WO2014042197A1 - 有機電界発光素子用材料、有機電界発光素子、表示装置、及び照明装置 - Google Patents
有機電界発光素子用材料、有機電界発光素子、表示装置、及び照明装置 Download PDFInfo
- Publication number
- WO2014042197A1 WO2014042197A1 PCT/JP2013/074561 JP2013074561W WO2014042197A1 WO 2014042197 A1 WO2014042197 A1 WO 2014042197A1 JP 2013074561 W JP2013074561 W JP 2013074561W WO 2014042197 A1 WO2014042197 A1 WO 2014042197A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- ring
- formula
- optionally substituted
- organic electroluminescent
- Prior art date
Links
- 0 c(cc1)cc(*(*23)c(cccc4)c4-c4c2cccc4)c1-c1c3cccc1 Chemical compound c(cc1)cc(*(*23)c(cccc4)c4-c4c2cccc4)c1-c1c3cccc1 0.000 description 30
- QUQKNEYWULBERV-RNAASMMVSA-N C[C@H](/C=C\C=C)N(c(cccc1)c1-c1c2cccc1)[P+]2(C1=CC=CCC=C1)=O Chemical compound C[C@H](/C=C\C=C)N(c(cccc1)c1-c1c2cccc1)[P+]2(C1=CC=CCC=C1)=O QUQKNEYWULBERV-RNAASMMVSA-N 0.000 description 1
- XVXJIRFYIRBDRS-UHFFFAOYSA-N C[Si](C)(C)c(cc1)ccc1N(c1ccccc1)c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1 Chemical compound C[Si](C)(C)c(cc1)ccc1N(c1ccccc1)c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1 XVXJIRFYIRBDRS-UHFFFAOYSA-N 0.000 description 1
- OGHUXULKMCHEBP-UHFFFAOYSA-N Cc(cc1c2c3ccc(-c4ccccc4)c2)ccc1[n]3-c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1 Chemical compound Cc(cc1c2c3ccc(-c4ccccc4)c2)ccc1[n]3-c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1 OGHUXULKMCHEBP-UHFFFAOYSA-N 0.000 description 1
- IOKAPZIBFLPOFO-UHFFFAOYSA-N Cc(cc1c2c3ccc(C)c2)ccc1[n]3-c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1 Chemical compound Cc(cc1c2c3ccc(C)c2)ccc1[n]3-c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1 IOKAPZIBFLPOFO-UHFFFAOYSA-N 0.000 description 1
- XQNRDLPDGWHLGV-UHFFFAOYSA-N Cc(ccnc1)c1-c(cc1)ccc1-c(cc1)cc(-c2ccccc22)c1N(c(ccc(-c1ccccc1)c1)c1-c1c3cccc1)P23=O Chemical compound Cc(ccnc1)c1-c(cc1)ccc1-c(cc1)cc(-c2ccccc22)c1N(c(ccc(-c1ccccc1)c1)c1-c1c3cccc1)P23=O XQNRDLPDGWHLGV-UHFFFAOYSA-N 0.000 description 1
- AOTZLDUIJYNHEK-UHFFFAOYSA-N Cc(ccnc1)c1-c(cc1)ccc1-c(cc1-c2c3cccc2)ccc1N(c(c(-c1c2cccc1)c1)ccc1-c(cc1)ccc1-c1cnccc1C)P32=O Chemical compound Cc(ccnc1)c1-c(cc1)ccc1-c(cc1-c2c3cccc2)ccc1N(c(c(-c1c2cccc1)c1)ccc1-c(cc1)ccc1-c1cnccc1C)P32=O AOTZLDUIJYNHEK-UHFFFAOYSA-N 0.000 description 1
- KDPQWSJVVFHPAT-UHFFFAOYSA-N Cc(ccnc1)c1-c(cc1-c2ccccc22)ccc1N(c(c(-c1c3cccc1)c1)ccc1-c1ccccc1)P23=O Chemical compound Cc(ccnc1)c1-c(cc1-c2ccccc22)ccc1N(c(c(-c1c3cccc1)c1)ccc1-c1ccccc1)P23=O KDPQWSJVVFHPAT-UHFFFAOYSA-N 0.000 description 1
- JTGMTYWYUZDRBK-UHFFFAOYSA-N Cc1c(cccc2)c2c(C)c2c1cccc2 Chemical compound Cc1c(cccc2)c2c(C)c2c1cccc2 JTGMTYWYUZDRBK-UHFFFAOYSA-N 0.000 description 1
- SMSUQGYWACFERA-UHFFFAOYSA-N Cc1cc(-c(cc2)cc(-c3ccccc33)c2N(c(c(-c2c4cccc2)c2)ccc2-c2ccccc2)P34=O)ccn1 Chemical compound Cc1cc(-c(cc2)cc(-c3ccccc33)c2N(c(c(-c2c4cccc2)c2)ccc2-c2ccccc2)P34=O)ccn1 SMSUQGYWACFERA-UHFFFAOYSA-N 0.000 description 1
- MSIMBJVWXKPVLA-UHFFFAOYSA-N Cc1cc(-c(cc2)ccc2-c(cc2)cc(-c3c4cccc3)c2N(c(c(-c2c3cccc2)c2)ccc2-c(cc2)ccc2-c2cc(C)ncc2)P43=O)ccn1 Chemical compound Cc1cc(-c(cc2)ccc2-c(cc2)cc(-c3c4cccc3)c2N(c(c(-c2c3cccc2)c2)ccc2-c(cc2)ccc2-c2cc(C)ncc2)P43=O)ccn1 MSIMBJVWXKPVLA-UHFFFAOYSA-N 0.000 description 1
- WFNHNCASZBFYGG-UHFFFAOYSA-N Cc1cncc(-c(cc2)ccc2-c(cc2)cc(-c3ccccc33)c2N(c(c(-c2c4cccc2)c2)ccc2-c(cc2)ccc2-c2cc(C)cnc2)P34=O)c1 Chemical compound Cc1cncc(-c(cc2)ccc2-c(cc2)cc(-c3ccccc33)c2N(c(c(-c2c4cccc2)c2)ccc2-c(cc2)ccc2-c2cc(C)cnc2)P34=O)c1 WFNHNCASZBFYGG-UHFFFAOYSA-N 0.000 description 1
- NRFXJBQPRYCOKX-UHFFFAOYSA-N Cc1cncc(-c(cc2)ccc2-c(cc2)cc(-c3ccccc33)c2N(c(c(-c2c4cccc2)c2)ccc2-c2ccccc2)P34=O)c1 Chemical compound Cc1cncc(-c(cc2)ccc2-c(cc2)cc(-c3ccccc33)c2N(c(c(-c2c4cccc2)c2)ccc2-c2ccccc2)P34=O)c1 NRFXJBQPRYCOKX-UHFFFAOYSA-N 0.000 description 1
- SPCBYHDVWYWPDD-UHFFFAOYSA-N Cc1cncc(-c(cc2)ccc2-c(cc2-c3c4cccc3)ccc2N(c(c(-c2c3cccc2)c2)ccc2-c2cc5ccccc5cc2)P43=O)c1 Chemical compound Cc1cncc(-c(cc2)ccc2-c(cc2-c3c4cccc3)ccc2N(c(c(-c2c3cccc2)c2)ccc2-c2cc5ccccc5cc2)P43=O)c1 SPCBYHDVWYWPDD-UHFFFAOYSA-N 0.000 description 1
- KMDWHKYXDGIQNM-UHFFFAOYSA-N Cc1cncc(-c(cc2-c3ccccc33)ccc2N(c(c(-c2c4cccc2)c2)ccc2-c2ccccc2)P34=O)c1 Chemical compound Cc1cncc(-c(cc2-c3ccccc33)ccc2N(c(c(-c2c4cccc2)c2)ccc2-c2ccccc2)P34=O)c1 KMDWHKYXDGIQNM-UHFFFAOYSA-N 0.000 description 1
- ISCCWNHTJZTJJC-UHFFFAOYSA-N Cc1nccc(-c(cc2)ccc2-c(cc2-c3ccccc33)ccc2N(c(c(-c2c4cccc2)c2)ccc2-c2cccc5c2cccc5)P34=O)c1 Chemical compound Cc1nccc(-c(cc2)ccc2-c(cc2-c3ccccc33)ccc2N(c(c(-c2c4cccc2)c2)ccc2-c2cccc5c2cccc5)P34=O)c1 ISCCWNHTJZTJJC-UHFFFAOYSA-N 0.000 description 1
- GETTZEONDQJALK-UHFFFAOYSA-N FC(c1ccccc1)(F)F Chemical compound FC(c1ccccc1)(F)F GETTZEONDQJALK-UHFFFAOYSA-N 0.000 description 1
- JSVRQXARBXFDQM-UHFFFAOYSA-N O=P(c(cccc1)c1-c1cc(-c2ccccc2)ccc11)(c(cccc2)c2-c2c3)N1c2ccc3-c1cccnc1 Chemical compound O=P(c(cccc1)c1-c1cc(-c2ccccc2)ccc11)(c(cccc2)c2-c2c3)N1c2ccc3-c1cccnc1 JSVRQXARBXFDQM-UHFFFAOYSA-N 0.000 description 1
- PEMNSYREHYCMRL-UHFFFAOYSA-N O=P(c(cccc1)c1-c1cc(-c2ccccc2)ccc11)(c2ccccc2-c2c3)N1c2ccc3-c1ncccc1 Chemical compound O=P(c(cccc1)c1-c1cc(-c2ccccc2)ccc11)(c2ccccc2-c2c3)N1c2ccc3-c1ncccc1 PEMNSYREHYCMRL-UHFFFAOYSA-N 0.000 description 1
- QVCIRWLEVPZGLG-UHFFFAOYSA-N O=P1(c(cccc2)c2-c2c3ccc(-c4ccccc4)c2)N3c(ccc(-c2ccncc2)c2)c2-c2ccccc12 Chemical compound O=P1(c(cccc2)c2-c2c3ccc(-c4ccccc4)c2)N3c(ccc(-c2ccncc2)c2)c2-c2ccccc12 QVCIRWLEVPZGLG-UHFFFAOYSA-N 0.000 description 1
- PEWXHFZXSRBDRB-UHFFFAOYSA-N c(cc1)cc(c2ccccc22)c1[n]2-c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1 Chemical compound c(cc1)cc(c2ccccc22)c1[n]2-c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1 PEWXHFZXSRBDRB-UHFFFAOYSA-N 0.000 description 1
- GYRJNZBGIVSDEJ-UHFFFAOYSA-N c(cc1)cc(c2ccccc22)c1[n]2-c1cc(c2cc(-[n]3c4ccccc4c4c3cccc4)c3)c4[n]5c2c3-c2ccccc2B5c(cccc2)c2-c4c1 Chemical compound c(cc1)cc(c2ccccc22)c1[n]2-c1cc(c2cc(-[n]3c4ccccc4c4c3cccc4)c3)c4[n]5c2c3-c2ccccc2B5c(cccc2)c2-c4c1 GYRJNZBGIVSDEJ-UHFFFAOYSA-N 0.000 description 1
- DJEOVJZHZZOGPB-UHFFFAOYSA-N c(cc1)cc2c1-c1cccc(c3ccc4)c1[n]1c3c4-c3ccccc3B21 Chemical compound c(cc1)cc2c1-c1cccc(c3ccc4)c1[n]1c3c4-c3ccccc3B21 DJEOVJZHZZOGPB-UHFFFAOYSA-N 0.000 description 1
- GXTQYZXNSXUFKQ-UHFFFAOYSA-N c(cc1)ccc1-c(cc1c2c3ccc(-c4ccccc4)c2)ccc1[n]3-c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1 Chemical compound c(cc1)ccc1-c(cc1c2c3ccc(-c4ccccc4)c2)ccc1[n]3-c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1 GXTQYZXNSXUFKQ-UHFFFAOYSA-N 0.000 description 1
- IZZSDYPKMXAUCN-UHFFFAOYSA-N c(cc1)ccc1N(c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1)c1c(cccc2)c2ccc1 Chemical compound c(cc1)ccc1N(c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1)c1c(cccc2)c2ccc1 IZZSDYPKMXAUCN-UHFFFAOYSA-N 0.000 description 1
- JKGYJPIAMMPBIN-UHFFFAOYSA-N c(cc1)ccc1N(c1ccccc1)c(cc1)ccc1-c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1 Chemical compound c(cc1)ccc1N(c1ccccc1)c(cc1)ccc1-c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1 JKGYJPIAMMPBIN-UHFFFAOYSA-N 0.000 description 1
- IEAYDXZLAJZLDP-UHFFFAOYSA-N c(cc1)ccc1N(c1ccccc1)c(cc1)ccc1-c1cc(c2cc(-c(cc3)ccc3N(c3ccccc3)c3ccccc3)c3)c4[n]5c2c3-c2ccccc2B5c(cccc2)c2-c4c1 Chemical compound c(cc1)ccc1N(c1ccccc1)c(cc1)ccc1-c1cc(c2cc(-c(cc3)ccc3N(c3ccccc3)c3ccccc3)c3)c4[n]5c2c3-c2ccccc2B5c(cccc2)c2-c4c1 IEAYDXZLAJZLDP-UHFFFAOYSA-N 0.000 description 1
- ZMZUYMBBLIOHIS-UHFFFAOYSA-N c(cc1)ccc1N(c1ccccc1)c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1 Chemical compound c(cc1)ccc1N(c1ccccc1)c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1 ZMZUYMBBLIOHIS-UHFFFAOYSA-N 0.000 description 1
- GHOWJPZHEJTXDN-UHFFFAOYSA-N c(cc1)ccc1N(c1ccccc1)c1cc(c2cc(N(c3ccccc3)c3ccccc3)c3)c4[n]5c2c3-c2ccccc2B5c(cccc2)c2-c4c1 Chemical compound c(cc1)ccc1N(c1ccccc1)c1cc(c2cc(N(c3ccccc3)c3ccccc3)c3)c4[n]5c2c3-c2ccccc2B5c(cccc2)c2-c4c1 GHOWJPZHEJTXDN-UHFFFAOYSA-N 0.000 description 1
- XCPHGYNSJGQQGX-UHFFFAOYSA-N c(cc1c2ccccc22)ccc1[n]2-c(cc1)ccc1-c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1 Chemical compound c(cc1c2ccccc22)ccc1[n]2-c(cc1)ccc1-c1cc(-c2ccccc2B2c(cccc3)c3-c3ccc4)c5[n]2c3c4c5c1 XCPHGYNSJGQQGX-UHFFFAOYSA-N 0.000 description 1
Images

Classifications
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/649—Aromatic compounds comprising a hetero atom
- H10K85/657—Polycyclic condensed heteroaromatic hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic System
- C07F5/02—Boron compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic System
- C07F7/22—Tin compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6564—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms
- C07F9/6581—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus and nitrogen atoms with or without oxygen or sulfur atoms, as ring hetero atoms
- C07F9/6584—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus and nitrogen atoms with or without oxygen or sulfur atoms, as ring hetero atoms having one phosphorus atom as ring hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6564—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms
- C07F9/6581—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus and nitrogen atoms with or without oxygen or sulfur atoms, as ring hetero atoms
- C07F9/6584—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus and nitrogen atoms with or without oxygen or sulfur atoms, as ring hetero atoms having one phosphorus atom as ring hetero atom
- C07F9/65842—Cyclic amide derivatives of acids of phosphorus, in which one nitrogen atom belongs to the ring
- C07F9/65846—Cyclic amide derivatives of acids of phosphorus, in which one nitrogen atom belongs to the ring the phosphorus atom being part of a six-membered ring which may be condensed with another ring system
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K11/00—Luminescent, e.g. electroluminescent, chemiluminescent materials
- C09K11/06—Luminescent, e.g. electroluminescent, chemiluminescent materials containing organic luminescent materials
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05B—ELECTRIC HEATING; ELECTRIC LIGHT SOURCES NOT OTHERWISE PROVIDED FOR; CIRCUIT ARRANGEMENTS FOR ELECTRIC LIGHT SOURCES, IN GENERAL
- H05B33/00—Electroluminescent light sources
- H05B33/12—Light sources with substantially two-dimensional radiating surfaces
- H05B33/14—Light sources with substantially two-dimensional radiating surfaces characterised by the chemical or physical composition or the arrangement of the electroluminescent material, or by the simultaneous addition of the electroluminescent material in or onto the light source
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
- H10K50/10—OLEDs or polymer light-emitting diodes [PLED]
- H10K50/11—OLEDs or polymer light-emitting diodes [PLED] characterised by the electroluminescent [EL] layers
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
- H10K50/10—OLEDs or polymer light-emitting diodes [PLED]
- H10K50/14—Carrier transporting layers
- H10K50/15—Hole transporting layers
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
- H10K50/10—OLEDs or polymer light-emitting diodes [PLED]
- H10K50/17—Carrier injection layers
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
- H10K50/10—OLEDs or polymer light-emitting diodes [PLED]
- H10K50/17—Carrier injection layers
- H10K50/171—Electron injection layers
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
- H10K50/10—OLEDs or polymer light-emitting diodes [PLED]
- H10K50/18—Carrier blocking layers
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/30—Coordination compounds
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/30—Coordination compounds
- H10K85/321—Metal complexes comprising a group IIIA element, e.g. Tris (8-hydroxyquinoline) gallium [Gaq3]
- H10K85/322—Metal complexes comprising a group IIIA element, e.g. Tris (8-hydroxyquinoline) gallium [Gaq3] comprising boron
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/40—Organosilicon compounds, e.g. TIPS pentacene
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/649—Aromatic compounds comprising a hetero atom
- H10K85/657—Polycyclic condensed heteroaromatic hydrocarbons
- H10K85/6572—Polycyclic condensed heteroaromatic hydrocarbons comprising only nitrogen in the heteroaromatic polycondensed ring system, e.g. phenanthroline or carbazole
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1003—Carbocyclic compounds
- C09K2211/1007—Non-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1003—Carbocyclic compounds
- C09K2211/1011—Condensed systems
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1003—Carbocyclic compounds
- C09K2211/1014—Carbocyclic compounds bridged by heteroatoms, e.g. N, P, Si or B
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1018—Heterocyclic compounds
- C09K2211/1025—Heterocyclic compounds characterised by ligands
- C09K2211/1029—Heterocyclic compounds characterised by ligands containing one nitrogen atom as the heteroatom
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1018—Heterocyclic compounds
- C09K2211/1025—Heterocyclic compounds characterised by ligands
- C09K2211/1029—Heterocyclic compounds characterised by ligands containing one nitrogen atom as the heteroatom
- C09K2211/104—Heterocyclic compounds characterised by ligands containing one nitrogen atom as the heteroatom with other heteroatoms
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K2101/00—Properties of the organic materials covered by group H10K85/00
- H10K2101/10—Triplet emission
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
- H10K50/10—OLEDs or polymer light-emitting diodes [PLED]
- H10K50/14—Carrier transporting layers
- H10K50/16—Electron transporting layers
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
- H10K50/10—OLEDs or polymer light-emitting diodes [PLED]
- H10K50/14—Carrier transporting layers
- H10K50/16—Electron transporting layers
- H10K50/166—Electron transporting layers comprising a multilayered structure
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/615—Polycyclic condensed aromatic hydrocarbons, e.g. anthracene
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/615—Polycyclic condensed aromatic hydrocarbons, e.g. anthracene
- H10K85/622—Polycyclic condensed aromatic hydrocarbons, e.g. anthracene containing four rings, e.g. pyrene
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/631—Amine compounds having at least two aryl rest on at least one amine-nitrogen atom, e.g. triphenylamine
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/649—Aromatic compounds comprising a hetero atom
- H10K85/654—Aromatic compounds comprising a hetero atom comprising only nitrogen as heteroatom
Definitions
- the present invention relates to an organic electroluminescent element, a display device and a lighting device using a polycyclic aromatic compound.
- the organic electroluminescent element has a structure composed of a pair of electrodes composed of an anode and a cathode, and one or a plurality of layers including an organic compound disposed between the pair of electrodes.
- the layer containing an organic compound include a light-emitting layer and a charge transport / injection layer that transports or injects charges such as holes and electrons.
- Various organic materials suitable for these layers have been developed.
- benzofluorene compounds and chrysene compounds have been developed as light emitting layer materials (International Publication No. 2004/061047 and International Publication No. 2008/147721).
- hole transport materials for example, triphenylamine compounds and carbazole compounds have been developed (JP 2001-172232 A, JP 2006-199679 A, JP 2005-268199 A). JP 2007-088433 A, International Publication No. 2003/078541, International Publication No. 2003/080760).
- anthracene compounds and compounds in which a central skeleton is bianthracene, binaphthalene, or a combination of naphthalene and anthracene have been developed (JP 2005-170911, JP 2003). No. -146951, JP 08-12600, JP 2003-123983, JP 11-297473).
- PAHs polycyclic aromatic hydrocarbons
- Non-Patent Document 1 a dibenzochrysene compound having a BN bonding site as reported in Non-Patent Document 1 is used for the device. It is not yet known how much performance it will have when applied.
- the present inventors have found a novel polycyclic aromatic compound in which a nitrogen atom and another heteroatom or metal atom (X) are adjacent in a non-aromatic ring, and the production thereof succeeded in. Further, it has been found that an organic electroluminescent device with improved driving voltage and current efficiency can be obtained by arranging an organic electroluminescent device by arranging a layer containing this polycyclic aromatic compound between a pair of electrodes.
- the present invention has been completed. That is, the present invention provides a material for an organic electroluminescence device containing the following polycyclic aromatic compound or salt thereof, and further including the following polycyclic aromatic compound or salt thereof.
- a material for an organic electroluminescence device comprising a polycyclic aromatic compound having a partial structure represented by the following general formula (I) or a salt thereof.
- a ring, B ring, C ring and D ring are each independently an aromatic ring which may be substituted or a heteroaromatic ring which may be substituted, and two adjacent rings are a linking group or A single bond may form a ring between them, and
- the partial structure represented by the above formula (I) has at least one hydrogen, and at least one hydrogen in the partial structure may be substituted with deuterium.
- the benzene ring and the five-membered ring in each of the above formulas may be independently substituted, and adjacent substituents in the same ring may be bonded to form a cyclohexane ring, a benzene ring or a pyridine ring
- the benzene ring and the five-membered ring in each of the above formulas may be independently substituted, and adjacent substituents in the same ring may be bonded to form a cyclohexane ring, a benzene ring or a pyridine ring, Two adjacent benzene rings in each of the above formulas may form a ring between them by a linking group or a single bond, and
- the partial structures represented by the above formulas have at least one hydrogen, and at least one hydrogen in the partial structure may be substituted with deuterium.
- the benzene ring and the five-membered ring in each of the above formulas may be independently substituted, and adjacent substituents in the same ring may be bonded to form a cyclohexane ring, a benzene ring or a pyridine ring, Two adjacent benzene rings in each of the above formulas may form a ring between them by a linking group or a single bond, and
- the partial structures represented by the above formulas have at least one hydrogen, and at least one hydrogen in the partial structure may be substituted with deuterium.
- Two adjacent R's in the same ring may combine to form a cyclohexane ring, a benzene ring or a pyridine ring;
- Two adjacent benzene rings in each of the above formulas are a single bond, CH 2 , CHR a , C (R a ) 2 , NR a , Si (R a ) 2 , BR a (where R a is as defined above). May be linked by a bond through Se, S, or O to form a ring between them, n is an integer from 0 to 4, m is an integer from 0 to 3, and At least one hydrogen in the compound represented by each of the above formulas or a salt thereof may be substituted with deuterium.
- Two adjacent R's in the same ring may combine to form a cyclohexane ring, a benzene ring or a pyridine ring;
- Two adjacent benzene rings in each of the above formulas are a single bond, CH 2 , CHR a , C (R a ) 2 , NR a , Si (R a ) 2 , BR a (where R a is as defined above). May be linked by a bond through Se, S, or O to form a ring between them, n is an integer from 0 to 4, h is an integer from 0 to 3, and At least one hydrogen in the compound represented by each of the above formulas or a salt thereof may be substituted with deuterium.
- Two adjacent R's in the same ring may combine to form a cyclohexane ring, a benzene ring or a pyridine ring;
- Two adjacent benzene rings in each of the above formulas are a single bond, CH 2 , CHR a , C (R a ) 2 , NR a , Si (R a ) 2 , BR a (where R a is as defined above). May be linked by a bond through Se, S, or O to form a ring between them, n is an integer from 0 to 4, h is an integer from 0 to 3, and At least one hydrogen in the compound represented by each of the above formulas or a salt thereof may be substituted with deuterium.
- R is fluorine-substituted or unsubstituted C 1-20 alkyl, C 3-8 cycloalkyl, C 2-20 alkenyl, mono- or diaryl-substituted C 2-12 alkenyl, mono- or diheteroaryl- substituted C 2-12 alkenyl, Fluorine-substituted or unsubstituted C 1-20 alkoxy, C 1-20 alkylcarbonyl, cyano, nitro, diarylamino, optionally substituted aryl, optionally substituted heteroaryl, B (R a ) 2 , Or Si (R a ) 3, wherein each R a is independently an optionally substituted alkyl, an optionally substituted aryl, or an optionally substituted
- Two adjacent R's in the same ring may combine to form a cyclohexane ring, a benzene ring or a pyridine ring; n is an integer from 0 to 4, m is an integer from 0 to 3, and At least one hydrogen in the compound represented by each of the above formulas or a salt thereof may be substituted with deuterium.
- R is fluorine-substituted or unsubstituted C 1-20 alkyl, C 3-8 cycloalkyl, C 2-20 alkenyl, mono- or diaryl-substituted C 2-12 alkenyl, mono- or diheteroaryl- substituted C 2-12 alkenyl, Fluorine-substituted or unsubstituted C 1-20 alkoxy, C 1-20 alkylcarbonyl, cyano, nitro, diarylamino, optionally substituted aryl, optionally substituted heteroaryl, B (R a ) 2 , Or Si (R a ) 3, wherein each R a is independently an optionally substituted alkyl, an optionally substituted aryl, or an optionally substituted heteroaryl.
- Two adjacent R's in the same ring may combine to form a cyclohexane ring, a benzene ring or a pyridine ring; n is an integer from 0 to 4, h is an integer from 0 to 3, and At least one hydrogen in the compound represented by the above formula or a salt thereof may be substituted with deuterium.
- R is fluorine-substituted or unsubstituted C 1-20 alkyl, C 3-8 cycloalkyl, C 2-20 alkenyl, mono- or diaryl-substituted C 2-12 alkenyl, mono- or diheteroaryl- substituted C 2-12 alkenyl, Fluorine-substituted or unsubstituted C 1-20 alkoxy, C 1-20 alkylcarbonyl, cyano, nitro, diarylamino, optionally substituted aryl, optionally substituted heteroaryl, B (R a ) 2 , Or Si (R a ) 3, wherein each R a is independently an optionally substituted alkyl, an optionally substituted aryl, or an optionally substituted heteroaryl.
- Two adjacent R's in the same ring may combine to form a cyclohexane ring, a benzene ring or a pyridine ring; n is an integer from 0 to 4, h is an integer from 0 to 3, and At least one hydrogen in the compound represented by the above formula or a salt thereof may be substituted with deuterium.
- An organic electroluminescent element comprising a pair of electrodes composed of an anode and a cathode, and a light emitting layer disposed between the pair of electrodes and containing the light emitting layer material described in [17] above.
- An organic electroluminescent device having a hole injection layer and / or a hole transport layer containing
- a pair of electrodes composed of an anode and a cathode, a light emitting layer disposed between the pair of electrodes, and a hole blocking layer disposed between the cathode and the light emitting layer, according to the above [19]
- An organic electroluminescence device comprising a hole blocking layer and / or an electron transport layer containing a material or a material for an electron transport layer.
- the electron transport layer and / or the electron injection layer further disposed between the cathode and the light emitting layer, wherein at least one of the electron transport layer and the electron injection layer is a quinolinol-based metal complex,
- At least one of the hole blocking layer and the electron transport layer contains at least one selected from the group consisting of a quinolinol-based metal complex, a pyridine derivative, a phenanthroline derivative, a borane derivative, and a benzimidazole derivative,
- a quinolinol-based metal complex a quinolinol-based metal complex
- a pyridine derivative a phenanthroline derivative
- a borane derivative a benzimidazole derivative
- the hole blocking layer, the electron transport layer, and / or the electron injection layer are further made of an alkali metal, an alkaline earth metal, a rare earth metal, an alkali metal oxide, an alkali metal halide, or an alkaline earth metal. At least selected from the group consisting of oxides, halides of alkaline earth metals, oxides of rare earth metals, halides of rare earth metals, organic complexes of alkali metals, organic complexes of alkaline earth metals and organic complexes of rare earth metals.
- the organic electroluminescent element according to the above [23] or [24], containing one.
- a display device comprising the organic electroluminescent element as described in any one of [20] to [25].
- An illumination device including the organic electroluminescent element according to any one of [20] to [25].
- an excellent polycyclic aromatic compound as an organic electroluminescent element material can be provided, and driving voltage and current efficiency are improved by using this polycyclic aromatic compound.
- An organic electroluminescent element can be provided.
- Partial structure constituting polycyclic aromatic compound The polycyclic aromatic compound (and its salt) of the present invention has a partial structure represented by the following general formula (I) and is useful as a material for an organic electroluminescence device. is there. Each symbol in the formula is as described above.
- partial structure represented by the formula (I) include a partial structure represented by the following general formula (II) or general formula (II ′). Each symbol in the formula is as described above.
- More specific examples of the partial structure represented by the above formula (II) or formula (II ′) include, for example, the following general formulas (III-1) to (III-54) and (III-55): ) To a partial structure represented by formula (III-60).
- the benzene ring and the five-membered ring in each formula may be independently substituted, and adjacent substituents in the same ring may be bonded to form a cyclohexane ring, a benzene ring or a pyridine ring.
- two adjacent benzene rings in each formula may form a ring between them by a linking group or a single bond, each partial structure has at least one hydrogen, and in the partial structure At least one hydrogen may be replaced with deuterium.
- the Z can see "and two Y a adjacent the bond between them together in the same ring, N, O, becomes S or Se" below the defined description of.
- the polycyclic aromatic compound (and its salt) of the present invention is a compound containing the above-mentioned partial structure (for example, consisting of repetition of the partial structure). Examples thereof include compounds represented by the following general formulas (IV-1) to (IV-22).
- each Y is independently CR (R will be described later) or N, and two Ys adjacent to each other in the same ring and a bond between them are combined.
- NR R will be described later
- a metal element of Group 3 to 11 of the periodic table that may be substituted, or a metal element or metalloid element of Groups 13 to 14 of the periodic table that may be substituted is shown.
- R in the above formulas (IV-1) to (IV-22) is hydrogen, halogen, C 1-20 alkyl, hydroxy C 1-20 alkyl, trifluoromethyl C 2-12 perfluoroalkyl, C 3-8 cycloalkyl, C 2-20 alkenyl, C 2-20 alkynyl, mono- or diaryl-substituted alkenyl, mono- or diheteroaryl-substituted alkenyl, arylethynyl, heteroarylethynyl, Hydroxy, C 1-20 alkoxy, aryloxy, trifluoromethoxy, trifluoroethoxy, C 2-12 perfluoroalkoxy, C 1-20 alkylcarbonyl, C 1-20 alkylsulfonyl, cyano, nitro, amino, monoalkylamino , Monoarylamino, monoheteroarylamino , Diarylamino, carbazolyl,
- alkyl, alkenyl, alkynyl and alkoxy are halogen, hydroxy, C 1-20 alkoxy, aryloxy, amino, carbazolyl, N (R a ) 2 (R a is as defined above), trifluoro Optionally substituted by 1 to 3 groups selected from methyl, C 2-12 perfluoroalkyl, C 3-8 cycloalkyl, aryl and heteroaryl, and the above aryl group, aryl moiety, heteroaryl Groups, heteroaryl moieties, carbazole groups are halogen, C 1-20 alkyl, hydroxy C 1-20 alkyl, trifluoromethyl, C 2-12 perfluoroalkyl, C 3-8 cycloalkyl, C 2-20 alkenyl, C 2-20 alkynyl, mono- or diaryl-substituted alkenyl, Roh or heteroaryl substituted alkenyl into di, aryl ethynyl, hetero aryl eth
- Two adjacent R's may be a monocyclic group, a bicyclic group or a tricyclic group which may have a 5-membered or 6-membered heteroatom together with the carbon atom to which they are bonded.
- a cyclic group may be formed, and examples thereof include a cyclohexane ring, a benzene ring or a pyridine ring.
- three adjacent Rs may form a bicyclic group or a tricyclic group which may have a hetero atom together with the carbon atom to which they are bonded.
- the two R's are a single bond, CH 2 , CHR a , CR a 2 , NR a , Si (R a ) 2 , BR a (R a is as defined above) and may be Se, S, or O to join two adjacent rings.
- at least one hydrogen in the entire structure may be replaced with deuterium.
- M is an integer of 0 to 3, preferably an integer of 0 to 2, more preferably an integer of 0 to 1, and still more preferably 0.
- k is an integer of 0 to 2, preferably an integer of 0 to 1, and more preferably 0.
- polycyclic aromatic compound (and its salt) of the present invention include, for example, the following general formulas (V-1) to (V-26) and (V-27) to ( And a compound represented by V-34).
- R represents hydrogen, fluorine-substituted or unsubstituted C 1-20 alkyl, C 3- 8 cycloalkyl, C 2-20 alkenyl, mono or diaryl substituted C 2-12 alkenyl, mono or diheteroaryl substituted C 2-12 alkenyl, fluorine substituted or unsubstituted C 1-20 alkoxy, C 1-20 alkylcarbonyl , Cyano, nitro, diarylamino, optionally substituted aryl, optionally substituted heteroaryl, B (R a ) 2 , or Si (R a ) 3 (wherein R a is independently Or an optionally substituted alkyl, an optionally substituted aryl or an optionally substituted heteroaryl.
- pyrrole rings in formulas (V-27) to (V-34) pyrrole rings other than those in which N is involved in the condensation (eg pyrrole ring in formula (V-27))
- hydrogen is basically bonded to N (> N—H), but a substituent R may be bonded (> N—R).
- Detailed description with reference to the figures may refer to the description of "the two Y a adjacent in the same ring and coupling therebetween together, comprising N, O, an S or Se" later.
- two adjacent Rs in the same ring may be bonded to form a cyclohexane ring, a benzene ring or a pyridine ring.
- two adjacent benzene rings in each of the above formulas are a single bond, CH 2 , CHR a , C (R a ) 2 , NR a , Si (R a ) 2 , BR a (where R a is And may be linked by a bond via Se, S, or O to form a ring between them.
- at least one hydrogen in the entire structure may be replaced with deuterium.
- N is an integer of 0 to 4, preferably an integer of 0 to 2, more preferably an integer of 0 to 1, and still more preferably 0.
- m is an integer of 0 to 3, preferably an integer of 0 to 2, more preferably an integer of 0 to 1, and still more preferably 0.
- k is an integer of 0 to 2, preferably an integer of 0 to 1, and more preferably 0.
- h is an integer of 0 to 3, preferably an integer of 0 to 2, more preferably an integer of 0 to 1, and still more preferably 0.
- polycyclic aromatic compound (and its salt) of the present invention include, for example, the following general formula (V-1 ′), formula (V-2 ′), or formula (V-3 ′): And a compound represented by the following formula (V-27 ′) or formula (V-32 ′).
- V-1 formula
- V-2 formula
- V-3 formula
- V-27 ′ formula
- V-32 ′ a compound represented by the following formula (V-27 ′) or formula (V-32 ′).
- These compounds are represented by the above formula (V-1), formula (V-2) or formula (V-3) and formula (V-27) or formula (V-32) as X as element B Corresponds to the selected one.
- R, n, m and h in the formula are as defined above.
- R is aryl.
- R include, for example, phenyl , (2-, 3-, 4-) biphenylyl, terphenylyl (m-terphenyl-2'-yl, m-terphenyl-4'-yl, m-terphenyl-5'-yl, o-terphenyl- 3'-yl, o-terphenyl-4'-yl, p-terphenyl-2'-yl, m-terphenyl-2-yl, m-terphenyl-3-yl, m-terphenyl-4- Yl, o-terphenyl-2-yl, o-terphenyl-3-yl, o-terphenyl-4-yl, p-terphenyl-2-yl, p-terphenyl-3-yl, p-terphenyl-4-yl, p-terphenyl-2-yl, p-terphenyl-3-yl, p-
- substitution position of R is the benzene ring bonded to N in formula (V-1 ′), formula (V-27 ′) and formula (V-32 ′) (the B ring of formula (I) and / or (Corresponding to the D ring), substitution at the para position is preferred based on the position of the carbon bonded to N, and the para position of one ring of the B ring or D ring is substituted, or the paras of both rings The position may be substituted, and it is preferred that the para positions of both rings are substituted.
- the benzene ring bonded to B (corresponding to the A ring and / or the C ring of formula (I)) is the carbon bonded to B.
- the substitution at the ortho position is preferred with respect to the position of, and the ortho position of one of the rings A or C may be substituted, or the ortho positions of both rings may be substituted.
- compounds represented by formulas (51) to (86) described later are preferable, compounds represented by formulas (66) to (83) and (86) are more preferable, and formulas (66) to (86) 74) is more preferable.
- the substituent R (aryl) may be further substituted.
- the example substituted by the phenyl group, the diarylamino group, the carbazolyl group which may be substituted, etc. are mentioned.
- Examples of the “aryl” in the diarylamino group include an aryl described below (eg, phenyl or naphthyl), and examples of the substituent on the carbazolyl group include an alkyl described below (eg, C 1-3 alkyl) and an aryl described below (eg, phenyl and biphenylyl). And naphthyl).
- compounds represented by the following formulas (192), (196), (199), (205) and (209) are preferable.
- the compound in which the substituent R is an N-containing structure includes, for example, diaryl.
- examples thereof include an amino group and an optionally substituted carbazolyl group.
- Examples of the “aryl” in the diarylamino group include an aryl described below (eg, phenyl or naphthyl), and examples of the substituent on the carbazolyl group include an alkyl described below (eg, C 1-3 alkyl) and an aryl described below (eg, phenyl and biphenylyl). And naphthyl).
- substitution position of R is the benzene ring bonded to N in formula (V-1 ′), formula (V-27 ′) and formula (V-32 ′) (the B ring of formula (I) and / or (Corresponding to the D ring), substitution at the para position is preferred based on the position of the carbon bonded to N, and the para position of one ring of the B ring or D ring is substituted, or the paras of both rings The position may be substituted. Specifically, formulas (188) to (191), formulas (193) to (195), formula (197), formula (198), formulas (200) to (204), and formula (206) to The compound represented by (208) is preferred.
- polycyclic aromatic compound (and its salt) of the present invention include, for example, compounds represented by the following general formulas (VI-1) to (VI-149) (these compounds are further These may be substituted, and these substituents may be bonded to each other to form a cyclohexane ring, a benzene ring or a pyridine ring).
- X and Z are as defined above.
- Examples of the metal element of Group 3 to 11 of the periodic table, the metal element or metalloid of Group 13 to 14 of the periodic table represented by X include the following.
- the metal element of Group 3 to 11 of the periodic table, the metal element of Group 13 to 14 of the periodic table, or the metalloid element represented by X may each be substituted.
- these metal elements or metalloid elements are “substituted” by 1 to 3 substituents R (R is as defined above), or 1 to 3 neutrals. It meant having a ligand R 1.
- the neutral ligand R 1 include aromatic compounds having a nitrogen atom as a ring atom, such as pyridine, bipyridine, phenanthroline, terpyridine, imidazole, pyrimidine, pyrazine, quinoline, isoquinoline, and acridine, and derivatives thereof.
- R and R 1 may be formed by a single compound (8-hydroxyquinoline) as follows Case (3).
- the compound having a neutral ligand R 1 can be produced, for example, as follows. (In the following formula, (R) indicates that R 1 is R as defined above, and (R 1 ) indicates that R 1 is a neutral ligand.)
- Case (1) represents a case where a neutral ligand (R 1 ) is bonded to X (metal element or metalloid element) of formula (I) to obtain (I ′) compound.
- a compound having a neutral ligand can be easily produced by those skilled in the art with reference to the above Case (1) to Case (3).
- X 1 and X 2 can be changed when the electronegativity is approximately the same or in a combination where X 1 ⁇ X 2 .
- X 1 Ge—R
- X 2 may be B
- P, P O
- P S
- As S
- Sb It can be changed to O
- Solvents include anhydrous ether solvents such as anhydrous diethyl ether, anhydrous THF, and anhydrous dibutyl ether, aromatic hydrocarbon solvents such as benzene, toluene, xylene, mesitylene, and aromatics such as chlorobenzene and 1,2-dichlorobenzene.
- Anhydrous ether solvents such as anhydrous diethyl ether, anhydrous THF, and anhydrous dibutyl ether
- aromatic hydrocarbon solvents such as benzene, toluene, xylene, mesitylene
- aromatics such as chlorobenzene and 1,2-dichlorobenzene.
- a halide solvent is used.
- Lewis acids examples include AlCl 3 , AlBr 3 , BF 3 .OEt 2 , BCl 3 , BBr 3 , GaCl 3 , GaBr 3 , InCl 3 , InBr 3 , In (OTf) 3 , SnCl 4 , SnBr 4 f, AgT (OTf) 3 , ZnCl 2 , ZnBr 2 , Zn (OTf) 2 , MgCl 2 , MgBr 2 , Mg (OTf) 2, or the like is used.
- Bases include diisopropylethylamine, 2,2,6,6-tetramethylpiperidine, 1,2,2,6,6-pentamethylpiperidine, 2,4,6-collidine, 2,6-lutidine, triethylamine, triethylamine Isobutylamine or the like is used.
- a compound having a sulfur atom bonded thereto can be similarly obtained when X 2 is other elements such as As and Sb.
- Preferred X includes B, P, P ⁇ O, P ⁇ S, Si—R, Ge—R, Ga, Pt, Ru, Ir, Au, and the like.
- the partial structure has at least one hydrogen means that the atoms forming ring A, ring B, ring C, and ring D are all other atoms when the above general formula (I) is used. Means that at least one atom is necessarily bonded to hydrogen and terminates, and includes a partial structure represented by the general formula (I) (for example, repetition of the partial structure). This means that the polycyclic aromatic compound or a salt thereof does not include, for example, heterofullerene or heterocarbon nanotube in which a part of the carbon skeleton of fullerene or carbon nanotube is substituted with boron or nitrogen.
- adjacent R may be an adjacent group of the same ring, or may be the closest Rs of adjacent rings.
- aromatic ring of “optionally substituted aromatic ring” examples include benzene ring, naphthalene ring, azulene ring, biphenylene ring, fluorene ring, anthracene ring, indacene ring, phenanthrene ring, phenalene ring, pyrene ring, chrysene ring , Triphenylene ring, fluoranthene ring, acephenanthrylene ring, acanthrylene ring, picene ring, naphthacene ring, perylene ring, acenaphthylene ring, acenaphthene ring, indane ring, indene ring, and tetrahydronaphthalene ring.
- heteroaromatic ring of the “optionally substituted heteroaromatic ring” examples include a furan ring, a thiophene ring, a selenophene ring, a pyrrole ring, an imidazole ring, a thiazole ring, an isothiazole ring, an oxazole ring, an isoxazole ring, and a triazole.
- the number of substituents of the optionally substituted aromatic ring or optionally substituted heteroaromatic ring is 1 to 4, preferably 1, 2 or 3.
- substituents of the aromatic ring which may be substituted or the heteroaromatic ring which may be substituted include a group represented by R.
- Examples of the “monocyclic group, bicyclic group or tricyclic group optionally having a 5-membered or 6-membered hetero atom” include benzene, naphthalene, azulene, biphenylene, fluorene, anthracene, indacene, Phenanthrene, phenalene, acenaphthylene, acenaphthene, indane, indene, tetrahydronaphthalene, cyclopentadiene, cyclohexadiene, furan, thiophene, selenophene, pyrrole, imidazole, thiazole, isothiazole, oxazole, isoxazole, triazole, borole, phosphorol, silole, azaborine Pyridine, pyrimidine, triazine, pyran, indole, isoindole, quinoline, isoquinoline, qui
- Examples of the ⁇ bicyclic group or tricyclic group optionally having a hetero atom '' include naphthalene, azulene, biphenylene, fluorene, anthracene, indacene, phenanthrene, phenalene, acenaphthylene, acenaphthene, indane, indene, tetrahydronaphthalene, Indole, isoindole, quinoline, isoquinoline, quinoxaline, benzoxazole, benzothiazole, benzoisoxazole, benzoisothiazole, benzofuran, benzothiophene, benzopyran, benzimidazole, benzoborol, benzophosphole, benzosilol, benzoazaborine, indolizine, acridine, Phenazine, phenanthridine, phenanthroline, benzoselenophene, naphthofur
- the number of carbon atoms is defined as “C 1-20 alkylcarbonyl”, but this number of carbon atoms modifies only the immediately following group or moiety. Therefore, in the above case, since C 1-20 modifies only alkyl, “C 1 alkylcarbonyl” corresponds to acetyl.
- the alkyl group and the alkyl moiety may be linear or branched.
- alkyl may be substituted, C 1-20 alkylsulfonyl, C 1-20 alkylsulfonylamino, each alkyl such as in C 1-20 alkylcarbonylamino and C 1-20 alkylcarbonyl It includes not only a group but also an alkyl group which is a substituent such as monoalkylamino, mono- or di-alkylsulfamoyl, mono- or di-alkylcarbamoyl.
- the aryl moiety means an aryl group such as mono- or diaryl-substituted alkenyl, arylethynyl, aryloxy, monoarylamino, and optionally substituted aryl.
- the heteroaryl moiety means a heteroaryl group such as monoheteroarylamino, mono- or heteroaryl-substituted alkenyl, heteroarylethynyl, and optionally substituted heteroaryl.
- Halogen means fluorine, chlorine, bromine or iodine, with fluorine, chlorine or bromine being preferred.
- the “C 1-20 alkyl” may be linear, branched or cyclic, for example, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, t-butyl, n-pentyl. , Isopentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, tetradecyl, hexadecyl, octadecyl, eicosyl, etc., C 1-20 alkyl, preferably C 1-10 alkyl, more preferably C 1-6 alkyl It is done.
- C 3-8 cycloalkyl includes cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl, cyclooctyl.
- C 2-20 alkenyl may be linear, branched or cyclic, and means at least one double bond, such as vinyl, allyl, 1-propenyl, 2-methyl. -2-propenyl, isopropenyl, 1-, 2- or 3-butenyl, 2-, 3- or 4-pentenyl, 2-methyl-2-butenyl, 3-methyl-2-butenyl, 5-hexenyl, 1- Examples thereof include cyclopentenyl, 1-cyclohexenyl and 3-methyl-3-butenyl, preferably C 2-12 alkenyl, more preferably C 2-6 alkenyl.
- C 2-20 alkynyl may be linear, branched or cyclic, and has at least one triple bond, such as ethynyl, 1- or 2-propynyl, 1-, 2- or 3-butynyl, 1-methyl-2-propynyl, 1-pentynyl, 1-hexynyl, 1-heptynyl, 1-octynyl, 1-nonenyl, 1-decynyl, 1-undecynyl, 1-dodecynyl C 2-10 alkynyl is preferable, and C 2-6 alkynyl is more preferable.
- “Hydroxy C 1-20 alkyl” may be linear or branched. For example, hydroxymethyl, hydroxyethyl, hydroxy n-propyl, hydroxyisopropyl, hydroxy n-butyl, hydroxyisobutyl, hydroxy t -Butyl, hydroxy n-pentyl, hydroxyisopentyl, hydroxyhexyl, hydroxyheptyl, hydroxyoctyl, hydroxynonyl, hydroxydecyl, hydroxyundecyl, hydroxydodecyl, hydroxytetradecyl, hydroxyhexadecyl, hydroxyoctadecyl, hydroxyeicosyl, etc. Examples thereof include hydroxy C 1-20 alkyl, preferably hydroxy C 1-10 alkyl, more preferably hydroxy C 1-6 alkyl.
- C 1-20 alkoxy may be linear or branched, and includes, for example, methoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy, t-butoxy, pentyloxy, isopentyloxy, hexyl.
- C 1-20 alkoxy such as oxy, heptyloxy, octyloxy, nonyloxy, decyloxy, undecyloxy, dodecyloxy, tetradecyloxy, hexadecyloxy, octadecyloxy, eicosyloxy, preferably C 1-10 alkoxy, and more
- C 1-6 alkoxy is used.
- CF 3 CH 2 O— is preferable.
- C 2-12 perfluoroalkyl may be either linear or branched, for example, perfluoroethyl, perfluoro n-propyl, perfluoroisopropyl, perfluoro n-butyl, perfluoroisobutyl.
- C 2-12 perfluoro such as perfluoro t-butyl, perfluoro n-pentyl, perfluoroisopentyl, perfluorohexyl, perfluoroheptyl, perfluorooctyl, perfluorononyl, perfluorodecyl, perfluoroundecyl
- alkyl preferably C 2-10 perfluoroalkyl, more preferably C 2-6 perfluoroalkyl.
- C 2-12 perfluoroalkoxy may be linear or branched, and examples thereof include perfluoroethoxy, perfluoro n-propyloxy, perfluoroisopropyloxy, perfluoro n-butoxy, perfluoro Fluoroisobutoxy, perfluoro t-butoxy, perfluoro n-pentyloxy, perfluoroisopentyloxy, perfluorohexyloxy, perfluoroheptyloxy, perfluorooctyloxy, perfluorononyloxy, perfluorodecyloxy, perfluoro Examples thereof include C 2-12 perfluoroalkoxy such as undecyloxy , preferably C 2-10 perfluoroalkoxy, more preferably C 2-6 perfluoroalkoxy.
- “Monoalkyl” in monoalkylamino, mono or dialkylcarbamoyl or mono or dialkylsulfamoyl has one of the hydrogen atoms bonded to the nitrogen atom of amino, carbamoyl or sulfamoyl substituted with C 1-20 alkyl
- dialkyl means that two of the hydrogen atoms bonded to the nitrogen atom of amino, carbamoyl or sulfamoyl are substituted with the same or different C 1-20 alkyl, or preferably 3 to 8 members, preferably It is substituted with a 5- or 6-membered nitrogen-containing cyclic group.
- Nitrogen-containing cyclic groups include morpholino, 1-pyrrolidinyl, piperidino and 4-methyl-1-piperazinyl.
- Examples of monoalkylamino include C 1-20 such as methylamino, ethylamino, n-propylamino, isopropylamino, n-butylamino, isobutylamino, t-butylamino, n-pentylamino, isopentylamino, hexylamino and the like.
- Examples include amino monosubstituted by alkyl, preferably C 1-10 alkyl, more preferably C 1-6 alkyl.
- Examples of the monoalkylcarbamoyl include C 1-20 such as methylcarbamoyl, ethylcarbamoyl, n-propylcarbamoyl, isopropylcarbamoyl, n-butylcarbamoyl, isobutylcarbamoyl, t-butylcarbamoyl, n-pentylcarbamoyl, isopentylcarbamoyl, hexylcarbamoyl and the like. Mention may be made of carbamoyl monosubstituted by alkyl, preferably C 1-10 alkyl, more preferably C 1-6 alkyl.
- dialkylcarbamoyl examples include dimethylcarbamoyl, diethylcarbamoyl, di-n-propylcarbamoyl, diisopropylcarbamoyl, din-butylcarbamoyl, diisobutylcarbamoyl, dit-butylcarbamoyl, din-pentylcarbamoyl, diisopentylcarbamoyl, dihexylcarbamoyl, etc.
- Examples thereof include carbamoyl disubstituted with C 1-20 alkyl, preferably C 1-10 alkyl, more preferably C 1-6 alkyl.
- monoalkylsulfamoyl examples include methylsulfamoyl, ethylsulfamoyl, n-propylsulfamoyl, isopropylsulfamoyl, n-butylsulfamoyl, isobutylsulfamoyl, t-butylsulfamoyl, n -Sulfamoyl monosubstituted by C 1-20 alkyl, preferably C 1-10 alkyl, more preferably C 1-6 alkyl, such as pentyl sulfamoyl, isopentyl sulfamoyl, hexyl sulfamoyl and the like.
- Dialkylsulfamoyl includes dimethylsulfamoyl, diethylsulfamoyl, di-n-propylsulfamoyl, diisopropylsulfamoyl, di-n-butylsulfamoyl, diisobutylsulfamoyl, di-t-butylsulfamoyl Sulfamoyl disubstituted with C 1-20 alkyl, preferably C 1-10 alkyl, more preferably C 1-6 alkyl, such as di-n-pentylsulfamoyl, diisopentylsulfamoyl, dihexylsulfamoyl, etc. Is mentioned.
- Aryl means a monocyclic or polycyclic group consisting of a 5- or 6-membered aromatic hydrocarbon ring. Specific examples include phenyl, (1-, 2-) naphthyl, fluorenyl, anthryl, (2-, 3-, 4-) biphenylyl, tetrahydronaphthyl, 2,3-dihydro-1,4-dioxanaphthalenyl, terphenylyl (m-terphenyl-2'-yl, m-terphenyl-4 ' -Yl, m-terphenyl-5'-yl, o-terphenyl-3'-yl, o-terphenyl-4'-yl, p-terphenyl-2'-yl, m-terphenyl-2- Yl, m-terphenyl-3-yl, m-terphenyl-4-yl, o-terphenyl-2-yl, o-terphenyl-3-yl,
- Heteroaryl means a monocyclic or polycyclic group of 5 or 6 membered aromatic rings containing 1 to 3 heteroatoms selected from N, O, S, Se and Si In the case of a polycyclic system, at least one ring may be an aromatic ring.
- Monoarylamino includes monoarylamino, where aryl is as defined above.
- Diarylamino includes diarylamino where aryl is as defined above.
- Monoheteroarylamino includes monoheteroarylamino wherein heteroaryl is as defined above.
- the “C 1-20 alkylsulfonyl” may be linear, branched or cyclic, and examples thereof include methylsulfonyl, ethylsulfonyl, n-propylsulfonyl, isopropylsulfonyl, n-butylsulfonyl, isobutylsulfonyl, t-butylsulfonyl, n-pentylsulfonyl, isopentylsulfonyl, hexylsulfonyl, heptylsulfonyl, octylsulfonyl, nonylsulfonyl, decylsulfonyl, undecylsulfonyl, dodecylsulfonyl, tetradecylsulfonyl, hexadecylsulfonyl, oc
- C 1-20 alkylcarbonylamino may be any of linear, branched or cyclic, for example, methylcarbonylamino, ethylcarbonylamino, n-propylcarbonylamino, isopropylcarbonylamino, n-butyl.
- Carbonylamino isobutylcarbonylamino, t-butylcarbonylamino, n-pentylcarbonylamino, isopentylcarbonylamino, hexylcarbonylamino, heptylcarbonylamino, octylcarbonylamino, nonylcarbonylamino, decylcarbonylamino, undecylcarbonylamino, dodecyl carbonylamino, tetradecyl carbonylamino, hexadecyl carbonylamino, octadecyl carbonylamino, C 1-20 aralkyl, such as eicosyl carbonylamino Le carbonylamino, preferably C 1-10 alkylcarbonylamino, or more preferably C 1-6 alkylcarbonylamino.
- C 1-20 alkoxycarbonylamino (for example, C 1-12 alkoxycarbonylamino, C 1-6 alkoxycarbonylamino) includes methoxycarbonylamino, ethoxycarbonylamino, propoxycarbonylamino, isopropoxycarbonylamino, butoxycarbonylamino, iso Examples include butoxycarbonylamino, t-butoxycarbonylamino, pentyloxycarbonylamino, isopentyloxycarbonylamino and hexyloxycarbonylamino.
- C 1-20 alkylsulfonylamino (eg, C 1-10 alkylsulfonylamino, C 1-6 alkylsulfonylamino) includes methylsulfonylamino, ethylsulfonylamino, n-propylsulfonylamino, isopropylsulfonylamino, n-butylsulfonyl Amino, isobutylsulfonylamino, t-butylsulfonylamino, n-pentylsulfonylamino, isopentylsulfonylamino, hexylsulfonylamino, octylsulfonylamino, nonylsulfonylamino, decylsulfonylamino, undecylsulfonylamino, dodecylsulfon
- C 1-20 alkoxycarbonyl (eg C 1-10 alkoxycarbonyl, C 1-6 alkoxycarbonyl) includes methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, isopropoxycarbonyl, butoxycarbonyl, isobutoxycarbonyl, t-butoxycarbonyl, Examples include pentyloxycarbonyl, isopentyloxycarbonyl and hexyloxycarbonyl.
- C 1-20 alkylcarbonyl (eg C 1-10 alkylcarbonyl, C 1-6 alkylcarbonyl) includes acetyl, propionyl, butyryl, pentylcarbonyl, hexylcarbonyl, heptylcarbonyl, octylcarbonyl, nonylcarbonyl, decylcarbonyl. It is done.
- Monoaryl substituted alkenyl (eg monoaryl substituted C 2-12 alkenyl, monoaryl substituted C 2-6 alkenyl) includes monoaryl substituted alkenyl wherein aryl is as defined above, eg styryl.
- Diaryl-substituted alkenyl includes diaryl-substituted alkenyl wherein aryl is as defined above, eg diphenylvinyl.
- Monoheteroaryl-substituted alkenyl eg mono-heteroaryl-substituted C 2-12 alkenyl, mono-heteroaryl-substituted C 2-6 alkenyl
- heteroaryl eg thienyl Vinyl
- Diheteroaryl substituted alkenyl (eg diheteroaryl substituted C 2-12 alkenyl, diheteroaryl substituted C 2-6 alkenyl) includes diheteroaryl substituted alkenyl wherein heteroaryl is as defined above, eg diheteroaryl substituted alkenyl Examples include thienyl vinyl.
- Arylethynyl includes arylethynyl, where aryl is as defined above.
- Heteroarylethynyl includes heteroarylethynyl where heteroaryl is as defined above.
- Aryloxy includes aryloxy in which aryl is as defined above.
- R a represents an optionally substituted alkyl, an optionally substituted aryl, or an optionally substituted heteroaryl.
- alkyl of the optionally substituted alkyl include the above C 1-20 alkyl
- aryl of the optionally substituted aryl includes the above aryl.
- heteroaryl of the optionally substituted heteroaryl include the above heteroaryl.
- polycyclic aromatic compound of the present invention include compounds represented by the following formulas (1) to (709).
- the compound of the present invention is a polycyclic aromatic compound (and a salt thereof) and has a partial structure represented by the above general formula (I), more specifically, the above general formula (II) or general formula (II). And a partial structure represented by the general formula (III-1) to the formula (III-54) and the general formula (III-55) to the formula (III-60). It is a polycyclic aromatic compound.
- the overall structure is, for example, a polycyclic aromatic compound represented by the above general formula (IV-1) to formula (IV-22), and more specifically, the above general formula (V-1) to formula ( V-26) and the polycyclic aromatic compounds represented by the general formulas (V-27) to (V-34), the general formula (V-1 ′), the formula (V-2 ′) and the formula (V V-3 ′) and a polycyclic aromatic compound represented by the above general formula (V-27 ′) or formula (V-32 ′), the above general formula (VI-1) A polycyclic aromatic compound represented by the formula (VI-149), and a polycyclic aromatic compound represented by the above formulas (1) to (709).
- an alkyl lithium such as n-BuLi
- a Grignard reagent such as n-BuMgBr
- an alkali metal hydride such as NaH or KH
- an alkali such as NaO t Bu or KO t Bu
- a base such as an alkali metal carbonate such as metal alkoxide, Na 2 CO 3 , NaHCO 3 , K 2 CO 3 , Cs 2 CO 3, etc.
- the compound (a3) is obtained by reacting with Pd (dba) 2 and P t Bu 3 while stirring in a solvent at a temperature of about ⁇ 78 ° C. to about room temperature for 30 minutes to 24 hours.
- a solvent an anhydrous ether solvent such as anhydrous diethyl ether, anhydrous THF, or anhydrous dibutyl ether or an aromatic hydrocarbon solvent such as benzene, toluene, xylene, mesitylene or the like is used.
- Step 2 the compound (a3) is deprotonated with a deprotonating agent such as n-BuLi, and then a compound containing X (a halide of X, an alkoxy derivative, an aryloxy derivative, an acyloxy derivative, a haloamino derivative) ) Is introduced, and a Friedel-Crafts-type reaction is carried out in the presence of a Lewis acid such as AlCl 3 and a base such as diisopropylethylamine to obtain the compound (a4).
- a deprotonating agent such as n-BuLi
- the compound containing X is a halide such as PF 3 , PCl 3 , PBr 3 , PI 3 , P (OMe) 3 , P (OEt) 3 , P (OnPr) 3 , Alkoxy derivatives such as P (O-iPr) 3 , P (O-nBu) 3 , P (O-iBu) 3 , P (O-secBu) 3 , P (Ot-Bu) 3 , P (OPh) 3 , aryloxy derivatives such as P (O-naphthyl) 3 , P (OAc) 3 , P (O-trifluoroacetyl) 3 , P (O-propionyl) 3 , P (O-butyryl) 3 , P (O -Benzoyl) 3 and other acyloxy derivatives, PCl (NMe 2 ) 2 , PCl (NEt 2 ) 2 , PCl (NPr 2 )
- Solvents include anhydrous ether solvents such as anhydrous diethyl ether, anhydrous THF, and anhydrous dibutyl ether, aromatic hydrocarbon solvents such as benzene, toluene, xylene, and mesitylene, and aromatic substances such as chlorobenzene and 1,2-dichlorobenzene.
- anhydrous ether solvents such as anhydrous diethyl ether, anhydrous THF, and anhydrous dibutyl ether
- aromatic hydrocarbon solvents such as benzene, toluene, xylene, and mesitylene
- aromatic substances such as chlorobenzene and 1,2-dichlorobenzene.
- Group halide solvents are used.
- deprotonating agents examples include n-BuLi, alkyllithium such as MeLi, t-BuLi, and PhLi, Grignard reagents such as MeMgBr, EtMgBr, and n-BuMgBr, or alkali metal hydrides such as NaH and KH. Used.
- Lewis acids examples include AlCl 3 , AlBr 3 , BF 3 .OEt 2 , BCl 3 , BBr 3 , GaCl 3 , GaBr 3 , InCl 3 , InBr 3 , In (OTf) 3 , SnCl 4 , SnBr 4 f, AgT (OTf) 3 , ZnCl 2 , ZnBr 2 , Zn (OTf) 2 , MgCl 2 , MgBr 2 , Mg (OTf) 2, or the like is used.
- Bases include diisopropylethylamine, 2,2,6,6-tetramethylpiperidine, 1,2,2,6,6-pentamethylpiperidine, 2,4,6-collidine, 2,6-lutidine, triethylamine, triethylamine Isobutylamine or the like is used.
- Step 2 ′ reaction compound (a3 ′) is used instead of compound (a3), and compound (a4 ′) is obtained by performing Friedel-Crafts type reaction and Scholl type reaction under the same conditions as in Step 2 reaction. be able to.
- the compound (a4 ′) can be obtained by performing the Friedel-Crafts type reaction under the same conditions as in the step 2 reaction using the compound (a3 ′′) instead of the compound (a3).
- Step 1 'of the following scheme 1-3 can also be used. That is, it is a step of producing a diarylamine (a3) by reacting an aromatic halide (a1 ') with an aromatic amine (a2) using a palladium catalyst in the presence of a base.
- palladium catalyst used in Step 1 ′ are [1,1-bis (diphenylphosphino) ferrocene] palladium (II) dichloride: Pd (dppf) Cl 2 , tetrakis (triphenylphosphine) palladium (0): Pd (PPh 3 ) 4 , bis (triphenylphosphine) palladium (II) dichloride: PdCl 2 (PPh 3 ) 2 , palladium (II) acetate: Pd (OAc) 2 , tris (dibenzylideneacetone) dipalladium (0): Pd 2 (dba) 3 , tris (dibenzylideneacetone) dipalladium (0) chloroform complex: Pd 2 (dba) 3 ⁇ CHCl 3 , bis (dibenzylideneacetone) palladium (0): Pd (dba) 2 , PdCl 2 ⁇ P
- a phosphine compound may be added to these palladium compounds in some cases.
- the phosphine compound include tri (t-butyl) phosphine, tricyclohexylphosphine, 1- (N, N-dimethylaminomethyl) -2- (di-t-butylphosphino) ferrocene, 1- (N, N -Dibutylaminomethyl) -2- (di-t-butylphosphino) ferrocene, 1- (methoxymethyl) -2- (di-t-butylphosphino) ferrocene, 1,1'-bis (di-t-butylphosphino) ) Ferrocene, 2,2′-bis (di-t-butylphosphino) -1,1′-binaphthyl, 2-methoxy-2 ′-(di-t-butylphosphino) -1,
- base used in Step 1 ′ are sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogen carbonate, sodium hydroxide, potassium hydroxide, barium hydroxide, sodium ethoxide, sodium t-butoxide, sodium acetate, phosphoric acid. Tripotassium, potassium fluoride, etc.
- solvent used in Step 1 ′ are benzene, 1,2,4-trimethylbenzene, toluene, xylene, N, N-dimethylformamide, tetrahydrofuran, diethyl ether, t-butyl methyl ether, 1,4- Dioxane, methanol, ethanol, isopropyl alcohol and the like.
- solvents can be appropriately selected according to the structure of the aromatic halide to be reacted.
- a solvent may be used independently and may be used as a mixed solvent.
- the target compound can be obtained in the same manner as in Scheme 1 except that the compound to be reacted is changed.
- the target compound can be obtained in the same manner as in Scheme 1 except that the compound to be reacted is changed.
- the target compound can be obtained in the same manner as in Scheme 1 except that the compound to be reacted is changed.
- the target compound can be obtained in the same manner as in Scheme 1 except that the compound to be reacted is changed.
- the target compound can be obtained in the same manner as in Scheme 1 except that the compound to be reacted is changed.
- the target compound can be obtained in the same manner as in Scheme 1 except that the compound to be reacted is changed.
- the target compound can be obtained in the same manner as in Scheme 1 except that the compound to be reacted is changed.
- FIG. 1 is a schematic cross-sectional view showing an organic electroluminescent element according to this embodiment.
- An organic electroluminescent device 100 shown in FIG. 1 includes a substrate 101, an anode 102 provided on the substrate 101, a hole injection layer 103 provided on the anode 102, and a hole injection layer 103.
- the cathode 108 provided on the electron injection layer 107.
- the organic electroluminescent element 100 is manufactured in the reverse order, for example, the substrate 101, the cathode 108 provided on the substrate 101, the electron injection layer 107 provided on the cathode 108, and the electron injection layer.
- a structure including the hole injection layer 103 provided above and the anode 102 provided on the hole injection layer 103 may be employed.
- each said layer may consist of a single layer, respectively, and may consist of multiple layers.
- the substrate 101 serves as a support for the organic electroluminescent device 100, and usually quartz, glass, metal, plastic, or the like is used.
- the substrate 101 is formed into a plate shape, a film shape, or a sheet shape according to the purpose.
- a glass plate, a metal plate, a metal foil, a plastic film, a plastic sheet, or the like is used.
- glass plates and transparent synthetic resin plates such as polyester, polymethacrylate, polycarbonate, polysulfone and the like are preferable.
- soda lime glass, non-alkali glass, or the like is used, and the thickness only needs to be sufficient to maintain the mechanical strength.
- the upper limit value of the thickness is, for example, 2 mm or less, preferably 1 mm or less.
- the glass material is preferably alkali-free glass because it is better to have less ions eluted from the glass.
- soda lime glass with a barrier coat such as SiO 2 is also commercially available, so it can be used. it can.
- the substrate 101 may be provided with a gas barrier film such as a dense silicon oxide film on at least one surface in order to improve the gas barrier property, and a synthetic resin plate, film or sheet having a low gas barrier property is used as the substrate 101. When used, it is preferable to provide a gas barrier film.
- the anode 102 serves to inject holes into the light emitting layer 105.
- the hole injection layer 103 and / or the hole transport layer 104 are provided between the anode 102 and the light emitting layer 105, holes are injected into the light emitting layer 105 through these layers. .
- Examples of the material for forming the anode 102 include inorganic compounds and organic compounds.
- Examples of inorganic compounds include metals (aluminum, gold, silver, nickel, palladium, chromium, etc.), metal oxides (indium oxide, tin oxide, indium-tin oxide (ITO), indium-zinc oxide) Products (IZO), metal halides (copper iodide, etc.), copper sulfide, carbon black, ITO glass, Nesa glass, and the like.
- Examples of the organic compound include polythiophene such as poly (3-methylthiophene), conductive polymer such as polypyrrole and polyaniline, and the like. In addition, it can select suitably from the substances currently used as an anode of an organic electroluminescent element, and can use it.
- the resistance of the transparent electrode is not limited as long as it can supply a sufficient current for light emission of the light emitting element, but is preferably low resistance from the viewpoint of power consumption of the light emitting element.
- an ITO substrate of 300 ⁇ / ⁇ or less functions as an element electrode, but at present, since it is possible to supply a substrate of about 10 ⁇ / ⁇ , for example, 100 to 5 ⁇ / ⁇ , preferably 50 to 5 ⁇ . It is particularly desirable to use a low resistance product of / ⁇ .
- the thickness of ITO can be arbitrarily selected according to the resistance value, but is usually used in a range of 50 to 200 nm.
- the hole injection layer 103 plays a role of efficiently injecting holes moving from the anode 102 into the light emitting layer 105 or the hole transport layer 104.
- the hole transport layer 104 plays a role of efficiently transporting holes injected from the anode 102 or holes injected from the anode 102 through the hole injection layer 103 to the light emitting layer 105.
- the hole injection layer 103 and the hole transport layer 104 are each formed by laminating and mixing one kind or two or more kinds of hole injection / transport materials or a mixture of the hole injection / transport material and the polymer binder. Is done.
- an inorganic salt such as iron (III) chloride may be added to the hole injection / transport material to form a layer.
- a hole injection / transport material As a hole injection / transport material, it is necessary to efficiently inject and transport holes from the positive electrode between electrodes to which an electric field is applied. The hole injection efficiency is high, and the injected holes are transported efficiently. It is desirable to do. For this purpose, it is preferable to use a substance that has a low ionization potential, a high hole mobility, excellent stability, and is less likely to generate trapping impurities during production and use.
- a polycyclic aromatic compound having a partial structure represented by the general formula (I) or a salt thereof may be used as a material (hole layer material) for forming the hole injection layer 103 or the hole transport layer 104. It can.
- the content of the polycyclic aromatic compound having a partial structure represented by the above general formula (I) or a salt thereof in the hole injection layer 103 or the hole transport layer 104 varies depending on the type of the compound, and is adjusted to the characteristics. You can decide.
- the standard for the content of the polycyclic aromatic compound having a partial structure represented by the general formula (I) or a salt thereof is preferably 1 to 100% by weight of the whole hole layer material, more preferably It is 10 to 100% by weight, more preferably 50 to 100% by weight, and particularly preferably 80 to 100% by weight.
- the polycyclic aromatic compound having a partial structure represented by the general formula (I) or a salt thereof is not used alone (100% by weight), other materials described in detail below may be mixed.
- hole injection layer 103 and the hole transport layer 104 include photoconductive materials, compounds conventionally used as charge transport materials for holes, p-type semiconductors, and organic electroluminescent devices. Any one of known materials used for the hole injection layer and the hole transport layer can be selected and used. Specific examples thereof include carbazole derivatives (N-phenylcarbazole, polyvinylcarbazole, etc.), biscarbazole derivatives such as bis (N-arylcarbazole) or bis (N-alkylcarbazole), triarylamine derivatives (aromatic tertiary class).
- polycarbonate having the above monomers in the side chain And styrene derivatives, polyvinyl carbazole, and polysilane are preferable, but the compound is not particularly limited as long as it is a compound that can form a thin film necessary for manufacturing a light-emitting element, inject holes from the anode, and further transport holes. Absent.
- organic semiconductors are strongly influenced by the doping.
- Such an organic semiconductor matrix material is composed of a compound having a good electron donating property or a compound having a good electron accepting property.
- Strong electron acceptors such as tetracyanoquinone dimethane (TCNQ) or 2,3,5,6-tetrafluorotetracyano-1,4-benzoquinone dimethane (F4TCNQ) are known for doping of electron donor materials.
- TCNQ tetracyanoquinone dimethane
- F4TCNQ 2,3,5,6-tetrafluorotetracyano-1,4-benzoquinone dimethane
- the light emitting layer 105 emits light by recombining holes injected from the anode 102 and electrons injected from the cathode 108 between electrodes to which an electric field is applied.
- the material for forming the light-emitting layer 105 may be a compound that emits light by being excited by recombination of holes and electrons (a light-emitting compound), can form a stable thin film shape, and is in a solid state It is preferable that the compound exhibits a high emission (fluorescence and / or phosphorescence) efficiency.
- the light emitting material of the light emitting element according to the present embodiment may be either fluorescent or phosphorescent.
- the light emitting layer may be either a single layer or a plurality of layers, each formed of a light emitting material (host material, dopant material). Each of the host material and the dopant material may be one kind or a plurality of combinations.
- the dopant material may be included in the host material as a whole, or may be included partially. As a doping method, it can be formed by a co-evaporation method with a host material, but it may be pre-mixed with the host material and then simultaneously deposited.
- the amount of host material used depends on the type of host material and can be determined according to the characteristics of the host material.
- the amount of the host material used is preferably 50 to 99.999% by weight of the entire light emitting material, more preferably 80 to 99.95% by weight, and still more preferably 90 to 99.9% by weight. .
- the amount of dopant material used varies depending on the type of dopant material, and can be determined according to the characteristics of the dopant material (for example, if the amount used is too large, there is a risk of concentration quenching).
- the standard of the amount of dopant used is preferably 0.001 to 50% by weight of the entire light emitting material, more preferably 0.05 to 20% by weight, and still more preferably 0.1 to 10% by weight.
- the polycyclic aromatic compound having a partial structure represented by the general formula (I) or a salt thereof can also be used as a host material or a dopant material.
- the content of the polycyclic aromatic compound having a partial structure represented by the above general formula (I) or a salt thereof in each material varies depending on the type thereof, and may be determined according to the characteristics thereof.
- the standard of the content of the polycyclic aromatic compound having a partial structure represented by the above general formula (I) or a salt thereof is preferably 1 to 100% by weight of the entire host material (or dopant material), and more The amount is preferably 10 to 100% by weight, more preferably 50 to 100% by weight, and particularly preferably 80 to 100% by weight.
- the polycyclic aromatic compound having a partial structure represented by the general formula (I) or a salt thereof is not used alone (100 wt%), other host materials (or dopant materials) described in detail below Can be mixed.
- the host material is not particularly limited, but has previously been known as a phosphor, fused ring derivatives such as anthracene and pyrene, metal chelated oxinoid compounds such as tris (8-quinolinolato) aluminum, bis Bisstyryl derivatives such as styryl anthracene derivatives and distyrylbenzene derivatives, tetraphenylbutadiene derivatives, coumarin derivatives, oxadiazole derivatives, pyrrolopyridine derivatives, perinone derivatives, cyclopentadiene derivatives, thiadiazolopyridine derivatives, pyrrolopyrrole derivatives, fluorene derivatives, For benzofluorene derivatives and polymer systems, polyphenylene vinylene derivatives, polyparaphenylene derivatives, and polythiophene derivatives are preferably used.
- metal chelated oxinoid compounds such as tris (8-quinolinolato) aluminum
- the host material can be appropriately selected from the compounds described in Chemical Industry, June 2004, page 13, and references cited therein.
- the dopant material is not particularly limited, and a known compound can be used, and can be selected from various materials according to a desired emission color.
- condensed ring derivatives such as phenanthrene, anthracene, pyrene, tetracene, pentacene, perylene, naphthopylene, dibenzopyrene, rubrene and chrysene, benzoxazole derivatives, benzthiazole derivatives, benzimidazole derivatives, benztriazole derivatives, oxazoles Derivatives, oxadiazole derivatives, thiazole derivatives, imidazole derivatives, thiadiazole derivatives, triazole derivatives, pyrazoline derivatives, stilbene derivatives, thiophene derivatives, tetraphenylbutadiene derivatives, cyclopentadiene derivatives, bisstyrylanthracene derivatives, distyrylbenzene derivative
- blue to blue-green dopant materials include naphthalene, anthracene, phenanthrene, pyrene, triphenylene, perylene, fluorene, indene, chrysene and other aromatic hydrocarbon compounds and derivatives thereof, furan, pyrrole, thiophene, Aromatic complex such as silole, 9-silafluorene, 9,9'-spirobisilafluorene, benzothiophene, benzofuran, indole, dibenzothiophene, dibenzofuran, imidazopyridine, phenanthroline, pyrazine, naphthyridine, quinoxaline, pyrrolopyridine, thioxanthene Ring compounds and their derivatives, distyrylbenzene derivatives, tetraphenylbutadiene derivatives, stilbene derivatives, aldazine derivatives, coumarin derivatives, imidazo
- examples of the green to yellow dopant material include coumarin derivatives, phthalimide derivatives, naphthalimide derivatives, perinone derivatives, pyrrolopyrrole derivatives, cyclopentadiene derivatives, acridone derivatives, quinacridone derivatives, and naphthacene derivatives such as rubrene.
- a compound in which a substituent capable of increasing the wavelength such as aryl, heteroaryl, arylvinyl, amino and cyano is introduced into the compound exemplified as a blue-green dopant material is also a suitable example.
- orange to red dopant materials include naphthalimide derivatives such as bis (diisopropylphenyl) perylenetetracarboxylic imide, perinone derivatives, rare earth complexes such as Eu complexes having acetylacetone, benzoylacetone and phenanthroline as ligands, 4 -(Dicyanomethylene) -2-methyl-6- (p-dimethylaminostyryl) -4H-pyran and its analogs, metal phthalocyanine derivatives such as magnesium phthalocyanine and aluminum chlorophthalocyanine, rhodamine compounds, deazaflavin derivatives, coumarin derivatives, quinacridone Derivatives, phenoxazine derivatives, oxazine derivatives, quinazoline derivatives, pyrrolopyridine derivatives, squarylium derivatives, violanthrone derivatives, phenazine derivatives, phenoxazo Derivatives, thi
- the dopant can be appropriately selected from the compounds described in Chemical Industry, June 2004, page 13, and references cited therein.
- perylene derivatives perylene derivatives, borane derivatives, amine-containing styryl derivatives, aromatic amine derivatives, coumarin derivatives, pyran derivatives, iridium complexes, or platinum complexes are particularly preferable.
- perylene derivatives examples include 3,10-bis (2,6-dimethylphenyl) perylene, 3,10-bis (2,4,6-trimethylphenyl) perylene, 3,10-diphenylperylene, 3,4- Diphenylperylene, 2,5,8,11-tetra-t-butylperylene, 3,4,9,10-tetraphenylperylene, 3- (1'-pyrenyl) -8,11-di (t-butyl) perylene 3- (9′-anthryl) -8,11-di (t-butyl) perylene, 3,3′-bis (8,11-di (t-butyl) perylenyl), and the like.
- JP-A-11-97178, JP-A-2000-133457, JP-A-2000-26324, JP-A-2001-267079, JP-A-2001-267078, JP-A-2001-267076, Perylene derivatives described in JP-A No. 2000-34234, JP-A No. 2001-267075, JP-A No. 2001-217077 and the like may be used.
- borane derivatives examples include 1,8-diphenyl-10- (dimesitylboryl) anthracene, 9-phenyl-10- (dimesitylboryl) anthracene, 4- (9′-anthryl) dimesitylborylnaphthalene, 4- (10 ′ -Phenyl-9'-anthryl) dimesitylborylnaphthalene, 9- (dimesitylboryl) anthracene, 9- (4'-biphenylyl) -10- (dimesitylboryl) anthracene, 9- (4 '-(N-carbazolyl) phenyl) And -10- (dimesitylboryl) anthracene. Further, borane derivatives described in International Publication No. 2000/40586 and the like may be used.
- amine-containing styryl derivatives include N, N, N ′, N′-tetra (4-biphenylyl) -4,4′-diaminostilbene, N, N, N ′, N′-tetra (1-naphthyl).
- aromatic amine derivative examples include N, N, N, N-tetraphenylanthracene-9,10-diamine, 9,10-bis (4-diphenylamino-phenyl) anthracene, and 9,10-bis (4- Di (1-naphthylamino) phenyl) anthracene, 9,10-bis (4-di (2-naphthylamino) phenyl) anthracene, 10-di-p-tolylamino-9- (4-di-p-tolylamino-1) -Naphthyl) anthracene, 10-diphenylamino-9- (4-diphenylamino-1-naphthyl) anthracene, 10-diphenylamino-9- (6-diphenylamino-2-naphthyl) anthracene, [4- (4-diphenyl Amino-phenyl) naphthalen-1-yl]
- Examples of coumarin derivatives include coumarin-6 and coumarin-334. Moreover, you may use the coumarin derivative described in Unexamined-Japanese-Patent No. 2004-43646, Unexamined-Japanese-Patent No. 2001-76876, and Unexamined-Japanese-Patent No. 6-298758.
- Examples of the pyran derivative include the following DCM and DCJTB. Also, JP 2005-126399, JP 2005-097283, JP 2002-234892, JP 2001-220577, JP 2001-081090, and JP 2001-052869. Alternatively, pyran derivatives described in the above may be used.
- iridium complex examples include Ir (ppy) 3 described below. Further, the iridium complexes described in JP-A-2006-089398, JP-A-2006-080419, JP-A-2005-298483, JP-A-2005-097263, JP-A-2004-111379, etc. It may be used.
- platinum complex examples include the following PtOEP. Further, the platinum complexes described in JP-A-2006-190718, JP-A-2006-128634, JP-A-2006-093542, JP-A-2004-335122, JP-A-2004-331508, etc. It may be used.
- the electron injection layer 107 plays a role of efficiently injecting electrons moving from the cathode 108 into the light emitting layer 105 or the electron transport layer 106.
- the electron transport layer 106 plays a role of efficiently transporting electrons injected from the cathode 108 or electrons injected from the cathode 108 through the electron injection layer 107 to the light emitting layer 105.
- the electron transport layer 106 and the electron injection layer 107 are each formed by laminating and mixing one or more electron transport / injection materials or a mixture of the electron transport / injection material and the polymer binder.
- the electron injection / transport layer is a layer that is responsible for injecting electrons from the cathode and further transporting the electrons. It is desirable that the electron injection efficiency is high and the injected electrons are transported efficiently. For this purpose, it is preferable to use a substance that has a high electron affinity, a high electron mobility, excellent stability, and is unlikely to generate trapping impurities during production and use. However, considering the transport balance between holes and electrons, if the role of effectively preventing the holes from the anode from flowing to the cathode side without recombination is mainly played, the electron transport capability is much higher. Even if it is not high, the effect of improving the luminous efficiency is equivalent to that of a material having a high electron transport capability. Therefore, the electron injection / transport layer in this embodiment may include a function of a layer that can efficiently block the movement of holes.
- a polycyclic aromatic compound having a partial structure represented by the general formula (I) or a salt thereof can be used as the material (electron layer material) for forming the electron transport layer 106 or the electron injection layer 107.
- the content of the polycyclic aromatic compound having a partial structure represented by the above general formula (I) or the salt thereof in the electron transport layer 106 or the electron injection layer 107 differs depending on the type of the compound, and is determined according to the characteristics thereof. That's fine.
- the standard of the content of the polycyclic aromatic compound having a partial structure represented by the above general formula (I) or a salt thereof is preferably 1 to 100 of the entire electron transport layer material (or electron injection layer material).
- % By weight, more preferably 10 to 100% by weight, still more preferably 50 to 100% by weight, and particularly preferably 80 to 100% by weight.
- polycyclic aromatic compound having a partial structure represented by the general formula (I) or a salt thereof is not used alone (100% by weight), other materials described in detail below may be mixed.
- Other materials for forming the electron transport layer or electron injection layer include compounds conventionally used as electron transport compounds in photoconductive materials, and known materials used for electron injection layers and electron transport layers of organic electroluminescent devices. Any of these compounds can be selected and used.
- Materials used for the electron transport layer or the electron injection layer include compounds composed of aromatic rings or heteroaromatic rings composed of one or more atoms selected from carbon, hydrogen, oxygen, sulfur, silicon, and phosphorus, and pyrrole derivatives. And at least one selected from the condensed ring derivatives thereof and metal complexes having electron-accepting nitrogen.
- condensed ring aromatic ring derivatives such as naphthalene and anthracene, styryl aromatic ring derivatives represented by 4,4′-bis (diphenylethenyl) biphenyl, perinone derivatives, coumarin derivatives, naphthalimide derivatives, anthraquinones And quinone derivatives such as diphenoquinone, phosphorus oxide derivatives, carbazole derivatives, and indole derivatives.
- metal complexes having electron-accepting nitrogen include hydroxyazole complexes such as hydroxyphenyloxazole complexes, azomethine complexes, tropolone metal complexes, flavonol metal complexes, and benzoquinoline metal complexes.
- anthracene derivatives such as 9,10-bis (2-naphthyl) anthracene, styryl aromatic ring derivatives such as 4,4′-bis (diphenylethenyl) biphenyl, 4,4′-bis (N-carbazolyl) biphenyl
- a carbazole derivative such as 1,3,5-tris (N-carbazolyl) benzene is preferably used from the viewpoint of durability.
- electron transfer compounds include pyridine derivatives, naphthalene derivatives, anthracene derivatives, phenanthroline derivatives, perinone derivatives, coumarin derivatives, naphthalimide derivatives, anthraquinone derivatives, diphenoquinone derivatives, diphenylquinone derivatives, perylene derivatives, oxadiazoles.
- metal complexes having electron-accepting nitrogen can also be used, such as hydroxyazole complexes such as quinolinol-based metal complexes and hydroxyphenyloxazole complexes, azomethine complexes, tropolone metal complexes, flavonol metal complexes, and benzoquinoline metal complexes. can give.
- the above-mentioned materials can be used alone, but they may be mixed with different materials.
- quinolinol metal complexes bipyridine derivatives, phenanthroline derivatives, borane derivatives or benzimidazole derivatives are preferable.
- the quinolinol-based metal complex is a compound represented by the following general formula (E-1).
- R 1 to R 6 are hydrogen or a substituent
- M is Li, Al, Ga, Be or Zn
- n is an integer of 1 to 3.
- quinolinol metal complexes include 8-quinolinol lithium, tris (8-quinolinolato) aluminum, tris (4-methyl-8-quinolinolato) aluminum, tris (5-methyl-8-quinolinolato) aluminum, tris (3 , 4-dimethyl-8-quinolinolato) aluminum, tris (4,5-dimethyl-8-quinolinolato) aluminum, tris (4,6-dimethyl-8-quinolinolato) aluminum, bis (2-methyl-8-quinolinolato) ( Phenolate) aluminum, bis (2-methyl-8-quinolinolato) (2-methylphenolate) aluminum, bis (2-methyl-8-quinolinolato) (3-methylphenolato) aluminum, bis (2-methyl-8- Quinolinolato) (4- Tylphenolate) aluminum, bis (2-methyl-8-quinolinolato) (2-phenylphenolate) aluminum, bis (2-methyl-8-quinolinolato) (3-phenylphenolate)
- the bipyridine derivative is a compound represented by the following general formula (E-2).
- G represents a simple bond or an n-valent linking group, and n is an integer of 2 to 8. Carbon atoms that are not used for the bond of pyridine-pyridine or pyridine-G may be substituted.
- G in the general formula (E-2) examples include the following structural formulas.
- each R is independently hydrogen, methyl, ethyl, isopropyl, cyclohexyl, phenyl, 1-naphthyl, 2-naphthyl, biphenylyl or terphenylyl.
- pyridine derivative examples include 2,5-bis (2,2′-bipyridin-6-yl) -1,1-dimethyl-3,4-diphenylsilole, 2,5-bis (2,2′- Bipyridin-6-yl) -1,1-dimethyl-3,4-dimesitylsilole, 2,5-bis (2,2′-bipyridin-5-yl) -1,1-dimethyl-3,4 Diphenylsilole, 2,5-bis (2,2′-bipyridin-5-yl) -1,1-dimethyl-3,4-dimesitylsilole, 9,10-di (2,2′-bipyridine-6) -Yl) anthracene, 9,10-di (2,2′-bipyridin-5-yl) anthracene, 9,10-di (2,3′-bipyridin-6-yl) anthracene, 9,10-di (2,3′
- the phenanthroline derivative is a compound represented by the following general formula (E-3-1) or (E-3-2).
- R 1 to R 8 are hydrogen or a substituent, adjacent groups may be bonded to each other to form a condensed ring, G represents a simple bond or an n-valent linking group, and n represents 2 It is an integer of ⁇ 8.
- Examples of G in the general formula (E-3-2) include the same ones as described in the bipyridine derivative column.
- phenanthroline derivatives include 4,7-diphenyl-1,10-phenanthroline, 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline, 9,10-di (1,10-phenanthroline- 2-yl) anthracene, 2,6-di (1,10-phenanthroline-5-yl) pyridine, 1,3,5-tri (1,10-phenanthroline-5-yl) benzene, 9,9′-difluor -Bis (1,10-phenanthroline-5-yl), bathocuproin, 1,3-bis (2-phenyl-1,10-phenanthroline-9-yl) benzene and the like.
- a phenanthroline derivative is used for the electron transport layer and the electron injection layer.
- the substituent itself has a three-dimensional structure, or a phenanthroline skeleton or Those having a three-dimensional structure by steric repulsion with an adjacent substituent or those having a plurality of phenanthroline skeletons linked to each other are preferred.
- a compound containing a conjugated bond, a substituted or unsubstituted aromatic hydrocarbon, or a substituted or unsubstituted aromatic heterocycle in the linking unit is more preferable.
- the borane derivative is a compound represented by the following general formula (E-4), and is disclosed in detail in JP-A-2007-27587.
- R 11 and R 12 are each independently at least one of hydrogen, alkyl, optionally substituted aryl, substituted silyl, optionally substituted nitrogen-containing heterocycle, or cyano
- R 13 to R 16 are each independently an optionally substituted alkyl or an optionally substituted aryl
- X is an optionally substituted arylene
- Y is a substituted And optionally substituted aryl having 16 or less carbon atoms, substituted boryl, or optionally substituted carbazolyl
- each n is independently an integer of 0 to 3.
- the compound represented by -1-4) is preferred. Specific examples include 9- [4- (4-Dimesitylborylnaphthalen-1-yl) phenyl] carbazole, 9- [4- (4-Dimesitylborylnaphthalen-1-yl) naphthalen-1-yl. Carbazole and the like.
- R 11 and R 12 are each independently at least one of hydrogen, alkyl, optionally substituted aryl, substituted silyl, optionally substituted nitrogen-containing heterocycle, or cyano
- R 13 to R 16 are each independently an optionally substituted alkyl or an optionally substituted aryl
- R 21 and R 22 are each independently hydrogen, alkyl, or substituted.
- X 1 is an optionally substituted arylene having 20 or less carbon atoms
- n is each Each independently represents an integer of 0 to 3, and each m independently represents an integer of 0 to 4;
- R 31 to R 34 are each independently methyl, isopropyl or phenyl
- R 35 and R 36 are each independently hydrogen, methyl, isopropyl or phenyl. It is.
- R 11 and R 12 are each independently at least one of hydrogen, alkyl, optionally substituted aryl, substituted silyl, optionally substituted nitrogen-containing heterocycle, or cyano
- R 13 to R 16 are each independently an optionally substituted alkyl or an optionally substituted aryl
- X 1 is an optionally substituted arylene having 20 or less carbon atoms
- N is an integer of 0 to 3 independently.
- R 31 to R 34 are each independently any of methyl, isopropyl or phenyl
- R 35 and R 36 are each independently any of hydrogen, methyl, isopropyl or phenyl It is.
- R 11 and R 12 are each independently at least one of hydrogen, alkyl, optionally substituted aryl, substituted silyl, optionally substituted nitrogen-containing heterocycle, or cyano
- R 13 to R 16 are each independently an optionally substituted alkyl or an optionally substituted aryl
- X 1 is an optionally substituted arylene having 10 or less carbon atoms
- Y 1 is an optionally substituted aryl having 14 or less carbon atoms
- n is each independently an integer of 0 to 3.
- R 31 to R 34 are each independently methyl, isopropyl or phenyl
- R 35 and R 36 are each independently hydrogen, methyl, isopropyl or phenyl. It is.
- the benzimidazole derivative is a compound represented by the following general formula (E-5).
- Ar 1 to Ar 3 are each independently hydrogen or aryl having 6 to 30 carbon atoms which may be substituted.
- a benzimidazole derivative which is anthryl optionally substituted with Ar 1 is preferable.
- aryl having 6 to 30 carbon atoms include phenyl, 1-naphthyl, 2-naphthyl, acenaphthylene-1-yl, acenaphthylene-3-yl, acenaphthylene-4-yl, acenaphthylene-5-yl, and fluorene-1- Yl, fluoren-2-yl, fluoren-3-yl, fluoren-4-yl, fluoren-9-yl, phenalen-1-yl, phenalen-2-yl, 1-phenanthryl, 2-phenanthryl, 3-phenanthryl, 4-phenanthryl, 9-phenanthryl, 1-anthryl, 2-anthryl, 9-anthryl, fluoranthen-1-yl, fluoranthen-2-yl, fluoranthen-3-yl, fluoranthen-7-yl, fluoranthen-8-yl, Triphenylene-1-yl, 2-
- benzimidazole derivative examples include 1-phenyl-2- (4- (10-phenylanthracen-9-yl) phenyl) -1H-benzo [d] imidazole, 2- (4- (10- (naphthalene-2) -Yl) anthracen-9-yl) phenyl) -1-phenyl-1H-benzo [d] imidazole, 2- (3- (10- (naphthalen-2-yl) anthracen-9-yl) phenyl) -1- Phenyl-1H-benzo [d] imidazole, 5- (10- (naphthalen-2-yl) anthracen-9-yl) -1,2-diphenyl-1H-benzo [d] imidazole, 1- (4- (10 -(Naphthalen-2-yl) anthracen-9-yl) phenyl) -2-phenyl-1H-benzo [d] imidazole, 2- (4- (9,10-di (n)-
- the electron transport layer or the electron injection layer may further contain a substance capable of reducing the material forming the electron transport layer or the electron injection layer.
- a substance capable of reducing the material forming the electron transport layer or the electron injection layer various substances can be used as long as they have a certain reducing ability.
- Preferred reducing substances include alkali metals such as Na (work function 2.36 eV), K (2.28 eV), Rb (2.16 eV) or Cs (1.95 eV), and Ca (2. 9eV), Sr (2.0 to 2.5 eV) or Ba (2.52 eV), and alkaline earth metals such as those having a work function of 2.9 eV or less are particularly preferable.
- a more preferable reducing substance is an alkali metal of K, Rb or Cs, more preferably Rb or Cs, and most preferably Cs.
- alkali metals have particularly high reducing ability, and by adding a relatively small amount to the material forming the electron transport layer or the electron injection layer, the luminance of the organic EL element can be improved and the lifetime can be extended.
- a reducing substance having a work function of 2.9 eV or less a combination of two or more alkali metals is also preferable.
- a combination containing Cs such as Cs and Na, Cs and K, Cs and Rb, or A combination of Cs, Na and K is preferred.
- Cs such as Cs and Na, Cs and K, Cs and Rb, or A combination of Cs, Na and K is preferred.
- the cathode 108 serves to inject electrons into the light emitting layer 105 through the electron injection layer 107 and the electron transport layer 106.
- the material for forming the cathode 108 is not particularly limited as long as it is a substance that can efficiently inject electrons into the organic layer, but the same material as that for forming the anode 102 can be used.
- metals such as tin, indium, calcium, aluminum, silver, copper, nickel, chromium, gold, platinum, iron, zinc, lithium, sodium, potassium, cesium and magnesium or alloys thereof (magnesium-silver alloy, magnesium -Indium alloys, aluminum-lithium alloys such as lithium fluoride / aluminum, etc.) are preferred.
- Lithium, sodium, potassium, cesium, calcium, magnesium, or alloys containing these low work function metals are effective for increasing the electron injection efficiency and improving device characteristics.
- metals such as platinum, gold, silver, copper, iron, tin, aluminum and indium, or alloys using these metals, and inorganic materials such as silica, titania and silicon nitride, polyvinyl alcohol, vinyl chloride Lamination of hydrocarbon polymer compounds and the like is a preferred example.
- the method for producing these electrodes is not particularly limited as long as conduction can be achieved, such as resistance heating, electron beam, sputtering, ion plating, and coating.
- the materials used for the hole injection layer, hole transport layer, light emitting layer, electron transport layer and electron injection layer can form each layer alone, but as a polymer binder, polyvinyl chloride, polycarbonate, Polystyrene, poly (N-vinylcarbazole), polymethyl methacrylate, polybutyl methacrylate, polyester, polysulfone, polyphenylene oxide, polybutadiene, hydrocarbon resin, ketone resin, phenoxy resin, polyamide, ethyl cellulose, vinyl acetate resin, ABS resin, polyurethane resin It can also be used by dispersing it in solvent-soluble resins such as phenol resins, xylene resins, petroleum resins, urea resins, melamine resins, unsaturated polyester resins, alkyd resins, epoxy resins, silicone resins, etc. is there.
- solvent-soluble resins such as phenol resins, xylene resins, petroleum resins, urea resins, melamine resins,
- Each layer constituting the organic electroluminescent element is formed by a method such as vapor deposition, resistance heating vapor deposition, electron beam vapor deposition, sputtering, molecular lamination method, printing method, spin coating method or cast method, coating method, etc. It can be formed by using a thin film.
- the film thickness of each layer thus formed is not particularly limited and can be appropriately set according to the properties of the material, but is usually in the range of 2 nm to 5000 nm. The film thickness can usually be measured with a crystal oscillation type film thickness measuring device or the like.
- the vapor deposition conditions vary depending on the type of material, the target crystal structure and association structure of the film, and the like.
- Deposition conditions generally include boat heating temperature of 50 to 400 ° C., vacuum degree of 10 ⁇ 6 to 10 ⁇ 3 Pa, deposition rate of 0.01 to 50 nm / second, substrate temperature of ⁇ 150 to + 300 ° C., film thickness of 2 nm to 5 ⁇ m. It is preferable to set appropriately within the range.
- an organic electric field composed of an anode / hole injection layer / hole transport layer / a light emitting layer composed of a host material and a dopant material / electron transport layer / electron injection layer / cathode.
- a method for manufacturing a light-emitting element will be described.
- a thin film of an anode material is formed on a suitable substrate by vapor deposition or the like to produce an anode, and then a thin film of a hole injection layer and a hole transport layer is formed on the anode.
- a host material and a dopant material are co-evaporated to form a thin film to form a light emitting layer.
- An electron transport layer and an electron injection layer are formed on the light emitting layer, and a thin film made of a cathode material is formed by vapor deposition. By forming it as a cathode, a desired organic electroluminescent element can be obtained.
- the order of preparation may be reversed, and the cathode, electron injection layer, electron transport layer, light emitting layer, hole transport layer, hole injection layer, and anode may be fabricated in this order. Is possible.
- the anode When a DC voltage is applied to the organic electroluminescent device thus obtained, the anode may be applied with a positive polarity and the cathode with a negative polarity. When a voltage of about 2 to 40 V is applied, the organic electroluminescent device is transparent or translucent. Luminescence can be observed from the electrode side (anode or cathode, and both). The organic electroluminescence device emits light when a pulse current or an alternating current is applied. The alternating current waveform to be applied may be arbitrary.
- the present invention can also be applied to a display device provided with an organic electroluminescent element or a lighting device provided with an organic electroluminescent element.
- a display device or an illuminating device including an organic electroluminescent element can be manufactured by a known method such as connecting the organic electroluminescent element according to the present embodiment and a known driving device, such as direct current driving, pulse driving, or alternating current. It can be driven by appropriately using a known driving method such as driving.
- Examples of the display device include a panel display such as a color flat panel display, and a flexible display such as a flexible color organic electroluminescence (EL) display (for example, JP-A-10-335066 and JP-A-2003-321546). Gazette, JP-A-2004-281086, etc.).
- Examples of the display method of the display include a matrix and / or segment method. Note that the matrix display and the segment display may coexist in the same panel.
- a matrix is a pixel in which pixels for display are arranged two-dimensionally, such as a grid or mosaic, and displays characters and images as a set of pixels.
- the shape and size of the pixel are determined by the application. For example, a square pixel with a side of 300 ⁇ m or less is usually used for displaying images and characters on a personal computer, monitor, TV, and a pixel with a side of mm order for a large display such as a display panel. become.
- monochrome display pixels of the same color may be arranged. However, in color display, red, green, and blue pixels are displayed side by side. In this case, there are typically a delta type and a stripe type.
- the matrix driving method may be either a line sequential driving method or an active matrix.
- the line-sequential driving has an advantage that the structure is simple. However, the active matrix may be superior in consideration of the operation characteristics, so that it is necessary to properly use it depending on the application.
- a pattern is formed so as to display predetermined information, and a predetermined region is caused to emit light.
- a predetermined region is caused to emit light.
- the time and temperature display in a digital clock or a thermometer the operation state display of an audio device or an electromagnetic cooker, the panel display of an automobile, and the like can be mentioned.
- the illuminating device examples include an illuminating device such as indoor lighting, a backlight of a liquid crystal display device, and the like (for example, JP 2003-257621 A, JP 2003-277741 A, JP 2004-119211 A).
- the backlight is used mainly for the purpose of improving the visibility of a display device that does not emit light, and is used for a liquid crystal display device, a clock, an audio device, an automobile panel, a display panel, a sign, and the like.
- a backlight for liquid crystal display devices especially personal computers for which thinning is an issue, considering that conventional methods are made of fluorescent lamps and light guide plates, it is difficult to reduce the thickness.
- the backlight using the light emitting element according to the embodiment is thin and lightweight.
- Synthesis example (1) Synthesis of 4b-aza-12b-thiophosphadibenzo [g, p] chrysene
- Synthesis example (2) Synthesis of 4b-aza-12b-phenyl-12b-siladibenzo [g, p] chrysene
- N-bromosuccinimide (19.9 g) was added to a solution of 4b-aza-12b-boradibenzo [g, p] chrysene (18.0 g) in THF (180 ml) and stirred at room temperature for 1 hour. did. After completion of the reaction, an aqueous sodium sulfite solution was added, THF was distilled off under reduced pressure, and toluene was added to separate the layers.
- Synthesis example (8) Synthesis of 6,9-dichloro-14b 1 -aza-14b-borabenzo [p] indeno [1,2,3,4-defg] chrysene
- Synthesis example (11) Synthesis of 8b, 19b-diaza-11b, 22b-dithiophosphahexabenzo [a, c, fg, j, l, op] tetracene
- N, N′-bis (biphenyl-2-yl) -2,6-diaminobiphenyl (0.977 g, 2.00 mmol) and toluene (20 mL) in hexane and butyllithium solution at ⁇ 78 ° C. under argon atmosphere (2.45 mL, 1.63 M, 4.0 mmol) was added and stirred. After 1 hour, phosphorus trichloride (0.549 g, 4.0 mmol) was added and stirred for 1 hour, then the temperature was raised to 0 ° C. and the mixture was further stirred for 1 hour.
- Synthesis example (12) Synthesis of 8b, 19b-diaza-11b, 22b-diborahexabenzo [a, c, fg, j, l, op] tetracene
- N, N′-bis (biphenyl-2-yl) -2,6-diaminobiphenyl (0.977 g, 2.00 mmol) and toluene (20 mL) in hexane and butyllithium solution at ⁇ 78 ° C. under argon atmosphere (2.45 mL, 1.63 M, 4.0 mmol) was added and stirred. After 1 hour, the temperature was raised to 0 ° C. and the mixture was further stirred for 1 hour.
- N-[(2-thienyl) phenyl] -N- (biphenyl-2-yl) amine (0.655 g, 2.00 mmol) and toluene (10 mL) in hexane of butyllithium at ⁇ 78 ° C. under argon atmosphere.
- the solution (1.25 mL, 1.60 M, 2.00 mmol) was added and stirred. After 1 hour, the temperature was raised to 0 ° C. and the mixture was further stirred for 1 hour. Then, a heptane solution of boron trichloride (2.00 mL, 1.00 M, 2.00 mmol) was added at ⁇ 78 ° C., and the mixture was stirred at room temperature for 12 hours.
- N-([1,1′-biphenyl] -2-yl) -2-phenylthiophen-3-amine (0.655 g, 2.00 mmol) and toluene (10 mL) at ⁇ 78 ° C. under argon atmosphere.
- a solution of butyl lithium in hexane (1.25 mL, 1.60 M, 2.00 mmol) was added and stirred. After 1 hour, the temperature was raised to 0 ° C. and the mixture was further stirred for 1 hour. Then, a heptane solution of boron trichloride (2.00 mL, 1.00 M, 2.00 mmol) was added at ⁇ 78 ° C., and the mixture was stirred at room temperature for 12 hours.
- N-([1,1′-biphenyl] -2-yl) -3-phenylpyridin-2-amine (0.645 g, 2.00 mmol) and toluene (10 mL) at ⁇ 78 ° C. under argon atmosphere.
- a solution of butyl lithium in hexane (1.25 mL, 1.60 M, 2.00 mmol) was added and stirred. After 1 hour, the temperature was raised to 0 ° C. and the mixture was further stirred for 1 hour. Then, a heptane solution of boron trichloride (2.00 mL, 1.00 M, 2.00 mmol) was added at ⁇ 78 ° C., and the mixture was stirred at room temperature for 12 hours.
- Synthesis example (25) Synthesis of 8b, 11b, 14b-triaza-22b, 25b, 28b-triboraoctabenzo [a, c, fg, jk, n, p, st, wx] hexacene
- N 2 -([1,1′-biphenyl] -2-yl) -N 6- (6-([1,1′-biphenyl] -2-ylamino)-[1,1′-biphenyl] -2- Yl)-[1,1′-biphenyl] -2,6-diamine (1.31 g, 2.00 mmol) and toluene (20 mL) at ⁇ 78 ° C. in an argon atmosphere at ⁇ 78 ° C. 68 mL, 1.63 M, 6.00 mmol) was added and stirred. After 1 hour, the temperature was raised to 0 ° C. and the mixture was further stirred for 1 hour.
- 2-bromo-1,1 ′ 4 ′, 1 ′′ -terphenyl (35.0 g), sodium-t-butoxide (10.9 g), Pd (dba) 2 (0.65 g), 4- ( A flask containing di-t-butylphosphino) -N, N-dimethylaniline (0.60 g), xylene (100 ml) and lithium amide (1.3 g) was stirred at 90 ° C. for 2 hours under a nitrogen atmosphere. After cooling the reaction solution to room temperature, water and ethyl acetate were added for liquid separation, and then the solvent was distilled off under reduced pressure and then purified by activated alumina column chromatography (developing solution: toluene).
- Synthesis example (31) Synthesis of 2,7-dibromo-11,14-diphenyl-4b-aza-12b-boradibenzo [g, p] chrysene
- N-bromosuccinimide (NBS) was added to a THF (50 ml) solution of 2,7,11,14-tetraphenyl-4b-aza-12b-boradibenzo [g, p] chrysene (4.8 g) in a nitrogen atmosphere. (3.7 g) was added and stirred at room temperature for 1 hour. After completion of the reaction, an aqueous sodium nitrite solution was added and the deposited precipitate was collected by suction filtration.
- Synthesis example (32) Synthesis of 10,15-diphenyl-4b-aza-12b-boradibenzo [g, p] chrysene
- the obtained oily substance was purified with a silica gel short column (developing solution: toluene), the solvent was distilled off under reduced pressure, and heptane was added to the obtained oily substance for reprecipitation, and [1,1 ′: 3 ′, 1 ” -Terphenyl] -2-amine (33.0 g) was obtained.
- the obtained oily substance was purified by silica gel column chromatography (developing solution: toluene / heptane mixed solution), and di ([1,1 ′: 2 ′, 1 ′′ -terphenyl] -2-yl) amine (32.
- silica gel column chromatography developing solution: toluene / heptane mixed solution
- di ([1,1 ′: 2 ′, 1 ′′ -terphenyl] -2-yl) amine 32.
- the target product was eluted by gradually increasing the ratio of toluene.
- Di ([1,1 ′: 2 ′, 1 ′′ -terphenyl] -2-yl) amine (20.1 g) was obtained.
- N-bromosuccinimide (13.6 g) was added to a THF (150 ml) solution of 4b-aza-12b-oxaphospha-dibenzo [g, p] chrysene (3.5 g) in a nitrogen atmosphere and refluxed. Stir at temperature for 2 hours. After completion of the reaction, an aqueous sodium nitrite solution and toluene were added for liquid separation, and the solvent was distilled off under reduced pressure. The resulting oily substance was reprecipitated by adding ethanol to obtain 2,7-dibromo-4b-aza-12b-oxaphospha-dibenzo [g, p] chrysene (4.5 g).
- the obtained solid was washed with warm water and purified by silica gel column chromatography (developing solution: toluene / ethyl acetate mixed solution). At this time, the target product was eluted by gradually increasing the ratio of ethyl acetate in the developing solution. Moreover, when charging a sample on silica gel, a solution dissolved in heated chlorobenzene was used. Further, recrystallization from ethyl acetate gave a compound (1.3 g) represented by the formula (366).
- the residue was purified by silica gel column chromatography (developing solution: toluene / ethyl acetate mixed solution). At this time, the target product was eluted by gradually increasing the ratio of ethyl acetate in the developing solution. After the solvent was distilled off under reduced pressure, the residue was washed with ethyl acetate and reprecipitated with a mixed solvent of chlorobenzene / ethyl acetate to obtain a compound (0.9 g) represented by the formula (424).
- [1,1 ′: 4 ′, 1 ′′ -terphenyl] -2-amine (11.0 g), 2-bromobiphenyl (10.5 g), sodium-t-butoxide (5.2 g), Pd
- a flask containing (dba) 2 (0.06 g), 4- (di-t-butylphosphino) -N, N-dimethylaniline (0.06 g) and toluene was placed in a nitrogen atmosphere and stirred at reflux temperature for 3 hours.
- 6-iodo-14b 1 -aza-14b-borabenzo [p] indeno [1,2,3,4-defg] chrysene (0.4 g), carbazole (0.2 g), sodium-t-butoxide ( 0.1 g), Pd (dba) 2 (0.03 g), 1M tri-t-butylphosphine toluene solution (0.13 ml) and 1,2,4-trimethylbenzene “Me 3 Ph” (10 ml)
- the flask was placed in a nitrogen atmosphere and stirred at reflux temperature for 3 hours. After cooling the reaction solution to room temperature, water was added and the precipitated solid was collected by suction filtration.
- EDTA ⁇ 4Na aqueous solution a solution prepared by dissolving ethylenediaminetetraacetic acid / tetrasodium salt dihydrate in an appropriate amount of water (hereinafter abbreviated as EDTA ⁇ 4Na aqueous solution) and toluene were added to separate the layers. After evaporating the solvent under reduced pressure, the residue was purified by silica gel column chromatography (developing solution: heptane) to obtain 5,5′-dimethyl-2,2′-bithiophene (20.3 g).
- N-bromosuccinimide “NBS” (6.9 g) was slowly added thereto, and the temperature was raised to room temperature. After completion of the reaction, water was added for liquid separation, and the organic layer was washed with an aqueous sodium carbonate solution.
- N- (diphenylmethylene) -5,5'-dimethyl- [2,2'-bithiophene] -3-amine (11.4 g) was dissolved in THF (165 ml). 6M hydrochloric acid (98 ml) was added thereto, and the mixture was stirred at room temperature for 10 minutes. The solvent was distilled off under reduced pressure, and the precipitated solid was collected by suction filtration and washed with heptane to obtain 5,5′-dimethyl- [2,2′-bithiophene] -3-amine hydrochloride (10.0 g). Got.
- 2- (1-phenyl-1H-indolo-2-yl) aniline (25.0 g), 2-bromobiphenyl (20.5 g), sodium-t-butoxide (13.0 g), Pd (dba) 2 (0.13 g), 4- (di-t-butylphosphino) -N, N-dimethylaniline (0.12 g) and xylene (120 ml) were placed in a nitrogen atmosphere and stirred at 90 ° C. for 1 hour. . After cooling the reaction solution to room temperature, water and chlorobenzene were added for analysis, and purified with an activated alumina short column (developing solution: chlorobenzene).
- polycyclic aromatic compounds of the present invention can be synthesized by a method according to the synthesis example described above by appropriately changing the raw material compound.
- the electroluminescent devices according to Examples 1 to 4 and Comparative Example 1 were manufactured, and the driving start voltage (V) and the current efficiency (cd / A) at the time of constant current driving at a current density at which a luminance of 1000 cd / m 2 was obtained, respectively. ) was measured.
- V driving start voltage
- cd / A current efficiency
- Table 1 below shows the material structure of each layer in the organic electroluminescent elements according to Examples 1 to 4 and Comparative Example 1.
- HI refers to N 4 , N 4 ′ -diphenyl-N 4 , N 4 ′ -bis (9-phenyl-9H-carbazol-3-yl)-[1,1′-biphenyl] -4, 4′-diamine
- NPD means N 4 , N 4 ′ -di (naphthalen-1-yl) -N 4 , N 4 ′ -diphenyl- [1,1′-biphenyl] -4,4′-diamine
- CBP is 4,4′-di (9H-carbazolyl-9-yl) -1,1′-biphenyl
- Ir (PPy) 3 is tris (2-phenylpyridine) iridium (III)
- BCP Is 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline
- ET1 is 2,5-bis- (2 ′, 2 ′′ -bipyridin
- Example 1 ⁇ Element Using Compound (1) as Host Material for Light-Emitting Layer> A glass substrate of 26 mm ⁇ 28 mm ⁇ 0.7 mm (manufactured by Optoscience Co., Ltd.) obtained by polishing ITO deposited to a thickness of 180 nm by sputtering to 150 nm was used as a transparent support substrate.
- the transparent support substrate is fixed to a substrate holder of a commercially available vapor deposition apparatus (manufactured by Showa Vacuum Co., Ltd.), a molybdenum vapor deposition boat containing HI, a molybdenum vapor deposition boat containing NPD, and the compound of the present invention (1 ) Molybdenum vapor deposition boat, Ir (PPy) 3 molybdenum vapor deposition boat, BCP molybdenum vapor deposition boat, ET1 molybdenum vapor deposition boat, LiF A vapor deposition boat and a tungsten vapor deposition boat containing aluminum were mounted.
- a commercially available vapor deposition apparatus manufactured by Showa Vacuum Co., Ltd.
- a molybdenum vapor deposition boat containing HI a molybdenum vapor deposition boat containing NPD
- the compound of the present invention (1 ) Molybdenum vapor deposition boat, Ir (PPy) 3 moly
- the following layers were sequentially formed on the ITO film of the transparent support substrate.
- the vacuum chamber was depressurized to 5 ⁇ 10 ⁇ 4 Pa, first, a vapor deposition boat containing HI was heated and vapor-deposited to a film thickness of 40 nm to form a hole injection layer, and then NPD was contained. The vapor deposition boat was heated and vapor-deposited to a film thickness of 10 nm to form a hole transport layer. Next, the vapor deposition boat containing the compound (1) and the vapor deposition boat containing Ir (PPy) 3 were simultaneously heated to vapor-deposit so as to have a film thickness of 35 nm, thereby forming a light emitting layer.
- the deposition rate was adjusted so that the weight ratio of compound (1) to Ir (PPy) 3 was approximately 95: 5.
- a vapor deposition boat containing BCP was heated and vapor-deposited to a thickness of 5 nm to form a hole blocking layer.
- the evaporation boat containing ET1 was heated and evaporated to a film thickness of 15 nm to form an electron transport layer.
- the deposition rate of each layer was 0.01 to 1 nm / second.
- the deposition boat containing LiF was heated to deposit at a deposition rate of 0.01 to 0.1 nm / second so as to have a film thickness of 1 nm.
- a vapor deposition boat containing aluminum was heated, and aluminum was deposited at a deposition rate of 0.01 to 2 nm / second so as to have a film thickness of 100 nm to form a cathode, thereby obtaining an organic EL device.
- Example 2 ⁇ Element Using Compound (66) as Host Material for Light-Emitting Layer> An organic EL device was obtained by the method according to Example 1 except that the compound (1) as the host material of the light emitting layer was changed to the compound (66). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining the initial luminance of 1000 cd / m 2 was 5.7 V, and the current efficiency at that time was 36.4 cd / A.
- Example 3 ⁇ Element Using Compound (197) as Host Material for Light-Emitting Layer> An organic EL device was obtained by the method according to Example 1 except that the compound (1) which is the host material of the light emitting layer was changed to the compound (197). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 5.6 V, and the current efficiency at that time was 28.1 cd / A.
- Example 4 ⁇ Element Using Compound (198) for Hole Transport Layer> An organic EL device was obtained by the same method as in Example 1 except that NPD as the hole transport material was changed to compound (198) and compound (1) as the host material of the light emitting layer was changed to CBP. When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 5.9 V, and the current efficiency at that time was 26.4 cd / A.
- Example 1 An organic EL device was obtained by the method according to Example 1 except that the compound (1) as the host material of the light emitting layer was changed to CBP. When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining the initial luminance of 1000 cd / m 2 was 6.7 V, and the current efficiency at that time was 24.6 cd / A.
- Example 5 the electroluminescent elements according to Example 5 and Comparative Example 2 were manufactured, and the driving start voltage (V) and the current efficiency (cd /) when driven at a constant current at a current density at which a luminance of 1000 cd / m 2 was obtained, respectively. A) was measured.
- V driving start voltage
- cd / current efficiency
- Table 3 below shows the material structure of each layer in the organic electroluminescent elements according to the manufactured Example 5 and Comparative Example 2.
- HT is N-([1,1′-biphenyl] -4-yl) -9,9-dimethyl-N- (4- (9-phenyl-9H-carbazol-3-yl) phenyl. ) -9H-fluoren-2-amine (the same applies to the following tables).
- the chemical structure is shown below.
- Example 5 ⁇ Element Using Compound (251) as Host Material for Light-Emitting Layer> A glass substrate of 26 mm ⁇ 28 mm ⁇ 0.7 mm (manufactured by Optoscience Co., Ltd.) obtained by polishing ITO deposited to a thickness of 180 nm by sputtering to 150 nm was used as a transparent support substrate.
- This transparent support substrate is fixed to a substrate holder of a commercially available vapor deposition apparatus (manufactured by Showa Vacuum Co., Ltd.), a molybdenum vapor deposition boat containing HI, a molybdenum vapor deposition boat containing HT, the compound of the present invention (251 ) Molybdenum vapor deposition boat, Ir (PPy) 3 molybdenum vapor deposition boat, BCP molybdenum vapor deposition boat, ET1 molybdenum vapor deposition boat, LiF A vapor deposition boat and a tungsten vapor deposition boat containing aluminum were mounted.
- a commercially available vapor deposition apparatus manufactured by Showa Vacuum Co., Ltd.
- a molybdenum vapor deposition boat containing HI a molybdenum vapor deposition boat containing HT
- the compound of the present invention (251 ) Molybdenum vapor deposition boat, Ir (PPy) 3 mo
- the following layers were sequentially formed on the ITO film of the transparent support substrate.
- the vacuum chamber was depressurized to 5 ⁇ 10 ⁇ 4 Pa, first, a vapor deposition boat containing HI was heated and vapor-deposited to a film thickness of 30 nm to form a hole injection layer, and then HT was contained.
- the vapor deposition boat was heated and vapor-deposited to a film thickness of 20 nm to form a hole transport layer.
- the vapor deposition boat containing the compound (251) and the vapor deposition boat containing Ir (PPy) 3 were simultaneously heated to vapor-deposit so as to have a film thickness of 35 nm, thereby forming a light emitting layer.
- the deposition rate was adjusted so that the weight ratio of compound (251) to Ir (PPy) 3 was approximately 95: 5.
- a vapor deposition boat containing BCP was heated and vapor-deposited to a thickness of 5 nm to form a hole blocking layer.
- the evaporation boat containing ET1 was heated and evaporated to a film thickness of 15 nm to form an electron transport layer.
- the deposition rate of each layer was 0.01 to 1 nm / second.
- the deposition boat containing LiF was heated to deposit at a deposition rate of 0.01 to 0.1 nm / second so as to have a film thickness of 1 nm.
- a vapor deposition boat containing aluminum was heated, and aluminum was deposited at a deposition rate of 0.01 to 2 nm / second so as to have a film thickness of 100 nm to form a cathode, thereby obtaining an organic EL device.
- Example 2 An organic EL device was obtained by the method according to Example 5 except that the compound (251) as the host material of the light emitting layer was changed to CBP. When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 5.9 V, and the current efficiency at that time was 31.8 cd / A.
- Table 5 below shows the material structure of each layer in the organic electroluminescent elements according to the produced Examples 6 to 14 and Comparative Examples 3 to 4.
- HB1 is 9- (4 ′-(dimesitylboryl)-[1,1′-binaphthalene] -4-yl) -9H-carbazole
- ET2 is 5,5 ”-(2-phenylanthracene -9,10-diyl) di-2,2'-bipyridine (the same applies to the following tables)
- the chemical structure is shown below.
- Example 6 ⁇ Element Using Compound (1) as Host Material for Light-Emitting Layer, Part 2> A glass substrate of 26 mm ⁇ 28 mm ⁇ 0.7 mm (manufactured by Optoscience Co., Ltd.) obtained by polishing ITO deposited to a thickness of 180 nm by sputtering to 150 nm was used as a transparent support substrate.
- This transparent support substrate is fixed to a substrate holder of a commercially available vapor deposition apparatus (manufactured by Showa Vacuum Co., Ltd.), a molybdenum vapor deposition boat containing HI, a molybdenum vapor deposition boat containing HT, the compound of the present invention (1 ) Molybdenum vapor deposition boat, Ir (PPy) 3 molybdenum vapor deposition boat, HB1 molybdenum vapor deposition boat, ET2 molybdenum vapor deposition boat, LiF A vapor deposition boat and a tungsten vapor deposition boat containing aluminum were mounted.
- a commercially available vapor deposition apparatus manufactured by Showa Vacuum Co., Ltd.
- a molybdenum vapor deposition boat containing HI a molybdenum vapor deposition boat containing HT
- the compound of the present invention (1 ) Molybdenum vapor deposition boat, Ir (PPy) 3 moly
- the following layers were sequentially formed on the ITO film of the transparent support substrate.
- the vacuum chamber was depressurized to 5 ⁇ 10 ⁇ 4 Pa, first, a vapor deposition boat containing HI was heated and vapor-deposited to a film thickness of 30 nm to form a hole injection layer, and then HT was contained. The vapor deposition boat was heated and vapor-deposited to a film thickness of 10 nm to form a hole transport layer. Next, the vapor deposition boat containing the compound (1) and the vapor deposition boat containing Ir (PPy) 3 were heated at the same time to form a light emitting layer by vapor deposition to a film thickness of 30 nm.
- the deposition rate was adjusted so that the weight ratio of compound (1) to Ir (PPy) 3 was approximately 95: 5.
- the evaporation boat containing HB1 was heated and evaporated to a film thickness of 10 nm to form a hole blocking layer.
- the evaporation boat containing ET2 was heated and evaporated to a thickness of 20 nm to form an electron transport layer.
- the deposition rate of each layer was 0.01 to 1 nm / second.
- the deposition boat containing LiF was heated to deposit at a deposition rate of 0.01 to 0.1 nm / second so as to have a film thickness of 1 nm.
- a vapor deposition boat containing aluminum was heated, and aluminum was deposited at a deposition rate of 0.01 to 2 nm / second so as to have a film thickness of 100 nm to form a cathode, thereby obtaining an organic EL device.
- Example 7 ⁇ Element Using Compound (501) as Host Material for Light-Emitting Layer> An organic EL device was obtained by the method according to Example 6 except that the compound (1) as the host material of the light emitting layer was changed to the compound (501). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 6.2 V, and the current efficiency at that time was 29.0 cd / A.
- Example 8 ⁇ Element Using Compound (551) as Host Material for Light-Emitting Layer> An organic EL device was obtained by the method according to Example 6 except that the compound (1) which is the host material of the light emitting layer was changed to the compound (551). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 4.8 V, and the current efficiency at that time was 31.7 cd / A.
- Example 9 ⁇ Element Using Compound (687) as Host Material for Light-Emitting Layer> An organic EL device was obtained by the method according to Example 6 except that the compound (1) which is the host material of the light emitting layer was changed to the compound (687). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 4.0 V, and the current efficiency at that time was 28.3 cd / A.
- Example 10 ⁇ Element Using Compound (301) for Hole-Blocking and Electron Transport Layer (Used in One Layer)> A glass substrate of 26 mm ⁇ 28 mm ⁇ 0.7 mm (manufactured by Optoscience Co., Ltd.) obtained by polishing ITO deposited to a thickness of 180 nm by sputtering to 150 nm was used as a transparent support substrate.
- This transparent support substrate is fixed to a substrate holder of a commercially available vapor deposition apparatus (manufactured by Showa Vacuum Co., Ltd.), and a molybdenum vapor deposition boat containing HI, a molybdenum vapor deposition boat containing HT, and the CBP of the present invention are placed.
- Molybdenum vapor deposition boat, molybdenum vapor deposition boat with Ir (PPy) 3 molybdenum vapor deposition boat with compound (301), molybdenum vapor deposition boat with LiF, and tungsten with aluminum A vapor deposition boat was installed.
- the following layers were sequentially formed on the ITO film of the transparent support substrate.
- the vacuum chamber was depressurized to 5 ⁇ 10 ⁇ 4 Pa, first, a vapor deposition boat containing HI was heated and vapor-deposited to a film thickness of 30 nm to form a hole injection layer, and then HT was contained. The vapor deposition boat was heated and vapor-deposited to a film thickness of 10 nm to form a hole transport layer. Next, the vapor deposition boat containing CBP and the vapor deposition boat containing Ir (PPy) 3 were heated at the same time to form a light emitting layer by vapor deposition to a film thickness of 30 nm.
- the deposition rate was adjusted so that the weight ratio of CBP to Ir (PPy) 3 was approximately 95: 5.
- the evaporation boat containing the compound (301) was heated and evaporated to a thickness of 30 nm to form a hole blocking layer / electron transport layer.
- the deposition rate of each layer was 0.01 to 1 nm / second.
- the deposition boat containing LiF was heated to deposit at a deposition rate of 0.01 to 0.1 nm / second so as to have a film thickness of 1 nm.
- a vapor deposition boat containing aluminum was heated, and aluminum was deposited at a deposition rate of 0.01 to 2 nm / second so as to have a film thickness of 100 nm to form a cathode, thereby obtaining an organic EL device.
- Example 11 ⁇ Element Using Compound (391) for Hole Blocking and Electron Transport Layer (Used in One Layer)> An organic EL device was obtained by a method according to Example 10 except that the compound (301) which was the hole blocking layer and electron transport layer was replaced with the compound (391). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining the initial luminance of 1000 cd / m 2 was 6.0 V, and the current efficiency at that time was 28.0 cd / A.
- Example 12 ⁇ Element Using Compound (392) for Hole-Blocking and Electron Transport Layer (Used in One Layer)> An organic EL device was obtained by a method according to Example 10 except that the compound (301), which was a hole blocking layer / electron transport layer, was replaced with the compound (392). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 6.2 V, and the current efficiency at that time was 26.2 cd / A.
- Example 13 ⁇ Element Using Compound (391) for Hole Blocking Layer> A glass substrate of 26 mm ⁇ 28 mm ⁇ 0.7 mm (manufactured by Optoscience Co., Ltd.) obtained by polishing ITO deposited to a thickness of 180 nm by sputtering to 150 nm was used as a transparent support substrate.
- This transparent support substrate is fixed to a substrate holder of a commercially available vapor deposition apparatus (manufactured by Showa Vacuum Co., Ltd.), and a molybdenum vapor deposition boat containing HI, a molybdenum vapor deposition boat containing HT, and the CBP of the present invention are placed.
- Molybdenum vapor deposition boat, molybdenum vapor deposition boat containing Ir (PPy) 3 , molybdenum vapor deposition boat containing compound (391), molybdenum vapor deposition boat containing ET2, molybdenum product containing LiF A vapor deposition boat and a tungsten vapor deposition boat containing aluminum were mounted.
- the following layers were sequentially formed on the ITO film of the transparent support substrate.
- the vacuum chamber was depressurized to 5 ⁇ 10 ⁇ 4 Pa, first, a vapor deposition boat containing HI was heated and vapor-deposited to a film thickness of 30 nm to form a hole injection layer, and then HT was contained. The vapor deposition boat was heated and vapor-deposited to a film thickness of 10 nm to form a hole transport layer. Next, the vapor deposition boat containing CBP and the vapor deposition boat containing Ir (PPy) 3 were heated at the same time to form a light emitting layer by vapor deposition to a film thickness of 30 nm.
- the deposition rate was adjusted so that the weight ratio of CBP to Ir (PPy) 3 was approximately 95: 5.
- the evaporation boat containing the compound (391) was heated and evaporated to a thickness of 10 nm to form a hole blocking layer.
- the evaporation boat containing ET2 was heated and evaporated to a thickness of 20 nm to form an electron transport layer.
- the deposition rate of each layer was 0.01 to 1 nm / second.
- the deposition boat containing LiF was heated to deposit at a deposition rate of 0.01 to 0.1 nm / second so as to have a film thickness of 1 nm.
- a vapor deposition boat containing aluminum was heated, and aluminum was deposited at a deposition rate of 0.01 to 2 nm / second so as to have a film thickness of 100 nm to form a cathode, thereby obtaining an organic EL device.
- Example 14 ⁇ Device Using Compound (392) for Hole Blocking Layer> An organic EL device was obtained by the method according to Example 13 except that the compound (391) as the hole blocking layer was changed to the compound (392). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 4.9 V, and the current efficiency at that time was 32.7 cd / A.
- Table 8 below shows the material configuration of each layer in the organic electroluminescent elements according to Examples 15 to 29 and Comparative Example 5 that were produced.
- HAT-CN is 1,4,5,8,9,12-hexaazatriphenylenehexacarbonitrile
- TBB is N 4 , N 4 , N 4 ′, N 4 ′ -tetra ([[ 1,1′-biphenyl] -4-yl)-[1,1′-biphenyl] -4,4′-diamine
- TBi is 1,3,5-tris (1-phenyl-1H-benzo [d ] Imidazol-2-yl) benzene (the same applies to the following tables). The chemical structure is shown below.
- Example 15 ⁇ Element Using Compound (1) as Host Material for Light-Emitting Layer, Part 3> A glass substrate of 26 mm ⁇ 28 mm ⁇ 0.7 mm (manufactured by Optoscience Co., Ltd.) obtained by polishing ITO deposited to a thickness of 180 nm by sputtering to 150 nm was used as a transparent support substrate.
- This transparent support substrate is fixed to a substrate holder of a commercially available vapor deposition apparatus (manufactured by Showa Vacuum Co., Ltd.), a molybdenum vapor deposition boat containing HAT-CN, a molybdenum vapor deposition boat containing TBB, and a compound of the present invention Molybdenum deposition boat containing (1), molybdenum deposition boat containing Ir (PPy) 3 , molybdenum deposition boat containing TPBi, molybdenum deposition boat containing LiF, and aluminum A tungsten evaporation boat was attached.
- a commercially available vapor deposition apparatus manufactured by Showa Vacuum Co., Ltd.
- Molybdenum deposition boat containing (1) molybdenum deposition boat containing Ir (PPy) 3
- molybdenum deposition boat containing TPBi molybdenum deposition boat containing LiF
- aluminum A tungsten evaporation boat was attached.
- the following layers were sequentially formed on the ITO film of the transparent support substrate. Depressurize the vacuum chamber to 5 ⁇ 10 ⁇ 4 Pa, first heat the evaporation boat containing HAT-CN to form a hole injection layer by vapor deposition to a film thickness of 10 nm, and then TBB The vapor deposition boat was heated and vapor deposited to a film thickness of 30 nm to form a hole transport layer. Next, the vapor deposition boat containing the compound (1) and the vapor deposition boat containing Ir (PPy) 3 were heated at the same time to form a light emitting layer by vapor deposition to a film thickness of 30 nm.
- the deposition rate was adjusted so that the weight ratio of compound (1) to Ir (PPy) 3 was approximately 95: 5.
- the evaporation boat containing TPBi was heated and evaporated to a thickness of 50 nm to form an electron transport layer.
- the deposition rate of each layer was 0.01 to 1 nm / second.
- the deposition boat containing LiF was heated to deposit at a deposition rate of 0.01 to 0.1 nm / second so as to have a film thickness of 1 nm.
- a vapor deposition boat containing aluminum was heated, and aluminum was deposited at a deposition rate of 0.01 to 2 nm / second so as to have a film thickness of 100 nm to form a cathode, thereby obtaining an organic EL device.
- Example 16 ⁇ Element Using Compound (66) as Host Material for Light-Emitting Layer> An organic EL device was obtained by the method according to Example 15 except that the compound (1) as the host material for the light emitting layer was changed to the compound (66). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 4.8 V, and the current efficiency at that time was 37.0 cd / A.
- Example 17 ⁇ Element Using Compound (84) as Host Material for Light-Emitting Layer> An organic EL device was obtained by the method according to Example 15 except that the compound (1) as the host material for the light emitting layer was changed to the compound (84). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 5.6 V, and the current efficiency at that time was 35.9 cd / A.
- Example 18 ⁇ Element Using Compound (86) as Host Material for Light-Emitting Layer> An organic EL device was obtained by the method according to Example 15 except that the compound (1) as the host material for the light emitting layer was changed to the compound (86). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 5.0 V, and the current efficiency at that time was 29.0 cd / A.
- Example 19 ⁇ Element Using Compound (197) as Host Material for Light-Emitting Layer> An organic EL device was obtained by the method according to Example 15 except that the compound (1), which is the host material of the light emitting layer, was changed to the compound (197). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining the initial luminance of 1000 cd / m 2 was 4.9 V, and the current efficiency at that time was 32.4 cd / A.
- Example 20> ⁇ Element Using Compound (51) as Host Material for Light-Emitting Layer> An organic EL device was obtained by the method according to Example 15 except that the compound (1), which is the host material of the light emitting layer, was changed to the compound (51). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 5.1 V, and the current efficiency at that time was 39.2 cd / A.
- Example 21 ⁇ Element Using Compound (214) as Host Material for Light-Emitting Layer> An organic EL device was obtained by the method according to Example 15 except that the compound (1), which is the host material of the light emitting layer, was changed to the compound (214). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 4.2 V, and the current efficiency at that time was 35.2 cd / A.
- Example 22> ⁇ Element Using Compound (26) as Host Material for Light-Emitting Layer> An organic EL device was obtained by the method according to Example 15 except that the compound (1) as the host material of the light emitting layer was changed to the compound (26). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 4.7 V, and the current efficiency at that time was 42.2 cd / A.
- Example 23> ⁇ Device Using Compound (210) as Host Material for Light-Emitting Layer>
- An organic EL device was obtained by the method according to Example 15 except that the compound (1), which is the host material of the light emitting layer, was changed to the compound (210).
- a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained.
- the driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 5.1 V, and the current efficiency at that time was 32.7 cd / A.
- Example 24 ⁇ Element Using Compound (212) as Host Material for Light-Emitting Layer> An organic EL device was obtained by the method according to Example 15 except that the compound (1), which is the host material of the light emitting layer, was changed to the compound (212). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining the initial luminance of 1000 cd / m 2 was 5.3 V, and the current efficiency at that time was 27.0 cd / A.
- Example 25 ⁇ Element Using Compound (215) as Host Material for Light-Emitting Layer> An organic EL device was obtained by the method according to Example 15 except that the compound (1) which is the host material of the light emitting layer was changed to the compound (215). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 5.5 V, and the current efficiency at that time was 27.7 cd / A.
- Example 26 ⁇ Element Using Compound (48) as Host Material for Light-Emitting Layer> An organic EL device was obtained by the method according to Example 15 except that the compound (1) which is the host material of the light emitting layer was changed to the compound (48). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 5.5 V, and the current efficiency at that time was 29.2 cd / A.
- Example 27 ⁇ Element Using Compound (209) for Hole Transport Layer>
- a glass substrate of 26 mm ⁇ 28 mm ⁇ 0.7 mm obtained by polishing ITO deposited to a thickness of 180 nm by sputtering to 150 nm was used as a transparent support substrate.
- This transparent support substrate is fixed to a substrate holder of a commercially available vapor deposition apparatus (made by Showa Vacuum Co., Ltd.), a molybdenum vapor deposition boat containing HAT-CN, and a molybdenum vapor deposition containing the compound (209) of the present invention.
- the following layers were sequentially formed on the ITO film of the transparent support substrate.
- the vacuum chamber is depressurized to 5 ⁇ 10 ⁇ 4 Pa, and first, a vapor deposition boat containing HAT-CN is heated to deposit to a thickness of 10 nm to form a hole injection layer. 209) was heated and evaporated to a thickness of 30 nm to form a hole transport layer.
- the vapor deposition boat containing CBP and the vapor deposition boat containing Ir (PPy) 3 were heated at the same time to form a light emitting layer by vapor deposition to a film thickness of 30 nm.
- the deposition rate was adjusted so that the weight ratio of CBP to Ir (PPy) 3 was approximately 95: 5.
- the evaporation boat containing TPBi was heated and evaporated to a film thickness of 50 nm to form an electron transport layer.
- the deposition rate of each layer was 0.01 to 1 nm / second.
- the deposition boat containing LiF was heated to deposit at a deposition rate of 0.01 to 0.1 nm / second so as to have a film thickness of 1 nm.
- a vapor deposition boat containing aluminum was heated, and aluminum was deposited at a deposition rate of 0.01 to 2 nm / second so as to have a film thickness of 100 nm to form a cathode, thereby obtaining an organic EL device.
- Example 28 ⁇ Element Using Compound (366) for Electron Transport Layer>
- a glass substrate of 26 mm ⁇ 28 mm ⁇ 0.7 mm obtained by polishing ITO deposited to a thickness of 180 nm by sputtering to 150 nm was used as a transparent support substrate.
- This transparent support substrate was fixed to a substrate holder of a commercially available vapor deposition apparatus (manufactured by Showa Vacuum Co., Ltd.), and a molybdenum vapor deposition boat containing HAT-CN, a molybdenum vapor deposition boat containing TBB, and CBP were introduced.
- Molybdenum deposition boat molybdenum deposition boat containing Ir (PPy) 3 , molybdenum deposition boat containing the compound of the present invention (366), molybdenum deposition boat containing LiF, and aluminum A tungsten evaporation boat was attached.
- the following layers were sequentially formed on the ITO film of the transparent support substrate.
- Depressurize the vacuum chamber to 5 ⁇ 10 ⁇ 4 Pa first heat the evaporation boat containing HAT-CN to form a hole injection layer by vapor deposition to a film thickness of 10 nm, and then TBB The vapor deposition boat was heated and vapor deposited to a film thickness of 30 nm to form a hole transport layer.
- the vapor deposition boat containing CBP and the vapor deposition boat containing Ir (PPy) 3 were heated at the same time to form a light emitting layer by vapor deposition to a film thickness of 30 nm.
- the deposition rate was adjusted so that the weight ratio of CBP to Ir (PPy) 3 was approximately 95: 5.
- the evaporation boat containing the compound (366) was heated and evaporated to a film thickness of 50 nm to form an electron transport layer.
- the deposition rate of each layer was 0.01 to 1 nm / second.
- the deposition boat containing LiF was heated to deposit at a deposition rate of 0.01 to 0.1 nm / second so as to have a film thickness of 1 nm.
- a vapor deposition boat containing aluminum was heated, and aluminum was deposited at a deposition rate of 0.01 to 2 nm / second so as to have a film thickness of 100 nm to form a cathode, thereby obtaining an organic EL device.
- Example 29 ⁇ Element Using Compound (424) for Electron Transport Layer> An organic EL device was obtained by the method according to Example 28 except that the compound (366) as the electron transport material was changed to the compound (424). When a DC voltage was applied to both electrodes, green light emission with a wavelength of about 515 nm was obtained. The driving voltage for obtaining an initial luminance of 1000 cd / m 2 was 6.4 V, and the current efficiency at that time was 27.5 cd / A.
- a glass substrate of 26 mm ⁇ 28 mm ⁇ 0.5 mm (manufactured by Nippon Sheet Glass Co., Ltd.) was used as a transparent support substrate.
- This transparent support substrate was mounted on a substrate holder of a commercially available vapor deposition apparatus at the same time as a metal mask for obtaining a lower aluminum electrode having a width of 2 mm.
- a tungsten vapor deposition boat on which aluminum was placed was set in a vapor deposition apparatus.
- the vacuum chamber was depressurized to 5 ⁇ 10 ⁇ 3 Pa or less, and the evaporation boat was heated to form a translucent lower aluminum electrode so as to have a film thickness of 10 nm.
- the deposition rate was 0.05 to 1 nm / second.
- a metal mask for forming an organic layer designed to cover the lower aluminum electrode is mounted on the substrate holder, and set in the vapor deposition apparatus together with the molybdenum vapor deposition boat containing the compound represented by the formula (1).
- the vacuum tank was depressurized to 5 ⁇ 10 ⁇ 3 Pa or less, and the evaporation boat was heated to deposit the compound represented by the formula (1).
- the film thickness was 6 ⁇ m, and the deposition rate was 0.1 to 10 nm / second.
- a metal mask for forming the upper aluminum electrode was mounted on the substrate holder, and set in a vapor deposition apparatus together with a tungsten vapor deposition boat on which aluminum was placed.
- This metal mask is designed so that the overlapping area between the organic layers of the upper and lower aluminum electrodes is 4 mm 2 .
- the vacuum chamber was depressurized to 5 ⁇ 10 ⁇ 3 Pa or less, and the deposition boat was heated to form an upper aluminum electrode so as to have a film thickness of 50 nm.
- the deposition rate was 0.05 to 1 nm / second.
- the mobility was measured using the Time Of Flight method.
- the measurement was performed using a commercially available measuring device, TOF-401 (manufactured by Sumitomo Heavy Industries Advanced Machinery Co., Ltd.).
- a nitrogen gas laser was used as the excitation light source.
- With a moderate voltage applied between the upper aluminum electrode and lower aluminum electrode light was irradiated from the translucent lower aluminum electrode side, and the transient photocurrent was observed to determine the mobility.
- a method for deriving the mobility from the analysis of the transient photocurrent waveform is described in P69-70 of the document “Organic EL materials and displays” (published by CMC Co., Ltd.).
- an organic electroluminescent element with improved driving voltage and current efficiency, a display device including the same, a lighting device including the same, and the like.
Abstract
Description
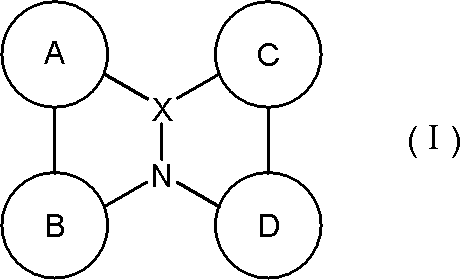
(上記式(I)中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
A環、B環、C環およびD環は、それぞれ独立して、置換されていてもよい芳香族環または置換されていてもよいヘテロ芳香族環であり、隣接する2つの環は連結基または単結合によりそれらの間に環を形成していてもよく、そして、
上記式(I)で表される部分構造は少なくとも1つの水素を有し、そして、該部分構造における少なくとも1つの水素が重水素で置換されていてもよい。)
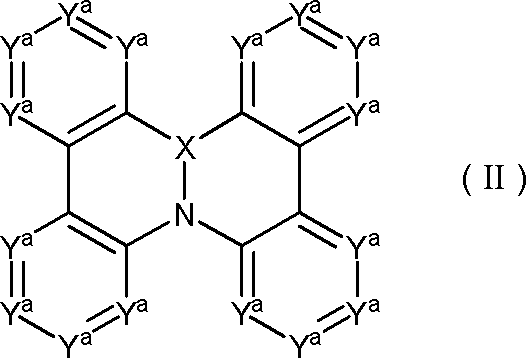
(上記式(II)中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
Yaは、それぞれ独立して、CまたはNであり、同じ環で隣接する2つのYaとそれらの間の結合とが一緒になって、N、O、SまたはSeになってもよく、環はそれぞれ独立して置換されていてもよく、同じ環における隣接する置換基が結合してシクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、隣接する2つの環は連結基または単結合によりそれらの間に環を形成していてもよく、そして、
上記式(II)で表される部分構造は少なくとも1つの水素を有し、そして、該部分構造における少なくとも1つの水素が重水素で置換されていてもよい。)

(上記式(III-1)中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
上記式中のベンゼン環はそれぞれ独立して置換されていてもよく、同じ環における隣接する置換基が結合してシクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
上記式中の隣接する2つのベンゼン環は連結基または単結合によりそれらの間に環を形成していてもよく、そして、
上記式(III-1)で表される部分構造は少なくとも1つの水素を有し、そして、該部分構造における少なくとも1つの水素が重水素で置換されていてもよい。)

(上記各式中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
Zは、N、O、SまたはSeであり、
上記各式中のベンゼン環および五員環はそれぞれ独立して置換されていてもよく、同じ環における隣接する置換基が結合してシクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
上記各式中の隣接する2つのベンゼン環は連結基または単結合によりそれらの間に環を形成していてもよく、そして、
上記各式で表される部分構造は少なくとも1つの水素を有し、そして、該部分構造における少なくとも1つの水素が重水素で置換されていてもよい。)

(上記各式中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
上記各式中のベンゼン環および五員環はそれぞれ独立して置換されていてもよく、同じ環における隣接する置換基が結合してシクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
上記各式中の隣接する2つのベンゼン環は連結基または単結合によりそれらの間に環を形成していてもよく、そして、
上記各式で表される部分構造は少なくとも1つの水素を有し、そして、該部分構造における少なくとも1つの水素が重水素で置換されていてもよい。)
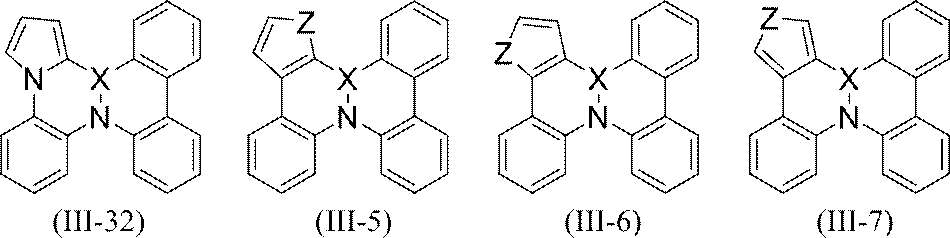
(上記各式中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
Zは、N、O、SまたはSeであり、
上記各式中のベンゼン環および五員環はそれぞれ独立して置換されていてもよく、同じ環における隣接する置換基が結合してシクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
上記各式中の隣接する2つのベンゼン環は連結基または単結合によりそれらの間に環を形成していてもよく、そして、
上記各式で表される部分構造は少なくとも1つの水素を有し、そして、該部分構造における少なくとも1つの水素が重水素で置換されていてもよい。)

(上記各式中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
上記各式中のベンゼン環および五員環はそれぞれ独立して置換されていてもよく、同じ環における隣接する置換基が結合してシクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
上記各式中の隣接する2つのベンゼン環は連結基または単結合によりそれらの間に環を形成していてもよく、そして、
上記各式で表される部分構造は少なくとも1つの水素を有し、そして、該部分構造における少なくとも1つの水素が重水素で置換されていてもよい。)

(上記各式中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
Rは、水素、フッ素置換または無置換のC1-20アルキル、C3-8シクロアルキル、C2-20アルケニル、モノもしくはジアリール置換C2-12アルケニル、モノもしくはジヘテロアリール置換C2-12アルケニル、フッ素置換または無置換のC1-20アルコキシ、C1-20アルキルカルボニル、シアノ、ニトロ、ジアリールアミノ、置換されていてもよいアリール、置換されていてもよいヘテロアリール、B(Ra)2、またはSi(Ra)3(ここでRaは、それぞれ独立して、置換されていてもよいアルキル、置換されていてもよいアリールまたは置換されていてもよいヘテロアリールである。)であり、
同じ環で隣接する2つのRが結合して、シクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
上記各式中の隣接する2つのベンゼン環は、単結合、CH2、CHRa、C(Ra)2、NRa、Si(Ra)2、BRa(ここでRaは前記に定義されるとおりである)、Se、S、またはOを介した結合により結合されて、それらの間に環を形成していてもよく、
nは0~4の整数であり、mは0~3の整数であり、そして、
上記各式で表される化合物またはその塩における少なくとも1つの水素が重水素で置換されていてもよい。)

(上記各式中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
Rは、水素、フッ素置換または無置換のC1-20アルキル、C3-8シクロアルキル、C2-20アルケニル、モノもしくはジアリール置換C2-12アルケニル、モノもしくはジヘテロアリール置換C2-12アルケニル、フッ素置換または無置換のC1-20アルコキシ、C1-20アルキルカルボニル、シアノ、ニトロ、ジアリールアミノ、置換されていてもよいアリール、置換されていてもよいヘテロアリール、B(Ra)2、またはSi(Ra)3(ここでRaは、それぞれ独立して、置換されていてもよいアルキル、置換されていてもよいアリールまたは置換されていてもよいヘテロアリールである。)であり、
同じ環で隣接する2つのRが結合して、シクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
上記各式中の隣接する2つのベンゼン環は、単結合、CH2、CHRa、C(Ra)2、NRa、Si(Ra)2、BRa(ここでRaは前記に定義されるとおりである)、Se、S、またはOを介した結合により結合されて、それらの間に環を形成していてもよく、
nは0~4の整数であり、hは0~3の整数であり、そして、
上記各式で表される化合物またはその塩における少なくとも1つの水素が重水素で置換されていてもよい。)
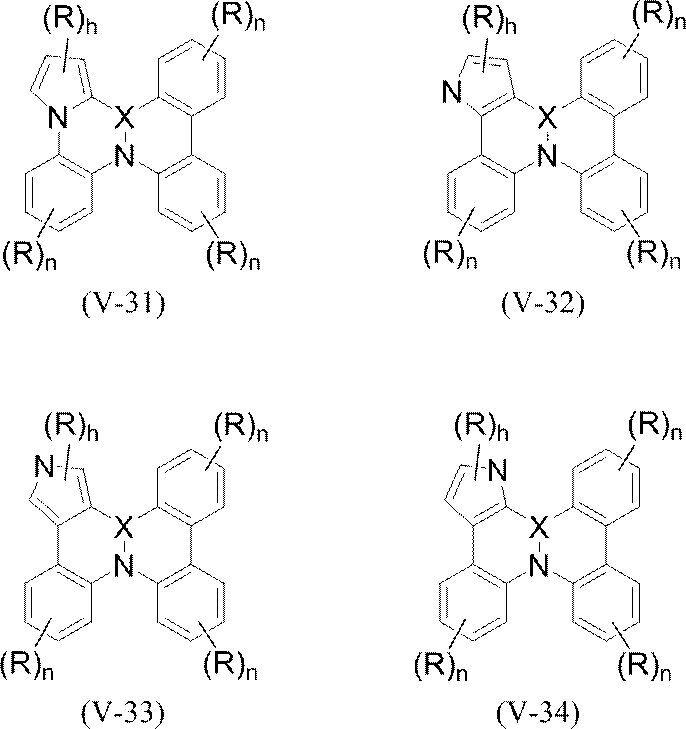
(上記各式中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
Rは、水素、フッ素置換または無置換のC1-20アルキル、C3-8シクロアルキル、C2-20アルケニル、モノもしくはジアリール置換C2-12アルケニル、モノもしくはジヘテロアリール置換C2-12アルケニル、フッ素置換または無置換のC1-20アルコキシ、C1-20アルキルカルボニル、シアノ、ニトロ、ジアリールアミノ、置換されていてもよいアリール、置換されていてもよいヘテロアリール、B(Ra)2、またはSi(Ra)3(ここでRaは、それぞれ独立して、置換されていてもよいアルキル、置換されていてもよいアリールまたは置換されていてもよいヘテロアリールである。)であり、
同じ環で隣接する2つのRが結合して、シクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
上記各式中の隣接する2つのベンゼン環は、単結合、CH2、CHRa、C(Ra)2、NRa、Si(Ra)2、BRa(ここでRaは前記に定義されるとおりである)、Se、S、またはOを介した結合により結合されて、それらの間に環を形成していてもよく、
nは0~4の整数であり、hは0~3の整数であり、そして、
上記各式で表される化合物またはその塩における少なくとも1つの水素が重水素で置換されていてもよい。)

(上記各式中、
Rは、フッ素置換または無置換のC1-20アルキル、C3-8シクロアルキル、C2-20アルケニル、モノもしくはジアリール置換C2-12アルケニル、モノもしくはジヘテロアリール置換C2-12アルケニル、フッ素置換または無置換のC1-20アルコキシ、C1-20アルキルカルボニル、シアノ、ニトロ、ジアリールアミノ、置換されていてもよいアリール、置換されていてもよいヘテロアリール、B(Ra)2、またはSi(Ra)3(ここでRaは、それぞれ独立して、置換されていてもよいアルキル、置換されていてもよいアリールまたは置換されていてもよいヘテロアリールである。)であり、
同じ環で隣接する2つのRが結合して、シクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
nは0~4の整数であり、mは0~3の整数であり、そして、
上記各式で表される化合物またはその塩における少なくとも1つの水素が重水素で置換されていてもよい。)
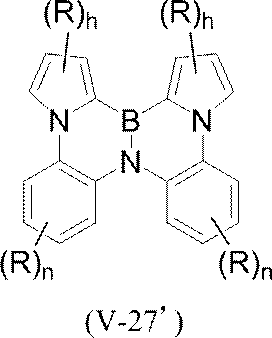
(上記式中、
Rは、フッ素置換または無置換のC1-20アルキル、C3-8シクロアルキル、C2-20アルケニル、モノもしくはジアリール置換C2-12アルケニル、モノもしくはジヘテロアリール置換C2-12アルケニル、フッ素置換または無置換のC1-20アルコキシ、C1-20アルキルカルボニル、シアノ、ニトロ、ジアリールアミノ、置換されていてもよいアリール、置換されていてもよいヘテロアリール、B(Ra)2、またはSi(Ra)3(ここでRaは、それぞれ独立して、置換されていてもよいアルキル、置換されていてもよいアリールまたは置換されていてもよいヘテロアリールである。)であり、
同じ環で隣接する2つのRが結合して、シクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
nは0~4の整数であり、hは0~3の整数であり、そして、
上記式で表される化合物またはその塩における少なくとも1つの水素が重水素で置換されていてもよい。)

(上記式中、
Rは、フッ素置換または無置換のC1-20アルキル、C3-8シクロアルキル、C2-20アルケニル、モノもしくはジアリール置換C2-12アルケニル、モノもしくはジヘテロアリール置換C2-12アルケニル、フッ素置換または無置換のC1-20アルコキシ、C1-20アルキルカルボニル、シアノ、ニトロ、ジアリールアミノ、置換されていてもよいアリール、置換されていてもよいヘテロアリール、B(Ra)2、またはSi(Ra)3(ここでRaは、それぞれ独立して、置換されていてもよいアルキル、置換されていてもよいアリールまたは置換されていてもよいヘテロアリールである。)であり、
同じ環で隣接する2つのRが結合して、シクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
nは0~4の整数であり、hは0~3の整数であり、そして、
上記式で表される化合物またはその塩における少なくとも1つの水素が重水素で置換されていてもよい。)
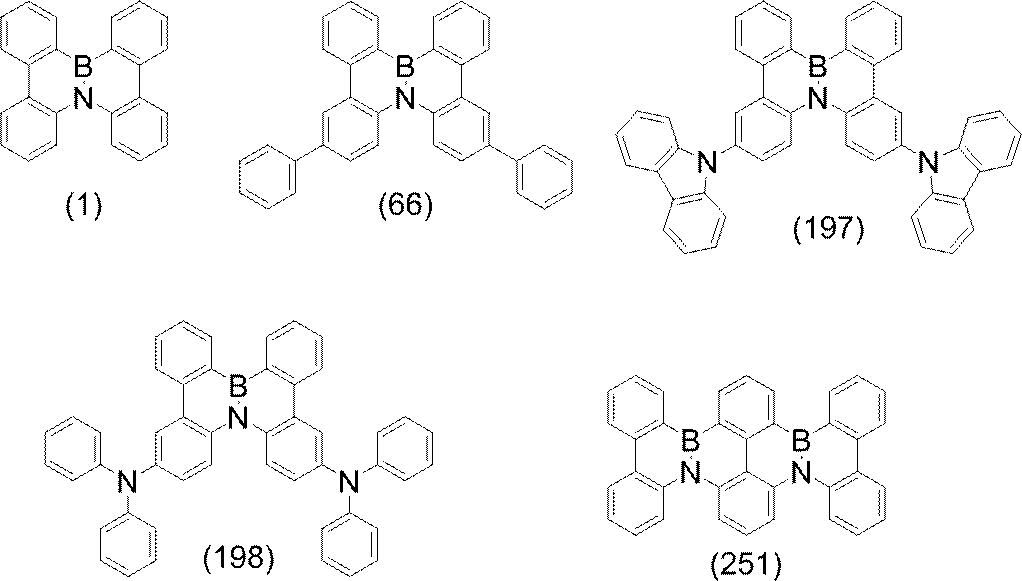
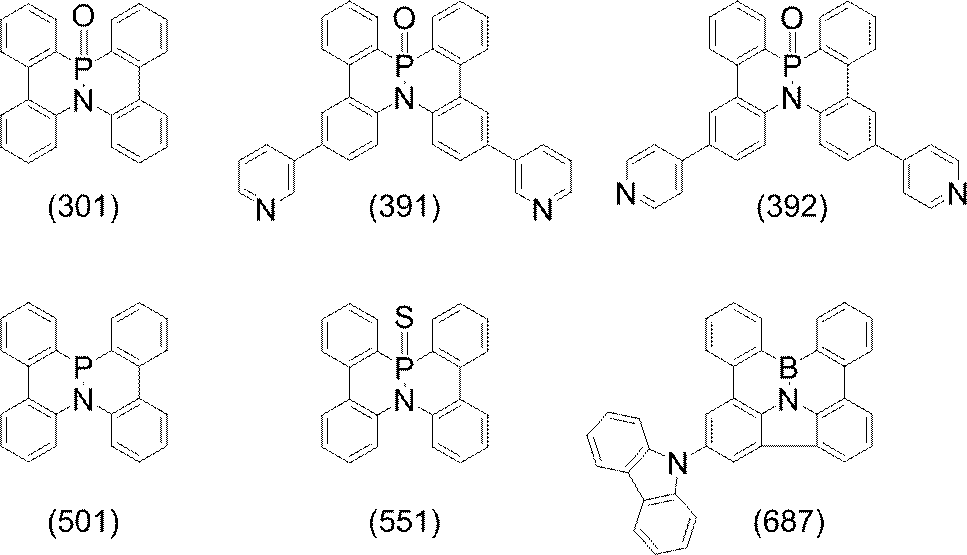

本発明の多環芳香族化合物(およびその塩)は、下記一般式(I)で表される部分構造を有し、有機電界発光素子用材料として有用である。なお、式中の各記号は上述するとおりである。
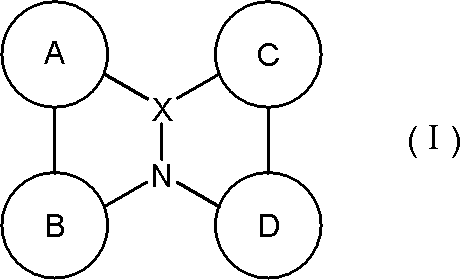



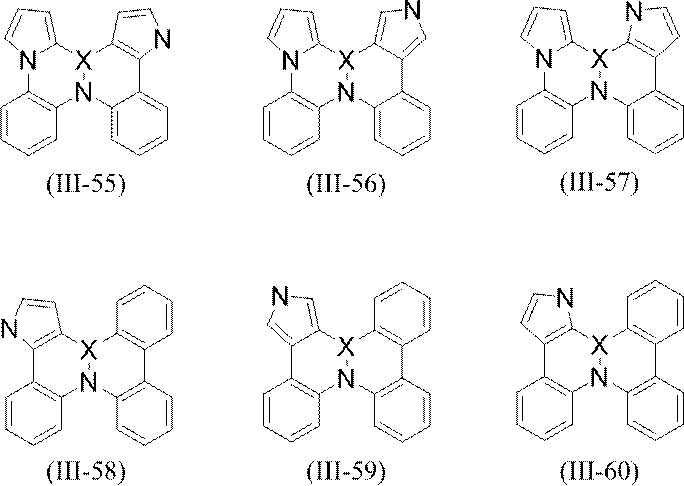
本発明の多環芳香族化合物(およびその塩)は、上述した部分構造を含む(例えば該部分構造の繰り返しからなる)化合物であり、具体的なものとしては、例えば、下記一般式(IV-1)~式(IV-22)で表される化合物が挙げられる。

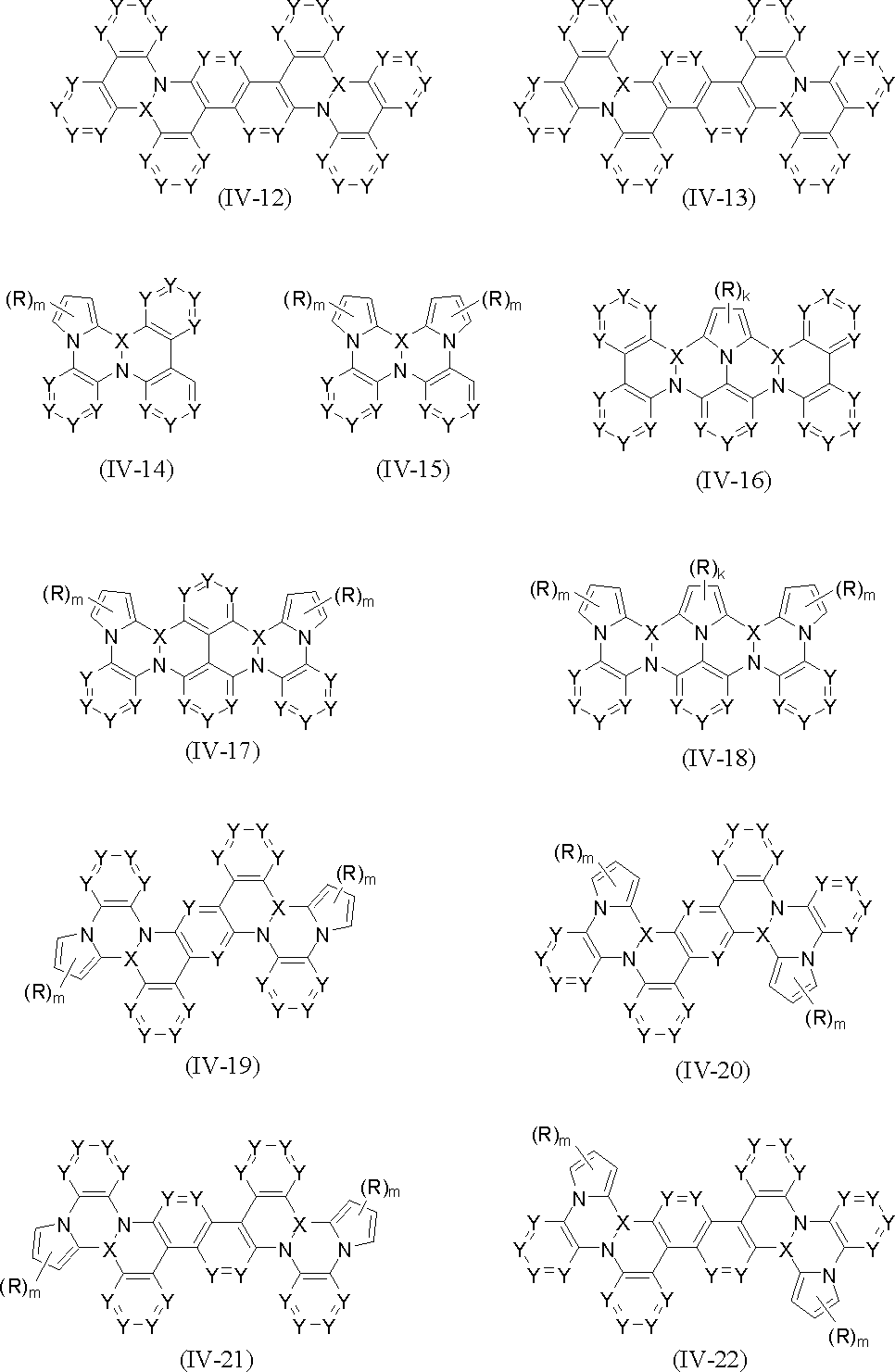
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素を示す。






Rの置換位置は、上記一般式(V-1’)、一般式(V-27’)および式(V-32’)においてNに結合するベンゼン環(式(I)のB環および/またはD環に相当)については、Nに結合する炭素の位置を基準としてパラ位への置換が好ましく、B環またはD環の一方の環のパラ位が置換されているか、または両方の環のパラ位が置換されていてもよく、両方の環のパラ位が置換されていることが好ましい。また、上記一般式(V-1’)および式(V-32’)においてBに結合するベンゼン環(式(I)のA環および/またはC環に相当)については、Bに結合する炭素の位置を基準としてオルト位への置換が好ましく、A環またはC環の一方の環のオルト位が置換されているか、または両方の環のオルト位が置換されていてもよい。
具体的には、後述する式(51)~(86)で表される化合物が好ましく、式(66)~(83)および(86)で表される化合物がより好ましく、式(66)~(74)で表される化合物がさらに好ましい。
Rの置換位置は、上記一般式(V-1’)、一般式(V-27’)および式(V-32’)においてNに結合するベンゼン環(式(I)のB環および/またはD環に相当)については、Nに結合する炭素の位置を基準としてパラ位への置換が好ましく、B環またはD環の一方の環のパラ位が置換されているか、または両方の環のパラ位が置換されていてもよい。
具体的には、後述する式(188)~(191)、式(193)~(195)、式(197)、式(198)、式(200)~(204)、および式(206)~(208)で表される化合物が好ましい。




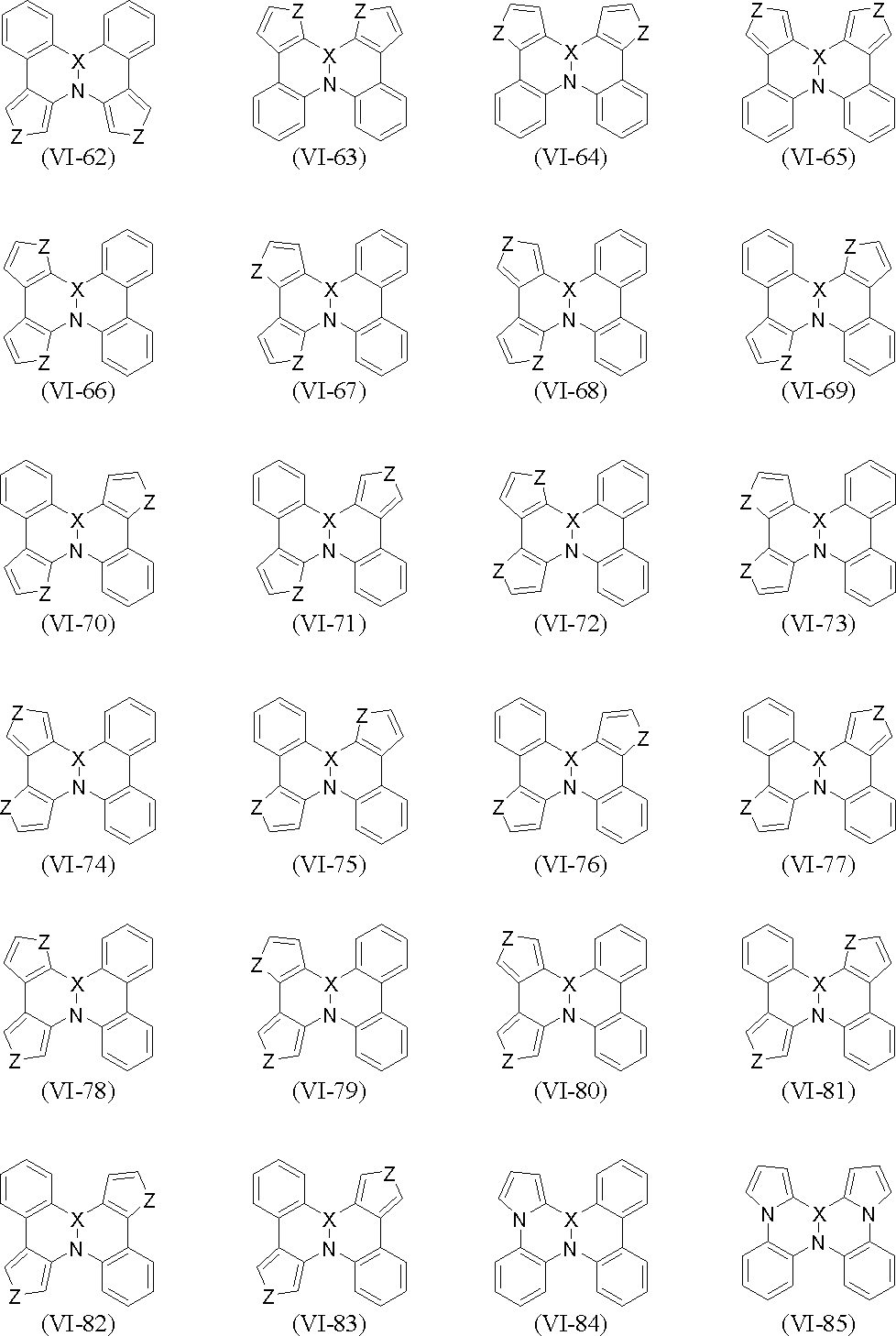

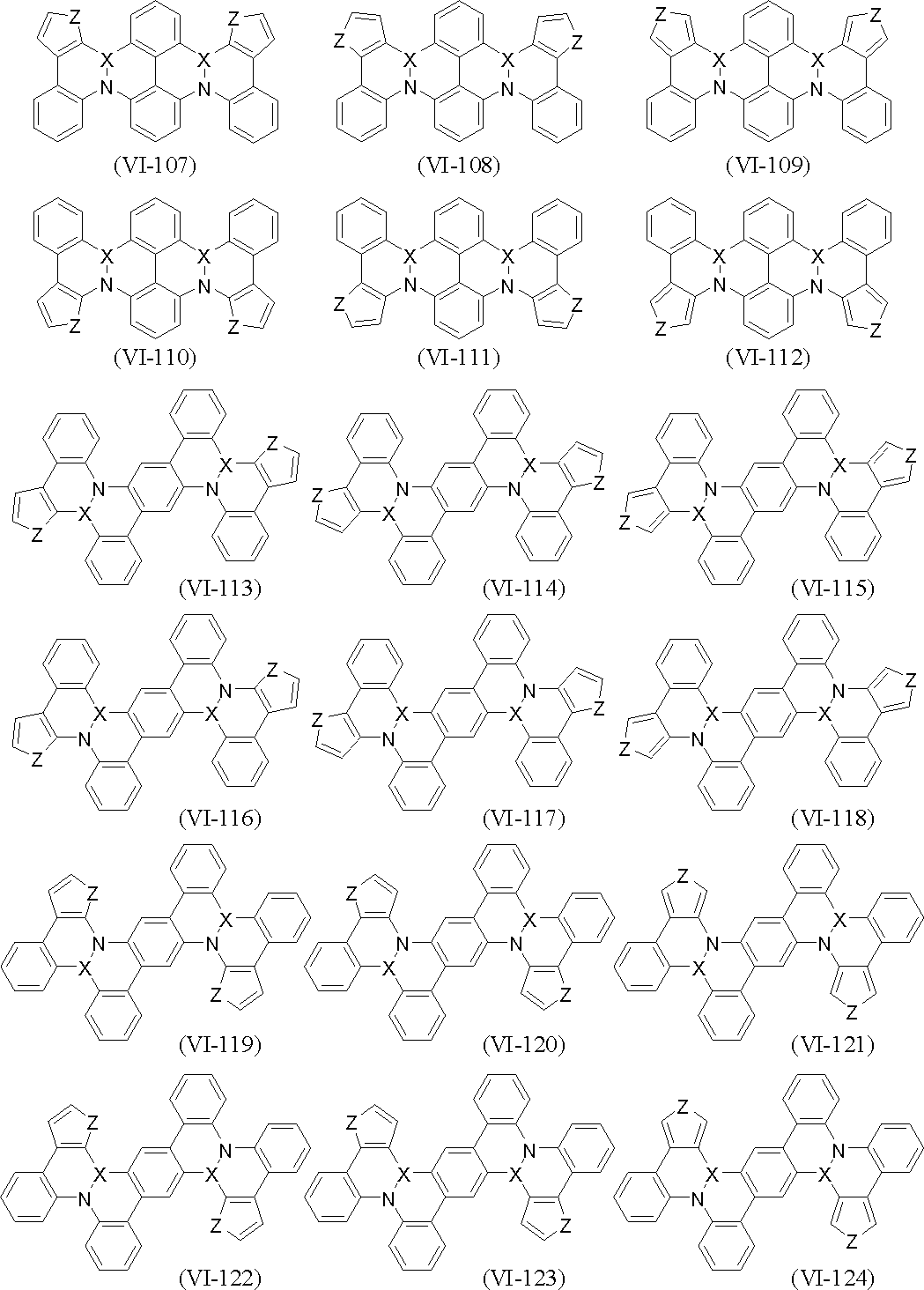
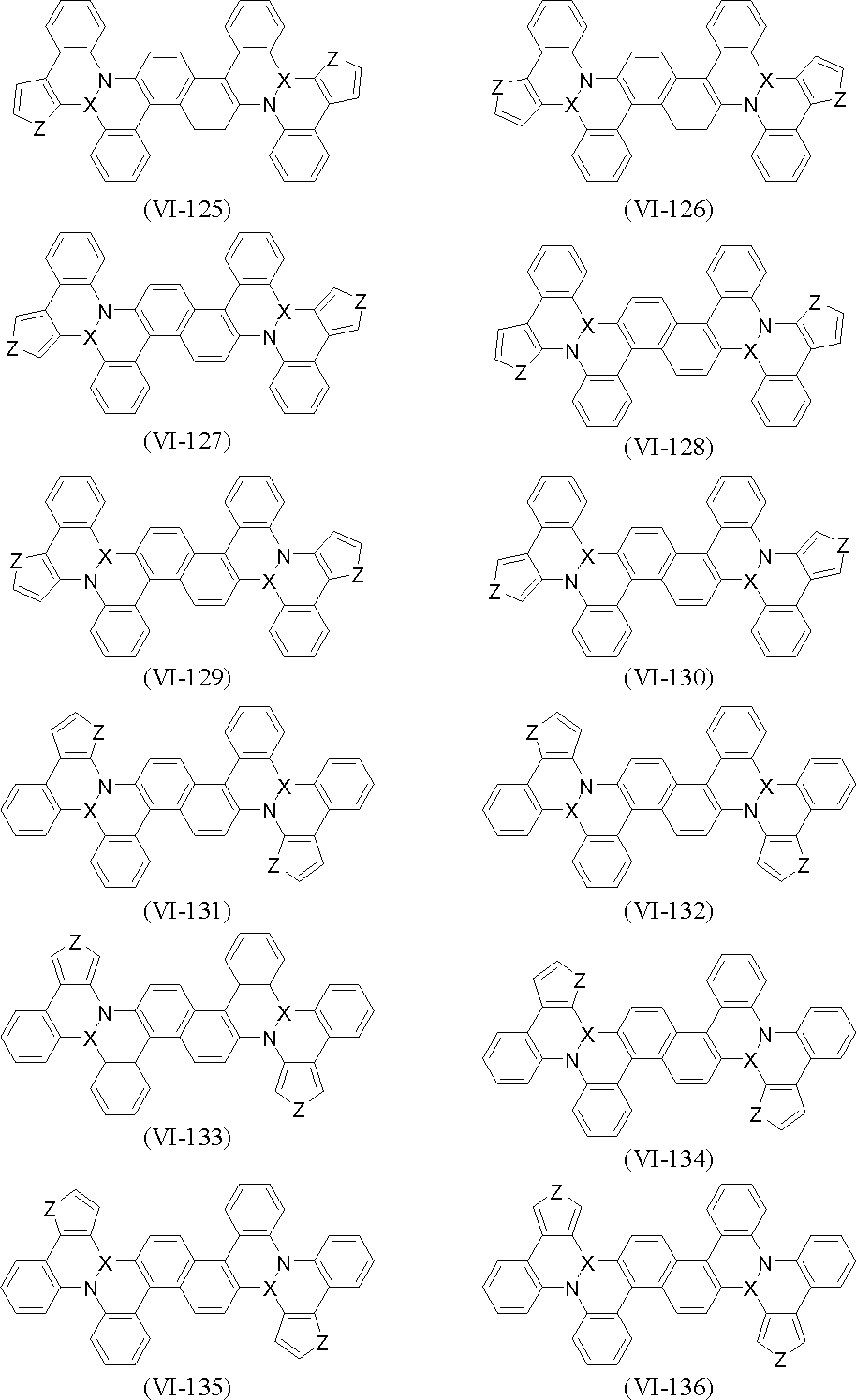


第3族:Sc、Y、ランタノイド
第4族:Ti、Zr、Hf
第5族:V、Nb、Ta
第6族:Cr、Mo、W
第7族:Mn、Tc、Re
第8族:Fe、Ru、Os
第9族:Co、Rh、Ir
第10族:Ni、Pd、Pt
第11族:Cu、Ag、Au
第13族:Al、Ga、In、Tl
第14族:Si、Ge、Sn、Pb
(下記式中、(R)はR1が上記に定義されるRであることを示し、(R1)は、R1が中性の配位子であることを示す。)


本明細書において「部分構造は少なくとも1つの水素を有する」とは、上記一般式(I)を用いて説明すると、環A、環B、環Cおよび環Dを形成する原子の全てが他の構造と連結し得るわけではなく、少なくとも1つの原子は必ず水素と結合して終端していることを意味し、上記一般式(I)で表される部分構造を含む(例えば該部分構造の繰り返しからなる)多環芳香族化合物またはその塩には、例えば、フラーレンやカーボンナノチューブの炭素骨格の一部をホウ素及び窒素などで置換したヘテロフラーレンやヘテロカーボンナノチューブなどは含まれないことを意味する。

「同じ環で隣接する2つのYとそれらの間の結合とが一緒になって、NR、O、SまたはSeになる」も同様の意味である。


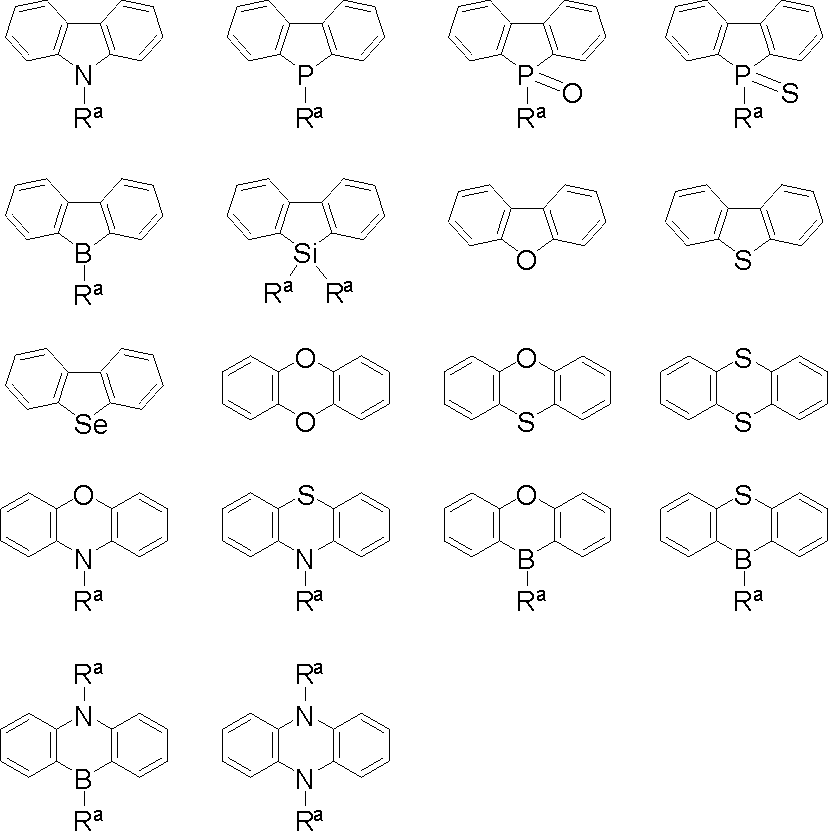


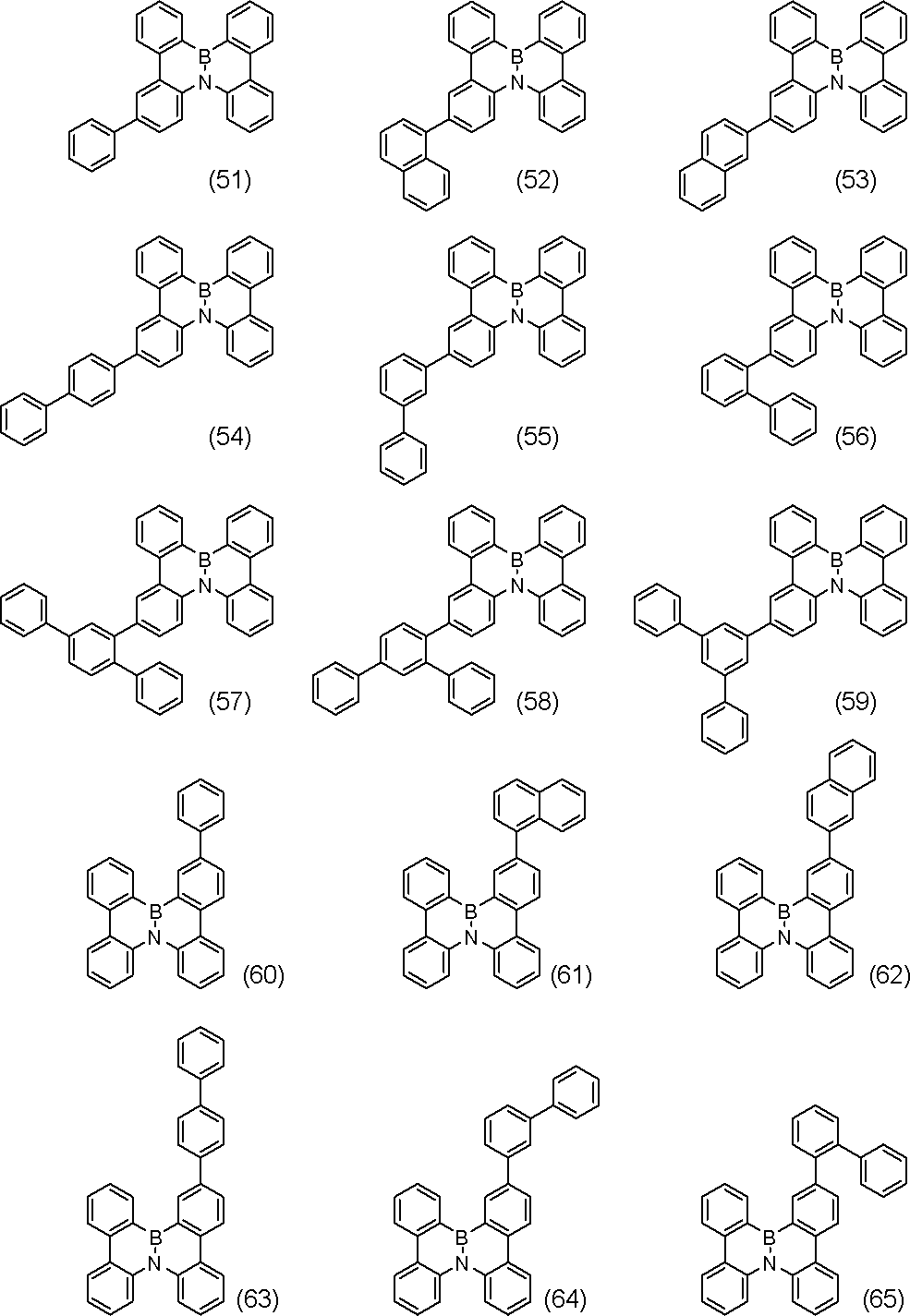



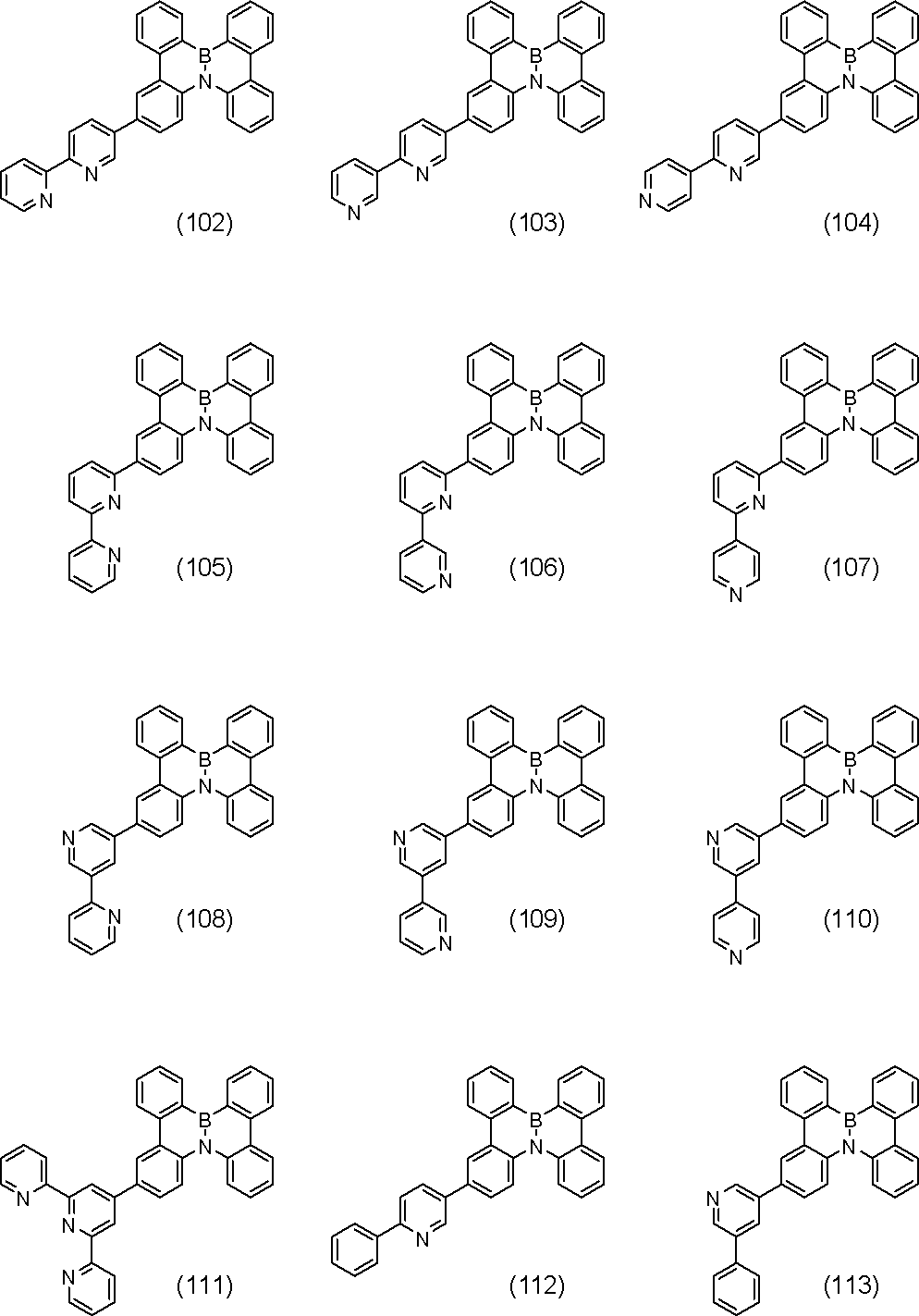

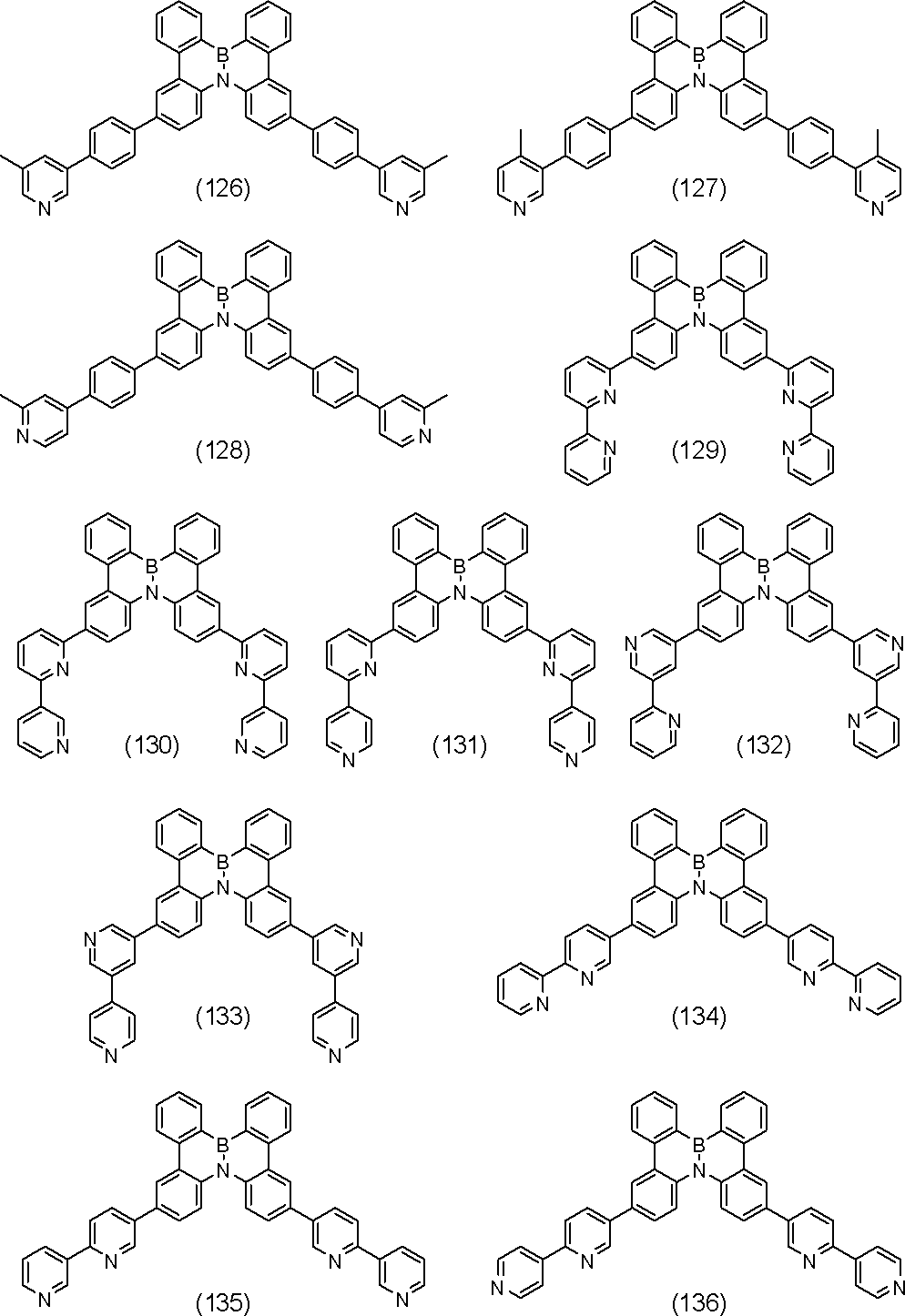



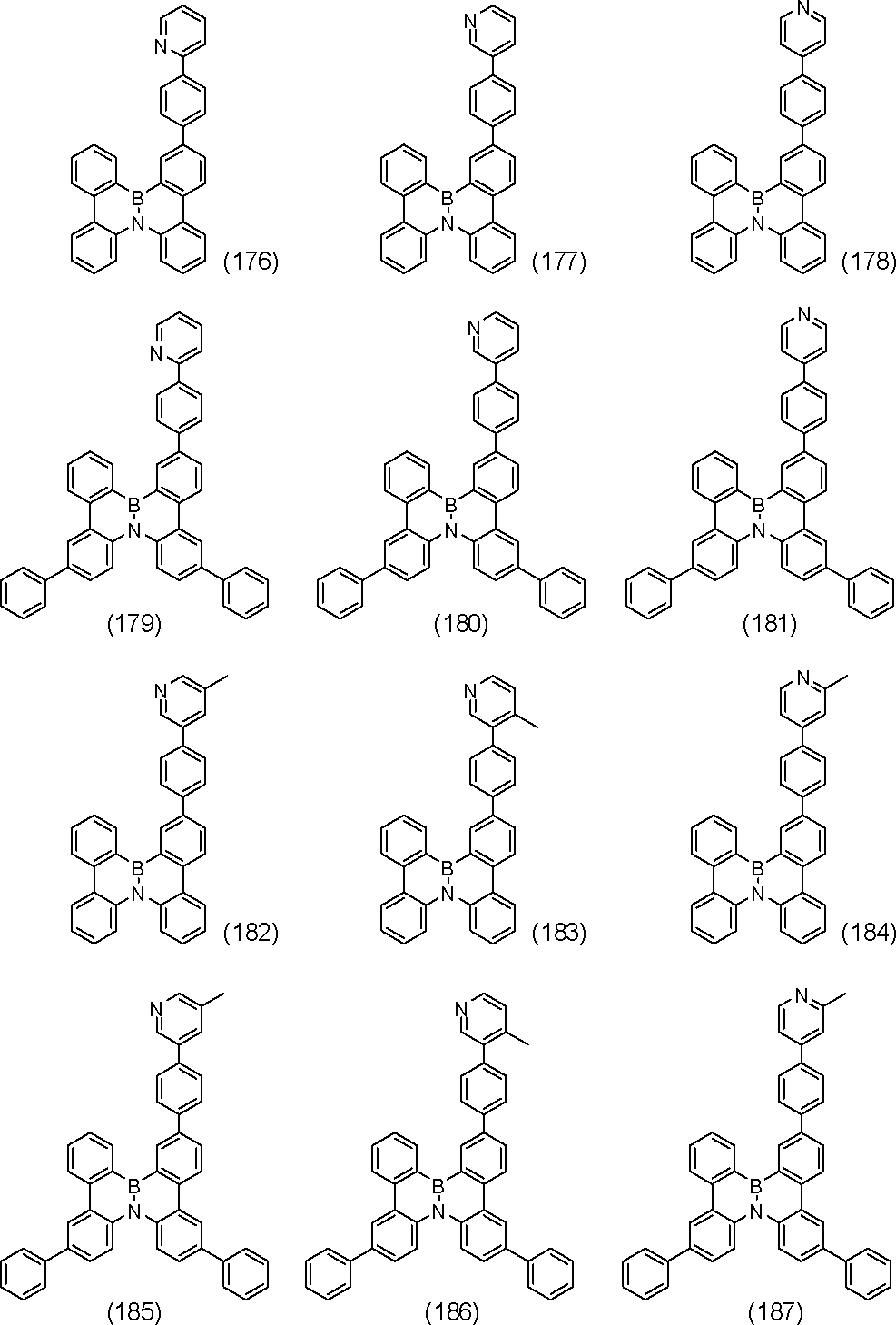






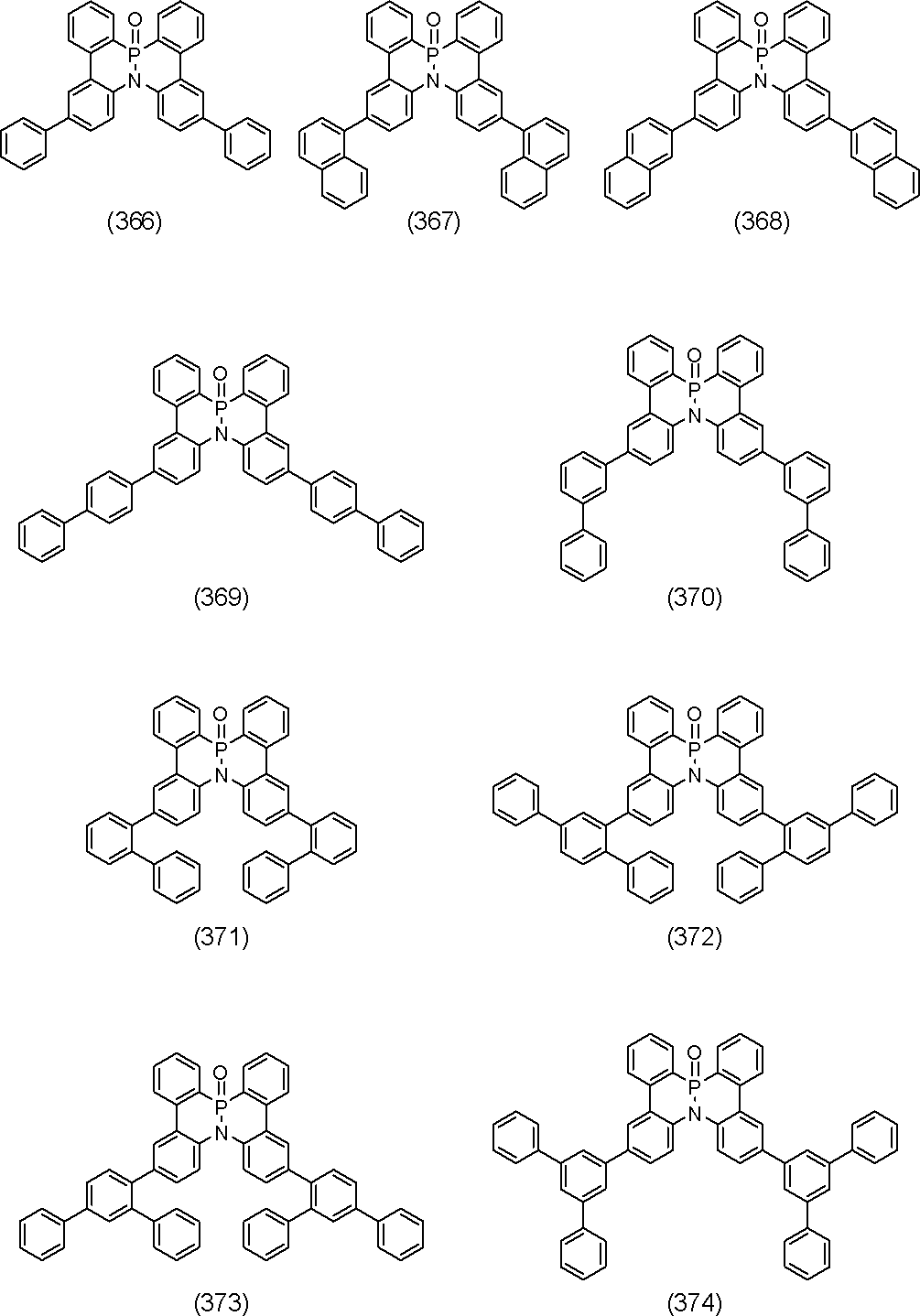




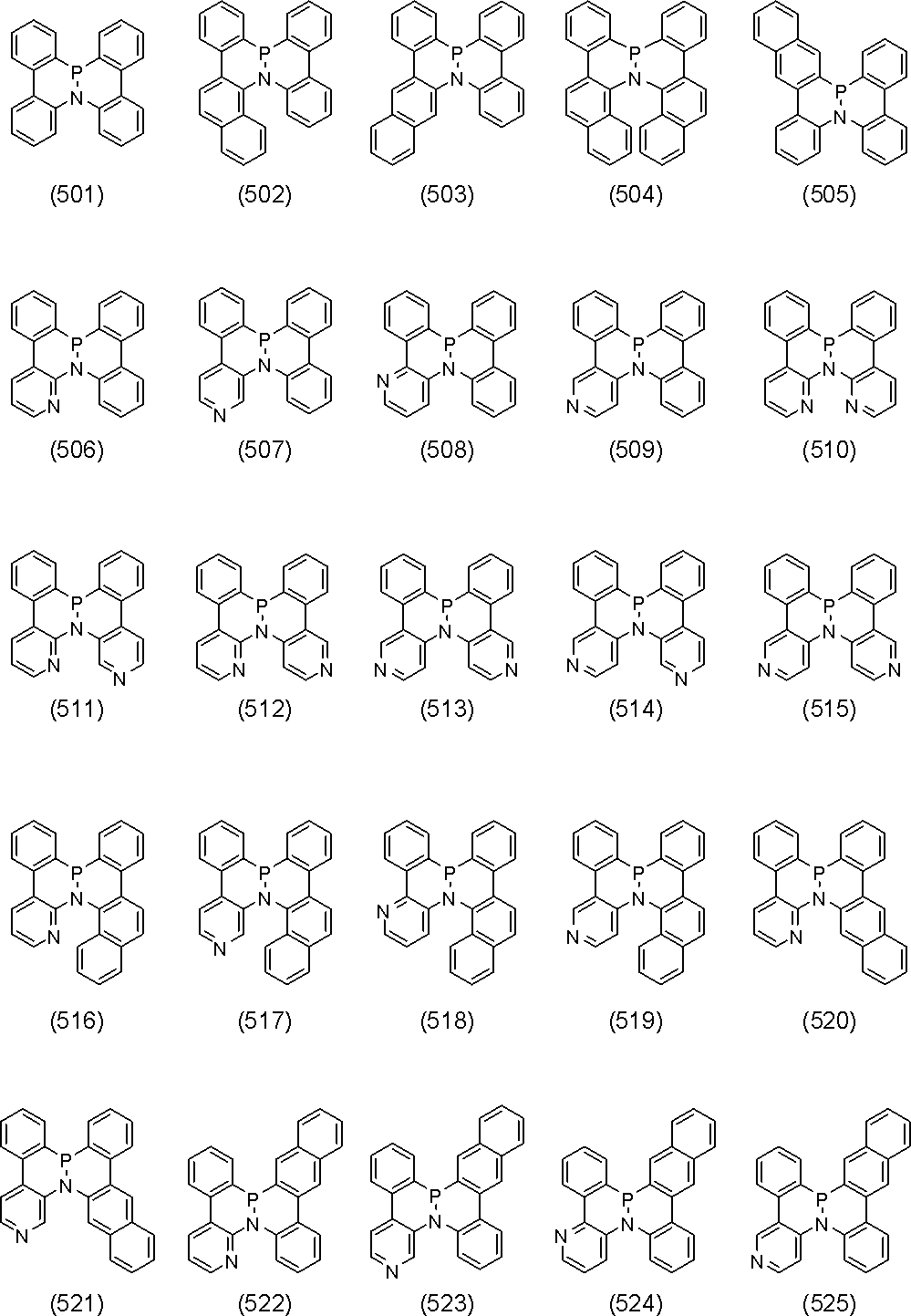







次に、本発明の化合物の製造方法について説明する。本発明の化合物は多環芳香族化合物(およびその塩)であり、上記一般式(I)で表される部分構造を有し、より具体的には上記一般式(II)または一般式(II’)で表される部分構造、さらには上記一般式(III-1)~式(III-54)および上記一般式(III-55)~式(III-60)などで表される部分構造を有する多環芳香族化合物である。全体構造としては、例えば、上記一般式(IV-1)~式(IV-22)で表される多環芳香族化合物であり、より具体的には上記一般式(V-1)~式(V-26)および上記一般式(V-27)~式(V-34)で表される多環芳香族化合物、上記一般式(V-1’)、式(V-2’)および式(V-3’)で表される多環芳香族化合物および上記一般式(V-27’)または式(V-32’)で表される多環芳香族化合物、上記一般式(VI-1)~式(VI-149)で表される多環芳香族化合物、上記式(1)~(709)で表される多環芳香族化合物である。









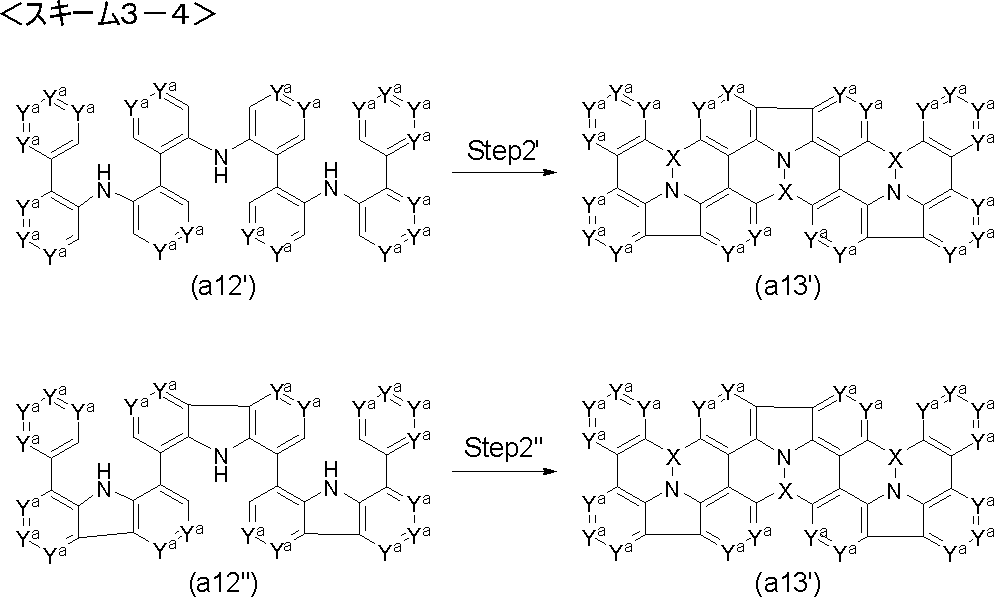

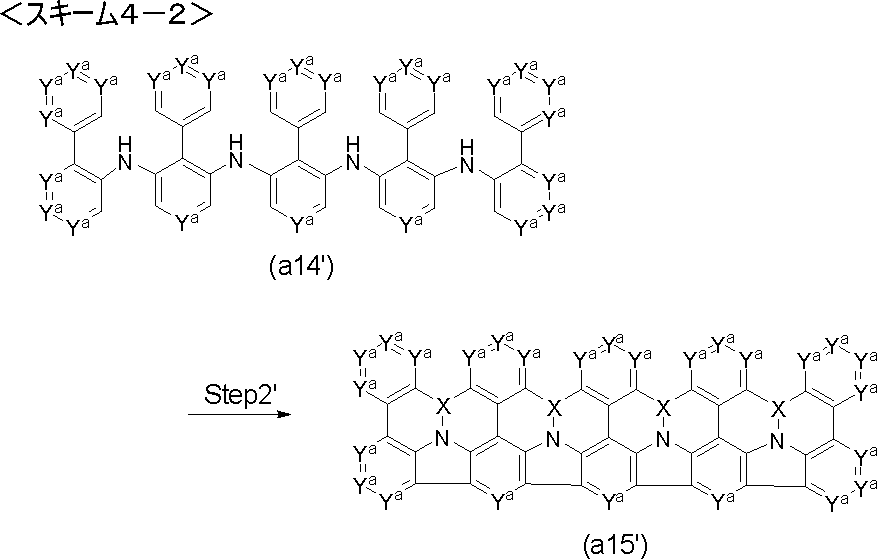




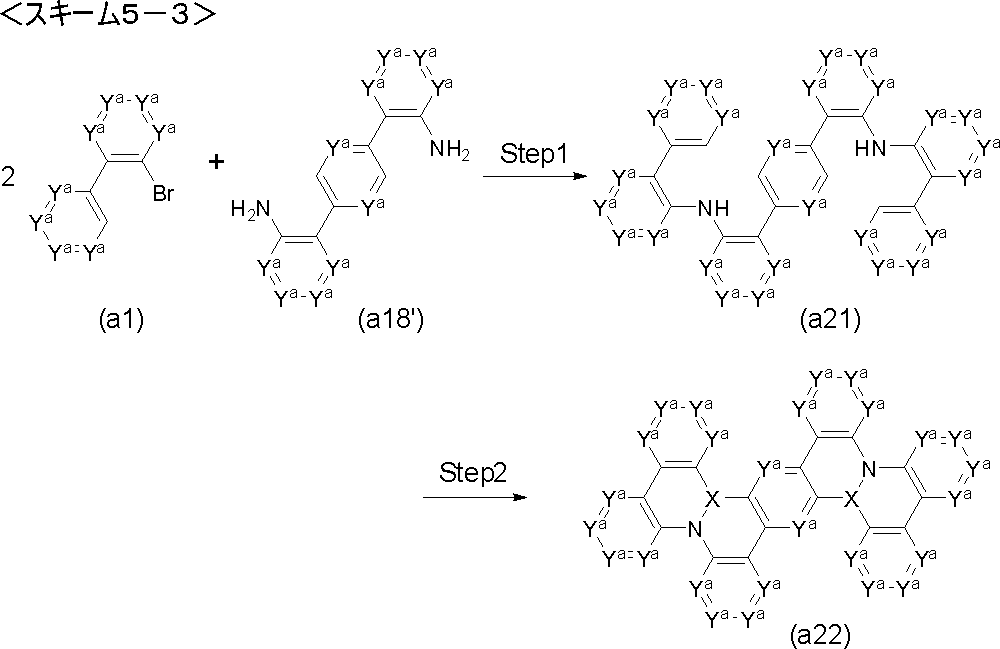




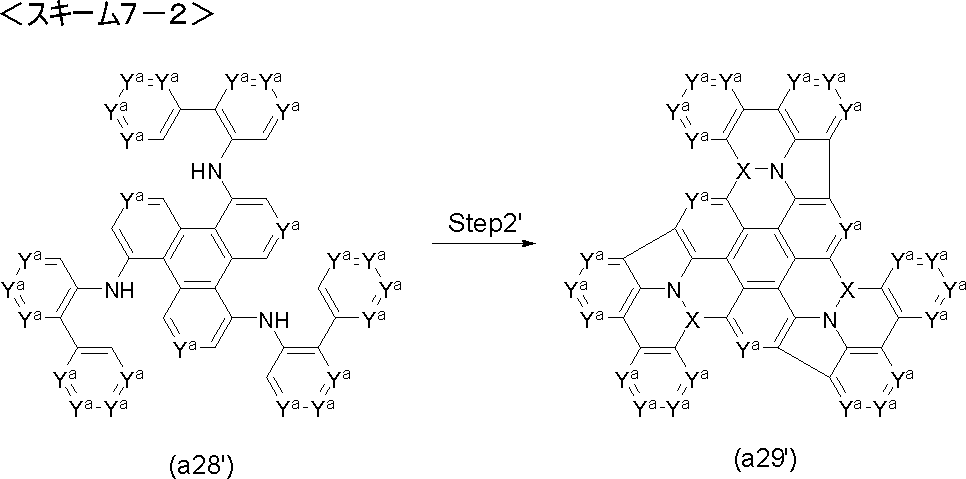

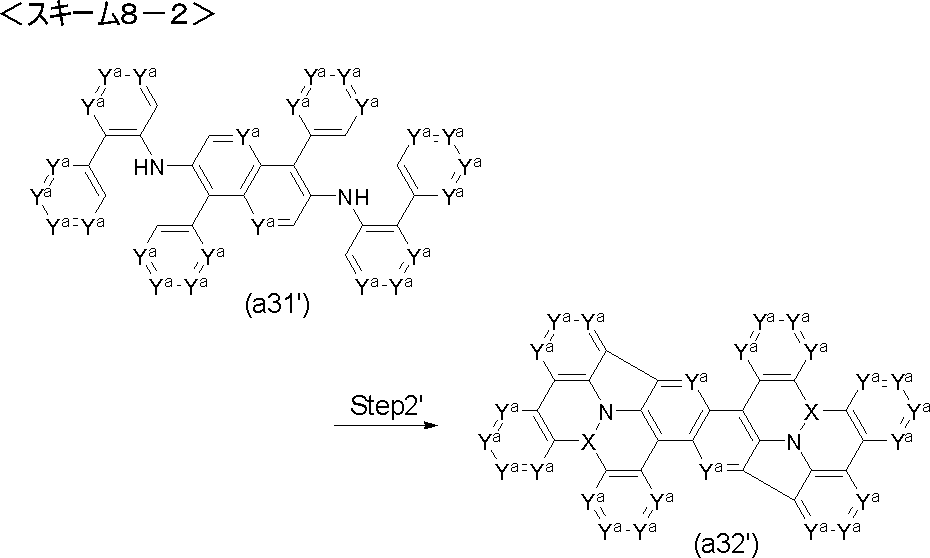



本発明に係る多環芳香族化合物は、例えば、有機電界発光素子の材料として用いることができる。以下に、本実施形態に係る有機電界発光素子について図面に基づいて詳細に説明する。図1は、本実施形態に係る有機電界発光素子を示す概略断面図である。
図1に示された有機電界発光素子100は、基板101と、基板101上に設けられた陽極102と、陽極102の上に設けられた正孔注入層103と、正孔注入層103の上に設けられた正孔輸送層104と、正孔輸送層104の上に設けられた発光層105と、発光層105の上に設けられた電子輸送層106と、電子輸送層106の上に設けられた電子注入層107と、電子注入層107の上に設けられた陰極108とを有する。
基板101は、有機電界発光素子100の支持体となるものであり、通常、石英、ガラス、金属、プラスチックなどが用いられる。基板101は、目的に応じて板状、フィルム状、またはシート状に形成され、例えば、ガラス板、金属板、金属箔、プラスチックフィルム、プラスチックシートなどが用いられる。なかでも、ガラス板、および、ポリエステル、ポリメタクリレート、ポリカーボネート、ポリスルホンなどの透明な合成樹脂製の板が好ましい。ガラス基板であれば、ソーダライムガラスや無アルカリガラスなどが用いられ、また、厚みも機械的強度を保つのに十分な厚みがあればよいので、例えば、0.2mm以上あればよい。厚さの上限値としては、例えば、2mm以下、好ましくは1mm以下である。ガラスの材質については、ガラスからの溶出イオンが少ない方がよいので無アルカリガラスの方が好ましいが、SiO2などのバリアコートを施したソーダライムガラスも市販されているのでこれを使用することができる。また、基板101には、ガスバリア性を高めるために、少なくとも片面に緻密なシリコン酸化膜などのガスバリア膜を設けてもよく、特にガスバリア性が低い合成樹脂製の板、フィルムまたはシートを基板101として用いる場合にはガスバリア膜を設けるのが好ましい。
陽極102は、発光層105へ正孔を注入する役割を果たすものである。なお、陽極102と発光層105との間に正孔注入層103および/または正孔輸送層104が設けられている場合には、これらを介して発光層105へ正孔を注入することになる。
正孔注入層103は、陽極102から移動してくる正孔を、効率よく発光層105内または正孔輸送層104内に注入する役割を果たすものである。正孔輸送層104は、陽極102から注入された正孔または陽極102から正孔注入層103を介して注入された正孔を、効率よく発光層105に輸送する役割を果たすものである。正孔注入層103および正孔輸送層104は、それぞれ、正孔注入・輸送材料の一種または二種以上を積層、混合するか、正孔注入・輸送材料と高分子結着剤の混合物により形成される。また、正孔注入・輸送材料に塩化鉄(III)のような無機塩を添加して層を形成してもよい。
発光層105は、電界を与えられた電極間において、陽極102から注入された正孔と、陰極108から注入された電子とを再結合させることにより発光するものである。発光層105を形成する材料としては、正孔と電子との再結合によって励起されて発光する化合物(発光性化合物)であればよく、安定な薄膜形状を形成することができ、かつ、固体状態で強い発光(蛍光および/または燐光)効率を示す化合物であるのが好ましい。本実施形態に係る発光素子の発光材料は蛍光性であっても燐光性であってもどちらでもかまわない。
また、特開平11-97178号公報、特開2000-133457号公報、特開2000-26324号公報、特開2001-267079号公報、特開2001-267078号公報、特開2001-267076号公報、特開2000-34234号公報、特開2001-267075号公報、および特開2001-217077号公報などに記載されたペリレン誘導体を用いてもよい。
また、国際公開第2000/40586号などに記載されたボラン誘導体を用いてもよい。
また、特開2006-156888号公報などに記載された芳香族アミン誘導体を用いてもよい。
また、特開2004-43646号公報、特開2001-76876号公報、および特開平6-298758号公報などに記載されたクマリン誘導体を用いてもよい。

また、特開2005-126399号公報、特開2005-097283号公報、特開2002-234892号公報、特開2001-220577号公報、特開2001-081090号公報、および特開2001-052869号公報などに記載されたピラン誘導体を用いてもよい。

また、特開2006-089398号公報、特開2006-080419号公報、特開2005-298483号公報、特開2005-097263号公報、および特開2004-111379号公報などに記載されたイリジウム錯体を用いてもよい。
また、特開2006-190718号公報、特開2006-128634号公報、特開2006-093542号公報、特開2004-335122号公報、および特開2004-331508号公報などに記載された白金錯体を用いてもよい。
電子注入層107は、陰極108から移動してくる電子を、効率よく発光層105内または電子輸送層106内に注入する役割を果たすものである。電子輸送層106は、陰極108から注入された電子または陰極108から電子注入層107を介して注入された電子を、効率よく発光層105に輸送する役割を果たすものである。電子輸送層106および電子注入層107は、それぞれ、電子輸送・注入材料の一種または二種以上を積層、混合するか、電子輸送・注入材料と高分子結着剤の混合物により形成される。
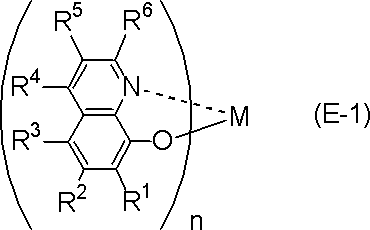
式中、R1~R6は水素または置換基であり、MはLi、Al、Ga、BeまたはZnであり、nは1~3の整数である。

式中、Gは単なる結合手またはn価の連結基を表し、nは2~8の整数である。また、ピリジン-ピリジンまたはピリジン-Gの結合に用いられない炭素原子は置換されていてもよい。

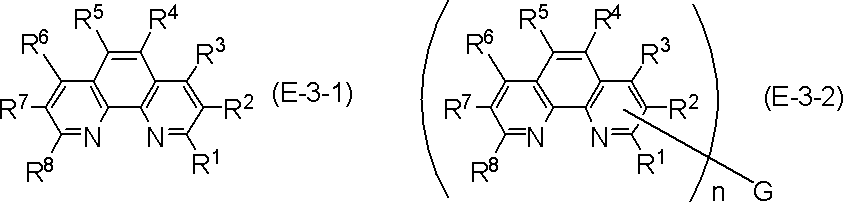
式中、R1~R8は水素または置換基であり、隣接する基は互いに結合して縮合環を形成してもよく、Gは単なる結合手またはn価の連結基を表し、nは2~8の整数である。また、一般式(E-3-2)のGとしては、例えば、ビピリジン誘導体の欄で説明したものと同じものがあげられる。

式中、R11およびR12は、それぞれ独立して、水素、アルキル、置換されていてもよいアリール、置換シリル、置換されていてもよい窒素含有複素環、またはシアノの少なくとも一つであり、R13~R16は、それぞれ独立して、置換されていてもよいアルキル、または置換されていてもよいアリールであり、Xは、置換されていてもよいアリーレンであり、Yは、置換されていてもよい炭素数16以下のアリール、置換ボリル、または置換されていてもよいカルバゾリルであり、そして、nはそれぞれ独立して0~3の整数である。

式中、R11およびR12は、それぞれ独立して、水素、アルキル、置換されていてもよいアリール、置換シリル、置換されていてもよい窒素含有複素環、またはシアノの少なくとも一つであり、R13~R16は、それぞれ独立して、置換されていてもよいアルキル、または置換されていてもよいアリールであり、R21およびR22は、それぞれ独立して、水素、アルキル、置換されていてもよいアリール、置換シリル、置換されていてもよい窒素含有複素環、またはシアノの少なくとも一つであり、X1は、置換されていてもよい炭素数20以下のアリーレンであり、nはそれぞれ独立して0~3の整数であり、そして、mはそれぞれ独立して0~4の整数である。
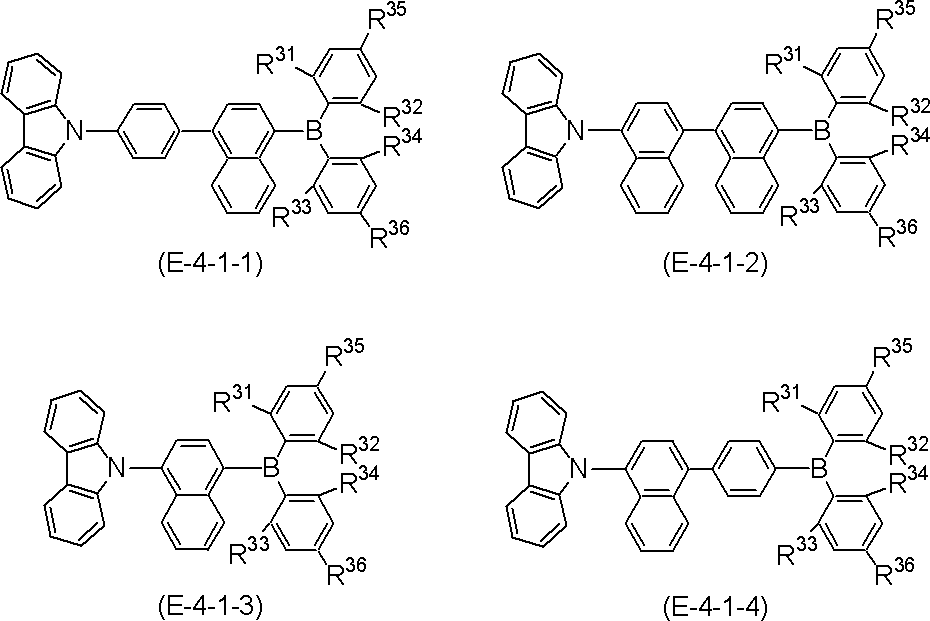
各式中、R31~R34は、それぞれ独立して、メチル、イソプロピルまたはフェニルのいずれかであり、そして、R35およびR36は、それぞれ独立して、水素、メチル、イソプロピルまたはフェニルのいずれかである。

式中、R11およびR12は、それぞれ独立して、水素、アルキル、置換されていてもよいアリール、置換シリル、置換されていてもよい窒素含有複素環、またはシアノの少なくとも一つであり、R13~R16は、それぞれ独立して、置換されていてもよいアルキル、または置換されていてもよいアリールであり、X1は、置換されていてもよい炭素数20以下のアリーレンであり、そして、nはそれぞれ独立して0~3の整数である。
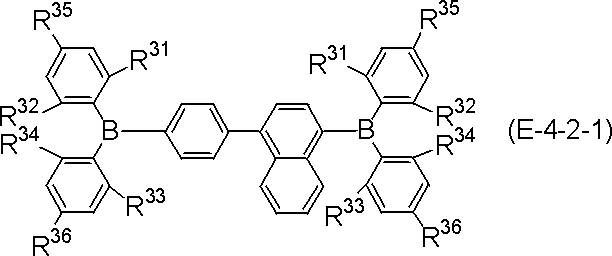
式中、R31~R34は、それぞれ独立して、メチル、イソプロピルまたはフェニルのいずれかであり、そして、R35およびR36は、それぞれ独立して、水素、メチル、イソプロピルまたはフェニルのいずれかである。
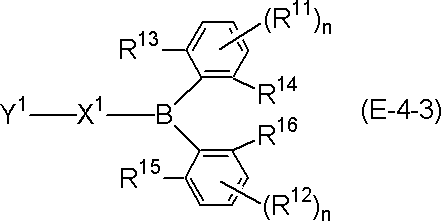
式中、R11およびR12は、それぞれ独立して、水素、アルキル、置換されていてもよいアリール、置換シリル、置換されていてもよい窒素含有複素環、またはシアノの少なくとも一つであり、R13~R16は、それぞれ独立して、置換されていてもよいアルキル、または置換されていてもよいアリールであり、X1は、置換されていてもよい炭素数10以下のアリーレンであり、Y1は、置換されていてもよい炭素数14以下のアリールであり、そして、nはそれぞれ独立して0~3の整数である。
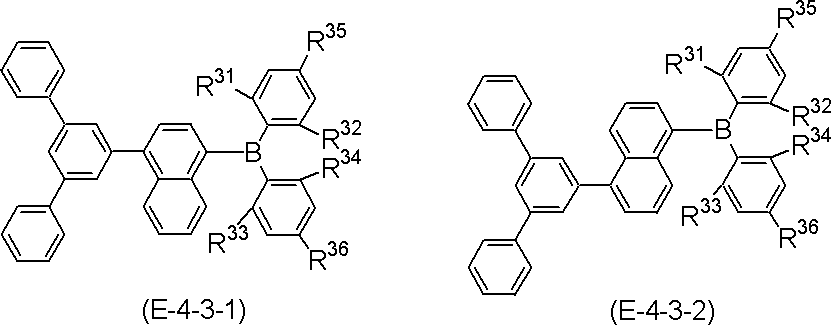
各式中、R31~R34は、それぞれ独立して、メチル、イソプロピルまたはフェニルのいずれかであり、そして、R35およびR36は、それぞれ独立して、水素、メチル、イソプロピルまたはフェニルのいずれかである。

式中、Ar1~Ar3はそれぞれ独立に水素または置換されてもよい炭素数6~30のアリールである。特に、Ar1が置換されてもよいアントリルであるベンゾイミダゾール誘導体が好ましい。
陰極108は、電子注入層107および電子輸送層106を介して、発光層105に電子を注入する役割を果たすものである。
以上の正孔注入層、正孔輸送層、発光層、電子輸送層および電子注入層に用いられる材料は単独で各層を形成することができるが、高分子結着剤としてポリ塩化ビニル、ポリカーボネート、ポリスチレン、ポリ(N-ビニルカルバゾール)、ポリメチルメタクリレート、ポリブチルメタクリレート、ポリエステル、ポリスルホン、ポリフェニレンオキサイド、ポリブタジエン、炭化水素樹脂、ケトン樹脂、フェノキシ樹脂、ポリアミド、エチルセルロース、酢酸ビニル樹脂、ABS樹脂、ポリウレタン樹脂などの溶剤可溶性樹脂や、フェノール樹脂、キシレン樹脂、石油樹脂、ユリア樹脂、メラミン樹脂、不飽和ポリエステル樹脂、アルキド樹脂、エポキシ樹脂、シリコーン樹脂などの硬化性樹脂などに分散させて用いることも可能である。
有機電界発光素子を構成する各層は、各層を構成すべき材料を蒸着法、抵抗加熱蒸着、電子ビーム蒸着、スパッタリング、分子積層法、印刷法、スピンコート法またはキャスト法、コーティング法などの方法で薄膜とすることにより、形成することができる。このようにして形成された各層の膜厚については特に限定はなく、材料の性質に応じて適宜設定することができるが、通常2nm~5000nmの範囲である。膜厚は通常、水晶発振式膜厚測定装置などで測定できる。蒸着法を用いて薄膜化する場合、その蒸着条件は、材料の種類、膜の目的とする結晶構造および会合構造などにより異なる。蒸着条件は一般的に、ボート加熱温度50~400℃、真空度10-6~10-3Pa、蒸着速度0.01~50nm/秒、基板温度-150~+300℃、膜厚2nm~5μmの範囲で適宜設定することが好ましい。
また、本発明は、有機電界発光素子を備えた表示装置または有機電界発光素子を備えた照明装置などにも応用することができる。
有機電界発光素子を備えた表示装置または照明装置は、本実施形態にかかる有機電界発光素子と公知の駆動装置とを接続するなど公知の方法によって製造することができ、直流駆動、パルス駆動、交流駆動など公知の駆動方法を適宜用いて駆動することができる。
4b-アザ-12b-チオホスファジベンゾ[g,p]クリセンの合成

13C NMR (δppm in CDCl3) 117.0, 120.8, 127.2, 128.1, 128.7, 129.0, 130.6, 132.0, 138.9, 140.1.

1H NMR (δppm in CD2Cl2at -40 ℃); 6.65(d, 1H, J = 8.4 Hz), 7.01(t, 1H, J = 7.2 Hz), 7.09(t, 1H, J = 7.8 Hz), 7.19 (dd, 1H, J = 7.8, 13.8 Hz), 7.31 (td, 1H, J = 3.0, 7.8 Hz), 7.54 (t, 1H, J = 7.8 Hz), 7.62 (d, 1H, J = 7.2 Hz), 7.65-7.69 (m, 2H), 7.75 (td, 1H, J = 3.0, 7.8 Hz), 7.84-7.91 (m, 3H), 8.05 (d, 2H, J = 7.2 Hz), 8.09 (t, 1H, J = 7.2 Hz), 8.58 (dd, 1H, J = 7.8, 15.6 Hz)
13C NMR (δppm in CD2Cl2at -40 ℃); 118.1, 120.8, 121.2, 122.3, 124.4, 126.5, 128.1, 128.5, 128.6, 128.7, 128.9, 129.3, 130.2 (2C), 131.6, 132.1, 132.8, 132.9, 134.4, 134.5, 135.2, 135.3, 136.2, 141.5
4b-アザ-12b-フェニル-12b-シラジベンゾ[g,p]クリセンの合成

また式(601)で表される化合物はヘキサンから再結晶することで無色針状結晶が得られ、X線結晶構造解析により構造を決定した。
X-ray crystal structure
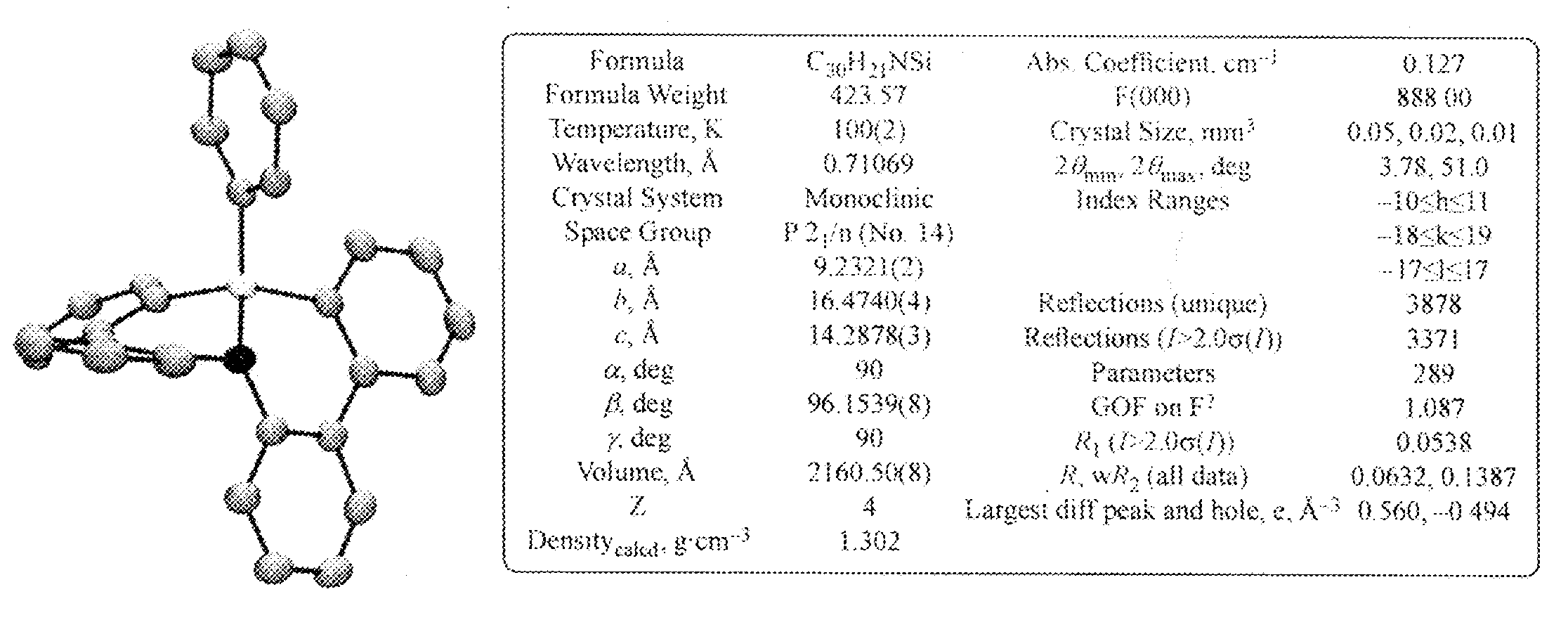
4b-アザ-12b-ゲルマ-12b-フェニルジベンゾ[g,p]クリセンの合成

また標題化合物はヘキサンから再結晶することで無色柱状結晶が得られ、X線結晶構造解析により構造を決定した。
X-ray crystal structure
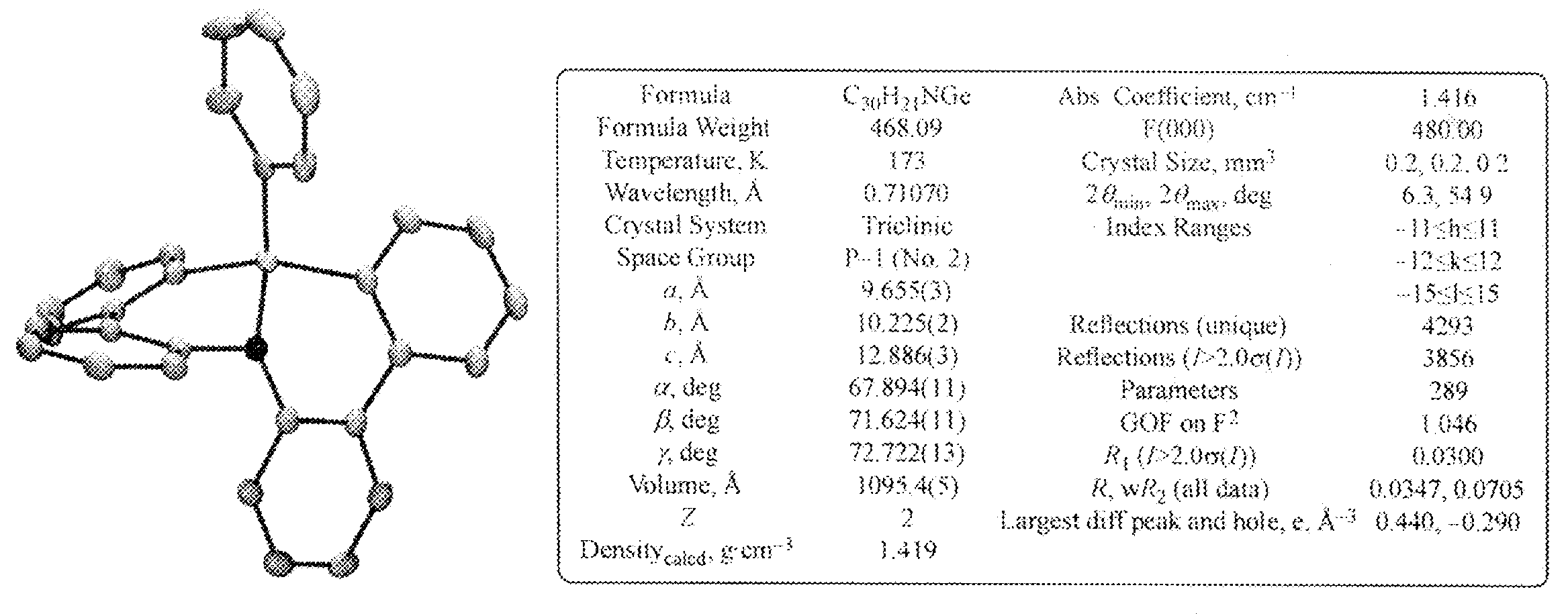
4b-アザ-12b-ボラジベンゾ[g,p]クリセンの合成


2,7-ジブロモ-4b-アザ-12b-ボラジベンゾ[g,p]クリセンの合成

2,7-ジメチル-4b-アザ-12b-ボラジベンゾ[g,p]クリセンの合成

11B NMR (δppm in C6D6) 34.0.
14b1-アザ-14b-ボラベンゾ[p]インデノ[1,2,3,4-defg]クリセンの合成
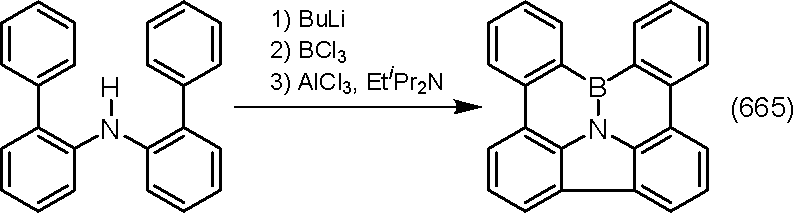
1H NMR (δppm in CDCl3); 7.66-7.72 (m, 4H), 7.84 (td, 2H, J = 1.4, 8.2 Hz), 8.21 (d, 2H, J = 7.8 Hz), 8.43 (d, 2H, J = 7.8 Hz), 8.67 (d, 2H, J = 7.8 Hz), 9.18 (d, 2H, J = 7.8 Hz).
6,9-ジクロロ-14b1-アザ-14b-ボラベンゾ[p]インデノ[1,2,3,4-defg]クリセンの合成
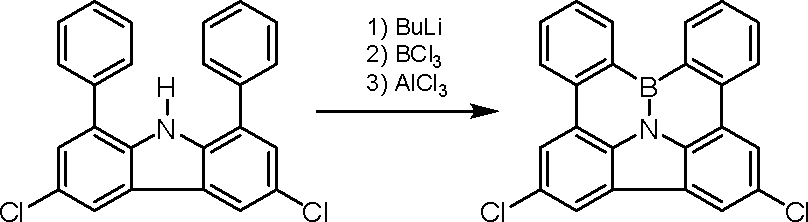
4b-アザ-12b-ホスファジベンゾ[g,p]クリセンの合成

31P NMR (δppm in C6D6) 12.7.
4b-アザ-12b-オキサホスファ-ジベンゾ[g,p]クリセンの合成

31P NMR (δppm in C6D6) 6.6.
8b,19b-ジアザ-11b,22b-ジチオホスファヘキサベンゾ[a,c,fg,j,l,op]テトラセンの合成
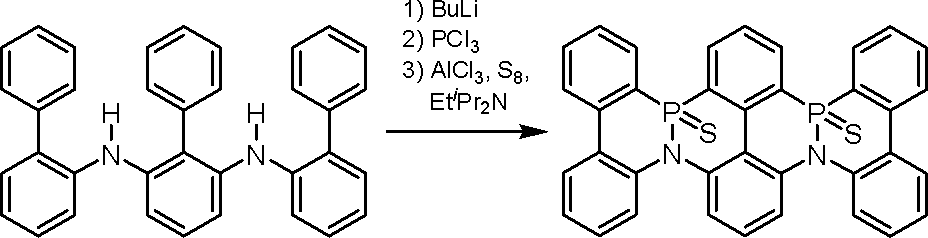
8b,19b-ジアザ-11b,22b-ジボラヘキサベンゾ[a,c,fg,j,l,op]テトラセンの合成

1H NMR (δppm in CS2/CD2Cl2=2/1, 600 MHz) 7.31-7.34 (m, 4H, NCCCHCH), 7.55 (t, J = 8.4 Hz, 1H, NCCHCHCHCN), 7.61 (td, J = 1.2, 7.2 Hz, 2H, BCCHCHCHCH), 7.78 (td, J = 1.2, 7.2 Hz, 2H, BCCHCHCHCH), 7.91 (t, J = 7.2 Hz, 1H, BCCHCHCHCB), 8.05 (d, J = 8.4 Hz, 2H, NCCHCHCHCN), 8.11-8.13 (m, 2H, NCCHCHCHCH), 8.32-8.35 (m, 2H, NCCCH), 8.40 (d, J = 7.2 Hz, 2H, BCCCH), 8.71 (d, J = 7.2 Hz, 2H, BCCHCHCHCH), 8.96 (d, J = 7.2 Hz, 2H, BCCHCHCHCB)
13C NMR (δppm in CS2/CD2Cl2=2/1, 151 MHz) 114.3 (2C), 119.2, 121.8 (2C), 123.1 (2C), 123.4 (2C), 125.7, 125.8 (2C), 126.2, 126.7 (2C), 127.1 (2C), 128.1 (2C), 130.5 (br, 2C, CBCCCBC), 131.4 (2C), 133.0 (br, 2C, CBCCCBC), 135.8 (2C), 137.5 (4C), 137.6 (2C), 138.9, 139.0 (2C)
11B NMR (δppm in CS2/CD2Cl2=2/1, 193 MHz) 36.5.
4b,17b-ジアザ-9b,22b-ジボラテトラベンゾ[a,c,f,m]フェナントロ[9,10-k]テトラフェンの合成

11B NMR (δppm in CS2/C6D6=2/1, 126 MHz) 35.7.
11b-アザ-3b-ボラベンゾ[11,12]クリセノ[6,5-b]チオフェンの合成

1H NMR (δppm in C6D6, 392 MHz) 6.92-7.11 (m, 5H), 7.39 (td, J = 0.9, 7.6 Hz, 1H), 7.50 (td, J = 1.8, 7.2 Hz, 1H), 7.91-8.00 (m, 4H), 8.11-8.16 (m, 2H), 8.63 (dd, J = 0.9, 7.6 Hz, 1H).
11b-アザ-3b-ボラベンゾ[11,12]クリセノ[5,6-b]チオフェンの合成

1H NMR (δppm in C6D6, 392 MHz) 7.00-7.05 (m, 2H), 7.07-7.12 (m, 2H), 7.40-7.50 (m, 3H), 7.63 (d, J = 4.9 Hz, 1H), 7.94 (dd, J = 1.8, 8.1 Hz, 1H), 8.03 (dd, J = 1.3, 8.5 Hz, 1H), 8.08-8.15 (m, 3H), 8.95 (dd, J = 1.4, 7.6 Hz, 1H).
1-メチル-11b-アザ-3b-ボラベンゾ[11,12]クリセノ[5,6-c]チオフェンの合成
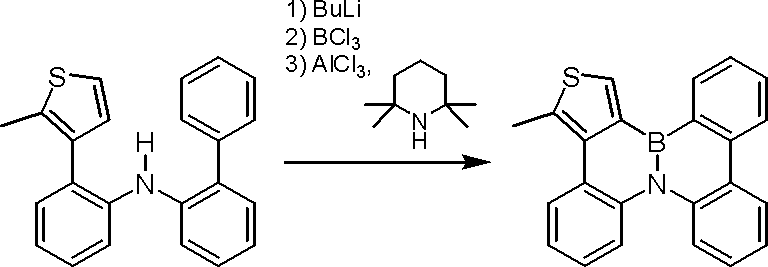
HRMS(MALDI) m/z; calcd. 349.1091[M]+; found 349.1088.
11B NMR (δppm in C6D6, 126 MHz) 32.5.
3b-アザ-11b-ボラベンゾ[11,12]クリセノ[6,5-b]チオフェンの合成

11B NMR (δppm in C6D6, 126 MHz) 34.5.
12b-アザ-4b-ボラジベンゾ[l,k]ピロロ[1,2-f]フェナントリジンの合成

1H NMR (δppm in C6D6, 392 MHz) 6.72-6.73 (m, 1H), 6.76-6.80 (m, 1H), 6.85-6.89 (m, 1H), 7.01-7.09 (m, 2H), 7.28 (dd, J = 1.3, 8.5 Hz, 1H), 7.35-7.39 (m, 1H), 7.46-7.51 (m, 2H), 7.55 (dd, J = 1.3, 3.6 Hz, 1H), 7.80 (dd, J = 1.3, 8.5 Hz, 1H), 7.86-7.89 (m, 1H), 8.09-8.13 (m, 2H), 8.71 (dd, J = 1.3, 7.6 Hz, 1H).
4b-アザ-12b-ボラベンゾ[f]フェナントロ[9,10-h]キノリンの合成

4b-アザ-12b-フェニル-12b-スタンナジベンゾ[g,p]クリセンの合成

6c-アザ-16b-ボラジベンゾ[c,p]ナフト[1,2-g]クリセンの合成

1H NMR (δppm in CDCl3); 6.65-6.69 (m, 2H), 7.11 (t, 2H, J = 7.4 Hz), 7.16 (d, 2H, J = 8.9 Hz), 7.64-7.70 (m, 4H), 7.79 (d, 2H, J = 8.9 Hz), 7.86 (dd, 2H, J = 0.9, 7.6 Hz), 8.48 (d, 2H, J = 8.9 Hz), 8.60 (d, 2H, J = 8.1 Hz), 8.84 (d, 2H, J = 7.1 Hz).
6c-アザ-14b-ボラトリベンゾ[c,g,p]クリセンの合成
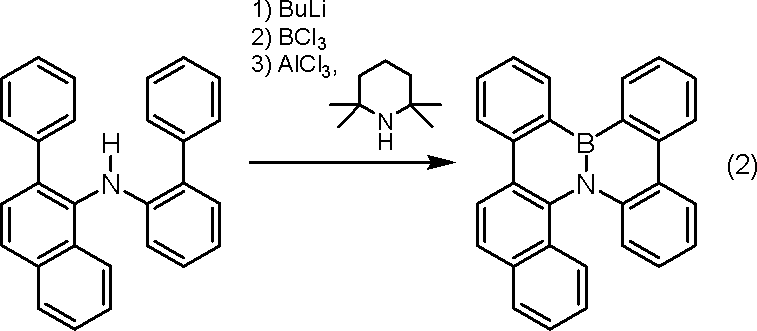
1H NMR (δppm in CDCl3); 7.04-7.06 (m, 2H), 7.10-7.15 (m, 1H), 7.18-7.22 (m, 1H), 7.37-7.41 (m, 1H), 7.58-7.62 (m, 2H), 7.63-7.67 (m, 1H), 7.77-7.89 (m, 4H), 8.30 (d, 1H, J = 7.6 Hz), 8.44 (d, 1H, J = 8.5 Hz), 8.46 (d, 1H, J = 8.0 Hz), 8.51 (d, 1H, J = 8.0 Hz), 8.74-8.77 (m, 2H).
4b-アザ-14b-ボラトリベンゾ[a,c,f]テトラフェンの合成

1H NMR (δppm in CDCl3); 7.26-7.38 (m, 4H), 7.49-7.53 (m, 1H), 7.54-7.60 (m, 1H), 7.61-7.65 (m, 1H), 7.75-7.79 (m, 1H), 7.97-8.07 (m, 4H), 8.32 (dd, 1H, J = 1.6, 7.8 Hz), 8.38-8.43 (m, 2H), 8.73 (s, 1H), 8.78 (dd, 1H, J = 1.4, 7.6 Hz), 9.10 (s, 1H).
2,11-ジブロモ-6c-アザ-16b-ボラジベンゾ[c,p]ナフト[1,2-g]クリセンの合成

1H NMR (δppm in CDCl3); 6.79 (dt, 2H, J = 1.4, 7.7 Hz), 7.19-7.24 (m, 4H), 7.70 (t, 2H, J = 6.7 Hz), 7.90 (dt, 2H, J = 1.3, 7.4 Hz), 8.12 (d, 2H, J = 8.5 Hz), 8.55 (d, 2H, J = 8.1 Hz), 8.80 (s, 2H), 8.84 (d, 2H, J = 6.7 Hz).
8b,11b,14b-トリアザ-22b,25b,28b-トリボラオクタベンゾ[a,c,fg,jk,n,p,st,wx]ヘキサセンの合成

9b,22b-ジアザ-4b,17b-ジボラテトラベンゾ[a,c,f,m]フェナントロ[9,10-k]テトラフェンの合成

2,7-ジフェニル-4b-アザ-12b-ボラジベンゾ[g,p]クリセンの合成

1H-NMR(CDCl3):δ=8.73(d,2H)、8.60(m,2H)、8.51(d,2H)、8.23(d,2H)、7.82(t,2H)、7.75(d,4H)、7.64(m,4H)、7.51(t,4H)、7.40(t,2H).
N2,N2,N7,N7-テトラフェニル-4b-アザ-12b-ボラジベンゾ[g,p]クリセン-2,7-ジアミンの合成

1H-NMR(CDCl3):δ=8.67(d,2H)、8.12(m,4H)、8.03(d,2H)、7.67(t,2H)、7.57(t,2H)、7.26(m,8H)、7.16(m,8H)、7.13(dd,2H)、7.02(t,4H).
2,7-ジカルバゾリル-4b-アザ-12b-ボラジベンゾ[g,p]クリセンの合成

1H-NMR(CDCl3):δ=8.80(d,2H)、8.60(m,2H)、8.48(d,2H)、8.37(d,2H)、8.20(d,4H)、7.81(t,2H)、7.70(t,2H)、7.65(dd,2H)、7.40-7.60(m,8H)、7.33(t,4H).
2,7,11,14-テトラフェニル-4b-アザ-12b-ボラジベンゾ[g,p]クリセンの合成
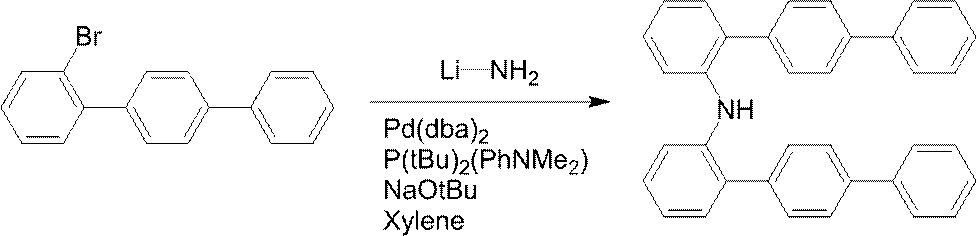

1H-NMR(CDCl3):δ=9.00(m,2H)、8.50(d,2H)、8.41(d,2H)、8.15(d,2H)、8.04(d,2H)、7.77(d,4H)、7.50(t,4H)、7.40(m,6H).
2,7-ジブロモ-11,14-ジフェニル-4b-アザ-12b-ボラジベンゾ[g,p]クリセンの合成


1H-NMR(CDCl3):δ=9.03(s,2H)、8.64(s,2H)、8.59(d,2H)、8.26(d,2H)、8.06(d,2H)、7.79(m,8H)、7.67(d,2H)、7.52(m,8H)、7.40(m,4H).
10,15-ジフェニル-4b-アザ-12b-ボラジベンゾ[g,p]クリセンの合成
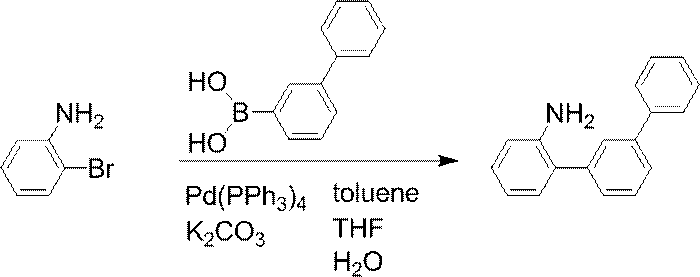
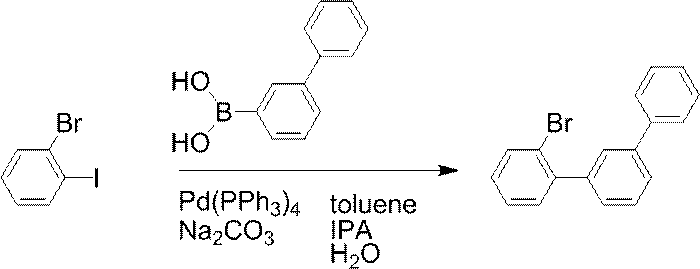


1H-NMR(CDCl3):δ=8.80(d,2H)、8.64(m,2H)、8.47(d,2H)、8.16(d,2H)、7.87(d,2H)、7.82(d,4H)、7.55(t,4H)、7.34-7.50(m,6H).
2,7,10,15-テトラフェニル-4b-アザ-12b-ボラジベンゾ[g,p]クリセンの合成


1H-NMR(CDCl3):δ=8.82(d,2H)、8.69(m,2H)、8.65(m,2H)、8.25(d,2H)、7.88(dd,2H)、7.82(d,4H)、7.76(d,4H)、7.66(dd,2H)、7.49-7.59(m,8H)、7.46(t,2H)、7.40(t,2H).
9,16-ジフェニル-4b-アザ-12b-ボラジベンゾ[g,p]クリセン化合物の合成

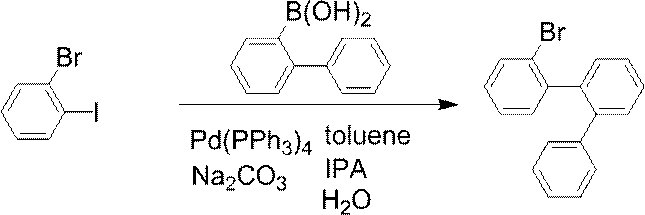
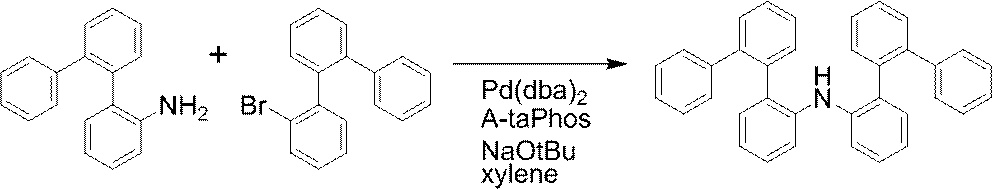

1H-NMR(CDCl3):δ=8.74(d,2H)、8.70(m,1H)、8.06(d,2H)、7.26-7.70(m,16H)、7.21(t,2H)、6.77(t,2H).
2-フェニル-4b-アザ-12b-ボラジベンゾ[g,p]クリセン化合物の合成
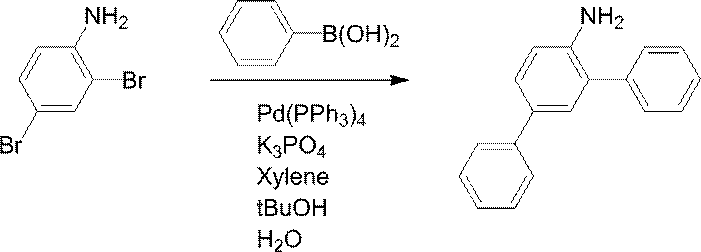

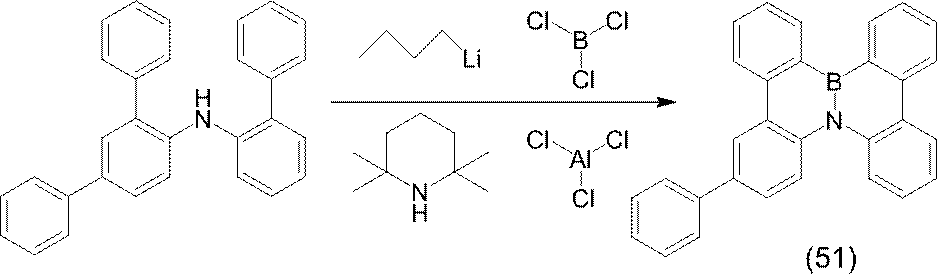
1H-NMR(CDCl3):δ=8.71(m,2H)、8.58(m,1H)、8.50(d,1H)、8.43(d,1H)、8.38(d,1H)、8.19(d,1H)、8.16(d,1H)、7.80(m,2H)、7.74(d,2H)、7.63(m,3H)、7.50(t,2H)、7.33-7.43(m,3H).
9-(4-(7-フェニル-4b-アザ-12b-ボラジベンゾ[g,p]クリセン-2-イル)フェニル)-9H-カルバゾールの合成
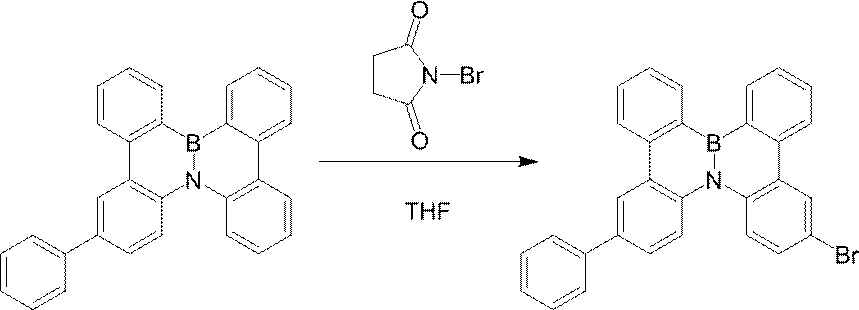
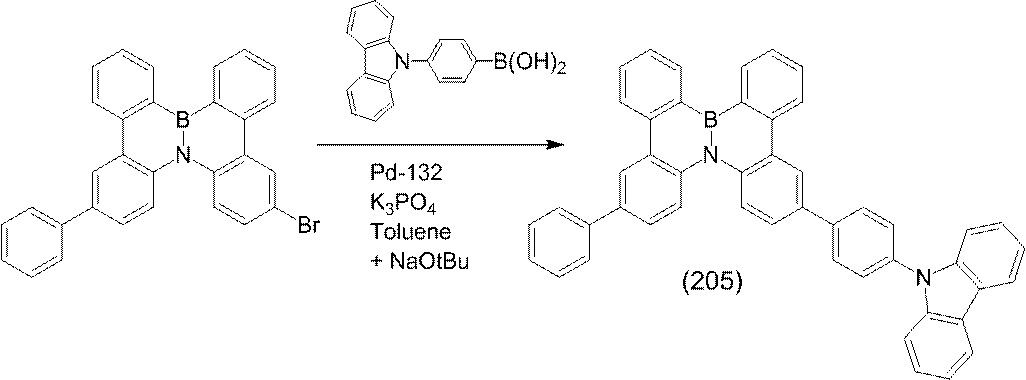
1H-NMR(CDCl3):δ=8.76(m,2H)、8.70(m,1H)、8.62(m,1H)、8.58(d,1H)、8.54(d,1H)、8.30(d,1H)、8.26(d,1H)、8.18(d,2H)、7.99(d,2H)、7.84(m,2H)、7.64-7.79(m,8H)、7.53(m,4H)、7.45(t,2H)、7.40(t,1H)、7.32(t,2H).
N,N-ジフェニル-4-(7-フェニル-4b-アザ-12b-ボラジベンゾ[g,p]クリセン-2-イル)アニリンの合成

1H-NMR(CDCl3):δ=8.73(d,2H)、8.60(m,1H)、8.57(m,1H)、8.51(m,2H)、8.22(m,2H)、7.81(m,2H)、7.75(d,2H)7.63(m,6H)、7.52(t,2H)、7.40(t,1H)、7.29(m,4H)、7.20(d,2H)、7.18(m,4H)、7.05(t,2H).
9-フェニル-3-(7-フェニル-4b-アザ-12b-ボラジベンゾ[g,p]クリセン-2-イル)-2-イル)-9H-カルバゾールの合成

1H-NMR(CDCl3):δ=8.75(d,2H)、8.71(m,1H)、8.61(m,1H)、8.59(d,1H)、8.51(d,1H)、8.50(m,1H)、8.26(m,3H)、7.79-7.87(m,3H)、7.77(m,3H)、7.60-7.70(m,7H)、7.47-7.57(m,4H)、7.45(m,2H)、7.40(t,1H)、7.33(m,1H).
2,7-ジフェニル-4b-アザ-12b-オキサホスファ-ジベンゾ[g,p]クリセンの合成


2,7-ジ(ピリジン-3-イル)-4b-アザ-12b-オキサホスファ-ジベンゾ[g,p]クリセンの合成

2,7-ジ(ピリジン-4-イル)-4b-アザ-12b-オキサホスファ-ジベンゾ[g,p]クリセンの合成

2,7-ビス(3-(ピリジン-2-イル)フェニル)-4b-アザ-12b-オキサホスファ-ジベンゾ[g,p]クリセンの合成

2,7-ジ(カルバゾール-9-イル)-4b-アザ-12b-オキサホスファ-ジベンゾ[g,p]クリセンの合成

2-フェニル-14b1-アザ-14b-ボラベンゾ[p]インデノ[1,2,3,4-defg]クリセンの合成



1H-NMR(CDCl3):δ=9.42(m,1H)、9.27(d,1H)、8.77(d,1H)、8.73(d,1H)、8.50(dd,2H)、8.27(dd,2H)、8.10(dd,1H)、7.88(m,3H)、7.73(m,3H)、7.60(t,2H)、7.47(t,1H).
2-(14b1-アザ-14b-ボラベンゾ[p]インデノ[1,2,3,4-defg]クリセン-6-イル)-9H-カルバゾールの合成


1H-NMR(CDCl3):δ=9.27(m,2H)、8.74(d,1H)、8.61(m,2H)、8.53(d,1H)、8.40(m,1H)、8.24(m,3H)、7.91(t,1H)、7.85(t,1H)、7.72-7.80(m,3H)、7.42-7.51(m,4H)、7.35(t,2H).
2,5,8,11-テトラメチル-3b-アザ-9b-ボラ-ナフト[2,1-b:3,-b’:6,5-b”:7,8-b''']テトラチオフェンの合成






1H-NMR(CDCl3):δ=7.73(s,2H)、7.71(s,2H)、2.69(s,6H)、2.66(s,6H).
11b-アザ-3b-ボラジベンゾ[c,f]ジピロロ[2,1-a:1’,2’-h][2,7]ナフチリジンの合成

1H-NMR(CDCl3):δ=8.01(dd,2H)、7.84(dd,2H)、7.74(dd,2H)、7.31(dd,2H)、7.16-7.23(m,4H)、6.73(dd,2H).
2,7-ジシアノ-4b-アザ-12b-ボラジベンゾ[g,p]クリセンの合成
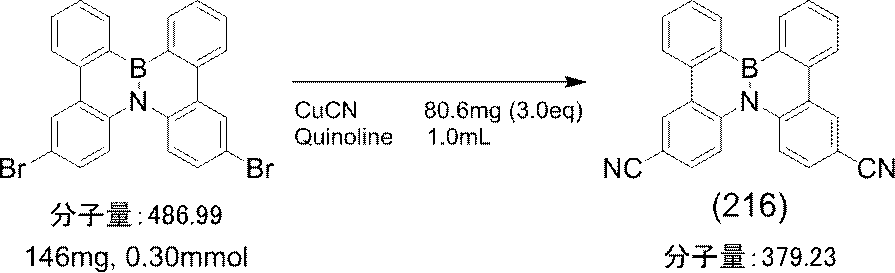
1H-NMR(CDCl3):δ=8.66-8.70(m,4H)、8.38(d,2H)、8.05(d,2H)、7.89(d,2H)、7.73(d,2H)、7.65(dd,2H).
3,6-ジフルオロ-4b-アザ-12b-ボラジベンゾ[g,p]クリセンの合成

1H-NMR(CDCl3):δ=8.63(dd,2H)、8.30(d,2H)、8.29(d,2H)、7.78(dd,2H)、7.76(dd,2H)、7.59(dd,2H)、7.08(dd,2H)、7.06(dd,2H).
2,7,9,16-テトラフェニル-4b-アザ-12b-ボラジベンゾ[g,p]クリセンの合成
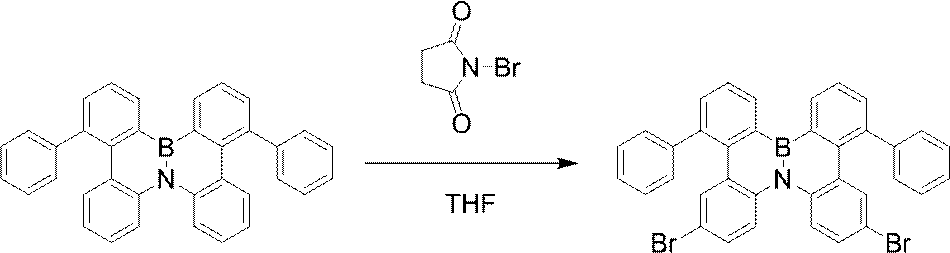
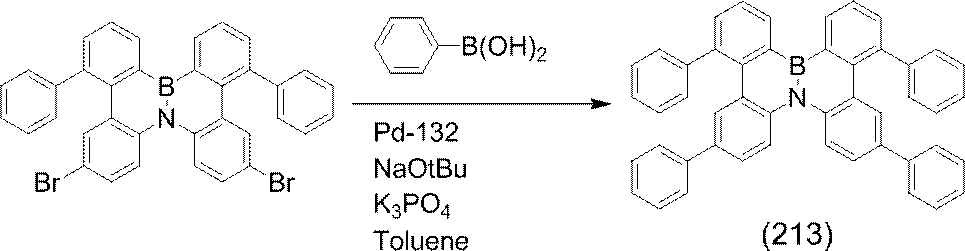
1H-NMR(CDCl3):δ=8.77(d,2H)、8.16(d,2H)、7.81(m,2H)、7.66-7.74(m,4H)、7.46-7.55(m,4H)、7.20-7.30(m,14H)、7.01(m,4H).
2-フェニル-7-(トリフェニレン-2-イル)-4b-アザ-12b-ボラジベンゾ[g,p]クリセンの合成

1H-NMR(CDCl3):δ=9.00(m,1H)、8.83(m,1H)、8.75-8.80(m,4H)、8.68-8.74(m,3H)、8.61(m,2H)、8.53(d,1H)、8.31(d,1H)、8.27(d,1H)、8.05(d,1H)、7.81-7.89(m,3H)、7.77(m,2H)、7.64-7.75(m,7H)、7.52(t,2H)、7.40(t,1H).
13c-アザ-4b-ボラ-9-フェニル-9,13c-ジヒドロ-4bH-ベンゾ[a]フェナントロ[9,10-c]カルバゾールの合成


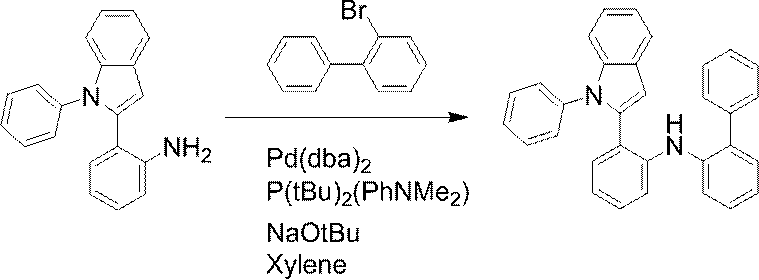

1H-NMR(CDCl3):δ=8.97(d,1H)、8.55(m,1H)、8.40(d,1H)、8.38(d,1H)、8.23(d,1H)、8.11(d,1H)、7.73-7.90(m,3H)、7.54-7.68(m,3H)、7.21-7.39(m,8H)、6.92(t,1H).


<化合物(1)を発光層のホスト材料に用いた素子>
スパッタリングにより180nmの厚さに製膜したITOを150nmまで研磨した、26mm×28mm×0.7mmのガラス基板((株)オプトサイエンス製)を透明支持基板とした。この透明支持基板を市販の蒸着装置((株)昭和真空製)の基板ホルダーに固定し、HIを入れたモリブデン製蒸着用ボート、NPDを入れたモリブデン製蒸着用ボート、本発明の化合物(1)を入れたモリブデン製蒸着用ボート、Ir(PPy)3を入れたモリブデン製蒸着用ボート、BCPを入れたモリブデン製蒸着用ボート、ET1を入れたモリブデン製蒸着用ボート、LiFを入れたモリブデン製蒸着用ボートおよびアルミニウムを入れたタングステン製蒸着用ボートを装着した。
<化合物(66)を発光層のホスト材料に用いた素子>
発光層のホスト材料である化合物(1)を化合物(66)に替えた以外は実施例1に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は5.7Vであり、その時の電流効率は36.4cd/Aであった。
<化合物(197)を発光層のホスト材料に用いた素子>
発光層のホスト材料である化合物(1)を化合物(197)に替えた以外は実施例1に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は5.6Vであり、その時の電流効率は28.1cd/Aであった。
<化合物(198)を正孔輸送層に用いた素子>
正孔輸送材料であるNPDを化合物(198)に発光層のホスト材料である化合物(1)をCBPに替えた以外は実施例1に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は5.9Vであり、その時の電流効率は26.4cd/Aであった。
発光層のホスト材料である化合物(1)をCBPに替えた以外は実施例1に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は6.7Vであり、その時の電流効率は24.6cd/Aであった。

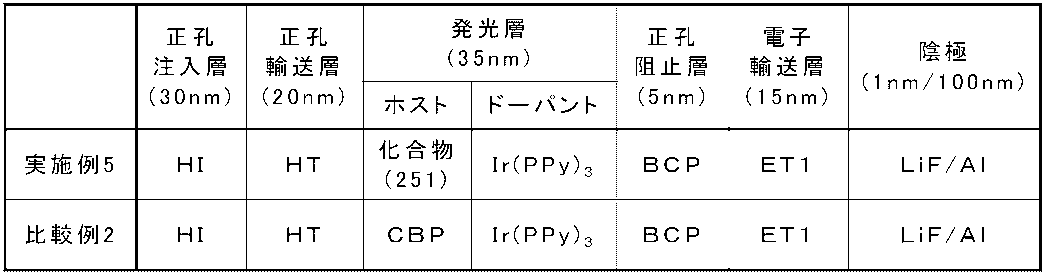
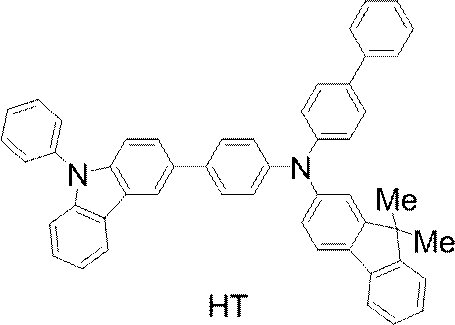
<化合物(251)を発光層のホスト材料に用いた素子>
スパッタリングにより180nmの厚さに製膜したITOを150nmまで研磨した、26mm×28mm×0.7mmのガラス基板((株)オプトサイエンス製)を透明支持基板とした。この透明支持基板を市販の蒸着装置((株)昭和真空製)の基板ホルダーに固定し、HIを入れたモリブデン製蒸着用ボート、HTを入れたモリブデン製蒸着用ボート、本発明の化合物(251)を入れたモリブデン製蒸着用ボート、Ir(PPy)3を入れたモリブデン製蒸着用ボート、BCPを入れたモリブデン製蒸着用ボート、ET1を入れたモリブデン製蒸着用ボート、LiFを入れたモリブデン製蒸着用ボートおよびアルミニウムを入れたタングステン製蒸着用ボートを装着した。
発光層のホスト材料である化合物(251)をCBPに替えた以外は実施例5に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は5.9Vであり、その時の電流効率は31.8cd/Aであった。



<化合物(1)を発光層のホスト材料に用いた素子 その2>
スパッタリングにより180nmの厚さに製膜したITOを150nmまで研磨した、26mm×28mm×0.7mmのガラス基板((株)オプトサイエンス製)を透明支持基板とした。この透明支持基板を市販の蒸着装置((株)昭和真空製)の基板ホルダーに固定し、HIを入れたモリブデン製蒸着用ボート、HTを入れたモリブデン製蒸着用ボート、本発明の化合物(1)を入れたモリブデン製蒸着用ボート、Ir(PPy)3を入れたモリブデン製蒸着用ボート、HB1を入れたモリブデン製蒸着用ボート、ET2を入れたモリブデン製蒸着用ボート、LiFを入れたモリブデン製蒸着用ボートおよびアルミニウムを入れたタングステン製蒸着用ボートを装着した。
<化合物(501)を発光層のホスト材料に用いた素子>
発光層のホスト材料である化合物(1)を化合物(501)に替えた以外は実施例6に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は6.2Vであり、その時の電流効率は29.0cd/Aであった。
<化合物(551)を発光層のホスト材料に用いた素子>
発光層のホスト材料である化合物(1)を化合物(551)に替えた以外は実施例6に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は4.8Vであり、その時の電流効率は31.7cd/Aであった。
<化合物(687)を発光層のホスト材料に用いた素子>
発光層のホスト材料である化合物(1)を化合物(687)に替えた以外は実施例6に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は4.0Vであり、その時の電流効率は28.3cd/Aであった。
<CBPを発光層のホスト材料に用いた素子>
発光層のホスト材料である化合物(1)をCBPに替えた以外は実施例6に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は5.4Vであり、その時の電流効率は24.2cd/Aであった。
<化合物(301)を正孔阻止層兼電子輸送層(1層で使用)に用いた素子>
スパッタリングにより180nmの厚さに製膜したITOを150nmまで研磨した、26mm×28mm×0.7mmのガラス基板((株)オプトサイエンス製)を透明支持基板とした。この透明支持基板を市販の蒸着装置((株)昭和真空製)の基板ホルダーに固定し、HIを入れたモリブデン製蒸着用ボート、HTを入れたモリブデン製蒸着用ボート、本発明のCBPを入れたモリブデン製蒸着用ボート、Ir(PPy)3を入れたモリブデン製蒸着用ボート、化合物(301)を入れたモリブデン製蒸着用ボート、LiFを入れたモリブデン製蒸着用ボートおよびアルミニウムを入れたタングステン製蒸着用ボートを装着した。
<化合物(391)を正孔阻止層兼電子輸送層(1層で使用)に用いた素子>
正孔阻止層兼電子輸送層である化合物(301)を化合物(391)に替えた以外は実施例10に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は6.0Vであり、その時の電流効率は28.0cd/Aであった。
<化合物(392)を正孔阻止層兼電子輸送層(1層で使用)に用いた素子>
正孔阻止層兼電子輸送層である化合物(301)を化合物(392)に替えた以外は実施例10に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は6.2Vであり、その時の電流効率は26.2cd/Aであった。
<化合物(391)を正孔阻止層に用いた素子>
スパッタリングにより180nmの厚さに製膜したITOを150nmまで研磨した、26mm×28mm×0.7mmのガラス基板((株)オプトサイエンス製)を透明支持基板とした。この透明支持基板を市販の蒸着装置((株)昭和真空製)の基板ホルダーに固定し、HIを入れたモリブデン製蒸着用ボート、HTを入れたモリブデン製蒸着用ボート、本発明のCBPを入れたモリブデン製蒸着用ボート、Ir(PPy)3を入れたモリブデン製蒸着用ボート、化合物(391)を入れたモリブデン製蒸着用ボート、ET2を入れたモリブデン製蒸着用ボート、LiFを入れたモリブデン製蒸着用ボートおよびアルミニウムを入れたタングステン製蒸着用ボートを装着した。
<化合物(392)を正孔阻止層に用いた素子>
正孔阻止層である化合物(391)を化合物(392)に替えた以外は実施例13に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は4.9Vであり、その時の電流効率は32.7cd/Aであった。
<BCPを正孔阻止層に用いた素子>
正孔阻止層である化合物(391)をBCPに替えた以外は実施例13に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は5.7Vであり、その時の電流効率は28.7cd/Aであった。


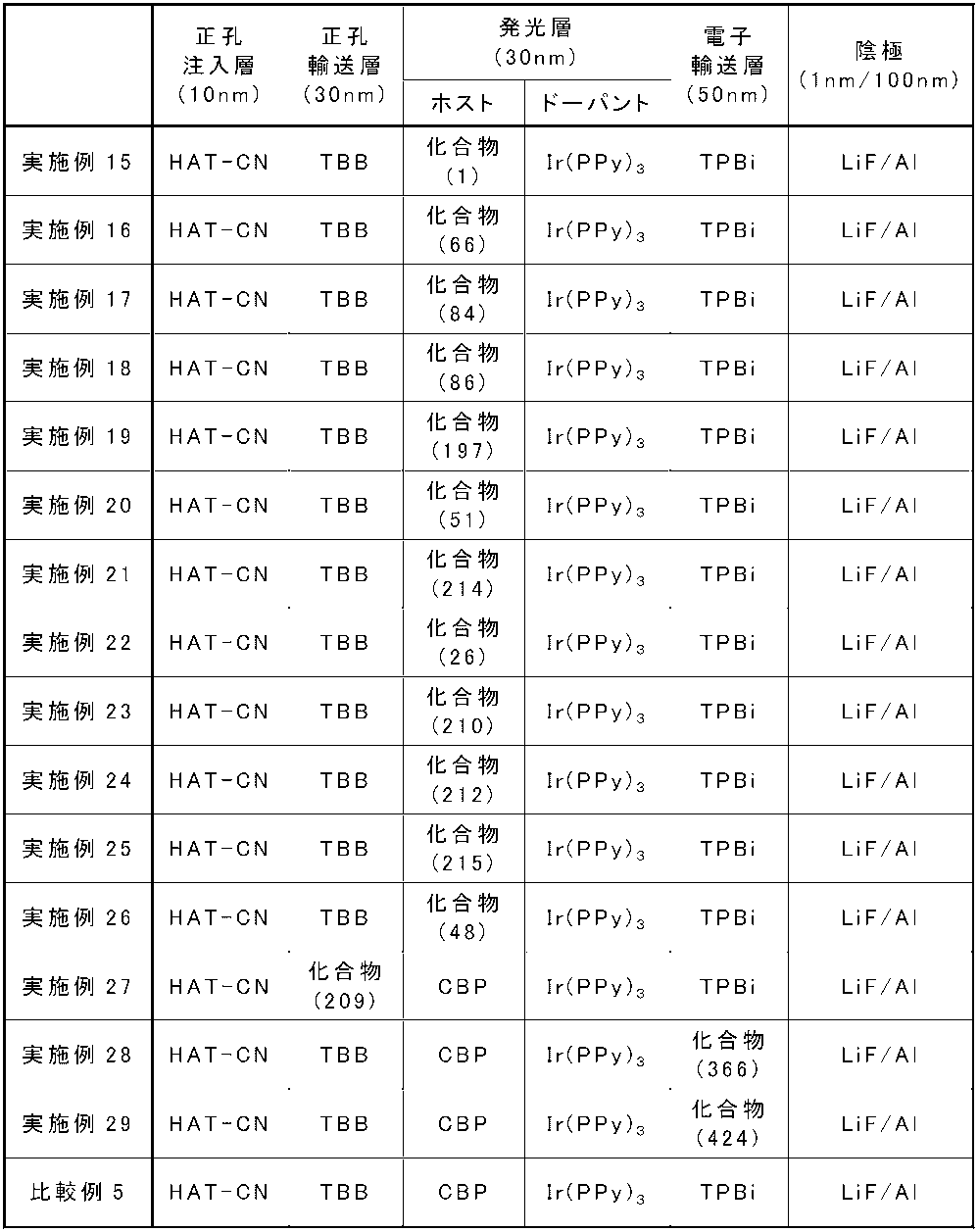
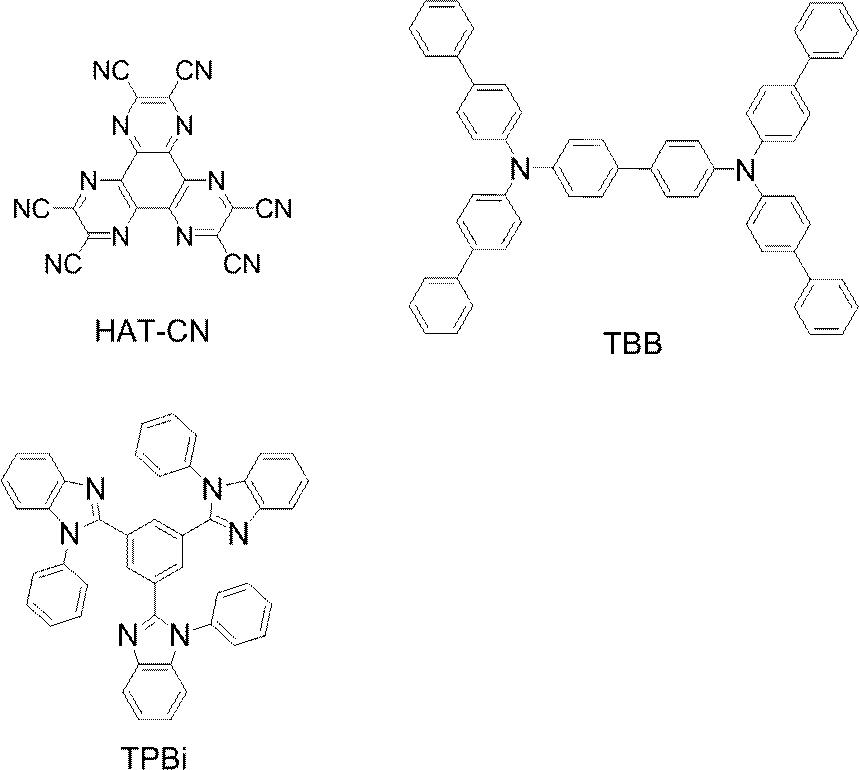
<化合物(1)を発光層のホスト材料に用いた素子 その3>
スパッタリングにより180nmの厚さに製膜したITOを150nmまで研磨した、26mm×28mm×0.7mmのガラス基板((株)オプトサイエンス製)を透明支持基板とした。この透明支持基板を市販の蒸着装置((株)昭和真空製)の基板ホルダーに固定し、HAT-CNを入れたモリブデン製蒸着用ボート、TBBを入れたモリブデン製蒸着用ボート、本発明の化合物(1)を入れたモリブデン製蒸着用ボート、Ir(PPy)3を入れたモリブデン製蒸着用ボート、TPBiを入れたモリブデン製蒸着用ボート、LiFを入れたモリブデン製蒸着用ボートおよびアルミニウムを入れたタングステン製蒸着用ボートを装着した。
<化合物(66)を発光層のホスト材料に用いた素子>
発光層のホスト材料である化合物(1)を化合物(66)に替えた以外は実施例15に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は4.8Vであり、その時の電流効率は37.0cd/Aであった。
<化合物(84)を発光層のホスト材料に用いた素子>
発光層のホスト材料である化合物(1)を化合物(84)に替えた以外は実施例15に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は5.6Vであり、その時の電流効率は35.9cd/Aであった。
<化合物(86)を発光層のホスト材料に用いた素子>
発光層のホスト材料である化合物(1)を化合物(86)に替えた以外は実施例15に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は5.0Vであり、その時の電流効率は29.0cd/Aであった。
<化合物(197)を発光層のホスト材料に用いた素子>
発光層のホスト材料である化合物(1)を化合物(197)に替えた以外は実施例15に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は4.9Vであり、その時の電流効率は32.4cd/Aであった。
<化合物(51)を発光層のホスト材料に用いた素子>
発光層のホスト材料である化合物(1)を化合物(51)に替えた以外は実施例15に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は5.1Vであり、その時の電流効率は39.2cd/Aであった。
<化合物(214)を発光層のホスト材料に用いた素子>
発光層のホスト材料である化合物(1)を化合物(214)に替えた以外は実施例15に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は4.2Vであり、その時の電流効率は35.2cd/Aであった。
<化合物(26)を発光層のホスト材料に用いた素子>
発光層のホスト材料である化合物(1)を化合物(26)に替えた以外は実施例15に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は4.7Vであり、その時の電流効率は42.2cd/Aであった。
<化合物(210)を発光層のホスト材料に用いた素子>
発光層のホスト材料である化合物(1)を化合物(210)に替えた以外は実施例15に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は5.1Vであり、その時の電流効率は32.7cd/Aであった。
<化合物(212)を発光層のホスト材料に用いた素子>
発光層のホスト材料である化合物(1)を化合物(212)に替えた以外は実施例15に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は5.3Vであり、その時の電流効率は27.0cd/Aであった。
<化合物(215)を発光層のホスト材料に用いた素子>
発光層のホスト材料である化合物(1)を化合物(215)に替えた以外は実施例15に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は5.5Vであり、その時の電流効率は27.7cd/Aであった。
<化合物(48)を発光層のホスト材料に用いた素子>
発光層のホスト材料である化合物(1)を化合物(48)に替えた以外は実施例15に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は5.5Vであり、その時の電流効率は29.2cd/Aであった。
<化合物(209)を正孔輸送層に用いた素子>
スパッタリングにより180nmの厚さに製膜したITOを150nmまで研磨した、26mm×28mm×0.7mmのガラス基板((株)オプトサイエンス製)を透明支持基板とした。この透明支持基板を市販の蒸着装置((株)昭和真空製)の基板ホルダーに固定し、HAT-CNを入れたモリブデン製蒸着用ボート、本発明の化合物(209)を入れたモリブデン製蒸着用ボート、CBPを入れたモリブデン製蒸着用ボート、Ir(PPy)3を入れたモリブデン製蒸着用ボート、TPBiを入れたモリブデン製蒸着用ボート、LiFを入れたモリブデン製蒸着用ボートおよびアルミニウムを入れたタングステン製蒸着用ボートを装着した。
<化合物(366)を電子輸送層に用いた素子>
スパッタリングにより180nmの厚さに製膜したITOを150nmまで研磨した、26mm×28mm×0.7mmのガラス基板((株)オプトサイエンス製)を透明支持基板とした。この透明支持基板を市販の蒸着装置((株)昭和真空製)の基板ホルダーに固定し、HAT-CNを入れたモリブデン製蒸着用ボート、TBBを入れたモリブデン製蒸着用ボート、CBPを入れたモリブデン製蒸着用ボート、Ir(PPy)3を入れたモリブデン製蒸着用ボート、本発明の化合物(366)を入れたモリブデン製蒸着用ボート、LiFを入れたモリブデン製蒸着用ボートおよびアルミニウムを入れたタングステン製蒸着用ボートを装着した。
<化合物(424)を電子輸送層に用いた素子>
電子輸送材料である化合物(366)を化合物(424)に替えた以外は実施例28に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は6.4Vであり、その時の電流効率は27.5cd/Aであった。
電子輸送材料である化合物(366)をTPBiに替えた以外は実施例28に準じた方法で有機EL素子を得た。両電極に直流電圧を印加すると、波長約515nmの緑色発光が得られた。また、初期輝度1000cd/m2を得るための駆動電圧は5.5Vであり、その時の電流効率は27.1cd/Aであった。

<式(1)で表される化合物のキャリア移動度測定>
26mm×28mm×0.5mmのガラス基板(日本板硝子(株)製)を透明支持基板とした。この透明支持基板を市販の蒸着装置の基板ホルダーに、2mm幅の下部アルミ電極を得るためのメタルマスクと同時に装着した。次いで、アルミニウムをのせたタングステン製の蒸着ボートを蒸着装置にセットした。真空槽を5×10-3Pa以下まで減圧し、蒸着用ボートを加熱して膜厚10nmになるように半透明の下部アルミ電極を形成した。蒸着速度は、0.05~1nm/秒であった。
式(1)で表される化合物を式(4)で表される化合物に変更し、蒸着した有機層の膜厚が8.2μmになった以外は同様の手法でサンプルを作製し、同様の方法で移動度の観測を行った。
101 基板
102 陽極
103 正孔注入層
104 正孔輸送層
105 発光層
106 電子輸送層
107 電子注入層
108 陰極
Claims (27)
- 下記一般式(I)で表される部分構造を有する多環芳香族化合物またはその塩を含む、有機電界発光素子用材料。
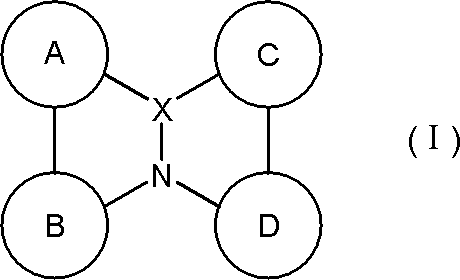
(上記式(I)中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
A環、B環、C環およびD環は、それぞれ独立して、置換されていてもよい芳香族環または置換されていてもよいヘテロ芳香族環であり、隣接する2つの環は連結基または単結合によりそれらの間に環を形成していてもよく、そして、
上記式(I)で表される部分構造は少なくとも1つの水素を有し、そして、該部分構造における少なくとも1つの水素が重水素で置換されていてもよい。) - 下記一般式(II)で表される部分構造を有する多環芳香族化合物またはその塩を含む、請求項1に記載の有機電界発光素子用材料。

(上記式(II)中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
Yaは、それぞれ独立して、CまたはNであり、同じ環で隣接する2つのYaとそれらの間の結合とが一緒になって、N、O、SまたはSeになってもよく、環はそれぞれ独立して置換されていてもよく、同じ環における隣接する置換基が結合してシクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、隣接する2つの環は連結基または単結合によりそれらの間に環を形成していてもよく、そして、
上記式(II)で表される部分構造は少なくとも1つの水素を有し、そして、該部分構造における少なくとも1つの水素が重水素で置換されていてもよい。) - 下記一般式(III-1)で表される部分構造を有する多環芳香族化合物またはその塩を含む、請求項1に記載の有機電界発光素子用材料。

(上記式(III-1)中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
上記式中のベンゼン環はそれぞれ独立して置換されていてもよく、同じ環における隣接する置換基が結合してシクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
上記式中の隣接する2つのベンゼン環は連結基または単結合によりそれらの間に環を形成していてもよく、そして、
上記式(III-1)で表される部分構造は少なくとも1つの水素を有し、そして、該部分構造における少なくとも1つの水素が重水素で置換されていてもよい。) - 下記一般式(III-11)~(III-13)および一般式(III-33)~(III-36)のいずれかで表される部分構造を有する多環芳香族化合物またはその塩を含む、請求項1に記載の有機電界発光素子用材料。

(上記各式中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
Zは、N、O、SまたはSeであり、
上記各式中のベンゼン環および五員環はそれぞれ独立して置換されていてもよく、同じ環における隣接する置換基が結合してシクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
上記各式中の隣接する2つのベンゼン環は連結基または単結合によりそれらの間に環を形成していてもよく、そして、
上記各式で表される部分構造は少なくとも1つの水素を有し、そして、該部分構造における少なくとも1つの水素が重水素で置換されていてもよい。) - 下記一般式(III-33)および一般式(III-55)~(III-57)のいずれかで表される部分構造を有する多環芳香族化合物またはその塩を含む、請求項1に記載の有機電界発光素子用材料。

(上記各式中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
上記各式中のベンゼン環および五員環はそれぞれ独立して置換されていてもよく、同じ環における隣接する置換基が結合してシクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
上記各式中の隣接する2つのベンゼン環は連結基または単結合によりそれらの間に環を形成していてもよく、そして、
上記各式で表される部分構造は少なくとも1つの水素を有し、そして、該部分構造における少なくとも1つの水素が重水素で置換されていてもよい。) - 下記一般式(III-32)および一般式(III-5)~(III-7)のいずれかで表される部分構造を有する多環芳香族化合物またはその塩を含む、請求項1に記載の有機電界発光素子用材料。

(上記各式中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
Zは、N、O、SまたはSeであり、
上記各式中のベンゼン環および五員環はそれぞれ独立して置換されていてもよく、同じ環における隣接する置換基が結合してシクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
上記各式中の隣接する2つのベンゼン環は連結基または単結合によりそれらの間に環を形成していてもよく、そして、
上記各式で表される部分構造は少なくとも1つの水素を有し、そして、該部分構造における少なくとも1つの水素が重水素で置換されていてもよい。) - 下記一般式(III-32)および一般式(III-58)~(III-60)のいずれかで表される部分構造を有する多環芳香族化合物またはその塩を含む、請求項1に記載の有機電界発光素子用材料。

(上記各式中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
上記各式中のベンゼン環および五員環はそれぞれ独立して置換されていてもよく、同じ環における隣接する置換基が結合してシクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
上記各式中の隣接する2つのベンゼン環は連結基または単結合によりそれらの間に環を形成していてもよく、そして、
上記各式で表される部分構造は少なくとも1つの水素を有し、そして、該部分構造における少なくとも1つの水素が重水素で置換されていてもよい。) - 下記一般式(V-1)、一般式(V-3)、一般式(V-5)、一般式(V-15)、または一般式(V-16)で表される多環芳香族化合物またはその塩を含む、請求項1に記載の有機電界発光素子用材料。

(上記各式中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
Rは、水素、フッ素置換または無置換のC1-20アルキル、C3-8シクロアルキル、C2-20アルケニル、モノもしくはジアリール置換C2-12アルケニル、モノもしくはジヘテロアリール置換C2-12アルケニル、フッ素置換または無置換のC1-20アルコキシ、C1-20アルキルカルボニル、シアノ、ニトロ、ジアリールアミノ、置換されていてもよいアリール、置換されていてもよいヘテロアリール、B(Ra)2、またはSi(Ra)3(ここでRaは、それぞれ独立して、置換されていてもよいアルキル、置換されていてもよいアリールまたは置換されていてもよいヘテロアリールである。)であり、
同じ環で隣接する2つのRが結合して、シクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
上記各式中の隣接する2つのベンゼン環は、単結合、CH2、CHRa、C(Ra)2、NRa、Si(Ra)2、BRa(ここでRaは前記に定義されるとおりである)、Se、S、またはOを介した結合により結合されて、それらの間に環を形成していてもよく、
nは0~4の整数であり、mは0~3の整数であり、そして、
上記各式で表される化合物またはその塩における少なくとも1つの水素が重水素で置換されていてもよい。) - 下記一般式(V-27)~(V-30)で表される多環芳香族化合物またはその塩を含む、請求項1に記載の有機電界発光素子用材料。
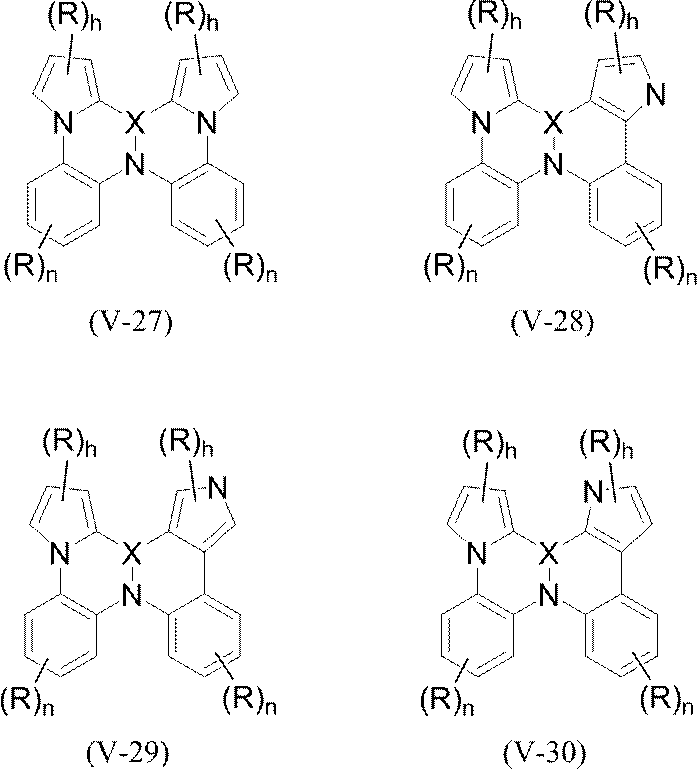
(上記各式中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
Rは、水素、フッ素置換または無置換のC1-20アルキル、C3-8シクロアルキル、C2-20アルケニル、モノもしくはジアリール置換C2-12アルケニル、モノもしくはジヘテロアリール置換C2-12アルケニル、フッ素置換または無置換のC1-20アルコキシ、C1-20アルキルカルボニル、シアノ、ニトロ、ジアリールアミノ、置換されていてもよいアリール、置換されていてもよいヘテロアリール、B(Ra)2、またはSi(Ra)3(ここでRaは、それぞれ独立して、置換されていてもよいアルキル、置換されていてもよいアリールまたは置換されていてもよいヘテロアリールである。)であり、
同じ環で隣接する2つのRが結合して、シクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
上記各式中の隣接する2つのベンゼン環は、単結合、CH2、CHRa、C(Ra)2、NRa、Si(Ra)2、BRa(ここでRaは前記に定義されるとおりである)、Se、S、またはOを介した結合により結合されて、それらの間に環を形成していてもよく、
nは0~4の整数であり、hは0~3の整数であり、そして、
上記各式で表される化合物またはその塩における少なくとも1つの水素が重水素で置換されていてもよい。) - 下記一般式(V-31)~(V-34)で表される多環芳香族化合物またはその塩を含む、請求項1に記載の有機電界発光素子用材料。

(上記各式中、
Xは、B、P、P=O、P=S、P=Se、As、As=O、As=S、As=Se、Sb、Sb=O、Sb=S、Sb=Se、置換されていてもよい周期表第3~11族の金属元素、または置換されていてもよい周期表第13~14族の金属元素もしくは半金属元素であり、
Rは、水素、フッ素置換または無置換のC1-20アルキル、C3-8シクロアルキル、C2-20アルケニル、モノもしくはジアリール置換C2-12アルケニル、モノもしくはジヘテロアリール置換C2-12アルケニル、フッ素置換または無置換のC1-20アルコキシ、C1-20アルキルカルボニル、シアノ、ニトロ、ジアリールアミノ、置換されていてもよいアリール、置換されていてもよいヘテロアリール、B(Ra)2、またはSi(Ra)3(ここでRaは、それぞれ独立して、置換されていてもよいアルキル、置換されていてもよいアリールまたは置換されていてもよいヘテロアリールである。)であり、
同じ環で隣接する2つのRが結合して、シクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
上記各式中の隣接する2つのベンゼン環は、単結合、CH2、CHRa、C(Ra)2、NRa、Si(Ra)2、BRa(ここでRaは前記に定義されるとおりである)、Se、S、またはOを介した結合により結合されて、それらの間に環を形成していてもよく、
nは0~4の整数であり、hは0~3の整数であり、そして、
上記各式で表される化合物またはその塩における少なくとも1つの水素が重水素で置換されていてもよい。) - 下記一般式(V-1’)~(V-3’)で表される多環芳香族化合物またはその塩を含む、請求項1に記載の有機電界発光素子用材料。

(上記各式中、
Rは、フッ素置換または無置換のC1-20アルキル、C3-8シクロアルキル、C2-20アルケニル、モノもしくはジアリール置換C2-12アルケニル、モノもしくはジヘテロアリール置換C2-12アルケニル、フッ素置換または無置換のC1-20アルコキシ、C1-20アルキルカルボニル、シアノ、ニトロ、ジアリールアミノ、置換されていてもよいアリール、置換されていてもよいヘテロアリール、B(Ra)2、またはSi(Ra)3(ここでRaは、それぞれ独立して、置換されていてもよいアルキル、置換されていてもよいアリールまたは置換されていてもよいヘテロアリールである。)であり、
同じ環で隣接する2つのRが結合して、シクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
nは0~4の整数であり、mは0~3の整数であり、そして、
上記各式で表される化合物またはその塩における少なくとも1つの水素が重水素で置換されていてもよい。) - 下記一般式(V-27’)で表される多環芳香族化合物またはその塩を含む、請求項1に記載の有機電界発光素子用材料。

(上記式中、
Rは、フッ素置換または無置換のC1-20アルキル、C3-8シクロアルキル、C2-20アルケニル、モノもしくはジアリール置換C2-12アルケニル、モノもしくはジヘテロアリール置換C2-12アルケニル、フッ素置換または無置換のC1-20アルコキシ、C1-20アルキルカルボニル、シアノ、ニトロ、ジアリールアミノ、置換されていてもよいアリール、置換されていてもよいヘテロアリール、B(Ra)2、またはSi(Ra)3(ここでRaは、それぞれ独立して、置換されていてもよいアルキル、置換されていてもよいアリールまたは置換されていてもよいヘテロアリールである。)であり、
同じ環で隣接する2つのRが結合して、シクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
nは0~4の整数であり、hは0~3の整数であり、そして、
上記式で表される化合物またはその塩における少なくとも1つの水素が重水素で置換されていてもよい。) - 下記一般式(V-32’)で表される多環芳香族化合物またはその塩を含む、請求項1に記載の有機電界発光素子用材料。

(上記式中、
Rは、フッ素置換または無置換のC1-20アルキル、C3-8シクロアルキル、C2-20アルケニル、モノもしくはジアリール置換C2-12アルケニル、モノもしくはジヘテロアリール置換C2-12アルケニル、フッ素置換または無置換のC1-20アルコキシ、C1-20アルキルカルボニル、シアノ、ニトロ、ジアリールアミノ、置換されていてもよいアリール、置換されていてもよいヘテロアリール、B(Ra)2、またはSi(Ra)3(ここでRaは、それぞれ独立して、置換されていてもよいアルキル、置換されていてもよいアリールまたは置換されていてもよいヘテロアリールである。)であり、
同じ環で隣接する2つのRが結合して、シクロヘキサン環、ベンゼン環またはピリジン環を形成してもよく、
nは0~4の整数であり、hは0~3の整数であり、そして、
上記式で表される化合物またはその塩における少なくとも1つの水素が重水素で置換されていてもよい。) - 下記式(1)、式(66)、式(197)、式(198)、または式(251)で表される多環芳香族化合物またはその塩を含む、請求項1に記載の有機電界発光素子用材料。

- 下記式(301)、式(391)、式(392)、式(501)、式(551)、または式(687)で表される多環芳香族化合物またはその塩を含む、請求項1に記載の有機電界発光素子用材料。

- 下記式(26)、式(48)、式(51)、式(84)、式(86)、式(209)、式(210)、式(212)、式(214)、式(215)、式(366)または式(424)で表される多環芳香族化合物またはその塩を含む、請求項1に記載の有機電界発光素子用材料。

- 発光層用材料である、請求項1~16のいずれかに記載する有機電界発光素子用材料。
- 正孔注入層用材料または正孔輸送層用材料である、請求項1~16のいずれかに記載する有機電界発光素子用材料。
- 正孔阻止層用材料または電子輸送層用材料である、請求項1~16のいずれかに記載する有機電界発光素子用材料。
- 陽極および陰極からなる一対の電極と、該一対の電極間に配置され、請求項17に記載する発光層用材料を含有する発光層とを有する、有機電界発光素子。
- 陽極および陰極からなる一対の電極と、該一対の電極間に配置された発光層と、前記陽極および前記発光層の間に配置され、請求項18に記載する正孔層用材料を含有する正孔注入層および/または正孔輸送層とを有する、有機電界発光素子。
- 陽極および陰極からなる一対の電極と、該一対の電極間に配置された発光層と、前記陰極および前記発光層の間に配置され、請求項19に記載する正孔阻止層用材料または電子輸送層用材料を含有する正孔阻止層および/または電子輸送層とを有する、有機電界発光素子。
- さらに、前記陰極と該発光層との間に配置される電子輸送層および/または電子注入層を有し、該電子輸送層および電子注入層の少なくとも1つは、キノリノール系金属錯体、ピリジン誘導体、フェナントロリン誘導体、ボラン誘導体およびベンゾイミダゾール誘導体からなる群から選択される少なくとも1つを含有する、請求項20または21に記載する有機電界発光素子。
- 前記正孔阻止層および電子輸送層の少なくとも1つは、キノリノール系金属錯体、ピリジン誘導体、フェナントロリン誘導体、ボラン誘導体およびベンゾイミダゾール誘導体からなる群から選択される少なくとも1つを含有する、請求項22に記載する有機電界発光素子。
- 前記正孔阻止層、電子輸送層および/または電子注入層が、さらに、アルカリ金属、アルカリ土類金属、希土類金属、アルカリ金属の酸化物、アルカリ金属のハロゲン化物、アルカリ土類金属の酸化物、アルカリ土類金属のハロゲン化物、希土類金属の酸化物、希土類金属のハロゲン化物、アルカリ金属の有機錯体、アルカリ土類金属の有機錯体および希土類金属の有機錯体からなる群から選択される少なくとも1つを含有する、請求項23または24に記載の有機電界発光素子。
- 請求項20~25のいずれかに記載する有機電界発光素子を備えた表示装置。
- 請求項20~25のいずれかに記載する有機電界発光素子を備えた照明装置。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201380047231.1A CN104641483B (zh) | 2012-09-11 | 2013-09-11 | 有机电场发光元件用材料、有机电场发光元件、显示装置、以及照明装置 |
| US14/386,153 US20150097162A1 (en) | 2012-09-11 | 2013-09-11 | Material for organic electroluminescent elements, organic electroluminescent element, display device, and lighting device |
| EP13836272.8A EP2897184A4 (en) | 2012-09-11 | 2013-09-11 | MATERIAL FOR ORGANIC ELECTROLUMINESCENE ELEMENTS, ORGANIC ELECTROLUMINESCENT ELEMENT, DISPLAY DEVICE AND LIGHTING DEVICE |
| JP2014535578A JP5819534B2 (ja) | 2012-09-11 | 2013-09-11 | 有機電界発光素子用材料、有機電界発光素子、表示装置、及び照明装置 |
| KR1020157008326A KR102157994B1 (ko) | 2012-09-11 | 2013-09-11 | 유기 전계 발광 소자용 재료, 유기 전계 발광 소자, 표시 장치 및 조명 장치 |
| US15/782,065 US20180047913A1 (en) | 2012-09-11 | 2017-10-12 | Material for organic electroluminescent elements, organic electroluminescent element, display device, and lighting device |
| US16/354,164 US20190214575A1 (en) | 2012-09-11 | 2019-03-14 | Material for organic electroluminescent elements, organic electroluminescent element, display device, and lighting device |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-199232 | 2012-09-11 | ||
| JP2012199232 | 2012-09-11 | ||
| JP2013140007 | 2013-07-03 | ||
| JP2013-140007 | 2013-07-03 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/386,153 A-371-Of-International US20150097162A1 (en) | 2012-09-11 | 2013-09-11 | Material for organic electroluminescent elements, organic electroluminescent element, display device, and lighting device |
| US15/782,065 Continuation US20180047913A1 (en) | 2012-09-11 | 2017-10-12 | Material for organic electroluminescent elements, organic electroluminescent element, display device, and lighting device |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014042197A1 true WO2014042197A1 (ja) | 2014-03-20 |
Family
ID=50278307
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/074561 WO2014042197A1 (ja) | 2012-09-11 | 2013-09-11 | 有機電界発光素子用材料、有機電界発光素子、表示装置、及び照明装置 |
Country Status (7)
| Country | Link |
|---|---|
| US (3) | US20150097162A1 (ja) |
| EP (1) | EP2897184A4 (ja) |
| JP (3) | JP5819534B2 (ja) |
| KR (1) | KR102157994B1 (ja) |
| CN (2) | CN107266481B (ja) |
| TW (1) | TWI612054B (ja) |
| WO (1) | WO2014042197A1 (ja) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016143624A1 (ja) * | 2015-03-09 | 2016-09-15 | 学校法人関西学院 | 多環芳香族化合物および発光層形成用組成物 |
| JP2017028266A (ja) * | 2015-07-21 | 2017-02-02 | ▲いく▼▲雷▼光電科技股▲分▼有限公司 | 有機発光素子 |
| WO2017195669A1 (ja) * | 2016-05-13 | 2017-11-16 | コニカミノルタ株式会社 | 有機エレクトロルミネッセンス素子用材料、有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| US10033004B2 (en) | 2015-06-01 | 2018-07-24 | Universal Display Corporation | Organic electroluminescent materials and devices |
| JP2018125387A (ja) * | 2017-01-31 | 2018-08-09 | 日本放送協会 | 有機エレクトロルミネッセンス素子 |
| US10418568B2 (en) | 2015-06-01 | 2019-09-17 | Universal Display Corporation | Organic electroluminescent materials and devices |
| US10468609B2 (en) | 2016-06-02 | 2019-11-05 | Universal Display Corporation | Organic electroluminescent materials and devices |
| US10913758B2 (en) | 2017-03-27 | 2021-02-09 | Samsung Electronics Co., Ltd. | Organometallic compound, organic light-emitting device including the organometallic compound, and diagnostic compound including the organometallic compound |
Families Citing this family (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9882151B2 (en) * | 2014-11-14 | 2018-01-30 | Universal Display Corporation | Organic electroluminescent materials and devices |
| US9871212B2 (en) * | 2014-11-14 | 2018-01-16 | Universal Display Corporation | Organic electroluminescent materials and devices |
| KR102523099B1 (ko) * | 2015-06-18 | 2023-04-18 | 엘지디스플레이 주식회사 | 유기전계발광소자 |
| KR102491790B1 (ko) * | 2015-09-25 | 2023-01-26 | 엘지디스플레이 주식회사 | 유기전계발광소자 |
| CN106986894A (zh) * | 2016-01-20 | 2017-07-28 | 中国科学院宁波材料技术与工程研究所 | 硫、磷杂原子取代二苯并[g,p]稠二萘型衍生物、其制备方法与应用 |
| CN116096201A (zh) * | 2016-02-10 | 2023-05-09 | 学校法人关西学院 | 延迟荧光有机电场发光元件、显示装置及照明装置 |
| CN107459528A (zh) * | 2016-06-03 | 2017-12-12 | 上海和辉光电有限公司 | 一种有机电致发光化合物 |
| KR102600472B1 (ko) * | 2016-09-13 | 2023-11-13 | 삼성디스플레이 주식회사 | 축합환 화합물 및 이를 포함한 유기 발광 소자 |
| US20190372023A1 (en) * | 2017-02-09 | 2019-12-05 | Kwansei Gakuin Education Foundation | Organic electroluminescent element |
| US10998506B2 (en) | 2017-08-22 | 2021-05-04 | Beijing Summer Sprout Technology Co., Ltd. | Boron containing heterocyclic compound for OLEDs, an organic light-emitting device, and a formulation comprising the boron-containing heterocyclic compound |
| EP3681972B1 (en) * | 2017-09-12 | 2021-07-28 | cynora GmbH | Organic molecules, in particular for use in optoelectronic devices |
| KR102608283B1 (ko) * | 2017-11-24 | 2023-11-29 | 가꼬우 호징 관세이 가쿠잉 | 유기 디바이스용 재료 및 이것을 사용한 유기 전계 발광 소자 |
| CN108503657B (zh) * | 2017-12-20 | 2022-08-16 | 天津理工大学 | 含五元杂环的硼氮掺杂稠环芳香烃及其合成方法及应用 |
| CN108299481B (zh) * | 2018-01-12 | 2020-10-09 | 华南协同创新研究院 | 电致发光聚合单体、聚合物及其制备方法和应用 |
| CN108912157A (zh) * | 2018-10-08 | 2018-11-30 | 天津理工大学 | 一种简单高效合成硼氮杂芳烃的方法 |
| JP7468857B2 (ja) * | 2018-12-27 | 2024-04-16 | 学校法人関西学院 | 多環芳香族化合物、有機デバイス用材料、有機el素子、表示装置および照明装置 |
| US20220052265A1 (en) * | 2018-12-28 | 2022-02-17 | Cynora Gmbh | Organic molecules for optoelectronic devices |
| KR20200086782A (ko) * | 2019-01-09 | 2020-07-20 | 삼성디스플레이 주식회사 | 유기 전계 발광 소자 및 이를 포함하는 표시 장치 |
| CN110003260B (zh) * | 2019-04-30 | 2021-06-01 | 武汉天马微电子有限公司 | 硼杂环化合物、显示面板以及显示装置 |
| KR102653917B1 (ko) * | 2019-06-27 | 2024-04-01 | 주식회사 엘지화학 | 신규한 이미노티오펜계 화합물, 및 이미노티오펜계 공액계 고분자 |
| KR20210043054A (ko) * | 2019-10-10 | 2021-04-21 | 삼성디스플레이 주식회사 | 유기 전계 발광 소자 및 유기 전계 발광 소자용 다환 화합물 |
| CN115023429A (zh) * | 2019-12-20 | 2022-09-06 | 杜邦电子公司 | 电活性化合物 |
| KR20210083464A (ko) * | 2019-12-26 | 2021-07-07 | 삼성디스플레이 주식회사 | 유기 전계 발광 소자 및 유기 전계 발광 소자용 다환 화합물 |
| CN113185541A (zh) * | 2021-02-07 | 2021-07-30 | 上海蓝骋光电科技有限公司 | 一种有机化合物及含有该化合物的有机光电元件与应用 |
| CN114835736B (zh) * | 2022-03-31 | 2024-04-05 | 武汉天马微电子有限公司 | 化合物、显示面板及显示装置 |
| CN115010734A (zh) * | 2022-05-26 | 2022-09-06 | 武汉天马微电子有限公司 | 化合物、显示面板及显示装置 |
Citations (57)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01245087A (ja) | 1987-12-11 | 1989-09-29 | Idemitsu Kosan Co Ltd | 有機エレクトロルミネッセンス素子 |
| JPH02247278A (ja) | 1989-03-20 | 1990-10-03 | Idemitsu Kosan Co Ltd | エレクトロルミネッセンス素子 |
| JPH06298758A (ja) | 1993-04-16 | 1994-10-25 | Chisso Corp | クマリン誘導体 |
| JPH0812600A (ja) | 1994-04-26 | 1996-01-16 | Tdk Corp | フェニルアントラセン誘導体および有機el素子 |
| JPH10335066A (ja) | 1997-06-02 | 1998-12-18 | Sony Corp | 有機電界発光素子およびこれを用いたフラットパネルディスプレイ |
| JPH1197178A (ja) | 1997-09-24 | 1999-04-09 | Mitsui Chem Inc | 有機電界発光素子 |
| JPH11297473A (ja) | 1998-04-15 | 1999-10-29 | Nec Corp | 有機エレクトロルミネッセンス素子 |
| JP2000026324A (ja) | 1998-07-02 | 2000-01-25 | Mitsui Chemicals Inc | 炭化水素化合物および有機電界発光素子 |
| JP2000034234A (ja) | 1998-07-15 | 2000-02-02 | Mitsui Chemicals Inc | 炭化水素化合物および有機電界発光素子 |
| JP2000133457A (ja) | 1998-10-30 | 2000-05-12 | Mitsui Chemicals Inc | 炭化水素化合物および有機電界発光素子 |
| WO2000040586A1 (fr) | 1999-01-08 | 2000-07-13 | Chisso Corporation | Derives de borane et composes organiques electroluminescents |
| JP2001052869A (ja) | 1999-08-11 | 2001-02-23 | Hayashibara Biochem Lab Inc | 有機電界発光素子 |
| JP2001076876A (ja) | 1999-09-01 | 2001-03-23 | Hayashibara Biochem Lab Inc | 有機電界発光素子 |
| JP2001081090A (ja) | 1999-03-09 | 2001-03-27 | Hayashibara Biochem Lab Inc | ピラン誘導体 |
| JP2001172232A (ja) | 1999-12-21 | 2001-06-26 | Univ Osaka | エレクトロルミネッセンス素子 |
| JP2001217077A (ja) | 2000-02-01 | 2001-08-10 | Mitsui Chemicals Inc | 有機電界発光素子 |
| JP2001220577A (ja) | 1999-03-09 | 2001-08-14 | Hayashibara Biochem Lab Inc | ピラン誘導体 |
| JP2001267076A (ja) | 2000-03-16 | 2001-09-28 | Mitsui Chemicals Inc | 炭化水素化合物および有機電界発光素子 |
| JP2001267078A (ja) | 2000-03-23 | 2001-09-28 | Mitsui Chemicals Inc | 炭化水素化合物および有機電界発光素子 |
| JP2001267079A (ja) | 2000-03-23 | 2001-09-28 | Mitsui Chemicals Inc | 炭化水素化合物および有機電界発光素子 |
| JP2001267075A (ja) | 2000-03-16 | 2001-09-28 | Mitsui Chemicals Inc | 炭化水素化合物および有機電界発光素子 |
| JP2001307884A (ja) | 2000-04-26 | 2001-11-02 | Toray Ind Inc | 発光素子 |
| JP2002234892A (ja) | 2000-11-07 | 2002-08-23 | Hayashibara Biochem Lab Inc | ピラン誘導体 |
| JP2003123983A (ja) | 2001-10-10 | 2003-04-25 | Konica Corp | 有機エレクトロルミネッセンス素子 |
| JP2003146951A (ja) | 2001-08-06 | 2003-05-21 | Mitsubishi Chemicals Corp | アントラセン系化合物、その製造方法および有機電界発光素子 |
| JP2003257621A (ja) | 2002-02-27 | 2003-09-12 | Seiko Epson Corp | 有機el素子とその製造方法ならびに表示装置 |
| WO2003078541A1 (en) | 2002-03-15 | 2003-09-25 | Idemitsu Kosan Co., Ltd. | Material for organic electroluminescent devices and organic electroluminescent devices made by using the same |
| JP2003277741A (ja) | 2002-03-26 | 2003-10-02 | Sharp Corp | 有機el発光素子およびそれを用いた液晶表示装置 |
| WO2003080760A1 (fr) | 2002-03-22 | 2003-10-02 | Idemitsu Kosan Co., Ltd. | Materiau pour dispositifs electroluminescents organiques et dispositifs electroluminescents organiques produits avec ce materiau |
| JP2003321546A (ja) | 2002-04-26 | 2003-11-14 | Nippon Hoso Kyokai <Nhk> | 燐光発光性高分子化合物ならびにこれを用いた発光材料および有機el素子 |
| JP2003347056A (ja) | 2002-05-30 | 2003-12-05 | Fuji Photo Film Co Ltd | 発光素子 |
| JP2004043646A (ja) | 2002-07-11 | 2004-02-12 | Hayashibara Biochem Lab Inc | 有機電界発光素子 |
| JP2004111379A (ja) | 2002-08-29 | 2004-04-08 | Fuji Photo Film Co Ltd | 発光素子及びイリジウム錯体 |
| JP2004119211A (ja) | 2002-09-26 | 2004-04-15 | Toyota Industries Corp | El素子用透明基板及びel装置並びに液晶表示装置 |
| WO2004061047A2 (en) | 2002-12-31 | 2004-07-22 | Eastman Kodak Company | Complex fluorene-containing compounds for use in organic light emitting devices |
| JP2004281086A (ja) | 2003-03-12 | 2004-10-07 | Nippon Hoso Kyokai <Nhk> | フレキシブルフルカラー有機elディスプレイおよびその製造方法 |
| JP2004335122A (ja) | 2003-04-30 | 2004-11-25 | Takasago Internatl Corp | 発光素子 |
| JP2004331508A (ja) | 2003-04-30 | 2004-11-25 | Takasago Internatl Corp | 白金錯体 |
| JP2005097283A (ja) | 2003-08-29 | 2005-04-14 | Semiconductor Energy Lab Co Ltd | ピラン誘導体とその製造方法、並びにピラン誘導体を用いた発光素子及び発光装置。 |
| JP2005097263A (ja) | 2003-08-22 | 2005-04-14 | National Institute Of Advanced Industrial & Technology | 新規イリジウム錯体およびこれを用いた発光材料 |
| JP2005126399A (ja) | 2003-10-27 | 2005-05-19 | Semiconductor Energy Lab Co Ltd | ピラン誘導体 |
| JP2005167175A (ja) | 2003-12-04 | 2005-06-23 | Novaled Gmbh | 有機半導体をキノンジイミン誘導体によってドーピングする方法 |
| JP2005170911A (ja) | 2003-12-15 | 2005-06-30 | Idemitsu Kosan Co Ltd | 芳香族化合物およびそれを用いた有機エレクトロルミネッセンス素子 |
| JP2005268199A (ja) | 2003-07-31 | 2005-09-29 | Mitsubishi Chemicals Corp | 化合物、電荷輸送材料および有機電界発光素子 |
| JP2005298483A (ja) | 2004-03-17 | 2005-10-27 | National Institute Of Advanced Industrial & Technology | イリジウム錯体およびこれを用いた発光材料 |
| JP2006080419A (ja) | 2004-09-13 | 2006-03-23 | Takasago Internatl Corp | イリジウム錯体を含有する発光素子 |
| JP2006089398A (ja) | 2004-09-22 | 2006-04-06 | Takasago Internatl Corp | イリジウム錯体を含有する発光素子 |
| JP2006093542A (ja) | 2004-09-27 | 2006-04-06 | Fuji Photo Film Co Ltd | 発光素子 |
| JP2006128634A (ja) | 2004-09-28 | 2006-05-18 | Fuji Photo Film Co Ltd | 有機電界発光素子 |
| JP2006156888A (ja) | 2004-12-01 | 2006-06-15 | Idemitsu Kosan Co Ltd | 有機電界発光素子 |
| JP2006190718A (ja) | 2004-12-28 | 2006-07-20 | Fuji Photo Film Co Ltd | 有機電界発光素子 |
| JP2006199679A (ja) | 2004-12-24 | 2006-08-03 | Pioneer Electronic Corp | 有機化合物、電荷輸送材料および有機電界発光素子 |
| JP2007027587A (ja) | 2005-07-20 | 2007-02-01 | Chisso Corp | 有機電界発光素子 |
| JP2007088433A (ja) | 2005-08-23 | 2007-04-05 | Mitsubishi Chemicals Corp | 電荷輸送材料、電荷輸送材料組成物及び有機電界発光素子 |
| WO2008147721A1 (en) | 2007-06-01 | 2008-12-04 | E. I. Du Pont De Nemours And Company | Chrysenes for blue luminescent applications |
| WO2010104047A1 (ja) * | 2009-03-11 | 2010-09-16 | 国立大学法人京都大学 | 多環芳香族化合物 |
| WO2012121398A1 (ja) * | 2011-03-10 | 2012-09-13 | 国立大学法人京都大学 | 多環芳香族化合物 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2004028217A1 (ja) * | 2002-09-20 | 2006-01-19 | 出光興産株式会社 | 有機エレクトロルミネッセンス素子 |
| JP2006004721A (ja) * | 2004-06-16 | 2006-01-05 | Fuji Electric Holdings Co Ltd | トップエミッション型有機el素子 |
| JP5799772B2 (ja) * | 2010-11-25 | 2015-10-28 | Jnc株式会社 | 電子輸送材料およびこれを用いた有機電界発光素子 |
| WO2012073541A1 (ja) * | 2010-12-03 | 2012-06-07 | Jnc株式会社 | ピリジンを含む置換基を有するベンゾ[c]カルバゾール化合物および有機電界発光素子 |
| JP5865715B2 (ja) * | 2012-01-25 | 2016-02-17 | 三菱電機株式会社 | モータ制御装置 |
| US9318710B2 (en) * | 2012-07-30 | 2016-04-19 | Universal Display Corporation | Organic electroluminescent materials and devices |
-
2013
- 2013-09-11 KR KR1020157008326A patent/KR102157994B1/ko active IP Right Grant
- 2013-09-11 US US14/386,153 patent/US20150097162A1/en not_active Abandoned
- 2013-09-11 WO PCT/JP2013/074561 patent/WO2014042197A1/ja active Application Filing
- 2013-09-11 CN CN201710301330.8A patent/CN107266481B/zh active Active
- 2013-09-11 EP EP13836272.8A patent/EP2897184A4/en not_active Withdrawn
- 2013-09-11 TW TW102132851A patent/TWI612054B/zh active
- 2013-09-11 CN CN201380047231.1A patent/CN104641483B/zh active Active
- 2013-09-11 JP JP2014535578A patent/JP5819534B2/ja active Active
-
2015
- 2015-05-25 JP JP2015105421A patent/JP6298424B2/ja active Active
- 2015-05-25 JP JP2015105422A patent/JP6393657B2/ja active Active
-
2017
- 2017-10-12 US US15/782,065 patent/US20180047913A1/en not_active Abandoned
-
2019
- 2019-03-14 US US16/354,164 patent/US20190214575A1/en not_active Abandoned
Patent Citations (57)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01245087A (ja) | 1987-12-11 | 1989-09-29 | Idemitsu Kosan Co Ltd | 有機エレクトロルミネッセンス素子 |
| JPH02247278A (ja) | 1989-03-20 | 1990-10-03 | Idemitsu Kosan Co Ltd | エレクトロルミネッセンス素子 |
| JPH06298758A (ja) | 1993-04-16 | 1994-10-25 | Chisso Corp | クマリン誘導体 |
| JPH0812600A (ja) | 1994-04-26 | 1996-01-16 | Tdk Corp | フェニルアントラセン誘導体および有機el素子 |
| JPH10335066A (ja) | 1997-06-02 | 1998-12-18 | Sony Corp | 有機電界発光素子およびこれを用いたフラットパネルディスプレイ |
| JPH1197178A (ja) | 1997-09-24 | 1999-04-09 | Mitsui Chem Inc | 有機電界発光素子 |
| JPH11297473A (ja) | 1998-04-15 | 1999-10-29 | Nec Corp | 有機エレクトロルミネッセンス素子 |
| JP2000026324A (ja) | 1998-07-02 | 2000-01-25 | Mitsui Chemicals Inc | 炭化水素化合物および有機電界発光素子 |
| JP2000034234A (ja) | 1998-07-15 | 2000-02-02 | Mitsui Chemicals Inc | 炭化水素化合物および有機電界発光素子 |
| JP2000133457A (ja) | 1998-10-30 | 2000-05-12 | Mitsui Chemicals Inc | 炭化水素化合物および有機電界発光素子 |
| WO2000040586A1 (fr) | 1999-01-08 | 2000-07-13 | Chisso Corporation | Derives de borane et composes organiques electroluminescents |
| JP2001081090A (ja) | 1999-03-09 | 2001-03-27 | Hayashibara Biochem Lab Inc | ピラン誘導体 |
| JP2001220577A (ja) | 1999-03-09 | 2001-08-14 | Hayashibara Biochem Lab Inc | ピラン誘導体 |
| JP2001052869A (ja) | 1999-08-11 | 2001-02-23 | Hayashibara Biochem Lab Inc | 有機電界発光素子 |
| JP2001076876A (ja) | 1999-09-01 | 2001-03-23 | Hayashibara Biochem Lab Inc | 有機電界発光素子 |
| JP2001172232A (ja) | 1999-12-21 | 2001-06-26 | Univ Osaka | エレクトロルミネッセンス素子 |
| JP2001217077A (ja) | 2000-02-01 | 2001-08-10 | Mitsui Chemicals Inc | 有機電界発光素子 |
| JP2001267076A (ja) | 2000-03-16 | 2001-09-28 | Mitsui Chemicals Inc | 炭化水素化合物および有機電界発光素子 |
| JP2001267075A (ja) | 2000-03-16 | 2001-09-28 | Mitsui Chemicals Inc | 炭化水素化合物および有機電界発光素子 |
| JP2001267078A (ja) | 2000-03-23 | 2001-09-28 | Mitsui Chemicals Inc | 炭化水素化合物および有機電界発光素子 |
| JP2001267079A (ja) | 2000-03-23 | 2001-09-28 | Mitsui Chemicals Inc | 炭化水素化合物および有機電界発光素子 |
| JP2001307884A (ja) | 2000-04-26 | 2001-11-02 | Toray Ind Inc | 発光素子 |
| JP2002234892A (ja) | 2000-11-07 | 2002-08-23 | Hayashibara Biochem Lab Inc | ピラン誘導体 |
| JP2003146951A (ja) | 2001-08-06 | 2003-05-21 | Mitsubishi Chemicals Corp | アントラセン系化合物、その製造方法および有機電界発光素子 |
| JP2003123983A (ja) | 2001-10-10 | 2003-04-25 | Konica Corp | 有機エレクトロルミネッセンス素子 |
| JP2003257621A (ja) | 2002-02-27 | 2003-09-12 | Seiko Epson Corp | 有機el素子とその製造方法ならびに表示装置 |
| WO2003078541A1 (en) | 2002-03-15 | 2003-09-25 | Idemitsu Kosan Co., Ltd. | Material for organic electroluminescent devices and organic electroluminescent devices made by using the same |
| WO2003080760A1 (fr) | 2002-03-22 | 2003-10-02 | Idemitsu Kosan Co., Ltd. | Materiau pour dispositifs electroluminescents organiques et dispositifs electroluminescents organiques produits avec ce materiau |
| JP2003277741A (ja) | 2002-03-26 | 2003-10-02 | Sharp Corp | 有機el発光素子およびそれを用いた液晶表示装置 |
| JP2003321546A (ja) | 2002-04-26 | 2003-11-14 | Nippon Hoso Kyokai <Nhk> | 燐光発光性高分子化合物ならびにこれを用いた発光材料および有機el素子 |
| JP2003347056A (ja) | 2002-05-30 | 2003-12-05 | Fuji Photo Film Co Ltd | 発光素子 |
| JP2004043646A (ja) | 2002-07-11 | 2004-02-12 | Hayashibara Biochem Lab Inc | 有機電界発光素子 |
| JP2004111379A (ja) | 2002-08-29 | 2004-04-08 | Fuji Photo Film Co Ltd | 発光素子及びイリジウム錯体 |
| JP2004119211A (ja) | 2002-09-26 | 2004-04-15 | Toyota Industries Corp | El素子用透明基板及びel装置並びに液晶表示装置 |
| WO2004061047A2 (en) | 2002-12-31 | 2004-07-22 | Eastman Kodak Company | Complex fluorene-containing compounds for use in organic light emitting devices |
| JP2004281086A (ja) | 2003-03-12 | 2004-10-07 | Nippon Hoso Kyokai <Nhk> | フレキシブルフルカラー有機elディスプレイおよびその製造方法 |
| JP2004335122A (ja) | 2003-04-30 | 2004-11-25 | Takasago Internatl Corp | 発光素子 |
| JP2004331508A (ja) | 2003-04-30 | 2004-11-25 | Takasago Internatl Corp | 白金錯体 |
| JP2005268199A (ja) | 2003-07-31 | 2005-09-29 | Mitsubishi Chemicals Corp | 化合物、電荷輸送材料および有機電界発光素子 |
| JP2005097263A (ja) | 2003-08-22 | 2005-04-14 | National Institute Of Advanced Industrial & Technology | 新規イリジウム錯体およびこれを用いた発光材料 |
| JP2005097283A (ja) | 2003-08-29 | 2005-04-14 | Semiconductor Energy Lab Co Ltd | ピラン誘導体とその製造方法、並びにピラン誘導体を用いた発光素子及び発光装置。 |
| JP2005126399A (ja) | 2003-10-27 | 2005-05-19 | Semiconductor Energy Lab Co Ltd | ピラン誘導体 |
| JP2005167175A (ja) | 2003-12-04 | 2005-06-23 | Novaled Gmbh | 有機半導体をキノンジイミン誘導体によってドーピングする方法 |
| JP2005170911A (ja) | 2003-12-15 | 2005-06-30 | Idemitsu Kosan Co Ltd | 芳香族化合物およびそれを用いた有機エレクトロルミネッセンス素子 |
| JP2005298483A (ja) | 2004-03-17 | 2005-10-27 | National Institute Of Advanced Industrial & Technology | イリジウム錯体およびこれを用いた発光材料 |
| JP2006080419A (ja) | 2004-09-13 | 2006-03-23 | Takasago Internatl Corp | イリジウム錯体を含有する発光素子 |
| JP2006089398A (ja) | 2004-09-22 | 2006-04-06 | Takasago Internatl Corp | イリジウム錯体を含有する発光素子 |
| JP2006093542A (ja) | 2004-09-27 | 2006-04-06 | Fuji Photo Film Co Ltd | 発光素子 |
| JP2006128634A (ja) | 2004-09-28 | 2006-05-18 | Fuji Photo Film Co Ltd | 有機電界発光素子 |
| JP2006156888A (ja) | 2004-12-01 | 2006-06-15 | Idemitsu Kosan Co Ltd | 有機電界発光素子 |
| JP2006199679A (ja) | 2004-12-24 | 2006-08-03 | Pioneer Electronic Corp | 有機化合物、電荷輸送材料および有機電界発光素子 |
| JP2006190718A (ja) | 2004-12-28 | 2006-07-20 | Fuji Photo Film Co Ltd | 有機電界発光素子 |
| JP2007027587A (ja) | 2005-07-20 | 2007-02-01 | Chisso Corp | 有機電界発光素子 |
| JP2007088433A (ja) | 2005-08-23 | 2007-04-05 | Mitsubishi Chemicals Corp | 電荷輸送材料、電荷輸送材料組成物及び有機電界発光素子 |
| WO2008147721A1 (en) | 2007-06-01 | 2008-12-04 | E. I. Du Pont De Nemours And Company | Chrysenes for blue luminescent applications |
| WO2010104047A1 (ja) * | 2009-03-11 | 2010-09-16 | 国立大学法人京都大学 | 多環芳香族化合物 |
| WO2012121398A1 (ja) * | 2011-03-10 | 2012-09-13 | 国立大学法人京都大学 | 多環芳香族化合物 |
Non-Patent Citations (7)
| Title |
|---|
| CHEMICAL INDUSTRY, June 2004 (2004-06-01), pages 13 |
| J. AM. CHEM. SOC., vol. 133, 2011, pages 18614 - 18617 |
| J. BLOCHWITZ; M. PHEIFFER; T. FRITZ; K. LEO, APPL. PHYS. LETT., vol. 73, no. 6, 1998, pages 729 - 731 |
| M. PFEIFFER; A. BEYER; T. FRITZ; K. LEO, APPL. PHYS. LETT., vol. 73, no. 22, 1998, pages 3202 - 3204 |
| RUI-HUA XIE ET AL.: "Tuning spectral properties of fullerenes by substitutional doping", PHYSICAL REVIEW B, vol. 69, no. 20, 20 May 2004 (2004-05-20), pages 201403-1 - 201403-4, XP002636359 * |
| See also references of EP2897184A4 |
| TAKUJI HATAKEYAMA ET AL.: "Synthesis of BN- Fused Polycyclic Aromatics via Tandem Intramolecular Electrophilic Arene Borylation", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 133, no. 46, 25 October 2011 (2011-10-25), pages 18614 - 18617, XP055128034 * |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102595325B1 (ko) * | 2015-03-09 | 2023-10-26 | 가꼬우 호징 관세이 가쿠잉 | 다환 방향족 화합물 및 발광층 형성용 조성물 |
| KR20170126888A (ko) * | 2015-03-09 | 2017-11-20 | 가꼬우 호징 관세이 가쿠잉 | 다환 방향족 화합물 및 발광층 형성용 조성물 |
| CN107406759A (zh) * | 2015-03-09 | 2017-11-28 | 学校法人关西学院 | 多环芳香族化合物及发光层形成用组合物 |
| JPWO2016143624A1 (ja) * | 2015-03-09 | 2017-12-21 | 学校法人関西学院 | 多環芳香族化合物および発光層形成用組成物 |
| CN107406759B (zh) * | 2015-03-09 | 2020-10-30 | 学校法人关西学院 | 多环芳香族化合物及发光层形成用组合物与其用途 |
| WO2016143624A1 (ja) * | 2015-03-09 | 2016-09-15 | 学校法人関西学院 | 多環芳香族化合物および発光層形成用組成物 |
| US10418568B2 (en) | 2015-06-01 | 2019-09-17 | Universal Display Corporation | Organic electroluminescent materials and devices |
| US10033004B2 (en) | 2015-06-01 | 2018-07-24 | Universal Display Corporation | Organic electroluminescent materials and devices |
| JP2017028266A (ja) * | 2015-07-21 | 2017-02-02 | ▲いく▼▲雷▼光電科技股▲分▼有限公司 | 有機発光素子 |
| US10243147B2 (en) | 2015-07-21 | 2019-03-26 | E-Ray Optoelectronics Technology Co., Ltd. | Organic light-emitting element |
| KR20180132129A (ko) * | 2016-05-13 | 2018-12-11 | 코니카 미놀타 가부시키가이샤 | 유기 일렉트로루미네센스 소자용 재료, 유기 일렉트로루미네센스 소자, 표시 장치 및 조명 장치 |
| JPWO2017195669A1 (ja) * | 2016-05-13 | 2019-04-04 | コニカミノルタ株式会社 | 有機エレクトロルミネッセンス素子用材料、有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| KR102237305B1 (ko) | 2016-05-13 | 2021-04-06 | 코니카 미놀타 가부시키가이샤 | 유기 일렉트로루미네센스 소자용 재료, 유기 일렉트로루미네센스 소자, 표시 장치 및 조명 장치 |
| WO2017195669A1 (ja) * | 2016-05-13 | 2017-11-16 | コニカミノルタ株式会社 | 有機エレクトロルミネッセンス素子用材料、有機エレクトロルミネッセンス素子、表示装置及び照明装置 |
| US10468609B2 (en) | 2016-06-02 | 2019-11-05 | Universal Display Corporation | Organic electroluminescent materials and devices |
| JP2018125387A (ja) * | 2017-01-31 | 2018-08-09 | 日本放送協会 | 有機エレクトロルミネッセンス素子 |
| US10913758B2 (en) | 2017-03-27 | 2021-02-09 | Samsung Electronics Co., Ltd. | Organometallic compound, organic light-emitting device including the organometallic compound, and diagnostic compound including the organometallic compound |
Also Published As
| Publication number | Publication date |
|---|---|
| TW201412760A (zh) | 2014-04-01 |
| CN104641483A (zh) | 2015-05-20 |
| CN107266481A (zh) | 2017-10-20 |
| CN107266481B (zh) | 2020-04-14 |
| US20180047913A1 (en) | 2018-02-15 |
| KR102157994B1 (ko) | 2020-09-21 |
| JP2015216380A (ja) | 2015-12-03 |
| CN104641483B (zh) | 2017-06-06 |
| EP2897184A4 (en) | 2016-05-11 |
| TWI612054B (zh) | 2018-01-21 |
| JPWO2014042197A1 (ja) | 2016-08-18 |
| JP6298424B2 (ja) | 2018-03-20 |
| JP5819534B2 (ja) | 2015-11-24 |
| KR20150056567A (ko) | 2015-05-26 |
| JP6393657B2 (ja) | 2018-09-19 |
| JP2015188099A (ja) | 2015-10-29 |
| US20150097162A1 (en) | 2015-04-09 |
| US20190214575A1 (en) | 2019-07-11 |
| EP2897184A1 (en) | 2015-07-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6393657B2 (ja) | 有機電界発光素子用材料、有機電界発光素子、表示装置、及び照明装置 | |
| TWI688137B (zh) | 有機電場發光元件、顯示裝置以及照明裝置 | |
| JP5353233B2 (ja) | ピリジルフェニル基を有するアントラセン誘導体化合物及び有機電界発光素子 | |
| JP5556168B2 (ja) | ピリジルナフチル基を有するアントラセン誘導体及び有機電界発光素子 | |
| KR20190116976A (ko) | 유기 전계 발광 소자 | |
| JP5780132B2 (ja) | ベンゾフルオレン化合物、該化合物を用いた発光層用材料および有機電界発光素子 | |
| JP6156389B2 (ja) | ベンゾフルオレン化合物、該化合物を用いた発光層用材料および有機電界発光素子 | |
| TW202017225A (zh) | 有機電場發光元件、顯示裝置以及照明裝置 | |
| WO2013150674A1 (ja) | ベンゾフルオレン化合物、該化合物を用いた発光層用材料および有機電界発光素子 | |
| JP2011037838A (ja) | ベンゾフルオレン化合物、該化合物を用いた発光層用材料および有機電界発光素子 | |
| JP5699581B2 (ja) | 縮合ピロール多環化合物、発光層用材料およびこれを用いた有機電界発光素子 | |
| JP5949779B2 (ja) | ベンゾフルオレン化合物、該化合物を用いた発光層用材料および有機電界発光素子 | |
| JP6221560B2 (ja) | 有機電界発光素子 | |
| WO2013114941A1 (ja) | アントラセン誘導体およびこれを用いた有機電界発光素子 | |
| JP6750623B2 (ja) | アゾリン環含有化合物、これを含有する電子輸送/注入層用材料およびこれを用いた有機電界発光素子 | |
| JP2012094823A (ja) | ピリジルフェニル置換アントラセン化合物および有機電界発光素子 | |
| JP5783173B2 (ja) | 電子受容性窒素含有へテロアリールを含む置換基を有するカルバゾール化合物および有機電界発光素子 | |
| JP7113455B2 (ja) | 有機電界発光素子 | |
| JP5807601B2 (ja) | アントラセン誘導体およびこれを用いた有機電界発光素子 | |
| JP5867269B2 (ja) | ベンゾフルオレン化合物、該化合物を用いた発光層用材料および有機電界発光素子 | |
| JP5949354B2 (ja) | 電子受容性窒素含有へテロアリールを含む置換基を有するカルバゾール化合物および有機電界発光素子 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13836272 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014535578 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14386153 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20157008326 Country of ref document: KR Kind code of ref document: A |