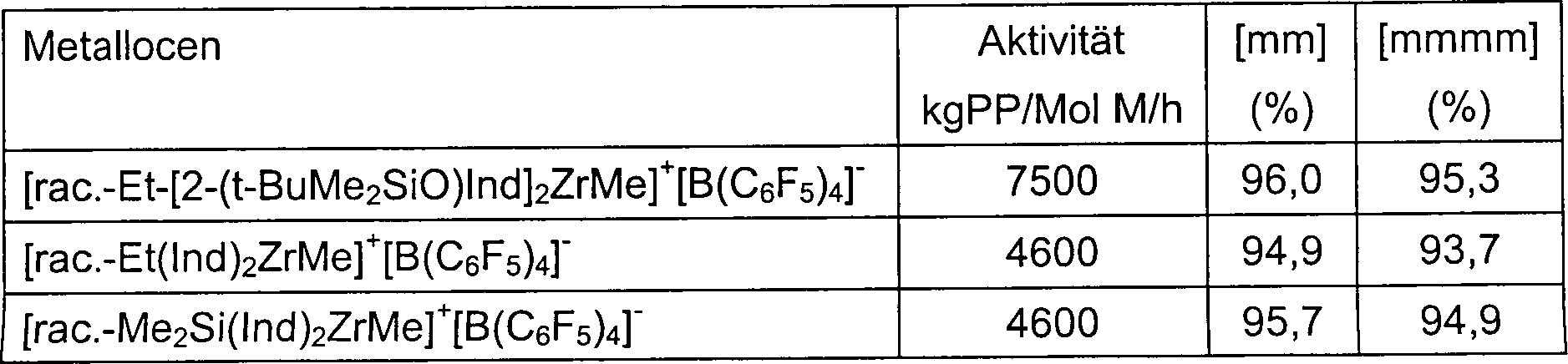-
Die
vorliegende Erfindung betrifft neue Metallocen-Katalysatorsysteme
für die
Homo- und Copolymerisation
von Olefinen, insbesondere Propylen, Ethylen und höheren alpha-Olefinen,
in der Gegenwart eines Cokatalysators wie etwa Methylalumoxan (MAO).
Insbesondere betrifft die Erfindung Metallocene mit Heteroatomsubstituierten
Indenyl- und Indenylderivat-Liganden, ein Verfahren zu deren Herstellung
und deren Verwendung bei der Polymerisation von Olefinen, insbesondere
Propylen und Ethylen.
-
Chirale
C2-symmetrische Bis(indenyl)-ansa-Metallocene
sind Vorläufer
für hoch
aktive Katalysatoren für
die stereoselektive Polymerisation von alpha-Olefinen. Die Leistungscharakteristika
dieser Systeme sind unterschiedlich, wobei die Variationen durch
Größe und Position
der Substituenten induziert werden. Beispielsweise erzeugen Dimethylsilylen-verbrückte 2,2'-Dimethyl-4,4'-diaryl-substituierte
Bis(indenyl)zirkonocene, entwickelt von Brintzinger und Mitarbeitern
(Organometallics 1994, 13, 964) und Spaleck et al. (Organometallics 1994,
13, 954) isotaktische Polypropylene, mit Katalysatoraktivitäten und
Polymereigenschaften, welche mit den mit heterogenen Ziegler-Natta
Katalysatoren erhaltenen vergleichbar sind.
-
Das
Gebiet der elektronisch veränderten
Bis(indenyl)-Metallocene ist relativ unerforscht geblieben. Es war
früher
berichtet worden, dass Halogen- oder Alkoxysubstitution in den sechsgliedrigen
Ringen von Indenen die Aktivität
des Katalysatorsystems und das Molekulargewicht des erzeugten Polymers
verringert (Consiglio et al., Organometallics 1990, 9, 3098; Collins
et al., Organometallics 1992, 11, 2115). Bis(indenyl)zirkonocene
mit 2-Amino-funktionalisierten Liganden wurden vor kurzem von mehreren
Gruppen berichtet (Luttikhedde et al., Organometallics 1996, 15,
3092; Plenio and Burth, J. Organomet. Chem. 1996, 519, 269; Brintzinger
et al., J. Organomet. Chem. 1996, 520, 63). Die verbrückten Komplexe
zeigen im Vergleich mit ihren unsubstituierten Bis(indenyl)zirkonocen-Analoga
etwas niedrigere Katalysatoraktivitäten. WO 94/28034,
EP 485 822 und
EP 584 609 offenbaren 2-substituierte
Indenylmetallocene im Allgemeinen und insbesondere 2-Methyl-Derivate.
Die Katalysatoren waren trägergestützt und
für ataktische
Polymerisation, um verbesserte Polymermorphologie zu ergeben, vorgesehen.
-
Die
vorliegende Erfindung betrifft neue Metallocenkomplexe (I) mit einem
Sauerstoffatom direkt an die 2-Position einer Pentahaptoindenyl-Gruppe
gebunden, z. B. racemisches [Ethylenbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid,
und deren Anwendung bei der Polymerisation von Olefinen. Diese Komplexe (I),
in Kombination mit MAO oder anderen Aktivatoren, bilden hoch aktive
Katalysatorsysteme für
die Homo- und Copolymerisation von Olefinen. Beispielsweise polymerisieren
die I/MAO Katalysatorsysteme Propylen zu hoch isotaktischem Polypropylen.
Die Propylen- und Ethylenpolymerisationsaktivitäten dieser (I)/MAO-Systeme übersteigen
unter ähnlichen
Polymerisationsbedingungen die von mehreren herkömmlichen ansa-Metallocen/MAO
Katalysatorsystemen, wie etwa von Dimethylsilylenbis(4,5,6,7-tetrahydroindenyl)zirkoniumdichlorid/MAO.
Die neuen Katalysatorsysteme sind sehr stabil und behalten ihre
hohen Aktivitäten
bei außergewöhnlich niedrigen
[Al] : [Zr] Verhältnissen.
-
Erfindungsgemäß wird ein
neuer Katalysatorvorläufer
erhalten, bei dem eine Siloxysubstitution in der 2-Position des
fünfgliedrigen
Rings einer Verbindung vom Indenyl-Typ ausgeführt worden ist. Dadurch ist
es möglich,
Metallocenverbindungen herzustellen, bei denen ein Sauerstoffatom
direkt an die 2-Position einer Pentahaptoindenylgruppe gebunden
ist.
-
Der
erfindungsgemäße Katalysatorvorläufer betrifft
somit eine Indenylverbindung mit der Formel (I):
(IndY)mMRnBo (I)worin: jedes
IndY gleich oder verschieden ist, und ein mono- oder polysubstituierter,
kondensierter oder nicht-kondensierter, homo- oder heterozyklischer
Indenylligand, Dihydroindenylligand oder Tetrahydroindenylligand
ist, der Ligand an 2-Position seiner Indenylstruktur durch die Gruppe
Y substituiert ist, wobei die Gruppe Y die nachfolgende Struktur
(II) hat:
worin D Silizium oder Germanium
ist; R
3, R
3' und R
4 gleich oder verschieden sind und ein Wasserstoffatom, eine
C
1-C
10 Hydrocarbylgruppe
oder eine C
1-C
10 Hydrocarbyloxygruppe
sind, oder mindestens zwei aus R
3, R
3' und
R
4 zusammen eine C
2-C
20 Ringstruktur bilden; M ein Übergangsmetall
der Gruppe 4 des Periodensystems ist und an den Liganden IndY in
mindestens einem η5-Bindungsmodus gebunden
ist; jedes R gleich oder verschieden ist und ein Wasserstoffatom,
ein Halogenatom, eine C
1-C
10 Hydrocarbylgruppe,
eine C
1-C
10 Hydrocarbyloxygruppe,
eine Trihydrocarbylsilylgruppe ist, oder zwei R zusammen eine C
2-C
20 Ringstruktur
bilden; B ein Brückenatom
oder ein Brückengruppe
zwischen zwei IndY-Liganden oder zwischen einem IndY-Liganden und
dem Übergangsmetall
M ist; m gleich 1 oder 2 ist; o gleich 0 oder 1 ist; und n gleich
4-m ist, wenn keine Brücke
B vorhanden ist oder B ein Brückenatom
oder ein Brückengruppe
zwischen zwei IndY-Liganden ist, oder n gleich 4-m-o ist, wenn B
ein Brückenatom
oder ein Brückengruppe
zwischen einem IndY-Liganden und dem Übergangsmetall M ist.
-
Unter
mono- oder polysubstituiert wird verstanden, dass zusätzlich zum
Substituenten Y gegebenenfalls andere Substituenten an den Ringen
des Liganden IndY vorhanden sein können.
-
Unter
kondensiert oder nicht-kondensiert wird verstanden, dass jeder Ring
des Liganden IndY kondensiert oder nicht-kondensiert sein kann,
d. h. mit mindestens einem weiteren Ring mindestens zwei Atome gemeinsam
haben kann.
-
Unter
homo- und heterozyklisch wird verstanden, dass jeder Ring des Liganden
IndY nur Kohlenstoffringatome (homo- oder isozyklisch) haben kann
oder andere Ringatome als Kohlenstoff (heterozyklisch) haben kann.
-
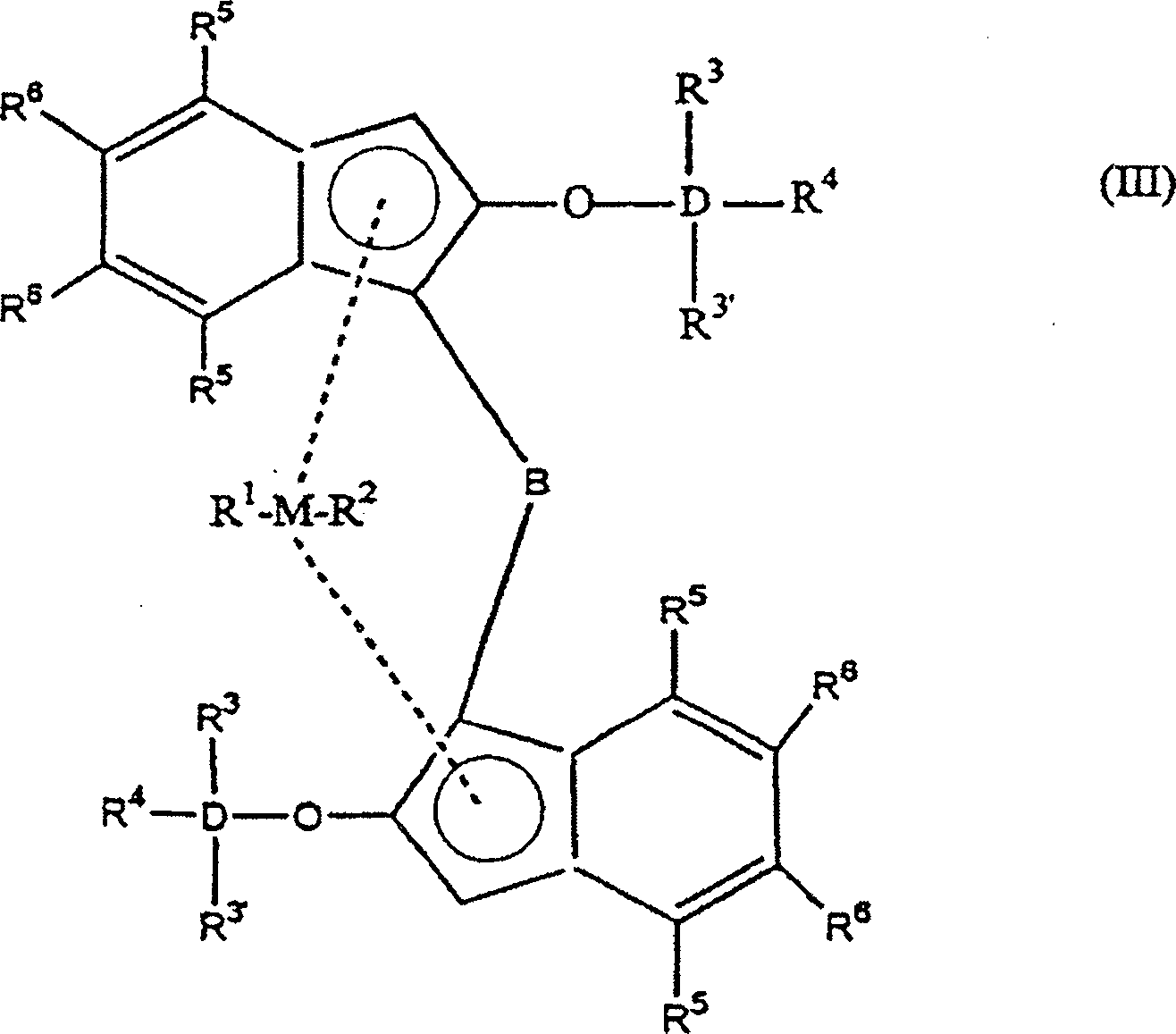
Bevorzugt
hat die Indenylverbindung nach Formel (I) die nachfolgende Formel
(III)
worin
M
ein Metall, ausgewählt
aus Zirkonium, Titan oder Hafnium ist,
D ein Element, ausgewählt aus
Silizium (Si) oder Germanium (Ge) ist,
B eine Brücke ist,
umfassend mindestens eine Gruppe aus -(CH
2)
n-, -Si(R
3)
2-, oder -Ge(R
3)
2-,
R
1 und R
2 die gleichen oder verschiedene Gruppen
sind, ausgewählt
aus einem Wasserstoffatom, einer C
1-C
10 Alkylgruppe, C
1-C
10 Alkoxygruppe, C
6-C
10 Arylgruppe, C
6-C
10 Aryloxygruppe, C
2-C
10 Alkenylgruppe, C
2-C
10 Arylalkylgruppe, C
2-C
10 Alkylarylgruppe, C
8-C
40 Arylalkenylgruppe oder einem Halogenatom,
bevorzugt eine C
1-C
10 Alkylgruppe
und/oder ein Halogenatom
R
3', R
3–R
6 die gleichen oder verschiedene Gruppen
sind, ausgewählt
aus Wasserstoff, einer C
1-C
10 Alkylgruppe,
C
1-C
10 Alkoxygruppe,
C
6-C
10 Arylgruppe,
C
6-C
10 Aryloxygruppe, C
2-C
10 Alkenylgruppe, C
2-C
10 Arylalkylgruppe, C
2-C
10 Alkylarylgruppe, C
8-C
40 Arylalkenylgruppe und einem Halogenatom.
Die R
3'-
und R
3-Gruppen
können
auch miteinander verknüpft
sein um eine Ringstruktur zu bilden, und R
4 kann
auch Teil einer Ringstruktur sein.
-
Bevorzugt
ist M Zirkonium.
-
R1 und R2 sind bevorzugt
gleich und am meisten bevorzugt sind sie Halogenatome, beispielsweise Chloratome.
-
R3 ist bevorzugt eine C1-C10 Alkyl- oder Arylgruppe und am meisten
bevorzugt ist es eine Methylgruppe. Gleichermaßen ist R4 bevorzugt
eine C1-C10 Alkyl-
oder Arylgruppe, beispielsweise eine tert.-Butylgruppe, t-Hexylgruppe
oder Cyclohexylgruppe.
-
Am
meisten bevorzugt ist D gleich Si, ist B gleich -CH2CH2-, ist R1 = R2 und gleich Chlor, ist R3 gleich CH3 und ist R5-R6 aromatisch oder kondensiert aromatisch
oder Alkyl oder Wasserstoff.
-
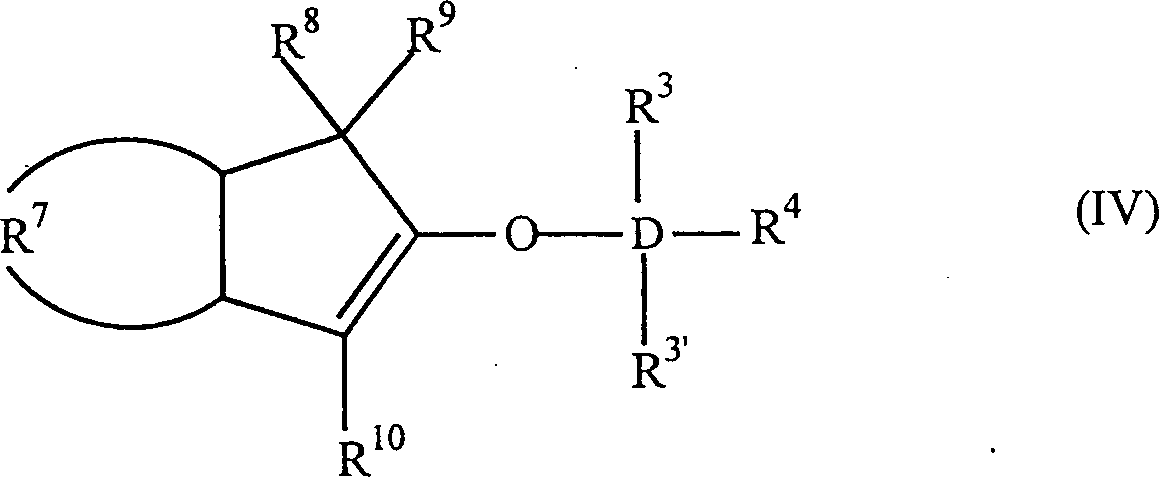
Die
Erfindung betrifft auch 2-Trihydracarbyl- und 2-Trihydrocarbyloxysiloxyinden-
und -germyloxyindenverbindungen mit der allgemeinen Formel:
worin:
D Silizium oder Germanium ist; R
3, R
3' und
R
4 gleich oder verschieden sind und ein
Wasserstoffatom, eine C
1-C
10 Hydrocarbylgruppe
oder eine C
1-C
10 Hydrocarbyloxygruppe
sind, oder mindestens zwei aus R
3, R
3' und
R
4 zusammen eine C
2-C
20 Ringstruktur bilden; R
7 eine
vieratomige Kette ist, welche einen unsubstituierten oder substituierten,
weiter nicht-kondensierten oder weiter kondensierten, homozyklischen
(= isozyklischen) oder heterozyklischen, ungesättigten oder gesättigten,
aliphatischen oder aromatischen sechsgliedrigen Ring bildet; und
R
8, R
9 und R
10 gleich oder verschieden sind und ein Wasserstoffatom,
eine C
1-C
10 Hydrocarbylgruppe,
eine C
1-C
10 Hydrocarbyloxygruppe,
eine Tri-C
1-C
10-hydrocarbylsilylgruppe
oder eine Tri-C
1-C
10-hydrocarbyloxysilylgruppe
sind, oder einer aus R
8 und R
9 eine
Gruppe B sein kann, welche ein Brückenatom oder eine Brückengruppe
zu einer Cyclopentadienyl-, Fluorenyl- oder Indenylgruppe ist, mit
der Maßgabe,
dass wenn R
7 einen unsubstituierten Benzolring
bildet, D Silizium ist, R
3, R
3' und R
4 Methylgruppen sind und R
8 und
R
10 Wasserstoffe sind, R
9 kein
Wasserstoff ist. Bevorzugt haben sie die Formel:
worin:
R
3',
R
3–R
6 gleich oder verschieden sind und ein Wasserstoffatom,
eine C
1-C
10 Alkylgruppe,
eine C
1-C
10 Alkoxygruppe,
eine C
6-C
10 Arylgruppe,
eine C
2-C
10 Alkenylgruppe,
eine C
7-C
22 Arylalkylgruppe,
eine C
7-C
22 Alkylarylgruppe,
eine C
8-C
23 Arylalkenylgruppe
oder ein Halogenatom sind, oder mindestens zwei aus R
3,
R
3' und R
4 zusammen eine C
2-C
10 Ringstruktur bilden, und R
8,
R
9 und R
10 die gleichen
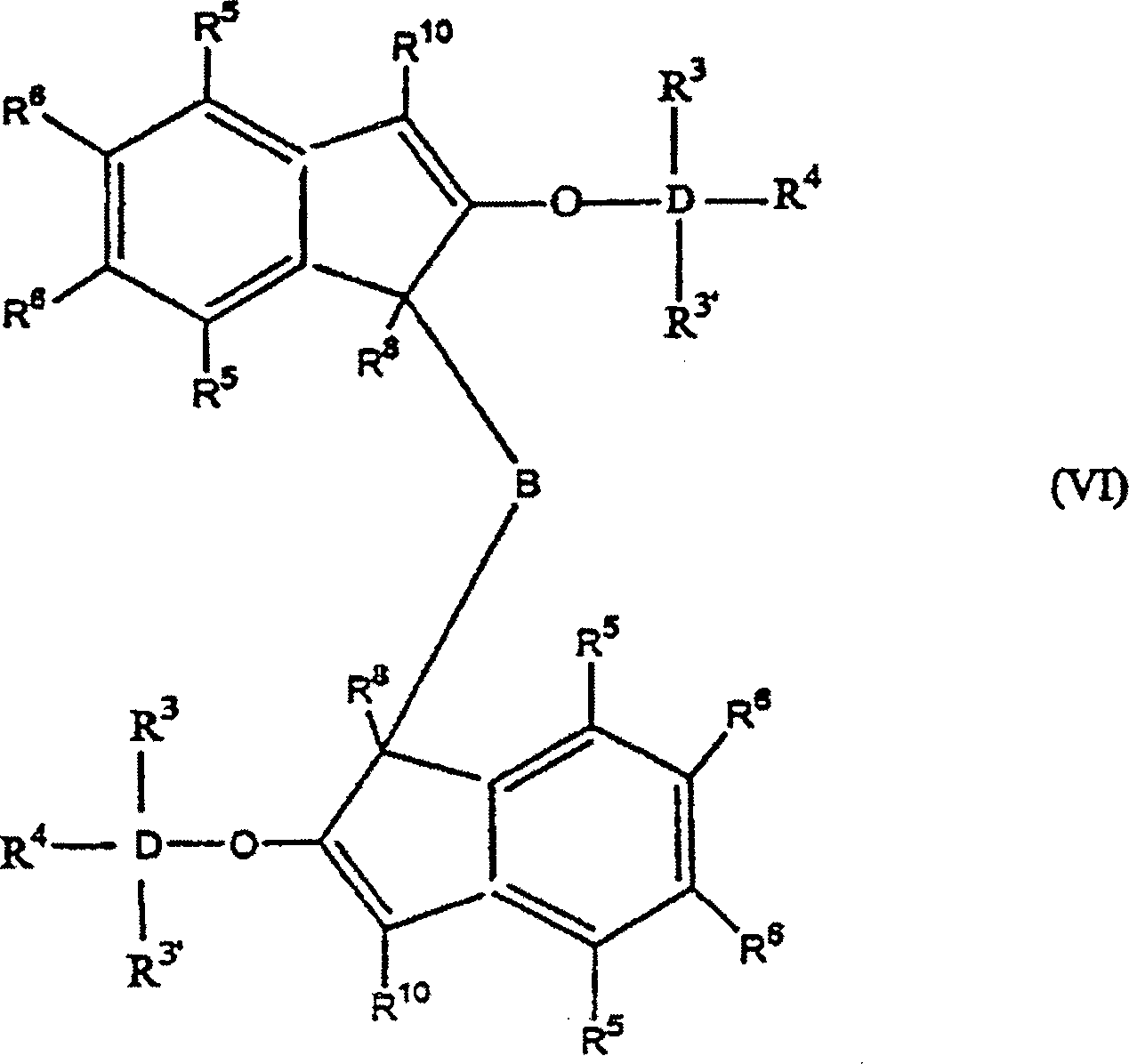
wie vorstehend sind, oder alternativ die Formel:
worin:
R
3, R
3', R
4-R
6, R
8, R
10 und
D die gleichen wie vorstehend sind, und B eine Brücke von
mindestens einer der Gruppen -(CH
2)
n-, -Si(R
3)
2-, oder -Ge(R
3)
2- ist, wobei n gleich 1–8 ist und R
3 gleich
wie vorstehend ist. D ist bevorzugt Si.
-
Die
erfindungsgemäßen Katalysatorverbindungen
können
aus 2-Indanon hergestellt werden. Diese Verbindung kann in einem
geeigneten Lösungsmittel
mit einer Base und einem Chlorsilan umgesetzt werden, wobei 2-Siloxyinden
in einer Ausbeute von über
80% gebildet wird. Geeignete Lösungsmittel
sind beispielsweise Dimethylformamid (DMF) und Tetrahydrofuran (THF).
Geeignete Basen sind beispielsweise Imidazol, Triethylamin (TEA)
und 1,8-Diazabicyclo[5.4.0]undec-7-en. Geeignete Chlorsilane sind
beispielsweise tert.-Butyldimethylchlorsilan, t-Hexyldimethylchlorsilan und Cyclohexyldimethylchlorsilan.
Die Reaktion verläuft
nach dem folgenden Reaktionsschema (VII):
-
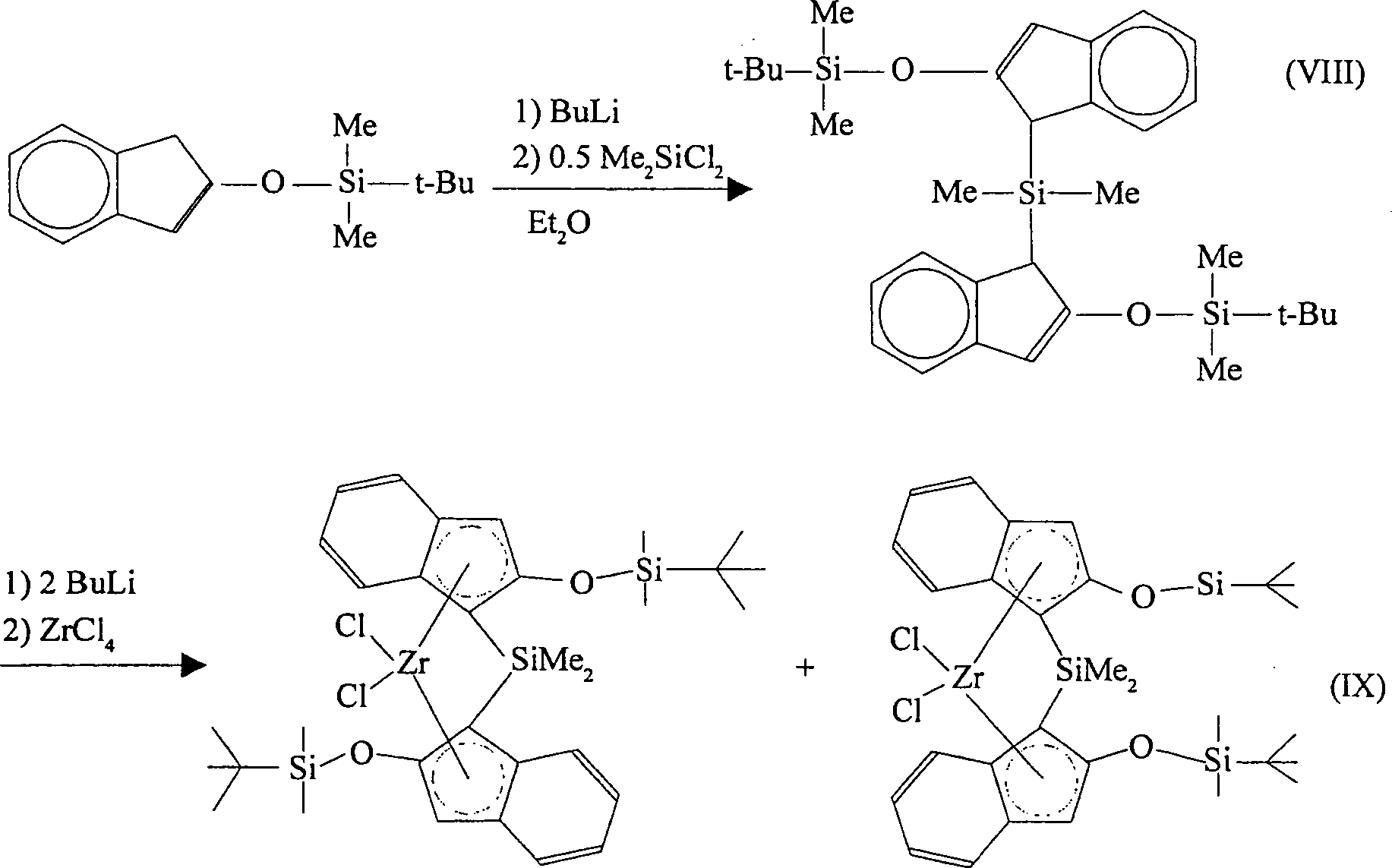
-
Gemäß einer
Ausführungsform
der Erfindung wird 2-(tert-Butyldimethylsiloxy)inden zunächst mit
Butyllithium reagiert und danach mit Dimethyldichlorsilan (Me2SiCl2), wobei Dimethylsilylbis(2-(tert-butyldimethylsiloxy)indenyl)silan
gebildet wird.
-
Butyllithium
kann durch Methyllithium, Natriumhydrid oder Kaliumhydrid ersetzt
werden. Gleichermaßen
kann Dimethyldichlorsilan durch ein beliebiges Dialkyl- oder Diarylsilan
ersetzt werden. Silizium kann durch Germanium ersetzt werden.
-
Dimethylsilylbis(2-(tert-butyldimethylsiloxy)indenyl)silan
kann mit Butyllithium reagiert werden, was das entsprechende Bislithiumsalz
ergibt. Dieses Produkt kann mit Zirkoniumtetrachlorid reagiert werden,
wobei [Dimethylsilylbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid
als ein Gemisch der racemischen und meso Diastereomere erhalten
wird. Butyllithium kann ersetzt werden wie vorstehend beschrieben.
Zirkoniumtetrachlorid kann durch Titantetrachlorid oder Hafniumtetrachlorid
ersetzt werden, wobei die entsprechenden Titan-. und Hafniumkomplexe
erhalten werden. Die Reaktionen verlaufen gemäß den nachfolgenden Reaktionsschemata
(VIII) und (IX):
-
-
Gemäß einer
anderen Ausführungsform
der Erfindung wird 2-(tert-Butyldimethylsiloxy)inden
zunächst mit
Butyllithium und danach mit Dibromethan reagiert, wobei Bis(2-(tert-butyldimethylsiloxy)indenyl)ethan
gebildet wird. Diese Verbindung kann mit zwei Äquivalenten Butyllithium reagiert
werden, was das entsprechende Bislithiumsalz ergibt. Dieses kann
danach mit Zirkoniumtetrachlorid reagiert werden, wobei [Ethylenbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid
erhalten wird. Das racemische Diastereomer des letzteren wird in
großem Überschuss
gebildet und wird vom meso-Isomer
durch fraktionierte Kristallisierung leicht abgetrennt. Katalytische
Hydrierung von racemischem [Ethylenbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid
ergibt den entsprechenden Tetrahydroindenylkomplex. Diese Reaktionen
verlaufen gemäß dem nachfolgenden
Reaktionsschema (X):
-
-
In
den vorstehenden Reaktionen kann Butyllithium ersetzt werden wie
vorstehend beschrieben. Zirkoniumtetrachlorid kann durch Titantetrachlorid
oder Hafniumtetrachlorid ersetzt werden, wobei die entsprechenden
Titan-. und Hafniumkomplexe erhalten werden.
-
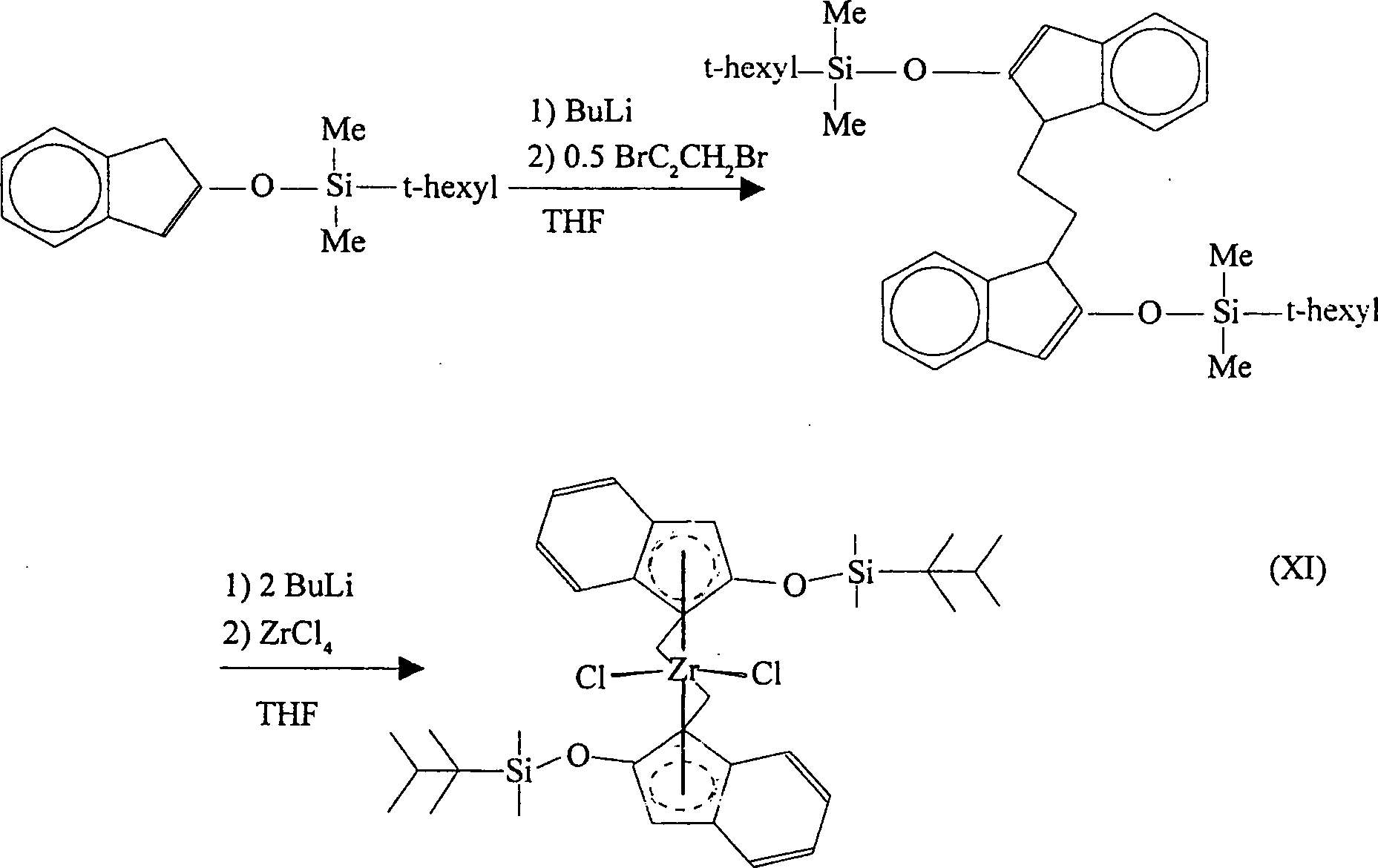
Gemäß einer
nochmals weiteren Ausführungsform
der Erfindung wird 2-(t-Hexyldimethylsiloxy)inden zunächst mit
Butyllithium und danach mit Dibromethan reagiert, wobei Bis(2-(t-hexyldimethylsiloxy)indenyl)ethan
gebildet wird. Diese Verbindung kann mit zwei Äquivalenten Butyllithium reagiert
werden, was das entsprechende Bislithiumsalz ergibt. Dieses kann
danach mit Zirkoniumtetrachlorid reagiert werden, wobei [Ethylenbis(2-(t-hexyldimethylsiloxy)indenyl)]zirkoniumdichlorid
erhalten wird. Das racemische Diastereomer des letzteren wird in
großem Überschuss
gebildet und wird vom meso-Isomer
durch fraktionierte Kristallisierung leicht abgetrennt. Die Reaktionen
verlaufen gemäß dem nachfolgenden
Reaktionsschema (XI):
-
-
In
den vorstehenden Reaktionen kann Butyllithium ersetzt werden wie
vorstehend beschrieben. Zirkoniumtetrachlorid kann durch Titantetrachlorid
oder Hafniumtetrachlorid ersetzt werden, wobei die entsprechenden
Titan-. und Hafniumkomplexe erhalten werden. Hydrierung von [Ethylenbis(2-(t-hexyldimethylsiloxy)indenyl)]zirkoniumdichlorid
ergibt den entsprechenden Tetrahydroindenylkomplex.
-
Veranschaulichende,
aber nicht einschränkende
Beispiele der erfindungsgemäßen Verbindungen
sind unter anderem 2-(tert-Butyldimethylsiloxy)inden, 2-(t-Hexyldimethylsiloxy)inden,
2-(Cyclohexyldimethylsiloxy)inden, 2-(tert-Butyldiphenylsiloxy)inden, Dimethylsilylbis(2-(tert-butyldimethylsiloxy)inden),
Diphenylsilylbis(2-(tert-butyldimethylsiloxy)inden), Dimethylsilylbis(2-(t- hexyldimethylsiloxy)inden),
Diphenylsilylbis(2-(t-hexyldimethylsiloxy)inden), Dimethylsilylbis(2-(cyclohexyldimethylsiloxy)inden),
Diphenylsilylbis(2-(cyclohexyldimethylsiloxy)inden),
Dimethylsilylbis(2-(tert-butyldiphenylsiloxy)inden), Diphenylsilylbis(2-(tert-butyldiphenylsiloxy)inden),
racemisches und meso [Dimethylsilylbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid, racemisches
und meso [Diphenylsilylbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid,
racemisches und meso [Dimethylsilylbis(2-(t-hexyldimethylsiloxy)indenyl)]zirkoniumdichlorid,
racemisches und meso [Diphenylsilylbis(2-(t-hexyldimethylsiloxy)indenyl)]zirkoniumdichlorid,
racemisches und meso [Dimethylsilylbis(2-(cyclohexyldimethylsiloxy)indenyl)]zirkoniumdichlorid,
racemisches und meso [Dimethylsilylbis(2-(cyclohexyldimethylsiloxy)indenyl)]zirkoniumdichlorid,
racemisches und meso [Dimethylsilylbis(2-2-(tert-butyldiphenylsiloxy)indenyl)]zirkoniumdichlorid,
racemisches und meso [Diphenylsilylbis(2-(tert-butyldiphenylsiloxy)indenyl)]zirkoniumdichlorid,
racemisches und meso [Dimethylsilylbis(2-(tert-butyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid,
racemisches und meso [Diphenylsilylbis(2-(tert-butyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid,
racemisches und meso [Dimethylsilylbis(2-(t-hexyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid,
racemisches und meso [Diphenylsilylbis(2-(t-hexyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid,
racemisches und meso [Dimethylsilylbis(2-(cyclohexyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid,
racemisches und meso [Diphenylsilylbis(2-(cyclohexyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid,
racemisches und meso [Dimethylsilylbis(2-(tert-butyldiphenylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid,
racemisches und meso [Diphenylsilylbis(2-(tert-butyldiphenylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid,
Bis(2-(tert-butyldimethylsiloxy)indenyl)ethan,
Bis(2-(t-hexyldimethylsiloxy)indenyl)ethan, Bis(2-(tert-butyldiphenylsiloxy)indenyl)ethan, Bis(2-(cyclohexyldimethylsiloxy)indenyl)ethan,
[rac-Ethylenbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid,
racemisches und meso [Ethylenbis(2-(t-hexyldimethylsiloxy)indenyl)]zirkoniumdichlorid,
racemisches und meso [Ethylenbis(2-(cyclohexyldimethylsiloxy)indenyl)]zirkoniumdichlorid,
racemisches und meso [Ethylenbis(2-(tert-butyldiphenylsiloxy)indenyl)]zirkoniumdichlorid,
[rac-Ethylenbis(2- (tert-butyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid,
racemisches und meso [Ethylenbis(2-(cyclohexyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid,
racemisches und meso [Ethylenbis(2-(tert-butyldiphenylsiloxy )-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid,
Bis(2-(t-hexyldimethylsiloxy)indenyl)ethan
und [rac-Ethylenbis(2-(t-hexyldimethylsiloxy)indenyl)]zirkoniumdichlorid.
Titan oder Hafnium können
anstelle von Zirkonium in entsprechenden Komplexen verwendet werden.
-
Die
erfindungsgemäßen Metallocenverbindungen
können
als Katalysatorkomponenten für
die Polymerisation oder Copolymerisation von Olefinen verwendet
werden. Die Olefine können
ausgewählt
werden aus Ethylen, Propylen, Buten, Penten, Hexen, Hepten und Octen
oder Gemischen davon. Insbesondere können die Olefine aus Ethylen
und Propylen und Gemischen davon oder zusammen mit anderen α-Olefinen
ausgewählt
werden.
-
Die
Katalysatoren können
entweder trägergestützt oder
ungestützt
sein. Die trägergestützten Katalysatoren
werden hauptsächlich
für Suspensions-
und Gasphaseverfahren verwendet. Der Träger kann jeder Träger sein,
der für
Metallocene und andere Typen von Katalysatoren bekannt ist. Bevorzugte
Träger
sind Siliziumoxid und Aluminiumoxid.
-
Die
erfindungsgemäßen Verbindungen
können
in Kombination mit Aktivatorverbindungen, was organometallische
Verbindungen sind, verwendet werden oder allen anderen Verbindungen,
die in Kombination mit Metallocenverbindungen verwendet werden.
Geeignete Cokatalysatoren sind beispielsweise Alkylaluminiumverbindungen,
Aluminoxane, Methylaluminoxan oder modifiziertes Methylaluminoxan.
Der bevorzugte Cokatalysator ist Methylalumoxan (MAO). Andere Cokatalysatoren,
die verwendet werden können,
umfassen Lewis-Säuren oder
prolische Säuren,
wie etwa (B(C6F5)3 oder [PhNMe2H]1B(C6F5)4, welche kationische Metallocene mit kompatiblen
nicht-koordinierenden Anionen erzeugen, in der Gegenwart oder Abwesenheit
von Alkylaluminiumverbindungen. Besonders geeignete Aktivatoren
sind beispielsweise Alumoxanverbindungen mit der Formel R-(Al(R)-O-)m-AlR2, wobei n gleich
1–40 ist,
m gleich 3–40
ist und R eine C1-C8 Alkylgruppe
ist. Bevorzugt ist R eine Methylgruppe, und der bevorzugte Aktivator
ist Methylalumoxan (MAO). Der Aktivator kann gemäß den in der Technik bekannten
Methoden angewandt werden.
-
Experimenteller
Teil
-
Alle
Arbeitsschritte wurden in Argon- oder Stickstoffatmosphäre unter
Verwendung von Schlenk-, Vakuum- oder Handschuhkasten-Standardtechniken
durchgeführt.
Lösungsmittel
wurden vor Verwendung getrocknet und unter Argon destilliert. Die 1H und 13C NMR-Spektren
wurden in CDCl3 oder CD2Cl2 Lösung
aufgezeichnet, unter Verwendung der NMR-Spektrometer JEOL JNM-LA400
oder JEOL JNM-A500, und gegen Tetramethylsilan (TMS) oder die restlichen
Protonen der deuterierten Lösungsmittel
abgeglichen. Direkte Elektronenionisation-Massenspektren (EIMS)
wurden an einem Varian VG-7070E oder einem Varian-8000 Massenspektrometer
erhalten.
-
KATALYSATORHERSTELLUNG
-
Beispiel 1
-
2-(tert-Butyldimethylsiloxy)inden
-
Eine
Lösung
von tert-Butyldimethylchlorsilan (248,69 g, 1,65 Mol) und Imidazol
(112,33 g, 1,65 Mol) in DMF (900 ml) wurde mit 2-Indanon (198,24
g, 1,50 Mol) reagiert und danach über Nacht bei Raumtemperatur gerührt. Das
Reaktionsgemisch wurde mit Wasser (800 ml) behandelt und mit Diethylether
(3 × 400
ml) extrahiert. Die kombinierten organischen Phasen wurden mit Wasser
(2 × 400
ml) gewaschen und über
Natriumsulfat getrocknet. Die Lösungsmittel
wurden unter verringertem Druck entfernt, wobei ein orangefarbenes Öl zurück blieb.
Destillation unter verringertem Druck ergab 331,2 g (89,6%) der
Titelverbindung als ein gelbes Öl (Sdp.
105–107°C/0,1 mbar). 1H NMR (CDCl3, δ): 7,19-7,07
(m, 3H); 6,97 (td, 3J = 7,3 Hz, 4J = 1,4 Hz, 1H); 5,72 (dd, 4J
= 1,9 Hz, 1,1 Hz, 1H); 3,24 (dd, 4J = 1,7
Hz, 1,1 Hz, 2H); 0,96 (s, 9H); 0,23 (s, 6H). 13C
NMR (CDCl3, δ): 162,44; 145,14; 136,53; 126,44;
123,01; 122,39; 118,92; 106,58; 39,46; 25,59; 18,14; –4,68.
-
Beispiel 2
-
2-(t-Hexyldimethylsiloxy)inden
-
Eine
Lösung
von t-Hexyldimethylchlorsilan (100,0 g, 559,3 mMol) und Imidazol
(38,08 g, 559,3 mMol) in DMF (350 ml) wurde mit 2-Indanon (67,40
g, 510,0 mMol) reagiert und danach zwei Tage bei Raumtemperatur
gerührt.
Das Reaktionsgemisch wurde mit Wasser (300 ml) behandelt und mit
Et2O (3 × 200 ml) extrahiert. Die kombinierten
organischen Phasen wurden mit Wasser (2 × 200 ml) gewaschen und über Natriumsulfat
getrocknet. Abdampfen der Lösungsmittel
ergab ein rotes Öl.
Destillation unter verringertem Druck ergab 116,89 g (83,5%) der
Titelverbindung als ein gelbes Öl
(Sdp. 128–130°C/0,4 mbar). 1H NMR (CDCl3, δ): 7,24–7,10 (m, 3H);
7,00 (td, 3J = 7,3 Hz, 4J
= 1,3 Hz, 1H); 5,74 (d, 4J = 0,6 Hz, 1H);
3,28 (s, 2H); 1,70 (sept, 3J = 6,8 Hz, 1H); 0,93
(s, 6H); 0,92 (d, 3J = 6,8 Hz, 6H); 0,29
(s, 6H). 13C NMR (CDCl3, δ): 162,82;
145,71; 137,03; 126,86; 123,45; 122,73; 119,29; 106,92; 40,00; 34,49;
25,51; 20,51; 18,89; –2,26.
-
Beispiel 3
-
2-(Cyclohexyldimethylsiloxy)inden
-
Eine
Lösung
von Cyclohexyldimethylchlorsilan (84,62 g, 478,7 mMol) und Imidazol
(32,59 g, 478,7 mMol) in DMF (300 ml) wurde mit 2-Indanon (57,62
g, 436,0 mMol) reagiert. Das Reaktionsgemisch wurde über Nacht
bei Raumtemperatur gerührt,
mit Wasser (300 ml) behandelt und mit Et2O
(3 × 200
ml) extrahiert. Die kombinierten organischen Phasen wurden mit Wasser
(3 × 300
ml) gewaschen und über
Natriumsulfat getrocknet. Die Lösungsmittel
wurden entfernt, wobei ein orangefarbenes Öl zurück blieb. Destillation unter
verringertem Druck ergab 81,67 g (68,8%) der Titelverbindung als
ein gelbes Öl
(Sdp. 140–142°C/0,2 mbar). 1H NMR (CDCl3, δ): 7,25–7,11 (m,
3H); 7,01 (td, 3J = 7,3 Hz, 4J
= 1,4 Hz, 1H); 5,76 (d, 4J = 0,7 Hz, 1H);
3,29 (s, 2H); 1,77–1,74
(m, 5H); 1,27–1,13
(m, 5H); 0,90 (s, 1H); 0,24 (s, 6H). 13C
NMR (CDCl3, δ): 162,44; 145,24; 136,60; 126,47;
123,07; 122,37; 118,91; 106,32; 39,52; 27,69; 26,77; 26,46; 26,33; –3,55.
-
Beispiel 4
-
2-(tert-Butyldiphenylsiloxy)inden
-
Zu
einer Lösung
von tert-Butyldiphenylchlorsilan (42,43 g, 154,4 mMol) und DBU (25,64
g, 168,4 mMol) in Benzol (200 ml) wurde 2-Indanon (18,38 g, 139,1
mMol) auf ein Mal zugegeben. Das Reaktionsgemisch wurde über Nacht
gerührt,
mit Et2O (200 ml) verdünnt, mit 10% HCl (2 × 200 ml)
und Wasser (2 × 200 ml)
gewaschen und über
Natriumsulfat getrocknet. Die Lösungsmittel
wurden abgedampft, wobei ein dunkelbraunes Öl zurück blieb. Destillation unter
verringertem Druck ergab 38,22 g (74,1%) der Titelverbindung als ein
gelbes Öl
(Sdp. 172–175°C/0,05 mbar). 1H NMR (CDCl3, δ): 7,77–7,74 (m,
4H); 7,46–7,36
(m, 6H); 7,18–7,16
(m. 1H); 7,11–7,07
(m. 1H); 6,98–6,94
(m. 2H); 5,48 (d, 4J = 0,7 Hz, 1H); 3,29
(s, 2H); 1,09 (s, 9H). 13C NMR (CDCl3, δ):
162,13; 145,17; 136,75; 135,53; 132,43; 130,20; 127,99; 126,51;
123,15; 122,49; 119,24; 107,91; 39,43; 26,60; 19,46.
-
Beispiel 5
-
2-(tert-Butyldimethylsiloxy)bisbenz[e,g]inden
-
Zu
einer Suspension von Bisbenz[e,g]indanon (35,90 g, 154,5 mMol) und
tert-Butyldimethylchlorsilan (28,00
g, 185,5 mMol) in Benzol (300 ml) wurde DBU (30,60 g, 201,0 mMol)
zugetropft, während
das Reaktionsgemisch mit einem Eisbad kalt gehalten wurde. Das Rühren wurde
1 Stunde bei Raumtemperatur fortgesetzt. Die organische Phase wurde
mit Wasser (200 ml), 5% HCl (2 × 200
ml), Wasser (200 ml) gewaschen und über Natriumsulfat getrocknet.
Nach Verdampfen der Lösungsmittel
blieb ein Rückstand
zurück,
der mit MeOH (3 × 200
ml) gewaschen wurde, wobei die Titelverbindung (38,35 g, 110,7 mMol,
71,6%) als ein leicht rosafarbenes Pulver erhalten wurde. EIMS (berechnet/gefunden):
346,1753/346,1744. 1H NMR (CDCl3, δ): 8,72–8,67 (m,
1H); 8,66–8,62
(m, 1H); 8,02–7,96
(m, 1H); 7,82–7,99
(m, 1H); 7,62–7,57
(m, 2H); 7,56–7,50
(m, 1H); 7,49–7,44
(m, 1H); 6,32 (m, 1H); 3,73 (m, 2H); 1,03 (s, 9H); 0,33 (s, 6H). 13C NMR (CDCl3, δ): 163,16; 139,79;
130,11; 129,41; 128,34; 127,73; 127,21; 126,67; 126,07; 125,53;
124,42; 123,91; 123,38; 123,24; 122,96; 104,55; 39,81; 25,68; 18,29; –4,50.
-
Beispiel 6
-
2-(tert-Butyldimethylsiloxy)-4,7-dimethylinden
-
Zu
einer Lösung
von tert-Butyldimethylchlorsilan (2,85 g, 18,9 mMol) und 4,7-Dimethyl-2-indanon (2,53
g, 15,8 mMol, erhalten durch Oxidation von 4,7-Dimethylinden) in Benzol (30 ml) wurde
DBU (3,13 g, 20,5 mMol) zugegeben und das Reaktionsgemisch wurde
2 Stunden bei Raumtemperatur gerührt.
Das Gemisch wurde mit Et2O (50 ml) verdünnt, mit
Wasser (2 × 50
ml), 5% HCl (50 ml), Wasser (2 × 50
ml) gewaschen und über
Natriumsulfat getrocknet. Nach Verdampfen der Lösungsmittel blieb ein dunkles Öl zurück, das
in Pentan (30 ml) aufgelöst
wurde. Die nicht-abreagierten Ausgangsmaterialien wurden bei –15°C kristallisiert und
durch Filtration entfernt. Verdampfen des Lösungsmittels ergab 3,03 g (69,9%)
der ziemlich reinen Titelverbindung als ein orangefarbenes Öl. 1H NMR (CDCl3, δ): 6,94 (dq, 3J = 7,7 Hz, 4J =
0,3 Hz, 1H); 6,78 (dq, 3J = 7,7 Hz, 4J = 0,3 Hz, 1H); 5,84 (t, 4J
= 1,1 Hz, 1H); 3,20 (m, 2H); 2,30 (s, 3H); 2,25 (s, 3H); 1,00 (s,
9H); 0,27 (s, 6H). 13C NMR (CDCl3, δ):
162,08; 143,39; 134,89; 129,45; 127,78; 125,49; 123,84; 105,28;
38,80; 25,65; 18,29; 18,21; –4,57.
-
Beispiel 7
-
Bis(2-(tert-butyldimethylsiloxy)indenyl)ethan
-
Zu
einer Lösung
von 2-(tert-Butyldimethylsiloxy)inden (36,96 g, 150,0 mMol) in THF
(150 ml) bei 0°C wurde
n-BuLi (60,0 ml einer 2,5 M Lösung
in Hexanen, 150,0 mMol) zugetropft und das Reaktionsgemisch wurde über Nacht
bei Raumtemperatur gerührt.
Die resultierende Lösung
wurde danach auf –80°C gekühlt und tropfenweise
mit einer Lösung
von Dibromethan (14,09 g, 75,0 mMol) in THF (50 ml) behandelt. Nach
Beendigung der Zugabe wurde das Reaktionsgemisch über Nacht
bei Raumtemperatur gerührt
und mit gesättigter Ammoniumchloridlösung (300
ml) gewaschen. Lösungsmittel
aus der organischen Phase wurden abgedampft und das Produkt wurde
in Et2O (300 ml) aufgelöst, mit Wasser (2 × 200 ml)
gewaschen und über
Natriumsulfat getrocknet. Wiederholte Kristallisationen bei –15°C ergaben
22,54 g (57,9%) der Titelverbindung als einen schmutzig weißen Feststoff.
Die erste Kristallfraktion bestand aus diastereomer reinem Material
(Smp. 108–110°C). EIMS
(berechnet/gefunden): 518,3036/518,3028. 1H
NMR (CDCl3, δ, Hauptdiastereomer): 7,18–7,07 (m,
6H); 6,97 (td, 3J = 7,4 Hz, 4J
= 1,3 Hz, 2H); 5,66 (s, 2H); 3,17 (m, 2H); 1,89–1,84 (m, AA', 2H); 1,59–1,54 (m,
BB', 2H); 0,94 (s,
18H); 0,23 (s, 6H); 0,21 (s, 6H). 13C NMR
(CDCl3, δ,
Hauptdiastereomer): 164,96; 144,39; 140,62; 126,50; 122,58; 122,41;
118,74; 104,97; 49,18; 25,67; 24,34; 18,12; –4,68; –4,88. 1H NMR
(CDCl3, δ,
Nebendiastereomer): 7,17–7,05
(m, 6H); 6,97 (td, 3J = 7,4 Hz, 4J = 1,2 Hz, 2H); 5,63 (s, 2H); 3,21 (m,
2H); 1,76–1,75
(m, 4H); 0,94 (s, 18H); 0,22 (s, 6H); 0,20 (s, 6H). 13C
NMR (CDCl3, δ, Nebendiastereomer): 165,18;
144,35; 140,68; 126,53; 122,71; 122,35; 118,74; 104,87; 49,04; 25,67;
25,30; 18,14; –4,77.
-
Beispiel 8
-
Bis(2-(t-hexyldimethylsiloxy)indenyl)ethan
-
Zu
einer Lösung
von 2-(t-Hexyldimethylsiloxy)inden (68,62 g, 250,0 mMol) in THF
(250 ml) bei 0°C wurde
n-BuLi (100,0 ml einer 2,5 M Lösung
in Hexanen, 250,0 mMol) zugetropft und das Reaktionsgemisch wurde über Nacht
bei Raumtemperatur gerührt.
Die resultierende Lösung
wurde danach auf –80°C gekühlt und tropfenweise
mit einer Lösung
von Dibromethan (23,48 g, 125,0 mMol) in THF (100 ml) behandelt.
Nach Beendigung der Zugabe wurde das Reaktionsgemisch über Nacht
bei Raumtemperatur gerührt
und mit gesättigter
Ammoniumchloridlösung
(350 ml) gewaschen. Lösungsmittel
aus der organischen Phase wurden abgedampft und das Produkt wurde
in Et2O (350 ml) aufgelöst, mit Wasser (2 × 300 ml)
gewaschen und über
Natriumsulfat getrocknet. Wiederholte Kristallisationen bei –15°C ergaben
37,48 g (52,2%) der Titelverbindung als schmutzig weiße Kristalle.
Die erste Kristallfraktion bestand aus diastereomer reinem Material,
das zur spektralen Charakterisierung verwendet wurde. EIMS (berechnet/gefunden):
m/e 574,3662/574,3659. 1H NMR (CDCl3, δ):
7,22–7,07
(m, 6H); 6,97 (td, 3J = 7,4 Hz, 4J = 1,2 Hz, 2H); 5,65 (s, 2H); 3,15 (m,
2H); 1,91–1,84
(m, AA', 2H); 1,67
(sept, 3J = 6,8 Hz, 2H); 1,57–1,50 (m,
BB', 2H); 0,89 (m,
24H); 0,27 (s, 6H); 0,24 (s, 6H). 13C NMR
(CDCl3, δ):
164,75; 144,45; 140,58; 126,46; 122,60; 122,23; 118,68; 104,96;
49,24; 33,93; 25,03; 24,32; 20,14; 20,02; 18,51; 18,47; –2,66; –2,95.
-
Beispiel 9
-
Bis(2-(cyclohexyldimethylsiloxy)indenyl)ethan
-
Zu
einer eisgekühlten
Lösung
von 2-(Cyclohexyldimethylsiloxyinden (13,62 g, 50,0 mMol) in THF
(50 ml) wurde n-BuLi (20,0 ml einer 2,5 M Lösung in Hexanen, 50,0 mMol)
zugetropft und das Reaktionsgemisch wurde über Nacht bei Raumtemperatur
gerührt.
Die resultierende Lösung
wurde danach auf –80°C gekühlt und tropfenweise
mit einer Lösung
von Dibromethan (4,70 g, 25,0 mMol) in THF (30 ml) behandelt. Das
Reaktionsgemisch wurde allmählich
auf Raumtemperatur erwärmt, über Nacht
gerührt
und mit gesättigter
Ammoniumchloridlösung
(150 ml) gewaschen. Die organische Phase wurde über Natriumsulfat getrocknet.
Lösungsmittel wurden
abgedampft und das zurückbleibende Öl wurde
in Et2O (100 ml) aufgelöst. Wiederholte Kristallisationen
ergaben eine Gesamtausbeute von 4,03 g (28,2%) der Titelverbindung
als schmutzig weiße
Kristalle. Die erste Kristallfraktion wurde für die spektrale Charakterisierung
verwendet. EIMS (berechnet/gefunden): m/e 570,3349/570,3342. 1H NMR (CDCl3, δ): 7,19–7,07 (m,
6H); 6,99 (td, 3J = 7,3 Hz, 4J
= 1,3 Hz, 2H); 5,65 (s, 2H); 3,17 (m, 2H); 1,84–1,79 (m, AA', 2H); 1,73 (m, 10H);
1,59–1,54
(m, BB', 2H); 1,20
(m, 10H); 0,84 (m, 2H); 0,22 (s, 6H); 0,22 (s, 6H). 13C
NMR (CDCl3, δ): 164,98; 144,47; 140,65; 126,51;
122,52; 122,33; 118,72; 104,64; 49,09; 27,72; 26,80; 26,48; 26,37;
24,29; –3,60.
-
Beispiel 10
-
Bis(2-(tert-butyldiphenylsiloxy)indenyl)ethan
-
Zu
einer Lösung
von 2-(tert-Butyldiphenylsiloxy)inden (32,6 g, 88,0 mMol) in THF
(200 ml) bei 0°C
wurde n-BuLi (35,5 ml einer 2,5 M Lösung in Hexan, 88,8 mMol) zugetropft
und das Reaktionsgemisch wurde über Nacht
bei Raumtemperatur gerührt.
Die resultierende Lösung
wurde danach auf –80°C gekühlt und
tropfenweise mit einer Lösung
von Dibromethan (8,26 g, 44,0 mMol) in THF (50 ml) behandelt. Das
Reaktionsgemisch wurde allmählich
auf Raumtemperatur erwärmt, über Nacht
gerührt
und mit gesättigter
Ammoniumchloridlösung
(2 × 300
ml) gewaschen. Die organische Phase wurde über Natriumsulfat getrocknet.
Abdampfen der Lösungsmittel
ergab ein braunes Öl,
das in siedendem Et2O (300 ml) aufgelöst wurde.
Abkühlen
auf –15°C ergab 13,7
g (40,7%) der Titelverbindung als ein diastereomer reines, weißes mikrokristallines
Pulver. EIMS (berechnet/gefunden): m/e 766,3662/766,3641. 1H NMR (CDCl3, δ): 7,78–7,75 (m,
4H); 7,71–7,68
(m, 4H); 7,48–7,33
(m, 12H); 7,22–7,20
(m, 2H); 7,09–7,05
(m, 2H); 6,93–6,88
(m, 4H); 5,28 (s, 2H); 3,36 (m, 2H); 2,21–2,15 (m, AA', 2H); 1,85–1,79 (m,
BB', 2H); 1,06 (s,
18H). 13C NMR (CDCl3, δ): 164,06;
144,19; 140,54; 135,45; 132,28; 131,91; 130,06; 129,99; 127,84;
127,79; 126,45; 122,65; 122,54; 119,06; 106,81; 49,25; 26,61; 25,10;
19,38. Die Mutterlauge wurde zur Trockene eingedampft und in siedendem
Pentan (150 ml) aufgelöst.
Aufkonzentrieren und Abkühlen
auf –15°C ergab die
zweite Ernte von 1,62 g (4,8%) der Titelverbindung als ein braunes
Pulver, das mit dem Nebendiastereomer angereichert war (Gesamtausbeute
45,5%).
-
Beispiel 11
-
Bis(2-(tert-butyldimethylsiloxy)bisbenz[e,g]indenyl)ethan
-
Zu
einer eisgekühlten
Lösung
von 2-(tert-Butyldimethylsiloxybisbenzyl[e,g]inden (20,00 g, 63,48 mMol)
in THF (80 ml) wurde n-BuLi (25,4 ml einer 2,5 M Lösung in
Hexanen, 63,50 mMol) zugetropft und das Reaktionsgemisch wurde 1
Stunde bei Raumtemperatur gerührt.
Die Lösungsmittel
wurden unter Vakuum entfernt und Toluol (200 ml) wurde zugegeben.
Die resultierende Lösung
wurde auf –80°C gekühlt und
Dibromethan (5,95 g, 31,67 mMol) in Toluol (20 ml) wurde zugetropft.
Das Reaktionsgemisch wurde allmählich
auf Raumtemperatur erwärmt, über Nacht
gerührt
und mit gesättigter
Ammoniumchloridlösung
(2 × 100
ml) gewaschen. Die organische Phase wurde über Natriumsulfat getrocknet.
Aufkonzentrieren und Abkühlen
auf –15°C ergab die
Titelverbindung (900 mg, 1,25 mMol, 3,9%) als einen schmutzig weißen Feststoff.
EIMS (berechnet/gefunden): m/e 718,3662/718,3659. 1H
NMR (CDCl3, δ): 8,73–8,69 (m, 2H); 8,66 (dt, 3J = 8,2 Hz, 4J =
0,6 Hz, 2H); 8,02–7,98
(m, 2H); 7,63–7,58
(m, 6H); 7,42 (ddd, 3J = 8,2 Hz, 3J = 6,9 Hz, 4J =
1,3 Hz, 2H); 7,34 (ddd, 3J = 8,2 Hz, 3J = 6,9 Hz, 4J =
1,2 Hz, 2H); 6,17 (d, 4J = 0,4 Hz, 2H);
3,57–3,54
(m, 2H); 2,09–2,06
(m, 2H); 1,76–1,73
(m, 2H); 0,72 (s, 18H); 0,11 (s, 6H); 0,03 (s, 6H). 13C
NMR (CDCl3, δ): 166,68; 139,37; 131,67; 130,32;
129,25; 127,93; 127,04; 126,70; 125,91; 125,46; 124,54; 123,61;
123,43; 123,20; 122,97; 102,67; 49,93; 25,48; 23,68; 18,04; –4,82; –5,03.
-
Beispiel 12
-
Bis(1-(trimethylsilyl)-2-(tert-butyldimethylsiloxy)-3-indenyl)ethan
-
Zu
einer Lösung
von Bis(2-tert-butyldimethylsiloxy)indenyl)ethan (10,38 g, 20,0
mMol) in THF (100 ml) bei –20°C wurde n-Buli
(16,1 ml einer 2,5 M Lösung
in Hexan, 40,2 mMol) zugetropft und das Reaktionsgemisch wurde 3
Stunden bei Raumtemperatur gerührt.
Zu der resultierenden Lösung
wurde Chlortrimethylsilan (6,85 g, 63,0 mMol, 8,0 ml) bei 0°C zugetropft.
Nach Beendigung der Zugabe wurde das Reaktionsgemisch über Nacht
bei Raumtemperatur gerührt
und die Lösungsmittel
wurden abgedampft. Der zurückbleibende orangefarbene
Feststoff wurde mit CH2Cl2 extrahiert
und durch Celite filtriert. Das Lösungsmittel wurde abgedampft
und das Produkt wurde in Et2O (150 ml) aufgelöst. Aufkonzentrieren
und Abkühlen
auf 0°C
ergab 10,29 g (77,6%) eines 2 : 1 Diastereomerengemisches der Titelverbindung
als schmutzig weiße
Kristalle. EIMS (berechnet/gefunden): m/e 662,3827/662,3834. 1H NMR (CD2Cl2, δ,
Haupt/Nebendiastereomer): 7,51–7,49
(m, 2H, Haupt); 7,42–7,40
(m, 2H, Neben); 7,29–7,24
(m, 4 + 2H, Haupt/Neben); 7,22–7,17
(m, 2H, Neben); 7,08–7,02
(m, 2 + 2H, Haupt/Neben); 3,29 (s, 1H, Neben); 3,27 (s, 1H, Haupt);
3,00–2,96
(m, AA', 2H, Neben); 2,72–2,65 (m,
AA'BB', 4H, Haupt); 2,44–2,40 (m,
BB', 2H, Neben);
1,02 (s, 18H, Haupt); 1,01 (s, 18H, Neben); 0,20 (s, 6H, Neben);
0,19 (s, 6H, Haupt); 0,10 (s, 6H, Neben); 0,05 (s, 6H, Haupt); 0,02
(s, 18H, Neben); 0,01 (s, 18H, Haupt). 13C
NMR (CD2Cl2, δ, Hauptdiastereomer):
158,32; 144,56; 139,24; 125,09; 122,74; 121,98; 119,89; 118,08;
45,08; 26,14; 24,06; 18,52; –2,39; –3,40; –4,66. 13C NMR (CDCl3, δ, Nebendiastereomer): 158,08;
144,50; 139,14; 124,97; 122,62; 121,95; 119,95; 118,50; 45,13; 26,14;
23,86; 18,47; –2,29; –3,40; –4,24.
-
Beispiel 13
-
Dimethylbis(2-(tert-butyldimethylsiloxy)indenyl)silan
-
Zu
einer Lösung
von 2-(tert-Butyldimethylsiloxy)inden (12,32 g, 50,0 mMol) in Et2O (50 ml) bei 0°C wurde n-BuLi (20,0 ml einer
2,5 M Lösung
in Hexan, 50,0 mMol) zugetropft und das Reaktionsgemisch wurde über Nacht
bei Raumtemperatur gerührt.
Die resultierende Lösung
wurde danach zu einer Lösung
von Dimethyldichlorsilan (3,03 ml, 25,0 mMol) in Et2O
(25 ml) bei 0°C
zugetropft. Das Reaktionsgemisch wurde über Nacht bei Raumtemperatur
gerührt
und mit gesättigter
Ammoniumchloridlösung
(150 ml), Wasser (3 × 100
ml) gewaschen und über
Natriumsulfat getrocknet. Abdampfen der Lösungsmittel ergab ein rotes Öl, das in
einem 1 : 1 Gemisch von MeOH und Aceton aufgelöst wurde. Aufkonzentrieren
und Abkühlen
auf –30°C ergab 5,78 g
(42,1%) der Titelverbindung als leicht gelbe Kristalle. Die erste
Ernte bestand ausschließlich
aus dem racemischen Diastereomer. EIMS (berechnet/gefunden): m/e
548,2962/548,2958. Smp. 92–94°C. 1H NMR (CDCl3, δ): 7,22–7,14 (m,
6H); 6,98 (td, 3J = 7,3 Hz, 4J
= 1,4 Hz, 2H); 5,85 (s, 2H); 3,99 (s, 2H); 0,97 (s, 18H); 0,34 (s, 6H);
0,28 (s, 6H); –0,23
(s, 6H). 13C NMR (CDCl3, δ): 164,96;
144,44; 137,55; 125,13; 122,75; 121,41; 118,83; 104,00; 43,06; 25,77;
18,20; –4,34; –4,88; –6,99. Der ölige Rückstand
wurde ausgiebig mit kaltem MeOH (3 × 50 ml), kaltem Pentan (50
ml) gewaschen und unter Vakuum getrocknet, wobei 3,47 g (24,8%)
des ziemlich reinen meso-Diasteromers als ein rotes Öl erhalten
wurden (Gesamtausbeute 66,9%).
-
Beispiel 14
-
Dimethylbis(2-(t-hexyldimethylsiloxy)indenyl)silan
-
Zu
einer Lösung
von 2-(t-Hexyldimethylsiloxy)inden (35,58 g, 129,6 mMol) in Et2O (120 ml) bei 0°C wurde n-BuLi (52,4 ml einer
2,5 M Lösung
in Hexan, 130,9 mMol) zugetropft und das Reaktionsgemisch wurde über Nacht
bei Raumtemperatur gerührt.
Die resultierende Lösung
wurde danach zu einer Lösung
von Dimethyldichlorsilan (7,9 ml, 64,8 mMol) in Et2O
(50 ml) bei 0°C
zugetropft. Das Reaktionsgemisch wurde über Nacht bei Raumtemperatur
gerührt
und mit gesättigter
Ammoniumchloridlösung
(300 ml), Wasser (2 × 200
ml) gewaschen und über
Natriumsulfat getrocknet. Abdampfen der Lösungsmittel ergab ein rotes Öl, das in
einem 4 : 1 Gemisch von MeOH und Aceton aufgelöst wurde. Aufkonzentrieren
und Abkühlen
auf –30°C ergab 11,67 g
(29,7%) der Titelverbindung als ein weißes Pulver. Die kristalline
erste Ernte bestand ausschließlich
aus dem racemischen Diastereomer. EIMS (berechnet/gefunden): m/e
604,3588/604,3585. 1H NMR (CDCl3, δ): 7,22–7,13 (m,
6H); 6,98 (td, 3J = 7,3 Hz, 4J
= 1,4 Hz, 2H); 5,85 (s, 2H); 3,99 (s, 2H); 1,72 (sept, 3J
= 6,9 Hz, 2H); 0,94 (s, 6H); 0,94 (s, 6H); 0,90 (d, 3J
= 6,9 Hz, 6H); 0,89 (d, 3J = 6,9 Hz, 6H);
0,38 (s, 6H); 0,34 (s, 6H); –0,23
(s, 6H). 13C NMR (CDCl3, δ): 164,87;
144,49; 137,53; 125,13; 122,86; 121,38; 118,84; 104,26; 43,15; 33,76;
25,19; 20,17; 20,08; 18,54; 18,45; –2,30; –2,75; –6,73. Der ölige Rückstand wurde mit kaltem MeOH
(3 × 100
ml) gewaschen und unter Vakuum getrocknet, wobei 14,02 g (35,8%)
des ziemlich reinen meso-Diasteromers als ein rotes Öl erhalten
wurden (Gesamtausbeute 65,5%).
-
Beispiel 15
-
[Bis(2-(tert-butyldimethylsiloxy)bisbenz[e,g]indenyl)]zirkoniumdichlorid
-
Zu
einer Lösung
von 2-tert-Butyldimethylsiloxy)bisbenz[e,g]inden (6,83 g, 19,7 mMol)
in THF (50 ml) wurde bei –20°C n-Buli
(7,9 ml einer 2,5 M Lösung
in Hexanen, 19,7 mMol) zugetropft. Die resultierende rote Lösung wurde
auf Raumtemperatur erwärmen
gelassen und wurde mittels einer Kanüle einer Lösung von ZrCl4 (2,24
g, 9,6 mMol) in THF (50 ml) zugegeben. Das Reaktionsgemisch wurde
20 h unter Rückfluss
erwärmt. Nach
Verdampfen des THF blieb ein Feststoff zurück, der mit CH2Cl2 (50 ml) extrahiert wurde und durch Celite filtriert
wurde, um Lithiumchlorid zu entfernen. Nach Verdampfen des CH2Cl2 blieb ein roter öliger Feststoff
zurück,
der mit Et2O (2 × 50 ml) gewaschen wurde, wobei
2,97 g (34,0%) der Titelverbindung als ein blassgrünes Pulver
zurück
blieben. Im EIMS Massenspektrum der Titelverbindung wurden Stammionen
der Zusammensetzung C46H50Si2O2ZrCl2 + in den entsprechenden Isotopenverhältnissen
bei m/e = 850–859
beobachtet. 1H NMR (CDCl3, δ): 8,45 (m,
4H); 7,66 (m, 4H); 7,50–7,41
(m, 8H); 6,24 (s, 4H); 1,08 (s, 18H); 0,34 (s, 12H); 13C
NMR (CDCl3, δ): 153,11; 129,88; 129,02; 128,21;
128,13; 126,95; 126,82; 124,25; 123,37; 118,32; 97,22; 25,80; 18,48; –4,12.
-
Beispiel 16
-
rac-[Ethylenbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid
-
Zu
einer eisgekühlten
Lösung
von Bis(2-tert-butyldimethylsiloxy)indenyl)ethan (5,37 g, 10,3 mMol)
in THF (50 ml) wurde n-Buli (8,3 ml einer 2,5 M Lösung in
Hexanen, 20,7 mMol) zugetropft und das Reaktionsgemisch wurde über Nacht
bei Raumtemperatur gerührt.
Die resultierende schmutzig gelbe Suspension wurde danach mittels
einer Kanüle
einer Suspension von ZrCl4 (2,41 g, 10,3
mMol) in THF (20 ml) bei –80°C zugegeben.
Das Reaktionsgemisch wurde langsam auf Raumtemperatur erwärmt und über Nacht
gerührt.
Nach Verdampfen der Lösungsmittel
blieb ein gelber Feststoff zurück,
der mit CH2Cl2 (150
ml) extrahiert wurde und durch Celite filtriert wurde, um Lithiumchlorid
zu entfernen. Aufkonzentrieren und Abkühlen auf –30°C ergab 1,47 g (21,0%) der Titelverbindung
als einen gelben mikrokristallinen Feststoff. Im EIMS Massenspektrum
der Titelverbindung wurden Stammionen der Zusammensetzung C32H44Si2O2ZrCl2 + in
den entsprechenden Isotopenverhältnissen
bei m/e = 676–684
beobachtet. 1H NMR (CD2Cl2, δ):
7,64 (dq, J = 8,6 Hz, 1,9 Hz, 0,9 Hz, 2H); 7,31–7,27 (m, 4H); 7,07–7,03 (m,
2H); 5,93 (d, J = 0,8 Hz, 2H); 4,01–3,90 (m, AA', 2H); 3,58–3,47 (m,
BB', 2H); 1,00 (s,
18H); 0,20 (s, 6H); 0,19 (s, 6H). 13C NMR
(CD2Cl2, δ): 150,12;
126,17; 125,14; 124,86; 124,79; 123,35; 116,99; 108,54; 98,61; 26,30;
25,80; 18,61; –3,94; –4,27.
-
Beispiel 17
-
rac-[Ethylenbis(2-(tert-butyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)1-zirkoniumdichlorid
-
Ein
Gemisch von rac-[Ethylenbis((2-tert-butyldimethylsiloxy)indenyl)]-zirkoniumdichlorid
(1,00 g, 1,47 mMol) und PtO2 (20 mg) in
CH2Cl2 (150 ml)
wurde 16 h bei 70 bar in einem gerührten Reaktor hydriert. Die hellgrüne Suspension
wurde durch Celite filtriert und das Lösungsmittel abgedampft. Der
Rückstand
wurde in Hexan (50 ml) aufgelöst
und auf 0°C
abgekühlt,
wobei 0,80 g (79,2%) der Titelverbindung als ein hellgrüner mikrokristalliner
Feststoff erhalten wurden. Im EIMS Massenspektrum der Titelverbindung
wurden Stammionen der Zusammensetzung C32H52Si2O2ZrCl2 + in den entsprechenden
Isotopenverhältnissen
bei m/e = 684–692
beobachtet. 1H NMR (CD2Cl2, δ):
5,69 (s, 2H); 3,39–3,29
(m, AA', 2H); 3,04–2,97 (m,
2H); 2,85–2,77 (m,
2H); 2,73–2,64
(m, BB', 2H); 2,48–2,34 (m,
4H); 1,90–1,70
(m, 4H); 1,59–1,42
(m, 4H); 0,92 (s, 18H); 0,20 (s, 6H); 0,16 (s, 6H). 13C
NMR (CD2Cl2, δ): 142,24;
127,66; 117,29; 114,18; 106,45; 25,46; 24,46; 23,80; 22,05; 21,92;
21,75; 18,10; –4,09; –4,65.
-
Beispiel 18
-
rac-Ethylenbis(2-tert-butyldimethylsiloxy-1-indenyl)hafniumdichlorid
-
Zu
einer Lösung
von Bis(2-(tert-butyldimethylsiloxy)indenyl)ethan (10,38 g, 20,0
mMol) in THF (80 ml) bei 0°C
wurde n-Buli (16,1 ml einer 2,5 M Lösung in Hexan, 40,2 mMol) zugetropft
und das Reaktionsgemisch wurde über
Nacht bei Raumtemperatur gerührt.
Die Lösungsmittel
wurden unter Vakuum entfernt und das resultierende schmutzig weiße Pulver
wurde mit HfCl4 (6,41 g, 20,0 mMol) gemischt.
Das Gemisch wurde auf –80°C gekühlt und
vorgekühltes
CH2Cl2 (150 ml)
wurde zugegeben. Die hellgelbe Suspension wurde langsam auf Raumtemperatur
erwärmt, über Nacht
gerührt
und durch Celite filtriert, um Lithiumchlorid zu entfernen. Aufkonzentrieren
und Abkühlen
auf –30°C ergab 1,96
g (12,8%) der Titelverbindung als einen hellgelben mikrokristallinen
Feststoff. Im EIMS Massenspektrum der Titelverbindung wurden Stammionen
der Zusammensetzung C32H44Si2O2NfCl2 + in den entsprechenden Isotopenverhältnissen
bei m/e = 760–774
beobachtet. 1H NMR (CD2Cl2, δ):
7,64 (dq, J = 8,5 Hz, 1,8 Hz, 1,0 Hz, 2H); 7,34–7,25 (m, 4H); 7,05–7,01 (m,
2H); 5,86 (d, 4J = 0,7 Hz, 2H); 4,02–3,92 (AA', 2H); 3,71–3,61 (BB', 2H); 1,02 (s, 18H);
0,22 (s, 6H); 0,21 (s, 6H). 13C NMR (CD2Cl2, δ): 148,64;
126,19; 124,97; 124,35; 124,30; 123,49; 115,47; 105,37; 96,46; 25,84;
25,52; 18,62; –3,92; –4,26.
-
Beispiel 19
-
rac-[Ethylenbis(2-(t-hexyldimethylsiloxy)indenyl)]zirkoniumdichlorid
-
Zu
einer eisgekühlten
Lösung
von Bis(2-(t-hexyldimethylsiloxy)indenyl)ethan (11,50 g, 20,0 mMol)
in THF (100 ml) wurde n-Buli (16,0 ml einer 2,5 M Lösung in
Hexan, 40,0 mMol) zugetropft und das Reaktionsgemisch wurde über Nacht
bei Raumtemperatur gerührt.
Die resultierende schmutzig gelbe Suspension wurde danach mittels
einer Kanüle
einer Suspension von ZrCl4 (4,66 g, 20,0
mMol) in THF (25 ml) bei –80°C zugegeben.
Das Reaktionsgemisch wurde langsam auf Raumtemperatur erwärmt und über Nacht
gerührt.
Nach Verdampfen der Lösungsmittel
blieb ein gelber Feststoff zurück,
der mit CH2Cl2 (150
ml) extrahiert wurde und durch Celite filtriert wurde, um Lithiumchlorid
zu entfernen. Aufkonzentrieren und Abkühlen auf
–30°C ergab 3,25
g (22,1%) der Titelverbindung als einen gelben mikrokristallinen
Feststoff. Einkristalle für Röntgenbeugung
wurden aus einer gesättigten
Toluollösung
bei Raumtemperatur erhalten. Im EIMS Massenspektrum der Titelverbindung
wurden Stammionen der Zusammensetzung C36H52Si2O2ZrCl2 + in den entsprechenden
Isotopenverhältnissen
bei m/e = 732–740
beobachtet. 1H NMR (CD2Cl2, δ):
7,66 (dq, J = 8,6 Hz, 1,8 Hz, 1,0 Hz, 2H); 7,35–7,29 (m, 4H); 7,08–7,04 (m,
2H); 5,95 (d, 4J = 0,6 Hz, 2H); 4,04–3,94 (m,
AA', 2H); 3,56–3,46 (m,
BB', 2H); 1,74 (sept, 3J = 6,8 Hz, 2H); 0,99 (s, 6H); 0,97 (s,
6H); 0,95 (d, 3J = 6,8 Hz, 6H); 0,94 (d, 3J = 6,8 Hz, 6H); 0,26 (s, 6H); 0,25 (s,
6H). 13C NMR (CD2Cl2, δ):
150,48; 126,13; 125,18; 125,10; 124,85; 123,32; 116,94; 98,53; 34,22;
26,41; 25,84; 20,35; 20,14; 18,80; 18,64; –1,62; –2,01.
-
Beispiel 20
-
rac- und meso-[Dimethylsilylenbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid
-
Zu
einer Lösung
von Dimethylbis(2-(tert-butyldimethylsiloxy)indenyl)silan (10,98
g, 20,0 mMol) in Et2O (40 ml) bei 0°C wurde n-Buli
(16,0 ml einer 2,5 M Lösung
in Hexan, 40,0 mMol) zugetropft und das Reaktionsgemisch wurde über Nacht
bei Raumtemperatur gerührt.
Die Lösungsmittel
wurden unter Vakuum entfernt, wobei ein schmutzig weißes Pulver
zurück
blieb, das mit ZrCl4 (4,66 g, 20 mMol) gemischt
wurde. Das Gemisch wurde auf –80°C gekühlt und
vorgekühltes
CH2Cl2 (150 ml)
wurde zugegeben. Die hellgelbe Suspension wurde langsam auf Raumtemperatur
erwärmt, über Nacht
gerührt
und durch Celite filtriert, um Lithiumchlorid zu entfernen. Aufkonzentrieren
und Abkühlen
auf –30°C ergab 7,95
g (56,0%) eines annähernd
2 : 1 rac/meso-Gemisches der ziemlich reinen Titelverbindung als
ein hellgelbes Pulver. Im EIMS Massenspektrum der Titelverbindung
wurden Stammionen der Zusammensetzung C32H46Si3O2ZrCl2 + in den entsprechenden
Isotopenverhältnissen
bei m/e = 706–714
beobachtet. Kleine Mengen des ziemlich reinen rac-Diastereomers der
Titelverbindung wurden erhalten durch Ausführen der Reaktion in THF-Lösung, gefolgt
von Entfernen von LiCl und Kristallisation aus Et2O. 1H NMR (CD2Cl2, δ,
rac): 7,47–7,44
(m, 2H); 7,37–7,27
(m, 4H); 6,91–6,87
(m, 2H); 6,22 (d, 4J = 0,8 Hz, 2H); 1,19
(s, 6H); 0,92 (s, 18H); 0,27 (s, 6H); 0,21 (s, 6H). 13C
NMR (CD2Cl2, δ, rac): 155,89; 128,69;
127,13; 126,84; 125,07; 124,94; 119,69; 104,50; 73,22; 26,12; 19,23;
1,04; –3,36; –3,87.
-
POLYMERISATIONSTESTS
-
Beispiel 21
-
Die
Polymerisationen wurden in einem 0,5 l Büchi-Glasautoklav in 200 ml
Toluol durchgeführt.
In einem typischen Lauf wurde die Hälfte der MAO/Toluol-Lösung dem Reaktor
zugegeben und 5 Minuten gerührt, um
Verunreinigungen im Reaktor zu reduzieren. In einer parallelen Prozedur
wurden 10–15 μmMol des
Metallocens in der restlichen Hälfte
der MAO/Toluol-Lösung
aufgelöst
und 5 min bei 25°C
präaktiviert.
Das Katalysator/Aktivator-Gemisch wurde in den Reaktor gegeben und
die Polymerisation wurde durch Einleiten des Olefinmonomers gestartet.
Die Polymerisation wurde nach 20 oder 60 Minuten durch Zugabe von
Ethanol oder Methanol unterbrochen. Das Polymer wurde nach Waschen
mit Ethanol/HCl oder Methanol/HCl analysiert.
-
Methylalumoxan
(MAO) wurde als eine 29,3 Gew.-% Toluollösung verwendet (Al gesamt 13,1%
w/w, Al als TMA 3,50% w/w). Ethylen und Propylen mit hoher Reinheit
(99,5%) wurden als Monomere verwendet.
-
Die
Ergebnisse sind nachstehend in Tabelle 1 angegeben.
-
-
Beispiel 22
-
Propylen
wurde gemäß Beispiel
21 polymerisiert. Die Bedingungen und die Ergebnisse sind nachstehend
in Tabelle 2 angegeben. Tabelle
2
- Tpol = 20°C; p = 2,0
bar; Polymerisationsdauer = 60 min; Al : Zr = 3000 : 1
-
Beispiel 23
-
Die
Polymerisationen mit dem Katalysatorsystem [Metallocen]+[B(C6F6)4]– wurden
bei –20°C und 2,0 bar
Propylendruck ausgeführt,
unter Verwendung von Triethylaluminium (TEA) als einen Abfänger für Verunreinigungen.
In einem typischen Lauf wurden 0,3 g TEA mit 50 ml Toluol gerührt, gefolgt
von der Zugabe von 5 μMol
des Metallocens. Das Kation-bildende Mittel (14 μMol) wurde über eine Spritze zugegeben.
Die Polymerisation wurde nach 60 Minuten durch Zugabe von Ethanol
oder Methanol unterbrochen. Das Polymer wurde nach Waschen mit Ethanol/HCl
oder Methanol/HCl analysiert.
-
Die
Bedingungen und die Ergebnisse sind nachstehend in Tabelle 3 angegeben. Tabelle
3
- Temperatur = –20°C; Polymerisationsdauer = 20
min
-
Beispiel 24
-
Ethylen
wurde wie vorstehend polymerisiert. Die Ergebnisse sind nachstehend
in Tabelle 4 angegeben.
-
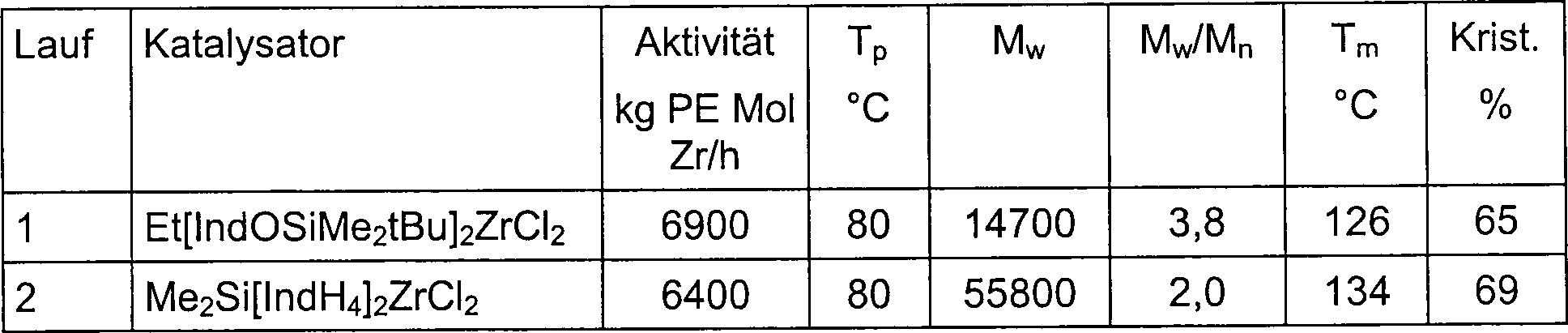
Tabelle
4: Ethylenpolymerisation mit den Katalysatorsystemen
Et[IndOSiMe
2tBu]
2ZrCl
2/MAO und Me
2Si[IndH
4]
2ZrCl
2/MAO/(Referenz)
in Toluol (Al : Zr = 3000 : 1; Monomerdruck = 2 bar)
-
Beispiel 25
-
Die
Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan
durchgeführt.
Eine Komplexlösung
von ÅBO3
(rac-Et[2-(t-BuMe2SiO)Ind]2ZrCl2) und MAO in Toluol, mit Al/Zr = 200, wurde
in den Reaktor eingebracht. Die Metallocenmenge betrug 2,5 μMol. Nach
dem Zuführen
der Katalysatorkomplexlösung
wurde der Polymerisationsreaktor auf die Polymerisationstemperatur
von 70°C
erwärmt.
Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar
in den Polymerisationsreaktor eingeführt und die Reaktion wurde
gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des
Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
-
Nach
30 min Polymerisation betrug die Polymerausbeute 120 g Polymer,
was eine Katalysatoraktivität von
526 kgHD-PE/g·Zr·h ergibt.
-
Beispiel 26
-
Die
Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan
durchgeführt.
Eine Komplexlösung
von ÅBO3
(rac-Et[2-(t-BuMe2SiO)Ind]2ZrCl2) und MAO in Toluol, mit Al/Zr = 200, wurde
in den Reaktor eingebracht. Die Metallocenmenge betrug 1,25 μMol. Nach
dem Zuführen
der Katalysatorkomplexlösung
wurde der Polymerisationsreaktor auf die Polymerisationstemperatur
von 70°C
erwärmt.
Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar
in den Polymerisationsreaktor eingeführt und die Reaktion wurde
gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des
Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
-
Nach
30 min Polymerisation betrug die Polymerausbeute 103 g Polymer,
was eine Katalysatoraktivität von
903 kgHD-PE/g·Zr·h ergibt.
-
Beispiel 27
-
Die
Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan
durchgeführt.
Eine Komplexlösung
von ÅBO3
(rac-Et[2-(t-BuMe2SiO)Ind]2ZrCl2) und MAO in Toluol, mit Al/Zr = 100, wurde
in den Reaktor eingebracht. Die Metallocenmenge betrug 0,63 μMol. Nach
dem Zuführen
der Katalysatorkomplexlösung
wurde der Polymerisationsreaktor auf die Polymerisationstemperatur
von 70°C
erwärmt.
Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar
in den Polymerisationsreaktor eingeführt und die Reaktion wurde
gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des
Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
-
Nach
30 min Polymerisation betrug die Polymerausbeute 96 g Polymer, was
eine Katalysatoraktivität von
1670 kgHD-PE/g·Zr·h ergibt.
-
Beispiel 28
-
Die
Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan
durchgeführt.
Eine Komplexlösung
von ÅBO3
(rac-Et[2-(t-BuMe2SiO)Ind]2ZrCl2) und MAO in Toluol, mit Al/Zr = 100, wurde
in den Reaktor eingebracht. Die Metallocenmenge betrug 0,6 μMol. Nach
dem Zuführen
der Katalysatorkomplexlösung
wurde der Polymerisationsreaktor auf die Polymerisationstemperatur
von 70°C
erwärmt.
Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar
in den Polymerisationsreaktor eingeführt und die Reaktion wurde
gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des
Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
-
Nach
30 min Polymerisation betrug die Polymerausbeute 12 g Polymer, was
eine Katalysatoraktivität von
435 kgHD-PE/g·Zr·h ergibt.
-
Beispiel 29
-
Die
Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan
durchgeführt.
Eine Komplexlösung
von ÅBO3
(rac-Et[2-(t-BuMe2SiO)Ind]2ZrCl2) und MAO in Toluol, mit Al/Zr = 200, wurde
in den Reaktor eingebracht. Die Metallocenmenge betrug 0,6 μMol. Nach
dem Zuführen
der Katalysatorkomplexlösung
wurde der Polymerisationsreaktor auf die Polymerisationstemperatur
von 70°C
erwärmt.
Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar
in den Polymerisationsreaktor eingeführt und die Reaktion wurde
gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des
Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
-
Nach
30 min Polymerisation betrug die Polymerausbeute 27 g Polymer, was
eine Katalysatoraktivität von
936 kgHD-PE/g·Zr·h ergibt.
-
Beispiel 30
-
Die
Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan
durchgeführt.
Eine Komplexlösung
von ÅBO3
(rac-Et[2-(t-BuMe2SiO)Ind]2ZrCl2) und MAO in Toluol, mit Al/Zr = 500, wurde
in den Reaktor eingebracht. Die Metallocenmenge betrug 0,6 μMol. Nach
dem Zuführen
der Katalysatorkomplexlösung
wurde der Polymerisationsreaktor auf die Polymerisationstemperatur
von 70°C
erwärmt.
Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar
in den Polymerisationsreaktor eingeführt und die Reaktion wurde
gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des
Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
-
Nach
30 min Polymerisation betrug die Polymerausbeute 73 g Polymer, was
eine Katalysatoraktivität von
2534 kgHD-PE/g·Zr·h ergibt.
-
Vergleichsbeispiel 1
-
Die
Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan
durchgeführt.
Eine Komplexlösung
von TA2677 (Et[1-Ind]2ZrCl2)
und MAO in Toluol, mit Al/Zr = 100, wurde in den Reaktor eingebracht.
Die Metallocenmenge betrug 0,65 μMol.
Nach dem Zuführen
der Katalysatorkomplexlösung
wurde der Polymerisationsreaktor auf die Polymerisationstemperatur
von 70°C
erwärmt.
Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar
in den Polymerisationsreaktor eingeführt und die Reaktion wurde
gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des
Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
-
Nach
30 min Polymerisation betrug die Polymerausbeute 9 g Polymer, was
eine Katalysatoraktivität von
151 kgHD-PE/g·Zr·h ergibt.
-
Vergleichsbeispiel 2
-
Die
Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan
durchgeführt.
Eine Komplexlösung
von TA2677 (Et[1-Ind]2ZrCl2)
und MAO in Toluol, mit Al/Zr = 200, wurde in den Reaktor eingebracht.
Die Metallocenmenge betrug 0,65 μMol.
Nach dem Zuführen
der Katalysatorkomplexlösung
wurde der Polymerisationsreaktor auf die Polymerisationstemperatur
von 70°C
erwärmt.
Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar
in den Polymerisationsreaktor eingeführt und die Reaktion wurde
gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des
Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
-
Nach
30 min Polymerisation betrug die Polymerausbeute 27 g Polymer, was
eine Katalysatoraktivität von
455 kgHD-PE/g·Zr·h ergibt.
-
Vergleichsbeispiel 3
-
Die
Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan
durchgeführt.
Eine Komplexlösung
von TA2677 (Et[1-Ind]2ZrCl2)
und MAO in Toluol, mit Al/Zr = 500, wurde in den Reaktor eingebracht.
Die Metallocenmenge betrug 0,65 μMol.
Nach dem Zuführen
der Katalysatorkomplexlösung
wurde der Polymerisationsreaktor auf die Polymerisationstemperatur
von 70°C
erwärmt.
Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar
in den Polymerisationsreaktor eingeführt und die Reaktion wurde
gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des
Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
-
Nach
30 min Polymerisation betrug die Polymerausbeute 25 g Polymer, was
eine Katalysatoraktivität von
421 kgHD-PE/g·Zr·h ergibt. Erklärungen von
Chemikalien die in den Experimenten verwendet wurden
- MAO
- 30 Gew.-% Lösung von
Methylalumoxan in Toluol, hergestellt von Albemarle
- Pentan
- Ultrareines Pentan
in Polymerisationsgüte,
H2O und O2 << 1,0 ppm
- Ethylen
- Polymerisationsgüte, H2O und 02 << 1,0 ppm
-
Die
Polymerisationen 28–30
wurden mit den Vergleichspolymerisationen 1–3 gemäß dem bekannten Stand der Technik
verglichen. Die Ergebnisse sind in Tabelle 5 angegeben.
-
Tabelle
5 Vergleich von ÅBO3
und TA2677
-
Figur
1: Vergleich der Wirkung einer Siloxysubstitution auf die Aktivität des Metallzentrums
-
Beschreibung
von Beispielen
-
Die
Beispiele 25–27
beschreiben den Einfluss der Metallocenkonzentration auf die Polymerisation.
Bei Verwendung einer zu hohen Konzentration wird die Metallaktivität zu niedrig.
Aus diesen Beispielen ist ersichtlich, dass die Metallaktivität erhöht wird,
wenn die Katalysatormenge von 2,5 μMol herab auf 0,6 μMol erniedrigt wird.
-
Die
Beispiele 28–30
und die Vergleichsbeispiele 1–3
zeigen die Bedeutung eines 2-Siloxysubstituenten
auf die Katalysatoraktivität.
Wenn Ethylenbisindenylzirkoniumdichlorid einen Siloxysubstituenten
aufweist, wird die Katalysatoraktivität insbesondere bei niedrigen
Al/Zr-Verhältnissen
erhöht,
was auf eine leichte Komplexbildung zwischen Metallocen und MAO
und auch auf eine erhöhte
Aktivität
des Zirkoniums hinweist. Der Unterschied kann am besten in der vorstehenden 1 erkannt werden.

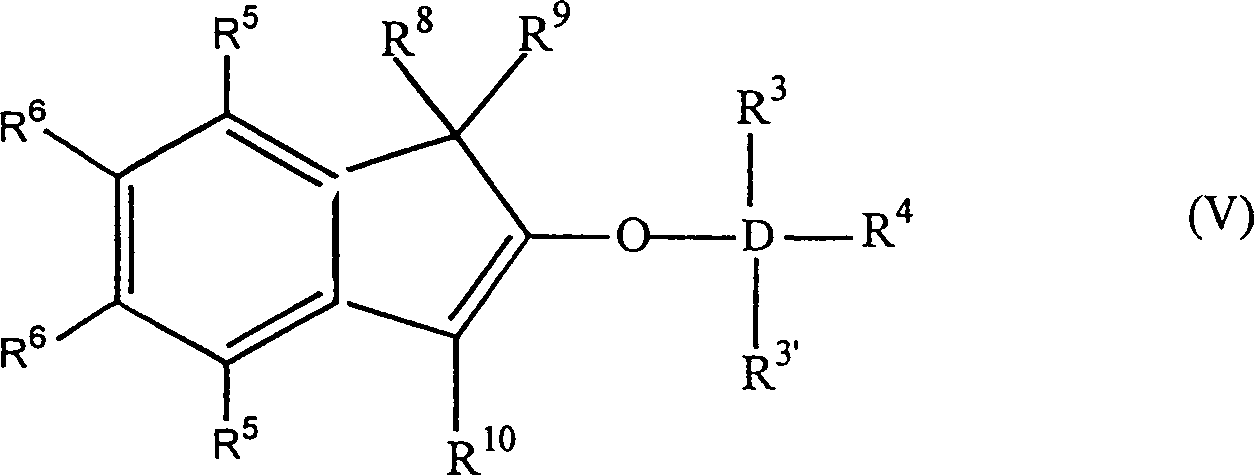
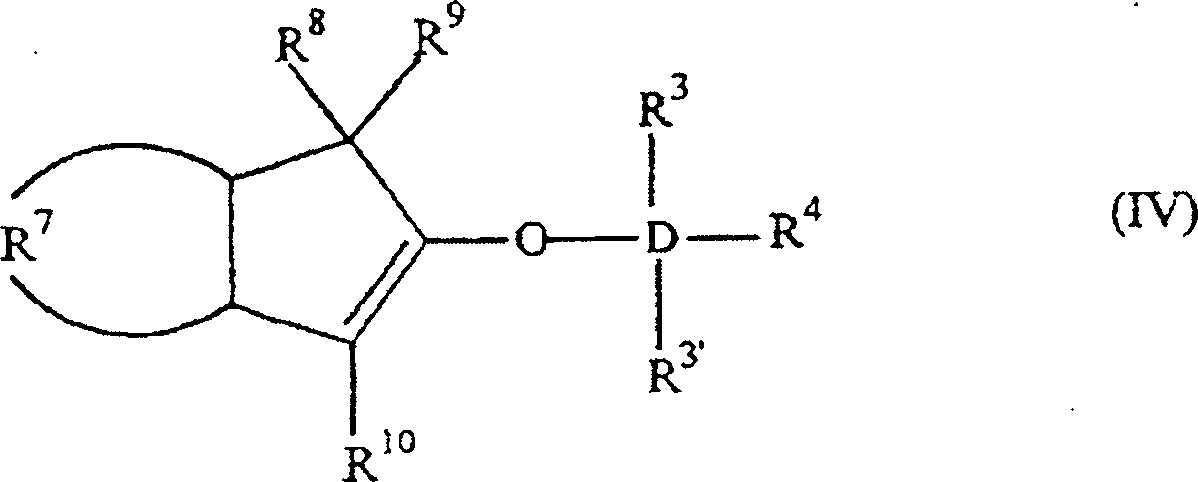 worin: R3', R3–R6 gleich oder verschieden sind und ein Wasserstoffatom, eine C1-C10 Alkylgruppe, eine C1-C10 Alkoxygruppe, eine C6-C10 Arylgruppe, eine C2-C10 Alkenylgruppe, eine C7-C22 Arylalkylgruppe, eine C7-C22 Alkylarylgruppe, eine C8-C23 Arylalkenylgruppe oder ein Halogenatom sind, oder mindestens zwei aus R3, R3' und R4 zusammen eine C2-C10 Ringstruktur bilden, und R8, R9 und R10 die gleichen wie vorstehend sind, oder alternativ die Formel:
worin: R3', R3–R6 gleich oder verschieden sind und ein Wasserstoffatom, eine C1-C10 Alkylgruppe, eine C1-C10 Alkoxygruppe, eine C6-C10 Arylgruppe, eine C2-C10 Alkenylgruppe, eine C7-C22 Arylalkylgruppe, eine C7-C22 Alkylarylgruppe, eine C8-C23 Arylalkenylgruppe oder ein Halogenatom sind, oder mindestens zwei aus R3, R3' und R4 zusammen eine C2-C10 Ringstruktur bilden, und R8, R9 und R10 die gleichen wie vorstehend sind, oder alternativ die Formel: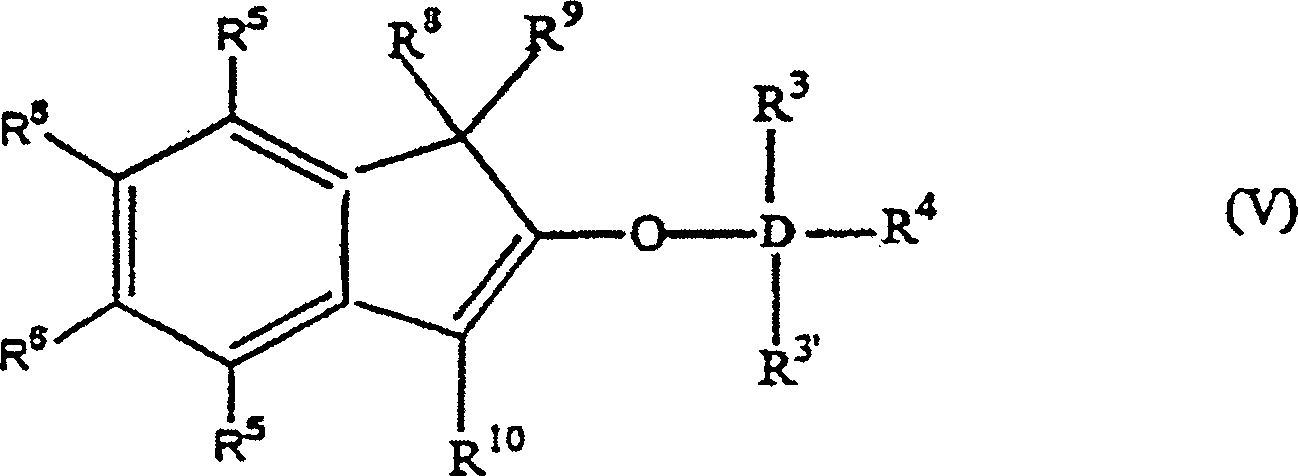 worin: R3, R3', R4-R6, R8, R10 und D die gleichen wie vorstehend sind, und B eine Brücke von mindestens einer der Gruppen -(CH2)n-, -Si(R3)2-, oder -Ge(R3)2- ist, wobei n gleich 1–8 ist und R3 gleich wie vorstehend ist. D ist bevorzugt Si.
worin: R3, R3', R4-R6, R8, R10 und D die gleichen wie vorstehend sind, und B eine Brücke von mindestens einer der Gruppen -(CH2)n-, -Si(R3)2-, oder -Ge(R3)2- ist, wobei n gleich 1–8 ist und R3 gleich wie vorstehend ist. D ist bevorzugt Si.