WO2012019496A1 - C-芳基葡萄糖苷衍生物、其制备方法及其在医药上的应用 - Google Patents
C-芳基葡萄糖苷衍生物、其制备方法及其在医药上的应用 Download PDFInfo
- Publication number
- WO2012019496A1 WO2012019496A1 PCT/CN2011/076680 CN2011076680W WO2012019496A1 WO 2012019496 A1 WO2012019496 A1 WO 2012019496A1 CN 2011076680 W CN2011076680 W CN 2011076680W WO 2012019496 A1 WO2012019496 A1 WO 2012019496A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- phenyl
- methyl
- chloro
- mmol
- Prior art date
Links
- 238000002360 preparation method Methods 0.000 title claims abstract description 10
- 229930182478 glucoside Natural products 0.000 title abstract description 6
- 125000001424 substituent group Chemical group 0.000 claims abstract description 42
- 150000003839 salts Chemical class 0.000 claims abstract description 38
- 239000003814 drug Substances 0.000 claims abstract description 13
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 12
- 239000003112 inhibitor Substances 0.000 claims abstract description 6
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 418
- -1 hydroxy, amino Chemical group 0.000 claims description 153
- 150000001875 compounds Chemical class 0.000 claims description 83
- 125000000217 alkyl group Chemical group 0.000 claims description 82
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 79
- 125000003118 aryl group Chemical group 0.000 claims description 78
- 125000001072 heteroaryl group Chemical group 0.000 claims description 67
- 125000000623 heterocyclic group Chemical group 0.000 claims description 65
- 229910052736 halogen Inorganic materials 0.000 claims description 50
- 150000002367 halogens Chemical class 0.000 claims description 49
- 125000003545 alkoxy group Chemical group 0.000 claims description 36
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 33
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 claims description 30
- 125000003342 alkenyl group Chemical group 0.000 claims description 29
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 24
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 23
- 125000005843 halogen group Chemical group 0.000 claims description 20
- 238000000034 method Methods 0.000 claims description 20
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 17
- 206010012601 diabetes mellitus Diseases 0.000 claims description 16
- 238000011282 treatment Methods 0.000 claims description 15
- 125000000000 cycloalkoxy group Chemical group 0.000 claims description 14
- 206010022489 Insulin Resistance Diseases 0.000 claims description 12
- 208000001072 type 2 diabetes mellitus Diseases 0.000 claims description 12
- 150000001733 carboxylic acid esters Chemical class 0.000 claims description 11
- 208000002249 Diabetes Complications Diseases 0.000 claims description 10
- 206010060378 Hyperinsulinaemia Diseases 0.000 claims description 10
- 208000031226 Hyperlipidaemia Diseases 0.000 claims description 10
- 208000008589 Obesity Diseases 0.000 claims description 10
- 230000003451 hyperinsulinaemic effect Effects 0.000 claims description 10
- 201000008980 hyperinsulinism Diseases 0.000 claims description 10
- 229910052757 nitrogen Inorganic materials 0.000 claims description 10
- 235000020824 obesity Nutrition 0.000 claims description 10
- 208000011580 syndromic disease Diseases 0.000 claims description 10
- 201000001320 Atherosclerosis Diseases 0.000 claims description 9
- 208000007342 Diabetic Nephropathies Diseases 0.000 claims description 9
- 208000032131 Diabetic Neuropathies Diseases 0.000 claims description 9
- 206010012655 Diabetic complications Diseases 0.000 claims description 9
- 206010020772 Hypertension Diseases 0.000 claims description 9
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims description 9
- 208000033679 diabetic kidney disease Diseases 0.000 claims description 9
- 235000014113 dietary fatty acids Nutrition 0.000 claims description 9
- 239000000194 fatty acid Substances 0.000 claims description 9
- 229930195729 fatty acid Natural products 0.000 claims description 9
- 150000004665 fatty acids Chemical class 0.000 claims description 9
- 201000001421 hyperglycemia Diseases 0.000 claims description 9
- 206010012689 Diabetic retinopathy Diseases 0.000 claims description 8
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 claims description 8
- 125000003277 amino group Chemical group 0.000 claims description 8
- 201000010099 disease Diseases 0.000 claims description 8
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 8
- 239000011734 sodium Substances 0.000 claims description 8
- 229910052708 sodium Inorganic materials 0.000 claims description 8
- 238000011161 development Methods 0.000 claims description 7
- 125000005842 heteroatom Chemical group 0.000 claims description 7
- 208000006575 hypertriglyceridemia Diseases 0.000 claims description 7
- 102000018711 Facilitative Glucose Transport Proteins Human genes 0.000 claims description 6
- 108091052347 Glucose transporter family Proteins 0.000 claims description 6
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 6
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 4
- 229910052801 chlorine Inorganic materials 0.000 claims description 4
- 230000001419 dependent effect Effects 0.000 claims description 4
- 229910052731 fluorine Inorganic materials 0.000 claims description 4
- 239000003937 drug carrier Substances 0.000 claims description 3
- 125000006239 protecting group Chemical group 0.000 claims description 3
- 239000002253 acid Substances 0.000 claims description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 2
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 claims description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 2
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical group [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 claims 1
- 229940124597 therapeutic agent Drugs 0.000 abstract description 2
- 102100037202 Sodium/myo-inositol cotransporter 2 Human genes 0.000 abstract 2
- 101710090560 Sodium/myo-inositol cotransporter 2 Proteins 0.000 abstract 2
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 361
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 314
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 271
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 222
- 235000019439 ethyl acetate Nutrition 0.000 description 143
- 239000000243 solution Substances 0.000 description 113
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 96
- 238000006243 chemical reaction Methods 0.000 description 90
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 89
- 230000002829 reductive effect Effects 0.000 description 76
- 239000011541 reaction mixture Substances 0.000 description 70
- 239000000203 mixture Substances 0.000 description 65
- 239000007787 solid Substances 0.000 description 54
- 238000005160 1H NMR spectroscopy Methods 0.000 description 51
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 50
- 239000012074 organic phase Substances 0.000 description 50
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 48
- 239000000706 filtrate Substances 0.000 description 46
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 46
- 239000000047 product Substances 0.000 description 43
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 41
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 39
- 239000003921 oil Substances 0.000 description 39
- 235000019198 oils Nutrition 0.000 description 39
- 238000004949 mass spectrometry Methods 0.000 description 35
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 34
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 34
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 33
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 31
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 30
- 238000002955 isolation Methods 0.000 description 28
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 27
- 239000007788 liquid Substances 0.000 description 26
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 24
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 24
- RFFLAFLAYFXFSW-UHFFFAOYSA-N 1,2-dichlorobenzene Chemical compound ClC1=CC=CC=C1Cl RFFLAFLAYFXFSW-UHFFFAOYSA-N 0.000 description 23
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 22
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 21
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 21
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 20
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 20
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 20
- 239000008103 glucose Substances 0.000 description 20
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 20
- 239000001257 hydrogen Substances 0.000 description 19
- 229910052739 hydrogen Inorganic materials 0.000 description 19
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 19
- 239000012046 mixed solvent Substances 0.000 description 18
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 17
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 17
- 229910052799 carbon Inorganic materials 0.000 description 15
- 239000012279 sodium borohydride Substances 0.000 description 15
- 229910000033 sodium borohydride Inorganic materials 0.000 description 15
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 14
- 238000010898 silica gel chromatography Methods 0.000 description 14
- 229910000104 sodium hydride Inorganic materials 0.000 description 14
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 13
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 13
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 13
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 13
- 239000012230 colorless oil Substances 0.000 description 13
- 239000012312 sodium hydride Substances 0.000 description 13
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 12
- 125000003367 polycyclic group Chemical group 0.000 description 12
- 125000006413 ring segment Chemical group 0.000 description 12
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 12
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 11
- 239000008098 formaldehyde solution Substances 0.000 description 11
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 11
- 239000000741 silica gel Substances 0.000 description 11
- 229910002027 silica gel Inorganic materials 0.000 description 11
- OZJPLYNZGCXSJM-UHFFFAOYSA-N 5-valerolactone Chemical compound O=C1CCCCO1 OZJPLYNZGCXSJM-UHFFFAOYSA-N 0.000 description 10
- 108091006269 SLC5A2 Proteins 0.000 description 10
- 102000058081 Sodium-Glucose Transporter 2 Human genes 0.000 description 10
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 10
- 239000012346 acetyl chloride Substances 0.000 description 10
- 125000002619 bicyclic group Chemical group 0.000 description 10
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 10
- 229940098779 methanesulfonic acid Drugs 0.000 description 10
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 10
- 238000000926 separation method Methods 0.000 description 10
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 10
- 150000003573 thiols Chemical class 0.000 description 10
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 9
- 239000008346 aqueous phase Substances 0.000 description 9
- 239000008280 blood Substances 0.000 description 9
- 210000004369 blood Anatomy 0.000 description 9
- 125000004432 carbon atom Chemical group C* 0.000 description 9
- 239000013078 crystal Substances 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- 150000001335 aliphatic alkanes Chemical class 0.000 description 8
- 210000004027 cell Anatomy 0.000 description 8
- 238000001914 filtration Methods 0.000 description 8
- 238000004809 thin layer chromatography Methods 0.000 description 8
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical class [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 7
- 239000007983 Tris buffer Substances 0.000 description 7
- 125000004414 alkyl thio group Chemical group 0.000 description 7
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 7
- 239000010410 layer Substances 0.000 description 7
- WSFSSNUMVMOOMR-BJUDXGSMSA-N methanone Chemical compound O=[11CH2] WSFSSNUMVMOOMR-BJUDXGSMSA-N 0.000 description 7
- 239000011259 mixed solution Substances 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical compound C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 6
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 6
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 6
- 238000005481 NMR spectroscopy Methods 0.000 description 6
- 125000003282 alkyl amino group Chemical group 0.000 description 6
- 238000004440 column chromatography Methods 0.000 description 6
- 125000005366 cycloalkylthio group Chemical group 0.000 description 6
- 238000001035 drying Methods 0.000 description 6
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 6
- 235000019341 magnesium sulphate Nutrition 0.000 description 6
- 125000002950 monocyclic group Chemical group 0.000 description 6
- 229910052760 oxygen Inorganic materials 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- VNGTZLYNGGLPIZ-WCXIOVBPSA-N (3r,4s,5r,6r)-3,4,5-tris(trimethylsilyloxy)-6-(trimethylsilyloxymethyl)oxan-2-one Chemical compound C[Si](C)(C)OC[C@H]1OC(=O)[C@H](O[Si](C)(C)C)[C@@H](O[Si](C)(C)C)[C@@H]1O[Si](C)(C)C VNGTZLYNGGLPIZ-WCXIOVBPSA-N 0.000 description 5
- SQSJADMOVKSAQY-UHFFFAOYSA-N 1-ethoxy-2-fluorobenzene Chemical compound CCOC1=CC=CC=C1F SQSJADMOVKSAQY-UHFFFAOYSA-N 0.000 description 5
- 108091006277 SLC5A1 Proteins 0.000 description 5
- 102000058090 Sodium-Glucose Transporter 1 Human genes 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 5
- 150000001721 carbon Chemical group 0.000 description 5
- 238000005984 hydrogenation reaction Methods 0.000 description 5
- 230000002218 hypoglycaemic effect Effects 0.000 description 5
- 230000000670 limiting effect Effects 0.000 description 5
- 239000001301 oxygen Substances 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 229940121649 protein inhibitor Drugs 0.000 description 5
- 239000012268 protein inhibitor Substances 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- AVOGLGBKOFOSBN-UHFFFAOYSA-N 1-ethoxy-2,3-difluorobenzene Chemical compound CCOC1=CC=CC(F)=C1F AVOGLGBKOFOSBN-UHFFFAOYSA-N 0.000 description 4
- HBEDSQVIWPRPAY-UHFFFAOYSA-N 2,3-dihydrobenzofuran Chemical compound C1=CC=C2OCCC2=C1 HBEDSQVIWPRPAY-UHFFFAOYSA-N 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 4
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 4
- ARJLOKZKUREWCI-UHFFFAOYSA-N OCC1(OC(CCC1)OC)C=O Chemical compound OCC1(OC(CCC1)OC)C=O ARJLOKZKUREWCI-UHFFFAOYSA-N 0.000 description 4
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 4
- 239000012300 argon atmosphere Substances 0.000 description 4
- 239000012141 concentrate Substances 0.000 description 4
- 239000000470 constituent Substances 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 4
- 210000003734 kidney Anatomy 0.000 description 4
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 4
- WAFOSDMKSJGJBX-UHFFFAOYSA-N octane-2,3,4-triol Chemical compound CCCCC(O)C(O)C(C)O WAFOSDMKSJGJBX-UHFFFAOYSA-N 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- BTRYMXMKDSWQEU-UHFFFAOYSA-N (5-bromo-2-chlorophenyl)-(4-ethoxy-3-fluorophenyl)methanone Chemical compound C1=C(F)C(OCC)=CC=C1C(=O)C1=CC(Br)=CC=C1Cl BTRYMXMKDSWQEU-UHFFFAOYSA-N 0.000 description 3
- LEWYCBRUDUVOLG-UHFFFAOYSA-N 1,1,2,2-tetrafluoro-2-(1,1,2,2,3,3,4,4-octafluoro-4-iodobutoxy)ethanesulfonyl fluoride Chemical compound FC(F)(I)C(F)(F)C(F)(F)C(F)(F)OC(F)(F)C(F)(F)S(F)(=O)=O LEWYCBRUDUVOLG-UHFFFAOYSA-N 0.000 description 3
- GGQCXRHQDDSHAE-UHFFFAOYSA-N 4-[(5-bromo-2-chlorophenyl)methyl]-2-fluorophenol Chemical compound C1=C(F)C(O)=CC=C1CC1=CC(Br)=CC=C1Cl GGQCXRHQDDSHAE-UHFFFAOYSA-N 0.000 description 3
- ABWOXAUTADIJBN-UHFFFAOYSA-N 4-bromo-1-chloro-2-[(4-ethoxy-3-fluorophenyl)methyl]benzene Chemical compound C1=C(F)C(OCC)=CC=C1CC1=CC(Br)=CC=C1Cl ABWOXAUTADIJBN-UHFFFAOYSA-N 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- 230000009102 absorption Effects 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 229910052786 argon Inorganic materials 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 239000003480 eluent Substances 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 230000000968 intestinal effect Effects 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 125000001544 thienyl group Chemical group 0.000 description 3
- 125000003944 tolyl group Chemical group 0.000 description 3
- KXGPXDJIBZLZDX-UHFFFAOYSA-N trifluoromethoxybenzene hydrobromide Chemical compound FC(OC1=CC=CC=C1)(F)F.Br KXGPXDJIBZLZDX-UHFFFAOYSA-N 0.000 description 3
- PQDJYEQOELDLCP-UHFFFAOYSA-N trimethylsilane Chemical compound C[SiH](C)C PQDJYEQOELDLCP-UHFFFAOYSA-N 0.000 description 3
- XZPAIXSIFQRATD-UHFFFAOYSA-N (5-bromo-2-chlorophenyl)-(2,3-difluoro-4-methoxyphenyl)methanol Chemical compound FC1=C(F)C(OC)=CC=C1C(O)C1=CC(Br)=CC=C1Cl XZPAIXSIFQRATD-UHFFFAOYSA-N 0.000 description 2
- FXVTVFDIAOTIBG-UHFFFAOYSA-N (5-bromo-2-chlorophenyl)-(2,3-dihydro-1-benzofuran-5-yl)methanol Chemical compound C=1C=C2OCCC2=CC=1C(O)C1=CC(Br)=CC=C1Cl FXVTVFDIAOTIBG-UHFFFAOYSA-N 0.000 description 2
- XYUCCVUAJNUOSY-UHFFFAOYSA-N (5-bromo-2-chlorophenyl)-[5-(4-fluorophenyl)thiophen-2-yl]methanone Chemical compound C1=CC(F)=CC=C1C1=CC=C(C(=O)C=2C(=CC=C(Br)C=2)Cl)S1 XYUCCVUAJNUOSY-UHFFFAOYSA-N 0.000 description 2
- YKYIFUROKBDHCY-ONEGZZNKSA-N (e)-4-ethoxy-1,1,1-trifluorobut-3-en-2-one Chemical group CCO\C=C\C(=O)C(F)(F)F YKYIFUROKBDHCY-ONEGZZNKSA-N 0.000 description 2
- RDOGTTNFVLSBKG-UHFFFAOYSA-N 1,2-difluoro-3-methoxybenzene Chemical compound COC1=CC=CC(F)=C1F RDOGTTNFVLSBKG-UHFFFAOYSA-N 0.000 description 2
- 125000005918 1,2-dimethylbutyl group Chemical group 0.000 description 2
- KRJNOLMPEBOGPS-UHFFFAOYSA-N 1-[(5-bromo-2-chlorophenyl)methyl]-2,3-difluoro-4-methoxybenzene Chemical compound FC1=C(F)C(OC)=CC=C1CC1=CC(Br)=CC=C1Cl KRJNOLMPEBOGPS-UHFFFAOYSA-N 0.000 description 2
- GMMNZXZMQUIXDM-UHFFFAOYSA-N 1-[(5-bromo-2-chlorophenyl)methyl]-4-ethoxy-2,3-difluorobenzene Chemical compound FC1=C(F)C(OCC)=CC=C1CC1=CC(Br)=CC=C1Cl GMMNZXZMQUIXDM-UHFFFAOYSA-N 0.000 description 2
- IANQTJSKSUMEQM-UHFFFAOYSA-N 1-benzofuran Chemical compound C1=CC=C2OC=CC2=C1 IANQTJSKSUMEQM-UHFFFAOYSA-N 0.000 description 2
- SEAOBYFQWJFORM-UHFFFAOYSA-N 1-bromo-4-(trifluoromethoxy)benzene Chemical compound FC(F)(F)OC1=CC=C(Br)C=C1 SEAOBYFQWJFORM-UHFFFAOYSA-N 0.000 description 2
- 125000004793 2,2,2-trifluoroethoxy group Chemical group FC(CO*)(F)F 0.000 description 2
- HOKWHGYUNYFNTP-UHFFFAOYSA-N 2,2-difluoroethoxybenzene Chemical compound FC(F)COC1=CC=CC=C1 HOKWHGYUNYFNTP-UHFFFAOYSA-N 0.000 description 2
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 2
- 125000006176 2-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])*)C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 2
- 125000005916 2-methylpentyl group Chemical group 0.000 description 2
- 125000003542 3-methylbutan-2-yl group Chemical group [H]C([H])([H])C([H])(*)C([H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000005917 3-methylpentyl group Chemical group 0.000 description 2
- UQQPCVZSHCSXAT-UHFFFAOYSA-N 4-bromo-1-chloro-2-[[4-(2,2,2-trifluoroethoxy)phenyl]methyl]benzene Chemical compound C1=CC(OCC(F)(F)F)=CC=C1CC1=CC(Br)=CC=C1Cl UQQPCVZSHCSXAT-UHFFFAOYSA-N 0.000 description 2
- ZDKHNIYNAYJURM-UHFFFAOYSA-N 4-bromo-1-chloro-2-[[4-(trifluoromethoxy)phenyl]methyl]benzene Chemical compound C1=CC(OC(F)(F)F)=CC=C1CC1=CC(Br)=CC=C1Cl ZDKHNIYNAYJURM-UHFFFAOYSA-N 0.000 description 2
- UQLOKDSDWXQUBM-UHFFFAOYSA-N 5-(hydroxymethyl)-7,8-dioxabicyclo[3.2.1]octane-2,3,4-triol Chemical compound O1C2OCC1(CO)C(O)C(O)C2O UQLOKDSDWXQUBM-UHFFFAOYSA-N 0.000 description 2
- YEWZKUKCZHEMSI-UHFFFAOYSA-N 5-[(5-bromo-2-chlorophenyl)methyl]-2,3-dihydro-1-benzofuran Chemical compound ClC1=CC=C(Br)C=C1CC1=CC=C(OCC2)C2=C1 YEWZKUKCZHEMSI-UHFFFAOYSA-N 0.000 description 2
- VUXHGIZYRNNOSE-UHFFFAOYSA-N 5-bromo-2-chloro-n-methoxy-n-methylbenzamide Chemical compound CON(C)C(=O)C1=CC(Br)=CC=C1Cl VUXHGIZYRNNOSE-UHFFFAOYSA-N 0.000 description 2
- FGERXQWKKIVFQG-UHFFFAOYSA-N 5-bromo-2-chlorobenzoic acid Chemical compound OC(=O)C1=CC(Br)=CC=C1Cl FGERXQWKKIVFQG-UHFFFAOYSA-N 0.000 description 2
- TZIQQJRRMJWMDI-UHFFFAOYSA-N 5-bromo-2-chlorobenzoyl chloride Chemical compound ClC(=O)C1=CC(Br)=CC=C1Cl TZIQQJRRMJWMDI-UHFFFAOYSA-N 0.000 description 2
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 2
- 102000004877 Insulin Human genes 0.000 description 2
- 108090001061 Insulin Proteins 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- 235000019502 Orange oil Nutrition 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 208000017442 Retinal disease Diseases 0.000 description 2
- 206010038923 Retinopathy Diseases 0.000 description 2
- 229940100389 Sulfonylurea Drugs 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- 239000003472 antidiabetic agent Substances 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 150000004283 biguanides Chemical class 0.000 description 2
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 2
- QARVLSVVCXYDNA-UHFFFAOYSA-N bromobenzene Chemical compound BrC1=CC=CC=C1 QARVLSVVCXYDNA-UHFFFAOYSA-N 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- NEHMKBQYUWJMIP-UHFFFAOYSA-N chloromethane Chemical compound ClC NEHMKBQYUWJMIP-UHFFFAOYSA-N 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- LFQSCWFLJHTTHZ-LIDOUZCJSA-N ethanol-d6 Chemical compound [2H]OC([2H])([2H])C([2H])([2H])[2H] LFQSCWFLJHTTHZ-LIDOUZCJSA-N 0.000 description 2
- 238000003810 ethyl acetate extraction Methods 0.000 description 2
- 125000004494 ethyl ester group Chemical group 0.000 description 2
- QEWYKACRFQMRMB-UHFFFAOYSA-N fluoroacetic acid Chemical compound OC(=O)CF QEWYKACRFQMRMB-UHFFFAOYSA-N 0.000 description 2
- 125000004785 fluoromethoxy group Chemical group [H]C([H])(F)O* 0.000 description 2
- 230000006377 glucose transport Effects 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N hydrazine group Chemical group NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 229940125396 insulin Drugs 0.000 description 2
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 2
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- HOVAGTYPODGVJG-UHFFFAOYSA-N methyl beta-galactoside Natural products COC1OC(CO)C(O)C(O)C1O HOVAGTYPODGVJG-UHFFFAOYSA-N 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- 239000010502 orange oil Substances 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000003208 petroleum Substances 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 2
- BBEAQIROQSPTKN-UHFFFAOYSA-N pyrene Chemical compound C1=CC=C2C=CC3=CC=CC4=CC=C1C2=C43 BBEAQIROQSPTKN-UHFFFAOYSA-N 0.000 description 2
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 2
- 235000019345 sodium thiosulphate Nutrition 0.000 description 2
- 125000003003 spiro group Chemical group 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 229940126585 therapeutic drug Drugs 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 125000004149 thio group Chemical group *S* 0.000 description 2
- 210000002700 urine Anatomy 0.000 description 2
- WXEVXMHLWGMBSY-UHFFFAOYSA-N (5-bromo-2-chlorophenyl)-(4-ethoxy-2,3-difluorophenyl)methanol Chemical compound FC1=C(F)C(OCC)=CC=C1C(O)C1=CC(Br)=CC=C1Cl WXEVXMHLWGMBSY-UHFFFAOYSA-N 0.000 description 1
- NBXDEJGLPKMAHF-UHFFFAOYSA-N (5-bromo-2-chlorophenyl)-(4-ethoxy-3-fluorophenyl)methanol Chemical compound C1=C(F)C(OCC)=CC=C1C(O)C1=CC(Br)=CC=C1Cl NBXDEJGLPKMAHF-UHFFFAOYSA-N 0.000 description 1
- BRJOSBFTUMJFBU-UHFFFAOYSA-N (5-bromo-2-chlorophenyl)-[4-(2,2-difluoroethoxy)phenyl]methanone Chemical compound C1=CC(OCC(F)F)=CC=C1C(=O)C1=CC(Br)=CC=C1Cl BRJOSBFTUMJFBU-UHFFFAOYSA-N 0.000 description 1
- RTIMCJWREDZAPM-UHFFFAOYSA-N (5-bromo-2-chlorophenyl)-[4-(trifluoromethoxy)phenyl]methanol Chemical compound C=1C(Br)=CC=C(Cl)C=1C(O)C1=CC=C(OC(F)(F)F)C=C1 RTIMCJWREDZAPM-UHFFFAOYSA-N 0.000 description 1
- ZMAUUXSVHSHELR-UHFFFAOYSA-N (5-bromo-2-chlorophenyl)-[4-(trifluoromethoxy)phenyl]methanol (5-bromo-2-chlorophenyl)-[4-(trifluoromethoxy)phenyl]methanone Chemical compound BrC=1C=CC(=C(C1)C(=O)C1=CC=C(C=C1)OC(F)(F)F)Cl.BrC=1C=CC(=C(C1)C(O)C1=CC=C(C=C1)OC(F)(F)F)Cl ZMAUUXSVHSHELR-UHFFFAOYSA-N 0.000 description 1
- NBXDEJGLPKMAHF-RWFJLFJASA-N (5-bromo-2-chlorophenyl)-deuterio-(4-ethoxy-3-fluorophenyl)methanol Chemical compound C=1C(Br)=CC=C(Cl)C=1C(O)([2H])C1=CC=C(OCC)C(F)=C1 NBXDEJGLPKMAHF-RWFJLFJASA-N 0.000 description 1
- ZOBPZXTWZATXDG-UHFFFAOYSA-N 1,3-thiazolidine-2,4-dione Chemical compound O=C1CSC(=O)N1 ZOBPZXTWZATXDG-UHFFFAOYSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 1
- WRWPPGUCZBJXKX-UHFFFAOYSA-N 1-fluoro-4-methylbenzene Chemical compound CC1=CC=C(F)C=C1 WRWPPGUCZBJXKX-UHFFFAOYSA-N 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- VOGSDFLJZPNWHY-UHFFFAOYSA-N 2,2-difluoroethanol Chemical compound OCC(F)F VOGSDFLJZPNWHY-UHFFFAOYSA-N 0.000 description 1
- 125000003562 2,2-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- RPEPGIOVXBBUMJ-UHFFFAOYSA-N 2,3-difluorophenol Chemical compound OC1=CC=CC(F)=C1F RPEPGIOVXBBUMJ-UHFFFAOYSA-N 0.000 description 1
- 125000003660 2,3-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003764 2,4-dimethylpentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- PURJRGMZIKXDMW-UHFFFAOYSA-N 2-(4-fluorophenyl)thiophene Chemical compound C1=CC(F)=CC=C1C1=CC=CS1 PURJRGMZIKXDMW-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- HFHFGHLXUCOHLN-UHFFFAOYSA-N 2-fluorophenol Chemical compound OC1=CC=CC=C1F HFHFGHLXUCOHLN-UHFFFAOYSA-N 0.000 description 1
- 125000003229 2-methylhexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- WXWQJUNPKFWAEX-UHFFFAOYSA-N 2h-pyran-2-carbaldehyde Chemical compound O=CC1OC=CC=C1 WXWQJUNPKFWAEX-UHFFFAOYSA-N 0.000 description 1
- 125000004336 3,3-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- HNNQYHFROJDYHQ-UHFFFAOYSA-N 3-(4-ethylcyclohexyl)propanoic acid 3-(3-ethylcyclopentyl)propanoic acid Chemical compound CCC1CCC(CCC(O)=O)C1.CCC1CCC(CCC(O)=O)CC1 HNNQYHFROJDYHQ-UHFFFAOYSA-N 0.000 description 1
- KLDLRDSRCMJKGM-UHFFFAOYSA-N 3-[chloro-(2-oxo-1,3-oxazolidin-3-yl)phosphoryl]-1,3-oxazolidin-2-one Chemical compound C1COC(=O)N1P(=O)(Cl)N1CCOC1=O KLDLRDSRCMJKGM-UHFFFAOYSA-N 0.000 description 1
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 1
- 125000004080 3-carboxypropanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C(O[H])=O 0.000 description 1
- 125000004337 3-ethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- BIHOEWPVHXJXQM-UHFFFAOYSA-N 3-fluoro-2,2-bis(fluoromethyl)propan-1-ol Chemical compound OCC(CF)(CF)CF BIHOEWPVHXJXQM-UHFFFAOYSA-N 0.000 description 1
- 125000004180 3-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(F)=C1[H] 0.000 description 1
- KFPMLWUKHQMEBU-UHFFFAOYSA-N 3-methylbenzenesulfonyl chloride Chemical compound CC1=CC=CC(S(Cl)(=O)=O)=C1 KFPMLWUKHQMEBU-UHFFFAOYSA-N 0.000 description 1
- 125000003469 3-methylhexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- CSQCYSDEAYXXTN-UHFFFAOYSA-N 4-[(5-bromo-2-chlorophenyl)methyl]phenol Chemical compound C1=CC(O)=CC=C1CC1=CC(Br)=CC=C1Cl CSQCYSDEAYXXTN-UHFFFAOYSA-N 0.000 description 1
- RXWWLXYBRSNHSH-UHFFFAOYSA-N 4-bromo-1-chloro-2-[[4-(2,2-difluoroethoxy)phenyl]methyl]benzene Chemical compound C1=CC(OCC(F)F)=CC=C1CC1=CC(Br)=CC=C1Cl RXWWLXYBRSNHSH-UHFFFAOYSA-N 0.000 description 1
- DYXJGWLSYLPWMO-UHFFFAOYSA-N 6,8-dioxabicyclo[3.2.1]octane Chemical compound C1CCC2COC1O2 DYXJGWLSYLPWMO-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- 208000004998 Abdominal Pain Diseases 0.000 description 1
- 206010000060 Abdominal distension Diseases 0.000 description 1
- 206010067484 Adverse reaction Diseases 0.000 description 1
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 1
- 229940123208 Biguanide Drugs 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- IVPNHKIPGCALCW-UHFFFAOYSA-N BrC=1C=CC(=C(C(=O)Cl)C1)Cl.BrC=1C=CC(=C(C1)C(=O)C1=C(C(=C(C=C1)OCC)F)F)Cl Chemical compound BrC=1C=CC(=C(C(=O)Cl)C1)Cl.BrC=1C=CC(=C(C1)C(=O)C1=C(C(=C(C=C1)OCC)F)F)Cl IVPNHKIPGCALCW-UHFFFAOYSA-N 0.000 description 1
- RZQFOCOXAWQBQR-UHFFFAOYSA-N BrC=1C=CC(=C(C(=O)Cl)C=1)Cl.BrC=1C=CC(=C(C=1)C(=O)C=1C=CC2=C(CCO2)C=1)Cl Chemical compound BrC=1C=CC(=C(C(=O)Cl)C=1)Cl.BrC=1C=CC(=C(C=1)C(=O)C=1C=CC2=C(CCO2)C=1)Cl RZQFOCOXAWQBQR-UHFFFAOYSA-N 0.000 description 1
- HHNSVQSNVHWGDZ-UHFFFAOYSA-N BrC=1C=CC(=C(C1)C(=O)C1=CC(=C(C=C1)OCC)F)Cl.BrC=1C=CC(=C(C1)C(O)C1=CC(=C(C=C1)OCC)F)Cl Chemical compound BrC=1C=CC(=C(C1)C(=O)C1=CC(=C(C=C1)OCC)F)Cl.BrC=1C=CC(=C(C1)C(O)C1=CC(=C(C=C1)OCC)F)Cl HHNSVQSNVHWGDZ-UHFFFAOYSA-N 0.000 description 1
- DBAHUJRFCNAWPK-UHFFFAOYSA-N BrC=1C=CC(=C(C1)C(O)C1=C(C(=C(C=C1)OC)F)F)Cl.BrC=1C=CC(=C(C1)CC1=C(C(=C(C=C1)OC)F)F)Cl Chemical compound BrC=1C=CC(=C(C1)C(O)C1=C(C(=C(C=C1)OC)F)F)Cl.BrC=1C=CC(=C(C1)CC1=C(C(=C(C=C1)OC)F)F)Cl DBAHUJRFCNAWPK-UHFFFAOYSA-N 0.000 description 1
- HEOIHLLFSFXOBC-UHFFFAOYSA-N BrC=1C=CC(=C(C1)C(O)C1=C(C(=C(C=C1)OCC)F)F)Cl.BrC=1C=CC(=C(C1)CC1=C(C(=C(C=C1)OCC)F)F)Cl Chemical compound BrC=1C=CC(=C(C1)C(O)C1=C(C(=C(C=C1)OCC)F)F)Cl.BrC=1C=CC(=C(C1)CC1=C(C(=C(C=C1)OCC)F)F)Cl HEOIHLLFSFXOBC-UHFFFAOYSA-N 0.000 description 1
- ALOZRHQHMDKITR-UHFFFAOYSA-N BrC=1C=CC(=C(C1)CC1=CC(=C(C=C1)OCCCCCCCCCCCCCC)F)Cl Chemical compound BrC=1C=CC(=C(C1)CC1=CC(=C(C=C1)OCCCCCCCCCCCCCC)F)Cl ALOZRHQHMDKITR-UHFFFAOYSA-N 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 108020004635 Complementary DNA Proteins 0.000 description 1
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 1
- 206010012735 Diarrhoea Diseases 0.000 description 1
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- 101000801643 Homo sapiens Retinal-specific phospholipid-transporting ATPase ABCA4 Proteins 0.000 description 1
- 208000013016 Hypoglycemia Diseases 0.000 description 1
- JSBAXHVMHXNCSX-UHFFFAOYSA-N IC=1SC=CC1.FC1=CC=C(C=C1)C=1SC=CC1 Chemical compound IC=1SC=CC1.FC1=CC=C(C=C1)C=1SC=CC1 JSBAXHVMHXNCSX-UHFFFAOYSA-N 0.000 description 1
- 208000001145 Metabolic Syndrome Diseases 0.000 description 1
- JCXJVPUVTGWSNB-UHFFFAOYSA-N Nitrogen dioxide Chemical compound O=[N]=O JCXJVPUVTGWSNB-UHFFFAOYSA-N 0.000 description 1
- ILTSVMJFHXTYHY-QXCZDIPSSA-N OC[C@]1([C@H]([C@@H]([C@H]2O)O)O)O[C@]2(c(cc2Cc(cc3)ccc3OC(F)(F)F)ccc2Cl)O1 Chemical compound OC[C@]1([C@H]([C@@H]([C@H]2O)O)O)O[C@]2(c(cc2Cc(cc3)ccc3OC(F)(F)F)ccc2Cl)O1 ILTSVMJFHXTYHY-QXCZDIPSSA-N 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 102100033617 Retinal-specific phospholipid-transporting ATPase ABCA4 Human genes 0.000 description 1
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- 102000003673 Symporters Human genes 0.000 description 1
- 108090000088 Symporters Proteins 0.000 description 1
- 229940123464 Thiazolidinedione Drugs 0.000 description 1
- CFDZJBMBSJSDDI-FRQUIGDTSA-N [(1r,2r,3s,4s,5s)-5-[4-chloro-3-[(2,3-difluoro-4-methoxyphenyl)methyl]phenyl]-2,3,4-tris(phenylmethoxy)-6,8-dioxabicyclo[3.2.1]octan-1-yl]methanol Chemical compound FC1=C(F)C(OC)=CC=C1CC1=CC([C@@]23O[C@](CO)(CO2)[C@H](OCC=2C=CC=CC=2)[C@H](OCC=2C=CC=CC=2)[C@@H]3OCC=2C=CC=CC=2)=CC=C1Cl CFDZJBMBSJSDDI-FRQUIGDTSA-N 0.000 description 1
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 description 1
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 1
- 239000012445 acidic reagent Substances 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 230000009056 active transport Effects 0.000 description 1
- 230000006838 adverse reaction Effects 0.000 description 1
- 230000004520 agglutination Effects 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 208000024330 bloating Diseases 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- MXOSTENCGSDMRE-UHFFFAOYSA-N butyl-chloro-dimethylsilane Chemical compound CCCC[Si](C)(C)Cl MXOSTENCGSDMRE-UHFFFAOYSA-N 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- PBAYDYUZOSNJGU-UHFFFAOYSA-N chelidonic acid Natural products OC(=O)C1=CC(=O)C=C(C(O)=O)O1 PBAYDYUZOSNJGU-UHFFFAOYSA-N 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 230000001054 cortical effect Effects 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000003678 cyclohexadienyl group Chemical group C1(=CC=CCC1)* 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 125000000131 cyclopropyloxy group Chemical group C1(CC1)O* 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 125000003963 dichloro group Chemical group Cl* 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- CZZYITDELCSZES-UHFFFAOYSA-N diphenylmethane Chemical group C=1C=CC=CC=1CC1=CC=CC=C1 CZZYITDELCSZES-UHFFFAOYSA-N 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000009207 exercise therapy Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- GVEPBJHOBDJJJI-UHFFFAOYSA-N fluoranthrene Natural products C1=CC(C2=CC=CC=C22)=C3C2=CC=CC3=C1 GVEPBJHOBDJJJI-UHFFFAOYSA-N 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 125000006005 fluoroethoxy group Chemical group 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 208000018914 glucose metabolism disease Diseases 0.000 description 1
- 230000002641 glycemic effect Effects 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 1
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 1
- USZLCYNVCCDPLQ-UHFFFAOYSA-N hydron;n-methoxymethanamine;chloride Chemical compound Cl.CNOC USZLCYNVCCDPLQ-UHFFFAOYSA-N 0.000 description 1
- 150000002440 hydroxy compounds Chemical class 0.000 description 1
- 229940126904 hypoglycaemic agent Drugs 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 230000031891 intestinal absorption Effects 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- 208000006443 lactic acidosis Diseases 0.000 description 1
- 238000011866 long-term treatment Methods 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- HOVAGTYPODGVJG-ZFYZTMLRSA-N methyl alpha-D-glucopyranoside Chemical compound CO[C@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O HOVAGTYPODGVJG-ZFYZTMLRSA-N 0.000 description 1
- 229940050176 methyl chloride Drugs 0.000 description 1
- KWKAKUADMBZCLK-UHFFFAOYSA-N methyl heptene Natural products CCCCCCC=C KWKAKUADMBZCLK-UHFFFAOYSA-N 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 210000004165 myocardium Anatomy 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N n-Octanol Natural products CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 201000001119 neuropathy Diseases 0.000 description 1
- 230000007823 neuropathy Effects 0.000 description 1
- 239000003538 oral antidiabetic agent Substances 0.000 description 1
- 229940127209 oral hypoglycaemic agent Drugs 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- 125000001037 p-tolyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 1
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 1
- AZQWKYJCGOJGHM-UHFFFAOYSA-N para-benzoquinone Natural products O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 description 1
- 230000009057 passive transport Effects 0.000 description 1
- 208000033808 peripheral neuropathy Diseases 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- DLRJIFUOBPOJNS-UHFFFAOYSA-N phenetole Chemical compound CCOC1=CC=CC=C1 DLRJIFUOBPOJNS-UHFFFAOYSA-N 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 210000000512 proximal kidney tubule Anatomy 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 230000009103 reabsorption Effects 0.000 description 1
- 230000026304 regulation of glucose transport Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229910000077 silane Inorganic materials 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 229940079827 sodium hydrogen sulfite Drugs 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 238000005063 solubilization Methods 0.000 description 1
- 230000007928 solubilization Effects 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- GQHWSLKNULCZGI-UHFFFAOYSA-N trifluoromethoxybenzene Chemical compound FC(F)(F)OC1=CC=CC=C1 GQHWSLKNULCZGI-UHFFFAOYSA-N 0.000 description 1
- BKZSHWBRUJTUIK-TVFCKZIOSA-N trimethyl-[[(2r,3r,4s,5r)-3,4,5-tris(trimethylsilyloxy)oxan-2-yl]methoxy]silane Chemical compound C[Si](C)(C)OC[C@H]1OC[C@@H](O[Si](C)(C)C)[C@H](O[Si](C)(C)C)[C@@H]1O[Si](C)(C)C BKZSHWBRUJTUIK-TVFCKZIOSA-N 0.000 description 1
- 239000005051 trimethylchlorosilane Substances 0.000 description 1
- 210000004926 tubular epithelial cell Anatomy 0.000 description 1
- 210000003934 vacuole Anatomy 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/04—Heterocyclic radicals containing only oxygen as ring hetero atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/10—Spiro-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/18—Acyclic radicals, substituted by carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/357—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having two or more oxygen atoms in the same ring, e.g. crown ethers, guanadrel
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H7/00—Compounds containing non-saccharide radicals linked to saccharide radicals by a carbon-to-carbon bond
- C07H7/04—Carbocyclic radicals
Definitions
- the present invention relates to a C-aryl glucoside derivative represented by the formula (I), or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof, a process for producing the same, and a pharmaceutical composition containing the same, and It is used as a therapeutic agent, particularly as a sodium-dependent glucose transporter (SGLT) inhibitor.
- SGLT sodium-dependent glucose transporter
- hypoglycemic drugs have been used in clinical treatment, including biguanides, sulfonylureas, insulin resistance improvers, and (X-glucosidase inhibitors, etc., but these drugs have different side effects, Unable to meet the needs of long-term treatment.
- biguanide compounds are prone to cause lactic acidosis; sulfonylureas cause hypoglycemia; insulin resistance improvers are prone to edema and heart failure, and (X-glucosidase inhibition)
- the agent can cause symptoms such as abdominal pain, bloating, diarrhea, etc.
- people are eager to develop a new safe and effective hypoglycemic agent to meet the therapeutic needs of diabetes.
- glucose transporter GLUTs
- SGLTs sodium-dependent glucose co-transporters
- the members of the SGLTs family with glucose transport function are mainly distributed in the proximal tubules of the intestines and kidneys, and it is inferred that they play a key role in the absorption of intestinal glucose and the reuptake of renal glucose, thus It is one of the ideal potential targets for the treatment of diabetes.
- the family member SGLT-1 protein is mainly distributed in the intestinal mucosal cells of the small intestine, and is also expressed in a small amount in the myocardium and kidney. It mainly cooperates with GLUTs protein to regulate the intestinal absorption process of glucose.
- Another member, SGLT-2 is responsible for the regulation of the glucose kidney reuptake process because of its high level of expression in the kidney. That is, the glucose in the urine can actively attach to the renal tubular epithelial cells when it is filtered through the glomerulus. It is re-utilized by SGLT-2 protein transport into the cell. In this process, SGLT-2 is responsible for 90% of the reabsorption process, and the remaining 10% is done by SGLT-1.
- SGLT-2 is the major transporter
- Inhibition of renal glucose reuptake in rats was achieved by inhibiting SGLT-2 mR A levels in rat renal cortical cells using specific SGLT-2 antisense oligonucleotides.
- SGLTs protein inhibitors can be used in the treatment of diabetes-related complications. Such as retinopathy, neuropathy, kidney disease, insulin resistance caused by glucose metabolism disorders, hyperinsulinemia, hyperlipidemia, obesity and the like.
- SGLTs protein inhibitors can also be used in combination with existing therapeutic drugs, such as sulphonic
- amide, thiazolidinedione, metformin and insulin can reduce the dosage of the drug without affecting the efficacy, thereby avoiding or reducing the occurrence of adverse reactions and improving the patient's compliance with the treatment.
- SGLTs protein inhibitors especially SGLT-2 protein inhibitors
- a series of patents relating to the use of C-aryl glucoside and its derivatives as inhibitors of SGLT-2 protein such as CN1989132A, CN1671682A, CN1829729A and WO2010023594A1 have been disclosed, there is still a need to develop therapeutic effects and pharmacokinetic properties. Better, safer compounds are used in the treatment of diabetes and related metabolic disorders.
- the present invention discloses a compound having a structure represented by the general formula (I), and has found that a compound having such a structure exhibits an excellent SGLT-2 protein inhibitory effect and a hypoglycemic action.
- Ring A is selected from aryl or heteroaryl, wherein each aryl or heteroaryl is independently further optionally further selected from one or more selected from the group consisting of halogen, alkyl, alkenyl, block, cycloalkyl, heterocyclyl, aromatic Substituents, heteroaryl, -OR 7 , -S(0) m R ⁇ -C(0)R 7 -C(0)OR 7 -NR 8 R 9 or -C(0)NR 8 R 9 substituent Substituted, wherein the alkyl, cycloalkyl, heterocyclyl, aryl or heteroaryl group may each independently be further optionally further selected from one or more selected from the group consisting of a halogen atom, a halogen, an alkenyl group, a block group, and a nitro group.
- RR 2 , R 3 or R 4 are each independently selected from a hydrogen atom, a halogen, a cyano group, a hydroxyl group, an amino group, an alkyl group, an alkoxy group, a cycloalkyl group, a heterocyclic group, an aryl group or a heteroaryl group, wherein The alkyl, alkoxy, cycloalkyl, heterocyclyl, aryl or heteroaryl groups are each independently optionally further selected from one or more selected from the group consisting of a halogen atom, a halogen, a hydroxyl group, an amino group, an alkyl group, an alkoxy group. Substituted by a substituent of a carboxylic acid or a carboxylic acid ester;
- R 2 and R 3 may be fused to the attached phenyl group to form a ring, which ring is optionally a cycloalkyl group, a heterocyclic group, an aryl group or a heteroaryl group, wherein a cycloalkyl group, a heterocyclic group, or a aryl group Or a heteroaryl group, each of which is optionally independently selected from one or more selected from the group consisting of halogen, hydroxy, amino, alkyl, alkoxy, alkenyl, aryl, cycloalkyl, heterocyclyl, aryl, heteroaryl Substituted by a substituent of a carboxylic acid or a carboxylic acid ester;
- R 1 is selected from a hydrogen atom, d- 4 alkyl group
- ring A cannot be selected from d- 4 alkyl, F, Cl, cyano, hydroxy, -OR N F substituted C 1-2 alkyl, -SC ⁇ R ⁇ 6 cycloalkyl, substituted with 1-2 substituents of C 5-6 saturated heterocyclic group of N, O or S;
- R 5 and R 6 are each independently selected from a hydrogen atom or a halogen atom
- R 7 is selected from a hydrogen atom, a halogen atom, an alkyl group, a cycloalkyl group, a heterocyclic group, an aryl group or a heteroaryl group, wherein the alkyl group, cycloalkyl group, heterocyclic group, aryl group or heteroaryl group may be Each of them is optionally further further selected from one or more selected from the group consisting of a halogen atom, an alkyl group, a halogen, a hydroxyl group, an amino group, an alkoxy group, a cycloalkyl group, a heterocyclic group, a cycloalkoxy group, an aryl group, a heteroaryl group, a carboxy group. Substituted by a substituent of an acid or a carboxylic acid ester;
- R 8 or R 9 are each independently selected from a hydrogen atom, an alkyl group, a cycloalkyl group, a heterocyclic group, an aryl group or a heteroaryl group, wherein said alkyl group, cycloalkyl group, heterocyclic group, aryl group or heteroaryl group
- the groups are each independently optionally further substituted by one or more selected from the group consisting of alkyl, halogen, hydroxy, amino, alkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl, carboxylic acid or carboxylic acid esters. Substituted by
- R 8 or R 9 forms a heterocyclic group with a nitrogen atom to which the ring is bonded, wherein the heterocyclic group contains one or more N, 0 or S(0) M heteroatoms, and the heterocyclic group is Further substituted with one or more substituents selected from alkyl, halogen, hydroxy, alkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl, carboxylic acid or carboxylic acid esters;
- R 10 is a Ci-4 fluorenyl group
- R 11 is selected from CM alkyl, ;
- n 0, 1 or 2.
- a preferred embodiment of the invention a compound of the formula (I), a pharmaceutically acceptable salt thereof or a stereoisomer thereof, including the formula of the formula ( ⁇ )
- a preferred embodiment of the invention a compound of the formula (I), a pharmaceutically acceptable salt thereof or a stereoisomer thereof, wherein:
- Ring A is an aryl group, wherein the aryl group is optionally further further selected from one or more selected from the group consisting of halogen, alkyl, alkenyl, aryl, cycloalkyl, heterocyclyl, aryl, heteroaryl, -OR 7 , S(0) M R 7 , -C(0)R 7 , -C(0)OR 7 , -NR 8 R 9 or -C(0)NR 8 R 9 , wherein the alkyl group, cycloalkyl group Or a heterocyclic group, an aryl group or a heteroaryl group, each independently optionally further selected from one or more selected from the group consisting of a halogen atom, a halogen, an alkenyl group, a aryl group, a nitro group, a cyano group, an alkoxy group, a cycloalkyl group, -OR 7 , -S(0) M R 7 -C(0)R 7 -C(0)OR 7 -NR
- a preferred embodiment of the invention a compound of the formula (I), a pharmaceutically acceptable salt thereof or a stereoisomer thereof, wherein:
- Ring A is an aryl group, wherein the aryl group is optionally further further selected from one or more selected from the group consisting of halogen, alkyl, alkenyl, aryl, cycloalkyl, heterocyclyl, aryl, heteroaryl, -OR 7 , S(0) m R 7 , -C(0)R 7 , -C(0)OR 7 , -NR 8 R 9 or -C(0)NR 8 R 9 , wherein the alkyl group, cycloalkyl group Or a heterocyclic group, an aryl group or a heteroaryl group, each independently optionally further selected from one or more selected from the group consisting of a halogen atom, a halogen, an alkenyl group, a aryl group, a nitro group, a cyano group, an alkoxy group, a cycloalkyl group, -OR 7, -S (0) m R 7 -C (0) R 7 -C (0) o
- R 2 , R 3 or R 4 are each independently a hydrogen atom
- R 1 is a halogen
- a preferred embodiment of the invention a compound of the formula (I), a pharmaceutically acceptable salt thereof or a stereoisomer thereof, wherein:
- Ring A is phenyl, wherein the phenyl group optionally further substituted selected from halogen or ⁇ 5 being substituted with -OR 7;
- R 7 is an alkyl group, wherein the alkyl group may be further substituted with 1 to 3 substituents selected from a halogen atom, a halogen, an alkoxy group or a cycloalkoxy group.
- a preferred embodiment of the invention a compound of the formula (A), a pharmaceutically acceptable salt thereof or a stereoisomer thereof, wherein:
- Ring A is a heteroaryl group, wherein the heteroaryl group may be optionally selected from one or more selected from the group consisting of halogen, alkyl, alkenyl, aryl, cycloalkyl, heterocyclyl, aryl, heteroaryl, -OR 7 -S(0) m R 7 , -C(0)R 7 , -C(0)OR 7 , -NR 8 R 9 or -C(0)NR 8 R 9 , wherein the alkyl group, the ring
- the alkyl, heterocyclyl, aryl or heteroaryl are each independently optionally further selected from one or more selected from the group consisting of a halogen atom, a halogen, an alkenyl group, a aryl group, a nitro group, a cyano group, an alkoxy group, a cycloalkyl group, Substituents of -OR 7 , -S(0) m R 7 , -C(0)R 7 ,
- a preferred embodiment of the invention a compound of the formula (A), a pharmaceutically acceptable salt thereof or a stereoisomer thereof, wherein:
- Ring A is a heteroaryl group, wherein the heteroaryl group is optionally selected from one or more selected from the group consisting of halogen, alkyl, alkenyl, block, cycloalkyl, heterocyclyl, aryl, heteroaryl, -OR 7 , -S(0) m R 7 , -C(0)R 7 , -C(0)OR 7 , -NR 8 R 9 or -C(0)NR 8 R 9 , wherein the alkyl group, naphthenic acid
- the radical, heterocyclyl, aryl or heteroaryl are each independently optionally further selected from one or more selected from the group consisting of a halogen atom, a halogen, an alkenyl group, a aryl group, a nitro group, a cyano group, an alkoxy group, a cycloalkyl group, Substituted by a substituent of OR 7 , -S(0) m R 7 -C(0)R 7 -C(0)
- R 2 , R 3 or R 4 are each independently a hydrogen atom
- R 1 is a halogen
- a preferred embodiment of the invention a compound of the formula (I), a pharmaceutically acceptable salt thereof or a stereoisomer thereof Wherein ring A is Y or thienyl.
- a preferred embodiment of the invention a compound of the formula (I), a pharmaceutically acceptable salt thereof or a stereoisomer thereof, wherein:
- the ring oxime is optionally substituted by one or more substituents selected from aryl halogen or -OR 7 , wherein said aryl group is optionally further substituted with one or more halogens; provided that when ring A is Substituted by a substituent of -OR 7 wherein, when R 7 is d- 4 alkyl, then ring A- is simultaneously substituted with one or more halogens.
- a preferred embodiment of the invention a compound of the formula (I), a pharmaceutically acceptable salt thereof or a stereoisomer thereof, wherein R 5 or R 6 is a halogen atom.
- a preferred embodiment of the invention a compound of the formula (I), a pharmaceutically acceptable salt thereof or a stereoisomer thereof, wherein R 7 is an alkyl group, wherein the alkyl group is further selected from one or more selected from the group consisting of hydrazine Substituted by an atomic substituent.
- the compound of the formula (I) may contain an asymmetric carbon atom and may therefore exist in the form of an optically pure diastereomer, a mixture of diastereomers, a diastereomeric racemate, a mixture of diastereomeric racemates. Or exist as a meso compound.
- the invention includes all of these forms. Mixtures of diastereomeric mixtures, diastereomeric racemates or diastereomeric racemates can be separated by conventional methods, for example by column chromatography, thin layer chromatography and HPLC.
- Preferred compounds of the formula (I) of the present invention include, but are not limited to:
- the invention relates to a compound of the formula (I):
- ⁇ 11 6 and ring A are as defined in the general formula (; I);
- X, Y is a hydroxy protecting group, preferably an alkyl group or a benzyl group.
- the present invention relates to a compound of the formula (I) or a pharmaceutically acceptable salt thereof or all stereoisomers thereof Use in the preparation of a sodium-dependent glucose transporter inhibitor.
- the present invention relates to the use of a compound of the formula (I), or a pharmaceutically acceptable salt thereof, or all stereoisomers thereof, for the manufacture of a medicament for the treatment or delay of the development or onset of the following diseases, wherein
- the disease is selected from the group consisting of diabetes, diabetic retinopathy, diabetic neuropathy, diabetic nephropathy, insulin resistance, hyperglycemia, hyperinsulinemia, elevated levels of fatty acids or glycerol, hyperlipidemia, obesity, high glycerol Triesteremia, X syndrome, diabetic complications or atherosclerosis or hypertension.
- the invention also relates to a treatment for diabetes, diabetic retinopathy, diabetic neuropathy, diabetic nephropathy, insulin resistance, hyperglycemia, hyperinsulinemia, elevated levels of fatty acids or glycerol, hyperlipidemia, obesity a method of hypertriglyceridemia, X syndrome, diabetic complications, or atherosclerosis or hypertension, the method comprising administering to a patient in need of treatment a therapeutically effective amount of a compound of formula (I) or A pharmaceutically acceptable salt or a stereoisomer thereof.
- the present invention also relates to a compound of the above formula (I), or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof, as a medicament for treating or delaying the development or onset of a disease selected from the group consisting of diabetes and diabetes Retinopathy, diabetic neuropathy, diabetic nephropathy, insulin resistance, hyperglycemia, hyperinsulinemia, elevated levels of fatty acids or glycerol, hyperlipidemia, obesity, hypertriglyceridemia, X syndrome , diabetic complications or atherosclerosis or high blood pressure.
- a disease selected from the group consisting of diabetes and diabetes Retinopathy, diabetic neuropathy, diabetic nephropathy, insulin resistance, hyperglycemia, hyperinsulinemia, elevated levels of fatty acids or glycerol, hyperlipidemia, obesity, hypertriglyceridemia, X syndrome , diabetic complications or atherosclerosis or high blood pressure.
- the present invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising an effective amount of a formula
- composition for the preparation of a medicament for treating or delaying the development or onset of a disease selected from the group consisting of diabetes, diabetic retinopathy, diabetic neuropathy, diabetic nephropathy, insulin resistance, hyperglycemia , hyperinsulinemia, elevated levels of fatty acids or glycerol, hyperlipidemia, obesity, hypertriglyceridemia, X syndrome, diabetic complications or atherosclerosis or hypertension.
- a disease selected from the group consisting of diabetes, diabetic retinopathy, diabetic neuropathy, diabetic nephropathy, insulin resistance, hyperglycemia , hyperinsulinemia, elevated levels of fatty acids or glycerol, hyperlipidemia, obesity, hypertriglyceridemia, X syndrome, diabetic complications or atherosclerosis or hypertension.
- the invention also relates to a treatment for diabetes, diabetic retinopathy, diabetic neuropathy, diabetic nephropathy, insulin resistance, hyperglycemia, hyperinsulinemia, elevated levels of fatty acids or glycerol, hyperlipidemia, obesity
- a method of hypertriglyceridemia, X syndrome, diabetic complications, or atherosclerosis or hypertension comprising administering to a patient in need of treatment a therapeutically effective amount of a pharmaceutical composition comprising the formula (I) Things.
- the present invention also relates to a general formula of a medicament for treating or delaying the development or onset of the following diseases
- composition wherein the disease is selected from the group consisting of diabetes, diabetic retinopathy, diabetic neuropathy, diabetic nephropathy, insulin resistance, hyperglycemia, hyperinsulinemia, elevation of fatty acids or glycerol Level, hyperlipidemia, obesity, hypertriglyceridemia, X syndrome, diabetic complications or atherosclerosis or hypertension.
- diabetes diabetic retinopathy, diabetic neuropathy, diabetic nephropathy, insulin resistance, hyperglycemia, hyperinsulinemia, elevation of fatty acids or glycerol Level
- hyperlipidemia obesity, hypertriglyceridemia, X syndrome, diabetic complications or atherosclerosis or hypertension.
- 'Alkyl means a saturated aliphatic hydrocarbon group, including straight-chain and branched-chain groups of 1 to 20 carbon atoms.
- An alkyl group of 1 to 12 carbon atoms non-limiting examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, sec-butyl, n-pentyl, U-dimethylpropyl, 1,2-dimethylpropyl, 2,2-dimethylpropyl, 1-ethylpropyl, 2-methylbutyl, 3-methylbutyl, hexyl Base, 1-ethyl-2-methylpropyl, 1,1,2-trimethylpropyl, 1,1-dimethylbutyl, 1,2-dimethylbutyl, 2,2- Dimethylbutyl, 1,3-dimethylbutyl, 2-ethylbutyl, 2-methylpentyl, 3-methylpentyl, 4-methylp
- lower alkyl groups having 1 to 6 carbon atoms More preferred are lower alkyl groups having 1 to 6 carbon atoms, and non-limiting examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, t-butyl, sec-butyl Base, n-pentyl, 1,1-dimethylpropyl, 1,2-dimethylpropyl, 2,2-dimethylpropyl, 1-ethylpropyl, 2-methylbutyl, 3-methylbutyl, n-hexyl, 1-ethyl-2-methylpropyl, 1,1,2-trimethylpropyl, 1,1-dimethylbutyl, 1,2-dimethyl Butyl, 2,2-dimethylbutyl, 1,3-dimethylbutyl, 2-ethylbutyl, 2-methylpentyl, 3-methylpentyl, 4-methylpentyl Base, 2,3-dimethylbutyl and the like.
- the alkyl group may be substituted or unsubstituted, and when substituted, the substituent may be substituted at any available point of attachment, preferably one or more of the following groups, independently selected from alkyl, alkenyl, Block group, alkoxy group, alkylthio group, alkylamino group, halogen, thiol, hydroxy group, nitro group, cyano group, cycloalkyl group, heterocycloalkyl group, aryl group, heteroaryl group, cycloalkoxy group, hetero Cycloalkoxy, cycloalkylthio, heterocycloalkylthio, oxo, -OR 7 , -S(0) m R 7 , -C(0)R 7 , -C(0)OR 7 , -NR 8 R 9 or -C(0)NR 8 R 9 .
- Cycloalkyl means a saturated or partially unsaturated monocyclic or polycyclic cyclic hydrocarbon substituent comprising from 3 to 20 carbon atoms, preferably from 3 to 12 carbon atoms, more preferably the cycloalkyl ring comprises from 3 to 10 One carbon atom.
- monocyclic cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, cyclohexadienyl, cycloheptyl, cycloheptatriene Alkenyl, cyclooctyl and the like.
- Polycyclic cycloalkyl groups include spiro, fused, and bridged cycloalkyl groups.
- Spirocycloalkyl means a polycyclic group of 5 to 20 members which shares a carbon atom (referred to as a spiro atom) between the monocyclic rings. These may contain one or more double bonds, but none of the rings are fully conjugated. ⁇ electronic system. It is preferably 6 to 14 members, more preferably 7 to 10 members.
- the spirocycloalkyl group is classified into a monospirocycloalkyl group, a bispirocycloalkyl group or a polyspirocycloalkyl group, preferably a monospirocycloalkyl group and a bispirocycloalkyl group, depending on the number of common spiro atoms between the ring and the ring.
- “Fused cycloalkyl” refers to 5 to 20 members, and each ring in the system shares an adjacent pair with other rings in the system.
- Bridge cycloalkyl means 5 to 20 members, any two rings sharing two carbon-free all-carbon polycyclic groups, which may contain one or more double bonds, but none of the rings have a total The ⁇ -electron system of the yoke. It is preferably 6 to 14 members, more preferably 7 to 10 members. Depending on the number of constituent rings, it may be classified into a bicyclic, tricyclic, tetracyclic or polycyclic bridged cycloalkyl group, preferably a bicyclic ring, a tricyclic ring or a tetracyclic ring, and more preferably a bicyclic ring or a tricyclic ring. Bridged cycloalkyl
- the cycloalkyl ring may be fused to an aryl, heteroaryl or heterocycloalkyl ring, wherein the ring to which the parent structure is attached is a cycloalkyl group, non-limiting examples include indanyl, tetrahydrogen Naphthyl, benzocycloheptyl and the like.
- the cycloalkyl group may be optionally substituted or unsubstituted, and when substituted, the substituent is preferably one or more of the following groups independently selected from the group consisting of alkyl, alkenyl, block, alkoxy, alkylthio Base, alkylamino, halogen, thiol, hydroxy, nitro, cyano, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, cycloalkoxy, heterocycloalkoxy, cycloalkylthio ,heterocycloalkylthio,oxo, -OR 7 , -S(0) m R 7 -C(0)R 7 -C(0)OR 7 -NR 8 R 9 or -C(0)NR 8 R 9 .
- alkenyl refers to an alkyl group as defined above consisting of at least two carbon atoms and at least one carbon-carbon double bond. For example, vinyl, 1-propenyl, 2-propenyl, 1-, 2- or 3-butenyl, and the like.
- the alkenyl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more of the following groups, independently selected from the group consisting of alkyl, alkenyl, block, alkoxy, alkylthio, alkane.
- Block group refers to an alkyl group as defined above consisting of at least two carbon atoms and at least one carbon-carbon triple bond. For example, an ethyl group, a 1-propyl block group, a 2-propyl block group, a 1-, 2- or 3-butyl block group, and the like.
- the block group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more of the following groups, independently selected from the group consisting of alkyl, alkenyl, block, alkoxy, alkylthio, alkane.
- Heterocyclyl means a saturated or partially unsaturated monocyclic or polycyclic cyclic hydrocarbon substituent comprising from 3 to 20 ring atoms wherein one or more of the ring atoms are selected from nitrogen, oxygen or S(0) m ( Wherein m is a hetero atom of the integer 0 to 2), but does not include a ring moiety of -0-0-, -0-S- or -SS-, and the remaining ring atoms are carbon. It preferably comprises from 3 to 12 ring atoms, wherein from 1 to 4 are heteroatoms, more preferably the cycloalkyl ring contains from 3 to 10 ring atoms.
- Non-limiting examples of monocyclic cycloalkyl groups include pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, thiomorpholinyl, homopiperazinyl and the like.
- Polycyclic cycloalkyl groups include spiro, fused, and bridged heterocyclic groups.
- spiroheterocyclyl means a polycyclic heterocyclic group of 5 to 20 members in which one atom (called a spiro atom) is shared between monocyclic rings, wherein one or more ring atoms are selected from nitrogen, oxygen or S(0) p The hetero atom (where p is an integer from 0 to 2) and the remaining ring atoms are carbon.
- the spirocycloalkyl group is classified into a monospiroheterocyclic group, a dispiroheterocyclic group or a polyspirocyclic group according to the number of common spiro atoms between the rings, preferably a monospirocycloalkyl group and a bispirocycloalkyl group.
- spirocycloalkyl groups include
- “Fused heterocyclic group” means 5 to 20 members, each ring in the system shares an adjacent pair of atomic polycyclic heterocyclic groups with other rings in the system, and one or more rings may contain one or more a bond, but none of the rings have a fully conjugated ⁇ -electron system in which one or more ring atoms are selected from nitrogen, oxygen or S(0) p (where p is an integer from 0 to 2), and the remaining ring atoms are carbon. It is preferably 6 to 14 members, more preferably 7 to 10 members.
- fused heterocyclic groups include
- “Bridge heterocyclyl” refers to a polycyclic heterocyclic group of 5 to 14 members, and any two rings share two atoms which are not directly bonded. Groups, these may contain one or more double bonds, but none of the rings have a fully conjugated ⁇ -electron system in which one or more ring atoms are selected from nitrogen, oxygen or S(0) m (where m is an integer 0 to 2) Heteroatoms, the remaining ring atoms are carbon. It is preferably 6 to 14 members, more preferably 7 to 10 members. 7 to 10 yuan.
- bridged cycloalkyl groups include:
- the heterocyclyl ring may be fused to an aryl, heteroaryl or cycloalkyl ring wherein the ring to which the parent structure is attached is a heterocyclic group, non-limiting examples comprising:
- the heterocyclic group may be optionally substituted or unsubstituted, and when substituted, the substituent is preferably one or more of the following groups, independently selected from the group consisting of alkyl, alkenyl, block, alkoxy, alkylthio Base, alkylamino, halogen, thiol, hydroxy, nitro, cyano, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, cycloalkoxy, heterocycloalkoxy, cycloalkylthio , heterocycloalkylthio, oxo, -OR 7 , -S(0) m R 7 -C(0)R 7 -C(0)OR 7 -NR 8 R 9 or -C(0)NR 8 R 9 .
- Aryl means a 6 to 14 membered all-carbon monocyclic or fused polycyclic ring (ie, a ring that shares a pair of adjacent carbon atoms), a polycyclic ring having a conjugated ⁇ -electron system (ie, having adjacent pairs)
- the ring of a carbon atom is preferably 6 to 10 members such as a phenyl group and a naphthyl group.
- the aryl ring may be fused to a heteroaryl, heterocyclyl or cycloalkyl ring, wherein the ring to which the parent structure is attached is an aryl ring, non-limiting examples comprising:
- the aryl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more of the following groups, independently selected from the group consisting of alkyl, alkenyl, block, alkoxy, alkylthio, alkane.
- Heteroaryl means a heteroaromatic system containing from 1 to 4 heteroatoms, from 5 to 14 ring atoms, wherein the heterogeneous Subsoil includes oxygen, sulfur and nitrogen. It is preferably 5 to 10 yuan.
- the heteroaryl group is preferably 5- or 6-membered, such as furyl, thienyl, pyridyl, pyrrolyl, N-alkylpyrrolyl, pyrimidinyl, pyrazinyl, imidazolyl, tetrazolyl, and the like.
- the heteroaryl ring can be fused to an aryl, heterocyclyl or cycloalkyl ring, wherein the parent structure is attached:
- the heteroaryl group may be optionally substituted or unsubstituted, and when substituted, the substituent is preferably one or more of the following groups independently selected from the group consisting of alkyl, alkenyl, block, alkoxy, alkylthio Base, alkylamino, halogen, thiol, hydroxy, nitro, cyano, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, cycloalkoxy, heterocycloalkoxy, cycloalkylthio ,heterocycloalkylthio,oxo, -OR 7 , -S(0) m R 7 -C(0)R 7 -C(0)OR 7 -NR 8 R 9 or -C(0)NR 8 R 9 .
- Alkoxy means -O-(indenyl), wherein alkyl is as defined above. Non-limiting examples include methoxy, ethoxy, propoxy, butoxy, and the like.
- the alkoxy group may be optionally substituted or unsubstituted, and when substituted, the substituent is preferably one or more of the following groups, independently selected from the group consisting of an alkyl group, an alkenyl group, a block group, an alkoxy group, and an alkane group.
- Cycloalkoxy refers to -O-(unsubstituted cycloalkyl), wherein cycloalkyl is as defined above. Non-limiting examples include cyclopropoxy, cyclobutoxy, cyclopentyloxy, cyclohexyloxy, and the like.
- the alkoxy group may be optionally substituted or unsubstituted, and when substituted, the substituent is preferably one or more of the following groups, independently selected from the group consisting of an alkyl group, an alkenyl group, a block group, an alkoxy group, and an alkane group.
- Haldroxy means an -OH group.
- Halogen means fluoro, chloro, bromo or iodo.
- Amino means -NH 2 .
- Neitro means -N0 2 .
- Network group means -CH 2 -phenyl.
- Carboxylic acid means -C(0)OH.
- Carboxylic ester means -C(O)0(alkyl) or (cycloalkyl). r
- heterocyclic group optionally substituted by an alkyl group means that an alkyl group may be, but not necessarily, present, including the case where the heterocyclic group is substituted by an alkyl group and the case where the heterocyclic group is not substituted by an alkyl group.
- Substituted means one or more hydrogen atoms in the group, preferably up to 5, more preferably 1 to 3, hydrogen atoms are independently substituted with each other by a corresponding number of substituents. It goes without saying that the substituents are only in their possible chemical positions, and those skilled in the art will be able to determine (by experiment or theory) substitutions that may or may not be possible without undue effort. For example, an amino group or a hydroxyl group having a free hydrogen may be unstable when combined with a carbon atom having an unsaturated (e.g., olefinic) bond.
- “Pharmaceutical composition” means a mixture containing one or more of the compounds described herein, or a physiologically/pharmaceutically acceptable salt or prodrug thereof, and other chemical components, as well as other components such as physiological/pharmaceutically acceptable carriers. And excipients.
- the purpose of the pharmaceutical composition is to promote administration to an organism, to facilitate absorption of the active ingredient and to exert biological activity.
- the preparation method of the compound of the formula (I) or a pharmaceutically acceptable salt thereof or all the stereoisomers thereof of the present invention comprises the following steps:
- R ⁇ R 6 is as defined in the general formula (I);
- X and Y are a protecting group for a hydroxyl group, preferably an alkyl group or a benzyl group. detailed description
- the structure of the compound is determined by nuclear magnetic resonance (NMR) or/and mass spectrometry (MS). NMR shifts ([delta]) are given in units of 10- 6 (ppm) a.
- the NMR was measured by a Bruker AVANCE-400 nuclear magnetic apparatus, and the solvent was deuterated dimethyl sulfoxide deuterated chloroform (CDC1 3 ), deuterated methanol (CD 3 OD), and the internal standard was tetramethylsilane (TMS).
- the MS was assayed using a FINMGAN LCQAd (ESI) mass spectrometer (manufacturer: Thermo, model: Finnigan LCQ advantage MAX).
- ESI FINMGAN LCQAd
- HPLC HPLC was determined using an Agilent 1200 DAD high pressure liquid chromatograph (Sunfire C18 150 x 4.6 mm) Column) and Waters 2695-2996 high pressure liquid chromatograph (Gimini C18 150 x 4.6 mm column).
- the thin layer chromatography silica gel plate uses Yantai Yellow Sea HSGF254 or Qingdao GF254 silica gel plate.
- the silica gel plate used for thin layer chromatography (TLC) has a specification of 0.15 mm ⁇ 0.2 mm, and the thin layer chromatography separation and purification product adopts a specification of 0.4 mm. ⁇ 0.5 mm.
- the known starting materials of the present invention may be synthesized by or according to methods known in the art, or may be purchased from ABCR GmbH & Co. KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc, Companies such as Dare Chemicals.
- An argon atmosphere or a nitrogen atmosphere means that the reaction flask is connected to an argon or nitrogen balloon having a volume of about 1 L.
- the hydrogen atmosphere means that the reaction flask is connected to a hydrogen balloon of about 1 L volume.
- the pressurized hydrogenation reaction was carried out using a Parr Model 3916EKX hydrogenation apparatus and a clear blue QL-500 type hydrogen generator or a HC2-SS type hydrogenation apparatus.
- the hydrogenation reaction is usually evacuated, charged with hydrogen, and operated three times.
- the microwave reaction was carried out using a CEM Discover-S Model 908860 microwave reactor.
- reaction is carried out under a nitrogen atmosphere or an argon atmosphere.
- the solution means an aqueous solution.
- reaction temperature is room temperature.
- the optimum temperature for the reaction at room temperature is from 20 ° C to 30 ° C.
- the reaction progress in the examples was monitored by thin layer chromatography (TLC).
- TLC thin layer chromatography
- the system used for the reaction was: dichloromethane and methanol systems, n-hexane and ethyl acetate systems, and the volume ratio of the solvent was based on the polar group of the compound. Adjust for sex.
- the column chromatography eluent system and the thin layer chromatography developer system include: A: dichloromethane and methanol systems, B: n-hexane and ethyl acetate systems, the volume ratio of the solvent according to the compound
- the polarity is adjusted to adjust, and a small amount of an alkaline or acidic reagent such as triethylamine or acetic acid may be added for adjustment.
- A dichloromethane and methanol systems
- B n-hexane and ethyl acetate systems
- the polarity is adjusted to adjust, and a small amount of an alkaline or acidic reagent such as triethylamine or acetic acid may be added for adjustment.
- an alkaline or acidic reagent such as triethylamine or acetic acid
- the oxalyl chloride was dissolved in 5 mL of dichloromethane, cooled to -78 ° C, and 3 mL of dimethyl sulfoxide (0.26 mL, 3.59 mmol) in dichloromethane was added dropwise, and reacted for 15 minutes, and 5 mL was added dropwise.
- 2,3-Difluorophenol 3a (4 g, 30.7 mmol) was dissolved in 60 mL of acetone, and potassium carbonate was added in sequence.
- Oxalyl chloride (0.37 mL, 4.37 mmol) was dissolved in 20 mL of dichloromethane, cooled to -78 ° C, and then a solution of B 10 mL dimethyl sulfoxide (0.5 mL, 7.05 mmol) in dichloromethane.
- EtOAc EtOAc m. 4S,5 6R-6-[(tert-Butyl(dimethyl)silyl)oxymethyl]-2-[4-chloro-3-[(4-ethoxy-3-fluoro-phenyl) Methyl]phenyl]-2-methoxy-tetrahydropyran-3,4,5-triol 4 g (7.14 g, colorless oil) was used in the next step without isolation.
- Oxalyl chloride (1.17 mL, 13.6 mmol) was dissolved in 20 mL of dichloromethane, cooled to -78 °C, then 20 mL of dimethyl sulfoxide (1.56 mL, 21.9 mmol) in dichloromethane and 50 mL [(2R,3R,4S,5R,6-3,4,5-tribenzyloxy-6-[4-chloro-3-[(4-ethoxy-3-fluoro-phenyl)methyl]] a solution of phenyl]-6-methoxy-tetrahydropyran-2-yl]methanol 4i (7.6 g, 10.45 mmol) in dichloromethane, mp.
- EtOAc EtOAc
- EtOAcjjjjjjjj 6-[4-Chloro-3-[(4-ethoxy-3-fluoro-phenyl)methyl]phenyl]-2-(hydroxymethyl)-6-methoxy-tetrahydropyran- 2-Formaldehyde 4k (7.9 g, colorless oil) was used in the next step without isolation.
- the reaction mixture was evaporatedjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjj 6 -3,4,5-tribenzyloxy-6-[4-chloro-3-(2,3-dihydrobenzofuran-5-ylmethyl)phenyl]-6-methoxy-tetra Hydropyran-2-yl]methanol 5h (9.0 g, yellow liquid yield: 88.1%.
- N-Methyl-N-methoxyamine hydrochloride (3.41 g, 35 mmol) was dissolved in 140 mL of dichloromethane, and then triethylamine (14.6 mL, 105 mmol) was added and reacted for 10 minutes, and then added 5 Bromo-2-chloro-benzoic acid 6a (8.24 g, 35 mmol) and bis(2-oxo-3-oxazolidinyl)phosphoryl chloride (10.69 g, 42 mmol) were reacted for 16 hours.
- Trifluoromethoxybenzene 6c (11.35 g, 70 mmol) was dissolved in 3.57 mL of liquid bromine, and 0.24 g of iron was added thereto, and the mixture was reacted at 100 ° C for 16 hours. 450 mL of dichloromethane was added, and the organic phase was washed successively with 6 M hydrochloric acid (140 mL), 10% sodium hydrogensulfite solution (140 mL) and saturated sodium chloride solution (140 mL). Drying over anhydrous magnesium sulfate, EtOAc ⁇
- Bromo-2-chloro-phenyl)-[4-(trifluoromethoxy)phenyl]methanone 5-bromo-2-chloro-N-methoxy-N-methyl-benzamide 6b ( 5.16 g, 18.5 mmol) was dissolved in 50 mL of tetrahydrofuran, and 1-bromomagnesium bromide-4-(trifluoromethoxy)benzene 6e (13.21 g, 49.8 mmol) was added dropwise.
- the reaction mixture was evaporatedjjjjjjjjjjj -3,4,5-tribenzyloxy-6-[4-chloro-3-[[4-(trifluoromethoxy)phenyl]methyl]phenyl]-6-methoxy-tetrahydro Pyran-2-yl]methanol 6 m (1.22 g, pale yellow oil), yield: 60.7 %.
- Oxalyl chloride (0.21 mL, 2.45 mmol) was dissolved in 5 mL of dichloromethane, cooled to -78 ° C, and 5 mL of dimethyl sulfoxide (0.24 mL, 3.26 mmol) in dichloromethane was added dropwise for 15 min.
- Oxalyl chloride C 0.16 mL, 1.87 mmol) was dissolved in 5 mL of dichloromethane, cooled to -78 ° C, and 3 mL of dimethyl sulfoxide (0.2 mL, 2.88 mmol) in dichloromethane was added dropwise.
- reaction solution was concentrated under reduced pressure, and then crystallised from petroleum ether, filtered, filtered, and filtered, and then taken in 50 ml of methanol, and added to 2 mL of hydrogen peroxide. After stirring for 1 minute, 5 g of sodium thiosulfate, 40 mL of water and 50 mL of acetic acid were added. Ethyl ester, liquid separation, water phase with B Ethyl acetate extraction (30 mL ⁇ 3), the organic phase was washed with a saturated sodium chloride solution (30 mL), and the organic phase was combined, dried over anhydrous magnesium sulfate, filtered and evaporated.
- Oxalyl chloride (0.34 mL, 3.93 mmol) was dissolved in 5 mL of dichloromethane, cooled to -78 ° C, and 10 mL of dimethyl sulfoxide (0.43 mL, 6.04 mmol) in dichloromethane was added dropwise for 15 min.
- Oxidyl chloride C 0.27 mL, 3.12 mmol) was dissolved in 8 mL of dichloromethane, cooled to -78 ° C, and 4 mL of dimethyl sulfoxide (0.36 mL, 5.04 mmol) in dichloromethane was added dropwise.
- Benzyl bromide (1.90 mL, 14 mmol) was reacted for 3 hours. Add 5 mL of methanol and 100 mL of water, and extract with ethyl acetate (50 mL ⁇ 3). The organic phase is washed with saturated sodium chloride (30 mL).
- Oxalyl chloride (0.36 mL, 3.93 mmol) was dissolved in 5 mL of dichloromethane, cooled to -78 ° C, and 10 mL of dimethyl sulfoxide (0.35 mL, 4.92 mmol) in dichloromethane was added dropwise for 15 min.
- EtOAc EtOAc m. 4-(1,1,2,2,2-pentamethoxy)phenyl]methyl]phenyl]-6-methoxy-tetrahydropyran-2-carbaldehyde 10f (1.2 g, Yellow liquid), used directly in the next reaction without separation.
- Oxalyl chloride (0.3 mL, 3.57 mmol) was dissolved in 20 mL of dichloromethane, cooled to -78 ° C, then 10 mL of dimethyl sulfoxide (0.39 mL, 5.48 mmol) in dichloromethane and 15 mL [(2R,3R,4S,5R,6 -3,4,5-tribenzyloxy-6-[4-chloro-3-[di-deutero-(4-ethoxy-3-fluoro-benzene) a solution of methyl]phenyl]-6-methoxy-tetrahydropyran-2-yl]methanol llf (2 g, 2.74 mmol) in methylene chloride, react at -78 ° C for 30 min, add three Ethylamine (1.9 mL, 13.7 mmol), react at room temperature for 2 hours.
- Inoculate SGLT1 or SGLT2 transient transfectants in 96-well plates prepared according to the existing literature "Dibetes, 57, 1723-1729, 2008", in which the cDNAs for SGLT1 and SGLT2 were purchased from Origene), with a cell density of 1-1.5. ⁇ 10 4 .
- the cells were cultured for 48 hours at 37 ° C, 5 % CO 2 , and washed twice with 200 ⁇ L of sodium-free buffer; 90 sodium-containing buffers containing different concentrations of the test compound were added to the wells, each test compound Make 3 duplicate wells at the corresponding concentration.
- Methyl ⁇ -D-glucopyranoside (methyl ⁇ -D-glucopyranoside) was added to each well of a 96-well plate. Incubate for 2 hours at 37 ° C, discard the supernatant, wash the cells twice with pre-cooled sodium-free buffer, dissolve the cells with 100 L of 200 mM NaOH, add 100 scintillation fluid, and mix.
- the IC 5Q value of the compound for 14 C using a liquid scintillation meter can be calculated from the agglutination rate at different concentrations.
- the compounds of the present invention have high selectivity to SGLT2 and have significant inhibitory effects on SGLT2.
- the glucose content in the tail blood of the mice at different times within 2 hours after the administration of the sugar was measured and analyzed by the blood glucose meter, and the hypoglycemic effect in the body was preliminarily evaluated. .
- the dose was 1 mg/kg, and the Blank group was given water (both containing 5% DMSO).
- the blood glucose level (-15 minutes) was measured by dose administration.
- the compound of the present invention showed a significant decrease in blood glucose after 15 minutes of administration.
Description






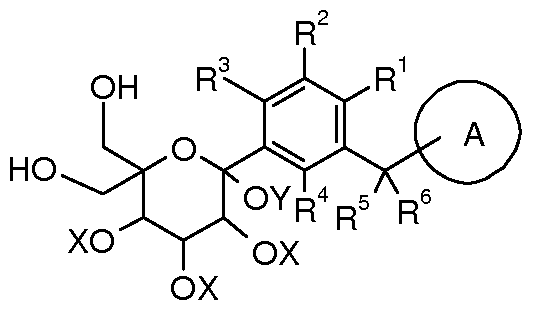

















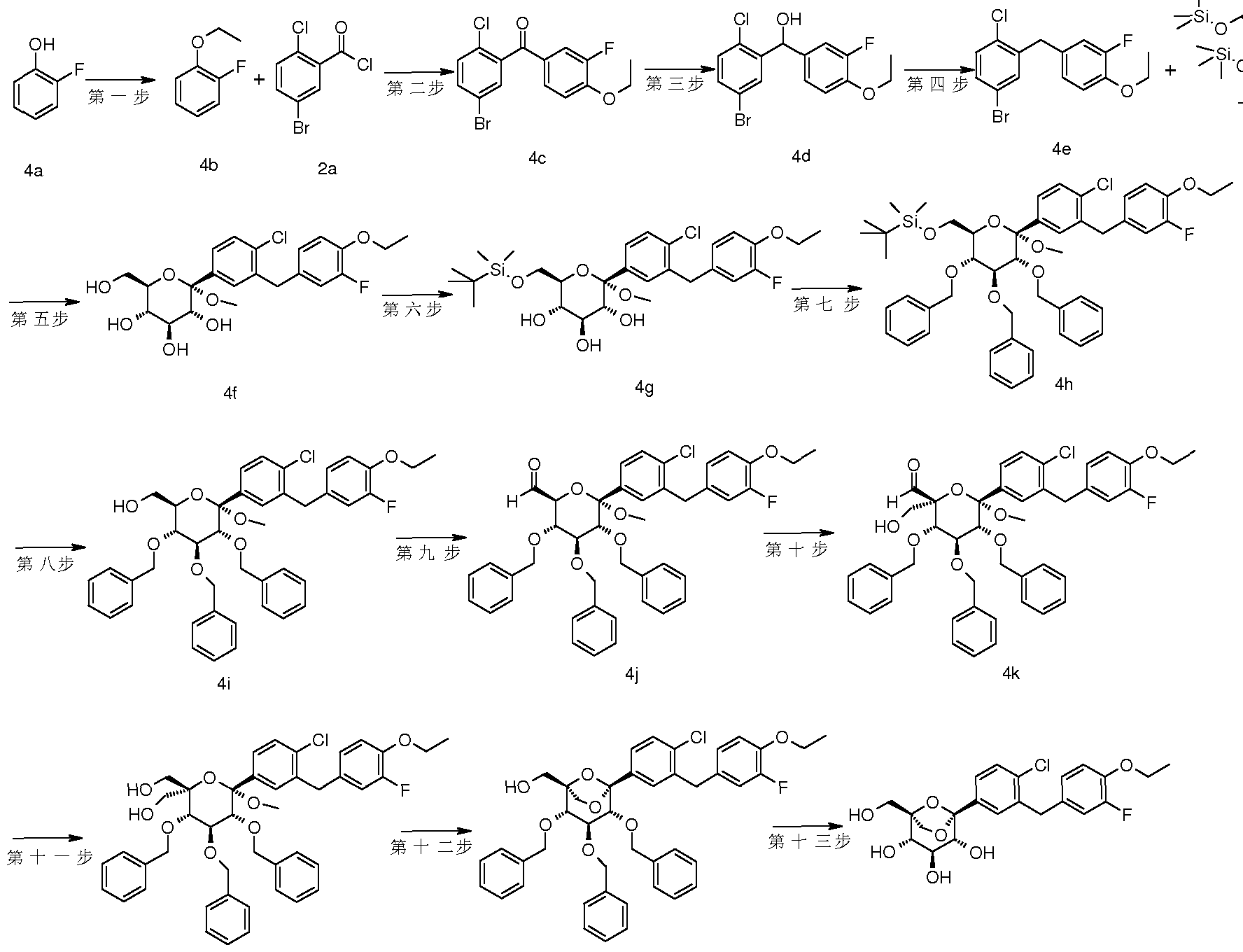




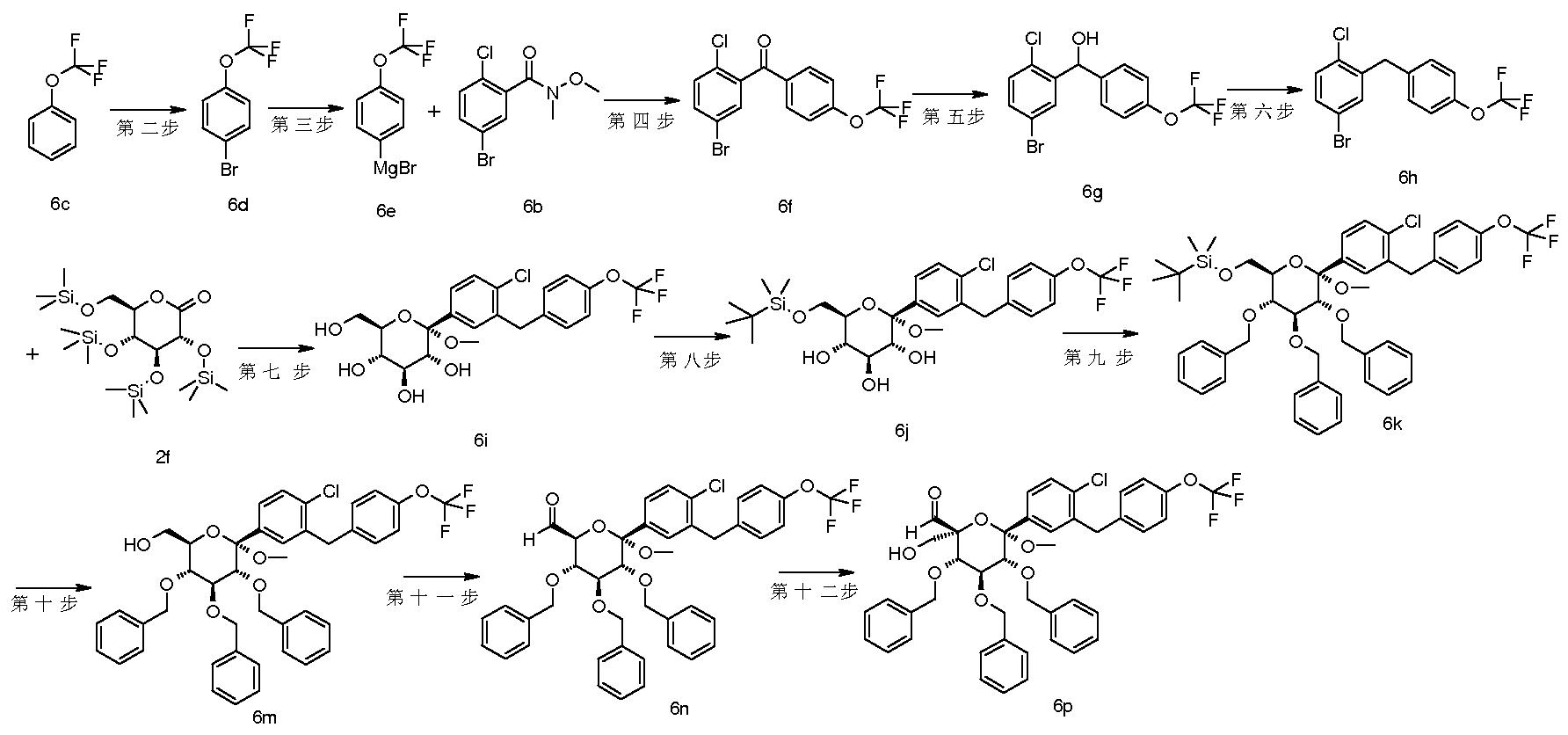




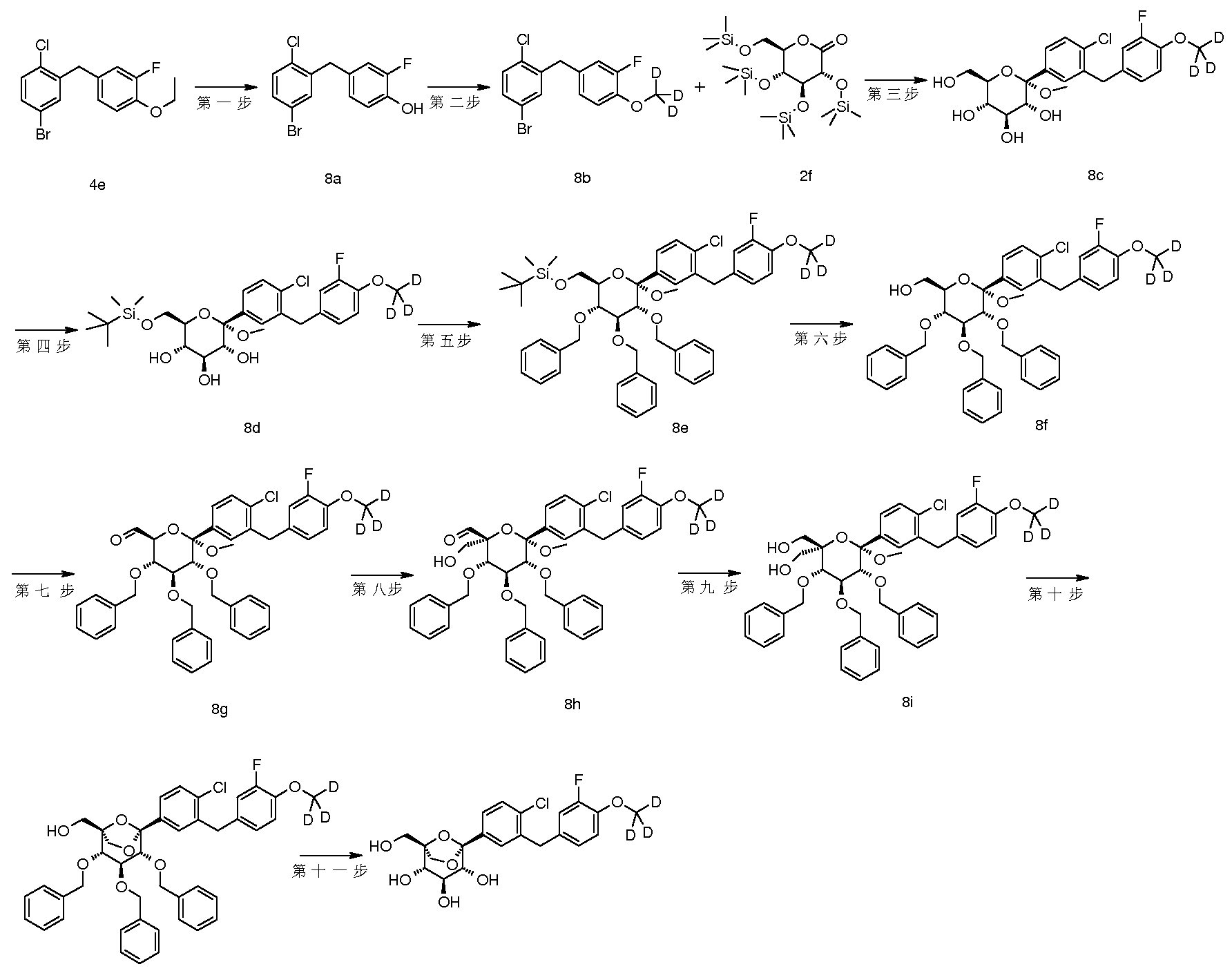





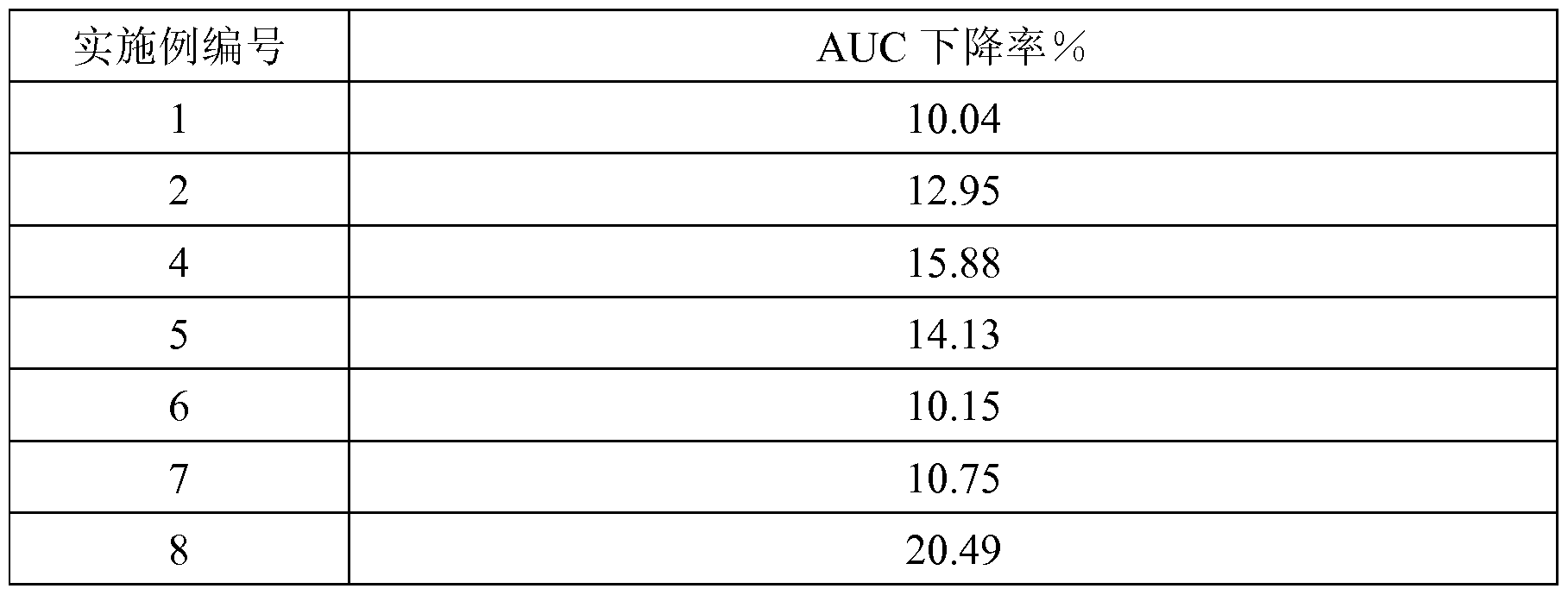
Claims





Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020137005058A KR101862891B1 (ko) | 2010-08-10 | 2011-06-30 | C-아릴 글루코시드 유도체, 그 제조 방법 및 약제학적 용도 |
| CA2807034A CA2807034C (en) | 2010-08-10 | 2011-06-30 | C-aryl glucoside derivatives, preparation process and pharmaceutical use thereof |
| EP11816055.5A EP2604612B1 (en) | 2010-08-10 | 2011-06-30 | C-aryl glucoside derivatives, preparation rpocess and pharmaceutical use thereof |
| BR112013002125A BR112013002125A2 (pt) | 2010-08-10 | 2011-06-30 | derivados de c-aril glicosídeo, seus usos e seu processo de preparação, bem como composição farmacêutica |
| ES11816055.5T ES2581728T3 (es) | 2010-08-10 | 2011-06-30 | Derivados de C-aril-glucósido, procedimiento de preparación y uso farmacéutico de los mismos |
| JP2013523477A JP5984808B2 (ja) | 2010-08-10 | 2011-06-30 | C−アリールグルコシド誘導体、製造法およびその医薬用途 |
| US13/813,045 US8609622B2 (en) | 2010-08-10 | 2011-06-30 | C-aryl glucoside derivatives, preparation process and pharmaceutical use thereof |
| MX2013001098A MX2013001098A (es) | 2010-08-10 | 2011-06-30 | Derivados de c-aril glucosido, proceso para su preparacion y farmaceutico de los mismos. |
| RU2013107748A RU2606501C2 (ru) | 2010-08-10 | 2011-06-30 | Производные с-арилглюкозидов, способ их получения и фармацевтическое применение |
| CN201180003767.4A CN102482290B (zh) | 2010-08-10 | 2011-06-30 | C-芳基葡萄糖苷衍生物、其制备方法及其在医药上的应用 |
| TW100134532A TWI510491B (zh) | 2011-06-30 | 2011-09-26 | C-芳基葡萄糖苷衍生物、其製備方法及其在醫藥上的應用 |
| HK12108091.7A HK1167395A1 (zh) | 2010-08-10 | 2012-08-17 | -芳基葡萄糖苷衍生物、其製備方法及其在醫藥上的應用 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201010249618.3 | 2010-08-10 | ||
| CN201010249618 | 2010-08-10 | ||
| CN2010105896065A CN102372722A (zh) | 2010-08-10 | 2010-12-06 | C-芳基葡萄糖苷衍生物、其制备方法及其在医药上的应用 |
| CN201010589606.5 | 2010-12-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012019496A1 true WO2012019496A1 (zh) | 2012-02-16 |
Family
ID=45567352
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2011/076680 WO2012019496A1 (zh) | 2010-08-10 | 2011-06-30 | C-芳基葡萄糖苷衍生物、其制备方法及其在医药上的应用 |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US8609622B2 (zh) |
| EP (1) | EP2604612B1 (zh) |
| JP (1) | JP5984808B2 (zh) |
| KR (1) | KR101862891B1 (zh) |
| CN (2) | CN102372722A (zh) |
| BR (1) | BR112013002125A2 (zh) |
| CA (1) | CA2807034C (zh) |
| ES (1) | ES2581728T3 (zh) |
| HK (1) | HK1167395A1 (zh) |
| MX (1) | MX2013001098A (zh) |
| RU (1) | RU2606501C2 (zh) |
| WO (1) | WO2012019496A1 (zh) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013038429A2 (en) | 2011-09-13 | 2013-03-21 | Panacea Biotec Ltd. | Novel sglt inhibitors |
| WO2013178064A1 (zh) * | 2012-05-29 | 2013-12-05 | 广东东阳光药业有限公司 | 吡喃葡萄糖基衍生物、其制备方法及其在医药上的应用 |
| JP2014512399A (ja) * | 2011-04-25 | 2014-05-22 | ベイジン・プレリュード・ファーム・サイ・アンド・テック・カンパニー・リミテッド | ナトリウム依存性グルコース輸送タンパク質阻害剤並びにその調製方法及び使用 |
| WO2014159151A1 (en) * | 2013-03-14 | 2014-10-02 | Msd International Gmbh | Methods for preparing sglt2 inhibitors |
| CN104151306A (zh) * | 2013-05-13 | 2014-11-19 | 北京新天宇科技开发有限公司 | 一种坎格列净的新的制备方法 |
| WO2015032272A1 (zh) * | 2013-09-09 | 2015-03-12 | 江苏豪森药业股份有限公司 | C-芳基葡糖苷衍生物、其制备方法及其在医药上的应用 |
| WO2015043511A1 (en) * | 2013-09-27 | 2015-04-02 | Sunshine Lake Pharma Co., Ltd. | Glucopyranosyl derivatives and their uses in medicine |
| WO2015043473A1 (en) * | 2013-09-25 | 2015-04-02 | Sunshine Lake Pharma Co., Ltd. | Glucopyranosyl derivatives and their uses in medicine |
| US9340553B2 (en) | 2014-05-19 | 2016-05-17 | Pfizer Inc. | Substituted-6,8-dioxabicyclo[3.2.1]octane-2,3-diol compounds as targeting agents of ASGPR |
| US10301344B2 (en) | 2014-09-30 | 2019-05-28 | Jiangsu Hengrui Medicine Co., Ltd. | L-proline complex of sodium-glucose cotransporter 2 inhibitor, monohydrate and crystal form thereof |
| WO2021176096A1 (en) | 2020-03-05 | 2021-09-10 | Krka, D.D., Novo Mesto | Pharmaceutical composition comprising sglt2 inhibitor |
| WO2021245253A1 (en) | 2020-06-05 | 2021-12-09 | Krka, D.D., Novo Mesto | Preparation of highly pure amorphous dapagliflozin |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102627535A (zh) * | 2012-03-31 | 2012-08-08 | 天津药物研究院 | 含1,1-二苯基环丙基结构的化合物的制备方法 |
| WO2014187365A1 (zh) * | 2013-05-24 | 2014-11-27 | 四川海思科制药有限公司 | 氧杂双环衍生物、制备方法及其应用 |
| WO2014206349A1 (zh) * | 2013-06-28 | 2014-12-31 | 四川海思科制药有限公司 | 氧杂-硫杂-双环[3.2.1]辛烷衍生物、制备方法及其用途 |
| WO2015101670A2 (en) * | 2014-01-03 | 2015-07-09 | Elexopharm Gmbh | Inhibitors of 17beta-hydroxysteroid dehydrogenases type 1 and type 2 |
| CN104017031A (zh) * | 2014-06-21 | 2014-09-03 | 李友香 | 降血糖药物和组合物 |
| CN104031098A (zh) * | 2014-06-21 | 2014-09-10 | 李友香 | 降糖药物 |
| CN105294785A (zh) * | 2014-07-02 | 2016-02-03 | 上海阳帆医药科技有限公司 | C-苯并五元杂芳环类芳基葡萄糖苷衍生物及其制备方法与用途 |
| US20160002275A1 (en) * | 2014-07-03 | 2016-01-07 | Cadila Healthcare Limited | Process for preparation and purification of canagliflozin |
| CN104119324B (zh) * | 2014-07-23 | 2016-03-30 | 齐鲁天和惠世制药有限公司 | 一种卡格列净的制备方法 |
| CN105461762B (zh) * | 2014-09-27 | 2018-12-04 | 广东东阳光药业有限公司 | 吡喃葡萄糖基衍生物及其在医药上的应用 |
| EP3226874A1 (en) * | 2014-12-03 | 2017-10-11 | Sun Pharmaceutical Industries Ltd | Processes for the preparation of ertugliflozin |
| EP3303345A4 (en) | 2015-05-25 | 2019-01-30 | Sun Pharmaceutical Industries Ltd | ERTUGLIFOZINE CO-CRYSTALS AND PROCESS FOR PREPARING THEM |
| BR112018013408B1 (pt) * | 2016-01-04 | 2024-01-30 | Je Il Pharmaceutical Co., Ltd | Derivados de c-glicosídeo que têm anel fenila fusionado ou sais farmaceuticamente aceitáveis dos mesmos, método para preparar os mesmos e composição farmacêutica compreendendo os mesmos |
| CN106955273B (zh) * | 2016-01-11 | 2020-10-20 | 江苏恒瑞医药股份有限公司 | 一种含有钠-葡萄糖协同转运蛋白2抑制剂的药物组合物 |
| CN107686496B (zh) * | 2016-08-05 | 2021-01-19 | 江苏恒瑞医药股份有限公司 | 一种钠-葡萄糖协同转运蛋白2抑制剂的制备方法 |
| CN106674245B (zh) * | 2017-01-10 | 2019-07-26 | 北京中海康医药科技发展有限公司 | 吡喃葡萄糖基衍生物的制备及医药上的应用 |
| CN109549939A (zh) * | 2017-09-26 | 2019-04-02 | 江苏恒瑞医药股份有限公司 | Sglt2抑制剂和dpp-4抑制剂联合在制备治疗糖尿病的药物中的用途 |
| CN109806397A (zh) * | 2017-11-20 | 2019-05-28 | 江苏恒瑞医药股份有限公司 | Sglt2抑制剂与arb联合在制备治疗高血压等疾病的药物中的用途 |
| WO2020039394A1 (en) | 2018-08-24 | 2020-02-27 | Novartis Ag | New drug combinations |
| CN113004349A (zh) * | 2019-12-19 | 2021-06-22 | 上海研健新药研发有限公司 | 一种SGLTs抑制剂的制备方法及其关键中间体 |
| CN113330017B (zh) * | 2019-12-19 | 2023-01-31 | 上海研健新药研发有限公司 | 一种SGLTs抑制剂的纯化方法及其应用 |
| CN114195748B (zh) * | 2020-09-17 | 2023-11-14 | 上海森辉医药有限公司 | 一种钠-葡萄糖协同转运蛋白2抑制剂的制备方法 |
| CN112375087A (zh) * | 2020-11-27 | 2021-02-19 | 浙江天宇药业股份有限公司 | 一种脯氨酸恒格列净的合成方法 |
| CN113880701A (zh) * | 2021-10-10 | 2022-01-04 | 浙江司太立制药股份有限公司 | 一种抗糖尿病药物中间体及其制备方法 |
| CN116785268A (zh) * | 2022-03-14 | 2023-09-22 | 江苏万邦生化医药集团有限责任公司 | 一种sglt-2抑制剂的药物组合物 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1671682A (zh) | 2002-08-05 | 2005-09-21 | 山之内制药株式会社 | 甘菊环衍生物及其盐 |
| CN1829729A (zh) | 2003-08-01 | 2006-09-06 | 田边制药株式会社 | 新颖化合物 |
| CN1989132A (zh) | 2004-07-27 | 2007-06-27 | 中外制药株式会社 | 新的葡萄糖醇衍生物、其前体药物及其盐、以及含有它的糖尿病治疗药 |
| WO2009026537A1 (en) | 2007-08-23 | 2009-02-26 | Theracos, Inc. | Benzylbenzene derivatives and methods of use |
| WO2010022313A2 (en) | 2008-08-22 | 2010-02-25 | Theracos, Inc. | Processes for the preparation of sglt2 inhibitors |
| WO2010023594A1 (en) | 2008-08-28 | 2010-03-04 | Pfizer Inc. | Dioxa-bicyclo[3.2.1.]octane-2,3,4-triol derivatives |
| WO2010048358A2 (en) | 2008-10-22 | 2010-04-29 | Auspex Pharmaceutical, Inc. | Ethoxyphenylmethyl inhibitors of sglt2 |
| WO2011051864A1 (en) * | 2009-11-02 | 2011-05-05 | Pfizer Inc. | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002083066A2 (en) * | 2001-04-11 | 2002-10-24 | Bristol-Myers Squibb Company | Amino acid complexes of c-aryl glucosides for treatment of diabetes and method |
| PT1980560E (pt) * | 2003-03-14 | 2011-06-29 | Astellas Pharma Inc | Derivados de c-glicósido para o tratamento da diabetes |
| JP5372759B2 (ja) * | 2006-09-21 | 2013-12-18 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | グルコピラノシル−置換ジフルオロベンジル−ベンゼン誘導体、該化合物を含有する医薬品及びその使用と調製方法 |
| BRPI0916769A2 (pt) * | 2008-07-15 | 2017-09-26 | Theracos Inc | derivados de benzilbenzeno deuterados e métodos de uso |
| WO2010074219A1 (ja) * | 2008-12-26 | 2010-07-01 | アステラス製薬株式会社 | ベンゾチオフェン化合物 |
| JP2014517032A (ja) * | 2011-06-13 | 2014-07-17 | パナセア バイオテック リミテッド | 新規のsglt阻害剤 |
-
2010
- 2010-12-06 CN CN2010105896065A patent/CN102372722A/zh active Pending
-
2011
- 2011-06-30 EP EP11816055.5A patent/EP2604612B1/en active Active
- 2011-06-30 ES ES11816055.5T patent/ES2581728T3/es active Active
- 2011-06-30 JP JP2013523477A patent/JP5984808B2/ja active Active
- 2011-06-30 CN CN201180003767.4A patent/CN102482290B/zh active Active
- 2011-06-30 MX MX2013001098A patent/MX2013001098A/es unknown
- 2011-06-30 US US13/813,045 patent/US8609622B2/en active Active
- 2011-06-30 CA CA2807034A patent/CA2807034C/en active Active
- 2011-06-30 WO PCT/CN2011/076680 patent/WO2012019496A1/zh active Application Filing
- 2011-06-30 BR BR112013002125A patent/BR112013002125A2/pt not_active Application Discontinuation
- 2011-06-30 RU RU2013107748A patent/RU2606501C2/ru active
- 2011-06-30 KR KR1020137005058A patent/KR101862891B1/ko active IP Right Grant
-
2012
- 2012-08-17 HK HK12108091.7A patent/HK1167395A1/zh unknown
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1671682A (zh) | 2002-08-05 | 2005-09-21 | 山之内制药株式会社 | 甘菊环衍生物及其盐 |
| CN1829729A (zh) | 2003-08-01 | 2006-09-06 | 田边制药株式会社 | 新颖化合物 |
| CN1989132A (zh) | 2004-07-27 | 2007-06-27 | 中外制药株式会社 | 新的葡萄糖醇衍生物、其前体药物及其盐、以及含有它的糖尿病治疗药 |
| WO2009026537A1 (en) | 2007-08-23 | 2009-02-26 | Theracos, Inc. | Benzylbenzene derivatives and methods of use |
| WO2010022313A2 (en) | 2008-08-22 | 2010-02-25 | Theracos, Inc. | Processes for the preparation of sglt2 inhibitors |
| WO2010023594A1 (en) | 2008-08-28 | 2010-03-04 | Pfizer Inc. | Dioxa-bicyclo[3.2.1.]octane-2,3,4-triol derivatives |
| WO2010048358A2 (en) | 2008-10-22 | 2010-04-29 | Auspex Pharmaceutical, Inc. | Ethoxyphenylmethyl inhibitors of sglt2 |
| WO2011051864A1 (en) * | 2009-11-02 | 2011-05-05 | Pfizer Inc. | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
Non-Patent Citations (4)
| Title |
|---|
| DIABETES, vol. 57, 2008, pages 1723 - 1729 |
| JOHANNSSON, J.CLIN. ENDOCRINOL. METAB., vol. 82, 1997, pages 727 - 734 |
| See also references of EP2604612A4 |
| VINCENT MASCITTI ET AL.: "Stereoselective synthesis of a dioxa-bicyclo[3.2.1]octane SGLT2 inhibitor", ORGANIC LETTERS, vol. 12, no. 13, 8 June 2010 (2010-06-08), pages 2940 - 2943, XP002614054, DOI: doi:10.1021/OL100940W * |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014512399A (ja) * | 2011-04-25 | 2014-05-22 | ベイジン・プレリュード・ファーム・サイ・アンド・テック・カンパニー・リミテッド | ナトリウム依存性グルコース輸送タンパク質阻害剤並びにその調製方法及び使用 |
| WO2013038429A2 (en) | 2011-09-13 | 2013-03-21 | Panacea Biotec Ltd. | Novel sglt inhibitors |
| WO2013178064A1 (zh) * | 2012-05-29 | 2013-12-05 | 广东东阳光药业有限公司 | 吡喃葡萄糖基衍生物、其制备方法及其在医药上的应用 |
| US9573959B2 (en) | 2013-03-14 | 2017-02-21 | Msd International Gmbh | Methods for preparing SGLT2 inhibitors |
| WO2014159151A1 (en) * | 2013-03-14 | 2014-10-02 | Msd International Gmbh | Methods for preparing sglt2 inhibitors |
| CN104151306A (zh) * | 2013-05-13 | 2014-11-19 | 北京新天宇科技开发有限公司 | 一种坎格列净的新的制备方法 |
| WO2015032272A1 (zh) * | 2013-09-09 | 2015-03-12 | 江苏豪森药业股份有限公司 | C-芳基葡糖苷衍生物、其制备方法及其在医药上的应用 |
| CN105518014B (zh) * | 2013-09-09 | 2018-03-23 | 上海研健新药研发有限公司 | C‑芳基葡糖苷衍生物、其制备方法及其在医药上的应用 |
| CN105518014A (zh) * | 2013-09-09 | 2016-04-20 | 江苏豪森药业集团有限公司 | C-芳基葡糖苷衍生物、其制备方法及其在医药上的应用 |
| US10011627B2 (en) | 2013-09-09 | 2018-07-03 | Youngene Therapeutics Co., Ltd | C-aryl glucoside derivative, preparation methods thereof, and medical applications thereof |
| WO2015043473A1 (en) * | 2013-09-25 | 2015-04-02 | Sunshine Lake Pharma Co., Ltd. | Glucopyranosyl derivatives and their uses in medicine |
| WO2015043511A1 (en) * | 2013-09-27 | 2015-04-02 | Sunshine Lake Pharma Co., Ltd. | Glucopyranosyl derivatives and their uses in medicine |
| US9617293B2 (en) | 2014-05-19 | 2017-04-11 | Pfizer Inc. | Substituted-6,8-dioxabicyclo[3.2.1]octane-2,3-diol compounds as targeting agents of ASGPR |
| US9340553B2 (en) | 2014-05-19 | 2016-05-17 | Pfizer Inc. | Substituted-6,8-dioxabicyclo[3.2.1]octane-2,3-diol compounds as targeting agents of ASGPR |
| US10039778B2 (en) | 2014-05-19 | 2018-08-07 | Pfizer Inc. | Substituted-6,8-dioxabicyclo[3.2.1]octane-2,3-diol compounds as targeting agents of ASGPR |
| US10376531B2 (en) | 2014-05-19 | 2019-08-13 | Pfizer Inc. | Substituted-6,8-dioxabicyclo[3.2.1]octane-2,3-diol compounds as targeting agents of ASGPR |
| US10813942B2 (en) | 2014-05-19 | 2020-10-27 | Pfizer Inc. | Substituted-6,8-dioxabicyclo[3.2.1]octane-2,3-diol compounds as targeting agents of ASGPR |
| US10301344B2 (en) | 2014-09-30 | 2019-05-28 | Jiangsu Hengrui Medicine Co., Ltd. | L-proline complex of sodium-glucose cotransporter 2 inhibitor, monohydrate and crystal form thereof |
| WO2021176096A1 (en) | 2020-03-05 | 2021-09-10 | Krka, D.D., Novo Mesto | Pharmaceutical composition comprising sglt2 inhibitor |
| WO2021245253A1 (en) | 2020-06-05 | 2021-12-09 | Krka, D.D., Novo Mesto | Preparation of highly pure amorphous dapagliflozin |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102372722A (zh) | 2012-03-14 |
| KR101862891B1 (ko) | 2018-05-30 |
| ES2581728T3 (es) | 2016-09-07 |
| EP2604612A1 (en) | 2013-06-19 |
| RU2606501C2 (ru) | 2017-01-10 |
| HK1167395A1 (zh) | 2012-11-30 |
| EP2604612A4 (en) | 2013-07-24 |
| KR20130095741A (ko) | 2013-08-28 |
| CA2807034C (en) | 2019-05-07 |
| RU2013107748A (ru) | 2014-09-20 |
| JP5984808B2 (ja) | 2016-09-06 |
| CA2807034A1 (en) | 2012-02-16 |
| BR112013002125A2 (pt) | 2016-05-24 |
| CN102482290A (zh) | 2012-05-30 |
| US8609622B2 (en) | 2013-12-17 |
| MX2013001098A (es) | 2013-06-05 |
| JP2013533291A (ja) | 2013-08-22 |
| EP2604612B1 (en) | 2016-04-20 |
| US20130130997A1 (en) | 2013-05-23 |
| CN102482290B (zh) | 2014-01-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2012019496A1 (zh) | C-芳基葡萄糖苷衍生物、其制备方法及其在医药上的应用 | |
| JP4809931B2 (ja) | ベンジルベンゼン誘導体およびその使用方法 | |
| ES2422863T3 (es) | Derivados de benzonitrilo sustituidos con glucopiranosilo, composiciones farmacéuticas que contienen compuestos de este tipo, su uso y procedimiento para su fabricación | |
| AU2009270936B2 (en) | Deuterated benzylbenzene derivatives and methods of use | |
| EP3056507B1 (en) | C-aryl glucoside derivative, preparation method for same, and medical applications thereof | |
| JP6450769B2 (ja) | グルコピラノシル誘導体及びその医薬品における使用 | |
| BRPI0717156B1 (pt) | inibidores do co-transportador 2 da glicose de sódio e composição farmacêutica que os compreende | |
| WO2013038429A2 (en) | Novel sglt inhibitors | |
| WO2012025857A1 (en) | Cycloalkyl methoxybenzyl phenyl pyran derivatives as sodium dependent glucose co transporter (sglt2) inhibitors | |
| KR20140057527A (ko) | 씨 글루코사이드 유도체 | |
| JP2014517032A (ja) | 新規のsglt阻害剤 | |
| JP6373352B2 (ja) | 細菌感染を処置するためのマンノース誘導体 | |
| KR101513234B1 (ko) | Sglt2 억제제로서의 신규 티오펜 유도체 및 이를 포함하는 약학 조성물 | |
| TWI510491B (zh) | C-芳基葡萄糖苷衍生物、其製備方法及其在醫藥上的應用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180003767.4 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11816055 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2013523477 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2013/001098 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2807034 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13813045 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011816055 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20137005058 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2013107748 Country of ref document: RU Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112013002125 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112013002125 Country of ref document: BR Kind code of ref document: A2 Effective date: 20130128 |