明細書
末端にエポキシ基および/またはォキセタン基含有ケィ素基を有する有機重合 体を含む光硬化性組成物、 それから得られる硬化物、 及ぴ製造方法
技術分野
本発明は、 各種の有機重合体の末端に選択的にエポキシ基および/またはォキ セタン基を含有したケィ素基が導入された、 新規な末端にエポキシ基および/ま たはォキセタン基含有ケィ素基を有する有機重合体およぴカチオン系光開始剤を 含有する光硬化性組成物に関する。
背景技術
従来エポキシ基の良好な反応性、 接着性から、 エポキシ基を様々な重合体に導 入したエポキシ基含有重合体が開発されている。 し し、 エポキシ基を導入する 際にォレフィンを過酸化物等によりエポキシ化する方法では、 酸化等による重合 体の劣化を伴ったり、 重合体末端への選択的なエポキシ基の導入や多官能化が困 難であるという問題があった。 また製造法によっては副生成物の除去が必要であ る場合がある。 特に上記の方法として、 エポキシ基含有ポリイソブテンを重合す る方法、 およびそれから得られるエポキシ基含有重合体の構造では、 上記のよう な酸化劣化等の懸念や、 得られる重合体のエポキシ基周辺の立体的障害による反 応性への懸念があった (特開平 3 _ 5 6 5 0 5号) 。 このため、 従来法によるェ ポキシ基含有重合体では、 種々の用途に対し必ずしも十分に満足できる物性を示 すに至っていなかった。
一方、 各種の有機重合体は、 それぞれに独特の特徵を有することが広く知られ ており、 特に主鎖骨格がポリイソプチレン、 水素添カロポリイソプレン、 水素添加 ポリブタジエン及びその共重合体からなる群から選ばれる飽和炭化水素系重合体 は、 高耐候性、 高耐熱性、 低透湿性、 低気体透過性、 優れた可とう性等の特徴を 有している。一方、ォキシアルキレン系重合体は、他の重合体との優れた相溶性、 可とう性、 更には優れた低温特性を有している。
また前記の飽和炭化水素系重合体やォキシアルキレン系重合体等の末端に加水
分解性基、 不飽和基、 ヒ ドロシリル基等を導入した重合体は種々開発されている 力 それらの硬化においては水分や加熱が必要であり、 十分な硬化までにある程 度の時間や高温状態を要した。 また貯蔵安定性の点でも問題を有している。 特に 近年、 電子部品周りの接着、 シーリング用途において、 工程時間の短縮を目的と した従来の熱硬化系から光硬化系への変更、 また有機 E L封止剤等の熱に弱い部 品への光硬化系の利用が求められており、 エポキシ基を有する重合体は光力チォ ン硬化等の新しい用途に使用され、 このような電子材料等の分野での使用が期待 されている。
以上のことから、 各種の有機重合体の末端に選択的にエポキシ基が導入された 重合体への要求は高く、 また硬化工程の時間短縮が可能な光硬化への適用の必要 性が非常に高いと考えられる。
発明の開示
本発明は、 各種の有機重合体の末端に選択的にエポキシ基および/またはォキ セタン基を含有したケィ素基が導入された、 新規な末端にエポキシ基おょぴ Zま たはォキセタン基含有ケィ素基を有する有機重合体およぴカチオン系光開始剤を 含有する光硬化性組成物に関する。 本組成物においては、 基材に負担のかかる高 温や湿分等の硬化条件を必要とせず、 光エネルギーの照射により短時間で十分な 硬化が可能である。
本発明者らは、 上記課題を解決するため鋭意検討を重ねた結果、 特定のェポキ シ基含有ケィ素基を有する重合体が優れた物性を有することを見い出し、 本発明 を完成するに至った。
すなわち、
1 ) 末端に一般式 (1 ) 、 一般式 (2 ) あるいは一般式 (3 ) で表される構造 を有する有機重合体 (A) 、 及び、 カチオン系光開始剤 (B ) を含むこと を特徴とする光硬化性組成物に関する。
(式中、 R1および R2は、 同一または異なった炭素数 1から 20のアルキル基、 炭素数 6から 20のァリール基、炭素数 7から 20のァラルキル基若しくは(R') 3S i 0_で示される トリオルガノシロキシ基を示し、 R1または R2が 2個以上存 在するとき、 それらは同一であってもよく、 異なっていてもよい。 ここで R' は 炭素数 1から 20の 1価の炭化水素基であり 3個の R, は同一であってもよく、 異なっていてもよい。 Xはエポキシ基および/またはォキセタン基を含有する 1 価の有機基、 mは 0以上 20以下の整数、 ηは 1、 2または 3の整数を示す。 )
(式中、 X、 R2は上記と同じ、 R3、 R4はメチル基もしくは Xあるいは R2と同一 のものであるか、 いずれかが有機重合体への結合部である。 ここで 1 ' は平均 1 で有機重合体末端への結合部を表すが R3、 R4いずれかが有機重合体末端への結 合部の場合 1 ' = 0である。 1≤m, + n, ≤ 50 1≤m' 、 0≤ n ' であり、 各ュニットの位置は特定されたものではなく、 それぞれ複数個含有される場合に 交互あるいはランダムに配置されていて良い。 )
(式中、 X、 R2は上記と同一である。 ここで 1 ' ' は平均 1で有機重合体末端へ の結合部を表す。 l≤m' ' + n ' ≤ 20 1 ^m' ' 、 0≤η' ' であり、 各ュ-ットの位置は特定されたものではなく、 それぞれ複数個含有される場合に 交互あるいはランダムに配置されていて良い。 )
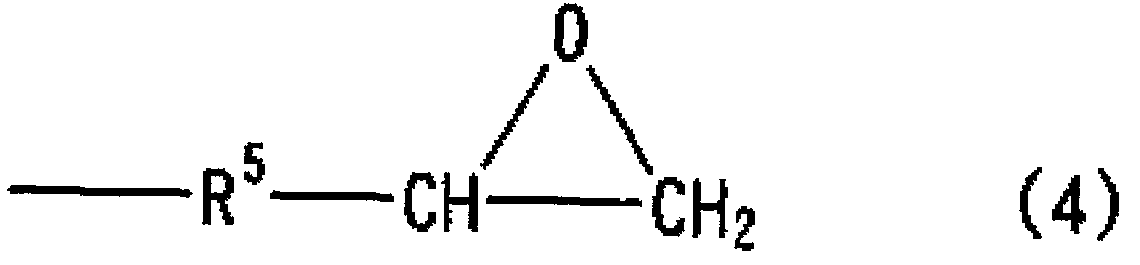
2) 有機重合体 (Α) の Xが一般式 (4) で表される構造を有する 1) に記載 の光硬化性組成物に関する。
(式中、 R5は水素、 酸素、 及び窒素からなる群より選択される 1種以上を構成原 子として含有する炭素数 1から 20の 2価の有機基を示す。 )
3) 有機重合体 (A) の Xが一般式 (5) で表される構造を有する 1 ) に記載 の光硬化性組成物に関する。
(式中、 R6は水素、 酸素、 及び窒素からなる群より選択される 1種以上を構成原 子として含有する炭素数 1から 20の 2価の有機基を示す。 )
4) 有機重合体 (A) の Xが一般式 (6) で表される構造を有する 1) に記載 の光硬化性組成物に関する。
(式中、 R5は上記と同じ)
5) 有機重合体 (A) の主鎖骨格が、 飽和炭化水素系重合体、 ォキシアルキレ ン系重合体、 又はビュル系重合体から選ばれることを特徴とする 1 ) 〜4) のい ずれかに記載の光硬化性組成物に関する。
6) 飽和炭化水素系重合体が、 ポリイソプチレン、 水素添加ポリブタジエン、 水素添加ポリィソプレン及ぴその共重合体からなる群から選ばれることを特徴と する 5) に記載の光硬化性組成物に関する。
7) 有機重合体 (A) 力 末端に不飽和基を有する有機重合体とヒ ドロシラン 化合物との反応により製造されるものであり、 前記のヒ ドロシラン化合物がェポ キシ基および/またはォキセタン基を有するヒ ドロシラン化合物である、 1) 〜 6) のいずれかに記載の光硬化性組成物に関する。
8) 有機重合体 (A) 力 末端に加水分解性シリル基を有する有機重合体と 1 分子中に 1つの水酸基を有する化合物との加水分解性基の交換反応により製造さ れるものであり、 前記の 1分子中に 1つの水酸基を有する化合物が少なく とも 1 つ以上のエポキシ基および Zまたはォキセタン基を有する化合物である、 1) 〜
6) のいずれかに記載の光硬化性組成物に関する。
9 ) カチオン系光開始剤 (B) 力 一般式 (7) で表される構造であることを 特徴とする 1) 〜8) のいずれかに記載の光硬化性組成物に関する。
[R7 aR8 bR9 cR10 dW] U+ [MZV+U] u— (7) (式中、 Wは、 S、 S e、 T e、 P、 A s、 S b、 B i、 0、 I、 B r、 C l、
T i、 Z r、 F e、 R u、 O sまたは N≡Nであり、 R7、 R8、 R9、 および R10 は同一または異なる有機基であり、 a、 b、 cおよび dはそれぞれ 0〜 3の整数 であって、 (a + b + c + d) は Wの価数に等しい。 Mは、 錯体 [MZV+U] の中 心原子を構成する金属またはメタロイ ドであり、 B、 P、 A s、 S b、 F e、 S n、 B i、 A l、 C a、 I n、 T i、 Z n、 S c、 V、 C r、 Mnおよび C oか ら選ばれる金属またはメタロイドである。 Zは、 Mに配位する配位子で、 ハロゲ ン原子または有機基である。 uは錯体イオンの正味の電荷である。 Vは Mの原子 価である。 )
1 0) カチオン系光開始剤 (B) 力 S、 ォ -ゥム塩、 スルホン酸のジァリールョ 一ドニゥム塩、 スルホン酸のトリァリールスルホ -ゥム塩、 ボロン酸のジァリー ノレョードニゥム塩又はボロン酸のトリァリ一ルスルホニゥム塩から選ばれるカチ オン系光開始剤であることを特徴とする 1) 〜8) のいずれかに記載の光硬化性 組成物に関する。
1 1) (C)エポキシ基含有化合物および/またはエポキシ基含有オリゴマー、 (D) ォキセタン基含有化合物および またはォキセタン基含有オリゴマーから 選ばれる少なく とも一種以上をさらに含むことを特徴とする 1 ) 〜1 0) のいず れかに記載の光硬化性組成物に関する。
1 2) 1) から 1 1) のいずれかに記載の光硬化性組成物に、 光エネルギー源 を照射して硬化物を得る、 硬化物の製造方法に関する。
1 3) 1 2) 記載の製造方法で得られる硬化物に関する。
1 4) 1 3) 記載の硬化物を構成要素として含む製品に関する。
本発明は、 新規な末端にエポキシ基および Zまたはェポキシ基含有ケィ素基を 有する有機重合体およぴカチオン系光開始剤を含有する光硬化性組成物に関する ものであり、 光エネルギー源の照射により短時間で優れた硬化性を発現すること が可能である。 当該硬化性組成物は、 コーティング剤、 接着剤、 封止剤等の各ェ 業的用途において非常に有用である。
発明を実施するための最良の形態
本発明の光硬化性組成物は、 新規な末端にエポキシ基および zまたはエポキシ 基含有ケィ素基を有する有機重合体 (A ) およぴカチオン系光開始剤 (B ) を含 有する光硬化性組成物であり、 場合によってはェポキシ基含有化合物および Zま たはエポキシ基含有オリゴマー (C ) 、 ォキセタン基含有化合物および/または ォキセタン基含有オリゴマー(D )を含有するものである。さらに必要に応じて、 本発明の効果を損なわない範囲で、 他の光力チオン重合性化合物、 シランカップ リング剤、 充填剤、 改質剤、 安定剤、 他の樹脂成分等のその他の成分を含有する ことができる。
本発明の光硬化性組成物は優れた硬化性を発現するとともに、 その骨格となる 有機重合体主鎖の種類によって、その重合体独特の特性を発現することができる。 上記有機重合体の主鎖骨格には特に限定はなく、 例えば、 一般的に知られている アクリル系重合体、 ポリエステル系重合体、 飽和炭化水素系重合体、 ォキシアル キレン系重合体等の有機重合体を使用することができる。
本発明における有機重合体 (A) の少なくとも一つの末端部分の構造は、 以下 の一般式 (1 ) 、 一般式 (2 ) あるいは一般式 (3 ) で示される。
ここで式中、 R
1および は、 同一または異なった炭素数 1から 2 0のアルキ ル基、 炭素数 6から 2 0のァリール基、 炭素数 7から 2 0のァラルキル基若しく は (R ' )
3 S i O—で示される トリオルガノシロキシ基を示し、 R
1または R
2 が 2個以上存在するとき、 それらは同一であってもよく、 異なっていてもよい。 ここで R ' は炭素数 1から 2 0の 1価の炭化水素基であり 3個の R, は同一であ つてもよく、 異なっていてもよい。 Xはエポキシ基および/またはォキセタン基
を含有する 1価の有機基、 mは 0以上 20以下の整数、 nは 1、 2または 3の整 数を示す。
一般式 (1) での mは、 原料の入手性の点から 0以上 1 0以下のものが好まし い。
ここで式中、 Xはエポキシ基を含有する 1価の有機基、 R
2は炭素数 1から 20 の炭化水素基で 1個以上のフヱニル基を含有していもよい、 R
3、 R
4はメチル基 もしくは Xあるいは R
2と同一のものである力 \いずれかが有機重合体への結合部 である。 ここで 1 ' は平均 1で有機重合体末端への結合部を表すが R
3、 R
4いず れかが有機重合体末端への結合部の場合 =0である。 l≤m' +n '≤ 50、 1≤m' 、 0≤ n ' であり、 各ユニッ トの位置は特定されたものではなく、 それ ぞれ複数個含有される場合に交互あるいはランダムに配置されていて良い。
ここで式中、 X、 R2は上記と同一である。 ここで ' は平均 1で有機重合体 末端への結合部を表す。 l≤m' ' + ' ≤ 20 1 ^m' ' 、 0≤ η' ' で あり、 各ユニッ トの位置は特定されたものではなく、 それぞれ複数個含有される
場合に交互あるいはランダムに配置されていて良い。
一般式 (2 ) での m ' + n, は 1以上 5 0以下であるが、 重合体とヒ ドロシラ ン化合物との相溶性、 得られるエポキシ基および/またはォキセタン基含有有機 重合体を含む光硬化性組成物の反応性の点から、 好ましくは 3以上 3 0以下であ り、 特に好ましくは 4以上 2 0以下である。
m ' の数は、 1以上であるが、 m ' の数により有機重合体 (A ) の反応性が調 整可能である。 得られるエポキシ基および/またはォキセタン基含有有機重合体 の反応性の点より、 m ' は 2以上が好ましい。
n ' の数は、 0以上であるが、 n, の数によりヒ ドロシラン化合物と有機重合 体との相溶性が調整可能である。 特に有機重合体 (A ) の主鎖骨格が飽和炭化水 素有機重合体である場合は、 n ' は 1以上が好ましく、 m ' が 2以上のときには n ' も 2以上がより好ましい。 また極性の高い主鎖骨格の場合は、 例えばォキシ アルキレン重合体のような場合は、 n, は 1が好ましい。
一般式 (3 ) での ' - η ' ' は 1以上 2 0以下であるが、 重合体末端のェ ポキシ基数の増加の点および重合体とヒ ドロシラン化合物との相溶性の点から、 好ましくは 3以上 2 0以下である。
m' ' の数は、 1以上であるが、 m' ' の数により有機重合体 (A ) の反応性 が調整可能である。 得られるエポキシ基および/またはォキセタン基含有有機重 合体の反応性の点より、 m' ' は 2以上が好ましい。
' の数は、 0以上であるが、 ' の数によりヒ ドロシラン化合物と有機 重合体との相溶性が調整可能である。 特に有機重合体 (Α ) の主鎖骨格が飽和炭 化水素有機重合体である場合は、 η ' ' 1以上が好ましい。 また極性の高い主鎖 骨格の場合は、 例えばォキシアルキレン重合体のような場合は、 η ' ' は 1が好 ましい。
本発明の有機重合体の末端部分の構造は、 エポキシ基の反応性の点から、 Xは 一般式 (4 ) で表される構造であることが好ましく、 更には、 一般式 (5 ) で表 される構造であることが、 製造の容易さ、 原料の入手の点から、 より好ましい。
ここで式中、 R
&は水素、 酸素、 及ぴ窒素からなる群より選択される 1種以上を 構成原子として含有する炭素数 1から 2 0の 2価の有機基を示す。
ここで式中、 R
¾は水素、 酸素、 及び窒素からなる群より選択される 1種以上を 構成原子として含有する炭素数 1から 2 0の 2価の有機基を示す。
また一般式 (6 ) で表される構造であることが、 硬化性の点から好ましい。
本発明における有機重合体の主鎖骨格は限定されるものではないが、 主鎖骨格 がポリイソプチレン、 水素添加ポリイソプレン、 水素添加ポリブタジエン及びそ の共重合体からなる群から選ばれる飽和炭化水素系重合体あるいはォキシアルキ レン系重合体である場合、 それから得られる硬化物がゴム状の弾性を示す特徴を 有する。
前記飽和炭化水素系重合体は、 芳香族環以外の炭素一炭素不飽和結合を実質的 に含有しない重合体であり、 たとえば、 ポリエチレン、 ポリプロピレン、 ポリイ ソブチレン、 水素添加ポリブタジエン、 水素添加ポリイソプレンなどがあげられ る。
本発明に用いる飽和炭化水素系重合体の主鎖骨格をなす重合体は、 ( 1 ) ェチ
レン、 プロピレン、 1ーブテン、 ィソブチレンなどのような炭素数 1〜 6のォレ フィン系化合物を主成分として単独重合もしくは共重合させるか、 (2 ) ブタジ ェン、 ィソプレンなどのようなジェン系化合物を単独重合もしくは共重合させ、 あるいは、 上記ォレフィン系化合物とを共重合させた後、 水素添加するなどの方 法により得ることができる。 中でも、 イソプチレン系重合体や水添ポリブタジェ ン系重合体は、 末端に官能基を導入しやすく、 分子量を制御しやすく、 また、 末 端官能基の数を多くすることができるので好ましい。 さらに、 イソブチレン系重 合体は液状または流動性を有するので取り扱いやすく、 主鎖に芳香族環以外の炭 素一炭素不飽和結合を含まないため水添の必要が無く、 耐候性に極めて優れてい るので特に好ましい。
ィソプチレン系重合体は、 単量体単位のすべてがィソブチレン単位から形成さ れていてもよいし、 ィソブチレンと共重合可能な単量体単位をィソブチレン系重 合体中に、 好ましくは 5 0重量%以下、 さらに好ましくは 3 0重量%以下、 とく に好ましくは 1 0重量%以下の範囲で含有してもよい。 この中でも、 単独重合体 が最も好ましい。
このような単量体成分としては、 たとえば、 炭素数 4〜 1 2のォレフィン、 ビ ニルエーテル、 芳香族ビュル化合物、 ビュルシラン類、 了リルシラン類などがあ げられる。このような共重合体成分としては、たとえば 1ーブテン、 2—ブテン、 2—メチル一 1 ーブテン、 3—メチル一 1 —ブテン、 ペンテン、 4ーメチルー 1 一ペンテン、 へキセン、 ビニノレシク口へキセン、 メチノレビニノレエーテノレ、 ェチノレ ビニルエーテル、 イソプチ/レビニノレエーテノレ、 スチレン、 一メチレスチレン、 ジメチノレスチレン、 モノクロロスチレン、 ジクロロスチレン、 β―ピネン、 イン デン、 ビュルトリ クロロシラン、 ビニノレメチノレジクロロシラン、 ビュルジメチノレ クロロシラン、 ビニノレジメチノレメ トキシシラン、 ビニノレトリ メチルシラン、 ジビ ニノレジクロロシラン、 ジビニノレジメ トキシシラン、 ジビニノレジメチルシラン、 1 , 3—ジビニルー 1, 1, 3, 3—テ トラメチルジシロキサン、 トリ ビニルメチル シラン、 テ トラビュルシラン、 ァリルト リ クロロシラン、 ァリルメチルジクロ口
シラン、 ァリルジメチルクロロシラン、 ァリルジメチルメ トキシシラン、 ァリル トリメチルシラン、 ジァリルジクロロシラン、 ジァリルジメ トキシシラン、 ジァ リルジメチルシラン、 γ—メタタリロイルォキシプロピルトリメ トキシシラン、 γ—メタタリロイルォキシプロピルメチルジメ トキシシランなどがあげられる。 水添ポリブタジエン系重合体や他の飽和炭化水素系重合体においても、 上記ィ ソブチレン系重合体の場合と同様に、 主成分となる単量体単位の他に他の単量体 単位を含有させてもよい。
飽和炭化水素系重合体、 好ましくはィソプチレン系重合体または水添ポリブタ ジェン系重合体の数平均分子量は 5 00〜 5 0, 00 0程度であるのが好ましく、 とくに 1, 000〜 20, 000程度の液状ないし流動性を有するものが取扱い やすいなどの点から、 好ましい。
ォキシアルキレン系重合体の主鎖構造としては、 一R11— Ο—で示される構造 を繰り返し単位とする重合体であればよく、 このとき、 R 11は炭素数 1から 20 の 2価の有機基であればよい。 また、 繰り返し単位の全てが同一である単独重合 体であっても良く、 2つ以上の種類の繰り返し単位を含む共重合体であっても良 い。 さらに、 主鎖中に分岐構造を有していても良い。
R11の具体例としては、 一 CH2CH2—、 一 CH (CH3) CH2_、 - CH (C2 H5) CH2—、 一 C (CH3) 2CH2—、 一 CH2CH2CH2CH2—等が挙げられる。 R11としては特に一 CH (CH3) CH2—が好ましい。
ォキシアルキレン系重合体の主鎖骨格は、 例えば開始剤と触媒の存在下、 モノ エポキシドを開環重合することによって得られる。
開始剤の具体例としては、 エチレングリコール、 プロピレングリコール、 プタ ンジォ一ノレ、 へキサメチレングリコーノレ、 メタリノレアノレコーノレ、 ビスフエノーノレ A、 水素化ビスフエノール A、 ネオペンチルグリコール、 ポリブタジエンジォー ル、ジエチレングリコール、 トリエチレングリコール、ポリエチレングリコール、 ポリプロピレングリコール、 ポリプロピレントリオール、 ポリプロピレンテトラ ォーノレ、 ジプロピレングリコール、 グリセリン、 トリメチローノレメタン、 トリメ
チロールプロノ ン、ペンタエリスリ トール等の 2価ァノレコーノレや多価ァノレコール、 水酸基を有する各種のオリゴマ一等が挙げられる。
モノエポキシドの具体例としては、 エチレンォキサイ ド、 プロピレンォキサイ ド、 α -ブチレンォキサイ ド、 β -プチレンォキサイ ド、 へキセンォキサイ ド、 シ ク口へキセンォキサイ ド、 スチレンォキサイ ド、 α—メチルスチレンオキサイ ド 等のァノレキレンォキサイ ド類や、 メチルダリシジノレエーテル、 ェチルダリシジル エーテル、 イソプロピルグリシジルエーテル、 プチルグリシジルエーテル等のァ ルキルグリシジルエーテル類、 ァリルグリシジルェ一テル類、 ァリールグリシジ ルエーテル類等が挙げられる。
ポリオキシアルキレン系重合体の合成法としては、 たとえば ΚΟΗのようなァ ルカリ触媒による重合法、 たとえば特開昭 6 1 - 2 1 5 6 2 3号公報に示される 有機アルミニウム化合物とポルフィリンとを反応させて得られる錯体のような遷 移金属化合物一ポルフィ リ ン錯体触媒による重合法、 たとえば特公昭 4 6 - 2 7 2 5 0号公報おょぴ特公昭 5 9— 1 5 336号公報などに示される複合金属シァ ン化物錯体触媒による重合法、 セシウム触媒による重合法、 ホスファゼン触媒に よる重合法等があげられるが、 特に限定されるものではない。 中でも、 高分子量 でかつ着色の少ない重合体が容易に得られる点からは、 複合金属シアン化物錯体 触媒による重合法が好ましい。
この他、 ォキシアルキレン系重合体の主鎖骨格は、 水酸基末端ォキシアルキレ ン重合体を塩基性化合物、 例えば KOH、 N a OH、 KOCH3、 N a OCH3等 の存在下、 2官能以上のハロゲン化アルキル、 例えば CH2C 12、 CH2B r2等に よる鎖延長等によっても得ることができる。
さらに、 上記ォキシアルキレン系重合体の主鎖骨格中にはォキシアルキレン系 重合体の特性を大きく損なわない範囲でウレタン結合成分等の他の成分を含んで .いてもよレヽ。
次に主鎖骨格がビュル系重合体の場合について説明する。
本発明のビュル系重合体の主鎖を構成するビュル系モノマーとしては特に限定
されず、 各種のものを用いることができる。 例示するならば、 (メタ) アクリル 酸、 (メタ) ァクリル酸メチル、 (メタ) アクリル酸ェチル、 (メタ) アクリル 酸 _ n—プロピル、 (メタ) アクリル酸イソプロピル、 (メタ) アクリル酸一 n 一プチル、 (メタ) アクリル酸イソブチル、 (メタ) アクリル酸一 t e r t—ブ チル、 (メタ) アタリル酸一 n—ペンチル、 (メタ) アクリル酸一 n—へキシル、 (メタ) アクリル酸シクロへキシル、 (メタ) アクリル酸一 n—ヘプチル、 (メ タ) アクリル酸一 n—ォクチル、 (メタ) アクリル酸 _ 2—ェチルへキシル、 (メ タ) アクリル酸ノニル、 (メタ) アクリル酸デシル、 (メタ) アクリル酸ドデシ ル、 (メタ) アクリル酸フエニル、 (メタ) アクリル酸トルィル、 (メタ) ァク リル酸ベンジル、 (メタ) アクリル酸一 2—メ トキシェチル、 (メタ) アクリル 酸 _ 3—メ トキシプチル、 (メタ) アクリル酸一 2—ヒ ドロキシェチル、 (メタ) アクリル酸 _ 2—ヒ ドロキシプロピル、 (メタ) アクリル酸ステアリル、 (メタ) アクリル酸グリシジル、 (メタ) アクリル酸 2—アミノエチル、 γ— (メタタリ ロイルォキシプロピル) トリメ トキシシラン、 (メタ) アクリル酸のエチレンォ キサイ ド付加物、 (メタ) アクリル酸トリフルォロメチルメチル、 (メタ) ァク リル酸 2— トリフルォロメチルェチル、 (メタ) アクリル酸 2—パーフルォロェ チルェチル、 (メタ) アクリル酸 2—パーフルォロェチルー 2—パーフルォロブ チルェチル、 (メタ) アクリル酸 2—パーフルォロェチル、 (メタ) アクリル酸 パーフルォロメチル、 (メタ) アクリル酸ジパーフルォロメチルメチル、 (メタ) アタリノレ酸 2—パーフルォロメチル一 2—パーフルォロェチルメチル、 (メタ) アクリル酸 2 _パーフルォ口へキシルェチル、 (メタ) アクリル酸 2—パーフル ォロデシルェチル、 (メタ) アクリル酸 2—パーフルォ口へキサデシルェチル等 の (メタ) アク リル系モノマー ; スチレン、 ビニノレトノレェン、 α—メチノレスチレ ン、 クロノレスチレン、 スチレンスノレホン酸及びその塩等の芳香族ビニノレ系モノマ 一;パーフノレオ口エチレン、 パーフノレオ口プロピレン、 フッ化ビユリデン等のフ ッ素含有ビュル系モノマー; ビエルトリメ トキシシラン、 ビュルトリエトキシシ ラン等のケィ素含有ビュル系モノマー ;無水マレイン酸、 マレイン酸、 マレイン
酸のモノアルキルエステル及ぴジアルキルエステル ; フマル酸、 フマル酸のモノ アルキルエステル及ぴジアルキルエステル; マレイミ ド、 メチルマレイ ミ ド、 ェ チルマレイミ ド、プロピルマレイ ミ ド、ブチルマレイミ ド、へキシノレマレイミ ド、 ォクチルマレイ ミ ド、 ドデシノレマレイ ミ ド、 ステアリノレマレイ ミ ド、 フエ-ノレマ レイミ ド、 シク口へキシルマレイ ミ ド等のマレイミ ド系モノマー ; ァク リ ロ- ト リル、 メタタ リ ロニ 卜リル等のァク リ ロニトリル系モノマー ; ァク リルアミ ド、 メタクリルアミ ド等のアミ ド基含有ビュル系モノマー ;酢酸ビュル、 プロピオン 酸ビュル、 ピパリ ン酸ビュル、 安息香酸ビュル、 桂皮酸ビニル等のビニノレエステ ル類;エチレン、 プロピレン等のアルケン類; ブタジエン、 イソプレン等の共役 ジェン類 ; 塩化ビニル、 塩化ビニリデン、 塩化ァリル、 ァリルアルコール等が挙 げられる。 これらは、 単独で用いても良いし、 複数を共重合させても構わない。 ビュル系重合体の主鎖が、 (メタ) アク リル系モノマー、 アク リ ロニ ト リル系 モノマー、 芳香族ビュル系モノマー、 フッ素含有ビニル系モノマー及ぴケィ素含 有ビュル系モノマーからなる群より選ばれる 1種類のモノマー 1 0 0モル%を重 合して製造されること、 あるいはそれらの少なく とも 1つのモノマーを主として 重合して製造されるものであることが好ましい。 ここで 「主として」 とは、 ビエ ル系重合体を構成するモノマー単位のうち 5 0モル%以上、 好ましくは 7 0モ ル%以上が、 上記モノマーであることを意味する。 なかでも、 生成物の物性等か ら、 スチレン系モノマー及ぴ (メタ) アクリル酸系モノマーが好ましい。 より好 ましくは、 アクリル酸エステルモノマー及びメタクリル酸エステルモノマーであ り、 特に好ましくはアクリル酸エステルモノマーであり、 更に好ましくは、 ァク リル酸ブチルである。 本発明においては、 これらの好ましいモノマーを他のモノ マーと共重合、 更にはブロック共重合させても構わなく、 その際は、 これらの好 ましいモノマーが重量比で 4 0 %以上含まれていることが好ましい。 なお上記表 現形式で例えば (メタ) アクリル酸とは、 アクリル酸およぴ あるいはメタタリ ル酸を表す。
なお、 限定はされないが、 ゴム弾性を要求する用途には本ビニル系重合体のガ
ラス転移温度が室温ないしは使用温度よりも低いことが好ましい。
本発明における、 ビュル系重合体の合成法は、 フリーラジカル重合、 制御ラジ カル重合等の公知の方法が好適に使用できる。 中でも末端に本発明の構造を導入 し易い点より、 制御ラジカル重合を用いることが好ましく、 また制御ラジカル重 合を用いた場合は、 リ ビングラジカル重合が好ましく、 原子移動ラジカル重合が より好ましい。
原子移動ラジカル重合では、 有機ハロゲン化物、 特に反応性の高い炭素—ハロ ゲン結合を有する有機ハロゲン化物 (例えば、 α位にハロゲンを有するカルボ二 ル化合物や、 ベンジル位にハロゲンを有する化合物) 、 あるいはハロゲン化スノレ ホニル化合物等が開始剤として用いられる。
原子移動ラジカル重合で重合した場合の本発明のビュル系重合体の分子量分布、 すなわち、 ゲルパーミエーションクロマトグラフィ一で測定した重量平均分子量
(Mw) と数平均分子量 (Mn) との比 (Mw/Mn) は、 特に限定されないが、 好ましくは 1. 8未満であり、 より好ましくは 1. 7以下であり、 さらに好まし くは 1. 6以下であり、 なお好ましくは 1. 5以下であり、 特に好ましくは 1. 4以下であり、 最も好ましくは 1. 3以下である。 本発明での G P C測定におい ては、 通常、 移動相としてクロ口ホルムを用い、 測定はポリスチレンゲルカラム にておこない、 数平均分子量等はポリスチレン換算で求めることができる。
本発明におけるビュル系重合体の数平均分子量は特に制限はないが、 ゲルパー ミエーシヨンクロマトグラフィーで測定した場合、 500〜 1, 000, 000 の範囲が好ましく、 1, 000〜: L 00, 000がより好ましく、 5, 000〜 50, 000がさらに好ましい。
フリーラジカル重合法により重合する場合は、 上記のモノマーが使用可能であ り、 例えばラジカル反応による溶液重合法が利用できる。 重合は、 通常、 前記の 単量体おょぴラジカル開始剤や連鎖移動剤等を加えて 50〜 1 50°Cで反応させ ることにより行われる。
前記ラジカル開始剤の例としては、 2, 2, ーァゾビスイソプチ口-トリル、
2, 2, ーァゾビス (2—メチルブチロニト リル) 、 4 , 4, ーァゾビス ( 4一 シァノバレリ ック) アシッ ド、 1, 1 , ーァゾビス ( 1ーシク口へキサンカルボ 二 トリル) 、 ァゾビスイソ酪酸アミジン塩酸塩 、 2, 2, 一ァゾビス ( 2, 4 ージメチルパレロニトリル) などのァゾ系開始剤、 過酸化べンゾィル、 過酸化ジ - t e r t 一ブチルなどの有機過酸化物系開始剤があげられるが、 重合に使用す る溶媒の影響を受けない、 爆発等の危険性が低いなどの点から、 ァゾ系開始剤の 使用が好ましい。
連鎖移動剤の例としては、 n _ドデシルメルカプタン、 t e r t —ドデシルメ ルカブタン、 ラウリルメルカプタン、 y一メルカプトプロピノレト リメ トキシシラ ン、 γ—メルカプトプロピルメチルジメ トキシシラン、 γ—メルカプトプロピル ト リエトキシシラン、 y—メルカプトプロピルメチルジェトキシシラン等のメノレ カプタン類ゃ含ハロゲン化合物等があげられる。
重合は溶剤中で行なってもよい。溶剤の例としては、エーテル類、炭化水素類、 エステル類などの非反応性の溶剤が好ましい。 フリ一ラジカル重合法で重合し た場合の数平均分子量は特に制限はないが、 ゲルパーミエーシヨンクロマトグラ フィ一で測定した場合、 5 0 0〜1 0 0, 0 0 0のものが取り扱いの容易さの点 から好ましい。 さらに 5 , 0 0 0〜3 0, 0 0 0のものが硬化物の耐候性、 作業 性が良好であることからより好ましい。
本発明における有機重合体の末端へ一般式 (1 ) 、 (2 ) または (3 ) の構造 を有するエポキシ基および/またはォキセタン基含有ケィ素基を導入する方法に は特に限定はないが、 導入時の酸化等による劣化や導入後の脱酸等の精製の必要 性のないことから、 エポキシ基および/またはォキセタン基を有するヒ ドロシラ ン化合物の不飽和基への付加反応による導入、 あるいは末端に加水分解性シリル 基を有する有機重合体と 1分子中に少なく とも 1つ以上のエポキシ基および/ま たはォキセタン基および 1つの水酸基を有する化合物との加水分解性基の交換反 応による導入が好ましい。
ヒ ドロシラン化合物の付加反応による導入は、 ( I ) 末端に不飽和基を有する
有機重合体を合成し、 その後エポキシ基および/またはォキセタン基を有する平 均 1個のヒ ドロシリル基を有するヒ ドロシラン化合物を付加反応させる方法、 あ るいは (II) ヒ ドロシリル基を分子内に 2個以上有するヒ ドロシラン化合物の有 機重合体末端への付加反応後、 ァリル基等の不飽和基を有するエポキシ化合物に よる未反応ヒ ドロシリル基への付加反応による方法のいずれでも可能である。 前者 ( I ) の方法では、 ヒ ドロシラン化合物のヒ ドロシリル基が平均 1個のた め選択的に重合体末端に容易に導入でき、 重合体の高分子量化が抑制できる。 こ の場合、 反応物の仕込み順序等に限定はないが、 反応系の発熱、 有機重合体の粘 度等を考慮すると、 ヒ ドロシリル化触媒と末端に不飽和基を有する有機重合体と の混合物に、 エポキシ基および/またはォキセタン基を有する平均 1個のヒ ドロ シリル基を有するヒ ドロシラン化合物を滴下する方法が好ましい。
有機重合体中の末端不飽和基とヒ ドロシリル基のモル比は、特に限定はない力 s、
0 . 5≤ヒ ドロシリル基 Z有機重合体中の末端不飽和基≤ 2 . 0の範囲であれば 良く、 エポキシ基おょぴ Zまたはォキセタン基の導入率を高くする点から 0 . 8 ≤ヒ ドロシリル基 Z有機重合体中の末端不飽和基≤ 1 . 5が好ましく、 残存する ヒ ドロシラン化合物の除去性から 0 . 8≤ヒ ドロシリル基/有機重合体中の末端 不飽和基≤ 1 . 2がより好ましい。
また後者 (II) の方法では、 重合体末端の未反応ヒ ドロシリル基に対し、 十分 にエポキシ基および Zまたはォキセタン基含有化合物を反応させることができ、 重合体末端に複数のエポキシ基および/またはォキセタン基を導入することが可 能である。 この場合も、 反応物の仕込み順序等に限定はないが、 高分子量化を抑 える点からヒ ドロシラン化合物へ有機重合体と触媒の混合物をゆつく り滴下する ことが好ましい。
有機重合体中の末端不飽和基とヒ ドロシリル基のモル比は、特に限定はないが、 2 . 0≤ヒ ドロシリル基 Z有機重合体中の末端不飽和基の範囲であれば良く、 有 機重合体末端へ複数個のエポキシ基および/またはォキセタン基を導入する点か ら 3 . 0≤ヒ ドロシリル基 有機重合体中の末端不飽和基であることが好ましく、
高分子量化の抑制の点から 3. 0≤ヒ ドロシリル基/有機重合体中の末端不飽和 基≤ 5. 0であることがより好ましい。
本発明において、 特定の末端構造を有する重合体を得るためには、 以下の一般 式 (8) 、 一般式 (9) あるいは一般式 (10) で示されるヒ ドロシラン化合物 を使用することができる。
ここで式中、 R1および R2は、 同一または異なった炭素数 1から 20のアルキル 基、 炭素数 6から 20のァリール基、 炭素数 7から 20のァラルキル基若しくは
(R, ) 3S i O—で示されるトリオルガノシロキシ基を示し、 R1または R2が 2 個以上存在するとき、 それらは同一であってもよく、 異なっていてもよい。 ここ で R, は炭素数 1から 20の 1価の炭化水素基であり 3個の R' は同一であって もよく、 異なっていてもよい。 Xはエポキシ基および/またはォキセタン基を含 有する 1価の有機基、 mは 0以上 20以下の整数、 nは 1、 2または 3の整数を 示す。
ここで式中、 Xはエポキシ基および Zまたはォキセタン基を含有する 1価の有機 基、 R2は炭素数 1から 20の炭化水素基で 1個以上のフエ-ル基を含有していも よい、 R3、 R4は水素、 メチル基もしくは Xあるいは R2と同一のものである。 1 ' は平均 1であるが R3、 R4いずれかが水素の場合 1 ' = 0である, ≤≥ m + n
50、 m, 、 0≤ n ' であり、 各ユニットの位置は特定されたものではな
く、 それぞれ複数個含有される場合に交互あるいはランダムに配置されていて良 い。
ここで式中、 X、 R は上記と同一である。 ここで 1 ' ' は平均 1である。 1≤ m' ' + η ' ' ≤ 2 0 , l≤m' ' 、 0≤ ' であり、 各ユニッ トの位置は特 定されたものではなく、 それぞれ複数個含有される場合に交互あるいはランダム に配置されていて良い。
有機重合体の末端部分の構造は、エポキシ基の反応性の点から、 Xは一般式( 4 ) で表される構造であることが好ましく、 更には、 一般式 (5 ) で表される構造で あることが、 製造の容易さ、 原料の入手の点から、 より好ましい。
ここで式中、 R 5は水素、 酸素、 及び窒素からなる群より選択される 1種以上を 構成原子として含有する炭素数 1から 2 0の 2価の有機基を示す。
(5) で式中、 R
6は水素、 酸素、 及び窒素からなる群より選択される 1種以上を
構成原子として含有する炭素数 1から 20の 2価の有機基を示す。 また一般式 (6) で表される構造であることが、 硬化性の点から好ましい。
ここで式中、 R5は上記と同じ。 一般式 (9) での m, +n, は 1以上 5 0以 下であるが、 重合体とヒ ドロシラン化合物との相溶性、 得られるエポキシ基およ び/またはォキセタン基含有有機重合体を含む光硬化性組成物の反応性の点から、 好ましくは 3以上 3 0以下であり、 特に好ましくは 4以上 20以下である。
m' の数は、 1以上であるが、 m' の数により有機重合体 (A) の反応性が調 整可能である。 得られるエポキシ基および Zまたはォキセタン基含有有機重合体 の反応性の点より、 m' は 2以上が好ましい。
n ' の数は、 0以上であるが、 n' の数によりヒ ドロシラン化合物と不飽和基 含有有機重合体との相溶性が調整可能である。 特に有機重合体 (A) の主鎖骨格 が飽和炭化水素有機重合体である場合は、 n' は 1以上が好ましく、 m' が 2以 上のときには n' も 2以上がより好ましい。 また極性の高い主鎖骨格の場合は、 例えばォキシアルキレン重合体のような場合は、 n, は 1が好ましい。
一般式 (1 0) での in' ' +η' ' は 1以上 20以下であるが、 重合体末端の エポキシ基数の増加の点および重合体とヒ ドロシラン化合物との相溶性の点から、 好ましくは 3以上 2 0以下である。
mf ' の数は、 1以上であるが、 m' ' の数により有機重合体 (A) の反応性 が調整可能である。 得られるエポキシ基および/またはォキセタン基含有有機重 合体の反応性の点より、 ' は 2以上が好ましい。
η' ' の数は、 0以上であるが、 η' ' の数により ヒ ドロシラン化合物と有機 重合体との相溶性が調整可能である。 特に有機重合体 (Α) の主鎖骨格が飽和炭
化水素有機重合体である場合は、 n ' ' 1以上が好ましい。 また極性の高い主鎖 骨格の場合は、 例えばォキシアルキレン重合体のような場合は、 n ' ' は 1が好 ま,しい。
なお、有機重合体とヒ ドロシラン化合物の相溶性は、上記の如く、 n 'や η ' ' の増減により調整できるが、 その他の相溶性調整手段として置換基 R 2を適切に 選択する方法がある。 例えば、 有機重合体が飽和炭化水素系重合体の場合には、 R 2は炭素数 2〜 2 0のアルキル基や炭素数 6〜 2 0のァリール基、 炭素数 Ί〜 2 0のァラルキル基が好ましい。
これらのヒ ドロシラン化合物は、公知の合成方法により合成することができる。 例えば、 S i原子上に炭化水素および水素原子を有するポリシロキサン化合物の ヒ ドロシリル基を、 ァリル基等の末端に不飽和基を有する化合物でヒ ドロシリル 化反応させることにより、 上記のヒ ドロシラン化合物を得ることができる。
すなわちエポキシ基おょぴ zまたはォキセタン基を導入する場合は、 例えばァ リルダリシジルエーテルなどの化合物とヒ ドロシリル基を有するポリシロキサン 化合物とをヒ ドロシリル化することで導入可能である。
同様に上記一般式 (2 ) 、 ( 3 ) での R 2の導入方法は、 末端にァリル基等の不 飽和基を有する炭化水素あるいは α—メチルスチレン等をヒ ドロシリル化させる ことで導入可能である。
一般式 ( 1 0 ) の化合物としては、 1, 3, 5, 7—テトラメチルシクロテト ラシロキサンを上記のように変成し、 エポキシ基および/またはォキセタン基お ょぴ R 2を導入する方法が、 原料の 1, 3, 5, 7—テトラメチルシクロテトラシ ロキサンの入手性、 導入時の選択性が高く好ましい。
末端に不飽和基を有する有機重合体の合成方法としては、 一般的に知られてい る方法で問題はなく、 例えばリ ビングカチオン重合等により末端がハロゲン基で あるようなものは、 金属アルコキシドにより脱ハロゲン化水素する方法、 あるい は四塩化チタン等の存在下、 ァリルトリメチルシラン等を反応させることにより 不飽和基を導入することができる。 また、 水酸基末端に不飽和結合を有する化合
物を反応させて、 エーテル結合、 エステル結合、 ウレタン結合、 カーボネート結 合などにより導入させる方法等が挙げられる。
例えば、 水酸基末端を有する重合体を不飽和基末端にする場合は、 水酸基末端 を一 ON aや一 OKなどのォキシメタル基にした後、 一般式 (1 1 )
CH2= C H— R12— Y —般式 (1 1 )
または一般式 (1 2 )
CH2= C (R13) 一 R12— Y —般式 (1 2 )
(式中、 R12は炭素数 1から 2 0の 2価の有機基、 R13は炭素数 1 0以下の炭化水 素基、 Yはハロゲン原子。 ) で示される不飽和基含有化合物を反応させる方法が 挙げられる。
末端水酸基をォキシメタル基にする方法としては、 N a Kのごときアルカリ 金属; N a Hのごとき金属水素化物; N a O C 113のごとき金属アルコキシド; N a OH KOHなどのアル力リ水酸化物などと反応させる方法があげられる。 一般式 (1 0 ) または (1 1 ) で示される不飽和基含有化合物の具体例として は、
CH2= C H- CH2- C 1 C H2= CH- CH2- B r C H2= C H— C 2H4_ C
1 G ri o— C H― C 2H4 ~ £> Γ Ii_2 h"― 3H g― 1 Crig^ CH— C 3
H6- B r CH2= C (CH3) 一 CH2_ C 1 CH2= C (CH3) _ CH2_ B r CH2= C (CH2C H3) — C H2— C l CH2= C (CH2C H3) — CH2— B r CH2= C (CH2CH (CH3) 2) — CH2— C l CH2= C (CH2CH (CH3) 2) - C H2- B r
等が挙げられ、 特に反応性の点から、 CH2= CH— CH2— Cし CH2= C (C H3) _ CH2— C 1が好ましい。
不飽和基の導入方法としては、 これ以外に CH2= CH— C H2—基や CH2= C (C H3) — CH2_基等を有するイソシァネート化合物、 カルボン酸、 エポキシ 化合物等を用いることもできる。
上記ヒ ドロシリル化の反応は、 末端に不飽和基を有する有機重合体とヒ ドロシ
ラン化合物を、 V I I I族遷移金属触媒の存在下で反応させる方法が好ましい。
V I I I族遷移金属触媒としては、 白金、 ロジウム、 コバルト、 パラジウム及 ぴニッケル等の V I I I族遷移金属元素から選ばれた金属錯体触媒等が有効に使 用される。 例えば、 H2 P t C 1 6 · 6 H20、 白金一ビュルシロキサン錯体、 白金 —ォレフィン錯体、 P tメタル、 R h C 1 ( P P h 3) 3、 R h C 1 3、 R h /A 1 203、 R u C 1 3、 I r C 1 3、 F e C 1 3、 P d C 1 2 · 2 H20、 N i C 1 2等のよ うな化合物が使用できるが、 ヒ ドロシリル化の反応性の点から、 H2 P t C 1 6 · 6 H20、 白金一ビニルシロキサン錯体、 白金ーォレフイン錯体のいずれかである ことが好ましい。 特に白金—ビュルシロキサン錯体が反応誘導期が短い等の点で 好ましい。
ヒ ドロシリル化反応の触媒としては、 これら以外にも A 1 C 1 3、 T i C l 4等 やペンゾィルパーオキサイ ドなどのラジカル開始剤等も使用することができる。
ヒ ドロシリル化反応は、 重合体が劣化等の好ましくない副反応が起こらない温 度であれば、 反応速度等の点から好ましい温度を選択すればよいが、 通常 1 0〜 1 5 0 °C、 好ましくは 2 0〜 1 2 0 °C、 さらに好ましくは 4 0〜: L O 0 °Cの範囲 とするのが好適であり、反応温度の調節、反応系の粘度の調整など必要に応じて、 ベンゼン、 トルエン、 キシレン、 テ トラヒ ドロフラン、塩化メチレン、 ペンタン、 へキサン、 ヘプタン等の溶剤を用いることができる。
ヒ ドロシリル化反応の反応促進には、 特開平 8— 2 8 3 3 3 9号公報で開示さ れる酸素の使用による触媒の再活性化や硫黄添加などの方法を用いることができ る。
さらにヒ ドロシリル化反応において有機重合体、 反応溶媒、 系中の可塑剤等が 酸素により酸化されることを抑制するために、 酸化防止剤の存在下でヒ ドロシリ ル化反応を行うことができる。
エポキシ基および/またはォキセタン基含有ケィ素基の導入率を測定する方法 としては種々の方法が考えられるが、 現在のところ NM Rスペクトルにより、 ェ ポキシ基および/またはォキセタン基含有ケィ素基の導入された末端と導入され
なかった末端の積分値を比較することで正確な値を得ることができる。 次に、 本発明における末端にエポキシ基および/またはォキセタン基含有ケィ 素基を有する飽和炭化水素系重合体の製法について詳しく説明する。
本発明の末端にエポキシ基および Zまたはォキセタン基を有するィソブチレン 系重合体は、 ィニファー法と呼ばれる重合法 (ィニファーと呼ばれる開始剤と連 鎖移動剤を兼用する特定の化合物を用いるカチオン重合法) で得られた末端官能 型、 好ましくは、 全末端官能型イソブチレン系重合体を用いて製造することがで きる。 例えば、 この重合体の脱ハロゲン化水素反応や特開昭 6 3— 1 0 5 0 0 5 号公報に記載されているような重合体への不飽和基導入反応等により末端に不飽 和基を有するポリイソプチレンを得た後、 一般式 (8 ) 、 ( 9 ) あるいは (1 0 ) で示されるようなエポキシ基含有ヒ ドロシラン化合物を白金触媒を用いてヒ ドロ シリル化反応で付加反応をさせることによりエポキシ基含有ケィ素基を重合体に 導入する方法があげられる。
水添ポリブタジエン系重合体では、 たとえば、 まず、 末端ヒ ドロキシ水添ポリ ブタジエン系重合体の水酸基末端を一 O N aや _ O Kなどのォキシメタル基にし た後、 一般式 (1 1 ) または一般式 (1 2 ) で表される不飽和基含有化合物を反 応させる方法により、 同様に末端に不飽和基を含有する水添ポリブタジエン系重 合体を得ることが可能である。
上記方法では、 出発原料として使用した末端ヒ ドロキシ水添ポリブタジエン系 重合体とほぼ同じ分子量をもつ末端不飽和基含有水添ポリブタジェン系重合体が 得られるが、 より高分子量の重合体を得たい場合には、 一般式 (1 1 ) あるいは 一般式(1 2 ) の有機ハロゲン化合物を反応させる前に、塩化メチレン、 ビス (ク 口ロメチル) ベンゼン、 ビス (クロロメチル) エーテノレなどのごとき、 1分子中 にハロゲンを 2個以上含む多価有機ハ口ゲン化合物と反応させれば分子量を増大 させることができ、 その後一般式 (1 1 ) あるいは一般式 (1 2 ) で示される有 機ハロゲン化合物と反応させれば、 より高分子量でかつ末端にォレフィン基を有
する水添ポリブタジエン系重合体を得ることができる。
末端不飽和基含有水添ポリブタジエン系重合体へのエポキシ基および/または ォキセタン基含有ケィ素基の導入は、 前記ィソブチレン系重合体の場合と同様に ヒ ドロシラン化合物を白金系触媒を用いて付加反応をさせることにより製造され る。
飽和炭化水素系重合体が、 芳香族環でない不飽和結合を分子中に実質的に含有 しない場合には、 不飽和結合を有する有機系重合体のような従来のゴム系重合体 から形成される被膜と比べて耐候性がよい。 また、 該重合体は炭化水素系重合体 であるので低気体透過性や耐水性がよく、 低気体透過性の高い被膜を形成する。 本発明のエポキシ基および/またはォキセタン基含有ケィ素基を末端に有する ォキシアルキレン系重合体の製造法としては、 特に限定されず、 例えば末端に不 飽和基を有するォキシアルキレン系重合体と一般式 (8 ) 、 (9 ) あるいは一般 式 (1 0 ) で示されるエポキシ基含有モノヒ ドロシラン化合物によるヒ ドロシリ ル化反応により得ることができる。
末端に不飽和基を有するォキシアルキレン系重合体の製造法としては、 例えば エーテル結合により不飽和基を導入する場合は、 ォキシアルキレン系重合体の水 酸基末端のメタルォキシ化により— O M (Mは N aまたは K等) を生成した後、 一般式.(1 1 ) または一般式 (1 2 ) で表される不飽和基含有化合物を反応させ る方法が同様に利用できる。
本発明のエポキシ基および/またはォキセタン基含有ケィ素基を末端に有する ビニル系重合体の製造法としては、 特に限定されず、 例えば末端に不飽和基を有 するォキシアルキレン系重合体と一般式 (8 ) 、 ( 9 ) あるいは一般式 (1 0 ) で示されるエポキシ基含有モノヒ ドロシラン化合物によるヒ ドロシリル化反応に より得ることができる。
加水分解性基の交換反応によるエポキシ基および/またはォキセタン基の導入 は、 末端に加水分解性シリル基を有する有機重合体と 1分子中に少なく とも 1つ 以上のェポキシ基および/またはォキセタン基おょぴ 1つの水酸基を有する化合
物との加水分解性基の交換反応により末端にェポキシ基および またはォキセタ ン基が導入可能となる。
上記の末端に加水分解性シリル基を有する有機重合体の加水分解性シリル基は、 特に限定されるものではないが、 代表的なものを示すと、 例えば一般式 (1 3) で表わされる基が挙げられる。
_[S i R^O]^ i (R2 3_n) Qn (1 3)
(式中 R R2、 mおよび nは前記一般式 (1) と同じである。 Qは水酸基また は加水分解性基を示し、 Qが二価以上存在する時、それらは同一であってもよく、 異なっていてもよい。 )
上記 Qのうちの加水分解性基は特に限定されず、 従来公知の加水分解性基であ れば良い。 具体的には例えば水素原子、 ハロゲン原子、 アルコキシ基、 ァシルォ キシ基、 ケトキシメート基、 アミノ基、 アミ ド基、酸アミ ド基、 アミノォキシ基、 メルカプト基、 アルケニルォキシ基等が挙げられる。 これらの内では、 加水分解 性が穏やかで取扱やすいという点でメ トキシ基、 エトキシ基、 プロポキシ基、 ィ ソプロポキシ基等のアルコキシ基が好ましい。
水酸基や加水分解性基が反応性ケィ素基中に 2個以上存在する場合には、 それ らは同一であっても良く、 異なっていてもよい。
なお、 下記一般式 (1 4) で表される反応性ケィ素基が入手が容易であるため 好ましい。
—S i (R2 3-n) Qn ( 1 4)
(式中 R2、 Q、 nは前記と同じ。 )
上記の末端に加水分解性シリル基を有する有機重合体の製造法としては、 特に 限定されず、前述した不飽和基を末端に有する有機重合体と、 下記一般式 (1 5) で表されるヒ ドロシラン化合物とを前述した付加反応の方法を用いることで得る ことができる。
H-[S i R^]^ i (R23_n) Qn ( 1 5)
(式中 R 1, R 2, Q , mおよび nは前記と同じ。 )
上記ヒ ドロシラン化合物は、 特に一般式 (1 6 )
H - S i ( R 23-n) Q n ( 1 6 )
で表わされる化合物が入手性の点から好ましい。
(式中 R 2, Q, mおよび nは前記と同じ。 )
一般式 (1 5 ) または ( 1 6 ) の化合物を具体的に例示するならば、 トリ クロ ノレシラン、 メチノレジクロノレシラン、 ジメチノレクロノレシラン、 フエニノレジクロノレシ ラン、 ト リ メチノレシ口キシメチノレク口ルシラン、 1, 1, 3, 3 —テ トラメチル - 1 —ブロモジシロキサンの如きハロゲン化シラン類; トリメ トキシシラン、 ト リエ トキシシラン、 メチルジェ トキシシラン、 メチノレジメ トキシシラン、 フエ二 ルジメ トキシシラン、 ト リ メチルシロキシメチルメ トキシシラン、 ト リメチルシ 口キシジェ トキシシランの如きアルコキシシラン類;メチルジァセ トキシシラン、 フエニルジァセ トキシシラン、 ト リァセ トキシシラン、 ト リ メチルシ口キシメチ ルァセ トキシシラン、 トリメチルシロキシジァセ トキシシランの如きァシロキシ シラン類; ビス (ジメチルケ トキシメー ト) メチルシラン、 ビス (シクロへキシ ルケトキシメー ト) メチルシラン、 ビス (ジェチルケトキシメ一ト) トリメチル シロキシシラン、 ビス (メチノレエチルケ トキシメート) メチルシラン、 トリス (ァ セ トキシメート) シランの如きケ トキシメートシラン類 ; メチルイソプロぺニル 才キシシランの如きアルケニルォキシシラン類などが挙げられる。 これらのなか で、 特にアルコキシシラン類が好ましく、 アルコキシ基の中でもメ トキシ基、 ェ トキシ基、 プロポキシ基、 イソプロポキシ基が特に好ましい。
前記末端に加水分解性シリル基を有する有機重合体と反応せしめる 1分子中に 少なく とも 1つ以上のエポキシ基および/またはォキセタン基おょぴ 1つの水酸 基を有する化合物としては、 特に限定はないが反応性の点で 2級あるいは 1級の 水酸基を有する化合物が好ましい。
上記 1分子中に少なく とも 1つ以上のエポキシ基および Zまたはォキセタン基 および 1つの水酸基を有する化合物の具体的例としては、 下記一般式 (1 7 ) で
表される化合物が使用できる。
W - O H ( 1 7 )
(W はエポキシ基および/またはォキセタン基を含有する 1価の有機基) これらの化合物の具体的な化合物としては、 入手性の点より 2 , 3—エポキシ 一 1—プロパノール、 3—ェチノレ _ 3—ヒ ドロキシメチノレオキセタン、 グリセリ ンジグリ シジルエーテル等の化合物が挙げられる。
これら化合物の使用量に特に限定はないが、 交換反応を速やかに進行させるた めに末端に加水分解性シリル基を有する有機重合体の加水分解性基に対し、 1当 量以上使用することが好ましい。
加水分解性基の交換反応は、 上記の末端に加水分解性シリル基を有する有機重 合体と上記の 1分子中に少なくとも 1つ以上のエポキシ基および/またはォキセ タン基および 1つの水酸基を有する化合物にエステル交換反応触媒を加え反応さ せることにより実施することができる。
上記エステル交換反応触媒は、 アルカリ金属アルコキシド、 S n化合物、 T i 化合物、 Z n化合物、 B a化合物、 及び慣用的な強アルカリ化合物により例示さ れる。適切なエステル交換反応触媒の例としては、ジメチル錫ネオデカノエート、 ジプチル錫ジァセテ一ト、ジブチル錫ジラゥレート、ジォクチル錫ジラゥレー ト、 ジブチル錫ジォクテート、 ナフテン酸亜鉛、 ナフテン酸コバルト、 ォクチル酸亜 鉛、ォクチル酸錫、ォクチル酸コバルト、ジィソォクチルメルカプトァセテ一ト、 ナフテン酸ジノレコニゥム、 ォクチノレ酸ジノレコニゥム、 テトラブチルチタネー ト、 テトライソプロピルチタネート、 水酸化バリゥム一水和物及ぴ他の有機金属触媒 が挙げられる。 特に、 エステル交換反応触媒がテトライソプロピルチタネート、 水酸化バリゥム一水和物及びナトリウムメ トキシド等のアルコキシドから選ばれ ることが好ましい。
上記エステル交換反応触媒の量は、 特に限定はないが、 通常上記有機重合体に 対し 5 0 p p m〜 1 0 0 , 0 0 0 p p m、 好ましくは 5 0 p p m〜 3 0 0 0 p p mの範囲で使用される。
この反応は溶剤をさらに含むことができる。 この溶剤に特に限定はないが、 例 えばペンタン、 シクロペンタン、 へキサン、 シク口へキサン、 ヘプタン、 ォクタ ン及ぴノナン等の脂肪族炭化水素;例えばベンゼン、 トルエン及びキシレン等の 芳香族炭化水素;並びに例えばペルクロ口エチレン及ぴブロモベンゼン等のフッ 素置換、 塩素置換及び臭素置換された脂肪族又は芳香族炭化水素により例示され る。 また 2種以上の無極性溶剤を併用することもできる。
溶剤の量に限定はないが、 ポリマー 1 0 0重量部当たり 0〜 1 0 0重量部の溶 剤を含むことができる。
この方法は、 生成物から揮発分を除去することで反応を促進することが可能で ある。 揮発性成分を除去する方法は当該技術分野で公知であるものが使用可能で ある。 本発明において任意の揮発性成分除去方法を使用することができる。 その ような方法は、 加熱、 加熱し減圧すること、 ロータリーエバポレーター、 薄膜ス トリ、フ / 一、 ! ィプ式フイノレムェ/ ポレー夕一 (wiped fi lm evaporator)又 fまこ れらの組み合わせにより例示される。 好ましくは、 揮発分は、 生成物を約 2 6 0 0〜1 3 3 0 0 P aの減圧下で 5 0〜 1 5 0 °Cの温度に加熱することにより除去 される。
本発明の末端にエポキシ基および/またはォキセタン基含有ケィ素基を有する 有機重合体は、 選択的に末端にエポキシ基および/またはォキセタン基含有ケィ 素基を導入した新規な重合体であり、 製造の際にも重合体主鎖の劣化等を回避し 合成することが可能である。 このようにして得られた重合体は、 それ単独でェポ キシ基の公知な反応を利用して硬化させることも可能であり、 また従来使用され ているェポキシ系硬化物への改質剤的な使用法によっても重合体主鎖由来の特徴 を発現することが期待される。
本発明の末端にエポキシ基および/またはォキセタン基含有ケィ素基を有する 有機重合体硬化方法としては、 エポキシ基および/またはォキセタン基含有化合 物の一般的な硬化剤により、 エポキシ基および/またはォキセタン基を反応させ 硬化させることができる。 硬化剤としては、 アミン系硬化剤、 酸系硬化剤、 3フ
ッ化ホウ素ァミンコンプレックス系硬化剤、 カチオン系光硬化剤等が一般的な方 法で使用可能である。
特に光硬化反応を用いた場合は、 短時間で硬化させることが可能となり好まし レ、。
本発明の光力チオン開始剤 (B) は、 光により、 (A) 成分の樹脂のカチオン 重合を開始する化合物であれば特に限定はなく、 いずれでも使用することができ る。 例えば光力チオン開始剤の好ましい例として下記一般式 (7) で表される構 造が挙げられる。
[R7 aR8 bR9 cR10 dW] u+ [MZV+U] u- (7)
(式中、 Wは、 S、 S e、 T e、 P、 A s、 S b、 B i、 0、 I、 B r、 C l、
T i、 Z r、 F e、 Ru、 〇 sまたは N≡Nであり、 R7、 R8、 R9、 および R10 は同一または異なる有機基であり、 a、 b、 cおよび dはそれぞれ 0〜 3の整数 であって、 (a + b + c + d) は Wの価数に等しい。 Mは、 錯体 [MZV+U] の中 心原子を構成する金属またはメタロイ ドであり、 例えば、 B、 P、 A s、 S b、 F e、 S n、 B i、 A l、 C a、 I n、 T i、 Z n、 S c、 V、 C r、 Mn、 C o等である。 Zは例えば、 Mに配位する配位子で、 F、 C l、 B r等のハロゲン 原子や有機基である。 uは錯体イオンの正味の電荷である。 Vは Mの原子価であ る。 )
またカチオン系光開始剤 (B) 力 ォニゥム塩、 スルホン酸のジァリールョー ドニゥム塩、 スルホン酸のトリァリールスルホニゥム塩、 ボロン酸のジァリール ョ一ドニゥム塩又はボロン酸のトリァリ一ルスルホ -ゥム塩から選ばれるカチォ ン系光開始剤であることが、 入手性が容易なことから好ましい。
これらのォニゥム塩の具体例としては、 ジフエ二ルョードニゥム、 4ーメ トキ シジフエニノレョー ドニゥム、 ビス (4—メチノレフェェノレ) ョー ドニゥム、 ビス ( 4 - t e r t一プチノレフエニル) ョ一ドニゥム、 ビス (ドデシルフェニル) ョード 二ゥム、 ト リノレク ミノレョードニゥム、 ト リ フエニノレスノレホニゥム、 ト リ フエ二ノレ スノレホニゥム、 ジフエニノレー 4—チオフエノキシフエニノレスノレホニゥム、 ビス 〔 4
- (ジフヱ-ノレスルフォニォ) 一フエ二ノレ〕 スノレフイ ド、 ビス 〔4一 (ジ (4— ( 2—ヒ ドロキシェチノレ) フエ二ノレ) スノレホニォ) 一フエ二ノレ〕 スノレフイ ド、 η 5 - 2 , 4 - (シク口ペンタジェニル) 〔1, 2, 3, 4, 5, 6— η — (メチ ルェチル) ベンゼン〕 一鉄 (1 +) 等が挙げられる。 一般式 (7) において陰ィ オンの具体例としては、 テトラフルォロポレート、 テトラキス (ペンタフルォロ フエ-ノレ) ポレート、 へキサフノレオ口ホスフェー ト、 へキサフノレオ口アンチモネ ート、 へキサフルォロアルセネート、 へキサクロ口アンチモネート等が挙げられ る。 これらの光力チオン開始剤は、 1種単独であるいは 2種以上を組み合わせて 使用することができる。
また上記のォニゥム塩以外にも、 デカメチルフエ口セン/テトラキス ( 3, 5 ージフルオロフェエル) ポラート、 デカメチルフエ口セン/テトラキス (3, 5 一ジブルォロメチルフエニル)ポラート、デカメチルフエ口セン/テトラキス [ 4 — (トリフルォロメチル) フエニル] ポラートなど特開平 1 1 一 4 9 7 9 1、 特 開 2 0 0 0— 2 2 6 3 9 6等に記載の開始剤を使用することが可能であり、 組成 物の安定性向上等の効果がある。
本発明の樹脂組成物における (Β) 成分の含有割合は、 通常 0. 1〜1 0重量 部であり、 好ましくは、 0. 3〜3重量部である。 (Β) 成分の含有割合 0. 1 重量部以上であるとより樹脂組成物の硬化状況が良好となり好ましく、 又硬化後 に光力チオン開始剤が溶出の予防の観点から 1 0重量部以下が好ましい。
本発明の光硬化性組成物には、 (Β) 成分に加え增感剤を使用することが可能 である。 増感剤としては、 特に限定はなく、 一般のカチオン系光開始剤に用いら れる增感剤なら問題なく使用できる。 具体例としては、 ジァリールョードユウム やトリァリ一ルスルホニゥム塩の增感にはアントラセン、 ピレン、 ペリレンなど の芳香族炭化水素が、 ジァリールョードニゥム塩の增感にはべンゾフエノン、 キ サントン、 チォキサントン、 ミヒラーケトン、 9, 1 0—フエナントラキノンな どの芳香族ケトン、 ェォシン、 ケトクマリン、 アタリジン染料などが、 トリァリ 一ルスルホニゥム塩の増感には芳香族ァミン、 芳香族 3級ァミン、 クマリン、 ィ
ソベンゾフランなどが挙げられるが、 これらに限定されるものではない。 . 本発明の光硬化性組成物には、 必要に応じてエポキシ基を有する化合物 (C ) および/またはォキセタン基を有する化合物 (D ) を含有することができる。 ェ ポキシ基を有する化合物 (C ) は、 硬化物の硬化性や機械的強度を向上すること ができ、 以下のものが例示できる。 例えば、 エポキシ基を 1個有する化合物とし ては、 フヱ-ルグリシジルエーテル、 プチルグリシジルエーテル等があり、 ェポ キシ基を 2個以上有する化合物としては、 へキサンジオールジグリシジルエーテ ル、 テトラエチレンダリコールジグリシジルエーテル、 トリメチローノレプロパン トリグリシジルエーテル、 ビスフエノール Aジグリシジルエーテル、 水添ビスフ ェノール Aジグリシジルエーテル、ノボラック型エポキシ化合物等が挙げられる。 また、 脂環式エポキシ基を有する化合物も問題なく使用できる。
これらの (C ) 成分は、 1種単独であるいは 2種以上を組み合わせて使用する ことができる。 また (A) 成分の主鎖骨格の種類により相溶性等が異なるため、 ( A) 成分に適した化合物を選択することが好ましい。 本発明の樹脂組成物にお ける (C ) 成分の含有割合は、 通常 1〜 7 0重量部であり、 好ましくは、 1〜5 0重量部である。 (C ) 成分の添加は、 組成物の硬化性、 接着性、 耐熱性を改良 させるのに有効である。
本発明におけるォキセタン環を有する化合物 (D ) は、 一般式 (1 8 ) で表さ れるォキセタン環を少なく とも 1つ有する化合物であればいずれでも使用するこ とができる。 5 (18)
ここで式中、 R 14、 R 15は、 同一または異なった水素、 酸素、 及び窒素からな
る群より選択される 1種以上を構成原子として含有する炭素数 1から 2 0の 2価 の有機基を示す。
これらォキセタン環を有する化合物としては、 3—ェチルー 3 - ヒ ドロキシメ チルォキセタン、 3ーェチルー 3― (フエノキシメチル) 才キセタン、 3—ェチ ノレ一 3 —へキシロキシメチノレオキセタン、 3—ェチルー 3― ( 2—ェチノレへキシ 口キシメチル) 才キセタン、 3—ェチルー 3— { [ 3 - (ト リエ トキシシリル) プロポキシ] メチル } 才キセタン、 ジ [ 1一ェチル ( 3—ォキセタニル) ] メチ ルエーテル、 1 , 4一ビス { [ 3 - ェチル一 ( 3 - ォキセタニル) メ トキシ] メ チル } ベンゼン、 3 , 3 '一ジメチノレー 2 - ( ρ—メ トキシフヱ二ノレ) —ォキセ タン等の化合物が挙げられる。
これらの (D ) 成分は、 1種単独であるいは 2種以上を組み合わせて使用する ことができる。 また (A) 成分の主鎖骨格の種類により相溶性等が異なるため、
(A) 成分に適した化合物を選択することが好ましい。 本発明の樹脂組成物にお ける (D ) 成分の含有割合は、 通常 1 〜 7 0重量部であり、 好ましくは、 1 〜 5 0重量部である。 (D ) 成分の添加は、 組成物の高速硬化性、 高分子量化に有効 である。
本発明の硬化性組成物には、 本発明の効果を損なわない範囲で、 他の光力チォ ン重合性化合物、 シランカップリング剤、 充填剤、 改質剤、 安定剤、 他の樹脂成 分等のその他の成分を含有することができる。
他の光力チオン重合性化合物としては、 例えば、 ォキソラン化合物、 環状ァセ タール化合物、 環状ラク トン化合物、 チイラン化合物、 チェタン化合物、 スピロ オルソエステル化合物、 ビュルエーテル化合物、 エチレン性不飽和化合物、 環状 エーテル化合物、 環状チォエーテル化合物、 ビ二ル化合物等が挙げられる。 これ らは、 1種単独でも複数種を組み合わせて使用してもよい。
シランカツプリング剤とは、エポキシ基、力ルポキシル基、メタクリロイル基、 イソシァネート基等の反応性基を有するシラン化合物が挙げられる。具体的には、 トリメ トキシシリル安息香酸、 γ—メタクリロキシプロピルトリメ トキシシラン、
ビュルト リァセ トキシシラン、 ビュルト リメ トキシシラン、 γ —イソシアナトプ 口ピルト リエ トキシシラン、 γ —グリ シドキシプロピルトリ メ トシキシラン、 β 一 (3, 4 _エポキシシクロへキシル) ェチルトリメ トシキシラン等が挙げられ る。 これらの成分は、 1種単独であるいは 2種以上を組み合わせて使用すること ができる。本発明の樹脂組成物におけるシランカツプリング剤成分の含有割合は、 特に限定はないが通常 0 . 1〜 2 0重量部であり、 好ましくは、 0 . 3〜 1 0重 量部である。 0 . 1〜 2 0重量部の範囲では、 接着性向上の効果と経済性のバラ ンスの点で優れている。
充填剤としては、 例えば、 微粒子シリ力、 ガラスビーズ、 タルク、 スチレン系 ポリマー粒子、 メタクリレート系ポリマー粒子、 エチレン系ポリマー粒子、 プロ ピレン系ポリマー粒子等が挙げられ、 中でも無機充填剤が好ましく使用でき、 特 に微粒子シリカが好ましい。 これらは、 1種単独でも複数種を組み合わせて使用 してもよい。
微粒子シリ力は、 一次粒子の平均系が 5〜 1 0 0 n mのシリ力が好ましい。 こ れらは、 表面処理、 未処理のものいずれも使用できる。
これらの無機充填剤を使用することにより、 高強度化、 耐透湿性や接着性を向 上させることができる。
改質剤としては、 例えば重合開始助剤、 レべリング剤、 濡れ性改良剤、 界面活 性剤、 可塑剤等が挙げられる。 これらは、 1種単独でも複数種を組み合わせて使 用してもよレ、。
安定剤としては、 老化防止剤、 酸化防止剤、 光安定剤、 紫外線吸収剤等が挙げ られる。 これらは 1種単独でも複数種を組み合わせて使用してもよい。
他の樹脂成分としては、 例えばポリアミ ド、 ポリウレタン、 ポリブタジエン、 ポリエーテル、 ポリエステル、 アク リル樹脂、 シリ コン樹脂、 フッ素系樹脂等の 樹脂成分が挙げられる。
本発明の硬化性組成物は、 各成分を均一に混合することにより調製される。 混 合する方法に特に限定はないが、 まず (B ) 成分のカチオン系光開始剤を除くそ
の他の成分を十分に混合した後に、 (B ) 成分のカチオン系光開始剤を混合する ことが組成物の安定性の点で好ましい。 特に水分を多く含有するような成分につ いては、 事前に脱水処理を施し、 混合することが好ましい。 混合する方法、 装置 には特に限定はないが、 手攪拌、 機械的攪拌装置、 ロールミル等を用い適宜混合 することにより調整される。
本発明の硬化物は、 硬化性組成物に光エネルギー源を照射することにより得ら れる。 光エネルギー源としては、 一般に光硬化反応に用いられるものを特に制限 なく使用できるが、 紫外線、 電子線、 可視光等を挙げることができる。 例えば、 光硬化性組成物自体の硬化は、 塗布された基材を望ましい光エネルギー源、 例え ば紫外線ランプの下を所定の速度で通過させ、 そして必要なエネルギー源を出力 状態に所定の時間おくことによりその塗布された基材を完全に光エネルギーに暴 露することを含む公知の方法のうちのいずれかにより達成される。
硬化物を得る際に施す光硬化性組成物の塗布としては、 例えば、 はけ塗り、 押 出、 吹付け、 グラビア、 キスロール、 ディスペンサー及びエアーナイフによるよ うな当該技術分野で公知の任意の適切な手法が適用できる。
本発明の光硬化性組成物を塗布する固体基材は、 例えば紙、 ポリオレフインフ イルム、 ポリオレフイン被覆紙、 箔、 木材、 厚紙及び綿等の柔軟なシート材料; 例えばアルミニウム、 銅、 スチール及び銀等の金属材料;例えばガラス及び石等 のケィ質材料;並びに例えばポリオレフィン、 ポリアミ ド、 ポリエステル及びポ リアタリ レート等の合成ポリマー等が挙げられる。
また必要に応じて光エネルギーの照射の後に、 さらに加熱等の後硬化させるこ とにより、 より十分に硬化させることができる。
本発明の光硬化性組成物は、 接着剤、 塗料、 シーリング剤組成物、 防水剤、 吹 き付け剤、 型取り用材料、 注入型ゴム材料等として有用である。 具体的には、 U V硬化型塗料 ' コーティング ·インキ、 液状ソルダレジスト、 液晶用レジスト、 光ファイバ一コーティング剤、 U V ·可視光硬化型接着剤、 光ディスクコーティ ング剤、 電子部品用封止剤等が挙げられる。 中でも、 加熱工程の低減、 生産性向
上が求められる電子部品用途への使用が好適である。
本発明の光硬化性組成物を含む硬化物を構成要素としてなる製品は、 本発明の 光硬化性組成物、 あるいは必要に応じて他の光力チオン重合性化合物、 シラン力 ップリング剤、 充填剤、 改質剤、 他の樹脂成分等混合した光硬化性組成物を、 各 用途の使用部位に本発明の光硬化性組成物を塗布し、 光エネルギーを照射するこ とによりその部位を接着、 シール、 封止したものである。
これら製品の製造方法としては特に限定はなく、 各用途に適した工程が使用で きる。 例えば、 光ファイバ一コーティングでは、 光ファイバ一の紡糸装置中に本 発明の光硬化性組成物を満たした力ップおよび U V照射装置を設置し、 ファイバ 一線を引きカップを通過することで光硬化性組成物をコーティングし、 その後 U V照射により硬化し、 本発明の硬化性組成物からなる硬化物で被覆された光ファ ィバーを得ることができる。
その他、 有機 E L素子の封止剤として使用した場合は、 基材上の素子周辺を本 発明の光硬化性組成物でシールし、 キャップした後 U V照射し封止することによ り、 本発明の硬化性組成物からなる硬化物で本発明の硬化物で封止された有機 E L素子が得られる。
本発明の光硬化性組成物は、 (A) 成分の主鎖骨格の種類を変更することでそ の主鎖骨格独特の性能が期待できる。
特に本発明の (A) 成分の主鎖骨格が飽和炭化水素系重合体の場合は、 本発明 の光硬化性組成物おょぴそれを含む硬化物に優れた耐熱性、低透湿性、低吸湿性、 低気体透過性等を付与することができる。 このような硬化性組成物は、 電子材料 周辺での接着剤あるいはシーリング剤として好適であり、 特に熱およぴ湿気等に 弱い部品等、 例えば有機 E L等に対する接着剤、 シーリング剤に好適である。 また (A) 成分の主鎖骨格がォキシアルキレン系重合体の場合は、 本発明の光 硬化性組成物おょぴそれを含む硬化物に優れた低温特性、 可とう性、 他成分との 優れた相溶性等を付与することができる。
また (A) 成分の主鎖骨格がアクリル系重合体の場合は、 そのモノマー種の調
整により本発明の光硬化性組成物およびそれを含む硬化物に優れた耐候性、 可と う性、 他成分との優れた相溶性等を付与することができる。
これら (A) 成分の主鎖骨格は、 単一であっても良く、 2種以上を組み合わせ ることで上記の特徴を併せ持つ光硬化性組成物およびそれを含む硬化物を得るこ とが可能である。
実施例
以下、 実施例に基づき本発明を更に詳細に説明するが、 本発明はこれらにより なんら制限を受けるものではない。
(合成例 1 )
(エポキシ基含有ヒ ドロシランの合成)
シロキサン結合の繰返し単位が平均 5個のメチルハイ ドロジエンポリシロキサ ン 23. 3 g、 トルエン 20 gを 20 Om 1三口フラスコに計量し、 冷却管およ ぴ滴下管を取り付け、 90°Cに昇温した。 続いて α—メチルスチレン 7. 7 g、 了リルグリシジルエーテル 7. 44 g、 トルエン 1 5 gおよび白金— 1, 1, 3, 3—テトラメチルー 1, 3—ジビュルジシロキサン錯体(白金換算で 0. 3重量% の トルエン溶液) 6 μ 1の混合物をゆっく り滴下し、 2時間攪拌した。 ヒ ドロシ ランのモル数は、 アルコー/レに溶解させたメチルハイ ドロジエンポリシロキサン にアルカリ水溶液を滴下したときの水素発生量により算出した。 反応の進行は、 1 H— NMRにて α—メチルスチレンの不飽和基のピーク ( 5. 0 p m付近、 5. 3 ρ p m付近) の減少、 ァリルグリシジルエーテルの不飽和基のピーク (5. 3 p pm付近、 5. 9 p pm付近) の減少およびヒ ドロシランのピーク (4. 4 p pm付近) の減少により追跡した。 反応終了後、 — NMRで確認の結果下記式 (14) で表すような α—メチルスチレン基平均 2個、 エポキシ含有基平均 2個 が導入され、 ヒ ドロシリル基が平均 1個残存するヒ ドロシラン (SH— 1) が得 られた。
(合成例 2)
(エポキシ基含有環状ヒ ドロシランの合成)
1, 3, 5, 7—テトラメチルシクロテトラシロキサン 1 O O gを 3 0 0m l 三ッロフラスコに計量し、 9 0°Cに昇温した。 続いて上記ヒ ドロシランに対しァ リルグリシジルエーテル 1 04. 5 g、 1 -へキサデセン 1 02. 6 gおよび白 金一 1, 1 , 3, 3—テトラメチル— 1, 3—ジビ -ルジシロキサン錯体 (白金 換算で 0. 3重量0 /0のトルエン溶液) 1 0 μ 1の混合物をゆっく り と滴下した。 1時間反応後、 1 H— NMRでァリル基のピーク (5. 3 p p m付近、 5. 9 p p m付近) を確認した結果、 反応物のァリル基の消滅が確認でき、 下記式 (1 5) で示される平均 1個のヒ ドロシリル基を有する化合物 (SH— 2) が得られた。
(ァリル末端イソプチレン系重合体の合成)
2 Lの耐圧ガラス製容器に、 三方コックを取り付け、 容器内を窒素置換した後、 注射器を用いて容器内に、 ェチルシクロへキサン (モレキュラーシーブス 3 Aと ともに 1夜間以上放置することにより乾燥したもの) 1 38m lおよびトルエン (モレキュラーシーブス 3 Aとともに 1夜間以上放置することにより乾燥したも の) 1 0 1 2m l、 1, 4—ビス (ひ一クロロイソプロピル) ベンゼン 8. 1 4 g (3 5. 2 mm o 1 ) を加えた。
次にィソブチレンモノマー 2 54m l (2. 9 9 m o 1 ) が入っているニード ルバルブ付耐圧ガラス製液化採取管を、 三方コックに接続して、 重合容器を一 7 o°cの ドライアイス/エタノールバス中につけて冷却した後、 真空ポンプを用い て容器内を減圧にした。 ニードルバルブを開け、 イソブチレンモノマーを液化ガ ス採取管から重合容器内に導入した後、 三方コック内の一方から窒素を導入する ことにより容器内を常圧に戻した。次に、 2—メチルピリジン 0. 38 7 g (4. 1 5 mm o 1 ) を加えた。 次に、 四塩化チタン 4. 90m l (44. 7 mm o 1 ) を加えて重合を開始した。 反応時間 70分後に、 ァリルトリメチルシラン 9. 6 5 g ( 1 3. 4 mm o 1 )を加えてポリマー末端にァリル基の導入反応を行った。 反応時間 1 20分後に、 反応溶液を水 20 0m lで 4回洗浄した後、 溶剤を留去 することによりァリル末端イソブチレン系重合体 (P— 1) を得た。
こう して得られたポリマーの収量より収率を算出するとともに、 Mn及ぴ Mw /Mnを G P C法により、また末端構造を 3 00MH z 1H_NMR分析により各 構造に帰属するプロ トン (開始剤由来のプロ トン : 6. 5〜 7. 5 p p m、 ポリ マー末端由来のァリル基のピーク (4. 9 7 p p m : =CH2、 5. 7 9 p p m : _CH=C) ) の共鳴信号の強度を測定、 比較することにより求めた。 — NM Rは、 V a r i a n G e m i n i 300 (30 OMH z f o r ipi)を用い、 四塩化炭素 Z重ァセ トン中で測定した。
なお、 G P Cは送液システムとして Wa t e r s L C Mo d u l e l、 力
ラムは S h o d e x K— 804を用いて行った。 分子量はポリスチレンスタン ダードに対する相対分子量で与えられる。ポリマーの分析値は、 Μη = 5800、 Mw/Mn = 1. 3 9、
F n (v) = 1. 88 (NMR分析において、 開始剤残基となる芳香族環 1分 子当たりに対するァリル基の数) であった。
(合成例 4 )
(末端ァリル基含有ォキシプロピレン系重合体の合成)
数平均分子量が 2000のポリプロピレンダリコールを開始剤とし、 亜鉛へキ サシァノコパルテー トグライム錯体触媒の存在下、 プロピレンォキサイ ドの重合 を行い、 数平均分子量が 10000のポリプロピレングリコールを得た。 続いて このポリプロピレングリコールの末端水酸基に対して 1. 2倍当量の CH3ON a (メタノール溶液) を添加し、 減圧下でメタノールを除去しながら、 末端をメタ ルォキシ化した。 ここに 1. 3倍当量の 3 _クロロー 1—プロペンを添加し、 反 応させた後、 副生した塩を脱塩精製により除き、 末端にァリル基を有するォキシ プロピレン系重合体 (P— 2) を得た。
得られた重合体の末端ァリル基濃度を測定したところ、 0. 223mmo l / gでめつ 7こ。
(合成例 5 )
(重合体末端へのエポキシ基含有ケィ素基の導入一 1 )
上記の末端にァリル基を含有するポリイソブチレン系重合体 (P— 1 ) 1 00 g、 1, 4, 一 t e r t—ブチノレー 4—ヒ ドロキシトノレエン 0. 05 gおよびト ルェン 100 gを 500m lの三ッ口フラスコに計量し、真空シール付き攪拌機、 冷却管およぴ玉栓を取り付けた。
続いて 1 00°Cに昇温後、 6 %酸素含有空気雰囲気化で硫黄の 1 %トルエン溶 液を 1 1. 1 μ 1滴下、 攪拌し、 続いて白金一 1, 1, 3, 3—テトラメチルー 1, 3—ジビニルジシロキサン錯体のトルエン溶液 21. 6 μ 1 (白金換算で 3 重量%のトルエン溶液) を滴下、攪拌し、上記で合成したヒ ドロシラン化合物 (S
H— 2) 23. 9 gをゆっく り と滴下し 2時間反応させた。
反応の進行は、 1 H— NMRにより末端ァリル基のピーク (5. 1 p p m: =C H2、 5. 9 p p m: -CH=C) の減少、 消滅おょぴ滴下したエポキシ基含有ヒ ドロシランのヒ ドロシリル基 (s i— H) のピーク (4. 8 p p m) の減少によ り確認した。
得られた反応物の1 H— NMRを測定したところ、初期末端ァリル基含有重合体 に対し、 上記記載のァリル基を示すピークおよびヒ ドロシランを示すピークが完 全に消滅していることが判明し、 目的の末端に下記の構造のエポキシ基含有ケィ 素基を含有するイソブチレン系重合体 (A— 1) が得られた。
(合成例 6 )
(重合体末端へのエポキシ基含有ケィ素基の導入一 2)
上記の末端にァリル基を含有するォキシプロピレン系重合体 100 gおよびへ キサン 2 gを 30 Om 1の三ッロフラスコに計量し、 真空シール付き攪拌機、 三 方コックおよぴ玉栓を取り付けた。 これを 90°Cに昇温、 攪拌し、 真空ポンプに より 2時間共沸脱水を行った。
続いて白金一 1, 1, 3, 3—テトラメチル一 1 , 3—ジビュルジシロキサン 錯体(白金換算で 3重量%のトルエン溶液) 4. 10 1滴下し、 よく攪拌した。 続いてエポキシ基含有ヒ ドロシラン(S H— 1 ) 50. 5 gを窒素雰囲気下ゆつく りと滴下し、 その後 6時間攪拌した。
反応の進行は、 1 H— NMRにより末端ァリル基のピーク (4. 97 p p m: = CH2、 5. 79 p p m:— CH= C) の減少、 消滅および滴下したエポキシ基含 有モノ ヒ ドロシランのヒ ドロシリル基 (S i -H) のピーク (4. 6 p pm付近) の減少により確認した。
得られた反応物の1 H— NMRを測定したところ、初期末端ァリル基含有重合体 に対し、 上記記載のァリル基を示すピークおょぴヒ ドロシランを示すピークが完 全に消滅していることが判明し、 末端に下記の構造のエポキシ基含有ケィ素基を 有するォキシアルキレン系重合体 (A— 2) が得られた。
(合成例 7)
(加水分解性シリル基含有重合体の合成)
上記の末端にァリル基を含有するポリイソブチレン系重合体 1 00 gおよぴト ルェン 2 gを 300 m 1の三ッロフラスコに計量し、 真空シール付き攪拌機、 三 方コックおよぴ玉栓を取り付けた。 これを 1 80°Cに昇温、 攪拌し、 真空ポンプ により 2時間脱水おょぴ脱塩酸を行った。
続いて 100°Cに冷却後、 1, 4, 一 t e r t—ブチル一 4—ヒ ドロキシトル ェン 0. 05 g、 白金一 1, 1, 3, 3—テトラメチルー 1, 3ージビュルジシ ロキサン錯体 21. 6 μ 1 (白金換算で 3重量%のトルエン溶液) 、 硫黄の 1 % トルエン溶液を 1 1. 滴下し、 よく攪拌した。
さらに、メチルジメ トキシシラン 5.86 gを滴下管によりゆっく り と滴下し、 その後 6%酸素含有空気中で 2時間攪拌した。 その後、 過剰のメチルジメ トキシ シランを減圧除去し、 下記の構造の末端に加水分解性基を有するイソブチレン系 重合体を得た。
(合成例 8 )
(加水分解性基交換反応)
上記合成例 7で得られた、 末端にアルコキシシリル基を有するポリイソブチレ ン系重合体 100 g、 ぉょぴトノレェン 1 00 gをディーンスタークセパレーター を備えた 5 O Om lのフラスコに計量した。 続いて 3—ェチル一 3—ヒ ドロキシ メチルォキセタン 14. 4 gおよびテトライソプロポキシチタネート 200 1 を加え、 攪拌しながら 70°Cに昇温し、 1 6時間反応させた。 反応後、 トルエン および過剰の 3—ェチルー 3—ヒ ドロキシメチルォキセタンを減圧除去した。 反応の進行は、 1 H— NMRにより末端メ トキシ基のピーク (3. 5 p p m: - CH3) の減少、 消滅により確認した。
得られた反応物の1 H— NMRを測定したところ、 3—ェチル一 3—ヒ ドロキシ メチルォキセタンが末端に平均 1. 5個導入されたことが判明し、 目的の末端に 下記の構造のォキセタン基含有ケィ素基を含有するイソブチレン系重合体 (A_ 3) が得られた。
(光硬化性組成物およびそれらを含む硬化物の作製)
下記実施例に従い光硬化性組成物を調整し、 フィルムを塗布し、 UV照射する ことでそれらの硬化物を得た。 さらに硬化物の良溶媒で抽出することで、 不溶分 のゲル分率を測定算出し、 硬化性を判断した。
ゲル分率の評価方法:得られた硬化物フィルムを 200メッシュ金網に適量計 量し、 不溶分が流出しないよう包み込んだ。 これらを十分量のへキサンに 1 5時 間浸漬し、 溶出分を抽出し、 その後 8 0°C2時間乾燥した。 その時の不溶分の初 期重量に対する割合をもってゲル分率 (%) とした。
光照射の方法:市販の UVランプ (4 00W) を用い、 サンプル位置での照度 を 2 800 0〜30 000 · c m 2 (測定波長 : 3 1 0〜 400 n m) に調 整した。 照射時間を調整し、 積算光量を調整した。 本実施例の条件では、 1分間 の照射で 1. 5 Jの積算光量となった。 また、 本実施例でのサンプル表面の温度 は約 50°Cとなった。
(実施例 1 )
上記で合成例 5で合成した有機重合体 (A— 1 ) 1 00重量部に対し、 トリア リルスルフォニゥムへキサフルォロアンチモネ一ト系のカチオン系光開始剤 (ァ デカオブトマー S P— 1 7 2 :旭電化工業(株)製) 1重量部を十分に混合し、 光 硬化性組成物を調整した。 この混合物をアプリケーターを用いテフロン (R) 製 のシート上に均一に 1 00 mの厚みで成膜し、 その後 U Vランプにより U V照 射した。
積算光量 1. 5 Jの UV照射により硬化したフィルムを剥し、 へキサンにより
溶出分を抽出し、不溶分のゲル分率を計算することで硬化性を判断した(表 1 )。 (実施例 2)
上記で合成例 5で合成した有機重合体 (A— 1) 100重量部に対し、 (D) 成分のォキセタン化合物 (ジ [1一ェチル ( 3ーォキセタニル) ] メチルエーテ ル) 10重量部を十分混合し、 更にトリァリルスルフォ -ゥムへキサフルォロア ンチモネート系のカチオン系光開始剤 (アデカオプトマー S P— 1 72 :旭電化 工業(株)製) 1重量部を十分に混合し、 光硬化性組成物を調整した。 この混合物 をアプリケーターを用いテフロン (R) 製のシート上に均一に 1 00 μ mの厚み で成膜し、 その後 UVランプにより UV照射した。
積算光量 1. 5 Jの UV照射により硬化したフィルムを剥し、 へキサンにより 溶出分を抽出し、不溶分のゲル分率を計算することで硬化性を判断した(表 1 )。
(実施例 3)
上記で合成例 6で合成した有機重合体 (A— 2) 100重量部に対し、 トリア リルスルフォニゥムへキサフルオロフォスフエート系のカチオン系光開始剤 (ァ デカオブトマー S P_ 1 52 :旭電化工業(株)製) 1重量部を十分に混合し、 光 硬化性組成物を調整した。 この混合物をアプリケーターを用いテフロン (R) 製 のシート上に均一に 1 00 μ mの厚みで成膜し、 その後 UVランプにより UV照 射した。
積算光量 1. 5 Jの UV照射により硬化したフィルムを剥し、 へキサンにより 溶出分を抽出し、不溶分のゲル分率を計算することで硬化性を判断した(表 1 )。
(実施例 4)
上記で合成例 6で合成した有機重合体 (A— 2) 100重量部に対し、 (C) 成分のエポキシ化合物 (1, 6一へキサンジオールジグリシジルエーテル) 20重 量部を十分混合し、 更にトリァリルスルフォニゥムへキサフルオロフォスフエー ト系のカチオン系光開始剤 (アデカオブトマー S P - 1 5 2 :旭電化工業(株)製) 1重量部を十分に混合し、 光硬化性組成物を調整した。 この混合物をアプリケー ターを用いテフロン (R) 製のシート上に均一に 1 00 μ mの厚みで成膜し、 そ
の後 UVランプにより UV照射した。
積算光量 1. 5 Jの UV照射により硬化したフィルムを剥し、 へキサンにより 溶出分を抽出し、不溶分のゲル分率を計算することで硬化性を判断した(表 1 )。
(実施例 5)
上記で合成例 8で合成した有機重合体 (A— 3) 100重量部に対し、 (D) 成分のォキセタン化合物 (ジ [1一ェチル (3—ォキセタ -ル) ] メチルエーテ ル) 1 0重量部を十分混合し、 更にトリァリルスルフォニゥムへキサフルォロア ンチモネート系のカチオン系光開始剤 (アデカオプトマー S P— 1 72 :旭電化 工業(株)製) 1重量部を十分に混合し、 光硬化性組成物を調整した。 この混合物 をアプリケーターを用いテフロン (R) 製のシート上に均一に 100 μ mの厚み で成膜し、 その後 UVランプにより UV照射した。
積算光量 1. 5 Jの UV照射により硬化したフィルムを剥し、 へキサンにより 溶出分を抽出し、不溶分のゲル分率を計算することで硬化性を判断した(表 1 )。
(実施例 6 )
上記で合成例 5で合成した有機重合体 (A— 1) 1 00重量部に対し、 (C) 成分のエポキシ化合物 (脂環式エポキシ希釈剤 : セロキサイ ド 3000 : ダイセ ル化学工業 (株) 製) 1 0重量部、 (D) 成分のォキセタン化合物 (ジ [1ーェ チル (3—ォキセタニル) ] メチルエーテル) 1 0重量部を十分混合し、 更にト リアリルスルフォニゥムへキサフルォロアンチモネ一ト系の力チオン系光開始剤 (アデカオブトマー S P— 1 72:旭電化工業(株)製) 1重量部を十分に混合し、 光硬化性組成物を調整した。 この混合物をアプリケーターを用いテフロン (R) 製のシート上に均一に 1 00 μ mの厚みで成膜し、 その後 UVランプにより UV 照射した。
積算光量 1. 5 Jの UV照射により硬化したフィルムを剥し、 へキサンにより 溶出分を抽出し、不溶分のゲル分率を計算することで硬化性を判断した(表 1 )。
(比較例 1 )
本発明の末端構造を有しない、 加水分解性シリル基を分子内に有するポリオキ
シプロピレン重合体 (MSポリマー S AT 350 :鐘淵化学工業 (株) ) 100 重量部に対し、 ジブチルスズジァセチルァセトナート (ネオスタン U— 220 : 日東化成 (株) ) を 0. 5部混合し、 アプリケーターを用いテフロン (R) 製の' シート上に均一に 1 00 μ mの厚みで成膜し、 その後 UVランプにより UV照射 した。 硬化性を確認しょう としたが、 フィルムは形成されず、 硬化しなかった。 別途作製したサンプルを実施例のサンプル表面の温度と同等の温度(約 50°C) で実施例の UV照射と同等の時間 (本試験では 1分) での硬化性を確認したが、 フィルムは形成されず、 ほとんど硬化していない状態であった。 さらに同温度で 2時間放置した後にゲル分率が実施例と同等となった (表 2) 。
(比較例 2)
本発明の末端構造を有しない、 分子量約 10000のァリル基末端ポリイソブ チレン (鐘淵化学工業 (株) 製、 EP 40 OA) 1 00重量部に α-メチルスチレ ン変性メチルハイ ドロジヱンポリシロキサン (鐘淵化学工業 (株) 製、 CR 1 0 0) を 7. 3重量部を混合し、 手混ぜした後, さらに保存性改良剤としてマレイ ン酸ジメチル (和光純薬 (株) 製、 特級試薬) を 90 ^i L、 白金ビニルシロキサ ン錯体触媒 (鐘淵化学工業 (株) 製、 HS— KA) を 60 / L加え、 手混ぜ混合 した。
この混合物をアプリケーターを用いテフロン (R) 製のシート上に均一に 10 0 mの厚みで成膜し、 その後 UVランプにより UV照射した。 硬化性を確認し ようとしたが、 フィルムは形成されず、 硬化しなかった。
別途作製したサンプルを 1 0 o°cおよび実施例のサンプル表面の温度と同等の 温度 (約 50°C) で実施例の UV照射と同等の時間 (本試験では 1分) 加熱した 後の硬化性を確認したが、 ほとんど硬化していない状態であった。 さらに 50°C で、 2時間放置した後でもゲル分率が実施例と同等にならず、より高温の 100 °C 1時間硬化させた場合に実施例と同等のゲル分率が得られた (表 2) 。
表 1、 表 2より、 いずれの実施例の組成物も短時間の UV照射のみで良好な硬 化性を有し、 かつ硬化物はゴム弾性を有する比較的柔らかい硬化物であった。 ま
た比較例の湿分硬化性あるいは熱硬化性の組成物では、 このような短時間では十 分な硬化性が得られなかった。
(実施例 7 )
実施例 1のサンプルを別途作製し、 U V照射に加え、 さらに 8 0 °C 1時間カロ熱 硬化した。 硬化したフィルムを剥し、 へキサンにより溶出分を抽出し、 不溶分の ゲル分率を計算することで硬化性を確認した。 その結果、 実施例 1での U V照射 のみに比べ、 ゲル分率が向上していることが確認された (表 3 ) 。
(実施例 8 )
実施例 5のサンプルを別途作製し、 U V照射に加え、 さらに 8 0 °C 1時間カロ熱 硬化した。 硬化したフィルムを剥し、 へキサンにより溶出分を抽出し、 不溶分の ゲル分率を計算することで硬化性を確認した。 その結果、 実施例 5での U V照射 のみに比べ、 ゲル分率が向上していることが確認された (表 3 ) 。
以上の結果より、 本発明の光硬化性組成物は、 良好な光硬化性を有し、 (A) 成分の特徴であるゴム弾性を併せ持つ硬化物が得られることが判明した。
«1
注 3: セロキサ仆' 3000:脂 3式エポキシ希釈剤 (ダイセル化学工業 (株)製)
表 2
比 1 比較'列 2
MSホリマー SAT350 100
EP400A 100 ネオスタン U220 0.5
CR100 7.3 マレイン酸シ'メチル 90 l 白金ビニルシロキサン eojLi i
UV照射のみ 0 0
硬化性 (ゲル分率 (お)) 50°C1min 0 0
50°C2h 92 60
100°C1 h ― 93
表 3
, :
注 3 : セロキサイド 3000:脂環式エポキシ希釈剤 (ダイセル化学工業(株)製) 産業上の利用可能性
本発明の光硬化性組成物は光硬化性と硬化後のゴム弾性の両面において優れた 組成物である。