA unit / DMT-MM / Phe-OEt ⁇ HCl 1/3/5. It is shown in Fig. 6-3. Integral value of acetyl-derived peak (-COCH 3 , 2.0 ppm; 3H) of glucosamine and peak derived from aromatic ring (-C 6 H 4 F, 7.1, 7.3 ppm; 4H) in pFPhe From the value, the modification rate by pFPhe in the HA unit was calculated in the same manner as in Example 1-4-1 (Table 5).
- the reaction solution was reprecipitated with diethyl ether, and the precipitate was collected by filtration, dissolved again in ultrapure water, and then dialyzed against a large excess of ultrapure water (Spectrapore 4, molecular weight cut off (MWCO): 12 k). To 14 kDa) and lyophilized to obtain the target substance (PEG-FL) as a yellow solid.
- MWCO molecular weight cut off
- FIG. 7 shows the 1 H-NMR spectrum of the product measured under the same conditions as described in Example 1-3.
- a peak derived from one end of PEG (—OCH 3 , 3.4 ppm, 3H) and a peak derived from fluorescein (7-8 ppm) were observed, confirming that the PEG end was modified with FL. .
- Example 1-4 and Comparative Example 1-1 A part of the product obtained in Example 1-4 and Comparative Example 1-1 was measured. A 5 mg / mL or 20 mg / mL aqueous solution of the product was used as a measurement target sample, and a 5 mg / mL aqueous solution of HA-Na was used as a standard sample. Samples were diluted 10-fold with PBS or rat plasma, PBS diluted samples were incubated at 37 ° C. for 0 hours, 24 hours, 1 week, plasma diluted samples for 24 hours.
- Hyase-treated sample Hyase untreated sample, and calibration curve sample obtained by the above method
- sample: 0.8M borate buffer (pH 9.1): 1N KOH was mixed at a mixing ratio of 25: 5: 2. Heated in a boiling water bath for 3 minutes and immediately placed on ice.
- a reagent (10 mg / mL) in which p-dimethylaminobenzaldehyde is dissolved in a 10N HCl / acetic acid mixed solution (1:70) is mixed so that the mixing ratio of the above-mentioned sample: reagent becomes 32: 150, and 20 ° C. at 37 ° C. Incubated for minutes and immediately placed on ice.
- HA-EDOBEA-Ac / FL (Comparative Example 1-1-2) is biodegradable because the low molecular weight administration compound is not observed in urine in the size exclusion chromatography analysis of Example 3-3-2. Although not confirmed, even in the results of this test, the degradability by hyase was not confirmed. On the other hand, all other HA derivatives showed degradability.
- samples treated with rat plasma for 1 day showed improved degradability compared to samples treated with PBS for 1 day. It is considered that the release of glycine has changed to a form more susceptible to Hyase degradation.
- Example 3 Confirmation of blood retention and biodegradability in vivo (Example 3-1) Rat biological sample administered with HA derivative 20 mg of the compound obtained in Example 1-4 and Comparative Example 1-1 The compound obtained in Comparative Example 1-3 was administered into a rat intravenously at a dose of 10 mg / kg at a dose of / kg, and 5 minutes after administration, 2, 7, 24, 48, 72, 168, 240 Blood was collected from the jugular vein using a syringe treated with sodium heparin at 336 hours, and plasma was obtained by centrifugation. This plasma sample was stored frozen at ⁇ 20 ° C. or lower until measurement.
- urine was collected using a metabolic cage at 0 to 24 hours, 24 to 48 hours, 48 to 72 hours, 72 to 96 hours, 168 to 192 hours, 240 to 264 hours, and 336 to 360 hours after administration. A portion of the sample was stored frozen at -20 ° C. or lower until measurement.
- Example 3-2 Analysis of rat plasma treated with HA derivative
- Example 3-2-2 Concentration measurement The fluorescence intensity of the sample prepared in Example 3-2-1 was measured using a plate reader (SPECTRAmax GEMINI, manufactured by Molecular Devices) (FL label body: excitation wavelength) : 495 nm / fluorescence wavelength: 520 nm, Rh label: excitation wavelength: 552 nm / fluorescence wavelength: 595 nm).
- a calibration curve is prepared for each compound from the measurement values of the calibration curve sample, the plasma concentration at each time point of each individual is calculated, the average value is shown in FIG. 8, and the graph of plasma concentration transition is shown in FIG.
- Example 3-2-2 Pharmacokinetic parameter calculation Pharmacokinetic parameters were calculated using WinNonlin Ver 4.0.1 / Ver 6.1 (Pharsight). Model-independent analysis was performed on the average data of each group obtained in Example 3-2-2, and clearance (CL) was calculated (Table 7). As a result, as compared with PEG having practical blood retention, the HA derivatives of Examples 1-4-1 to 1-4-19 all had a smaller CL value, and practical blood retention. It was shown to have sex. In addition, CL of some of the comparative HA derivatives showed a larger value than that of PEG, and it was clarified that HA derivatives exhibiting various retention in blood can be synthesized depending on the type of compound to be modified.
- Example 3-3 Analysis of rat urine administered with HA derivative (Example 3-3-1)
- Example 3-3-2 Size Exclusion Chromatography Analysis The sample prepared in Example 3-3-1 was subjected to size exclusion chromatography, and changes in molecular weight distribution were observed. Size exclusion chromatography analysis was performed using an Alliance HPLC system (manufactured by Nippon Waters Co., Ltd.). The analysis conditions are shown below.
- the administration compound reduced in molecular weight was not observed in the urine, whereas in the HA derivatives of the examples, the administration compound reduced in molecular weight was detected in the urine. It was done. This indicates that the HA derivative of the present invention is biodegradable and is excreted outside the body after administration.
- Example 4 Verification of membrane damage using liposomes (Example 4-1) Preparation of liposome containing fluorescent substance and quenching substance 8-hydroxypyrene-1,3,6-trisulfonic acid trisodium salt (Pyramine) ) (SIGMA-ALDRICH) was dissolved in distilled water at 60 ⁇ mol / mL, and p-xylene-bis (N-pyridinium bromide) (DPX) (SIGMA-ALDRICH) was dissolved in distilled water at 85.7 ⁇ mol / mL. Thereafter, the solution was mixed in an equal amount to prepare a Pyramine / DPX aqueous solution.
- Pyramine 8-hydroxypyrene-1,3,6-trisulfonic acid trisodium salt
- DPX p-xylene-bis (N-pyridinium bromide)
- a liposome solution was prepared by adding and mixing the Pyramine / DPX solution (2 mL) to COATSOME EL-11A vial (NOF Corporation).
- PD-10 column purification and dialysis purification (Spectrapore 4, fractional molecular weight (MWCO): 12 k-14 kDa) against 500 mL of physiological saline were performed three times to obtain a liposome solution.
- the liposome solution was diluted 5-fold with a phosphate buffer solution having an ionic strength of 0.15, pH 6 or 7.4 to prepare a liposome solution at each pH.
- Example 4-2 Liposome membrane damage evaluation
- the HA derivative obtained in Example 1-5 and Comparative Example 1-2 was dissolved in physiological saline so as to be 10 mg / mL as HA-Na. It diluted with the salt solution sequentially, and adjusted so that it might become 10, 5, 1, 0.5, 0.1, 0.05, 0.01 mg / mL.
- 10 mg / mL physiological saline solution of Triton X was prepared as a positive target. Saline was used as a negative subject.
- poly-L-lysine hydrobromide (PLL, MW 15 kDa to 30 kDa, manufactured by Wako Pure Chemical Industries, Ltd.) is selected as a positive control substance having membrane damage, and dissolved in physiological saline so as to be 10 mg / mL. I let you.
- the index indicating membrane damage was calculated as the liposome degradation rate according to the following formula.
- HA-Nle, HA-tLeu, and HA-pFPhe used as comparative examples did not show any membrane damage at any pH, but the samples used in the examples had more membrane damage than pH 7.4. It was shown that the membrane hindrance at pH 6.0 was high.
- HA-pcACHCA, HA-Nal, HA-APBA, HA-Cha, HA-AMBA, HA-3AMBA, HA-APhPA, HA-AEB, HA-AMClBA, HA-AMSA, HA-4AMCHCA, HA-Chg and With HA-pHPhg no membrane damage was observed at pH 7.4, and membrane failure was first observed at pH 6.0.
- the degradation rate of the liposome at pH 7.4 of PLL was 6.6%, it can be judged that if it exhibits a degradation rate of several percent, it can function sufficiently as a substance having membrane damage, and the HA derivative of the present invention was shown to have membrane damage at pH 6.0.
- Example 5 Synthesis of HA-Ala Derivative-PTH Analog Conjugate
- Example 5-1 Synthesis of HA-Ala TBA Salt (HA-Ala-TBA) HA-Na Synthesized in Example 1-2
- An anhydrous DMSO solution (5 mg / mL) of HA-TBA starting from (99 kDa) was prepared. Thereafter, L-alanine ethyl ester hydrochloride was added in a 3-fold molar equivalent with respect to the HA unit.
- DMT-MM was added in a 3-fold molar equivalent with respect to the HA unit and stirred overnight at room temperature.
- reaction solution was dialyzed in the order of 0.3 M NaCl aqueous solution and distilled water (Spectrapore 4, fractional molecular weight (MWCO): 12 k-14 kDa).
- a 5N NaOH solution was added to the resulting dialysate to adjust the pH to 12.5 or more, and the mixture was stirred for 1 hour to hydrolyze the ethyl ester, and carboxy was deprotected. Thereafter, neutralization was performed using a 5N HCl solution, and dialysis was further performed with distilled water and ultrapure water.
- Example 1-1 To the resulting dialysate, the suspension of the cation exchange resin TBA-chlorinated in Example 1-1 was added to the HA unit in an amount of 5 times the molar equivalent in terms of the ion exchange capacity of the resin. After stirring for 15 minutes, filtration was performed using a 0.45 ⁇ m filter, and the filtrate was freeze-dried to obtain HA-Ala-TBA as a white solid. A part of the product was taken, dialyzed in the order of 0.3 M NaCl aqueous solution and distilled water, and freeze-dried to obtain a sample for calculating the alanine modification rate.
- FIG. 12-1 shows the 1 H-NMR spectra of the sample for calculating the alanine modification rate and the product, measured under the same conditions as described in Example 1-3.
- the modification rate with alanine in the HA unit was calculated in the same manner as in Example 1-4-1 and found to be 98%.
- the HA unit content per weight of HA-Ala-TBA was determined in the same manner as in Example 1-3.
- Example 5-2 Synthesis of HA-Ala-EDOBEA
- HA-Ala-TBA 5 mg / mL
- 50 mol equivalent and 0.15 mol equivalent respectively were added, and it stirred at room temperature for 6 hours.
- the reaction solution was purified by dialysis in the order of 0.3 M NaCl aqueous solution and distilled water (Spectrapore 4, fractional molecular weight (MWCO): 12 k-14 kDa) and lyophilized to obtain HA-Ala-EDOBEA as a white solid. It was.
- Example 5-3 Synthesis of HA-Ala-EDOBEA-Rh
- HA-Ala-EDOBEA 10 mg / mL
- phosphate buffer pH 7.4
- 0.07-fold molar equivalent of NHS-rhodamine with respect to HA unit was added in 5 mg / mL DMSO solution, and the mixture was stirred at room temperature for 2 hours.
- the reaction solution is purified by applying it to a desalting column (PD-10, manufactured by GE Healthcare) equilibrated with ultrapure water, and the HA fraction is lyophilized to convert HA-Ala-EDOBEA-Rh into a red solid. Obtained.
- a desalting column PD-10, manufactured by GE Healthcare
- FIG. 12-3 shows the 1 H-NMR spectrum of a product using D 2 O as a measurement solvent.
- the HA unit content per weight was quantified in the same manner as in Example 1-3. Further, the product was dissolved at 0.042 mg / mL in 100 mM carbonate buffer (pH 9.0), the Rh content per weight was determined from the absorbance at 552 nm, and the modification rate by Rh in the HA unit was calculated. It was 1%. Further, the molar extinction coefficient of the obtained product was calculated to be 2690 M ⁇ 1 cm ⁇ 1 .
- Example 5-4 Synthesis of bromoacetyl derivative of HA (HA-Ala-EDOBEA-BA / Rh / Ac)
- An aqueous solution (20 mg / mL) of HA-Ala-EDOBEA-Rh synthesized in Example 5-3 was used. Used as starting material.
- the aqueous solution was diluted 2-fold with 100 mM phosphate buffer (pH 7.4), and NHS-bromoacetic acid (NHS-BA, Sigma-Aldrich) 0.5-fold molar equivalent to HA unit was 50 mg / mL.
- NHS-bromoacetic acid NHS-bromoacetic acid
- acetic anhydride was added to the HA unit and further stirred for 1 hour to acetylate excess terminal amino acid of EDOBEA.
- the reaction solution was purified by subjecting it to a desalting column (PD-10) equilibrated with ultrapure water, and the fractionated HA fraction was subjected to centrifugal ultrafiltration (MacroSep, MWCO3000, manufactured by Pole Life Sciences) about 2
- the resultant solution was concentrated twice to obtain HA-Ala-EDOBEA-BA / Rh / Ac as an aqueous solution.
- the aqueous solution was frozen and stored at ⁇ 20 ° C. until next use, and a certain fixed volume was weighed after lyophilization to calculate the solid content concentration in the solution.
- Example 5-5 Synthesis of HA-Ala-PTH Analog Conjugate (HA-Ala-PTH / Rh)
- HA-Ala-EDOBEA-BA / Rh / Ac synthesized in Example 5-4 was used as a starting material. It was.
- PTH analog PTH-Cys (SVSEIQLMMHNLKHLNSMERVEWLRKKLQDVHNFC, manufactured by American Peptide Company) obtained by introducing cysteine into the C-terminus of human PTH (1-34) was used.
- PTH-Cys SVSEIQLMMHNLKHLNSMERVEWLRKKLQDVHNFC, manufactured by American Peptide Company
- HA unit After dissolving PTH-Cys in 10 mM citrate buffer (pH 4.5) at a concentration of 5 mg / mL, 0.03-fold molar equivalent of HA unit is HA-Ala-EDOBEA-BA / Rh / Ac solution. And incubated overnight at 37 ° C. 1-fold molar equivalent of cysteine hydrochloride monohydrate (manufactured by Wako Pure Chemical Industries, Ltd.) is added as a 20 mg / mL aqueous solution to the HA unit, and further incubated at 37 ° C. for 3 hours, and then with a 1N sodium hydroxide aqueous solution. The pH was 4.5. The HA unit content per weight was set to 1.5 ⁇ mol / mg and used for the calculation of HA units.
- reaction solution is purified by centrifugal ultrafiltration (Macrosep, MWCO10000) by repeating dilution and concentration four times with 2 mM citrate buffer (pH 4.5), and concentrating HA-Ala-PTH / Rh.
- a liquid was prepared.
- the concentrated solution was diluted 100-fold with 100 mM carbonate buffer (pH 9.0), the absorbance at 552 nm was measured, and the HA concentration was determined using the HA-Ala-EDOBEA-Rh extinction coefficient calculated in Example 5-3. The calculated value was 24.3 ⁇ mol / mL.
- amino acid analysis was performed by the AccQ ⁇ Tag method (manufactured by Waters), and the PTH concentration was calculated to be 141.75 nmol / mL and the PTH modification rate was 0.58%.
- Hydrolysis and derivatization operations in amino acid analysis were performed using a Pico Tag workstation and AccQ Tag chemistry package according to the manual accompanying the package.
- the HPLC analysis used a Waters 2690 separation module and a Waters 474 fluorescence detector, and the analysis for calculating the amino acid concentration was based on the concentration of lysine residues.
- Example 6 Confirmation of in vivo blood retention and biodegradability of HA-Ala derivative-PTH analog conjugate (Example 6-1) HA derivative-administered rat biological sample obtained in Example 5-5 Single dose administration of compound at a dose of 10 mg / kg into rat vein and jugular vein blood collection using syringes treated with sodium heparin at 5 minutes, 2, 7, 24, 48, 72, 168, 240 and 312 hours after administration Plasma was obtained by centrifugation. This plasma sample was stored frozen at ⁇ 20 ° C. or lower until measurement.
- urine was collected using a metabolic cage at 0-24 hours, 24-48 hours, 48-72 hours, 72-96 hours, 144-168 hours, 216-240 hours, and 288-312 hours after administration. A portion of the sample was stored frozen at -20 ° C. or lower until measurement.
- Example 6-2-1 Analysis of rat plasma treated with HA derivative
- Example 6-2-1 Preparation of measurement sample A sample for measurement was prepared by the method described in Example 3-2-1.
- Example 6-2-2 Concentration measurement Using the sample prepared in Example 6-2-1, the plasma concentration of each individual at each time point was calculated by the method described in Example 3-2-2. . The average value is shown in FIG. 13, and the graph of plasma concentration transition is shown in FIG.
- Example 6-2-3 Calculation of pharmacokinetic parameters With respect to the average data obtained in Example 6-2-2, the clearance (CL) was calculated by the method described in Example 3-2-2. 0. Practical blood retention was also demonstrated in drug conjugates of biodegradable hyaluronic acid derivatives.
- Example 6-3 Analysis of HA Derived Rat Urine (Example 6-3-1) Measurement Sample Preparation A measurement sample was prepared by the method described in Example 3-3-1.
- FIG. 15 shows the result of analyzing the sample prepared in Example 6-3-1 by the method described in Example 3-3-2.
- the chromatogram at each time point is shown in the same range on the left, and the chromatogram at each time point is normalized with the strongest peak on the right.
- Example 5-5 In the urine of an individual administered with Example 5-5, a low molecular weight administration compound was detected. This indicates that the biodegradable hyaluronic acid derivative drug conjugate maintains biodegradability and is excreted outside the body after administration.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Pharmacology & Pharmacy (AREA)
- Materials Engineering (AREA)
- Organic Chemistry (AREA)
- Polymers & Plastics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biochemistry (AREA)
- Dermatology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medicinal Preparation (AREA)
- Polysaccharides And Polysaccharide Derivatives (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Materials For Medical Uses (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Description
(1)式(I):

R8は、水素原子、ホルミルまたはC1-6アルキルカルボニルであり;
Xは、-NRx-A-B-Zで表される基であり;
Aは、-CRaRb-、およびC3-8シクロアルキレンから選択され、Bは直接結合であり、Zは-COORyまたは-CONRyaRybであり;または
-A-B-Zは、C1-6アルキルを表し;または
Aは-CH2-または-CH2-CH2-であり、Bはフェニレン(ここでフェニレンは、ヒドロキシおよびハロゲン原子から選択される1以上の基で置換されていてもよい)、C3-8シクロアルキレンおよびフェニルメタン-1,1-ジイルから選択され、Zは-COORyであり;
RxおよびRyは、それぞれ独立に、水素原子およびC1-6アルキルから選択され;
Raは、水素原子およびC1-6アルキルから選択され;
Rbは、水素原子、C1-6アルキル(ここでC1-6アルキルは、ヒドロキシ、カルボキシ、C3-8シクロアルキル、アリールおよびカルバモイルから選択される1以上の基で置換されていてもよく、ここでアリールはヒドロキシにより置換されていてもよい)、C3-8シクロアルキル、およびアリール(ここでアリールはヒドロキシにより置換されていてもよい)から選択され;
RyaおよびRybは、それぞれ独立して、水素原子、C1-6アルキル、ホルミルおよびC1-6アルキルカルボニルから選択される]
で表される二糖単位を含み、前記式(I)のAが-CRaRb-またはC3-8シクロアルキレンの場合、存在する二糖単位に対する式(I)の二糖単位の割合は70%以上であるヒアルロン酸誘導体。
(2)Xが-NHCH3、-NH(CH2)2CH3、-NRx-A-Zであり;Aが-CRaRb-またはC3-8シクロアルキレンである、上記(1)に記載のヒアルロン酸誘導体。
(3)AがC3-8シクロアルキレンであり、Bが直接結合である;
Aが-CH2-または-CH2-CH2-であり、Bがフェニレン(ここでフェニレンは、ヒドロキシおよびハロゲン原子から選択される1以上の基で置換されていてもよい)、C3-8シクロアルキレンおよびフェニルメタン-1,1-ジイルから選択される;または
Aが2-シクロヘキシルエタン-1,1-ジイル、2-(2-ナフチル)エタン-1,1-ジイル、3-フェニルプロパン-1,1-ジイル、シクロヘキシルメタン-1,1-ジイルおよび4-ヒドロキシフェニルメタン-1,1-ジイルから選択され、Bが直接結合である、上記(1)に記載のヒアルロン酸誘導体。
(4)Aが-CH2-であり、Bがシクロヘキサン-1,1-ジイル、ベンゼン-1,4-ジイル、ベンゼン-1,3-ジイル、2-クロロベンゼン-1,4-ジイルおよびフェニルメタン-1,1-ジイルから選択される;
Aが-CH2CH2-であり、Bがベンゼン-1,4-ジイルである;または、
Aが2-シクロヘキシルエタン-1,1-ジイル、2-(2-ナフチル)エタン-1,1-ジイルおよび3-フェニルプロパン-1,1-ジイルから選択され;Bが直接結合である、上記(1)または(3)に記載のヒアルロン酸誘導体。
(5)Zが-COORyである、上記(1)~(4)のいずれかに記載のヒアルロン酸誘導体。
(6)式(I):

R8は、水素原子、ホルミルまたはC1-6アルキルカルボニルであり;
Xは、-NHCH3、-NH(CH2)2CH3、-NRx-A-B-COORyまたは-NRx-CHRc-CONRyaRybで表される基であり;
RxおよびRyは、それぞれ独立に、水素原子およびC1-6アルキルから選択され;
Aは、-CRaRb-、C3-8シクロアルキレン、2-シクロヘキシルエタン-1,1-ジイル、2-(2-ナフチル)エタン-1,1-ジイル、3-フェニルプロパン-1,1-ジイル、シクロヘキシルメタン-1,1-ジイルおよび4-ヒドロキシフェニルメタン-1,1-ジイルから選択され、Bは直接結合であり;または
Aは-CH2-または-CH2-CH2-であり、Bはフェニレン(ここでフェニレンは、ヒドロキシおよびハロゲン原子から選択される1以上の基で置換されていてもよい)、C3-8シクロアルキレンおよびフェニルメタン-1,1-ジイルから選択され;
Raは、水素原子およびC1-6アルキルから選択され;
Rbは、水素原子およびC1-6アルキル(ここでC1-6アルキルは、ヒドロキシ、カルボキシおよびカルバモイルから選択される1以上の基で置換されていてもよい)から選択され;
Rcは、カルバモイルで置換されていてもよいC1-6アルキルを表し;
RyaおよびRybは、それぞれ独立して、水素原子、C1-6アルキル、ホルミルおよびC1-6アルキルカルボニルから選択される]
で表される二糖単位を含み、前記式(I)のAが-CRaRb-またはC3-8シクロアルキレンの場合、存在する二糖単位に対する式(I)の二糖単位の割合は70%以上であるヒアルロン酸誘導体。
(7)Xが-NHCH3、-NH(CH2)2CH3または-NRx-CHRc-CONRyaRybであるか、あるいは、Aが-CRaRb-またはC3-8シクロアルキレンである、上記(6)に記載のヒアルロン酸誘導体。
(8)AがC3-8シクロアルキレンであり、Bが直接結合である;
Aが-CH2-または-CH2-CH2-であり、Bがフェニレン(ここでフェニレンは、ヒドロキシおよびハロゲン原子から選択される1以上の基で置換されていてもよい)、C3-8シクロアルキレンおよびフェニルメタン-1,1-ジイルから選択される;または
Aが2-シクロヘキシルエタン-1,1-ジイル、2-(2-ナフチル)エタン-1,1-ジイル、3-フェニルプロパン-1,1-ジイル、シクロヘキシルメタン-1,1-ジイルおよび4-ヒドロキシフェニルメタン-1,1-ジイルから選択され、Bが直接結合である、上記(6)に記載のヒアルロン酸誘導体。
(9)Aが-CH2-であり、Bがシクロヘキサン-1,1-ジイル、ベンゼン-1,4-ジイル、ベンゼン-1,3-ジイル、2-クロロベンゼン-1,4-ジイルおよびフェニルメタン-1,1-ジイルから選択される;
Aが-CH2CH2-であり、Bがベンゼン-1,4-ジイルである;または、
Aが2-シクロヘキシルエタン-1,1-ジイル、2-(2-ナフチル)エタン-1,1-ジイルおよび3-フェニルプロパン-1,1-ジイルから選択され;Bが直接結合である、上記(6)または(8)に記載のヒアルロン酸誘導体。
(10)式(I):

R8は、水素原子、ホルミルまたはC1-6アルキルカルボニルであり;
Xは、-NRx-A-B-COORyで表される基であり;
RxおよびRyは、それぞれ独立に、水素原子およびC1-6アルキルから選択され;
Aは、-CRaRb-、C3-8シクロアルキレン、2-シクロヘキシルエタン-1,1-ジイル、2-(2-ナフチル)エタン-1,1-ジイル、および3-フェニルプロパン-1,1-ジイルから選択され、Bは直接結合であり;または
Aは-CH2-であり、BはフェニレンおよびC3-8シクロアルキレンから選択され;
Raは、水素原子およびC1-6アルキルから選択され;
Rbは、水素原子およびC1-6アルキル(ここでC1-6アルキルは、ヒドロキシおよびカルボキシから選択される1以上の基で置換されていてもよい)から選択される]
で表される二糖単位を含み、前記式(I)のAが-CRaRb-またはC3-8シクロアルキレンの場合、存在する二糖単位に対する式(I)の二糖単位の割合が70%以上である、ヒアルロン酸誘導体。
(11)Aが-CRaRb-またはC3-8シクロアルキレンである、上記(10)に記載のヒアルロン酸誘導体。
(12)AがC3-8シクロアルキレンであり、Bが直接結合である;
Aが-CH2-であり、BがフェニレンおよびC3-8シクロアルキレンから選択される;または
Aが2-シクロヘキシルエタン-1,1-ジイル、2-(2-ナフチル)エタン-1,1-ジイルおよび3-フェニルプロパン-1,1-ジイルから選択され、Bが直接結合である、上記(10)に記載のヒアルロン酸誘導体。
(13)Aが-CH2-であり、Bがシクロヘキサン-1,1-ジイルおよびベンゼン-1,4-ジイルから選択される;または、
Aが2-シクロヘキシルエタン-1,1-ジイル、2-(2-ナフチル)エタン-1,1-ジイルおよび3-フェニルプロパン-1,1-ジイルから選択され、Bが直接結合である、上記(10)または(12)に記載のヒアルロン酸誘導体。
(14)式(II):

R8aは、水素原子、ホルミルまたはC1-6アルキルカルボニルであり;
Xaは、ヒドロキシおよび-O-Q+、から選択され;ここで、Q+は、カウンターカチオンを表す]
で表される二糖単位をさらに含む、上記(1)~(13)のいずれかに記載のヒアルロン酸誘導体。
(15)式(III):

R8bは、水素原子、ホルミルまたはC1-6アルキルカルボニルであり;
Xbは、-NRe-Yb-Rdで表される基であり;ここで、
Reは、水素原子またはC1-6アルキルであり;
Rdは、水素原子、C1-6アルキル、-CO-C(R7)=CH2、または-CO-G4-Xcであり;
R7は、水素原子またはメチルであり;
G4は、フェニレン、C3-8シクロアルキレン、または-G5-(C1-10アルキレン)-G6-から選択され、ここでC1-10アルキレン部分は、1~3のフェニレンまたはC3-8シクロアルキレンが挿入されていてもよく;
G5およびG6は、それぞれ独立に、直接結合、フェニレン、またはC3-8シクロアルキレンから選択され;
Xcは、メルカプト、ハロゲン原子または
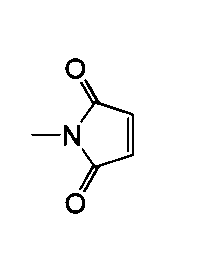
Ybは、-CH2-(CHR5)l-2-CH2-NH-、-CH2-(CHR6)p-2-CH2-O-、-(CH2)j-S-または-(CH2)a-(Y1-(CH2)b)c-G-であり;
l、p、およびjは、それぞれ独立に2~10から選択される整数であり、R5およびR6は、それぞれ独立に、水素原子またはヒドロキシであり;
aは、2~10から選択される整数であり;
bは、それぞれ独立に2~10から選択される整数であり;
cは、1~200から選択される整数であり;
Y1は、酸素原子または-NRn-であり;
Gは、酸素原子、硫黄原子または-NH-であり;
Rnは、水素原子、C1-6アルキル、-CO-(CH2)d-Ro、-(CH2)e-Rpまたは-(CH2)f-(Y2-(CH2)g)h-Rqであり;
gは、それぞれ独立に、2~10から選択される整数であり;
d、e、fおよびhは、それぞれ独立に、2~10から選択される整数であり;
Ro,RpおよびRqは、それぞれ独立に、水素原子、ヒドロキシ、カルボキシまたは-NHRrであり;
Y2は、酸素原子または-NH-であり;
Rrは、水素原子、ホルミルまたはC1-6アルキルカルボニルである]
で表される二糖単位をさらに含む、上記(1)~(14)のいずれかに記載のヒアルロン酸誘導体。
(16)R1a、R2a、R3a、およびR4aの全てを水素原子とし、R8aをアセチルとし、かつ、Xaを-O-Na+とした場合の重量平均分子量が20~120キロダルトンとなる、上記(14)に記載の式(II)で表される二糖単位のみから構成されるヒアルロン酸を用いて製造される、上記(1)~(15)のいずれかに記載のヒアルロン酸誘導体。
(17)前記ヒアルロン酸誘導体に主鎖構造が対応する誘導体化されていないヒアルロン酸の重量平均分子量が20~120キロダルトンであり、ここで誘導体化されていないヒアルロン酸は、前記式(II)においてR1a、R2a、R3aおよびR4aが水素原子であり、R8aがアセチルであり、Xaが-O-Na+である二糖単位のみからなるヒアルロン酸である、上記(1)~(15)のいずれかに記載のヒアルロン酸誘導体。
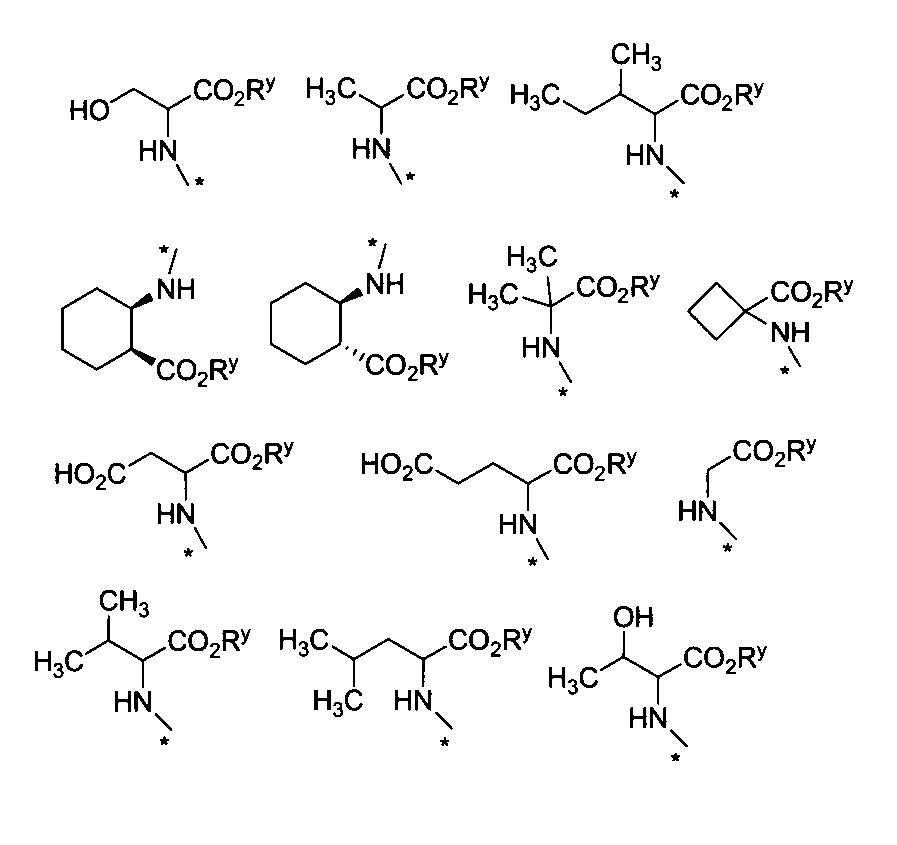

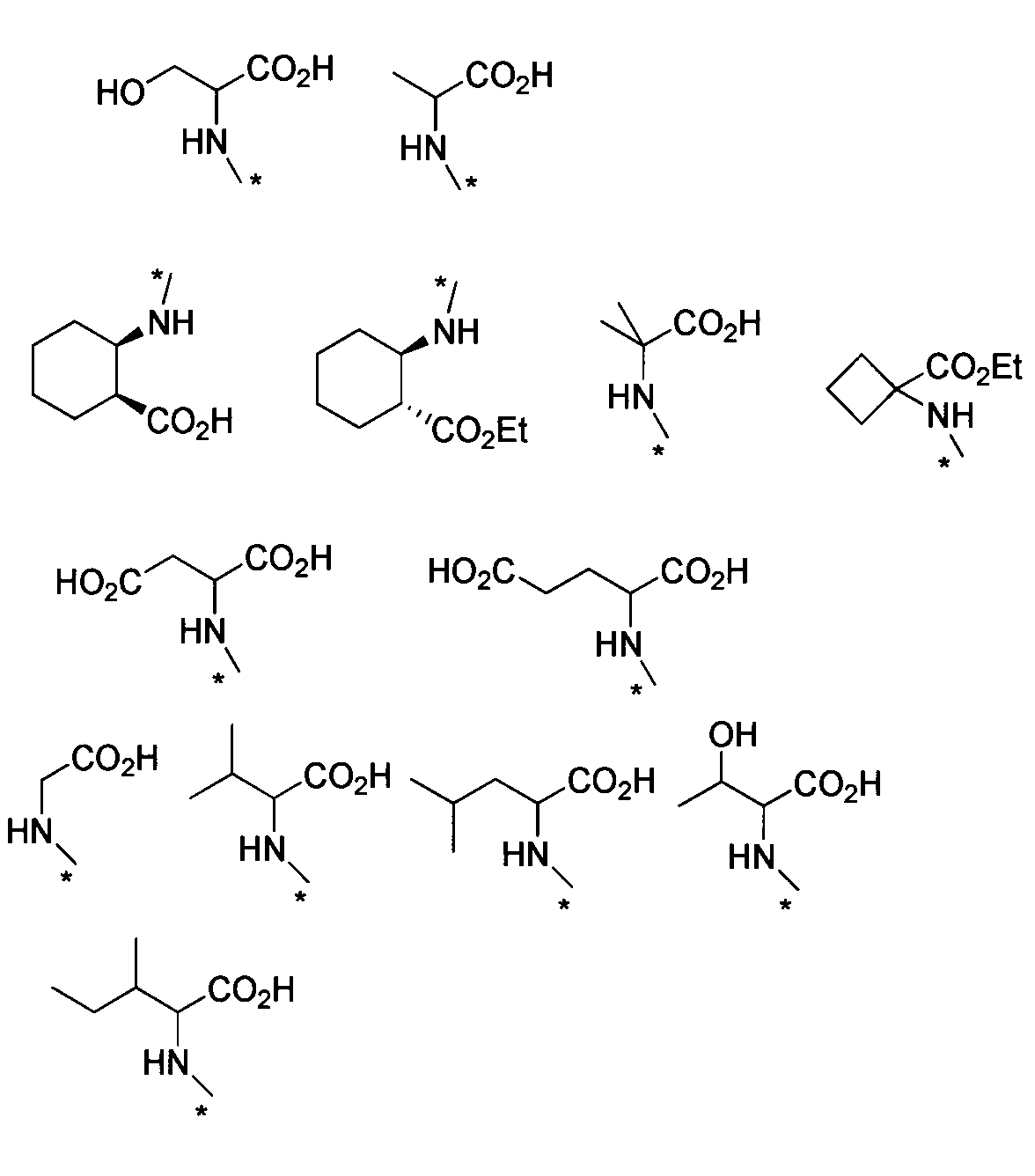







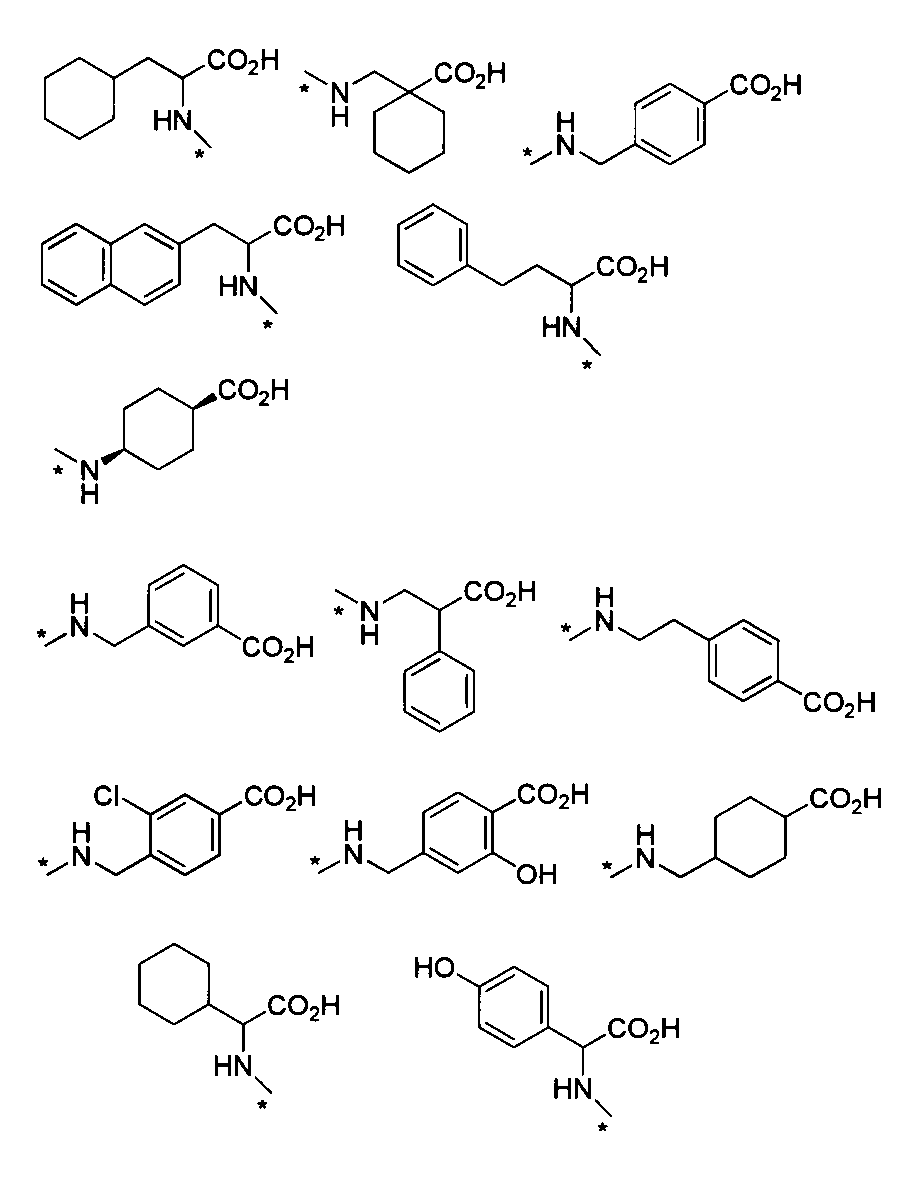

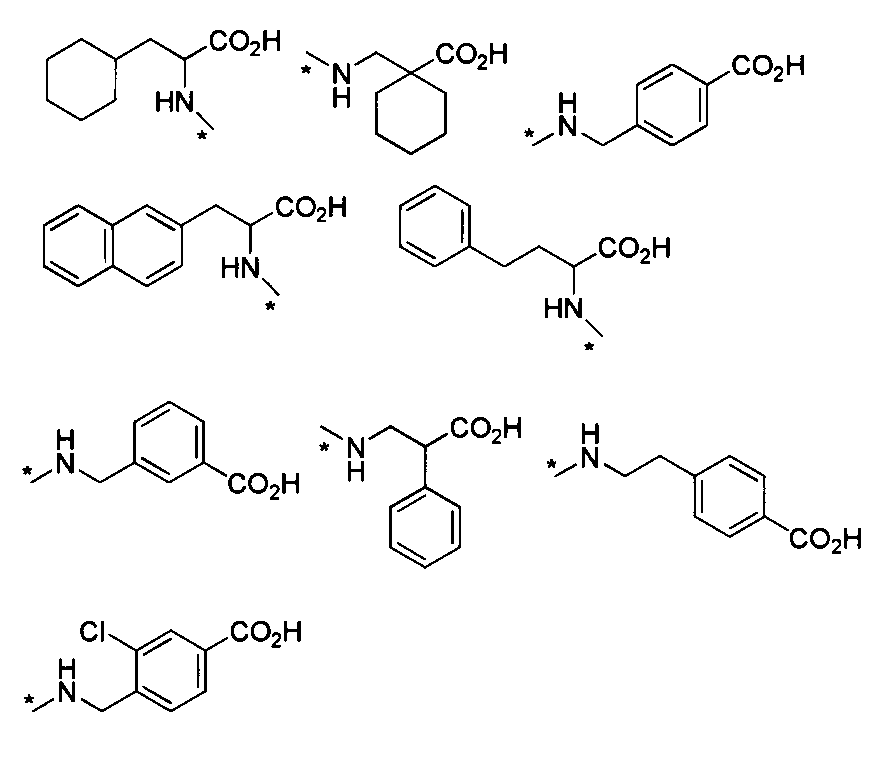
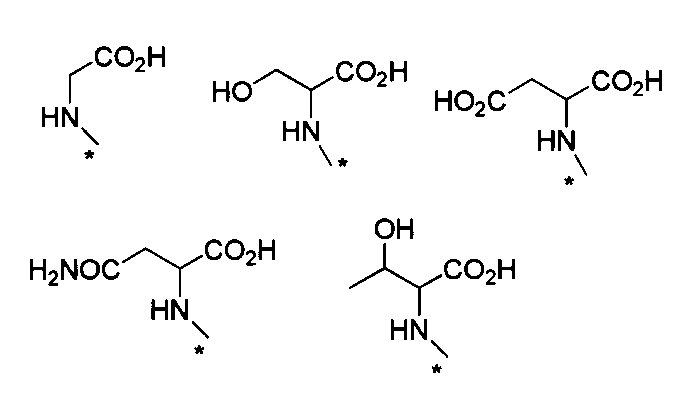

-HN-(CH2)j-SH;
-HN-CH2-CH2-(Y1-CH2-CH2)c-SH;
-HN-(CH2)p-O-CO-C(R7)=CH2;
-HN-(CH2)l-NHCO-C(R7)=CH2;
-HN-CH2-CH2-(Y1-CH2-CH2)c-NHCO-C(R7)=CH2;または
-HN-CH2-CH2-(Y1-CH2-CH2)c-O-CO-C(R7)=CH2
[式中、j、Yl、c、p、R7、およびlは、本明細書において既に定義されたとおりである]。
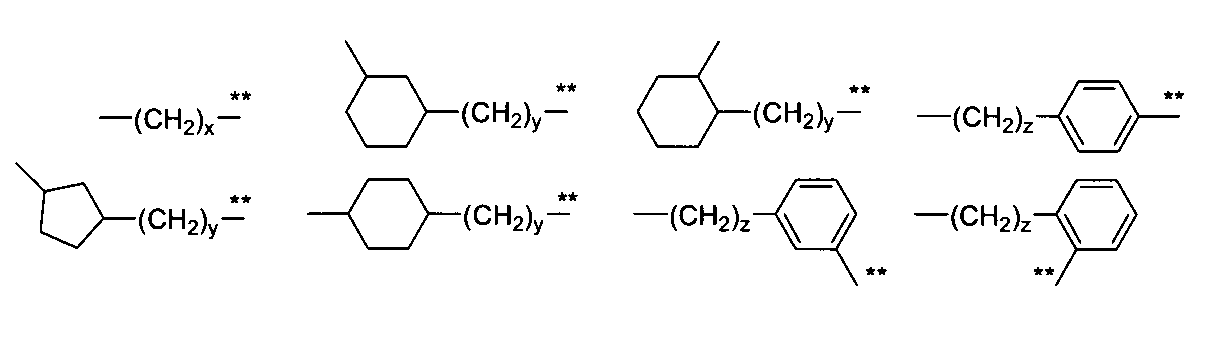
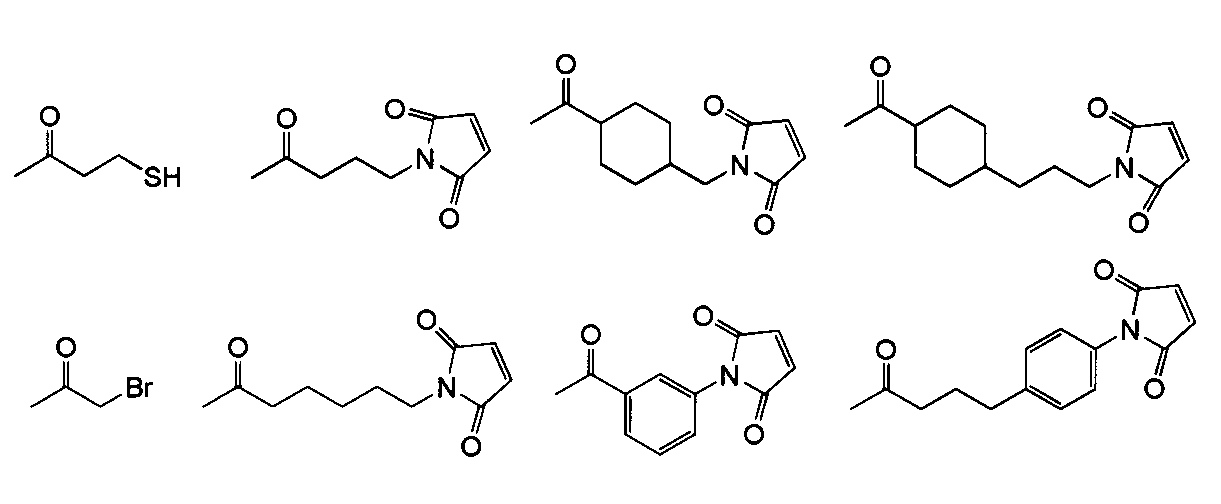
-NH-CH2-(CHR5)l-2-CH2-NH2;
-NH-CH2-CH2-(Y1-CH2-CH2)c-NH2;
-NH-CH2-(CHR6)p-2-CH2-OH;
-NH-CH2-CH2-(Y1-CH2-CH2)c-OH;
-NH-(CH2)j-SH;
-NH-CH2-CH2-(Y1-CH2-CH2)c-SH;
-NH-(CH2)p-O-CO-C(R7)=CH2;
-NH-(CH2)l-NHCO-C(R7)=CH2;
-NH-CH2-(CHR5)l-2-CH2-NH-CO-CH2-SH;
-NH-CH2-CH2-(Y1-CH2-CH2)c-NH-CO-CH2-SH;
-NH-(CH2)p-O-CO-CH2-CH2-SH;
-NH-(CH2)l-NHCO-CH2-CH2-SH;
-NH-CH2-CH2-(Y1-CH2-CH2)c-O-CO-CH2-CH2-SH;
-NH-CH2-CH2-(Y1-CH2-CH2)c-NHCO-CH2-CH2-SH;
-NH-CH2-(CHR5)l-2-CH2-NH-CO-CH2-Br;
-NH-CH2-CH2-(Y1-CH2-CH2)c-NH-CO-CH2-I;
-NH-CH2-CH2-(Y1-CH2-CH2)c-NHCO-C(R7)=CH2;または
-NH-CH2-CH2-(Y1-CH2-CH2)c-O-CO-C(R7)=CH2
[式中、R5、R6、R7、Y1、c、j、l、およびpは、本明細書で定義されるとおりである]
で表される基が挙げられる。
(式中、***は、薬物との結合部位を表し、
G1は、直接結合、-C(=O)-、-NRs-および-S-から選択され、
G2は、-(CH2)i-および-(CH2)q-(O-CH2-CH2)k-から選択され、
G3は、直接結合、-C(=O)-NRt-(CH2)r-および-NRu-C(=O)-(CH2)m-から選択され、
Jは、以下の式で表される基を表し、
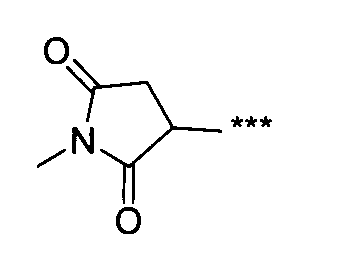
で表されるスペーサーが挿入されていてもよい。

Data point:16384
Spectral width(X sweep):15ppm
Acquisition time(X acq time):1.749s
Pulse delay(Relaxation delay):30s
Transients(Scans):64
(実施例1-1)カチオン交換樹脂のテトラブチルアンモニウム(TBA)塩化
DOWEX(登録商標)50WX-8-400(シグマ-アルドリッチ社製)を超純水に懸濁させ、デカンテーションにより樹脂を超純水で3回程度洗浄した。40重量%テトラブチルアンモニウムヒドロキシド水溶液(TBA-OH)(シグマ-アルドリッチ社製)を樹脂のカチオン交換能に対し約1.5倍モル等量を加え、30分間攪拌した。余剰のTBA-OH溶液をデカンテーションにより除去した後、さらに過剰の超純水で洗浄を繰り返し、最後に0.45μmのフィルターを用いて濾過することによりTBA塩化したカチオン交換樹脂を得た。
分子量25kDaおよび99kDaのヒアルロン酸ナトリウム塩(HA-Na、資生堂株式会社製)をそれぞれ15mg/mLの濃度で超純水に溶解した。実施例1-1で得たTBA塩化したカチオン交換樹脂をHAユニット(ユニット分子量401.3)のモル数に対し樹脂のイオン交換能換算で5倍モル等量添加した。15分間攪拌した後、0.45μmのフィルターを用いて濾過を行い、濾液を凍結乾燥し、HA-TBAを白色固体として得た。
実施例1-2で合成した、HA-Na(99kDa)を出発原料とするHA-TBAの無水DMSO溶液(10mg/mL)を調製した。その後、5-(アミノメチル)フルオロセインの塩酸塩(invitrogen社製)をHAユニットに対して0.05倍モル等量添加した。次に、4-(4,6-ジメトキシ-1,3,5-トリアジン-2-イル)-4-メチルモルホリニウムクロリド(DMT-MM、国産化学株式会社製)をHAユニットに対して0.2倍モル等量を添加し、室温で6時間以上撹拌した。反応溶液は、0.3M NaCl水溶液、超純水の順で透析(スペクトラポア4、分画分子量(MWCO):12k~14kDa)した。得られた透析液に、実施例1-1でTBA塩化したカチオン交換樹脂の懸濁液をHAユニットに対し樹脂のイオン交換能換算で5倍モル等量添加した。15分間撹拌した後、0.45μmのフィルターを用いて濾過を行い、濾液を凍結乾燥し、目的物(HA-FL/TBA)を黄色固体として得た。
(実施例1-4-1)L-アラニン(Ala)により修飾したHA誘導体(HA-Ala/FL)の合成
実施例1-2で合成した、HA-Na(99kDa)を出発原料とするHA-TBAの無水DMSO溶液(5mg/mL)を調製した。その後、L-アラニンエチルエステル塩酸塩(アルドリッチ社製)をHAユニットに対して3倍モル等量と、5-(アミノメチル)フルオロセインの塩酸塩を0.15倍モル等量添加した。DMT-MMをHAユニットに対して6倍モル等量を添加し、室温で一晩撹拌した。反応溶液は、DMSO、0.3M NaCl水溶液、超純水の順で透析(スペクトラポア4、分画分子量(MWCO):12k~14kDa)した。得られた透析液に2N NaOHを添加しpH12.5以上として、1時間攪拌してエチルエステルを加水分解し、カルボキシの脱保護を行った。その後、2N HClを用いて中和を行い、さらに透析を行った後、凍結乾燥してHA-Ala/FLを黄色固体として得た。
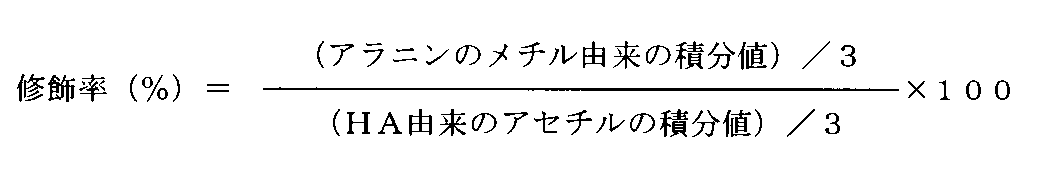
L-アラニンエチルエステル塩酸塩の代わりにL-セリンエチルエステル塩酸塩(アルドリッチ社製)を用いたこと以外は、実施例1-4-1と同様の方法で行い、HA-Ser/FLを黄色固体として得た。また、カルボキシを脱保護する前の透析溶液の一部を採り凍結乾燥して修飾率算出用サンプルとした(HA-Ser-OEt/FL)。
L-アラニンエチルエステル塩酸塩の代わりにL-グルタミン酸ジエチルエステル塩酸塩(東京化成工業株式会社製)を用いたこと以外は、実施例1-4-1と同様の方法で行い、HA-Glu/FLを黄色固体として得た。
実施例1-2で合成したHA-TBAの無水DMSO溶液(5mg/mL)もしくは、実施例1-3で合成したHA-FL/TBAを超純水/DMSO混液(1:3)溶液(約4mg/mL)を出発原料とした。グリシンエチルエステル塩酸塩(和光純薬株式会社製)をHAユニットに対して5倍モル等量添加した。次に、DMT-MMをHAユニットに対して3倍モル等量を添加し、室温で一晩撹拌した。反応溶液は、0.3M NaCl水溶液、超純水の順で透析(スペクトラポア4、分画分子量(MWCO):12k~14kDa)した。得られた透析液に2N NaOHを添加しpH12.5以上として、1時間攪拌してエステルを加水分解し、カルボキシの脱保護を行った。その後、2N HClを用いて中和を行い、さらに透析を行った後、凍結乾燥して、HA-Glyを白色個体、HA-Gly/FLを黄色固体として得た。また、カルボキシを脱保護する前の透析溶液の一部を採り凍結乾燥して修飾率算出用サンプルとした(HA-Gly-OEt/FL)。
グリシンエチルエステル塩酸塩の代わりにL-バリンエチルエステル塩酸塩(渡辺化学工業株式会社製)を用いたこと以外は、実施例1-4-4と同様の方法で行い、HA-Valを白色個体、HA-Val/FLを黄色固体として得た。
グリシンエチルエステル塩酸塩の代わりにL-ロイシンエチルエステル塩酸塩(東京化成工業株式会社製)を用いたこと以外は、実施例1-4-4と同様の方法で行い、HA-Leuを白色個体、HA-Leu/FLを黄色固体として得た。
溶媒として無水DMSOを用いたことと、グリシンエチルエステル塩酸塩の代わりにL-イソロイシンメチルエステル塩酸塩(渡辺化学工業株式会社製)を用いたこと以外は、実施例1-4-4と同様の方法で行い、HA-Ileを白色個体、HA-Ile/FLを黄色固体として得た。
グリシンエチルエステル塩酸塩の代わりにL-スレオニンメチルエステルの塩酸塩(Bachem社製)を用いたこと以外は、実施例1-4-4と同様の方法で行い、HA-Thrを白色個体、HA-Thr/FLを黄色固体として得た。
グリシンエチルエステル塩酸塩の代わりにL-アスパラギン酸ジエチルエステル塩酸塩(渡辺化学工業株式会社製)を用いたこと以外は、実施例1-4-4と同様の方法で行い、HA-Aspを白色固体、HA-Asp/FLを黄色固体として得た。
グリシンエチルエステル塩酸塩の代わりにcACHCAエチルエステルの塩酸塩(Acros社製)を用い、カルボキシの脱保護を5N NaOHを用いてpH13.2で行ったこと以外は、実施例1-4-4と同様の方法で行い、HA-cACHCA/FLを黄色固体として得た。
グリシンエチルエステル塩酸塩の代わりにtACHCA-OEtの塩酸塩(Acros社製)を用い、カルボキシの脱保護を行わなかったこと以外は実施例1-4-4と同様の方法で行い、HA-tACHCA-OEt/FLを黄色固体として得た。
グリシンエチルエステル塩酸塩の代わりにAibエチルエステルの塩酸塩(渡辺化学工業株式会社製)を用い、カルボキシの脱保護を5N NaOHを用いてpH13.2で行ったこと以外は、実施例1-4-4と同様の方法で行い、HA-Aib/FLを黄色固体として得た。
グリシンエチルエステル塩酸塩の代わりにACBuCA-OEtの塩酸塩(シグマーアルドリッチ社製)を用い、カルボキシの脱保護を行わなかったこと以外は、実施例1-4-4と同様の方法で行い、HA-ACBuCA-OEt/FLを黄色固体として得た。
HA-Asnは以下のように合成した。実施例1-2で合成した、HA-Na(99kDa)を出発原料とするHA-TBAの無水DMSO溶液(5mg/mL)に、L-アスパラギンメチルエステル塩酸塩(Bachem社製)をHAユニットに対して5倍モル等量添加した。次に、DMT-MMをHAユニットに対して3~6倍モル等量添加し、室温で一晩撹拌した。反応溶液はジエチルエーテルにより再沈殿を行い、沈殿を超純水に溶解させた後、5N NaOH溶液を添加してpH12.5以上として、1時間攪拌してエステルを加水分解し、カルボキシの脱保護を行った。その後、5N HCl溶液を用いて中和を行い、さらに0.3M NaCl水溶液、蒸留水、超純水の順で透析精製し、凍結乾燥して、HA-Asnを白色固体として得た。 HA-Asn/Rhは以下のように合成した。実施例1-2で合成した、HA-Na(99kDa)を出発原料とするHA-TBAの無水DMSO溶液(5mg/mL)に、1-(9-フルオレニルメチルオキシカルボニル-アミノ)-3、6-ジオキサ-8-オクタンアミン塩酸塩(Fmoc-EDOBEA、Iris Biotech GmbH社製)をHAユニットに対して0.1倍モル等量添加した。次に、DMT-MMをHAユニットに対して0.2倍モル等量を添加し、室温で6時間撹拌した。続いて、L-アスパラギンメチルエステル塩酸塩およびDMT-MMを順に、HAユニットに対してそれぞれ5倍モル等量、3~6倍モル等量添加し、室温で一晩撹拌した。反応溶液は、DMSOに対して透析精製を行った。得られた透析液にピペリジン(和光純薬株式会社製)を20%の濃度になるよう添加し2時間攪拌してFmoc基の除去を行った。その後、DMSO、0.3M NaCl水溶液、蒸留水の順で透析精製した。得られた透析液に5N NaOH溶液を添加しpH12.5以上として、1時間攪拌してエステルを加水分解し、カルボキシの脱保護を行った。その後、5N HCl溶液を用いて中和を行い、さらに蒸留水、超純水の順で透析精製し、凍結乾燥して、HA-Asn/EDOBEAを得た。
L-アスパラギンメチルエステル塩酸塩の代わりにL-アラニンアミド塩酸塩(東京化成工業株式会社製)を用いたこと、再沈殿およびカルボキシの脱保護に関わる操作を行わなかったこと以外は、実施例1-4-14と同様の方法で行い、HA-Ala-NH2を白色固体、HA-Ala-NH2/Rhを赤色固体として得た。
L-アラニンアミド塩酸塩の代わりにL-バリンアミド塩酸塩(東京化成工業株式会社製)を用いたこと以外は、実施例1-4-15と同様の方法で行い、HA-Val-NH2を白色固体、HA-Val-NH2/Rhを赤色固体として得た。
L-アラニンアミド塩酸塩の代わりにL-アスパラギンアミド塩酸塩(国産化学株式会社製)を用いたこと以外は、実施例1-4-15と同様の方法で行い、HA-Asn-NH2を白色固体、HA-Asn-NH2/Rhを赤色固体として得た。
HA-Meは以下のように合成した。実施例1-2で合成した、HA-Na(99kDa)を出発原料とするHA-TBAの無水DMSO溶液(5mg/mL)を調製した。その後、HAユニット/BOP(和光純薬株式会社製)/メチルアミン(40%メタノール溶液、東京化成工業株式会社製)=1/3/50(mol/mol/mol)の等量比でメチルアミン、BOPの順で添加し、室温で一晩攪拌した。反応溶液は、0.3M NaCl水溶液、超純水の順で透析(スペクトラポア4、分画分子量(MWCO):12k~14kDa)した後、凍結乾燥してHA-Meを白色個体として得た。
HA-Prは以下のように合成した。実施例1-2で合成した、HA-Na(99kDa)を出発原料とするHA-TBAの無水DMSO溶液(5mg/mL)を調製した。その後、HAユニット/BOP/プロピルアミン(シグマ-アルドリッチ社製)=1/3/50(mol/mol/mol)の等量比でプロピルアミン、BOPの順で添加し、室温で一晩攪拌した。反応溶液は、0.3M NaCl水溶液、超純水の順で透析(スペクトラポア4、分画分子量(MWCO):12k~14kDa)した後、凍結乾燥してHA-Prを白色個体として得た。

(実施例1-5-1)1-アミノメチル-1-シクロヘキサン酸(AMCHCA)により修飾したHA誘導体(HA-AMCHCA)の合成
実施例1-2で調製したHA-Na(25kDaおよび99kDa)を出発原料とするHA-TBAの無水DMSO溶液(10mg/mL)を調製した後、1-アミノメチル-1-シクロヘキサン酸メチルエステル塩酸塩(AMCHCA-OMe・HCl、Bionet Research社製)をHA-TBAユニットに対して表2に示す比率で各溶液に添加した。次に、DMT-MMをHA-TBAユニットに対して表2に示す比率で加え、室温で一晩攪拌した。反応溶液は大過剰の0.3M NaCl水溶液に対して透析精製し(スペクトラポア4、分画分子量(MWCO):12k~14kDa)、得られた透析液を5N NaOH水溶液を加えてpHを13.2にして1時間室温で攪拌し、さらに5N HClを加えて中和を行った。次に蒸留水、超純水の順で透析精製し(スペクトラポア4、MWCO:12k~14kDa)、得られた透析液を凍結乾燥して目的物(HA-AMCHCA)を白色固体として得た。
AMCHCA-OMe・HClの代わりにシス-4-アミノ-1-シクロヘキシルカルボン酸メチルエステル塩酸塩(pcACHCA-OMe・HCl、Iris Biotech社製)を用いたこと以外は実施例1-5-1と同様の方法で調製を行い、目的物(HA-pcACHCA)を白色固体として得た。試薬の添加量は表2に示す。
AMCHCA-OMe・HClの代わりにL-2-ナフチルアラニンベンジルエステルパラトシレート塩(Nal-Obzl・p-Ts、Bachem製)を用いたこと以外は実施例1-5-1と同様の方法で調製を行い、目的物(HA-Nal)を白色固体として得た。試薬の添加量は表2に示す。
AMCHCA-OMe・HClの代わりに2-アミノ-4-フェニルブタン酸エチルエステル塩酸塩(APBA-OEt・HCl、シグマ-アルドリッチ社製)を用いたこと以外は実施例1-5-1と同様の方法で調製を行い、目的物(HA-APBA)を白色固体として得た。試薬の添加量は表2に示す。
AMCHCA-OMe・HClの代わりにシクロヘキシル-L-アラニンメチルエステル塩酸塩(Cha-OMe・HCl、渡辺化学工業株式会社製)を用いたこと以外は実施例1-5-1と同様の方法で調製を行い、目的物(HA-Cha)を白色固体として得た。試薬の添加量は表2に示す。
AMCHCA-OMe・HClの代わりに4-アミノメチル安息香酸メチルエステル塩酸塩(AMBA-OMe・HCl、シグマ-アルドリッチ社製)を用いたこと以外は実施例1-5-1と同様の方法で調製を行い、目的物(HA-AMBA)を白色固体として得た。試薬の添加量は表2に示す。このHA-AMBAの1H-NMRスペクトルを図4-6に示す。グルコサミンのアセチル由来のピーク(-COCH3、2.0ppm;3H)の積分値と、AMBAに属するフェニル由来のピーク(-CH2C6H4COOH、7.4、7.9ppm;4H)の積分値より、実施例1-4-1と同様にしてHAユニットにおけるAMBAによる修飾率を算出した(表2)。
AMCHCA-OMe・HClの代わりに3-アミノメチル安息香酸メチルエステル塩酸塩(AMBA-OMe・HCl、Fluorochem社製)を用いたこと以外は実施例1-5-1と同様の方法で調製を行い、目的物(HA-3AMBA)を白色固体として得た。試薬の添加量は表2に示す。
AMCHCA-OMe・HClの代わりに3-アミノ-2-フェニルプロピオン酸エチルエステル塩酸塩(APhPA-OEt・HCl、Tyger社製)を用いたこと以外は実施例1-5-1と同様の方法で調製を行い、目的物(HA-APhPA)を白色固体として得た。試薬の添加量は表2に示す。
AMCHCA-OMe・HClの代わりに4-(2-アミノエチル)安息香酸メチルエステル塩酸塩(AEBA-OMe・HCl、Enamine社製)を用いたこと以外は実施例1-5-1と同様の方法で調製を行い、目的物(HA-AEBA)を白色固体として得た。試薬の添加量は表2に示す。
AMCHCA-OMe・HClの代わりに4-アミノメチル-3-クロロ安息香酸メチルエステル塩酸塩(AMClBA-OMe・HCl、Anichem社製)を用いたこと以外は実施例1-5-1と同様の方法で調製を行い、目的物(HA-AMClBA)を白色固体として得た。試薬の添加量は表2に示す。
AMCHCA-OMe・HClの代わりに5-アミノメチルサリチル酸メチルエステル塩酸塩(AMSA-OMe・HCl、Oakwood社製)を用いたこと以外は実施例1-5-1と同様の方法で調製を行い、目的物(HA-AMSA)を白色固体として得た。試薬の添加量は表2に示す。
AMCHCA-OMe・HClの代わりにトランス-4-アミノメチルシクロヘキサン酸メチルエステル塩酸塩(4AMCHCA-OMe・HCl、AK scientific社製)を用いたこと以外は実施例1-5-1と同様の方法で調製を行い、目的物(HA-4AMCHCA)を白色固体として得た。試薬の添加量は表2に示す。
AMCHCA-OMe・HClの代わりにL-シクロヘキシルグリシンメチルエステル塩酸塩(Chg-OMe・HCl、INDOFINE Chemical Company社製)を用いたこと以外は実施例1-5-1と同様の方法で調製を行い、目的物(HA-Chg)を白色固体として得た。試薬の添加量は表2に示す。
AMCHCA-OMe・HClの代わりに(R)-アミノ-(4-ヒドロキシフェニル)酢酸メチルエステル塩酸塩(pHPhg-OMe・HCl、シグマ-アルドリッチ社製)を用いたこと以外は実施例1-5-1と同様の方法で調製を行い、目的物(HA-pHPhg)を白色固体として得た。試薬の添加量は表2に示す。
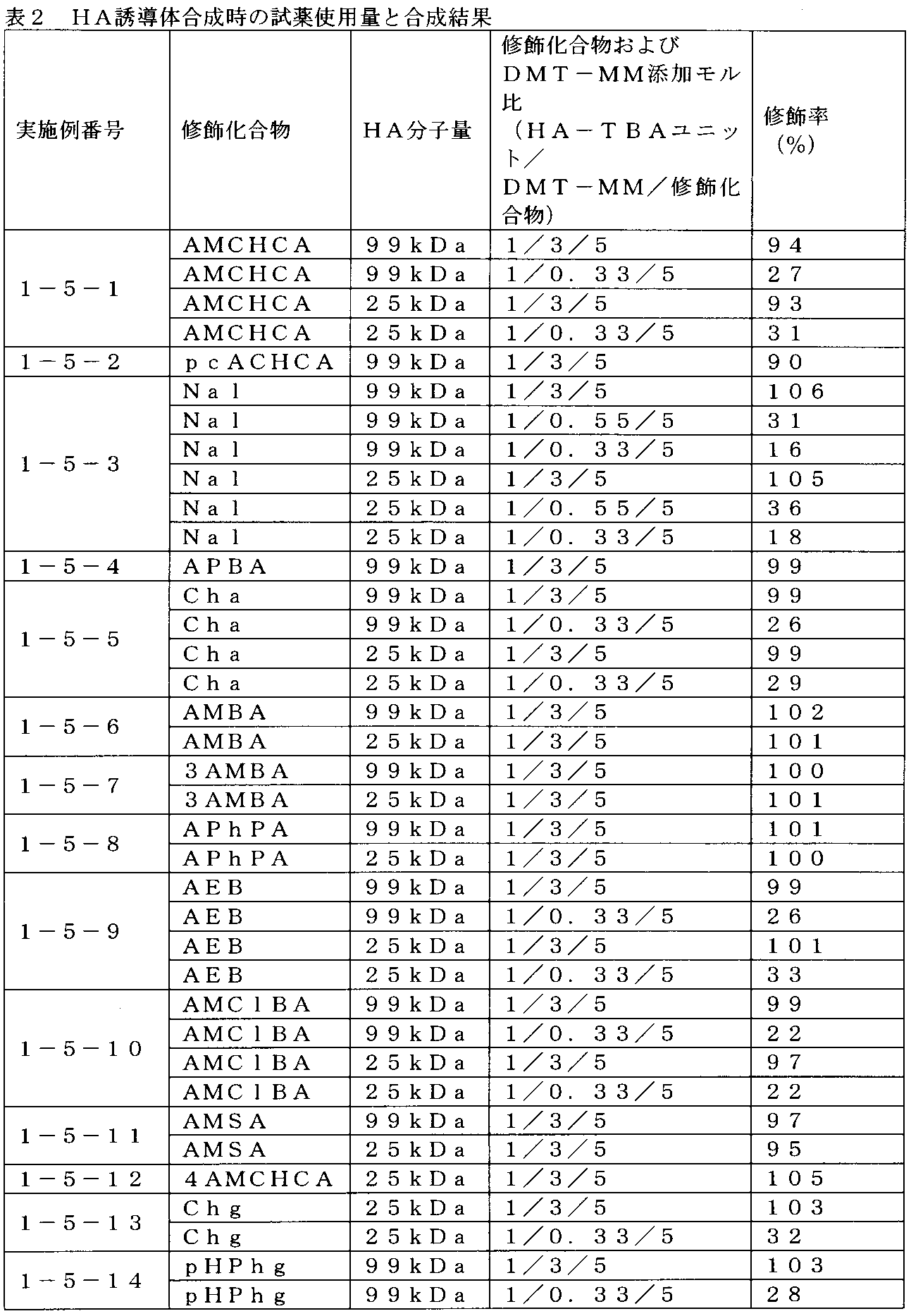
(比較例1-1-1)HAのFLラベル体(HA-FL)の合成
実施例1-2で合成した、HA-Na(99kDa)を出発原料とするHA-TBAの無水DMSO溶液(5mg/mL)を調製した。その後、HAユニット/BOP(和光純薬株株式会社製)/2,2’-(エチレンジオキシ)ビス(エチルアミン)(EDOBEA、シグマ-アルドリッチ社製)=1/0.2/50(mol/mol/mol)の等量比でEDOBEA、BOPの順で添加し、室温で一晩攪拌した。反応溶液は、0.3M NaCl水溶液、超純水の順で透析(スペクトラポア4、分画分子量(MWCO):12k~14kDa)した後、凍結乾燥して低修飾率のHA-EDOBEAを得た。
試薬の添加量比をHAユニット/BOP/EDOBEA=1/2.5/50(mol/mol/mol)としたこと以外は比較例1-1-1と同様に行い、高修飾率のHA-EDOBEAを得た。

L-アラニンエチルエステル塩酸塩の代わりにL-フェニルアラニンエチルエステル塩酸塩(アルドリッチ社製)を用いたこと以外は、実施例1-4-1と同様の方法で行い、HA-Phe/FLを黄色固体として得た。
グリシンエチルエステル塩酸塩の代わりにL-チロシンエチルエステル塩酸塩(和光純薬株式会社製)を用いたこと以外は、実施例1-4-4と同様の方法で行い、HA-Tyr/FLを黄色固体として得た。
グリシンエチルエステル塩酸塩の代わりにMePheメチルエステル塩酸塩(渡辺化学工業社製)を用い、カルボキシの脱保護を5N NaOHを用いてpH13.2で行ったこと以外は、実施例1-4-4と同様の方法で行い、HA-MePhe/FLを黄色固体として得た。
グリシンエチルエステル塩酸塩の代わりにPro-OMeの塩酸塩(シグマ-アルドリッチ社製)を用い、カルボキシの脱保護を行わなかったこと以外は、実施例1-4-4と同様の方法で行い、HA-Pro-OMe/FLを黄色固体として得た。
L-アラニンアミド塩酸塩の代わりにグリシンアミド塩酸塩(渡辺化学工業株式会社製)を用いたこと以外は、実施例1-4-15と同様の方法で行い、HA-Gly-NH2を白色固体、HA-Gly-NH2/Rhを赤色固体として得た。
L-アラニンアミド塩酸塩の代わりにL-セリンアミド塩酸塩(渡辺化学工業株式会社製)を用いたこと以外は、実施例1-4-15と同様の方法で行い、HA-Ser-NH2を白色固体、HA-Ser-NH2/Rhを赤色固体として得た。
L-アラニンアミド塩酸塩の代わりにL-ロイシンアミド塩酸塩(東京化成工業株式会社製)を用いたこと以外は、実施例1-4-15と同様の方法で行い、HA-Leu-NH2を白色固体、HA-Leu-NH2/Rhを赤色固体として得た。
L-アラニンアミド塩酸塩の代わりにL-イソロイシンアミド塩酸塩(東京化成工業株式会社製)を用いたこと以外は、実施例1-4-15と同様の方法で行い、HA-Ile-NH2を白色固体、HA-Ile-NH2/Rhを赤色固体として得た。
L-アラニンアミド塩酸塩の代わりにL-スレオニンアミド塩酸塩(渡辺化学工業株式会社製)を用いたこと以外は、実施例1-4-15と同様の方法で行い、HA-Thr-NH2を白色固体、HA-Thr-NH2/Rhを赤色固体として得た。
L-アラニンアミド塩酸塩の代わりにL-グルタミンアミド塩酸塩(国産化学株式会社製)を用いたこと以外は、実施例1-4-15と同様の方法で行い、HA-Gln-NH2を白色固体、HA-Gln-NH2/Rhを赤色固体として得た。

(比較例1-2-1)L-ノルロイシン(Nle)により修飾したHA誘導体(HA-Nle)の合成
AMCHCA-OMe・HClの代わりにL-ノルロイシンメチルエステル塩酸塩(Nle-OMe・HCl、Bachem社製)を用いたこと以外は実施例1-5-1と同様の方法で調製を行い、目的物(HA-Nle)を白色固体として得た。試薬の添加量は表5に示す。
AMCHCA-OMe・HClの代わりにL-ターシャリーロイシンメチルエステル塩酸塩(tLeu-OMe・HCl、Fluka社製)を用いたこと以外は、実施例1-5-1と同様の方法で行い、目的物(HA-tLeu)を白色固体として得た。試薬の添加量は表5に示す。
AMCHCA-OMe・HClの代わりにパラフルオロフェニルアラニンエチルエステル塩酸塩(pFPhe-OEt・HCl、Bachem社製)を用い、カルボキシの脱保護を5N NaOHを用いてpH13.2で行ったこと以外は、実施例1-5-1と同様の方法で行い、目的物(HA-pFPhe)を白色固体として得た。
AMCHCA-OMe・HClの代わりに(s)-(+)-2-フェニルグリシンメチルエステル塩酸塩(Phg-OMe・HCl、シグマ-アルドリッチ社製)を用いたこと以外は、実施例1-5-1と同様の方法で行い、目的物(HA-Phg)を白色固体として得た。

PEG(MEPA-30T、日油株式会社製)を超純水で10mg/mlとなるように溶解させた後、100mMのリン酸緩衝溶液(pH7.4)で2倍希釈して5mg/ml溶液を調製した。この溶液にPEG末端に対して15倍モル等量のNHS-フルオレセイン(NHS-FL、PIERCE社製)をDMSO溶液として加えて室温で終夜攪拌した。反応溶液は、ジエチルエーテルにより再沈殿を行って、沈殿をろ過により回収し、再度超純水に溶解させた後に大過剰の超純水で透析(スペクトラポア4、分画分子量(MWCO):12k~14kDa)し、凍結乾燥して目的物質(PEG-FL)を黄色固体として得た。
一定条件でのインキュベート後のHyaluronidase SD(Hyase、生化学工業株式会社製)に対する耐性を確認した。


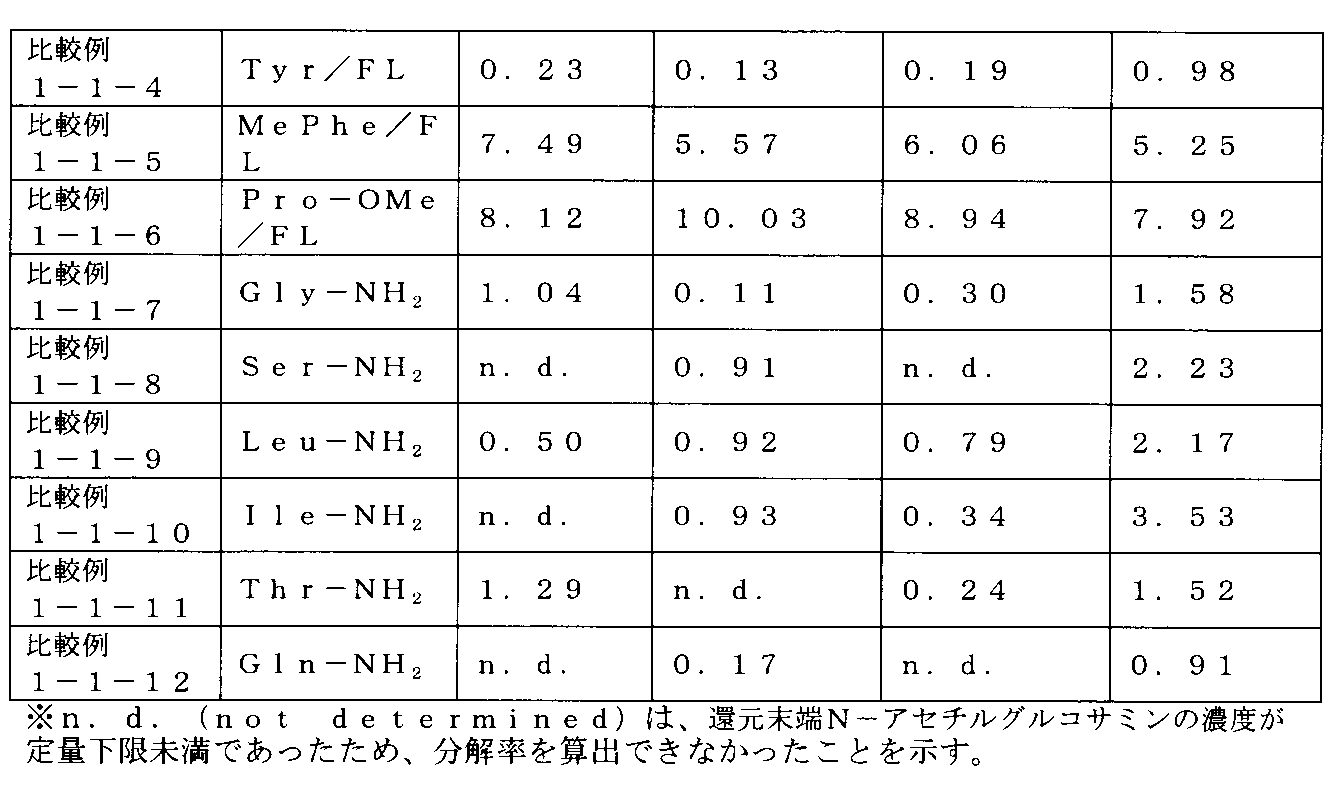
(実施例3-1)HA誘導体投与ラット生体サンプル
実施例1-4および比較例1-1で得られた化合物を20mg/kgの用量で、比較例1-3で得られた化合物を10mg/kgの用量でラット静脈内に単回投与し、投与後5分、2、7、24、48、72、168、240および336時間でヘパリンナトリウム処理したシリンジを用いて頸静脈採血し、遠心分離により血漿を得た。この血漿サンプルは測定まで-20℃以下で凍結保存した。また、投与後0~24時間、24~48時間、48~72時間、72~96時間、168~192時間、240~264時間、および336~360時間で代謝ケージを用いて採尿し、尿量を量り、その一部を測定まで-20℃以下で凍結保存した。
(実施例3-2-1)測定試料調製
血漿サンプルを融解後、攪拌、遠心分離し、上清を採取した。バッファー(FLラベル体:リン酸バッファー(pH8.0,I=0.15)、Rhラベル体:PBS)を用いて希釈を行った。投与液をバッファーとラットブランク血漿の混合液(3:1)で希釈し、検量線試料を調製した。
実施例3-2-1で調製したサンプルについて、プレートリーダー(SPECTRAmax GEMINI、Molecular Devices社製)を用いて蛍光強度を測定した(FLラベル体:励起波長:495nm/蛍光波長:520nm、Rhラベル体:励起波長:552nm/蛍光波長:595nm)。検量線試料の測定値から、化合物ごとに検量線を作成し、各個体の各時点の血漿中濃度を算出し、平均値を図8に、血漿中濃度推移のグラフを図9に示す。
薬物動態学的パラメーターを、WinNonlin Ver 4.0.1/Ver 6.1(Pharsight社製)を用いて算出した。実施例3-2-2で求めた各群の平均データについてモデル非依存的解析を行い、クリアランス(CL)を算出した(表7)。この結果、実用的な血中滞留性を有するPEGと比較して、実施例1-4-1~1-4-19のHA誘導体はいずれもCLの値が小さくなり、実用的な血中滞留性を有することが示された。また、比較例の一部のHA誘導体のCLはPEGよりも大きな値を示しており、修飾する化合物の種類によって様々な血中滞留性を示すHA誘導体が合成可能であることを明らかにした。

(実施例3-3-1)測定試料調製
尿サンプルを融解後、攪拌し、0.22μmのフィルターでろ過し、ろ液を採取した。バッファー(FLラベル体:リン酸バッファー(pH6.5,I=0.15)、Rhラベル体:PBS)を用いて希釈を行った。投与液をバッファーとラットブランク尿の混合液(1:1)で希釈し、標準試料を調製した。
実施例3-3-1で調製したサンプルをサイズ排除クロマトグラフィーに供し、分子量分布の変化を観察した。サイズ排除クロマトグラフィー分析は、Alliance HPLCシステム(日本ウォーターズ株式会社製)を用いて行った。分析条件を以下に示す。
サイズ排除クロマトグラフィー分析条件
分析カラム:TSKgel G5000PWXL(東ソー株式会社)
カラム温度:25℃
移動相:リン酸バッファー(pH6.5,I=0.15)(FLラベル体)
PBS(Rhラベル体)
流速:0.5mL/min
検出:Ex 495nm/Em 520nm(FLラベル体)
Ex 552nm/Em 595nm(Rhラベル体)
(実施例4-1)蛍光物質および消光物質内包リポソームの調製
8-ヒドロキシピレン-1,3,6-トリスルホン酸トリナトリウム塩(Pyramine)(SIGMA-ALDRICH)を蒸留水に60μmol/mLで溶解させ、またp-キシレン-ビス(N-ピリジニウムブロミド)(DPX)(SIGMA-ALDRICH)を蒸留水に85.7μmol/mLで溶解させた後、溶液を等量で混合させ、Pyramine/DPX水溶液を調製した。
実施例1-5および比較例1-2で得られたHA誘導体を、HA-Naとして10mg/mLとなるように生理食塩水に溶解させ、生理食塩水で順次希釈し、10、5、1、0.5、0.1、0.05、0.01mg/mLとなるように調整した。また、陽性対象としてTritonXの10mg/mL生理食塩水溶液の調製を行った。陰性対象としては生理食塩水を用いた。さらに、膜障害性を有する陽性対照物質としてポリ-L-リジン臭化水素塩(PLL、MW15kDa~30kDa、和光純薬株式会社製)を選択し、10mg/mLとなるように生理食塩水に溶解させた。
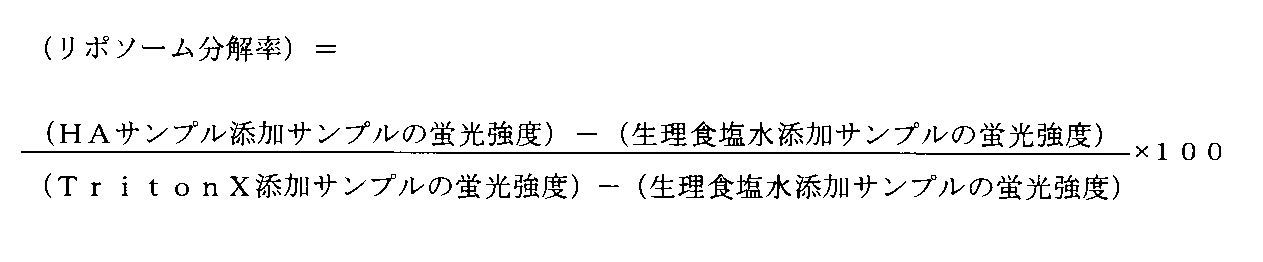
(実施例5-1)HA-AlaのTBA塩(HA-Ala-TBA)の合成
実施例1-2で合成した、HA-Na(99kDa)を出発原料とするHA-TBAの無水DMSO溶液(5mg/mL)を調製した。その後、L-アラニンエチルエステル塩酸塩をHAユニットに対して3倍モル等量添加した。次に、DMT-MMをHAユニットに対して3倍モル等量添加し、室温で一晩撹拌した。反応溶液は、0.3M NaCl水溶液、蒸留水の順で透析(スペクトラポア4、分画分子量(MWCO):12k~14kDa)した。得られた透析液に5N NaOH溶液を添加しpH12.5以上として、1時間攪拌してエチルエステルを加水分解し、カルボキシの脱保護を行った。その後、5N HCl溶液を用いて中和を行い、蒸留水、超純水でさらに透析を行った。得られた透析液に、実施例1-1でTBA塩化したカチオン交換樹脂の懸濁液をHAユニットに対し樹脂のイオン交換能換算で5倍モル等量添加した。15分間撹拌した後、0.45μmのフィルターを用いて濾過を行い、濾液を凍結乾燥し、HA-Ala-TBAを白色固体として得た。生成物の一部をとり、0.3M NaCl水溶液、蒸留水の順で透析を行い、凍結乾燥して、アラニン修飾率算出用サンプルとした。
実施例x-1で合成したHA-Ala-TBAの無水DMSO溶液(5mg/mL)に、EDOBEA、BOPの順で、HAユニットに対してそれぞれ50倍モル等量、0.15倍モル等量添加し、室温で6時間攪拌した。反応溶液は0.3M NaCl水溶液、蒸留水の順で透析精製を行い(スペクトラポア4、分画分子量(MWCO):12k~14kDa)、凍結乾燥して、HA-Ala-EDOBEAを白色固体として得た。
実施例5-2で合成したHA-Ala-EDOBEAの水溶液(10mg/mL)を100mMリン酸緩衝液(pH7.4)で2倍希釈し、NHS-ローダミンをHAユニットに対して0.07倍モル等量を5mg/mL DMSO溶液で添加し、室温で2時間攪拌した。反応溶液を超純水で平衡化した脱塩カラム(PD-10、GEヘルスケア社製)に供して精製を行い、HA画分を凍結乾燥してHA-Ala-EDOBEA-Rhを赤色固体として得た。
実施例5-3で合成したHA-Ala-EDOBEA-Rhの水溶液(20mg/mL)を出発原料とした。水溶液を100mMリン酸緩衝液(pH7.4)で2倍希釈し、NHS-ブロモ酢酸(NHS-BA、シグマ-アルドリッチ社製)をHAユニットに対して0.5倍モル等量を50mg/mLアセトニトリル溶液で添加し、室温で2時間攪拌した後、HAユニットに対して20倍モル等量の無水酢酸を添加してさらに1時間攪拌することにより、余分なEDOBEAの末端アミノをアセチル化した。反応溶液を超純水で平衡化した脱塩カラム(PD-10)に供して精製を行い、分取したHA画分を遠心限外濾過(マクロセップ、MWCO3000、ポールライフサイエンス社製)で約2倍に濃縮してHA-Ala-EDOBEA-BA/Rh/Acを水溶液として得た。水溶液は次の使用まで-20℃で凍結して保存し、一部の一定容量を凍結乾燥後に重量測定して、溶液中の固形分濃度を算出した。
実施例5-4で合成したHA-Ala-EDOBEA-BA/Rh/Acの水溶液を出発原料とした。PTHアナログはヒトPTH(1-34)のC末端にシステインを導入して得たPTH-Cys(SVSEIQLMHNLGKHLNSMERVEWLRKKLQDVHNFC、American Peptide Company社製)を用いた。HA-Ala-EDOBEA-BA/Rh/Ac溶液に3/10容量のアセトニトリルおよび1/10容量の1Mリン酸緩衝液(pH7.4)を加えた。PTH-Cysを、10mMクエン酸緩衝液(pH4.5)に5mg/mLの濃度で溶解後、HAユニットに対して0.03倍モル等量をHA-Ala-EDOBEA-BA/Rh/Ac溶液に添加し37℃で終夜インキュベートした。HAユニットに対して1倍モル等量のシステイン塩酸塩一水和物(和光純薬(株)製)を20mg/mL水溶液で加え、更に37℃で3時間インキュベートした後に1N水酸化ナトリウム水溶液でpH4.5とした。なお、重量あたりのHAユニット含量を1.5μmol/mgとしてHAユニットの計算に使用した。
(実施例6-1)HA誘導体投与ラット生体サンプル
実施例5-5で得られた化合物を10mg/kgの用量でラット静脈内に単回投与し、投与後5分、2、7、24、48、72、168、240および312時間でヘパリンナトリウム処理したシリンジを用いて頸静脈採血し、遠心分離により血漿を得た。この血漿サンプルは測定まで-20℃以下で凍結保存した。また、投与後0~24時間、24~48時間、48~72時間、72~96時間、144~168時間、216~240時間、および288~312時間で代謝ケージを用いて採尿し、尿量を量り、その一部を測定まで-20℃以下で凍結保存した。
(実施例6-2-1)測定試料調製
実施例3-2-1に記載の方法で、測定用の試料を調製した。
実施例6-2-1で調製したサンプルを用いて、実施例3-2-2に記載の方法で各個体の各時点の血漿中濃度を算出した。平均値を図13に、血漿中濃度推移のグラフを図14に示す。
実施例6-2-2で求めた平均データについて、実施例3-2-3に記載の方法でクリアランス(CL)を算出したところ、1.0であった。生分解性を有するヒアルロン酸誘導体の薬物結合体においても実用的な血中滞留性が示された。
(実施例6-3-1)測定試料調製
実施例3-3-1に記載の方法で、測定用の試料を調製した。
実施例6-3-1で調製したサンプルを実施例3-3-2に記載の方法で分析した結果を図15に示す。左に各時点のクロマトグラムを同じレンジで示し、右に各時点のクロマトグラムを最強ピークでノーマライズした図を示す。
Claims (11)
- 式(I):

R8は、水素原子、ホルミルまたはC1-6アルキルカルボニルであり;
Xは、-NHCH3、-NH(CH2)2CH3、-NRx-A-B-COORyまたは-NRx-CHRc-CONRyaRybで表される基であり;
RxおよびRyは、それぞれ独立に、水素原子およびC1-6アルキルから選択され;
Aは、-CRaRb-、C3-8シクロアルキレン、2-シクロヘキシルエタン-1,1-ジイル、2-(2-ナフチル)エタン-1,1-ジイル、3-フェニルプロパン-1,1-ジイル、シクロヘキシルメタン-1,1-ジイルおよび4-ヒドロキシフェニルメタン-1,1-ジイルから選択され、Bは直接結合であり;または
Aは-CH2-または-CH2-CH2-であり、Bはフェニレン(ここでフェニレンは、ヒドロキシおよびハロゲン原子から選択される1以上の基で置換されていてもよい)、C3-8シクロアルキレンおよびフェニルメタン-1,1-ジイルから選択され;
Raは、水素原子およびC1-6アルキルから選択され;
Rbは、水素原子およびC1-6アルキル(ここでC1-6アルキルは、ヒドロキシ、カルボキシおよびカルバモイルから選択される1以上の基で置換されていてもよい)から選択され;
Rcは、カルバモイルで置換されていてもよいC1-6アルキルを表し;
RyaおよびRybは、それぞれ独立して、水素原子、C1-6アルキル、ホルミルおよびC1-6アルキルカルボニルから選択される]
で表される二糖単位を含み、前記式(I)のAが-CRaRb-またはC3-8シクロアルキレンの場合、存在する二糖単位に対する式(I)の二糖単位の割合は70%以上であるヒアルロン酸誘導体。 - Xが-NHCH3、-NH(CH2)2CH3または-NRx-CHRc-CONRyaRybであるか、あるいは、Aが-CRaRb-またはC3-8シクロアルキレンである、請求項1に記載のヒアルロン酸誘導体。
- AがC3-8シクロアルキレンであり、Bが直接結合である;
Aが-CH2-または-CH2-CH2-であり、Bがフェニレン(ここでフェニレンは、ヒドロキシおよびハロゲン原子から選択される1以上の基で置換されていてもよい)、C3-8シクロアルキレンおよびフェニルメタン-1,1-ジイルから選択される;または
Aが2-シクロヘキシルエタン-1,1-ジイル、2-(2-ナフチル)エタン-1,1-ジイル、3-フェニルプロパン-1,1-ジイル、シクロヘキシルメタン-1,1-ジイルおよび4-ヒドロキシフェニルメタン-1,1-ジイルから選択され、Bが直接結合である、請求項1に記載のヒアルロン酸誘導体。 - Aが-CH2-であり、Bがシクロヘキサン-1,1-ジイル、ベンゼン-1,4-ジイル、ベンゼン-1,3-ジイル、2-クロロベンゼン-1,4-ジイルおよびフェニルメタン-1,1-ジイルから選択され;
Aが-CH2CH2-であり、Bがベンゼン-1,4-ジイルであり;
または、
Aが2-シクロヘキシルエタン-1,1-ジイル、2-(2-ナフチル)エタン-1,1-ジイルおよび3-フェニルプロパン-1,1-ジイルから選択され;Bが直接結合である、請求項1または3に記載のヒアルロン酸誘導体。 - 式(II):
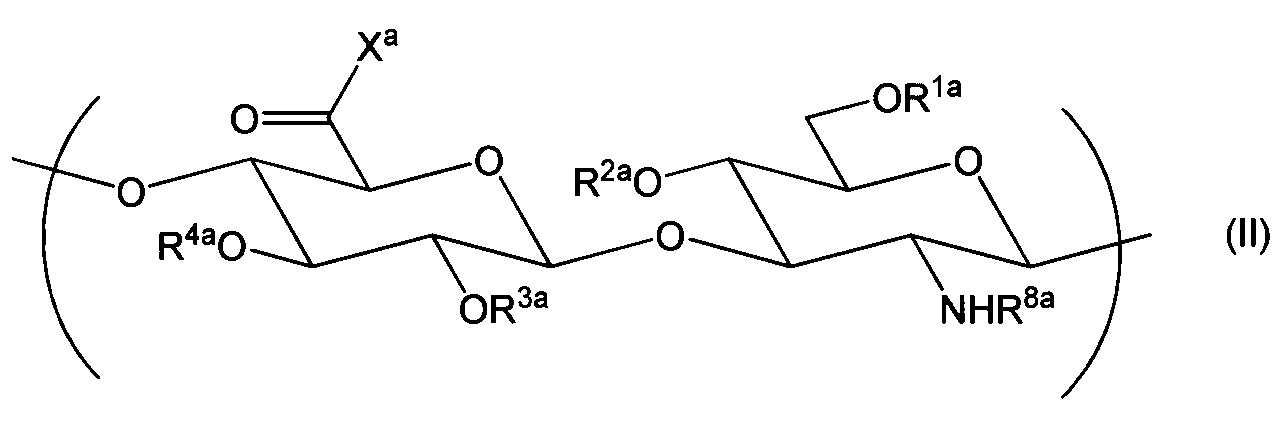
R8aは、水素原子、ホルミルまたはC1-6アルキルカルボニルであり;
Xaは、ヒドロキシおよび-O-Q+、から選択され;ここで、Q+は、カウンターカチオンを表す]
で表される二糖単位をさらに含む、請求項1~4のいずれか1項に記載のヒアルロン酸誘導体。 - 式(III):

R8bは、水素原子、ホルミルまたはC1-6アルキルカルボニルであり;
Xbは、-NRe-Yb-Rdで表される基であり;ここで、
Reは、水素原子またはC1-6アルキルであり;
Rdは、水素原子、C1-6アルキル、-CO-C(R7)=CH2、または-CO-G4-Xcであり;
R7は、水素原子またはメチルであり;
G4は、フェニレン、C3-8シクロアルキレン、または-G5-(C1-10アルキレン)-G6-から選択され、ここでC1-10アルキレン部分は、1~3のフェニレンまたはC3-8シクロアルキレンが挿入されていてもよく;
G5およびG6は、それぞれ独立に、直接結合、フェニレン、またはC3-8シクロアルキレンから選択され;
Xcは、メルカプト、ハロゲン原子または式:
Ybは、-CH2-(CHR5)l-2-CH2-NH-、-CH2-(CHR6)p-2-CH2-O-、-(CH2)j-S-または-(CH2)a-(Y1-(CH2)b)c-G-であり;
l、p、およびjは、それぞれ独立に2~10から選択される整数であり、R5およびR6は、それぞれ独立に、水素原子またはヒドロキシであり;
aは、2~10から選択される整数であり;
bは、それぞれ独立に2~10から選択される整数であり;
cは、1~200から選択される整数であり;
Y1は、酸素原子または-NRn-であり;
Gは、酸素原子、硫黄原子または-NH-であり;
Rnは、水素原子、C1-6アルキル、-CO-(CH2)d-Ro、-(CH2)e-Rpまたは-(CH2)f-(Y2-(CH2)g)h-Rqであり;
gは、それぞれ独立に、2~10から選択される整数であり;
d、e、fおよびhは、それぞれ独立に、2~10から選択される整数であり;
Ro,RpおよびRqは、それぞれ独立に、水素原子、ヒドロキシ、カルボキシまたは-NHRrであり;
Y2は、酸素原子または-NH-であり;
Rrは、水素原子、ホルミルまたはC1-6アルキルカルボニルである]
で表される二糖単位を含む、請求項1~5のいずれか1項に記載のヒアルロン酸誘導体。 - R1a、R2a、R3a、およびR4aの全てを水素原子とし、R8aをアセチルとし、かつ、Xaを-O-Na+とした場合の重量平均分子量が20~120キロダルトンとなる、請求項5に定義した式(II)で表される二糖単位のみから構成されるヒアルロン酸を用いて製造される、請求項1~6のいずれか1項に記載のヒアルロン酸誘導体。
- 前記ヒアルロン酸誘導体に主鎖構造が対応する誘導体化されていないヒアルロン酸の重量平均分子量が20~120キロダルトンであり、ここで誘導体化されていないヒアルロン酸は、請求項5に定義した式(II)においてR1a、R2a、R3aおよびR4aが水素原子であり、R8aがアセチルであり、Xaが-O-Na+である繰り返し単位のみからなるヒアルロン酸である、請求項1~6のいずれか1項に記載のヒアルロン酸誘導体。
- 請求項1~8のいずれか1項に記載のヒアルロン酸誘導体を担体として含む、医薬組成物。
- 請求項1~8のいずれか1項に記載のヒアルロン酸誘導体に、1以上の薬物が結合した、ヒアルロン酸誘導体-薬物結合体。
- 請求項1、2および5~8のいずれか1項に記載のヒアルロン酸誘導体を含む、生分解性の薬物担体。
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DK12752403.1T DK2682409T3 (en) | 2011-03-03 | 2012-03-02 | Hyaluronic acid derivative modified with aminocarboxylic acid |
| EP17152228.7A EP3184553B1 (en) | 2011-03-03 | 2012-03-02 | Derivative of hyaluronic acid modified with amino-carboxylic acid |
| US14/002,919 US9243077B2 (en) | 2011-03-03 | 2012-03-02 | Derivative of hyaluronic acid modified with amino-carboxylic acid |
| SI201231008T SI2682409T1 (sl) | 2011-03-03 | 2012-03-02 | Derivat hialuronske kisline modificiran z amino-karboksilno kislino |
| EP12752403.1A EP2682409B1 (en) | 2011-03-03 | 2012-03-02 | Derivative of hyaluronic acid modified with amino-carboxylic acid |
| ES12752403.1T ES2635087T3 (es) | 2011-03-03 | 2012-03-02 | Derivado de ácido hialurónico modificado con ácido amino-carboxilico |
| PL12752403T PL2682409T3 (pl) | 2011-03-03 | 2012-03-02 | Pochodna kwasu hialuronowego zmodyfikowana kwasem aminokarboksylowym |
| JP2013502420A JP6141180B2 (ja) | 2011-03-03 | 2012-03-02 | アミノ−カルボン酸により修飾されたヒアルロン酸誘導体 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011046749 | 2011-03-03 | ||
| JP2011-046749 | 2011-03-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012118189A1 true WO2012118189A1 (ja) | 2012-09-07 |
Family
ID=46758114
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/055421 WO2012118189A1 (ja) | 2011-03-03 | 2012-03-02 | アミノ-カルボン酸により修飾されたヒアルロン酸誘導体 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US9243077B2 (ja) |
| EP (2) | EP3184553B1 (ja) |
| JP (2) | JP6141180B2 (ja) |
| DK (1) | DK2682409T3 (ja) |
| ES (1) | ES2635087T3 (ja) |
| HU (1) | HUE033169T2 (ja) |
| PL (1) | PL2682409T3 (ja) |
| SI (1) | SI2682409T1 (ja) |
| WO (1) | WO2012118189A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014038641A1 (ja) * | 2012-09-05 | 2014-03-13 | 中外製薬株式会社 | アミノ酸およびステリル基が導入されたヒアルロン酸誘導体 |
| WO2016136707A1 (ja) * | 2015-02-27 | 2016-09-01 | 学校法人常翔学園 | 膜透過性ペプチド鎖を有する多糖誘導体 |
| JP2016529362A (ja) * | 2013-08-29 | 2016-09-23 | ホーリー ストーン バイオテック カンパニー リミテッド | グリコサミノグリカン化合物、その調製方法および使用 |
| JPWO2016159159A1 (ja) * | 2015-03-31 | 2018-01-25 | キユーピー株式会社 | ヒアルロン酸誘導体およびその製造方法、ならびにヒアルロン酸誘導体を含む化粧料、食品組成物および医薬組成物 |
| WO2019098393A1 (ja) * | 2017-11-15 | 2019-05-23 | 中外製薬株式会社 | ポリエチレングリコールにより修飾されたヒアルロン酸誘導体 |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3252080A1 (en) * | 2016-05-31 | 2017-12-06 | Galderma S.A. | Method for preparing acylated crosslinked glycosaminoglycans |
| KR20180120142A (ko) * | 2015-12-29 | 2018-11-05 | 갈데르마 소시에떼아노님 | 탄수화물 가교제 |
| PT3623390T (pt) | 2016-05-31 | 2023-10-27 | Galderma Sa | Reticulador de hidrato de carbono |
| CN110891611B (zh) | 2017-03-22 | 2024-03-29 | 阿森迪斯制药公司 | 水凝胶交联透明质酸前药组合物和方法 |
| CN109206537B (zh) * | 2018-10-10 | 2021-04-09 | 华熙生物科技股份有限公司 | 一种乙酰化透明质酸钠的制备方法及其应用 |
| IL293540A (en) | 2019-12-02 | 2022-08-01 | Galderma Holding SA | High molecular weight aesthetic preparations |
| JP7343058B2 (ja) | 2020-11-12 | 2023-09-12 | 三菱電機株式会社 | 乗客コンベアの欄干 |
Citations (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02273176A (ja) | 1989-04-13 | 1990-11-07 | Seikagaku Kogyo Co Ltd | グリコサミノグリカンで修飾されたスーパーオキシドジスムターゼ及びその製造法 |
| WO1992006714A1 (en) | 1990-10-18 | 1992-04-30 | Shiseido Co., Ltd. | Combination of hyaluronic acid with medicinal ingredient and production thereof |
| WO1992020349A1 (en) | 1991-05-20 | 1992-11-26 | Genzyme Corporation | Water insoluble derivatives of polyanionic polysaccharides |
| JPH0585942A (ja) | 1991-02-26 | 1993-04-06 | Denki Kagaku Kogyo Kk | インターフエロン−ヒアルロン酸及び/又はその塩の結合体 |
| WO1999029839A1 (en) | 1997-12-12 | 1999-06-17 | Expression Genetics, Inc. | Grafted copolymers as gene carriers |
| WO2000001733A1 (en) | 1998-07-06 | 2000-01-13 | Fidia Advanced Biopolymers S.R.L. | Amides of hyaluronic acid and the derivatives thereof and a process for their preparation |
| WO2001005434A2 (en) | 1999-07-20 | 2001-01-25 | Amgen Inc. | Hyaluronic acid-protein conjugates |
| JP2001081103A (ja) | 1999-09-13 | 2001-03-27 | Denki Kagaku Kogyo Kk | ヒアルロン酸結合薬剤 |
| WO2001060412A2 (en) | 2000-02-15 | 2001-08-23 | Genzyme Corporation | Modification of biopolymers for improved drug delivery |
| JP2003518167A (ja) * | 1999-12-22 | 2003-06-03 | ジェンザイム、コーポレーション | ポリアニオン性多糖類の水不溶性誘導体 |
| WO2005023906A1 (ja) * | 2003-09-08 | 2005-03-17 | Chugai Seiyaku Kabushiki Kaisha | ヒアルロン酸修飾物、及びそれを用いた薬物担体 |
| WO2005054301A1 (ja) * | 2003-11-14 | 2005-06-16 | Chugai Seiyaku Kabushiki Kaisha | 架橋多糖微粒子およびその製造方法 |
| WO2005054302A1 (ja) * | 2003-12-05 | 2005-06-16 | Chugai Seiyaku Kabushiki Kaisha | 薬物担体及びその製造方法 |
| WO2006028110A1 (ja) | 2004-09-07 | 2006-03-16 | Chugai Seiyaku Kabushiki Kaisha | 水溶性ヒアルロン酸修飾物の製造方法 |
| WO2006095775A1 (ja) * | 2005-03-08 | 2006-09-14 | Chugai Seiyaku Kabushiki Kaisha | 水溶性ヒアルロン酸修飾物とglp-1アナログの結合体 |
| WO2010053140A1 (ja) * | 2008-11-05 | 2010-05-14 | 国立大学法人 東京医科歯科大学 | ヒアルロン酸誘導体、およびその医薬組成物 |
| WO2011148116A2 (fr) | 2010-05-27 | 2011-12-01 | Laboratoire Idenov | Acide hyaluronique modifie, procede de fabrication et utilisations |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4937270A (en) * | 1987-09-18 | 1990-06-26 | Genzyme Corporation | Water insoluble derivatives of hyaluronic acid |
| JP4018181B2 (ja) * | 1995-11-07 | 2007-12-05 | 生化学工業株式会社 | グリコサミノグリカン誘導体およびその製造法 |
| US6630457B1 (en) * | 1998-09-18 | 2003-10-07 | Orthogene Llc | Functionalized derivatives of hyaluronic acid, formation of hydrogels in situ using same, and methods for making and using same |
| JP5450381B2 (ja) * | 2007-04-30 | 2014-03-26 | プロメティック・バイオサイエンスィズ・インコーポレーテッド | 化合物、そのような化合物を含有する組成物、及びそのような化合物を用いるがん及び自己免疫疾患の治療法 |
| IT1399351B1 (it) | 2009-06-16 | 2013-04-16 | Fidia Farmaceutici | Procedimento per la sintesi di coniugati di glicosamminoglicani (gag) con molecole biologicamente attive, coniugati polimerici e usi relativi |
-
2012
- 2012-03-02 WO PCT/JP2012/055421 patent/WO2012118189A1/ja active Application Filing
- 2012-03-02 JP JP2013502420A patent/JP6141180B2/ja active Active
- 2012-03-02 HU HUE12752403A patent/HUE033169T2/en unknown
- 2012-03-02 EP EP17152228.7A patent/EP3184553B1/en active Active
- 2012-03-02 US US14/002,919 patent/US9243077B2/en active Active
- 2012-03-02 SI SI201231008T patent/SI2682409T1/sl unknown
- 2012-03-02 EP EP12752403.1A patent/EP2682409B1/en active Active
- 2012-03-02 PL PL12752403T patent/PL2682409T3/pl unknown
- 2012-03-02 ES ES12752403.1T patent/ES2635087T3/es active Active
- 2012-03-02 DK DK12752403.1T patent/DK2682409T3/en active
-
2017
- 2017-05-01 JP JP2017091352A patent/JP2017179374A/ja active Pending
Patent Citations (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02273176A (ja) | 1989-04-13 | 1990-11-07 | Seikagaku Kogyo Co Ltd | グリコサミノグリカンで修飾されたスーパーオキシドジスムターゼ及びその製造法 |
| WO1992006714A1 (en) | 1990-10-18 | 1992-04-30 | Shiseido Co., Ltd. | Combination of hyaluronic acid with medicinal ingredient and production thereof |
| JPH0585942A (ja) | 1991-02-26 | 1993-04-06 | Denki Kagaku Kogyo Kk | インターフエロン−ヒアルロン酸及び/又はその塩の結合体 |
| WO1992020349A1 (en) | 1991-05-20 | 1992-11-26 | Genzyme Corporation | Water insoluble derivatives of polyanionic polysaccharides |
| WO1999029839A1 (en) | 1997-12-12 | 1999-06-17 | Expression Genetics, Inc. | Grafted copolymers as gene carriers |
| WO2000001733A1 (en) | 1998-07-06 | 2000-01-13 | Fidia Advanced Biopolymers S.R.L. | Amides of hyaluronic acid and the derivatives thereof and a process for their preparation |
| JP2002519481A (ja) * | 1998-07-06 | 2002-07-02 | フィディア・アドバンスト・バイオポリマーズ・ソシエタ・ア・レスポンサビリタ・リミタータ | ヒアルロン酸アミドおよびそれらの誘導体、並びにそれらの製造法 |
| WO2001005434A2 (en) | 1999-07-20 | 2001-01-25 | Amgen Inc. | Hyaluronic acid-protein conjugates |
| JP2001081103A (ja) | 1999-09-13 | 2001-03-27 | Denki Kagaku Kogyo Kk | ヒアルロン酸結合薬剤 |
| JP2003518167A (ja) * | 1999-12-22 | 2003-06-03 | ジェンザイム、コーポレーション | ポリアニオン性多糖類の水不溶性誘導体 |
| WO2001060412A2 (en) | 2000-02-15 | 2001-08-23 | Genzyme Corporation | Modification of biopolymers for improved drug delivery |
| WO2005023906A1 (ja) * | 2003-09-08 | 2005-03-17 | Chugai Seiyaku Kabushiki Kaisha | ヒアルロン酸修飾物、及びそれを用いた薬物担体 |
| WO2005054301A1 (ja) * | 2003-11-14 | 2005-06-16 | Chugai Seiyaku Kabushiki Kaisha | 架橋多糖微粒子およびその製造方法 |
| WO2005054302A1 (ja) * | 2003-12-05 | 2005-06-16 | Chugai Seiyaku Kabushiki Kaisha | 薬物担体及びその製造方法 |
| WO2006028110A1 (ja) | 2004-09-07 | 2006-03-16 | Chugai Seiyaku Kabushiki Kaisha | 水溶性ヒアルロン酸修飾物の製造方法 |
| WO2006095775A1 (ja) * | 2005-03-08 | 2006-09-14 | Chugai Seiyaku Kabushiki Kaisha | 水溶性ヒアルロン酸修飾物とglp-1アナログの結合体 |
| WO2010053140A1 (ja) * | 2008-11-05 | 2010-05-14 | 国立大学法人 東京医科歯科大学 | ヒアルロン酸誘導体、およびその医薬組成物 |
| WO2011148116A2 (fr) | 2010-05-27 | 2011-12-01 | Laboratoire Idenov | Acide hyaluronique modifie, procede de fabrication et utilisations |
Non-Patent Citations (22)
| Title |
|---|
| BERGMAN K ET AL.: "Hyaluronic Acid Derivatives Prepared in Aqueous Media by Triazine-Activated Amidation", BIOMACROMOLECULES, vol. 8, no. 7, 2007, pages 2190 - 2195, XP002566505 * |
| BIOCONJUGATE CHEM., vol. 10, 1999, pages 406 - 411 |
| BIOCONJUGATE CHEM., vol. 14, 2003, pages 51 - 57 |
| BIOCONJUGATE CHEM., vol. 19, 2008, pages 1040 - 1048 |
| BIOCONJUGATE CHEM., vol. 4, 1993, pages 372 - 379 |
| BIOMACROMOLECULES, vol. 8, 2007, pages 2190 - 2195 |
| BIOMATERIALS, vol. 30, 2009, pages 1954 - 1961 |
| BIOPOLYMERS, vol. 89, 2008, pages 635 - 642 |
| CARBOHYDRATE POLYMERS, vol. 86, 2011, pages 747 - 752 |
| CARBOHYDRATE POLYMERS, vol. 87, 2012, pages 2211 - 2216 |
| CHEM. RES., vol. 25, 1992, pages 336 - 342 |
| J CONTROL. REL., vol. 61, 1999, pages 137 - 143 |
| J CONTROL. REL., vol. 88, 2003, pages 35 - 42 |
| J. BIOL. CHEM., vol. 262, 1987, pages 4429 - 4432 |
| J. BIOL. CHEM., vol. 272, 1997, pages 16010 - 16017 |
| J. BIOL. CHEM., vol. 276, 2001, pages 35103 - 35110 |
| J. CONTROL. REL., vol. 119, 2007, pages 245 - 252 |
| J. CONTROL. REL., vol. 56, 1998, pages 259 - 272 |
| J. CONTROL. REL., vol. 60, 1999, pages 149 - 160 |
| PHARM., vol. 255, 2003, pages 167 - 174 |
| See also references of EP2682409A4 * |
| T. W. GREENE; P. G. M. WUTS: "Protective Groups in Organic Synthesis, Third Edition,", 1999, JOHN WILEY & SONS, INC. |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014038641A1 (ja) * | 2012-09-05 | 2014-03-13 | 中外製薬株式会社 | アミノ酸およびステリル基が導入されたヒアルロン酸誘導体 |
| KR20150048238A (ko) | 2012-09-05 | 2015-05-06 | 추가이 세이야쿠 가부시키가이샤 | 아미노산 및 스테릴기가 도입된 히알루론산 유도체 |
| JPWO2014038641A1 (ja) * | 2012-09-05 | 2016-08-12 | 中外製薬株式会社 | アミノ酸およびステリル基が導入されたヒアルロン酸誘導体 |
| US11564971B2 (en) | 2012-09-05 | 2023-01-31 | Chugai Seiyaku Kabushiki Kaisha | Hyaluronic acid derivative having amino acid and steryl group introduced thereinto |
| JP2016529362A (ja) * | 2013-08-29 | 2016-09-23 | ホーリー ストーン バイオテック カンパニー リミテッド | グリコサミノグリカン化合物、その調製方法および使用 |
| WO2016136707A1 (ja) * | 2015-02-27 | 2016-09-01 | 学校法人常翔学園 | 膜透過性ペプチド鎖を有する多糖誘導体 |
| US10793603B2 (en) | 2015-02-27 | 2020-10-06 | Josho Gakuen Educational Foundation | Polysaccharide derivative having membrane-permeable peptide chain |
| JPWO2016159159A1 (ja) * | 2015-03-31 | 2018-01-25 | キユーピー株式会社 | ヒアルロン酸誘導体およびその製造方法、ならびにヒアルロン酸誘導体を含む化粧料、食品組成物および医薬組成物 |
| WO2019098393A1 (ja) * | 2017-11-15 | 2019-05-23 | 中外製薬株式会社 | ポリエチレングリコールにより修飾されたヒアルロン酸誘導体 |
| JPWO2019098393A1 (ja) * | 2017-11-15 | 2020-11-19 | 中外製薬株式会社 | ポリエチレングリコールにより修飾されたヒアルロン酸誘導体 |
| US11512147B2 (en) | 2017-11-15 | 2022-11-29 | Chugai Seiyaku Kabushiki Kaisha | Hyaluronic acid derivative modified with polyethylene glycol |
| JP7221211B2 (ja) | 2017-11-15 | 2023-02-13 | 中外製薬株式会社 | ポリエチレングリコールにより修飾されたヒアルロン酸誘導体 |
Also Published As
| Publication number | Publication date |
|---|---|
| US9243077B2 (en) | 2016-01-26 |
| EP3184553A1 (en) | 2017-06-28 |
| HUE033169T2 (en) | 2017-11-28 |
| JP6141180B2 (ja) | 2017-06-07 |
| US20130338352A1 (en) | 2013-12-19 |
| EP2682409B1 (en) | 2017-07-05 |
| EP2682409A4 (en) | 2014-11-19 |
| ES2635087T3 (es) | 2017-10-02 |
| SI2682409T1 (sl) | 2017-08-31 |
| JPWO2012118189A1 (ja) | 2014-07-07 |
| EP2682409A1 (en) | 2014-01-08 |
| EP3184553B1 (en) | 2019-05-01 |
| JP2017179374A (ja) | 2017-10-05 |
| PL2682409T3 (pl) | 2017-12-29 |
| DK2682409T3 (en) | 2017-09-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6141180B2 (ja) | アミノ−カルボン酸により修飾されたヒアルロン酸誘導体 | |
| JP5060131B2 (ja) | 水溶性ヒアルロン酸修飾物の製造方法 | |
| TWI359665B (en) | Hyaluronic acid/methotrexate compound | |
| RU2674003C2 (ru) | Производное гиалуроновой кислоты, содержащее аминокислотную и стерильную группы, введенные в него | |
| JP5542687B2 (ja) | ヒアルロン酸誘導体、およびその医薬組成物 | |
| CN109789188B (zh) | 具有低峰-谷比的pth化合物 | |
| Zhang et al. | Stimuli-responsive polypeptides for controlled drug delivery | |
| JPWO2006095775A1 (ja) | 水溶性ヒアルロン酸修飾物とglp−1アナログの結合体 | |
| KR101818705B1 (ko) | 약물이 충전된 히알루론산 가교물 하이드로겔 및 이의 이용 | |
| WO2005095464A1 (ja) | ヒアルロン酸-メトトレキサート結合体 | |
| JP6893918B2 (ja) | カチオン性基および疎水性基が導入されたヒアルロン酸誘導体 | |
| US8975079B2 (en) | Reducible polymers for nonviral gene delivery | |
| Pogostin et al. | Dynamic imine bonding facilitates mannan release from a nanofibrous peptide hydrogel | |
| Diao et al. | Peptide-based Self-assembly: Design, Bioactive Properties, and Its Applications | |
| US20170067012A1 (en) | Hyaluronic acid derivatives and composition for cell-surface engineering using the same | |
| Xiao et al. | pH-activatable lactam-stapled peptide-based nanoassemblies for enhanced chemo-photothermal therapy | |
| Simpson | Synthesis, Characterization, and Assessment of Cationic Polypeptoids Toward Gene Delivery and Development of Air Stable N-Substituted N-Thiocarboxyanhydrides | |
| WO2010100781A1 (ja) | 核酸デリバリー用組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12752403 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2013502420 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012752403 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14002919 Country of ref document: US |