RU2201418C2 - Производные аминофенилкетона и способ их получения - Google Patents
Производные аминофенилкетона и способ их получения Download PDFInfo
- Publication number
- RU2201418C2 RU2201418C2 RU97122130/04A RU97122130A RU2201418C2 RU 2201418 C2 RU2201418 C2 RU 2201418C2 RU 97122130/04 A RU97122130/04 A RU 97122130/04A RU 97122130 A RU97122130 A RU 97122130A RU 2201418 C2 RU2201418 C2 RU 2201418C2
- Authority
- RU
- Russia
- Prior art keywords
- compound
- formula
- organic solvent
- compounds
- isolation
- Prior art date
Links
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/26—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D307/30—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D307/32—Oxygen atoms
- C07D307/33—Oxygen atoms in position 2, the oxygen atom being in its keto or unsubstituted enol form
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C307/00—Amides of sulfuric acids, i.e. compounds having singly-bound oxygen atoms of sulfate groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C307/04—Diamides of sulfuric acids
- C07C307/10—Diamides of sulfuric acids having nitrogen atoms of the sulfamide groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C303/00—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides
- C07C303/02—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of sulfonic acids or halides thereof
- C07C303/04—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of sulfonic acids or halides thereof by substitution of hydrogen atoms by sulfo or halosulfonyl groups
- C07C303/08—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of sulfonic acids or halides thereof by substitution of hydrogen atoms by sulfo or halosulfonyl groups by reaction with halogenosulfonic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/15—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings
- C07C311/21—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
- Plural Heterocyclic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Изобретение относится к производным аминофенилкетона формулы I
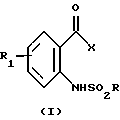
где R представляет фенил, замещенный С1-С6 алкилом; R1 представляет водород; Х представляет -(CH2)3-Y, циклопропил или тетрагидро-2-оксо-3-фуроил; Y представляет хлор, бром или гидрокси,
или к их кислотно-аддитивным солям. Соединения I являются промежуточными для полуряда соединений, например, способ получения соединения формулы А

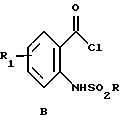
включает взаимодействие соединения формулы В
с соединением формулы С

в присутствии основания и органического растворителя с образованием смеси, включающей соединение формулы Е

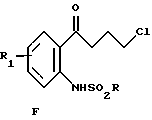
выделение соединения Е путем гидролиза или кристаллизации, взаимодействие соединения Е с концентрированной НСl в присутствии органического растворителя с образованием соединения формулы F
обработку соединения F водным раствором основания при повышенной температуре, выделение соединения формулы G

где в формулах Е, F, G радикалы R и R1 - такие, как определено в п.1,
обработку соединения G сильной кислотой, выделение соединения формулы Н

и взаимодействие соединения формулы Н с HCl с образованием соединения формулы А. Полученные промежуточные соединения используют для получения гербицидных сульфамоилмочевин. 2 с. и 5 з.п.ф-лы.
где R представляет фенил, замещенный С1-С6 алкилом; R1 представляет водород; Х представляет -(CH2)3-Y, циклопропил или тетрагидро-2-оксо-3-фуроил; Y представляет хлор, бром или гидрокси,
или к их кислотно-аддитивным солям. Соединения I являются промежуточными для полуряда соединений, например, способ получения соединения формулы А
включает взаимодействие соединения формулы В
с соединением формулы С
в присутствии основания и органического растворителя с образованием смеси, включающей соединение формулы Е
выделение соединения Е путем гидролиза или кристаллизации, взаимодействие соединения Е с концентрированной НСl в присутствии органического растворителя с образованием соединения формулы F
обработку соединения F водным раствором основания при повышенной температуре, выделение соединения формулы G
где в формулах Е, F, G радикалы R и R1 - такие, как определено в п.1,
обработку соединения G сильной кислотой, выделение соединения формулы Н
и взаимодействие соединения формулы Н с HCl с образованием соединения формулы А. Полученные промежуточные соединения используют для получения гербицидных сульфамоилмочевин. 2 с. и 5 з.п.ф-лы.
Description
Изобретение относится к производным о-аминофенилкетона формулы I

где R, R1 и X - такие, как указано ниже.
где R, R1 и X - такие, как указано ниже.
Соединения формулы I полезны в качестве промежуточных продуктов при получении широкого спектра гербицидных производных сульфамоилмочевины и, в частности, при производстве избирательного по отношению к сельскохозяйственным культурам гербицида 1-{[о-(циклопропилкарбонил)фенил]сульфамоил}-3-(4,6-диметокси-2-пиримидинил)мочевины. Предлагается также способ получения указанного промежуточного соединения формулы I.
В соответствии с настоящим изобретением предлагается соединение формулы I

где R представляет неразветвленный или разветвленный C1-C6 алкил или фенил, необязательно замещенный C1-С3 алкилом, C1-С3 алкокси, хлором или бромом;
R1 представляет водород, циано, нитро, галоген, формил, C1-C4 алкил, необязательно замещенный одной или несколькими группами, представляющими собой галоген, C1-С3 алкокси, C1-С3 алкилтио, C1-С3 алкилсульфинил или C1-С3 алкилсульфонил,
C1-С4 алкокси, необязательно замещенный одной или несколькими группами, представляющими собой галоген, C1-С3 алкокси, C1-С3 алкилтио, C1-С3 алкилсульфинил или C1-С3 алкилсульфонил,
C1-С4 алкилтио, необязательно замещенный одной или несколькими группами, представляющими собой галоген, C1-С3 алкокси, C1-С3 алкилтио, C1-С3 алкилсульфинил или C1-С3 алкилсульфонил,
C1-С4 алкилсульфинил, необязательно замещенный одной или несколькими группами, представляющими собой: галоген, C1-С3 алкокси, C1-С3 алкилтио, C1-С3 алкилсульфинил или C1-С3 алкилсульфонил,
C1-С4 алкилсульфонил, необязательно замещенный одной или несколькими группами, представляющими собой галоген, C1-С3 алкокси, C1-С3 алкилтио, C1-С3 алкилсульфинил или C1-С3 алкилсульфонил,
C1-С4 алкилкарбонил, необязательно замещенный одной или несколькими группами, представляющими собой галоген, C1-С3 алкокси, C1-С3 алкилтио, C1-С3 алкилсульфинил или C1-С3 алкилсульфонил,
C1-С4 алкоксикарбонил, необязательно замещенный одной или несколькими группами, представляющими собой галоген или C1-С3 алкокси,
ди(C1-С4 алкил)амино, необязательно замещенный одной или несколькими группами, представляющими собой галоген или C1-С3 алкокси,
ди(C1-С4 алкил)аминокарбонил, необязательно замещенный одной или несколькими группами, представляющими собой галоген или C1-С3 алкокси,
ди(C1-С4 алкил)аминосульфонил, необязательно замещенный одной или несколькими группами, представляющими собой галоген или C1-С3 алкокси, или
гетероциклическое кольцо, имеющее 2-6 атомов углерода и 1-3 атома азота, кислорода или серы и являющееся необязательно замещенным у атомов углерода одной или несколькими группами, представляющими собой галоген, C1-С4 алкил или C1-С4 галогеналкил;
Х представляет -(СН2)3-Y, циклопропил или тетрагидро-2-оксо-3-фуроил и
Y представляет хлор, бром или гидрокси,
или его кислотно-аддитивная соль.
где R представляет неразветвленный или разветвленный C1-C6 алкил или фенил, необязательно замещенный C1-С3 алкилом, C1-С3 алкокси, хлором или бромом;
R1 представляет водород, циано, нитро, галоген, формил, C1-C4 алкил, необязательно замещенный одной или несколькими группами, представляющими собой галоген, C1-С3 алкокси, C1-С3 алкилтио, C1-С3 алкилсульфинил или C1-С3 алкилсульфонил,
C1-С4 алкокси, необязательно замещенный одной или несколькими группами, представляющими собой галоген, C1-С3 алкокси, C1-С3 алкилтио, C1-С3 алкилсульфинил или C1-С3 алкилсульфонил,
C1-С4 алкилтио, необязательно замещенный одной или несколькими группами, представляющими собой галоген, C1-С3 алкокси, C1-С3 алкилтио, C1-С3 алкилсульфинил или C1-С3 алкилсульфонил,
C1-С4 алкилсульфинил, необязательно замещенный одной или несколькими группами, представляющими собой: галоген, C1-С3 алкокси, C1-С3 алкилтио, C1-С3 алкилсульфинил или C1-С3 алкилсульфонил,
C1-С4 алкилсульфонил, необязательно замещенный одной или несколькими группами, представляющими собой галоген, C1-С3 алкокси, C1-С3 алкилтио, C1-С3 алкилсульфинил или C1-С3 алкилсульфонил,
C1-С4 алкилкарбонил, необязательно замещенный одной или несколькими группами, представляющими собой галоген, C1-С3 алкокси, C1-С3 алкилтио, C1-С3 алкилсульфинил или C1-С3 алкилсульфонил,
C1-С4 алкоксикарбонил, необязательно замещенный одной или несколькими группами, представляющими собой галоген или C1-С3 алкокси,
ди(C1-С4 алкил)амино, необязательно замещенный одной или несколькими группами, представляющими собой галоген или C1-С3 алкокси,
ди(C1-С4 алкил)аминокарбонил, необязательно замещенный одной или несколькими группами, представляющими собой галоген или C1-С3 алкокси,
ди(C1-С4 алкил)аминосульфонил, необязательно замещенный одной или несколькими группами, представляющими собой галоген или C1-С3 алкокси, или
гетероциклическое кольцо, имеющее 2-6 атомов углерода и 1-3 атома азота, кислорода или серы и являющееся необязательно замещенным у атомов углерода одной или несколькими группами, представляющими собой галоген, C1-С4 алкил или C1-С4 галогеналкил;
Х представляет -(СН2)3-Y, циклопропил или тетрагидро-2-оксо-3-фуроил и
Y представляет хлор, бром или гидрокси,
или его кислотно-аддитивная соль.
В соответствии с настоящим изобретением предлагается также способ получения соединения формулы А

где R1 - такой, как определено в п.1, включающий следующие стадии:
i) взаимодействия соединения формулы В

где R и R1 - такие, как определено в п.1,
с соединением формулы С

в присутствии основания и органического растворителя с образованием смеси, содержащей соединение формулы Е

ii) выделения соединения Е путем гидролиза или кристаллизации;
iii) взаимодействия соединения Е с концентрированной НСl в присутствии органического растворителя с образованием соединения формулы F

iv) обработки соединения F водным раствором основания при повышенной температуре;
v) выделения соединения формулы G

vi) обработки соединения G сильной кислотой;
vii) выделения соединения формулы Н

viii) и взаимодействия соединения формулы Н с НСl с образованием соединения формулы А.
где R1 - такой, как определено в п.1, включающий следующие стадии:
i) взаимодействия соединения формулы В
где R и R1 - такие, как определено в п.1,
с соединением формулы С
в присутствии основания и органического растворителя с образованием смеси, содержащей соединение формулы Е
ii) выделения соединения Е путем гидролиза или кристаллизации;
iii) взаимодействия соединения Е с концентрированной НСl в присутствии органического растворителя с образованием соединения формулы F
iv) обработки соединения F водным раствором основания при повышенной температуре;
v) выделения соединения формулы G
vi) обработки соединения G сильной кислотой;
vii) выделения соединения формулы Н
viii) и взаимодействия соединения формулы Н с НСl с образованием соединения формулы А.
Соединение А, где R1 представляет водород, т.е. 1-(о-аминофенил)-4-хлор-1-бутанонгидрохлорид, используют для получения гербицидного промежуточного продукта - о-(аминофенил)циклопропилкетона. Описание о-(аминофенил)циклопропилкетона и применения его в производстве гербицида 1-{ [о-(циклопропилкарбонил)фенил] сульфамоил} -3-(4,6-диметокси-2-пиримидинил)мочевины приведено в известном уровне техники. Настоящее изобретение позволяет избежать использования в качестве промежуточного соединения о-нитробензоилхлорида, которое является взрывчатым веществом.
Основанием, используемым на стадии i), может быть C1-C4 алкоксид (алкоголят) магния, предпочтительно легко доступный, такой как метилат магния или этилат магния. Органическим растворителем, используемым на стадии i), может быть ароматический углеводород или диалкиловый эфир, такой как толуол, ксилол или тетрагидрофуран. Органическим растворителем, используемым на стадии iii) для получения соединения F, может быть инертный органический растворитель, такой как толуол или ксилол, а кислотой, используемой на стадии iii), может быть минеральная кислота, такая как концентрированная НСl. Основанием, используемым на стадии iv) для получения соединения G, может быть гидроксид щелочного металла, такой как гидроксид натрия или гидроксид калия. Повышенная температура на стадии iv) может представлять собой любую температуру выше 25oС, предпочтительно примерно 90-130oС. В качестве сильной кислоты, используемой на стадии vi) для получения соединения Н, может служить серная кислота. Кислотой, используемой для получения соединения А на стадии viii), может быть минеральная кислота, такая как концентрированная НСl.
Соединения по настоящему изобретению могут быть использованы для получения гербицидных соединений (К) сульфамоилмочевины путем использования способа по настоящему изобретению для получения соединения формулы А и превращения этих соединений формулы А в соответствующие о-(аминофенил)циклопропилкетоны (J) традиционными способами, описанными в уровне техники, с превращением указанных фенилкетонов в целевые гербицидные продукты сульфамоилмочевины, предпочтительно в избирательную по отношению к зерновым культурам гербицидную сульфамоилмочевину, 1-{[о-(циклопропилкарбонил)фенил] сульфамоил} -3-(4,6-диметокси-2-пиримидинил)мочевину. Превращение производных фенилкетона в сульфамоилмочевинные гербициды может быть осуществлено известными способами.
Получение показано на схеме технологического процесса, приведенной в конце описания.
Способом по настоящему изобретению соединение формулы А получают так, как описано выше, и обычными методами дегидрогалогенирования преобразуют в о-(аминофенил)циклопропилкетон формулы J, который в свою очередь может быть подвергнут взаимодействию с 2-аминоарилом формулы L и хлорсульфонилизоцианатом в присутствии триэтиламина и растворителя с получением целевой гербицидной сульфамоилмочевины формулы К.
Далее изобретение проиллюстрировано примерами, которые не следует рассматривать как ограничивающие изобретение. Термины ЯМР и МС означают спектроскопию магнитного резонанса на ядрах протона и масс-спектрометрию соответственно.
ПРИМЕР 1
Получение -2'-(тетрагидро-2-оксо-3-фуроил)-п-толуолсульфонанилида (см. схему к примеру 1).
Получение -2'-(тетрагидро-2-оксо-3-фуроил)-п-толуолсульфонанилида (см. схему к примеру 1).
К смеси 40 мл толуола и 2,03 г (178 ммоль) этоксида (этилата) магния в колбе под азотом при 5-10oС добавляют 4,6 г (36 ммоль) 2-ацетилбутиролактона в течение 2 минут. Полученную суспензию перемешивают 10 минут при 5-10oС и еще примерно 1,5 часа при 20oС. Реакционную смесь обрабатывают раствором 10,0 г (32 ммоль) N-п-толилсульфонилантраноилхлорида в 20 мл толуола, перемешивают несколько часов при температуре окружающей среды и примерно 2 часа при 45-50oС. Добавляют воду (120 мл) и серую суспензию перемешивают в течение примерно 4 часов при 65-70oС. Концентрированной серной кислотой доводят рН до 1. Разделяют фазы и органический слой фильтруют с получением 7,6 г 2'-(тетрагидро-2-оксо-3-фуроил)-п-толуолсульфонанилида. Остальной продукт извлекают из фильтрата органического слоя путем концентрирования в вакууме, в результате которого получают еще 2,1 г продукта с общим (суммарным) выходом 83% (т. пл. 138-141oС). Продукт идентифицируют методами ЯМР- и МС-анализов.
ПРИМЕР 2
Получение 2'-(циклопропилкарбонил)-п-толуолсульфонанилида (см. схему к примеру 2).
Получение 2'-(циклопропилкарбонил)-п-толуолсульфонанилида (см. схему к примеру 2).
Двухфазную суспензионную смесь 3,56 г (1,0 ммоль) продукта из примера 1, 25 мл толуола и 20 мл 37%-ной НСl нагревают с обратным холодильником в течение примерно 12 часов, охлаждают и полученную суспензию фильтруют с получением 1,98 г 4-хлор-1-(2-N-тозиламинофенил)-1-бутанона. Разделяют фазы фильтрата и водную фазу экстрагируют толуолом. Органические фазы объединяют и концентрируют в вакууме с получением остального продукта - 4-хлор-1- (2-N-тозиламинофенил)-1-бутанона (1,15 г) при общем выходе 90% (т.пл. 108-113oС). Продукт идентифицируют методами ЯМР- и МС-анализов.
В раствор 1,62 г (4,6 ммоль) 4-хлор-1-(2-N-тозиламинофенил)-1-бутанона в 10 мл толуола загружают 17,3 г (28,7 ммоль) 6,6%-ного раствора гидроксида натрия. Полученную двухфазную смесь нагревают с обратным холодильником в течение примерно 1 часа, охлаждают и доводят до рН 1 посредством концентрированной серной кислоты. Органический слой отделяют и концентрируют в вакууме с получением 1,50 г 2'-(циклопропилкарбонил)-п-толуолсульфонанилида при 100%-ном выходе (т.пл. 92-100oС). Продукт идентифицируют методами ЯМР- и МС- анализов.
ПРИМЕР 3
Получение 1-(о-аминофенил)-4-хлор-1-бутанонгидрохлорида (см. схему к примеру 3).
Получение 1-(о-аминофенил)-4-хлор-1-бутанонгидрохлорида (см. схему к примеру 3).
Продукт примера 2 (1,5 г, 4,7 ммоль) обрабатывают 96%-ной серной кислотой и нагревают до 90oС в течение 15 минут. Раствор охлаждают, доводят рН до 9 посредством гидроксида аммония и экстрагируют метиленхлоридом. Объединенные экстракты концентрируют в вакууме с получением 1-(о-аминофенил)-4-гидрокси-1-бутанона (выход 80%, т.пл. 58-61oС). Продукт идентифицируют методами ЯМР- и МС-анализов.
Смесь 9,3 г (5,1 ммоль) 1-(о-аминофенил)-4-гидрокси-1-бутанона, 26 мл воды и 90 мл 37%-ной НС1 нагревают с обратным холодильником в течение примерно 6,5 часов, охлаждают и фильтруют с получением 8,0 г гидрохлорида 1-(о-аминофенил)-4-хлор-1-бутанона. Экстрагирование водного маточного раствора метиленхлоридом дает еще 1,10 г указанного в заголовке продукта с общим выходом 73% (т.пл.142-145oС). Продукт идентифицируют методами ЯМР- и МС-анализов.
ПРИМЕР 4
Получение о-аминофенилциклопропилкетона (см. схему к примеру 4).
Получение о-аминофенилциклопропилкетона (см. схему к примеру 4).
Раствор 0,30 г (1,3 ммоль) гидрохлорида 1-(о-аминофенил)-4-хлор-1-бутанона в 3 мл метиленхлорида и 3 мл этилендихлорида обрабатывают 1,2 г (3 ммоль) 10%-ного раствора гидроксида натрия и 0,05 г (0,2 ммоль) 75%-ного водного раствора метилтрибутиламмонийхлорида и нагревают до 50oС в течение примерно 5 часов. После охлаждения до комнатной температуры разделяют фазы. Водный слой экстрагируют метиленхлоридом. Объединенные органические экстракты промывают водой и концентрируют в вакууме с получением 0,14 г (выход 70%) о-аминофенилциклопропилкетона (т.пл. 46-48oС). Продукт идентифицируют методами ЯМР- и МС-анализов.
Claims (7)
1. Производные аминофенилкетона общей формулы

где R представляет фенил, замещенный C1-C6 алкилом;
R1 представляет водород;
Х представляет -(СН2)3-Y, циклопропил или тетрагидро-2-оксо-3-фуроил;
Y представляет хлор, бром или гидрокси,
или его кислотно-аддитивная соль.
где R представляет фенил, замещенный C1-C6 алкилом;
R1 представляет водород;
Х представляет -(СН2)3-Y, циклопропил или тетрагидро-2-оксо-3-фуроил;
Y представляет хлор, бром или гидрокси,
или его кислотно-аддитивная соль.
2. Соединение по п. 1, в котором R представляет п-толил.
3. Соединение по п. 2, в котором Х представляет циклопропил, -(СН2)3-Y или тетрагидро-2-оксо-3-фуроил.
4. Способ получения соединения формулы А

отличающийся тем, что включает: i) взаимодействие соединения формулы В

с соединением формулы С

в присутствии основания и органического растворителя с образованием смеси, содержащей соединение формулы Е

ii) выделение соединения Е путем гидролиза или кристаллизации; iii) взаимодействие соединения Е с концентрированной НСl в присутствии органического растворителя с образованием соединения формулы F

iv) обработку соединения F водным раствором основания при повышенной температуре; v) выделение соединения формулы G

vi) обработку соединения G сильной кислотой; vii) выделение соединения формулы Н

где в формулах Е-Н радикалы R и R1 такие, как определено в п. 1; viii) взаимодействие соединения формулы Н с НСl с образованием соединения формулы А.
отличающийся тем, что включает: i) взаимодействие соединения формулы В
с соединением формулы С
в присутствии основания и органического растворителя с образованием смеси, содержащей соединение формулы Е
ii) выделение соединения Е путем гидролиза или кристаллизации; iii) взаимодействие соединения Е с концентрированной НСl в присутствии органического растворителя с образованием соединения формулы F
iv) обработку соединения F водным раствором основания при повышенной температуре; v) выделение соединения формулы G
vi) обработку соединения G сильной кислотой; vii) выделение соединения формулы Н
где в формулах Е-Н радикалы R и R1 такие, как определено в п. 1; viii) взаимодействие соединения формулы Н с НСl с образованием соединения формулы А.
5. Способ по п. 4, в котором основанием на стадии i) является этилат магния, а органическим растворителем - толуол; органическим растворителем на стадии iii) является толуол; основанием на стадии v) - NaOH и сильной кислотой на стадии vi) - серная кислота.
6. Способ по п. 5, в котором используют соединение формулы В, где R1 представляет собой водород.
7. Способ по п. 5, в котором используют соединение формулы В, где R представляет п-толил.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US77131896A | 1996-12-20 | 1996-12-20 | |
| US08/771,318 | 1996-12-20 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU97122130A RU97122130A (ru) | 1999-09-20 |
| RU2201418C2 true RU2201418C2 (ru) | 2003-03-27 |
Family
ID=25091429
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU97122130/04A RU2201418C2 (ru) | 1996-12-20 | 1997-12-19 | Производные аминофенилкетона и способ их получения |
Country Status (29)
| Country | Link |
|---|---|
| EP (1) | EP0849262B1 (ru) |
| JP (1) | JP3958851B2 (ru) |
| KR (1) | KR100555052B1 (ru) |
| CN (1) | CN1100755C (ru) |
| AR (1) | AR011771A1 (ru) |
| AT (1) | ATE207066T1 (ru) |
| AU (1) | AU721862B2 (ru) |
| BG (1) | BG63390B1 (ru) |
| BR (1) | BR9706365A (ru) |
| CA (1) | CA2225206C (ru) |
| CO (1) | CO5021225A1 (ru) |
| CZ (1) | CZ393797A3 (ru) |
| DE (1) | DE69707408T2 (ru) |
| EG (1) | EG21392A (ru) |
| ES (1) | ES2166052T3 (ru) |
| HK (1) | HK1012504A1 (ru) |
| HR (1) | HRP970613B1 (ru) |
| HU (1) | HU219612B (ru) |
| IL (1) | IL122672A (ru) |
| NO (1) | NO309934B1 (ru) |
| NZ (1) | NZ329428A (ru) |
| PL (1) | PL190620B1 (ru) |
| RU (1) | RU2201418C2 (ru) |
| SG (1) | SG64466A1 (ru) |
| SK (1) | SK169397A3 (ru) |
| TR (1) | TR199701597A2 (ru) |
| UA (1) | UA43896C2 (ru) |
| YU (1) | YU48897A (ru) |
| ZA (1) | ZA9711397B (ru) |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4559081A (en) * | 1984-12-03 | 1985-12-17 | Ppg Industries, Inc. | Sulfamoyl urea derivatives |
| EP0184122B1 (en) * | 1984-12-03 | 1991-07-17 | Ppg Industries, Inc. | Sulfamoyl urea derivatives |
| US5009699A (en) * | 1990-06-22 | 1991-04-23 | American Cyanamid Company | 1-{[O-(cyclopropylcarbonyl)phenyl]sulfamoyl}-3-(4,6-dimethoxy-2-pyrimidinyl)urea herbicidal composition and use |
| CZ289916B6 (cs) * | 1993-11-30 | 2002-04-17 | American Cyanamid Company | Způsob výroby o-aminofenylketonů |
-
1997
- 1997-11-14 HR HR970613A patent/HRP970613B1/xx not_active IP Right Cessation
- 1997-12-05 CZ CZ973937A patent/CZ393797A3/cs unknown
- 1997-12-09 SK SK1693-97A patent/SK169397A3/sk unknown
- 1997-12-11 BG BG102110A patent/BG63390B1/bg unknown
- 1997-12-11 TR TR97/01597A patent/TR199701597A2/xx unknown
- 1997-12-15 ES ES97310120T patent/ES2166052T3/es not_active Expired - Lifetime
- 1997-12-15 AT AT97310120T patent/ATE207066T1/de not_active IP Right Cessation
- 1997-12-15 DE DE69707408T patent/DE69707408T2/de not_active Expired - Lifetime
- 1997-12-15 EP EP97310120A patent/EP0849262B1/en not_active Expired - Lifetime
- 1997-12-16 JP JP36342597A patent/JP3958851B2/ja not_active Expired - Fee Related
- 1997-12-17 BR BR9706365A patent/BR9706365A/pt not_active Application Discontinuation
- 1997-12-18 CA CA002225206A patent/CA2225206C/en not_active Expired - Fee Related
- 1997-12-18 SG SG1997004560A patent/SG64466A1/en unknown
- 1997-12-18 ZA ZA9711397A patent/ZA9711397B/xx unknown
- 1997-12-18 UA UA97126152A patent/UA43896C2/uk unknown
- 1997-12-18 YU YU48897A patent/YU48897A/sr unknown
- 1997-12-18 CO CO97073946A patent/CO5021225A1/es unknown
- 1997-12-18 AU AU48459/97A patent/AU721862B2/en not_active Ceased
- 1997-12-18 EG EG135297A patent/EG21392A/xx active
- 1997-12-18 NZ NZ329428A patent/NZ329428A/en unknown
- 1997-12-18 IL IL12267297A patent/IL122672A/xx not_active IP Right Cessation
- 1997-12-19 PL PL97323892A patent/PL190620B1/pl not_active IP Right Cessation
- 1997-12-19 RU RU97122130/04A patent/RU2201418C2/ru not_active IP Right Cessation
- 1997-12-19 AR ARP970106066A patent/AR011771A1/es active IP Right Grant
- 1997-12-19 KR KR1019970070780A patent/KR100555052B1/ko not_active IP Right Cessation
- 1997-12-19 HU HU9702511A patent/HU219612B/hu not_active IP Right Cessation
- 1997-12-19 NO NO975983A patent/NO309934B1/no not_active IP Right Cessation
- 1997-12-19 CN CN97108762A patent/CN1100755C/zh not_active Expired - Fee Related
-
1998
- 1998-12-15 HK HK98113419A patent/HK1012504A1/xx not_active IP Right Cessation
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3166927B1 (en) | Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids | |
| EP0738713B1 (en) | Process for the preparation of pesticidal 1-(chloroaryl)heterocyclic compounds | |
| RU2201418C2 (ru) | Производные аминофенилкетона и способ их получения | |
| US6127576A (en) | Aminophenyl ketone derivatives and a method for the preparation thereof | |
| KR100548807B1 (ko) | 아린 중간체 및 이의 제조 방법 | |
| JP2020537680A (ja) | 除草性ピリダジノン化合物を製造するプロセス | |
| JP3499595B2 (ja) | 2−シアノイミダゾール系化合物の製造方法 | |
| KR100415520B1 (ko) | 1-(2-클로로페닐)-5(4h)-테트라졸리논의제조방법 | |
| JP2003506421A (ja) | アシル化1,3−ジカルボニル化合物の製法 | |
| JP3804078B2 (ja) | β−ニトロエナミンの製造方法 | |
| JP3646225B2 (ja) | 芳香族エステル誘導体及びその中間体並びにそれらの製造方法 | |
| TW202423902A (zh) | 製藥方法和中間體 | |
| JPS58152857A (ja) | フルオロアシル尿素類又はフルオロアシルチオ尿素類 | |
| JPS61134378A (ja) | ピリミジン誘導体の製法 | |
| CZ287105B6 (en) | Process for preparing l-{[2-(cyclopropylcarbonyl)phenyl]sulfamoyl}-3-(4,6-dialkoxy-2-pyrimidinyl)urea and intermediates for such preparation process | |
| JPH05294931A (ja) | 新規中間体及び2−クロロ−5−(アミノメチル)ピリジンの製造方法 | |
| DE2920941A1 (de) | Verfahren zur herstellung von pyrazol- derivaten | |
| JP2003505379A (ja) | 置換ベンゾイソチアゾール化合物 | |
| JPH07165707A (ja) | 5−アミノ−3,4−ジヒドロ−1−オキシド−2h−ピロール類およびその中間体 | |
| JPH0572381B2 (ru) | ||
| JPH05286906A (ja) | 炭酸エステル誘導体およびその製造法 | |
| JPH0499761A (ja) | フェニルスルホンアミドの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MM4A | The patent is invalid due to non-payment of fees |
Effective date: 20101220 |