ES2875899T3 - Compuestos inhibidores de autotaxina - Google Patents
Compuestos inhibidores de autotaxina Download PDFInfo
- Publication number
- ES2875899T3 ES2875899T3 ES14864293T ES14864293T ES2875899T3 ES 2875899 T3 ES2875899 T3 ES 2875899T3 ES 14864293 T ES14864293 T ES 14864293T ES 14864293 T ES14864293 T ES 14864293T ES 2875899 T3 ES2875899 T3 ES 2875899T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- thio
- acid
- fluoro
- substituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 Cc(cc1)c(*)c2c1c(*)c(*)[n]2 Chemical compound Cc(cc1)c(*)c2c1c(*)c(*)[n]2 0.000 description 9
- ODBMLKPFRGSZSG-UHFFFAOYSA-N FC(C[n]1ncc(Br)c1)(F)F Chemical compound FC(C[n]1ncc(Br)c1)(F)F ODBMLKPFRGSZSG-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/4155—1,2-Diazoles non condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0014—Skin, i.e. galenical aspects of topical compositions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/06—Ointments; Bases therefor; Other semi-solid forms, e.g. creams, sticks, gels
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/08—Solutions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/10—Spiro-condensed systems
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Dermatology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Physiology (AREA)
- Nutrition Science (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
Un compuesto de Fórmula (III), o una sal o un solvato farmacéuticamente aceptables del mismo: **(Ver fórmula)** en la que, R1 es -Cl, -Br o -CN; R2 es Cl; R3 es H, F o Cl; W es CH, CF o N; RA es H; L1 está ausente, es alquileno C1-C6 o cicloalquileno C3-C6; Q es -CO2H o -CO2(alquilo C1-C6); el Anillo B es furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo, tiadiazolilo, piridinilo, pirimidinilo, pirazinilo, piridazinilo o triazinilo; cada RB es independientemente H, halógeno, -CN, -NO2, -OH, -OR9, -SR9, -S(=O)R9, -S(=O)2R9, -S(=O)2N(R10)2, alquilo C1-C6, fluoroalquilo C1-C6, deuteroalquilo C1-C6, heteroalquilo C1-C6, cicloalquilo C3-C10 sustituido o sin sustituir, heterocicloalquilo C2-C10 sustituido o sin sustituir, fenilo sustituido o sin sustituir, alquileno C1-C4-(fenilo sustituido o sin sustituir), heteroarilo monocíclico sustituido o sin sustituir, alquileno C1-C4-(heteroarilo monocíclico sustituido o sin sustituir), un heteroarilo bicíclico sustituido o sin sustituir o alquileno C1-C4- (heteroarilo bicíclico sustituido o sin sustituir); n es 0, 1 o 2; R9 es alquilo C1-C6, fluoroalquilo C1-C6, deuteroalquilo C1-C6, cicloalquilo C3-C6, un fenilo sustituido o sin sustituir, un heteroarilo monocíclico sustituido o sin sustituir o un heteroarilo bicíclico sustituido o sin sustituir; cada R10 es independientemente H, alquilo C1-C6, fluoroalquilo C1-C6, deuteroalquilo C1-C6, cicloalquilo C3-C6, un fenilo sustituido o sin sustituir o un heteroarilo monocíclico sustituido o sin sustituir; o dos grupos R10 unidos al mismo átomo de N se toman junto con el átomo de N al que están unidos para formar un heterociclo sustituido o sin sustituir; en donde los grupos sustituidos están sustituidos por uno o más grupos seleccionados individual e independientemente de halógeno, -CN, -NH2, -OH, -NH(CH3), -N(CH3)2, -CH3, -CH2CH3, -CF3, -OCH3 y -OCF3; y en donde el término "alquilo" solo o en "heteroalquilo" también se refiere a un tipo de grupos alquilo en el que está presente al menos un doble enlace carbono-carbono.
Description
DESCRIPCIÓN
Compuestos inhibidores de autotaxina
Campo de la invención
En el presente documento se describen compuestos que son inhibidores de autotaxina, métodos para preparar dichos compuestos, composiciones farmacéuticas y medicamentos que comprenden dichos compuestos y métodos para usar dichos compuestos en el tratamiento de afecciones, enfermedades o trastornos asociados a la actividad de la autotaxina.
Antecedentes
El ácido lisofosfatídico (LPA) es un mediador lipídico que funciona, por ejemplo, como mitógeno, quimioatrayente y factor de supervivencia para muchos tipos de células. La señalización de LPA está implicada, por ejemplo, en el cáncer y enfermedades fibróticas.
El documento WO 2012/166415 A1 (Amira Pharmaceuticals, Inc., 6 de diciembre de 2012) describe determinados compuestos de la siguiente fórmula que supuestamente inhiben la autotaxina y que supuestamente pueden usarse para tratar, prevenir o diagnosticar enfermedades, trastornos y afecciones asociados a la actividad de la autotaxina.

El documento WO 2012/024620 A2 (Amira Pharmaceuticals, Inc., 23 de febrero de 2012) describe determinados compuestos de la siguiente fórmula que supuestamente inhiben la autotaxina y que supuestamente pueden usarse para tratar, prevenir o diagnosticar enfermedades, trastornos y afecciones asociados a la actividad de la autotaxina.
Barbayianni et al., "Autotaxin inhibitors: a patent review", Expert Opin. Ther. Patents, 2013, Vol. 23, N.° 9, págs.
1123-1132, proporciona una revisión que resume los inhibidores de ATX informados en las patentes de 2006 a 2013, que describe su descubrimiento y evaluación biológica.
Albers et al., "Chemical Evolution of Autotaxin Inhibitors", Chem. Rev., 2012, Vol. 112, págs. 2593-2603, proporciona una revisión de la autotaxina y sus propiedades y función, y algunos inhibidores de la misma.
Sumario de la invención
Un primer aspecto de la invención es un compuesto de Fórmula (III), o una sal o solvato farmacéuticamente aceptable del mismo:

en la que,
R1 es -Cl, -Br o -CN;
R2 es Cl;
R3 es H, F o Cl;
W es CH, CF o N;
RA es H;
L1 está ausente, alquileno C1-C6 o cicloalquileno C3-C6 ;
Q es -CO2H o -CO2(alquilo C1-C6);
el Anillo B es furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo, tiadiazolilo, piridinilo, pirimidinilo, pirazinilo, piridazinilo o triazinilo;
cada RB es independientemente H, halógeno, -CN, -NO2, -OH, -OR9, -SR9, -S(=O)R9, -S(=O)2R9, -S(=O)2N(R10)2, alquilo C1-C6 , fluoroalquilo C1-C6, deuteroalquilo C1-C6, heteroalquilo C1-C6, cicloalquilo C3-C10 sustituido o sin sustituir, heterocicloalquilo C2-C10 sustituido o sin sustituir, fenilo sustituido o sin sustituir, alquileno C1-C4-(fenilo sustituido o sin sustituir), heteroarilo monocíclico sustituido o sin sustituir, alquileno C1-C4-(heteroarilo monocíclico sustituido o sin sustituir), un heteroarilo bicíclico sustituido o sin sustituir o alquileno C1-C4-(heteroarilo bicíclico sustituido o sin sustituir);
n es 0, 1 o 2;
R9 es alquilo C1-C6, fluoroalquilo C1-C6, deuteroalquilo C1-C6 , cicloalquilo C3-C6 , un fenilo sustituido o sin sustituir, un heteroarilo monocíclico sustituido o sin sustituir o un heteroarilo bicíclico sustituido o sin sustituir;
cada R10 es independientemente H, alquilo C1-C6 , fluoroalquilo C1-C6, deuteroalquilo C1-C6, cicloalquilo C3-C6, un fenilo sustituido o sin sustituir o un heteroarilo monocíclico sustituido o sin sustituir; o
dos grupos R10 unidos al mismo átomo de N se toman junto con el átomo de N al que están unidos para formar un heterociclo sustituido o sin sustituir;
en la que los grupos sustituidos están sustituidos por uno o más grupos seleccionados individual e independientemente de halógeno, halógeno, -CN, -NH2 , - O h , -NH(CH3), -N(CH3)2, -CH3, -CH2CH3 , -CF3, -OCH3 y -OCF3 ; y
en la que el término "alquilo" solo o en "heteroalquilo" también se refiere a un tipo de grupos alquilo en el que está presente al menos un doble enlace carbono-carbono.
En una realización, L1 está ausente, -CH2-, -CH(CH3)-, -C(CH3)2- o ciclopropil-1,1-diilo.
En una realización, el compuesto tiene la siguiente estructura:

en la que,
RA es H;
RB es H, alquilo C1-C6 , fluoroalquilo C1-C6 o deuteroalquilo C1-C6 ;
R1 es -Cl, -Br o -CN;
R2 es Cl; y
R3 es H, F o Cl;
o una sal o solvato farmacéuticamente aceptable del mismo.
En una realización, el compuesto tiene la siguiente estructura:

o una sal o solvato farmacéuticamente aceptable del mismo.
En una realización, RB es alquilo C1-C6.
En una realización, L1 está ausente, -CH2-, -CH(CH3)-, -C(CH3)2- o ciclopropil-1,1-diilo.
En una realización, L1 está ausente.
En una realización, el compuesto es:
ácido 3-((2,6-dicloro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico;
ácido 3-((6-cloro-2-ciano-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico;
ácido 3-((2,6-dicloro-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico;
ácido 3-((2-bromo-6-cloro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico;
ácido 3-((2,6-dicloro-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico;
ácido 3-((2,6-dicloro-7-fluoro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico;
ácido 3-((2,6-dicloro-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico;
ácido 3-((2,6-dicloro-7-fluoro-1-(piridin-3-il)-1H-indol-3-il)tio)-2-fluorobenzoico;
ácido 3-((2-bromo-6-cloro-7-fluoro-1-(piridin-3-il)-1H-indol-3-il)tio)-2-fluorobenzoico;
o una sal o solvato farmacéuticamente aceptable de los mismos.
En una realización, el compuesto es:
ácido 3-((2,6-dicloro-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico;
ácido 3-((2,6-dicloro-7-fluoro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico;
ácido 3-((2,6-dicloro-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico;
o una sal o solvato farmacéuticamente aceptable de los mismos.
En una realización, el compuesto es:
ácido 3-((2,6-dicloro-7-fluoro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico;
o una sal o solvato farmacéuticamente aceptable del mismo.
En una realización, el compuesto es:
ácido 3-((2,6-dicloro-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico;
o una sal o solvato farmacéuticamente aceptable del mismo.
Un segundo aspecto de la invención es una composición farmacéutica que comprende un compuesto, sal o solvato del primer aspecto y al menos un excipiente farmacéuticamente aceptable.
En una realización, la composición farmacéutica se encuentra en forma de un comprimido, una píldora, una cápsula, un líquido, una suspensión, un gel, una dispersión, una solución, una emulsión, un ungüento o una loción.
Un tercer aspecto de la invención es un compuesto, sal o solvato del primer aspecto para su uso en un método para el tratamiento de un mamífero mediante terapia, o para su uso en un método de tratamiento de fibrosis o cáncer en un mamífero.
Descripción detallada
Los compuestos descritos en el presente documento son inhibidores de autotaxina (ATX). En algunas realizaciones, los inhibidores de autotaxina descritos en el presente documento son útiles como agentes para el tratamiento o la prevención de enfermedades o afecciones en las que ATX y/o LPA participan, están implicadas en la etiología o patología de la enfermedad o están asociadas de otro modo a al menos un síntoma de la enfermedad. La inhibición de la actividad fisiológica de ATX y/o LPA es útil en diversas enfermedades o afecciones. La vía de señalización ATX-LPA se ha relacionado con enfermedades fibróticas y cáncer.
Los compuestos descritos en el presente documento se usan en el tratamiento de enfermedades o afecciones en las que la actividad de la autotaxina contribuye a la sintomatología o progresión de la enfermedad, trastorno o afección. En algunas realizaciones, los métodos, compuestos, composiciones farmacéuticas y medicamentos descritos en el presente documento comprenden inhibidores de autotaxina.
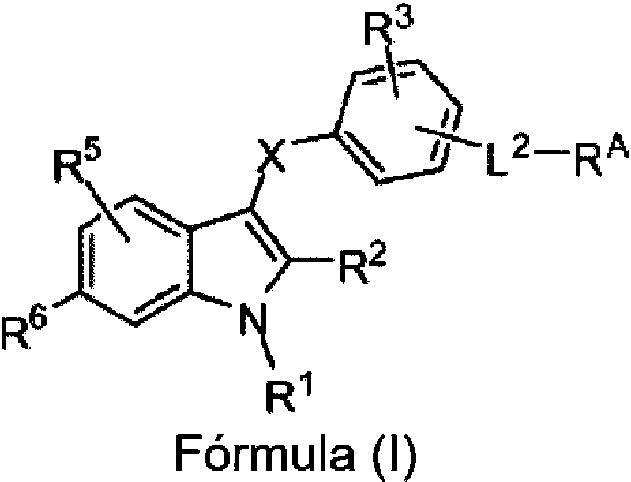
En el presente documento se describe un compuesto de Fórmula (I), o una sal o solvato farmacéuticamente aceptable del mismo:

en la que,
R1 es -F, -Cl, -Br, -CN, vinilo, cicloalquilo C3-C6, -NH2 , -NH(alquilo C1-C4), -N(alquilo C1-C4)2 , -O-alquilo C1-C4 o -S-alquilo C1-C4 ;
R2 es H, halógeno, -CN, -NO2 , -OH, -OR9, -SR9, -S(=O)R9, -S(=O)2R9, -S(=O)2N(R10)2, -NR10S(=O)2R9, -C(=O)R9, -OC(=O)R9, -CO2R10, -OCO2R9 , -N(R10)2 , -C(=O)N(R10)2, -OC(=O)N(R10)2, -NHC(=O)R9, -NHC(=O)OR9, alquilo C1-C4 , fluoroalquilo C1-C4, deuteroalquilo C1-C4 , hidroxialquilo C1-C4, heteroalquilo C1-C4, cicloalquilo C3-C6 , fenilo sustituido o sin sustituir o heteroarilo monocíclico sustituido o sin sustituir;
el Anillo A es un arilo monocíclico, arilo bicíclico, heterocicloalquilo monocíclico, heteroarilo monocíclico o heteroarilo bicíclico;
cada RA es H, halógeno, -CN, -NO2, -OH, -OR9, -SR9, -S(=O)R9, -S(=O)2R9, -S(=O)2N(R10)2 , -NR10S(=O)2R9, -C(=O)R9, -OC(=O)R9, -CO2R10, -OCO2R9, -N(R10)2, -C(=O)N(R10)2, -OC(=O)N(R10)2, -NHC(=O)R9, -NHC(=O)OR9, alquilo C1-C6, fluoroalquilo C1-C6 , deuteroalquilo C1-C6, heteroalquilo C1-C6, cicloalquilo C3-C10 sustituido o sin sustituir, heterocicloalquilo C2-C10 sustituido o sin sustituir, fenilo sustituido o sin sustituir o heteroarilo monocíclico sustituido o sin sustituir;
m es 0, 1 o 2;
L1 está ausente, alquileno C1-C6, fluoroalquileno C1-C6 o cicloalquileno C3-C6;
Q es -CO2H, -CO2(alquilo C1-C6), -OH, -CN, -B(OH)2 , -C(=O)NHSO2R9, -C(=O)N(R10)2, -SO2NHC(=O)R9, -CN, tetrazolilo, -OP(=O)(o H)2 , -P(=O)(OH)2 o bioisóstero de ácido carboxílico;
L2 está ausente, alquileno C1-C4 o cicloalquileno C3-C7 ;
L3 es -S-, S(=O), S(=O)2 u -O-;
el Anillo B es un arilo monocíclico, arilo bicíclico, heteroarilo monocíclico o heteroarilo bicíclico;
cada RB es independientemente H, halógeno, -CN, -NO2, -OH, -OR9, -SR9, -S(=O)R9, -S(=O)2R9, -S(=O)2N(R10)2, -NR10S(=O)2R9, -C(=O)R9, -OC(=O)R9, -CO2R10, -OCO2R9, -N(R10)2, -C(=O)N(R10)2 , -OC(=O)N(R10)2 , -NHC(=O)R9, -NHC(=O)OR9, alquilo C1-C6 , fluoroalquilo C1-C6, deuteroalquilo C1-C6, heteroalquilo C1-C6 , cicloalquilo C3-C10 sustituido o sin sustituir, heterocicloalquilo C2-C10 sustituido o sin sustituir, fenilo sustituido o sin sustituir, alquileno C1-C4-(fenilo sustituido o sin sustituir), heteroarilo monocíclico sustituido o sin sustituir, alquileno C1-C4-(heteroarilo monocíclico sustituido o sin sustituir), un heteroarilo bicíclico sustituido o sin sustituir o alquileno C1-C4-(heteroarilo bicíclico sustituido o sin sustituir); n es 0, 1 o 2;
R9 es alquilo C1-C6, fluoroalquilo C1-C6, deuteroalquilo C1-C6, cicloalquilo C3-C6 , un fenilo sustituido o sin sustituir, un heteroarilo monocíclico sustituido o sin sustituir o un heteroarilo bicíclico sustituido o sin sustituir;
cada R10 se selecciona independientemente de H, alquilo C1-C6, fluoroalquilo C1-C6, deuteroalquilo C1-C6, cicloalquilo C3-C6 , un fenilo sustituido o sin sustituir o un heteroarilo monocíclico sustituido o sin sustituir; o dos grupos R10 unidos al mismo átomo de N se toman junto con el átomo de N al que están unidos para formar un heterociclo sustituido o sin sustituir.
Para cualquiera y todas las realizaciones, los sustituyentes se seleccionan de entre un subconjunto de las alternativas enumeradas. Por ejemplo, en algunas realizaciones, X es -O-, -S-, -S(=O)- o -S(=O)2-. En otras realizaciones, X es -O- o -S-. En otras realizaciones, X es -S-, -S(=O)- o -S(=O)2-. En algunas realizaciones, X es -S-
En algunas realizaciones, R1 es -F, -Cl, -Br, -CN, vinilo, ciclopropilo, ciclobutilo, -NH2 , -NH(CH3), -N(CH3)2 , -O-CH3 o -S-CH3.
En algunas realizaciones, R1 es vinilo, ciclopropilo o ciclobutilo
En algunas realizaciones, R1 es ciclopropilo o ciclobutilo
En algunas realizaciones, R1 es -F, -Cl o -Br.
En algunas realizaciones, L2 está ausente o es alquileno C1-C4 ; L3 es -S-, S(=O) o S(=O)2.
En algunas realizaciones, L2 está ausente, -CH2-, -CH2CH2-, -CH2CH2CH2- o -CH(CH3)-.
En algunas realizaciones, L1 está ausente, -CH2-, -CH2CH2-, -CH2CH2CH2-, -CH(CH3)-, -CH(CH2CH3)-, -C(CH3)2-, -C(CH2CH3)2-, ciclopropil-1,1-diilo, ciclobutil-1,1-diilo, ciclopentil-1,1-diilo o ciclohexil-1,1-diilo; Q es -CO2H, -CO2(alquilo C1-C6), -C(=O)NHSO2R9 o tetrazolilo.
En algunas realizaciones, L1 está ausente o -CH2-; Q es -CO2H, o -CO2(alquilo C1-C6).
En algunas realizaciones, el compuesto de Fórmula (I) tiene la siguiente estructura de Fórmula (II):

o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, el Anillo A es fenilo, naftilo, heteroarilo monocíclico que contiene 1-4 átomos de N y 0 o 1 átomos de O o S, heteroarilo monocíclico que contiene 0-4 átomos de N y 1 átomo de O o S, heteroarilo bicíclico que contiene 1-4 átomos de N y 0 o 1 átomos de O o S, o heteroarilo bicíclico que contiene 0-4 átomos de N y 1 átomo de O o S; el Anillo B es fenilo, naftilo, heteroarilo monocíclico que contiene 1-4 átomos de N y 0 o 1 átomos de O o S, heteroarilo monocíclico que contiene 0-4 átomos de N y 1 átomo de O o S, heteroarilo bicíclico que contiene 1-4
átomos de N y 0 o 1 átomos de O o S, o heteroarilo bicíclico que contiene 0-4 átomos de N y 1 átomo de O o S. En algunas realizaciones, el Anillo A es fenilo, naftilo, furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo, tiadiazolilo, piridinilo, pirimidinilo, pirazinilo, piridazinilo, triazinilo, quinolinilo, isoquinolinilo, quinazolinilo, quinoxalinilo, naftiridinilo, indolilo, indazolilo, benzoxazolilo, benzoisoxazolilo, benzofuranilo, benzotienilo, benzotiazolilo, benzoimidazolilo, purinilo, cinolinilo, ftalazinilo, pteridinilo, piridopirimidinilo, pirazolopirimidinilo o azaindolilo.
En algunas realizaciones, el Anillo A es fenilo o naftilo.
En algunas realizaciones, el Anillo A es furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo, tiadiazolilo, piridinilo, pirimidinilo, pirazinilo, piridazinilo o triazinilo. En algunas realizaciones, el Anillo A es piridinilo, pirimidinilo, pirazinilo, piridazinilo o triazinilo.
En algunas realizaciones, el Anillo A es furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo o tiadiazolilo.
En algunas realizaciones, el Anillo A es quinolinilo, isoquinolinilo, quinazolinilo, quinoxalinilo, naftiridinilo, indolilo, indazolilo, benzoxazolilo, benzoisoxazolilo, benzofuranilo, benzotienilo, benzotiazolilo, benzoimidazolilo, purinilo, cinolinilo, ftalazinilo, pteridinilo, piridopirimidinilo, pirazolopirimidinilo o azaindolilo.
En algunas realizaciones, cada RA es H, halógeno, -CN, -OH, -OR9, -SR9, alquilo C1-C6, fluoroalquilo C1-C6, deuteroalquilo C1-C6 , heteroalquilo C1-C6.
En algunas realizaciones, L3 es -S-.
En algunas realizaciones, L2 está ausente.
En algunas realizaciones, el compuesto de Fórmula (I) o Fórmula (II) tiene la siguiente estructura de Fórmula (III):

en la que,
W es CH, CF o N;
o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, L1 está ausente; y Q es -CO2H.
En algunas realizaciones, en el presente documento se describe un compuesto de Fórmula (III), o una sal o solvato farmacéuticamente aceptable del mismo:

en la que,
R1 es -Cl, -Br, -CN o cicloalquilo C3-C6;
R2 es H, halógeno, -CN, -NO2, -OH, -OR9, -SR9, -S(=O)R9, -S(=O)2R9, -S(=O)2N(R10)2, alquilo C1-C4, fluoroalquilo C1-C4 , deuteroalquilo C1-C4 o cicloalquilo C3-C6;
R3 es H, halógeno, -CN, -OH, alquilo C1-C4 , fluoroalquilo C1-C4 , deuteroalquilo C1-C4, alcoxi C1-C4 o fluoroalcoxi C1-C4;
W es CH, CF o N;
cada RA es H, halógeno, -CN, -NO2, -OH, -OR9, -SR9, -S(=O)R9, -S(=O)2R9, -S(=O)2N(R10)2, alquilo C1-C6 o fluoroalquilo C1-C6;
L1 está ausente, alquileno C1-C6 o cicloalquileno C3-C6 ;
Q es -CO2H, -CO2(alquilo C1-C6), -OH, -CN, -B(OH)2 , -C(=O)NHSO2R9, -C(=O)N(R10)2, -SO2NHC(=O)R9, -CN, tetrazolilo, -OP(=O)(o H)2 , -P(=O)(OH)2 o bioisóstero de ácido carboxílico;
el Anillo B es un heteroarilo monocíclico;
cada RB es independientemente H, halógeno, -CN, -NO2, -OH, -OR9, -SR9, -S(=O)R9, -S(=O)2R9, -S(=O)2N(R10)2, alquilo C1-C6, fluoroalquilo C1-C6 , deuteroalquilo C1-C6 , heteroalquilo C1-C6 , cicloalquilo C3-C10 sustituido o sin sustituir, heterocicloalquilo C2-C10 sustituido o sin sustituir, fenilo sustituido o sin sustituir, alquileno C1-C4-(fenilo sustituido o sin sustituir), heteroarilo monocíclico sustituido o sin sustituir, alquileno C1-C4-(heteroarilo monocíclico sustituido o sin sustituir), un heteroarilo bicíclico sustituido o sin sustituir o alquileno C1-C4-(heteroarilo bicíclico sustituido o sin sustituir);
n es 0, 1 o 2;
R9 es alquilo C1-C6, fluoroalquilo C1-C6, deuteroalquilo C1-C6, cicloalquilo C3-C6 , un fenilo sustituido o sin sustituir, un heteroarilo monocíclico sustituido o sin sustituir o un heteroarilo bicíclico sustituido o sin sustituir;
cada R10 es independientemente H, alquilo C1-C6 , fluoroalquilo C1-C6, deuteroalquilo C1-C6, cicloalquilo C3-C6, un fenilo sustituido o sin sustituir o un heteroarilo monocíclico sustituido o sin sustituir; o
dos grupos R10 unidos al mismo átomo de N se toman junto con el átomo de N al que están unidos para formar un heterociclo sustituido o sin sustituir.
En algunas realizaciones, R1 es -Cl, -Br, -CN o ciclopropilo. En algunas realizaciones, R1 es ciclopropilo. En algunas realizaciones, R1 es -Cl.
En algunas realizaciones, L1 está ausente, -CH2-, -CH2CH2-, -CH2CH2CH2-, -CH(CH3)-, -CH(CH2CH3)-, -C(CH3)2-, -C(CH2CH3)2-, ciclopropil-1,1-diilo, ciclobutil-1,1-diilo, ciclopentil-1,1-diilo o ciclohexil-1,1 -diilo; y Q es -CO2H, -CO2(alquilo C1-C6), -C(=O)NHSO2R9 o tetrazolilo.
En algunas realizaciones, L1 está ausente, -CH2-, -CH(CH3)-, -C(CH3)2- o ciclopropil-1,1-diilo; y Q es -CO2H, o -CO2(alquilo C1-C6).
En algunas realizaciones, L1 está ausente o -CH2-; y Q es -CO2H, o -CO2(alquilo C1-C6).
En algunas realizaciones, el Anillo B es heteroarilo monocíclico que contiene 1-4 átomos de N y 0 o 1 átomos de O o S, o heteroarilo monocíclico que contiene 0-4 átomos de N y 1 átomo de O o S.
En algunas realizaciones, el Anillo B es furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo, tiadiazolilo, piridinilo, pirimidinilo, pirazinilo, piridazinilo o triazinilo.
En algunas realizaciones, cada RA es H, halógeno, -CN, -OH, -OR9, -SR9, alquilo C1-C6 o fluoroalquilo C1-C6.
En algunas realizaciones, L1 está ausente, -CH2-, -CH(CH3)-, -C(CH3)2- o ciclopropil-1,1-diilo; y Q es -CO2H.
En algunas realizaciones, L1 está ausente; y Q es -CO2H.
En algunas realizaciones, el compuesto de Fórmula (III) tiene la siguiente estructura:

o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, R2 es H, F, Cl, Br, I, -CN, -OH, -CH3 , -CF3, -CD3 , -OCH3, -OCH2CH3 , -OCF3 u -OCH2CF3 ;
R3 es H, F, Cl, Br, I, -CN, -OH, -CH3 , -CF3, -CD3, -OCH3, -OCH2CH3 , -OCF3 u -OCH2CF3.
En algunas realizaciones, R2 es Cl; R3 es H, F o Cl.
En algunas realizaciones, el Anillo B es furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo, tiadiazolilo, piridinilo, pirimidinilo, pirazinilo, piridazinilo o triazinilo.
En algunas realizaciones, el Anillo B es pirazolilo.
En algunas realizaciones, el compuesto de Fórmula (III) tiene la siguiente estructura:

o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, RA es H, halógeno, -CN, -OH, -OR9, -SR9, alquilo C1-C6 o fluoroalquilo C1-C6;
RB es H, alquilo C1-C6 , fluoroalquilo C1-C6 o deuteroalquilo C1-C6; R1 es -Cl, -Br, -CN o ciclopropilo; R2 es H, halógeno, -CN, -OH, alquilo C1-C4, fluoroalquilo C1-C4, deuteroalquilo C1-C4, alcoxi C1-C4 o fluoroalcoxi C1-C es H, halógeno, -CN, -OH, alquilo C1-C4 , fluoroalquilo C1-C4 , deuteroalquilo C1-C4 , alcoxi C1-C4 o fluoroalcoxi C1-C4.
En algunas realizaciones, R1 es -Cl o -Br. En algunas realizaciones, R1 es ciclopropilo.
En algunas realizaciones, el compuesto de Fórmula (III) tiene la siguiente estructura:

o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, el compuesto de Fórmula (III) tiene la siguiente estructura:

o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, RA es H, F, Cl, Br, I, -CN, -OH, -OCH3 , -OCH2CH3 , -OCF3, -OCH2CF3, -CH3 , -CH2CH3, -CF3 o -CD3.
En algunas realizaciones, RB es alquilo C1-C6.
En algunas realizaciones, R2 es H, F, Cl, Br, I, -CN, -OH, -CH3 , -CF3, -CD3 , -OCH3, -OCH2CH3 , -OCF3 u -OCH2CF3 ; R3 es H, F, Cl, Br, I, -CN, -OH, -CH3 , -CF3, -CD3, -OCH3, -OCH2CH3 , -OCF3 u -OCH2CF3.
En algunas realizaciones, R2 es Cl; R3 es H, F o Cl.
En algunas realizaciones, L1 está ausente, -CH2-, -CH(CH3)-, -C(CH3)2- o ciclopropil-1,1 -diilo.
En algunas realizaciones, L1 está ausente.
En algunas realizaciones, el compuesto de Fórmula (I), Fórmula (II) o Fórmula (III) tiene la siguiente estructura de Fórmula (IV):

en la que,
W es CH, CF o N;
o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, R2 es H, halógeno, -CN, -OH, alquilo C1-C4 , fluoroalquilo C1-C4 , deuteroalquilo C1-C4 , alcoxi C1-C4, fluoroalcoxi C1-C4 o hidroxialquilo C1-C4.
En algunas realizaciones, R2 es H, F, Cl, Br, I, -CN, -OH, -CH3 , -CF3, -CD3, -OCH3, -OCH2CH3 , -OCF3 , -OCH2CF3 o -CH2OH.
En algunas realizaciones, R2 es Cl.
En algunas realizaciones, R3 es H, halógeno, -CN, -OH, alquilo C1-C4 , fluoroalquilo C1-C4 , deuteroalquilo C1-C4 , alcoxi C1-C4, fluoroalcoxi C1-C4 o hidroxialquilo C1-C4.
En algunas realizaciones, R3 es H, F, Cl, Br, I, -CN, -OH, -CH3 , -CF3, -CD3, -OCH3, -OCH2CH3 , -OCF3 , -OCH2CF3 o -CH2OH.
En algunas realizaciones, R3 es H, F o Cl.
En algunas realizaciones, el Anillo B es fenilo, naftilo, furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo, tiadiazolilo, piridinilo, pirimidinilo, pirazinilo, piridazinilo, triazinilo, quinolinilo, isoquinolinilo, quinazolinilo, quinoxalinilo, naftiridinilo, indolilo, indazolilo, benzoxazolilo, benzoisoxazolilo, benzofuranilo, benzotienilo, benzotiazolilo, benzoimidazolilo, purinilo, cinolinilo, ftalazinilo, pteridinilo, piridopirimidinilo, pirazolopirimidinilo o azaindolilo.
En algunas realizaciones, el Anillo B es fenilo o naftilo.
En algunas realizaciones, el Anillo B es furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo, tiadiazolilo, piridinilo, pirimidinilo, pirazinilo, piridazinilo o triazinilo. En algunas realizaciones, el Anillo B es furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo o tiadiazolilo.
En algunas realizaciones, el Anillo B es pirazolilo.
En algunas realizaciones, el Anillo B es pirazolilo; y cada RB es independientemente H, alquilo C1-C6, fluoroalquilo C1-C6 o deuteroalquilo C1-C6; n es 1.
En algunas realizaciones, el Anillo B es piridinilo, pirimidinilo, pirazinilo, piridazinilo o triazinilo.
En algunas realizaciones, el Anillo B es quinolinilo, isoquinolinilo, quinazolinilo, quinoxalinilo, naftiridinilo, indolilo, indazolilo, benzoxazolilo, benzoisoxazolilo, benzofuranilo, benzotienilo, benzotiazolilo, benzoimidazolilo, purinilo, cinolinilo, ftalazinilo, pteridinilo, piridopirimidinilo, pirazolopirimidinilo o azaindolilo.
En algunas realizaciones, el compuesto de Fórmula (I) tiene la siguiente estructura de Fórmula (V):

en la que,
W es CH, CF o N;
o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, RA es H, halógeno, -CN, -OH, -OR9, -SR9, alquilo C1-C6, fluoroalquilo C1-C6 , deuteroalquilo C1-C6 , heteroalquilo C1-C6 ; RB es H, alquilo C1-C6, fluoroalquilo C1-C6 o deuteroalquilo C1-C6; R1 es -F, -Cl, -Br, -CN, cicloalquilo C3-C6, -NH2 u -O-alquilo C1-C4; R2 es H, halógeno, -CN, -OH, alquilo C1-C4, fluoroalquilo C1-C4 , deuteroalquilo C1-C4 , alcoxi C1-C4 , fluoroalcoxi C1-C4 o hidroxialquilo C1-C4; R3 es H, halógeno, -CN, -OH, alquilo C1-C4 , fluoroalquilo C1-C4, deuteroalquilo C1-C4 , alcoxi C1-C4, fluoroalcoxi C1-C4 o hidroxialquilo C1-C4.
En algunas realizaciones, R1 es -F, -Cl, -Br, -CN, ciclopropilo, -NH2 u -O-CH3. En algunas realizaciones, R1 es -F, -Cl o -Br. En algunas realizaciones, R1 es cicloalquilo C3-C6. En algunas realizaciones, R1 es ciclopropilo.
En algunas realizaciones, el compuesto de Fórmula (I) o Fórmula (V) tiene la siguiente estructura de Fórmula (VI):
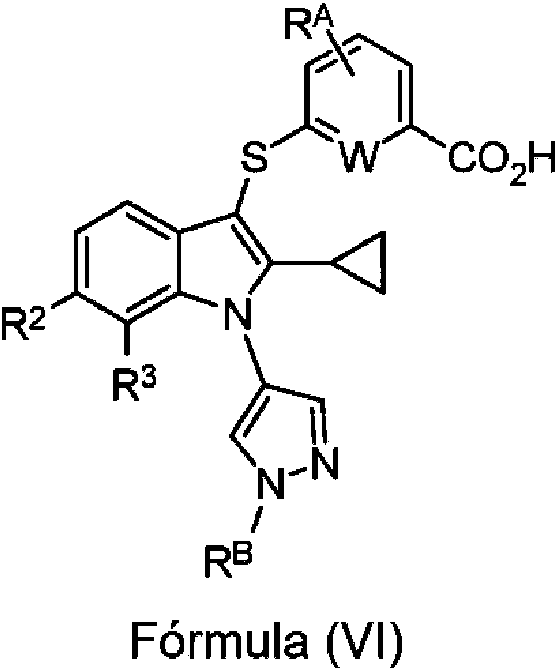
en la que,
W es CH, CF o N;
o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, RA es H, F, Cl, Br, I, -CN, -OH, -OCH3 , -OCH2CH3 , -OCF3, -OCH2CF3, -CH3 , -CH2CH3, -CF3 o -CD3. En algunas realizaciones, RA es H.
En algunas realizaciones, RB es alquilo C1-C6. En algunas realizaciones, RB es -CH3 , -CH2CH3, o -CH2CH2CH3 o -CH(CH3)2.
En algunas realizaciones, R2 es H, F, Cl, Br, I, -CN, -OH, -CH3 , -CF3, -CD3, -OCH3, -OCH2CH3 , -OCF3 , -OCH2CF3 o -CH2OH. En algunas realizaciones, R2 es Cl.
En algunas realizaciones, R3 es H, F, Cl, Br, I, -CN, -OH, -CH3 , -CF3, -CD3, -OCH3, -OCH2CH3 , -OCF3 , -OCH2CF3 o -CH2OH. En algunas realizaciones, R3 es H, F o Cl.
En algunas realizaciones, un compuesto de Fórmula (I), o una sal o solvato farmacéuticamente aceptable del mismo, es:
ácido 3-((2,6-dicloro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (Compuesto n.° 1-1);
ácido 3-((6-cloro-2-ciano-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (Compuesto n.° 1-3);
ácido 3-((6-cloro-2-ciclopropil-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (Compuesto n.° 1-4); ácido 3-((2,6-dicloro-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (Compuesto n.° 1-7); ácido 3-((2-bromo-6-cloro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (Compuesto n.° 1-2);
ácido 3-((6-cloro-2-ciclopropil-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (Compuesto n.° 1-10); ácido 3-((6-cloro-2-ciclopropil-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto n.° 1-16);
ácido 3-((2,6-dicloro-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto n.° 1-13); ácido 3-((6-Cloro-2-ciclopropil-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto n.° 1-34);
ácido 6-((6-cloro-2-ciclopropil-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)picolínico (Compuesto n.° 1-92); ácido 3-((6-cloro-2-ciclopropil-7-fluoro-1-(1H-pirazol-4-il)-7H-indol-3-il)tio)-2-fluorobenzoico (Compuesto n.° 1 119);
ácido 3-((6-cloro-2-ciclopropil-7-fluoro-1-(1-(2-hidroxietil)-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto n.° 1-120);
ácido 3-((1-(1-(2-(carbamo¡lox¡)et¡l)-1H-p¡razol-4-¡l)-6-cloro-2-c¡cloprop¡l-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto n.° 1-121);
ác¡do 3-((1-(1-(2-am¡noet¡l)-1H-p¡razol-4-¡l)-6-cloro-2-c¡cloprop¡l-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto n.° 1-122);
ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-(2-ure¡doet¡l)-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto n.° 1-123);
ác¡do 3-((1-(1-(3-carbox¡prop¡l)-1H-p¡razol-4-¡l)-6-cloro-2-c¡cloprop¡l-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto n.° 1-124);
ác¡do 3-((1-(1-(4-am¡no-4-oxobut¡l)-1H-p¡razol-4-¡l)-6-cloro-2-c¡cloprop¡l-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto n.° 1-125);
ác¡do 3-((2,6-d¡cloro-7-fluoro-1-(1-met¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto n.° 1-49);
ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-(2,2,2-tr¡fluoroet¡l)-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto n.° 1-126);
ác¡do 3-((6-cloro-2-c¡cloprop¡l-1-(1-(et¡l-d5)-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto n.° 1-127);
ác¡do 3-((2,6-d¡cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto n.° 1-31);
ác¡do 3-((2,6-d¡cloro-7-fluoro-1-(p¡r¡d¡n-3-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto n.° 2-1);
ác¡do 3-((2-bromo-6-cloro-7-fluoro-1-(p¡r¡d¡n-3-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto n.° 2-2);
ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(p¡r¡d¡n-3-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto n.° 2-3);
ác¡do 3-((1-(1-(6-am¡noet¡l)-1H-p¡razol-4-¡l)-6-cloro-2-c¡cloprop¡l-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto n.° 1-128);
ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-(hex-5-¡n-1-¡l)-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto n.° 1-129);
ác¡do 3-((1-(1-(3-h¡drox¡-2,2-d¡met¡lprop¡l)-1H-p¡razol-4-¡l)-6-cloro-2-c¡cloprop¡l-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto n.° 1-130); o
ác¡do 3-((6-cloro-2-c¡cloprop¡l-1-(1-(6-(3-(3',6'-d¡h¡drox¡-3-oxo-3H-esp¡ro[¡sobenzofuran-1,9'-xanten]-5-¡l)ure¡do)hex¡l)-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto 1-131).
En el presente documento se contempla cualqu¡er comb¡nac¡ón de los grupos descr¡tos anter¡ormente para las d¡versas var¡ables. A lo largo de la memor¡a descr¡pt¡va, los grupos y sust¡tuyentes de los m¡smos se selecc¡onan por un experto en el campo para proporc¡onar compuestos y restos estables.
En el presente documento tamb¡én se descr¡be una compos¡c¡ón farmacéut¡ca que comprende un compuesto descr¡to en el presente documento, o una sal o solvato farmacéut¡camente aceptable del m¡smo, y al menos un exc¡p¡ente farmacéut¡camente aceptable. En algunas real¡zac¡ones, la compos¡c¡ón farmacéut¡ca se formula para su adm¡n¡strac¡ón a un mamífero med¡ante adm¡n¡strac¡ón ¡ntravenosa, adm¡n¡strac¡ón subcutánea, adm¡n¡strac¡ón oral, ¡nhalac¡ón, adm¡n¡strac¡ón nasal, adm¡n¡strac¡ón dérm¡ca o adm¡n¡strac¡ón oftálm¡ca. En algunas real¡zac¡ones, la compos¡c¡ón farmacéut¡ca se encuentra en forma de un compr¡m¡do, una píldora, una cápsula, un líqu¡do, una suspens¡ón, un gel, una d¡spers¡ón, una soluc¡ón, una emuls¡ón, un ungüento o una loc¡ón.
En el presente documento tamb¡én se descr¡be un método para tratar o preven¡r una cualqu¡era de las enfermedades o afecc¡ones descr¡tas en el presente documento que comprende adm¡n¡strar una cant¡dad terapéut¡camente ef¡caz de un compuesto descr¡to en el presente documento, o una sal o solvato farmacéut¡camente aceptable del m¡smo, a un mamífero que lo neces¡te.
En el presente documento tamb¡én se descr¡be un método para tratar o preven¡r el cáncer o la f¡bros¡s, o comb¡nac¡ones de los m¡smos en un mamífero que comprende adm¡n¡strar una cant¡dad terapéut¡camente ef¡caz de un compuesto descr¡to en el presente documento, o una sal o solvato farmacéut¡camente aceptable del m¡smo, al mamífero que lo neces¡te.
En el presente documento tamb¡én se descr¡be un método para tratar o preven¡r el cáncer en un mamífero que comprende adm¡n¡strar una cant¡dad terapéut¡camente ef¡caz de un compuesto descr¡to en el presente documento, o una sal o solvato farmacéut¡camente aceptable del mismo, al mamífero que lo neces¡te. En algunas real¡zac¡ones, el cáncer es suscept¡ble de tratam¡ento con un ¡nh¡b¡dor de autotax¡na. En algunas real¡zac¡ones, el método comprende además adm¡n¡strar un segundo agente terapéut¡co al mamífero además del compuesto descr¡to en el presente documento, o una sal o solvato farmacéut¡camente aceptable del m¡smo.
En el presente documento tamb¡én se descr¡be un método para el tratam¡ento o prevenc¡ón de la f¡bros¡s en un mamífero que comprende adm¡n¡strar una cant¡dad terapéut¡camente ef¡caz de un compuesto descr¡to en el presente documento, o una sal o solvato farmacéut¡camente aceptable del m¡smo, al mamífero que lo neces¡te. En otras real¡zac¡ones, la f¡bros¡s es suscept¡ble de tratam¡ento con un ¡nh¡b¡dor de autotax¡na. En algunas real¡zac¡ones, el método comprende además adm¡n¡strar un segundo agente terapéut¡co al mamífero además del compuesto descr¡to en el presente documento, o una sal o solvato farmacéut¡camente aceptable del m¡smo.
En real¡zac¡ones ad¡c¡onales, la cant¡dad ef¡caz del compuesto descr¡to en el presente documento, o una sal farmacéut¡camente aceptable del mismo, es: (a) adm¡n¡strada s¡stém¡camente al mamífero; y/o (b) adm¡n¡strada por
vía oral al mamífero; y/o (c) administrada por vía intravenosa al mamífero; y/o (d) administrada por inhalación; y/o (e) administrada por administración nasal; o y/o (f) administrada mediante inyección al mamífero; y/o (g) administrada por vía tópica al mamífero; y/o (h) administrada por administración oftálmica; y/o (i) administrada por vía rectal al mamífero; y/o (j) administrada de forma no sistémica o local al mamífero.
Las realizaciones adicionales comprenden administraciones únicas de la cantidad eficaz del compuesto, incluidas realizaciones adicionales en las que el compuesto se administra una vez al día al mamífero o el compuesto se administra al mamífero varias veces durante el lapso de un día. En algunas realizaciones, el compuesto se administra según un programa de dosificación continuo. En algunas realizaciones, el compuesto se administra según un programa de dosificación diario continuo.
Las realizaciones adicionales que implican el tratamiento de enfermedades o afecciones dependientes de ATX comprenden la administración de al menos un agente adicional además de la administración de un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo. En diversas realizaciones, cada agente se administra en cualquier orden, incluyendo simultáneamente.
En cualquiera de las realizaciones desveladas en el presente documento, el mamífero es un ser humano.
En algunas realizaciones, los compuestos proporcionados en el presente documento se administran a un ser humano.
En algunas realizaciones, los compuestos proporcionados en el presente documento se administran por vía oral.
Se proporcionan artículos de fabricación, que incluyen material de envasado, un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, dentro del material de envasado, y una etiqueta que indica que el compuesto o composición, o sal farmacéuticamente aceptable, tautómeros, N-óxido farmacéuticamente aceptable, metabolito farmacéuticamente activo, profármaco farmacéuticamente aceptable o solvato farmacéuticamente aceptable del mismo, se usa para inhibir la actividad de la autotaxina, o para el tratamiento, prevención o mejora de uno o más síntomas de una enfermedad o afección que se beneficiará de la inhibición de la actividad de la autotaxina.
Otros objetos, características y ventajas de los compuestos, métodos y composiciones que se describen en el presente documento resultarán evidentes a partir de esta descripción detallada. Debe entenderse, sin embargo, que la descripción detallada y los ejemplos específicos, aunque indican realizaciones específicas, se proporcionan solamente a modo de ilustración, ya que diversos cambios y modificaciones resultarán evidentes para los expertos en la técnica a partir de esta descripción detallada.
Descripción detallada
Autotaxina y LPA
La autotaxina (ATX, NPP2 o E-NPP2), una glucoproteína de aproximadamente 120 kDa, es un nucleótido pirofosfatasa/fosfodiesterasa (NPP) secretado con actividad de lisofosfolipasa D que convierte la lisofosfatidilcolina extracelular (LPC) y otros lisofosfolípidos en ácido lisofosfatídico (LPA). Se considera que ATX es responsable de la mayor parte de la producción de LPA circulante.
El LPA actúa a través de conjuntos de receptores acoplados a la proteína G específicos (GPCR, por sus siglas en inglés), tales como LPA1, l Pa 2, LPA3, l Pa 4, LPA5, LPA6, LpA7, LPA8, de forma autocrina y paracrina para producir diversas respuestas biológicas. Por ejemplo, se sabe que los lisofosfolípidos, tal como ácido lisofosfatídico (LPA), afectan a funciones biológicas tales como la proliferación, diferenciación, supervivencia, migración, adhesión, invasión y morfogénesis celular. Además, se sabe que el LPA desempeña un papel en procesos como la activación plaquetaria, la contracción del músculo liso, la formación de fibras de estrés de actina y la migración celular.
Se han detectado ATX y LPA en diversos fluidos biológicos tales como suero, plasma, líquido cefalorraquídeo, fluido seminal, orina y saliva, tanto en animales como en seres humanos, lo que sugiere que son biomarcadores potenciales para predecir determinadas enfermedades. Por ejemplo, la concentración y la actividad de ATX en suero están elevadas en pacientes con enfermedades hepáticas crónicas y en mujeres embarazadas. Además, se ha encontrado que la concentración de ATX es menor en pacientes con cáncer posoperatorio como resultado del daño posoperatorio o un estado nutricional deficiente. Además, se sabe que ATX es esencial para el desarrollo normal. Por ejemplo, los ratones deficientes en ATX mueren el día embrionario 9,5 con profundos defectos vasculares tanto en el saco vitelino como en el embrión. Además, el día embrionario 8,5 se encontró que los embriones deficientes en ATX tenían alantoides malformados, defectos del tubo neural y pliegues de cabeza asimétricos.
Cáncer
Se ha demostrado que la ATX aumenta la motilidad celular, la neovascularización, la proliferación y la agresividad de
los tumores. Está regulada positivamente en numerosos linajes tumorales, tales como cáncer de mama, renal, de hígado, glioblastoma, de ovario y próstata.
En algunas realizaciones, en el presente documento se desvelan métodos para tratar el cáncer con un compuesto desvelado en el presente documento.
La ATX es una enzima prometastásica aislada inicialmente del medio acondicionado de células de melanoma humano. Además, la sobreexpresión de ATX se observa con frecuencia en tejidos tumorales neoplásicos tales como cáncer de mama, cáncer renal, linfoma de Hodgkin, carcinoma hepatocelular, cáncer de páncreas y glioblastoma. El LPA también contribuye a la tumorigénesis al aumentar la motilidad y la invasividad de las células.
El término "cáncer", como se usa en el presente documento, se refiere a un crecimiento anormal de células que tienden a proliferar de manera incontrolada y, en algunos casos, a metastatizar (diseminarse). Los tipos de cáncer incluyen, pero sin limitación, tumores sólidos (tales como los de la vejiga, intestino, cerebro, mama, endometrio, corazón, riñón, pulmón, hígado, útero, tejido linfático (linfoma), ovario, páncreas u otro órgano endocrino (tiroides), próstata, piel (melanoma o cáncer de células basales) o hematológicos (tales como leucemias y linfomas) en cualquier estadio de la enfermedad con o sin metástasis.
Fibrosis
En algunas realizaciones, en el presente documento se desvelan métodos para tratar la fibrosis con un compuesto desvelado en el presente documento.
"Fibrosis", como se usa en el presente documento, se refiere a la acumulación de constituyentes de la matriz extracelular que se produce después de un traumatismo, inflamación, reparación tisular, reacciones inmunitarias, hiperplasia celular y neoplasia.
En algunas realizaciones, en el presente documento se desvela un método para reducir la fibrosis en un tejido que comprende poner en contacto una célula o tejido fibrótico con un compuesto desvelado en el presente documento, en una cantidad suficiente para disminuir o inhibir la fibrosis. En algunas realizaciones, la fibrosis incluye una afección fibrótica.
En algunas realizaciones, la reducción de la fibrosis o el tratamiento de una afección fibrótica incluye reducir o inhibir uno o más de: la formación o deposición de proteínas de la matriz extracelular; el número de tipos de células profibróticas (por ejemplo, número de fibroblastos o de células inmunitarias); el contenido de colágeno celular o hidroxiprolina dentro de una lesión fibrótica; la expresión o actividad de una proteína fibrinógena; o la reducción de la fibrosis asociada a una respuesta inflamatoria.
En algunas realizaciones, la afección fibrótica es fibrosis primaria. En algunas realizaciones, la afección fibrótica es idiopática. En algunas realizaciones, la afección fibrótica está asociada a (por ejemplo, es secundaria a) una enfermedad; una toxina; un insulto (por ejemplo, un peligro ambiental); un tratamiento médico o una combinación de los mismos.
En algunas realizaciones, la afección fibrótica es una afección fibrótica del pulmón (fibrosis pulmonar), una afección fibrótica del hígado (fibrosis renal), una afección fibrótica del corazón o la vasculatura (fibrosis cardíaca), una afección fibrótica del riñón (fibrosis renal), una afección fibrótica de la piel, una afección fibrótica del tracto gastrointestinal o una combinación de las mismas.
En algunas realizaciones, la afección fibrótica es una afección fibrótica del pulmón. En algunas realizaciones, la afección fibrótica del pulmón se elige entre una o más de: fibrosis pulmonar, fibrosis pulmonar idiopática (FPI), neumonitis intersticial habitual (NIU), enfermedad pulmonar intersticial, alveolitis fibrosante criptogénica (AFC), bronquiolitis obliterante o bronquiectasia. En algunas realizaciones, la afección fibrótica del pulmón tratada con los métodos descritos en el presente documento está asociada a (por ejemplo, es secundaria a) un tratamiento contra el cáncer.
En algunas realizaciones, la afección fibrótica es una afección fibrótica del hígado.
En algunas realizaciones, la afección fibrótica es una afección fibrótica del corazón.
En algunas realizaciones, la afección fibrótica es una afección fibrótica del riñón.
En algunas realizaciones, la afección fibrótica es una afección fibrótica de la piel.
En algunas realizaciones, la afección fibrótica es una afección fibrótica del tracto gastrointestinal.
Compuestos
Los compuestos descritos en el presente documento, incluyendo sales farmacéuticamente aceptables, profármacos, metabolitos activos y solvatos farmacéuticamente aceptables de los mismos, son inhibidores de autotaxina.
En el presente documento se describe un compuesto de Fórmula (I), o una sal o solvato farmacéuticamente aceptable del mismo:

en la que,
R1 es -F, -Cl, -Br, -CN, vinilo, cicloalquilo C3-C6, -NH2, -NH(alquilo C1-C4), -N(alquilo Ct C4)2 , -O-alquilo C1-C4 o -S-alquilo C1-C4;
R2 es H, halógeno, -CN, -NO2, -OH, -OR9, -SR9, -S(=O)R9, -S(=O)2R9, -S(=O)2N(R10)2, -NR10S(=O)2R9, -C(=O)R9, -OC(=O)R9, -CO2R10, -OCO2R9, -N(R10)2, -C(=O)N(R10)2, -OC(=O)N(R10)2 , -NHC(=O)R9, -NHC(=O)OR9, alquilo C1-C4 , fluoroalquilo C1-C4, deuteroalquilo C1-C4 , hidroxialquilo C1-C4, heteroalquilo C1-C4, cicloalquilo C3-C6, fenilo sustituido o sin sustituir o heteroarilo monocíclico sustituido o sin sustituir;
el Anillo A es un arilo monocíclico, arilo bicíclico, heterocicloalquilo monocíclico, heteroarilo monocíclico o heteroarilo bicíclico;
cada RA es H, halógeno, -CN, -NO2, -OH, -OR9, -SR9, -S(=O)R9, -S(=O)2R9, -S(=O)2N(R10)2 , -NR10S(=O)2R9, -C(=O)R9, -OC(=O)R9, -CO2R10, -OCO2R9, -N(R10)2, -C(=O)N(R10)2, -OC(=O)N(R10)2, -NHC(=O)R9, -NHC(=O)OR9, alquilo C1-C6, fluoroalquilo C1-C6 , deuteroalquilo C1-C6, heteroalquilo C1-C6, cicloalquilo C3-C10 sustituido o sin sustituir, heterocicloalquilo C2-C10 sustituido o sin sustituir, fenilo sustituido o sin sustituir o heteroarilo monocíclico sustituido o sin sustituir;
m es 0, 1 o 2;
L1 está ausente, alquileno C1-C6, fluoroalquileno C1-C6 o cicloalquileno C3-C6;
Q es -CO2H, -CO2(alquilo C1-C6), -OH, -CN, -B(OH)2 , -C(=O)NHSO2R9, -C(=O)N(R10)2, -SO2NHC(=O)R9, -CN, tetrazolilo, -OP(=O)(o H)2 , -P(=O)(OH)2 o bioisóstero de ácido carboxílico;
L2 está ausente, alquileno C1-C4 o cicloalquileno C3-C7 ;
L3 es -S-, S(=O), S(=O)2 u -O-;
el Anillo B es un arilo monocíclico, arilo bicíclico, heteroarilo monocíclico o heteroarilo bicíclico;
cada RB es independientemente H, halógeno, -CN, -NO2, -OH, -OR9, -SR9, -S(=O)R9, -S(=O)2R9, -S(=O)2N(R10)2, -NR10S(=O)2R9, -C(=O)R9, -OC(=O)R9, -CO2R10, -OCO2R9, -N(R10)2, -C(=O)N(R10)2, -OC(=O)N(R10)2 , -NHC(=O)R9, -NHC(=O)OR9, alquilo C1-C6 , fluoroalquilo C1-C6, deuteroalquilo C1-C6, heteroalquilo C1-C6 , cicloalquilo C3-C10 sustituido o sin sustituir, heterocicloalquilo C2-C10 sustituido o sin sustituir, fenilo sustituido o sin sustituir, alquileno C1-C4-(fenilo sustituido o sin sustituir), heteroarilo monocíclico sustituido o sin sustituir, alquileno C1-C4-(heteroarilo monocíclico sustituido o sin sustituir), un heteroarilo bicíclico sustituido o sin sustituir o alquileno C1-C4-(heteroarilo bicíclico sustituido o sin sustituir); n es 0, 1 o 2;
R9 es alquilo C1-C6, fluoroalquilo C1-C6, deuteroalquilo C1-C6, cicloalquilo C3-C6 , un fenilo sustituido o sin sustituir, un heteroarilo monocíclico sustituido o sin sustituir o un heteroarilo bicíclico sustituido o sin sustituir;
cada R10 se selecciona independientemente de H, alquilo C1-C6, fluoroalquilo C1-C6, deuteroalquilo C1-C6, cicloalquilo C3-C6 , un fenilo sustituido o sin sustituir o un heteroarilo monocíclico sustituido o sin sustituir; o dos grupos R10 unidos al mismo átomo de N se toman junto con el átomo de N al que están unidos para formar un heterociclo sustituido o sin sustituir.
Para cualquiera y todas las realizaciones, los sustituyentes se seleccionan de entre un subconjunto de las alternativas enumeradas. Por ejemplo, en algunas realizaciones, X es -O-, -S-, -S(=O)- o -S(=O)2-. En otras
realizaciones, X es -O- o -S-. En otras realizaciones, X es -S-, -S(=O)- o -S(=O)2-. En algunas realizaciones, X es -S-
En algunas realizaciones, R1 es -F, -Cl, -Br, -CN, vinilo, ciclopropilo, ciclobutilo, -NH2 , -NH(CH3), -N(CH3)2 , -O-CH3 o -S-CH3.
En algunas realizaciones, R1 es vinilo, ciclopropilo o ciclobutilo.
En algunas realizaciones, R1 es ciclopropilo o ciclobutilo.
En algunas realizaciones, R1 es -F, -Cl o -Br.
En algunas realizaciones, L2 está ausente o es alquileno C1-C4 ; L3 es -S-, S(=O) o S(=O)2.
En algunas realizaciones, L2 está ausente, -CH2-, -CH2CH2-, -CH2CH2CH2- o -CH(CH3)-.
En algunas realizaciones, L1 está ausente, -CH2-, -CH2CH2-, -CH2CH2CH2-, -CH(CH3)-, -CH(CH2CH3)-, -C(CH3)2-, -C(CH2CH3)2-, ciclopropil-1,1-diilo, ciclobutil-1,1-diilo, ciclopentil-1,1-diilo o ciclohexil-1,1-diilo; Q es -CO2H, -CO2(alquilo C1-C6), -C(=O)NHSO2R9 o tetrazolilo.
En algunas realizaciones, L1 está ausente o -CH2-; Q es -CO2H, o -CO2(alquilo C1-C6).
En algunas realizaciones, el compuesto de Fórmula (I) tiene la siguiente estructura de Fórmula (II):

o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, el Anillo A es fenilo, naftilo, heteroarilo monocíclico que contiene 1-4 átomos de N y 0 o 1 átomos de O o S, heteroarilo monocíclico que contiene 0-4 átomos de N y 1 átomo de O o S, heteroarilo bicíclico que contiene 1-4 átomos de N y 0 o 1 átomos de O o S, o heteroarilo bicíclico que contiene 0-4 átomos de N y 1 átomo de O o S; el Anillo B es fenilo, naftilo, heteroarilo monocíclico que contiene 1-4 átomos de N y 0 o 1 átomos de O o S, heteroarilo monocíclico que contiene 0-4 átomos de N y 1 átomo de O o S, heteroarilo bicíclico que contiene 1-4 átomos de N y 0 o 1 átomos de O o S, o heteroarilo bicíclico que contiene 0-4 átomos de N y 1 átomo de O o S. En algunas realizaciones, el Anillo A es fenilo, naftilo, furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo, tiadiazolilo, piridinilo, pirimidinilo, pirazinilo, piridazinilo, triazinilo, quinolinilo, isoquinolinilo, quinazolinilo, quinoxalinilo, naftiridinilo, indolilo, indazolilo, benzoxazolilo, benzoisoxazolilo, benzofuranilo, benzotienilo, benzotiazolilo, benzoimidazolilo, purinilo, cinolinilo, ftalazinilo, pteridinilo, piridopirimidinilo, pirazolopirimidinilo o azaindolilo.
En algunas realizaciones, el Anillo A es fenilo o naftilo.
En algunas realizaciones, el Anillo A es furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo, tiadiazolilo, piridinilo, pirimidinilo, pirazinilo, piridazinilo o triazinilo. En algunas realizaciones, el Anillo A es piridinilo, pirimidinilo, pirazinilo, piridazinilo o triazinilo.
En algunas realizaciones, el Anillo A es furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo o tiadiazolilo.
En algunas realizaciones, el Anillo A es quinolinilo, isoquinolinilo, quinazolinilo, quinoxalinilo, naftiridinilo, indolilo,
indazolilo, benzoxazolilo, benzoisoxazolilo, benzofuranilo, benzotienilo, benzotiazolilo, benzoimidazolilo, purinilo, cinolinilo, ftalazinilo, pteridinilo, piridopirimidinilo, pirazolopirimidinilo o azaindolilo.
En algunas realizaciones, cada RA es H, halógeno, -CN, -OH, -OR9, -SR9, alquilo C1-C6, fluoroalquilo C1-C6, deuteroalquilo C1-C6 , heteroalquilo C1-C6.
En algunas realizaciones, L3 es -S-.
En algunas realizaciones, L2 está ausente.
En algunas realizaciones, el compuesto de Fórmula (I) o Fórmula (II) tiene la siguiente estructura de Fórmula (III):

en la que,
W es CH, CF o N;
o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, L1 está ausente; y Q es -CO2H.
En algunas realizaciones, en el presente documento se describe un compuesto de Fórmula (III), o una sal o solvato farmacéuticamente aceptable del mismo:

en la que,
R1 es -Cl, -Br, -CN o cicloalquilo C3-C6;
R2 es H, halógeno, -CN, -NO2, -OH, -OR9, -SR9, -S(=O)R9, -S(=O)2R9, -S(=O)2N(R10)2, alquilo C1-C4, fluoroalquilo
C1-C4 , deuteroalquilo C1-C4 o cicloalquilo C3-C6;
R3 es H, halógeno, -CN, -OH, alquilo C1-C4 , fluoroalquilo C1-C4 , deuteroalquilo C1-C4, alcoxi C1-C4 o fluoroalcoxi
C1-C4;
W es CH, CF o N;
cada RA es H, halógeno, - -S(=O)2N( fluoroalquilo C1-C6;
-S(=O)2N( fluoroalquilo C1-C6;
L1 está ausente, alquileno C1-C6 o cicloalquileno C3-C6 ;
Q es -CO2H, -CO2(alquilo C1-C6), -OH, -CN, -B(OH)2 , -C(=O)NHSO2R9, -C(=O)N(R10)2, -SO2NHC(=O)R9, -CN, tetrazolilo, -OP(=O)(o H)2 , -P(=O)(OH)2 o bioisóstero de ácido carboxílico;
el Anillo B es un heteroarilo monocíclico;
cada RB es independientemente H, halógeno, -CN, -NO2, -OH, -OR9, -SR9, -S(=O)R9, -S(=O)2R9, -S(=O)2N(R10)2, alquilo C1-C6, fluoroalquilo C1-C6 , deuteroalquilo C1-C6 , heteroalquilo C1-C6 , cicloalquilo C3-C10 sustituido o sin sustituir, heterocicloalquilo C2-C10 sustituido o sin sustituir, fenilo sustituido o sin sustituir, alquileno C1-C4-(fenilo sustituido o sin sustituir), heteroarilo monocíclico sustituido o sin sustituir, alquileno C1-C4-(heteroarilo monocíclico sustituido o sin sustituir), un heteroarilo bicíclico sustituido o sin sustituir o alquileno C1-C4-(heteroarilo bicíclico sustituido o sin sustituir);
n es 0, 1 o 2;
R9 es alquilo C1-C6, fluoroalquilo C1-C6, deuteroalquilo C1-C6, cicloalquilo C3-C6 , un fenilo sustituido o sin sustituir, un heteroarilo monocíclico sustituido o sin sustituir o un heteroarilo bicíclico sustituido o sin sustituir;
cada R10 es independientemente H, alquilo C1-C6 , fluoroalquilo C1-C6, deuteroalquilo C1-C6, cicloalquilo C3-C6, un fenilo sustituido o sin sustituir o un heteroarilo monocíclico sustituido o sin sustituir; o
dos grupos R10 unidos al mismo átomo de N se toman junto con el átomo de N al que están unidos para formar un heterociclo sustituido o sin sustituir.
En algunas realizaciones, R1 es -Cl, -Br, -CN o ciclopropilo. En algunas realizaciones, R1 es ciclopropilo. En algunas realizaciones, R1 es -Cl.
En algunas realizaciones, L1 está ausente, -CH2-, -CH2CH2-, -CH2CH2CH2-, -CH(CH3)-, -CH(CH2CH3)-, -C(CH3)2-, -C(CH2CH3)2-, ciclopropil-1,1-diilo, ciclobutil-1,1-diilo, ciclopentil-1,1-diilo o ciclohexil-1,1 -diilo; y Q es -CO2H, -CO2(alquilo C1-C6), -C(=O)NHSO2R9 o tetrazolilo.
En algunas realizaciones, L1 está ausente, -CH2-, -CH(CH3)-, -C(CH3)2- o ciclopropil-1,1-diilo; y Q es -CO2H, o -CO2(alquilo C1-C6).
En algunas realizaciones, L1 está ausente, -CH2-, -CH2CH2-, -CH2CH2CH2-, -CH(CH3)-, -CH(CH2CH3)-, -C(CH3)2- o -C(CH2CH3)2-. En algunas realizaciones, L1 está ausente, -CH2-, -CH(CH3)-, -CH(CH2CH3)-, -C(CH3)2- o -C(CH2CH3)2-. En algunas realizaciones, L1 está ausente, -CH2-, -CH(CH3)- o -C(CH3)2-. En algunas realizaciones, L1 está ausente o -CH2-.
En algunas realizaciones, L1 es -CH2-, -CH2CH2-, -CH2CH2CH2-, -CH(CH3)-, -CH(CH2CH3)-, -C(CH3)2- o -C(CH2CH3)2-. En algunas realizaciones, L1 es -CH2-, -CH(CH3)-, -CH(CH2CH3)-, -C(CH3)2- o -C(CH2CH3)2-. En algunas realizaciones, L1 es -CH2-, -CH(CH3)- o -C(CH3)2-. En algunas realizaciones, L1 es -CH2-.
En algunas realizaciones, L1 está ausente o -CH2-; y Q es -CO2H o -CO2(alquilo C1-C6).
En algunas realizaciones, el Anillo B es heteroarilo monocíclico que contiene 1-4 átomos de N y 0 o 1 átomos de O o S, o heteroarilo monocíclico que contiene 0-4 átomos de N y 1 átomo de O o S.
En algunas realizaciones, el Anillo B es furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo, tiadiazolilo, piridinilo, pirimidinilo, pirazinilo, piridazinilo o triazinilo. En algunas realizaciones, cada RA es H, halógeno, -CN, -OH, -OR9, -SR9, alquilo C1-C6 o fluoroalquilo C1-C6.
En algunas realizaciones, L1 está ausente, -CH2-, -CH(CH3)-, -C(CH3)2- o ciclopropil-1,1-diilo; y Q es -CO2H.
En algunas realizaciones, L1 está ausente; y Q es -CO2H.
En algunas realizaciones, el compuesto de Fórmula (III) tiene la siguiente estructura:

o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, R2 es H, F, Cl, Br, I, -CN, -OH, -CH3 , -CF3, -CD3 , -OCH3, -OCH2CH3 , -OCF3 u -OCH2CF3 ;
R3 es H, F, Cl, Br, I, -CN, -OH, -CH3 , -CF3, -CD3, -OCH3, -OCH2CH3 , -OCF3 u -OCH2CF3.
En algunas realizaciones, R2 es Cl; R3 es H, F o Cl.
En algunas realizaciones, el Anillo B es furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo, tiadiazolilo, piridinilo, pirimidinilo, pirazinilo, piridazinilo o triazinilo.
En algunas realizaciones, el Anillo B es pirazolilo.
En algunas realizaciones, el compuesto de Fórmula (III) tiene la siguiente estructura:

o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, RA es H, halógeno, -CN, -OH, -OR9, -SR9, alquilo C1-C6 o fluoroalquilo C1-C6;
RB es H, alquilo C1-C6 , fluoroalquilo C1-C6 o deuteroalquilo C1-C6; R1 es -Cl, -Br, -CN o ciclopropilo; R2 es H, halógeno, -CN, -OH, alquilo C1-C4, fluoroalquilo C1-C4, deuteroalquilo C1-C4, alcoxi C1-C4 o fluoroalcoxi C1-C es H, halógeno, -CN, -OH, alquilo C1-C4 , fluoroalquilo C1-C4 , deuteroalquilo C1-C4 , alcoxi C1-C4 o fluoroalcoxi C1-C4.
En algunas realizaciones, R1 es -Cl o -Br. En algunas realizaciones, R1 es ciclopropilo.
En algunas realizaciones, el compuesto de Fórmula (III) tiene la siguiente estructura:

o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, el compuesto de Fórmula (III) tiene la siguiente estructura:

o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, RA es H, F, Cl, Br, I, -CN, -OH, -OCH3 , -OCH2CH3 , -OCF3, -OCH2CF3, -CH3 , -CH2CH3, -CF3 o -CD3.
En algunas realizaciones, RB es alquilo C1-C6.
En algunas realizaciones, R2 es H, F, Cl, Br, I, -CN, -OH, -CH3 , -CF3, -CD3 , -OCH3, -OCH2CH3 , -OCF3 u -OCH2CF3 ; R3 es H, F, Cl, Br, I, -CN, -OH, -CH3 , -CF3, -CD3, -OCH3, -OCH2CH3 , -OCF3 u -OCH2CF3.
En algunas realizaciones, R2 es Cl; R3 es H, F o Cl.
En algunas realizaciones, L1 está ausente, -CH2-, -CH(CH3)-, -C(CH3)2- o ciclopropil-1,1 -diilo.
En algunas realizaciones, L1 está ausente.
En algunas realizaciones, el compuesto de Fórmula (I), Fórmula (II) o Fórmula (III) tiene la siguiente estructura de Fórmula (IV):

en la que,
W es CH, CF o N;
o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, R2 es H, halógeno, -CN, -OH, alquilo C1-C4 , fluoroalquilo C1-C4 , deuteroalquilo C1-C4 , alcoxi C1-C4, fluoroalcoxi C1-C4 o hidroxialquilo C1-C4.
En algunas realizaciones, R2 es H, F, Cl, Br, I, -CN, -OH, -CH3 , -CF3, -CD3, -OCH3, -OCH2CH3 , -OCF3 , -OCH2CF3 o -CH2OH.
En algunas realizaciones, R2 es Cl.
En algunas realizaciones, R3 es H, halógeno, -CN, -OH, alquilo C1-C4 , fluoroalquilo C1-C4 , deuteroalquilo C1-C4 , alcoxi C1-C4, fluoroalcoxi C1-C4 o hidroxialquilo C1-C4.
En algunas realizaciones, R3 es H, F, Cl, Br, I, -CN, -OH, -CH3 , -CF3, -CD3, -OCH3, -OCH2CH3 , -OCF3 , -OCH2CF3 o -CH2OH.
En algunas realizaciones, R3 es H, F o Cl.
En algunas realizaciones, el Anillo B es fenilo, naftilo, furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo, tiadiazolilo, piridinilo, pirimidinilo, pirazinilo, piridazinilo, triazinilo, quinolinilo, isoquinolinilo, quinazolinilo, quinoxalinilo, naftiridinilo, indolilo, indazolilo, benzoxazolilo, benzoisoxazolilo, benzofuranilo, benzotienilo, benzotiazolilo, benzoimidazolilo, purinilo, cinolinilo, ftalazinilo, pteridinilo, piridopirimidinilo, pirazolopirimidinilo o azaindolilo.
En algunas realizaciones, el Anillo B es fenilo o naftilo.
En algunas realizaciones, el Anillo B es furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo, tiadiazolilo, piridinilo, pirimidinilo, pirazinilo, piridazinilo o triazinilo. En algunas realizaciones, el Anillo B es furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo o tiadiazolilo.
En algunas realizaciones, el Anillo B es pirazolilo.
En algunas realizaciones, el Anillo B es pirazolilo; y cada RB es independientemente H, alquilo C1-C6, fluoroalquilo C1-C6 o deuteroalquilo C1-C6; n es 1.
En algunas realizaciones, el Anillo B es piridinilo, pirimidinilo, pirazinilo, piridazinilo o triazinilo.
En algunas realizaciones, el Anillo B es quinolinilo, isoquinolinilo, quinazolinilo, quinoxalinilo, naftiridinilo, indolilo, indazolilo, benzoxazolilo, benzoisoxazolilo, benzofuranilo, benzotienilo, benzotiazolilo, benzoimidazolilo, purinilo, cinolinilo, ftalazinilo, pteridinilo, piridopirimidinilo, pirazolopirimidinilo o azaindolilo.
En algunas realizaciones, el compuesto de Fórmula (I) tiene la siguiente estructura de Fórmula (V):

en la que,
W es CH, CF o N;
o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, RA es H, halógeno, -CN, -OH, -OR9, -SR9, alquilo C1-C6, fluoroalquilo C1-C6 , deuteroalquilo C1-C6 , heteroalquilo C1-C6 ; RB es H, alquilo C1-C6, fluoroalquilo C1-C6 o deuteroalquilo C1-C6; R1 es -F, -Cl, -Br, -CN, cicloalquilo C3-C6, -NH2 u -O-alquilo C1-C4; R2 es H, halógeno, -CN, -OH, alquilo C1-C4, fluoroalquilo C1-C4 , deuteroalquilo C1-C4 , alcoxi C1-C4 , fluoroalcoxi C1-C4 o hidroxialquilo C1-C4; R3 es H, halógeno, -CN, -OH, alquilo C1-C4 , fluoroalquilo C1-C4, deuteroalquilo C1-C4 , alcoxi C1-C4, fluoroalcoxi C1-C4 o hidroxialquilo C1-C4.
En algunas realizaciones, R1 es -F, -Cl, -Br, -CN, ciclopropilo, -NH2 u -O-CH3. En algunas realizaciones, R1 es -F, -Cl o -Br. En algunas realizaciones, R1 es -Cl. En algunas realizaciones, R1 es cicloalquilo C3-C6. En algunas realizaciones, R1 es ciclopropilo.
En algunas realizaciones, el compuesto de Fórmula (I) o Fórmula (V) tiene la siguiente estructura de Fórmula (VI):

en la que,
W es CH, CF o N;
o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, el compuesto de Fórmula (I) o Fórmula (V) tiene la siguiente estructura de Fórmula (VII):

en la que,
W es CH, CF o N;
o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, RA es H, F, Cl, Br, I, -CN, -OH, -OCH3 , -OCH2CH3 , -OCF3, -OCH2CF3, -CH3 , -CH2CH3, -CF3 o -CD3. En algunas realizaciones, RA es H.
En algunas realizaciones, RB es alquilo C1-C6. En algunas realizaciones, RB es -CH3 , -CH2CH3, o -CH2CH2CH3 o -CH(CH3)2.
En algunas realizaciones, R2 es H, F, Cl, Br, I, -CN, -OH, -CH3 , -CF3, -CD3, -OCH3, -OCH2CH3 , -OCF3 , -OCH2CF3 o -CH2OH. En algunas realizaciones, R2 es Cl.
En algunas realizaciones, R3 es H, F, Cl, Br, I, -CN, -OH, -CH3 , -CF3, -CD3, -OCH3, -OCH2CH3 , -OCF3 , -OCH2CF3 o -CH2OH. En algunas realizaciones, R3 es H, F o Cl.
En algunas realizaciones, W es CH, CF o N. En algunas realizaciones, W es CH. En algunas realizaciones, W es CH o CF. En algunas realizaciones, W es CF. En algunas realizaciones, W es N.
En algunas realizaciones, el compuesto de Fórmula (I) tiene la siguiente estructura:
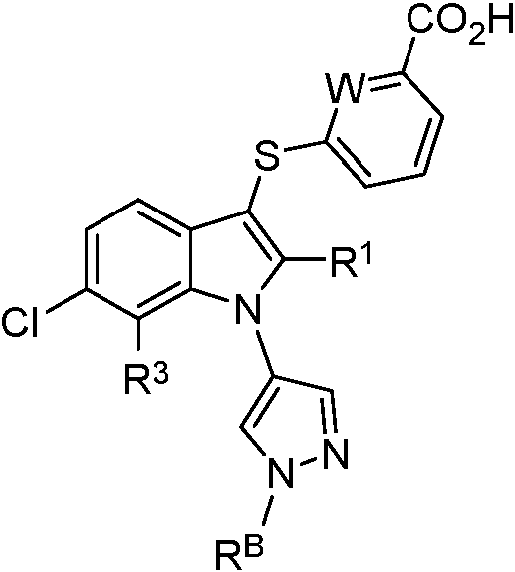
en la que,
W es CH, CF o N;
o una sal o solvato farmacéuticamente aceptable del mismo.
En algunas realizaciones, R1 es -Cl o ciclopropilo. En algunas realizaciones, R1 es -Cl. En algunas realizaciones, R1 es ciclopropilo.
En algunas realizaciones, R1 es como se describe en las Tablas 1 y 2. En algunas realizaciones, R3 es como se describe en las Tablas 1 y 2. En algunas realizaciones, RB es como se describe en las Tablas 1 y 2. En algunas realizaciones, R1, R3 y RB son como se describen en las Tablas 1 y 2. En algunas realizaciones, L1 es como se describe en la Tabla 2.
En el presente documento se contempla cualquier combinación de los grupos descritos anteriormente para las diversas variables. A lo largo de la memoria descriptiva, los grupos y sustituyentes de los mismos se seleccionan por un experto en el campo para proporcionar compuestos y restos estables.
Los compuestos de ejemplo incluyen los siguientes compuestos. Los compuestos mencionados a continuación, que no están incluidos en las reivindicaciones adjuntas, se incluyen en el presente documento como ejemplos de referencia.
T l 1

continuación

continuación

continuación

Los compuestos en la Tabla 1 se denominan:
ácido 3-((2,6-dicloro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-1);
ácido 3-((2-bromo-6-cloro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-2);
ácido 3-((6-cloro-2-ciano-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-3);
ácido 3-((6-cloro-2-ciclopropil-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-4); ácido 3-((2-amino-6-cloro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-5);
ácido 3-((6-cloro-2-metoxi-1-(1-propil-1H-pirazol-4-ii)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-6);
ácido 3-((2,6-dicloro-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-7); ácido 3-((2-bromo-6-cloro-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-8); ácido 3-((6-cloro-2-ciano-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-9); ácido 3-((6-cloro-2-ciclopropil-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-10); ácido 3-((2-amino-6-cloro-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-11); ácido 3-((6-cloro-7-fluoro-2-metoxi-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-12); ácido 3-((2,6-dicloro-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico (compuesto n.° 1-13); ácido 3-((2-bromo-6-cloro-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico (compuesto n.° 1 14);
ácido 3-((6-cloro-2-ciano-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico (compuesto n.° 1 15);
ácido 3-((6-cloro-2-ciclopropil-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico (compuesto n.° 1-16);
ácido 3-((2-amino-6-cloro-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico (compuesto n.° 1 17);
ácido 3-((6-cloro-7-fluoro-2-metoxi-1-(1-propil-1H-pirazol-4-il)-1H-indol-3 -il)tio)-2-fluorobenzoico (compuesto n.° 1-18);
ácido 3-((2,6-dicloro-1-(1-etil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-19);
ácido 3-((2-bromo-6-cloro-1-(1-etil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-20);
ácido 3-((6-cloro-2-ciano-1-(1-etil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-21);
ácido 3-((6-cloro-2-ciclopropil-1-(1-etil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-22);
ácido 3-((2-amino-6-cloro-1-(1-etil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-23);
ácido 3-((6-cloro-2-metoxi-1-(1-etil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-24);
ácido 3-((2,6-dicloro-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)benzoico (compuesto n.° 1-25);
ácido 3-((2-bromo-6-cloro-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)benzoico (compuesto n.° 1-26); ácido 3-((6-cloro-2-ciano-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)benzoico (compuesto n.° 1-27); ácido 3-((6-cloro-2-ciclopropil-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)benzoico (compuesto n.° 1-28); ácido 3-((2-amino-6-cloro-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)benzoico (compuesto n.° 1-29); ácido 3-((6-cloro-1-(1-etil-1H-pirazol-4-il)-7-fluoro-2-metoxi-1H-indol-3-il)tio)benzoico (compuesto n.° 1-30); ácido 3-((2,6-dicloro-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico (compuesto n.° 1-31); ácido 3-((2-bromo-6-cloro-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico (compuesto n.° 1 32);
ácido 3-((6-cloro-2-ciano-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico (compuesto n.° 1-33); ácido 3-((6-cloro-2-ciclopropil-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico (compuesto n.° 1-34);
ácido 3-((2-amino-6-cloro-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico (compuesto n.° 1 35);
ácido 3-((6-cloro-2-metoxi-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico (compuesto n.° 1 36);
ácido 3-((2,6-dicloro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-37);
ácido 3-((2-bromo-6-cloro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-38);
ácido 3-((6-cloro-2-ciano-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-39);
ácido 3-((6-cloro-2-ciclopropil-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-40); ácido 3-((2-amino-6-cloro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-41);
ácido 3-((6-cloro-2-metoxi-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-42);
ácido 3-((2,6-dicloro-7-fluoro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-43); ácido 3-((2-bromo-6-cloro-7-fluoro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-44); ácido 3-((6-cloro-2-ciano-7-fluoro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-45); ácido 3-((6-cloro-2-ciclopropil-7-fluoro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-46); ácido 3-((2-amino-6-cloro-7-fluoro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico (compuesto n.° 1-47); ácido 3-((6-cloro-7-fluoro-1-(1-metil-1H-pirazol-4-il)-2-metoxi-1H-indol-3-il)tio)benzoico (compuesto n.° 1-48); ácido 3-((2,6-dicloro-7-fluoro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico (compuesto n.° 1-49); ácido 3-((2-bromo-6-cloro-7-fluoro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico (compuesto n.° 1
50);
ácido 3-((6-cloro-2-c¡ano-7-fluoro-1-(1-met¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1 51);
ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-met¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-52);
ác¡do 3-((2-am¡no-6-cloro-7-fluoro-1-(1-met¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1 53);
ác¡do 3-((2,6-d¡cloro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)benzo¡co (compuesto n.° 1-54);
ác¡do 3-((2-bromo-6-cloro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)benzo¡co (compuesto n.° 1-55); ác¡do 3-((6-cloro-2-c¡ano-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)benzo¡co (compuesto n.° 1-56); ác¡do 3-((6-cloro-2-c¡cloprop¡l-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)benzo¡co (compuesto n.° 1-57); ác¡do 3-((2-am¡no-6-cloro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)benzo¡co (compuesto n.° 1-58); ác¡do 3-((6-cloro-2-metox¡-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)benzo¡co (compuesto n.° 1-59); ác¡do 3-((2,6-d¡cloro-7-fluoro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)benzo¡co (compuesto n.° 1-60); ác¡do 3-((2-bromo-6-cloro-7-fluoro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)benzo¡co (compuesto n.° 1-61); ác¡do 3-((6-cloro-2-c¡ano-7-fluoro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)benzo¡co (compuesto n.° 1-62); ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)benzo¡co (compuesto n.° 1 63);
ác¡do 3-((2-am¡no-6-cloro-7-fluoro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)benzo¡co (compuesto n.° 1-64); ác¡do 3-((6-cloro-7-fluoro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-2-metox¡-1H-¡ndol-3-¡l)t¡o)benzo¡co (compuesto n.° 1-65); ác¡do 3-((2,6-d¡cloro-7-fluoro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1 66);
ác¡do 3-((2-bromo-6-cloro-7-fluoro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-67);
ác¡do 3-((6-cloro-2-c¡ano-7-fluoro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-68);
ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-69);
ác¡do 3-((2-am¡no-6-cloro-7-fluoro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-70);
ác¡do 6-((2,6-d¡cloro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-71);
ác¡do 6-((2-bromo-6-cloro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-72);
ác¡do 6-((6-cloro-2-c¡ano-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-73);
ác¡do 6-((6-cloro-2-c¡cloprop¡l-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-74); ác¡do 6-((2-am¡no-6-cloro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-75);
ác¡do 6-((6-cloro-2-metox¡-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-76);
ác¡do 6-((2,6-d¡cloro-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-77); ác¡do 6-((2-bromo-6-cloro-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-78); ác¡do 6-((6-cloro-2-c¡ano-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-79); ác¡do 6-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-80); ác¡do 6-((2-am¡no-6-cloro-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-81); ác¡do 6-((6-cloro-7-fluoro-2-metox¡-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-82); ác¡do 6-((2,6-d¡cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-83);
ác¡do 6-((2-bromo-6-cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-84);
ác¡do 6-((6-cloro-2-c¡ano-1-(1-et¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-85);
ác¡do 6-((6-cloro-2-c¡cloprop¡l-1-(1-et¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-86); ác¡do 6-((2-am¡no-6-cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-87);
ác¡do 6-((6-cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-2-metox¡-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-88);
ác¡do 6-((2,6-d¡cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-89);
ác¡do 6-((2-bromo-6-cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-90); ác¡do 6-((6-cloro-2-c¡ano-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-91); ác¡do 6-((6-cloro-2-c¡cloprop¡l-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-92); ác¡do 6-((2-am¡no-6-cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-93); ác¡do 6-((6-cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-2-metox¡-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-94); ác¡do 6-((2,6-d¡cloro-1-(1-met¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-95);
ác¡do 6-((2-bromo-6-cloro-1-(1-met¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-96);
ác¡do 6-((6-cloro-2-c¡ano-1-(1-met¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-97);
ác¡do 6-((6-cloro-2-c¡cloprop¡l-1-(1-met¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-98); ác¡do 6-((2-am¡no-6-cloro-1-(1-met¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-99);
ác¡do 6-((6-cloro-2-metox¡-1-(1-met¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-100); ác¡do 6-((2,6-d¡cloro-7-fluoro-1-(1-met¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-101); ác¡do 6-((2-bromo-6-cloro-7-fluoro-1-(1-met¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-102); ác¡do 6-((6-cloro-2-c¡ano-7-fluoro-1-(1-met¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-103); ác¡do 6-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-met¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-104); ác¡do 6-((2-am¡no-6-cloro-7-fluoro-1-(1-met¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-105); ác¡do 6-((6-cloro-7-fluoro-2-metox¡-1-(1-met¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-106);
ácido 6-((2,6-d¡cloro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-107);
ácido 6-((2-bromo-6-cloro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-108); ác¡do 6-((6-cloro-2-c¡ano-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-109); ác¡do 6-((6-cloro-2-c¡cloprop¡l-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-110; ác¡do 6-((2-am¡no-6-cloro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-111); ác¡do 6-((6-cloro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-2-metox¡-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-112); ác¡do 6-((2,6-d¡cloro-7-fluoro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-113); ác¡do 6-((2-bromo-6-cloro-7-fluoro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-114); ác¡do 6-((6-cloro-2-c¡ano-7-fluoro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-115); ác¡do 6-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1 116);
ác¡do 6-((2-am¡no-6-cloro-7-fluoro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-117); ác¡do 6-((6-cloro-7-fluoro-1-(1-¡soprop¡l-1H-p¡razol-4-¡l)-2-metox¡-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-118); ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-119); ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-(2-h¡drox¡et¡l)-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-120);
ác¡do 3-(1-(1-(2-(carbamo¡lox¡)et¡l)-1H-p¡razol-4-¡l)-(6-cloro-2-c¡cloprop¡l-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-121);
ác¡do 3-(1-(1-(2-ammoet¡l)-1H-p¡razol-4-¡l)-(6-cloro-2-c¡cloprop¡l-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-122);
ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-(2-ure¡doet¡l)-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-123);
ác¡do 3-(1-(1-(3-carbox¡prop¡l)-1H-p¡razol-4-¡l)-(6-cloro-2-c¡cloprop¡l-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-124);
ác¡do 3-(1-(1-(4-am¡no-4-oxobut¡l)-1H-p¡razol-4-¡l)-(6-cloro-2-c¡cloprop¡l-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-125);
ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-(2,2,2-tnfluoroet¡l)-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-126);
ác¡do 3-((6-cloro-2-c¡cloprop¡l-1-(1-(2H5)et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co;
(compuesto n.° 1-127);
ác¡do 3-(1-(1-(6-am¡nohex¡l)-1H-p¡razol-4-¡l)-(6-cloro-2-c¡cloprop¡l-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-128);
ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-(hex-5-¡ml)-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-129);
ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-(3-h¡drox¡-2,2-d¡met¡lprop¡l)-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-130);
ác¡do 3-((6-cloro-2-c¡cloprop¡l-1-(1-(6-(3-(3',6'-d¡h¡drox¡-3-oxo-3H-esp¡ro[¡sobenzofuran-1,9'-xanten]-5-¡l)ure¡do)hex¡l)-1 H-p¡razol-4-¡l)-7-fluoro-1 H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-131);
ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-(4-(met¡lam¡no)-4-oxobut¡l)-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 1-132);
ác¡do 3-(6-cloro-2-c¡cloprop¡l-1-(1-(4-(d¡met¡lam¡no)-4-oxobut¡l)-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡lt¡o)-2-fluorobenzo¡co (compuesto n.° 1-133);
ác¡do 3-(1-(1-(4-am¡no-3,3-d¡met¡l-4-oxobut¡l)-1H-p¡razol-4-¡l)-6-cloro-2-c¡cloprop¡l-7-fluoro-1H-¡ndol-3-¡lt¡o)-2-fluorobenzo¡co (compuesto n.° 1-134);
ác¡do 6-(1-(1-(4-am¡no-4-oxobut¡l)-1H-p¡razol-4-¡l)-(6-cloro-2-c¡cloprop¡l-7-fluoro-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-135);
ác¡do 6-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-(4-(met¡lam¡no)-4-oxobut¡l)-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-136);
ác¡do 6-((6-cloro-2-c¡cloprop¡l-1-(1-(4-(d¡met¡lam¡no)-4-oxobut¡l)-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)p¡colm¡co (compuesto n.° 1-137);
ác¡do 6-(1-(1-(4-am¡no-3,3-d¡met¡l-4-oxobut¡l)-1H-p¡razol-4-¡l)-(6-cloro-2-c¡cloprop¡l-7-fluoro-1H-¡ndol-3-¡l)t¡o)p¡colín¡co (compuesto n.° 1-138).


Los compuestos en la Tabla 2 se denominan:
ácido 3-((2,6-d¡cloro-7-fluoro-1-(p¡rid¡n-3-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 2-1);
ácido 3-((2-bromo-6-cloro-7-fluoro-1-(p¡rid¡n-3-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 2-2);
ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(p¡rid¡n-3-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 2-3);
ác¡do 3-((6-cloro-2-c¡ano-7-fluoro-1-(p¡rid¡n-3-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (compuesto n.° 2-4);
ác¡do 2-(3-((2,6-d¡doro-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l)acét¡co (compuesto n.° 2-5); ác¡do 2-(3-((2-bromo-6-cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l)acét¡co (compuesto n.° 2-6);
ác¡do 2-(3-((6-cloro-2-c¡cloprop¡l-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofeml)acét¡co (compuesto n.° 2-7);
ác¡do 2-(3-((6-cloro-2-c¡ano-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofeml)acét¡co (compuesto n.° 2-8);
ác¡do 2-(3-((2,6-d¡doro-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l)-2-met¡lpropano¡co (compuesto n.° 2-9);
ác¡do 2-(3-((2-bromo-6-doro-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l)-2-met¡lpropano¡co (compuesto n.° 2-10);
ác¡do 2-(3-((6-cloro-2-c¡cloprop¡l-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofeml)-2-met¡lpropano¡co (compuesto n.° 2-11);
ác¡do 2-(3-((6-cloro-2-c¡ano-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofeml)-2-met¡lpropano¡co (compuesto n.° 2-12)
ác¡do 1-(3-((2,6-d¡cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofeml)c¡clopropanocarboxíl¡co (compuesto n.° 2-13);
ác¡do 1-(3-((2-bromo-6-cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l) c¡clopropanocarboxíl¡co (compuesto n.°2-14);
ác¡do 1-(3-((6-cloro-2-c¡cloprop¡l-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2
fluorofen¡l)c¡clopropanocarboxíl¡co (compuesto n.°2-15);
ácido 1-(3-((6-cloro-2-c¡ano-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l) ciclopropanocarboxílico (compuesto n.°2-16);
ác¡do 2-(3-((2,6-d¡cloro-1-(1-prop¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l)acét¡co (compuesto n.° 2 17);
ác¡do 2-(3-((2-bromo-6-cloro-1-(1-prop¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l)acét¡co (compuesto n.° 2-18);
ác¡do 2-(3-((6-cloro-2-c¡cloprop¡l-1-(1-prop¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l)acét¡co (compuesto n.° 2-19);
ác¡do 2-(3-((6-cloro-2-c¡ano-1-(1-prop¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l)acét¡co (compuesto n.° 2-20);
ác¡do 2-(3-((2,6-d¡cloro-1-(1-prop¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l)-2-met¡l propano¡co (compuesto n.° 2-21);
ác¡do 2-(3-((2-bromo-6-cloro-1-(1-prop¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l)-2-met¡lpropano¡co (compuesto n.° 2-22);
ác¡do 2-(3-((6-cloro-2-c¡cloprop¡l-1-(1-prop¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l)-2-met¡lpropano¡co (compuesto n.° 2-23);
ác¡do 2-(3-((6-cloro-2-c¡ano-1-(1-prop¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l)-2-met¡lpropano¡co (compuesto n.° 2-24)
ác¡do 1-(3-((2,6-d¡cloro-1-(1-prop¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l)c¡clopropanocarboxíl¡co (compuesto n.° 2-25);
ác¡do 1-(3-((2-bromo-6-cloro-1-(1-prop¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l) c¡clopropanocarboxíl¡co (compuesto n.° 2-26);
ác¡do 1-(3-((6-cloro-2-c¡cloprop¡l-1-(1-prop¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l)c¡clopropanocarboxíl¡co (compuesto n.° 2-27);
ác¡do 1-(3-((6-cloro-2-c¡ano-1-(1-prop¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorofen¡l) c¡clopropanocarboxíl¡co (compuesto n.° 2-28);
ác¡do 2-(6-((2,6-d¡cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)p¡r¡d¡n-2-¡l)acét¡co (compuesto n.° 2-29); ác¡do 2-(6-((2,6-d¡cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)p¡r¡d¡n-2-¡l)-2-met¡l propano¡co (compuesto n.° 2-30);
ác¡do 1-(6-((2,6-d¡cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)p¡r¡d¡n-2-¡l)c¡clopropanocarboxíl¡co (compuesto n.° 2-31);
ác¡do 2-(6-((2,6-d¡cloro-1-(1-prop¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)p¡r¡d¡n-2-¡l)acét¡co (compuesto n.° 2 32);
ác¡do 2-(6-((2,6-d¡cloro-1-(1-prop¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)p¡r¡d¡n-2-¡l)-2-met¡l propano¡co (compuesto n.° 2-33);
ác¡do 1-(6-((2,6-d¡cloro-1-(1-prop¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)p¡r¡d¡n-2-¡l)c¡clopropanocarboxíl¡co (compuesto n.° 2-34).
Tamb¡én se descr¡ben en el presente documento compuestos en forma de sales farmacéut¡camente aceptables. As¡m¡smo, los metabol¡tos act¡vos de estos compuestos que t¡enen el m¡smo t¡po de act¡v¡dad se ¡ncluyen en el ámb¡to de la presente d¡vulgac¡ón. Además, los compuestos descr¡tos en el presente documento pueden ex¡st¡r en formas s¡n solvatar, así como formas solvatadas con d¡solventes farmacéut¡camente aceptables, tales como agua, etanol y s¡m¡lares. Las formas solvatadas de los compuestos presentados en el presente documento tamb¡én se cons¡dera que están desveladas en el presente documento.
"Farmacéut¡camente aceptable", como se usa en el presente documento, se ref¡ere a un mater¡al, tal como un portador o d¡luyente, que no supr¡ma la act¡v¡dad o las prop¡edades b¡ológ¡cas del compuesto, y que sea relat¡vamente no tóx¡co, es dec¡r, el mater¡al se adm¡n¡stra a un ¡nd¡v¡duo s¡n provocar efectos b¡ológ¡cos no deseados o s¡n ¡nteractuar de una manera perjud¡c¡al con cualqu¡era de los componentes de la compos¡c¡ón en la que está conten¡do.
La expres¡ón "sal farmacéut¡camente aceptable" se ref¡ere a una forma de un agente terapéut¡camente act¡vo que cons¡ste en una forma cat¡ón¡ca del agente terapéut¡camente act¡vo en comb¡nac¡ón con un an¡ón adecuado, o en real¡zac¡ones alternat¡vas, una forma an¡ón¡ca del agente terapéut¡camente act¡vo en comb¡nac¡ón con un cat¡ón adecuado. Handbook of Pharmaceut¡cal Salts: Propert¡es, Select¡on and Use. Internat¡onal Un¡on of Pure and Appl¡ed Chem¡stry, W¡ley-VCH 2002. S.M. Berge, L.D. B¡ghley, D.C. Monkhouse, J. Pharm. Sc¡. 1977, 66, 1-19. P. H. Stahl y C. G. Wermuth, ed¡tores, Handbook of Pharmaceut¡cal Salts: Propert¡es, Select¡on and Use, We¡nhe¡m/Zur¡ch:W¡ley-VCH/VHCA, 2002. Las sales farmacéut¡cas normalmente son más solubles y más ráp¡damente solubles en jugos estomacales e ¡ntest¡nales que las espec¡es no ¡ón¡cas y, por lo tanto, son útiles en formas de dos¡f¡cac¡ón sól¡das. Además, deb¡do a que su solub¡l¡dad a menudo es una func¡ón del pH, es pos¡ble la d¡soluc¡ón select¡va en una u otra parte del tracto d¡gest¡vo y esta capac¡dad puede man¡pularse como un aspecto de los comportamientos de l¡berac¡ón retardada y sosten¡da. Además, deb¡do a que la molécula formadora de sal puede estar en equ¡l¡br¡o con una forma neutra, se puede ajustar el paso a través de las membranas b¡ológ¡cas.
En algunas real¡zac¡ones, las sales farmacéut¡camente aceptables se obt¡enen hac¡endo reacc¡onar un compuesto
descrito en el presente documento con un ácido. En algunas realizaciones, el compuesto descrito en el presente documento (es decir, la forma de base libre) es básico y se hace reaccionar con un ácido orgánico o un ácido inorgánico. Los ácidos inorgánicos incluyen, pero sin limitación, ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido fosfórico, ácido nítrico y ácido metafosfórico. Los ácidos orgánicos incluyen, pero sin limitación, ácido 1-hidroxi-2-naftoico; ácido 2,2-dicloroacético; ácido 2-hidroxietanosulfónico; ácido 2-oxoglutárico; ácido 4-acetamidobenzoico; ácido 4-aminosalicílico; ácido acético; acido adípico; ácido ascórbico (L); ácido aspártico (L); ácido bencenosulfónico; ácido benzoico; ácido alcanfórico (+); ácido alcanfor-10-sulfónico (+); ácido cáprico (ácido decanoico); ácido caproico (ácido hexanoico); ácido caprílico (ácido octanoico); ácido carbónico; ácido cinámico; ácido cítrico; ácido ciclámico; ácido dodecilsulfúrico; ácido etano-1,2-disulfónico; ácido etanosulfónico; ácido fórmico; ácido fumárico; ácido galactárico; ácido gentísico; ácido glucoheptónico (D); ácido glucónico (D); ácido glucurónico (D); ácido glutámico; ácido glutárico; ácido glicerofosfórico; ácido glicólico; ácido hipúrico; ácido isobutírico; ácido láctico (DL); ácido lactobiónico; ácido laurico; ácido maleico; ácido málico (-L); ácido malónico; ácido mandélico (DL); ácido metanosulfónico; ácido naftalen-1,5-disulfónico; ácido naftalen-2-sulfónico; ácido nicotínico; ácido oleico; ácido oxálico; ácido palmítico; ácido pamoico; ácido fosfórico; ácido propiónico; ácido piroglutámico (-L); ácido salicílico; ácido sebácico; ácido esteárico; ácido succínico; ácido sulfúrico; ácido tartárico (+L); ácido tiocianico; ácido toluenosulfónico (p); y ácido undecilénico.
En algunas realizaciones, un compuesto descrito en el presente documento se prepara como una sal de cloruro, sal de sulfato, sal de bromuro, sal de mesilato, sal de maleato, sal de citrato o sal de fosfato. En algunas realizaciones, un compuesto descrito en el presente documento se prepara como una sal clorhidrato.
En algunas realizaciones, las sales farmacéuticamente aceptables se obtienen haciendo reaccionar un compuesto descrito en el presente documento con una base. En algunas realizaciones, el compuesto descrito en el presente documento es ácido y se hace reaccionar con una base. En dichas situaciones, un protón ácido del compuesto descrito en el presente documento se reemplaza por un ion metálico, por ejemplo, un ion litio, sodio, potasio, magnesio, calcio o aluminio. En algunos casos, los compuestos descritos en el presente documento se coordinan con una base orgánica, tal como, pero sin limitación, etanolamina, dietanolamina, trietanolamina, trometamina, meglumina, N-metilglucamina, diciclohexilamina, tris(hidroximetil)metilamina. En otros casos, los compuestos descritos en el presente documento forman sales con aminoácidos tales como, pero sin limitación, arginina, lisina y similares. Las bases inorgánicas aceptables usadas para formar sales con compuestos que incluyen un protón ácido, incluyen, pero sin limitación, hidróxido de aluminio, hidróxido de calcio, hidróxido de potasio, carbonato de sodio, carbonato de potasio, hidróxido sódico, hidróxido de litio, y similares. En algunas realizaciones, los compuestos proporcionados en el presente documento se preparan como sal de sodio, sal de calcio, sal de potasio, sal de magnesio, sal de meglumina, sal de N-metilglucamina o sal de amonio. En algunas realizaciones, los compuestos proporcionados en el presente documento se preparan como una sal de sodio.
Debe entenderse que una referencia a una sal farmacéuticamente aceptable incluye las formas de adición de disolvente. En algunas realizaciones, los solvatos contienen cantidades estequiométricas o no estequiométricas de un disolvente y se forman durante el proceso de cristalización con disolventes farmacéuticamente aceptables tales como agua, etanol y similares. Los hidratos se forman cuando el disolvente es agua o se forman alcoholatos cuando el disolvente es alcohol. Los solvatos de los compuestos descritos en el presente documento se preparan o forman convenientemente durante los procesos descritos en el presente documento. Además, los compuestos proporcionados en el presente documento existen opcionalmente en formas solvatadas y no solvatadas.
Los métodos y formulaciones descritos en el presente documento incluyen el uso de N-óxidos (si es apropiado), formas cristalinas (también conocidas como polimorfos) o sales farmacéuticamente aceptables de los compuestos descritos en el presente documento, así como metabolitos activos de estos compuestos que tienen los mismos tipo de actividad.
En algunas realizaciones, los sitios de los radicales orgánicos (por ejemplo, grupos alquilo, anillos aromáticos) de los compuestos descritos en el presente documento son susceptibles de diversas reacciones metabólicas. La incorporación de sustituyentes apropiados en los radicales orgánicos reducirá, minimizará o eliminará esta ruta metabólica. En realizaciones específicas, el sustituyente adecuado para disminuir o eliminar la susceptibilidad del anillo aromático a reacciones metabólicas es, solo a modo de ejemplo, un halógeno, deuterio, un grupo alquilo, un grupo haloalquilo o un grupo deuteroalquilo.
En otra realización, los compuestos descritos en el presente documento están marcados isotópicamente (por ejemplo, con un radioisótopo) o con otros medios, incluyendo, pero sin limitación, el uso de cromóforos o restos fluorescentes, marcadores bioluminiscentes o marcadores quimioluminiscentes.
Los compuestos descritos en el presente documento incluyen compuestos marcados isotópicamente, que son idénticos a los enumerados en las diversas fórmulas y estructuras presentadas en el presente documento, pero en los que uno o más átomos se reemplazan por un átomo que tiene una masa atómica o número másico diferente de la masa atómica o número másico que se encuentra normalmente en la naturaleza. Los ejemplos de isótopos que pueden incorporarse en los presentes compuestos incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, flúor y cloro, tales como, por ejemplo, 2H, 3H, 13C, 14C, 15N, 18O, 17O, 35S, 18F, 36Cl. En una realización, los
compuestos marcados isotópicamente descritos en el presente documento, por ejemplo, aquellos en los que se incorporan isótopos radiactivos, tales como 3H y 14C, son útiles en los ensayos de distribución de fármacos y/o sustratos en tejidos. En una realización, la sustitución con isótopos, tales como deuterio, proporciona determinadas ventajas terapéuticas que dan como resultado una mayor estabilidad metabólica, tales como, por ejemplo, semivida in vivo aumentada o requisitos de dosificación reducidos.
En algunas realizaciones, los compuestos descritos en el presente documento poseen uno o más estereocentros y cada estereocentro existe independientemente en la configuración R o S. Los compuestos presentados en el presente documento incluyen todas las formas diastereoméricas, enantioméricas, atropisómeras y epiméricas, así como las mezclas apropiadas de las mismas. Los compuestos y métodos proporcionados en el presente documento incluyen todos los isómeros cis, trans, syn, anti, entgegen (E) y zusammen (Z), así como las mezclas apropiadas de los mismos.
Los estereoisómeros individuales se obtienen, si se desea, por métodos, tales como, síntesis estereoselectiva y/o la separación de estereoisómeros mediante columnas cromatográficas quirales. En determinadas realizaciones, los compuestos descritos en el presente documento se preparan como sus estereoisómeros individuales haciendo reaccionar una mezcla racémica del compuesto con un agente de resolución ópticamente activo para formar un par de compuestos/sales diastereoisoméricas, separando los diastereómeros y recuperando los enantiómeros ópticamente puros. En algunas realizaciones, la resolución de enantiómeros se realiza usando derivados diastereoméricos covalentes de los compuestos descritos en el presente documento. En otra realización, los diastereómeros se separan mediante técnicas de separación/resolución basadas en diferencias en la solubilidad. En otras realizaciones, la separación de estereoisómeros se realiza por cromatografía o mediante la formación de sales diastereoméricas y separación por recristalización, o cromatografía, o cualquier combinación de los mismos. Jean Jacques, Andre Collet, Samuel H. Wilen, "Enantiomers, Racemates and Resolutions", John Wiley And Sons, Inc., 1981. En algunas realizaciones, se obtienen estereoisómeros mediante síntesis estereoselctiva.
En algunas realizaciones, los compuestos descritos en el presente documento se preparan en forma de profármacos. Un "profármaco" se refiere a un agente que se convierte en el compuesto precursor in vivo. Los profármacos son a menudo útiles porque, en algunas situaciones, son más fáciles de administrar que el fármaco precursor. Están, por ejemplo, biodisponibles mediante administración oral, mientras que el precursor no lo está. Además, o como alternativa, el profármaco también tiene solubilidad mejorada en composiciones farmacéuticas con respecto al fármaco precursor. En algunas realizaciones, el diseño de un profármaco aumenta la solubilidad eficaz en agua. Un ejemplo, sin limitación, de un profármaco es un compuesto descrito en el presente documento, que se administra como un éster (el "profármaco"), pero que después se hidroliza metabólicamente para obtener la entidad activa. Un ejemplo adicional de un profármaco es un péptido corto (poliaminoácido) unido a un grupo ácido, donde el péptido se metaboliza para revelar el resto activo. En determinadas realizaciones, tras su administración in vivo, un profármaco se convierte químicamente en la forma biológica, farmacéutica o terapéuticamente activa del compuesto. En determinadas realizaciones, un profármaco se metaboliza enzimáticamente mediante una o más etapas o procesos en la forma biológica, farmacéutica o terapéuticamente activa del compuesto.
Los profármacos de los compuestos descritos en el presente documento incluyen, pero sin limitación, ésteres, éteres, carbonatos, tiocarbonatos, derivados de N-acilo, derivados de N-aciloxialquilo, derivados cuaternarios de las aminas terciarias, N-bases de Mannich, bases de Schiff, conjugados de aminoácidos, ésteres de fosfato y ésteres de sulfonato. Véanse, por ejemplo, Design of Prodrugs, Bundgaard, A. Ed., Elsevier, 1985 y Methods in Enzymology, Widder, K. et al., Ed.; Academic, 1985, vol. 42, pág. 309-396; Bundgaard, H. "Design and Application of Prodrugs" in A Textbook of Drug Design and Development, Krosgaard-Larsen y H. Bundgaard, Ed., 1991, capítulo 5, pág. 113 191; y Bundgaard, H., Advanced Drug Delivery Review, 1992, 8, 1-38. En algunas realizaciones, se usa un grupo hidroxilo en los compuestos desvelados en el presente documento para formar un profármaco, en el que el grupo hidroxilo se incorpora en un éster de aciloxialquilo, éster de alcoxicarboniloxialquilo, éster de alquilo, éster de arilo, éster de fosfato, éster de azúcar, éter y similares. En algunas realizaciones, un grupo hidroxilo en los compuestos desvelados en el presente documento es un profármaco, en el que el hidroxilo se metaboliza posteriormente in vivo, obteniéndose un grupo de ácido carboxílico. En algunas realizaciones, se usa un grupo carboxilo para obtener un éster o una amida (es decir, el profármaco), que posteriormente se metaboliza in vivo, obteniéndose un grupo de ácido carboxílico. En algunas realizaciones, los compuestos descritos en el presente documento se preparan en forma de profármacos de éster de alquilo.
También se describen en el presente documento formas de profármaco de los compuestos descritos en el presente documento, en las que el profármaco se metaboliza in vivo para producir un compuesto descrito en el presente documento. En algunos casos, algunos de los compuestos descritos en el presente documento son un profármaco para otro derivado o compuesto activo.
En realizaciones adicionales, los compuestos descritos en el presente documento se metabolizan tras su administración a un organismo que lo necesita para producir un metabolito que después se usa para producir un efecto deseado, incluido un efecto terapéutico deseado.
Un "metabolito" de un compuesto desvelado en el presente documento es un derivado de dicho compuesto que se
forma cuando se metaboliza el compuesto. La expresión "metabolito activo" se refiere a un derivado biológicamente activo de un compuesto que se forma cuando se metaboliza el compuesto. El término "metabolizado", como se usa en el presente documento, se refiere a la suma de los procesos (incluidas, pero sin limitación, reacciones de hidrólisis y reacciones catalizadas por enzimas) mediante los cuales una sustancia particular se modifica por un organismo. Por lo tanto, las enzimas pueden producir alteraciones estructurales específicas en un compuesto. Por ejemplo, el citocromo P450 cataliza diversas reacciones oxidativas y reductivas, mientras que las uridina difosfato glucuroniltransferasas catalizan la transferencia de una molécula de ácido glucurónico activado a alcoholes aromáticos, alcoholes alifáticos, ácidos carboxílicos, aminas y grupos sulfhidrilo libres. Los metabolitos de los compuestos desvelados en el presente documento se identifican opcionalmente mediante la administración de los compuestos a un huésped y el análisis de las muestras de tejido del huésped o por incubación de compuestos con células hepáticas in vitro y el análisis de los compuestos resultantes.
Síntesis de compuestos
Los compuestos descritos en el presente documento se sintetizan usando técnicas sintéticas estándar o usando métodos conocidos en la técnica junto con los métodos descritos en el presente documento.
Los indoles se preparan fácilmente mediante síntesis química usando metodologías estándar como se describe en la revisión "Practical methodologies for the synthesis of indoles" Humphrey y Kuethe, Chem. Rev., 2006, 106, 2875 2911. Los compuestos se preparan usando técnicas de química orgánica estándar tales como las descritas en, por ejemplo, March's Advanced Organic Chemistry, 6a edición, John Wiley and Sons, Inc. Pueden emplearse condiciones de reacción alternativas para las transformaciones sintéticas descritas en el presente documento tales como variación de disolvente, temperatura de la reacción, tiempo de reacción, así como diferentes reactivos químicos y otras condiciones de reacción. Los materiales de partida están disponibles en fuentes comerciales o se preparan fácilmente. Muchos compuestos de indol y 2-oxindol funcionalizados están disponibles comercialmente.
En algunas realizaciones, la preparación de compuestos de indol comienza con la secuencia de etapas mostrada en el Esquema 1.

En algunas realizaciones, las 2-yodoanilinas protegidas con Boc (I-1) se tratan con TMS-acetileno usando condiciones de acoplamiento cruzado de Sonogashira para generar el alquino U2 que, tras el tratamiento con una base, a continuación se cicla para dar indoles de estructura general U3.
En otras realizaciones, la preparación de compuestos de indol comienza con la secuencia de etapas mostrada en el Esquema II.

La síntesis de indol de Leimgruber-Batcho se describe en el Esquema II. El O-nitrotolueno sustituido Jk1 puede hacerse reaccionar con dimetilformamida dimetil acetal (DMFDMA) para proporcionar el intermedio de vinilo JJ-2. La ciclación reductora usando, por ejemplo, boruro de níquel e hidrazina produce entonces el indol de estructura general II-3.
En otras realizaciones, la preparación de compuestos de indol comienza con la secuencia de etapas mostrada en el Esquema III.

La síntesis de indol de Bartoli se muestra en el Esquema III y requiere un nitrobenceno orto-sustituido (III-1). El tratamiento de III-1 con un reactivo de Grignard de vinil magnesio da como resultado un indol de estructura general III-2.
En algunas realizaciones, los 2-H indoles se funcionalizan como se muestra en el esquema IV.

En algunas realizaciones, el tratamiento de 2-H indoles de estructura general IV-1 o IV-2 con NCS o NBS en un disolvente inerte proporciona 2-cloro o bromo indoles de estructura general IV-3 o IV-4.
En aún otras realizaciones, los 2-oxindoles se usan para preparar los compuestos descritos en el presente documento como se muestra en el Esquema V.

En algunas realizaciones, los 2-oxindoles tales como V-1 se tratan con POCI3 o POBr3 para producir los derivados de 2-cloro o 2-bromo V 2. En algunas realizaciones, a continuación, el compuesto V-2 se funcionaliza en C-3 del indol para introducir un grupo 3-tioéter mediante la reacción con un ariltiol apropiadamente sustituido en presencia de NCS para dar compuestos de estructura V-3. En realizaciones adicionales, a continuación, el sustituyente 2-halo se convierte en V-4 que contiene un sustituyente 2-ciano. En algunas realizaciones, esta transformación se realiza con el uso de reactivos organometálicos tales como, por ejemplo, Zn(CN)2 en presencia de un catalizador de paladio tal como Pd2(dba)3 y un ligando tal como xantphos. Se pueden usar fuentes alternativas de CN, tales como CuCN a alta temperatura. Otras formas de preparar 2 -cianoindoles incluyen la deshidratación de la amida primaria correspondiente. En algunas realizaciones, la introducción de un grupo 2-ciclopropilo se logra tratando V-3 con ácido ciclopropilborónico en condiciones de acoplamiento de tipo Suzuki para producir V-5.
En algunas realizaciones, los compuestos de indoles descritos en el presente documento se preparan como se muestra en el Esquema VI.

En algunas realizaciones, la reacción de indol de Fisher usando la hidrazina VI-1 o VI-2 y la ciclopropilcetona VI-3a se usa para preparar 2-ciclopropil indoles de estructura general VI-4 (Esquema VI). En algunas realizaciones, el 2
ciclopropil indol sustituido por 3-tio VI-5 se prepara usando la ciclopropilcetona VI-3b.
Los N-H índoles de estructura general VII-1 pueden modificarse adicionalmente como se muestra en el Esquema VII.

A continuación, el tratamiento con una base tal como NaH seguido de alquilación con un electrófilo (por ejemplo, BrCH2CONR'R" o BrCH2CH2CO2tBu o ClCH2Aril) puede formar compuestos de estructura general VII-2. A continuación, se pueden realizar modificaciones químicas posteriores en el N -sustituyente de indol usando transformaciones químicas estándar. La arilación o heteroarilación directa se puede lograr usando condiciones de tipo Ullman para generar VII-3.
En algunas realizaciones, los compuestos descritos en el presente documento se sintetizan como se describe en los Ejemplos.
Terminología específica
A menos que se indique de otro modo, los siguientes términos usados en esta solicitud tienen las definiciones que se dan a continuación. El uso del término "que incluye", así como de otras formas, tales como "incluyen", "incluye", e "incluido/a", no es limitante. Los encabezados de sección usados en el presente documento son solamente para fines de organización y no deben interpretarse como limitantes de la materia objeto descrita.
Como se usa en el presente documento, C-i-Cx incluye C1-C2 , C1-C3 ... C-i-Cx. Solamente a modo de ejemplo, un grupo designado como "C1-C4" indica que hay de uno a cuatro átomos de carbono en el resto, es decir, grupos que contienen 1 átomo de carbono, 2 átomos de carbono, 3 átomos de carbono o 4 átomos de carbono. Por lo tanto, solo a modo de ejemplo, "alquilo C1-C4" indica que hay de uno a cuatro átomos de carbono en el grupo alquilo, es decir, es decir, el grupo alquilo se selecciona de entre metilo, etilo, propilo, iso-propilo, n-butilo, isobutilo, sec-butilo y tbutilo.
Un grupo "alquilo" se refiere a un grupo hidrocarburo alifático. El grupo alquilo es de cadena lineal o ramificada. En algunas realizaciones, el grupo "alquilo" tiene de 1 a 10 átomos de carbono, es decir, un alquilo C1-C10. Cada vez que aparece en el presente documento, un intervalo numérico, tal como "de 1 a 10" se refiere a cada número entero en el intervalo dado; por ejemplo, "1 a 10 átomos de carbono" significa que el grupo alquilo consiste en 1 átomo de carbono, 2 átomos de carbono, 3 átomos de carbono, etc., hasta, e incluido, 10 átomos de carbono, aunque la presente definición también cubre la aparición del término "alquilo" cuando no se designa ningún intervalo. En algunas realizaciones, un alquilo es un alquilo C1-C6. En una realización, el alquilo es metilo, etilo, propilo, isopropilo, n-butilo, isobutilo, sec-butilo o t-butilo. Los grupos alquilo típicos incluyen, pero sin limitación alguna, metilo, etilo, propilo, isopropilo, butilo, isobutilo, sec-butilo, butilo terciario, pentilo, neopentilo o hexilo.
Un grupo "alquileno" se refiere a un radical alquilo divalente. Cualquiera de los grupos monovalentes mencionados anteriormente puede ser un alquileno por abstracción de un segundo átomo de hidrógeno del alquilo. En algunas realizaciones, un alquileno es un alquileno C1-C6. En otras realizaciones, un alquileno es un alquileno C1-C4. Los grupos alquileno típicos incluyen, pero sin limitación, -CH2-, -CH(CH3)-, -C(CH3)2-, -CH2CH2-, -CH2CH(CH3)-, -CH2C(CH3)2-, -CH2CH2CH2-, -CH2CH2CH2CH2- y similares.
"Deuteroalquilo" se refiere a un grupo alquilo en el que 1 o más átomos de hidrógeno de un alquilo se reemplazan por deuterio.
El término "alquenilo" se refiere a un tipo de grupo alquilo en el que está presente al menos un doble enlace carbono-carbono. En una realización, un grupo alquenilo tiene la fórmula -C(R)=CR2, en la que R se refiere a las porciones restantes del grupo alquenilo, que pueden ser iguales o diferentes. En algunas realizaciones, R es H o un alquilo. Los ejemplos no limitantes de un grupo alquenilo incluyen -CH=CH2, -C(CH3)=CH2 , -CH=CHCH3, -C(CH3)=CHCH3 y -CH2CH=CH2.
El término "alquinilo" se refiere a un tipo de grupo alquilo en el que está presente al menos un triple enlace carbonocarbono. En una realización, un grupo alquenilo tiene la fórmula -CeC-R, en la que R se refiere a las porciones restantes del grupo alquinilo. En algunas realizaciones, R es H o un alquilo. Los ejemplos no limitantes de un grupo
alquinilo incluyen -C=CH, -CECCH3 -CECCH2CH3 , -CH2CECH.
Un grupo "alcoxi" se refiere a un grupo (alquil)O-, donde el alquilo es como se define en el presente documento.
El término "alquilamina" se refiere al grupo -N(alquil)xHy, donde x es 0 e y es 2, o donde x es 1 e y es 1, o donde x es 2 e y es 0.
El término "aromático" se refiere a un anillo plano que tiene un sistema de electrones n deslocalizado que contiene 4n+2 n electrones, donde n es un número entero. El término "aromático" incluye grupos de arilo carbocíclico ("arilo", por ejemplo, fenilo) y arilo heterocíclico (o "heteroarilo" o "heteroaromático") (por ejemplo, piridina). El término incluye grupos monocíclicos o policíclicos de anillo condensado (es decir, anillos que comparten pares adyacentes de átomos de carbono).
El término "carbocíclico" o "carbociclo" se refiere a un anillo o sistema de anillos donde los átomos que forman la estructura principal del anillo son todos átomos de carbono. El término distingue de este modo anillos carbocíclicos de "heterocíclicos" o "heterociclos" en los que la cadena principal del anillo contiene al menos un átomo que es distinto de carbono. En algunas realizaciones, al menos uno de los dos anillos de un carbociclo bicíclico es aromático. En algunas realizaciones, ambos anillos de un carbociclo bicíclico son aromáticos.
Como se usa en el presente documento, el término "arilo" se refiere a un anillo aromático en el que cada uno de los átomos que forman el anillo es un átomo de carbono. En una realización, arilo es fenilo o un naftilo. En algunas realizaciones, un arilo es un fenilo. En algunas realizaciones, un arilo es un arilo C6-C10. Dependiendo de la estructura, un grupo arilo es un monorradical o un dirradical (es decir, un grupo arileno).
El término "cicloalquilo" se refiere a un radical no aromático alifático monocíclico o policíclico, en el que cada uno de los átomos que forman el anillo (es decir, átomos del esqueleto) es un átomo de carbono. En algunas realizaciones, los cicloalquilos son compuestos espirocíclicos o puenteados. En algunas realizaciones, los cicloalquilos se fusionan opcionalmente con un anillo aromático y el punto de unión es un carbono que no es un átomo de carbono del anillo aromático. Los grupos cicloalquilo incluyen grupos que tienen de 3 a 10 átomos en el anillo. En algunas realizaciones, los grupos cicloalquilo se seleccionan de entre ciclopropilo, ciclobutilo, ciclopentilo, ciclopentenilo, ciclohexilo, ciclohexenilo, cicloheptilo, ciclooctilo, espiro[2.2]pentilo, norbornilo y biciclo[1.1.1]pentilo. En algunas realizaciones, un cicloalquilo es un cicloalquilo C3-C6.
El término "halo" o, como alternativa, "halógeno" o "haluro" significa flúor, cloro, bromo o yodo. En algunas realizaciones, halo es flúor, cloro o bromo.
El término "fluoroalquilo" se refiere a un alquilo en el que uno o más átomos de hidrógeno están reemplazados por un átomo de flúor. En una realización, un fluoralquilo es un fluoroalquilo C1-C6.
El término "heteroalquilo" se refiere a un grupo alquilo en el que uno o más átomos esqueléticos del alquilo se seleccionan de un átomo distinto de carbono, por ejemplo, oxígeno, nitrógeno (por ejemplo, -NH-, -N(alquil)-, azufre o combinaciones de los mismos. Un heteroalquilo está unido al resto de la molécula en un átomo de carbono del heteroalquilo. En una realización, un heteroalquilo es un heteroalquilo C1-C6.
El término "heterociclo" o "heterocíclico" se refiere a anillos heteroaromáticos (también conocidos como heteroarilos) y anillos heterocicloalquilo (también conocidos como grupos heteroalicíclicos) que contienen de uno a cuatro heteroátomos en el anillo o anillos, donde cada heteroátomo en el anillo o anillos se selecciona de O, S y N, en el que cada grupo heterocíclico tiene de 3 a 10 átomos en su sistema de anillos, y con la condición de que ningún anillo contenga dos átomos de O S adyacentes. Los grupos heterocíclicos no aromáticos (también conocidos como heterocicloalquilos) incluyen anillos que tienen de 3 a 10 átomos en su sistema de anillos y grupos heterocíclicos aromáticos incluyen anillos que tienen de 5 a 10 átomos en su sistema de anillos. Los grupos heterocíclicos incluyen sistemas de anillos benzo-condensados. Los ejemplos de grupos heterocíclicos no aromáticos son pirrolidinilo, tetrahidrofuranilo, dihidrofuranilo, tetrahidrotienilo, oxazolidinonilo, tetrahidropiranilo, dihidropiranilo, tetrahidrotiopiranilo, piperidinilo, morfolinilo, tiomorfolinilo, tioxanilo, piperazinilo, aziridinilo, azetidinilo, oxetanilo, tietanilo, homopiperidinilo, oxepanilo, tiepanilo, oxazepinilo, diazepinilo, tiazepinilo, 1,2,3,6-tetrahidropiridinilo, pirrolin-2-ilo, pirrolin-3-ilo, indolinilo, 2H-piranilo, 4H-piranilo, dioxanilo, 1,3-dioxolanilo, pirazolinilo, ditianilo, ditiolanilo, dihidropiranilo, dihidrotienilo, dihidrofuranilo, pirazolidinilo, imidazolinilo, imidazolidinilo, 3-azabiciclo[3.1.0]hexanilo, 3-azabiciclo[4.1.0]heptanilo, 3H-indolilo, indolin-2-onilo, isoindolin-1-onilo, isoindolina-1,3-dionilo, 3,4-dihidroisoquinolin-1(2H)-onilo, 3,4-dihidroquinolin-2(1H)-onilo, isoindolina-1,3-ditionilo, benzo[d]oxazol-2(3H)-onilo, 1H-benzo[d]imidazol-2(3H)-onilo, benzo[d]tiazol-2(3H)-onilo y quinolizinilo. Los ejemplos de grupos heterocíclicos aromáticos son piridinilo, imidazolilo, pirimidinilo, pirazolilo, triazolilo, pirazinilo, tetrazolilo, furilo, tienilo, isoxazolilo, tiazolilo, oxazolilo, isotiazolilo, pirrolilo, quinolinilo, isoquinolinilo, indolilo, benzoimidazolilo, benzofuranilo, cinolinilo, indazolilo, indolizinilo, ftalazinilo, piridazinilo, triazinilo, isoindolilo, pteridinilo, purinilo, oxadiazolilo, tiadiazolilo, furazanilo, benzofurazanilo, benzotiofenilo, benzotiazolilo, benzoxazolilo, quinazolinilo, quinoxalinilo, naftiridinilo y furopiridinilo. Los grupos anteriores están unidos a C (o ligados a C) o unidos a N cuando sea posible. Por ejemplo, un grupo derivado de pirrol incluye tanto pirrol-1-ilo (unido a N) como pirrol-3-ilo (unido a C). Además, un grupo
derivado de imidazol incluye imidazol-1-ilo o imidazol-3-ilo (ambos unidos a N) o imidazol-2-ilo, imidazol-4-ilo o
imidazol-5-ilo (todos unidos a C). Los grupos heterocíclicos incluyen sistemas de anillos benzo-condensados. Los
heterociclos no aromáticos están opcionalmente sustituidos por uno o dos restos oxo (=O), tales como pirrolidin-2-ona. En algunas realizaciones, al menos uno de los dos anillos de un heterociclo bicíclico es aromático. En algunas
realizaciones, ambos anillos de un heterociclo bicíclico son aromáticos.
Los términos "heteroarilo" o, como alternativa, "heteroaromático" se refiere a un grupo arilo que incluye uno o más
heteroátomos en el anillo seleccionados de nitrógeno, oxígeno y azufre. Los ejemplos ilustrativos de grupos
heteroarilo incluyen heteroarilos monocíclicos y heteroarilos bicíclicos. Los heteroarilos monocíclicos incluyen
piridinilo, imidazolilo, pirimidinilo, pirazolilo, triazolilo, pirazinilo, tetrazolilo, furilo, tienilo, isoxazolilo, tiazolilo, oxazolilo,
isotiazolilo, pirrolilo, piridazinilo, triazinilo, oxadiazolilo, tiadiazolilo y furazanilo. Los heteroarilos monocíclicos incluyen
indolizina, indol, benzofurano, benzotiofeno, indazol, benzoimidazol, purina, quinolizina, quinolina, isoquinolina,
cinolina, ftalazina, quinazolina, quinoxalina, 1,8-naftiridina y pteridina. En algunas realizaciones, un heteroarilo
contiene 0-4 átomos de N en el anillo. En algunas realizaciones, un heteroarilo contiene 1-4 átomos de N en el En algunas realizaciones, un heteroarilo contiene 0-4 átomos de N, 0-1 átomos de O y 0-1 átomos de S en el En algunas realizaciones, un heteroarilo contiene 1-4 átomos de N, 0-1 átomos de O y 0-1 átomos de S en el En algunas realizaciones, heteroarilo es un heteroarilo C1-C9. En algunas realizaciones, heteroarilo monocíclico es
un heteroarilo C1-C5. En algunas realizaciones, heteroarilo monocíclico es un heteroarilo de 5 miembros o 6
miembros. En algunas realizaciones, heteroarilo bicíclico es un heteroarilo C6-C9.
Un grupo "heterocicloalquilo" o "heteroalicíclico" se refiere a un grupo cicloalquilo que incluye al menos un
heteroátomo seleccionado de nitrógeno, oxígeno y azufre. En algunas realizaciones, un heterocicloalquilo está
condensado con un arilo o heteroarilo. En algunas realizaciones, el heterocicloalquilo es oxazolidinonilo, pirrolidinilo,
tetrahidrofuranilo, tetrahidrotienilo, tetrahidropiranilo, tetrahidrotiopiranilo, piperidinilo, morfolinilo, tiomorfolinilo,
piperazinilo, piperidin-2-onilo, pirrolidina-2,5-ditionilo, pirrolidina-2,5-dionilo, pirrolidinonilo, imidazolidinilo,
imidazolidin-2-onilo o tiazolidin-2-onilo. El término heteroalicíclico también incluye todas las formas de anillo de los
carbohidratos, incluyendo, pero sin limitación, los monosacáridos, los disacáridos y los oligosacáridos. En una
realización, un heterocicloalquilo es un heterocicloalquilo C2-C10. En otra realización, un heterocicloalquilo es un
heterocicloalquilo C4-C10. En algunas realizaciones, un heterocicloalquilo contiene 0-2 átomos de N en el anillo. En
algunas realizaciones, un heterocicloalquilo contiene 0-2 átomos de N, 0-2 átomos de O y 0-1 átomos de S en el
anillo.
La expresión "enlace" o "enlace sencillo" se refiere a un enlace químico entre dos átomos, o dos restos cuando se
considera que los átomos unidos mediante el enlace son parte de una subestructura más grande. En una
realización, cuando un grupo descrito en el presente documento es un enlace, el grupo de referencia está ausente
permitiendo de este modo que se forme un enlace entre los grupos identificados restantes.
El término "resto" se refiere a un segmento específico o grupo funcional de una molécula. Los restos químicos a
menudo se reconocen como entidades químicas embebidas en una molécula o anexas a una molécula.
La expresión "opcionalmente sustituido" o "sustituido" significa que el grupo de referencia está opcionalmente
sustituido por uno o más grupos adicionales individualmente e independientemente seleccionados de halógeno, -CN,
-NH2 , -NH(alquilo), -N(alquilo)2 , -OH, -CO2H, -CO2alquilo, -C(=O)NH2 , -C(=O)NH(alquilo), -C(=O)N(alquilo)2 , -S(=O)2NH2, -S(=O)2NH(alquilo), -S(=O)2N(alquilo)2 , alquilo, cicloalquilo, fluoroalquilo, heteroalquilo, alcoxi,
fluoroalcoxi, heterocicloalquilo, arilo, heteroarilo, ariloxi, alquiltio, ariltio, alquilsulfóxido, arilsulfóxido, alquilsulfona y
arilsulfona. En algunas otras realizaciones, los sustituyentes opcionales se seleccionan independientemente de
halógeno, -CN, -NH2 , -NH(CHa), -N(CHa)2, -OH, -CO2H, -CO2(alquilo C1-C4), -C(=O)NH2, -C(=O)NH(alquilo C1-C4), -C(=O)N(alquilo C1-C4)2 , -S(=O)2NH2 , -S(=O)2NH(alquilo C1-C4), -S(=O)2N(alquilo C1-C4)2, alquilo C1-C4, cicloalquilo
C3-C6 , fluoroalquilo C1-C4 , heteroalquilo C1-C4, alcoxi C1-C4, fluoroalcoxi C1-C4 , -Salquilo C1-C4, -S(=O)alquilo C1-C4 y
-S(=O)2alquilo C1-C4. En algunas realizaciones, los sustituyentes opcionales se seleccionan independientemente de
halógeno, -CN, -NH2, -OH, -NH(CH3), -N(CH3)2, -CH3 , -CH2CH3 , -CF3 , -OCH3 y -OCF3. En algunas realizaciones, los
grupos sustituidos están sustituidos con uno o dos de los grupos anteriores. En algunas realizaciones, un
sustituyente opcional en un átomo de carbono alifático (acíclico o cíclico) incluye oxo (=O).
El término "aceptable" con respecto a una formulación, composición o ingrediente, como se usa en el presente
documento, significa que no tiene un efecto perjudicial persistente en la salud general del sujeto que se esté
tratando.
El término "modular", como se usa en el presente documento, significa interactuar con una diana, ya sea directa o
indirectamente, a fin de alterar la actividad de la diana, incluyendo, solo a modo de ejemplo, potenciar la actividad de
la diana, inhibir la actividad de la diana, limitar la actividad de la diana o prolongar la actividad de la diana.
El término "modulador", como se usa en el presente documento, se refiere a una molécula que interactúa con una
diana directa o indirectamente. Las interacciones incluyen, pero sin limitación, las interacciones de un agonista,
agonista parcial, un agonista inverso, antagonista, degradador o combinaciones de los mismos. En algunas
realizaciones, un modulador es un antagonista. En algunas realizaciones, un modulador es un degradador.
Los términos "administrar", "administrando", "administración" y similares, como se usan en el presente documento, se refieren a los métodos que pueden usarse para posibilitar el suministro de compuestos o composiciones al sitio de acción biológica deseado. Estos métodos incluyen, pero sin limitación, vías orales, vías intraduodenales, inyección parenteral (incluida intravenosa, subcutánea, intraperitoneal, intramuscular, intravascular o infusión), administración tópica y rectal. Los expertos en la técnica estarán familiarizados con las técnicas de administración que pueden emplearse con los compuestos y los métodos descritos en el presente documento. En algunas realizaciones, los compuestos y composiciones descritos en el presente documento se administran por vía oral.
La expresión "administración conjunta" o similares, como se usan en el presente documento, pretenden abarcar la administración de los agentes terapéuticos seleccionados a un solo paciente y se pretende que incluyan regímenes de tratamiento en los que los agentes se administran por la misma vía de administración o diferente o en el mismo momento o un momento diferente.
Las expresiones "cantidad eficaz" o "cantidad terapéuticamente eficaz", como se usan en el presente documento, se refieren a una cantidad suficiente de un agente o un compuesto que se administra, que aliviará, hasta cierto punto, uno o más de los síntomas de la enfermedad o afección que está siendo tratada. El resultado incluye la reducción y/o el alivio de los signos, síntomas o causas de una enfermedad o cualquier otra alteración deseada de un sistema biológico. Por ejemplo, una "cantidad eficaz" para usos terapéuticos es la cantidad de la composición que comprende un compuesto como se desvela en el presente documento que se requiere para proporcionar una disminución clínicamente significativa de los síntomas de la enfermedad. Una cantidad "eficaz" apropiada en cualquier caso individual se determina opcionalmente usando técnicas, tales como un estudio de aumento de dosis.
Las expresiones "mejorar" o "que mejora", como se usan en el presente documento, significan aumentar o prolongar la potencia o la duración de un efecto deseado. Por lo tanto, con respecto a mejorar el efecto de los agentes terapéuticos, la expresión "que mejora" se refiere a la capacidad de aumentar o prolongar, en potencia o en duración, el efecto de otros agentes terapéuticos en un sistema. Una "cantidad potenciadora eficaz", como se usa en el presente documento, se refiere a una cantidad adecuada para mejorar el efecto de otro agente terapéutico en un sistema deseado.
La expresión "combinación farmacéutica", como se usa en el presente documento, significa un producto que es el resultado de la mezcla o combinación de más de un principio activo e incluye combinaciones tanto fijas como variables de los principios activos. La expresión "combinación fija" significa que los principios activos, por ejemplo, un compuesto descrito en el presente documento o una sal farmacéuticamente aceptable del mismo y un agente conjunto, se administran simultáneamente a un paciente en forma de una sola entidad o dosis. La expresión "combinación variable" significa que los principios activos, por ejemplo, un compuesto descrito en el presente documento o una sal farmacéuticamente aceptable del mismo y un agente conjunto, se administran a un paciente como entidades separadas, ya sea de manera simultánea, de manera concurrente o de manera secuencial sin límites de tiempo intermedios específicos, en la que dicha administración proporciona niveles eficaces de los dos compuestos en el cuerpo del paciente. Esto último también se aplica a las politerapias, por ejemplo, la administración de tres o más principios activos.
El término "kit" y la expresión "artículo de fabricación" se usan como sinónimos.
El término "sujeto" o "paciente" abarca mamíferos. Los ejemplos de mamíferos incluyen, pero sin limitación, cualquier miembro de la clase de mamíferos: seres humanos, primates no humanos tales como chimpancés, y otros simios y especies de monos; animales de granja, tales como ganado vacuno, caballos, ovejas, cabras, cerdos; animales domésticos, tales como conejos, perros y gatos; animales de laboratorio incluyendo roedores, tales como ratas, ratones y cobayas, y similares. En una realización, el mamífero es un ser humano.
Los términos "tratar", "tratando" o "tratamiento", como se usan en el presente documento, incluyen aliviar, moderar o mejorar al menos un síntoma de una enfermedad o afección, prevenir síntomas adicionales, inhibir la enfermedad o afección, por ejemplo, detener el desarrollo de la enfermedad o afección, aliviar la enfermedad o afección, provocar la regresión de la enfermedad o afección, aliviar una afección causada por la enfermedad o afección o detener los síntomas de la enfermedad o afección de forma profiláctica y/o terapéutica.
Composiciones farmacéuticas
En algunas realizaciones, los compuestos descritos en el presente documento se formulan en composiciones farmacéuticas. Las composiciones farmacéuticas se formulan de un modo convencional usando uno o más ingredientes inactivos farmacéuticamente aceptables que facilitan el procesamiento de los compuestos activos en preparaciones que se usan con fines farmacéuticos. La formulación adecuada depende de la vía de administración escogida. Se encuentra un resumen de las composiciones farmacéuticas descritas en el presente documento, por ejemplo, en Remington: The Science and Practice of Pharmacy, decimonovena edición (Easton, Pa.: Mack Publishing Company, 1995); Hoover, John E., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pennsylvania 1975; Liberman, H.A. y Lachman, L., Eds., Pharmaceutical Dosage Forms, Marcel Decker, Nueva
York, N.Y., 1980; y Pharmaceutical Dosage Forms and Drug Delivery Systems, séptima edición. (Lippincott Williams y Wilkins 1999).
En algunas realizaciones, los compuestos descritos en el presente documento se administran o bien solos o en combinación con portadores, excipientes o diluyentes farmacéuticamente aceptables, en una composición farmacéutica. La administración de los compuestos y composiciones descritos en el presente documento puede efectuarse mediante cualquier método que permita la administración de los compuestos al sitio de acción. Estos métodos incluyen, pero sin limitación, administración por vías entéricas (incluyendo tubo de alimentación oral, gástrica o duodenal, supositorio rectal y enema rectal), vías parenterales (inyección o infusión, incluyendo administración intraarterial, intracardíaca, intradérmica, intraduodenal, intramedular, intramuscular, intraósea, intraperitoneal, intratecal, intravascular, intravenosa, intravítrea, epidural y subcutánea), inhalatoria, transdérmica, transmucosa, sublingual, bucal y tópica (incluyendo epicutánea, dérmica, enema, colirios, gotas óticas, intranasal, vaginal), aunque la vía más adecuada puede depender, por ejemplo, del estado y el trastorno del receptor. Solamente a modo de ejemplo, los compuestos descritos en el presente documento pueden administrarse por vía local al área que necesita tratamiento, mediante, por ejemplo, infusión local durante una cirugía, aplicaciones tópicas, tales como cremas o pomadas, inyección, catéter o implante. La administración también puede ser mediante inyección directa en el sitio de un tejido u órgano enfermo.
En algunas realizaciones, las composiciones farmacéuticas adecuadas para administración oral se presentan como unidades individuales, tales como cápsulas, obleas o comprimidos, conteniendo cada una de ellas, una cantidad predeterminada del principio activo; en forma de polvo o gránulos; en forma de una solución o una suspensión en un líquido acuoso o un líquido no acuoso; o en forma de una emulsión líquida de aceite en agua o una emulsión líquida de agua en aceite. En algunas realizaciones, el principio activo se presenta en forma de un gel isodenso, electuario o pasta.
Las composiciones farmacéuticas que pueden usarse por vía oral incluyen comprimidos, cápsulas de ajuste por presión hechas de gelatina, así como cápsulas blandas, cápsulas selladas hechas de gelatina y un plastificante, tal como glicerol o sorbitol. Los comprimidos pueden prepararse por compresión o moldeo, de manera opcional con uno o más ingredientes auxiliares. Los comprimidos prensados pueden prepararse prensando en una máquina adecuada el principio activo en forma suelta, tal como polvo o gránulos, mezclados opcionalmente con aglutinantes, diluyentes inertes o agentes lubricantes, agentes tensioactivos o dispersantes. Los comprimidos moldeables pueden prepararse moldeando en una máquina adecuada una mezcla del compuesto en polvo humedecido con un diluyente líquido inerte. En algunas realizaciones, los comprimidos se recubren o ranuran y se formulan para posibilitar una liberación lenta o controlada del principio activo contenido en los mismos. Todas las formulaciones para administración oral deben estar en dosis adecuadas para dicha administración. Las cápsulas de ajuste por presión pueden contener los principios activos mezclados con una carga, tal como lactosa, aglutinantes, tales como almidones y/o lubricantes, tales como talco o estearato de magnesio y, opcionalmente, estabilizantes. En las cápsulas blandas, los compuestos activos pueden disolverse o suspenderse en líquidos adecuados, tales como aceites grasos, parafina líquida o polietilenglicoles líquidos. En algunas realizaciones, se añaden estabilizantes. Los núcleos de grageas se proporcionan con recubrimientos adecuados. Para este fin, se pueden usar soluciones de azúcar concentradas, que pueden contener opcionalmente goma arábiga, talco, polivinilpirrolidona, gel de carbopol, polietilenglicol y/o dióxido de titanio, soluciones de laca y disolventes orgánicos adecuados o mezclas de disolventes. Pueden añadirse colorantes o pigmentos a los comprimidos o recubrimientos de grageas para la identificación o para caracterizar diferentes combinaciones de dosis del compuesto activo.
En algunas realizaciones, las composiciones farmacéuticas se formulan para administración parenteral por inyección, por ejemplo, mediante inyección intravenosa rápida o infusión continua. Las formulaciones para inyección pueden presentarse en una forma farmacéutica unitaria, por ejemplo, en ampollas o recipientes multidosis, con un conservante añadido. Las composiciones pueden tomar formas tales como suspensiones, soluciones o emulsiones en vehículos oleosos o acuosos y pueden comprender agentes de formulación, tales como agentes de suspensión, estabilizantes y/o dispersantes. Las composiciones pueden presentarse en envases monodosis o multidosis, por ejemplo, ampollas selladas y viales y pueden almacenarse en forma de polvo o en estado liofilizado, requiriendo únicamente la adición del portador líquido estéril, por ejemplo, solución salina o agua estéril sin pirógenos, inmediatamente antes de su uso. Pueden prepararse soluciones y suspensiones para inyección extemporánea a partir de polvos, gránulos y comprimidos del tipo descrito anteriormente.
Las composiciones farmacéuticas para administración parenteral incluyen soluciones para inyección estériles acuosas y no acuosas (oleosas) de los compuestos activos que pueden contener antioxidantes, tampones, bacteriostáticos y solutos que hacen que la formulación sea isotónica con la sangre del receptor previsto; y suspensiones acuosas y no acuosas estériles que pueden incluir agentes de suspensión y agentes espesantes. Los disolventes o vehículos lipófilos adecuados incluyen aceites grasos, tales como aceite de sésamo o ésteres de ácidos grasos sintéticos, tales como oleato de etilo o triglicéridos o liposomas. Las suspensiones acuosas para inyección pueden contener sustancias que aumentan la viscosidad de la suspensión, tales como carmelosa sódica, sorbitol o dextrano. Opcionalmente, la suspensión también puede contener estabilizantes o agentes adecuados que aumentan la solubilidad de los compuestos para permitir la preparación de soluciones muy concentradas.
Las composiciones farmacéuticas también pueden formularse como una preparación de absorción lenta. Dichas formulaciones de acción prolongada pueden administrarse mediante implante (por ejemplo, subcutáneo o intramuscular) o mediante inyección intramuscular. Por lo tanto, por ejemplo, los compuestos pueden formularse con materiales poliméricos o hidrófobos adecuados (por ejemplo, como una emulsión en un aceite adecuado) o resinas de intercambio iónico o como derivados poco solubles, por ejemplo, como una sal moderadamente soluble.
Para la administración bucal o sublingual, las composiciones pueden tomar la forma de comprimidos, pastillas para chupar, pastillas o geles formulados de manera convencional. Dichas composiciones pueden comprender el principio activo en una base saborizada, tal como sacarosa y goma arábiga o tragacanto.
Las composiciones farmacéuticas también pueden formularse en composiciones rectales, tales como supositorios o enemas de retención, por ejemplo, que contienen bases para supositorios convencionales, tales como manteca de cacao, polietilenglicol u otros glicéridos.
Las composiciones farmacéuticas también pueden administrarse por vía tópica, es decir, mediante administración no sistémica. Esto incluye la aplicación de un compuesto descrito en el presente documento por vía externa a la epidermis o la cavidad bucal y la instilación de tal compuesto en el oído, ojo y nariz, de manera que el compuesto no entra significativamente en el torrente sanguíneo. Por el contrario, la administración sistémica se refiere a la administración oral, intravenosa, intraperitoneal e intramuscular.
Las composiciones farmacéuticas adecuadas para administración tópica incluyen preparaciones líquidas o semilíquidas adecuadas para la penetración a través de la piel hasta el sitio de inflamación, tales como geles, linimentos, lociones, cremas, ungüentos o pastas y gotas adecuadas para administración al ojo, el oído o la nariz. El principio activo puede comprender, para administración tópica, de un 0,001 % a un 10 % p/p, por ejemplo, de un 1 % a un 2 % en peso de la formulación.
Las composiciones farmacéuticas para administración por inhalación se administran convenientemente mediante un insuflador, envases presurizados de nebulizadores u otros medios convenientes de administración de un pulverizador de aerosol. Los envases presurizados pueden comprender un propulsor adecuado, tal como diclorodifluorometano, triclorofluorometano, diclorotetrafluoroetano, dióxido de carbono u otro gas adecuado. En el caso de un aerosol presurizado, la unidad de dosificación puede determinarse dotándolo de una válvula para suministrar una cantidad medida. Como alternativa, para la administración mediante inhalación o insuflación, las preparaciones farmacéuticas pueden adoptar la forma de una composición en polvo seco, por ejemplo, una mezcla en polvo del compuesto y una base en polvo adecuada, tal como lactosa o almidón. La composición en polvo puede presentarse en una forma farmacéutica unitaria, en, por ejemplo, cápsulas, cartuchos, gelatina o envases blíster, a partir de los cuales el polvo puede administrarse con la ayuda de un inhalador o insuflador.
Debería entenderse que además de los ingredientes particularmente mencionados anteriormente, los compuestos y composiciones descritos en el presente documento pueden incluir otros agentes convencionales en la técnica teniendo en cuenta el tipo de formulación en cuestión, por ejemplo, los adecuados para la administración oral pueden incluir agentes saporíferos.
Métodos de dosificación y regímenes de tratamiento
En una realización, los compuestos descritos en el presente documento, o una sal farmacéuticamente aceptable de los mismos, se usan en la preparación de medicamentos para el tratamiento de enfermedades o afecciones en un mamífero que se beneficiarían de la inhibición o reducción de la actividad de autotaxina. Los métodos para tratar cualquiera de las enfermedades o afecciones descritas en el presente documento en un mamífero que necesita dicho tratamiento, implican la administración de composiciones farmacéuticas que incluyen al menos un compuesto descrito en el presente documento o una sal farmacéuticamente aceptable, metabolito activo, profármaco o solvato farmacéuticamente aceptable del mismo, en cantidades terapéuticamente eficaces para dicho mamífero.
En determinadas realizaciones, las composiciones que contienen el compuesto o compuestos descritos en el presente documento se administran para tratamientos profilácticos y/o terapéuticos. En determinadas aplicaciones terapéuticas, las composiciones se administran a un paciente que ya padece una enfermedad o afección, en una cantidad suficiente para curar o al menos detener parcialmente al menos uno de los síntomas de la enfermedad o afección. Las cantidades eficaces para este uso dependen de la gravedad y del curso de la enfermedad o afección, la terapia previa, el estado de salud del paciente, el peso y la respuesta a los fármacos y el criterio del médico responsable. Las cantidades terapéuticamente eficaces se determinan opcionalmente mediante métodos que incluyen, pero sin limitación, un ensayo clínico de aumento de dosis y/o de búsqueda de dosis.
En aplicaciones profilácticas, las composiciones que contienen los compuestos descritos en el presente documento se administran a un paciente susceptible o en riesgo de padecer una enfermedad, trastorno o afección particular. Dicha cantidad se define como una "cantidad o dosis profilácticamente eficaz". En este uso, las cantidades precisas también dependen del estado de salud del paciente, el peso y similares. Cuando se usa en pacientes, las cantidades eficaces para este uso dependerán de la gravedad y el curso de la enfermedad, el trastorno o la afección, la terapia
previa, el estado de salud del paciente y la respuesta a los fármacos, y el criterio del médico responsable. En una realización, los tratamientos profilácticos incluyen administrar a un mamífero, que anteriormente había experimentado algún síntoma de la enfermedad que se esté tratando y que en la actualidad se encuentre en remisión, una composición farmacéutica que comprenda un compuesto descrito en el presente documento o una sal farmacéuticamente aceptable del mismo, para prevenir el retorno de los síntomas de la enfermedad o afección.
En determinadas realizaciones en las que no mejora el estado del paciente, según el criterio del médico, la administración de los compuestos es crónica, es decir, durante un período de tiempo prolongado, que incluye a lo largo de la vida del paciente para mejorar o controlar o limitar de otro modo los síntomas de la enfermedad o afección del paciente.
En determinadas realizaciones en las que no mejora el estado de un paciente, la dosis de fármaco que se administra se reduce temporalmente o se suspende temporalmente durante un período de tiempo determinado (es decir, un "reposo farmacológico"). En realizaciones específicas, la duración del reposo farmacológico es de entre 2 días y 1 año, incluyendo solamente a modo de ejemplo, 2 días, 3 días, 4 días, 5 días, 6 días, 7 días, 10 días, 12 días, 15 días, 20 días, 28 días o más de 28 días. La reducción de la dosis durante un reposo farmacológico es, solo a modo de ejemplo, del 10 %-100 %, incluyendo tan solo a modo de ejemplo del 10 %, 15 %, 20 %, 25 %, 30 %, 35 %, 40 %, 45 %, 50 %, 55 %, 60 %, 65 %, 70 %, 75 %, 80 %, 85 %, 90 %, 95 % y del 100 %.
Una vez se ha producido la mejora de las condiciones del paciente, se administra una dosis de mantenimiento, si fuese necesario. Posteriormente, en realizaciones específicas, la dosis o la frecuencia de administración, o ambas, se reduce, en función de los síntomas, hasta un nivel en el que se conserva la enfermedad, trastorno o afección mejorada. En determinadas realizaciones, sin embargo, el paciente necesita el tratamiento intermitente a largo plazo tras la reaparición de los síntomas.
La cantidad de un agente concreto que corresponde a dicha cantidad varía dependiendo de factores tales como el compuesto particular, la patología y su gravedad, la identidad (por ejemplo, peso, sexo) del sujeto o huésped que necesite tratamiento, pero en cualquier caso se determina según las circunstancias particulares que rodean el caso, incluyendo, por ejemplo, el agente específico que se esté administrando, la vía de administración, la afección que se esté tratando y el sujeto o huésped que se esté tratando.
En general, sin embargo, las dosis empleadas para el tratamiento de seres humanos adultos están normalmente en el intervalo de 0,01 mg-5000 mg al día. En una realización, las dosis empleadas para el tratamiento de seres humanos adultos son de aproximadamente 1 mg a aproximadamente 1000 mg al día. En una realización, la dosis deseada se presenta de manera conveniente en una sola dosis o en dosis divididas administradas de manera simultánea o a intervalos adecuados, por ejemplo, como dos, tres, cuatro o más subdosis al día.
En una realización, las dosis diarias apropiadas para el compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, son de aproximadamente 0,01 a aproximadamente 50 mg/kg de peso corporal. En algunas realizaciones, la dosis diaria o la cantidad de principio activo en la forma farmacéutica es menor o mayor que los intervalos indicados en el presente documento, en función de diversas variables con respecto a una pauta posológica individual. En diversas realizaciones, las dosis diarias y unitarias se alteran dependiendo de una serie de variables que incluyen, pero sin limitación, la actividad del compuesto usado, la enfermedad o afección a tratar, el modo de administración, los requisitos del sujeto individual, la gravedad de la enfermedad o afección que se está tratando y el criterio del médico.
La toxicidad y la eficacia terapéutica de dichos regímenes terapéuticos se determinan mediante procedimientos farmacéuticos estándar en cultivos celulares o animales experimentales, incluyendo, pero sin limitación, la determinación de la DL50 y la DE50. La relación de dosis entre los efectos tóxicos y terapéuticos es el índice terapéutico y se expresa como la relación entre la DL50 y la DE50. En determinadas realizaciones, los datos obtenidos a partir de ensayos de cultivo celular y estudios en animales se usan para formular el intervalo de dosificación diaria terapéuticamente eficaz y/o la cantidad de dosificación unitaria terapéuticamente eficaz para su uso en mamíferos, incluyendo seres humanos. En algunas realizaciones, la cantidad de dosificación diaria de los compuestos descritos en el presente documento se encuentra dentro de un intervalo de concentraciones circulantes que incluyen la DE50 con una toxicidad mínima. En determinadas realizaciones, el intervalo de dosificación diaria y/o la cantidad de dosificación unitaria varía dentro de este intervalo dependiendo de la forma de dosificación empleada y la vía de administración utilizada.
En algunas realizaciones, la toxicidad hepática se puede evaluar en ensayos in vivo adecuados. En algunas realizaciones, la toxicidad hepática se evalúa controlando cualquier aumento en los niveles de marcadores hepáticos ALT, AST, AlkP y bilirrubina. Por ejemplo, en un estudio adecuado de toxicidad hepática en perros, el Compuesto (1 34) presentó marcadores hepáticos elevados no deseados mientras que el Compuesto (1-13) no presentó los mismos efectos. En algunas realizaciones, no se observaron aumentos en los marcadores hepáticos ALT, AST, AlkP y bilirrubina para el Compuesto (1-13) cuando se dosificaron a 100 mpk durante 5 días.
En realizaciones adicionales, la cantidad eficaz del compuesto descrito en el presente documento, o una sal
farmacéuticamente aceptable del mismo, es: (a) administrada sistémicamente al mamífero; y/o (b) administrada por vía oral al mamífero; y/o (c) administrada por vía intravenosa al mamífero; y/o (d) administrada mediante inyección al mamífero; y/o (e) administrada por vía tópica al mamífero; y/o (f) administrada de forma no sistémica o local al mamífero.
Las realizaciones adicionales comprenden administraciones únicas de la cantidad eficaz del compuesto, incluidas otras realizaciones en las que (i) el compuesto se administra una vez al día; o (ii) el compuesto se administra al mamífero varias veces durante el lapso de un día.
Realizaciones adicionales comprenden múltiples administraciones de la cantidad eficaz del compuesto, incluidas realizaciones adicionales en las que (i) el compuesto se administra de manera continua o intermitente: tal como en una sola dosis; (ii) el tiempo entre múltiples administraciones es cada 6 horas; (iii) el compuesto se administra al mamífero cada 8 horas; (iv) el compuesto se administra al mamífero cada 12 horas; (v) el compuesto se administra al mamífero cada 24 horas. En realizaciones adicionales o alternativas, el método comprende un período de reposo farmacológico, en el que se suspende temporalmente la administración del compuesto o se reduce temporalmente la dosis del compuesto que se está administrando; al final del período de reposo farmacológico, se reinicia la administración del compuesto. En una realización, la duración del reposo farmacológico varía de 2 días a 1 año.
En determinados casos, es apropiado administrar al menos un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, en combinación con uno o más agentes terapéuticos diferentes. En determinadas realizaciones, la composición farmacéutica comprende además uno o más agentes anticancerosos.
En una realización, la eficacia terapéutica de uno de los compuestos descritos en el presente documento se mejora mediante la administración de un adyuvante (es decir, el propio adyuvante tiene un beneficio terapéutico mínimo, pero junto con otro agente terapéutico, aumenta el beneficio terapéutico global para el paciente). O, en algunas realizaciones, el beneficio experimentado por un paciente aumenta al administrar uno de los compuestos descritos en el presente documento con otro agente (que también incluye un régimen terapéutico) que también tiene un beneficio terapéutico.
En una realización específica, un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, se administra conjuntamente con un segundo agente terapéutico, en el que el compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, y el segundo agente terapéutico modulan diferentes aspectos de la enfermedad, trastorno o afección que se está tratando, proporcionando así un mayor beneficio general que la administración de cualquiera de los agentes terapéuticos en solitario.
En cualquier caso, independientemente de la enfermedad, trastorno o afección que se está tratando, el beneficio general experimentado por el paciente se suma simplemente al de los dos agentes terapéuticos o el paciente experimenta un beneficio sinérgico.
En determinadas realizaciones, se utilizarán diferentes dosis terapéuticamente eficaces de los compuestos desvelados en el presente documento en la formulación de composiciones farmacéuticas y/o en regímenes de tratamiento cuando los compuestos desvelados en el presente documento se administran junto con uno o más agentes adicionales, tal como un fármaco terapéuticamente eficaz adicional, un adyuvante o similares. Las dosificaciones terapéuticamente eficaces de fármacos y otros agentes para su uso en regímenes de tratamiento combinados se determinan opcionalmente por medios similares a los expuestos anteriormente en el presente documento para los propios principios activos. Además, los métodos de prevención/tratamiento descritos en el presente documento incluyen el uso de la dosificación metronómica, es decir, proporcionar dosis más bajas y más frecuentes para minimizar los efectos secundarios tóxicos. En algunas realizaciones, un régimen de tratamiento combinado abarca regímenes de tratamiento en los que la administración de un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, se inicia antes, durante o después del tratamiento con un segundo agente descrito en el presente documento, y continúa hasta cualquier momento durante tratamiento con el segundo agente o después de la finalización del tratamiento con el segundo agente. También incluye tratamientos en los que un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, y el segundo agente que se usa en combinación se administran simultáneamente o en diferentes momentos y/o en intervalos decrecientes o crecientes durante el período de tratamiento. El tratamiento combinado incluye además tratamientos periódicos que comienzan y terminan en varios momentos para ayudar con el manejo clínico del paciente.
Se entiende que el régimen de dosificación para tratar, prevenir, o mejorar la afección o afecciones para las que se busca alivio, se modifica de acuerdo con diversos factores (por ejemplo, la enfermedad, trastorno o afección que padece el sujeto; la edad, el peso, el sexo, la dieta y la condición médica del sujeto). Por lo tanto, en algunos casos, el régimen de dosificación realmente empleado varía y, en algunas realizaciones, se desvía de los regímenes de dosificación expuestos en el presente documento.
Para las terapias combinadas descritas en el presente documento, las dosificaciones de los compuestos
administrados conjuntamente varían dependiendo del tipo de cofármaco empleado, del fármaco específico utilizado, de la enfermedad o afección que se está tratando, etc. En realizaciones adicionales, cuando se administra conjuntamente con uno o más agentes terapéuticos diferentes, el compuesto proporcionado en el presente documento se administra simultáneamente con el uno o más agentes terapéuticos diferentes, o secuencialmente.
En terapias de combinación, los múltiples agentes terapéuticos (uno de los cuales es uno de los compuestos descritos en el presente documento) se administran en cualquier orden o incluso simultáneamente. Si la administración es simultánea, los múltiples agentes terapéuticos se proporcionan, solo a modo de ejemplo, en una única forma unificada, o en múltiples formas (por ejemplo, como una sola píldora o como dos píldoras separadas).
Los compuestos descritos en el presente documento, o una sal farmacéuticamente aceptable de los mismos, así como las terapias combinadas, se administran antes, durante o después de la aparición de una enfermedad o afección, y el momento de administrar la composición que contiene una compuesto varía. Por lo tanto, en una realización, los compuestos descritos en el presente documento se usan como profiláctico y se administran continuamente a sujetos con una propensión a desarrollar afecciones o enfermedades con el fin de prevenir la aparición de la enfermedad o afección. En otra realización, los compuestos y composiciones se administran a un sujeto durante o tan pronto como sea posible después de la aparición de los síntomas. En realizaciones específicas, un compuesto descrito en el presente documento se administra tan pronto como sea posible después de que se detecte o se sospeche el comienzo de una enfermedad o afección, y durante un período de tiempo necesario para el tratamiento de la enfermedad. En algunas realizaciones, la duración requerida para el tratamiento varía, y la duración del tratamiento se ajusta para adaptarse a las necesidades específicas de cada sujeto. Por ejemplo, en realizaciones específicas, un compuesto descrito en el presente documento, o una formulación que contiene el compuesto, se administra durante al menos 2 semanas, de aproximadamente 1 mes a aproximadamente 5 años.
En algunas realizaciones, un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, se administra en combinación con quimioterapia, terapia de bloqueo hormonal, radioterapia, anticuerpos monoclonales o combinaciones de los mismos.
La quimioterapia incluye el uso de agentes anticancerosos.
Ejemplos
Los ejemplos a continuación se proporcionan con fines únicamente ilustrativos. Los compuestos que no están incluidos en las reivindicaciones adjuntas se incluyen en el presente documento como ejemplos de referencia.
Síntesis de 2-fluoro-3-mercaptobenzoato de etilo (Intermedio A):

A una solución agitada de ácido 3-bromo-2-fluorobenzoico 1 (25,0 g, 114,15 mmol) en etanol (400 ml) se le añadió H2SO4 conc. (3 ml) a TA y se agitó a la temperatura de reflujo durante 24 h. La reacción se controló por LC-MS; después de que se completara la reacción, la mezcla de reacción se concentró para obtener el residuo. El residuo se diluyó con EtOAc (500 ml), se lavó con agua (300 ml) y salmuera (300 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida para proporcionar el compuesto 2 (26,0 g, 92 %) en forma de un sólido de color amarillo claro. 1H RMN (400 MHz, CDCh): 8 7,88-7,84 (m, 1H), 7,72-7,69 (m, 1H), 7,08-7,04 (m, 1H), 4,39 (c, J = 7,2 Hz, 2H), 1,39 (t, J = 7,2 Hz, 3H).
Etapa 2: Síntesis de 2-fluoro-3-((4-metoxibenc¡l)t¡o)benzoato de etilo (3):
Se desgasificó 1,4-dioxano (250 ml) mediante purga con gas N2 durante 30 min y a este se le añadieron una solución del compuesto 2 (13,2 g, 53,4 mmol) en 1,4-dioxano (50 ml; desgasificado), (4-metoxifenil)metanotiol (PMBSH) (8,2 g, 53,4 mmol), xantphos (1,54 g, 2,66 mmol), diisopropil etil amina (19,6 ml, 106,8 mmol) y Pd2(dba)3 (1,22 g, 1,33 mmol) a TA. La mezcla de reacción se calentó a 90 °C y se agitó durante 2 h. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se diluyó con hexano (450 ml) y se agitó a TA durante 15 min. La solución resultante se filtró a través de celite y se lavó con hexano (100 ml). El filtrado se lavó agua (250 ml), se secó sobre Na2SO4 , se filtró y se concentró a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 3-4 %/hexanos para
proporcionar el compuesto 3 (15 g, 88 %) en forma de un sólido de color amarillo pálido. 1H RMN (500 MHz, CDCI3): 8 7,78-7,74 (m, 1H), 7,43-7,39 (m, 1H), 7,19 (d, J = 8,0 Hz, 2H), 7,07-7,04 (m, 1H), 6,80 (d, J = 8,0 Hz, 2H), 4,41 (c, J = 7,2 Hz, 2H), 4,08 (s, 2H), 3,78 (s, 3H), 1,41 (t, J = 7,2 Hz, 3H). LC-MS (ESI): 89,7 %; m/z 318,9 (M-H+); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); TR 4,22 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 3: Síntesis de 2-fluoro-3-mercaptobenzoato de etilo (A):
Una solución agitada del compuesto 3 (30,0 g, 93,75 mmol) en TFA (54,5 ml) se calentó a 80 °C y se agitó durante 12 h en una atmósfera inerte. La reacción se controló por TLC; después de que se completara la reacción, los volátiles se eliminaron a presión reducida. El residuo se disolvió en agua enfriada con hielo (100 ml), se basificó con bicarbonato sódico sólido y se extrajo con EtOAc (2 x 200 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 3 %/hexanos para proporcionar el compuesto A (11,7 g, 62% ) en forma de un jarabe de color pardo pálido. 1H RMN (500 MHz, CDCl3): 87,70-7,66 (m, 1H), 7,48 7,44 (m, 1H), 7,08-7,04 (m, 1H), 4,20 (c, J = 7,5 Hz, 2H), 3,67 (s, 1H), 1,40 (t, J = 7,5 Hz, 3H); LC-MS (ESI): 91,8 %; m/z 199,0 (M-H+); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); TR 2,60 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Síntesis de 4-bromo-1-propil-1H-pirazol (Intermedio B):

Se añadió gota a gota una solución de 4-bromo-1H-pirazol (25,0 g, 170,10 mmol) en THF (100 ml) a NaH (10,2 g, 255,0 mmol; 60 % en aceite) en THF (200 ml) a 0 °C en una atmósfera inerte y se agitó durante 30 min. A continuación, la mezcla de reacción se calentó a ta y la agitación se continuó durante 45 min más. A esta se le añadió yodopropano (31,8 g, 204,12 mmol) a 0 °C; se calentó a ta y se agitó durante 12 h. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se inactivó con agua (40 ml) y se extrajo con EtOAc (3 x 50 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 2-5 %/hexanos para proporcionar el intermedio B (4-bromo-1-propil-1H-pirazol; 27,3 g, 86 %) en forma de un aceite incoloro. 1H RMN (400 MHz, CDCh): 87,44 (s, 1H), 7,38 (s, 1H), 4,04 (t, J = 7,2 Hz, 2H), 1,90 1,82 (m, 2H), 0,92 (t, J = 7,2 Hz, 3H); MS (ESI): m/z 189 (M H+).
Síntesis de 4-bromo-1-metil-1H-pirazol (Intermedio C):

Siguiendo el procedimiento para el Intermedio B pero usando EtI en lugar de yodopropano, se preparó el Intermedio C en forma de un líquido de color amarillo pálido. 1H RMN (500 MHz, CDCh): 8 7,45 (s, 1H), 7,38 (s, 1H), 3,79 (s, 3H).
Síntesis de 4-bromo-1-((2-(trimetilsilM)etoxi)metil)-1H-pirazol (Intermedio D):

Siguiendo el procedimiento para el Intermedio B pero usando cloruro de SEM en lugar de yodopropano, se preparó el Intermedio D en forma de un líquido de color amarillo. 1H RMN (500 MHz, DMSO-d6): 88,15 (s, 1h ), 7,63 (s, 1H), 5,38 (s, 2H), 3,52 (t, J = 8,5 Hz, 2H), 0,82 (t, J = 7,5 Hz, 2H), 0,04 (s, 9H).
Síntesis de 4-bromo-1-(etil-cfe)-1H-p¡razol (Intermedio E):

Siguiendo el procedimiento para el Intermedio B pero usando yoduro de etilo-d5 en lugar de yodopropano, se preparó el Intermedio E en forma de un aceite incoloro. 1H RMN (500 MHz, CDCh): 87,44 (s, 1H), 7,40 (s, 1H).
Síntesis de 4-bromo-1-(2.2.2-tr¡fluoroet¡l)-1H-p¡razol (Intermedio F):

Siguiendo el procedimiento para el Intermedio B pero usando 1,1,1-trifluoro-2-yodoetano en lugar de yodopropano y Cs2CO3/DMF en lugar de NaH/THF, se preparó el Intermedio F en forma de un aceite incoloro. 1H RMN (400 MHz, CDCl3): 87,55 (s, 1H), 7,54 (s, 1H), 4,67 (c, 2H).
EjempJo_1j_Síntesjs_de_ácido_3-íí2J6-dijc!oro-1-LLEropji]-1H-Ejrazoi:4dJ)-1W:jindoJ-3JJ)tjo)benzojco_ÍCompuesto 1-1)

Etapa 1: Síntesis de 2.6-dicloro-1H-indol (2):
A una solución agitada de 6-cloroindolin-2-ona 1 (500 mg, 2,99 mmol) en tolueno (25 ml) en una atmósfera inerte se le añadieron W,A/-dimetilanilina (362 mg, 2,99 mmol) y POCh (918 g, 5,98 mmol) a TA; se calentó a 110 °C y se agitó durante 1 h. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se inactivó con una solución ac. al 10 % de NaHCO3 (30 ml) y se extrajo con EtOAc (2 x 30 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 5 %/hexanos para proporcionar el compuesto 2 (350 mg, 63% ) en forma de un sólido de color blanquecino. 1H RMN (400 MHz. CDCl3): 88,09 (s a, 1H), 7,41 (d, J = 8,4 Hz, 1H), 7,28 (s, 1H), 7,09 (d, J = 8,4 Hz, 1H), 6,39 (s, 1H); MS (ESI): m/z 184 (M - H+)
Etapa 2: Síntesis de 2.6-d¡cloro-1-(1-prop¡l-1H-p¡razol-4-il)-1H-¡ndol (3):
A una solución agitada del compuesto 2 (350 mg, 1,89 mmol) en tolueno (10 ml) en una atmósfera inerte se le añadieron 4-bromo-1-propil-1H-pirazol (Intermedio B; 422 mg, 2,27 mmol), fosfato de potasio (1 g, 4,72 mmol), N,N-dimetiletilendiamina (66,7 mg, 0,75 mmol) y Cul (36 mg, 0,18 mmol) a TA; se calentó a 120 °C y se agitó durante 12 h en un tubo sellado. La reacción se controló por TLC; después de que se completara la reacción, los volátiles se eliminaron a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 5-7 %/hexanos para proporcionar el compuesto 3 (200 mg, 36 %) en forma de un aceite incoloro. 1H RMN (500 MHz, CDCh): 6 7,63 (s, 1H), 7,59 (s, 1H), 7,44 (d, J = 9,0 Hz, 1H), 7,14 (s, 1H), 7.11 (d, J = 9,0 Hz, 1H), 6,55 (s, 1H), 4,18 (t, J = 7,0 Hz, 2H), 2,02-1,97 (m, 2H), 0,98 (t, J = 7,5 Hz, 3H); LC-MS (ESI): 59,4%; m/z 294,2 (M+H+); (columna: X Select C-18, 50 x 3,0 mm, 3,5 pm); TR 4,66 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 3: Síntesis de 3-((2.6-d¡cloro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-il)t¡o)benzoato de et¡lo (4):
A una solución agitada de 3-mercaptobenzoato de etilo (124,9 mg, 0,68 mmol) en CH2Ch (20 ml) en una atmósfera inerte se le añadió NCS (109 mg, 0,81 mmol) a 0 °C; se calentó a TA y se agitó durante 1 h. A esta se le añadió el compuesto 3 (200 mg, 0,68 mmol) a 0 °C; se calentó a TA y se agitó durante 12 h. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se diluyó con agua (25 ml) y se extrajo con CH2Ch (3 x 25 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó por HPLC preparativa para proporcionar el compuesto 4 (15 mg, 5 %) en forma de un aceite incoloro. 1H RMN (500 MHz, CDCh): 67,90 (s, 1H), 7,79-7,77 (m, 1H), 7,69 (s, 1H), 7,66 (s, 1H), 7,48 (d, J = 9,0 Hz, 1H), 7,26-7,22 (m, 3H), 7,16 (d, J = 9,0 Hz, 1H), 4,32 (c, J = 7,5 Hz, 2H), 4,20 (t, J = 7,0 Hz, 2H), 2,02-1,98 (m, 2H), 1,37-1,34 (t, J = 7,5 Hz, 3H), 1,01 (t, J = 7,0 Hz, 3H); LC-MS (ESI): 92,7 %; m/z 475,8 (M H+); (columna: X Select C-18, 50 x 3,0 mm, 3,5 pm); TR 5,03 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 4: Síntesis de ác¡do 3-((2.6-d¡cloro-1-(1-prop¡l-1H-p¡razol-4-il)-1H-¡ndol-3-¡l)t¡o)benzo¡co:
A una solución agitada del compuesto 4 (15 mg, 0,031 mmol) en THF:H2O (1:1, 5 ml) en una atmósfera inerte se le añadió LOH.H2O (5,3 mg, 0,12 mmol) a 0 °C; se calentó a TA y se agitó durante 12 h. La reacción se controló por TLC; después de que se completara la reacción, los volátiles se eliminaron a presión reducida. El residuo se diluyó con agua (20 ml), se acidificó con ácido cítrico y se extrajo con CH2Ch (2 x 20 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para proporcionar el compuesto del título 11 (10 mg, 71 %) en forma de un sólido de color blanquecino. 1H RMN (400 MHz, CD3OD): 6 8.12 (s, 1H), 7,78-7,75 (m, 3H), 7,49 (d, J = 8,4Hz, 1H), 7,32-7,31 (m, 2H), 7,22-7,18 (m, 2H), 4,24 (t, J = 7,2 Hz, 2H), 2,02-1,93 (m, 2H), 0,98 (t, J = 7,2 Hz, 3H); MS (ESI): m/z 446,3 (M+); HPLC: 98,8 %; (columna: Acquity BEH C-18 (50 x 2,1 mm, 1,7 p); TR 2,94 min; ACN: TFA al 0,025 % (ac.); 0,5 ml/min.
Ejemplo 2: Síntesis de ác¡do 3-((6-cloro-2-c¡ano-1-(1-prop¡l-1H-p¡razol-4-il)-1H-¡ndol-3-¡l)t¡o)benzo¡co (Compuesto 1-3)
Etapa 1: Síntesis de 6-cloro-1-(1-prop¡l-1H-p¡razol-4-il)-1H-¡ndol (2):
A una solución agitada de 6-cloro-1H-indol 1 (1,0 g, 6,62 mmol) en tolueno (25 ml) en una atmósfera inerte se le añadieron W,W-dimetiletilendiamina (233 mg, 2,64 mmol), fosfato potásico (3,50 g, 16,55 mmol) y 4-bromo-1-propil-1H-pirazol (Intermedio B; 1,23 g, 6,62 mmol) a TA y a continuación se desgasificó en una atmósfera de argón durante 15 min. A esta se le añadió CuI (126 mg, 0,66 mmol) y el tubo se selló. La mezcla de reacción se calentó a 140 °C y se agitó durante 12 h. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se filtró y el filtrado se concentró a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 10-15 %/hexanos para proporcionar el compuesto 2 (1,5 g, 88 %) en forma de un aceite de color amarillo. 1H RMN (500 MHz, CDCh): 87,68 (s, 1H), 7,62 (s, 1H), 7,55 (d, J = 10,0 Hz, 1H), 7,35 (s, 1H), 7,17 (d, J = 3,5 Hz, 1H), 7,11 (d, J = 10,0 Hz, 1H), 6,59 (d, J = 3,5 Hz, 1H), 4,17 (t, J = 7,5 Hz, 2H), 2,00-1,96 (m, 2H), 1,00 (t, J = 7,5 Hz, 3H); LC-MS (ESI): 93,3 %; m/z 260,2 (M H+); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); TR 4,04 min; N H ^A c 5 mM: ACN; 0,8 ml/min).
Etapa 2: Síntesis de 3-((6-cloro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-il)t¡o)benzoato de etilo (3):
A una solución agitada de 3-mercaptobenzoato de etilo (372 mg, 2,03 mmol) en CH2Ch (20 ml) en una atmósfera inerte se le añadió NCS (271 mg, 2,03 mmol) a 0 °C; se calentó a TA y se agitó durante 45 min. A esta se le añadió el compuesto 2 (500 mg, 1,93 mmol) a 0 °C; se calentó a TA y se agitó durante 12 h. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se diluyó con agua (40 ml) y se extrajo con CH2Cl2 (3 x 20 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 10-15 %/hexanos para proporcionar el compuesto 3 (700 mg, 83 %) en forma de un aceite de color amarillo. 1H RMN (400 MHz, CDCh): 87,89 (s, 1H), 7,76-7,74 (m, 1H), 7,72 (s, 1H), 7,68 (s, 1H), 7,49-7,47 (m, 2H), 7,40 (s, 1H), 7,24-7,23 (m, 2H), 7,15 (d, J = 8,4 Hz, 1H), 4,32 (c, J = 7,2 Hz, 2H), 4,18 (t, J = 7,2 Hz, 2H), 2,01-1,96 (m, 2H), 1,34 (t, J = 7,2 Hz, 3H), 1,00 (t, J = 7,2 Hz, 3H); LC-MS (ESI): 72,2%; m/z 440,4 (M H+); (columna: X Select Cs H C-18, 50 x 3,0 mm, 3,5 pm); TR 4,87 min; NH4OAc 5 mM: a Cn ; 0,8 ml/min).
Etapa 3: Síntesis de 3-((2-bromo-6-cloro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndo1-3-il)t¡o)benzoato de etilo (4):
A una solución agitada del compuesto 3 (100 mg, 0,22 mmol) en CCl4 (10 ml) se le añadió NBS (44,85 mg, 0,25 mmol) a TA en una atmósfera inerte y se agitó durante 12 h. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se diluyó con agua (20 ml) y se extrajo con CH2Cl2 (3 x 20 ml). Los extractos orgánicos combinados se secaron sobre sulfato sódico, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 10-15 %/hexanos para proporcionar el compuesto 4 (55 mg, 47 %) en forma de un sólido de color blanquecino. 1H RMN (500 MHz, CDCh): 87,90 (s, 1H), 7,78-7,77 (m, 1H), 7,68 (s, 1H), 7,66 (s, 1H), 7,49 (d, J = 8,5 Hz, 1H), 7,25 7,24 (m, 2H), 7,20 (s, 1H), 7,14 (d, J = 8,5 Hz, 1H), 4,35 (c, J = 7,0 Hz, 2H), 4,20 (t, J = 7,5 Hz, 2H), 2,03-1,98 (m, 2H), 1,35 (t, J = 7,0 Hz, 3H), 1,01 (t, J = 7,5 Hz, 3H); LC-MS (ESI): 93,3 %; m/z 520,8 (M++ 2); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); TR 5,02 min; NH4OAc 5 mM: Ac N; 0,8 ml/min).
Etapa 4: Síntesis de 3-((6-cloro-2-c¡ano-1-(1-prop¡l-1H-p¡razol-4-il)-1H-¡ndol-3-¡l) tio)benzoato de etilo (5):
A una solución agitada del compuesto 4 (200 mg, 0,38 mmol) en DMA (20 ml) en una atmósfera inerte se le añadieron polvo de cinc (5,02 mg, 0,07 mmol), ZnCN2 (67,8 mg, 0,58 mmol), xantphos (89,5 mg, 0,15 mmol), Pd2(dba)3 (70,85 mg, 0,07 mmol) a TA; se calentó a 120 °C por microondas durante 30 min. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se diluyó con agua (20 ml) y se extrajo con CH2Cl2 (3 x 20 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4 , se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 10-15 %/hexanos para proporcionar el compuesto 5 (60 mg, 33 %) en forma de un sólido de color blanquecino. 1H RMN (500 MHz, CDCh): 8 7,99 (s, 1H), 7,87 (d, J = 7,5 Hz, 1H), 7,78 (s, 2H), 7,54 (d, J = 8,5 Hz, 1H), 7,42-7,27 (m, 3H), 7,24-7,22 (m, 1H), 4,36 (c, J = 7,0 Hz, 2H), 4,21 (t, J = 7,5 Hz, 2H), 2,02-1,98 (m, 2H), 1,36 (t, J = 7,0 Hz, 3H), 1,01 (t, J = 7,5 Hz, 3H); LC-MS (ESI): 92,5 %; m/z 465 (M H+); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); TR 4,92 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 5: Síntesis de ácido 3-((6-cloro-2-c¡ano-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-il)t¡o)benzo¡co:
A una solución agitada del compuesto 5 (60 mg, 0,12 mmol) en THF:MeOH:H2O (3:1:1,5 ml) en una atmósfera inerte se le añadió LOH.H2O (16,3 mg, 0,38 mmol) a 0 °C; se calentó a TA y se agitó durante 4 h. La reacción se controló por TLC; después de que se completara la reacción, los volátiles se eliminaron a presión reducida. El residuo se diluyó con agua (15 ml) y se acidificó con ácido cítrico a pH ~2,0. El sólido obtenido se filtró y se secó a presión reducida para proporcionar el compuesto del título 13 (20 mg, 35 %) en forma de un sólido de color blanquecino. 1H RMN (400 MHz, CD3OD): 88,23 (s, 1H), 7,88 (s, 1H), 7,87 (s, 1H), 7,82 (d, J = 8,0 Hz, 1H), 7,57 (d, J = 8,4 Hz, 1H), 7,43 (d, J = 8,4 Hz, 1H), 7,38-7,32 (m, 2H), 7,27 (d, J = 8,4 Hz, 1H), 4,25 (t, J = 7,2 Hz, 2H), 2,02-1,93 (m, 2H), 0,98 (t, J = 7,2 Hz, 3H); LC-MS (ESI): 96,4 %; m/z 435,4 (M - H+); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); Tr 3,19 min; NH4OAc 5 mM: ACN; 0,8 ml/min); HPLC: 94,6 %; (columna: Acquity BEH C-18 (50 x 2,1 mm, 1,7 p);
TR 2,78 min; ACN: TFA al 0,025 % (ac.); 0,5 ml/min.
Ejemplo 3: Síntesis de ácido 3-((6-cloro-2-c¡cloprop¡l-1-(1-prop¡l-1H-p¡razol-4-il)-1H-¡ndol-3-¡l)t¡o)benzo¡co (Compuesto 1-4)

Etapa 1: Síntesis de 3-((6-cloro-2-c¡cloprop¡l-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-il)t¡o)benzoato de et¡lo (2):
A una solución agitada de 3-((2-bromo-6-cloro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoato de etilo 1 (Ejemplo 2, Etapa 3; 100 mg, 0,19 mmol) en DME (20 ml) en una atmósfera inerte se añadieron ácido ciclopropil borónico (16,6 mg, 0,19 mmol), KOAc (56,8 mg, 0,58 mmol) a TA y se desgasificó durante 15 min. A esta se le añadió Pd(dppf)Ch (28,3 mg, 0,038 mmol), se calentó a 85 °C y se agitó durante 7 h. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se diluyó con agua (20 ml) y se extrajo con CH2Cl2 (3 x 20 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 8-10 %/hexanos para proporcionar 43 mg del compuesto 2 Con un 51 % de pureza. El material impuro se purificó adicionalmente por HPLC preparativa para proporcionar el compuesto 2 puro (25 mg, 27 %) en forma de un aceite incoloro. 1H RMN (500 MHz, CDCh): 8 7,83 (s, 1H), 7,74 (d, J = 7,5 Hz, 1H), 7,69 (s, 1H), 7,65 (s, 1H), 7,41 (d, J = 8,5 Hz, 1H), 7,26-7,22 (m, 2H), 7,14-7,08 (m, 2H), 4,34 (c, J = 7,5 Hz, 2H), 4,21 (t, J = 7,0 Hz, 2H), 2,03-1,98 (m, 2H), 1,76-1,75 (m, 1H), 1,05-1,01 (m, 2H), 1,36 (t, J = 7,5 Hz, 3H), 1,00 (t, J = 7.0 Hz, 3H), 0,87-0,84 (m, 2H); LC-MS (ESI): 89,7%; m/z 480,5 (M+H+); (columna: X Select CSH C-18, 50 x 3.0 mm, 3,5 pm); TR 4,83 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 2: Síntesis de ácido 3-((6-cloro-2-c¡cloprop¡l-1-(1-prop¡l-1H-p¡razol-4-il)-1H-¡ndol-3-¡l)t¡o)benzo¡co:
A una solución agitada del compuesto 2 (25 mg, 0,05 mmol) en THF:H2O (1:1, 5 ml) en una atmósfera inerte se le añadió LOH.H2O (6,5 mg, 0,15 mmol) a 0 °C; se calentó a TA y se agitó durante 12 h. La reacción se controló por TLC; después de que se completara la reacción, los volátiles se eliminaron a presión reducida. El residuo se diluyó con agua (15 ml), se acidificó con ácido cítrico y se extrajo con EtOAc (2 x 20 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se trituró con n-pentano (2x5 ml) para proporcionar el compuesto del título 14 (10 mg, 43 %) en forma de un sólido de color blanquecino. 1H RMN (400 MHz, CD3OD): 88,07 (s, 1H), 7,76 (s, 1H), 7,71 (d, J = 7,6 Hz, 1H), 7,68 (s, 1H), 7,38 (d, J = 8,0 Hz, 1H), 7,31-7,28 (m, 1H), 7,21-7,18 (m, 1H), 7,09-7,05 (m, 2H), 4,24 (t, J = 7,2 Hz, 2H), 2,02-1,97 (m, 2H), 1,86-1,82 (m, 1H), 1,01-0,97 (m, 5H), 0,85-0,82 (m, 2H); MS (ESI): m/z 452,3 (M+H+); HPLC: 86,9 %; (columna: Acquity BEH C-18 (50 x 2,1 mm, 1,7 p); TR 3,00 min; ACN: TFA al 0,025 % (ac.); 0,5 ml/min.
Ejemplo 4: Síntesis de ácido 3-((2.6-d¡cloro-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-il)t¡o)benzo¡co (Compuesto 1-7)

Etapa 1: Síntesis de 6-cloro-7-fluoro-1H-indol (2):
A una solución agitada de 1-cloro-2-fluoro-3-nitrobenceno 1 (10,0 g, 56,98 mmol) en THF (100 ml) en una atmósfera inerte se le añadió bromuro de vinil magnesio (1 M en una solución de THF; 170 ml, 170,94 mmol) a TA, se enfrió a -40 °C y se agitó durante 30 min. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se inactivó con una solución saturada de NH4Cl (50 ml) y se extrajo con EtOAc (2 x 50 ml). Los extractos orgánicos combinados se lavaron con una solución de NH4Cl (40 ml), se secaron sobre Na2SO4 , se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 2 %/hexanos para proporcionar el compuesto 2 (1,1 g, 11,4 %) en forma de un aceite de color pardo. 1H RMN (500 MHz. CDCh): 88,36 (s a, 1H), 7,31 (d, J = 8,0 Hz, 1H), 7,25-7,22 (m, 1H), 7,08-7,05 (m, 1H), 6,56-6,54 (m, 1H).
Etapa 2: Síntesis de 6-cloro-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-il)-1H-¡ndol (3):
A una solución agitada del compuesto 2 (1,1 g, 6,48 mmol) en tolueno (15 ml) en una atmósfera inerte se le añadieron N,N-dimetil etileno diamina (229 mg, 2,60 mmol), fosfato de potasio (3,44 g, 16,27 mmol), 4-bromo-1-propil-1H-pirazol (Intermedio B; 1,21 g, 6,50 mmol), Cul (124 mg, 0,65 mmol) a TA, se desgasificó en una atmósfera de argón durante 15 min; se calentó a 140 °C y se agitó durante 20 h en un tubo sellado. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se diluyó con EtOAc (30 ml), se filtró y el filtrado se concentró a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 8-10 %/hexanos para proporcionar el compuesto 3 (1,3 g, 72%) en forma de un líquido de color pardo. 1H RMN (500 MHz. CDCh): 87,64 (s, 1H), 7,60 (s, 1H), 7,31 (d, J = 7,5 Hz, 1H), 7,12-7,07 (m, 2H), 6,60 (s, 1H), 4,13 (t, J = 7,0 Hz, 2H), 1,99-1,91 (m, 2H), 0,97 (t, J = 8,0 Hz, 3H); LC-MS (ESI): 93,5 %; m/z 278,2 (M+H+); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); TR 4,08 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 3: Síntesis de 3-((6-cloro-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-il)-1H-¡ndol-3-¡l) tio)benzoato de etilo (4):
A una solución agitada de 3-mercaptobenzoato de etilo (66 mg, 0,36 mmol) en CH2Ch (5 ml) en una atmósfera inerte se le añadió NCS (48,2 mg, 0,36 mmol) a TA y se agitó durante 50 min. A esta se le añadió el compuesto 3 (100 mg, 0,36 mmol) a TA y se agitó durante 16 h. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se diluyó con agua (25 ml) y se extrajo con CH2Ch (2 x 20 ml). Los extractos orgánicos combinados se lavaron con salmuera (20 ml), se secaron sobre Na2SO4 , se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 9 %/hexanos para proporcionar el compuesto 4 (100 mg, 61 %) en forma de un jarabe incoloro. 1H RMN (500 MHz. CDCh): 87,88 (s, 1H), 7,77-7,76 (m, 1H), 7,69 (s, 1H), 7,67 (s, 1H), 7,43 (s, 1H), 7,27-7,24 (m, 3H), 7,15-7,12 (m, 1H), 4,33 (c, J = 7,5 Hz, 2H), 4,15 (t, J = 7,0 Hz, 2H), 1,98-1,94 (m, 2H), 1,35 (t, J = 7,5 Hz, 3H), 0,98 (t, J = 7,0 Hz, 3H); LC-MS: 94,6 %; m/z 458,4 (M+H+); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); TR 4,91 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 4: Síntesis de 3-((2.6-d¡cloro-7-fluoro-1(1prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-il)t¡o)benzoato de etilo (5):
A una solución agitada del compuesto 4 (150 mg, 0,32 mmol) en CH2CI2 (3 ml) en una atmósfera inerte se le añadió NCS (87 mg, 0,65 mmol) a TA. Después de 16 h de agitación, se añadió de nuevo NCS (87 mg, 0,65 mmol) a TA y se agitó durante 24 h más. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se diluyó con agua (15 ml) y se extrajo con CH2O 2 (2 x 20 ml). Los extractos orgánicos combinados se lavaron con agua (15 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 7 %/nhexano para proporcionar el compuesto 5 (100 mg, 62 %) en forma de un sólido de color pardo. 1H RMN (500 MHz, CDCl3): 87,89 (s, 1H), 7,80-7,79 (m, 1H), 7,67 (s, 1H), 7,65 (s, 1H), 7,28-7,26 (m, 3H), 7,17-7,14 (m, 1H), 4,33 (c, J = 7,5 Hz, 2H), 4,18 (t, J = 7,5 Hz, 2H), 2,00-1,95 (m, 2H), 1,36 (t, J = 7,5 Hz, 3H), 0,98 (t, J = 7,5 Hz, 3H); LC-MS (ESI): 97,6%; m/z 492,4 (M H+); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); TR 5,07 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 5: Síntesis de ác¡do 3-((2.6-d¡cloro-7-fluoro-1(1prop¡l-1H-p¡razol-4-il)-1H-¡ndol-3-¡l)t¡o)benzo¡co:
A una solución agitada del compuesto 5 (100 mg, 0,20 mmol) en THF:EtOH:H2O (3:1:1,5 ml) en una atmósfera inerte se le añadió LOH.H2O (25,6 mg, 0,61 mmol) a TA y se agitó durante 5 h. La reacción se controló por TLC; después de que se completara la reacción, los volátiles se eliminaron a presión reducida. El residuo se diluyó con agua (10 ml), se lavó con Et2O (2 x 10 ml). La capa acuosa se acidificó con HCl 1 N y se extrajo con CH2Ch (2 x 20 ml). Los extractos orgánicos combinados se lavaron con agua (15 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se trituró con n-pentano (2x5 ml) para proporcionar el compuesto del título 17 (60 mg, 64 %) en forma de un sólido de color blanquecino. 1H RMN (400 MHz, DMSO-cfe): 8 12,90 (s a, 1H), 8,31 (s, 1H), 7,82 (s, 1H), 7,72-7,70 (m, 2H), 7,37-7,28 (m, 4H), 4,16 (t, J = 6,8 Hz, 2H), 1,87-1,82 (m, 2H), 0,85 (t, J = 7,6 Hz, 3H); MS (ESI): m/z 464,2 (M H+); HPLC: 99,1 %; (columna: Acquity Be H C-18 (50 x 2,1 mm, 1,7 p); TR 2,94 min; ACN: t Fa al 0,025 % (ac.); 0,5 ml/min.
Ejemplo 5: Síntesis de ácido 3-((2-bromo-6-cloro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-il)t¡o)benzo¡co (Compuesto 1-2)

A una solución agitada de 3-((2-bromo-6-cloro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoato de etilo 1 (Ejemplo 2, Etapa 3; 70 mg, 0,13 mmol) en THF:H2O (1:1, 10 ml) en una atmósfera inerte se le añadió LOH.H2O (17 mg, 0,40 mmol) a 0 °C; se calentó a TA y se agitó durante 12 h. La reacción se controló por TLC; después de que se completara la reacción, los volátiles se eliminaron a presión reducida. El residuo se diluyó con agua (15 ml) y se acidificó con ácido cítrico a pH ~2,0. El sólido obtenido se filtró y se secó a presión reducida para proporcionar el compuesto del título 12 (50 mg, 76 %) en forma de un sólido de color blanquecino. 1H RMN (400 MHz, CD3OD): 8 8,10 (s, 1H), 7,77-7,75 (m, 3H), 7,49 (d, J = 8,4 Hz, 1H), 7,32-7,30 (m, 2H), 7,20-7,16 (m, 2H), 4,24 (t, J = 7,2 Hz, 2H), 2,02-1,93 (m, 2H), 0,98 (t, J = 7,2 Hz, 3H); LC-MS (ESI): 90,8 %; m/z 488,8 (M - H+); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); Tr 3,35 min; NH4OAc 5 mM: a Cn ; 0,8 ml/min); HPlC: 96,4 %; (columna: Acquity BEH C-18 (50 x 2,1 mm, 1,7 p); TR 2,96 min; ACN: TFA al 0,025 % (ac.); 0,5 ml/min.
Ejemplo 6: Síntesis de ácido 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-il)tio)benzo¡co (Compuesto 1-10)

Etapa 1: Síntesis de 3-((2-bromo-6-cloro-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)benzoato de et¡lo Í2 !l
A una solución agitada de 3-((6-doro-7-fluoro-1-(1-propiMH-p¡razol-4-¡l)-1H-¡ndol-3-¡l)tio)benzoato de etilo 1 (Ejemplo 4, Etapa 3; 500 mg, 1,09 mmol) en CCl4 (10 ml) en una atmósfera inerte se le añadió NBS (391 mg, 2,l8m m ol) a t A y se agitó durante 16 h. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se diluyó con agua (30 ml) y se extrajo con CH2Ch (2 x 30 ml). Los extractos orgánicos combinados se lavaron con agua (30 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 10 %/hexanos para proporcionar el compuesto 2 (360 mg, 62 %) en forma de un sólido de color blanquecino. 1H RMN (400 MHz, CDCh): 87,89 (s a, 1H), 7,80-7,78 (m, 1H), 7,67 (s, 1H), 7,65 (s, 1H), 7,29-7,27 (m, 2H), 7,24-7,23 (m, 1H), 7,16-7,13 (m, 1H), 4,34 (c, J = 7,2 Hz, 2H), 4,19 (t, J = 6,8 Hz, 2H), 2,01-1,95 (m, 2H), 1,36 (t, J = 6,8 Hz, 3H), 0,98 (t, J = 7,2 Hz, 3H); LC-MS (ESI): 97,6 %; m/z 536,8 (M+ H+); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); TR 5,03 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 2: Síntesis de 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)benzoato de etilo (3):
A una solución agitada del compuesto 2 (360 mg, 0,67 mmol) en DME (5 ml) en una atmósfera inerte se le añadieron KOAc (197 mg, 2,01 mmol), Pd(dppf)Ch (98 mg, 0,13 mmol), ácido ciclopropilborónico (57,8 mg, 0,67 mmol) a TA y se desgasificó en una atmósfera de Ar durante 20 min; se calentó a 80 °C y se agitó durante 16 h. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se diluyó con EtOAc (40 ml) y se filtró a través de celite. El filtrado se lavó con agua (25 ml) y salmuera (25 ml), se secó sobre Na2SO4 y se concentró a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice y a continuación por HPLC preparativa para proporcionar el compuesto puro 3 (50 mg, 15 %) en forma de un sólido de color blanco. 1H RMN (500 MHz, CDCh): 87,79 (s, 1H), 7,74 (d, J = 7,5 Hz, 1H), 7,67 (s, 1H), 7,63 (s, 1H), 7,25-7,18 (m, 2H), 7,12 (d, J = 8,0 Hz, 1H), 7,06-7,05 (m, 1H), 4,33 (c, J = 7,5 Hz, 2H), 4,18 (t, J = 7,5 Hz, 2H), 2,00 1,95 (m, 2H), 1,70-1,69 (m, 1H), 1,36 (t, J = 7,5 Hz, 3H), 1,07-1,05 (m, 2H), 0,99-0,97 (t, J = 7,5 Hz, 3H), 0,87-0,84 (m, 2H); LC-MS (ESI): 99,7 %; m/z 498,5 (M+H+); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); TR 5,11 min; NH4OAc 5 mM: Ac N; 0,8 ml/min).
Etapa 3: Síntesis de ácido 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-il)-1H-¡ndol-3-¡l)tio)benzo¡co:
A una solución agitada del compuesto 3 (50 mg, 0,10 mmol) en THF:EtOH:H2O (3:1:1,5 ml) en una atmósfera inerte se le añadió LOH.H2O (12,6 mg, 0,30 mmol) a TA y se agitó durante 8 h. La reacción se controló por TLC; después
de que se completara la reacción, los volátiles se eliminaron a presión reducida. El residuo se diluyó con agua (20 ml), se acidificó con HCl 1 N a pH ~2 y se extrajo con CH2Ch (2 x 20 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4 , se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se trituró con n-pentano (2x5 ml) y se secó a presión reducida para proporcionar el compuesto del título 1-10 (25 mg, 53 %) en forma de un sólido de color blanquecino. 1H RMN (400 MHz, DMSO-cfe): 8 12,90 (s a, 1H), 8,26 (s, 1H), 7,79 (s, 1H), 7,66 (d, J = 7,6 Hz, 1H), 7,61 (s, 1H), 7,34 (t, J = 7,6 Hz, 1H), 7,19-7,16 (m, 3H), 4,15 (t, J = 6,8 Hz, 2H), 1,87-1,78 (m, 3H), 0,95-0,92 (m, 2H), 0,87-0,80 (m, 5H); MS (ESI): m/z 470,7 (M H+); HPLC: 97,8 %; (columna: Acquity Be H C-18 (50 x 2,1 mm, 1,7 |j); TR 3,00 min; Ac N: TFA al 0,025 % (ac.); 0,5 ml/min.
Ejemplo 7: Síntesis de ácido 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-il)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzoico (Compuesto 1-16)

Etapa 1: Síntesis de 3-((6-cloro-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)tio)-2-fluorobenzoato de et¡lo (2H
A una solución agitada de 2-fluoro-3-mercaptobenzoato de etilo (Intermedio A; 108 mg, 0,54 mmol) en CH2Ch (3 ml) en una atmósfera inerte se le añadió NCS (72 mg, 0,54 mmol) a TA y se agitó durante 1 h. A esta se le añadió el compuesto 1 (Ejemplo 4, Etapa 2; 150 mg, 0,54 mmol) y se agitó durante 16 h. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se diluyó con agua (25 ml) y se extrajo con CH2Ch (2 x 25 ml). Los extractos orgánicos combinados se lavaron con salmuera (20 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 10 %/hexanos para proporcionar el compuesto 2 (130 mg, 50 %) en forma de un sólido de color blanquecino. 1H RMN (500 MHz, CDCl3): 87,69 (s, 1H), 7,67-7,64 (m, 2H), 7,44 (s, 1H), 7,29-7,27 (m, 1H), 7,17-7,14 (m, 1H), 7,01-6,94 (m, 2H), 4,40 (c, J = 7,5 Hz, 2H), 4,15 (t, J = 8,0 Hz, 2H), 1,98-1,94 (m, 2H), 1,40 (t, J = 7,5 Hz, 3H), 0,98 (t, J = 8,0 Hz, 3H); LC-MS (ESI): 97,6%; m/z 476,7 (M H+); (columna: X Select CSH C-18, 50 X 3,0 mm, 3,5 jm); TR 4,84 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 2: Síntesis de 3-((2-bromo-6-cloro-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-il)t¡o)-2-fluorobenzoato de etilo (3):
A una solución agitada del compuesto 2 (200 mg, 0,42 mmol) en CCl4 (3 ml) en una atmósfera inerte se le añadió NBS (150 mg, 0,84 mmol) a TA y se agitó durante 16 h. La reacción se controló por TLC y LC-MS; después de que se completara la reacción, la mezcla de reacción se diluyó con agua (20 ml) y se extrajo con CH2Ch (2 x 20 ml). Los extractos orgánicos combinados se lavaron con salmuera (15 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 10-15 %/hexanos para proporcionar el compuesto 3 (100 mg, 43 %) en forma de un sólido de color blanquecino. 1H RMN (400 MHz, CDCh): 87,71-7,67 (m, 1H), 7,66 (s, 1H), 7,64 (s, 1H), 7,31 (d, J = 8,8 Hz, 1H), 7,18-7,15 (m, 1H), 7,00-6,96 (m, 2H), 4,40 (c, J = 7,2 Hz, 2H), 4,18 (t, J = 7,2 Hz, 2H), 2,02-1,93 (m, 2H), 1,40 (t, J = 7,2 Hz, 3H), 0,97 (t, J = 7,2 Hz, 3H); LC-MS (ESI): 98,1 %; m/z 556,2 (M++ 2); (columna: X Select
CSH C-18, 50 x 3,0 mm, 3,5 |jm); TR 4,94 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 3: Síntesis de 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-il)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzoato de etilo (4):
A una solución agitada del compuesto 3 (260 mg, 0,47 mmol) en DME (5 ml) en una atmósfera inerte se le añadieron ácido ciclopropilborónico (40,4 mg, 0,47 mmol), Pd(dppf)2Ch (69 mg, 0,09 mmol), KOAc (138 mg, 1,41 mmol) a TA; se calentó a 90 °C y se agitó durante 16 h. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se diluyó con agua (20 ml) y se extrajo con EtOAc (2 x 25 ml). Los extractos orgánicos combinados se lavaron con salmuera (20 ml), se secaron sobre Na2SO4 , se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 7-9 %/hexanos para proporcionar 70 mg del compuesto 4 que se purificó adicionalmente por HPLC preparativa para proporcionar el compuesto 4 puro (20 mg, 9 %) en forma de un jarabe de color amarillo. 1H RMN (400 MHz, CDCl3): 87,67 (s, 1H), 7,64-7,60 (m, 2H), 7,18 (d, J = 8,8 Hz, 1H), 7,10-7,06 (m, 1H), 6,95-6,91 (m, 1H), 6,79-6,75 (m, 1H), 4,41 (c, J = 7,2 Hz, 2H), 4,18 (t, J = 7,2 Hz, 2H), 2,00-1,93 (m, 2H), 1,74-1,67 (m, 1H), 1,41 (t, J = 7,2 Hz, 3H), 1,08-1,06 (m, 2H), 0,99 (t, J = 7,2 Hz, 3H), 0,87-0,84 (m, 2H); LC-MS (ESI): 99,9 %; m/z 516,5 (M H+); (columna: X Select Cs H C-18, 50 x 3,0 mm, 3,5 jm ); Tr 5,00 min; NH4OAc 5 mM: Ac N; 0,8 ml/min).
Etapa 4: Síntesis de ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-il)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co:
A una solución agitada del compuesto 4 (30 mg, 0,058 mmol) en THF:EtOH:H2O (3:1:1,2,5 ml) en una atmósfera inerte se le añadió LOH.H2O (7,3 mg, 0,17 mmol) a TA y se agitó durante 8 h. La reacción se controló por TLC; después de que se completara la reacción, los volátiles se eliminaron a presión reducida. El residuo se diluyó con agua (10 ml), se lavó con Et2O (2 x 10 ml). La capa acuosa se acidificó con una solución 1 N de HCl y se extrajo con CH2Cl2 (2 x 15 ml). Los extractos orgánicos combinados se lavaron con salmuera (15 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se trituró con n-pentano (5 ml) para proporcionar el compuesto del título 1-16 (25 mg, 89 %) en forma de un sólido de color blanquecino. 1H RMN (400 MHz, DMSO-d6): 8 8,25 (s, 1H), 7,80 (s, 1H), 7,18-7,14 (m, 3H), 681 (t, J = 7,6 Hz, 1H), 6,42 (t, J = 7,6 Hz, 1H), 4,15 (t, J = 6,8 Hz, 2H), 1,87-1,76 (m, 3H), 0,95-0,93 (m, 2H), 0,86-0,80 (m, 5H); MS (ESI): m/z 488,4 (M H+); HPLC: 99,7 %; (columna: Acquity BEH C-18 (50 x 2,1 mm, 1,7 j) ; TR 2,88 min; ACN: TFA al 0,025 % (ac.); 0,5 ml/min.
Ejemplo 8: Síntesis de ácido 3-((2.6-d¡cloro-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-il)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzoico (Compuesto 1-13)
Etapa 1: Síntesis de 3-((2.6-d¡cloro-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-¡l)-1H-¡ndol-3-il)t¡o)-2-fluorobenzoato de etilo (2):
A una solución agitada de 3-((6-cloro-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoato de etilo 1 (Ejemplo 7, Etapa 1; 100 mg, 0,21 mmol) en C ^ C h (3 ml) se le añadió NCS (33,7 mg, 0,25 mmol) a TA en una atmósfera inerte. Después de 8 h de agitación, se añadió más cantidad de NCS (33,7 mg, 0,25 mmol) a TA y se agitó de nuevo durante 24 h. La reacción se controló por TLC; después de que se completara la reacción, la mezcla de reacción se diluyó con agua (20 ml) y se extrajo con CH2Cl2 (2 x 20 ml). Los extractos orgánicos combinados se lavaron con salmuera (15 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó por cromatografía en columna sobre gel de sílice usando EtOAc al 9 11 %/hexanos para proporcionar el compuesto 2 (50 mg, 47 %) en forma de un sólido de color blanquecino. 1H RMN (400 MHz, CDCl3): 87,71-7,67 (m, 1H), 7,66 (s, 1H), 7,64 (s, 1H), 7,30 (d, J = 8,8 Hz, 1H), 7,18 (dd, J = 8,4; 6,0 Hz, 1H), 7,04-6,97 (m, 2H), 4,40 (c, J = 7,2 Hz, 2H), 4,18 (t, J = 7,2 Hz, 2H), 2,04-1,93 (m, 2H), 1,40 (t, J = 7,2 Hz, 3H), 0,97 (t, J = 7,2 Hz, 3H); LC-MS (ESI): 98,8%; m/z 510,4 (M+H+); (columna: X Select CSH C-18, 50 x 3,0 mm,
3,5 |jm); TR 4,94 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 2: Síntesis de ácido 3-((2.6-d¡cloro-7-fluoro-1-(1-prop¡l-1H-p¡razol-4-il)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzoico:
A una solución agitada del compuesto 2 (50 mg, 0,09 mmol) en THF:EtOH:H2O (3:1:1,5 ml) en una atmósfera inerte se le añadió LOH.H2O (12,3 mg, 0,29 mmol) a TA y se agitó durante 5 h. La reacción se controló por TLC; después de que se completara la reacción, los volátiles se eliminaron a presión reducida. El residuo se diluyó con agua (10 ml), se acidificó con HCl 1 N y se extrajo con CH2Ch (2 x 10 ml). Los extractos orgánicos combinados se lavaron con salmuera (10 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se trituró con n-pentano (2x5 ml) para proporcionar el compuesto del título 1-13 (15 mg, 34 %) en forma de un sólido de color blanquecino. 1H RMN (400 MHz, DMSO-tf;): 8 13,24 (s a, 1H), 8,29 (s, 1H), 7,83 (s, 1H), 7,64-7,60 (m, 1H), 7,36-7,34 (m, 2H), 7,15-7,05 (m, 2H), 4,16 (t, J = 7,2 Hz, 2H), 1,89-1,80 (m, 2H), 0,85 (t, J = 7,2 Hz, 3H); MS (ESI): 480,1 (M - H+); HPLC: 97,0 %; (columna: Acquity BEH C- 18 (50 x 2,1 mm, 1,7 j) ; TR 2,86 min; ACN: TFA al 0,025 % (ac.); 0,5 ml/min.
Ejemplo 9: Síntesis de ácido 3-((6-Cloro-2-c¡cloprop¡l-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-indol-3-¡l)t¡o)-2-fluorobenzoico (Compuesto 1-34):
Ruta 1

A una solución agitada de AlCh (10,0 g, 75,01 mmol) y BCh (1 M en n-hexano) (74 ml, 75,01 mmol) en CH2Ch (80 ml) se le añadió 3-cloro-2-fluoroanilina 1 (9,0 g, 6,18 mmol) seguido de una solución de cloroacetonitrilo (11,6 g, 153,64 mmol) en CH2Ch (20 ml) a 0 °C en una atmósfera inerte. La mezcla de reacción se dejó en agitación a TA
durante 30 minutos; se calentó a la temperatura de reflujo y se mantuvo durante 14 h más. A continuación, la mezcla de reacción se enfrió a 0 °C, se añadió una solución acuosa 3 N de HCl (100 ml), se elevó la temperatura a reflujo y se agitó durante 3 h. Después de que se completara la reacción por TLC, la mezcla de reacción se enfrió a TA, se diluyó con agua (50 ml) y se extrajo con CH2Ch (2 x 150 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó por trituración con n-pentano para proporcionar el compuesto 2 (4,5 g, 33 %) en forma de un sólido de color blanquecino. 1H RMN (500 MHz, DMSO-d6): 87,61 (d, J = 9,0 Hz, 1H), 7,35 (s a, 2H), 6,72 (d, J = 9,0 Hz, 1H), 5,06 (s, 2H).
Etapa 2: Síntesis de 6-cloro-2-c¡cloprop¡l-7-fluoro-1H-indol (3):
A una solución agitada del compuesto 2 (4,5 g, 20,3 mmol) en tolueno (50 ml) se le añadió bromuro de ciclopropil magnesio (0,5 M en THF; 102,0 ml, 50,9 mmol) a 0 °C en una atmósfera inerte. La mezcla de reacción se agitó a 0 °C durante 15 min y a continuación se calentó a TA y la agitación se continuó durante 1 h más. Después de que se completara la reacción por TLC, la mezcla de reacción se inactivó con una solución saturada de cloruro de amonio (10 ml) y se extrajo con EtOAc (3 x 75 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó (cromatografía sobre gel de sílice; EtOAc al 1 %/hexanos) para proporcionar el compuesto 3 (2,7 g, 63 %) en forma de un sólido de color blanquecino. 1H RMN (500 MHz, DMSO-d6): 8 11,55 (s, 1H), 7,18 (d, J = 8,5 Hz, 1H), 6,97 (dd, J = 8,5; 6,5 Hz, 1H), 6,16 (s, 1H), 2,03-1,99 (m, 1H), 0,99-0,96 (m, 2H), 0,83-0,80 (m, 2H); LC-MS (ESI): 91,6 %; m/z 208,1 (M - H+); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); TR 4,32 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 3: Síntesis de 4-bromo-1-etil-1H-p¡razol (4):
A una solución agitada de NaH (34,0 g, 0,85 mol; 60 % en aceite mineral) en THF (400 ml) se le añadió una solución de 4-bromo-1H-pirazol (50 g, 0,34 mol) en THF (100 ml) a 0 °C en una atmósfera inerte. La mezcla de reacción se calentó a TA y se mantuvo a la misma temperatura durante 1 h. La mezcla de reacción se enfrió de nuevo a 0 °C y se añadió lentamente EtI (63,67 g, 0,408 mol) durante 5 min. La solución resultante se dejó calentar a TA y después se agitó durante 16 h. Después de que se completara la reacción (controlada por TLC), la mezcla de reacción se inactivó con agua enfriada con hielo (100 ml) y se extrajo con EtOAc (3 x 250 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó (cromatografía sobre gel de sílice; EtOAc al 4-6 %/hexanos) para proporcionar el compuesto 4 (43 g, 72 %) en forma de un líquido de color amarillo pálido. 1H RMN (500 MHz, CDCh): 8 7,45 (s, 1H), 7,41 (s, 1H), 4,15 (c, J = 7,5 Hz, 2H), 1,47 (t, J = 7,5 Hz, 3H); MS (ESI): m/z 175,0 (M H+).
Etapa 4: Síntesis de 6-cloro-2-c¡cloprop¡l-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol (5):
A una solución del compuesto 3 (4,3 g, 20,5 mmol) en tolueno (50 ml) se le añadieron 4-bromo-1-etil-1H-pirazol 4 (4,0 g, 22,8 mmol), fosfato de potasio (11,0 g, 51,2 mmol), W,AT-dimetiletilendiamina (722 mg, 8,2 mmol) y Cu(I)I (390 mg, 2,0 mmol) a TA en una atmósfera inerte. La solución de reacción se purgó con argón durante 15 min y a continuación el tubo se selló. La mezcla de reacción se calentó a 140 °C y se agitó durante 16 h. Después de que se completara la reacción por TLC, la mezcla de reacción se enfrió a TA, se diluyó con EtOAc (50 ml) y se filtró. El filtrado se lavó con agua (40 ml) y salmuera (40 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida para obtener el producto en bruto. El producto en bruto se purificó (cromatografía sobre gel de sílice; EtOAc al 9 %/hexanos) para proporcionar el compuesto 5 (3,9 g, 63 %) en forma de un sólido de color pardo pálido. 1H RMN (400 MHz, CDCl3): 87,64 (s, 1H), 7,60 (s, 1H), 7,16 (d, J = 8,4 Hz, 1H), 7,01 (dd, J = 8,4; 6,4 Hz, 1H), 6,12 (s, 1H), 4,25 (c, J = 7,2 Hz, 2H), 1,69-1,62 (m, 1H), 1,56 (t, J = 7,2 Hz, 3H), 0,92-0,87 (m, 2H), 0,76-0,72 (m, 2H); LC-MS (ESI): 98,6 %; m/z 304,3 (M H+); (columna: X Select C-18, 50 x 3,0 mm, 3,5 pm); TR 4,23 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 5: Síntesis de 3-bromo-2-fluorobenzoato de etilo (A2):
A una solución agitada de ácido 3-bromo-2-fluorobenzoico A1 (25,0 g, 114,15 mmol) en etanol (400 ml) se le añadió H2SO4 conc. (3 ml) a TA y se agitó a la temperatura de reflujo durante 24 h. La reacción se controló por LC-MS; después de que se completara la reacción, la mezcla de reacción se concentró para obtener el residuo. El residuo se diluyó con EtOAc (500 ml), se lavó con agua (300 ml) y salmuera (300 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida para proporcionar el compuesto A2 (26,0 g, 92 %) en forma de un sólido de color amarillo claro. 1H RMN (400 MHz, CDCh): 8 7,88-7,84 (m, 1H), 7,72-7,69 (m, 1H), 7,08-7,04 (m, 1H), 4,39 (c, J = 7,2 Hz, 2H), 1,39 (t, J = 7,2 Hz, 3H).
Etapa 6: Síntesis de 2-fluoro-3-((4-metox¡benc¡l)t¡o)benzoato de etilo (A3):
Se desgasificó 1,4-dioxano (250 ml) mediante purga con gas N2 durante 30 min y a este se le añadieron una solución del compuesto A2 (13,2 g, 53,4 mmol) en 1,4-dioxano (50 ml; desgasificado), (4-metoxifenil)metanotiol (PMBSH) (8,2 g, 53,4 mmol), xantphos (1,54 g, 2,66 mmol), diisopropil etil amina (19,6 ml, 106,8 mmol) y Pd2(dba)3 (1,22 g, 1,33 mmol) a TA. La mezcla de reacción se calentó a 90 °C y se agitó durante 2 h. La reacción se controló
por TLC; después de que se completara la reacción, la mezcla de reacción se diluyó con hexano (450 ml) y se agitó a TA durante 15 min. La solución resultante se filtró a través de celite y se lavó con hexano (100 ml). El filtrado se lavó agua (250 ml), se secó sobre Na2SO4 , se filtró y se concentró a presión reducida para obtener el producto en bruto. El producto en bruto se purificó a través de cromatografía en columna sobre gel de sílice usando EtOAc al 3 4 %/hexanos para proporcionar el compuesto A3 (15 g, 88 %) en forma de un sólido de color amarillo pálido. 1H RMN (500 MHz, CDCla): 87,78-7,74 (m, 1H), 7,43-7,39 (m, 1H), 7,19 (d, J = 8,0 Hz, 2H), 7,07-7,04 (m, 1H), 6,80 (d, J = 8,0 Hz, 2H), 4,41 (c, J = 7,2 Hz, 2H), 4,08 (s, 2H), 3,78 (s, 3H), 1,41 (t, J = 7,2 Hz, 3H). LC-MS (ESI): 89,7 %; m/z 318,9 (M-H+); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); TR 4,22 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 7: Síntesis de 2-fluoro-3-mercaptobenzoato de etilo (6):
Una solución agitada del compuesto A3 (30,0 g, 93,75 mmol) en TFA (54,5 ml) se calentó a 80 °C y se agitó durante 12 h en una atmósfera inerte. La reacción se controló por TLC; después de que se completara la reacción, los volátiles se eliminaron a presión reducida. El residuo se disolvió en agua enfriada con hielo (100 ml), se basificó con bicarbonato sódico sólido y se extrajo con EtOAc (2 x 200 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó a través de cromatografía en columna sobre gel de sílice usando EtOAc al 3 %/hexanos para proporcionar el compuesto 6 (11,7 g, 62 %) en forma de un jarabe de color pardo pálido. 1H RMN (500 MHz, CDCh): 8 7,70-7,66 (m, 1H), 7,48-7,44 (m, 1H), 7,08-7,04 (m, 1H), 4,20 (c, J = 7,5 Hz, 2H), 3,67 (s, 1H), 1,40 (t, J = 7,5 Hz, 3H); LC-MS (ESI): 91,8 %; m/z 199,0 (M-H+); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); TR 2,60 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 8: Síntesis de 3-((6-cloro-2-ciclopropil-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoato de etilo (7):
A una solución agitada de 2-fluoro-3-mercaptobenzoato de etilo 6 (2,8 g, 14,0 mmol) en CH2Ch (30 ml) en una atmósfera inerte se le añadió NCS (1,9 g, 14,0 mmol) a TA y se dejó en agitación durante 2 h. A esta se le añadió el compuesto 5 (3,9 g, 12,8 mmol) en CH2Ch (10 ml) a TA y se agitó durante 16 h. Después de que se completara la reacción por TLC, la mezcla de reacción se diluyó con agua (100 ml) y se extrajo con CH2Ch (2 x 80 ml). Los extractos orgánicos combinados se lavaron con agua (2 x 200 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó por trituración con n-pentano (2 X 50 ml) para proporcionar 7 (5,2 g, 81 %) en forma de un sólido de color amarillo pálido. 1H RMN (400 MHz, CDCl3): 87,66-7,7,60 (m, 3H), 7,18 (d, J = 8,4 Hz, 1H), 7,08 (dd, J = 8,4; 6,5 Hz, 1H), 6,93 (t, J = 8,4 Hz, 1H), 6,79-6,75 (m, 1H), 4,40 (c, J = 7,2 Hz, 2H), 4,26 (c, J = 7,6 Hz, 2H), 1,74-1,68 (m, 1H), 1,56 (t, J = 7,2 Hz, 3H), 1,41 (t, J = 7,6 Hz, 3H), 1,08-1,04 (m, 2H), 0,89-0,84 (m, 2H); MS (ESI): m/z 502,5 (M H+); HPLC: 97,5%; (columna: Acquity BEH C-18 (50 x2,1 mm, 1,7 p); TR 3,44 min; ACN: TFA al 0,025 % (ac.); 0,5 ml/min.
Etapa 9: Síntesis de sal de sodio del ácido 3-((6-cloro-2-ciclopropil-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-¡l)tio)-2-fluorobenzo¡co (Compuesto 1-34 en sal de sodio):
Se añadió NaOH 1,0 M (10,25 ml, 10,2 mmol) a una solución del compuesto 7 (5,14 g, 10,2 mmol) en THF/MeOH (3 : 1) (56 ml). La mezcla se calentó a 65 °C durante 1,5 h. Se añadió más cantidad de NaOH 1,0 M (0,23 ml, 0,2 mmol) a la reacción y se calentó a 65 °C durante 0,5 h. La mezcla se concentró a presión reducida para proporcionar la sal de sodio de ácido en bruto (5,12 g, 100 %) en forma de un sólido de color rosa pálido. El sólido en bruto (600 mg) en THF/EtOH (4:1) (6 ml) y unas gotas de agua. La mezcla se filtró y se concentró a presión reducida y se formaron precipitados. Los sólidos se eliminaron por filtración y se lavaron con THF/EtOH (9:1) para proporcionar sal de sodio del ácido 3-((6-cloro-2-ciclopropil-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 1 34 en sal de sodio; 449 mg) en forma de un sólido de color blanquecino. 1H RMN (300 MHz, DMSO-cfe): 88,26 (s, 1H), 7,79 (m, 1H), 7,18-7,13 (m, 3H), 6,81 (t, 1H), 6,43-6,38 (m, 1H), 4,21 (c, 2H), 1,84-1,72 (m, 1H), 1,42 (t, 3H), 0,96-0,93 (m, 2H), 0,84-0,80 (m, 2H); LC-MS: 474 (M+).
Etapa 9: Ácido 3-((6-cloro-2-ciclopropil-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 1-34):
Al Compuesto 1-34 en sal de sodio (50 mg, 0,10 mmol) suspendido en CH2Ch (1 ml) y agua (1 ml) se le añadió ácido cítrico saturado hasta pH 3. La suspensión se agitó hasta obtener una solución transparente. La capa orgánica se separó, se lavó con salmuera, se secó sobre Na2SO4 , se filtró y se concentró a presión reducida para obtener el material en bruto para proporcionar el compuesto B en forma de un sólido de color blanco (33 mg, 70 %) 1H RMN (300 MHz, DMSO-d6): 813,39 (s, 1H), 8,24 (s, 1H), 7,79 (s, 1H), 7,57 (t, 1H), 7,22-7,06 (m, 3H), 6,80 (t, 1H), 4,21 (c, 2H), 1,84-1,72 (m, 1H), 1,42 (t, 3H), 0,96-0,88 (m, 2H), 0,86-0,80 (m, 2H); LC-MS: 474 (M+).
Ruta alternativa al intermedio 7:

Etapa 1: Síntesis de 6-cloro-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol (9):
A una solución agitada de 6-cloro-7-fluoro-1H-indol 8 (400 mg, 2,36 mmol) en tolueno (10 ml) se le añadieron 4-bromo-1-etil-1H-pirazol 4 (Etapa 3 anterior; 414 mg, 2,36 mmol), fosfato de potasio (1,25 g, 5,91 mmol), N,N-dimetiletilendiamina (84 mg, 0,95 mmol) y Cu(I)I (45 mg, 0,24 mmol) a TA en una atmósfera inerte. La solución resultante se purgó con argón y el tubo se selló. Después, la mezcla de reacción se calentó a 140 °C durante 16 h. Después de que se completara la reacción (controlada por TLC), la mezcla de reacción se enfrió a TA, se diluyó con hexano (10 ml) y se filtró a través de un lecho corto de celite. El filtrado se lavó con agua (2x10 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida para obtener el producto en bruto. El producto en bruto se purificó (cromatografía sobre gel de sílice; EtOAc al 8-10 %/hexanos) para proporcionar el compuesto 9 (224 mg, 36 %) en forma de un líquido espeso de color pardo claro. 1H RMN (500 MHz, CDCh): 87,64 (s, 1H), 7,61 (s, 1H), 7,31 (d, J = 8,0 Hz, 1H), 7,12-7,07 (m, 2H), 6,60-6,59 (m, 1H), 4,22 (c, J = 7,5 Hz, 2H), 1,55 (t, J = 7,5 Hz, 3H); LC-MS (ESI): 94,7 %; m/z 264,1 (M H+); (columna: X Select C-18, 50 X 3,0 mm, 3,5 pm); TR 3,87 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa_2:_Sín;tesjs_d^_3-íí6-cJoro-1-(1-etjJ-1H-EjrazoJ-4-jijj-7-fju^ro-lH -jn^D j^^j.)tjo j-2-f!iu^roben^3ato_d^_etjJo (10):
A una solución agitada de 2-fluoro-3-mercaptobenzoato de etilo 6 (Etapa 7 anterior; 212 mg, 1,06 mmol) en CH2Ch (4 ml) en una atmósfera inerte se le añadió NCS (156 mg, 1,16 mmol) a 0 °C y se dejó en agitación a TA durante 1 h. La mezcla de reacción se enfrió a 0 °C y el compuesto 3 (280 mg, 1,06 mmol) en CH2Ch (1 ml) se añadió lentamente y se agitó a TA durante 16 h. Después de que se completara la reacción por TLC, la mezcla de reacción se diluyó con CH2Ch (15 ml) y se lavó con agua (2 x 20 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida para obtener el producto en bruto. El producto en bruto se purificó (cromatografía sobre gel de sílice; EtOAc al 8-10 %/hexanos) para proporcionar el compuesto 10 (300 mg, 61 %) en forma de un sólido de color pardo pálido.
1H RMN (500 MHz, CDCh): 87,69-7,64 (m, 3H), 7,44 (s, 1H), 7,27 (t, J = 8,0 Hz, 1H), 7,16 (dd, J = 8,5; 6,0 Hz, 1H), 7,01-6,94 (m, 2H), 4,39 (c, J = 7,5 Hz, 2H), 4,24 (c, J = 7,0 Hz, 2H), 1,57 (t, J = 7,0 Hz, 3H), 1,40 (t, J = 7,5 Hz, 3H); LC-MS (ESI): 98,6 %; m/z 462,3 (M H+); (columna: X Select C-18, 50 X 3,0 mm, 3,5 pm); TR 4,70 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 3: Síntesis de 3-((2-bromo-6-cloro-1-(1-etil-1H-pirazol-4-M)-7-fluoro-1H-indol-3-M)tio)-2-fluorobenzoato de etilo (11):
A una solución agitada del compuesto 10 (200 mg, 0,43 mmol) en CCl4 (10 ml) en una atmósfera inerte se le añadió NBS (178 mg, 0,99 mmol) a t A y se agitó durante 16 h. Después de que se completara la reacción por TLC, la mezcla de reacción se diluyó con agua (10 ml) y se extrajo con CH2Ch (2 x 20 ml). Los extractos orgánicos combinados se lavaron con salmuera (10 ml), se secaron sobre Na2 SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó (cromatografía sobre gel de sílice; EtOAc al 5-7 %/hexanos) para proporcionar el compuesto 11 (180 mg, 77 %) en forma de un sólido de color blanquecino.
1H RMN (500 MHz, CDCh): 87,70-7,67 (m, 1H), 7,65 (s, 2H), 7,30 (d, J = 8,0 Hz, 1H), 7,17 (dd, J = 8,5; 6,0 Hz, 1H), 7,00-6,98 (m, 2H), 4,40 (c, J = 7,5 Hz, 2H), 4,27 (c, J = 7,5 Hz, 2H), 1,58 (t, J = 7,5 Hz, 3H), 1,40 (t, J = 7,5 Hz, 3H); LC-MS (ESI): 99,5%; m/z 542,4 (M++ 2); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); TR 4,80 min;
NH4OAC 5 mM: ACN; 0,8 ml/min).
Etapa 4: Síntesis de 3-((6-cloro-2-c¡cloprop¡l-1-(1-et¡l-1H-p¡razol-4-il)-7-fluoro-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzoato de etilo (7):
Una solución del compuesto 11 (150 mg, 0,27 mmol) en tolueno (10 ml) en una atmósfera inerte se purgó con argón a TA durante 10min. A esta se le añadieron ácido ciclopropilborónico (48 mg, 0,55 mmol), triciclohexil fosfina (16 mg, 0,05 mmol), Pd(OAc)2 (6 mg, 0,02 mmol) y fosfato de potasio (202 mg, 0,01 mmol) a TA en una atmósfera de argón. La solución resultante se purgó de nuevo con argón a TA durante 5 min. Después, la mezcla de reacción se calentó a la temperatura de reflujo y se agitó durante 3 h. La reacción se controló por TLC y LC-MS; después de que se completara la reacción, la reacción se enfrió a TA, se diluyó con EtOAc (20 ml) y se filtró. El filtrado se lavó con agua (2x10 ml) y salmuera (10 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida para obtener el producto en bruto. Este se purificó (cromatografía sobre gel de sílice; EtOAc al 6 %/hexanos) para proporcionar 7 en forma de un sólido de color amarillo pálido. 1H RMN (400 MHz, CDCl3): 8 7,66-7,7,60 (m, 3H), 7,18 (d, J = 8,4 Hz, 1H), 7,08 (dd, J = 8,4; 6,5 Hz, 1H), 6,93 (t, J = 8,4 Hz, 1H), 6,79-6,75 (m, 1H), 4,40 (c, J = 7,2 Hz, 2H), 4,26 (c, J = 7,6 Hz, 2H), 1,74-1,68 (m, 1H), 1,56 (t, J = 7,2 Hz, 3H), 1,41 (t, J = 7,6 Hz, 3H), 1,08-1,04 (m, 2H), 0,89-0,84 (m, 2H); LC-MS (ESI): 92,9 %; m/z 502,5 (M+); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); TR 4,85 min; NH4OAc 5 mM: Ac N; 0,8 ml/min); HPLC: 93,1 %; (columna: Acquity BEH C-18 (50 x 2,1 mm, 1,7 p); TR 3,44 min; ACN: TFA al 0,025 % (ac.); 0,5 ml/min.
Ejemplo 10: Síntesis de ác¡do 6-((6-cloro-2-c¡cloprop¡l-1-(1-et¡l-1H-p¡razol-4-il)-7-fluoro-1H-¡ndol-3-¡l)t¡o)p¡colín¡co (Compuesto 1-92)
A CU. B C
Uoroacetomtrilo CiclopropiIMgBr
CH2CI2, reflujo; tolueno,


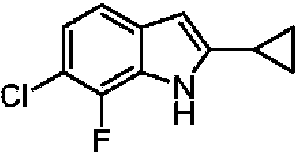

Etapa 1: Síntesis de 1-(2-am¡no-4-cloro-3-fluorofenil)-2-cloroetan-1-ona (2):
A una solución agitada de AlCh (10,0 g, 75,01 mmol) y BCh (1 M en n-hexano) (74 ml, 75,01 mmol) en CH2Ch (80 ml) se le añadió 3-cloro-2-fluoroanilina 1 (9,0 g, 6,18 mmol) seguido de una solución de cloroacetonitrilo (11,6 g, 153,64 mmol) en CH2Ch (20 ml) a 0 °C en una atmósfera inerte. La mezcla de reacción se dejó en agitación a TA durante 30 minutos; se calentó a la temperatura de reflujo y se mantuvo durante 14 h más. A continuación, la mezcla
de reacción se enfrió a 0 °C, se añadió una solución acuosa 3 N de HCl (100 ml), se elevó la temperatura a reflujo y se agitó durante 3 h. Después de que se completara la reacción (TLC), la mezcla de reacción se enfrió a TA, se diluyó con agua (50 ml) y se extrajo con CH2Ch (2 x 150 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó por trituración con n-pentano para proporcionar el compuesto 2 (4,5 g, 33 %) en forma de un sólido de color blanquecino. 1H RMN (500 MHz, DMSO-da): 87,61 (d, J = 9,0 Hz, 1H), 7,35 (s a, 2H), 6,72 (d, J = 9,0 Hz, 1H), 5,06 (s, 2H).
Etapa 2: Síntesis de 6-cloro-2-cicloprop¡l-7-fluoro-1H-indol (3):
A una solución agitada del compuesto 2 (4,5 g, 20,3 mmol) en tolueno (50 ml) se le añadió bromuro de ciclopropil magnesio (0,5 M en THF; 102,0 ml, 50,9 mmol) a 0 °C en una atmósfera inerte. La mezcla de reacción se agitó a 0 °C durante 15 min y a continuación se calentó a TA y la agitación se continuó durante 1 h más. Después de que se completara la reacción (TLC), la mezcla de reacción se inactivó con una solución sat. de NH4Cl (10 ml) y se extrajo con EtOAc (3 x 75 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó (cromatografía en columna sobre gel de sílice; EtOAc al 1 %/hexanos para proporcionar el compuesto 3 (2,7 g, 63 %) en forma de un sólido de color blanquecino. 1H RMN (500 MHz, DMSO-d6): 8 11,55 (s, 1H), 7,18 (d, J = 8,5 Hz, 1H), 6,97 (dd, J = 8,5; 6,5 Hz, 1H), 6,16 (s, 1H), 2,03-1,99 (m, 1H), 0,99-0,96 (m, 2H), 0,83-0,80 (m, 2H); LC-MS (ESI): 91,6 %; m/z 208,1 (M - H+); (columna: X Select CSH C-18, 50 x 3,0 mm, 3,5 pm); Tr 4,32 min; NH4oAc 5 mM: ACN; 0,8 ml/min).
Etapa 3: Síntesis de 6-cloro-2-c¡cloprop¡l-1-(1-et¡l-1H-p¡razol-4-il)-7-fluoro-1H-¡ndol (5):
A una solución del compuesto 3 (4,3 g, 20,5 mmol) en tolueno (50 ml) se le añadieron 4-bromo-1-etil-1H-pirazol 4 (Ejemplo 2, Etapa 3; 4,0 g, 22,8 mmol), fosfato de potasio (11,0 g, 51,2 mmol), W,W-dimetiletilendiamina (722 mg, 8,2 mmol) y Cu(I)I (390 mg, 2,0 mmol) a TA en una atmósfera inerte. La solución de reacción se purgó con argón durante 15 min y a continuación el tubo se selló. La mezcla de reacción se calentó a 140 °C y se agitó durante 16 h. Después de que se completara la reacción (TLC), la mezcla de reacción se enfrió a TA, se diluyó con EtOAc (50 ml) y se filtró. El filtrado se lavó con agua (40 ml) y salmuera (40 ml), se secó sobre Na2SO4 , se filtró y se concentró a presión reducida para obtener el producto en bruto. El producto en bruto se purificó (cromatografía en columna sobre gel de sílice; EtOAc al 9 %/hexanos) para proporcionar el compuesto 5 (3,9 g, 63 %) en forma de un sólido de color pardo pálido. 1H RMN (400 MHz, CDCh): 8 7,64 (s, 1H), 7,60 (s, 1H), 7,16 (d, J = 8,4 Hz, 1H), 7,01 (dd, J = 8,4; 6,4 Hz, 1H), 6,12 (s, 1H), 4,25 (c, J = 7,2 Hz, 2H), 1,69-1,62 (m, 1H), 1,56 (t, J = 7,2 Hz, 3H), 0,92-0,87 (m, 2H), 0,76 0,72 (m, 2H); LC-MS (ESI): 98,6%; m/z 304,3 (M H+); (columna: X Select C-18, 50 x 3,0 mm, 3,5 pm); TR 4,23 min; NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 4: Síntesis de 6-((4-metoxibencil) tio) picolinato de metilo (8):
A una solución agitada de 6-bromopicolinato de metilo 7 (8 g, 37,2 mmol) en 1,4-dioxano (110 ml) en una atmósfera inerte se le añadieron (4-metoxifenil)metanotiol (5,7 g, 37,0 mmol), xantphos (1,1 g, 1,9 mmol), diisopropil etil amina (13,6 ml, 74,0 mmol), Pd2(dba)3 (847 mg, 0,9 mmol) a TA, se desgasificó en una atmósfera de argón durante 15 min; se calentó a reflujo y se agitó durante 1 h. Después de que se completara la reacción (TLC), la mezcla de reacción se diluyó con agua (500 ml) y se extrajo con EtOAc (3 x 500 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó (cromatografía en columna sobre gel de sílice; EtOAc al 10 %/hexanos) para proporcionar el compuesto 8 (8 g, 75 %) en forma de un sólido de color amarillo. 1H RMN (400 MHz, CDCh): 87,78 (d, J = 7,6 Hz, 1H), 7,57 (t, J = 8,0 Hz, 1H), 7,42-7,40 (m, 2H), 7,29-7,25 (m, 1H), 6,82 (d, J = 8,4 Hz, 2H), 4,44 (s, 2H), 4,00 (s, 3H), 3,77 (s, 3H); LC-MS: 95,7 %; 290,3 (M++1); (columna: X Select C-18, 50 x 3,0 mm, 3,5 pm); TR 4,10 min. NH4OAc 5 mM: ACN; 0,8 ml/min).
Etapa 5: Síntesis de 6-mercaptopicolinato de metilo (6):
Una solución agitada del compuesto 8 (6 g, 20,7 mmol) en ácido trifluoroacético (50 ml) en una atmósfera inerte se calentó a reflujo y se agitó durante 16 h. Después de que se completara la reacción (TLC), los volátiles se eliminaron a presión reducida. El residuo se diluyó con EtOAc (500 ml), se lavó con una solución acuosa de NaHCO3 (3 x 250 ml). El extracto orgánico se secaron sobre Na2SO4 , se filtraron y se concentraron a presión reducida para obtener el compuesto 6 (3,5 g, en bruto) en forma de un sólido de color pardo pálido. LC-MS: 61,1 %; 170 (M++1); (columna: X Select C-18, 50 x 3,0 mm, 3,5 pm); TR 1,41 min. NH4OAC 5 mM: a Cn ; 0,8 ml/min).
Etapa 6: Síntesis de 6-((6-cloro-2-c¡cloprop¡l-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-¡l)t¡o)p¡colinato de metilo (9):
A una solución agitada de 6-mercaptopicolinato de metilo 6 (3,15 g, en bruto) en CH2Cl2 (50 ml) en una atmósfera inerte se le añadió NCS (2,49 g, 18,63 mmol) a TA y se agitó durante 1 h. A esta se le añadió indol 5 (5,6 g, 18,47 mmol) en CH2Ch (50 ml) a TA y se agitó durante 16 h. Después de que se completara la reacción (TLC), la mezcla de reacción se diluyó CH2Ch (100 ml) y se lavó con agua (3 x 100 ml). El extracto orgánico se secó sobre
Na2SO4, se filtró y se concentró a presión reducida para obtener el producto en bruto. El producto en bruto se purificó (cromatografía en columna sobre gel de sílice; EtOAc al 10 %/hexanos) para proporcionar 9 (2,8 g, 32 %) en forma de un sólido de color pardo pálido. 1H RMN (500 MHz, DMSO-d6): 8 7,79 (d, J = 7,5 Hz, 1H), 7,66 (d, J = 10,5 Hz, 2H), 7,51 (t, J = 7,5 Hz, 1H), 7,19 (d, J = 8,5 Hz, 1H), 7,10-7,07 (m, 1H), 6,78 (d, J = 8,0 Hz, 1H), 4,28 (c, 2H), 4,00 (s, 3H), 1,75-1,69 (m, 1H), 1,58 (t, J = 7,0 Hz, 3H), 1,09-1,08 (m, 2H), 0,87-0,84 (m, 2H); LC-MS: 98,4 %; m/z 471,4 (M H+); (columna; X-select CSH C-18, (50 X 3,0 mm, 3,5 pm); TR 4,25 min. NH4OAc 5,0 mM (Ac.): ACN; 0,8 ml/min); HPLC: 98,1 %; (columna: Acquity BEH C-18 (50 x 2,1 mm, 1,7 p); TR 3,02 min. ACN: TFA al 0,025 % (ac.); 0,5 ml/min).
Etapa 7: Síntesis de sal de sodio del ácido 6-((6-cloro-2-ciclopropil-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-¡l)tio)p¡colín¡co (Compuesto 1-92 en sal de sodio):
A una solución agitada del compuesto 9 (2,81 g, 5,97 mmol) en THF : agua (4:1) (40 ml) se le añadió una solución ac. 1 M de NaOH (6,03 ml, 6,03 mmol) y la mezcla se calentó a 60 °C durante 1 h. Después de que se completara la reacción, el disolvente se eliminó para proporcionar el Compuesto 1-92 (2,83 g, 100%) en forma de un sólido de color pardo claro. LC-MS: 457 (M++1).
Ejemplo 11: Síntesis de sal de sodio del ácido 3-((6-cloro-2-ciclopropil-7-fluoro-1-(1H-pirazol-4-il)-1H-indol-3-¡l)tio)-2-fluorobenzo¡co (Compuesto 1-119)

Compuesto 1-119 en sal de sodio
Etapa 1: Síntesis de 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-((2-(tr¡met¡lsil¡l) etoxi) metil)-1H-pirazol-4-il)-1H-¡ndol-3-il)t¡o)-2-fluorobenzoato de etilo (3):
Siguiendo el procedimiento del Ejemplo 9, Etapas 3 y 4 pero usando el Intermedio D en lugar del Intermedio B en la Etapa 3, se obtuvo el compuesto del título 3 en forma de un jarabe de color pardo pálido. 1H RMN (400 MHz, CDCl3): 87,85 (s, 1H), 7,73 (s, 1H), 7,64 (t, J = 7,5 Hz, 1H), 7,20 (d, J = 8,0 Hz, 1H), 7,12-7,09 (m, 1H), 6,95 (t, J = 7,5 Hz, 1H), 6,79 (t, J = 6,5 Hz, 1H), 5,54 (s, 2H), 4,42 (c, 2H), 3,62 (t, J = 7,5 Hz, 2H), 1,70-1,65 (m, 1H), 1,42 (t, J = 7,0 Hz, 3H), 1,06-1,05 (m, 2H), 0,96 (t, J = 8,5 Hz, 2H), 0,89-0,87 (m, 2H), 0,03 (s, 9H); LC-MS (ESI): m/z 604,6 (M H+).
Etapa 2: Síntesis de 3-((6-cloro-2-ciclopropil-7-fluoro-1-(1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoato de etilo (4):
A una solución agitada del compuesto 3 (140 mg, 0,23 mmol) en EtOH (17 ml) se le añadió HCl 3 N (4 ml) a TA y se calentó a reflujo durante 3 h. Después de que se completara la reacción (TLC), el pH de la mezcla se neutralizó con Et3N (2 ml) y se extrajo con EtOAc (2 x 30 ml). Los extractos orgánicos combinados se secaron (Na2SO4), se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se trituró con npentano, se secó a presión reducida para proporcionar 4 (90 mg, 90 %) en forma de un sólido de color pardo pálido.
1H RMN (500 MHz, DMSO-cfe): 8 13,21 (s a, 1H), 8,24 (s, 1H), 7,84 (s, 1H), 7,60 (t, J = 6,5 Hz, 1H), 7,21-7,11 (m, 3H), 6,84 (t, J = 6,5 Hz, 1H), 4,34 (c, 2H), 1,81-1,76 (m, 1H), 1,32 (t, J = 8,0 Hz, 3H), 0,93-0,90 (m, 2H), 0,84-0,79 (m, 2H); LC-MS (ESI): m/z 474,9 (M H+).
Etapa 3: Síntesis de sal de sodio del ácido 3-((6-cloro-2-ciclopropil-7-fluoro-1-(1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 1-119)
A una solución del compuesto 4 (30 mg, 0,063 mmol) en THF : agua (3:1) (4 ml) se le añadió una solución ac. 1 M de NaOH (0,063 ml, 0,063 mmol) a TA y a continuación se calentó a 60 °C durante una noche. Después de que la reacción se completara, el disolvente se eliminó para proporcionar el Compuesto 1-119 en sal de sodio (29 mg, 100 %) en forma de un sólido de color blanquecino. LC-MS: m/z 446 (M+1).
Ejemplo 12: Síntesis de sal de sodio del ácido 3-((6-cloro-2-ciclopropil-7-fluoro-1-(1-(2-hidroxietil)-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 1-120)

Etapa 1: Síntesis de 3-((6-cloro-2-ciclopropil-7-fluoro-1-(1-(2-hidroxietil)-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoato de etilo (2):
A una solución agitada de indol 1 (Ejemplo 11, Etapa 3; 480 mg, 1,01 mmol) en DMF (10 ml) en una atmósfera inerte se le añadieron Cs2CO3 (1,32 g, 4,05 mmol) y 2-bromoetan-1-ol (152 mg, 1,27 mmol) a TA y se agitó durante 16 h. La mezcla se diluyó con agua (20 ml) y se extrajo con EtOAc (3 x 20 ml). Los extractos orgánicos combinados se secaron (Na2SO4), se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó (gel de sílice; EtOAc al 55 %/hexanos) para obtener el compuesto 2 (100 mg, 19 %) en forma de un jarabe incoloro. 1H RMN (400 MHz, CDCh): 87,72 (d, J = 6,8 Hz, 2H), 7,62 (dt, J = 8,0 Hz, 1,6 Hz, 1H), 7,18 (d, J = 8,4 Hz, 1H), 7,10-7,07 (m, 1H), 6,94 (t, J = 8,0 Hz, 1H), 6,78 (dt, J = 8,0, 1,6 Hz, 1H), 4,41 (c, 2H), 4,36-4,34 (m, 2H), 4,10 (t, J = 4,8 Hz, 2H), 2,72 (s a, 1H), 1,74-1,67 (m, 1H), 1,41 (t, J = 7,2Hz, 3H), 1,08-1,04 (m, 2H), 0,90-0,85 (m, 2H); LC-MS: m/z 518,7 (M H+).
Etapa 2: Síntesis de sal de sodio del ácido 3-((6-cloro-2-ciclopropil-7-fluoro-1-(1-(2-hidroxietil)-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 1-120)
Siguiendo el procedimiento del Ejemplo 11, Etapa 3 pero usando el Intermedio 2 en lugar del Intermedio 4 en la Etapa 3, se obtuvo el Compuesto 1-120 en sal de sodio del título en forma de un sólido de color blanco. LC-MS: m/z 490 (M+1).
Ejemplo 13: Síntesis de sal de sodio del ácido 3-((1-(1-(2-(carbamoiloxi)etil)-1H-pirazol-4-il)-6-cloro-2-ciclopropil-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 1-121)

Etapa 1: Síntesis de 3-((6-cloro-2-ciclopropil-7-fluoro-1-(1-(2-fenoxicarbonil) oxi)etil)-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoato de etilo (2):
A una solución agitada de indol 1 (Ejemplo 12, Etapa 1; 50 mg, 0,096 mmol) en piridina (2 ml) en una atmósfera inerte se le añadió cloroformiato de fenilo (18 mg, 0,11 mmol) a 0 °C; se calentó a 60 °C y se agitó durante 1 h. La mezcla se diluyó con agua (20 ml), se acidificó con HCl ac. 1 N (5 ml) y se extrajo con EtOAc (2 x 10 ml). Los extractos orgánicos combinados se lavaron con salmuera (10 ml), se secaron (Na2SO4), se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó (gel de sílice; EtOAc al 30 %/hexanos) para proporcionar el compuesto 2 (20 mg, 32 %) en forma de un aceite de color amarillo. LC-MS (ESI): m/z 638,5 (M H+).
Etapa 2: Síntesis de 3-((1-(1-(2-(carbamoiloxi)etil)-1H-pirazol-4-il)-6-cloro-2-ciclopropil-7-fluoro-1H-indol-3-¡l)tio)-2-fluorobenzoato de etilo (3):
A una solución agitada del compuesto 2 (20 mg, 0,031 mmol) en THF (3 ml) en una atmósfera inerte se le pasó gas amoniaco a 0 °C durante 15 min; se calentó a TA y se agitó durante 30 min. Los volátiles se eliminaron a presión reducida y el producto en bruto se purificó por trituración con n-pentano (2 x 5 ml) y se secó a presión reducida para proporcionar 3 (6 mg, 35 %) en forma de un aceite de color pardo pálido. 1H RMN (500 MHz, CDCh): 87,73 (s, 1H), 7,70 (s, 1H), 7,64 (t, J = 8,0 Hz, 1H), 7,20 (d, J = 8,5 Hz, 1H), 7,11-7,08 (m, 1H), 6,95 (t, J = 8,0 Hz, 1H), 6,79 (t, J = 8,0 Hz, 1H), 4,65 (s a, 2H), 4,54-4,47 (m, 4H), 4,42 (c, 2H), 1,74-1,70 (m, 1H), 1,43 (t, J = 7,5 Hz, 3H), 1,07-1,05 (m, 2H), 0,91-0,88 (m, 2H); LC-MS: m/z 561,7 (M H+).
Etapa 3: Síntesis de sal de sodio del ácido 3-((1-(1-(2-(carbamoiloxi)etil)-1H-pirazol-4-il)-6-cloro-2-ciclopropil-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 1-121)
A una solución del compuesto 3 (5 mg, 0,009 mmol) en THF : agua (3:1) (4 ml) se le añadió una solución ac. 1 M de NaOH (0,009 ml, 0,009 mmol) a TA durante una noche. Después de que la reacción se completara, el disolvente se eliminó para proporcionar el Compuesto 1-121 en sal de sodio (5 mg, 100%) en forma de un sólido de color blanquecino. Lc-MS: m/z 533 (M+1).
Ejemplo 14: Síntesis de sal de sodio del ácido 3-((1-(1-(2-aminoetil)-1H-pirazol-4-il)-6-cloro-2-ciclopropil-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 1-122)

Etapa 1: Síntesis de 3-((1-(1-(2-((terc-butoxicarbonil)amino)etil)-1H-pirazol-4-il)-6-cloro-2-ciclopropil-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoato de etilo (3):
A una solución agitada de indol 1 (Ejemplo 11, Etapa 3; 300 mg, 0,63 mmol) en DMF (5 ml) en una atmósfera inerte se le añadieron Cs2CO3 (310 mg, 0,95 mmol) y (2-bromo etil)carbamato de tere-butilo 2 (213 mg, 0,95 mmol) a TA y se agitó durante 16 h. La mezcla se inactivó con NH4Cl ac. sat. (10 ml) y se extrajo con EtOAc (2 x 30 ml). Los extractos orgánicos combinados se secaron (Na2SO4), se filtraron y se concentraron a presión reducida para obtener el producto en bruto que se purificó (gel de sílice; EtOAc al 30 %/hexanos) para proporcionar el compuesto 3 (200 mg, 51 %) en forma de un jarabe incoloro. 1H RMN (500 MHz, CDCh): 67,71 (s, 1H), 7,65-7,62 (m, 2H), 7,19 (d, J = 8,5 Hz, 1H), 7,11-7,08 (m, 1H), 6,95 (t, J = 8,0 Hz, 1H), 6,79 (t, J = 6,5 Hz, 1H), 4,82 (s a, 1H), 4,41 (c, 2H), 4,34 (t, J = 5,0 Hz, 2H), 3,65-2,04 (m, 2H), 1,72-1,68 (m, 1H), 1,43 (t, J = 7,5 Hz, 3H), 1,29 (s, 9H), 1,06-1,03 (m, 2H), 0,89-0,85 (m, 2H); LC-MS: 517,4 (Des-Boc) (M+ H+).
Etapa 2: Síntesis de 3-((1-(1-(2-aminoetil)-1H-pirazol-4-il)-6-cloro-2-ciclopropil-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoato de etilo (4):
Una solución del compuesto 3 (200 mg, 0,32 mmol) en HCl 4,0 M en 1,4-dioxano (5 ml) en una atmósfera inerte se agitó a 0 °C-TA durante 2 h. Los volátiles se eliminaron a presión reducida. El residuo se diluyó con agua (5 ml), se basificó con NaHCO3 ac. (5 ml) y se extrajo con EtOAc (2 x 20 ml). Los extractos orgánicos combinados se secaron (Na2SO4), se filtraron y se concentraron a presión reducida para proporcionar 4 (130 mg, 81 %) en forma de un sólido de color blanquecino. 1H RMN (400 MHz, DMSO-cfe): 6 8,24 (s, 1H), 7,82 (s, 1H), 7,60 (t, J = 6,8 Hz, 1H), 7,22-7,11 (m, 3H), 6,84 (t, J = 6,8 Hz, 1H), 4,33 (c, 2H), 4,17 (t, J = 6,0 Hz, 2H), 2,98 (t, J = 6,0 Hz, 2H), 1,84-1,80 (m, 3H), 1,32 (t, J = 7,2 Hz, 3H), 0,92-0,91 (m, 2H), 0,84-0,82 (m, 2H); MS: m/z 517,6 (M H+).
Etapa 3: Síntesis de sal de sodio del ácido 3-((1-(1-(2-aminoetil)-1H-pirazol-4-il)-6-cloro-2-ciclopropil-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 1-122)
Siguiendo el procedimiento del Ejemplo 11, Etapa 3 pero usando el Intermedio 4 en lugar del Intermedio 4 en la Etapa 3, se obtuvo el Compuesto 1-122 en sal de sodio del título en forma de un sólido de color blanquecino. LC-MS: m/z 489 (M+1).
Ejemplo 15: Síntesis de sal de sodio del ácido 3-((6-cloro-2-ciclopropil-7-fluoro-1-(1-(2-ureidoetil)-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 1-123)

Etapa 1: Síntesis de 3-((6-cloro-2-ciclopropil-7-fluoro-1-(1-(2-ureidoetil)-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoato de etilo (2)
A una solución agitada de indol 1 (Ejemplo 14, Etapa 2; 80 mg, 0,15 mmol) en THF (5 ml) en una atmósfera inerte se le añadieron Et3N (0,04 ml, 0,31 mmol) y trifosgeno (18,3 mg, 0,06 mmol) a 0 °C, se agitó durante 1 h, se calentó a TA y se agitó durante 2 h. A esta solución de isocianato en bruto se le pasó gas amoniaco a -78 °C durante 10 min; se calentó a 0 °C y se agitó durante 1 h. La mezcla se diluyó con agua y se extrajo con EtOAc (2 x 20 ml). Los extractos orgánicos combinados se secaron (Na2SO4), se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó por trituración con Et2O (2 x 5 ml) y se secó a presión reducida para proporcionar 2 (30 mg, 34 %) en forma de un sólido de color blanquecino. 1H RMN (400 MHz, DMSO-d6): 88,19 (s, 1H), 7,82 (s, 1H), 7,60 (t, J = 7,2 Hz, 1H), 7,22-7,11 (m, 3H), 6,84 (t, J = 7,2 Hz, 1H), 6,04-6,03 (m, 1H), 5,53 (s a, 2H), 4,33 (c, 2H), 4,21 (t, J = 6,0 Hz, 2H), 3,43 (t, J = 6,0 Hz, 2H), 1,84-1,78 (m, 1H), 1,32 (t, J = 7,2 Hz, 3H), 0,92 0,91 (m, 2H), 0,87-0,85 (m, 2H); MS: m/z 560,6 (M H+).
Etapa 2: Síntesis de sal de sodio del ácido 3-((6-cloro-2-ciclopropil-7-fluoro-1-(1-(2-ureidoetil)-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 1-123)
Siguiendo el procedimiento del Ejemplo 13, Etapa 3 pero usando el Intermedio 2 en lugar del Intermedio 3 en la Etapa 3, se obtuvo el Compuesto 1-123 en sal de sodio en forma de un sólido de color blanquecino. LC-MS: m/z 532 (M+1).
Ejemplo 16: Síntesis de sal de sodio del ácido 3-((1-(1-(3-carboxipropil)-1H-pirazol-4-il)-6-cloro-2-ciclopropil-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 1-124)

Etapa 1: Síntesis de 3-((1-(1-(4-(terc-butoxi)-4-oxobutil)-1H-pirazol-4-il)-6-cloro-2-ciclopropil-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoato de etilo (2):
A una solución agitada de indol 1 (Ejemplo 11, Etapa 3; 200 mg, 0,42 mmol) en DMF (5 ml) en una atmósfera inerte se le añadieron Cs2CO3 (206 mg, 0,63 mmol) y 4-bromobutanoato de tere-butilo (141 mg, 0,63 mmol) a TA y se agitó durante 16 h. La mezcla se diluyó con agua enfriada con hielo (20 ml) y se extrajo con EtOAc (3 x 30 ml). Los extractos orgánicos combinados se secaron (Na2SO4), se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó (cromatografía sobre gel de sílice; EtOAc al 20 %/hexanos) para proporcionar el compuesto 2 (180 mg, 70 %) en forma de un aceite de color pardo pálido. 1H RMN (400 MHz, CdCU): 87,67 (s, 1H), 7,64 (s, 1H), 7,63-7,60 (m, 1H), 7,18 (d, J = 8,4 Hz, 1H), 7,10-7,06 (m, 1H), 6,93 (t, J = 8,0 Hz, 1H), 6,77 (t, J = 7,6 Hz, 1H), 4,41 (c, 2H), 4,28 (t, J = 6,8 Hz, 2H), 2,28-2,19 (m, 4H), 1,74-1,66 (m, 1H), 1,46 (s, 9H), 1,41 (t, J = 7,2 Hz, 3H), 1,06-1,02 (m, 2H), 0,89-0,84 (m, 2H); LC-MS (ESI): m/z 618,6 (M H+).
Etapa 2: Síntesis de ácido 4-(4-(6-cloro-2-ciclopropil-3-((3-(etoxicarbonil)-2-fluorofenil)tio)-7-fluoro-1H-indol-1-il)-1H-pirazol-1-il)butanoico (3):
Una solución del compuesto 2 (100 mg, 0,29 mmol) en HCl 4,0 M en 1,4-dioxano (2 ml) en una atmósfera inerte se agitó a 0 °C-TA durante 16 h. Los volátiles se eliminaron al vacío y el residuo se diluyó con agua (5 ml), se basificó con NaHCO3 ac. (5 ml) y se extrajo con EtOAc (2 x 20 ml). Los extractos orgánicos combinados se secaron (Na2SO4), se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó por tratamiento con ácido-base para proporcionar 3 (50 mg, 56 %) en forma de un sólido de color blanco. 1H RMN (500 MHz, DMSO-d6): 812,75 (s a, 1H), 8,24 (s, 1H), 7,82 (s, 1H), 7,59 (t, J = 7,0 Hz, 1H), 7,22-7,10 (m, 3H), 6,84 (t, J = 7,5 Hz, 1H), 4,33 (c, 2H), 4,22 (t, J = 7,0 Hz, 2H), 2,21 (t, J = 7,5 Hz, 2H), 2,06-2,03 (m, 2H), 1,81-1,78 (m, 1H), 1,32 (t, J = 7,0 Hz, 3H), 0,89-0,88 (m, 2H), 0,82-0,81 (m, 2H); MS: m/z 560,7 (M H+).
Etapa 3: Síntesis de sal de sodio del ácido 3-((1-(1-(3-carboxipropil)-1H-pirazol-4-il)-6-cloro-2-ciclopropil-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 1-124)
A una solución del compuesto 3 (10 mg, 0,018 mmol) en THF : agua (3:1) (4 ml) se le añadió una solución ac. 1 M de NaOH (0,036 ml, 0,036 mmol) a TA y después se calentó a 60 °C durante una noche. Después de que la reacción se completara, el disolvente se eliminó para proporcionar el Compuesto 1-124 en sal de sodio (10 mg, 100 %) en forma de un sólido de color blanco. LC-MS: m/z 532 (M+1).
Ejemplo 17: Síntesis de sal de sodio del ácido 3-((1-(1-(4-amino-4-oxobutil)-1H-pirazol-4-il)-6-cloro-2-ciclopropil-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 1-125)

Etapa 1: Síntesis de 3-((1-(1-(4-amino-4-oxobutil)-1H-pirazol-4-il)-6-cloro-2-ciclopropil-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoato de etilo (2):
A una solución agitada de indol 1 (Ejemplo 16, Etapa 2; 150 mg, 0,26 mmol) en DMF (3 ml) en una atmósfera inerte se le añadieron EDCl.HCl (36 mg, 0,40 mmol), HOBt (61,5 mg, 0,40 mmol), NMM (0,07 ml, 0,67 mmol) a TA y se agitó durante 10 min. A esta se le añadió NH4Cl (17,1 mg, 0,32 mmol) a TA y se agitó durante 16 h. La mezcla se diluyó con agua (30 ml) y se extrajo con EtOAc (2 x 30 ml). Los extractos orgánicos combinados se lavaron con salmuera (20 ml), se secaron (Na2SO4), se filtraron y se secaron a presión reducida para obtener el producto en bruto. Este se purificó (gel de sílice; MeOH al 2 %/CH2Ch) para proporcionar el compuesto 2 (15 mg, 10 %) en forma de un sólido de color blanquecino. 1H RMN (400 MHz, DMSO-d6): 8 8,23 (s, 1H), 7,81 (s, 1H), 7,60 (dt, J = 8,0, 1,6 Hz, 1H), 7,29 (s a, 1H), 7,22-7,11 (m, 3H), 6,84 (dt, J = 8,0, 1,6 Hz, 1H), 6,78 (s a, 1H), 4,33 (c, 2H), 4,20 (t, J = 6,4 Hz, 2H), 2,08-2,02 (m, 4H), 1,82-1,77 (m, 1H), 1,32 (t, J = 7,2 Hz, 3H), 0,92-0,88 (m,2H), 0,86-0,80 (m, 2H); MS: m/z 559,6 (M H+).
Etapa 2: Síntesis de sal de sodio del ácido 3-((1-(1-(4-amino-4-oxobutil)-1H-pirazol-4-il)-6-cloro-2-ciclopropil-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 1-125)
Siguiendo el procedimiento del Ejemplo 11, Etapa 3 pero usando el Intermedio 2 en lugar del Intermedio 4 en la Etapa 3, se obtuvo el Compuesto 1-125 en sal de sodio del título en forma de un sólido de color blanco. LC-MS: m/z 531 (M+1).
Ejemplo 18: Síntesis de sal de sodio del ácido 3-((2.6-dicloro-7-fluoro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 1-49)

ompuesto - en sa e so o
Etapa 1: Síntesis de 3-((6-cloro-7-fluoro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoato de etilo Í3H
Siguiendo el procedimiento del Ejemplo 9, Etapas 3 y 4 pero usando el Intermedio A en lugar del Intermedio B en la Etapa 3, se obtuvo el compuesto del título 3 en forma de un sólido de color pardo claro. 1H RMN (400 MHz, CDCh): 8 7,68 (s, 1H), 7,67-7,64 (m, 2H), 7,43 (s, 1H), 7,28 (d J = 8,4 Hz, 1H), 7,16 (dd, J = 8,4, 6,0 Hz, 1H), 7,00-6,93 (m, 2H), 4,40 (c, J = 6,8 Hz, 2H), 3,99 (s, 3H), 1,40 (t, J = 6,8 Hz, 3H); LC-MS (ESI): m/z 448,4 (M H+).
Etapa 2: Síntesis de 3-((2.6-dicloro-7-fluoro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoato de etilo (4):
A una solución agitada del compuesto 3 (150 mg, 0,33 mmol) en Et2O (10 ml) en una atmósfera inerte se le añadió SO2Cl2 (87 mg, 0,65 mmol) a 0 °C y se agitó durante 1 h. La mezcla de reacción se inactivó con agua (10 ml) y se extrajo con EtOAc (2 x 20 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4 y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó (gel de sílice; EtOAc al 15 %/n-Hexano) para proporcionar 4 (35 mg, 22 %) en forma de un sólido de color blanquecino. 1H RMN (500 MHz, CDCh): 8 7,72-7,68 (m, 1H), 7,62 (s, 1H), 7,58 (s, 1H), 7,36 (d, J = 8,5 Hz, 1H), 7,24-7,19 (m, 1H), 7,08-6,79 (m, 2H), 4,42 (c, J = 7,0 Hz, 2H), 4,08 (s, 3H), 1,42 (t, J = 7,0 Hz, 3H); LC-MS: m/z 482,4 (M+).
Etapa 3: Síntesis de sal de sodio del ácido 3-((2.6-dicloro-7-fluoro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 1-49):
Siguiendo el procedimiento del Ejemplo 11, Etapa 3 pero usando el Intermedio 4 en lugar del Intermedio 4 en la Etapa 3, se obtuvo el Compuesto 1-49 en sal de sodio en forma de un sólido de color castaño. LC-MS: m/z 454 (M+1).


Etapa 1: Síntesis de 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-(2.2.2-tr¡fluoroet¡l)-1H-p¡razol-4-¡l)-1H-indol-3-¡l)t¡o)-2-fluorobenzoato de etilo (3):
Siguiendo el procedimiento del Ejemplo 9, Etapas 3 y 4 pero usando el Intermedio F en lugar del Intermedio B en la Etapa 3, se obtuvo el compuesto del título 3 en forma de un sólido de color blanquecino. 1H RMN (400 MHz, CD3OD): 88,23 (s, 1H), 7,88 (s, 1H), 7,61 (dt, J = 8,0, 1,6 Hz, 1H), 7,20 (d, J = 8,4 Hz, 1H), 7,15-7,11 (m, 1H), 7,02 (t, J = 8,0 Hz, 1H), 6,84 (dt, J = 8,4, 1,6 Hz, 1H), 5,04 (c, 2H), 4,39 (c, 2H), 1,81-1,73 (m, 1H), 1,41-1,38 (m, 3H), 0,98-0,90 (m, 2H), 0,88-0,84 (m, 2H); MS: m/z 556,5 (M H+).
Etapa 2: Síntesis de sal de sod¡o del ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-(2.2.2-trifluoroet¡l)-1H-p¡razol-4-il)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto 1-126)
Siguiendo el procedimiento del Ejemplo 11, Etapa 3 pero usando el Intermedio 3 en lugar del Intermedio 4 en la Etapa 3, se obtuvo el Compuesto 1-126 en sal de sodio en forma de un sólido de color blanquecino. LC-MS: m/z 528 (M+1).
Ejemplo 20: Síntesis de ácido 3-((6-cloro-2-c¡cloprop¡l-1-(1-(et¡l-cfe)-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-il)t¡o)-2-fluorobenzoico (Compuesto 1-127)
Etapa 1: Síntesis de 3-((6-cloro-2-c¡cloprop¡l-1-(1-(et¡l-d5)-1H-p¡razol-4-¡l)-7-fluoro-1H-indol-3-¡l)t¡o)-2-fluorobenzoato de etilo (3):
Siguiendo el procedimiento del Ejemplo 9, Etapas 3 y 4 pero usando el Intermedio E en lugar del Intermedio B en la Etapa 3, se obtuvo el compuesto del título 3 en forma de un sólido de color rojo. 1H RMN (500 MHz CDCh): 87,67 7,61 (m, 3H), 7,18 (d, J = 8,0 Hz, 1H), 7,09-7,07 (m, 1H), 6,94 (t, J = 8,0 Hz, 1H), 6,92-6,75 (m, 1H), 4,41 (c, 2H), 1,72-1,68 (m, 1H), 1,41 (t, J = 7,5 Hz, 3H), 1,08-1,05 (m, 2H), 0,89-0,85 (m, 2H); LC-MS (ESI): 509,5 (M H+).
Etapa 2: Síntesis de ác¡do 3-((6-cloro-2-c¡cloprop¡l-1-(1-(et¡l-d5)-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-il)t¡o)-2-fluorobenzo¡co (Compuesto 1-127):
A una solución agitada de indol 3 (200 mg, 0,39 mmol) en THF:MeOH:H2O (2:2:1, 5 ml) se le añadió LOH.H2O (66 mg, 1,57 mmol) a TA y se agitó durante 8 h. Los volátiles se eliminaron al vacío. El residuo se diluyó con agua (5 ml), se acidificó con HCl ac. 2 M (5 ml); el sólido obtenido se filtró, se lavó con agua (25 ml), se trituró con npentano (2 x 5 ml) y se secó a presión reducida para proporcionar el Compuesto 1-127 del título (110 mg, 59 %) en forma de un sólido de color blanquecino. 1H RMN (400 MHz, DMSO-d6): 8 13,49 (s a, 1H), 8,22 (s, 1H), 7,73 (s, 1H), 7,51 (t, J = 6,8 Hz, 1H), 7,19-7,13 (m, 2H), 7,04 (t, J =8,0 Hz, 1H), 6,74 (t, J = 7,2 Hz, 1H), 1,80-1,73 (m, 1H), 0,91 0,90 (m, 2H), 0,82-0,80 (m, 2H); MS (ESI): m/z 480,8 (M H+).
Ejemplo 21: Síntesis de sal de sodio del ácido 3-((2.6-d¡cloro-1-(1-et¡l-1H-p¡razol-4-il)-7-fluoro-1H-¡ndol-3-¡l)tio)-2-fluorobenzo¡co (Compuesto 1-31)

Etapa 1: Síntesis de 3-((6-cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-il)t¡o)-2-fluorobenzoato de etilo (3):
Siguiendo el procedimiento del Ejemplo 9, Etapas 3 y 4 pero usando indol 1 (Ejemplo 4, Etapa 1) en lugar de indol 3 en la Etapa 3, se obtuvo el compuesto del título 3 en forma de un sólido de color pardo claro. 1H RMN (500 MHz, CDCl3): 87,69-7,64 (m, 3H), 7,44 (s, 1H), 7,27 (t, J = 8,0 Hz, 1H), 7,16 (dd, J = 8,5; 6,0 Hz, 1H), 7,01-6,94 (m, 2H), 4,40 (c, J = 7,5 Hz, 2H), 4,26 (c, J = 8,0 Hz, 2H), 1,57 (t, J = 8,0 Hz, 3H), 1,57 (t, J = 7,5 Hz, 3H); LC-MS (ESI): m/z 462,5 (M+H+).
Etapa 2: S íntesis de 3-((2.6-d¡cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-il)t¡o)-2-fluorobenzoato de etilo Í41l
A una solución del compuesto 3 (100 mg, 0,21 mmol) en CH2Ch (3 ml) en una atmósfera inerte se le añadió NCS (58 mg, 0,43 mmol) a t A y se agitó durante 16 h. La mezcla de reacción se diluyó con agua (10 ml) y se extrajo con CH2Cl2 (2 x 15 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4 y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó (gel de sílice; EtOAc al 14 17 %/hexanos) para proporcionar 5 (35 mg, 33 %) en forma de un sólido de color blanquecino. 1H RMN (400 MHz, CDCl3): 87,71-7,66 (m, 3H), 7,30 (d, J = 7,6 Hz, 1H), 7,19 (dd, J = 8,8; 6,4 Hz, 1H), 7,04-6,97 (m, 2H), 4,40 (c, J = 7,2 Hz, 2H), 4,27 (c, J = 7,6 Hz, 2H), 1,58 (t, J = 7,6 Hz, 3H), 1,49 (t, J = 7,2 Hz, 3H); LC-MS (ESI): m/z 496,7 (M+H+).
Etapa 3: Síntesis de ácido 3-((2.6-d¡cloro-1-(1-et¡l-1H-p¡razol-4-¡l)-7-fluoro-1H-¡ndol-3-il)t¡o)-2-fluorobenzo¡co (Compuesto 1-31)
Siguiendo el procedimiento del Ejemplo 11, Etapa 3 pero usando el Intermedio 4 en lugar del Intermedio 4 en la Etapa 3, se obtuvo el Compuesto 1-31 en sal de sod¡o en forma de un sólido de color blanquecino. LC-MS: m/z 468 (M+1).
Ejemplo 22: Síntesis de sal de sod¡o del ác¡do 3-((2.6-d¡cloro-7-fluoro-1-(p¡r¡d¡n-3-¡l)-1H-¡ndol-3-il)t¡o)-2-fluorobenzo¡co (Compuesto 2-1)

Etapa 1: Síntesis de 6-cloro-7-fluoro-1-(p¡r¡d¡n-3-il)-1H-¡ndol (2):
A una solución agitada de indol 1 (Ejemplo 4, Etapa 1; 2,0 g, 11,8 mmol) en tolueno (50 ml) se le añadieron 3-bromopiridina (2,9 g, 17,7 mmol), W,AT-dimetiletilendiamina (418 mg, 4,73 mmol), KpPÜ4 (6,3 g, 29,5 mmol), Cul (225 mg, 1,18 mmol) a TA en una atmósfera inerte. La mezcla se purgó con argón durante 15 min y se calentó a 140 °C en un tubo cerrado herméticamente durante 16 h. La mezcla de reacción se enfrió a TA, se le añadió nhexano (20 ml), se agitó durante 5 minutos y después se filtró. El filtrado se diluyó con agua (20 ml) y se extrajo con EtOAc (2 x 50 ml). Los extractos orgánicos combinados se lavaron con una solución de agua y salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó (gel de sílice; EtOAc al 10 %/hexanos) para proporcionar el compuesto 2 (2,0 g, 69 %) en forma de un sólido de color pardo claro. 1H RMN (500 MHz, DMSO-d6): 88,84 (s, 1H), 8,66 (d, J = 5,0 Hz, 1H), 8,06-8,02 (m, 1H), 7,74-7,72 (m, 1H), 7,64-7,58 (m, 1H), 7,52 (d, J = 8,0 Hz, 1H), 7,28-7,24 (m, 1H), 6,82 (m, 1H).
Etapa 2: Síntesis de 3-((6-cloro-7-fluoro-1-(p¡r¡d¡n-3-il)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzoato de etilo (3):
A una solución agitada del Intermedio A (243 mg, 1,21 mmol) en 1,2-dicloroetano (8 ml) en una atmósfera inerte se le añadió NCS (163 mg, 1,21 mmol) a TA y se agitó durante 1 h. A esta se le añadieron el compuesto 2 (200 mg, 0,81 mmol) en 1,2-dicloroetano (2 ml) y NaHCO3 (204 mg, 2,45 mmol) a TA. Después de 24 h de agitación a TA, la mezcla de reacción se diluyó con agua (15 ml) y se extrajo con EtOAc (2 x 30 ml). Los extractos orgánicos combinados se lavaron con agua (30 ml) y salmuera (30 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó (gel de sílice; EtOAc al 10 %/hexanos) para proporcionar el compuesto 3 (50 mg, 9 %) en forma de un sólido de color blanquecino.
1H RMN (500 MHz, DMSO-d6): 8 8,91 (s, 1H), 8,70 (d, J = 5,0 Hz, 1H), 8,30 (s, 1H), 8,18-8,15 (m, 1H), 7,66-7,762 (m, 2H), 7,36-7,34 (m, 2H), 7,17-7,13 (m, 2H), 4,34 (c, J = 7,5 Hz, 2H), 1,32 (t, J = 7,5 Hz, 3H).
Etapa 3: Síntesis de 3-((2.6-d¡cloro-7-fluoro-1-(p¡r¡d¡n-3-¡l)-1H-¡ndol-3-il)t¡o)-2-fluorobenzoato de etilo (4):
A una solución agitada del compuesto 3 (50 mg, 0,11 mmol) en Et2O (10 ml) en una atmósfera inerte se le añadió lentamente SO2Cl2 (18 mg, 0,13 mmol) a 0 °C y se agitó durante 1 h. Después de que se completara la reacción por TLC, la mezcla de reacción se inactivó con agua (10 ml) y se extrajo con EtOAc (2 x 20 ml). Los extractos orgánicos combinados se lavaron con una solución de agua y salmuera, se secaron sobre Na2SO4 y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó por HPLC preparativa para proporcionar 4 (17 mg, 32 %) en forma de un sólido de color blanquecino. 1H r Mn (400 MHz, DMSO-d6): 8 8,93 (d, J = 2,4 Hz, 1H), 8,80 (dd, J = 4,8, 1,6 Hz, 1H), 8,23-8,21 (m, 1H), 7,71-7,66 (m, 2H), 7,39-7,38 (m, 2H), 7,20-7,18 (m, 2H), 4,34 (c, J = 7,2 Hz, 2H), 1,32 (t, J = 7,2 Hz, 3H); LC-MS: m/z 479,4 (M+).
Etapa 4: Síntesis de sal de sodio del ácido 3-((2.6-dicloro-7-fluoro-1-(piridin-3-il)-1H-indol-3-il)tio)-2-fluorobenzoico (Compuesto 2-1):
A una solución del compuesto 4 (16 mg, 0,033 mmol) en THF : agua (3:1) (4 ml) se le añadió una solución ac. 1 M de NaOH (0,033 ml, 0,033 mmol) a TA y después se calentó a 60 °C durante 3 horas. Después de que la reacción se completara, el disolvente se eliminó para proporcionar el Compuesto 2-1 en sal de sodio (16 mg, 100 %) en forma de un sólido de color blanquecino. LC-MS: m/z 451 (M+1).
Ruta alternativa para la preparación del Compuesto 4:

Etapa 1: Síntesis de 3-((6-cloro-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoato de etilo (2):
A una solución agitada del Intermedio A (1,18 g, 5,91 mmol) en CH2Ch (30 ml) en una atmósfera inerte se le añadió NCS (792 mg, 5,91 mmol) a TA y se agitó durante 1 h. A esta se le añadió indol 1 (Ejemplo 4, Etapa 1; 1,0 g, 5,91 mmol) en CH2Ch (20 ml) a TA y se agitó durante 4 h. Después de que se completara la reacción por TLC, la mezcla de reacción se diluyó con agua (40 ml) y se extrajo con C ^ C h (2 x 60 ml). Los extractos orgánicos combinados se lavaron con agua (50 ml) y salmuera (50 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó (gel de sílice; EtOAc al 10 %/hexanos) para proporcionar el compuesto 2 (1,2 g, 55 %) en forma de un sólido de color pardo claro.
1H RMN (500 MHz, DMSO-d6): 8 12,60 (s a, 1H), 8,00 (d, J = 4,0 Hz, 1H), 7,61-7,57 (m, 1H), 7,24-7,17 (m, 2H), 7,09 (t, J = 8,0 Hz, 1H), 6,90-6,86 (m, 1H), 4,34 (c, J = 7,5 Hz, 2H), 1,32 (t, J = 7,5 Hz, 3H); MS: m/z 368,6 (M H+).
Síntesis de 3-((6-cloro-7-fluoro-1-(piridin-3-il)-1H-indol-3-il)tio)-2-fluorobenzoato de etilo (3):
A una solución agitada del compuesto 2 (200 mg, 0,54 mmol) en tolueno (5 ml) se le añadieron 3-bromopiridina (131 mg, 0,81 mmol), frans-1,2-diaminociclohexano (24,8 mg, 0,21 mmol), K3PO4 (288 mg, 1,35 mmol), Cu(I)I (10,3 mg, 0,05 mmol) a TA en una atmósfera inerte. La mezcla se purgó con argón durante 15 min y se calentó a 140 °C en un tubo cerrado herméticamente durante 16 h. La mezcla de reacción se enfrió a TA, se le añadió nhexano ( 6 ml), se agitó durante 5 minutos y después se filtró. El filtrado se diluyó con agua (20 ml) y se extrajo con EtOAc (2 x 50 ml). Los extractos orgánicos combinados se lavaron con una solución de agua y salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó (gel de sílice; EtOAc al 10-12 %/hexanos) para proporcionar el compuesto 3 (130 mg, 54% ) en forma de un sólido de color blanquecino.
Ejemplo 23: Síntesis de ácido 3-((2-bromo-6-cloro-7-fluoro-1-(p¡r¡d¡n-3-¡l)-1H-¡ndol-3-il)t¡o)-2-fluorobenzo¡co (Compuesto 2-2)

Etapa 1: Síntesis de 3-((2-bromo-6-cloro-7-fluoro-1-(p¡r¡d¡n-3-¡l)-1H-¡ndol-3-il)t¡o)-2-fluorobenzoato de et¡lo (2):
A una solución agitada de indol 1 (Ejemplo 22, Etapa 2; 60 mg, 0,13 mmol) en CH2CI2 (5 ml) en una atmósfera inerte se le añadió NBS (60 mg, 0,33 mmol) a TA y se agitó durante 16 h. La mezcla se diluyó con agua (10 ml) y se extrajo con CH2Ch (2 x 25 ml). Los extractos orgánicos combinados se lavaron con salmuera (10 ml), se secaron (Na2SO4), se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó (gel de sílice; EtOAc al 10%/hexanos) para proporcionar el compuesto 2 (35 mg, 50%) en forma de un sólido de color pardo pálido. 1H RMN (500 MHz, DMSO-d6): 8 8,91 (s, 1H), 8,81-8,80 (m, 1H), 8,20 (d, J = 8,5 Hz, 1H), 7,70-7,66 (m, 2H), 7,40-7,37 (m, 2H), 7,18 (t, J = 8,0 Hz, 1H), 7,16-7,13 (m, 1H), 4,33 (c, 2H), 1,32 (t, J = 7,5 Hz, 3H); MS: m/z 525,3 (M++2).
Etapa 2: Síntesis de ácido 3-((2-bromo-6-cloro-7-fluoro-1-(p¡r¡d¡n-3-il)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto 2-2):
A una solución agitada del compuesto 2 (35 mg, 0,06 mmol) en THF: H2O (1:1, 4 ml) se le añadió LOH.H2O (11,2 mg, 0,26 mmol) a TA y se agitó durante 5 h. Los volátiles se eliminaron a presión reducida y el residuo se diluyó con agua (5 ml), se acidificó con ácido cítrico y se extrajo con EtOAc (2 x 25 ml). Los extractos orgánicos combinados se lavaron con salmuera (10 ml), se secaron (Na2SO4), se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se trituró con n-pentano (2 x 5 ml) y se secó al vacío para proporcionar el Compuesto 2-2 del título (25 mg, 76 %) en forma de un sólido de color pardo pálido. 1H RMN (500 MHz, DMSO-d6): 8 13,41 (s a, 1H), 8,93 (s, 1H), 8,81-8,80 (m, 1H), 8,20 (d, J = 7,5 Hz, 1H), 7,70-7,64 (m, 2H), 7,40-7,35 (m, 2H), 7,17-7,11 (m, 2H); LC-MS (ESI): m/z 497,3 (M++2).
Ejemplo 24: Síntesis de sal de sodio del ácido 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(p¡r¡d¡n-3-il)-1H-¡ndol-3-¡l)tio)-2-fluorobenzo¡co (Compuesto 2-3)

Etapa 1: Síntesis de 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1H-¡ndol-3-il)t¡o)-2-fluorobenzoato de etilo (3):
A una solución agitada del Intermedio A (190 mg, 0,95 mmol) en CH2Ch (10 ml) en una atmósfera inerte se le añadió
NCS (128 mg, 0,95 mmol) a TA y se agitó durante 1 h. A esta se le añadió indol 1 (Ejemplo 9, Etapa 2; 200 mg, 0,95 mmol) en CH2Ch (5 ml) a tA y se agitó durante 12 h. La mezcla se diluyó con agua (50 ml) y se extrajo con CH2Cl2 (3 x 50 ml). Los extractos orgánicos combinados se lavaron con salmuera (100 ml), se secaron (Na2SÜ4), se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó (cromatografía sobre gel de sílice; EtOAc al 5-10 %/hexanos) para obtener el compuesto 2 (300 mg, 77 %) en forma de un sólido de color rosa pálido. 1H RMN (400 MHz, DMSO-d6): 8 11,91 (s, 1H), 7,89-7,84 (m, 1H), 7,57 (t, J = 8,0 Hz, 1H), 7,14 7,07 (m, 2H), 6,78 (t, J = 8,0 Hz, 1H), 4,35-4,29 (m, 2H), 2,32-2,25 (m, 1H), 1,33-1,28 (m, 3H), 1,15-1,10 (m, 2H), 1,08-1,03 (m, 2H); LC-MS (ESI): m/z 406,3 (M - H+).
Etapa 2: Síntesis de 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(p¡r¡d¡n-3-¡l)-1H-¡ndol-3-¡l)tio)-2-fluorobenzoato de etilo Í3H
A una solución agitada del compuesto 2 (100 mg, 0,24 mmol) en tolueno (5 ml) se le añadieron 3-bromopiridina (59,3 mg, 0,36 mmol), frans-1,2-diaminociclohexano (11,2 mg, 0,098 mmol), K3PO4 (130 mg, 0,65 mmol), Cul (4,6 mg, 0,024 mmol) a TA en una atmósfera de argón en un tubo sellado. La solución se purgó con argón; se calentó a 140 °C y se agitó durante 40 h. La mezcla se enfrió a TA, se le añadió n-hexano (6 ml), se agitó durante 5 minutos y después se filtró. El filtrado se diluyó con agua (20 ml) y se extrajo con EtOAc (2 x 50 ml). Los extractos orgánicos combinados se lavaron con una solución de agua y salmuera, se secaron (Na2SO4), se filtraron y se concentraron a presión reducida para obtener el producto en bruto. Este se purificó (gel de sílice; EtOAc al 5 10 %/hexanos) para proporcionar el compuesto 3 (10 mg, 8,4 %) en forma de un sólido de color pardo pálido. 1H RMN (400 MHz, CDCh): 8 8,77 (s a, 2H), 7,81 (d, J = 8,0 Hz, 1H), 7,68-7,62 (m, 1H), 7,53-7,50 (m, 1H), 7,25-7,23 (m, 1H), 7,17-7,11 (m, 1H), 6,96 (t, J = 7,6 Hz, 1H), 6,82 (dt, J = 8,4, 1,6 Hz, 1H), 4,40 (c, 2H), 1,65-1,60 (m, 1H), 1,41 (t, J = 7,2 Hz, 3H), 0,91-0,82 (m, 4H); LC-MS (ESI): m/z 485,5 (M H+).
Etapa 3: Síntesis de sal de sod¡o del ác¡do 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(p¡r¡d¡n-3-il)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzo¡co (Compuesto 2-3)
Siguiendo el procedimiento del Ejemplo 11, Etapa 3 pero usando el Intermedio 3 en lugar del Intermedio 4 en la Etapa 3, se obtuvo el Compuesto 2-3 en sal de sod¡o en forma de un sólido de color blanquecino. LC-MS: m/z 457 (M+1).
Ejemplo 25: Síntesis de sal de sod¡o del ác¡do 3-((1-(1-(6-am¡noet¡l)-1H-p¡razol-4-il)-6-cloro-2-c¡cloprop¡l-7-fluoro-1H-indol-3-¡l)tio)-2-fluorobenzo¡co (Compuesto 1-128)

A una solución agitada de 6-aminohexan-1-ol (1 g, 8,55 mmol) en THF (20 ml) se le añadió Boc2O (1,86 g, 8,55 mmol) a TA en una atmósfera inerte y se agitó durante 6 h. La mezcla se diluyó con agua (50 ml) y se extrajo con EtOAc (3 x 60 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para obtener el producto en bruto. El producto en bruto se purificó (cromatografía sobre gel de sílice; EtOAc al 30 %/hexanos) para proporcionar el compuesto 2 (1 g, 54% ) en forma de un jarabe incoloro. 1H RMN (400 MHz, CDCh): 8 4,51 (s a, 1H), 3,63 (t, J = 6,4 Hz, 2H), 3,14-3,09 (m, 2H), 1,60-1,53 (m, 2H), 1,52-1,48 (m, 2H), 1,47 (s, 9H), 1,41-1,30 (m, 4H); LC-MS (ESI): m/z 118,1 (M+ -Boc).
Etapa 2: Síntesis de (6-bromohexil)carbamato de tere-butilo (3):
A una solución agitada del compuesto 2 (1 g, 4,61 mmol) en tolueno (30 ml) se le añadieron Ph3P (1,81 g, 6,91 mmol) y CBr4 (2,29 g, 6,91 mmol) a TA en una atmósfera inerte y se agitó durante 3 h. La mezcla se diluyó con agua (50 ml) y se extrajo con EtOAc (3 x 60 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron al vacío para obtener el producto en bruto. El producto en bruto se purificó (gel de sílice; EtOAc al 20 %/hexanos) para proporcionar el compuesto 3 (1 g, 78%) en forma de un aceite incoloro. 1H RMN (400 MHz, CDCI3): 8 4,51 (s a, 1H), 3,40 (t, J = 6,8 Hz, 2H), 3,13-3,08 (m, 2H), 1,89-1,82 (m, 2H), 1,52-1,42 (m, 13H), 1,37-1,31 (m, 2H).
Etapa 3: Síntesis de sal de sodio del ácido 3-((1-(1-(6-am¡nohex¡l)-1H-p¡razol-4-il)-6-cloro-2-c¡cloprop¡l-7-fluoro-1H-indol-3-¡l)tio)-2-fluorobenzo¡co (Compuesto 1-128)
Siguiendo el procedimiento del Ejemplo 14 pero usando el compuesto 3 en lugar de (2-bromo etil)carbamato de terc-butilo, se obtuvo el Compuesto 1-128 en sal de sodio en forma de un sólido de color blanquecino. LC-MS: m/z 545 (M+1).
Ejemplo 26: Síntesis de sal de sodio del ácido 3-((6-cloro-2-c¡cloprop¡l-7-fluoro-1-(1-(hex-5-in-1-¡l)-1H-p¡razol-4-¡l)-1H-¡ndol-3-¡l)t¡o)-2-fluorobenzoico (Compuesto 1-129)

Siguiendo el procedimiento del Ejemplo 12 pero usando 6-bromohex-1-ina en lugar de 2-bromoetan-1-ol en la Etapa 1, se obtuvo el Compuesto 1-129 en sal de sodio del título en forma de un sólido de color blanco. LC-MS: m/z 526 (M+1).
Ejemplo 27: Síntesis de sal de sodio del ácido 3-((1-(1-(3-h¡drox¡-2.2-d¡met¡lprop¡l)-1H-p¡razol-4-il)-6-cloro-2-c¡cloprop¡l-7-fluoro-1H-¡ndol-3-il)t¡o)-2-fluorobenzo¡co (Compuesto 1-130)

Siguiendo el procedimiento del Ejemplo 12 pero usando 3-bromo-2,2-dimetil-propan-1-ol en lugar de 2-bromoetan-1-ol en la Etapa 1, se obtuvo el Compuesto 1-130 en sal de sodio del título en forma de un sólido de color blanco. LC-MS: m/z 481 (M+1).
Ejemplo 28: S ntesis de sal de sodio del cido 3-((6-cloro-2-ciclo ro il-1- 1- 6- 3- 3'.6'-dihidroxi-3-oxo-3H-espirorisobenzofuran-1.9'-xanten1-5-il)ureido)hexil)-1H-pirazol-4-il) )tio)-2-fluorobenzoico (Compuesto 1-131)
)tio)-2-fluorobenzoico (Compuesto 1-131)

A una solución agitada de amina 1 (Ejemplo 25; 11,1 mg, 0,020 mmol) en CH2CI2 (2 ml) en una atmosfera inerte se le añadió isotiocianato de fluoresceína (7,6 mg, 0,020 mmol) seguido de DIEA (3,4 pl, 0,020 mmol). A continuación, la mezcla se agitó a temperatura ambiente en una atmósfera de N2 durante una noche. Después de que se completara la reacción, la mezcla se evaporó a sequedad. A continuación, el producto en bruto se purificó por HPLC prep. para proporcionar 3 mg del Compuesto 1-131 del título en forma de un sólido de color naranja. LC-MS: m/z 934 (M+1).
Ejemplo 29: Síntesis de ácido 2-(3-((6-cloro-2-ciclopropil-7-fluoro-1-(1-etil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorofenil)acético (compuesto 2-7):

A una solución agitada de ácido 3-((6-cloro-2-ciclopropil-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico compuesto 1-34 (Ejemplo 9; 30 mg, 0,058 mmol) en tolueno (2,0 ml) en una atmósfera inerte se le añadió cloruro de tionilo (0,22 ml, 3,0 mmol) y la mezcla se calentó a 85 °C durante 2 h. Los volátiles se eliminaron a presión reducida y el residuo se suspendió de nuevo en una solución de tolueno (2,0 ml) y (trimetilsilil)diazometano (2,0 M en hexanos, 0,88 ml, 0,88 mmol). Después del desprendimiento de gas, se añadió alcohol t-butílico (2,0 ml) y la mezcla se calentó durante 0,5 h a 50 °C. El residuo se purificó por cromatografía sobre gel de sílice (Hx al 0 20 %/EtOAC). Después de la evaporación de las fracciones, el residuo se disolvió en HCl 4,0 M en 1,4-dioxano (0,5 ml) y la solución se agitó a TA durante 2 h. El disolvente se eliminó y el residuo se purificó por HPLC para dar el compuesto diana en forma de una película transparente. MS (ESI): m/z 488,0 (M+H+).
Ejemplo 30: Composición farmacéutica parenteral
Para preparar una composición farmacéutica parenteral adecuada para la administración por inyección (subcutánea, intravenosa), se disuelven en agua estéril 1-100 mg de una sal soluble en agua de un compuesto descrito en el presente documento, o una sal o solvato farmacéuticamente aceptable del mismo, y continuación se mezclan con 10 ml de una solución salina estéril al 0,9 %. Se añade opcionalmente un tampón adecuado, así como un ácido o una base opcionales para ajustar el pH. La mezcla se incorpora en una forma unitaria de dosificación adecuada para la administración por inyección.
Ejemplo 31: Solución oral
Para preparar una composición farmacéutica para administración oral, se añade una cantidad suficiente de un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, al agua (con uno o más solubilizadores opcionales, uno o más tampones opcionales y excipientes enmascaradores del sabor) para proporcionar una solución de 20 mg/ml.
Ejemplo 32: Comprimido oral
Se prepara un comprimido mezclando el 20-50 % en peso de un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, el 20-50 % en peso de celulosa microcristalina, el 1-10 % en peso de hidroxipropilcelulosa de baja sustitución y el 1-10% en peso de estearato de magnesio u otros excipientes apropiados. Los comprimidos se preparan mediante compresión directa. El peso total de los comprimidos se mantiene en 100-500 mg.
Ejemplo 33: Cápsula oral
Para preparar una composición farmacéutica para administración oral, se mezclan 10-500 mg de un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, con almidón u otra mezcla en polvo adecuada. La mezcla se incorpora a una unidad de dosificación oral, tal como una cápsula de gelatina dura, que es adecuada para la administración oral.
En otra realización, se colocan 10-500 mg de un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, en una cápsula de tamaño 4 o una cápsula de tamaño 1 (hipromelosa o gelatina dura) y la cápsula se cierra.
Ejemplo 34: Composición de gel tópico
Para preparar una composición farmacéutica de gel tópico, un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, se mezcla con hidroxipropilcelulosa, propilenglicol, miristato de isopropilo y alcohol purificado USP. A continuación, la mezcla de gel resultante se incorpora a recipientes, tales como tubos, que son adecuados para la administración tópica.
Ejemplo 35: Ensayo de autotaxina humana
La actividad de ATX se ensaya en medio acondicionado concentrado de células de carcinoma hepatocelular humano Hep3B midiendo la cantidad de colina liberada del sustrato, lisofosfatidilcolina (LPC) a medida que se escinde a LPA. El medio acondicionado se recoge de las células Hep3B confluentes y se concentra 20 veces usando dispositivos de filtrado Centriprep-30 (Millipore). Para ensayar la inhibición de la autotaxina, se incuban 10-20 pl del medio acondicionado concentrado con 2,5 pl de un compuesto de prueba en DMSO y 72,5-82,5 pl de tampón liso-PLD (Tris 100 mM a pH 9, NaCl 500 mM, MgCh 5 mM, CaCh 5mM, Triton X-100 al 0,05% en presencia o ausencia de albúmina sérica humana libre de ácidos grasos al 0,2 %) durante 15 min a 37 °C. Después de 15 min de incubación, se añaden 5 ul de LPC 2 mM (14:0; Avanti Polar Lipids Cat. N.° 855575C) diluido en tampón liso-PLD para una concentración final de 100 uM y la incubación continúa durante 1,5-3 horas a 37 °C. Se añaden 100 pl de una mezcla de color que contiene 4-aminoantipirina 4,5 mM, N-etil-N-(2-hidroxi-3-sulfopropil)-m-toluidina 2,7 mM, 21 unidades/ml de peroxidasa de rábano picante y 3 unidades/ml de colina oxidasa en Tris 50 mM, pH 8, MgCh 4,5 mM y la incubación continúa durante 15 minutos a temperatura ambiente antes de leer la absorbancia a 555 nm.
La actividad biológica ilustrativa de compuestos representativos en el ensayo de autotaxina humana descrito en el presente documento se presenta en la siguiente tabla. Los compuestos mencionados a continuación, que no están incluidos en las reivindicaciones adjuntas, se incluyen en el presente documento como ejemplos de referencia.

continuación

Ejemplo 36: Ensayo de autotaxina en sangre completa humana
La inhibición de la actividad de ATX en sangre completa humana se ensaya midiendo la concentración de LPA a 20:4 en plasma después de una incubación prolongada a 37 °C. Se extrae sangre de voluntarios humanos con su consentimiento en tubos vacutainer de heparina y se añaden 200 j l de alícuotas a 2 j l de compuesto de prueba en DMSO o DMSO solo. Varios de los tubos de vehículo se centrifugan inmediatamente a 800 x g durante 10 minutos a 4 °C y se extrae el plasma para su procesamiento para determinar la concentración inicial de LPA a 20:4. Las muestras de sangre restantes que contienen vehículo o compuesto de prueba se incuban a 37 °C durante 4 horas antes de la centrifugación a 800 x g durante 10 minutos a 4 °C para obtener plasma. El plasma se procesa para el análisis por LCMS de la siguiente manera: Se extraen 40 ul de plasma y se añaden 5 volúmenes de metanol que contienen 125 ng/ml de 17:0 de LPA como patrón interno y la mezcla se incuba a -20 °C durante 10 min antes la centrifugación a 4000 xg durante 10 minutos a 4 °C. Se transfieren 150 j l del sobrenadante a una placa de 96 pocillos y se diluyen con 100 j l de una solución orgánica (90:10:0,1 de agua/acetonitrilo/hidróxido de amonio) para el análisis de concentraciones de LPA a 20:4 por LCMS. El LPA a 20:4 y el patrón interno (LPA a 17:0) se analizaron en un espectrómetro de masas de cuadrupolo (ABI Sciex 4000QTrap) en el modo de ion negativo (ESI) mediante monitorización de reacciones múltiples (MRM). Las fases móviles contienen hidróxido de amonio al 0,1 % en agua al 90 %/acetonitrilo al 10% (disolvente A) e hidróxido de amonio al 0,1% en acetonitrilo al 90 %/agua al 10% (disolvente B). El caudal se mantuvo a 0,8 ml/min y el tiempo de ciclo total fue de 3 min. Los analitos se separaron usando un gradiente lineal de la siguiente manera: 1) la fase móvil se mantuvo durante 0,5 min al 10 % de B; 2) B se aumentó del 10 % al 90 % durante el minuto siguiente; 3) B se mantuvo constante durante 0,5 min al 90 %; y 4) B volvió a las condiciones de gradiente iniciales.
Los ejemplos y realizaciones que se describen en el presente documento son solo para fines ilustrativos.
Claims (14)
1. Un compuesto de Fórmula (III), o una sal o un solvato farmacéuticamente aceptables del mismo:

en la que,
R1 es -Cl, -Br o -CN;
R2 es Cl;
R3 es H, F o Cl;
W es CH, CF o N;
RA es H;
L1 está ausente, es alquileno C1-C6 o cicloalquileno C3-C6 ;
Q es -CO2H o -CO2(alquilo C1-C6);
el Anillo B es furanilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, isoxazolilo, isotiazolilo, oxadiazolilo, tiadiazolilo, piridinilo, pirimidinilo, pirazinilo, piridazinilo o triazinilo;
cada RB es independientemente H, halógeno, -CN, -NO2, -OH, -OR9, -SR9, -S(=O)R9, -S(=O)2R9, -S(=O)2N(R10)2, alquilo C1-C6 , fluoroalquilo C1-C6, deuteroalquilo C1-C6, heteroalquilo C1-C6, cicloalquilo C3-C10 sustituido o sin sustituir, heterocicloalquilo C2-C10 sustituido o sin sustituir, fenilo sustituido o sin sustituir, alquileno C1-C4-(fenilo sustituido o sin sustituir), heteroarilo monocíclico sustituido o sin sustituir, alquileno C1-C4-(heteroarilo monocíclico sustituido o sin sustituir), un heteroarilo bicíclico sustituido o sin sustituir o alquileno C1-C4-(heteroarilo bicíclico sustituido o sin sustituir);
n es 0, 1 o 2;
R9 es alquilo C1-C6, fluoroalquilo C1-C6, deuteroalquilo C1-C6, cicloalquilo C3-C6 , un fenilo sustituido o sin sustituir, un heteroarilo monocíclico sustituido o sin sustituir o un heteroarilo bicíclico sustituido o sin sustituir;
cada R10 es independientemente H, alquilo C1-C6 , fluoroalquilo C1-C6, deuteroalquilo C1-C6, cicloalquilo C3-C6, un fenilo sustituido o sin sustituir o un heteroarilo monocíclico sustituido o sin sustituir; o
dos grupos R10 unidos al mismo átomo de N se toman junto con el átomo de N al que están unidos para formar un heterociclo sustituido o sin sustituir;
en donde los grupos sustituidos están sustituidos por uno o más grupos seleccionados individual e independientemente de halógeno, -CN, -NH2 , -OH, -NH(CH3), -N(CH3)2 , -CH3, -CH2CH3 , -CF3 , -OCH3 y -OCF3; y en donde el término "alquilo" solo o en "heteroalquilo" también se refiere a un tipo de grupos alquilo en el que está presente al menos un doble enlace carbono-carbono.
2. El compuesto de la reivindicación 1, o una sal o un solvato farmacéuticamente aceptables del mismo, en el que:
L1 está ausente, es -CH2-, -CH(CH3)-, -C(CH3)2-o ciclopropil-1,1-diilo.
3. El compuesto de la reivindicación 1, en donde el compuesto tiene la siguiente estructura:

en la que,
RA es H;
RB es H, alquilo C1-C6 , fluoroalquilo C1-C6 o deuteroalquilo C1-C6 ;
R1 es -Cl, -Br o -CN;
R2 es Cl; y
R3 es H, F o Cl;
o una sal o un solvato farmacéuticamente aceptables del mismo.
4. El compuesto de la reivindicación 3, en donde el compuesto tiene la siguiente estructura:

o una sal o un solvato farmacéuticamente aceptables del mismo.
5. El compuesto de una cualquiera de las reivindicaciones 1-4, o una sal o un solvato farmacéuticamente aceptables del mismo, en el que:
RB es alquilo C1-C6.
6. El compuesto de una cualquiera de las reivindicaciones 3-5, o una sal o un solvato farmacéuticamente aceptables del mismo, en el que:
L1 está ausente, es -CH2-, -CH(CH3)-, -C(CH3)2- o ciclopropil-1,1 -diilo.78
7. El compuesto de una cualquiera de las reivindicaciones 1-5, o una sal o un solvato farmacéuticamente aceptable del mismo, en el que:
L1 está ausente.
8. El compuesto de la reivindicación 1, en donde el compuesto es:
ácido 3-((2,6-dicloro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico;
ácido 3-((6-cloro-2-ciano-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico;
ácido 3-((2,6-dicloro-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico;
ácido 3-((2-bromo-6-cloro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)benzoico;
ácido 3-((2,6-dicloro-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico;
ácido 3-((2,6-dicloro-7-fluoro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico;
ácido 3-((2,6-dicloro-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico;
ácido 3-((2,6-dicloro-7-fluoro-1-(piridin-3-il)-1H-indol-3-il)tio)-2-fluorobenzoico;
ácido 3-((2-bromo-6-cloro-7-fluoro-1-(piridin-3-il)-1H-indol-3-il)tio)-2-fluorobenzoico;
o una sal o un solvato farmacéuticamente aceptables de los mismos.
9. El compuesto de la reivindicación 1, en donde el compuesto es:
ácido 3-((2,6-dicloro-7-fluoro-1-(1-propil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico;
ácido 3-((2,6-dicloro-7-fluoro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico;
ácido 3-((2,6-dicloro-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico;
o una sal o un solvato farmacéuticamente aceptables de los mismos.
10. El compuesto de la reivindicación 1, en donde el compuesto es:
ácido 3-((2,6-dicloro-7-fluoro-1-(1-metil-1H-pirazol-4-il)-1H-indol-3-il)tio)-2-fluorobenzoico;
o una sal o un solvato farmacéuticamente aceptables del mismo.
11. El compuesto de la reivindicación 1, en donde el compuesto es:
ácido 3-((2,6-dicloro-1-(1-etil-1H-pirazol-4-il)-7-fluoro-1H-indol-3-il)tio)-2-fluorobenzoico;
o una sal o un solvato farmacéuticamente aceptables del mismo.
12. Una composición farmacéutica que comprende un compuesto de una cualquiera de las reivindicaciones 1-11, o una sal o un solvato farmacéuticamente aceptables del mismo, y al menos un excipiente farmacéuticamente aceptable.
13. La composición farmacéutica de la reivindicación 12, en donde la composición farmacéutica está en forma de un comprimido, una píldora, una cápsula, un líquido, una suspensión, un gel, una dispersión, una solución, una emulsión, un ungüento o una loción.
14. Un compuesto de una cualquiera de las reivindicaciones 1-11, o una sal o un solvato farmacéuticamente aceptables del mismo, para su uso en un método para el tratamiento de un mamífero mediante terapia, o para su uso en un método de tratamiento de fibrosis o cáncer en un mamífero.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361907965P | 2013-11-22 | 2013-11-22 | |
| US201462038121P | 2014-08-15 | 2014-08-15 | |
| PCT/US2014/066706 WO2015077503A1 (en) | 2013-11-22 | 2014-11-20 | Autotaxin inhibitor compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2875899T3 true ES2875899T3 (es) | 2021-11-11 |
Family
ID=53180152
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14864293T Active ES2875899T3 (es) | 2013-11-22 | 2014-11-20 | Compuestos inhibidores de autotaxina |
Country Status (31)
| Country | Link |
|---|---|
| US (7) | US9334261B2 (es) |
| EP (2) | EP3878848A1 (es) |
| JP (1) | JP6501367B2 (es) |
| KR (2) | KR102367876B1 (es) |
| CN (2) | CN110372671B (es) |
| AU (2) | AU2014352888B2 (es) |
| BR (1) | BR112016010220B1 (es) |
| CA (1) | CA2930737C (es) |
| CL (1) | CL2016001231A1 (es) |
| CR (2) | CR20160289A (es) |
| CY (1) | CY1124407T1 (es) |
| DK (1) | DK3071561T3 (es) |
| EA (1) | EA030068B1 (es) |
| ES (1) | ES2875899T3 (es) |
| HK (1) | HK1226075A1 (es) |
| HR (1) | HRP20210957T1 (es) |
| HU (1) | HUE055213T2 (es) |
| IL (2) | IL245383B (es) |
| LT (1) | LT3071561T (es) |
| MX (2) | MX2016006623A (es) |
| MY (1) | MY182095A (es) |
| NI (1) | NI201600071A (es) |
| NZ (1) | NZ720478A (es) |
| PE (1) | PE20160654A1 (es) |
| PH (1) | PH12016500862A1 (es) |
| PL (1) | PL3071561T3 (es) |
| PT (1) | PT3071561T (es) |
| RS (1) | RS62027B1 (es) |
| SI (1) | SI3071561T1 (es) |
| WO (1) | WO2015077503A1 (es) |
| ZA (2) | ZA201603083B (es) |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EA201492223A1 (ru) | 2012-06-13 | 2015-03-31 | Ф. Хоффманн-Ля Рош Аг | Новые диазаспироциклоалканы и азаспироциклоалканы |
| EP2900669B1 (en) | 2012-09-25 | 2019-09-04 | F.Hoffmann-La Roche Ag | Hexahydropyrrolo[3,4-c]pyrrole derivatives and related compounds as autotaxin (atx) inhibitors and as inhibitors of the lysophosphatidic acid (lpa) production for treating e.g. renal diseases |
| AR095079A1 (es) | 2013-03-12 | 2015-09-16 | Hoffmann La Roche | Derivados de octahidro-pirrolo[3,4-c]-pirrol y piridina-fenilo |
| WO2015042052A1 (en) | 2013-09-17 | 2015-03-26 | Pharmakea, Inc. | Heterocyclic vinyl autotaxin inhibitor compounds |
| US9714240B2 (en) | 2013-09-17 | 2017-07-25 | Pharmakea, Inc. | Vinyl autotaxin inhibitor compounds |
| US9850203B2 (en) | 2013-09-26 | 2017-12-26 | Pharmakea, Inc. | Autotaxin inhibitor compounds |
| KR20160088878A (ko) | 2013-11-22 | 2016-07-26 | 파마케아, 인크. | 사환식 오토탁신 억제제 |
| CA2930737C (en) | 2013-11-22 | 2023-02-21 | Pharmakea, Inc. | Autotaxin inhibitor compounds |
| KR20160087900A (ko) | 2013-11-26 | 2016-07-22 | 에프. 호프만-라 로슈 아게 | 신규한 옥타하이드로-사이클로부타[1,2-c;3,4-c'']다이피롤-2-일 |
| UA119347C2 (uk) | 2014-03-26 | 2019-06-10 | Ф. Хоффманн-Ля Рош Аг | Конденсовані [1,4]діазепінові сполуки як інгібітори продукції аутотаксину (atх) та лізофосфатидилової кислоти (lpa) |
| CN106103446B (zh) | 2014-03-26 | 2019-07-30 | 豪夫迈·罗氏有限公司 | 作为自分泌运动因子(atx)和溶血磷脂酸(lpa)生产抑制剂的二环化合物 |
| MA41898A (fr) | 2015-04-10 | 2018-02-13 | Hoffmann La Roche | Dérivés de quinazolinone bicyclique |
| KR20240060887A (ko) * | 2015-05-27 | 2024-05-08 | 사브레 테라퓨틱스 엘엘씨 | 오토탁신 억제제 및 이의 용도 |
| AU2016316717B2 (en) | 2015-09-04 | 2021-02-18 | F. Hoffmann-La Roche Ag | Phenoxymethyl derivatives |
| MX2018001430A (es) | 2015-09-24 | 2018-04-20 | Hoffmann La Roche | Nuevos compuestos biciclicos como inhibidores duales de autotaxina (atx)/anhidrasa carbonica (ca). |
| WO2017050792A1 (en) | 2015-09-24 | 2017-03-30 | F. Hoffmann-La Roche Ag | Bicyclic compounds as atx inhibitors |
| KR20180053408A (ko) | 2015-09-24 | 2018-05-21 | 에프. 호프만-라 로슈 아게 | 이중 오토탁신(atx)/탄산 무수화효소(ca) 억제제로서의 신규한 이환형 화합물 |
| EP3353180B1 (en) | 2015-09-24 | 2022-03-16 | F. Hoffmann-La Roche AG | Bicyclic compounds as atx inhibitors |
| US11000142B2 (en) | 2016-12-06 | 2021-05-11 | John Joseph Girard | Flexible floor mat incorporating LED lighting |
| CN110382484B (zh) | 2017-03-16 | 2022-12-06 | 豪夫迈·罗氏有限公司 | 新的作为atx抑制剂的二环化合物 |
| RU2019132254A (ru) | 2017-03-16 | 2021-04-16 | Ф. Хоффманн-Ля Рош Аг | Гетероциклические соединения, пригодные в качестве дуальных ингибиторов atx/ca |
| CN111565750B (zh) | 2017-08-28 | 2023-11-03 | 安吉克公司 | 抗tm4sf1抗体及其使用方法 |
| WO2023215196A1 (en) * | 2022-05-03 | 2023-11-09 | Sabre Therapeutics Llc | Polymorphic and salt forms of the autotaxin inhibitor cudetaxestat |
| CN116239577A (zh) * | 2023-03-27 | 2023-06-09 | 沈阳药科大学 | 一种制备Cudetaxestat的方法 |
| CN116396205B (zh) * | 2023-04-20 | 2024-10-01 | 沈阳药科大学 | 一种n-芳基吲哚衍生物及其制备方法和应用 |
Family Cites Families (80)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5210574A (en) | 1991-03-08 | 1993-05-11 | Mita Industrial Co., Ltd. | Photosensitive drum body-mounting mechanism including a drive coupling member with a coupling protrusion adapted to bite into the inner surface of the mechanism's photosensitive drum |
| US5186522A (en) | 1991-09-24 | 1993-02-16 | Midland Brake, Inc. | Air dryer purge line |
| AU663990B2 (en) | 1992-11-10 | 1995-10-26 | Caterpillar Inc. | Solenoid valve assembly |
| US5407368A (en) | 1992-12-15 | 1995-04-18 | Minnesota Mining And Manufacturing Company | Electrode connector |
| US5761473A (en) | 1993-01-08 | 1998-06-02 | International Business Machines Corporation | Method and system for increased instruction synchronization efficiency in a superscalar processsor system utilizing partial data dependency interlocking |
| US5564949A (en) | 1995-01-05 | 1996-10-15 | Thomas & Betts Corporation | Shielded compact data connector |
| JP2664048B2 (ja) | 1995-03-27 | 1997-10-15 | 科学技術庁金属材料技術研究所長 | 六フッ化二ケイ素の合成法 |
| GB9514473D0 (en) | 1995-07-14 | 1995-09-13 | Glaxo Lab Sa | Chemical compounds |
| WO2001030343A1 (en) | 1999-10-22 | 2001-05-03 | Merck & Co., Inc. | Pharmaceuticals for treating obesity |
| US6890933B1 (en) | 2000-02-24 | 2005-05-10 | President And Fellows Of Harvard College | Kinesin inhibitors |
| WO2002028865A2 (en) | 2000-10-03 | 2002-04-11 | Lilly Icos Llc | Condensed pyridoindole derivatives |
| WO2002083126A1 (en) | 2001-04-11 | 2002-10-24 | Idenix (Cayman) Limited | Phenylindoles for the treatment of hiv |
| MXPA04002824A (es) * | 2001-09-27 | 2004-07-05 | Hoffmann La Roche | Derivados de indol como inhibidores de cox ii. |
| US7528165B2 (en) | 2001-12-13 | 2009-05-05 | National Health Research Institutes | Indole compounds |
| PT1537078E (pt) | 2002-08-29 | 2010-06-18 | Merck Sharp & Dohme | Indoles que possuem actividade antidiabética |
| JP4340232B2 (ja) | 2002-08-29 | 2009-10-07 | メルク エンド カムパニー インコーポレーテッド | 抗糖尿病活性を有するインドール類 |
| US7060697B2 (en) | 2003-05-19 | 2006-06-13 | Irm Llc | Immunosuppressant compounds and compositions |
| RS50589B (sr) | 2003-12-23 | 2010-05-07 | H. Lundbeck A/S. | 2-(1h-indolilsulfanil)-benzil amin derivati kao ssri |
| JP4714143B2 (ja) | 2004-05-19 | 2011-06-29 | 住友電気工業株式会社 | Iii族窒化物半導体結晶の製造方法 |
| KR101141650B1 (ko) | 2004-09-30 | 2012-05-17 | 엘지전자 주식회사 | 매체접속제어 계층에서의 데이터 처리 방법 및 이동통신용단말 |
| WO2006041961A1 (en) | 2004-10-05 | 2006-04-20 | GOVERNMENT OF THE UNITED STATES OF AMERICA, as represented by THE SECRETARY, DEPARTMENT OF HEALTHAND HUMAN SERVICES | Arylthioindole tubulin polymerization inhibitors and methods of treating or preventing cancer using same |
| WO2007134169A2 (en) | 2006-05-10 | 2007-11-22 | Nuada, Llc | Indole, benzimidazole, and benzolactam boronic acid compounds, analogs thereof and methods of use thereof |
| WO2006050236A2 (en) | 2004-11-01 | 2006-05-11 | Nuada, Llc | Compounds and methods of use thereof |
| US20060270634A1 (en) | 2005-05-06 | 2006-11-30 | Miller Duane D | Acetal phosphate-derived LPA mimics, PPARgamma activators, and autotaxin inhibitors |
| AR054394A1 (es) | 2005-06-17 | 2007-06-20 | Lundbeck & Co As H | Derivados de 2- (1h-indolilsulfanil)-aril amina |
| WO2007041600A2 (en) | 2005-10-03 | 2007-04-12 | Luminescent Technologies, Inc. | Mask-pattern determination using topology types |
| JP5081161B2 (ja) | 2005-12-19 | 2012-11-21 | スミスクライン ビーチャム コーポレーション | ファルネソイドx受容体アゴニスト |
| CA2690762C (en) | 2007-06-15 | 2016-06-14 | Glenn Prestwich | .alpha.-chloro and .alpha.-bromo phosphonate analogs of lysophosphatidic acid and methods of making and using thereof |
| DE102007047737A1 (de) | 2007-10-05 | 2009-04-30 | Merck Patent Gmbh | Piperidin- und Piperazinderivate |
| DE102007047735A1 (de) | 2007-10-05 | 2009-04-09 | Merck Patent Gmbh | Thiazolderivate |
| DE102007047738A1 (de) | 2007-10-05 | 2009-04-09 | Merck Patent Gmbh | Imidazolderivate |
| US8268891B1 (en) | 2007-11-13 | 2012-09-18 | University Of Memphis Research Foundation | Autotaxin inhibitors |
| US8378100B2 (en) | 2008-01-09 | 2013-02-19 | University Of Virginia Patent Foundation | Phosphonate derivatives as autotaxin inhibitors |
| WO2009151644A2 (en) | 2008-06-13 | 2009-12-17 | Yale University | Small molecule inhibitors of autotaxin methods of use |
| US8022239B2 (en) | 2008-10-03 | 2011-09-20 | The University Of Memphis Research Foundation | Mechanism-based inactivators of autotaxin |
| DE102008059578A1 (de) | 2008-11-28 | 2010-06-10 | Merck Patent Gmbh | Benzo-Naphtyridin Verbindungen |
| EA201100879A1 (ru) | 2008-12-01 | 2012-01-30 | Мерк Патент Гмбх | Производные пиридопиримидина в качестве ингибиторов аутотаксина |
| US8329907B2 (en) * | 2009-04-02 | 2012-12-11 | Merck Patent Gmbh | Autotaxin inhibitors |
| KR20120027177A (ko) | 2009-04-02 | 2012-03-21 | 메르크 파텐트 게엠베하 | 오토탁신 저해제로서의 피페리딘 및 피라진 유도체 |
| EP2414327B1 (de) * | 2009-04-02 | 2014-11-19 | Merck Patent GmbH | Heterocyclische verbindungen als autotaxin-inhibitoren |
| US8445200B2 (en) | 2009-04-15 | 2013-05-21 | The Regents Of The University Of California | Genotoxicity as a biomarker for inflammation |
| EA034552B1 (ru) | 2009-05-11 | 2020-02-19 | БЕРГ ЭлЭлСи | Способ лечения или предотвращения прогрессирования онкологических заболеваний |
| US8592388B2 (en) | 2009-05-20 | 2013-11-26 | Eth Zurich | Targeting MicroRNAs for metabolic disorders |
| US8343934B2 (en) | 2009-06-30 | 2013-01-01 | University Of Memphis | Diverse lead compound autotaxin inhibitors |
| DE102009033392A1 (de) | 2009-07-16 | 2011-01-20 | Merck Patent Gmbh | Heterocyclische Verbindungen als Autotaxin-Inhibitoren II |
| DE102009049211A1 (de) | 2009-10-13 | 2011-04-28 | Merck Patent Gmbh | Sulfoxide |
| WO2011049433A1 (en) | 2009-10-23 | 2011-04-28 | Academisch Ziekenhuis Bij De Universiteit Van Amsterdam | Diagnosis and treatment of pruritis |
| US8497371B2 (en) | 2009-10-26 | 2013-07-30 | University Of Memphis Research Foundation | Pipemidic acid derivative autotaxin inhibitors |
| WO2011053597A1 (en) | 2009-10-26 | 2011-05-05 | The University Of Memphis Research Foundation | Pipemidic acid derivative autotaxin inhibitors |
| WO2011103430A1 (en) | 2010-02-19 | 2011-08-25 | Medivation Technologies, Inc. | Pyrido[4,3-b]indole and pyrido[3,4-b]indole derivatives and methods of use |
| EP2552914B1 (de) * | 2010-03-26 | 2015-11-11 | Merck Patent GmbH | Benzonaphthyridinamine als autotaxin-inhibitoren |
| EP2558115B1 (en) | 2010-04-16 | 2019-07-31 | The Salk Institute for Biological Studies | Methods for treating metabolic disorders using fgf |
| ES2646834T3 (es) | 2010-08-20 | 2017-12-18 | Amira Pharmaceuticals, Inc. | Inhibidores de autotaxina y usos de los mismos |
| US8771696B2 (en) | 2010-11-23 | 2014-07-08 | Regeneron Pharmaceuticals, Inc. | Method of reducing the severity of stress hyperglycemia with human antibodies to the glucagon receptor |
| WO2012100018A1 (en) | 2011-01-20 | 2012-07-26 | University Of Tennessee Research Foundation | Inhibitors of autotaxin |
| WO2012112964A2 (en) | 2011-02-18 | 2012-08-23 | Medivation Technologies, Inc. | PYRIDO[4,3-b]INDOLE AND PYRIDO[3,4-b]INDOLE DERIVATIVES AND METHODS OF USE |
| CA2827642A1 (en) | 2011-02-18 | 2012-11-15 | Medivation Technologies, Inc. | Compounds and methods of treating diabetes |
| EP2685976B1 (en) | 2011-03-17 | 2017-12-27 | Tel HaShomer Medical Research Infrastructure and Services Ltd. | Quinolone analogs for treating autoimmune diseases |
| US9260416B2 (en) | 2011-05-27 | 2016-02-16 | Amira Pharmaceuticals, Inc. | Heterocyclic autotaxin inhibitors and uses thereof |
| GB2494154B (en) | 2011-08-31 | 2017-11-29 | Metaswitch Networks Ltd | Conditional telecommunications |
| WO2013054185A1 (en) | 2011-10-13 | 2013-04-18 | Pfizer, Inc. | Pyrimidine and pyridine derivatives useful in therapy |
| US9273011B2 (en) | 2011-10-28 | 2016-03-01 | Inhibitaxin Limited | Substituted pyridazines for the treatment of pain |
| JP2013129632A (ja) | 2011-12-22 | 2013-07-04 | Ono Pharmaceut Co Ltd | Enpp2阻害化合物 |
| US20130270634A1 (en) | 2012-04-12 | 2013-10-17 | Richtek Technology Corporation | High voltage device and manufacturing method thereof |
| EA201492223A1 (ru) | 2012-06-13 | 2015-03-31 | Ф. Хоффманн-Ля Рош Аг | Новые диазаспироциклоалканы и азаспироциклоалканы |
| WO2014031170A1 (en) | 2012-08-22 | 2014-02-27 | Medivation Technologies, Inc. | Pyrido[4,3-b]indole and pyrido[3,4-b]indole derivatives and methods of use |
| EP2900669B1 (en) | 2012-09-25 | 2019-09-04 | F.Hoffmann-La Roche Ag | Hexahydropyrrolo[3,4-c]pyrrole derivatives and related compounds as autotaxin (atx) inhibitors and as inhibitors of the lysophosphatidic acid (lpa) production for treating e.g. renal diseases |
| US9409895B2 (en) | 2012-12-19 | 2016-08-09 | Novartis Ag | Autotaxin inhibitors |
| JP6363616B2 (ja) | 2012-12-19 | 2018-07-25 | ノバルティス アーゲー | オートタキシン阻害剤 |
| TWI499591B (zh) | 2013-01-11 | 2015-09-11 | Lilly Co Eli | 雙環嘧啶化合物 |
| US9714240B2 (en) | 2013-09-17 | 2017-07-25 | Pharmakea, Inc. | Vinyl autotaxin inhibitor compounds |
| WO2015042052A1 (en) | 2013-09-17 | 2015-03-26 | Pharmakea, Inc. | Heterocyclic vinyl autotaxin inhibitor compounds |
| US9850203B2 (en) | 2013-09-26 | 2017-12-26 | Pharmakea, Inc. | Autotaxin inhibitor compounds |
| SI3066079T1 (sl) | 2013-11-05 | 2018-09-28 | Bayer Cropscience Ag | Nove spojine za boj proti členonožcem |
| CL2016001118A1 (es) | 2013-11-22 | 2016-12-02 | Basf Se | Compuestos heterocíclicos de n-acilimino |
| KR20160088878A (ko) | 2013-11-22 | 2016-07-26 | 파마케아, 인크. | 사환식 오토탁신 억제제 |
| AU2014351870B2 (en) | 2013-11-22 | 2018-07-05 | Basf Se | N-acylimino heterocyclic compounds |
| CA2930737C (en) | 2013-11-22 | 2023-02-21 | Pharmakea, Inc. | Autotaxin inhibitor compounds |
| US9051320B1 (en) * | 2014-08-18 | 2015-06-09 | Pharmakea, Inc. | Methods for the treatment of metabolic disorders by a selective small molecule autotaxin inhibitor |
| KR20240060887A (ko) | 2015-05-27 | 2024-05-08 | 사브레 테라퓨틱스 엘엘씨 | 오토탁신 억제제 및 이의 용도 |
-
2014
- 2014-11-20 CA CA2930737A patent/CA2930737C/en active Active
- 2014-11-20 PE PE2016000669A patent/PE20160654A1/es unknown
- 2014-11-20 KR KR1020167014496A patent/KR102367876B1/ko active IP Right Grant
- 2014-11-20 KR KR1020227005531A patent/KR102515248B1/ko active IP Right Grant
- 2014-11-20 DK DK14864293.7T patent/DK3071561T3/da active
- 2014-11-20 BR BR112016010220-7A patent/BR112016010220B1/pt active IP Right Grant
- 2014-11-20 CR CR20160289A patent/CR20160289A/es unknown
- 2014-11-20 MX MX2016006623A patent/MX2016006623A/es active IP Right Grant
- 2014-11-20 RS RS20210786A patent/RS62027B1/sr unknown
- 2014-11-20 MY MYPI2016000959A patent/MY182095A/en unknown
- 2014-11-20 HU HUE14864293A patent/HUE055213T2/hu unknown
- 2014-11-20 EP EP21156420.8A patent/EP3878848A1/en active Pending
- 2014-11-20 NZ NZ720478A patent/NZ720478A/en unknown
- 2014-11-20 WO PCT/US2014/066706 patent/WO2015077503A1/en active Application Filing
- 2014-11-20 ES ES14864293T patent/ES2875899T3/es active Active
- 2014-11-20 AU AU2014352888A patent/AU2014352888B2/en active Active
- 2014-11-20 EA EA201690881A patent/EA030068B1/ru unknown
- 2014-11-20 SI SI201431843T patent/SI3071561T1/sl unknown
- 2014-11-20 LT LTEP14864293.7T patent/LT3071561T/lt unknown
- 2014-11-20 EP EP14864293.7A patent/EP3071561B1/en active Active
- 2014-11-20 PT PT148642937T patent/PT3071561T/pt unknown
- 2014-11-20 JP JP2016533532A patent/JP6501367B2/ja active Active
- 2014-11-20 CN CN201910686732.3A patent/CN110372671B/zh active Active
- 2014-11-20 PL PL14864293T patent/PL3071561T3/pl unknown
- 2014-11-20 CN CN201480063881.XA patent/CN105793253B/zh active Active
- 2014-11-20 CR CR20190394A patent/CR20190394A/es unknown
-
2015
- 2015-10-29 US US14/926,877 patent/US9334261B2/en active Active
-
2016
- 2016-03-03 US US15/060,398 patent/US9468628B2/en active Active
- 2016-05-02 IL IL24538316A patent/IL245383B/en active IP Right Grant
- 2016-05-09 ZA ZA2016/03083A patent/ZA201603083B/en unknown
- 2016-05-10 PH PH12016500862A patent/PH12016500862A1/en unknown
- 2016-05-20 NI NI201600071A patent/NI201600071A/es unknown
- 2016-05-20 CL CL2016001231A patent/CL2016001231A1/es unknown
- 2016-05-20 MX MX2020009780A patent/MX2020009780A/es unknown
- 2016-09-09 US US15/261,659 patent/US9999615B2/en active Active
- 2016-12-21 HK HK16114544A patent/HK1226075A1/zh unknown
-
2018
- 2018-05-03 US US15/970,337 patent/US10688081B2/en active Active
-
2019
- 2019-08-09 AU AU2019213425A patent/AU2019213425B2/en active Active
- 2019-10-24 IL IL270168A patent/IL270168B/en unknown
-
2020
- 2020-05-14 US US16/874,073 patent/US11344533B2/en active Active
- 2020-08-05 ZA ZA2020/05029A patent/ZA202005029B/en unknown
-
2021
- 2021-06-15 HR HRP20210957TT patent/HRP20210957T1/hr unknown
- 2021-06-23 CY CY20211100562T patent/CY1124407T1/el unknown
-
2022
- 2022-05-06 US US17/662,403 patent/US11779568B2/en active Active
-
2023
- 2023-10-04 US US18/481,141 patent/US20240189281A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2875899T3 (es) | Compuestos inhibidores de autotaxina | |
| ES2872553T3 (es) | Inhibidores de lisil oxidasa tipo-2 y usos de los mismos | |
| JP6905530B2 (ja) | ファルネソイドx受容体アゴニストとその使用 | |
| ES2290538T3 (es) | Nuevas imidazopiridinas y su uso. | |
| ES2544289T3 (es) | Derivados de quinolina y quinoxalina como inhibidores de cinasa | |
| ES2294046T3 (es) | Inhibidores de girasa bacteriana y usos de los mismos. | |
| ES2900815T3 (es) | Compuestos y métodos para inducir la condrogénesis | |
| ES2880766T3 (es) | Inhibidores de quinolinona lisil oxidasa similar al tipo 2 y usos de los mismos | |
| KR20060015283A (ko) | 신규한 피리도피라진 및 키나제 억제제로서의 이의 용도 | |
| ES2889100T3 (es) | Métodos de uso de los derivados de benzotriazol trisustituidos como inhibidores de dihidroorotato oxigenasa | |
| EP2036906A1 (de) | Azaindole als Inhibitoren der löslichen Adenylatzyklase | |
| KR100747106B1 (ko) | 퓨린 유도체 이수화물, 이를 유효성분으로 함유하는 의약및 이의 제조 중간체 | |
| CN103012274A (zh) | 异羟肟酸类化合物,制备方法和用途 |