KR930004609B1 - 폴리카보네이트 및 그 제조방법 - Google Patents
폴리카보네이트 및 그 제조방법 Download PDFInfo
- Publication number
- KR930004609B1 KR930004609B1 KR1019890013660A KR890013660A KR930004609B1 KR 930004609 B1 KR930004609 B1 KR 930004609B1 KR 1019890013660 A KR1019890013660 A KR 1019890013660A KR 890013660 A KR890013660 A KR 890013660A KR 930004609 B1 KR930004609 B1 KR 930004609B1
- Authority
- KR
- South Korea
- Prior art keywords
- polycarbonate
- less
- ppm
- groups
- phenol
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
- C08G64/04—Aromatic polycarbonates
- C08G64/06—Aromatic polycarbonates not containing aliphatic unsaturation
- C08G64/14—Aromatic polycarbonates not containing aliphatic unsaturation containing a chain-terminating or -crosslinking agent
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
- C08G64/20—General preparatory processes
- C08G64/30—General preparatory processes using carbonates
- C08G64/307—General preparatory processes using carbonates and phenols
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Polyesters Or Polycarbonates (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Processes Of Treating Macromolecular Substances (AREA)
Abstract
내용 없음.
Description
본 발명은 폴리카보네이트 및 그 제조방법에 관한것으로 구체적으로는 우수한 색조, 내열성 및 내수성을 갖는 폴리카보네이트 및 그 제조방법에 관한 것이다.
폴리카보네이트는 충격강도 등의 우수한 기계적 성질 및 우수한 내열성 및 투명성을 갖기 때문에 다양한 용도에 널리 쓰이고 있다. 기존의 제조방법으로는 비스페놀류 등의 방향족 디히드록시 화합물을 포스겐과 직접 반응시키는 방법(인터페이스법)과 비스페놀류 등의 방향족 디히드록시 화합물과 카본산 디페닐 등의 카본산 디에스테르류를 용융상태에서 에스테르 교환반응(중축합 반응)시키는 방법 등이 있다.
카본산 디에스테르류와 방향족 디히드록시 화합물의 에스테르 교환반응에 의한 폴리카보네이트의 제조에서는 반응물들을 통상 유기염류, 무기염류, 금속의 산화물, 수산화물, 수소화물 및 알콜레이트 등의 촉매 존재하에서 감압하 250-330℃로 가열하여 용융상태에서 에스테르 교환반응한다. 이 방법은 비교적 적은 비용으로 폴라카보네이트가 제조된다는 점에서 상기 인터페이스법보다 유리하다. 그러나, 카본산 디에스테르류와 방향족 디히드록시 화합물과의 반응이 용융상태에서 행해지므로 상기 방법들로 생성된 폴리카보네이트는 통상 색조, 내열성 또는 내수성이 불량하게 되는 문제가 있다.
상기 문제점의 해결을 위해 본 발명자들은 광범위한 연구를 한 결과, 생성되는 폴리카보네이트의 말단을 특정 화합물로 캐핑(capping)하여 말단의 히드록시기를 환원시키므로써, 우수한 내열성 및 내수성을 제공할 수 있음을 기초로 하여 본 발명을 완성하였다.
본 발명은 상술한 종래 기술의 문제점을 해결하려는 것이며, 본 발명의 한 목적은 그 색조, 내열성 및 내수성이 우수한 폴리카보네이트의 제조방법을 제공하는데 있다.
본 발명에 의한 폴리카보네이트의 제1제조 방법은 방향족 디히드록시 화합물 1몰에 대하여 탄소수가 10~40인 페놀 0.05~15몰% 존재하에
(a) 질소 함유 염기성 화합물과
(b) 방향족 디히드록시 화합물 1몰에 대하여, 알칼리금속 또는 알칼리 토금속 화합물 10-8~10-3몰과
(c) 임의적으로 붕산 또는 붕산 에스테르를 함유하는 촉매를 사용하여 중축합을 행하여 그 말단기 전체의 30% 이하가 히드록시 말단기이고, 20℃ 메틸레 클로라이드 중에서 측정한 고유점도[η]가 0.3~1.0dℓ/g인 폴라카보네이트를 제조하는 것을 특징으로 하는 방향족 디히드록시 화합물과 카본산디에스테르의 용융 중축합 반응을 포함한다.
본 발명에 의한 폴리카보네이트의 제2제조 방법은 방향족 디히드록시 화합물 1몰에 대하여 탄소수가 17~50인 카본산디에스테르 0.05~15몰% 존재하에
(a) 질소 함유 염기성 화합물과
(b) 방향족 디히드록시 화합물 1몰에 대하여, 알칼리금속 또는 알칼리 토금속 화합물 10-8~10-3몰과
(c) 임의적으로 붕산 또는 붕산 에스테르를 함유하는 촉매를 사용하여 중축합을 행하여 그 말단기 전체의 30% 이하가 히드록시 말단기이고, 20℃ 메틸렌 클로라이드 중에서 측정한 고유점도[η]가 0.3~1.0dℓ/g인 폴라카보네이트를 제조하는 것을 특징으로 하는 방향족 디히드록시 화합물과 카본산 디페닐 화합물의 용융 중축합 반응을 포함한다.
본 발명에 의한 폴리카보네이트의 제3제조방법은 방향족 디히드록시 화합물 1몰을 기준하여 탄소수가 13~16인 카본산 디에스테르 0.05~15몰% 존재하에서,
(a) 질소함유 염기성 화합물과
(b) 방향족 디히드록시 화합물 1몰을 기준으로 알칼리금속 또는 알칼리 토금속 화합물 10-8~10-3몰과
(c) 임의적으로 붕산 또는 붕산 에스테르를 함유하는 촉매를 사용하여 중축합을 행하여 그 말단기 전체의 30% 이하가 히드록시 말단기이고, 20℃ 메틸렌 클로라이드 중에서 측정한 고유점도[η]가 0.3~1.0dℓ/g인 폴리카보네이트를 제조하는 것을 특징으로 하는 방향족 디히드록시 화합물과 카본산디페닐 혼합물의 용융 중축합 반응을 포함한다.
본 발명에 의한 폴리카보네이트는 그 말단기의 10~30%가 히드록시기인 것을 특징으로 한다.
또, 본 발명에 의한 폴리카보네이트는 그 말단기의 30% 이하가 히드록시기이며, 그 나트륨 함량이 1ppm이하, 염소 함량이 20ppm이하인 것을 특징으로 한다.
본 발명에 의한 폴리카보네이트의 제조방법을 상세히 설명한다.
본 발명에 의한 제1제조방법에서, 상기 폴리카보네이트는 탄소수가 10~40인 페놀 존재하에서 방향족 디히드록시화합물과 카본산 디에스테르의 용융 중축합 반응에 의해 제조된다.
본 발명에서 사용되는 상기 방향족 디히드록시 화합물들은 하기 일반식(Ⅰ)으로 표시되는 것들이다.
식에서 X는 또는 -SO2-이며, R1및 R2는 각각 수소 또는 1가의 탄화수소기이며, R3는 2가의 탄화수소기이며, 상기 방향성 핵은 1가의 탄화수소기 하나 또는 그 이상의 갯수로 지환될 수 있다.
또는 -SO2-이며, R1및 R2는 각각 수소 또는 1가의 탄화수소기이며, R3는 2가의 탄화수소기이며, 상기 방향성 핵은 1가의 탄화수소기 하나 또는 그 이상의 갯수로 지환될 수 있다.
상기에서 언급한 유용한 방향족 디히드록시 화합물은 구체적으로 비스(4-히드록시페닐)메탄, 1,1-비스(4-히드록시페닐)에탄, 2,2-비스(4-히드록시페닐)프로판, 2,2-비스(4-히드록시페닐)부탄, 2,2-비스(4-히드록시페닐)옥탄, 비스(4-히드록시페닐)페닐메탄, 2,2-비스(4-히드록시-1-메틸페닐)프로판, 1,1-비스(4-히드록시-t-부틸페닐)프로판 및 2,2-비스(4-히드록시-3-브로모페닐)프로판과 같은 비스(히드록시아릴)알칸류와 ; 1,1-비스(4-히드록시페닐)시클로펜탄, 1,1-비스(4-히드록시페닐)시클로헥산과 같은 비스(히드록시아릴)시클로알칸류와 ; 4,4′-디히드록시디페닐 에테르 및 4,4′-디히드록시-3,3′-디메틸디페닐 에테르와 같은 디히드록시아릴에테르류와, 4,4′-디히드록시디페닐 설파이드 및 4,4′-디히드록시-3,3′-디메틸 디페닐설파이드와 같은 디히드록시디아릴 설파이드류와 ; 4,4′-디히드록시디페닐 설폭사이드 및 4,4′-디히드록시-3,3′-디메틸디페닐설폭사이드와 같은 디히드록시디아릴설폭사이드류와 ; 4,4′-디히드록시디페닐 설폰 및 4,4′-디히드록시-3,3′-디메틸디페닐설폰과 같은 디히드록시아릴 설폰류 등이 있다.
상기에 예로 들은 화합물중에서 2,2-비스(4-히드록시페닐)프로판이 특히 양호하다.
본 발명에 유용한 카본산 디에스테르의 구체적인 예를 들면 디페닐카보네이트, 디톨릴 카보네이트, 비스(클로로페닐)카보네이트, m-크리실카보네이트, 디나프틸카보네이트, 비스(디페닐)카보네이트, 디에틸카보네이트, 디메틸카보네이트, 디부틸카보네이트 및 디시클로 헥실카보네이트 등이 있다.
상기에 예로들은 디에스테르중에서 디페닐 카보네이트가 특히 양호하다. 여기에서 사용된 카본산디에스테르는 50몰% 이하의 바람직하게는 30몰% 이하의 디카르복시산 또는 그의 에스테르를 소량 포함할 수도 있다. 상기 디카르복시산 및 그의 에스테르의 탄소수에 특별한 제한은 없으며 디카르복시산 및 그의 에스테르의 예로서는 테레프탈산, 이소프탈산, 테레프탈산 디페닐 및 이소프탈산디페닐 등이 있다. 이와같은 디카르복시산 또는 그의 에스테르를 조합사용하였을 경우에는 폴리카보네이트 대신에 폴리에스테르 폴리카보네이트가 얻어진다.
본 발명은 이 폴리에스테르 폴리카보네이트의 생성을 주목한다.
본 발명에 의한 제1제조방법으로 폴리카보네이트를 제조하는데 있어서 상기 카본산 디에스테르의 양을 상기 방향족 디히드록시 화합물 1몰에 대해 1.01~1.30몰, 바람직하게는 1.02~1.20몰의 양을 사용하는 것이 바람직하다.
본 발명에 의한 폴리카보네이트의 제1제조 방법에서, 방향족 디히드록시 화합물과 카본산디에스테르의 용융중축합 반응은 방향족 디히드록시 화합물 1몰에 대해 탄소수가 10~40, 바람직하게는 15~40인 페놀 0.05~15몰%, 바람직하게는 0.5~7몰%, 더욱 바람직하게는 1.5몰% 존재하에서 행한다.
본 발명에 의한 제1제조방법에 유용한 페놀류의 예로는 o-n-부틸페놀, m-n-부틸페놀, p-n-부틸페놀, o-이소부틸펜, m-이소부틸페놀, p-이소부틸페놀, o-t-부틸페놀, m-t-부틸페놀, p-t-부틸페놀, o-n-펜틸페놀, m-n-펜틸페놀, p-n-펜틸페놀, o-n-헥실페놀, m-n-헥실페놀, p-n-헥실페놀, o-시클로헥실페놀, m-시클로헥실페놀, p-시클로헥실페놀, o-페닐페놀, m-페닐페놀, p-페닐페놀, o-n-노닐페놀, m-n-노닐페놀, p-n-노닐페놀, o-큐밀페놀, m-큐밀페놀, p-큐밀페놀, o-나프틸페놀, m-나프틸페놀, p-나프틸페놀, 2,6-디-t-부틸페놀, 2,5-디-t-부틸페놀, 2,4-디-t-부틸페놀, 3,5-디-t-부틸페놀, 2,5-디큐밀페놀, 3,5-디큐밀페놀, 하기와 같은 크로마닐(chromanyl)화합물.

및 하기식을 갖는 1가 페노류 등이 있다.

상기 예시한 페놀류중에서 p-큐밀페놀 및 p-페닐페놀 등의 두개의 방향환을 갖는 2핵페놀류가 특히 바람직하다.
본 발명에 의한 폴리카보네이트의 제1제조 방법에서는 상기 방향족 디히드록시화합물 1몰에 대해 상기 페놀 0.05~15몰% 존재하에서 방향족 디히드록시 화합물과 카본산 디에스테르의 용융 중축합 반응을 행하는 것이 필수적이다. 이렇게 하므로서, 폴리카보네이트 생성물의 히드록시 말단기의 이온 봉쇄가 충분히 되어 만족스런 내열성과 내수성을 갖는 생성물이 얻어지며, 용융 중축합 반응의 속도가 현저히 빨라진다.
사용하는 페놀의 전량을 반응 전에 반응계에 가할 수도 있으며 사용하는 페놀의 일부량을 반응 전에 가하고 나머지 양은 반응중에 가할 수도 있다. 몇몇의 경우에서는 방향족 디히드록시 화합물과 카본산 디에스테르와의 반응을 페놀없이도 개시할 수도 있으며 반응의 어느정도 진행된 후에 페놀 전량을 반응계에 가할 수도 있다.
본 발명에 의한 폴리카보네이트의 제2제조방법을 상세히 설명한다.
본 발명에 의한 폴리카보네이트의 제2제조방법에서는 방향족 디히드록시 화합물과 카본산 디페닐 화합물의 용융 중축합 반응을 탄소수가 17~50인 카본산 디에스테르의 존재하에서 행한다.
상기 방향족 디히드록시 화합물로서 제1제조방법에서 쓰인 것을 사용할 수 있다.
상기 카본산 디페닐 화합물로는 탄소수가 13~16인 카본산 2핵 방향족 에스테르를 사용할 수 있다. 이러한 화합물의 예로는 카본산 디페닐, 카본산 트리페닐, 카본산 디톨릴, 카본산 비스(클로로페닐) 및 카본산 m-크리실 등이 있으며 이들중에서 카본산 디페닐이 특히 바람직하다.
본 발명에서 사용가능한 카본산 디페닐 화합물은 디카르복시산 및 2 에스테르를 50몰% 이하, 바람직하게는 30몰% 이하의 양을 함유하기도 한다. 상기 디카르복시산 및 그의 에스테르의 탄소수에 특별한 제한은 없으며, 디카르복시산 및 그 에스테르의 예로는 테레프날산, 이소프탈산, 테레프탈산 디페닐 및 이소프탈산 디페닐 등이 있다. 이와같은 디카르복시산 또는 그의 에스테르를 조합 사용하였을 경우에는 폴리카보네이트 대신에 폴리에스테르 카보네이트가 얻어진다. 본 발명은 이 폴리에스테르 폴리카보네이트의 생성을 주목한다.
본 발명에 의한 제2제조방법에서는 상기 카본산 디페닐 화합물을 상기 방향족 디히드록시 화합물 1몰당 1.01~1.30몰, 바람직하게는 1.02~1.20몰의 양으로 사용한다.
본 발명의 제2제조방법에서는 상기 방향족 디히드록시 화합물은 하기식으로 표시되는 C17~C50카본산 디에스테르 존재하에서 카본산 디페닐 화합물과 함께 용융 중축합 된다.
식에서 A는 탄소수가 6~25인 유기 기이며, B는 분자내의 총 탄소 원자수가 50이하인 탄소수가 10~25인 유기 기이다.
더욱 구체적으로는 본 발명에서 사용 가능한 적합한 C17~C50카본산 디에스테르류는 하기식들로 표시할 수 있다.

식에서 R1는 탄소수가 3~36인 탄화수소기이다.

식에서, R2는 탄소수가 1~19인 탄화수소기이며, R3는 분자내의 총 탄소수가 3~19인 탄화수소기이다.

식에서, R4는 탄소수가 1~30인 탄화수소기이며, R5는 분자내의 총 탄소수가 17~50의 범위내인 탄소수가 1~20인 탄화수소기이다.

식에서, R6는 탄소수가 4~37인 탄화수소기이다.

식에서, R7은 탄소수가 1~30인 탄화수소기이며, R8은 분자내의 총 탄소수가 50이하인 탄소수가 2~20인 탄화수소기이다.
본 발명에 의한 제2제조방법에 사용 가능한 양호한 C17~C50카본산 디에스테르의 예로는 카보부톡시페닐 페닐 카본산, 부틸페닐 메틸페닐 카본산, 부틸페닐 에틸페닐 카본산, 디-부톡시 페닐카본산, 비페닐 페닐 카본산, 디비페닐카본산, 큐밀페닐페닐카본산, 디-큐밀페닐카본산, 나프틸페닐카본산, 디-나프틸카본산, 카보르폭시 페닐페닐 카본산, 카보헵톡시페닐페닐카본산, 카보메틱시-t-부틸페닐페닐카본산, 크로마닐페닐카본산 및 디크로마닐 카본산 등이 잇다.
본 발명에 의한 폴리카보네이트의 제2제조 방법에서는 상기 방향족 디히드록시 화합물 1몰에 대해 상기 C17~C50카본산 디에스테르 0.05~15몰% 존재하에서 방향족 디히드록시 화합물과 카본산 디페닐 화합물의 용융 중축합 반응을 행하는 것이 필수적이다. 이렇게하므로써 폴리카보네이트 생성물의 히드록시 말단기의 이온봉쇄가 충분히 되어 만족스런 내열성과 내수성을 갖는 생성물이 얻어지며, 용융 중축합의 속도가 현저히 빨라진다.
사용되는 상기 C17~C50카본산 디에스테르의 전량을 반응 전에 반응계에 가할 수도 있으며, 사용하는 C17~C50카본산 디에스테르의 일부량을 반응 전에 가하고 나머지량은 반응중에 가할 수도 있다. 몇몇의 경우에서는 방향족 디히드록시 화합물과 카본산 디페닐 화합물과의 반응을 C17~C50카본산 디에스테르 없이도 개시할 수 있으며 반응이 어느정도 진행된후에 C17~C50카본산 디에스테르 전량을 반응계에 가할 수도 있다.
본 발명에 의한 폴리카보네이트의 제3제조방법을 상세히 설명한다.
본 발명에 의한 폴리카보네이트의 제3제조방법에서는 방향족 디히드록시 화합물과 카본산 디페닐화합물과의 용융 중축합 반응을 탄소수가 13~16인 카본산 디에스테르 존재하에 행한다.
상기의 방향족 디히드록시 화합물의 예로는 상기 제1제조방법에서 쓰인 것들을 사용할 수 있다.
상기의 카본산 디페닐 화합물의 예로는, 제2제조방법에서 쓰인것들을 사용할 수 있다.
본 발명에 의한 제3제조방법에서는 상기 카본산디페닐 화합물을 상기 방향족 디히드록시 화합물 1몰당 1.01~1.30몰, 바람직하게는 1.02~1.20몰의 양으로 사용하는 것이 바람직하다.
본 방법에서 이온봉쇄제(sequestering agent)로서 사용할 수 있는 C13~D16카본산 디에스테르의 예로는 카본산 디페닐, 페닐톨릴카본산 및 카본산 디톨릴등이 있다. 따라서, 본 발명에서는 상기 이온봉쇄제와 카본산 디페닐 반응물이 동일한 경우가 고려된다.
상기 방향족 디히드록시 화합물 1몰에 대해 상기 카본산 디페닐 화합물과 같거나 다른 C13~C16카본산 디에스테르 0.05~15몰% 존재하에서 방향족 디히드록시 화합물과 카본산 디페닐화합물의 용융 중축합 반응을 행하므로써 폴라카보네이트 생성물의 히드록시 말단기의 이온분쇄가 바라는 바대로 충분히 되어, 만족스런 내열성 및 내수성을 갖는 생성물이 얻어진다.
사용되는 상기 C13~C16카본산 디에스테르의 전량을 반응전에 반응계에 가할 수도 있으며, 사용하는 C13~C16카본산 디에스테르의 일부량을 반응전에 가하고 나머지량은 반응중에 가할 수도 있다. 몇몇의 경우에서는 방향족 디히드록시 화합물과 카본산 디페닐 화합물과의 반응을 C13~C16카본산 디에스테르 없이도 개시할 수 있으며 반응이 어느정도 진행된후에 C13~C16카본산 디에스테르 전량을 반응계에 가할 수도 있다.
본 발명에 의한 제1, 제2 및 제3제조방법에서 원료물질들에 함유된 염소의 총량은 바람직하게는 20ppm이하, 더욱 바람직하게는 10ppm이하가 좋다.
상기 “염소함량”은 염화나트륨과 염화칼륨등의 염상태로 존재하는 염소와 페닐클로로 포르메이트 및 메틸렌클로라이드 등의 유기 화합물의 형태로 존재하는 염소를 의미한다. 염소함량은 이온크로마토그래피등의 분석법으로 정량을 할 수 있다.
원료물질의 염소함량이 20ppm이하인 경우, 폴리카보네이트 생성물의 색조가 우수해진다.
원료물질들을 pH가 6.0~9.0, 바람직하게는 7.0~8.5, 더욱 바람직하게는 7.0~8.0인 온수로 세척하고 78~105℃, 바람직하게는 80~100℃, 더욱 바람직하게는 80~90℃의 온도하에 방치시켜 그 총 염소함량을 쉽게 감소시킬 수 있다.
상기 방법에서 사용가능한 약알칼리 수용액의 예로는 수산화나트륨, 수산화칼륨, 수산화리튬, 탄산나트륨, 탄산칼륨, 수산화암모늄, 탄산수소나트륨, 탄산수소칼륨 및 수산화 테트라메틸 암모늄 등의 수용액이 있다. 이들중에서, 탄산수소나트륨, 탄산나트륨, 탄산수소칼륨 및 탄산칼륨의 수용액이 더욱 바람직하다.
상기와 같이 뜨거운 약알칼리 수용액으로 세척된 카본산 디에스테르 또는 카본산디페닐 및 카본산 디페닐 화합물들은 상기 용융중축합 반응에 직접 또는 바람직하게는 증류후 사용된다.
또한, 본 발명에서는 나트륨이온의 총합계 함량을 바람직하게는 1.0ppm이하, 더욱 바람직하게는 0.5ppm이하로 감소시킬 원료물질들을 사용하는 것이 유리하다. 이렇게 하므로써 더욱 훌륭한 색조를 갖는 폴리카보네이트가 제조된다.
상기 각 원료물질내의 나트륨 이온 함량은 원자흡수 분광계 또는 유도 커플링(induced coupling)플라즈마 방전분광계로 정량할 수 있다.
상기 각 원료물질내의 나트륨 이온 함량은 증류 또는 재결정등의 정제법에 의해 감속시킬 수 있다. 구체적으로는 상기 방향족 디히드록시 화합물의 나트륨 이온 함량은 방향족 디히드록시 화합물과 1가 페놀과의 부가반응 생성물을 생성시켜 이 부가반응 생성물을 분리한후, 이 생성물에서 1가페놀을 제거하는 과정을 포함하는 방법에 의해 감소될 수 있다.
방향족 디히드록시화합물과 카본산 디에스테르를 용융 중축합시키는 또는 방향족 디히드록시 화합물과 카본산 디페닐을 용융 중축합시키는 본 발명에 의한 폴리카보네이트의 제조방법에서 폴리카보네이트의 제조는
(a) 질소함유 염기성 화합물과
(b) 알칼리토금속 또는 알칼리토금속 화합물을 함유하는 촉매를 사용하여 행해진다.
상기 촉매성분(a)로서 유용한 질소함유 염기성 화합물의 예로는, 구체적으로는 수산화 테트라메틸암모늄(Me4NOH), 수산화 테트라에틸 암모늄(Et4NOH), 수산화 테트라부틸암모늄(Bu4NOH) 및 수산화트리메틸벤질암모늄 ( Ch2(ME)3ㆍNOH)와 같은 수산화 테트라 알킬-, 아릴- 또는 알카릴암모늄류와 ; 트리메틸아민, 트리에틸아민, 디메틸벤질아민 및 트리페닐아민 등과 같은 3급 아민류와 ; R2NH(이 식에서 R는 메틸 또는 에틸과 같은 알킬기이거나, 페닐 또는 톨루일과 같은 아릴기임)로 표시되는 2급아민류와 ; RNH2(식에서 R은 상기와 동일함)로 표시되는 1급아민류와 ; 또는 암모니아, 테트라메틸암모늄 보로히드라이드(Me4NBH4), 테트라부틸암모늄 보로히드라이드(Bu4NBH4), 테트라부틸암모늄 테트라 페닐보레이트(Bu4NBph4) 및 테트라메틸암모늄 테트라페닐보레이트(Me4NBph4)등과 같은 염기성 염등이 있다.
Ch2(ME)3ㆍNOH)와 같은 수산화 테트라 알킬-, 아릴- 또는 알카릴암모늄류와 ; 트리메틸아민, 트리에틸아민, 디메틸벤질아민 및 트리페닐아민 등과 같은 3급 아민류와 ; R2NH(이 식에서 R는 메틸 또는 에틸과 같은 알킬기이거나, 페닐 또는 톨루일과 같은 아릴기임)로 표시되는 2급아민류와 ; RNH2(식에서 R은 상기와 동일함)로 표시되는 1급아민류와 ; 또는 암모니아, 테트라메틸암모늄 보로히드라이드(Me4NBH4), 테트라부틸암모늄 보로히드라이드(Bu4NBH4), 테트라부틸암모늄 테트라 페닐보레이트(Bu4NBph4) 및 테트라메틸암모늄 테트라페닐보레이트(Me4NBph4)등과 같은 염기성 염등이 있다.
상기에 예로 들은 염기성 화합물중에서 수산화 테트라알킬암모늄이 특히 양호하다.
상기 촉매성분(b)으로서 유용한 알칼리금속의 예로서는 구체적으로는 수산화나트륨, 수산화칼륨, 수산화리튬, 탄산수소나트륨, 탄산수소칼륨, 탄산수소리튬, 탄산나트륨, 탄산칼륨, 탄산리튬, 초산나트륨, 초산칼륨, 초산리튬, 스테아린산나트륨, 스테아린칼륨, 스테아린산리튬, 나트륨보로히드라이드, 리튬보로히드라이드 및 나트륨보로페닐레이트, 벤조산나트륨, 벤조산칼륨, 벤조산리튬, 디나트륨하이드로겐포스페이트, 디칼륨하이드로겐포스페이트, 디리튬하이드로겐포스페이트, BPA의 디나트륨염, BPA의 디리튬염, BPA의 디칼륨염, 나트륨페닐레이트, 칼륨페닐레이트 및 리듐페닐레이트가 있다.
또한, 상기 촉매성분(b)로서 융용한 알칼리 토금속 화합물로는 구체적으로 수산화칼륨, 수산화바륨, 수산화마그네슘, 수산화스트론튬, 탄산수소칼슘, 탄산수소스트론튬, 카본산칼슘, 카본산바륨, 카본산마그네슘, 카본산스트론튬, 초산칼슘, 초산바륨, 초산마그네슘, 초산스트론튬, 스테아린산칼슘, 스테아린산바륨, 스테아린산마그네슘 및 스테아린산스트론튬 등이 있다.
상기에 예로들은 염기성 화합물을 함유하는 질소(a)는 전술한 방향족 디히드록시 화합물 1몰에 대해 10-6~10-1, 바람직하게는 10-5~10-2몰 정도의 양으로 사용하며, 상기에 예로들은 알칼리금속 또는 알칼리토금속 화합물(b)은 방향족 디히드록시 화합물 1몰당 10-8~10-3몰, 바람직하게는 10-7~10-5몰의 양을 사용한다.
상기 질소함유 염기성 화합물의 사용량은 방향족 디히드록시 화합물 1몰당 10-6~10-1몰의 양으로 하는 것이 바람직하다. 왜냐하면 에스테르 교환반응과 중축합 반응의 속도가 빨라져서 생성되는 폴리카보네이트의 색조와 내열성 및 내수성이 우수해지기 때문이다.
또한, 알칼리금속 또는 알칼리토금속 화합물은 방향족 디히드록시화합물 1몰당 10-8~10-3몰의 양으로 사용하는 것이 바람직하다. 왜냐하면, 중합반응 활성, 특히 중합반응 속도가 현저히 빨라져서 생성되는 폴리카보네이트의 색조, 내열성 및 내수성이 우수해지기 때문이다.
본 발명에 의한 방법에서 사용된 질소함유 염기성 화합물(a)과 알칼리 또는 알칼리토금속 화합물(b)의 조합을 포함하는 촉매는 높은 중합반응 활성을 가짐이 밝혀졌다.
이들은 우수한 내열성 및 내수성과 향상된 색조 및 투명성을 갖는 고분자 폴리카보네이트를 형성시킬 수 있다.
본 발명에 사용 가능한 바람직한 촉매는,
(a) 질소함유 염기성 화합물과,
(b) 알칼리금속 또는 알칼리토금속 화합물과,
(c) 붕산 또는 붕산에스테르
를 함유하는 것을 특징으로 한다.
상기 촉매성분(a)로서 유용한 질소함유 염기성 화합물과 상기 촉매성분(b)로서 유용한 알칼리 또는 알칼리토금속 화합물들은 앞서 예시한 것들과 같다.
상기 촉매성분(c)로서 유용한 붕산 또는 붕산 에스테르에는 붕산과 일반식 B(OR)n(OH)3-n(식에서 R는 메틸 및 에틸과 같은 알킬이거나 페닐과 같은 아릴이며, n은 1, 2 또는 3이다)로 표시되는 붕산에스테르 등이 있다.
구체적으로는 상기와 같은 붕산에스테르에는 붕산 트리메틸, 붕산트리에틸, 붕산트리부틸, 붕산트리헥실, 붕산트리헵틸, 붕산트리페닐, 붕산트리톨릴 및 붕산트리나프틸등이 있다.
상기 바림직한 촉매는 방향족 디히드록시 화합물 1몰당 10-6~10×1몰, 바람직하게는 10-5~10-2몰의 질소함유 염기성 화합물(a)과, 방향족 디히드록시 화합물 1몰당 10-8~10-3몰, 바람직하게는 10-7~10-4몰, 더욱 바람직하게는 10-7~10-5몰의 알칼리금속 또는 알칼리토금속 화합물(b)과, 방향족 디히드록시 화합물 1몰당 10-8~10-1몰, 바람직하게는 10-7~10-2몰, 더욱 바람직하게는 10-6~10-4몰의 붕산 또는 붕산에스테르(c)를 함유하는 것이 특징이다.
질소함유 염기성 화합물(a)과 알칼리금속 또는 알칼리토금속 화합물(b)는 방향족 디히드록시 화합물 1몰당 각각 10-6~10-1몰, 10-8~10-3몰의 양으로 사용하는 것이 상기와 같은 이유에서 바람직하다.
붕산 또는 붕산에스테르(c)는 방향족디히드록시 화합물 1몰당 10-8~10-1몰의 양으로 사용한다. 왜냐하면 생성되는 폴리카보네이트의 색조, 내수성 및 내열성이 우수하고, 열노화시 그 분자량의 감소가 방지되기 때문이다.
본 발명에 의한 제조방법에서 방향족 디히드록시화합물과 카본산디에스테르의 또는 방향족 디히드록시 화합물과 카본산 디페닐화합물의 중축합반응은 통상적으로 행하는 조건하에서도 수행할 수 있는데 구체적으로는 방향족 디히드록시화합물과 카본산디에스테르의 1단계 반응은 80~250℃, 바람직하게는 100~230℃, 더욱 바람직하게는 120~190℃하에서 상압하 0~5시간, 바람직하게는 0~4시간, 더욱 바람직하게는 0.25~3시간동안 수행한다. 제2단계에서는 감압하 반응온도를 높여 반응계를 지속시키고 최종적으로 240~320℃하에서 1mmHg 또는 더 낮은 and의 감압하에서 수행한다.
상기 중축합반응은 연속식 또는 배치식으로 행할 수 있으며 반응장치는 탱크, 튜브 또는 컬럼을 사용할 수도 있다.
본 발명에 의한 폴리카보네이트를 상세히 설명한다.
본 발명에 의한 폴리카보네이트는 그 말단기의 10~30%가 히드록시기인 것이 특징이다.
또한, 본 발명에 의한 폴리카보네이트는 그 말단기의 30%이하, 바람직하게는 5~25%, 더욱 바람직하게는 10~20%가 히드록시기이고, 그 나트륨함량이 1ppm이하, 바림직하게는 0.5ppm이하이고, 염소함량이 20ppm이하, 바람직하게는 10ppm이하인 것을 특징으로 한다.
상기와 같은 폴리카보네이트는 색조, 내열성 및 내수성이 특히 우수하다. 폴리카보네이트의 히드록시 말단기가 반응성 기를 제공하는 한편, 전술한 감량된 염소 및 나트륨함량이 히드록시 말단의 바람직하지 못한 반응을 억제하여 폴리카보네이트의 탈색을 방지하는 것으로 보여진다.
또한, 본 발명에 의한 폴리카보네이트가 리튬, 칼륨 및 세슘등의 알칼리 금속과 베릴륨, 마그네슘 및 칼슘등의 알칼리토금속의 합계 함량을 1ppm이하로 함유하는 경우, 그 폴리카보네이트의 색조, 내수성 및 내열성이 우수하다.
본 발명에 의한 폴리카보네이트는 [η]가 20℃메틸렌 클로라이드내에서 측정한 고유점도 0.3~1.0 dℓ/g이다.
본 발명에 의한 폴리카본이트는 그 제조방법에 결코 구애되지 않는다. 이들은 차후의 실시예에서 설명되듯이 원료물질들의 나트륨 및 염소함량과 이온분쇄제의 양 등의 조건을 적절하게 선택한 본 발명에 의한 제조방법에 의해 제조될 수 있다. 또한 이들은 특히 포스겐과 반응하는 원료 방향족 디히드록시 화합물의 나트륨 및 염소함량과 이온봉쇄제의 양이 적절하게 선택된 조건인 경우, 소위 인터페이스법에 의해 제조된다.
방향족 디히드록시화합물과 카본산 디에스테르 또는 카본산 디페닐화합물과의 용융 중축합반응이 선택된 이온봉쇄제의 특정량 존재하에서 특정 촉매가 사용되어 행해지는 본 발명의 제1, 제2 및 제3제조방법에 의하면, 전체말단기의 30%이하, 바람직하게는 20%이하, 더욱 바람직하게는 10~20%가 히드록시 말단기이고, 20℃메틸렌 클로라이드내에서 측정한 그 고유점도[η]가 0.3~1.0dℓ/g인 폴리카보네이트를 제조할 수 있다. 본 발명에 의한 제1, 제2 및 제3제조방법에 의해 얻어지는 폴리카보네이트는 그 내열성 및 내수성(내가수분해성)이 우수하다. 더우기 이들은 비등수 침지후에도 색조와 인장강도가 우수하다.
그 말단이 보호된 소정량의 히드록시말단기와 감소된 염소 및 타트륨함량을 갖는 본 발명에 의한 폴리카보네이트는 색조와 내열성과 내수성(내가수분해성)이 우수하다.
본 발명을 하기의 실시예들을 참조하여 상세히 설명한다. 그러나, 본 발명의 범위는 이들 실시예들에 한정되지는 않는다.
물성의 측정을 위해 사용한 방법들은 하기와 같다.
고유점도(Ⅳ) ; 유베로데스 점도계를 사용하여 20℃의 시료의 메틸렌클로라이드(0.5dℓ/g)용액으로 시료의 점도를 측정했다.
Hue(b 값) ; 니뽄덴쇼꾸 고오교오 K.K에서 생산 시판되는 색차측정계 ND-1001Dp를 사용한 투광법으로 2mm두께의 압축쉬트로 된 시험편의 랩(Lab)값을 특정했다. 측정된 b값은 황색도 측정시 이용했다.
열노화테스트(1) ; 시료의 펠릿을 400mmHg하, 120℃에서 12시간동안 건조시키고, 이 건조된 펠릿 4.5g을 테플론으로 만든 직경 40nm의 페트리접시에 계량하여 기어오븐(공기치환 비율이 71.6/시간인 디바이셰이사구제 GHPS-211)내에서 250℃하 16시간동안 방치시킨후 실온으로 냉각했다. 이렇게 처리한 시료를 2mm두께의 압축쉬트로 만들어 hue(b값) 및 Ⅳ을 측정하였다.
열노화테스트(2) ; 두께가 3mm인 시료의 압축쉬트를 기어오븐(공기치환비율이 71.6/시간인 디바이세이사구쇼 K.K에서 제조, 시판되는 GHPS-212)내에서 140℃하에, 1,000시간 동안 방치시킨후 실온에서 냉각했다. 이렇게 열노화된 시료의 hue(b값)을 측정하고, 하기식에 따라 황색도지수를 측정하였다.
YI=(71.53a+178.82b)/L=(100/Y)×(1.277X-1.60Z)
내수성테스트(1) ; 너비 5mm, 길이 5cm의 담벨로 0.5mm두께의 압축쉬트에 구멍이 나게 박은것을 비등수에 침지시키고 그 경과시간이 각각 1일인것, 3일인것, 7일인것을 회수했다. 회수후 1시간이내에 상기 처리된 담벨을 지퍼간격 30mm, 당김속도 50mm/min 측정범위 50kg의 조건하에서 인스트론 1132로 인장테스트를 하여 연신율(%)을 측정했다.
내수성테스트(2) ; 두께가 3mm인 시료의 압축 쉬트를 125℃에서 5일간 유지시킨 고압솔(autoclave)의 물에 침지시켰다. 처리된 압축쉬트의 헤이즈(haze)를 니뽄 덴쇼구 고오교오 K.K제 NDH-200으로 측정하였다.
압축쉬트 제조조건 ; 압축쉬트를 400mmHg하, 120℃에서 12시간동안 건조시키고, 질소공기에서 10분간 방치했다. 이후, 펠릿을 280℃하 100kg/㎠의 압력으로 5분간 압축한후, 실온에서 5분간 압축했다.
말단기 구조 ; 3ml 클로로포름에 시료 0.4g을 용해시킨 용액으로 40℃에서13C-NMR(니뽄 일렉트릭사제 GX-270)을 사용하여 말단 OH기와 말단기 구조를 측정하였다. 말단 OH기의 농도는 전체말단기의 양을 기준으로 %로 나타냈다.
말단 OH기의 농도 ; 메틸렌클로라이드 10ml에 시료 0.25g을 녹인 용액으로 FT-IR(시마쭈 세이사꾸쇼 K.K제 FT-IR 4300)을 사용하여 3580cm-1부근에서의 OH기의 흡수도를 측정하여 말단 OH기의 농도를 측정한후, 결과를 산출하였다.
나트륨함량 ; 중합체의 나트륨함량을 원자흡수 분광법(히다찌 세이사꾸쇼 K.K제 히다찌 180-80사용)에 의해 상기 중합체 20g의 애쉬(ash)에 대해 0.05ppm의 임계치로 측정하였다.
염소함량 ; 쉐니거법(Schoniger′s method)으로 중합체 50g을 기체화시켜 물에 녹인후, 이온크로마토그래피법(디오넥스 K.K사제 이온크로마토그래피 2000i 사용)에 의해 임계치 0.05ppm으로 염소함량을 측정하였다.
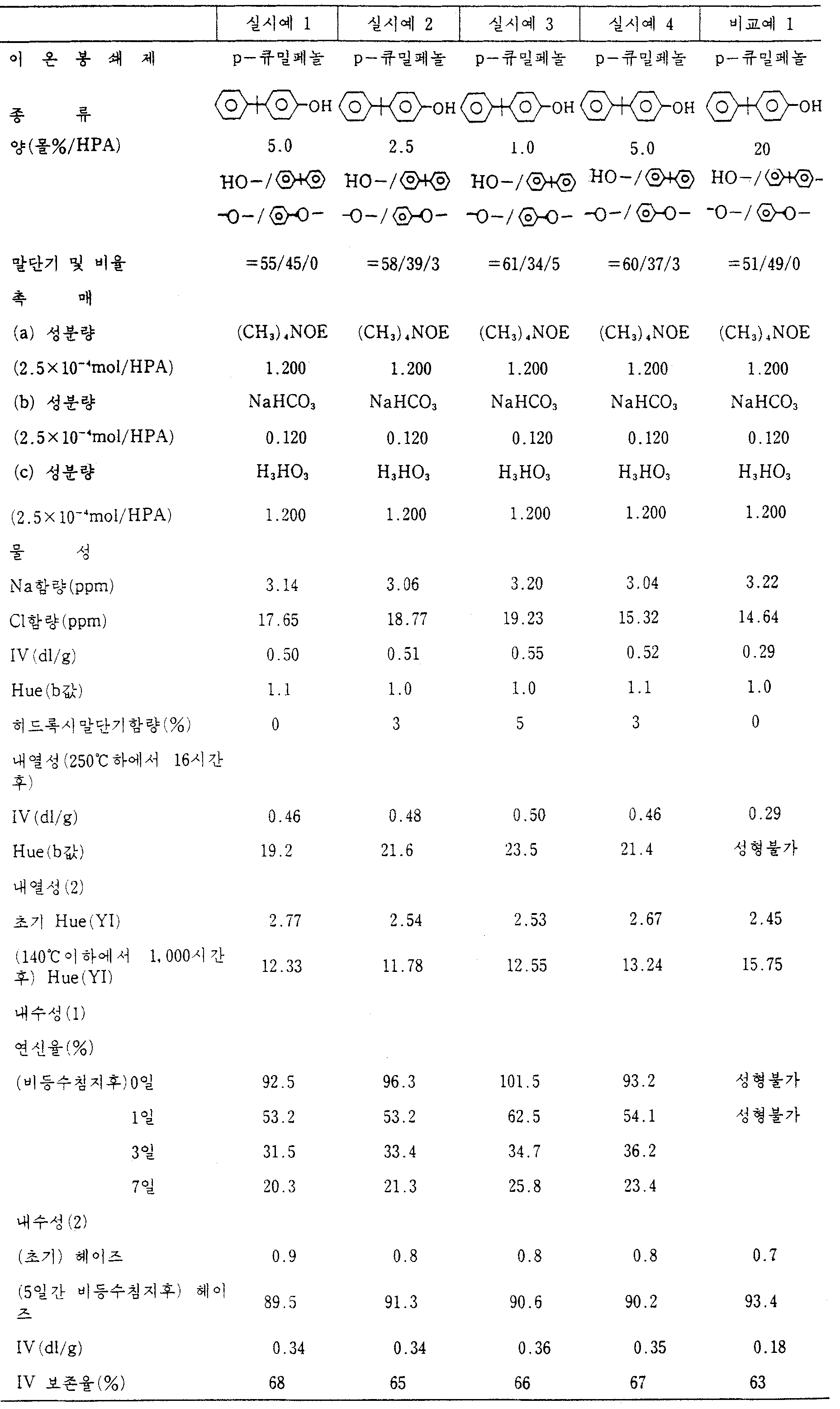
[실시예 1]
카본산디페닐(화르벤화브리켄 바이엘 A.G사제 0.05ppm이하의 나트륨함량과 24.0ppm의 염소 함량을 갖는) 47.08g(0.220몰)과, 비스페놀A(G.E사제 0.05ppm이하의 나트륨함량과 16.4ppm의 염소함량을 갖는) 45.600g(0.200몰)과 p-큐밀페놀 OH 2.12g(0.01몰, 즉 5몰%/비스페놀A) 및 붕산 H3BO3(와꼬 K.K제 보증부시약) 3.72mg(3×10-4몰/비스페놀A몰)을 100ml유리반응기내에 질소분위기하에서 충전시키고 이 혼합물을 온도 180℃로 가열한후, 니켈로 만든 교반기로 30분간 교반했다. 시간이 종료되는 순간에 도요고세이 K.K제 수산화 테트라메틸암모늄 Me4NOH 15% 수용성 36.48mg(3×10-4Me4NOH몰/비스페놀A)과 탄산수소나트륨 NaHCO3(와꼬 K.K제 보증부시약) 0.50mg(3×10-4Me4NOH몰/비스페놀A)을 반응기에 넣고 그 혼합액을 180℃, 질소공기하에서 30분 더 교반하여 에스테르 교환반응을 촉진시켰다.
OH 2.12g(0.01몰, 즉 5몰%/비스페놀A) 및 붕산 H3BO3(와꼬 K.K제 보증부시약) 3.72mg(3×10-4몰/비스페놀A몰)을 100ml유리반응기내에 질소분위기하에서 충전시키고 이 혼합물을 온도 180℃로 가열한후, 니켈로 만든 교반기로 30분간 교반했다. 시간이 종료되는 순간에 도요고세이 K.K제 수산화 테트라메틸암모늄 Me4NOH 15% 수용성 36.48mg(3×10-4Me4NOH몰/비스페놀A)과 탄산수소나트륨 NaHCO3(와꼬 K.K제 보증부시약) 0.50mg(3×10-4Me4NOH몰/비스페놀A)을 반응기에 넣고 그 혼합액을 180℃, 질소공기하에서 30분 더 교반하여 에스테르 교환반응을 촉진시켰다.
이후, 반응기를 210℃까지 가열하고 압력을 200mmHg까지 서서히 감압했다. 이 상태에서 1시간 유지시킨후, 240℃, 200mmHg에서 20분 더 유지시켰다. 이후, 압력을 150mmHg까지 서서히 감압하고 이 압력하에서 혼합액을 240℃하 20분간 유지시킨후, 100mmHg에서 20분간 더 유지시킨후 이후 15mmHg에서 반시간동안 더 유지시켰다. 최종적으로 온도는 270℃로 상승시키고 압력은 0.5mmHg로 감압시켜, 이 상태에서 반응계를 2.0시간 동안 유지시켰다.
0.50의 고유점도를 갖는 폴리카보네이트를 얻었다. 이 생성물의 말단 OH기들의 함량은 0%였다.
상기의 결과들을 표 1에 나타냈다.
[실시예 2~4 및 비교예 1]
표 1의 각 이온봉쇄제들을 표 1에 나타낸 양으로 사용한것만을 제외하고는 실시예 1을 반복했다.
그 결과들을 표 1에 나타냈다.
[표 1]
[실시예 5]
카본산디페닐(실시예 1에서 사용된 것과 동일) 45.368g(0.212몰)과 비스페놀A(실시예 1에서 사용된 것과 동일) 45.600g(0.200몰)과, 2-카보메톡시-5-t-부틸페닐페닐카본산(0.01몰, 즉 5몰%/비스페놀A) 3.280g 및 붕산 H3BO3(와꼬 K.K제 보증부시약) 3.72mg(3×10-4몰/비스페놀A몰)을 100ml유리반응기내에 질소분위기하에서 충전시키고 이 혼합물을 온도 180℃로 가열한 후, 니켈로 만든 교반기로 30분간 교반했다. 시간이 종료되는 순간에 도요고세이 K.K제 수산화 테트라메틸암모늄 Me4NOH 15% 수용액 36.48mg(3×10-4Me4NOH몰/비스페놀A)과 탄산수소나트륨 NaHCO3(와꼬 K.K제 보증부시약) 0.50mg(3×10-4Me4NOH몰/비스페놀A)을 반응기에 넣고 그 혼합액을 180℃, 질소공기하에서 30분 더 교반하여 에스테르 교환반응을 촉진시켰다. 이후, 반응기를 210℃까지 가열하고 압력을 200mmHg까지 서서히 감압했다. 이 상태에서 1시간 유지시킨후, 240℃, 200mmHg에서 20분 더 유지시켰다. 이후, 압력을 150mmHg까지 서서히 감압하고 이 압력하에서 혼합액을 240℃하 20분간 유지시킨후, 100mmHg에서 20분간 더 유지시킨후 이후 15mmHg에서 반시간동안 더 유지시켰다. 최종적으로 온도는 270℃로 상승시키고 압력은 0.5mmHg로 감압시켜, 이 상태에서 반응계를 2.0시간 동안 유지시켰다.
Ⅳ값이 0.51인 폴리카보네이트를 얻었다. 생성물의 말단 OH기들의 함량은 1%였다.
그 결과들을 표 2에 나타냈다.
[실시예 6~8 및 비교예 2]
표 2의 각 이온봉쇄제를 표 2에 나타낸 양으로 사용한 것만을 제외하고는 실시예 5와 동일하게 실시하였다.
그 결과를 표 2에 나타냈다.
[실시예 9]
상기 2-카보메톡시-5-t-부틸페닐 페닐 카본산 대신에 카본산 디페닐 0.01몰(5몰%/비스페놀A)를 부가적으로 사용한 것만을 제외하고는 실시예 5와 동일하게 실시하였다.
그 결과들을 표 2에 나타냈다.
[표 2]

[실시예 10]
카본산 디페닐(에니 K.K사제 카본산 디페닐을 pH7, 80℃의 물로 세척한후 90%수율로 증류시켜 얻어지며 0.05ppm이하의 나트륨 함량과 15.0ppm의 염소함량을 갖는) 141.24g(0.660몰)과, 비스페놀A(G.E사제, 0.10ppm이하의 나트륨함량과 0.8ppm의 염소함량을 갖는) 136.8g(0.600몰)과,

6.36g(0.03몰, 즉 5몰%/비스페놀A) 및 붕산 H3BO3(와꼬 K.K제 보증부시약) 의 3% 수용액 30mg(0.25×10-4몰/비스페놀A의 몰)을 500ml 유리반응기내에 질소분위기하에서 충전시키고 이 혼합물을 온도 180℃로 가열한후, 니켈로 만든 교반기로 30분간 교반했다. 시간이 종료되는 순간에 도요고세이 K.K제 수산화 테트라 메틸암모늄 Me4NOH 15% 수용액 91.2mg Me4NOH(2.5×10-4몰/비스페놀A)과 수산화나트륨 12.0mg(2.5×10-4몰/비스페놀A)을 반응기에 넣고, 그 혼합액을 180℃, 질소공기하에서 30분 더 교반하여 에스테르 교환반응을 촉진시켰다. 이후, 반응기를 210℃까지 가열하고 압력을 200mmHg까지 서서히 감압했다. 이 상태에서 1시간 유지시킨후, 240℃, 200mmHg에서 20분 더 유지시켰다. 이후 압력을 150mmHg까지 서서히 감압하고 이 압력하에서 혼합액을 240℃하 20분간 유지시킨후, 100mmHg에서 20분간 더 유지시킨후, 이후 15mmHg에서 반시간동안 더 유지시켰다. 최종적으로 온도는 270℃로 상승시키고 압력은 0.5mmHg로 감압시켜 이 상태에서 반응을 2.0시간동안 지속시켰다. Ⅳ값이 0.50인 폴리카보네이트가 얻어졌다. 이 생성물의 말단 OH기의 함량은 14%였다.
그 결과들을 표 3에 나타냈다.
[실시예 11]
카본산디페닐(실시예 10의 것과 동일) 136.1g(0.636몰)과 비스페놀A(실시예 10의 것과 동일) 136.8g(0.600몰)과, P-큐밀페닐페닐카본산 9.960g(0.03몰, 즉 5몰%/비스페놀A) 및 붕산 H3BO33% 수용액 3.0mg(0.025×10-4몰/비스페놀A 몰)을 500ml 유리반응기내에 질소분위기하에서 충전시키고 이 혼합물을 온도 180℃로 가열한후, 니켈로 만든 교반기로 30분간 교반했다. 시간이 종료되는 순간에 도요고세이 K.K제 수산화 테트라메틸암모늄 Me4NOH 15% 수용액 91.2mg (Me4NOH 2.5×10-4/비스페놀A)과 수산화나트륨 12.0mg(Me4NOH 2.5×10-4몰/비스페놀A)을 반응기에 넣고 그 혼합액을 180℃, 질소공기하에서 30분 더 교반하여 에스테르 교환반응을 촉진시켰다. 이후 반응기를 210℃까지 가열하고 압력을 200mmHg까지 서서히 감압했다. 이 상태에서 1시간 유지시킨후 240℃, 200mmHg에서 20분 더 유지시켰다. 이후, 압력을 150mmHg까지 서서히 감압하고 이 압력하에서 혼합액을 240℃하 20분간 유지시킨후 100mmHg에서 20분간 더 유지시킨후, 이후 15mmHg에서 반시간동안 더 유지시켰다. 최종적으로 온도는 270℃로 상승시키고 압력은 0.5mmHg로 감압시켜 이 상태에서 반응계를 2.0시간 동안 유지시켰다.
9.960g(0.03몰, 즉 5몰%/비스페놀A) 및 붕산 H3BO33% 수용액 3.0mg(0.025×10-4몰/비스페놀A 몰)을 500ml 유리반응기내에 질소분위기하에서 충전시키고 이 혼합물을 온도 180℃로 가열한후, 니켈로 만든 교반기로 30분간 교반했다. 시간이 종료되는 순간에 도요고세이 K.K제 수산화 테트라메틸암모늄 Me4NOH 15% 수용액 91.2mg (Me4NOH 2.5×10-4/비스페놀A)과 수산화나트륨 12.0mg(Me4NOH 2.5×10-4몰/비스페놀A)을 반응기에 넣고 그 혼합액을 180℃, 질소공기하에서 30분 더 교반하여 에스테르 교환반응을 촉진시켰다. 이후 반응기를 210℃까지 가열하고 압력을 200mmHg까지 서서히 감압했다. 이 상태에서 1시간 유지시킨후 240℃, 200mmHg에서 20분 더 유지시켰다. 이후, 압력을 150mmHg까지 서서히 감압하고 이 압력하에서 혼합액을 240℃하 20분간 유지시킨후 100mmHg에서 20분간 더 유지시킨후, 이후 15mmHg에서 반시간동안 더 유지시켰다. 최종적으로 온도는 270℃로 상승시키고 압력은 0.5mmHg로 감압시켜 이 상태에서 반응계를 2.0시간 동안 유지시켰다.
Ⅳ값이 0.50인 폴리카보네이트를 얻었다. 그 생성물의 말단 OH기의 함량은 14%였다.
그 결과들을 표 3에 나타냈다.
[실시예 12]
상기 P-큐밀페닐 페닐카본산 대신에 카본산 디페닐을 표 3에 나타낸 양으로 부가하여 사용한 것을 제외하고는 실시예 11과 동일하게 실시하였다. Ⅳ값이 0.52인 폴리카보네이트가 얻어졌다. 이 생성물의 말단 OH기의 함량은 14%였다.
그 결과들을 표 3에 나타냈다.
[실시예 13]
상기 270℃, 0.5mmHg하에서의 반응을 2.2시간동안 행한 것을 제외하고는 실시예 10과 동일하게 실시하였다. Ⅳ값이 0.52인 폴리카보네이트가 얻어졌다. 이 생성물의 말단 OH기의 함량은 7%였다.
[실시예 14]
상기 270℃, 0.5mmHg하에서의 반응을 2.2시간동안 행한 것을 제외하고는 실시예 12와 동일하게 실시하였다. Ⅳ값이 0.52인 폴리카보네이트가 얻어졌으며 그의 말단 OH기의 함량은 7%였다.
그 결과들을 표 3에 나타냈다.
[실시예 15]
상기 270℃, 0.5mmHg하에서의 반응을 2.5시간동안 행한 것을 제외하고는 실시예 10과 동일하게 실시하였다. Ⅳ가 0.53인 폴리카보네이트가 얻어졌으며 그의 말단 OH기의 함량은 2%였다.
그 결과들을 표 3에 나타냈다.
[실시예 16]
상기 270℃, 0.5mmHg하에서의 반응을 2.5시간동안 행한 것을 제외하고는 실시예 12와 동일하게 실시하였다. Ⅳ가 0.56인 폴리카보네이트가 얻어졌으며 그의 말단 OH기의 함량은 2%였다.
그 결과들을 표 3에 나타냈다.
[실시예 17]
상기 NaOH를 표 3에 나타낸 양으로 사용하고, 270℃ 0.5mmHg하에서의 반응을 2.2시간동안 행한 것을 제외하고는 실시예 12와 동일하게 실시하였다.
그 결과들을 표 3에 나타냈다.
[실시예 18]
상기 NaOH를 표 3에 나타낸 양으로 사용하고, 270℃ 0.5mmHg하에서의 반응을 1.8시간동안 행한 것을 제외하고는 실시예 12와 동일하게 실시하였다.
그 결과들을 표 3에 나타냈다.
[실시예 19~22]
상기 NaOH대신에 표 3의 나트륨 화합물을 표 3에 나타낸 양으로 사용한 것을 제외하고는 실시예 12와 동일하게 실시하였다.
그 결과들을 표 3에 나타냈다.
[비교예 4 및 비교예 5]
상기 카본산 디페닐을 표 3에 나타난 양으로 부가하여 사용한 것을 제외하고는 실시예 12와 동일하게 실시하였다.
그 결과들을 표 3에 나타냈다.
[비교예 6]
비스페놀A와 포스겐으로부터 인터페이스법에 의해 제조된 것으로서 C.E.사제 폴리카보네이트(Ⅳ가 0.50dℓ/g이며, 말단 히드록시기 함량이 0%이고, 나트륨 함량은 0.5ppm이하, 염소함량이 30ppm)의 성질을 표 3에 나타냈다.
[실시예 23~25]
상기 NaOH 대신에 표 4에 나타낸 화합물들을 표 4에 나타낸 양으로 사용한 것을 제외하고는 실시예 12와 동일하게 실시하였다.
그 결과들을 표 4에 나타냈다.
[표 3a]

[표 3b]

[표 4]

Claims (30)
- 방향족 디히드록시화합물과 카본산 디에스테르의 용융중축합 반응에 의하여 폴리카보네이트를 제조함에 있어서, 방향족 디히드록시화합물 1몰에 대해 탄소수가 10~40인 페놀과 탄소수가 약 13~50인 카본산 디에스테르로 구성된 군에서 선택한 화합물 0.05~15몰%를 존재시켜 (a) 질소함유 염기성 화합물과 (b) 방향족 디히드록시 화합물 1몰에 대해 알칼리금속 또는 알칼리토금속 화합물 10-8~10-3몰과 (c) 임의적으로 붕산 또는 붕산에스테르로 구성된 촉매를 사용하여 중축합을 행하여 그 말단기 전체의 30% 이하가 히드록시 말단기이고 20℃, 메틸렌 클로라이드중에서 측정한 고유점도[η]가 0.3~1.0dl/g인 폴리카보네이트를 제조함을 특징으로 하는 폴리카보네이트의 제조방법.
- 제1항에 있어서, 상기 붕산 및 붕산 에스테르를 사용하는 것이 특징인 폴리카보네이트의 제조방법.
- 제1항에 있어서, 상기 페놀이 10~25개의 탄소원자를 갖는 것이 특징인 폴리카보네이트의 제조방법.
- 제2항에 있어서, 상기 페놀이 10~25개의 탄소원자를 갖는 것이 특징인 폴리카보네이트의 제조방법.
- 제1항에 있어서, 상기 페놀이 2핵페놀인 것이 특징인 폴리카보네이트의 제조방법.
- 제2항에 있어서, 상기 페놀이 2핵페놀인 것이 특징인 폴리카보네이트의 제조방법.
- 제1항에 있어서, 상기 페놀이 P-큐밀 페놀인 것이 특징인 폴리카보네이트의 제조방법.
- 제2항에 있어서, 상기 페놀이 P-큐밀 페놀인 것이 특징인 폴리카보네이트의 제조방법.
- 제1항에 있어서, 상기 화합물이 탄소수가 10~40인 페놀인 것이 특징인 폴리카보네이트의 제조방법.
- 제1항에 있어서, 상기 화합물이 탄소수가 10~16인 카본산 디에스테르인 것이 특징인 폴리카보네이트의 제조방법.
- 제1항에 있어서, 상기 화합물이 탄소수가 17~50인 카본산 디에스테르인 것이 특징인 폴리카보네이트의 제조방법.
- 제1항에 있어서, 제조된 폴리카보네이트의 전체 말단기의 30%이하가 히드록시기이고, 그 폴리카보네이트는 그 나트륨 함량이 0.5ppm이하이고, 염소함량이 10ppm이하인 것이 특징인 폴리카보네이트의 제조방법.
- 제1항에 있어서, 제조된 폴리카보네이트의 전체 말단기의 5~25%이하가 히드록시기이고, 그 폴리카보네이트는 그 나트륨 함량이 0.5ppm이하이고, 염소함량이 10ppm이하인 것이 특징인 폴리카보네이트의 제조방법.
- 제2항에 있어서, 제조된 폴리카보네이트의 전체 말단기의 30%이하가 히드록시기이고, 그 폴리카보네이트는 그 나트륨 함량이 0.5ppm이하이고, 염소함량이 10ppm이하인 것이 특징인 폴리카보네이트의 제조방법.
- 제2항에 있어서, 제조된 폴리카보네이트의 전체 말단기의 5~25%이하가 히드록시기이고, 그 폴리카보네이트는 그 나트륨 함량이 0.5ppm이하이고, 염소함량이 10ppm이하인 것이 특징인 폴리카보네이트의 제조방법.
- 제9항에 있어서, 제조된 폴리카보네이트의 전체 말단기의 30%이하가 히드록시기이고, 그 폴리카보네이트는 그 나트륨 함량이 0.5ppm이하이고, 염소함량이 10ppm이하인 것이 특징인 폴리카보네이트의 제조방법.
- 제9항에 있어서, 제조된 폴리카보네이트의 전체 말단기의 5~25%이하가 히드록시기이고, 그 폴리카보네이트는 그 나트륨 함량이 0.5ppm이하이고, 염소함량이 10ppm이하인 것이 특징인 폴리카보네이트의 제조방법.
- 제10항에 있어서, 제조된 폴리카보네이트의 전체 말단기의 30%이하가 히드록시기이고, 그 폴리카보네이트는 그 나트륨 함량이 0.5ppm이하이고, 염소함량이 10ppm이하인 것이 특징인 폴리카보네이트의 제조방법.
- 제10항에 있어서, 제조된 폴리카보네이트의 전체 말단기의 5~25%가 히드록시기이고, 그 폴리카보네이트는 그 나트륨 함량이 0.5ppm이하이고, 염소함량이 10ppm이하인 것이 특징인 폴리카보네이트의 제조방법.
- 제11항에 있어서, 제조된 폴리카보네이트의 전체 말단기의 30%이하가 히드록시기이고, 그 폴리카보네이트는 그 나트륨 함량이 0.5ppm이하이고, 염소함량이 10ppm이하인 것이 특징인 폴리카보네이트의 제조방법.
- 제11항에 있어서, 제조된 폴리카보네이트의 총 말단기의 5~25%가 히드록시기이고, 그 폴리카보네이트는 그 나트륨 함량이 0.5ppm이하이고, 염소함량이 10ppm이하인 것이 특징인 폴리카보네이트의 제조방법.
- 말단기의 10~30%가 히드록시기인 것이 특징인 폴리카보네이트.
- 말단기의 30%이하가 히드록시기이며 나트륨 함량이 1ppm이하이고, 염소함량이 20ppm이하인 것이 특징인 폴리카보네이트.
- 제23항에 있어서, 말단기의 5~25%가 히드록시기인 것이 특징인 폴리카보네이트.
- 제23항에 있어서, 말단기의 10~20%가 히드록시기인 것이 특징인 폴리카보네이트.
- 제23항에 있어서, 나트륨 함량이 0.5ppm이하이고 염소함량이 10ppm이하인 것이 특징인 폴리카보네이트.
- 제24항에 있어서, 나트륨 함량이 0.5ppm이하이고 염소함량이 10ppm이하인 것이 특징인 폴리카보네이트.
- 제25항에 있어서, 나트륨 함량이 0.5ppm이하이고 염소함량이 10ppm이하인 것이 특징인 폴리카보네이트.
- 제23~25항중 어느 한 항에 있어서, 용융 중축합 반응에 의해 제조된 것이 특징인 폴리카보네이트.
- 제26~28항중 어느 한 항에 있어서, 용융 중축합 반응에 의해 제조된 것이 특징인 폴리카보네이트.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP63-238429 | 1988-09-22 | ||
| JP23842888 | 1988-09-22 | ||
| JP23842988 | 1988-09-22 | ||
| JP63-238428 | 1988-09-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR900004799A KR900004799A (ko) | 1990-04-13 |
| KR930004609B1 true KR930004609B1 (ko) | 1993-06-01 |
Family
ID=26533688
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1019890013660A KR930004609B1 (ko) | 1988-09-22 | 1989-09-22 | 폴리카보네이트 및 그 제조방법 |
Country Status (9)
| Country | Link |
|---|---|
| US (2) | US5151491A (ko) |
| EP (1) | EP0360578B1 (ko) |
| JP (1) | JPH0739483B2 (ko) |
| KR (1) | KR930004609B1 (ko) |
| AT (1) | ATE169316T1 (ko) |
| BR (1) | BR8904803A (ko) |
| DE (1) | DE68928766T2 (ko) |
| ES (1) | ES2121739T3 (ko) |
| SG (1) | SG73353A1 (ko) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7369789B2 (en) | 2004-12-07 | 2008-05-06 | Samsung Electronics Co., Ltd. | Door status switch operation mechanism for image forming apparatus |
Families Citing this family (108)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE68928766T2 (de) * | 1988-09-22 | 1999-03-25 | Ge Plastics Japan Ltd., Tokio/Tokyo | Verfahren zur Herstellung von Polycarbonaten |
| US5055523A (en) * | 1989-03-20 | 1991-10-08 | Ce Plastics Japan Limited | Aromatic polycarbonate resin composition |
| JP2662049B2 (ja) * | 1989-09-14 | 1997-10-08 | 出光石油化学株式会社 | 光学式ディスク基板及び光学式情報記録媒体 |
| JP2554775B2 (ja) * | 1990-11-29 | 1996-11-13 | 出光石油化学株式会社 | ポリカーボネートの製造方法 |
| JPH04222829A (ja) * | 1990-12-25 | 1992-08-12 | Teijin Chem Ltd | 燈光カバー成形用材料 |
| US5548041A (en) * | 1991-05-08 | 1996-08-20 | Daicel Chemical Industries, Ltd. | Process for producing polycarbonate |
| US5434227A (en) * | 1991-05-08 | 1995-07-18 | Daicel Chemical Industries, Ltd. | Process for producing polycarbonate |
| JP2924985B2 (ja) * | 1991-06-28 | 1999-07-26 | 日本ジーイープラスチックス株式会社 | ポリカーボネートの製造方法 |
| US5276109A (en) * | 1991-06-28 | 1994-01-04 | General Electric Company | Optical polycarbonate compositions |
| JPH059287A (ja) * | 1991-06-28 | 1993-01-19 | Nippon G Ii Plast Kk | ポリカーボネートの製造方法 |
| JP2922337B2 (ja) * | 1991-06-28 | 1999-07-19 | 日本ジーイープラスチックス株式会社 | 共重合ポリカーボネートの製造方法 |
| JPH05117382A (ja) * | 1991-10-29 | 1993-05-14 | Nippon G Ii Plast Kk | 共重合ポリカーボネート、その製造方法およびそれからなる組成物 |
| US5468836A (en) * | 1992-01-29 | 1995-11-21 | Daicel Chemical Industries, Ltd. | Process for the preparation of polycarbonate |
| JPH05262872A (ja) * | 1992-03-17 | 1993-10-12 | Idemitsu Petrochem Co Ltd | ポリカーボネートの製造方法 |
| DE4217775A1 (de) * | 1992-05-29 | 1993-12-02 | Bayer Ag | Verfahren zur Herstellung von Polycarbonaten |
| DE69333628T2 (de) * | 1992-10-14 | 2005-02-03 | Daicel Chemical Industries, Ltd., Sakai | Verfahren zur Darstellung von Polycarbonaten durch Polykondensation in der Schmelze |
| EP0592900A3 (en) * | 1992-10-14 | 1995-02-22 | Daicel Chem | Process for the preparation of polycarbonates by polycondensation in the melt. |
| DE4238123C2 (de) * | 1992-11-12 | 2000-03-09 | Bayer Ag | Verfahren zur Herstellung von thermoplastischen Polycarbonaten |
| DE4240588A1 (de) * | 1992-12-03 | 1994-06-09 | Bayer Ag | Verfahren zur Herstellung von Polycarbonaten |
| JPH06228300A (ja) * | 1992-12-11 | 1994-08-16 | Daicel Chem Ind Ltd | ポリカーボネートの製造法 |
| EP0892010A3 (en) * | 1993-01-29 | 1999-03-31 | Daicel Chemical Industries, Ltd. | Boron containing polycarbonate composition |
| DE4312390A1 (de) * | 1993-04-16 | 1994-10-20 | Bayer Ag | Zweistufen-Verfahren zur Herstellung von thermoplastischem Polycarbonat |
| DE4320156A1 (de) * | 1993-06-18 | 1994-12-22 | Bayer Ag | Verfahren zur Herstellung von thermoplastischen Polycarbonaten |
| JP3396942B2 (ja) * | 1994-02-21 | 2003-04-14 | 三菱化学株式会社 | 芳香族ポリカーボネートの製造方法 |
| EP0677545B1 (en) * | 1994-04-14 | 2000-07-05 | Daicel Chemical Industries, Ltd. | Process for the preparation of (co)-polycarbonate |
| JP3390277B2 (ja) | 1994-12-28 | 2003-03-24 | 日本ジーイープラスチックス株式会社 | ポリカーボネートの製造方法 |
| JP3327308B2 (ja) * | 1994-12-28 | 2002-09-24 | 日本ジーイープラスチックス株式会社 | ポリカーボネートの製造方法 |
| US5717057A (en) * | 1994-12-28 | 1998-02-10 | General Electric Company | Method of manufacturing polycarbonate |
| JP3327307B2 (ja) | 1994-12-28 | 2002-09-24 | 日本ジーイープラスチックス株式会社 | ポリカーボネートの製造方法 |
| DE19511467A1 (de) * | 1995-03-29 | 1996-10-02 | Bayer Ag | Verfahren zur Herstellung von thermoplastischem Polycarbonat |
| JP3613355B2 (ja) * | 1995-09-19 | 2005-01-26 | 出光興産株式会社 | ポリカーボネート及びその製造方法 |
| DE69605167T2 (de) * | 1995-09-19 | 2000-09-14 | Teijin Ltd., Osaka | Verfahren zur Herstellung von Polycarbonat |
| EP0774491A3 (en) | 1995-11-20 | 1997-12-03 | General Electric Company | Films, sheets and molded products made of a polyester/polycarbonate composition |
| JPH1087969A (ja) | 1996-09-11 | 1998-04-07 | Nippon G Ii Plast Kk | 透明なポリエステル/ポリカーボネート組成物の製造方法 |
| EP0893476A1 (en) * | 1997-07-23 | 1999-01-27 | Daicel Chemical Industries, Ltd. | Thermoplastic resin composition |
| US6486241B2 (en) | 1997-08-29 | 2002-11-26 | General Electric Company | Polycarbonate resin composition |
| US6613820B2 (en) | 1997-08-29 | 2003-09-02 | General Electric Company | Polycarbonate resin composition |
| MY121010A (en) * | 1997-08-29 | 2005-12-30 | Sabic Innovative Plastics Ip | Polycarbonate resin composition |
| US5973101A (en) * | 1997-09-30 | 1999-10-26 | General Electric Company | Aromatic polycarbonate resin composition |
| US6323302B1 (en) | 1998-04-27 | 2001-11-27 | Teijin Limited | Carbonic acid diester, aromatic polycarbonate and facilities, and preparation thereof |
| JP3621972B2 (ja) * | 1998-09-18 | 2005-02-23 | 出光興産株式会社 | ポリカーボネート樹脂組成物及びそのシート成形品 |
| US6339138B1 (en) | 1998-11-04 | 2002-01-15 | General Electric Company | Method of manufacturing polycarbonates |
| US6140457A (en) * | 1999-02-26 | 2000-10-31 | General Electric Company | Static-resistant polycarbonates having DI-t-alkylphenyl end groups |
| US6022943A (en) * | 1999-04-07 | 2000-02-08 | General Electric Company | Optical quality polycarbonates with reduced static charge and method for making same |
| DE19933132A1 (de) | 1999-07-19 | 2001-01-25 | Bayer Ag | Verfahren zur Herstellung von modifizierten Polycarbonaten |
| DE19933128A1 (de) | 1999-07-19 | 2001-01-25 | Bayer Ag | Polycarbonat und dessen Formkörper |
| DE19954787A1 (de) * | 1999-11-15 | 2001-05-17 | Bayer Ag | Verfahren zur Herstellung von Polycarbonat |
| JP4322376B2 (ja) | 1999-11-26 | 2009-08-26 | Sabicイノベーティブプラスチックスジャパン合同会社 | 難燃性樹脂組成物およびその成型品 |
| JP2002194073A (ja) | 2000-12-26 | 2002-07-10 | Ge Plastics Japan Ltd | 新規なポリカーボネート系樹脂および該ポリカーボネート系樹脂の製造方法 |
| US6653434B2 (en) | 2000-12-29 | 2003-11-25 | General Electric Company | Process for the production of polycarbonate |
| WO2002060855A2 (en) * | 2000-12-28 | 2002-08-08 | General Electric Company | Process for the production of polycarbonate |
| US6608163B2 (en) * | 2001-01-17 | 2003-08-19 | General Electric Company | Polycarbonate copolymers having improved hydrolytic stability |
| JP2002348368A (ja) | 2001-05-28 | 2002-12-04 | Ge Plastics Japan Ltd | 光学用ポリカーボネートおよびその用途 |
| JP3866934B2 (ja) * | 2001-05-28 | 2007-01-10 | 日本ジーイープラスチックス株式会社 | 光学用ポリカーボネートおよびその用途 |
| JP4606660B2 (ja) | 2001-07-17 | 2011-01-05 | Sabicイノベーティブプラスチックスジャパン合同会社 | ポリカーボネートの製造方法およびポリカーボネート製造装置 |
| US6870025B2 (en) | 2001-07-24 | 2005-03-22 | General Electric Company | Method of polycarbonate preparation |
| US6548623B2 (en) * | 2001-07-24 | 2003-04-15 | General Electric Company | Method of polycarbonate preparation |
| US6809817B2 (en) | 2001-08-24 | 2004-10-26 | General Electric Company | Method and apparatus for in situ determination of molten polycarbonate composition using electronic absorption spectroscopy |
| US20030120025A1 (en) * | 2001-09-07 | 2003-06-26 | Brack Hans Peter | Process for the production of polycarbonate |
| US6683689B2 (en) | 2001-10-02 | 2004-01-27 | General Electric Company | Method for rapid determination of composition of polycarbonate resin |
| US6500914B1 (en) | 2001-10-10 | 2002-12-31 | General Electric Company | Method for end-capping polycarbonate resins and composition for use in same |
| KR20050035139A (ko) * | 2001-10-10 | 2005-04-15 | 제너럴 일렉트릭 캄파니 | 폴리카보네이트 수지를 말단 캡핑하는 방법 및 상기방법에서 사용하기 위한 조성물 |
| US6706846B2 (en) | 2001-10-10 | 2004-03-16 | General Electric Company | Method for end-capping polycarbonate resins and composition for use in same |
| US6504002B1 (en) | 2001-12-21 | 2003-01-07 | General Electric Company | Process for the production of branched melt polycarbonate by late addition of fries-inducing catalyst |
| US7488764B2 (en) * | 2003-01-23 | 2009-02-10 | Sabic Innovative Plastics Ip B.V. | Polymer encapsulation of high aspect ratio materials and methods of making same |
| US7312257B2 (en) * | 2003-01-23 | 2007-12-25 | General Electric Company | Polymer encapsulation of high aspect ratio materials and methods of making same |
| US20050085580A1 (en) * | 2003-10-16 | 2005-04-21 | General Electric Company | Light-Colored Polycarbonate Compositions and Methods |
| US7496938B2 (en) * | 2003-11-24 | 2009-02-24 | Sabic Innovative Plastics Ip B.V. | Media drive with a luminescence detector and methods of detecting an authentic article |
| US7175086B2 (en) * | 2004-04-21 | 2007-02-13 | General Electric Company | Authentication system, data device, and methods for using the same |
| US20050110978A1 (en) * | 2003-11-26 | 2005-05-26 | Radislav Potyrailo | Method of authenticating articles, authenticatable polymers, and authenticatable articles |
| US20050112768A1 (en) * | 2003-11-26 | 2005-05-26 | Thomas Evans | Method of authenticating tagged polymers |
| US7169615B2 (en) * | 2003-11-26 | 2007-01-30 | General Electric Company | Method of authenticating polymers, authenticatable polymers, methods of making authenticatable polymers and authenticatable articles, and articles made there from |
| US7094364B2 (en) * | 2003-11-26 | 2006-08-22 | General Electric Company | Method of authenticating polymers, authenticatable polymers, methods of making authenticatable polymers and authenticatable articles, and articles made there from |
| US20090266991A1 (en) * | 2003-11-26 | 2009-10-29 | Sabic Innovative Plastics Ip B.V. | Method of authenticating tagged polymers |
| US7112645B2 (en) * | 2004-03-19 | 2006-09-26 | General Electric Company | Polycarbonate composition and method of making thereof |
| US7041775B2 (en) * | 2004-04-20 | 2006-05-09 | General Electric Company | Method for preparing a polycarbonate oligomer mixture at low temperature for manufacturing polycarbonate |
| US20050277710A1 (en) * | 2004-06-14 | 2005-12-15 | Joyce Richard P | Tagged resin, method of making a tagged resin, and articles made therefrom |
| US7597961B2 (en) * | 2004-07-13 | 2009-10-06 | Sabic Innovative Plastics Ip B.V. | Authenticatable article and method of authenticating |
| US7312352B2 (en) * | 2004-08-02 | 2007-12-25 | Paul William Buckley | Method of preparing ester-substituted diaryl carbonates |
| US7105626B2 (en) * | 2004-09-10 | 2006-09-12 | General Electric Company | Method for incorporating alkyl ester endgroups to improve the release properties of melt polycarbonate |
| US7682696B2 (en) * | 2004-09-13 | 2010-03-23 | Sabic Innovative Plastics Ip B.V. | Medical article and method of making and using the same |
| US7132498B2 (en) * | 2004-09-27 | 2006-11-07 | General Electric Company | Process to make polycarbonate from bismethylsalicylcarbonate (BMSC) |
| US7230066B2 (en) * | 2004-12-16 | 2007-06-12 | General Electric Company | Polycarbonate—ultem block copolymers |
| US7635778B2 (en) * | 2004-12-17 | 2009-12-22 | Sabic Innovative Plastics Ip B.V. | Composition, method of authenticating, methods of making authenticatable compositions, authenticatable articles made there from |
| KR100713099B1 (ko) * | 2005-03-07 | 2007-05-02 | 주식회사 엘지화학 | 폴리카보네이트 수지의 제조 방법 |
| US7365149B2 (en) * | 2005-12-12 | 2008-04-29 | Hans-Peter Brack | Equipment cleaning in the manufacture of polycarbonates |
| US7485694B2 (en) * | 2005-12-21 | 2009-02-03 | Sabic Innovative Plastics Ip B.V. | Polycarbonates containing low levels of methyl salicylate prepared by a melt polymerization in a reactive extruder |
| US7485695B2 (en) * | 2005-12-21 | 2009-02-03 | Sabic Innovative Plastics Ip B.V | Polycarbonates containing low levels of methyl salicylate prepared by a melt polymerization in a reactive extruder |
| US7498399B2 (en) * | 2006-05-31 | 2009-03-03 | Sabic Innovative Plastics Ip B.V. | Method of preparing ester-substituted diaryl carbonates |
| US7495064B2 (en) * | 2006-06-26 | 2009-02-24 | Sabic Innovative Plastics Ip Bv | Manufacture of polycarbonates |
| US7482423B2 (en) * | 2006-06-30 | 2009-01-27 | Sabic Innovative Plastics Ip B.V. | Polycarbonates and method of preparing same |
| US7645851B2 (en) * | 2006-06-30 | 2010-01-12 | Sabic Innovative Plastics Ip B.V. | Polycarbonate with reduced color |
| US7541420B2 (en) * | 2006-06-30 | 2009-06-02 | Sabic Innovative Plastics Ip B.V. | Method for making molded polycarbonate articles with improved color |
| US7498400B2 (en) * | 2006-06-30 | 2009-03-03 | Sabic Innovative Plastics Ip B.V. | Method of preparing polycarbonate |
| US7601794B2 (en) * | 2007-09-28 | 2009-10-13 | Sabic Innovative Plastics Ip B.V. | Monomer solution for producing polycarbonate |
| US7632913B2 (en) * | 2007-09-28 | 2009-12-15 | Sabic Innovative Plastics Ip B.V. | Method of producing polycarbonate in a flash devolatilization system |
| US7619053B2 (en) * | 2007-09-28 | 2009-11-17 | Sabic Innovative Plastics Ip B.V. | Monomer solution for producing polycarbonate |
| US7615605B2 (en) * | 2008-03-26 | 2009-11-10 | Sabic Innovative Plastics Ip B.V. | Monomer solution for producing polycarbonate |
| US7671165B2 (en) * | 2008-05-16 | 2010-03-02 | Sabic Innovative Plastics Ip B.V. | Method of forming polycarbonate |
| US7674872B2 (en) * | 2008-06-17 | 2010-03-09 | Sabic Innovative Plastics Ip B.V. | Method of producing high molecular weight polymer |
| US7547799B1 (en) | 2008-06-20 | 2009-06-16 | Sabic Innovative Plastics Ip B.V. | Method for producing phenolic compound |
| US9403939B2 (en) | 2012-02-28 | 2016-08-02 | Sabic Global Technologies B.V. | Processes for preparing polycarbonates with enhanced optical properties |
| US20140179855A1 (en) | 2012-12-20 | 2014-06-26 | Sabic Innovative Plastics Ip B.V. | Thermoplastic compositions, methods of manufacture, and articles thereof |
| EP3152257A1 (en) | 2014-06-05 | 2017-04-12 | SABIC Global Technologies B.V. | Thermoplastic composition and laser-welded article |
| CN108368296B (zh) | 2015-11-13 | 2020-07-03 | 沙特基础工业全球技术公司 | 冲击性能改进的高透明熔融聚合聚碳酸酯 |
| WO2017145076A1 (en) | 2016-02-26 | 2017-08-31 | Sabic Global Technologies B.V. | Articles of manufacture using an impact performance modified melt polycarbonate |
| WO2018122140A1 (de) * | 2016-12-28 | 2018-07-05 | Covestro Deutschland Ag | Zusammensetzung und thermoplastische formmasse mit guter tieftemperaturzähigkeit, hohem glanzgrad und hoher verarbeitungsstabilität |
| EP4141073A1 (en) | 2021-08-31 | 2023-03-01 | SHPP Global Technologies B.V. | Filled conductive compositions with improved conductivity |
Family Cites Families (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3177179A (en) * | 1960-05-31 | 1965-04-06 | Dow Chemical Co | Chain terminated polycarbonates |
| US3240756A (en) * | 1961-03-02 | 1966-03-15 | Allied Chem | Continuous addition of phenolic chain terminator in bisphenol polycarbonate production by emulsion polymerization |
| DE1570533C3 (de) * | 1965-01-15 | 1975-05-28 | Bayer Ag, 5090 Leverkusen | Verfahren zum Herstellen von Polycarbonaten |
| US3544514A (en) * | 1965-01-15 | 1970-12-01 | Bayer Ag | Process for the production of thermoplastic polycarbonates |
| DE1570546A1 (de) * | 1965-03-09 | 1970-01-29 | Bayer Ag | Verfahren zum Herstellen hochmolekularer thermoplastischer Polycarbonate nach dem Umesterungsverfahren |
| US3697481A (en) * | 1971-03-08 | 1972-10-10 | Gen Electric | Chromanyl terminated polycarbonate |
| JPS5120993A (ja) * | 1974-08-14 | 1976-02-19 | Mitsubishi Gas Chemical Co | Tainetsuseihorikaaboneetojushino seizohoho |
| DE2439552A1 (de) * | 1974-08-17 | 1976-02-26 | Bayer Ag | Verfahren zur herstellung von bisphenol-a-polycarbonat nach dem schmelzumesterungsverfahren |
| DE2837526A1 (de) * | 1978-08-28 | 1980-03-20 | Bayer Ag | Verfahren zur herstellung von polymeren mit diphenolcarbonat-endgruppen |
| DE2842005A1 (de) * | 1978-09-27 | 1980-04-10 | Bayer Ag | Polycarbonate mit alkylphenyl-endgruppen, ihre herstellung und ihre verwendung |
| US4330664A (en) * | 1979-12-26 | 1982-05-18 | General Electric Company | Polycarbonate transesterification with catalyst containing aluminum hydride or borohydride group |
| USRE31262E (en) * | 1980-03-27 | 1983-05-31 | General Electric Company | Polycarbonate transesterification |
| US4469860A (en) * | 1981-09-28 | 1984-09-04 | General Electric Company | Aromatic polycarbonate resin end capped with hydroxy arylene sulfonate |
| US4590257A (en) * | 1983-07-05 | 1986-05-20 | General Electric Company | Boron- and nitrogen-containing compositions and their use in polycarbonate and polyester-polycarbonate synthesis |
| JPS6289723A (ja) * | 1985-10-15 | 1987-04-24 | Teijin Chem Ltd | ポリカ−ボネ−トの製造方法 |
| US4677184A (en) * | 1985-11-12 | 1987-06-30 | General Electric Company | Polycarbonate containing cycloalkenylphenyl terminal groups |
| US4699971A (en) * | 1985-11-12 | 1987-10-13 | General Electric Company | Polycarbonate with cycloalkylphenyl end group |
| US4788276A (en) * | 1985-11-12 | 1988-11-29 | General Electric Company | Copolyester carbonate with cycloalkylphenyl end groups |
| US4775739A (en) * | 1986-06-18 | 1988-10-04 | Mitsubishi Chemical Industries Limited | Process for producing polycarbonate resin with tertiary alryl phenol molecular weight modifier |
| US4839458A (en) * | 1986-06-18 | 1989-06-13 | Mitsubishi Chemical Industries Ltd. | Preparation with controlled amounts of polycarbonate carbon tetrachloride |
| JPS6312338A (ja) * | 1987-04-30 | 1988-01-19 | Nippon Steel Chem Co Ltd | 高温溶融物の定量排出調整方法 |
| US4880896A (en) * | 1987-05-30 | 1989-11-14 | Idemitsu Petrochemical Co., Ltd. | Polycarbonate for disc substrate having low bisphenol content |
| JPH0727662B2 (ja) * | 1987-08-27 | 1995-03-29 | 出光石油化学株式会社 | 光ディスク基板 |
| DE68911180T2 (de) * | 1988-03-28 | 1994-04-21 | Idemitsu Petrochemical Co | Polycarbonat. |
| DE68928766T2 (de) * | 1988-09-22 | 1999-03-25 | Ge Plastics Japan Ltd., Tokio/Tokyo | Verfahren zur Herstellung von Polycarbonaten |
-
1989
- 1989-09-20 DE DE68928766T patent/DE68928766T2/de not_active Expired - Lifetime
- 1989-09-20 AT AT89309548T patent/ATE169316T1/de not_active IP Right Cessation
- 1989-09-20 ES ES89309548T patent/ES2121739T3/es not_active Expired - Lifetime
- 1989-09-20 EP EP89309548A patent/EP0360578B1/en not_active Expired - Lifetime
- 1989-09-20 SG SG1996000063A patent/SG73353A1/en unknown
- 1989-09-21 US US07/410,464 patent/US5151491A/en not_active Expired - Lifetime
- 1989-09-22 KR KR1019890013660A patent/KR930004609B1/ko not_active IP Right Cessation
- 1989-09-22 JP JP1247533A patent/JPH0739483B2/ja not_active Expired - Fee Related
- 1989-09-22 BR BR898904803A patent/BR8904803A/pt not_active IP Right Cessation
-
1992
- 1992-06-19 US US07/901,012 patent/US5276129A/en not_active Expired - Lifetime
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7369789B2 (en) | 2004-12-07 | 2008-05-06 | Samsung Electronics Co., Ltd. | Door status switch operation mechanism for image forming apparatus |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0360578A3 (en) | 1990-10-31 |
| ATE169316T1 (de) | 1998-08-15 |
| JPH02175723A (ja) | 1990-07-09 |
| EP0360578A2 (en) | 1990-03-28 |
| JPH0739483B2 (ja) | 1995-05-01 |
| US5151491A (en) | 1992-09-29 |
| KR900004799A (ko) | 1990-04-13 |
| EP0360578B1 (en) | 1998-08-05 |
| US5276129A (en) | 1994-01-04 |
| DE68928766T2 (de) | 1999-03-25 |
| DE68928766D1 (de) | 1998-09-10 |
| BR8904803A (pt) | 1990-05-01 |
| ES2121739T3 (es) | 1998-12-16 |
| SG73353A1 (en) | 2000-06-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR930004609B1 (ko) | 폴리카보네이트 및 그 제조방법 | |
| KR920010147B1 (ko) | 폴리카보네이트의 제조방법 | |
| KR100681372B1 (ko) | 폴리카르보네이트 및 그의 성형품 | |
| JP3122721B2 (ja) | ポリカーボネート組成物およびその製造方法 | |
| JP2924985B2 (ja) | ポリカーボネートの製造方法 | |
| JPH0618868B2 (ja) | ポリカーボネートの製造方法 | |
| KR20020010939A (ko) | 개질된 폴리카르보네이트의 제조 방법 | |
| KR100671618B1 (ko) | 폴리카보네이트의 제조방법 | |
| US9458290B2 (en) | Process for preparing highly polymerized aromatic polycarbonate resin | |
| JPH0627190B2 (ja) | ポリカーボネートの製造方法 | |
| JP2628905B2 (ja) | ポリカーボネートの製造方法 | |
| JP3729681B2 (ja) | ポリカーボネートの製造方法 | |
| JPH11106630A (ja) | 芳香族ポリカーボネート樹脂組成物 | |
| JP2996880B2 (ja) | ポリカーボネート | |
| JPH11106631A (ja) | 芳香族ポリカーボネート樹脂組成物 | |
| JP2984289B2 (ja) | コポリエステルカーボネートの製造方法 | |
| JP3281077B2 (ja) | ポリカーボネート及びポリアリーレートを含む樹脂組成物 | |
| JP2000136239A (ja) | ポリカーボネートの製造方法 | |
| JPH1067850A (ja) | ポリカーボネートの製造方法 | |
| JPH0649195A (ja) | 食品用プラスチック容器 | |
| JPH11106634A (ja) | 芳香族ポリカーボネート樹脂組成物 | |
| JP3037721B2 (ja) | ポリカーボネートおよびその製造方法ならびにポリカーボネート樹脂組成物 | |
| JP2000136238A (ja) | ポリカーボネートの製造方法 | |
| JP2830945B2 (ja) | ポリカーボネート組成物およびその製造方法 | |
| JPH11106632A (ja) | 芳香族ポリカーボネート樹脂組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A201 | Request for examination | ||
| E902 | Notification of reason for refusal | ||
| E701 | Decision to grant or registration of patent right | ||
| GRNT | Written decision to grant | ||
| FPAY | Annual fee payment |
Payment date: 20080529 Year of fee payment: 16 |
|
| LAPS | Lapse due to unpaid annual fee |