RU2702631C2 - Ингибитор egfr и его получение и применение - Google Patents
Ингибитор egfr и его получение и применение Download PDFInfo
- Publication number
- RU2702631C2 RU2702631C2 RU2017114687A RU2017114687A RU2702631C2 RU 2702631 C2 RU2702631 C2 RU 2702631C2 RU 2017114687 A RU2017114687 A RU 2017114687A RU 2017114687 A RU2017114687 A RU 2017114687A RU 2702631 C2 RU2702631 C2 RU 2702631C2
- Authority
- RU
- Russia
- Prior art keywords
- methyl
- compound
- group
- amino
- formula
- Prior art date
Links
- 238000004519 manufacturing process Methods 0.000 title claims description 8
- 108060006698 EGF receptor Proteins 0.000 title description 3
- 239000003112 inhibitor Substances 0.000 title description 3
- 150000001875 compounds Chemical class 0.000 claims abstract description 273
- 238000000034 method Methods 0.000 claims abstract description 129
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 77
- 239000001257 hydrogen Substances 0.000 claims abstract description 77
- 125000000623 heterocyclic group Chemical group 0.000 claims abstract description 61
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims abstract description 58
- 150000002431 hydrogen Chemical class 0.000 claims abstract description 56
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims abstract description 39
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 38
- 150000002367 halogens Chemical class 0.000 claims abstract description 38
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 28
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 25
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims abstract description 19
- 229910052799 carbon Inorganic materials 0.000 claims abstract description 19
- 230000000694 effects Effects 0.000 claims abstract description 17
- 102200048955 rs121434569 Human genes 0.000 claims abstract description 15
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims abstract description 11
- 201000005202 lung cancer Diseases 0.000 claims abstract description 11
- 208000020816 lung neoplasm Diseases 0.000 claims abstract description 11
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 10
- 201000011510 cancer Diseases 0.000 claims abstract description 9
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims abstract description 9
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims abstract description 9
- 230000004663 cell proliferation Effects 0.000 claims abstract description 8
- 201000010099 disease Diseases 0.000 claims abstract description 8
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 8
- 230000001404 mediated effect Effects 0.000 claims abstract description 8
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims abstract description 8
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims abstract description 6
- 208000005718 Stomach Neoplasms Diseases 0.000 claims abstract description 6
- 206010017758 gastric cancer Diseases 0.000 claims abstract description 6
- 201000001441 melanoma Diseases 0.000 claims abstract description 6
- 201000011549 stomach cancer Diseases 0.000 claims abstract description 6
- 125000000714 pyrimidinyl group Chemical group 0.000 claims abstract description 5
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims abstract description 4
- 206010073478 Anaplastic large-cell lymphoma Diseases 0.000 claims abstract description 3
- 206010006187 Breast cancer Diseases 0.000 claims abstract description 3
- 208000026310 Breast neoplasm Diseases 0.000 claims abstract description 3
- 206010008342 Cervix carcinoma Diseases 0.000 claims abstract description 3
- 206010009944 Colon cancer Diseases 0.000 claims abstract description 3
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims abstract description 3
- 206010014733 Endometrial cancer Diseases 0.000 claims abstract description 3
- 206010014759 Endometrial neoplasm Diseases 0.000 claims abstract description 3
- 208000032612 Glial tumor Diseases 0.000 claims abstract description 3
- 206010018338 Glioma Diseases 0.000 claims abstract description 3
- 208000008839 Kidney Neoplasms Diseases 0.000 claims abstract description 3
- 208000032004 Large-Cell Anaplastic Lymphoma Diseases 0.000 claims abstract description 3
- 206010025323 Lymphomas Diseases 0.000 claims abstract description 3
- 206010027406 Mesothelioma Diseases 0.000 claims abstract description 3
- 208000034578 Multiple myelomas Diseases 0.000 claims abstract description 3
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 claims abstract description 3
- 206010033128 Ovarian cancer Diseases 0.000 claims abstract description 3
- 206010061535 Ovarian neoplasm Diseases 0.000 claims abstract description 3
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims abstract description 3
- 206010060862 Prostate cancer Diseases 0.000 claims abstract description 3
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims abstract description 3
- 206010038389 Renal cancer Diseases 0.000 claims abstract description 3
- 208000024770 Thyroid neoplasm Diseases 0.000 claims abstract description 3
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 claims abstract description 3
- 125000003282 alkyl amino group Chemical group 0.000 claims abstract description 3
- 201000010881 cervical cancer Diseases 0.000 claims abstract description 3
- 208000006990 cholangiocarcinoma Diseases 0.000 claims abstract description 3
- 201000011243 gastrointestinal stromal tumor Diseases 0.000 claims abstract description 3
- 208000005017 glioblastoma Diseases 0.000 claims abstract description 3
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims abstract description 3
- 231100000844 hepatocellular carcinoma Toxicity 0.000 claims abstract description 3
- 201000010982 kidney cancer Diseases 0.000 claims abstract description 3
- 208000032839 leukemia Diseases 0.000 claims abstract description 3
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims abstract description 3
- 201000002528 pancreatic cancer Diseases 0.000 claims abstract description 3
- 208000008443 pancreatic carcinoma Diseases 0.000 claims abstract description 3
- 201000002510 thyroid cancer Diseases 0.000 claims abstract description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract 4
- 125000004430 oxygen atom Chemical group O* 0.000 claims abstract 4
- 125000004433 nitrogen atom Chemical group N* 0.000 claims abstract 2
- -1 4-substituted 2-chloropyrimidine Chemical class 0.000 claims description 208
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 127
- 150000003839 salts Chemical class 0.000 claims description 87
- 125000000217 alkyl group Chemical group 0.000 claims description 72
- 239000000460 chlorine Substances 0.000 claims description 42
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 32
- 125000003342 alkenyl group Chemical group 0.000 claims description 28
- 125000000304 alkynyl group Chemical group 0.000 claims description 27
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 claims description 26
- HFBMWMNUJJDEQZ-UHFFFAOYSA-N acryloyl chloride Chemical compound ClC(=O)C=C HFBMWMNUJJDEQZ-UHFFFAOYSA-N 0.000 claims description 24
- 239000011737 fluorine Substances 0.000 claims description 24
- 229910052731 fluorine Inorganic materials 0.000 claims description 24
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 23
- 229910052801 chlorine Inorganic materials 0.000 claims description 23
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 21
- 125000004432 carbon atom Chemical group C* 0.000 claims description 18
- 125000001424 substituent group Chemical group 0.000 claims description 15
- 239000003814 drug Substances 0.000 claims description 11
- 125000001153 fluoro group Chemical group F* 0.000 claims description 11
- 238000011282 treatment Methods 0.000 claims description 9
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 5
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 claims description 3
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 claims 2
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 claims 2
- LLIOADBCFIXIEU-UHFFFAOYSA-N 4-fluoro-3-nitroaniline Chemical compound NC1=CC=C(F)C([N+]([O-])=O)=C1 LLIOADBCFIXIEU-UHFFFAOYSA-N 0.000 claims 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 abstract description 21
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 abstract description 10
- 229940121647 egfr inhibitor Drugs 0.000 abstract description 10
- 239000000126 substance Substances 0.000 abstract description 4
- 206010035226 Plasma cell myeloma Diseases 0.000 abstract description 2
- 150000001721 carbon Chemical group 0.000 abstract description 2
- 125000004648 C2-C8 alkenyl group Chemical group 0.000 abstract 3
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 abstract 2
- 125000004649 C2-C8 alkynyl group Chemical group 0.000 abstract 2
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 abstract 1
- 238000006243 chemical reaction Methods 0.000 description 295
- 238000002360 preparation method Methods 0.000 description 273
- 239000000243 solution Substances 0.000 description 201
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 165
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 121
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 118
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 113
- 238000005481 NMR spectroscopy Methods 0.000 description 113
- 239000011541 reaction mixture Substances 0.000 description 97
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 96
- 239000000203 mixture Substances 0.000 description 96
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 92
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 72
- 239000012043 crude product Substances 0.000 description 60
- 238000004440 column chromatography Methods 0.000 description 59
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 58
- 239000012074 organic phase Substances 0.000 description 53
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 49
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 47
- 235000019439 ethyl acetate Nutrition 0.000 description 46
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 46
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 44
- 239000000047 product Substances 0.000 description 43
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 42
- 125000003118 aryl group Chemical group 0.000 description 42
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 39
- 238000012746 preparative thin layer chromatography Methods 0.000 description 38
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 34
- 229920006395 saturated elastomer Polymers 0.000 description 31
- 239000000706 filtrate Substances 0.000 description 30
- 238000001816 cooling Methods 0.000 description 28
- 239000007787 solid Substances 0.000 description 28
- 239000000741 silica gel Substances 0.000 description 27
- 229910002027 silica gel Inorganic materials 0.000 description 27
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 26
- JYVLIDXNZAXMDK-UHFFFAOYSA-N pentan-2-ol Chemical compound CCCC(C)O JYVLIDXNZAXMDK-UHFFFAOYSA-N 0.000 description 26
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 26
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 description 25
- 239000007864 aqueous solution Substances 0.000 description 25
- 238000003818 flash chromatography Methods 0.000 description 25
- 239000012071 phase Substances 0.000 description 25
- 229910000104 sodium hydride Inorganic materials 0.000 description 23
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 22
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 22
- 125000000753 cycloalkyl group Chemical group 0.000 description 22
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 22
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 22
- 239000012267 brine Substances 0.000 description 20
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 20
- 125000004104 aryloxy group Chemical group 0.000 description 19
- 125000005553 heteroaryloxy group Chemical group 0.000 description 19
- 125000005844 heterocyclyloxy group Chemical group 0.000 description 19
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- 125000005110 aryl thio group Chemical group 0.000 description 18
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 description 18
- 125000001072 heteroaryl group Chemical group 0.000 description 18
- 125000005368 heteroarylthio group Chemical group 0.000 description 18
- 230000002401 inhibitory effect Effects 0.000 description 18
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 18
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 17
- 125000004468 heterocyclylthio group Chemical group 0.000 description 17
- 239000005457 ice water Substances 0.000 description 17
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 16
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 15
- 230000002829 reductive effect Effects 0.000 description 15
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 14
- 239000012298 atmosphere Substances 0.000 description 14
- 239000003480 eluent Substances 0.000 description 14
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 14
- HVOYZOQVDYHUPF-UHFFFAOYSA-N n,n',n'-trimethylethane-1,2-diamine Chemical compound CNCCN(C)C HVOYZOQVDYHUPF-UHFFFAOYSA-N 0.000 description 14
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 14
- 230000005855 radiation Effects 0.000 description 14
- 125000003545 alkoxy group Chemical group 0.000 description 13
- 235000019270 ammonium chloride Nutrition 0.000 description 13
- 238000012360 testing method Methods 0.000 description 13
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 12
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 12
- 229910052805 deuterium Inorganic materials 0.000 description 12
- 125000004786 difluoromethoxy group Chemical group [H]C(F)(F)O* 0.000 description 12
- 239000003208 petroleum Substances 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 12
- 210000004027 cell Anatomy 0.000 description 11
- 238000002474 experimental method Methods 0.000 description 11
- 230000005764 inhibitory process Effects 0.000 description 11
- 230000035772 mutation Effects 0.000 description 11
- 229910052760 oxygen Inorganic materials 0.000 description 11
- FYSIGSQCZXQTIH-UHFFFAOYSA-N 4-fluoro-2-methoxy-5-nitroaniline Chemical compound COC1=CC(F)=C([N+]([O-])=O)C=C1N FYSIGSQCZXQTIH-UHFFFAOYSA-N 0.000 description 10
- VTJXLVGZNOINAD-UHFFFAOYSA-N 6-methoxy-1-methylindole Chemical compound COC1=CC=C2C=CN(C)C2=C1 VTJXLVGZNOINAD-UHFFFAOYSA-N 0.000 description 10
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 10
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 10
- 239000001301 oxygen Chemical group 0.000 description 10
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 10
- 235000017557 sodium bicarbonate Nutrition 0.000 description 10
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 9
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 125000004429 atom Chemical group 0.000 description 9
- 239000012065 filter cake Substances 0.000 description 9
- 230000000670 limiting effect Effects 0.000 description 9
- BTTNYQZNBZNDOR-UHFFFAOYSA-N 2,4-dichloropyrimidine Chemical compound ClC1=CC=NC(Cl)=N1 BTTNYQZNBZNDOR-UHFFFAOYSA-N 0.000 description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- DVBFIBOOYOMVOL-UHFFFAOYSA-N FC(OC1=C(C=C(C(=C1)F)[N+](=O)[O-])N)F Chemical compound FC(OC1=C(C=C(C(=C1)F)[N+](=O)[O-])N)F DVBFIBOOYOMVOL-UHFFFAOYSA-N 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- 241000700159 Rattus Species 0.000 description 8
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 8
- 239000008346 aqueous phase Substances 0.000 description 8
- GTCAXTIRRLKXRU-UHFFFAOYSA-N carbamic acid methyl ester Natural products COC(N)=O GTCAXTIRRLKXRU-UHFFFAOYSA-N 0.000 description 8
- 229960003278 osimertinib Drugs 0.000 description 8
- DUYJMQONPNNFPI-UHFFFAOYSA-N osimertinib Chemical compound COC1=CC(N(C)CCN(C)C)=C(NC(=O)C=C)C=C1NC1=NC=CC(C=2C3=CC=CC=C3N(C)C=2)=N1 DUYJMQONPNNFPI-UHFFFAOYSA-N 0.000 description 8
- 125000003367 polycyclic group Chemical group 0.000 description 8
- 238000010992 reflux Methods 0.000 description 8
- 230000002441 reversible effect Effects 0.000 description 8
- 239000002904 solvent Substances 0.000 description 8
- 238000004809 thin layer chromatography Methods 0.000 description 8
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 7
- 102000004190 Enzymes Human genes 0.000 description 7
- 108090000790 Enzymes Proteins 0.000 description 7
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 7
- 125000000000 cycloalkoxy group Chemical group 0.000 description 7
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 7
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 7
- 239000013641 positive control Substances 0.000 description 7
- 239000007858 starting material Substances 0.000 description 7
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 7
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 6
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 6
- QFDKCJQODJFZID-UHFFFAOYSA-N 4-fluoro-2-(trifluoromethoxy)aniline Chemical compound NC1=CC=C(F)C=C1OC(F)(F)F QFDKCJQODJFZID-UHFFFAOYSA-N 0.000 description 6
- GBXPTJGVHFNUJH-UHFFFAOYSA-N 4-n-[2-(dimethylamino)ethyl]-1-n-[4-(1h-indol-3-yl)pyrimidin-2-yl]-2-methoxy-4-n-methyl-5-nitrobenzene-1,4-diamine Chemical compound COC1=CC(N(C)CCN(C)C)=C([N+]([O-])=O)C=C1NC1=NC=CC(C=2C3=CC=CC=C3NC=2)=N1 GBXPTJGVHFNUJH-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- 241000282472 Canis lupus familiaris Species 0.000 description 6
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- 235000011089 carbon dioxide Nutrition 0.000 description 6
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 6
- 125000005842 heteroatom Chemical group 0.000 description 6
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 6
- 238000004811 liquid chromatography Methods 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- VBQCXPDCHJSVEM-UHFFFAOYSA-N n-(4-fluoro-2-methoxy-5-nitrophenyl)-4-(1h-indol-3-yl)pyrimidin-2-amine Chemical compound COC1=CC(F)=C([N+]([O-])=O)C=C1NC1=NC=CC(C=2C3=CC=CC=C3NC=2)=N1 VBQCXPDCHJSVEM-UHFFFAOYSA-N 0.000 description 6
- 239000012299 nitrogen atmosphere Substances 0.000 description 6
- 239000008194 pharmaceutical composition Substances 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- FGIUAXJPYTZDNR-UHFFFAOYSA-N potassium nitrate Chemical compound [K+].[O-][N+]([O-])=O FGIUAXJPYTZDNR-UHFFFAOYSA-N 0.000 description 6
- 238000010926 purge Methods 0.000 description 6
- 125000006413 ring segment Chemical group 0.000 description 6
- HCDMJFOHIXMBOV-UHFFFAOYSA-N 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-4,7-dihydropyrrolo[4,5]pyrido[1,2-d]pyrimidin-2-one Chemical compound C=1C2=C3N(CC)C(=O)N(C=4C(=C(OC)C=C(OC)C=4F)F)CC3=CN=C2NC=1CN1CCOCC1 HCDMJFOHIXMBOV-UHFFFAOYSA-N 0.000 description 5
- YJGKIMCJZOZIOQ-UHFFFAOYSA-N 3-(2-chloropyrimidin-4-yl)-1h-indole Chemical compound ClC1=NC=CC(C=2C3=CC=CC=C3NC=2)=N1 YJGKIMCJZOZIOQ-UHFFFAOYSA-N 0.000 description 5
- MYKUATQRVMDZPQ-UHFFFAOYSA-N 3-(2-chloropyrimidin-4-yl)-6-methoxy-1-methylindole Chemical compound ClC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)OC)C MYKUATQRVMDZPQ-UHFFFAOYSA-N 0.000 description 5
- VBATUITURYFZIU-UHFFFAOYSA-N 4-fluoro-1-nitro-2-(trifluoromethoxy)benzene Chemical compound [O-][N+](=O)C1=CC=C(F)C=C1OC(F)(F)F VBATUITURYFZIU-UHFFFAOYSA-N 0.000 description 5
- SDGKIZRFIPVYPZ-UHFFFAOYSA-N 4-fluoro-5-nitro-2-(trifluoromethoxy)aniline Chemical compound FC1=CC(=C(N)C=C1[N+](=O)[O-])OC(F)(F)F SDGKIZRFIPVYPZ-UHFFFAOYSA-N 0.000 description 5
- PXWCHDJQVIOXKN-UHFFFAOYSA-N N-[2-(difluoromethoxy)-4-fluoro-5-nitrophenyl]-4-(1-methylindol-3-yl)pyrimidin-2-amine Chemical compound FC(OC1=C(C=C(C(=C1)F)[N+](=O)[O-])NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C)F PXWCHDJQVIOXKN-UHFFFAOYSA-N 0.000 description 5
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- JFCHSQDLLFJHOA-UHFFFAOYSA-N n,n-dimethylsulfamoyl chloride Chemical compound CN(C)S(Cl)(=O)=O JFCHSQDLLFJHOA-UHFFFAOYSA-N 0.000 description 5
- GYGYDOOXVKDZCF-UHFFFAOYSA-N n-methyl-2-pyrrolidin-1-ylethanamine Chemical compound CNCCN1CCCC1 GYGYDOOXVKDZCF-UHFFFAOYSA-N 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 239000012312 sodium hydride Substances 0.000 description 5
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid Substances OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 5
- IIBWXYHPBMUNJP-UHFFFAOYSA-N 3-(2-chloropyrimidin-4-yl)-1-methylindole Chemical compound C12=CC=CC=C2N(C)C=C1C1=CC=NC(Cl)=N1 IIBWXYHPBMUNJP-UHFFFAOYSA-N 0.000 description 4
- GLLRJTMXNVQJSU-UHFFFAOYSA-N 3-cyclopropyl-5-methoxy-2h-indazole Chemical compound C=12C=C(OC)C=CC2=NNC=1C1CC1 GLLRJTMXNVQJSU-UHFFFAOYSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- IDNOPHPTLHVPJP-UHFFFAOYSA-N 4-N-[2-(dimethylamino)ethyl]-2-methoxy-1-N-[4-(6-methoxy-1H-indol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]-4-N-methyl-5-nitrobenzene-1,4-diamine Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=C(C(=N1)C1=CNC2=CC(=CC=C12)OC)C(F)(F)F)[N+](=O)[O-])C)C IDNOPHPTLHVPJP-UHFFFAOYSA-N 0.000 description 4
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 4
- ZKHQWZAMYRWXGA-KQYNXXCUSA-J ATP(4-) Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-J 0.000 description 4
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 description 4
- 206010059866 Drug resistance Diseases 0.000 description 4
- 229910021529 ammonia Inorganic materials 0.000 description 4
- 125000002619 bicyclic group Chemical group 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 229910002091 carbon monoxide Inorganic materials 0.000 description 4
- 238000012054 celltiter-glo Methods 0.000 description 4
- 238000012217 deletion Methods 0.000 description 4
- 230000037430 deletion Effects 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 239000002207 metabolite Substances 0.000 description 4
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 4
- 230000035755 proliferation Effects 0.000 description 4
- LEHBURLTIWGHEM-UHFFFAOYSA-N pyridinium chlorochromate Chemical compound [O-][Cr](Cl)(=O)=O.C1=CC=[NH+]C=C1 LEHBURLTIWGHEM-UHFFFAOYSA-N 0.000 description 4
- 102200048928 rs121434568 Human genes 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 4
- JIJGCIDINLEGCZ-UHFFFAOYSA-N (2-fluorophenyl)-(2-methylsulfanylpyrimidin-4-yl)methanone Chemical compound CSC1=NC=CC(C(=O)C=2C(=CC=CC=2)F)=N1 JIJGCIDINLEGCZ-UHFFFAOYSA-N 0.000 description 3
- CBCKQZAAMUWICA-UHFFFAOYSA-N 1,4-phenylenediamine Chemical compound NC1=CC=C(N)C=C1 CBCKQZAAMUWICA-UHFFFAOYSA-N 0.000 description 3
- KGKWXEGYKGTMAK-UHFFFAOYSA-N 1-(2-amino-4,5-dimethoxyphenyl)ethanone Chemical compound COC1=CC(N)=C(C(C)=O)C=C1OC KGKWXEGYKGTMAK-UHFFFAOYSA-N 0.000 description 3
- IZOXSYNKYHJCTD-UHFFFAOYSA-N 1-(2-chloropyrimidin-4-yl)-5-methoxybenzimidazole Chemical compound ClC1=NC=CC(=N1)N1C=NC2=C1C=CC(=C2)OC IZOXSYNKYHJCTD-UHFFFAOYSA-N 0.000 description 3
- XXCIUEHDGFHCTH-UHFFFAOYSA-N 1-(2-chloropyrimidin-4-yl)indazole Chemical compound ClC1=NC=CC(N2C3=CC=CC=C3C=N2)=N1 XXCIUEHDGFHCTH-UHFFFAOYSA-N 0.000 description 3
- ZVLFGESHYMKNQP-UHFFFAOYSA-N 1-(4,5-dimethoxy-2-nitrophenyl)ethanone Chemical compound COC1=CC(C(C)=O)=C([N+]([O-])=O)C=C1OC ZVLFGESHYMKNQP-UHFFFAOYSA-N 0.000 description 3
- UVAGGEUONBIJCX-UHFFFAOYSA-N 1-(5-bromo-2-fluorophenyl)butan-1-ol Chemical compound CCCC(O)C1=CC(Br)=CC=C1F UVAGGEUONBIJCX-UHFFFAOYSA-N 0.000 description 3
- YFIKSXUBLVNPSA-UHFFFAOYSA-N 1-(5-bromo-2-fluorophenyl)butan-1-one Chemical compound CCCC(=O)C1=CC(Br)=CC=C1F YFIKSXUBLVNPSA-UHFFFAOYSA-N 0.000 description 3
- AYDIISBDADEEKC-UHFFFAOYSA-N 1-(5-bromo-2-fluorophenyl)butylidenehydrazine Chemical compound BrC=1C=CC(=C(C=1)C(CCC)=NN)F AYDIISBDADEEKC-UHFFFAOYSA-N 0.000 description 3
- QIXJGFDVTVREMC-UHFFFAOYSA-N 1-N-(2-chloropyrimidin-4-yl)-4-methoxybenzene-1,2-diamine Chemical compound ClC1=NC=CC(=N1)NC=1C(=CC(=CC=1)OC)N QIXJGFDVTVREMC-UHFFFAOYSA-N 0.000 description 3
- QEMJRFKFYOSMOE-UHFFFAOYSA-N 1-N-[2-(dimethylamino)ethyl]-1-N-methyl-4-N-[4-(1-methylindol-3-yl)pyrimidin-2-yl]-2-nitro-5-(trifluoromethoxy)benzene-1,4-diamine Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC(F)(F)F)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C)[N+](=O)[O-])C)C QEMJRFKFYOSMOE-UHFFFAOYSA-N 0.000 description 3
- JMZOJQYCHGDMOS-UHFFFAOYSA-N 1-N-[2-(dimethylamino)ethyl]-4-N-(4-indazol-1-ylpyrimidin-2-yl)-5-methoxy-1-N-methylbenzene-1,2,4-triamine Chemical compound N1(N=CC2=CC=CC=C12)C1=NC(=NC=C1)NC=1C=C(C(=CC=1OC)N(C)CCN(C)C)N JMZOJQYCHGDMOS-UHFFFAOYSA-N 0.000 description 3
- HFMDCOJPBOJBJA-UHFFFAOYSA-N 1-N-[2-(dimethylamino)ethyl]-4-N-[4-(6-ethynyl-1-methylindol-3-yl)pyrimidin-2-yl]-5-methoxy-1-N-methylbenzene-1,2,4-triamine Chemical compound CN(CCN(C=1C(=CC(=C(C=1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)C#C)C)N)C)C HFMDCOJPBOJBJA-UHFFFAOYSA-N 0.000 description 3
- OGYUDMSSKOQYQA-UHFFFAOYSA-N 1-N-[2-(dimethylamino)ethyl]-5-methoxy-1-N-methyl-4-N-[4-(1-prop-2-ynylindol-3-yl)pyrimidin-2-yl]benzene-1,2,4-triamine Chemical compound CN(CCN(C=1C(=CC(=C(C=1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)CC#C)N)C)C OGYUDMSSKOQYQA-UHFFFAOYSA-N 0.000 description 3
- IVHIAYNISQWWCF-UHFFFAOYSA-N 1-N-[2-(dimethylamino)ethyl]-5-methoxy-4-N-[4-(5-methoxybenzimidazol-1-yl)pyrimidin-2-yl]-1-N-methylbenzene-1,2,4-triamine Chemical compound CN(CCN(C=1C(=CC(=C(C=1)OC)NC1=NC=CC(=N1)N1C=NC2=C1C=CC(=C2)OC)N)C)C IVHIAYNISQWWCF-UHFFFAOYSA-N 0.000 description 3
- NYRKYIAGVRUJAN-UHFFFAOYSA-N 1-N-[2-(dimethylamino)ethyl]-5-methoxy-4-N-[4-(6-methoxy-1-methylindol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]-1-N-methylbenzene-1,2,4-triamine Chemical compound CN(CCN(C=1C(=CC(=C(C=1)OC)NC1=NC=C(C(=N1)C1=CN(C2=CC(=CC=C12)OC)C)C(F)(F)F)N)C)C NYRKYIAGVRUJAN-UHFFFAOYSA-N 0.000 description 3
- SRPGCBOEGMXTHE-UHFFFAOYSA-N 1-N-[4-(1-cyclopropylindazol-3-yl)pyrimidin-2-yl]-4-N-[2-(dimethylamino)ethyl]-2-methoxy-4-N-methyl-5-nitrobenzene-1,4-diamine Chemical compound C1(CC1)N1N=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC1=C(C=C(C(=C1)[N+](=O)[O-])N(C)CCN(C)C)OC SRPGCBOEGMXTHE-UHFFFAOYSA-N 0.000 description 3
- VGVRETPAUJJWIY-UHFFFAOYSA-N 1-N-[4-(1-cyclopropylindol-3-yl)pyrimidin-2-yl]-2-(difluoromethoxy)-4-N-[2-(dimethylamino)ethyl]-4-N-methyl-5-nitrobenzene-1,4-diamine Chemical compound C1(CC1)N1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC1=C(C=C(C(=C1)[N+](=O)[O-])N(C)CCN(C)C)OC(F)F VGVRETPAUJJWIY-UHFFFAOYSA-N 0.000 description 3
- LSPWKZNDBUPNMK-UHFFFAOYSA-N 1-N-[4-(1H-indol-3-yl)pyrimidin-2-yl]-2-methoxy-4-N-methyl-5-nitro-4-N-(2-pyrrolidin-1-ylethyl)benzene-1,4-diamine Chemical compound N1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC1=C(C=C(C(=C1)[N+](=O)[O-])N(CCN1CCCC1)C)OC LSPWKZNDBUPNMK-UHFFFAOYSA-N 0.000 description 3
- WGDPFHMPSPQKNU-UHFFFAOYSA-N 1-N-[4-(6-cyclopropyl-1-methylindol-3-yl)pyrimidin-2-yl]-4-N-[2-(dimethylamino)ethyl]-2-methoxy-4-N-methyl-5-nitrobenzene-1,4-diamine Chemical compound C1(CC1)C1=CC=C2C(=CN(C2=C1)C)C1=NC(=NC=C1)NC1=C(C=C(C(=C1)[N+](=O)[O-])N(C)CCN(C)C)OC WGDPFHMPSPQKNU-UHFFFAOYSA-N 0.000 description 3
- ZHWPYOBVPHJQDX-UHFFFAOYSA-N 1-cyclopropyl-3-(2-methylsulfanylpyrimidin-4-yl)indazole Chemical compound C1(CC1)N1N=C(C2=CC=CC=C12)C1=NC(=NC=C1)SC ZHWPYOBVPHJQDX-UHFFFAOYSA-N 0.000 description 3
- JSAAIHWEYKWHCM-UHFFFAOYSA-N 1-cyclopropyl-3-(2-methylsulfonylpyrimidin-4-yl)indazole Chemical compound C1(CC1)N1N=C(C2=CC=CC=C12)C1=NC(=NC=C1)S(=O)(=O)C JSAAIHWEYKWHCM-UHFFFAOYSA-N 0.000 description 3
- YGBYVNQFNMXDJM-UHFFFAOYSA-N 1h-indazole-5-carbonitrile Chemical compound N#CC1=CC=C2NN=CC2=C1 YGBYVNQFNMXDJM-UHFFFAOYSA-N 0.000 description 3
- VUZSIOOEQLAKRM-UHFFFAOYSA-N 2-(difluoromethoxy)-4-N-[2-(dimethylamino)ethyl]-4-N-methyl-1-N-[4-(1-methylindol-3-yl)pyrimidin-2-yl]-5-nitrobenzene-1,4-diamine Chemical compound FC(OC1=C(C=C(C(=C1)N(C)CCN(C)C)[N+](=O)[O-])NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C)F VUZSIOOEQLAKRM-UHFFFAOYSA-N 0.000 description 3
- ZVZMBGQCIYWPBE-UHFFFAOYSA-N 2-(difluoromethoxy)-4-fluoro-1-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=C(F)C=C1OC(F)F ZVZMBGQCIYWPBE-UHFFFAOYSA-N 0.000 description 3
- SDBGBQSQKWGACK-UHFFFAOYSA-N 2-[(5-bromoindazol-1-yl)methoxy]ethyl-trimethylsilane Chemical compound BrC1=CC=C2N(COCC[Si](C)(C)C)N=CC2=C1 SDBGBQSQKWGACK-UHFFFAOYSA-N 0.000 description 3
- GCWPPMJTUOGKNE-UHFFFAOYSA-N 2-[3-[2-[2-(difluoromethoxy)-4-fluoro-5-nitroanilino]pyrimidin-4-yl]indol-1-yl]ethanol Chemical compound FC(OC1=C(C=C(C(=C1)F)[N+](=O)[O-])NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)CCO)F GCWPPMJTUOGKNE-UHFFFAOYSA-N 0.000 description 3
- VUOMQUOUTJEOMS-UHFFFAOYSA-N 2-chloro-N-(4-methoxy-2-nitrophenyl)pyrimidin-4-amine Chemical compound ClC1=NC=CC(=N1)NC1=C(C=C(C=C1)OC)[N+](=O)[O-] VUOMQUOUTJEOMS-UHFFFAOYSA-N 0.000 description 3
- KPWFDJPAMDASRN-UHFFFAOYSA-N 3-(2-chloropyrimidin-4-yl)-1-(oxetan-3-yl)indole Chemical compound ClC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C1COC1 KPWFDJPAMDASRN-UHFFFAOYSA-N 0.000 description 3
- JIXRPHPWRVLUAV-UHFFFAOYSA-N 3-(2-chloropyrimidin-4-yl)-1-cyclopropylindole Chemical compound ClC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C1CC1 JIXRPHPWRVLUAV-UHFFFAOYSA-N 0.000 description 3
- FFYIZIPKFBGCAF-UHFFFAOYSA-N 3-[2-[4-[2-(diethylamino)ethyl-methylamino]-2-methoxy-5-nitroanilino]pyrimidin-4-yl]-N,N-dimethylindole-1-sulfonamide Chemical compound C(C)N(CCN(C1=CC(=C(C=C1[N+](=O)[O-])NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)S(=O)(=O)N(C)C)OC)C)CC FFYIZIPKFBGCAF-UHFFFAOYSA-N 0.000 description 3
- CPXVIGLNNNWJFQ-UHFFFAOYSA-N 3-[2-[5-amino-2-methoxy-4-[methyl(2-pyrrolidin-1-ylethyl)amino]anilino]pyrimidin-4-yl]-N,N-dimethylindole-1-sulfonamide Chemical compound NC=1C(=CC(=C(C=1)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)S(=O)(=O)N(C)C)OC)N(CCN1CCCC1)C CPXVIGLNNNWJFQ-UHFFFAOYSA-N 0.000 description 3
- LURSLIIBGOXRBR-UHFFFAOYSA-N 3-[2-[5-amino-4-[2-(diethylamino)ethyl-methylamino]-2-methoxyanilino]pyrimidin-4-yl]-N,N-dimethylindole-1-sulfonamide Chemical compound NC=1C(=CC(=C(C=1)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)S(=O)(=O)N(C)C)OC)N(C)CCN(CC)CC LURSLIIBGOXRBR-UHFFFAOYSA-N 0.000 description 3
- DUFMXQXETWANPY-UHFFFAOYSA-N 3-[2-[5-amino-4-[2-(dimethylamino)ethyl-methylamino]-2-methoxyanilino]-5-(trifluoromethyl)pyrimidin-4-yl]-6-methoxy-N,N-dimethylindole-1-sulfonamide Chemical compound NC=1C(=CC(=C(C=1)NC1=NC=C(C(=N1)C1=CN(C2=CC(=CC=C12)OC)S(=O)(=O)N(C)C)C(F)(F)F)OC)N(C)CCN(C)C DUFMXQXETWANPY-UHFFFAOYSA-N 0.000 description 3
- AJZJRHWSUZDDIU-UHFFFAOYSA-N 3-[2-[5-amino-4-[2-(dimethylamino)ethyl-methylamino]-2-methoxyanilino]pyrimidin-4-yl]-N,N-dimethylindole-1-sulfonamide Chemical compound NC=1C(=CC(=C(C=1)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)S(=O)(=O)N(C)C)OC)N(C)CCN(C)C AJZJRHWSUZDDIU-UHFFFAOYSA-N 0.000 description 3
- YSUICPSNOIPQKE-UHFFFAOYSA-N 3-[2-chloro-5-(trifluoromethyl)pyrimidin-4-yl]-6-methoxy-1H-indole Chemical compound ClC1=NC=C(C(=N1)C1=CNC2=CC(=CC=C12)OC)C(F)(F)F YSUICPSNOIPQKE-UHFFFAOYSA-N 0.000 description 3
- WVHZCDQLDZQYPH-UHFFFAOYSA-N 3-bromo-2h-indazole-5-carbonitrile Chemical compound C1=C(C#N)C=C2C(Br)=NNC2=C1 WVHZCDQLDZQYPH-UHFFFAOYSA-N 0.000 description 3
- DGIJTFJIIQKZEN-UHFFFAOYSA-N 4-(1-cyclopropylindazol-3-yl)-N-(4-fluoro-2-methoxy-5-nitrophenyl)pyrimidin-2-amine Chemical compound C1(CC1)N1N=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC1=C(C=C(C(=C1)[N+](=O)[O-])F)OC DGIJTFJIIQKZEN-UHFFFAOYSA-N 0.000 description 3
- QHRLLZJKCZYZOV-UHFFFAOYSA-N 4-(1-cyclopropylindol-3-yl)-N-[2-(difluoromethoxy)-4-fluoro-5-nitrophenyl]pyrimidin-2-amine Chemical compound C1(CC1)N1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC1=C(C=C(C(=C1)[N+](=O)[O-])F)OC(F)F QHRLLZJKCZYZOV-UHFFFAOYSA-N 0.000 description 3
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 3
- BQHBYVSBVYNTOD-UHFFFAOYSA-N 4-(6-cyclopropyl-1-methylindol-3-yl)-N-(4-fluoro-2-methoxy-5-nitrophenyl)pyrimidin-2-amine Chemical compound C1(CC1)C1=CC=C2C(=CN(C2=C1)C)C1=NC(=NC=C1)NC1=C(C=C(C(=C1)[N+](=O)[O-])F)OC BQHBYVSBVYNTOD-UHFFFAOYSA-N 0.000 description 3
- VJOLHJBSBZNMJO-UHFFFAOYSA-N 4-N-[2-(diethylamino)ethyl]-1-N-[4-(1H-indol-3-yl)pyrimidin-2-yl]-2-methoxy-4-N-methyl-5-nitrobenzene-1,4-diamine Chemical compound N1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC1=C(C=C(C(=C1)[N+](=O)[O-])N(C)CCN(CC)CC)OC VJOLHJBSBZNMJO-UHFFFAOYSA-N 0.000 description 3
- MEIYUYKFTUHJBG-UHFFFAOYSA-N 4-N-[2-(dimethylamino)ethyl]-1-N-(4-indazol-1-ylpyrimidin-2-yl)-2-methoxy-4-N-methyl-5-nitrobenzene-1,4-diamine Chemical compound N1(N=CC2=CC=CC=C12)C1=NC(=NC=C1)NC1=C(C=C(C(=C1)[N+](=O)[O-])N(C)CCN(C)C)OC MEIYUYKFTUHJBG-UHFFFAOYSA-N 0.000 description 3
- OXLFMUDQCYQPPJ-UHFFFAOYSA-N 4-N-[2-(dimethylamino)ethyl]-1-N-[4-(6-iodo-1-methylindol-3-yl)pyrimidin-2-yl]-2-methoxy-4-N-methyl-5-nitrobenzene-1,4-diamine Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)I)C)[N+](=O)[O-])C)C OXLFMUDQCYQPPJ-UHFFFAOYSA-N 0.000 description 3
- FGYXNYSTFSOHTD-UHFFFAOYSA-N 4-N-[2-(dimethylamino)ethyl]-2-methoxy-1-N-[4-(5-methoxybenzimidazol-1-yl)pyrimidin-2-yl]-4-N-methyl-5-nitrobenzene-1,4-diamine Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)N1C=NC2=C1C=CC(=C2)OC)[N+](=O)[O-])C)C FGYXNYSTFSOHTD-UHFFFAOYSA-N 0.000 description 3
- MKHPCLMGVJNJCL-UHFFFAOYSA-N 4-N-[2-(dimethylamino)ethyl]-2-methoxy-1-N-[4-(6-methoxy-1-methylindol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]-4-N-methyl-5-nitrobenzene-1,4-diamine Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=C(C(=N1)C1=CN(C2=CC(=CC=C12)OC)C)C(F)(F)F)[N+](=O)[O-])C)C MKHPCLMGVJNJCL-UHFFFAOYSA-N 0.000 description 3
- GTXJKTWDKWOYCR-UHFFFAOYSA-N 4-N-[2-(dimethylamino)ethyl]-2-methoxy-4-N-methyl-5-nitro-1-N-[4-[1-(oxetan-3-yl)indol-3-yl]pyrimidin-2-yl]benzene-1,4-diamine Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C1COC1)[N+](=O)[O-])C)C GTXJKTWDKWOYCR-UHFFFAOYSA-N 0.000 description 3
- SMRDKAQTLJJBQU-UHFFFAOYSA-N 4-N-[4-(1-cyclopropylindazol-3-yl)pyrimidin-2-yl]-1-N-[2-(dimethylamino)ethyl]-5-methoxy-1-N-methylbenzene-1,2,4-triamine Chemical compound C1(CC1)N1N=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC=1C=C(C(=CC=1OC)N(C)CCN(C)C)N SMRDKAQTLJJBQU-UHFFFAOYSA-N 0.000 description 3
- YHWPCGNTHYIMDP-UHFFFAOYSA-N 4-N-[4-(6-cyclopropyl-1-methylindol-3-yl)pyrimidin-2-yl]-1-N-[2-(dimethylamino)ethyl]-5-methoxy-1-N-methylbenzene-1,2,4-triamine Chemical compound C1(CC1)C1=CC=C2C(=CN(C2=C1)C)C1=NC(=NC=C1)NC=1C=C(C(=CC=1OC)N(C)CCN(C)C)N YHWPCGNTHYIMDP-UHFFFAOYSA-N 0.000 description 3
- HLLLJXYDSHIJAB-UHFFFAOYSA-N 4-[2-(methylamino)ethyl]morpholin-3-one Chemical compound CNCCN1CCOCC1=O HLLLJXYDSHIJAB-UHFFFAOYSA-N 0.000 description 3
- PXAJTMZHRYJMEQ-UHFFFAOYSA-N 4-[2-[5-(difluoromethoxy)-N-methyl-4-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]-2-nitroanilino]ethyl]morpholin-3-one Chemical compound FC(OC=1C(=CC(=C(C=1)N(CCN1C(COCC1)=O)C)[N+](=O)[O-])NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C)F PXAJTMZHRYJMEQ-UHFFFAOYSA-N 0.000 description 3
- AMARRLPHSCFJJY-UHFFFAOYSA-N 5-(difluoromethoxy)-1-N-[2-(dimethylamino)ethyl]-1-N-methyl-4-N-[4-(1-methylindol-3-yl)pyrimidin-2-yl]benzene-1,2,4-triamine Chemical compound FC(OC1=C(C=C(C(=C1)N(C)CCN(C)C)N)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C)F AMARRLPHSCFJJY-UHFFFAOYSA-N 0.000 description 3
- JBELPQSCYLPMMS-UHFFFAOYSA-N 5-(difluoromethoxy)-1-N-[2-(dimethylamino)ethyl]-4-N-[4-(6-dimethylphosphoryl-1-methylindol-3-yl)pyrimidin-2-yl]-1-N-methylbenzene-1,2,4-triamine Chemical compound NC=1C(=CC(=C(C=1)NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)P(C)(C)=O)C)OC(F)F)N(C)CCN(C)C JBELPQSCYLPMMS-UHFFFAOYSA-N 0.000 description 3
- RYPKWHPCFQSDEO-UHFFFAOYSA-N 5-(difluoromethoxy)-1-N-[2-(dimethylamino)ethyl]-4-N-[4-(6-methoxy-1-methylindol-3-yl)pyrimidin-2-yl]-1-N-methylbenzene-1,2,4-triamine Chemical compound FC(OC1=C(C=C(C(=C1)N(C)CCN(C)C)N)NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)OC)C)F RYPKWHPCFQSDEO-UHFFFAOYSA-N 0.000 description 3
- XDJNHYAQZWCIAH-UHFFFAOYSA-N 5-bromo-3-methyl-2h-indazole Chemical compound C1=CC(Br)=CC2=C(C)NN=C21 XDJNHYAQZWCIAH-UHFFFAOYSA-N 0.000 description 3
- JLJODUUBUCVUKP-UHFFFAOYSA-N 5-bromo-3-propyl-2h-indazole Chemical compound C1=C(Br)C=C2C(CCC)=NNC2=C1 JLJODUUBUCVUKP-UHFFFAOYSA-N 0.000 description 3
- QENJUGGVVMREAK-UHFFFAOYSA-N 5-chloro-1,2-dihydroindazol-3-one Chemical compound ClC1=CC=C2NNC(=O)C2=C1 QENJUGGVVMREAK-UHFFFAOYSA-N 0.000 description 3
- FWGSYXZLPCUQDP-UHFFFAOYSA-N 5-chloro-2-hydrazinylbenzoic acid Chemical compound NNC1=CC=C(Cl)C=C1C(O)=O FWGSYXZLPCUQDP-UHFFFAOYSA-N 0.000 description 3
- HFBCAEIFXHJBHB-UHFFFAOYSA-N 5-ethoxy-1-(2-methylsulfonylpyrimidin-4-yl)indazole Chemical compound C(C)OC=1C=C2C=NN(C2=CC=1)C1=NC(=NC=C1)S(=O)(=O)C HFBCAEIFXHJBHB-UHFFFAOYSA-N 0.000 description 3
- WRTDLNBUBANYHX-UHFFFAOYSA-N 5-methoxy-1-(2-methylsulfanylpyrimidin-4-yl)indazole Chemical compound COC=1C=C2C=NN(C2=CC=1)C1=NC(=NC=C1)SC WRTDLNBUBANYHX-UHFFFAOYSA-N 0.000 description 3
- PTRWUQJPKSFSOS-UHFFFAOYSA-N 5-methoxy-1-(2-methylsulfonylpyrimidin-4-yl)indazole Chemical compound COC=1C=C2C=NN(C2=CC=1)C1=NC(=NC=C1)S(=O)(=O)C PTRWUQJPKSFSOS-UHFFFAOYSA-N 0.000 description 3
- PXHJDPPKNUGKPM-UHFFFAOYSA-N 6-bromo-1-methylindole Chemical compound C1=C(Br)C=C2N(C)C=CC2=C1 PXHJDPPKNUGKPM-UHFFFAOYSA-N 0.000 description 3
- SZQVUHDYDJTERQ-UHFFFAOYSA-N 6-cyclopropyl-1-methylindole Chemical compound C1(CC1)C1=CC=C2C=CN(C2=C1)C SZQVUHDYDJTERQ-UHFFFAOYSA-N 0.000 description 3
- QJRWYBIKLXNYLF-UHFFFAOYSA-N 6-methoxy-1h-indole Chemical compound COC1=CC=C2C=CNC2=C1 QJRWYBIKLXNYLF-UHFFFAOYSA-N 0.000 description 3
- CHYHFXULCIXGHB-UHFFFAOYSA-N BrC1=NN(C2=CC=C(C=C12)C#N)COCC[Si](C)(C)C Chemical compound BrC1=NN(C2=CC=C(C=C12)C#N)COCC[Si](C)(C)C CHYHFXULCIXGHB-UHFFFAOYSA-N 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- SLMNIGBMLGXWPK-UHFFFAOYSA-N C(C)C1=NN(C2=CC=C(C=C12)C#N)COCC[Si](C)(C)C Chemical compound C(C)C1=NN(C2=CC=C(C=C12)C#N)COCC[Si](C)(C)C SLMNIGBMLGXWPK-UHFFFAOYSA-N 0.000 description 3
- RTGANCFIOXXGGB-UHFFFAOYSA-N CN1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)C1=C(C(=CC(=C1N)OC(F)(F)F)N)N Chemical compound CN1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)C1=C(C(=CC(=C1N)OC(F)(F)F)N)N RTGANCFIOXXGGB-UHFFFAOYSA-N 0.000 description 3
- UQMKSFZBAUPBMJ-UHFFFAOYSA-N COC1=CC(=NC(=N1)N)C1=CN(C2=CC=CC=C12)C Chemical compound COC1=CC(=NC(=N1)N)C1=CN(C2=CC=CC=C12)C UQMKSFZBAUPBMJ-UHFFFAOYSA-N 0.000 description 3
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 3
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 3
- ZCKHDTSPFZCICJ-UHFFFAOYSA-N N-(4-fluoro-2-methoxy-5-nitrophenyl)-4-(5-methoxybenzimidazol-1-yl)pyrimidin-2-amine Chemical compound FC1=CC(=C(C=C1[N+](=O)[O-])NC1=NC=CC(=N1)N1C=NC2=C1C=CC(=C2)OC)OC ZCKHDTSPFZCICJ-UHFFFAOYSA-N 0.000 description 3
- GNNLPUIHYDTTCF-UHFFFAOYSA-N N-(4-fluoro-2-methoxy-5-nitrophenyl)-4-(6-iodo-1-methylindol-3-yl)pyrimidin-2-amine Chemical compound FC1=CC(=C(C=C1[N+](=O)[O-])NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)I)C)OC GNNLPUIHYDTTCF-UHFFFAOYSA-N 0.000 description 3
- BIUFUUSJVLSTQP-UHFFFAOYSA-N N-(4-fluoro-2-methoxy-5-nitrophenyl)-4-(6-methoxy-1-methylindol-3-yl)-5-(trifluoromethyl)pyrimidin-2-amine Chemical compound FC1=CC(=C(C=C1[N+](=O)[O-])NC1=NC=C(C(=N1)C1=CN(C2=CC(=CC=C12)OC)C)C(F)(F)F)OC BIUFUUSJVLSTQP-UHFFFAOYSA-N 0.000 description 3
- IZEUKRXCWCYTCT-UHFFFAOYSA-N N-(4-fluoro-2-methoxy-5-nitrophenyl)-4-(6-methoxy-1H-indol-3-yl)-5-(trifluoromethyl)pyrimidin-2-amine Chemical compound FC1=CC(=C(C=C1[N+](=O)[O-])NC1=NC=C(C(=N1)C1=CNC2=CC(=CC=C12)OC)C(F)(F)F)OC IZEUKRXCWCYTCT-UHFFFAOYSA-N 0.000 description 3
- YFYIJODEMBLXEW-UHFFFAOYSA-N N-(4-fluoro-2-methoxy-5-nitrophenyl)-4-[1-(oxetan-3-yl)indol-3-yl]pyrimidin-2-amine Chemical compound FC1=CC(=C(C=C1[N+](=O)[O-])NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C1COC1)OC YFYIJODEMBLXEW-UHFFFAOYSA-N 0.000 description 3
- YVONWLRXQKUEBA-UHFFFAOYSA-N N-(4-fluoro-2-methoxy-5-nitrophenyl)-4-indazol-1-ylpyrimidin-2-amine Chemical compound FC1=CC(=C(C=C1[N+](=O)[O-])NC1=NC=CC(=N1)N1N=CC2=CC=CC=C12)OC YVONWLRXQKUEBA-UHFFFAOYSA-N 0.000 description 3
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 3
- SLDUSGJHDQMBFI-UHFFFAOYSA-N N-[2-(difluoromethoxy)-4-(4-methylpiperazin-1-yl)-5-nitrophenyl]-4-(1-methylindol-3-yl)pyrimidin-2-amine Chemical compound FC(OC1=C(C=C(C(=C1)N1CCN(CC1)C)[N+](=O)[O-])NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C)F SLDUSGJHDQMBFI-UHFFFAOYSA-N 0.000 description 3
- PJPKRCZHCWXBFW-UHFFFAOYSA-N N-[2-(difluoromethoxy)-4-fluoro-5-nitrophenyl]-4-(1H-indol-3-yl)pyrimidin-2-amine Chemical compound FC(OC1=C(C=C(C(=C1)F)[N+](=O)[O-])NC1=NC=CC(=N1)C1=CNC2=CC=CC=C12)F PJPKRCZHCWXBFW-UHFFFAOYSA-N 0.000 description 3
- KAHMTVBLFXQQDR-UHFFFAOYSA-N N-[2-[2-(diethylamino)ethyl-methylamino]-5-[[4-[1-(dimethylsulfamoyl)indol-3-yl]pyrimidin-2-yl]amino]-4-methoxyphenyl]prop-2-enamide Chemical compound C(C)N(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)S(N(C)C)(=O)=O)NC(C=C)=O)C)CC KAHMTVBLFXQQDR-UHFFFAOYSA-N 0.000 description 3
- MRVXYPXROPDGCF-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(6-methoxy-1-methylindol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=C(C(=N1)C1=CN(C2=CC(=CC=C12)OC)C)C(F)(F)F)NC(C=C)=O)C)C MRVXYPXROPDGCF-UHFFFAOYSA-N 0.000 description 3
- JKTPCFSUDWANJF-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(6-methoxy-3-methylindazol-1-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)N1N=C(C2=CC=C(C=C12)OC)C)NC(C=C)=O)C)C JKTPCFSUDWANJF-UHFFFAOYSA-N 0.000 description 3
- QHMWSARDRXHJRH-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-[1-(oxetan-3-yl)indol-3-yl]pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C1COC1)NC(C=C)=O)C)C QHMWSARDRXHJRH-UHFFFAOYSA-N 0.000 description 3
- FGZYQNJCGKLJDH-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-5-[(4-indazol-1-ylpyrimidin-2-yl)amino]-4-methoxyphenyl]prop-2-enamide Chemical compound N1(N=CC2=CC=CC=C12)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC FGZYQNJCGKLJDH-UHFFFAOYSA-N 0.000 description 3
- NZRVVOPTFQTXGU-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]-4-(trifluoromethoxy)phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC(F)(F)F)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C)NC(C=C)=O)C)C NZRVVOPTFQTXGU-UHFFFAOYSA-N 0.000 description 3
- CLQQESZFBCCZBE-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-(5-ethoxyindazol-1-yl)pyrimidin-2-yl]amino]-4-methoxyphenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)N1N=CC2=CC(=CC=C12)OCC)NC(C=C)=O)C)C CLQQESZFBCCZBE-UHFFFAOYSA-N 0.000 description 3
- GVTZPYYVNWZLQS-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-(6-ethynyl-1-methylindol-3-yl)pyrimidin-2-yl]amino]-4-methoxyphenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)C#C)C)NC(C=C)=O)C)C GVTZPYYVNWZLQS-UHFFFAOYSA-N 0.000 description 3
- QASDTIFXGLFUKQ-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-[1-(dimethylsulfamoyl)-6-methoxyindol-3-yl]-5-(trifluoromethyl)pyrimidin-2-yl]amino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(NC2=NC(=C(C=N2)C(F)(F)F)C=2C3=CC=C(OC)C=C3N(C=2)S(=O)(=O)N(C)C)=CC(NC(=O)C=C)=C(N(CCN(C)C)C)C=C1OC QASDTIFXGLFUKQ-UHFFFAOYSA-N 0.000 description 3
- VLMFINHKLSUJLY-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-[1-(dimethylsulfamoyl)indol-3-yl]pyrimidin-2-yl]amino]-4-methoxyphenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)S(N(C)C)(=O)=O)NC(C=C)=O)C)C VLMFINHKLSUJLY-UHFFFAOYSA-N 0.000 description 3
- SNYMWPGTTMOZHR-UHFFFAOYSA-N N-[4-(difluoromethoxy)-2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-(6-dimethylphosphoryl-1-methylindol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound FC(OC1=CC(=C(C=C1NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)P(=O)(C)C)C)NC(C=C)=O)N(C)CCN(C)C)F SNYMWPGTTMOZHR-UHFFFAOYSA-N 0.000 description 3
- PLWNWKKLVWMANB-UHFFFAOYSA-N N-[4-(difluoromethoxy)-2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-[1-(2-hydroxyethyl)indol-3-yl]pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound FC(OC1=CC(=C(C=C1NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)CCO)NC(C=C)=O)N(C)CCN(C)C)F PLWNWKKLVWMANB-UHFFFAOYSA-N 0.000 description 3
- XGJHJTXTONGMIR-UHFFFAOYSA-N N-[4-(difluoromethoxy)-5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]-2-[methyl-[2-[3-(oxomethylidene)morpholin-4-yl]ethyl]amino]phenyl]prop-2-enamide Chemical compound FC(OC1=CC(=C(C=C1NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C)NC(C=C)=O)N(CCN1C(COCC1)=C=O)C)F XGJHJTXTONGMIR-UHFFFAOYSA-N 0.000 description 3
- AEMAXSXBRJLYRK-UHFFFAOYSA-N N-[4-methoxy-5-[[4-(6-methoxy-3-methylindazol-1-yl)pyrimidin-2-yl]amino]-2-[methyl(2-pyrrolidin-1-ylethyl)amino]phenyl]prop-2-enamide Chemical compound COC1=CC(=C(C=C1NC1=NC=CC(=N1)N1N=C(C2=CC=C(C=C12)OC)C)NC(C=C)=O)N(CCN1CCCC1)C AEMAXSXBRJLYRK-UHFFFAOYSA-N 0.000 description 3
- AKCRNFBYRAEUPF-UHFFFAOYSA-N N-[5-[[4-(1-cyclopropyl-6-methoxyindol-3-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)N1C=C(C2=CC=C(C=C12)OC)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC AKCRNFBYRAEUPF-UHFFFAOYSA-N 0.000 description 3
- KKCZLKXAVRLESI-UHFFFAOYSA-N N-[5-[[4-(1-cyclopropylindazol-3-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)N1N=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC KKCZLKXAVRLESI-UHFFFAOYSA-N 0.000 description 3
- ZQGNFVVEEQCIPF-UHFFFAOYSA-N N-[5-[[4-(3-cyclopropylindazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)C1=NN(C2=CC=CC=C12)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC ZQGNFVVEEQCIPF-UHFFFAOYSA-N 0.000 description 3
- FJBBUUXKFWLWBM-UHFFFAOYSA-N N-[5-[[4-(5-cyano-3-ethylindazol-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C(#N)C=1C=C2C(=NN(C2=CC=1)C1=NC(=NC=C1C(F)(F)F)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)CC FJBBUUXKFWLWBM-UHFFFAOYSA-N 0.000 description 3
- XEBWRFVEGMHYEV-UHFFFAOYSA-N N-[5-[[4-(5-cyano-3-methylindazol-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C(#N)C=1C=C2C(=NN(C2=CC=1)C1=NC(=NC=C1C(F)(F)F)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)C XEBWRFVEGMHYEV-UHFFFAOYSA-N 0.000 description 3
- BSRNIVMPUIXBAP-UHFFFAOYSA-N N-[5-[[4-(5-cyanoindazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C(#N)C=1C=C2C=NN(C2=CC=1)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC BSRNIVMPUIXBAP-UHFFFAOYSA-N 0.000 description 3
- DGLJXDDEYLHVJE-UHFFFAOYSA-N N-[5-[[4-(6-cyclopropyl-1-methylindol-3-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)C1=CC=C2C(=CN(C2=C1)C)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC DGLJXDDEYLHVJE-UHFFFAOYSA-N 0.000 description 3
- DDLYKIVCAWTCLZ-UHFFFAOYSA-N N-[5-[[4-[1-(dimethylsulfamoyl)indol-3-yl]pyrimidin-2-yl]amino]-4-methoxy-2-[methyl(2-pyrrolidin-1-ylethyl)amino]phenyl]prop-2-enamide Chemical compound CN(S(=O)(=O)N1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(CCN1CCCC1)C)OC)C DDLYKIVCAWTCLZ-UHFFFAOYSA-N 0.000 description 3
- KHPUUNWGNTXYJB-UHFFFAOYSA-N N-[5-[[5-chloro-4-(3-cyclopropyl-5-methoxyindazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound ClC=1C(=NC(=NC=1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)N1N=C(C2=CC(=CC=C12)OC)C1CC1 KHPUUNWGNTXYJB-UHFFFAOYSA-N 0.000 description 3
- DWZUHGUAOPMEQZ-UHFFFAOYSA-N N-[5-[[5-chloro-4-(5-cyano-3-propylindazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound ClC=1C(=NC(=NC=1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)N1N=C(C2=CC(=CC=C12)C#N)CCC DWZUHGUAOPMEQZ-UHFFFAOYSA-N 0.000 description 3
- PHKARQYNDLNWDL-UHFFFAOYSA-N N-[5-[[5-chloro-4-(6-methoxy-1-methylindol-3-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound ClC=1C(=NC(=NC=1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)C1=CN(C2=CC(=CC=C12)OC)C PHKARQYNDLNWDL-UHFFFAOYSA-N 0.000 description 3
- 229960001686 afatinib Drugs 0.000 description 3
- ULXXDDBFHOBEHA-CWDCEQMOSA-N afatinib Chemical compound N1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC1=CC=C(F)C(Cl)=C1 ULXXDDBFHOBEHA-CWDCEQMOSA-N 0.000 description 3
- JSYBAZQQYCNZJE-UHFFFAOYSA-N benzene-1,2,4-triamine Chemical compound NC1=CC=C(N)C(N)=C1 JSYBAZQQYCNZJE-UHFFFAOYSA-N 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical group BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 3
- 229910000024 caesium carbonate Inorganic materials 0.000 description 3
- 125000001309 chloro group Chemical group Cl* 0.000 description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- GVXZWEHGCLOGAI-UHFFFAOYSA-N cyclopropyl-(2-fluorophenyl)methanol Chemical compound C=1C=CC=C(F)C=1C(O)C1CC1 GVXZWEHGCLOGAI-UHFFFAOYSA-N 0.000 description 3
- QUTQPZKZGGQBET-UHFFFAOYSA-N cyclopropyl-(2-fluorophenyl)methanone Chemical compound FC1=CC=CC=C1C(=O)C1CC1 QUTQPZKZGGQBET-UHFFFAOYSA-N 0.000 description 3
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 3
- FJQXASRLNDBWET-UHFFFAOYSA-N ethyl 5-chloro-3-methoxyindazole-1-carboxylate Chemical compound ClC=1C=C2C(=NN(C2=CC=1)C(=O)OCC)OC FJQXASRLNDBWET-UHFFFAOYSA-N 0.000 description 3
- OJSWBXRXQATKBH-UHFFFAOYSA-N ethyl 5-chloro-3-oxo-2h-indazole-1-carboxylate Chemical compound ClC1=CC=C2N(C(=O)OCC)NC(=O)C2=C1 OJSWBXRXQATKBH-UHFFFAOYSA-N 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 238000005984 hydrogenation reaction Methods 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 description 3
- 235000010333 potassium nitrate Nutrition 0.000 description 3
- 239000004323 potassium nitrate Substances 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 238000010898 silica gel chromatography Methods 0.000 description 3
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- DYOVUMBZNAUSRN-UHFFFAOYSA-N tert-butyl-(2-indol-1-ylethoxy)-dimethylsilane Chemical compound C1=CC=C2N(CCO[Si](C)(C)C(C)(C)C)C=CC2=C1 DYOVUMBZNAUSRN-UHFFFAOYSA-N 0.000 description 3
- KIWFWUBDIONBFK-UHFFFAOYSA-N 1-N-[2-(dimethylamino)ethyl]-5-methoxy-1-N-methyl-4-N-[4-[1-(oxetan-3-yl)indol-3-yl]pyrimidin-2-yl]benzene-1,2,4-triamine Chemical compound CN(CCN(C=1C(=CC(=C(C=1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C1COC1)N)C)C KIWFWUBDIONBFK-UHFFFAOYSA-N 0.000 description 2
- JFQRDYXFHVOZHD-UHFFFAOYSA-N 1-N-[2-(dimethylamino)ethyl]-5-methoxy-4-N-[4-(5-methoxyindazol-1-yl)pyrimidin-2-yl]-1-N-methylbenzene-1,2,4-triamine Chemical compound CN(CCN(C=1C(=CC(=C(C=1)OC)NC1=NC=CC(=N1)N1N=CC2=CC(=CC=C12)OC)N)C)C JFQRDYXFHVOZHD-UHFFFAOYSA-N 0.000 description 2
- UKVUVAMKXKTRIS-UHFFFAOYSA-N 1-[3-[2-[5-amino-2-(difluoromethoxy)-4-[2-(dimethylamino)ethyl-methylamino]anilino]pyrimidin-4-yl]indol-1-yl]ethanone Chemical compound NC=1C(=CC(=C(C=1)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C(C)=O)OC(F)F)N(C)CCN(C)C UKVUVAMKXKTRIS-UHFFFAOYSA-N 0.000 description 2
- AUKDFDQPJWJEDH-UHFFFAOYSA-N 1-fluoro-3-(trifluoromethoxy)benzene Chemical compound FC1=CC=CC(OC(F)(F)F)=C1 AUKDFDQPJWJEDH-UHFFFAOYSA-N 0.000 description 2
- 238000005160 1H NMR spectroscopy Methods 0.000 description 2
- IDRUEHMBFUJKAK-UHFFFAOYSA-N 2,4-dichloro-5-(trifluoromethyl)pyrimidine Chemical compound FC(F)(F)C1=CN=C(Cl)N=C1Cl IDRUEHMBFUJKAK-UHFFFAOYSA-N 0.000 description 2
- BWSFIURINWWNHK-UHFFFAOYSA-N 2-(difluoromethoxy)-4-N-[2-(dimethylamino)ethyl]-1-N-[4-(6-dimethylphosphoryl-1-methylindol-3-yl)pyrimidin-2-yl]-4-N-methyl-5-nitrobenzene-1,4-diamine Chemical compound FC(OC1=C(C=C(C(=C1)N(C)CCN(C)C)[N+](=O)[O-])NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)P(C)(C)=O)C)F BWSFIURINWWNHK-UHFFFAOYSA-N 0.000 description 2
- VZCRDZHFCBXDOM-UHFFFAOYSA-N 2-(difluoromethoxy)-6-[2-(dimethylamino)ethyl]-1-N-[4-(1H-indol-3-yl)pyrimidin-2-yl]-3-methyl-5-nitrobenzene-1,4-diamine Chemical compound N1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC1=C(C(=C(C(=C1CCN(C)C)[N+](=O)[O-])N)C)OC(F)F VZCRDZHFCBXDOM-UHFFFAOYSA-N 0.000 description 2
- LJKWFJDRHUWIGG-UHFFFAOYSA-N 2-[(5-cyclopropylindazol-1-yl)methoxy]ethyl-trimethylsilane Chemical compound C=1C=C2N(COCC[Si](C)(C)C)N=CC2=CC=1C1CC1 LJKWFJDRHUWIGG-UHFFFAOYSA-N 0.000 description 2
- RGYMAKIBNCJPRS-UHFFFAOYSA-N 2-[3-[2-[5-amino-2-(difluoromethoxy)-4-[2-(dimethylamino)ethyl-methylamino]anilino]pyrimidin-4-yl]indol-1-yl]ethanol Chemical compound NC=1C(=CC(=C(C=1)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)CCO)OC(F)F)N(C)CCN(C)C RGYMAKIBNCJPRS-UHFFFAOYSA-N 0.000 description 2
- LHPTXGAYGSHWTC-UHFFFAOYSA-N 2-[methyl-[(2-methylpropan-2-yl)oxycarbonyl]amino]ethyl 4-methylbenzenesulfonate Chemical compound CC(C)(C)OC(=O)N(C)CCOS(=O)(=O)C1=CC=C(C)C=C1 LHPTXGAYGSHWTC-UHFFFAOYSA-N 0.000 description 2
- ZWDVQMVZZYIAHO-UHFFFAOYSA-N 2-fluorobenzaldehyde Chemical compound FC1=CC=CC=C1C=O ZWDVQMVZZYIAHO-UHFFFAOYSA-N 0.000 description 2
- LSFFHEHCBFEPIV-UHFFFAOYSA-N 3-(2,5-dichloropyrimidin-4-yl)-6-methoxy-1-methylindole Chemical compound ClC1=NC=C(C(=N1)C1=CN(C2=CC(=CC=C12)OC)C)Cl LSFFHEHCBFEPIV-UHFFFAOYSA-N 0.000 description 2
- AWCWSZLNUOEZDH-UHFFFAOYSA-N 3-(2-chloropyrimidin-4-yl)-6-cyclopropyl-1-methylindole Chemical compound ClC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)C1CC1)C AWCWSZLNUOEZDH-UHFFFAOYSA-N 0.000 description 2
- CIUUSAREQXFCHS-UHFFFAOYSA-N 3-(2-chloropyrimidin-4-yl)-6-iodo-1-methylindole Chemical compound ClC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)I)C CIUUSAREQXFCHS-UHFFFAOYSA-N 0.000 description 2
- BYHQTRFJOGIQAO-GOSISDBHSA-N 3-(4-bromophenyl)-8-[(2R)-2-hydroxypropyl]-1-[(3-methoxyphenyl)methyl]-1,3,8-triazaspiro[4.5]decan-2-one Chemical compound C[C@H](CN1CCC2(CC1)CN(C(=O)N2CC3=CC(=CC=C3)OC)C4=CC=C(C=C4)Br)O BYHQTRFJOGIQAO-GOSISDBHSA-N 0.000 description 2
- SKCLPLPDZKCVMB-UHFFFAOYSA-N 3-[2-[2-methoxy-4-[methyl(2-pyrrolidin-1-ylethyl)amino]-5-nitroanilino]pyrimidin-4-yl]-N,N-dimethylindole-1-sulfonamide Chemical compound C1(=C(C=C(C(=C1)N(=O)=O)N(CCN1CCCC1)C)OC)NC1=NC(C=2C3=CC=CC=C3N(C=2)S(=O)(=O)N(C)C)=CC=N1 SKCLPLPDZKCVMB-UHFFFAOYSA-N 0.000 description 2
- MSOSATXFWUHSCD-UHFFFAOYSA-N 3-[2-[4-[2-(dimethylamino)ethyl-methylamino]-2-methoxy-5-nitroanilino]-5-(trifluoromethyl)pyrimidin-4-yl]-6-methoxy-N,N-dimethylindole-1-sulfonamide Chemical compound CN(CCN(C1=CC(=C(C=C1[N+](=O)[O-])NC1=NC=C(C(=N1)C1=CN(C2=CC(=CC=C12)OC)S(=O)(=O)N(C)C)C(F)(F)F)OC)C)C MSOSATXFWUHSCD-UHFFFAOYSA-N 0.000 description 2
- IEQXVKLTBRMZLG-UHFFFAOYSA-N 3-[2-[4-[2-(dimethylamino)ethyl-methylamino]-2-methoxy-5-nitroanilino]pyrimidin-4-yl]-N,N-dimethylindole-1-sulfonamide Chemical compound CN(CCN(C1=CC(=C(C=C1[N+](=O)[O-])NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)S(=O)(=O)N(C)C)OC)C)C IEQXVKLTBRMZLG-UHFFFAOYSA-N 0.000 description 2
- LRCVYCFQSZNJIB-UHFFFAOYSA-N 3-[2-[5-amino-2-(difluoromethoxy)-4-[2-(dimethylamino)ethyl-methylamino]anilino]pyrimidin-4-yl]-N,N-dimethylindole-1-sulfonamide Chemical compound NC=1C(=CC(=C(C=1)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)S(=O)(=O)N(C)C)OC(F)F)N(C)CCN(C)C LRCVYCFQSZNJIB-UHFFFAOYSA-N 0.000 description 2
- FDJNHURABSSGSF-UHFFFAOYSA-N 3-[2-chloro-5-(trifluoromethyl)pyrimidin-4-yl]-6-methoxy-1-methylindole Chemical compound ClC1=NC=C(C(=N1)C1=CN(C2=CC(=CC=C12)OC)C)C(F)(F)F FDJNHURABSSGSF-UHFFFAOYSA-N 0.000 description 2
- WNEODWDFDXWOLU-QHCPKHFHSA-N 3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[(2s)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one Chemical compound C([C@@H](N(CC1)C=2C=NC(NC=3C(N(C)C=C(C=3)C=3C(=C(N4C(C5=CC=6CC(C)(C)CC=6N5CC4)=O)N=CC=3)CO)=O)=CC=2)C)N1C1COC1 WNEODWDFDXWOLU-QHCPKHFHSA-N 0.000 description 2
- JAVBTLDJPYBQEW-UHFFFAOYSA-N 3-cyclopropyl-2h-indazole Chemical compound C1CC1C1=NNC2=CC=CC=C12 JAVBTLDJPYBQEW-UHFFFAOYSA-N 0.000 description 2
- LQEQPYISQMDCII-UHFFFAOYSA-N 3-ethyl-2h-indazole-5-carbonitrile Chemical compound C1=C(C#N)C=C2C(CC)=NNC2=C1 LQEQPYISQMDCII-UHFFFAOYSA-N 0.000 description 2
- USZDSFDKLKSIGM-UHFFFAOYSA-N 3-methyl-2h-indazole-5-carbonitrile Chemical compound C1=C(C#N)C=C2C(C)=NNC2=C1 USZDSFDKLKSIGM-UHFFFAOYSA-N 0.000 description 2
- YKSFNKWDISPIFQ-UHFFFAOYSA-N 3-propyl-2h-indazole-5-carbonitrile Chemical compound C1=CC(C#N)=CC2=C(CCC)NN=C21 YKSFNKWDISPIFQ-UHFFFAOYSA-N 0.000 description 2
- KTGLBACKKNVPHQ-UHFFFAOYSA-N 4-(5-ethoxyindazol-1-yl)-N-(4-fluoro-2-methoxy-5-nitrophenyl)pyrimidin-2-amine Chemical compound C(C)OC=1C=C2C=NN(C2=CC=1)C1=NC(=NC=C1)NC1=C(C=C(C(=C1)[N+](=O)[O-])F)OC KTGLBACKKNVPHQ-UHFFFAOYSA-N 0.000 description 2
- HGAIUIVBVKMVQL-UHFFFAOYSA-N 4-(difluoromethoxy)-3-N-[4-(1-methylindol-3-yl)pyrimidin-2-yl]-6-(4-methylpiperazin-1-yl)benzene-1,3-diamine Chemical compound C(OC1=C(NC2=NC=CC(C=3C4=C(N(C=3)C)C=CC=C4)=N2)C=C(N)C(N2CCN(CC2)C)=C1)(F)F HGAIUIVBVKMVQL-UHFFFAOYSA-N 0.000 description 2
- RPFHZZLWJHXTRO-UHFFFAOYSA-N 4-N-[4-(1-cyclopropylindol-3-yl)pyrimidin-2-yl]-5-(difluoromethoxy)-1-N-[2-(dimethylamino)ethyl]-1-N-methylbenzene-1,2,4-triamine Chemical compound C1(CC1)N1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC=1C=C(C(=CC=1OC(F)F)N(C)CCN(C)C)N RPFHZZLWJHXTRO-UHFFFAOYSA-N 0.000 description 2
- AHRGCNDJWBEXJH-UHFFFAOYSA-N 4-N-[4-(5-ethoxyindazol-1-yl)pyrimidin-2-yl]-5-methoxy-1-N-methyl-1-N-(2-pyrrolidin-1-ylethyl)benzene-1,2,4-triamine Chemical compound C(C)OC=1C=C2C=NN(C2=CC=1)C1=NC(=NC=C1)NC=1C=C(C(=CC=1OC)N(CCN1CCCC1)C)N AHRGCNDJWBEXJH-UHFFFAOYSA-N 0.000 description 2
- PBZUCAMKUHYGEG-UHFFFAOYSA-N 4-[2-[2-amino-5-(difluoromethoxy)-N-methyl-4-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]anilino]ethyl]morpholin-3-one Chemical compound NC1=C(C=C(C(=C1)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C)OC(F)F)N(CCN1C(COCC1)=O)C PBZUCAMKUHYGEG-UHFFFAOYSA-N 0.000 description 2
- DFOHHQRGDOQMKG-UHFFFAOYSA-N 4-chloro-2-methylsulfanylpyrimidine Chemical compound CSC1=NC=CC(Cl)=N1 DFOHHQRGDOQMKG-UHFFFAOYSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- ZOQBOJGXVDTPEJ-UHFFFAOYSA-N 5,6-dimethoxy-3-methyl-2h-indazole Chemical compound C1=C(OC)C(OC)=CC2=NNC(C)=C21 ZOQBOJGXVDTPEJ-UHFFFAOYSA-N 0.000 description 2
- VQOMHYLIIWQHCS-UHFFFAOYSA-N 5-chloro-3-methoxy-1h-indazole Chemical compound C1=C(Cl)C=C2C(OC)=NNC2=C1 VQOMHYLIIWQHCS-UHFFFAOYSA-N 0.000 description 2
- LTBUIGRDYXEGEY-UHFFFAOYSA-N 5-cyclopropyl-1h-indazole Chemical compound C1CC1C1=CC=C(NN=C2)C2=C1 LTBUIGRDYXEGEY-UHFFFAOYSA-N 0.000 description 2
- BAAOULLTESKBQV-UHFFFAOYSA-N 5-ethoxy-1h-indazole Chemical compound CCOC1=CC=C2NN=CC2=C1 BAAOULLTESKBQV-UHFFFAOYSA-N 0.000 description 2
- HQNPKVBTBJUMTR-UHFFFAOYSA-N 5-methoxy-1-methylindole Chemical compound COC1=CC=C2N(C)C=CC2=C1 HQNPKVBTBJUMTR-UHFFFAOYSA-N 0.000 description 2
- MAWGHOPSCKCTPA-UHFFFAOYSA-N 6-bromo-1h-indole Chemical compound BrC1=CC=C2C=CNC2=C1 MAWGHOPSCKCTPA-UHFFFAOYSA-N 0.000 description 2
- PRGVQKFCKJOOCV-UHFFFAOYSA-N 6-iodo-1-methylindole Chemical compound C1=C(I)C=C2N(C)C=CC2=C1 PRGVQKFCKJOOCV-UHFFFAOYSA-N 0.000 description 2
- UYFYWUXBZHYQRM-UHFFFAOYSA-N 6-iodo-1h-indole Chemical compound IC1=CC=C2C=CNC2=C1 UYFYWUXBZHYQRM-UHFFFAOYSA-N 0.000 description 2
- VDRWFBGSDLRPLK-UHFFFAOYSA-N 6-methoxy-3-methyl-2h-indazole Chemical compound COC1=CC=C2C(C)=NNC2=C1 VDRWFBGSDLRPLK-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 102000001301 EGF receptor Human genes 0.000 description 2
- 101150039808 Egfr gene Proteins 0.000 description 2
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 2
- XZKGCKLLBHVTQT-UHFFFAOYSA-N FC(OC1=CC(=C(C=C1NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C)NC(C=C)=O)N(C)CCN(C)C)F Chemical compound FC(OC1=CC(=C(C=C1NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C)NC(C=C)=O)N(C)CCN(C)C)F XZKGCKLLBHVTQT-UHFFFAOYSA-N 0.000 description 2
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- HJCSILVWWQJEIH-UHFFFAOYSA-N N-(4-fluoro-2-methoxy-5-nitrophenyl)-4-(5-methoxyindazol-1-yl)pyrimidin-2-amine Chemical compound FC1=CC(=C(C=C1[N+](=O)[O-])NC1=NC=CC(=N1)N1N=CC2=CC(=CC=C12)OC)OC HJCSILVWWQJEIH-UHFFFAOYSA-N 0.000 description 2
- PNKCYZLTHHRLLO-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(1,5,6-trimethylindol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC(=C(C=C12)C)C)C)NC(C=C)=O)C)C PNKCYZLTHHRLLO-UHFFFAOYSA-N 0.000 description 2
- XGPVBWBHUUBXEF-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(4,5,6,7-tetrafluoro-1-methylindol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CN(C2=C(C(=C(C(=C12)F)F)F)F)C)NC(C=C)=O)C)C XGPVBWBHUUBXEF-UHFFFAOYSA-N 0.000 description 2
- AZTVPXDFXOCPAT-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(4,5,6,7-tetrafluoro-1H-indol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CNC2=C(C(=C(C(=C12)F)F)F)F)NC(C=C)=O)C)C AZTVPXDFXOCPAT-UHFFFAOYSA-N 0.000 description 2
- UFYRAUUYGCRDGV-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(4-methoxyindazol-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=C(C(=N1)N1N=CC2=C(C=CC=C12)OC)C(F)(F)F)NC(C=C)=O)C)C UFYRAUUYGCRDGV-UHFFFAOYSA-N 0.000 description 2
- SARZSTXOSQRROD-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(4-methoxyindazol-1-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)N1N=CC2=C(C=CC=C12)OC)NC(C=C)=O)C)C SARZSTXOSQRROD-UHFFFAOYSA-N 0.000 description 2
- HCMAFFQONYSYPP-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(5-methoxy-1-methylindol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC=C(C=C12)OC)C)NC(C=C)=O)C)C HCMAFFQONYSYPP-UHFFFAOYSA-N 0.000 description 2
- BFBHPXBFJQYYSN-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(5-methoxy-2-methylbenzimidazol-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=C(C(=N1)N1C(=NC2=C1C=CC(=C2)OC)C)C(F)(F)F)NC(C=C)=O)C)C BFBHPXBFJQYYSN-UHFFFAOYSA-N 0.000 description 2
- ZSZPCMFLPJNBIF-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(5-methoxy-2-methylbenzimidazol-1-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)N1C(=NC2=C1C=CC(=C2)OC)C)NC(C=C)=O)C)C ZSZPCMFLPJNBIF-UHFFFAOYSA-N 0.000 description 2
- YKOCAQICAQGCRU-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(5-methoxybenzimidazol-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=C(C(=N1)N1C=NC2=C1C=CC(=C2)OC)C(F)(F)F)NC(C=C)=O)C)C YKOCAQICAQGCRU-UHFFFAOYSA-N 0.000 description 2
- UHFATHRGLQJDIM-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(5-methoxybenzimidazol-1-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)N1C=NC2=C1C=CC(=C2)OC)NC(C=C)=O)C)C UHFATHRGLQJDIM-UHFFFAOYSA-N 0.000 description 2
- DMLKSDLIZAGJMX-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(5-methoxyindazol-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=C(C(=N1)N1N=CC2=CC(=CC=C12)OC)C(F)(F)F)NC(C=C)=O)C)C DMLKSDLIZAGJMX-UHFFFAOYSA-N 0.000 description 2
- VMUUWFVISMYKHT-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(5-methoxyindazol-1-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)N1N=CC2=CC(=CC=C12)OC)NC(C=C)=O)C)C VMUUWFVISMYKHT-UHFFFAOYSA-N 0.000 description 2
- ZCMUSPYEKLRMED-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(6-methoxy-1-methylindol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)OC)C)NC(C=C)=O)C)C ZCMUSPYEKLRMED-UHFFFAOYSA-N 0.000 description 2
- XJCCSTFNFNZFKN-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(6-methoxy-1H-indol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CNC2=CC(=CC=C12)OC)NC(C=C)=O)C)C XJCCSTFNFNZFKN-UHFFFAOYSA-N 0.000 description 2
- JMWPJKHDHWHPGK-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(6-methoxy-2-methylbenzimidazol-1-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)N1C(=NC2=C1C=C(C=C2)OC)C)NC(C=C)=O)C)C JMWPJKHDHWHPGK-UHFFFAOYSA-N 0.000 description 2
- GLGXIUOIGBXFOS-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(6-methoxybenzimidazol-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=C(C(=N1)N1C=NC2=C1C=C(C=C2)OC)C(F)(F)F)NC(C=C)=O)C)C GLGXIUOIGBXFOS-UHFFFAOYSA-N 0.000 description 2
- NZAXEWDSWUQLQD-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(6-methoxybenzimidazol-1-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)N1C=NC2=C1C=C(C=C2)OC)NC(C=C)=O)C)C NZAXEWDSWUQLQD-UHFFFAOYSA-N 0.000 description 2
- RCVOHKQKIRPXEP-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(6-methoxyindazol-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=C(C(=N1)N1N=CC2=CC=C(C=C12)OC)C(F)(F)F)NC(C=C)=O)C)C RCVOHKQKIRPXEP-UHFFFAOYSA-N 0.000 description 2
- NNNDJEYHZURYDV-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(6-methoxyindazol-1-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)N1N=CC2=CC=C(C=C12)OC)NC(C=C)=O)C)C NNNDJEYHZURYDV-UHFFFAOYSA-N 0.000 description 2
- YVTYISANKJEPRR-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(7-methoxy-1H-indol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CNC2=C(C=CC=C12)OC)NC(C=C)=O)C)C YVTYISANKJEPRR-UHFFFAOYSA-N 0.000 description 2
- CKXXVJHURHIMOT-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-[1-(oxetan-3-yl)indol-3-yl]-5-(trifluoromethyl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=C(C(=N1)C1=CN(C2=CC=CC=C12)C1COC1)C(F)(F)F)NC(C=C)=O)C)C CKXXVJHURHIMOT-UHFFFAOYSA-N 0.000 description 2
- YSVCHXCTWCTWAX-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-[2-methyl-5-(trifluoromethoxy)benzimidazol-1-yl]-5-(trifluoromethyl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=C(C(=N1)N1C(=NC2=C1C=CC(=C2)OC(F)(F)F)C)C(F)(F)F)NC(C=C)=O)C)C YSVCHXCTWCTWAX-UHFFFAOYSA-N 0.000 description 2
- CECHMLGUWQDDHZ-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-[2-methyl-5-(trifluoromethyl)benzimidazol-1-yl]pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)N1C(=NC2=C1C=CC(=C2)C(F)(F)F)C)NC(C=C)=O)C)C CECHMLGUWQDDHZ-UHFFFAOYSA-N 0.000 description 2
- MDFMYJBHTVYJCF-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-[2-methyl-6-(trifluoromethyl)benzimidazol-1-yl]pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)N1C(=NC2=C1C=C(C=C2)C(F)(F)F)C)NC(C=C)=O)C)C MDFMYJBHTVYJCF-UHFFFAOYSA-N 0.000 description 2
- BIIPKIXRXFHSMP-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-[5-(2-methoxyethoxy)indazol-1-yl]pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)N1N=CC2=CC(=CC=C12)OCCOC)NC(C=C)=O)C)C BIIPKIXRXFHSMP-UHFFFAOYSA-N 0.000 description 2
- SVJGJKROAVWLAP-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-(5-fluoro-4,6-dimethoxy-1-methylindol-3-yl)pyrimidin-2-yl]amino]-4-methoxyphenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC(=C(C(=C12)OC)F)OC)C)NC(C=C)=O)C)C SVJGJKROAVWLAP-UHFFFAOYSA-N 0.000 description 2
- CRRXPSJSHJNEIQ-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-(6-ethenyl-1-methylindol-3-yl)pyrimidin-2-yl]amino]-4-methoxyphenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)C=C)C)NC(C=C)=O)C)C CRRXPSJSHJNEIQ-UHFFFAOYSA-N 0.000 description 2
- UCIQBSMWHFVGPO-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-(6-methoxy-1-methylindol-3-yl)pyrimidin-2-yl]amino]-4-(trifluoromethoxy)phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC(F)(F)F)NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)OC)C)NC(C=C)=O)C)C UCIQBSMWHFVGPO-UHFFFAOYSA-N 0.000 description 2
- JWPUYBBTIGXYHG-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-[1-(2-hydroxyethyl)indol-3-yl]pyrimidin-2-yl]amino]-4-(trifluoromethoxy)phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC(F)(F)F)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)CCO)NC(C=C)=O)C)C JWPUYBBTIGXYHG-UHFFFAOYSA-N 0.000 description 2
- HMVAYSNSOMAAEU-UHFFFAOYSA-N N-[4-(difluoromethoxy)-2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-(1-methyl-6-propan-2-ylsulfonylindol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound FC(OC1=CC(=C(C=C1NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)S(=O)(=O)C(C)C)C)NC(C=C)=O)N(C)CCN(C)C)F HMVAYSNSOMAAEU-UHFFFAOYSA-N 0.000 description 2
- NBDYYNQHNPFOPW-UHFFFAOYSA-N N-[4-(difluoromethoxy)-2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-(1H-indol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound N1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC(F)F NBDYYNQHNPFOPW-UHFFFAOYSA-N 0.000 description 2
- OEPUEQMRBURIEZ-UHFFFAOYSA-N N-[4-(difluoromethoxy)-2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-(6-methoxy-1-methylindol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound FC(OC1=CC(=C(C=C1NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)OC)C)NC(C=C)=O)N(C)CCN(C)C)F OEPUEQMRBURIEZ-UHFFFAOYSA-N 0.000 description 2
- MGDZXPZQKQSRJF-UHFFFAOYSA-N N-[4-(difluoromethoxy)-5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]-2-(4-methylpiperazin-1-yl)phenyl]prop-2-enamide Chemical compound FC(OC1=CC(=C(C=C1NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C)NC(C=C)=O)N1CCN(CC1)C)F MGDZXPZQKQSRJF-UHFFFAOYSA-N 0.000 description 2
- JQMYCVABZBALLM-UHFFFAOYSA-N N-[4-fluoro-5-nitro-2-(trifluoromethoxy)phenyl]-4-(1-methylindol-3-yl)pyrimidin-2-amine Chemical compound FC1=CC(=C(C=C1[N+](=O)[O-])NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C)OC(F)(F)F JQMYCVABZBALLM-UHFFFAOYSA-N 0.000 description 2
- WRUNJHNZMVMEFU-UHFFFAOYSA-N N-[4-methoxy-5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]-2-(2-oxa-6-azaspiro[3.3]heptan-6-yl)phenyl]prop-2-enamide Chemical compound COC1=CC(=C(C=C1NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C)NC(C=C)=O)N1CC2(COC2)C1 WRUNJHNZMVMEFU-UHFFFAOYSA-N 0.000 description 2
- GCQNVAFPZJWTNX-UHFFFAOYSA-N N-[4-methoxy-5-[[4-(4-methoxyindazol-1-yl)pyrimidin-2-yl]amino]-2-[methyl(2-pyrrolidin-1-ylethyl)amino]phenyl]prop-2-enamide Chemical compound COC1=CC(=C(C=C1NC1=NC=CC(=N1)N1N=CC2=C(C=CC=C12)OC)NC(C=C)=O)N(CCN1CCCC1)C GCQNVAFPZJWTNX-UHFFFAOYSA-N 0.000 description 2
- CGMJLUGKHPAPFO-UHFFFAOYSA-N N-[4-methoxy-5-[[4-(5-methoxybenzimidazol-1-yl)pyrimidin-2-yl]amino]-2-[methyl(2-pyrrolidin-1-ylethyl)amino]phenyl]prop-2-enamide Chemical compound COC1=CC(=C(C=C1NC1=NC=CC(=N1)N1C=NC2=C1C=CC(=C2)OC)NC(C=C)=O)N(CCN1CCCC1)C CGMJLUGKHPAPFO-UHFFFAOYSA-N 0.000 description 2
- YTYJGRDUSPZQAT-UHFFFAOYSA-N N-[4-methoxy-5-[[4-(5-methoxyindazol-1-yl)pyrimidin-2-yl]amino]-2-[methyl(2-pyrrolidin-1-ylethyl)amino]phenyl]prop-2-enamide Chemical compound COC1=CC(=C(C=C1NC1=NC=CC(=N1)N1N=CC2=CC(=CC=C12)OC)NC(C=C)=O)N(CCN1CCCC1)C YTYJGRDUSPZQAT-UHFFFAOYSA-N 0.000 description 2
- MLCCFGUIJDYKSS-UHFFFAOYSA-N N-[4-methoxy-5-[[4-(6-methoxy-1-methylindol-3-yl)pyrimidin-2-yl]amino]-2-[methyl-[2-(methylamino)ethyl]amino]phenyl]prop-2-enamide Chemical compound COC1=CC(=C(C=C1NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)OC)C)NC(C=C)=O)N(CCNC)C MLCCFGUIJDYKSS-UHFFFAOYSA-N 0.000 description 2
- HXWVNJFHGBWKDD-UHFFFAOYSA-N N-[4-methoxy-5-[[4-(6-methoxybenzimidazol-1-yl)pyrimidin-2-yl]amino]-2-[methyl(2-pyrrolidin-1-ylethyl)amino]phenyl]prop-2-enamide Chemical compound COC1=CC(=C(C=C1NC1=NC=CC(=N1)N1C=NC2=C1C=C(C=C2)OC)NC(C=C)=O)N(CCN1CCCC1)C HXWVNJFHGBWKDD-UHFFFAOYSA-N 0.000 description 2
- RJOFECTYAMDDFY-UHFFFAOYSA-N N-[4-methoxy-5-[[4-(6-methoxyindazol-1-yl)pyrimidin-2-yl]amino]-2-[methyl(2-pyrrolidin-1-ylethyl)amino]phenyl]prop-2-enamide Chemical compound COC1=CC(=C(C=C1NC1=NC=CC(=N1)N1N=CC2=CC=C(C=C12)OC)NC(C=C)=O)N(CCN1CCCC1)C RJOFECTYAMDDFY-UHFFFAOYSA-N 0.000 description 2
- QQMCBHWQUUEABX-UHFFFAOYSA-N N-[4-methoxy-5-[[4-[5-(2-methoxyethoxy)indazol-1-yl]pyrimidin-2-yl]amino]-2-[methyl(2-pyrrolidin-1-ylethyl)amino]phenyl]prop-2-enamide Chemical compound COC1=CC(=C(C=C1NC1=NC=CC(=N1)N1N=CC2=CC(=CC=C12)OCCOC)NC(C=C)=O)N(CCN1CCCC1)C QQMCBHWQUUEABX-UHFFFAOYSA-N 0.000 description 2
- GXTXDTVSLUNVOX-UHFFFAOYSA-N N-[5-[[4-(1,5-dicyclopropyl-4,6-difluoroindol-3-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)N1C=C(C2=C(C(=C(C=C12)F)C1CC1)F)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC GXTXDTVSLUNVOX-UHFFFAOYSA-N 0.000 description 2
- KKVKWEUIXYCLDZ-UHFFFAOYSA-N N-[5-[[4-(1-acetylindol-3-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-(trifluoromethoxy)phenyl]prop-2-enamide Chemical compound C(C)(=O)N1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC(F)(F)F KKVKWEUIXYCLDZ-UHFFFAOYSA-N 0.000 description 2
- CLXOEYRLLVMPJB-UHFFFAOYSA-N N-[5-[[4-(1-azatricyclo[6.3.1.04,12]dodeca-2,4,6,8(12)-tetraen-3-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(=CN2CCCC3=CC=CC1=C23)C2=NC(=NC=C2)NC=2C(=CC(=C(C2)NC(C=C)=O)N(C)CCN(C)C)OC CLXOEYRLLVMPJB-UHFFFAOYSA-N 0.000 description 2
- UCVOLEZKCQQMFX-UHFFFAOYSA-N N-[5-[[4-(1-cyclopropyl-4,6-dimethyl-5-methylsulfonylindol-3-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)N1C=C(C2=C(C(=C(C=C12)C)S(=O)(=O)C)C)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC UCVOLEZKCQQMFX-UHFFFAOYSA-N 0.000 description 2
- WCWIMRZNJKECIS-UHFFFAOYSA-N N-[5-[[4-(1-cyclopropylindol-3-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-(trifluoromethoxy)phenyl]prop-2-enamide Chemical compound C1(CC1)N1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC(F)(F)F WCWIMRZNJKECIS-UHFFFAOYSA-N 0.000 description 2
- RDGHBEFODFJNRC-UHFFFAOYSA-N N-[5-[[4-(1-cyclopropylindol-3-yl)pyrimidin-2-yl]amino]-4-methoxy-2-[methyl(2-morpholin-4-ylethyl)amino]phenyl]prop-2-enamide Chemical compound C1(CC1)N1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(CCN1CCOCC1)C)OC RDGHBEFODFJNRC-UHFFFAOYSA-N 0.000 description 2
- YZACRBFHOGQIME-UHFFFAOYSA-N N-[5-[[4-(1-cyclopropylindol-3-yl)pyrimidin-2-yl]amino]-4-methoxy-2-[methyl(2-pyrrolidin-1-ylethyl)amino]phenyl]prop-2-enamide Chemical compound C1(CC1)N1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(CCN1CCCC1)C)OC YZACRBFHOGQIME-UHFFFAOYSA-N 0.000 description 2
- BQRQLWNUMIUJBY-UHFFFAOYSA-N N-[5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]-2-(4-methylpiperazin-1-yl)-4-(trifluoromethoxy)phenyl]prop-2-enamide Chemical compound CN1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N1CCN(CC1)C)OC(F)(F)F BQRQLWNUMIUJBY-UHFFFAOYSA-N 0.000 description 2
- UWXUKAUOQOODCD-UHFFFAOYSA-N N-[5-[[4-(2-cyclopropyl-5-methoxybenzimidazol-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)C1=NC2=C(N1C1=NC(=NC=C1C(F)(F)F)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)C=CC(=C2)OC UWXUKAUOQOODCD-UHFFFAOYSA-N 0.000 description 2
- UVHCCKVMKXKADO-UHFFFAOYSA-N N-[5-[[4-(2-cyclopropyl-5-methoxybenzimidazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)C1=NC2=C(N1C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)C=CC(=C2)OC UVHCCKVMKXKADO-UHFFFAOYSA-N 0.000 description 2
- WIEXYOWCSHIRKH-UHFFFAOYSA-N N-[5-[[4-(2-cyclopropylbenzimidazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)C1=NC2=C(N1C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)C=CC=C2 WIEXYOWCSHIRKH-UHFFFAOYSA-N 0.000 description 2
- SNZHDQDMZYTOOJ-UHFFFAOYSA-N N-[5-[[4-(3-cyclopropyl-5-methoxyindazol-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)C1=NN(C2=CC=C(C=C12)OC)C1=NC(=NC=C1C(F)(F)F)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC SNZHDQDMZYTOOJ-UHFFFAOYSA-N 0.000 description 2
- DGOLNLWTOGMDCN-UHFFFAOYSA-N N-[5-[[4-(3-cyclopropyl-5-methoxyindazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)C1=NN(C2=CC=C(C=C12)OC)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC DGOLNLWTOGMDCN-UHFFFAOYSA-N 0.000 description 2
- GRYCHRAWTKNYQX-UHFFFAOYSA-N N-[5-[[4-(3-cyclopropylindazol-2-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)C=1N(N=C2C=CC=CC=12)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC GRYCHRAWTKNYQX-UHFFFAOYSA-N 0.000 description 2
- GEBOQXFAIYZHDN-UHFFFAOYSA-N N-[5-[[4-(4,6-difluoro-1,7-dimethylindol-3-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound FC1=C2C(=CN(C2=C(C(=C1)F)C)C)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC GEBOQXFAIYZHDN-UHFFFAOYSA-N 0.000 description 2
- MVWZAKYJXYBSSU-UHFFFAOYSA-N N-[5-[[4-(5,6-dimethoxy-3-methylindazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound COC=1C=C2C(=NN(C2=CC=1OC)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)C MVWZAKYJXYBSSU-UHFFFAOYSA-N 0.000 description 2
- MLZINPMOMYICIS-UHFFFAOYSA-N N-[5-[[4-(5,7-difluoro-3-methylindazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound FC=1C=C2C(=NN(C2=C(C=1)F)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)C MLZINPMOMYICIS-UHFFFAOYSA-N 0.000 description 2
- RCJFAZQEZGOTHC-UHFFFAOYSA-N N-[5-[[4-(5-chloro-3-methoxyindazol-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound ClC=1C=C2C(=NN(C2=CC=1)C1=NC(=NC=C1C(F)(F)F)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)OC RCJFAZQEZGOTHC-UHFFFAOYSA-N 0.000 description 2
- JOAPLDLXNGRNFG-UHFFFAOYSA-N N-[5-[[4-(5-chloro-3-methoxyindazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound ClC=1C=C2C(=NN(C2=CC=1)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)OC JOAPLDLXNGRNFG-UHFFFAOYSA-N 0.000 description 2
- XHVWVTYAEVUUHQ-UHFFFAOYSA-N N-[5-[[4-(5-cyano-2-methylbenzimidazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C(#N)C1=CC2=C(N(C(=N2)C)C2=NC(=NC=C2)NC=2C(=CC(=C(C=2)NC(C=C)=O)N(C)CCN(C)C)OC)C=C1 XHVWVTYAEVUUHQ-UHFFFAOYSA-N 0.000 description 2
- YYJQSTMSTWTKNC-UHFFFAOYSA-N N-[5-[[4-(5-cyano-3-methylindazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C(#N)C=1C=C2C(=NN(C2=CC=1)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)C YYJQSTMSTWTKNC-UHFFFAOYSA-N 0.000 description 2
- RSVZCMWSWGRPEZ-UHFFFAOYSA-N N-[5-[[4-(5-cyanobenzimidazol-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C(#N)C1=CC2=C(N(C=N2)C2=NC(=NC=C2C(F)(F)F)NC=2C(=CC(=C(C=2)NC(C=C)=O)N(C)CCN(C)C)OC)C=C1 RSVZCMWSWGRPEZ-UHFFFAOYSA-N 0.000 description 2
- BDIZVJFGMRZTNT-UHFFFAOYSA-N N-[5-[[4-(5-cyanobenzimidazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C(#N)C1=CC2=C(N(C=N2)C2=NC(=NC=C2)NC=2C(=CC(=C(C=2)NC(C=C)=O)N(C)CCN(C)C)OC)C=C1 BDIZVJFGMRZTNT-UHFFFAOYSA-N 0.000 description 2
- GKZFPRARIPNBDB-UHFFFAOYSA-N N-[5-[[4-(5-cyclopropyl-1-methylindol-3-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)C=1C=C2C(=CN(C2=CC=1)C)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC GKZFPRARIPNBDB-UHFFFAOYSA-N 0.000 description 2
- CXULGWINTHUMAI-UHFFFAOYSA-N N-[5-[[4-(5-cyclopropyl-2-methylbenzimidazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)C1=CC2=C(N(C(=N2)C)C2=NC(=NC=C2)NC=2C(=CC(=C(C=2)NC(C=C)=O)N(C)CCN(C)C)OC)C=C1 CXULGWINTHUMAI-UHFFFAOYSA-N 0.000 description 2
- UEPOXHGONZEAGI-UHFFFAOYSA-N N-[5-[[4-(5-cyclopropylindazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)C=1C=C2C=NN(C2=CC=1)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC UEPOXHGONZEAGI-UHFFFAOYSA-N 0.000 description 2
- BVTIINSKKGWJOM-UHFFFAOYSA-N N-[5-[[4-(6-cyano-1-methylindol-3-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-(trifluoromethoxy)phenyl]prop-2-enamide Chemical compound C(#N)C1=CC=C2C(=CN(C2=C1)C)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC(F)(F)F BVTIINSKKGWJOM-UHFFFAOYSA-N 0.000 description 2
- MQWUBMAPJRYPHW-UHFFFAOYSA-N N-[5-[[4-(6-cyano-1-methylindol-3-yl)pyrimidin-2-yl]amino]-4-(difluoromethoxy)-2-[2-(dimethylamino)ethyl-methylamino]phenyl]prop-2-enamide Chemical compound C(#N)C1=CC=C2C(=CN(C2=C1)C)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC(F)F MQWUBMAPJRYPHW-UHFFFAOYSA-N 0.000 description 2
- WGRAAGCXGASPQO-UHFFFAOYSA-N N-[5-[[4-(6-cyano-2-methylbenzimidazol-1-yl)-5-fluoropyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C(#N)C=1C=CC2=C(N(C(=N2)C)C2=NC(=NC=C2F)NC=2C(=CC(=C(C=2)NC(C=C)=O)N(C)CCN(C)C)OC)C=1 WGRAAGCXGASPQO-UHFFFAOYSA-N 0.000 description 2
- SVQGWIGFYILFRC-UHFFFAOYSA-N N-[5-[[4-(6-cyano-2-methylbenzimidazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C(#N)C=1C=CC2=C(N(C(=N2)C)C2=NC(=NC=C2)NC=2C(=CC(=C(C=2)NC(C=C)=O)N(C)CCN(C)C)OC)C=1 SVQGWIGFYILFRC-UHFFFAOYSA-N 0.000 description 2
- QEYBJMRKEPNLKW-UHFFFAOYSA-N N-[5-[[4-(6-cyano-3-methylindazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C(#N)C1=CC=C2C(=NN(C2=C1)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)C QEYBJMRKEPNLKW-UHFFFAOYSA-N 0.000 description 2
- LYIQMUUIHIAJCW-UHFFFAOYSA-N N-[5-[[4-(6-cyanobenzimidazol-1-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C(#N)C=1C=CC2=C(N(C=N2)C2=NC(=NC=C2C(F)(F)F)NC=2C(=CC(=C(C=2)NC(C=C)=O)N(C)CCN(C)C)OC)C=1 LYIQMUUIHIAJCW-UHFFFAOYSA-N 0.000 description 2
- QZIYTJGZZRZVTO-UHFFFAOYSA-N N-[5-[[4-(6-cyanobenzimidazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C(#N)C=1C=CC2=C(N(C=N2)C2=NC(=NC=C2)NC=2C(=CC(=C(C=2)NC(C=C)=O)N(C)CCN(C)C)OC)C=1 QZIYTJGZZRZVTO-UHFFFAOYSA-N 0.000 description 2
- NCHNEMQDHXJLIG-UHFFFAOYSA-N N-[5-[[4-(6-cyanoindazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C(#N)C1=CC=C2C=NN(C2=C1)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC NCHNEMQDHXJLIG-UHFFFAOYSA-N 0.000 description 2
- KVVHIARIIWQMRC-UHFFFAOYSA-N N-[5-[[4-(7-cyclopropyl-1-methylindol-3-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)C=1C=CC=C2C(=CN(C=12)C)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC KVVHIARIIWQMRC-UHFFFAOYSA-N 0.000 description 2
- QYZJRGLWXOESGS-UHFFFAOYSA-N N-[5-[[4-(7-cyclopropyl-1H-indol-3-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)C=1C=CC=C2C(=CNC=12)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC QYZJRGLWXOESGS-UHFFFAOYSA-N 0.000 description 2
- MDMRSEASTLZMHQ-UHFFFAOYSA-N N-[5-[[4-(benzimidazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound N1(C=NC2=C1C=CC=C2)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC MDMRSEASTLZMHQ-UHFFFAOYSA-N 0.000 description 2
- KANWFQREODXBNQ-UHFFFAOYSA-N N-[5-[[4-[1-cyclopropyl-5,7-difluoro-6-(oxetan-3-yl)indol-3-yl]pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)N1C=C(C2=CC(=C(C(=C12)F)C1COC1)F)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC KANWFQREODXBNQ-UHFFFAOYSA-N 0.000 description 2
- XQCUQKUBLFYJIW-UHFFFAOYSA-N N-[5-[[4-[2-cyclopropyl-5-(trifluoromethyl)benzimidazol-1-yl]pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)C1=NC2=C(N1C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)C=CC(=C2)C(F)(F)F XQCUQKUBLFYJIW-UHFFFAOYSA-N 0.000 description 2
- KFVVTNLWFINXNH-UHFFFAOYSA-N N-[5-[[4-[2-cyclopropyl-6-(trifluoromethyl)benzimidazol-1-yl]pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)C1=NC2=C(N1C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)C=C(C=C2)C(F)(F)F KFVVTNLWFINXNH-UHFFFAOYSA-N 0.000 description 2
- YYFPPUNLDSRYAK-UHFFFAOYSA-N N-[5-[[4-[5,7-difluoro-6-(trifluoromethyl)-1H-indol-3-yl]pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound FC=1C=C2C(=CNC2=C(C=1C(F)(F)F)F)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC YYFPPUNLDSRYAK-UHFFFAOYSA-N 0.000 description 2
- PQQGPGAQPJEOIN-UHFFFAOYSA-N N-[5-[[5-chloro-4-(1-cyclopropylindol-3-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound ClC=1C(=NC(=NC=1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)C1=CN(C2=CC=CC=C12)C1CC1 PQQGPGAQPJEOIN-UHFFFAOYSA-N 0.000 description 2
- JKBPSAHZVCFEGW-UHFFFAOYSA-N N-[5-[[5-chloro-4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]-4-(difluoromethoxy)-2-[2-(dimethylamino)ethyl-methylamino]phenyl]prop-2-enamide Chemical compound ClC=1C(=NC(=NC=1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC(F)F)C1=CN(C2=CC=CC=C12)C JKBPSAHZVCFEGW-UHFFFAOYSA-N 0.000 description 2
- QJFWVXYAXQHNEL-UHFFFAOYSA-N N-[5-[[5-chloro-4-(5-chloro-3-methoxyindazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound ClC=1C(=NC(=NC=1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)N1N=C(C2=CC(=CC=C12)Cl)OC QJFWVXYAXQHNEL-UHFFFAOYSA-N 0.000 description 2
- CLOGGCQBGNAECJ-UHFFFAOYSA-N N-[5-[[5-chloro-4-(5-cyano-2-cyclopropylbenzimidazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound ClC=1C(=NC(=NC=1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)N1C(=NC2=C1C=CC(=C2)C#N)C1CC1 CLOGGCQBGNAECJ-UHFFFAOYSA-N 0.000 description 2
- KQVRDVNKFJJSAP-UHFFFAOYSA-N N-[5-[[5-chloro-4-(5-cyanoindazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound ClC=1C(=NC(=NC=1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)N1N=CC2=CC(=CC=C12)C#N KQVRDVNKFJJSAP-UHFFFAOYSA-N 0.000 description 2
- IJPAAYSLMRRAAF-UHFFFAOYSA-N N-[5-[[5-chloro-4-(5-methoxybenzimidazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound ClC=1C(=NC(=NC=1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)N1C=NC2=C1C=CC(=C2)OC IJPAAYSLMRRAAF-UHFFFAOYSA-N 0.000 description 2
- DUDCIJQMAKDOBU-UHFFFAOYSA-N N-[5-[[5-chloro-4-(5-methoxyindazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound ClC=1C(=NC(=NC=1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)N1N=CC2=CC(=CC=C12)OC DUDCIJQMAKDOBU-UHFFFAOYSA-N 0.000 description 2
- WERRTTYKOPFPSS-UHFFFAOYSA-N N-[5-[[5-chloro-4-(6-cyanobenzimidazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound ClC=1C(=NC(=NC=1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)N1C=NC2=C1C=C(C=C2)C#N WERRTTYKOPFPSS-UHFFFAOYSA-N 0.000 description 2
- VOLBSHLEXQQPSA-UHFFFAOYSA-N N-[5-[[5-chloro-4-(6-methoxybenzimidazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound ClC=1C(=NC(=NC=1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)N1C=NC2=C1C=C(C=C2)OC VOLBSHLEXQQPSA-UHFFFAOYSA-N 0.000 description 2
- RIRTYAAQUXGTMC-UHFFFAOYSA-N N-[5-[[5-chloro-4-[1-(oxetan-3-yl)indol-3-yl]pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound ClC=1C(=NC(=NC=1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)C1=CN(C2=CC=CC=C12)C1COC1 RIRTYAAQUXGTMC-UHFFFAOYSA-N 0.000 description 2
- MYGVAIOTJYJZNJ-UHFFFAOYSA-N N-[5-[[5-chloro-4-[2-cyclopropyl-5-(trifluoromethyl)benzimidazol-1-yl]pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C(C=C)(=O)NC1=C(C=C(C(=C1)NC1=NC=C(C(=N1)N1C(=NC2=C1C=CC(=C2)C(F)(F)F)C1CC1)Cl)OC)N(C)CCN(C)C MYGVAIOTJYJZNJ-UHFFFAOYSA-N 0.000 description 2
- WYGNJMCAJWJTLK-UHFFFAOYSA-N NC1=C(C=CC(=C1OC)OC)C(C)=NO Chemical compound NC1=C(C=CC(=C1OC)OC)C(C)=NO WYGNJMCAJWJTLK-UHFFFAOYSA-N 0.000 description 2
- 102000010410 Nogo Proteins Human genes 0.000 description 2
- 108010077641 Nogo Proteins Proteins 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 2
- IQZLUWLMQNGTIW-UHFFFAOYSA-N acetoveratrone Chemical compound COC1=CC=C(C(C)=O)C=C1OC IQZLUWLMQNGTIW-UHFFFAOYSA-N 0.000 description 2
- 239000008186 active pharmaceutical agent Substances 0.000 description 2
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 238000004891 communication Methods 0.000 description 2
- 239000013058 crude material Substances 0.000 description 2
- WLVKDFJTYKELLQ-UHFFFAOYSA-N cyclopropylboronic acid Chemical compound OB(O)C1CC1 WLVKDFJTYKELLQ-UHFFFAOYSA-N 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- WQAWEUZTDVWTDB-UHFFFAOYSA-N dimethyl(oxo)phosphanium Chemical compound C[P+](C)=O WQAWEUZTDVWTDB-UHFFFAOYSA-N 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 238000000132 electrospray ionisation Methods 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 229960001433 erlotinib Drugs 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 2
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 2
- 210000001035 gastrointestinal tract Anatomy 0.000 description 2
- 229960002584 gefitinib Drugs 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- VSEAAEQOQBMPQF-UHFFFAOYSA-N morpholin-3-one Chemical compound O=C1COCCN1 VSEAAEQOQBMPQF-UHFFFAOYSA-N 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 239000007800 oxidant agent Substances 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- 229910000160 potassium phosphate Inorganic materials 0.000 description 2
- 235000011009 potassium phosphates Nutrition 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 230000000171 quenching effect Effects 0.000 description 2
- 230000019491 signal transduction Effects 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 125000003003 spiro group Chemical group 0.000 description 2
- 239000012089 stop solution Substances 0.000 description 2
- GKYVIEZZLURHRE-UHFFFAOYSA-N tert-butyl-[2-[3-(2-chloropyrimidin-4-yl)indol-1-yl]ethoxy]-dimethylsilane Chemical compound [Si](C)(C)(C(C)(C)C)OCCN1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)Cl GKYVIEZZLURHRE-UHFFFAOYSA-N 0.000 description 2
- 239000012085 test solution Substances 0.000 description 2
- VDZOOKBUILJEDG-UHFFFAOYSA-M tetrabutylammonium hydroxide Chemical compound [OH-].CCCC[N+](CCCC)(CCCC)CCCC VDZOOKBUILJEDG-UHFFFAOYSA-M 0.000 description 2
- 238000002877 time resolved fluorescence resonance energy transfer Methods 0.000 description 2
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 2
- PYOKUURKVVELLB-UHFFFAOYSA-N trimethyl orthoformate Chemical compound COC(OC)OC PYOKUURKVVELLB-UHFFFAOYSA-N 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 2
- GTLDTDOJJJZVBW-UHFFFAOYSA-N zinc cyanide Chemical compound [Zn+2].N#[C-].N#[C-] GTLDTDOJJJZVBW-UHFFFAOYSA-N 0.000 description 2
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 1
- MOWXJLUYGFNTAL-DEOSSOPVSA-N (s)-[2-chloro-4-fluoro-5-(7-morpholin-4-ylquinazolin-4-yl)phenyl]-(6-methoxypyridazin-3-yl)methanol Chemical compound N1=NC(OC)=CC=C1[C@@H](O)C1=CC(C=2C3=CC=C(C=C3N=CN=2)N2CCOCC2)=C(F)C=C1Cl MOWXJLUYGFNTAL-DEOSSOPVSA-N 0.000 description 1
- 125000005918 1,2-dimethylbutyl group Chemical group 0.000 description 1
- VJXLBTQOYLNGRO-UHFFFAOYSA-N 1-(2-amino-3,4-dimethoxyphenyl)ethanone Chemical compound COC1=CC=C(C(C)=O)C(N)=C1OC VJXLBTQOYLNGRO-UHFFFAOYSA-N 0.000 description 1
- FSNGFFWICFYWQC-UHFFFAOYSA-N 1-(2-chloroethyl)pyrrolidine;hydron;chloride Chemical compound Cl.ClCCN1CCCC1 FSNGFFWICFYWQC-UHFFFAOYSA-N 0.000 description 1
- PIRRWUMTIBFCCW-UHFFFAOYSA-N 1-(2-fluoro-4-methoxyphenyl)ethanone Chemical compound COC1=CC=C(C(C)=O)C(F)=C1 PIRRWUMTIBFCCW-UHFFFAOYSA-N 0.000 description 1
- XNRQIHIOKXQSPG-UHFFFAOYSA-N 1-(5-bromo-2-fluorophenyl)ethanone Chemical compound CC(=O)C1=CC(Br)=CC=C1F XNRQIHIOKXQSPG-UHFFFAOYSA-N 0.000 description 1
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 1
- DFYYOSJPAYTCFL-UHFFFAOYSA-N 1-N-[4-(6-bromo-1-methylindol-3-yl)pyrimidin-2-yl]-2-(difluoromethoxy)-4-N-[2-(dimethylamino)ethyl]-4-N-methyl-5-nitrobenzene-1,4-diamine Chemical compound BrC1=CC=C2C(=CN(C2=C1)C)C1=NC(=NC=C1)NC1=C(C=C(C(=C1)[N+](=O)[O-])N(C)CCN(C)C)OC(F)F DFYYOSJPAYTCFL-UHFFFAOYSA-N 0.000 description 1
- ABDDQTDRAHXHOC-QMMMGPOBSA-N 1-[(7s)-5,7-dihydro-4h-thieno[2,3-c]pyran-7-yl]-n-methylmethanamine Chemical compound CNC[C@@H]1OCCC2=C1SC=C2 ABDDQTDRAHXHOC-QMMMGPOBSA-N 0.000 description 1
- MLYOYUYYQBQIHW-UHFFFAOYSA-N 1-[3-(2-chloropyrimidin-4-yl)indol-1-yl]ethanone Chemical compound ClC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C(C)=O MLYOYUYYQBQIHW-UHFFFAOYSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000004972 1-butynyl group Chemical group [H]C([H])([H])C([H])([H])C#C* 0.000 description 1
- VHSUHSBTEGHGTG-UHFFFAOYSA-N 1-cyclopropylindole Chemical compound C1CC1N1C2=CC=CC=C2C=C1 VHSUHSBTEGHGTG-UHFFFAOYSA-N 0.000 description 1
- ZRIKJXDEJYMBEJ-UHFFFAOYSA-N 1-fluoro-4-methoxy-2-nitrobenzene Chemical compound COC1=CC=C(F)C([N+]([O-])=O)=C1 ZRIKJXDEJYMBEJ-UHFFFAOYSA-N 0.000 description 1
- BLRHMMGNCXNXJL-UHFFFAOYSA-N 1-methylindole Chemical compound C1=CC=C2N(C)C=CC2=C1 BLRHMMGNCXNXJL-UHFFFAOYSA-N 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- 125000000530 1-propynyl group Chemical group [H]C([H])([H])C#C* 0.000 description 1
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 1
- ZHDXWEPRYNHNDC-UHFFFAOYSA-N 1h-indazol-5-ol Chemical compound OC1=CC=C2NN=CC2=C1 ZHDXWEPRYNHNDC-UHFFFAOYSA-N 0.000 description 1
- XAWPKHNOFIWWNZ-UHFFFAOYSA-N 1h-indol-6-ol Chemical compound OC1=CC=C2C=CNC2=C1 XAWPKHNOFIWWNZ-UHFFFAOYSA-N 0.000 description 1
- 125000003562 2,2-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003660 2,3-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- GIKMWFAAEIACRF-UHFFFAOYSA-N 2,4,5-trichloropyrimidine Chemical compound ClC1=NC=C(Cl)C(Cl)=N1 GIKMWFAAEIACRF-UHFFFAOYSA-N 0.000 description 1
- 125000003764 2,4-dimethylpentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- BPXKZEMBEZGUAH-UHFFFAOYSA-N 2-(chloromethoxy)ethyl-trimethylsilane Chemical compound C[Si](C)(C)CCOCCl BPXKZEMBEZGUAH-UHFFFAOYSA-N 0.000 description 1
- USZCDSGRLOKEDU-UHFFFAOYSA-N 2-(difluoromethoxy)-4-N-[2-(dimethylamino)ethyl]-1-N-[4-(6-methoxy-1-methylindol-3-yl)pyrimidin-2-yl]-4-N-methyl-5-nitrobenzene-1,4-diamine Chemical compound FC(OC1=C(C=C(C(=C1)N(C)CCN(C)C)[N+](=O)[O-])NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)OC)C)F USZCDSGRLOKEDU-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 1
- LPBDZVNGCNTELM-UHFFFAOYSA-N 2-chloropyrimidin-4-amine Chemical compound NC1=CC=NC(Cl)=N1 LPBDZVNGCNTELM-UHFFFAOYSA-N 0.000 description 1
- 125000006176 2-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- 125000003229 2-methylhexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000005916 2-methylpentyl group Chemical group 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 125000004336 3,3-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- PSGUTOJJBLVCKB-UHFFFAOYSA-N 3-[2-[2-(difluoromethoxy)-4-[2-(dimethylamino)ethyl-methylamino]-5-nitroanilino]pyrimidin-4-yl]-N,N-dimethylindole-1-sulfonamide Chemical compound FC(OC1=C(C=C(C(=C1)N(C)CCN(C)C)[N+](=O)[O-])NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)S(=O)(=O)N(C)C)F PSGUTOJJBLVCKB-UHFFFAOYSA-N 0.000 description 1
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 1
- 125000000474 3-butynyl group Chemical group [H]C#CC([H])([H])C([H])([H])* 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- 125000004337 3-ethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- KBEIFKMKVCDETC-UHFFFAOYSA-N 3-iodooxetane Chemical compound IC1COC1 KBEIFKMKVCDETC-UHFFFAOYSA-N 0.000 description 1
- 125000003542 3-methylbutan-2-yl group Chemical group [H]C([H])([H])C([H])(*)C([H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000003469 3-methylhexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005917 3-methylpentyl group Chemical group 0.000 description 1
- QGLQEYPASZYGBM-UHFFFAOYSA-N 4-N-[2-(dimethylamino)ethyl]-2-methoxy-1-N-[4-(5-methoxyindazol-1-yl)pyrimidin-2-yl]-4-N-methyl-5-nitrobenzene-1,4-diamine Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)N1N=CC2=CC(=CC=C12)OC)[N+](=O)[O-])C)C QGLQEYPASZYGBM-UHFFFAOYSA-N 0.000 description 1
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 1
- QVBHRCAJZGMNFX-UHFFFAOYSA-N 4-fluoro-3-formylbenzonitrile Chemical compound FC1=CC=C(C#N)C=C1C=O QVBHRCAJZGMNFX-UHFFFAOYSA-N 0.000 description 1
- 125000000590 4-methylphenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 1
- STVHMYNPQCLUNJ-UHFFFAOYSA-N 5-bromo-1h-indazole Chemical compound BrC1=CC=C2NN=CC2=C1 STVHMYNPQCLUNJ-UHFFFAOYSA-N 0.000 description 1
- MMFGGDVQLQQQRX-UHFFFAOYSA-N 5-bromo-2-fluorobenzaldehyde Chemical compound FC1=CC=C(Br)C=C1C=O MMFGGDVQLQQQRX-UHFFFAOYSA-N 0.000 description 1
- WGAVMKXCDMQVNF-UHFFFAOYSA-N 5-chloro-2-fluorobenzoic acid Chemical compound OC(=O)C1=CC(Cl)=CC=C1F WGAVMKXCDMQVNF-UHFFFAOYSA-N 0.000 description 1
- QQURWFRNETXFTN-UHFFFAOYSA-N 5-fluoro-2-nitrophenol Chemical compound OC1=CC(F)=CC=C1[N+]([O-])=O QQURWFRNETXFTN-UHFFFAOYSA-N 0.000 description 1
- GZWWDKIVVTXLFL-UHFFFAOYSA-N 5-methoxy-1h-indazole Chemical compound COC1=CC=C2NN=CC2=C1 GZWWDKIVVTXLFL-UHFFFAOYSA-N 0.000 description 1
- DWAQDRSOVMLGRQ-UHFFFAOYSA-N 5-methoxyindole Chemical compound COC1=CC=C2NC=CC2=C1 DWAQDRSOVMLGRQ-UHFFFAOYSA-N 0.000 description 1
- KCBWAFJCKVKYHO-UHFFFAOYSA-N 6-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-[[4-[1-propan-2-yl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]pyrazolo[3,4-d]pyrimidine Chemical compound C1(CC1)C1=NC=NC(=C1C1=NC=C2C(=N1)N(N=C2)CC1=CC=C(C=C1)C=1N(C=C(N=1)C(F)(F)F)C(C)C)OC KCBWAFJCKVKYHO-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 1
- UQMUVKHIRWJXSS-UHFFFAOYSA-N CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)C#C[Si](C)(C)C)C)[N+](=O)[O-])C)C Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)C#C[Si](C)(C)C)C)[N+](=O)[O-])C)C UQMUVKHIRWJXSS-UHFFFAOYSA-N 0.000 description 1
- LDCMIZRJIVPHDE-UHFFFAOYSA-N CN(CCN(C=1C(=CC(=C(C=1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)C#C[Si](C)(C)C)C)N)C)C Chemical compound CN(CCN(C=1C(=CC(=C(C=1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)C#C[Si](C)(C)C)C)N)C)C LDCMIZRJIVPHDE-UHFFFAOYSA-N 0.000 description 1
- POLHPQCMCYRWNT-UHFFFAOYSA-N COC1=CC(=NC(=N1)C1(CC=C(C=C1[N+](=O)[O-])N)NC)C1=CN(C2=CC=CC=C12)C Chemical compound COC1=CC(=NC(=N1)C1(CC=C(C=C1[N+](=O)[O-])N)NC)C1=CN(C2=CC=CC=C12)C POLHPQCMCYRWNT-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- 238000006646 Dess-Martin oxidation reaction Methods 0.000 description 1
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 229910004373 HOAc Inorganic materials 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 1
- ITYYAINPUOKSDE-UHFFFAOYSA-N N-(4-fluoro-2-methoxy-5-nitrophenyl)formamide Chemical compound FC1=CC(=C(C=C1[N+](=O)[O-])NC=O)OC ITYYAINPUOKSDE-UHFFFAOYSA-N 0.000 description 1
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 1
- UVKIJHIAPURYHP-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(1-prop-2-ynylindol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC)NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)CC#C)NC(C=C)=O)C)C UVKIJHIAPURYHP-UHFFFAOYSA-N 0.000 description 1
- MNIPZYVGDRHCDH-UHFFFAOYSA-N N-[2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-(6-dimethylphosphoryl-1-methylindol-3-yl)pyrimidin-2-yl]amino]-4-(trifluoromethoxy)phenyl]prop-2-enamide Chemical compound CN(CCN(C1=C(C=C(C(=C1)OC(F)(F)F)NC1=NC=CC(=N1)C1=CN(C2=CC(=CC=C12)P(=O)(C)C)C)NC(C=C)=O)C)C MNIPZYVGDRHCDH-UHFFFAOYSA-N 0.000 description 1
- SNYMAKDKMCAXHS-UHFFFAOYSA-N N-[4-(difluoromethoxy)-2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-[1-(dimethylsulfamoyl)indol-3-yl]pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound FC(OC1=CC(=C(C=C1NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)S(N(C)C)(=O)=O)NC(C=C)=O)N(C)CCN(C)C)F SNYMAKDKMCAXHS-UHFFFAOYSA-N 0.000 description 1
- UENMWNYZTHFWCY-UHFFFAOYSA-N N-[4-methoxy-2-[methyl-[2-(methylamino)ethyl]amino]-5-[[4-(1-prop-2-ynylindol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide Chemical compound COC1=CC(=C(C=C1NC1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)CC#C)NC(C=C)=O)N(CCNC)C UENMWNYZTHFWCY-UHFFFAOYSA-N 0.000 description 1
- HYGKWMZHAGPMDO-UHFFFAOYSA-N N-[5-[[4-(1-cyclopropylindol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)N1C=C(C2=CC=CC=C12)C1=NC(=NC=C1C(F)(F)F)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC HYGKWMZHAGPMDO-UHFFFAOYSA-N 0.000 description 1
- DOEOECWDNSEFDN-UHFFFAOYSA-N N-[5-[[4-(1-cyclopropylindol-3-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C1(CC1)N1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC DOEOECWDNSEFDN-UHFFFAOYSA-N 0.000 description 1
- KLFANBQCAOJEEQ-UHFFFAOYSA-N N-[5-[[4-(1-cyclopropylindol-3-yl)pyrimidin-2-yl]amino]-4-methoxy-2-[methyl-[2-(methylamino)ethyl]amino]phenyl]prop-2-enamide Chemical compound C1(CC1)N1C=C(C2=CC=CC=C12)C1=NC(=NC=C1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(CCNC)C)OC KLFANBQCAOJEEQ-UHFFFAOYSA-N 0.000 description 1
- HEHARNBGGJOPGM-UHFFFAOYSA-N N-[5-[[4-(5-cyano-2-cyclopropylbenzimidazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound C(#N)C1=CC2=C(N(C(=N2)C2CC2)C2=NC(=NC=C2)NC=2C(=CC(=C(C=2)NC(C=C)=O)N(C)CCN(C)C)OC)C=C1 HEHARNBGGJOPGM-UHFFFAOYSA-N 0.000 description 1
- SWQKTZYLQAOZRV-UHFFFAOYSA-N N-[5-[[5-chloro-4-(5-cyano-3-ethylindazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound ClC=1C(=NC(=NC=1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)N1N=C(C2=CC(=CC=C12)C#N)CC SWQKTZYLQAOZRV-UHFFFAOYSA-N 0.000 description 1
- SSCPYYYMXXPSBY-UHFFFAOYSA-N N-[5-[[5-chloro-4-(5-cyanobenzimidazol-1-yl)pyrimidin-2-yl]amino]-2-[2-(dimethylamino)ethyl-methylamino]-4-methoxyphenyl]prop-2-enamide Chemical compound ClC=1C(=NC(=NC=1)NC=1C(=CC(=C(C=1)NC(C=C)=O)N(C)CCN(C)C)OC)N1C=NC2=C1C=CC(=C2)C#N SSCPYYYMXXPSBY-UHFFFAOYSA-N 0.000 description 1
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 102000004278 Receptor Protein-Tyrosine Kinases Human genes 0.000 description 1
- 108090000873 Receptor Protein-Tyrosine Kinases Proteins 0.000 description 1
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical class [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 1
- 241001660687 Xantho Species 0.000 description 1
- LXRZVMYMQHNYJB-UNXOBOICSA-N [(1R,2S,4R)-4-[[5-[4-[(1R)-7-chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxycyclopentyl]methyl sulfamate Chemical compound CC1=C(C=C(S1)C(=O)C1=C(N[C@H]2C[C@H](O)[C@@H](COS(N)(=O)=O)C2)N=CN=C1)[C@@H]1NCCC2=C1C=C(Cl)C=C2 LXRZVMYMQHNYJB-UNXOBOICSA-N 0.000 description 1
- DBMWEYKCQJHBKQ-UHFFFAOYSA-N [Si](C)(C)(C)C[Si](C)(C)C.C[Si](C)(C)C Chemical compound [Si](C)(C)(C)C[Si](C)(C)C.C[Si](C)(C)C DBMWEYKCQJHBKQ-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- PMZXXNPJQYDFJX-UHFFFAOYSA-N acetonitrile;2,2,2-trifluoroacetic acid Chemical compound CC#N.OC(=O)C(F)(F)F PMZXXNPJQYDFJX-UHFFFAOYSA-N 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 230000006909 anti-apoptosis Effects 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- INLLPKCGLOXCIV-UHFFFAOYSA-N bromoethene Chemical compound BrC=C INLLPKCGLOXCIV-UHFFFAOYSA-N 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 230000010307 cell transformation Effects 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000002188 cycloheptatrienyl group Chemical group C1(=CC=CC=CC1)* 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000003678 cyclohexadienyl group Chemical group C1(=CC=CCC1)* 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- SDYFTMROHZCYSK-UHFFFAOYSA-N cyclopropylhydrazine;hydrochloride Chemical compound Cl.NNC1CC1 SDYFTMROHZCYSK-UHFFFAOYSA-N 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- WGLUMOCWFMKWIL-UHFFFAOYSA-N dichloromethane;methanol Chemical compound OC.ClCCl WGLUMOCWFMKWIL-UHFFFAOYSA-N 0.000 description 1
- 125000004772 dichloromethyl group Chemical group [H]C(Cl)(Cl)* 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- QKIUAMUSENSFQQ-UHFFFAOYSA-N dimethylazanide Chemical compound C[N-]C QKIUAMUSENSFQQ-UHFFFAOYSA-N 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 108700021358 erbB-1 Genes Proteins 0.000 description 1
- IDGUHHHQCWSQLU-UHFFFAOYSA-N ethanol;hydrate Chemical compound O.CCO IDGUHHHQCWSQLU-UHFFFAOYSA-N 0.000 description 1
- PAVZHTXVORCEHP-UHFFFAOYSA-N ethylboronic acid Chemical compound CCB(O)O PAVZHTXVORCEHP-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 238000009093 first-line therapy Methods 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- 239000000710 homodimer Substances 0.000 description 1
- WQYVRQLZKVEZGA-UHFFFAOYSA-N hypochlorite Inorganic materials Cl[O-] WQYVRQLZKVEZGA-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- 238000011221 initial treatment Methods 0.000 description 1
- 230000031146 intracellular signal transduction Effects 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical group II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- 229940084651 iressa Drugs 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- VFZXMEQGIIWBFJ-UHFFFAOYSA-M magnesium;cyclopropane;bromide Chemical compound [Mg+2].[Br-].C1C[CH-]1 VFZXMEQGIIWBFJ-UHFFFAOYSA-M 0.000 description 1
- UGVPKMAWLOMPRS-UHFFFAOYSA-M magnesium;propane;bromide Chemical compound [Mg+2].[Br-].CC[CH2-] UGVPKMAWLOMPRS-UHFFFAOYSA-M 0.000 description 1
- 238000003760 magnetic stirring Methods 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- GBMDVOWEEQVZKZ-UHFFFAOYSA-N methanol;hydrate Chemical compound O.OC GBMDVOWEEQVZKZ-UHFFFAOYSA-N 0.000 description 1
- XMJHPCRAQCTCFT-UHFFFAOYSA-N methyl chloroformate Chemical compound COC(Cl)=O XMJHPCRAQCTCFT-UHFFFAOYSA-N 0.000 description 1
- SURZCVYFPAXNGN-UHFFFAOYSA-N methyl-carbamic acid ethyl ester Chemical compound CCOC(=O)NC SURZCVYFPAXNGN-UHFFFAOYSA-N 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- KVKFRMCSXWQSNT-UHFFFAOYSA-N n,n'-dimethylethane-1,2-diamine Chemical compound CNCCNC KVKFRMCSXWQSNT-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 125000006299 oxetan-3-yl group Chemical group [H]C1([H])OC([H])([H])C1([H])* 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- YORCIIVHUBAYBQ-UHFFFAOYSA-N propargyl bromide Chemical compound BrCC#C YORCIIVHUBAYBQ-UHFFFAOYSA-N 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 229940083082 pyrimidine derivative acting on arteriolar smooth muscle Drugs 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000008261 resistance mechanism Effects 0.000 description 1
- 238000002390 rotary evaporation Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- MRTAVLDNYYEJHK-UHFFFAOYSA-M sodium;2-chloro-2,2-difluoroacetate Chemical compound [Na+].[O-]C(=O)C(F)(F)Cl MRTAVLDNYYEJHK-UHFFFAOYSA-M 0.000 description 1
- VNFWTIYUKDMAOP-UHFFFAOYSA-N sphos Chemical compound COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 VNFWTIYUKDMAOP-UHFFFAOYSA-N 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229940120982 tarceva Drugs 0.000 description 1
- 238000002626 targeted therapy Methods 0.000 description 1
- VSFIVFVGTPNMMI-UHFFFAOYSA-N tert-butyl 3-[2-[2-methoxy-4-[methyl-[2-[methyl-[(2-methylpropan-2-yl)oxycarbonyl]amino]ethyl]amino]-N-[(2-methylpropan-2-yl)oxycarbonyl]-5-nitroanilino]pyrimidin-4-yl]indole-1-carboxylate Chemical compound C(C)(C)(C)OC(=O)N(C1=NC=CC(=N1)C1=CN(C2=CC=CC=C12)C(=O)OC(C)(C)C)C1=C(C=C(C(=C1)[N+](=O)[O-])N(C)CCN(C)C(=O)OC(C)(C)C)OC VSFIVFVGTPNMMI-UHFFFAOYSA-N 0.000 description 1
- RFDSJHHLGFFVHD-UHFFFAOYSA-N tert-butyl n-(2-hydroxyethyl)-n-methylcarbamate Chemical compound OCCN(C)C(=O)OC(C)(C)C RFDSJHHLGFFVHD-UHFFFAOYSA-N 0.000 description 1
- DFVRUHANEXOZGT-UHFFFAOYSA-N tert-butyl n-methyl-n-[2-(methylamino)ethyl]carbamate Chemical compound CNCCN(C)C(=O)OC(C)(C)C DFVRUHANEXOZGT-UHFFFAOYSA-N 0.000 description 1
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- YJBKVPRVZAQTPY-UHFFFAOYSA-J tetrachlorostannane;dihydrate Chemical compound O.O.Cl[Sn](Cl)(Cl)Cl YJBKVPRVZAQTPY-UHFFFAOYSA-J 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 102000035160 transmembrane proteins Human genes 0.000 description 1
- 108091005703 transmembrane proteins Proteins 0.000 description 1
- 125000004784 trichloromethoxy group Chemical group ClC(O*)(Cl)Cl 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- CWMFRHBXRUITQE-UHFFFAOYSA-N trimethylsilylacetylene Chemical group C[Si](C)(C)C#C CWMFRHBXRUITQE-UHFFFAOYSA-N 0.000 description 1
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/662—Phosphorus acids or esters thereof having P—C bonds, e.g. foscarnet, trichlorfon
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/48—Two nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/06—Peri-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/50—Organo-phosphines
- C07F9/53—Organo-phosphine oxides; Organo-phosphine thioxides
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pulmonology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Изобретение относится к новому производному 4-замещенного-2-(N-(5-замещенного-аллиламида)фенил)-амино)пиримидина, представленному формулой (I). Соединения обладают свойствами ингибитора EGFR мутантного типа, в частности мутанта EGFR-T790M, и могут найти применение для лечения заболевания, связанного с клеточной пролиферацией, опосредованного активностью мутанта EGFR-T790M. Таким заболеванием может быть рак, который может быть выбран из группы, состоящей из рака яичника, рака шейки матки, колоректального рака, рака молочной железы, рака поджелудочной железы, глиомы, глиобластомы, меланомы, рака предстательной железы, лейкемии, лимфомы, неходжкинской лимфомы, рака желудка, рака легкого, гепатоцеллюлярной карциномы, рака желудка, гастроинтестинальной стромальной опухоли (GIST), рака щитовидной железы, холангиокарциномы, рака эндометрия, рака почки, анапластической крупноклеточной лимфомы, острого миелоидного лейкоза (AML), множественной миеломы, меланомы и мезотелиомы; предпочтительно не мелкоклеточного рака легкого. В формуле (I)

кольцо А выбирают из группы, состоящей из

где Q представляет собой связь; R выбирают из группы, состоящей из водорода; X1, Х2 и Х3 каждый независимо выбирают из группы, состоящей из NR7 и CR8, где один или два из X1, Х2 и Х3 представляют собой NR7, причем, в случае когда Х2 представляет собой NR7, другие группы представляют собой CR8, и кольцо с указанной группой Х2 присоединяется атомом углерода к пиримидиновому кольцу, или когда два из X1, Х2 и Х3 представляют собой NR7, то кольцо присоединяется атомом азота указанной группы к пиримидиновому кольцу; R1 выбирают из группы, состоящей из:


причем три R6 в  представляют собой необязательно одинаковые или разные заместители; R2 выбирают из группы, состоящей из C1-8 алкила, где С1-8 алкил необязательно замещен одной или более группами, выбранными из группы, состоящей из галогена; R3 выбирают из группы, состоящей из водорода, галогена, трифторметила, трифторметокси и P(O)R11R12, где R11R12 представляют собой C1-6 алкил; R4 независимо выбирают из группы, состоящей из водорода, галогена, циано, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 4-членного насыщенного гетероциклила, содержащего атом кислорода в цикле, -P(O)R11R12, где R11R12 представляют собой С1-6 алкил, -S(O)rR9, -O-R10, -C(O)R10, где C1-8 алкил необязательно замещен одной или более группами, выбранными из группы, состоящей из галогена, гидрокси и -O-R10; R6 выбирают из группы, состоящей из водорода и C1-8 алкила; R7 выбирают из группы, состоящей из C1-8 алкила, С2-8 алкенила, С3-6 циклоалкила, -S(O)rR9, -O-R10, -C(O)R10 и 4-членного насыщенного гетероциклила, содержащего атом кислорода в цикле; где C1-8 алкил замещен одной или более группами, выбранными из группы, состоящей из гидрокси, С2-8 алкенила и С2-8 алкинила; R8 выбирают из группы, состоящей из водорода, С1-8 алкила, С3-8 циклоалкила и -O-R10; R9 выбирают из группы, состоящей из бис-C1-8 алкиламино; R10 выбирают из группы, состоящей из C1-8 алкила и С3-8 циклоалкила; m равно 0, 1, 2; r равно 2; о равно 0; р равно 1; q равно 1. Способ получения соединения I осуществляют согласно следующей схеме
представляют собой необязательно одинаковые или разные заместители; R2 выбирают из группы, состоящей из C1-8 алкила, где С1-8 алкил необязательно замещен одной или более группами, выбранными из группы, состоящей из галогена; R3 выбирают из группы, состоящей из водорода, галогена, трифторметила, трифторметокси и P(O)R11R12, где R11R12 представляют собой C1-6 алкил; R4 независимо выбирают из группы, состоящей из водорода, галогена, циано, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 4-членного насыщенного гетероциклила, содержащего атом кислорода в цикле, -P(O)R11R12, где R11R12 представляют собой С1-6 алкил, -S(O)rR9, -O-R10, -C(O)R10, где C1-8 алкил необязательно замещен одной или более группами, выбранными из группы, состоящей из галогена, гидрокси и -O-R10; R6 выбирают из группы, состоящей из водорода и C1-8 алкила; R7 выбирают из группы, состоящей из C1-8 алкила, С2-8 алкенила, С3-6 циклоалкила, -S(O)rR9, -O-R10, -C(O)R10 и 4-членного насыщенного гетероциклила, содержащего атом кислорода в цикле; где C1-8 алкил замещен одной или более группами, выбранными из группы, состоящей из гидрокси, С2-8 алкенила и С2-8 алкинила; R8 выбирают из группы, состоящей из водорода, С1-8 алкила, С3-8 циклоалкила и -O-R10; R9 выбирают из группы, состоящей из бис-C1-8 алкиламино; R10 выбирают из группы, состоящей из C1-8 алкила и С3-8 циклоалкила; m равно 0, 1, 2; r равно 2; о равно 0; р равно 1; q равно 1. Способ получения соединения I осуществляют согласно следующей схеме

где кольцо A, Q, X1, X2, X3, R, R1, R2, R3, R4, R6, R7, R8, R9, R10, R11, R12, m, r, о, p и q являются такими, как определено выше. 16 н. и 20 з.п. ф-лы, 6 табл., 143 пр.
Description
Область техники
Настоящее изобретение относится к области фармацевтического синтеза, в частности, относится к ингибитору EGFR, способу его получения и его применению.
Предшествующий уровень техники
EGFR (Epidermal Growth Factor Receptor, рецептор эпидермального фактора роста) является членом семейства erbB рецепторов, которое относится к трансмембранному белковому тирозинкиназному рецептору. При связывании с его лигандом, таким как эпидермальный фактор роста (EGF), EGFR может образовывать гомодимер на клеточной мембране, или образовывать гетеродимер с другими рецепторами в семействе, такими как erbB2, erbB3 или erbB4. Образование этих димеров может вызвать фосфорилирование ключевых тирозиновых остатков в клетках EGFR, тем самым активируя ряд нижележащих сигнальных путей в клетках. Эти внутриклеточные сигнальные пути играют важную роль в клеточной пролиферации, выживании и антиапоптозе. Нарушения EGFR-зависимых путей трансдукции сигнала, включая повышение экспрессии лигандов и рецепторов, амплификацию и мутацию гена EGFR и т.п., могут способствовать злокачественной трансформации клеток и играть важную роль в пролиферации, инвазии, метастазировании и ангиогенезе опухолевых клеток. Таким образом, EGFR является обоснованной мишенью для развития противоопухолевых препаратов.
Первое поколение низкомолекулярных ингибиторов EGFR, включая гефитиниб (Iressa™) и эрлотиниб (Tarceva™), показало хорошую эффективность при лечении рака легких и использовалось в качестве препаратов первой линии для лечения немелкоклеточного рака легкого (НМРЛ), связанного с EGFR-активирующей мутацией (New England Journal of Medicine (2008), том 358, 1160-74, Biochemical and Biophysical Communications Communications (2004), том 319, 1-11).
В отличие от EGFR дикого типа (WT wild-type), активированный EGFR мутантного типа (включая L858R и delE746_A750 с делецией в экзоне 19) имеет более низкое сродство к аденозинтрифосфату (АТФ), но обладает более высоким сродством к низкомолекулярным ингибиторам, что приводит к повышению восприимчивости опухолевых клеток к первому поколению ингибиторов EGFR, таких как гефитиниб или эрлотиниб, тем самым достигая таргетной терапии [Science, 2004, No. 304, 1497-500], New England Journalof Medicine [2004] No. 350, 2129-39).
Однако через 10-12 месяцев лечения первым поколением низкомолекулярных ингибиторов EGFR, резистентность к этим низкомолекулярным ингибиторам наблюдалась почти у всех пациентов с НМРЛ. Механизмы устойчивости включают вторичные мутации EGFR, обходную активацию и тому подобное. Таким образом, половина лекарственной устойчивости связана с вторичными мутациями Т790М, которая является остатком гена-привратника EGFR. Вторичные мутации уменьшают сродство лекарственного средства к мишени, тем самым вызывая резистентность к лекарству, и приводя к рецидиву опухоли или прогрессированию заболевания. Ввиду важности и универсальности этой мутации для резистентности к лекарственным средствам, развивающейся в ходе терапии направленной на EGFR рака легкого, ряд фармацевтических научно-исследовательских компаний (Pfizer, BI, AZ и др.) попытались разработать второе поколение низкомолекулярных ингибиторов EGFR для лечения этих пациентов с лекарственно-устойчивым раком легких, путем ингибирования мутанта EGFR-T790M. Однако, все попытки терпели неудачу из-за плохой избирательности. Несмотря на то, что афатиниб (afatinib) был одобрен FDA для лечения рака легких, он использовался только в терапии первой линии у пациентов с раком, связанным с EGFR-активирующей мутацией. Однако афатиниб не проявлял терапевтического эффекта у пациентов с раком, связанным с EGFR-T790M мутацией, поскольку афатиниб оказывает более сильное ингибирующее действие на EGFR дикого типа, что вызывает серьезную токсичность для кожи и желудочно-кишечного тракта, тем самым ограничивая дозу введения.
Таким образом, необходимо разработать третье поколение низкомолекулярных ингибиторов EGFR, которые могут с высокой селективностью ингибировать мутант EGFR Т790М и не обладают, или обладают низкой активностью в отношении EGFR дикого типа.
Из-за такой высокой селективности, повреждение кожи и желудочно-кишечного тракта, вызванное ингибированием EGFR дикого типа, может быть значительно уменьшено, и лекарственно-резистентную опухоль, вызванную вторичной EGFR-T790M мутацией, можно лечить. Кроме того, имеет смысл поддерживать ингибирующую активность в отношении EGFR-активирующего мутанта (включая EGFR-L858R и delE746_A750 с делецией в 19-м экзоне).
Из-за более низкого ингибирования EGFR дикого типа, третье поколение ингибиторов EGFR обладает лучшей безопасностью, чем первое поколение ингибиторов EGFR, и прогнозируется в качестве терапии первой линии при лечении НМРЛ, связаного с EGFR-активирующей мутацией, в то же время, устраняющей небольшое количество мутанта EGFR-T790T, которое может присутствовать у пациентов с начальным лечением, чтобы задержать лекарственную устойчивость.
Рак легких - это серьезное заболевание, которое угрожает здоровью человека, а смертность от рака легких стоит на первом месте среди всех злокачественных опухолей. В Китае заболеваемость раком легких увеличивается из года в год, и ежегодно регистрируется приблизительно 700000 новых случаев заболевания. В Европе и Америке на рак легкого, связанный с EGFR-активирующей мутацией, приходится приблизительно 10% всех случаев НМРЛ; в то время как в Китае это соотношение составляет до 30%. Таким образом, Китай имеет больший рынок для ингибиторов EGFR.
Краткое описание изобретения
В ходе исследования, изобретатели обнаружили серию производных 4-замещенного-2-(N-(5-аллиламидо)фенил)амино)пиримидина, представленного формулой (I), которые обладают ингибирующей активностью в отношении мутанта EGFR-L858R, мутанта EGFR-T790M и мутанта, активированного делецией 19 экзона, и могут быть использованы отдельно или частично для лечения заболеваний, опосредованных активностью мутанта EGFR. Например, эти производные предназначены для широкого использования в профилактике и лечении рака, в частности, немелкоклеточного рака легкого.
В одном аспекте, настоящее изобретение относится к соединению формулы (I), его стереоизомеру или его фармацевтически приемлемой соли:

где
кольцо А выбрано из группы, состоящей из

Q выбран из группы, состоящей из связи, О, S, NR7 и CR7R8; R выбран из группы, состоящей из водорода и бис-C1-8 алкиламинометила;
X1, Х2 и Х3 каждый независимо выбраны из группы, состоящей из NR7 и CR8, где, по меньшей мере, один из X1, Х2 и Х3 представляет собой NR7;
R1 выбран из группы, состоящей из:


где три R6 в  представляют собой, необязательно, одинаковые или разные заместители;
представляют собой, необязательно, одинаковые или разные заместители;
R2 выбран из группы, состоящей из C1-8 алкила и С3-8 циклоалкила, где C1-8 алкил и С3-8 циклоалкил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из галогена, гидрокси, C1-8 алкила, C1-8 алкокси, галоген C1-8 алкокси, С3-8 циклоалкила и С3-8циклоалкокси;
R3 выбран из группы, состоящей из водорода, дейтерия, галогена, циано, нитро, C1-8 алкила, C1-8 алкокси, С3-8 циклоалкила, трифторметила, трифторметокси, SO2R9, C(O)R10, C(O)OR10, P(O)R11R12;
R4 и R5 каждый независимо выбраны из группы, состоящей из водорода, дейтерия, галогена, гидрокси, сульфгидрила, циано, нитро, азидо, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арил, С5-10 арилокси, С5-10 арилтио, 5-10-членного гетероарила, 5-10- членного гетероарилокси, 5-10-членного гетероарилтио, -C0-8-P(O)R11R12, -C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10;
или,
два R4 или два R5 вместе с атомами углерода присоединенного бензольного кольца образуют 5-7-членный карбоцикл, 5-7-членный гетероцикл, С5-7 арил или 5-7-членный гетероарил;
где C1-8 алкил, С3-8 циклоалкил, 3-8-членный гетероциклил, С5-10 арил, 5-10-членный гетероарил, 5-7-членный карбоцикл, 5-7-членный гетероцикл, С5-7 арил и 5-7-членный гетероарил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из галогена, гидрокси, сульфгидрила, циано, нитро, азидо, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арила, С5-10 арилокси, С5-10 арилтио, 5-10-членного гетероарила, 5-10-членного гетероарилокси, 5-10-членного гетероарилтио, -C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10;
R6 выбран из группы, состоящей из водорода, дейтерия, C1-8 алкила, галоген C1-8 алкила и C(O)R10;
R7 выбран из группы, состоящей из водорода, дейтерия, C1-8 алкила С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, С5-10 арила, 5-10-членного гетероарила, C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8 и -C0-8-C(O)NR7R8;
где C1-8 алкил, С3-8 циклоалкил, 3-8-членный гетероциклил, С5-10 арил и 5-10-членный гетероарил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из галогена, гидрокси, сульфгидрила, циано, нитро, азидо, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арил, С5-10 арилокси, С5-10 арилтио, 5-10-членный гетероарил, 5-10-членный гетероарилокси, 5-10-членный гетероарилтио, C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10;
R8 выбран из группы, состоящей из водорода, дейтерия, галогена, гидрокси, сульфгидрила, циано, нитро, азидо, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арила, С5-10 арилокси, С5-10 арилтио, 5-10-членного гетероарила, 5-10-членного гетероарилокси, 5-10-членного гетероарилтио, -C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10;
где C1-8 алкил, С3-8 циклоалкил, 3-8-членный гетероциклил, С5-10 арил и 5-10-членный гетероарил, каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из галогена, гидрокси, сульфгидрила, циано, нитро, азидо, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арила, С5-10 арилокси, С5-10 арилтио, 5-10-членного гетероарила, 5-10-членного гетероарилокси, 5-10-членного гетероарилтио, -C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10;
R9 выбран из группы, состоящей из водорода, дейтерия, C1-8 алкила, С3-8 циклоалкила, галоген C1-8 алкила, бис-C1-8 алкиламино, фенила и п-метилфенила;
R10, R11 и R12 каждый независимо выбраны из группы, состоящей из водорода, дейтерия, C1-8 алкила, С3-8 циклоалкила, галоген C1-8 алкила и гидрокси C1-8 алкила;
m равно 0, 1, 2, 3 или 4;
n равно 0, 1, 2, 3, 4 или 5;
r равно 0, 1 или 2;
о равно 0, 1, 2, 3 или 4;
р равно 0, 1, 2 или 3;
q равно 0, 1, 2, 3 или 4;
В предпочтительном варианте осуществления, в соединении формулы (I) его стереоизомере или его фармацевтически приемлемой соли, R2 выбран из группы, состоящей из С1-4 алкила и С3-6 циклоалкила, где С1-4 алкил и С3-6 циклоалкил, необязательно, каждый замещен одной или более группами, выбранными из группы, состоящей из галогена, гидрокси, С1-8 алкила, C1-8 алкокси, галогена C1-8 алкокси, С3-8 циклоалкила и С3-8 циклоалкокси; кольцо A, Q, R, X1, Х2, Х3, R1, R3, R4, R5, R6, R7, R8, R9, R10, R11, R12, m, n, r, о, p и q являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления в соединении формулы (I) его стереоизомере или его фармацевтически приемлемой соли, R2 выбран из группы, состоящей из С1-4 алкила и С3-6 циклоалкила, где С1-4 алкил и С3-6 циклоалкил, необязательно, замещены одной или более группами, выбранными из группы, состоящей из галогена и гидрокси; кольцо A, Q, R, Х1, Х2, Х3, R1, R3, R4, R5, R6, R7, R8, R9, R10, R11, R12, m, n, r, о, p и q являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, в соединении формулы (I) его стереоизомере или его фармацевтически приемлемой соли, R2 представляет собой С1-4 алкил, необязательно замещенный одной или более группами, выбранными из группы, состоящей из фтора и гидроксигруппы; кольцо A, Q, R, Х1, Х2, Х3, R1, R3, R4, R5, R6, R7, R8, R9, R10, R11, R12, m, n, r, о, p и q являются такими, как определено в соединение формулы (I).
В более предпочтительном варианте осуществления, в соединении формулы (I), его стереоизомере или его фармацевтически приемлемой соли, R2 выбран из группы, состоящей из метила, дифторметила и трифторметила; кольцо A, Q, R, Х1, Х2, Х3, R1, R3, R4, R5, R6, R7, R8, R9, R10, R11, R12, m, n и r являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль представляют собой соединение формулы (IA):

где R2 выбран из группы, состоящей из метила, дифторметила и трифторметила; кольцо A, R, Х1, Х2, Х3, R1, R3, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, соединение формулы (I), стереоизомер или его фармацевтически приемлемую соль выбирают из группы, состоящей из соединения формулы (IIA1) и соединения формулы (IIA2):


где R2 выбран из группы, состоящей из метила, дифторметила и трифторметила; кольцо A, R, X1, Х2, Х3, R1, R3, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль выбраны из группы, состоящей из соединения формулы (IIIA1-1), соединения формулы (IIIA1-2), соединения формулы (IIIA1-3), соединения формулы (IIIA1-4), соединения формулы (IIIA1-5) и соединения формулы (IIIA1-6):


где R2 выбран из группы, состоящей из метила, дифторметила и трифторметила; R, R1, R3, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, соединение формулы (I), стереоизомер или его фармацевтически приемлемая соль выбран из группы, состоящей из соединения формулы (IVA1-1) и соединения формулы (IVA1-2):

где R, R1, R3, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, в соединении формулы (I) его стереоизомере или его фармацевтически приемлемой соли, R3 выбран из группы, состоящей из водорода, дейтерия, галогена, C1-8 алкила, C1-8 алкокси, С3-8 циклоалкила, трифторметила и трифторметокси; R, R1, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления в соединении формулы (I) его стереоизомере или его фармацевтически приемлемой соли, R3 выбрана из группы, состоящей из водорода, фтора, хлора, метила, этила, изопропила, метокси, этокси, изопропокси, циклопропила, циклобутила, трифторметила и трифторметокси; R, R1, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления в соединении формулы (I) его стереоизомере или его фармацевтически приемлемой соли, R3 выбран из группы, состоящей из водорода, фтора, хлора, метила, циклопропила и трифторметила; R, R1, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в соединении формулы (I).
В наиболее предпочтительном варианте осуществления соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль, выбраны из группы, состоящей из:







В более предпочтительном варианте осуществления, соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль, представляют собой соединение формулы (IVA1-3):

где R, R1, R3, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления изобретения, в соединении формулы (I) его стереоизомере или его фармацевтически приемлемой соли, R, R1, R3, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в соединении формулы (I), при условии, что, когда оба из R7 и R8 представляют собой водород, m равно 3 или 4; или, когда один из R7 и R8 представляет собой водород, m равно 2, 3 или 4; или когда оба из R7 и R8 не являются водородом, m равно 1, 2, 3 или 4.
В более предпочтительном варианте осуществления, в соединении формулы (I) его стереоизомере или его фармацевтически приемлемой соли, R3 выбран из группы, состоящей из водорода, фтора, хлора, метила, циклопропила и трифторметила; и R, R1, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в соединении формулы (I), при условии, что когда оба R7 и R8 представляют собой водород, m равно 3 или 4; или, когда один из R7 и R8 представляет собой водород, m равно 2, 3 или 4; или когда оба из R7 и R8 не являются водородом, m равно 1, 2, 3 или 4.
В наиболее предпочтительном варианте осуществления, соединение формулы (I), стереоизомер или его фармацевтически приемлемая соль выбраны из группы, состоящей из:





В более предпочтительном варианте осуществления, в соединении формулы (I) его стереоизомере или его фармацевтически приемлемой соли, где m равно 2, 3 или 4, когда R3 выбран из группы, состоящей из водорода, фтора, хлора, метила, циклопропила и трифторметила; два R4 вместе с атомами углерода присоединенного бензольного кольца образуют 5-7-членный карбоцикл, 5-7-членный гетероцикл, С5-7 арил или 5-7-членный гетероарил, 5-7-членный карбоцикл, 5-7-членный гетероцикл, С5-7 арил и 5-7-членный гетероарил, выбранный из группы, состоящей из:





где 5-7-членный карбоцикл, 5-7-членный гетероцикл, С5-7 арил и 5-7-членный гетероарил, каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из галогена, гидрокси, сульфгидрила, циано, нитро, азидо, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арила, С5-10 арилокси, С5-10 арилтио, 5-10-членного гетероарила, 5-10-членного гетероарилокси, 5-10-членного гетероарилтио, -C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10;
R, R1, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, в соединении формулы (I) его стереоизомере или его фармацевтически приемлемой соли, когда оба из R7 и R8 предстапвляют собой водород, m равно 1 или 2; или когда один из R7 и R8 представляет собой водород, m равно 0 или 1; или когда оба из R7 и R8 не являются водородом, m равно 0, 1 или 2; R3 выбран из водорода, фтора, хлора, метила, циклопропила и трифторметила;
R4 выбран из группы, состоящей из водорода, дейтерия, галогена, гидрокси, сульфгидрила, циано, нитро, азидо, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арила, С5-10 арилокси, С5-10 арилтио, 5-10-членного гетероарила, 5-10-членного гетероарилокси, 5-10-членного гетероарилтио, -C0-8-P(O)R11R12, -C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10;
где C1-8 алкил, С3-8 циклоалкил, 3-8-членный гетероциклил, С5-10 арил, 5-10-членный гетероарил, 5-7-членный карбоцикл, 5-7-членный гетероцикл, С5-7 арил и 5-7-членный гетероарил каждый необязательно замещен одной или более группами, выбранными из группы, состоящей из галогена, гидрокси, сульфгидрила, циано, нитро, азидо, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арила, С5-10 арилокси, С5-10 арилтио, 5-10-членного гетероарила, 5-10-членного гетероарилокси, 5-10-членного гетероарилтио, -C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10;
R, R1, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления в соединении формулы (I) его стереоизомере или его фармацевтически приемлемой соли, R3 выбран из группы, состоящей из водорода, фтора, хлора и трифторметила; R4 выбран из группы, состоящей из водорода, дейтерия, галогена, гидрокси, сульфгидрила, циано, нитро, азидо, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арила, С5-10 арилокси, С5-10 арилтио, 5-10-членного гетероарила, 5-10-членного гетероарилокси, 5-10-членного гетероарилтио, -C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10;
R, R1, R6, R7, R8, R9, R10, R11, R12, r и q являются такими, как определено в соединении формулы (I); и m является таким, как определено выше.
В более предпочтительном варианте осуществления в соединении формулы (I) его стереоизомере или его фармацевтически приемлемой соли, R3 выбран из группы, состоящей из водорода, фтора, хлора и трифторметила; R4 выбран из группы, состоящей из водорода, дейтерия, гидрокси, циано, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, С5-10 арила, С5-10 арилокси, 5-10-членного гетероарила, 5-10-членного гетероарилокси, -C0-8-O-R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8;
R, R1, R6, R7, R8, R9, R10, R11, R12 и q являются такими, как определено в соединении формулы (I); и m является таким, как определено выше.
В более предпочтительном варианте осуществления, в соединении формулы (I) его стереоизомере или его фармацевтически приемлемой соли, R3 выбран из группы, состоящей из водорода, фтора, хлора и трифторметила; R4 выбран из группы, состоящей из водорода, дейтерия, гидрокси, циано, этенила, этинила, циклопропила, циклобутила, оксетан-3-ила, N-R6-азетидин-3-ила, циклопропилокси, циклобутилокси, фенила, фенокси, -C0-8-O-R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8 и -C0-8-C(O)NR7R8;
R, R1, R6, R7, R8, R9, R10, R11, R12 и q являются такими, как определено в соединении формулы (I); и m является таким, как определено выше.
В наиболее предпочтительном варианте осуществления, соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль выбраны из группы, состоящей из:















В более предпочтительном варианте осуществления, соединение формулы (I), стереоизомер или его фармацевтически приемлемая соль выбраны из группы, состоящей из соединения формулы (IIIA1-7), соединения формулы (IIIA1-8) и соединения формулы (IIIA1-9):

где R2 выбран из группы, состоящей из метила, дифторметила и трифторметила; R, R1, R3, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, в соединении формулы (I) его стереоизомере или его фармацевтически приемлемой соли, R2 выбран из группы, состоящей из метила, дифторметила и трифторметила; R3 выбран из группы, состоящей из водорода, фтора, хлора и трифторметила; R, R1, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, в соединении формулы (I), его стереоизомере или его фармацевтически приемлемой соли, R1 выбран из группы, состоящей из  и
и  ; R2 выбран из группы, состоящей из метила, дифторметила и трифторметила; R3 выбран из группы, состоящей из водорода, фтора, хлора и трифторметила; R, R4, R6, R7, R8, R9, R10, R11, R12, m и r являются такими, как определено в соединении формулы (I).
; R2 выбран из группы, состоящей из метила, дифторметила и трифторметила; R3 выбран из группы, состоящей из водорода, фтора, хлора и трифторметила; R, R4, R6, R7, R8, R9, R10, R11, R12, m и r являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, соединение формулы (I), его стереоизомер или его фармацевтически приемлемую соль выбирают из группы, состоящей из соединения формулы (IVA1-4) и соединения формулы (IVA1-5):

где R3 выбран из группы, состоящей из водорода, фтора, хлора и трифторметила; R, R4, R6, R7, R8, R9, R10, R11, R12, m и r являются такими, как определено в соединении формулы (I).
В наиболее предпочтительном варианте осуществления соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль выбраны из группы, состоящей из:












В более предпочтительном варианте осуществления, соединение формулы (I), его стереоизомер или его фармацевтически приемлемую соль выбирают из группы, состоящей из соединения формулы (IVA1-6) и соединения формулы (IVA1-7):

где R3 выбран из группы, состоящей из водорода, фтора, хлора и трифторметила; R, R4, R6, R7, R8, R9, R10, R11, R12, m и r являются такими, как определено в соединении формулы (I).
В наиболее предпочтительном варианте осуществления, соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль выбраны из группы, состоящей из:











В более предпочтительном варианте осуществления, соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль выбраны из группы, состоящей из соединения формулы (IIIA2-1), соединения формулы (IIIA2-2), соединения формулы (IIIA2-3), соединения формулы (IIIA2-4), соединения формулы (IIIA2-5) и соединения формулы (IIIA2-6):


где R2 выбран из группы, состоящей из метила, дифторметила и трифторметила; R, R1, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, в соединении формулы (I) его стереоизомере или его фармацевтически приемлемой соли, R2 выбран из группы, состоящей из метила, дифторметила и трифторметила; R3 выбран из группы, состоящей из водорода, фтора, хлора и трифторметила; R, R1, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, в соединении формулы (I), его стереоизомере или его фармацевтически приемлемой соли, R1 представляет собой  ; R2 выбран из группы, состоящей из метила, дифторметила и трифторметила; R3 выбран из группы, состоящей из водорода, фтора, хлора и трифторметила; R4, R6, R7, R8, R9, R10, R11, R12, m и r такие, как определено в соединении формулы (I).
; R2 выбран из группы, состоящей из метила, дифторметила и трифторметила; R3 выбран из группы, состоящей из водорода, фтора, хлора и трифторметила; R4, R6, R7, R8, R9, R10, R11, R12, m и r такие, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль представляют собой соединение формулы (IB):

где Q, R, R1, R2, R3, R5, R6, R7, R8, R9, R10, R11, R12, n, r и q являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, в соединении формулы (I) его стереоизомере или его фармацевтически приемлемой соли, R2 выбирают из группы, состоящей из метила, дифторметила и трифторметила; R3 выбирают из группы, состоящей из водорода, фтора, хлора и трифторметила; Q, R, R1, R5, R6, R7, R8, R9, R10, R11, R12, n, r и q являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, в соединении формулы (I), его стереоизомере или его фармацевтически приемлемой соли, R1 представляет собой  ; R2 выбран из группы, состоящей из метила, дифторметила и трифторметила; R3 выбран из группы, состоящей из водорода, фтора, хлора и трифторметила; Q, R5, R6, R7, R8, R9, R10, R11, R12, n и r являются такими, как определено в соединении формулы (I).
; R2 выбран из группы, состоящей из метила, дифторметила и трифторметила; R3 выбран из группы, состоящей из водорода, фтора, хлора и трифторметила; Q, R5, R6, R7, R8, R9, R10, R11, R12, n и r являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, соединение формулы (I), стереоизомер или его фармацевтически приемлемая соль представляют собой соединение формулы (IIB):

где R2 выбран из группы, состоящей из метила, дифторметила и трифторметила; Q, R5, R6, R7, R8, R9, R10, R11, R12, n и r являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль представляют собой соединение формулы (IIIB):

где R2 выбран из группы, состоящей из метила, дифторметила и трифторметила; R5, R6, R7, R8, R9, R10, R11, R12, n и r являются такими, как определено в соединении формулы (I).
В наиболее предпочтительном варианте осуществления, соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль выбрано из группы, состоящей из:






В более предпочтительном варианте осуществления, соединение формулы (I), его стереоизомер или его фармацевтически приемлемая соль представляет собой соединение формулы (IC):

где Q, R, R1, R2, R3, R4, R6, R7, R8, R9, R10, R11, R12, r, p, о и q являются такими, как определено в п. 1.
В более предпочтительном варианте осуществления, в соединении формулы (I) его стереоизомере или его фармацевтически приемлемой соли R2 выбирают из группы, состоящей из дифторметила, трифторметила и метила; R3 выбирают из группы, состоящей из водорода, фтора, хлора, метила, этила, трифторметила, циано и нитро; Q, R, R1, R4, R6, R7, R8, R9, R10, r, p и о являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, соединение формулы (I), его стереоизомер или его фармацевтически приемлемую соль выбирают из группы, состоящей из соединения формулы (IIC1) и соединения формулы (IIC2):

где R2 выбран из группы, состоящей из дифторметила, трифторметила и метила; R3 выбран из группы, состоящей из водорода, фтора, хлора, метила, этила, трифторметила, циано и нитро; Q, R, R4, R6, R7, R8, R9, R10, R11, R12, о и r являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, соединение формулы (I), его стереоизомер или его фармацевтически приемлемую соль выбирают из группы, состоящей из соединения формулы (IIIC1) и соединения формулы (IIIC2):

где R2 выбран из группы, состоящей из дифторметила, трифторметила и метила; R3 выбран из группы, состоящей из водорода, фтора, хлора, метила, этила, трифторметила, циано и нитро; Q, R, R4, R6, R7, R8, R9, R10, r и о являются такими, как определено в соединении формулы (I).
В более предпочтительном варианте осуществления, соединение формулы (I), его стереоизомер или его фармацевтически приемлемую соль выбирают из группы, состоящей из:





В другом аспекте, настоящее изобретение относится к способу получения соединения формулы (I), его стереоизомера или его фармацевтически приемлемой соли, включающему стадии:

где кольцо A, Q, X1, Х2, Х3, R, R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R11, R12, m, n, r, о, p и q является таким как определено в соединении формулы (I).
В другом аспекте, настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество вышеуказанного соединения формулы (I), его стереоизомера или его фармацевтически приемлемой соли, и фармацевтически приемлемый носитель.
В другом аспекте, настоящее изобретение относится к применению вышеуказанного соединения формулы (I), его стереоизомера или его фармацевтически приемлемой соли или вышеуказанной фармацевтической композиции для приготовления лекарственного средства для лечения заболевания, опосредованного активностью мутанта EGFR, в частности мутанта EGFR-L858R, мутанта EGFR-T790M и активностью мутанта, активированного делецией в 19 экзоне.
В другом аспекте, настоящее изобретение относится к применению вышеуказанного соединения формулы (I), стереоизомера или его фармацевтически приемлемой соли, или вышеуказанной фармацевтической композиции для приготовления лекарственного средства для лечения заболевания, опосредованного только или частично активностью мутанта EGFR.
В другом аспекте, настоящее изобретение относится к применению вышеуказанного соединения формулы (I), стереоизомера или его фармацевтически приемлемой соли или вышеуказанной фармацевтической композиции для приготовления лекарственного средства для лечения рака.
В более предпочтительном варианте осуществления, рак выбран из группы, состоящей из рака яичника, рака шейки матки, колоректального рака, рака молочной железы, рака поджелудочной железы, глиомы, глиобластомы, меланомы, рака предстательной железы, лейкемии, лимфомы, неходжкинской лимфомы, рака желудка, рака легкого, гепатоцеллюлярной карциномы, рака желудка, гастроинтестинальной стромальной опухоли (GIST), рака щитовидной железы, холангиокарциномы, рака эндометрия, рака почки, анапластической крупноклеточной лимфомы, острого миелоидного лейкоза (AML), множественной миеломы, меланомы или мезотелиомы; предпочтительно немелкоклеточного рака легкого.
Подробное описание изобретения
Подробное описание: если не указано иное, следующие термины, которые используются в описании и формуле изобретения, имеют следующие значения.
«C1-8 алкил» относится к алкильной группе с прямой или разветвленной цепью, имеющей от 1 до 8 атомов углерода, «алкил» относится к насыщенной алифатической углеводородной группе, С0-8 относится к группе, не содержащей углерод, и C1-8 алкильной группе, такой как метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил и их различные изомеры с разветвленной цепью и тому подобное.
Алкил может быть замещенным или незамещенным. Когда алкил замещен, заместитель может быть замещен в любой доступной точке соединения и, предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из галогена, гидрокси, сульфгидрила, циано, нитро, азидо, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арила, С5-10 арилокси, С5-10 арилтио, 5-10-членного гетероарила, 5-10-членного гетероарилокси, 5-10-членного гетероарилтио, -C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10.
«Циклоалкил» относится к насыщенному или частично ненасыщенному моноциклическому или полициклическому углеводородному заместителю, «С3-8 циклоалкил» относится к циклоалкильной группе, имеющей от 3 до 8 атомов углерода, «5-10-членный циклоалкил» относится к циклоалкильной группе, имеющей от 5 до 10 атомов углерода, например:
неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и тому подобное.
Полициклический циклоалкил включает циклоалкил, имеющий спиро-кольцо, конденсированное кольцо и мостиковое кольцо. «Спироциклоалкил» относится к полициклической группе с кольцами, соединенными через один общий атом углерода (называемый спироатомом), где данные кольца могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы. Спироциклоалкил может быть разделен на моноспироциклоалкил, ди-спироциклоалкил или полиспироциклоалкил в соответствии с числом спироатомов, общих для колец. Неограничивающие примеры спироциклоалкила включают в себя:

«Конденсированный циклоалкил» относится к полностью углеродной полициклической группе, в которой каждое кольцо в системе разделяет соседнюю пару атомов углерода с другим кольцом, причем одно или более колец могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы. Согласно числу членных колец, конденсированный циклоалкил может быть разделен на бициклический, трициклический, тетрациклический и полициклический конденсированный циклоалкил. Неограничивающие примеры конденсированного циклоалкила включают:

«Мостиковый циклоалкил» относится к полностью углеродной полициклической группе, в которой любые два кольца в системе имеют два общих несвязанных атома углерода, причем эти кольца могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы. Согласно числу членных колец мостиковый циклоалкил может быть разделен на бициклический, трициклический, тетрациклический и полициклический мостиковый циклоалкил. Неограничивающие примеры мостикового циклоалкила включают:

Циклоалкил может быть конденсирован с кольцом арила, гетероарила или гетероциклила, где кольцо, связанное с исходной структурой, является циклоалкилом, и неограничивающие примеры включают инданил, тетрагидронафтил, бензоциклогептилалкил и тому подобное.
Циклоалкил может быть необязательно замещенным или незамещенным. Когда циклоалкил замещен, заместителем предпочтительно является одна или более групп, независимо выбранных из группы, состоящей из галогена, гидрокси, сульфгидрила, циано, нитро, азидо, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арила, С5-10 арилокси, С5-10 арилтио, 5-10-членного гетероарила, 5-10-членного гетероарилокси, 5-10-членного гетероарилтио, -C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10.
«Гетероциклил» относится к насыщенному или частично ненасыщенному моноциклическому или полициклическому циклическому углеводородному заместителю, где один или более кольцевых атомов являются гетероатомами, выбранными из группы, состоящей из азота, кислорода и S(O)r (где r представляет собой целое число от 0 до 2), но циклическая часть не включает -O-O-, -O-S- или -S-S-, и остальные атомы в кольце представляют собой углерод. «5-10-членный гетероциклил» относится к гетероциклильной группе, имеющей от 5 до 10 атомов в кольце, «3-8-членный гетероциклил» относится к гетероциклильной группе, имеющей от 3 до 8 кольцевых атомов.
Неограничивающие примеры моноциклического гетероциклила включают пирролидинил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил и тому подобное.
Полициклический гетероциклил включает гетероциклил, имеющий спиро кольцо, конденсированное кольцо и мостиковое кольцо. «Спирогетероциклил» относится к полициклической гетероциклильной группе с кольцами, соединенными через один общий атом (называемый спироатом), где один или более кольцевых атомов являются гетероатомами, выбранными из азота, кислорода или S(O)r (где r представляет собой целое число от 0 до 2), а остальные атомы в кольце представляют собой углерод. Эти кольца могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы. Спироциклоалкил может быть разделен на моноспиро гетероциклил, ди-спиро гетероциклил и полиспиро гетероциклил по числу спироатомов, общих для колец. Неограничивающие примеры спиро-гетероциклила включают:

Термин «конденсированный гетероциклил» относится к полициклической группе, в которой каждое кольцо в системе имеет общую соседнюю пару атомов с другим кольцом, где одно или более колец могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы, и где один или более кольцевых атомов являются гетероатомами, выбранными из группы, состоящей из азота, кислорода и S(O)r (где r представляет собой целое число от 0 до 2), а остальные атомы в кольце представляют собой углерод. Согласно числу членных колец конденсированный гетероциклил может быть разделен на бициклический, трициклический, тетрациклический и полициклический конденсированный гетероциклил. Неограничивающие примеры конденсированного гетероциклила включают:


«Мостиковый гетероциклил» относится к полициклической гетероциклической группе, в которой любые два кольца в системе имеют два общих несвязанных атома, кольца могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы, и одно или более кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода и S(O)r (где r представляет собой целое число от 0 до 2), а остальные атомы в кольце представляют собой углерод. По числу членных колец мостиковый гетероциклил может быть разделен на бициклический, трициклический, тетрациклический и полициклический мостиковый гетероциклил. Неограничивающие примеры мостикового гетероциклила включают:

Гетероциклил может быть конденсирован с кольцом арила, гетероарила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой гетероциклил, и не ограничивающие примеры включают:

Гетероциклил может быть необязательно замещенным или незамещенным. Когда гетероциклил замещен, заместителем предпочтительно является одна или более групп, независимо выбранных из группы, состоящей из галогена, гидрокси, сульфгидрила, циано, нитро, азидо, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арила, С5-10 арилокси, С5-10 арилтио, 5-10-членного гетероарила, 5-10-членного гетероарилокси, 5-10-членного гетероарилтио, -C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10.
«Арил» относится к полностью углеродному моноциклу или сконденсированному полициклу (то есть, кольцо в системе имеет общую соседнюю пару углеродных атомов с другим кольцом) с сопряженной системой π-электронов. «С5-10 арил» относится к арильной полностью углеродной группе, содержащей от 5 до 10 атомов углерода, «5-10-членный арил» относится к арильной группе, включающей только углерод, содержащей от 5 до 10 атомов углерода, такой как фенил и нафтил. Арил может быть конденсирован с кольцом гетероарила, гетероциклила или циклоалкила, где кольцо, связанное с исходной структурой, является арилом, и неограничивающие примеры включают:



Арил может быть замещенным или незамещенным. Когда арил замещен, заместителем предпочтительно является одна или более групп, независимо выбранных из группы, состоящей из галогена, гидрокси, сульфгидрила, циано, нитро, азидо, С1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арила, С5-10 арилокси, С5-10 арилтио, 5-10-членного гетероарила, 5-10-членного гетероарилокси, 5-10-членного гетероарилтио, -C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10.
«Гетероарил» относится к гетероароматической системе, содержащей от 1 до 4 гетероатомов, где гетероатомы включают азот, кислород или S(O)r (где r представляет собой целое число от 0 до 2). 5-7-членный гетероарил относится к гетероароматической системе с 5-7 атомами в кольце, 5-10-членный гетероарил относится к гетероароматической системе, имеющей от 5 до 10 кольцевых атомов, такой как фурил, тиенил, пиридил, пирролил, N-алкилпирролил, пиримидинил, пиразинил, имидазолил, тетразолил и тому подобное. Гетероарил может быть конденсирован с кольцом арила, гетероциклила или циклоалкила, где кольцо, связанное с исходной структурой, является гетероарилом, и неограничивающие примеры включают:


Гетероарил может быть необязательно замещенным или незамещенным. Когда гетероарил замещен, заместителем предпочтительно является одна или более групп, независимо выбранных из группы, состоящей из галогена, гидрокси, сульфгидрила, циано, нитро, азидо, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арила, С5-10 арилокси, С5-10 арилтио, 5-10-членного гетероарила, 5-10-членного гетероарилокси, 5-10-членного гетероарилтио, -C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10.
«Алкенил» относится к алкильной группе, как определено выше, которая имеет, по меньшей мере, два атома углерода и, по меньшей мере, одну углерод-углеродную двойную связь; С2-8 алкенил относится к алкенильной группе с прямой или разветвленной цепью, имеющей от 2 до 8 атомов углерода, для например, винил, 1-пропенил, 2-пропенил, 1-, 2- или 3-бутенил и тому подобное.
Алкенил может быть замещенным или незамещенным. Когда алкенил замещен, заместителем, предпочтительно является одна или более групп, независимо выбранных из группы, состоящей из галогена, гидрокси, сульфгидрила, циано, нитро, азидо, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арила, С5-10 арилокси, С5-10 арилтио, 5-10-членного гетероарила, 5-10-членного гетероарилокси, 5-10-членного гетероарилтио, -C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10.
«Алкинил» относится к алкильной группе, как определено выше, которая имеет, по меньшей мере, два атома углерода и, по меньшей мере, одну тройную связь углерод-углерод; С2-8 алкинил относится к алкинильной группе с прямой или разветвленной цепью, имеющей от 2 до 8 атомов углерода, например, этинил, 1-пропинил, 2-пропинил, 1-, 2- или 3-бутинил и тому подобное.
Алкинил может быть замещенным или незамещенным. Когда алкинил замещен, заместителем предпочтительно является одна или более групп, независимо выбранных из группы, состоящей из галогена, гидрокси, сульфгидрила, циано, нитро, азидо, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арила, С5-10 арилокси, С5-10 арилтио, 5-10-членного гетероарила, 5-10-членного гетероарилокси, 5-10-членного гетероарилтио, -C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10.
«Алкокси» относится к -О- (алкилу), где алкил является таким, как определено выше. C1-8 алкокси означает алкокси, содержащий от 1 до 8 атомов углерода, а неограничивающие примеры включают метокси, этокси, пропокси, бутокси и тому подобное.
Алкокси может быть необязательно замещенным или незамещенным. Когда алкокси замещен, заместителем предпочтительно является одна или более групп, независимо выбранных из группы, состоящей из галогена, гидрокси, сульфгидрила, циано, нитро, азидо, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арила, С5-10 арилокси, С5-10 арилтио, 5-10-членного гетероарила, 5-10-членного гетероарилокси, 5-10-членного гетероарилтио, -C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10.
«Циклоалкокси» относится к -О- (незамещенному циклоалкилу), где циклоалкил является таким, как определено выше. С3-8 циклоалкокси относится к циклоалкоксигруппе, имеющей от 3 до 8 атомов углерода, и неограничивающие примеры включают циклопропокси, циклобутилокси, циклопентилокси, циклогексилокси и тому подобное.
Циклоалкокси может быть необязательно замещенным или незамещенным. Когда циклоалкокси замещен, заместителем, предпочтительно является одна или более групп, независимо выбранных из группы, состоящей из галогена, гидрокси, сульфгидрила, циано, нитро, азидо, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 3-8-членного гетероциклила, 3-8-членного гетероциклилокси, 3-8-членного гетероциклилтио, С5-10 арила, С5-10 арилокси, С5-10 арилтио, 5-10-членного гетероарила, 5-10-членного гетероарилокси, 5-10-членного гетероарилтио, -C0-8-S(O)rR9, -C0-8-O-R10, -C0-8-C(O)R10, -C0-8-C(O)OR10, -C0-8-O-C(O)R10, -C0-8-NR7R8, -C0-8-C(O)NR7R8, -N(R7)-C(O)R10 и -N(R7)-C(O)OR10.
«Галоген C1-8 алкил» относится к C1-8 алкильной группе, где водороды в алкиле замещены атомами фтора, хлора, брома и йода, например дифторметилом, дихлорметилом, дибромметилом, трифторметилом, трихлорметилом, трибромметилом и тому подобное.
«Галоген C1-8 алкокси» относится к C1-8 алкоксигруппе, в которой водород в алкиле замещен фтором, хлором, бромом и йодом, например дифторметокси, дихлорметокси, дибромметокси, трифторметокси, трихлорметокси, трибромметокси и тому подобными.
«Галоген» относится к фтору, хлору, брому или йоду.
«C(O)R10» относится к карбонильной группе, замещенной R10.
«-C0-8-P(O)R11R12» относится к фосфорильной С0-8 алкильной группе, замещенной R11 и R12, где R11 и R12 каждый необязательно являются одинаковыми или разными заместителями.
«THF » относится к тетрагидрофурану (tetrahydrofuran).
«DCM» относится к дихлорметану (dichloromethane).
«DMF» относится к N,N-диметилформамиду (N,N-dimethylformamide).
«DIPEA» относится к диизопропилэтиламину (diisopropylethylamine).
«Необязательно» или «факультативно» означает, что описанное впоследствии событие или обстоятельство могут, но, необязательно, иметь место. Его значение включает случаи, когда событие или обстоятельство имеет место, или не имеет места. Например, «гетероциклил, необязательно, замещенный алкилом», означает, что алкильная группа может присутствовать, но не обязательно должна присутствовать. Его значение включает примеры, в которых гетероциклил является замещенным или незамещенным алкилом.
«Замещенный» означает, что один или более атомов водорода, предпочтительно до 5 и более предпочтительно от 1 до 3 атомов водорода в группе, каждый независимо замещен соответствующим числом заместителей. По-видимому, заместители расположены только в возможных для них химических положениях, и возможные или невозможные замены могут быть определены (посредством экспериментов или теоретически) специалистами в данной области техники без особых усилий. Например, комбинация амино или гидрокси, имеющего свободный водород и атомы углерода, имеющие ненасыщенные связи (такие как олефины), может быть нестабильной.
«Фармацевтическая композиция» относится к смеси, содержащей одно или более соединений, описанных здесь, или физиологические/фармацевтические соли или пролекарства и другие химические компоненты, такие как физиологические/фармацевтические носители и эксципиенты. Целью фармацевтической композиции является облегчение введения соединения в организм, которое поможет абсорбции активного ингредиента, тем самым реализуя биологическую активность.
Следующие примеры служат для иллюстрации настоящего изобретения подробно и полностью, но эти примеры не следует рассматривать как ограничивающие объем настоящего изобретения, и настоящее изобретение не ограничено примерами.
Структуры соединений в настоящем изобретении идентифицировали с помощью ядерного магнитного резонанса (ЯМР) и/или жидкостной хроматографии-масс-спектрометрии (ЖХ-МС). Химический сдвиг ЯМР дан в 10-6 (м.д.). ЯМР определяли с помощью устройства Bruker AVANCE-400, растворителями для определения являлись дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный метанол (CD3OD) и дейтерированный хлороформ (CDCl3), а внутренний стандарт представлял собой тетраметилсилан (TMS, tetramethylsilane).
Жидкостную хроматографию-масс-спектрометрию (ЖХ-МС) проводили с использованием масс-спектрометра Agilent 1200 Infinity Series. ВЭЖХ осуществляли на приборе Agilent 1200DAD для жидкостной хроматографии высокого давления (хроматографическая колонка Sunfire С18 150×4,6 мм) и приборе Waters 2695-2996 для жидкостной хроматографии высокого давления (хроматографическая колонка Gimini С18 150×4,6 мм).
Для тонкослойной хроматографии на силикагеле (ТСХ) использовали пластинку силикагеля Yantai Huanghai HSGF254 или Qingdao GF254. Размер пластин, используемых в ТСХ, составлял от 0,15 до 0,2 мм, а размер пластин, используемых при очистке продукта, составлял от 0,4 мм до 0,5 мм. Колоночная хроматография обычно использовала силикагель Yantai Huanghai 200-300 меш в качестве носителя.
Исходные вещества, используемые в примерах настоящего изобретения, являются известными и коммерчески доступными, или могут быть синтезированы с использованием или в соответствии с известным способом в данной области техники.
Если не указано иное, все реакции настоящего изобретения проводят при непрерывном магнитном перемешивании в атмосфере сухого азота или аргона, растворитель является сухим и температура реакции указана в градусах Цельсия.
Получение промежуточного соединения
1. Промежуточное соединение 1: получение 2-(дифторметокси)-4-фторнитробензола

5-фтор-2-нитрофенол (3,0 г, 19,1 ммоль) и карбонат калия (5,28 г, 38,2 ммоль) растворяли в DMF с последующим добавлением хлордифторацетата натрия (4,37 г, 28,6 ммоль). Реакционный раствор нагревали до 100°С в атмосфере азота и перемешивали в течение 16 часов, затем концентрировали. К полученному остатку для экстракции добавляли H2O (50 мл) и метил-трет-бутиловый эфир (50 мл). Органическую фазу промывали три раза водой, сушили над безводным сульфатом магния, фильтровали и фильтрат концентрировали. Полученный остаток очищали колоночной флэш-хроматографией на силикагеле с получением 2-(дифторметокси)-4-фторнитробензола (3,0 г, 75%).
2. Промежуточное соединение 2: получение 2-(дифторметокси)-4-фтор-5-нитроанилина

2-(дифторметокси)-4-фторнитробензол (3,0 г, 14,5 ммоль) растворяли в метаноле (30 мл) с последующим добавлением Pd/C (500 мг) и проводили реакцию в атмосфере водорода при комнатной температуре в течение 2 часов. После того, как ТСХ показывал завершение реакции, реакционный раствор фильтровали через целит и фильтрат концентрировали с получением неочищенного продукта (1,7 г, 66%).
Сырой продукт тщательно растворяли в концентрированной серной кислоте (5 мл) на ледяной бане. После перемешивания реакционной смеси для получения прозрачного раствора на ледяной бане, медленно частями добавляли нитрат калия (1,1 г, 9,5 ммоль), и затем реакционную смесь перемешивали в течение 3 часов на ледяной бане. После того как ЖХМС показывала завершение реакции, реакционный раствор медленно добавляли к насыщенному водному раствору карбоната натрия (100 мл). После гашения реакции, водную фазу экстрагировали метил-трет-бутиловым эфиром (3×20 мл), затем органическую фазу сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали и полученный остаток очищали флэш-хроматографией на силикагеле с получением 2-(дифторметокси)-4-фтор-5-нитроанилина (2,0 г, 90%).
3. Промежуточное соединение 3: получение 4-фтор-1-нитро-2-(трифторметокси)бензола

3-фтор-трифторметоксибензол (20 г) растворяли в 40 мл концентрированной серной кислоты при охлаждении со льдом. Нитрат калия (28 г) добавляли частями при быстром перемешивании. Реакционную смесь перемешивали при 0°С в течение 3 часов и затем перемешивали при комнатной температуре в течение ночи. Реакционный раствор осторожно выливали в 1 кг измельченного льда, перемешивали в течение 30 минут, экстрагировали этилацетатом, сушили над сульфатом натрия, фильтровали и фильтрат выпаривали. Полученный остаток очищали колоночной хроматографией с получением 12 г неочищенного продукта в виде желтоватой жидкости.
4. Промежуточное соединение 4: получение 4-фтор-2-(трифторметокси)анилина

Сырой продукт (12 г) 4-фтор-1-нитро-2-(трифторметокси)бензол, полученный на предыдущей стадии, растворяли в 100 мл безводного этанола и затем добавляли дигидрат хлорида олова (25 г) при охлаждении на ледяной воде. Реакционный раствор перемешивали при комнатной температуре в течение ночи. Добавляли 1 Н водный раствор гидроксида натрия для доведения рН до 12. Реакционный раствор фильтровали и фильтрат экстрагировали этилацетатом. Экстракт сушили над безводным сульфатом натрия, фильтровали и выпаривали. Полученный остаток очищали колоночной хроматографией с получением 4-фтор-2-(трифторметокси)анилина в виде желтоватой маслянистой жидкости (4,78 г, 46%).
1Н ЯМР (400 МГц, CDCl3) δ 6,94 (d, J=8,8 Гц, 1Н), 6,83 (m, 1Н), 6,76 (dd, J=5,4, 8,8 Гц, 1Н), 3,87-3,59 (br, 2Н).
5. Промежуточное соединение 5: получение 4-фтор-5-нитро-2-(трифторметокси)анилина

4-фтор-2-(трифторметокси)анилин (2,5 г) растворяли в концентрированной серной кислоте (10 мл) при охлаждении льдом с последующим добавлением нитрата калия (3 г) и затем реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реактивный раствор выливали в ледяную воду и добавляли 3 N водный раствор гидроксида натрия для доведения рН до 10. Реакционную смесь экстрагировали с этилацетатом и экстракт сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали колоночной хроматографией с получением 4-фтор-5-нитро-2-(трифторметокси)анилина (1,79 г, 58%).
6. Промежуточное соединение 6: получение 6-метокси-1-метил-1Н-индола

1Н-индол-6-ол(1 г, 7,51 ммоль) растворяли в безводном DMF (20 мл) и NaH (900 мг, 22,53 ммоль) частями добавляли на ледяной бане. Реакционную смесь перемешивали на ледяной бане в течение 20 минут, затем медленно добавляли по каплям метилйодид (2,67 г, 18,78 ммоль). Реакционную смесь перемешивали в течение 2 часов на ледяной бане. После того как ЖХМС показывала завершение реакции, реакцию гасили насыщенным NH4Cl (40 мл) на ледяной бане и экстрагировали метил-трет-бутиловым эфиром (3×30 мл). Органические фазы объединяли, промывали водой (20 мл × 2), сушили над безводным сульфатом магния, фильтровали и концентрировали. Полученный остаток очищали флэш-хроматографией на силикагеле с получением продукта 6-метокси-1-метил-1Н-индол (1,1 г, 90%).
7. Промежуточное соединение 7: получение 3-(2-хлорпиримидин-4-ил)-1-метил-1Н-индола

N-метилиндол (300 мг, 2,29 ммоль), 2,4-дихлорпиримидин (340 мг, 2,30 ммоль) и безводный трихлорид алюминия (460 мг, 3,43 ммоль) растворяли в диметиловом эфире этиленгликоля (12 мл). Реакционную смесь нагревали до 60°С в атмосфере азота и перемешивали в течение 3 часов. По окончании реакции, реакционный раствор выливали в смесь льда и воды (приблизительно 50 мл) и экстрагировали метил-трет-бутиловым эфиром (20 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом магния, фильтровали и концентрировали. Полученный остаток очищали колоночной хроматографией с получением продукта 3-(2-хлорпиримидин-4-ил)-1-метил-1Н-индола (400 мг, 72%).
8. Промежуточное соединение 8: получение 3-(2-хлорпиримидин-4-ил)-6-метокси-1-метил-1Н-индола

6-метокси-1-метил-1Н-индол (300 мг, 1,86 ммоль) и 2,4-дихлорпиримидин (330 мг, 2,23 ммоль) растворяли в диметиловом эфире этиленгликоля (10 мл) с последующим добавлением безводного трихлорида алюминия 500 мг, 3,72 ммоль). Реакционную смесь нагревали до 60°С в атмосфере азота и перемешивали в течение 3 часов. После того, как ЖХМС показывала завершение реакции, реакционный раствор выливали в смесь льда и воды (приблизительно 50 мл) и экстрагировали метил-трет-бутиловым эфиром (50 мл × 3). Органические фазы объединяли, промывали последовательно насыщенным бикарбонатом натрия (30 мл × 2) и H2O (30 мл), сушили, фильтровали и концентрировали. Полученный остаток очищали флэш-хроматографией на силикагеле с получением продукта 3-(2-хлорпиримидин-4-ил)-6-метокси-1-метил-1Н-индола (120 мг, 24%).
9. Промежуточное соединение 9: получение 4-(2-(метиламино)этил)морфолин-3-она

Трет-бутил(2-гидроксиэтил)(метил)карбамат (300 мг, 1,71 ммоль) и триэтиламин (350 мг, 3,42 ммоль) растворяли в безводном дихлорметане (10 мл), п-толуолсульфонилхлорид (490 мг, 2,57 ммоль) частями добавляли при комнатной температуре. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре. После того как ЖХМС показывала завершение реакции, реакционный раствор последовательно промывали насыщенным водным раствором бикарбоната натрия (10 мл), 1N HCl (10 мл) и H2O (10 мл × 2), сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали с получением сырого продукта 2-((трет-бутоксикарбонил)(метил)амино)этил 4-метилбензолсульфоната (560 мг, 99%), который использовали непосредственно на следующей стадии без дальнейшей обработки.
Морфолин-3-он (180,5 мг, 1,79 ммоль) растворяли в безводном DMF, добавляли NaH (136 мг, 3,4 ммоль) при 0°С и реакционную смесь перемешивали в течение 10 минут на ледяной бане. К реакционному раствору добавляли 2-((трет-бутоксикарбонил)(метил)амино)этил-4-метилбензолсульфонат (560 мг, 1,7 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После того как ЖХМС показывала завершение реакции, реакционный раствор гасили насыщенным водным раствором NH4Cl (20 мл) и экстрагировали дихлорметаном (10 мл × 2). Органические фазы объединяли, сушили над безводным сульфатом магния, фильтровали и концентрировали. Полученный остаток растворяли в растворе 4N соляной кислоты в диоксане (10 мл) и перемешивали при комнатной температуре в течение 1 часа. После того как ЖХМС показывала завершение реакции, реакционный раствор концентрировали с получением неочищенного продукта 4-(2-(метиламино)этил)морфолин-3-она (150 мг, 98%), который использовали непосредственно на следующей стадии без дальнейшей очистки.
10. Промежуточное соединение 10: получение 1-(2-((трет-бутилдиметилсилил)окси)этил)-1Н-индола

Индол (4,45 г, 38 ммоль) растворяли в 100 мл DMF, а затем добавляли 60% гидрида натрия (4,6 г, 113,9 ммоль). После перемешивания реакционного раствора при комнатной температуре в течение 15 минут по каплям добавляли ((трет-бутилдиметилсилил)окси)-2-бромэтил (10 г, 41,81 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционный раствор выливали в воду и экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали, концентрировали и получали неочищенный продукт, который очищали флэш-хроматографией на силикагеле, с получением 1-(2-((трет-бутилдиметилсилил)окси)этил)-1Н-индола (9,54, 90%).
11. Промежуточное соединение 11: получение 1-(2-((трет-бутилдиметилсилил)окси)этил)-3-(2-хлорпиримидин-4-ил)-1Н-индола

1-(2-((трет-бутилдиметилсилил)окси)этил)-1Н-индол (2 г, 7,26 ммоль), 2,4-дихлорпиримидин (1,2 г, 8,00 ммоль) и трихлорид алюминия (1,45 г, 10,89 ммоль) растворяли в 30 мл DME и реакционную смесь перемешивали при 75°С в течение ночи. По окончании реакции реакционный раствор выливали в ледяную воду и экстрагировали три раза метил-трет-бутиловым эфиром. Органические фазы объединяли, промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта, который затем очищали флэш-хроматографией на силикагеле, с получением продукта (1,1, 39%).
12. Промежуточное соединение 12: получение 5-метокси-1-метил-1Н-индола

5-метокси-1Н-индол (2,2 г, 15 ммоль) растворяли в THF (30 мл) и реакционный раствор охлаждали до 0°С перед добавлением NaH (0,9 г, 32 ммоль) при перемешивании. Реакционную смесь перемешивали при 0°С в течение 1 часа и при той же температуре добавляли метилйодид (4,2 г, 30 ммоль) и затем реакционную смесь перемешивали при комнатной температуре в течение ночи. После того, как исчезновение исходного материала определяли с помощью ЖХ-МС, раствор доводили до рН 3 с помощью HCl (1 N водн.). THF удаляли при пониженном давлении, затем добавляли CH2Cl2 (60 мл). Органическую фазу промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией (элюент: петролейный эфир-петролейный эфир: EtOAc = 10:1) с получением 5-метокси-1-метил-1Н-индола (0,9 Г, 35%).
1Н ЯМР (400 МГц, CD3OD): δ 7,25 (d, J=8,4 Гц, 1Н), 7,10 (d, J=2,8 Гц, 1Н), 7,06 (d, J=2,0 Гц, 1Н), 6,83 (d, J=2,4 Гц, 1Н), 6,34 (m, 1Н), 3,82 (s, 3Н), 3,77 (s, 3Н);
MS m/z(ESI): 162,2 [М+Н]+.
Примеры получения
Пример 1: получение N-(4-(дифторметокси)-5-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-(4-метилпиперазин-1-ил)фенил)акриламида

Стадия 1: получение 2-(дифторметокси)-N4-(2-(диметиламино)этил)-N4-метил-N1-(4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)-5-нитробензол-1,4-диамина

N-(2-(дифторметокси)-4-фтор-5-нитрофенил)-4-(1-метил-1Н-индол-3-ил)пиримидин-2-амин (250 мг, 0,58 ммоль) растворяли в DMF с последующим добавлением диизопропилэтиламина (150 мг, 1,16 ммоль) и триметилэтилендиамина (120 мг, 1,16 ммоль). Реакционную смесь нагревали до 120°С микроволновым излучением и проводили реакцию в течение 30 мин. После того как ЖХМС показывала завершение реакции, реакционный раствор концентрировали досуха. Полученный остаток экстрагировали дихлорметаном (10 мл) и водой (10 мл). Органическую фазу очищали препаративной тонкослойной хроматографией с получением продукта 2-(дифторметокси)-N4-(2-(диметиламино)этил)-N4-метил-N1-(4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)-5-нитробензол-1,4-диамина (150 мг, 50%).
Стадия 2: 5-(дифторметокси)-N1-(2-(диметиламино)этил)-N1-метил-N4-(4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,2,4-триамин

2-(дифторметокси)-N4-(2-(диметиламино)этил)-N4-метил-N1-(4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)-5-нитробензол-1,4-диамин (60 мг, 0,12 ммоль) растворяли в метаноле с последующим добавлением Pd/C (10 мг) и затем реакционную смесь перемешивали при комнатной температуре в атмосфере водорода в течение 2 часов. После того, как ЖХМС показывала завершение реакции, раствор фильтровали и фильтрат концентрировали, с получением продукта 5-(дифторметокси)-N1-(2-(диметиламино)этил)-N1-метил-N4-(4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,2,4-триамина (55 мг, 95%), который непосредственно использовали в следующей реакции.
Стадия 3: N-(4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламид

5-(дифторметокси)-N1-(2-(диметиламино)этил)-N1-метил-N4-(4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,2,4-триамин (55 мг, 0,11 ммоль) и триэтиламин (58 мг, 0,57 ммоль) растворяли в безводном тетрагидрофуране (15 мл). После того, как реакционный раствор перемешивали в течение 10 минут на бане с ледяной водой, по каплям медленно добавляли акрилоилхлорид (0,17 мл, 1 M в THF). Реакционную смесь перемешивали в течение 30 минут на ледяной бане. После того, как ЖХМС показывала завершение реакции, реакцию гасили насыщенным водным раствором NH4Cl (3 мл) и концентрировали. Полученный остаток очищали препаративной тонкослойной хроматографией с получением продукта N-(4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-Метил-1Н-индол-3-ил) пиримидин-2-ил) амино)фенил)акриламида (12,3 мг, 20%).
1Н ЯМР (400 МГц, CDCl3) δ 10,17 (s, 1Н), 9,81 (s, 1Н), 8,89 (s, 1Н), 8,33 (d, J=5,3 Гц, 1Н), 8,00 (dd, J=6,7, 2,0 Гц, 1Н), 7,40 (s, 1Н), 7,32 (dd, J=6,8, 1,9 Гц, 1Н), 7,25-7,13 (m, 1Н), 6,98 (s, 1Н), 6,66-6,20 (m, 3Н), 5,74-5,58 (m, 1Н), 3,90 (s, 3Н), 2,92-2,77 (m, 2Н), 2,62 (s, 3Н), 2,27 (s, 2Н), 2,22 (s, 6Н);
MS m/z (ESI): 536,2 [М+Н]+.
Пример 2: получение N-(4-(дифторметокси)-5-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-(4-метилпиперазин-1-ил)фенил)акриламида

Стадия 1: получение N-(2-(дифторметокси)-4-фтор-5-нитрофенил)-4-(1-метил-1Н-индол-3-ил)пиримидин-2-амина

3-(2-хлорпиримидин-4-ил)-1-метил-1Н-индол (250 мг, 1,0 ммоль), 2-(дифторметокси)-4-фтор-5-нитроанилина (230 мг, 1,0 ммоль) и моногидрат п-толуолсульфокислоты (200 мг, 1,1 ммоль) растворяли в 2-пентаноле (2 мл). Реакционную смесь нагревали до 120°С микроволновым излучением и проводили реакцию в течение 1 часа. После того как ЖХМС показывала завершение реакции, реакционный раствор естественным образом охлаждали до комнатной температуры и осаждали темное твердое вещество. Твердое вещество фильтровали и осадок на фильтре промывали метанолом (1 мл) и метил-трет-бутиловым эфиром (1 мл), с получением сырого продукта N-(2-(дифторметокси)-4-фтор-5-нитрофенил)-4-(1-метил-1Н-индол-3-ил)пиримидин-2-амина (250 мг).
Стадия 2: получение N-(2-(дифторметокси)-4-(4-метилпиперазин-1-ил)-5-нитрофенил)-4-(1-метил-1Н-индол-3-ил)пиримидин-2-амина

N-(2-(дифторметокси)-4-фтор-5-нитрофенил)-4-(1-метил-1Н-индол-3-ил)пиримидин-2-амин (150 мг, 0,35 ммоль) и метилпиперазин (105 мг, 1,05 ммоль) растворяли в DMF (2 мл). Реакционную смесь нагревали до 120°С микроволновым излучением и проводили реакцию течение 30 мин. После того как ЖХМС показывала завершение реакции, реакционный раствор концентрировали. Полученный остаток очищали препаративной тонкослойной хроматографией с получением
N-(2-(дифторметокси)-4-(4-метилпиперазин-1-ил)-5-нитрофенил)-4-(1-метил-1Н-индол-3-ил)пиримидин-2-амина (50 мг, 28%).
Стадия 3: получение 6-(дифторметокси)-N1-(4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)-4-(4-метилпиперазин-1-ил)бензол-1,3-диамина

N-(2-(дифторметокси)-4-(4-метилпиперазин-1-ил)-5-нитрофенил)-4-(1-метил-1Н-индол-3-ил)пиримидин-2-амин (50 мг, 98,0 мкмоль) растворяли в метаноле (10 мл) и затем добавляли Pd/C (10 мг). Реакцию гидрогенизации проводили при комнатной температуре в течение 2 часов. После того, как ЖХМС показывала завершение реакции, реакционный раствор фильтровали через целит, и фильтрат концентрировали с получением неочищенного продукта 6-(дифторметокси)-N1-(4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)-4-(4-метилпиперазин-1-ил)бензол-1,3-диамина (40 мг, 85%), который использовали непосредственно в следующей реакции.
Стадия 4: получение N-(4-(дифторметокси)-5-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-(4-метилпиперазин-1ил)фенил)акриламида

6-(дифторметокси)-N1-(4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)-4-(4-метилпиперазин-1-ил)бензол-1,3-диамин (40 мг, 83,4 мкмоль) и триэтиламин (50 мг, 0,50 ммоль) растворяли в безводном тетрагидрофуране (15 мл). Реакционную смесь перемешивали в течение 10 минут на ледяной бане и медленно добавляли акрилоилхлорид (0,15 мл, 0,15 ммоль, 1 М в THF). Реакционную смесь перемешивали в течение 2 часов на ледяной бане и гасили насыщенным NH4Cl (5 мл), после того как ЖХМС показывала завершение реакции. Реакционный раствор концентрировали и оставшийся водный раствор экстрагировали дихлорметаном (10 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом магния, фильтровали и концентрировали. Полученный остаток очищали препаративной тонкослойной хроматографией с получением N-(4-(дифторметокси)-5-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-(4-метилпиперазин-1-ил)фенил)акриламида (12 мг, 27%).
1Н ЯМР (400 МГц, CDCl3) δ 9,76 (s, 1Н), 8,82 (s, 1Н), 8,66 (s, 1Н), 8,33 (d, J=5,3 Гц, 1Н), 8,05-7,97 (m, 1Н), 7,44 (s, 1Н), 7,33 (dd, J=6,9, 1,8 Гц, 1H), 7,23 (dd, J=7,1, 1.4 Гц, 2H), 7,18 (d, J=5,3 Гц, 1H), 7,00 (s, 1Н), 6,64-6,20 (m, 3Н), 5,75 (dd, J=10,0, 1.5 Гц, 1H), 3,90 (s, 3Н), 2,90 (s, 4Н), 2,68 (s, 4Н), 2,41 (s, 3Н);
MS m/z (ESI): 534,3 [M+H]+.
Пример 3: получение N-(4-(дифторметокси)-2-(метил(2-(3-карбонилморфолино)этил)амино)-5-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида

Стадия 1: получение 4-(2-((5-(дифторметокси)-4-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-нитрофенил)(метил)амино)этил)морфолин-3-она

N-(2-(дифторметокси)-4-фтор-5-нитрофенил)-4-(1-метил-1Н-индол-3-ил)пиримидин-2-амин (200 мг, 0,46 ммоль), 4-(2-(метиламино)этил)морфолин-3-он (110 мг, 0,69 ммоль) и диизопропилэтиламин (180 мг, 1,4 ммоль) растворяли в DMF. Реакционную смесь нагревали до 120°С при микроволновом излучении в течение 30 мин. После того как ЖХМС показывала завершение реакции, реакционный раствор концентрировали. Полученный остаток очищали препаративной тонкослойной хроматографией с получением продукта 4-(2-((5-(дифторметокси)-4-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-нитрофенил)(метил)амино)этил)морфолин-3-она (100 мг, 38%).
Стадия 2: получение 4-(2-((2-амино-5-(дифторметокси)-4-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)(метил)амино)этил) морфолин-3-она

4-(2-((5-(дифторметокси)-4-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-нитрофенил)(метил)амино)этил)морфолин-3-он (100 мг, 0,17 ммоль) растворяли в метаноле (5 мл) с последующим добавлением Pd/C (10 мг). Реакцию гидрогенизации проводили при комнатной температуре в течение 2 часов. После того, как ЖХМС показывала завершение реакции, реакционный раствор фильтровали через целит и фильтрат концентрировали с получением продукта 4-(2-((2-амино-5-(дифторметокси)-4-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)(метил)амино)этил)морфолин-3-она (40 мг, 40%).
Стадия 3: получение N-(4-(дифторметокси)-2-(метил(2-(3-карбонилморфолино)этил)амино)-5-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида

4-(2-(2-амино-5-(дифторметокси)-4-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)(метил)амино)этил)морфолин-3-он (40 мг, 74,4 ммоль) и триэтиламин (40 мг, 0,37 ммоль) растворяли в безводном тетрагидрофуране (15 мл). Реакционную смесь перемешивали в течение 10 минут на ледяной бане и медленно добавляли на ледяной бане акрилоилхлорид (0,1 мл, 100 мкмоль, 1 М в THF). Реакционную смесь перемешивали в течение 30 минут на ледяной бане, после того как ЖХМС показывала завершение реакции, реакцию гасили насыщенным NH4Cl (5 мл). Реакционный раствор концентрировали, затем оставшийся водный раствор экстрагировали дихлорметаном (5 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом магния, фильтровали и концентрировали. Полученный остаток очищали препаративной тонкослойной хроматографией с получением продукта N-(4-(дифторметокси)-2-(метил(2-(3-карбонилморфолино)этил)амино)-5-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида (10 мг, 23%).
1Н ЯМР (400 МГц, CDCl3) δ 9,79 (s, 1Н), 8,83 (d, J=44,6 Гц, 2Н), 8,30 (d, J=5,4 Гц, 1Н), 7,99 (dd, J=6,7, 1,8 Гц, 1Н), 7,33 (dd, J=6,9, 1,8 Гц, 1Н), 7,25-7,20 (m, 2Н), 7,18 (s, 1Н), 6,96 (s, 1Н), 6,71-6,30 (m, 3Н), 5,74 (dd, J=9,5, 2,2 Гц, 1Н), 4,08 (s, 2Н), 3,90 (s, 3Н), 3,73 (dd, J=5,7, 4,5 Гц, 2Н), 3,49 (t, J=6,1 Гц, 2Н), 3,24-3,19 (m, 2Н), 3,05 (t, J=6,1 Гц, 2Н),2,61 (s, 3Н);
MS m/z (ESI): 592,3 [М+Н]+.
Пример 4: получение N-(4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)-5-((4-(6-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида

Стадия 1: получение 3-(2-хлорпиримидин-4-ил)-6-метокси-1-метил-1Н-индола

6-метокси-1-метил-1Н-индол (300 мг, 1,86 ммоль) и 2,4-дихлорпиримидин (330 мг, 2,23 ммоль) растворяли в диметиловом эфире этиленгликоля (10 мл), затем добавляли безводный трихлорид алюминия (500 мг, 3,72 ммоль). Реакционную смесь нагревали до 60°С в атмосфере азота и перемешивали в течение 3 ч. После того, как ЖХМС показывала завершение реакции, реакционный раствор выливали в приблизительно 50 мл ледяной воды и экстрагировали метил-трет-бутиловым эфиром (50 мл × 3). Органические фазы объединяли, промывали последовательно насыщенным бикарбонатом натрия (30 мл) × 2) и H2O (30 мл), сушили, фильтровали и концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле, с получением продукта 3-(2-хлорпиримидин-4-ил)-6-метокси-1-метил-1Н-индола (120 мг, 24%).
Стадия 2: получение N-(2-(дифторметокси)-4-фтор-5-нитрофенил)-4-(6-метокси-(1-метил-1Н-индол-3-ил)пиримидин-2-амина

3-(2-хлорпиримидин-4-ил)-6-метокси-1-метил-1Н-индол (120 мг, 0,44 ммоль), 2-(дифторметокси)-4-фтор-5-нитроанилин (120 мг, 0,53 ммоль) и п-толуолсульфонилхлорид (110 мг, 0,57 ммоль) растворяли в 2-пентаноле (5 мл). Реакционную смесь нагревали до 120°С и перемешивали в течение 16 часов. После того как ЖХМС показывала завершение реакции, реакционный раствор концентрировали и экстрагировали DCM (10 мл) и насыщенным водным раствором бикарбоната натрия (10 мл). Органическую фазу сушили и фильтровали. Фильтрат концентрировали, с получением неочищенного продукта N-(2-(дифторметокси)-4-фтор-5-нитрофенил)-4-(6-метокси-(1-метил-1Н-индол-3-ил)пиримидин-2-амина (200 мг, 98%), который непосредственно использовали на следующей стадии.
Стадия 3: получение 2-(дифторметокси)-N4-(2-(диметиламино)этил)-N1-(4-(6-метокси-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)-N4-метил-5-нитробензол-1,4-диамина

N-(2-(дифторметокси)-4-фтор-5-нитрофенил)-4-(6-метокси-(1-метил-1Н-индол-3-ил)пиримидин-2-амин (100 мг, 0,21 ммоль), триэтиламин (100 мг, 0,98 ммоль) и триметилэтилендиамин (60 мг, 0,59 ммоль) растворяли в DMF (1 мл). Реакционную смесь нагревали до 120°С микроволновым излучением и перемешивали в течение 30 минут. Полученный остаток очищали препаративной тонкослойной хроматографией с получением продукта 2-(дифторметокси)-N4-(2-(диметиламино)этил)-N1-(4-(6-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-N4-метил-5-нитробензол-1,4-диамина (20 мг, 18%).
Стадия 4: получение 5-(дифторметокси)-N1-(2-(диметиламино)этил)-N4-(4-(6-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-N1-метилбензол-1,2,4-триамина

2-(дифторметокси)-N4-(2-(диметиламино)этил)-N1-(4-(6-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-N4-метил-5-нитробензол-1,4-диамин (20 мг, 36,9 мкмоль) растворяли в метаноле (5 мл), затем добавляли Pd/C (10 мг). Реакцию гидрогенизации проводили при комнатной температуре в течение 2 часов. После того как ЖХМС показывала завершение реакции, реакционную смесь фильтровали через целит и фильтрат концентрировали с получением продукта
5-(дифторметокси)-N1-(2-(диметиламино)этил)-N4-(4-(6-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-N1-метилбензол-1,2,4-триамина (15 мг, 80%), который использовали непосредственно в следующей реакции.
Стадия 5: получение N-(4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)-5-((4-(6-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида

5-(дифторметокси)-N1-(2-(диметиламино)этил)-N4-(4-(6-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-N1-метилбензол-1,2,4-триамин (15 мг, 29,4 мкмоль) и триэтиламин (50 мг) растворяли в безводном тетрагидрофуране (15 мл). Реакционную смесь перемешивали в течение 10 минут на ледяной бане и затем медленно добавляли акрилоилхлорид (0,1 мл, 100 мкмоль, 1 М раствор в THF) на ледяной бане. Реакционную смесь перемешивали в течение 30 минут на ледяной бане и гасили насыщенным NH4Cl (5 мл), после того как ЖХМС показывала завершение реакции. Реакционный раствор концентрировали и оставшийся водный раствор экстрагировали дихлорметаном (5 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали и полученный остаток очищали препаративной тонкослойной хроматографией с получением продукта N-(4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)-5-((4-(6-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида (5,0 мг, 30%).
1Н ЯМР (400 МГц, CDCl3) δ 10,01 (s, 1Н), 9,70 (s, 1Н), 8,63 (s, 1Н), 8,25 (d, J=5,3 Гц, 1Н), 7,83 (d, J=8,8 Гц, 1Н), 7,33 (s, 1Н), 7,06 (d, J=5,3 Гц, 1Н), 6,91 (s, 1Н), 6,79 (dd, J=8,8, 2,3 Гц, 1Н), 6,72 (d, J=2,2 Гц, 1Н), 6,37 (dd, J=83,2, 64,3 Гц, 3Н), 5,63 (d, J=11,9 Гц, 1Н), 3,78 (d, J=3,4 Гц, 3Н), 3,36 (s, 3Н), 2,82 (s, 2Н), 2,57 (s, 3Н), 2,24 (s, 6Н), 1,89 (d, J=5,9 Гц, 2Н);
МС m/z (ESI): 566,2 [М+Н]+.
Пример 5: получение N-(5-((5-хлор-4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)фенил)акриламида

Способ получения N-(5-((5-хлор-4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(дифторметокси)-2-((2-(диметиламино)этил)(метил) амино)фенил)акриламид был аналогичен Примеру 1.
1Н ЯМР (400 МГц, CDCl3) δ 9,41 (s, 1Н), 8,39 (s, 1Н), 8,33 (d, J=5,3 Гц, 2Н), 7,60-7,18 (m, 3Н), 6,98 (s, 1Н), 6,66-6,20 (m, 3Н), 5,74-5,58 (m, 1Н), 3,90 (s, 3Н), 2,92-2,77 (m, 2Н), 2,62 (s, 3Н), 2,27 (s, 2Н), 2,22 (s, 6Н);
MS m/z (ESI): 571,0 [М+Н]+.
Пример 6: получение N-(4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-(2-гидроксиэтил)-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида

Стадия 1: получение 2-(3-(2-((2-(дифторметокси)-4-фтор-5-нитрофенил)амино)пиримидин-4-ил)-1Н-индол-1-ил)этан-1-ола

1-(2-((трет-бутилдиметилсилил)окси)этил)-3-(2-хлорпиримидин-4-ил)-1Н-индол (619 мг, 1,595 ммоль), 2-(дифторметокси)-4-фтор-5-нитроанилин (322 мг, 1,45 ммоль) и моногидрат п-толуолсульфоновой кислоты (276 мг, 1,45 ммоль) растворяли в 2-пентаноле (5 мл). Реакционную смесь нагревали до 120°С в течение ночи. После того как ЖХМС показывала завершение реакции, реакционный раствор естественным образом охлаждали до комнатной температуры и осаждали темное твердое вещество. Твердое вещество фильтровали и осадок на фильтре промывали метанолом (1 мл) и метил-трет-бутиловым эфиром (1 мл), с получением продукта 2-(3-(2-((2-(дифторметокси)-4-фтор-5-нитрофенил)амино)пиримидин-4-ил)-1Н-индол-1-ил)этан-1-ола (135 мг, 20%).
Стадия 2. получение 2-(3-(2-((2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)-5-нитрофенил)амино)пиримидин-4-ил)-1Н-индол-этан-1-ола

2-(3-(2-((2-(дифторметокси)-4-фтор-5-нитрофенил)амино)пиримидин-4-ил)-1Н-индол-1-ил)этан-1-ол (130 мг, 0,283 ммоль) растворяли в 2 мл DMF, затем добавляли триэтиламин (87 мг, 0,849 ммоль) и триметилэтилендиамин (87 мг, 0,849 ммоль). Реакционную смесь нагревали до 120°С микроволновым излучением и перемешивали в течение 30 мин. После того как ЖХМС показывала завершение реакции, реакционный раствор концентрировали досуха. Неочищенный продукт очищали препаративной тонкослойной хроматографией с получением продукта (131 мг, 90%).
Стадия 3: получение 2-(3-(2-((5-амино-2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)фенил)амино)пиримидин-4-ил)-1Н-индол-1-ил)этан-1-ола

2-(3-(2-((2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)-5-нитрофенил)амино)пиримидин-4-ил)-1Н-индол-этан-1-ол (130 мг, 0,24 ммоль) растворяли в метаноле (5 мл) и затем добавляли Pd/C (10 мг). Реакционную смесь перемешивали в течение 1 часа в атмосфере водорода при комнатной температуре. После того как ЖХМС показывала завершение реакции, реакционный раствор фильтровали и фильтрат концентрировали, с получением продукта (104 мг, 85%), который непосредственно использовали на следующей стадии.
Стадия 4. получение N-(4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-(2-гидроксиэтил)-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил) акриламида

2-(3-(2-((5-амино-2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)фенил)амино)пиримидин-4-ил)-1Н-индол-1-ил)этан-1-ол (97 мг, 0,19 ммоль) и триэтиламин (19 мг, 0,19 ммоль) растворяли в безводном тетрагидрофуране (50 мл) и реакционную смесь перемешивали при -78°С в течение 10 минут. Медленно и по каплям добавляли акрилоилхлорид (0,6 мл, 1 M в THF). Реакционную смесь перемешивали при этой температуре в течение 30 минут и гасили метанолом, после того как ЖХМС показывала завершение реакции. Реакционный раствор концентрировали и полученный остаток очищали препаративной тонкослойной хроматографией с получением продукта N-(4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-(2-гидроксиэтил)-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида (15 мг, 14%).
1Н ЯМР (400 МГц, CD3OD) δ 8,59 (s, 1Н), 8,45 (s, 1Н), 8,25 (s, 1Н), 8,07 (d, J=6,7 Гц, 1Н), 7,58 (d, J=8,2 Гц, 1Н), 7,53 (s, 1Н), 7,44 (d, J=6,7 Гц, 1Н), 7,41 (s, 1Н), 7,35-7,16 (m, 5Н), 7,01 (dd, J=17,2, 10,0 Гц, 2Н), 6,42 (d, J=16,9 Гц, 1Н), 5,83 (d, J=10,3 Гц, 1Н), 4,41 (t, J=5,1 Гц, 2Н), 3,95 (t, J=5,1 Гц, 2Н), 3,50 (d, J=5,3 Гц, 2Н), 3,44 (d, J=5,2 Гц, 2Н), 3,37 (s, 1Н), 2,93 (s, 6Н), 2,81 (s, 3Н);
MS m/z (ESI): 566 М+Н]+.
Пример 7: получение N-(5-((4-(1-ацетил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(дифторметокси)-2-((2-(диметиламино))этил)(метил)амино)фенил)акриламида

Стадия 1: получение N-(2-(дифторметокси)-4-фтор-5-нитрофенил)-4-(1Н-индол-3-ил)пиримидин-2-амина

1-(3-(2-хлорпиримидин-4-ил)-1Н-индол-1-ил)этан-1-он (735 мг, 2,71 ммоль), 2-(дифторметокси)-4-фтор-5-нитроанилин (600 мг, 2,71 ммоль) и моногидрат п-толуолсульфоновой кислоты (514 мг, 2,71 ммоль) растворяли в 2-пентаноле (20 мл) и реакционную смесь нагревали до 120°С в течение ночи. После того как ЖХМС показывала завершение реакции, реакционный раствор естественным образом охлаждали до комнатной температуры и осаждали темное твердое вещество. Твердое вещество фильтровали, и осадок на фильтре промывали метанолом (1 мл) и метил-трет-бутиловым эфиром (1 мл), с получением N-(2-(дифторметокси)-4-фтор-5-нитрофенил)-4-(1Н-индол-3-ил)пиримидин-2-амина (250 мг, 20%).
Стадия 2: получение N-(4-(1Н-индол-3-ил)пиримидин-2-ил)2-(дифторметокси)-(2-(диметиламино)этил)-метил-5-нитробензол-1,4-диамина

N-(2-(дифторметокси)-4-фтор-5-нитрофенил)-4-(1Н-индол-3-ил)пиримидин-2-амин (100 мг, 0,241 ммоль) растворяли в DMF (2 мл) и затем добавляли триэтиламин (73 мг, 0,72 ммоль) и триметилэтилендиамин (74 мг, 0,72 ммоль). Реакционную смесь нагревали до 120°С микроволновым излучением и перемешивали в течение 30 мин. После того как ЖХМС показывала завершение реакции, реакционный раствор концентрировали. Неочищенный продукт очищали препаративной тонкослойной хроматографией с получением продукта (100 мг, 83%).
Стадия 3: получение N-(4-(1-ацетил-1Н-индол-3-ил)пиримидин-2-ил)-N-(2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)-5-нитрофенил)ацетамида

N-(4-(1Н-индол-3-ил)пиримидин-2-ил)-2-(дифторметокси)-(2-(диметиламино)этил)-метил-5-нитробензол-1,4-диамин (100 мг, 0,20 ммоль) растворяли в уксусном ангидриде (4 мл) и затем добавляли триэтиламин (0,5 мл) и DMAP (3 мг, 0,02 ммоль). Реакционную смесь перемешивали при 120°С в течение 30 минут. Затем реакционный раствор концентрировали и экстрагировали три раза этилацетатом и водой. Органические фазы объединяли, промывали последовательно насыщенным водным раствором бикарбоната натрия, водой и насыщенным солевым раствором, сушили, фильтровали и концентрировали, с получением неочищенного продукта, который непосредственно использовали на следующей стадии.
Стадия 4: получение 1-(3-(2-((5-амино-2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)фенил)амино)пиримидин-4-ил)-1Н-индол-1-ил)этан-1-она

N-(4-(1-ацетил-1Н-индол-3-ил)пиримидин-2-ил)-N-(2-(дифторметокси)-4-((2-(диметиламино)этил)-5-нитрофенил)ацетамид, который был получен в предыдущей реакции, растворяли в метаноле (5 мл) и добавляли Pd/C (15 мг) и затем реакционную смесь перемешивали при 24°С в атмосфере водорода в течение 1 часа. После того как ЖХМС показывала завершение реакции, реакционный раствор фильтровали и фильтрат концентрировали с получением неочищенного продукта, который затем очищали колоночной флэш-хроматографией с получением продукта 1-(3-(2-((5-амино-2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)фенил)амино)пиримидин-4-ил)-1Н-индол-1-ил)этан-1-она (30 мг, 32%).
Стадия 5: получение N-(5-(4-(1-ацетил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)фенил)-акриламида

1-(3-(2-((5-амино-2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)фенил)амино)пиримидин-4-ил)-1Н-индол-1-ил)этан-1-он (30 мг, 0,059 ммоль) и триэтиламин (6 мг, 0,19 ммоль) растворяли в безводном тетрагидрофуране (30 мл). Реакционную смесь перемешивали при -78°С в течение 10 минут и медленно добавляли акрилоилхлорид (0,2 мл, 1 М в THF). Реакционную смесь перемешивали при этой температуре в течение 30 минут и реакцию гасили метанолом после того, как ЖХМС показывала завершение реакции. Реакционный раствор концентрировали и полученный остаток очищали препаративной тонкослойной хроматографией с получением продукта N-(5-(4-(1-ацетил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)фенил)акриламида (15 мг, 45%).
1Н ЯМР (400 МГц, CDCl3) δ 9,81 (s, 1Н), 9,34 (s, 1Н), 8,61 (s, 1Н), 8,40 (d, J=5,4 Гц, 1Н), 7,90 (d, J=8,0 Гц, 1Н), 7,78 (d, J=2,7 Гц, 1Н), 7,35 (d, J=8,2 Гц, 1Н), 7,20 (d, J=5,4 Гц, 1Н), 7,08 (d, J=3,2 Гц, 1Н), 6,54-6,33 (m, 3Н), 6,22-6,04 (m, 2Н), 5,82 (dd, J=4,0 Гц, 1Н), 5,69 (dd, J=4,0 Гц, 1Н), 3,07-3,02 (m, 2Н), 2,84 (s, 2Н), 2,92-2,76 (m, 3Н), 2,59 (s, 3Н), 2,53 (s, 6Н);
MS m/z (ESI): 564 [М+Н]+.
Пример 8: получение N-(5-((4-(1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)фенил)-акриламида

N-(5-((4-(1-ацетил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(дифторметокси)-2-((2-(диметиламино)этил))амино)фенил)акриламид (12 мг, 0,021 ммоль) растворяли в метаноле (2 мл) и затем добавляли водный раствор 1 Н карбоната натрия (1 мл). Раствор подвергали взаимодействию при комнатной температуре в течение 3 часов и концентрировали с получением неочищенного продукта, который затем очищали колоночной флэш-хроматографией на силикагеле, с получением N-(5-((4-(1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)фенил)акриламида (4 мг, 36,4%).
1Н ЯМР (400 МГц, CD3OD) δ 8,54 (d, J=3,4 Гц, 1Н), 8,38-8,04 (m, 3Н), 7,51 (d, J=7,3 Гц, 2Н), 7,39 (t, J=9,9 Гц, 1Н), 7,31-7,15 (m, 3Н), 6,87 (ddd, J=40,9, 27,6, 6,5 Гц, 2Н), 6,45 (d, J=17,0 Гц, 1Н), 5,85 (d, J=10,3 Гц, 1Н), 5,36 (t, J=4,7 Гц, 1Н), 3,56-3,47 (m, 2Н), 3,45-3,38 (m, 2Н), 2,93 (s, 6Н), 2,82 (s, 3Н);
MS m/z (ESI): 522 [М+Н]+.
Пример 9: получение N-(5-((4-(6-циано-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)фенил)-акриламида

Способ получения N-(5-((4-(6-циано-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)фенил)-акриламида был аналогичен Примеру 4.
Пример 10: получение N-(4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-метил-6-(изопропилсульфонил)-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-метил-6-(изопропилсульфонил)-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 4.
Пример 11: получение N-(4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)-5-((4-(6-(диметилфосфорил)-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида

Стадия 1: получение (3-(2-((2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)-5-нитрофенил)амино)пиримидин-4-ил)-1-метил-1Н-индол-6-ил)диметилфосфиноксида

N1-(4-(6-бром-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-2-(дифторметокси)-N4-(2-(диметиламино)этил)-N4-метил-5-нитробензол-1,4-диамин (50 мг, 84,6 мкмоль), диметилфосфиноксид (66,1 мг, 0,85 ммоль), ацетат палладия (10 мг), триэтиламин (0,25 мл) и X-Phos (20 мг) растворяли в DMF (2 мл). Смесь продували азотом для удаления кислорода в течение 10 минут и нагревали до 130°С при микроволновом излучении в течение 1 часа. После того как ЖХМС показывала завершение реакции, реакционный раствор фильтровали и фильтрат выпаривали досуха. Полученный остаток очищали препаративной тонкослойной хроматографией с получением (3-(2-((2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)-5-нитрофенил)амино)пиримидин-4-ил)-1-метил-1Н-индол-6-ил)диметилфосфиноксида (40 мг, 80%).
Стадия 2: получение (3-(2-((5-амино-2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)фенил)амино)пиримидин-4-ил)-1-метил-1Н-индол-6-ил)диметилфосфиноксида

3-(2-((2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)-5-нитрофенил)амино)пиримидин-4-ил)-1-метил-1Н-индол-6-ил)диметилфосфиноксид (40 мг, 68,1 мкмоль) растворяли в метаноле (5 мл) и затем добавляли Pd/C (10 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода в течение 10 минут. После того как ЖХМС показывала завершение реакции, реакционный раствор фильтровали и фильтрат концентрировали. Полученный остаток очищали колоночной хроматографией с обращенной фазой с получением (3-(2-((5-амино-2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)фенил)амино)пиримидин-4-ил)-1-метил-1Н-индол-6-ил)диметилфосфиноксида (15 мг, 27%).
Стадия 3: получение N-(4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)-5-((4-(6-(диметилфосфорил)-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида

(3-(2-((5-амино-2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)-фенил)амино)пиримидин-4-ил)-1-метил-1Н-индол-6-ил)диметилфосфиноксид (15 мг, 26,9 мкмоль) и триэтиламин (0,1 мл) растворяли в тетрагидрофуране (10 мл) и реакционный раствор охлаждали до -10 до -5°С. Медленно добавляли акрилоилхлорид (0,1 мл, 1 М в THF) в атмосфере азота. Реакционную смесь перемешивали при температуре от -10 до -5°С в течение 30 минут. После завершения реакции добавляли метанол (3 мл) и реакционный раствор дополнительно перемешивали в течение 10 минут, затем концентрировали. Полученный остаток очищали препаративной тонкослойной хроматографией с последующей колоночной хроматографией с обращенной фазой с получением N-(4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)-5-((4-(6-(диметилфосфорил)-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида (3,5 мг, 21%).
1Н ЯМР (400 МГц, CD3OD) δ 8,67 (s, 1Н), 8,39 (d, J=7,4 Гц, 1Н), 8,25 (s, 1Н), 8,20 (d, J=6,6 Гц, 1Н), 8,00 (d, J=13,1 Гц, 1Н), 7,58 (dd, J=10,6, 8,9 Гц, 1Н), 7,48 (d, J=6,7 Гц, 1Н), 7,32 (s, 1Н), 6,97 (t, J=73,2 Гц, 2Н), 6,62 (dd, J=16,9, 10,0 Гц, 1Н), 6,50 (dd, J=16,9, 1,7 Гц, 1Н), 5,89 (dd, J=10,0, 1,7 Гц, 1Н), 4,05 (s, 3Н), 3,53 (t, J=5,9 Гц, 2Н), 3,38 (t, J=5,9 Гц, 2Н), 2,96 (s, 6Н), 2,82 (s, 3Н), 1,86 (d, J=13,3 Гц, 6Н);
MS m/z (ESI): 612,3 [М+Н]+.
Пример 12: получение N-(5-((4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(дифторметокси-2-((2-(диметиламино))этил)(метил)-амино)фенил)акриламида

Стадия 1: получение 3-(2-хлорпиримидин-4-ил)-1-циклопропил-1Н-индола

1-циклопропил-1Н-индол (140 мг, 0,89 ммоль) и 2,4-дихлорпиримидин (170 мг, 1,14 ммоль) растворяли в диметиловом эфире этиленгликоля (10 мл) и затем добавляли безводный хлорид алюминия (180 мг, 1,35 ммоль). Реакционную смесь нагревали до 100°С в течение ночи. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток растворяли в дихлорметане (30 мл) и органическую фазу дважды промывали водой, сушили и концентрировали. Полученный остаток очищали препаративной тонкослойной хроматографией (петролейный эфир : этилацетат = 8:1) с получением 3-(2-хлорпиримидин-4-ил)-1-циклопропил-1Н-индола (80 мг, 80%).
MS m/z (ESI): 270,1 [М+Н]+.
Стадия 2: получение 4-(1-циклопропил-1Н-индол-3-ил)-N-(2-(дифторметокси)-4-фтор-5-нитрофенил)пиримидин-2-амина

3-(2-хлорпиримидин-4-ил)-1-циклопропил-1Н-индол (80 мг, 0,29 ммоль) и 2-(дифторметокси)-4-фтор-5-нитроанилин (64 мг, 0,29 ммоль) растворяли в 2-пентаноле. Смесь нагревали в течение 1 часа микроволновым излучением и охлаждали до комнатной температуры. Растворитель выпаривали и полученный остаток очищали препаративной тонкослойной хроматографией, с получением 4-(1-циклопропил-1Н-индол-3-ил)-N-(2-(дифторметокси)-4-фтор-5-нитрофенил)-пиримидин-2-амина (76 мг).
MS m/z (ESI): 456,1 [М+Н]+.
Стадия 3: получение N1-(4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)-2-(дифторметокси)-N4-(2-(диметиламино)этил)-N4-метил-5-нитробензол-1,4-диамина

4-(1-циклопропил-1Н-индол-3-ил)-N-(2-(дифторметокси)-4-фтор-5-нитрофенил)пиримидин-2-амин (76 мг) растворяли в N,N-диметилацетамиде, а затем добавляли триметилэтилендиамин (0,1 г). Смесь нагревали до температуры дефлегмации в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры и растворитель выпаривали с получением N1-(4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)-2-(дифторметокси)-N4-(2-(диметиламино)этил)-N4-метил-5-нитробензол-1,4-диамина (50 мг).
MS m/z (ESI): 538,3 [M+H]+.
Стадия 4: получение N4-(4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)-5-(дифторметокси)-N1-(2-(диметиламино)этил)-N1-метил-бензол-1,2,4-триамина

N1-(4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)-2-(дифторметокси)-N4-(2-(диметиламино)этил)-N4-метил-5-нитробензол-1,4-диамин (50 мг) растворяли в 6 мл смешанного растворителя этанол-вода (5:1), затем добавляли 65 мг порошка железа и 50 мг хлорида аммония. Смесь кипятили до температуры дефлегмации в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры, фильтровали и собирали фильтрат. Фильтрат концентрировали при пониженном давлении для удаления этанола с последующим добавлением воды и дихлорметан-метанола (20:1). Органическую фазу отделяли и концентрировали с получением неочищенного продукта (20 мг).
MS m/z (ESI): 508,3 [М+Н]+.
Стадия 5: получение N-(5-((4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(дифторметокси)-2-((2-(диметиламино))этил)(метил)амино)фенил)-акриламида

N4-(4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)-5-(дифторметокси)-N1-(2-(диметиламино)этил)-N1-метилбензол-1,2,4-триамин (20 мг) растворяли в безводном тетрагидрофуране. В атмосфере азота добавляли DIPEA (0,1 мл) при °С и по каплям добавляли раствор 1 М акрилоилхлорида в тетрагидрофуране (0,2 мл). Реакцию проводили при 0°С в течение 1 часа. К реакционному раствору добавляли воду и дихлорметан, и водную фазу и органическую фазу отделяли. Водную фазу экстрагировали три раза дихлорметаном. Затем органические фазы объединяли, сушили и концентрировали. Неочищенный продукт получали тонкослойной хроматографией. Неочищенный продукт далее очищали колоночной хроматографией с обращенной фазой (вода : метанол = 25:75) с получением конечного продукта (6,2 мг).
1Н ЯМР (400 МГц, CD3OD) δ 8,56 (s, 1Н), 8,26 (m, 2Н), 8,08 (d, 1Н), 7,71 (d, 1Н), 7,50 (d, 1Н), 7,32 (m, 3Н), 6,96 (m, 1Н), 6,79 (m, 1Н), 6,44 (dd, 1Н), 5,85 (d, 1H), 3,62 (m, 1Н), 3,52 (m, 2Н), 3,40 (m, 2Н), 2,94 (s, 6Н), 2,82 (s, 3Н), 1,24 (m, 2Н), 1,14 (m, 2Н);
19F ЯМР (376 МГц, CD3OD) δ -83,26;
MS m/z (ESI): 562,2 [M+H]+.
Пример 13: получение N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(трифторметокси)фенил)-акриламида

Стадия 1: получение 4-фтор-1-нитро-2-(трифторметокси)бензола

1-фтор-3-(трифторметокси)бензол (7,5 г, 41,6 ммоль) растворяли в концентрированной серной кислоте (30 мл) и смесь охлаждали до 0°С. Медленно частями добавляли KNO3 (1,04 г, 10,25 ммоль). Внутреннюю температуру поддерживали ниже 5°С. По завершении добавления смесь перемешивали в течение 2 часов. Добавляли смесь лед-вода (приблизительно 50 мл). Реакционный раствор экстрагировали метил-трет-бутиловым эфиром (20×3 мл) и органические фазы объединяли, сушили и фильтровали. Фильтрат концентрировали и очищали колоночной флэш-хроматографией на силикагеле, с получением 4-фтор-1-нитро-2-(трифторметокси)бензола (4,0 г, 42%).
Стадия 2: получение 4-фтор-2-(трифторметокси)анилина

4-фтор-1-нитро-2-(трифторметокси)бензол (4,0 г, 17,8 ммоль) растворяли в метаноле (50 мл) и затем добавляли Pd/C (200 мг). Реакционную смесь перемешивали в течение 2 часов в атмосфере водорода. После того как ЖХМС показывал завершение реакции, реакционный раствор фильтровали и фильтрат концентрировали. Полученный остаток очищали колоночной хроматографией с обращенной фазой с получением 4-фтор-2-(трифторметокси)анилина (3,0 г, 86%).
Стадия 3: получение 4-фтор-5-нитро-2-(трифторметокси)анилина

4-фтор-2-(трифторметокси)анилин (2,0 г, 10,25 ммоль) растворяли в концентрированной серной кислоте (10 мл) и смесь охлаждали до -20°С. KNO3 (1,04 г, 10,25 ммоль) добавляли медленно частями. Внутреннюю температуру поддерживали ниже -10°С. По завершении добавления, смесь перемешивали в течение 1 часа. Добавляли смесь лед-вода (приблизительно 50 мл). Реакционный раствор экстрагировали метил-трет-бутиловым эфиром (20 мл × 3) и органические фазы объединяли, сушили и фильтровали. Фильтрат концентрировали и очищали колоночной флэш-хроматографией на силикагеле, с получением 4-фтор-5-нитро-2-(трифторметокси)анилина (500 мг, 20%).
Стадия 4: получение N-(4-фтор-5-нитро-2-(трифторметокси)фенил)-4-(1-метил-1Н-индол-3-ил)пиримидин-2-амина

4-фтор-5-нитро-2-(трифторметокси)анилин (500 мг, 2,08 ммоль), 3-(2-хлорпиримидин-4-ил)-1-метил-1Н-индол (508 мг, 2,08 ммоль) и моногидрат п-толуолсульфокислоты (400 мг, 2,08 ммоль) растворяли в 1,4-диоксане (10 мл). Реакционную смесь нагревали до 110°С и перемешивали в течение 16 часов. После того как ЖХМС показывала завершение реакции, добавляли насыщенный водный раствор NaHCO3 (20 мл) и смесь перемешивали в течение 20 минут и фильтровали. Осадок на фильтре промывали метил-трет-бутиловым эфиром с получением неочищенного продукта N-(4-фтор-5-нитро-2-(трифторметокси)-фенил)-4-(1-метил-1Н-индол-3-пиримидин-2-амина (400 мг, 43%).
Стадия 5: получение N1-(2-(диметиламино)этил)-N1-метил-N4-(4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)-2-нитро-5-(трифторметокси)бензол-1,4-диамина

N-(4-фтор-5-нитро-2-(трифторметокси)фенил)-4-(1-метил-1Н-индол-3-ил)пиримидин-2-амин (400 мг, 0,89 ммоль), N,N,N-триметилэтилендиамин (180 мг, 1,79 ммоль) и триэтиламин (1 мл) растворяли в DMF (5 мл). Реакционную смесь нагревали до 110°С в течение 2 часов. После того как ЖХМС показывал завершение реакции, добавляли дихлорметан (10 мл) и воду (10 мл). Органическую фазу промывали три раза водой, сушили над безводным сульфатом магния, фильтровали и концентрировали. Полученный остаток очищали колоночной флэш-хроматографией на силикагеле, с получением N1-(2-(диметиламино)этил)-N1-метил-N4-(4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)-2-нитро-5-(трифторметокси)бензол-1,4-диамина (80 мг, 17%).
Стадия 6: получение N1-(2-(диметиламино)этил)-N1-метил-N4-(4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил-5-(трифторметокси)бензола-1,2,4-триамина

N1-(2-(диметиламино)этил)-N1-метил-N4-(4-(1-метил-1Н-индол-3-ил)-пиримидин-2-ил)-2-нитро-5-(трифторметокси)бензол-1,4-диамин (80 мг, 0,15 ммоль) растворяли в метаноле (5 мл) и затем добавляли Pd/C (10 мг). Реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение 10 минут. После того как ЖХМС показывала завершение реакции, реакционный раствор фильтровали и фильтрат концентрировали, с получением N1-(2-(диметиламино)этил)-N1-метил-N4-(4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил-5-(трифторметокси)бензол-1,2,4-триамина (50 мг, 70%), который использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 7: получение N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(трифторметокси)фенил) акриламида

N1-(2-(диметиламино)этил)-N1-метил-N4-(4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил-5-(трифторметокси)бензол-1,2,4-триамин (50 мг, 0,24 ммоль) и триэтиламин (0,2 мл) растворяли в тетрагидрофуране (10 мл). Реакционный раствор охлаждали до от -10 до -5°С. Акрилоилхлорид (0,35 мл, 1 М в THF) медленно добавляли в атмосфере азота, реакционную смесь перемешивали при температуре от -10 до -5°С в течение 30 минут, после завершения реакции добавляли метанол (3 мл) и реакционный раствор дополнительно перемешивали в течение 10 минут, затем концентрировали при пониженном давлении. Полученный остаток очищали препаративной тонкослойной хроматографией с последующей колоночной хроматографией с обращенной фазой с получением N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(трифторметокси)фенил)акриламида (19,0 мг, 14%).
1Н ЯМР (400 МГц, CD3OD) δ 8,53 (s, 1Н), 8,47 (s, 1Н), 8,14 (d, J=7,8 Гц, 1Н), 8,07 (d, J=6,8 Гц, 1Н), 7,51 (d, J=8,2 Гц, 1Н), 7,47 (d, J=0,9 Гц, 1Н), 7,42 (d, J=6,9 Гц, 1Н), 7,19 (t, J=7,5 Гц, 1Н), 6,72 (dd, J=16,9, 10,2 Гц, 1Н), 6,47 (dd, J=16,9, 1,5 Гц, 1Н), 5,88 (dd, J=10,3, 1,5 Гц, 1Н), 3,93 (s, 3Н), 3,51 (t, J=5,9 Гц, 2Н), 3,40 (t, J=5,8 Гц, 2Н), 2,95 (s, 6Н), 2,82 (s, 3Н);
MS m/z (ESI): 554,2 [М+Н]+.
Пример 14: получение N-(5-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-(4-метилпиперазин-1-ил)-4-(трифторметокси)фенил)акриламида

Способ получения N-(5-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-(4-метилпиперазин-1-ил)-4-(трифторметокси)фенил)акриламида был аналогичен Примеру 2.
Пример 15: получение N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(6-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(трифторметокси)-фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(6-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(трифторметокси)фенил)акриламида был аналогичен Примеру 4.
Пример 16: получение N-(5-((4-(6-циано-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-(трифторметокси)фенил)-акриламида

Способ получения N-(5-((4-(6-циано-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-(трифторметокси)фенил)-акриламид был аналогичен Примеру 4.
Пример 17: получение N-(5-((4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-(трифторметокси)фенил)акриламида

Способ получения N-(5-((4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-(трифторметокси)фенил)-акриламида был аналогичен Примеру 1.
Пример 18: получение N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-(2-гидроксиэтил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(трифторметокси)-фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-(2-гидроксиэтил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(трифторметокси)фенил) акриламида был аналогичен Примеру 6.
Пример 19: получение N-(5-((4-(1-ацетил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-(трифторметокси)фенил)-акриламида

Способ получения N-(5-((4-(1-ацетил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-(трифторметокси)фенил)-акриламида был аналогичен Примеру 4.
Пример 20: получение N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(6-(диметилфосфорил)-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(трифторметокси)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(6-диметилфосфорил)-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-(трифторметокси)фенил)акриламид был аналогичен Примеру 4.
Пример 21: получение N-(4-(дифторметокси)-2-(2-(диметиламино)этил)(метил)амино)-5-((4-(1-(N,N-диметилсульфамоил)-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида

Стадия 1: получение 3-(2-((2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)-5-нитрофенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамида

N-(4-(1Н-индол-3-ил)пиримидин-2-ил)-2-(дифторметокси)-(2-(диметиламино)-этил)-метил-5-нитрофенил-1,4-диамин (100 мг, 0,201 ммоль) растворяли в 10 мл DMF и смесь охлаждали до 0°С на ледяной бане с последующим добавлением гидрида натрия (24 мг, 0,603 ммоль).
После того, как реакцию проводили в течение 10 минут при 0°С, по каплям добавляли диметилсульфамоилхлорид (35 мг, 0,241 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 30 минут. После гашения, в реакцию добавляли дихлорметан и воду и экстрагировали три раза. Органические фазы объединяли, промывали последовательно насыщенным водным раствором бикарбоната натрия, водой и насыщенным солевым раствором, сушили и концентрировали, с получением сырого продукта, который затем очищали колоночной флэш-хроматографией на силикагеле с получением продукта (95 мг, 78%).
Стадия 2: получение 3-(2-((5-амино-2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)фенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамида

3-(2-((2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)-5-нитро-фенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамид растворяли в 5 мл метанола, затем добавляли Pd/C (15 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода в течение 1 часа. После того, как ЖХМС показывала завершение реакции, реакционную смесь фильтровали и фильтрат концентрировали с получением неочищенного продукта, который затем очищали колоночной флэш-хроматографией на силикагеле, с получением продукта (35 мг, 39%).
Стадия 3: получение N-(4-(дифторметокси)-2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-(N,N-диметилсульфамоил)-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида

3-(2-((5-амино-2-(дифторметокси)-4-((2-(диметиламино)этил)(метил)амино)-фенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамид (35 мг, 0,061 ммоль) и триэтиламин (18 мг, 0,183 ммоль) растворяли в безводном тетрагидрофуране (30 мл). Реакционный раствор перемешивали при -78°С в течение 10 минут. Медленно и по каплям добавляли акрилоилхлорид (0,2 мл, 1 М в THF). Реакционную смесь перемешивали при этой температуре в течение 30 минут. После того как ЖХМС показывала завершение реакции, реакцию гасили метанолом. Реакционный раствор концентрировали и полученный остаток очищали препаративной тонкослойной хроматографией с получением продукта (25 мг, 65%).
1Н ЯМР (400 МГц, CD3OD) δ 8,57 (s, 1Н), 8,39 (s, 1Н), 8,36-8,18 (m, 2Н), 7,97 (d, J=8,4 Гц, 1Н), 7,52 (d, J=6,2 Гц, 1Н), 7,37 (t, J=7,7 Гц, 1Н), 7,33-7,18 (m, 2Н), 7,16-6,58 (m, 2Н), 6,45 (d, J=16,0 Гц, 1Н), 5,96-5,77 (m, 1Н), 3,49 (t, J=5,6 Гц, 2Н), 3,37 (t, J=5,6 Гц, 2Н), 2,93 (s, 6Н), 2,89 (s, 6Н), 2,78 (s, 3Н);
MS m/z (ESI): 629,2 [М+Н]+.
Пример 22: получение N-(5-((4-(6-циклопропил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Стадия 1: получение 6-бром-1-метил-1Н-индола

К раствору 6-бром-1Н-индола (3,00 г, 15,3 ммоль) в DMF (30 мл) добавляли NaH (60%, 734 мг, 18,4 ммоль) на бане с ледяной водой и смесь перемешивали при этой температуре в течение 20 минут. Затем по каплям добавляли раствор MeI (1,14 мл, 18,4 ммоль) в DMF (10 мл) и смесь перемешивали при этой температуре в течение 30 минут. Добавляли 100 мл воды и реакционный раствор экстрагировали EtOAc. Фазу EtOAc промывали несколько раз насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией (элюент: чистый петролейный эфир) с получением указанного в заголовке соединения 6-бром-1-метил-1Н-индола (2,70 г, 84%).
Стадия 2: получение 6-циклопропил-1-метил-1Н-индола

6-бром-1-метил-1Н-индол (1,44 г, 6,85 ммоль) и циклопропилбороновую кислоту (1,18 г, 13,7 ммоль) смешивали в смешанном растворителе из толуола (20 мл) и воды (3 мл). Смесь продували азотом для удаления кислорода в атмосфере азота в течение 5 минут. Затем добавляли Pd(OAc)2 (231 мг, 1,03 ммоль) и безводный фосфат калия (4,37 г, 20,6 ммоль), и смесь снова продували азотом в течение 10 минут. Наконец, добавляли трициклогексилфосфин (769 мг, 2,74 ммоль) и смесь продували азотом в течение 5 минут. В атмосфере азота реакционный раствор перемешивали в течение ночи при 100°С на масляной бане. После охлаждения, органический растворитель удаляли выпариванием. Добавляли EtOAc и воду, и разделяли две фазы. Фазу EtOAc промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией (элюент: чистый петролейный эфир) с получением указанного в заголовке соединения 6-циклопропил-1-метил-1Н-индола (770 мг, 66%).
1Н ЯМР (400 МГц, CDCl3): δ 7,55 (d, J=8,0 Гц, 1Н), 7,09 (d, J=0,8 Гц, 1Н), 7,01 (d, J=3,2 Гц, 1Н), 6,93 (m, 1Н), 6,46 (dd, J=3,2, 0,8 Гц, 1Н), 3,79 (s, 3Н), 2,09 (m, 1Н), 1,01 (m, 2Н), 0,80 (m, 2Н).
Стадия 3: получение 3-(2-хлорпиримидин-4-ил)-6-циклопропил-1-метил-1Н-индола

FeCl3 (864 мг, 5,33 ммоль) добавляли к смешанному раствору 6-циклопропил-1-метил-1Н-индола (760 мг, 4,44 ммоль) и 2,4-дихлорпиримидина (674 мг, 4,44 ммоль) в диметиленэтиленгликолевом эфире (10 мл), и смесь перемешивали в течение ночи при 60°С. После охлаждения добавляли большое количество EtOAc и воду и разделяли две фазы. Нерастворенное вещество удаляли через целит, а затем водную фазу удаляли. Органическую фазу промывали последовательно насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали. Полученный остаток очищали колоночной хроматографией (элюент: петролейный эфир : EtOAc = 3:1) с получением указанного в заголовке соединения (533 мг, 42%).
MS m/z (ESI): 284,2 [М+Н]+.
Стадия 4: получение 4-(6-циклопропил-1-метил-1Н-индол-3-ил)-N-(4-фтор-2-метокси-5-нитрофенил)пиримидин-2-амина

3-(2-хлорпиримидин-4-ил)-6-циклопропил-1-метил-1Н-индол (533 мг, 1,88 ммоль), 4-фтор-2-метокси-5-нитроанилин (350 мг, 1,88 ммоль) и TsOH⋅H2O (429 мг, 2,25 ммоль) смешивали в 2-пентаноле (10 мл) и реакцию проводили при 125°С в течение 3 часов. После охлаждения смеси и фильтрования, полученное твердое вещество растворяли в CH2Cl2, промывали насыщенным водным раствором бикарбоната натрия и насыщенным соленым раствором, сушили над безводным сульфатом натрия и концентрировали с получением указанного в заголовке соединения.
4-(6-циклопропил-1-метил-1Н-индол-3-ил)-N-(4-фтор-2-метокси-5-нитрофенил)-пиримидин-2-амина (750 мг, 92%).
MS m/z (ESI): 434,2 М+Н]+.
Стадия 5: получение N1-(4-(6-циклопропил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамина

4-(6-циклопропил-1-метил-1Н-индол-3-ил)-N-(4-фтор-2-метокси-5-нитрофенил)пиримидин-2-амин (647 мг, 1,49 ммоль), N1,N1,N2-триметилэтан-1,2-диамин (229 мг, 2,24 ммоль) и DIPEA (0,740 мл, 4,48 ммоль) растворяли в DMA (10 мл) и реакцию проводили при 85°С в течение 3 часов. После охлаждения добавляли воду и EtOAc и разделяли две фазы. Органическую фазу несколько раз промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали с получением указанного в заголовке соединения.
N1-(4-(6-циклопропил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамина в виде неочищенного продукта, который непосредственно использовали на следующей стадии.
MS m/z (ESI): 516,3 [М+Н]+.
Стадия 6: получение N4-(4-(6-циклопропил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-N1-(2-(диметиламино)этил)-5-метокси-N1-метилбензол-1,2,4-триамина

N1-(4-(6-циклопропил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамин (1,4 г, неочищенный продукт), восстановленный порошок железа (1,22 г, 21,7 ммоль) и хлорид аммония (100 мг, 1,90 ммоль) смешивали в растворе EtOH (30 мл) и воды (10 мл). Смесь нагревали до температуры дефлегмации в течение трех часов. После охлаждения добавляли большое количество EtOH и нерастворенное вещество удаляли фильтрованием через целит. EtOH удаляли при пониженном давлении. Затем водную фазу экстрагировали EtOAc и EtOAc-фазу промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией [элюент : CH2Cl2 → CH2Cl2 : МеОН (содержащий 10% концентрированного аммиака) = 17:1] с получением указанного в заголовке соединения N4-(4-(6-циклопропил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-N1-(2-(диметиламино)этил)-5-метокси-N1-метилбензол-1,2,4-триамина (490 мг, выход двух стадий: 68%).
MS m/z (ESI): 486,3 [М+Н]+.
Стадия 7: получение N-(5-((4-(6-циклопропил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Раствор акрилоилхлорида (22,0 мг, 0,247 ммоль) в THF (1 мл) добавляли по каплям к раствору N4-(4-(6-циклопропил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-N1-(2-(диметиламино)этил)-5-метокси-N1-метилбензол-1,2,4-триамина (80,0 мг, 0,164 ммоль) и TEA (50,0 мг, 0,492 ммоль) в THF (2 мл) на бане со льдом и водой. По завершении добавления, смесь перемешивали при той же температуре в течение 15 минут. Реакцию гасили метанолом, концентрировали при пониженном давлении и очищали препаративной тонкослойной хроматографией (CH2Cl2 : МеОН : концентрированный аммиак = 100:10:1) с получением указанного в заголовке соединения N-(5-((4-(6-циклопропил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида (45 мг, 51%).
1Н ЯМР (400 МГц, CDCl3): δ 10,1 (br s, 1Н), 9,83 (s, 1Н), 9,00 (s, 1Н), 8,36 (d, J=5,2 Гц, 1Н), 7,93 (d, J=8,4 Гц, 1Н), 7,71 (s, 1Н), 7,16 (d, J=5,2 Гц, 1Н), 7,10 (s, 1Н), 7,00 (dd, J=8,4, 1,6 Гц, 1Н), 6,79 (s, 1Н), 6,39-6,44 (m, 2Н), 5,70 (dd, J=9,6, 2,0 Гц, 1Н), 3,95 (s, 3Н), 3,87 (s, 3Н), 2,89 (t, J=5,6 Гц, 2Н), 2,69 (s, 3Н), 2,27 (t, J=5,6 Гц, 2Н), 2,25 (s, 6Н), 2,06 (m, 1Н), 1,00 (m, 2Н), 0,78 (m, 2Н);
МС m/z (ESI): 540,3 [М+Н]+.
Пример 23: получение N-(5-((4-(5-циклопропил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5-циклопропил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 22.
1Н ЯМР (400 МГц, CDCl3): δ 10,0 (brs, 1Н), 9,76 (s, 1Н), 8,99 (s, 1Н), 8,31 (d, J=5,2 Гц, 1Н), 7,72 (s, 1Н), 7,65 (s, 1Н), 7,21 (s, 1Н), 7,13 (d, J=5,2 Гц, 1Н), 6,94 (dd, J=8,4, 1,6 Гц, 1Н), 6,72 (s, 1Н), 6,36 (m, 2Н), 5,63 (dd, J=8,8, 2,8 Гц, 1Н), 3,89 (s, 3Н), 3,82 (s, 3Н), 2,84 (t, J=6,0 Гц, 2Н), 2,62 (s, 3Н), 2,25 (t, J=6,0 Гц, 2Н), 2,20 (s, 6Н), 2,00 (m, 1Н), 0,93 (m, 2Н), 0,75 (m, 2Н);
MS m/z (ESI): 540,4 [M+H]+.
Пример 24: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 22.
1Н ЯМР (400 МГц, CDCl3): δ 10,09 (s, 1Н), 9,76 (s, 1Н), 8,97 (s, 1Н), 8,30 (d, J=5,2 Гц, 1Н), 7,64 (s, 1Н), 7,44 (d, J=2,4 Гц, 1Н), 7,20 (d, J=9,2 Гц, 1Н), 7,05 (d, J=5,2 Гц, 1Н), 6,85 (m, 1Н), 6,72 (s, 1Н), 6,36 (m, 2Н), 5,63 (m, 1Н), 3,89 (s, 3Н), 3,85 (s, 3Н), 3,81 (s, 3Н), 2,81 (t, J=5,6 Гц, 2Н), 2,63 (s, 3Н), 2,20 (m, 8Н);
MS m/z (ESI): 530,2 [М+Н]+.
Пример 25: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 22.
1Н-ЯМР (400 МГц, CDCl3): δ 9,99 (d, J=29,8 Гц, 1Н), 9,75 (s, 1Н), 8,85 (s, 1Н), 8,29 (d, J=5,3 Гц, 1Н), 7,87 (d, J=8,8 Гц, 1Н), 7,64 (s, 1Н), 7,06 (d, J=5,3 Гц, 1Н), 6,83 (dd, J=8,7, 2,3 Гц, 1Н), 6,75 (t, J=8,0 Гц, 1Н), 6,70 (s, 1Н), 6,50-6,24 (m, 2Н), 5,76-5,53 (m, 1Н), 3,85 (s, 3Н), 3,82 (d, J=4,9 Гц, 3Н), 3,80 (s, 3Н), 2,94-2,74 (m, 2Н), 2,62 (s, 3Н), 2,24 (d, J=4,8 Гц, 2Н), 2,20 (s, 6Н);
MS m/z (ESI): 530,3 [М+Н]+.
Пример 26: получение N-(5-((4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил(метил)амино)-4-метоксифенил)акриламид был аналогичен Примеру 22.
1Н ЯМР (400 МГц, CDCl3): δ 9,78 (s, 1Н), 9,74 (s, 1Н), 8,55 (s, 1Н), 8,39 (d, J=5,3 Гц, 1Н), 8,11 (d, J=7,0 Гц, 1Н), 7,74-7,55 (m, 2Н), 7,18 (d, J=5,3 Гц, 1Н), 6,76 (s, 1Н), 6,62 (dd, J=16,8, 10,1 Гц, 1Н), 6,46 (dd, J=16,9, 1,9 Гц, 1Н), 6,24 (m, 1Н), 5,80-5,59 (m, 1Н), 3,88 (s, 3Н), 3,55-3,34 (m, 1Н), 3,02 (t, J=5,8 Гц, 2Н), 2,68 (s, 3Н), 2,57 (t, J=5,7 Гц, 2Н), 2,42 (s, 6Н), 1,24-1,17 (m, 2Н), 1,14-1,04 (m, 2Н);
MS m/z (ESI): 526,3 [М+Н]+.
Пример 27: Получение N-(4-метокси-5-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-(2-окса-6-азаспиро[3.3]гептан-6-ил)фенил)акриламида

Способ получения N-(4-метокси-5-((4-(1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-(2-окса-6-азаспиро[3.3]гептан-6-ил)фенил)акриламида был аналогичен Примеру 22.
1Н ЯМР (400 МГц, CDCl3): δ 8,76 (s, 1Н), 8,32 (d, J=26,6 Гц, 1Н), 8,20 (dd, J=20,7, 6,3 Гц, 1Н), 8,11-7,97 (m, 1Н), 7,48-7,36 (m, 1Н), 7,36-7,24 (m, 2Н), 6,97 (dd, J=25,1, 11,6 Гц, 1Н), 6,44-6,30 (m, 1Н), 6,24 (dd, J=16,9, 10,0 Гц, 1Н), 6,13-6,01 (m, 1Н), 5,66 (dd, J=9,9, 1,5 Гц, 1Н), 4,69 (s, 4Н), 3,95-3,67 (m, 10Н);
MS m/z (ESI): 497,2 [М+Н]+.
Пример 28: получение N-(5-((4-(5,6-дифтор-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино))этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5,6-дифтор-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино))этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 22.
1Н ЯМР (400 МГц, CDCl3): δ 10,06 (s, 1Н), 9,83-9,46 (m, 1Н), 8,86 (s, 1Н), 8,30 (d, J=5,3 Гц, 1Н), 7,78 (dd, J=11,4 Гц, 1Н), 7,62 (s, 1Н), 7,11-6,99 (m, 1Н), 6,93 (t, J=6,2 Гц, 1Н), 6,72 (s, 1Н), 6,34 (d, J=5,6 Гц, 2Н), 5,72-5,51 (m, 1Н), 3,84 (d, J=6,4 Гц, 3Н), 3,81 (d, J=4,4 Гц, 3Н), 2,92-2,76 (m, 2Н), 2,72-2,54 (m, 3Н), 2,21 (s, 6Н), 1,36-1,07 (m, 2Н);
MS m/z (ESI): 536,2 [М+Н]+.
Пример 29: получение N-(5-((4-(4,6-дифтор-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино))этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(4,6-дифтор-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино))этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 22.
1Н ЯМР (400 МГц, CDCl3): δ 9,97 (s, 1Н), 9,71 (s, 1Н), 9,00 (s, 1Н), 8,53-8,19 (m, 1Н), 7,71-7,56 (m, 1Н), 7,33 (d, J=5,4 Гц, 1Н), 6,79 (dd, J=8,8, 2,1 Гц, 1Н), 6,71 (s, 1Н), 6,65 (ddd, J=11,9, 9,7, 2,1 Гц, 1Н), 6,42-6,22 (m, 2Н), 5,68-5,53 (m, 1Н), 3,87 (s, 3Н), 3,81 (s, 3Н), 2,94-2,77 (m, 2Н), 2,62 (d, J=9,2 Гц, 3Н), 2,28 (s, 2Н), 2,23 (s, 6Н);
MS m/z (ESI): 536,2 [М+Н]+.
Пример 30: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4,5,6,7-тетрафтор-1-метил-1Н-индол-3-ил)пиримидин-2-ил) амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4,5,6,7-тетрафтор-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 22.
Пример 31: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1,5,6-триметил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1,5,6-триметил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 22.
Пример 32: получение N-(5-((4-(4,6-дифтор-1,7-диметил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(4,6-дифтор-1,7-диметил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 22.
Пример 33: получение N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(5-фтор-4,6-диметокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метоксифенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(5-фтор-4,6-диметокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 22.
Пример 34: получение N-(5-((4-(5,7-дифтор-6-(трифторметил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5,7-дифтор-6-(трифторметил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 22.
Пример 35: получение N-(5-((4-(4,6-дифтор-5-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино))этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(4,6-дифтор-5-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино))этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 22.
Пример 36: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4,5,6,7-тетрафтор-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4,5,6,7-тетрафтор-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 22.
Пример 37: получение N-(5-((4-(1-циклопропил-4,6-диметил-5-(метилсульфонил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(1-циклопропил-4,6-диметил-5-(метилсульфонил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 22.
Пример 38: получение N-(5-((4-(1,5-дициклопропил-4,6-дифтор-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(1,5-дициклопропил-4,6-дифтор-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 22.
Пример 39: получение N-(5-((4-(1-циклопропил-5,7-дифтор-6-(оксетан-3-ил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(1-циклопропил-5,7-дифтор-6-(оксетан-3-ил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 22.
Пример 40: получение N-(5-((4-(1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Стадия 1: получение 1-(2-хлорпиримидин-4-ил)-1Н-индазола

2,4-дихлорпиримидин (1,18 г, 8 ммоль) и DMF (40 мл) добавляли в 100-мл одногорлую колбу. NaH (0,4 г, 10 ммоль) добавляли к раствору частями и смесь перемешивали при комнатной температуре в течение получаса. После охлаждения до 0°С добавляли индазол (1,49 г, 12,6 ммоль). Реакционную смесь медленно нагревали до комнатной температуры при медленном помешивании и проводили реакцию в течение 4 часов. Реакцию гасили водой, экстрагировали этилацетатом и очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 20/1) с получением указанного в заголовке продукта 1-(2-хлорпиримидин-4-ил)-1Н-индазола (450 мг, 26%).
Стадия 2: получение N-(4-фтор-2-метокси-5-нитрофенил)-4-(1Н-индазол-1-ил)пиримидин-2-амина

1-(2-хлорпиримидин-4-ил)-1Н-индазол (450 мг, 1,96 ммоль), 4-фтор-2-метокси-5-нитроанилин (363 мг, 1,96 ммоль), п-толуолсульфокислоту (336 мг, 1,96 ммоль) и 2-пентанол (20 мл) последовательно добавляли в 50 мл одногорлую колбу. Смесь перемешивали при 120°С в течение 5 часов и концентрировали, с получением черной смеси, которую очищали колоночной хроматографией на силикагеле (1% MeOH/DCM) с получением указанного в заголовке соединения N-(4-фтор-2-метокси-5-нитрофенил)-4-(1Н-индазол-1-ил)пиримидин-2-амина (200 мг, 27%).
Стадия 3: получение N1-(4-(1Н-индазол-1-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамина

N-(4-фтор-2-метокси-5-нитрофенил)-4-(1Н-индазол-1-ил)пиримидин-2-амин (200 мг, 0,53 ммоль), триметилэтилендиамин (107 мг, 1,05 ммоль) DIPEA (203 мг, 1,57 ммоль) и DMF (8 мл) последовательно добавляли в 50 мл одногорлую колбу. Реакционный раствор перемешивали при 100°С в течение 1 часа, концентрировали и очищали препаративной тонкослойной хроматографией (5% MeOH/DCM) с получением указанного в заголовке соединения. N1-(4-(1Н-индазол-1-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамина (300 мг, 60%).
Стадия 4: получение N4-(4-(1Н-индазол-1-ил)пиримидин-2-ил)-N1-(2-(диметиламино)этил)-5-метокси-N1-метилбензол-1,2,4-триамина

N1-(4-(1Н-индазол-1-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамин (300 мг, 0,65 ммоль), 5% Pd/C (100 мг) и метанол (50 мл) последовательно добавляли в 50 мл одногорлую колбу. Смесь перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали и очищали тонкослойной хроматографией (5% MeOH/DCM) с получением указанного в заголовке соединения N4-(4-(1Н-индазол-1-ил)пиримидин-2-ил)-N1-(2-(диметиламино)этил)-5-метокси-N1-метилбензол-1,2,4-триамина (110 мг, 30%).
Стадия 5: получение N-(5-((4-(1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

N4-(4-(1Н-индазол-1-ил)пиримидин-2-ил)-N1-(2-(диметиламино)этил)-5-метокси-N1-метилбензол-1,2,4-триамин (110 мг, 0,25 ммоль), DIPEA (109 мг, 0,84 ммоль) и THF (30 мл) добавляли в 100 мл одногорлую колбу. После охлаждения до 0°С добавляли по каплям 0,5 мл акрилоилхлорида (1 M в THF) и реакционный раствор перемешивали при 0°С в течение 2 часов. Реакцию гасили метанолом, концентрировали и очищали тонкослойной хроматографией (10% MeOH/DCM) с получением указанного в заголовке соединения N-(5-((4-(1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида (20 мг, 16%).
1Н ЯМР (400 МГц, CD3OD) δ 8,52 (d, J=8,6 Гц, 1Н), 8,43 (s, 1Н), 8,29 (dd, J=5,6, 3,9 Гц, 1Н), 8,19 (d, J=3,5 Гц, 1Н), 7,70 (d, J=8,0 Гц, 1Н), 7,45-7,23 (m, 2Н), 7,17 (t, J=7,5 Гц, 1Н), 6,90 (s, 1Н), 6,42 (dd, J=17,0, 10,2 Гц, 1Н), 6,24 (d, J=16,9 Гц, 1Н), 5,68 (d, J=10,3 Гц, 1Н), 3,83 (s, 3Н), 3,16 (s, 2Н), 2,79-2,61 (m, 5Н), 2,66 (d, J=12,3 Гц, 3Н), 2,44 (s, 5Н);
MS m/z (ESI): 487 [M+H]+.
Пример 41: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 40.
1Н ЯМР (400 МГц, CD3OD) δ 8,46 (s, 1Н), 8,33 (d, J=7,3 Гц, 1Н), 8,05 (s, 1Н), 7,85 (s, 1Н), 7,63 (d, J=6,4 Гц, 1Н), 7,46 (t, J=8,0 Гц, 1Н), 7,08 (s, 1Н), 6,87 (d, J=7,9 Гц, 1Н), 6,59 (dd, J=16,9, 10,0 Гц, 1Н), 6,53-6,42 (m, 1Н), 5,87 (d, J=9,9 Гц, 1Н), 4,00 (s, 3Н), 3,96 (s, 3Н), 3,57 (t, J=5,4 Гц, 2Н), 3,40-3,35 (m, 2Н), 2,93 (s, 6Н), 2,81 (s, 3Н);
MS m/z (ESI): 517,3 [M+H]+.
Пример 42: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)фенил)-акриламида был аналогичен Примеру 40.
1Н ЯМР (400 МГц, CD3OD) δ 8,35 (s, 1Н), 8,23 (d, J=6,3 Гц, 1Н), 8,08 (s, 1Н), 7,87 (s, 1Н), 7,74 (d, J=8,7 Гц, 1Н), 7,61 (d, J=6,8 Гц, 1Н), 7,12-7,04 (m, 2Н), 6,63 (dd, J=16,9, 10,2 Гц, 1Н), 6,41 (dd, J=16,9, 1,2 Гц, 1Н), 5,87-5,79 (m, 1Н), 3,94 (s, 3Н), 3,86 (s, 3Н), 3,54 (t, J=5,6 Гц, 2Н), 3,37 (t, J=5,6 Гц, 2Н), 2,91 (s, 6Н), 2,79 (s, 3Н);
MS m/z (ESI): 517,3 [M+H]+.
Пример 43: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида

Стадия 1: получение 5-метокси-1-(2-(метилтио)пиримидин-4-ил)-1Н-индазола

5-метокси-1Н-индазол (500 мг, 3,38 ммоль) растворяли в DMF (10 мл). Добавляли NaH (148 мг, 3,72 ммоль) при 0°С и затем добавляли 4-хлор-2-(метилтио)пиримидин (542 мг, 3,38 ммоль). После перемешивания при этой температуре в течение 2 часов добавляли 30 мл воды. Реакционный раствор фильтровали, экстрагировали и сушили, с получением 5-метокси-1-(2-(метилтио)пиримидин-4-ил)-1Н-индазола (850 мг, 92%) в виде белого твердого вещества.
Стадия 2: получение 5-метокси-1-(2-(метилсульфонил)пиримидин-4-ил)-1Н-индазола

5-метокси-1-(2-(метилтио)пиримидин-4-ил)-1Н-индазол (850 мг, 3,125 ммоль) растворяли в DCM (50 мл), затем добавляли 3-хлорпероксибензойную кислоту (1,68 г, 7,8125 ммоль). Смесь перемешивали при 50°С в течение 3 часов, экстрагировали DCM и очищали колоночной хроматографией с получением 5-метокси-1-(2-(метилсульфонил)пиримидин-4-ил)-1Н-индазола (400 мг, 42%).
Стадия 3: получение N-(4-фтор-2-метокси-5-нитрофенил)-4-(5-метокси-1Н-индазол-1-ил)пиримидин-2-амина

5-метокси-1-(2-(метилсульфонил)пиримидин-4-ил)-1Н-индазол (100 мг, 0,329 ммоль), 4-фтор-2-метокси-5-нитроанилин (73 мг, 0,395 ммоль) и п-толуолсульфокислоту (57 мг, 0,329 ммоль) растворяли в 1,4-диоксане (5 мл). Реакционный раствор нагревали до температуры дефлегмации в течение ночи. После охлаждения добавляли соответствующее количество водного раствора бикарбоната натрия. Реакционный раствор фильтровали, экстрагировали и сушили, с получением твердого вещества серого цвета (200 мг), которое непосредственно использовали на следующей стадии.
Стадия 4: N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(5-метокси-1Н-индазол-1-ил)пиримидин-2-ил)-N1-метил-2-нитробензол-1,4-диамин

N-(4-фтор-2-метокси-5-нитрофенил)-4-(5-метокси-1Н-индазол-1-ил)пиримидин-2-амин (120 мг, 0,122 ммоль), DIPEA (47 мг, 0,658 моль), триметилэтилендиамин (37 мг, 0,366 ммоль) растворяли в DMF (5 мл). Реакционный раствор нагревали при 100°С в течение 1 часа, концентрировали и очищали колоночной хроматографией с получением 30 мг твердого вещества желтого цвета, которое использовали непосредственно на следующей стадии.
Стадия 5: получение N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(5-метокси-1Н-индазол-1-ил)пиримидин-2-ил)-N1-метилбензол-1,2,4-триамина

Способ получения N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(5-метокси-1Н-индазол-1-ил)пиримидин-2-ил)-N1-метилбензол-1,2,4-триамина был аналогичен Примеру 40.
Стадия 6: N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)фенил)-акриламид

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)фенил)-акриламида был аналогичен примеру 40.
1Н ЯМР (400 МГц, CD3OD) δ 8,38 (s, 2Н), 8,31 (d, J=6,6 Гц, 1Н), 7,86 (s, 1Н), 7,60 (d, J=6,7 Гц, 1Н), 7,33 (d, J=2,4 Гц, 1Н), 7,15 (dd, J=9,1, 2,2 Гц, 1Н), 7,08 (s, 1Н), 6,53 (dd, J=8,9, 5,9 Гц, 2Н), 5,88 (dd, J=9,2, 2,6 Гц, 1Н), 3,98 (s, 3Н), 3,89 (s, 3Н), 3,58 (t, J=5,7 Гц, 2Н), 3,38-3,34 (m, 2Н), 2,93 (s, 6Н), 2,81 (s, 3Н);
MS m/z (ESI): 517[M+H]+.
Пример 44: получение N-(5-((4-(3-циклопропил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)-акриламида

Способ получения N-(5-((4-(3-циклопропил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 40.
1Н ЯМР (400 МГц, CD3OD) δ 8,39 (s, 1Н), 8,18 (d, J=5,2 Гц, 1Н), 7,87 (d, J=7,3 Гц, 2Н), 7,48 (dd, J=17,0, 7,1 Гц, 2Н), 7,40 (t, J=7,4 Гц, 1Н), 7,09 (s, 1Н), 6,64 (dd, J=16,9, 10,2 Гц, 1Н), 6,45 (d, J=16,8 Гц, 1Н), 5,84 (d, J=10,2 Гц, 1Н), 3,94 (s, 3Н), 3,55 (d, J=5,2 Гц, 2Н), 3,38 (t, J=5,3 Гц, 2Н), 2,93 (s, 6Н), 2,81 (s, 3Н), 2,39-2,29 (m, 1Н), 1,19 (d, J=6,5 Гц, 4Н);
MS m/z (ESI): 527,3 [М+Н]+.
Пример 45: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-метокси-1Н-индазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(4-метокси-1Н-индазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 40.
Пример 46: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-1Н-индазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-1Н-индазол-1-ил)(трифторметил)пиримидин-2-ил)амино)фенил)-акриламида был аналогичен Примеру 40.
Пример 47: получение N-(5-((4-(5-циклопропил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)амино)-4-метоксифенил)акриламида

Стадия 1: получение 5-бром-1-((2-(триметилсилил)этокси)метил)-1Н-индазола

5-бром-1Н-индазол (2,0 г, 10 ммоль) растворяли в DMF (15 мл) и добавляли NaH (480 мг, 12 ммоль) при 0°С. Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 30 мин, а затем охлаждали до 0°С. После добавляли 2-(триметилсилил)этилгипохлорит (2,0 г, 12 ммоль), реакционный раствор перемешивали в течение 2 часов, гасили 30 мл воды и экстрагировали метил-трет-бутиловым эфиром (30 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта, который затем очищали колоночной хроматографией с получением 5-бром-1-((2-(триметилсилил)этокси)метил)-1Н-индазола (2,0 г, 63%).
Стадия 2: получение 5-циклопропил-1-((2-(триметилсилил)этокси)метил)-1Н-индазола

5-бром-1-((2-(триметилсилил)этокси)метил)-1Н-индазол (1,5 г, 4,6 ммоль), циклопропилбороновую кислоту (790 мг, 9,2 ммоль) и фосфат калия (3,0 г, 13,8 ммоль) растворяли в смеси толуола и воды (30/10 мл). После трехкратной продувки смеси азотом, добавляли ацетат палладия (103 мг, 0,46 ммоль) и трициклогексилфосфин (258 мг, 0,92 ммоль). Реакционную смесь перемешивали при 100°С в течение 16 часов, гасили 30 мл воды и экстрагировали этилацетатом (50 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта, который затем очищали колоночной хроматографией с получением 5-циклопропил-1-((2-(триметилсилил)этокси)метил)-1Н-индазола (1,2 г, 89%).
Стадия 3: получение 5-циклопропил-1Н-индазола

5-циклопропил-1-((2-(триметилсилил)этокси)-)метил)-1Н-индазол (1,2 г, 4,2 ммоль) растворяли в дихлорметане (30 мл). Добавляли трифторуксусную кислоту (12 мл) и проводили реакцию при комнатной температуре в течение 2,5 ч., и концентрировали досуха. Неочищенный продукт растворяли в смеси дихлорметана (50 мл) и этилендиамина (18 мл), и смесь перемешивали в течение 1 часа. После добавления 30 мл воды реакционный раствор экстрагировали дихлорметаном (50 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта, который затем очищали колоночной хроматографией с получением 5-циклопропил-1Н-индазола (280 мг, 42%).
Стадии 4-9: получение N-(5-((4-(5-циклопропил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5-циклопропил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)-акриламида был аналогичен Примеру 43.
1Н ЯМР (400 МГц, CD3OD) δ 8,26 (s, 2Н), 8,19 (s, 1Н), 7,78 (s, 1Н), 7,49 (d, J=6,7 Гц, 1Н), 7,44 (s, 1Н), 7,15 (d, J=8,6 Гц, 1Н), 6,98 (s, 1Н), 6,49 (dd, J=16,9, 9,9 Гц, 1Н), 6,38 (dd, J=16,9, 1,9 Гц, 1Н), 5,77 (dd, J=9,9, 1,9 Гц, 1Н), 3,86 (s, 3Н), 3,46 (t, J=5,6 Гц, 2H), 3,26 (t, J=5,6 Гц, 2H), 2,82 (s, 6H), 2,69 (s, 3Н), 2,00-1,88 (m, 1H), 0,97-0,89 (m, 2H), 0,67-0,59 (m, 2H);
MS m/z (ESI): 527,3 [M+H]+.
Пример 48: получение N-(5-((5-хлор-4-(5-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((5-хлор-4-(5-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 40.
1Н ЯМР (400 МГц, CD3OD) δ 8,49 (s, 1Н), 8,20 (d, J=10,0 Гц, 2Н), 8,00 (d, J=9,0 Гц, 1Н), 7,21 (s, 1Н), 7,03 (d, J=9,0 Гц, 1Н), 6,94 (s, 1Н), 6,42 (s, 2Н), 5,83 (s, 1Н) 3,95 (s, 3Н), 3,84 (s, 3Н), 3,47 (s, 2Н), 3,28 (s, 2Н), 2,86 (s, 6Н), 2,69 (s, 3Н);
MS m/z (ESI): 551 [М+Н]+.
Пример 49: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-1Н-индазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения
N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-1Н-индазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 40.
1Н ЯМР (400 МГц, CD3OD) δ 8,79 (s, 1Н), 8,23 (s, 1Н), 8,11-8,03 (m, 1Н), 7,27 (s, 1Н), 7,12-7,04 (m, 1Н), 7,01 (s, 1Н), 6,47 (s, 2Н), 5,92-5,80 (m, 1Н), 3,99 (s, 3Н), 3,88 (s, 3Н), 3,54 (s, 2Н), 3,32-3,29 (m, 2Н), 2,90 (s, 6Н), 2,76 (s, 3Н);
MS m/z (ESI): 585 [M+H]+.
Пример 50: получение N-(5-((4-(5-циано-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Стадия 1: получение 1Н-индазол-5-карбонитрила

4-фтор-3-формилбензонитрил (25 г, 16,78 ммоль) растворяли в 100 мл гидразин гидрата (85%). Смесь перемешивали при комнатной температуре в течение 24 часов и очищали колоночной хроматографией с получением 1Н-индазол-5-карбонитрила (2,1 г, 87%).
1Н ЯМР (400 МГц, ДМСО) δ 13,60 (s, 1Н), 8,42 (s, 1Н), 8,27 (s, 1Н), 7,73 (d, J=8,6 Гц, 1Н), 7,67 (d, J=8,6 Гц, 1Н);
MS m/z (ESI): 144 [М+Н]+.
Стадии 2-6: получение N-(5-((4-(5-циано-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5-циано-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 40.
1Н ЯМР (400 МГц, CD3OD) δ 8,77-8,68 (m, 1Н), 8,53 (s, 1Н), 8,47-8,41 (m, 1Н), 8,33 (s, 1Н), 7,99 (s, 1Н), 7,75 (s, 1Н), 7,56 (d, J=6,2 Гц, 1Н), 7,06 (s, 1Н), 6,55 (s, 1Н), 6,53 (s, 1Н), 5,92 (s, 1Н), 4,00 (s, 3Н), 3,56 (s, 2Н), 3,36 (s, 2Н), 2,94 (s, 6Н), 2,80 (s, 3Н);
MS m/z (ESI): 512 [М+Н]+.
Пример 51: получение N-(5-((5-хлор-4-(5-циано-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((5-хлор-4-(5-циано-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 50.
1Н ЯМР (400 МГц, CD3OD) δ 8,60 (s, 1Н), 8,46 (s, 1Н), 8,33 (s, 1Н), 8,25 (d, J=8,8 Гц, 1Н), 8,20 (s, 1Н), 7,63 (d, J=8,8 Гц, 1Н), 6,98 (s, 1Н), 6,59-6,37 (m, 2Н), 5,96-5,86 (m, 1Н), 4,00 (s, 3Н), 3,50 (t, J=5,7 Гц, 2Н), 3,32-3,28 (m, 2Н), 2,90 (s, 6Н), 2,72 (s, 3Н);
MS m/z (ESI): 546 [M+H]+.
Пример 52: получение N-(5-((4-(5-циано-1Н-индазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино))этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5-циано-1Н-индазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино))этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 50.
1Н ЯМР (400 МГц, CD3OD) δ 8,88 (s, 1Н), 8,48 (s, 1Н), 8,35 (s, 1Н), 8,10 (s, 1Н), 7,75-7,60 (m, 1Н), 7,02 (s, 1Н), 6,51 (s, 2Н), 5,91 (d, J=11,7 Гц, 1Н), 4,00 (s, 3Н), 3,54 (s, 2Н), 3,32 (s, 2Н), 2,91 (s, 6Н), 2,75 (s, 3Н);
MS m/z (ESI): 580 [М+Н]+.
Пример 53. получение N-(5-((4-(5,6-диметокси-3-метил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино))этил)(метил)амино)-4-метоксифенил)акриламида

Стадия 1: получение 1-(4,5-диметокси-2-нитрофенил)этан-1-она

К уксусному ангидриду (30 мл) добавляли 1-(3,4-диметоксифенил)этан-1-он (10 г). После охлаждения раствора до 0°С добавляли по каплям смесь азотной кислоты (200 мл) и уксусного ангидрида (10 мл). По завершении добавления, реакционный раствор перемешивали в течение 4 часов, выливали в 1 л ледяной воды, фильтровали, промывали водой и сушили с получением 1-(4,5-диметокси-2-нитрофенил)этан-1-она (8 г, 67%).
Стадия 2: получение 1-(4,5-диметокси-2-аминофенил)этан-1-она

1-(4,5-диметокси-2-нитрофенил)этан-1-он (8 г) и порошок железа (20 г) добавляли к смеси НОАс (70 мл), воды (100 мл) и EtOAc (20 мл). Реакцию проводили при 100°С в течение 2 часов и доводили рН до 7 водным раствором бикарбоната натрия. В реакционный раствор добавляли 400 мл этилацетата, фильтровали, концентрировали. Полученный остаток перекристаллизовывали из смеси этилацетат-петролейного эфира, с получением 1-(4,5-диметокси-2-аминофенил)этан-1-она (1,37 г, 30%).
Стадия 3: получение оксима 1-(2-аминодиметоксифенил)этан-1-она

К 6 мл этанола добавляли 1-(4,5-диметокси-2-аминофенил)этан-1-он (800 мг, 4,1 ммоль), гидрохлорид гидроксиламина (880 мг, 12,3 ммоль) и NaOH (1,31 г, 32,8 ммоль) (85%). Реакционный раствор нагревали при 60°С в течение 1 часа, концентрировали, экстрагировали этилацетатом и перекристаллизовывали из смеси этилацетат-петролейного эфира с получением оксим 1-(2-аминодиметоксифенил)этан-1-она (500 мг, 58%).
Стадия 4: получение 5,6-диметокси-3-метил-1Н-индазола

1-(2-амино-диметоксифенил)этан-1-он оксим (450 мг, 2,14 ммоль) и триэтиламин (432 мг, 4,28 ммоль) добавляли в DCM (15 мл), и по каплям добавляли 0,2 мл метансульфонилхлорида при 0°С. Смесь перемешивали при комнатной температуре в течение одного часа, концентрировали и очищали колоночной хроматографией с получением 5,6-диметокси-3-метил-1Н-индазола (200 мг, 37%).
Стадии 5-10: получение N-(5-((4-(5,6-диметокси-3-метил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5,6-диметокси-3-метил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 40.
1Н ЯМР (400 МГц, CD3OD) δ 8,00 (s, 1Н), 7,91-7,80 (m, 1Н), 7,75 (s, 1Н), 7,37 (d, J=7,0 Гц, 1Н), 6,98 (d, J=12,7 Гц, 2Н), 6,53 (s, 1Н), 6,32 (d, J=16,7 Гц, 1Н), 5,74 (s, 1Н), 3,83 (s, 3Н), 3,78 (s, 3Н), 3,77-3,67 (m, 3Н), 3,42 (s, 2Н), 3,27 (s, 2Н), 2,82 (s, 6Н), 2,67 (s, 2Н), 2,41 (s, 3Н);
MS m/z (ESI): 561[М+1]+.
Пример 54. получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-3-метил-1Н-индазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида

Стадия 1: получение 6-метокси-3-метил-1Н-индазола

1-(2-фтор-4-метоксифенил)этан-1-он (2 г, 11,9 ммоль) растворяли в 5 мл смеси гидразингидрата (85%) и NMP (15 мл). Смесь перемешивали при 120°С в течение 24 ч и очищали колоночной хроматографией с получением 6-метокси-3-метил-1Н-индазола (1,5 г, 78%).
Стадии 2-7: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-3-метил-1Н-индазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-3-метил-1Н-индазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 47.
1Н ЯМР (400 МГц, CD3OD) δ 8,25-8,13 (m, 1Н), 8,11-8,00 (m, 1Н), 7,81 (s, 1Н), 7,70 (d, J=8,7 Гц, 1Н), 7,59 (d, J=7,0 Гц, 1Н), 7,09 (s, 2Н), 6,69-6,56 (m, 1Н), 6,45 (s, 1Н), 5,86 (s, 1Н), 3,95 (s, 3Н), 3,87 (s, 3Н), 3,56 (s, 2Н), 3,38 (s, 2Н), 2,92 (s, 6Н), 2,81 (s, 3Н), 2,58 (s, 3Н);
MS m/z (ESI): 531 [М+1]+.
Пример 55: получение N-(5-((4-(6-циано-3-метил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(6-циано-3-метил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 40.
Пример 56: получение N-(5-((4-(5-циано-3-метил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5-циано-3-метил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 40.
1Н ЯМР (400 МГц, CD3OD) δ 8,67-8,55 (m, 1Н), 8,36 (d, J=6,4 Гц, 1Н), 8,28 (s, 1Н), 7,94 (s, 1Н), 7,73 (d, J=8,7 Гц, 1Н), 7,51 (d, J=6,5 Гц, 1Н), 7,08 (s, 1Н), 6,55 (dd, J=7,9, 5,9 Гц, 2Н), 5,91 (d, J=11,8 Гц, 1Н), 4,00 (s, 3Н), 3,57 (s, 2Н), 3,38 (d, J=5,9 Гц, 2Н), 2,95 (s, 6Н), 2,80 (s, 3Н), 2,64 (s, 3Н);
MS m/z (ESI): 526 [М+Н]+.
Пример 57. получение N-(5-((4-(5,6-дифтор-3-метил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино))этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5,6-дифтор-3-метил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино))этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 40.
Пример 58: получение N-(5-((4-(5,7-дифтор-3-метил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5,7-дифтор-3-метил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 40.
Пример 59: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида

Стадия 1: получение 2-хлор-N-(4-метокси-2-нитрофенил)пиримидин-4-амина

2-хлорпиримидин-4-амин (518,2 мг, 4 ммоль) и DMF (10 мл) добавляли в 100 мл круглодонную колбу. Под защитой N2 смесь охлаждали до 0°С на бане со льдом и солью, и затем добавляли NaH (295 мг, 8 ммоль). После перемешивания в течение 30 минут добавляли 1-фтор-4-метокси-2-нитробензол (684,5 мг, 4 ммоль) и реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 1 часа. На бане со льдом и солью добавляли 30 мл воды и осаждали твердое вещество. Твердое вещество фильтровали и осадок на фильтре растворяли в дихлорметане. Раствор сушили над безводным сульфатом натрия и концентрировали, с получением 2-хлор-N-(4-метокси-2-нитрофенил)пиримидин-4-амина (1 г, 89%).
MS m/z (ESI): 281,0 [М+Н]+.
Стадия 2: получение N1-(2-хлорпиримидин-4-ил)-4-метоксибензол-1,2-диамина

2-хлор-N-(4-метокси-2-нитрофенил)пиримидин-4-амин, этанол (15 мл) и воду (5 мл) добавляли в 100 мл круглодонную колбу с последующим добавлением порошка железа (1,37 г, 24,5 ммоль) и хлорида аммония (131,5 мг, 2,5 ммоль). Реакцию проводили при 80°С в течение 3 ч, перед тем как смесь фильтровали и концентрировали. Полученный остаток растворяли в этилацетате (50 мл) и добавляли 30 мл воды, затем разделяли две фазы. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали, с получением N1-(2-хлорпиримидин-4-ил)-4-метоксибензол-1,2-диамина (676,9 мг, 77%).
MS m/z (ESI): 251,1 [М+Н]+.
Стадия 3: получение 1-(2-хлорпиримидин-4-ил)-5-метокси-1Н-бензо[d]имидазола

N1-(2-хлорпиримидин-4-ил)-4-метоксибензол-1,2-диамин (300 мг, 1,2 ммоль), этанол (10 мл) и триметилортоформиат (1,0 г, 9,6 ммоль) и п-толуолсульфокислоту (20 мг, 0,12 ммоль) добавляли в 100 мл круглодонную колбу и проводили реакцию при 80°С в течение 1 часа. После охлаждения до комнатной температуры реакционный раствор концентрировали и полученный остаток подвергали колоночной хроматографии, с получением 1-(2-хлорпиримидин-4-ил)-5-метокси-1Н-бензо[d]имидазола (195 мг, 62%).
MS m/z (ESI): 261,1 [М+Н]+.
Стадия 4: получение N-(4-фтор-2-метокси-5-нитрофенил)-4-(5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-амина

1-(2-хлорпиримидин-4-ил)-5-метокси-1Н-бензо[d]имидазол (195 мг, 0,75 ммоль), 4-фтор-2-метокси-5-нитроанилин (140 мг, 0,75 ммоль), п-толуолсульфокислоту (129 мг, 0,75 ммоль) и 2-пентанол (5 мл) добавляли в 100 мл круглодонную колбу и реакцию проводили при 100°С в течение 4 часов. После охлаждения до комнатной температуры реакционный раствор концентрировали и остаток подвергали колоночной хроматографии с получением N-(4-фтор-2-метокси-5-нитрофенил)-4-(5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-амина (62 мг, 20%).
MS m/z (ESI): 411,1 [М+Н]+.
Стадия 5: получение N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)-N1-метил-2-нитробензол-1,4-диамина

N-(4-фтор-2-метокси-5-нитрофенил)-4-(5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-амин (62 мг, 0,15 ммоль) N1,N1,N2,N2-тетраметилэтан-1,2-диамин (30,6 мг, 0,3 ммоль), DIPEA (58 мг, 0,45 ммоль) и DMF (5 мл) добавляли в 100 мл колбу с круглым дном, Проводили реакцию при 80°С в течение 1 часа. После охлаждения до комнатной температуры реакционный раствор концентрировали. Полученный остаток подвергали колоночной хроматографии с получением N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)-N1-метил-2-нитробензол-1,4-диамина (40 мг, 54%).
MS m/z (ESI): 493,3 [М+Н]+.
Стадия 6: получение N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)-N1-метилбензол-1,2,4-триамина

N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)-N1-метил-2-нитробензол-1,4-диамин (39,4 мг, 0,08 ммоль) и метанол (20 мл) добавляли в 100 мл круглодонную колбу. В атмосфере водорода реакционный раствор подвергали реакции при комнатной температуре в течение 30 минут. Затем реакционный раствор фильтровали и концентрировали с получением N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)-N1-метилбензол-1,2,4-триамина (32 мг, 85%).
MS m/z (ESI): 463,1 [М+Н]+.
Стадия 7: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил-амино)фенил)акриламида

N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)-N1-метилбензол-1,2,4-триамин (32 мг, 0,07 ммоль) и безводный тетрагидрофуран (20 мл) добавляли в 100 мл круглодонную колбу. Под защитой N2 реакционный раствор охлаждали до 0°С на бане со льдом и солью с последующим добавлением DIPEA (18 мг, 0,14 ммоль) и акрилоилхлорида (0,2 мл, 0,1 ммоль). Реакцию проводили при 0°С в течение 30 минут и прекращали добавлением 0,5 мл воды, а затем реакционный раствор концентрировали. Полученный остаток подвергали колоночной хроматографии с получением N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида (10 мг, 28%).
1Н ЯМР (400 МГц, CD3OD) δ 9,36 (s, 1Н), 8,57 (d, J=5,4 Гц, 1Н), 8,26 (s, 2Н), 7,39-7,24 (m, 2Н), 7,12-6,97 (m, 2Н), 6,52 (qd, J=17,0, 5,8 Гц, 2Н), 5,86 (dd, J=9,7, 1,8 Гц, 1Н), 3,99 (s, 3Н), 3,89 (s, 3Н), 3,51 (d, J=5,5 Гц, 2Н), 3,33 (s, 2Н), 2,90 (d, J=5,1 Гц, 6Н), 2,75 (s, 3Н);
MS m/z (ESI): 517, 2 [М+Н]+.
Пример 60: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 59.
1Н ЯМР (400 МГц, CD3OD) δ 9,21 (s, 1Н), 8,58 (d, J=5,6 Гц, 1Н), 8,34 (s, 1Н), 7,86 (s, 1Н), 7,69 (d, J=8,9 Гц, 1Н), 7,30 (d, J=5,6 Гц, 1Н), 7,11 (dd, J=8,7, 2,1 Гц, 1Н), 7,00 (s, 1Н), 6,57 (dd, J=16,9, 10,0 Гц, 1Н), 6,44 (dd, J=16,9, 1,6 Гц, 1Н), 5,84 (dd, J=10,1, 1,6 Гц, 1Н), 3,98 (s, 3Н), 3,86 (s, 3Н), 3,50 (t, J=5,7 Гц, 2Н), 3,32 (s, 2Н), 2,89 (s, 6Н), 2,74 (s, 3Н);
MS m/z (ESI): 517,3 [M+H]+.
Пример 61: получение N-(5-((4-(5-циано-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5-циано-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
Пример 62: получение N-(5-((4-(6-циано-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(6-циано-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
1Н ЯМР (400 МГц, CD3OD) δ 9,19 (s, 1Н), 8,78 (s, 1Н), 8,52 (d, J=5,7 Гц, 1Н), 8,24 (s, 1Н), 7,86 (d, J=8,3 Гц, 1Н), 7,66 (dd, J=8,3, 1,2 Гц, 1Н), 7,31 (d, J=5,8 Гц, 1Н), 7,07 (s, 1Н), 6,59 (dd, J=16,9, 10,2 Гц, 1Н), 6,35 (d, J=16,8 Гц, 1Н), 5,81 (dd, J=10,3, 1,2 Гц, 1Н), 3,97 (s, 3Н), 3,53 (t, J=5,8 Гц, 2Н), 3,36 (t, J=5,8 Гц, 2Н), 2,92 (s, 6Н), 2,77 (s, 3Н);
MS m/z (ESI): 512,2 [М+Н]+.
Пример 63: получение N-(5-((5-хлор-4-(5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((5-хлор-4-(5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
1Н ЯМР (400 МГц, CD3OD) δ 9,25 (s, 1Н), 8,74 (s, 1Н), 8,25 (s, 1Н), 7,85 (d, J=8,2 Гц, 1Н), 7,33 (s, 1Н), 7,08 (d, J=9,0 Гц, 1Н), 6,98 (s, 1Н), 6,47-6,41 (m, 2Н), 5,85 (dd, J=7,8, 3,9 Гц, 1Н), 4,00 (s, 3Н), 3,91 (s, 3Н), 3,49 (t, J=5,7 Гц, 2Н), 3,28 (t, J=5,6 Гц, 2Н), 2,86 (s, 6Н), 2,71 (s, 3Н);
MS m/z (ESI): 551,2 [М+Н]+.
Пример 64: получение N-(5-((5-хлор-4-(6-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((5-хлор-4-(6-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
Пример 65: получение N-(5-((5-хлор-4-(5-циано-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((5-хлор-4-(5-циано-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид был аналогичен Примеру 59.
Пример 66: получение N-(5-((5-хлор-4-(6-циано-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((5-хлор-4-(6-циано-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
Пример 67: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-1Н-бензо[d]имидазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-1Н-бензо[d]имидазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламидбыл аналогичен Примеру 59.
1Н ЯМР (400 МГц, CD3OD) δ 8,98 (s, 1Н), 8,87 (s, 1Н), 8,15 (s, 1Н), 7,73 (d, J=8,7 Гц, 1Н), 7,32 (s, 1Н), 7,10 (s, 1Н), 6,98 (s, 1Н), 6,43 (s, 2Н), 5,84 (d, J=11,1 Гц, 1Н), 3,98 (s, 3Н), 3,91 (s, 3Н), 3,49 (t, J=5,3 Гц, 2Н), 3,29 (d, J=5,4 Гц, 2Н), 2,85 (s, 6Н), 2,71 (s, 3Н);
MS m/z (ESI): 585,3 [М+Н]+.
Пример 68: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-1Н-бензо[d]имидазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-1Н-бензо[d]имидазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 59.
Пример 69: получение N-(5-((4-(5-циано-1Н-бензо[d]имидазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5-циано-1Н-бензо[d]имидазол-1-ил)-5- (трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
Пример 70: получение N-(5-((4-(6-циано-1Н-бензо[d]имидазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(6-циано-1Н-бензо[d]имидазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
Пример 71: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-2-метил-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-2-метил-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 59.
Пример 72: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-2-метил-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-2-метил-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 59.
Пример 73: получение N-(5-((4-(5-циано-2-метил-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5-циано-2-метил-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
Пример 74. Получение N-(5-((4-(6-циано-2-метил-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(6-циано-2-метил-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
Пример 75: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-2-метил-1Н-бензо[d]имидазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-2-метил-1Н-бензо[d]имидазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 59.
Пример 76: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(2-метил-5-(трифторметил)-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(2-метил-5-(трифторметил)-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 59.
Пример 77: получение N-(5-((4-(6-циано-2-метил-1Н-бензо[d]имидазол-1-ил)-5-фторпиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(6-циано-2-метил-1Н-бензо[d]имидазол-1-ил)-5-фторпиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
Пример 78: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(2-метил-5-(трифторметокси)-1Н-бензо[d]имидазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(2-метил-5-(трифторметокси)-1Н-бензо[d]имидазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 59.
Пример 79: получение N-(5-((4-(5-циклопропил-2-метил-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5-циклопропил-2-метил-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
Пример 80: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(2-метил-6-(трифторметил)-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(2-метил-6-(трифторметил)-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 59.
Пример 81: получение N-(5-((4-(2-циклопропил-5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)-амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(2-циклопропил-5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
Пример 82: получение N-(5-((4-(2-циклопропил-5-метокси-1Н-бензо[d]имидазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(2-циклопропил-5-метокси-1Н-бензо[d]имидазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
Пример 83: получение N-(5-((4-(5-циано-2-циклопропил-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5-циано-2-циклопропил-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид был аналогичен Примеру 59.
Пример 84: получение N-(5-((5-хлор-4-(5-циано-2-циклопропил-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((5-хлор-4-(5-циано-2-циклопропил-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
Пример 85: получение N-(5-((4-(2-циклопропил-5-(трифторметил)-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)-амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(2-циклопропил-5-(трифторметил)-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
Пример 86: получение N-(5-((4-(2-циклопропил-6-(трифторметил)-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(2-циклопропил-6-(трифторметил)-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)-амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
Пример 87: получение N-(5-((5-хлор-4-(2-циклопропил-5-(трифторметил)-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)-амино)-4-метоксифенил)акриламида

Способ получения N-(5-((5-хлор-4-(2-циклопропил-5-(трифторметил)-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)-(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
Пример 88: получение N-(5-((4-(2-циклопропил-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(2-циклопропил-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
1Н ЯМР (400 МГц, CDCl3): δ 10,3 (br s, 1Н), 8,75 (s, 1Н), 8,46 (d, J=6,0 Гц, 1Н), 8,00 (dd, J=4,8, 1,6 Гц, 1Н), 7,64 (dd, J=6,0, 1,6 Гц, 1Н), 7,21 (m, 3Н), 6,96 (d, J=6,4 Гц, 1Н), 6,82 (s, 1Н), 6,45 (m, 2Н), 5,73 (d, J=9,2 Гц, 1Н), 3,84 (s, 3Н), 2,99 (m, 1Н), 2,89 (m, 2Н), 2,73 (s, 3Н), 2,36 (m, 2Н), 2,31 (s, 6Н), 1,04 (m, 2Н), 0,87 (m, 2Н);
MS m/z (ESI): 527,2 [М+Н]+.
Пример 89: получение N-(5-((4-(5,7-дифтор-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино))этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5,7-дифтор-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино))этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 22.
1Н ЯМР (400 МГц, CDCl3): δ 10,2 (brs, 1Н), 9,79 (s, 1Н), 9,14 (s, 1Н), 8,37 (d, J=5,2 Гц, 1Н), 7,72 (s, 1Н), 7,40 (d, J=5,2 Гц, 1Н), 6,87 (dd, J=8,8, 1,6 Гц, 1Н), 6,79 (s, 1Н), 6,74 (m, 1Н), 6,41 (m, 2Н), 5,70 (m, 1Н), 3,95 (s, 3Н), 3,88 (s, 3Н), 2,91 (m, 2Н), 2,57 (s, 3Н), 2,22 (m, 8Н);
MS m/z (ESI): 527,3 [М+Н]+.
Пример 90: получение N-(5-((4-(4,5-дифтор-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино))этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(4,5-дифтор-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино))этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 22.
1Н ЯМР (400 МГц, CD3OD): δ 9,20 (s, 1Н), 8,36 (s, 1Н), 8,18 (d, J=5,6 Гц, 1Н), 7,99 (m, 1Н), 7,26 (m, 1Н), 7,04 (d, J=5,6 Гц, 1Н), 6,97 (s, 1Н), 6,56 (m, 1Н), 6,29 (dd, J=17,2 Гц, 2,0 Гц, 1Н), 5,74 (dd, J=10,4 Гц, 1,6 Гц, 1Н), 3,91 (s, 3Н), 3,80 (s, 3Н), 3,06 (t, J=6,4 Гц, 2Н), 2,70 (s, 3Н), 2,46 (t, J=6,0 Гц, 1Н), 2,31 (m, 6Н);
MS m/z (ESI): 536,2 [М+Н]+.
Пример 91: получение N-(5-((4-(7-циклопропил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(7-циклопропил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 22.
1Н ЯМР (400 МГц, CDCl3): δ 10,08 (s, 1Н), 9,83 (s, 1Н), 8,96 (s, 1Н), 8,36 (d, J=5,2 Гц, 1Н), 7,91 (d, J=8,0 Гц, 1Н), 7,73 (s, 1Н), 7,17 (d, J=5,2 Гц, 1Н), 7,10 (t, J=7,6 Гц, 1Н), 6,99 (d, J=7,2 Гц, 1Н), 6,79 (s, 1Н), 6,44 (m, 2Н), 5,71 (m, 1Н), 4,45 (s, 3Н), 3,88 (s, 3Н), 2,91 (t, J=5,6 Гц, 2Н), 2,71 (s, 3Н), 2,46 (m, 1Н), 2,28 (m, 8Н), 1,01 (m, 2Н), 0,90 (m, 2Н);
MS m/z (ESI): 540, 2 [М+Н]+.
Пример 92: N-(5-((4-(7-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(7-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 22.
1Н ЯМР (400 МГц, CDCl3) δ 10,27 (s, 1Н), 10,13 (s, 1Н), 9,83 (s, 1Н), 9,04 (s, 1Н), 8,36 (d, J=5,3 Гц, 1Н), 7,89 (d, J=8,0 Гц, 1Н), 7,72 (s, 1Н), 7,16 (d, J=5,3 Гц, 1Н), 7,09 (t, J=7,7 Гц, 1Н), 6,79 (d, J=5,6 Гц, 2Н), 6,71-6,50 (m, 1Н), 6,39 (s, 1Н), 5,82-5,58 (m, 1Н), 3,88 (s, 3Н), 3,08-2,83 (m, 2Н), 2,70 (s, 3Н), 2,39-2,20 (m, 8Н), 2,16 (t, J=5,1 Гц, 1Н), 0,96-0,73 (m, 2Н), 0,73-0,55 (m, 2Н);
MS m/z (ESI): 526, 7 [М+Н]+.
Пример 93: Получение N-(5-((4-(1-циклопропил-6-метокси-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(1-циклопропил-6-метокси-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 22.
Соль TFA N-(5-((4-(1-циклопропил-6-метокси-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида.
1Н ЯМР (400 МГц, CD3OD): δ 8,41 (s, 1Н), 8,15 (br, 1Н), 7,98 (d, J=6,8 Гц, 1Н), 7,89 (s, 1Н), 7,40 (d, J=6,8 Гц, 1Н), 7,17 (d, J=2,4 Гц, 1Н), 7,06 (s, 1Н), 6,87 (m, 1Н), 6,50 (m, 2Н), 5,87 (m, 1Н), 3,95 (s, 3Н), 3,88 (s, 3Н), 3,55 (m, 3Н), 3,35 (m, 2Н), 2,92 (s, 6Н), 2,80 (s, 3Н), 1,22 (m, 2Н), 0,90 (m, 2Н);
MS m/z (ESI): 556, 2 [М+Н]+.
Пример 94: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(7-метокси-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)-акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(7-метокси-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)-акриламида был аналогичен Примеру 22.
1Н ЯМР (400 МГц, CD3OD): δ 8,40 (s, 1Н), 8,03 (d, J=6,8 Гц, 1Н), 7,46 (d, J=6,8 Гц, 1Н), 7,12 (m, 1Н), 7,09 (s, 1Н), 6,77 (d, J=8,0 Гц, 1Н), 6,51 (m, 2Н), 5,87 (m, 1Н), 3,98 (s, 3Н), 3,87 (s, 3Н), 3,57 (m, 2Н), 3,36 (m, 2Н), 2,92 (m, 6Н), 2,80 (s, 3Н);
MS m/z (ESI): 516,2 [М+Н]+.
Пример 95: получение N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(6-этинил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метоксифенил)акриламида

Стадия 1: получение 6-йод-1Н-индола

NaI (4,59, 30,6 ммоль), CuI (290 мг, 1,53 ммоль) и N,N'-диметилэтилендиамин (0,35 мл) добавляли к раствору 6-бром-1Н-индола (3,00 г, 15,3 ммоль) в диоксане (30 мл) при комнатной температуре. Смесь продували азотом для удаления кислорода в течение 5 минут. Смесь перемешивали при 110°С на масляной бане в течение ночи в атмосфере азота. После охлаждения, органический растворитель удаляли концентрированием при пониженном давлении. Добавляли EtOAc и воду и разделяли две фазы. Фазу EtOAc промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией (элюент: чистый петролейный эфир) с получением указанного в заголовке соединения 6-йод-1Н-индола (2,1 г, 57%).
MS m/z (ESI): 244,0[М+Н]+.
Стадия 2: получение 6-йод-1-метил-1Н-индола

На бане с ледяной водой, NaH (60%, 734 мг, 18,4 ммоль) добавляли к раствору 6-йод-1Н-индола (2,00 г, 8,23 ммоль) в DMF (30 мл). Смесь перемешивали в течение 20 минут при этой температуре с последующим добавлением раствора MeI (1,14 мл, 18,4 ммоль) в DMF (10 мл) и дополнительно перемешивали при этой температуре в течение 30 минут. Добавляли приблизительно 100 мл воды и реакционный раствор экстрагировали EtOAc. Фазу EtOAc промывали несколько раз насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией (элюент: чистый петролейный эфир) с получением указанного в заголовке соединения 6-йод-1-метил-1Н-индола (1,98 г, 94%).
MS m/z (ESI): 258,1 [М+Н]+.
Стадия 3: получение 3-(2-хлорпиримидин-4-ил)-6-йод-1-метил-1Н-индола

FeCl3 (441 мг, 2,72 ммоль) добавляли к раствору 6-йод-1-метил-1Н-индола (700 мг, 2,72 ммоль) и 2,4-дихлорпиримидина (405 мг, 2,72 ммоль) в диметиловом эфире этиленгликоля (10 мл). Смесь перемешивали при 60°С в течение ночи. После охлаждения добавляли большое количество EtOAc и воду, и разделяли две фазы. Нерастворенное вещество удаляли через целит, и водную фазу удаляли. Органическую фазу промывали последовательно насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией (элюент : EtOAc = 3: 1) с получением указанного в заголовке соединения 3-(2-хлорпиримидин-4-ил)-6-йод-1-метил-1Н-индола (533 мг, 53%).
MS m/z (ESI): 370,59[М+Н]+.
Стадия 4: получение N-(4-фтор-2-метокси-5-нитрофенил)-4-(6-йод-1-метил-1Н-индол-3-ил)пиримидин-2-амина

3-(2-хлорпиримидин-4-ил)-6-йод-1-метил-1Н-индол (283 мг, 0,765 ммоль), 4-фтор-2-метокси-5-нитроанилин (142 мг, 0,765 ммоль) и TsOH.Н2О (175 мг, 0,918 ммоль) смешивали в 2-пентаноле (10 мл) и реакцию проводили при 125°С в течение 3 часов. После охлаждения смеси и фильтрования, полученное твердое вещество растворяли в CH2Cl2. Затем раствор промывали последовательно насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали с получением указанного в заголовке соединения N-(4-фтор-2-метокси-5-нитрофенил)-4-(6-йод-1-метил-1Н-индол-3-ил)пиримидин-2-амина (350 мг, 88%).
MS m/z (ESI): 520,2[М+Н]+.
Стадия 5: получение N1-(2-(диметиламино)этил)-N4-(4-(6-йод-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-5-метокси-N1-метил-2-нитробензол-1,4-диамина

N-(4-фтор-2-метокси-5-нитрофенил)-4-(6-йод-1-метил-1Н-индол-3-ил)пиримидин-2-амин (100 мг, 0,192 ммоль), N1,N1,N2-триметилэтан-1,2-диамин (39 мг, 0,385 ммоль) и DIPEA (42 мг, 0,385 ммоль) растворяли в DMA (10 мл) и реакцию проводили при 85°С в течение 3 часов. После охлаждения добавляли EtOAc и воду и разделяли две фазы. Органическую фазу несколько раз промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали с получением 105 мг неочищенного указанного в заголовке соединения N1-(2-(диметиламино)этил)-N4-(4-(6-йод-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-5-метокси-N1-метил-2-нитробензол-1,4-диамин, который непосредственно использовали для следующей стадии.
MS m/z (ESI): 602,4[М+Н]+.
Стадия 6: получение N1-(2-(диметиламино)этил)-5-метокси-N1-метил-N4-(4-(1-метил-6-((триметилсилил)этинил)-1Н-индол-3-ил)пиримидин-2-ил)-2-нитробензол-1,4-диамина

N1-(2-(диметиламино)этил)-N4-(4-(6-йод-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-5-метокси-N1-метил-2-нитробензол-1,4-диамин (100 мг, 0,166 ммоль), (триметилсилил)ацетилен (48 мг, 0,498 ммоль) и триэтиламин (51 мг, 0,498 ммоль) смешивали в смеси THF (10 мл) и DMF (5 мл) с последующим добавлением CuI (16 мг, 0,083 ммоль) и тетракис(трифенилфосфин)палладия (40 мг, 0,041 ммоль). После трехкратной продувки азотом, реакционный раствор нагревали до 70°С в течение ночи на масляной бане. Растворитель удаляли при пониженном давлении и водную фазу экстрагировали EtOAc. Фазу EtOAc промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией [элюент : CH2Cl2 → CH2Cl2 : МеОН = 20:1] с получением указанного в заголовке соединения N1-(2-(диметиламино)этил)-5-метокси-N1-метил-N4-(4-(1-метил-6-((триметилсилил)этинил)-1Н-индол-3-ил)пиримидин-2-ил)-2-нитробензол-1,4-диамина (65 мг, 68%).
MS m/z (ESI): 572,7 [М+Н]+.
Стадия 7: получение N1-(2-(диметиламино)этил)-5-метокси-N1-метил-N4-(4-(1-метил-6-((триметилсилил)этинил)-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,2,4-триамина

N1-(2-(диметиламино)этил)-5-метокси-N1-метил-N4-(4-(1-метил-6-((триметилсилил)этинил)-1Н-индол-3-ил)пиримидин-2-ил)-2-нитробензол-1,4-диамин (200 мг, 0,35 ммоль), восстановленный порошок железа (136 мг, 2,45 ммоль) и хлорид аммония (20,6 мг, 0,386 ммоль) смешивали в смеси EtOH (30 мл) и воды (10 мл) и смесь до температуры дефлегмации в течение трех часов. После охлаждения добавляли большое количество EtOH и нерастворенное вещество удаляли фильтрованием через целит. EtOH удаляли при пониженном давлении и водную фазу экстрагировали EtOAc. Фазу EtOAc промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией (элюент : CH2Cl2 → CH2Cl2 : MeOH (содержащий 10% концентрированного аммиака) = 17:1] с получением указанного в заголовке соединения N1-(2-(диметиламино)этил)-5-метокси-N1-метил-N4-(4-(1-метил-6-((триметилсилил)этинил)-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,2,4-триамина (166 мг, 88%).
MS m/z (ESI): 542,3 [М+Н]+.
Стадия 8: получение N1-(2-(диметиламино)этил)-N4-(4-(6-этинил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-5-метокси-N1-метилбензол-1,2,4-триамина

N1-(2-(диметиламино)этил)-5-метокси-N1-метил-N4-(4-(1-метил-6-((триметилсилил)этинил)-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,2,4-триамин (90 мг, 0,166 ммоль) растворяли в смеси THF (10 мл) и МеОН (10 мл), затем добавляли карбонат калия (69 мг, 0,50 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 3 часов. Растворитель удаляли при пониженном давлении и добавляли воду и EtOAc и разделяли две фазы. Органическую фазу несколько раз промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали с получением указанного в заголовке соединения N1-(2-(диметиламино)этил)-N4-(4-(6-этинил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-5-метокси-N1-метилбензол-1,2,4-триамина (71 мг, 91%).
MS m/z (ESI): 470,26 [М+Н]+.
Стадия 9: получение N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(6-этинил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метоксифенил)акриламида

Раствор акрилоилхлорида (22,0 мг, 0,247 ммоль) в THF (1 мл) добавляли по каплям в раствор N1-(2-(диметиламино)этил)-N4-(4-(6-этинил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)-5-метокси-N1-метилбензол-1,2,4-триамина (80 мг, 0,169 ммоль) и TEA (50 мг, 0,492 ммоль) в THF (2 мл) на бане со льдом и водой. По завершении добавления, смесь перемешивали в течение 15 минут при этой температуре. Реакцию гасили метанолом. Реакционный раствор концентрировали при пониженном давлении и очищали препаративной тонкослойной хроматографией (CH2Cl2 : МеОН: концентрированный аммиак = 100:10:1) с получением указанного в заголовке соединения N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(6-этинил-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метоксифенил)акриламида (45 мг, 51%).
1Н ЯМР (400 МГц, CDCl3) δ 10,03 (s, 1Н), 9,74 (d, J=6,2 Гц, 1Н), 9,07 (s, 1Н), 8,31 (d, J=5,3 Гц, 1Н), 7,91 (d, J=8,3 Гц, 1Н), 7,65 (s, 1Н), 7,48 (s, 1Н), 7,29 (dd, J=8,3, 1,2 Гц, 1Н), 7,08 (d, J=5,3 Гц, 1Н), 6,70 (s, 1Н), 6,37 (d, J=16,3 Гц, 2Н), 5,81-5,57 (m, 1Н), 3,89 (d, J=13,0 Гц, 3Н), 3,81 (s, 3Н), 3,02 (s, 1Н), 2,85 (s, 2Н), 2,62 (s, 3Н), 2,24 (m, 8Н);
MS m/z (ESI): 524,6 [М+Н]+.
Пример 96: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1-метил-6-винил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)-акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1-метил-6-винил-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 95.
1Н ЯМР (400 МГц, CDCl3) δ 9,96 (s, 1Н), 9,76 (d, J=4,7 Гц, 1Н), 8,99 (s, 1Н), 8,30 (d, J=5,3 Гц, 1Н), 7,92 (d, J=8,7 Гц, 1Н), 7,64 (s, 1Н), 7,30 (dd, J=4,3, 2,8 Гц, 2Н), 7,11 (t, J=5,1 Гц, 1Н), 6,80 (dd, J=17,5, 10,9 Гц, 1Н), 6,69 (s, 1Н), 6,38 (d, J=16,7 Гц, 2Н), 5,90-5,52 (m, 2Н), 5,19-5,06 (m, 1Н), 3,91 (s, 3Н), 3,80 (s, 3Н), 2,88 (s, 2Н), 2,63 (s, 3Н), 2,28 (m, 8Н);
MS m/z (ESI): 526,6 [М+Н]+.
Пример 97: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 22.
1Н ЯМР (400 МГц, CDCl3) δ 10,29 (s, 1Н), 9,85 (s, 1Н), 9,70 (d, J=14,2 Гц, 1Н), 8,29 (s, 1Н), 8,16 (s, 1Н), 7,88 (d, J=9,4 Гц, 1Н), 7,56 (s, 1Н), 6,88 (s, 1Н), 6,79-6,60 (m, 3Н), 6,43 (d, J=15,5 Гц, 2Н), 5,62 (d, J=10,3 Гц, 1Н), 3,81 (s, 3Н), 3,62 (s, 3Н), 2,84 (s, 2Н), 2,64 (s, 3Н), 2,21 (m, 8Н);
MS m/z (ESI): 516,6[М+Н]+.
Пример 98: получение N-(5-((5-хлор-4-(6-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Стадия 1: получение 6-метокси-1-метил-1Н-индола

6-метокси-1Н-индол (500 мг, 3,4 ммоль) растворяли в N,N-диметилформамиде (16 мл). Раствор охлаждали на ледяной бане и добавляли гидрид натрия (320 мг, 6,8 ммоль). После перемешивания реакционного раствора в течение 15 минут, по каплям добавляли метилйодид (0,25 мл, 3,7 ммоль). Реакционный раствор естественным образом нагревали до комнатной температуры и перемешивали в течение 2 ч, а затем гасили насыщенным водным раствором хлорида аммония (20 мл) и экстрагировали этилацетатом (30 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта, который затем очищали колоночной хроматографией с получением 6-метокси-1-метил-1Н-индола (480 мг, 88%).
Стадия 2: получение 3-(2,5-дихлорпиримидин-4-ил)-6-метокси-1-метил-1Н-индола

6-метокси-1-метил-1Н-индол (480 мг, 3,0 ммоль) и 2,4,5-трихлорпиримидин (660 мг, 3,6 ммоль) растворяли в диметиловом эфире этиленгликоля (20 мл). Реакционную смесь нагревали до 80°С в течение 20 мин и добавляли безводный хлорид алюминия (720 мг, 5,4 ммоль). Реакционную смесь перемешивали в течение 1 часа в атмосфере азота. Реакционную смесь гасили смесью льда с водой (приблизительно 50 мл) и смесь экстрагировали метил-трет-бутиловым эфиром (20 мл × 3). Органические фазы объединяли, сушили над сульфатом магния, фильтровали и концентрировали с получением сырого продукта 3-(2,5-дихлорпиримидин-4-ил)-6-метокси-1-метил-1Н-индола (320 мг, 30%), который использовали непосредственно на следующей стадии.
Стадии 3-6: получение N-(5-((5-хлор-4-(6-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((5-хлор-4-(6-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 22.
1Н ЯМР (400 МГц, CD3OD) δ 8,48 (s, 1Н), 8,29-8,21 (m, 2Н), 8,19 (s, 1Н), 7,01 (s, 1Н), 6,96 (d, J=2,2 Гц, 1Н), 6,78 (dd, J=8,9, 2,2 Гц, 1Н), 6,49-6,44 (m, 2Н), 5,84 (dd, J=7,7, 4,1 Гц, 1Н), 3,97 (s, 3Н), 3,86 (d, J=10,8 Гц, 6Н), 3,53 (t, J=5,7 Гц, 2Н), 3,33-3,31 (m, 2Н), 2,91 (s, 6Н), 2,76 (s, 3Н);
MS m/z (ESI): 564,3 [М+1]+.
Пример 99: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-1-метил-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида

Стадия 1: получение 6-метокси-1-метил-1Н-индола

Исходный материал 6-метокси-1Н-индол (1 г, 6,73 ммоль) растворяли в DMF (20 мл) и смесь охлаждали до 0°С, а затем добавляли NaH (815 мг, 20,38 ммоль). Реакционный раствор перемешивали при 0°С в течение десяти минут. Затем в реакционную систему добавляли йодметан (1,447 г, 10,19 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. Реакционный раствор выливали в ледяную воду и экстрагировали этилацетатом. Органические фазы объединяли, промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением продукта, который затем очищали колоночной флэш-хроматографией на силикагеле с получением продукта 6-метокси-1-метил-1Н-индола (850 мг, 77,3%).
Стадия 2: получение 3-(2-хлор-5-(трифторметил)пиримидин-4-ил)-6-метокси-1-метил-1Н-индола

Исходный материал 6-метокси-1-метил-1Н-индол (850 мг, 5,27 ммоль), 2,4-дихлор-5-(трифторметил)пиримидин (1,26 г, 5,8 ммоль) и трихлорид алюминия (1,05 г, 7,91 ммоль) растворяли в DME (30 мл) и реакционную смесь перемешивали в течение ночи при 70°С.По окончании реакции реакционный раствор выливали в ледяную воду и экстрагировали три раза метил-трет-бутиловым эфиром. Органические фазы объединяли, промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с получением сырого продукта, который затем очищали колоночной флэш-хроматографией на силикагеле, с получением продукта 3-(2-хлор-5-(трифторметил)пиримидин-4-ил)-6-метокси-1-метил-1Н-индола (700 мг, 39%).
Стадия 3: получение N-(4-фтор-2-метокси-5-нитрофенил)-4-(6-метокси-1-метил-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-амина

3-(2-хлор-5-(трифторметил)пиримидин-4-ил)-6-метокси-1-метил-1Н-индол (700 мг, 2,05 ммоль), исходный материал 4-фтор-2-метокси-5-нитроанилин (419 мг, 2,25 ммоль) и моногидрат п-толуолсульфоновой кислоты (390 мг, 2,05 ммоль) растворяли в 2-пентаноле (10 мл). Реакционную смесь нагревали до 120°С и проводили реакцию в течение ночи. После того как ЖХМС показывала завершение реакции, реакционный раствор естественным образом охлаждали до комнатной температуры и осаждали темное твердое вещество. Твердое вещество фильтровали и осадок на фильтре промывали метанолом (1 мл) и метил-трет-бутиловым эфиром (1 мл) с получением продукта N-(4-фтор-2-метокси-5-нитрофенил)-4-(6-метокси-1-метил-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-амина (600 мг, 60%)
Стадия 4: получение N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(6-метокси-1-метил-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)-N1-метил-2-нитробензол-1,4-диамина

N-(4-фтор-2-метокси-5-нитрофенил)-4-(6-метокси-1-метил-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-амин (100 мг, 0,204 ммоль) растворяли в DMF (5 мл). Затем добавляли триэтиламин (31 мг, 0,305 ммоль) и N1,N1,N2-триметилэтан-1,2-диамин (42 мг, 0,407 ммоль). Реакционную смесь нагревали до 120°С с помощью микроволнового излучения в течение 30 минут. После того как ЖХМС показал завершение реакции, реакционный раствор концентрировали досуха, чтобы получить сырой продукт, который затем очищали препаративной тонкослойной хроматографией с получением продукта N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(6-метокси-1-метил-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)-N1-метил-2-нитробензол-1,4-диамина (90 мг, 77%).
Стадия 5: получение N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(6-метокси-1-метил-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)-N1-метилбензол-1,2,4-триамина

N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(6-метокси-1-метил-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)-N1-метил-2-нитробензол-1,4-диамин (90 мг, 0,157 ммоль) растворяли в 10 мл метанола и добавляли Pd/C (15 мг). Реакционную смесь перемешивали в атмосфере водорода при 24°С в течение 1 часа. После того как ЖХМС показывала завершение реакции, реакционный раствор фильтровали и фильтрат концентрировали. Полученный остаток очищали колоночной флэш-хроматографией на силикагеле с получением 80 мг неочищенного продукта N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(6-метокси-1-метил-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)-N1-метилбензол-1,2,4-триамина.
Стадия 6: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-1-метил-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида

N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(6-метокси-1-метил-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)-N1-метилбензол-1,2,4-триамин (80 мг, 0,147 ммоль) и триэтиламин (45 мг, 0,442 ммоль) растворяли в безводном тетрагидрофуране (20 мл). Реакционный раствор перемешивали при -78°С в течение 10 минут и затем медленно и по каплям добавляли акрилоилхлорид (0,4 мл, 1 M в THF). Реакционную смесь перемешивали в течение 30 минут на бане с сухим льдом. После того как ЖХМС показывала завершение реакции, реакцию гасили метанолом. Реакционный раствор концентрировали и полученный остаток очищали препаративной тонкослойной хроматографией с получением продукта N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(6-метокси-1-метил-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида (20 мг, 25%).
1Н ЯМР (400 МГц, CD3OD) δ 8,64 (s, 1Н), 8,37 (s, 1Н), 8,09 (d, J=8,6 Гц, 1Н), 7,75 (s, 1Н), 7,02-6,95 (m, 2Н), 6,78 (dd, J=8,8, 1,8 Гц, 1Н), 6,46-6,34 (m, 2Н), 5,83 (dd, J=8,3, 3,5 Гц, 1Н), 4,00 (s, 3Н), 3,87 (d, J=8,4 Гц, 6Н), 3,51 (t, J=5,7 Гц, 2Н), 3,30 (t, J=5,7 Гц, 2Н), 2,88 (s, 6Н), 2,72 (s, 3Н);
MS m/z (ESI): 598, 4 [М+Н]+.
Пример 100: получение N-(5-((5-хлор-4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((5-хлор-4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)-акриламид был аналогичен Примеру 98.
1Н ЯМР (400 МГц, CD3OD) δ 8,44 (s, 1Н), 8,37 (d, J=8,0 Гц, 1Н), 8,31 (d, J=10,3 Гц, 2Н), 7,66 (d, J=8,2 Гц, 1Н), 7,31-7,23 (m, 1Н), 7,16 (dd, J=11,2, 4,0 Гц, 1Н), 6,99 (s, 1Н), 6,45 (d, J=6,2 Гц, 2Н), 5,88-5,80 (m, 1Н), 3,99 (d, J=2,8 Гц, 3Н), 3,52 (dt, J=7,1, 3,7 Гц, 3Н), 3,32-3,29 (m, 2Н), 2,89 (s, 6Н), 2,73 (s, 3Н), 1,20 (dt, J=7,2, 3,6 Гц, 2Н), 1,08-1,00 (m, 2Н);
MS m/z (ESI): 560,3 [М+Н]+.
Пример 101: получение N-(5-((4-(1-циклопропил-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино))этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(1-циклопропил-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид был аналогичен Примеру 98.
1Н ЯМР (400 МГц, CD3OD) δ 8,68 (s, 1Н), 8,41 (s, 1Н), 8,19 (d, J=7,8 Гц, 1Н), 7,82 (s, 1Н), 7,67 (d, J=8,2 Гц, 1Н), 7,26 (t, J=7,6 Гц, 1Н), 7,14 (t, J=7,5 Гц, 1Н), 6,99 (s, 1Н), 6,44 (dt, J=14,3, 7,1 Гц, 2Н), 5,85 (dd, J=9,2, 2,6 Гц, 1Н), 4,01 (s, 3Н), 3,60-3,44 (m, 3Н), 3,29 (t, J=5,6 Гц, 2Н), 2,87 (s, 6Н), 2,71 (s, 3Н), 1,25-1,18 (m, 2Н), 1,06-0,98 (m, 2Н);
MS m/z (ESI): 594, 3 [М+Н]+.
Пример 102: получение N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-(N,N-диметилсульфамоил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метоксифенил)акриламида

Стадия 1: получение 3-(2-хлорпиримидин-4-ил)-1Н-индола

3-(2-хлорпиримидин-4-ил)-1Н-индол (1 г, 4,37 ммоль), 2-(дифторметокси)-4-фтор-5-нитроанилин (810 мг, 4,37 ммоль) и п-толуолсульфокислоту (750 мг, 4,37 ммоль) растворяли в 2-пентаноле (40 мл) и затем реакционный раствор нагревали при 110°С в течение 3 часов. После того как ЖХМС показывала завершение реакции, реакционный раствор естественным образом охлаждали до комнатной температуры и осаждали темное твердое вещество. Твердое вещество фильтровали и осадок на фильтре промывали метанолом и метил-трет-бутиловым эфиром с получением 3-(2-хлорпиримидин-4-ил)-1Н-индола (1,3 г, 79%).
Стадия 2: получение N-(4-фтор-2-метокси-5-нитрофенил)-4-(1Н-индол-3-ил)пиримидин-2-амина

3-(2-хлорпиримидин-4-ил)-1Н-индол (500 мг, 2,177 ммоль), исходный материал 4-фтор-2-метокси-5-нитроанилин (445 мг, 2,394 ммоль) и моногидрат п-толуолсульфокислоты (414 мг, 2,177 ммоль) растворяли в 2-пентаноле (20 мл). Реакционную смесь нагревали до 120°С в течение ночи. После того как ЖХМС показывала завершение реакции, реакционный раствор естественным образом охлаждали до комнатной температуры и осаждали темное твердое вещество. Твердое вещество фильтровали и осадок на фильтре промывали метанолом (1 мл) и метил-трет-бутиловым эфиром (1 мл) с получением N-(4-фтор-2-метокси-5-нитрофенил)-4-(1Н-индол-3-ил)пиримидин-2-амина (180 мг, 22%).
Стадия 3: получение N1-(4-(1Н-индол-3-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамина

N-(4-фтор-2-метокси-5-нитрофенил)-4-(1Н-индол-3-ил)пиримидин-2-амин (178 мг, 0,469 ммоль) растворяли в DMF (2 мл), с последующим добавлением триэтиламина (142 мг, 1,41 ммоль) и триметилэтилендиамина (144 мг, 1,41 ммоль) Реакционную смесь нагревали до 120°С микроволновым излучением и затем проводили реакцию в течение 30 минут. После того как ЖХМС показывала завершение реакции, реакционный раствор концентрировали досуха, чтобы получить сырой продукт, который затем очищали препаративной тонкослойной хроматографией с получением продукта N1-(4-(1Н-индол-3-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамина (217 мг, 100%).
Стадия 4: получение 3-(2-((4-((2-(диметиламино)этил)(метил)амино)2-метокси-5-нитрофенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамида

N1-(4-(1Н-индол-3-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамин (217 мг, 0,47 ммоль) растворяли в DMF (10 мл) и смесь охлаждали до 0°С на ледяной бане, затем добавляли NaH (56 мг, 1,41 ммоль). После того, как реакцию проводили при 0°С в течение 10 минут, по каплям добавляли диметилсульфамоилхлорид (74 мг, 0,52 ммоль). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 30 минут. После прекращения реакции добавляли дихлорметан и воду. Реакционный раствор экстрагировали три раза. Органические фазы объединяли, промывали насыщенным бикарбонатом натрия, водой и насыщенным солевым раствором, фильтровали и концентрировали с получением неочищенного продукта, который затем очищали колоночной флэш-хроматографией на силикагеле, с получением продукта 3-(2-((4-(2-(диметиламино)этил)(метил)амино)-2-метокси-5-нитрофенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамида (160 мг, 60%).
Стадия 5: получение 3-(2-((5-амино-4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамида

3-(2-((4-((2-(диметиламино)этил)(метил)амино)-2-метокси-5-нитрофенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамид растворяли в метаноле (5 мл), затем добавляли Pd/C (15 мг) и реакционную смесь перемешивали при 24°С в течение 1 часа в атмосфере водорода. После того как ЖХМС показывала завершение реакции, реакционный раствор фильтровали и концентрировали с получением сырого продукта, который затем очищали колоночной флэш-хроматографией на силикагеле с получением продукта 3-(2-((5-амино-4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамида (90 мг, 59%).
Стадия 6: получение N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-(N,N-диметилсульфамоил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метоксифенил)акриламида

3-(2-((5-амино-4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамид (90 мг, 0,167 ммоль) и триэтиламин (51 мг, 0,501 ммоль) растворяли в безводном тетрагидрофуране (30 мл) и реакционный раствор перемешивали при -78°С в течение 10 минут. Медленно и по каплям добавляли акрилоилхлорид (0,5 мл, 1М в THF). Реакционную смесь перемешивали на бане с сухим льдом в течение 30 минут. После того как ЖХМС показывала завершение реакции, реакцию гасили метанолом. Реакционный раствор концентрировали и полученный остаток очищали препаративной тонкослойной хроматографией с получением продукта N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-(N,N-диметилсульфамоил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метоксифенил)акриламида (10 мг, 10%).
1Н ЯМР (400 МГц, CD3OD) δ 8,62 (s, 1Н), 8,44 (d, J=7,9 Гц, 1Н), 8,27 (d, J=6,2 Гц, 1Н), 8,10 (s, 1Н), 8,00 (d, J=8,3 Гц, 1Н), 7,54 (d, J=6,3 Гц, 1Н), 7,37 (dt, J=15,0, 7,3 Гц, 2Н), 7,06 (s, 1Н), 6,58 (dd, J=16,9, 10,0 Гц, 1Н), 6,46 (dd, J=16,9, 1,8 Гц, 1Н), 5,86 (dd, J=10,0, 1,7 Гц, 1Н), 3,98 (s, 3Н), 3,55 (t, J=5,7 Гц, 2Н), 3,36 (d, J=5,9 Гц, 2Н), 2,92 (d, J=3,7 Гц, 12Н), 2,79 (s, 3Н);
MS m/z (ESI): 593, 5 [M+H]+.
Пример 103: получение N-(2-((2-(диэтиламино)этил)(метил)амино)-5-((4-(1-(N,N-диметилсульфамоил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метоксифенил)акриламида

Стадия 1: получение N1-(4-(1Н-индол-3-ил)пиримидин-2-ил)-N4-(2-(диэтиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамина

N-(4-фтор-2-метокси-5-нитрофенил)-4-(1Н-индол-3-ил)пиримидин-2-амин (120 мг, 0,316 ммоль) растворяли в DMF (2 мл). Добавляли триэтиламин (96 мг, 0,949 ммоль) и N,N-диэтил-N-метилэтан-1,2-диамин (124 мг, 0,949 ммоль). Реакционную смесь нагревали до 120°С микроволновым излучением и проводили реакцию в течение 30 мин. После того как ЖХМС показывала завершение реакции, реакционный раствор концентрировали досуха, чтобы получить сырой продукт, который затем очищали препаративной тонкослойной хроматографией с получением продукта N1-(4-(1Н-индол-3-ил)пиримидин-2-ил)-N4-(2-(диэтиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамина (155 мг, 100%).
Стадия 2: получение 3-(2-((4-((2-(диэтиламино)этил)(метил)амино)-2-метокси-5-нитрофенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамида

N1-(4-(1Н-индол-3-ил)пиримидин-2-ил)-N4-(2-(диэтиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамин (155 мг, 0,316 ммоль) растворяли в DMF (10 мл). Реакционную смесь охлаждали до 0°С на ледяной бане, а затем добавляли NaH (38 мг, 0,945 ммоль). После того, как реакцию проводили в течение 10 минут при 0°С, по каплям добавляли диметилсульфамоилхлорид (55 мг, 0,38 ммоль). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 30 минут. После прекращения реакции добавляли дихлорметан и воду. Реакционный раствор экстрагировали три раза. Органические фазы объединяли, промывали насыщенным водным раствором бикарбоната натрия, водой и насыщенным солевым раствором, фильтровали и выпаривали досуха с получением сырого продукта, который затем очищали колоночной флэш-хроматографией на силикагеле с получением продукта 3-(2-((4-((2-(диэтиламино)этил)(метил)амино)-2-метокси-5-нитрофенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамида (130 мг, 19%).
Стадия 3: получение 3-(2-((5-амино-4-((2-(диэтиламино)этил)(метил)амино)-2-метоксифенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамида

3-(2-((4-((2-(диэтиламино)этил)(метил)амино)-2-метокси-5-нитрофенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамид растворяли в метаноле (5 мл) и добавляли Pd/C (15 мг). Реакционную смесь перемешивали в атмосфере водорода при 24°С в течение 1 часа. После того как ЖХМС показывала завершение реакции, реакционный раствор фильтровали и фильтрат концентрировали. Полученный остаток очищали колоночной флэш-хроматографией на силикагеле с получением 118 мг неочищенного продукта. 3-(2-((5-амино-4-((2-(диэтиламино)этил)(метил)амино)-2-метоксифенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамида.
Стадия 4: получение N-(2-((2-(диэтиламино)этил)(метил)амино)-5-((4-(1-(N,N-диметилсульфамоил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метоксифенил)акриламида

3-(2-((5-амино-4-((2-(диэтиламино)этил)(метил)амино)-2-метоксифенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамид (118 мг, 0,208 ммоль) и триэтиламин (63 мг, 0,624 ммоль) растворяли в безводном тетрагидрофуране (30 мл). Реакционный раствор перемешивали при -78°С в течение 10 минут, затем медленно и по каплям добавляли акрилоилхлорид (0,62 мл, 1 M в THF). Реакционную смесь перемешивали в течение 30 минут на бане с сухим льдом. После того как ЖХМС показывала завершение реакции, реакцию гасили метанолом. Реакционный раствор концентрировали и полученный остаток очищали препаративной тонкослойной хроматографией с получением продукта N-(2-((2-(диэтиламино)этил)(метил)амино)-5-((4-(1-(N,N-диметилсульфамоил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метоксифенил)акриламида (4,8 мг, 3,7%).
1Н ЯМР (400 МГц, CD3OD) δ 8,61 (s, 1Н), 8,46 (d, J=8,0 Гц, 1Н), 8,33 (d, J=6,1 Гц, 1Н), 8,08 (s, 1Н), 8,02 (d, J=8,3 Гц, 1Н), 7,55 (d, J=6,1 Гц, 1Н), 7,45-7,26 (m, 2Н), 7,04 (s, 1Н), 6,48 (qd, J=17,0, 5,9 Гц, 2Н), 5,87 (dd, J=9,4, 2,5 Гц, 1Н), 4,01 (s, 3Н), 3,57 (t, J=5,7 Гц, 2Н), 3,25 (dt, J=19,4, 7,2 Гц, 4Н), 2,93 (s, 6Н), 2,79 (s, 3Н), 1,29 (t, J=7,3 Гц, 6Н);
MS m/z (ESI): 621, 5 [М+Н]+.
Пример 104: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1-(оксетан-3-ил)-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)-акриламида

Стадия 1: получение 3-(2-хлорпиримидин-4-ил)-1-(оксетан-3-ил)-1Н-индола

3-(2-хлорпиримидин-4-ил)-1Н-индол (500 мг, 2,18 ммоль), 3-йод-оксетан (480 мг, 2,61 ммоль) и карбонат цезия (1,42 г, 4,36 ммоль) смешивали в DMF (5 мл). Реакцию проводили при 110°С в течение 1 часа при микроволновом излучении. После охлаждения реакционный раствор разбавляли CH2Cl2. Органическую фазу промывали три раза насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением указанного в заголовке соединения 3-(2-хлорпиримидин-4-ил)-1-(оксетан-3-ил)-1Н-индола (110 мг, 18%).
1Н ЯМР (400 МГц, CDCl3): δ 8,54 (d, J=5,2 Гц, 1Н), 8,38 (m, 1Н), 8,33 (s, 1Н), 7,60 (d, J=5,2 Гц, 1Н), 7,54 (m, 1Н), 7,38 (m, 2Н), 5,66 (m, 1Н), 5,26 (t, J=7,6 Гц, 2Н), 5,14 (t, J=7,6 Гц, 2Н);
MS m/z (ESI): 286,1 [М+Н]+.
Стадия 2: получение N-(4-фтор-2-метокси-5-нитрофенил)-4-(1-(оксетан-3-ил)-1Н-индол-3-ил)пиримидин-2-амина

3-(2-хлорпиримидин-4-ил)-1-(оксетан-3-ил)-1Н-индол (110 мг, 0,385 ммоль), 4-фтор-2-метокси-5-нитроанилин (86 мг, 0,462 ммоль), ацетат палладия (9 мг, 0,0385 ммоль) и карбонат цезия (376 мг, 1,16 ммоль) смешивали в смеси DMA (1 мл) и 1,4-диоксана (2 мл). Реакционную смесь продували азотом для удаления кислорода в течение 15 минут. Добавляли ксантфос (45 мг, 0,0770 ммоль), и реакционную смесь дополнительно продували азотом в течение 5 минут. Реакцию проводили при 160°С в течение 30 минут в микроволновом реакторе. После охлаждения, реакционную смесь разбавляли CH2Cl2, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали препаративной тонкослойной хроматографией с получением указанного в заголовке соединения N-(4-фтор-2-метокси-5-нитрофенил)-4-(1-(оксетан-3-ил)-1Н-индол-3-ил)пиримидин-2-амина (80 мг, 46%).
MS m/z (ESI): 436,1 [М+Н]+.
Стадия 3: получение N1-(2-(диметиламино)этил)-5-метокси-N1-метил-2-нитро-N4-(4-(1-(оксетан-3-ил)-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,4-диамина

Триметилэтилендиамин (0,1 мл) и DIPEA (0,1 мл) добавляли к раствору N-(4-фтор-2-метокси-5-нитрофенил)-4-(1-(оксетан-3-ил)-1Н-индол-3-ил)пиримидин-2-амина (80 мг, 0,18 ммоль) в DMA (1 мл). Реакционную смесь перемешивали при 85°С в течение 3 часов. После охлаждения добавляли воду и затем осаждали твердое вещество. Твердое вещество очищали препаративной тонкослойной хроматографией с получением соединения N1-(2-(диметиламино)этил)-5-метокси-N1-метил-2-нитро-N4-(4-(1-(оксетан-3-ил)-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,4-диамина (35 мг, 38%).
MS m/z (ESI): 518,2 [М+Н]+.
Стадия 4: получение N1-(2-(диметиламино)этил)-5-метокси-N1-метил-N4-(4-(1-(оксетан-3-ил)-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,2,4-триамина

N1-(2-(диметиламино)этил)-5-метокси-N1-метил-2-нитро-N4-(4-(1-(оксетан-3-ил)-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,4-диамин (28 мг, 0,054 ммоль), восстановленный порошок железа (30 мг, 0,54 ммоль) и хлорид аммония (2,0 мг, 0,032 ммоль) смешивали в смеси этанола (3 мл) и воды (1 мл), а затем реакционную смесь нагревали до температуры дефлегмации в течение одного часа, затем охлаждали и фильтровали через целит. Полученный фильтрат концентрировали и очищали препаративной тонкослойной хроматографией с получением указанного в заголовке соединения N1-(2-(диметиламино)этил)-5-метокси-N1-метил-N4-(4-(1-(оксетан-3-ил)-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,2,4-триамина (25 мг, 95%).
MS m/z (ESI): 488,3 [М+Н]+.
Стадия 5: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1-(оксетан-3-ил)-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)-акриламида

Раствор акрилоилхлорида (0,025 мл, 0,31 ммоль) в THF (0,5 мл) добавляли по каплям к раствору N1-(2-(диметиламино)этил)-5-метокси-N1-метил-N4-(4-1-(оксетан-3-ил)-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,2,4-триамина (25 мг, 0,051 ммоль) и триэтиламина (0,050 мл, 0,36 ммоль) в THF (2 мл) при -15°С. По завершении добавления, смесь перемешивали при этой температуре в течение 5 минут, гасили метанолом и очищали препаративной тонкослойной хроматографией с получением указанного в заголовке соединения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1-(оксетан-3-ил)-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида (7 мг, 25%).
1Н ЯМР (400 МГц, CDCl3): δ 10,2 (brs, 1Н), 9,80 (s, 1Н), 9,06 (s, 1Н), 8,42 (d, J=5,2 Гц, 1Н), 8,10 (m, 1Н), 7,73 (m, 2Н), 7,30 (m, 2Н), 7,23 (d, J=5,2 Гц, 1Н), 6,79 (s, 1Н), 6,44 (m, 2Н), 5,89 (m, 1Н), 5,74 (m, 1Н), 5,38 (t, J=6,8 Гц, 2Н), 5,15 (t, J=7,6 Гц, 2Н), 3,89 (s, 3Н), 2,92 (m, 2Н), 2,71 (s, 3Н), 2,29 (m, 8Н);
MS m/z (ESI): 542, 3 [М+Н]+.
Пример 105: получение N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(5-этокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-4-метоксифенил)-акриламида

Стадия 1: получение 5-этокси-1Н-индазола

5-гидрокси-1Н-индазол (2,68 г, 20 ммоль) растворяли в DMF (50 мл) и затем добавляли этилйодид (3,28 г, 21 ммоль) и карбонат калия (4,16 г, 30 ммоль). Смесь перемешивали при комнатной температуре в течение 24 часов, экстрагировали этилацетатом и очищали колоночной хроматографией с получением 5-этокси-1Н-индазола (1,5 г, 46%).
1Н ЯМР (400 МГц, CDCl3) δ 8,73-8,18 (m, 1Н), 8,03 (d, J=0,9 Гц, 1Н), 7,42 (d, J=8,6 Гц, 1Н), 7,15-7,06 (m, 2Н), 4,10 (q, J=7,0 Гц, 2Н), 1,48 (t, J=7,0 Гц, 3Н);
MS m/z (ESI): 163 [М+Н]+.
Стадии 2 и 3: получение 5-этокси-1-(2-(метилсульфонил)пиримидин-4-ил)-1Н-индазола

Способ получения 5-этокси-1-(2-(метилсульфонил)пиримидин-4-ил)-1Н-индазола был аналогичен Примеру 43.
Стадия 4: получение 4-(5-этокси-1Н-индазол-1-ил)-N-(4-фтор-2-метокси-5-нитрофенил)пиримидин-2-амина

N-(4-фтор-2-метокси-5-нитрофенил)формамид (134 мг, 0,63 ммоль) растворяли в THF (20 мл) и добавляли при 0°С гидрид натрия (50 мг, 1,26 ммоль). После перемешивания смеси в течение 10 минут добавляли 5-этокси-1-(2-(метилсульфонил)пиримидин-4-ил)-1Н-индазол (200 мг, 0,63 ммоль) и затем смесь перемешивали в течение ночи. После добавлени соответствующего количества 1N водного раствора гидроксида натрия, смесь перемешивали в течение 30 минут, экстрагировали DCM и очищали колоночной хроматографией с получением 4-(5-этокси-1Н-индазол-1-ил)-N-(4-фтор-2-метокси-5-нитрофенил)пиримидин-2-амина (210 мг, 78%).
Стадии 5-7: получение N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(5-этокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-4-метоксифенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(5-этокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-4-метоксифенил)-акриламида был аналогичен Примеру 43.
1Н ЯМР (400 МГц, CD3OD) δ 8,39 (s, 2Н), 8,33-8,22 (m, 1Н), 7,84 (s, 1Н), 7,62 (d, J=6,8 Гц, 1Н), 7,31 (s, 1Н), 7,19-7,11 (m, 1Н), 7,09 (s, 1Н), 6,52 (d, J=9,4 Гц, 2Н), 5,95-5,84 (m, 1Н), 4,12 (d, J=7,0 Гц, 2Н), 3,97 (s, 3Н), 3,58 (s, 2Н), 3,37 (s, 2Н), 2,94 (s, 6H), 2,82 (s, 3Н), 1,45 (t, J=7,0 Гц, 3Н);
MS m/z (ESI): 531 [M+H]+.
Пример 106: получение N1-(5-((4-(5-циано-3-метил-1Н-индазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Стадия 1: получение 5-бром-3-метил-1Н-индазола

1-(5-бром-2-фторфенил)этан-1-он (5 г, 23,04 ммоль) и гидразингидрат (20 мл) нагревали в течение 2 дней. Продукт подвергали колоночной хроматографии с получением 5-бром-3-метил-1Н-индазола (2,8 г, 58%).
Стадия 2: получение 5-циано-3-метил-1Н-индазола

5-бром-3-метил-1Н-индазол (500 мг, 2,38 моль), цианид цинка (418 мг, 3,57 ммоль), Pd2(dba)3 (194 мг, 0,238 ммоль) и X-Phos (227 мг, 0,476 моль) добавляли в пробирку для проведения реакций под воздействием микроволнового излучения. После продувки азотом для удаления кислорода, смесь нагревали в течение 1 часа и затем подвергали колоночной хроматографии с получением 5-циано-3-метил-1Н-индазола (430 мг, 98%).
1Н-ЯМР (400 МГц, CDCl3) δ 8,12 (s, 1Н), 7,61 (s, 1Н), 7,56 (d, J=8,7 Гц, 1Н), 2,66 (s, 3Н); MS m/z (ESI): 158 [М+Н]+.
Стадии 3-7: получение N-(5-((4-(5-циано-3-метил-1Н-индазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Стадии 3-7: способ получения N-(5-((4-(5-циано-3-метил-1Н-индазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)-амино)-4-метоксифенил)акриламида был аналогичен Примеру 40.
1Н ЯМР (400 МГц, CD3OD) δ 8,85 (s, 1Н), 8,48-8,35 (m, 1Н), 8,35-8,30 (m, 1Н), 8,06 (s, 1Н), 7,77-7,62 (m, 1Н), 7,01 (s, 1Н), 6,59-6,49 (m, 1Н), 6,47-6,37 (m, 1Н), 5,94-5,86 (m, 1Н), 4,01 (s, 3Н), 3,61-3,50 (m, 2Н), 3,32 (s, 2Н), 2,91 (s, 6Н), 2,75 (s, 3Н), 2,65 (s, 3Н);
MS m/z (ESI): 594 [М+Н]+.
Пример 107: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-3-метил-1Н-индазол-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-метокси-3-метил-1Н-индазол-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 40.
1Н ЯМР (400 МГц, CD3OD) δ 8,74 (s, 1Н), 8,25-8,10 (m, 1Н), 8,04 (s, 1Н), 7,18 (s, 1Н), 7,10-7,03 (m, 1Н), 7,00 (s, 1Н), 6,47 (s, 2Н), 5,89-5,83 (m, 1Н), 3,98 (s, 3Н), 3,89 (s, 3Н), 3,54 (s, 2Н), 3,01 (s, 1Н), 2,90 (s, 6Н), 2,88 (s, 1Н), 2,76 (s, 3Н), 2,57 (s, 3Н);
MS m/z (ESI): 599 [M+H]+.
Пример 108: получение N-(5-(5-хлор-4-(5-хлор-3-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Стадия 1: получение 5-хлор-2-гидразинилбензойной кислоты

5-хлор-2-фторбензойную кислоту (5 г, 0,0287 ммоль) и гидразингидрат (10 мл, 85%) нагревали при 100°С в течение ночи. Реакционный раствор концентрировали, подкисляли, фильтровали и сушили с получением 5-хлор-2-гидразинилбензойной кислоты (2,5 г, 50%).
Стадия 2: получение 5-хлор-1Н-индазол-3-ола

5-хлор-2-гидразинилбензойную кислоту (2,5 г), концентрированную соляную кислоту (10 мл) и воду (200 мл) нагревали при 100°С в течение 3 часов. Затем смесь концентрировали до 100 мл, доводили до рН 7,0 карбонатом натрия, фильтровали и сушили с получением 5-хлор-1Н-индазол-3-ола (1,7 г, 60%).
1Н ЯМР (400 МГц, ДМСО) δ 11,73 (s, 1Н), 10,67 (s, 1Н), 7,65 (d, J=1,3 Гц, 1Н), 7,30 (dt, J=8,9, 5,1 Гц, 2Н);
MS m/z (ESI): 169 [М+Н]+.
Стадия 3: Получение этил-5-хлор-3-гидрокси-1Н-индазол-1-карбоксилата

5-хлор-1Н-индазол-3-ол (1,7 г, 10,12 ммоль) растворяли в пиридине (10 мл) и затем добавляли метилхлорформиат (1,31 г, 12,14 ммоль). Смесь нагревали до 100°С в течение 2 часов. По окончании реакции смесь охлаждали и добавляли 150 мл воды. Реакционный раствор фильтровали и сушили с получением этил-5-хлор-3-гидрокси-1Н-индазол-1-карбоксилата (2,2 г, 90%).
Стадия 4: Получение этил-5-хлор-3-метокси-1Н-индазол-1-карбоксилата

К ацетону (20 мл) добавляли этил 5-хлор-3-гидрокси-1Н-индазол-1-карбоксилат (2,2 г, 9,17 ммоль) и карбонат цезия (3,6 г, 11,0 ммоль). Затем добавляли йодметан (1,56 г, 11,0 ммоль) и смесь нагревали при 70°С в течение 2 часов и подвергали колоночной хроматографии с получением этил-5-хлор-3-метокси-1Н-индазол-1-карбоксилата (0,8 г, 30%).
Стадия 5: получение 5-хлор-3-метокси-1Н-индазола

Этил 5-хлор-3-метокси-1Н-индазол-1-карбоксилат (610 мг, 2,40 ммоль), гидроксид натрия (3,6 мл, 1 N) и этанол (20 мл) перемешивали при комнатной температуре в течение 2 часов. рН доводили до концентрированной соляной кислоты и затем продукт подвергали колоночной хроматографии с получением 5-хлор-3-метокси-1Н-индазола (360 мг, 60%).
1Н ЯМР (400 МГц, ДМСО) δ 12,14 (s, 1Н), 7,62 (s, 1Н), 7,37 (dd, J=27,3, 8,9 Гц, 2Н), 3,99 (s, 3Н);
MS m/z (ESI): 183 [M+H]+.
Стадии 6-10: получение N-(5-((5-хлор-4-(5-хлор-3-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Стадии с 6 по 10: способ получения N-(5-((5-хлор-4-(5-хлор-3-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 40.
1Н ЯМР (400 МГц, CD3OD) δ 8,41 (s, 1Н), 8,13 (d, J=7,7 Гц, 2Н), 7,61 (d, J=1,8 Гц, 1Н), 7,35 (d, J=9,0 Гц, 1Н), 6,98 (s, 1Н), 6,57-6,37 (m, 2Н), 5,87 (dd, J=8,2, 3,5 Гц, 1Н), 4,13 (s, 3Н), 3,99 (s, 3Н), 3,51 (s, 2Н), 3,30 (s, 2Н), 2,90 (s, 6Н), 2,73 (s, 3Н);
MS m/z (ESI): 585 [M+H]+.
Пример 109: получение N-(5-((4-(5-хлор-3-метокси-1Н-индазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5-хлор-3-метокси-1Н-индазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 108
1Н ЯМР (400 МГц, CD3OD) δ 8,62 (s, 1Н), 8,38-8,16 (m, 1Н), 8,09 (s, 1Н), 7,50 (s, 1Н), 7,30 (s, 1Н), 6,99 (s, 1Н), 6,48 (t, J=15,4 Гц, 2Н), 5,85 (d, J=11,5 Гц, 1Н), 4,05 (s, 3Н), 3,96 (s, 3Н), 3,52 (s, 2Н), 2,91 (s, 6Н), 2,75 (s, 3Н);
MS m/z (ESI): 619 [М+Н]+.
Пример 110: получение N-(5-((4-(5-хлор-3-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5-хлор-3-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)-акриламида был аналогичен Примеру 108.
1Н ЯМР (400 МГц, CD3OD) δ 8,20 (s, 2Н), 7,88 (s, 1Н), 7,58 (s, 1Н), 7,41 (d, J=8,8 Гц, 1Н), 7,30 (d, J=6,9 Гц, 1Н), 7,10 (s, 1Н), 6,66 (dd, J=16,9, 10,1 Гц, 1Н), 6,49 (d, J=16,9 Гц, 1Н), 5,87 (d, J=11,7 Гц, 1Н), 4,15 (s, 3Н), 3,96 (s, 3Н), 3,56 (d, J=5,7 Гц, 2Н), 3,40 (d, J=5,6 Гц, 2Н), 2,95 (s, 6Н), 2,81 (s, 3Н);
MS m/z (ESI): 551 [М+Н]+.
Пример 111: получение N-(5-((4-(3-циклопропил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Стадия 1: получение циклопропил-(2-фторфенил)метанола

2-фторбензальдегид (1,0 г, 8 ммоль) растворяли в THF (20 мл) и реакционный раствор охлаждали на ледяной бане. При трехкратной продувке азотом добавляли по каплям циклопропилмагнийбромид (32 мл, 16 ммоль). По завершении добавления, реакционную смесь постепенно нагревали до комнатной температуры, проводили реакцию в течение 16 ч, гасили 20 мл насыщенного водного раствора хлорида аммония и экстрагировали этилацетатом (50 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта, который затем очищали колоночной хроматографией с получением циклопропил-(2-фторфенил)метанола (800 мг, 62%).
Стадия 2: Получение циклопропил-(2-фторфенил)метанона

Циклопропил-(2-фторфенил)метанол (800 мг, 4,8 ммоль) растворяли в дихлорметане (20 мл) и затем добавляли окислитель Десса-Мартина. Реакцию проводили при комнатной температуре в течение 5,5 ч и реакцию гасили 20 мл насыщенного водного раствора бикарбоната натрия и 20 мл 10% водного раствора сульфита натрия. После перемешивания в течение 15 минут, раствор экстрагировали дихлорметаном (30 мл × 4). Органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта, который затем очищали колоночной хроматографией с получением циклопропил(2-фторфенил)метанона (430 мг, 54%).
Стадия 3: получение 3-циклопропил-1Н-индазола

Циклопропил(2-фторфенил)метанон (430 мг, 2,6 ммоль) растворяли в 10 мл гидразингидрата. Реакцию проводили при 120°С в течение 1 ч при микроволновом излучении. Реакционный раствор концентрировали с получением неочищенного продукта, который затем очищали колоночной хроматографией с получением 3-циклопропил-1Н-индазола (250 мг, 61%).
Стадии 4-9: получение N-(5-((4-(3-циклопропил-2Н-индазол-2-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)-акриламида

Способ получения N-(5-((4-(3-циклопропил-2Н-индазол-2-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)-акриламида был аналогичен Примеру 43.
1Н ЯМР (400 МГц, CD3OD) δ 8,43 (s, 1Н), 8,19 (d, J=6,3 Гц, 1Н), 7,91 (d, J=7,9 Гц, 1Н), 7,83 (s, 1Н), 7,52 (d, J=7,0 Гц, 2Н), 7,43 (t, J=7,4 Гц, 1Н), 7,10 (s, 1Н), 6,63 (dd, J=16,9, 10,2 Гц, 1Н), 6,46 (dd, J=16,9, 1,5 Гц, 1Н), 5,85 (dd, J=10,2, 1,4 Гц, 1Н), 3,94 (s, 3Н), 3,57 (t, J=5,5 Гц, 2Н), 3,39 (t, J=5,5 Гц, 2Н), 2,93 (s, 6Н), 2,82 (s, 3Н), 2,42-2,33 (m, 1Н), 1,24-1,18 (m, 4Н);
MS m/z (ESI): 527, 2 [М+Н]+.
Пример 112: получение N-(5-((4-(6-циано-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(6-циано-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 43.
1Н ЯМР (400 МГц, CD3OD) δ 9,01 (s, 1Н), 8,59 (d, J=0,7 Гц, 1Н), 8,40 (d, J=6,5 Гц, 1Н), 8,10-8,02 (m, 2Н), 7,67 (dd, J=8,2, 1,2 Гц, 1Н), 7,62 (d, J=6,5 Гц, 1Н), 7,14 (s, 1Н), 6,60 (dd, J=16,9, 10,2 Гц, 1Н), 6,36 (dd, J=16,9, 1,3 Гц, 1Н), 5,82 (dd, J=10,3, 1,5 Гц, 1Н), 3,98 (s, 3Н), 3,57 (t, J=6,0 Гц, 2Н), 3,39 (dd, J=11,1, 5,2 Гц, 2Н), 2,94 (s, 6Н), 2,81 (s, 3Н);
MS m/z (ESI): 512, 2 [M+H]+.
Пример 113: получение N-(5-((4-(3-циклопропил-5-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(3-циклопропил-5-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 111.
1Н ЯМР (400 МГц, CD3OD) δ 8,18 (dd, J=40,0, 6,7 Гц, 2Н), 7,85 (s, 1Н), 7,40 (d, J=7,1 Гц, 1Н), 7,29 (d, J=2,3 Гц, 1Н), 7,08 (d, J=10,5 Гц, 2Н), 6,64 (dd, J=16,9, 10,1 Гц, 1Н), 6,47 (dd, J=16,9, 1,6 Гц, 1Н), 5,86 (dd, J=10,2, 1,6 Гц, 1Н), 3,95 (s, 3Н), 3,89 (s, 3Н), 3,57 (t, J=5,7 Гц, 2Н), 3,39 (t, J=5,6 Гц, 2Н), 2,94 (s, 6Н), 2,81 (s, 3Н), 2,35-2,26 (m, 1Н), 1,21-1,15 (m, 4Н);
MS m/z (ESI): 557, 3 [М+Н]+.
Пример 114: получение N-(5-((4-(3-циклопропил-5-метокси-1Н-индазол-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Стадии 1-3: Получение 3-циклопропил-5-метокси-1Н-индазола

Способ получения 3-циклопропил-5-метокси-1Н-индазола был аналогичен Примеру 111.
Стадии 4-8: получение N-(5-((4-(3-циклопропил-5-метокси-1Н-индазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(3-циклопропил-5-метокси-1Н-индазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 40.
1Н ЯМР (400 МГц, CD3OD) δ 8,70 (s, 1Н), 8,19 (s, 1Н), 8,03 (s, 1Н), 7,30 (d, J=2,3 Гц, 1Н), 7,09-6,97 (m, 2Н), 6,47 (d, J=5,6 Гц, 2Н), 5,91-5,81 (m, 1Н), 3,97 (s, 3Н), 3,90 (s, 3Н), 3,54 (t, J=5,6 Гц, 2Н), 3,31 (d, J=6,0 Гц, 2Н), 2,90 (s, 6Н), 2,76 (s, 3Н), 2,28 (ddd, J=13,2, 6,2, 3,8 Гц, 1Н), 1,11 (dt, J=4,0, 2,8 Гц, 4Н);
MS m/z (ESI): 625, 3 [М+Н]+.
Пример 115: получение N-(5-((5-хлор-4-(3-циклопропил-5-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Стадии 1-3: Получение 3-циклопропил-5-метокси-1Н-индазола

Способ получения 3-циклопропил-5-метокси-1Н-индазола был аналогичен Примеру 111.
Стадии 4-8: получение N-(5-((5-хлор-4-(3-циклопропил-5-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((5-хлор-4-(3-циклопропил-5-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 40.
1Н ЯМР (400 МГц, CD3OD) δ 8,47 (s, 1Н), 8,18 (s, 1Н), 8,05 (d, J=9,1 Гц, 1Н), 7,28 (d, J=2,3 Гц, 1Н), 7,05 (dd, J=9,1, 2,4 Гц, 1Н), 6,97 (s, 1Н), 6,44 (dd, J=5,8, 4,1 Гц, 2Н), 5,85 (dd, J=8,4, 3,4 Гц, 1Н), 3,99 (s, 3Н), 3,90 (s, 3Н), 3,51 (t, J=5,6 Гц, 2Н), 3,32-3,26 (m, 2Н), 2,88 (s, 6Н), 2,72 (s, 3Н), 2,35-2,26 (m, 1Н), 1,13 (dq, J=4,4, 2,4 Гц, 4Н);
MS m/z (ESI): 591, 3 [М+Н]+.
Пример 116:
получение N-(5-((5-хлор-4-(5-циано-3-пропил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)-акриламида

Стадия 1: получение 1-(5-бром-2-фторфенил)бутан-1-ола

5-бром-2-фторбензальдегид (5,0 г, 24,6 ммоль) растворяли в THF (30 мл) и реакционный раствор охлаждали на ледяной бане. При трехкратной продувке азотом, по каплям добавляли пропилмагнийбромид (25 мл, 49,3 ммоль). По завершении добавления, реакционную смесь постепенно нагревали до комнатной температуры и проводили реакцию в течение 16 ч, затем гасили 30 мл насыщенного водного раствора хлорида аммония и экстрагировали этилацетатом (50 мл × 4). Органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта, который затем очищали колоночной хроматографией с получением 1-(5-бром-2-фторфенил)бутан-1-ола (2,2 г, 36%).
Стадия 2: Получение 1-(5-бром-2-фторфенил)бутан-1-она

1-(5-бром-2-фторфенил)бутан-1-ол (2,2 г, 8,9 ммоль) растворяли в 50 мл дихлорметана и добавляли окислитель хлорхромат пиридиния (РСС) (3,8 г, 17,8 ммоль). После того, как реакцию проводили в течение 16 ч при комнатной температуре, реакционную смесь фильтровали через целит. Органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта, который затем очищали колоночной хроматографией с получением 1-(5-бром-2-фторфенил)бутан-1-она (1,5 г, 71%).
Стадия 3: получение (1-(5-бром-2-фторфенил)бутилиден)гидразина

1-(5-бром-2-фторфенил)бутан-1-он (1,0 г, 4,1 ммоль) растворяли в 20 мл гидразингидрата и реакцию проводили при температуре 130°С при микроволновом излучении в течение 5 часов. Реакционный раствор концентрировали и неочищенный продукт очищали колоночной хроматографией с получением (1-(5-бром-2-фторфенил)бутилиден)гидразина (800 мг, 80%).
Стадия 4: получение 5-бром-3-пропил-1Н-индазола

(1-(5-бром-2-фторфенил)бутилиден)гидразин (600 мг, 2,3 ммоль) растворяли в 10 мл N-метилпирролидона и проводили реакцию при 150° при микроволновом излучении в течение 1 часа. Добавляли 20 мл воды и реакционный раствор экстрагировали этилацетатом (30 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта, который затем очищали колоночной хроматографией с получением 5-бром-3-пропил-1Н-индазола (380 мг, 69%).
Стадия 5: получение 3-пропил-1Н-индазол-5-карбонитрила

5-бром-3-пропил-1Н-индазол (380 мг, 1,6 ммоль), цианид цинка (223 мг, 1,9 ммоль), трис(дибензилиденацетон)дипалладий (140 мг, 0,16 ммоль) и 2-дициклогексилфосфино-2'4',6'-триизопропилбифенил (150 мг, 0,32 ммоль) растворяли в N,N-диметилформамиде (10 мл). Реакцию проводили при 150°С при микроволновом излучении в течение 1 часа и затем добавляли 20 мл насыщенного водного раствора хлорида натрия. Реакционный раствор экстрагировали этилацетатом (30 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением сырого продукта, который затем очищали колоночной хроматографией с получением 3-пропил-1Н-индазол-5-карбонитрила (110 мг, 38%).
Стадии 6-10: получение N-(5-((5-хлор-4-(5-циано-3-пропил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Стадии 6-10: способ получения N-(5-((5-хлор-4-(5-циано-3-пропил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 40.
1Н ЯМР (400 МГц, CD3OD) δ 8,60 (s, 1Н), 8,33 (s, 1Н), 8,23 (d, J=8,8 Гц, 1Н), 8,19 (s, 1Н), 7,62 (d, J=8,7 Гц, 1Н), 6,98 (s, 1Н), 6,51 (dd, J=16,9, 1,5 Гц, 1Н), 6,40 (dd, J=17,0, 9,9 Гц, 1Н), 5,91 (dd, J=10,0, 1,5 Гц, 1Н), 4,01 (s, 3Н), 3,50 (t, J=5,5 Гц, 2Н), 3,31-3,24 (m, 2Н), 3,04 (t, J=7,4 Гц, 2Н), 2,89 (s, 6Н), 2,71 (s, 3Н), 1,91 (dd, J=14,8, 7,4 Гц, 2Н), 1,07 (t, J=7,4 Гц, 3Н);
MS m/z (ESI): 588, 3 [М+Н]+.
Пример 117: получение N-(5-((4-(5-циано-3-этил-1Н-индазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Стадия 1: получение 3-бром-1Н-индазол-5-карбонитрила

1Н-индазол-5-карбонитрил (544 мг, 3,8 ммоль), N-бромсукцинимид (NBS) (812 мг, 4,6 ммоль) и DMF (10 мл) добавляли в 100 мл круглодонную колбу. Под защитой N2 смесь перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали с получением сырого продукта, который растворяли в 100 мл DCM, промывали 50 мл насыщенного водного раствора бикарбоната натрия, воды и насыщенного солевого раствора, соответственно. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Полученный фильтрат концентрировали с получением 3-бром-1Н-индазол-5-карбонитрила (750 мг, 89%).
Стадия 2: получение 3-бром-1-((2-(триметилсилил)этокси)метил)-1Н-индазол-5-карбонитрила

3-бром-1Н-индазол-5-карбонитрил (710 мг, 3,2 ммоль) и THF (15 мл) добавляли в 100 мл круглодонную колбу, и по каплям добавляли SEM-Cl (640 мг, 3,8 ммоль) на бане с ледяной водой. Реакцию проводили при 0°С в течение 2 ч и затем гасили насыщенным водным раствором хлорида аммония (1 мл). Реакционный раствор концентрировали с получением неочищенного продукта, который затем очищали колоночной хроматографией (60% DCM/петролейный эфир) с получением 3-бром-1-((2-(триметилсилил)этокси)метил)-1Н-индазол-5-карбонитрила (680 мг, 61%).
Стадия 3: получение 3-этил-1-((2-(триметилсилил)этокси)метил)-1Н-индазол-5-карбонитрила

3-бром-1-((2-(триметилсилил)этокси)метил)-1Н-индазол-5-карбонитрил (669 мг, 1,9 ммоль), этилбороновую кислоту (281 мг, 3,8 ммоль), K3PO4 (1,2 г, 5,7 ммоль) и PCy3 (213 мг, 0,4 ммоль) добавляли в 100 мл круглодонную колбу. После трехкратной продувки азотом, добавляли Pd(OAc)2 (85 мг, 0,2 ммоль) и реакцию проводили при 100°С в течение 2 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли 100 мл этилацетата и промывали водой (50 мл × 2). Органическую фазу концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией с получением 3-этил-1-((2-(триметилсилил)этокси)метил)-1Н-индазол-5-карбонитрила (605 мг, 100%).
MS m/z (ESI): 302,2 [М+Н]+.
Стадия 4: получение 3-этил-1Н-индазол-5-карбонитрила

3-этил-1-((2-(триметилсилил)этокси)метил)-1Н-индазол-5-карбонитрил (603 мг, 1,9 ммоль), 1 М гидроксид тетра-бутиламмония/THF (30 мл) и этилендиамин (240 мг) добавляли в 100 мл круглодонную колбу и реакцию проводили при 70°С в течение 2 часов. Реакционный раствор концентрировали с получением неочищенного продукта. Добавляли 100 мл этилацетата и реакционный раствор промывали водой (50 мл × 2). Органическую фазу концентрировали и полученный остаток очищали колоночной хроматографией с обращенной фазой (25% ацетонитрил/вода) с получением 3-этил-1Н-индазол-5-карбонитрила (170 мг, 50%).
Стадии 5-8: получение N-(5-((4-(5-циано-3-этил-1Н-индазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Этапы 5-8: способ получения N-(5-((4-(5-циано-3-этил-1Н-индазол-1-ил)-5-(трифторметил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид был аналогичен Примеру 106.
1Н ЯМР (400 МГц, CD3OD) δ 8,80 (s, 1Н), 8,39 (s, 1Н), 8,27 (d, J=0,7 Гц, 1Н), 8,12 (s, 1Н), 7,63 (s, 1Н), 7,02 (s, 1Н), 6,56-6,44 (m, 2Н), 5,93-5,87 (m, 1Н), 4,00 (s, 3Н), 3,53 (t, J=5,7 Гц, 2Н), 3,36-3,33 (m, 2Н), 3,03 (q, J=7,5 Гц, 2Н), 2,92 (s, 6Н), 2,75 (s, 3Н), 1,44 (t, J=7,5 Гц, 3Н);
MS m/z (ESI): 608, 3 [М+Н]+.
Пример 118:
получение N-(5-((5-хлор-4-(5-циано-3-этил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)-акриламида

Способ получения N-(5-((5-хлор-4-(5-циано-3-этил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-((2-диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид был аналогичен Примеру 117.
1Н ЯМР (400 МГц, CD3OD) δ 8,44 (s, 1Н), 8,18 (s, 1Н), 8,09 (d, J=11,2 Гц, 2Н), 7,49 (d, J=8,7 Гц, 1Н), 6,86 (s, 1Н), 6,34 (qd, J=16,9, 5,7 Гц, 2Н), 5,79 (dd, J=9,9, 1,6 Гц, 1Н), 3,89 (s, 3Н), 3,39 (t, J=5,4 Гц, 2Н), 3,18 (d, J=5,3 Гц, 2Н), 2,95 (q, J=7,5 Гц, 2Н), 2,78 (s, 6Н), 2,60 (s, 3Н), 1,33 (t, J=7,5 Гц, 3Н);
MS m/z (ESI): 574, 3 [М+Н]+.
Пример 119:
получение N-(5-((4-(1-циклопропил-1Н-индазол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Стадия 1: получение (2-фторфенил)(2-(метилтио)пиримидин-4-ил)метанона

4-хлор-2-(метилтио)пиримидин (5,00 г, 31,1 ммоль), 2-фторбензальдегид (4,64 г, 37,4 ммоль) и [mmim] [I] (2,09 г, 9,33 ммоль) растворяли в 1,4-диоксане (70 мл), а затем добавляли частями NaH (1,74 г, 60%, 43,6 ммоль). Затем смесь перемешивали при 100°С в течение 1 часа. После охлаждения реакционный раствор разбавляли EtOAc. Органическую фазу промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали колоночной хроматографией с получением указанного в заголовке соединения (2-фторфенил)(2-(метилтио)пиримидин-4-ил)метанона (4,3 г, 56%).
1Н ЯМР (400 МГц, CDCl3): δ 8,71 (d, J=5,2 Гц, 1Н), 7,68 (m, 1Н), 7,51 (m, 1Н), 7,44 (d, J=5,2 Гц, 1Н), 7,20 (m, 1Н), 7,07 (m, 1Н), 2,38 (s, 3Н);
MS m/z (ESI): 249,1 [М+Н]+.
Стадия 2: получение 1-циклопропил-3-(2-(метилтио)пиримидин-4-ил)-1Н-индазола

(2-фторфенил)(2-(метилтио)пиримидин-4-ил)метанон (1,3 г, 5,24 ммоль) и гидрохлорид циклопропилгидразина (800 мг, 7,33 ммоль) смешивали в этаноле. Смесь нагревали до температуры дефлегмации в течение 2 часов, охлаждали и концентрировали. Полученный неочищенный продукт растворяли в 50 мл DMF и добавляли частями гидрид натрия (500 мг, 12,5 ммоль), а затем реакционный раствор перемешивали при 80°С в течение 2 часов. После охлаждения добавляли воду, затем осаждали твердое вещество. Твердое вещество очищали колоночной хроматографией с получением указанного в заголовке соединения 1-циклопропил-3-(2-(метилтио)пиримидин-4-ил)-1Н-индазола (140 мг, выход двух стадий: 10%).
MS m/z (ESI): 283,1 [М+Н]+.
Стадия 3: получение 1-циклопропил-3-(2-(метилсульфонил)пиримидин-4-ил)-1Н-индазола

mCPBA (231 мг, 70%, 1,00 ммоль) добавляли в одной части к раствору 1-циклопропил-3-(2-(метилтио)пиримидин-4-ил)-1Н-индазола (135 мг, 0,478 ммоль) в дихлорметане (3 мл) на бане с ледяной водой. Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 2 часов. Реакционный раствор дважды промывали насыщенным водным раствором бикарбоната натрия, промывали один раз насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали с получением указанного в заголовке соединения 1-циклопропил-3-(2-(метилсульфонил)пиримидин-4-ил)-1Н-индазола (185 мг, 100%).
MS m/z (ESI): 315,1 [М+Н]+.
Стадия 4: получение 4-(1-циклопропил-1Н-индазол-3-ил)-N-(4-фтор-2-метокси-5-нитрофенил)пиримидин-2-амина

Водный раствор муравьиной кислоты (0,5 мл, 85%) добавляли к раствору 4-фтор-2-метокси-5-нитроанилина (1,0 г, 5,4 ммоль) в толуоле (5 мл) и смесь нагревали до температуры дефлегмации в течение ночи. Реакционный раствор концентрировали путем ротационного испарения и использовали непосредственно в следующей реакции.
Вышеуказанный неочищенный продукт (180 мг, 0,840 ммоль) растворяли в 2 мл DMF и добавляли NaH (41 мг, 1,68 ммоль) на бане с ледяной водой. Смесь перемешивали при этой температуре в течение 30 минут. Затем добавляли раствор 1-циклопропил-3-(2-(метилсульфонил)пиримидин-4-ил)-1Н-индазола (184 мг, 0,588 ммоль) в DMF (2 мл) и смесь перемешивали при комнатной температуре в течение ночи. Реакционный раствор перемешивали в течение еще 30 минут после добавления 0,5 мл воды. Затем добавляли 10 мл воды и реакционный раствор фильтровали. Полученное твердое вещество очищали колоночной хроматографией с получением 4-(1-циклопропил-1Н-индазол-3-ил)-N-(4-фтор-2-метокси-5-нитрофенил)пиримидин-2-амина (200 мг, 81%).
MS m/z (ESI): 421,1 [М+Н]+.
Стадия 5: получение N1-(4-(1-циклопропил-1Н-индазол-3-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамина

4-(1-циклопропил-1Н-индазол-3-ил)-N-(4-фтор-2-метокси-5-нитрофенил)пиримидин-2-амин (200 мг, 0,48 ммоль), триметилэтилендиамин (58,0 мг, 0,57 Ммоль) и DIPEA (0,24 мл, 1,43 ммоль) растворяли в 2 мл DMA и смесь перемешивали при 90°С в течение 2 часов. После охлаждения реакционный раствор разбавляли EtOAc, промывали несколько раз насыщенным солевым раствором, сушили над безводным сульфатом натрия, концентрировали и очищали препаративной тонкослойной хроматографией с получением указанного в заголовке соединения N1-(4-(1-циклопропил-1Н-индазол-3-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамина (40 мг, 17%).
MS m/z (ESI): 503,2 [М+Н]+.
Стадия 6: получение N4-(4-(1-циклопропил-1Н-индазол-3-ил)пиримидин-2-ил)-N1-(2-(диметиламино)этил)-5-метокси-N1-метилбензол-1,2,4-триамина

N1-(4-(1-циклопропил-1Н-индазол-3-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамин (40 мг, 0,080 ммоль), восстановленный порошок железа (44 мг, 0,80 ммоль) и хлорид аммония (3,4 мг, 0,064 ммоль) смешивали в смеси 6 мл этанола и 2 мл воды, смесь перемешивали при 70°С в течение ночи. После охлаждения реакционный раствор фильтровали через целит, концентрировали и очищали препаративной тонкослойной хроматографией с получением указанного в заголовке соединения. N4-(4-(1-циклопропил-1Н-индазол-3-ил)пиримидин-2-ил)-N1-(2-(диметиламино)этил)-5-метокси-N1-метилбензол-1,2,4-триамина (19 мг, 50%).
MS m/z (ESI): 473,3 [M+H]+.
Стадия 7: получение N-(5-((4-(1-циклопропил-1Н-индазол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

В ацетоновой бане с сухим льдом, TEA (0,15 мл, 1,1 ммоль) и раствор акрилоилхлорида (0,045 мл, 0,56 ммоль) в THF (0,5 мл) последовательно добавляли по каплям к раствору N4-(4-(1-циклопропил-1Н-индазол-3-ил)пиримидин-2-ил)-N1-(2-(диметиламино)этил)-5-метокси-N1-метилбензол-1,2,4-триамина (19 мг, 0,040 ммоль) в THF (2 мл). Затем смесь перемешивали при этой температуре в течение 5 минут и реакцию гасили 1 мл метанола. После концентрирования растворителя, полученный остаток очищали препаративной тонкослойной хроматографией с получением указанного в заголовке соединения N-(5-((4-(1-циклопропил-1Н-индазол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида (10 мг, 47%).
1Н ЯМР (400 МГц, CDCl3): δ 10,1 (br s, 1Н), 9,51 (s, 1Н), 8,61 (d, J=8,0 Гц, 1Н), 8,54 (d, J=5,2 Гц, 1Н), 7,60 (m, 3Н), 7,41 (m, 1Н), 7,22 (m, 1Н), 6,79 (s, 1Н), 6,38 (m, 2Н), 5,66 (m, 1Н), 3,89 (s, 3Н), 3,68 (m, 1Н), 2,95 (m, 2Н), 2,72 (s, 3Н), 2,40 (m, 8Н), 0,88 (m, 4H);
MS m/z (ESI): 527, 3 [M+H]+.
Пример 120: получение N-(5-((4-(1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 59.
1Н ЯМР (400 МГц, CD3OD) δ 9,32 (s, 1Н), 8,47 (d, J=5,8 Гц, 1Н), 8,25 (s, 2Н), 7,71 (dd, J=6,2, 2,8 Гц, 1Н), 7,39 (dd, J=6,1, 3,2 Гц, 2Н), 7,27 (d, J=5,8 Гц, 1Н), 6,93 (s, 1Н), 6,52 (dd, J=16,9, 10,0 Гц, 1Н), 6,40 (dd, J=16,9, 1,7 Гц, 1Н), 5,78 (dd, J=10,0, 1,7 Гц, 1Н), 3,89 (s, 3Н), 3,42 (t, J=5,5 Гц, 2Н), 3,28-3,25 (m, 2Н), 2,83 (s, 6Н), 2,67 (s, 3Н);
MS m/z (ESI): 487, 3 [М+Н]+.
Пример 121: получение N-(2((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-(2-метоксиэтокси)-1Н-индазол-1-ил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(2((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(5-(2-метоксиэтокси)-1Н-индазол-1-ил)пиримидин-2-ил)амино)фенил)-акриламида был аналогичен Примеру 105.
1Н ЯМР (400 МГц, CD3OD) δ 8,37 (s, 1Н), 8,27 (s, 1Н), 7,84 (s, 1Н), 7,59 (s, 1Н), 7,32 (s, 1Н), 7,15 (s, 1Н), 7,09 (s, 1Н), 6,70-6,53 (m, 1Н), 6,50 (s, 1Н), 5,88 (s, 1Н), 4,18 (s, 2Н), 3,96 (s, 3Н), 3,80 (s, 2Н), 3,57 (s, 2Н), 3,46 (s, 3Н), 3,38 (s, 2Н), 2,94 (s, 6Н), 2,81 (s, 3Н);
MS m/z (ESI): 561 [М+Н]+.
Пример 122: получение (E)-N-(5-((4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)-4-(диметиламино)бут-2-енамида

Способ получения (E)-N-(5-((4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)-4-(диметиламино)бут-2-енамида был аналогичен Примеру 22.
1Н ЯМР (400 МГц, CDCl3) δ 9,70 (d, J=17,4 Гц, 2Н), 8,49 (d, J=19,3 Гц, 1Н), 8,32 (d, J=5,3 Гц, 1Н), 8,05 (m, 1Н), 7,59 (m, 2Н), 7,09 (d, J=5,3 Гц, 1Н), 6,90 (m, 1Н), 6,68 (s, 1Н), 6,46 (s, 1Н), 3,81 (s, 3Н), 3,39 (ddd, J=10,8, 7,1, 3,8 Гц, 1Н), 3,16 (d, J=6,0 Гц, 2Н), 2,89 (m, 2Н), 2,63 (s, 3Н), 2,46 (s, 2Н), 2,31 (d, J=23,0 Гц, 12Н), 1,16 (m, 2Н), 1,01 (m, 2Н);
MS m/z (ESI): 583, 7 [М+Н]+.
Пример 123: получение N-(5-((4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метокси-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)акриламида

Способ получения N-(5-((4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метокси-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)акриламида был аналогичен Примеру 22.
1Н ЯМР (400 МГц, CDCl3) δ 9,66 (s, 1Н), 9,49 (s, 1Н), 8,42 (s, 1Н), 8,32 (d, J=5,3 Гц, 1Н), 8,04 (d, J=6,9 Гц, 1Н), 7,28 (m, 1Н), 7,68-7,45 (m, 2Н), 7,10 (d, J=5,3 Гц, 1Н), 6,63 (s, 1Н), 6,37 (dd, J=16,8, 1,8 Гц, 1Н), 5,63 (dd, J=10,2, 1,8 Гц, 1Н), 3,80 (s, 3Н), 3,47-3,16 (m, 1Н), 3,07 (s, 2Н), 2,82 (s, 3Н), 2,62 (s, 4Н), 1,92 (s, 4Н), 1,30 (m, 2Н), 1,18 (m, 2Н), 1,06-0,96 (m, 2Н);
MS m/z (ESI): 552, 7 [М+Н]+.
Пример 124: получение N-(5-((4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метокси-2-(метил(2-морфолиноэтил)амино)фенил)акриламида

Способ получения N-(5-((4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метокси-2-(метил(2-морфолиноэтил)амино)фенил)акриламида был аналогичен Примеру 22.
1Н ЯМР (400 МГц, CDCl3) δ 9,72 (s, 1Н), 9,21 (s, 1Н), 8,49 (s, 1Н), 8,33 (d, J=5,3 Гц, 1Н), 8,04 (d, J=7,1 Гц, 1Н), 7,65-7,42 (m, 2Н), 7,34-7,13 (m, 2Н), 7,11 (d, J=5,3 Гц, 1Н), 6,70 (s, 1Н), 6,40 (s, 2Н), 5,77-5,51 (m, 1Н), 3,80 (s, 3Н), 3,65 (s, 4Н), 3,50-3,20 (m, 1Н), 3,02-2,79 (m, 2Н), 2,59 (s, 3Н), 2,32 (d, J=37,1 Гц, 6Н), 1,20-1,08 (m, 2Н), 1,08-0,95 (m, 2Н);
MS m/z (ESI): 568, 6 [М+Н]+.
Пример 125: получение N-(5-((4-(5,6-дигидро-4Н-пирроло[3,2,1-ij]хинолин-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(5,6-дигидро-4Н-пирроло[3,2,1-ij]хинолин-1-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 22.
1Н ЯМР (400 МГц, CD3OD): δ 8,47 (s, 1Н), 7,99 (m, 3Н), 7,36 (d, J=6,8 Гц, 1Н), 7,12 (t, J=6,8 Гц, 1Н), 7,07 (s, 1Н), 7,00 (d, J=7,2 Гц, 1Н), 6,58 (m, 1Н), 6,45 (m, 1Н), 5,85 (m, 1Н), 4,30 (t, J=6,0 Гц, 2Н), 3,95 (s, 3Н), 3,54 (t, J=6,0 Гц, 2Н), 3,36 (t, J=5,6 Гц, 2Н), 3,00 (t, J=5,6 Гц, 2Н), 2,90 (s, 6Н), 2,80 (s, 3Н), 2,25 (m, 2Н);
MS m/z (ESI): 526, 2 [М+Н]+.
Пример 126: получение N-(5-((4-(1-аллил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метокси-2-(метил(2-(метиламино)этил)амино)фенил)акриламида

Стадия 1: получение трет-бутил{2-[(4-{[4-(1Н-индол-3-ил)пиримидин-2-ил]амино}-5-метокси-2-нитрофенил)(метил)амино]этил}метилкарбамата

N-(4-фтор-2-метокси-5-нитрофенил)-4-(1Н-индол-3-ил)пиримидин-2-амин (1,3 г, 3,43 ммоль), DIPEA (2,2 г, 7,14 ммоль) и трет-бутил метил(2-(метиламино)этил)карбамат (0,77 г, 4,12 ммоль) растворяли в DMA (15 мл). После нагревания до 100°С в течение ночи реакционный раствор концентрировали и очищали колоночной хроматографией с получением трет-бутил{2-[(4-{[4-(1Н-индол-3-ил)пиримидин-2-ил]амино}-5-метокси-2-нитрофенил)(метил)амино]этил}метилкарбамата (1,9 г, 80%).
Стадия 2: получение трет-бутил 3-(2-((трет-бутоксикарбонил)(4-((2-((трет-бутоксикарбонил)(метил)амино)этил)(метил)амино)-2-метокси-5-нитрофенил)амино)пиримидин-4-ил)-1Н-индол-1-карбоксилата

Трет-бутил{2-[(4-{[4-(1Н-индол-3-ил)пиримидин-2-ил]амино}-5-метокси-2-нитрофенил)(метил)амино]этил}метилкарбамат (1,9 г, 3,34 моль), Boc2O (1,87 г, 8,58 ммоль) и DMAP (84 мг, 0,69 ммоль) добавляли к тетрагидрофурану (30 мл). Реакционный раствор перемешивали при 50°С в течение ночи и концентрировали с получением 2,0 г твердого вещества желтого цвета, которое использовали непосредственно в следующей реакции.
Стадия 3: получение трет-бутил(2-((4-((4-(1Н-индол-3-ил)пиримидин-2-ил)(трет-бутоксикарбонил)амино)-5-метокси-2-нитрофенил)(метил)амино)этил)-(метил)карбамата

Трет-бутил-3-(2-((трет-бутоксикарбонил)(4-((2-((трет-бутоксикарбонил)(метил)амино)этил)(метил)амино)-2-метокси-5-нитрофенил)пиримидин-4-ил)-1Н-индол-1-карбоксилата (2,0 г, 2,678 ммоль) растворяли в метаноле (20 мл). Затем добавляли метоксид натрия (29 мг, 0,535 ммоль). После нагревания смеси до 50°С в течение приблизительно 2 часов, реакцию гасили водой. Реакционный раствор концентрировали, экстрагировали дихлорметаном, концентрировали и сушили с получением трет-бутил (2-((4-((4-(1Н-индол-3-ил)пиримидин-2-ил)(трет-бутоксикарбонил)амино)-5-метокси-2-нитрофенил)(метил)амино)этил)(метил)-карбамата (2,3 г, 90%).
Стадия 4: получение трет-бутил 2-((4-((4-(1-аллил-1Н-индол-3-ил)пиримидин-2-ил)(трет-бутоксикарбонил)амино)-5-метокси-2-нитрофенил)(метил)амино)этил(метил)карбамата

Трет-бутил (2-((4-((4-(1Н-индол-3-ил)пиримидин-2-ил)(трет-бутоксикарбонил)амино)-5-метокси-2-нитрофенил)(метил)амино)этил)(метил)карбамат (200 мг, 0,309 ммоль) и NaH (11 мг, 0,46 ммоль) добавляли к THF (10 мл). Затем добавляли винилбромид (55 мг, 0,46 ммоль) и смесь перемешивали при комнатной температуре в течение 3 часов. Реакцию гасили водой. Реакционный раствор экстрагировали дихлорметаном, концентрировали и сушили с получением 200 мг трет-бутил-2-((4-((4-(1-аллил-1Н-индол-3-ил)пиримидин-2-ил)(трет-бутоксикарбонил)амино)-5-метокси-2-нитрофенил)-(метил)амино)этил)(метил)карбамата в виде твердого вещества желтого цвета, которое непосредственно использовали на следующей стадии.
Стадия 5: получение трет-бутил (2-((4-((4-(1-аллил-1Н-индол-3-ил)пиримидин-2-ил)(трет-бутоксикарбонил)амино-2-амино-5-метоксифенил)(метил)амино)этил)(метил)карбамата

Сырой продукт, полученный на предыдущей стадии (2,4 г, 3,45 ммоль), порошок железа (2 г, 34,5 ммоль) и хлорид аммония (3,7 г, 70 ммоль) добавляли в смесь этанола (60 мл) и воды (20 мл). После нагревания при 60°С в течение ночи реакционный раствор фильтровали, концентрировали, экстрагировали дихлорметаном и очищали колоночной хроматографией с получением трет-бутил(2-((4-((4-(1-аллил-1Н-индол-3-ил)пиримидин-2-ил)(трет-бутоксикарбонил)амино)-2-амино-5-метоксифенил)(метил)амино)этил)метил)карбамата (1 г, 50%).
1Н ЯМР (400 МГц, ДМСО) δ 8,45 (d, J=5,4 Гц, 1Н), 8,36 (s, 1Н), 8,08 (d, J=8,0 Гц, 1Н), 7,48 (dd, J=9,3, 6,9 Гц, 2Н), 7,17 (t, J=7,1 Гц, 1Н), 7,00 (t, J=7,4 Гц, 1Н), 6,79 (s, 1H), 6,47 (s, 1H), 6,09-5,97 (m, 1H), 5,19 (dd, J=10,3, 1,4 Гц, 1H), 5,06 (dd, J=17,1, 1,5 Гц, 1H), 4,90 (d, J=5,3 Гц, 2H), 4,37 (s, 2H), 3,65 (s, 3Н), 3,36 (d, J=6,6 Гц, 2H), 2,96 (t, J=6,7 Гц, 2H), 2,75 (d, J=12,4 Гц, 3Н), 2,66 (d, J=11,3 Гц, 3Н), 1,41 (s, 18H);
MS m/z (ESI): 658 [M+H]+.
Стадия 6: получение трет-бутил(2-((2-акриламидо-4-((4-(1-аллил-1Н-индол-3-ил)пиримидин-2-ил)(трет-бутоксикарбонил)амино)-5-метоксифенил)(метил)-амино)этил)(метил)карбамата

Исходный материал, полученный на предыдущей стадии (1 г, 1,52 ммоль), и DIPEA (0,56 г, 4,56 ммоль) растворяли в тетрагидрофуране (100 мл). Реакционную систему охлаждали до -10°С и в колбу по каплям добавляли 2,3 мл раствора акрилоилхлорида в тетрагидрофуране (1 М). Реакционную смесь перемешивали в течение 30 минут и гасили 1 мл метанола. Реакционный раствор концентрировали, экстрагировали дихлорметаном и очищали колоночной хроматографией с получением трет-бутил(2-((2-акриламидо-4-((4-(1-аллил-1Н-индол-3-ил)-пиримидин-2-ил)(трет-бутоксикарбонил)амино)-5-метоксифенил)(метил)амино)-этил)(метил)карбамата (0,9 г, 90%).
1Н ЯМР (400 МГц, CDCl3) δ 8,73-8,39 (m, 3Н), 7,91 (s, 2Н), 7,29 (d, J=8,3 Гц, 2Н), 7,19 (t, J=7,6 Гц, 1Н), 7,01 (t, J=7,5 Гц, 1Н), 6,87 (s, 1Н), 6,36 (s, 2Н), 6,00 (m, 1Н), 5,69 (s, 1Н), 5,26 (dd, J=10,3, 1,0 Гц, 1Н), 5,19-5,10 (m, 1Н), 4,77 (d, J=5,4 Гц, 2Н), 3,80 (s, 3Н), 3,44 (s, 2Н), 3,00 (d, J=25,1 Гц, 2Н), 2,84 (s, 3Н), 2,78 (s, 3Н), 1,48 (s, 9Н), 1,47 (s, 9Н);
MS m/z (ESI): 712[М+Н]+.
Стадия 7: получение N-(5-((4-(1-аллил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метокси-2-(метил(2-(метиламино)этил)амино)фенил)акриламида

Сырой материал, полученный на предыдущей стадии (0,9 г), растворяли в растворе дихлорметана (50 мл), содержащем 20% трифторуксусной кислоты по объему, и смесь перемешивали при комнатной температуре в течение 6 часов. После того, как ТСХ показывала завершение реакции, рН доводили до щелочной насыщенным водным раствором бикарбоната натрия. Затем реакционный раствор экстрагировали дихлорметаном и концентрировали с получением N-(5-((4-(1-аллил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метокси-2-(метил(2-(метиламино)этил)амино)фенил)акриламида (535 мг, 83%).
1Н ЯМР (400 МГц, CDCl3) δ 9,74 (s, 2Н), 8,99-8,92 (m, 1Н), 8,38 (d, J=5,3 Гц, 1Н), 8,11 (s, 1Н), 7,68 (s, 1Н), 7,38 (s, 1Н), 7,26-7,23 (m, 1Н), 7,19 (d, J=5,3 Гц, 1Н), 6,71 (s, 1Н), 6,67-6,57 (m, 1Н), 6,40 (d, J=16,9 Гц, 1Н), 6,12-5,98 (m, 1Н), 5,69 (d, J=11,9 Гц, 1Н), 5,23-5,13 (m, 2Н), 4,97 (s, 2Н), 3,87 (s, 3Н), 2,96 (s, 2Н), 2,70 (s, 2Н), 2,66 (s, 3Н), 2,45 (s, 3Н);
MS m/z (ESI): 512 [M+H]+.
Пример 127: получение N-(4-метокси-5-((4-(6-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-(метил-(2-(метиламино)этил)амино)фенил)-акриламида

Способ получения N-(4-метокси-5-((4-(6-метокси-1-метил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-(метил-(2-(метиламино)этил)амино)фенил)акриламида был аналогичен Примеру 126.
1Н ЯМР (400 МГц, CD3OD) δ 8,32 (s, 1Н), 8,07 (s, 1Н), 7,87 (d, J=6,9 Гц, 2Н), 7,25 (d, J=7,0 Гц, 1Н), 7,01-6,90 (m, 2Н), 6,77 (dd, J=8,8, 2,0 Гц, 1Н), 6,48 (dd, J=16,9, 10,1 Гц, 1Н), 6,34 (dd, J=17,0, 1,8 Гц, 1Н), 5,75 (dd, J=10,1, 1,8 Гц, 1Н), 3,92-3,70 (m, 9Н), 3,43-3,30 (m, 2Н), 3,17-3,05 (m, 2Н), 2,67 (d, J=9,0 Гц, 6Н);
MS m/z (ESI): 516, 2 [М+Н]+.
Пример 128: получение N-(5-((4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метокси-2-(метил(2-(метиламино)этил)амино)фенил)акриламида

Способ получения N-(5-((4-(1-циклопропил-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метокси-2-(метил(2-(метиламино)этил))амино)фенил)акриламида был аналогичен Примеру 126.
1Н ЯМР (400 МГц, CDCl3) δ 9,37 (s, 1Н), 9,29 (s, 1Н), 8,29 (d, J=5,3 Гц, 1Н), 8,18 (s, 1Н), 8,07 (d, J=7,2 Гц, 1Н), 7,61-7,40 (m, 2Н), 7,24-7,15 (m, 2Н), 7,06 (d, J=5,3 Гц, 1Н), 6,94 (dd, J=15,9, 9,9 Гц, 1Н), 6,45 (s, 1Н), 6,17 (d, J=16,9 Гц, 1Н), 5,58 (d, J=10,2 Гц, 1Н), 3,79 (s, 3Н), 3,39-3,15 (m, 1Н), 2,93 (s, 2Н), 2,65 (s, 2Н), 2,44 (s, 3Н), 2,26 (s, 3Н), 1,01 (d, J=5,2 Гц, 4Н);
MS m/z (ESI): 512, 6 [М+Н]+.
Пример 129: получение N-(4-метокси-2-(метил(2-(метиламино)этил)амино)-5-((4-(1-(проп-2-ин-1-ил)-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида

Способ получения N-(4-метокси-2-(метил(2-(метиламино)этил)амино)-5-((4-(1-(проп-2-ин-1-ил)-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 126.
1Н ЯМР (400 МГц, CDCl3) δ 9,90-9,83 (m, 1Н), 9,81-9,73 (m, 1Н), 9,25-9,12 (m, 1Н), 8,39 (d, J=5,3 Гц, 1H), 8,12-8,05 (m, 1Н), 7,70 (s, 1Н), 7,61-7,53 (m, 1Н), 7,30 (s, 2Н), 7,20 (d, J=5,3 Гц, 1H), 6,75 (s, 1Н), 6,62-6,44 (m, 2Н), 5,81-5,63 (m, 1Н), 5,30 (s, 1Н), 5,18 (s, 2Н), 3,88 (s, 3Н), 2,97-2,87 (m, 2Н), 2,69 (s, 3Н), 2,68-2,63 (m, 2Н), 2,47 (s, 3Н), 2,38 (s, 1Н);
MS m/z (ESI): 510[M+H]+.
Пример 130: получение N-(5-((5-хлор-4-(1-(оксетан-3-ил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((5-хлор-4-(1-(оксетан-3-ил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида был аналогичен Примеру 126.

1Н ЯМР (400 МГц, CD3OD) δ 8,74 (s, 1Н), 8,53 (s, 1Н), 8,30-8,23 (m, 2Н), 7,50 (d, J=8,3 Гц, 1Н), 7,17-7,10 (m, 1Н), 7,03 (t, J=7,2 Гц, 1Н), 6,86 (s, 1Н), 6,38 (dd, J=17,0, 10,2 Гц, 1Н), 6,17 (d, J=17,0 Гц, 1Н), 5,69-5,59 (m, 2Н), 5,10 (t, J=7,4 Гц, 2Н), 4,99-4,93 (m, 2H), 3,81 (s, 3Н), 3,02 (s, 2H), 2,59 (s, 3Н), 2,47 (s, 2H), 2,27 (s, 6H);
MS m/z (ESI): 576, 3 [M+H]+.
Пример 131: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1-(оксетан-3-ил)-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)-амино)фенил)акриламида

Способ получения N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1-(оксетан-3-ил)-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламида был аналогичен Примеру 130.
1Н ЯМР (400 МГц, CD3OD) δ 8,61 (s, 1Н), 8,33 (s, 1Н), 8,09 (d, J=8,0 Гц, 1Н), 7,98 (s, 1Н), 7,55 (d, J=8,3 Гц, 1Н), 7,15 (t, J=7,4 Гц, 1Н), 7,05 (t, J=7,5 Гц, 1Н), 6,87 (s, 1Н), 6,37-6,27 (m, 2Н), 5,76-5,64 (m, 2Н), 5,13 (t, J=7,4 Гц, 2Н), 4,95-4,86 (m, 2Н), 3,90 (s, 3Н), 3,39 (t, J=5,5 Гц, 2Н), 3,17 (t, J=5,5 Гц, 2Н), 2,75 (s, 6Н), 2,59 (s, 3Н);
MS m/z (ESI): 610,3 [М+Н]+.
Пример 132: получение N-(4-метокси-5-((4-(6-метокси-3-метил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-(метил-(2-(пирролидин-1-ил)этил)амино)фенил)-акриламида

Стадия 1: получение N-метил-2-(пирролидин-1-ил)этан-1-амина

Водный раствор (50 мл) 1-(2-хлорэтил)пирролидингидрохлорида (25 г, 0,147 ммоль) медленно, по каплям добавляли к водному раствору метиламина (114 мл). По завершении добавления, смесь перемешивали в течение 30 минут с последующим добавлением гидроксида натрия (46,25 г, 1,15 ммоль). Появлялся желтый супернатант. Реакционный раствор экстрагировали метил-трет-бутиловым эфиром, концентрировали при комнатной температуре и сушили в вакууме с получением N-метил-2-(пирролидин-1-ил)этан-1-амина (17 г, 90%).
Стадии 2-7: получение N-(4-метокси-5-((4-(6-метокси-3-метил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)-акриламида

Стадии 2-7: способ получения N-(4-метокси-5-((4-(6-метокси-3-метил-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)акриламида был аналогичен Примеру 54.
1Н ЯМР (400 МГц, CD3OD) δ 8,19 (d, J=7,0 Гц, 1Н), 8,05 (s, 1Н), 7,78-7,66 (m, 2Н), 7,58 (d, J=7,0 Гц, 1Н), 7,09 (dd, J=8,5, 2,4 Гц, 2Н), 6,62 (dd, J=16,9, 10,2 Гц, 1Н), 6,39 (dd, J=16,9, 1,5 Гц, 1Н), 5,85 (dd, J=10,2, 1,5 Гц, 1Н), 3,95 (s, 3Н), 3,87 (s, 3Н), 3,68-3,60 (m, 2Н), 3,56 (t, J=5,7 Гц, 2Н), 3,43 (t, J=5,7 Гц, 2Н), 3,13 (d, J=8,6 Гц, 2Н), 2,82 (s, 3Н), 2,58 (s, 3Н), 2,17 (d, J=6,0 Гц, 4Н);
MS m/z (ESI): 557 [М+Н]+.
Пример 133: получение N4-(4-(5-этокси-1Н-индазол-1-ил)пиримидин-2-ил)-5-метокси-N1-метил-N1-(2-(пирролидин-1-ил)этил)бензол-1,2,4-триамина

Способ получения N4-(4-(5-этокси-1Н-индазол-1-ил)пиримидин-2-ил)-5-метокси-N1-метил-N1-(2-(пирролидин-1-ил)этил)бензол-1,2,4-триамина был аналогичен Примеру 105.
1Н ЯМР (400 МГц, CD3OD) δ 8,38 (s, 1Н), 8,27 (d, J=6,6 Гц, 2Н), 7,76 (s, 1Н), 7,60 (d, J=6,9 Гц, 1Н), 7,29 (d, J=2,2 Гц, 1Н), 7,12 (d, J=8,9 Гц, 1Н), 7,08 (s, 1Н), 6,63-6,53 (m, 1Н), 6,43 (dd, J=16,9, 1,6 Гц, 1Н), 5,88 (dd, J=10,1, 1,5 Гц, 1Н), 4,11 (q, J=6,9 Гц, 2Н), 3,96 (s, 3Н), 3,65 (d, J=9,9 Гц, 2Н), 3,58 (t, J=5,6 Гц, 2Н), 3,43 (t, J=5,4 Гц, 2Н), 3,14 (d, J=9,9 Гц, 2Н), 2,83 (s, 3Н), 2,21 (t, J=6,6 Гц, 4Н), 1,45 (t, J=7,0 Гц, 3Н);
MS m/z (ESI): 557 [М+Н]+.
Пример 134: получение N-(4-метокси-5-((4-(5-(2-метоксиэтокси)-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)-акриламида

Способ получения N-(4-метокси-5-((4-(5-(2-метоксиэтокси)-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)-акриламид был аналогичен Примеру 105.
1Н ЯМР (400 МГц, CD3OD) δ 8,35 (s, 1Н), 8,28 (m, 2Н), 7,79 (s, 1Н), 7,55 (d, J=6,6 Гц, 1Н), 7,30 (s, 1Н), 7,13 (d, J=8,7 Гц, 1Н), 7,06 (s, 1Н), 6,58 (dd, J=16,8, 10,1 Гц, 1Н), 6,42 (d, J=16,8 Гц, 1Н), 5,86 (d, J=10,1 Гц, 1Н), 4,17 (m, 2Н), 3,95 (s, 3Н), 3,78 (m, 2Н), 3,62 (m, 2Н), 3,56 (m, 2Н), 3,45 (s, 3Н), 3,42 (m, 2Н), 3,13 (m, 2Н), 2,81 (s, 3Н), 2,19 (m, 4Н);
MS m/z (ESI): 587 [M+H]+.
Пример 135: получение N-(4-метокси-5-((4-(4-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)акриламида

Стадия 1: получение N-метил-2-(пирролидин-1-ил)этан-1-амина

Способ получения N-метил-2-(пирролидин-1-ил)этан-1-амина был аналогичен Примеру 132.
Стадии 2-7: получение N-(4-метокси-5-((4-(4-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-(метил(2-пирролидин-1-ил)этил)амино)фенил)акриламида

Способ получения N-(4-метокси-5-((4-(4-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)акриламида был аналогичен Примеру 43.
1Н ЯМР (400 МГц, CD3OD) δ 8,43 (s, 1Н), 8,27 (d, J=6,9 Гц, 1Н), 7,99 (s, 1Н), 7,82 (s, 1Н), 7,59 (d, J=6,9 Гц, 1Н), 7,43 (t, J=8,1 Гц, 1Н), 7,07 (s, 1Н), 6,85 (d, J=8,0 Гц, 1Н), 6,61 (dd, J=16,9, 10,1 Гц, 1Н), 6,42 (dd, J=16,9, 1,6 Гц, 1Н), 5,87 (dd, J=10,2, 1,6 Гц, 1Н), 3,99 (s, 3Н), 3,96 (d, J=5,1 Гц, 3Н), 3,64 (d, J=10,0 Гц, 2Н), 3,56 (t, J=5,6 Гц, 2Н), 3,43 (t, J=5,6 Гц, 2Н), 3,13 (d, J=8,9 Гц, 2Н), 2,82 (s, 3Н), 2,19 (s, 4Н);
MS m/z (ESI): 543, 3 [М+Н]+.
Пример 136: получение N-(4-метокси-5-((4-(6-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)-акриламида

Способ получения N-(4-метокси-5-((4-(6-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)акриламида был аналогичен Примеру 135.
1Н ЯМР (400 МГц, CD3OD) δ 8,24 (s, 1Н), 8,17 (d, J=5,5 Гц, 1Н), 8,04 (s, 1Н), 7,70 (s, 1Н), 7,65 (d, J=8,7 Гц, 1Н), 7,50 (d, J=6,5 Гц, 1Н), 7,02-6,92 (m, 2Н), 6,47 (dd, J=16,9, 10,2 Гц, 1Н), 6,27 (d, J=16,9 Гц, 1Н), 5,74 (d, J=10,1 Гц, 1Н), 3,84 (s, 3Н), 3,76 (s, 3Н), 3,49 (s, 2Н), 3,46-3,42 (m, 2Н), 3,31 (d, J=5,2 Гц, 2Н), 3,01 (s, 2Н), 2,69 (s, 3Н), 2,06 (s, 4Н);
MS m/z (ESI): 543, 3 [М+Н]+.
Пример 137: получение N-(4-метокси-5-((4-(5-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)акриламида

Способ получения N-(4-метокси-5-((4-(5-метокси-1Н-индазол-1-ил)пиримидин-2-ил)амино)-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)акриламида был аналогичен Примеру 135.
1Н ЯМР (400 МГц, CD3OD) δ 8,27 (s, 1Н), 8,16 (d, J=6,2 Гц, 2Н), 7,66 (s, 1Н), 7,48 (d, J=6,9 Гц, 1Н), 7,19 (d, J=2,3 Гц, 1Н), 7,01 (d, J=9,1 Гц, 1Н), 6,96 (s, 1Н), 6,48 (dd, J=16,9, 10,1 Гц, 1Н), 6,31 (dd, J=16,9, 1,6 Гц, 1Н), 5,75 (dd, J=10,1, 1,6 Гц, 1Н), 3,84 (s, 3Н), 3,76 (s, 3Н), 3,56-3,49 (m, 2Н), 3,49-3,43 (m, 2Н), 3,31 (t, J=5,6 Гц, 2Н), 3,01 (d, J=9,6 Гц, 2Н), 2,71 (s, 3Н), 2,09 (d, J=6,8 Гц, 4Н);
MS m/z (ESI): 543, 3 [М+Н]+.
Пример 138: получение N-(4-метокси-5-((4-(5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)акриламида

Способ получения N-(4-метокси-5-((4-(5-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)акриламида был аналогичен Примеру 59.
1Н ЯМР (400 МГц, CD3OD) δ 9,49 (s, 1Н), 8,56 (d, J=5,8 Гц, 1Н), 8,22 (d, J=10,4 Гц, 2Н), 7,34 (d, J=5,8 Гц, 1Н), 7,27 (d, J=2,1 Гц, 1Н), 7,08 (dd, J=9,2, 1,9 Гц, 1Н), 7,01 (s, 1Н), 6,59 (dd, J=16,9, 10,1 Гц, 1Н), 6,43 (dd, J=16,9, 1,6 Гц, 1Н), 5,86 (dd, J=10,1, 1,6 Гц, 1Н), 3,97 (s, 3Н), 3,89 (s, 3Н), 3,62 (s, 2Н), 3,51 (t, J=5,5 Гц, 2Н) 3,39 (t, J=5,5 Гц, 2Н), 3,11 (d, J=8,1 Гц, 2Н), 2,77 (s, 3Н), 2,18 (s, 4Н);
MS m/z (ESI): 543, 3 [М+Н]+.
Пример 139: получение N-(4-метокси-5-((4-(6-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)акриламида

Способ получения N-(4-метокси-5-((4-(6-метокси-1Н-бензо[d]имидазол-1-ил)пиримидин-2-ил)амино)-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)акриламида был аналогичен Примеру 59.
1Н ЯМР (400 МГц, CD3OD) δ 9,44 (s, 1Н), 8,58 (d, J=5,2 Гц, 1Н), 8,29 (s, 1Н), 7,88 (s, 1Н), 7,72 (s, 1Н), 7,33 (d, J=5,7 Гц, 1Н), 7,14 (d, J=5,3 Гц, 1Н), 7,00 (s, 1Н), 6,60 (dd, J=16,9, 10,2 Гц, 1Н), 6,39 (dd, J=16,9, 1,4 Гц, 1Н), 5,84 (dd, J=10,2, 1,3 Гц, 1Н), 3,97 (s, 3Н), 3,85 (s, 3Н), 3,62 (s, 2Н), 3,49 (t, J=5,5 Гц, 2Н), 3,38 (dd, J=9,6, 4,1 Гц, 2Н), 3,10 (s, 2Н), 2,75 (s, 3Н), 2,16 (d, J=2,9 Гц, 4Н);
MS m/z (ESI): 543, 3 [М+Н]+.
Пример 140: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1-(проп-2-ин-1-ил)-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)-акриламида

Стадия 1:
получение N1-(4-(1Н-индол-3-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамина

Способ получения N1-(4-(1Н-индол-3-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамина был аналогичен Примеру 102.
Стадия 2: получение N1-(2-(диметиламино)этил)-5-метокси-N1-метил-2-нитро-N4-(4-(1-(проп-2-ин-1-ил)-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,4-диамина

N1-(4-(1Н-индол-3-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамин (0,51 г, 1,1 ммоль) растворяли в безводном DMF (20 мл) с последующим добавлением NaH (47 мг, 1,16 ммоль) при комнатной температуре при перемешивании. После перемешивания в течение 30 минут реакционную смесь охлаждали до 0°С и добавляли пропаргилбромид (137 мг, 1,16 ммоль) при 0°С. Реакционный раствор перемешивали в течение 20 минут. После того, как ЖХ-МС показывала образование продукта, реакцию гасили насыщенным водным раствором хлорида аммония. Продукт очищали колоночной хроматографией с обращенной фазой (элюент: 0,1% водный раствор TFA - ацетонитрил) с получением соли TFA N1-(2-(диметиламино)этил)-5-метокси-N1-метил-2-нитро-N4-4-(1-(проп-2-ин-1-ил)-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,4-диамин (0,25 г, 28%).
1Н ЯМР (400 МГц, CD3OD): δ 8,62 (s, 1Н), 8,57 (s, 1Н), 8,28 (d, J=8,0 Гц, 1Н),8,17 (d, J=6,8 Гц, 1Н), 7,64 (d, J=8,0 Гц, 1Н), 7,48 (d, J=6,4 Гц, 1Н), 7,37 (t, J=7,6 Гц, 1Н), 7,24 (t, J=7,2 Гц, 1Н), 7,09 (s, 1Н), 5,19 (d, J=2,4 Гц, 2Н), 4,06 (s, 3Н), 3,62 (t, J=6,0 Гц, 2Н), 3,52 (t, J=6,0 Гц, 2Н), 3,36 (s, 1Н), 3,01 (s, 6Н), 2,98 (s, 3Н);
MS m/z (ESI): 500, 2 [М+Н]+.
Стадия 3: получение N1-(2-(диметиламино)этил)-5-метокси-N1-метил-N4-(4-(1-(проп-2-ин-1-ил)-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,2,4-триамина

Соль TFA N1-(2-(диметиламино)этил)-5-метокси-N1-метил-2-нитро-N4-(4-(1-(проп-2-ин-1-ил)-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,4-диамина (0,25 г, 0,30 ммоль), восстановленный порошок железа (112 мг, 2,0 ммоль) и хлорид аммония (0,015 г, 0,3 ммоль) добавляли в смесь этанола (8 мл) и воды (2 мл). Реакционную смесь перемешивали в атмосфере азота при 75°С в течение ночи. После охлаждения реакционного раствора до комнатной температуры на следующий день добавляли этанол (60 мл). Реакционный раствор фильтровали через целит и промывали этанолом (10 мл). Фильтрат концентрировали при пониженном давлении и добавляли DCM (60 мл). Слой DCM промывали насыщенным солевым раствором (30 мл), сушили над безводным сульфатом натрия и концентрировали с получением N1-(2-(диметиламино)этил)-5-метокси-N1-метил-N4-(4-(1-(проп-2-ин-1-ил)-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,2,4-триамина (130 мг, 90%), который непосредственно использовали на следующей стадии без дополнительной очистки.
MS m/z (ESI): 470,2 [М+Н]+.
Стадия 4: получение N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1-(проп-2-ин-1-ил)1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида

N1-(2-(диметиламино)этил)-5-метокси-N1-метил-N4-(4-(1-(проп-2-ин-1-ил)-1Н-индол-3-ил)пиримидин-2-ил)бензол-1,2,4-триамин (130 мг, 0,28 ммоль) и триэтиламин (170 мг, 1,7 ммоль) растворяли в THF (20 мл). Раствор охлаждали до -78°С. К реакционной смеси по каплям добавляли раствор акрилоилхлорида (75 мг, 0,84 ммоль) в THF (4 мл). Реакцию проводили в течение 5 минут при этой температуре, гасили метанолом (1 мл). После добавления TFA (200 мг), реакционный раствор концентрировали при пониженном давлении и очищали колоночной хроматографией с обращенной фазой (элюент: 0,1% водный раствор TFA-ацетонитрил) с получением соли TFA N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1-(проп-2-ин-1-ил)-1Н-индол-3-ил)пиримидин-2-ил)амино)фенил)акриламида (105 мг, 45%).
1Н ЯМР (400 МГц, CD3OD): δ 8,62 (s, 1Н), 8,37 (br, 1Н), 8,10 (d, J=6,8 Гц, 1Н), 7,96 (s, 1Н), 7,64 (d, J=8,0 Гц, 1Н), 7,46 (d, J=7,2 Гц, 1Н), 7,36 (m, 1Н), 7,27 (t, J=7,2 Гц, 1Н), 7,10 (s, 1Н), 6,54 (m, 2Н), 5,90 (m, 1Н), 5,20 (d, J=2,8 Гц, 2Н), 3,99 (s, 3Н), 3,57 (t, J=6,0 Гц, 2Н), 3,34 (t, J=6,0 Гц, 2Н), 3,03 (t, J=2,8 Гц, 1Н), 2,93 (s, 6Н), 2,82 (s, 3Н);
MS m/z (ESI): 524,2 [M+H]+.
Пример 141:
получение N-(5-((4-(1-аллил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида

Способ получения N-(5-((4-(1-аллил-1Н-индол-3-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)-акриламида был аналогичен Примеру 140.
1Н ЯМР (400 МГц, CD3OD): δ 8,53 (s, 1Н), 8,31 (b, 1Н), 8,00 (d, J=6,8 Гц, 1Н),7,94 (s, 1Н), 7,49 (d, J=8,0 Гц, 1Н), 7,39 (d, J=6,8 Гц, 1Н), 7,28 (t, J=7,2 Гц, 1Н), 7,19 (t, J=8,0 Гц, 1Н), 7,05 (s, 1Н), 6,57 (m, 1Н), 6,42 (m, 1Н), 6,07 (m, 1Н), 5,82 (m, 1Н), 5,25 (m, 1Н), 5,23 (m, 1Н), 4,92 (d, J=5,2 Гц, 2Н), 3,93 (s, 3Н), 3,86 (m, 2Н), 3,52 (t, J=4,4 Гц, 2Н), 3,32 (t, J=5,6 Гц, 2Н), 2,88 (s, 6Н), 2,76 (s, 3Н);
MS m/z (ESI): 526,2 [М+Н]+.
Пример 142: получение N-(5-((4-(1-(N,N-диметилсульфамоил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метокси-2-(метил(2-(пирролидин-1-ил)этил)-амино)фенил)акриламида

Стадия 1: получение N-(4-(1Н-индол-3-ил)пиримидин-2-ил)-2-метокси-N4-метил-5-нитро-N4-(2-(пирролидин-1-ил)этил)бензол-1,4-диамина

N-(4-фтор-2-метокси-5-нитрофенил)-4-(1Н-индол-3-ил)пиримидин-2-амин (118 мг, 0,312 ммоль) растворяли в DMF (2 мл) и затем добавляли триэтиламин (95 мг, 0,936 ммоль) и N-метил-2-(пирролидин-1-ил)этан-1-амин (60 мг, 0,468 ммоль). Реакционную смесь нагревали до 120°С микроволновым излучением и проводили реакцию в течение 30 мин. После того как ЖХМС показывала завершение реакции реакционный раствор концентрировали досуха. Неочищенный продукт очищали препаративной тонкослойной хроматографией с получением N-(4-(1Н-индол-3-ил)пиримидин-2-ил)-2-метокси-N4-метил-5-нитро-N4-(2-(пирролидин-1-ил)этил)бензол-1,4-диамина (122 мг, 100%).
Стадия 2: получение 3-(2-((2-метокси-4-(метил(2-(пирролидин-1-ил)этил)амино)-5-нитрофенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамида

N-(4-(1Н-индол-3-ил)пиримидин-2-ил)-2-метокси-N4-метил-5-нитро-N4-(2-(пирролидин-1-ил)этил)бензол-1,4-диамин (122 мг, 0,25 ммоль) растворяли в DMF (10 мл) и смесь охлаждали до 0°С на ледяной бане. Затем добавляли NaH (30 мг, 0,75 ммоль) и реакцию проводили при 0°С в течение десяти минут, и затем по каплям добавляли диметилсульфамоилхлорид (54 мг, 0,374 ммоль). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 30 минут. После прекращения реакции добавляли дихлорметан и воду, и реакционный раствор экстрагировали три раза. Органические фазы объединяли, промывали насыщенным водным раствором бикарбоната натрия, водой и насыщенным солевым раствором, фильтровали и концентрировали с получением неочищенного продукта, который затем очищали колоночной флэш-хроматографией на силикагеле с получением 3-(2-((2-метокси-4-(метил(2-(пирролидин-1-ил)этил)амино)-5-нитрофенил)амино)-пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамида (60 мг, 40%).
Стадия 3: получение 3-(2-((5-амино-2-метокси-4-(метил(2-(пирролидин-1-ил)этил)амино)фенил)амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамида

Вышеуказанное соединение 3-(2-((2-метокси-4-(метил(2-(пирролидин-1-ил)этил)амино)-5-нитрофенил)амино)-пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамид растворяли в метаноле (5 мл) и затем добавляли Pd/C (15 мг). Реакционную смесь перемешивали в атмосфере водорода при 24°С в течение 1 часа. После того, как ЖХМС показывала завершение реакции, реакционный раствор фильтровали, и фильтрат концентрировали. Полученный остаток очищали колоночной флэш-хроматографией на силикагеле с получением 15 мг неочищенного продукта 3-(2-((5-амино-2-метокси-4-(метил(2-(пирролидин-1-ил)этил)амино)фенил)амино)-пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамида.
Стадия 4: получение N-(5-((4-(1-(N,N-диметилсульфамоил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метокси-2-(метил(2-(пирролидин-1-ил)этил)амино)-фенил)акриламида
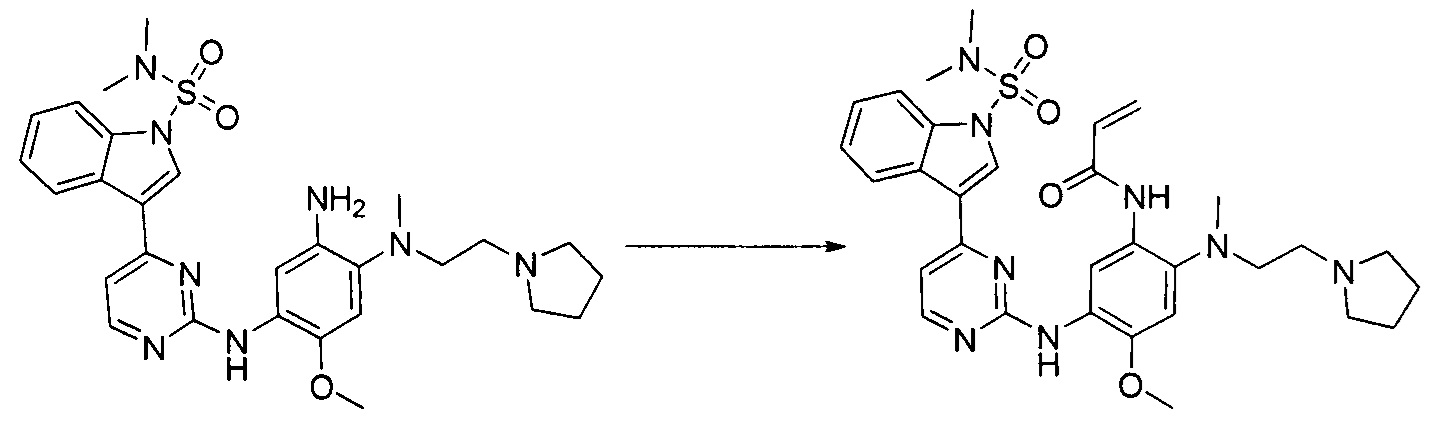
3-(2-((5-амино-2-метокси-4-(метил(2-(пирролидин-1-ил)этил)амино)фенил)-амино)пиримидин-4-ил)-N,N-диметил-1Н-индол-1-сульфонамид (15 мг, 0,027 ммоль) и триэтиламин (8 мг, 0,08 ммоль) растворяли в безводном тетрагидрофуране (20 мл). После перемешивания реакционного раствора при -78°С в течение 10 минут, медленно добавляли по каплям акрилоилхлорид (0,05 мл, 1 M в THF). Реакционную смесь перемешивали в течение 30 минут на бане с сухим льдом. После того как ЖХМС показывала завершение реакции, реакцию гасили метанолом. Реакционный раствор концентрировали и полученный остаток очищали препаративной тонкослойной хроматографией с получением N-(5-((4-(1-(N,N-диметилсульфамоил)-1Н-индол-3-ил)пиримидин-2-ил)амино)-4-метокси-2-(метил(2-(пирролидин-1-ил)этил)амино)фенил)акриламида (7 мг, 44%).
1Н ЯМР (400 МГц, CD3OD) δ 8,63 (s, 1Н), 8,46 (d, J=7,9 Гц, 1Н), 8,32 (s, 1Н), 8,11-7,92 (m, 2Н), 7,56 (d, J=5,9 Гц, 1Н), 7,37 (dt, J=15,2, 7,3 Гц, 2Н), 7,04 (s, 1Н), 6,55 (dd, J=16,8, 10,1 Гц, 1Н), 6,42 (dd, J=16,9, 1,7 Гц, 1Н), 5,87 (dd, J=10,0, 1,7 Гц, 1Н), 3,99 (s, 3Н), 3,60 (s, 2Н), 3,58-3,50 (m, 2Н), 3,43-3,36 (m, 2Н), 3,10 (s, 2Н), 2,93 (s, 6Н), 2,79 (s, 3Н), 2,27-2,09 (m, 4Н);
MS m/z (ESI): 619,6 [M+H]+.
Пример 143:
получение N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-(N,N-диметилсульфамоил)-6-метокси-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)амино)-4-метоксифенил)акриламида

Стадия 1:
получение 3-(2-хлор-5-(трифторметил)пиримидин-4-ил)-6-метокси-1Н-индола

6-метокси-1Н-индол (5 г, 33,97 ммоль), 2,4-дихлор-5-(трифторметил) пиримидин (8,1 г, 37,36 ммоль) и трихлорид алюминия (6,79 г, 50,95 ммоль) растворяли в DME (50 мл) и реакционную смесь перемешивали в течение ночи при 70°С. По окончании реакции реакционный раствор выливали в ледяную воду и экстрагировали три раза метил-трет-бутиловым эфиром. Органические фазы объединяли, промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с получением сырого продукта, который затем очищали колоночной флэш-хроматографией на силикагеле с получением 3-(2-хлор-5-(трифторметил)пиримидин-4-ил)-6-метокси-1Н-индола (4,3 г, 39%).
Стадия 2: получение N-(4-фтор-2-метокси-5-нитрофенил)-4-(6-метокси-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-амина

3-(2-хлор-5-(трифторметил)пиримидин-4-ил)-6-метокси-1Н-индол (165 мг, 0,504 ммоль), сырой материал 4-фтор-2-метокси-5-нитроанилин (103 мг, 0,554 ммоль) и моногидрат п-толуолсульфоновой кислоты (96 мг, 0,504 ммоль) растворяли в 2-пентаноле (20 мл) и реакционную смесь нагревали до 120°С в течение ночи. После того как ЖХМС показывала завершение реакции, реакционный раствор естественным образом охлаждали до комнатной температуры и осаждали темное твердое вещество. Твердое вещество фильтровали и осадок на фильтре промывали метанолом (1 мл) и метил-трет-бутиловым эфиром (1 мл), с получением N-(4-фтор-2-метокси-5-нитрофенил)-4-(6-метокси-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-амина (125 мг, 52%).
Стадия 3:
получение N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(6-метокси-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)-N1-метил-2-нитробензол-1,4-диамина

N-(4-фтор-2-метокси-5-нитрофенил)-4-(6-метокси-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-амин (125 мг, 0,262 ммоль) растворяли в 2 мл DMF, а затем добавляли триэтиламин (80 мг, 0,786 ммоль) и триметилэтилендиамин (80 мг, 0,786 ммоль). Реакционную смесь нагревали до 120°С микроволновым излучением и проводили реакцию в течение 30 мин. После того как ЖХМС показывала завершение реакции, реакционный раствор концентрировали досуха. Неочищенный продукт очищали препаративной тонкослойной хроматографией с получением N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(6-метокси-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)-N1-метил-2-нитробензол-1,4-диамина (146 мг, 99%).
Стадия 4: получение 3-(2-((4-((2-(диметиламино)этил)(метил)амино)-2-метокси-5-нитрофенил)амино)-5-(трифторметил)пиримидин-4-ила)-6-метокси-N,N-диметил-1Н-индол-1-сульфонамида

N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(6-метокси-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)-N1-метил-2-нитробензол-1,4-диамин (146 мг, 0,262 ммоль) растворяли в DMF (10 мл) и смесь охлаждали до 0°С на ледяной бане до добавления NaH (31 мг, 0,786 ммоль). Реакцию проводили при 0°С в течение десяти минут, а затем по каплям добавляли диметилсульфамоилхлорид (41 мг, 0,288 ммоль). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 30 минут. После прекращения реакции добавляли дихлорметан и воду, и реакционный раствор экстрагировали три раза. Органические фазы объединяли, промывали насыщенным водным раствором бикарбоната натрия, водой и насыщенным солевым раствором последовательно, фильтровали и выпаривали досуха с получением сырого продукта, который затем очищали колоночной флэш-хроматографией на силикагеле с получением 3-(2-((4-((2-(диметиламино)этил)(метил)амино)-2-метокси-5-нитрофенил)амино)-5-(трифторметил)пиримидин-4-ил)-6-метокси-N,N-диметил-1Н-индол-1-сульфонамида (80 мг, 46%).
Стадия 5:
получение 3-(2-((5-амино-4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)амино)-5-(трифторметил)пиримидин-4-ил)-6-метокси-N,N-диметил-1Н-индол-1-сульфонамида

Вышеуказанное соединение N1-(2-(диметиламино)этил)-5-метокси-N4-(4-(6-метокси-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)-N1-метил-2-нитробензол-1,4-диамин растворяли в метаноле (10 мл) и добавляли Pd/C (20 мг). Реакционную смесь перемешивали в атмосфере водорода при 24°С в течение 1 часа. После того как ЖХМС показывала завершение реакции, реакционный раствор фильтровали и фильтрат концентрировали с получением неочищенного продукта, который затем очищали колоночной флэш-хроматографией на силикагеле с получением 44 мг 3-(2-((5-амино-4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)амино)-5-(трифторметил)пиримидин-4-ил)-6-метокси-N,N-диметил-1Н-индол-1-сульфонамида.
Стадия 6:
получение N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-(N,N-диметилсульфамоил)-6-метокси-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)амино)-4-метоксифенил)акриламида

3-(2-((5-амино-4-((2-(диметиламино)этил)(метил)амино)-2-метоксифенил)амино)-5-(трифторметил)пиримидин-4-ил)-6-метокси-N,N-диметил-1Н-индол-1-сульфонамид (44 мг, 0,069 ммоль) и триэтиламин (21 мг, 0,207 ммоль) растворяли в безводном тетрагидрофуране (20 мл). Реакционную смесь перемешивали при -78°С в течение 10 минут, перед тем как медленно и по каплям добавляли акрилоилхлорид (0,2 мл, 1 M в THF). Затем реакционную смесь перемешивали в течение 30 минут на бане с сухим льдом. После того как ЖХМС показывала завершение реакции, реакцию гасили метанолом. Реакционный раствор концентрировали и полученный остаток очищали препаративной тонкослойной хроматографией с получением N-(2-((2-(диметиламино)этил)(метил)амино)-5-((4-(1-(N,N-диметилсульфамоил)-6-метокси-1Н-индол-3-ил)-5-(трифторметил)пиримидин-2-ил)амино)-4-метоксифенил)акриламида (16 мг, 33%).
1Н ЯМР (400 МГц, CD3OD) δ 8,78 (s, 1Н), 8,39 (s, 1Н), 7,91 (d, J=8,8 Гц, 1Н), 7,82 (s, 1Н), 7,50 (d, J=2,1 Гц, 1Н), 6,97 (s, 1Н), 6,90 (d, J=8,2 Гц, 1Н), 6,41 (d, J=5,3 Гц, 2Н), 5,83 (t, J=5,9 Гц, 1Н), 4,00 (s, 3Н), 3,88 (s, 3Н), 3,49 (t, J=5,5 Гц, 2Н), 3,28 (t, J=5,5 Гц, 2Н), 2,89 (s, 6Н), 2,86 (s, 6Н), 2,70 (s, 3Н);
MS m/z (ESI): 691,5 [M+H]+.
Биологические испытания и оценка
1. Энзимологический эксперимент с EGFR T790M-мутантного типа
В этом эксперименте ингибирующее действие соединений на фермент EGFR-экзон 20 T790M-мутантного типа тестировали методом флуоресцентного резонансного переноса энергии (TR-FRET) и определяли полумаксимальную ингибирующую концентрацию (IC50) соединений в отношении активности фермента.
1) в 384-луночный планшет добавляли 1-5 мкл раствора фермента Т790М EGFR, и конечная концентрация фермента составляла 0,1-1 нМ.
2) добавляли 1-5 мкл разбавленного раствора в градиенте соединения.
3) смесь инкубировали в течение 10 минут при комнатной температуре.
4) добавляли 1-5 мкл смеси субстрата, содержащей субстратный полипептид, с 5-50 нМ конечной концентрации, и добавляли АТФ с конечной концентрацией 1-10 мкМ.
5) Смесь инкубировали при комнатной температуре в течение 0,5-2 часов.
6) Для прекращения реакции добавляли 5 мкл стоп-раствора EDTA в течение 5 минут.
7) Добавляли 5 мкл тестового раствора, содержащего меченое антитело, и смесь инкубировали при комнатной температуре в течение 1 часа.
8) Значения сигнала флуоресценции для каждой пластины определяли с помощью микропланшет-ридера при 665 нм.
9) Скорости ингибирования рассчитывали в соответствии со значениями сигнала флуоресценции.
10) IC50 соединения получали путем аппроксимации кривой в соответствии со скоростями ингибирования в разных концентрациях.
2. Энзимологический эксперимент с EGFR дикого типа (WT)
В этом эксперименте ингибирующее действие соединений на фермент EGFR дикого типа тестировали методом флуоресцентного резонансного переноса энергии (TR-FRET) и определяли полумаксимальную ингибирующую концентрацию IC50 соединений в отношении ферментативной активности.
1) в 384-луночный планшет добавляли 1-5 мкл раствора фермента EGFR дикого типа, и конечная концентрация фермента составляла 0,1-1 нМ.
2) Добавляли 1-5 мкл разбавленного раствора в градиенте соединения.
3) Смесь инкубировали в течение 10 минут при комнатной температуре.
4) Добавляли 1-5 мкл субстратной смеси, содержащей субстратный полипептид, с конечной концентрацией 5-50 нМ и АТФ с конечной концентрацией 0,1-5 мкМ.
5) Смесь инкубировали при комнатной температуре в течение 0,5-2 часов.
6) Для прекращения реакции в течение 5 минут добавляли 5 мкл стоп-раствора EDTA.
7) Добавляли 5 мкл тестового раствора, содержащего меченое антитело, и смесь инкубировали при комнатной температуре в течение 1 часа.
8) Значения сигнала флуоресценции для каждой пластины определяли с помощью микропланшет-ридера при 665 нм.
9) Скорости ингибирования рассчитывали по значениям сигнала флуоресценции.
10) IC50 соединения получали путем аппроксимации кривой в соответствии со скоростями ингибирования в разных концентрациях.
Биохимическую активность соединений по настоящему изобретению определяли в соответствии с вышеупомянутым экспериментом, и значения IC50 были показаны в Таблице ниже.




Где NT означает, что активность не определена.
Величины IC50 в отношении EGFR у других примерных соединений по настоящему изобретению были подобны эффектам вышеуказанных примеров, и эти соединения проявляли сходную ингибирующую активность и закономерность.
Вывод: примерные соединения по настоящему изобретению обладали сильной ингибирующей активностью в отношении EGFR-киназы мутантного типа, но в то же время имели слабую ингибирующую активность в отношении киназы дикого типа. Следовательно, соединения по настоящему изобретению обладали очень хорошей селективностью.
3. Эксперимент по ингибированию пролиферации клеток NCI-H1975
В этом эксперименте ингибирующее действие соединений на пролиферацию клеток NCI-H1975 тестировали методом CellTiter-Glo и определяли полумаксимальную ингибирующую концентрацию (IC50) соединений в отношении активности клеточной пролиферации.
1) в 96-луночный культуральный планшет клеток высевали 90 мкл суспензии клеток Н1975 с плотностью 1-5×103 клеток/мл. Культуральный планшет инкубировали в инкубаторе в течение 16-24 часов (37°С, 5% CO2).
2) Различные концентрации тестируемого соединения в градиентном разбавлении добавляли к клеткам в культуральном планшете. Культуральный планшет инкубировали в инкубаторе в течение 72 часов (37°С, 5% CO2).
3) В каждую лунку добавляли 50-100 мкл реагента CellTiter-Glo. Затем культуральный планшет встряхивали в течение 10 минут и оставляли стоять при комнатной температуре в течение 10 минут.
4) Значения сигнала хемилюминесценции каждой пластины определяли с помощью микропланшетного ридера.
5) Скорость ингибирования рассчитывали в соответствии со значениями сигнала хемилюминесценции.
6) IC50 соединения получали путем аппроксимации кривой в зависимости от скоростей ингибирования при разных концентрациях.
4. Эксперимент по ингибированию пролиферации клеток А431
В этом эксперименте ингибирующее действие соединений на пролиферацию клеток А431 тестировали методом CellTiter-Glo и определяли полумаксимальную ингибирующую концентрацию (IC50) соединений в отношении активности клеточной пролиферации.
1) 96-луночный культуральный планшет клеток высевали с 90 мкл суспензии клеток А431 с плотностью 1-5×103 клеток/мл. Культуральный планшет инкубировали в инкубаторе в течение 16-24 часов (37°С, 5% СО2).
2) Различные концентрации тестируемого соединения в градиентном разбавлении добавляли к клеткам в культуральном планшете. Культуральный планшет инкубировали в инкубаторе в течение 72 часов (37°С, 5% СО2).
3) В каждую лунку добавляли 50-100 мкл реагента CellTiter-Glo. Затем культуральный планшет встряхивали в течение 10 минут и оставляли стоять при комнатной температуре в течение 10 минут.
4) Значения сигнала хемилюминесценции каждой пластины определяли с помощью микропланшетного ридера.
5) Скорости ингибирования рассчитывали в соответствии со значениями сигнала хемилюминесценции.
6) IC50 соединения получали путем аппроксимации кривой в зависимости от скоростей ингибирования при разных концентрациях.
Биохимическую активность соединений по настоящему изобретению определяли в соответствии с вышеупомянутым экспериментом, и значения IC50 были показаны в Таблице ниже.




Значения IC50 EGFR других примерных соединений по настоящему изобретению были подобны действию вышеуказанных примеров, и эти соединения проявляли сходную ингибирующую активность и закономерность.
Вывод: примерные соединения по настоящему изобретению обладали сильной ингибирующей активностью в отношении пролиферации клеток Н1975 мутантного типа с EGFR мутантного типа, но обладали слабой ингибирующей активностью в отношении пролиферации клеток А431 дикого типа, так что примерные соединения обладали очень хорошей избирательностью для клеток дикого типа/мутантного типа.
Фармакокинетическое (ФК) испытание примерных соединений
I. ФК анализ на крысах
Фармакокинетическое испытание на крысах предпочтительного соединения из Примера 26 настоящего изобретения и положительного контрольного соединения AZD-9291 проводили на крысах SD (Shanghai Slac Laboratory Animal Co., LTD).
1. 1,0 мл внутривенной крови собирали и помещали в пробирку с K2EDTA. Кровь центрифугировали при КТ при 6000 об/мин в течение 5 минут для выделения плазмы, которую хранили при -80°С.
2. 160 мкл ацетонитрила добавляли к 40 мкл образцов плазмы для осаждения, а затем смесь центрифугировали при 3500 об/мин в течение 5 минут.
3. Отбирали 100 мкл обработанного раствора и анализировали концентрацию испытуемого соединения с помощью ЖХ/МС/МС. Аналитическим инструментом ЖХ/МС/МС являлся АВ Sciex API 4000.
Жидкостная хроматография:

Масс-спектрометрия:
Состояние масс-спектрометра: режим ионизации электрораспылением с регистрацией положительных ионов (ESI).
Результаты анализа жидкостной хроматографии и масс-спектрометрии:
1. Соединение Примера 26:
1Н ЯМР (400 МГц, CDCl3): δ 9,78 (s, 1Н), 9,74 (s, 1Н), 8,55 (s, 1Н), 8,39 (d, J=5,3 Гц, 1Н), 8,11 (d, J=7,0 Гц, 1Н), 7,74-7,55 (m, 2Н), 7,18 (d, J=5,3 Гц, 1Н), 6,76 (s, 1Н), 6,62 (dd, J=16,8, 10,1 Гц, 1Н), 6,46 (dd, J=16,9, 1,9 Гц, 1Н), 6,24 (m, 1Н), 5,80-5,59 (m, 1Н), 3,88 (s, 3Н), 3,55-3,34 (m, 1Н), 3,02 (t, J=5,8 Гц, 2Н), 2,68 (s, 3Н), 2,57 (t, J=5,7 Гц, 2Н), 2,42 (s, 6Н), 1,24-1,17 (m, 2Н), 1,14-1,04 (m, 2Н);
MS m/z (ESI): 526,3 [М+Н]+.
Метаболит:
1Н ЯМР (400 МГц, CDCl3) δ 9,37 (s, 1Н), 9,29 (s, 1Н), 8,29 (d, J=5,3 Гц, 1Н), 8,18 (s, 1Н), 8,07 (d, J=7,2 Гц, 1Н), 7,61-7,40 (m, 2Н), 7,24-7,15 (m, 2Н), 7,06 (d, J=5,3 Гц, 1Н), 6,94 (dd, J=15,9, 9,9 Гц, 1Н), 6,45 (s, 1Н), 6,17 (d, J=16,9 Гц, 1Н), 5,58 (d, J=10,2 Гц, 1Н), 3,79 (s, 3Н), 3,39-3,15 (m, 1Н), 2,93 (s, 2Н), 2,65 (s, 2Н), 2,44 (s, 3Н), 2,26 (s, 3Н), 1,01 (d, J=5,2 Гц, 4Н);
MS m/z (ESI): 512,6 [М+Н]+.
Структура была определена следующим образом:

2. Структура метаболита положительного контрольного соединения AZD-9291 была идентифицирована следующим образом:

Данные в значительной степени согласовались с данными, описанными в Journal of Medicinal Chemistry (2014), 57 (20), 8249-8267).
Основные параметры были рассчитаны с помощью WinNonlin 6.1, и экспериментальные результаты фармакокинетического теста на крысах приведены в Таблице 11 ниже:

Из результатов фармакокинетического испытания на крысах в Таблице 11 видно, что:
1. Положительное контрольное соединение AZD-9291 содержало два метаболита в плазме у крысы; тогда как соединение Примера 26 по настоящему изобретению содержало только один метаболит у крысы.
2. Соединение Примера 26 по настоящему изобретению не продуцировало Метаболит-2 положительного контрольного соединения AZD-9291, тем самым избегая проблемы, связанной с низкой селективностью Метаболита-2 AZD-9291 в отношении Т790М мутантного типа/дикого типа белка-мишени, и преодолевая недостатки предшествующего уровня техники.
II. ФК анализ у собак
Фармакокинетическое испытание на собаках предпочтительного соединения из Примера 26 по настоящему изобретению и положительного контрольного соединения AZD-9291 проводили на собаках породы бигль.
1. 1,0 мл внутривенной крови собирали и помещали в пробирку с гепарином. Кровь центрифугировали при RT 6000 об/мин в течение 5 минут для выделения плазмы, которую хранили при -80°С.
2. 160 мкл ацетонитрила добавляли к 40 мкл образцов плазмы для осаждения, а затем смесь центрифугировали при 3500 об/мин в течение 5 минут.
3. Отбирали 100 мкл обработанного раствора и анализировали концентрацию испытуемого соединения с помощью ЖХ/МС/МС. Аналитическим инструментом ЖХ/МС/МС являлся АВ Sciex API 4000.
Жидкостная хроматография:

Состояние масс-спектрометра: режим ионизации электрораспылением с регистрацией положительных ионов (ESI).
Основные параметры были рассчитаны с помощью WinNonlin 6.1, и экспериментальные результаты фармакокинетического теста у собак были показаны в Таблице 12 ниже:

Из результатов фармакокинетического испытания у собак в Таблице 12 видно, что: фармакокинетические параметры у собак предпочтительного соединения из Примера 26 по настоящему изобретению, превосходят фармакокинетические показатели положительного контрольного соединения AZD-9291. Количество воздействия соединением Примера 26 может превышать более чем в 6 раз количество воздействия положительного контрольного соединения AZD-9291. Между тем период полураспада соединения Примера 26 также значительно дольше, поэтому он больше соответствует медицинским требованиям введения.
Claims (112)
1. Соединение формулы (I) или его фармацевтически приемлемая соль:

где:
кольцо А выбирают из группы, состоящей из:

Q представляет собой связь;
R выбирают из группы, состоящей из водорода;
X1, Х2 и Х3 каждый независимо выбирают из группы, состоящей из NR7 и CR8, где один или два из X1, Х2 и Х3 представляют собой NR7, причем, в случае когда Х2 представляет собой NR7, другие группы представляют собой CR8, и кольцо с указанной группой Х2 присоединяется атомом углерода к пиримидиновому кольцу, или когда два из X1, Х2 и Х3 представляют собой NR7, то кольцо присоединяется атомом азота указанной группы к пиримидиновому кольцу;
R1 выбирают из группы, состоящей из:


где три R6 в  представляют собой необязательно одинаковые или разные заместители;
представляют собой необязательно одинаковые или разные заместители;
R2 выбирают из группы, состоящей из C1-8 алкила, где С1-8 алкил необязательно замещен одной или более группами, выбранными из группы, состоящей из галогена;
R3 выбирают из группы, состоящей из водорода, галогена, трифторметила, трифторметокси и P(O)R11R12, где R11R12 представляют собой C1-6 алкил;
R4 независимо выбирают из группы, состоящей из водорода, галогена, циано, C1-8 алкила, С2-8 алкенила, С2-8 алкинила, С3-8 циклоалкила, 4-членного насыщенного гетероциклила, содержащего атом кислорода в цикле, -P(O)R11R12, где R11R12 представляют собой С1-6 алкил, -S(O)rR9, -O-R10, -C(O)R10,
где C1-8 алкил необязательно замещен одной или более группами, выбранными из группы, состоящей из галогена, гидрокси и -O-R10;
R6 выбирают из группы, состоящей из водорода и C1-8 алкила;
R7 выбирают из группы, состоящей из C1-8 алкила, С2-8 алкенила, С3-6 циклоалкила, -S(O)rR9, -O-R10, -C(O)R10 и 4-членного насыщенного гетероциклила, содержащего атом кислорода в цикле;
где C1-8 алкил замещен одной или более группами, выбранными из группы, состоящей из гидрокси, С2-8 алкенила и С2-8 алкинила;
R8 выбирают из группы, состоящей из водорода, С1-8 алкила, С3-8 циклоалкила и -O-R10;
R9 выбирают из группы, состоящей из бис-C1-8 алкиламино;
R10 выбирают из группы, состоящей из C1-8 алкила и С3-8 циклоалкила;
m равно 0, 1, 2;
r равно 2;
о равно 0;
р равно 1;
q равно 1.
2. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где R2 выбирают из группы, состоящей из С1-4 алкила, где С1-4 алкил необязательно замещен одной или более группами, выбранными из группы, состоящей из галогена; кольцо A, Q, R, X1, Х2, Х3, R1, R3, R4, R6, R7, R8, R9, R10, R11, R12, m, r, о, p и q являются такими, как определено в п. 1.
3. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 2, где R2 представляет собой С1-4 алкил, необязательно замещенный одним или более фтором; кольцо A, Q, R, Х1, Х2, Х3, R1, R3, R4, R6, R7, R8, R9, R10, R11, R12, m, r, о, p и q являются такими, как определено в п. 1.
4. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 3, где R2 выбирают из группы, состоящей из метила, дифторметила и трифторметила; кольцо А, Q, R, X1, Х2, Х3, R1, R3, R4, R6, R7, R8, R9, R10, R11, R12, m, r, о, p и q являются такими, как определено в п. 1.
5. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп. 1-4, где соединение представляет собой соединение формулы (IA):

где R2 выбирают из группы, состоящей из метила, дифторметила и трифторметила; кольцо A, R, X1, Х2, Х3, R1, R3, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в п. 1.
6. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 5, где соединение выбирают из группы, состоящей из соединения формулы (IIA1) и соединения формулы (IIA2):

где R2 выбирают из группы, состоящей из метила, дифторметила и трифторметила; кольцо A, R, X1, Х2, Х3, R1, R3, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в п. 1.
7. Соединение, выбранное из группы, состоящей из соединения формулы (IIIA1-1), соединения формулы (IIIA1-2), соединения формулы (IIIA1-3), соединения формулы (IIIA1-4), соединения формулы (IIIA1-5) и соединения формулы (IIIA1-6), или его фармацевтически приемлемая соль:


где R2 выбирают из группы, состоящей из метила, дифторметила и трифторметила; R, R1, R3, R4, R7, R8 и m являются такими, как определено в п. 1.
8. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 5, где соединение выбирают из группы, состоящей из соединения формулы (IVA1-1) и соединения формулы (IVA1-2):

где R, R1, R3, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в п. 1.
9. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 8, где R3 выбирают из группы, состоящей из водорода, галогена, трифторметила и трифторметокси; R, R1, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в п. 1.
10. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 9, где R3 выбирают из группы, состоящей из водорода, фтора, хлора, трифторметила и трифторметокси; R, R1, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в п. 1.
11. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 10, где R3 выбирают из группы, состоящей из водорода, фтора, хлора и трифторметила.
12. Соединение, выбранное из группы, состоящей из:



или его фармацевтически приемлемая соль.
13. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 5, где соединение представляет собой соединение формулы (IVA1-3):

где R, R1, R3, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в п. 1.
14. Соединение, выбранное из группы, состоящей из:


15. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 13, где, когда R8 не является водородом, m равно 2; R3 выбирают из водорода, фтора, хлора и трифторметила; R, R1, R4, R6, R7, R8, R9, R10, R11, R12, m, r и q являются такими, как определено в п. 1.
16. Соединение, выбранное из группы, состоящей из:





или его фармацевтически приемлемая соль.
17. Соединение, выбранное из группы, состоящей из соединения формулы (IIIA1-7), соединения формулы (IIIA1-8) и соединения формулы (IIIA1-9), или его фармацевтически приемлемая соль:

где R2 выбирают из группы, состоящей из метила, дифторметила и трифторметила; R, R1, R3, R4, R8 и m являются такими, как определено в п. 1.
18. Соединение или его фармацевтически приемлемая соль по п. 17, где R2 выбирают из группы, состоящей из метила, дифторметила и трифторметила; R3 выбирают из группы, состоящей из водорода, фтора, хлора и трифторметила; R, R1, R4, R8 и m являются такими, как определено в п. 1.
19. Соединение или его фармацевтически приемлемая соль по п. 18, где R1 выбирают из группы, состоящей из  .
.
20. Соединение, выбранное из группы, состоящей из соединения формулы (IVA1-4) и соединения формулы (IVA1-5), или его фармацевтически приемлемая соль:

где R3 выбирают из группы, состоящей из водорода, фтора, хлора и трифторметила; R, R4, R8 и m являются такими, как определено в п. 1.
21. Соединение, выбранное из группы, состоящей из:



или его фармацевтически приемлемая соль.
22. Соединение, выбранное из группы, состоящей из соединения формулы (IVA1-6) и соединения формулы (IVA1-7), или его фармацевтически приемлемая соль:

где R3 выбирают из группы, состоящей из водорода, фтора, хлора и трифторметила; R, R4, R8 и m являются такими, как определено в п. 1.
23. Соединение, выбранное из группы, состоящей из:


или его фармацевтически приемлемая соль.
24. Соединение, выбранное из группы, состоящей из соединения формулы (IIIA2-1), соединения формулы (IIIA2-2), соединения формулы (IIIA2-3), соединения формулы (IIIA2-4), соединения формулы (IIIA2-5) и соединения формулы (IIIA2-6):


где R2 выбирают из группы, состоящей из метила, дифторметила и трифторметила; R, R1, R3, R4, R7, R8 и m являются такими, как определено в п. 1.
25. Соединение или его фармацевтически приемлемая соль по п. 24, где R3 выбирают из группы, состоящей из водорода, фтора, хлора и трифторметила; R, R1, R4, R7, R8 и m являются такими, как определено в п. 1.
26. Соединение или его фармацевтически приемлемая соль по п. 25, где R1 представляет собой  .
.
27. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 1, где соединение представляет собой соединение формулы (IC):

где Q, R, R1, R2, R3, R4, R6, R7, R8, R9, R10, R11, R12, r, о, p и q являются такими, как определено в п. 1.
28. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 27, где R2 выбирают из группы, состоящей из дифторметила, трифторметила и метила; R3 выбирают из группы, состоящей из водорода, фтора, хлора, трифторметила; Q, R, R1, R4, R6, R7, R8, R9, R10, r, о, р и q являются такими, как определено в п. 1.
29. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 28, где соединение выбирают из группы, состоящей из соединения формулы (IIC1) и соединения формулы (IIC2):

где Q, R, R4, R7, R8, R9, R10, R11, R12, r и о являются такими, как определено в п. 1.
30. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 29, где соединение выбирают из группы, состоящей из соединения формулы (IIIC1) и соединения формулы (IIIC2):

где R, R4, R7, R8, R9, R10, r и о являются такими, как определено в п. 1.
31. Соединение, представляющее собой:

или его фармацевтически приемлемая соль.
32. Способ получения соединения формулы (I) или его фармацевтически приемлемой соли по п. 1, включающий следующие стадии:
соответствующий 4-замещенный 2-хлорпиримидин подвергают реакции с соответствующим 4-фтор-5-нитроанилином с получением соответствующего N-(2-замещенного-4-фтор-5-нитрофенил)пиримидин-2-амина, соответствующий N-(2-замещенный-4-фтор-5-нитрофенил)пиримидин-2-амин подвергают реакции с R1-H с получением соответствующего N-(2-замещенного-4-замещенного-5-нитрофенил)пиримидин-2-амина, который подвергают гидрогенизации с получением N-(2-замещенного-4-замещенного-5-аминофенил)пиримидин-2-амина, и затем N-(2-замещенный-4-замещенный-5-аминофенил)пиримидин-2-амин подвергают реакции с акрилоилхлоридом, согласно следующей схеме:

где кольцо A, Q, X1, X2, X3, R, R1, R2, R3, R4, R6, R7, R8, R9, R10, R11, R12, m, r, о, p и q являются такими, как определено в п. 1, с получением соответствующего соединения формулы (I) или его фармацевтически приемлемой соли.
33. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-31 для получения лекарственного средства для лечения заболевания, опосредованного активностью мутанта EGFR, где мутант EGFR выбирают из мутанта EGFR-T790M.
34. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-31 в качестве лекарственного средства для лечения заболевания, связанного с клеточной пролиферацией, опосредованного активностью мутанта EGFR-T790M.
35. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-31 в качестве лекарственного средства для лечения рака, опосредованного активностью мутанта EGFR-T790M.
36. Применение по п. 35, где рак выбирают из группы, состоящей из рака яичника, рака шейки матки, колоректального рака, рака молочной железы, рака поджелудочной железы, глиомы, глиобластомы, меланомы, рака предстательной железы, лейкемии, лимфомы, неходжкинской лимфомы, рака желудка, рака легкого, гепатоцеллюлярной карциномы, рака желудка, гастроинтестинальной стромальной опухоли (GIST), рака щитовидной железы, холангиокарциномы, рака эндометрия, рака почки, анапластической крупноклеточной лимфомы, острого миелоидного лейкоза (AML), множественной миеломы, меланомы и мезотелиомы; предпочтительно немелкоклеточного рака легкого.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201410534203 | 2014-10-11 | ||
| CN201410534203.9 | 2014-10-11 | ||
| PCT/CN2015/091189 WO2016054987A1 (zh) | 2014-10-11 | 2015-09-30 | Egfr抑制剂及其制备和应用 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| RU2017114687A RU2017114687A (ru) | 2018-11-20 |
| RU2017114687A3 RU2017114687A3 (ru) | 2019-01-31 |
| RU2702631C2 true RU2702631C2 (ru) | 2019-10-09 |
Family
ID=55652583
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2017114687A RU2702631C2 (ru) | 2014-10-11 | 2015-09-30 | Ингибитор egfr и его получение и применение |
Country Status (23)
| Country | Link |
|---|---|
| US (2) | US10259820B2 (ru) |
| EP (1) | EP3205650B1 (ru) |
| JP (1) | JP6691281B2 (ru) |
| KR (1) | KR102589982B1 (ru) |
| CN (7) | CN111187221B (ru) |
| AU (1) | AU2015330506B2 (ru) |
| BR (1) | BR112017006713B1 (ru) |
| CA (1) | CA2959194A1 (ru) |
| CY (1) | CY1124476T1 (ru) |
| DK (1) | DK3205650T3 (ru) |
| ES (1) | ES2886887T3 (ru) |
| HR (1) | HRP20211392T1 (ru) |
| HU (1) | HUE055233T2 (ru) |
| LT (1) | LT3205650T (ru) |
| MX (1) | MX2017004234A (ru) |
| PL (1) | PL3205650T3 (ru) |
| PT (1) | PT3205650T (ru) |
| RS (1) | RS62350B1 (ru) |
| RU (1) | RU2702631C2 (ru) |
| SI (1) | SI3205650T1 (ru) |
| TW (1) | TWI704138B (ru) |
| WO (1) | WO2016054987A1 (ru) |
| ZA (1) | ZA201701465B (ru) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2733412C2 (ru) * | 2016-03-22 | 2020-10-01 | Цзянсу Хансох Фармасьютикал Груп Ко., Лтд. | Поликристаллическая форма свободного основания или соли присоединения кислоты ингибитора egfr, способ её получения и применение |
Families Citing this family (74)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10227342B2 (en) | 2014-06-19 | 2019-03-12 | Ariad Pharmaceuticals, Inc. | Heteroaryl compounds for kinase inhibition |
| CN105384694B (zh) * | 2014-08-22 | 2020-09-04 | 四川海思科制药有限公司 | 一种取代的氨基嘧啶衍生物及其制备方法和药物用途 |
| CN111170998B (zh) * | 2014-11-05 | 2023-04-11 | 益方生物科技(上海)股份有限公司 | 嘧啶或吡啶类化合物、其制备方法和医药用途 |
| CN113563332A (zh) * | 2014-12-23 | 2021-10-29 | 达纳-法伯癌症研究所公司 | 作为egfr抑制剂的新的嘧啶和治疗病症的方法 |
| CN106117185B (zh) * | 2015-08-31 | 2017-11-07 | 广州必贝特医药技术有限公司 | 2,4‑二含氮基团取代嘧啶类化合物及其制备方法和应用 |
| CN106749193B (zh) * | 2015-11-23 | 2020-11-20 | 南京圣和药业股份有限公司 | 吲唑取代的表皮生长因子受体抑制剂及其应用 |
| CN106928150B (zh) * | 2015-12-31 | 2020-07-31 | 恩瑞生物医药科技(上海)有限公司 | 丙烯酰胺苯胺衍生物及其药学上的应用 |
| TWI726968B (zh) * | 2016-01-07 | 2021-05-11 | 開曼群島商Cs醫藥技術公司 | Egfr酪胺酸激酶之臨床重要突變體之選擇性抑制劑 |
| WO2017119732A1 (en) | 2016-01-08 | 2017-07-13 | Samsung Electronics Co., Ltd. | Electronic device and operating method thereof |
| JP7112957B2 (ja) | 2016-03-09 | 2022-08-04 | ベイジン パーカンズ オンコロジー カンパニー リミテッド | 腫瘍細胞懸濁培養及び関連方法 |
| CN107344934B (zh) * | 2016-05-04 | 2019-08-23 | 江苏正大丰海制药有限公司 | 药物活性物质的固体形式 |
| CN107417626B (zh) * | 2016-05-23 | 2020-01-24 | 江苏奥赛康药业有限公司 | 一种2-氨基嘧啶类化合物的多晶型形式 |
| AR110038A1 (es) * | 2016-05-26 | 2019-02-20 | Kalyra Pharmaceuticals Inc | Compuestos inhibidores de egfr; composición farmacéutica que lo comprende; métodos para mejorar o tratar un cáncer; método para inhibir la replicación de un crecimiento maligno o un tumor; métodos para inhibir la actividad del egfr; y usos de los compuestos |
| CN106083736A (zh) * | 2016-06-21 | 2016-11-09 | 郑州泰基鸿诺医药股份有限公司 | 一种嘧啶类化合物、egfr抑制剂及其应用 |
| JP6740452B2 (ja) | 2016-07-26 | 2020-08-12 | 深▲セン▼市塔吉瑞生物医▲薬▼有限公司Shenzhen TargetRx,Inc. | プロテインチロシンキナーゼの活性を阻害するためのアミノピリミジン系化合物 |
| CN107793413B (zh) * | 2016-09-05 | 2021-09-28 | 上海科州药物研发有限公司 | 嘧啶杂环化合物及其制备方法和应用 |
| AU2017326029B2 (en) * | 2016-09-19 | 2019-11-21 | Nanjing Chuangte Pharmaceutical Technology Co., Ltd | Deuterated 3-(4,5-substituted pyrimidinamine) phenyl derivatives and applications thereof |
| CN107973783B (zh) * | 2016-10-21 | 2024-09-06 | 正大天晴药业集团股份有限公司 | 作为erk抑制剂的苯胺嘧啶衍生物 |
| CN108057036B (zh) * | 2016-11-07 | 2023-06-13 | 正大天晴药业集团股份有限公司 | 一种egfr抑制剂的固体药物组合物 |
| CN108503627A (zh) * | 2017-04-19 | 2018-09-07 | 郑州泰基鸿诺医药股份有限公司 | 用作egfr抑制剂的2,4-二取代苯-1,5-二胺衍生物及其应用 |
| CN110753691B (zh) * | 2017-06-02 | 2024-02-02 | 豪夫迈·罗氏有限公司 | 用于治疗性和/或预防性治疗癌症的化合物 |
| EP3659307A4 (en) * | 2017-07-28 | 2021-09-22 | Yale University | ANTI-CANCER MEDICINAL PRODUCTS AND THEIR MANUFACTURING AND USE PROCEDURES |
| CN108947974B (zh) * | 2017-08-30 | 2020-06-05 | 深圳市塔吉瑞生物医药有限公司 | 一种氨基嘧啶类化合物及包含该化合物的组合物及其用途 |
| CN109503573A (zh) * | 2017-09-14 | 2019-03-22 | 昆明圣加南生物科技有限公司 | 2-取代苯胺基嘧啶衍生物及其用途 |
| TWI702205B (zh) * | 2017-10-06 | 2020-08-21 | 俄羅斯聯邦商拜奧卡德聯合股份公司 | 表皮生長因子受體抑制劑 |
| CN108017633A (zh) * | 2018-01-30 | 2018-05-11 | 天津大学 | N-[5-(嘧啶-2-氨基)-2,4-二取代苯基]-2-氟代丙烯酰胺衍生物及应用 |
| WO2019177375A1 (ko) * | 2018-03-13 | 2019-09-19 | 포로노이바이오 주식회사 | 2, 4, 5-치환된 피리미딘 유도체, 이의 제조방법 및 이를 유효성분으로 포함하는 암의 예방 또는 치료용 약학적 조성물 |
| CN110272420A (zh) * | 2018-03-16 | 2019-09-24 | 江苏正大丰海制药有限公司 | 氘代3-(4,5-取代氨基嘧啶)苯基化合物单甲磺酸盐晶型及其制备方法 |
| EP4234547A3 (en) | 2018-05-14 | 2023-11-01 | Takeda Pharmaceutical Company Limited | Pharmaceutical salts of pyrimidine derivatives and method of treating disorders |
| AU2019269815B2 (en) * | 2018-05-15 | 2024-08-01 | Jiangsu Hansoh Pharmaceutical Group Co., Ltd. | Pharmaceutical composition comprising small molecule EGFR inhibitor and preparation method therefor |
| MX2020012540A (es) * | 2018-05-23 | 2021-02-16 | Jiangsu Hengrui Medicine Co | Uso de inhibidor de cdk4/6 en combinacion con inhibidor del egfr en la preparacion de medicamento para tratar enfermedades tumorales. |
| WO2019228330A1 (zh) * | 2018-05-29 | 2019-12-05 | 深圳市塔吉瑞生物医药有限公司 | 取代的苯并[d]咪唑类化合物及其药物组合物 |
| CN110652514A (zh) * | 2018-06-29 | 2020-01-07 | 上海翰森生物医药科技有限公司 | 第三代egfr抑制剂的制药用途 |
| CN110698461B (zh) * | 2018-07-09 | 2024-04-05 | 上海翰森生物医药科技有限公司 | 第三代egfr抑制剂的制备方法 |
| WO2020052575A1 (zh) * | 2018-09-12 | 2020-03-19 | 江苏恒瑞医药股份有限公司 | Jak激酶抑制剂与egfr抑制剂联合在制备治疗肿瘤疾病的药物中的用途 |
| CN109761960B (zh) * | 2019-02-25 | 2021-08-31 | 江苏豪森药业集团有限公司 | 抗耐药抗肿瘤egfr抑制剂的制备方法 |
| CN111606889B (zh) * | 2019-02-25 | 2023-03-07 | 上海翰森生物医药科技有限公司 | 4-(1-环丙基-1h-吲哚-3-基)-n-苯基嘧啶-2-胺衍生物的制备方法 |
| CN110041302B (zh) * | 2019-03-01 | 2021-11-30 | 南方医科大学 | 2-氨基-4-取代吡啶衍生物及其合成方法和应用 |
| KR102220624B1 (ko) * | 2019-03-15 | 2021-02-25 | 김민서 | 드라이 플라워의 처리 방법 및 드라이 플라워의 처리제 |
| EP3943491A4 (en) | 2019-03-19 | 2022-11-23 | Voronoi Inc. | HETEROARYL DERIVATIVE, PROCESS FOR THE PRODUCTION THEREOF AND PHARMACEUTICAL COMPOSITION THEREFORE AS AN ACTIVE INGREDIENT |
| EP3942045A1 (en) | 2019-03-21 | 2022-01-26 | Onxeo | A dbait molecule in combination with kinase inhibitor for the treatment of cancer |
| CN111747950B (zh) * | 2019-03-29 | 2024-01-23 | 深圳福沃药业有限公司 | 用于治疗癌症的嘧啶衍生物 |
| WO2020233669A1 (zh) * | 2019-05-22 | 2020-11-26 | 上海翰森生物医药科技有限公司 | 含吲哚类衍生物抑制剂、其制备方法和应用 |
| US20220229072A1 (en) | 2019-06-04 | 2022-07-21 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of cd9 as a biomarker and as a biotarget in glomerulonephritis or glomerulosclerosis |
| CN110183399A (zh) * | 2019-06-05 | 2019-08-30 | 辽宁科隆精细化工股份有限公司 | 一种合成4-(2-(n,n-二甲氨基)乙基)吗啉的方法 |
| US20220227781A1 (en) * | 2019-06-20 | 2022-07-21 | Oncobix Co., Ltd. | Pyrimidine derivative inhibiting growth of cancer cell and medicinal use thereof |
| CN110684016A (zh) * | 2019-09-27 | 2020-01-14 | 上海应用技术大学 | 一种含氟的azd9291衍生物及其制备方法和应用 |
| KR102383200B1 (ko) * | 2019-10-18 | 2022-04-06 | 한국화학연구원 | 피리미딘 유도체, 이의 제조방법 및 이를 유효성분으로 함유하는 암의 예방 또는 치료용 약학적 조성물 |
| EP4054579A1 (en) | 2019-11-08 | 2022-09-14 | Institut National de la Santé et de la Recherche Médicale (INSERM) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| WO2021094379A1 (en) | 2019-11-12 | 2021-05-20 | Astrazeneca Ab | Epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of cancer |
| WO2021104305A1 (zh) * | 2019-11-26 | 2021-06-03 | 上海翰森生物医药科技有限公司 | 含氮多环类衍生物抑制剂、其制备方法和应用 |
| KR20220130190A (ko) | 2020-01-20 | 2022-09-26 | 아스트라제네카 아베 | 암 치료를 위한 표피성장인자 수용체 티로신 키나제 억제제 |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
| WO2021180238A1 (zh) * | 2020-03-13 | 2021-09-16 | 郑州同源康医药有限公司 | 一类用作激酶抑制剂的化合物及其应用 |
| CN113493420A (zh) * | 2020-03-18 | 2021-10-12 | 南京药石科技股份有限公司 | Egfr酪氨酸激酶抑制剂及其用途 |
| US20210369709A1 (en) | 2020-05-27 | 2021-12-02 | Astrazeneca Ab | EGFR TKIs FOR USE IN THE TREATMENT OF NON-SMALL CELL LUNG CANCER |
| WO2021243596A1 (en) * | 2020-06-03 | 2021-12-09 | InventisBio Co., Ltd. | Aminopyrimidine compounds, preparation methods and uses thereof |
| TW202227425A (zh) * | 2020-09-18 | 2022-07-16 | 南韓商沃若諾伊生物公司 | 雜芳基衍生物、彼之製法、及包含彼作為活性成份之藥學組成物 |
| WO2022105882A1 (zh) * | 2020-11-19 | 2022-05-27 | 上海翰森生物医药科技有限公司 | 一种含吲哚类衍生物的盐、晶型及其制备方法和应用 |
| CN112457299B (zh) * | 2020-12-14 | 2021-12-17 | 江苏豪森药业集团有限公司 | Egfr抑制剂的纯化方法 |
| KR20220085735A (ko) * | 2020-12-14 | 2022-06-22 | 보로노이바이오 주식회사 | 아이소옥사졸리딘 유도체 화합물 및 이의 용도 |
| TW202241890A (zh) * | 2020-12-30 | 2022-11-01 | 俄羅斯聯邦商拜奧卡德聯合股份公司 | 表皮生長因子受體之抑制劑 |
| CN112679448B (zh) * | 2020-12-31 | 2022-08-19 | 苏州昊帆生物股份有限公司 | N-(2-氨基乙基)吗啉的制备方法 |
| EP4335847A1 (en) * | 2021-05-07 | 2024-03-13 | Voronoi Inc. | Heteroaryl derivative, method for preparing same, and pharmaceutical composition comprising same as active ingredient |
| CA3222537A1 (en) * | 2021-05-14 | 2022-11-17 | Nanjing Chuangte Pharmaceutical Technology Co., Ltd | Pharmaceutical formulations |
| IL298474A (en) * | 2021-05-17 | 2023-01-01 | Voronoibio Inc | Compounds of heteroaryl derivatives and uses thereof |
| CN113387935B (zh) * | 2021-07-23 | 2022-06-10 | 苏州雅深智慧科技有限公司 | 抑制三突变表皮生长因子受体酪氨酸激酶的化合物及用途 |
| WO2023187037A1 (en) | 2022-03-31 | 2023-10-05 | Astrazeneca Ab | Epidermal growth factor receptor (egfr) tyrosine kinase inhibitors in combination with an akt inhibitor for the treatment of cancer |
| WO2023209084A1 (en) | 2022-04-28 | 2023-11-02 | Astrazeneca Ab | Condensed bicyclic heteroaromatic compounds and their use in the treatment of cancer |
| US20240116926A1 (en) | 2022-04-28 | 2024-04-11 | Astrazeneca Ab | Heteroaromatic compounds |
| WO2023209088A1 (en) | 2022-04-28 | 2023-11-02 | Astrazeneca Ab | Bicyclic heteroaromatic compounds and their use in the treatment of cancer |
| WO2023209090A1 (en) | 2022-04-28 | 2023-11-02 | Astrazeneca Ab | Bicyclic heteroaromatic compounds and their application in the treatment of cancer |
| TW202417040A (zh) | 2022-06-27 | 2024-05-01 | 瑞典商阿斯特捷利康公司 | 涉及用於治療癌症的上皮生長因子受體酪胺酸激酶抑制劑之組合 |
| TW202421146A (zh) | 2022-07-08 | 2024-06-01 | 瑞典商阿斯特捷利康公司 | 用於治療癌症的上皮生長因子受體酪胺酸激酶抑制劑與hgf受體抑制劑的組合 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011140338A1 (en) * | 2010-05-05 | 2011-11-10 | Gatekeeper Pharmaceuticals, Inc. | Compounds that modulate egfr activity and methods for treating or preventing conditions therewith |
| WO2013169401A1 (en) * | 2012-05-05 | 2013-11-14 | Ariad Pharmaceuticals, Inc. | Compounds for inhibiting cell proliferation in egfr-driven cancers |
| CN103501612A (zh) * | 2011-05-04 | 2014-01-08 | 阿里亚德医药股份有限公司 | 抑制表皮生长因子受体导致的癌症中细胞增殖的化合物 |
| CN103702990A (zh) * | 2011-07-27 | 2014-04-02 | 阿斯利康(瑞典)有限公司 | 2-(2,4,5-取代苯胺)嘧啶衍生物作为egfr调谐子用于治疗癌症 |
| CN104140418A (zh) * | 2014-08-15 | 2014-11-12 | 朱孝云 | 新的2-(2,4,5-取代苯胺)嘧啶衍生物及其用途 |
| CN104761544A (zh) * | 2014-01-03 | 2015-07-08 | 南京波尔泰药业科技有限公司 | Egfr酪氨酸激酶的临床重要突变体的选择性抑制剂 |
| CN104860941A (zh) * | 2014-02-25 | 2015-08-26 | 上海海雁医药科技有限公司 | 2,4-二取代苯-1,5-二胺衍生物及其应用以及由其制备的药物组合物和药用组合物 |
| RU2636584C2 (ru) * | 2009-12-29 | 2017-11-24 | Селджен Авиломикс Рисерч,Инк. | Гетероарильные соединения и их применение |
| RU2677653C2 (ru) * | 2013-07-11 | 2019-01-18 | Ацея Байосайенсиз Инк. | Производные пиримидина в качестве ингибиторов киназы |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011526299A (ja) * | 2008-06-27 | 2011-10-06 | アビラ セラピューティクス, インコーポレイテッド | ヘテロアリール化合物およびそれらの使用 |
| CA2760794C (en) * | 2009-05-05 | 2017-07-25 | Dana Farber Cancer Institute | Egfr inhibitors and methods of treating disorders |
| US20170166598A1 (en) * | 2014-05-13 | 2017-06-15 | Ariad Pharmaceuticals, Inc. | Heteroaryl compounds for kinase inhibition |
| CN111875585B (zh) * | 2014-06-12 | 2023-06-23 | 上海艾力斯医药科技股份有限公司 | 一类激酶抑制剂 |
| US10227342B2 (en) * | 2014-06-19 | 2019-03-12 | Ariad Pharmaceuticals, Inc. | Heteroaryl compounds for kinase inhibition |
| WO2016029839A1 (zh) * | 2014-08-25 | 2016-03-03 | 四川海思科制药有限公司 | 一种(取代的苯基)(取代的嘧啶)胺基衍生物及其制备方法和药物用途 |
| CN105461695B (zh) * | 2014-09-29 | 2018-03-27 | 齐鲁制药有限公司 | 嘧啶或三嗪衍生物及其制备方法和用途 |
| CN111170998B (zh) * | 2014-11-05 | 2023-04-11 | 益方生物科技(上海)股份有限公司 | 嘧啶或吡啶类化合物、其制备方法和医药用途 |
| CN113563332A (zh) * | 2014-12-23 | 2021-10-29 | 达纳-法伯癌症研究所公司 | 作为egfr抑制剂的新的嘧啶和治疗病症的方法 |
| CN107835811B (zh) * | 2015-07-16 | 2019-11-08 | 正大天晴药业集团股份有限公司 | 苯胺嘧啶衍生物及其用途 |
| CN106117185B (zh) * | 2015-08-31 | 2017-11-07 | 广州必贝特医药技术有限公司 | 2,4‑二含氮基团取代嘧啶类化合物及其制备方法和应用 |
-
2015
- 2015-09-30 EP EP15849629.9A patent/EP3205650B1/en active Active
- 2015-09-30 MX MX2017004234A patent/MX2017004234A/es unknown
- 2015-09-30 CN CN201910210387.6A patent/CN111187221B/zh active Active
- 2015-09-30 SI SI201531690T patent/SI3205650T1/sl unknown
- 2015-09-30 ES ES15849629T patent/ES2886887T3/es active Active
- 2015-09-30 PL PL15849629T patent/PL3205650T3/pl unknown
- 2015-09-30 DK DK15849629.9T patent/DK3205650T3/da active
- 2015-09-30 AU AU2015330506A patent/AU2015330506B2/en active Active
- 2015-09-30 JP JP2017517797A patent/JP6691281B2/ja active Active
- 2015-09-30 CN CN201910208542.0A patent/CN111170999B/zh active Active
- 2015-09-30 PT PT158496299T patent/PT3205650T/pt unknown
- 2015-09-30 RS RS20211144A patent/RS62350B1/sr unknown
- 2015-09-30 HU HUE15849629A patent/HUE055233T2/hu unknown
- 2015-09-30 CN CN201910210386.1A patent/CN111171000B/zh active Active
- 2015-09-30 CN CN201580045311.2A patent/CN106661000B/zh active Active
- 2015-09-30 CA CA2959194A patent/CA2959194A1/en active Pending
- 2015-09-30 US US15/517,193 patent/US10259820B2/en active Active
- 2015-09-30 CN CN202311030017.7A patent/CN117050062A/zh active Pending
- 2015-09-30 KR KR1020177012005A patent/KR102589982B1/ko active IP Right Grant
- 2015-09-30 BR BR112017006713-7A patent/BR112017006713B1/pt active IP Right Grant
- 2015-09-30 LT LTEPPCT/CN2015/091189T patent/LT3205650T/lt unknown
- 2015-09-30 RU RU2017114687A patent/RU2702631C2/ru active
- 2015-09-30 CN CN202311029815.8A patent/CN117069700A/zh active Pending
- 2015-09-30 WO PCT/CN2015/091189 patent/WO2016054987A1/zh active Application Filing
- 2015-09-30 CN CN202311339167.6A patent/CN117402143A/zh active Pending
- 2015-10-07 TW TW104133007A patent/TWI704138B/zh active
-
2017
- 2017-02-27 ZA ZA2017/01465A patent/ZA201701465B/en unknown
-
2018
- 2018-12-03 US US16/207,968 patent/US10428081B2/en active Active
-
2021
- 2021-09-01 HR HRP20211392TT patent/HRP20211392T1/hr unknown
- 2021-09-07 CY CY20211100794T patent/CY1124476T1/el unknown
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2636584C2 (ru) * | 2009-12-29 | 2017-11-24 | Селджен Авиломикс Рисерч,Инк. | Гетероарильные соединения и их применение |
| WO2011140338A1 (en) * | 2010-05-05 | 2011-11-10 | Gatekeeper Pharmaceuticals, Inc. | Compounds that modulate egfr activity and methods for treating or preventing conditions therewith |
| CN103501612A (zh) * | 2011-05-04 | 2014-01-08 | 阿里亚德医药股份有限公司 | 抑制表皮生长因子受体导致的癌症中细胞增殖的化合物 |
| CN103702990A (zh) * | 2011-07-27 | 2014-04-02 | 阿斯利康(瑞典)有限公司 | 2-(2,4,5-取代苯胺)嘧啶衍生物作为egfr调谐子用于治疗癌症 |
| EA201391491A1 (ru) * | 2011-07-27 | 2014-08-29 | Астразенека Аб | Производные 2-(2,4,5-замещенного анилино)пиримидина в качестве модуляторов egfr, полезных для лечения рака |
| EA201690328A1 (ru) * | 2011-07-27 | 2016-11-30 | Астразенека Аб | Производные 2-(2,4,5-замещенного анилино)пиримидина в качестве модуляторов egfr, полезных для лечения рака |
| EA201792394A2 (ru) * | 2011-07-27 | 2018-03-30 | Астразенека Аб | Производные 2-(2,4,5-замещенного анилино)пиримидина в качестве модуляторов egfr, полезных для лечения рака |
| WO2013169401A1 (en) * | 2012-05-05 | 2013-11-14 | Ariad Pharmaceuticals, Inc. | Compounds for inhibiting cell proliferation in egfr-driven cancers |
| RU2677653C2 (ru) * | 2013-07-11 | 2019-01-18 | Ацея Байосайенсиз Инк. | Производные пиримидина в качестве ингибиторов киназы |
| CN104761544A (zh) * | 2014-01-03 | 2015-07-08 | 南京波尔泰药业科技有限公司 | Egfr酪氨酸激酶的临床重要突变体的选择性抑制剂 |
| CN104860941A (zh) * | 2014-02-25 | 2015-08-26 | 上海海雁医药科技有限公司 | 2,4-二取代苯-1,5-二胺衍生物及其应用以及由其制备的药物组合物和药用组合物 |
| RU2649001C1 (ru) * | 2014-02-25 | 2018-03-29 | Шанхай Хайянь Фармасьютикал Текнолоджи Ко., Лтд. | Производные 2,4-дизамещенного фенилен-1,5-диамина и их применения, фармацевтические композиции и фармацевтически приемлемые композиции, полученные из них |
| CN104140418A (zh) * | 2014-08-15 | 2014-11-12 | 朱孝云 | 新的2-(2,4,5-取代苯胺)嘧啶衍生物及其用途 |
| EP3181559B1 (en) * | 2014-08-15 | 2018-10-03 | Changzhou Runnor Biological Technology Co., Ltd | 2-(2,4,5-substituted aniline) pyrimidine derivative, pharmaceutical composition and use thereof |
Non-Patent Citations (2)
| Title |
|---|
| приоритет 11.07.2013 и 02.01.2014. * |
| приоритет 25.02.2014. * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2733412C2 (ru) * | 2016-03-22 | 2020-10-01 | Цзянсу Хансох Фармасьютикал Груп Ко., Лтд. | Поликристаллическая форма свободного основания или соли присоединения кислоты ингибитора egfr, способ её получения и применение |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2702631C2 (ru) | Ингибитор egfr и его получение и применение | |
| CA2583737C (en) | Compounds and compositions as protein kinase inhibitors | |
| CN105461695B (zh) | 嘧啶或三嗪衍生物及其制备方法和用途 | |
| JP2022020860A (ja) | サイクリン依存性キナーゼ7(cdk7)の阻害剤 | |
| AU2015407300B2 (en) | 2-Arylamino pyridine, pyridine or triazine derivative, preparation method and use thereof | |
| WO2018045957A1 (zh) | 一种cdk4/6抑制剂及其制备方法和应用 | |
| KR20200115583A (ko) | Cdk4 및 cdk6 억제제로서의 2h-인다졸 유도체 및 그의 치료 용도 | |
| PT1899329E (pt) | Derivados de benzimidazole substituídos por pirimidina como inibidores de proteína quinase | |
| EP2933248B1 (en) | Novel renin inhibitor | |
| JP2018528193A (ja) | インドール誘導体、その調製方法および医薬におけるその使用 | |
| JP2023022230A (ja) | Ehmt2阻害剤としての置換された融合二環式または三環式複素環化合物 | |
| TWI819470B (zh) | Fgfr激酶抑制劑及其應用 | |
| CN112313207B (zh) | 一种氰基取代吡啶及氰基取代嘧啶类化合物、制备方法及其应用 | |
| CN115803325B (zh) | 一种egfr抑制剂及其制备方法和应用 | |
| WO2017008708A1 (zh) | 吲唑类化合物的制备方法和用途 | |
| CN115894381A (zh) | 一种2,4,5-三取代嘧啶类化合物及其制备方法和用途 | |
| CN114539226A (zh) | 一种含吲哚类衍生物自由碱的晶型及其制备方法和应用 | |
| WO2024194460A2 (en) | Atm kinase inhibitors | |
| AU2013204962B2 (en) | Polymorphic form of a mesylate salt of n-(2-{2-dimethylaminoethyl-methylamino}-4-methoxy-5-{[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino}phenyl)prop-2-enamide | |
| CN118359627A (zh) | 具有四环并环结构的mta协同性prmt5抑制剂化合物 | |
| EA041747B1 (ru) | Ингибиторы g12c kras, содержащая их фармацевтическая композиция и использующий их способ лечения рака |