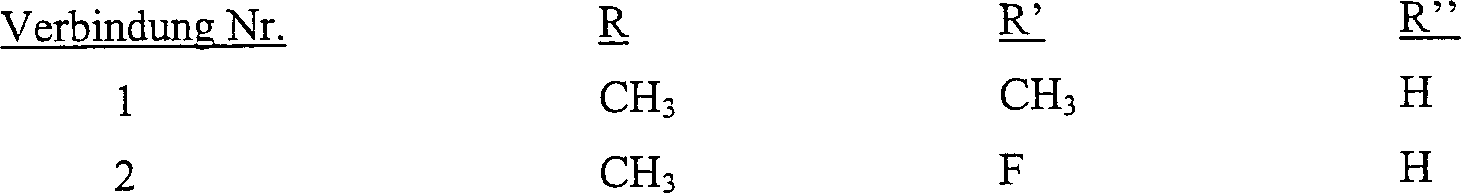-
TECHNISCHES
GEBIET
-
Die vorliegende Erfindung betrifft
bestimmte substituierte 5-(2-Imidazolinnylamino)benzimidazol-Verbindungen. Es
wurde gefunden, dass die Verbindungen alpha-Adrenoceptor-Agonisten
darstellen und nützlich sind
für die
Behandlung einer oder mehrerer Atmungs-Funktionsstörungen,
insbesondere nasale Kongestion; Augen-Funktionsstörungen,
insbesondere Glaukom; und gastrointestinale Funktionsstörungen,
insbesondere Diarrhöe.
-
HINTERGRUND
DER ERFINDUNG
-
Allgemeine Informationen betreffend
alpha-adrenerge Rezeptoren, Agonisten und Antagonisten, und betreffend
Verbindungen, die eine ähnliche
Struktur aufweisen wie die der vorliegenden Erfindung, sind in den folgenden
Literaturstellen offenbart: Timmermans, P:B:M:W:M., A. T. Chiu & M. J. M. C. Thoolen, „12.1 α-Adrenergic
Receptors", Comprehensive
Medicinal Chemistry, Bd. 3, Membranes & Receptors, P. G. Sammes & J. B. Taylor,
Hrg., Pergamon Press (1990), S. 133– 185; Timmermans, P.B.M.W.M. & P.A. van Zwieten, „α-Adrenoceptor
Agonists and Antagonists",
Drugs of the Future, Bd. 9, Nr. 1, (Januar 1984), S. 41–55; Megens, A.A.H.P.,
J. E. Leysen, F. H. L. Awouters & C.
J. E. Niemegeers, „Further
Validation of in vivo and in vitro Pharmacological Procedures for
Assessing the α1 and α2-Selectivity of Test Compounds: (2) α-Adrenoceptor Agonists", European Journal
of Pharmacology, Bd. 129 (1986), S. 57–64; Timmermans, P.B.M.W.M.,
A. de Jonge, M. J. M. C. Thoolen, B. Wilffert, H. Batink & P.A. van Zwieten, „Quantitative
Relationships between α-Adrenergic
Activity and Binding Affinity of α-Adrenoceptor Agonists
and Antagonists",
Journal of Medicinal Chemistry, Bd. 27 (1984), S. 495– 503; van
Meel, J.C.A., A. de Jonge, P.B.M.W.M. Timmermans & P.A. van Zwieten, "Selectivity of Some
Alpha Adrenoceptor Agonists for Peropheral Alpha-1 and Alpha-2 Adrenoceptors
in the Normotensive Rat",
The Journal of Pharmacology and Experimental Therapeutics, Bd. 219,
Nr. 3 (1981), S. 760–767;
Chapleo, C. B., J. C. Doxey, P. L. Myers, M. Myers, C. F. C. Smith & M. R. Stillings, "Effect of 1,4-Dioxanyl
Substitution on the Adrenergic Activity of Some Standard α-Adrenoreceptor Agents", European Journal of
Medicinal Chemistry, Bd. 24 (1989), S. 619–622; Chapleo, C. B., R. C.
M. Butler, D. C. England, P. L. Myers, A. G. Roach, C. F. C. Smith,
M. R. Stillings & I.
F. Tulloch, "Heteroaromatic
Analogues of the α-2-Adrenoreceptor
Partial Agonist Clonidine",
J. Med. Chem., Bd. 32 (1989), S. 1627–1630; Clare, K. A., M. C.
Scutton & N.
T. Thompson, "Effects
of α2-Adrenoceprtor Agonists and of Related Compounds
on Aggregation of and on Adenylate Cyclase Activity in, Human Platelets", Br. J. Pharmac.,
Bd. 82 (1984), S. 467– 476;
U.S.-Patente 3,890,319 und 5,180,721; WO 92/02515, WO 92/04345 (=
U.S. 5,091,528) und WO 92/13855. Viele Verbindungen jedoch, die
strukturell solchen der vorliegenden Erfindung ähneln, stellen nicht die gewünschte Aktivität und Spezifität zur Verfügung, wenn
Atmungs-, Augen- und gastrointestinale Funktionsstörungen behandelt werden.
-
US
4,398,028 , die als anti-hypertensive Wirkstoffe verwendbare
Verbindungen betrifft, und EP-A-399,791,
die die Potenzierung von alpha-2-Agonisten mit einem alpha-3-Antagonisten
betrifft, offenbaren beide eine generische Formel, die einige der
Verbindungen der vorliegenden Erfindung umfassen kann.
-
WO 96/04270, deren frühestes Prioritätsdatum
der zweiten Priorität
der vorliegenden Anmeldung vorausgeht, und die nur unter Art. 54(3)
EPÜ relevant
ist, offenbart ebenfalls 5-(2-Imidazolinylamino)benzimidazol-Derivate.
-
Es ist für die vorliegende Erfindung
insbesondere relevant, dass bei Verbindungen, von denen gefunden
wurde, dass sie wirksame nasale Abschweller darstellen, oft unerwünschte Nebenwirkungen
auftreten, wie zum Beispiel Bluthochdruck und Schlaflosigkeit verursachend,
insbesondere wenn sie systemisch verabreicht werden. Es gibt eine
Notwendigkeit für
neue Arzneimittel, die eine Entlastung von nasaler Kongestion bereit
stellen, ohne diese unerwünschten
Nebenwirkungen zu verursachen.
-
Es ist eine Aufgabe der vorliegenden
Erfindung, neue Verbindungen mit umfangreicher Aktivität bei der
Vermeidung oder Behandlung von nasaler Kongestion bereit zu stellen.
-
Es ist eine weitere Aufgabe der vorliegenden
Erfindung, solche Verbindungen zur Verfügung zu stellen, die keine
Hypotonie, Benommenheit, Bluthochdruck, Schlaflosigkeit oder andere
unerwünschte
Nebenwirkungen verursachen, insbesondere, wenn sie systemisch verabreicht
werden.
-
Es ist außerdem eine Aufgabe der vorliegenden
Erfindung, neue Verbindungen zur Behandlung von Husten, chronischer
Bronchitis (chronic obstructive pulmonary disease – COPD)
und/oder Asthma zur Verfügung
zu stellen.
-
Es ist außerdem eine Aufgabe der vorliegenden
Erfindung, neue Verbindungen zur Behandlung von Glaukom und/oder
Diarrhöe
zur Verfügung
zu stellen.
-
Es ist noch eine weitere Aufgabe
der vorliegenden Erfindung, solche Verbindungen zur Verfügung zu stellen,
die eine gute Aktivität
aus einer peroralen und/oder topischen Dosierung aufweisen.
-
ZUSAMMENFASSUNG DER ERFINDUNG
-
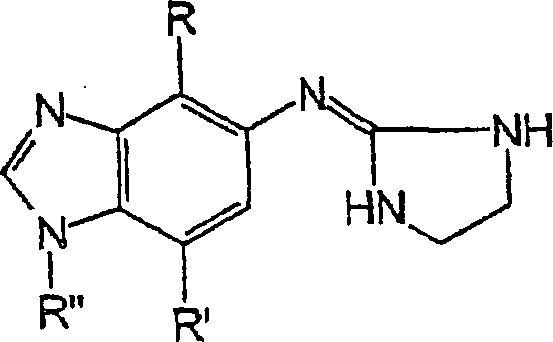
Die vorliegende Erfindung betrifft
Verbindungen mit der folgenden Struktur:
worin
(a) R unsubstituiertes
Alkanyl oder Alkenyl mit bis zu 3 Kohlenstoffatomen ist;
(b)
R' gewählt ist
aus Methyl, Cyano- und Fluor-; und
(c) R'' Wasserstoff
ist
pharmazeutische Zusammensetzungen, die solche neuen Verbindungen
enthalten, und die Verwendung solcher Verbindungen zur Vermeidung
oder Behandlung anderer Atmungs-, Augen- und/oder gastrointestinaler Funktionsstörungen.
-
DETAILLIERTE BESCHREIBUNG
DER ERFINDUNG
-
Wie hierin verwendet, meint „Alkanyl" einen gesättigten
Kohlenwasserstoffsubstituenten, gerad- oder verzweigtkettig.
-
Wie hierin verwendet, meint „Alkenyl" einen Kohlenwasserstoffsubstituenten
mit einer Doppelbindung, gerad- oder verzweigtkettig.
-
Verbindungen
-
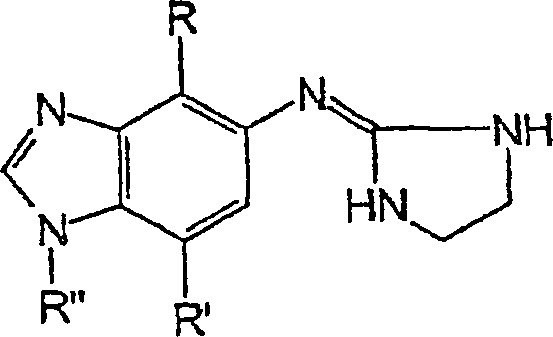
Die vorliegende Erfindung umfasst
Verbindungen mit der folgenden Struktur:
-
-
In der obigen Struktur ist R unsubstituiertes
Alkanyl oder Alkenyl mit bis zu 3 Kohlenstoffatomen. R ist vorzugsweise
Alkanyl. R ist höchst
vorzugsweise Methyl oder Ethyl.
-
In der obigen Struktur ist R' gewählt aus
Methyl, Cyano- und Fluor-.
-
In der obigen Struktur ist R'' Wasserstoff.
-
Bevorzugte Verbindungen der vorliegenden
Erfindung weisen die folgende Struktur auf:
worin R, R' und R'' wie in der folgenden Tabelle angegeben
sind:
-
-
Die Verbindungen der vorliegenden
Erfindung sind insbesondere nützlich
für die
Behandlung von nasaler Kongestion, verbunden mit Allergien, Erkältungen
und anderen nasalen Funktionsstörungen,
als auch ihrer Folgekrankheiten (zum Beispiel Sinusitis und Otitis).
Gleichzeitig wurde gefunden, dass unerwünschte Nebenwirkungen, wie
zum Beispiel Hypotonie, Benommenheit, Bluthochdruck oder Schlaflosigkeit
oftmals vermieden werden können.
Ohne einen speziellen Wirkmechanismus beschränkt zu sein wird von den vorliegenden
Verbindungen angenommen, dass sie gegenüber ähnlichen Verbindungen Vorteile
bei der Behandlung von nasaler Abschwellung durch ihre Fähigkeit,
mit alpha-2-Adrenozeptoren zusammenzuwirken, bieten. Bei den vorliegenden
Verbindungen wurde gefunden, dass sie alpha-2-Adrenozeptor-Agonisten
darstellen, die ein Zusammenziehen von peripheren Gefäßbahnen
in den Nasenmuscheln verursachen.
-
Bestimmte vorliegende Verbindungen
weisen keine oder nur schwache alpha-1-Agonistenaktivität auf und
besitzen eine geringe oder keine Wirkung auf das zentrale Nervensystem,
selbst wenn sie systemisch dosiert werden.
-
Die Verbindungen der vorliegenden
Erfindung sind ebenfalls nützlich
für die
Behandlung von Augen-Funktionsstörungen,
verbunden mit erhöhtem
Augeninnendruck, wie zum Beispiel Glaukom. Die Verbindungen werden
entweder peroral oder topisch als Tropfen, Gels oder Cremes direkt
auf die Oberfläche
des Säugetierauges
verabreicht.
-
Die Verbindungen der vorliegenden
Erfindung sind ebenfalls nützlich
zur Kontrolle von gastrointestinalen Bewegungs-Funktionsstörungen,
wie zum Beispiel Diarrhöe,
durch antimotilitale und antisekretorische Wirkungen auf den Gastrointestinaltrakt.
-
Die pharmakologische Aktivität und Selektivität der vorliegenden
Verbindungen kann unter Verwendung veröffentlichter Testverfahren
bestimmt werden. Die alpha-2-Selektivität der Verbindungen wird bestimmt durch
Messung von Rezeptor-Bindungsaffinitäten und in vitro-funktionalen Wirkstoffgehalten
in einer Vielzahl von Geweben, von denen bekannt ist, dass sie alpha-2-
und/oder alpha-1-Rezeptoren besitzen. (Vgl., z. B., The Alpha-2
Adrenergnic Receptors, L. E. Limbird, Hrg., Humana Press, Clifton,
NJ.) Die folgenden in vivo-Untersuchungen werden typischerweise
in Nagern oder anderen Spezies durchgeführt. Die Zentralnervensystem-Aktivität wird bestimmt
durch Messung der Bewegungsaktivität als dem Sedierungsindex.
(Vgl., z. B., Spyraki, C. & H.
Fibiger, „Clonidine-induced
Sedation in Rats: Evidence for Mediation by Postsynaptic Alpha-2
Adrenoreceptors",
J. Neural. Trans., Bd. 54 (1982), S. 153–163). Die nasale Abschwellaktivität wird unter
Verwendung der Rhinomanometrie als Abschätzung der nasalen Atemwegswiderstandsfähigkeit
gemessen. (Vgl., z. B., Salem, S. & E. Clemente, „A New Experimantal Method
for Evaluating Drugs in the Nasal Cavity", Arch. Otolarynng, Bd. 96 (1972), S.
524–529).
Die Antiglaukomaktivität
wird durch Messung des Augeninnendrucks bestimmt. (Vgl., z. B.,
Potter, D., „Adrenergic
Pharmacology of Aqueous Human Dynamics", Pharmacol. Rev., Bd. 13 (1981), S.
133–153).
Die Antidiarrhöe-Aktivität wird durch
Messung der Fähigkeit
der Verbindungen, prostaglandin-induzierte Diarrhöe zu verhindern,
bestimmt. (Vgl., z. B., Thollander, M., P. Hellstrom & T. Svensson, „Suppression
of Castor Oil-Induced Diarrhea by Alpha-2 Adrenoceptor Agonists", Aliment. Pharmacol. Therap.,
Bd. 5 (1991), S. 255–262).
Die Antiasthma-Aktivität wird durch
Messung des Wirkung der Verbindung auf Bronchokonstriktion, verbunden
mit pulmonalen Schwierigkeiten, wie zum Beispiel eingeatmeten Antigenen,
bestimmt. (Vgl., z. B., Chang, J. J. Musser & J. Hind, § "Effects of a Novel Leukotriene D4 Antagonist
with 5-Lipoxygenase
and Cyclooxygenase Inhibitory Activity, Wy-45,911, on Leukotriene-D4-
and Antigen-Induced Bronchoconstriction in Guinea Pig", Int. Arch. Allegry
Appl. Immun., Bd. 86 (1988), S. 48–54; und Delehunt, J., A. Perruchound,
L. Yerger, B. Marchette, J. Stevenson & W. Abraham, „The Role of Slow-Reacting
Substance of Anaphylaxis in the Late Bronchial Response After Antigen
Challenge in Allergic Sheep",
Am. Rev. Respir. Dis., Bd. 130 (1984), S. 748–754). Die Aktivität bei Husten
wird bestimmt durch Messung der Anzahl und der Latenzzeit der Hustenreaktion
auf Atemschwierigkeiten, wie zum Beispiel eingeatmete Zitronensäure. (Vgl.,
z. B., Callaway, J. & R.
King, "Effects of
Inhaled Alpha-2-Adrenoceptor and GABAB Receptor Agonists on Citric Acid-Induced
Cough and Tidal Volume Changes in Guinea Pig", Eur. J. Pharmacol., Bd. 220 (1992),
S. 187–185).
-
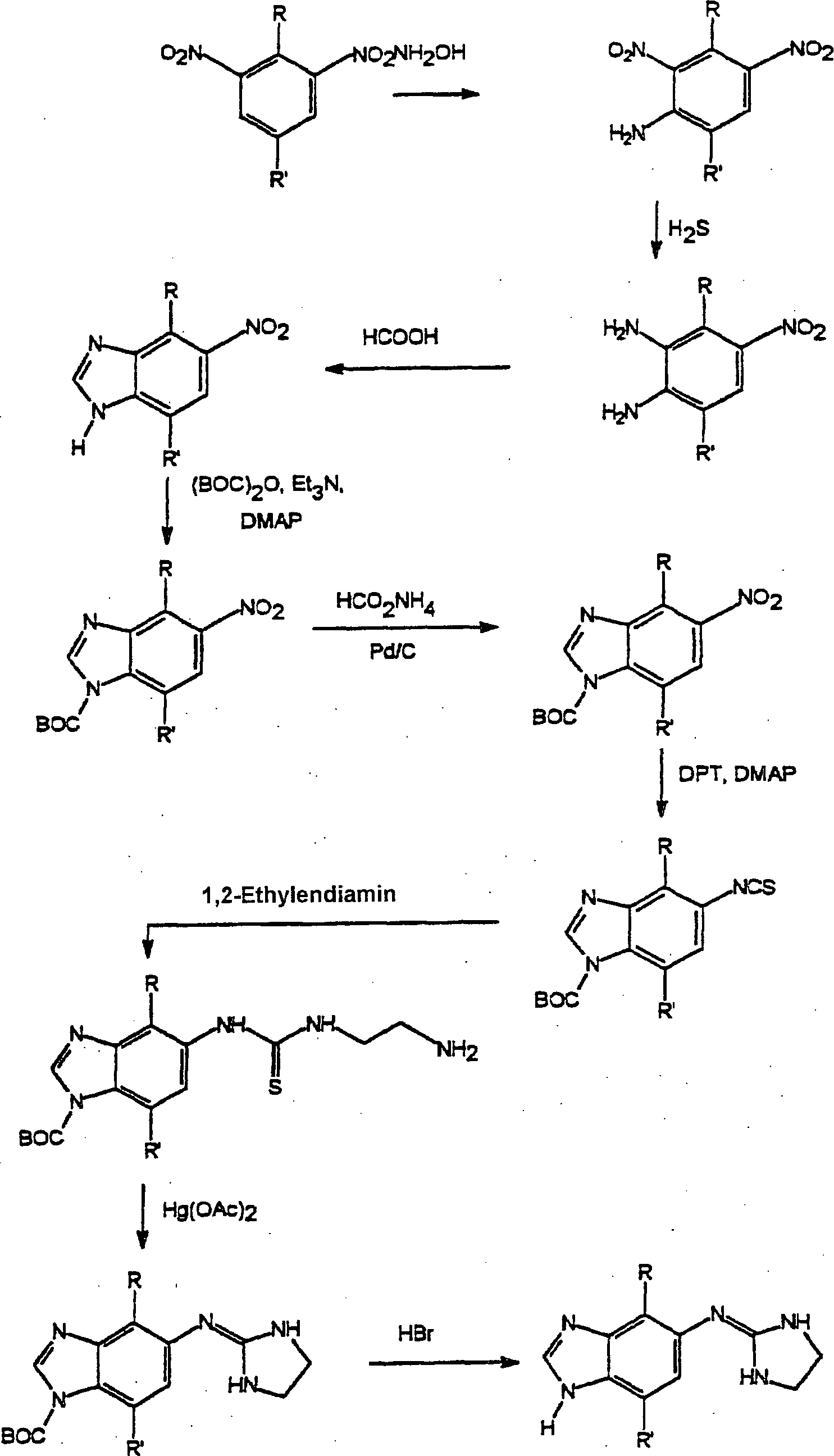
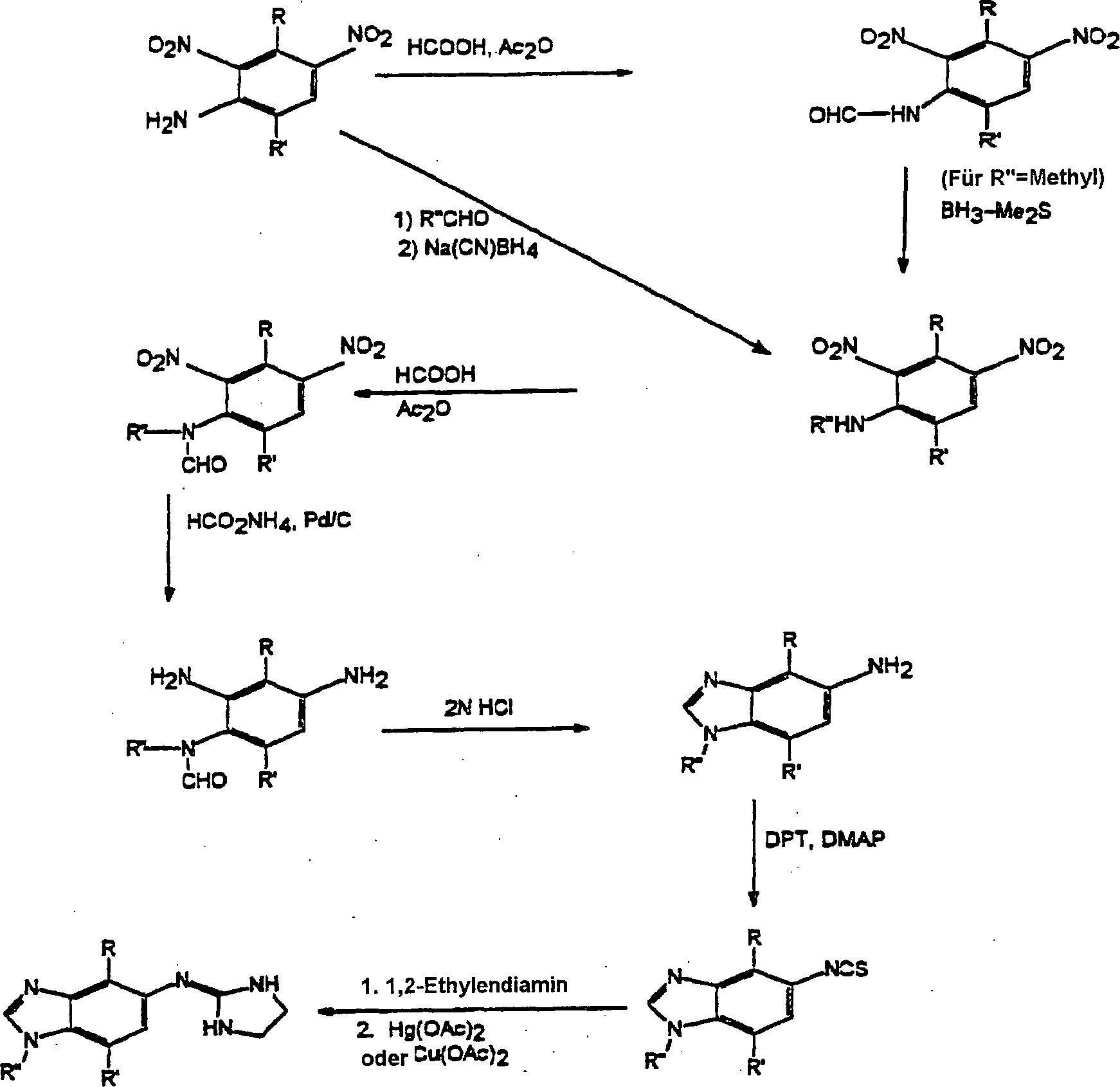
Die Verbindungen der vorliegenden
Erfindung werden synthetisiert unter Verwendung der folgenden allgemeinen
Arbeitsverfahren:
-
-
-
-
Beispiel
1
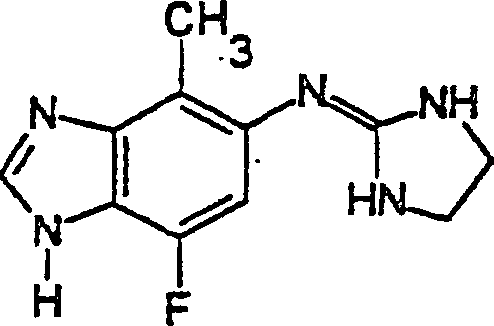
Synthese von 7-Fluor-4-methyl-5-(2-imidazolinylamino)benzimidazol:
-
2,3-Dinitro-4-fluortoluol.
-
Rauchende Schwefelsäure (180
ml) wird unter Argonatmosphäre
tropfenweise zu 4-Fluor-2-nitrotoluol (50,21 g) gegeben. Die interne
Temperatur der Mischung wird unter Verwendung eines Eis/Natriumchloridbades
bei 0–5°C gehalten.
Eine vorgebildete (Eisbad) Mischung aus rauchender Salpetersäure (30
ml) und rauchender Schwefelsäure
(90 ml) wird tropfenweise über
drei Stunden zu der vorhergehenden Lösung gegeben. Die Reaktion
wird dann auf Raumtemperatur aufwärmen gelassen. Nach zweistündigem Rühren bei
Raumtemperatur wird die Mischung langsam in Eis eingegossen und
die Produkte werden mit Methylenchlorid (4 × 500 ml) extrahiert. Die kombinierten
Extrakte werden über
Magnesiumsulfat getrocknet, filtriert und rotationsverdampft. Das
Rohprodukt wird durch Flash-Chromatographie über Silicagel mittels Eluieren
mit 5% Ethylacetat/Hexan gereinigt, um 2,3-Dinitro-4-fluortoluol
als blassgelben Feststoff hervorzubringen.
-
4-Fluor-7-methylbenzimidazol.
-
Eine Suspension von 2,3-Dimethyl-4-fluortoluol
(1 g), Eisenpulver (1,95 g) und Palladium-auf-Kohlenstoff (10%,
150 mg) in Ameisensäure
(995, 25 ml) wird am Rückfluss
für 2,5
Stunden erhitzt. Die resultierende Mischung wird durch Celite gefiltert
und die Feststoffe mit Methanol gewaschen. Das Filtrat wird rotationsverdampft
und der Rückstand
zwischen Wasser und Ethylacetat verteilt. Die organische Phase wird über Magnesiumsulfat
getrocknet, filtriert und rotationsverdampft, um 4-Fluor-7-methylbenzimidazol
als cremefarbenen Feststoff hervorzubringen.
-
7-Fluor-4-methyl-5-nitrobenzimidazol.
-
Zu einer kalten (Eisbad) Lösung von
4-Fluor-7-methylbenzimidazol
(724 g) in konzentrierter Schwefelsäure (10 ml) wird über 1 Stunde
tropfenweise konzentrierte Salpetersäure (0,22 ml) gegeben. Die
Mischung wird zusätzlich
15 Minuten im Eisbad gerührt,
dann in eine Mischung aus gestoßenem
Eis (20 ml) und Ammoniumhydroxid (20 ml) gegossen. Die resultierende
Mischung wird über
Magnesiumsulfat getrocknet, filtriert und rotationsverdampft, um
7-Fluor-4-methyl-5-nitrobenzimidazol als blassgelben Feststoff hervorzubringen.
-
1-tert-Butoxycarbonyl-7-fluor-4-methyl-5-nitrobenzimidazol.
-
Eine Suspension von 7-Fluor-4-methyl-5-nitrobenzimidazol
(0,556 g), di-tert-Butyl-dicarbonat (0,870 g), Triethylenamin (0,475
ml) und 4-Dimethylaminopyridin (0,01 g) in Ethylacetat (100 ml)
wird bei Raumtemperatur über
Nacht gerührt.
Die Mischung wird rotationsverdampft und der Rückstand durch Flash-Chromatographie über Silicagel
mittels Eluieren mit 10% Ethylacetat/Hexan gereinigt, um 1-tert-butoxycarbonyl-7-fluor-5-nitrobenzimidazol
als cremefarbenen Feststoff hervorzubringen.
-
5-Amino-1-tert-butoxycarbonyl-7-fluor-4-methylbenzimidazol.
-
Zu einer Lösung von 1-tert-butoxycarbonyl-7-fluor-5-nitrobenzimidazol
(0,776 g) in Methanol (100 ml)/Ethylacetat (50 ml) wird Palladium-auf-Kohlenstoff
(10%, 0,1 g) und Ammoniumformiat (0,663 g) gegeben. Die Mischung
wird bei Raumtemperatur 5 Stunden lang gerührt, dann über Celite® filtriert,
und die Feststoffe mit Methanol gewaschen. Das Filtrat wird rotationsverdampft
und der Rückstand
zwischen Wasser und Ethylacetat verteilt. Die organische Phase wird über Magnesiumsulfat
getrocknet, filtriert und rotationsverdampft. Der Rückstand
wird durch Flash-Chromatographie über Silicagel mittels Eluieren
mit 25% Ethylacetat/Hexan gereinigt, um 5-Amino-1-tert-butoxycarbonyl-7-fluor-4-methylbenzimidazol
als gelbes Öl
hervorzubringen.
-
1-tert-butoxycarbonyl-7-fluor-4-methyl-5-benzimidazolylisothiocyanoat.
-
Zu einer Lösung von di-2-Pyridylthioncarbonat
(0,393 mg) und 4-Dimethylaminopyridin (0,01 g) in Methylenchlorid
(100 ml) wird tropfenweise über
30 Minuten eine Lösung
von 5-Amino-1-tert-butoxycarbonyl-7-fluor-4-methylbenzimidazol (0,409 g)
in Methylenchlorid (70 ml) gegeben. Die Mischung wird bei Raumtemperatur
3 Stunden lang gerührt,
dann rotationsverdampft. Der Rückstand
wird durch Flash-Chromatographie über Silicagel mittels Eluieren
mit 10% Ethylacetat/Hexan gereinigt, um 1-tert-butoxycarbonyl-7-fluor-4-methyl-5-benzimidazolylisothiocyanoat
als weißen
Feststoff hervorzubringen.
-
N-(1-tert-butoxvcarbonvl-7-fluor-4-methyl-5-benzimidazolyl-N'-2-aminoethylthioharnstoff
-
Eine Lösung von 1-tert-butoxycarbonyl-7-fluor-4-methyl-5-benzimidazolylisothiocyanoat
(0,42 g) in Methylenchlorid (50 ml) wird tropfenweise über 15 Minuten
zu 1,2-Ethylendiamitt (0,45 ml) in Lösung in Methylenchlorid (100
ml) gegeben. Die Mischung wird bei Raumtemperatur 10 Minuten lang
gerührt,
dann rotationsverdampft. Der Rückstand
wird 30 Minuten lang mit Ether (100 ml) zerrieben. Die resultierende
weiße
Suspension wird filtriert und der Feststoff wird im Vakuum über Nacht
getrocknet.
-
7-Fluor-4-methyl-5-(2-imidazolinylamino)benzimidazol.
-
Eine Mischung von N-(1-tert-butoxycarbonyl-7-fluor-4-methyl-5-benzimidazolyl-N'-2-aminoethylthioharnstoff
(0,5 g) und Quecksilberacetat (0,52 g) in methanol (150 ml) wird
bei Raumtemperatur 1 Stunde lang gerührt. Die resultierende schwarze
Mischung wird über
Celite filtriert, und die Feststoffe mit Methanol gewaschen. Das
Filtrat wird rotationsverdampft und der Rückstand durch einen kurzen
Block Silicagel mittels Eluieren mit 25% Methanol in Chloroform,
enthaltend 1% Ammoniumhydroxid, filtriert. Die produktenthaltenden Fraktionen
werden rotationsverdampft, der Rückstand
mit Wasser (15 ml) verdünnt,
durch einen Block Glaswolle filtriert und gefriergetrocknet, um
7-Fluor-4-methyl-5-(2-imidazolinylamino)benzimidazol
als blassgelben Feststoff hervorzubringen.
-
Zusammensetzungen
-
Ein anderer Aspekt der vorliegenden
Erfindung sind Zusammensetzungen, die eine sichere und wirksame
Menge einer betreffenden Verbindung, oder eines pharmazeutisch annehmbaren
Salzes davon, und einen pharmazeutisch annehmbaren Träger umfassen.
Wie hierin verwendet, meint „sichere
und wirksame Menge" eine
Menge der betreffenden Verbindung, die ausreichend ist, um bedeutsam
eine positive Veränderung in
dem zu behandelnden Zustand zu induzieren, aber niedrig genug ist,
um ernsthafte Nebenwirkungen (bei einem vernünftigen Vorteil/Risiko-Verhältnis) innerhalb
des Rahmens eines vernünftigen
medizinischen Urteilsvermögens
zu vermeiden. Eine sichere und wirksame Menge der betreffenden Verbindung
wird mit dem Alter und dem physischen Zustand des zu behandelnden
Patienten variieren, wobei die Natur der gleichzeitigen Therapie,
des verwendeten speziellen pharmazeutisch annehmbaren Trägers, und ähnliche
Faktoren innerhalb des Wissens und der Fachkenntnis des behandelnden
Arztes liegen.
-
Zusammensetzungen der vorliegenden
Erfindung umfassen vorzugsweise 0,0001 bis 99 Gew.-% der betreffenden
Verbindung, mehr vorzugsweise 0,01 bis 90%; ebenfalls vorzugsweise
10 bis 50%, ebenfalls vorzugsweise 5 bis 10%; ebenfalls vorzugsweise
1 bis 5%, und ebenfalls vorzugsweise 0,1 bis 1%.
-
Zusätzlich zu der betreffenden
Verbindung enthalten die Zusammensetzung der vorliegenden Erfindung
einen pharmazeutisch annehmbaren Träger. Der Begriff „pharmazeutisch
annehmbarer Träger", wie hierin verwendet,
meint einen oder mehrere kompatible feste oder flüssige Füllverdünnungsmittel
oder einkapselnde Substanzen, die zur Gabe an einen Menschen oder
ein niederes Tier geeignet sind. Der begriff „kompatibel", wie hierin verwendet,
meint, dass die Komponenten der Zusammensetzung in der Lage sind,
in einer An und Weise mit der betreffenden Verbindung und miteinander
zusammen gemischt zu werden, dass keine Wechselwirkung auftritt,
die die pharmazeutische Wirkung der Zusammensetzung unter normalen
Gebrauchsumständen
wesentlich reduzieren würde.
Pharmazeutisch annehmbare Träger
müssen
natürlich
von ausreichend hoher Reinheit und ausreichend niedriger Toxizität sein,
um sie für
die Gabe an einen zu behandelnden Menschen oder ein niederes Tier
geeignet zu machen.
-
Einige Beispiele von Substanzen,
die als pharmazeutisch annehmbare Träger oder deren Bestandteile dienen
können,
sind Zucker, wie zum Beispiel Lactose, Glucose und Sucrose; Stärken, wie
zum Beispiel Maisstärke
und Kartoffelstärke;
Cellulose und ihre Derivate, wie zum Beispiel Natriumcarboxymethylcellulose, Ethylcellulose,
und Methylcellulose; gepulverter Tragantgummi; Malz; Gelatine; Talg;
feste Schmierstoffe, wie zum Beispiel Stearinsäure und Magnesiumstearat; Calciumsulfat;
Pflanzenöle,
wie zum Beispiel Erdnussöl, Baumwollsamenöl, Sesamöl, Olivenöl, Maisöl und Kakaobutter;
Polyole, wie zum Beispiel Propylenglykol, Glyzerin, Sorbitol, Mannitol, und
Polyethylenglykol; Alginsäure;
Emulgatoren, wie zum Beispiel das Tweens®; Benetzungsmittel,
wie zum Beispiel Natriumlaurylsulfat; Färbemittel; Aromastoffe; Tablettierungsmittel,
Stabilisatoren; Antioxidanzien; Konservierungsmittel; Pyrogen-freies
Wasser; isotonische Kochsalzlösung;
und Phosphatpufferlösungen.
-
Die Wahl eines in Verbindung mit
der betreffenden Verbindung zu verwendenden pharmazeutisch annehmbaren
Trägers
wird grundsätzlich
bestimmt durch den Weg, auf dem die Verbindung verabreicht werden soll.
-
Wenn die betreffende Verbindung injiziert
werden soll, ist der bevorzugte pharmazeutisch annehmbare Träger eine
sterile physiologische Kochsalzlösung,
mit einem blut-kompatiblen Lösungsmittel,
deren pH auf ungefähr
7,4 eingestellt wurde.
-
Die bevorzugte Art, die betreffenden
Verbindungen zu verabreichen, ist peroral. Die bevorzugte Dosierungsform
sind daher Tabletten, Kapseln, Pastillen, Kautabletten und dergleichen.
Solche Dosierungsformen umfassen eine sichere und wirksame Menge
der betreffenden Verbindung, die vorzugsweise zwischen 0,01 mg und
200 mg, mehr vorzugsweise zwischen 0,1 mg und 50 mg, noch mehr vorzugsweise
zwischen 0,5 mg und 25 mg, ebenfalls vorzugsweise zwischen 1 mg
und 10 mg beträgt.
Die für
die Herstellung von Dosierungsformen zur peroralen Gabe geeigneten
pharmazeutisch annehmbaren Träger
sind in der Technik wohlbekannt. Tabletten umfassen herkömmlicherweise
pharmazeutisch annehmbare Zusatzstoffe als inerte Verdünnungsmittel,
wie zum Beispiel Calciumcarbonat, Natriumcarbonat, Mannitol, Lactose
und Cellulose; Bindemittel, wie zum Beispiel Stärke, Gelatine und Sucrose;
Desintegrationsmittel, wie zum Beispiel Stärke, Alginsäure und Croscarmelose; Schmiermittel,
wie zum Beispiel Magnesiumstearat, Stearinsäure und Talg. Gleitmittel,
wie zum Beispiel Siliziumdioxid können verwendet werden, um die
Fließeigenschaften
der Pulvermischung zu verbessern. Färbemittel, wie zum Beispiel
die FD&C-Farbstoffe können für das Aussehen
zugegeben werden. Süßstoffe
und Aromastoffe, wie zum Beispiel Aspartam, Saccharin, Menthol,
Pfefferminze, und Fruchtaromen sind nützliche Zusatzstoffe für Kautabletten.
Kapseln umfassen typischerweise eine oder mehrere oben offenbarte
feste Verdünnungsstoffe.
Die Auswahl von Trägerkomponenten
hängt von
untergeordneten Erwägungen
ab wie Geschmack, Kosten, und Lagerstabilität, die für die Zwecke der vorliegenden
Erfindung nicht entscheidend sind, und kann leicht von einem Fachmann
durchgeführt
werden.
-
Perorale Zusammensetzungen beinhalten
auch flüssige
Lösungen,
Emulsionen, Suspensionen und dergleichen. Die für die Herstellung solcher Zusammensetzungen
geeigneten pharmazeutisch annehmbaren Träger sind in der Technik wohlbekannt.
Solche flüssigen
oralen Zusammensetzungen umfassen vorzugsweise 0,001% der betreffenden
Verbindung, mehr vorzugsweise 0,01 bis 0,5%. Typische Bestandteile
von Trägern für Sirupe,
Elixiere, Emulsionen und Suspensionen beinhalten Ethanol, Glycerol,
Propylenglykol, Polyethylenglykol, flüssige Sucrose, Sorbitol und
Wasser. Typische Suspendiermittel für eine Suspension beinhalten
Methylcellulose, Natriumcarboxymethylcellulose, Avicel® RC-591,
Tragantgummi und Natriumalginat; typische Benetzungsmittel beinhalten
Lecithin und Polysorbat 80; und typische Konservierungsmittel beinhalten
Methylparaben und Natriumbenzoat. Perorale flüssige Zusammensetzungen können auch
eine oder mehrere oben offenbarten Komponenten wie zum Beispiel
Süßstoffe,
Aromastoffe und Färbemittel
enthalten.
-
Andere für das Erzielen einer systemischen
Abgabe der betreffenden Verbindungen beinhalten sublinguale und
buccale Dosierungsformen. Solche Zusammensetzungen umfassen typischerweise
eine oder mehrere von löslichen
Füllsubstanzen
wie zum Beispiel Sucrose, Sorbitol und Mannitol; und Bindemittel
wie zum Beispiel Akazie, mikrokristalline Cellulose, Carboxymethylcellulose
und Hydroxypropylmethylcellulose. Gleitmittel, Schmiermittel, Süßstoffe,
Färbemittel,
Antioxidanzien und Aromastoffe, die oben offenbart sind, können ebenfalls
enthalten sein.
-
Ein bevorzugtes Verfahren der Gabe
der betreffenden Verbindungen ist topisch an die Stelle, an der eine
Aktivität
gewünscht
ist: intranasale Dosen für
nasale Abschwellung, Inhalationsmittel für Asthma, Augentropfen, Gele
und Cremes für
Augen-Funktionsstörungen,
und perorale Dosen für
gastrointestinale Funktionsstörungen.
-
Bevorzugte Zusammensetzungen der
vorliegenden Erfindung beinhalten wässrige Lösungen, umfassend eine sichere
und wirksame Menge einer betreffenden Verbindung, beabsichtigt für die topische
nasale Gabe. Solche Zusammensetzungen enthalten vorzugsweise 0,001
bis 5% einer betreffenden Verbindung, mehr vorzugsweise 0,01 bis
0,5%. Solche Zusammensetzungen beinhalten ebenfalls typischerweise
sichere und wirksame Mengen an Konservierungsmitteln, wie zum Beispiel
Benzalkoniumchlorid und Thimerosal; Puffer, wie zum Beispiel Phosphat
und Acetat; Tonizitätsmittel,
wie zum Beispiel Natriumchlorid; Antioxidanzien, wie zum Beispiel
Ascorbinsäure;
aromatische Stoffe; und Säuren
und Basen, um den pH dieser wässrigen
Zusammensetzungen wie benötigt
einzustellen.
-
Bevorzugte Zusammensetzungen der
vorliegenden Erfindung beinhalten wässrige Lösungen, Suspensionen und trockene
Pulver, umfassend eine sichere und wirksame Menge einer betreffenden
Verbindung, gedacht für
Atomisierung und topische Inhalations-Gabe. Solche Zusammensetzungen
umfassen vorzugsweise 0,1 bis 50% einer betreffenden Verbindung,
mehr vorzugsweise 1 bis 20%. Solche Zusammensetzungen sind typischerweise
in einem Behälter
mit aufgesetzten Atomisierungsmitteln enthalten. Solche Zusammensetzungen
beinhalten typischerweise auch Treibgase wie zum Beispiel Chlorfluorkohlenwasserstoffe
12/11 und 12/114; Lösungsmittel
wie zum Beispiel Wasser, Glycerol und Ethanol; Stabilisatoren wie
zum Beispiel Ascorbinsäure,
Natriummetabisulfit; Konservierungsmittel wie zum Beispiel Cetylpyridiniumchlorid
und Benzalkoniumchlorid; Tonizitätseinstellmittel
wie zum Beispiel Natriumchlorid; uns Aromastoffe wie zum Beispiel
Natriumsaccharin.
-
Bevorzugte Zusammensetzungen der
vorliegenden Erfindung beinhalten wässrige Lösungen umfassend eine sichere
und wirksame Menge einer betreffenden Verbindung, gedacht für eine topische
intraokulare Gabe. Solche Zusammensetzungen umfassen vorzugsweise
0,0001 bis 5% einer betreffenden Verbindung, mehr vorzugsweise 0,01
bis 0,5%. Solche Zusammensetzungen beinhalten typischerweise ebenfalls
ein oder mehrere Konservierungsmittel, wie zum Beispiel Benzalkoniumchlorid,
Thimerosal, Phenylquecksilberacetat; Vehikel, wie zum Beispiel Poloxamere,
modifizierte Cellulosen, Povidon und gereinigtes Wasser; Tonizitätseinstellmittel,
wie zum Beispiel Natriumchlorid, Mannitol und Glycerin; Puffer,
wie zum Beispiel Acetat, Citrat, Phosphat und Borat; Antioxidanzien,
wie zum Beispiel Natriummetabisulfit, butyliertes Hydroxytoluol
und Acetylcystein; Säuren
und Basen können
verwendet werden, um den pH dieser Formulierungen wie nötig einzustellen.
-
Bevorzugte Zusammensetzungen der
vorliegenden Erfindung beinhalten Feststoffe, wie zum Beispiel Tabletten
und Kapseln, und Flüssigkeiten,
wie zum Beispiel Lösungen,
Suspensionen und Emulsionen (vorzugsweise in Weichgelatinekapseln),
umfassend eine sichere und wirksame Menge einer betreffenden Verbindung,
gedacht für
topische Gabe in den Gastrointestinaltrakt durch perorale Gabe.
Solche Zusammensetzungen umfassen vorzugsweise 0,01 bis 100 mg pro
Dosis, mehr vorzugsweise 0,1 bis 5 mg pro Dosis. Solche Zusammensetzungen
können
durch herkömmliche
Verfahren beschichtet werden, typischerweise mit pH- oder zeitabhängigen Beschichtungen,
so dass die betreffende Verbindung im Gastrointestinaltrakt in der
Nähe der gewünschten
topischen Anwendung oder zu unterschiedlichen Zeiten freigesetzt
wird, um die gewünschte
Wirkung zu erweitern. Solche Dosierungsformen beinhalten typischerweise,
sind aber nicht darauf beschränkt, eins
oder mehrere aus Celluloseacetatphthalat, Polyvinylacetatphthalat,
Hydroxypropylmethylcellulosephthalat, Ethylcellulose, Eudragit®-Beschichtungen,
Wachsen und Schelllack.
-
Zusammensetzungen der vorliegenden
Erfindung können
wahlweise andere Arzneimittelwirkstoffe enthalten. Nicht beschränkende Beispiele
von Arzneimittelwirkstoffen, die in die betreffenden Zusammensetzungen
eingebaut werden können,
und typische Dosierungsmengen davon, beinhalten: Atmungs-Arzneiwirkstoffe:
klassische Antihistaminika, z. B., Chlorpheniramin von 1 mg bis
4 mg pro Dosis, und Diphenhydramin von 10 mg bis 50 mg pro Dosis;
nicht sedierende. Antihistaminika, z. B., Terfenadin von 30 mg bis
60 mg pro Dosis, Lorantadin von 5 mg pro Dosis bis 10 mg pro Dosis,
und Ceterizin von 5 mg pro Dosis bis 10 mg pro Dosis; Schleimlöser, z.
B., Guaifenesin von 100 mg bis 200 mg pro Dosis; Antitussiva, z.
B., Dextromethorphan von 5 mg bis 30 mg pro Dosis; und Analgetika,
z. B., Ibuprofen von 100 mg bis 800 mg pro Dosis; und Acetaminophen
von 80 mg bis 1000 mg pro Dosis; Augen-Arzneimittelwirkstoffe: Acetylcholinesterase-Hemmer,
z. B., Echothiophat von 0,03% bis 0,25% in topischer Lösung; und
gastrointestinale Wirkstoffe: Anti-Diarrhöe-Mittel, z. B., Loperamid
von 0,1 mg bis 1,0 mg pro Dosis, und Wismutsubsalicylat von 25 mg
bis 300 mg pro Dosis.
-
Verfahren
-
Ein anderer Aspekt der vorliegenden
Erfindung umfasst die Herstellung von Medikamenten zur Vermeidung
oder Behandlung nasaler Kongestion durch Gabe einer sicheren und
wirksamen Menge einer betreffenden Verbindung an einen Menschen
oder ein niederes Tier, die an nasaler Kongestion leiden oder dadurch gefährdet sind.
Eine solche nasale Kongestion kann einhergehen mit menschlichen
Krankheiten oder Funktionsstörungen,
die beinhalten, aber nicht beschränkt sind auf, saisonalen Heuschnupfen,
akute Virusinfektionen der oberen Atemwege, Sinusitis, chronische
Rhinitis, und vasomotorische Rhinitis. Jede Gabe einer Dosis der
betreffenden Verbindung appliziert vorzugsweise eine Dosis innerhalb
des Bereichs von 0,001 mg/kg und 10 mg/kg einer Verbindung, mehr
vorzugsweise von 0,01 mg/kg und 5 mg/kg. Noch mehr vorzugsweise
von 0,1 mg/kg und 1 mg/kg. Eine perorale Gabe solcher Dosen ist
bevorzugt. Die Häufigkeit
der Gabe einer betreffenden Verbindung gemäß der vorliegenden Erfindung
liegt vorzugsweise zwischen einmal und sechsmal am Tag, mehr vorzugsweise
zwischen zweimal und viermal am Tag. Solche Dosen und Häufigkeiten
sind auch bevorzugt für
die Behandlung anderer Atemzustände,
wie zum Beispiel Otitis media, Husten, COPD und Asthma.
-
Ein anderer Aspekt der vorliegenden
Erfindung umfasst die Herstellung von Medikamenten zur Vermeidung
oder Behandlung von Glaukom durch Gabe einer sicheren und wirksamen
Menge einer betreffenden Verbindung an einen Menschen oder ein niederes
Tier, die an Glaukom leiden oder dadurch gefährdet sind. Jede Gabe einer
Dosis der betreffenden Verbindung appliziert vorzugsweise eine Dosis
innerhalb des Bereichs von 0,01 μg/kg
und 10 mg/kg einer Verbindung, mehr vorzugsweise von 0,001 mg/kg
bis 1 mg/kg, noch mehr vorzugsweise von 0,01 mg/kg bis 0,1 mg/kg.
Eine intraokulare Gabe solcher Dosen ist bevorzugt. Die Häufigkeit
der Gabe einer betreffenden Verbindung gemäß der vorliegenden Erfindung
liegt vorzugsweise zwischen einmal und sechsmal am Tag, mehr vorzugsweise
zwischen zweimal und viermal am Tag.
-
Ein anderer Aspekt der vorliegenden
Erfindung umfasst die Herstellung von Medikamenten zur Vermeidung
oder Behandlung von funktionellen Darm-Funktionsstörungen,
wie zum Beispiel Diarrhöe,
durch Gabe einer sicheren und wirksamen Menge einer betreffenden
Verbindung an einen Menschen oder ein niederes Tier, die an Diarrhöe leiden
oder dadurch gefährdet
sind. Jede Gabe einer Dosis der betreffenden Verbindung appliziert
vorzugsweise eine Dosis innerhalb des Bereichs von 0,001 mg/kg und
10 mg/kg einer Verbindung, mehr vorzugsweise von 0,01 mg/kg bis
5 mg/kg, noch mehr vorzugsweise von 0,1 mg/kg bis 1 mg/kg. Eine
perorale Gabe solcher Dosen ist bevorzugt. Die Häufigkeit der Gabe einer betreffenden
Verbindung gemäß der vorliegenden
Erfindung liegt vorzugsweise zwischen einmal und sechsmal am Tag,
mehr vorzugsweise zwischen zweimal und viermal am Tag.
-
Zusammensetzungs-
und Verfahrensbeispiele
-
Die folgenden Beispiele illustrieren
die Zusammensetzungen und Anwendungsverfahren der vorliegenden Erfindung,
in denen „betreffende
Verbindung" eine
5-(2-Imidazolinylamino)benzimidazol-Verbindung gemäß der Erfindung
bezeichnet.
-
Beispiel
A
Orale Tabletten-Zusammensetzung
| Bestandteil | Menge
pro Tablette (mg) |
| Betreffende
Verbindung | 20,0 |
| Mikrokrstalline
Cellulose (Avicel PH 102®) | 80,0 |
| Dicalciumphosphat | 96,0 |
| Pyrogenes
Silizium (Cab-O-Sil®) | 1,0 |
| Magnesiumstearat | 3,0 |
| Gesamt
= | 200,0 |
-
Eine Tablette wird von einem Patienten
mit nasaler Kongestion geschluckt. Die Kongestion wird wesentlich
verringert.
-
Beispiel
B
Kautabletten-Zusammensetzung
| Bestandteil | Menge
pro Tablette (mg) |
| Betreffende
Verbindung | 15,0 |
| Mannitol | 255,6 |
| Mikrokristalline
Cellulose (Avicel PH 101®) | 100,8 |
| Dextrinierte
Sucrose (Di-Pac®) | 199,5 |
| Künstliches
Orangenaroma | 4,2 |
| Natriumsaccharin | 1,2 |
| Stearinsäure | 15,0 |
| Magnesiumstearat | 3,0 |
| FD&C Gelb #6-Farbstoff | 3,0 |
| Pyrogenes
Silizium (Cab-O-Sil®) | 2,7 |
| Gesamt
= | 600,0 |
-
Eine Tablette wird von einem Patienten
mit nasaler Kongestion gekaut und geschluckt, Die Kongestion wird
wesentlich verringert.
-
Beispiel
C
Sublinguale Tabletten-Zusammensetzung
| Bestandteil | Menge
pro Tablette (mg) |
| Betreffende
Verbindung | 2,00 |
| Mannitol | 2,00 |
| Mikrokristalline
Cellulose (Avicel PH 101®) | 29,00 |
| Minzaromastoffe | 0,25 |
| Natriumsaccharin | 0,08 |
| Gesamt
= | 33,33 |
-
Eine Tablette wird unter die Zunge
eines Patienten mit nasaler Kongestion platziert und sich auflösen gelassen.
Die Kongestion wird schnell und wesentlich reduziert.
-
Beispiel
D
Intranasale Lösungs-Zusammensetzung
| Bestandteil | Zusammensetzung
(% Gew./Vol.) |
| Betreffende
Verbindung | 0,20 |
| Benzalkoniumchlorid | 0,002 |
| d-Sorbitol | 5,0 |
| Glycin | 0,35 |
| Aromatische
Stoffe | 0,075 |
| Gereinigtes
Wasser | q.
s. |
| Gesamt
= | 100,00 |
-
1/10 ml der Zusammensetzung wurde
aus einem Pumpaktuator in jedes Nasenloch eines Patienten mit nasaler
Kongestion gesprüht.
Die Kongestion wird wesentlich reduziert.
-
Beispiel
E
Intranasale Gel-Zusammensetzung
| Bestandteil | Zusammensetzung
(% Gew./Vol.) |
| Betreffende
Verbindung | 0,10 |
| Benzalkoniumchlorid | 0,02 |
| Thimerosal | 0,002 |
| Hydroxypropylmethylcellulose | 1,00 |
| (Metolose
65SH4000®)
Aromatische Stoffe | 0,06 |
| Natriumchlorid
(0,65%) | q.
s. |
| Gesamt
= | 100,00 |
-
1/5 ml der Zusammensetzung werden
als Tropfen aus einem Tropfer in jedes Nasenloch eines Patienten
mit nasaler Kongestion appliziert. Die Kongestion wird wesentlich
verringert.
-
Beispiel
F
Inhalations-Aerosol-Zusammensetzung
| Bestandteil | Zusammensetzung
(% Gew./Vol.) |
| Betreffende
Verbindung | 5,0 |
| Alkohol | 33,0 |
| Ascorbinsäure | 0,1 |
| Menthol | 0,1 |
| Natriumsaccharin | 0,2 |
| Treibgas
(F12, F114) | q.
s. |
| Gesamt
= | 100,0 |
-
Zwei Stöße der Aerosol-Zusammensetzung
werden aus einem Inhalator mit Dosierungsmessung durch einen Patienten
mit Asthma inhaliert. Der asthmatische Zustand wird wirksam abgebaut.
-
Beispiel
G
Topische ophtalmische Zusammensetzung
| Bestandteil | Zusammensetzung
(% Gew./Vol.) |
| Betreffende
Verbindung | 0,10 |
| Benzalkoniumchlorid | 0,01 |
| EDTA | 0,05 |
| Hydroxylethylcellulose
(Natrosol M®) | 0,50 |
| Natriumunetabisulfit | 0,10 |
| Natriumchlorid
(0,9%) | q.
s. |
| Gesamt
= | 100,0 |
-
1/10 ml der Zusammensetzung wird
direkt in jedes Auge eines Patienten mit Glaukom verabreicht.
-
Der Augeninnendruck wird wesentlich
reduziert.
-
Beispiel
H
Orale flüssige
Zusammensetzung
| Bestandteil | Menge/15
ml Dosis |
| Betreffende
Verbindung | 15
mg |
| Chlorpheniraminmaleat | 4
mg |
| Propylenglykol | 1,8
g |
| Ethanol
(95%) | 1,5
ml |
| Methanol | 12,5
ml |
| Eukalyptusöl | 7,55
mg |
| Aromastoffe | 0,05
ml |
| Sucrose | 7,65
g |
| Cyrboxymethylcellulose
(CMC) | 7,5
mg |
| Mikrokristalline
Cellulose und | 187,5
mg |
| Natrium-CMC
(Avicel RC 591®)
Polysorbat 80 | 3,0
mg |
| Glycerin | 300
mg |
| Sorbitol | 300
mg |
| FD&C Rot #40 Farbstoff | 3
mg |
| Natriumsaccharin | 22,5
mg |
| Natriumphosphat
monobasisch | 44
mg |
| Natriumcitrat-Monohydrat | 28
mg |
| Gereinigtes
Wasser | q.
s. |
| Gesamt
= | 15 ml |
-
1/15 ml der flüssigen Zusammensetzung wird
von einem Patienten mit nasaler Kongestion und laufender Nase aufgrund
von Heuschnupfen geschluckt. Die Kongestion und die laufende Nase
werden wirksam verringert.
-
Beispiel
7
Orale flüssige
Zusammensetzung
| Bestandteil | Menge/15
ml Dosis |
| Betreffende
Verbindung | 30
mg |
| Sucrose | 8,16
g |
| Glycerin | 300
mg |
| Sorbitol | 300
mg |
| Methylparaben | 19,5
mg |
| Propylparaben | 4,5
mg |
| Menthol | 22,5
mg |
| Eukalyptusöl | 7,5
mg |
| Aromastoffe | 0,07
ml |
| FD&C Rot #40 Farbstoff | 3,0
mg |
| Natriumsaccharin | 30
mg |
| Gereinigtes
Wasser | q.
s. |
| Gesamt
= | 15 ml |
-
1/15 ml der alkoholfreien flüssigen Medikation
wird von einem Patienten mit nasaler Kongestion geschluckt. Die
Kongestion wird wesentlich verringert.