WO2010038676A1 - アクリル酸製造用の触媒および該触媒を用いたアクリル酸の製造方法 - Google Patents
アクリル酸製造用の触媒および該触媒を用いたアクリル酸の製造方法 Download PDFInfo
- Publication number
- WO2010038676A1 WO2010038676A1 PCT/JP2009/066655 JP2009066655W WO2010038676A1 WO 2010038676 A1 WO2010038676 A1 WO 2010038676A1 JP 2009066655 W JP2009066655 W JP 2009066655W WO 2010038676 A1 WO2010038676 A1 WO 2010038676A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- catalyst
- acrylic acid
- crystallinity
- experimental example
- reaction
- Prior art date
Links
- 239000003054 catalyst Substances 0.000 title claims abstract description 232
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 title claims abstract description 89
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 title claims abstract description 89
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 26
- 238000000034 method Methods 0.000 title abstract description 35
- HGINCPLSRVDWNT-UHFFFAOYSA-N Acrolein Chemical compound C=CC=O HGINCPLSRVDWNT-UHFFFAOYSA-N 0.000 claims abstract description 92
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 claims abstract description 44
- 239000007789 gas Substances 0.000 claims abstract description 42
- 238000007254 oxidation reaction Methods 0.000 claims abstract description 38
- 239000001294 propane Substances 0.000 claims abstract description 22
- 230000003197 catalytic effect Effects 0.000 claims abstract description 21
- 230000003647 oxidation Effects 0.000 claims abstract description 21
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 claims abstract description 19
- 238000002441 X-ray diffraction Methods 0.000 claims abstract description 19
- 229910001882 dioxygen Inorganic materials 0.000 claims abstract description 19
- 229910052750 molybdenum Inorganic materials 0.000 claims abstract description 14
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 claims abstract description 13
- 239000011733 molybdenum Substances 0.000 claims abstract description 13
- 229910052720 vanadium Inorganic materials 0.000 claims abstract description 13
- LEONUFNNVUYDNQ-UHFFFAOYSA-N vanadium atom Chemical compound [V] LEONUFNNVUYDNQ-UHFFFAOYSA-N 0.000 claims abstract description 12
- 239000012808 vapor phase Substances 0.000 claims abstract description 8
- 238000006243 chemical reaction Methods 0.000 claims description 80
- 239000002994 raw material Substances 0.000 claims description 22
- 239000012071 phase Substances 0.000 claims description 14
- 230000007423 decrease Effects 0.000 claims description 4
- 230000001590 oxidative effect Effects 0.000 abstract description 3
- 239000004615 ingredient Substances 0.000 abstract 2
- 239000007788 liquid Substances 0.000 description 50
- 239000000203 mixture Substances 0.000 description 30
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 19
- 239000001301 oxygen Substances 0.000 description 19
- 229910052760 oxygen Inorganic materials 0.000 description 19
- 229910052751 metal Inorganic materials 0.000 description 15
- 239000002184 metal Substances 0.000 description 15
- 239000012018 catalyst precursor Substances 0.000 description 13
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 12
- 238000000465 moulding Methods 0.000 description 10
- 239000000725 suspension Substances 0.000 description 10
- 239000002245 particle Substances 0.000 description 9
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 8
- ADCOVFLJGNWWNZ-UHFFFAOYSA-N antimony trioxide Chemical compound O=[Sb]O[Sb]=O ADCOVFLJGNWWNZ-UHFFFAOYSA-N 0.000 description 8
- 239000010949 copper Substances 0.000 description 7
- 238000010304 firing Methods 0.000 description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- 239000007864 aqueous solution Substances 0.000 description 6
- XTVVROIMIGLXTD-UHFFFAOYSA-N copper(II) nitrate Chemical compound [Cu+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O XTVVROIMIGLXTD-UHFFFAOYSA-N 0.000 description 6
- 238000011068 loading method Methods 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
- 239000007858 starting material Substances 0.000 description 6
- 230000003247 decreasing effect Effects 0.000 description 5
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 4
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- UNTBPXHCXVWYOI-UHFFFAOYSA-O azanium;oxido(dioxo)vanadium Chemical compound [NH4+].[O-][V](=O)=O UNTBPXHCXVWYOI-UHFFFAOYSA-O 0.000 description 4
- 238000001035 drying Methods 0.000 description 4
- 238000011156 evaluation Methods 0.000 description 4
- 239000011261 inert gas Substances 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 4
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 4
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- CPLXHLVBOLITMK-UHFFFAOYSA-N Magnesium oxide Chemical compound [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 3
- 238000011000 absolute method Methods 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 239000011259 mixed solution Substances 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- PAWQVTBBRAZDMG-UHFFFAOYSA-N 2-(3-bromo-2-fluorophenyl)acetic acid Chemical compound OC(=O)CC1=CC=CC(Br)=C1F PAWQVTBBRAZDMG-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 229910052581 Si3N4 Inorganic materials 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 239000012153 distilled water Substances 0.000 description 2
- 238000001125 extrusion Methods 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 238000005469 granulation Methods 0.000 description 2
- 230000003179 granulation Effects 0.000 description 2
- 125000001475 halogen functional group Chemical group 0.000 description 2
- 239000000395 magnesium oxide Substances 0.000 description 2
- ALTWGIIQPLQAAM-UHFFFAOYSA-N metavanadate Chemical compound [O-][V](=O)=O ALTWGIIQPLQAAM-UHFFFAOYSA-N 0.000 description 2
- 239000011148 porous material Substances 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- 239000012744 reinforcing agent Substances 0.000 description 2
- 229920005989 resin Polymers 0.000 description 2
- 239000011347 resin Substances 0.000 description 2
- HBMJWWWQQXIZIP-UHFFFAOYSA-N silicon carbide Chemical compound [Si+]#[C-] HBMJWWWQQXIZIP-UHFFFAOYSA-N 0.000 description 2
- 229910010271 silicon carbide Inorganic materials 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- HQVNEWCFYHHQES-UHFFFAOYSA-N silicon nitride Chemical compound N12[Si]34N5[Si]62N3[Si]51N64 HQVNEWCFYHHQES-UHFFFAOYSA-N 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- -1 steatite Chemical compound 0.000 description 2
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 1
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- 229910021536 Zeolite Inorganic materials 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 239000001099 ammonium carbonate Substances 0.000 description 1
- 235000012501 ammonium carbonate Nutrition 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 229910052787 antimony Inorganic materials 0.000 description 1
- WATWJIUSRGPENY-UHFFFAOYSA-N antimony atom Chemical compound [Sb] WATWJIUSRGPENY-UHFFFAOYSA-N 0.000 description 1
- 229910052797 bismuth Inorganic materials 0.000 description 1
- JCXGWMGPZLAOME-UHFFFAOYSA-N bismuth atom Chemical compound [Bi] JCXGWMGPZLAOME-UHFFFAOYSA-N 0.000 description 1
- 238000001354 calcination Methods 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 229910052804 chromium Inorganic materials 0.000 description 1
- 239000011651 chromium Substances 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 229910052878 cordierite Inorganic materials 0.000 description 1
- XAYGUHUYDMLJJV-UHFFFAOYSA-Z decaazanium;dioxido(dioxo)tungsten;hydron;trioxotungsten Chemical compound [H+].[H+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].[NH4+].O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.[O-][W]([O-])(=O)=O.[O-][W]([O-])(=O)=O.[O-][W]([O-])(=O)=O.[O-][W]([O-])(=O)=O.[O-][W]([O-])(=O)=O.[O-][W]([O-])(=O)=O XAYGUHUYDMLJJV-UHFFFAOYSA-Z 0.000 description 1
- 230000006837 decompression Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- JSKIRARMQDRGJZ-UHFFFAOYSA-N dimagnesium dioxido-bis[(1-oxido-3-oxo-2,4,6,8,9-pentaoxa-1,3-disila-5,7-dialuminabicyclo[3.3.1]nonan-7-yl)oxy]silane Chemical compound [Mg++].[Mg++].[O-][Si]([O-])(O[Al]1O[Al]2O[Si](=O)O[Si]([O-])(O1)O2)O[Al]1O[Al]2O[Si](=O)O[Si]([O-])(O1)O2 JSKIRARMQDRGJZ-UHFFFAOYSA-N 0.000 description 1
- HNPSIPDUKPIQMN-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical compound O=[Si]=O.O=[Al]O[Al]=O HNPSIPDUKPIQMN-UHFFFAOYSA-N 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000003365 glass fiber Substances 0.000 description 1
- 238000010335 hydrothermal treatment Methods 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- WUJISAYEUPRJOG-UHFFFAOYSA-N molybdenum vanadium Chemical compound [V].[Mo] WUJISAYEUPRJOG-UHFFFAOYSA-N 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 229910052758 niobium Inorganic materials 0.000 description 1
- 239000010955 niobium Substances 0.000 description 1
- GUCVJGMIXFAOAE-UHFFFAOYSA-N niobium atom Chemical compound [Nb] GUCVJGMIXFAOAE-UHFFFAOYSA-N 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 239000003973 paint Substances 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 238000010298 pulverizing process Methods 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 239000012495 reaction gas Substances 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000007873 sieving Methods 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 229920003002 synthetic resin Polymers 0.000 description 1
- 239000000057 synthetic resin Substances 0.000 description 1
- 229910052714 tellurium Inorganic materials 0.000 description 1
- PORWMNRCUJJQNO-UHFFFAOYSA-N tellurium atom Chemical compound [Te] PORWMNRCUJJQNO-UHFFFAOYSA-N 0.000 description 1
- 229910052718 tin Inorganic materials 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 239000010937 tungsten Substances 0.000 description 1
- 239000010457 zeolite Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052726 zirconium Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/16—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation
- C07C51/21—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen
- C07C51/215—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen of saturated hydrocarbyl groups
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/002—Mixed oxides other than spinels, e.g. perovskite
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/24—Chromium, molybdenum or tungsten
- B01J23/28—Molybdenum
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/85—Chromium, molybdenum or tungsten
- B01J23/888—Tungsten
- B01J23/8885—Tungsten containing also molybdenum
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/30—Catalysts, in general, characterised by their form or physical properties characterised by their physical properties
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/40—Catalysts, in general, characterised by their form or physical properties characterised by dimensions, e.g. grain size
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/0009—Use of binding agents; Moulding; Pressing; Powdering; Granulating; Addition of materials ameliorating the mechanical properties of the product catalyst
- B01J37/0027—Powdering
- B01J37/0036—Grinding
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/0215—Coating
- B01J37/0221—Coating of particles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/16—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation
- C07C51/21—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen
- C07C51/25—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen of unsaturated compounds containing no six-membered aromatic ring
- C07C51/252—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen of unsaturated compounds containing no six-membered aromatic ring of propene, butenes, acrolein or methacrolein
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/0009—Use of binding agents; Moulding; Pressing; Powdering; Granulating; Addition of materials ameliorating the mechanical properties of the product catalyst
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/08—Heat treatment
Definitions
- the present invention relates to a catalyst suitable for producing acrylic acid by catalytic gas phase oxidation of propane and / or acrolein with molecular oxygen and a method for producing acrylic acid using the catalyst.
- Acrylic acid is industrially important as a raw material for various synthetic resins, paints, and plasticizers, and in recent years, its importance is increasing as a raw material for water-absorbing resins.
- the most common method for producing acrylic acid is a two-stage oxidation method in which acrolein is obtained by catalytic vapor phase oxidation of propylene and acrylic acid is obtained by catalytic vapor phase oxidation of the obtained acrolein.
- Patent Document 1 discloses a catalyst having a peak intensity ratio between a peak caused by VMo 3 O 11 and a peak caused by V 2 O 5 measured by X-ray diffraction analysis using Cu—K ⁇ ray in a specific range. Is disclosed. Patent Document 2 discloses a catalyst that maximizes a specific peak in X-ray diffraction analysis using Cu—K ⁇ rays.
- Patent Document 3 discloses a catalyst having a specific peak in the range of 2 ⁇ of 5 ° to 50 ° in X-ray diffraction analysis using Cu—K ⁇ rays.
- Patent Document 4 discloses a catalyst having a peak showing a maximum intensity at a specific reflection angle or a peak at a specific reflection angle in X-ray diffraction analysis. Catalysts that are not present are disclosed.
- JP 2002-233757 A JP-A-8-299797 JP-A-9-194213 JP-T-2004-504288
- Acrylic acid is currently produced on a scale of several million tons / year worldwide, and the demand for it as a raw material for water-absorbing resins is growing further. Considering the recent increase in raw material prices, if the acrylic acid yield is improved even at an industrial scale of 0.1%, it has a very significant economic significance. Although all of the catalysts disclosed in Patent Documents 1 to 4 show some improvement in the target catalyst performance such as acrylic acid yield and life, there is still room for improvement from the industrial scale. It is something to leave.
- Patent Document 2 In the catalyst described in Patent Document 1, although the yield of acrylic acid at the beginning of the reaction is relatively high, the yield of acrylic acid was 94.4 mol% at the beginning of the reaction even under a low raw material acrolein concentration of 4.5%. However, when it continues after 8000 hours of reaction, it is reduced to 92.9 mol%, which is still not sufficient in terms of the life of the catalyst. Patent Document 2 certainly discloses a high acrylic acid yield, but is a laboratory level evaluation with a catalyst amount of 30 mL at most. Moreover, the evaluation regarding the lifetime of the catalyst is not performed.
- Patent Document 3 although the acrylic acid selectivity at the initial stage of the reaction is relatively high, as in Patent Document 1, the evaluation is performed under the condition that the raw material acrolein concentration is as low as 5.0%, and the initial stage of the reaction is only It is a performance and has not been evaluated for catalyst life. Questions remain about the catalyst performance when acrylic acid is produced by continuing the reaction for a long period of time.
- Patent Document 4 although a relatively high acrylic acid yield is shown as a method by vapor phase oxidation of propane, it is a performance at the initial stage of the reaction to the end, and evaluation on the catalyst life is not performed. Questions remain about the catalyst performance when the reaction is continued for a long time. Furthermore, Patent Documents 1 to 4 do not disclose any technical idea of increasing the acrylic acid yield over a long period of time by controlling the crystallinity of the catalytically active component within an appropriate range.
- the present invention has been made in view of the circumstances described above, and its purpose is to produce an acrylic acid from propane and / or acrolein, which is excellent in catalyst performance such as catalytic activity and selectivity and catalyst life and stable over a long period of time.
- the object is to provide a catalyst exhibiting performance.
- the catalyst of the present invention has excellent catalyst performance such as catalyst activity and selectivity and catalyst life, and has stable performance over a long period of time.
- the crystallinity ratio R calculated as the ratio of the crystallinity M to the crystallinity T is preferably in the range of 0.06 to 0.30.
- the method for producing acrylic acid according to the present invention is a method for producing acrylic acid by catalytic gas phase oxidation of propane and / or acrolein with a molecular oxygen-containing gas, and in the presence of the catalyst of the present invention, It is characterized by having a step of performing phase oxidation. According to the method for producing acrylic acid of the present invention, acrylic acid can be produced stably in a high yield over a long period of time.
- acrylic acid production catalyst and acrylic acid production method of the present invention when producing acrylic acid by catalytic gas phase oxidation of propane and / or acrolein with molecular oxygen, a stable and high yield is obtained over a long period of time. Acrylic acid can be produced at a rate.
- the catalyst for producing acrylic acid of the present invention is a catalyst for producing acrylic acid by catalytic gas phase oxidation of propane and / or acrolein with a molecular oxygen-containing gas.
- the raw material gas for producing acrylic acid only needs to contain at least propane and / or propylene together with the molecular oxygen-containing gas.
- the molecular oxygen-containing gas is not particularly limited as long as it contains molecular oxygen.
- a gas composed only of molecular oxygen may be used, or air may be used.
- the crystallinity T is in the range of 5% to 20%.
- the crystallinity T is preferably 7% or more and 15% or less.
- the catalyst performance and catalyst life such as catalyst activity and selectivity are excellent and the catalyst has stable performance over a long period of time.
- the crystallinity T is less than 5%, the catalyst has low acrylic acid selectivity from the beginning of the reaction, and if the crystallinity T exceeds 20%, the catalyst is from the beginning of the reaction.
- the life of the catalyst is short, leading to early deterioration in performance over time.
- the total diffracted X-ray intensity in the following range is obtained, and is calculated from the ratio of the X-ray diffracted intensity of the entire crystalline portion to the total diffracted X-ray intensity.
- T (diffracted X-ray intensity from all crystalline parts) / (total diffracted X-ray intensity) ⁇ 100 (2)
- a specific method for obtaining the crystallinity T is as follows. After removing the background from the diffraction profile obtained by X-ray diffraction analysis, the halo pattern due to the amorphous part is separated, and the resulting diffraction line of the crystalline part is separated for each peak.
- the integrated intensity (S n ) of the halo pattern due to the amorphous part and the integrated intensity (S c1 , S c2 , S c3 ...) Of each peak of the crystalline part are calculated, and each peak of the crystalline part is calculated.
- the diffracted X-ray intensity S c of the entire crystalline portion is calculated from the total sum of the integrated intensities (S c1 + S c2 + S c3 +).
- the crystallinity ratio R calculated as a ratio of the peak crystallinity M to the crystallinity T is preferably in the range of 0.06 to 0.30.
- the crystallinity ratio R is more preferably in the range of 0.07 to 0.25.
- R M / T (1)
- the crystallinity ratio R the ratio of a specific crystalline portion in the total crystalline portion is defined, and the catalyst performance and the life of the catalyst are excellent.
- acrylic acid production that exhibits stable performance over a long period of time It becomes easy to obtain a catalyst.
- the catalyst for producing acrylic acid of the present invention contains a catalytically active component containing molybdenum and vanadium as essential components. It is important that the acrylic acid production catalyst of the present invention has a crystallinity T satisfying the above range, and the present invention is applied to a catalyst containing molybdenum and vanadium as essential components as catalytic active components.
- a catalyst containing molybdenum and vanadium as essential components various companies have proposed various compositions and preparation methods for the catalytically active component.
- the composition of the catalytically active component is represented by the following general formula (4). Preferred is a catalyst.
- Mo molybdenum
- V vanadium
- A is niobium and / or tungsten
- B is at least one element selected from the group consisting of chromium, manganese, iron, cobalt, nickel, copper, zinc, and bismuth
- C represents at least one element selected from the group consisting of tin, antimony and tellurium
- D represents at least one element selected from titanium, aluminum, silicon and zirconium
- O represents oxygen
- the catalyst of the present invention may have an inert carrier for supporting the catalytically active component in addition to the catalytically active component containing molybdenum and vanadium as essential components.
- an inert carrier for supporting the catalytically active component in addition to the catalytically active component containing molybdenum and vanadium as essential components.
- the shape of the carrier is not particularly limited, and those having a spherical shape, a cylindrical shape, a ring shape, or the like can be used.
- raw materials for the catalytically active component oxides, hydroxides, ammonium salts, nitrates, carbonates, sulfates or organic acid salts of the elements constituting the catalytically active component, aqueous solutions thereof, sols thereof
- a compound containing a plurality of elements is mixed with water to obtain an aqueous solution or an aqueous slurry (hereinafter referred to as “starting material mixture”).
- the obtained starting material mixture is dried by a method such as heating or decompression as necessary to obtain a catalyst precursor.
- a drying method by heating for example, a spray dryer, a drum dryer or the like may be used, and as a result, a powdery catalyst precursor is obtained.
- the starting raw material mixture can be heated in an air stream using a box dryer, a tunnel dryer, or the like to obtain a block or flake catalyst precursor.
- a method of further heating the solid can be employed.
- a drying method by reduced pressure for example, a block or powdery catalyst precursor may be obtained using a vacuum dryer.
- the obtained catalyst precursor is sent to a subsequent molding step through a pulverization step and a classification step for obtaining a powder having an appropriate particle size, if necessary.
- the catalyst precursor may be once calcined and then sent to the molding step.
- the particle size of the catalyst precursor before being sent to the molding step is not particularly limited, but is preferably a particle size in which the passing particle portion of the sieve having an opening of 500 ⁇ m is 90% by mass or more from the viewpoint of excellent moldability.
- the catalyst precursor may be molded into a fixed shape by an extrusion molding method, a tableting molding method, or the like to obtain a molded body.
- a starting material mixture or catalyst precursor as a catalytically active component may be supported on an arbitrary inert carrier having a certain shape to obtain a support (supporting method).
- the shape of the molded body obtained by the extrusion molding method or tableting molding method there is no particular limitation on the shape of the molded body obtained by the extrusion molding method or tableting molding method, and any shape such as a spherical shape, a cylindrical shape, a ring shape, or an indefinite shape may be used.
- a spherical shape it does not need to be a true sphere, and may be substantially spherical, and the same applies to a cylindrical shape and a ring shape.
- an evaporative drying method in which a starting material mixed solution is applied to or adhered to an inert carrier having a certain shape as it is without being dried in an aqueous solution or an aqueous slurry while being heated and dried.
- a solid method or a granulation method in which the catalyst precursor is supported in powder form on an inert carrier can be employed.
- the centrifugal fluid coating method described in JP-A No. 63-200839 and the granulation method using the rocking mixer method described in JP-A No. 2004-136267 are particularly preferable.
- the material and shape of the inert carrier that can be used in the loading method are as described above.
- a molding aid or binder for improving moldability, a pore forming agent for forming appropriate pores in the catalyst, or the like may be used.
- organic compounds such as ethylene glycol, glycerin, propionic acid, maleic acid, benzyl alcohol, propyl alcohol, butyl alcohol, and phenols; water; inorganic salts such as nitric acid, ammonium nitrate, and ammonium carbonate. It is done.
- a reinforcing agent such as silica, alumina, glass fiber, silicon carbide, or silicon nitride can be used.
- the reinforcing agent may be added to the starting raw material mixture or may be blended with the catalyst precursor.
- the molded body or carrier obtained in the molding process is sent to the subsequent firing process and fired.
- the firing temperature is preferably in the range of 350 ° C to 450 ° C, more preferably in the range of 380 ° C to 420 ° C.
- the firing time is preferably 1 hour to 10 hours.
- the firing furnace used in the firing step is not particularly limited, and a generally used box-type firing furnace, tunnel-type firing furnace, or the like may be used.
- the catalyst of the present invention has a specific crystallinity T, and more preferably has a specific crystallinity ratio R.
- the following method is preferably adopted. That is, in the above catalyst preparation method, when preparing the starting raw material mixed solution, the raw materials are dividedly added, the raw material charging time and the temperature of the mixed liquid when mixing the raw materials are adjusted; The catalyst may be hydrothermally treated under specific conditions. Specifically, when preparing the starting raw material mixed liquid, a raw material such as molybdenum is divided into two or more times, or B in the above formula (4) is added to an aqueous solution (liquid A) containing molybdenum and vanadium.
- liquid B When the aqueous solution containing the component (liquid B) is added, liquid B may be added over a period of 30 seconds to 10 minutes, preferably 1 minute to 5 minutes. What is necessary is just to set suitably the temperature of the liquid mixture at the time of mixing a raw material with the raw material to supply.
- the catalyst after calcination is charged into the autoclave together with moisture that becomes saturated steam, and under pressure, the temperature is 150 ° C. to 250 ° C., more preferably 190 ° C. to 240 ° C. for 2 hours to What is necessary is just to process for 48 hours.
- the crystallinity ratio R can be changed by appropriately adjusting the composition ratio of molybdenum and vanadium in the catalytically active component.
- the method for producing acrylic acid of the present invention is a method for producing acrylic acid by catalytic gas phase oxidation of propane and / or acrolein with a molecular oxygen-containing gas, and catalytic gas phase oxidation in the presence of the catalyst of the present invention. It has the process of performing. According to the method for producing acrylic acid of the present invention, acrylic acid can be produced stably in a high yield over a long period of time.
- the reactor used is not particularly limited as long as the catalytic gas phase oxidation reaction is performed using the catalyst of the present invention.
- the reactor any of a fixed bed reactor, a fluidized bed reactor, and a moving bed reactor can be used.
- a fixed bed reactor is used as the reactor.
- the fixed bed reactor As the fixed bed reactor, a tubular type is preferable, and a multi-tube type is more preferable.
- the fixed bed reactor has one or more reaction tubes, and the catalyst is filled in the reaction tubes.
- the reaction tubes are generally arranged vertically in a fixed bed reactor.
- the inner diameter of the reaction tube is not particularly limited as long as it can be filled with the catalyst, but is usually 15 to 50 mm, more preferably 20 to 40 mm, and further preferably 22 to 38 mm.
- the catalyst filled in the reaction tube may be a single catalyst or two or more types, but the catalyst filled in the reaction tube is divided into two layers in the tube axis direction in the catalyst layer in the reaction tube. It is preferable to fill the plurality of reaction zones divided as described above so that the crystallinity ratio R of the catalyst charged in each reaction zone is different. That is, the reaction tube preferably has a plurality of reaction zones divided in the tube axis direction, and each reaction zone is preferably filled with catalysts having different crystallinity ratios R.
- the reaction tube is configured so that the crystallinity ratio R of the plurality of reaction zones decreases sequentially from the inlet side to the outlet side of the source gas containing propane and / or acrolein and the molecular oxygen-containing gas. Is filled with a catalyst.
- the catalyst charged in any of the reaction zones preferably has a crystallinity ratio R in the range of 0.06 to 0.30.
- the above-mentioned catalyst filling method is combined with a method of filling the catalyst so that the occupied volume of the catalyst decreases from the inlet side to the outlet side of the raw material gas, a method of diluting a part of the catalyst with an inert carrier, etc. May be.
- the number of reaction zones is appropriately determined depending on the reaction conditions and the scale of the reactor. However, if the number of reaction zones is too large, problems such as complicated packing operation of the catalyst occur. Therefore, the number of reaction zones is preferably about 2 to 6 industrially.
- the reaction conditions of the method for producing acrylic acid of the present invention are not particularly limited, and any conditions can be used as long as they are generally used for this kind of reaction.
- the source gas may be 1 to 15% by volume (preferably 4 to 12% by volume) propane and / or acrolein, 0.5 to 25% by volume (preferably 2 to 20% by volume) molecular oxygen, 0
- a gas mixture of up to 30% by volume (preferably 0-25% by volume) of water vapor and the balance of an inert gas such as nitrogen may be used.
- the raw material gas may be brought into contact with the catalyst at a space velocity of 300 to 5,000 h ⁇ 1 (STP) under a pressure of 0.1 to 1.0 MPa in a temperature range of 200 to 400 ° C.
- reaction gas not only a mixed gas composed of propane and / or acrolein, molecular oxygen and an inert gas, but also a mixed gas containing acrolein obtained by a dehydration reaction of glycerin or an oxidation reaction of propylene can be used. is there. Moreover, you may add air or oxygen etc. to this mixed gas as needed.
- X-ray diffraction measurement was performed using Spectris Co., Ltd. X'PertPro.
- a sample for X-ray diffraction measurement was prepared by sieving the catalytically active component, and compressing and molding about 0.5 g of the powder having an opening of 150 ⁇ m into a tablet with a diameter of 16 mm and a thickness of 2 mm.
- the diffraction intensity was measured. From the obtained X-ray diffraction profile, the crystallinity T and the crystallinity ratio R were calculated according to the method described in JIS ⁇ K0131 (absolute method).
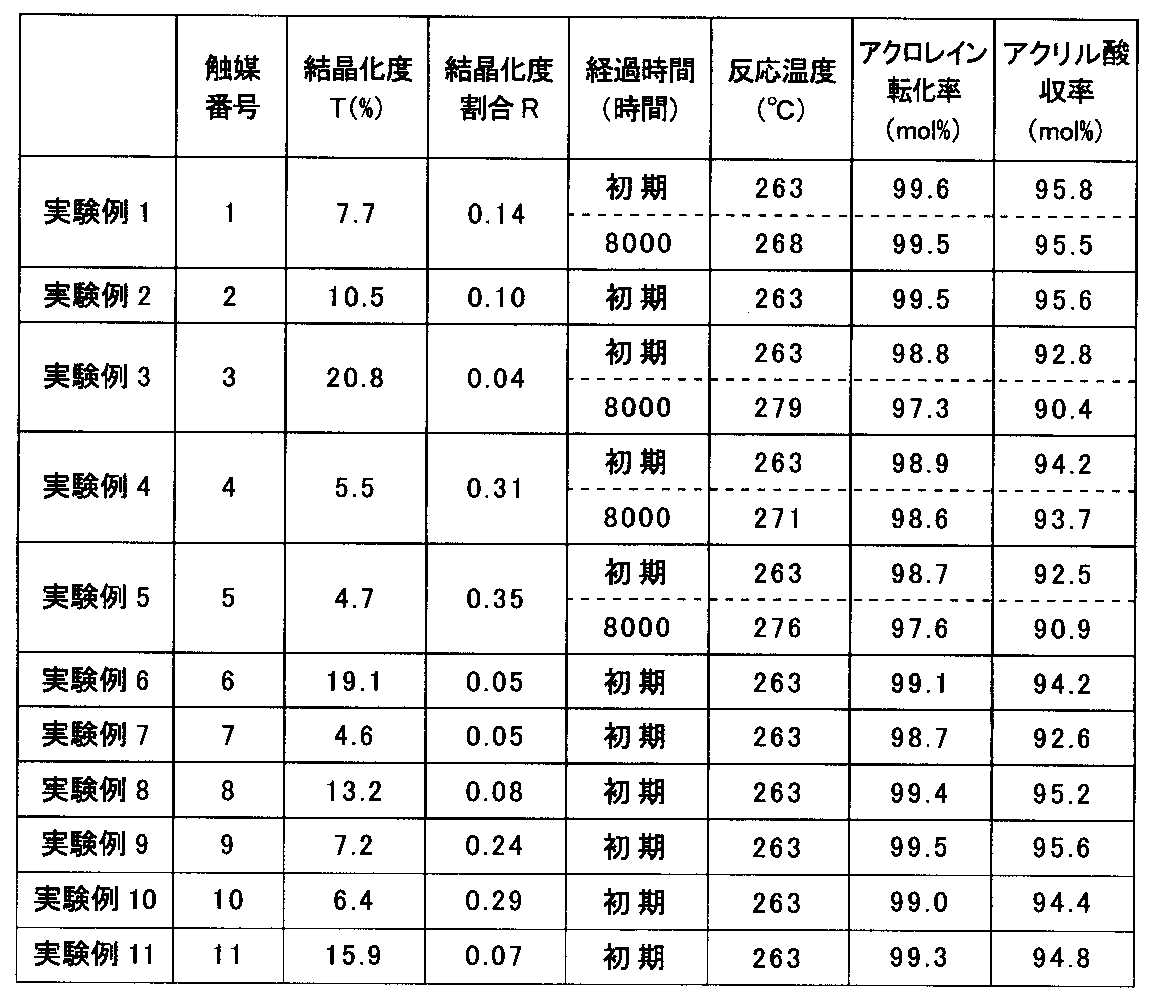
- Catalyst 1 Mo 12 V 4.2 Cu 2.5 W 1.2 Sb 0.7
- the loading rate was determined by the following formula.
- Support rate (mass%) (mass of catalyst (g) ⁇ mass of carrier used (g)) / mass of carrier used (g) ⁇ 100
- the crystallinity T in the range of 2 ⁇ 5 ° to 90 ° by X-ray diffraction measurement of the catalytically active component of catalyst 1 was 7.7%, and the crystallinity ratio R of catalyst 1 was 0.14. .
- (1-2) Reaction A reactor comprising a SUS reaction tube having a total length of 3000 mm and an inner diameter of 25 mm and a shell for flowing a heat medium covering the reaction tube was prepared in the vertical direction.
- the catalyst 1 was dropped from the upper part of the reaction tube and filled so that the layer length was 2900 mm.
- Mixing consisting of 7% by volume of acrolein, 8.5% by volume of oxygen, 10% by volume of water vapor, and the balance of an inert gas such as nitrogen from the bottom of the reaction tube filled with the catalyst while maintaining the heat medium temperature (reaction temperature) at 263 ° C.
- Gas was introduced at a space velocity of 1650 hr ⁇ 1 (STP) to carry out acrolein oxidation reaction.
- the obtained catalyst 2 was charged in the same manner as in Experimental Example 1, and an acrolein oxidation reaction was performed under the same conditions. The results are shown in Table 1. Also in Experimental Example 2, the initial acrylic acid yield was as high as 95.0 mol% or more.
- the obtained catalyst 3 was charged in the same manner as in Experimental Example 1, and an acrolein oxidation reaction was performed under the same conditions. The results are shown in Table 1. The results of continuing the reaction for 8000 hours while appropriately changing the reaction temperature are also shown in Table 1.
- the crystallinity T of the catalyst 3 exceeded 20%, and the initial acrylic acid yield was as low as 92.8 mol%. After 8000 hours, the acrylic acid yield further decreased, and the acrylic acid yield decreased by 2.4 mol% compared to the initial stage.
- the obtained catalyst 4 was packed in the same manner as in Experimental Example 1, and an acrolein oxidation reaction was performed under the same conditions. The results are shown in Table 1. The results of continuing the reaction for 8000 hours while appropriately changing the reaction temperature are also shown in Table 1.
- the crystallinity T of the catalyst 4 was slightly low as 5.5%, and the crystallinity ratio R was as high as 0.31, so that the acrylic acid yield was lower than in Experimental Examples 1 and 2.
- the initial value was 94.2 mol% and 8000 hours later was 93.7 mol%.
- Experimental example 5 In Experimental Example 4, the holding temperature of liquid A and liquid B was changed to 80 ° C., the amount of ammonium paramolybdate added to liquid A and the suspension was changed to 0 parts and 500 parts, respectively, liquid A A catalyst was prepared in the same manner as in Experimental Example 4 except that the time for adding the liquid B to 20 minutes was changed to 20 minutes.
- the supporting rate of the catalyst 5 was about 30% by mass, and the metal element composition excluding oxygen was the same as that of the catalyst 4.
- the crystallinity T of the catalyst 5 was 4.7%, and the crystallinity ratio R was 0.35.
- the obtained catalyst 5 was filled in the same manner as in Experimental Example 1, and an acrolein oxidation reaction was performed under the same conditions. The results are shown in Table 1. The results of continuing the reaction for 8000 hours while appropriately changing the reaction temperature are also shown in Table 1.
- the crystallinity T of the catalyst 5 was less than 5.0%, and the initial acrylic acid yield was as low as 92.5 mol%. After 8000 hours, the acrylic acid yield further decreased, and the acrylic acid yield decreased by 1.6 mol% compared to the initial stage.
- the obtained catalyst 6 was filled in the same manner as in Experimental Example 1, and an acrolein oxidation reaction was performed under the same conditions.
- the results are shown in Table 1.
- the crystallinity T of the catalyst 6 was slightly high as 19.1%, and the crystallinity ratio R was as low as 0.05. Also decreased to 94.2 mol%.
- Experimental example 7 In Experimental Example 6, except that the holding temperature of the liquid A and the liquid B was changed to 80 ° C., and the amount of ammonium paramolybdate added to the liquid A and the suspension was changed to 0 parts and 500 parts, respectively.
- a catalyst was prepared in the same manner as in Experimental Example 6 to obtain Catalyst 7.
- the supporting rate of the catalyst 7 was about 30% by mass, and the metal element composition excluding oxygen was the same as that of the catalyst 6.
- the crystallinity T of the catalyst 7 was 4.6%, and the crystallinity ratio R was 0.05.
- the obtained catalyst 7 was packed in the same manner as in Experimental Example 1, and an acrolein oxidation reaction was performed under the same conditions.
- the results are shown in Table 1.
- the crystallinity T of the catalyst 7 was less than 5.0%, and the initial acrylic acid yield was as low as 92.6 mol%.
- Experimental Example 8 In Experimental Example 1, the amount of ammonium metavanadate was changed to 110 parts, the amount of copper nitrate was changed to 200 parts, the amount of antimony trioxide was changed to 34.4 parts, and the time for adding the B liquid to the A liquid A catalyst was prepared in the same manner as in Experimental Example 1 except that the catalyst was changed to 30 seconds.
- the supporting rate of the catalyst 8 was about 32% by mass, and the metal element composition excluding oxygen was as follows.
- the crystallinity T of the catalyst 8 was 13.2%, and the crystallinity ratio R was 0.08.
- the obtained catalyst 8 was filled in the same manner as in Experimental Example 1, and an acrolein oxidation reaction was performed under the same conditions.
- the results are shown in Table 1.
- the initial acrylic acid yield was as high as 95.0 mol% or more.
- the obtained catalyst 9 was filled in the same manner as in Experimental Example 1, and an acrolein oxidation reaction was performed under the same conditions. The results are shown in Table 1.
- the initial acrylic acid yield was as high as 95.0 mol% or more.
- the obtained catalyst 10 was filled in the same manner as in Experimental Example 1, and an acrolein oxidation reaction was performed under the same conditions. The results are shown in Table 1.
- the initial acrylic acid yield showed a relatively high value of 94.4 mol%.
- the obtained catalyst 11 was filled in the same manner as in Experimental Example 1, and an acrolein oxidation reaction was performed under the same conditions.
- the results are shown in Table 1.
- the initial acrylic acid yield showed a relatively high value of 94.8 mol%.
- a reactor composed of a SUS reaction tube having a total length of 3000 mm and an inner diameter of 25 mm and a shell for flowing a heat medium covering the SUS reaction tube was prepared in the vertical direction.
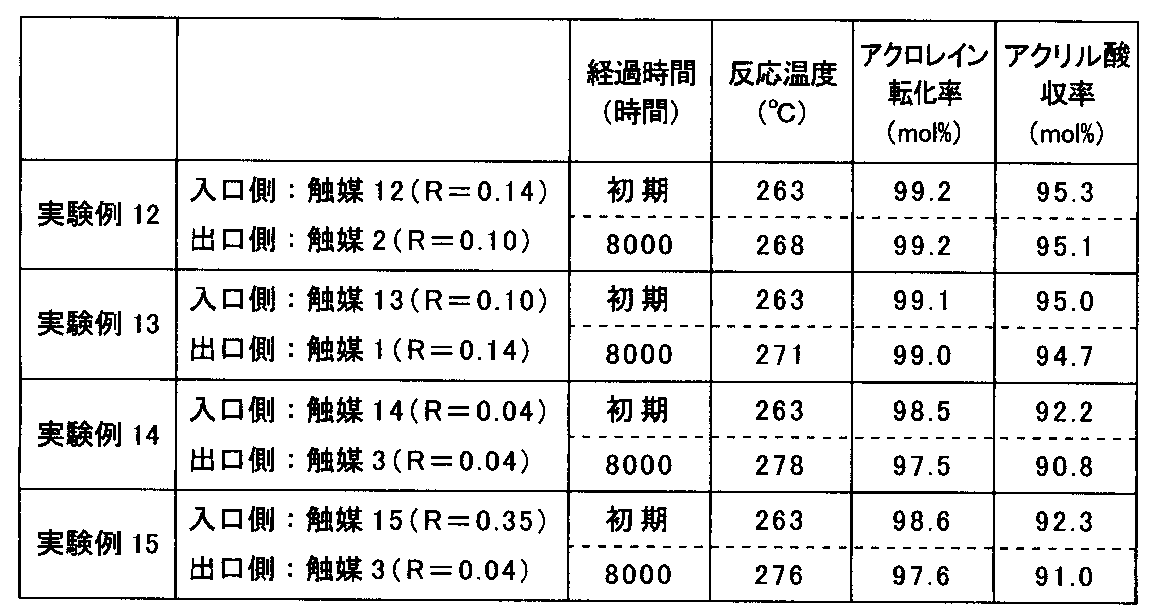
- the catalyst 12 and the catalyst 2 were sequentially dropped from the upper part of the reaction tube, and were filled so that the layer length of the catalyst 12 was 900 mm and the layer length of the catalyst 2 was 2000 mm, respectively.
- reaction temperature Maintaining the heat medium temperature (reaction temperature) at 263 ° C., from the bottom of the reaction tube filled with the catalyst, 8.5% by volume of acrolein, 10.2% by volume of oxygen, 8% by volume of water vapor, and the remainder from an inert gas such as nitrogen
- the mixed gas was introduced at a space velocity of 1700 hr ⁇ 1 (STP) to perform an acrolein oxidation reaction.
- STP space velocity
- Experimental Example 13 A catalyst was prepared in the same manner as in Experimental Example 2 except that the average particle diameter of the silica-alumina spherical carrier used in Experimental Example 2 was changed to 7.5 mm.
- the supported rate of the catalyst 13 was about 30% by mass, and the metal element composition excluding oxygen was the same as that of the catalyst 2.
- the crystallinity T and the crystallinity ratio R were the same as those of the catalyst 2.
- the catalyst 13 and the catalyst 1 are sequentially dropped from the upper part of the reaction tube into the same reaction tube as in Experimental Example 12, and filled so that the layer length of the catalyst 13 is 900 mm and the layer length of the catalyst 1 is 2000 mm, respectively.
- the acrolein oxidation reaction was performed under the same conditions as in No. 12.
- the catalyst 13 was disposed on the inlet side of the reaction tube and the catalyst 1 was disposed on the outlet side of the reaction tube with respect to the mixed gas flow.
- the results are shown in Table 2.
- two types of catalyst 1 and catalyst 13 having a crystallinity ratio R in the range of 0.06 to 0.30 are filled in a reaction tube, and a catalyst 13 having a low crystallinity ratio R is provided on the inlet side.
- the catalyst 1 having a high crystallinity ratio R was disposed on the outlet side.
- a high acrylic acid yield was maintained both at the initial stage and after 8000 hours, but the performance was lower than in Experimental Example 12.
- Experimental Example 14 A catalyst was prepared in the same manner as in Experimental Example 3 except that the average particle diameter of the silica-alumina spherical carrier used in Experimental Example 3 was changed to 7.5 mm.
- the loading ratio of the catalyst 14 was about 30% by mass, and the metal element composition excluding oxygen was the same as that of the catalyst 3.
- the crystallinity T and the crystallinity ratio R were the same as those of the catalyst 3.
- the catalyst 14 and the catalyst 3 are dropped sequentially from the upper part of the reaction tube and filled so that the layer length of the catalyst 14 is 900 mm and the layer length of the catalyst 3 is 2000 mm, respectively.
- the acrolein oxidation reaction was performed under the same conditions as in No. 12.
- the catalyst 14 was arranged on the inlet side of the reaction tube and the catalyst 3 was arranged on the outlet side of the reaction tube with respect to the mixed gas flow.
- the results are shown in Table 2.
- the catalyst 3 and the catalyst 14 having the same crystallinity ratio R when the crystallinity ratio R was outside the range of 0.06 to 0.30 were charged in the reaction tube.
- the acrylic acid yield was lower than in Experimental Examples 12 and 13.
- Experimental Example 15 A catalyst was prepared in the same manner as in Experimental Example 5 except that the average particle diameter of the silica-alumina spherical carrier used in Experimental Example 5 was changed to 7.5 mm.
- the supporting rate of the catalyst 15 was about 30% by mass, and the metal element composition excluding oxygen was the same as that of the catalyst 5.
- the crystallinity T and the crystallinity ratio R were the same as those of the catalyst 5.
- the catalyst 15 and the catalyst 3 are dropped sequentially from the upper part of the reaction tube into the same reaction tube as in Experiment Example 12 and filled so that the catalyst 15 has a layer length of 900 mm and the catalyst 3 has a layer length of 2000 mm.
- the acrolein oxidation reaction was performed under the same conditions as in No. 12.
- the catalyst 15 was disposed on the inlet side of the reaction tube and the catalyst 3 was disposed on the outlet side of the reaction tube with respect to the flow of the mixed gas.
- the results are shown in Table 2.
- two types of catalyst 3 and catalyst 15 having a crystallinity ratio R outside the range of 0.06 to 0.30 are filled in the reaction tube, and the catalyst 15 having a high crystallinity ratio R on the inlet side.
- a catalyst 3 having a low crystallinity ratio R was arranged on the outlet side.
- the acrylic acid yield was lower than in Experimental Examples 12 and 13.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Catalysts (AREA)
Abstract
Description
R=M/T (1)
T=(全結晶性部分からの回折X線強度)/(全体の回折X線強度)×100 (2)
R=M/T (1)
M=(ピークの結晶性部分からの回折X線強度)/(全体の回折X線強度)×100 (3)
MoaVbAcBdCeDfOz (4)
(ここで、Moはモリブデン、Vはバナジウム、Aはニオブおよび/またはタングステン、Bはクロム、マンガン、鉄、コバルト、ニッケル、銅、亜鉛、およびビスマスからなる群より選ばれる少なくとも1種の元素、Cはスズ、アンチモン、テルルからなる群より選ばれる少なくとも1種の元素、Dはチタン、アルミニウム、ケイ素およびジルコニウムから選ばれる少なくとも1種の元素、Oは酸素を表し、またa、b、c、d、e、fおよびzはそれぞれMo、V、A、B、C、DおよびOの原子比を表し、a=12のとき、1≦b≦14、0≦c≦12、0≦d≦10、0≦e≦6、0≦f≦40であり、zは各元素の酸化状態によって定まる数値である。)
アクロレイン転化率(モル%)=(反応したアクロレインのモル数)/(供給したアクロレインのモル数)×100
アクリル酸収率(モル%)=(生成したアクリル酸のモル数)/(供給したアクロレインのモル数)×100
(1-1)触媒の調製
蒸留水3500部を80℃に維持しながら攪拌し、そこにパラモリブデン酸アンモニウム400部、メタバナジン酸アンモニウム116部、およびパラタングステン酸アンモニウム76.5部を添加した(A液)。別に蒸留水400部を80℃に維持しながら攪拌し、そこに硝酸銅143部を溶解した(B液)。A液を撹拌しながら、A液にB液を2分間かけて添加して、混合液を得た。得られた混合液に、さらに三酸化アンチモン24.1部を添加し、1時間攪拌し懸濁液を得た。得られた懸濁液に再度パラモリブデン酸アンモニウム100部を添加し、さらに1時間攪拌し出発原料混合液を得た。この出発原料混合液をドラムドライヤーにて乾燥を行い、顆粒状粉体を得た。得られた顆粒状粉体を、空気雰囲気下210℃で約2時間乾燥を行った。乾燥後の顆粒状粉体を250μm以下に粉砕し、粉体の触媒前駆体を得た。遠心流動コーティング装置に平均粒径4.5mmのシリカ-アルミナ球形担体1500部を投入し、次いで、結合剤として15質量%の硝酸アンモニウム水溶液と共に触媒前駆体を、90℃の熱風を通しながら投入して、担体に担持させた。それを引き続き、空気雰囲気下390℃で6時間焼成して、触媒1を得た。触媒1の担持率は、約30質量%であり、酸素を除く金属元素組成は次のとおりであった。
触媒1:Mo12V4.2Cu2.5W1.2Sb0.7
担持率(質量%)=(触媒の質量(g)-用いた担体の質量(g))/用いた担体の質量(g)×100
触媒1の触媒活性成分のX線回折測定による2θ=5°以上90°以下の範囲の結晶化度Tは7.7%であり、触媒1の結晶化度割合Rは0.14であった。
全長3000mm、内径25mmのSUS製反応管、およびこれを覆う熱媒体を流すためのシェルからなる反応器を鉛直方向に用意した。反応管上部より触媒1を落下させて、層長が2900mmとなるように充填した。熱媒体温度(反応温度)を263℃に保ち、触媒を充填した反応管下部より、アクロレイン7容量%、酸素8.5容量%、水蒸気10容量%、残部が窒素等の不活性ガスからなる混合ガスを、空間速度1650hr-1(STP)で導入し、アクロレイン酸化反応を行った。アクロレイン転化率およびアクリル酸収率の結果を表1に示す。また、反応温度を適宜変更しながら8000時間反応を継続した結果も表1に示す。実験例1では、初期のアクリル酸収率が95.0mol%以上と高い値を示し、8000時間経過後もアクリル酸収率はわずか0.3mol%の低下に抑えられた。実験例1では、長期間にわたり安定して高収率でアクリル酸を製造できた。
実験例1において、A液および懸濁液に添加するパラモリブデン酸アンモニウムの量をそれぞれ500部、0部に変更したこと、A液にB液を添加する際の時間を1分に変更したこと以外は、実験例1と同様に触媒を調製し、触媒2を得た。この触媒2の担持率は、約30質量%であり、酸素を除く金属元素組成は触媒1と同様であった。触媒2の結晶化度Tは10.5%であり、結晶化度割合Rは0.10であった。
実験例2において、A液およびB液の保持温度を90℃に変更したこと、A液にB液を添加する際の時間を10秒以内に添加したこと以外は、実験例2と同様に触媒を調製し、触媒3を得た。この触媒3の担持率は、約30質量%であり、酸素を除く金属元素組成は触媒2と同様であった。触媒3の結晶化度Tは20.8%であり、結晶化度割合Rは0.04であった。
実験例1において、A液およびB液の保持温度を70℃に変更したこと、A液および懸濁液に添加するパラモリブデン酸アンモニウムの量をそれぞれ100部、400部に、メタバナジン酸アンモニウムの量を138部に変更したこと、A液にB液を添加する際の時間を12分に変更したこと以外は、実験例1と同様に触媒を調製し、触媒4を得た。この触媒4の担持率は、約30質量%であり、酸素を除く金属元素組成は次のとおりであった。
触媒4:Mo12V5Cu2.5W1.2Sb0.7
触媒4の結晶化度Tは5.5%であり、結晶化度割合Rは0.31であった。
実験例4において、A液およびB液の保持温度を80℃に変更したこと、A液および懸濁液に添加するパラモリブデン酸アンモニウムの量をそれぞれ0部、500部に変更したこと、A液にB液を添加する際の時間を20分に変更したこと以外は、実験例4と同様に触媒を調製し、触媒5を得た。この触媒5の担持率は、約30質量%であり、酸素を除く金属元素組成は触媒4と同様であった。触媒5の結晶化度Tは4.7%であり、結晶化度割合Rは0.35であった。
実験例1において、A液およびB液の保持温度を90℃に変更したこと、A液および懸濁液に添加するパラモリブデン酸アンモニウムの量をそれぞれ450部、50部に、メタバナジン酸アンモニウムの量を82.8部に、硝酸銅の量を228部に変更したこと、A液にB液を添加する際一気に添加したこと以外は、実験例1と同様に触媒を調製し、触媒6を得た。この触媒6の担持率は、約30質量%であり、酸素を除く金属元素組成は次のとおりであった。
触媒6:Mo12V3Cu4W1.2Sb0.7
触媒6の結晶化度Tは19.1%であり、結晶化度割合Rは0.05であった。
実験例6において、A液およびB液の保持温度を80℃に変更したこと、A液および懸濁液に添加するパラモリブデン酸アンモニウムの量をそれぞれ0部、500部に変更したこと以外は、実験例6と同様に触媒を調製し、触媒7を得た。この触媒7の担持率は、約30質量%であり、酸素を除く金属元素組成は触媒6と同様であった。触媒7の結晶化度Tは4.6%であり、結晶化度割合Rは0.05であった。
実験例1において、メタバナジン酸アンモニウムの量を110部に、硝酸銅の量を200部に、三酸化アンチモンの量を34.4部に変更したこと、A液にB液を添加する際の時間を30秒に変更したこと以外は、実験例1と同様に触媒を調製し、触媒8を得た。この触媒8の担持率は、約32質量%であり、酸素を除く金属元素組成は次のとおりであった。
触媒8:Mo12V4Cu3.5W1.2Sb1
触媒8の結晶化度Tは13.2%であり、結晶化度割合Rは0.08であった。
実験例1において、A液およびB液の保持温度を70℃に変更したこと、A液および懸濁液に添加するパラモリブデン酸アンモニウムの量をそれぞれ300部、200部に変更したこと、メタバナジン酸アンモニウムの量を138部に、硝酸銅の量を171部に、三酸化アンチモンの量を34.4部に変更したこと、A液にB液を添加する際の時間を5分に変更したこと以外は、実験例1と同様に触媒を調製し、触媒9を得た。この触媒9の担持率は、約32質量%であり、酸素を除く金属元素組成は次のとおりであった。
触媒9:Mo12V5Cu3W1.2Sb1
触媒9の結晶化度Tは7.2%であり、結晶化度割合Rは0.24であった。
実験例9において、A液および懸濁液に添加するパラモリブデン酸アンモニウムの量をそれぞれ150部、350部に変更したこと、A液にB液を添加する際の時間を8分に変更したこと以外は、実験例9と同様に触媒を調製し、触媒10を得た。この触媒10の担持率は、約32質量%であり、酸素を除く金属元素組成は触媒9と同様であった。触媒10の結晶化度Tは6.4%であり、結晶化度割合Rは0.29であった。
実験例1において、A液およびB液の保持温度を90℃に変更したこと、A液および懸濁液に添加するパラモリブデン酸アンモニウムの量をそれぞれ350部、150部に変更したこと、メタバナジン酸アンモニウムの量を82.8部に、硝酸銅の量を228部に、三酸化アンチモンの量を34.4部に変更したこと、A液にB液を添加する際の時間を30秒に変更したこと以外は、実験例1と同様に触媒を調製し、触媒11を得た。この触媒11の担持率は、約31質量%であり、酸素を除く金属元素組成は次のとおりであった。触媒11:Mo12V3Cu4W1.2Sb1
触媒11の結晶化度Tは15.9%であり、結晶化度割合Rは0.07であった。
(12-1)触媒の調製
実験例1において用いたシリカ-アルミナ球状担体の平均粒径を7.5mmのものに変更した以外は、実験例1と同様に触媒を調製し、触媒12を得た。この触媒12の担持率は、約30質量%であり、酸素を除く金属元素組成は触媒1と同様であった。結晶化度Tおよび結晶化度割合Rは触媒1と同様であった。
全長3000mm、内径25mmのSUS製反応管、およびこれを覆う熱媒体を流すためのシェルからなる反応器を鉛直方向に用意した。反応管上部より触媒12、触媒2を順次落下させて、それぞれ触媒12の層長が900mm、触媒2の層長が2000mmとなるように充填した。
実験例2において用いたシリカ-アルミナ球状担体の平均粒径を7.5mmのものに変更した以外は、実験例2と同様に触媒を調製し、触媒13を得た。この触媒13の担持率は、約30質量%であり、酸素を除く金属元素組成は触媒2と同様であった。結晶化度Tおよび結晶化度割合Rは触媒2と同様であった。
実験例3において用いたシリカ-アルミナ球状担体の平均粒径を7.5mmのものに変更した以外は、実験例3と同様に触媒を調製し、触媒14を得た。この触媒14の担持率は、約30質量%であり、酸素を除く金属元素組成は触媒3と同様であった。結晶化度Tおよび結晶化度割合Rも触媒3と同様であった。
実験例5において用いたシリカ-アルミナ球状担体の平均粒径を7.5mmのものに変更した以外は、実験例5と同様に触媒を調製し、触媒15を得た。この触媒15の担持率は、約30質量%であり、酸素を除く金属元素組成は触媒5と同様であった。結晶化度Tおよび結晶化度割合Rも触媒5と同様であった。
Claims (7)
- プロパンおよび/またはアクロレインを分子状酸素含有ガスにより接触気相酸化してアクリル酸を製造するための触媒であって、
モリブデンおよびバナジウムを必須成分として含む触媒活性成分を含有し、
触媒活性成分のCu-Kα線を用いたX線回折分析によって測定される2θ=5°以上90°以下の範囲の結晶化度Tが5%以上20%以下の範囲であることを特徴とするアクリル酸製造用の触媒。 - 下記式(1)で表される結晶化度割合Rが0.06以上0.30以下の範囲である請求項1に記載のアクリル酸製造用の触媒。
R=M/T (1)
(Mは、触媒活性成分のCu-Kα線を用いたX線回折分析によって測定される2θ=22.2±0.5°におけるピークの結晶化度を表す。) - 前記触媒は、さらに、前記触媒活性成分を担持するための不活性担体を有する請求項1または2に記載のアクリル酸製造用の触媒。
- プロパンおよび/またはアクロレインを分子状酸素含有ガスにより接触気相酸化してアクリル酸を製造する方法であって、
請求項1~3のいずれか1項に記載の触媒の存在下に接触気相酸化を行う工程を有することを特徴とするアクリル酸の製造方法。 - 前記触媒が充填された反応管を有する固定床反応器を用いる請求項4に記載のアクリル酸の製造方法。
- 前記反応管は、管軸方向に分割された複数個の反応帯を有し、
各反応帯は、前記結晶化度割合Rが互いに異なる触媒が充填されている請求項5に記載のアクリル酸の製造方法。 - 前記複数個の反応帯の結晶化度割合Rは、プロパンおよび/またはアクロレインと分子状酸素含有ガスとを含む原料ガスの入口側から出口側に向かって順次小さくなる請求項6に記載のアクリル酸の製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP09817709.0A EP2347823B1 (en) | 2008-09-30 | 2009-09-25 | Catalyst for producing acrylic acid and process for producing acrylic acid using the catalyst |
| US12/999,789 US20110166384A1 (en) | 2008-09-30 | 2009-09-25 | Catalyst for producing acrylic acid and process for producing acrylic acid using the catalyst |
| CN2009801231284A CN102066000A (zh) | 2008-09-30 | 2009-09-25 | 用于制备丙烯酸的催化剂以及使用了该催化剂的丙烯酸的制备方法 |
| JP2010531831A JP5548132B2 (ja) | 2008-09-30 | 2009-09-25 | アクリル酸製造用の触媒および該触媒を用いたアクリル酸の製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008252081 | 2008-09-30 | ||
| JP2008-252081 | 2008-09-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010038676A1 true WO2010038676A1 (ja) | 2010-04-08 |
Family
ID=42073440
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2009/066655 WO2010038676A1 (ja) | 2008-09-30 | 2009-09-25 | アクリル酸製造用の触媒および該触媒を用いたアクリル酸の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20110166384A1 (ja) |
| EP (1) | EP2347823B1 (ja) |
| JP (1) | JP5548132B2 (ja) |
| CN (1) | CN102066000A (ja) |
| WO (1) | WO2010038676A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140323291A1 (en) * | 2011-03-30 | 2014-10-30 | Eni S.P.A. | Mixed oxides of transition metals, hydrotreatment catalysts obtained therefrom and preparation process |
| JP2015034154A (ja) * | 2013-08-09 | 2015-02-19 | 日本化薬株式会社 | プロピオン酸を選択的に低減させる触媒の充填方法 |
| JP2021531961A (ja) * | 2018-07-19 | 2021-11-25 | ノヴァ ケミカルズ(アンテルナショナル)ソシエテ アノニム | アルカンの酸化的脱水素化のための触媒 |
| WO2022050110A1 (ja) * | 2020-09-03 | 2022-03-10 | 株式会社日本触媒 | アクリル酸製造用触媒とその製造方法およびアクリル酸の製造方法 |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63200839A (ja) | 1987-02-17 | 1988-08-19 | Nippon Shokubai Kagaku Kogyo Co Ltd | プロピレン酸化用触媒および再現性に優れたその製造方法 |
| JPH03218334A (ja) * | 1989-11-06 | 1991-09-25 | Nippon Shokubai Kagaku Kogyo Co Ltd | アクリル酸の製造方法 |
| JPH0710802A (ja) * | 1993-06-28 | 1995-01-13 | Sumitomo Chem Co Ltd | アクリル酸の製造方法 |
| JPH08299797A (ja) | 1995-03-03 | 1996-11-19 | Nippon Kayaku Co Ltd | 触媒及びその製造方法 |
| JPH09194213A (ja) | 1995-11-16 | 1997-07-29 | Basf Ag | 多金属酸化物 |
| JPH09241209A (ja) * | 1996-03-06 | 1997-09-16 | Nippon Shokubai Co Ltd | アクリル酸の製造方法 |
| JP2001162173A (ja) * | 1999-12-08 | 2001-06-19 | Nippon Shokubai Co Ltd | 複合酸化物触媒およびアクリル酸の製造方法 |
| JP2001354612A (ja) * | 2000-06-12 | 2001-12-25 | Nippon Shokubai Co Ltd | アクリル酸の製造方法 |
| JP2002233757A (ja) | 1994-11-14 | 2002-08-20 | Nippon Shokubai Co Ltd | アクリル酸製造用触媒 |
| JP2003089671A (ja) * | 2001-09-19 | 2003-03-28 | Nippon Shokubai Co Ltd | アクリル酸の製造方法 |
| JP2003171340A (ja) * | 2001-12-06 | 2003-06-20 | Mitsubishi Chemicals Corp | アクリル酸の製造方法 |
| JP2004504288A (ja) | 2000-07-18 | 2004-02-12 | ビーエーエスエフ アクチェンゲゼルシャフト | 不均一系触媒作用によりプロパンを気相酸化することによるアクリル酸の製造方法 |
| JP2004136267A (ja) | 2002-08-20 | 2004-05-13 | Nippon Shokubai Co Ltd | 触媒の製造方法 |
Family Cites Families (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2532756A (en) * | 1946-02-20 | 1950-12-05 | Lummus Co | Tower reactor |
| US2451485A (en) * | 1947-09-27 | 1948-10-19 | Shell Dev | Production of unsaturated carbonylic compounds |
| DE2238851B2 (de) * | 1972-08-07 | 1979-08-30 | Hoechst Ag, 6000 Frankfurt | Verfahren zur Herstellung von Acrolein und/oder Acrylsäure unter Vermeidung von Nachreaktionen bei der katalytischen Oxidation von Propylen und/oder Acrolein |
| US4014927A (en) * | 1972-09-07 | 1977-03-29 | Mitsubishi Petrochemical Company Limited | Process for production of unsaturated acids from corresponding unsaturated aldehydes |
| JPS52108917A (en) * | 1976-03-11 | 1977-09-12 | Nippon Shokubai Kagaku Kogyo Co Ltd | Preparation of acrylic acid by vapor-phase catalytic oxidation of prop ylene |
| FR2364061A1 (fr) * | 1976-09-14 | 1978-04-07 | Rhone Poulenc Ind | Nouveau catalyseur pour la preparation d'aldehydes a-b insatures par oxydation d'olefines en phase gazeuse et son procede de preparation |
| AU529228B2 (en) * | 1977-07-13 | 1983-06-02 | Nippon Shokubai Kagaku Kogyo Co. Ltd. | Catalytic vapour phase oxidation |
| JPS55102536A (en) * | 1979-01-30 | 1980-08-05 | Mitsubishi Petrochem Co Ltd | Preparation of acrylic acid |
| JPS63137755A (ja) * | 1986-11-28 | 1988-06-09 | Nippon Shokubai Kagaku Kogyo Co Ltd | 触媒の再活性化法 |
| DE3867249D1 (de) * | 1987-02-17 | 1992-02-13 | Nippon Catalytic Chem Ind | Katalysator zur oxydation von olefin oder tertiaerem alkohol und verfahren zu seiner herstellung. |
| DE3740271A1 (de) * | 1987-11-27 | 1989-06-01 | Basf Ag | Verfahren zur herstellung einer fuer die gasphasenoxidation von propylen zu acrolein und acrylsaeure katalytisch aktiven masse |
| DE3813863A1 (de) * | 1988-04-23 | 1989-11-02 | Uhde Gmbh | Einrichtung zur aufnahme von katalysatoren, insbesondere bei der erzeugung von synthesegas |
| EP0383224B1 (en) * | 1989-02-17 | 1992-09-16 | Jgc Corporation | Shell-and-tube apparatus having an intermediate tube plate |
| US5123123A (en) * | 1991-01-17 | 1992-06-23 | American Standard Inc. | Bathtub overflow control device |
| DE4132263A1 (de) * | 1991-09-27 | 1993-04-01 | Basf Ag | Verfahren zur katalytischen gasphasenoxidation von acrolein zu acrylsaeure |
| JP2702864B2 (ja) * | 1993-03-12 | 1998-01-26 | 株式会社日本触媒 | 触媒の再生方法 |
| ES2148885T3 (es) * | 1994-11-14 | 2000-10-16 | Nippon Catalytic Chem Ind | Procedimiento para la produccion de acido acrilico. |
| JP3948798B2 (ja) * | 1997-10-27 | 2007-07-25 | 株式会社日本触媒 | アクリル酸の製造方法 |
| DE19855913A1 (de) * | 1998-12-03 | 2000-06-08 | Basf Ag | Multimetalloxidmasse zur gasphasenkatalytischen Oxidation organischer Verbindungen |
| EP1080780B1 (en) * | 1999-08-31 | 2007-08-01 | Nippon Shokubai Co., Ltd. | Reactor for catalytic gas phase oxidation |
| EP1090684A1 (en) * | 1999-10-01 | 2001-04-11 | Rohm And Haas Company | A catalyst useful for the gas phase oxidation of alkanes, alkenes or alcohols to unsaturated aldehydes or carboxylic acids |
| EP1166864A1 (en) * | 2000-06-30 | 2002-01-02 | Casale Chemicals SA | Method for the production of formaldehyde |
| US7115776B2 (en) * | 2002-07-18 | 2006-10-03 | Basf Aktiengesellschaft | Heterogeneously catalyzed gas-phase partial oxidation of at least one organic compound |
| US7022643B2 (en) * | 2002-08-20 | 2006-04-04 | Nippon Shokubai Co., Ltd. | Production process for catalyst |
| US20060205978A1 (en) * | 2002-08-20 | 2006-09-14 | Nippon Shokubai Co., Ltd. | Production process for catalyst |
| US20040038820A1 (en) * | 2002-08-20 | 2004-02-26 | Nippon Shokubai Co., Ltd. | Production process for catalyst |
| DE10258180A1 (de) * | 2002-12-12 | 2004-06-24 | Basf Ag | Verfahren zur Herstellung von Chlor durch Gasphasenoxidation von Chlorwasserstoff |
| DE10313209A1 (de) * | 2003-03-25 | 2004-03-04 | Basf Ag | Verfahren der heterogen katalysierten partiellen Gasphasenoxidation von Propen zu Acrylsäure |
| US20060135346A1 (en) * | 2003-08-22 | 2006-06-22 | Hiroya Nakamura | Method of regenerating catalyst |
| RU2365577C2 (ru) * | 2003-10-29 | 2009-08-27 | Басф Акциенгезельшафт | Способ проведения гетерогенного каталитического частичного окисления в газовой фазе акролеина в акриловую кислоту |
| DE10350812A1 (de) * | 2003-10-29 | 2005-06-02 | Basf Ag | Verfahren zum Langzeitbetrieb einer heterogen katalysierten Gasphasenpartialoxidation von Propen zu Acrolein |
| DE10351269A1 (de) * | 2003-10-31 | 2005-06-02 | Basf Ag | Verfahren zum Langzeitbetrieb einer heterogen katalysierten Gasphasenpartialoxidation von Propen zu Acrylsäure |
| JP2005224660A (ja) * | 2004-02-10 | 2005-08-25 | Nippon Shokubai Co Ltd | アクロレインの接触気相酸化反応用触媒、及び該触媒を用いた接触気相酸化方法によるアクリル酸の製造方法 |
| JP4947917B2 (ja) * | 2005-04-18 | 2012-06-06 | 株式会社日本触媒 | 気相接触酸化用の固定床反応器およびアクロレインまたはアクリル酸の製造方法 |
| JP4650354B2 (ja) * | 2006-06-28 | 2011-03-16 | 住友化学株式会社 | 不飽和アルデヒド及び/又は不飽和カルボン酸製造用触媒の再生方法、並びに不飽和アルデヒド及び/又は不飽和カルボン酸の製造方法 |
| US7897813B2 (en) * | 2006-07-19 | 2011-03-01 | Nippon Shokubai Co., Ltd. | Reactor for gas phase catalytic oxidation and a process for producing acrylic acid using it |
| WO2009017074A1 (ja) * | 2007-07-27 | 2009-02-05 | Nippon Shokubai Co., Ltd. | 二段接触気相酸化によるアクリル酸の製造方法 |
-
2009
- 2009-09-25 WO PCT/JP2009/066655 patent/WO2010038676A1/ja active Application Filing
- 2009-09-25 EP EP09817709.0A patent/EP2347823B1/en active Active
- 2009-09-25 US US12/999,789 patent/US20110166384A1/en not_active Abandoned
- 2009-09-25 CN CN2009801231284A patent/CN102066000A/zh active Pending
- 2009-09-25 JP JP2010531831A patent/JP5548132B2/ja active Active
Patent Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63200839A (ja) | 1987-02-17 | 1988-08-19 | Nippon Shokubai Kagaku Kogyo Co Ltd | プロピレン酸化用触媒および再現性に優れたその製造方法 |
| JPH03218334A (ja) * | 1989-11-06 | 1991-09-25 | Nippon Shokubai Kagaku Kogyo Co Ltd | アクリル酸の製造方法 |
| JPH0710802A (ja) * | 1993-06-28 | 1995-01-13 | Sumitomo Chem Co Ltd | アクリル酸の製造方法 |
| JP2002233757A (ja) | 1994-11-14 | 2002-08-20 | Nippon Shokubai Co Ltd | アクリル酸製造用触媒 |
| JPH08299797A (ja) | 1995-03-03 | 1996-11-19 | Nippon Kayaku Co Ltd | 触媒及びその製造方法 |
| JPH09194213A (ja) | 1995-11-16 | 1997-07-29 | Basf Ag | 多金属酸化物 |
| JPH09241209A (ja) * | 1996-03-06 | 1997-09-16 | Nippon Shokubai Co Ltd | アクリル酸の製造方法 |
| JP2001162173A (ja) * | 1999-12-08 | 2001-06-19 | Nippon Shokubai Co Ltd | 複合酸化物触媒およびアクリル酸の製造方法 |
| JP2001354612A (ja) * | 2000-06-12 | 2001-12-25 | Nippon Shokubai Co Ltd | アクリル酸の製造方法 |
| JP2004504288A (ja) | 2000-07-18 | 2004-02-12 | ビーエーエスエフ アクチェンゲゼルシャフト | 不均一系触媒作用によりプロパンを気相酸化することによるアクリル酸の製造方法 |
| JP2003089671A (ja) * | 2001-09-19 | 2003-03-28 | Nippon Shokubai Co Ltd | アクリル酸の製造方法 |
| JP2003171340A (ja) * | 2001-12-06 | 2003-06-20 | Mitsubishi Chemicals Corp | アクリル酸の製造方法 |
| JP2004136267A (ja) | 2002-08-20 | 2004-05-13 | Nippon Shokubai Co Ltd | 触媒の製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2347823A4 |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140323291A1 (en) * | 2011-03-30 | 2014-10-30 | Eni S.P.A. | Mixed oxides of transition metals, hydrotreatment catalysts obtained therefrom and preparation process |
| US9522394B2 (en) * | 2011-03-30 | 2016-12-20 | Eni S.P.A. | Mixed oxides of transition metals, hydrotreatment catalysts obtained therefrom and preparation process |
| US10005071B2 (en) | 2011-03-30 | 2018-06-26 | Eni S.P.A. | Mixed oxides of transition metals, hydrotreatment catalysts obtained therefrom and preparation process |
| JP2015034154A (ja) * | 2013-08-09 | 2015-02-19 | 日本化薬株式会社 | プロピオン酸を選択的に低減させる触媒の充填方法 |
| JP2021531961A (ja) * | 2018-07-19 | 2021-11-25 | ノヴァ ケミカルズ(アンテルナショナル)ソシエテ アノニム | アルカンの酸化的脱水素化のための触媒 |
| JP7364659B2 (ja) | 2018-07-19 | 2023-10-18 | ノヴァ ケミカルズ(アンテルナショナル)ソシエテ アノニム | アルカンの酸化的脱水素化のための触媒 |
| WO2022050110A1 (ja) * | 2020-09-03 | 2022-03-10 | 株式会社日本触媒 | アクリル酸製造用触媒とその製造方法およびアクリル酸の製造方法 |
| JPWO2022050110A1 (ja) * | 2020-09-03 | 2022-03-10 | ||
| JP7360557B2 (ja) | 2020-09-03 | 2023-10-12 | 株式会社日本触媒 | アクリル酸製造用触媒とその製造方法およびアクリル酸の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2347823B1 (en) | 2016-08-31 |
| EP2347823A1 (en) | 2011-07-27 |
| EP2347823A4 (en) | 2013-08-14 |
| JPWO2010038676A1 (ja) | 2012-03-01 |
| CN102066000A (zh) | 2011-05-18 |
| US20110166384A1 (en) | 2011-07-07 |
| JP5548132B2 (ja) | 2014-07-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5845337B2 (ja) | 固定床多管式反応器を用いてのアクリル酸の製造方法 | |
| KR101642163B1 (ko) | 기하학적 촉매 성형체의 제조 방법 | |
| KR101821023B1 (ko) | 불포화 알데히드 및/또는 불포화 카르복실산 제조용 촉매의 제조 방법, 및 불포화 알데히드 및/또는 불포화 카르복실산의 제조 방법 | |
| US7414008B2 (en) | Catalyst for synthesis of unsaturated aldehyde, production process for said catalyst, and production process for unsaturated aldehyde using said catalyst | |
| JP5628930B2 (ja) | 不飽和アルデヒドおよび/または不飽和カルボン酸製造用触媒および該触媒を用いる不飽和アルデヒドおよび/または不飽和カルボン酸の製造方法 | |
| TWI444230B (zh) | 用於製備丙烯酸之觸媒、使用該觸媒製備丙烯酸之方法及使用該丙烯酸製備吸水樹脂之方法 | |
| JP5420556B2 (ja) | アクロレインおよび/またはアクリル酸製造用の触媒および該触媒を用いたアクロレインおよび/またはアクリル酸の製造方法 | |
| JP5548132B2 (ja) | アクリル酸製造用の触媒および該触媒を用いたアクリル酸の製造方法 | |
| JP5628936B2 (ja) | 不飽和カルボン酸製造用触媒および該触媒を用いる不飽和カルボン酸の製造方法 | |
| JP5845338B2 (ja) | 固定床多管式反応器を用いてのアクロレインおよびアクリル酸の製造方法 | |
| JP5448331B2 (ja) | アクリル酸製造用触媒および該触媒を用いたアクリル酸の製造方法 | |
| JP6504774B2 (ja) | アクリル酸製造用の触媒および該触媒を用いたアクリル酸の製造方法 | |
| JP2015120133A (ja) | アクリル酸製造用の触媒および該触媒を用いたアクリル酸の製造方法 | |
| JP4970986B2 (ja) | 複合酸化物触媒の製造方法および該触媒を用いた不飽和アルデヒドおよび/または不飽和カルボン酸の製造方法 | |
| JP5582709B2 (ja) | アクリル酸製造用の触媒および該触媒を用いたアクリル酸の製造方法 | |
| JP6487242B2 (ja) | アクリル酸製造用触媒の製造方法とその触媒、ならびに該触媒を用いたアクリル酸の製造方法 | |
| JP2004130261A (ja) | 不飽和アルデヒドおよび不飽和カルボン酸の合成用触媒の製造方法 | |
| JP2011102247A (ja) | アクロレインおよび/またはアクリル酸の製造方法 | |
| JP2004002323A (ja) | 不飽和アルデヒドの製造法 | |
| JP2008149263A (ja) | モリブデン、ビスマス、及び鉄含有酸化物触媒の製造方法 | |
| WO2022050110A1 (ja) | アクリル酸製造用触媒とその製造方法およびアクリル酸の製造方法 | |
| JP2011102248A (ja) | アクリル酸の製造方法 | |
| JPH1071333A (ja) | 不飽和アルデヒドおよび不飽和カルボン酸合成用触媒の製造法 | |
| JP2011102249A (ja) | アクリル酸の製造方法 | |
| JP2004002208A (ja) | 不飽和アルデヒドの製造法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980123128.4 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09817709 Country of ref document: EP Kind code of ref document: A1 |
|
| REEP | Request for entry into the european phase |
Ref document number: 2009817709 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009817709 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010531831 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |