[0001] O presente pedido de patente diz respeito a um novo processo para a preparação de derivados de aminoácido.
[0002] Em particular, o presente pedido diz respeito a um processo melhorado para a fabricação de Lacosamida (LCM), (R)-2-acetamido-N-benzil-3-metoxipropion- amida, que é útil como um medicamento anticonvulsivo.
[0003] A LCM tem demonstrado eficácia antiepiléptica em modelos de convulsão de roedor diferentes e potencial antinociceptivo em modelos de animal experimentais que refletem tipos e sintomas distintos de dor neuropática assim como dor inflamatória crônica.
[0004] A Patente US 5.378.729 descreve a preparação de aminoácidos funcionalizados pela reação de aminas com derivados de acetilação de um ácido carboxílico sob as condições de formação de amida. A Patente US 5.378.729 é entretanto silenciosa sobre a preparação direta de um enantiômero único de aminoácidos funcionalizados, tais como Lacosamida.
[0005] A Patente US 5.773.475 diz respeito a métodos de preparação de Lacosamida ‘opticamente pura de modo substancial’, como aí definida, partindo de D-Serina. O dito método de preparação envolve o uso de iodeto de metila e óxido de prata (I) como agente de O-metilação que apresenta as desvantagens de ser caro e leva à racemização parcial do produto que passa pela O-metilação. Esta é uma desvantagem principal em termos de produtividade industrial do processo.
[0006] A Patente US 6.048.899 descreve variantes do processo descrito na Patente US 5.773.475.
[0007] O pedido de patente internacional publicado como WO 2006/037574 diz respeito a uma via de síntese melhorada para a Lacosamida em que um agente de O-metilação alternativo ao iodeto de metila e óxido de prata (I) é usado, em particular sulfato de dimetila.
[0008] Entretanto o uso de um excesso de sulfato de dimetila como descrito na WO 2006/037574 pode levar a problemas de segurança ou ambientais quando da produção de Lacosamida em uma larga escala. Além disso, o uso de etapas de proteção de N/desproteção de N da porção de amina pode levar a problemas de custo e produtividade para a produção industrial do processo global.
[0009] Existe portanto uma necessidade para encontrar um processo alternativo e melhorado para a fabricação de Lacosamida que seja competitivo, mais eficiente em custo, leve a uma produtividade aumentada e não apresente as desvantagens maiores em termos de segurança e/ou ambiente.
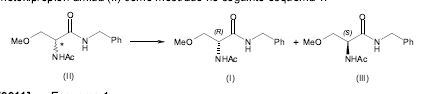
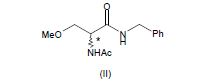
[0010] Em um primeiro aspecto, a presente invenção diz respeito a um processo para a fabricação de (R)-2-acetamido-N-benzil-3-metoxipropion-amida (I) opticamente enriquecido que compreende a resolução da 2-acetamido-N-benzil-3- metoxipropion-amida (II) como mostrado no seguinte esquema 1.
[0011] Esquema 1
[0012] O termo “opticamente enriquecido” como aqui usado quando da alusão a um composto particular significa que mais do que 50 %, preferivelmente mais do que 75 %, mais preferivelmente mais do que 85 %, o mais preferivelmente mais do que 94 % do composto tem o centro estereogênico indicado por (*) em uma dada configuração (R) ou (S).
[0013] Portanto, a expressão “(R)-2-acetamido-N-benzil-3-metóxi-propion-amida opticamente enriquecido” significa que mais do que 50 %, preferivelmente mais do que 75 %, mais preferivelmente mais do que 85 %, o mais preferivelmente mais do que 94 % do composto tem o centro estereogênico indicado por (*) na configuração(R).
[0014] Em um segundo aspecto, a presente invenção diz respeito a um processo para a fabricação de (R)-2-acetamido-N-benzil-3-metoxipropion-amida (I) de modo substancial opticamente puro que compreende a resolução de 2-acetamido-N-benzil- 3-metoxipropion-amida (II) como mostrado no esquema 1.
[0015] O termo “de modo substancial opticamente puro” como aqui usado quando da alusão a um composto particular significa que pelo menos 95 %, preferivelmente pelo menos 96 %, mais preferivelmente pelo menos 97 %, o mais preferivelmente pelo menos 98 %, ainda o mais preferivelmente pelo menos 99 % do composto tem o centro estereogênico indicado por (*) em uma dada configuração (R) ou (S).
[0016] Portanto, a expressão “(R)-2-acetamido-N-benzil-3-metóxi-propion-amida de modo substancial opticamente puro” significa que pelo menos 95 %, preferivelmente pelo menos 96 %, mais preferivelmente pelo menos 97 %, o mais preferivelmente pelo menos 98 %, ainda o mais preferivelmente pelo menos 99 % do composto tem o centro estereogênico indicado por (*) na configuração (R).
[0017] O termo “resolução” como aqui usado refere-se à separação de uma mistura de enantiômeros nos seus enantiômeros individuais correspondentes. Os enantiômeros podem estar presentes na mistura em várias razões de enantiômero versus o outro. As razões típicas de enantiômeros de acordo com a presente invenção variam de cerca de 3/97 a 97/3, preferivelmente de cerca de 5/95 a 95/5, mais preferivelmente de cerca de 30/70 a 70/30, o mais preferivelmente de cerca de 40/60 a 60/40, ainda mais preferivelmente de cerca de 45/55 a 55/45.
[0018] Particularmente, a mistura é uma mistura racêmica. Uma mistura racêmica como aqui definida é uma mistura que compreende 50 % de um enantiômero e 50 % do outro enantiômero.
[0019] A resolução pode ser obtida por vários métodos incluindo a conversão aos diastereoisômeros, absorção diferencial, reconhecimento quiral, processos bioquímicos, separação mecânica, resolução cinética e desracemização como detalhado em Jerry March “Advanced Organic Chemistry”, quarta edição, Capítulo 4, páginas 120 a 125.
[0020] Preferivelmente, a resolução de acordo com a presente invenção é obtida pelo método de separação diferencial, mais preferivelmente pela separação cromatográfica quiral usando colunas empacotadas com uma fase estacionária quiral (CSP) e uma fase móvel.A separação cromatográfica quiral pode ser realizada em lote ou pela Cromatografia em Coluna Múltipla (MCC).
[0021] O termo “Cromatografia em Coluna Múltipla” (MCC) como aqui usado refere- se a uma tecnologia de separação cromatográfica contínua com base na injeção controlada contínua de misturas em uma série de colunas ligadas empacotada com uma fase estacionária. Os componentes separados da mistura são depois retirados continuamente do sistema. Este método incluiria, mas não é limitado ao, modo de cromatografia de Leite Movediço Simulado (modo SMB), ou modo onde os orifícios de entrada e saída são mudados assincronicamente (tal como o modo Varicol) ou modo em que as taxas de fluxo e/ou concentrações de entrada e saída são trocadas no tempo durante o período de comutação.
[0022] A aplicação da técnica de SMB para a resolução enantiomérica de misturas racêmicas, por exemplo, foi descrita no artigo “Lit mobile simulé. Application à la séparation d’isomères optiques [Leito móvel simulado.Aplicação para a separação de isômeros ópticos]” por R. M. Nicoud, Information Chimie N° 368 (Maio de 1995), pp. 113-115.
[0023] O sistema Varicol é descrito no pedido de patente internacional WO 00/25885 e o modo no qual as taxas de fluxo de fluido são mudadas no tempo durante o período de comutação é descrito na Pat. U.S. N° 5.102.553.
[0024] Consequentemente, em uma forma de realização particular de acordo com a presente invenção, a resolução de 2-acetamido-N-benzil-3-metoxipropion- amida (II) é realizada pela separação cromatográfica quiral. Em uma outra forma de realização particular, a resolução de 2-acetamido-N-benzil-3-metoxipropion-amida (II) é realizada pela MCC.
[0025] As colunas usadas de acordo com a presente invenção são no geral empacotadas com um CSP que compreende uma cadeia principal de sílica na qual um seletor quiral polimérico é revestido de acordo com técnicas bem conhecidas no ramo.
[0026] O seletor quiral polimérico pode ser adicionalmente imobilizado sobre a cadeia principal de sílica que fornece à coluna, entre outras vantagens, uma melhor resistência aos solventes.
[0027] Em uma forma de realização particular de acordo com a presente invenção, a separação cromatográfica quiral de 2-acetamido-N-benzil-3- metoxipropion-amida (II) é realizada usando uma coluna que compreende um seletor quiral polimérico que é imobilizado sobre a cadeia principal de sílica.
[0028] O seletor quiral polimérico de acordo com a presente invenção no geral compreende um polissacarídeo, por exemplo amilose ou celulose.
[0029] Os exemplos de seletor quiral polimérico que podem ser usados de acordo com a presente invenção são tris(4-metilbenzoato) de celulose, tribenzoato de celulose, tris(3,5-dimetilfenilcarbamato) de amilose, tris(3,5-dimetilfenilcarbamato) de celulose, tris(4-metilfenil-carbamato) de celulose, tris (3,5-diclorofenilcarbamato) de celulose, tris [(S)-a-metilbenzilcarbamato] de amilose e tris(3-cloro-4-metilfenil-carbamato) de celulose.
[0030] Em uma forma de realização particular de acordo com a presente invenção, a separação cromatográfica quiral de 2-acetamido-N-benzil-3- metoxipropion-amida (II) nos seus enantiômeros é realizada usando tris (3,5- diclorofenilcarbamato) de celulose imobilizado sobre uma cadeia principal de sílica como fase estacionária quiral.
[0031] Os exemplos de fase móvel que pode ser usada de acordo com a presente invenção são alcanos, tais como heptano, hexano, alcoóis, tais como metanol, etanol, iso-propanol, n-propanol, acetonitrila, acetato de isopropila, acetato de etila, diclorometano, clorofórmio, éteres, tais como éter metil t-butílico (MTBE), ou misturas dos mesmos.
[0032] Se misturas de solventes são usadas, a razão dependerá do tipo de solventes que constituem a mistura, do tipo de coluna que é usado e da solubilidade naquelas misturas do composto a ser separado.
[0033] Os exemplos de misturas de solventes de acordo com a presente invenção são misturas de diclorometano e um álcool ou misturas que compreendem acetonitrila e um álcool ou misturas que compreendem acetato de etila e um álcool.
[0034] Preferivelmente, as misturas de diclorometano e um álcool compreendem entre 90 % e 99 % de diclorometano.
[0035] Preferivelmente as misturas de acetonitrila e um álcool compreendem entre 90 % e 99 % de acetonitrila.
[0036] Preferivelmente, as misturas de acetato de etila e álcool compreendem entre 90 % e 99 % de acetato de etila.
[0037] Os solventes preferidos de acordo com a presente invenção são etanol, metanol n-propanol, iso-propanol, acetonitrila, diclorometano e acetato de etila.
[0038] Em uma forma de realização particular de acordo com a presente invenção, uma mistura de acetato de etila e metanol é usada como fase móvel. Em uma outra forma de realização de acordo com a presente invenção, uma mistura de acetato de etila e metanol em uma razão de 90/10 v:v é usada como fase móvel.
[0039] Em uma outra forma de realização particular de acordo com a presente invenção, acetonitrila é usada como fase móvel.
[0040] De acordo com a presente invenção, uma produtividade da separação cromatográfica quiral maior do que 1 Kg de mistura racêmica separada por Kg de fase estacionária quiral por dia é obtida.
[0041] Em uma forma de realização particular de acordo com a presente invenção, uma produtividade da resolução cromatográfica quiral maior do que 2 Kg de mistura racêmica separada por Kg de fase estacionária quiral por dia é obtida.
[0042] Como mostrado no esquema 2, em um segundo aspecto, a presente invenção diz respeito a um processo para a fabricação de (R)-2-acetamido-N-benzil- 3-metoxipropion-amida (I) de modo substancial opticamente puro que compreende: (a)resolução de 2-acetamido-N-benzil-3-metoxipropion-amida (II) em (R)-2-acetamido-N-benzil-3-metoxipropion-amida (I) e (S)-2- acetamido-N-benzil-3-metoxipropion-amida (III); (b)Racemização de (S)-2-acetamido-N-benzil-3-metoxipropion- amida (III); e (c)resolução adicional da 2-acetamido-N-benzil-3-metoxipropion- amida (II) resultante.
[0043] Esquema 2
[0044] As etapas (a) e (c) são no geral realizadas pela separação cromatográfica quiral, preferivelmente pela MCC.
[0045] A (R)-2-acetamido-N-benzil-3-metoxipropion-amida (I) obtida nas etapas (a) e (c) pode ser opticamente enriquecida ou opticamente pura de modo substancial. No geral, a (R)-2-acetamido-N-benzil-3-metoxipropion-amida (I) obtida nas etapas (a) e (c) é opticamente pura de modo substancial. Isto é particularmente vantajoso, visto que evita o uso de etapas de purificação iterativas, tais como cristalização que impactaria na produtividade do processo global.
[0046] A (S)-2-acetamido-N-benzil-3-metoxipropion-amida (III) obtida na etapa (a) pode ser opticamente enriquecida ou opticamente pura de modo substancial.
[0047] O termo “Racemização” como aqui usado refere-se à transformação de um enantiômero opticamente enriquecido ou um enantiômero de modo substancial opticamente puro em uma mistura consistindo do dito enantiômero e do outro enantiômero, até uma mistura racêmica.
[0048] A etapa (b) pode ser tipicamente obtida pela reação da (S)-2-acetamido- N-benzil-3-metoxipropion-amida (III) opticamente enriquecida ou opticamente pura de modo substancial com uma base, com ou sem acidificação do meio.
[0049] Os exemplos de bases que podem ser usadas de acordo com a presente invenção são metóxido de sódio, hidróxido de potássio, hidróxido de sódio, carbonato de potássio, carbonato de sódio, aminas terciárias, tais como trietilamina, 1,8-Diazabiciclo[5,4,0]undec-7-eno e resinas de troca aniônica forte ou fracamente básicas, tais como AMBERLYST® A21, AMBERLITE® IRA400 ou IRA410, e outros.
[0050] As bases preferidas de acordo com a presente invenção são metóxido de sódio, hidróxido de potássio, hidróxido de sódio, carbonato de potássio e carbonato de sódio.
[0051] Quando a acid if i cação do meio é feita, a mesma é preferivelmente realizada usando uma quantidade estequiométrica de ácido com respeito à base para evitar a formação de quaisquer produtos de degradação.
[0052] O dito processo global é particularmente vantajoso visto que o mesmo evita qualquer perda de produtividade que pode algumas vezes ocorrer quando do uso de uma etapa de resolução. De fato, frequentemente o enantiômero indesejado é um subproduto do processo que necessita ser eliminado do meio de reação. Pela reciclagem do enantiômero indesejado e realização de uma resolução adicional, por exemplo via separação pela cromatografia quiral, o rendimento e a produtividade globais do processo são aumentados.
[0053] As etapas (b) e (c) podem ser repetidas para aumentar ainda mais o rendimento global do processo.
[0054] Em uma forma de realização particular de acordo com a invenção, metóxido de sódio é usado como a base. A Racemização é no geral realizada em um solvente em uma temperatura compreendida entre 20°C e 80°C, preferivelmente em uma temperatura compreendida entre 40°C e 60°C. Mais preferivelmente, a reação é realizada em uma temperatura mais baixa do que 60°C de modo a evitar a formação de produtos de degradação.
[0055] Os exemplos de solventes que podem ser usados para a etapa (b) são álcool, tal como metanol, etanol, éteres, tais como tetra-hidrofurano, 2-metil-tetra- hidrofurano ou acetonitrila. Em uma forma de realização particular de acordo com a presente invenção, o solvente é metanol.
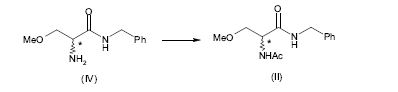
[0056] A 2-acetamido-N-benzil-3-metoxipropion-amida (II) pode ser preparada pela acetilação de 2-amino-N-benzil-3-metoxipropion-amida (IV) de acordo com métodos conhecidos pela pessoa habilitada na técnica, como mostrado no seguinte esquema 3.
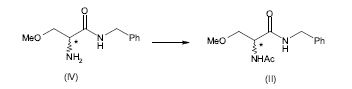
[0057] Esquema 3
[0058] Preferivelmente, a acetilação pode ser realizada usando um agente de acetilação tal como anidrido acético ou cloreto de acetila.
[0059] Um agente de acetilação particularmente preferido de acordo com a presente invenção é o anidrido acético.
[0060] A acetilação é no geral realizada em um solvente em uma temperatura compreendida entre 20°C e 70°C. Os exemplos de solventes que podem ser usados para a acetilação são diclorometano, tetra-hidrofurano, acetato de etila, acetato de isopropila e acetato de isobutila.
[0061]Os solventes preferidos são acetato de isopropila e acetato de isobutila.
[0062] A acetilação é preferivelmente realizada em uma temperatura compreendida entre 50°C e 70°C. Mais preferivelmente, a acetilação é realizada em uma temperatura de cerca de 60°C.
[0063] A 2-amino-N-benzil-3-metoxipropion-amida (IV) pode ser preparada de acordo com o método descrito no esquema 1 da Patente US 6.048.899, aqui incorporada como referência, partindo de serina racêmica, ou de acordo com qualquer outro método conhecido pela pessoa habilitada na técnica.
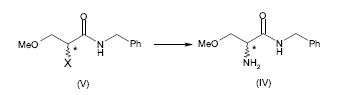
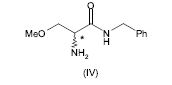
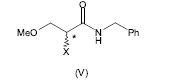
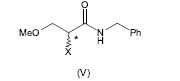
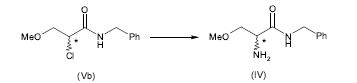
[0064] Alternativamente, a 2-amino-N-benzil-3-metoxipropion-amida (IV) pode ser preparada pela amonólise do composto (V), em que X é um grupo de partida, de acordo com o seguinte esquema 4.
[0065] Esquema 4
[0066] O termo “grupo de partida” como aqui usado tem o mesmo significado como descrito na técnica (Advanced Organic Chemistry: reactions, mechanisms and structure - Terceira Edição por Jerry March, John Wiley and Sons Ed.; 1985 página 179), isto é, o mesmo representa um grupo que é parte de e ligado a uma molécula de substrato e que em uma reação onde a molécula de substrato sofre uma reação de deslocamento (com por exemplo um nucleófilo), o mesmo (o grupo de partida) é depois deslocado.
[0067] Os exemplos de um grupo de partida de acordo com a presente invenção são grupos halogênio ou sulfonato.
[0068] O termo “grupo sulfonato” como aqui usado representa um grupo da fórmula -O-SO2-Ra em que Ra é um alquila ou um arila. Os grupos sulfonato preferidos são metanossulfonato, grupo para-toluenossulfonato ou trifluorometanossulfonato.
[0069] Preferivelmente, o grupo de partida X no composto da fórmula (V) é um halogênio, mais preferivelmente bromo ou cloro. O mais preferivelmente, X é bromo.
[0070] A reação de amonólise pode ser realizada de acordo com métodos conhecidos na técnica. Por exemplo, a amonólise pode ser realizada de acordo com o método descrito no pedido de patente internacional publicado sob a WO 03/014080.
[0071] A amonólise de acordo com a presente invenção é preferivelmente realizada com amónia aquosa na presença de metanol.
[0072] Em uma forma de realização particular de acordo com a presente invenção um excesso de amónia com respeito ao composto (V) é usado de modo na evitar a formação de impurezas de amina secundária. Por exemplo, de 20 a 25 equivalentes molares de amónia com respeito ao composto (V) são usados.
[0073] Depois da reação de amonólise, a 2-amino-N-benzil-3-metoxipropion- amida (IV) é tipicamente extraída do meio de reação com um solvente. Os exemplos de solventes que podem ser usados para a dita extração são acetato de isobutila, acetato de isopropila, acetato de propila, acetato de etila, 2-metil-tetra-hidrofurano, diclorometano e tolueno.
[0074] Preferivelmente acetato de isobutila é usado como solvente para a extração de 2-amino-N-benzil-3-metoxipropion-amida (IV). A extração com acetato de isobutila é preferivelmente realizada em um pH mais alto do que 10 e mais baixo do que 12, mais preferivelmente em um pH entre 11 e 12 de modo a aumentar o rendimento da isolação de 2-amino-N-benzil-3-metoxipropion-amida (IV).
[0075] Em uma outra forma de realização de acordo com a presente invenção, 2-amino-N-benzil-3-metoxipropion-amida (IV) pode ser obtida pela realização da síntese de Gabriel no composto (V), em que o grupo de partida é um halogênio. As condições de reação típicas de acordo com esta forma de realização compreendem reagir o composto da fórmula (V) com ftalimida de potássio e reagir ainda o intermediário deste modo formado com hidrazina em etanol como mostrado noseguinte esquema 5.
[0076] Esquema 5
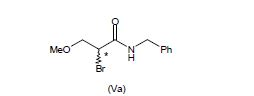
[0077] O composto da fórmula (V) pode ser sintetizada de acordo com vários métodos conhecidos na técnica. Por exemplo, a 2-bromo-N-benzil-3-metoxipropion- amida (Va) pode ser obtida pela aplicação do método descrito em Bioorganic and Medicinal Chemistry 2004, 3079.
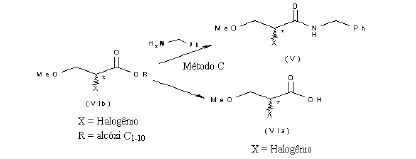
[0078] Alternativamente, os compostos da fórmula (V) de acordo com a presente invenção pode ser preparada pela reação do composto da fórmula (VI), em que X é um grupo de partida como acima definido e Y é hidróxi ou alcóxi C1-10, com benzilamina, de acordo com o seguinte esquema 6.
[0079] Esquema 6
[0080]O termo “hidróxi”, como aqui usado, representa um grupo da fórmula -OH.
[0081]O termo “alcóxi”, como aqui usado, representa um grupo da fórmula -ORb em que Rb é alquila C1-10 como definido acima.
[0082] O termo “alquila”, como aqui usado, é um grupo que representa radicais de hidrocarboneto saturados, monovalentes tendo porções retas (não ramificadas), ramificadas ou cíclicas, ou combinações destas.
[0083]As condições de reação dependem da natureza dos grupos X e Y.
[0084] O composto da fórmula (VI), em que X é um halogênio e Y é um grupo hidróxi, aqui depois aludido como composto da fórmula (Via), pode ser convertido in situ a um anidrido misto (Villa) (Método A), em que R é alquila C1-10 ou a um cloreto ácido (Vlllb) (Método B), cujos intermediários depois reagem com benzilamina, como mostrado no seguinte esquema 7.
[0085] De acordo com o Método A, o composto da fórmula (Via), em que X é cloro ou bromo, é reagido com um halo formiato de alquila, por exemplo cloroformiato de etila ou cloroformiato de isobutila, a uma temperatura compreendida entre -10°C e 10°C, seguido pela adição de uma base, para produzir o anidrido misto correspondente (Villa) que não é isolado do meio de reação. O anidrido misto (Villa) é reagido com benzilamina em um solvente em uma temperatura compreendida entre -10°C e 10°C. Preferivelmente a dita temperatura é compreendida entre -5°C e 0°C.Os exemplos de solvente que podem ser usados de acordo com esta forma de realização são diclorometano, acetato de etila, acetato de isobutila, tetra-hidrofurano, tolueno, acetato de propila, acetato de isopropila. Em uma forma de realização particular da presente invenção, acetato de isobutila ou tolueno são usados como um solvente.
[0086] Os exemplos de bases adequadas de acordo com a presente forma de realização são trietilamina, piridina, N-metilmorfolina, base de hüning. Em uma forma de realização preferida da invenção, a base é N-metilmorfolina.
[0087] Os compostos da fórmula (Via) que podem ser usado como material de partida no Método A são o ácido 2-cloro-3-metóxi-propiônico (Vlc) ou o ácido 2- bromo-3-metóxi-propiônico (Vld).
[0088] O ácido 2-cloro-3-metóxi-propiônico (Vlc) é comercialmente disponível de vários fornecedores.
[0089] O ácido 2-bromo-3-metóxi-propiônico (Vld) é facilmente obtenível pela reação do ácido 2,3-bromo-propiônico, ou ésteres de alquila correspondentes, com metóxido de sódio em metanol, em rendimentos de mais do que 85 %. Um método para produzir o ácido 2-bromo-3-metóxi-propiônico (Vld) é por exemplo descrito por L. L. Wood & V. du Vigneaud, J. Biol. Chemistry, 1940, 134, 413.
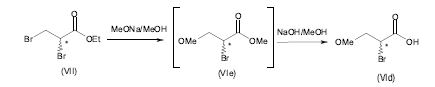
[0090] Em uma forma de realização particular de acordo com a presente invenção, o ácido 2-bromo-3-metóxi-propiônico (Vld) pode ser obtido partindo de propionato de 2,3-dibromo etila comercialmente disponível (Vila) ou propionato de 2,3-dibromo metila (Vllb). O propionato de 2,3-dibromo etila (Vila) ou propionato de 2,3-dibromo metila (Vllb) é reagido com metóxido de sódio em um solvente orgânico, preferivelmente metanol, a uma temperatura mais baixa do que 10°C, preferivelmente mais baixa do que 0°C, produzindo o propionato de 2-bromo-3- metóxi-metila (Vle) que é reagido ainda in situ com hidróxido de sódio, em uma temperatura compreendida entre 0°C e 25°C, preferivelmente em uma temperatura compreendida entre 20°C e 25°C, para produzir o ácido 2-bromo-3-metóxi- propiônico (Vld), depois da acidificação da mistura com ácido clorídrico aquoso, em um rendimento compreendido entre 80 % e 90 %, de acordo com o seguinte esquema 8.
[0091] Esquema 8
[0092] Preferivelmente uma quantidade estequiométrica de hidróxido de sódio com respeito ao propionato de 2-bromo-3-metóxi-metila (Vle) é usado de modo a evitar a formação de produtos de degradação.
[0093] Consequentemente, tanto o ácido 2-cloro-3-metóxi-propiônico (Vle) quanto o ácido 2-bromo-3-metóxi-propiônico (Vld) são facilmente obteníveis a partir de materiais de partida baratos que podem ser usados no processo de acordo com a invenção.
[0094] O ácido 2-bromo-3-metóxi propiônico (Vld) é particularmente vantajoso visto que ele produz o composto correspondente da fórmula (V) em rendimento mais alto e pureza mais alta do que o ácido 2-cloro-3-metóxi-propiônico (Vle) correspondente.
[0095] De acordo com o Método B, os compostos da fórmula (Via), em que X é cloro ou bromo, pode ser convertido no seu cloreto ácido correspondente de acordo com métodos padrão conhecidos pela pessoa habilitada na técnica, ou pela reação com um composto selecionado do grupo que consiste de cloreto de tionila, cloreto de oxalila, tricloreto de fósforo, pentacloreto de fósforo, oxicloreto de fósforo, dicloreto fenilfosfônico e N-clorossuccinimida, preferivelmente com cloreto de tionila.
[0096] O dito Método B pode ser realizado em um solvente selecionado de diclorometano, tolueno, acetato de isobutila ou acetato de isopropila em uma temperatura que compreende entre 20°C e 60°C.
[0097] O exemplo de composto (Via) que pode ser usado para o Método B é o ácido 2-cloro-3-metóxi-propiônico (Vlc).
[0098] O composto da fórmula (VI), em que X é um halogênio e Y é um grupo alcóxi Ci-10, aqui depois aludido como composto da fórmula (Vlb), pode ser convertido no composto correspondente da fórmula (V) pela reação com benzilamina (Método C), saponificado no ácido correspondente (Via) que pode depois sofrer qualquer uma das transformações (Métodos A & B) mencionados no esquema 7 acima, como mostrado aqui abaixo no esquema 9.
[0099] Esquema 9
[00100] O Método A é particularmente preferido em relação aos Métodos B e C visto que o mesmo produz o composto (V) em rendimentos mais altos e com uma qualidade melhorada.
[00101] Consequentemente, em uma forma de realização, a 2-acetamido-N- benzil-3-metoxipropion-amida (II) de acordo com a presente invenção pode ser fabricada por um processo que compreende as seguintes etapas:reagir um composto da fórmula (Via)
em que X é um grupo de partida, com um haloformiato de alquila na presença de uma base e benzilamina; (II)realizar a amonólise do composto (V) que resulta da etapa (i), em que X é como definido no composto (Via);
(III)acetilar o composto da fórmula (IV) deste modo obtido o
com anidrido acético em um solvente; (iv) isolar o composto da fórmula (II).
[00102] A Etapa (i) é preferivelmente realizada pela reação do composto (Via) com cloroformiato de etila ou cloroformiato de isobutila, seguido pela adição de N- Metil-Morfolina, que resulta na formação in situ do composto (Villa).
[00103] Consequentemente em uma forma de realização particular de acordo com a presente invenção, o composto da fórmula (Villa), em que X é halogênio e R é um alquila C1-10 é formado in situ, como mostrado no esquema 7 acima.
[00104] O composto da fórmula (Villa) formado in situ reage então com benzilamina para produzir o composto da fórmula (V).
[00105] A reação da etapa (i) é realizada em uma temperatura compreendida entre -10°C e 10°C.Preferivelmente a dita temperatura é compreendida entre -5°C e 0°C.
[00106] A Etapa (ii) é preferivelmente realizada tratando-se o composto (V) com um excesso molar de amónia aquosa na presença de metanol. Mais preferivelmente, de 20 a 25 equivalentes molares de amónia com respeito ao composto (V) são usados.
[00107] A Etapa (iii) é preferivelmente realizada usando anidrido acético como agente de acetilação e está em uma temperatura compreendida entre 50°C e 70°C. Mais preferivelmente, a acet ilação é realizada em uma temperatura de cerca de 60°C.
[00108] Alternativamente, o composto da fórmula (V) pode ser sintetizada pela reação do composto da fórmula (Vila) ou (Vllb) sob as condições mencionadas acima para a conversão de (Vila) ou (Vllb) em (Vld), seguido pela reação com benzilamina, sem a isolação do composto intermediário (VI).
[00109] Isto fornece uma vantagem em termos de rendimento e produtividade visto que o processo global tem uma etapa menos. Os exemplos de compostos da fórmula (V) de acordo com a presente invenção são 2-bromo-N-benzil-3- metoxipropion-amida (Va) e 2-cloro-N-benzil-3-metoxipropion-amida (Vb).
[00110] Os exemplos de compostos da fórmula (VI) de acordo com a presente invenção são o ácido 2-bromo-3-metóxi propiônico (Vld), o éster metílico do ácido 2- bromo-3-metóxi propiônico (Vle), éster etílico do ácido 2-bromo-3-metóxi propiônico, ácido 2-cloro-3-metóxi propiônico (Vle), éster metílico do ácido 2-cloro-3-metóxi propiônico, éster etílico do ácido 2-cloro-3-metóxi propiônico e cloreto de 2-bromo-3- metóxi propionila.
[00111] Preferivelmente os compostos (V), (IV) e (II) são respectivamente isolados do meio de reação antes de sofrerem qualquer outra transformação química. A dita isolação pode ser realizada por quaisquer métodos conhecidos pela pessoa habilitada na técnica.
[00112] Preferivelmente o composto da fórmula (V) é isolado pela cristalização em uma mistura de solventes selecionados de heptano, tolueno, acetato de isobutila, acetato de propila, éter metil t-butílico.Em uma forma de realização particular, a dita mistura compreende heptano.
[00113] O composto da fórmula (VI) pode ser isolado do meio de reação ou não.
[00114] Preferivelmente o composto da fórmula (II) é isolado pela cristalização em uma mistura de solventes selecionada de acetato de isobutila, acetato de isopropila, acetato de propila, 2-Me-tetra-hidrofurano e acetonitrila.Em uma forma de realização particular de acordo com a presente invenção, a dita mistura compreende acetato de isobutila ou acetato de etila.
[00115] Em uma forma de realização particular, a 2-acetamido-N-benzil-3- metoxipropion-amida (II) de acordo com a presente invenção é fabricada por um processo que compreende as seguintes etapas:
[00116] reagir o ácido 2-bromo-3-metóxi metil propiônico (Vld),
com um cloroformiato de alquila na presença de uma base seguido pela benzilamina; (II)reagir a 2-bromo-N-benzil-3-metoxipropion-amida (Va) resultante com amónia aquosa;
(III)acetilar a 2-cloro-N-benzil-3-metoxipropion-amida (IV) resultante com anidrido acético em um solvente;
(iv) isolar o composto da fórmula (II) deste modo obtido.
[00117] Nesta forma de realização particular o ácido 2-bromo-3-metóxi metil propiônico (Vld) é preparado a partir do propionato de 2,3-dibromo etila (Vila) ou propionato de 2,3-dibromo metila (Vllb) comercialmente disponíveis como mostrado no esquema 8 do presente pedido.
[00118] As condições de reação para as etapas (i), (ii), (iii) e (iv) são como descritas aqui acima para o Método A do Esquema 7. Em uma forma de realização particular de acordo com a presente invenção, o propionato de 2,3-dibromo etila (Vila) pode ser transformado em 2-bromo-N-benzil-3-metoxipropion-amida (Va) sem a isolação do ácido 2-bromo-3-metoxi metil propiônico (Vld), que é formado in situ.
[00119] Consequentemente em uma forma de realização particular, a presente invenção diz respeito a um processo para a fabricação de 2-acetamido-N-benzil-3- metoxipropion-amida (II) que compreende as seguintes etapas:
[00120] (iii) reagir propionato de 2,3-dibromo etila (Vila) ou propionato de 2,3- dibromo metila (Vllb)
com metóxido de sódio em metanol na presença de um cloroformiato de alquila seguido pela benzilamina; (II)reagir a 2-bromo-N-benzil-3-metoxipropion-amida (Va) resultante com amónia aquosa;
(III)acetilar a 2-cloro-N-benzil-3-metoxipropion-amida (IV) resultante com anidrido acético em um solvente;
(iv) isolar o composto da fórmula (II) deste modo obtido.
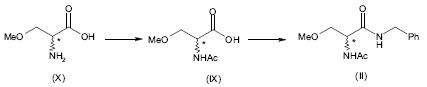
[00121] Em uma outra forma de realização de acordo com a presente invenção, a 2-acetamido-N-benzil-3-metoxipropion-amida (II) é fabricada pela reação do ácido 2- acetamido-3-metoxipropiônico (IX) com um cloroformiato de alquila, preferivelmente cloroformiato de etila ou isobutila, formando deste modo um anidrido misto que é depois reagido com benzilamina.
[00122] O ácido 2-acetamido-3-metoxipropiônico (IX) pode ser obtido pela acetilação de O-Metil-D,L-serina (X) comercialmente disponível de acordo com o seguinte esquema 10.
[00123] Esquema 10
[00124] Consequentemente, a presente invenção também diz respeito a um processo para a fabricação de Lacosamida que compreende as seguintes etapas:acetilação de O-Metil-D,L-Serina (X); (ii)reagir o ácido 2-acetamido-3-metoxipropiônico (IX) deste modo obtido com um cloroformiato de alquila seguido pela benzil amina; (iii)isolar o composto da fórmula (II); (iv)resolução de 2-acetamido-N-benzil-3-metoxipropion-amida (II) em (R)- 2-acetamido-N-benzil-3-metoxipropion-amida (I) e (S)-2-acetamido-N-benzil-3- metoxipropion-amida (III); (v)racemização da (S)-2-acetamido-N-benzil-3-metoxipropion-amida (III) deste modo obtida.
[00125] A Etapa (i) é no geral realizada usando anidrido acético como agente de acetilação em ácido acético, tolueno, tetra-hidrofurano, 2-metil-tetra-hidrofurano, acetato de isobutila, diclorometano ou água, ou misturas dos mesmos. Em uma forma de realização particular de acordo com a presente invenção a dita etapa é obtida em uma mistura de tetra-hidrofurano e água.
[00126] A Etapa (ii) é no geral realizada na presença de cloroformiato de etila ou cloroformiato de isobutila e N-metilmorfolina ou trietilamina em tetra-hidrofurano, 2- metil-tetra-hidrofurano, tolueno, acetato de etila ou diclorometano.
[00127] Alternativamente, a Etapa (ii) pode ser realizada na presença de um catalisador selecionado do grupo que consiste de ácido bórico, ácido fenil borônico, ácido 3,4,5-trifluorofenilborônico, ácido 2-(N,N-di-isopropilaminometil)fenilborônico e ácido 2-(N,N-dimetilaminometil)fenil borônico pelo refluxo de um solvente selecionado do grupo que consiste de tolueno, N-metilpirrolidona e misturas dos mesmos, tetra-hidrofurano, 2-metil-tetra-hidrofurano, éter ciclopentilmetílico, éter di n-butílico, fluorobenzeno usando um aparelho de Dean-Stark, peneiras moleculares ou sulfato de sódio para remover continuamente a água.
[00128] O catalisador de acordo com a presente invenção pode ser solúvel no meio de reação ou pode ser sustentado em sólido.
[00129] Em uma forma de realização particular de acordo com a presente invenção, uma mistura de tolueno e N-metilpirrolidona é usada como um solvente. A razão do volume de tolueno com respeito à N-metilpirrolidona é por exemplo de 80/20 ou 99/1.
[00130] Em uma outra forma de realização, a benzilação do composto da fórmula (IX) para produzir o composto da fórmula (II) pode ser realizada com benzilamina na presença de dicarbonato de di-terc-butila (BOC2O) na presença de piridina, trietilamina ou base de Hunig em um solvente selecionado do grupo que consiste de tetra-hidrofurano, 2-metil-tetra-hidrofurano, acetato de etila e diclorometano.
[00131] Em uma outra forma de realização, a benzilação do composto da fórmula (IX) para produzir o composto da fórmula (II) pode ser realizada com benzilamina na presença de anidrido do ácido n-propanofosfônico (T3P®) na presença de trietilamina ou base de Hunig em um solvente selecionado de acetato de etila, tetra- hidrofurano, diclorometano e 2-metil-tetra-hidrofurano.
[00132] Já em uma outra forma de realização, a benzilação do composto da fórmula (IX) para produzir o composto da fórmula (II) pode ser realizada com benzilamina na presença de dicicloexil- (DCC) ou diisopropil-carbodiimida (DIC) em um solvente selecionado do grupo que consiste de tetra-hidrofurano, acetato de etila e diclorometano.
[00133] Já em uma outra forma de realização, a benzilação do composto da fórmula (IX) para produzir o composto da fórmula (II) pode ser realizada em benzilamina pura na presença de hexametildissalazano (HMDS).
[00134] Já em uma outra forma de realização, a benzilação do composto da fórmula (IX) para produzir o composto da fórmula (II) pode ser realizada pelo aquecimento de uma mistura 1:1 sólida de (IX) com benzilamina acima de 130°C.
[00135] Os catalisadores usados para a etapa de benzilação de acordo com a presente forma de realização da invenção pode ser solúvel na reação ou sustentado em um sólido.
[00136] Este método é particularmente vantajoso visto que o mesmo compreende poucas etapas químicas do material de partida até o composto (II).
[00137] Em um aspecto particular, a presente invenção diz respeito a um processo para a preparação de Lacosamida que compreende as seguintes etapas:
[00138] reagir um composto da fórmula (Via),
em que X é um grupo de partida, com um haloformiato de alquila na presença de uma base e benzilamina; (II)realizar a amonólise do composto (V) que resulta da etapa (i), em que X é como definido no composto (Via);
(III)acetilar o composto da fórmula (IV) deste modo obtido
com anidrido acético em um solvente; (iv) isolar o composto da fórmula (II); e (v) realizar a resolução de 2-acetamido-N-benzil-3-metoxipropion-amida (II) em (R)-2-acetamido-N-benzil-3-metoxipropion-amida (I) e (S)-2-acetamido-N- benzil-3-metoxipropion-amida (III).
[00139] Em um aspecto ainda mais particular, a presente invenção diz respeito a um processo para a fabricação de (R)-2-acetamido-N-benzil-3-metoxipropion-amida (I) que compreende as seguintes etapas: reagir um composto da fórmula (Via),
em que X é um grupo de partida, com um haloformiato de alquila na presença de uma base e benzilamina; (ii)realizar a amonólise do composto (V) que resulta da etapa (i), em que X é como definido no composto (Via);
(iii)acetilar o composto da fórmula (IV) deste modo obtido
com anidrido acético em um solvente; (iv)isolar o composto da fórmula (II); (v)resolução de 2-acetamido-N-benzil-3-metoxipropion-amida (II) em (R)-2-acetamido-N-benzil-3-metoxipropion-amida (I) e (S)-2-acetamido-N-benzil-3- metoxipropion-amida (III); (vi)racemização de (S)-2-acetamido-N-benzil-3-metoxipropion-amida (III) desse modo obtida; e (vii)resolução adicional da 2-acetamido-N-benzil-3-metoxipropion-amida (II) resultante.
[00140] A Etapa (iv) é preferivelmente realizada pela cristalização em um solvente selecionado do grupo que consiste de tolueno, acetato de etila, acetato de isobutila, acetato de isopropila, acetonitrila, 2-metil-tetra-hidrofurano e misturas dos mesmos.
[00141] O composto (II) é deste modo preferivelmente obtido com uma pureza de pelo menos cerca de 98 % medida pela HPLC, mais preferivelmente com uma pureza de pelo menos cerca de 99 %, o mais preferivelmente com uma pureza de pelo menos cerca de 99,5 %.
[00142] A Etapa (v) nas duas formas de realização detalhadas aqui acima é preferivelmente realizada pela separação cromatográfica quiral.Em uma forma de realização de acordo com a presente invenção, a separação cromatográfica quiral é realizada pela MCC.
[00143] A dita separação é preferivelmente realizada usando um CSP que compreende um seletor de polissacarídeo revestido ou imobilizado em uma cadeia principal de sílica de acordo com técnicas bem conhecidas no ramo e uma fase móvel, como aqui detalhada acima no relatório descritivo.
[00144] Em uma forma de realização particular de acordo com a invenção, o seletor quiral polimérico é selecionado de tris(4-metilbenzoato) de celulose, tribenzoato de celulose, tris(3,5-dimetilfenilcarbamato) de celulose, tris(3,5- dimetilfenilcarbamato) de amilose, tris(4-metilfenil-carbamato) de celulose, tris (3,5- diclorofenilcarbamato) de celulose, tris [(S)-a-metilbenzilcarbamato] de amilose e tris(3-cloro-4-metilfenil-carbamato) de celulose e o solvente é selecionado de alcanos, tais como heptano, hexano, alcoóis, tais como metanol, etanol, isopropanol, n-propanol, acetonitrila, acetato de isopropila, acetato de etila, diclorometano, clorofórmio, éteres, tais como éter metil t-butílico (MTBE) ou misturas dos mesmos.
[00145] Em uma forma de realização preferida de acordo com a presente invenção, a separação na etapa (v) é realizada usando tris (3,5- diclorofenilcarbamato) de celulose imobilizada em uma cadeia principal de sílica como seletor quiral polimérico e uma mistura de acetato de etila e metanol em uma razão de 90/10 v:v foi usada como fase móvel.
[00146] A Etapa (vi) é preferivelmente realizada pela reação do composto (III) com metóxido de sódio, seguido pela acidificação estequiométrica.
[00147] A temperatura da racemização é preferivelmente mais baixa do que 60°C.
[00148] No processo para a fabricação de Lacosamida, a (R)-2-acetamido-N- benzil-3-metoxipropion-amida (I) opticamente pura de modo substancial obtida depois da etapa de resolução pode ser cristalizada ainda com propósitos de purificação.Esta cristalização é preferivelmente realizada em acetato de etila.
[00149] Em uma forma de realização particular de acordo com a presente invenção, A cristalização é iniciada pela semeadura do meio de cristalização com (R)-2-acetamido-N-benzil-3-metoxipropion-amida (I) opticamente pura de modo substancial. Nesta forma de realização particular a semeadura é realizada a uma temperatura compreendida entre 60°C e 80°C, preferivelmente em uma temperatura compreendida entre 65°C e 75°C.
[00150] O processo de acordo com a presente invenção é particularmente vantajoso em relação aos processos conhecidos de fabricação de Lacosamida porque: -Os materiais de partida são facilmente disponíveis; -O mesmo não requer o uso de agente de proteção para as funções amina presentes nos intermediários sintéticos que gera etapas de processo de proteção-desproteção adicionais e assim aumenta os custos de produção; -O mesmo não usa reagentes que sejam nocivos para o ambiente; -O enantiômero (III) indesejado pode ser no final das contas reciclado na Lacosamida, aumentando deste modo a produtividade global do processo. Em uma outra forma de realização, a presente invenção diz respeito a um processo para a fabricação de (R)-2-acetamido-N-benzil-3-metoxipropion-amida (I) opticamente pura de modo substancial que compreende as seguintes etapas: (i)resolução da 2-amino-N-benzil-3-metoxipropion-amida (IV); (ii)acetilação da (R)-2-amino-N-benzil-3-metoxipropion-amida (IVa) deste modo obtida com anidrido acético; e (iii)cristalização da (R)-2-acetamido-N-benzil-3-metóxi-propion-amida deste modo obtida.
[00151] Na presente forma de realização, a resolução é preferivelmente realizada pela formação de sal diastereomérico pela reação do composto da fórmula (IV) com um ácido selecionado do grupo que consiste do ácido (R)-(-)-mandélico, ácido (S)- (+)-mandélico, ácido (D)-(+)-málico, ácido (L)-(-)-málico, ácido (+)-O,O‘-dibenzoil tartárico, (L)-N-acetil-alanina e (D)-N-acetil-leucina, isto é, a “Conversão para diastereoisômeros” método descrito em Jerry March em “Advanced Organic Chemistry”, quarta edição, Capítulo 4, páginas 120 a 125.
[00152] A resolução é preferivelmente realizada em um solvente selecionado do grupo que consiste de acetona, metanol, etanol, 1-propanol, éter metil-terc-butílico, heptano, cicloexano, metiletilcetona, acetato de isopropila e misturas dos mesmos.
[00153] A resolução é preferivelmente realizada em uma temperatura compreendida entre 20°C e 60°C seguida pelo resfriamento a uma temperatura compreendida entre 0°C e 20°C.
[00154] A (R)-2-acetamido-N-benzil-3-metoxipropion-amida é cristalizada ainda para produzir a (R)-2-acetamido-N-benzil-3-metoxipropion-amida opticamente pura de modo substancial. A dita cristalização é repetida até que a (R)-2-acetamido-N- benzil-3-metóxi-propion-amida seja obtida na pureza óptica desejada.
[00155] Alternativamente, a etapa de resolução pode ser realizada pela separação cromatográfica quiral usando métodos de absorção diferentes, mais preferivelmente usando separação cromatográfica quiral realizada em lote ou em MCC (Cromatografia de Coluna Múltipla) incluindo o modo de SMB (leito movediço simulado) ou modo onde os orifícios de entrada e saída são mudados assincronamente ou modo em que as taxas de fluxo e/ou as concentrações de entrada e saída e são mudadas no tempo durante o período de comutação, como aqui detalhado acima na especificação para 2-acetamido-N-benzil-3-metoxipropion- amida (II).
[00156] A 2-amino-N-benzil-3-metoxipropion-amida (IV) usada na presente forma de realização pode ser preparada de acordo com qualquer um dos métodos aqui descritos acima no relatório descritivo.
[00157] Todas as etapas de processo aqui mencionadas acima, e particularmente os processos de fabricação do composto (II), incluindo a síntese e extração dos materiais podem ser individual ou coletivamente realizadas no modo de batelada ou de acordo com um processo contínuo, usando, por exemplo, micro-reatores.
[00158] Exemplos
[00159] Os seguintes exemplos são fornecidos apenas para os propósitos ilustrativos e não são intencionados, nem devem ser interpretados, como limitando a invenção de nenhuma maneira. Aqueles habilitados na técnica avaliarão que as variações e modificações de rotina dos seguintes exemplos podem ser feitas sem exceder o espírito ou escopo da invenção.
[00160] Os espectros de RMN são registrados em um espectrômetro Bruker 400 MHz como soluções em clorofórmio deuterado (CDCh). As mudanças químicas são expressadas em partes por milhão (ppm, õ) a jusante de tetrametilsilano e são referenciadas para o solvente deuterado (CDCh).
[00161] Os dados de 1H RMN foram relatados na ordem de mudança química, multiplicidade (s, singleto; d, dupleto; t, tripleto; q, quarteto; m, multipleto; app, ressonância aparente e/ou múltipla), a constante de ligação (J) em hertz (Hz) e o número de prótons.
[00162] Os espectros de Cromatografia Líquida de Alto Desempenho (HPLC) são registrados em um Alliance Waters 2695 equipado com os espectros de HPLC foram registrados em um Alliance Waters 2695 equipado com uma coluna Atlantis T3 3 microns (4,6 X 100 mm), detectando a 200 nm - composição do solvente de partida = água: 90 % vol/água + 1 % de H3PO4: 10 % vol; composição do solvente final = água + 1 % de H3PO4 :10 % vol/ acetonitrila : 90 % vol em 6 minutos seguido pelo equilíbrio de re-equilíbrio de 1 min para a composição de solvente inicial.
[00163] A HPLC quiral é registrada em um Merck-Hitachi L-7100 equipado com urn Daicel Chiralpak OJ-H® 5 μm. O eluente é uma mistura de heptano/etanol 96/4 com um fluxo de 2 ml/min.
[00164] Os espectros de cromatografia gasosa (GC) são registrados em um Agilent série 6890 equipado com uma coluna de GC DB-5MS da Altech (15 m x 0,25 mm). A estufa é aquecida a 50°C com um fluxo de hélio de 1,5 ml/min e um detector FID aquecido a 300°C.
[00165] Espectroscopia de massa (MS): Os espectros API foram realizados usando um espectrômetro de massa de aprisionamento de ion LCQ da FINNIGAN (San Jose, CA, USA). A fonte de APCI operada a 450°C e o aquecedor capilar a 160°C. A fonte de ESI operada a 3,5 kV e o aquecedor capilar a 210°C.
[00166] Exemplo 1 - Preparação de Lacosamida partindo de propionato de 2,3- dibromo etila (VII)
[00167] Exemplo 1a - Preparação do ácido 2-bromo-3-metóxi propiônico (Vld) a partir do propionato de 2,3-dibromo etila (VII)
[00168] Em um equipamento seco sob nitrogênio, propionato de 2,3-dibromo etila (VII) comercialmente disponível (1 equiv) é dissolvido em 4 volumes de metanol seco e a solução é esfriada a -10°C sob agitação. Uma solução a 25 % p/p de metóxido de sódio em metanol (1,1 equiv) é lentamente adicionada de modo que a temperatura seja mantida abaixo de -5°C. Depois da adição, a mistura é deixada aquecer até a temperatura ambiente e a reação é pós-agitada a 20°C por 1 hora.
[00169] Hidróxido de sódio a 20 % aquoso (1 equiv) é adicionado lentamente a uma temperatura mantida abaixo de 23°C. A mistura de reação é depois agitada a 20°C por 1 hora.
[00170] HCI a 37 % aquoso é lentamente adicionado até o pH 5-6 e a mistura de reação concentrada até o volume de agitação mínimo, isto é, 1 volume vs composto (VII), sob vácuo a uma temperatura de no máximo 40°C.
[00171] HCI a 37 % aquoso é adicionado até que o pH seja 2 e o resíduo salgado é coletado em uma quantidade mínima de água (~0,6 volumes) para se obter uma solução e o composto (Vld) é extraído com acetato de isobutila (3X2 volumes). A camada orgânica é evaporada a cerca de 4 volumes produzindo uma solução do ácido 2-bromo-3-metóxi propiônico (Vld).
[00172] Exemplo 1b - Preparação de 2-bromo-N-benzil-3-metoxipropion-amida (Va) a partir do ácido 2-bromo-3-metóxi propiônico (Vld)
[00174] A solução do ácido 2-bromo-3-metóxi propiônico (Vld) (1 equiv, quantidade calculada pelo ensaio p/p) em acetato de isobutila da etapa anterior é esfriada a -5°C sob agitação. Cloroformiato de etila (1,1 equiv) é adicionado de modo a se obter uma temperatura de massa entre -5 e 0°C. O funil de adição é enxaguado com 0,1 volume de acetato de isobutila. N-metilmorfolina (1,1 equiv) é adicionado lentamente de modo a se obter uma temperatura de massa entre -5 e 0°C. O funil de adição é enxaguado com 0,1 volume de acetato de isobutila. A solução é pós agitada por 30 min a -5/0° C. Benzilamina (1,1 equiv) é adicionada de modo a se obter uma temperatura de massa entre -5°C e 0°C. O funil de adição é enxaguado com 0,25 volume de acetato de isobutila. A mistura de reação é deixada aquecer até 25-30°C e pós agitada por cerca de 1 hora (área de HPLC de (Vld) mais baixa do que 0,2 %).
[00175] 1 volume de água com respeito à solução inicial de (Vld) é adicionada e a mistura agitada por 15 min. A camada aquosa é separada e a camada orgânica lavada com água (0,5 volume). A solução é concentrada a cerca de 2,5 a 3 volumes de acetato de isobutila a 40°C sob vácuo e semeada com composto (Va) a 30-35° e a suspensão esfriada até a temperatura ambiente até que a cristalização esteja bem iniciada. Na mesma temperatura, cerca de 5 volumes de heptano são lentamente adicionados.Depois a suspensão é progressivamente esfriada a -10°C. Os cristais são filtrados, lavados com iButOAc/heptano 1:2 gelado (1 volume) e heptano (2 volumes) e secos a 40°C sob vácuo. A 2-bromo-N-benzil-3-metoxipropion-amida (Va) é isolada com um rendimento de 70 % (vs dibromopropionato de etila usado na Etapa 1).
[00176] (Va): pf 74-75°C; 1H RMN (CDCh) δ 3,43 (s, OCH3), 3,87 (dd, J = 5,0, 10,7 Hz, CHH’0), 3,95 (dd, J = 4,6, 10,7 Hz, CHH’O), 4,43 - 4,50 (m, CH e CH2Ph), 6,93 (br s, NH), 7,24 - 7,37 (m, 5 PhH); 13C RMN (CDCh) 44,0, 47,9, 59,2, 73,6, 127,4, 127,5, 128,7, 137,4, 167,0 ppm.
[00178] Em um vaso de reação, equipado com um agitador mecânico, um funil de adição e uma sonda termométrica, adicionar a 20°C, 1 equiv de (Vld), 8 volumes de tolueno e 0,1 equiv de dimetilformamida. A 20°C, adicionar às gotas 1,1 equiv de SOCI2 dentro de 15 minutos. Agitar por 20 minutos a 20°C depois da adição.A mistura de reação é depois esfriada a -10°C e 2 equiv de trietilamina são adicionados às gotas dentro de 15 minutos.Aquecer a 20°C e agitar por 20 minutos a 20°C. Esfriar a mistura de reação a -10°C, adicionar às gotas 1 equiv de benzilamina e deixar a mistura de reação aquecer até 20°C. 1 volume de água com respeito à solução inicial de Vld é adicionado e a mistura agitada por 15 min. A camada aquosa é separada e a camada orgânica lavada com água (0,5 volume). A solução é concentrada a cerca de 2,5 a 3 volumes de acetato de isobutila a 40°C sob vácuo e semeada com composto (Va) de 30 a 35° e a suspensão esfriada até a temperatura ambiente até que a cristalização esteja bem iniciada. Na mesma temperatura, cerca de 5 volumes de heptano são lentamente adicionados. Depois a suspensão é progressivamente esfriada a -10°C. Os cristais são filtrados, lavados com iButOAc/heptano 1:2 (1 volume) e heptano (2 volumes) gelados e secos a 40°C sob vácuo.
[00179] Exemplo 1c - Preparação de 2-bromo-N-benzil-3-metoxipropion-amida (Va) a partir do propionato de 2,3-dibromo etila (VII)
[00180] 5,2 g de propionato de 2,3-dibromo etila (VII) foram dissolvidos com 20 ml de Metanol. A uma temperatura entre -10°C e -5°C dentro de 5 minutos, 4,08 ml de uma solução de metanolato de sódio a 30 % em Metanol foram adicionados. A solução foi agitada por 10 minutos de 0°C a 5°C. 10,9 ml de benzil amina foi depois adicionada à mistura que foi agitada por 2 h na temperatura ambiente. Metanol foi evaporado e o resíduo foi coletado em 100 ml de ácido clorídrico 1 M.A formação de precipitado foi extraída com 50 ml de diclorometano. O diclorometano foi depois evaporado do meio até a secura.
[00181] Resíduo: 4,2 g de bruto incolor
[00182] O resíduo foi recristalizado a partir de uma mistura de 6 ml de éter metil terc-butílico e 3 ml de n-heptano e 2,79 foram isolados.
[00183] Rendimento = 51,26 %; LCMS: 97,9 % [M + H] = 272
[00184] Exemplo 1d - Preparação de 2-amino-N-benzil-3-metoxipropion-amida (IV)pela amonólise de 2-bromo-N-benzil-3-metoxipropion-amida (Va)
[00185] 2-bromo-N-benzil-3-metoxipropion-amida (Va) (1 equiv) é dissolvida em 6 volumes de amónia aquosa a 28 % e 2 volumes de metanol. A solução é aquecida a 100°C sob agitação por cerca de 2 horas a uma pressão de 7 bar. A mistura de reação é concentrada sob vácuo de 50 a 60°C até a precipitação dos sais. Na temperatura ambiente quantidade mínima de água é adicionada de modo a solubilizar os sais. O pH da solução é ajustada a 12 com hidróxido de sódio a 50 % aquoso e a 2-amino-N-benzil-3-metoxipropion-amida (IV) é extraída com acetato de isobutila (3 a 4 x 2 volumes). As camadas orgânicas combinadas são azeotropicamente secas. Os sais que precipitam depois da secagem são eliminados pela filtração e o composto (IV) é usado diretamente na etapa seguinte sem outra purificação.
[00186] A reação pode ser monitorada pela HPLC.LC-MS (+CI) (intensidade rei) 210(12), 209 (M++ 1, 100).
[00187] Uma amostra é evaporada até a secura (óleo): 1H RMN (CDCh) δ 1,74 (br, s, NH2), 3,34 (s, OCH3), 3,53 - 3,61 (m, CHCH2), 4,36 - 4,48 (m, CH2NH), 7,22 - 7,33 (m, 5 PhH), 7,75 - 7,86 (m, NH).
[00188] Exemplo 1e - Preparação de 2-acetamido-N-benzil-3-metoxipropion-amida (II) pela acetilação de 2-amino-N-benzil-3-metoxipropion-amida (IV)
[00189] As camadas orgânicas combinadas na etapa precedente são ajustadas a 7 volumes com respeito à quantidade inicial de 2-bromo-N-benzil-3-metoxipropion- amida (Va) usada na etapa precedente e a temperatura da solução é ajustada a 60°C. 1 equivalente (com respeito à quantidade inicial de 2-bromo-N-benzil-3- metoxipropion-amida (Va) usada na etapa precedente) de anidrido acético é adicionado às gotas enquanto que a temperatura é mantida abaixo de 70°C.
[00190] 0,5 volume de água é adicionado e a mistura agitada por 15 minutos. A camada aquosa é decantada e descartada e a mesma operação é repetida.
[00191] A solução é depois semeada com 2-acetamido-N-benzil-3-metoxipropion- amida (II) e lentamente esfriada a 0°C. Os cristais são filtrados, lavados com 2 volumes de acetato de isobutila a 0°C e secos sob vácuo a 40°C.
[00192] A 2-acetamido-N-benzil-3-metoxipropion-amida (II) é deste modo isolada em 80 % de rendimento.
[00193] (II): pf 122 a 123°C; 1H RMN (CDCh): δ 2,02 (s, C(O)CH3), 3,37 (s, OCH3), 3,42 (dd, J= 7,8, 9,0 Hz, CHH’OCHs), 3,80 (dd, J = 4,0, 9,0 Hz, CHH’OCHs), 4,47 (d, J = 6,0 Hz, NHCH2), 4,49 - 4,56 (m, CH), 6,41 (br d, J = 6,0 Hz, NH), 6,73 (br s, NH), 7,22 - 7,37 (m, 5 PhH)
[00194] Exemplo 1f - Resolução de 2-acetamido-N-benzil-3-metoxipropion-amida (II) em (R)-2-acetamido-N-benzil-3-metoxipropion-amida (I) e (S)-2-acetamido-N- benzil-3-metoxipropion-amida (III)
[00195] Método A
[00196] Uma solução alimentada de 2,1 kg de 2-acetamido-N-benzil-3- metoxipropion-amida (II) em acetonitrila é preparada e agitada sob nitrogênio até que a dissolução completa seja obtida. A solução é continuamente injetada em um sistema SMB que é equipado com seis colunas idênticas de 12,46 cm de comprimento e 4,8 cm de diâmetro interno, em uma configuração 1-2-2-1. Cada coluna contém 125 g de uma fase estacionária quiral que compreende tris(3,5- dimetilfenilcarbamato) de celulose revestida sobre a cadeia principal de sílica e os enantiômeros são separados usando acetonitrila como a fase móvel.
[00197] A (R)-2-acetamido-N-benzil-3-metoxipropion-amida (I) opticamente pura de modo substancial é extraída da corrente e obtida com um excesso enantiomérico maior do que 99 %.
[00198] Método B
[00199] Uma solução alimentada de 12 kg de 2-acetamido-N-benzil-3- metoxipropion-amida (II) em Acetato de etila-MeOH (90/10) é preparada e agitada sob nitrogênio até que a dissolução completa seja obtida. A solução é continuamente injetada em um sistema SMB que é equipado com seis colunas idênticas de 12,4 cm de comprimento e 4,8 cm de diâmetro interno, em uma configuração 1-2-2-1. Cada coluna contém 125 g de uma fase estacionária quiral que compreende tris(3,5-diclorofenilcarbamato) de celulose imobilizada sobre a cadeia principal de sílica e os enantiômeros são separados usando acetato de etila- MeOH (90/10) como a fase móvel.
[00200] A (R)-2-acetamido-N-benzil-3-metoxipropion-amida (I) opticamente pura de modo substancial é extraída da corrente e obtida com um excesso enantiomérico maior do que 99 %.
[00201] Exemplo 1g - Racemização de (S)-2-acetamido-N-benzil-3-metoxipropion- amida (III) em 2-acetamido-N-benzil-3-metoxipropion-amida (II)
[00202] A uma solução de (S)-2-acetamido-N-benzil-3-metoxipropion-amida (III) isolada na etapa precedente, em 5 volumes de metanol, 0,05 equivalente de metóxido de sódio é adicionado sob fluxo de nitrogênio. A mistura é aquecida até 60°C por 8 h seguida por um esfriamento a 10°C.
[00203] A mistura é extinta com 0,05 equivalente de uma solução aquosa de HCI enquanto se mantém a temperatura da massa em cerca de 20°C. O metanol é destilado sob pressão atmosférica até que cerca de 1 volume de solvente permaneça. Uma destilação azeotrópica com 9 volumes de acetato de isopropila é depois realizada. A destilação é realizada pela adição contínua de acetato de isopropila de modo a manter um total de 10 volumes (cerca de 5 volumes são destilados para se obter um nível residual de MeOH < 0,1 % pela GC).
[00204] A mistura é esfriada abaixo de 60°C e é lavada com água. A água residual na camada orgânica é removida pela destilação azeotrópica com acetato de isopropila de acordo com o mesmo método como mencionado aqui acima.
[00205] A solução é esfriada abaixo de 0°C para cristalização.A suspensão é filtrada e a torta é lavada com acetato de isopropila. O sólido é seco sob pressão reduzida a 40°C.
[00206] A 2-acetamido-N-benzil-3-metoxipropion-amida (II) é obtida com um rendimento de 74 %.
[00207] As condições do exemplo 1e são mais uma vez aplicadas à 2-acetamido- N-benzil-3-metoxipropion-amida (II) aqui obtida.
[00208] Exemplo 2 - Preparação de 2-amino-N-benzil-3-metoxipropion-amida (IV) a partir do ácido 2-cloro-3-metóxi propiônico (Vlc)
[00209] Exemplo 2.1. Preparação da N-benzil-2-cloro-3-metóxi-propionamida (Vb) a partir do ácido 2-cloro-3-metóxi propiônico (Vic)
[00210] 5,54 g (0,04 mol) do ácido 2-cloro-3-metóxi propiônico (Vic) são dissolvidos em 50 ml de diclorometano. A solução é esfriada a -5°C. 6,24 ml (0,048Mol) de cloroformiato de isobutila são adicionados dentro de 5 minutos. A solução é esfriada a -10°C. Dentro de 5 minutos 5,28 ml (0,048 Mol) de N-Metil morfolina são gotejados na solução, a temperatura elevada até -2°C (Temperatura de banho: - 15°C). Para completar a formação de anidrido, a suspensão de fluido fino foi agitada por 30 minutos a -5°C. Depois a suspensão é esfriada a -10°C, seguida pela adição de uma solução preparada a partir de 5,24 ml (0,048 Mol) de benzil amina e 10 ml de diclorometano dentro de 20 minutos em uma faixa de temperatura de -10°C a -5°C. A suspensão de fluido fino é agitada por mais 1 h sem banho de esfriamento Tende: 11 °C A suspensão é extraída quatro vezes com: •10 ml de Água •10 ml de hidrogeno carbonato de sódio a 5 % •10 ml de ácido clorídrico a 1 M •10 ml de Água
[00211] A camada de produto remanescente é evaporada até a secura.
[00212] Resíduo: 9,36 g de produto oleoso (80 %)
[00213] Exemplo 2.2. Preparação de 2-amino-N-benzil-3-metóxi-propionamida (IV) a partir da N-Benzil-2-cloro-3-metóxi-propionamida (Vb)
[00214] 12,07 g de N-Benzil-2-cloro-3-metóxi-propionamida (Vb) são dissolvidos em 66 ml de metanol. A solução é transferida para uma autoclave e tratada com 212 ml de amónia aquosa a 28 %.A autoclave é fechada e aquecida 2 h a 120°C. Pressão final: 5,3 bar.
[00215] A solução é evaporado até a secura. O resíduo é coletado em 200 ml de água e extraída duas vezes com diclorometano, para remover impurezas menos polares. A camada de produto aquosa é tratada com 35 % de solução de hidróxido de sódio e extraída duas vezes com diclorometano. A camada de diclorometano é separada e evaporada até a secura.
[00216] Resíduo: 6,52 g de óleo amarronzado
[00217] Rendimento: 59 %
[00218] LCMS: 91,6 % [M + H] 209
[00219] Exemplo 3. Preparação de Lacosamida a partir da 2-amino-N-benzil-3- metóxi-propionamida (IV)
[00220] Exemplo 3.1. Resolução diastereoisomérica de 2-amino-N-benzil-3- metóxi-propionamida (IV) com sais
[00221] 3.1.1. Resolução com o ácido (+)-(+)-O,O‘-dibenzoil tartárico
[00222] 500 mg de 2-amino-N-benzil-3-metóxi-propionamida (IV) são dissolvidos na temperatura ambiente em 2,5 ml de 1-propanol. Em paralelo, 430 mg do ácido (+)-(+)-O,O‘-dibenzoil tartárico são dissolvidos na temperatura ambiente em 2,5 ml de 1-propanol. A solução salina é adicionada na temperatura ambiente à solução de 2-amino-N-benzil-3-metóxi-propionamida (IV)/1-propanol. 10 ml da mistura são semeados na temperatura ambiente com cristais de (R)-2-amino-N-benzil-3-metóxi- propionamida (IVa) e agitados por cerca de 5 minutos. Deste modo cristais brancos, em flocos precipitaram.Estes cristais são extraídos.
[00223] 320 mg de produto sólido são obtidos a partir da solução (rendimento: 23,5 %, pureza quiral: 84 %, pureza quiral: 68 % do enantiômero R e 32 % do enantiômero S).
[00224] 160 mg do dito produto sólido são adicionados a 4 ml de 1-propanol e dissolvidos em um extintor de água a uma temperatura de cerca de 85°C. A mistura é esfriada e semeada como descrito antes. Cristais brancos, em flocos são extraídos depois de cerca de 5 minutos na temperatura ambiente (rendimento: 55 mg, 34 %, pureza química 96 %, pureza quiral 26 % do enantiômero S; 74 % do enantiômero R (IVa)). Os 55 mg são adicionados a 1 ml de 1-propanol e dissolvidos em um extintor de água em uma temperatura de cerca de 85°C. A mistura é esfriada até a temperatura ambiente. Nenhuma semeadura é requerida. Os cristais precipitados, brancos podem ser extraídos depois de 5 a 10 minutos agitando (rendimento: 30 mg, 75 %, pureza química 98,3 %, pureza quiral 15 % do enantiômero S e 85 % do enantiômero R (IVa).
[00225] 3.1.2. Resolução com N-acetil-D-leucina
[00226] A 6 g de 2-amino-N-benzil-3-metóxi-propionamida (IV) são adicionados 4,99 g de N-acetil-D-leucina em 30 ml de acetato de isopropila. A suspensão é aquecida a 85°C por 1 hora e depois filtrada a 80°C (rendimento: 5,06 g, 46,5 %; pureza quiral: 80,5 % do enantiômero R (IVa), 19,5 % do enantiômero S). 2 g deste sal são aquecidos a 85°C em 10 ml de acetato de isopropila por 1 hora e depois filtrado a 80°C (rendimento: 1,14 g, 57,25 %, pureza quiral: 97,5 % do enantiômero R (IVa), 2,5 % do enantiômero S)
[00227] Exemplo 3.2. Preparação da (R)-2-acetamido-N-benzil-3-metóxi- propionamida (I) a partir da (R)-2-amino-N-benzil-3-metóxi-propionamida (IVa)
[00228] 1,12 g da (R)-2-amino-N-benzil-3-metóxi-propionamida (IVa) (0,0054 mol) é dissolvido em 25 ml de diclorometano. Depois de adicionar 0,756 ml de anidrido acético (0,8167 g, 0,08 mol), a mistura foi agitada por 2 horas na temperatura ambiente. Subsequentemente, a mistura foi consecutivamente extraída com 5 ml de água, 5 ml de ácido clorídrico 1 M, 5 ml de solução a 5 % de hidrogeno carbonato de sódio e 5 ml de água. A fase de produto orgânico é evaporada (HPLC: Isômero R: 93,46 % (I), Isômero S: 6,54 % (III)).
[00229] Exemplo 4. Preparação de 2-acetamido-N-benzil-3-metoxipropionamida(11)a partir de N-acetil-O-metil-D,L-serina
[00230] Exemplo 4.1. Preparação de N-acetil-O-metil-D,L-serina (IX)
[00231] Em um vaso, equipado com um agitador mecânico, adicionar a 20°C, 1,0 eq. de O-metil-D,L-serina. Adicionar 6 volumes de tetra-hidrofurano e 0,65 volume de água. Adicionar às gotas em 5 minutos 1,2 eq. de anidrido acético. Agitar mecanicamente a suspensão branca até conversão completa (monitoramento pela HPLC, tempo de reação típico: 16 horas). No final da reação a mistura torna-se homogênea. Remover o tetra-hidrofurano assim como a água azeotropicamente. Adicionar o tolueno de modo a remover completamente a água e o ácido acético (controle KF e GC). Cristalizar o sólido bruto obtido em 6 volumes de acetona. Secar o sólido úmido a 40°C sob vácuo durante a noite. A N-acetil-O-metil-D,L-serina (IX) é obtida como um sólido branco em 80 % de rendimento com uma pureza química de mais do que 99 % medida pela HPLC.
[00232] O espectro de RMN obtido é compatível com as características descritas para o composto (IX) na literatura (S. V. Andurkar et al., Tetrahedron: Asymmetry 9, 3841-3854).
[00233] Exemplo 4.2. Preparação de 2-acetamido-N-benzil-3-metoxipropion-amida (II) a partir da N-acetil-O-metil-D,L-serina (IX)
[00234] Exemplo 4.2.1.Uso de cloroformiato de isobutila e N-metilmorfolina
[00235] Em um vaso de reação equipado com um agitador mecânico foram carregados 1,0 eq. de N-acetil-O-metil-D,L-serina (IX) e 10 volumes de THF anidro. Depois de esfriar a -20°C, 1,15 eq. de cloroformiato de isobutila e depois 1,15 eq. de N-metilmorfolina foram sucessivamente adicionados às gotas mantendo a temperatura abaixo de -15°C. A mistura de reação foi agitada mais 15 min. a -20°C e depois 1,15 eq. de benzilamina foi adicionado às gotas mantendo a temperatura da massa abaixo de -15°C. Depois de mais 15 min agitando, a mistura foi deixada aquecer até a temperatura ambiente e os sais foram removidos pela filtração. O filtrado foi concentrado até a secura sob vácuo e o sólido residual foi recristalizado em acetato de etila. Os cristais obtidos são secos durante a noite sob vácuo em uma temperatura de 50°C.
[00236] O Composto (II) é obtido em 62 % de rendimento e uma pureza de 99,6 %.
[00237] Exemplo 4.2.2.Uso de ácido bórico anidro
[00238] Em um vaso de reação equipado com um aparelho de Dean-Stark, um agitador mecânico e uma sonda termométrica, adicionar 1,0 eq. de N-acetil-O-metil- D,L-serina (IX) e 8 volumes de tolueno. Adicionar 0,1 eq. de ácido bórico anidro e 1 eq. de benzilamina. Aquecer a suspensão ao refluxo por 16 horas com remoção contínua de água com o aparelho de Dean-Stark. No final da reação, a mistura de reação é homogênea. Adicionar 1 volume de água e gradualmente esfriar a mistura de reação a 0°C. Filtrar sob vácuo em um filtro de vidro sinterizado. Enxaguar com o mínimo de água e tolueno. Secar o sólido úmido a 40°C sob vácuo durante a noite.
[00239] A 2-acetamido-N-benzil-3-metoxipropion-amida (II) é obtida em um rendimento de 65 % com uma pureza química maior do que 99 % medida pela HPLC. Os espectros de RMN são compatíveis com aquele obtido com uma amostra de referência de Lacosamida.
[00240] 4.2.3. Pelo uso de trifenilboroxina como catalisador
[00241] Em um vaso de reação equipado com um aparelho de Dean-Stark, um agitador mecânico e sonda termométrica, foram carregados 1,0 eq. de N-acetil-O- metil-D,L-serina (IX) e 8 volumes de tolueno. 0,6 eq. de ácido fenil borônico (ou 0,2 eq. da trifenilboroxina correspondente) foi adicionado e a mistura de reação foi aquecida ao refluxo. 1 eq. de benzilamina foi depois adicionado continuamente em 2 horas e a mistura de reação foi mantida mais 22 horas no refluxo. Depois da conversão completa, a mistura foi esfriada a 75°C, e 3 volumes de acetato de etila foram adicionados. A mistura homogênea foi esfriada a 60°C e semeada com 1 % p:p de (II). A cristalização foi deixada desenvolver nesta T° e a mistura foi filtrada a 0°C, enxaguada com uma mistura de tolueno/acetato de etila (70:30) fresca e seca sob vácuo por 24 horas.
[00242] O Composto (II) é obtido em 70 % de rendimento e uma pureza química maior do que 94,5 %.
[00243] 4.2.4.Uso de diisopropilcarbodiimida
[00244] Em uma suspensão de 1,0 eq. de N-acetil-O-metil-D,L-serina em 10 volumes de diclorometano foi adicionada às gotas a 15°C uma solução de 1,2 eq. de diispropilcarbodiimida (DIC) em 1 volume de diclorometano. A mistura foi agitada 2 horas a 20°C e depois esfriada a 0°C e agitada mais 1 hora. A 1,3-diisopropiluréia foi separada por filtração e 1,05 eq. de benzilamina foi adicionado às gotas ao filtrado resultante mantendo a temperatura abaixo de 20°C. Depois da conversão completa, a solução foi depois evaporada até a secura levando ao material bruto que foi depois cristalizado ainda em acetato de etila.
[00245] O Composto (II) foi obtido com 78,2 % de rendimento e pureza maior do que 98,8 %.