ES2951829T3 - Formas sólidas de la sal de tartrato de 3-((1R,3R)-1-(2,6-difluoro-4-((1-(3-fluoropropil)acetidin-3-il)amino)fenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol, proceso para su preparación y procedimientos de su uso en el tratamiento de cánceres - Google Patents
Formas sólidas de la sal de tartrato de 3-((1R,3R)-1-(2,6-difluoro-4-((1-(3-fluoropropil)acetidin-3-il)amino)fenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol, proceso para su preparación y procedimientos de su uso en el tratamiento de cánceres Download PDFInfo
- Publication number
- ES2951829T3 ES2951829T3 ES19745335T ES19745335T ES2951829T3 ES 2951829 T3 ES2951829 T3 ES 2951829T3 ES 19745335 T ES19745335 T ES 19745335T ES 19745335 T ES19745335 T ES 19745335T ES 2951829 T3 ES2951829 T3 ES 2951829T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- formula
- solid
- acid
- crystalline form
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000007787 solid Substances 0.000 title claims abstract description 389
- 238000000034 method Methods 0.000 title claims abstract description 130
- 230000008569 process Effects 0.000 title claims abstract description 41
- 238000011282 treatment Methods 0.000 title claims abstract description 33
- 206010028980 Neoplasm Diseases 0.000 title claims abstract description 29
- 201000011510 cancer Diseases 0.000 title claims abstract description 28
- 238000002360 preparation method Methods 0.000 title claims description 62
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 title abstract description 15
- 150000001875 compounds Chemical class 0.000 claims abstract description 715
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 169
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 160
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 103
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 96
- 206010006187 Breast cancer Diseases 0.000 claims description 80
- 208000026310 Breast neoplasm Diseases 0.000 claims description 79
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 claims description 35
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 30
- 239000008194 pharmaceutical composition Substances 0.000 claims description 22
- 206010033128 Ovarian cancer Diseases 0.000 claims description 15
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 15
- 206010014733 Endometrial cancer Diseases 0.000 claims description 14
- 206010014759 Endometrial neoplasm Diseases 0.000 claims description 14
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 14
- 206010060862 Prostate cancer Diseases 0.000 claims description 14
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 14
- 208000002495 Uterine Neoplasms Diseases 0.000 claims description 14
- 201000005202 lung cancer Diseases 0.000 claims description 14
- 208000020816 lung neoplasm Diseases 0.000 claims description 14
- 206010046766 uterine cancer Diseases 0.000 claims description 14
- 125000001041 indolyl group Chemical group 0.000 claims description 13
- 238000001953 recrystallisation Methods 0.000 claims description 8
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 5
- 229940034982 antineoplastic agent Drugs 0.000 claims description 4
- 239000002246 antineoplastic agent Substances 0.000 claims description 4
- 239000000543 intermediate Substances 0.000 claims description 4
- 206010055113 Breast cancer metastatic Diseases 0.000 claims description 3
- 208000003721 Triple Negative Breast Neoplasms Diseases 0.000 claims description 3
- 208000022679 triple-negative breast carcinoma Diseases 0.000 claims description 3
- 210000004291 uterus Anatomy 0.000 claims description 3
- 208000027706 hormone receptor-positive breast cancer Diseases 0.000 claims description 2
- 238000001959 radiotherapy Methods 0.000 claims description 2
- 208000036907 triple-positive breast carcinoma Diseases 0.000 claims 1
- 239000000203 mixture Substances 0.000 abstract description 164
- 150000003839 salts Chemical class 0.000 abstract description 104
- 230000015572 biosynthetic process Effects 0.000 abstract description 22
- 238000003786 synthesis reaction Methods 0.000 abstract description 11
- 238000009472 formulation Methods 0.000 abstract description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 193
- 238000006243 chemical reaction Methods 0.000 description 174
- 239000000243 solution Substances 0.000 description 134
- 239000002904 solvent Substances 0.000 description 125
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 96
- -1 hydrocarbon radical Chemical class 0.000 description 92
- 238000005481 NMR spectroscopy Methods 0.000 description 87
- 239000011541 reaction mixture Substances 0.000 description 85
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 81
- 239000000725 suspension Substances 0.000 description 75
- 229940126062 Compound A Drugs 0.000 description 66
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 66
- 239000003960 organic solvent Substances 0.000 description 61
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 58
- 239000002253 acid Substances 0.000 description 58
- 238000002474 experimental method Methods 0.000 description 55
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 54
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 45
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 44
- 239000012458 free base Substances 0.000 description 44
- 239000012074 organic phase Substances 0.000 description 42
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical class OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 38
- 239000002585 base Substances 0.000 description 38
- 239000003054 catalyst Substances 0.000 description 38
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 36
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 35
- 238000002411 thermogravimetry Methods 0.000 description 35
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 33
- 125000000217 alkyl group Chemical group 0.000 description 33
- 229910052739 hydrogen Inorganic materials 0.000 description 33
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 33
- 102100038595 Estrogen receptor Human genes 0.000 description 32
- 239000001257 hydrogen Substances 0.000 description 32
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 29
- 238000001914 filtration Methods 0.000 description 28
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 28
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 27
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 27
- 238000005160 1H NMR spectroscopy Methods 0.000 description 26
- 239000012296 anti-solvent Substances 0.000 description 26
- 238000000113 differential scanning calorimetry Methods 0.000 description 26
- 108010038795 estrogen receptors Proteins 0.000 description 26
- LVTJOONKWUXEFR-FZRMHRINSA-N protoneodioscin Natural products O(C[C@@H](CC[C@]1(O)[C@H](C)[C@@H]2[C@]3(C)[C@H]([C@H]4[C@@H]([C@]5(C)C(=CC4)C[C@@H](O[C@@H]4[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@@H](O)[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@H](CO)O4)CC5)CC3)C[C@@H]2O1)C)[C@H]1[C@H](O)[C@H](O)[C@H](O)[C@@H](CO)O1 LVTJOONKWUXEFR-FZRMHRINSA-N 0.000 description 26
- 238000003756 stirring Methods 0.000 description 26
- 239000007795 chemical reaction product Substances 0.000 description 25
- 238000004519 manufacturing process Methods 0.000 description 25
- 229910052731 fluorine Inorganic materials 0.000 description 24
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 23
- 239000008346 aqueous phase Substances 0.000 description 23
- 238000002425 crystallisation Methods 0.000 description 23
- 230000008025 crystallization Effects 0.000 description 23
- 238000002844 melting Methods 0.000 description 23
- 230000008018 melting Effects 0.000 description 23
- AHJRHEGDXFFMBM-UHFFFAOYSA-N palbociclib Chemical compound N1=C2N(C3CCCC3)C(=O)C(C(=O)C)=C(C)C2=CN=C1NC(N=C1)=CC=C1N1CCNCC1 AHJRHEGDXFFMBM-UHFFFAOYSA-N 0.000 description 23
- 239000012453 solvate Substances 0.000 description 23
- 229910052736 halogen Inorganic materials 0.000 description 22
- 150000002367 halogens Chemical class 0.000 description 22
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 22
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 22
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 21
- 102000003929 Transaminases Human genes 0.000 description 21
- 108090000340 Transaminases Proteins 0.000 description 21
- 239000000460 chlorine Substances 0.000 description 21
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 21
- 239000011734 sodium Substances 0.000 description 21
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 20
- 125000004093 cyano group Chemical group *C#N 0.000 description 20
- 238000004128 high performance liquid chromatography Methods 0.000 description 20
- 239000011975 tartaric acid Substances 0.000 description 20
- 229960001367 tartaric acid Drugs 0.000 description 20
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 20
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical group N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 19
- 238000005804 alkylation reaction Methods 0.000 description 19
- 239000012267 brine Substances 0.000 description 19
- 238000005259 measurement Methods 0.000 description 19
- 150000003892 tartrate salts Chemical class 0.000 description 19
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 19
- 238000012216 screening Methods 0.000 description 18
- 235000002906 tartaric acid Nutrition 0.000 description 18
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 17
- 239000003377 acid catalyst Substances 0.000 description 17
- 238000001704 evaporation Methods 0.000 description 17
- 230000008020 evaporation Effects 0.000 description 16
- 239000011521 glass Substances 0.000 description 16
- 239000012454 non-polar solvent Substances 0.000 description 16
- 238000010992 reflux Methods 0.000 description 16
- 238000001757 thermogravimetry curve Methods 0.000 description 16
- 230000004580 weight loss Effects 0.000 description 16
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 15
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 15
- 238000004458 analytical method Methods 0.000 description 15
- 229910052799 carbon Inorganic materials 0.000 description 15
- 238000005119 centrifugation Methods 0.000 description 15
- 238000012512 characterization method Methods 0.000 description 15
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 15
- 150000002431 hydrogen Chemical class 0.000 description 15
- 229910052757 nitrogen Inorganic materials 0.000 description 15
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 14
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 14
- 125000003545 alkoxy group Chemical group 0.000 description 14
- 238000004587 chromatography analysis Methods 0.000 description 14
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 14
- 230000000670 limiting effect Effects 0.000 description 14
- 238000004809 thin layer chromatography Methods 0.000 description 14
- 238000005406 washing Methods 0.000 description 14
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 13
- 230000029936 alkylation Effects 0.000 description 13
- 150000001412 amines Chemical class 0.000 description 13
- 125000003118 aryl group Chemical group 0.000 description 13
- 230000000694 effects Effects 0.000 description 13
- 125000000623 heterocyclic group Chemical group 0.000 description 13
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 13
- 230000036961 partial effect Effects 0.000 description 13
- 239000012071 phase Substances 0.000 description 13
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 12
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 12
- 125000004429 atom Chemical group 0.000 description 12
- 238000001816 cooling Methods 0.000 description 12
- 239000002274 desiccant Substances 0.000 description 12
- 238000001938 differential scanning calorimetry curve Methods 0.000 description 12
- 238000009792 diffusion process Methods 0.000 description 12
- 125000006583 (C1-C3) haloalkyl group Chemical group 0.000 description 11
- NQVQUJRBCVAMIL-UHFFFAOYSA-N 2,3-dibromopropan-1-amine;hydrobromide Chemical compound Br.NCC(Br)CBr NQVQUJRBCVAMIL-UHFFFAOYSA-N 0.000 description 11
- OCKGFTQIICXDQW-ZEQRLZLVSA-N 5-[(1r)-1-hydroxy-2-[4-[(2r)-2-hydroxy-2-(4-methyl-1-oxo-3h-2-benzofuran-5-yl)ethyl]piperazin-1-yl]ethyl]-4-methyl-3h-2-benzofuran-1-one Chemical compound C1=C2C(=O)OCC2=C(C)C([C@@H](O)CN2CCN(CC2)C[C@H](O)C2=CC=C3C(=O)OCC3=C2C)=C1 OCKGFTQIICXDQW-ZEQRLZLVSA-N 0.000 description 11
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 11
- 239000000706 filtrate Substances 0.000 description 11
- 125000001072 heteroaryl group Chemical group 0.000 description 11
- 239000007800 oxidant agent Substances 0.000 description 11
- 238000001907 polarising light microscopy Methods 0.000 description 11
- 125000001424 substituent group Chemical group 0.000 description 11
- 239000003826 tablet Substances 0.000 description 11
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 10
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 10
- RGSFGYAAUTVSQA-UHFFFAOYSA-N Cyclopentane Chemical compound C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 10
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 10
- 150000001299 aldehydes Chemical group 0.000 description 10
- 239000001530 fumaric acid Substances 0.000 description 10
- 235000011087 fumaric acid Nutrition 0.000 description 10
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 10
- 239000000843 powder Substances 0.000 description 10
- 229960004622 raloxifene Drugs 0.000 description 10
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 10
- 239000007858 starting material Substances 0.000 description 10
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 9
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 9
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 9
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- 229950001573 abemaciclib Drugs 0.000 description 9
- 229960000583 acetic acid Drugs 0.000 description 9
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 9
- 239000002775 capsule Substances 0.000 description 9
- 125000004432 carbon atom Chemical group C* 0.000 description 9
- 125000004122 cyclic group Chemical group 0.000 description 9
- 201000010099 disease Diseases 0.000 description 9
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 9
- 239000007788 liquid Substances 0.000 description 9
- UZWDCWONPYILKI-UHFFFAOYSA-N n-[5-[(4-ethylpiperazin-1-yl)methyl]pyridin-2-yl]-5-fluoro-4-(7-fluoro-2-methyl-3-propan-2-ylbenzimidazol-5-yl)pyrimidin-2-amine Chemical group C1CN(CC)CCN1CC(C=N1)=CC=C1NC1=NC=C(F)C(C=2C=C3N(C(C)C)C(C)=NC3=C(F)C=2)=N1 UZWDCWONPYILKI-UHFFFAOYSA-N 0.000 description 9
- 229960004390 palbociclib Drugs 0.000 description 9
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 9
- 238000001556 precipitation Methods 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- 239000003586 protic polar solvent Substances 0.000 description 9
- 235000011121 sodium hydroxide Nutrition 0.000 description 9
- 229910021591 Copper(I) chloride Inorganic materials 0.000 description 8
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 8
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 8
- 235000011054 acetic acid Nutrition 0.000 description 8
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 8
- 230000008859 change Effects 0.000 description 8
- 239000010949 copper Substances 0.000 description 8
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical compound [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 description 8
- 125000000753 cycloalkyl group Chemical group 0.000 description 8
- 229940011871 estrogen Drugs 0.000 description 8
- 239000000262 estrogen Substances 0.000 description 8
- 238000000605 extraction Methods 0.000 description 8
- 239000011737 fluorine Substances 0.000 description 8
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 8
- 230000001590 oxidative effect Effects 0.000 description 8
- 230000035484 reaction time Effects 0.000 description 8
- 230000002829 reductive effect Effects 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- 229940095064 tartrate Drugs 0.000 description 8
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 8
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 7
- RHXHGRAEPCAFML-UHFFFAOYSA-N 7-cyclopentyl-n,n-dimethyl-2-[(5-piperazin-1-ylpyridin-2-yl)amino]pyrrolo[2,3-d]pyrimidine-6-carboxamide Chemical compound N1=C2N(C3CCCC3)C(C(=O)N(C)C)=CC2=CN=C1NC(N=C1)=CC=C1N1CCNCC1 RHXHGRAEPCAFML-UHFFFAOYSA-N 0.000 description 7
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 description 7
- 239000012388 BrettPhos 3rd generation precatalyst Substances 0.000 description 7
- 101710196141 Estrogen receptor Proteins 0.000 description 7
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 7
- FEWJPZIEWOKRBE-LWMBPPNESA-N L-(+)-Tartaric acid Natural products OC(=O)[C@@H](O)[C@H](O)C(O)=O FEWJPZIEWOKRBE-LWMBPPNESA-N 0.000 description 7
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 7
- 150000007513 acids Chemical class 0.000 description 7
- 229960002932 anastrozole Drugs 0.000 description 7
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 7
- 239000003886 aromatase inhibitor Substances 0.000 description 7
- 125000002619 bicyclic group Chemical group 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 229910052801 chlorine Inorganic materials 0.000 description 7
- 235000015165 citric acid Nutrition 0.000 description 7
- 229960004106 citric acid Drugs 0.000 description 7
- 238000007906 compression Methods 0.000 description 7
- 230000006835 compression Effects 0.000 description 7
- 229960000255 exemestane Drugs 0.000 description 7
- 229960003881 letrozole Drugs 0.000 description 7
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 7
- 150000007530 organic bases Chemical class 0.000 description 7
- 239000003880 polar aprotic solvent Substances 0.000 description 7
- 239000002798 polar solvent Substances 0.000 description 7
- 229950003687 ribociclib Drugs 0.000 description 7
- 229920006395 saturated elastomer Polymers 0.000 description 7
- 238000004626 scanning electron microscopy Methods 0.000 description 7
- 229960001603 tamoxifen Drugs 0.000 description 7
- 229960005026 toremifene Drugs 0.000 description 7
- XFCLJVABOIYOMF-QPLCGJKRSA-N toremifene Chemical compound C1=CC(OCCN(C)C)=CC=C1C(\C=1C=CC=CC=1)=C(\CCCl)C1=CC=CC=C1 XFCLJVABOIYOMF-QPLCGJKRSA-N 0.000 description 7
- 229910052723 transition metal Inorganic materials 0.000 description 7
- 150000003624 transition metals Chemical class 0.000 description 7
- WQADWIOXOXRPLN-UHFFFAOYSA-N 1,3-dithiane Chemical compound C1CSCSC1 WQADWIOXOXRPLN-UHFFFAOYSA-N 0.000 description 6
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 6
- ZQZFYGIXNQKOAV-OCEACIFDSA-N Droloxifene Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=C(O)C=CC=1)\C1=CC=C(OCCN(C)C)C=C1 ZQZFYGIXNQKOAV-OCEACIFDSA-N 0.000 description 6
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- 229930012538 Paclitaxel Natural products 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 6
- 125000005334 azaindolyl group Chemical group N1N=C(C2=CC=CC=C12)* 0.000 description 6
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 6
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 6
- 238000000354 decomposition reaction Methods 0.000 description 6
- 208000035475 disorder Diseases 0.000 description 6
- 229950004203 droloxifene Drugs 0.000 description 6
- 238000010438 heat treatment Methods 0.000 description 6
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 6
- 150000007524 organic acids Chemical class 0.000 description 6
- 229960001592 paclitaxel Drugs 0.000 description 6
- 125000004076 pyridyl group Chemical group 0.000 description 6
- 239000013557 residual solvent Substances 0.000 description 6
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 6
- XRRYOHBVVKHARG-LLVKDONJSA-N tert-butyl n-[(2r)-1-(1h-indol-3-yl)propan-2-yl]carbamate Chemical compound C1=CC=C2C(C[C@@H](C)NC(=O)OC(C)(C)C)=CNC2=C1 XRRYOHBVVKHARG-LLVKDONJSA-N 0.000 description 6
- 230000009466 transformation Effects 0.000 description 6
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 6
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 5
- 229940124297 CDK 4/6 inhibitor Drugs 0.000 description 5
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 5
- 238000010521 absorption reaction Methods 0.000 description 5
- 125000006242 amine protecting group Chemical group 0.000 description 5
- 238000013459 approach Methods 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- 239000006227 byproduct Substances 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 239000002131 composite material Substances 0.000 description 5
- 229910052802 copper Inorganic materials 0.000 description 5
- 238000010511 deprotection reaction Methods 0.000 description 5
- 239000003814 drug Substances 0.000 description 5
- 229960002258 fulvestrant Drugs 0.000 description 5
- DMEGYFMYUHOHGS-UHFFFAOYSA-N heptamethylene Natural products C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 5
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 5
- 239000003446 ligand Substances 0.000 description 5
- CCERQOYLJJULMD-UHFFFAOYSA-M magnesium;carbanide;chloride Chemical compound [CH3-].[Mg+2].[Cl-] CCERQOYLJJULMD-UHFFFAOYSA-M 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 229910052751 metal Inorganic materials 0.000 description 5
- 239000002184 metal Substances 0.000 description 5
- 150000007522 mineralic acids Chemical class 0.000 description 5
- 239000012038 nucleophile Substances 0.000 description 5
- 239000012044 organic layer Substances 0.000 description 5
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 5
- 229910052708 sodium Inorganic materials 0.000 description 5
- 238000001179 sorption measurement Methods 0.000 description 5
- 230000008685 targeting Effects 0.000 description 5
- 230000007704 transition Effects 0.000 description 5
- APJYDQYYACXCRM-UHFFFAOYSA-N tryptamine Chemical compound C1=CC=C2C(CCN)=CNC2=C1 APJYDQYYACXCRM-UHFFFAOYSA-N 0.000 description 5
- SPEUIVXLLWOEMJ-UHFFFAOYSA-N 1,1-dimethoxyethane Chemical compound COC(C)OC SPEUIVXLLWOEMJ-UHFFFAOYSA-N 0.000 description 4
- NUMQSSSVVADVQJ-SNVBAGLBSA-N 2,2-difluoro-3-[[(2R)-1-(1H-indol-3-yl)propan-2-yl]amino]propan-1-ol Chemical compound N1C=C(C2=CC=CC=C12)C[C@@H](C)NCC(CO)(F)F NUMQSSSVVADVQJ-SNVBAGLBSA-N 0.000 description 4
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 4
- DODQJNMQWMSYGS-QPLCGJKRSA-N 4-[(z)-1-[4-[2-(dimethylamino)ethoxy]phenyl]-1-phenylbut-1-en-2-yl]phenol Chemical compound C=1C=C(O)C=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 DODQJNMQWMSYGS-QPLCGJKRSA-N 0.000 description 4
- 108010012934 Albumin-Bound Paclitaxel Proteins 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- 229940122815 Aromatase inhibitor Drugs 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 4
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 4
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 4
- BLCLNMBMMGCOAS-URPVMXJPSA-N Goserelin Chemical compound C([C@@H](C(=O)N[C@H](COC(C)(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NNC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 BLCLNMBMMGCOAS-URPVMXJPSA-N 0.000 description 4
- 108010069236 Goserelin Proteins 0.000 description 4
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 4
- FEWJPZIEWOKRBE-JCYAYHJZSA-L L-tartrate(2-) Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O FEWJPZIEWOKRBE-JCYAYHJZSA-L 0.000 description 4
- 108010000817 Leuprolide Proteins 0.000 description 4
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 4
- 238000002441 X-ray diffraction Methods 0.000 description 4
- IHGLINDYFMDHJG-UHFFFAOYSA-N [2-(4-methoxyphenyl)-3,4-dihydronaphthalen-1-yl]-[4-(2-pyrrolidin-1-ylethoxy)phenyl]methanone Chemical compound C1=CC(OC)=CC=C1C(CCC1=CC=CC=C11)=C1C(=O)C(C=C1)=CC=C1OCCN1CCCC1 IHGLINDYFMDHJG-UHFFFAOYSA-N 0.000 description 4
- 230000000118 anti-neoplastic effect Effects 0.000 description 4
- 239000012062 aqueous buffer Substances 0.000 description 4
- 229960003852 atezolizumab Drugs 0.000 description 4
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 4
- 210000004027 cell Anatomy 0.000 description 4
- 125000001309 chloro group Chemical group Cl* 0.000 description 4
- 239000006184 cosolvent Substances 0.000 description 4
- 239000013078 crystal Substances 0.000 description 4
- 238000004090 dissolution Methods 0.000 description 4
- 238000004821 distillation Methods 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 229960004679 doxorubicin Drugs 0.000 description 4
- 239000012039 electrophile Substances 0.000 description 4
- 230000002124 endocrine Effects 0.000 description 4
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 4
- 125000000524 functional group Chemical group 0.000 description 4
- 125000004612 furopyridinyl group Chemical group O1C(=CC2=C1C=CC=N2)* 0.000 description 4
- 239000000499 gel Substances 0.000 description 4
- 229960002913 goserelin Drugs 0.000 description 4
- 150000004795 grignard reagents Chemical class 0.000 description 4
- 125000001188 haloalkyl group Chemical group 0.000 description 4
- 125000005842 heteroatom Chemical group 0.000 description 4
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 4
- 150000002475 indoles Chemical class 0.000 description 4
- 229940011051 isopropyl acetate Drugs 0.000 description 4
- GWYFCOCPABKNJV-UHFFFAOYSA-M isovalerate Chemical compound CC(C)CC([O-])=O GWYFCOCPABKNJV-UHFFFAOYSA-M 0.000 description 4
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 4
- 229960004338 leuprorelin Drugs 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 229960000485 methotrexate Drugs 0.000 description 4
- 150000004682 monohydrates Chemical class 0.000 description 4
- 125000004433 nitrogen atom Chemical group N* 0.000 description 4
- 229910052760 oxygen Inorganic materials 0.000 description 4
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 4
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 4
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 4
- 125000006239 protecting group Chemical group 0.000 description 4
- 125000006413 ring segment Chemical group 0.000 description 4
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 239000011550 stock solution Substances 0.000 description 4
- 229910052717 sulfur Inorganic materials 0.000 description 4
- PXLWDYOUWTYABH-LLVKDONJSA-N tert-butyl n-[(1s)-1-(3-fluorophenyl)-2-hydroxyethyl]carbamate Chemical compound CC(C)(C)OC(=O)N[C@H](CO)C1=CC=CC(F)=C1 PXLWDYOUWTYABH-LLVKDONJSA-N 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 4
- 238000002076 thermal analysis method Methods 0.000 description 4
- 150000003573 thiols Chemical class 0.000 description 4
- 229950000212 trioxifene Drugs 0.000 description 4
- 238000001195 ultra high performance liquid chromatography Methods 0.000 description 4
- 229960001183 venetoclax Drugs 0.000 description 4
- LQBVNQSMGBZMKD-UHFFFAOYSA-N venetoclax Chemical compound C=1C=C(Cl)C=CC=1C=1CC(C)(C)CCC=1CN(CC1)CCN1C(C=C1OC=2C=C3C=CNC3=NC=2)=CC=C1C(=O)NS(=O)(=O)C(C=C1[N+]([O-])=O)=CC=C1NCC1CCOCC1 LQBVNQSMGBZMKD-UHFFFAOYSA-N 0.000 description 4
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 4
- QSQQQURBVYWZKJ-MRVPVSSYSA-N (2r)-1-(1h-indol-3-yl)propan-2-amine Chemical compound C1=CC=C2C(C[C@H](N)C)=CNC2=C1 QSQQQURBVYWZKJ-MRVPVSSYSA-N 0.000 description 3
- LKJPYSCBVHEWIU-KRWDZBQOSA-N (R)-bicalutamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)S(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-KRWDZBQOSA-N 0.000 description 3
- BURHGPHDEVGCEZ-KJGLQBJMSA-N (e)-3-[4-[(e)-2-(2-chloro-4-fluorophenyl)-1-(1h-indazol-5-yl)but-1-enyl]phenyl]prop-2-enoic acid Chemical compound C=1C=C(F)C=C(Cl)C=1C(/CC)=C(C=1C=C2C=NNC2=CC=1)\C1=CC=C(\C=C\C(O)=O)C=C1 BURHGPHDEVGCEZ-KJGLQBJMSA-N 0.000 description 3
- LJCZNYWLQZZIOS-UHFFFAOYSA-N 2,2,2-trichlorethoxycarbonyl chloride Chemical compound ClC(=O)OCC(Cl)(Cl)Cl LJCZNYWLQZZIOS-UHFFFAOYSA-N 0.000 description 3
- AMCKYDINHDOOCB-UHFFFAOYSA-N 2,2-difluoropropane-1,3-diol Chemical compound OCC(F)(F)CO AMCKYDINHDOOCB-UHFFFAOYSA-N 0.000 description 3
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 3
- CZGVAISJIQNQEJ-UHFFFAOYSA-N 4-bromo-2,6-difluorobenzaldehyde Chemical compound FC1=CC(Br)=CC(F)=C1C=O CZGVAISJIQNQEJ-UHFFFAOYSA-N 0.000 description 3
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 3
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 3
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 3
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 3
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 3
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 3
- 239000005977 Ethylene Substances 0.000 description 3
- 108010029961 Filgrastim Proteins 0.000 description 3
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 3
- 102100039619 Granulocyte colony-stimulating factor Human genes 0.000 description 3
- 239000007818 Grignard reagent Substances 0.000 description 3
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 3
- XNRVGTHNYCNCFF-UHFFFAOYSA-N Lapatinib ditosylate monohydrate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1.CC1=CC=C(S(O)(=O)=O)C=C1.O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 XNRVGTHNYCNCFF-UHFFFAOYSA-N 0.000 description 3
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 3
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 3
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 3
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 3
- PRXRUNOAOLTIEF-ADSICKODSA-N Sorbitan trioleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCC\C=C/CCCCCCCC)[C@H]1OC[C@H](O)[C@H]1OC(=O)CCCCCCC\C=C/CCCCCCCC PRXRUNOAOLTIEF-ADSICKODSA-N 0.000 description 3
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 3
- 239000000370 acceptor Substances 0.000 description 3
- 229910052782 aluminium Inorganic materials 0.000 description 3
- 239000000908 ammonium hydroxide Substances 0.000 description 3
- 235000011114 ammonium hydroxide Nutrition 0.000 description 3
- 239000011260 aqueous acid Substances 0.000 description 3
- 229940046844 aromatase inhibitors Drugs 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 3
- 229960000997 bicalutamide Drugs 0.000 description 3
- 230000004071 biological effect Effects 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 229960004117 capecitabine Drugs 0.000 description 3
- 150000001721 carbon Chemical group 0.000 description 3
- 229960004562 carboplatin Drugs 0.000 description 3
- YAYRGNWWLMLWJE-UHFFFAOYSA-L carboplatin Chemical compound O=C1O[Pt](N)(N)OC(=O)C11CCC1 YAYRGNWWLMLWJE-UHFFFAOYSA-L 0.000 description 3
- HODHLQXRQOHAAZ-UHFFFAOYSA-M chloropalladium(1+);dicyclohexyl-[2-(2,6-dimethoxyphenyl)phenyl]phosphane;2-phenylaniline Chemical compound [Pd+]Cl.NC1=CC=CC=C1C1=CC=CC=[C-]1.COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 HODHLQXRQOHAAZ-UHFFFAOYSA-M 0.000 description 3
- GITFHTZGVMIBGS-UHFFFAOYSA-M chloropalladium(1+);ditert-butyl-[2-[2,4,6-tri(propan-2-yl)phenyl]phenyl]phosphane;2-phenylethanamine Chemical compound [Pd+]Cl.NCCC1=CC=CC=[C-]1.CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C(C)(C)C)C(C)(C)C GITFHTZGVMIBGS-UHFFFAOYSA-M 0.000 description 3
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 3
- 229960004316 cisplatin Drugs 0.000 description 3
- 230000000052 comparative effect Effects 0.000 description 3
- 230000002860 competitive effect Effects 0.000 description 3
- 229960004397 cyclophosphamide Drugs 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 238000002050 diffraction method Methods 0.000 description 3
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 3
- 238000009826 distribution Methods 0.000 description 3
- 229960003668 docetaxel Drugs 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 229960001904 epirubicin Drugs 0.000 description 3
- 229960003649 eribulin Drugs 0.000 description 3
- UFNVPOGXISZXJD-XJPMSQCNSA-N eribulin Chemical compound C([C@H]1CC[C@@H]2O[C@@H]3[C@H]4O[C@H]5C[C@](O[C@H]4[C@H]2O1)(O[C@@H]53)CC[C@@H]1O[C@H](C(C1)=C)CC1)C(=O)C[C@@H]2[C@@H](OC)[C@@H](C[C@H](O)CN)O[C@H]2C[C@@H]2C(=C)[C@H](C)C[C@H]1O2 UFNVPOGXISZXJD-XJPMSQCNSA-N 0.000 description 3
- DSGUSEBCDAKBCM-UHFFFAOYSA-N ethane-1,2-disulfonic acid;dihydrate Chemical compound O.O.OS(=O)(=O)CCS(O)(=O)=O DSGUSEBCDAKBCM-UHFFFAOYSA-N 0.000 description 3
- 229960004177 filgrastim Drugs 0.000 description 3
- 229960002949 fluorouracil Drugs 0.000 description 3
- 238000004817 gas chromatography Methods 0.000 description 3
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 3
- 229960005277 gemcitabine Drugs 0.000 description 3
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 3
- 229910052737 gold Inorganic materials 0.000 description 3
- 239000010931 gold Substances 0.000 description 3
- 125000005843 halogen group Chemical group 0.000 description 3
- 238000004896 high resolution mass spectrometry Methods 0.000 description 3
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- FABUFPQFXZVHFB-CFWQTKTJSA-N ixabepilone Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@H](C)C(=O)C(C)(C)[C@H](O)CC(=O)N1)O)C)=C\C1=CSC(C)=N1 FABUFPQFXZVHFB-CFWQTKTJSA-N 0.000 description 3
- 229960002014 ixabepilone Drugs 0.000 description 3
- 150000002576 ketones Chemical class 0.000 description 3
- 229960004891 lapatinib Drugs 0.000 description 3
- 108020001756 ligand binding domains Proteins 0.000 description 3
- 238000004811 liquid chromatography Methods 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- 235000012054 meals Nutrition 0.000 description 3
- 229940098779 methanesulfonic acid Drugs 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- YKYONYBAUNKHLG-UHFFFAOYSA-N n-Propyl acetate Natural products CCCOC(C)=O YKYONYBAUNKHLG-UHFFFAOYSA-N 0.000 description 3
- 229960002653 nilutamide Drugs 0.000 description 3
- XWXYUMMDTVBTOU-UHFFFAOYSA-N nilutamide Chemical compound O=C1C(C)(C)NC(=O)N1C1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 XWXYUMMDTVBTOU-UHFFFAOYSA-N 0.000 description 3
- 229960003301 nivolumab Drugs 0.000 description 3
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 3
- 229960000572 olaparib Drugs 0.000 description 3
- FAQDUNYVKQKNLD-UHFFFAOYSA-N olaparib Chemical compound FC1=CC=C(CC2=C3[CH]C=CC=C3C(=O)N=N2)C=C1C(=O)N(CC1)CCN1C(=O)C1CC1 FAQDUNYVKQKNLD-UHFFFAOYSA-N 0.000 description 3
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 238000003921 particle size analysis Methods 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- HQQSBEDKMRHYME-UHFFFAOYSA-N pefloxacin mesylate Chemical compound [H+].CS([O-])(=O)=O.C1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC(F)=C1N1CCN(C)CC1 HQQSBEDKMRHYME-UHFFFAOYSA-N 0.000 description 3
- 229960001373 pegfilgrastim Drugs 0.000 description 3
- 108010044644 pegfilgrastim Proteins 0.000 description 3
- 229960002621 pembrolizumab Drugs 0.000 description 3
- 229960002087 pertuzumab Drugs 0.000 description 3
- 230000000704 physical effect Effects 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 3
- 238000010926 purge Methods 0.000 description 3
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 3
- 238000006268 reductive amination reaction Methods 0.000 description 3
- 238000010583 slow cooling Methods 0.000 description 3
- 229940083542 sodium Drugs 0.000 description 3
- 235000015424 sodium Nutrition 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- 235000017550 sodium carbonate Nutrition 0.000 description 3
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 description 3
- 239000007790 solid phase Substances 0.000 description 3
- 238000000527 sonication Methods 0.000 description 3
- 238000002336 sorption--desorption measurement Methods 0.000 description 3
- 238000004611 spectroscopical analysis Methods 0.000 description 3
- VNFWTIYUKDMAOP-UHFFFAOYSA-N sphos Chemical compound COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 VNFWTIYUKDMAOP-UHFFFAOYSA-N 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- ROUONLKDWVQKNB-NWDGAFQWSA-N tert-butyl n-[(1r,2s)-2-hydroxy-2,3-dihydro-1h-inden-1-yl]carbamate Chemical compound C1=CC=C2[C@@H](NC(=O)OC(C)(C)C)[C@@H](O)CC2=C1 ROUONLKDWVQKNB-NWDGAFQWSA-N 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- 238000005891 transamination reaction Methods 0.000 description 3
- 229960000575 trastuzumab Drugs 0.000 description 3
- 229960001612 trastuzumab emtansine Drugs 0.000 description 3
- GKASDNZWUGIAMG-UHFFFAOYSA-N triethyl orthoformate Chemical compound CCOC(OCC)OCC GKASDNZWUGIAMG-UHFFFAOYSA-N 0.000 description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 3
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 3
- 229960003048 vinblastine Drugs 0.000 description 3
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 3
- 229960002066 vinorelbine Drugs 0.000 description 3
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 3
- 229910052725 zinc Inorganic materials 0.000 description 3
- 239000011701 zinc Substances 0.000 description 3
- GRZXWCHAXNAUHY-NSISKUIASA-N (2S)-2-(4-chlorophenyl)-1-[4-[(5R,7R)-7-hydroxy-5-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-yl]-1-piperazinyl]-3-(propan-2-ylamino)-1-propanone Chemical group C1([C@H](C(=O)N2CCN(CC2)C=2C=3[C@H](C)C[C@@H](O)C=3N=CN=2)CNC(C)C)=CC=C(Cl)C=C1 GRZXWCHAXNAUHY-NSISKUIASA-N 0.000 description 2
- KJAAPZIFCQQQKX-NDEPHWFRSA-N (2s)-2-[4-[2-[3-(fluoromethyl)azetidin-1-yl]ethoxy]phenyl]-3-(3-hydroxyphenyl)-4-methyl-2h-chromen-6-ol Chemical compound C1=CC([C@H]2C(=C(C3=CC(O)=CC=C3O2)C)C=2C=C(O)C=CC=2)=CC=C1OCCN1CC(CF)C1 KJAAPZIFCQQQKX-NDEPHWFRSA-N 0.000 description 2
- SIFNOOUKXBRGGB-AREMUKBSSA-N (6r)-6-[2-[ethyl-[[4-[2-(ethylamino)ethyl]phenyl]methyl]amino]-4-methoxyphenyl]-5,6,7,8-tetrahydronaphthalen-2-ol Chemical compound C1=CC(CCNCC)=CC=C1CN(CC)C1=CC(OC)=CC=C1[C@H]1CC2=CC=C(O)C=C2CC1 SIFNOOUKXBRGGB-AREMUKBSSA-N 0.000 description 2
- HJQQVNIORAQATK-LXFXVVTESA-N (e)-3-[4-[(e)-1,2-diphenylbut-1-enyl]phenyl]prop-2-enoic acid Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(\C=C\C(O)=O)=CC=1)\C1=CC=CC=C1 HJQQVNIORAQATK-LXFXVVTESA-N 0.000 description 2
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 2
- DHKHKXVYLBGOIT-UHFFFAOYSA-N 1,1-Diethoxyethane Chemical compound CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 2
- IMLSAISZLJGWPP-UHFFFAOYSA-N 1,3-dithiolane Chemical compound C1CSCS1 IMLSAISZLJGWPP-UHFFFAOYSA-N 0.000 description 2
- OQCYTSHIQNPJIC-QURGRASLSA-N 1-[(e)-2-bromo-1,2-diphenylethenyl]-4-ethylbenzene Chemical compound C1=CC(CC)=CC=C1C(\C=1C=CC=CC=1)=C(\Br)C1=CC=CC=C1 OQCYTSHIQNPJIC-QURGRASLSA-N 0.000 description 2
- VNHWPVLQRKKKRY-UHFFFAOYSA-N 1-bromo-3-fluoropropane Chemical compound FCCCBr VNHWPVLQRKKKRY-UHFFFAOYSA-N 0.000 description 2
- JIMZJGNNRSCEFH-UHFFFAOYSA-N 1-chloroethyl carbamate Chemical compound CC(Cl)OC(N)=O JIMZJGNNRSCEFH-UHFFFAOYSA-N 0.000 description 2
- IHWDSEPNZDYMNF-UHFFFAOYSA-N 1H-indol-2-amine Chemical compound C1=CC=C2NC(N)=CC2=C1 IHWDSEPNZDYMNF-UHFFFAOYSA-N 0.000 description 2
- MVXVYAKCVDQRLW-UHFFFAOYSA-N 1h-pyrrolo[2,3-b]pyridine Chemical compound C1=CN=C2NC=CC2=C1 MVXVYAKCVDQRLW-UHFFFAOYSA-N 0.000 description 2
- XLKDJOPOOHHZAN-UHFFFAOYSA-N 1h-pyrrolo[2,3-c]pyridine Chemical compound C1=NC=C2NC=CC2=C1 XLKDJOPOOHHZAN-UHFFFAOYSA-N 0.000 description 2
- NRKYWOKHZRQRJR-UHFFFAOYSA-N 2,2,2-trifluoroacetamide Chemical compound NC(=O)C(F)(F)F NRKYWOKHZRQRJR-UHFFFAOYSA-N 0.000 description 2
- PGHWCZITRQNIPM-QPLCGJKRSA-N 2-[4-[(z)-2-chloro-1,2-diphenylethenyl]phenoxy]-n,n-diethylethanamine oxide Chemical compound C1=CC(OCC[N+]([O-])(CC)CC)=CC=C1C(\C=1C=CC=CC=1)=C(/Cl)C1=CC=CC=C1 PGHWCZITRQNIPM-QPLCGJKRSA-N 0.000 description 2
- BHNHHSOHWZKFOX-UHFFFAOYSA-N 2-methyl-1H-indole Chemical compound C1=CC=C2NC(C)=CC2=C1 BHNHHSOHWZKFOX-UHFFFAOYSA-N 0.000 description 2
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 2
- REFMCNOVXIVZAX-BIBXISHDSA-N 3-[(1R,3R)-1-(4-bromo-2,6-difluorophenyl)-3-methyl-1,3,4,9-tetrahydropyrido[3,4-b]indol-2-yl]-2,2-difluoropropan-1-ol Chemical compound BrC1=CC(=C(C(=C1)F)[C@H]1N([C@@H](CC2=C1NC1=CC=CC=C21)C)CC(CO)(F)F)F REFMCNOVXIVZAX-BIBXISHDSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- LAXZKWVVCYMNSN-UHFFFAOYSA-N 5-bromo-2-(diethoxymethyl)-1,3-difluorobenzene Chemical compound CCOC(OCC)C1=C(F)C=C(Br)C=C1F LAXZKWVVCYMNSN-UHFFFAOYSA-N 0.000 description 2
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- 229940126638 Akt inhibitor Drugs 0.000 description 2
- 238000012935 Averaging Methods 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- HVOLVGXBUGAQJF-GSVOUGTGSA-N C[C@@H]1COS(=O)(=O)N1C(O)=O Chemical compound C[C@@H]1COS(=O)(=O)N1C(O)=O HVOLVGXBUGAQJF-GSVOUGTGSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 101000709520 Chlamydia trachomatis serovar L2 (strain 434/Bu / ATCC VR-902B) Atypical response regulator protein ChxR Proteins 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- GVOUFPWUYJWQSK-UHFFFAOYSA-N Cyclofenil Chemical compound C1=CC(OC(=O)C)=CC=C1C(C=1C=CC(OC(C)=O)=CC=1)=C1CCCCC1 GVOUFPWUYJWQSK-UHFFFAOYSA-N 0.000 description 2
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 2
- 108010041356 Estrogen Receptor beta Proteins 0.000 description 2
- 102100029951 Estrogen receptor beta Human genes 0.000 description 2
- 229920013685 Estron Polymers 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 229910003771 Gold(I) chloride Inorganic materials 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 241000872931 Myoporum sandwicense Species 0.000 description 2
- 229910019093 NaOCl Inorganic materials 0.000 description 2
- 239000012661 PARP inhibitor Substances 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 241000286209 Phasianidae Species 0.000 description 2
- 229940121906 Poly ADP ribose polymerase inhibitor Drugs 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 102000001708 Protein Isoforms Human genes 0.000 description 2
- 108010029485 Protein Isoforms Proteins 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical group CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- 238000001069 Raman spectroscopy Methods 0.000 description 2
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 2
- 229910004298 SiO 2 Inorganic materials 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- GEPYJRACPVWVMX-MRVPVSSYSA-N [(1S)-1-(3-fluorophenyl)-2-hydroxyethyl]carbamic acid Chemical compound OC(=O)N[C@H](CO)C1=CC=CC(F)=C1 GEPYJRACPVWVMX-MRVPVSSYSA-N 0.000 description 2
- NOECSYBNZHIVHW-LKADTRSGSA-N [(2r,3as,3bs,5as,6r,8as,8br,10as)-2,6-diethynyl-3a,5a-dimethyl-2-propanoyloxy-1,3,3b,4,5,7,8,8a,8b,9,10,10a-dodecahydroindeno[5,4-e]inden-6-yl] propanoate Chemical compound C([C@]1(C)[C@](OC(=O)CC)(C#C)CC[C@H]1[C@@H]1CC2)C[C@@H]1[C@]1(C)[C@@H]2C[C@@](C#C)(OC(=O)CC)C1 NOECSYBNZHIVHW-LKADTRSGSA-N 0.000 description 2
- DUYNJNWVGIWJRI-LJAQVGFWSA-N acolbifene Chemical compound C1=CC([C@H]2C(=C(C3=CC=C(O)C=C3O2)C)C=2C=CC(O)=CC=2)=CC=C1OCCN1CCCCC1 DUYNJNWVGIWJRI-LJAQVGFWSA-N 0.000 description 2
- 229950002421 acolbifene Drugs 0.000 description 2
- 235000004279 alanine Nutrition 0.000 description 2
- 125000003295 alanine group Chemical group N[C@@H](C)C(=O)* 0.000 description 2
- 230000001476 alcoholic effect Effects 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 230000002152 alkylating effect Effects 0.000 description 2
- 125000002947 alkylene group Chemical group 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 235000001014 amino acid Nutrition 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 229950002916 avelumab Drugs 0.000 description 2
- 239000003637 basic solution Substances 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- SVXPPSFKJQWISH-UHFFFAOYSA-N benzyl carbamoperoxoate Chemical compound NC(=O)OOCC1=CC=CC=C1 SVXPPSFKJQWISH-UHFFFAOYSA-N 0.000 description 2
- GMVGZINKTXAKDT-UHFFFAOYSA-N bis(trifluoromethylsulfonyl)azanide;dicyclohexyl-[2-[2,4,6-tri(propan-2-yl)phenyl]phenyl]phosphane;gold(1+) Chemical compound [Au+].FC(F)(F)S(=O)(=O)[N-]S(=O)(=O)C(F)(F)F.CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 GMVGZINKTXAKDT-UHFFFAOYSA-N 0.000 description 2
- 229950004948 brilanestrant Drugs 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 229950003738 broparestrol Drugs 0.000 description 2
- XUPYJHCZDLZNFP-UHFFFAOYSA-N butyl butanoate Chemical compound CCCCOC(=O)CCC XUPYJHCZDLZNFP-UHFFFAOYSA-N 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 238000002619 cancer immunotherapy Methods 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 238000001311 chemical methods and process Methods 0.000 description 2
- NMMPMZWIIQCZBA-UHFFFAOYSA-M chloropalladium(1+);dicyclohexyl-[2-[2,4,6-tri(propan-2-yl)phenyl]phenyl]phosphane;2-phenylethanamine Chemical compound [Pd+]Cl.NCCC1=CC=CC=[C-]1.CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 NMMPMZWIIQCZBA-UHFFFAOYSA-M 0.000 description 2
- GKIRPKYJQBWNGO-OCEACIFDSA-N clomifene Chemical compound C1=CC(OCCN(CC)CC)=CC=C1C(\C=1C=CC=CC=1)=C(\Cl)C1=CC=CC=C1 GKIRPKYJQBWNGO-OCEACIFDSA-N 0.000 description 2
- 229960003608 clomifene Drugs 0.000 description 2
- 229950008864 clomifenoxide Drugs 0.000 description 2
- 208000029742 colonic neoplasm Diseases 0.000 description 2
- 238000009838 combustion analysis Methods 0.000 description 2
- 238000011217 control strategy Methods 0.000 description 2
- 239000013256 coordination polymer Substances 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 239000002178 crystalline material Substances 0.000 description 2
- 229960002944 cyclofenil Drugs 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 230000018044 dehydration Effects 0.000 description 2
- 238000006297 dehydration reaction Methods 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- MXFYYFVVIIWKFE-UHFFFAOYSA-N dicyclohexyl-[2-[2,6-di(propan-2-yloxy)phenyl]phenyl]phosphane Chemical compound CC(C)OC1=CC=CC(OC(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 MXFYYFVVIIWKFE-UHFFFAOYSA-N 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- IEJIGPNLZYLLBP-UHFFFAOYSA-N dimethyl carbonate Chemical compound COC(=O)OC IEJIGPNLZYLLBP-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 150000002009 diols Chemical class 0.000 description 2
- 238000006073 displacement reaction Methods 0.000 description 2
- CNXMDTWQWLGCPE-UHFFFAOYSA-N ditert-butyl-(2-phenylphenyl)phosphane Chemical compound CC(C)(C)P(C(C)(C)C)C1=CC=CC=C1C1=CC=CC=C1 CNXMDTWQWLGCPE-UHFFFAOYSA-N 0.000 description 2
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 229950009791 durvalumab Drugs 0.000 description 2
- 229950005473 elacestrant Drugs 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 229960005309 estradiol Drugs 0.000 description 2
- 102000015694 estrogen receptors Human genes 0.000 description 2
- 150000002167 estrones Chemical class 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 230000004927 fusion Effects 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 2
- FDWREHZXQUYJFJ-UHFFFAOYSA-M gold monochloride Chemical compound [Cl-].[Au+] FDWREHZXQUYJFJ-UHFFFAOYSA-M 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- 125000004404 heteroalkyl group Chemical group 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- 238000013537 high throughput screening Methods 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 229910052741 iridium Inorganic materials 0.000 description 2
- GKOZUEZYRPOHIO-UHFFFAOYSA-N iridium atom Chemical compound [Ir] GKOZUEZYRPOHIO-UHFFFAOYSA-N 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- PHTQWCKDNZKARW-UHFFFAOYSA-N isoamylol Chemical compound CC(C)CCO PHTQWCKDNZKARW-UHFFFAOYSA-N 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- GXESHMAMLJKROZ-IAPPQJPRSA-N lasofoxifene Chemical compound C1([C@@H]2[C@@H](C3=CC=C(C=C3CC2)O)C=2C=CC(OCCN3CCCC3)=CC=2)=CC=CC=C1 GXESHMAMLJKROZ-IAPPQJPRSA-N 0.000 description 2
- 229960002367 lasofoxifene Drugs 0.000 description 2
- XZEUAXYWNKYKPL-WDYNHAJCSA-N levormeloxifene Chemical compound C1([C@H]2[C@@H](C3=CC=C(C=C3OC2(C)C)OC)C=2C=CC(OCCN3CCCC3)=CC=2)=CC=CC=C1 XZEUAXYWNKYKPL-WDYNHAJCSA-N 0.000 description 2
- IHLVCKWPAMTVTG-UHFFFAOYSA-N lithium;carbanide Chemical compound [Li+].[CH3-] IHLVCKWPAMTVTG-UHFFFAOYSA-N 0.000 description 2
- 238000011068 loading method Methods 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 150000002690 malonic acid derivatives Chemical class 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 206010061289 metastatic neoplasm Diseases 0.000 description 2
- GPKUICFDWYEPTK-UHFFFAOYSA-N methoxycyclohexatriene Chemical compound COC1=CC=C=C[CH]1 GPKUICFDWYEPTK-UHFFFAOYSA-N 0.000 description 2
- SKTCDJAMAYNROS-UHFFFAOYSA-N methoxycyclopentane Chemical compound COC1CCCC1 SKTCDJAMAYNROS-UHFFFAOYSA-N 0.000 description 2
- GTCAXTIRRLKXRU-UHFFFAOYSA-N methyl carbamate Chemical compound COC(N)=O GTCAXTIRRLKXRU-UHFFFAOYSA-N 0.000 description 2
- UAEPNZWRGJTJPN-UHFFFAOYSA-N methylcyclohexane Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 2
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- 238000000386 microscopy Methods 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 229910052759 nickel Inorganic materials 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 2
- 238000010899 nucleation Methods 0.000 description 2
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 239000008203 oral pharmaceutical composition Substances 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 150000002894 organic compounds Chemical class 0.000 description 2
- 229960003327 ormeloxifene Drugs 0.000 description 2
- 229960003969 ospemifene Drugs 0.000 description 2
- LUMKNAVTFCDUIE-VHXPQNKSSA-N ospemifene Chemical compound C1=CC(OCCO)=CC=C1C(\C=1C=CC=CC=1)=C(\CCCl)C1=CC=CC=C1 LUMKNAVTFCDUIE-VHXPQNKSSA-N 0.000 description 2
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 238000010979 pH adjustment Methods 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 2
- 229940046159 pegylated liposomal doxorubicin Drugs 0.000 description 2
- 238000005191 phase separation Methods 0.000 description 2
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 229910052697 platinum Inorganic materials 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011736 potassium bicarbonate Substances 0.000 description 2
- 235000015497 potassium bicarbonate Nutrition 0.000 description 2
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 235000011181 potassium carbonates Nutrition 0.000 description 2
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 2
- 150000003141 primary amines Chemical class 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- RUOJZAUFBMNUDX-UHFFFAOYSA-N propylene carbonate Chemical compound CC1COC(=O)O1 RUOJZAUFBMNUDX-UHFFFAOYSA-N 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 239000003197 protein kinase B inhibitor Substances 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 229940076788 pyruvate Drugs 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 239000002516 radical scavenger Substances 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000006722 reduction reaction Methods 0.000 description 2
- 229920005989 resin Polymers 0.000 description 2
- 239000011347 resin Substances 0.000 description 2
- 229910052703 rhodium Inorganic materials 0.000 description 2
- 239000010948 rhodium Substances 0.000 description 2
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 2
- 229910052707 ruthenium Inorganic materials 0.000 description 2
- 229960004889 salicylic acid Drugs 0.000 description 2
- 229940125944 selective estrogen receptor degrader Drugs 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- 239000011877 solvent mixture Substances 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 239000011593 sulfur Chemical group 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- XMMLCGSWFIUFKX-LLVKDONJSA-N tert-butyl (4S)-2,2-dioxo-4-[3-(trifluoromethyl)phenyl]oxathiazolidine-3-carboxylate Chemical compound CC(C)(C)OC(=O)N1[C@H](COS1(=O)=O)c1cccc(c1)C(F)(F)F XMMLCGSWFIUFKX-LLVKDONJSA-N 0.000 description 2
- DJEIJXAMTDRFKA-INIZCTEOSA-N tert-butyl N-[(1S)-1-cyclopropyl-2-(1H-indol-3-yl)ethyl]carbamate Chemical compound C1(CC1)[C@H](CC1=CNC2=CC=CC=C12)NC(OC(C)(C)C)=O DJEIJXAMTDRFKA-INIZCTEOSA-N 0.000 description 2
- BKWOGZLRUOWJKP-GFCCVEGCSA-N tert-butyl N-[(1S)-2-hydroxy-1-(4-methoxyphenyl)ethyl]carbamate Chemical compound C1(OC)=CC=C([C@@H](CO)NC(=O)OC(C)(C)C)C=C1 BKWOGZLRUOWJKP-GFCCVEGCSA-N 0.000 description 2
- FRRCSAZNGYCTLR-UHFFFAOYSA-N tert-butyl n-(1-cyclopropyl-2-hydroxyethyl)carbamate Chemical compound CC(C)(C)OC(=O)NC(CO)C1CC1 FRRCSAZNGYCTLR-UHFFFAOYSA-N 0.000 description 2
- PDAFIZPRSXHMCO-LURJTMIESA-N tert-butyl n-[(2s)-1-hydroxypropan-2-yl]carbamate Chemical compound OC[C@H](C)NC(=O)OC(C)(C)C PDAFIZPRSXHMCO-LURJTMIESA-N 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 2
- 125000003944 tolyl group Chemical group 0.000 description 2
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 description 2
- 238000005292 vacuum distillation Methods 0.000 description 2
- 238000009834 vaporization Methods 0.000 description 2
- 230000008016 vaporization Effects 0.000 description 2
- LOPKSXMQWBYUOI-DTWKUNHWSA-N (1r,2s)-1-amino-2,3-dihydro-1h-inden-2-ol Chemical compound C1=CC=C2[C@@H](N)[C@@H](O)CC2=C1 LOPKSXMQWBYUOI-DTWKUNHWSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- ZPXAEESIPIUPTJ-MRVPVSSYSA-N (2s)-2-amino-2-(3-fluorophenyl)ethanol Chemical compound OC[C@@H](N)C1=CC=CC(F)=C1 ZPXAEESIPIUPTJ-MRVPVSSYSA-N 0.000 description 1
- MAXXCANBKVBAQQ-SBSPUUFOSA-N (2s)-2-amino-2-(4-methoxyphenyl)ethanol;hydrochloride Chemical compound Cl.COC1=CC=C([C@H](N)CO)C=C1 MAXXCANBKVBAQQ-SBSPUUFOSA-N 0.000 description 1
- ZGPPQPPTLTYYKV-DDWIOCJRSA-N (2s)-2-amino-2-[3-(trifluoromethyl)phenyl]ethanol;hydrochloride Chemical compound Cl.OC[C@@H](N)C1=CC=CC(C(F)(F)F)=C1 ZGPPQPPTLTYYKV-DDWIOCJRSA-N 0.000 description 1
- COXVPYKZDDKVRF-ULQDDVLXSA-N (4,4-difluorocyclohexyl)methyl N-[(2S)-4-methyl-1-oxo-1-[[(2S)-1-oxo-3-[(3S)-2-oxopyrrolidin-3-yl]propan-2-yl]amino]pentan-2-yl]carbamate Chemical compound CC(C)C[C@@H](C(=O)N[C@@H](C[C@@H]1CCNC1=O)C=O)NC(=O)OCC2CCC(CC2)(F)F COXVPYKZDDKVRF-ULQDDVLXSA-N 0.000 description 1
- TVCSALYWDGDRBY-GSVOUGTGSA-N (4R)-4-methyloxathiazolidine 2,2-dioxide Chemical compound C[C@@H]1COS(=O)(=O)N1 TVCSALYWDGDRBY-GSVOUGTGSA-N 0.000 description 1
- LEYGSKCNBPBVSJ-VKHMYHEASA-N (5s)-5-methyl-2,2-dioxooxathiazolidine-3-carboxylic acid Chemical compound C[C@H]1CN(C(O)=O)S(=O)(=O)O1 LEYGSKCNBPBVSJ-VKHMYHEASA-N 0.000 description 1
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N (e)-2-hydroxybut-2-enedioic acid Chemical compound OC(=O)\C=C(\O)C(O)=O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- ZXMGHDIOOHOAAE-UHFFFAOYSA-N 1,1,1-trifluoro-n-(trifluoromethylsulfonyl)methanesulfonamide Chemical compound FC(F)(F)S(=O)(=O)NS(=O)(=O)C(F)(F)F ZXMGHDIOOHOAAE-UHFFFAOYSA-N 0.000 description 1
- SMWUDAKKCDQTPV-UHFFFAOYSA-N 1,3-dimethylimidazolidine Chemical compound CN1CCN(C)C1 SMWUDAKKCDQTPV-UHFFFAOYSA-N 0.000 description 1
- HNAGHMKIPMKKBB-UHFFFAOYSA-N 1-benzylpyrrolidine-3-carboxamide Chemical compound C1C(C(=O)N)CCN1CC1=CC=CC=C1 HNAGHMKIPMKKBB-UHFFFAOYSA-N 0.000 description 1
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 1
- LDMOEFOXLIZJOW-UHFFFAOYSA-N 1-dodecanesulfonic acid Chemical compound CCCCCCCCCCCCS(O)(=O)=O LDMOEFOXLIZJOW-UHFFFAOYSA-N 0.000 description 1
- RTBFRGCFXZNCOE-UHFFFAOYSA-N 1-methylsulfonylpiperidin-4-one Chemical compound CS(=O)(=O)N1CCC(=O)CC1 RTBFRGCFXZNCOE-UHFFFAOYSA-N 0.000 description 1
- LNETULKMXZVUST-UHFFFAOYSA-N 1-naphthoic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=CC2=C1 LNETULKMXZVUST-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- 238000004293 19F NMR spectroscopy Methods 0.000 description 1
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Chemical group C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- CFTOTSJVQRFXOF-UHFFFAOYSA-N 2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole Chemical compound N1C2=CC=CC=C2C2=C1CNCC2 CFTOTSJVQRFXOF-UHFFFAOYSA-N 0.000 description 1
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 1
- KTVKQTNGWVJHFL-UHFFFAOYSA-N 2-ethylchromen-4-one Chemical compound C1=CC=C2OC(CC)=CC(=O)C2=C1 KTVKQTNGWVJHFL-UHFFFAOYSA-N 0.000 description 1
- 125000004777 2-fluoroethyl group Chemical group [H]C([H])(F)C([H])([H])* 0.000 description 1
- SDTMFDGELKWGFT-UHFFFAOYSA-N 2-methylpropan-2-olate Chemical compound CC(C)(C)[O-] SDTMFDGELKWGFT-UHFFFAOYSA-N 0.000 description 1
- KLLLJCACIRKBDT-UHFFFAOYSA-N 2-phenyl-1H-indole Chemical compound N1C2=CC=CC=C2C=C1C1=CC=CC=C1 KLLLJCACIRKBDT-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- 125000002774 3,4-dimethoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C(OC([H])([H])[H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- ADVPTQAUNPRNPO-REOHCLBHSA-N 3-sulfino-L-alanine Chemical compound OC(=O)[C@@H](N)C[S@@](O)=O ADVPTQAUNPRNPO-REOHCLBHSA-N 0.000 description 1
- MSFQEZBRFPAFEX-UHFFFAOYSA-N 4-methoxybenzenesulfonamide Chemical compound COC1=CC=C(S(N)(=O)=O)C=C1 MSFQEZBRFPAFEX-UHFFFAOYSA-N 0.000 description 1
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 1
- LCFDJWUYKUPBJM-UHFFFAOYSA-N 5-(trifluoromethyl)-1h-indole Chemical compound FC(F)(F)C1=CC=C2NC=CC2=C1 LCFDJWUYKUPBJM-UHFFFAOYSA-N 0.000 description 1
- MYTGFBZJLDLWQG-UHFFFAOYSA-N 5-chloro-1h-indole Chemical compound ClC1=CC=C2NC=CC2=C1 MYTGFBZJLDLWQG-UHFFFAOYSA-N 0.000 description 1
- DWAQDRSOVMLGRQ-UHFFFAOYSA-N 5-methoxyindole Chemical compound COC1=CC=C2NC=CC2=C1 DWAQDRSOVMLGRQ-UHFFFAOYSA-N 0.000 description 1
- YPKBCLZFIYBSHK-UHFFFAOYSA-N 5-methylindole Chemical compound CC1=CC=C2NC=CC2=C1 YPKBCLZFIYBSHK-UHFFFAOYSA-N 0.000 description 1
- LPYXADUZSWBHCT-UHFFFAOYSA-N 5-phenyl-1h-indole Chemical compound C=1C=C2NC=CC2=CC=1C1=CC=CC=C1 LPYXADUZSWBHCT-UHFFFAOYSA-N 0.000 description 1
- ONYNOPPOVKYGRS-UHFFFAOYSA-N 6-methylindole Natural products CC1=CC=C2C=CNC2=C1 ONYNOPPOVKYGRS-UHFFFAOYSA-N 0.000 description 1
- KGWPHCDTOLQQEP-UHFFFAOYSA-N 7-methylindole Chemical compound CC1=CC=CC2=C1NC=C2 KGWPHCDTOLQQEP-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 241000349731 Afzelia bipindensis Species 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 229910000809 Alumel Inorganic materials 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 102100029361 Aromatase Human genes 0.000 description 1
- 108010078554 Aromatase Proteins 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 1
- ZGXOMSXYLSVVSM-JOCHJYFZSA-N C1(=CC=CC=C1)C[C@H](CC1=C(NC2=CC=CC=C12)C1=CC=CC=C1)NC(OC(C)(C)C)=O Chemical compound C1(=CC=CC=C1)C[C@H](CC1=C(NC2=CC=CC=C12)C1=CC=CC=C1)NC(OC(C)(C)C)=O ZGXOMSXYLSVVSM-JOCHJYFZSA-N 0.000 description 1
- KBWMGFXBXOTZLO-LJQANCHMSA-N CC=1C=CC=C2C(=CNC=12)C[C@@H](CC1=CC=CC=C1)NC(OC(C)(C)C)=O Chemical compound CC=1C=CC=C2C(=CNC=12)C[C@@H](CC1=CC=CC=C1)NC(OC(C)(C)C)=O KBWMGFXBXOTZLO-LJQANCHMSA-N 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 1
- 229940083347 Cyclin-dependent kinase 4 inhibitor Drugs 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- DSLZVSRJTYRBFB-LLEIAEIESA-N D-glucaric acid Chemical compound OC(=O)[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O DSLZVSRJTYRBFB-LLEIAEIESA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- 230000004544 DNA amplification Effects 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 102220550450 Dihydropyrimidinase-related protein 2_Y59F_mutation Human genes 0.000 description 1
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 1
- ASMQGLCHMVWBQR-UHFFFAOYSA-N Diphenyl phosphate Chemical compound C=1C=CC=CC=1OP(=O)(O)OC1=CC=CC=C1 ASMQGLCHMVWBQR-UHFFFAOYSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 101150054472 HER2 gene Proteins 0.000 description 1
- 208000017891 HER2 positive breast carcinoma Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000882584 Homo sapiens Estrogen receptor Proteins 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- 238000004566 IR spectroscopy Methods 0.000 description 1
- QZRGKCOWNLSUDK-UHFFFAOYSA-N Iodochlorine Chemical compound ICl QZRGKCOWNLSUDK-UHFFFAOYSA-N 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- NHTMVDHEPJAVLT-UHFFFAOYSA-N Isooctane Chemical compound CC(C)CC(C)(C)C NHTMVDHEPJAVLT-UHFFFAOYSA-N 0.000 description 1
- 239000001358 L(+)-tartaric acid Substances 0.000 description 1
- 235000011002 L(+)-tartaric acid Nutrition 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- XVOYSCVBGLVSOL-REOHCLBHSA-N L-cysteic acid Chemical compound OC(=O)[C@@H](N)CS(O)(=O)=O XVOYSCVBGLVSOL-REOHCLBHSA-N 0.000 description 1
- XVOYSCVBGLVSOL-UHFFFAOYSA-N L-cysteine sulfonic acid Natural products OC(=O)C(N)CS(O)(=O)=O XVOYSCVBGLVSOL-UHFFFAOYSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 239000005639 Lauric acid Substances 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 239000012270 PD-1 inhibitor Substances 0.000 description 1
- 239000012668 PD-1-inhibitor Substances 0.000 description 1
- 239000012271 PD-L1 inhibitor Substances 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 238000006929 Pictet-Spengler synthesis reaction Methods 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 102220468699 Protein arginine N-methyltransferase 3_Y87F_mutation Human genes 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 238000004474 TOSS sequence Methods 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 1
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 1
- 238000000441 X-ray spectroscopy Methods 0.000 description 1
- 239000012391 XPhos Pd G2 Substances 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- WUEMMAGKFRXZGJ-UHFFFAOYSA-K [Au](Cl)(Cl)Cl.C1(CCCCC1)P(C1=C(C=CC=C1)C1=C(C=C(C=C1C(C)C)C(C)C)C(C)C)C1CCCCC1 Chemical compound [Au](Cl)(Cl)Cl.C1(CCCCC1)P(C1=C(C=CC=C1)C1=C(C=C(C=C1C(C)C)C(C)C)C(C)C)C1CCCCC1 WUEMMAGKFRXZGJ-UHFFFAOYSA-K 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 238000009098 adjuvant therapy Methods 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 238000007605 air drying Methods 0.000 description 1
- 229940050528 albumin Drugs 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 125000003172 aldehyde group Chemical group 0.000 description 1
- IAJILQKETJEXLJ-RSJOWCBRSA-N aldehydo-D-galacturonic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-RSJOWCBRSA-N 0.000 description 1
- 229960000548 alemtuzumab Drugs 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 125000005137 alkenylsulfonyl group Chemical group 0.000 description 1
- 150000003973 alkyl amines Chemical group 0.000 description 1
- 150000005215 alkyl ethers Chemical class 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 229940061720 alpha hydroxy acid Drugs 0.000 description 1
- 150000001280 alpha hydroxy acids Chemical class 0.000 description 1
- ADVPTQAUNPRNPO-UHFFFAOYSA-N alpha-amino-beta-sulfino-propionic acid Natural products OC(=O)C(N)CS(O)=O ADVPTQAUNPRNPO-UHFFFAOYSA-N 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 150000001408 amides Chemical group 0.000 description 1
- 150000001414 amino alcohols Chemical class 0.000 description 1
- JFCQEDHGNNZCLN-UHFFFAOYSA-N anhydrous glutaric acid Natural products OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- 239000000010 aprotic solvent Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 125000003828 azulenyl group Chemical group 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 238000003287 bathing Methods 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000005874 benzothiadiazolyl group Chemical group 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 125000006269 biphenyl-2-yl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C1=C(*)C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 239000007844 bleaching agent Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 125000003865 brosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1Br)S(*)(=O)=O 0.000 description 1
- OBNCKNCVKJNDBV-UHFFFAOYSA-N butanoic acid ethyl ester Natural products CCCC(=O)OCC OBNCKNCVKJNDBV-UHFFFAOYSA-N 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- FNAQSUUGMSOBHW-UHFFFAOYSA-H calcium citrate Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O.[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O FNAQSUUGMSOBHW-UHFFFAOYSA-H 0.000 description 1
- 239000001354 calcium citrate Substances 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 238000001460 carbon-13 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 229920001429 chelating resin Polymers 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 235000013330 chicken meat Nutrition 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- NVVXJVMOIGXUGC-UHFFFAOYSA-M chloropalladium(1+);dicyclohexyl-[2-[2,6-di(propan-2-yloxy)phenyl]phenyl]phosphane;2-phenylaniline Chemical compound [Pd+]Cl.NC1=CC=CC=C1C1=CC=CC=[C-]1.CC(C)OC1=CC=CC(OC(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 NVVXJVMOIGXUGC-UHFFFAOYSA-M 0.000 description 1
- RSLSVURFMXHEEU-UHFFFAOYSA-M chloropalladium(1+);dicyclohexyl-[3-[2,4,6-tri(propan-2-yl)phenyl]phenyl]phosphane;2-phenylaniline Chemical compound [Pd+]Cl.NC1=CC=CC=C1C1=CC=CC=[C-]1.CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC(P(C2CCCCC2)C2CCCCC2)=C1 RSLSVURFMXHEEU-UHFFFAOYSA-M 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 238000002591 computed tomography Methods 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 150000001879 copper Chemical class 0.000 description 1
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 1
- SFJMFSWCBVEHBA-UHFFFAOYSA-M copper(i)-thiophene-2-carboxylate Chemical compound [Cu+].[O-]C(=O)C1=CC=CS1 SFJMFSWCBVEHBA-UHFFFAOYSA-M 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 238000013480 data collection Methods 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 230000009849 deactivation Effects 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 231100000517 death Toxicity 0.000 description 1
- 125000004856 decahydroquinolinyl group Chemical group N1(CCCC2CCCCC12)* 0.000 description 1
- 239000008380 degradant Substances 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 239000001064 degrader Substances 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000004807 desolvation Methods 0.000 description 1
- 238000003795 desorption Methods 0.000 description 1
- 239000011903 deuterated solvents Substances 0.000 description 1
- GUJOJGAPFQRJSV-UHFFFAOYSA-N dialuminum;dioxosilane;oxygen(2-);hydrate Chemical compound O.[O-2].[O-2].[O-2].[Al+3].[Al+3].O=[Si]=O.O=[Si]=O.O=[Si]=O.O=[Si]=O GUJOJGAPFQRJSV-UHFFFAOYSA-N 0.000 description 1
- 125000002576 diazepinyl group Chemical group N1N=C(C=CC=C1)* 0.000 description 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-O diazynium Chemical group [NH+]#N IJGRMHOSHXDMSA-UHFFFAOYSA-O 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- JVSWJIKNEAIKJW-UHFFFAOYSA-N dimethyl-hexane Natural products CCCCCC(C)C JVSWJIKNEAIKJW-UHFFFAOYSA-N 0.000 description 1
- 125000000532 dioxanyl group Chemical group 0.000 description 1
- 125000005879 dioxolanyl group Chemical group 0.000 description 1
- 238000007907 direct compression Methods 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 150000004252 dithioacetals Chemical class 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 238000007908 dry granulation Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 238000002003 electron diffraction Methods 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000009261 endocrine therapy Methods 0.000 description 1
- 229940034984 endocrine therapy antineoplastic and immunomodulating agent Drugs 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 238000006345 epimerization reaction Methods 0.000 description 1
- 108700020302 erbB-2 Genes Proteins 0.000 description 1
- 201000007281 estrogen-receptor positive breast cancer Diseases 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-L ethanedisulfonate group Chemical group C(CS(=O)(=O)[O-])S(=O)(=O)[O-] AFAXGSQYZLGZPG-UHFFFAOYSA-L 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-N ethanedisulfonic acid Chemical compound OS(=O)(=O)CCS(O)(=O)=O AFAXGSQYZLGZPG-UHFFFAOYSA-N 0.000 description 1
- OCLXJTCGWSSVOE-UHFFFAOYSA-N ethanol etoh Chemical compound CCO.CCO OCLXJTCGWSSVOE-UHFFFAOYSA-N 0.000 description 1
- IDGUHHHQCWSQLU-UHFFFAOYSA-N ethanol;hydrate Chemical class O.CCO IDGUHHHQCWSQLU-UHFFFAOYSA-N 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 229960005167 everolimus Drugs 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 238000002509 fluorescent in situ hybridization Methods 0.000 description 1
- 238000000806 fluorine-19 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 229940083124 ganglion-blocking antiadrenergic secondary and tertiary amines Drugs 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 230000009477 glass transition Effects 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 125000004438 haloalkoxy group Chemical group 0.000 description 1
- 231100001261 hazardous Toxicity 0.000 description 1
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-M hexanoate Chemical compound CCCCCC([O-])=O FUZZWVXGSFPDMH-UHFFFAOYSA-M 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 150000007857 hydrazones Chemical class 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 239000001341 hydroxy propyl starch Substances 0.000 description 1
- 229920003063 hydroxymethyl cellulose Polymers 0.000 description 1
- 229940031574 hydroxymethyl cellulose Drugs 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 235000013828 hydroxypropyl starch Nutrition 0.000 description 1
- AUONNNVJUCSETH-UHFFFAOYSA-N icosanoyl icosanoate Chemical compound CCCCCCCCCCCCCCCCCCCC(=O)OC(=O)CCCCCCCCCCCCCCCCCCC AUONNNVJUCSETH-UHFFFAOYSA-N 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 238000009169 immunotherapy Methods 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 1
- 229930005303 indole alkaloid Natural products 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 230000002601 intratumoral effect Effects 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 229950006331 ipatasertib Drugs 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 229940045996 isethionic acid Drugs 0.000 description 1
- GJRQTCIYDGXPES-UHFFFAOYSA-N iso-butyl acetate Natural products CC(C)COC(C)=O GJRQTCIYDGXPES-UHFFFAOYSA-N 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- FGKJLKRYENPLQH-UHFFFAOYSA-M isocaproate Chemical compound CC(C)CCC([O-])=O FGKJLKRYENPLQH-UHFFFAOYSA-M 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- TWBYWOBDOCUKOW-UHFFFAOYSA-M isonicotinate Chemical compound [O-]C(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-M 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- OQAGVSWESNCJJT-UHFFFAOYSA-N isovaleric acid methyl ester Natural products COC(=O)CC(C)C OQAGVSWESNCJJT-UHFFFAOYSA-N 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 229910052743 krypton Inorganic materials 0.000 description 1
- DNNSSWSSYDEUBZ-UHFFFAOYSA-N krypton atom Chemical compound [Kr] DNNSSWSSYDEUBZ-UHFFFAOYSA-N 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000004973 liquid crystal related substance Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 1
- RYEXTBOQKFUPOE-UHFFFAOYSA-M magnesium;propane;chloride Chemical compound [Mg+2].[Cl-].CC[CH2-] RYEXTBOQKFUPOE-UHFFFAOYSA-M 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 229910052748 manganese Inorganic materials 0.000 description 1
- 239000011572 manganese Substances 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 229960001786 megestrol Drugs 0.000 description 1
- JBVNBBXAMBZTMQ-CEGNMAFCSA-N megestrol Chemical compound C1=CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)C)(O)[C@@]1(C)CC2 JBVNBBXAMBZTMQ-CEGNMAFCSA-N 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- GYNNXHKOJHMOHS-UHFFFAOYSA-N methyl-cycloheptane Natural products CC1CCCCCC1 GYNNXHKOJHMOHS-UHFFFAOYSA-N 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 238000001000 micrograph Methods 0.000 description 1
- 238000003801 milling Methods 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 150000004250 monothioacetals Chemical class 0.000 description 1
- 229910052901 montmorillonite Inorganic materials 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- LNOPIUAQISRISI-UHFFFAOYSA-N n'-hydroxy-2-propan-2-ylsulfonylethanimidamide Chemical compound CC(C)S(=O)(=O)CC(N)=NO LNOPIUAQISRISI-UHFFFAOYSA-N 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- XBXCNNQPRYLIDE-UHFFFAOYSA-M n-tert-butylcarbamate Chemical compound CC(C)(C)NC([O-])=O XBXCNNQPRYLIDE-UHFFFAOYSA-M 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 238000009099 neoadjuvant therapy Methods 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000004971 nitroalkyl group Chemical group 0.000 description 1
- 125000001736 nosyl group Chemical group S(=O)(=O)(C1=CC=C([N+](=O)[O-])C=C1)* 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 125000005060 octahydroindolyl group Chemical group N1(CCC2CCCCC12)* 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 201000004228 ovarian endometrial cancer Diseases 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 238000007833 oxidative deamination reaction Methods 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 125000005489 p-toluenesulfonic acid group Chemical group 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- 229960001972 panitumumab Drugs 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 229940121655 pd-1 inhibitor Drugs 0.000 description 1
- 229940121656 pd-l1 inhibitor Drugs 0.000 description 1
- 230000010412 perfusion Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 125000005561 phenanthryl group Chemical group 0.000 description 1
- 229960003424 phenylacetic acid Drugs 0.000 description 1
- 239000003279 phenylacetic acid Substances 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- LMRCKXYHPYNEJV-UHFFFAOYSA-N piperazine;piperidine Chemical compound C1CCNCC1.C1CNCCN1 LMRCKXYHPYNEJV-UHFFFAOYSA-N 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 238000007747 plating Methods 0.000 description 1
- 239000003495 polar organic solvent Substances 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 230000007425 progressive decline Effects 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 229940090181 propyl acetate Drugs 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000004929 pyrrolidonyl group Chemical group N1(C(CCC1)=O)* 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 238000006798 ring closing metathesis reaction Methods 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 229910052594 sapphire Inorganic materials 0.000 description 1
- 239000010980 sapphire Substances 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 238000013341 scale-up Methods 0.000 description 1
- 238000001878 scanning electron micrograph Methods 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 150000007659 semicarbazones Chemical class 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 229920002545 silicone oil Polymers 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000004332 silver Substances 0.000 description 1
- 238000004467 single crystal X-ray diffraction Methods 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 229960003885 sodium benzoate Drugs 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229940001607 sodium bisulfite Drugs 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 229960001790 sodium citrate Drugs 0.000 description 1
- 235000011083 sodium citrates Nutrition 0.000 description 1
- JVBXVOWTABLYPX-UHFFFAOYSA-L sodium dithionite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])=O JVBXVOWTABLYPX-UHFFFAOYSA-L 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 238000000371 solid-state nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- 238000007614 solvation Methods 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003456 sulfonamides Chemical class 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 238000004808 supercritical fluid chromatography Methods 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- UBECIPPDKSGYCX-LJQANCHMSA-N tert-butyl N-[(1R)-1-(3-fluorophenyl)-2-(1H-indol-3-yl)ethyl]carbamate Chemical compound FC=1C=C(C=CC=1)[C@@H](CC1=CNC2=CC=CC=C12)NC(OC(C)(C)C)=O UBECIPPDKSGYCX-LJQANCHMSA-N 0.000 description 1
- SHWIWXHVNDRTOQ-FQEVSTJZSA-N tert-butyl N-[(1S)-2-(1H-indol-3-yl)-1-(4-methoxyphenyl)ethyl]carbamate Chemical compound N1C=C(C2=CC=CC=C12)C[C@@H](C1=CC=C(C=C1)OC)NC(OC(C)(C)C)=O SHWIWXHVNDRTOQ-FQEVSTJZSA-N 0.000 description 1
- LEFGYJLWBARLFL-IBGZPJMESA-N tert-butyl N-[(1S)-2-(1H-indol-3-yl)-1-phenylethyl]carbamate Chemical compound N1C=C(C2=CC=CC=C12)C[C@@H](C1=CC=CC=C1)NC(OC(C)(C)C)=O LEFGYJLWBARLFL-IBGZPJMESA-N 0.000 description 1
- ISIHNHCDZFNAEN-IBGZPJMESA-N tert-butyl N-[(1S)-3-(1H-indol-3-yl)-1-phenylpropyl]carbamate Chemical compound N1C=C(C2=CC=CC=C12)CC[C@@H](C1=CC=CC=C1)NC(OC(C)(C)C)=O ISIHNHCDZFNAEN-IBGZPJMESA-N 0.000 description 1
- ZAAJZKJFFQVEOW-FXAWDEMLSA-N tert-butyl N-[(1S,2S)-2-(1H-indol-3-yl)-2,3-dihydro-1H-inden-1-yl]carbamate Chemical compound N1C=C(C2=CC=CC=C12)[C@H]1[C@@H](C2=CC=CC=C2C1)NC(OC(C)(C)C)=O ZAAJZKJFFQVEOW-FXAWDEMLSA-N 0.000 description 1
- UHWHJJHCBXGFAB-MRXNPFEDSA-N tert-butyl N-[(2R)-1-(1H-indol-3-yl)-3-methylbutan-2-yl]carbamate Chemical compound N1C=C(C2=CC=CC=C12)C[C@H](C(C)C)NC(OC(C)(C)C)=O UHWHJJHCBXGFAB-MRXNPFEDSA-N 0.000 description 1
- BCMQDTCTNOTZAM-GOSISDBHSA-N tert-butyl N-[(2R)-1-(1H-indol-3-yl)-3-phenylpropan-2-yl]carbamate Chemical compound N1C=C(C2=CC=CC=C12)C[C@@H](CC1=CC=CC=C1)NC(OC(C)(C)C)=O BCMQDTCTNOTZAM-GOSISDBHSA-N 0.000 description 1
- DPKWKRYAEWDPDW-GOSISDBHSA-N tert-butyl N-[(2R)-1-(2-methyl-1H-indol-3-yl)-3-phenylpropan-2-yl]carbamate Chemical compound CC=1NC2=CC=CC=C2C=1C[C@@H](CC1=CC=CC=C1)NC(OC(C)(C)C)=O DPKWKRYAEWDPDW-GOSISDBHSA-N 0.000 description 1
- XSNORKSVMMOZDI-XMMPIXPASA-N tert-butyl N-[(2R)-1-phenyl-3-(5-phenyl-1H-indol-3-yl)propan-2-yl]carbamate Chemical compound C1(=CC=CC=C1)C[C@H](CC1=CNC2=CC=C(C=C12)C1=CC=CC=C1)NC(OC(C)(C)C)=O XSNORKSVMMOZDI-XMMPIXPASA-N 0.000 description 1
- ZUUOLYIXQOXXIT-LLVKDONJSA-N tert-butyl N-[(2S)-2-(1H-indol-3-yl)propyl]carbamate Chemical compound N1C=C(C2=CC=CC=C12)[C@@H](CNC(OC(C)(C)C)=O)C ZUUOLYIXQOXXIT-LLVKDONJSA-N 0.000 description 1
- QSLMSOBCWZNNCH-UHFFFAOYSA-N tert-butyl carbamoperoxoate Chemical compound CC(C)(C)OOC(N)=O QSLMSOBCWZNNCH-UHFFFAOYSA-N 0.000 description 1
- YNJCFDAODGKHAV-UHFFFAOYSA-N tert-butyl n-(2-hydroxypropyl)carbamate Chemical compound CC(O)CNC(=O)OC(C)(C)C YNJCFDAODGKHAV-UHFFFAOYSA-N 0.000 description 1
- IBDIOGYTZBKRGI-LLVKDONJSA-N tert-butyl n-[(1s)-2-hydroxy-1-phenylethyl]carbamate Chemical compound CC(C)(C)OC(=O)N[C@H](CO)C1=CC=CC=C1 IBDIOGYTZBKRGI-LLVKDONJSA-N 0.000 description 1
- SMEMODSNLZWKBF-LBPRGKRZSA-N tert-butyl n-[(1s)-3-hydroxy-1-phenylpropyl]carbamate Chemical compound CC(C)(C)OC(=O)N[C@@H](CCO)C1=CC=CC=C1 SMEMODSNLZWKBF-LBPRGKRZSA-N 0.000 description 1
- PDAFIZPRSXHMCO-ZCFIWIBFSA-N tert-butyl n-[(2r)-1-hydroxypropan-2-yl]carbamate Chemical compound OC[C@@H](C)NC(=O)OC(C)(C)C PDAFIZPRSXHMCO-ZCFIWIBFSA-N 0.000 description 1
- BRGKFDMYQYKVHU-UHFFFAOYSA-N tert-butyl oxathiazolidine-3-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCOS1 BRGKFDMYQYKVHU-UHFFFAOYSA-N 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- BPEWUONYVDABNZ-DZBHQSCQSA-N testolactone Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(OC(=O)CC4)[C@@H]4[C@@H]3CCC2=C1 BPEWUONYVDABNZ-DZBHQSCQSA-N 0.000 description 1
- 229960005353 testolactone Drugs 0.000 description 1
- ZXLDQJLIBNPEFJ-UHFFFAOYSA-N tetrahydro-beta-carboline Natural products C1CNC(C)C2=C1C1=CC=C(OC)C=C1N2 ZXLDQJLIBNPEFJ-UHFFFAOYSA-N 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000005958 tetrahydrothienyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 238000006177 thiolation reaction Methods 0.000 description 1
- 229960005267 tositumomab Drugs 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 235000013337 tricalcium citrate Nutrition 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 230000005751 tumor progression Effects 0.000 description 1
- 229940005605 valeric acid Drugs 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 238000002460 vibrational spectroscopy Methods 0.000 description 1
- 238000005550 wet granulation Methods 0.000 description 1
- 238000010947 wet-dispersion method Methods 0.000 description 1
- 238000003963 x-ray microscopy Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D205/00—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom
- C07D205/02—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D205/04—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/14—Radicals substituted by nitrogen atoms, not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/14—Radicals substituted by nitrogen atoms, not forming part of a nitro radical
- C07D209/16—Tryptamines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
En el presente documento se proporcionan formas sólidas, sales tales como el compuesto B y formulaciones de 3-((1R,3R)-1-(2,6-difluoro-4-((1-(3-fluoropropil)azetidin-3-il)). amino)fenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol, procesos y síntesis de los mismos y métodos de su uso en el tratamiento del cáncer. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Formas sólidas de la sal de tartrato de 3-((1R,3R)-1-(2,6-difluoro-4-((1-(3-fluoropropil)acetidin-3-il)amino)fenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol, proceso para su preparación y procedimientos de su uso en el tratamiento de cánceres
Listado de secuencias
Esta solicitud incorpora por referencia un listado de secuencias enviado con esta solicitud como archivo de texto titulado P34807-WO_SL.txt creado el 12 de junio de 2019 y que tiene un tamaño de 16,4 kilobytes.
Campo de la invención
Se proporcionan en el presente documento formas sólidas de 3-((1R,3R)-1-(2,6-difluoro-4-((1-(3-fluoropropil)acetidin-3-il)amino)fenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol, y procedimientos de su uso en el tratamiento del cáncer. En el presente documento se describen además procesos para preparar compuestos tricíclicos fusionados que comprenden un resto fenilo o piridinilo sustituido.
Antecedentes
Los compuestos tricíclicos fusionados que comprenden un resto fenilo o piridinilo sustituido dentro del alcance de la presente divulgación son útiles como agentes dirigidos al receptor estrogénico ("RE").
El RE es una proteína reguladora transcripcional activada por ligando que media en la inducción de una variedad de efectos biológicos a través de su interacción con estrógenos endógenos. Los estrógenos endógenos incluyen 17β (beta)-estradiol y estronas. Se ha descubierto que el RE tiene dos isoformas, RE-α (alfa) y RE-β (beta). Los estrógenos y los receptores estrogénicos están implicados en una serie de enfermedades o afecciones, tales como cáncer de mama, cáncer de pulmón, cáncer ovárico, cáncer de colon, cáncer de próstata, cáncer endometrial, cáncer uterino, así como otras enfermedades o afecciones. Los agentes dirigidos a RE-α tienen actividad particular en el contexto de la enfermedad metastásica y la resistencia adquirida. Los agentes dirigidos a RE-α se divulgan en la publicación de EE. UU. número 2016/0175289.
Los procesos útiles para preparar compuestos tricíclicos fusionados que comprenden un resto fenilo o piridinilo sustituido se divulgan en la publicación de EE. UU. número 2016/0175289. Sin embargo, existe la necesidad de procesos mejorados para preparar agentes dirigidos a RE-α.
Existe una complejidad significativa que rodea la identificación y selección de una forma sólida de un compuesto farmacéutico. Las diferencias en las formas sólidas de dichos compuestos afectan a propiedades tanto físicas como químicas y pueden alterar el procesamiento, estabilidad, biodisponibilidad, formulación y almacenamiento de los compuestos farmacéuticos. No existe una predictibilidad fiable de la forma sólida y su utilidad como sólido cristalino o sólido amorfo. Los sólidos cristalinos se pueden considerar útiles, por ejemplo, para la estabilidad física o química, mientras que los sólidos amorfos se pueden considerar útiles, por ejemplo, para una disolución potenciada y una biodisponibilidad incrementada.
Las mezclas de materiales cristalinos surgen del polimorfismo. No es posible predecir, a priori, si existen formas cristalinas de un compuesto, mucho menos si se pueden preparar o aislar formas cristalinas. Jones et al., 2006, Pharmaceutical Cocrystals: An Emerging Approach to Physical Property Enhancement", MRS Bulletin 31: 875-879 (en la actualidad, en general no es posible predecir computacionalmente el número de polimorfos observables incluso de las moléculas más simples). El número de formas sólidas posibles da como resultado diferentes propiedades químicas y físicas para un compuesto farmacéutico y puede afectar en gran medida al desarrollo, estabilidad y comercialización del producto.
El receptor estrogénico ("RE") es una proteína reguladora transcripcional activada por ligando que media en la inducción de una variedad de efectos biológicos a través de su interacción con estrógenos endógenos. Los estrógenos endógenos incluyen 17β (beta)-estradiol y estronas. Se ha descubierto que el RE tiene dos isoformas, RE-α (alfa) y RE-β (beta). Los estrógenos y los receptores estrogénicos están implicados en una serie de enfermedades o afecciones, tales como cáncer de mama, cáncer de pulmón, cáncer ovárico, cáncer de colon, cáncer de próstata, cáncer endometrial, cáncer uterino, así como otras enfermedades o afecciones. Existe la necesidad de obtener nuevos agentes dirigidos a RE-α que tengan actividad en el contexto de una enfermedad metastásica y resistencia adquirida. En consecuencia, sigue existiendo la necesidad de tratamientos contra el cáncer que tengan formas sólidas particulares.
Sumario
Se proporcionan en el presente documento soluciones a los problemas anteriores y otros problemas en la técnica.
En un aspecto se proporciona en el presente documento un compuesto, compuesto B, que tiene el nombre tartrato de 3-((1R,3R)-1-(2,6-difluoro-4-((1-(3-fluoropropil)acetidin-3-il)amino)fenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol, como se describe en el presente documento.
En otro aspecto se proporciona en el presente documento una forma cristalina del compuesto B que tiene un patrón de difracción de rayos X en polvo que comprende picos en 19,32, 20,26, 21,63, 23,28 o 24,81 ± 0,1° 2θ (± 0,1° 2θ).
En otro aspecto se proporciona en el presente documento una forma cristalina del compuesto B que tiene un patrón de difracción de rayos X en polvo que comprende picos en 11,49, 12,54, 19,16, 19,42 o 24,67 ± 0,1° 2θ (± 0,1° 2θ).
En otro aspecto se proporciona en el presente documento una forma cristalina del compuesto B que tiene un patrón de difracción de rayos X en polvo sustancialmente como se muestra en la FIG.10 o la FIG.14.
En otro aspecto se proporciona en el presente documento una forma cristalina del compuesto B que tiene un patrón de difracción de rayos X en polvo que comprende picos en 11,31, 15,70, 16,54, 19,10 o 22,76 ± 0,1° 2θ.
En otro aspecto se proporciona en el presente documento una forma cristalina del compuesto B que tiene un patrón de difracción de rayos X en polvo que comprende picos en 12,52, 15,90, 19,66, 20,65 o 24,99 ± 0,1° 2θ.
En otro aspecto se proporciona en el presente documento una forma cristalina del compuesto B que tiene un patrón de difracción de rayos X en polvo que comprende picos en 11,46, 12,51, 19,29, 19,42 o 20,23 ± 0,1° 2θ.
También se divulga en el presente documento un sólido amorfo que comprende el compuesto A.
Se proporcionan además en el presente documento composiciones farmacéuticas que comprenden el compuesto B o una sal cristalina del mismo. Dichos compuestos y composiciones farmacéuticas se pueden usar en procedimientos para tratar cáncer como se expone en el presente documento.
También se divulga en el presente documento un proceso para preparar un compuesto de fórmula (IV) o una sal del mismo como se expone en el presente documento. El proceso comprende (1) hacer reaccionar una mezcla de reacción que comprende un compuesto de fórmula (I) como se describe en el presente documento, un disolvente orgánico y cloruro de tionilo para formar un compuesto de fórmula (IIa) como se describe en el presente documento y (2) hacer reaccionar una mezcla de reacción que comprende el compuesto de fórmula (IIa), un catalizador, un oxidante y un disolvente para formar un compuesto de fórmula (II) como se describe en el presente documento. El proceso comprende además hacer reaccionar una mezcla de reacción que comprende el compuesto de fórmula (II) y un compuesto de fórmula (III) como se describe en el presente documento en un disolvente orgánico para formar un compuesto de fórmula (IV) o una sal del mismo como se describe en el presente documento.
También se divulga en el presente documento un proceso para preparar un compuesto de fórmula (VIII) o una sal farmacéuticamente aceptable del mismo como se describe en el presente documento. El proceso comprende hacer reaccionar una mezcla de reacción que comprende un compuesto de fórmula (IV) como se describe en el presente documento, un compuesto de fórmula (V) como se describe en el presente documento o un compuesto de fórmula (X) como se describe en el presente documento, y un disolvente orgánico para formar un compuesto de fórmula (VI) como se describe en el presente documento. El proceso comprende además hacer reaccionar una mezcla de reacción que comprende el compuesto de fórmula (VI), un disolvente orgánico y un compuesto de fórmula (VII) como se describe en el presente documento o una sal del mismo para formar un compuesto de fórmula (VIII) o una sal del mismo.
También se divulga en el presente documento un proceso para preparar un compuesto de fórmula (VIII) o una sal farmacéuticamente aceptable del mismo como se describe en el presente documento. El proceso comprende hacer reaccionar una mezcla de reacción que comprende un compuesto de fórmula (IX) como se describe en el presente documento o un compuesto de fórmula (X) como se describe en el presente documento, un compuesto de fórmula (IV) como se describe en el presente documento y un disolvente orgánico para formar el compuesto de fórmula (VIII) o una sal del mismo como se describe en el presente documento.
También se divulga en el presente documento un proceso para preparar un compuesto de fórmula (IX) o una sal del mismo como se describe en el presente documento. El proceso comprende hacer reaccionar una mezcla de reacción que comprende un compuesto de fórmula (X) como se describe en el presente documento, un compuesto de fórmula (VII) o una sal del mismo, como se describe en el presente documento, un disolvente orgánico y un catalizador para formar un compuesto de fórmula (XI) o una sal del mismo como se describe en el presente documento.
También se divulga en el presente documento un proceso para preparar un compuesto de fórmula (III) o una sal del mismo como se describe en el presente documento. El proceso comprende hacer reaccionar una mezcla de
reacción que comprende un compuesto de fórmula (XII) como se describe en el presente documento, un compuesto B y un disolvente orgánico para formar el compuesto de fórmula (XIII) como se describe en el presente documento.
También se divulga en el presente documento un compuesto de fórmula (XVI) como se describe en el presente documento.
También se divulga en el presente documento un proceso para preparar un compuesto que tiene la fórmula (XX) que se proporciona, donde el proceso comprende poner en contacto un compuesto de fórmula (XXI) como se describe en el presente documento con una proteína transaminasa para formar un compuesto de fórmula (3). El compuesto de fórmula (3) se pone en contacto con un compuesto de fórmula (II) como se describe en el presente documento para formar el compuesto de fórmula (XX).
Breve descripción de las figuras
La FIG.1 representa el patrón de XRPD para el compuesto B forma A.
La FIG.2 representa el TGA y DSC para el compuesto B forma A.
La FIG.3 representa una imagen de PLM para el compuesto B forma A.
La FIG.4 representa el patrón de XRPD para el compuesto B forma B.
La FIG.5 representa el TGA y DSC para el compuesto B forma B.
La FIG.6 representa la RMNes de 13C para el compuesto B forma B.
La FIG.7 representa la RMNes de 19F para el compuesto B forma B.
La FIG.8 representa la curva de sorción/desorción de agua para el compuesto B forma B.
La FIG.9a representa la imagen de SEM; la FIG.9b representa la imagen de PLM para el compuesto B forma B; la FIG.9c representa la distribución del tamaño de partícula (PSD) para el compuesto B forma B.
La FIG. 10 representa un patrón de XRPD comparativo para el compuesto B forma C frente a la forma A y forma B del compuesto B. Se descubrió que la forma C era una mezcla de la forma A y la forma B.
La FIG.11 representa el TGA y DSC para el compuesto B forma C.
La FIG.12 representa el patrón de XRPD para el compuesto B forma D.
La FIG.13 representa el TGA y DSC para el compuesto B forma D.
La FIG.14 representa el patrón de XRPD para el compuesto B forma E.
La FIG.15 representa el TGA y DSC para el compuesto B forma E.
La FIG.16 representa el patrón de XRPD para el compuesto B forma F.
La FIG.17 representa la curva de DVS para el compuesto B forma F.
La FIG.18 representa la DSC para el compuesto B forma F.
La FIG.19 representa el patrón de XRPD para el compuesto B forma G.
La FIG.20 representa el TGA y DSC para el compuesto B forma G.
La FIG.21 representa la superposición del patrón de XRPD para el compuesto B forma A, forma B, forma C, forma D, forma F y forma G.
La FIG.22 representa la XRPD para las propiedades de compresibilidad del compuesto B forma B.
La FIG.23 representa la RMNes de 19F para las propiedades de compresibilidad del compuesto B forma B. La FIG.24 representa la DSC para las propiedades de compresibilidad para el compuesto B forma B.
La FIG.25 representa vías de transformación de fase para el compuesto B formas A, B, D y F.
La FIG.26 representa vías de transformación de fase para obtener el compuesto B de forma B a forma F.
La FIG.27a representa el patrón de XRPD para el compuesto C forma 1; la FIG.27b representa el patrón de XRPD para el compuesto C forma 2.
La FIG.28 representa el TGA y DSC para el compuesto C forma 1.
La FIG.29 representa una imagen de PLM para el compuesto C forma 1.
La FIG.30 representa el patrón de XRPD para el compuesto D forma M.
La FIG.31 representa el TGA y DSC para el compuesto D forma M.
La FIG.32 representa el patrón de XRPD para la forma amorfa del compuesto A.
La FIG.33 representa el TGA y DSC para la forma amorfa del compuesto A.
La FIG. 34 representa la viabilidad celular de la línea celular de cáncer de mama RE+ para el compuesto B en comparación con GDC-0810 y GDC-0927.
La FIG.35 representa el efecto sobre el volumen tumoral del compuesto B a 0,1 mg/kg y 1 mg/kg en comparación con GDC-0927 a 100 mg/kg.
La FIG.36a representa una TAC y la FIG.36b muestra el barrido FES-PET de una paciente con cáncer de mama tratada con el compuesto B.
La FIG.37a representa una TAC y la FIG. 37b muestra el barrido FES-PET de una segunda paciente con cáncer de mama tratada con el compuesto B.
Descripción detallada
A menos que se defina de otro modo, todos los términos técnicos y científicos usados en el presente documento tienen el mismo significado que se entiende comúnmente por los expertos en la técnica a la que pertenece la invención. Véanse, por ejemplo, Singleton et al., DICTIONARY OF MICROBIOLOGY AND MOLECULAR BIOLOGY 2.ª ed., J. Wiley & Sons (New York, NY 1994); Sambrook et al., MOLECULAR CLONING, A LABORATORY MANUAL, Cold Springs Harbor Press (Cold Springs Harbor, NY 1989). Cualquier procedimiento, dispositivo y material similar o equivalente a los descritos en el presente documento se puede usar en la práctica de la presente invención.
Las siguientes definiciones se proporcionan para facilitar el entendimiento de determinados términos usados con frecuencia en el presente documento y no están destinadas a limitar el alcance de la presente divulgación.
Como se usa en el presente documento, y a menos que se especifique de otro modo, el término "aproximadamente", cuando se refiere a dosis, cantidades o porcentajes en peso de ingredientes de una composición o una forma de dosificación, quiere decir una dosis, cantidad o porcentaje en peso que se reconoce por un experto en la técnica que proporciona un efecto farmacológico equivalente al obtenido a partir de la dosis, cantidad o porcentaje en peso especificado. La dosis, cantidad o porcentaje en peso equivalente puede estar dentro de un 30 %, 20 %, 15 %, 10 %, 5 %, 1 % o menos de la dosis, cantidad o porcentaje en peso especificado. Como se usa en el presente documento, y a menos que se especifique de otro modo, el término "aproximadamente", cuando se refiere a un valor numérico o intervalo de valores usado para la caracterización de una forma sólida particular descrita en el presente documento (por ejemplo, valores de picos de XRPD) indica que el valor o intervalo de valores se puede desviar de un valor dado en una medida que se considere razonable para un experto en la técnica mientras todavía describe la forma sólida. En un modo de realización, el valor de una posición de pico de XRPD puede variar hasta ± 0,1° 2θ (o ± 0,05 grados 2θ) mientras todavía describe el pico de XRPD particular.
Como se usa en el presente documento, y a menos que se especifique de otro modo, un cristalino que es "puro", es decir, sustancialmente libre de otros sólidos cristalinos o amorfos u otros compuestos químicos, y contiene menos de aproximadamente un 10 %, 9 %, 8 %, 7 %, 6 %, 5 %, 4 %, 3 %, 2 %, 1 %, 0,5 %, 0,4 %, 0,3 %, 0,2 %, 0,1 %, 0,05 % o 0,01 % de una o más de otras formas sólidas en una base en peso. La detección de otras formas sólidas se puede lograr, por ejemplo, por análisis de difracción, análisis térmico, análisis de combustión elemental y/o análisis espectroscópico. La detección de otros compuestos químicos se puede lograr, por ejemplo, por análisis de espectrometría de masas, análisis espectroscópico, análisis térmico, análisis de combustión elemental y/o
análisis cromatográfico.
A menos que se especifique de otro modo, los términos "solvato" y "solvatado", como se usa en el presente documento, se refieren a una forma sólida de una sustancia que contiene disolvente. Los términos "hidrato" e "hidratado" se refieren a un solvato en el que el disolvente es agua. Los términos "solvato" y "solvatado", como se usa en el presente documento, también se pueden referir a un solvato de una sal, cocristal o complejo molecular. Los términos "hidrato" e "hidratado", como se usa en el presente documento, también se pueden referir a un hidrato de una sal, cocristal o complejo molecular.
El término "farmacéuticamente aceptable" se refiere a un diluyente, excipiente o vehículo en una formulación compatible con el/los otro(s) ingrediente(s) de la formulación y no perjudicial para el receptor de la misma.
El compuesto A se refiere al compuesto que tiene la estructura:

y que tiene el nombre 3-((1R,3R)-1-(2,6-difluoro-4-((1-(3-fluoropropil)acetidin-3-il)amino)fenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol, incluyendo una sal farmacéuticamente aceptable del mismo. El compuesto A puede ser una sal de ácido tartárico como se describe en el presente documento (por ejemplo, compuesto B). El compuesto A puede ser una sal de ácido fumárico como se describe en el presente documento (por ejemplo, compuesto C). El compuesto A puede ser una sal de malonato como se describe en el presente documento (por ejemplo, compuesto D).
El término "forma sólida" se refiere a una forma física que no está predominantemente en estado líquido o gaseoso. Una forma sólida puede ser una forma cristalina o una mezcla de las mismas. En determinados modos de realización, una forma sólida puede ser un cristal líquido. En determinados modos de realización, la forma sólida del compuesto A es la forma A, forma B, forma C, forma D, forma E, forma F, forma G, forma 1 o forma 2, un sólido amorfo o una mezcla de los mismos. En un modo de realización, la forma sólida del compuesto A es una sal de tartrato. En otro modo de realización, la forma sólida del compuesto A es una sal de fumarato o una mezcla de las mismas. Una forma sólida puede ser una forma cristalina como se define en el presente documento.
El término "forma cristalina" se refiere a una forma sólida que es cristalina. En determinados modos de realización, una forma cristalina de un compuesto descrito en el presente documento puede estar sustancialmente libre de sólidos amorfos y/u otras formas cristalinas. En determinados modos de realización, una forma cristalina de un compuesto descrito en el presente documento puede contener menos de aproximadamente un 1 %, menos de aproximadamente un 2 %, menos de aproximadamente un 3 %, menos de aproximadamente un 4 %, menos de aproximadamente un 5 %, menos de aproximadamente un 6 %, menos de aproximadamente un 7 %, menos de aproximadamente un 8 %, menos de aproximadamente un 9 %, menos de aproximadamente un 10 %, menos de aproximadamente un 15 %, menos de aproximadamente un 20 %, menos de aproximadamente un 25 %, menos de aproximadamente un 30 %, menos de aproximadamente un 35 %, menos de aproximadamente un 40 %, menos de aproximadamente un 45 % o menos de aproximadamente un 50 % en peso de uno o más sólidos amorfos y/u otras formas cristalinas. En determinados modos de realización, una forma cristalina descrita en el presente documento es pura. En determinados modos de realización, una forma cristalina de un compuesto descrito en el presente documento puede ser aproximadamente un 99 %, 98 %, 97 %, 96 %, 95 %, 94 %, 93 %, 92 %, 91 % o 90 % pura.
El término "amorfo" o "sólido amorfo" se refiere a una forma sólida que no es sustancialmente cristalina como se determina por difracción de rayos X. En particular, el término "sólido amorfo" describe una forma sólida desordenada, es decir, una forma sólida que carece de orden cristalino de largo alcance. En determinados modos de realización, un sólido amorfo de un compuesto descrito en el presente documento puede estar sustancialmente libre de otros sólidos amorfos y/o formas cristalinas. En determinados modos de realización, un sólido amorfo puede ser puro. En determinados modos de realización, un sólido amorfo de un compuesto descrito en el presente documento puede ser aproximadamente un 99 %, 98 %, 97 %, 96 %, 95 %, 94 %, 93 %, 92 %, 91 % o 90 % puro. "Tratar", como se usa en el presente documento, quiere decir un alivio, en su totalidad o en parte, de un trastorno, enfermedad o afección, o uno o más de los síntomas asociados con un trastorno, enfermedad o afección, o
ralentización o detención de la progresión o empeoramiento adicional de esos síntomas, o aliviar o erradicar la(s) causa(s) del trastorno, enfermedad o afección en sí. En un modo de realización, el trastorno es un cáncer.
El término "cantidad eficaz" o "cantidad terapéuticamente eficaz" se refiere a una cantidad de un compuesto descrito en el presente documento que puede tratar o prevenir un trastorno, enfermedad o afección, o síntomas de los mismos, divulgados en el presente documento.
"Paciente" o "sujeto" se define en el presente documento para incluir animales, tales como mamíferos, incluyendo pero sin limitarse a, primates (por ejemplo, seres humanos), vacas, ovejas, cabras, caballos, perros, gatos, conejos, ratas, ratones, monos, pollos, pavos, codornices o cobayas y similares, en un modo de realización un mamífero, en otro modo de realización un ser humano. En un modo de realización, un sujeto es un ser humano que tiene o está en riesgo de tener cáncer.
Como se usa en el presente documento, los términos "resto" y "sustituyente" se refieren a un átomo o grupo de átomos enlazados químicamente que se une a otro átomo o molécula por uno o más enlaces químicos formando de este modo parte de una molécula.
Como se usa en el presente documento, el término "alquilo" se refiere a un resto de hidrocarburo saturado de cadena lineal o de cadena ramificada alifático que tiene de 1 a 20 átomos de carbono. En modos de realización particulares, el alquilo tiene de 1 a 10 átomos de carbono. En modos de realización particulares, el alquilo tiene de 1 a 6 átomos de carbono. Los grupos alquilo pueden estar opcionalmente sustituidos independientemente con uno o más sustituyentes descritos en el presente documento.
Como se usa en el presente documento, el término "sustituido" se refiere al reemplazo de al menos uno de los átomos de hidrógeno de un compuesto o resto con otro sustituyente o resto. Los ejemplos de dichos sustituyentes incluyen, sin limitación, halógeno, -OH, -CN, oxo, alcoxi, alquilo, alquileno, arilo, heteroarilo, haloalquilo, haloalcoxi, cicloalquilo y heterociclo. Por ejemplo, el término "haloalquilo" se refiere al hecho de que uno o más átomos de hidrógeno de un alquilo (como se define a continuación) se reemplazan por uno o más átomos de halógeno (por ejemplo, trifluorometilo, difluorometilo, fluorometilo, clorometilo, etc.). En un modo de realización, sustituido como se usa en el presente documento se puede referir al reemplazo de al menos un átomo de hidrógeno de un compuesto o resto descrito en el presente documento por halógeno o alquilo.
Como se usa en el presente documento, el término "alquileno" como se usa en el presente documento se refiere a un radical de hidrocarburo divalente saturado lineal o ramificado de uno a doce átomos de carbono, y en otro aspecto de uno a seis átomos de carbono, en el que el radical alquileno puede estar opcionalmente sustituido independientemente con uno o más sustituyentes descritos en el presente documento. Los ejemplos incluyen, pero no se limitan a, metileno, etileno, propileno, 2-metilpropileno, pentileno y similares.
Como se usa en el presente documento, el término "alcoxi" se refiere a un grupo de fórmula -O-R', en la que R' es un grupo alquilo. Los grupos alcoxi pueden estar opcionalmente sustituidos independientemente con uno o más sustituyentes descritos en el presente documento. Los ejemplos de restos alcoxi incluyen metoxi, etoxi, isopropoxi y terc-butoxi.
Como se usa en el presente documento, el término "arilo" se refiere a un resto de hidrocarburo aromático cíclico que tiene un anillo aromático mono, bi o tricíclico de 5 a 16 átomos de anillo de carbono. Los sistemas de anillo de arilo bicíclico incluyen bicíclicos fusionados que tienen dos anillos de arilo de cinco miembros fusionados (indicados como 5-5), que tienen un anillo de arilo de cinco miembros y un anillo de arilo de seis miembros fusionado (indicados como 5-6 y como 6-5), y que tiene dos anillos de arilo de seis miembros fusionados (indicados como 66). El grupo arilo puede estar opcionalmente sustituido como se define en el presente documento. Los ejemplos de restos arilo incluyen, pero no se limitan a, fenilo, naftilo, fenantrilo, fluorenilo, indenilo, pentalenilo, azulenilo y similares. El término "arilo" también incluye derivados parcialmente hidrogenados del resto de hidrocarburo aromático cíclico siempre que al menos un anillo del resto de hidrocarburo aromático cíclico sea aromático, estando cada uno opcionalmente sustituido.
Como se usa en el presente documento, el término "heteroarilo" se refiere a un sistema de anillo mono, bi o tricíclico heterocíclico aromático de 5 a 16 átomos de anillo, que comprende 1, 2, 3 o 4 heteroátomos seleccionados de N, O y S, siendo los átomos de anillo restantes carbono. En algunos modos de realización, los anillos heteroarilo monocíclicos pueden tener 5-6 miembros. Los sistemas de anillo heteroarilo bicíclico incluyen bicíclicos condensados que tienen dos anillos heteroarilo de cinco miembros fusionados (indicados como 5-5), que tienen un anillo heteroarilo de cinco miembros y un anillo heteroarilo de seis miembros fusionado (indicados como 5-6 y 6-5) y que tienen dos anillos heteroarilo de seis miembros fusionados (indicados como 6-6). El grupo heteroarilo puede estar opcionalmente sustituido como se define en el presente documento. Los ejemplos de restos heteroarilo incluyen pirrolilo, furanilo, tienilo, imidazolilo, oxazolilo, tiazolilo, triazolilo, oxadiazolilo, tiadiazolilo, tetrazolilo, piridinilo, piracinilo, pirazolilo, piridacinilo, pirimidinilo, triacinilo, isoxazolilo, benzofuranilo, isotiazolilo, benzotienilo, benzotiofenilo, indolilo, aza-indolilo, isoindolilo, isobenzofuranilo, bencimidazolilo, benzoxazolilo, benzoisoxazolilo, benzotiazolilo, benzoisotiazolilo, benzooxadiazolilo, benzotiadiazolilo, benzotriazolilo, purinilo, quinolinilo,
isoquinolinilo, quinazolinilo, quinoxalinilo, pirrolopiridinilo, furopiridinilo, tienopiridinilo, pirrolopiridacinilo, pirrolopirimidinilo, pirrolopiracinilo, tienopiridacinilo, tienopirimidinilo, tienopiracinilo, furopiridacinilo, furopirimidinilo y furopiracinilo.
Como se usa en el presente documento, los términos "halo", "halógeno" y "haluro", que se pueden usar de manera intercambiable, se refieren a un sustituyente flúor, cloro, bromo o yodo.
Como se usa en el presente documento, el término "haloalquilo" se refiere a un grupo alquilo en el que uno o más de los átomos de hidrógeno del grupo alquilo se han reemplazado por átomos de halógeno iguales o diferentes, en particular átomos de flúor y/o cloro. Los ejemplos de haloalquilo incluyen monofluoro-, difluoro- o trifluoro-metilo, -etilo o -propilo, por ejemplo, 3,3,3-trifluoropropilo, 2-fluoroetilo, 2,2,2-trifluoroetilo, fluorometilo, difluorometilo o trifluorometilo.
Como se usa en el presente documento, el término "hidroxialquilo" se refiere a un grupo alquilo en el que uno o más de los átomos de hidrógeno del grupo alquilo se han reemplazado por un resto hidroxilo. Los ejemplos incluyen alcoholes y dioles
Como se usa en el presente documento, el término "heteroalquilo" se refiere a un alquilo de cadena lineal o ramificada como se define en el presente documento que tiene de 2 a 14 carbonos, de 2 a 10 carbonos o de 2 a 6 carbonos en la cadena, de los que uno o más se ha reemplazado por un heteroátomo seleccionado de S, O, P y N. Los ejemplos no limitantes de heteroalquilos incluyen éteres de alquilo, aminas de alquilo secundarias y terciarias, amidas y sulfuros de alquilo.
Como se usa en el presente documento, el término "cicloalquilo" quiere decir un resto carbocíclico saturado o parcialmente insaturado que tiene anillos mono, bi (incluyendo bicíclicos con puente) o tricíclicos y de 3 a 10 átomos de carbono en el anillo. El resto cicloalquilo puede estar opcionalmente sustituido con uno o más sustituyentes. En modos de realización particulares, el cicloalquilo contiene de 3 a 8 átomos de carbono (es decir, cicloalquilo (C3C8)). En otros modos de realización particulares, el cicloalquilo contiene de 3 a 6 átomos de carbono (es decir, cicloalquilo (C3-C6)). Los ejemplos de restos cicloalquilo incluyen, pero no se limitan a, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo y derivados parcialmente insaturados (cicloalquenilo) de los mismos (por ejemplo, ciclopentenilo, ciclohexenilo y cicloheptenilo), biciclo[3.1.0]hexanilo, biciclo[3.1.0]hexenilo, biciclo[3.1.1]heptanilo y biciclo[3.1.1]heptenilo. El resto cicloalquilo se puede unir en forma de "espirocicloalquilo" tal como "espirociclopropilo":

Como se usa en el presente documento, los términos "heterociclo" o "heterociclilo" se refieren a un resto heterocíclico monocíclico de 4, 5, 6 y 7 miembros, bicíclico de 7, 8, 9 y 10 miembros (incluyendo bicíclico con puente) o bicíclico de 10, 11, 12, 13, 14 y 15 miembros que está saturado o parcialmente insaturado y tiene uno o más (por ejemplo, 1, 2, 3 o 4 heteroátomos seleccionados de oxígeno, nitrógeno y azufre en el anillo, siendo los átomos de anillo restantes carbono. En algunos modos de realización, el heterociclo es un heterocicloalquilo. En modos de realización particulares, heterociclo o heterociclilo se refiere a un heterociclo de 4, 5, 6 o 7 miembros. Cuando se usa en referencia a un átomo de anillo de un heterociclo, un nitrógeno o azufre también pueden estar en forma oxidada, y un nitrógeno puede estar sustituido con uno o más alquilo (C1−C6) o grupos. El heterociclo se puede unir a su grupo lateral en cualquier heteroátomo o átomo de carbono que dé como resultado una estructura estable. Cualquiera de los átomos de anillo heterociclo puede estar opcionalmente sustituido con uno o más sustituyentes descritos en el presente documento. Los ejemplos de dichos heterociclos saturados o parcialmente insaturados incluyen, sin limitación, tetrahidrofuranilo, tetrahidrotienilo, pirrolidinilo, pirrolidonilo, piperidinilo, pirrolinilo, tetrahidroquinolinilo, tetrahidroisoquinolinilo, decahidroquinolinilo, oxazolidinilo, piperacinilo, dioxanilo, dioxolanilo, diacepinilo, oxacepinilo, tiacepinilo, morfolinilo y quinuclidinilo. El término heterociclo también incluye grupos en los que un heterociclo está fusionado a uno o más anillos arilo, heteroarilo o cicloalquilo, tales como indolinilo, 3H-indolilo, cromanilo, azabiciclo[2.2.1]heptanilo, azabiciclo[3.1.0]hexanilo, azabiciclo[3.1.1]heptanilo, octahidroindolilo o tetrahidroquinolinilo.
A menos que se indique de otro modo, el término "hidrógeno" o "hidro" se refiere al resto de un átomo de hidrógeno (-H) y no a H2.
Como se usa en el presente documento, el término "disolvente orgánico" se refiere a cualquier disolvente aprótico polar no acuoso, disolvente prótico polar y disolvente apolar.
Como se usa en el presente documento, el término "disolvente orgánico polar" se refiere tanto a disolventes apróticos polares como a disolventes próticos polares, excluyendo el agua.
Como se usa en el presente documento, el término "disolvente aprótico polar" se refiere a cualquier disolvente polar que no tiene capacidad de donar protones. Los ejemplos incluyen, sin ninguna limitación, 2metiltetrahidrofurano, tetrahidrofurano, acetato de etilo, acetato de propilo (por ejemplo, acetato de isopropilo), acetona, dimetilsulfóxido, N,N-dimetilformamida, acetonitrilo, N,N-dimetilacetamida, N-metilpirrolidona, hexametilfosforamida y carbonato de propileno.
Como se usa en el presente documento, el término "disolvente prótico polar" se refiere a cualquier disolvente polar que tiene capacidad de donar protones. Los ejemplos incluyen, sin limitación, metanol, etanol, 1-propanol, 2-propanol, 1-butanol, ácido fórmico, nitrometano y ácido acético. Un disolvente prótico polar orgánico excluye cualquier cantidad eficaz de agua.
Como se usa en el presente documento, el término "disolvente apolar" se refiere a disolventes que contienen enlaces entre átomos con electronegatividades similares, tales como carbono e hidrógeno, de modo que la carga eléctrica en la molécula se distribuye uniformemente. Los disolventes apolares se caracterizan por tener una constante dieléctrica baja. Los ejemplos incluyen, sin limitación, pentano, hexano, heptano, ciclopentano, éter metil-terc-butílico (MTBE), éter dietílico, tolueno, benceno, 1,4-dioxano, tetracloruro de carbono, cloroformo y diclorometano (DCM). En algunos modos de realización, el disolvente apolar tiene una constante dieléctrica menor de 2, del que sus ejemplos incluyen, sin limitación, pentano, hexano y heptano. En comparación con otros disolventes apolares, el DCM presenta cierto grado de polaridad a nivel de enlace (es decir, entre carbono y cloro), pero solo un pequeño grado de polaridad a nivel molecular debido a la cancelación de la polaridad basada en la simetría.
Como se usa en el presente documento, el término "antidisolvente" se refiere a un disolvente en el que el compuesto de referencia es poco soluble y que induce la precipitación o cristalización de dicho compuesto de la solución.
Como se usa en el presente documento, el término "catalizador ácido" se refiere a un catalizador ácido tal como, pero sin limitarse a, un ácido de Brönsted, un ácido de Lewis o un catalizador de Brönsted-Lowry. Los ejemplos no limitantes de catalizadores ácidos incluyen ácido acético, ácido acético glacial, ácido trifluoroacético, ácido benzoico, ácido piválico, ácido difenilfosfórico, ácido tríflico, ácido fórmico, ácido tartárico, ácido fumárico, ácido malónico, ácido salicílico, ácido p-toluenosulfónico, ácido sulfúrico, ácido clorhídrico, ácido fosfórico, ácido metanosulfónico, ácido alcanforsulfónico, ácido naftalenosulfónico, montmorillonita K-10 a base de arcilla y amberlyst a base de resina, y combinaciones de los mismos.
Como se usa en el presente documento, el término "grupo protector de amina" se refiere a cualquier grupo protector conocido que bloquea o protege la funcionalidad de las aminas. Los grupos protectores de amina dentro del alcance de la divulgación incluyen, sin limitación, carbamato de 1-cloroetilo (ACD); 4-metoxibencenosulfonamida; acetamida (Ac); bencilamina (Bn); carbamato de benciloxi (CBz); formamida; carbamato de metilo; trifluoroacetamida; carbamato de terc-butoxi (Boc); p-metoxibencilcarbonilo (MeOZ); 9-fluorenilmetoxicarbonilo (FMOC); bezoílo (Bz); p-metoxibencilo (PMB); 3,4-dimetoxibencilo (DMPM); p-metoxifenilo (PMP); tosilo (Ts); y cloroformiato de tricloroetilo (Troc). Para obtener una descripción de los grupos protectores de amina y su uso, véase P. G. M. Wuts y T. W. Greene, Greene's Protective Groups in Organic Synthesis 4.a edición, Wiley-Interscience, New York, 2006.
Como se usa en el presente documento, el término "grupo protector de aldehído" se refiere a cualquier sustituyente conocido unido a un grupo aldehído que bloquea o protege el grupo carbonilo de la funcionalidad aldehído. Los grupos protectores adecuados de la funcionalidad aldehído incluyen, pero no se limitan a (a) acetales y cetales cíclicos, (b) mono o ditioacetales o cetales cíclicos u otros derivados tales como iminas, hidrazonas, cianohidrina, oximas o semicarbazonas, por ejemplo, dialquil o diarilacetales o 1,3-ditiano, (c) iminas cíclicas tales como derivados de metileno sustituidos o N,N'-dimetilimidazolidina. Algunos ejemplos no limitantes de grupos protectores de aldehído incluyen 1,3-ditiano, 1,3-ditiolano, dietilacetal, dimetilacetal, etilenglicolacetal, neopentilglicolacetal, trimetilsililcianhidrina y ortoformiatos de trialquilo tales como ortoformiato de trietilo. Para una descripción de los grupos protectores de aldehído y su uso, véase Wuts y Greene.
Como se usa en el presente documento, "grupo saliente" se refiere a un átomo o un grupo de átomos que se desplaza en una reacción química como especie estable. Los grupos salientes adecuados son bien conocidos en la técnica, por ejemplo, véanse, March's Advanced Organic Chemistry, 5.ª ed., Ed.: Smith, M. B. y March, J., John Wiley & Sons, New York: 2001 y T. W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991. Dichos grupos salientes incluyen, pero no se limitan a, restos halógeno, alcoxi, sulfoniloxi, alquilsulfonilo opcionalmente sustituido, alquenilsulfonilo opcionalmente sustituido, arilsulfonilo opcionalmente sustituido y diazonio. Los ejemplos de algunos grupos salientes incluyen cloro, yodo, bromo, fluoro, metanosulfonilo (mesilo), tosilo, triflato, nitro-fenilsulfonilo (nosilo) y bromo-fenilsulfonilo (brosilo).
Los "catalizadores de metales de transición" dentro del alcance de la divulgación incluyen, sin limitación, catalizadores de paladio, platino, oro, rutenio, rodio e iridio. Los ejemplos no limitantes de catalizadores adecuados incluyen: cloruro de (2-bifenil)di-terc-butilfosfina-oro(I) ("JohnPhos"), cloruro de 2-diciclohexilfosfino-2',4',6'-triisopropilbifenil-oro(I) ("XPhos AuCl"), bis(trifluorometanosulfonil)imida de 2-diciclohexilfosfino-2',4',6'-triisopropilbifenil-oro(I) ("XPhos AuNTf2"), cloro(2-diciclohexilfosfino-2',4',6'-triisopropil-1,1'-bifenil)[2-(2-aminoetil)fenil)]paladio(II) ("XPhos Palladacicle"), aducto de éter metil-t-butílico y cloro(2-diciclohexilfosfino-2',6'-dimetoxi-1,1'-bifenil)[2-(2-aminoetilfenil)]paladio(II) ("SPhos Palladacicle"), cloruro de fenetilamina de t-BuXPhospaladio(II) ("tBuXPhos Pd G1"), cloro(2-diciclohexilfosfino-2',4',6'-triisopropil-1,1'-bifenil)[2-(2'-amino-1,1'-bifenil)]paladio(II) ("Xphos Pd G2"), cloro(2-diciclohexilfosfino-2',6'-dimetoxi-1,1'-bifenil)[2-(2'-amino-1,1'-bifenil)]paladio(II) ("SPhos Pd G2"), cloro(2-diciclohexilfosfino-2',6'-diisopropoxi-1,1'-bifenil)[2-(2'-amino-1,1'-bifenil)]paladio(II) ("RuPhos Pd G2"), cloro[(2-diciclohexilfosfino-2',6'-bis(N,N-dimetilamino)-1,1'-bifenil)-2-(2'-amino-1,1'-bifenil)]paladio(II) ("CPhos-Pd-G2"), metanosulfonato de [(2-diciclohexilfosfino-2',6'-bis(N,N-dimetilamino)-1,1'-bifenil)-2-(2'-amino-1,1'-bifenil)]paladio(II) ("CPhos-Pd-G3"), metanosulfonato de [(2-di-terc-butilfosfino-2',4',6'-triisopropil-1,1'-bifenil)-2-(2'-amino-1,1'-bifenil)]paladio(II) ("tBuXPhos-Pd-G3"), metanosulfonato de (2-diciclohexilfosfino-2',6'-diisopropoxi-1,1'-bifenil)[2-(2'-amino-1,1'-bifenil)]paladio(II) ("RuPhos-Pd-G3"), metanosulfonato de (2-diciclohexilfosfino-2',4',6'-triisopropil-1,1'-bifenil)[2-(2'-amino-1,1'-bifenil)]paladio(II) ("XPhos-Pd-G3"), metanosulfonato de [(2-di-ciclohexilfosfino-3,6-dimetoxi-2',4',6'-triisopropil-1,1'-bifenil)-2-(2'-amino-1,1'-bifenil)]paladio(II) ("BrettPhos-Pd-G3"), metanosulfonato de [(2-{bis[3,5-bis(trifluorometil)fenil]fosfina-3,6-dimetoxi-2',4',6'-triisopropil-1,1'-bifenil)-2-(2'-amino-1,1'-bifenil)]paladio(II) ("JackiePhos-Pd-G3"), terc-butil-BrettPhos-Pd-G3, [terc-butil-BrettPhos-Pd (alil)]OTf) y combinaciones de los mismos.
Como se usa en el presente documento, "ácidos inorgánicos" se refiere a ácidos tales como, pero sin limitarse a, ácido clorhídrico, ácido bromhídrico, ácido yodhídrico, ácido sulfúrico, ácido sulfámico, ácido nítrico, ácido bórico, ácido fosfórico y combinaciones de los mismos.
Como se usa en el presente documento, "ácidos orgánicos" se refiere a ácidos tales como, pero sin limitarse a: ácido acético; ácido trifluoroacético; ácido fenilacético; ácido propiónico; ácido esteárico; ácido láctico; ácido ascórbico; ácido maleico; ácido hidroximaleico; ácido isetiónico; ácido succínico; ácido valérico; ácido fumárico; ácido malónico; ácido pirúvico; ácido oxálico; ácido glicólico; ácido salicílico; ácido oleico; ácido palmítico; ácido láurico; un ácido piranosidilo, tal como ácido glucurónico o ácido galacturónico; un ácido alfa-hidroxi, tal como ácido mandélico, ácido cítrico o ácido tartárico; ácido cisteína-sulfínico; un aminoácido, tal como ácido aspártico, ácido glutárico o ácido glutámico; un ácido aromático, tal como ácido benzoico, ácido 2-acetoxibenzoico, ácido naftoico o ácido cinámico; un ácido sulfónico, tal como ácido laurilsulfónico, ácido p-toluenosulfónico, ácido metanosulfónico, ácido bencenosulfónico o ácido etanosulfónico; ácido cisteína-sulfónico; y combinaciones de los mismos.
Como se usa en el presente documento, "bases inorgánicas" se refiere a bases tales como, pero sin limitarse a, hidróxido de sodio, hidróxido de potasio, hidróxido de litio, hidróxido de amonio, hidróxido de magnesio, carbonato de sodio, carbonato de potasio y combinaciones de los mismos.
Como se usa en el presente documento, el término "base orgánica" se refiere a un compuesto orgánico que contiene uno o más átomos de nitrógeno y que actúa como una base. Los ejemplos de bases orgánicas incluyen, pero no se limitan a, bases de amina terciaria. Los ejemplos de bases orgánicas incluyen, pero no se limitan a, 1,8-diazabiciclo[5.4.0]undec-7-eno ("DBU"), N-metilmorfolina (NMM), diisopropiletilamina (DIPEA), trietilamina (TEA), un t-butóxido (por ejemplo, terc-butóxido de sodio, potasio, calcio o magnesio).
Los compuestos de la presente divulgación se pueden presentar en forma de sal que engloba sales farmacéuticamente aceptables y sales no farmacéuticamente aceptables. Como se usa en el presente documento, el término "sales farmacéuticamente aceptables" se refiere a aquellas sales que conservan la eficacia biológica y las propiedades de las bases libres o ácidos libres, que no son indeseables biológicamente o de otro modo. Además de las sales farmacéuticamente aceptables, los compuestos de la presente divulgación pueden estar en forma de sales no farmacéuticamente aceptables que pueden ser útiles como intermedio para aislar o purificar dichos compuestos.
Las sales de ácido ejemplares de los compuestos de la presente divulgación incluyen, pero no se limitan a, sales sulfato, citrato, acetato, oxalato, cloruro, bromuro, yoduro, nitrato, bisulfato, fosfato, fosfato ácido, isonicotinato, lactato, salicilato, citrato ácido, tartrato, oleato, tanato, pantotenato, bitartrato, ascorbato, succinato, maleato, gentisinato, fumarato, gluconato, glucuronato, sacarato, formiato, benzoato, glutamato, metanosulfonato "mesilato", etanosulfonato, bencenosulfonato, p-toluenosulfonato y pamoato (es decir, 1,1'-metileno-bis-(2-hidroxi-3-naftoato)). Una sal farmacéuticamente aceptable puede implicar la inclusión de otra molécula tal como un ion acetato, un ion succinato u otro contraión. El contraión puede ser cualquier resto orgánico o inorgánico que estabilice la carga en el compuesto original. Además, una sal farmacéuticamente aceptable puede tener más de un átomo cargado en su estructura. Los casos donde múltiples átomos cargados son parte de la sal farmacéuticamente aceptable pueden tener múltiples contraiones. Por lo tanto, una sal farmacéuticamente
aceptable puede tener uno o más átomos cargados y/o uno o más contraiones.
Las sales de base ejemplares de los compuestos de la presente divulgación incluyen, pero no se limitan a, sales inorgánicas formadas a partir de cationes de sodio, potasio, amonio, calcio, magnesio, hierro, cinc, cobre, manganeso y aluminio. Las sales orgánicas formadas a partir de cationes que incluyen aminas primarias, secundarias y terciarias; aminas sustituidas que incluyen aminas sustituidas naturales; aminas cíclicas; resinas de intercambio iónico básicas; isopropilamina; trimetilamina; dietilamina; trimetilamina; tripropilamina; etanolamina; 2-dietilaminoetanol; trimetamina; diciclohexilamina; lisina; arginina; histidina; cafeína; procaína; hidrabamina; colina; betaína; etilendiamina; glucosamina; metilglucamina; teobromina; purinas; piperacina; piperidina; N-etilpiperidina; y resinas de poliamina.
Los compuestos de la presente divulgación también se pueden solvatar, es decir, hidratar. La solvatación se puede efectuar en el transcurso del proceso de fabricación o puede tener lugar, por ejemplo, como consecuencia de propiedades higroscópicas de compuestos inicialmente anhidros. Como en el presente documento, "solvato" se refiere a una asociación o complejo de una o más moléculas de disolvente y un compuesto de la invención. Los ejemplos no limitantes de disolventes que forman solvatos incluyen, pero no se limitan a, agua, isopropanol, etanol, metanol, DMSO, acetato de etilo (EtOAc), ácido acético (AcOH) y etanolamina.
Los compuestos que tienen la misma fórmula molecular pero difieren en la naturaleza o secuencia de enlace de sus átomos o la disposición de sus átomos en el espacio se denominan "isómeros". Los isómeros que difieren en la disposición de sus átomos en el espacio se denominan "estereoisómeros". Los diastereómeros son estereoisómeros con configuración opuesta en uno o más centros quirales que no son enantiómeros. Los estereoisómeros que portan uno o más centros asimétricos que son imágenes especulares no superponibles entre sí se denominan "enantiómeros". Cuando un compuesto tiene un centro asimétrico, por ejemplo, si un átomo de carbono se une a cuatro grupos diferentes, es posible un par de enantiómeros. Un enantiómero se puede caracterizar por la configuración absoluta de su centro o centros asimétricos y se describe por las reglas de secuenciación R y S de Cahn, Ingold y Prelog, o por la manera en que la molécula gira el plano de luz polarizada y se designa como dextrógiro o levógiro (es decir, como isómeros (+) o (-) respectivamente). Un compuesto quiral puede existir como enantiómero individual o bien como una mezcla de los mismos. Una mezcla que contiene proporciones iguales de los enantiómeros se llama "mezcla racémica". En determinados modos de realización, el compuesto se enriquece en al menos aproximadamente un 90 % en peso con un solo diastereómero o enantiómero. En otros modos de realización, el compuesto se enriquece en al menos aproximadamente un 95 %, 98 % o 99 % en peso con un solo diastereómero o enantiómero.
Determinados compuestos de la presente divulgación poseen átomos de carbono asimétricos (centros ópticos) o enlaces dobles; se pretende que todos los racematos, diastereómeros, regioisómeros e isómeros individuales (por ejemplo, enantiómeros separados) estén englobados dentro del alcance de la presente divulgación.
Los compuestos de la invención pueden contener centros asimétricos o quirales y, por lo tanto, existir en diferentes formas estereoisómeras. Se pretende que todas las formas estereoisómeras de los compuestos de la invención, incluyendo, pero sin limitarse a, diastereómeros, enantiómeros y atropisómeros, así como mezclas de los mismos, tales como mezclas racémicas, formen parte de la presente invención. En algunos casos, la estereoquímica no se ha determinado o se ha asignado provisionalmente. Muchos compuestos orgánicos existen en formas ópticamente activas, es decir, tienen la capacidad de girar el plano de luz polarizada en el plano. Al describir un compuesto ópticamente activo, se usan los prefijos D y L, o R y S, para indicar la configuración absoluta de la molécula sobre su(s) centro(s) quiral(es). Los prefijos d y l o (+) y (-) se emplean para designar el signo de giro de la luz polarizada en el plano por el compuesto, queriendo decir (-) o l que el compuesto es levógiro. Un compuesto con el prefijo (+) o d es dextrógiro. Para una estructura química dada, estos estereoisómeros son idénticos excepto porque son imágenes especulares entre sí. Un estereoisómero específico también se puede denominar enantiómero, y una mezcla de dichos isómeros a menudo se llama mezcla enantiómera. Una mezcla 50:50 de enantiómeros se denomina mezcla racémica o un racemato, lo que se puede producir cuando no haya existido ninguna estereoselección o estereoespecificidad en una reacción o proceso químico. Los términos "mezcla racémica" y "racemato" se refieren a una mezcla equimolar de dos especies enantiómeras carentes de actividad óptica. Los enantiómeros se pueden separar de una mezcla racémica por un procedimiento de separación quiral, tal como cromatografía de fluidos supercríticos (SFC). La asignación de configuración en centros quirales en enantiómeros separados puede ser tentativa mientras se establece definitivamente la estereoquímica, tal como a partir de datos cristalográficos de rayos X.
Como se usa en el presente documento, "esencialmente" se refiere a al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 %.
En la descripción en el presente documento, si existe una discrepancia entre una estructura representada y un nombre dado a esa estructura, entonces prevalece la estructura representada. Adicionalmente, si no se indica la estereoquímica de una estructura o una parte de una estructura con, por ejemplo, cuña en negrita o líneas discontinuas, se debe interpretar que la estructura o parte de la estructura engloba todos los estereoisómeros de la misma. En algunos casos, sin embargo, donde existe más de un centro quiral, las estructuras y nombres se
pueden representar como enantiómeros individuales para ayudar a describir la estereoquímica relativa.
A menos que se indique de otro modo, los términos "un compuesto de fórmula" o "compuestos de fórmula" se refieren a cualquier compuesto seleccionado del género de compuestos como de define por la fórmula (incluyendo cualquier sal farmacéuticamente aceptable de cualquiera de dichos compuestos si no se indica de otro modo). El alcance patentable de la invención se define por las reivindicaciones. Cualquier materia objeto que quede fuera del alcance de las reivindicaciones se proporciona solo con propósitos informativos. Las referencias a procedimientos de tratamiento en los párrafos posteriores de la presente descripción se deben interpretar como referencia a los compuestos, composiciones farmacéuticas y medicamentos de la presente invención para su uso en un procedimiento para el tratamiento del cuerpo humano por tratamiento.
En un aspecto se proporciona en el presente documento una sal de tartrato cristalina que tiene la estructura:

También se divulga un proceso para preparar un compuesto de fórmula (IV), o una sal del mismo:

donde el proceso comprende las etapas:
(a) hacer reaccionar una mezcla de reacción que comprende un compuesto de fórmula (I), un disolvente orgánico y cloruro de tionilo para formar un compuesto de fórmula (IIa) de acuerdo con la etapa 1 a continuación, y hacer reaccionar una mezcla de reacción que comprende el compuesto de fórmula (IIa), un catalizador, un oxidante y un disolvente para formar un compuesto de fórmula (II) de acuerdo con la etapa 2 a continuación

en la que
cada uno de R1a y R1b es independientemente hidrógeno, halógeno, alquilo C1−3, haloalquilo C1−3, alcoxi C1−3, −CN, cicloalquilo C3−6 o espirocicloalquilo C3−6, y
n es un número entero de 2 o 3; y
(b) hacer reaccionar una mezcla de reacción que comprende el compuesto de fórmula (II) y un compuesto de fórmula (III) en un disolvente orgánico para formar un compuesto de fórmula (IV) o una sal del mismo de acuerdo con la etapa 3 a continuación

en la que B es indolilo, benzofuranilo, benzotiofenilo, indazolilo, aza-indolilo, bencimidazolilo, pirrolopiridinilo, furopiridinilo, tienopiridinilo, pirrolopiridacinilo, pirrolopirimidinilo, pirrolopiracinilo, tienopiridacinilo, tienopirimidinilo, tienopiracinilo, furopiridacinilo, furopirimidinilo o furopiracinilo sustituido o no sustituido,
cada uno de R2a y R2b es independientemente hidrógeno, halógeno, −OH, alquilo C1−3, haloalquilo C1−3, alcoxi C1−3, hidroxialquilo C1−3, −CN, cicloalquilo C3−6 o espirocicloalquilo C3−6,
R3a y R3b son independientemente hidrógeno, alquilo C1−3, haloalquilo C1−3, hidroxialquilo C1−3, −CN, cicloalquilo C3−6, heterocicloalquilo C3−6, fenilo, heteroarilo C3−6 o espirocicloalquilo C3−6, y el asterisco representa un centro quiral cuando R3a y R3b son diferentes.
En un modo de realización, B es indolilo, benzofuranilo, benzotiofenilo, indazolilo, aza-indolilo, bencimidazolilo, pirrolopiridinilo, furopiridinilo, tienopiridinilo, pirrolopiridacinilo, pirrolopirimidinilo, pirrolopiracinilo, tienopiridacinilo, tienopirimidinilo, tienopiracinilo, furopiridacinilo, furopirimidinilo o furopiracinilo sustituido.
En otro modo de realización, B es indolilo, benzofuranilo, benzotiofenilo, indazolilo, aza-indolilo, bencimidazolilo, pirrolopiridinilo, furopiridinilo, tienopiridinilo, pirrolopiridacinilo, pirrolopirimidinilo, pirrolopiracinilo, tienopiridacinilo, tienopirimidinilo, tienopiracinilo, furopiridacinilo, furopirimidinilo o furopiracinilo no sustituido.
En un modo de realización, B es un indolilo, benzofuranilo o benzotiofenilo sustituido o no sustituido. En otro modo de realización, B es un indolilo, benzofuranilo o benzotiofenilo sustituido con uno o más halógeno o alquilo C1-3 como se describe en el presente documento. Todavía en otro modo de realización, B es un pirrolopiridacinilo, pirrolopirimidinilo o pirrolopiracinilo sustituido o no sustituido. En otro modo de realización, B es un pirrolopiridacinilo, pirrolopirimidinilo o pirrolopiracinilo sustituido con uno o más halógeno o alquilo C1−3 como se describe en el presente documento. Aún en otro modo de realización, B es un indolilo sustituido o no sustituido. En un modo de realización preferente, B es indolilo no sustituido. En un modo de realización, B es indolilo sustituido (por ejemplo, sustituido con uno o más halógeno o alquilo C1-3 como se describe en el presente documento). En otro modo de realización preferente, B es indolilo sustituido que comprende la sustitución con al menos un resto seleccionado del grupo que consiste en metilo, Cl y F. Todavía en otro modo de realización, B es un benzofuranilo o un benzofuranilo sustituido que comprende la sustitución con al menos un resto seleccionado del grupo que comprende metilo, Cl y Fl.
B puede estar adecuadamente sustituido con uno o dos sustituyentes independientemente seleccionados del grupo que consiste en flúor, cloro, alquilo C1-3, haloalquilo C1-3, -CN, -OH, alcoxi C1-3 e hidroxialquilo C1-3. En un modo de realización, B es indolilo sustituido con halógeno (por ejemplo, F o Cl).
En un modo de realización, cada uno de R1a y R1b son independientemente hidrógeno, halógeno, alquilo C1-3, haloalquilo C1-3, alcoxi C1-3, -CN o cicloalquilo C3-6. En otro modo de realización, R1a y R1b son cada uno independientemente hidrógeno, -F, -Cl, -OH, -CN, -CH3, -CF3, -CHF2, -CH2F o espirociclopropilo. En un modo de realización, R1a y R1b son independientemente F o hidrógeno. En un modo de realización preferente, R1a y R1b son cada uno independientemente hidrógeno, -F o -CH3. En otro modo de realización preferente, R1a y R1b son cada uno independientemente hidrógeno, -F o ciclopropilo. En un modo de realización, n es 3.
En un modo de realización:

R2a y R2b son cada uno independientemente hidrógeno, halógeno, -OH, alquilo C1-3, haloalquilo C1-3, alcoxi C1-3, hidroxialquilo C1-3, -CN o cicloalquilo C3-6. En algunos modos de realización, R2a y R2b son cada uno hidrógeno. En un modo de realización, R1a y R1b son cada uno independientemente -F o CH3 y R2a y R2b son cada uno independientemente hidrógeno. En un modo de realización, B es un indolilo, benzofuranilo o benzotiofenilo, R1a y R1b son cada uno independientemente -F o CH3, y R2a y R2b son cada uno independientemente hidrógeno. R3a y R3b son cada uno independientemente hidrógeno, alquilo C1−3, haloalquilo C1−3, hidroxialquilo C1−3, −CN, cicloalquilo C3−6, heterocicloalquilo C3−6, fenilo o heteroarilo C3−6. En un modo de realización, R3a y R3b son cada uno independientemente hidrógeno o -CH3.
El asterisco en la fórmula (IV) representa un centro quiral cuando R3a y R3b son diferentes. En algunos modos de realización, por lo tanto, R3a y R3b son diferentes y son hidrógeno o -CH3.
En un modo de realización, el compuesto de fórmula (I) es:

incluyendo estereoisómeros del mismo.
En un modo de realización particular, el compuesto de fórmula (I) es:

En otro modo de realización, el compuesto de fórmula (I) es:

incluyendo estereoisómeros del mismo.
En un modo de realización, el compuesto de fórmula (II) es:

incluyendo estereoisómeros del mismo.
En un modo de realización particular, la fórmula (II) es:

En algunos modos de realización particulares, el compuesto de fórmula (II) es:

incluyendo estereoisómeros del mismo.
En un modo de realización, el compuesto de fórmula (III) es:

incluyendo estereoisómeros del mismo, donde X es -NH-, -N-alquilo no sustituido C1-C3, -O- o -S-. En un modo de realización particular, el compuesto de fórmula (III) es:

En otro modo de realización particular, el compuesto de fórmula (III) es:

En un modo de realización, el compuesto de fórmula (IV) es:


En un modo de realización particular, la fórmula (IV) es:

En otro modo de realización particular, la fórmula (IV) es:

En un modo de realización particular, el compuesto de fórmula (I) es:

el compuesto de fórmula (II) es

el compuesto de fórmula (III) es

; y
(3)
el compuesto de fórmula (IV) es

En la etapa 1, se hace reaccionar una mezcla de reacción que comprende un compuesto de fórmula (I), un disolvente orgánico y cloruro de tionilo para formar un compuesto de fórmula (IIa). En un modo de realización, el compuesto de fórmula (I) es el compuesto 1. En un modo de realización, el disolvente orgánico es un disolvente apolar o un disolvente polar. En un modo de realización, el disolvente es apolar. Los ejemplos no limitantes de disolventes apolares adecuados incluyen pentano, ciclopentano, hexano, ciclohexano, benceno, tolueno, 1,4dioxano, cloroformo, éter dietílico, diclorometano ("DCM") y combinaciones de los mismos. En un modo de realización, el disolvente es DCM. En un modo de realización, la concentración de fórmula (I) en el disolvente puede ser adecuadamente de aproximadamente 25 g/l, aproximadamente 50 g/l, aproximadamente 100 g/l,
aproximadamente 150 g/l, aproximadamente 200 g/l, aproximadamente 250 g/l y hasta una concentración cercana a la saturación a la temperatura de reacción, e intervalos construidos a partir de esas concentraciones, tales como de aproximadamente 100 g/l a aproximadamente 250 g/l. La proporción de equivalentes de cloruro de tionilo con respecto al compuesto de fórmula (I) es adecuadamente de aproximadamente 1:1, aproximadamente 1,1:1, aproximadamente 1,2:1, aproximadamente 1,3:1, aproximadamente 1,5:1 o aproximadamente 2:1, e intervalos construidos a partir de esas proporciones, tales como de aproximadamente 1,1:1 a aproximadamente 1,5:1. En un modo de realización, la temperatura de reacción está por debajo de la temperatura de reflujo de mezcla de reacción. En un modo de realización, la reacción se realiza a reflujo. Por ejemplo, cuando el disolvente es DCM, la temperatura de reacción puede ser adecuadamente de aproximadamente 25 °C a aproximadamente 40 °C. El tiempo de reacción no se limita estrictamente y la reacción continúa típicamente hasta que la conversión de la fórmula (I) a la fórmula (IIa) se finaliza esencialmente tal como se determina por cromatografía (por ejemplo, TLC, CG o HPLC).
Tras la finalización de la reacción, se puede desactivar la mezcla de reacción. En algunos de dichos modos de realización, la mezcla de reacción se puede desactivar con agua fría. En dichos modos de realización, las fases se pueden separar en una fase acuosa y una fase orgánica que comprende el compuesto de fórmula (IIa). La fase acuosa se puede extraer una o más veces con un disolvente orgánico para recuperar el compuesto de fórmula (IIa) adicional.
En la etapa 2, se hace reaccionar una mezcla de reacción que comprende el compuesto de fórmula (IIa), un catalizador, un oxidante y un disolvente para formar un compuesto de fórmula (II). En un modo de realización, la fase orgánica o fases orgánicas combinadas de la etapa 1, que comprende la fórmula (IIa), se usa como fuente de fórmula (IIa) para la etapa 2. En un modo de realización, el catalizador es un catalizador de metal activo redox. Los ejemplos no limitantes de catalizadores adecuados incluyen NiCl2, RuCl3, CoCl2, FeCl3, FeCl2, y MnCl2. Los ejemplos no limitantes de oxidantes adecuados incluyen NaIO4, NaOCl y Oxone. Los disolventes orgánicos adecuados incluyen disolventes apolares y polares, como se analiza en otra parte en el presente documento. En general, el oxidante está en exceso de equivalentes con respecto al compuesto de fórmula (IIa), por ejemplo, la proporción de oxidante con respecto al compuesto de fórmula (IIa) puede ser de 1,1:1, 1,5:1, 2:1, 2,5:1, 3:1, 3,5:1, 4:1, 4,5:1 o 5:1. La mezcla de reacción de la etapa 2 puede comprender además agua. En dichos modos de realización, la proporción de volumen de agua con respecto al disolvente orgánico usada en la mezcla de reacción de la etapa 1 puede ser de aproximadamente 9:1, aproximadamente 5:1, aproximadamente 3:1, aproximadamente 2:1, aproximadamente 1:1, aproximadamente 1:2, aproximadamente 1:3, aproximadamente 1:5 o aproximadamente 1:9, e intervalos construidos a partir de las mismas, tales como de aproximadamente 2:1 a aproximadamente 1:2. La temperatura de reacción de la etapa 2 puede ser adecuadamente de aproximadamente 25 °C, aproximadamente 15 °C, aproximadamente 5 °C, aproximadamente 0 °C, aproximadamente -5 °C o aproximadamente -10 °C, e intervalos construidos a partir de las mismas, tales como de aproximadamente -10 °C a aproximadamente 10 °C. En un modo de realización, la(s) fase(s) orgánica(s) que comprende(n) la fórmula (IIa), catalizador y agua se combinan y se enfrían hasta una temperatura de reacción. A continuación se añade el oxidante durante un período de tiempo mientras se mantiene la temperatura alrededor de la temperatura de reacción.
Tras la finalización de la reacción, se puede separar la mezcla de reacción de la etapa 2 en una fase acuosa y una fase orgánica que comprende el compuesto de fórmula (II) en solución. En algunos modos de realización opcionales, la mezcla de reacción se puede filtrar, tal como a través de un filtro auxiliar (por ejemplo, celite) antes de la separación de fases. La fase acuosa se puede extraer una o más veces con un disolvente orgánico para recuperar el compuesto de fórmula (II) adicional.
En otro modo de realización, la(s) fase(s) orgánica(s) de la etapa 2 se puede(n) procesar por procedimientos conocidos para los expertos en la técnica. Por ejemplo, las fases orgánicas se pueden lavar con una base, tal como con una solución acuosa de Na2SO3. Las fases orgánicas se pueden secar además opcionalmente, tal como con una solución de salmuera y/o por la adición de un agente secante sólido tal como CaCl2, MgSO4 o Na2SO4. Los desecantes sólidos se pueden retirar adecuadamente por filtración. En un modo de realización, la solución del compuesto de fórmula (II) se puede usar para la reacción posterior. En un modo de realización, el compuesto de fórmula (II) se puede aislar de la solución por procedimientos conocidos en la técnica tales como por destilación, concentración, precipitación (tal como por adición de un antidisolvente o ajuste de pH) y/o cristalización. En algunos de dichos modos de realización, la(s) fase(s) orgánica(s) se puede(n) concentrar por destilación o extracción para reducir el volumen, tal como en al menos un 25 %, 50 %, 100 % o más. A continuación, el compuesto de fórmula (II) se puede precipitar/cristalizar a partir de la solución por adición de un antidisolvente seguido de una concentración adicional opcional. En un modo de realización, el antidisolvente es un disolvente no iónico C4-8 tal como pentano, hexano o heptano. Los sólidos del compuesto de fórmula (II) se pueden recoger por procedimientos conocidos en la técnica tales como filtración o centrifugación. Los sólidos se pueden secar, tal como a vacío parcial, para proporcionar el compuesto sólido de fórmula (II). El rendimiento para el compuesto de fórmula (II) a partir del compuesto de fórmula (I) para las etapas 1 y 2 es de al menos un 60 %, al menos un 70 % o al menos un 75 %. En un modo de realización, el compuesto de fórmula (II) es el compuesto 2.
En la etapa 3, se hace reaccionar una mezcla de reacción que comprende el compuesto de fórmula (II), un
compuesto de fórmula (III) y un disolvente orgánico para formar un compuesto de fórmula (IV). En un modo de realización, el disolvente orgánico es un disolvente aprótico polar. Los ejemplos no limitantes de disolventes adecuados incluyen tetrahidrofurano, acetato de etilo, acetona, dimetilformamida, acetonitrilo ("ACN"), sulfóxido de dimetilo, nitrometano y carbonato de propileno. En un modo de realización, el disolvente es ACN. La proporción molar del compuesto de fórmula (II) con respecto al compuesto de fórmula (III) es adecuadamente de aproximadamente 1:1, aproximadamente 1,1:1, aproximadamente 1,2:1, aproximadamente 1,3:1, aproximadamente 1,4:1, aproximadamente 1,5:1 o mayor, e intervalos construidos a partir de esas proporciones, tales como entre 1:1 y 1,3:1. La concentración del compuesto de fórmula (II) en el disolvente es adecuadamente de aproximadamente 10 g/l, aproximadamente 25 g/l, aproximadamente 50 g/l, aproximadamente 75 g/l, aproximadamente 100 g/l, aproximadamente 125 g/l, aproximadamente 150 g/l y hasta una concentración cercana a la saturación a la temperatura de reacción, e intervalos construidos a partir de esas concentraciones, tales como de aproximadamente 50 g/l a aproximadamente 150 g/l. El catalizador ácido puede ser un catalizador ácido como se describe en otra parte en el presente documento. En algunos modos de realización, el catalizador ácido es ácido sulfúrico, ácido p-toluenosulfónico (p-TsOH) o ácido metanosulfónico, o combinaciones de los mismos. En un modo de realización, el catalizador ácido es ácido p-toluenosulfónico. La proporción de equivalentes del catalizador ácido con respecto al compuesto de fórmula (II) es adecuadamente de aproximadamente 0,75:1, aproximadamente 0,9:1, aproximadamente 1:1, aproximadamente 1,05:1, aproximadamente 1,1:1, aproximadamente 1,2:1, aproximadamente 1,3:1, aproximadamente 1,4:1, aproximadamente 1:5:1 o mayor, e intervalos construidos a partir de las mismas, tales como de aproximadamente 1:1 a aproximadamente 1,2:1. En un modo de realización, el compuesto de fórmula (III) es el compuesto 3.
En algunos modos de realización de la etapa 3, el compuesto de fórmula (II), el compuesto de fórmula (III), el disolvente orgánico y una base se combinan para formar una mezcla. La base puede ser adecuadamente una base moderada, con ejemplos no limitantes que incluyen terc-butóxido de potasio, trimetilamina, bicarbonato de sodio, bicarbonato de potasio, carbonato de sodio, carbonato de potasio, hidróxido de sodio, hidróxido de amonio y combinaciones de los mismos. La mezcla se puede calentar con agitación hasta una temperatura de reacción, típicamente una temperatura de desde 2 °C a aproximadamente 30 °C por debajo de la temperatura de reflujo hasta la temperatura de reflujo, y mantener durante un tiempo suficiente para finalizar esencialmente la formación de un producto de reacción que comprende el compuesto de fórmula (III). En el caso del disolvente ACN, la temperatura de reacción es adecuadamente de aproximadamente 65 °C, aproximadamente 70 °C, aproximadamente 75 °C o aproximadamente 80 °C. A continuación, la mezcla de productos de reacción se puede enfriar, por ejemplo a menos de 50 °C, de 40 °C o de 30 °C, y opcionalmente filtrar para retirar impurezas sólidas. Los sólidos se pueden lavar opcionalmente con el disolvente para recuperar producto de reacción adicional. A continuación, se añaden ácido (por ejemplo, p-TsOH) y agua. La proporción de volumen de disolvente orgánico con respecto al agua puede ser de 25:1, 15:1, 10:1, 5:1, 2:1 o 1:1, e intervalos construidos a partir de las mismas, tales como de aproximadamente 15:1 a aproximadamente 5:1. La mezcla se puede calentar con agitación hasta una temperatura de reacción, típicamente a una temperatura de desde 2 °C a aproximadamente 20 °C por debajo de la temperatura de reflujo hasta la temperatura de reflujo, y mantener durante un tiempo suficiente para finalizar esencialmente la formación del compuesto de fórmula (IV) tal como se determina por cromatografía (por ejemplo, TLC, CG o HPLC). Tras la finalización de la reacción, se puede desactivar la mezcla de reacción, por ejemplo con agua fría (por ejemplo, a menos de 10 °C o menos de 5 °C). El pH de la mezcla de reacción desactivada se puede ajustar a continuación con una base a más de 7, tal como aproximadamente pH 8, aproximadamente pH 9, aproximadamente pH 10 o aproximadamente pH 11. En un modo de realización, la base es una base acuosa tal como bicarbonato de sodio, bicarbonato de potasio, carbonato de sodio, carbonato de potasio, hidróxido de sodio o hidróxido de amonio.
Tras la finalización de la reacción, se puede separar la mezcla de reacción de la etapa 3 en una fase acuosa y una fase orgánica que comprende el compuesto de fórmula (IV) en solución. En algunos modos de realización opcionales, la mezcla de reacción se puede filtrar, tal como a través de un filtro auxiliar (por ejemplo, celite) antes de la separación de fases. La fase acuosa se puede extraer una o más veces con un disolvente orgánico para recuperar compuesto de fórmula (IV) adicional. En un modo de realización, el disolvente es aprótico. En un modo de realización particular, el disolvente de extracción es adecuadamente acetato de isopropilo ("i-PrOAc").
En algunos modos de realización, la(s) fase(s) orgánica(s) de la etapa 3 se puede(n) procesar, por ejemplo, lavando las fases orgánicas con agua. Las fases orgánicas se pueden secar opcionalmente, tal como con una solución de salmuera y/o por la adición de un agente secante sólido tal como CaCl2, MgSO4 o Na2SO4. Los desecantes sólidos se pueden retirar adecuadamente por filtración, y el desecante recogido se puede lavar opcionalmente con disolvente para recuperar el compuesto de fórmula (IV) del mismo. En dichos modos de realización, la(s) fase(s) orgánica(s) se puede(n) concentrar por destilación a vacío parcial o extracción para formar el residuo del compuesto de fórmula (IV). A continuación, el residuo del compuesto de fórmula (IV) se puede disolver en un disolvente orgánico a una temperatura por debajo de la temperatura de reflujo. A continuación, se puede añadir un antidisolvente, tal como un disolvente orgánico apolar como se describe en otra parte en el presente documento, a la solución del compuesto de fórmula (IV) mientras se enfría, tal como a menos de aproximadamente 10 °C, para precipitar/cristalizar el compuesto de fórmula (IV) de la solución. Los sólidos del compuesto de fórmula (IV) se pueden recoger por procedimientos conocidos en la técnica tales como filtración o centrifugación y, opcionalmente, lavar con antidisolvente. Los sólidos se pueden secar, tal como a vacío parcial, para proporcionar el compuesto
sólido de fórmula (IV). El rendimiento para el compuesto de fórmula (IV) a partir del compuesto de fórmula (II) para la etapa 3 es al menos un 80 %, al menos un 85 %, al menos un 90 %, al menos un 95 %, al menos un 96 % o al menos un 97 %. La pureza del compuesto de fórmula (IV) es de al menos un 95 %, al menos un 98 % o al menos un 99 %.
Un aspecto de la divulgación se refiere a un proceso para preparar un compuesto de fórmula (VIII) o una sal del mismo:

donde el proceso comprende las etapas de:
(a) hacer reaccionar una mezcla de reacción que comprende un compuesto de fórmula (IV), un compuesto de fórmula (V) o un compuesto de fórmula (X) y un disolvente orgánico para formar un compuesto de fórmula (VI) de acuerdo con la etapa 1 a continuación

en la que
B es indolilo, benzofuranilo, benzotiofenilo, aza-indolilo, indazolilo, bencimidazolilo, pirrolopiridinilo, furopiridinilo, tienopiridinilo, pirrolopiridacinilo, pirrolopirimidinilo, pirrolopiracinilo, tienopiridacinilo, tienopirimidinilo, tienopiracinilo, furopiridacinilo, furopirimidinilo o furopiracinilo sustituido o no sustituido;
cada uno de R1a y R1b es independientemente hidrógeno, flúor, cloro, −OH, alquilo C1−3, haloalquilo C1−3, alcoxi C1−3, hidroxialquilo C1−3 y −CN, cicloalquilo C3−6 o espirocicloalquilo C3−6,
n es un número entero de 2 o 3,
cada uno de R2a y R2b es independientemente hidrógeno, halógeno, −OH, alquilo C1−3, haloalquilo C1−3, alcoxi C1−3, hidroxialquilo C1−3, −CN, cicloalquilo C3−6 o espirocicloalquilo C3−6,
R3a y R3b son independientemente hidrógeno, alquilo C1−3, haloalquilo C1−3, alcoxi C1−3, −CN, cicloalquilo C3−6, heterocicloalquilo C3−6, fenilo, heteroarilo C3−6 o espirocicloalquilo C3−6,
J es fenilo o piridinilo;
cada R4 es independientemente hidrógeno, halógeno o alquilo C1-3,
s es un número entero de 0 a 2,
LG es un grupo saliente,
LG y CHO se localizan en la posición para entre sí en J en el compuesto de fórmula (V),
PG es un grupo protector de aldehído,
LG y CH-PG se localizan en la posición para entre sí en J en el compuesto de fórmula (X), y
cada asterisco representa independientemente un centro quiral en el que el carbono que porta R3a y R3b es un centro quiral cuando R3a y R3b son diferentes; y
(b) hacer reaccionar una mezcla de reacción que comprende el compuesto de fórmula (VI), un disolvente orgánico y un compuesto de fórmula (VII) o una sal del mismo para formar un compuesto de fórmula (VIII) o una sal del mismo de acuerdo con la etapa 2 a continuación

en la que
G es alquilo C1−3,
p es 0 o 1,
E es acetidinilo o pirrolidinilo sustituido o no sustituido,
cada R5 es independientemente hidrógeno, halógeno, -OH, -CN, alcoxi C1-5 o hidroxialquilo C1-5,
v es un número entero de 1 a 5,
R6 es halógeno o −CN; y
R10 es hidrógeno o alquilo C1-3.
B, R1a, R1b, n, R2a, R2b, R3a, R3b y el asterisco (*) son como se define en el presente documento.
En un modo de realización preferente, J es fenilo. En otro modo de realización, J es piridinilo.
En un modo de realización, cada R4 es independientemente hidrógeno o halógeno. En un modo de realización preferente, cada R4 es flúor. En un modo de realización, s es 1 o 2. En un modo de realización, s es 2. En un modo de realización preferente, cada R4 es flúor y s es 2.
En un modo de realización, G es metileno o etileno.
En un modo de realización, p es 0.
En un modo de realización, cada R5 es independientemente hidrógeno, halógeno, -OH o -CN. En un modo de realización preferente, cada R5 es hidrógeno. En un modo de realización, v es 2. En otro modo de realización, v es 3. En otro modo de realización, v es 5. En un modo de realización preferente, cada R5 es hidrógeno y v es 3. En un modo de realización preferente, R6 es halógeno. En un modo de realización, R6 es F. En otro modo de realización, R6 es -CN.
En un modo de realización, R10 es hidrógeno o metilo. En un modo de realización preferente, R10 es hidrógeno En un modo de realización, E es acetidinilo. En otro modo de realización, E es pirrolidinilo.
En un modo de realización, E tiene la siguiente estructura:

En un modo de realización, E es acetidinilo de la siguiente estructura:

En un modo de realización, E tiene la siguiente estructura:

donde R5 es H, v es 2 o 3 y R6 es halógeno.
En un modo de realización, E es acetidinilo de la siguiente estructura:

En un modo de realización, la fórmula (VIII) es una sal de ácido. Dicha sal de ácido puede ser una sal farmacéuticamente aceptable. En algunos de dichos modos de realización, la fórmula (VIII) es una sal de un ácido farmacéuticamente aceptable. En algunos modos de realización particulares, la fórmula (VIII) es una sal de un ácido orgánico farmacéuticamente aceptable. En un modo de realización preferente, la fórmula (VIII) es una sal farmacéuticamente aceptable de ácido tartárico. En un modo de realización, el compuesto de fórmula (VIII) es el compuesto A como se describe en el presente documento. En otro modo de realización, el compuesto de fórmula (VIII) es el compuesto A (sal de tartrato) del compuesto A descrito en el presente documento. En otro modo de realización, la fórmula (VIII) es una sal farmacéuticamente aceptable de ácido fumárico. Todavía en otro modo de realización, el compuesto de fórmula (VIII) es el compuesto B (sal de fumarato) del compuesto A como se describe en el presente documento.
En un modo de realización, la fórmula (VIII) tiene una cualquiera de las siguientes estructuras, o una sal farmacéuticamente aceptable del mismo:



aceptable del mismo e incluyendo estereoisómeros del mismo.
En un modo de realización, la fórmula (VIII) tiene la siguiente estructura, o una sal farmacéuticamente aceptable del mismo:

En un modo de realización, la fórmula (VIII) tiene la siguiente estructura:

En un modo de realización, la fórmula (VIII) tiene la siguiente estructura:

La etapa 1 del proceso para sintetizar compuestos de fórmula (VIII) comprende hacer reaccionar una mezcla de reacción que comprende un compuesto de fórmula (IV), un compuesto de fórmula (V) o un compuesto de fórmula (X) y un disolvente orgánico para formar un compuesto de fórmula (VI) como se expone en el presente documento. En un modo de realización, LG es bromo. En un modo de realización preferente, cuando se hacen reaccionar con compuestos de fórmula (IV) y fórmula (V), LG y CHO se localizan en J en la posición para entre sí. En un modo de realización preferente, cuando se hacen reaccionar los compuestos de fórmula (IV) y fórmula (X), LG y CH-PG se localizan en la posición para entre sí en J.
El compuesto de fórmula (IV) es como se describe en el presente documento.
En un modo de realización, el compuesto de fórmula (V) es uno cualquiera de los siguientes compuestos, o una sal del mismo:

o una sal del mismo.
En algunos modos de realización, la fórmula (X) corresponde a cualquiera de las estructuras de fórmula (V) anteriores, o una sal de la misma, pero donde el aldehído (-CHO) es un resto protegido de la estructura -CH-PG donde PG es un grupo protector de aldehído como se define en otra parte en el presente documento.
En algunos modos de realización, la fórmula (VI) tiene una cualquiera de las siguientes estructuras:



estereoisómeros del mismo.
En la etapa 2 de la síntesis de los compuestos de fórmula (VIII) expuestos en el presente documento, el compuesto de fórmula (VII) puede ser una cualquiera de las siguientes estructuras:

o una sa e msmo.
En un modo de realización, la sal es un etano-disulfonato (por ejemplo, una sal de etano-1,2-disulfonato).
En un modo de realización, el compuesto de fórmula (VII) tiene la estructura

El compuesto (7) se puede preparar de acuerdo con los ejemplos proporcionados en el presente documento, tales como, por ejemplo, ejemplo 4 o ejemplo 4a.
En un modo de realización, el compuesto 7 se prepara de acuerdo con el esquema a continuación:

En la etapa 1, se hace reaccionar una mezcla de reacción que comprende un compuesto de fórmula (IV), un compuesto de fórmula (V) o un compuesto de fórmula (X) y un disolvente orgánico para formar un compuesto de fórmula (VI). En un modo de realización, el disolvente orgánico es un disolvente prótico polar, un disolvente apolar, un disolvente aprótico polar o una combinación de los mismos, como se describe en otra parte en el presente documento. En algunos modos de realización no limitantes, el disolvente es tolueno. En un modo de realización, el disolvente es acetonitrilo, metiletilcetona o metiltetrahidrofurano. En un modo de realización, la mezcla de reacción de la etapa 1 comprende además un catalizador ácido como se describe en otra parte en el presente documento. En algunos de dichos modos de realización no limitantes, el catalizador ácido es ácido acético. La proporción molar del compuesto (IV) con respecto al compuesto (V) o al compuesto (X) es de aproximadamente 0,95:1 a aproximadamente 1,05:1, cantidades estequiométricas o, en algunos modos de realización, el compuesto (V) o el compuesto (X) está en ligero exceso molar. El catalizador ácido en general está presente en exceso estequiométrico, tal como en una proporción de equivalentes con respecto al compuesto de fórmula (IV) de aproximadamente 1,1:1, aproximadamente 1,2:1, aproximadamente 1,3:1, aproximadamente 1,4:1, aproximadamente 1,5:1, aproximadamente 1,6:1, aproximadamente 1,7:1, aproximadamente 1,8:1, aproximadamente 2:1 o mayor, e intervalos construidos a partir de las mismas, tales como de aproximadamente 1,2:1 a aproximadamente 1,8:1.
En algunos modos de realización de la etapa 1, la mezcla de reacción se puede calentar con agitación hasta una temperatura de reacción, de desde 2 °C a aproximadamente 30 °C por debajo de la temperatura de reflujo hasta la temperatura de reflujo, y mantener durante un tiempo suficiente para finalizar esencialmente la reacción para el compuesto de fórmula (VI) tal como se determina por cromatografía (por ejemplo, TLC, CG o HPLC). En el caso del disolvente tolueno, la temperatura de reacción es adecuadamente de aproximadamente 65 °C, aproximadamente 70 °C, aproximadamente 75 °C o aproximadamente 80 °C. A continuación, la mezcla de
productos de reacción se puede enfriar y, opcionalmente, diluir con disolvente adicional. A continuación, la mezcla de productos de reacción se puede desactivar con una base, tal como una solución acuosa de una base como se describe en otra parte en el presente documento. A continuación, una fase orgánica de mezcla de productos de reacción de la etapa 1 que comprende el compuesto de fórmula (VI) se puede aislar y, en algunos modos de realización, procesar por procedimientos conocidos en la técnica tales como lavado con agua y/o una solución de salmuera como se describe en otra parte en el presente documento seguido de aislamiento del producto como un sólido. En un modo de realización, la mezcla de productos de reacción se puede tratar con carbón activado, seguido de filtración y lavado opcional de la torta de filtro de carbón activado con disolvente. Como se describe en otra parte en el presente documento: (i) la(s) fase(s) orgánica(s) que contiene(n) el compuesto de fórmula (VI) se puede(n) concentrar por destilación o extracción para reducir el volumen, tal como en al menos un 25 %, 50 %, 100 % o más; y (ii) el compuesto de fórmula (IV) a continuación se puede precipitar/cristalizar a partir de la solución por adición de un antidisolvente seguido de una concentración adicional opcional.
En algunos otros modos de realización de la etapa 1, la mezcla de reacción de la etapa 1 se calienta a reflujo durante un tiempo para finalizar esencialmente la reacción tal como se determina por cromatografía (por ejemplo, TLC, CG o HPLC). A continuación, la mezcla de reacción se puede enfriar y ajustar el pH con una base, tal como una solución acuosa de una base, hasta un pH en el que el compuesto de fórmula (VI) precipita de la solución.
En cualquiera de los diversos modos de realización de la etapa 1, los sólidos del compuesto de fórmula (VI) se pueden recoger por procedimientos conocidos en la técnica tales como filtración o centrifugación. Los sólidos se pueden secar, tal como a vacío parcial, para proporcionar el compuesto sólido de fórmula (VI). El rendimiento para el compuesto de fórmula (VI) a partir del compuesto de fórmula (V) para las etapas 1 y 2 es de al menos un 65 %, al menos un 70 %, al menos un 75 %, al menos un 80 %, al menos un 85 %, al menos un 90 % o al menos un 95 %.
En la etapa 2, se hace reaccionar una mezcla de reacción que comprende un compuesto de fórmula (VI), un compuesto de fórmula (VII) y un disolvente orgánico para formar un compuesto de fórmula (VIII). En algunos modos de realización, el disolvente orgánico es un disolvente aprótico polar como se describe en otra parte en el presente documento. En algunos modos de realización no limitantes, el disolvente es ACN. La mezcla de reacción de la etapa 2 puede comprender además una base, tal como una base orgánica. Los ejemplos no limitantes de bases orgánicas incluyen DBU, NMM, DIPEA y TEA. En algunos de dichos modos de realización, la base es DBU. La mezcla de reacción de la etapa 2 puede comprender además un catalizador, tal como un catalizador de metal de transición. En algunos modos de realización, el catalizador es un catalizador de Pd. La proporción molar de compuesto (VII) con respecto al compuesto (VI) es de aproximadamente 0,95:1, aproximadamente 1:1, aproximadamente 1,05:1, aproximadamente 1,1:1, aproximadamente 1,2:1, aproximadamente 1,3:1, aproximadamente 1,4:1 o aproximadamente 1,5:1, e intervalos construidos a partir de las mismas, tales como de aproximadamente 1:1 a aproximadamente 1,4:1. La base en general está presente en exceso estequiométrico, tal como en una proporción de equivalentes con respecto al compuesto de fórmula (VI) de aproximadamente 1,1:1, aproximadamente 2:1, aproximadamente 3:1, aproximadamente 4:1, aproximadamente 5:1, aproximadamente 6:1, aproximadamente 7:1 o aproximadamente 8:1, e intervalos construidos a partir de las mismas, tales como de aproximadamente 3:1 a aproximadamente 7:1. La mezcla de reacción se puede calentar con agitación hasta una temperatura de reacción, típicamente una temperatura de desde 2 °C a aproximadamente 30 °C por debajo de la temperatura de reflujo hasta la temperatura de reflujo, y mantener durante un tiempo suficiente para finalizar esencialmente la reacción tal como se determina por cromatografía (por ejemplo, TLC, CG o HPLC). En el caso del disolvente ACN, la temperatura de reacción es adecuadamente de aproximadamente 65 °C, aproximadamente 70 °C, aproximadamente 75 °C o aproximadamente 80 °C.
Después de la finalización, la mezcla de productos de reacción se puede enfriar adecuadamente y opcionalmente diluirse con un disolvente orgánico. En un modo de realización, la mezcla de productos de reacción se diluye con un disolvente apolar como se describe en otra parte en el presente documento. Un ejemplo no limitante de un disolvente apolar adecuado es MTBE. La mezcla de productos de reacción de la etapa 2 se puede procesar por procedimientos conocidos para los expertos en la técnica incluyendo lavado con agua y lavado con salmuera. En algunos de dichos modos de realización no limitantes, el tratamiento puede incluir lavar con una solución acuosa de cloruro de amonio, salmuera y agua. En algunos modos de realización, la mezcla de productos de reacción de la etapa 2 se puede poner en contacto con un depurador de metales conocido en la técnica, tal como por ejemplo y sin limitación, SiliaMetS Thiol. A continuación, la mezcla de productos de reacción se puede filtrar para retirar sólidos antes del aislamiento del compuesto de fórmula (VIII) de la misma.
En algunos modos de realización, la mezcla de productos de reacción de la etapa 2 se puede concentrar, tal como por destilación a vacío o extracción, y diluir con un disolvente orgánico, tal como un alcohol (por ejemplo, etanol), tal como en una etapa de intercambio de disolvente. A continuación, se puede añadir un ácido a la solución diluida del compuesto de fórmula (VIII) seguido de enfriamiento para cristalizar el compuesto de fórmula (VIII) como una sal de ácido. En algunos modos de realización particulares, el ácido es ácido tartárico y el compuesto de fórmula (VIII) es la sal tartárica. En algunos de dichos modos de realización, el ácido es ácido (2R-3R)-tartárico (ácido L-(+)-tartárico). En otro aspecto, el ácido es ácido (2S-3S)-tartárico (ácido D-(-)-tartárico). En algunos de dichos modos de realización, el disolvente comprende un disolvente orgánico. El material cristalino se puede recoger por
centrifugación o filtración, opcionalmente lavar con disolvente y opcionalmente secar.
En algunos otros modos de realización, el compuesto de fórmula (VIII) se puede aislar de la mezcla de productos de reacción de la etapa 2 usando procedimientos descritos en otra parte en el presente documento, incluyendo: (i) destilación, concentración, precipitación (tal como por adición de un antidisolvente o ajuste de pH) y/o cristalización; (ii) recogida de sólidos por centrifugación o filtración; (iii) lavado opcional de los sólidos recogidos; (iv) y secado. El rendimiento de la etapa 2 del compuesto de fórmula (VIII), como base libre o bien como sal de ácido, es de al menos un 80 %, al menos un 85 % o al menos un 90 %.
Otro aspecto de la divulgación se refiere a un proceso para preparar un compuesto de fórmula (VIII) o una sal del mismo, en el que el compuesto de fórmula (VIII) es como se describe en otra parte en el presente documento. El proceso para preparar la fórmula (VIII) de acuerdo con este modo de realización comprende la etapa de reacción 1 como se representa a continuación:

Cada uno de B, R1a, R1b, n, R2a, R2b, R3a, R3b, R4, s, J, R5, v, R6, R10, G, p, E, PG y el asterisco son como se describe en otra parte en el presente documento. El resto CHO y el átomo de nitrógeno que enlaza J y G se localizan en la posición para entre sí en J. El resto CH-PG y el átomo de nitrógeno que enlaza J y G se localizan en la posición para entre sí en J.
En un modo de realización, el compuesto de fórmula (IX) es:

En un modo de realización preferente, el compuesto de fórmula (IX) es el compuesto (8a). En algunos modos de realización, la fórmula (XI) corresponde a cualquiera de las estructuras de fórmula (IX) anteriores, o una sal de la misma, pero donde el aldehído (-CHO) es un resto protegido de la estructura -CH-PG donde PG es un grupo protector de aldehído como se define en otra parte en el presente documento.
En la etapa 1, se hace reaccionar una mezcla de reacción que comprende un compuesto de fórmula (IX) o de fórmula (XI), un compuesto de fórmula (IV) y un disolvente orgánico para formar un compuesto de fórmula (VIII), o
una sal del mismo. En un modo de realización, el disolvente orgánico es un disolvente polar o es un disolvente prótico polar. La mezcla de reacción de la etapa 1 comprende además un catalizador ácido como se describe en otra parte en el presente documento. En un modo de realización particular, el catalizador ácido es ácido tartárico o ácido fumárico. En un modo de realización, el catalizador ácido es ácido tartárico. En otro modo de realización, el catalizador ácido es ácido fumárico. Los ejemplos no limitantes de disolventes adecuados incluyen n-butanol, alcohol isopropílico, n-propanol, i-propanol, etanol, metanol y combinaciones de los mismos. En algunos modos de realización particulares, el disolvente es etanol. En un modo de realización, la concentración de fórmula (IX) en el disolvente puede ser adecuadamente de aproximadamente 25 g/l, aproximadamente 50 g/l, aproximadamente 100 g/l, aproximadamente 150 g/l, aproximadamente 200 g/l, aproximadamente 250 g/l y hasta una concentración cercana a la saturación a la temperatura de reacción, e intervalos construidos a partir de esas concentraciones, tales como de aproximadamente 100 g/l a aproximadamente 250 g/l. La proporción molar de fórmula (IX) o de fórmula (XI) con respecto a la fórmula (IV) es adecuadamente de aproximadamente 0,25:1, aproximadamente 0,3:1, aproximadamente 0,4:1, aproximadamente 0,5:1, aproximadamente 0,6:1, aproximadamente 0,7:1, aproximadamente 0,8:1, aproximadamente 0,9:1, aproximadamente 0,95:1, aproximadamente 1:1, aproximadamente 1,05:1, aproximadamente 1,1:1 o aproximadamente 1,2:1, e intervalos construidos a partir de esas proporciones, como de aproximadamente 0,95:1 a aproximadamente 1,05:1. En un modo de realización, la fórmula (IX) o fórmula (XI) y fórmula (IV) están presentes en cantidades aproximadamente estequiométricas. En un modo de realización, la temperatura de reacción está por debajo de la temperatura de reflujo de mezcla de reacción. En algunos otros modos de realización, la reacción se realiza a reflujo. Por ejemplo, cuando el disolvente es etanol, la temperatura de reacción puede ser adecuadamente de aproximadamente 50 °C a aproximadamente 75 °C. El tiempo de reacción no se limita estrictamente y la reacción continúa típicamente hasta que la conversión de la fórmula (IX) o la fórmula (XI) a la fórmula (VIII) se finaliza esencialmente tal como se determina por cromatografía (por ejemplo, TLC, CG o HPLC).
En un modo de realización, la fórmula (VIII) se puede formar como una sal de un ácido. Los ácidos adecuados incluyen ácidos inorgánicos y ácidos inorgánicos como se describe en otra parte en el presente documento. En un modo de realización, el ácido es un ácido orgánico. En un modo de realización preferente, el ácido es ácido tartárico. En otro modo de realización, el ácido es ácido fumárico. En un modo de realización, la base libre de fórmula (VIII) se puede disolver en un disolvente adecuado, tal como un disolvente prótico polar (por ejemplo, un alcohol tal como metanol o etanol) a una temperatura elevada seguido de la adición de un ácido. En general, el ácido está en exceso estequiométrico en comparación con el compuesto de fórmula (VIII). En algunos de dichos modos de realización, la temperatura de la solución y/o la concentración de la fórmula (VIII) se ajusta para mantener la concentración por debajo de la saturación y evitar de este modo la precipitación y/o cristalización de la fórmula (VIII). Después de la adición de ácido, la solución se puede sembrar opcionalmente con una sal cristalina de fórmula (VIII) del ácido. En cualquiera de los diversos modos de realización, la solución se enfría con agitación para formar una sal cristalina de fórmula (VIII). A continuación, la sal se puede recoger por procedimientos conocidos en la técnica tales como por filtración o centrifugación. En un modo de realización, la sal se recoge por filtración. La sal de fórmula (VIII) recogida se puede lavar opcionalmente, tal como con el disolvente de disolución, y a continuación secar, tal como a vacío parcial.
En un modo de realización particular, la mezcla de reacción de la etapa 1 como se expone anteriormente para la síntesis de un compuesto de fórmula (VIII) comprende ácido tartárico como catalizador ácido, ácido tartárico cristalino de fórmula (VIII) y un disolvente alcohólico (por ejemplo, etanol); la mezcla de productos de reacción de la etapa 1 se diluye con el disolvente alcohólico; y la suspensión resultante se enfría con agitación para formar ácido tartárico cristalino de fórmula (VIII). En otro modo de realización, la mezcla de reacción de la etapa 1 comprende ácido fumárico como catalizador ácido, ácido fumárico cristalino de fórmula (VIII) y un disolvente. El rendimiento de la etapa 1 del compuesto de fórmula (VIII) como sal de ácido tartárico es de al menos un 70 %, al menos un 75 %, al menos un 80 %, al menos un 85 %, al menos un 90 % o al menos un 95 %. En dichos modos de realización, el esquema de reacción de la etapa 1 es como sigue:

Todavía otro aspecto de la divulgación se refiere a un proceso para preparar un compuesto de fórmula (IX) o una sal del mismo. El proceso para preparar el compuesto de fórmula (IX) comprende dos etapas de reacción como se representa a continuación:

R4, s, LG, PG, R5, v, R6, R10, G, p, J y E son como se describe en otra parte en el presente documento.
Los grupos protectores de aldehído se definen en el presente documento y los ejemplos no limitantes incluyen 1,3ditiano, 1,3-ditiolano, dietilacetal, dimetilacetal, etilenglicolacetal, neopentilglicolacetal, trimetilsililcianhidrina y ortoformiato de trietilo.
En la etapa 1, se hace reaccionar una mezcla de reacción que comprende un compuesto de fórmula (X), un compuesto de fórmula (VII) o una sal del mismo, un disolvente orgánico y un catalizador para formar un compuesto de fórmula (XI). En un modo de realización, el disolvente es un disolvente apolar como se describe en otra parte en el presente documento. Los ejemplos no limitantes de disolventes adecuados incluyen pentano, hexano, heptano, ciclopentano, MTBE, éter dietílico, tolueno, 2-metiltetrahidrofurano (2-MeTHF), benceno, 1,4-dioxano,
tetracloruro de carbono, cloroformo, diclorometano y combinaciones de los mismos. En algunos ejemplos, el disolvente es tolueno. En un modo de realización, el catalizador es un catalizador de metal de transición como se describe en el presente documento. En un modo de realización, los ejemplos no limitantes de catalizadores de metales de transición incluyen catalizadores de paladio, platino, oro, rutenio, rodio e iridio. Los ejemplos no limitantes de catalizadores adecuados incluyen JohnPhos, XPhos AuCl, XPhos AuNTf2, XPhos Palladacycle, SPhos Palladacycle, tBuXPhos Pd G1, Xphos Pd G2, SPhos Pd G2, RuPhos Pd G2, CPhos-Pd-G2, CPhos-Pd-G3, tBuXPhos-Pd-G3, RuPhos-Pd-G3, XPhos-Pd-G3, BrettPhos-Pd-G3, JackiePhos-Pd-G3, terc-butil-BrettPhos-Pd-G3, [terc-butil-BrettPhos-Pd (alil)]OTf) y combinaciones de los mismos. En algunos modos de realización, el catalizador es BrettPhos-Pd-G3. En algunos modos de realización, la mezcla de reacción de la etapa 1 comprende además una base orgánica como se describe en otra parte en el presente documento. En algunos de dichos modos de realización, la base es un terc-butóxido tal como terc-butóxido de sodio o potasio. La proporción molar del compuesto de fórmula (VII) con respecto al compuesto de fórmula (X) es adecuadamente de aproximadamente 1:1, aproximadamente 1,1:1, aproximadamente 1,2:1, aproximadamente 1,3:1, aproximadamente 1,5:1 o aproximadamente 2:1, e intervalos construidos a partir de esas proporciones, tales como de aproximadamente 1,1:1 a aproximadamente 1,5:1.
En algunos modos de realización de la etapa 1, la mezcla de reacción se puede calentar con agitación hasta una temperatura de reacción, de desde 2 °C a aproximadamente 30 °C por debajo de la temperatura de reflujo hasta la temperatura de reflujo, y mantener durante un tiempo suficiente para finalizar esencialmente la reacción para el compuesto de fórmula (XI) tal como se determina por cromatografía (por ejemplo, TLC, CG o HPLC). En el caso del disolvente tolueno, la temperatura de reacción es adecuadamente de aproximadamente 50 °C, aproximadamente 55 °C, aproximadamente 60 °C, aproximadamente 65 °C, aproximadamente 70 °C, aproximadamente 75 °C o aproximadamente 80 °C. El tiempo de reacción no se limita estrictamente y la reacción típicamente continúa hasta que la conversión de las fórmulas (VII) y (X) a la fórmula (XI) se finaliza esencialmente tal como se determina por cromatografía (por ejemplo, TLC, CG o HPLC). Después de que finaliza la reacción, la mezcla de productos de reacción se puede desactivar adecuadamente. En algunos modos de realización, la reacción de la etapa 1 se puede desactivar con agua. Cuando la reacción se desactiva con agua, la fase orgánica que comprende el compuesto de fórmula (XI) en solución se puede aislar y opcionalmente lavar al menos una vez con agua. En algunos modos de realización, la mezcla de productos de reacción de la etapa 1 se puede poner en contacto con un depurador de metales conocido en la técnica, tal como por ejemplo y sin limitación, SiliaMetS Thiol. A continuación, la mezcla de productos de reacción se puede filtrar para retirar sólidos.
En la etapa 2, el compuesto de fórmula (XI) se desprotege para formar el compuesto de fórmula (IX) combinando una solución del compuesto (XI) en un disolvente orgánico (por ejemplo, tolueno) con un ácido y agua. El ácido en general está presente en un exceso de equivalentes, tal como una proporción de equivalentes de ácido con respecto al compuesto (XI) de 1,01:1, 1,05:1, 1,1:1, 1,15:1, 1,2:1 o mayor. En un modo de realización, la temperatura de desprotección no es estrictamente crítica y puede ser adecuadamente la temperatura ambiente. Después de la desprotección, se separan la fase orgánica y la fase acuosa (que comprende el compuesto de fórmula (IX)). La fase orgánica se puede lavar opcionalmente con agua. La(s) fase(s) acuosa(s) se puede(n) tratar con una base, tal como una base inorgánica (por ejemplo, NaOH o KOH), combinada con cristales de siembra del compuesto de fórmula (IX). La base se puede añadir adecuadamente en un exceso equivalente. Se forma una suspensión del compuesto cristalino de fórmula (IX) con enfriamiento opcional. Los sólidos del compuesto de fórmula (IX) se pueden recoger por procedimientos conocidos en la técnica tales como filtración o centrifugación. Los sólidos se pueden secar, tal como a vacío parcial, para proporcionar el compuesto sólido de fórmula (IX). El rendimiento para el compuesto de fórmula (IX) a partir del compuesto de fórmula (XI) para las etapas 1 y 2 es de al menos un 65 %, al menos un 70 %, al menos un 75 %, al menos un 80 % o al menos un 85 %.
Un aspecto de la divulgación se refiere a un proceso para preparar un compuesto de fórmula (III) o una sal del mismo. El proceso para preparar el compuesto de fórmula (III) comprende dos etapas de reacción como se representa a continuación:
(1) hacer reaccionar una mezcla de reacción que comprende un compuesto de fórmula (XII), un compuesto B y un disolvente orgánico para formar el compuesto de fórmula (XIII) de acuerdo con la etapa 1 a continuación

donde PG es un grupo protector de amina; y
(2) desproteger el compuesto de fórmula (XIII) para formar el compuesto de fórmula (III) de acuerdo con la etapa 2 a continuación

donde R2a, R2b, R3a, R3b, B y el asterisco son como se describe en otra parte en el presente documento.
Los ejemplos no limitantes de grupos protectores de amina incluyen ACD, Ac, Bn, CBz, trifluoroacetamida, Boc, MeOZ, FMOC, Bz, PMB, DMPM, PMP, Ts y Troc. En un modo de realización, PG es Boc.
En un modo de realización, el compuesto de fórmula (III) tiene la estructura

o una sal del mismo como se describe en el presente documento.
En algunos otros modos de realización, el compuesto de fórmula (III) tiene la estructura

o una sal del mismo como se describe en otra parte en el presente documento.
En la etapa 1, se hace reaccionar una mezcla de reacción que comprende un compuesto de fórmula (XII), B y un disolvente orgánico para formar un compuesto de fórmula (XIII). En un modo de realización, el disolvente orgánico es un disolvente apolar o un disolvente polar. En un modo de realización, el disolvente es apolar. Los ejemplos no limitantes de disolventes adecuados incluyen pentano, ciclopentano, hexano, ciclohexano, benceno, tolueno, 1,4dioxano, cloroformo, éter dietílico, DCM y combinaciones de los mismos. En un modo de realización, el disolvente es DCM. En algunos modos de realización, la concentración de B en el disolvente puede ser adecuadamente de aproximadamente 10 g/l, aproximadamente 25 g/l, aproximadamente 50 g/l, aproximadamente 75 g/l, aproximadamente 100 g/l, aproximadamente 125 g/l, aproximadamente 150 g/l, aproximadamente 175 g/l o aproximadamente 200 g/l, y hasta una concentración cercana a la saturación a la temperatura de reacción, e intervalos construidos a partir de esas concentraciones, tales como de aproximadamente 25 g/l a aproximadamente 125 g/l. La proporción de equivalentes de B con respecto al compuesto de fórmula (XII) es de aproximadamente 0,75:1, aproximadamente 0,9:1, aproximadamente 1:1, aproximadamente 1,25:1, aproximadamente 1,5:1, aproximadamente 1,75:1 o aproximadamente 2:1, e intervalos de las mimas, tales como de aproximadamente 1,25:1 a aproximadamente 1,75:1. La mezcla de reacción de la etapa 1 puede comprender además un agente alquilante adecuado. Los ejemplos no limitantes de reactivos alquilantes incluyen alquillitio (por ejemplo, metillitio) tales como compuestos de haluro de organomagnesio, tales como cloruro de metilmagnesio (por ejemplo, en THF). La proporción de equivalentes de B con respecto al reactivo alquilante es adecuadamente de aproximadamente 0,75:1, aproximadamente 0,8:1, aproximadamente 0,85:1, aproximadamente 0,9:1,
aproximadamente 0,95:1, aproximadamente 1:1, aproximadamente 1,05:1, aproximadamente 1,1:1, aproximadamente 1,15:2, aproximadamente 1,2:1, aproximadamente 1,3:1, aproximadamente 1,4:1, aproximadamente 1,5:1 o mayor, e intervalos construidos a partir de las mismas, tales como de aproximadamente 1,1 a aproximadamente 1,3:1. En un modo de realización, la mezcla de reacción comprende además un catalizador de alquilación. En algunos de dichos modos de realización, el catalizador es un catalizador de metal de transición. En algunos de dichos modos de realización, el metal de transición es cobre. En un modo de realización particular, el catalizador es adecuadamente un haluro de metal de transición, tal como haluro de cobre (I) (por ejemplo, CuCl). En un modo de realización, la temperatura de reacción es de aproximadamente 25 °C, aproximadamente 20 °C, aproximadamente 15 °C, aproximadamente 10 °C, aproximadamente 5 °C, aproximadamente 0 °C, aproximadamente -5 °C, aproximadamente -10 °C, aproximadamente -15 °C, aproximadamente -20 °C, aproximadamente -25 °C, aproximadamente -30 °C, aproximadamente -35 °C, aproximadamente -40 °C, aproximadamente -45 °C o aproximadamente -50 °C, e intervalos construidos a partir de las mismas, tales como de aproximadamente -20 °C a aproximadamente 0 °C. En un modo de realización, el tiempo de reacción no se limita estrictamente y la reacción continúa típicamente hasta que la conversión de B y el compuesto de fórmula (XII) en el compuesto de fórmula (XIII) se finaliza esencialmente tal como se determina por cromatografía (por ejemplo, TLC, CG o HPLC).
Después de la finalización de la reacción, se puede desactivar la reacción, tal como por ejemplo por la adición de ácido acuoso. En un modo de realización, el ácido puede ser un ácido orgánico como se describe en otra parte en el presente documento, con un ejemplo no limitante que es ácido cítrico. A continuación, la fase orgánica que comprende el compuesto de fórmula (XIII) se puede procesar para secar el compuesto. En algunos de dichos modos de realización, la mezcla de productos de reacción desactivada se puede separar en una fase acuosa y una fase orgánica que comprende el compuesto de fórmula (XIII). La fase acuosa se puede lavar una o más veces con un disolvente orgánico, tal como el disolvente usado para formar la mezcla de reacción (por ejemplo, dos lavados, cada uno con un volumen de disolvente en comparación con el volumen de mezcla de reacción). Las fases orgánicas se pueden combinar y lavar una o más veces con salmuera (por ejemplo, dos lavados con salmuera, cada uno con un volumen de salmuera en comparación con el volumen de mezcla de reacción). A continuación, las fases orgánicas lavadas se pueden combinar con agitación con carbón activado y con un agente secante sólido (por ejemplo, Na2SO4). Cualquier carbón activado y agente secante sólido se pueden retirar por filtración o centrifugación. A continuación, cualquier sólido recogido se puede lavar opcionalmente con disolvente adicional para recuperar el compuesto de fórmula (XIII) del mismo.
En un modo de realización, la solución del compuesto de fórmula (XIII) se puede usar como material de partida para la etapa 2. En un modo de realización, se puede preparar el compuesto sólido de fórmula (XIII). En dichos modos de realización, la solución recogida del compuesto de fórmula (XIII) en un disolvente orgánico se puede concentrar a vacío parcial para formar el compuesto de fórmula (XIII) bruto. De forma alternativa, el compuesto sólido de fórmula (XIII) se puede precipitar/cristalizar de la solución por la adición de un antidisolvente, tal como un disolvente apolar (por ejemplo, 2 volúmenes de heptano en comparación con el volumen de disolvente en la mezcla de reacción). Los sólidos se pueden recoger por filtración o centrifugación y los sólidos recogidos se pueden lavar con antidisolvente. Los sólidos se pueden secar a vacío parcial, a una temperatura de menos de 40 °C para proporcionar el compuesto de fórmula (XIII) terminado. El rendimiento del compuesto de fórmula (XIII) basado en el compuesto (XII) es típicamente al menos un 50 %, al menos un 55 %, al menos un 60 % o al menos un 65 %. La pureza por HPLC (porcentaje de área) es de al menos un 90 %, al menos un 95 %, al menos un 98 % o al menos un 99 %.
En algunos modos de realización de la etapa 1, el compuesto B, un catalizador de metal y un agente alquilante se combinan en un primer volumen del disolvente a la temperatura de reacción indicada anteriormente. El volumen de disolvente es adecuadamente de aproximadamente un 30 % a aproximadamente un 80 % del volumen total de disolvente usado para la etapa 1. Después de esto, se añade una solución del compuesto de fórmula (XII) en el resto del disolvente a la temperatura de reacción durante un período de tiempo para formar la mezcla de reacción. A continuación, la mezcla de reacción se mantiene a una temperatura durante un tiempo para finalizar esencialmente la formación del compuesto de fórmula (XIII), seguido de desactivación y procesamiento hasta una solución del compuesto de fórmula (XIII) o compuesto seco de fórmula (XIII).
En la etapa 2, el compuesto de fórmula (XIII) se desprotege para formar el compuesto de fórmula (III). En cualquiera de los diversos modos de realización, una solución del compuesto de fórmula (XIII) se desprotege adecuadamente por la adición de un ácido. En un modo de realización, el compuesto sólido de fórmula (XIII) se disuelve en un disolvente prótico polar como se describe en otra parte en el presente documento (tal como metanol, etanol o i-propanol). La concentración del compuesto de fórmula (XIII) en el disolvente es de aproximadamente 25 g/l, aproximadamente 50 g/l, aproximadamente 75 g/l, aproximadamente 100 g/l, aproximadamente 125 g/l, aproximadamente 150 g/l, aproximadamente 175 g/l o aproximadamente 200 g/l, e intervalos construidos a partir de las mismas, tales como de aproximadamente 50 g/l a aproximadamente 150 g/l. La temperatura de adición de ácido no es estrictamente crítica.
En algunos modos de realización de la etapa 2, el ácido es un ácido inorgánico como se describe en otra parte en el presente documento. Un ejemplo no limitante de un ácido inorgánico adecuado es HCl. En un modo de
realización, la proporción de equivalentes de ácido con respecto al compuesto de fórmula (XIII) es de aproximadamente 1,5:1, aproximadamente 2,5:1, aproximadamente 5:1, aproximadamente 7,5:1, aproximadamente 10:1, aproximadamente 12,5:1 o aproximadamente 15:1, e intervalos construidos a partir de las mismas, tales como de aproximadamente 5:1 a aproximadamente 15:1. Después de la adición de ácido, la solución se mantiene a temperatura con agitación hasta que la desprotección del compuesto de fórmula (XIII) para formar el compuesto de fórmula (III) se finaliza esencialmente. En un modo de realización, se puede realizar un intercambio de disolvente del disolvente prótico polar a un disolvente apolar (como se describe en otra parte en el presente documento) o un disolvente aprótico polar (como se describe en otra parte en el presente documento). En algunos de dichos modos de realización, la solución del compuesto de fórmula (III) se puede concentrar a vacío parcial y extraer con el disolvente aprótico polar o apolar. En algunos de dichos modos de realización, el disolvente de extracción es DCM. Después de la extracción, se puede ajustar el pH de la fase acuosa a fuertemente básico (es decir, mayor de pH 11) con una base adecuada, tal como hidróxido de sodio acuoso. Después de la basificación, a continuación se puede extraer además la fase acuosa al menos una vez con el disolvente de extracción. A continuación, la(s) fase(s) orgánica(s) se puede(n) secar, por ejemplo, con salmuera y/o sobre un desecante sólido seguido de filtración para retirar cualquier sólido. Los sólidos recogidos se pueden lavar opcionalmente para recuperar el compuesto de fórmula (III) adicional. Se puede aislar el compuesto sólido de la solución en disolvente orgánico por procedimientos conocidos en la técnica. Por ejemplo, la solución se puede concentrar a vacío parcial hasta sequedad. De forma alternativa, la solución se puede concentrar seguido de la adición de un antidisolvente para formar el compuesto sólido (III) que se puede recoger por filtración o centrifugación, lavar y secar. El rendimiento del compuesto de fórmula (III) basado en el compuesto (XIII) es típicamente al menos un 85 %, al menos un 90 %, al menos un 95 % o al menos un 97 %. La pureza por HPLC (porcentaje de área) es de al menos un 90 %, al menos un 95 % o al menos un 96 %.
En algunos otros modos de realización de la etapa 2, el ácido es un ácido orgánico como se describe en otra parte en el presente documento. Los ejemplos no limitantes de ácidos orgánicos adecuados incluyen ácidos sulfónicos y ácido alcanforsulfónico (CSA) (por ejemplo, L-(-)-CSA). En el caso de CSA, la proporción de equivalentes de CSA con respecto al compuesto de fórmula (XIII) es de aproximadamente 1,5:1, aproximadamente 2:1, aproximadamente 2,5:1, aproximadamente 5:1, aproximadamente 7,5:1 o aproximadamente 10:1, e intervalos construidos a partir de las mismas, tales como de aproximadamente 2:1 a aproximadamente 4:1. Después de la adición de ácido, la solución se puede calentar y mantener a temperatura elevada (por ejemplo, de aproximadamente 35 °C a aproximadamente 60 °C) con agitación para finalizar la desprotección y formar la sal de ácido del compuesto de fórmula (XIII). A continuación, se puede enfriar la solución/suspensión de sal de ácido, tal como hasta menos de aproximadamente 5 °C, para formar una suspensión de la sal de ácido del compuesto de fórmula (XIII). La sal se puede recoger por filtración o centrifugación y, opcionalmente, lavar con disolvente. A continuación, se puede secar la sal a vacío parcial para proporcionar la sal de compuesto sólida del compuesto de fórmula (XIII), tal como la sal L-(-)-CSA del mismo. El rendimiento de sal del compuesto de fórmula (III) basado en el compuesto de fórmula (XIII) es típicamente al menos un 85 %, al menos un 90 %, al menos un 92 % o al menos un 95 %. La pureza por HPLC (porcentaje de área) es de al menos un 90 %, al menos un 95 %, al menos un 96 % o al menos un 97 %. La sal del compuesto de fórmula (XIII) se puede disolver en agua y ajustar el pH a más de 13, tal como aproximadamente 14, con una base fuerte formando de este modo una suspensión que comprende la base libre del compuesto sólido de fórmula (III). El material sólido se puede recoger por filtración o centrifugación y, opcionalmente, lavar con agua fría. A continuación, se pueden secar los sólidos a vacío parcial para proporcionar la base libre del compuesto seco de fórmula (III). El rendimiento del compuesto de fórmula (III) basado en el compuesto (XIII) es típicamente al menos un 80 %, al menos un 85 % o al menos un 90 %. La pureza por HPLC (porcentaje de área) es de al menos un 90 %, al menos un 95 %, al menos un 96 % o al menos un 97 %.
En un modo de realización, un compuesto de fórmula (VIII) como se prepara en el presente documento se recristaliza además. En un modo de realización, la recristalización comprende recristalizar el compuesto de fórmula (VIII) en un proceso de 2 etapas. El proceso comprende calentar una suspensión que comprende un compuesto de fórmula (VIII) en una mezcla de metanol/etanol, destilar con metanol y enfriar la mezcla. En un modo de realización, la mezcla de metanol/etanol es una mezcla 95:5, 90:10, 85:15 u 80:20 de metanol/etanol. En otro modo de realización, la mezcla de etanol/metanol es una mezcla 90:10 de metanol/etanol. La mezcla se puede calentar a una temperatura de > aproximadamente 50 °C, por ejemplo, aproximadamente 55 °C, 60 °C o 65 °C. El enfriamiento se puede reducir hasta aproximadamente la temperatura ambiente. En un modo de realización, el enfriamiento es hasta aproximadamente 20 °C, 25 °C o 30 °C. La solución se puede filtrar y secar.
En otro modo de realización, la recristalización comprende recristalizar el compuesto de fórmula (VIII) a partir de MTBE. Se añade una base, tal como NaOH o KOH, a la suspensión que comprende el compuesto de fórmula (VIII) en MTBE. Se agita la mezcla, por ejemplo, a 15 °C, 20 °C, 25 °C o 30 °C, opcionalmente se filtra y se destila con etanol.
En algunos modos de realización del proceso para preparar un compuesto de fórmula (III) o una sal del mismo, el proceso comprende además preparar el compuesto de fórmula (XII) o una sal del mismo.
El proceso para preparar el compuesto de fórmula (XII) comprende las etapas de reacción 3a y 3b representadas a continuación:
(1) hacer reaccionar una mezcla de reacción que comprende un compuesto de fórmula (XIV), cloruro de tionilo y un disolvente orgánico para formar un compuesto de fórmula (XV) de acuerdo con la etapa 3a a continuación

(2) hacer reaccionar una mezcla de reacción que comprende el compuesto de fórmula (XV), un catalizador, un agente oxidante y un disolvente orgánico para formar el compuesto de fórmula (XII) de acuerdo con la etapa 3b a continuación

R2a, R2b, R3a, R3b y el grupo protector de nitrógeno PG son como se describe en el presente documento.
En la etapa 3a, se hace reaccionar una mezcla de reacción que comprende un compuesto de fórmula (XIV), cloruro de tionilo y un disolvente orgánico para formar un compuesto de fórmula (XV). En un modo de realización, el disolvente orgánico es un disolvente apolar o un disolvente polar como se describe en otra parte en el presente documento. En un modo de realización, el disolvente es apolar, como se describe en otra parte en el presente documento. Los ejemplos no limitantes de disolventes adecuados incluyen pentano, ciclopentano, hexano, ciclohexano, benceno, tolueno, 1,4-dioxano, cloroformo, éter dietílico, DCM y combinaciones de los mismos. En algunos modos de realización particulares, el disolvente es DCM. En un modo de realización, la concentración del compuesto de fórmula (XIV) en el disolvente puede ser adecuadamente de aproximadamente 10 g/l, aproximadamente 25 g/l, aproximadamente 50 g/l, aproximadamente 75 g/l, aproximadamente 100 g/l, aproximadamente 125 g/l, aproximadamente 150 g/l, aproximadamente 175 g/l o aproximadamente 200 g/l y hasta una concentración cercana a la saturación a la temperatura de reacción, e intervalos construidos a partir de esas concentraciones, tales como de aproximadamente 25 g/l a aproximadamente 125 g/l. La proporción de equivalentes de cloruro de tionilo con respecto al compuesto de fórmula (XIV) es de aproximadamente 1:1, aproximadamente 1,25:1, aproximadamente 1,5:1, aproximadamente 1,75:1, aproximadamente 2:1, aproximadamente 2,25:1 o aproximadamente 2,5:1, e intervalos de las mimas, tales como de aproximadamente 1,25:1 a aproximadamente 1,75:1. En un modo de realización, la temperatura de reacción es de aproximadamente 10 °C, aproximadamente 5 °C, aproximadamente 0 °C, aproximadamente -5 °C o aproximadamente -10 °C, e intervalos construidos a partir de las mismas, tales como de aproximadamente -5 °C a aproximadamente 5 °C. En un modo de realización, el tiempo de reacción no se limita estrictamente y la reacción continúa típicamente hasta que la conversión del compuesto de fórmula (XIV) se finaliza esencialmente tal como se determina por cromatografía (por ejemplo, TLC, CG o HPLC). La mezcla de reacción de la etapa 3a puede comprender además una base, de la que un ejemplo no limitante es imidazol. En dichos modos de realización, la proporción de equivalentes de reactivo de tiolación con respecto a cloruro de tionilo puede ser de aproximadamente 1:1, aproximadamente 2:1, aproximadamente 3:1 o aproximadamente 4:1. La mezcla de reacción de la etapa 3a puede comprender además una base, tal como una base orgánica como se describe en otra parte en el presente documento. En algunos de dichos modos de realización, la base puede ser TEA. En dichos modos de realización, la proporción de equivalentes de la base con respecto al compuesto de fórmula (XIV) es de aproximadamente 1,25:1, aproximadamente 1,5:1, aproximadamente 1,75:1, aproximadamente 2:1, aproximadamente 2,25:1, aproximadamente 2,5:1, aproximadamente 3:1, aproximadamente 3,5:1 o aproximadamente 4:1, e intervalos
construidos a partir de las mismas, tales como de aproximadamente 1,5:1 a aproximadamente 2,5:1.
En algunos modos de realización de la etapa 3a particulares, se forma una solución de la base (por ejemplo, imidazol) en el disolvente a la que se añade cloruro de tionilo con agitación mientras se mantiene la temperatura de reacción. A continuación se puede añadir el compuesto de fórmula (XIV) en el disolvente con agitación mientras se mantiene la temperatura de reacción, seguido de la adición de la base con agitación mientras se mantiene la temperatura de reacción. A continuación, la mezcla de reacción se mantiene a la temperatura de reacción con agitación hasta que se finaliza esencialmente la conversión del compuesto de fórmula (XIV) para formar una mezcla de productos de reacción que comprende el compuesto de fórmula (XV).
En cualquiera de los diversos modos de realización de la etapa 3a, la mezcla de productos de reacción se puede procesar por procedimientos conocidos para los expertos en la técnica. Por ejemplo, la mezcla de reacción de la etapa 3a se puede desactivar con agua enfriada (por ejemplo, de 0,25 a 2 volúmenes de agua por volumen de disolvente orgánico en la mezcla de productos de reacción de la etapa 3a). Una fase orgánica que comprende el compuesto de fórmula (XV) en solución se puede aislar y la fase acuosa aislada se puede extraer con disolvente orgánico para recuperar compuesto de fórmula (XV) adicional. Las fases orgánicas se pueden combinar y lavar con una solución ácida acuosa y una solución básica acuosa y salmuera. Un ejemplo no limitante de una solución ácida acuosa es una solución de un ácido débil, tal como ácido cítrico. Un ejemplo no limitante de una solución básica es una solución de una base débil, tal como bicarbonato de sodio.
En la etapa 3b, una mezcla de reacción que comprende la fórmula (XV) en solución en el disolvente orgánico se combina con un catalizador y un agente oxidante, y se hace reaccionar para formar una mezcla de productos de reacción que comprende el compuesto de fórmula (XII). En un modo de realización, el catalizador es un catalizador de metal activo redox como se describe en otra parte en el presente documento. Los ejemplos no limitantes de catalizadores adecuados incluyen NiCl2, RuCl3, CoCl2, FeCl3, FeCl2, y MnCl2. Los ejemplos no limitantes de oxidantes adecuados incluyen NalO4, NaOCl y Oxone. En algunos modos de realización, la mezcla de reacción comprende además agua. La proporción de volumen de agua con respecto al disolvente orgánico es adecuadamente de aproximadamente 0,25:1, aproximadamente 0,5:1, aproximadamente 0,75:1, aproximadamente 1:1, aproximadamente 1,25:1, aproximadamente 1,50:1, aproximadamente 1,75:1, aproximadamente 2:1 o aproximadamente 2,5:1, e intervalos construidos a partir de las mismas, tales como de aproximadamente 0,5:1 a aproximadamente 1,5:1. La temperatura de reacción es adecuadamente de aproximadamente 5 °C a aproximadamente 50 °C. La mezcla de reacción se mantiene a la temperatura de reacción con agitación hasta que se finaliza esencialmente la conversión del compuesto de fórmula (XV) para formar una mezcla de productos de reacción que comprende el compuesto de fórmula (XII) tal como se determina por cromatografía (por ejemplo, TLC, CG o HPLC).
La mezcla de productos de reacción de la etapa 3b se puede procesar por procedimientos conocidos para los expertos en la técnica. Por ejemplo, la fase orgánica que comprende el compuesto de fórmula (XII) en solución se puede aislar y la fase acuosa se puede filtrar opcionalmente y a continuación extraer con un disolvente orgánico para recuperar el compuesto de fórmula (XII). La(s) fase(s) orgánica(s) se puede(n) lavar opcionalmente con un agente reductor (por ejemplo, tiosulfato de sodio) y con salmuera. La(s) fase(s) orgánica(s) se puede(n) secar con un desecante sólido (por ejemplo, sulfato de sodio) seguido de filtración para retirar sólidos y lavado opcional de los mismos con disolvente orgánico. Compuesto sólido de fórmula (XII) a partir de solución en disolvente orgánico por procedimientos conocidos en la técnica. Por ejemplo, la solución se puede concentrar a vacío parcial hasta sequedad. De forma alternativa, la solución se puede concentrar seguido de la adición de un antidisolvente para formar el compuesto sólido (XII) que se puede recoger por filtración o centrifugación, lavar y secar. El rendimiento del compuesto de fórmula (XII) basado en el compuesto (XIV) es típicamente al menos un 85 %, al menos un 90 % o al menos un 92 %. La pureza por HPLC (porcentaje de área) es de al menos un 95 %, al menos un 98 % o al menos un 99 %.
Se pueden producir aminas primarias quirales usando una transaminasa estereoespecífica por medio de una transaminación asimétrica de una cetona proquiral o bien resolución cinética de una amina racémica. La transaminación es un equilibrio de reacción que consiste en un par de aminas y cetonas (donantes y aceptores) y las condiciones de reacción aplicadas desplazan el equilibrio hacia la amina diana quiral. En uno de dichos aspectos, el donante de amina es alanina o 2-propilamina. En un modo de realización, el aceptor de amina es piruvato o acetona.
Por tanto, se proporciona además en el presente documento un proceso para preparar un compuesto de fórmula (XX) o una sal del mismo, en el que la conversión de la triptamina quiral comprende usar la transformación enzimática:

El proceso para preparar el compuesto de fórmula (XX) comprende poner en contacto un compuesto de fórmula (XXI)

con una proteína transaminasa para formar un compuesto de fórmula (3):

El proceso comprende además la presencia de uno o más donantes de amina. En un modo de realización, el donante de amina es alanina o isopropilamina.
El contacto descrito anteriormente se puede realizar en sistemas de disolventes orgánicos acuosos mixtos. Por ejemplo, la reacción de la transaminasa se puede realizar en un tampón acuoso con un codisolvente orgánico tal como ciclohexano, metilciclohexano, isooctano, DMSO, acetonitrilo o acetona. Dichos codisolventes pueden estar presentes al 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45 o 50 % v/v.
El sistema de disolventes orgánicos/acuosos mixto (sistema de reacción microacuoso) puede comprender, en determinados casos, uno o más disolventes orgánicos que comprenden una pequeña cantidad de tampón acuoso. En un modo de realización, el contacto se realiza en TBME, éter dibutílico, CPME, tolueno, acetato de etilo, acetato de butilo, acetato de isopropilo, butirato de butilo, butirato de etilo o acetato de isobutilo que comprende menos de un 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2 o 1 % v/v de tampón acuoso. Cuando se realiza en un disolvente orgánico, la transaminasa se puede inmovilizar en un soporte sólido.
En otro modo de realización, la conversión del compuesto (XXI) en el compuesto (3) está por encima de al menos un 70 %, 75 %, 80 %, 85 %, 90 %, 95 %, 96 %, 97 %, 98 % o 99 %. En otro aspecto, la conversión está por encima de un 90 %. En otro modo de realización, la conversión está por encima de un 95 %.
La aminación reductora finalizada por la transaminasa puede proporcionar el compuesto de fórmula (3) en al menos un 90, 91, 92, 93, 94, 95, 96, 97, 98, 99 o 100 % de exceso enantiomérico (EE). En determinados casos, la transaminasa convierte el compuesto de fórmula (XXI) en (3) con un EE mayor de un 99 %.
En un modo de realización, la transaminación asimétrica del compuesto de fórmula (XXI) se realiza usando el esquema a continuación:

El compuesto de fórmula (3) se pone en contacto con un compuesto de fórmula (II) como se describe en el presente documento:

para formar el compuesto de fórmula (XX).
R1a, R1b y n son como se describe en otra parte en el presente documento. En un modo de realización, R1a y R1b son independientemente flúor.
En un modo de realización, el compuesto de fórmula (XX) tiene la estructura:

La proteína transaminasa se puede seleccionar de la tabla 6. En uno de dichos aspectos, la transaminasa es (R)-selectiva. En un modo de realización, la transaminasa es TA-P2-A01. En un modo de realización, la proteína transaminasa se selecciona de una transaminasa (S)-enantioselectiva de SEQ ID NO:1, SEQ ID NO:2, SEQ ID NO:3 y SEQ ID NO:4.
Se proporcionan además en el presente documento procedimientos para sintetizar enzimáticamente un compuesto de fórmula (3) a través de resolución cinética de una amina racémica en presencia de un aceptor de amina y una transaminasa. La reacción se puede realizar en condiciones como se describe en el presente documento, por ejemplo, en sistemas de disolventes acuosos/orgánicos mixtos como se describe en el presente documento. En uno de dichos aspectos, la reacción se realiza en tampón acuoso que comprende acetonitrilo. Dicha resolución cinética puede proporcionar el compuesto de fórmula (3) en un EE de al menos un 99 %, requiriendo una tasa de conversión por encima de un 50 %. La resolución cinética comprende el uso de una transaminasa (S)-selectiva como se describe en el presente documento para realizar la resolución cinética. En un modo de realización, la transaminasa se selecciona de la tabla 6.
Un modo de realización de la divulgación se refiere a un compuesto de fórmula (XVI):

R4, s, R10, G y p son como se define en el presente documento.
cada uno de R7a, R7b, R8a y R8b es independientemente hidrógeno, halógeno, alquilo C1−3, haloalquilo C1−3, hidroxialquilo C1−3 o −CN. En un modo de realización, cada uno de R7a, R7b, R8a y R8b es independientemente hidrógeno, flúor, −CH3 o −CN. En un modo de realización, cada uno de R7a, R7b, R8a y R8b es hidrógeno.
y es un número entero de 1 o 2, x es un número entero de 1 o 2, y el total de x e y es 2 o 3.
M es alquilo C1−5;
r es 0 o 1; y
R9 es halógeno o -CN.
En un modo de realización, M es −CH2CH2CH2−, p es 1, y R9 es flúor.
En un modo de realización, M es −CH2CH2CH2−, p es 1, y R9 es −CN.
En un modo de realización, el compuesto de fórmula (XVI) es:


En un modo de realización particular, el compuesto de fórmula (XVI) es:

En otro aspecto se proporciona en el presente documento un compuesto que tiene la estructura:

En otro aspecto se proporciona en el presente documento un proceso para preparar un compuesto que tiene la fórmula:

en el que el proceso se realiza como se expone en el esquema A a continuación y de acuerdo con los modos de realización y modos de realización proporcionados en el presente documento.
Esquema A:

BrettPhos Pd G3 = metanosulfonato de [(2-di-ciclohexilfosfino-3,6-dimetoxi-2',4',6'-triisopropil-1,1'-bifenil)-2-(2'-amino-1,1'-bifenil)]paladio(II)
Todavía en otro modo de realización de la divulgación es un proceso para preparar un compuesto que tiene la fórmula:

en el que el proceso se realiza como se expone en el esquema B a continuación y de acuerdo con los modos de realización y modos de realización proporcionados en el presente documento.
Esquema B:

Los procesos para la síntesis del compuesto B anterior (por ejemplo, esquema A y B) pueden comprender además la recristalización de acuerdo con el esquema C o D a continuación:
Esquema C:

Esquema D:

En un modo de realización, los compuestos (3) y (4) como se describe en el presente documento y se proporciona en el esquema A y esquema B, y opcionalmente esquema C o D, anteriores se sintetizan de acuerdo con el esquema E a continuación y de acuerdo con los modos de realización y los modos de realización proporcionados en el presente documento.
Esquema E:

Se proporciona además en el presente documento un proceso para preparar un compuesto de fórmula (XXIII) o una sal del mismo:

B, R1a, R1b, n, R2a, R2b, R3a, R3b, J, R4, s, G, R5, v, R6, E y los asteriscos son como se define en el presente documento.
En un modo de realización, el compuesto de fórmula (XXIII) es una sal de ácido. En dichos modos de realización, el compuesto de fórmula (VIII) es una sal de un ácido. En un modo de realización preferente, el compuesto de fórmula (XXIII) es una sal de ácido tartárico. En algunos modos de realización, el compuesto de fórmula (XXIII) es una sal de ácido fumárico.
En un modo de realización, el compuesto de fórmula (XXIII) tiene una cualquiera de las siguientes estructuras, o una sal de ácido tartárico de las mismas:



En otro modo de realización, los compuestos anteriores son una sal de ácido fumárico.
En un modo de realización, el compuesto de fórmula (XXIII) tiene la siguiente estructura, o una sal farmacéuticamente aceptable del mismo:

El proceso para preparar el compuesto de fórmula (XXIII) comprende dos etapas de reacción como se representa a continuación:


LG es como se define en el presente documento.
Las variables de fórmulas (IV), (V) y (VI) son como se describe en el presente documento.
En un modo de realización, el compuesto de fórmula (XXIV) tiene una cualquiera de las siguientes estructuras, o una sal del mismo:

Un modo de realización de la divulgación se refiere a un proceso para preparar un compuesto de fórmula (XXIII) o una sal del mismo, en el que el compuesto de fórmula (XXIII) es como se describe en el presente documento. El proceso para preparar el compuesto de fórmula (XXIII) de acuerdo con este modo de realización comprende la etapa de reacción 1 como se representa a continuación:

Cada uno de B, R1a, R1b, n, R2a, R2b, R3a, R3b, R4, s, J, R5, v, R6, G, p, E y el asterisco son como se describe en otra parte en el presente documento. El resto CHO y el átomo de nitrógeno que enlaza J y G se localizan en la
posición para entre sí en J.
En algunos modos de realización, el compuesto de fórmula (XXIV) es:

Un modo de realización de la divulgación se refiere a un proceso para preparar un compuesto de fórmula (XXVII) o una sal del mismo.
El proceso para preparar el compuesto de fórmula (XXVII) comprende dos etapas de reacción como se representa a continuación:

R4, s, LG, R5, v, R6, G, p, E y PG son como se describe en el presente documento.
Se proporcionan además en el presente documento formas sólidas, formulaciones que comprenden dichas formas sólidas y procedimientos de uso de dichas formas sólidas del compuesto A:

y que tiene el nombre 3-((1R,3R)-1-(2,6-difluoro-4-((1-(3-fluoropropil)acetidin-3-il)amino)fenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol, incluyendo una sal farmacéuticamente aceptable del mismo.
En un modo de realización se proporcionan en el presente documento formas sólidas del compuesto A. El compuesto A puede ser una base libre como se describe en el presente documento que existe en una forma sólida amorfa. En otro modo de realización, el compuesto A es un sólido cristalino como se describe en el presente documento. Todavía en otro modo de realización, el compuesto A es una sal de tartrato cristalina que tiene la estructura:
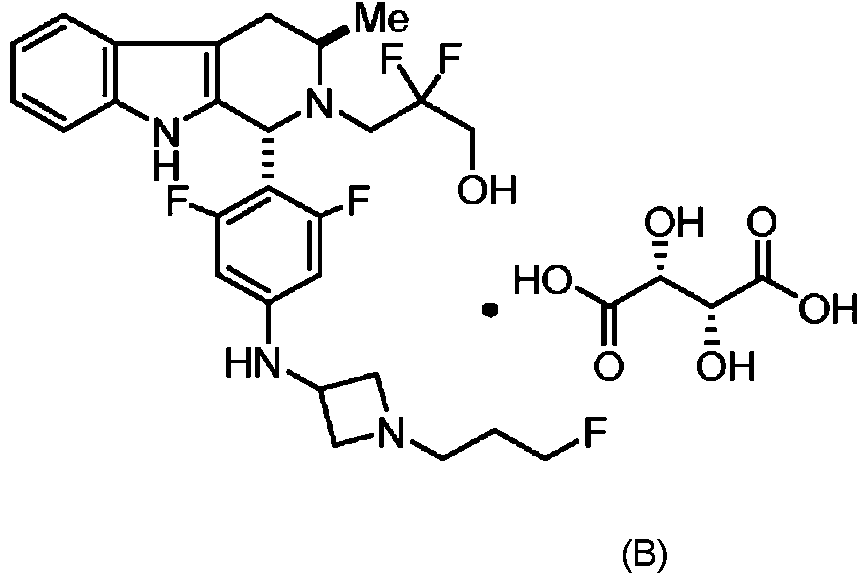
En otro aspecto se proporcionan en el presente documento formas sólidas cristalinas del compuesto A como una sal de fumarato que tiene la estructura:
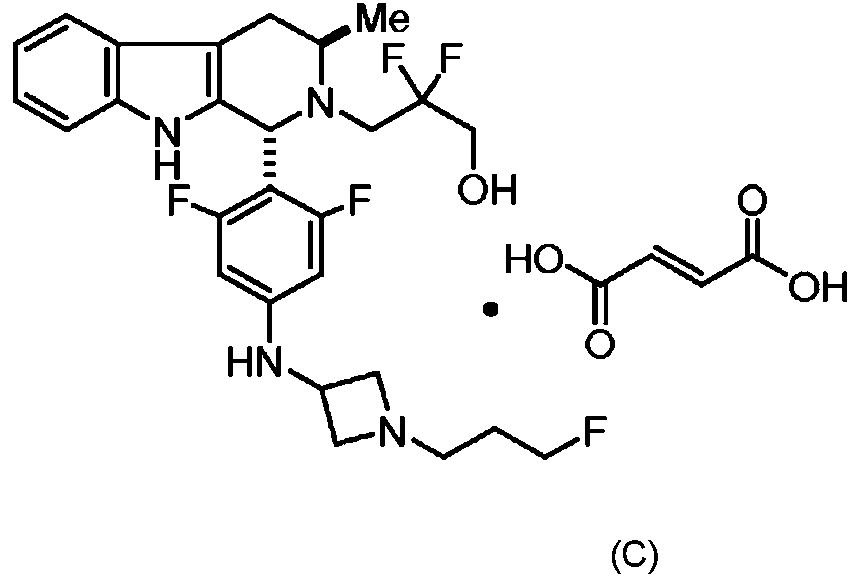
Todavía en otro aspecto, se proporcionan en el presente documento formas sólidas de sales de malonato del compuesto A que tiene la estructura:

Las formas sólidas descritas en el presente documento pueden ser cristalinas. En otro modo de realización, la forma sólida es una forma sólida de un solo componente. Las formas sólidas descritas en el presente documento pueden ser solvatos, hidratos, anhidratos o sales como se expone en el presente documento. En un modo de realización, las formas sólidas descritas en el presente documento comprenden sales de tartrato. En otro modo de realización, las formas sólidas descritas en el presente documento comprenden anhidratos del compuesto B. En otro modo de realización, las formas sólidas descritas en el presente documento comprenden sales de fumarato. Todavía en otro modo de realización, las formas sólidas descritas en el presente documento comprenden sales de fosfato u otras sales.
Aunque sin pretender limitarse por ninguna teoría particular, las formas sólidas se pueden caracterizar por propiedades físicas tales como, por ejemplo, estabilidad, solubilidad y tasa de disolución, densidad, compresibilidad, dureza, morfología, escisión, pegajosidad, solubilidad, absorción de agua, propiedades eléctricas, comportamiento térmico, reactividad en estado sólido, estabilidad física y estabilidad química) que afectan a procesos particulares (por ejemplo, rendimiento, filtración, lavado, secado, molienda, mezcla, formación de comprimidos, fluidez, disolución, formulación y liofilización) lo que hace que determinadas formas sólidas sean adecuadas para la fabricación de una forma de dosificación sólida. Dichas propiedades se pueden determinar usando técnicas químicas analíticas particulares, incluyendo técnicas analíticas de estado sólido (por ejemplo, difracción de rayos X, microscopía, espectroscopia y análisis térmico), como se describe en el presente documento. Las formas sólidas descritas en el presente documento, incluyendo formas de sal, formas cristalinas y sólidos amorfos, se pueden caracterizar por una serie de procedimientos incluyendo, por ejemplo, difracción de rayos X de monocristal, difracción de rayos X en polvo (XRPD), microscopía (por ejemplo, microscopía electrónica de barrido (SEM)), análisis térmico (por ejemplo, calorimetría diferencial de barrido (DSC), sorción dinámica de vapor (DVS), análisis termogravimétrico (TGA) y microscopía de etapa caliente), espectroscopia (por ejemplo, infrarrojo, Raman, y resonancia magnética nuclear de estado sólido), cromatografía de líquidos de ultraalto rendimiento (UHPLC), resonancia magnética nuclear de protones (espectro.
Las técnicas para caracterizar formas cristalinas y sólidos amorfos incluyen, por ejemplo, análisis termogravimétrico (TGA), calorimetría diferencial de barrido (DSC), difractometría de rayos X en polvo (XRPD), difractometría de rayos X de monocristal, espectroscopia vibratoria, por ejemplo, espectroscopia de infrarrojo (IR) y Raman, espectroscopia de resonancia magnética nuclear (RMN) de estado sólido y solución (incluyendo RMN de 1H y RMN de F), microscopía electrónica de barrido (SEM), cristalografía electrónica y análisis cuantitativo, análisis del tamaño de partícula (PSA), análisis del área de superficie, estudios de solubilidad y estudios de disolución.
La pureza de las formas sólidas proporcionadas en el presente documento se puede determinar por procedimientos analíticos estándar, tales como cromatografía en capa fina (TLC), electroforesis en gel, cromatografía de gases, cromatografía de líquidos de ultraalto rendimiento (UHPLC) y espectrometría de masas (EM).
Compuesto B, forma A:
En determinados modos de realización, se proporciona en el presente documento una forma sólida del compuesto B designada como forma A. La forma A es una forma sólida cristalina del compuesto B. En un modo de realización, la forma A es un solvato de acetona del compuesto B. En un modo de realización, la forma A es una sal de tartrato de solvato de acetona cristalina del compuesto A.
En otro modo de realización, la forma A del compuesto B se obtiene suspendiendo en acetona seguido de evaporación. La forma A se puede preparar de acuerdo con los procedimientos y ejemplos descritos en el presente documento.
En un modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma A, es
una sal de tartrato del compuesto A (es decir, compuesto B), y es cristalina, como se indica por mediciones del patrón de difracción de rayos X en polvo (XRPD). En un modo de realización, el XRPD de una forma sólida proporcionada en el presente documento, por ejemplo, forma A, es sustancialmente como se muestra en la FIG.
1. En otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma A, tiene uno o más picos de XRPD característicos a aproximadamente 4,64, 8,26, 9,28, 11,18, 11,49, 11,96, 12,54, 13,77, 14,22, 14,61, 15,09, 15,56, 16,01, 17,35, 18,55, 18,84, 19,32, 19,82, 20,26, 21,34, 21,63, 21,92, 22,52, 22,97, 23,28, 23,54, 23,94, 24,81 o 25,96 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG. 1 y como se encuentra en la tabla 16 en el presente documento. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma A, tiene al menos 3, al menos 5, al menos 7 o al menos 10 picos de XPRD a aproximadamente 4,64, 8,26, 9,28, 11,18, 11,49, 11,96, 12,54, 13,77, 14,22, 14,61, 15,09, 15,56, 16,01, 17,35, 18,55, 18,84, 19,32, 19,82, 20,26, 21,34, 21,63, 21,92, 22,52, 22,97, 23,28, 23,54, 23,94, 24,81 o 25,96 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG.1 y como se encuentra en la tabla 16 en el presente documento. Aún en otro modo de realización, una forma sólida descrita en el presente documento, por ejemplo, forma A, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve, veinte o todos los picos de XRPD característicos como se expone en la tabla 16.
Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma A, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve o diez picos de XRPD característicos a aproximadamente 12,54, 14,61, 16,01, 19,32, 20,26, 21,63, 23,28, 23,54, 23,94 o 24,81 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG.1 y como se encuentra en la tabla 16 en el presente documento. En otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma A, tiene uno, dos, tres, cuatro o cinco picos de XRPD característicos a aproximadamente 19,32, 20,26, 21,63, 23,28 o 24,81 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG. 1 y como se encuentra en la tabla 16 en el presente documento. En otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma A, tiene uno, dos, tres, cuatro o cinco picos de XRPD característicos a aproximadamente 19,32, 20,26, 21,63, 23,28 o 24,81 ± 0,05° 2θ, como se representa, por ejemplo, en la FIG.1 y como se encuentra en la tabla 16 en el presente documento.
En un modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma A, que tiene un termógrafo de TGA que corresponde sustancialmente al termograma de TGA representativo como se representa en la FIG. 2. En determinados modos de realización, la forma cristalina presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente un 7,2 % de la masa total de la muestra antes de aproximadamente 125 °C.
En otro modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma A, que tiene un termograma de DSC sustancialmente como se representa en la FIG.2 que comprende un acontecimiento de desolvatación a aproximadamente 124 °C y una temperatura de fusión que tiene una temperatura de aparición de aproximadamente 164 °C y una temperatura máxima de pico de aproximadamente 171 °C.
En otro modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma A, que tiene una imagen de microscopía de luz polarizada como se representa en la FIG.3.
Todavía en otro modo de realización, la forma A es pura. En determinados modos de realización, la forma A pura está sustancialmente libre de otras formas sólidas, por ejemplo, sólido amorfo. En determinados modos de realización, la pureza de la forma A no es menos de aproximadamente un 95 %, no menos de aproximadamente un 96 %, no menos de aproximadamente un 97 %, no menos de aproximadamente un 98 %, no menos de aproximadamente un 98,5 %, no menos de aproximadamente un 99 %, no menos de aproximadamente un 99,5 %, o no menos de aproximadamente un 99,9 %.
Compuesto B, forma B:
En determinados modos de realización, se proporciona en el presente documento una forma sólida del compuesto B designada como forma B. La forma B es una forma sólida cristalina del compuesto B. En un modo de realización, la forma B es un anhidrato del compuesto B. En otro modo de realización, la forma B es una sal de tartrato de anhidrato del compuesto A.
En un modo de realización, la forma B del compuesto B se obtiene suspendiendo el compuesto B en acetato de etilo a TA durante aproximadamente 24 horas. En otro modo de realización, la forma B del compuesto B se obtiene suspendiendo el compuesto B en acetona:agua (por ejemplo, 90:10, 95:5, 96:4, 97:3, 99:1 v/v) a aproximadamente 50 °C durante aproximadamente 6 horas. Todavía en otro modo de realización, la forma B del compuesto B se obtiene suspendiendo el compuesto B en etanol.
En determinados casos, la forma B del compuesto B se obtiene suspendiendo el compuesto B en un sistema de disolventes que comprende acetona al ≥ 95 % (por ejemplo, acetona:agua ≥95:5). A continuación, la forma B del compuesto B se aísla de la suspensión, por ejemplo, por centrifugación o filtración. En otro modo de realización,
la forma B del compuesto B se obtiene a partir de la forma A o bien la forma F como se describe en el presente documento. En un modo de realización, la forma A del compuesto B se resuspende en etanol (por ejemplo, etanol al 100 %) durante 10, 12, 16 o 24 horas (por ejemplo, durante la noche) para obtener la forma B. En un modo de realización, la forma F se convierte en la forma D como se describe en el presente documento en presencia de agua. A continuación, la mezcla se puede suspender en etanol puro (a, por ejemplo, aproximadamente 50 °C) o acetona:agua 95:5 o 97:3 (v/v) para formar la forma B. Las mezclas se pueden sembrar opcionalmente con cristales de forma B.
La forma B se puede preparar de acuerdo con los procedimientos y ejemplos descritos en el presente documento. Por tanto, se proporciona en el presente documento un procedimiento para preparar la forma B, donde el procedimiento comprende suspender el compuesto B en un sistema de disolventes que comprende acetona o mezclas acuosas de acetona y agua (por ejemplo, acetona al 50 %, 90 %, 95 %, 96 %, 97 %, 98 % y 99 % v/v). En un modo de realización, el sistema de disolventes para cristalizar la forma B comprende acetona al ≥ 95 %. En un modo de realización, el sistema de disolventes para la forma B comprende acetona:agua 96:4. Las mezclas se pueden suspender a TA durante aproximadamente 120 h. Las mezclas se pueden filtrar y analizar como se describe en el presente documento (por ejemplo, XRPD). En un modo de realización, la forma B se prepara de acuerdo con los procedimientos y/o ejemplos expuestos en el presente documento.
En un modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma B, es una sal de tartrato del compuesto A y es cristalina, como se indica por las mediciones del patrón de difracción de rayos X en polvo (XRPD). En un modo de realización, el XRPD de una forma sólida proporcionada en el presente documento, por ejemplo, forma B, es sustancialmente como se muestra en la FIG.4. En otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma B, tiene uno o más picos de XRPD característicos a aproximadamente 7,68, 11,49, 12,54, 14,24, 15,30, 15,55, 16,01, 16,63, 17,37, 18,24, 19,16, 19,42, 19,89, 20,24, 21,81, 22,52, 22,99, 23,25, 23,57, 24,67, 25,07, 25,91 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG. 4 y como se encuentra en la tabla 17 en el presente documento. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma B, tiene al menos 3, al menos 5, al menos 7 o al menos 10 picos de XPRD característicos a aproximadamente 7,68, 11,49, 12,54, 14,24, 15,30, 15,55, 16,01, 16,63, 17,37, 18,24, 19,16, 19,42, 19,89, 20,24, 21,81, 22,52, 22,99, 23,25, 23,57, 24,67, 25,07, 25,91 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG. 4 y como se encuentra en la tabla 17 en el presente documento. Aún en otro modo de realización, una forma sólida descrita en el presente documento, por ejemplo, forma B, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince o todos los picos de XRPD característicos como se expone en la tabla 17.
Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma B, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve o diez picos de XRPD característicos a aproximadamente 11,49, 12,54, 15,30, 15,55, 19,16, 19,42, 20,24, 23,25, 24,67 o 25,91 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG.4. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma B, tiene uno, dos, tres, cuatro o cinco picos de XRPD característicos a aproximadamente 11,49, 12,54, 19,16, 19,42 o 24,67 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG. 4. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma B, tiene uno, dos, tres, cuatro o cinco picos de XRPD característicos a aproximadamente 11,49, 12,54, 19,16, 19,42 o 24,67 ± 0,05° 2θ, como se representa, por ejemplo, en la FIG.4.
En un modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma B, que tiene un termógrafo de TGA que corresponde sustancialmente al termograma de TGA representativo como se representa en la FIG. 5. En determinados modos de realización, la forma cristalina presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente un 3,5 % de la masa total de la muestra. En otro modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma B, que tiene un termograma de DSC sustancialmente como se representa en la FIG.5 que comprende un acontecimiento endotérmico con una temperatura de aparición de aproximadamente 171 °C y una temperatura máxima de pico de aproximadamente 179 °C.
En otro modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma B, que tiene un espectro de RMN de 13C y 19F sustancialmente como se representa en la FIG.6 y FIG.7, respectivamente. En otro modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma B, que tiene un perfil de sorción-desorción de agua como se representa en la FIG.8. La forma sólida, por ejemplo, forma B del compuesto B, absorbe aproximadamente un 1,2 % p/p de humedad hasta un 90 % de humedad relativa (HR) a aproximadamente 25 °C.
En otro modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma B, que tiene una imagen de microscopio electrónico de barrido (SEM) y una imagen PLM como se representa en la FIG.
9a y FIG.9b, respectivamente. La muestra comprende agregados esféricos densos.
En otro modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma B, que tiene una distribución del tamaño de partícula (PSD) como se representa en la FIG.9c.
Todavía en otro modo de realización, una forma sólida, por ejemplo, forma B, permanece sustancialmente sin cambios después de la compresión como se describe en el presente documento. Las FIG.22, FIG. 23 y FIG. 24, muestran respectivamente XRPD, RMNes de 19F y DSC de la forma B del compuesto B y comparan el compuesto antes y después de la compresión como se describe en el presente documento.
Todavía en otro modo de realización, la forma B es sustancialmente pura. En determinados modos de realización, la forma B pura está sustancialmente libre de otras formas sólidas, por ejemplo, sólido amorfo. En determinados modos de realización, la forma B sustancialmente pura está sustancialmente libre de forma A, forma D o forma F. En determinados modos de realización, la pureza de la forma B no es menos de aproximadamente un 95 %, no menos de aproximadamente un 96 %, no menos de aproximadamente un 97 %, no menos de aproximadamente un 98 %, no menos de aproximadamente un 98,5 %, no menos de aproximadamente un 99 %, no menos de aproximadamente un 99,5 % o no menos de aproximadamente un 99,9 %.
Compuesto B, forma C:
En determinados modos de realización, se proporciona en el presente documento una forma sólida del compuesto B designada como forma C. La forma C es una forma sólida cristalina del compuesto B. En un modo de realización, la forma C es un solvato de THF del compuesto B.
En un modo de realización, la forma C del compuesto B se obtiene suspendiendo el compuesto B en THF. A continuación, se puede filtrar la mezcla. La forma C se puede preparar de acuerdo con los procedimientos y ejemplos descritos en el presente documento.
En un modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma C, es sal de tartrato del compuesto A y es sustancialmente cristalina, como se indica por las mediciones del patrón de difracción de rayos X en polvo (XRPD). En un modo de realización, el XRPD es sustancialmente como se muestra en la FIG.10.
En un modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma C, que tiene un termógrafo de TGA que corresponde sustancialmente al termograma de TGA representativo como se representa en la FIG. 11. En determinados modos de realización, la forma cristalina presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente un 6,8 % de la masa total de la muestra. En otro modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma C, que tiene un termograma de DSC sustancialmente como se representa en la FIG.11 que comprende un acontecimiento endotérmico con una temperatura de aparición de aproximadamente 118 °C y una temperatura máxima de pico de aproximadamente 125 °C.
Todavía en otro modo de realización, la forma C es pura. En determinados modos de realización, la pureza de la forma C no es menos de aproximadamente un 95 %, no menos de aproximadamente un 96 %, no menos de aproximadamente un 97 %, no menos de aproximadamente un 98 %, no menos de aproximadamente un 98,5 %, no menos de aproximadamente un 99 %, no menos de aproximadamente un 99,5 % o no menos de aproximadamente un 99,9 %.
Compuesto B, forma D:
En determinados modos de realización, se proporciona en el presente documento una forma sólida del compuesto B designada como forma D. La forma D es una forma sólida cristalina del compuesto B. En un modo de realización, la forma D es un hidrato del compuesto B. En otro modo de realización, la forma D es un monohidrato del compuesto B.
En un modo de realización, la forma D del compuesto B se obtiene suspendiendo el compuesto B en etanol al 100 % durante aproximadamente 48 horas. A continuación, se puede filtrar la mezcla.
En un modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma D, es una sal de tartrato del compuesto A y es sustancialmente cristalina, como se indica por las mediciones del patrón de difracción de rayos X en polvo (XRPD). En otro modo de realización, la forma D es un hidrato del compuesto B. En un modo de realización, el XRPD de una forma sólida proporcionada en el presente documento, por ejemplo, forma D, es sustancialmente como se muestra en la FIG. 12. En otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma D, tiene uno o más picos de XRPD característicos a aproximadamente 7,32, 10,99, 11,31, 12,18, 13,23, 13,48, 14,11, 14,66, 15,14, 15,70, 16,03, 16,21, 16,54, 17,24, 17,63, 18,11, 18,34, 19,10, 20,20, 20,58, 21,16, 21,47, 21,89, 22,76, 23,33 o 23,56 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG. 12 y como se encuentra en la tabla 19. Todavía en otro modo de realización, una forma
sólida proporcionada en el presente documento, por ejemplo, forma D, tiene al menos 3, al menos 5, al menos 7 o al menos 10 picos característicos de XRPD a aproximadamente 7,32, 10,99, 11,31, 12,18, 13,23, 13,48, 14,11, 14,66, 15,14, 15,70, 16,03, 16,21, 16,54, 17,24, 17,63, 18,11, 18,34, 19,10, 20,20, 20,58, 21,16, 21,47, 21,89, 22,76, 23,33 o 23,56 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG. 12 y como se encuentra en la tabla 19. Aún en otro modo de realización, una forma sólida descrita en el presente documento tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince o todos los picos de XRPD característicos como se expone en la tabla 19.
Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma D, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve o diez picos de XRPD característicos a aproximadamente 11,31, 15,70, 16,54, 19,10, 20,58, 21,16, 21,47, 21,89, 22,76 o 23,33 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG.12. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma D, tiene uno, dos, tres, cuatro o cinco picos de XRPD característicos a aproximadamente 11,31, 15,70, 16,54, 19,10 o 22,76 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG. 12. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma D, tiene uno, dos, tres, cuatro o cinco picos de XRPD característicos a aproximadamente 11,31, 15,70, 16,54, 19,10 o 22,76 ± 0,05° 2θ, como se representa, por ejemplo, en la FIG.12.
En un modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma D, que tiene un termógrafo de TGA que corresponde sustancialmente al termograma de TGA representativo como se representa en la FIG. 13. En determinados modos de realización, la forma cristalina presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente un 1,4 % de la masa total de la muestra antes de aproximadamente 150 °C.
En otro modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma D, que tiene un termograma de DSC sustancialmente como se representa en la FIG.13 que comprende un acontecimiento endotérmico que tiene una temperatura de aparición de aproximadamente 55 °C y una temperatura máxima de pico de aproximadamente 82 °C seguido de un segundo acontecimiento endotérmico con una temperatura de aparición de aproximadamente 165 °C y una temperatura máxima de pico de aproximadamente 172 °C.
Todavía en otro modo de realización, la forma D es pura. En determinados modos de realización, la forma D está sustancialmente libre de otras formas sólidas, por ejemplo, sólido amorfo. En determinados modos de realización, la forma D está sustancialmente libre de forma A, forma B o forma F. En determinados modos de realización, la pureza de la forma D no es menos de aproximadamente un 95 %, no menos de aproximadamente un 96 %, no menos de aproximadamente un 97 %, no menos de aproximadamente un 98 %, no menos de aproximadamente un 98,5 %, no menos de aproximadamente un 99 %, no menos de aproximadamente un 99,5 % o no menos de aproximadamente un 99,9 %.
Compuesto B, forma E:
En determinados modos de realización, se proporciona en el presente documento una forma sólida del compuesto B designada como forma E. La forma E es una forma sólida del compuesto B. En un modo de realización, la forma E es un solvato de DMSO del compuesto B.
En un modo de realización, la forma E del compuesto B se obtiene suspendiendo el compuesto B en DMSO y añadiendo IPAc, durante aproximadamente 24 horas. A continuación, se puede filtrar la mezcla. La forma E se puede preparar de acuerdo con los procedimientos y ejemplos descritos en el presente documento.
En un modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma E, es una sal de tartrato del compuesto A y es sustancialmente cristalina, como se indica por las mediciones del patrón de difracción de rayos X en polvo (XRPD). En un modo de realización, el XRPD es sustancialmente como se muestra en la FIG.14.
En un modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma E, que tiene un termógrafo de TGA que corresponde sustancialmente al termograma de TGA representativo como se representa en la FIG. 15. En determinados modos de realización, la forma cristalina presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente un 8,3 % de la masa total de la muestra. En otro modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma E, que tiene un termograma de DSC sustancialmente como se representa en la FIG. 15 que comprende una primera endotermia con una temperatura de aparición de aproximadamente 126 °C y una temperatura máxima de pico de aproximadamente 134 °C y un segundo acontecimiento endotérmico con una temperatura de aparición de aproximadamente 143 °C y una temperatura máxima de pico de aproximadamente 147 °C.
Todavía en otro modo de realización, la forma E es pura. En determinados modos de realización, la forma E está sustancialmente libre de otras formas sólidas, por ejemplo, sólido amorfo. En determinados modos de realización,
la pureza de la forma E no es menos de aproximadamente un 95 %, no menos de aproximadamente un 96 %, no menos de aproximadamente un 97 %, no menos de aproximadamente un 98 %, no menos de aproximadamente un 98,5 %, no menos de aproximadamente un 99 %, no menos de aproximadamente un 99,5 % o no menos de aproximadamente un 99,9 %.
Compuesto B, forma F:
En determinados modos de realización, se proporciona en el presente documento una forma sólida del compuesto B designada como forma F. La forma F es una forma sólida cristalina del compuesto B. En un modo de realización, la forma F es un anhidrato del compuesto B. En otro modo de realización, la forma F es una sal de tartrato de anhidrato del compuesto A.
En un modo de realización, la forma F del compuesto B se obtiene suspendiendo el compuesto B en etanol al 100 % a TA o a 50 °C durante aproximadamente 8, 10, 12, 15, 20 o 25 horas (por ejemplo, durante la noche). En otro modo de realización, la forma F se puede obtener suspendiendo el compuesto B en etanol/agua (por ejemplo, 65:35 v/v). La suspensión del compuesto B para obtener la forma F puede incluir opcionalmente sembrar con la forma B descrita en el presente documento. La suspensión se puede filtrar para obtener la forma F. La forma F también se puede obtener suspendiendo a TA en agua DI al 100 %, acetona/agua 1:1 o acetona al 100 %. Las suspensiones de la forma F en disolventes puros se pueden mantener a TA. En un modo de realización, la forma F se puede obtener a partir de una mezcla de acetona:agua 1:1 agitada a 5 °C. Todavía en otro modo de realización, la forma F se puede obtener suspendiendo el compuesto B en mezclas de acetona:agua 95:5 y acetona:agua 97:3 a 50 °C durante aproximadamente 2 horas y enfriando hasta TA. La forma F se puede preparar de acuerdo con los procedimientos y ejemplos descritos en el presente documento.
En un modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma F, es una sal de tartrato del compuesto A y es sustancialmente cristalina, como se indica por las mediciones del patrón de difracción de rayos X en polvo (XRPD). En un modo de realización, el XRPD de una forma sólida proporcionada en el presente documento, por ejemplo, forma F, es sustancialmente como se muestra en la FIG.16. En otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma F, tiene uno o más picos de XRPD característicos a aproximadamente 3,92, 10,54, 11,72, 12,52, 14,22, 15,40, 15,54, 15,90, 16,48, 16,84, 17,29, 18,26, 18,47, 19,39, 19,66, 20,00, 20,50, 20,65, 21,16, 21,28, 21,95, 22,97, 23,49, 23,70, 23,94, 24,31, 24,67 o 24,99 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG.16 y como se encuentra en la tabla 20 en el presente documento. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma F, tiene al menos 3, al menos 5, al menos 7 o al menos 10 picos de XPRD característicos a aproximadamente 3,92, 10,54, 11,72, 12,52, 14,22, 15,40, 15,54, 15,90, 16,48, 16,84, 17,29, 18,26, 18,47, 19,39, 19,66, 20,00, 20,50, 20,65, 21,16, 21,28, 21,95, 22,97, 23,49, 23,70, 23,94, 24,31, 24,67 o 24,99 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG.16 y como se encuentra en la tabla 20 en el presente documento. Aún en otro modo de realización, una forma sólida descrita en el presente documento, por ejemplo, forma F, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, veinte, veinticinco o todos los picos de XRPD característicos como se expone en la tabla 20.
Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma F, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve o diez picos de XRPD característicos a aproximadamente 12,52, 15,90, 19,66, 20,65 o 24,99 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG. 16. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma F, tiene uno, dos, tres, cuatro o cinco picos de XRPD característicos a aproximadamente 12,52, 15,90, 19,66, 20,65 o 24,99 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG.16. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma F, tiene uno, dos, tres, cuatro o cinco picos de XRPD característicos a aproximadamente 12,52, 15,90, 19,66, 20,65 o 24,99 ± 0,05° 2θ, como se representa, por ejemplo, en la FIG.16.
En un modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma F, que tiene una curva de isoterma DVS que corresponde sustancialmente a la curva de isoterma DVS representativa como se representa en la FIG.17.
En otro modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma F, que tiene un termograma de DSC sustancialmente como se representa en la FIG.18 que comprende un acontecimiento endotérmico con una temperatura de aparición de aproximadamente 162 °C y una temperatura máxima de pico de aproximadamente 167 °C.
Todavía en otro modo de realización, la forma F es pura. En determinados modos de realización, la forma F pura está sustancialmente libre de otras formas sólidas, por ejemplo, sólido amorfo. En determinados modos de realización, la pureza de la forma F no es menos de aproximadamente un 95 %, no menos de aproximadamente un 96 %, no menos de aproximadamente un 97 %, no menos de aproximadamente un 98 %, no menos de aproximadamente un 98,5 %, no menos de aproximadamente un 99 %, no menos de aproximadamente un 99,5 % o no menos de aproximadamente un 99,9 %.
Compuesto B, forma G
En determinados modos de realización, se proporciona en el presente documento una forma sólida del compuesto B designada como forma G. La forma G es una forma sólida del compuesto B. En un modo de realización, la forma G es un solvato de metanol del compuesto B. En otro modo de realización, la forma F es una sal de tartrato de solvato de metanol del compuesto A.
En un modo de realización, la forma G del compuesto B se obtiene suspendiendo el compuesto B en metanol y evaporando lentamente el metanol. La mezcla se puede filtrar. La forma G se puede preparar de acuerdo con los procedimientos y ejemplos descritos en el presente documento.
En un modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma G, es una sal de tartrato del compuesto A, y es sustancialmente cristalina, como se indica por las mediciones del patrón de difracción de rayos X en polvo (XRPD). En un modo de realización, el XRPD de una forma sólida proporcionada en el presente documento, por ejemplo, forma G, es sustancialmente como se muestra en la FIG.19. En otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma G, tiene uno o más picos de XRPD característicos a aproximadamente 7,65, 11,46, 12,51, 15,27, 15,51, 16,00, 17,34, 18,21, 19,11, 19,29, 19,42, 19,84, 20,23, 21,31, 21,57, 21,79, 22,49, 22,97, 23,22, 24,65, 25,04 o 25,88 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG. 19 y como se encuentra en la tabla 21 en el presente documento. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma G, tiene al menos 3, al menos 5, al menos 7 o al menos 10 picos de XPRD característicos a aproximadamente 7,65, 11,46, 12,51, 15,27, 15,51, 16,00, 17,34, 18,21, 19,11, 19,29, 19,42, 19,84, 20,23, 21,31, 21,57, 21,79, 22,49, 22,97, 23,22, 24,65, 25,04 o 25,88 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG. 19 y como se encuentra en la tabla 21 en el presente documento. Aún en otro modo de realización, una forma sólida descrita en el presente documento, por ejemplo, forma G, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, veinte o todos los picos de XRPD característicos como se expone en la tabla 21.
Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma G, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve o diez picos de XRPD característicos a aproximadamente 11,46, 12,51, 15,27, 16,00, 19,29, 19,42, 20,23, 22,49, 22,97 o 24,65 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG.19. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma G, tiene uno, dos, tres, cuatro o cinco picos de XRPD característicos a aproximadamente 11,46, 12,51, 19,29, 19,42 o 20,23 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG. 19. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma G, tiene uno, dos, tres, cuatro o cinco picos de XRPD característicos a aproximadamente 11,46, 12,51, 19,29, 19,42 o 20,23 ± 0,05° 2θ, como se representa, por ejemplo, en la FIG.19.
En un modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma G, que tiene un termógrafo de TGA que corresponde sustancialmente al termograma de TGA representativo como se representa en la FIG. 20. En determinados modos de realización, la forma cristalina presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente un 2 % de la masa total de la muestra. En otro modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma G, que tiene un termograma de DSC sustancialmente como se representa en la FIG.20 que comprende un acontecimiento endotérmico con una temperatura de aparición de aproximadamente 173 °C y una temperatura máxima de pico de aproximadamente 178 °C.
Todavía en otro modo de realización, la forma G es pura. En determinados modos de realización, la forma G pura está sustancialmente libre de otras formas sólidas, por ejemplo, sólido amorfo. En determinados modos de realización, la forma G pura está sustancialmente libre de forma B, forma D o forma E. En determinados modos de realización, la pureza de la forma G no es menos de aproximadamente un 95 %, no menos de aproximadamente un 96 %, no menos de aproximadamente un 97 %, no menos de aproximadamente un 98 %, no menos de aproximadamente un 98,5 %, no menos de aproximadamente un 99 %, no menos de aproximadamente un 99,5 % o no menos de aproximadamente un 99,9 %.
Compuesto C, forma 1
En determinados modos de realización, se proporciona en el presente documento una forma sólida del compuesto C designado como forma 1. La forma 1 es una forma sólida cristalina del compuesto C.
En un modo de realización, la forma 1 del compuesto C se obtiene suspendiendo el compuesto C en alcohol isoamílico/agua en una proporción de aproximadamente 3:1 durante aproximadamente 1,5 horas a aproximadamente 55 °C. A continuación, el líquido de la mezcla se evapora bajo flujo de nitrógeno y presión reducida. En otro modo de realización, la forma 1 del compuesto C se obtiene suspendiendo el compuesto C en etanol/heptano en una proporción de aproximadamente 3:8 a TA. Todavía en otro modo de realización, la forma 1
del compuesto C se obtiene suspendiendo el compuesto C en etanol/heptano en una proporción de aproximadamente 1:1 a TA. La forma 1 se puede preparar de acuerdo con los procedimientos y ejemplos descritos en el presente documento.
En un modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma 1, es sal de fumarato del compuesto A y es sustancialmente cristalina, como se indica por las mediciones del patrón de difracción de rayos X en polvo (XRPD). En un modo de realización, el XRPD es sustancialmente como se muestra en la FIG. 27a. En otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma 1, tiene uno o más picos de XRPD característicos a aproximadamente 7,58, 10,59, 11,44, 11,84, 12,5, 14,44, 15,45, 15,78, 16,09, 17,55, 18,92, 19,69, 19,86, 20,23, 21,35, 22,04, 23,16, 23,89, 24,23, 24,67, 25,23 o 25,93 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG. 27a y como se encuentra en la tabla 36 en el presente documento. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma 1, tiene al menos 3, al menos 5, al menos 7 o al menos 10 picos de XPRD característicos a aproximadamente 7,58, 10,59, 11,44, 11,84, 12,5, 14,44, 15,45, 15,78, 16,09, 17,55, 18,92, 19,69, 19,86, 20,23, 21,35, 22,04, 23,16, 23,89, 24,23, 24,67, 25,23 o 25,93 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG. 27a y como se encuentra en la tabla 36 en el presente documento. Aún en otro modo de realización, una forma sólida descrita en el presente documento, por ejemplo, forma 1, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, veinte o todos los picos de XRPD característicos como se expone en la tabla 36.
Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma 1, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve o diez picos de XRPD característicos a aproximadamente 10,59, 15,45, 15,78, 16,09, 18,92, 19,69, 19,86, 21,35, 23,16 o 24,23 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG. 27a. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma 1, tiene uno, dos, tres, cuatro o cinco picos de XRPD característicos a aproximadamente 16,09, 18,92, 19,69, 19,86 o 23,16 ± 0,1° 2θ como se representa, por ejemplo, en la FIG. 27a. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma 1, tiene uno, dos, tres, cuatro o cinco picos de XRPD característicos a aproximadamente 16,09, 18,92, 19,69, 19,86 o 23,16 ± 0,05° 2θ como se representa, por ejemplo, en la FIG.27a.
En un modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma 1, que tiene un termógrafo de TGA que corresponde sustancialmente al termograma de TGA representativo como se representa en la FIG. 28. En determinados modos de realización, la forma cristalina presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente un 2 % de la masa total de la muestra. En otro modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma 1, que tiene un termograma de DSC sustancialmente como se representa en la FIG. 28, que comprende un acontecimiento endotérmico con una temperatura de aparición de aproximadamente 167 °C y una temperatura máxima de pico de aproximadamente 172 °C.
Todavía en otro modo de realización, la forma 1 es pura. En determinados modos de realización, la forma 1 pura está sustancialmente libre de otras formas sólidas, por ejemplo, sólido amorfo. En determinados modos de realización, la pureza de la forma 1 no es menos de aproximadamente un 95 %, no menos de aproximadamente un 96 %, no menos de aproximadamente un 97 %, no menos de aproximadamente un 98 %, no menos de aproximadamente un 98,5 %, no menos de aproximadamente un 99 %, no menos de aproximadamente un 99,5 % o no menos de aproximadamente un 99,9 %.
En otro modo de realización descrito en el presente documento, es una forma sólida, por ejemplo, forma 1, que tiene una imagen PLM como se representa en la FIG.29.
Compuesto C, forma 2
En determinados modos de realización, se proporciona en el presente documento una forma sólida del compuesto C designado como forma 2. La forma 2 es una forma sólida cristalina del compuesto C.
En un modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma 2, es sal de fumarato del compuesto A y es sustancialmente cristalina, como se indica por las mediciones del patrón de difracción de rayos X en polvo (XRPD). En un modo de realización, el XRPD es sustancialmente como se muestra en la FIG. 27b. En otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma 2, tiene uno o más picos de XRPD característicos a aproximadamente 11,52, 11,87, 15,55, 16,04, 16,51, 17,32, 18,36, 19,00, 19,43, 19,87, 20,24, 21,35, 22,03, 23,23, 23,91, 25,43 o 26,03 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG.27b y como se encuentra en la tabla 37 en el presente documento. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma 2, tiene al menos 3, al menos 5, al menos 7 o al menos 10 picos de XPRD característicos a aproximadamente 11,52, 11,87, 15,55, 16,04, 16,51, 17,32, 18,36, 19,00, 19,43, 19,87, 20,24, 21,35, 22,03, 23,23, 23,91, 25,43 o 26,03 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG. 27b y como se encuentra en la tabla 37 en el presente documento.
Aún en otro modo de realización, una forma sólida descrita en el presente documento, por ejemplo, forma 1, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince o todos los picos de XRPD característicos como se expone en la tabla 37 en el presente documento.
Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma 1, tiene uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve o diez picos de XRPD característicos a aproximadamente 11,87, 15,55, 16,04, 16,51, 17,32, 19,43, 19,87, 20,24, 23,23 o 23,91 ± 0,1° 2θ, como se representa, por ejemplo, en la FIG.27b. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma 1, tiene uno, dos, tres, cuatro o cinco picos de XRPD característicos a aproximadamente 15,55, 19,43, 19,87, 20,24 o 23,23 ± 0,1° 2θ como se representa, por ejemplo, en la FIG. 27b. Todavía en otro modo de realización, una forma sólida proporcionada en el presente documento, por ejemplo, forma 1, tiene uno, dos, tres, cuatro o cinco picos de XRPD característicos a aproximadamente 15,55, 19,43, 19,87, 20,24 o 23,23 ± 0,05° 2θ como se representa, por ejemplo, en la FIG.27b.
Forma amorfa del compuesto A de base libre
En determinados modos de realización, se proporciona en el presente documento una forma sólida amorfa del compuesto A de base libre.
En un modo de realización, es una forma sólida amorfa del compuesto A de base libre como se indica por mediciones del patrón de difracción de rayos X en polvo (XRPD). En un modo de realización, el XRPD es sustancialmente como se muestra en la FIG.32.
En un modo de realización descrito en el presente documento, es una forma sólida amorfa del compuesto A de base libre que tiene un termógrafo de TGA que corresponde sustancialmente al termograma de TGA y DSC representativo como se representa en la FIG.33. En determinados modos de realización, la forma amorfa presenta un termograma de TGA que comprende una pérdida de masa total de aproximadamente un 9,3 % de la masa total de la muestra.
Todavía en otro modo de realización, la forma sólida amorfa del compuesto A es pura. En determinados modos de realización, la forma sólida amorfa pura del compuesto A está sustancialmente libre de otras formas sólidas, por ejemplo, sólidos cristalinos como se describe en el presente documento. En determinados modos de realización, la pureza no es menos de aproximadamente un 95 %, no menos de aproximadamente un 96 %, no menos de aproximadamente un 97 %, no menos de aproximadamente un 98 %, no menos de aproximadamente un 98,5 %, no menos de aproximadamente un 99 %, no menos de aproximadamente un 99,5 % o no menos de aproximadamente un 99,9 %.
Composiciones farmacéuticas
Los compuestos descritos en el presente documento se pueden administrar, por ejemplo, por vía oral, intramuscular, subcutánea, intravenosa, intradérmica, percutánea, intraarterial, intraperitoneal, intralesional, intracraneal, intraarticular, intraprostática, intrapleural, intratraqueal, intratecal, intranasal, intravaginal, intrarrectal, tópica, intratumoral, peritoneal, subconjuntival, intravesicular, mucosa, intrapericárdica, intraumbilical, intraocular, intraorbitaria, intravítrea (por ejemplo, por inyección intravítrea), por colirio, tópica, transdérmica, parenteral, por inhalación, por inyección, por implantación, por infusión, por infusión continua, por perfusión localizada bañando células diana directamente, por catéter, por lavado, en cremas o en composiciones lipídicas. Los compuestos descritos en el presente documento se pueden formular en composiciones farmacéuticas como se proporciona en el presente documento adecuadas para administración oral. En otro modo de realización, un compuesto descrito en el presente documento se puede administrar por vía intramuscular.
En un modo de realización, los compuestos descritos en el presente documento se administran como composiciones farmacéuticas que se pueden administrar a un sujeto por vía oral o parenteral. Las composiciones farmacéuticas de los compuestos descritos en el presente documento se pueden preparar como formas de dosificación oral tales como, por ejemplo, cápsulas, microcápsulas, comprimidos (comprimidos recubiertos y no recubiertos), gránulos, polvos, pastillas o supositorios. Los compuestos descritos en el presente documento se pueden formular para uso tópico o parenteral cuando el compuesto se disuelve o se suspende de otro modo en una solución adecuada para inyecciones, suspensiones, jarabes, cremas, pomadas, geles, aerosoles, soluciones y emulsiones.
Las composiciones farmacéuticas descritas en el presente documento incluyen uno o más excipientes farmacéuticamente aceptables tales como, pero sin limitarse a: sacarosa, almidón, manitol, sorbitol, lactosa, glucosa, celulosa, talco, fosfato de calcio o carbonato de calcio, celulosa, metilcelulosa, hidroximetilcelulosa, hidroxipropilcelulosa, carboximetilcelulosa, hidroxipropilalmidón, polipropilpirrolidona, polivinilpirrolidona, gelatina, goma arábiga, polietilenglicol (PEG), almidón, bicarbonato de sodio, citrato de calcio, estearato de magnesio, laurilsulfato de sodio, benzoato de sodio, bisulfito de sodio, metilparabeno, propilparabeno, ácido cítrico, citrato de sodio o ácido acético, polivinilpirrolidona, estearato de aluminio), agua y manteca de cacao. Los usos como, por
ejemplo, diluyentes, aglutinantes, lubricantes y disgregantes de dichos excipientes son bien conocidos en la técnica.
Las composiciones farmacéuticas descritas en el presente documento incluyen una cantidad eficaz del compuesto descrito en el presente documento (por ejemplo, compuesto A, compuesto B, compuesto C, compuesto D o una forma sólida de los mismos). La dosis del compuesto descrito en el presente documento puede ser una medida de una cantidad específica del compuesto (por ejemplo, una cantidad de dosis estándar) o se puede medir en función de, por ejemplo, el peso corporal de un paciente. En un modo de realización, un compuesto descrito en el presente documento se administra en una cantidad equivalente a aproximadamente 0,1, 0,5, 0,75, 1, 2, 3, 4, 5, 10, 15, 20, 30, 50, 75, 100, 200 o 250 mg/kg. En otro modo de realización, un compuesto descrito en el presente documento se administra en una cantidad de aproximadamente 0,1 mg/kg a aproximadamente 1 mg/kg; de aproximadamente 0,5 mg/kg a aproximadamente 2 mg/kg; de aproximadamente 1 mg/kg a aproximadamente 5 mg/kg; de aproximadamente 3 mg/kg a aproximadamente 10 mg/kg; de aproximadamente 8 mg/kg a aproximadamente 15 mg/kg; o de aproximadamente 15 mg/kg a aproximadamente 30 mg/kg. Todavía en otro modo de realización, un compuesto descrito en el presente documento se administra en una cantidad menor de aproximadamente 100 mg/kg, menor de aproximadamente 50 mg/kg, menor de aproximadamente 30 mg/kg, menor de aproximadamente 10 mg/kg o menor de aproximadamente 1 mg/kg.
En un modo de realización, un compuesto descrito en el presente documento se administra en una cantidad de aproximadamente 1, 5, 10, 20, 25, 30, 50, 60, 75, 90, 100, 120, 150 o 250 mg. En otro modo de realización, un compuesto descrito en el presente documento se administra en una cantidad de aproximadamente 10 mg. Todavía en otro modo de realización, un compuesto descrito en el presente documento se administra en una cantidad de aproximadamente 30 mg. Todavía en otro modo de realización, un compuesto descrito en el presente documento se administra en una cantidad de aproximadamente 90 mg. En un modo de realización, la composición farmacéutica que comprende el compuesto descrito en el presente documento se administra en una cantidad prescrita anteriormente una vez al día (qd). El compuesto puede ser el compuesto B, o una forma sólida del mismo (por ejemplo, forma A, forma B, forma C, forma D, forma E, forma F o forma G). En otro modo de realización, el compuesto es una forma sólida del compuesto B (por ejemplo, forma B, forma D o forma F). En un modo de realización, el compuesto es el compuesto C o compuesto C forma 1 o forma 2. En otro modo de realización, el compuesto es el compuesto D o una forma sólida descrita en el presente documento.
En otro modo de realización, un compuesto descrito en el presente documento se administra en una cantidad de aproximadamente 1 mg a aproximadamente 10 mg; de aproximadamente 10 mg a aproximadamente 30 mg; de aproximadamente 10 mg a aproximadamente 90 mg; de aproximadamente 30 mg a aproximadamente 90 mg; o de aproximadamente 90 mg a aproximadamente 250 mg. En un modo de realización, el compuesto administrado es el compuesto B en una cantidad de aproximadamente 1, 10, 30, 50, 90, 100 o 150 mg. Las dosis de un compuesto descrito en el presente documento se pueden proporcionar como una dosis individual (por ejemplo, un comprimido o cápsula individual de la cantidad de dosificación dada) o se pueden proporcionar como dosis múltiples dadas durante un período de tiempo (por ejemplo, 2 o más comprimidos o cápsulas equivalentes a la cantidad de dosificación). El compuesto puede ser el compuesto B, o una forma sólida del mismo (por ejemplo, forma A, forma B, forma C, forma D, forma E, forma F o forma G). En otro modo de realización, el compuesto es una forma sólida del compuesto B (por ejemplo, forma B, forma D o forma F). En un modo de realización, el compuesto es el compuesto C o compuesto C forma 1 o forma 2. En otro modo de realización, el compuesto es el compuesto D o una forma sólida descrita en el presente documento.
Las composiciones farmacéuticas descritas en el presente documento se pueden administrar una vez al día (qd); dos veces al día (bid), tres veces al día (tid), cada dos días (q2d), cada tres días (q3d) o una vez a la semana. Además, las dosis de las composiciones farmacéuticas proporcionadas en el presente documento que comprenden un compuesto descrito en el presente documento se pueden administrar antes de la comida (a.c.), después de la comida (d.c.) o con la comida. En un modo de realización, un compuesto descrito en el presente documento se administra qd durante un período de tratamiento (un período de tiempo donde el fármaco se administra a un paciente descrito en el presente documento) seguido de un período de descanso (un período de tiempo donde el fármaco no se administra a un paciente descrito en el presente documento). Los períodos de descanso pueden incluir la administración de agentes antineoplásicos distintos de un compuesto descrito en el presente documento. En un modo de realización, un compuesto descrito en el presente documento se formula para administración oral como se proporciona en el presente documento y se administra qd durante 20-28 días, seguido de un período de descanso de 3-10 días. En otro modo de realización, el compuesto se administra qd sin período de descanso.
Preferentemente, un compuesto descrito en el presente documento se formula para administración oral. La administración oral puede promover el cumplimiento por parte del paciente al tomar el compuesto (por ejemplo, formulado como una composición farmacéutica), incrementando de este modo el cumplimiento y la eficacia. Las composiciones farmacéuticas orales que comprenden un compuesto descrito en el presente documento incluyen, pero no se limitan a, comprimidos (por ejemplo, recubiertos, no recubiertos y masticables) y cápsulas (por ejemplo, cápsulas de gelatina dura, cápsulas de gelatina blanda, cápsulas con recubrimiento entérico y cápsulas de liberación mantenida). Los comprimidos se pueden preparar por compresión directa, por granulación en húmedo o por granulación en seco. Las composiciones farmacéuticas orales que comprenden un compuesto descrito en el
presente documento se pueden formular como se entiende en la técnica para liberación retardada o prolongada. En un modo de realización, el compuesto B o una forma sólida descrita en el presente documento (por ejemplo, forma B, forma D o forma F) se formula como un comprimido o una cápsula para administración oral en una cantidad expuesta en el presente documento.
Se proporcionan además en el presente documento compuestos que tienen las fórmulas:

(M3) (M4)
Los compuestos M1, M2, M3 y M4 se pueden considerar metabolitos y/o degradantes del compuesto B, incluyendo formas sólidas descritas en el presente documento. En determinados casos, dichos compuestos se pueden encontrar en las composiciones descritas en el presente documento donde dichas composiciones se han almacenado durante un período de tiempo dado a una humedad relativa (HR) de aproximadamente un 50 %, 55 %, 60 %, 65 %, 70 %, 75 %, 80 % o más. Dichos compuestos también se pueden encontrar a temperaturas elevadas de aproximadamente 30 °C, 35 °C, 40 °C, 45 °C o aproximadamente 50 °C. En un modo de realización, el compuesto M1, M2, M3 o M4 se encuentra en la composición descrita en el presente documento donde la composición comprende menos de aproximadamente 30 mg del compuesto B o una forma sólida del mismo. En determinados casos, dichos compuestos se encuentran en composiciones donde la composición comprende un comprimido no recubierto como se describe en el presente documento.
En determinados modos de realización, las composiciones descritas en el presente documento que comprenden el compuesto B o una forma sólida del compuesto B descrito en el presente documento (por ejemplo, forma A, forma B, forma C, forma D, forma E, forma F o forma G) comprenden menos de un 0,01 %, 0,02 %, 0,03 %, 0,04, 0,05, 0,1, 0,15, 0,75, 0,2, 0,225, 0,25, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9 o 1 % p/p de uno o más de M1, M2, M3 o M4. En un modo de realización, una composición descrita en el presente documento comprende menos de aproximadamente un 0,5 % p/p de uno o más de M1, M2, M3 o M4.
Procedimientos para tratar el cáncer: las referencias a procedimientos de tratamiento en los siguientes párrafos se deben interpretar como referencias al compuesto, la forma sólida o la composición farmacéutica del mismo para
su uso en un procedimiento para el tratamiento del cuerpo humano o animal.
Los compuestos y formas sólidas descritos en el presente documento se pueden administrar en una cantidad eficaz (por ejemplo, una cantidad como se describe en el presente documento) para tratar el cáncer. Se debe entender que los procedimientos descritos en el presente documento también incluyen el tratamiento con una composición farmacéutica como se describe en el presente documento que comprende un compuesto (por ejemplo, el compuesto B o una forma sólida del mismo) descrito en el presente documento y uno o más excipientes farmacéuticamente aceptables.
En un modo de realización se proporciona en el presente documento un procedimiento para tratar el cáncer administrando una cantidad eficaz del compuesto B como se describe en el presente documento a un paciente que tiene cáncer. En un modo de realización, el compuesto B es una forma sólida como se describe en el presente documento (por ejemplo, forma A, forma B, forma C, forma D, forma E, forma F o forma G).
En otro aspecto proporcionado en el presente documento, el compuesto B, forma A se puede administrar como se describe en el presente documento para tratar a un paciente que tiene un cáncer como se expone en el presente documento.
En otro aspecto proporcionado en el presente documento, el compuesto B, forma B se puede administrar como se describe en el presente documento para tratar a un paciente que tiene un cáncer como se expone en el presente documento.
En otro aspecto proporcionado en el presente documento, el compuesto B, forma C se puede administrar como se describe en el presente documento para tratar a un paciente que tiene un cáncer como se expone en el presente documento.
En otro aspecto proporcionado en el presente documento, el compuesto B, forma D se puede administrar como se describe en el presente documento para tratar a un paciente que tiene un cáncer como se expone en el presente documento.
En otro aspecto proporcionado en el presente documento, el compuesto B, forma E se puede administrar como se describe en el presente documento para tratar a un paciente que tiene un cáncer como se expone en el presente documento.
En otro aspecto proporcionado en el presente documento, el compuesto B, forma F se puede administrar como se describe en el presente documento para tratar a un paciente que tiene un cáncer como se expone en el presente documento.
En otro aspecto proporcionado en el presente documento, el compuesto B, forma G se puede administrar como se describe en el presente documento para tratar a un paciente que tiene cáncer de pulmón, cáncer de ovario, cáncer de endometrio, cáncer de próstata, cáncer de útero o cáncer de mama como se expone en el presente documento. En otro aspecto proporcionado en el presente documento, una forma no cristalina amorfa del compuesto A o compuesto B se puede administrar como se describe en el presente documento para tratar a un paciente que tiene cáncer de pulmón, cáncer de ovario, cáncer de endometrio, cáncer de próstata, cáncer de útero o cáncer de mama como se expone en el presente documento.
En otro aspecto se proporciona en el presente documento un procedimiento para tratar cáncer de pulmón, cáncer de ovario, cáncer de endometrio, cáncer de próstata, cáncer de útero o cáncer de mama administrando una cantidad eficaz del compuesto B o una forma sólida como se describe en el presente documento a un paciente que tiene dicho cáncer. En un modo de realización, el cáncer es cáncer de ovario o cáncer de endometrio. En un modo de realización, el cáncer es cáncer de mama. En un modo de realización de dichos procedimientos, el compuesto B es una forma sólida como se describe en el presente documento (por ejemplo, forma A, forma B, forma C, forma D, forma E, forma F o forma G).
En otro aspecto, el compuesto B, forma A, se puede administrar como se describe en el presente documento para tratar a un paciente que tiene cáncer de pulmón, cáncer de ovario, cáncer de endometrio, cáncer de próstata, cáncer de útero o cáncer de mama como se expone en el presente documento.
En otro aspecto, el compuesto B, forma B se puede administrar como se describe en el presente documento para tratar a un paciente que tiene cáncer de pulmón, cáncer de ovario, cáncer de endometrio, cáncer de próstata, cáncer de útero o cáncer de mama como se expone en el presente documento.
En otro aspecto, el compuesto B, forma C se puede administrar como se describe en el presente documento para tratar a un paciente que tiene cáncer de pulmón, cáncer de ovario, cáncer de endometrio, cáncer de próstata, cáncer de útero o cáncer de mama como se expone en el presente documento.
En otro aspecto, el compuesto B, forma D se puede administrar como se describe en el presente documento para tratar a un paciente que tiene cáncer de pulmón, cáncer de ovario, cáncer de endometrio, cáncer de próstata, cáncer de útero o cáncer de mama como se expone en el presente documento.
En otro aspecto, el compuesto B, forma E se puede administrar como se describe en el presente documento para tratar a un paciente que tiene cáncer de pulmón, cáncer de ovario, cáncer de endometrio, cáncer de próstata, cáncer de útero o cáncer de mama como se expone en el presente documento.
En otro aspecto, el compuesto B, forma F se puede administrar como se describe en el presente documento para tratar a un paciente que tiene cáncer de pulmón, cáncer de ovario, cáncer de endometrio, cáncer de próstata, cáncer de útero o cáncer de mama como se expone en el presente documento.
En otro aspecto, el compuesto B, forma G se puede administrar como se describe en el presente documento para tratar a un paciente que tiene cáncer de pulmón, cáncer de ovario, cáncer de endometrio, cáncer de próstata, cáncer de útero o cáncer de mama como se expone en el presente documento.
En otro aspecto, una forma no cristalina amorfa del compuesto A o compuesto B se puede administrar como se describe en el presente documento para tratar a un paciente que tiene cáncer de pulmón, cáncer de ovario, cáncer de endometrio, cáncer de próstata, cáncer de útero o cáncer de mama como se expone en el presente documento. Se proporcionan además en el presente documento procedimientos para tratar cáncer de mama en una paciente que tiene cáncer de mama administrando una cantidad eficaz del compuesto B o una forma sólida del compuesto B como se describe en el presente documento. En un modo de realización, es un procedimiento para tratar cáncer de mama en dicha paciente administrando una cantidad eficaz de una forma sólida del compuesto B como se describe en el presente documento. En un modo de realización, el procedimiento comprende tratar cáncer de mama en una paciente que tiene cáncer de mama administrando a la paciente una cantidad eficaz del compuesto B, forma B como se describe en el presente documento. En un modo de realización, el procedimiento comprende tratar cáncer de mama en un paciente que tiene cáncer de mama administrando a la paciente una cantidad eficaz del compuesto B puro (por ejemplo, sustancialmente libre de otra forma sólida descrita en el presente documento, por ejemplo, sustancialmente libre de la forma D y/o forma F). En otro aspecto, el procedimiento comprende tratar cáncer de mama en un paciente que tiene cáncer de mama administrando a la paciente una cantidad eficaz del compuesto B, forma D como se describe en el presente documento. En otro aspecto, el procedimiento comprende tratar cáncer de mama en una paciente que tiene cáncer de mama administrando a la paciente una cantidad eficaz del compuesto B, forma F, como se describe en el presente documento. Todavía en otro aspecto, el procedimiento comprende tratar cáncer de mama en una paciente que tiene cáncer de mama administrando a la paciente una cantidad eficaz del compuesto B, forma A, como se describe en el presente documento. Todavía en otro aspecto, el procedimiento comprende tratar cáncer de mama en una paciente que tiene cáncer de mama administrando a la paciente una cantidad eficaz del compuesto B, forma C, como se describe en el presente documento. Todavía en otro aspecto, el procedimiento comprende tratar cáncer de mama en una paciente que tiene cáncer de mama administrando a la paciente una cantidad eficaz del compuesto B, forma E, como se describe en el presente documento. Todavía en otro aspecto, el procedimiento comprende tratar cáncer de mama en una paciente que tiene cáncer de mama administrando a la paciente una cantidad eficaz del compuesto B, forma G, como se describe en el presente documento. Todavía en otro aspecto, el procedimiento comprende tratar cáncer de mama en una paciente que tiene cáncer de mama administrando a la paciente una cantidad eficaz de una forma no cristalina amorfa del compuesto B como se describe en el presente documento.
Los compuestos descritos en el presente documento (por ejemplo, compuesto B o una forma sólida del mismo como se describe en el presente documento) se pueden usar en la fabricación de un medicamento para su uso en el tratamiento de cáncer de mama como se describe en el presente documento.
Los procedimientos para tratar cáncer de mama proporcionados en el presente documento comprenden un tratamiento donde el cáncer de mama puede ser cáncer de mama con receptores hormonales (por ejemplo, cáncer de mama RE+), cáncer de mama HER2 positivo, cáncer de mama HER2 negativo o cáncer de mama triple negativo (TNBC).
En un modo de realización, el cáncer de mama es cáncer de mama HER2 negativo. El cáncer de mama HER2 negativo se puede definir en el presente documento como, por ejemplo, una puntuación IHQ de HER2 de 0 o 1+, o una puntuación IHQ de 2+ acompañada de una prueba de hibridación in situ con plata, cromogénica o fluorescente negativa que indica la ausencia de amplificación génica de HER2, o una proporción HER2/CEP17 de < 2,0, o guías clínicas locales. En un modo de realización, el cáncer de mama es cáncer de mama RE+/HER2-. El cáncer de mama puede estar en fase 0, I, II, III o IV como se entiende en la técnica.
En otro modo de realización, el cáncer de mama es cáncer de mama localmente avanzado o metastásico (mBC). En un modo de realización, el compuesto B o una forma sólida del mismo (por ejemplo, forma B, forma D o forma
F) se puede administrar como un componente del tratamiento adyuvante. En otro modo de realización, el compuesto B o una forma sólida del mismo (por ejemplo, forma B, forma D o forma F) se puede administrar como un componente del tratamiento neoadyuvante.
Las pacientes con cáncer de mama descritas en el presente documento pueden ser premenopáusicas antes del tratamiento con un compuesto o forma sólida como se describe en el presente documento. Las pacientes con cáncer de mama descritas en el presente documento pueden ser posmenopáusicas antes del tratamiento con un compuesto o forma sólida como se describe en el presente documento.
Los procedimientos proporcionados en el presente documento incluyen administrar una cantidad eficaz del compuesto B o una forma sólida como se describe en el presente documento al paciente en una cantidad como se expone en el presente documento. La cantidad eficaz puede ser, por ejemplo, una cantidad de aproximadamente 10 mg, 30 mg, 50 mg, 90 mg, 100 mg, 125 mg o 250 mg. En un modo de realización de los procedimientos proporcionados en el presente documento, el compuesto B o una forma sólida descrita en el presente documento se administra por vía oral. En un modo de realización, el compuesto B o una forma sólida del mismo se administra como un comprimido (por ejemplo, un comprimido recubierto o no recubierto). En otro modo de realización, el compuesto B o una forma sólida del mismo se administra como una cápsula. Por tanto, se proporcionan en el presente documento composiciones adecuadas para la administración a una paciente con cáncer de mama donde dichas composiciones comprenden una cantidad del compuesto B o una forma sólida descrita en el presente documento de aproximadamente 10 mg, 30 mg, 50 mg, 90 mg, 100 mg, 125 mg o 250 mg en un comprimido o cápsula como se expone en el presente documento. Cuando se administra de acuerdo con los procedimientos proporcionados en el presente documento, el compuesto B o una forma sólida del mismo puede ser puro como se describe en el presente documento.
Los pacientes de los procedimientos descritos anteriormente pueden haber recibido tratamiento previo con uno o más agentes antineoplásicos o radioterapia. Por ejemplo, en un modo de realización, un paciente se puede haber tratado previamente (por ejemplo, con un tratamiento de 1L, 2L, 3L o más líneas) con doxorrubicina, doxorrubicina liposomal pegilada, epirrubicina, paclitaxel, paclitaxel unido a albúmina, docetaxel, 5-fluorouracilo, ciclofosfamida, cisplatino, carboplatino, vinorelbina, capecitabina, gemcitabina, ixabepilona, eribulina, olaparib, metotrexato, anastrozol, exemestano, toremifeno, letrozol, tamoxifeno, 4-hidroxitamoxifeno, raloxifeno, droloxifeno, trioxifeno, keoxifeno, ftutamida, nilutamida, bicalutamida, lapatinib, vinblastina, goserelina, leuprolida, pegfilgrastim, filgrastim o venetoclax.
En otro modo de realización, un paciente se puede haber tratado previamente (por ejemplo, con un tratamiento de 1L, 2L, 3L o más líneas) con un inhibidor de AKT, un inhibidor de CDK4/6, un inhibidor de PARP o un inhibidor de aromatasa. En un modo de realización, el inhibidor de AKT es ipatasertib (GDC-0068). En un modo de realización, el inhibidor de CDK4/6 es abemaciclib, ribociclib o palbociclib. En determinados casos, un paciente se puede haber tratado previamente con: (1) abemaciclib, ribociclib o palbociclib; (2) ipatasertib; (3) everolimus o fulvestrant; (4) -trastuzumab emtansina, trastuzumab, pertuzumab o atezolizumab; o (5) alemtuzumab, bevacizumab, cetuximab, panitumumab, rituximab, tositumomab o una combinación de los mismos. Los pacientes descritos en el presente documento se pueden haber sometido a cirugía antes del tratamiento con el compuesto B o la forma sólida del mismo.
En otro modo de realización, un paciente en el presente documento puede ser resistente a uno o más tratamientos antineoplásicos. Por ejemplo, un paciente en el presente documento puede ser resistente a inhibidores de la aromatasa. En otro ejemplo, un paciente en el presente documento puede ser resistente a un degradador selectivo del receptor estrogénico (SERD) tal como, por ejemplo, fulvestrant. Todavía en otro ejemplo, un paciente puede ser resistente a uno o más tratamientos endocrinos tales como clomifeno, toremifeno, raloxifeno, anordrin, bacedoxifeno, broparestrol, ciclofenil, lasofoxifeno, ormeloxifeno, acolbifeno, elacestrant, brilanestrant, clomifenóxido, droloxifeno, etacstil u ospemifeno. En otro modo de realización, un paciente puede ser resistente a abemaciclib, anastrozol, exemestano, fulvestrant, goserelina, letrozol, leuprorelina, megestrol, palbociclib, tamoxifeno o toremifeno. En otro ejemplo, un paciente puede ser resistente al tratamiento con trastuzumab emtansina, trastuzumab, pertuzumab, atezolizumab, pembrolizumab, durvalumab, avelumab o nivolumab.
Los compuestos descritos en el presente documento también se pueden usar en procedimientos que comprenden inhibir RE-alfa en un paciente. Dichos procedimientos comprenden administrar una cantidad de un compuesto descrito en el presente documento (por ejemplo, compuesto A o compuesto B, incluyendo formas sólidas de los mismos como se describe en el presente documento) al paciente.
Politerapias
Los compuestos y formas sólidas descritos en el presente documento se pueden administrar en combinación con uno o más agentes antineoplásicos. La administración "en combinación" como se expone en el presente documento incluye administración secuencial (en cualquier orden) de un compuesto descrito en el presente documento y uno o más tratamientos antineoplásicos así como administración simultánea. En consecuencia, se proporcionan en el presente documento procedimientos para tratar cáncer de mama en una paciente que tiene
cáncer de mama, comprendiendo dichos procedimientos administrar el compuesto B o una forma sólida como se describe en el presente documento en combinación con uno o más tratamientos antineoplásicos adicionales. En un modo de realización, el tratamiento antineoplásico comprende doxorrubicina, doxorrubicina liposomal pegilada, epirrubicina, paclitaxel, paclitaxel unido a albúmina, docetaxel, 5-fluorouracilo, ciclofosfamida, cisplatino, carboplatino, vinorelbina, capecitabina, gemcitabina, ixabepilona, eribulina, olaparib, metotrexato, anastrozol, exemestano, toremifeno, letrozol, tamoxifeno, 4-hidroxitamoxifeno, raloxifeno, droloxifeno, trioxifeno, keoxifeno, ftutamida, nilutamida, bicalutamida, lapatinib, vinblastina, goserelina, leuprolida, pegfilgrastim, filgrastim o venetoclax.
En un modo de realización se proporciona en el presente documento un procedimiento para tratar cáncer de mama en una paciente que tiene cáncer de mama administrando una cantidad eficaz del compuesto B o una forma sólida como se describe en el presente documento en combinación con doxorrubicina, doxorrubicina liposomal pegilada, epirrubicina, paclitaxel, paclitaxel unido a albúmina, docetaxel, 5-fluorouracilo, ciclofosfamida, cisplatino, carboplatino, vinorelbina, capecitabina, gemcitabina, ixabepilona, eribulina, olaparib, metotrexato, anastrozol, exemestano, toremifeno, letrozol, tamoxifeno, 4-hidroxitamoxifeno, raloxifeno, droloxifeno, trioxifeno, keoxifeno, ftutamida, nilutamida, bicalutamida, lapatinib, vinblastina, goserelina, leuprolida, pegfilgrastim, filgrastim o venetoclax.
En otro aspecto se proporciona en el presente documento un procedimiento para tratar cáncer de mama en una paciente que tiene cáncer de mama administrando una cantidad eficaz del compuesto B o una forma sólida como se describe en el presente documento en combinación con paclitaxel, paclitaxel unido a albúmina, metotrexato, anastrozol, exemestano, toremifeno, letrozol, tamoxifeno, 4-hidroxitamoxifeno, raloxifeno, droloxifeno, trioxifeno, keoxifeno o venetoclax. Todavía en otro aspecto se proporciona en el presente documento un procedimiento para tratar cáncer de mama en una paciente que tiene cáncer de mama administrando una cantidad eficaz del compuesto B o una forma sólida como se describe en el presente documento en combinación con fulvestrant, paclitaxel, paclitaxel unido a albúmina, clomifeno, toremifeno, raloxifeno, anordrín, bacedoxifeno, broparestrol, ciclofenil, lasofoxifeno, ormeloxifeno, acolbifeno, elacestrant, brilanestrant, clomifenóxido, droloxifeno, etacstil u ospemifeno.
Todavía en otro aspecto se proporciona en el presente documento un procedimiento para tratar cáncer de mama en una paciente que tiene cáncer de mama administrando una cantidad eficaz del compuesto B o una forma sólida como se describe en el presente documento en combinación con un inhibidor de CDK4/6, un inhibidor de PARP o un inhibidor de aromatasa.
En otro aspecto se proporciona en el presente documento un procedimiento para tratar cáncer de mama en una paciente que tiene cáncer de mama como se describe en el presente documento donde el procedimiento comprende administrar una cantidad eficaz del compuesto B o una forma sólida como se describe en el presente documento en combinación con un inhibidor de CDK4/6 donde el inhibidor de CDK4/6 es abemaciclib, ribociclib o palbociclib. En un modo de realización preferente, el procedimiento comprende administrar el compuesto B o una forma sólida como se describe en el presente documento en combinación con palbociclib. Todavía en otro modo de realización, el procedimiento comprende administrar el compuesto B o una forma sólida como se describe en el presente documento en combinación con abemaciclib o ribociclib. En otro aspecto se proporciona en el presente documento un kit que comprende (i) el compuesto B o una forma sólida del mismo en una forma de dosificación unitaria; (ii) un inhibidor de CDK4/6 (por ejemplo, palbociclib) en una segunda forma de dosificación unitaria; y un recipiente que contiene cada forma de dosificación.
La dosis de abemaciclib puede ser de 50 mg a 500 mg al día, o de 150 mg a 450 mg al día y la dosificación puede ser diaria en ciclos de 28 días o menos de 28 días por ciclos de 28 días tal como 21 días por ciclo de 28 días o 14 días por ciclo de 28 días o 7 días por ciclos de 28 días. En un modo de realización, abemaciclib se dosifica una vez al día o preferentemente en una pauta bid donde la dosificación es oral. En el caso de dosificación bid, las dosis se pueden separar 4 horas, 8 horas o 12 horas. En determinados modos de realización, abemaciclib se dosifica a 150 mg por vía oral bid donde cada dosis se administra con aproximadamente 12 h de diferencia. En determinados modos de realización, la dosis de abemaciclib se administra de acuerdo con un prospecto del envase.
La dosis de ribociclib puede ser de 200 mg a 1000 mg al día; o de 250 mg a 750 mg al día y la dosificación puede ser diaria en ciclos de 28 días o menos de 28 días por ciclos de 28 días tal como 21 días por ciclo de 28 días o 14 días por ciclo de 28 días o 7 días por ciclos de 28 días. En un modo de realización, ribociclib se dosifica una vez al día cuando la dosificación es oral. En determinados modos de realización, la dosis de ribociclib se administra de acuerdo con un prospecto del envase.
La dosis de palbociclib puede ser de 25 mg a 250 mg al día o de 50 mg a 125 mg al día o de 75 mg a 125 mg al día o de 75 mg al día a 100 mg al día o 125 mg al día. La dosificación puede ser diaria en ciclos de 28 días o menos de 28 días por ciclos de 28 días tal como 21 días por ciclo de 28 días o 14 días por ciclo de 28 días o 7 días por ciclos de 28 días. En un modo de realización, palbociclib se dosifica una vez al día cuando la dosificación es oral. En determinados modos de realización, la dosis de palbociclib se administra de acuerdo con un prospecto
del envase.
En otro aspecto proporcionado en el presente documento, los procedimientos descritos en el presente documento comprenden administrar una cantidad eficaz del compuesto B o una forma sólida como se describe en el presente documento en combinación con un inhibidor de aromatasa (AI), donde AI es letrozol, anastrozol, exemestano o testolactona.
Aún en otro aspecto se proporciona en el presente documento un procedimiento para tratar cáncer de mama en una paciente que tiene cáncer de mama administrando una cantidad eficaz del compuesto B o una forma sólida como se describe en el presente documento en combinación con una inmunoterapia contra el cáncer (por ejemplo, un anticuerpo). En un modo de realización, el compuesto B o una forma sólida como se describe en el presente documento se administra en combinación con trastuzumab emtansina, trastuzumab, pertuzumab, atezolizumab, pembrolizumab, durvalumab, avelumab o nivolumab, o una combinación de los mismos. En un modo de realización, el compuesto B o una forma sólida como se describe en el presente documento se administra en combinación con una inmunoterapia contra el cáncer que comprende un inhibidor de PD-1 o PD-L1, donde la inmunoterapia contra el cáncer es atezolizumab, pembrolizumab o nivolumab.
La administración de un compuesto descrito en el presente documento (por ejemplo, el compuesto B o una forma sólida como se describe en el presente documento) da como resultado que los pacientes tengan efectos adversos (EA) caracterizados como de grado 2 o menor. En un modo de realización, un paciente al que se le administra el compuesto B o una forma sólida como se describe en el presente documento tiene EA de grado 2 o menor. Ejemplos:
Los siguientes ejemplos se presentan a modo de ilustración, y cualquier ejemplo que quede fuera del alcance de las reivindicaciones adjuntas se proporciona solo con propósitos informativos.
Síntesis de compuestos descritos en el presente documento. Todos los reactivos y disolventes se adquirieron de proveedores comerciales y se usaron sin purificación adicional. Se utilizó disolvente anhidro (diclorometano). Los disolventes disponibles comercialmente no se purificaron adicionalmente.
Todas las reacciones se llevaron a cabo en viales con tapón de rosca equipados con membranas de teflón bajo una atmósfera de nitrógeno.
Se realizó cromatografía en columna ultrarrápida usando un instrumento Teledyne Isco CombiFlash(R) Rf con cartuchos de sílice RediSepRf Gold de relleno previo.
A menos que se indique de otro modo, los rendimientos informados son para material aislado y se corrigen para disolventes residuales.
Se caracterizaron los compuestos por uno o más de RMN de 1H, RMN de 13C, punto de fusión y HRMS, y análisis HPLC (por ejemplo, para la confirmación de la pureza).
Los espectros de resonancia magnética nuclear de 1H y 13C se registraron en un instrumento Bruker 400 MHz a temperatura ambiente. Todos los espectros de RMN de 1H se midieron en partes por millón (ppm) en relación con la señal de cloroformo residual (7,26 ppm) o sulfóxido de dimetilo (2,50 ppm) en el disolvente deuterado a menos que se indique de otro modo. Se informa de los datos de RMN de 1H como sigue: desplazamiento químico, multiplicidad (a. = señal ancha, superpuesto = superposición, s = singlete, d = doblete, t = triplete, q = cuartete, p = pentete, m = multiplete), constantes de acoplamiento e integración. Todos los espectros de RMN de 13C se informan en ppm en relación con el deuterocloroformo (77,06 ppm) o dimetilsulfóxido deuterado (39,53 ppm), y se obtuvieron con desacoplamiento completo de 1H a menos que se indique de otro modo. Se realizaron análisis de HPLC en un sistema de HPLC Agilent 1260 Infinity con un detector UV a 220 nm usando una columna Ace Super C18. Se obtuvieron puntos de fusión usando un aparato de punto de fusión Buchi B-540 y están sin corregir. Se adquirieron datos de espectrometría de masas de alta resolución (HRMS) en un espectrómetro de masas Thermo Scientific Orbitrap Fusion.
Ejemplos 1-3: alquilación de indol.
Se realizó la alquilación de indol por la secuencia de protección Boc (ejemplo 1), formación de sulfamida (ejemplo 2) y alquilación de indol (ejemplo 3)
Ejemplo 1: protección Boc
Ejemplo 1: esquema de reacción general de protección Boc.
Se realizó la protección Boc de acuerdo con el siguiente esquema de reacción general donde RA y RB corresponden
a los diversos grupos funcionales en las siguientes reacciones de protección del ejemplo 1, y en el que los asteriscos representan un centro quiral:

Ejemplo 1A: preparación de (S)-(2-hidroxi-1-(4-metoxifenil)etil)carbamato de terc-butilo:

Se realizó el esquema de reacción general 1 anterior como sigue. A una suspensión de clorhidrato de (S)-2-amino-2-(4-metoxifenil)etan-1-ol (1,04 g, 5,10 mmol, 100 % mol) en THF (4,4 ml) se le añadió Boc2O (1,21 ml, 5,61 mmol, 110 % mol), NaHCO3 (451 mg, 5,10 mmol, 100 % mol) y agua (4,4 ml) a ta. Se agitó la solución a ta durante 18 h y se extrajo con iProAc (20 ml x 2). Se secó la capa orgánica con salmuera saturada (20 ml), se secó (Na2SO4), se filtró y se concentró a presión reducida para proporcionar el producto sin purificación adicional. Se informa del rendimiento del ejemplo 1A corregido en base al disolvente residual a partir de RMN de 1H. La reacción proporcionó (S)-(2-hidroxi-1-(4-metoxifenil)etil)carbamato de terc-butilo (1,36 g, 100 % de rendimiento) como un sólido blanco, p.f.: 139,0-139,9 °C; FTIR (neto, cm-1) 3370, 2984, 2837, 1681, 1613, 1512, 1461; RMN de 1H (400 MHz, CDCl3): δ 7,24-7,20 (m, 2H), 6,92-6,86 (m, 2H), 5,10 (d, J = 7,2 Hz, 1H), 4,72 (a., 1H), 3,82 (t, J = 5,6 Hz, 2H), 3,80 (s, 3H), 2,35 (a., 1H), 1,43 (s, 9H); RMN de 13C (100 MHz, CDCl3): δ 159,1, 156,2, 131,6, 127,7, 114,2, 80,0, 66,8, 56,4, 55,3, 28,4.
Ejemplo 1B: preparación de (R)-(1-ciclopropil-2-hidroxietil)carbamato de terc-butilo:

El esquema de reacción general según el ejemplo 1A anterior se realizó con (R)-2-amino-2-ciclopropiletano-1-ol (1,16 g, 11,5 mmol, 100 % mol) para proporcionar (R)-(1-ciclopropil-2-hidroxietil)carbamato de terc-butilo (2,31 g, 100 % de rendimiento) como un sólido blanco, p.f.: 70,0-70,8 °C; FTIR (neto, cm-1) 3358, 2974, 2937, 1682, 1521, 1366; RMN de 1H (400 MHz, CDCl3): δ 4,80 (a., 1H), 3,80 (ddd, J = 10,8, 6,8, 3,2 Hz, 1H), 3,67 (ddd, J = 10,8, 6,0, 4,8 Hz, 1H), 2,94 (dtd, J = 9,6, 6,4, 3,2 Hz, 1H), 2,81 (a., 1H), 1,45 (s, 9H), 0,85 (dtt, J = 9,6, 8,0, 4,8 Hz, 1H), 0,60-0,47 (m, 2H), 0,44-0,25 (m, 2H); RMN de 13C (100 MHz, CDCl3): δ 156,6, 79,7, 66,3, 57,9, 28,4, 13,0, 3,3, 2,9.
Ejemplo 1C: preparación de ((1R,2S)-2-hidroxi-2,3-dihidro-1H-inden-1-il)carbamato de terc-butilo:

El esquema de reacción general según el ejemplo 1A anterior se realizó con (1R,2S)-1-amino-2,3-dihidro-1H-inden-2-ol (5,15 g, 34,5 mmol, 100 % mol) para proporcionar ((1R,2S)-2-hidroxi-2,3-dihidro-1H-inden-1-il)carbamato de terc-butilo (8,61 g, 100 % de rendimiento) como un sólido blanco, p.f.: 67,3-68,4 °C; FTIR (neto, cm-1) 3428, 3350, 2983, 2933, 1688, 1509; RMN de 1H (400 MHz, CDCl3): δ 7,31-7,26 (m, 1H), 7,26-7,18 (m, 3H), 5,17 (a., 1H), 5,05 (a., 1H), 4,57 (ddd, J = 7,2, 4,8, 2,0 Hz, 1H), 3,12 (dd, J = 16,8, 5,2 Hz, 1H), 2,91 (dd, J =16,8, 2,4 Hz, 1H), 2,31 (d, J =4,8Hz, 1H), 1,50 (s, 9H); RMN de 13C (100 MHz, CDCl3): δ 156,3, 140,9, 139,9, 128,2, 127,1, 125,3, 124,5, 79,9, 73,6, 58,9, 39,4, 28,4.
Ejemplo 1D: preparación de (S)-(1-(3-fluorofenil)-2-hidroxietil)carbamato de terc-butilo:

El esquema de reacción general según el ejemplo 1A anterior se realizó con (S)-2-amino-2-(3-fluorofenil)etan-1-ol (1,36 g, 8,73 mmol, 100 % mol) para proporcionar (S)-(1-(3-fluorofenil)-2-hidroxietil)carbamato de terc-butilo (2,23 g, 100 % de rendimiento) como un sólido blanco, p.f.: 106,5-107,9 °C; FTIR (neto, cm-1) 3251, 3059, 2977, 2901, 1671, 1587, 1543; RMN de 1H (400 MHz, CDCl3): δ 7,33 (ddd, J = 7,6, 7,6, 6,0 Hz, 1H), 7,12-7,07 (m, 1H), 7,05-6,95 (m, 2H), 5,24 (a., 1H), 4,77 (a., 1H), 3,93-3,77 (m, 2H), 2,02 (a., 1H), 1,44 (s, 9H); RMN de 13C (100 MHz, CDCl3): δ 163,0 (d, 1 J CF = 246 Hz), 156,0, 142,5, 130,2 (d, 3 J CF = 9 Hz), 122,2 (d, 4 J CF = 3 Hz), 114,5 (d, 2 J CF = 21 Hz), 113,6 (d, 2 J CF = 21 Hz), 80,2, 66,2, 56,3, 28,3; RMN de 19F (CDCl3, 376 MHz): δ -112,4.
Ejemplo 1E: preparación de (S)-(1-(3-fluorofenil)-2-hidroxietil)carbamato

El esquema de reacción general según el ejemplo 1A anterior se realizó con clorhidrato de (S)-2-amino-2-(3-(trifluorometil)fenil)etan-1-ol (1,00 g, 4,12 mmol, 100 % mol) para proporcionar (S)-(1-(3-fluorofenil)-2-hidroxietil)carbamato (1,26 g, 100 % de rendimiento) como un sólido blanco, p.f.: 50,3-52,5 °C; FTIR (neto, cm-1) 3368, 3254, 2979, 2939, 1691, 1510, 1453, 1333; RMN de 1H (400 MHz, CDCl3): δ 7,59-7,54 (m, 2H), 7,53-7,45 (m, 2H), 5,31 (d, J = 6,4 Hz, 1H), 4,83 (a., 1H), 3,92 (ddd, J = 11,2, 6,8, 4,0 Hz, 1H), 3,88-3,79 (m, 1H), 1,94 (a., 1H), 1,43 (s, 9H); RMN de 13C (100 MHz, CDCl3): δ 156,1, 141,1, 130,9 (q, 2 J CF = 32 Hz), 130,1, 129,0, 124,3 (q, 3 J CF = 4 Hz), 124,1 (q, 1JCF = 270 Hz), 123,4 (q, 3 J CF = 4 Hz), 80,3, 65,8, 56,3, 28,2; RMN de 19F (CDCl3, 376 MHz): δ -62,6. Ejemplo 2: formación de sulfamidato
Ejemplo 2: esquema de reacción general de formación de sulfamidato
Se realizó la formación de sulfamidato de acuerdo con el siguiente esquema de reacción general, donde RA y RB corresponden a los diversos grupos funcionales en las siguientes reacciones del ejemplo 2, y en el que los asteriscos representan un centro quiral:

Ejemplo 2A: preparación de 2,2-dióxido de (R)-4-bencil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo:

El esquema de reacción general 2 anterior se realizó como sigue. A una solución fría (-40 °C) de SOCl2 (10,9 ml, 149 mmol, 250 % mol) en CH2Cl2 (60,0 ml) se le añadió una solución de (R)-(1-hidroxi-3-fenilpropan-2-il)carbamato de terc-butilo (15,0 g, 59,7 mmol, 100 % mol) en CH2Cl2 (60,0 ml) durante 60 min a -40 °C. A continuación, se añadió piridina (25,3 ml, 313 mmol, 525 % mol) a la mezcla de reacción durante 30 min a -40 °C. Se agitó la mezcla de reacción a -40 °C durante 2 h, se cambió el disolvente por una mezcla de CH2Cl2/iPrOAc (1:1), se filtró. Se lavó el filtrado con una solución de salmuera saturada (20 ml), se secó (Na2SO4), se filtró y se concentró a presión reducida. Se disolvió el residuo en CH3CN (60,0 ml) a 0 °C. Se añadieron NaIO4 (14,0 g, 65,7 mmol, 110 % mol), RuCl3 (61,9 mg, 0,298 mmol, 0,5 % mol) y agua (60,0 ml) a la mezcla de reacción a 0 °C y se agitó durante 15 min. A continuación, se calentó la mezcla de reacción hasta temperatura ambiente y se agitó a temperatura ambiente durante 2 h, se extrajo con iPrOAc (20 ml), se lavó con solución saturada de NaHCO3 (15 ml), solución de salmuera
saturada (15 ml), se secó (Na2SO4), se filtró, se purificó por cromatografía sobre SiO2. El gradiente específico usado para cada muestra se incluye en los datos de caracterización. Todos los rendimientos informados se corrigen en base al disolvente residual a partir de RMN de 1H. La reacción 2A proporcionó 2,2-dióxido de (R)-4-bencil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (18,7 g, 56 % de rendimiento) como un sólido blanco. Gradiente de columna: CH3OH del 0 al 5 % en CH2Cl2. p.f.: 134,4-135,0 °C; FTIR (neto, cm-1) 3261, 2979, 2903, 1712, 1673, 1540; RMN de 1H (400 MHz, CDCl3): δ 7,38-7,20 (m, 5H), 4,49-4,40 (m, 2H), 4,35-4,28 (m, 1H), 3,37 (dd, J = 14,0, 4,0 Hz, 1H), 2,98-2,87 (m, 1H), 1,56 (s, 9H); RMN de 13C (100 MHz, CDCl3): δ 148,5, 135,2, 129,5, 129,1, 127,5, 85,6, 68,8, 58,6, 37,9, 28,0.
Ejemplo 2B: preparación de 2,2-dióxido de (S)-4-fenil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo:

El esquema de reacción general según el ejemplo 2A anterior se realizó con (S)-(2-hidroxi-1-feniletil)carbamato de terc-butilo (10,0 g, 42,1 mmol, 100 % mol) para proporcionar el compuesto 2,2-dióxido de (S)-4-fenil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (5,23 g, 42 % de rendimiento) como un sólido blanco. Gradiente de columna: CH3OH del 0 al 5 % en CH2Cl2. p.f.: 144,3-145,0 °C; FTIR (neto, cm-1) 2976, 1722, 1458, 1377; RMN de 1H (400 MHz, CDCl3): δ 7,44-7,35 (m, 5H), 5,28 (dd, J = 6,4, 4,0 Hz, 1H), 4,87 (dd, J = 9,2, 6,4 Hz, 1H), 4,39 (dd, J = 9,2, 4,4 Hz, 1H), 1,42 (s, 9H); RMN de 13C (100 MHz, CDCl3): δ 148,3, 137,0, 129,2, 129,1, 126,2, 85,5, 71,8, 60,8, 27,8.
Ejemplo 2C: preparación de 2,2-dióxido de (S)-4-(4-metoxifenil)-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo:

El esquema de reacción general según el ejemplo 2A anterior se realizó con (S)-(2-hidroxi-1-(4-metoxifenil)etil)carbamato de terc-butilo (1,45 g, 5,42 mmol, 100 % mol) para proporcionar el compuesto 2,2-dióxido de (S)-4-(4-metoxifenil)-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (1,05 g, 59 % de rendimiento) como un sólido blanco. Gradiente de columna: CH3OH del 0 al 5 % en CH2Cl2. p.f.: 151,6-153,0 °C; FTIR (neto, cm-1) 2979, 2933, 2838, 1721, 1636, 1510, 1457; RMN de 1H (400 MHz, CDCl3): 57,37-7,32 (m, 2H), 6,95-6,90 (m, 2H), 5,24 (dd, J= 6,8, 4,4 Hz, 1H), 4,84 (dd, J= 9,2, 6,8 Hz, 1H), 4,39 (dd, J= 9,2, 4,4 Hz, 1H), 3,82 (s, 3H), 1,44 (s, 9H); RMN de 13C (100 MHz, CDCl3): δ 160,2, 148,3, 128,9, 127,7, 114,6, 85,5, 72,0, 60,5, 55,4, 27,9.
Ejemplo 2D: preparación de 2,2-dióxido de (R)-4-metil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo:

El esquema de reacción general según el ejemplo 2A anterior se realizó con (R)-(1-hidroxipropan-2-il(carbamato) de terc-butilo (5,00 g, 28,5 mmol, 100 % mol) para proporcionar el compuesto 2,2-dióxido de (R)-4-metil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (3,85 g, 57 % de rendimiento) como un sólido blanco. Gradiente de columna: CH3OH del 0 al 5 % en CH2Cl2. FTIR (neto, cm-1) 3245, 2982, 1719, 1402, 1329; RMN de 1H (400 MHz, CDCl3): δ 4,66 (dd, J = 9,2, 6,0 Hz, 1H), 4,41 (qdd, J = 6,4, 6,0, 2,8 Hz, 1H), 4,19 (dd, J = 9,2, 2,8 Hz, 1H), 1,54 (s, 9H), 1,50 (d, J = 6,4 Hz, 3 H); RMN de 13C (100 MHz, CDCl3): δ 148,5, 85,4, 71,4, 53,8, 28,0, 18,3.
Ejemplo 2E: preparación de 2,2-dióxido de (R)-4-isopropil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo:

El esquema de reacción general según el ejemplo 2A anterior se realizó con (R)-(1-hidroxi-3-metilbutan-2-il)carbamato de terc-butilo (5,00 g, 24,6 mmol, 100 % mol) para proporcionar el compuesto 2,2-dióxido de (R)-4-isopropil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (3,93 g, 60 % de rendimiento) como un sólido blanco. Gradiente de columna: CH3OH del 0 al 5 % en CH2Cl2. p.f.: 104,8-105,8 °C; RMN de 1H (400 MHz, CDCl3): δ 4,55 (dd, J= 9,6, 6,4 Hz, 1H), 4,38 (dd, J= 9,6, 2,0 Hz, 1H), 4,17 (ddd, J = 6,4, 5,2, 1,6 Hz, 1H), 2,24 (qqd, J = 6,8, 6,8, 5,2 Hz, 1H), 1,53 (s, 9H), 1,00 (d, J = 6,8 Hz, 3 H), 0,95 (d, J = 6,8 Hz, 3H); RMN de 13C (100 MHz, CDCl3): δ 149,1, 85,3, 67,0, 62,0, 30,0, 27,9, 18,0, 16,4.
Ejemplo 2F: preparación de 2,2-dióxido de (R)-4-ciclopropil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo:

El esquema de reacción general según el ejemplo 2A anterior se realizó con (R)-(1-ciclopropil-2-hidroxietil)carbamato de terc-butilo (2,31 g, 11,5 mmol, 100 % mol) para proporcionar 2,2-dióxido de (R)-4-ciclopropil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (1,36 g, 45 % de rendimiento) como un sólido blanco. Gradiente de columna: CH3OH del 0 al 5 % en CH2Cl2. p.f.: 52,7-55,7 °C; FTIR (neto, cm-1) 2977, 1734, 1460, 1363; RMN de 1H (400 MHz, CDCl3): δ 4,64 (dd, J =9,2, 6,0 Hz, 1H), 4,40 (dd, J = 8,8, 2,0 Hz, 1H), 3,77 (ddd, J = 9,2, 6,0, 2,0 Hz, 1H), 1,54 (s, 9H), 1,35-1,23 (m, 1H), 0,74-0,65 (m, 2H), 0,63-0,54 (m, 1H), 0,29-0,20 (m, 1H); RMN de 13C (100 MHz, CDCl3): δ 148,9, 85,3, 71,1, 61,6, 27,9, 14,3, 4,4, 1,7.
Ejemplo 2G: preparación de 2,2-dióxido de (3aR,8aS)-8,8a-dihidroindeno[1,2-d][1,2,3]oxatiazol-3(3aH)-carboxilato de terc-butilo:

El esquema de reacción general según el ejemplo 2A anterior se realizó con ((1R,2S)-2-hidroxi-2,3-dihidro-1H-inden-1-il)carbamato de terc-butilo (9,12 g, 36,6 mmol, 100 % mol) para proporcionar 2,2-dióxido de (3aR,8aS)-8,8a-dihidroindeno[1,2-d][1,2,3]oxatiazol-3(3aH)-carboxilato de terc-butilo (7,50 g, 66 % de rendimiento) como un sólido blanco. Gradiente de columna: CH3OH del 0 al 5 % en CH2Cl2. p.f.: 134,2-135,0 °C; FTIR (neto, cm-1) 2988, 2937, 1732, 1462, 1375; RMN de 1H (400 MHz, CDCl3): δ 7,62-7,57 (m, 1H), 7,39-7,24 (m, 3H), 5,71 (d, J = 5,6 Hz, 1H), 5,50 (dt, J = 6,0, 3,2 Hz, 1 H), 3,38 (d, J = 3,2 Hz, 1 H), 1,62 (s, 9H); RMN de 13C (100 MHz, CDCl3): δ 149,6, 138,4, 137,9, 129,9, 128,4, 126,2, 125,2, 85,7, 82,2, 65,0, 36,5, 28,0.
Ejemplo 2H: preparación de 2,2-dióxido de (S)-5-metil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo:

El esquema de reacción general según el ejemplo 2A anterior se realizó con (S)-(2-hidroxipropil)carbamato de terc-butilo (6,25 g, 35,7 mmol, 100 % mol) para proporcionar el compuesto 2,2-dióxido de (S)-5-metil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (6,10 g, 72 % de rendimiento) como un sólido blanco. Gradiente de columna: CH3OH del 0 al 5 % en CH2Cl2. p.f.: 116,9-118,2 °C; FTIR (neto, cm-1) 3370, 2956, 2938, 2837, 1681, 1512, 1461, 1366; RMN de 1H (400 MHz, CDCl3): δ 5,00-4,90 (m, 1H), 4,06 (dd, J= 9,6, 5,6 Hz, 1H), 3,63 (dd, J = 9,6, 9,2 Hz, 1H), 1,56 (d, J = 6,4 Hz, 3H), 1,53 (s, 9H); RMN de 13C (100 MHz, CDCl3): δ 148,6, 85,3, 76,2, 51,7, 27,9, 18,0.
Ejemplo 2I: preparación de 2,2-dióxido de (S)-4-(3-fluorofenil)-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo:

El esquema de reacción general según el ejemplo 2A anterior se realizó con (S)-(1-(3-fluorofenil)-2
hidroxietil)carbamato de terc-butilo (1,45 g, 5,68 mmol, 100 % mol) para proporcionar 2,2-dióxido de (S)-4-(3-fluorofenil)-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (0,702 g, 39 % de rendimiento) como un sólido blanco. Gradiente de columna: iProAc del 0 al 50 % en heptano, p.f.: 112,9-114,3 °C; FTIR (neto, cm-1) 2976, 1722, 1636, 1594, 1458; RMN de 1H (400 MHz, CDCl3): δ 7,44-7,36 (m, 1H), 7,24-7,18 (m, 1H), 7,17-7,11 (m, 1H), 7,11-7,05 (m, 1H), 5,28 (dd, 7=6,8, 3,6 Hz, 1H), 4,88 (dd, J = 9,2, 6,8 Hz, 1H), 4,39 (dd, J = 9,2, 3,6 Hz, 1H), 1,47 (s, 9H); RMN de 13C (100 MHz, CDCl3): δ 163,2 (d, 1 J CF = 246 Hz), 148,2, 139,6 (d, 3 J CF = 7 Hz), 131,1 (d, 3 J CF = 8 Hz), 121,8 (d, 4 J CF = 3 Hz), 116,2 (d, 2 J CF = 21 Hz), 113,4 (d, 2 J CF = 22 Hz), 86,0, 71,6, 60,2 (d, 4 J CF = 3 Hz), 27,9; RMN de 19F (CDCl3, 376 MHz): δ -111,0.
Ejemplo 2J: preparación de 2,2-dióxido de (S)-4-(3-(trifluorometil)fenil)-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo:

El esquema de reacción general según el ejemplo 2A anterior se realizó con (S)-(1-(3-fluorofenil)-2-hidroxietil)carbamato de terc-butilo (1,00 g, 3,28 mmol, 100 % mol) para proporcionar 2,2-dióxido de (S)-4-(3-(trifluorometil)fenil)-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (0,528 g, 44 % de rendimiento) como un sólido blanco. Gradiente de columna: iProAc del 0 al 50 % en heptano, p.f.: 92,0-92,6 °C; FTIR (neto, cm-1) 2989, 1720, 1463, 1373, 1325; RMN de 1H (400 MHz, CDCl3): δ 7,70-7,61 (m, 3H), 7,61-7,55 (m, 1H), 5,35 (dd, J = 6,8, 4,0 Hz, 1H), 4,92 (dd, J = 9,2, 6,8 Hz, 1H), 4,41 (dd, J = 9,2, 3,6 Hz, 1H), 1,46 (s, 9H); RMN de 13C (100 MHz, CDCl3): δ 148,2, 138,2, 131,8 (q, 2 J CF = 32 Hz), 130,1, 129,4, 126,1 (q, 3 J CF = 4 Hz), 123,7 (q, 1 J CF = 270 Hz), 123,4 (q, 3 J CF = 4 Hz), 86,2, 71,4, 60,2, 27,8; RMN de 19F (CDCl3, 376 MHz): δ -62,8.
Ejemplo 2K: preparación de 2,2-dióxido de (S)-4-fenil-1,2,3-oxatiacinano-3-carboxilato de terc-butilo:

El esquema de reacción general según el ejemplo 2A anterior se realizó con (S)-(3-hidroxi-1-fenilpropil)carbamato de terc-butilo (2,00 g, 7,96 mmol, 100 % mol) para proporcionar 2,2-dióxido de (S)-4-fenil-1,2,3-oxatiacinano-3-carboxilato de terc-butilo (1,26 g, 51 % de rendimiento) como un sólido blanco. Gradiente de columna: iProAc del 0 al 50 %. p.f.: 128,6-129,9 °C; FTIR (neto, cm-1) 2986, 1727, 1449, 1367; RMN de 1H (400 MHz, DMSO-d 6) (94:6 mezcla de rotámeros): δ 7,48-7,35 (m, 4H), 7,35-7,23 (m, 1H), 5,65 (dd, J = 4,4, 4,4 Hz, 0,94H), 5,53 (dd, J = 11,2, 4,4 Hz, 0,06H), 4,70 (ddd, J = 10,4, 7,2, 2,8 Hz, 0,94H), 4,51 (ddd, J = 8,8, 4,4, 4,4 Hz, 0,06H), 4,40 (ddd, J = 10,4, 10,4, 6,8 Hz, 1H), 2,80-2,68 (m, 1H), 2,66-2,56 (m, 1H), 1,41 (s, 8,46H), 1,12 (s, 0,54H); RMN de 13C (100 MHz, DMSO-d 6) (rotámeros): δ 155,1, 143,0, 128,4(128,6), 127,0(127,3), 126,3(125,4), 78,1(84,3), 73,6(70,9), 50,5(60,0), 36,0, 28,2(27,4).
Ejemplo 3: alquilación de indol
Las triptaminas quirales se encuentran con frecuencia en farmacología debido a sus actividades biológicas significativas en el sistema nervioso central. Además, el resto de triptamina quiral sirve como precursor sintético de muchos alcaloides de indol médicamente importantes y se encuentra en numerosos productos naturales y farmacéuticos biológicamente activos.
En particular, la síntesis estereocontrolada de compuestos que contienen tetrahidro-β-carbolina tales como los descritos en el presente documento a menudo se basa en la reacción diastereoselectiva de Pictet-Spengler, que a su vez requiere triptaminas enantioméricamente puras como material de partida. Este último típicamente se prepara de manera no estereoselectiva por medio de una secuencia de múltiples etapas que implica reactivos de nitroalcano peligrosos. Por lo tanto, el desarrollo de un enfoque de alquilación regioselectiva simple que implica indoles no protegidos fácilmente disponibles como nucleófilos y electrófilos derivados de aminas quirales, tales como aciridinas quirales y sulfamidatos cíclicos, proporcionó un acceso conveniente a estos valiosos andamios quirales.
Se han aplicado electrófilos de aciridina para proporcionar alquilación selectiva de C3 en condiciones ácidas de Lewis; sin embargo, este procedimiento solo es aplicable para la síntesis de triptaminas β-sustituidas ya que el desplazamiento se produce preferentemente en el carbono más sustituido. Descubierto en el presente documento,
el cuprato de indoles de bajo orden en combinación con sulfamidatos cíclicos quirales proporcionó con éxito un acceso práctico a ambas triptaminas quirales α y β-sustituidas que tienen alta regioselectividad.
Se descubrió que la reacción con aciridinas quirales como electrófilo siempre daba lugar a una mezcla de triptaminas α y β-sustituidas, mientras que la reacción con sulfamidatos cíclicos como electrófilo reaccionaba inequívocamente para desplazar el enlace C-O.
Contrariamente a la precedencia de la literatura que sugiere una alquilación preferencial en la posición C3 cuando se usaron reactivos de Grignard como base, los intentos iniciales dieron como resultado rendimientos menores a los esperados y poca selectividad de sitio. Sin quedar vinculado a ninguna teoría particular, se probaron nucleófilos de indol más blandos, ya que probablemente preferirían reaccionar como nucleófilo centrado en carbono. Se examinaron diversos aditivos incluyendo sales de Cu y Zn. De forma interesante, un sistema de haluro mixto tal como MeMgCl en combinación con CuBr o Cul, o MeMgBr en combinación con CuCl fue mucho menos eficaz que el sistema de solo cloruro. Otras sales de cobre, tales como CuCl2, CuCN, CuTC o Cu(SCN) también fueron inferiores a CuCl.

No se toleró una cantidad catalítica de CuCl y dio como resultado una disminución significativa en el rendimiento y la selectividad. De forma notable, se observó una inversión de la regioselectividad cuando se usaron haluros de cinc en lugar de CuCl (véase anteriormente). Lo mismo sucedió cuando se usó MeLi como base en lugar de MeMgCl, mientras que el uso de otro reactivo de Grignard fue bien tolerado. Se evaluó el efecto de la temperatura de reacción y se estableció que la reacción de desplazamiento se realizó de manera óptima a alrededor de -20 °C. Por debajo de -30 °C, se observó un descenso significativo en la regioselectividad, presumiblemente debido a la formación de cuprato incompleta.
La alquilación de indol mediada por Cu toleró una variedad de sustituciones tanto en el nucleófilo de indol como en el sulfamidato cíclico proporcionando los productos de indol alquilados en C3 con un rendimiento de moderado a bueno y excelente regioselectividad. Por ejemplo, los indoles con sustituyentes que donan electrones o bien que sustraen electrones participaron bien en la reacción (compuestos ejemplares 6b-6g anteriores), y los sustratos estéricamente exigentes también funcionaron razonablemente bien (compuestos ejemplares 6h y 6i anteriores). Por otra parte, los azaindoles parecen ser posibles sustratos insuficientes. En condiciones de reacción estándar, 6-azaindol proporcionó solo un 8 % del producto de alquilación, aunque con una regioselectividad comparable (véase, por ejemplo, el compuesto ejemplar 6j anterior). Otros azaindoles tales como indazol y 7-azaindol no lograron producir ningún producto alquilado deseado.
Se sometieron a prueba una variedad de sulfamidatos en la reacción con éxito. Los sulfamidatos tanto sustituidos con arilo como con alquilo, preparados en una secuencia de dos etapas a partir de los correspondientes aminoalcoholes, se convirtieron suavemente en las respectivas triptaminas quirales sustituidas en o. De forma similar, el sulfamidato cíclico de 6 miembros también participó en la alquilación y produjo una triptamina
homologada con buen rendimiento y regioselectividad. Este proceso de alquilación también se puede aplicar para obtener acceso a triptaminas β-sustituidas y α,β-disustituidas (véanse los ejemplos de compuestos 6s y 6t anteriores). En estos casos, el nucleófilo de indol se añadió al correspondiente sulfamidato cíclico con la inversión de la estereoquímica en el carbono que porta oxígeno con estereoespecificidad completa. La utilidad de este proceso de alquilación se demostró como se proporciona en el presente documento.
Ejemplo 3: esquema de reacción general de alquilación de indol
La alquilación de indol se realizó de acuerdo con el siguiente esquema de reacción general, donde RA y RB corresponden a los diversos grupos funcionales en las siguientes reacciones del ejemplo 3, y en el que los asteriscos representan un centro quiral:

Ejemplo 3A: preparación de (R)-(1-(1H-indol-3-il)-3-fenilpropan-2-il)carbamato de terc-butilo:

El esquema de reacción general 3 anterior se realizó como sigue. A una mezcla fría (0 °C) de indol (280 mg, 2,39 mmol, 150 % mol) y CuCl (193 mg, 1,95 mmol, 130 % mol) en CH2Cl2 (3,0 ml) se le añadió MeMgCl (3,0 M en THF, 0,65 ml, 1,95 mmol, 130 % mol) durante 10 min a 0 °C. Se agitó la mezcla de reacción a 0 °C durante 1 h y se enfrió hasta -20 °C. Se añadió una solución de 2,2-dióxido de (R)-4-bencil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (500 mg, 1,60 mmol, 100 % mol) en CH2Cl2 (2,0 ml) a la mezcla de reacción durante 30 min a -20 °C. A continuación, se agitó la mezcla de reacción a -20 °C durante 18 h, se desactivó con ácido cítrico acuoso al 10 % (5,0 ml) a 0 °C, se filtró, se extrajo con CH2Cl2 (10,0 ml x 2), se lavó con salmuera saturada (20,0 ml x 2), se secó (Na2SO4), se filtró, se purificó por cromatografía sobre SiO2. El gradiente específico usado para cada muestra se incluye en los datos de caracterización. Todos los rendimientos informados se corrigen en base al disolvente residual a partir de RMN de 1H. La reacción 3A proporcionó (R)-(1-(1H-indol-3-il)-3-fenilpropan-2-il)carbamato de terc-butilo (424 mg, 76 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 97:3. Gradiente de columna: iProAc del 0 al 50 % en heptano, p.f.: 152,1-153,2 °C; FTIR (neto, cm-1) 3418, 3402, 3376, 2974, 2911, 1684, 1522; RMN de 1H (400 MHz, DMSO-d 6) (85:15 mezcla de rotámeros): δ 10,78 (a., 1H), 7,46 (d, J = 8,0 Hz, 1H), 7,32 (d, J = 8,0 Hz, 1H), 7,28-7,21 (m, 2H), 7,19-7,10 (m, 4H), 7,05 (ddd, J = 8,4, 7,2, 1,2 Hz, 1H), 6,95 (ddd, J = 8,0, 6,8, 1,2 Hz, 1H), 6,76 (d, J = 8,4 Hz, 0,85H), 6,34 (d, J = 9,2 Hz, 0,15H), 3,97-3,83 (m, 1H), 2,90-2,65 (m, 4H), 1,29 (s, 7,65H), 1,12 (s, 1,35H); RMN de 13C (100 MHz, DMSO-d 6) (rotámeros): δ 155,1, 139,6, 136,1, 129,0, 128,0, 127,5, 125,8, 123,2, 120,8, 118,3, 118,1, 111,4, 111,3, 77,3, 52,6, 39,9, 30,4, 28,2(27,8).
Ejemplo 3B: preparación de (S)-(2-(1H-indol-3-il)-1-feniletil)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre indol (294 mg, 2,51 mmol, 150 % mol) y 2,2-dióxido de (S)-4-fenil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (500 mg, 1,67 mmol, 100 % mol) para proporcionar (S)-(2-(1H-indol-3-il)-1-feniletil)carbamato de terc-butilo (398 mg, 71 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 97:3. Gradiente de columna: iProAc del 0 al 50 % en heptano, p.f.: 134,6-135,2 °C; FTIR
(neto, cm-1) 3416, 3401, 3371, 2980, 2909, 1683, 1524; RMN de 1H (400 MHz, DMSO-d 6) (85:15 mezcla de rotámeros): δ 10,74 (a., 1H), 7,54 (d, J = 7,6 Hz, 1H), 7,41 (d, J = 8,4 Hz, 1H), 7,38-7,25 (m, 5H), 7,20 (dd, J = 6,8, 7,2 Hz, 1H), 7,06 (dd, J = 7,6, 7,2 Hz, 1H), 7,02 (s, 1H), 6,98 (dd, J = 7,6, 7,2 Hz, 1H), 4,89-4,74 (m, 1H), 3,08 (dd, J = 14,8, 8,8 Hz, 1H), 2,99 (dd, J = 14,4, 6,0 Hz, 1H), 1,31 (s, 7,65H), 1,08 (s, 1,35H); RMN de 13C (100 MHz, DMSO-d 6): δ 155,0, 144,5, 136,0, 128,0, 127,3, 126,5, 126,4, 123,2, 120,8, 118,3, 118,2, 111,3, 111,3, 77,6, 55,0, 32,8, 28,2.
Ejemplo 3C: preparación de (S)-(2-(1H-indol-3-il)-1-(4-metoxifenil)etil)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre indol (264 mg, 2,25 mmol, 150 % mol) y 2,2-dióxido de (S)-4-(4-metoxifenil)-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (495 mg, 1,50 mmol, 100 % mol) para proporcionar (S)-(2-(1H-indol-3-il)-1-(4-metoxifenil)etil)carbamato de terc-butilo (378 mg, 69 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 98:2. Gradiente de columna: iProAc del 0 al 50 % en heptano, p.f.: 173,3-176,5 °C; FTIR (neto, cm-1) 3402, 3326, 2979, 2925, 2904, 1690, 1611, 1506; RMN de 1H (400 MHz, DMSO-d 6) (85:15 mezcla de rotámeros): δ 10,72 (a., 1H), 7,54 (d, J = 8,0 Hz, 1H), 7,39-7,28 (m, 2H), 7,24 (d, J = 8,8 Hz, 2H), 7,05 (ddd, J = 8,4, 7,2, 1,2 Hz, 1H), 7,02-6,94 (m, 2H), 6,84 (d, J = 8,4 Hz, 2H), 4,88-4,57 (m, 1H), 3,72 (s, 3H), 3,06 (dd, J = 14,8, 8,4 Hz, 1H), 2,96 (dd, J = 14,8, 6,4 Hz, 1H), 1,31 (s, 7,65H), 1,11 (s, 1,35H); RMN de 13C (100 MHz, DMSO-d 6): δ 157,9, 154,9, 136,4, 136,0, 127,5, 127,3, 123,2, 120,7, 118,3, 118,1, 113,4, 111,4, 111,2, 77,5, 55,0, 54,4, 32,8, 28,2.
Ejemplo 3D: preparación de (R)-(1-(1H-indol-3-il)propan-2-il)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre indol (370 mg, 3,16 mmol, 150 % mol) y 2,2-dióxido de (R)-4-metil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (500 mg, 2,11 mmol, 100 % mol) para proporcionar (R)-(1-(1H-indol-3-il)propan-2-il)carbamato de terc-butilo (406 mg, 70 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 99:1. Gradiente de columna: iProAc del 0 al 50 % en heptano, p.f.: 82,2-84,3 °C; FTIR (neto, cm-1) 3416, 3401, 3366, 2974, 2963, 1684, 1524; RMN de 1H (400 MHz, DMSO-d 6): δ 10,77 (a., 1H), 7,56 (d, J = 7,6 Hz, 1H), 7,33 (ddd, J = 8,0, 1,2, 0,8 Hz, 1H), 7,10 (d, J = 2,4 Hz, 1H), 7,05 (ddd, J = 8,0, 6,8, 1,2 Hz, 1H), 6,97 (ddd, J = 8,0, 6,8, 1,2 Hz, 1H), 6,71 (d, J = 8,4 Hz, 1H), 3,82-3,66 (m, 1H), 2,87 (dd, J = 14,0, 6,0 Hz, 1H), 2,65 (dd, J= 14,0, 7,6 Hz, 1H), 1,38 (s, 9H), 1,01 (d, J =6,8Hz, 3H); RMN de 13C (100 MHz, DMSO-d 6): δ 155,0, 136,1, 127,5, 123,1, 120,7, 118,4, 118,1, 111,6, 111,2, 77,3, 46,8, 32,2, 28,3, 20,1.
Ejemplo 3E: preparación de (R)-(1-(1H-indol-3-il)-3-metilbutan-2-il)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre indol (331 mg, 2,83 mmol, 150 % mol) y 2,2-dióxido de (R)-4-isopropil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (500 mg, 1,88 mmol, 100 % mol) para proporcionar
(R)-(1-(1H-indol-3-il)-3-metilbutan-2-il)carbamato de terc-butilo (335 mg, 59 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 94:6. Gradiente de columna: iProAc del 0 al 50 % en heptano, p.f.: 145,1-146,9 °C; FTIR (neto, cm-1) 3417, 3402, 3362, 2978, 1686, 1526; RMN de 1H (400 MHz, DMSO-d 6) (85:15 mezcla de rotámeros): δ 10,72 (a., 1H), 7,51 (d, J = 8,0 Hz, 1H), 7,31 (ddd, J = 8,0, 1,2, 0,8 Hz, 1H), 7,07 (d, J = 2,0Hz, 1H), 7,04 (ddd, J = 8,0, 6,8, 1,2 Hz, 1H), 6,96 (ddd, J = 8,0, 6,8, 1,2 Hz, 1H), 6,59 (d, J = 9,2 Hz, 0,85H), 6,15 (d, J = 10,0 Hz, 0,15H), 3,64-3,53 (m, 1H), 2,80 (dd, J = 14,8, 5,2 Hz, 1H), 2,68 (dd, J = 14,8, 8,8 Hz, 1H), 1,77-1,65 (m, 1H), 1,32 (s, 7,65H), 1,12 (s, 1,35H), 0,95-0,82 (m, 6H); RMN de 13C (100 MHz, DMSO-d 6): δ 155,6, 136,1, 127,5, 122,7, 120,7, 118,3, 118,0, 112,0, 111,2, 77,1, 55,6, 31,4, 28,3, 27,1, 19,4, 17,7.
Ejemplo 3F: preparación de (S)-(1-ciclopropil-2-(1H-indol-3-il)etil)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre indol (264 mg, 2,25 mmol, 150 % mol) y 2,2-dióxido de (R)-4-ciclopropil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (395 mg, 1,50 mmol, 100 % mol) para proporcionar (S)-(1-ciclopropil-2-(1H-indol-3-il)etil)carbamato de terc-butilo (292 mg, 65 % de rendimiento) como un blanco sólido. La proporción C3/N1 fue de 99:1. Gradiente de columna: iProAc del 0 al 50 % en heptano, p.f.: 128,8-130,5 °C; FTIR (neto, cm-1) 3414, 3400, 3362, 2981, 2937, 1683, 1525; RMN de 1H (400 MHz, DMSO-d 6) (9:1 mezcla de rotámeros): δ 10,73 (a., 1H), 7,52 (d, J = 7,6 Hz, 1H), 7,31 (ddd, J = 8,0, 1,2, 0,8 Hz, 1H), 7,07 (d, J = 2,4 Hz, 1H), 7,04 (ddd, J = 8,0, 6,8, 1,2 Hz, 1H), 6,95 (ddd, J = 8,0, 6,8, 1,2 Hz, 1H), 6,66 (d, J = 8,4 Hz, 0,9H), 6,26 (s, 0,1H), 3,30-3,18 (m, 1H), 2,91 (dd, J = 14,4, 5,6 Hz, 1H), 2,85 (dd, J = 14,4, 8,0 Hz, 1H), 1,33 (s, 8,1H), 1,17(s, 0,9H), 0,96-0,83 (m, 1H), 0,42-0,32 (m, 1H), 0,32-0,24 (m, 2H), 0,15-0,01 (m, 1H); RMN de 13C (100 MHz, DMSO-d 6): δ 155,3, 136,0, 127,7, 122,9, 120,6, 118,4, 118,0, 111,6, 111,2, 77,2, 54,2, 30,4, 28,2, 16,0, 3,0, 1,9. Ejemplo 3G: preparación de ((1S,2S)-2-(1H-indol-3-il)-2,3-dihidro-1H-inden-1-il)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre indol (282 mg, 2,41 mmol, 150 % mol) y 2,2-dióxido de (3aR,8aS)-8,8a-dihidroindeno[1,2-d][1,2,3]oxatiazol-3(3aH)-carboxilato de terc-butilo (500 mg, 1,61 mmol, 100 % mol) para proporcionar ((1S,2S)-2-(1H-indol-3-il)-2,3-dihidro-1H-inden-1-il)carbamato de terc-butilo (304 mg, 54 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 95:5. Gradiente de columna: iProAc del 0 al 50 % en heptano, p.f.: 155,7-157,1 °C; FTIR (neto, cm-1) 3387, 3351, 2980, 2938, 1691, 1500; RMN de 1H (400 MHz, DMSO-d 6) (87:13 mezcla de rotámeros): 10,85 (a., 1H), 7,61 (d, J = 8,0 Hz, 1H), 7,42-7,32 (m, 2H), 7,30-7,20 (m, 4H), 7,17 (dd, J = 8,0, 4,0 Hz, 1H), 7,07 (ddd, J = 8,0, 6,8, 1,2 Hz, 1H), 6,97 (ddd, J = 8,0, 6,8, 1,2 Hz, 1H), 5,18 (dd, J =9,2, 9,2 Hz, 1H), 3,69 (dd, J = 18,4, 9,6 Hz, 0,87H), 3,64-3,53 (m, 0,13H), 3,37 (dd, J = 15,2, 8,0 Hz, 0,87H), 3,30-3,23 (m, 0,13H), 3,13-3,04 (m, 0,13H), 2,99 (dd, J = 15,2, 10,4 Hz, 0,87H), 1,38 (s, 7,83H), 1,12 (s, 1,17H); RMN de 13C (100 MHz, DMSO-d 6) (rotámeros): δ 155,9, 144,5, 141,3, 136,6, 127,2, 127,1, 126,4, 124,4, 123,3, 121,7, 120,9, 119,1, 118,2, 115,5, 111,4, 77,7, 60,8, 44,0, 37,2, 28,2 (27,7).
Ejemplo 3H: preparación de (S)-(2-(1H-indol-3-il)propil)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre indol (264 mg, 2,25 mmol, 150 % mol) y 2,2-dióxido de (S)-5-metil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (356 mg, 1,50 mmol, 100 % mol) para proporcionar (S)-(2-(1H-indol-3-il)propil)carbamato de terc-butilo (319 mg, 78 % de rendimiento) como un líquido incoloro. La proporción C3/N1 fue de 93:7. Gradiente de columna: iProAc del 0 al 50 % en heptano. FTIR (neto, cm-1) 3412, 3327, 2971, 2930, 1685, 1508, 1456; RMN de 1H (400 MHz, DMSO-d 6) (9:1 mezcla de rotámeros): δ 10,78 (a., 1H), 7,58 (d, J = 7,6 Hz, 1H), 7,33 (ddd, J = 8,0, 1,2, 0,8 Hz, 1H), 7,10 (d, J = 2,0 Hz, 1H), 7,05 (ddd, J = 8,0, 6,8, 1,2 Hz, 1H), 6,96 (ddd, J = 8,0, 6,8, 1,2 Hz, 1H), 6,86 (dd, J = 6,0, 6,0 Hz, 0,9H), 6,51 (s, 0,1H), 3,29 (ddd, J = 13,2, 5,6, 5,6 Hz, 1H), 3,18-3,05 (m, 1H), 2,97 (ddd, J = 13,2, 8,8, 6,0 Hz, 1H), 1,41 (s, 0,9H), 1,38 (s, 8,1H), 1,25 (d, J = 6,8 Hz, 3H); RMN de 13C (100 MHz, DMSO-d 6) (rotámeros): δ 155,7, 136,4, 126,6, 121,0, 120,8, 118,6, 118,1, 117,9, 111,4, 77,4, 46,8, 30,9, 28,3(28,2), 18,4(18,2).
Ejemplo 3I: preparación de (R)-(1-(3-fluorofenil)-2-(1H-indol-3-il)etil)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre indol (263 mg, 2,25 mmol, 150 % mol) y 2,2-dióxido de (S)-4-(3-fluorofenil)-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (475 mg, 1,50 mmol, 100 % mol) para proporcionar (R)-(1-(3-fluorofenil)-2-(1H-indol-3-il)etil)carbamato de terc-butilo (382 mg, 72 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 99:1. Gradiente de columna: iProAc del 0 al 50 % en heptano, p.f.: 148,6-151,2 °C; FTIR (neto, cm-1) 3414, 3398, 3363, 3055, 2981, 1682, 1591, 1527; RMN de 1H (400 MHz, DMSO-d 6) (9:1 mezcla de rotámeros): δ 10,75 (a., 1H), 7,53 (d, J = 7,6 Hz, 1H), 7,44 (d, J = 8,4 Hz, 1H), 7,37-7,26 (m, 2H), 7,20-7,11 (m, 2H), 7,08-6,93 (m, 4H), 4,85-4,65 (m, 1H), 3,06 (dd, J = 14,4, 8,4 Hz, 1H), 2,98 (dd, J = 14,4, 6,4 Hz, 1H), 1,30 (s, 8,1H), 1,08 (s, 0,9H); RMN de 13C (100 MHz, DMSO-d 6): δ 162,2 (d, 1 J CF = 243 Hz), 155,0, 147,5 (d, 3 J CF = 7 Hz), 136,0, 129,9 (d, 3 J CF = 8 Hz), 127,2, 123,3, 122,6, 120,8, 118,3, 118,2, 113,3 (d, 2 J CF = 21 Hz), 113,0 (d, 2 J CF = 21 Hz), 111,3, 111,0, 77,8, 54,7, 32,5, 28,2; RMN de 19F (DMSO-d 6, 376 MHz): δ -113,6. Ejemplo 3J: preparación de (R)-(2-(1H-indol-3-il)-1-(3-(trifluorometil)fenil)etil)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre indol (176 mg, 1,50 mmol, 150 % mol) y 2,2-dióxido de (S)-4-(3-(trifluorometil)fenil)-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (368 mg, 1,00 mmol, 100 % mol) para proporcionar (R)-(2-(1H-indol-3-il)-1-(3-(trifluorometil)fenil)etil)carbamato de terc-butilo (320 mg, 79 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 98:2. Gradiente de columna: iProAc del 0 al 50 % en heptano, p.f.: 99,3-101,4 °C; FTIR (neto, cm-1) 3414, 3399, 3361, 2982, 2936, 1683, 1523; RMN de 1H (400 MHz, DMSO-d 6) (88:12 mezcla de rotámeros): 510,76 (a., 1H), 7,80-7,45 (m, 6H), 7,31 (d J = 8,0 Hz, 1H), 7,08-7,00 (m, 2H), 6,96 (ddd, J = 8,0, 7,2, 1,2 Hz, 1H), 5,11-4,69 (m, 1H), 3,09 (dd, J = 14,4, 8,4 Hz, 1H), 3,00 (dd, J = 14,4, 6,4 Hz, 1H), 1,30 (s, 7,9H), 1,07 (s, 1,1H); RMN de 13C (100 MHz, DMSO-d 6): δ 155,1, 145,8, 136,1, 130,8, 129,0, 128,9 (q, 2 J CF = 32 Hz), 127,3, 124,4 (q, 1 J CF = 270 Hz), 123,3 (q, 3 J CF = 4 Hz), 123,0, 122,8 (q, 3 J CF = 4 Hz), 120,8, 118,3, 118,2, 111,3, 110,8, 77,9, 55,0, 32,5, 28,2; RMN de 19F (DMSO-d 6, 376 MHz): δ -61,0.
Ejemplo 3K: preparación de (S)-(3-(1H-indol-3-il)-1-fenilpropil)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre indol (264 mg, 2,25 mmol, 150 % mol) y 2,2-dióxido de (S)-4-fenil-1,2,3-oxatiacinano-3-carboxilato de terc-butilo (470 mg, 1,50 mmol, 100 % mol) para proporcionar (S)-(3-(1H-indol-3-il)-1-fenilpropil)carbamato de terc-butilo (401 mg, 73 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 98:2. Gradiente de columna: iProAc del 0 al 50 % en heptano, p.f.: 121,7-123,2 °C; FTIR (neto, cm-1) 3390, 2979, 2929, 2859, 1681, 1507, 1457, 1364; RMN de 1H (400 MHz, CDCl3) (80:20 mezcla de rotámeros): δ 8,04 (a., 0,2H), 7,96 (a., 0,8H), 7,66 (d, J = 8,0 Hz, 0,2H), 7,52 (d, J = 8,0 Hz, 0,8H), 7,40-7,22 (m, 6H), 7,22-7,15 (m, 1H), 7,15-7,06 (m, 1H), 7,00 (a., 1H), 4,88 (a., 0,8H), 4,75 (a., 1H), 4,53 (a., 0,2H), 3,52-3,35 (m, 0,4H), 3,32-3,20 (m, 0,4H), 2,87-2,67 (m, 1,6H), 2,16 (d, J = 8,8 Hz, 1,6 H), 1,42 (s, 9H); RMN de 13C (100 MHz, DMSO-d 6) (rotámeros): δ 155,4(156,2), 143,0, 136,4(136,6), 128,6, 127,3, 127,2, 126,4, 121,8(122,0), 121,5(120,8), 119,0(119,2), 118,7, 115,3, 111,2(111,3), 79,5, 54,7(46,7), 37,2(31,6), 28,4(29,7), 21,9(18,7). Ejemplo 3L: preparación de (R)-(1-(5-metil-1H-indol-3-il)-3-fenilpropan-2-il)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre 5-metil-1H-indol (295 mg, 2,25 mmol, 150 % mol) y 2,2-dióxido de (R)-4-bencil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (470 mg, 1,50 mmol, 100 % mol) para proporcionar (R)-(1-(5-metil-1H-indol-3-il)-3-fenilpropan-2-il)carbamato de terc-butilo (401 mg, 73 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 92:8. Gradiente de columna: iProAc del 0 al 50 % en heptano, p.f.: 123,0-123,8 °C; FTIR (neto, cm-1) 3413, 3368, 2974, 2927, 1685, 1524; RMN de 1H (400 MHz, DMSO-d 6) (85:15 mezcla de rotámeros): δ 10,63 (a., 1H), 7,30-7,13 (m, 7H), 7,07 (d, J = 2,0 Hz, 1H), 6,87 (dd, J = 8,4, 1,6 Hz, 1H), 6,74 (d, J = 8,8 Hz, 0,85H), 6,34 (d, J = 7,6 Hz, 0,15H), 3,96-3,80 (m, 1H), 2,86-2,64 (m, 4H), 2,36 (s, 3H), 1,30 (s, 7,65H), 1,16(s, 1,35H); RMN de 13C (100 MHz, DMSO-d 6) (rotámeros): δ 155,1, 139,6, 134,5, 129,1, 128,0, 127,7, 126,4, 125,8, 123,2, 122,3, 117,9, 111,0, 110,9, 77,2, 52,7, 40,0, 30,2, 28,2(27,8), 21,3. Ejemplo 3M: preparación de (R)-(1-(7-metil-1H-indol-3-il)-3-fenilpropan-2-il)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre 7-metil-1H-indol (295 mg, 2,25 mmol, 150 % mol) y 2,2-dióxido de (R)-4-bencil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (470 mg, 1,50 mmol, 100 % mol) para proporcionar (R)-(1-(7-metil-1H-indol-3-il)-3-fenilpropan-2-il)carbamato de terc-butilo (371 mg, 68 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 97:3. Gradiente de columna: iProAc del 0 al 50 % en heptano, p.f.: 124,4-125,9 °C; FTIR (neto, cm-1) 3417, 3407, 3370, 2974, 2926, 1683, 1524; RMN de 1H (400 MHz, DMSO-d 6) (85:15 mezcla de rotámeros): δ 10,75 (d a., J = 2,0 Hz, 1H), 7,34-7,21 (m, 3H), 7,21-7,09 (m, 4H), 6,90-6,83 (m, 2H), 6,74 (d, J = 8,8 Hz, 0,85H), 6,34 (d, J = 7,6 Hz, 0,15H), 3,98-3,84 (m, 1H), 2,91-2,65 (m, 4H), 2,44 (s, 3H), 1,31 (s, 7,65H), 1,14 (s, 1,35H); RMN de 13C (100 MHz, DMSO-d 6) (rotámeros): δ 155,2, 139,6, 135,7, 129,0, 128,0, 127,2, 125,8, 122,9, 121,3, 120,3, 118,4, 115,9, 111,9, 77,3, 52,6, 39,9, 30,5, 28,2(27,8), 16,7. Ejemplo 3N: preparación de (R)-(1-(5-cloro-1H-indol-3-il)-3-fenilpropan-2-il)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre 5-cloro-1H-indol (341 mg, 2,25 mmol, 150 % mol) y 2,2-dióxido de (R)-4-bencil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (470 mg, 1,50 mmol, 100 % mol) para proporcionar (R)-(1-(5-cloro-1H-indol-3-il)-3-fenilpropan-2-il)carbamato de terc-butilo (345 mg, 60 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 96:4. Gradiente de columna: iProAc del 0 al 50 % en heptano, p.f.: 70,5-73,9 °C; FTIR (neto, cm-1) 3417, 3368, 2980, 2928, 1684, 1518; RMN de 1H (400 MHz, DMSO-d 6) (85:15 mezcla de rotámeros): δ 10,99 (a., 1H), 7,49 (d, J = 2,0 Hz, 1H), 7,34 (d, J = 8,8 Hz, 1H), 7,30-7,22 (m, 2H), 7,22-7,13 (m, 4H), 7,04 (dd, J = 8,4, 2,0 Hz, 1H), 6,77 (d, J = 8,8 Hz, 0,85H), 6,34 (d, J = 8,8 Hz, 0,15H), 3,90-3,76 (m, 1H), 2,83-2,62 (m, 4H), 1,27 (s, 7,65H), 1,10 (s, 1,35H); RMN de 13C (100 MHz, DMSO-d 6) (rotámeros): δ 155,1, 139,5, 134,6, 129,0, 128,8, 128,0, 125,8, 125,2, 122,9, 120,6, 117,7, 112,8, 111,5, 77,2, 52,9, 40,0, 30,1, 28,2(27,7).
Ejemplo 3O: preparación de (R)-(1-fenil-3-(5-(trifluorometil)-1H-indol-3-il)propan-2-il)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre 5-(trifluorometil)-1H-indol (417 mg, 2,25 mmol, 150 % mol) y 2,2-dióxido de (R)-4-bencil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (470 mg, 1,50 mmol, 100 % mol) para proporcionar (R)-(1-fenil-3-(5-(trifluorometil)-1H-indol-3-il)propan-2-il)carbamato de terc-butilo (297 mg, 47 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 96:4. Gradiente de columna: iProAc del 0 al 50 % en heptano, p.f.: 130,9-131,8 °C; FTIR (neto, cm-1) 3417, 3368, 2980, 2929, 1684, 1519; RMN de 1H (400 MHz, DMSO-d 6) (85:15 mezcla de rotámeros): 11,26 (a., 1H), 7,84 (s, 1H), 7,50 (d, J = 8,4 Hz, 1H), 7,37-7,22 (m, 4H), 7,22-7,10 (m, 3H), 6,80 (d, J = 8,8 Hz, 0,85H), 6,36 (d, J = 9,2 Hz, 0,15H), 3,99-3,77 (m, 1H), 2,86 (d, J = 6,8 Hz, 2H), 2,76 (d, J = 6,8 Hz, 2H), 1,22 (s, 7,65H), 1,04 (s, 1,35H); RMN de 13C (100 MHz, DMSO-d 6) (rotámeros): δ 155,1(154,6), 139,5, 137,6(137,7), 129,1, 128,0, 126,9, 125,8, 125,7 (q, 1 J CF = 269 Hz), 125,6, 119,1 (q, 2 J CF = 32 Hz), 117,1 (q, 3 J CF = 4 Hz), 116,1 (q, 3 J CF = 4 Hz), 112,9, 111,9, 77,2, 53,0(53,7), 40,3(41,0), 29,9(30,9), 28,1(27,6); RMN de 19F (DMSO-d 6, 376 MHz): δ -58,1.
Ejemplo 3P: preparación de (R)-(1-(5-metoxi-1H-indol-3-il)-3-fenilpropan-2-il)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre 5-metoxi-1H-indol (331 mg, 2,25 mmol, 150 % mol) y 2,2-dióxido de (R)-4-bencil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (470 mg, 1,50 mmol, 100 % mol) para proporcionar (R)-(1-(5-metoxi-1H-indol-3-il)-3-fenilpropan-2-il)carbamato de terc-butilo (523 mg, 92 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 97:3. Gradiente de columna: iProAc del 0 al 50 % en heptano, p.f.: 123,2-123,9 °C; FTIR (neto, cm-1) 3368, 2975, 2933, 1692, 1680, 1516; RMN de 1H (400 MHz, DMSO-d 6) (85:15 mezcla de rotámeros): δ 10,60 (s, 1H), 7,32-7,24 (m, 2H), 7,24-7,13 (m, 4H), 7,08 (d, J = 2,4 Hz, 1H), 6,91 (d, J = 2,4 Hz, 0,85H), 6,83 (a., 0,15H), 6,77 (d, J = 8,8 Hz, 0,85H), 6,70 (dd, J = 8,8, 2,4 Hz, 1H), 6,34 (d, J = 9,2 Hz, 0,15H), 3,94-3,80 (m, 1H), 3,72 (s, 3H), 2,85-2,65 (m, 4H), 1,29 (s, 7,65H), 1,13 (s, 1,35H); RMN de 13C (100 MHz, DMSO-d 6) (rotámeros): δ 155,2, 152,9, 139,6, 131,3, 129,1, 128,0, 127,8, 125,8, 123,8, 111,9, 111,3, 110,8, 100,3, 77,3, 55,3, 52,8, 40,2, 30,2, 28,2(27,8).
Ejemplo 3Q: preparación de (R)-(1-fenil-3-(5-fenil-1H-indol-3-il)propan-2-il)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre 5-fenil-1H-indol (435 mg, 2,25 mmol, 150 % mol) y 2,2-dióxido de (R)-4-bencil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (470 mg, 1,50 mmol, 100 % mol) para proporcionar (R)-(1-fenil-3-(5-fenil-1H-indol-3-il)propan-2-il)carbamato de terc-butilo (523 mg, 82 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 97:3. Gradiente de columna: iProAc del 0 al 50 % en heptano. p.f.: 68,9-70,8 °C; FTIR (neto, cm-1) 3427, 3411, 3392, 3310, 2977, 2929, 1715, 1696, 1506; RMN de 1H (400 MHz, DMSO-d 6) (85:15 mezcla de rotámeros): δ 10,85 (a., 1H), 7,70 (s, 1H), 7,67-7,60 (m, 2H), 7,48-7,33 (m, 4H), 7,32-7,24 (m, 3H), 7,23-7,15 (m, 4H), 6,80 (d, J = 8,8 Hz, 0,85H), 6,39 (d, J = 9,2 Hz, 0,15H), 4,03-3,84 (m, 1H), 2,87 (d, J =6,8 Hz, 2H), 2,77 (d, J = 6,8Hz, 2H), 1,24 (s, 7,65H), 1,09 (s, 1,35H); RMN de 13C (100 MHz, DMSO-d 6) (rotámeros): δ 155,1, 142,0, 139,6, 135,8, 130,7, 129,1, 128,6, 128,1, 128,0, 126,6, 126,0, 125,8, 124,0, 120,1, 116,6, 112,2, 111,6, 77,2, 53,1, 40,4, 30,1, 28,2(27,8).
Ejemplo 3R: preparación de (R)-(1-(2-metil-1H-indol-3-il)-3-fenilpropan-2-il)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre 2-metil-1H-indol (295 mg, 2,25 mmol, 150 % mol) y 2,2-dióxido de (R)-4-bencil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (470 mg, 1,50 mmol, 100 % mol) para proporcionar (R)-(1-(2-metil-1H-indol-3-il)-3-fenilpropan-2-il)carbamato de terc-butilo (386 mg, 71 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 99:1. Gradiente de columna: iProAc del 0 al 50 % en heptano. p.f.: 117,6-119,0 °C; FTIR (neto, cm-1) 3440, 3385, 2984, 2930, 1684, 1524; RMN de 1H (400 MHz, DMSO-d 6) (83:17 mezcla de rotámeros): δ 10,67 (a., 1H), 7,39 (d, J = 7,6 Hz, 0,83H), 7,33 (d, J = 8,0 Hz, 0,17H), 7,28-7,18 (m, 3H), 7,18-7,07 (m, 3H), 6,96 (ddd, J = 8,0, 7,2, 1,2 Hz, 1H), 6,90 (ddd, J = 8,0, 8,0, 1,2 Hz, 1H), 6,73 (d, J = 8,8 Hz, 0,83H), 6,30 (d, J = 8,8 Hz, 0,17H), 3,94-3,74 (m, 1H), 2,85 (dd, J = 14,0, 6,4 Hz, 1H), 2,75-2,62 (m, 3H), 2,32 (s, 3H), 1,27 (s, 7,47H), 1,04 (s, 1,53H); RMN de 13C (100 MHz, DMSO-d 6) (rotámeros): 155,1, 139,8, 135,2, 132,4, 128,9, 128,7, 128,0, 125,7, 119,8, 118,0, 117,5, 110,2, 107,4, 77,2, 53,4, 39,4, 29,9, 28,2(27,7), 11,4.
Ejemplo 3S: preparación de (R)-(1-fenil-3-(2-fenil-1H-indol-3-il)propan-2-il)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre 2-fenil-1H-indol (435 mg, 2,25 mmol, 150 % mol) y 2,2-dióxido de (R)-4-bencil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (470 mg, 1,50 mmol, 100 % mol) para proporcionar (R)-(1-fenil-3-(2-fenil-1H-indol-3-il)propan-2-il)carbamato de terc-butilo (324 mg, 51 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 98:2. Gradiente de columna: iProAc del 0 al 50 % en heptano. p.f.: 178,5-178,8 °C; FTIR (neto, cm-1) 3380, 3354, 3376, 2981, 2932, 1683, 1508; RMN de 1H (400 MHz, DMSO-d 6) (82:18 mezcla de rotámeros): δ 11,14 (s, 1H), 7,69 (d, J = 7,6 Hz, 1H), 7,60 (d, J = 7,2 Hz, 2H), 7,44 (dd, J = 7,2, 7,2 Hz, 2H), 7,35 (dd, J = 7,2, 7,2 Hz, 2H), 7,21 (dd, J = 7,2, 7,2 Hz, 2H), 7,17-6,97 (m, 5H), 6,79 (d, J = 9,2 Hz, 0,82H), 6,38 (d, J = 9,6 Hz, 0,18H), 4,15-3,90 (m, 1H), 3,06 (dd, J =14,0, 6,8 Hz, 1H), 2,96 (dd, J = 14,4, 7,2 Hz, 1H), 2,64 (d, J = 6,8 Hz, 2H), 1,21 (s, 7,40H), 0,97 (s, 1,60H); RMN de 13C (100 MHz, DMSO-d 6) (rotámeros): δ 155,0, 139,5, 136,0, 134,7, 133,0, 129,2, 128,9, 128,6, 128,0, 127,8, 127,1, 125,8, 121,3, 119,2, 118,5, 111,0, 109,2, 77,2, 53,4, 40,2, 28,1(27,6).
Ejemplo 3T: preparación de (R)-(1-fenil-3-(1H-pirrolo[2,3-c]piridin-3-il)propan-2-il)carbamato de terc-butilo:

La reacción general según el ejemplo 3A se realizó entre 1H-pirrolo[2,3-c]piridina (266 mg, 2,25 mmol, 150 % mol) y 2,2-dióxido de (R)-4-bencil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo (470 mg, 1,50 mmol, 100 % mol) para proporcionar (R)-(1-fenil-3-(1H-pirrolo[2,3-c]piridin-3-il)propan-2-il)carbamato de terc-butilo (40 mg, 8 % de rendimiento) como un sólido blanco. La proporción C3/N1 fue de 98:2. Gradiente de columna: iProAc del 0 al 50 % en heptano. p.f.: 196,2-197,0 °C; FTIR (neto, cm-1) 3360, 2978, 2931, 1685, 1625, 1524; RMN de 1H (400 MHz, CDCl3) (90:10 mezcla de rotámeros): δ 8,56 (a., 1H), 8,28 (s, 1H), 7,57 (d, J = 6,4 Hz, 1H), 7,38-7,31 (m, 2H), 7,30-7,25 (m, 2H), 7,25-7,20 (m, 2H), 6,70 (a., 1H), 5,44 (a., 0,10H), 5,24 (d, J = 5,6 Hz, 0,90H), 4,52 (dd, J = 13,2, 8,4 Hz, 1H), 4,36 (dd, J = 13,6, 4,8 Hz, 1H), 4,26-4,14 (m, 1H), 3,00 (dd, J = 14,0, 7,6 Hz, 1H), 2,91 (dd, J = 14,0, 6,8 Hz, 1H), 1,27 (s, 8,10H), 1,11 (s, 0,90H).
Ejemplo 4
Preparación de acetidinil-aminas
La preparación de los compuestos de fórmula (VII) se realizó de acuerdo con el esquema general a continuación:

R5, v, R6 y R10 son como se describe en el presente documento.
Ejemplo 4A:
Preparación de etano-1,2-disulfonato de 1-(3-fluoropropil)acetidin-3-amina
Se preparó etano-1,2-disulfonato de 1-(3-fluoropropil)acetidin-3-amina de acuerdo con el siguiente esquema de reacción:

Se disolvió clorhidrato de acetidin-3-ilcarbamato de terc-butilo (109,7 kg, 1,0 eq.) en MTBE (793,4 kg) y se añadió 1-bromo-3-fluoropropano (82,3 kg). Se añadieron MTBE (14 kg) y agua (530 kg), seguido de LiOH•H2O (66,0 kg) a 15-25 °C seguido de agitación a 50-60 °C. Después de la finalización de la reacción, se separó una fase orgánica y a continuación se combinó solución acuosa de dihidrato de ácido 1,2-etanodisulfónico (247,8 kg) a 0-5 °C. Se agitó la mezcla resultante durante 30 min a 15-20 °C. Se separó una fase acuosa y se añadió dihidrato de ácido 1,2-etanodisulfónico (56,85 kg) a la fase orgánica. Se agitó la mezcla resultante durante 5 h a 35-40 °C hasta que se finalizó la desprotección. A continuación se añadió MeOH (884,1 kg) a 35-40 °C y se agitó la mezcla durante 2 h a 35-40 °C. Después de enfriar hasta temperatura ambiente, se agitó la mezcla de reacción durante 4 h. Se recogió el sólido por filtración y se aclaró con MeOH ac. Se secó el sólido lavado a 35-42 °C bajo presión reducida para proporcionar 106,8 kg de etano-1,2-disulfonato de 1-(3-fluoropropil)acetidin-3-amina (63 % de rendimiento). RMN de 1H (400 MHz, DMSO-d6) δ 9,92 (s, 0,7H), 9,63 (s, 0,3H), 8,35 (s, 3H), 4,52 (dt, J = 47,1, 5,6 Hz, 2H), 4,43-4,08 (m, 5H), 3,36 (s, 3H), 2,74 (s, 4H), 2,06-1,77 (m, 2H).
Ejemplo 4B:
Preparación de etano-1,2-disulfonato de 1-(3-fluoropropil)acetidin-3-amina por medio de química de liberación de tensión.

Preparación de bromhidrato de 2,3-dibromopropan-1-amina (3)
A 1 (20,0 g, 1,0 equiv, 50 % en peso en agua) se le añadió una solución de K2CO3 (29,55 g, 1,0 equiv) en agua (100 ml) y una solución de Boc2O (93,32 g, 2,0 equiv)) en EtOAc (100 ml) a 0 °C. Se agitó la mezcla de reacción a 20 °C durante 15 h. Se separó la capa orgánica y se cambió el disolvente a EtOH (100 ml) para proporcionar una solución de 2 en EtOH. A continuación se añadió en una solución fría (0 °C) de Br2 (71,74 g, 2,1 equiv) en EtOH (60 ml) a 0 °C. Se agitó la mezcla de reacción a 20 °C durante 16 h, se filtró, se lavó con MTBE (60 ml), se secó a vacío a 40 °C durante 15 h para proporcionar 3 como un sólido incoloro (43,01 g, 66 %): RMN de 1H (400 MHz, CD3OD) δ 4,55 - 4,47 (m, 1H), 4,01 (dd, J = 10,9, 4,6 Hz, 1H), 3,86 (dd, J = 10,9, 8,7 Hz, 1H), 3,71 (dd, J = 13,9, 3,2 Hz, 1H), 3,39 - 3,33 (m, 1H).
Preparación de etano-1,1-disulfonato de N,N-dibencilacetidin-3-amina.

A una solución de Bn2NH (33,12 g, 1,0 equiv) en THF (330 ml) se le añadió iPrMgCl•LiCl (1,3 M en THF, 130 ml, 1,0 equiv) a 20 °C y se agitó a 20 °C durante 5 h para proporcionar una solución de Bn2NMgCl•LiCl en THF. En un reactor separado se cargó 3 (50,0 g, 1,0 equiv) en THF (500 ml), se enfrió hasta -60 °C. Se añadió n-BuLi (201 ml, solución 2,5 M en n-hexano, 3,0 equiv) a la suspensión a -60 °C y se agitó a -60 °C durante 2 h para proporcionar una solución de 4 en THF. Se añadió una solución de Bn2NMgCl•LiCl a 4 a -60 °C, se calentó hasta 20 °C y se agitó a 20 °C durante 12 h para proporcionar una solución de 5 en THF. Se enfrió la mezcla de reacción hasta 0 °C, se añadió una solución de Boc2O (73,28 g, 2,0 equiv) en THF (200 ml) a 0 °C, se agitó a 20 °C durante 2 h, se enfrió hasta 0 °C, se desactivó con una solución de AcOH (20,16 g, 2,0 equiv) en H2O (383 ml). Se lavó la capa orgánica con Na2SO4 al 5 % (150 ml). Se volvió a extraer la capa acuosa combinada con MTBE (100 ml x 2) para proporcionar una solución de 6. Se añadió una solución de ácido etanodisulfónico (40,0 g, 1,5 equiv) en MeOH (225 ml) a la solución a 20 °C y se agitó a 40 °C durante 20 h. Se filtró la suspensión, se lavó con THF (100 ml), se secó a vacío (40 °C) durante 20 h para proporcionar 7 (69,21 g, 75 % de rendimiento) como un sólido blanco: RMN de 1H (400 MHz, D2O) δ 7,57 - 7,44 (m, 10H), 4,66 (t, J = 8,2 Hz, 1H), 4,36 (s, 4H), 4,10 - 3,95 (m, 4H), 3,22 (s, 4H). EM ([M+H]+) calculado para C17H21N2253,17, hallado 253,00.
Preparación de N,N-dibencil-1-(3-fluoropropil)acetidin-3-amina (8)

A una solución de 7 (40,0 g, 1,0 equiv) en MTBE (200 ml) y H2O (200 ml) se le añadió 1-bromo-3-fluoropropano (17,20 g, 1,5 equiv) y LiOH•H2O (15,18 g, 4,0 equiv). Se calentó la mezcla de reacción hasta 55 °C y se agitó a 55 °C durante 20 h, se enfrió hasta 20 °C. Se separó la capa orgánica y se lavó con Na2SO4 al 5 % (120 ml x 2).8 se puede obtener por concentración de solución de MTBE a vacío reducido como un sólido blanco (22,3 g, 83 % de rendimiento): RMN de 1H (400 MHz, DMSO-d 6) δ 7,35 - 7,22 (m, 10H), 4,45 (t, J = 6,0 Hz, 1H), 4,33 (t, J = 6,0 Hz, 1H), 3,45 - 3,39 (m, 4H), 3,28 - 3,17 (m, 3H), 2,67 - 2,56 (m, 2H), 2,35 (t, J = 7,0 Hz, 2H), 1,64 - 1,48 (m, 2H). EM ([M+H]+) calculado para C20H26FN2313,21, hallado 313,10.
Preparación de etano-1,2-disulfonato de 1-(3-fluoropropil)acetidin-3-amina

A una solución de 8 (2,00 g, 1,0 equiv) en MeOH (20,0 ml) se añadió una solución de dihidrato de ácido etanodisulfónico (1,23 g, 1,0 equiv) en H2O (10,0 ml) a 0 °C. Se calentó la mezcla de reacción hasta 30 °C, se pasó a través de un cartucho de carbón. A continuación se cargó Pd/C (0,40 g, 0,20 equiv) en la mezcla de reacción, se agitó a 45 °C con 40 psi de H2, se filtró, se concentró a 5 ml, se cargó con MeOH (20,0 ml), se agitó a 20 °C durante 3 h, se filtró, se lavó con MeOH (2 ml) para proporcionar el compuesto del título (1,36 g, 93 % de rendimiento) como un sólido blanco: RMN de 1H (400 MHz, D2O) δ 4,70 - 4,57 (m, 3H), 4,57 - 4,31 (m, 4H), 3,51 (t, J = 7,2 Hz, 2H), 3,24 (s, 4H), 2,11 - 1,97 (m, 2H); RMN de 19F (376 MHz, D2O) δ -219,59.
Ejemplo 5
Preparación de (R)-1-(1H-indol-3-il)propan-2-amina

A una solución de imidazol (102,8 g, 1,51 mol, 1,5 equiv.) en DCM (1,33 l) se le añadió gota a gota SOCl2 (179,3 g, 1,51 mol, 1,5 equiv.) a de -5 a 0 °C en N2 durante 30 min. Se agitó la mezcla de reacción durante 0,5 h a de -5 a 0 °C. Se añadió gota a gota (R)-(1-hidroxipropan-2-il)carbamato de terc-butilo (Boc-alaninol) (177,7 g, 1,01 mol, 1,0 equiv.) en DCM (1,33 l, 7,5 vol.) a de -5 °C a 0 °C durante 1 h. Se agitó la mezcla de reacción durante 0,5 h a de -2 a 0 °C seguido de la adición de trietilamina (204 g, 2,02 mol, 2 equiv.) gota a gota a de -5 a 0 °C. Se agitó la mezcla resultante durante 0,5 h o hasta que se consumió por completo N-Boc-alaninol como se determina por análisis de CG. Se añadió agua a la mezcla de reacción (1,3 l) a 0~20 °C. Se separaron las fases y se extrajo la
fase acuosa con DCM (1,3 l). Se combinaron las fases orgánicas y se lavó con ácido cítrico al 10 % en peso (1,3 l), NaHCO3 ac. (1,3 l) y salmuera (1 l) sucesivamente. Se enfrió la fase orgánica hasta 0~10 °C, seguido de la adición de agua (3,1 l) y RuCl3•xH2O (2,66 g), y a continuación seguido de la adición de Oxone (927,1 g, 1,51 mol, 1,5 equiv.). Se calentó la mezcla de reacción hasta 22 °C gradualmente y se mantuvo durante 3,5 h o hasta que el intermedio de sulfamidita se consumió por completo como se determina por análisis de CG. Se separaron las fases y se filtró la fase acuosa a través de una almohadilla de Celite (50 g) y seguido de aclarado con DCM (1,3 l). Se extrajo el filtrado con DCM (1,3 l) y se combinaron las fases orgánicas y a continuación se lavó con Na2S2O3 sat. (1,3 l) y salmuera (1 l x 2). Se secó la fase orgánica con Na2SO4 (50 g) y a continuación se filtró aclarando con DCM (200 ml). El filtrado combinado y los lavados se concentraron a vacío a 30 °C durante 1 h y a continuación se secaron además a alto vacío para proporcionar 220 g de 2,2-dióxido de (R)-4-metil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo con >99 % en peso con un 91,8 % de rendimiento aislado (corregido) en 2 etapas. RMN de 1H (400 MHz, CDCl3): δ 4,66 (dd, J = 9,2, 6,0 Hz, 1H), 4,41 (qdd, J = 6,4, 6,0, 2,8 Hz, 1H), 4,19 (dd, J = 9,2, 2,8 Hz, 1H), 1,54 (s, 9H), 1,50 (d, J = 6,4 Hz, 3H).
Se realizaron otras reacciones de oxidación en general de acuerdo con el procedimiento inmediato anterior donde se variaron el oxidante, catalizador y disolvente. Los resultados se informan en la tabla 1 a continuación, donde "Exp." se refiere al número de experimento, "Producto" se refiere a 2,2-dióxido de (R)-4-metil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo y "SM" se refiere al material de partida de Boc-alaninol, "% A CL" se refiere al porcentaje de área de pureza por cromatografía de líquidos, y "ND" se refiere a no detectado. La temperatura de reacción para los experimentos 1 y 3-9 fue de 0-25 °C y la temperatura de reacción para el experimento 2 fue de 0-40 °C. El tiempo de reacción para los experimentos 1-4 y 7 fue de 4 horas, el tiempo de reacción para los experimentos 5 y 6 fue de 8 horas y el tiempo de reacción para los experimentos 8 y 9 fue de 18 horas.
Tabla 1: condiciones y rendimientos del catalizador oxidante

A una mezcla de indol (7,4 g, 63,2 mmol, 1,5 equiv.) y CuCl (5,4 g, 54,7 mmol, 1,3 equiv.) en DCM (60 ml) se le añadió MeMgCl (3,0 M en THF, 18,7 ml, 56 mmol, 1,33 equiv.) a -15 °C bajo N2 durante 10 min. Se agitó la mezcla amarillo claro resultante a -20 °C durante 10 min y a continuación se añadió gota a gota una solución de 2,2-dióxido de (R)-4-metil-1,2,3-oxatiazolidin-3-carboxilato de terc-butilo (10,0 g, 42,1 mmol, 1,0 equiv.) en DCM (40 ml) a -10 °C bajo N2 durante 30 min. Se agitó la mezcla a -10 °C durante 2 horas o hasta que se finalizó la reacción según se juzgó por TLC (PE/EA = 5/1, desaparición de 2,2-dióxido de (R)-4-metil-1,2,3-oxatiazolidina-3-carboxilato de terc-butilo) o por CG. Se desactivó la reacción añadiendo ácido cítrico al 10 % (100 ml) mientras se mantenía la temperatura interna < 5 °C. Se separaron las fases y se extrajo la fase acuosa con DCM (100 ml x 2). Se combinaron las fases orgánicas y lavaron con salmuera (100 ml x 2), seguido de la adición a la fase orgánica de carbón activado (5 g) y Na2SO4 (10 g). Se agitó la mezcla resultante a temperatura ambiente durante 30 min, a continuación se filtró. Se lavó la torta con DCM (50 ml x 2). El filtrado y el lavado se combinaron y se concentraron a vacío a 30 °C para proporcionar (R)-(1-(1H-indol-3-il)propan-2-il)carbamato de terc-butilo bruto (16,9 g, 40,9 % A CL). Se añadió heptano (200 ml) al (R)-(1-(1H-indol-3-il)propan-2-il)carbamato de terc-butilo bruto, y se agitó la mezcla a temperatura ambiente durante 1 hora, momento durante el que precipitó gradualmente un sólido blanquecino. Se recogió el sólido por filtración y se lavó la torta con heptano (17 ml x 3). El sólido se secó a alto vacío a 25 °C para proporcionar 7,0 g de (R)-(1-(1H-indol-3-il)propan-2-il)carbamato de terc-butilo (25,5 mmol, 97 % A, 99 % en peso). RMN de 1H (400 MHz, DMSO-d 6) δ 10,76 (s, 1H), 7,55 (d, J = 7,9 Hz, 1H), 7,32 (dt, J = 8,1, 1,0 Hz, 1H), 7,09 (d, J = 2,3Hz, 1H), 7,05 (ddd, J = 8,2, 7,0, 1,2 Hz, 1H), 6,96 (ddd, J = 8,0, 6,9, 1,1 Hz, 1H),
6,71 (d, J = 8,0 Hz, 1H), 3,76-3,69 (m, 1H), 2,86 (dd, J = 13,9, 5,8 Hz, 1H), 2,64 (dd, J = 14,1, 7,7 Hz, 1H), 1,37 (s, 9H), 1,00 (d, J = 6,6 Hz, 3H).
Otras reacciones de alquilación de indol se realizaron en general de acuerdo con el procedimiento inmediato anterior donde se variaron las especies de catalizador de cobre y la estequiometría del reactivo de Grignard. Los resultados se informan en la tabla 2 a continuación, donde "Exp." se refiere al número de experimento, "CuX" se refiere a la especie de catalizador de cobre, "eq." se refiere a equivalentes, "Prod" se refiere a (R)-(1-(1H-indol-3-il)propan-2-il)carbamato de terc-butilo, "N1" se refiere a un subproducto en el que la indolamina está alquilada, "BA" se refiere a un subproducto de bisalquilo, y "AY" se refiere al rendimiento de ensayo de CL, "% A" se refiere al porcentaje de área de pureza por cromatografía de líquidos y "ND" se refiere a no detectado.
Tabla 2: condiciones y rendimientos de la reacción de alquilación de indol

Otras reacciones de alquilación de indol se realizaron en general de acuerdo con el procedimiento inmediato anterior donde se varió la temperatura de reacción. Los resultados se informan en la tabla 3 a continuación, donde "Exp." se refiere al número de experimento, "Temp G" se refiere a la temperatura de adición del reactivo de Grignard en °C, "Temp S" se refiere a la temperatura de adición de sulfamidato en °C, "Prod" se refiere a (R)-(1-(1H-indol-3-il)propan-2-il)carbamato de terc-butilo, "N1" se refiere a un subproducto en el que la indolamina está alquilada, "BA" se refiere a un subproducto de bisalquilo y "AY" se refiere al rendimiento de ensayo de CL, y "% A" se refiere al porcentaje de área de pureza por cromatografía de líquidos.
Tabla 3: experimentos de alquilación de indol a temperatura variable

A una solución de (R)-(1-(1H-indol-3-il)propan-2-il)carbamato de terc-butilo (0,5 g, 1,8 mmol, 92 % A) en MeOH (5 ml, 10 vol.) se le añadió gota a gota HCl en MeOH (10 M, 1,8 ml, 18 mmol) a 0 °C en atmósfera de N2 durante un período de 10 min. Se agitó la solución resultante a 0 °C durante 2 h, a continuación a 25-30 °C durante 1 h. Se concentró la solución a vacío a 30 °C, se diluyó con agua (10 ml) y se extrajo con DCM (10 ml x 2). Se ajustó
el pH de la fase acuosa a ~13 con NaOH ac. 1 M a 0-10 °C y a continuación se extrajo con DCM (20 ml x 4). Se secó la fase orgánica sobre Na2SO4 (10 g). Se filtró el desecante y se lavó la torta con DCM (10 ml). El filtrado y lavado combinados se concentró a vacío a 30 °C para dar 0,31 g de (R)-1-(1H-indol-3-il)propan-2-amina bruta con un 95,8 % A CL y un 99,2 % de pureza de ee en un 97 % de rendimiento aislado. RMN de 1H: 400 MHz (CDCl3): δ 1,20 (d, J = 6, 3H), 1,56 (s, a., 2H), 2,69 (dd, J = 14, 8, 1H), 2,91 (dd, J = 14, 4, 1H), 3,30-3,33 (m, 1H), 7,05 (s, 1H), 7,14 (t, J = 7, 1H), 7,22 (t, J = 7, 1H), 7,38 (d, J = 8, 1H), 7,64 (d, J = 8, 1H), 8,27 (s, 1H).
Ejemplo 6
Preparación de (R)-3-((1-(1H-indol-3-il)propan-2-il)amino)-2,2-difluoropropan-1-ol

A una solución de 2,2-difluoropropano-1,3-diol (51 kg, 90 % en peso, 409,5 mol, 1 eq.) en DCM (332 kg, 5 vol.) se le añadió cloruro de tionilo (58,4 kg, 491,4 mol, 1,2 eq.) a 20-25 °C durante 2 h. Se calentó la mezcla de reacción hasta 30-35 °C, se agitó durante 2 h y se añadió agua helada (255 kg) para desactivar la reacción. Se separaron las fases y se extrajo la fase acuosa con DCM. Se combinaron las fases orgánicas y se lavó con agua. A la fase orgánica bruta se le añadió agua (255 kg) y FeCl3 (1,6 kg) y se enfrió la mezcla bifásica hasta -3 °C. A continuación, se añadió gota a gota lejía [NaClO (8,1 % en peso), 680 kg, 1556 mol, 3,8 equiv.] durante un período de 5 h a de -5 a 1 °C, y se agitó la mezcla de reacción durante 1 h a 0 °C. Se filtró la mezcla de reacción a través de Celite. Se separaron las fases y se extrajo la fase acuosa con DCM. Se combinaron las fases orgánicas y se lavó con solución ac. de Na2SO3 y salmuera, a continuación se secó sobre Na2SO4. Se separó por filtración el desecante y se concentró el filtrado hasta aprox. 100 l. Se añadió heptano (80 kg) a la suspensión resultante y la mezcla se concentró además hasta aprox. 100 l. Se recogieron los sólidos por filtración y se secó a vacío para dar 52,01 kg de 2,2-dióxido de 5,5-difluoro-1,3,2-dioxatiano como un cristal blanquecino con (72 % de rendimiento, dos etapas de 2,2-difluoropropano-1,3-diol). RMN de 1H (400 MHz, DMSO-d 6) δ 5,14 (t, J = 10,8 Hz, 1H).
Se evaluaron diversos otros catalizadores para la preparación de 2,2-dióxido de 5,5-difluoro-1,3,2-dioxatiano en general de acuerdo con el procedimiento inmediato anterior. Se informa de los resultados en la tabla 4 a continuación donde "Exp." se refiere al número de experimento, "diol" se refiere a 2,2-difluoropropano-1,3-diol, "Prod" se refiere a un 90 % en peso de 2,2-dióxido de 5,5-difluoro-1,3,2-dioxatiano y "Rendimiento" se refiere al rendimiento del ensayo. Cada reacción se desactivó con 5 volúmenes de agua.
Tabla 4: condiciones para la preparación de 2,2-dióxido de 5,5-difluoro-1,3,2-dioxatiano


En un matraz de 2 l se colocaron (R)-1-(1H-indol-3-il)propan-2-amina (99 % en peso, 100 g, 568,2 mmol, 1 equiv.), 2,2-dióxido de 5,5-difluoro-1,3,2-dioxatiano (97,9 % en peso, 108 g, 608 mmol, 1,07 equiv.), K2CO3 (55 g, 398 mmol, 0,7 equiv.) y acetonitrilo (1 l, 10 vol.). Se calentó la mezcla resultante hasta 80 °C y se agitó durante 4 h. Se enfrió la reacción hasta 35 °C y se filtró. Se lavó la torta con acetonitrilo (100 ml x 2) y se añadieron monohidrato de p-TsOH (119 g, 625,6 mmol, 1,1 equiv.) y agua (100 ml, 1 vol) al filtrado combinado. Se calentó la mezcla bifásica resultante hasta 80 °C y se agitó durante 3 h. A continuación, se vertió la mezcla en 1 l de hieloagua y se ajustó el pH de la mezcla hasta 9 con Na2CO3 saturado (350 ml) a < 5 °C. Se separaron las fases y se extrajo la fase acuosa con i-PrOAc (500 ml x 2). Se combinaron las fases orgánicas y se lavó con agua (500 ml) y salmuera (500 ml x 2), a continuación se secó sobre Na2SO4 (30 g). Se separó por filtración el desecante y se lavó la torta con i-PrOAc (100 ml x 2). Se combinaron el filtrado y el lavado y se concentraron a vacío a 40 °C. Se disolvió el residuo en i-PrOAc (200 ml) y se calentó hasta 60 °C. Se añadió heptano (800 ml) gota a gota a 60 °C y a continuación se agitó la mezcla resultante durante 10 min. Se enfrió la mezcla hasta 30-35 °C lentamente durante 2 h y a continuación se enfrió además hasta 0 °C. Después de agitar durante 10 min, se recogieron los sólidos por filtración y se lavó la torta con heptano (100 ml x 2). Se secó el sólido así obtenido a vacío a 45 °C para dar (R)-3-((1-(1H-indol-3-il)propan-2-il)amino)-2,2-difluoropropan-1-ol (155 g, 99,2 % A CL, 96 % en peso, 97,6 % de rendimiento aislado). RMN de 1H (400 MHz, DMSO-d 6) δ 10,79 (s, 1H), 7,51 (dd, J = 7,9, 1,2 Hz, 1H), 7,33 (dt, J = 8,2, 1,0 Hz, 1H), 7,13 (d, J = 2,3 Hz, 1H), 7,05 (ddd, J = 8,2, 6,9, 1,3 Hz, 1H), 6,96 (ddd, J = 8,0, 6,9, 1,2 Hz, 1H), 5,35 (t, J = 6,3 Hz, 1H), 3,61 (td, J = 13,8, 5,2 Hz, 2H), 3,03-2,91 (m, 3H), 2,83 (dd, J = 14,0, 5,7 Hz, 1H), 2,59 (dd, J = 14,0, 7,2 Hz, 1H), 1,69 (s, 1H), 0,96 (d, J = 6,2 Hz, 3H).
Ejemplo 7
Preparación de (2R,3R)-2,3-dihidroxisuccinato de 3-((1R,3R)-1-(2,6-difluoro-4-((1-(3-fluoropropil)acetidin-3-il)amino)fenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol

En la etapa 1, en un matraz con aparato de agitación se combinó 4-bromo-2,6-difluorobenzaldehído (131,79 g, 596,35 mmol), (R)-3-((1-(1H-indol-3-il)propan-2-il)amino)-2,2-difluoropropan-1-ol (160 g, 596,35 mmol), ácido acético (51,26 ml, 894,52 mmol) y tolueno. Se calentó la reacción hasta 75 °C y se mantuvo durante la noche, se enfrió y a continuación se diluyó con tolueno. A continuación, se desactivó la solución resultante con una solución acuosa de carbonato de potasio, se lavó con salmuera y agua y se trató con carbón activado. Después de la
filtración, se concentró la solución y se cristalizó a partir de tolueno/heptano para dar 3-((1R,3R)-1-(4-bromo-2,6-difluorofenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol en un 81 % de rendimiento como un sólido amarillo pálido. RMN de 1H (400 MHz, DMSO-d 6) δ 10,59 (s, 1H), 7,45 - 7,36 (m, 3H), 7,20 (d, J = 8,1 Hz, 1H), 7,05 - 6,92 (m, 2H), 5,29 (t, J = 6,1 Hz, 1H), 5,21 (s, 1H), 3,71 - 3,55 (m, 1H), 3,53 - 3,38 (m, 2H), 3,17 (q, J = 15,2 Hz, 1H), 2,89 (ddd, J = 15,3, 4,8, 1,5 Hz, 1H), 2,71 - 2,55 (m, 2H), 1,08 (d, J = 6,5 Hz, 3H). EM ([M+H]+) calculado para C21H19BrF4N2O 470,06, hallado 470,80.
De forma alternativa, se combinaron 4-bromo-2,6-difluorobenzaldehído (67,2 g, 304 mmol), (R)-3-((1-(1H-indol-3-il)propan-2-il)amino)-2,2-difluoropropan-1-ol (80,0 g, 298 mmol), ácido acético (25,6 ml, 447 mmol) y metanol en un matraz agitado. Se sometió a reflujo la mezcla durante 24 horas, a continuación se enfrió. Se precipitaron los sólidos de la solución resultante por adición de una solución acuosa de carbonato de potasio. Se filtró la suspensión resultante y se lavó para dar 3-((1R,3R)-1-(4-bromo-2,6-difluorofenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol en un 97 % de rendimiento como un sólido amarillo pálido.
En las etapas 2 y 3, se combinaron 3-((1R,3R)-1-(4-bromo-2,6-difluorofenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol (100 g, 212,2 mmol), etano-1,2-disulfonato de 1-(3-fluoropropil)acetidin-3-amina (82,09 g, 254,6 mmol), acetonitrilo, DBU (1061 mmol 161,5 g 159,4 ml) en un matraz agitado por adición de Pd-175 (5,304 mmol 4,144 g). Se calentó la reacción hasta 75 °C durante 2 horas, se enfrió, se concentró y a continuación se diluyó con éter metil-terc-butílico. Esta solución resultante se procesó con una solución acuosa de cloruro de amonio, salmuera y agua, a continuación se depuró con SiliaMetS Thiol. Después de la filtración, se concentró la solución, se diluyó con etanol y se cristalizó con ácido tartárico para dar (2R,3R)-2,3-dihidroxisuccinato de 3-((1R,3R)-1-(2,6-difluoro-4-((1-(3-fluoropropil)acetidin-3-il)amino)fenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol en un 90 % de rendimiento como un sólido amarillo después de la filtración y el lavado. RMN de 1H (400 MHz, DMSO-d6 ) δ 10,52 (s, 1H), 7,38 (dd, J = 7,5, 1,3 Hz, 1H), 7,21 - 7,16 (m, 1H), 7,02 - 6,90 (m, 2H), 6,82 (d, J = 6,9 Hz, 1H), 6,18 - 6,08 (m, 2H), 5,07 (s, 1H), 4,53 (t, J = 5,8 Hz, 1H), 4,42 (t, J = 5,9 Hz, 1H), 4,18 (s, 2H), 4,14 - 4,05 (m, 1H), 3,93 (ddt, J = 9,1, 7,0, 3,5 Hz, 2H), 3,74 -3,60 (m, 1H), 3,51 - 3,32 (m, 2H), 3,21 - 3,02 (m, 3H), 2,88 - 2,73 (m, 3H), 2,70 - 2,51 (m, 2H), 1,83 - 1,66 (m, 2H), 1,09 - 1,05 (m, 3H). EM ([M+H]+) calculado para C27H31F5N4O 522,24, hallado 523,00.
Ejemplo 8
Preparación de (2R,3R)-2,3-dihidroxisuccinato de 3-((1R,3R)-1-(2,6-difluoro-4-((1-(3-fluoropropil)acetidin-3-il)amino)fenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol

En las etapas 1-4, se cargó un reactor con 4-bromo-2,6-difluorobenzaldehído (75,0 g, 0,339 mol, 100 % mol), monohidrato de ácido p-toluenosulfónico (162 mg, 0,000849 mol, 0,250 % mol) y tolueno (225 ml). Se cargó ortoformiato de trietilo (55,4 g, 62,1 ml, 0,373 mol, 110 % mol) durante 15 min a ta, se agitó la mezcla a ta durante 1 h y se destiló la mezcla para proporcionar una solución de tolueno de 5-bromo-2-(dietoximetil)-1,3-difluorobenceno. Se cargó otro reactor con 2-(trioxidaniltio)etano-1-sulfonato de 1-(3-fluoropropil)acetidin-3-amina (131,3 g, 0,407 mol, 120 % mol) y acetonitrilo (656 ml). Se cargó DBU (124,0 g, 122,8 ml, 0,814 mol, 240 % mol) durante 15 min a ta y se agitó la mezcla a ta durante 2 h, se destiló con tolueno y se filtró para proporcionar una solución de tolueno de 1-(3-fluoropropil)acetidin-3-amina que se cargó, junto con NaOtBu (39,14 g, 0,407 mol, 120 % mol), en la solución de tolueno de 5-bromo-2-(dietoximetil)-1,3-difluorobenceno. Se burbujeó la mezcla, se cargó con BrettPhos Pd G3 (3,076 g, 0,00309 mol, 1,00 % mol), se burbujeó, se calentó a 60 °C durante 18 h, se enfrió, se desactivó con agua y se lavó con agua dos veces. Se cargó SiliaMetS Thiol (20,0 g) y se calentó la mezcla a 50 °C durante 2 h, se enfrió y se filtró para proporcionar una solución de tolueno de N-(4-(dietoximetil)-3,5-difluorofenil)-1-(3-fluoropropil)acetidin-3-amina. Se añadieron ácido acético (21,4 ml, 0,373 mol, 110 % mol) y agua (300 ml) y se mantuvo la mezcla a ta durante 2 h. Se separó una fase acuosa, se trató con NaOH acuoso (50 % en peso, 21,5 ml, 0,407 mol, 120 % mol) y se sembró con 2,6-difluoro-4-((1-(3-fluoropropil)acetidin-3-il)amino)benzaldehído (923 mg, 0,00339 mol, 1,00 % mol). Se filtraron los sólidos resultantes, se lavó con agua y se secó para proporcionar 2,6-difluoro-4-((1-(3-fluoropropil)acetidin-3-il)amino)benzaldehído (77,9 g, 84,3 % de rendimiento) como un sólido blanco. RMN de 1H (400 MHz, CDCl3) δ 10,05 (dd, J = 1,2 Hz, 1H), 6,05-5,97 (m, 2H), 5,03 (d, J = 6,4 Hz, 1H), 4,54 (t, J = 6,0 Hz, 1H), 4,42 (t, J = 6,0 Hz, 1H), 4,14-4,03 (m, 1H), 3,70 (td, J = 6,8, 1,6 Hz, 2H), 2,99-2,93 (m, 2H), 2,59 (t, J = 7,2 Hz, 2H), 1,83-1,66 (m, 2H). EM: calc. para C13H15F3N2O [M+H]+ = 273,1, observado = 273,0.
En la etapa 5, se cargó un reactor con (R)-3-((1-(1H-indol-3-il)propan-2-il)amino)-2,2-difluoropropan-1-ol (6,60 kg, 24,60 mol, 100 % mol), 2,6-difluoro-4-((1-(3-fluoropropil)acetidin-3-il)amino)benzaldehído (6,70 kg, 24,60 mol, 100 % mol), ácido L-tartárico (5,54 kg, 36,90 mol, 150 % mol) y etanol (39,6 l). Se calentó la mezcla de reacción a 70 °C durante 2 h, se sembró con (2R,3R)-2,3-dihidroxisuccinato de 3-((1R,3R)-1-(2,6-difluoro-4-((1-(3-fluoropropil)acetidin-3-il)amino)fenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol (0,0827 kg, 0,123 mol, 0,5 % mol), se agitó a 70 °C durante 2 días, se desactivó con etanol (26,4 l), se enfrió y se filtró para recoger el sólido. Se lavó el sólido con etanol y se secó a vacío para proporcionar (2R,3R)-2,3-dihidroxisuccinato de 3-((1R,3R)-1-(2,6-difluoro-4-((1-(3-fluoropropil)acetidin-3-il)amino)fenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol (12,7 kg, 78 % de rendimiento) como un sólido blanquecino.
Se evaluaron diversos ácidos distintos de ácido tartárico, así como un ejemplo comparativo con ácido tartárico, para la reacción de cierre de anillo de la etapa 5 en general de acuerdo con el procedimiento inmediato anterior. Se informa de los resultados en la tabla 5 a continuación, donde "Exp." se refiere al número de experimento, y "Conv" se refiere a la conversión a la estructura de anillo tricíclico fusionado. La sal de ácido tartárico dio como resultado un incremento sustancial en el rendimiento final. Además, la sal de ácido tartárico minimizó la presencia de subproductos oligómeros formados durante la síntesis, dando como resultado un producto más puro y menos epimerización del producto de compuesto final.
Tabla 5: ácidos sometidos a prueba para el cierre de anillo para el compuesto A

Ejemplo 9:
Recristalización del compuesto B.

En un reactor de 250 ml se cargó el compuesto B bruto (4,00 g, 5,95 mmol) en una mezcla de MeOH (90,0 ml) y EtOH (10,0 ml). Se calentó la suspensión hasta 60 °C y se volvió homogénea. Se añadieron semillas del compuesto B (200 mg, 0,297 mmol, 5 % mol) a la solución. Se destiló la suspensión con EtOH, se enfrió hasta 25 °C durante 1 h, se mantuvo a 25 °C durante 1 h, se filtró, se secó para proporcionar el compuesto B (3,83 g, 91 % de rendimiento) como un sólido blanquecino.
Ejemplo 10:
Recristalización del compuesto B:

Ejemplo 10A: A un reactor de 30 l se le cargó el compuesto B bruto (1,26 kg, 1,88 mol, 100 % mol) y MTBE (6,30 l, 5,00 ml/g). Se añadió una solución de NaOH (154 g, 3,85 mol, 205 % mol) en agua (6,30 l, 5,00 ml/g) a la suspensión. Se agitó la mezcla de reacción a 20 °C durante 30 min. Se lavó la capa orgánica superior con agua (2,53 l, 2,00 ml/g) dos veces, se pasó a través de un cartucho de carbón vegetal, se destiló con EtOH para proporcionar la solución de base libre del compuesto B en EtOH.
Ejemplo 10B: a un reactor de 20 l se le cargó ácido L-tartárico (296 g, 1,97 mol, 105 % mol), EtOH (2,52 l, 2,00 ml/g) y se calentó hasta 70 °C. Se transfirió un 20 % de solución de base libre del compuesto B al reactor de 20 l. Se añadieron semillas del compuesto B (25,2 g, 0,0375 mol, 2 % mol) a la solución. El resto de la solución de base libre del compuesto B se transfirió al reactor de 20 l durante 1 h, se envejeció a 70 °C durante 30 min, se enfrió hasta 20 °C durante 5 h, se envejeció a 20 °C durante un mínimo de 2 h, se filtró, se lavó con EtOH (2,52 l, 2,00 ml/g) tres veces, se secó para proporcionar el compuesto B (1,12 kg, 89 %) como un sólido blanquecino. Ejemplo 11:
Se caracterizaron los compuestos proporcionados en el presente documento por técnicas de espectrometría de masas y/o RMN como se entiende en la técnica. Los compuestos de este ejemplo, a menos que se indique de otro modo, tienen estereoquímica verificada.
Compuesto (M1)

Determinado m/z: 672,64.
Compuesto (M2)

Determinado m/z: 672,64.
Compuesto (M3)

Determinado m/z: 652,63
Compuesto (M4)

Determinado m/z: 540,57 (mezcla estereomérica).
Ejemplo 12:
Transformaciones enzimáticas:
El cribado de transaminasas en el modo de resolución cinética de alpa-metiltriptamina rac. reveló enzimas que poseían suficiente enantioselectividad (R) para el enfoque de aminación reductora asimétrica y suficiente enantioselectividad (S) para el enfoque de resolución cinética. Las transaminasas ejemplares se enumeran en la tabla 6.
Tabla 6: transaminasas (TA) enantioselectivas:

Se realizó la reducción asimétrica aplicando transaminasa TA-P2-A07 en tampón; isopropilamina y codisolventes orgánicos tales como DMSO o acetonitrilo en una escala de 5 g. El compuesto (XXI) se convirtió en el compuesto (3) como se describe en el presente documento en un grado superior a un 95 % con un exceso enantiomérico (EE) superior a un 99 % después de 1 día.
También se realizó la aminación reductora en disolventes orgánicos que contenían pequeñas cantidades de tampón e isopropilamina donde se inmovilizó la transaminasa TA-P2-A07 sobre el soporte sólido. El compuesto (XXI) se convirtió en el compuesto (3) en un grado de conversión superior a un 95 % y un EE superior a un 99 % después de 4 semanas. El disolvente orgánico permitió cargas de sustratos mayores de hasta un 5 % [p/p]. Se realizó la resolución cinética en presencia de transaminasa ATA12 en (1) células completas; (2) lisado sin células o (3) liofilizado bruto en tampón que contiene piruvato y codisolvente orgánico tal como acetonitrilo. Se logró un exceso enantiomérico de >99 %. La conversión en el compuesto (3) se logró en un grado igual a o superior a un 50 %. Se redujo el enantiómero no deseado por desaminación oxidativa hacia la cetona.
Identificación de secuencia de TA seleccionadas:
3FCR Y59F/Y87F/T231G: aminoácido (SEQ ID NO: 1)

Los residuos en negrita y subrayados corresponden a mutaciones en la secuencia natural.
Ejemplo 13:
Caracterización de base libre, sal y polimorfo del compuesto A
Abreviaturas;
MIBK Metilisobutilcetona
ACN Acetonitrilo
EtOAc Acetato de etilo
EtOH Etanol
MTBE Éter metil-terc-butílico
IPAc Acetato de isopropilo
MeTHF Metiltetrahidrofurano
CPME Éter ciclofenilmetílico
MeOH Metanol
THF Tetrahidrofurano
IPA Alcohol isopropílico
DMSO Dimetilsulfóxido
DMC Carbonato de dimetilo
MEK Metiletilcetona
DCM Diclorometano
Cribado de polimorfos de base libre. Se realizó un cribado de alto rendimiento (HTS) automatizado en placa de 96 pocillos usando el sistema Symyx CM2 (Freeslate Inc., CA) para identificar formas polimórficas potenciales para la base libre del compuesto A. Se distribuyó el compuesto A en cada pocillo (8 mg/pocillo) usando un accesorio de distribución automática de polvo, después de esto se añadieron 800 µl de disolvente (puro o mezcla) y se agitó la suspensión durante 2 horas a 50 °C. Se distribuyó el compuesto A inicialmente en la placa de suspensión usando acetato de etilo (para tartrato) o bien MIBK (para fumarato) para mantener la forma sólida. A partir de esta placa maestra, se filtró el sobrenadante y se distribuyó en tres placas separadas para evaporación, precipitación por adición de antidisolvente y enfriamiento controlado durante 8-10 h a partir de 50-20 °C. En todos los casos, los disolventes residuales se evaporaron o bien se sifonaron, y se examinó el sólido usando microscopía de luz polarizada y XRPD.
Ejemplo 14:
Cribado de sal. De acuerdo con los datos de solubilidad aproximada de la base libre y la lista de ácidos deseados, se usaron cinco sistemas de disolventes en el cribado. Primero se dispersaron aproximadamente 20 mg de compuesto amorfo de base libre en 0,5 ml de disolvente seleccionado en un vial de vidrio y a continuación se añadió el correspondiente ácido con una proporción de 1:1 de carga molar. Se intentó una proporción molar adicional de 2:1 (ácido/base libre) para HCl, debido a los dos grupos funcionales básicos. Se agitaron las mezclas a TA durante la noche. Se analizaron los sólidos resultantes por XRPD. Las soluciones transparentes obtenidas después de agitar se agitaron a 5 °C durante 2 días, a continuación se añadieron 0,5 ml de H2O a cada una de las soluciones transparentes en ACN/H2O (19:1, v/v) mientras que se añadieron 0,5 ml de n-heptano a cada una de las soluciones transparentes en los otros sistemas de disolventes, seguido de agitación a 5 °C durante aproximadamente 3 días, y las soluciones transparentes finales se transfirieron a evaporación lenta a TA para identificar tantos resultados positivos cristalinos como fuera posible.
La primera ronda de cribado identificó los resultados positivos de forma cristalina como se proporciona en la tabla 7. Se aumentaron a escala tartrato y fumarato además hasta 50 mg-1,5 g para caracterización adicional. Someter a prueba en diferentes sistemas y condiciones de disolventes permitió a) una caracterización en profundidad de las diferentes formas polimórficas obtenidas como resultados positivos en el cribado y b) identificar condiciones para inhibir y controlar la formación de epímeros cis. El contenido en epímero varió de <1 %-22 % en diferentes lotes de base libre.
Tabla 7: cribado de sal y resultados positivos de forma cristalina para el compuesto A.


Procedimientos:
Difractometría de rayos X en polvo (XRPD) ambiente. Se recopilaron los patrones de XRPD usando el difractómetro de rayos X de polvo PANalytical Empyrean (PANalytical Inc., Lelyweg, Países Bajos). Se empaquetó la muestra de polvo en un soporte de silicona de fondo cero y se procesó en modo de reflexión (configuración Bragg Brentano). Se equipó el instrumento con una fuente de Cu Kα con voltaje de tubo y corriente de 45 kV y 40 mA respectivamente. Se recopilaron datos a temperatura ambiente de 3,0 a 40,0° 2θ usando un tamaño de etapa de 0,0263°, con una velocidad de revolución de 8 s. Se equipó la trayectoria del haz incidente con una rendija Soller de 0,02°, una rendija antidispersión fija de 1°, una máscara de haz incidente fija de 10 mm y una rendija de divergencia programable en modo automático. Se usó un cuchillo de haz para detectores lineales. Se equipó el haz difractado con una rendija Soller de 0,02°, una rendija antidispersión programable en modo automático y un filtro K-β de níquel. Se usó un detector PIXcel 1D en el modo detector de línea de barrido (1D). Se analizaron los datos usando un programa informático comercial (JADE®, versión 9, Materials Data Inc., Livermore, CA).
Análisis de sorción de agua. Se colocaron aproximadamente 5-6 mg de muestra de polvo en la bandeja de muestra de un analizador de sorción de agua automático (Q5000SA, TA Instruments, New Castle, DE) a 25 °C y un caudal de nitrógeno de 200 ml/min. Inicialmente se "secó" la muestra a 0 % de HR durante un total de 400 minutos (a 60 °C seguido de 25 °C), después de esto se sometió a un incremento progresivo en HR de 0-90 %, en incrementos de un 10 % con un tiempo de permanencia de 200 minutos en cada HR. Esto se siguió de una disminución progresiva en la HR en decrementos de un 10 % hasta un 0 % de HR, usando el mismo protocolo.
Calorimetría diferencial de barrido (DSC). Se analizaron aproximadamente 3-8 mg de muestra de polvo usando un DSC Q2000™ (TA Instruments, New Castle, DE) equipado con un accesorio de enfriamiento refrigerado. Se envasaron las muestras en bandejas no herméticas (Tzero™, bandejas de aluminio) y típicamente se calentaron de 0-200 °C a 10 °C/min bajo purga de nitrógeno seco. Se calibró el instrumento usando zafiro (valor de referencia) e indio (temperatura y constante de celda). Se analizaron los datos usando un programa informático comercial (Universal Analysis 2000, versión 4.7A, TA Instruments).
Termogravimetría (TGA). En un analizador termogravimétrico (Discovery TGA, TA instruments), se calentaron 3-5 mg de muestras de compuesto A en una bandeja de aluminio abierta de TA a 350 °C a una tasa de calentamiento de 10 °C/min bajo purga de nitrógeno seco. Se realizó la calibración de temperatura usando Alumel® y níquel. Se usaron pesas estándar de 100 mg y 1 g para la calibración de peso.
Microscopía de luz polarizada (PLM). Las muestras se dispersaron en aceite de silicona y se observaron bajo polarizadores cruzados de un microscopio Leica DM 4000B potenciado con video equipado con una cámara CCD de alta resolución y una platina motorizada (Clemex Technologies Inc., Longueuil, Quebec, Canadá) con un aumento de 200X. Se adquirieron fotomicrografías usando el programa informático Clemex Vision PE (Clemex Technologies Inc., Longueuil, Quebec, Canadá).
Microscopía electrónica de barrido (SEM). Se revistió por pulverización catódica la muestra de polvo en un trozo de SEM y a continuación se examinó usando un Phenom SEM de sobremesa (Nanoscience Instruments, Inc., AZ). Se adquirieron las micrografías a diferentes aumentos.
Distribución del tamaño de partícula (PSD). Se realizó el análisis del tamaño de partícula usando un instrumento Malvern Mastersizer 2000 equipado con un accesorio de dispersión húmeda Hydros 2000SM (Malvern Instruments Ltd., Malvern, Reino Unido). Se pesaron ~40 mg de API en un vial y se añadió 1 ml de Span 85 al 0,1 % en heptano.
Se sonicó el vial durante 10 segundos, se añadieron aproximadamente 0,5 ml al muestreador a una velocidad de agitación de 1500 rpm y se realizó una PSD con un oscurecimiento de un 10-20 %. Debido a la presencia de grandes agrupaciones en la muestra que se sedimentaban rápidamente fuera de la suspensión, se cambió el dispersante a Span 85 al 0,2 % en heptano para estabilizar la suspensión. Se aplicó sonicación externa de 2 seguido de dos de 5 minutos de duración para romper las agrupaciones y se adquirieron imágenes PLM antes y después de la sonicación. Se mezcló la alícuota en el muestreador durante 2 minutos antes de la recopilación de datos para garantizar la homogeneidad. Se aclaró el instrumento dos veces con alcohol isopropílico (IPA) y una vez con heptano antes de llenarlo con Span 85 al 0,1 % en heptano para cada muestra. Después de que se hubiera realizado la última muestra, se aclaró el instrumento una vez con IPA. Se ha informado de datos para réplicas con 5 minutos de sonicación.
Análisis de área de superficie BET. Se realizó la medición de área de superficie usando un Micromeritics ASAP 2460 con un accesorio Micromeritics Smart VacPrep (Micromeritics Instrument Corp., GA). Se pesó una muestra de 500 mg-1 g en un tubo ASAP 2460 vacío y se colocó en el Smart VacPrep, se desgasificó durante 24 horas en condiciones ambientales y a continuación se expuso a la adsorción de gas kriptón a 25 °C y una presión de mantenimiento de 100 mm Hg. Se realizó una medición de 11 puntos en el intervalo de presión relativa de 0,050-0,300 y se analizaron los datos usando el programa informático Micro Active proporcionado por el proveedor. Espectroscopia de resonancia magnética nuclear de estado sólido (RMNes). Todos los experimentos de RMNes de 13C (a velocidad de giro de 8 kHz) se realizaron usando el instrumento Bruker 500 MHz (Bruker BioSpin GmbH, Karlsruhe, Alemania), los datos de 13C se adquirieron usando una secuencia de CP/TOSS. Se recopilaron barridos de 1-2 K para promediar la señal. Se usó un tiempo de contacto de 2 ms y un retardo de reciclo de 5 segundos. Se usó la secuencia Spinal 64 para el desacoplamiento con una longitud de pulso de 5,3 microsegundos. Se empleó una longitud de pulso de 90 grados de 1H de 2,9 microsegundos. Todos los experimentos de RMNes de 19F (a velocidad de giro de 14 kHz) se realizaron usando el instrumento Bruker 500 Mhz. Se adquirieron datos de 19F usando un CP y secuencias de polarización directa. Se recopilaron barridos de 64-256 K para promediar la señal. Se usó un tiempo de contacto de 750 microsegundos y un retardo de reciclo de 7 segundos. Se empleó una longitud de pulso de 90 grados de 1H de 3,54 microsegundos.
Ejemplo 15:
Caracterización preliminar de la base libre del compuesto A. Se descubrió que la base libre del compuesto A era amorfa. El compuesto A de base libre del material de partida se caracterizó por XRPD, TGA y mDSC antes del cribado). Como se muestra por los resultados de caracterización en la FIG.32 y FIG.33, el material de partida del compuesto A era amorfo con una pérdida de peso de un 9,3 % hasta 220 °C en TGA y sin señal de transición vítrea sustancial en mDSC. Se obtuvieron sólidos blancos del compuesto A de base libre por adición de antidisolvente en MeOH/H2O (3:20, v/v) con agitación y secado al aire durante ~7 días.
Cribado de polimorfos. Se estimó la solubilidad del compuesto A de base libre en 16 disolventes a TA. (Tabla 8). Se realizaron experimentos de cribado de polimorfos usando diferentes procedimientos de transición de fase sólida o cristalización en solución. Los procedimientos usados y formas cristalinas identificadas se resumen en la tabla 9.
Tabla 8: solubilidad aproximada del compuesto A de base libre

Tabla 9: resumen de cribado de polimorfos del compuesto A de base libre

Difusión de vapor sólido. Se realizaron experimentos de difusión de vapor sólido usando 13 tipos diferentes de disolvente. Para cada experimento, se pesaron aproximadamente 15 mg de material de partida en un vial de 3 ml, que se colocó en un vial de 20 ml con 2 ml del correspondiente disolvente volátil. Se selló el vial de 20 ml con una tapa y se mantuvo a TA durante 11 días para permitir que el vapor del disolvente interactuara con la muestra sólida. Se sometieron a prueba los sólidos aislados por XRPD. Como se resume en la tabla 10, solo se obtuvo aceite o amorfo.
Tabla 10: resumen de experimentos de difusión de vapor sólido

Conversión de suspensión a TA. Se realizaron experimentos de conversión de suspensión a TA en diferentes sistemas de disolventes. Se suspendieron aproximadamente 40 mg de material de partida en 0,3 ml de disolvente en un vial de vidrio de 1,5 ml. Después de agitar, se transfirieron todas las soluciones transparentes hasta 5 °C, seguido de evaporación lenta a TA después de 3 días. Los resultados resumidos indicaron que solo se obtuvo amorfo, gel o aceite.
Tabla 11: resumen de experimentos de conversión de suspensión para el compuesto A de base libre

* no se aplicó evaporación lenta
** se centrifugaron los sólidos y se analizaron por XRPD después de agitar a TA durante 4 días
N/A: no se obtuvo ningún sólido
Adición de antidisolvente. Se llevaron a cabo un total de 15 experimentos de adición de antidisolvente. Se
pesaron aproximadamente 30 mg de material de partida en un vial de vidrio de 20 ml y se disolvieron en 0,15 ml del correspondiente disolvente a TA. Se añadió antidisolvente etapa a etapa hasta que apareció la precipitación o la cantidad total de antidisolvente alcanzó los 12 ml, y agitándose magnéticamente la muestra. Se aisló el precipitado para análisis XRPD. Si no se observó ningún sólido, se agitaron magnéticamente las soluciones transparentes a 5 °C durante la noche y a continuación se evaporaron a TA. Los resultados de la tabla 12 mostraron que solo se generó amorfo o gel.
Tabla 12: resumen de experimentos de adiciones de antidisolvente para el compuesto A de base libre

Difusión de vapor líquido. Se realizaron experimentos de difusión de vapor líquido en 14 condiciones (tabla 13). Se pesaron aproximadamente 30 mg de material de partida en cada vial de vidrio de 3 ml. Se añadió el correspondiente disolvente para obtener una solución. Se selló el vial en el vial de vidrio de 20 ml con 3 ml del correspondiente antidisolvente y se mantuvo a TA, lo que permitió que el vapor interactuara con la solución durante aproximadamente 11 días. Se aislaron los precipitados para análisis XRPD. Se transfirieron las soluciones transparentes para evaporación a TA.
Tabla 13: resumen de experimentos de difusión de vapor líquido para el compuesto A de base libre

Evaporación lenta. Se realizaron experimentos de evaporación lenta en 12 condiciones. Para cada experimento, se pesaron alrededor de 20 mg de material de partida en un vial de vidrio de 3 ml, seguido de la adición del correspondiente disolvente o mezcla de disolventes para obtener una solución transparente. Posteriormente, se cubrió el vial con Parafilm con 3~4 orificios y se mantuvo a 4 °C para permitir que la solución se evaporara lentamente. Solo se obtuvo gel, como se resume en la tabla 14.
Tabla 14: resumen de experimentos de evaporación lenta para el compuesto A de base libre


La tabla 15 resume las formas que se obtuvieron por medio de cribado manual como se describe en el presente documento.
Tabla 15: formas polimórficas del compuesto A

Ejemplo 16:
Caracterización del compuesto B forma A. Se preparó el compuesto B forma A a escala de 200 mg por medio de cristalización reactiva de solución en acetona, como se evidencia por XRPD (FIG. 1). Como se muestra en la FIG. 2a y FIG. 2b, se observó una pérdida de peso de aproximadamente un 7,2 % hasta 125 °C en TGA, un resultado de DSC mostró una endotermia a 124,3 °C (temperatura de aparición). La RMN de 1H mostró que su proporción molar fue de 0,98 (ácido/base libre) y se detectó un 5,6 % de acetona (una proporción molar de 0,69 con respecto a base libre). Se realizaron experimentos de calentamiento para identificar además la forma A. No se observó ningún cambio de forma después de que se calentara la forma A hasta 90 °C, pero la muestra se volvió amorfa después de calentarse hasta 140 °C. Todavía se observó una cantidad considerable de acetona (4,5 %) después de calentar la muestra de forma A hasta 90 °C. La muestra de forma A estaba compuesta principalmente de partículas finas y algunos agregados (FIG. 3). En base a los datos recopilados, el compuesto B forma A es un solvato de acetona y la pérdida de disolvente es concomitante con la fusión.
Tabla 16: picos de XRPD representativos para el compuesto B forma A:


Preparación del compuesto B forma A. Se pesaron aproximadamente 57,5 mg de ácido tartárico en un vial de vidrio de 5 ml y se añadieron 2,0 ml de acetona para obtener una solución transparente. Se añadió la solución ácida transparente a 2,0 ml de solución madre de base libre en acetona (~100 mg/ml) en el vial de 5 ml con una proporción molar de 1:1 y se agitó a TA. Se añadió aproximadamente 1 mg de semilla del compuesto B forma A y la solución se volvió turbia. La muestra se agitó durante la noche antes de tomar mediciones de XRPD como se describe en el presente documento. El patrón se ajustó al compuesto B forma A. A continuación, se agitó la suspensión a temperatura ambiente durante otras 24 horas y se secó la torta a 50 °C durante 1,5 h. Rendimiento: 170,4 mg, con un rendimiento de ~65,3 %.
Ejemplo 17:
Caracterización del compuesto B forma B. Se preparó el compuesto B forma B a escala de 200 mg por medio de cristalización reactiva de solución en EtOAc, como se evidencia por XRPD (FIG. 4). Como se muestra en la FIG. 5a y FIG. 5b, se observó una pérdida de peso limitada de aproximadamente un 1,3 % hasta 140 °C en TGA y el resultado de DSC mostró un pico endotérmico de fusión a 156,7 °C (temperatura de aparición). Se determinó que la estequiometría fue de 1,08 (ácido/base libre) y se detectó un 1,4 % de EtOAc (una proporción molar de 0,11 con respecto a base libre) por RMN de 1H. En base a los datos de caracterización recopilados, el compuesto B forma B es un anhidrato.
Los datos de caracterización de estado sólido indican que la forma B es sustancialmente cristalina (XRPD), y TGA indica la presencia de algo de disolvente superficial de un ~2 % por una aparición temprana de pérdida de peso de TA-100 °C) con una aparición de fusión de ~163 °C. La aparición de fusión, después de una endotermia poco profunda (vaporización de disolvente residual) es a 163 °C. En base a la curva de pérdida de peso derivada, se detectó una aparición muy temprana de pérdida de peso (2,4 % p/p) hasta 100 °C, lo que se puede atribuir al disolvente superficial. La pérdida de peso total, incluyendo la del fundido, es de un 3,5 % p/p. Se descubrió que la forma B era ligeramente higroscópica con una absorción de humedad de un 1,2 % p/p a un 90 % de HR, 25 °C, como se muestra en el perfil de sorción-desorción de agua en la FIG. 8. Los espectros de RMNes de 13C (FIG. 6) y 19F (FIG.7) indicaron además la formación de la forma B. Las imágenes de SEM (FIG. 9a, aumento de 500X) y PLM (FIG. 9b, aumentos de 200 X) del compuesto B muestran que la forma B comprende agregados esféricos densos.
Tabla 17: picos de XRPD representativos para el compuesto B forma B:


Análisis de estabilidad de tensión preliminar del compuesto B forma B. Los patrones de XRPD de sales del compuesto A (fumarato y tartrato) expuestas hasta 40 °C/75 % de HR durante un mes en condiciones abiertas. La forma sólida no cambió bajo estas condiciones.
Aunque no se observó correlación entre el contenido en epímero que varía de 0,56-0,72 % y las propiedades de estado sólido de las diferentes formas de sal, los datos de RMNes, XRPD y DSC indican que existía la posibilidad de obtener una mezcla de formas a partir de sales de fumarato y que ese control de forma en cualquier sal requirió una cantidad sustancial de esfuerzo posterior, dada la variabilidad en los datos estructurales y los puntos de fusión con el cambio en las condiciones de cristalización. La sal de tartrato carecía de las impurezas encontradas en la muestra de sal de fumarato en un 0,18 % tras la exposición hasta 40 °C/75 % de HR durante un mes.
Análisis de compresión del compuesto B forma B. Las FIG. 22, FIG. 23 y FIG. 24 demuestran el efecto de la compresión sobre el tartrato del compuesto B forma B. Se analizaron 250 mg de compactos del compuesto B forma B puro. Tanto RMNes como XRPD (FIG. 23 y FIG. 22, respectivamente) muestran que la forma permanece sin cambios tras la compresión. Se observó una reducción del tiempo de relajación T1 19F tras la compresión que es atribuible a la generación de trastorno, aunque muy menor como es evidente a partir de los espectros de RMNes de 19F del compuesto B forma B comprimido en la FIG. 23. Los valores de relajación de T1 19F se incluyen en la tabla 18 para el compuesto B forma B como tal y comprimido. La compresión no afectó el punto de fusión del compuesto B forma B, como se ve en el termograma DSC (FIG. 24). El XRPD comparativo recopilado antes y después de la exposición a condiciones de estabilidad acelerada de 30 °C/65 % de HR y 40 °C/75 % de HR durante hasta 6 meses (abierto) demostró que el compuesto A forma B permanece sin cambios tras la exposición a estas condiciones.
Tabla 18: tiempos de relajación T1 19F para el compuesto B forma B

XRPD de muestra de estabilidad de un mes. El XRPD del compuesto B forma B expuesto a 40 °C/75 % de HR durante un mes muestra que el compuesto B forma B permanece sin cambios en condiciones de estabilidad de tensión incluyendo temperatura y humedad elevadas.
Preparación del compuesto B forma B. Se pesaron aproximadamente 58,0 mg de ácido tartárico en un vial de vidrio de 5 ml y se añadieron 2,0 ml de EtOAc. El ácido permaneció sin disolver. Se añadieron aproximadamente 2,0 ml de solución madre de base libre en EtOAc (~100 mg/ml) al vial de 5 ml con una proporción molar de 1:1 y se agitó la solución a TA. Se añadió aproximadamente 1 mg de semilla de forma B y la solución permaneció transparente. Se agitó la solución durante la noche y se muestreó por XRPD. El patrón se ajustó al compuesto B forma B. Se agitó la suspensión a 50 °C durante 2 días más. Se centrifugó la suspensión y se secó la torta a 50 °C durante 2 h. Rendimiento: 144,5 mg, con un rendimiento de ~56,1 %.
Ejemplo 18:
Preparación del compuesto B forma C. Se preparó la forma C a escala de 200 mg por medio de cristalización reactiva de solución en THF, como se evidencia por XRPD (FIG.10). Como se muestra en la FIG.11a y FIG.11b, se observó una pérdida de peso de un 6,8 % hasta 130 °C en TGA y DSC mostró una endotermia a 118,1 °C (temperatura de aparición). Se determinó que la proporción estequiométrica fue de 1,02 (ácido/base libre) y 9,7 %.
Preparación del compuesto B forma C. Se añadieron aproximadamente 56,9 mg de ácido tartárico a un vial de vidrio de 3 ml y se añadió 1,0 ml de THF para obtener una solución transparente. Se añadió la solución ácida transparente a 2,0 ml de solución madre de base libre en THF (~100 mg/ml) con una proporción molar de 1:1 y se agitó a TA. Se añadió aproximadamente 1 mg de semilla de forma C y la solución se volvió un poco turbia. Se agitó
la suspensión durante la noche y se tomaron muestras por XRPD. El patrón se ajustó a la forma C. Se agitó la suspensión a TA durante otras 24 horas, se secó la torta a 50 °C durante 1,5 horas. Se centrifugó la suspensión para recoger los sólidos. Rendimiento: 161,4 mg, con un rendimiento de ~62,7 %.
Ejemplo 19:
Preparación del compuesto B forma D. Se obtuvieron muestras de forma D por medio de evaporación rápida en MeOH/DCM y suspensión en H2O a TA, respectivamente. El patrón de XRPD para la forma D se muestra en la FIG. 12. Los resultados de TGA y DSC de la muestra de forma D se proporcionan en la FIG. 13a y FIG. 13b, respectivamente, y muestran una pérdida de peso de un 3,5 % antes de 150 °C y una endotermia a 73,0 °C antes de la fusión/descomposición a 163,9 °C (temperatura de aparición). Se observó el cambio de forma a la forma F después de que se calentara la forma D a 150 °C, se enfriara a 30 °C bajo protección de nitrógeno y a continuación se expusiera a condiciones ambientales. No se detectó ninguna cantidad significativa de disolvente de proceso MeOH o DCM por RMN de 1H y se determinó además que la proporción estequiométrica de ácido L-tartárico con respecto a base libre fue de 1,0. La forma D es un hidrato.
Para evaluar la estabilidad física de la forma D bajo diferente humedad, se recopilaron datos de DVS de la muestra de forma D a 25 °C después de que se equilibrara la muestra a la humedad ambiental (80 % de HR). Se observó una plataforma de 20 % de HR (2,25 % de absorción de agua) a 80 % de HR (2,72 % de absorción de agua) durante la desorción en la prueba DVS de la forma D, lo que sugiere que se produjo la deshidratación de la forma D hidratada cuando el valor de humedad pertinente fue menor de un 20 %. Además, ya que el contenido en agua teórico de un monohidrato es de un 2,6 %. Por lo tanto, es probable que la forma D sea un monohidrato.
Tabla 19: picos de XRPD representativos para el compuesto B forma D:

Ejemplo 20:
Preparación del compuesto B forma E. Se obtuvo la forma E por medio de cristalización mediada por DMSO añadiendo IPAc a una solución de DMSO, y su XRPD se muestra en la FIG.14. Las curvas TGA y DSC (FIG.15a y FIG. 15b, respectivamente) mostraron una pérdida de peso considerable de un 8,3 % antes de 140 °C y una endotermia a 126,3 °C antes de la fusión/descomposición a 142,6 °C (temperatura de aparición). El espectro de RMN de 1H indicó 0,7 equivalentes de DMSO (~9,4 % en peso) y no se detectó ninguna cantidad significativa de IPAc. Se determinó que la proporción estequiométrica de ácido L-tartárico con respecto a base libre fue de 1,0. La forma E es un solvato de DMSO.
Ejemplo 21:
Preparación del compuesto B forma F. Se obtuvo la forma F por medio de calentamiento de una muestra de forma D hasta 150 °C, enfriamiento hasta 30 °C bajo protección de nitrógeno y exponiéndola a condiciones ambientales. El patrón de XRPD de la forma F se proporciona en la FIG. 16. El análisis de DSC (FIG. 18) indicó que la forma F era cristalina con un pico endotérmico a 164,2 °C (temperatura de aparición). La RMN de 1H determinó que la proporción estequiométrica de ácido L-tartárico con respecto a base libre fue de 1,0 y no se detectó ninguna señal de disolvente significativa.
Tabla 20: picos de XRPD representativos para el compuesto B forma F:

Para caracterizar además la forma F, se realizó XRPD de temperatura variable (VT-XRPD) en la forma D, donde la temperatura se incrementó hasta 150 °C y volvió hasta 30 °C bajo protección de nitrógeno. En dichas condiciones, se observó la forma F después de la deshidratación de la muestra de forma D a temperaturas elevadas, indicando que la forma F fue un anhidrato.
Para evaluar la estabilidad física de la forma F bajo diferente humedad, se recopilaron datos de sorción dinámica de vapor (DVS) de la forma F a 25 °C después de que se equilibrara la muestra a la humedad ambiental (80 % de HR). Se observó una absorción de agua de ~1,9 % hasta un 80 % de HR, lo que sugiere que la forma F es ligeramente higroscópica.
Preparación del compuesto B forma G. Se obtuvo la forma G por evaporación lenta en MeOH a TA. El XRPD de la forma G se proporciona en la FIG. 19. Los resultados de TGA y DSC se proporcionan en la FIG.20a y FIG.
20b, respectivamente, y demostraron una pérdida de peso de un 3,3 % antes de 150 °C y un pico de fusión/descomposición a 170,4 °C (temperatura de aparición). El espectro de RMN de 1H mostró 0,49 equivalentes de MeOH (igual a ~3,0 % en peso). Se determinó que la proporción estequiométrica de ácido L-tartárico con respecto a base libre fue de 1,0. La forma G es un solvato de MeOH.
Tabla 21: picos de XRPD representativos del compuesto B forma G;


Ejemplo 22:
Cribado de polimorfos de L-tartrato. La solubilidad del material del compuesto B forma B se estimó en 20 disolventes a TA. (Tabla 22). Se añadieron aproximadamente 2 mg de sólidos en un vial de vidrio de 3 ml. A continuación, se añadieron los disolventes en la tabla 6-7 a los viales etapa a etapa hasta que se disolvieron los sólidos o se alcanzó un volumen total de 1 ml. Se usaron los resultados de la tabla 22 para guiar la selección de disolvente en el cribado de polimorfos. Se realizaron experimentos de cribado de polimorfos usando diferentes procedimientos de transición de fase sólida o cristalización en solución. Los procedimientos utilizados y formas cristalinas identificadas se resumen en la tabla 23.
Tabla 22: solubilidades del compuesto B

Tabla 23: resumen de cribados de polimorfos

Ejemplo 23:
Adición de antidisolvente. Se llevaron a cabo un total de 14 experimentos de adición de antidisolvente. Para cada experimento, se pesaron aproximadamente 15 mg del compuesto B forma B en un vial de vidrio de 20 ml, seguido de la adición de 0,3-1,0 ml del correspondiente disolvente. A continuación, se agitó magnéticamente la mezcla a la velocidad de 500 rpm para obtener una solución transparente a TA. Posteriormente, se añadió el correspondiente antidisolvente a la solución para inducir la precipitación o hasta que la cantidad total de antidisolvente alcanzó 10,0 ml. Se transfirieron las soluciones transparentes a una suspensión a 5 °C. Si no se produce precipitación, a continuación se transfirió la solución para evaporación rápida a TA. Se aislaron los sólidos
para análisis XRPD. Los resultados resumidos en la tabla 24 mostraron que se obtuvieron las formas B, D y E del compuesto B.
Tabla 24: resumen de experimentos de adición de antidisolvente

* se obtuvo el sólido por medio de agitación a 5 °C
** se obtuvo el sólido por medio de evaporación rápida a TA
N/A: no se obtuvo ningún sólido
Ejemplo 24:
Conversión de suspensión a TA. Se realizaron experimentos de conversión de suspensión a TA en diferentes sistemas de disolventes. Para cada experimento, se suspendieron aproximadamente 20 mg del compuesto B forma B en 0,3 ml del disolvente correspondiente en un vial de vidrio de 1,5 ml. Después de que se agitara magnéticamente la suspensión durante 17 días a TA, se aislaron los sólidos restantes para el análisis XRPD. Los resultados resumidos en la tabla 25 mostraron que se obtuvieron las formas B, D y G del compuesto B.
Tabla 25: resumen de experimentos de conversión de suspensión a TA.

* Se realizó calentamiento-enfriamiento de la muestra después de agitarse durante 17 días.
Ejemplo 25:
Conversión de suspensión a temperaturas elevadas. Se realizaron experimentos de conversión de suspensión a 50 °C y 70 °C en diferentes sistemas de disolventes. Para cada experimento, se suspendieron aproximadamente 20 mg del compuesto B forma B en 0,3 ml del disolvente correspondiente en un vial de vidrio de 1,5 ml. Después
de que se agitara magnéticamente la suspensión durante 17 días a 50 °C y 70 °C, se aislaron los sólidos restantes para el análisis XRPD. Los resultados resumidos en la tabla 26 indicaron que se obtuvo el compuesto B forma B. Tabla 26: resumen de experimentos de conversión de suspensión a temperaturas elevadas

Ejemplo 26:
Evaporación lenta. Se realizaron experimentos de evaporación lenta en 9 condiciones. Para cada experimento, se pesaron alrededor de 15 mg del compuesto B forma B en un vial de vidrio de 3 ml, seguido de la adición del correspondiente disolvente o mezcla de disolventes para obtener una solución transparente. Posteriormente, se cubrió el vial con Parafilm con 3~4 orificios y se mantuvo a TA para permitir que la solución se evaporara lentamente. Se sometieron a prueba los sólidos aislados por XRPD. Como se resume en, se generó el compuesto B forma G.
Tabla 27: resumen de experimentos de evaporación lenta

Ejemplo 27:
Difusión de vapor sólido. Se realizaron experimentos de difusión de vapor sólido usando 11 disolventes. Para cada experimento, se pesaron aproximadamente 15 mg del compuesto B forma B en un vial de 3 ml, que se colocó en un vial de 20 ml con 4 ml del correspondiente disolvente. Se selló el vial de 20 ml con una tapa y se mantuvo a TA durante 14 días para permitir que el vapor del disolvente interactuara con la muestra sólida. Se sometieron a prueba los sólidos aislados por XRPD. Los resultados resumidos en la tabla 28 indicaron que se obtuvo el compuesto B forma B.
Tabla 28: resumen de experimentos de difusión de vapor sólido.


Ejemplo 28:
Difusión de vapor líquido. Se realizaron doce experimentos de difusión de vapor líquido. Para cada experimento, se disolvieron aproximadamente 15 mg del compuesto B forma B en 0,5-1,0 ml del correspondiente disolvente para obtener una solución transparente en un vial de 3 ml. Posteriormente, se colocó la solución en un vial de 20 ml con 4 ml del correspondiente antidisolvente. Se selló el vial de 20 ml con una tapa y se mantuvo a TA, lo que permitió suficiente tiempo para que el vapor del disolvente interactuara con la solución. Se aislaron los sólidos para el análisis XRPD. Los resultados resumidos en la tabla 29 mostraron que se obtuvieron el compuesto B forma B y una mezcla de forma B+G/B+D.
Tabla 29: resumen de experimentos de difusión de vapor líquido.

N/A: no se obtuvo ningún sólido
Ejemplo 29:
Enfriamiento lento. Se realizaron experimentos de enfriamiento lento en 10 sistemas de disolventes. Para cada experimento, se suspendieron aproximadamente 20 mg del compuesto B forma B en 1,0 ml del correspondiente disolvente en un vial de vidrio de 3 ml a TA. Se transfirió la suspensión a una suspensión a 50 °C con un agitador magnético a una velocidad de 500 rpm. Se equilibró la muestra a 50 °C durante 2 h y se filtró usando una membrana de nailon de 0,45 µm. Posteriormente, se enfrió lentamente el filtrado de 50 °C a 5 °C a una tasa de 0,1 °C/min. Los resultados resumidos en indicaron que no se observó ningún sólido.
Tabla 30: resumen de experimentos de enfriamiento lento.

Ejemplo 30:
Transformación de fase. La cristalización del compuesto B a partir de disolventes tales como acetato de etilo, etanol, acetona o una mezcla acuosa de estos puede dar como resultado la formación de una más de las siguientes formas: forma B anhidra, forma F anhidra, forma D monohidrato y forma A de solvato de acetona. Los patrones de XRPD de estas cuatro formas sólidas se muestran en la FIG.21. Por lo tanto, fue importante conocer la estabilidad física de estas formas para determinar el contexto de transformación de fase completo y controlar la cristalización para obtener la forma B deseada. Debido a la proximidad cercana de los puntos de fusión de las formas F y B
(160-164 °C de aparición para la forma F frente a 166-175 °C para la forma B), la conversión de forma fue lenta en experimentos de puenteo de suspensión competitivos a TA y se obtuvo una mezcla de ambas formas. Como resultado, se realizaron estudios de solubilidad en equilibrio para ambas formas en etanol durante 17 h a 25, 35 y 50 °C para determinar su relación de estabilidad termodinámica.
Se descubrió que la forma F tenía mayor solubilidad a estas tres temperaturas, confirmando que la forma B es la forma termodinámicamente estable de 25-50 °C. La curva de solubilidad de Van't Hoff de las formas B y F frente a la temperatura mostró que los dos anhidratos estaban relacionados enantiotrópicamente con una temperatura de transición de ~19 °C. En el caso de la estabilidad hidrato-anhidrato, se descubrió que la forma B permaneció estable a la actividad del agua (aw) aw<0,2 (TA), por encima de esta la forma D hidratada era estable. Sin embargo, se descubrió que la forma B era cinéticamente estable hasta una aw de 0,4 (TA) en experimentos de puenteo de suspensión hasta 36 días. Esto sugiere que aunque el valor de aw crítico para la conversión en hidrato es bajo, existe una gran barrera cinética para la conversión de la forma B a la forma D.
Muestras de suspensión y cristalización. Forma B forma A
Se prepararon mezclas 1:1 de lotes de compuesto B que comprendían (1) una mezcla de forma A/solvato de acetona y forma B) y (2) forma B y se añadieron a acetona al 100 % y mezclas acuosas de acetona y agua (acetona al 90 %, acetona al 95, 96, 97, 98 y 99 % v/v). Se suspendieron las muestras a TA durante 120 h, se filtraron y a continuación se analizaron por XRPD.
Forma F
Se suspendió la forma F en etanol al 100 % a TA y 50 °C durante la noche. Se sembró una de las suspensiones a TA con la forma B mientras que la otra se dejó sin sembrar. Se sembró la suspensión agitada a 50 °C con la forma B. Se filtraron las muestras y se analizaron por XRPD. Se suspendieron muestras de forma F por separado a TA en agua DI al 100 %, acetona/agua 1:1 y acetona al 100 %. Las suspensiones de la forma F en disolventes puros se mantuvieron a TA mientras que se obtuvo una solución de forma F en la mezcla de acetona:agua 1:1 que se agitó por tanto a 5 °C para enfriar la cristalización/precipitación. Después de 24 h, se filtraron todas las muestras y se analizaron por XRPD. También se suspendió la forma F en mezclas de acetona:agua 95:5 y acetona:agua 97:3 a 50 °C durante 2 h, después de esto se enfriaron las suspensiones hasta TA, se filtraron y se analizaron por XRPD. Finalmente, se recristalizó la forma F en acetona:agua 95:5 sembrada con forma B a 50 °C.
Forma D
Se suspendió la forma D a TA durante 48 h en etanol al 100 %. Posteriormente, se filtró la muestra y se analizó por XRPD.
Resultados
Desarrollo del procedimiento calorimétrico para estimar pureza. El cambio de etanol a acetona/agua como disolvente de cristalización mostró una mejora marcada en pureza, con un descenso de contenido en oligómero en un promedio de un 5 % p/p, con un rendimiento aceptable. El cambio en el contenido en oligómero así como el rendimiento fue una función de la composición del disolvente de suspensión. Un incremento en el contenido en agua mejoró la pureza pero redujo el rendimiento. Para una composición de disolvente dada, la aparición del punto de fusión y el % de contenido en oligómero mostraron consecuentemente resultados opuestos, indicando que es probable que la pureza afecte al punto de fusión, con mayor % de oligómero reduciendo el punto de fusión y viceversa. En base a esta observación, se analizaron varios lotes de suspensión obtenidos de acetona/agua usando DSC y se sometieron a prueba para determinar el contenido en oligómero por SEC y se desarrolló una curva de correlación. Se recopilaron datos de XRPD para todas estas muestras para garantizar que se ajustaran al compuesto B forma B. Usando el ajuste exponencial para los datos, se realizaron estimaciones de pureza para varios lotes, incluyendo un aumento de escala de 20 g y una muestra IPC. Se obtuvo un valor de R2 de 0,96 para el ajuste lineal entre el contenido en oligómero experimental y previsto. Dado que estas fueron muestras sólidas/en polvo con problemas inherentes de homogeneidad de la muestra, este alto valor de R2 proporcionó confianza en la solidez de la correlación determinada entre la aparición del punto de fusión y la pureza.
Consecuentemente se obtuvo el compuesto B forma B a partir de etanol al 100 % (suspensión o cristalización). Se realizaron otras condiciones incluyendo suspensión o cristalizaciones de etanol/agua y suspensión en acetona/agua (acetona al ≥ 95 %). Dependiendo de los parámetros de suspensión y cristalización, se obtuvieron varias otras formas sólidas del compuesto B, a saber, forma F (etanol/agua), forma A/disolvente de acetona (acetona al ≥ 95 %) y mezclas de forma A y B o forma A y F. En condiciones donde se obtuvo la forma F, se postuló la formación de un hidrato intermedio (Forma D) dado que los valores de aw de mezclas de etanol/agua empleadas para estas muestras estaban muy por encima de 0,2 (TA), donde el hidrato es termodinámicamente estable. La tabla 31 muestra las condiciones de cristalización con la forma obtenida. Dado que en varios casos se obtuvieron mezclas de formas, se implementó una estrategia de control de forma para obtener la forma B en la fase sólida final.
Tabla 31: condiciones de cristalización y correspondientes formas sólidas

Dado que el sistema de acetona/agua fue el más eficaz para purgar oligómeros del compuesto B, se realizó un experimento de puenteo de suspensión competitivo usando mezclas 1:1 de dos lotes separados del compuesto B: uno que era predominantemente forma A con algo de forma B y un segundo que se suspendió en mezclas de acetona/agua de diversas composiciones a TA durante 120 h. Se obtuvo solvato de acetona como forma estable con acetona al ≥95 %, mientras que se obtuvo una mezcla de hidrato de forma B y forma D con acetona al 90 %. Como el nivel de acetona deseado estuvo entre un 96 y un 95 % (a niveles de oligómero), existió la posibilidad de obtener cualquiera de las formas A, B o D de la cristalización final.
Estrategia de control de forma: conversión de formas A y F a forma B. La FIG. 2b muestra los termogramas de DSC del solvato de acetona de forma A. La primera endotermia a 124 °C indica pérdida de disolvente y vaporización, mientras que la segunda endotermia a 164 °C indica la fusión del correspondiente anhidrato. XRPD confirmó que este anhidrato era la forma B cuando se calentó la forma A hasta 152 °C. La forma A se resuspendió en etanol al 100 % durante la noche para su conversión en la forma B. Por tanto, se propuso un proceso de suspensión de dos etapas para ayudar al control de forma del compuesto B para obtener la forma B en caso de que la primera suspensión en acetona/agua produzca forma A o una mezcla de forma A y B, dependiendo de las condiciones de suspensión. Esto garantizaría que, independientemente de un cambio en la forma durante la primera suspensión, la forma B sería la forma del compuesto B final que se aísla después de la segunda etapa de suspensión.
A diferencia del solvato de acetona, la forma F obtenida a partir de una cristalización de etanol:agua 65:35 sembrada con la forma B mostró solo una endotermia de fusión a 162 °C. Debido a los valores de solubilidad casi idénticos de las formas F y B en etanol a 25 °C (0,23 mg/ml para la forma B frente a 0,26 mg/ml para la forma F), la fuerza impulsora termodinámica para la conversión de la forma F metaestable en la forma B estable es menos y por tanto la tasa de conversión forma F→forma B sería lenta, incluso en presencia de la forma B como semillas. Independientemente de la siembra y la temperatura, la conversión de forma F → forma B es lenta y se obtiene una mezcla de ambas formas después de 12 horas.
Para facilitar la conversión forma F → forma B en escalas de tiempo más cortas, se adoptó una vía de solvatos en la que la forma F se convertiría en la forma B por medio de la formación de solvato o hidrato de acetona, que posteriormente se desolvataría en la forma B. Se suspendió la forma F durante la noche en agua pura, acetona y mezcla de acetona:agua 1:1, sin siembra. La forma F permanece sin cambios en acetona al 100 % pero se convierte en hidrato (Forma D) en presencia de agua. Cuando se suspende el hidrato en etanol puro, da como resultado una mezcla de forma D y forma F, indicando por tanto la propensión del hidrato a desolvatar a la forma anhidra metaestable. La forma F suspendida en acetona:agua 95:5 y acetona:agua 97:3 a 50 °C demostró que aparece la forma B pero la conversión es incompleta. La recristalización de la forma F a partir de acetona:agua 95:5 usando semillas de la forma B a 50 °C dio como resultado una conversión completa de forma F → forma B. La FIG.25 y FIG.26 proporcionan esquemas de conversión de forma entre las formas A, B, D y F.
Relación termodinámica entre la forma F y forma B anhidra por competencia de suspensión entre la forma F y B anhidra. Para determinar la estabilidad termodinámica entre la forma B y F, se realizaron experimentos de suspensión competitiva en sistemas de disolventes de acetona/EtOH/EtOAc a TA (25 ± 3 °C) y 50 °C como se enumera en la tabla 32. Se suspendió una mezcla de forma B y F con igual proporción de masa en soluciones saturadas de acetona/EtOH/EtOAc de la forma B y a continuación se agitó magnéticamente a la temperatura seleccionada. Después de la suspensión durante aproximadamente 4-11 días, se aislaron los sólidos restantes para la caracterización de XRPD. Se observó una mezcla de forma B y F, indicando una transición lenta entre la forma B y F.
Tabla 32: condiciones de competencia de suspensión para la relación termodinámica entre el compuesto B, forma B y forma F.

( )
La relación de estabilidad termodinámica entre la forma F y B anhidra se determinó por medio de medición de solubilidad en el equilibrio y la competencia de suspensión. Como resultado, se observó una mezcla de forma B y F en todos los experimentos de competencia con suspensión, indicando transición lenta entre la forma B y F. Sin quedar vinculado a ninguna teoría particular, la baja solubilidad de la forma B y la forma F puede estar provocada por su baja solubilidad en disolventes sometidos a prueba (acetona/EtOH/EtOAc). Por lo tanto, se midió la solubilidad en el equilibrio (17 horas) en EtOH a 25 °C, 35 °C y 50 °C, respectivamente, para determinar su relación de estabilidad termodinámica. En comparación con la forma B, la forma F mostró mayor solubilidad bajo las tres temperaturas sometidas a prueba en EtOH, indicando que la forma B es termodinámicamente más estable que la forma F de 25 °C a 50 °C.
La aw entre la forma B anhidra y la forma D hidratada se determinó por medio de la competencia de suspensión bajo diversas condiciones de actividad de agua a TA. La forma D se observó en aw de 0,6 y 0,8 después de una semana y en agua después de 36 días. La forma B se observó en EtOH (aw <0,2) después de una semana. Se observó una mezcla de forma D y B en sistemas de aw de 0,2 y 0,4 después de agitar a TA durante 36 días. Para confirmar además la relación de estabilidad termodinámica entre la forma B y forma D en sistemas de aw de 0,2 y 0,4 a TA, se recopiló su solubilidad en el equilibrio (24 horas) bajo las correspondientes condiciones. En comparación con las muestras que comenzaron con la forma B, se observó menor solubilidad en las muestras que comenzaron con la forma D (no se comprobaron las formas cristalinas de los sólidos limitados finales) en ambos sistemas con aw de 0,2 y 0,4.
Medición de solubilidad en el equilibrio de forma F y B anhidra. Para determinar además la estabilidad termodinámica entre la forma B y F, se realizaron experimentos de medición de solubilidad en el equilibrio en EtOH a 25 °C, 35 °C y 50 °C, respectivamente. Los procedimientos detallados se resumen como sigue: se suspendieron los sólidos de la forma B y F en 0,4 ml de EtOH a las temperaturas seleccionadas y se agitaron magnéticamente durante 17 h (750 rpm). Después de la centrifugación, se sometió a prueba la concentración y pureza por HPLC de la base libre en el filtrado. La forma cristalina de los sólidos restantes se comprobó por XRPD.
En comparación con la forma B, la forma F mostró mayor solubilidad a 25 °C, 35 °C y 50 °C en EtOH (tabla 33). En base a los resultados de XRPD, no se observó ningún cambio de forma después de la prueba de solubilidad, indicando que la forma B probablemente sea termodinámicamente más estable que la forma F de 25 °C a 50 °C. Tabla 33: resumen de mediciones de solubilidad en el equilibrio de forma B y forma F en EtOH

Determinación de actividad de agua crítica entre la forma B y D. Para determinar la actividad de agua crítica entre la forma B anhidra y la forma D hidratada, se realizó una competencia de suspensión en diversas condiciones de actividad de agua a TA, como se enumera en la tabla 34. Se suspendió una mezcla de forma B y D con igual proporción de masa en soluciones saturadas de EtOH-agua (con diversas aw) de la forma B y a continuación se agitó magnéticamente a TA. Después de la suspensión durante aproximadamente 7~36 días, se aislaron los sólidos restantes para la caracterización de XRPD.
Tabla 34: resumen de competencia de suspensión de forma B y forma D bajo diversas condiciones de aw

La forma D se observó en aw de 0,6 y 0,8 después de la suspensión durante una semana y en agua después de 36 días. La forma B se observó en EtOH después de la suspensión durante una semana. Se observó una mezcla de forma D y B en sistemas de aw de 0,2 y 0,4 después de agitar a TA durante 36 días.
Medición de la solubilidad de la forma B y D. Para confirmar además la relación de estabilidad termodinámica entre la forma B y forma D en condiciones de aw de 0,2 y 0,4 a TA, se recopiló la solubilidad en el equilibrio bajo las condiciones establecidas en la tabla 35.
Tabla 35: resumen para la medición de la solubilidad en el equilibrio de la forma B y D en sistemas de aw=0,2/0,4

Se suspendieron los sólidos de la forma B anhidra y la forma D hidratada en 0,5 ml del sistema de disolventes diana (aw=0,2/0,4), respectivamente, y se agitaron magnéticamente durante 24 h (750 rpm). Se filtró la suspensión y se sometió a prueba la concentración de base libre en el filtrado. Se observó una solubilidad menor en las muestras que comenzaron con la forma D. La forma D hidratada parece termodinámicamente más estable que la forma B anhidra cuando aw ≥ 0,2 a TA (25±3 °C).
Ejemplo 31:
Malonato forma M
Se obtuvo un resultado positivo de malonato con baja cristalinidad, malonato forma M, a partir del cribado. Su patrón de XRPD se muestra en la FIG. 30. Se observó una pérdida de peso de un 4,1 % hasta 110 °C en TGA y el resultado de DSC mostró múltiples endotermias (FIG. 31). Se determinó que la estequiometría fue de 0,50 (ácido/base libre) y se detectó un 6,2 % de THF (una proporción molar de 0,58 con respecto a base libre) por RMN de 1H. Dado que se observan múltiples endotermias y una cantidad considerable de THF, malonato forma M es un solvato de THF, pero no se realizó caracterización adicional debido a su baja cristalinidad.
Ejemplo 32:
Fumarato forma 1
Se obtuvo un resultado positivo de fumarato cristalino, fumarato forma 1, a partir del cribado. Se observó una pérdida de peso de un 0,9 % hasta 150 °C en TGA y DSC (FIG. 28), el resultado mostró un pico endotérmico de fusión a 164,3 °C (temperatura de aparición). Se determinó que la estequiometría fue de 0,88 (ácido/base libre) y se detectó un 1,5 % de EtOAc (una proporción molar de 0,11 con respecto a base libre) por RMN de 1H.
Preparación de fumarato forma 1. Se pesaron aproximadamente 44,7 mg de ácido fumárico en un vial de vidrio de 5 ml y se añadieron 2,0 ml de EtOAc. El ácido permaneció sin disolver. Se añadieron aproximadamente 2,0 ml de solución madre de base libre en EtOAc (~100 mg/ml) al vial de 5 ml con una proporción molar de 1:1 y se agitó a TA. Se añadieron aproximadamente 3 mg de semilla de fumarato forma 1 y la solución se volvió turbia. Se agitó la suspensión durante la noche y se muestreó por XRPD (FIG. 27) con un patrón confirmado ajustado al fumarato forma 1. Se agitó la suspensión a 50 °C durante 3 días más para incrementar la cristalinidad seguido de centrifugación. Se secó la torta a 50 °C durante 4 h. Rendimiento: 186,6 mg, con un rendimiento de ~76,2 %.
Tabla 36. Picos de XRPD representativos para el compuesto C forma 1.

Tabla 37. Picos de XRPD representativos para el compuesto C forma 2.

Resumen. La base libre del compuesto A se identificó como una forma sólida amorfa con una pérdida de peso <0,5 % antes de la descomposición y un punto de fusión de aproximadamente 87 °C. Su higroscopicidad a un 95 % de HR fue de <1 % y tuvo un contenido en epímero de <1 %. Su solubilidad a 37 °C fue de aproximadamente 6,6 mg/ml. El patrón de XRPD para la forma amorfa se proporciona en la FIG.32. El compuesto C forma 1 fue un sólido cristalino con <0,5 % de pérdida de peso antes de la descomposición y un punto de fusión de aproximadamente 165-174 °C. Se obtuvieron múltiples formas del compuesto C dependiendo del disolvente. Su higroscopicidad a un 95 % de HR fue de <1,5 % y tuvo un contenido en epímero de <1 %. Su solubilidad a 37 °C fue de aproximadamente 6,6 mg/ml. El compuesto B forma B fue un sólido cristalino con <0,5 % de pérdida de peso antes de la descomposición y un punto de fusión de aproximadamente 168 °C. Una única forma estaba presente en la forma B anhidra. Su higroscopicidad a un 95 % de HR fue de <1 % y tuvo un contenido en epímero de <2,5 %. Su solubilidad a 37 °C fue de aproximadamente 5,9 mg/ml. Se descubrió que la forma anhidra del compuesto B forma B fue una forma cristalina pura estable con propiedades más favorables que las formas D, E y F como se describe en el presente documento.
Ejemplo 33:
Evaluación de seguridad, farmacocinética y actividad del compuesto B. El cáncer de mama es el cáncer más frecuente diagnosticado en mujeres, con una incidencia global estimada de 1,67 millones de casos nuevos informados en 2012 (Ferlay et al. 2013). El cáncer de mama representa aproximadamente un 15 %
(aproximadamente 522.000 casos) de todas las muertes por cáncer.
Aproximadamente un 80 % de todos los cánceres de mama expresan el factor del receptor estrogénico (RE), y la gran mayoría de estos son dependientes del RE para el crecimiento y la progresión tumoral. La modulación de la actividad y/o síntesis de estrógenos es el pilar de los enfoques terapéuticos en mujeres con cáncer de mama RE positivo. Sin embargo, a pesar de la eficacia de tratamientos endocrinos disponibles tales como los antagonistas de RE (por ejemplo, tamoxifeno), inhibidores de la aromatasa (por ejemplo, anastrozol, letrozol y exemestano) y antagonistas/degradadores de RE completos (por ejemplo, fulvestrant), muchas pacientes finalmente recaen o desarrollan resistencia a estos agentes y por lo tanto requieren tratamiento adicional para un control de enfermedad óptimo.
A pesar de volverse resistentes a los inhibidores de la aromatasa o al tamoxifeno, el crecimiento y supervivencia de las células tumorales resistentes siguen dependiendo de la señalización de RE; por lo tanto, las pacientes con cáncer de mama RE positivo todavía pueden responder al tratamiento endocrino de segunda o tercera línea después de la progresión sobre el tratamiento previo (Di Leo et al. 2010; Baselga et al. 2012). Cada vez hay más pruebas de que, en el estado de resistencia endocrina, el RE puede emitir señales de manera independiente del ligando por medio de la entrada de otras vías de señalización (Miller et al.2010; Van Tine et al.2011). Sin quedar vinculado a ninguna teoría particular, un agente con un mecanismo de acción doble (antagonismo de RE más degradación) tiene el potencial de dirigirse a la señalización de RE tanto dependiente de ligando como independiente de ligando y, en consecuencia, mejorar los resultados del tratamiento en el cánceres de mama RE positivos en fase tardía. Además, estudios recientes han identificado mutaciones en ESR1 (es decir, el gen que codifica REα) que afectan al dominio de unión a ligando (LBD) del RE (Segal y Dowsett 2014). En modelos no clínicos, el RE mutante puede impulsar la transcripción y proliferación en ausencia de estrógeno, lo que sugiere que las formas de RE mutantes en LBD pueden estar implicadas en mediar en la resistencia clínica a algunos tratamientos endocrinos (Li et al. 2013; Robinson et al. 2013; Toy et al. 2013). Los antagonistas de RE que son eficaces contra estos receptores mutados en RE constitutivamente activos independientes de ligando pueden poseer beneficio terapéutico sustancial.
Como tal, existe la necesidad de nuevos tratamientos dirigidos a RE con actividad antitumoral incrementada para retrasar además la progresión de la enfermedad y/o superar la resistencia a los tratamientos endocrinos actualmente disponibles y, en última instancia, prolongar la supervivencia en mujeres con cáncer de mama RE positivo.
El compuesto B es un agente terapéutico de molécula pequeña biodisponible por vía oral potente que se está desarrollando para el tratamiento de pacientes con cáncer de mama RE positivo. Como se proporciona en el presente documento, el compuesto B, incluyendo sus formas sólidas (por ejemplo, forma B) son compuestos estables con propiedades favorables para el desarrollo farmacéutico continuo. Sin quedar vinculado a ninguna teoría particular, el compuesto B parece antagonizar los efectos de los estrógenos por medio de unión competitiva al LBD de RE tanto natural como mutante con potencia nanomolar. Tras la unión, y sin quedar vinculado a ninguna teoría particular, el compuesto B induce una conformación inactiva al LBD de RE, como se mide por desplazamiento de péptidos coactivadores. Además de sus propiedades antagonistas directas, sin quedar vinculado a ninguna teoría particular, el mecanismo de acción del compuesto B incluye reducir niveles de proteína REα a través de la degradación mediada por proteasomas. Se supone que la degradación de RE permite la supresión total de la señalización de RE, lo que no se logra por tratamientos de RE de primera generación tales como tamoxifeno, que muestra agonismo parcial. El compuesto B inhibe potentemente la proliferación de múltiples líneas celulares de cáncer de mama RE positivo in vitro, incluyendo células genomanipuladas para expresar mutaciones clínicamente pertinentes en RE.
In vivo, el compuesto B presentó actividad antitumoral dependiente de la dosis en modelos de xenoinjerto de cáncer de mama RE positivo, incluyendo en un modelo de xenoinjerto derivado de paciente que alberga una mutación ESR1 (RE.Y537S). Se descubrió que el intervalo de dosis eficaz fue de 0,1-10 mg/kg/día, y todas las dosis se toleraron bien. Fulvestrant, cuando se dosificó de acuerdo con un esquema de dosificación clínicamente pertinente, fue menos eficaz que el compuesto B en los modelos de xenoinjerto evaluados. Por tanto, el compuesto B demostró actividad no clínica consistente en modelos de cáncer de mama RE positivo de enfermedad que porta la mutación de ESR1 y ESR1 natural.
Ejemplo 34:
Análisis de eficacia in vitro e in vivo del compuesto B. El compuesto B muestra degradación de RE y supresión de la vía de RE superiores en comparación con ambos GDC-0927 y GDC-0810. Además, el compuesto B tiene mejores propiedades de DMPK que ambos GDC-0927 y GDC-0810, lo que da como resultado la misma eficacia in vivo que GDC-0927 pero a dosis 100 veces menores (por ejemplo, dosis de 1 mg o 10 mg). (Véanse la FIG.34 y FIG.35).
Farmacocinética y metabolismo. Después de una sola administración i.v. a ratas, perros y monos, se descubrió que el compuesto B tenía un aclaramiento de bajo a moderado, un gran volumen de distribución y una semivida
de eliminación terminal de 7-24 horas. La biodisponibilidad oral fue moderada en ratas y perros (41 %-55 %) y baja (17 %) en monos. Los datos in vitro mostraron que la unión a proteína plasmática del compuesto B fue alta en todas las especies, variando de un 98 % a un 99 % de unión.
Los experimentos de identificación de metabolitos in vitro mostraron que la glucuronidación mediada por UGT1A4 fue la vía metabólica in vitro principal del compuesto B. La contribución de las isoformas CYP450 fue menor e incluyó tanto CYP3A4 como CYP2C9. Los estudios de inhibición de CYP in vitro en microsomas hepáticos humanos y los estudios de inducción en hepatocitos humanos sugirieron un potencial bajo a moderado de interacciones farmacológicas. El compuesto B inhibió directamente CYP3A4 con valores de concentración inhibidora (CI50) de un 50 % de 6,5 µM (midazolam 1'-hidroxilación) y 26 µM (testosterona 6β-hidroxilación); La CI50 para la inhibición de CYP2B6 y CYP2C8 fue de 13 µM y 21 µM, respectivamente. El compuesto B mostró una débil inhibición dependiente del metabolismo de CYP2C9.
Toxicología. Se realizaron estudios de toxicidad oral de dosis repetidas de prácticas correctas de laboratorio (BPL) de cuatro semanas en monos y ratas hembra con evaluaciones integradas de la función neurológica (ratas, monos), respiratoria (monos) y cardiovascular (monos) para caracterizar el perfil de seguridad no clínico del compuesto B. En el estudio con ratas, el compuesto B se toleró a los niveles de dosis ejemplares (10, 30 y 100 mg/kg) con efectos adversos predominantemente en los riñones y el hígado a 100 mg/kg. En el estudio con monos (20, 60 y 200 mg/kg), se consideró que la dosis tolerada máxima (DMT) fue de 60 mg/kg, ya que no se toleró la dosis alta de 200 mg/kg. Los efectos adversos se observaron principalmente en el nivel de dosis alta de 200 mg/kg, y la falta de tolerabilidad se atribuyó a daño renal y hepático e inanición.
Tanto en ratas como en monos, se observó PLD dependiente de la dosis en numerosos órganos a exposiciones que fueron mayores que las anticipadas con la dosis de partida humana en la fase I (al menos 44 veces y 6 veces en base al área bajo la curva [ABC] de concentración-tiempo, respectivamente), con efectos adversos en órganos limitados principalmente al riñón y al hígado. En ratas, no se observó PLD a 10 mg/kg (factor de exposición 18 veces mayor), pero se incrementó en incidencia y gravedad de 30 a 100 mg/kg. En monos, estuvo presente PLD sensible a la dosis en todas las dosis, pero se limitó a cambios mínimos en el pulmón a 20 mg/kg (factor de exposición 6 veces mayor). Estos múltiplos de exposición proporcionan pruebas de que el riesgo de toxicidad asociado a PLD para seres humanos en la dosis de partida de fase I es bajo.
La traducibilidad de PLD de especies no clínicas a pacientes no es segura, pero se puede razonar (Reasor et al.
2006). Fármacos tales como tamoxifeno y palbociclib no han demostrado ningún problema clínico a pesar de sus hallazgos de PLD en estudios no clínicos. Aunque el compuesto B se asoció con PLD en múltiples tejidos tanto en ratas como en monos, no hubo pruebas microscópicas ópticas de la afectación de órganos críticos tales como corazón, ojos o neuronas en estos estudios (Chatman et al.2009).
Después de una administración oral de 28 días a ratas y monos, los incrementos en la exposición sistémica del compuesto B fueron proporcionales a la dosis. En base a la naturaleza y reversibilidad de signos clínicos, la patología clínica y los hallazgos histopatológicos, la dosis extremadamente tóxica para un 10 % de los animales (STD10) para ratas se definió como 100 mg/kg, con la correspondiente concentración plasmática máxima (Cmáx) y valores de ABC de 0 a 24 horas (ABC0-24) de 6560 ng/ml y 143.000 ng ● h/ml, respectivamente. En monos, la mayor dosis no extremadamente tóxica se definió como 60 mg/kg/día, con correspondientes valores de Cmáx y ABC 0-24 de 841 ng/ml y 16.200 ng ● h/ml, respectivamente, debido a los signos clínicos y situaciones moribundas presentes a 200 mg/kg/día.
En resumen, los resultados de los estudios de farmacología de seguridad y toxicidad no clínica completados hasta la fecha proporcionan una caracterización sólida del perfil de toxicidad del compuesto B y respaldan la administración a pacientes con cáncer en un ensayo de fase I.
Administración del compuesto B como monoterapia de agente único. El compuesto B demostró actividad no clínica consistente en modelos de cáncer de mama RE positivo de enfermedad que porta la mutación de ESR1 y ESR1 natural. La actividad de seguridad, farmacocinética (FC), farmacodinámica (FD) y la actividad antitumoral preliminar del compuesto B como agente único se analizaron en un estudio abierto, multicéntrico, de fase Ia/Ib en pacientes con cáncer de mama RE positivo localmente avanzado o metastásico. Se incluyó a pacientes en una fase de aumento escalonado de dosis con inclusión en una fase de expansión a continuación. Durante el aumento escalonado de dosis de agente único, se evaluaron las cohortes a niveles de dosis con aumento escalonado para determinar MTD o dosis administrada máxima (MAD).
Una vez que se ha establecido MTD o MAD de agente único, se puede incluir una cohorte de aumento escalonado tratada con compuesto B (en o por debajo de MTD o MAD) o el compuesto B en combinación con palbociclib. Adicionalmente, se incluirán pacientes en la fase de expansión y se tratarán en o por debajo de MTD o MAD de agente único del compuesto B solo o en combinación con palbociclib y/o agonista de LHRH. Las expansiones de dosis de agente único pueden evaluar dos niveles de dosis diferentes del compuesto B con y sin un agonista de LHRH.
Se realizó un seguimiento de los pacientes para determinar acontecimientos adversos durante un margen de evaluación de toxicidad limitante de la dosis (DLT), definido como los días -7 a 28 del ciclo 1 (cohortes de agente único). Para la evaluación de DLT, se calificó la toxicidad de acuerdo con National Cancer Institute Common Terminology Criteria for Adverse Events, versión 4.0 (NCl CTCAE v4.0).
Pacientes incluidos en la fase de aumento escalonado de dosis del compuesto B como agente único, que consiste en un período de cribado, un período inicial de FC, un período de tratamiento y un período de seguimiento de seguridad. La dosificación continua una vez al día se inició durante el período de tratamiento que comenzó el día 1 del ciclo 1. La dosis de partida del compuesto B como agente único fue de 10 mg, administrada por vía oral a pacientes de la primera cohorte de forma continua en ciclos de 28 días. La dosis se incrementará en hasta un 200 % del nivel de dosis precedente para cada cohorte sucesiva, hasta que observe un hallazgo de seguridad preocupante (es decir, una DLT, cualquier paciente con toxicidad clínicamente significativa de grado ≥ 2 o bien un perfil global de acontecimientos adversos inapropiado para incrementos de un 200 %). Una vez que se observen hallazgos de seguridad preocupantes, el aumento escalonado de dosis no excederá de incrementos de un 100 %. El compuesto B tiene una semivida de aproximadamente 40 horas. Como se indica anteriormente, las exposiciones del compuesto B se incrementaron proporcionalmente de 10 a 30 mg con una variabilidad normal.
Se inició a seis pacientes en la dosis inicial de 10 mg qd en ciclos de 28 días como se describe en el presente documento. Como se indica en la tabla 38 a continuación, todos los pacientes tratados con FES-PET tuvieron respuestas cualitativas casi completas (NC) o completas (CR). No se observaron DLT, SAE, AESI o anomalías de laboratorio clínicamente significativas en los pacientes tratados. Todos los AA relacionados fueron acontecimientos de grado 1 o grado 2.
Tabla 38: respuesta del paciente al tratamiento con el compuesto B:

A una paciente tratada se le diagnosticó cáncer de mama RE+PR+. La paciente se había sometido a cirugía previa y tratamiento previo con agentes antineoplásicos, incluyendo tratamiento SERM y tratamiento AI antes de su inclusión y tratamiento. La paciente se trató con el compuesto B a 10 mg y mostró una respuesta al tratamiento después de 3 ciclos como se indica en la FIG.36a y FIG.36b.
A otra paciente se le diagnosticó cáncer de mama HR+ en fase temprana y había recibido cirugía previa y tratamiento con agentes antineoplásicos, incluyendo citotóxicos, inhibidores de CDK4/6 y AI. La paciente se trató con el compuesto B a 10 mg y mostró una respuesta al tratamiento después de 3 ciclos como se indica en la FIG.
37a y FIG.37b. se incluyó en este ensayo a 10 mg en marzo de 2018.
Las pacientes tratadas hasta la fecha demuestran que la administración del compuesto B se tolera bien a 10 mg y 30 mg, con solo AA de grado 1 y 2. En general, las exposiciones plasmáticas del compuesto B se incrementaron proporcionalmente con dosis de 10 a 30 mg después de una dosis individual. Las exposiciones en estado estacionario parecieron incrementar de manera mayor que la dosis proporcional de 10 mg a 30 mg. La semivida estimada de aproximadamente 40 horas respalda la dosificación una vez al día.
Claims (22)
1. Un compuesto B que tiene la estructura:

2. Una forma cristalina que comprende el compuesto B de la reivindicación 1

que tiene un patrón de difracción de rayos X en polvo que comprende picos en 19,32, 20,26, 21,63, 23,28 o 24,81 ± θ,1° 2θ (± 0,1°2θ).
3. La forma cristalina de la reivindicación 2, en la que la forma cristalina del compuesto B tiene un patrón de difracción de rayos X en polvo uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve o veinte picos de difracción de rayos X en polvo característicos seleccionados de:


4. La forma cristalina de la reivindicación 2, en la que la forma cristalina del compuesto B tiene un patrón de difracción de rayos X en polvo sustancialmente como se muestra en la FIG.1.
5. Una forma cristalina que comprende el compuesto B de la reivindicación 1

que tiene un patrón de difracción de rayos X en polvo que comprende picos en 11,49, 12,54, 19,16, 19,42 o 24,67 ± θ,1° 2θ (± 0,1°2θ).
6. La forma cristalina de la reivindicación 5, en la que la forma cristalina del compuesto B tiene un patrón de difracción de rayos X en polvo uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve o veinte picos de difracción de rayos X en polvo característicos seleccionados de:


7. La forma cristalina de la reivindicación 5, en la que la forma cristalina del compuesto B tiene un patrón de difracción de rayos X en polvo sustancialmente como se muestra en la FIG.4.
8. Una forma cristalina que comprende el compuesto B de la reivindicación 1

que tiene un patrón de difracción de rayos X en polvo que comprende picos en 11,31, 15,70, 16,54, 19,10 o 22,76 ± 0,1° 2θ.
9. La forma cristalina de la reivindicación 8, en la que la forma cristalina del compuesto B tiene un patrón de difracción de rayos X en polvo uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve o veinte picos de difracción de rayos X en polvo característicos seleccionados de:


10. La forma cristalina de la reivindicación 8, en la que la forma cristalina del compuesto B tiene un patrón de difracción de rayos X en polvo sustancialmente como se muestra en la FIG.12.
11. Una forma cristalina que comprende el compuesto B de la reivindicación 1

que tiene un patrón de difracción de rayos X en polvo que comprende picos en 12,52, 15,90, 19,66, 20,65 o 24,99 ± 0,1° 2θ.
12. La forma cristalina de la reivindicación 11, en la que la forma cristalina del compuesto B tiene un patrón de difracción de rayos X en polvo uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis, diecisiete, dieciocho, diecinueve o veinte picos de difracción de rayos X en polvo característicos seleccionados de:


13. La forma cristalina de la reivindicación 11, en la que la forma cristalina del compuesto B tiene un patrón de difracción de rayos X en polvo sustancialmente como se muestra en la FIG.16.
14. Una composición farmacéutica que comprende el compuesto de la reivindicación 1 y/o la forma sólida de la reivindicación 7 y al menos un excipiente farmacéuticamente aceptable.
15. El compuesto de la reivindicación 1, la forma sólida de la reivindicación 7 o la composición farmacéutica de la reivindicación 14 para su uso en un procedimiento para tratar cáncer de pulmón, cáncer de ovario, cáncer de endometrio, cáncer de próstata, cáncer de útero o cáncer de mama en un paciente que tiene dicho cáncer, comprendiendo dicho procedimiento administrar una cantidad eficaz de un compuesto de la reivindicación 1, una forma sólida de la reivindicación 7 o la composición farmacéutica de la reivindicación 14 al paciente con cáncer.
16. El compuesto de la reivindicación 1, la forma sólida de la reivindicación 7 o la composición farmacéutica de la reivindicación 14 para su uso en un procedimiento para tratar cáncer de mama en una paciente con cáncer de mama, comprendiendo dicho procedimiento administrar una cantidad eficaz de un compuesto de la reivindicación 1, una forma sólida de la reivindicación 7 o la composición farmacéutica de la reivindicación 14 a la paciente con cáncer, en el que el cáncer de mama es cáncer de mama positivo para receptores hormonales, cáncer de mama HER2 positivo o cáncer de mama triple negativo.
17. El compuesto, la forma sólida o la composición farmacéutica para su uso de acuerdo con la reivindicación 16, en el que el cáncer de mama es cáncer de mama metastásico.
18. El compuesto, la forma sólida o la composición farmacéutica para su uso de acuerdo con una cualquiera de las reivindicaciones 16-17, en el que el paciente ha recibido tratamiento previo con uno o más agentes antineoplásicos o radioterapia.
19. Un proceso para la preparación del compuesto B,

(B),
en el que el proceso comprende las etapas de:

20. El proceso de la reivindicación 19, en el que el proceso comprende además la recristalización del compuesto B en metanol y etanol;

21. El proceso de la reivindicación 19, en el que el proceso comprende además la recristalización del compuesto B en MTBE, agua, NaOH y etanol;

22. El proceso de una cualquiera de las reivindicaciones 19-21, en el que los intermedios indolilo se sintetizan de acuerdo con las etapas:

Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862687930P | 2018-06-21 | 2018-06-21 | |
| US201862719896P | 2018-08-20 | 2018-08-20 | |
| PCT/US2019/037492 WO2019245974A1 (en) | 2018-06-21 | 2019-06-17 | Solid forms of 3-((1r,3r)-1-(2,6-difluoro-4-((1-(3-fluoropropyl)azetidin-3-yl)amino)phenyl)-3-methyl-1,3,4,9-tetrahydro-2h-pyrido[3,4-b]indol-2-yl)-2,2-difluoropropan-1-ol and processes for preparing fused tricyclic compounds comprising a substituted phenyl or pyridinyl moiety, including methods of their use |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2951829T3 true ES2951829T3 (es) | 2023-10-25 |
Family
ID=67441580
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19745335T Active ES2951829T3 (es) | 2018-06-21 | 2019-06-17 | Formas sólidas de la sal de tartrato de 3-((1R,3R)-1-(2,6-difluoro-4-((1-(3-fluoropropil)acetidin-3-il)amino)fenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol, proceso para su preparación y procedimientos de su uso en el tratamiento de cánceres |
Country Status (29)
| Country | Link |
|---|---|
| US (3) | US10954234B2 (es) |
| EP (2) | EP4295845A3 (es) |
| JP (4) | JP6916969B1 (es) |
| KR (3) | KR102564647B1 (es) |
| CN (4) | CN118515596A (es) |
| AU (3) | AU2019290545B2 (es) |
| BR (1) | BR112020024073A2 (es) |
| CA (1) | CA3098203A1 (es) |
| CL (1) | CL2020003022A1 (es) |
| CO (1) | CO2020014604A2 (es) |
| CR (2) | CR20230409A (es) |
| DK (1) | DK3810283T3 (es) |
| ES (1) | ES2951829T3 (es) |
| FI (1) | FI3810283T3 (es) |
| HR (1) | HRP20230859T1 (es) |
| HU (1) | HUE064542T2 (es) |
| IL (1) | IL278063A (es) |
| LT (1) | LT3810283T (es) |
| MX (3) | MX2020011531A (es) |
| PE (1) | PE20211207A1 (es) |
| PH (1) | PH12020500676A1 (es) |
| PL (1) | PL3810283T3 (es) |
| PT (1) | PT3810283T (es) |
| RS (1) | RS64429B1 (es) |
| SG (1) | SG11202012171SA (es) |
| SI (1) | SI3810283T1 (es) |
| TW (1) | TWI846704B (es) |
| UA (1) | UA128114C2 (es) |
| WO (1) | WO2019245974A1 (es) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| UA128114C2 (uk) | 2018-06-21 | 2024-04-10 | Ф. Хоффманн-Ля Рош Аг | Тверді форми 3-((1r,3r)-1-(2,6-дифтор-4-((1-(3-фторпропіл)азетидин-3-іл)аміно)феніл)-3-метил-1,3,4,9-тетрагідро-2h-піридo[3,4-b]індол-2-іл)-2,2-дифторпропан-1-олу і способи одержання конденсованих трициклічних сполук, що містять заміщене фенільне або піридинільне угруповання, в тому числі способи їх застосування |
| CA3169679A1 (en) | 2020-03-06 | 2021-09-10 | Olema Pharmaceuticals, Inc. | Methods of treating estrogen receptor-associated diseases |
| WO2021226263A1 (en) * | 2020-05-07 | 2021-11-11 | Recurium Ip Holdings, Llc | Combinations |
| KR20230009390A (ko) * | 2020-05-12 | 2023-01-17 | 제넨테크, 인크. | Gdc-9545 및 cdk4/6 억제제를 포함하는 병용 요법을 사용한 유방암 치료 |
| AU2021269612A1 (en) * | 2020-05-15 | 2023-01-05 | Jiangsu Simcere Pharmaceutical Co., Ltd. | Pyrrolidine compound and use thereof |
| CN115916769B (zh) | 2020-06-28 | 2024-09-10 | 深圳扬厉医药技术有限公司 | 并环吲唑类化合物 |
| PE20230853A1 (es) * | 2020-06-30 | 2023-05-29 | Genentech Inc | Procesos para elaborar compuestos serd triciclicos que tienen un resto sustituido de fenilo o piridinilo |
| EP4267578A1 (en) | 2020-12-23 | 2023-11-01 | Recurium IP Holdings, LLC | Estrogen receptor modulators |
| EP4294394A1 (en) * | 2021-02-16 | 2023-12-27 | Genentech, Inc. | Treatment of breast cancer using combination therapies comprising gdc-9545 and abemaciclib or ribociclib |
| CN116847839A (zh) * | 2021-02-16 | 2023-10-03 | 基因泰克公司 | 使用包括gdc-9545和依帕他赛的组合疗法治疗乳腺癌 |
| CN117813288A (zh) | 2021-07-15 | 2024-04-02 | 西藏海思科制药有限公司 | 芳氨基衍生物雌激素受体调节剂及其用途 |
| WO2023125700A1 (zh) | 2021-12-28 | 2023-07-06 | 南京明德新药研发有限公司 | 四氢环庚吲唑化合物的盐型、晶型 |
| CN114656444A (zh) * | 2022-03-04 | 2022-06-24 | 河北松辰医药科技有限公司 | 一种2,2-二氟-1,3-丙基磺酸内酯的合成方法 |
| WO2023209062A1 (en) | 2022-04-28 | 2023-11-02 | F. Hoffmann-La Roche Ag | Solid form of 3-((1r,3r)-1-(2,6-difluoro-4-((1-(3- fluoropropyl)azetidin-3-yl)amino)phenyl)-3-methyl-1,3,4,9- tetrahydro-2h-pyrido[3,4-b]indol-2-yl)-2,2-difluoropropan-1-ol tartrate |
| CN115872996B (zh) * | 2023-02-21 | 2023-05-05 | 山东绿叶制药有限公司 | 一种雌激素受体降解剂化合物及其制备方法和应用 |
Family Cites Families (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SE7414572L (es) | 1973-12-06 | 1975-06-09 | Endo Lab | |
| SE455604B (sv) | 1982-06-29 | 1988-07-25 | Stiftelsen Ind Organisk Elektr | Sett att framstella fenetylaminer medelst elektrokemisk reduktion |
| DE3639329C1 (de) | 1986-11-18 | 1988-02-25 | Heesemann Karl Masch | Bandschleifmaschine |
| US5206377A (en) | 1991-12-05 | 1993-04-27 | Whitby Research, Inc. | Compounds useful as antiproliferative agents |
| EP0620222A3 (en) | 1993-04-14 | 1995-04-12 | Lilly Co Eli | Tetrahydro-beta-carbolines. |
| RU14873U1 (ru) | 2000-06-26 | 2000-09-10 | Закрытое акционерное общество "Патентные услуги" | Комбинированная машина непрерывного литья заготовок |
| WO2002062339A1 (en) | 2000-12-08 | 2002-08-15 | Smithkline Beecham Corporation | Antibacterial compounds |
| WO2002064590A2 (en) | 2001-02-12 | 2002-08-22 | Lilly Icos Llc | Carboline derivatives |
| US6951961B2 (en) | 2002-05-17 | 2005-10-04 | Marina Nikolaevna Protopopova | Methods of use and compositions for the diagnosis and treatment of infectious disease |
| TWI329111B (en) | 2002-05-24 | 2010-08-21 | X Ceptor Therapeutics Inc | Azepinoindole and pyridoindole derivatives as pharmaceutical agents |
| JP4629657B2 (ja) | 2003-04-11 | 2011-02-09 | ハイ・ポイント・ファーマスーティカルズ、エルエルシー | 11β−ヒドロキシステロイドデヒドロゲナーゼ1型化活性化合物 |
| WO2005034857A2 (en) | 2003-09-05 | 2005-04-21 | Sequella, Inc. | Methods and compositions comprising diamines as new anti-tubercular therapeutics |
| BRPI0508540B8 (pt) | 2004-03-11 | 2021-05-25 | Actelion Pharmaceuticals Ltd | composto, composição farmacêutica, e, uso de um composto |
| US8076352B2 (en) | 2004-03-15 | 2011-12-13 | Ptc Therapeutics, Inc. | Administration of carboline derivatives useful in the treatment of cancer and other diseases |
| SG184755A1 (en) | 2004-03-15 | 2012-10-30 | Ptc Therapeutics Inc | Carboline derivatives useful in the inhibition of angiogenesis |
| US7767689B2 (en) | 2004-03-15 | 2010-08-03 | Ptc Therapeutics, Inc. | Carboline derivatives useful in the treatment of cancer |
| US8076353B2 (en) | 2004-03-15 | 2011-12-13 | Ptc Therapeutics, Inc. | Inhibition of VEGF translation |
| WO2006015035A1 (en) | 2004-08-02 | 2006-02-09 | Smithkline Beecham Corporation | Useful compounds for hpv infection |
| WO2007002051A1 (en) | 2005-06-22 | 2007-01-04 | Smithkline Beecham Corporation | Carboline derivatives and their use as inhibitors of flaviviridae infections |
| WO2007077570A1 (en) | 2006-01-02 | 2007-07-12 | Hetero Drugs Limited | Process for obtaining pure oseltamivir |
| WO2008127714A1 (en) | 2007-04-13 | 2008-10-23 | Ptc Therapeutics, Inc. | Administration of carboline derivatives useful in the treatment of cancer and other diseases |
| BRPI0911577B8 (pt) | 2008-04-29 | 2021-05-25 | Novartis Ag | derivados espiro-indol, seus usos no tratamento de doenças parasíticas, e composição farmacêutica |
| US9292307B2 (en) | 2008-07-30 | 2016-03-22 | Kyocera Corporation | User interface generation apparatus |
| US20110195929A1 (en) | 2008-08-05 | 2011-08-11 | Summit Corporation Plc | Compounds for the treatment of flaviviral infections |
| WO2010015816A2 (en) | 2008-08-06 | 2010-02-11 | Summit Corporation Plc | Treatment of lysosomal storage disorders and other proteostatic diseases |
| WO2010029313A1 (en) | 2008-09-11 | 2010-03-18 | Summit Corporation Plc. | Antiinfective compounds |
| WO2010049678A2 (en) | 2008-10-31 | 2010-05-06 | Summit Corporation Plc | Treatment of energy utilization diseases |
| US20120040916A1 (en) | 2008-12-22 | 2012-02-16 | Massachusetts Institute Of Technology | Molecular inhibitors of the wnt/beta-catenin pathway |
| WO2010075286A1 (en) | 2008-12-24 | 2010-07-01 | University Of Washington | MOLECULAR ACTIVATORS OF THE Wnt/β-CATENIN PATHWAY |
| DE102009006545B4 (de) | 2009-01-29 | 2017-08-17 | Epcos Ag | Überspannungsableiter und Anordnung von mehreren Überspannungsableitern zu einem Array |
| US20120135089A1 (en) | 2009-03-17 | 2012-05-31 | Stockwell Brent R | E3 ligase inhibitors |
| EP2414044A1 (en) | 2009-03-31 | 2012-02-08 | ArQule, Inc. | Substituted indolo-piperidine compounds |
| WO2010138706A1 (en) | 2009-05-27 | 2010-12-02 | Ptc Therapeutics, Inc. | Methods for treating breast cancer |
| WO2010138659A1 (en) | 2009-05-27 | 2010-12-02 | Ptc Therapeutics, Inc. | Methods for treating brain tumors |
| US8697662B2 (en) | 2009-05-27 | 2014-04-15 | Ptc Therapeutics, Inc. | Methods for treating Kaposi sarcoma |
| WO2010138685A1 (en) | 2009-05-27 | 2010-12-02 | Ptc Therapeutics, Inc. | Methods for treating prostate conditions |
| CA3080983C (en) | 2009-05-27 | 2023-02-28 | Pct Therapeutics, Inc. | Method of inhibiting and reducing a viral infection |
| US20120157401A1 (en) | 2009-05-27 | 2012-06-21 | Ptc Therapeutics, Inc. | Methods for treating neurofibromatosis |
| US20130171103A1 (en) | 2010-05-27 | 2013-07-04 | Ptc Therapeutics, Inc. | Methods for treating viral conditions |
| ES2627692T3 (es) | 2010-06-10 | 2017-07-31 | Aragon Pharmaceuticals, Inc. | Moduladores de receptores de estrógenos y usos de los mismos |
| WO2011159769A2 (en) | 2010-06-17 | 2011-12-22 | Aragon Pharmaceuticals, Inc. | Indane estrogen receptor modulators and uses thereof |
| CA2819299A1 (en) | 2010-12-24 | 2012-06-28 | Merck Sharp & Dohme B.V. | N-substituted azetidine derivatives |
| WO2013090829A1 (en) | 2011-12-14 | 2013-06-20 | Aragon Pharmaceuticals, Inc. | Estrogen receptor modulators and uses thereof |
| CN104508126B (zh) | 2012-03-23 | 2017-06-30 | 科德克希思公司 | 用于合成色胺和色胺类似物的衍生物的生物催化剂和方法 |
| PL2828256T3 (pl) | 2012-03-23 | 2019-12-31 | Novartis Ag | Chemiczny sposób wytwarzania spiroindolonów i ich związków pośrednich |
| EP2682119A1 (en) | 2012-07-03 | 2014-01-08 | Université Libre de Bruxelles | Aromatic N-heterocycle derivatives for use as medicine |
| EP2738173A1 (en) | 2012-11-28 | 2014-06-04 | Commissariat A L'energie Atomique Et Aux Energies Alternatives | Heterocyclic compounds as inhibitors of the sodium iodide symporter |
| LT2997033T (lt) | 2013-05-17 | 2018-02-12 | Merck Sharp & Dohme Corp. | Kondensuoti tricikliniai heterocikliniai junginiai, kaip živ integrazės inhibitoriai |
| WO2014191726A1 (en) | 2013-05-28 | 2014-12-04 | Astrazeneca Ab | Chemical compounds |
| RU2015152171A (ru) | 2013-06-19 | 2017-07-25 | Серагон Фармасьютикалз, Инк. | Азетидиновые модуляторы эстрогеновых рецепторов и их применения |
| EA031077B1 (ru) | 2013-06-19 | 2018-11-30 | Серагон Фармасьютикалз, Инк. | Модулятор рецептора эстрогена и его применения |
| WO2015039348A1 (en) | 2013-09-23 | 2015-03-26 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds useful as hiv integrase inhibitors |
| AU2014342520B2 (en) | 2013-10-28 | 2019-08-08 | Drexel University | Novel treatments for attention and cognitive disorders, and for dementia associated with a neurodegenerative disorder |
| WO2015082990A1 (en) | 2013-12-06 | 2015-06-11 | Seragon Pharmaceuticals, Inc. | Estrogen receptor modulator for the treatment of locally advanced or metastatic estrogen receptor positive breast cancer |
| JP2017509631A (ja) | 2014-03-13 | 2017-04-06 | エフ・ホフマン−ラ・ロシュ・アクチェンゲゼルシャフト | エストロゲン受容体突然変異体を調節する方法及び組成物 |
| KR20160124909A (ko) | 2014-03-13 | 2016-10-28 | 에프. 호프만-라 로슈 아게 | 에스트로겐 수용체 조절제를 함유하는 치료 조합물 |
| EP3160964B1 (en) | 2014-06-27 | 2024-03-13 | Nogra Pharma Limited | Aryl receptor modulators and methods of making and using the same |
| EP3233818A1 (en) | 2014-12-18 | 2017-10-25 | F. Hoffmann-La Roche AG | Derivatives of 2,3-diphenylchromene useful for the treatment of cancer |
| BR112017007662B1 (pt) * | 2014-12-18 | 2021-11-03 | F. Hoffmann-La Roche Ag | Composto, composição farmacêutica e uso de um composto |
| JP6807841B2 (ja) | 2014-12-18 | 2021-01-06 | エフ・ホフマン−ラ・ロシュ・アクチェンゲゼルシャフト | エストロゲン受容体モジュレーター及びその使用 |
| UA122346C2 (uk) | 2015-10-01 | 2020-10-26 | Олема Фармасьютикалз, Інк. | ТЕТРАГІДРО-1H-ПІРИДО[3,4-b]ІНДОЛЬНІ АНТИЕСТРОГЕННІ ЛІКАРСЬКІ ПРЕПАРАТИ |
| EP3412667B1 (en) * | 2016-02-01 | 2021-11-03 | Takeda Pharmaceutical Company Limited | Cocrystal |
| MX2018009496A (es) | 2016-02-05 | 2018-12-11 | Inventisbio Inc | Degradadores selectivos del receptor de estrogeno y sus usos. |
| WO2017216279A1 (en) * | 2016-06-16 | 2017-12-21 | F. Hoffmann-La Roche Ag | Heteroaryl estrogen receptor modulators and uses thereof |
| JP2019521983A (ja) * | 2016-06-16 | 2019-08-08 | エフ・ホフマン−ラ・ロシュ・アクチェンゲゼルシャフト | テトラヒドロ−ピリド[3,4−b]インドールエストロゲン受容体モジュレーター及びその使用 |
| UA128114C2 (uk) | 2018-06-21 | 2024-04-10 | Ф. Хоффманн-Ля Рош Аг | Тверді форми 3-((1r,3r)-1-(2,6-дифтор-4-((1-(3-фторпропіл)азетидин-3-іл)аміно)феніл)-3-метил-1,3,4,9-тетрагідро-2h-піридo[3,4-b]індол-2-іл)-2,2-дифторпропан-1-олу і способи одержання конденсованих трициклічних сполук, що містять заміщене фенільне або піридинільне угруповання, в тому числі способи їх застосування |
-
2019
- 2019-06-17 UA UAA202100182A patent/UA128114C2/uk unknown
- 2019-06-17 AU AU2019290545A patent/AU2019290545B2/en active Active
- 2019-06-17 CN CN202410165685.9A patent/CN118515596A/zh active Pending
- 2019-06-17 DK DK19745335.0T patent/DK3810283T3/da active
- 2019-06-17 WO PCT/US2019/037492 patent/WO2019245974A1/en active Application Filing
- 2019-06-17 PT PT197453350T patent/PT3810283T/pt unknown
- 2019-06-17 CR CR20230409A patent/CR20230409A/es unknown
- 2019-06-17 KR KR1020217001924A patent/KR102564647B1/ko active IP Right Grant
- 2019-06-17 EP EP23178559.3A patent/EP4295845A3/en active Pending
- 2019-06-17 SI SI201930592T patent/SI3810283T1/sl unknown
- 2019-06-17 CR CR20200621A patent/CR20200621A/es unknown
- 2019-06-17 SG SG11202012171SA patent/SG11202012171SA/en unknown
- 2019-06-17 MX MX2020011531A patent/MX2020011531A/es unknown
- 2019-06-17 TW TW108120952A patent/TWI846704B/zh active
- 2019-06-17 EP EP19745335.0A patent/EP3810283B1/en active Active
- 2019-06-17 PE PE2020001888A patent/PE20211207A1/es unknown
- 2019-06-17 KR KR1020247022192A patent/KR20240113583A/ko active Search and Examination
- 2019-06-17 LT LTEPPCT/US2019/037492T patent/LT3810283T/lt unknown
- 2019-06-17 RS RS20230634A patent/RS64429B1/sr unknown
- 2019-06-17 CN CN202410165649.2A patent/CN117946107A/zh active Pending
- 2019-06-17 HU HUE19745335A patent/HUE064542T2/hu unknown
- 2019-06-17 JP JP2020570698A patent/JP6916969B1/ja active Active
- 2019-06-17 ES ES19745335T patent/ES2951829T3/es active Active
- 2019-06-17 KR KR1020237026161A patent/KR102682985B1/ko active IP Right Grant
- 2019-06-17 CN CN202410165668.5A patent/CN118239948A/zh active Pending
- 2019-06-17 PL PL19745335.0T patent/PL3810283T3/pl unknown
- 2019-06-17 FI FIEP19745335.0T patent/FI3810283T3/fi active
- 2019-06-17 US US16/443,515 patent/US10954234B2/en active Active
- 2019-06-17 BR BR112020024073-7A patent/BR112020024073A2/pt unknown
- 2019-06-17 HR HRP20230859TT patent/HRP20230859T1/hr unknown
- 2019-06-17 CA CA3098203A patent/CA3098203A1/en active Pending
- 2019-06-17 CN CN201980041221.4A patent/CN112437685B/zh active Active
-
2020
- 2020-10-15 IL IL278063A patent/IL278063A/en unknown
- 2020-10-29 MX MX2022016065A patent/MX2022016065A/es unknown
- 2020-10-29 MX MX2023006785A patent/MX2023006785A/es unknown
- 2020-11-20 CL CL2020003022A patent/CL2020003022A1/es unknown
- 2020-11-25 CO CONC2020/0014604A patent/CO2020014604A2/es unknown
- 2020-12-03 US US17/110,607 patent/US11780834B2/en active Active
- 2020-12-17 PH PH12020500676A patent/PH12020500676A1/en unknown
-
2021
- 2021-07-16 JP JP2021117638A patent/JP2021191751A/ja active Pending
- 2021-07-16 JP JP2021117633A patent/JP2021191750A/ja active Pending
-
2022
- 2022-05-03 AU AU2022202967A patent/AU2022202967B2/en active Active
-
2023
- 2023-08-25 US US18/455,783 patent/US20240317738A1/en active Pending
- 2023-09-13 AU AU2023229513A patent/AU2023229513A1/en active Pending
-
2024
- 2024-01-11 JP JP2024002374A patent/JP2024054111A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2951829T3 (es) | Formas sólidas de la sal de tartrato de 3-((1R,3R)-1-(2,6-difluoro-4-((1-(3-fluoropropil)acetidin-3-il)amino)fenil)-3-metil-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indol-2-il)-2,2-difluoropropan-1-ol, proceso para su preparación y procedimientos de su uso en el tratamiento de cánceres | |
| ES2944937T3 (es) | Tienopirroles en calidad de inhibidores de la histona desmetilasa | |
| KR102480074B1 (ko) | 설폰아마이드 화합물 및 이의 용도 | |
| WO2015196071A1 (en) | Compounds, compositions and methods of increasing cftr activity | |
| TW202341983A (zh) | 用於降解突變kras蛋白之化合物及其應用 | |
| US20170210769A1 (en) | Th-302 solid forms and methods related thereto | |
| RU2809220C2 (ru) | ТАРТРАТ 3-((1R,3R)-1-(2,6-ДИФТОР-4-((1-(3-ФТОРПРОПИЛ)АЗЕТИДИН-3-ИЛ)АМИНО)ФЕНИЛ)-3-МЕТИЛ-1,3,4,9-ТЕТРАГИДРО-2H-ПИРИДО[3,4-b]ИНДОЛ-2-ИЛ)-2,2-ДИФТОРПРОПАН-1-ОЛА, ЕГО ТВЕРДЫЕ ФОРМЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | |
| JP2009517483A (ja) | 一酸化窒素シンターゼの二量化の抑制剤としてのイミダゾール誘導体 | |
| BR122024013536A2 (pt) | Processos para preparar compostos de fórmula (viii) | |
| BR122024013557A2 (pt) | Processos para preparar compostos de fórmulas (iv), (iii), (xx) e (ix), e composto de fórmula (xvi) |