JP2014201698A - 硬化性樹脂組成物 - Google Patents
硬化性樹脂組成物 Download PDFInfo
- Publication number
- JP2014201698A JP2014201698A JP2013080551A JP2013080551A JP2014201698A JP 2014201698 A JP2014201698 A JP 2014201698A JP 2013080551 A JP2013080551 A JP 2013080551A JP 2013080551 A JP2013080551 A JP 2013080551A JP 2014201698 A JP2014201698 A JP 2014201698A
- Authority
- JP
- Japan
- Prior art keywords
- resin composition
- curable resin
- mass
- resin
- epoxy resin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 239000011342 resin composition Substances 0.000 title claims abstract description 107
- 239000003822 epoxy resin Substances 0.000 claims abstract description 126
- 229920000647 polyepoxide Polymers 0.000 claims abstract description 126
- 239000013034 phenoxy resin Substances 0.000 claims abstract description 78
- 229920006287 phenoxy resin Polymers 0.000 claims abstract description 78
- 239000003795 chemical substances by application Substances 0.000 claims abstract description 56
- 239000004593 Epoxy Substances 0.000 claims abstract description 31
- 125000005577 anthracene group Chemical group 0.000 claims abstract description 26
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical group C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 claims description 34
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical group O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 33
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical compound C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 claims description 31
- 150000002148 esters Chemical class 0.000 claims description 29
- 239000002648 laminated material Substances 0.000 claims description 28
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 claims description 24
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 claims description 22
- 239000011256 inorganic filler Substances 0.000 claims description 22
- 229910003475 inorganic filler Inorganic materials 0.000 claims description 22
- 239000004305 biphenyl Substances 0.000 claims description 17
- 235000010290 biphenyl Nutrition 0.000 claims description 17
- 239000004643 cyanate ester Substances 0.000 claims description 17
- 239000002245 particle Substances 0.000 claims description 17
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 claims description 13
- 239000000377 silicon dioxide Substances 0.000 claims description 13
- 239000004065 semiconductor Substances 0.000 claims description 12
- 229920001187 thermosetting polymer Polymers 0.000 claims description 12
- 239000000203 mixture Substances 0.000 claims description 11
- HECLRDQVFMWTQS-RGOKHQFPSA-N 1755-01-7 Chemical compound C1[C@H]2[C@@H]3CC=C[C@@H]3[C@@H]1C=C2 HECLRDQVFMWTQS-RGOKHQFPSA-N 0.000 claims description 9
- 230000001588 bifunctional effect Effects 0.000 claims description 9
- 229930185605 Bisphenol Natural products 0.000 claims description 8
- 238000000034 method Methods 0.000 abstract description 34
- 239000004020 conductor Substances 0.000 abstract description 20
- 238000007788 roughening Methods 0.000 abstract description 15
- 230000003746 surface roughness Effects 0.000 abstract description 5
- 239000012212 insulator Substances 0.000 abstract 1
- 229920005989 resin Polymers 0.000 description 60
- 239000011347 resin Substances 0.000 description 60
- 238000001723 curing Methods 0.000 description 57
- -1 hydrocarbon radicals Chemical class 0.000 description 57
- 239000000243 solution Substances 0.000 description 30
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 24
- 239000002313 adhesive film Substances 0.000 description 16
- PXKLMJQFEQBVLD-UHFFFAOYSA-N bisphenol F Chemical compound C1=CC(O)=CC=C1CC1=CC=C(O)C=C1 PXKLMJQFEQBVLD-UHFFFAOYSA-N 0.000 description 16
- 239000000758 substrate Substances 0.000 description 16
- 150000001875 compounds Chemical class 0.000 description 15
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 14
- 230000000052 comparative effect Effects 0.000 description 14
- 229920003986 novolac Polymers 0.000 description 14
- 239000000126 substance Substances 0.000 description 14
- 206010042674 Swelling Diseases 0.000 description 13
- 239000010408 film Substances 0.000 description 13
- 230000008961 swelling Effects 0.000 description 13
- 239000002966 varnish Substances 0.000 description 13
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 239000007788 liquid Substances 0.000 description 12
- 239000007822 coupling agent Substances 0.000 description 11
- 239000007787 solid Substances 0.000 description 11
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 10
- 238000010438 heat treatment Methods 0.000 description 10
- 150000002430 hydrocarbons Chemical group 0.000 description 10
- 238000005259 measurement Methods 0.000 description 10
- 229910052751 metal Inorganic materials 0.000 description 10
- 239000002184 metal Substances 0.000 description 10
- 239000003960 organic solvent Substances 0.000 description 10
- 239000005011 phenolic resin Substances 0.000 description 10
- 238000007747 plating Methods 0.000 description 10
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 9
- 125000003700 epoxy group Chemical group 0.000 description 8
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 8
- 239000002904 solvent Substances 0.000 description 8
- WBODDOZXDKQEFS-UHFFFAOYSA-N 1,2,3,4-tetramethyl-5-phenylbenzene Chemical group CC1=C(C)C(C)=CC(C=2C=CC=CC=2)=C1C WBODDOZXDKQEFS-UHFFFAOYSA-N 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 125000004185 ester group Chemical group 0.000 description 7
- 238000011156 evaluation Methods 0.000 description 7
- 229920006255 plastic film Polymers 0.000 description 7
- 239000002985 plastic film Substances 0.000 description 7
- 229920000139 polyethylene terephthalate Polymers 0.000 description 7
- 239000005020 polyethylene terephthalate Substances 0.000 description 7
- 230000001681 protective effect Effects 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- FRASJONUBLZVQX-UHFFFAOYSA-N 1,4-dioxonaphthalene Natural products C1=CC=C2C(=O)C=CC(=O)C2=C1 FRASJONUBLZVQX-UHFFFAOYSA-N 0.000 description 6
- BOKGTLAJQHTOKE-UHFFFAOYSA-N 1,5-dihydroxynaphthalene Chemical compound C1=CC=C2C(O)=CC=CC2=C1O BOKGTLAJQHTOKE-UHFFFAOYSA-N 0.000 description 6
- KJCVRFUGPWSIIH-UHFFFAOYSA-N 1-naphthol Chemical compound C1=CC=C2C(O)=CC=CC2=C1 KJCVRFUGPWSIIH-UHFFFAOYSA-N 0.000 description 6
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 6
- 229910000831 Steel Inorganic materials 0.000 description 6
- KKEYFWRCBNTPAC-UHFFFAOYSA-N Terephthalic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-N 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- MWPLVEDNUUSJAV-UHFFFAOYSA-N anthracene Chemical compound C1=CC=CC2=CC3=CC=CC=C3C=C21 MWPLVEDNUUSJAV-UHFFFAOYSA-N 0.000 description 6
- 125000003118 aryl group Chemical group 0.000 description 6
- YCIMNLLNPGFGHC-UHFFFAOYSA-N catechol Chemical compound OC1=CC=CC=C1O YCIMNLLNPGFGHC-UHFFFAOYSA-N 0.000 description 6
- 238000001035 drying Methods 0.000 description 6
- 229910052736 halogen Inorganic materials 0.000 description 6
- 150000002367 halogens Chemical class 0.000 description 6
- QQVIHTHCMHWDBS-UHFFFAOYSA-N isophthalic acid Chemical compound OC(=O)C1=CC=CC(C(O)=O)=C1 QQVIHTHCMHWDBS-UHFFFAOYSA-N 0.000 description 6
- 238000003475 lamination Methods 0.000 description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 6
- FZZQNEVOYIYFPF-UHFFFAOYSA-N naphthalene-1,6-diol Chemical compound OC1=CC=CC2=CC(O)=CC=C21 FZZQNEVOYIYFPF-UHFFFAOYSA-N 0.000 description 6
- 239000007800 oxidant agent Substances 0.000 description 6
- 239000010959 steel Substances 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical group C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 5
- VPWNQTHUCYMVMZ-UHFFFAOYSA-N 4,4'-sulfonyldiphenol Chemical compound C1=CC(O)=CC=C1S(=O)(=O)C1=CC=C(O)C=C1 VPWNQTHUCYMVMZ-UHFFFAOYSA-N 0.000 description 5
- 239000004793 Polystyrene Substances 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- 238000006243 chemical reaction Methods 0.000 description 5
- 238000005553 drilling Methods 0.000 description 5
- 238000005530 etching Methods 0.000 description 5
- 239000011888 foil Substances 0.000 description 5
- 238000010030 laminating Methods 0.000 description 5
- MNZMMCVIXORAQL-UHFFFAOYSA-N naphthalene-2,6-diol Chemical compound C1=C(O)C=CC2=CC(O)=CC=C21 MNZMMCVIXORAQL-UHFFFAOYSA-N 0.000 description 5
- 230000003472 neutralizing effect Effects 0.000 description 5
- 229920002223 polystyrene Polymers 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- 229920005992 thermoplastic resin Polymers 0.000 description 5
- DEQUKPCANKRTPZ-UHFFFAOYSA-N (2,3-dihydroxyphenyl)-phenylmethanone Chemical compound OC1=CC=CC(C(=O)C=2C=CC=CC=2)=C1O DEQUKPCANKRTPZ-UHFFFAOYSA-N 0.000 description 4
- JWAZRIHNYRIHIV-UHFFFAOYSA-N 2-naphthol Chemical compound C1=CC=CC2=CC(O)=CC=C21 JWAZRIHNYRIHIV-UHFFFAOYSA-N 0.000 description 4
- 229920002799 BoPET Polymers 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 238000001816 cooling Methods 0.000 description 4
- 229910052802 copper Inorganic materials 0.000 description 4
- 239000010949 copper Substances 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- 239000004744 fabric Substances 0.000 description 4
- 239000003063 flame retardant Substances 0.000 description 4
- 229910052731 fluorine Inorganic materials 0.000 description 4
- 239000011737 fluorine Substances 0.000 description 4
- 239000005350 fused silica glass Substances 0.000 description 4
- 238000005227 gel permeation chromatography Methods 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- GLTDLAUASUFHNK-UHFFFAOYSA-N n-silylaniline Chemical compound [SiH3]NC1=CC=CC=C1 GLTDLAUASUFHNK-UHFFFAOYSA-N 0.000 description 4
- 125000001624 naphthyl group Chemical group 0.000 description 4
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 4
- 238000003825 pressing Methods 0.000 description 4
- RNFJDJUURJAICM-UHFFFAOYSA-N 2,2,4,4,6,6-hexaphenoxy-1,3,5-triaza-2$l^{5},4$l^{5},6$l^{5}-triphosphacyclohexa-1,3,5-triene Chemical compound N=1P(OC=2C=CC=CC=2)(OC=2C=CC=CC=2)=NP(OC=2C=CC=CC=2)(OC=2C=CC=CC=2)=NP=1(OC=1C=CC=CC=1)OC1=CC=CC=C1 RNFJDJUURJAICM-UHFFFAOYSA-N 0.000 description 3
- PCFMUWBCZZUMRX-UHFFFAOYSA-N 9,10-Dihydroxyanthracene Chemical class C1=CC=C2C(O)=C(C=CC=C3)C3=C(O)C2=C1 PCFMUWBCZZUMRX-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 239000012670 alkaline solution Substances 0.000 description 3
- 125000000217 alkyl group Chemical group 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 238000004132 cross linking Methods 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 238000009826 distribution Methods 0.000 description 3
- 238000007772 electroless plating Methods 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 238000006386 neutralization reaction Methods 0.000 description 3
- 125000002524 organometallic group Chemical group 0.000 description 3
- QCDYQQDYXPDABM-UHFFFAOYSA-N phloroglucinol Chemical compound OC1=CC(O)=CC(O)=C1 QCDYQQDYXPDABM-UHFFFAOYSA-N 0.000 description 3
- 229960001553 phloroglucinol Drugs 0.000 description 3
- 229920001721 polyimide Polymers 0.000 description 3
- WQGWDDDVZFFDIG-UHFFFAOYSA-N pyrogallol Chemical compound OC1=CC=CC(O)=C1O WQGWDDDVZFFDIG-UHFFFAOYSA-N 0.000 description 3
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 2
- DHKHKXVYLBGOIT-UHFFFAOYSA-N 1,1-Diethoxyethane Chemical compound CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 2
- HTQNYBBTZSBWKL-UHFFFAOYSA-N 2,3,4-trihydroxbenzophenone Chemical compound OC1=C(O)C(O)=CC=C1C(=O)C1=CC=CC=C1 HTQNYBBTZSBWKL-UHFFFAOYSA-N 0.000 description 2
- JAHNSTQSQJOJLO-UHFFFAOYSA-N 2-(3-fluorophenyl)-1h-imidazole Chemical compound FC1=CC=CC(C=2NC=CN=2)=C1 JAHNSTQSQJOJLO-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- OKIZCWYLBDKLSU-UHFFFAOYSA-M N,N,N-Trimethylmethanaminium chloride Chemical compound [Cl-].C[N+](C)(C)C OKIZCWYLBDKLSU-UHFFFAOYSA-M 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 239000004642 Polyimide Substances 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 2
- 239000006087 Silane Coupling Agent Substances 0.000 description 2
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 2
- 239000011354 acetal resin Substances 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 229920000180 alkyd Polymers 0.000 description 2
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 2
- 238000000137 annealing Methods 0.000 description 2
- 150000001491 aromatic compounds Chemical class 0.000 description 2
- TZCXTZWJZNENPQ-UHFFFAOYSA-L barium sulfate Chemical compound [Ba+2].[O-]S([O-])(=O)=O TZCXTZWJZNENPQ-UHFFFAOYSA-L 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- KAKZBPTYRLMSJV-UHFFFAOYSA-N butadiene group Chemical group C=CC=C KAKZBPTYRLMSJV-UHFFFAOYSA-N 0.000 description 2
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 2
- 238000011088 calibration curve Methods 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- JUPWRUDTZGBNEX-UHFFFAOYSA-N cobalt;pentane-2,4-dione Chemical compound [Co].CC(=O)CC(C)=O.CC(=O)CC(C)=O.CC(=O)CC(C)=O JUPWRUDTZGBNEX-UHFFFAOYSA-N 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 239000011889 copper foil Substances 0.000 description 2
- 238000003851 corona treatment Methods 0.000 description 2
- XLJMAIOERFSOGZ-UHFFFAOYSA-M cyanate Chemical compound [O-]C#N XLJMAIOERFSOGZ-UHFFFAOYSA-M 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 229920001971 elastomer Polymers 0.000 description 2
- 238000009713 electroplating Methods 0.000 description 2
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- ZFSLODLOARCGLH-UHFFFAOYSA-N isocyanuric acid Chemical compound OC1=NC(O)=NC(O)=N1 ZFSLODLOARCGLH-UHFFFAOYSA-N 0.000 description 2
- 238000004898 kneading Methods 0.000 description 2
- RLSSMJSEOOYNOY-UHFFFAOYSA-N m-cresol Chemical compound CC1=CC=CC(O)=C1 RLSSMJSEOOYNOY-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 239000011976 maleic acid Substances 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 238000000691 measurement method Methods 0.000 description 2
- LVHBHZANLOWSRM-UHFFFAOYSA-N methylenebutanedioic acid Natural products OC(=O)CC(=C)C(O)=O LVHBHZANLOWSRM-UHFFFAOYSA-N 0.000 description 2
- 239000010445 mica Substances 0.000 description 2
- 229910052618 mica group Inorganic materials 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- IWDCLRJOBJJRNH-UHFFFAOYSA-N p-cresol Chemical compound CC1=CC=C(O)C=C1 IWDCLRJOBJJRNH-UHFFFAOYSA-N 0.000 description 2
- RMVRSNDYEFQCLF-UHFFFAOYSA-N phenyl mercaptan Natural products SC1=CC=CC=C1 RMVRSNDYEFQCLF-UHFFFAOYSA-N 0.000 description 2
- LYKRPDCJKSXAHS-UHFFFAOYSA-N phenyl-(2,3,4,5-tetrahydroxyphenyl)methanone Chemical compound OC1=C(O)C(O)=CC(C(=O)C=2C=CC=CC=2)=C1O LYKRPDCJKSXAHS-UHFFFAOYSA-N 0.000 description 2
- 229920000728 polyester Polymers 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 229920000098 polyolefin Polymers 0.000 description 2
- 229920006324 polyoxymethylene Polymers 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- CYIDZMCFTVVTJO-UHFFFAOYSA-N pyromellitic acid Chemical compound OC(=O)C1=CC(C(O)=O)=C(C(O)=O)C=C1C(O)=O CYIDZMCFTVVTJO-UHFFFAOYSA-N 0.000 description 2
- 230000007261 regionalization Effects 0.000 description 2
- 239000005060 rubber Substances 0.000 description 2
- FZHAPNGMFPVSLP-UHFFFAOYSA-N silanamine Chemical compound [SiH3]N FZHAPNGMFPVSLP-UHFFFAOYSA-N 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 238000004381 surface treatment Methods 0.000 description 2
- 230000000930 thermomechanical effect Effects 0.000 description 2
- 229910052718 tin Inorganic materials 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 229920002554 vinyl polymer Polymers 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- HJIAMFHSAAEUKR-UHFFFAOYSA-N (2-hydroxyphenyl)-phenylmethanone Chemical compound OC1=CC=CC=C1C(=O)C1=CC=CC=C1 HJIAMFHSAAEUKR-UHFFFAOYSA-N 0.000 description 1
- NAWXUBYGYWOOIX-SFHVURJKSA-N (2s)-2-[[4-[2-(2,4-diaminoquinazolin-6-yl)ethyl]benzoyl]amino]-4-methylidenepentanedioic acid Chemical compound C1=CC2=NC(N)=NC(N)=C2C=C1CCC1=CC=C(C(=O)N[C@@H](CC(=C)C(O)=O)C(O)=O)C=C1 NAWXUBYGYWOOIX-SFHVURJKSA-N 0.000 description 1
- WYTZZXDRDKSJID-UHFFFAOYSA-N (3-aminopropyl)triethoxysilane Chemical compound CCO[Si](OCC)(OCC)CCCN WYTZZXDRDKSJID-UHFFFAOYSA-N 0.000 description 1
- RUEBPOOTFCZRBC-UHFFFAOYSA-N (5-methyl-2-phenyl-1h-imidazol-4-yl)methanol Chemical compound OCC1=C(C)NC(C=2C=CC=CC=2)=N1 RUEBPOOTFCZRBC-UHFFFAOYSA-N 0.000 description 1
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 1
- 125000003837 (C1-C20) alkyl group Chemical group 0.000 description 1
- 125000006649 (C2-C20) alkynyl group Chemical group 0.000 description 1
- 125000006713 (C5-C10) cycloalkyl group Chemical group 0.000 description 1
- POILWHVDKZOXJZ-ARJAWSKDSA-M (z)-4-oxopent-2-en-2-olate Chemical compound C\C([O-])=C\C(C)=O POILWHVDKZOXJZ-ARJAWSKDSA-M 0.000 description 1
- SSUJUUNLZQVZMO-UHFFFAOYSA-N 1,2,3,4,8,9,10,10a-octahydropyrimido[1,2-a]azepine Chemical compound C1CCC=CN2CCCNC21 SSUJUUNLZQVZMO-UHFFFAOYSA-N 0.000 description 1
- JRNVQLOKVMWBFR-UHFFFAOYSA-N 1,2-benzenedithiol Chemical compound SC1=CC=CC=C1S JRNVQLOKVMWBFR-UHFFFAOYSA-N 0.000 description 1
- AYWSZYFQXSLSFY-UHFFFAOYSA-N 1,2-dihydrotriazine-5,6-dithione Chemical compound SC1=CN=NN=C1S AYWSZYFQXSLSFY-UHFFFAOYSA-N 0.000 description 1
- XZKLXPPYISZJCV-UHFFFAOYSA-N 1-benzyl-2-phenylimidazole Chemical compound C1=CN=C(C=2C=CC=CC=2)N1CC1=CC=CC=C1 XZKLXPPYISZJCV-UHFFFAOYSA-N 0.000 description 1
- AHDSRXYHVZECER-UHFFFAOYSA-N 2,4,6-tris[(dimethylamino)methyl]phenol Chemical compound CN(C)CC1=CC(CN(C)C)=C(O)C(CN(C)C)=C1 AHDSRXYHVZECER-UHFFFAOYSA-N 0.000 description 1
- WRBJZNAPDVBCQM-UHFFFAOYSA-N 2,6-dihydroxynaphthalene-1-carbaldehyde Chemical compound O=CC1=C(O)C=CC2=CC(O)=CC=C21 WRBJZNAPDVBCQM-UHFFFAOYSA-N 0.000 description 1
- OAYXUHPQHDHDDZ-UHFFFAOYSA-N 2-(2-butoxyethoxy)ethanol Chemical compound CCCCOCCOCCO OAYXUHPQHDHDDZ-UHFFFAOYSA-N 0.000 description 1
- FPZWZCWUIYYYBU-UHFFFAOYSA-N 2-(2-ethoxyethoxy)ethyl acetate Chemical compound CCOCCOCCOC(C)=O FPZWZCWUIYYYBU-UHFFFAOYSA-N 0.000 description 1
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 1
- SVONRAPFKPVNKG-UHFFFAOYSA-N 2-ethoxyethyl acetate Chemical compound CCOCCOC(C)=O SVONRAPFKPVNKG-UHFFFAOYSA-N 0.000 description 1
- QTWJRLJHJPIABL-UHFFFAOYSA-N 2-methylphenol;3-methylphenol;4-methylphenol Chemical compound CC1=CC=C(O)C=C1.CC1=CC=CC(O)=C1.CC1=CC=CC=C1O QTWJRLJHJPIABL-UHFFFAOYSA-N 0.000 description 1
- RJIQELZAIWFNTQ-UHFFFAOYSA-N 2-phenyl-1h-imidazole;1,3,5-triazinane-2,4,6-trione Chemical compound O=C1NC(=O)NC(=O)N1.C1=CNC(C=2C=CC=CC=2)=N1 RJIQELZAIWFNTQ-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- UIDDPPKZYZTEGS-UHFFFAOYSA-N 3-(2-ethyl-4-methylimidazol-1-yl)propanenitrile Chemical compound CCC1=NC(C)=CN1CCC#N UIDDPPKZYZTEGS-UHFFFAOYSA-N 0.000 description 1
- BVYPJEBKDLFIDL-UHFFFAOYSA-N 3-(2-phenylimidazol-1-yl)propanenitrile Chemical compound N#CCCN1C=CN=C1C1=CC=CC=C1 BVYPJEBKDLFIDL-UHFFFAOYSA-N 0.000 description 1
- SZUPZARBRLCVCB-UHFFFAOYSA-N 3-(2-undecylimidazol-1-yl)propanenitrile Chemical compound CCCCCCCCCCCC1=NC=CN1CCC#N SZUPZARBRLCVCB-UHFFFAOYSA-N 0.000 description 1
- JYLNVJYYQQXNEK-UHFFFAOYSA-N 3-amino-2-(4-chlorophenyl)-1-propanesulfonic acid Chemical compound OS(=O)(=O)CC(CN)C1=CC=C(Cl)C=C1 JYLNVJYYQQXNEK-UHFFFAOYSA-N 0.000 description 1
- JIGUICYYOYEXFS-UHFFFAOYSA-N 3-tert-butylbenzene-1,2-diol Chemical compound CC(C)(C)C1=CC=CC(O)=C1O JIGUICYYOYEXFS-UHFFFAOYSA-N 0.000 description 1
- UUEWCQRISZBELL-UHFFFAOYSA-N 3-trimethoxysilylpropane-1-thiol Chemical compound CO[Si](OC)(OC)CCCS UUEWCQRISZBELL-UHFFFAOYSA-N 0.000 description 1
- TYOXIFXYEIILLY-UHFFFAOYSA-N 5-methyl-2-phenyl-1h-imidazole Chemical compound N1C(C)=CN=C1C1=CC=CC=C1 TYOXIFXYEIILLY-UHFFFAOYSA-N 0.000 description 1
- ULKLGIFJWFIQFF-UHFFFAOYSA-N 5K8XI641G3 Chemical compound CCC1=NC=C(C)N1 ULKLGIFJWFIQFF-UHFFFAOYSA-N 0.000 description 1
- ZXLYUNPVVODNRE-UHFFFAOYSA-N 6-ethenyl-1,3,5-triazine-2,4-diamine Chemical compound NC1=NC(N)=NC(C=C)=N1 ZXLYUNPVVODNRE-UHFFFAOYSA-N 0.000 description 1
- 108010054404 Adenylyl-sulfate kinase Proteins 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 1
- 125000003358 C2-C20 alkenyl group Chemical group 0.000 description 1
- HFPOONLVDGDERK-UHFFFAOYSA-N CC(C)(c(cc1)ccc1N=C=O)c(cc1)ccc1Oc1nc(OC2C=CC(C(C)(C)c(cc3)ccc3N=C=O)=CC2)nc(Oc2ccc(C(C)(C)c(cc3)ccc3N=C=O)cc2)n1 Chemical compound CC(C)(c(cc1)ccc1N=C=O)c(cc1)ccc1Oc1nc(OC2C=CC(C(C)(C)c(cc3)ccc3N=C=O)=CC2)nc(Oc2ccc(C(C)(C)c(cc3)ccc3N=C=O)cc2)n1 HFPOONLVDGDERK-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 229920002284 Cellulose triacetate Polymers 0.000 description 1
- WAEMQWOKJMHJLA-UHFFFAOYSA-N Manganese(2+) Chemical compound [Mn+2] WAEMQWOKJMHJLA-UHFFFAOYSA-N 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- BPQQTUXANYXVAA-UHFFFAOYSA-N Orthosilicate Chemical compound [O-][Si]([O-])([O-])[O-] BPQQTUXANYXVAA-UHFFFAOYSA-N 0.000 description 1
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 1
- 101150003085 Pdcl gene Proteins 0.000 description 1
- FFFPYJTVNSSLBQ-UHFFFAOYSA-N Phenolphthalin Chemical compound OC(=O)C1=CC=CC=C1C(C=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 FFFPYJTVNSSLBQ-UHFFFAOYSA-N 0.000 description 1
- 239000004962 Polyamide-imide Substances 0.000 description 1
- 239000004695 Polyether sulfone Substances 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000004721 Polyphenylene oxide Substances 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 1
- 102100039024 Sphingosine kinase 1 Human genes 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- NNLVGZFZQQXQNW-ADJNRHBOSA-N [(2r,3r,4s,5r,6s)-4,5-diacetyloxy-3-[(2s,3r,4s,5r,6r)-3,4,5-triacetyloxy-6-(acetyloxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6s)-4,5,6-triacetyloxy-2-(acetyloxymethyl)oxan-3-yl]oxyoxan-2-yl]methyl acetate Chemical compound O([C@@H]1O[C@@H]([C@H]([C@H](OC(C)=O)[C@H]1OC(C)=O)O[C@H]1[C@@H]([C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(C)=O)O1)OC(C)=O)COC(=O)C)[C@@H]1[C@@H](COC(C)=O)O[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O NNLVGZFZQQXQNW-ADJNRHBOSA-N 0.000 description 1
- ORLQHILJRHBSAY-UHFFFAOYSA-N [1-(hydroxymethyl)cyclohexyl]methanol Chemical compound OCC1(CO)CCCCC1 ORLQHILJRHBSAY-UHFFFAOYSA-N 0.000 description 1
- UUQQGGWZVKUCBD-UHFFFAOYSA-N [4-(hydroxymethyl)-2-phenyl-1h-imidazol-5-yl]methanol Chemical compound N1C(CO)=C(CO)N=C1C1=CC=CC=C1 UUQQGGWZVKUCBD-UHFFFAOYSA-N 0.000 description 1
- AHZMUXQJTGRNHT-UHFFFAOYSA-N [4-[2-(4-cyanatophenyl)propan-2-yl]phenyl] cyanate Chemical compound C=1C=C(OC#N)C=CC=1C(C)(C)C1=CC=C(OC#N)C=C1 AHZMUXQJTGRNHT-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 125000002723 alicyclic group Chemical group 0.000 description 1
- 239000004844 aliphatic epoxy resin Substances 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 125000002877 alkyl aryl group Chemical group 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- ZRALSGWEFCBTJO-UHFFFAOYSA-N anhydrous guanidine Natural products NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 1
- 239000002518 antifoaming agent Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- UIJGNTRUPZPVNG-UHFFFAOYSA-N benzenecarbothioic s-acid Chemical compound SC(=O)C1=CC=CC=C1 UIJGNTRUPZPVNG-UHFFFAOYSA-N 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- RWCCWEUUXYIKHB-UHFFFAOYSA-N benzophenone Chemical compound C=1C=CC=CC=1C(=O)C1=CC=CC=C1 RWCCWEUUXYIKHB-UHFFFAOYSA-N 0.000 description 1
- 239000012965 benzophenone Substances 0.000 description 1
- ZFVMWEVVKGLCIJ-UHFFFAOYSA-N bisphenol AF Chemical compound C1=CC(O)=CC=C1C(C(F)(F)F)(C(F)(F)F)C1=CC=C(O)C=C1 ZFVMWEVVKGLCIJ-UHFFFAOYSA-N 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 238000007664 blowing Methods 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 239000006229 carbon black Substances 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- FCEOGYWNOSBEPV-FDGPNNRMSA-N cobalt;(z)-4-hydroxypent-3-en-2-one Chemical compound [Co].C\C(O)=C\C(C)=O.C\C(O)=C\C(C)=O FCEOGYWNOSBEPV-FDGPNNRMSA-N 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 238000011437 continuous method Methods 0.000 description 1
- 239000013256 coordination polymer Substances 0.000 description 1
- 150000001879 copper Chemical class 0.000 description 1
- 229910000365 copper sulfate Inorganic materials 0.000 description 1
- XCJYREBRNVKWGJ-UHFFFAOYSA-N copper(II) phthalocyanine Chemical compound [Cu+2].C12=CC=CC=C2C(N=C2[N-]C(C3=CC=CC=C32)=N2)=NC1=NC([C]1C=CC=CC1=1)=NC=1N=C1[C]3C=CC=CC3=C2[N-]1 XCJYREBRNVKWGJ-UHFFFAOYSA-N 0.000 description 1
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 1
- HCRZXNOSPPHATK-UHFFFAOYSA-L copper;3-oxobutanoate Chemical compound [Cu+2].CC(=O)CC([O-])=O.CC(=O)CC([O-])=O HCRZXNOSPPHATK-UHFFFAOYSA-L 0.000 description 1
- 229930003836 cresol Natural products 0.000 description 1
- 229910002026 crystalline silica Inorganic materials 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 150000001925 cycloalkenes Chemical class 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 125000000664 diazo group Chemical group [N-]=[N+]=[*] 0.000 description 1
- SOCTUWSJJQCPFX-UHFFFAOYSA-N dichromate(2-) Chemical compound [O-][Cr](=O)(=O)O[Cr]([O-])(=O)=O SOCTUWSJJQCPFX-UHFFFAOYSA-N 0.000 description 1
- 229940028356 diethylene glycol monobutyl ether Drugs 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- XXBDWLFCJWSEKW-UHFFFAOYSA-N dimethylbenzylamine Chemical compound CN(C)CC1=CC=CC=C1 XXBDWLFCJWSEKW-UHFFFAOYSA-N 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 125000003055 glycidyl group Chemical group C(C1CO1)* 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 238000013007 heat curing Methods 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 239000012943 hotmelt Substances 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002440 hydroxy compounds Chemical class 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 238000007733 ion plating Methods 0.000 description 1
- 150000002505 iron Chemical class 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- AQBLLJNPHDIAPN-LNTINUHCSA-K iron(3+);(z)-4-oxopent-2-en-2-olate Chemical compound [Fe+3].C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O AQBLLJNPHDIAPN-LNTINUHCSA-K 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 238000007561 laser diffraction method Methods 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 150000002696 manganese Chemical class 0.000 description 1
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 1
- 229910000000 metal hydroxide Inorganic materials 0.000 description 1
- 150000004692 metal hydroxides Chemical class 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- GEMHFKXPOCTAIP-UHFFFAOYSA-N n,n-dimethyl-n'-phenylcarbamimidoyl chloride Chemical compound CN(C)C(Cl)=NC1=CC=CC=C1 GEMHFKXPOCTAIP-UHFFFAOYSA-N 0.000 description 1
- KBJFYLLAMSZSOG-UHFFFAOYSA-N n-(3-trimethoxysilylpropyl)aniline Chemical compound CO[Si](OC)(OC)CCCNC1=CC=CC=C1 KBJFYLLAMSZSOG-UHFFFAOYSA-N 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- BMGNSKKZFQMGDH-FDGPNNRMSA-L nickel(2+);(z)-4-oxopent-2-en-2-olate Chemical compound [Ni+2].C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O BMGNSKKZFQMGDH-FDGPNNRMSA-L 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- QWVGKYWNOKOFNN-UHFFFAOYSA-N o-cresol Chemical compound CC1=CC=CC=C1O QWVGKYWNOKOFNN-UHFFFAOYSA-N 0.000 description 1
- JFOJYGMDZRCSPA-UHFFFAOYSA-J octadecanoate;tin(4+) Chemical compound [Sn+4].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O JFOJYGMDZRCSPA-UHFFFAOYSA-J 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 1
- 239000012766 organic filler Substances 0.000 description 1
- AFEQENGXSMURHA-UHFFFAOYSA-N oxiran-2-ylmethanamine Chemical compound NCC1CO1 AFEQENGXSMURHA-UHFFFAOYSA-N 0.000 description 1
- JCGNDDUYTRNOFT-UHFFFAOYSA-N oxolane-2,4-dione Chemical compound O=C1COC(=O)C1 JCGNDDUYTRNOFT-UHFFFAOYSA-N 0.000 description 1
- PRCNQQRRDGMPKS-UHFFFAOYSA-N pentane-2,4-dione;zinc Chemical compound [Zn].CC(=O)CC(C)=O.CC(=O)CC(C)=O PRCNQQRRDGMPKS-UHFFFAOYSA-N 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 150000004714 phosphonium salts Chemical class 0.000 description 1
- 150000003018 phosphorus compounds Chemical class 0.000 description 1
- IEQIEDJGQAUEQZ-UHFFFAOYSA-N phthalocyanine Chemical compound N1C(N=C2C3=CC=CC=C3C(N=C3C4=CC=CC=C4C(=N4)N3)=N2)=C(C=CC=C2)C2=C1N=C1C2=CC=CC=C2C4=N1 IEQIEDJGQAUEQZ-UHFFFAOYSA-N 0.000 description 1
- 238000009832 plasma treatment Methods 0.000 description 1
- 229920001643 poly(ether ketone) Polymers 0.000 description 1
- 229920003207 poly(ethylene-2,6-naphthalate) Polymers 0.000 description 1
- 229920002492 poly(sulfone) Polymers 0.000 description 1
- 229920002312 polyamide-imide Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 229920006393 polyether sulfone Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 239000011112 polyethylene naphthalate Substances 0.000 description 1
- 229920006290 polyethylene naphthalate film Polymers 0.000 description 1
- 239000009719 polyimide resin Substances 0.000 description 1
- 229920001955 polyphenylene ether Polymers 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 239000012286 potassium permanganate Substances 0.000 description 1
- LLHKCFNBLRBOGN-UHFFFAOYSA-N propylene glycol methyl ether acetate Chemical compound COCC(C)OC(C)=O LLHKCFNBLRBOGN-UHFFFAOYSA-N 0.000 description 1
- KOUKXHPPRFNWPP-UHFFFAOYSA-N pyrazine-2,5-dicarboxylic acid;hydrate Chemical compound O.OC(=O)C1=CN=C(C(O)=O)C=N1 KOUKXHPPRFNWPP-UHFFFAOYSA-N 0.000 description 1
- 230000003014 reinforcing effect Effects 0.000 description 1
- GHMLBKRAJCXXBS-UHFFFAOYSA-N resorcinol Chemical compound OC1=CC=CC(O)=C1 GHMLBKRAJCXXBS-UHFFFAOYSA-N 0.000 description 1
- 229960001755 resorcinol Drugs 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 239000004576 sand Substances 0.000 description 1
- 238000000790 scattering method Methods 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 239000011863 silicon-based powder Substances 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- WSFQLUVWDKCYSW-UHFFFAOYSA-M sodium;2-hydroxy-3-morpholin-4-ylpropane-1-sulfonate Chemical compound [Na+].[O-]S(=O)(=O)CC(O)CN1CCOCC1 WSFQLUVWDKCYSW-UHFFFAOYSA-M 0.000 description 1
- 125000003003 spiro group Chemical group 0.000 description 1
- 238000004544 sputter deposition Methods 0.000 description 1
- TXDNPSYEJHXKMK-UHFFFAOYSA-N sulfanylsilane Chemical compound S[SiH3] TXDNPSYEJHXKMK-UHFFFAOYSA-N 0.000 description 1
- 239000012756 surface treatment agent Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 229920002803 thermoplastic polyurethane Polymers 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 239000010409 thin film Substances 0.000 description 1
- 239000011135 tin Substances 0.000 description 1
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 1
- 125000005270 trialkylamine group Chemical group 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 1
- 239000013638 trimer Substances 0.000 description 1
- ZNOCGWVLWPVKAO-UHFFFAOYSA-N trimethoxy(phenyl)silane Chemical compound CO[Si](OC)(OC)C1=CC=CC=C1 ZNOCGWVLWPVKAO-UHFFFAOYSA-N 0.000 description 1
- BPSIOYPQMFLKFR-UHFFFAOYSA-N trimethoxy-[3-(oxiran-2-ylmethoxy)propyl]silane Chemical compound CO[Si](OC)(OC)CCCOCC1CO1 BPSIOYPQMFLKFR-UHFFFAOYSA-N 0.000 description 1
- 238000007740 vapor deposition Methods 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- XOOUIPVCVHRTMJ-UHFFFAOYSA-L zinc stearate Chemical compound [Zn+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O XOOUIPVCVHRTMJ-UHFFFAOYSA-L 0.000 description 1
- CHJMFFKHPHCQIJ-UHFFFAOYSA-L zinc;octanoate Chemical compound [Zn+2].CCCCCCCC([O-])=O.CCCCCCCC([O-])=O CHJMFFKHPHCQIJ-UHFFFAOYSA-L 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L71/00—Compositions of polyethers obtained by reactions forming an ether link in the main chain; Compositions of derivatives of such polymers
- C08L71/08—Polyethers derived from hydroxy compounds or from their metallic derivatives
- C08L71/10—Polyethers derived from hydroxy compounds or from their metallic derivatives from phenols
- C08L71/12—Polyphenylene oxides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/02—Polycondensates containing more than one epoxy group per molecule
- C08G59/04—Polycondensates containing more than one epoxy group per molecule of polyhydroxy compounds with epihalohydrins or precursors thereof
- C08G59/06—Polycondensates containing more than one epoxy group per molecule of polyhydroxy compounds with epihalohydrins or precursors thereof of polyhydric phenols
- C08G59/08—Polycondensates containing more than one epoxy group per molecule of polyhydroxy compounds with epihalohydrins or precursors thereof of polyhydric phenols from phenol-aldehyde condensates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/40—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the curing agents used
- C08G59/4007—Curing agents not provided for by the groups C08G59/42 - C08G59/66
- C08G59/4014—Nitrogen containing compounds
- C08G59/4028—Isocyanates; Thioisocyanates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/40—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the curing agents used
- C08G59/42—Polycarboxylic acids; Anhydrides, halides or low molecular weight esters thereof
- C08G59/4284—Polycarboxylic acids; Anhydrides, halides or low molecular weight esters thereof together with other curing agents
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/34—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from hydroxy compounds or their metallic derivatives
- C08G65/48—Polymers modified by chemical after-treatment
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/34—Silicon-containing compounds
- C08K3/36—Silica
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L63/00—Compositions of epoxy resins; Compositions of derivatives of epoxy resins
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05K—PRINTED CIRCUITS; CASINGS OR CONSTRUCTIONAL DETAILS OF ELECTRIC APPARATUS; MANUFACTURE OF ASSEMBLAGES OF ELECTRICAL COMPONENTS
- H05K1/00—Printed circuits
- H05K1/02—Details
- H05K1/03—Use of materials for the substrate
- H05K1/0313—Organic insulating material
- H05K1/032—Organic insulating material consisting of one material
- H05K1/0326—Organic insulating material consisting of one material containing O
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05K—PRINTED CIRCUITS; CASINGS OR CONSTRUCTIONAL DETAILS OF ELECTRIC APPARATUS; MANUFACTURE OF ASSEMBLAGES OF ELECTRICAL COMPONENTS
- H05K3/00—Apparatus or processes for manufacturing printed circuits
- H05K3/22—Secondary treatment of printed circuits
- H05K3/28—Applying non-metallic protective coatings
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K2201/00—Specific properties of additives
- C08K2201/002—Physical properties
- C08K2201/005—Additives being defined by their particle size in general
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Microelectronics & Electronic Packaging (AREA)
- General Chemical & Material Sciences (AREA)
- Manufacturing & Machinery (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Epoxy Resins (AREA)
Abstract
【解決手段】(A)アントラセン構造を有するフェノキシ樹脂、(B)エポキシ樹脂、及び(C)硬化剤を含有する硬化性樹脂組成物であって、前記(A)フェノキシ樹脂、(B)エポキシ樹脂、及び(C)硬化剤の合計を100質量%とした場合に、前記(A)フェノキシ樹脂が1〜15質量%であり、かつ前記(A)フェノキシ樹脂のエポキシ当量が5000以上であることを特徴とする硬化性樹脂組成物を提供する。
【選択図】なし
Description
すなわち、本発明は以下の態様を含むものである。
[1]
(A)アントラセン構造を有するフェノキシ樹脂、(B)エポキシ樹脂、及び(C)硬化剤を含有する硬化性樹脂組成物であって、前記(A)フェノキシ樹脂、(B)エポキシ樹脂、及び(C)硬化剤の合計を100質量%とした場合に、前記(A)フェノキシ樹脂が1〜15質量%であり、かつ前記(A)フェノキシ樹脂のエポキシ当量が5000以上であることを特徴とする硬化性樹脂組成物。
[2]
前記(A)フェノキシ樹脂の重量平均分子量が、8000〜100000である、[1]記載の硬化性樹脂組成物。
[3]
前記(A)フェノキシ樹脂が、無置換又は置換のビフェニル構造を更に有する、[1]又は[2]記載の硬化性樹脂組成物。
[4]
前記(B)エポキシ樹脂が、ビスフェノール型エポキシ樹脂、結晶性2官能エポキシ樹脂、ジシクロペンタジエン型エポキシ樹脂、ナフタレン型エポキシ樹脂、ビフェニル型エポキシ樹脂及びこれらのエポキシ樹脂の混合物からなる群から選択される、[1]〜[3]のいずれか1項記載の硬化性樹脂組成物。
[5]
前記(C)硬化剤が、フェノール硬化剤、シアネートエステル硬化剤、活性エステル硬化剤及びこれらの硬化剤の混合物からなる群から選択される、[1]〜[4]のいずれか1項記載の硬化性樹脂組成物。
[6]
前記(C)硬化剤が、シアネートエステル硬化剤又は活性エステル硬化剤である、[5]記載の硬化性樹脂組成物。
[7]
前記硬化性樹脂組成物中の不揮発成分を100質量%とした場合、前記(A)フェノキシ樹脂の含有量が0.3〜10質量%であり、前記(B)エポキシ樹脂の含有量が5〜30質量%であり、前記(C)硬化剤の含有量が3〜20質量%である、[1]〜[6]のいずれか1項記載の硬化性樹脂組成物。
[8]
さらに、(D)無機充填材を含む、[1]〜[7]のいずれか1項記載の硬化性樹脂組成物。
[9]
前記(D)無機充填材の平均粒径が、0.01〜5μmである、[8]記載の硬化性樹脂組成物。
[10]
前記硬化性樹脂組成物中の不揮発成分を100質量%とした場合の前記(D)無機充填材の含有量が30〜90質量%である、[8]又は[9]記載の硬化性樹脂組成物。
[11]
前記(D)無機充填材がシリカである、[8]〜[10]のいずれか1項記載の硬化性樹脂組成物。
[12]
[1]〜[11]のいずれか1項記載の硬化性樹脂組成物を含有することを特徴とする、多層プリント配線板の絶縁層用硬化性樹脂組成物。
[13]
[1]〜[11]のいずれか1項記載の硬化性樹脂組成物を含有することを特徴とする、多層プリント配線板のビルドアップ層用硬化性樹脂組成物。
[14]
[1]〜[13]のいずれか1項記載の硬化性樹脂組成物を含有することを特徴とする、シート状積層材料。
[15]
[1]〜[13]のいずれか1項記載の硬化性樹脂組成物若しくは[14]記載のシート状積層材料を熱硬化して得られた絶縁層を含むことを特徴とする、多層プリント配線板。
[16]
[15]記載の多層プリント配線板を含むことを特徴とする、半導体装置。
[I](A)25000〜40000の重量平均分子量を有し、アントラセン構造及びテトラメチルビフェニル構造を有するフェノキシ樹脂、
(B)ビスフェノール型エポキシ樹脂、結晶性2官能エポキシ樹脂、ジシクロペンタジエン型エポキシ樹脂、ナフタレン型エポキシ樹脂、ビフェニル型エポキシ樹脂及びこれらのエポキシ樹脂の混合物からなる群から選択されるエポキシ樹脂、
(C)シアネートエステル硬化剤及び活性エステル硬化剤から選択される1種以上を含む硬化剤、及び
(D)0.01〜5μmの平均粒径を有するシリカである無機充填材
を含有する硬化性樹脂組成物であって、
前記(A)フェノキシ樹脂、(B)エポキシ樹脂、及び(C)硬化剤の合計を100質量%とした場合に、前記(A)フェノキシ樹脂が1〜15質量%であり、
前記(A)フェノキシ樹脂のエポキシ当量が9000〜15000である、
ことを特徴とする硬化性樹脂組成物。
特に、本発明では、成分(A)の特定のアントラセン構造を有するフェノキシ樹脂を用いることで上記低粗度及び高ピール強度を達成できる。また、通常無機充填材を比較的多く含むエポキシ樹脂組成物は、薄膜での樹脂流れに劣り、積層する導体間にボイドが発生する場合があり、さらにピール強度が低下しやすいが、本発明のように成分(A)を用いることによりピール強度を向上させることが可能である。
本発明の態様の一つである硬化性樹脂組成物は、(A)アントラセン構造を有するフェノキシ樹脂、(B)エポキシ樹脂、及び(C)硬化剤を含有する硬化性樹脂組成物であって、前記(A)フェノキシ樹脂、(B)エポキシ樹脂、及び(C)硬化剤の合計を100質量%とした場合に、前記(A)フェノキシ樹脂が1〜15質量%であり、かつ前記(A)フェノキシ樹脂のエポキシ当量が5000以上であることを特徴とする硬化性樹脂組成物である。以下、本発明の硬化性樹脂組成物について、詳細に説明する。
本発明で使用し得るアントラセン構造を有するフェノキシ樹脂としては、アントラセン構造、例えば、以下に示すアントラセン構造を少なくとも1種以上有するフェノキシ樹脂であれば、あらゆるフェノキシ樹脂を使用することができる。
アントラセン構造:
好ましい態様としては、式(2)中、R2及びR3は、互いに同一であっても異なっていてもよく、水素原子又はC1〜8の炭化水素基、より好ましくは水素原子又はC1〜6のアルキル基、更に好ましくは水素原子又はメチル基、特に好ましくはテトラメチルビフェニル構造であり、mは0〜3、より好ましくは1〜2の整数である。
好ましい態様としては、式(3)中、R4は、互いに同一であっても異なっていてもよく、水素原子又はC1〜8の炭化水素基、より好ましくは水素原子又はC1〜6のアルキル基、更に好ましくは水素原子又はメチル基、特に好ましくは水素原子であり、R5は、水素原子又はC1〜8の炭化水素基、より好ましくは水素原子又はC1〜6のアルキル基、更に好ましくは水素原子又はメチル基、特に好ましくは水素原子であり、R6は、水素原子又はC1〜8の炭化水素基、より好ましくは水素原子又はC1〜6のアルキル基、更に好ましくは水素原子又はメチル基、特に好ましくはメチル基であり、mは0〜3、より好ましくは1〜2の整数である。
また、(A)フェノキシ樹脂と、後述する(B)エポキシ樹脂との合計を100質量%とした場合、(A)フェノキシ樹脂が好ましくは、2〜30質量%であり、より好ましくは、4〜20質量%である。
アントラセン構造を有するフェノキシ樹脂の重量平均分子量は、例えば、8000〜100000、好ましくは15000〜80000、より好ましくは20000〜60000、更に好ましくは25000〜40000である。フェノキシ樹脂の重量平均分子量が、8000以上であれば、優れた破壊伸びを有し、また、100000以下であれば樹脂組成物との相溶性が向上し、低い算術平均粗さ及び低い二乗平均平方根粗さとすることができる。当該フェノキシ樹脂の重量平均分子量は、ゲルパーミエーションクロマトグラフィー(GPC)法で測定することができる。具体的には、熱可塑性樹脂のポリスチレン換算の重量平均分子量は、測定装置として(株)島津製作所製LC−9A/RID−6Aを、カラムとして昭和電工(株)製Shodex K−800P/K−804L/K−804Lを、移動相としてクロロホルム等を用いて、カラム温度40℃にて測定し、標準ポリスチレンの検量線を用いて算出することができる。
(B)成分のエポキシ樹脂は、(A)成分のフェノキシ樹脂とは異なるエポキシ樹脂である。(B)成分のエポキシ樹脂として好ましくは、(A)成分のフェノキシ樹脂とは異なるエポキシ当量を有するものである。(B)成分のエポキシ樹脂のエポキシ当量(1当量のエポキシ基を含む樹脂の質量)は、好ましくは50〜3000(g/当量)、より好ましくは100〜2000(g/当量)、更に好ましくは150〜1000(g/当量)、特に好ましくは200〜500(g/当量)であることが適当である。これにより、硬化性樹脂組成物から得られる絶縁層の架橋密度が十分となり、低粗度化に有利となる。なお、エポキシ当量は、JIS K7236(2001)に従って測定することができる。また、好ましくは、エポキシ樹脂は、1分子中に2個以上のエポキシ基を有するエポキシ樹脂である。
具体的な(B)成分のエポキシ樹脂としては、例えば、ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、ビスフェノールS型エポキシ樹脂、ビスフェノールAF型エポキシ樹脂のようなビスフェノール型エポキシ樹脂、ジシクロペンタジエン型エポキシ樹脂、フェノールノボラック型エポキシ樹脂、tert-ブチル-カテコール型エポキシ樹脂、ナフトール型エポキシ樹脂、ナフタレン型エポキシ樹脂、ナフチレンエーテル型エポキシ樹脂、グリシジルアミン型エポキシ樹脂、グリシジルエステル型エポキシ樹脂、クレゾールノボラック型エポキシ樹脂、ビフェニル型エポキシ樹脂、アントラセン型エポキシ樹脂、線状脂肪族エポキシ樹脂、ブタジエン構造を有するエポキシ樹脂、脂環式エポキシ樹脂、複素環式エポキシ樹脂、スピロ環含有エポキシ樹脂、シクロヘキサンジメタノール型エポキシ樹脂、トリメチロール型エポキシ樹脂、ハロゲン化エポキシ樹脂、結晶性2官能エポキシ樹脂等が挙げられる。より好ましくは、(B)エポキシ樹脂は、ビスフェノール型エポキシ樹脂、結晶性2官能エポキシ樹脂、ジシクロペンタジエン型エポキシ樹脂、ナフタレン型エポキシ樹脂、ビフェニル型エポキシ樹脂及びこれらのエポキシ樹脂の混合物からなる群から選択される。これらは1種又は2種以上組み合わせて使用してもよい。
本発明の硬化性樹脂組成物中の(B)エポキシ樹脂の含有量は、特に限定されるものではないが、絶縁層の低粗度化と高ピール強度とを両立させるという観点から、当該硬化性樹脂組成物中の不揮発成分を100質量%とした場合、5〜30質量%が好ましく、7〜25質量%がより好ましく、10〜20質量%が更に好ましい。
本発明で使用される硬化剤は、上記フェノキシ樹脂及びエポキシ樹脂を架橋して硬化することができるものであれば特に限定されないが、フェノール硬化剤(フェノール樹脂)、シアネートエステル硬化剤(シアネートエステル樹脂)、活性エステル硬化剤(活性エステル樹脂)から選択される1種以上を含むことが好ましい。これらフェノール硬化剤、シアネートエステル硬化剤及び活性エステル硬化剤は、有意に絶縁層の表面粗度を低下することができる。
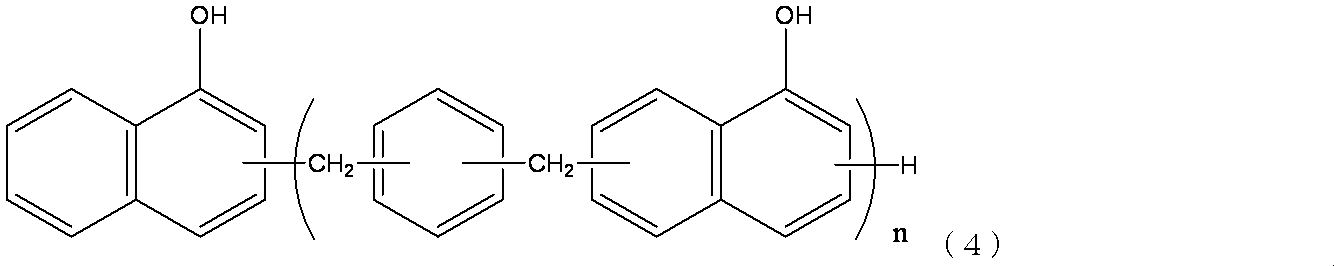
(nは1〜20までの整数。)
殊更好ましい活性エステル樹脂は、以下の式(10)で表されるジシクロペンタジエニルジフェノール構造を有し、末端にX−基及びXO−基(ここでXは置換基を有していてもよいナフチル基である)をそれぞれ有し、重量平均分子量が約2700の活性エステル樹脂であるHPC−8000−65Tである。
本発明で使用し得る無機充填材としては、例えば、シリカ、アルミナ、雲母、マイカ、珪酸塩、硫酸バリウム、水酸化マグネシウム、酸化チタン等が挙げられ、シリカ、アルミナが好ましく、特に無定形シリカ、溶融シリカ、結晶シリカ、合成シリカ、中空シリカ、球状シリカ等のシリカが好ましく、球状シリカ、溶融シリカがより好ましい。硬化性樹脂組成物を含む本発明のシート状積層材料に対する無機充填材の充填性向上の観点から、球状溶融シリカが更に好ましい。1種又は2種以上の無機充填材を使用することができる。市販されている球状溶融シリカとして、(株)アドマテックス製「SOC2」、「SOC1」が挙げられる。
本発明の硬化性樹脂組成物には、上述した成分の他、その他の成分として、硬化促進剤;熱可塑性樹脂;ビニルベンジル化合物、アクリル化合物、マレイミド化合物、ブロックイソシアネート化合物のような熱硬化性樹脂;リン系化合物、水酸化金属物等の難燃剤;シリコンパウダー、ナイロンパウダー、フッ素パウダー、ゴム粒子等の有機充填剤;有機溶媒;オルベン、ベントン等の増粘剤;シリコーン系、フッ素系、高分子系の消泡剤;イミダゾール系、チアゾール系、トリアゾール系、シランカップリング剤等の密着性付与剤;フタロシアニン・ブルー、フタロシアニン・グリーン、アイオジン・グリーン、ジスアゾイエロー、カーボンブラック等の着色剤;添加剤などを適宜配合することができる。
本発明の硬化性樹脂組成物は、上記成分を適宜混合し、また、必要に応じて三本ロール、ボールミル、ビーズミル、サンドミル等の混練手段、あるいは高速回転ミキサー、スーパーミキサー、プラネタリーミキサー等の撹拌手段により混練または混合することにより調製することができる。また、さらに上述した有機溶剤を加えることで樹脂ワニスとしても調製することができる。
本発明の硬化性樹脂組成物は、多層プリント配線板の絶縁層用硬化性樹脂組成物として用いることができる。本発明で使用され得る多層プリント配線板は、本発明の硬化性樹脂組成物やシート状積層材料を熱硬化して得られた絶縁層を含む、多層プリント配線板である。
ここで、熱硬化の条件は、硬化性樹脂組成物中のエポキシ樹脂の種類、含有量などに応じて適宜選択すればよいが、例えば硬化温度は90〜220℃、好ましくは160℃〜210℃であり、硬化時間は10分〜180分、好ましくは20〜120分として加熱されることによって行う。また、2段階に分けて熱硬化を行っても良い。
ここで絶縁層の線熱膨張係数(CTE)(JIS K7197)は、25〜150℃の平均の線熱膨張係数で測定して、20ppm/℃以下となるのが好ましく、19ppm/℃以下となるのがより好ましい。下限値に特に制限はないが、一般的に4ppm/℃となる。これにより、絶縁層(ビルドアップ層)と導体層(配線)とのひずみを防止し、信頼性の高い多層プリント配線板を得ることができる。
膨潤液による膨潤処理は、絶縁層を50〜80℃で5〜20分間(好ましくは55〜70℃で8〜15分間)、膨潤液に浸漬させることで行われる。膨潤液としては、例えばアルカリ溶液、界面活性剤溶液等が挙げられ、好ましくはアルカリ溶液である。該アルカリ溶液としては、例えば、水酸化ナトリウム溶液、水酸化カリウム溶液等が挙げられる。市販されている膨潤液としては、例えば、アトテックジャパン(株)製のスウェリング・ディップ・セキュリガントP(Swelling Dip Securiganth P)、スウェリング・ディップ・セキュリガントSBU(Swelling Dip Securiganth SBU)等を挙げることができる。
酸化剤による粗化処理は、絶縁層を60〜80℃で10〜30分間(好ましくは70〜80℃で15〜25分間)、酸化剤溶液に浸漬させることで行われる。酸化剤としては、例えば、水酸化ナトリウムの水溶液に過マンガン酸カリウムや過マンガン酸ナトリウムを溶解したアルカリ性過マンガン酸溶液、重クロム酸塩、オゾン、過酸化水素/硫酸、硝酸等を挙げることができる。また、アルカリ性過マンガン酸溶液における過マンガン酸塩の濃度は5〜10質量%とするのが好ましい。市販されている酸化剤としては、例えば、アトテックジャパン(株)製のコンセントレート・コンパクト CP、ドージングソリューション セキュリガントP等のアルカリ性過マンガン酸溶液が挙げられる。
中和液による中和処理は、30〜50℃で3〜10分間(好ましくは35〜45℃で3〜8分間)、中和液に浸漬させることで行われる。中和液としては、酸性の水溶液が好ましく、市販品としては、アトテックジャパン(株)製のリダクションソリューシン・セキュリガントPが挙げられる。
粗化処理後、絶縁層を50〜120℃で10〜60分間(好ましくは60〜100℃で20〜40分間)乾燥してもよい。
本発明で用いられるシート状積層材料は、上記硬化性樹脂組成物を層形成した、硬化前のシート状材料である。当該シート状積層材料は、当業者に公知の方法、例えば、上述した有機溶剤に樹脂組成物を溶解した樹脂ワニスを調製し、この樹脂ワニスを、ダイコーターなどを用いて、支持体に塗布し、更に加熱、あるいは熱風吹きつけ等により有機溶剤を乾燥させて支持体上に樹脂組成物層(シート状積層材料)を形成させることにより支持体付きシート状積層材料として製造することができる。また、樹脂ワニスをガラスクロス等のシート状補強基材にホットメルト法又はソルベント法により含浸、乾燥させることで、シート状積層材料をプリプレグとすることもできる。なお、支持体付きシート状積層材料を接着フィルムという場合もある。
得られたシート状積層材料の厚さは特に限定されないが、例えば1〜150μmの範囲が好ましく、2〜100μmの範囲がより好ましく、3〜50μmの範囲がさらに好ましく、5〜30μmの範囲が特に好ましい。
本発明で使用し得る支持体としては、プラスチックフィルムや金属箔が挙げられる。具体的に、プラスチックフィルムとしては、ポリエチレンテレフタレート(以下「PET 」と略称することがある。)、ポリエチレンナフタレート等のポリエステル、ポリカーボネート、ポリエチレン、ポリプロピレン、アクリル、環状ポリオレフィン、トリアセチルセルロース、ポリエーテルサルファイド、ポリエーテルケトン、ポリイミドなどが挙げられる。中でも、ポリエチレンテレフタレートフィルム、ポリエチレンナフタレートフィルムが好ましく、特に安価で入手容易なポリエチレンテレフタレートフィルムが好ましい。
金属箔としては、銅箔、アルミニウム箔などが挙げられる。
支持体の厚さは特に限定されないが、10〜150μmの範囲が好ましく、20〜50μmの範囲がより好ましく、25〜45μmの範囲がさらに好ましい。
次に、上記のようにして製造したシート状積層材料を用いて多層プリント配線板を製造する方法の一例を説明する。
まず、シート状積層材料を、真空ラミネーターを用いて回路基板の片面又は両面にラミネート(積層)する。回路基板に用いられる基板としては、例えば、ガラスエポキシ基板、金属基板、ポリエステル基板、ポリイミド基板、BTレジン基板、熱硬化型ポリフェニレンエーテル基板等が挙げられる。なお、ここで回路基板とは、上記のような基板の片面又は両面にパターン加工された導体層(回路)が形成されたものをいう。また導体層と絶縁層とを交互に積層してなる多層プリント配線板において、該多層プリント配線板の最外層の片面又は両面がパターン加工された導体層(回路)となっているものも、ここでいう回路基板に含まれる。なお導体層表面には、黒化処理、銅エッチング等により予め粗化処理が施されていてもよい。
その後、室温(25℃)付近に冷却してから、支持体を剥離する場合は剥離し、樹脂組成物を熱硬化して硬化物を形成することで、回路基板上に絶縁層を形成することができる。熱硬化の条件は、樹脂組成物中の樹脂成分の種類、含有量などに応じて適宜選択すればよいが、たとえば硬化温度は100〜220℃、好ましくは160℃〜210℃であり、硬化時間は20分〜180分、好ましくは30〜120分として加熱されることによって行う。また、2段階に分けて熱硬化を行っても良い。絶縁層を形成した後、硬化前に支持体を剥離しなかった場合は、必要によりここで剥離することもできる。
上述のようにして製造された多層プリント配線板を用いることで半導体装置を製造することができる。本発明で使用され得る多層プリント配線板の導通箇所に、半導体チップを実装することにより半導体装置を製造することができる。「導通箇所」とは、「多層プリント配線板における電気信号を伝える箇所」であって、その場所は表面であっても、埋め込まれた箇所であってもいずれでも構わない。また、導通するのであれば、導体層の一部であってもそれ以外のコネクタ等の導電部分であってもよい。「半導体チップ」とは、半導体を材料とする電気回路素子であれば特に限定されない。
まずは各種測定方法・評価方法について説明する。
(1)積層板の下地処理
ガラス布基材エポキシ樹脂両面銅張積層板(銅箔の厚さ18μm、基板厚み0.3mm、松下電工(株)製R5715ES)の両面をメック(株)製CZ8100にて1μmエッチングして銅表面の粗化処理をおこなった。
(2)接着フィルムのラミネート
実施例及び比較例で作成した接着フィルムを、バッチ式真空加圧ラミネーターMVLP-500(名機製作所製)を用いて、上記粗化処理したエポキシ樹脂両面銅張積層板の両面にラミネートした。ラミネートは、30秒間減圧して気圧を13hPa以下とし、その後30秒間、100℃、圧力0.74MPaで圧着することにより行った。
ラミネートされた接着フィルムから支持体であるポリエチレンテレフタレート(PET)フィルムを剥離した後、100℃、30分続けて180℃、30分の硬化条件で樹脂組成物を硬化して絶縁層を形成した。
絶縁層を形成した積層板を、膨潤液である、アトテックジャパン(株)のジエチレングリコールモノブチルエーテル含有のスウェリング・ディップ・セキュリガントP(グリコールエーテル類、水酸化ナトリウムの水溶液)に、60℃で5分間(実施例1、比較例14、5)又は10分間(実施例2、3、比較例2、3)浸漬した。次に粗化液として、アトテックジャパン(株)のコンセントレート・コンパクトP(KMnO4:60g/L、NaOH:40g/Lの水溶液)に、80℃で15分間(実施例1、比較例1、4、5)、20分間(実施例2、3、比較例2、3)浸漬した。最後に中和液として、アトテックジャパン(株)のリダクションショリューシン・セキュリガントP(硫酸の水溶液)に40℃で5分間浸漬した。80℃で30分乾燥後、この基板を評価基板Aとした。
評価基板Aをめっきして導体層を形成した。具体的には、評価基板Aを、PdCl2を含む無電解めっき用溶液に40℃で5分間浸漬し、次に無電解銅めっき液に25℃で20分間浸漬した。150℃にて30分間加熱してアニール処理を行った後に、エッチングレジストを形成し、エッチングによるパターン形成の後に、硫酸銅電解めっきを行い、30μmの厚さで導体層を形成した。次に、アニール処理を190℃にて60分間行った。この基板を評価基板Bとした。
評価基板Aを、非接触型表面粗さ計(ビーコインスツルメンツ社製WYKO NT3300)を用いて、VSIコンタクトモード、50倍レンズにより測定範囲を121μm×92μmとして得られる数値によりRa値、Rq値を求めた。それぞれ10点の平均値を求めることにより測定した。
評価基板Bの導体層に、幅10mm、長さ100mmの部分の切込みをいれ、この一端を剥がしてつかみ具(株式会社ティー・エス・イー、オートコム型試験機 AC−50C−SL)で掴み、室温(25℃)中にて、50mm/分の速度で垂直方向に35mmを引き剥がした時の荷重(kgf/cm(N/cm))を測定した。
実施例及び比較例において得られた接着フィルムを200℃で90分間加熱することで熱硬化させ、支持体であるPETフィルムから剥離することによりシート状の硬化物を得た。その硬化物を、幅5mm、長さ15mm、厚さ30mmの試験片に切断し、熱機械分析装置Thermo Plus TMA8310((株)リガク製)を使用して、引張加重法で熱機械分析を行った。試験片を前記装置に装着後、荷重1g、昇温速度5℃/分の測定条件にて連続して2回測定した。2回目の測定における25℃から150℃までの平均線熱膨張係数(ppm)を算出した。
実施例及び比較例において得られた接着フィルムを200℃で90分間加熱することで熱硬化させ、この硬化物をダンベル状に切り出し、PETフィルムを剥がして、試験片を得た。その試験片を、JIS K7127に準拠し、オリエンテック社製引張試験機RTC−1250Aを用いて引張強度測定を行い、23℃における破壊伸びを求めた。
アントラセン構造及びテトラメチルビフェニル構造を有するフェノキシ樹脂の合成
反応容器に、テトラメチルビフェニル型エポキシ樹脂(三菱化学(株)製YX4000、エポキシ当量185)191g、9,10−ジヒドロキシアントラセン(フェノール性水酸基当量210)210g、およびシクロヘキサノン150gを入れ、攪拌して溶解させた。次いで、テトラメチルアンモニウムクロライド溶液0.5gを滴下し、窒素雰囲気下、180℃5時間にて反応させた。反応終了後、濾布を用いて濾過して、溶剤により希釈することでフェノキシ樹脂Aを得た。
・エポキシ当量:11000
・重量平均分子量:35000
・固形分30質量%のMEKとシクロヘキサノンの1:1溶液
なお、フェノキシ樹脂Aは、以下の構造を有していた。
テトラメチルビフェニル構造及びビスフェノールアセトフェノン構造を有するフェノキシ樹脂の合成
反応容器に、テトラメチルビフェニル型エポキシ樹脂(三菱化学(株)製「YX4000」、エポキシ当量185)100g、ビスフェノールアセトフェノン(フェノール性水酸基当量145)80g、およびシクロヘキサノン150gを入れ攪拌して溶解させた。次いで、テトラメチルアンモニウムクロライド溶液0.5gを滴下し、窒素雰囲気下、180℃5時間にて反応させた。反応終了後、濾布を用いて濾過して、溶剤により希釈することでフェノキシ樹脂Bを得た。なお、フェノキシ樹脂Bは、アントラセン構造を有していないため、本発明の参考例である。
・エポキシ当量:13000
・重量平均分子量:38000
・固形分30質量%のMEKとシクロヘキサノンの1:1溶液とした。
なお、フェノキシ樹脂Bは、以下の構造を有していた。
ビスフェノール型エポキシ樹脂(新日鐵化学(株)製「ZX1059」、ビスフェノールA型とビスフェノールF型の1:1混合品、エポキシ当量169)10部、結晶性2官能エポキシ樹脂(三菱化学(株)製「YX4000HK」、エポキシ当量約185)10部、ジシクロペンタジエン型エポキシ樹脂(DIC(株)製「HP−7200H」、エポキシ当量275)20部を、ソルベントナフサ35部に撹拌しながら加熱溶解させた。室温(25℃)にまで冷却後、そこへ、フェノキシ樹脂Aを12部、トリアジン骨格含有フェノール系硬化剤(DIC(株)製「LA−7054」水酸基当量125の固形分60%のMEK溶液)12部、ナフタレン型硬化剤(新日鐵化学(株)製「SN−485」水酸基当量215の固形分60%のMEK溶液)15部、硬化促進剤(4−ジメチルアミノピリジン(DMAP)、固形分2質量%のMEK溶液)3部、難燃剤(三光(株)製「HCA−HQ」、10-(2,5-ジヒドロキシフェニル)-10-ヒドロ-9-オキサ-10-フォスファフェナンスレン-10-オキサイド、平均粒径2μm)2部、フェニルアミノシラン系カップリング剤(信越化学工業(株)製、「KBM573」)で表面処理された球形シリカ(平均粒径0.24μm、(株)アドマテックス製「SOC1」、単位面積当たりのカーボン量0.36mg/m2)150部、を混合し、高速回転ミキサーで均一に分散して、樹脂ワニスを作製した。次いで、離型処理付きポリエチレンテレフタレートフィルム(リンテック(株)製「AL5」、厚さ38μm)の離型面上に、乾燥後の樹脂組成物層の厚みが30μmとなるように樹脂ワニスを均一に塗布し、80〜120℃(平均100℃)で4分間乾燥させて、接着フィルムを作製した。
液状ナフタレン型エポキシ樹脂(エポキシ当量144、DIC(株)製「HP4032SS」)5部、結晶性2官能エポキシ樹脂(三菱化学(株)製「YX4000HK」、エポキシ当量約185)5部、ビフェニル型エポキシ樹脂(日本化薬(株)製「NC3000L」、エポキシ当量269)12部を、ソルベントナフサ30部に撹拌しながら加熱溶解させた。室温(25℃)にまで冷却後、そこへ、フェノキシ樹脂Aを5部、ビスフェノールAジシアネートのプレポリマー(ロンザジャパン(株)製「BA230S75」、シアネート当量約232、不揮発分75質量%のMEK溶液)20部、フェノールノボラック型多官能シアネートエステル樹脂(ロンザジャパン(株)製「PT30S」、シアネート当量約133、不揮発分85質量%のMEK溶液)6部、硬化促進剤(4−ジメチルアミノピリジン、固形分2質量%のMEK溶液)1部、硬化促進剤(東京化成(株)製、コバルト(III)アセチルアセトナート(Co(III)Ac))、固形分1質量%のMEK溶液)3部、ゴム粒子(ガンツ化成(株)製、スタフィロイドAC3816N)2部、難燃剤(三光(株)製「HCA−HQ」、10-(2,5-ジヒドロキシフェニル)-10-ヒドロ-9-オキサ-10-フォスファフェナンスレン-10-オキサイド、平均粒径2μm)2部、フェニルアミノシラン系カップリング剤(信越化学工業(株)製、「KBM573」)で表面処理された球形シリカ(平均粒径0.5μm、(株)アドマテックス製「SOC2」、単位面積当たりのカーボン量0.39mg/m2)100部を混合し、高速回転ミキサーで均一に分散して、樹脂ワニスを作製した。次いで、実施例1と同様にして、接着フィルムを作製した。
ビスフェノール型エポキシ樹脂(新日鐵化学(株)製「ZX1059」、ビスフェノールA型とビスフェノールF型の1:1混合品、エポキシ当量169)10部、ビフェニル型エポキシ樹脂(日本化薬(株)製「NC3000L」、エポキシ当量269)12部を、ソルベントナフサ30部に撹拌しながら加熱溶解させた。室温にまで冷却後、そこへ、フェノキシ樹脂Aを17部、活性エステル化合物(DIC(株)製「HPC8000−65T」、重量平均分子量が約2700、活性基当量約223の不揮発分65質量%のトルエン溶液)34部、硬化促進剤(4−ジメチルアミノピリジン、固形分2質量%のMEK溶液)6部、難燃剤(三光(株)製「HCA−HQ」、10-(2,5-ジヒドロキシフェニル)-10-ヒドロ-9-オキサ-10-フォスファフェナンスレン-10-オキサイド、平均粒径2μm)2部、フェニルアミノシラン系カップリング剤(信越化学工業(株)製、「KBM573」)で表面処理された球形シリカ(平均粒径0.5μm、(株)アドマテックス製「SOC2」、単位面積当たりのカーボン量0.39mg/m2)150部を混合し、高速回転ミキサーで均一に分散して、樹脂ワニスを作製した。次いで、実施例1と同様にして、接着フィルムを作製した。
実施例1のフェノキシ樹脂A12部を、ビスフェノールA型フェノキシ樹脂(三菱化学(株)製「E1256B40」、固形分40質量%のMEK溶液、エポキシ当量8000、重量平均分子量約50000)10部に変更する以外は、実施例1と全く同様にして接着フィルムを作製した。
<比較例2>
実施例2のフェノキシ樹脂A5部を、合成例2のフェノキシ樹脂B5部に変更する以外は、実施例2と全く同様にして接着フィルムを作製した。
実施例3のフェノキシ樹脂A17部を、合成例2のフェノキシ樹脂B17部に変更する以外は、実施例3と全く同様にして接着フィルムを作製した。
結果を表1及び表2に示す。
実施例1のフェノキシ樹脂A12部を、アントラセン型エポキシ樹脂(三菱化学(株)製「YX8800」、エポキシ当量181)3.6部に変更する以外は、実施例1と全く同様にして接着フィルムを作製した。なお、「YX8800」の構造式は以下の通りである。
実施例1のフェノキシ樹脂A12部を43部に変更する以外は、実施例1と全く同様にして接着フィルムを作製した。なお、比較例5では、(A)フェノキシ樹脂、(B)エポキシ樹脂、及び(C)硬化剤の合計を100質量%とした場合に、前記(A)フェノキシ樹脂が18.8質量%であるので、本発明の範囲外である。
結果を表1及び表2に示す。
Claims (16)
- (A)アントラセン構造を有するフェノキシ樹脂、(B)エポキシ樹脂、及び(C)硬化剤を含有する硬化性樹脂組成物であって、前記(A)フェノキシ樹脂、(B)エポキシ樹脂、及び(C)硬化剤の合計を100質量%とした場合に、前記(A)フェノキシ樹脂が1〜15質量%であり、かつ前記(A)フェノキシ樹脂のエポキシ当量が5000以上であることを特徴とする硬化性樹脂組成物。
- 前記(A)フェノキシ樹脂の重量平均分子量が、8000〜100000である、請求項1記載の硬化性樹脂組成物。
- 前記(A)フェノキシ樹脂が、無置換又は置換のビフェニル構造を更に有する、請求項1又は2記載の硬化性樹脂組成物。
- 前記(B)エポキシ樹脂が、ビスフェノール型エポキシ樹脂、結晶性2官能エポキシ樹脂、ジシクロペンタジエン型エポキシ樹脂、ナフタレン型エポキシ樹脂、ビフェニル型エポキシ樹脂及びこれらのエポキシ樹脂の混合物からなる群から選択される、請求項1〜3のいずれか1項記載の硬化性樹脂組成物。
- 前記(C)硬化剤が、フェノール硬化剤、シアネートエステル硬化剤、活性エステル硬化剤及びこれらの硬化剤の混合物からなる群から選択される、請求項1〜4のいずれか1項記載の硬化性樹脂組成物。
- 前記(C)硬化剤が、シアネートエステル硬化剤又は活性エステル硬化剤である、請求項5記載の硬化性樹脂組成物。
- 前記硬化性樹脂組成物中の不揮発成分を100質量%とした場合、前記(A)フェノキシ樹脂の含有量が0.3〜10質量%であり、前記(B)エポキシ樹脂の含有量が5〜30質量%であり、前記(C)硬化剤の含有量が3〜20質量%である、請求項1〜6のいずれか1項記載の硬化性樹脂組成物。
- さらに、(D)無機充填材を含む、請求項1〜7のいずれか1項記載の硬化性樹脂組成物。
- 前記(D)無機充填材の平均粒径が、0.01〜5μmである、請求項8記載の硬化性樹脂組成物。
- 前記硬化性樹脂組成物中の不揮発成分を100質量%とした場合の前記(D)無機充填材の含有量が30〜90質量%である、請求項8又は9記載の硬化性樹脂組成物。
- 前記(D)無機充填材がシリカである、請求項8〜10のいずれか1項記載の硬化性樹脂組成物。
- 請求項1〜11のいずれか1項記載の硬化性樹脂組成物を含有することを特徴とする、多層プリント配線板の絶縁層用硬化性樹脂組成物。
- 請求項1〜11のいずれか1項記載の硬化性樹脂組成物を含有することを特徴とする、多層プリント配線板のビルドアップ層用硬化性樹脂組成物。
- 請求項1〜13のいずれか1項記載の硬化性樹脂組成物を含有することを特徴とする、シート状積層材料。
- 請求項1〜13のいずれか1項記載の硬化性樹脂組成物若しくは請求項14記載のシート状積層材料を熱硬化して得られた絶縁層を含むことを特徴とする、多層プリント配線板。
- 請求項15記載の多層プリント配線板を含むことを特徴とする、半導体装置。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013080551A JP6308344B2 (ja) | 2013-04-08 | 2013-04-08 | 硬化性樹脂組成物 |
| TW103109641A TWI624508B (zh) | 2013-04-08 | 2014-03-14 | Curable resin composition |
| KR1020140039581A KR102129715B1 (ko) | 2013-04-08 | 2014-04-02 | 경화성 수지 조성물 |
| CN201810649524.1A CN108976709B (zh) | 2013-04-08 | 2014-04-04 | 固化性树脂组合物 |
| CN201410134634.6A CN104098871B (zh) | 2013-04-08 | 2014-04-04 | 固化性树脂组合物 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013080551A JP6308344B2 (ja) | 2013-04-08 | 2013-04-08 | 硬化性樹脂組成物 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018049034A Division JP6579500B2 (ja) | 2018-03-16 | 2018-03-16 | 硬化性樹脂組成物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2014201698A true JP2014201698A (ja) | 2014-10-27 |
| JP6308344B2 JP6308344B2 (ja) | 2018-04-11 |
Family
ID=51667440
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2013080551A Active JP6308344B2 (ja) | 2013-04-08 | 2013-04-08 | 硬化性樹脂組成物 |
Country Status (4)
| Country | Link |
|---|---|
| JP (1) | JP6308344B2 (ja) |
| KR (1) | KR102129715B1 (ja) |
| CN (2) | CN104098871B (ja) |
| TW (1) | TWI624508B (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015151483A (ja) * | 2014-02-17 | 2015-08-24 | 三菱瓦斯化学株式会社 | レジンシート、金属箔張積層板及びプリント配線板 |
| JP2016088050A (ja) * | 2014-11-11 | 2016-05-23 | 住友ベークライト株式会社 | 樹脂層付き金属膜 |
| JP2020041049A (ja) * | 2018-09-10 | 2020-03-19 | 日立化成株式会社 | エポキシ樹脂、エポキシ樹脂組成物、エポキシ樹脂硬化物及び複合材料 |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102334000B1 (ko) * | 2015-04-01 | 2021-12-01 | 에스케이이노베이션 주식회사 | 접착제 조성물 및 이로부터 제조된 커버레이 필름 |
| JP6620457B2 (ja) * | 2015-08-11 | 2019-12-18 | 味の素株式会社 | 樹脂組成物 |
| JP6852332B2 (ja) * | 2015-10-28 | 2021-03-31 | 味の素株式会社 | 接着フィルム |
| JPWO2017183722A1 (ja) * | 2016-04-22 | 2019-02-28 | 日立化成株式会社 | 多層プリント配線板用の接着フィルム |
| JP7203013B2 (ja) * | 2017-03-31 | 2023-01-12 | 太陽ホールディングス株式会社 | 硬化性樹脂組成物、ドライフィルム、硬化物および電子部品 |
| TWI620763B (zh) * | 2017-04-27 | 2018-04-11 | Taiwan Union Technology Corporation | 樹脂組成物、及使用該樹脂組成物所製得之預浸漬片、金屬箔積層板及印刷電路板 |
| TWI808973B (zh) * | 2017-06-28 | 2023-07-21 | 日商迪愛生股份有限公司 | 活性酯化合物及硬化性組成物 |
| US20200118716A1 (en) * | 2018-10-12 | 2020-04-16 | Te Connectivity Corporation | Electrical cable |
| CN109749440B (zh) * | 2018-12-29 | 2021-08-27 | 广东生益科技股份有限公司 | 氰酸酯树脂组合物及其用途 |
| JP7135970B2 (ja) * | 2019-03-27 | 2022-09-13 | 味の素株式会社 | 樹脂組成物 |
| CN111849122B (zh) * | 2019-04-25 | 2022-06-14 | 常熟生益科技有限公司 | 一种树脂组合物及其应用 |
| US20220260910A1 (en) * | 2019-05-27 | 2022-08-18 | Mitsubishi Gas Chemical Company, Inc. | Underlayer film forming composition for lithography, underlayer film for lithography, and pattern formation method and purification method |
| CN113088039A (zh) * | 2021-05-26 | 2021-07-09 | 深圳市纽菲斯新材料科技有限公司 | 一种绝缘胶膜及其制备方法和应用 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005255813A (ja) * | 2004-03-11 | 2005-09-22 | Japan Epoxy Resin Kk | エポキシ樹脂組成物及びその硬化体 |
| WO2007129662A1 (ja) * | 2006-05-08 | 2007-11-15 | Sekisui Chemical Co., Ltd. | 絶縁材料、電子部品装置の製造方法及び電子部品装置 |
| JP2011068779A (ja) * | 2009-09-25 | 2011-04-07 | Panasonic Electric Works Co Ltd | 液状熱硬化性樹脂組成物、プリプレグ、金属箔張積層板、及びプリント配線板 |
| JP2011089038A (ja) * | 2009-10-22 | 2011-05-06 | Ajinomoto Co Inc | 樹脂組成物 |
| JP2011174082A (ja) * | 2008-07-31 | 2011-09-08 | Sekisui Chem Co Ltd | エポキシ樹脂組成物、プリプレグ、硬化体、シート状成形体、積層板及び多層積層板 |
| WO2012029690A1 (ja) * | 2010-08-31 | 2012-03-08 | 三菱瓦斯化学株式会社 | 樹脂組成物、プリプレグ、および積層板 |
| JP2012064952A (ja) * | 2011-10-11 | 2012-03-29 | Sumitomo Bakelite Co Ltd | 多層プリント配線板用絶縁樹脂組成物、基材付き絶縁シート、多層プリント配線板及び半導体装置 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3668463B2 (ja) | 2002-03-06 | 2005-07-06 | ジャパンエポキシレジン株式会社 | 高分子量エポキシ樹脂とその製造方法、該エポキシ樹脂を用いた電気積層板用樹脂組成物及び電気積層板 |
| US20090166060A1 (en) * | 2006-03-20 | 2009-07-02 | Iji Onozuka | Insulating Resin Layer, Insulating Resin Layer With Carrier And Multiple-Layered Printed Wiring Board |
| US20080036097A1 (en) * | 2006-08-10 | 2008-02-14 | Teppei Ito | Semiconductor package, method of production thereof and encapsulation resin |
| KR101174971B1 (ko) * | 2007-09-05 | 2012-08-17 | 세키스이가가쿠 고교가부시키가이샤 | 절연 시트 및 적층 구조체 |
| TWI621638B (zh) * | 2008-11-28 | 2018-04-21 | 味之素股份有限公司 | Resin composition |
| TWI494364B (zh) * | 2009-01-30 | 2015-08-01 | Ajinomoto Kk | Resin composition |
-
2013
- 2013-04-08 JP JP2013080551A patent/JP6308344B2/ja active Active
-
2014
- 2014-03-14 TW TW103109641A patent/TWI624508B/zh active
- 2014-04-02 KR KR1020140039581A patent/KR102129715B1/ko active IP Right Grant
- 2014-04-04 CN CN201410134634.6A patent/CN104098871B/zh active Active
- 2014-04-04 CN CN201810649524.1A patent/CN108976709B/zh active Active
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005255813A (ja) * | 2004-03-11 | 2005-09-22 | Japan Epoxy Resin Kk | エポキシ樹脂組成物及びその硬化体 |
| WO2007129662A1 (ja) * | 2006-05-08 | 2007-11-15 | Sekisui Chemical Co., Ltd. | 絶縁材料、電子部品装置の製造方法及び電子部品装置 |
| JP2011174082A (ja) * | 2008-07-31 | 2011-09-08 | Sekisui Chem Co Ltd | エポキシ樹脂組成物、プリプレグ、硬化体、シート状成形体、積層板及び多層積層板 |
| JP2011068779A (ja) * | 2009-09-25 | 2011-04-07 | Panasonic Electric Works Co Ltd | 液状熱硬化性樹脂組成物、プリプレグ、金属箔張積層板、及びプリント配線板 |
| JP2011089038A (ja) * | 2009-10-22 | 2011-05-06 | Ajinomoto Co Inc | 樹脂組成物 |
| WO2012029690A1 (ja) * | 2010-08-31 | 2012-03-08 | 三菱瓦斯化学株式会社 | 樹脂組成物、プリプレグ、および積層板 |
| JP2012064952A (ja) * | 2011-10-11 | 2012-03-29 | Sumitomo Bakelite Co Ltd | 多層プリント配線板用絶縁樹脂組成物、基材付き絶縁シート、多層プリント配線板及び半導体装置 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015151483A (ja) * | 2014-02-17 | 2015-08-24 | 三菱瓦斯化学株式会社 | レジンシート、金属箔張積層板及びプリント配線板 |
| JP2016088050A (ja) * | 2014-11-11 | 2016-05-23 | 住友ベークライト株式会社 | 樹脂層付き金属膜 |
| JP2020041049A (ja) * | 2018-09-10 | 2020-03-19 | 日立化成株式会社 | エポキシ樹脂、エポキシ樹脂組成物、エポキシ樹脂硬化物及び複合材料 |
| JP7243093B2 (ja) | 2018-09-10 | 2023-03-22 | 株式会社レゾナック | エポキシ樹脂、エポキシ樹脂組成物、エポキシ樹脂硬化物及び複合材料 |
Also Published As
| Publication number | Publication date |
|---|---|
| TWI624508B (zh) | 2018-05-21 |
| CN108976709B (zh) | 2021-07-06 |
| CN104098871B (zh) | 2018-07-10 |
| JP6308344B2 (ja) | 2018-04-11 |
| KR20140121783A (ko) | 2014-10-16 |
| CN108976709A (zh) | 2018-12-11 |
| TW201500452A (zh) | 2015-01-01 |
| KR102129715B1 (ko) | 2020-07-03 |
| CN104098871A (zh) | 2014-10-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6308344B2 (ja) | 硬化性樹脂組成物 | |
| JP6183583B2 (ja) | 硬化性樹脂組成物 | |
| JP6163803B2 (ja) | 樹脂組成物 | |
| JP6582366B2 (ja) | 樹脂組成物、接着フィルム、硬化物、多層プリント配線板、半導体装置及び絶縁層用樹脂組成物 | |
| JP6098988B2 (ja) | 支持体含有プレポリマーシート | |
| JP6164113B2 (ja) | 支持体付き樹脂シート | |
| JP6728760B2 (ja) | 支持体付き樹脂シート | |
| JP6428147B2 (ja) | 樹脂組成物 | |
| JP6428153B2 (ja) | 樹脂組成物 | |
| JP2011089038A (ja) | 樹脂組成物 | |
| JP2014028880A (ja) | 樹脂組成物 | |
| JP6232762B2 (ja) | 樹脂組成物 | |
| JP2014152306A (ja) | 樹脂組成物 | |
| JP6418273B2 (ja) | 樹脂組成物 | |
| JP2014012763A (ja) | 樹脂組成物 | |
| JP6007663B2 (ja) | 樹脂組成物 | |
| KR20220091448A (ko) | 수지 조성물 | |
| JP2017048400A (ja) | 樹脂組成物 | |
| JP6452080B2 (ja) | 硬化性樹脂組成物 | |
| JP6217895B2 (ja) | 硬化性樹脂組成物 | |
| JP6217832B2 (ja) | 樹脂組成物 | |
| JP6135846B2 (ja) | 硬化性樹脂組成物 | |
| JP6579500B2 (ja) | 硬化性樹脂組成物 | |
| JP6337917B2 (ja) | 樹脂組成物 | |
| JP2019011481A (ja) | 樹脂組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20160317 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20170112 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20170118 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20170321 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20170718 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20170914 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20171115 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20180214 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20180227 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6308344 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |