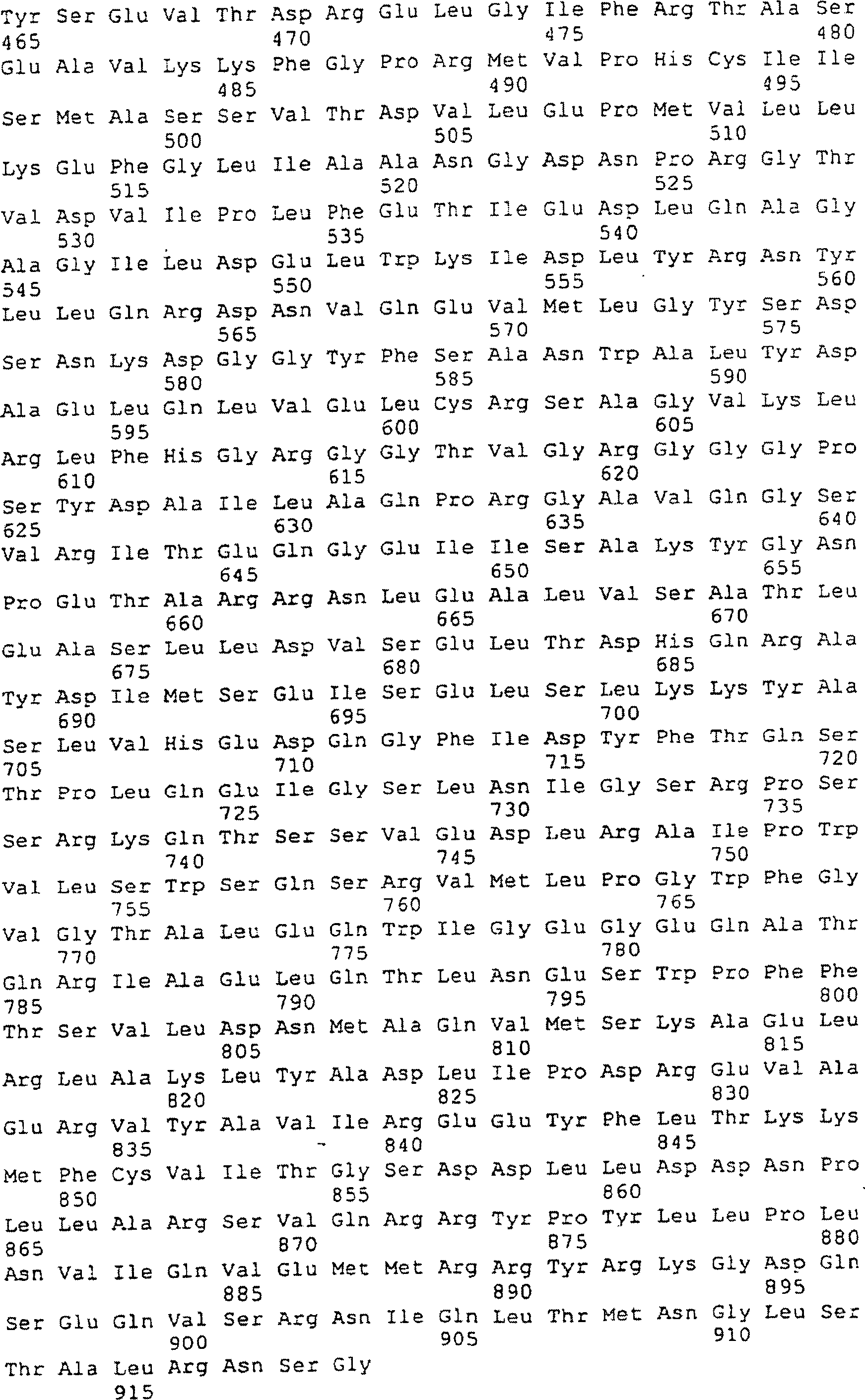-
Technischer
Hintergrund
-
Die
vorliegende Erfindung betrifft ein Verfahren zum Herstellen von
L-Lysin durch Kultivieren eines Microorganismus, der durch Modifizieren
eines coryneformen Bakteriums erhalten wird, das für die fermentative Produktion
der Aminosäure
oder ähnliche
mit Hilfe einer auf Gentechnik basierenden Technik.
-
L-Lysin,
welches als Futterzusatzmittel verwendet wird, wird für gewöhnlich durch
ein fermentatives Verfahren unter Verwendung eines L-Lysin-produzierenden Mutantenstamms
hergestellt, der zu den corynoformen Bakterien gehört. Zahlreiche
L-Lysin-herstellende Bakterien, die gegenwärtig bekannt sind, sind solche,
die durch eine artifizielle Mutation, ausgehend von Wildtypstämmen, die
zu den coryneformen Bakterien gehören, erzeugt wurden.
-
Was
das coryneforme Bakterium angeht, ist ein Vektorplasmid offenbart
worden, das autonom in Bakterienzellen replizierbar ist, und ein
Medikamenten-Resistenz-Markergen hat (siehe US Patent Nr. 4,514,502) sowie
ein Verfahren zum Einschleusen eines Gens in Bakterienzellen (z.
B. Japanische Patentanmeldung Offenlegungsschrift Nr. 2-207791).
Es ist ebenfalls eine Möglichkeit
zum Züchten
eines L-Threonin- oder L-Isoleucin-produzierenden Bakteriums offenbart
worden, in dem die oben beschriebenen Techniken verwendet werden
(siehe US Patent Nr: 4,452,890 und 4,442,208). Was das Züchten eines
L-Lysin produzierenden Bakteriums angeht, ist eine Technik bekannt,
bei der ein Gen, das an der Lysin-Biosynthese teilhat in ein Vektorplasmid
eingeschlossen wurde, um das Gen in Bakterienzellen zu amplifizieren
(siehe z. B. Japanische Patentanmeldung Offenlegungsschrift Nr.
56-160997).
-
Bekannte
Gene der L-Lysin-Biosynthese schließen beispielsweise ein Dihydrodipicolinatreductasegen (Japanische
Patentanmeldung Offenlegungsschrift Nr. 7-75578) und ein Diaminopimelatdehydrogenasegen (Ishino,
S. et al., Nucleic Acids Res., 15, 3917 (1987)) ein, bei denen ein
Gen, das an der L-Lysin-Biosynthese teilhat, kloniert worden ist,
wie auch ein Phosphoenolpyruvatcarboxylasegen (Japanische Patentanmeldung Offenlegungsschrift
Nr. 60-87788), ein Dihydrodipicolinatsynthasegen (Japanische Patentanmel dung
Offenlegungsschrift Nr. 6-55149), und ein Diaminopimelatdecarboxylasegen
(Japanische Patentanmeldung Offenlegungsschrift Nr. 60-62994), bei
denen eine Amplifikation eines Gens die L-Lysin-Produktionsfähigkeit
beeinflusst.
-
Bezüglich der
Enzyme, die an der L-Lysin-Biosynthese teilhaben, ist der Fall bekannt,
dass ein Enzym Rückkoppelungsinhibierung
unterworfen ist, wenn es als Wildtyp verwendet wird. In diesem Fall
wird die L-Lysin-Produktionsfähigkeit
durch Einschleusen eines Enzymgens verbessert, das eine solche Mutation
hat, dass die Rückkoppelungsinhibierung
desensibilisiert ist. Als solche bekannte Gene schließen beispielsweise
spezifisch ein Aspartokinasegen ein (Internationale Veröffentlichung
Pamphlet von WO 94/25605).
-
Wie
oben beschrieben wurde, sind einige erfolgreiche Ergebnisse mit
Hilfe einer Amplifizierung von Genen für das L-Lysin-Biosynthesesystem,
oder die Einschleusung von mutierten Genen erzielt worden. Beispielsweise
stellt ein coryneformes Bakterium, das ein mutiertes Aspartokinasegen
mit desensibilisierter konzertierter Inhibierung durch Lysin und
Threonin beherbergt, eine beträchtliche
Menge an L-Lysin (ungefähr
25 g/l) her. Jedoch leidet dieses Bakterium am Nachteil der Wachstumsgeschwindigkeit,
verglichen mit einem Bakterium, das kein mutiertes Aspartokinasegen
beherbergt. Es ist ebenfalls berichtet worden, dass die L-Lysin-Produktionsfähigkeit
weiter durch das Einschleusen eines Dihydrodipicolinasesynthasegens,
zusätzlich
zu einem mutierten Aspartokinasegen, verbessert werden kann (Applied
and Environmental Microbiology, 57 (6), 1746–1752 (1991)). Jedoch leidet
ein solches Bakterium weiter an einer Abnahme der Wachstumsgeschwindigkeit.
-
Es
ist noch kein Fall berichtet worden, bei dem beabsichtigt wird,
das Wachstum zu verbessern, in dem ein Gen für die L-Lysin-Biosynthese ebenfalls
verstärkt
wurde. Unter den vorliegenden Umständen ist kein Fall für coryneforme
Bakterien bekannt, bei denen irgend jemand es geschafft hat, eine
beträchtliche
Verbesserung der L-Lysin-Ausbeute ohne die Beeinträchtigung
des Wachstums zu erreichen, indem eine Vielzahl an Genen für die L-Lysin-Biosynthese
kombiniert wurden.
-
Zusammenfassung der Erfindung
-
Ein
Ziel der vorliegenden Erfindung liegt darin, die L-Lysin-Ausbeute
ohne die Beeinträchtigung
des Wachstums eines coryneformen Bakteriums zu verbessern, indem
eine Vielzahl an Genen der L-Lysin-Biosynthese in Kombination in
den coryneformen Bakterien verstärkt
werden.
-
Wenn
die gewünschte
Substanz fermentativ unter Verwendung eines Microorganismus hergestellt wird,
sind die Produktionsgeschwindigkeit wie auch die Ausbeute der gewünschten
Substanz bezogen auf das hinzugefügte Material ein außerordentlich
wichtiger Faktor. Eine gewünschte
Substanz kann merklich günstiger
durch Erhöhen
der Produktionsgeschwindigkeit pro Einheit der Fermentationsausrüstung hergestellt
werden. Daher ist es industriell äußerst bedeutsam, dass die fermentative
Ausbeute und die Produktionsgeschwindigkeit miteinander kompatibel
sind. Die vorliegende Erfindung stellt eine Lösung des Problems, das oben
beschrieben worden ist, bereit, um L-Lysin fermentativ unter Verwendung
eines coryneformen Bakteriums herzustellen.
-
Das
Prinzip der vorliegenden Erfindung beruht auf der Tatsache, dass
das Wachstum eines coryneformen Bakteriums verbessert werden kann,
und dass die L-Lysin-Produktionsgeschwindigkeit durch Verstärken einer
DNA-Sequenz, die für
ein Aspartokinase kodiert, bei der die Rückkoppelungsinhibierung durch
L-Lysin und L-Threonin im Wesentlichen desensiblisiert ist, und
einer DNA-Sequenz, die für
eine Diaminopimelatdecarboxylase, verglichen mit dem Fall, in dem
diese DNA-Sequenzen jeweils einzeln verstärkt sind, verbessert werden
kann.
-
Nach
einem ersten Aspekt der vorliegenden Erfindung wird eine rekombinante
DNA bereitgestellt, die autonom in Zellen coryneformer Bakterien
replizierbar ist, umfassend eine DNA-Sequenz, die für eine Aspartikinase
kodiert, bei der die Rückkoppelungsinhibierung
durch L-Lysin und L-Threonin im Wesentlichen desensibilisiert ist,
und eine DNA-Sequenz, die für
eine Diaminopimelatdecarboxylase kodiert. Die rekombinante DNA,
die weiter eine DNA-Sequenz umfasst, die für eine Phosphoenolpyruvatcarboxylase
kodiert, wird ebenfalls bereitgestellt.
-
Nach
einen zweiten Aspekt der vorliegenden Erfindung wird ein coryneformes
Bakterium bereitgestellt, das eine Aspartokinase beherbergt, in
der die Rückkoppelungsinhibierung
durch L-Lysin und L-Threonin im Wesentlichen desensibilisiert ist,
und die eine verstärkte
DNA-Sequenz umfasst, die für
eine Diaminopimelatdecarboxylase kodiert. Ein coryneformes Bakterium,
das weiter eine verstärkte
DNA-Sequenz umfasst, die für
eine Phosphoenolpyruvatcarboxylase kodiert, wird ebenfalls bereitgestellt.
-
Nach
einem dritten Aspekt der vorliegenden Erfindung wird ein Verfahren
zur Herstellung von L-Lysin bereitgestellt, das die Schritte der
Kultivierung irgendeines coryneformen Bakteriums, das oben beschrieben worden
ist, in einem geeigneten Medium, um zu ermöglichen, dass L-Lysin herge stellt
wird, und in einer Kultur des Bakteriums akkumuliert, und das Einsammeln
des L-Lysins aus der Kultur umfasst.
-
Nachfolgend
wird die Aspartokinase, falls notwendig, hier als „AK" bezeichnet, ein
Gen, das für
AK kodiert wird bezeichnet als „lysC", AK, die hinsichtlich der Rückkoppelungsinhibierung
durch L-Lysin und L-Threonin desensibiliert ist, wird als „mutierte
AK" bezeichnet,
und ein Gen, das für
eine mutierte AK kodiert, wird als „mutierte lysC" bezeichnet. Eine
Diaminopimelatdecarboxylase wird, falls notwendig, als „DDC" bezeichnet, ein
Gen, das für
die DDC kodiert wird als „lysA" bezeichnet, eine
Phosphoenolpyruvatcarboxylase wird als „PEPC" bezeichnet, und ein Gen, das für PEPC kodiert,
wird als „ppc" bezeichnet.
-
Coryneforme
Bakterien, auf die in der vorliegenden Erfindung Bezug genommen
wird, sind eine Gruppe von Mikroorganismen, wie in Bergey's Manual of Determinative
Bacteriology, 8. Ausgabe, Seite 599 (1974) definiert, welche aerobe
Gram-positive nicht-acid-fast-Stäbchen
sind, die keine Sporenbildende Fähigkeit
aufweisen. Die coryneformen Bakterien schließen Bakterien ein, die zum
Genus Corynebacterium gehören,
Bakterien, die zum Genus Brevibacterium gehören, die zuvor in den Genus
Brevibacterium klassifiziert worden sind, aber als Bakterien zusammengefasst
worden sind, die zum Genus Corynebacterium gehören, und Bakterien, die zum
Genus Brevibacterium gehören,
welche Bakterien, die zum Genus Corynebacterium gehören, nahe
verwandt sind.
-
Erfindungsgemäß kann die
Produktionsmenge und die Produktionsgeschwindigkeit von L-Lysin
in coryneformen Bakterien verbessert werden.
-
Kurze Beschreibung der
Zeichnungen
-
1 stellt
ein Verfahren der Herstellung der Plasmide p399AK9B und p399AKYB
dar, umfassend das mutierte lysC.
-
2 stellt
ein Verfahren der Herstellung des Plasmids p299LASA, umfassend lysA,
dar.
-
3 stellt
ein Verfahren zur Herstellung des Plasmids pLASAB, umfassend lysA
und Brevi.-ori, dar.
-
4 stellt
ein Verfahren zum Herstellen eines Plasmids pAKPFds, umfassend das
PEPC-Strukturgen, dar.
-
5 stellt
ein Verfahren zum Konstruieren von neuen Klonierungsvektoren, pVK6
und pVK7, für
coryneforme Bakterien dar.
-
6 stellt
ein Verfahren zur Herstellung des Plasmids pPwm, umfassend eine
Wildtyp-Hochexpressions-ppc, dar.
-
7 stellt
ein Verfahren zur Herstellung des Plasmids pCL, umfassend mutiertes
lysC, lysA und Brevi.-ori, dar.
-
8 stellt
ein Verfahren zur Herstellung eines Plasmids pDPSB, umfassend dapA
und Brevi.-ori, dar.
-
9 stellt
das Verfahren zur Herstellung des Plasmids pDPRB, umfassend dapB
und Brevi.-ori, dar.
-
10 stellt
das Verfahren zur Konstruktion des Plasmids pPK4D, umfassend ddh
und Brevi.-ori, dar.
-
11 stellt
ein Verfahren zur Konstruktion des Plasmids pCRCAB, umfassend lysC,
dapA und Brevi.-ori, dar.
-
12 stellt
das Verfahren zur Herstellung des Plasmids pCB, umfassend mutiertes
lysC, dapB und Brevi.-ori, dar.
-
13 stellt
das Verfahren zur Konstruktion des Plasmids pCD, umfassend mutiertes
lysC und ddh, dar.
-
Detaillierte Beschreibung
der Erfindung
-
<1> Herstellung
von Genen für
die L-Lysin-Biosynthese, die in der vorliegenden Erfindung verwendet
werden
-
Die
Gene für
die L-Lysin-Biosynthese, die in der vorliegenden Erfindung verwendet
werden, werden jeweils durch Herstellen chromosomaler DNA aus einem
Bakterium als DNA-Donor, Konstruieren einer chromosomalen DNA-Bibliothek
unter Verwendung eines Plasmidvektors oder ähnlicher, das Auswählen eines Stammes,
der das gewünschte
Gen beherbergt, und das Gewinnen aus dem ausgewählten Stamm von rekombinanter
DNA, in die das Gen inseriert worden ist, gewonnen. Der DNA-Donor
für das
Gen für
die L-Lysin-Biosynthese, das erfindungsgemäß verwendet wird, ist nicht
spezifisch limitiert, unter der Voraussetzung, dass das gewünschte Gen
für die
L-Lysin-Biosynthese
ein Enzymprotein exprimiert, das in Zellen coryneformer Bakterien
funktioniert. Jedoch ist der DNA-Donor bevorzugterweise ein coryneformes
Bakterium.
-
Alle
Gene lysC, dapA und ppc, die aus einem coryneformen Bakterium stammen,
haben bekannte Sequenzen. Daher können sie durch das Durchführen einer
Amplifikation gemäß dem Verfahren
der Polymerase-Kettenreaktion gewonnen werden (PCR, siehe White,
T. J. et al., Trends Genet., 5, 185 (1989)).
-
Jedes
der Gene für
die L-Lysin-Biosynthese, das erfindungsgemäß verwendet wird, ist in Übereinstimmung
mit bestimmten Verfahren, die unten beispielhaft dargestellt werden,
zu gewinnen.
-
(1) Herstellung von mutiertem
lysC
-
Ein
DNA-Fragment, das mutiertes lysC enthält, kann aus einem mutierten
Stamm hergestellt werden, in dem die synergistische Rückkoppelungsinhibierung
der AK-Aktivität
durch L-Lysin und L-Threonin im Wesentlichen desensibilisiert ist
(Internationale Veröffentlichung
Pamphlet der WO 94/25605). Ein solcher mutierter Stamm kann beispielsweise
aus einer Gruppe von Zellen gewonnen werden, die aus einem Wildtypstamm des
coryneformen Bakteriums hervorgehen, welcher einer Mutationsbehandlung
unterworfen worden ist, indem eine gewöhnliche Mutationsbehandlung
wie eine ultraviolette Bestrahlung und die Behandlung mit einem Mutationsmittel
wie N-Methyl-N'-nitro-N-nitrosoguanidin
(NTG) durchgeführt
worden ist. Die AK-Aktivität
kann unter Verwendung eines durch Miyajima, R. et al. in The Journal
of Biochemistry (1968), 63 (2), 139–148, beschriebenen Verfahrens
gemessen werden. Der am meisten bevorzugte derartige Mutantenstamm
wird durch das L-Lysin-produzierende Bakterium AJ3445 (FERM P-1944)
dargestellt, das durch eine Mutationsbehandlung aus einem Wildtypstamm
von Brevibacterium lactofermentum ATCC 13869 (mit dem jetzt geänderten
Namen Corynebacterium glutamicum) abstammt.
-
Alternativ
kann mutiertes lysC ebenfalls durch eine in vitro Mutationsbehandlung
von Plasmid-DNA, die Wildtyp-lysC enthält, erhältlich sein. In einem weiteren
Aspekt ist Information, die über
die Mutation speziell bekannt ist, um eine synergistische Rückkoppelungsinhibierung
von AK durch L-Lysin und L-Threonin zu desensibilisieren, vorhanden
(Internationale Veröffentlichung
Pamphlet der WO 94/25605). Daher kann mutiertes lysC auf Grundlage
dieser Information gemäß, beispielsweise
nach dem positionsabhängigen
Mutageneseverfahren ebenfalls aus Wildtyp-lysC hergestellt werden.
-
Ein
Fragment, das lysC umfasst, kann aus einem coryneformen Bakterium
durch Herstellen chromosomaler DNA, beispielsweise gemäß dem Verfahren
von Saito und Miura (H. Saito und K. Miura, Biochem. Biophys. Acta,
72, 619 (1963)) isoliert werden, und indem lysC gemäß dem Polymerase-Kettenreaktionsverfahren
amplifiziert wird (PCR; siehe White, T. J. et al., Trends Genet.,
5, 185 (1989)).
-
DNA-Primer
werden beispielhaft durch Einzelstränge der DNAs als 23-mer und
21-mer mit den Nucleotidsequenzen, die in den SEQ-Nummern 1 und
2 in der Sequenzliste gezeigt sind, dargestellt, um beispielsweise
einen Bereich von ungefähr
1.643 Basenpaaren zu amplifizieren, der für lysC auf Grundlage von Corynebacterium
glutamicum bekannten Sequenz kodiert (siehe Molecular Microbiology
(1991), 5 (5), 1197–1204; Mol.
Gen. Genet. (1990), 224, 317–324).
DNA kann in Übereinstimmung
mit einem gewöhnlichen
Verfahren un ter Verwendung eines DNA-Synthesegeräts vom Modells 380B synthetisiert
werden, das von Applied Biosystems hergestellt wird, und unter Verwendung
des Phosphoamiditverfahrens (siehe Tetrahedron Letters (1981), 22,
1859). Eine PCR kann unter Verwendung eines DNA-Thermo-Cyclers vom
Modell PJ2000 hergestellt werden, das von Takara Shuzo produziert
wird, und unter Verwendung von Taq DNA-Polymerase gemäß dem von
dem Lieferanten beschriebenen Verfahren.
-
Es
wird bevorzugt, dass lysC, welches durch PCR amplifiziert wird,
in Vektor-DNA ligiert wird, welche in E.coli-Zellen und/oder coryneformen
Bakterien autonom replizierbar ist, um rekombinante DNA herzustellen, und
dass die rekombinante DNA zuvor in E.coli-Zellen eingeschleust wurde.
Eine solche Vorgehensweise macht die folgenden Arbeitsschritte einfach.
Der autonom in E.coli-Zellen replizierbare Vektor ist bevorzugterweise
ein Plasmidvektor, welcher bevorzugterweise autonom in den Wirtszellen
replizierbar ist, einschließlich beispielsweise
pUC19, pUC18, pBR322, pHSG299, pHSG399, pHSG398 und RSF1010.
-
Wenn
ein DNA-Fragment mit der Fähigkeit,
dem Plasmid zu ermöglichen,
autonom in coryneformen Bakterien replizierbar zu sein, in diese
Vektoren inseriert ist, können
sie als sogenannte Shuttle-Vektoren verwendet werden, die autonom
sowohl in E.coli als auch in coryneformen Bakterien replizierbar
sind.
-
Ein
solcher Shuttle-Vektor schließt
die folgenden ein. Mikroorganismen, die jeden dieser Vektoren beherbergen,
und die Zugangsnummern in internationalen Hinterlegungsstellen (in
Klammern) sind angegeben.
pHC4: Escherichia coli AJ12617 (FERM
BP-3532)
pAJ655: Escherichia coli AJ11882 (FERM BP-136) Corynebacterium
glutamicum SR 8201 (ATCC 39135)
pAJ1844: Escherichia coli AJ11883
(FERM BP-137) Corynebacterium glutamicum SR8202 (ATCC 39136)
pAJ611:
Escherichia coli AJ11884 (FERM BP-138)
paJ3148: Corynebacterium
glutamicum SR8203 (ATCC 39137)
pAJ440: Bacillus subtilis AJ11901
(FERM BP-140)
-
Diese
Vektoren sind aus den hinterlegten Mikroorganismen wie folgt zu
gewinnen. Zellen, die sich in der logarithmischen Wachstumsphase
befinden, wurden unter Verwendung von Lysozym und SDS lysiert, gefolgt
von einer Auftrennung des Lysats durch Zentrifugieren bei 30.000
X g, um einen Überstand
zu gewinnen. Zu dem Überstand
wird Polyethylenglycol gegeben, gefolgt von der Fraktionierung und
Aufreiniung mit Hilfe einer Cäsiumchlorid-Ethidiumbromid-Gleichgewichts-Dichtegradienten-Zentrifugation.
-
E.coli
kann durch Einschleusen eines Plasmids, beispielsweise in Übereinstimmung
mit dem Verfahren von D. M. Morrison (Methods in Enzymology, 68,
326 (1979)), oder einem Verfahren, bei dem die Empfängerzellen
mit Calciumchlorid behandelt werden, um die Permeabilität der DNA
zu erhöhen
(Mandel, M. und Higa, A., J. Mol. Biol., 53, 159 (1970)) transformiert
werden.
-
Wildtyp-lysC
wird gewonnen, wenn lysC aus einem AK-Wildtypstamm isoliert wird,
während
mutierte lysC gewonnen wird, wenn lysC gemäß dem oben beschriebenen Verfahren
aus einem mutierten AK-Mutantenstamm isoliert wird.
-
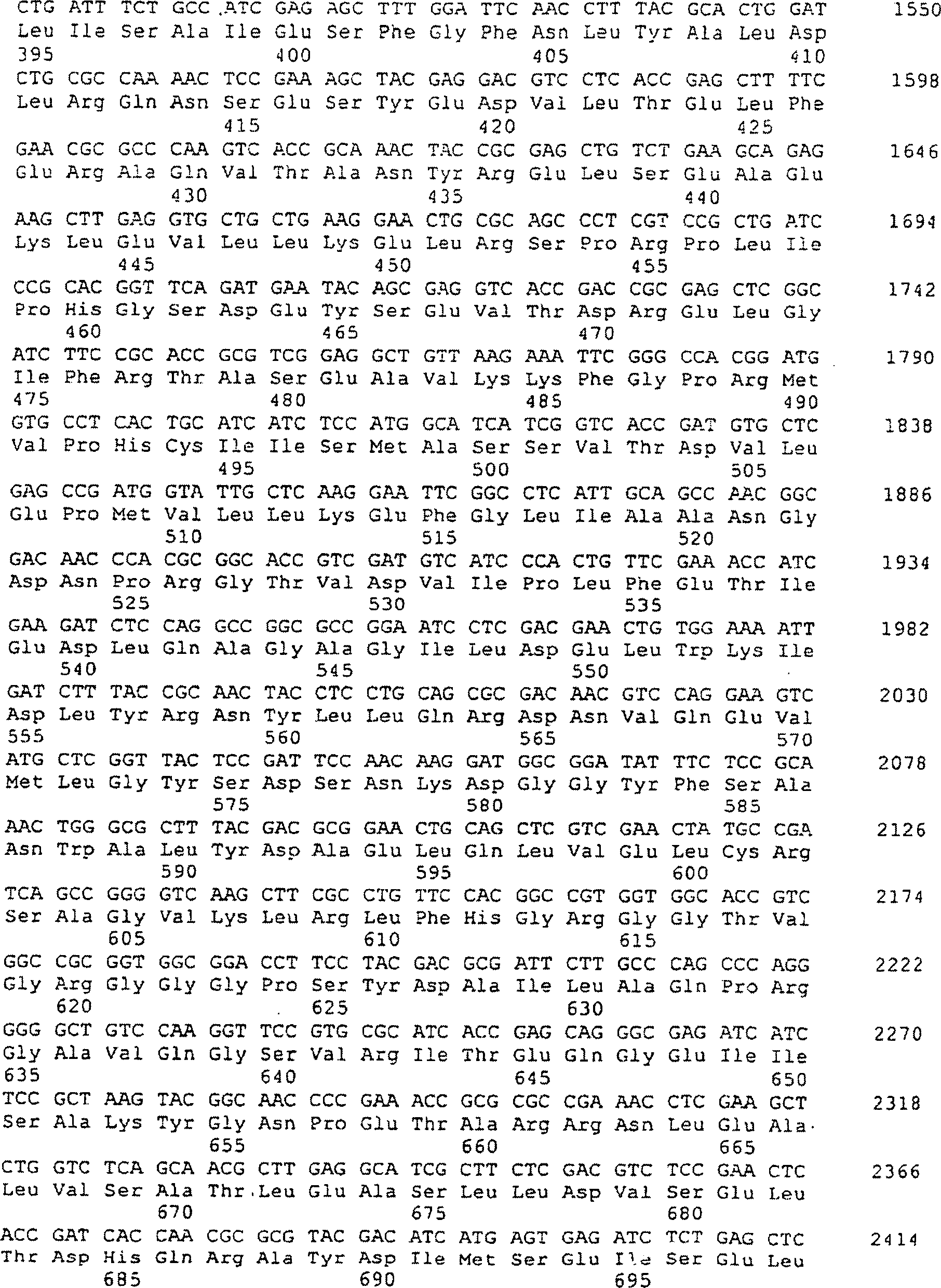
Ein
Beispiel einer Nucleotidsequenz eines DNA-Fragments, das WildtyplysC
enthält,
wird in SEQ-ID Nr. 3 in der Sequenzliste gezeigt. Eine Aminosäuresequenz
der α-Untereinheit
des Wildtyp-AK-Proteins wird von der Nucleotidsequenz abgeleitet,
und ist in SEQ-ID Nr. 4 in der Sequenzliste zusammen mit der DNA-Sequenz
gezeigt. Die Aminosäuresequenz
allein ist in SEQ-ID Nr. 5 gezeigt. Eine Aminosäuresequenz der β-Untereinheit
des Wildtyp-AK-Proteins wird von der Nucleotidsequenz der DNA abgeleitet,
und ist in SEQ-ID Nr. 6 in der Sequenzliste zusammen mit der DNA-Sequenz
gezeigt. Die Aminosäuresequenz
alleine ist in SEQ-ID Nr. 7 gezeigt. In jeder der Untereinheiten
wird GTG als Initiationscodon verwendet, und die entsprechende Aminosäure wird
durch Methionin dargestellt. Jedoch bezieht sich diese Darstellung
auf Methionin, Valin oder Formylmethionin.
-
Das
in der vorliegenden Erfindung verwendete mutierte lysC ist nicht
spezifisch beschränkt,
vorausgesetzt, dass es für
AK kodiert, in der die synergistische Rückkoppelungsinhibierung durch
L-Lysin und L-Thrionin desensibilisiert ist. Jedoch wird die mutierte
lysC beispielhaft durch eine dargestellt, die eine Mutation einschließt, in der
der Aminosäurerest,
der dem Alaninrest an Position 279, vom N-Terminus aus gezählt, in
einen Aminosäurerest
geändert
ist, der nicht Alanin ist, und anders ist als eine saure Aminosäure in der α-Untereinheit,
sowie einen Aminosäurerest,
der dem Alaninrest an Position 30 vom N-Terminus entspricht, wird
in einen anderen Aminosäurerest
geändert
als Alanin, und in eine andere als eine saure Aminosäure in der β-Untereinheit
in der Aminosäuresequenz
der Wildtyp-AK. Die Aminosäuresequenz
der Wildtyp-AK schließt
spezifisch die in SEQ-ID Nr. 5 der Sequenzliste als α-Untereinheit
gezeigte Aminosäuresequenz
ein, sowie die Aminosäuresequenz,
die in SEQ-ID Nr. 7 der Sequenzliste gezeigt ist, als β-Untereinheit
ein.
-
Solche,
die als Aminosäurerest
bevorzugt sind, die nicht Alanin ist, und keine saure Aminosäure sind, schließen Threonin-,
Arginin-, Cystein-, Phenylalanin-, Prolin-, Serin-, Tyrosin- und
Valinreste ein.
-
Der
Codon, der einem Aminosäurerest,
der zu substituieren ist, entspricht, ist hinsichtlich seiner Art nicht
besonders beschränkt,
unter der Voraussetzung, dass er für einen Aminosäurerest
kodiert. Es wird vorhergesagt, dass die Aminosäuresequenz der Wildtyp-AK sich
in Abhängigkeit
von dem Unterschied der Bakterienart und den Bakterienstämmen leicht
unterscheiden kann. AKs, die eine Mutation haben, welche beispielsweise
auf einer Substitution, Deletion oder Insertion einer oder mehrerer
Aminosäurereste
an einer oder mehreren Positionen beruht, die für die Enzymaktivität nicht
relevant sind, wie oben beschrieben wurde, können ebenfalls in der vorliegenden
Erfindung verwendet werden. Eine DNA, die für eine AK kodiert, die eine spontane
Mutation aufweist, kann durch Isolieren einer DNA gewonnen werden,
welche beispielsweise mit der DNA mit einem Teil der in SEQ-ID Nr.
3 gezeigten Nucleinsäuresequenz
unter stringenten Bedingungen hybridisierbar ist. Unter den hier
genannten „stringenten
Bedingungen" wird
eine Bedingung verstanden, bei der ein spezifisches Hybrid gebildet
wird, und ein nicht-spezifisches Hybrid nicht gebildet wird. Es
ist schwierig, die Bedingung in numerischen Werten klar auszudrücken. Jedoch
wird diese Bedingung exemplarisch dargestellt durch eine Bedingung,
unter der sich Nucleinsäuren
mit hoher Homologie, z. B. DNAs mit einer Homologie von nicht weniger
als 90 % miteinander hybridisieren, und Nucleinsäuren mit einer Homologie von
weniger als den oben angegebenen Wert nicht miteinander hybridisieren,
oder eine Bedingung mit einer Temperatur, ausgehend von der Schmelzpunkttemperatur
(Tm) eines vollständig
passenden Hybrids bis (Tm – 30)°C, bevorzugt von
Tm bis (Tm – 20)°C und einer
Salzkonzentration, die 1 × SSC,
bevorzugt 0,1 × SSC
entspricht.
-
Andere
AKs, die eine künstliche
Mutation, beispielsweise auf Grundlage einer Substitution, Deletion oder
Insertion von einer oder mehreren Aminosäureresten haben, können, unter
der Voraussetzung, dass im Wesentlich kein Einfluss auf die AK-Wirkung
und auf die Desensiblisierung der synergistischen Rückkoppelungsinhibierung
durch L-Lysin und L-Threonin ausgeübt wird, ebenfalls verwendet
werden. Eine DNA, die für eine
AK mit der künstlichen
Mutation kodiert, kann durch Modifizieren der Nucleotidsequenz gewonnen
werden, um eine Substitution, Deletion oder Insertion einer spezifischen
Position durch, beispielsweise, positionsspezifische Mutagenese
gewonnen werden. Auch lysC mit einer Mutation kann durch eine Behandlung
mit einem bekannten Mutagen gewonnen werden. Die Mutagenbehandlung
schließt
eine in vitro-Behandlung von DNA, die lysC enthält, mit Hydroxylamin oder ähnlichem
ein, und die Behandlung von Mikroorganismen, die eine DNA beherbergen,
welche lysC mit einer Mutation enthält, wie beispielsweise durch
ultraviolette Bestrahlung oder ein Mutationsmittel, das für gewöhnlich für die artifizielle
Mutagenese verwendet wird, beispielsweise N-Methyl-N'-nitro-N-nitrosoguanidin
(NTG) oder Salpetersäure.
Nach der Mutagenbehandlung kann eine Position, in der die Mutation
eingeschleust worden ist, oder an der eine Mutation aufgetreten
ist durch Auswählen einer
DNA oder eines Mikroorganismus selektioniert werden, welcher für eine AK
kodiert oder diese herstellt, die AK-Aktivität besitzt, und deren Aminosäuresequenz
mutiert ist anhand von DNA, die einer Mutagenbehandlung unterworfen
worden ist, oder des Mikroorganismus, der der Mutagenbehandlung
unterworfen worden ist. Die Position der eingeschleusten Mutationen
ist nicht spezifisch beschränkt,
unter der Voraussetzung, dass im Wesentlichen kein Einfluss auf
die AK-Aktivität
und auf die Desensiblisierung der Rückkoppelungsinhibierung ausgeübt wird.
Die Anzahl der eingeschleusten Mutationen varriiert in Abhängigkeit
von der Position oder der Art der mutierten Aminosäure in der
sterischen Struktur des Proteins, und ist nicht spezifisch beschränkt, mit
der Maßgabe,
dass im Wesentlichen kein Einfluss auf die AK-Aktivität und auf
die Desensibilisierung der Rückkoppelungsinhibierung
ausgeübt
wird. Die Anzahl beträgt
für gewöhnlich 1–20, bevorzugt
1–10.
-
Der
durch das Einschleusen eines mutierten lysC-Plasmids p399AK9B in
den AJ12036-Stamm (FERM BP-734) als Wildtypstamm von Brevibacterium
lactofermentum gewonnene AJ12691, ist am 10. April 1992, unter Zugangsnr.
FERM P-12918 beim National Institute of Bioscience and Human Technology
des Ministeriums für
Internationalen Handel und Industrie (1–3, Higashi 1-chome, Tsukuba-shi,
Ibaraki-ken, 305 Japan) hinterlegt worden, und in einer internationale
Hinterlegung, beruhend auf dem Budapester Vertrag am 10. Februar
1995 überführt worden,
und unter der Zugangsnr. FERM BP-4999 hinterlegt worden.
-
(2) Herstellung von lysA
-
Ein
DNA-Fragment, das lysA enthält,
kann vom Chromosom eines coryneformen Bakteriums mit Hilfe von PCR
hergestellt werden. Der DNA-Donor ist nicht besonders beschränkt, jedoch
wird er beispielhaft durch den Brevibacterium lactofermentum ATCC
13869-Stamm wiedergegeben.
-
In
coryneformen Bakterien bildet lysA zusammen mit argS (arginyl-tRNA
Synthasegen) ein Operon, und lysA liegt stromabwärts von argS. Die Expres sion
von lysA wird durch einen Promotor reguliert, der stromaufwärts von
argS vorhanden ist (siehe Journal of Bacteriology, Nov., 7356–7362 (1993)).
DNA-Sequenzen dieser Gene sind bei Corynebacterium glutanicum bekannt
(siehe Molecular Microbiology, 4 (11), 1819–1830 (1990); Molecular and
General Genetics, 212, 112–119
(1988)), auf deren Grundlage DNA-Primer für die PCR hergestellt werden
können.
Solche DNA-Primer werden beispielhaft wiedergegeben durch 23-mer-DNAs,
die jeweils die Nucleotidsequenzen haben, die in SEQ-ID Nr. 8 der
Sequenzliste (entsprechend den Nucleotidnummern 11–33 in der
Nucleotidsequenz, die in Molecular Microbiology, 4 (11), 1819–1830 (1990))
beschrieben ist und SEQ-ID Nr. 9 (entsprechend den Nucleotidpositionen
1370 bis 1392 der Nucleotidsequenz, die in Molecular and General
Genetics, 212, 112–119
(1988) beschrieben ist. Die Synthese von DNA, die PCR und die Herstellung
eines Plasmids, das das gewonnene lysA enthält, kann auf gleiche Weise
wie die für
lysC, welche oben beschrieben worden ist, durchgeführt werden.
-
Im
später
beschriebenen Beispiel wurde ein DNA-Fragment, das einen Promotor,
argS und lysA enthält,
verwendet, um lysA zu verstärken.
Jedoch ist argS für
die vorliegende Erfindung nicht wesentlich. Es ist erlaubt, ein
DNA-Fragment zu verwenden, in dem lysA genau stromabwärts des
Promotors ligiert ist.
-
Eine
Nucleotidsequenz eines DNA-Fragments, das argS und lysA enthält, und
eine Aminosäuresequenz,
die abgeleitet davon durch die Nucleotidsequenz kodiert wird, werden
in SEQ-ID Nr. 10 beispielhaft dargestellt. Ein Beispiel einer Aminosäuresequenz,
die durch argS kodiert wird, ist in SEQ-ID Nr. 11 gezeigt, und ein
Beispiel einer Aminosäuresequenz,
das durch lysA kodiert wird, ist in SEQ-ID Nr. 12 gezeigt. Zusätzlich zu DNA-Fragmenten,
die für
diese Aminosäuresequenzen
kodieren, kann die vorliegende Erfindung gleichermaßen DNA-Fragmente
verwenden, die für
Aminosäuresequenzen
kodieren, die im Wesentlichen die gleichen sind, wie die Aminosäuresequenz,
die in SEQ-ID Nr. 12 gezeigt ist, d. h. Aminosäuresequenzen mit einer Mutation,
die darauf beruhen, beispielsweise eine Substitution, Deletion oder
Insertion von einer oder mehreren Aminosäuren, vorausgesetzt, dass es
keinen wesentlichen Einfluss auf die DDC-Aktivität gibt. Das lysA mit spontaner
oder artifizieller Mutation kann auf gleiche Weise wie die DNA gewonnen
werden, die für
die AK mit einer Mutation, welche keinen Einfluss auf die AK-Aktivität und auf
die Desensibilisierung der synergistischen Rückkoppelungsinhibierung durch
L-Lysin und L-Threonin ausübt.
-
(3) Herstellung von PPC
-
Ein
DNA-Fragment, das PPC enthält,
kann vom Chromosom eines coryneformen Bakteriums mit Hilfe von PCR
gewonnen werden. Der DNA-Donor ist nicht besonders limitiert, jedoch
wird er beispielhaft wiedergegeben durch Brevibacterium lactofermentum
ATCC 13869-Stamm.
-
Die
DNA-Sequenzen des ppc-Gens sind bei Corynebacterium glutamicum bekannt
(siehe O'Regan, M.
et al., Gene, 77, 237–251
(1989), und auf Grundlage davon können DNA-Primer für die PCR
hergestellt werden. Solche DNA-Primer sind spezifisch beispielhaft
dargegeben durch 23-mer-DNAs, die jeweils die Nucleotidsequenzen
haben, die in SEQ-ID Nr. 13 und 14 der Sequenzliste gezeigt sind.
Die Synthese der DNA, die PCR, und die Herstellung eines Plasmids,
das das hergestellte ppc enthält,
kann auf gleiche Weise durchgeführt
werden, wie bei lysC, das oben beschrieben worden ist.
-
Eine
Nucleotidsequenz des DNA-Fragments, das ppc enthält, und eine Aminosäuresequenz,
die nach Ableitung durch die Nucleotidsequenz kodiert wird, wird
in SEQ-ID Nr. 15 gezeigt. Die Aminosäuresequenz allein ist in SEQ-ID
Nr. 16 gezeigt.
-
Zusätzlich zu
DNA-Fragmenten, die für
diese Aminosäuresequenzen
kodieren, kann die vorliegende Erfindung gleichermaßen DNA-Fragmente
verwenden, die Aminosäuresequenzen
kodieren, die im Wesentlichen die gleichen sind, wie die Aminosäuresequenz,
die in SEQ-ID Nr. 16 gezeigt ist, d. h. Aminosäuresequenzen mit einer Mutation,
die beispielsweise auf einer Substitution, Deletion oder Insertion
von einer oder mehreren Aminosäuren
beruht, vorausgesetzt, dass es keinen wesentlichen Einfluss auf
die PEPC-Aktivität
gibt. ppc mit spontaner oder artifizieller Mutation kann auf gleiche
Weise gewonnen werden, wie die DNA, die für eine AK mit einer Mutation
kodiert, die keinen Einfluss auf die AK-Aktivität und auf die Desibilisierung
der synergistischen Rückkoppelungsinhibierung
durch L-Lysin und L-Threonin ausübt.
-
Die
ppc des coryneformen Bakteriums bildet zusammen mit gap (Glyceraldehyd-3-phosphatdehydrogenase-Gen),
pgk (Phosphoglyceratkinase-Gen) und tpi (Triosephosphatisomerase-Gen)
ein Operon, und ppc liegt stromabwärts von tpi vor. Die Expression
von ppc wird durch einen Promotor reguliert, der stromaufwärts von
pgk vorhanden ist (siehe Schwinde, J. W. et al., J. Bacteriol.,
175 (12), 3905–3908
(1993). Daher, wie beim oben erwähnten
lysA, kann ppc zusammen mit pgk und tpi durch PCR amplifiziert werden,
um ein DNA-Fragment zu verwenden, das pgk, tpi und ppc enthält. wie
im später
beschriebenen Beispiel gezeigt wird, ist es erlaubt, ein DNA-Fragment
zu verwenden, bei dem ein geeigneter Promotor direkt stromaufwärts der
kodieren den Region von PEPC ligiert ist. Der Promotor schließt einen
Promotor für
lysC, einen tac-Promotor, der von E.coli stammt, und einen trc-Promotor
ein.
-
<2> Rekombinante
DNA und coryneformes Bakterium der vorliegenden Erfindung
-
Die
rekombinante DNA umfasst eine DNA-Sequenz, die eine Aspartokinase
kodiert, in der die Rückkoppelungsinhibierung
durch L-Lysin und L-Threonin im Wesentlichen desensibilisiert ist,
und eine DNA-Sequenz, die eine Diaminopimelatdecarboxylase kodiert,
und autonom replizierbar ist in coryneformen Bakterienzellen. In
einer bevorzugten Ausführungsform
umfasst die rekombinante DNA weiter eine DNA-Sequenz, die für eine Phosphoenolpyruvatcarboxylase
zusätzlich
zu den oben erwähnten
DNA-Sequenzen kodiert.
-
Das
erfindungsgemäße coryneforme
Bakterium beherbergt eine Aspartokinase (mutierte AK), in der die
Rückkoppelungsinhibierung
durch L-Lysin und L-Threonin im Wesentlichen desensibilisiert ist,
wobei DNA (lysA), die für
eine Diaminopimelatdecarboxylase kodiert, verstärkt ist. In einer bevorzugten
Ausführungsform ist
das coryneforme Bakterium der vorliegenden Erfindung ein coryneformes
Bakterium, bei dem DNA (ppc), die für eine Phosphoenolpyruvatcarboxalase
kodiert, weiter verstärkt
ist.
-
Der
Begriff „verstärkt" bezieht sich hier
auf die Tatsache, dass die intrazelluläre Aktivität eines Enzyms, das durch die
DNA kodiert wird, beispielsweise durch Erhöhung der Kopienanzahl eines
Gens, durch Verwendung eines starken Promotors, durch Verwendung
eines Gens, das ein Enzym kodiert, das eine hohe spezifische Aktivität hat, oder
durch eine Kombination dieser Mittel erhöht wird.
-
Das
coryneforme Bakterium, das die mutierte AK beherbergt, kann eines
sein, das die mutierte Aspartokinase als Ergebnis einer Mutation
herstellt, oder ein solches, das durch Einschleusen mutierten lysCs
transformiert ist.
-
Beispiele
des coryneformen Bakteriums, das verwendet wird, um die oben beschriebene
DNA einzuschleusen, schließen
beispielsweise die folgenden Lysin-produzierenden Wildtyp-Stämme ein:
Corynebacterium
acetoacidophilum ATCC 13870;
Corynebacterium acetoglutamicum
ATCC 15806;
Corynebacterium callunae ATCC 15991;
Corynebacterium
glutamicum ATCC 13032;
(Brevibacterium divaricatium) ATCC 14020;
(Brevibacterium
lactofermentum) ATCC 13869;
(Corynebacterium lilium) ATCC 15990;
(Brevibacterium
flavum) ATCC 14067;
Corynebacterium melassecola ATCC 17965;
Brevibacterium
saccharolyticum ATCC 14066;
Brevibacterium immariophilum ATCC
14068;
Brevibacterium roseum ATCC 13825;
Brevibacterium
thiogenitalis ATCC 19240;
Microbacterium ammoniaphilum ATCC
15354;
Corynebacterium thermoaminogenes AJ12340 (FERM BP-1539).
-
Anders
als die Bakterienstämme,
die oben beschrieben sind, schließen solche, die als Wirt verwendbar
sind, beispielsweise Mutantenstämme
mit L-Lysin-Produktionsfähigkeit
ein, die von den oben erwähnten Stämmen abstammen.
Solche artifiziellen mutierten Stämme schließen folgende ein: S-(2-Aminoethyl)-cystein (nachfolgend
abgekürzt
als „AEC"), resistente Mutantenstämme (z.
B. Brevibacterium lactofermentum AJ11082 (NRRL B-1147), Japanische
Patentveröffentlichungs-Nr.
56-1914, 56-1915, 57-14157, 57-14158, 57-30474, 58-10075, 59-4993,
61-35840, 62-24074, 62-36673, 5-11958, 7-112437 und 7-112438); Mutantenstämme, die
eine Aminosäure,
wie L-Homoserin für
Wachstum benötigen
(Japansiche Patentveröffentlichungs-Nr.
48-28078 und 56-6499); Mutantenstämme, die eine Resistenz gegenüber AEC
aufweisen und Aminosäuren
wie L-Leucin, L-Homoserin, L-Prolin, L-Serin, L-Arginin, L-Alanin
und L-Valin benötigen
(United States Patente Nr. 3,708,395 und 3,825,472); L-Lysin-produzierende
Mutantenstämme,
die eine Resistenz gegenüber
DL-α-Amino-ε-Caprocaltam, α-Amino-lauryllactam,
Aspartat-analoga, Sulfamedikamenten, Chinoid und N-Lauroylleucin
aufweisen; L-Lysin-herstellende Mutantenstämme, die eine Resistenz gegenüber Inhibitoren der
Oxyaloacetatdecarboxylase oder von Enzymen des Respirationssystems
aufweisen (Japanische Patentanmeldung Veröffentlichungs-Nr. 50-53588;
50-31093, 52-102498, 53-9394, 53-86089, 55-9783, 55-9759, 56-32995
und 56-39778 und Japanische Patentveröffentlichungs-Nr. 53-43591
und 53-1833); L-Lysin-herstellende Mutantenstämme, die Inositol und Essigsäure benötigen (Japanische
Patentanmeldung Offenlegungs-Nr. 55-9784 und 56-8692); L-Lysin-herstellende Mutantenstämme, die
eine Empfindlichkeit gegenüber Fluoropyrovatsäure oder
einer Temperatur von nicht weniger als 34°C aufweisen (Japanische Patentanmeldungs-Offenlegungs-Nr.
55-9783 und 53-86090); und herstellende Mutantenstämme, die
zum Genus Brevibacterium oder Corynebacterium gehören, welche
eine Resistenz gegenüber
Ethylenglycol aufweisen und L-Lysin herstellen (United States Patent
Nr. 4,411,997).
-
In
einer spezifischen Ausführungsform,
um, wie oben beschrieben, die Gene für die L-Lysin-Biosynthese in
dem Wirtzu verstärken,
werden die Gene in den Wirt unter Verwendung eines Plasmidvektors,
eines Transposons oder eines Phagenvektors oder ähnlicher eingeschleust. Nach
der Einschleusung wird erwartet, dass in gewissem Ausmaß selbst
bei der Verwendung eines Vektors mit niedriger Kopienzahl eine Verstärkung auftritt.
Jedoch wird es bevorzugt, einen Vektor vom Typ für mehrfache Kopien zu verwenden.
Solche Vektoren schließen
beispielsweise die Plasmidvektoren pAJ655, pAJ1844, pAJ611, pAJ3148
und pAJ440, die oben beschrieben wurden, ein. Daneben sind Transposons,
die von coryneformen Bakterien stammen, in den Internationalen Veröffentlichungspamphleten
der WO 02/02627 und WO 93/18151, Europäische Patentveröffentlichungs-Nr.
445385, Japanische Patenanmeldungs-Offenlegungs-Nr. 6-46867, Vertes,
A. A. et al., Mol. Microbiol., 11, 739–746 (1994), Bonamy, C., et
al., Mol. Microbiol., 14, 571–581
(1994), Vertes, A. A. et al., Mol. Gen. Genet., 245, 397–405 (1994),
Jagar, W. et al., FEMS Microbiology Letters, 126, 1–6 (1995),
Japanische Patentanmeldungs-Offenlegungs-Nr. 7-107976, Japanische
Patentanmeldungs-Offenlegungs-Nr. 7-327680 und ähnlichen beschrieben.
-
In
der vorliegenden Erfindung ist es nicht unverzichtbar, dass das
mutierte lysC notwendigerweise verstärkt ist. Es ist erlaubt, solche
zu verwenden, die eine Mutation in lysC der chromosomalen DNA haben,
oder in denen das mutierte lysC in chromosomale DNA eingeschleust
ist. Alternativ kann das mutierte lysC unter Verwendung eines Plasmidvektors
eingeschleust werden. Andererseits sind lysA und ppc vorzugsweise
verstärkt,
um L-Lysin wirksam herzustellen.
-
Jedes
der Gene lysC, lysA und ppc kann jeweils nacheinander unter Verwendung
verschiedener Vektoren in den Wirt eingeschleust werden. Alternativ
dazu können
zwei oder drei Arten an Genen zusammen unter Verwendung eines einzelnen
Vektors eingeschleust werden. Wenn verschiedene Vektoren verwendet
werden, können
die Gene in irgendeiner Reihenfolge eingeschleust werden, jedoch
ist es bevorzugt, Vektoren zu verwenden, die eine stabile Gemeinschaft
und einen Beherbungsmechanismus im Wirt haben, und die in der Lage
sind, gemeinsam vorzuliegen.
-
Ein
coryneformes Bakterium, das die mutierte AK beherbergt und außerdem verstärktes lysA
umfasst, wird beispielsweise durch Einschleusen einer rekombinanten
DNA in das Wirts-Corynebacterium gewonnen, die mutierte lysC, lysA
und ppc enthält,
welche in den Zellen coryneformer Bakterien autonom replizierbar
sind.
-
Ein
coryneformes Bakterium, das außerdem
zusätzlich
zu einer mutierten lysC und lysA eine verstärkte ppc umfasst, wird beispielsweise
durch Einschleusen einer rekombinanten DNA, die mutiertes lysC,
lysA und ppc enthält,
die autonom in Zellen coryneformer Bakterien replizierbar sind,
in das Wirts-Corynebacterium gewonnen. Ein coryneformes Bakterium,
das ein verstärktes
mutiertes lysC, lysA und ppc umfasst, wird außerdem durch Einschleusen einer
rekombinanten DNA, die ppc enthält,
die autonom in Zellen coryneformer Bakterien replizierbar ist, sowie
in ein coryneformes Bakterium, das ein verstärktes mutiertes lysC und lysA
umfasst, gewonnen.
-
Die
oben erwähnten
rekombinanten DNAs können
beispielsweise durch Inserieren jedes dieser Gene, die an der L-Lysin-Biosynthese
teilhaben in einen Vektor, wie einen Plasmidvektor, ein Transposon
oder Phagenvektor, wie oben beschrieben worden ist, gewonnen werden.
-
In
dem Fall, dass ein Plasmid als Vektor verwendet wird, kann die rekombinante
DNA in den Wirt gemäß dem elektrischen
Pulsverfahren (Sugimoto et al., Japanische Patentanmeldungs-Offenlegungs-Nr. 2-207791)
eingeschleust werden. Die Amplifikation eines Gens unter Verwendung
eines Transposons kann durch Einschleusen eines Plasmids in die
Wirtszelle durchgeführt
werden, das ein Transposon trägt,
und durch die Induktion der Transposition des Transposons.
-
<3> Verfahren
zum Produzieren von L-Lysin
-
L-Lysin
kann effizient durch Kultivieren des coryneformen Bakteriums, das
die verstärkten
Gene für
die L-Lysin-Biosynthese, die oben beschrieben worden sind, umfasst,
in einem geeigneten Medium hergestellt werden, um zu ermöglichen,
dass L-Lysin hergestellt und in der Kultur des Bakteriums akkumuliert
wird, und indem L-Lysin aus der Kultur eingesammelt wird.
-
Das
zu verwendende Medium wird beispielhaft dargestellt durch ein gewöhnliches
Medium, das eine Kohlenstoffquelle, eine Stickstoffquelle, anorganische
Ionen und gegebenenfalls andere organische Bestandteile enthält.
-
Als
Kohlenstoffquelle ist es möglich,
Zucker wie Glucose, Fructose, Saccharose, Molassen und Stärkehydrolysat,
und organische Säuren
wie Fumarsäure,
Zitronensäure
und Bernsteinsäure
zu verwenden.
-
Als
Stickstoffquelle ist es möglich,
anorganische Ammoniumsalze, wie Ammoniumsulfat, Ammoniumchlorid
und Ammoniumphosphat, organischen Stickstoff wie Sojabohnenhydrolysat,
Stickstoffgas und wässrigen
Harnstoff zu verwenden.
-
Als
organische Spurenelementquellen ist es wünschenswert, dass die benötigten Substanzen,
wie Vitamin B1 und L-Homoserin oder Hefeextrakt
oder ähnliche
in geeigneten Mengen enthalten sind. Zusätzlich zu den oben genannten
werden Kaliumphosphat, Magnesiumsulfat, Eisenionen, Manganionen
usw., falls notwendig, in geringen Mengen hinzugegeben.
-
Die
Kultivierung wird vorzugsweise unter aeroben Bedingungen für ungefähr 30 bis
90 Stunden durchgeführt.
Die Kultivierungstemperatur wird bevorzugterweise auf 25°C bis 37°C kontrolliert,
und der pH wird während
der Kultivierung vorzugsweise auf 5 bis 8 kontrolliert. Anorganische
oder organische, saure oder alkalische Substanzen, oder Stickstoffgas
oder ähnliche
können
zur Einstellung des pH-Werts verwendet werden. L-Lysin kann aus
einer Kultur durch Kombinieren eines gewöhnlichen Ionen-Austauschharzverfahrens, eines
Präzipitationsverfahrens
oder andere bekannte Verfahren eingesammelt werden.
-
Beispiele
-
Die
vorliegende Erfindung wird genauer unten unter Bezugnahme auf die
Beispiele geklärt.
-
Beispiel 1: Herstellung
des Wildtyp-lysC-Gens und eines mutierten lysC-Gens aus Brevibacterium
lactofermentum
-
<1> Herstellung
von Wildtyp-lysC und mutierten lysC's und Herstellung von Plasmiden, die
diese enthalten
-
Der
Brevibacterium lactofermentum ATCC 13869-Stamm und ein L-Lysin-produzierender Mutantenstamm
AJ3445 (FERM P-1944), der von dem ATCC 13869-Stamm durch eine Mutationsbehandlung
erhalten wurde, wurden als chromosomale DNA-Donoren verwendet. Der
AJ3445-Stamm ist einer Mutation unterworfen worden, so dass lysC
verändert
wurde, um eine wesentliche Desensibilisierung der konzertierten
Inhibierung durch Lysin und Threonin zu enthalten (Journal of Biochemistry,
68, 701–710
(1970)).
-
Ein
DNA-Fragment, das lysC enthält,
wurde aus chromosomaler DNA in Übereinstimmung
mit dem PCR-Verfahren (Polymerase-Kettenreaktion; siehe White, T.
J. et al., Trends Genet., 5, 185 (1989)) amplifiziert. Im Hinblick
auf die DNA-Primer, die für
die Amplifikation verwendet wurden, wurden Einzelstränge von 23-mer
und 21-mer DNAs mit den in SEQ-ID Nr. 1 und 2 gezeigten Nucleotidsequenzen
auf Grundlage einer Sequenz, die für Corynebacterium glutamicum
bekannt ist (siehe Molecular Microbiology (1991), 5 (5), 1197–1204, und
Mol. Gen. Genet. (1990), 224, 317–324)synthetisiert, um einen
Bereich von ungefähr
1.643 Basenpaaren zu amplifizieren, der für lysC kodiert. Die DNA wurde
nach einem allgemeinen Verfahren unter Verwendung eines DNA-Synthesegeräts vom Modell
380B synthetisiert, das von Applied Biosystems hergestellt wird,
und in dem das Phosphoramiditverfahren verwendet wurde (siehe Tetrahedron
Letters (1981), 22, 1859).
-
Das
Gen wurde mittels PCR unter Verwendung eines DNA-Thermocyclers vom
Modell PJ200 amplifiziert, der von Takara Shuzo hergestellt wird,
und in dem Taq DNA-Polymerase in Übereinstimmung mit dem Verfahren
verwendet wurde, das von dem Lieferanten angegeben worden ist. Das
amplifizerte Genfragment von 1.643 Kilobasen wurde durch Agarosegel-Electrophorese
bestätigt.
Danach wurde das Fragment aus dem Gel ausgeschnitten und nach einem
gewöhnlichen
Verfahren aufgereinigt, und es wurde mit den Restriktionsenzymen
NruI (hergestellt von Takara Shuzo) und EcoRI (hergestellt von Takara
Shuzo) verdaut.
-
pHSG399
(siehe Takeshita, 5. et al., Gene (1987), 61, 63–74) wurde als Klonierungsvektor
für das Genfragment
verwendet. pHS399 wurde mit den Restriktionsenzymen SmaI (hergestellt
von Takara Shuzo) und EcoRI verdaut, und es wurde mit dem amplifizierten
lysC-Fragment ligiert. DNA wurde unter Verwendung eines DNA-Ligations-Kits
(hergestellt von Takara Shuzo) nach dem angegebenen Verfahren verwendet.
So wurden Plasmide hergestellt, in denen die lysC-Fragmente von
den Chromosomen von Brevibacterium lactofermentum amplifiziert waren,
welche jeweils mit pHSG399 ligiert worden waren. Das Plasmid, das
lysC aus ATCC 13689 (Wildtyp-Stamm) umfasst, wurde als p399AKY bezeichnet,
und ein Plasmid, das lysC aus AJ3463 umfasst (L-Lysin herstellendes
Bakterium) wurde als p399AK9 bezeichnet.
-
Ein
DNA-Fragment (nachfolgend bezeichnet als „Brevi.-ori") mit der Fähigkeit,
das Plasmid autonom in Bakterien replizierbar zu machen, die zu
dem Genus Corynebacterium gehören,
wurde jeweils in p399AKY und p399AK9 eingeschleust, um Plasmide
herzustellen, die lysC tragen, welches in Bakterien autonom replizierbar
ist, die zu dem Genus Corynebacterium gehören. Brevi.-ori wurde vom Plasmidvektor
pHK4 ausgehend hergestellt, der den Brevi.-ori enthält und sowohl
in Escherichia coli-Zellen als auch in Bakterien, die zum Genus
Corynebacterium gehören,
autonom replizierbar ist. pHK4 wurde durch Verdauen von pHC4 mit
KpnI (hergestellt von Takara Shuzo) und BamHI (hergestellt von Takara
Shuzo) hergestellt, wobei ein Brevi.-ori-Fragment extrahiert wurde, und dieses
mit pHSG298 ligiert wurde, der zuvor ebenfalls mit KpnI und BamHI
verdaut worden ist (siehe Japanische Patentanmeldung Offenlegungs-Nr.
5-7491). pHK4 verleiht dem Wirt eine Kanamycin-Resistenz. Escherichia
coli, das pHK4 beherbergt, wurde als Escherichi coli AJ13136 bezeichnet
und am 1. August 1995 unter der Zugangs-Nr. FERM BP-5186 beim National
Institute of Bioscience and Human Technology der Agency of Industrial
Science and Technology des Ministerium für internationalen Handel und
Industrie (1–3,
Higashi 1-chome, Tsukuba-shi, Ibaraki-ken, 305 Japan) hinterlegt.
-
pHK4
wurde mit den Restriktionsenzymen KpnI und BamHI verdaut, und die
geschnittenen Enden wurden geglättet.
Die Glättungsprozedur
wurde unter Verwendung eines DNA-Blunting-Kits (hergestellt von Takara
Shuzo) nach dem angegebenen Verfahren durchgeführt. Nach der Bildung der glatten
Enden wurde ein phophorylierter BamHI-Linker (hergestellt von Takara
Shuzo) ligiert, um eine Modifizierung zu ergeben, damit das DNA-Fragment,
das dem Brevi.-ori-Teil entspricht, aus dem pHK4 alleine durch Verdau
mit BamHI ausgeschnitten werden kann. Dieses Plasmid wurde mit BamHI
verdaut, und das hergestellte Brevi.-ori DNA-Fragment wurde mit
p399AKY und p399AK9 ligiert, die ebenfalls mit BamHI verdaut worden
sind, um Plasmide herzustellen, die jeweils das lysC-Gen enthalten,
welches in Bakterien, die zum Genus Corynebacterium gehören, autonom
replizierbar ist.
-
Das
Plasmid, das das Wildtyp-lysC-Gen enthält, das aus p399AKY stammt,
wurde als p399AKYB bezeichnet, und das Plasmid, das das mutierte
lysC enthält,
das aus p399AK9 stammt, wurde als p399AK9B bezeichnet. Das Verfahren
zur Herstellung von p399AK9B und p399AKYB ist in 1 gezeigt.
Der Stamm AJ12691, der durch Einschleusen des mutierten lysC-Plasmids
p399AK9B in den Wildtyp-Stamm von Brevibacterium lactofermentum
(AJ12036-Stamm, FERM BP-734) gewonnen wurde, wurde am 10. April
1992 unter der Zugangs-Nr. FERM P-12918 beim National Institute
of Bioscience and Human Technology der Agency of Industrial Science
and Technology des Ministeriums für internationalen Handel und
Industrie (1–3,
Hiashi 1-chome, Tsukuba-shi, Ibaraki-ken, 305 Japan) hinterlegt,
in eine internationale Hinterlegung auf Grundlage des Budapester
Vertrags am 10. Februar 1995 transferiert, und unter der Zugangs-Nr.
FERM BP-4999 hinterlegt.
-
<2> Bestimmung
der Nucleotidsequenzen von Wildtyp-lysC und mutiertem lysC aus Brevibacterium
lactofermentum
-
Das
Plasmid p399AKY, das das Wildtyp-Gen lysC enthält, und das Plasmid p399AK9,
das das mutierte lysC enthält,
wurden aus den entsprechenden Transformanten hergestellt, um die
Nucleotidsequenzen des Wildtyps und des mutierten lysC zu bestimmen.
Die Nucleotidsequenz-Bestimmung wurde in Übereinstimmung mit dem Verfahren
von Sanger et al. (siehe z. B. F. Sanger et al., Proc. Natl. Acad.
Sci., 74, 5463 (1977)) durchgeführt.
-
Die
Nucleotidsequenz von Wildtyp-lysC, welches durch p399AKY kodiert
wird, ist in SEQ-ID Nr. 3 der Sequenzliste gezeigt. Andererseits
hatte die Nucleotidsequenz des mutierten lysC, das durch p399AK9
kodiert wird, nur eine Mutation eines Nucleotids, so dass das G
an Position 1051 in A in der SEQ-ID Nr. 3 geändert wurde, wenn es mit Wildtyp-lysC
verglichen wurde. Es ist bekannt, dass lysC von Corynebacterium
glutamicum zwei Untereinheiten (α, β) hat, welche
in einem identischen Leseraster auf einem DNA-Strang kodiert sind
(siehe Kalinowski, J. et al., Molecular Microbiology (1991) 5 (5),
1197–1204).
Wenn die Homologie beurteilt wird, wird vermutet, dass das Gen,
das hier sequenziert wurde, auch zwei Untereinheiten (α, β) hat, die
in einem identischen Leseraster auf einem identischen DNA-Strang
kodiert werden.
-
Die
Aminosäuresequenz
der α-Untereinheit
des Wildtyp-AK-Proteins, das von der Nucleotidsequenz der DNA abgeleitet
wird, ist zusammen mit der DNA-Sequenz in SEQ-ID Nr. 4 gezeigt.
Die Aminosäuresequenz
alleine ist in SEQ-ID Nr. 5 gezeigt. Die Aminosäuresequenz der β-Untereinheit
des Wildtyp-AK-Proteins, die von der Nucleotidsequenz der DNA abgeleitet
wird, ist zusammen mit der DNA-Sequenz in SEQ-ID Nr. 6 gezeigt.
Die Aminosäuresequenz
allein ist in SEQ-ID Nr. 7 gezeigt. In jeder der Untereinheiten
wird GTG als Initiationscodon verwendet, und die entsprechende Aminosäure ist
durch Methionin wiedergegeben. Jedoch bezieht sich diese Darstellung
auf Methionin, Valin oder Formylmethionin.
-
Andererseits
bedeutet die Mutation der Sequenz des mutierten lysC das Auftreten
der Aminosäurerestsubstitution,
bei der der Alaninrest an Position 279 der α-Untereinheit in einen Threoninrest
geändert
ist, und der Alaninrest an der Position 30 der β-Untereinheit in einen Threoninrest
in der Aminosäuresequenz
des Wildtyp-AK-Proteins (SEQ-ID Nr. 5, 7) geändert ist.
-
Beispiel 2: Herstellung
von lysA aus Brevibacterium lactofermentum
-
<1> Herstellung
von lysA und Herstellung eines Plasmids, das lysA enthält
-
Der
Wildtyp-Stamm von Brevibacterium lactofermentum ATCC 13869 wurde
als Chromosomen-DNA-Donor verwendet. Chromosomale DNA wurde aus
ATCC 13869-Stamm
in Übereinstimmung
mit einem gewöhnlichen
Verfahren hergestellt. Ein DNA-Fragment, das argS, lysA und einen
Promotor eines Operons enthält,
das diese enthält,
wurde von chromosomaler DNA mittels PCR amplifiziert. Im Hinblick
auf die DNA-Primer, die für
diese Amplifikation verwendet wurden, wurden auf Grundlage einer
Sequenz, die für
Corynebacterium glutamicum bekannt ist (siehe Molecular Microbiology,
4 (11), 1819–1830
(1990); Molecular and General Genetics, 212, 112–119 (1988)), synthetische
23-mer DNAs mit den in SEQ-ID Nr. 8 und 9 in der Sequenzliste gezeigten
Nucleotidsequenzen jeweils verwendet, um einen Bereich von ungefähr 3,6 Kilobasen
zu amplifizieren, der für
eine Arginyl-tRNA-Synthase und DDC kodiert. Die Synthese von DNA
und PCR wurde auf gleiche Weise, wie in Beispiel 1 beschrieben,
durchgeführt.
pHSG399 wurde als Klonierungsvektor für das amplifizierte Genfragment
von 3.579 Basenpaaren verwendet. pHSG399 wurde mit dem Restriktionsenzym SmaI
(hergestellt von Takara Shuzo) verdaut, und wurde mit dem DNA-Fragment
ligiert, das das amplifizierte lysA enthält. Das Plasmid, das wie oben
beschrieben gewonnen wurde, welches lysA enthält, das aus ATTC 13869 stammt,
wurde als p399LYSA bezeichnet.
-
Das
DNA-Fragment, das lysA enthält,
wurde durch Verdau von p399LYSA mit KpnI (hergestellt von Takara
Shuzo) und BamHI (hergestellt von Takara Shuzo) extrahiert. Dieses
DNA-Fragment wurde mit pHSG299 ligiert, das mit KpnI und BamHI verdaut
worden ist. Das gewonnene Plasmid wurde als p299LYSA bezeichnet.
Das Verfahren zur Herstellung von p299LYSA ist in 2 gezeigt.
-
Brevi.-ori
wurde in das erhaltene p299LYSA eingeschleust, um ein Plasmid herzustellen,
das lysA trägt,
welches autonom in coryneformen Bakterien replizierbar ist. pHK4
wurde mit den Restriktionsenzymen KpnI und BamHI verdaut, und die
geschnittenen Enden wurden geglättet.
Die Herstellung glatter Enden wurde unter Verwendung eines DNA-Blunting-Kits
(herstellt von Takara Shuzo) nach dem angegebenen Verfahren durchgeführt. Nach
der Bildung der glatten Enden wurde ein phosphorylierter KpnI-Linker
(hergestellt von Takara Shuzo) ligiert, um eine Modifizierung zu
ergeben, damit das DNA-Fragment, das dem Brevi.-ori entspricht, allein
durch Verdau mit KpnI aus pHK4 ausgeschnitten werden kann. Dieses
Plasmid wurde mit KpnI verdaut, und das erzeugte Brevi.-ori-DNA-Fragment
wurde mit p299LYSA ligiert, das ebenfalls mit KpnI verdaut worden ist,
um ein Plasmid herzustellen, das lysA enthält, welches autonom in coryneformen
Bakterien replizierbar ist. Das hergestellte Plasmid wurde als pLYSAB
bezeichnet. Das Verfahren zur Konstruktion von pLYSAB ist in 3 gezeigt.
-
<2> Bestimmung
der Nucleotidsequenz von lysA aus Brevibacterium lactofermentum
-
Plasmid-DNA
von p299LYSA wurde hergestellt, und dessen Nucleotidsequenz wurde
auf gleiche Weise, wie in Beispiel 1 beschrieben, bestimmt. Die
bestimmte Nucleotidsequenz und eine Aminosäuresequenz, die davon abgeleitet
wurde, und welche die Nucleotidsequenz kodiert, ist in SEQ-ID Nr.
10 gezeigt. Was die Nucleotidsequenz angeht, wurde eine Aminosäuresequenz,
die durch argS kodiert ist, und eine Aminosäuresequenz, die durch lysA
kodiert ist, jeweils in SEQ-ID Nr. 11 und 12 gezeigt.
-
Beispiel 3: Herstellung
von ppc aus Brevibacterium lactofermentum
-
<1> Herstellung
von ppc
-
Der
Wildtyp-Stamm von Brevibacterium lactofermentum ATCC 13869 wurde
als chromosomaler DNA-Donor verwendet. Chromosomale DNA wurde aus
dem ATCC-13869-Stamm nach einem gewöhnlichen Verfahren hergestellt.
Das DNA-Fragment, das ppc enthält,
wurde von der chromosomalen DNA mit dem PCR-Verfahren amplifiziert. Bezüglich der
DNA-Primer, die für
die Amplifizierung verwendet wurden, wurden basierend auf einer
bekannten Sequenz aus Corynebacterium glutamicum (siehe O'Regan, M. et al.,
Gene, 77, 237–251
(1989)) synthetische 23-mer-DNAs mit den in den SEQ-ID Nr. 13 bzw.
14 in der Sequenzliste gezeigten Nucleotidsequenzen verwendet, um
eine Region von ungefähr
3,3 Kilobasen zu amplifizieren, welche für PEPC kodiert. Die Synthese
von DNA und PCR wurde auf gleiche Weise, wie in Beispiel 1 beschrieben, durchgeführt.
-
Das
amplifizierte Genfragment von ungefähr 3.300 Basenpaaren wurde
durch Agarosegel-Elektrophorese identifiziert, und dann wurde das
aus dem Gel extrahierte Fragment durch ein gewöhnliches Verfahren gereinigt,
und mit dem Restriktionsenzym SalI verdaut (hergestellt von Takara
Shuzo). pHSG399 wurde als Klonierungsvektor für PPC verwendet. pHSG399 wurde
mit dem Restriktionsenzym SalI (hergestellt von Takara Shuzo) verdaut,
und wurde mit dem DNA-Fragment ligiert, das das amplifizierte ppc
enthält.
Ein Plasmid, welches ppc trägt,
das aus ATCC 13869 stammt, das wie oben beschrieben gewonnen wurde,
wurde als pPCF bezeichnet.
-
<2> Ligierung
des ppc-Gens mit dem lysC-Promotor
-
Das
wie oben beschrieben gewonnene pPCF, wurde mit dem Restriktionsenzym
DraI (hergestellt von Takara Shuzo) verdaut. Nachdem ein DNA-Fragment
von ungefähr
150 Basenpaaren stromaufwärts
des PEPC Strukturgens entfernt worden war, wurde eine Selbstligierung
durchgeführt,
um das Plasmid pPCFds zu gewinnen. pPCFds wurde mit dem Restriktionsenzym
SalI (hergestellt von Takara Shuzo) verdaut, und die geschnittenen
Enden wurden geglättet.
Die Bildung der glatten Enden wurde unter Verwendung eines DNA-Blunting-Kits
(hergestellt von Takara Shuzo) gemäß dem angegebenen Verfahren
durchgeführt.
-
p399AKYB,
das das Wildtyp-lysC enthält,
welches in Beispiel 1 gewonnen wurde, wurde mit den Restriktionsenzymen
ApaLI und PstI (beide hergestellt von Takara Shuzo) verdaut, und
die geschnittenen Enden wurden auf gleiche Weise wie oben beschrieben,
geglättet.
Ein kleineres Fragment, aus den zwei gewonnenen DNA-Fragmenten,
enthält
Brevi.-ori und einen Promotor für
lysC. Dieses Fragment wurde mit dem oben erwähnten Fragment ligiert, das
durch Verdauen von pPCFds mit SalI gewonnen wurde, und unter Verwendung des
DNA-Ligations-Kits geglättet
(hergestellt von Takara Shuzo).
-
Eine
DNA in einer Ligationslösung
wurde in Brevibacterium lactofermentum ATCC 13869 gemäß dem elektrischen
Pulsverfahren (Sugimoto et al., Japanische Patentanmeldung Veröffentlichungs-Nr.
2-207791) eingeschleust. Transformanten wurden auf Komplettmedium,
enthaltend 5 μg/ml
Chloramphenicol selektiert. Plasmid-DNA wurde aus den Transformanten
eingesammelt und mit EcoRI verdaut, um ein Plasmid zu gewinnen,
in dem der lysC-Promotor mit dem ppc-Strukturgen in normaler Richtung
ligiert war. Das erhaltene Plasmid wurde als pAKPFds bezeichnet.
Das Verfahren zur Konstruktion von pAKPFds ist in 4 gezeigt.
Das mit dem lysC-Promotor ligierte ppc wird nachfolgend als „Wildtyp-Hochexpressions-ppc" bezeichnet.
-
<3> Insertion
des Wildtyp-Hochexpressions-ppc in den Vektor
-
Das
Wildtyp-Hochexpressions-ppc, das oben gewonnen wurde, wurde mittels
PCR amplifiziert, um es in einen Vektor mit einem Replikationsursprung
einzubauen, der autonom in coryneformen Bakterien replizierbar ist,
der nicht Brevi.-ori ist. Bezüglich
der DNA-Primer wurde ein Oligonucleotid, das dem lysC-Promotorteil (SEQ-ID
Nr. 7) entspricht, welches von einer Sequenz für lysC, das für Corynebacterium
glutamicum bekannt ist (siehe Molecular Microbiology, 5 (5), 1197–1204 (1991);
Mol. Gen. Genet., 224, 317–324
(1990)), synthetisiert, und ein Oligonucleotid, das dem ppc-Teil
(SEQ-ID Nr. 8) entspricht, welches auf Grundlage einer Sequenz von
ppc, das für
Corynebacterium glutamicum bekannt ist (siehe O'Regan, M. et al., Gene, 77, 237–251 (1989))
wurde synthetisiert. Diese Primer wurden so entworfen, dass ein
Fragment von ungefähr
3.150 Basenpaaren, das das Wildtyp-Hochexpressions-ppc enthält, ampliziert
werden kann, und ein Ende des amplifizierten DNA-Fragments mit dem
Restriktionsenzym KpnI verdaut werden kann. Die Synthese von DNA
und die PCR wurden auf gleiche Weise, wie in Beispiel 1 beschrieben,
durchgeführt.
-
Der
Klonierungsvektor für
coryneforme Bakterien, pVK7, welcher neu hergestellt wurde, wurde
als Vektor zum Einschleusen des Wildtyp-Hochexpressions-ppc in coryneforme
Bakterien konstruiert. pVK7 wurde durch Ligieren von pHSG299, einen
Vektor für
E.coli (Kmr; Takeshita, S. et al., Gene,
61, 63–74
(1987)) mit pAM330, einem kryptischen Plasmid von Brevibacterium
lactofermentum konstruiert, wie unten beschrieben wird. pHSG299
wurde mit dem Restriktionsenzym AvaII (hergestellt von Takara Shuzo)
verdaut, das zu einer Spaltstelle führte, an den Enden geglättet, in
dem T4 DNA-Polymerase verwendet wurde, und mit pAM330 ligiert, das
mit HindIII (hergestellt von Takara Shuzo) verdaut worden ist, und
unter Verwendung von T4 DNA-Polymerase an den Enden geglättet. Abhängig von
der Orientierung des inserierten pAM330 in pHSG299 wurden die zwei
erhaltenen Plasmide als pVK6 und pVK7 bezeichnet, und pVK7 wurde
für die
folgenden Experimente verwendet. pVK7 ist sowohl in E.coli als auch
in Brevibacterium lactofermentum autonom replizierbar und hat eine
multiple Klonierungsstelle, die aus pHSG299 stammt, sowie lacZ'. Das Verfahren zur
Konstruktion von pVK6 und pVK7 ist in 5 gezeigt.
-
Ein
amplifiziertes Genfragment von ungefähr 3.150 Basenpaaren wurde
durch Agarosegel-Elektrophorese identifiziert, und dann wurde das
aus dem Gel extrahierte Fragment durch ein gewöhnliches Verfahren gereinigt
und mit dem Restriktionsenzym KpnI (hergestellt von Takara Shuzo)
verdaut. Das DNA-Fragment wurde mit pVK7 ligiert, das mit dem Restriktionsenzym
KpnI verdaut worden ist. Das hergestellte Plasmid wurde als pPwm
bezeichnet. Das Konstruktionsverfahren von pPwm ist in 6 gezeigt.
-
Beispiel 4: Herstellung
eines Plasmids, das eine Kombination aus mutiertem lysC und lysA
umfasst
-
Ein
Plasmid, das mutiertes lysC, lysA und einen Replikationsursprung
für coryneforme
Bakterien enthält,
wurde aus dem Plasmid p399AK9B, das mutiertes lysC und einen Brevi.-ori
enthält
und Plasmid p299LYSA, das lysA enthält, hergestellt. p299LYSA wurde
mit den Restriktionsenzymen BamHI und KpnI (beide hergestellt von
Takara Shuzo) verdaut und an den Enden geglättet. Die Bildung der glatten
Enden wurde unter Verwendung eines DNA-Blunting-Kits (hergestellt
von Takara Shuzo) nach dem angegebenen Verfahren durchgeführt. Das
gewonnene DNA-Fragment wurde mit p399AK9B ligiert, das mit SalI
verdaut worden ist und an den Enden geglättet wurde. Auf diese Weise
wurde ein Plasmid, das mutiertes lysC und lysA enthält, das in
coryneformen Bakterien autonom replizierbar ist, hergestellt und
als pCL bezeichnet. Das Konstruktionsverfahren von pCL ist in 7 gezeigt.
-
Vergleichsbeispiel 1:
Herstellung von dapA, dapB und ddh aus Brevibacterium lactofermentum
-
Als
Gene, die mit L-Lysin-Biosynthese neben lysC, lysA und ppc assoziiert
sind, wurden dapA (Dihydrodipicolinatsynthasegen), dapB (Dihydrodipicolinatreductasegen)
und ddh (Diaminopimelatdehydrogenasegen) wie folgt gewonnen.
-
<1> Herstellung
von dapA und Konstruktion eines Plasmids, das dapA enthält
-
Ein
Wildtyp-Stamm von Brevibacterium lactofermentum ATCC 13869 wurde
als chromosomaler DNA-Donor verwendet. Chromosomale DNA wurde aus
dem ATCC 13869-Stamm mit einem gewöhnlichen Verfahren hergestellt.
Ein DNA-Fragment, das dapA enthält,
wurde von chromosomaler DNA mittels PCR amplifiziert. Hinsichtlich
der DNA-Primer, die für
diese Amplifizierung verwendet wurden, wurden 23-mer DNAs mit den
Nucleotidsequenzen, die in den SEQ-ID Nr. 21 bzw. 22 der Sequenzliste
gezeigt sind, auf Grundlage einer bekannten Sequenz für Corynebacterium
glutamicum (siehe Nucleic Acids Research, 18 (21, 6421 (1990); EMBL
Zugangs-Nr. X53993) synthetisiert, um einen Bereich von ungefähr 1,5 Kilobasen
zu amplifizieren, welcher für
DDPS kodiert. Die Synthese der DNA und PCR wurden auf gleiche Weise,
wie in Beispiel 1 beschrieben, durchgeführt. pCR1000 (hergestellt von
Invitrogen, siehe Bio/Technology, 9, 657–663 (1991)) wurde als Klonierungsvektor
für das
amplifizierte Genfragment von 1.411 Basenpaaren verwendet und mit
dem amplifizierten dapA-Fragment ligiert. Die Ligierung der DNA
wurde unter Verwendung eines DNA-Ligations-Kits (hergestellt von
Takara Shuzo) durchgeführt,
die Waschschritte mit dem angegebenen Verfahren. Auf diese Weise
wurde ein Plasmid hergestellt, bei dem das dapA-Fragment von 1.411
Basenpaaren vom Chromosom von Brevibacterium lactofermentum amplifiziert
worden war, mit pCR1000 ligiert wurde. Das Plasmid, das wie oben
beschrieben gewonnen wurde, welches dapA enthält, das aus ATCC 13869 stammt,
wurde als pCRDAPA bezeichnet.
-
Ein
Transformantenstamm AJ13106, der durch Einschleusen von pCRDAPA
in dem E.coli JM109-Stamm gewonnen wurde, ist international seit
dem 26. Mai 1995 unter der Zugangs-Nr. FERM BP-5113 beim National
Institute of Biscience and Human Technology der Agency of Industrial
Science and Technology des Ministeriums für internationalen Handel und
Industrie (1–3,
Higashi 1-chome, Tsukuba-shi, Ibaraki-ken, 305 Japan) auf Grundlage
des Budapester Vertrages hinterlegt worden.
-
Brevi.-ori
wurde in das hergestellte pCRDAPA eingeschleust, um ein Plasmid
herzustellen, das dapA trägt,
das autonom in coryneformen Bakterien replizierbar ist. pHK4 wurde
mit den Restriktionsenzymen KpnI und BamHI (hergestellt von Takara
Shuzo) verdaut, und die gespaltenen Enden wurden geglättet. Die
Bildung der glatten Enden wurde unter Verwendung eines DNA-Blunting-Kits
(hergestellt von Takara Shuzo), nach dem angegebenen Verfahren hergestellt.
Nach der Bildung der glatten Enden wurde ein phosphory lierter SmaI-Linker
(hergestellt von Takara Shuzo) ligiert, um eine Modifizierung durchzuführen, damit
das DNA-Fragment, das dem Brevi.-ori-Teil entspricht, allein aus
pHK4 durch Verdau mit SmaI ausgeschnitten werden kann. Dieses Plasmid
wurde mit SmaI verdaut, und das hergestellte Brevi.-ori-DNA-Fragment wurde
mit pCRDAPA ligiert, das ebenfalls mit SmaI verdaut worden ist,
um ein Plasmid herzustellen, das dapA enthält, das autonom in coryneformen
Bakterien replizierbar ist. Dieses Plasmid wurde als pDPSB bezeichnet.
Das Verfahren zur Herstellung von pDPSB (Kmr)
ist in 8 gezeigt.
-
<2> Herstellung
von dapB und Konstruktion eines Plasmids, das dapB enthält
-
Ein
Wildtyp-Stamm von Brevibacterium lactofermentum ATCC 13869 wurde
als chromosomaler DNA-Donor verwendet. Chromosomale DNA wurde aus
ATCC 13869-Stamm durch ein gewöhnliches
Verfahren hergestellt. Ein DNA-Fragment, das dapB enthält, wurde
von der chromosomalen DNA mittels PCR amplifiziert. Hinsichtlich
der DNA-Primer, die für
die Amplifikation verwendet wurden, wurden 23-mer-DNAs mit den Nucleotidsequenzen,
die in den SEQ-ID Nr. 19 bzw. 20 in der Sequenzliste wiedergegeben
sind, basierend auf der Sequenz, die für Brevibacterium lactofermentum
bekannt ist (siehe Journal of Bacteriology, 175 (9), 2743–2749 (1993))
synthetisiert, um eine Region von ungefähr 2,0 kb zu amplifizieren,
die für
die DDPR kodiert. Die Synthese der DNA und PCR wurden auf gleiche
Weise, wie in Beispiel 1 beschrieben, durchgeführt. pCR-Script (hergestellt
von Invitrogen) wurde als Klonierungsvektor für das amplifizierte Genfragment
von 2.001 Basenpaaren verwendet, und mit dem amplifizierten dapB-Fragment
ligiert. Auf diese Weise wurde ein Plasmid hergestellt, in dem das
dapB-Fragment von 2.001 Basenpaaren, das aus dem Chromosom von Brevibacterium
lactofermentum amplifiziert wurde, mit pCR-Script ligiert. Das Plasmid,
das, wie oben beschrieben, gewonnen wurde, welches dapB enthielt,
das aus ATCC 13869 stammt, wurde als pCRDAPB bezeichnet. Der Transformantenstamm
AJ13107, der durch das Einschleusen von pCRDAPB in E.coli JM109-Stamm
gewonnen wurde, ist international seit dem 26. Mai 1995 unter der
Zugangs-Nr. FERM BP-5114 beim National Institute of Bioscience and
Human Technology der Agency of Industrial Science and Technology
des Ministeriums für
internationalen Handel und Industrie (1–3, Higashi 1-chome, Tsukuba-shi,
Ibaraki-ken, 305 Japan) auf Grundlage des Budapester Vertrags hinterlegt
worden.
-
Ein
Fragment von 1.101 Basenpaaren, das ein Strukturgen von DDPR enthält, wurde
durch Verdau von pCRDAPB mit EcoRV und SphI extrahiert. Dieses Fragment
wurde mit pHSG399 ligiert, das mit HincII und SphI verdaut worden
ist, um ein Plasmid herzustellen. Das hergestellte Plasmid wurde
als p399DPR bezeichnet.
-
Brevi.-ori
wurde in das hergestellte p399DPR eingeschleust, um ein Plasmid
herzustellen, das dapB trägt,
das autonom in coryneformen Bakterien replizierbar ist. pHK4 wurde
mit dem Restriktionsenzym KpnI (hergestellt von Takara Shuzo) verdaut,
und die gespaltenen Enden wurden geglättet. Die Bildung der glatten Enden
wurde unter Verwendung eines DNA-Blunting-Kits (hergestellt von
Takara Shuzo) nach dem angegebenen Verfahren durchgeführt. Nach
der Bildung der glatten Endung wurde ein phosphorylierter BamHI-Linker (hergestellt
von Takara Shuzo) ligiert, um eine Modifizierung zu ergeben, damit
das DNA-Fragment, das dem Brevi.-ori-Teil entspricht, aus pHK4 allein
durch den Verdau mit BamHI ausgeschnitten werden kann. Dieses Plasmid
wurde mit BamHI verdaut, und das hergestellte Brevi.-ori-DNA-Fragment
wurde mit p399DPR ligiert, das ebenfalls mit BamHI verdaut worden
ist, um ein Plasmid herzustellen, das dapB enthält, das autonom in coryneformen
Bakterien replizierbar ist. Das hergestellte Plasmid wurde als pDPRB
bezeichnet. Das verfahren zur Konstruktion von pDPRB ist in 9 gezeigt.
-
<3> Herstellung
von ddh und Konstruktion eines Plasmids, das ddh enthält.
-
Das
ddh-Gen wurde durch Amplifizieren des ddh-Gens von chromosomaler
DNA aus Brevibacterium lactofermentum ATCC 13869 in Übereinstimmung
mit dem PCR-Verfahren unter Verwendung von zwei Oligonucleotid-Primern
(SEQ-ID Nr. 23, 24) auf Grundlage einer bekannten Nucleotidsequenz
des ddh-Gens für
Corynebacterium glutamicum (Ishino, S. et al., Nucleic Acids Res.,
15, 3917 (1987)) hergestellt. Das gewonnene amplifizierte DNA-Fragment
wurde mit EcoT22I und mit AvaI verdaut, und die gespaltenen Enden
wurden geglättet.
Danach wurde das Fragment in eine SmaI-Schnittstelle von pMW119
inseriert, um das Plasmid pDDH zu gewinnen.
-
Als
nächstes
wurde pDDH mit SalI und EcoRI verdaut, gefolgt von der Bildung glatter
Enden. Danach wurde das gewonnene Fragment mit pUC18 ligiert, das
mit SmaI verdaut worden ist. Das Plasmid, das auf diese Weise gewonnen
wurde, wurde als pUC18DDH bezeichnet.
-
Der
Brevi.-ori wurde in pUC18DDH eingeschleust, um ein Plasmid herzustellen,
das ddh trägt,
und das autonom in coryneformen Bakterien replizierbar ist. pHK4
wurde mit den Restriktionsenzymen KpnI und BamHI verdaut, und die
geschnittenen Enden wurden geglättet.
Die Bildung der glatten Enden wurde durch Verwendung eines DNA-Blunting-Kits
(hergestellt von Takara Shuzo) nach dem angegebenen Verfahren hergestellt.
Nach der Bildung der glatten Enden wurde ein phosphorylierter PstI-Linker
(hergestellt von Takara Shuzo) ligiert, so dass es in die PstI-Schnittstelle
von pHSG299 inseriert wurde. Ein Plasmid, das, wie oben beschrieben,
konstruiert wurde, wurde als pPK4 bezeichnet. Als nächstes wurde
pUC18DDH mit XbaI und KpnI verdaut, und das erzeugte Fragment wurde
mit pPK4 ligiert, welches mit KpnI und XbaI verdaut worden ist.
Auf diese Weise wurde ein Plasmid, welches autonom in coryneformen
Bakterien replizierbar ist, das ddh enthält, hergestellt. Dieses Plasmid
wurde als pPK4D bezeichnet. Das Verfahren zur Konstruktion von pPK4D
ist in 10 gezeigt.
-
Vergleichsbeispiel 2:
Herstellung eines Plasmids, das die Kombination von mutiertem lysC
und dapA, dapB oder ddh umfasst
-
<1> Konstruktion
einer Kombinationen des mutierten lysC und dapA
-
Ein
Plasmid, das mutiertes lysC, dapA und den Replikationsursprung für coryneforme
Bakterien umfasst, wurde vom Plasmid pCRDAPA ausgehend hergestellt,
das dapA umfasst, und das Plasmid p399AK9B, das mutiertes lysC umfasst
und den Brevi.-ori. p399AK9B wurde vollständig mit SalI verdaut, und
dann wurden die Enden geglättet.
Ein EcoRI-Linker wurde daran ligiert, um ein Plasmid herzustellen,
bei dem die SalI-Schnittstelle in eine EcoRI modifiziert worden
ist. Das gewonnene Plasmid wurde als p399AK9BSE bezeichnet. Das
mutierte lysC und Brevi.-ori wurden als ein Fragment durch teilweises
Verdauen durch p399AK9BSE mit EcoRI ausgeschnitten. Dieses Fragment
wurde mit pCRDAPA ligiert, das mit EcoRI verdaut worden ist. Das
gewonnene Plasmid wurde als pCRCAB bezeichnet. Dieses Plasmid ist
in E.coli und coryneformen Bakterien autonom replizierbar und verleiht
dem Wirt eine Kanamycinresistenz, wobei das Plasmid eine Kombination
aus mutiertem lysC und dapA umfasst. Das Verfahren zur Konstruktion
von pCRCAB ist in 11 gezeigt.
-
<2> Konstruktion
eines Plasmids, das eine Kombination aus mutiertem lysC und dapB
umfasst
-
Ein
Plasmid, das mutiertes lysC und dapB umfasst, wurde vom Plasmid
p399AK9 mit mutiertem lysC und dem Plasmid p399DPR mit dapB hergestellt.
Ein Fragment von 1.101 Basenpaaren, das ein Strukturgen von DDPR
enthält,
wurde durch Verdau von p399DPR mit EcoRV und SphI extrahiert. Dieses
Fragment wurde mit p399AK9 ligiert, das mit SalI verdaut worden
ist, und dann an den Enden geglättet
worden ist, und dann weiter mit SphI verdaut worden ist, um ein
Plasmid herzustellen, das eine Kombination aus mutiertem lysC und dapB
umfasst. Dieses Plasmid wurde als p399AKDDPR bezeichnet.
-
Als
nächstes
wurde ein Brevi.-ori in das gewonnene p399AKDDPR eingeschleust.
Das Plasmid pHK4, das den Brevi.-ori enthält, wurde mit dem Restriktionsenzym
KpnI verdaut (hergestellt von Takara Shuzo), und die gespaltenen
Enden wurden geglättet.
Die Bildung glatter Enden wurde unter Verwendung eines DNA-Blunting-Kits
(hergestellt von Takara Shuzo) in Übereinstimmung mit dem angegebenen
Verfahren durchgeführt. Nach
der Bildung der glatten Enden wurde ein phosphorylierter BamHI-Linker
(hergestellt von Takara Shuzo) ligiert, um eine Modifizierung durchzuführen, damit
das DNA-Fragment, das dem Brevi.-ori-Teil entspricht, aus pHK4 allein
durch Verdau mit BamHI ausgeschnitten werden kann. Dieses Plasmid
wurde mit BamHI verdaut, und das hergestellte Brevi.-ori DNA-Fragment
wurde mit p399AKDDPR ligiert, das ebenfalls mit BamHI geschnitten
worden ist, um ein Plasmid herzustellen, das mutiertes lysC und
dapB enthält,
das in coryneformen Bakterien autonom replizierbar ist. Das konstruierte
Plasmid wurde als pCB bezeichnet. Das Verfahren zur Konstruktion
von pCB ist in 12 gezeigt.
-
<3> Konstruktion
eines Plasmids, das eine Kombination aus mutiertem lysC und ddh
umfasst
-
Ein
Plasmid, das mutiertes lysC, ddh und einen Replikationsursprung
für coryneforme
Bakterien enthält,
wurde aus dem Plasmid pUC18DDH hergestellt, das ddh enthält, und
dem Plasmid p399AK9B, das mutiertes lysC und den Brevi.-ori enthält. pUC18DDH
wurde mit dem Restriktionsenzym EcoRI verdaut (hergestellt von Takara
Shuzo), an den Enden geglättet
und mit einem SalI-Polylinker an einem Ende ligiert, um eine EcoRI-Schnittstelle
in eine SalI-Schnittstelle zu ändern.
Das erhaltene Plasmid wurde mit SalI verdaut, um ein DNA-Fragment
zu gewinnen, das ddh enthält.
-
Dann
wurde p399AK9B mit dem Restriktionsenzym SalI geschnitten, und mit
dem DNA-Fragment ligiert, das ddh enthält. Auf diese Weise wurde ein
Plasmid, das mutiertes lysC, ddh und einen Brevi.-ori enthält, das
in coryneformen Bakterien autonom replizierbar ist, hergestellt,
und als pCD bezeichnet. Das Konstruktionsverfahren für pCD ist
in 13 gezeigt.
-
Beispiel 5: Einschleusung
von Plasmiden, die Gene für
die L-Lysin-Biosynthese umfassen, in ein L-Lysin-produzierendes
Brevibacterium lactofermentum Bakterium
-
Die
Plasmide, die gene für
L-Lysin-Biosynthese umfassen, die, wie oben beschrieben, hergestellt
wurden, d. h. p399AK9B (Cmr), pLYSAB (Cmr), pPwm (Kmr), pCRCAB
(Kmr), pCB (Cmr),
pCD (Cmr) und pCL (Cmr) wurden
jeweils in das L-Lysin-produzierende Bakterium AJ11082 (NRRL B-11470)
von Brevibacterium lactofermentum eingeschleust. Der AJ11082-Stamm
hat eine AEC-Resistenz. Die Plasmide wurden nach dem elektrischen
Pulsverfahren (Sugimoto et al., Japanische Patentanmeldung Veröffentlichungs-Nr.
2-207791) eingeschleust. Die Transformanten wurden auf Grundlage
von Medikamentenresistenz-Markern ausgewählt, die die jeweiligen Plasmide
aufweisen. Transformanten wurden auf Komplettmedium selektiert,
das 5 μg/ml
Chloramphenicol enthält,
wenn ein Plasmid, das eine Chloramphenicolresistenz umfasst, eingeschleust
wurde, oder Transformanten wurden auf Komplettmedium, enthaltend
25 μg/ml
Kanmycin selektiert, wenn ein Plasmid eingeschleust wurde, das ein
Kanmycin-Resistenzgen umfasst.
-
In
einen Stamm aus den oben genannten Transformanten, in dem mutiertes
lysC und lysA verstärkt waren,
wurde pPwm (Kmr) eingeschleust, um einen
Stamm zu gewinnen, in dem die drei mutierten lysC, lysA und ppc
verstärkt
waren (AJ11082/pCL/pPwm). Die Transformanten wurden auf Komplettmedium,
enthaltend 5 μg/ml
Chloramphenicol und 25 μg/ml
Kanamycin selektiert.
-
Beispiel 6: Produktion
von L-Lysin
-
Jede
der Transformanten, die in Beispiel 5 gewonnen wurden, wurde in
einem L-Lysin-Produktionsmedium kultiviert, um dessen L-Lysin-Produktionsfähigkeit
zu untersuchen.
-
[L-Lysin-Produktionsmedium]
-
Neben
Calciumcarbonat wurden die folgenden Bestandteile (in 1 l) gelöst, und
der pH wurde auf 8,0 mit KOH eingestellt. Das Medium wurde bei 115°C für 15 min
sterilisiert, und Calciumcarbonat (50 g), das mit heißer Luft
in trockenem Zustand separat sterilisiert worden ist, wurde danach
hinzugegeben.
| Glucose | 100
g |
| (NH4)2SO4 | 55
g |
| KH2PO4 | 1
g |
| MgSO4 7H2O | 1
g |
| Biotin | 500 μg |
| Thiamin | 2.000 μg |
| FeSO4 7H2O | 0,01
g |
| MnSO4 7H2O | 0,01
g |
| Nicotinamid | 5
mg |
| Proteinhydrolysat
(Mamenou) | 30
ml |
| Calciumcarbonat | 50
g |
-
Jede
der zahlreichen Typen an Transformanten und der Parentalstämme wurden
in das Medium mit der oben beschriebenen Zusammensetzung inokuliert, um
eine Kultivierung bei 31,5°C
bei umgekehrtem Schütteln
durchzuführen.
Die Menge des hergestellten L-Lysins nach 40 ode72 Stunden Kultivierung
wird in Tabelle 1 gezeigt. In der Tabelle stellt lysC* mutiertes
lysC dar.
-
Tabelle
1 Akkumulierung
von L-Lysin nach Kultivierung über
40 oder 72 Stunden
-
Wie
oben gezeigt, war die Menge produzierten L-Lysins, wenn mutiertes
lysC, lysA oder ppc leicht erhöht
war, oder wenn mutiertes lysC in Kombination mit dapA oder ddh verstärkt war,
größer als
oder äquivalent der,
die durch den Parentalstamm nach 72 h Kultivierung hergestellt wurde,
jedoch war die Menge hergestellten L-Lysins geringer als die, welche
nach 40 h Kultivierung vom Parentalstamm hergestellt wurde. Das
bedeutet, dass die L-Lysin-Produktionsgeschwindigkeit durch Kultivierung über einen
kürzeren
Zeitraum verringert wurde. Gleichermaßen wurde, wenn mutiertes lysC
und ddh verstärkt
wurden, die Menge hergestellten L-Lysins kleiner als die, die durch
den Parentalstamm nach 40 h und 72 h Kultivierung hergestellt wurde.
Im Gegensatz dazu war in dem Fall, dass der Stamm, bei dem dapB
zusammen mit dem mutierten lysC verstärkt war, das Wachstum verbessert,
die L-Lysin-Produktionsgeschwindigkeit
wurde erfolgreich in der kurzen Kultivierungsdauer wiederhergestellt,
und die akkumulierte Menge an L-Lysin wurde über den langen Zeitraum der Kultivierung
ebenfalls verbessert. Im Fall eines Stamms, in dem drei mutierte
lysC, lysA und ppc gleichzeitig verstärkt waren, war die L-Lysin-Produktivität weiter
verbessert.
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-