ES2701051T3 - Conjugados de anticuerpo-fármaco - Google Patents
Conjugados de anticuerpo-fármaco Download PDFInfo
- Publication number
- ES2701051T3 ES2701051T3 ES14723915T ES14723915T ES2701051T3 ES 2701051 T3 ES2701051 T3 ES 2701051T3 ES 14723915 T ES14723915 T ES 14723915T ES 14723915 T ES14723915 T ES 14723915T ES 2701051 T3 ES2701051 T3 ES 2701051T3
- Authority
- ES
- Spain
- Prior art keywords
- seq
- antibody
- ckit
- variable region
- chain variable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- NFJWAGOKMMRHHY-UHFFFAOYSA-N O=C(CCCN(C(C=C1)=O)C1=O)NCC(NCC(NCC(NCCC(ON(C(CC1)=O)C1=O)=O)=O)=O)=O Chemical compound O=C(CCCN(C(C=C1)=O)C1=O)NCC(NCC(NCC(NCCC(ON(C(CC1)=O)C1=O)=O)=O)=O)=O NFJWAGOKMMRHHY-UHFFFAOYSA-N 0.000 description 1
- JSHOVKSMJRQOGY-UHFFFAOYSA-N O=C(CCCSSc1ncccc1)ON(C(CC1)=O)C1=O Chemical compound O=C(CCCSSc1ncccc1)ON(C(CC1)=O)C1=O JSHOVKSMJRQOGY-UHFFFAOYSA-N 0.000 description 1
- JTNCSFYQBRKCRP-UHFFFAOYSA-N O[S](C(CC(N1OC(CCCSSc2ncccc2)=O)=O)C1=O)(C1=NC1)(=O)=O Chemical compound O[S](C(CC(N1OC(CCCSSc2ncccc2)=O)=O)C1=O)(C1=NC1)(=O)=O JTNCSFYQBRKCRP-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/537—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines spiro-condensed or forming part of bridged ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39591—Stabilisation, fragmentation
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/12—Carboxylic acids; Salts or anhydrides thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6855—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from breast cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6857—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from lung cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6865—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from skin, nerves or brain cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6867—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from a cell of a blood cancer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/69—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit
- A61K47/6921—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a particulate, a powder, an adsorbate, a bead or a sphere
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/08—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins
- A61K51/10—Antibodies or immunoglobulins; Fragments thereof, the carrier being an antibody, an immunoglobulin or a fragment thereof, e.g. a camelised human single domain antibody or the Fc fragment of an antibody
- A61K51/1027—Antibodies or immunoglobulins; Fragments thereof, the carrier being an antibody, an immunoglobulin or a fragment thereof, e.g. a camelised human single domain antibody or the Fc fragment of an antibody against receptors, cell-surface antigens or cell-surface determinants
- A61K51/103—Antibodies or immunoglobulins; Fragments thereof, the carrier being an antibody, an immunoglobulin or a fragment thereof, e.g. a camelised human single domain antibody or the Fc fragment of an antibody against receptors, cell-surface antigens or cell-surface determinants against receptors for growth factors or receptors for growth regulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/08—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins
- A61K51/10—Antibodies or immunoglobulins; Fragments thereof, the carrier being an antibody, an immunoglobulin or a fragment thereof, e.g. a camelised human single domain antibody or the Fc fragment of an antibody
- A61K51/1093—Antibodies or immunoglobulins; Fragments thereof, the carrier being an antibody, an immunoglobulin or a fragment thereof, e.g. a camelised human single domain antibody or the Fc fragment of an antibody conjugates with carriers being antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/19—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles lyophilised, i.e. freeze-dried, solutions or dispersions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2863—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for growth factors, growth regulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/32—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against translation products of oncogenes
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/531—Production of immunochemical test materials
- G01N33/532—Production of labelled immunochemicals
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/574—Immunoassay; Biospecific binding assay; Materials therefor for cancer
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/574—Immunoassay; Biospecific binding assay; Materials therefor for cancer
- G01N33/57484—Immunoassay; Biospecific binding assay; Materials therefor for cancer involving compounds serving as markers for tumor, cancer, neoplasia, e.g. cellular determinants, receptors, heat shock/stress proteins, A-protein, oligosaccharides, metabolites
- G01N33/57492—Immunoassay; Biospecific binding assay; Materials therefor for cancer involving compounds serving as markers for tumor, cancer, neoplasia, e.g. cellular determinants, receptors, heat shock/stress proteins, A-protein, oligosaccharides, metabolites involving compounds localized on the membrane of tumor or cancer cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/10—Immunoglobulins specific features characterized by their source of isolation or production
- C07K2317/14—Specific host cells or culture conditions, e.g. components, pH or temperature
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/21—Immunoglobulins specific features characterized by taxonomic origin from primates, e.g. man
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/33—Crossreactivity, e.g. for species or epitope, or lack of said crossreactivity
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/34—Identification of a linear epitope shorter than 20 amino acid residues or of a conformational epitope defined by amino acid residues
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/55—Fab or Fab'
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/565—Complementarity determining region [CDR]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
- C07K2317/732—Antibody-dependent cellular cytotoxicity [ADCC]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/77—Internalization into the cell
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
Abstract
Un conjugado de anticuerpo-fármaco de la fórmula: Ab-(L-(D)m)n o una sal farmacéuticamente aceptable del mismo; en donde: Ab es un anticuerpo o fragmento de unión a antígeno del mismo que comprende: (i) una región variable de cadena pesada que comprende (a) una HCDR1 (CDR- Región Determinante de Complementariedad) de SEQ ID NO: 76, (b) una HCDR2 de SEQ ID NO: 77, (c) una HCDR3 de SEQ ID NO: 78; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 85, (e) una LCDR2 de SEQ ID NO: 86, y (f) una LCDR3 de SEQ ID NO: 87; (ii) una región variable de cadena pesada que comprende (a) una HCDR1 de SEQ ID NO: 22, (b) una HCDR2 de SEQ ID NO: 23, (c) una HCDR3 de SEQ ID NO: 24; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 31, (e) una LCDR2 de SEQ ID NO: 32, y (f) una LCDR3 de SEQ ID NO: 33; (iii) una región variable de cadena pesada que comprende (a) una HCDR1 de SEQ ID NO: 130, (b) una HCDR2 de SEQ ID NO: 131, (c) una HCDR3 de SEQ ID NO: 132; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 139, (e) una LCDR2 de SEQ ID NO: 140, y (f) una LCDR3 de SEQ ID NO: 141; (iv) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 58, (b) una HCDR2 de SEQ ID NO: 59, (c) una HCDR3 de SEQ ID NO: 60; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 67, (e) una LCDR2 de SEQ ID NO: 68, y (f) una LCDR3 de SEQ ID NO: 69; (v) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 40, (b) una HCDR2 de SEQ ID NO: 41, (c) una HCDR3 de SEQ ID NO: 42; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 49, (e) una LCDR2 de SEQ ID NO: 50, y (f) una LCDR3 de SEQ ID NO: 51; (vi) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 94, (b) una HCDR2 de SEQ ID NO: 95, (c) una HCDR3 de SEQ ID NO: 96; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 103, (d) una LCDR2 de SEQ ID NO: 104, y (f) una LCDR3 de SEQ ID NO: 105; (vii) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 112, (b) una HCDR2 de SEQ ID NO: 113, (c) una HCDR3 de SEQ ID NO: 114; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 121, (e) una LCDR2 de SEQ ID NO: 122, y (f) una LCDR3 de SEQ ID NO: 123; o (viii) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 3, (b) una HCDR2 de SEQ ID NO: 4, (c) una HCDR3 de SEQ ID NO: 5; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 12, (e) una LCDR2 de SEQ ID NO: 13, y (f) una LCDR3 de SEQ ID NO: 14. L es un enlazador; D es un maitansinoide; m es un entero de 1 a 8; y n es un entero de 1 a 10.
Description
DESCRIPCIÓN
Conjugados de anticuerpo-fármaco
CAMPO DE LA INVENCIÓN
La presente divulgación está dirigida a anticuerpos anti-cKIT, fragmentos de anticuerpo, conjugados anticuerpo-fármaco, y sus usos para el tratamiento del cáncer.
ANTECEDENTES DE LA INVENCIÓN
La cKIT es una tirosina quinasa receptora sencilla de transmembrana que se une al ligando factor de células madre (SCF). El SCF induce la homodimerización de cKIT que activa su actividad de tirosina quinasa y señaliza a través de las rutas PI3-AKT y MAPK (Kindblom etal., Am J. Path. 1998, 152(5):1259). La cKIT fue descubierta inicialmente como un oncogén como una forma truncada expresada por un retrovirus felino (Besmer et a l, J Virol. 1986; 60(1): 194-203). La clonación del gen humano correspondiente mostró que cKIT es un miembro de la clase de tirosina quinasas receptoras de tipo III, que incluye entre los miembros de la familia a FLT3, receptor de CSF-1 y receptor de PDGF.
Los ratones que son mutantes para cKIT han mostrado que cKIT es requerido para el desarrollo de células hematopoyéticas, células germinales, mastocitos y melanocitos. En el humano, la pérdida de la función de cKIT puede resultar en sordera y despigmentación de la piel y el cabello. Varias mutaciones de ganancia de función para cKIT se han descrito en varios cánceres. Estos cánceres incluyen tumores estromales gastrointestinales (GIST), leucemia mieloide aguda (AML), cáncer de pulmón de célula pequeña (SCLC), leucemia de mastocitos (MCL) y cáncer pancreático (Hirota et a l, Science. 1998 (279):577; Esposito et a l. Lab. Invets. 200282(11):1481).
Debido a estas indicaciones preliminares de que cKIT era un oncogén, se generó un anticuerpo que identificó a cKIT como un marcador de AML (Gadd et a l, Leuk. Res., 1985 (9):1329). Este anticuerpo monoclonal murino, conocido como YB5.B8, fue generado usando blastocitos leucémicos de un paciente humano, y se enlazó a cKIT, que fue expresado abundantemente sobre la superficie de las células de AML, pero no detectó cKIT en células normales de la sangre o médula ósea (Gadd et a l, supra). Se generó un segundo anticuerpo de cKIT (SR-1) que bloqueó la unión de SCF a cKIT y por lo tanto bloqueó la señalización de cKIT (Broudy et a l, Blood, 199279(2):338). El efecto biológico del anticuerpo SR-1 fue inhibir el crecimiento de BFU-E y CFU-g M, y basándose en esta evidencia se sugirió usarlo para estudios ulteriores sobre hematopoyesis o crecimiento de célula de tumor (Broudy et a l, supra).
En estudios de cáncer ulteriores, los investigadores encontraron que el tratamiento con imatinib, un inhibidor de molécula pequeña de cKIT, reduciría significativamente la proliferación de líneas celulares de GIST. Sin embargo, las células tratadas con imatinib se hacen resistentes con el tiempo debido a mutaciones secundarias en cKIT (Edris et a l, Proc. Nat. Acad. Sci. USA, edición inicial en línea 2013). Sin embargo, si las células de GIST fueron tratadas con el anticuerpo SR-1 como terapia secundaria, hubo una disminución significativa de la proliferación celular, y una disminución en la expresión de cKIT sobre la superficie celular (Edris et a l, supra). De esta manera, un anticuerpo SR-1 desnudo fue eficaz para manejar el problema de la resistencia al imatinib en líneas GIST humanas, sugiriendo que puede ser de utilidad una combinación imatinib/anticuerpo anti-cKIT.
Conjugados anticuerpo-fármaco
Se han utilizado conjugados de anticuerpo-fármaco (“ADC”) para el suministro local de agentes citotóxicos en el tratamiento del cáncer (véase, por ejemplo, Lambert, Curr. Opinion In Pharmacology5:543-549, 2005). Los ADC permiten un suministro dirigido de la porción de fármaco en donde se puede lograr una eficacia máxima con mínima toxicidad. Como más ADC muestran resultados clínicos prometedores, existe una mayor necesidad de desarrollar agentes terapéuticos novedosos para la terapia del cáncer.
BREVE DESCRIPCIÓN DE LA INVENCIÓN
La presente divulgación está dirigida a un conjugado anticuerpo-fármaco de la fórmula Ab-(L-(D)m)n o una sal farmacéuticamente aceptable del mismo, en donde Ab es un anticuerpo o fragmento de unión a antígeno del mismo que se une específicamente a un epítopo de cKIT humano; L es un enlazador; D es una porción de fármaco; m es un entero de 1 a 8; y n es un entero de 1 a 10.
El conjugado anticuerpo-fármaco, en donde dicho n es 3 ó 4.
El conjugado anticuerpo-fármaco, en donde dicho anticuerpo o fragmento de unión a antígeno del mismo se une específicamente al dominio extracelular de cKIT (SEQ ID NO: 160).
El conjugado anticuerpo-fármaco, en donde dicho anticuerpo o fragmento de unión a antígeno se une específicamente a un epítopo de cKIT de humano en los dominios 1-3 (SEQ ID NO: 155).
El conjugado anticuerpo-fármaco en donde dicho anticuerpo o fragmento de unión a antígeno del mismo se une específicamente a cKlT de humano en SEQ ID NO: 161 y SEQ ID NO: 162.
El conjugado anticuerpo-fármaco en donde dicho anticuerpo o fragmento de unión a antígeno del mismo se une específicamente a cKIT de humano en SEQ ID NO: 163 y SEQ ID NO: 164.
El conjugado anticuerpo-fármaco, en donde dicho anticuerpo o fragmento de unión a antígeno del mismo comprende: (i) una región variable de cadena pesada que comprende (a) una HCDR1 (CDR-Región Determinante de Complementariedad) de SEQ ID NO: 76, (b) una HCDR2 de SEQ iD NO: 77, (c) una HCDR3 de SEQ ID NO: 78; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 85, (e) una LCDR2 de SEQ ID n O: 86, y (f) una LCDR3 de SEQ ID NO: 87;
(ii) una región variable de cadena pesada que comprende (a) una HCDR1 de SEQ ID NO: 22, (b) una HCDR2 de SEQ ID NO: 23, (c) una HCDR3 de SEQ ID NO: 24; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 31, (e) una LCDR2 de SEQ ID NO: 32, y (f) una LCDR3 de SEQ ID NO: 33;
(iii) una región variable de cadena pesada que comprende (a) una HCDR1 de SEQ ID NO: 130, (b) una HCDR2 de SEQ ID NO: 131, (c) una HCDR3 de SEQ ID NO: 132; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 139, (e) una LCDR2 de SEQ ID NO: 140, y (f) una LCDR3 de SEQ ID NO: 141;
(iv) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 58, (b) una HCDR2 de SEQ ID NO: 59, (c) una HCDR3 de SEQ ID NO: 60; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 67, (e) una LCDR2 de SEQ ID NO: 68, y (f) una LCDR3 de SEQ ID NO: 69;
(v) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 40, (b) una HCDR2 de SEQ ID NO: 41, (c) una HCDR3 de SEQ ID NO: 42; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 49, (e) una LCDR2 de SEQ ID NO: 50, y (f) una LCDR3 de SEQ ID NO: 51;
(vi) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 94, (b) una HCDR2 de SEQ ID NO: 95, (c) una HCDR3 de SEQ ID NO: 96; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 103, (d) una LCDR2 de SEQ ID NO: 104, y (f) una LCDR3 de SEQ ID NO: 105;
(vii) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 112, (b) una HCDR2 de SEQ ID NO: 113, (c) una HCDR3 de SEQ ID NO: 114; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 121, (e) una LCDR2 de SEQ ID NO: 122, y (f) una LCDR3 de SEQ ID NO: 123; o
(viii) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 3, (b) una HCDR2 de SEQ ID NO: 4, (c) una HCDR3 de SEQ ID NO: 5; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 12, (e) una LCDR2 de SEQ ID NO: 13, y (f) una LCDR3 de SEQ ID NO: 14.
El conjugado anticuerpo-fármaco en el que por lo menos un aminoácido dentro de una CDR es sustituido con un residuo correspondiente de una CDR correspondiente de otro anticuerpo anti-cKIT de la tabla 1.
El conjugado anticuerpo-fármaco en el que uno o dos aminoácidos dentro de una CDR han sido modificados, suprimidos o sustituidos.
El conjugado anticuerpo-fármaco que retiene por lo menos 90, 91, 92, 93, 94, 95, 96, 97, 98 o 99 % de identidad sobre la región variable ligera o la variable pesada.
El conjugado anticuerpo-fármaco en donde el anticuerpo es un anticuerpo monoclonal, un anticuerpo quimérico, un anticuerpo humanizado, un anticuerpo humano modificado por ingeniería, un anticuerpo humano, un anticuerpo de cadena simple (scFv) o un fragmento de anticuerpo.
El conjugado anticuerpo-fármaco, en donde dicho enlazador (L) se selecciona del grupo que consiste en un enlazador
escindible, un enlazador no escindible, un enlazador hidrófilo, un enlazador procargado y un enlazador a base de ácido dicarboxílico.
El conjugado anticuerpo-fármaco, en donde el enlazador se deriva de un reactivo de entrelazamiento seleccionado del grupo que consiste en N-succinimidil-3-(2-piridilditio)propionato (SPDP), N-succinimidil-4-(2-piridilditio)pentanoato (SPP), N-succinimidil-4-(2-piridilditio)butanoato (SPDb), N-succinimidil-4-(2-piridilditio)2-sulfo-butanoato (sulfo-SPDB), N-succinimidil-yodoacetato (SIA), N-succinimidil(4-yodoacetil)aminobenzoato (SIAB), maleimida-PEG-NHS, N-succinimidil-4-(maleimidometilo)ciclohexancarboxilato (SMCC), N-sulfosuccinimidil-4-(maleimidometil)ciclohexancarboxilato (sulfo-SMCC), o 2,5-dioxopirrolidin-1-il-17-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-5,8,11,14-tetraoxo-4,7,10,13-tetraazaheptadecan-1-oato (CX1-1).
El conjugado anticuerpo-fármaco, en donde dicho enlazador se deriva del reactivo de entrelazamiento N-succinimidil-4-(maleimidometil)ciclohexancarboxilato (SMCC).
El conjugado anticuerpo-fármaco, en donde dicha porción de fármaco (D) se selecciona del grupo que consiste en un inhibidor de V-ATPasa, un agente pro-apoptótico, un inhibidor de Bcl2, un inhibidor de MCL1, un inhibidor de HSP90, un inhibidor de IAP, un inhibidor de mTor, un estabilizador de microtúbulos, un desestabilizador de microtúbulos, una auristatina, una dolastatina, un maitansinoide, una MetAP (metionina aminopeptidasa), un inhibidor de la exportación nuclear de proteínas CRM1, un inhibidor de DPPIV, inhibidores de proteasoma, inhibidores de reacciones de transferencia de fosforilo en la mitocondria, un inhibidor de la síntesis de proteína, un inhibidor de quinasa, un inhibidor de CDK2, un inhibidor de CDK9, un inhibidor de kinesina, un inhibidor de HDAC, un agente perjudicial para el ADN, un agente alquilante de ADN, un intercalador de ADN, un enlazador de la ranura menor de ADN y un inhibidor de DHFR.
El conjugado anticuerpo-fármaco, en donde la porción de fármaco es un maitansinoide.
El conjugado anticuerpo-fármaco, en donde el maitansinoide es N(2')-desacetil-N(2')-(3-mercapto-1-oxopropil)-maitansina (DM1) o N(2')-desacetil-N2-(4-mercapto-4-metil-1-oxopentil)-maitansina (DM4).
El conjugado anticuerpo-fármaco en combinación con otro agente terapéutico.
El conjugado anticuerpo-fármaco en combinación con un agente terapéutico indicado en la tabla 16.
Un conjugado anticuerpo-fármaco de la fórmula:

o una sal farmacéuticamente aceptable del mismo; en donde; Ab es un anticuerpo o fragmento de unión a antígeno del mismo que se une específicamente a cKIT de humano, y por lo menos n número de aminas primarias; y n es un entero de 1 a 10.
El conjugado anticuerpo-fármaco, en donde dicho anticuerpo o fragmento de unión a antígeno se une específicamente a
un epítopo de cKIT de humano en los dominios 1-3 (SEQ ID NO: 155).
El conjugado anticuerpo-fármaco, en donde dicho anticuerpo o fragmento de unión a antígeno del mismo se une específicamente a cKIT de humano en SEQ ID NO: 161 y Se Q ID NO: 162.
El conjugado anticuerpo-fármaco en donde dicho anticuerpo o fragmento de unión a antígeno del mismo se une específicamente a cKIT de humano en SEQ ID NO: 163 y SEQ ID NO: 164.
El conjugado anticuerpo-fármaco, en donde dicho Ab es un anticuerpo o fragmento de unión a antígeno del mismo, comprende: (i) una región variable de cadena pesada que comprende (a) una HCDR1 (CDR-Región Determinante de Complementariedad) de SEQ ID NO: 76, (b) una HCDR2 de SEQ ID n O: 77, (c) una HCDR3 de SEQ ID NO: 78; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 85, (e) una LCDR2 de SEQ ID n O: 86, y (f) una LCDR3 de SEQ ID NO: 87;
(ii) una región variable de cadena pesada que comprende (a) una HCDR1 de SEQ ID NO: 22, (b) una HCDR2 de SEQ ID NO: 23, (c) una HCDR3 de SEQ ID NO: 24; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 31, (e) una LCDR2 de SEQ ID NO: 32, y (f) una LCDR3 de SEQ ID NO: 33;
(iii) una región variable de cadena pesada que comprende (a) una HCDR1 de SEQ ID NO: 130, (b) una HCDR2 de SEQ ID NO: 131, (c) una HCDR3 de SEQ ID NO: 132; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 139, (e) una LCDR2 de SEQ ID NO: 140, y (f) una LCDR3 de SEQ ID NO: 141;
(iv) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 58, (b) una HCDR2 de SEQ ID NO: 59, (c) una HCDR3 de SEQ ID NO: 60; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 67, (e) una LCDR2 de SEQ ID NO: 68, y (f) una LCDR3 de SEQ ID NO: 69;
(v) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 40, (b) una HCDR2 de SEQ ID NO: 41, (c) una HCDR3 de SEQ ID NO: 42; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 49, (e) una LCDR2 de SEQ ID NO: 50, y (f) una LCDR3 de SEQ ID NO: 51;
(vi) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 94, (b) una HCDR2 de SEQ ID NO: 95, (c) una HCDR3 de SEQ ID NO: 96; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 103, (d) una LCDR2 de SEQ ID NO: 104, y (f) una LCDR3 de SEQ ID NO: 105;
(vii) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 112, (b) una HCDR2 de SEQ ID NO: 113, (c) una HCDR3 de SEQ ID NO: 114; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 121, (e) una LCDR2 de SEQ ID NO: 122, y (f) una LCDR3 de SEQ ID NO: 123; o
(viii) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 3, (b) una HCDR2 de SEQ ID NO: 4, (c) una HCDR3 de SEQ ID NO: 5; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 12, (e) una LCDR2 de SEQ ID NO: 13, y (f) una LCDR3 de SEQ ID NO: 14.
El conjugado anticuerpo-fármaco en el que por lo menos un aminoácido dentro de una CDR es sustituido con un residuo correspondiente de una CDR correspondiente de otro anticuerpo anti-cKIT de la tabla 1.
El conjugado anticuerpo-fármaco en el que uno o dos aminoácidos dentro de una CDR han sido modificados, suprimidos o sustituidos.
El conjugado anticuerpo-fármaco que retiene por lo menos 90, 91, 92, 93, 94, 95, 96, 97, 98 o 99 % de identidad sobre la región variable ligera o variable pesada.
El conjugado anticuerpo-fármaco en donde el anticuerpo es un anticuerpo monoclonal, un anticuerpo quimérico, un anticuerpo humanizado, un anticuerpo humano modificado por ingeniería, un anticuerpo humano, un anticuerpo de cadena simple (scFv) o un fragmento de anticuerpo.
El conjugado anticuerpo-fármaco, en donde dicho n es un entero de 2 a 8.
El conjugado anticuerpo-fármaco, en donde dicho n es un entero de 3 a 4.
El conjugado anticuerpo-fármaco en combinación con otro agente terapéutico.
El conjugado anticuerpo-fármaco en combinación con un agente terapéutico indicado en la tabla 16.
Una composición farmacéutica que comprende el conjugado anticuerpo-fármaco y un vehículo farmacéuticamente aceptable.
La composición farmacéutica, en donde dicha composición se prepara como un liofilizado.
La composición farmacéutica, en donde dicho liofilizado comprende el conjugado anticuerpo-fármaco, succinato de sodio y polisorbato 20.
Un método de tratamiento de un cáncer cKIT positivo en un paciente en necesidad del mismo, que comprende administrarle a dicho paciente el conjugado anticuerpo-fármaco o la composición farmacéutica.
El método de tratamiento en donde dicho cáncer se selecciona del grupo que consiste en tumores estromales gastrointestinales (GIST), cáncer de pulmón de célula pequeña (SCLC), leucemia mieloide aguda (AML), melanoma, leucemia de mastocitos (MCL), mastocitosis, neurofibromatosis, cáncer de mama, cáncer de pulmón de célula no pequeña (NSCLC) y cáncer pancreático.
El método en donde el conjugado anticuerpo-fármaco o la composición farmacéutica es administrada en combinación con otro agente terapéutico.
El método en donde el conjugado anticuerpo-fármaco o la composición farmacéutica es administrada en combinación con un agente terapéutico indicado en la tabla 16.
El conjugado anticuerpo-fármaco para usarse como un medicamento.
El conjugado anticuerpo-fármaco, o la composición farmacéutica para usarse en el tratamiento de un cáncer cKIT positivo. El conjugado anticuerpo-fármaco administrado en combinación con otro agente terapéutico.
El conjugado anticuerpo-fármaco administrado en combinación con un agente terapéutico indicado en la tabla 16. Un ácido nucleico que codifica el anticuerpo o fragmento de unión a antígeno.
Un vector que comprende el ácido nucleico.
Una célula hospedera que comprende el vector.
Un proceso para producir un anticuerpo o fragmento de unión a antígeno que comprende cultivar la célula hospedera y recuperar el anticuerpo del cultivo.
Un proceso para producir un conjugado de anticuerpo anti-cKIT-fármaco, el proceso comprendiendo: (a) enlazar químicamente SMCC a una porción de fármaco d M-1; (b) conjugar dicho enlazador-fármaco con el anticuerpo recuperado del cultivo celular; y (c) purificar el conjugado anticuerpo-fármaco.
El conjugado anticuerpo-fármaco que tiene una proporción de maitansinoide a anticuerpo (MAR) promedio, medida con un espectrofotómetro UV, de aproximadamente3,5.
Un anticuerpo o fragmento de unión a antígeno del mismo que comprende:
(i) una región variable de cadena pesada que comprende (a) una HCDR1 (CDR-Región Determinante de Complementariedad) de SEQ ID NO: 76, (b) una HCDR2 de SEQ ID NO: 77, (c) una HCDR3 de SEQ ID NO: 78; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 85, (e) una LCDR2 de SEQ ID n O: 86, y (f) una LCDR3 de SEQ ID NO: 87;
(ii) una región variable de cadena pesada que comprende (a) una HCDR1 de SEQ ID NO: 22, (b) una HCDR2 de SEQ ID NO: 23, (c) una HCDR3 de SEQ ID NO: 24; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 31, (e) una LCDR2 de SEQ ID NO: 32, y (f) una LCDR3 de SEQ ID NO: 33;
(iii) una región variable de cadena pesada que comprende (a) una HCDR1 de SEQ ID NO: 130, (b) una HCDR2 de SEQ ID NO: 131, (c) una HCDR3 de SEQ ID NO: 132; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 139, (e) una LCDR2 de SEQ ID NO: 140, y (f) una LCDR3 de SEQ ID NO: 141;
(iv) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 58, (b) una HCDR2 de SEQ ID NO: 59, (c) una HCDR3 de SEQ ID NO: 60; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 67, (e) una LCDR2 de SEQ ID NO: 68, y (f) una LCDR3 de SEQ ID NO: 69;
(v) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 40, (b) una HCDR2 de SEQ ID NO: 41, (c) una HCDR3 de SEQ ID NO: 42; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 49, (e) una LCDR2 de SEQ ID NO: 50, y (f) una LCDR3 de SEQ ID NO: 51;
(vi) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 94, (b) una HCDR2 de SEQ ID NO: 95, (c) una HCDR3 de SEQ ID NO: 96; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 103, (d) una LCDR2 de SEQ ID NO: 104, y (f) una LCDR3 de SEQ ID NO: 105;
(vii) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 112, (b) una HCDR2 de SEQ ID NO: 113, (c) una HCDR3 de SEQ ID NO: 114; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 121, (e) una LCDR2 de SEQ ID NO: 122, y (f) una LCDR3 de SEQ ID NO: 123; o
(viii) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 3, (b) una HCDR2 de SEQ ID NO: 4, (c) una HCDR3 de SEQ ID NO: 5; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 12, (e) una LCDR2 de SEQ ID NO: 13, y (f) una LCDR3 de SEQ ID NO: 14.
Un reactivo de diagnóstico que comprende el anticuerpo o fragmento de unión a antígeno del mismo que está marcado.
El reactivo de diagnóstico, en donde la marca se selecciona del grupo que consiste en una radiomarca, un fluoróforo, un cromóforo, un agente de imagenología, y un ion de metal.
Definiciones
A menos que se indique de otra manera, se entiende que los siguientes términos y frases usadas en la presente tienen los siguientes significados:
El término “alquilo” se refiere a una cadena de hidrocarburo saturado monovalente que tiene el número especificado de átomos de carbono. Por ejemplo, alquilo de C1 -6 se refiere a un grupo alquilo que tiene de 1 a 6 átomos de carbono. Los grupos alquilo pueden ser rectos o ramificados. Los grupos alquilo ramificados representativos tienen una, dos o tres ramificaciones. Los ejemplos de grupos alquilo incluyen, sin limitación, metilo, etilo, propilo (n-propilo e isopropilo), butilo (n-butilo, isobutilo, sec-butilo y t-butilo), pentilo (n-pentilo, isopentilo y neopentilo) y hexilo.
El término “anticuerpo” usado en la presente se refiere a un polipéptido de la familia de inmunoglobulinas que es capaz de unirse no covalentemente a un antígeno correspondiente, reversiblemente y de manera específica. Por ejemplo, un anticuerpo IgG que ocurre naturalmente es un tetrámero que comprende por lo menos dos cadenas pesadas (H) y dos cadenas ligeras (L) interconectadas por enlaces disulfuro. Cada cadena pesada está comprendida de una región variable de cadena pesada (abreviada aquí como VH), y una región constante de cadena pesada. La región constante de cadena pesada está comprendida de tres dominios, CH1, CH2 y CH3. Cada cadena ligera está comprendida de una región variable de cadena ligera (abreviada aquí como VL), y una región constante de cadena ligera. La región constante de cadena ligera está comprendida de un dominio, CL. Las regiones VH y VL pueden subdividirse además en regiones de hipervariabilidad, llamadas regiones determinantes de complementariedad (CDR), intercaladas con regiones que están más conservadas, llamadas regiones de armazón (FR). Cada VH y VL está compuesta de tres CDR y cuatro FR, dispuestas del extremo amino al extremo carboxi en el siguiente orden: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. Las regiones variables de las cadenas pesadas y ligeras contienen un dominio de unión que interacciona con un antígeno. Las regiones constantes de los anticuerpos pueden mediar la unión de la inmunoglobulina a tejidos o factores del hospedero, que incluyen varias células del sistema inmune (por ejemplo, células efectoras) y el primer componente (C1q) del sistema de complemento clásico.
El término “anticuerpo” incluye, sin limitación, anticuerpos monoclonales, anticuerpos humanos, anticuerpos humanizados, anticuerpos camélidos, anticuerpos quiméricos y anticuerpos anti-idiotípicos (anti-Id) (que incluyen por ejemplo anticuerpos anti-Id para los anticuerpos de la presente divulgación). Los anticuerpos pueden ser de cualquier isotipo/clase (por ejemplo, IgG, IgE, IgM, IgD, IgA e IgY), o subclase (por ejemplo, IgG 1, IgG2, IgG3, IgG4, IgA1 e IgA2).
Los “dominios determinantes de complementariedad” o “regiones determinantes de complementariedad” (“CDR”) se refieren intercambiablemente a las regiones hipervariables de VL y VH. Las CDR son el sitio de unión de la proteína objetivo de las cadenas de anticuerpo que alojan especificidad para tal proteína objetivo. Existen tres CDR (CDR1-3, numeradas secuencialmente desde el extremo N) en cada VL o VH humana, constituyendo aproximadamente 15-20% de los dominios variables. Las CDR pueden ser referidas por su región y orden. Por ejemplo, “VHCDR1” o “HCDR1” se refieren ambas a la primera CDR de la región variable de cadena pesada. Las CDR son estructuralmente complementarias al epítopo de la proteína objetivo y por lo tanto son directamente responsables de la especificidad de unión. Los tramos restantes de VL o VH, las denominadas regiones de armazón, exhiben menos variación en la secuencia de aminoácidos (Kuby, “Immunology”, 4a. ed., capítulo 4. W.H. Freeman & Co., Nueva York, 2000).
Las posiciones de las CDR y las regiones de armazón pueden ser determinadas usando varias definiciones muy conocidas en la técnica, por ejemplo Kabat, Chothia, y AbM (véase, por ejemplo, Johnson et al., Nucleic Acids Res., 29:205-206 (2001); Chothia y Lesk, J. Mol. Biol., 196:901-917 (1987); Chothia et al., Nature, 342:877-883 (1989); Chothia et al., J. Mol. Biol., 227:799-817 (1992); Al-Lazikani et al., J.Mol.Biol., 273:927-748 (1997)). Las definiciones de sitios de combinación de antígeno también son descritas en las siguientes referencias: Ruiz et al., Nucleic Acids Res., 28:219-221 (2000); y Lefranc, M.P., Nucleic Acids Res., 29:207-209 (2001); MacCallum et al., J. Mol. Biol., 262:732-745 (1996); y Martin et al., Proc. Natl. Acad. Sci. USA, 86:9268-9272 (1989); Martin et al., Methods Enzymol., 203:121-153 (1991); y Rees et al., en Sternberg M.J.E. (ed.), “Protein Structure Prediction”, Oxford University Press, Oxford, 141-172 (1996).
Tanto las cadenas ligeras como pesadas se dividen en regiones de homología estructural y funcional. Los términos “constante” y “variable” se usan funcionalmente. A este respecto, se apreciará que los dominios variables tanto de las porciones de cadena ligera (VL) como pesada (VH) determinan el reconocimiento y especificidad del antígeno. Inversamente, los dominios constantes de la cadena ligera (CL) y la cadena pesada (CH1, CH2 o CH3) confieren importantes propiedades biológicas, tales como secreción, movilidad transplacentaria, unión al receptor de Fc, unión de complemento y similares. Por convención, la numeración de los dominios de la región constante aumenta conforme se hacen más distantes del sitio de unión de antígeno o el extremo amino del anticuerpo. El extremo N es una región variable y en el extremo C está una región constante; los dominios CH3 y CL realmente comprenden los dominios carboxiterminales de la cadena pesada y ligera, respectivamente.
El término “fragmento de unión a antígeno”, como se usa en la presente, se refiere a una o más porciones de un anticuerpo que retienen la capacidad para interaccionar específicamente (por ejemplo, por unión, impedimento estérico, estabilización/desestabilización, distribución espacial) con un epítopo de un antígeno. Los ejemplos de fragmentos de unión incluyen, sin limitación, Fvs de cadena simple (scFv), Fvs enlazados por disulfuro (sdFv), fragmentos Fab, fragmentos F(ab'), un fragmento monovalente que consiste en los dominios VL, Vh , CL y CH1; un fragmento F(ab)2, un fragmento bivalente que comprende 2 fragmentos Fab enlazados por un puente disulfuro en la región bisagra; un fragmento Fd que consiste en los dominios VH y CH1; un fragmento Fv que consiste en los dominios VL y VH de un solo brazo de un anticuerpo; un fragmento dAb (Ward et al., Nature 341:544-546, 1989), que consiste en un dominio VH; y una región determinante de complementariedad (CDR) aislada, u otros fragmentos de unión de epítopo de un anticuerpo.
Además, aunque los dos dominios, VL y VH, del fragmento Fv son codificados por genes separados, se pueden unir usando métodos recombinantes, por medio de un enlazador sintético que permite hacerlos como una sola cadena de proteína en la que las regiones VL y VH se aparean para formar moléculas monovalentes (conocidas como Fv de cadena simple (“scFv”); véase por ejemplo Bird et al., Science 242:423-426, 1988; y Huston et al., Proc. Natl. Acad. Sci.85:5879-5883, 1988). Estos anticuerpos de cadena simple también se consideran abarcados dentro del término “fragmento de unión a antígeno”. Estos fragmentos de unión de antígeno se obtienen usando las técnicas convencionales conocidas para los expertos en la materia, y los fragmentos se tamizan por su utilidad de la misma manera que los anticuerpos intactos.
Los fragmentos de unión de antígeno también se pueden incorporar en anticuerpos de un solo dominio, maxianticuerpos, minianticuerpos, nanoanticuerpos, intraanticuerpos, dianticuerpos, trianticuerpos, tetraanticuerpos, v-NAR y bis-scFv (véase por ejemplo Hollinger y Hudson, Nature Biotechnology 23:1126-1136, 2005). Los fragmentos de unión de antígeno se pueden injertar en andamios basados en polipéptidos, tales como fibronectina de tipo III (Fn3) (véase la patente de EE. UU. No. 6.703.199, que describe monocuerpos de polipéptido de fibronectina).
Los fragmentos de unión de antígeno se pueden incorporar en moléculas de cadena simple que comprenden un par de segmentos Fv en tándem (VH-CH1-VH-CH1) que, junto con polipéptidos de cadena ligera complementarios, forman un par de regiones de unión de antígeno (Zapata et al., Protein Eng.8:1057-1062, 1995; y patente de EE. UU. No.5.641.870).
El término “anticuerpo monoclonal” o “composición de anticuerpo monoclonal”, como se usa en la presente, se refiere a
polipéptidos que incluyen anticuerpos y fragmentos de unión de antígeno que tienen una secuencia de aminoácidos sustancialmente idéntica, o se derivan de la misma fuente genética. Este término también incluye preparaciones de moléculas de anticuerpo de composición molecular única. Una composición de anticuerpo monoclonal exhibe una especificidad de unión y afinidad únicas para un epítopo particular.
El término “anticuerpo humano” usado en la presente incluye anticuerpos que tienen regiones variables en las que tanto las regiones de armazón como de CDR se derivan de secuencias de origen humano. Además, si el anticuerpo contiene una región constante, la región constante también se deriva de tales secuencias humanas, por ejemplo secuencias de línea germinal humanas, o versiones mutadas de secuencias de línea germinal humanas, o anticuerpo que contiene secuencias de armazón de consenso derivadas del análisis de secuencias de armazón humanas, por ejemplo como lo describen Knappik etal., J. Mol. Biol. 296:57-86, 2000).
Los anticuerpos humanos de la presente divulgación pueden incluir residuos de aminoácido no codificados por secuencias humanas (por ejemplo, mutaciones introducidas por mutagénesis aleatoria o específica de sitio in vitro, o por mutación somática in vivo, o una sustitución conservativa para promover estabilidad o facilitar la fabricación).
El término “reconocer” usado en la presente se refiere a un anticuerpo o fragmento de unión a antígeno del mismo que se une con su epítopo e interacciona (por ejemplo, se une) con el mismo, ya sea que el epítopo sea lineal o conformacional. El término “epítopo” se refiere a un sitio sobre un antígeno al que se une específicamente un anticuerpo o fragmento de unión a antígeno de la divulgación. Los epítopos se pueden formar de aminoácidos contiguos o aminoácidos no contiguos yuxtapuestos por pliegue terciario de una proteína. Los epítopos formados de aminoácidos contiguos típicamente son retenidos por exposición a disolventes desnaturalizantes, mientras que los epítopos formados por pliegue terciario típicamente se pierden por tratamiento con disolventes desnaturalizantes. Típicamente un epítopo incluye por lo menos 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 o 15 aminoácidos en una conformación espacial única. Los métodos para determinar la conformación espacial de los epítopos incluyen técnicas como por ejemplo cristalografía de rayos X y resonancia magnética nuclear bidimensional (véase, por ejemplo, “Epitope Mapping Protocols in Methods in Molecular Biology”, Vol.
66, G. E. Morris, Ed. (1996)). Un “parátopo” es la parte del anticuerpo que reconoce el epítopo del antígeno.
La frase “se une específicamente” o “se une selectivamente”, usada en el contexto de descripción de la interacción entre un antígeno (por ejemplo, una proteína) y un anticuerpo, fragmento de anticuerpo, o agente de unión derivado de anticuerpo, se refiere a una reacción de unión que es determinante de la presencia del antígeno en una población heterogénea de proteínas y otros agentes biológicos, por ejemplo en una muestra biológica, por ejemplo una muestra de sangre, suero, plasma o tejido. De esta manera, bajo ciertas condiciones de inmunoensayo designadas, los anticuerpos o agentes de unión con una especificidad de unión particular se unen a un antígeno particular por lo menos dos veces sobre el fondo, y no se unen sustancialmente en una cantidad significativa a otros antígenos presentes en la muestra. En un aspecto, bajo las condiciones de inmunoensayo designadas, el anticuerpo o agente de unión con una especificidad de unión particular se une a un antígeno particular por lo menos diez (10) veces sobre el fondo, y no se une sustancialmente en una cantidad significativa a otros antígenos presentes en la muestra. La unión específica a un anticuerpo o agente de unión bajo tales condiciones puede requerir que el anticuerpo o agente haya sido seleccionado por su especificidad para una proteína particular. Si se desea o es apropiado, esta selección se puede lograr sustrayendo anticuerpos que reaccionan cruzadamente con moléculas de otras especies (por ejemplo, ratón o rata) u otros subtipos. Alternativamente, en algunos aspectos se seleccionan anticuerpos o fragmentos de anticuerpo que reaccionan cruzadamente con ciertas moléculas deseadas.
El término “afinidad” usado en la presente se refiere a la fuerza de interacción entre anticuerpo y antígeno en los sitios antigénicos individuales. Dentro de cada sitio antigénico, la región variable del “brazo” de anticuerpo interacciona por medio de fuerzas débiles no covalentes con el antígeno en muchos sitios; a más interacciones la afinidad será más fuerte.
El término “anticuerpo aislado” se refiere a un anticuerpo que está sustancialmente libre de otros anticuerpos que tienen diferentes especificidades antigénicas. Sin embargo, un anticuerpo aislado que se une específicamente a un antígeno puede tener reactividad cruzada con otros antígenos. Además, un anticuerpo aislado puede estar sustancialmente libre de otro material celular o de agentes químicos.
El término “secuencia de línea germinal humana correspondiente” se refiere a la secuencia de ácido nucleico que codifica una secuencia o subsecuencia de aminoácidos de la región variable humana que comparte la más alta identidad de secuencia de aminoácidos, determinada con una secuencia o subsecuencia de aminoácidos de la región variable de referencia, en comparación con todas las otras secuencias de aminoácidos conocidas de la región variable codificadas por secuencias de la región variable de inmunoglobulina de línea germinal humana. La secuencia de línea germinal humana correspondiente también se puede referir a la secuencia o subsecuencia de aminoácidos de la región variable de humano con la más alta identidad de secuencia de aminoácidos con una secuencia o subsecuencia de aminoácidos
de la región variable de referencia, en comparación con todas las otras secuencias de aminoácidos de la región variable evaluadas. La secuencia de línea germinal humana correspondiente puede ser solo regiones de armazón, solo regiones determinantes de complementariedad, regiones de armazón y determinantes de complementariedad, un segmento variable (como se define arriba), u otras combinaciones de secuencias o subsecuencias que comprenden una región variable. La identidad de secuencias se puede determinar usando los métodos descritos en la presente, por ejemplo alineando dos secuencias usando BLAST, ALIGN, u otro algoritmo de alineación conocido en la técnica. La secuencia de ácido nucleico o aminoácido de línea germinal humana correspondiente puede tener por lo menos aproximadamente 90%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, o 100% de identidad de secuencia con la secuencia de ácido nucleico o aminoácido de la región variable de referencia.
Se pueden usar una variedad de formatos de inmunoensayo para seleccionar anticuerpos específicamente inmunorreactivos con una proteína particular. Por ejemplo, se usan rutinariamente inmunoensayos de ELISA de fase sólida para seleccionar anticuerpos específicamente inmunorreactivos con una proteína (véase por ejemplo Harlow y Lane, “Using Antibodies, A Laboratory Manual” (1998), para una descripción de formatos y condiciones de inmunoensayo que se pueden usar para determinar la inmunorreactividad específica). Típicamente una reacción de unión específica o selectiva producirá una señal de por lo menos dos veces la señal de fondo, y más típicamente por lo menos 10 a 100 veces sobre el fondo.
El término “constante de disociación en el equilibrio (KD, M)” se refiere a la constante de la velocidad de disociación (kd, tiempo-1) dividida entre la constante de la velocidad de asociación (ka, tiempo-1, M-1). Las constantes de disociación en el equilibrio se pueden medir usando cualquier método de la técnica. Los anticuerpos de la presente divulgación por lo general tendrán una constante de disociación en equilibrio menor de aproximadamente 10-7 o 10-8 M, por ejemplo menor de aproximadamente 10-9 M o 10-10 M, en algunos aspectos menor de aproximadamente 10-11 M, 10-12 M o 10-13 M.
El término “biodisponibilidad” se refiere a la disponibilidad sistémica (esto es, los niveles en sangre/plasma) de una cantidad dada de fármaco administrado a un paciente. La biodisponibilidad es un término absoluto que indica la medición tanto del tiempo (velocidad) como de la cantidad total (extensión) de fármaco que alcanza la circulación general a partir de una forma farmacéutica administrada.
Como se usa en la presente, la frase “que consiste esencialmente en” se refiere a los géneros o especies de cualquier agente farmacéutico activo incluido en un método o composición, así como también cualquier excipiente inactivo para el fin deseado de los métodos o composiciones. En algunos aspectos, la frase “que consiste esencialmente en” excluye expresamente la inclusión de uno o más agentes activos adicionales aparte de un conjugado anticuerpo-fármaco de la presente divulgación. En algunos aspectos, la frase “que consiste esencialmente en” excluye expresamente la inclusión de uno más agentes activos adicionales aparte de un conjugado anticuerpo-fármaco de la presente divulgación y un segundo agente coadministrado.
El término “aminoácido” se refiere a aminoácidos naturales, sintéticos y no naturales, y también análogos de aminoácido y miméticos de aminoácido que funcionan de manera similar a los aminoácidos naturales. Los aminoácidos naturales son aquellos codificados por el código genético, y también los aminoácidos que son modificados después, por ejemplo, hidroxiprolina, g-carboxiglutamato y O-fosfoserina. Los análogos de aminoácido se refieren a los compuestos que tienen la misma estructura química básica que un aminoácido natural, esto es, un carbono alfa que está unido a un hidrógeno, un grupo carboxilo, un grupo amino y un grupo R, por ejemplo, homoserina, norleucina, sulfóxido de metionina, metilsulfonio de metionina. Tales análogos tienen grupos R modificados (por ejemplo, norleucina) o esqueletos de péptido modificados, pero retienen la misma estructura química básica de un aminoácido natural. Los miméticos de aminoácido se refieren a compuestos químicos que tienen una estructura que es diferente de la estructura química general de un aminoácido, pero que funcionan de una manera similar a un aminoácido natural.
El término “variante modificada conservativamente” se aplica a secuencias tanto de aminoácidos como de ácidos nucleicos. Con respecto a las secuencias de ácido nucleico particulares, las variantes modificadas conservativamente se refieren a los ácidos nucleicos que codifican secuencias de aminoácidos idénticas o esencialmente idénticas o, cuando el ácido nucleico no codifica una secuencia de aminoácidos, a secuencias esencialmente idénticas. Debido a la degeneración del código genético, una gran cantidad de ácidos nucleicos funcionalmente idénticos codifican cualquier proteína dada. Por ejemplo, los codones GCA, GCC, GCG y GCU codifican todos el aminoácido alanina. De esta manera, en cada posición en donde una alanina es especificada por un codón, el codón puede ser alterado a cualquiera de los codones correspondientes descritos sin alterar el polipéptido codificado. Tales variaciones de ácido nucleico son “variaciones silenciosas”, que son una especie de variaciones modificadas conservativamente. Cada secuencia de ácido nucleico de la presente que codifica un polipéptido también describe toda variación silenciosa posible del ácido nucleico. El experto en la materia reconocerá que cada codón de un ácido nucleico (excepto AUG, que comúnmente es el único codón para metionina, y TGG, que comúnmente es el único codón para triptófano) puede ser modificado para producir
una molécula funcionalmente idéntica. Por consiguiente, cada variación silenciosa de un ácido nucleico que codifica un polipéptido está implícita en cada secuencia descrita.
Para las secuencias de polipéptidos, las “variantes modificadas conservativamente” incluyen sustituciones, supresiones o adiciones individuales a una secuencia de polipéptido que resultan en la sustitución de un aminoácido con un aminoácido químicamente similar. Las tablas de sustitución conservativa que proveen aminoácidos funcionalmente similares son muy conocidas. Tales variantes modificadas conservativamente son adicionales a las variantes polimórficas, homólogos entre especies y alelos, y no los excluyen. Los siguientes ocho grupos contienen aminoácidos que son sustituciones conservativas entre sí: 1) Alanina (A), Glicina (G); 2) Acido aspártico (D), Acido glutámico (E); 3) Asparagina (N), Glutamina (Q); 4) Arginina (R), Lisina (K); 5) Isoleucina (I), Leucina (L), Metionina (M), Valina (V); 6) Fenilalanina (F), Tirosina (Y), Triptófano (W); 7) Serina (S), Treonina (T); y 8) Cisteína (C), Metionina (M) (véase, por ejemplo, Creighton, “Proteins” (1984)). En algunos aspectos, el término “modificaciones de secuencia conservativas” se usa para referirse a modificaciones de aminoácido que no afectan o alteran significativamente las características de unión del anticuerpo que contiene la secuencia de aminoácidos.
Como se usa en la presente, el término “optimizado” se refiere a una secuencia de nucleótidos que ha sido alterada para codificar una secuencia de aminoácidos usando los codones que son preferidos en la célula u organismo de producción, generalmente una célula eucariota, por ejemplo, una célula de levadura, una célula de Pichia, una célula de hongo, una célula de Trichoderma, una célula de ovario de hámster chino (CHO) o una célula de humano. La secuencia de nucleótidos optimizada se modifica por ingeniería para retener completamente, o tanto como sea posible, la secuencia de aminoácidos codificada originalmente por la secuencia de nucleótidos inicial, que también es conocida como la secuencia “progenitora”.
Los términos “por ciento idéntico” o “porcentaje de identidad”, en el contexto de dos o más ácidos nucleicos o secuencias de polipéptido, se refieren a la extensión en la que dos o más secuencias o subsecuencias son iguales. Dos secuencias son “idénticas” si tienen la misma secuencia de aminoácidos o nucleótidos sobre la región comparada. Dos secuencias son “sustancialmente idénticas” si las dos secuencias tienen un porcentaje especificado de residuos de aminoácido o nucleótidos que son iguales (esto es, 60% de identidad, opcionalmente 65%, 70%, 75%, 80%, 85%, 90%, 95% o 99% de identidad sobre una región especificada o, cuando no se especifica, sobre toda la secuencia), cuando se comparan y alinean para correspondencia máxima sobre una ventana de comparación o región designada, medida usando uno de los siguientes algoritmos de comparación de secuencias, o por medio de alineación manual e inspección visual. Opcionalmente, la identidad existe sobre una región que es de por lo menos aproximadamente 30 nucleótidos (o 10 aminoácidos) de longitud, o muy preferiblemente sobre una región que es de 100 a 500 o 1000 o más nucleótidos (o 20, 50, 200 o más aminoácidos) de longitud.
Para comparación de secuencias, típicamente una secuencia actúa como secuencia de referencia con la que se comparan las secuencias de prueba. Cuando se usa un algoritmo de comparación de secuencias, se introducen en una computadora las secuencias de prueba y de referencia, se designan coordenadas de subsecuencia, si es necesario, y se designan los parámetros del programa del algoritmo de secuencia. Se pueden usar los parámetros por omisión del programa, o se pueden diseñar parámetros alternativos. El algoritmo de comparación de secuencias calcula entonces el porcentaje de identidad de secuencia de las secuencias de prueba con respecto a la secuencia de referencia, a base de los parámetros del programa.
Una “ventana de comparación”, como se usa aquí, incluye la referencia a un segmento de cualquiera de varias posiciones contiguas seleccionadas del grupo que consiste en 20 a 600, usualmente de aproximadamente 50 a aproximadamente 200, más usualmente de aproximadamente 100 a aproximadamente 150, en el que una secuencia puede ser comparada con una secuencia de referencia del mismo número de posiciones contiguas después de alinear de manera óptima las dos secuencias. Los métodos de alineación de secuencias para comparación son muy conocidos. La alineación óptima de secuencias para comparación se puede realizar, por ejemplo, por medio del algoritmo de homología local de Smith y Waterman, Adv. Appl. Math. 2:482c (1970); por medio del algoritmo de alineación de homología de Needleman y Wunsch, J. Mol. Biol. 48:443 (1970); por medio del método de búsqueda de similitud de Pearson y Lipman, Proc. Natl. Acad. Sci. USA 85:2444 (1988); por medio de implementaciones computarizadas de estos algoritmos (GAP, BESTFIT, FASTA y TFASTA del paquete de software Wisconsin Genetics Software, Genetics Computer Group, 575 Science Dr., Madison, WI); o por medio de alineación manual e inspección visual (véase, por ejemplo, Brent etal., “Current Protocols in Molecular Biology”, 2003).
Dos ejemplos de algoritmos que son adecuados para determinar el porcentaje de identidad de secuencia y la similitud de secuencia son los algoritmos BLAST y BLAST 2.0, descritos por Altschul et al. Nuc. Acids Res. 25:3389-3402, 1977; y Altschul et al., J. Mol. Biol. 215:403-410, 1990, respectivamente. El software para realizar los análisis BLAST está disponible públicamente a través del National Center for Biotechnology Information. Este algoritmo incluye identificar primero pares de secuencia de alta puntuación (HSP), identificando palabras cortas de longitud W en la secuencia de
interrogación que igualan o satisfacen alguna puntuación T de umbral de valor positivo cuando se alinean con una palabra de la misma longitud en una secuencia de la base de datos. T es referida como el umbral de puntuación de palabra vecina (Altschul et al., supra). Estos aciertos iniciales de palabra vecina actúan como semillas para iniciar búsquedas para encontrar HSP más largos que los contengan. Los aciertos de palabra son extendidos en ambas direcciones a lo largo de cada secuencia, tanto como pueda ser incrementada la puntuación de alineación acumulativa. Las puntuaciones acumulativas se calculan usando, para secuencias de nucleótidos, los parámetros M (puntuación de recompensa para un par de residuos concordantes; siempre >0) y N (puntuación de penalización para residuos discordantes; siempre <0). Para secuencias de aminoácidos se usa una matriz de puntuación para calcular la puntuación acumulativa. La extensión de los aciertos de palabra en cada dirección se detiene cuando: la puntuación de alineación acumulativa desciende en la cantidad X de su valor máximo alcanzado; la puntuación acumulativa tiende a cero o más abajo, debido a la acumulación de una o más alineaciones de residuo de puntuación negativa; o se alcanza el extremo de cada secuencia. Los parámetros W, T, y X del algoritmo BLAST determinan la sensibilidad y velocidad de la alineación. El programa BLASTN (para secuencias de nucleótidos) usa como valores por omisión una longitud de palabra (W) de 11, una expectativa (E) de 10, M=5, N=4, y una comparación de ambas cadenas. Para secuencias de aminoácidos, el programa BLASTP usa como valores por omisión una longitud de palabra de 3 y una expectativa (E) de 10, y la matriz de puntuación BLOSUM62 (véase Henikoff y Henikoff (1989), Proc. Natl Acad. Sci. USA 89:10915) alineaciones (B) de 50, expectativa (E) de 10, M=5, N=4, y una comparación de ambas cadenas.
El algoritmo BLAST también realiza un análisis estadístico de la similitud entre dos secuencias (véase, por ejemplo, Karlin y Altschul, Proc. Natl Acad. Sci. USA 90:5873-5787, 1993). Una medida de similitud provista por el algoritmo BLAST es la probabilidad de la suma más pequeña (P(N)), que provee una indicación de la probabilidad por medio de la cual ocurriría por azar una concordancia entre dos secuencias de nucleótidos o aminoácidos. Por ejemplo, un ácido nucleico se considera similar a una secuencia de referencia si la probabilidad de la suma más pequeña en una comparación del ácido nucleico de prueba con el ácido nucleico de referencia es menor de aproximadamente 0,2, de preferencia menor de aproximadamente 0,01, y muy de preferencia menor de aproximadamente 0,001.
El porcentaje de identidad entre dos secuencias de aminoácidos también se puede determinar usando el algoritmo de E. Meyers y W. Miller (Comput. Appl. Biosci, 4: 11-17, 1988), que ha sido incorporado en el programa ALIGN (versión 2.0) usando una tabla de residuo de ponderación PAM120, una penalización de longitud de hueco de 12 y una penalización de hueco de 4. Además, el porcentaje de identidad entre dos secuencias de aminoácidos se puede determinar usando el algoritmo de Needleman y Wunsch (J. MoI. Biol. 48:444-453, 1970), que ha sido incorporado en el programa GAP en el paquete de software GCG (disponible en www.gcg.com), usando una matriz Blossum 62 o una matriz PAM250, y una ponderación de hueco de 16, 14, 12, 10, 8, 6, o 4, y una ponderación de longitud de 1, 2, 3, 4, 5, o 6.
Aparte del porcentaje de identidad de secuencia indicado arriba, otra indicación de que dos secuencias de ácido nucleico o polipéptidos son sustancialmente idénticas es que el polipéptido codificado por el primer ácido nucleico es inmunológicamente reactivo de manera cruzada con los anticuerpos desarrollados contra el polipéptido codificado por el segundo ácido nucleico, como se describe más abajo. De esta manera, típicamente un polipéptido es sustancialmente idéntico a un segundo polipéptido, por ejemplo, cuando los dos péptidos difieren únicamente por sustituciones conservativas. Otra indicación de que dos secuencias de ácido nucleico son sustancialmente idénticas es que las dos moléculas o sus complementos se hibridan uno con otro bajo condiciones severas como se describen más abajo. Una indicación más de que dos secuencias de ácido nucleico son sustancialmente idénticas, es que se pueden usar los mismos cebadores para amplificar las secuencias.
El término “ácido nucleico” se usa aquí intercambiablemente con el término “polinucleótido” y se refiere a desoxirribonucleótidos o ribonucleótidos y polímeros de los mismos, ya sea en forma de cadena simple o doble. El término abarca ácidos nucleicos que contienen análogos de nucleótido conocidos o residuos de esqueleto o enlaces modificados, que son sintéticos, naturales y no naturales, que tienen propiedades de unión similares al ácido nucleico de referencia, y que son metabolizados de una manera similar a los nucleótidos de referencia. Los ejemplos de estos análogos incluyen, sin limitación, fosforotioatos, fosforamidatos, fosfonatos de metilo, fosfonatos de metilo quiral, 2-O-metil-ribonucleótidos, ácidos péptido-nucleicos (PNA).
A menos que se indique de otra manera, una secuencia de ácido nucleico particular también abarca implícitamente las variantes modificadas conservativamente de la misma (por ejemplo, sustituciones de codón degenerado) y secuencias complementarias, así como también la secuencia indicada explícitamente. Específicamente, como se detalla más abajo, se pueden obtener sustituciones de codón degenerado generando secuencias en las que la tercera posición de uno o más codones seleccionados (o todos) es sustituida con residuos de base mixta y/o desoxiinosina (Batzer et al. (1991), Nucleic Acid Res. 19:5081; Ohtsuka et al. (1985), J. Biol. Chem. 260:2605-2608; y Rossolini et al. (1994), Mol. Cell. Probes 8:91-98).
El término “enlazado operativamente” en el contexto de ácidos nucleicos se refiere a una relación funcional entre dos o más segmentos de polinucleótido (por ejemplo, ADN). Típicamente, se refiere a la relación funcional entre una secuencia reguladora de transcripción y una secuencia transcrita. Por ejemplo, una secuencia promotora o potenciadora está enlazada operativamente a una secuencia codificante si estimula o modula la transcripción de la secuencia codificante en una célula hospedera apropiada u otro sistema de expresión. Generalmente, las secuencias reguladoras de transcripción promotoras que están enlazadas operativamente a una secuencia transcrita son físicamente contiguas a la secuencia transcrita, es decir, están en modo de acción cis. Sin embargo, algunas secuencias reguladoras de transcripción, tales como potenciadores, no requieren ser físicamente contiguas o estar localizadas en estrecha cercanía a las secuencias codificantes cuya transcripción potencian.
Los términos “polipéptido” y “proteína” se usan aquí intercambiablemente para referirse a un polímero de residuos de aminoácidos. Los términos se aplican a polímeros de aminoácido en los que uno o más residuos de aminoácido son un mimético químico artificial de un aminoácido natural correspondiente, así como también polímeros de aminoácido naturales y polímeros aminoácido no naturales. A menos que se indique de otra manera, una secuencia de polipéptido particular también abarca implícitamente variantes modificadas conservativamente de la misma.
El término “inmunoconjugado” o “conjugado anticuerpo-fármaco”, como se usa en la presente, se refiere al enlace de un anticuerpo o un fragmento de unión a antígeno del mismo con otro agente, tal como un agente quimioterapéutico, una toxina, un agente inmunoterapéutico, una sonda de imagenología, y similares. El enlace puede ser enlaces covalentes, o interacciones no covalentes por ejemplo por medio de fuerzas electroestáticas. Se pueden utilizar varios enlazadores conocidos para formar el inmunoconjugado. Adicionalmente el inmunoconjugado se puede proveer en forma de una proteína de fusión que se puede expresar a partir de un polinucleótido que codifica el inmunoconjugado. Como se usa en la presente, “proteína de fusión” se refiere a proteínas creadas por medio de la unión de dos o más genes o fragmentos de gen que originalmente codificaron proteínas separadas (incluso péptidos y polipéptidos). La traducción del gen de fusión resulta en una sola proteína con propiedades funcionales derivadas de cada una de las proteínas originales.
El término “sujeto” incluye animales humanos y no humanos. Los animales no humanos incluyen todos los vertebrados (por ejemplo, mamíferos y no mamíferos) tales como primates no humanos, ovejas, perros, vacas, pollos, anfibios y reptiles. Excepto cuando se aclare, los términos “paciente” o “sujeto” se usan aquí intercambiablemente.
El término “toxina”, “citotoxina” o “agente citotóxico”, como se usa en la presente, se refiere a cualquier agente que es perjudicial para el desarrollo y proliferación de células y puede actuar para reducir, inhibir o destruir una célula o malignidad.
El término “agente anticanceroso” usado en la presente se refiere a cualquier agente que se puede usar para tratar un trastorno proliferativo de células, tal como cáncer, que incluye sin limitación agentes citotóxicos, agentes quimioterapéuticos, radioterapia y agentes radioterapéuticos, agentes anticancerosos dirigidos y agentes inmunoterapéuticos.
El término “porción de fármaco” o “carga útil” usado en la presente se refiere a una porción química que está conjugada con un anticuerpo o fragmento de unión a antígeno, y puede incluir cualquier agente terapéutico o diagnóstico, por ejemplo un agente anticanceroso, antiinflamatorio, antiinfeccioso (por ejemplo antimicótico, antibacteriano, antiparasitario, antiviral) o anestésico. En ciertos aspectos, una porción de fármaco se selecciona de un inhibidor de V-ATPasa, un inhibidor de HSP90, un inhibidor de lAP, un inhibidor de mTor, un estabilizador de microtúbulos, un desestabilizador de microtúbulos, una auristatina, una dolastatina, un maitansinoide, una MetAP (metionina aminopeptidasa), un inhibidor de la exportación nuclear de proteínas CRM1, un inhibidor de DPPIV, un inhibidor de las reacciones de transferencia de fosforilo en mitocondrias, un inhibidor de la síntesis de proteína, un inhibidor de quinasa, un inhibidor de CDK2 , un inhibidor de CDK9, un inhibidor del proteasoma, un inhibidor de cinesina, un inhibidor de HDAC, un agente perjudicial para el ADN, un agente alquilante de ADN, un intercalador de ADN, un enlazador de la ranura menor del ADN y un inhibidor de DHFR. Los métodos para unir cada uno de estos a un enlazador compatible con los anticuerpos y el método de la presente divulgación son conocidos; véase por ejemplo Singh et al. (2009) “Therapeutic Antibodies: Methods and Protocols”, vol. 525, 445-457. Además, una carga útil puede ser una sonda biofísica, un fluoróforo, una etiqueta de espín, una sonda infrarroja, una sonda de afinidad, un quelante, una sonda espectroscópica, una sonda radioactiva, una molécula de lípido, un polietilenglicol, un polímero, una etiqueta de espín, ADN, ARN, una proteína, un péptido, una superficie, un anticuerpo, un fragmento de anticuerpo, una nanopartícula, un punto cuántico, un liposoma, una partícula de PLGA, un sacárido o un polisacárido.
El término “porción de fármaco maitansinoide” significa la subestructura de un conjugado anticuerpo-fármaco que tiene la estructura de un compuesto maitansinoide. La maitansina fue aislada por primera vez del arbusto africano Maytenus serrata (patente de EE. UU. No. 3.896.111). Subsiguientemente se descubrió que ciertos microbios también producen
maitansinoides tales como maitansinol y ásteres de C-3 de maitansinol (patente de EE. UU. No. 4.151.042). Se han reportado maitansinoles sintéticos y análogos de maitansinol; véanse las patentes de EE. UU. Nos.4.137.230; 4.248.870; 4.256.746; 4.260.608; 4.265.814; 4.294.757; 4.307.016; 4.308.268; 4.308.269; 4.309.428; 4.313.946; 4.315.929; 4.317.821; 4.322.348; 4.331.598; 4.361.650; 4.364.866; 4.424.219; 4.450.254; 4.362.663; y 4.371.533; y Kawai et al. (1984) Chem. Pharm. Bull. 3441-3451), cada una de las cuales se incorpora expresamente como referencia. Ejemplos específicos de maitansinoides útiles para la conjugación incluyen DM1, DM3 y DM4.
“Tumor” se refiere al crecimiento y proliferación de células neoplásicas, ya sea malignas o benignas, y todas las células y tejidos precancerosos y cancerosos.
El término “actividad antitumoral” significa una reducción en la tasa de proliferación, viabilidad o actividad metastásica de la célula de tumor. Una forma posible de mostrar la actividad antitumoral es mostrar una declinación en la velocidad de crecimiento de las células de tumor, estasis del tamaño del tumor, o reducción del tamaño del tumor. Tal actividad se puede evaluar usando los modelos de tumor in vitro o in vivo aceptados, que incluyen, sin limitación modelos de xenoinjerto, modelos de aloinjerto, modelos de MMTV, y otros modelos conocidos en la técnica para investigar la actividad antitumoral.
El término “malignidad” se refiere a un tumor no benigno o un cáncer. Como se usa en la presente, el término “cáncer” incluye una malignidad caracterizada por crecimiento celular no regulado o descontrolado. Los ejemplos de cáncer incluyen: carcinomas, sarcomas, leucemias y linfomas.
El término “cáncer” incluye tumores malignos primarios (por ejemplo, aquellos cuyas células no han emigrado a sitios del cuerpo del sujeto distantes del sitio del tumor original), y tumores malignos secundarios (por ejemplo, los que se originan de metástasis, la emigración de las células de tumor a sitios secundarios que son diferentes del sitio del tumor original).
El término “cKIT” se refiere a un receptor tirosina quinasa que es un miembro de la familia III de tirosinas quinasas receptoras. Las secuencias de ácidos nucleicos y aminoácidos de cKIT son conocidas y han sido publicadas con los números de registro GenBank Nos. X06182.1, EU826594.1, GU983671.1, HM015525.1, HM015526.1, AK304031.1 y BC071593.1. Véase también SEQ ID NO:1 para la secuencia de ADNc de cKIT de humano, y SEQ ID NO:2 para la secuencia de proteína de cKIT de humano. Estructuralmente, el receptor cKIT es una proteína de transmembrana de tipo I y contiene un péptido señal, 5 dominios C2 similares a Ig en el dominio extracelular, y tiene un dominio de proteína quinasa en su dominio intracelular, y tiene sobre toda su longitud por lo menos aproximadamente 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, o 100% de identidad de secuencia con la secuencia de aminoácidos de SEQ ID NO:2. Estructuralmente, una secuencia de ácido nucleico de cKIT tiene sobre toda su longitud por lo menos aproximadamente 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, o 100% de identidad de secuencia con la secuencia de ácido nucleico de SEQ ID NO:1.
Los términos “cáncer que expresa cKIT” o “cáncer cKIT positivo” se refieren a un cáncer que expresa cKIT o una forma mutante de cKIT sobre la superficie de las células cancerosas.
Como se usa en la presente, los términos “tratar” o “tratamiento” de cualquier enfermedad o trastorno se refieren en un aspecto a mitigar la enfermedad o trastorno (esto es, retardar o detener o reducir el desarrollo de la enfermedad o por lo menos uno de sus síntomas clínicos). En otro aspecto, “tratar” o “tratamiento” se refiere a aliviar o mejorar por lo menos un parámetro físico que incluye los que pueden no ser discernibles por el paciente. En otro aspecto, “tratar” o “tratamiento” se refiere a modular la enfermedad o trastorno, ya sea físicamente (por ejemplo, la estabilización de un síntoma discernible), fisiológicamente (por ejemplo, estabilización de un parámetro físico), o ambos. En otro aspecto más, “tratar” o ‘tratamiento” se refiere a prevenir o retrasar el inicio, desarrollo o avance de la enfermedad o trastorno.
El término “cantidad terapéuticamente aceptable” o “dosis terapéuticamente eficaz” se refiere intercambiablemente a una cantidad suficiente para efectuar el resultado deseado (es decir, una reducción del tamaño del tumor, inhibición del crecimiento del tumor, prevención de metástasis, inhibición o prevención de infección viral, bacteriana, micótica o parasitaria). En algunos aspectos, una cantidad terapéuticamente aceptable no induce o causa efectos secundarios indeseables. Una cantidad terapéuticamente aceptable puede ser determinada administrando primero una dosis baja y después aumentando incrementalmente esa dosis hasta obtener el efecto deseado. Una “dosis profilácticamente eficaz” y una “dosis terapéuticamente eficaz” de las moléculas de la presente divulgación pueden prevenir el inicio o pueden producir una disminución de la severidad de los síntomas de enfermedad que incluyen los síntomas asociados con el cáncer, respectivamente.
El término “coadministrar” se refiere a la presencia simultánea de dos agentes activos en la sangre de un individuo. Los agentes activos que se coadministran se pueden suministrar concurrente o secuencialmente.
BREVE DESCRIPCIÓN DE LOS DIBUJOS
La figura 1 muestra la actividad de los ADC cKIT-MCC-DM1 en un subgrupo de líneas de células de cáncer.
La figura 2 representa la actividad de 9P3-MCC-DM1, 9P3-SPDB-DM4 y 9P3-CX1-1-DM1 en un subgrupo de líneas de células de cáncer.
La figura 3 muestra la actividad de 9P3-MCC-DM1 en un panel de líneas celulares AML, GIST, melanoma y SCLC, con grados variables de expresión de superficie del receptor cKlT.
La figura 4 muestra la capacidad de los ADC cKIT-MCC-DM1 para inhibir la proliferación de células GIST-T1 (sensibles a imatinib).
La figura 5 muestra la capacidad de los ADC cKIT-MCC-DM1 para inhibir la proliferación de células GIST430 (resistentes a imatinib).
La figura 6 muestra la capacidad de los ADC cKIT-MCC-DM1 para inhibir la proliferación de células NCI-H526 (SCLC que expresa altos niveles de cKIT).
La figura 7 muestra la capacidad de los ADC cKIT-MCC-DM1 para inhibir la proliferación de células NCI-H1048 (SCLC que expresa bajos niveles de cKIT).
La figura 8 muestra la capacidad de los ADC cKIT-MCC-DM1 para inhibir la proliferación de células CMK11-5 (AML que expresa altos niveles de cKIT).
La figura 9 muestra la capacidad de los ADC cKIT-MCC-DM1 para inhibir la proliferación de células Uke-1 (AML que expresa bajos niveles de cKIT).
La figura 10 son datos crudos de HDx-MS graficados como la diferencia corregida sobre el error estándar en la medición. Un valor más negativo indica más protección del intercambio de deuterio tras la unión de 9P3 al antígeno cKIT. Las dos regiones más significativas de protección son denotadas como región 1 y región 2.
La figura 11 muestra regiones de protección de HDx-MS mapeadas usando llenado de superficie: región 1 (negro) y región 2 (gris obscuro). Los sitios de unión de SCF son denotados como sitio I (esferas grises claras), sitio II (esferas grises medias) y sitio III (esferas grises más obscuras).
La figura 12 es un Western blot que muestra la capacidad de SCF, NEG085-MCC-DM1, NEG024-MCC-DM1 y 20376-MCC-DM1 para modular la fosforilazción de cKIT en una línea celular de cKIT de tipo silvestre Mo7e Figura 12(A), o línea celular de cKIT mutante GIST-T1 Figura (12B), después de 15 minutos.
La figura 13 muestra que los Ab NEG085 y 20376 median un rápido internamiento de cKIT de superficie en las células GIST-T1 (A), y en células de medula ósea humana (B).
La figura 14 muestra Western blots que muestran la capacidad de SCF o NEG085-MCC-DM1 para acelerar la degradación de cKIT en una línea celular de cKIT mutante, GIST-T1 (Figura 14A) y línea celular de cKIT de tipo silvestre NCI-H526 (Figura 14B) durante el curso del tiempo.
La figura 15 muestra la capacidad de NEG085, NEG024, 20376, NEG085-MCC-DM1 para inhibir la proliferación dependiente de SCF de las células Mo7e.
La figura 16 muestra la capacidad de NEG085 y NEG085-MCC-DM1 para inhibir la proliferación independiente de SCF de células Mo7e.
La figura 17 muestra la evaluación de la capacidad de los anticuerpos Campath (Ab anti-CD52), NEG085 o 20376 para no inducir ADCC in vitro en células Uke-1.
La figura 18 muestra que NEG085 y 20376 no median apoptosis de mastocitos humanos primarios.
La figura 19 muestra que NEG085 y 20376 no median la desgranulación de mastocitos humanos primarios.
La figura 20 muestra la co-localización de IgG1 y detención mitótica de NEG027-MCC-DM1 en el modelo de xenoinjerto de GIST T1.
La figura 21 muestra secciones de tejidos de detención mitótica (p-histona H3) y apoptosis (caspasa 3) después de una sola dosis de ADC de cKIT.
La figura 22 representa gráficamente la detención mitótica e inducción de apoptosis 8 días después de la dosis única de ADC de cKIT.
La figura 23 muestra (A) la eficacia a la respuesta a la dosis en el xenoinjerto de ratón GIST T1, y (B) el cambio en peso corporal durante el curso del tratamiento.
La figura 24 representa gráficamente (A) una ELISA anti-DM1 después de la administración en un modelo de xenoinjerto GIST T1, y (B) representa una ELISA anti-IgG1 humana después de la administración en un modelo de xenoinjerto GIST T1.
La figura 25 es una tabla de la dosis-respuesta de NEG027-MCC-DM1 en un modelo de ratón de xenoinjerto GIST T1. La figuras 26 muestra secciones de histología de la eficacia de la respuesta a la dosis de NEG027-MCC-DM1 en GIST T1. (A) son tumores reunidos del grupo 4; (B) tumores reunidos del grupo 5.
La figura 27 representa (A) la eficacia con 0,625 mg/kg en un modelo de xenoinjerto de ratón GIST T1; (B) el cambio del volumen de tumor contra el control (% T/C); y (C) el cambio en el peso corporal durante el curso del tratamiento.
La figura 28 muestra la agrupación del día 41 después de la administración de una sola dosis de ADC anti-cKIT a un ratón de xenoinjerto GIST T1.
La figura 29 es una tabla de la eficacia de ADC de cKIT a una dosis eficaz baja en un modelo de xenoinjerto GIST T1. La figura 30 muestra (A) la PK del anti-cKIT en un modelo de ratón de xenoinjerto GIST T1 (el panel izquierdo es una ELISA anti-DM1; (B) panel derecho es una ELISA anti-IgG1 humana.
La figura 31 A-C muestra (A) la actividad de NEG085-MCC-DM1, NEG024MCC-DM1 y NEG086-MCC-DM1 en un modelo de SCLC; (B) el cambio en el peso corporal durante el curso del tratamiento; (C) la expresión de cKIT sobre la muestra del tumor.
La figura 32 es una tabla de un estudio de la eficacia de anti-cKIT-ADC en SCLC NCI-H1048.
La figura 33A-B muestra (A) la respuesta a la dosis de NEG085-MCC-DM1 en el modelo de xenoinjerto NCI-H1048 (SCLC); (B) el cambio en el peso corporal durante el curso del tratamiento.
La figura 34 es una tabla que muestra un estudio de la eficacia de NEG085-MCC-DM1 en un modelo de ratón de xenoinjerto NCI-1048 (SCLC).
La figura 35A-C muestra (A) la eficacia de 20376 y NEG024 en el modelo de ratón de xenoinjerto NCI-H526 (SCLC); (B) la concentración de anticuerpo en el suero después de la administración; y (C) la IHC para cKIT mostrando la expresión de los niveles de cKIT en el tumor H526.
La figura 36 muestra el ADC anti-cKIT en un modelo de xenoinjerto de cáncer de pulmón de célula pequeña (SCLC). La figura 37 muestra la eficacia del ADC anti-cKIT en un modelo de xenoinjerto de AML (Kasumi-1).
La figura 38 muestra la eficacia de ADC anti-cKIT en un modelo de xenoinjerto de ratón de mastocitosis HMC-1.
La figura 39A/B muestra la eficacia de 20376-MCC-DM1 reactivo cruzadamente en el ratón, en el modelo de ratón de xenoinjerto GIST T1 con (A) la dosificación y volumen de tumor; y (B) el cambio en el peso corporal durante el curso de tratamiento.
La figura 40A/B muestra (A) la eficacia de 20376-MCC-DM1 reactivo cruzadamente en el ratón en el modelo de ratón de
xenoinjerto GIST T1 —PK; y (B) la concentración de anticuerpo en el suero después de la administración.
La figura 41 muestra un estudio de la eficacia de respuesta a la dosis en ratones SCID-beige GIST T1.
La figura 42A/B muestra (A) la eficacia en el modelo de ratón de xenoinjerto GIST T1 (no hay eficacia cuando no está conjugado); y (B) el cambio en el peso corporal durante el curso del tratamiento.
La figura 43 es una comparación de la eficacia en un modelo de xenoinjerto de ratón GIST T1 (MCC-DM1/SPDB-DM4 no marcado).
La figura 44A/B muestra (A) la eficacia en un modelo de xenoinjerto GIST 430 que compara SPDB-DM4 y MCC-DM1; y (B) el cambio en el peso corporal durante el curso del tratamiento.
La figura 45 muestra la eficacia en el modelo de ratón SCID-beige GIST 430.
La figura 46 son fotografías de la inmunotinción de la p-Histona H3 después de tratamiento con NEG085-MCC-DM1.
La figura 47 es una gráfica de detención mitótica mostrada por tinción de la p-Histona H3 después de la administración de NEG085-MCC-DM1.
La figura 48A muestra la tinción de cKIT de un tumor GIST T1; la figura 48B muestra la respuesta a la dosis de NEG085-MCC-DM1 en un modelo de xenoinjerto GIST T1; la figura 48C muestra el cambio en el peso corporal de los ratones tratados con NEG085-MCC-DM1.
La figura 49A muestra la tinción de cKIT de un tumor GIST 430; la figura 49B muestra la respuesta a la dosis de NEG085-MCC-DM1 en un modelo de xenoinjerto GIST 430; la figura 49C muestra el cambio en el peso corporal de los ratones tratados con NEG085-MCC-DM1.
La figura 50A muestra la tinción de cKIT de un tumor NCI-H526 (cáncer de pulmón de célula pequeña) (SCLC); la figura 50B muestra la respuesta a la dosis de NEG085-MCC-DM1 en un modelo de xenoinjerto NCI-H526; la figura 50C muestra el cambio en el peso corporal de los ratones tratados con NEG085-MCC-DM1.
La figura 51A muestra la cantidad de IgG1 después de la administración de NEG085-MCC-DM1 en un modelo de xenoinjerto NCI-H526; la figura 51B es una gráfica de una ELISA anti-DM1 en el modelo de xenoinjerto NCI-H526 después de la administración de NEG085-MCC-DM1.
La figura 52A es una gráfica que muestra la eficacia de NEG085-MCC-DM1 en un modelo de ratón de xenoinjerto de AML primaria; la figura 52B muestra el cambio en el peso corporal de los ratones tratados con NEG085-MCC-DM1.
La figura 53 es una representación de la estructura de cristal del Fab NEG085 en complejo con los dominios 1 y 2 de cKIT. Las cadenas pesadas de Fab están en gris obscuro; las cadenas ligeras de Fab están en blanco; y los dominios de cKIT están en gris claro. Los epítopos y parátopos están en negro.
DESCRIPCIÓN DETALLADA
La presente divulgación provee anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno), y conjugados anticuerpo-fármaco que se unen a cKIT. En particular, la presente divulgación está dirigida a anticuerpos y fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) que se unen a cKIT y se internan después de dicha unión. Los anticuerpos y fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) de la presente divulgación se pueden usar para producir conjugados de anticuerpo-fármaco. Además, la presente divulgación provee conjugados anticuerpo-fármaco que tienen características farmacocinéticas deseables y otros atributos deseables, y por lo tanto se pueden usar para tratar el cáncer que expresa cKIT, por ejemplo, sin limitación: tumores estromales gastrointestinales (GIST), cáncer de pulmón de célula pequeña (SCLC), leucemia mieloide aguda (AML), melanoma, leucemia de mastocitos (MCL), mastocitosis, neurofibromatosis, cáncer de mama, cáncer de pulmón de célula no pequeña (NSCLC) y cáncer pancreático. La presente divulgación provee además composiciones farmacéuticas que comprenden los conjugados anticuerpo-fármaco y métodos de preparación y uso de dichas composiciones farmacéuticas para el tratamiento del cáncer.
Conjugados anticuerpo-fármaco
La presente divulgación provee conjugados anticuerpo-fármaco, en donde un anticuerpo, fragmento de unión a antígeno o su equivalente funcional que se une específicamente a cKIT, está enlazado a una porción de fármaco. En un aspecto, los anticuerpos, fragmentos de unión de antígeno o sus equivalentes funcionales son enlazados, mediante unión covalente por medio de un enlazador, a una porción de fármaco que es un agente anticanceroso. Los conjugados anticuerpofármaco pueden suministrar selectivamente una dosis eficaz de un agente anticanceroso (por ejemplo, un agente citotóxico) a tejidos de tumor que expresan cKIT, con lo cual se puede lograr una mayor selectividad (y reducir la dosis eficaz).
En un aspecto, la divulgación provee un inmunoconjugado de fórmula (I):

en donde Ab representa un anticuerpo o fragmento de anticuerpo que se une a cKIT (por ejemplo, fragmento de unión a antígeno) descrito en la presente;
L es un enlazador;
D es una porción de fármaco;
m es un entero de 1-8; y
n es un entero de 1-20. En un aspecto, n es un entero de 1 a 10, de 2 a 8, o de 2 a 5. En un aspecto específico, n es de 3 a 4. En algunos aspectos, m es 1. En algunos aspectos, m es 2, 3 o 4.
Aunque la proporción de fármaco a anticuerpo tiene un valor entero exacto para una molécula conjugada específica (por ejemplo, n multiplicado por m en la fórmula (I)), se entiende que el valor frecuentemente será un valor promedio cuando se utiliza para describir una muestra que contiene muchas moléculas, debido en cierto grado a la falta de homogeneidad asociada típicamente con el paso de conjugación. La carga promedio para una muestra de un inmunoconjugado es referida aquí como la proporción de fármaco a anticuerpo, o “DAR”. En el aspecto de maitansinoides, esto se puede referir como proporción de maitansinoide a anticuerpo o “MAR”. En algunos aspectos, la DAR es entre aproximadamente 1 y aproximadamente 5, y típicamente es de aproximadamente 3, 3,5, 4, 4,5 o 5. En algunos aspectos, por lo menos 50% en peso de una muestra es un compuesto que tiene la DAR promedio, más o menos 2, y de preferencia por lo menos 50% de la muestra es un conjugado que contiene la DAR promedio más o menos 1. Otros aspectos incluyen inmunoconjugados en donde la DAR es aproximadamente 3,5. En algunos aspectos, una DAR de “aproximadamente n” significa que el valor medido para DAR está dentro del 20% de n.
La presente divulgación provee inmunoconjugados que comprenden los anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) y sus equivalentes funcionales divulgados en la presente, enlazados o conjugados con una porción de fármaco. En un aspecto, la porción de fármaco D es una porción de fármaco maitansinoide, que incluye los que contienen la estructura:

en donde la línea ondulada indica unión covalente del átomo de azufre del maitansinoide con un enlazador de un conjugado anticuerpo-fármaco. R en cada ocurrencia es independientemente H o un alquilo de C1-C 6. La cadena de alquileno que une el grupo amida al átomo de azufre puede ser metanilo, etanilo o propanilo, esto es, m es 1, 2 o 3 (patente de EE. UU. No. 633.410, patente de EE. UU. No. 5.208.020, Chari et al. (1992), Cáncer Res. 52;127-131, Lui et al. (1996) Proc. Natl. Acad. Sel. 93:8618-8623).
Todos los estereoisómeros de la porción de fármaco maitansinoide están contemplados por los inmunoconjugados divulgados: esto es, cualquier combinación de configuraciones R y S en los carbonos quirales del maitansinoide. En un aspecto, la porción de fármaco maitansinoide tiene la siguiente estereoquímica.

En otro aspecto, la porción de fármaco maitansinoide es N2-desacetil-N2-(3-mercapto-1-oxopropil)-maitansina (conocido también como DM1). DM1 se representa con la siguiente fórmula estructural.

En otro aspecto, la porción de fármaco maitansinoide es N2-desacetil-N2'-(4-mercapto-1-oxopentil)-maitansina (también conocido como DM3). DM3 se representa con la siguiente fórmula estructural.

En otro aspecto, la porción de fármaco maitansinoide es N2’-desacetil-N2'-(4-metil-4-mercapto-1 -oxopentil)-maitansina (conocida también como DM4). DM4 se representa con la siguiente fórmula estructural.

La porción de fármaco D puede estar enlazada al anticuerpo por medio de un enlazador L. L es cualquier porción química que sea capaz de enlazar el anticuerpo Ab a la porción de fármaco D. El enlazador L une el anticuerpo Ab al fármaco D por medio de enlaces covalentes. El reactivo enlazador es una porción bifuncional o multifuncional que se puede usar para enlazar una porción de fármaco D y un anticuerpo Ab para formar conjugados anticuerpo-fármaco. Los conjugados anticuerpo-fármaco se pueden preparar usando un enlazador que tiene una funcionalidad reactiva para unir la porción de fármaco D al anticuerpo Ab. Una cisteína, tiol o una amina, por ejemplo extremo N o cadena lateral de aminoácido tal como lisina del anticuerpo, puede formar un enlace con un grupo funcional de un reactivo enlazador.
En un aspecto, L es un enlazador escindible. En otro aspecto, L es un enlazador no escindible. En algunos aspectos, L es un enlazador lábil al ácido, enlazador foto-lábil, enlazador escindible por peptidasa, enlazador escindible por esterasa, un enlazador de enlace disulfuro reducible, un enlazador hidrófilo, un enlazador procargado, o un enlazador a base de ácido dicarboxílico.
Los reactivos de entrelazamiento adecuados que forman un enlazador no escindible entre la porción de fármaco D, por ejemplo maitansinoide, y el anticuerpo Ab, son muy conocidos en la técnica, y pueden formar enlazadores no escindibles que comprenden un átomo de azufre (SMCC) o los que están sin un átomo de azufre. Los reactivos de entrelazamiento preferidos que forman enlazadores no escindibles entre la porción de fármaco D, por ejemplo maitansinoide, y el anticuerpo Ab, comprenden una porción basada en maleimido o haloacetilo. De acuerdo con la presente divulgación, estos enlazadores no escindibles se dice que son derivados de porciones basadas en maleimido o haloacetilo.
Los reactivos de entrelazamiento que comprenden una porción basada en maleimido incluyen, sin limitación, N-succinimidil-4-(maleimidometil)ciclohexancarboxilato (SMCC), sulfosuccinimidil-4-(N-maleimidometil)ciclohexano-1-carboxilato (sulfo-SMCC), N-succinimidil-4-(maleimidometil)ciclohexano-1-carboxi-(6-amidocaproato), que es un análogo de “cadena larga” de SMCC (LC-SMCC), éster de N-succinimidilo de ácido K-maleimidoundeconoico (KMUA), éster de N-succinimidilo de ácido g-maleimidobutírico (GMBS), éster de N-succinimidilo de ácido £-maleimidocaproico (EMCS), éster de m-maleimidobenzoil-N-hidroxisuccinimida (MBS), éster de N-(a-maleimidoacetoxi)-succinimida (AMSA), succinimidil-6-(p-maleimidopropionamido)hexanoato (SMPH), N-succinimidil-4-(p-maleimidofenil)-butirato (SMPB), N-(-p-maleomidofenil)isocianato (PMIP) y reactivos de entrelazamiento basados en maleimido que contienen un espaciador de polietilenglicol, tal como MAL-PEG-NHS. Estos reactivos de entrelazamiento forman enlazadores no escindibles derivados de porciones basadas en maleimido. Más abajo se muestran estructuras representativas de reactivos de entrelazamiento basados en maleimido.

En otro aspecto, el enlazador L es derivado de AJ-succinimidil-4-(maleimidometil)ciclohexancarboxilato (SMCC), sulfosuccinimidil-4-(N-maleimidometil)ciclohexano-1-carboxilato (sulfo-SMCC) o MAL-PEG-NHS.
Los reactivos de entrelazamiento que comprenden una porción basada en haloacetilo incluyen N-succinimidilyodoacetato (SIA), N-succinimidil(4-yodoacetil)aminobenzoato (SIAB), N-succinimidil-bromoacetato (SBA) y N-succinimidil-3-(bromoacetamido)propionato (SBAP). Estos reactivos de entrelazamiento forman un enlazador no escindible derivado de porciones basadas en haloacetilo. Más abajo se muestran estructuras representativas de reactivos de entrelazamiento basados en haloacetilo.

En un aspecto, el enlazador L se deriva de N-succinimidil-yodoacetato (SIA) o N-succinimidil(4-yodoacetil)aminobenzoato (SIAB).
Los reactivos de entrelazamiento adecuados que forman un enlazador escindible entre la porción de fármaco D, por ejemplo maitansinoide, y el anticuerpo Ab, son muy conocidos en la técnica. Los enlazadores que contienen disulfuro son enlazadores escindibles por medio de intercambio de disulfuro, lo que puede ocurrir bajo condiciones fisiológicas. De acuerdo con la presente divulgación, tales enlazadores escindibles se dice que derivan de porciones basadas en disulfuro. Los reactivos de entrelazamiento de disulfuro adecuados incluyen N-succinimidil-3-(2-piridilditio)propionato (SPDP), N-succinimidil-4-(2-piridilditio)pentanoato (SPP), N-succinimidil-4-(2-piridilditio)butanoato (SPDB) y W-succinimidil-4-(2-piridilditio)2-sulfo-butanoato (sulfo-SPDB), cuyas estructuras se muestran más abajo. Estos reactivos de entrelazamiento de disulfuro forman un enlazador escindible derivado de porciones basadas en disulfuro.

N-succinimidil-3-(2-piridilditio)propionato (SPDP)

N-succinimidil-4-(2-piridilditio)pentanoato (SPP)

En un aspecto, el enlazador L es derivado de N-succinimidil-4-(2-piridilditio)butanoato (SPDB).
Los reactivos de entrelazamiento adecuados que forman un enlazador cargado entre la porción de fármaco D, por ejemplo maitansinoide, y el anticuerpo Ab, son conocidos como reactivos de entrelazamiento procargados. En un aspecto, el enlazador L que se deriva del reactivo de entrelazamiento procargado es CX1-1. La estructura de CX1-1 es la siguiente.

2,5-dioxopirrolidin-1-il-17-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-5,8,11,14-tetraoxo-4,7,10,13-tetraazaheptadecan-1-oato (CX1-1)
En un aspecto provisto por la divulgación, el conjugado se representa con cualquiera de las siguientes fórmulas estructurales:


Ab es un anticuerpo o fragmento de unión a antígeno del mismo que se une específicamente a cKIT humano;
n, que indica el número de grupos D-L unidos al Ab por medio de la formación de un enlace de amida con una amina primaria del Ab, es un entero de 1 a 20. En un aspecto, n es un entero de 1 a 10, de 2 a 8, o de 2 a 5. En un aspecto específico, n es 3 o 4.
En un aspecto, la proporción molar promedio de fármaco (por ejemplo, DM1 o DM4) a anticuerpo en el conjugado (es decir, el valor w promedio, conocido también como proporción de maitansinoide a anticuerpo (MAR)) es de aproximadamente 1 a aproximadamente 10, de aproximadamente 2 a aproximadamente 8 (por ejemplo, 1,9, 2,0, 2,1,2,2, 2,3, 2,4, 2,5, 2,6, 2,7, 2,8, 2,9, 3,0, 3,1,3,2, 3,3, 3,4, 3,5, 3,6, 3,7, 3,8, 3,9, 4,0, 4,1,4,2, 4,3, 4,4, 4,5, 4,6, 4,7, 4,8, 4,9, 5,0, 5,1,5,2, 5,3, 5,4, 5,5, 5,6, 5,7, 5,8, 5,9, 6,0, 6,1,6,2, 6,3, 6,4, 6,5, 6,6, 6,7, 6,8, 6,9, 7,0, 7,1,7,2, 7,3, 7,4, 7,5, 7,6, 7,7, 7,8, 7,9, 8,0, o 8,1), de aproximadamente 2,5 a aproximadamente 7, de aproximadamente 3 a aproximadamente 5, de aproximadamente 2,5 a aproximadamente 4,5 (por ejemplo, aproximadamente 2,5, aproximadamente 2,6, aproximadamente 2,7, aproximadamente 2,8, aproximadamente 2,9, aproximadamente 3,0, aproximadamente 3,1, aproximadamente 3,3, aproximadamente 3,4, aproximadamente 3,5, aproximadamente 3,6, aproximadamente 3,7, aproximadamente 3,8, aproximadamente 3,9, aproximadamente 4,0, aproximadamente 4,1, aproximadamente 4,2, aproximadamente 4,3, aproximadamente 4,4, aproximadamente 4,5), de aproximadamente 3,0 a aproximadamente 4,0, de aproximadamente 3,2 a aproximadamente 4,2, o de aproximadamente 4,5 a 5,5 (por ejemplo, aproximadamente 4,5, aproximadamente 4,6, aproximadamente 4,7, aproximadamente 4,8, aproximadamente 4,9, aproximadamente 5,0, aproximadamente 5,1, aproximadamente 5,2, aproximadamente 5,3, aproximadamente 5,4, o aproximadamente 5,5).
En un aspecto provisto por la divulgación, el conjugado tiene una pureza sustancialmente alta y tiene una o más de las siguientes características: (a) más de aproximadamente 90% (por ejemplo, mayor o igual que aproximadamente 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, o 100%), de preferencia más de aproximadamente 95% de las especies conjugadas son monoméricas; (b) el porcentaje de enlazador no conjugado en la preparación del conjugado es menor de aproximadamente 10% (por ejemplo, menor o igual que aproximadamente 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, o 0%) (con respecto al total del enlazador); (c) menos de 10% de las especies conjugadas están entrelazadas (por ejemplo, menor o igual que aproximadamente 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, o 0%); (d) el porcentaje de fármaco libre (por ejemplo, DM1 o DM4) en la preparación del conjugado es menor de aproximadamente 2% (por ejemplo, menor o igual que aproximadamente 1,5%, 1,4%, 1,3%, 1,2%, 1,1%, 1,0%, 0,9%, 0,8%, 0,7%, 0,6%, 0,5%, 0,4%, 0,3%, 0,2%, 0. 1% o 0%) (mol/mol con respecto al total de agente citotóxico).
Como se usa en la presente, el término “enlazador no conjugado” se refiere al anticuerpo que está unido covalentemente a un enlazador derivado de un reactivo de entrelazamiento (por ejemplo, SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB o CX1-1), en donde el anticuerpo no está acoplado covalentemente con el fármaco (por ejemplo, DM1 o DM4) por medio de un enlazador (esto es, el “enlazador no conjugado” puede ser representado por Ab-SMCC, Ab-SPDB, o Ab-CX1-1).
1. Porción de fármaco
La presente divulgación provee inmunoconjugados que se unen específicamente a cKIT. Los inmunoconjugados de la presente divulgación comprenden anticuerpos anti-cKIT, fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) o equivalentes funcionales que son conjugados con una porción de fármaco, por ejemplo, un agente anticanceroso, un agente contra trastornos hematológicos, un agente de tratamiento autoinmune, un agente antiinflamatorio, un agente antifúngico, un agente antibacteriano, un agente antiparasitario, un agente antiviral o un agente anestésico. Los anticuerpos, fragmentos de anticuerpo (por ej., fragmentos de unión de antígeno) o equivalentes funcionales pueden conjugarse con varias porciones de fármaco iguales o diferentes utilizando cualquiera de los métodos conocidos en la técnica.
En ciertos aspectos, la porción de fármaco de los inmunoconjugados de la presente divulgación es seleccionada de un grupo que consiste en un inhibidor de V-ATPasa, un agente pro-apoptótico, un inhibidor de Bcl2, un inhibidor de MCL1, un inhibidor de HSP90, un inhibidor de IAP, un inhibidor de mTor, un estabilizador de microtúbulos, un desestabilizador de microtúbulos, una auristatina, una dolastatina, un maitansinoide, una MetAP (metionina aminopeptidasa), un inhibidor de la exportación nuclear de proteínas CRM1, un inhibidor de DPPIV, inhibidores del proteasoma, un inhibidor de reacciones de transferencia de fosforilo en las mitocondrias, un inhibidor de la síntesis de proteínas, un inhibidor de quinasa, un inhibidor de CDK2, un inhibidor de CDK9, un inhibidor de cinesina, un inhibidor de HDAC, un agente perjudicial para el ADN, un agente alquilante de ADN, un intercalador de ADN, un enlazador de la ranura menor de ADN y un inhibidor de DHFR.
En un aspecto, la porción de fármaco de los inmunoconjugados de la presente divulgación es una porción de fármaco de maitansinoide, tal como por ejemplo, pero sin limitación, DM1, DM3 o DM4.
Además, los anticuerpos, fragmentos de anticuerpos (por ejemplo, fragmentos de unión de antígeno) o equivalentes funcionales de la presente divulgación pueden ser conjugados con una porción de fármaco que modifica una determinada respuesta biológica. Las porciones de fármaco no se consideran limitadas a los agentes terapéuticos químicos clásicos. Por ejemplo, la porción de fármaco puede ser una proteína, péptido, o polipéptido que posee una actividad biológica deseada. Estas proteínas pueden incluir, por ejemplo, una toxina como la abrina, ricina A, exotoxina de pseudomonas, toxina del cólera, o toxina de la difteria, una proteína como el factor de necrosis tumoral, a-interferón, p-interferón, factor de crecimiento de nervio, factor de crecimiento derivado de plaquetas, activador de plasminógeno de tejido, una citocina, un agente apoptótico, un agente anti-angiogénico, o un modificador de la respuesta biológica, tal como por ejemplo una linfocina.
En un aspecto, los anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) o equivalentes funcionales de la presente divulgación se conjugan con una porción de fármaco tal como una citotoxina, un fármaco (por ejemplo, un inmunosupresor) o una radiotoxina. Los ejemplos de citotoxina incluyen, sin limitación, taxanos (véanse, por ejemplo, las solicitudes de patente internacionales (PCT) Nos. WO 01/38318 y pCt /Us 03/02675), agentes alquilantes de ADN (por ejemplo, análogos de CC-1065), antraciclinas, análogos de tubulisina, análogos de duocarmicina, auristatina E, auristatina F, maitansinoides, y agentes citotóxicos que comprenden una porción reactiva de polietilenglicol (véase, por ejemplo, Sasse et al., J. Antibiot. (Tokio), 53, 879-85 (2000), Suzawa et al., Bioorg. Med. Chem., 8, 2175-84 (2000), Ichimura et al., J. Antibiot. (Tokio), 44, 1045-53 (1991), Francisco et al., Blood 200315;102(4): 1458-65), patentes de EE. UU. Nos. 5.475.092, 6.340.701, 6.372.738, y 6.436.931; publicación de la solicitud de patente de EE. UU. No.
2001/0036923 A1, solicitudes pendientes de patente de EE. UU. Nos. 10/024.290 y 10/116.053, y solicitud de patente internacional (PCT) No. WO 01/49698), taxon, citocalasina B, gramicidina D, bromuro de etidio, emetina, mitomicina, etopósido, tenopósido, vincristina, vinblastina, t. colchicina, doxorrubicina, daunorrubicina, dihidroxiantracindiona, mitoxantrona, mitramicina, actinomicina D, 1-deshidrotestosterona, glucocorticoides, procaína, tetracaína, lidocaína, propranolol, y puromicina y análogos u homólogas de la misma. Los agentes terapéuticos también incluyen, por ejemplo, anti-metabolitos (por ejemplo, metotrexato, 6-mercaptopurina, 6-tioguanina, citarabina, 5-fluorouracil decarbazina), agentes de ablación (por ejemplo, mecloretamina, tiotepa clorambucilo, meifalan, carmustina (BSNU) y lomustina (CCNU), ciclofosfamida, busulfan, dibromomannitol, estreptozotocina, mitomicina C, y cis-diclorodiamina platino (II) (DDP) cisplatino, antraciclinas (por ejemplo, daunorrubicina (anteriormente daunomicina) y doxorrubicina), antibióticos (por ejemplo, dactinomicina (anteriormente actinomicina), bleomicina, mitramicina y antramicina (AMC)), y agentes antimitóticos (por ejemplo, vincristina y vinblastina) (véase por ejemplo, Seattle Genetics, US20090304721).
Otros ejemplos de citotoxinas que se pueden conjugar con los anticuerpos, fragmentos de anticuerpo (fragmentos de unión de antígeno) o equivalentes funcionales de la presente divulgación incluyen duocarmicinas, caliqueamicinas, maitansinas y auristatinas, y derivados de los mismos.
Varios tipos de citotoxinas, enlazadores y los métodos para conjugar agentes terapéuticos con anticuerpos son conocidos en la técnica; véase, por ejemplo, Saito et al., (2003) Adv. Drug Deliv. Rev. 55:199-215; Trail et al., (2003) Cancer Immunol.
Immunother. 52:328-337; Payne, (2003) Cáncer Cell 3:207-212; Alien, (2002) Nat. Rev. Cáncer 2:750-763; Pastan y Kreitman, (2002) Curr. Opin. Investig. Drugs 3:1089-1091; Senter y Springer, (2001) Adv. Drug Deliv. Rev. 53:247-264.
Los anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) o equivalentes funcionales de la presente divulgación también se pueden conjugar con un isótopo radioactivo para generar agentes radiofarmacéuticos citotóxicos llamados radioinmunoconjugados. Los ejemplos de isótopos radioactivos que se pueden conjugar con los anticuerpos para su uso diagnóstico o terapéutico incluyen, sin limitación, yodo-131, indio-111, itrio-90 y lutecio-177. Los métodos para preparar radioinmunoconjugados están establecidos en la técnica. Están disponibles comercialmente radioinmunoconjugados ejemplares que incluyen Zevalin™ (IDEC Pharmaceuticals) y Bexxar™ (Corixa Pharmaceuticals), y se pueden usar métodos similares para preparar radioinmunoconjugados usando los anticuerpos divulgados en la presente. En ciertos aspectos, el quelante macrocíclico es el ácido 1,4,7,10-tetraazaciclododecano-N,N’,N” ,N” ’-tetraacético (DOTA), que se puede unir al anticuerpo por medio de una molécula enlazadora. Tales moléculas enlazadoras son conocidas comúnmente y se describen en Denardo et al., (1998) Clin Cancer Res. 4(10):2483-90; Peterson et al., (1999) Bioconjug. Chem. 10(4):553-7; y Zimmerman et al., (1999) Nucl. Med. Biol. 26(8):943-50, cada una incorporada como referencia en su totalidad.
Los anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) o equivalentes funcionales de la presente divulgación también se pueden conjugar con una proteína o polipéptido heterólogo (o fragmento del mismo, preferiblemente un polipéptido de por lo menos 10, por lo menos 20, por lo menos 30, por lo menos 40, por lo menos 50, por lo menos 60, por lo menos 70, por lo menos 80, por lo menos 90 o por lo menos 100 aminoácidos) para generar proteínas de fusión. En particular, la presente divulgación provee proteínas de fusión que comprenden un fragmento de anticuerpo (por ejemplo, fragmento de unión a antígeno) descrito en la presente (por ejemplo, un fragmento Fab, un fragmento Fd, fragmento Fv, fragmento F(ab)2, un dominio VH, una CDR VH, un dominio VL o una CDR VL) y una proteína, polipéptido o péptido heterólogo.
Se pueden generar proteínas de fusión adicionales por medio de las técnicas de barajado de genes, barajado de motivos, barajado de exones, o barajado de codones (referidos colectivamente como “barajado de ADN”). Se puede usar barajado de ADN para alterar la actividad de los anticuerpos de la presente divulgación o fragmentos de los mismos (por ejemplo, anticuerpos o fragmentos de los mismos con afinidades más altas y velocidades de disociación más bajas); véase en general las patentes de EE. UU. Nos. 5.605.793, 5.811.238, 5.830.721,5.834.252 y 5.837.458; Patten et al. (1997) Curr. Opinion Biotechnol. 8:724-33; Harayama (1998), Trends Biotechnol. 16(2):76-82; Hansson, et al. (1999), J. Mol. Biol.
287:265-76; y Lorenzo y Blasco (1998), Biotechniques 24(2):308-313 (cada una de estas patentes y publicaciones se incorporan aquí como referencia en su totalidad). Los anticuerpos o fragmentos de los mismos, o los anticuerpos o fragmentos de los mismos codificados, se pueden alterar antes de la recombinación sometiéndolos a mutagénesis aleatoria por PCR de propensión de error, inserción de nucleótido al azar u otros métodos. Un polinucleótido que codifica un anticuerpo o fragmento del mismo que se une específicamente a un antígeno se puede recombinar con uno o más componentes, motivos, secciones, partes, dominios, fragmentos, etc., de una o más moléculas heterólogas.
Más aún, los anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno), o equivalentes funcionales de la presente divulgación, se pueden conjugar con secuencias marcadoras, tales como un péptido, para facilitar la purificación. En aspectos preferidos, la secuencia de aminoácidos del marcador es un péptido de hexa-histidina, tal como la etiqueta provista en un vector püE (QIAGEN®, Inc., 9259 Eton Avenue, Chatsworth, Ca , 91311), entre otros, muchos de los cuales están disponibles comercialmente. Como se describe en Gentz et al. (1989), Proc Natl Acad. Sci. USA 86:821-824, por ejemplo, la hexa-histidina permite una purificación conveniente de la proteína de fusión. Otras etiquetas de péptido útiles para la purificación incluyen, sin limitación, la etiqueta de hemaglutinina (“HA”), que corresponde a un epítopo derivado de la proteína hemaglutinina de la influenza (Wilson et al. (1984), Cell 37:767), y la etiqueta “FLAG” (A. Einhauer et al., J. Biochem. Biophys. Methods 49: 455-465, 2001). Como se describe en la presente divulgación, los anticuerpos o fragmentos de unión de antígeno también se pueden conjugar con péptidos penetrantes de tumor para intensificar su eficacia.
En otros aspectos, los anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno), o equivalentes funcionales de la presente divulgación se conjugan con un agente diagnóstico o detectable. Tales inmunoconjugados pueden ser útiles para monitorear o pronosticar el inicio, desarrollo, avance o severidad de una enfermedad o trastorno como parte de un procedimiento de análisis clínico, tal como la determinación de la eficacia de una terapia particular. Tal diagnóstico y detección puede lograrse acoplando el anticuerpo con sustancias detectables que incluyen, sin limitación, varias enzimas tales como por ejemplo, sin limitación, peroxidasa de rábano, fosfatasa alcalina, beta-galactosidasa o acetilcolinesterasa; grupos prostéticos tales como por ejemplo, sin limitación, estreptavidina/biotina y avidina/biotina; materiales fluorescentes tales como por ejemplo, sin limitación, Alexa Fluor 350, Alexa Fluor 405, Alexa Fluor 430, Alexa Fluor 488, Alexa Fluor 500, Alexa Fluor 514, Alexa Fluor 532, Alexa Fluor 546, Alexa Fluor 555, Alexa Fluor 568, Alexa Fluor 594, Alexa Fluor 610, Alexa Fluor 633, Alexa Fluor 647, Alexa Fluor 660, Alexa Fluor 680, Alexa
Fluor 700, Alexa Fluor 750, umbeliferona, fluoresceína, isotiocianato de fluoresceína, rodamina, diclorotriazinilamina fluoresceína, cloruro de dansilo o ficoeritrina; materiales luminiscentes tales como por ejemplo, sin limitación, luminol; materiales bioluminiscentes tales como por ejemplo, sin limitación, luciferasa, luciferina y aequorina; materiales radioactivos tales como por ejemplo, sin limitación, yodo (131I, 125I, 123I y 121I), carbono (14C), azufre (35S), tritio (3H), indio (115In, 113In, 112In y 111In), tecnecio (99Tc), talio (201Tl), galio (68Ga, 67Ga), paladio (103Pd), molibdeno (99Mo), xenón (133Xe), flúor (18F), 153Sm, 177Lu, 159Gd, 149Pm, 140La, 175Yb, 166Ho, 90Y, 47Sc, 186Re, 188Re, 142 Pr, 105Rh, 97Ru, 68Ge, 57Co, 65Zn, 85Sr, 32P, 153Gd, 169Yb, 51Cr, 54Mn, 75Se, 64Cu, 113Sn y 117Sn; y metales que emiten positrones usando varias tomografías de emisión de positrones, y iones de metales paramagnéticos no radioactivos.
Los anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) o equivalentes funcionales de la presente divulgación también se pueden adherir a soportes sólidos, que son particularmente útiles para inmunoensayos o purificación del antígeno objetivo. Estos soportes sólidos incluyen, sin limitación, vidrio, celulosa, poliacrilamida, nylon, poliestireno, cloruro de polivinilo o polipropileno.
2. Enlazador
Como se usa en la presente, un “enlazador” es cualquier porción química que es capaz de enlazar un anticuerpo, fragmento de anticuerpo (por ejemplo, fragmentos de unión de antígeno) o equivalente funcional, con otra porción, tal como una porción de fármaco. Los enlazadores pueden ser susceptibles de escisión (enlazador escindible), tal como escisión inducida por ácido, escisión foto-inducida, escisión inducida por peptidasa, escisión inducida por esterasa, y escisión de enlace disulfuro, en condiciones en las que el compuesto o el anticuerpo permanece activo. Alternativamente, los enlazadores pueden ser sustancialmente resistentes a la escisión (por ejemplo, enlazador estable o enlazador no escindible). En algunos aspectos, el enlazador es un enlazador procargado, un enlazador hidrófilo, o un enlazador a base de ácido dicarboxílico.
En un aspecto, el enlazador usado se deriva de un reactivo de entrelazamiento tal como N-succinimidil-3-(2-piridilditio)propionato (SPDP), N-succinimidil-4-(2-piridilditio)pentanoato (SPP), N-succinimidil-4-(2-piridilditio)butanoato (SPDB), N-succinimidil-4-(2-piridilditio)-2-sulfo-butanoato (sulfo-SPDB), N-succinimidilyodoacetato (SIA), N-succinimidil(4-yodoacetil)aminobenzoato (SIAB), maleimida PEG NHS, N-succinimidil-4-(maleimidometil)ciclohexancarboxilato (SMCC), N-sulfosuccinimidil-4-(maleimidometil)ciclohexancarboxilato (sulfo-SMCC) o 2,5-dioxopirrolidin-1-il-17-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-5,8,11,14-tetraoxo-4,7,10,13-tetraazaheptadecan-1-oato (CX1-1). En otro aspecto, el enlazador usado se deriva de un agente de entrelazamiento tal como N-succinimidil-3-(2-piridilditio)propionato (SPDP), N-succinimidil-4-(maleimidometil)ciclohexancarboxilato (SMCC), N-sulfosuccinimidil-4-(maleimidometil)ciclohexancarboxilato (sulfo-SMCc), N-succinimidil-4-(2-piridilditio)-2-sulfo-butanoato (sulfo-SPDB) o 2,5-dioxopirrolidin-1-il-17-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-5,8,11,14-tetraoxo-4,7,10,13-tetraazaheptadecan-1-oato (CX1-1).
Los enlazadores no escindibles son cualquier porción química capaz de enlazar un fármaco, tal como un maitansinoide, a un anticuerpo, de una manera covalente, estable, y que no entran dentro de las categorías arriba mencionadas de enlazadores escindibles. De esta manera, los enlazadores no escindibles son sustancialmente resistentes a la escisión inducida por ácido, escisión foto-inducida, escisión inducida por peptidasa, escisión inducida por esterasa, y escisión de enlace disulfuro. Además, no escindible se refiere a la capacidad del enlace químico en el enlazador o adjunto al enlazador para resistir la escisión inducida por un ácido, agente de escisión fotolábil, una peptidasa, una esterasa, o un compuesto químico o fisiológico que escinde un enlace disulfuro, en condiciones en las cuales el fármaco, tal como maitansionoide o el anticuerpo, no pierde su actividad.
Los enlazadores lábiles al ácido son enlazadores escindibles a pH ácido. Por ejemplo, ciertos compartimentos intracelulares tales como los endosomas y lisosomas tienen un pH ácido (pH 4-5), y proveen las condiciones adecuadas para escindir los enlazadores lábiles al ácido.
Los enlazadores foto-lábiles son enlazadores que son útiles en la superficie del cuerpo y en muchas cavidades del cuerpo que son accesibles para la luz. Por otra parte, la luz infrarroja puede penetrar el tejido.
Algunos enlazadores pueden ser escindidos por peptidasas, es decir, enlazadores escindibles por peptidasa. Sólo ciertos péptidos se escinden fácilmente dentro o fuera de las células, por ejemplo, véase Trout et al., 79 Proc. Natl. Acad.Sci. USA, 626-629 (1982), y Umemoto et al. 43 Int. J. Cáncer, 677-684 (l989). Además, los péptidos están compuestos de a-aminoácidos y enlaces peptídicos, que son químicamente enlaces de amida entre el carboxilato de un aminoácido y el grupo amino de un segundo aminoácido. Se entiende que otros enlaces de amida, tales como el enlace entre un carboxilato y el grupo £-amino de la lisina, no son enlaces peptídicos y se consideran no escindibles.
Algunos enlazadores pueden ser escindidos por esterasas, es decir, enlazadores escindibles por esterasa. Una vez más, sólo ciertos ésteres pueden ser escindidos por esterasas presentes dentro o fuera de las células. Los ésteres se forman por la condensación de un ácido carboxílico y un alcohol. Los ésteres simples son ésteres producidos con alcoholes simples, tales como alcoholes alifáticos, y alcoholes aromáticos pequeños y cíclicos pequeños.
Los enlazadores procargados se derivan de reactivos de entrelazamiento cargados que conservan su carga después de su incorporación en un conjugado de anticuerpo-fármaco. Ejemplos de enlazadores procargados se pueden encontrar en el documento US 2009/0274713.
3. Conjugación y preparación de ADC
Los conjugados de la presente divulgación se pueden preparar mediante cualquiera de los métodos conocidos en la técnica, tales como los descritos en las patentes de EE. UU. Nos. 7.811.572, 6.411.163, 7.368.565 y 8.163.888, y las publicaciones de las solicitudes de EE. UU. 2011/0003969, 2011/0166319, 2012/0253021 y 2012/0259100. La totalidad de las enseñanzas de estas patentes y publicaciones de solicitudes de patente se incorporan aquí como referencia.
Proceso de un solo paso
En un aspecto, los conjugados de la presente divulgación se pueden preparar mediante un proceso de un solo paso. El proceso comprende la combinación del anticuerpo, el fármaco y el agente de entrelazamiento en un medio sustancialmente acuoso, que contiene opcionalmente uno o más codisolventes, a un pH adecuado. En un aspecto, el proceso comprende el paso de poner en contacto el anticuerpo de la presente divulgación con un fármaco (por ejemplo, DM1 o DM4) para formar una primera mezcla que comprende el anticuerpo y el fármaco, y luego poner en contacto la primera mezcla que comprende el anticuerpo y el fármaco con un agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1) en una solución que tiene un pH de aproximadamente 4 a aproximadamente 9, para proveer una mezcla que comprende (i) el conjugado (por ejemplo, Ab-MCC-DM1, Ab-SPDB-DM4 o Ab-CX1-1-DM1), (ii) fármaco libre (por ejemplo, DM1 o DM4), y (iii) subproductos de la reacción.
En un aspecto, el proceso de un paso comprende poner en contacto el anticuerpo con el fármaco (por ejemplo, DM1 o DM4) y luego el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1), en una solución que tiene un pH de aproximadamente 6 o mayor (por ejemplo, de aproximadamente 6 a aproximadamente 9, aproximadamente 6 a aproximadamente 7, aproximadamente 7 a aproximadamente 9, aproximadamente 7 a aproximadamente 8,5, aproximadamente 7,5 a aproximadamente 8,5, aproximadamente 7,5 a aproximadamente 8,0, aproximadamente 8,0 a aproximadamente 9,0, o aproximadamente 8,5 a aproximadamente 9,0), Por ejemplo, el proceso comprende poner en contacto un agente de unión a células con el fármaco (DM1 o DM4) y luego el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1) en una solución que tiene un pH de aproximadamente 6,0, aproximadamente 6,1, aproximadamente 6,2, aproximadamente 6,3, aproximadamente 6,4, aproximadamente 6,5, aproximadamente 6,6, aproximadamente 6,7, aproximadamente 6,8, aproximadamente 6,9, aproximadamente 7,0, aproximadamente 7,1, aproximadamente 7,2, aproximadamente 7,3, aproximadamente 7,4, aproximadamente 7,5, aproximadamente 7,6, aproximadamente 7,7, aproximadamente 7,8, aproximadamente 7,9, aproximadamente 8,0, aproximadamente 8,1, aproximadamente 8,2, aproximadamente 8,3, aproximadamente 8,4, aproximadamente 8,5, aproximadamente 8,6, aproximadamente 8,7, aproximadamente 8,8, aproximadamente 8,9, o aproximadamente 9,0. En otro aspecto, el proceso comprende poner en contacto un agente de unión a células con el fármaco (por ejemplo, DM1 o DM4) y luego el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1), en una solución que tiene un pH de aproximadamente 7,8 (por ejemplo, un pH de 7,6 a 8,0 o un pH de 7,7 a 7,9).
El proceso de un solo paso (es decir, el contacto del anticuerpo con el fármaco (por ejemplo, DM1 o DM4) y luego el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1) se puede llevar cabo a cualquier temperatura adecuada conocida en la técnica. Por ejemplo, el proceso de un solo paso puede ocurrir a aproximadamente 20 °C o menos (por ejemplo, de aproximadamente -10 °C (a condición de que se evite la congelación de la solución, por ejemplo, por la presencia del disolvente orgánico utilizado para disolver el agente citotóxico y el reactivo de entrelazamiento bifuncional) a aproximadamente 20 °C, de aproximadamente 0 °C a aproximadamente 18 °C, de aproximadamente 4 °C a aproximadamente 16 °C), a temperatura ambiente (por ejemplo, de aproximadamente 20 °C a aproximadamente 30 °C, o de aproximadamente 20 °C a aproximadamente 25 °C), o a una temperatura elevada (por ejemplo, de aproximadamente 30 °C a aproximadamente 37 °C). En un aspecto, el proceso de un solo paso se produce a una temperatura de aproximadamente 16 °C a aproximadamente 24 °C (por ejemplo, aproximadamente 16 °C, aproximadamente 17 °C, aproximadamente 18 °C, aproximadamente 19 °C, aproximadamente 20 °C, aproximadamente 21 °C, aproximadamente 22 °C, aproximadamente 23 °C, aproximadamente 24 °C, o aproximadamente 25 °C). En otro aspecto, el proceso de un solo paso se lleva a cabo a una temperatura de aproximadamente 15 °C o menor (por ejemplo,
de aproximadamente -10 °C a aproximadamente 15 °C, o de aproximadamente 0 °C a aproximadamente 15 °C). Por ejemplo, el proceso comprende poner en contacto el anticuerpo con el fármaco (por ejemplo, DM1 o DM4) y luego el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1) a una temperatura de aproximadamente 15 °C, aproximadamente 14 °C, aproximadamente 13 °C, aproximadamente 12 °C, aproximadamente 11 °C, aproximadamente 10 °C, aproximadamente 9 °C, aproximadamente 8 °C, aproximadamente 7 °C, aproximadamente 6 °C, aproximadamente 5 °C, aproximadamente 4 °C, aproximadamente 3 °C, aproximadamente 2 °C, aproximadamente 1 °C, aproximadamente 0 °C, aproximadamente -1 °C, aproximadamente -2 °C, aproximadamente -3 °C, aproximadamente -4 °C, aproximadamente -5 °C, aproximadamente -6 °C, aproximadamente -7 °C, aproximadamente -8 °C, aproximadamente -9 °C, o aproximadamente -10 °C, a condición de que se evite la congelación de la solución, por ejemplo, por la presencia de los disolventes orgánicos utilizados para disolver el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, sulfo-SPDB SPDB, o CX1-1). En un aspecto, el proceso comprende poner en contacto el anticuerpo con el fármaco (por ejemplo, DM1 o DM4) y luego el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o c X1-1) a una temperatura de aproximadamente -10 °C a aproximadamente 15 °C, de aproximadamente 0 °C a aproximadamente 15 °C, de aproximadamente 0 °C a aproximadamente 10 °C, de aproximadamente 0 °C a aproximadamente 5 °C, de aproximadamente 5 °C a aproximadamente 15 °C, de aproximadamente 10 °C a aproximadamente 15 °C, o de aproximadamente 5 °C a aproximadamente 10 °C. En otro aspecto, el proceso comprende poner en contacto el anticuerpo con el fármaco (por ejemplo, DM1 o DM4) y luego el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1) a una temperatura de aproximadamente 10 °C (por ejemplo, una temperatura de 8 °C a 12 °C o una temperatura de 9 °C a 11 °C).
En un aspecto, el contacto descrito anteriormente se efectúa proporcionando el anticuerpo, después poniendo en contacto el anticuerpo con el fármaco (por ejemplo, DM1 o DM4) para formar una primera mezcla que comprende el anticuerpo y el fármaco (por ejemplo, DM1 o DM4), y luego poner en contacto la primera mezcla que comprende el anticuerpo y el fármaco (por ejemplo, DM1 o DM4) con el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1). Por ejemplo, en un aspecto, el anticuerpo se provee en un recipiente de reacción, el fármaco (por ejemplo, DM1 o DM4) se añade al recipiente de reacción (poniéndolo en contacto así con el anticuerpo), y luego el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1) se añade a la mezcla que comprende el anticuerpo y el fármaco (por ejemplo, DM1 o DM4) (poniéndolo en contacto así con la mezcla que comprende el anticuerpo y el fármaco). En un aspecto, el anticuerpo se provee en un recipiente de reacción, y el fármaco (por ejemplo, DM1 o DM4) se añade al recipiente de reacción inmediatamente después de proveer el anticuerpo al recipiente. En otro aspecto, el anticuerpo se provee en un recipiente de reacción, y el fármaco (por ejemplo, DM1 o DM4) se añade al recipiente de reacción después de un intervalo de tiempo, después suministrando el anticuerpo al recipiente (por ejemplo, aproximadamente 5 minutos, aproximadamente 10 minutos, aproximadamente 20 minutos, aproximadamente 30 minutos, aproximadamente 40 minutos, aproximadamente 50 minutos, aproximadamente 1 hora, aproximadamente 1 día, o más, después de proveer el agente de unión celular al espacio). El fármaco (por ejemplo, DM1 o DM4) puede añadirse rápidamente (es decir, dentro de un intervalo de tiempo corto, tal como aproximadamente 5 minutos, aproximadamente 10 minutos) o lentamente (por ejemplo, mediante el uso de una bomba).
La mezcla que comprende el anticuerpo y el fármaco (por ejemplo, DM1 o DM4) puede entonces ponerse en contacto con el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1), ya sea inmediatamente después de poner en contacto con el anticuerpo con el fármaco (por ejemplo, DM1 o DM4), o en algún momento posterior (por ejemplo, aproximadamente 5 minutos a aproximadamente 8 horas o más) después de poner en contacto el anticuerpo con el fármaco (por ejemplo, DM1 o DM4). Por ejemplo, en un aspecto, el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1) se añade a la mezcla que comprende el anticuerpo y el fármaco (por ejemplo, DM1 o DM4) inmediatamente después de la adición del fármaco (por ejemplo, DM1 o DM4) al recipiente de reacción que comprende el anticuerpo. Alternativamente, la mezcla que comprende el anticuerpo y el fármaco (por ejemplo, DM1 o DM4) puede ponerse en contacto con el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1) en aproximadamente 5 minutos, aproximadamente 10 minutos, aproximadamente 20 minutos, aproximadamente 30 minutos, aproximadamente 1 hora, aproximadamente 2 horas, aproximadamente 3 horas, aproximadamente 4 horas, aproximadamente 5 horas, aproximadamente 6 horas, aproximadamente 7 horas, aproximadamente 8 horas o más, después de poner en contacto el anticuerpo con el fármaco (por ejemplo, DM1 o DM4).
Después de que la mezcla que comprende el anticuerpo y el fármaco (por ejemplo, DM1 o DM4) se pone en contacto con el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1), la reacción se deja proceder durante aproximadamente 1 hora, aproximadamente 2 horas, aproximadamente 3 horas, aproximadamente 4 horas, aproximadamente 5 horas, aproximadamente 6 horas, aproximadamente 7 horas, aproximadamente 8 horas, aproximadamente 9 horas, aproximadamente 10 horas, aproximadamente 11 horas, aproximadamente 12 horas, aproximadamente 13 horas, aproximadamente 14 horas, aproximadamente 15 horas, aproximadamente 16 horas, aproximadamente 17 horas, aproximadamente 18 horas, aproximadamente 19 horas, aproximadamente 20 horas,
aproximadamente 21 horas, aproximadamente 22 horas, aproximadamente 23 horas, aproximadamente 24 horas, o más (por ejemplo, aproximadamente 30 horas, aproximadamente 35 horas, aproximadamente 40 horas, aproximadamente 45 horas, o aproximadamente 48 h).
En un aspecto, el proceso de un solo paso comprende además un paso de apagado para apagar cualquier fármaco que no ha reaccionado (por ejemplo, DM1 o DM4) o agente de entrelazamiento que no ha reaccionado (por ejemplo, SMCc , sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1). El paso de apagado se realiza típicamente antes de la purificación del conjugado. En un aspecto, la mezcla se apaga poniendo en contacto la mezcla con un reactivo de apagado. Tal como se usa en la presente, el "reactivo de apagado" se refiere a un reactivo que reacciona con el fármaco (por ejemplo, DM1 o DM4) o agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1) libres. En un aspecto, se pueden usar reactivos de apagado de maleimida o haloacetamida, tales como ácido 4-maleimidobutírico, ácido 3 -maleimidopropiónico, N-etil-maleimida, yodoacetamida, o ácido yodoacetamidopropiónico, para asegurar que cualquier grupo que no ha reaccionado (por ejemplo, tiol) en el fármaco (por ejemplo, DM1 o DM4) sea apagado. El paso de apagado puede ayudar a prevenir la dimerización del fármaco (por ejemplo, DM1). El DM1 dimerizado puede ser difícil de separar. Tras el apagado con reactivos cargados polares de apagado de tiol (tales como ácido 4 - maleimidobutírico o ácido 3 -maleimidopropiónico), el exceso, DM1 que no ha reaccionado, es convertido en un aducto polar cargado soluble en agua, que se puede separar fácilmente del conjugado enlazado covalentemente durante el paso de purificación. También se puede utilizar apagado con reactivos de apagado de tiol no polares y neutros. En un aspecto, la mezcla se apaga poniendo en contacto la mezcla con un reactivo de apagado que reacciona con el agente de entrelazamiento que no ha reaccionado (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1). Por ejemplo, se pueden añadir a la mezcla nucleófilos para apagar cualquier SMCC que no ha reaccionado. El nucleófilo es preferiblemente un nucleófilo que contiene un grupo amino, tal como lisina, taurina e hidroxilamina.
En otro aspecto, la reacción (es decir, el contacto del anticuerpo con el fármaco (por ejemplo, DM1 o DM4) y luego el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1)) se deja para proceder hasta la terminación antes de poner en contacto la mezcla con un reactivo de apagado. A este respecto, el reactivo de apagado se añade a la mezcla en aproximadamente 1 hora a aproximadamente 48 horas (por ejemplo, aproximadamente 1 hora, aproximadamente 2 horas, aproximadamente 3 horas, aproximadamente 4 horas, aproximadamente 5 horas, aproximadamente 6 horas, aproximadamente 7 horas, aproximadamente 8 horas, aproximadamente 9 horas, aproximadamente 10 horas, aproximadamente 11 horas, aproximadamente 12 horas, aproximadamente 13 horas, aproximadamente 14 horas, aproximadamente 15 horas, aproximadamente 16 horas, aproximadamente 17 horas, aproximadamente 18 horas, aproximadamente 19 horas, aproximadamente 20 horas, aproximadamente 21 horas, aproximadamente 22 horas, aproximadamente 23 horas, aproximadamente 24 horas, o de aproximadamente 25 horas a aproximadamente 48 horas) después de que la mezcla que comprende el anticuerpo y el fármaco (por ejemplo, DM1 o DM4) se pone en contacto con el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1).
Alternativamente, la mezcla se apaga reduciendo el pH de la mezcla a aproximadamente 5,0 (por ejemplo, 4,8, 4,9, 5,0, 5,1 o 5,2). En otro aspecto, la mezcla se apaga reduciendo el pH a menos de 6,0, menos de 5,5, menos de 5,0, menos de 4,8, menos de 4,6, menos de 4,4, menos de 4,2, menos de 4,0. Alternativamente, el pH se reduce de aproximadamente 4.0 (por ejemplo, 3,8, 3,9, 4,0, 4,1 o 4,2) a aproximadamente 6,0 (por ejemplo, 5,8, 5,9, 6,0, 6,1 o 6,2), de aproximadamente 4,0 a aproximadamente 5,0, de aproximadamente 4,5 (por ejemplo, 4,3, 4,4, 4,5, 4,6 o 4,7) a aproximadamente 5,0, En un aspecto, la mezcla se apaga reduciendo el pH de la mezcla a 4,8. En otro aspecto, la mezcla se apaga reduciendo el pH de la mezcla a 5,5.
En un aspecto, el proceso de un solo paso comprende además un paso de retención para liberar del anticuerpo los enlazadores unidos de forma inestable. El paso de retención comprende retener la mezcla antes de la purificación del conjugado (por ejemplo, después del paso de reacción, entre el paso de reacción y el paso de apagado, o después del paso de apagado). Por ejemplo, el proceso comprende (a) poner en contacto el anticuerpo con el fármaco (por ejemplo, DM1 o DM4) para formar una mezcla que comprende el anticuerpo y el fármaco (por ejemplo, DM1 o DM4); y luego poner en contacto la mezcla que comprende el anticuerpo y fármaco (por ejemplo, DM1 o DM4) con el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1), en una solución que tiene un pH de aproximadamente 4 a aproximadamente 9, para proveer una mezcla que comprende (i) el conjugado (por ejemplo, Ab-MCC-DM1, Ab-SPDB-DM4 o Ab-CX1-1-DM1), (ii) el fármaco libre (por ejemplo, DM1 o DM4 ), y (iii) subproductos de la reacción; (b) retener la mezcla preparada en el paso (a) para liberar los enlazadores unidos de forma inestable del agente de unión a células, y (c) purificar la mezcla para proveer un conjugado purificado.
En otro aspecto, el proceso comprende (a) poner en contacto el anticuerpo con el fármaco (por ejemplo, DM1 o DM4) para formar una mezcla que comprende el anticuerpo y el fármaco (por ejemplo, DM1 o DM4); y luego poner en contacto la mezcla que comprende el anticuerpo y el fármaco (por ejemplo, DM1 o DM4) con el agente de entrelazamiento (por
ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1), en una solución que tiene un pH de aproximadamente 4 a aproximadamente 9, para proveer una mezcla que comprende (i) el conjugado, (ii) el fármaco libre (por ejemplo, DM1 o DM4), y (iii) subproductos de la reacción; (b) apagar la mezcla preparada en el paso (a) para apagar cualquier fármaco que no ha reaccionado (por ejemplo, DM1 o DM4) o agente de entrelazamiento que no ha reaccionado (por ejemplo, SMCC, sulfo-SMCC, SpDB, sulfo-SPDB o CX1-1); (c) retener la mezcla preparada en el paso (b) para liberar los enlazadores unidos de forma inestable desde el agente de unión a células; y (d) purificar la mezcla para proveer un conjugado purificado (por ejemplo, Ab-MCC-DM1, Ab-SPDB-DM4 o Ab-CX1-1-DM1).
Alternativamente, el paso de retención se puede realizar después de la purificación del conjugado, seguido por un paso de purificación adicional.
En otro aspecto se deja que la reacción prosiga hasta su finalización antes del paso de retención. En este sentido, el paso de retención se puede realizar en aproximadamente 1 hora a aproximadamente 48 horas (por ejemplo, aproximadamente 1 hora, aproximadamente 2 horas, aproximadamente 3 horas, aproximadamente 4 horas, aproximadamente 5 horas, aproximadamente 6 horas, aproximadamente 7 horas, aproximadamente 8 horas, aproximadamente 9 horas, aproximadamente 10 horas, aproximadamente 11 horas, aproximadamente 12 horas, aproximadamente 13 horas, aproximadamente 14 horas, aproximadamente 15 horas, aproximadamente 16 horas, aproximadamente 17 horas, aproximadamente 18 horas, aproximadamente 19 horas, aproximadamente 20 horas, aproximadamente 21 horas, aproximadamente 22 horas, aproximadamente 23 horas, aproximadamente 24 horas, o de aproximadamente 24 horas a aproximadamente 48 horas) después de que la mezcla que comprende el anticuerpo y el fármaco (por ejemplo, DM1 o DM4) se pone en contacto con el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1).
El paso de retención comprende mantener la solución a una temperatura adecuada (por ejemplo, de aproximadamente 0 °C a aproximadamente 37 °C) durante un periodo adecuado (por ejemplo, aproximadamente 1 hora a aproximadamente 1 semana, de aproximadamente 1 hora a aproximadamente 24 horas, de aproximadamente 1 hora a aproximadamente 8 horas, o de aproximadamente 1 hora a aproximadamente 4 horas) para liberar los enlazadores unidos de forma inestable del anticuerpo pero sin liberar sustancialmente los enlazadores unidos establemente del anticuerpo. En un aspecto, el paso de retención comprende mantener la solución a aproximadamente 20 °C o menos (por ejemplo, de aproximadamente 0 °C a aproximadamente 18 °C, de aproximadamente 4 °C a aproximadamente 16 °C), a temperatura ambiente (por ejemplo, de aproximadamente 20 °C a aproximadamente 30 °C, o de aproximadamente 20 °C a aproximadamente 25 °C), 0 a una temperatura elevada (por ejemplo, de aproximadamente 30 °C a aproximadamente 37 °C). En un aspecto, el paso de retención comprende mantener la solución a una temperatura de aproximadamente 16 °C a aproximadamente 24 °C (por ejemplo, aproximadamente 15 °C, aproximadamente 16 °C, aproximadamente 17 °C, aproximadamente 18 °C, aproximadamente 19 °C, aproximadamente 20 °C, aproximadamente 21 °C, aproximadamente 22 °C, aproximadamente 23 °C, aproximadamente 24 °C, o aproximadamente 25 °C). En otro aspecto, el paso de retención comprende mantener la solución a una temperatura de aproximadamente 2 °C a aproximadamente 8 °C (por ejemplo, aproximadamente 0 °C, aproximadamente 1 °C, aproximadamente 2 °C, aproximadamente 3 °C, aproximadamente 4 °C, aproximadamente 5 °C, aproximadamente 6 °C, aproximadamente 7 °C, aproximadamente 8 °C, aproximadamente 9 °C, o aproximadamente 10 °C). En otro aspecto, el paso de retención comprende mantener la solución a una temperatura de aproximadamente 37 °C (por ejemplo, aproximadamente 34 °C, aproximadamente 35 °C, aproximadamente 36 °C, aproximadamente 37 °C, aproximadamente 38 °C, aproximadamente 39 °C, o aproximadamente 40 °C).
La duración del paso de retención depende de la temperatura y el pH al que se efectúa el paso de retención. Por ejemplo, la duración del paso de retención se puede reducir sustancialmente efectuando el paso de retención a temperatura elevada; la temperatura máxima será limitada por la estabilidad del conjugado de agente de unión a célula-agente citotóxico. El paso de retención puede comprender mantener la solución por aproximadamente 1 hora a aproximadamente 1 día (por ejemplo, aproximadamente 1 hora, aproximadamente 2 horas, aproximadamente 3 horas, aproximadamente 4 horas, aproximadamente 5 horas, aproximadamente 6 horas, aproximadamente 7 horas, aproximadamente 8 horas, aproximadamente 9 horas, aproximadamente 10 horas, aproximadamente 12 horas, aproximadamente 14 horas, aproximadamente 16 horas, aproximadamente 18 horas, aproximadamente 20 horas, aproximadamente 22 horas, o aproximadamente 24 horas), de aproximadamente 10 horas a aproximadamente 24 horas, de aproximadamente 12 horas a aproximadamente 24 horas, de aproximadamente 14 horas a aproximadamente 24 horas, de aproximadamente 16 horas a aproximadamente 24 horas, de aproximadamente 18 horas a aproximadamente 24 horas, de aproximadamente 20 horas a aproximadamente 24 horas, de aproximadamente 5 horas a aproximadamente 1 semana, de aproximadamente 20 horas a aproximadamente 1 semana, de aproximadamente 12 horas a aproximadamente 1 semana (por ejemplo, aproximadamente 12 horas, aproximadamente 16 horas, aproximadamente 20 horas, aproximadamente 24 horas, aproximadamente 2 días, aproximadamente 3 días, aproximadamente 4 días, aproximadamente 5 días, aproximadamente 6 días, o aproximadamente 7 días), o de aproximadamente 1 día a aproximadamente 1 semana.
En un aspecto, el paso de retención comprende mantener la solución a una temperatura de aproximadamente 2 °C a aproximadamente 8 °C durante un periodo de por lo menos aproximadamente 12 horas hasta una semana. En otro aspecto, el paso de retención comprende mantener la solución a una temperatura de aproximadamente 2 °C a aproximadamente 8 °C durante la noche (por ejemplo, de aproximadamente 12 a aproximadamente 24 horas, de preferencia aproximadamente 20 horas).
El valor de pH para el paso de retención es, de preferencia, de aproximadamente 4 a aproximadamente 10. En un aspecto, el valor de pH para el paso de retención es aproximadamente 4 o mayor, pero menor de aproximadamente 6 (por ejemplo, de 4 a 5,9) o aproximadamente 5 o mayor, pero menor de aproximadamente 6 (por ejemplo, de 5 a 5,9). En otro aspecto, los valores de pH para el paso de retención varían de aproximadamente 6 a aproximadamente 10 (por ejemplo, de aproximadamente 6,5 a aproximadamente 9, de aproximadamente 6 a aproximadamente 8). Por ejemplo, los valores de pH para el paso de retención pueden ser de aproximadamente 6, aproximadamente 6,5, aproximadamente 7, aproximadamente 7,5, aproximadamente 8, aproximadamente 8,5, aproximadamente 9, aproximadamente 9,5, o aproximadamente 10.
En otros aspectos, el paso de retención puede comprender incubar la mezcla a 25 °C a un pH de aproximadamente 6 7,5 durante aproximadamente 12 horas a aproximadamente 1 semana; incubar la mezcla a 4 °C a un pH de aproximadamente 4.5-5,9 durante aproximadamente 5 horas a aproximadamente 5 días; o incubar la mezcla a 25 °C a un pH de aproximadamente 4.5-5,9 durante aproximadamente 5 horas a aproximadamente 1 día.
El proceso de un solo paso puede incluir, opcionalmente, la adición de sacarosa al paso de reacción para aumentar la solubilidad y la recuperación de los conjugados. De manera deseable, la sacarosa se añade a una concentración de aproximadamente 0,1% (p/v) a aproximadamente 20% (p/v) (por ejemplo, aproximadamente 0,1% (p/v), 1% (p/v), 5% (p/v), 10% (p/v), 15% (p/v), o 20% (p/v)). Preferiblemente, la sacarosa se agrega a una concentración de aproximadamente 1% (p/v) a aproximadamente 10% (p/v) (por ejemplo, aproximadamente 0,5% (p/v), aproximadamente 1% (p/v), aproximadamente 1,5% (p/v), aproximadamente 2% (p/v), aproximadamente 3% (p/v), aproximadamente 4% (p/v), aproximadamente 5% (p/v), aproximadamente 6% (p/v), aproximadamente 7% (p/v), aproximadamente 8% (p/v), aproximadamente 9% (p/v), aproximadamente 10% (p/v), o aproximadamente 11% (p/v)). Además, el paso de reacción puede comprender también la adición de un agente amortiguador. Cualquier agente amortiguador adecuado conocido en la técnica se puede utilizar. Agentes amortiguadores adecuados incluyen, por ejemplo, un amortiguador de citrato, un amortiguador de acetato, un amortiguador de succinato y un amortiguador de fosfato. En un aspecto, el agente amortiguador se selecciona del grupo que consiste en HEPPSO (ácido N-(2-hidroxietil)piperazin-N'-(2-hidroxipropansulfónico)), POPSO (ácido piperazin-1,4-bis-(2-hidroxi-propan-sulfónico) deshidratado), HEPES (ácido 4-(2-hidroxietil)piperazin-1-etansulfónico), HEPPS (EPPS) (ácido 4-(2-hidroxietil)piperazin-1-propansulfónico), TES (ácido N-[tris(hidroximetil)metil]-2-aminoetansulfónico), y una combinación de los mismos.
El proceso de un solo paso puede comprender además el paso de purificar la mezcla para proveer el conjugado purificado (por ejemplo, Ab-MCC-DM1, Ab-SPDB-DM4 o Ab-CX1-1-DM1). Se puede usar cualquier método de purificación conocido en la técnica para purificar los conjugados de la presente divulgación. En un aspecto, los conjugados de la presente divulgación utilizan filtración de flujo tangencial (TFF), cromatografía no adsorbente, cromatografía de adsorción, filtración de adsorción, precipitación selectiva, o cualquier otro proceso de purificación adecuado, así como combinaciones de los mismos. En otro aspecto, antes de someter los conjugados al proceso de purificación descrito anteriormente, los conjugados se filtran primero a través de una o más membranas de PVDF. Alternativamente, los conjugados se filtran a través de una o más membranas de PVDF después de someter los conjugados al proceso de purificación descrito anteriormente. Por ejemplo, en un aspecto, los conjugados se filtran a través de una o más membranas de PVDF y después se purifican usando filtración de flujo tangencial. Alternativamente, los conjugados se purifican usando filtración de flujo tangencial y luego se filtran a través de una o más membranas de PVDF.
Para la purificación se puede utilizar cualquier sistema de TFF adecuado, incluyendo: un sistema de tipo Pellicon® (Millipore, Billerica, MA), un sistema Sartocon® Cassette (Sartorius AG, Edgewood, NY), y un sistema de tipo Centrasette® (Pall Corp., East Hills, NY).
Para la purificación se puede utilizar cualquier resina de cromatografía de adsorción adecuada. Resinas de cromatografía de adsorción preferidas incluyen cromatografía de hidroxiapatita, cromatografía de inducción de carga hidrófoba (HCIC), cromatografía de interacción hidrófoba (HIC), cromatografía de intercambio iónico, cromatografía de intercambio iónico de modo mixto, cromatografía de afinidad con metal inmovilizado (IMAC), cromatografía de ligando de colorante, cromatografía de afinidad, cromatografía de fase inversa, y combinaciones de las mismas. Ejemplos de resinas de hidroxiapatita adecuadas incluyen hidroxiapatita de cerámica (CHT Tipo I y Tipo II, Bio-Rad Laboratories, Hercules, CA), hidroxiapatita HA Ultrogel® (Pall Corp., East Hills, NY), y fluoroapatita de cerámica (CFT Tipo I y Tipo II, Bio-Rad Laboratories, Hercules, CA). Un ejemplo de una resina HCIC adecuada es la resina MEP Hypercel® (Pall Corp., East
Hills, NY). Ejemplos de resinas HIC adecuadas incluyen las resinas Butyl-Sepharose, Hexyl-Sepaharose, Phenyl-Sepharose y Octyl Sepharose (todas de GE Healthcare, Piscataway, NJ), y también las resinas Macro-prep® Metil y Macro-Prep® t-Butyl (Biorad Laboratories, Hercules, CA). Ejemplos de resinas de intercambio iónico adecuadas incluyen las resinas SP-Sepharose®, CM-Sepharose®, y Q-Sepharose® (todas de GE Healthcare, Piscataway, NJ), y la resina Unosphere® S (Bio-Rad Laboratories, Hercules, CA). Ejemplos de intercambioderes iónicos de modo mixto adecuados incluyen la resina Bakerbond® ABx (JT Baker, Phillipsburg NJ). Ejemplos de resinas IMAC adecuadas incluyen la resina Chelating Sepharose ® (GE Healthcare, Piscataway, NJ) y la resina Profinity® IMAC (Bio-Rad Laboratories, Hercules, CA). Ejemplos de resinas de ligando de colorante adecuadas incluyen la resina Blue Sepharose (GE Healthcare, Piscataway, NJ) y la resina Affi-gel Blue (Bio-Rad Laboratories, Hercules, CA). Ejemplos de resinas de afinidad adecuadas incluyen la resina Protein A Sepharose (por ejemplo, MabSelect, GE Healthcare, Piscataway, NJ) y las resinas de afinidad de lectina, por ejemplo la resina Lentil Lectin Sepharose® (GE Healthcare, Piscataway, NJ), en donde el anticuerpo lleva sitios de unión de lectina apropiados. Ejemplos de resinas de fase inversa adecuadas incluyen las resinas C4, C8 y C18 (Grace Vydac, Hesperia, CA).
Para la purificación se puede utilizar cualquier resina de cromatografía no adsorbente adecuada. Ejemplos de resinas de cromatografía no adsorbente adecuadas incluyen, pero no se limitan a: SEPHADEX™ G-25, G-50, G-100, resinas SEPHACRYL™ (por ejemplo, S-200 y S-300), resinas SUPERDEX™ (por ejemplo, SUPERDEX™ 75 y SUPERDEX™ 200), resinas BIO-GEL® (por ejemplo, P-6, P-10, P-30, P-60, y P-100), y otras conocidas por los expertos en la técnica.
Proceso de dos pasos y proceso en una marmita
En un aspecto, los conjugados de la presente divulgación se pueden preparar como se describe en la patente de EE. UU.
7.811.572 y la publicación de la solicitud de patente de EE. Uu . No. 2006/0182750. El proceso comprende las pasos de: (a) poner en contacto el anticuerpo de la presente divulgación con el agente de entrelazamiento (por ejemplo, SMCC, sulfo-SMCC, SPDB, sulfo-SPDB o CX1-1) para unir covalentemente el enlazador (es decir, Ab-SMCC, Ab-SPDB o Ab-CX1-1) al anticuerpo y de este modo preparar una primera mezcla que comprende el anticuerpo que tiene el enlazador unido al mismo; (b) opcionalmente someter la primera mezcla a un proceso de purificación para preparar una primera mezcla purificada del anticuerpo que tiene el enlazador unido al mismo; (c) conjugar el fármaco (por ejemplo, DM1 o DM4) con el anticuerpo que tiene el enlazador unido al mismo en la primera mezcla haciendo reaccionar el anticuerpo que tiene el enlazador unido al mismo con el fármaco (por ejemplo, DM1 o DM4) en una solución que tiene un pH de aproximadamente 4 a aproximadamente 9, para preparar una segunda mezcla que comprende (i) el conjugado (por ejemplo, Ab-MCC-DM1, Ab-SPDB-DM4 o Ab-CX1-1-DM1), (ii) el fármaco libre (por ejemplo, DM1 o DM4), y (iii) subproductos de la reacción; y (d) someter la segunda mezcla a un proceso de purificación para purificar el conjugado de los otros componentes de la segunda mezcla. Alternativamente, el paso de purificación (b) puede omitirse. Cualquier método de purificación descrito en la presente puede ser usado para los pasos (b) y (d). En una modalidad, se usa TFF para ambos pasos (b) y (d). En otra modalidad se usa TFF para el paso (b) y cromatografía de absorción (por ejemplo, CHT) para el paso (d).
Reactivo de un paso y proceso in situ
En un aspecto, los conjugados de la presente divulgación se pueden preparar conjugando el compuesto fármacoenlazador preformado (por ejemplo, SMCC-DM1, Sulfo-SMCC-DM1, SPDB-DM4 o CX1-1-DM1) con el anticuerpo de la presente divulgación, como se describe en la patente de EE. UU. 6.441.163 y las publicaciones de solicitud de patente de EE. UU. Nos. 2011/0003969 y 2008/0145374, seguido por un paso de purificación. Se puede usar cualquier método de purificación descrito en la presente. El compuesto fármaco-enlazador se prepara haciendo reaccionar el fármaco (por ejemplo, DM1 o DM4) con el agente de entrelazamiento (por ejemplo, SMCC, Sulfo-SMCC, SPDB, Sulfo-SPDB o c X1-1). El compuesto fármaco-enlazador (por ejemplo, SMCC-DM1, Sulfo-SMCC-DM1, SPDB-DM4 o CX1-1-DM1) opcionalmente se somete a purificación antes de ser conjugado con el anticuerpo.
4. Caracterización y selección de anticuerpos deseables y conjugados anticuerpo-fármaco
Los anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmentos de unión a antígeno) o conjugados de anticuerpofármaco de la presente divulgación pueden ser caracterizados y seleccionados por sus propiedades físicas/químicas o sus actividades biológicas mediante varios ensayos conocidos en la técnica.
Por ejemplo, la actividad de unión de un anticuerpo de la presente divulgación a su antígeno se puede probar por medio de los métodos conocidos, tales como ELISA, FACS, Biacore o Western blot.
Los animales transgénicos y las líneas celulares son particularmente útiles para tamizar los conjugados de anticuerpofármaco (ADC) que tienen potencial como tratamientos profilácticos o terapéuticos de la sobreexpresión de antígenos
asociados a tumores y los receptores de superficie celular del cáncer. El tamizado de un ADC útil puede implicar la administración de un ADC candidato en una escala de dosis al animal transgénico, y ensayar en varios puntos de tiempo los efectos del ADC sobre la enfermedad o trastorno que está siendo evaluado. Alternativamente, o adicionalmente, el fármaco puede administrarse antes de o simultáneamente con la exposición a un inductor de la enfermedad, si es aplicable. El ADC candidato puede ser tamizado en serie y de forma individual, o en paralelo bajo un formato de tamizado de medio o alto rendimiento.
Un aspecto es un método de tamizado que comprende: (a) trasplantar células de una línea celular de cáncer estable o tumor de paciente humano que expresa cKIT (por ejemplo, una línea de células o fragmento de tumor GIST, una línea celular o fragmento de tumor de melanoma, células de AML primarias) a un animal no humano; (b) administrar un ADC fármaco candidato al animal no humano; y (c) determinar la capacidad del candidato para inhibir el crecimiento de tumores de la línea celular trasplantada. La presente divulgación también abarca un método de tamizado de candidatos de ADC para el tratamiento de una enfermedad o trastorno caracterizado por la sobreexpresión de cKit, que comprende (a) poner en contacto células de una línea celular de cáncer estable que expresa cKit, con un fármaco candidato; y (b) evaluar la capacidad del ADC candidato para inhibir el crecimiento de la línea celular estable.
Otro aspecto es un método de tamizado que comprende (a) poner en contacto células de una línea celular de cáncer estable que expresa cKit, con un ADC fármaco candidato; y (b) evaluar la capacidad del ADC candidato para bloquear la activación de ligando de cKit. En otro aspecto se evalúa la capacidad del ADC candidato para bloquear la fosforilación de tirosina estimulada por ligando.
Un aspecto adicional es un método de tamizado que comprende (a) poner en contacto células de una línea celular de cáncer estable que expresa cKit con un ADC fármaco candidato; y (b) evaluar la capacidad del ADC candidato para inducir la muerte celular. En un aspecto se evalúa la capacidad del ADC candidato para inducir la apoptosis.
El ADC candidato puede ser tamizado al ser administrado al animal transgénico en una escala de dosis, y evaluar la respuesta fisiológica del animal a los compuestos con el tiempo. En algunos casos, puede ser apropiado administrar el compuesto conjuntamente con cofactores que aumentarían la eficacia del compuesto. Si las líneas celulares derivadas de los animales transgénicos sujeto se utilizan para tamizar ADC útiles en el tratamiento de diversos trastornos asociados con la sobreexpresión de cKit, los ADC de prueba se añaden al medio de cultivo celular en un momento apropiado, y se evalúa la respuesta celular a los ADC con el tiempo utilizando los ensayos bioquímicos o histológicos apropiados.
De esta manera, la presente divulgación provee ensayos para la identificación de ADC que hacen blanco y se unen específicamente a cKit, y la sobreexpresión de cKit en las células tumorales.
Anticuerpos de cKIT
La presente divulgación provee anticuerpos o fragmentos de anticuerpo (por ejemplo, fragmentos de unión a antígeno) que se unen específicamente a cKit humano. Los anticuerpos o fragmentos de anticuerpo (por ejemplo, fragmentos de unión a antígeno) de la presente divulgación incluyen, pero sin limitación, los anticuerpos monoclonales humanos o fragmentos de los mismos, aislados como se describe en los ejemplos más abajo.
La presente divulgación, en ciertos aspectos, provee anticuerpos o fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) que se unen específicamente a cKit, dichos anticuerpos o fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) comprenden un dominio VH que tiene una secuencia de aminoácidos de SEQ ID NO: 9, 28, 46, 64, 82, 100, 118 o 136 (tabla 1). La presente divulgación también provee anticuerpos o fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) que se unen específicamente a cKit, dichos anticuerpos o fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) comprenden una CDR de VH que tiene una secuencia de aminoácidos de cualquiera de las CDR de VH mostradas en la tabla 1. En aspectos particulares, la presente divulgación provee anticuerpos o fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) que se unen específicamente a cKit, dichos anticuerpos comprendiendo (o, alternativamente, consisten en) uno, dos, tres, cuatro, cinco o más CDR de VH que tienen una secuencia de aminoácidos de cualquiera de las CDR de VH mostradas en la tabla 1, más abajo.
La presente divulgación provee anticuerpos o fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) que se unen específicamente a cKit, dichos anticuerpos o fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) comprenden un dominio VL que tiene una secuencia de aminoácidos de SEQ ID n O: 18, 37, 55, 73, 91, 109, 127 o 145 (tabla 1). La presente divulgación también provee anticuerpos o fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) que se unen específicamente a cKit, dichos anticuerpos o fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) comprenden una CDR de VL que tiene una secuencia de aminoácidos
de cualquiera de las CDR de VL mostradas en la Tabla 1, más abajo. En particular, la divulgación provee anticuerpos o fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) que se unen específicamente a cKit, dichos anticuerpos o fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) comprenden (o alternativamente, consisten en) uno, dos, tres o más CDR de VL que tienen una secuencia de aminoácidos de cualquiera de las CDR de VL mostradas en la tabla 1.
Otros anticuerpos o fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) de la presente divulgación incluyen aminoácidos que han sido mutados, sin embargo, tienen por lo menos 60, 70, 80, 90 o 95 por ciento de identidad en las regiones CDR con las regiones CDR representadas en las secuencias descritas en la tabla 1. En algunos aspectos incluye secuencias de aminoácidos mutantes en donde no más de 1, 2, 3, 4 o 5 aminoácidos han sido mutados en las regiones CDR en comparación con las regiones CDR representadas en la secuencia descrita en la tabla 1.
La presente divulgación también provee secuencias de ácidos nucleicos que codifican VH, VL, la cadena pesada de longitud completa y la cadena ligera de longitud completa de los anticuerpos que se unen específicamente a cKit. Tales secuencias de ácido nucleico pueden ser optimizadas para su expresión en células de mamífero.
Tabla 1: Ejemplos de anticuerpos anti-cKIT

















Otros anticuerpos de la presente divulgación incluyen aquellos en los que los aminoácidos o los ácidos nucleicos que codifican los aminoácidos han sido mutados, pero tienen por lo menos 60, 70, 80, 90 o 95 por ciento de identidad con las secuencias descritas en la tabla 1. En algunos aspectos, incluye secuencias de aminoácidos mutantes en donde no más de 1, 2, 3, 4 o 5 aminoácidos han sido mutados en las regiones variables en comparación con las regiones variables representadas en la secuencia descrita en la tabla 1, mientras que conservan sustancialmente la misma actividad terapéutica.
Puesto que cada uno de estos anticuerpos se puede unir a cKIT, las secuencias de VH, VL, cadena ligera de longitud completa y cadena pesada de longitud completa (secuencias de aminoácidos y las secuencias de nucleótidos que codifican las secuencia de aminoácidos) se pueden “mezclar y emparejar” para crear otros anticuerpos de unión a cKIT. Tales anticuerpos de unión a cKIT “mezclados y emparejados” se pueden probar usando los ensayos de unión conocidos (p.ej., ELISA y otros ensayos descritos en la sección de ejemplos). Cuando estas cadenas se mezclan y emparejan, una secuencia de VH de un par VH/VL particular puede ser reemplazada con una secuencia de VH estructuralmente similar. Asimismo, una secuencia de cadena pesada de longitud completa de un par particular de cadena pesada de longitud completa / cadena ligera de longitud completa, sería reemplazada con una secuencia de cadena pesada de longitud completa estructuralmente similar. Asimismo, una secuencia de VL de un par VH/VL particular sería reemplazada con una secuencia de VL estructuralmente similar. Asimismo, una secuencia de cadena ligera de longitud completa de un par particular de cadena pesada de longitud completa / cadena ligera de longitud completa, sería reemplazada con una secuencia de cadena ligera de longitud completa estructuralmente similar. Por consiguiente, en un aspecto, la divulgación provee un anticuerpo monoclonal aislado o región de unión de antígeno del mismo que tiene: una región variable de cadena pesada que comprende una secuencia de aminoácidos seleccionada del grupo que consiste en SEQ ID NOs: 9, 28, 46, 64, 82, 100, 118 o 136 (tabla 1); y una región variable de cadena ligera que comprende una secuencia de aminoácidos seleccionada del grupo que consiste en SEQ ID NOs: 18, 37, 55, 73, 91, 109, 127 o 145 (tabla 1); en donde el anticuerpo se une específicamente a cKit.
En otro aspecto, la divulgación provee (i) un anticuerpo monoclonal aislado que tiene: una cadena pesada de longitud completa que comprende una secuencia de aminoácidos que se ha optimizado para su expresión en la célula de un mamífero, seleccionada del grupo que consiste en SEQ ID NOs: 11, 29, 47, 65, 83, 101, 119 o 137; y una cadena ligera de longitud completa que comprende una secuencia de aminoácidos que se ha optimizado para su expresión en la célula de un mamífero, seleccionada del grupo que consiste en SEQ ID NOs: 20, 21,38, 56, 74, 92, 110, 128, o 146; o (ii) una proteína funcional que comprende una porción de unión de antígeno de la misma.
En otro aspecto, la presente divulgación provee anticuerpos de unión de cKIT que comprenden las CDR1, CDR2 y CDR3 de cadena pesada y cadena ligera que se describen en la tabla 1, o combinaciones de las mismas. Las secuencias de aminoácidos de las CDR1 de VH de los anticuerpos se muestran en SEQ ID NOs: 3, 22, 40, 58, 76, 94, 112 y 130. Las secuencias de aminoácidos de las CDR2 de VH de los anticuerpos se muestran en SEQ ID NOs: 4, 23, 41, 59, 77, 95, 113 y 131. Las secuencias de aminoácidos de las CDR3 de VH de los anticuerpos se muestran en SEQ ID NOs: 5, 24, 42, 60, 78, 96, 114 y 132. Las secuencias de aminoácidos de las CDR1 de VL de los anticuerpos se muestran en SEQ ID NOs: 12, 31, 49, 67, 85, 103, 121 y 139. Las secuencias de aminoácidos de las CDR2 de VL de los anticuerpos se muestran en SEQ ID NOs 13, 32, 50, 68, 86, 104, 122 y 140. Las secuencias de aminoácidos de las CDR3 de VL de los anticuerpos se muestran en SEQ ID NOs:14, 33, 51, 69, 87, 105, 123 y 141.
Dado que cada uno de estos anticuerpos se puede unir a cKIT y que la especificidad de unión al antígeno es provista principalmente por las regiones CDR1, 2 y 3, las secuencias de CDR1, 2 y 3 de VH y las secuencias de CDR1, 2 y 3 de VL se pueden “mezclar y emparejar” (esto es, las CDR de diferentes anticuerpos se pueden mezclar y emparejar, aunque cada anticuerpo debe contener una CDR1, 2 y 3 de VH y una CDR1,2 y 3 de VL para crear otras moléculas de unión a C5. Tales anticuerpos de unión de cKIT “mezclados y emparejados” se pueden probar usando los ensayos de unión conocidos y los que se describen en la sección de ejemplos (por ejemplo, ELISA). Cuando las secuencias de CDR de VH se mezclan y emparejan, la secuencia de CDR1, CDR2 o c DR3 de una secuencia VH particular sería reemplazada con una secuencia de CDR estructuralmente similar. Asimismo, cuando las secuencias de CDR de VL se mezclan y emparejan, la secuencia de CDR1, CDR2 o CDR3 de una secuencia VL particular sería reemplazada con una secuencia de CDR estructuralmente similar. Será evidente para el experto en la materia que se pueden crear secuencias VH y VL novedosas sustituyendo una o más secuencias de la región CDR de VH o VL con secuencias estructuralmente similares de las secuencias de CDR mostradas aquí para los anticuerpos monoclonales de la presente divulgación.
Por consiguiente, la presente divulgación provee un anticuerpo monoclonal aislado o región de unión de antígeno del mismo que comprende una CDR1 de cadena pesada que comprende una secuencia de aminoácidos seleccionada del grupo que consiste en SEQ ID NOs: 3, 22, 40, 58, 76, 94, 112 y 130; una CDR2 de cadena pesada que comprende una secuencia de aminoácidos seleccionada del grupo que consiste en SEQ ID NOs: 4, 23, 41, 59, 77, 95, 113 y 131; una
CDR3 de cadena pesada que comprende una secuencia de aminoácidos seleccionada del grupo que consiste en SEQ ID NOs: 5, 24, 42, 60, 78, 96, 114 y 132; una CDR1 de cadena ligera que comprende una secuencia de aminoácidos seleccionada del grupo que consiste en SEQ ID NOs: 12, 31,49, 67, 85, 103, 121 y 139; una CDR2 de cadena ligera que comprende una secuencia de aminoácidos seleccionada del grupo que consiste en SEQ ID NOs: 13, 32, 50, 68, 86, 104, 122 y 140; y una CDR3 de cadena ligera que comprende una secuencia de aminoácidos seleccionada del grupo que consiste en SEQ ID NOs: 14, 33, 51, 69, 87, 105, 123 y 141; en donde el anticuerpo se une específicamente a cKIT.
En un aspecto específico, un anticuerpo o fragmento de anticuerpo (por ejemplo, fragmentos de unión de antígeno) que se une específicamente a cKIT comprende una CDR1 de cadena pesada de SEQ ID NO:3, una CDR2 de cadena pesada de SEQ ID NO: 4; una CDR3 de cadena pesada de SEQ ID NO:5; una CDR1 de cadena ligera de SEQ ID NO:12; una CDR2 de cadena ligera de SEQ ID NO: 13; y una CDR3 de cadena ligera de SEQ ID NO: 14.
En un aspecto específico, un anticuerpo o fragmento de anticuerpo (por ejemplo, fragmentos de unión de antígeno) que se une específicamente a cKIT comprende una CDR1 de cadena pesada de s Eq ID NO:22, una CDR2 de cadena pesada de SEQ iD NO: 23; una CDR3 de cadena pesada de SEQ ID NO:24; una CDR1 de cadena ligera de SEQ ID NO:31; una CDR2 de cadena ligera de SEQ ID NO: 32; y una CDR3 de cadena ligera de SEQ ID NO: 33.
En un aspecto específico, un anticuerpo o fragmento de anticuerpo (por ejemplo, fragmentos de unión de antígeno) que se une específicamente a cKIT comprende una CDR1 de cadena pesada de s Eq ID NO:40, una CDR2 de cadena pesada de SEQ iD NO: 41; una CDR3 de cadena pesada de SEQ ID NO:42; una CDR1 de cadena ligera de SEQ ID NO:49; una CDR2 de cadena ligera de SEQ ID NO: 50; y una CDR3 de cadena ligera de SEQ ID NO: 51.
En un aspecto específico, un anticuerpo o fragmento de anticuerpo (por ejemplo, fragmentos de unión de antígeno) que se une específicamente a cKIT comprende una CDR1 de cadena pesada de s Eq ID NO:58, una CDR2 de cadena pesada de SEQ iD NO: 59; una CDR3 de cadena pesada de SEQ ID NO:60; una CDR1 de cadena ligera de SEQ ID NO:67; una CDR2 de cadena ligera de SEQ ID NO: 68; y una CDR3 de cadena ligera de SEQ ID NO: 69.
En un aspecto específico, un anticuerpo o fragmento de anticuerpo (por ejemplo, fragmentos de unión de antígeno) que se une específicamente a cKIT comprende una CDR1 de cadena pesada de s Eq ID NO:76, una CDR2 de cadena pesada de SEQ iD NO: 77; una CDR3 de cadena pesada de SEQ ID NO:78; una CDR1 de cadena ligera de SEQ ID NO:85; una CDR2 de cadena ligera de SEQ ID NO: 86; y una CDR3 de cadena ligera de SEQ ID NO: 87.
En un aspecto específico, un anticuerpo o fragmento de anticuerpo (por ejemplo, fragmentos de unión de antígeno) que se une específicamente a cKIT comprende una CDR1 de cadena pesada de s Eq ID NO:94, una CDR2 de cadena pesada de SEQ iD NO: 95; una CDR3 de cadena pesada de SEQ ID NO:96; una CDR1 de cadena ligera de SEQ ID NO:103; una CDR2 de cadena ligera de SEQ ID NO: 104; y una CDR3 de cadena ligera de SEQ ID NO: 105.
En un aspecto específico, un anticuerpo o fragmento de anticuerpo (por ejemplo, fragmentos de unión de antígeno) que se une específicamente a cKIT comprende una CDR1 de cadena pesada de SEQ ID NO:112, una CDR2 de cadena pesada de SEQ ID NO: 113; una CDR3 de cadena pesada de SEQ ID NO:114; una CDR1 de cadena ligera de SEQ ID NO:121; una CDR2 de cadena ligera de SEQ ID NO: 122; y una CDR3 de cadena ligera de SEQ ID NO: 123.
En un aspecto específico, un anticuerpo o fragmento de anticuerpo (por ejemplo, fragmentos de unión de antígeno) que se une específicamente a cKIT comprende una CDR1 de cadena pesada de SEQ ID NO:130, una CDR2 de cadena pesada de SEQ ID NO: 131; una CDR3 de cadena pesada de SEQ ID NO:132; una CDR1 de cadena ligera de SEQ ID NO:139; una CDR2 de cadena ligera de SEQ ID NO: 140; y una CDR3 de cadena ligera de SEQ ID NO: 141.
En ciertos aspectos, un anticuerpo que se une específicamente a cKIT es un anticuerpo o fragmento de anticuerpo (por ejemplo, fragmento de unión a antígeno) que se describe en la tabla 1.
1. identificación de epítopos y anticuerpos que se unen al mismo epítopo
La presente divulgación provee anticuerpos y fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) que se unen a un epítopo dentro del dominio extracelular del receptor cKit. En ciertos aspectos, los anticuerpos y fragmentos de anticuerpo se pueden unir a los epítopos con los dominios 1-3 del dominio extracelular de cKit.
La presente divulgación también provee anticuerpos y fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) que se unen al mismo epítopo que los anticuerpos anti-cKit descritos en la tabla 1. Por lo tanto se pueden identificar anticuerpos y fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) adicionales sobre la base de su capacidad para competir cruzadamente (por ejemplo, para inhibir competitivamente la unión, de una manera
estadísticamente significativa) con otros anticuerpos en los ensayos de unión de cKit. La capacidad de un anticuerpo de prueba para inhibir la unión de los anticuerpos y fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) de la presente divulgación a una proteína cKit (por ejemplo, cKit humana), demuestra que el anticuerpo de prueba puede competir con ese anticuerpo o fragmento de anticuerpo (por ejemplo, fragmentos de unión a antígeno) por la unión a cKit; un anticuerpo de este tipo, de acuerdo con una teoría no limitativa, se puede unir al mismo epítopo o uno relacionado (por ejemplo, un epítopo estructuramente similar o espacialmente proximal) en la proteína cKit que el anticuerpo o fragmento de anticuerpo (por ejemplo, fragmentos de unión al antígeno) con el que compite. En un aspecto determinado, el anticuerpo que se une al mismo epítopo en cKit que los anticuerpos o fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) de la presente divulgación, es un anticuerpo monoclonal humano o humanizado. Tales anticuerpos monoclonales humanos o humanizados pueden prepararse y aislarse como se describe aquí.
2. Alteración adicional del armazón de la región Fc
La presente divulgación provee inmunoconjugados marcados específicos de sitio. Estos inmunoconjugados pueden comprender anticuerpos modificados o fragmentos de unión de antígeno de los mismos que comprenden modificaciones adicionales en los residuos de armazón dentro de VH o VL, por ejemplo, para mejorar las propiedades del anticuerpo. Típicamente, tales modificaciones de armazón se realizan para disminuir la inmunogenicidad del anticuerpo. Por ejemplo, un enfoque consiste en "retromutar" uno o más residuos de armazón de vuelta a la secuencia de línea germinal correspondiente. Más específicamente, un anticuerpo que ha sido objeto de mutación somática puede contener residuos de armazón que difieren de la secuencia de línea germinal de la que deriva el anticuerpo. Tales residuos se pueden identificar mediante la comparación de las secuencias de armazón del anticuerpo con las secuencias de la línea germinal de la que se deriva el anticuerpo. Para retornar las secuencias de la región de armazón a su configuración de línea germinal, las mutaciones somáticas pueden ser "retromutadas" a la secuencia de la línea germinal, por ejemplo, por mutagénesis dirigida al sitio. Tales anticuerpos "retromutados" también se consideran abarcados.
Otro tipo de modificación de armazón implica la mutación de uno o más residuos dentro de la región de armazón, o incluso dentro de una o más regiones CDR, para remover los epítopos de células T y reducir así la posible inmunogenicidad del anticuerpo. Este enfoque también es referido como “desinmunización” y se describe en más detalle en la publicación de patente de EE. UU. No. 2003/0153043, de Carr et al.
Adicional o alternativamente a las modificaciones hechas dentro de las regiones de armazón o CDR, los anticuerpos se pueden modificar por ingeniería para incluir modificaciones dentro de la región Fc, típicamente para alterar una o más propiedades funcionales del anticuerpo, tales como la vida media en el suero, fijación del complemento, unión del receptor de Fc, o citotoxicidad celular dependiente de antígeno. Además, un anticuerpo se puede modificar químicamente (por ejemplo, se le pueden adherir una o más porciones químicas al anticuerpo), o se puede modificar para alterar su glicosilación, nuevamente para alterar una o más propiedades funcionales del anticuerpo. Cada uno de estos aspectos se describe más abajo en mayor detalle.
En un aspecto, la región bisagra de CH1 se modifica de tal manera que el número de residuos de cisteína en la región bisagra se altera, por ejemplo, se aumenta o disminuye. Este enfoque se describe adicionalmente en la patente de EE. UU. No. 5.677.425 de Bodmer et al. El número de residuos de cisteína en la región bisagra de CH1 se altera para, por ejemplo, facilitar el montaje de las cadenas ligeras y pesadas o para aumentar o disminuir la estabilidad del anticuerpo.
En otro aspecto, la región bisagra de Fc de un anticuerpo se muta para disminuir la vida media biológica del anticuerpo. Más específicamente, se introducen una o más mutaciones de aminoácidos en la región de interfaz del dominio CH2-CH3 del fragmento Fc-bisagra, de tal manera que el anticuerpo tiene unión deteriorada de la proteína A estafilocócica (SpA) con respecto a la unión de SpA del dominio Fc-bisagra nativo. Este enfoque se describe en más detalle en la patente de EE.UU. No. 6.165.745, de Ward et al.
En otros aspectos, la región Fc se altera reemplazando por lo menos un residuo de aminoácido con un residuo de aminoácido diferente para alterar las funciones efectoras del anticuerpo. Por ejemplo, uno o más aminoácidos pueden ser reemplazados con un residuo de aminoácido diferente, de tal manera que el anticuerpo tiene una afinidad alterada por un ligando efector, pero conserva la capacidad de unión al antígeno del anticuerpo progenitor. El ligando efector para el que se altera la afinidad puede ser, por ejemplo, un receptor de Fc o el componente C1 del complemento. Este enfoque se describe, por ejemplo, en las patentes de EE. UU. Nos. 5.624.821 y 5.648.260, ambas de Winter et al.
En otro aspecto, uno o más aminoácidos seleccionados de los residuos de aminoácido pueden ser reemplazados con un residuo de aminoácido diferente, de tal que manera que el anticuerpo tiene unión a C1q alterada o citotoxicidad dependiente del complemento (CDC) reducida o anulada. Este enfoque se describe, por ejemplo, en la patente de EE. UU. No. 6.194.551, de Idusogie et al.
En otro aspecto, uno o más residuos de aminoácido se alteran para así alterar la capacidad del anticuerpo para fijar el complemento. Este enfoque se describe, por ejemplo, en la publicación de PCT WO 94/29351 de Bodmer et al. En un aspecto específico, uno o más aminoácidos de un anticuerpo o fragmento de unión a antígeno del mismo de la presente divulgación se reemplazan con uno o más residuos de aminoácido alotípicos, para la subclase IgG1 y el isotipo kappa. Los residuos de aminoácido alotípicos también incluyen, sin limitación, la región constante de la cadena pesada de las subclases IgG1, IgG2, e IgG3, y también la región constante de la cadena ligera del isotipo kappa, como lo describen Jefferis et al., MAbs. 1:332-338 (2009).
En otro aspecto más, la región Fc se modifica para aumentar la capacidad del anticuerpo para mediar la citotoxicidad celular dependiente de anticuerpo (ADCC), o para aumentar la afinidad del anticuerpo por un receptor Fcy, modificando uno o más aminoácidos. Este enfoque se describe, por ejemplo, en la publicación PCT Wo 00/42072 de Presta. Además, los sitios de unión en la IgG 1 humana para FcyRI, FcyRII, FcyRIII y FcRn se han mapeado, y se han descrito variantes con unión mejorada (véase Shields et al., J. Biol Chem 276:6591-6604, 2001).
En otro aspecto más, la glicosilación de un anticuerpo se modifica. Por ejemplo, se puede hacer un anticuerpo aglicosilado (esto es, el anticuerpo carece de glicosilación). La glicosilación se puede alterar, por ejemplo, para aumentar la afinidad del anticuerpo por el “antígeno”. Tales modificaciones de carbohidrato se pueden efectuar, por ejemplo, alterando uno o más sitios de glicosilación dentro de la secuencia de anticuerpo. Por ejemplo, se pueden hacer una o más sustituciones de aminoácido que resultan en la eliminación de uno o más sitios de glicosilación del armazón de la región variable, para eliminar así la glicosilación en ese sitio. Tal aglicosilación puede aumentar la afinidad del anticuerpo por el antígeno. Tal enfoque se describe, por ejemplo, en las patentes de EE. UU. Nos. 5.714.350 y 6.350.861, de Co et al.
Adicional o alternativamente, se puede hacer un anticuerpo que tiene un tipo de glicosilación alterado, tal como un anticuerpo hipofucosilado que tiene cantidades reducidas de residuos de fucosilo, o un anticuerpo que tiene un aumento en las estructuras GlcNac bisecantes. Se ha mostrado que estos patrones de glicosilación alterados aumentan la capacidad de ADCC de los anticuerpos. Tales modificaciones de carbohidrato se pueden realizar, por ejemplo, expresando el anticuerpo en una célula hospedera con maquinaria de glicosilación alterada. Las células con maquinaria de glicosilación alterada se han descrito en la literatura y se pueden usar como células hospederas para expresar anticuerpos recombinantes para producir así un anticuerpo con glicosilación alterada. Por ejemplo, EP 1.176.195 de Hang et al. describe una línea celular con un gen FUT8 funcionalmente interrumpido que codifica una fucosil transferasa, de tal manera que los anticuerpos expresados en dicha línea celular exhiben hipofucosilación. La publicación de PCT WO 03/035835 de Presta describe una línea celular CHO variante, células Lecl3, con capacidad reducida para unir fucosa a carbohidratos ligados a Asn(297), resultando también en la hipofucosilación de los anticuerpos expresados en esa célula hospedera (véase también Shields et al., (2002), J. Biol. Chem. 277:26733-26740). La publicación de PCT WO 99/54342 de Umana et al. describe líneas celulares modificadas por ingeniería para expresar glicosil transferasas modificantes de glicoproteína (por ejemplo, beta(1,4)-N acetilglucosaminil-transferasa III (GnTIII)), de tal manera que los anticuerpos expresados en las líneas celulares modificadas exhiben un aumento de las estructuras de GlcNac bisecantes que resulta en un aumento de la actividad de ADCC de los anticuerpos (véase también Umana et al., Nat. Biotech. 17:176-180, 1999).
En otro aspecto, el anticuerpo se modifica para aumentar su vida media biológica. Son posibles varios enfoques. Por ejemplo, se pueden introducir una o más de las siguientes mutaciones: T252L, T254S, T256F, como se describe en la patente de Ee . UU. No. 6.277.375 de Ward. Alternativamente, para aumentar la vida media biológica, el anticuerpo se puede alterar dentro de la región CH1 o CL para contener un epítopo de unión de receptor de rescate tomado de dos lazos de un dominio CH2 de una región Fc de una IgG, como se describe en las patentes de EE. UU. Nos. 5.869.046 y 6.121.022, de Presta et al.
Para minimizar la actividad de ADCC de un anticuerpo, mutaciones específicas en la región Fc resultan en anticuerpos “Fc silentes” que tienen mínima interacción con células efectoras. En general, la “región Fc de IgG” se usa para definir la región C-terminal de una cadena pesada de inmunoglobulina, que incluye la región Fc de secuencia nativa y regiones Fc variantes. La región Fc de cadena pesada de IgG humana generalmente se define porque comprende el residuo de aminoácido de la posición C226 o desde P230 hasta el extremo carboxilo del anticuerpo IgG. La numeración de residuos en la región Fc es la del índice EU de Kabat. La lisina C-terminal (residuo K447) de la región Fc puede ser removida, por ejemplo, durante la producción o purificación del anticuerpo.
Se pueden obtener funciones efectoras silenciadas por mutación en la región Fc de los anticuerpos y se han descrito en la literatura: LALA y N297A (Strohl, W., 2009, Curr. Opin. Biotechnol. vol. 20(6):685-691); y D265A (Baudino et al., 2008, J. Immunol. 181: 6664-69) véase también Heusser et al., WO2012065950. Ejemplos de anticuerpos de IgG 1 Fc silentes son el mutante LALA que comprende la mutación L234A y L235A en la secuencia de aminoácidos de Fc de IgG1. Otro ejemplo de un anticuerpo IgG1 silente es la mutación DApA (D265A, P329A) (US 6.737.056). Otro anticuerpo IgG 1 silente
com p ren de la m utación N 297A, que resu lta en an ticue rpos ag licos ilados /no g licos ilados.
Los anticuerpos Fc silentes resultan en poco o nada de actividad de ADCC, lo que significa que un anticuerpo Fc silente exhibe una actividad de ADCC que es menor de 50% de lisis celular específica. Nada de actividad de ADCC significa que el anticuerpo Fc silente exhibe una actividad de ADCC (lisis específica de célula) que es menor de 1%.
3. Producción de los anticuerpos de cKIT
Los anticuerpos anti-cKIT y fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) de los mismos se pueden producir mediante cualquier método conocido que incluye, sin limitación, expresión recombinante, síntesis química y digestión enzimática de tetrámeros de anticuerpos, mientras que se pueden obtener anticuerpos monoclonales de longitud completa, por ejemplo, por hibridoma o producción recombinante. La expresión recombinante puede ser de cualquier célula hospedera apropiada conocida, por ejemplo, células hospederas de mamífero, células hospederas bacterianas, células hospederas de levadura, células hospederas de insecto, etc.
La divulgación provee además polinucleótidos que codifican los anticuerpos descritos en la presente, por ejemplo polinucleótidos que codifican regiones variables de cadena pesada o ligera o segmentos que comprenden las regiones determinantes de complementariedad como se describe en la presente. En algunos aspectos, el polinucleótido que codifica las regiones variables de cadena pesada tiene por lo menos 85%, 89%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, o 100% de identidad de secuencia de ácido nucleico con un polinucleótido seleccionado del grupo que consiste en SEQ ID NOs: 30, 48, 66, 84, 102, 120 y 137. En algunos aspectos, el polinucleótido que codifica las regiones variables de cadena ligera tienen por lo menos 85%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, o 100% de identidad de secuencia de ácido nucleico con un polinucleótido seleccionado del grupo que consiste en SEQ ID NOs: 39, 57, 75, 93, 111, 129 y 147.
En algunos aspectos, el polinucleótido que codifica la cadena pesada tiene por lo menos 85%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, o 100% de identidad de secuencia de ácido nucleico con un polinucleótido de SEQ ID NO: 30, 48, 66, 84, 102, 120. En algunos aspectos, el polinucleótido que codifica la cadena ligera tiene por lo menos 85%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, o 100% de identidad de secuencia de ácido nucleico con un polinucleótido de SEQ ID NO: 39, 57, 75, 93, 111, 129 y 147.
Los polinucleótidos de la presente divulgación pueden codificar solo la secuencia de la región variable de un anticuerpo anti-cKIT. También pueden codificar tanto una región variable como una región constante del anticuerpo. Algunas de las secuencias de polinucleótido codifican un polipéptido que comprende regiones variables tanto de la cadena pesada como de la cadena ligera de un anticuerpo anti-cKIT ejemplificado. Algunos otros polinucleótidos codifican dos segmentos de polipéptido que respectivamente son sustancialmente idénticos a las regiones variables de la cadena pesada y la cadena ligera de uno de los anticuerpos de ratón.
Las secuencias de polinucleótido pueden ser producidas por medio de síntesis de ADN de novo en fase sólida o por medio de mutagénesis de PCR de una secuencia existente (por ejemplo, secuencias como las que se describen en los ejemplos más abajo) que codifican un anticuerpo anti-cKIT o su fragmento de unión. La síntesis química directa de ácidos nucleicos se puede lograr mediante los métodos conocidos, tales como el método de fosfotriéster de Narang et al, Meth. Enzymol.
68:90, 1979; el método de fosfodiéster de Brown et al, Meth. Enzymol. 68:109, 1979; el método de dietilfosforamidita de Beaucage et al, Tetra. Lett., 22:1859, 1981; y el método de soporte sólido de la patente de EE. UU. No. 4.458.066. La introducción de mutaciones en una secuencia de polinucleótidos por PCR se puede realizar por ejemplo como se describe en “PCR Technology: Principles and Applications for DNA Amplification”, H.A. Erlich (Ed.), Freeman Press, NY, NY, 1992; “PCR Protocols: A Guide to Methods and Applications”, Innis et al. (Ed.), Academic Press, San Diego, CA, 1990; Mattila et al., Nucleic Acids Res. 19:967, 1991; y Eckert et al., PCR Methods and Applications 1:17, 1991.
También se proveen en la presente divulgación vectores de expresión y células hospederas para producir los anticuerpos anti-cKIT arriba descritos. Se puede utilizar varios vectores de expresión para expresar los polinucleótidos que codifican las cadenas de anticuerpo anti-cKIT o fragmentos de unión. Se pueden usar vectores de expresión tanto virales como no virales para producir los anticuerpos en una célula hospedera de mamífero. Los vectores y sistemas no virales incluyen plásmidos, vectores episómicos, típicamente con un casete de expresión para expresar una proteína o ARN, y cromosomas artificiales humanos (Harrington et al., Nat Genet 15:345, 1997). Por ejemplo, los vectores no virales útiles para la expresión de los polinucleótidos y polipéptidos anti-cKIT en células de mamífero (por ejemplo, de humano) incluyen pThioHis A, B y C, pcDNA3.1/His, pEBVHis A, B y C (Invitrogen, San Diego, CA), vectores MPSV y muchos otros vectores conocidos para expresar otras proteínas. Los vectores virales útiles incluyen vectores basados en retrovirus, adenovirus, virus adenoasociados, virus de herpes, vectores basados en SV40, virus de papiloma, virus de HBP de Epstein Barr, vectores de virus vaccinia y virus Semliki Forest (SFV). Veáse Brent et al, supra; Smith, Annu. Rev. Microbiol.
49:807, 1995; y Rosenfeld etal., Cell 68:143, 1992.
La elección del vector de expresión depende de las células hospederas deseadas en las que se va a expresar el vector. Típicamente, los vectores de expresión contienen un promotor y otras secuencias reguladoras (por ejemplo, potenciadores) que están enlazados operativamente con los polinucleótidos que codifican una cadena o fragmento de anticuerpo anti-cKIT. En algunos aspectos se utiliza un promotor inducible para prevenir la expresión de secuencias insertadas excepto bajo condiciones de inducción. Los promotores inducibles incluyen, por ejemplo, arabinosa, lacZ, promotor de metalotioneína o promotor de choque de calor. Los cultivos de organismos transformados se pueden expandir bajo condiciones no inductoras sin desviar la población para codificar secuencias cuyos productos de expresión son mejor tolerados por las células hospederas. Además de los promotores, también se pueden requerir o desear otros elementos reguladores para la expresión eficiente de una cadena o fragmento de anticuerpo anti-cKIT. Típicamente estos elementos incluyen un codón de iniciación ATG y un sitio de unión de ribosoma adyacente u otras secuencias. Además, la eficiencia de expresión puede ser incrementada por la inclusión de potenciadores apropiados para el sistema celular en uso (véase, por ejemplo, Scharf et al., Results Probl. Cell Differ. 20:125, 1994; y Bittner et al., Meth. Enzymol., 153:516, 1987). Por ejemplo, el potenciador de SV40 o potenciador CMV se pueden usar para aumentar la expresión en células hospederas de mamífero.
Los vectores de expresión también pueden proveer una posición de secuencia de señal de secreción para formar una proteína de fusión con polipéptidos codificados por secuencias insertadas del anticuerpo anti-cKIT. Más a menudo las secuencias de anticuerpo anti-cKIT insertadas son enlazadas con una secuencia señal antes de la inclusión en el vector. Los vectores por utilizar para recibir secuencias que codifican dominios variables de cadena ligera y cadena pesada de anticuerpo anti-cKIT algunas veces también codifican regiones constantes o partes de las mismas. Estos vectores permiten la expresión de las regiones variables como proteínas de fusión con las regiones constantes, llevando así a la producción de anticuerpos intactos o fragmentos de los mismos. Típicamente estas regiones constantes son humanas.
Las células hospederas para alojar y expresar las cadenas de anticuerpo anti-cKIT pueden ser procariotas o eucariotas. La E. coli es un hospedero procariota útil para clonar y expresar los polinucleótidos de la presente divulgación. Otros hospederos microbianos adecuados para usarse incluyen bacilos, tales como Bacillus subtilis, y otras Enterobacteriaceae, tales como Salmonella, Serratia, y varias especies de Pseudomonas. En estos hospederos procariotas también se pueden hacer vectores de expresión que contienen típicamente secuencias de control de expresión compatibles con la célula hospedera (por ejemplo, un origen de replicación). Además, estarán presentes cualquier cantidad de una variedad de promotores muy conocidos, tales como el sistema promotor de lactosa, un sistema promotor de triptófano (trp), un sistema promotor de beta-lactamasa, o un sistema promotor de fago lambda. Típicamente, los promotores controlan la expresión, opcionalmente con una secuencia operadora, y tienen secuencias de sitio de unión de ribosoma y similares para iniciar y terminar la transcripción y traducción. También se pueden utilizar otros microbios tales como levaduras, para expresar los polipéptidos anti-cKIT. También se pueden usar células de insecto en combinación con vectores de baculovirus.
En otros aspectos se usan células hospederas de mamífero para expresar y producir los polipéptidos anti-cKIT de la presente divulgación. Por ejemplo, pueden ser una línea celular de hibridoma que expresa genes endógenos de inmunoglobulina (por ejemplo, los clones de hibridoma de mieloma que se describen en los ejemplos), o una línea celular de mamífero que aloja un vector de expresión exógeno (por ejemplo, las células de mieloma SP2/0 ejemplificadas más abajo. Estos incluyen cualquier célula mortal normal, o inmortal normal o anormal, de animal o de humano. Por ejemplo, se han desarrollado varias líneas de células hospederas adecuadas capaces de secretar inmunoglobulinas intactas, que incluyen las líneas celulares CHO, varias líneas celulares COS, células HeLa, líneas de células de mieloma, células B transformadas e hibridomas. El uso del cultivo de células de tejido de mamífero para expresar polipéptidos se expone en general, por ejemplo, en Winnacker, “From Genes to Clones”, VCH Publishers, N.Y., N.Y., 1987. Los vectores de expresión para células hospederas de mamífero pueden incluir secuencias de control de expresión, tales como un origen de replicación, un promotor y un potenciador (veáse, por ejemplo, Queen et al., Immunol. Rev. 89:49-68, 1986), y sitios de información de procesamiento necesarios, tales como sitios de unión de ribosoma, sitios de empalme de ARN, sitios de poliadenilacion y secuencias de terminador de transcripción. Estos vectores de expresión usualmente contienen promotores derivados de genes de mamífero o de virus de mamífero. Los promotores adecuados pueden ser constitutivos, específicos del tipo de célula, específicos de etapa, o modulables o regulables. Los promotores útiles incluyen, sin limitación, el promotor de metalotioneína, el promotor tardío mayor constitutivo de adenovirus, el promotor de MMTV inducible por dexametasona, el promotor de SV40, el promotor de MRP polIII, el promotor MPSV constitutivo, el promotor CMV inducible por tetraciclina (tal como el promotor CMV inmediato - temprano de humanos), el promotor CMV constitutivo, y combinaciones promotor - potenciador conocidas.
Los métodos para introducir vectores de expresión que contienen las secuencias de polinucleótido de interés varían dependiendo del tipo de hospedero celular. Por ejemplo, comúnmente se utiliza tranfeccion con cloruro de calcio para células procariotas, mientras que se puede usar tratamiento con fosfato de calcio o electroporacion para otros hospederos
celulares (véase en general Sambrook et al, supra). Otros métodos incluyen, por ejemplo, electroporación, tratamiento con fosfato de calcio, transformación mediada por liposoma, inyección y microinyección, métodos balísticos, virosomas, inmunoliposomas, conjugados policatión: ácido nucleico, ADN desnudo, viriones artificiales, fusión con la proteína estructural del virus del Herpes VP22 (Elliot y O'Hare, Cell 88:223, 1997), captación de ADN intensificada por agente, y transducción ex vivo. Para la producción de alto rendimiento a largo plazo de proteínas recombinantes frecuentemente se desea la expresión estable. Por ejemplo, se pueden preparar líneas celulares que expresan establemente cadenas de anticuerpo anti-cKIT o fragmentos de unión usando vectores de expresión que contienen orígenes de replicación virales o elementos de expresión endógenos y un gen marcador seleccionable. Después de la introducción del vector, las células se dejan crecer por 1-2 días en un medio enriquecido antes de cambiarlas a un medio selectivo. La finalidad del marcador seleccionable es conferir resistencia a la selección, y su presencia permite el crecimiento de células que expresan exitosamente las secuencias introducidas en medio selectivo. Las células resistentes transfectadas establemente pueden proliferar usando técnicas de cultivo de tejido apropiadas para el tipo celular.
Usos terapéuticos y diagnósticos
Los anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno), y conjugados anticuerpofármaco de la presente divulgación son útiles en una variedad de aplicaciones que incluyen, sin limitación, tratamiento del cáncer, tal como cáncer sólido. En ciertos aspectos, los anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) y conjugados anticuerpo-fármaco son útiles para inhibir el crecimiento de tumor, inducir diferenciación, reducir el volumen del tumor, o reducir la tumorigenicidad de un tumor. Los métodos de uso pueden ser métodos in vitro, ex vivo, o in vivo.
En un aspecto, los anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) y conjugados anticuerpo-farmaco son útiles para detectar la presencia de cKlT en una muestra biológica. El término ‘'detectar’’ usado en la presente abarca detección cuantitativa o cualitativa. En ciertos aspectos, una muestra biológica comprende una célula o tejido. En ciertos aspectos, estos tejidos incluyen tejidos normales o cancerosos que expresan cKIT a niveles más altos con respecto a otros tejidos.
En un aspecto, la presente divulgación provee un método para detectar la presencia de cKIT en una muestra biológica. En ciertos aspectos, el método comprende poner en contacto la muestra biológica con un anticuerpo anti-cKIT bajo condiciones permisivas para la unión del anticuerpo al antígeno, y detectar si se forma un complejo entre el anticuerpo y el antígeno.
También se incluye un método para diagnosticar un trastorno asociado con aumento de la expresión de cKIT. En ciertos aspectos, el método comprende poner en contacto una célula de prueba con un anticuerpo anti-cKIT; determinar el grado de expresión (ya sea cuantitativa o cualitativamente) de cKIT en la célula de prueba detectando la unión del anticuerpo anti-cKIT al antígeno cKIT; y comparar el grado de expresión cKIT en la célula de prueba con el grado de expresión de cKIT en una célula de control (por ejemplo, una célula normal del mismo origen de tejido que la célula de prueba o una célula que expresa cKIT a niveles comparables a dicha célula normal), en donde un grado de expresión más alto de cKIT en la célula de prueba en comparación de la célula de control indica la presencia de un trastorno asociado con el aumento de expresión de cKIT. En ciertos aspectos, la célula de prueba se obtiene de un individuo sospechoso de tener un trastorno asociado con aumento de la expresión de cKIT. En ciertos aspectos, el trastorno es un trastorno proliferativo de células, tal como un cáncer o un tumor.
En ciertos aspectos, un método de diagnóstico o detección, como los que se describen arriba, comprende detectar la unión de un anticuerpo anti-cKIT a cKIT expresado sobre la superficie de una célula o en una preparación de membrana obtenida de una célula que expresa cKIT sobre su superficie. Un ensayo ejemplar para detectar la unión de un anticuerpo anti-cKIT a cKIT expresado sobre la superficie de una célula es un ensayo ‘’FACS’’.
Se pueden usar otros métodos para detectar la unión de anticuerpos anti-cKIT a cKIT. Estos métodos incluyen, sin limitación, ensayos de unión de antígeno que son muy conocidos, tales como western blots, radioinmunoensayos, ELISA (ensayo de inmunoabsorbente enlazado a enzima), inmunoensayos de ‘'sándwich’’, ensayos de inmunoprecipitación, inmunoensayos fluorescentes, inmunoensayos de proteína A, e inmunohistoquímica (IHC).
En ciertos aspectos, los anticuerpos anti-cKIT se marcan. Las marcas incluyen, sin limitación, marcas o porciones que son detectadas directamente (tales como marcas fluorescentes, cromofóricas, de densidad electrónica, quimioluminiscentes y radioactivas) y también porciones tales como enzimas o ligandos que son detectados indirectamente, por ejemplo por medio de una reacción enzimática o interacción molecular.
En ciertos aspectos, los anticuerpos anti-cKIT se inmovilizan sobre una matriz insoluble. La inmovilización abarca separar
el anticuerpo anti-cKIT de cualquier proteína cKIT que permanece libre en su solución. Esto se realiza convencionalmente insolubilizando el anticuerpo anti-cKIT antes del procedimiento de ensayo, tal como por adsorción en una matriz o superficie insoluble en agua (Bennich et al, patente de EE. UU. No.3.720.760), o por acoplamiento covalente (por ejemplo, usando entrelazamiento de glutaraldehídos, o insolubilizando el anticuerpo anti-cKIT después de la formación de un complejo entre el anticuerpo anti-cKIT y la proteína cKIT, por ejemplo por inmunoprecipitación.
Cualquiera de los aspectos anteriores de diagnóstico o detección se pueden efectuar usando un inmunoconjugado de la presente divulgación en lugar de, o además de, un anticuerpo anti-cKIT.
En un aspecto la divulgación provee un método de tratamiento, prevención, o alivio de una enfermedad, que comprende administrar los anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmento de unión a antígeno), y conjugados anticuerpo-fármaco a un paciente, tratando así la enfermedad. En ciertos aspectos, la enfermedad tratada con los anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) y conjugados anticuerpo-fármaco, es un cáncer. Los ejemplos de enfermedades que pueden ser tratadas o prevenidas incluyen, sin limitación, tumores estromales gastrointestinales (GIST), cáncer de pulmón de célula pequeña (SCLC), leucemia mieloide aguda (AML), melanoma, leucemia de mastocitos (MCL), mastocitosis, neurofibromatosis, cáncer de mama, cáncer de pulmón de célula no pequeña (NSCLC), y cáncer pancreático. En ciertos aspectos, el cáncer se caracteriza por células que expresan cKIT a las que los anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) y conjugados anticuerpo-fármaco se pueden unir específicamente.
La presente divulgación provee métodos de tratamiento del cáncer que comprenden adminisitrar una cantidad terapéuticamente eficaz de los anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígenos) o conjugados anticuerpo-fármaco. En ciertos aspectos, el cáncer es un cáncer sólido. En ciertos aspectos, el sujeto es un humano.
En ciertos aspectos, el método de inhibición de crecimiento de tumor comprende administrar a un sujeto una cantidad terapéuticamente eficaz de los anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) o conjugados anticuerpo-fármaco. En ciertos aspectos, el sujeto es un humano. En ciertos aspectos, el sujeto tiene un tumor o se le ha extirpado un tumor.
En ciertos aspectos, el tumor expresa la cKIT a la que se une el anticuerpo anti-cKIT. En ciertos aspectos, el tumor sobreexpresa la cKIT humana.
Para el tratamiento de la enfermedad, la dosis apropiada de los anticuerpos, fragmentos de anticuerpo (por ejemplo, fragmentos de unión de antígeno) o conjugados anticuerpo-fármaco depende de varios factores tales como el tipo de enfermedad por tratar, la severidad y curso de la enfermedad, la responsividad de la enfermedad, la terapia previa, la historia clínica del paciente, etc. El anticuerpo o agente se puede administrar una vez o durante una serie de tratamientos que duran de varios días a varios meses, o hasta que se efectúa una curación o una disminución del estado patológico (por ejemplo, reducción del tamaño del tumor). Los regímenes de administración óptimos se pueden calcular de mediciones de la acumulación de fármaco del cuerpo del paciente y variarán dependiendo de la potencia relativa de un anticuerpo, fragmento de anticuerpo (por ejemplo, fragmento de unión a antígeno) o conjugado anticuerpo-fármaco individual. En ciertos aspectos, la dosis es de 0,01 mg a 10 mg (por ejemplo, 0,01 mg, 0,05 mg, 0,1 mg, 0,5 mg, 1 mg, 2 mg, 3 mg, 4 mg, 5 mg, 7 mg, 8 mg, 9 mg, o 10 mg) por kg de peso corporal, y se pueden dar una vez o más al día, a la semana, al mes, o al año. En ciertos aspectos, el anticuerpo, fragmento de anticuerpo (por ejemplo, fragmento de unión a antígeno) o conjugado anticuerpo-fármaco de la presente divulgación, se administra una vez cada dos semanas o una vez cada tres semanas. El médico a cargo puede calcular las frecuencias de repetición para la administración de las dosis basándose en los tiempos de residencia medidos y las concentraciones del fármaco en los fluidos o tejidos corporales.
Terapia de combinación
En ciertos casos, un anticuerpo, fragmento de anticuerpo (por ejemplo, fragmento de unión de antígeno) o conjugado anticuerpo-fármaco de la presente divulgación se combina con otros agentes terapéuticos, tales como otros gentes anticancerosos, agentes antialérgicos, agentes contra la náusea (o antieméticos), aliviadores del dolor, agentes citoprotectores, y combinaciones de los mismos.
Los agentes quimioterapéuticos generales considerados para usarse en las terapias de combinación incluyen anastrozol (Arimidex®), bicalutamida (Casodex®), sulfato de bleomicina (Blenoxane®), busulfan (Myleran®), busulfan inyección (Busulfex®), capecitabina (Xeloda®), N4-pentoxicarbonil-5-desoxi-5-fluorocitidina, carboplatino (Paraplatin®), carmustina (BiCNU®), clorambucilo (Leukeran®), cisplatino (Platinol®), cladribina (Leustatin®), ciclofosfamida (Cytoxan®
o Neosar®), citarabina, citosina arabinósido (Cytosar-U®), inyección de liposoma de citarabina (DepoCyt®), dacarbazina (DTIC-Dome®), dactinomicina (Actinomicina D, Cosmegan), clorhidrato de daunorrubicina (Cerubidine®), inyección de liposoma de citrato de daunorrubicina (DaunoXome®), dexametasona, docetaxel (Taxotere®), clorhidrato de doxorrubicina (Adriamycin®, Rubex®), etopósido (Vepesid®), fosfato de fludarabina (Fludara®), 5-fluorouracilo (Adrucil®, Efudex®), flutamida (Eulexin®), tezacitibina, Gemcitabina (difluorodesoxicitidina), hidroxiurea (Hydrea®), idarrubicina (Idamycin®), ifosfamida (IFEX®), irinotecan (Camptosar®), L-asparaginasa (ELSPAR®), leucovorin calcio, melfalan (Alkeran®), 6-mercaptopurina (Purinethol®), metotrexato (Folex®), mitoxantrona (Novantrone®), mylotarg, paclitaxel (Taxol®), phoenix (Itrio90/MX-DTPA), pentostatina, polifeprosan 20 con implante de carmustina (Gliadel®), citrato de tamoxifeno (Nolvadex®), tenipósido (Vumon®), 6-tioguanina, tiotepa, tirapazamina (Tirazone®), topotecan clorhidrato para inyección (Hycamptin®), vinblastina (Velban®), vincristina (Oncovin®), y vinorelbina (Navelbine®).
En un aspecto, un anticuerpo, fragmento de anticuerpo (por ejemplo, fragmentos de unión de antígeno) o conjugado anticuerpo-fármaco de la presente divulgación se combina en una formulación de combinación farmacéutica, o régimen de dosificación como terapia de combinación con un segundo compuesto que tiene propiedades anticancerosas. El segundo compuesto de la formulación de combinación farmacéutica o régimen de dosificación puede tener actividades complementarias al anticuerpo o inmunoconjugado de la combinación, de tal manera que no afecte uno adversamente al otro. Por ejemplo, un anticuerpo, fragmento de anticuerpo (por ejemplo, fragmento de unión de antígeno) o conjugado anticuerpo-fármaco de la presente divulgación, se puede administrar en combinación con, sin limitación, un agente quimioterapéutico, un inhibidor de tirosina quinasa por ejemplo, imatinib, y otros inhibidores de la ruta de cKIT.
El término ‘'combinación farmacéutica” usado en la presente se refiere a una combinación fija en una forma farmacéutica unitaria, o una combinación no fija o un kit de partes para la administración combinada, en donde dos o más agentes terapéuticos se pueden administrar independientemente al mismo tiempo o separadamente en intervalos de tiempo, especialmente cuando estos intervalos permiten que los socios de combinación muestren un efecto cooperativo, por ejemplo, sinérgico.
El término ‘'terapia de combinación'' se refiere a la administración de dos o más agentes terapéuticos para tratar una condición terapéutica o trastorno descrito en la presente divulgación. Esta administración abarca la coadministración de estos agentes terapéuticos de manera sustancialmente simultánea, tal como en una sola cápsula que tiene una proporción fija de ingredientes activos. Alternativamente, esta administración abarca la coadministración en recipientes múltiples o separados (por ejemplo, cápsulas, polvos y líquidos) para cada ingrediente activo. Los polvos o líquidos pueden ser reconstituidos o diluidos a una dosis deseada antes de su administración. Además, esta administración también abarca el uso de cada tipo de agente terapéutico de manera secuencial, ya sea aproximadamente al mismo tiempo o en tiempos dieferentes. En cualquier caso, el régimen de tratamiento proveerá efectos beneficiosos de la combinación de fármaco en el tratamiento de las afecciones o trastornos descritos en la presente.
La terapia de combinación puede proveer ‘'sinergia'' y resultar ‘'sinérgica'', esto es, el efecto logrado cuando los ingredientes activos usados juntos es mayor que la suma de los efectos que resultan del uso de los compuestos separadamente. Un efecto sinérgico se puede obtener cuando los ingredientes activos: (1) se coformulan y administran o suministran simultáneamente en una formulación de dosis unitaria combinada; (2) son suministrados por alternación o en paralelo como formulaciones separadas; o (3) por medio de algún otro régimen. Cuando se administran en una terapia de alternación, se puede obtener un efecto sinérgico cuando los compuestos se administran o suministran secuencialmente, por ejemplo por medio de diferentes inyecciones en jeringas separadas. En general, durante la terapia de alternación, una dosis eficaz de cada ingrediente activo se administra secuencialmente, esto es, en serie, mientras que en la terapia de combinación las dosis eficaces de dos o más ingredientes activos se administran juntas.
En un aspecto, la presente divulgación provee un método de tratamiento del cáncer, administrando a un sujeto en necesidad del mismo un conjugado de anticuerpo-fármaco en combianción con uno o mas inhibidores de tirosina quinasa que incluyen, sin limitación, inhibidores de EGFR, inhibidores de Her2, inhibidores de Her3, inhibidores de IGFR, e inhibidores de Met.
Por ejemplo, los inhibidores de tirosina quinasa incluyen, sin limitación, clorhidrato de erlotinib (Tarceva®); linifanib (N-[4-(3-amino-1H-indazol-4-il)fenil]-N'-(2-fluoro-5-metilfenil)urea, conocido también como ABT 869, disponible de Genentech); malato de sunitinib (Sutent®); bosutinib (4-[(2,4-dicloro-5-metoxifenil)amino]-6-metoxi-7-[3-(4-metilpiperazin-1-il)propoxi]quinolin-3-carbonitrilo, conocido también como SKI-606, y descrito en la patente de e E. UU. No. 6.780.996); dasatinib (Sprycel®); pazopanib (Votrient®); sorafenib (Nexavar®); zactima (ZD6474); nilotinib (Tasigna®); regorafenib (Stivarga®), e imatinib o mesilato de imatinib (Gilvec® y Gleevec®).
Los inhibidores del receptor del factor de crecimiento epidérmico (EGFR) incluyen, sin limitación, clorhidrato de erlotinib (Tarceva®), Gefitnib (Iressa®); N-[4-[(3-cloro-4-fluorofenil)amino]-7-[[(3''S'')-tetrahidro-3-furanil]oxi]-6-quinazolinil]-4(dimetilamino)-2-butenamida, Tovok®); Vandetanib (Caprelsa®); Lapatinib (Tykerb®); (3R,4R)-4-amino-1-((4-((3-metoxifenil)amino)pirrolo[2,1-f][1,2,4]triazin—5—il)metil)piperidin—3—ol (BMS690514); diclorhidrato de canertinib (CI-1033); 6—[4—[(4—etil—1—piperazinil)metil]fenil]—N—[(1 R)—1—feniletil]—7H—pirrolo[2,3—d]pirimidin—4—amina (AEE788, CAS 497839 62-0); Mubritinib (TAK165); Pelitinib (EKB569); Afatinib (BIBW2992); Neratinib (HKI-272); ácido N -[4-[[1-[(3-fluorofenil)metil]-1 H-indazol-5-il]amino]-5-metilpirrolo[2,1-f][1,2,4]triazin-6-il]-carbámico, éster de (3S)-3-morfolinilmetilo (BMS599626); N-(3,4-dicloro-2-fluorofenil)-6-metoxi-7-[[(3aa,5|3,6aa)-octahidro-2-metilciclopenta[c]pirrol-5-il]metoxi]-4-quinazolinamina (XL647, CAS 781613-23-8); y 4-[4-[[(1R)-1-feniletil]amino]-7H-pirrolo[2,3-d]pirimidin -6 -il]-fenol (PKI166, CAS 187724-61-4).
Los anticuerpos de EGFR incluyen, sin limitación, Cetuximab (Erbitux®); Panitumumab (Vectibix®); Matuzumab (EMD-72000); Nimotuzumab (hR3); Zalutumumab; TheraCIM h-R3; MDX0447 (CAS 339151-96-1); y ch806 (mAb-806, CAS 946414-09-1).
Los inhibidores del receptor del factor de crecimiento epidérmico humano 2 (receptor HER2) (conocido también como Neu, ErbB-2, CD340, o p185) incluyen, sin limitación, Trastuzumab (Herceptin®); Pertuzumab (Omnitarg®); Neratinib (HKI-272, (2E)-N-[4-[[3-cloro-4-[(piridin-2-il)metoxi]fenil]amino]-3-ciano-7-etoxiquinolin-6-il]-4-(dimetilamino)but-2-enamida, y se describen en la publicación PCT No. WO 05/028443); Lapatinib o ditosilato de Lapatinib (Tykerb®); (3R,4R)-4-amino-1-((4-((3-metoxifenil)amino)pirrolo[2,1-f][1,2,4]triazin-5-il)metil)piperidin-3-ol (BMS690514); (2E)-N-[4-[(3-cloro-4-fluorofenil)amino]-7-[[(3S)-tetrahidro-3-furanil]oxi]-6-quinazolinil]-4-(dimetilamino)-2-butenamida (BIBW-2992, CAS 850140-72-6); ácido N-[4-[[1-[(3-fluorofenil)metil]-1H-indazol-5-il]amino]-5-metilpirrolo[2,1-f][1,2,4]triazin-6-il]-carbámico, éster de (3S)-3-morfolinilmetilo (BMS 599626, CAS 714971-09-2); diclorhidrato de Canertinib (PD183805 o CI-1033); y N-(3,4-dicloro-2-fluorofenil)-6-metoxi-7-[[(3aa,5|3,6aa)-octahidro-2-metilciclopenta[c]pirrol-5-il]metoxi]-4-quinazolinamina (XL647, CAS 781613-23-8).
Los inhibidores de HER3 incluyen, sin limitación, LJM716, MM-121, AMG-888, RG7116, REGN-1400, AV-203, MP-RM-1, MM-111, y MEHD-7945A.
Los inhibidores de MET incluyen, sin limitación, Cabozantinib (XL184, CAS 849217-68-1); Foretinib (GSK1363089, anteriormente XL880, CAS 849217-64-7); Tivantinib (ARQ197, CAS 1000873-98-2); 1-(2-hidroxi-2-metilpropil)-N-(5-(7-metoxiquinolin-4-iloxi)piridin-2-il)-5-metil-3-oxo-2-fenil-2,3-dihidro-1H-pirazol-4-carboxamida (AMG 458); Cryzotinib (Xalkori®, PF-02341066); (3Z)-5-(2,3-dihidro-1 H-indol-1-ilsulfonil)-3-({3,5-dimetil-4-[(4-metilpiperazin-1-il)carbonil]-1H-pirrol-2-il}metilen)-1,3-dihidro-2H-indol-2-ona (SU11271); (3Z)-N-(3-clorofenil)-3-({3,5-dimetil-4-[(4-metilpiperazin-1-il)carbonil]-1H-pirrol-2-il}metilen)-N-metil-2-oxoindolin-5-sulfonamida (SU11274); (3Z)-N-(3-clorofenil)-3-{[3,5-dimetil-4-(3-morfolin-4-ilpropil)-1H-pirrol-2-il]metilen}-N-metil-2-oxoindolin-5-sulfonamida (SU11606); 6-[difluoro[6-(1-metil-1H-pirazol-4-il)-1,2,4-triazolo[4,3-b]piridazin-3-il]metil]-quinolina (JNJ38877605, Ca s 943540-75-8); 2-[4-[1-(quinolin-6-ilmetil)-1H-[1,2,3]triazolo[4,5-b]pirazin-6-il]-1H-pirazol-1-il]etanol (PF04217903, CAS 956905-27-4); N-((2R)-1,4-dioxan-2-ilmetil)-N-metil-N '-[3-(1-metil-1H-pirazol-4-il)-5-oxo-5H-benzo[4,5]ciclohepta[1,2-b]piridin-7-il]sulfamida (MK2461, CAS 917879-39-1); 6-[[6-(1-Metil-1 H-pirazol-4-il)-1,2,4-triazolo[4,3-£>]piridazin-3-il]tio]-quinolina (SGX523, CAS 1022150-57-7); y (3Z)-5-[[(2,6-diclorofenil)metil]sulfonil]-3-[[3,5-dimetil-4-[[(2R)-2-(1-pirrolidinilmetil)-1-pirrolidinil]carbonil]-1H-pirrol-2-il]metilen]-1,3-dihidro-2H-indol-2-ona (PHA665752, CAS 477575-56-7).
Los inhibidores de IGF1R incluyen, sin limitación, BMS-754807, XL-228, OSI-906, GSK0904529A, A-928605, AXL1717, KW-2450, MK0646, AMG479, IMCA12, MEDI-573, y BI836845; véase, por ejemplo, Yee, JNCI, 104; 975 (2012) para una revisión.
En otro aspecto, la presente divulgación provee un método de tratamiento del cáncer, administrándole a un sujeto en necesidad del mismo un conjugado anticuerpo-fármaco en combinación con uno o más inhibidores de la ruta de señalización secuencia debajo de FGF, que incluyen, sin limitación, inhibidores de MEK, inhibidores de Braf, inhibidores de PI3K/Akt, inhibidores de SHP2, y también inhibidores de mTor.
Por ejemplo, los inhibidores de la proteína quinasa activada por mitógeno (MEK) incluyen, sin limitación, XL-518 (conocido también como GDC-0973, Cas No. 1029872-29-4, disponible de a Cc Corp.); 2-[(2-cloro-4-yodofenil)amino]-N-(ciclopropilmetoxi)-3,4-difluoro-benzamida (conocida también como CI-1040 o PD184352 y descrita en la publicación de PCT No. WO2000035436); N-[(2R)-2,3-Dihidroxipropoxi]-3,4-difluoro-2-[(2-fluoro-4-yodofenil)amino]-benzamida (conocida también como PD0325901 y descrita en la publicación de PCT No. WO2002006213); 2,3-Bis[amino[(2-aminofenil)tio]metilen]-butanodinitrilo (conocido también como U0126 y descrito en la patente de EE. UU. No. 2.779.780);
N-[3,4-Difluoro-2-[(2-fluoro-4-yodofenil)amino]-6-metoxifenil]-1-[(2R)-2,3-dihidroxipropil]-ciclopropanosulfonamida (conocida también como RDEA119 o BAY869766 y descrita en la publicación de Pc T No. WO2007014011); (3S,4R,5Z,8S,9S,11E)-14-(Etilamino)-8,9,16-trihidroxi-3,4-dimetil-3,4,9,19-tetrahidro-1H-2-benzoxaciclotetradecin-1,7(8H)-diona] (conocida también como E6201 y descrita en la publicación de PCT No. WO2003076424); 2’-Am ino-3’-metoxiflavona (conocida también como PD98059, disponible de Biaffin GmbH & Co., KG, Alemania); Vemurafenib (PLX-4032, CAS 918504-65-1); (R)-3-(2,3-Dihidroxipropil)-6-fluoro-5-(2-fluoro-4-yodofenilamino)-8-metilpirido[2,3-d]pirimidin-4,7(3H,8H)-diona (TAK-733, CAS 1035555-63-5); Pimasertib (AS-703026, CAS 1204531-26-9); y dimetilsulfóxido de trametinib (Gs K-1120212, CAS 1204531-25-80).
Los inhibidores de fosfatidilinositol-3-quinasa (PI3K) incluyen, sin limitación, 4-[2-(1H-Indazol-4-il)-6-[[4-(metilsulfonil)piperazin-1-il]metil]tieno[3,2-d]pirimidin-4-il]morfolina (conocida también como GDC 0941 y descrita en las publicaciones de PCT Nos. WO 09/036082 y WO 09/055730); 2-M etil-2-[4-[3-metil-2-oxo-8-(quinolin-3-il)-2,3-dihidroimidazo[4,5-c]quinolin-1 -il]fenil]propionitrilo (conocido también como BEZ 235 o NVP-BEZ 235, y descrito en la publicación de PCT No. WO 06/122806); 4-(trifluorometil)-5-(2,6-dimorfolinopirimidin-4-il)piridin-2-amina (conocida también como BKM120 o NVP-BKM120, y descrita en la publicación de PCT No. WO2007/084786); Tozasertib (VX680 o MK-0457, CAS 639089-54-6); (5Z)-5-[[4-(4-Piridinil)-6-quinolinil]metilen]-2,4-tiazolidindiona (GSK1059615, CAS 958852-01-2); (1 E,4S,4aR,5R,6aS,9aR)-5-(Acetiloxi)-1-[(di-2-propenilamino)metilen]-4,4a,5,6,6a,8,9,9a-octahidro-11-hidroxi-4-(metoximetil)-4a,6a-dimetil-ciclopenta[5,6]nafto[1,2-c]piran-2,7,10(1 H)-triona (PX866, CAS 502632-66 8); y 8-fenil-2-(morfolin-4-il)-cromen-4-ona (LY294002, CAS 154447-36-6).
Los inhibidores de mTor incluyen, sin limitación, Temsirolimus (Torisel®); Ridaforolimus (anteriormente conocido como deferolimus, (1fí,2fí,4S)-4-[(2fi)-2[(1fí,9S,12S,15fí,16E,18fí,19fí,21fí,23S,24E,26E,28Z,30S,32S,35fí)-1,18-dihidroxi-19,30-dimetoxi-15,17,21,23,29,35-hexametil-2,3,10,14,20-pentaoxo-11,36-dioxa-4-azatriciclo[30.3.1.049]hexatriaconta-16,24,26,28-tetraen-12-il]propil]-2-metoxiciclohexil dimetilfosfinato, conocido también como AP23573 y MK8669, y descrito en la publicación de PCT No. WO 03/064383); Everolimus (Afinitor® o RAD001); Rapamicina (AY22989, Sirolimus®); Simapimod (CAS 164301-51-3); (5-{2,4-Bis[(3S)-3-metilmorfolin-4-il]pirido[2,3-d]pirimidin-7-il}-2-metoxifenil)metanol (AZD8055); 2-Amino-8-[trans-4-(2-hidroxietoxi)ciclohexil]-6-(6-metoxi-3-piridinil)-4-metil-pirido[2,3-c/]pirimidin-7(8H)-ona (PF04691502, CAS 1013101-36-4); y N2-[1,4-dioxo-4-[[4-(4-oxo-8-fenil-4E-1-benzopiran-2-il)morfolinio-4-il]metoxi]butil]-L-arginilglicil-L-a-aspartil-L-serina, sal interna (SF1126, CAS 936487-67-1).
En otro aspecto, la presente divulgación provee un método de tratamiento del cáncer administrándole a un sujeto en necesidad del mismo un conjugado anticuerpo-fármaco en combinación con uno o más agentes pro-apoptóticos, que incluyen, sin limitación, inhibidores de IAP, inhibidores de Bcl2, inhibidores de MCl1, agentes de Trail, inhibidores de Chk.
Por ejemplo, los inhibidores de IAP incluyen, sin limitación, NVP-LCL161, GDC-0917, AEG-35156, AT406, y TL32711. Otros ejemplos de inhibidores de IAP incluyen, sin limitación, los que se describen en WO04/005284, WO 04/007529, WO05/097791, WO 05/069894, WO 05/069888, WO 05/094818, US2006/0014700, US2006/0025347, WO 06/069063, WO 06/010118, WO 06/017295, y WO08/134679, todas las cuales se incorporan aquí como referencia.
Los inhibidores de BCL-2 incluyen, sin limitación, 4-[4-[[2-(4-clorofenil)-5,5-dimetil-1-ciclohexen-1-il]metil]-1-piperazini l]-N-[[4-[[(1 R)-3-(4-morfolinil)-1-[(feniltio)metil]propil]amino]-3-[(trifluorometil)sulfonil]fenil]sulfonil]benzamida (conocida también como ABT-263 y descrita en la publicación de PCT No. Wo 09/155386); Tetrocarcin A; Antimicina; Gossypol ((-)BL-193); Obatoclax; Etil-2-amino-6-ciclopentil-4-(1-ciano-2-etoxi-2-oxoetil)-4Hcromeno-3-carboxilato (HA14-1); Oblimersen (G3139, Genasense®); péptido Bak BH3; ácido (-) -Gossypol acético (AT-101); 4-[4-[(4'-cloro[1,1'-bifenil]-2-il)metil]-1-piperazinil]-N-[[4-[[(1 R)-3-(dimetilamino)-1-[(feniltio)metil]propil]amino]-3-nitrofenil]sulfonil]-benzamida (ABT-737, CAS 852808-04-9); y Navitoclax (ABT-263, CAS 923564-51-6).
Los agonistas del receptor proapoptótico (PARA) que incluyen DR4 (TRAILR1) y DR5 (TRAILR2), incluyen, sin limitación, Dulanermin (AMG-951, RhApo2L/TRAIL); Mapatumumab (HRS-ETR1, CAS 658052-09-6); Lexatumumab (HGS-ETR2, CAS 845816-02-6); Apomab (Apomab®); Conatumumab (AMG655, CAS 896731-82-1); y Tigatuzumab (CS1008, CAS 946415-34-5, disponible de Daiichi Sankyo).
Los inhibidores de la quinasa de checkpoint (CHK) incluyen, sin limitación, 7-hidroxiestaurosporina (UCN-01); 6-Bromo-3-(1 -metil-1 E-pirazol-4-il)-5-(3fí)-3-piperidinil-pirazolo[1,5-a]pirimidin-7-amina (SCH900776, CAS 891494-63-6); N-[(S)-piperidin-3-il]amida del ácido 5-(3-fluorofenil)-3-ureidotiofen-2-carboxílico (AZD7762, CAS 860352-01-8); 4 -[((3S)-1-Azabiciclo[2.2.2]oct-3-il)amino]-3-(1H-bencimidazol-2-il)-6-cloroquinolin-2(1H)-ona (CHIR 124, CAS 405168-58-3); 7-Aminodactinomicina (7-AAD), Isogranulatimida, debromohimenialdisina; N-[5-Bromo-4-metil-2-[(2S)-2-morfolinilmetoxi]-fenil]-N'-(5-metil-2-pirazinil)urea (LY2603618, CAS 911222-45-2); Sulforafano (CAS 4478
93-7, isotiocianato de 4-metilsulfinilbutilo); 9,10,11,12-Tetrahidro-9,12-epoxi-1H-diindolo[1,2,3-/y:3',2',1'-W]pirrolo[3,4-/|[1,6]benzodiazocin-1,3(2H)-diona (SB-218078, CAS 135897-06-2); y TAT-S216A (Sha et al., Mol. Cáncer. Ther2007; 6(1):147-153), y CBP501 ((d-Bpa)sws(d-Phe-F5)(d-Cha)rrrqrr).
En un aspecto, la presente divulgación provee un método de tratamiento del cáncer, administrándole a un sujeto en necesidad del mismo un conjugado anticuerpo-fármaco en combinación con uno o más inhibidores de FGFR. Por ejemplo, los inhibidores de FGFR incluyen, sin limitación, Brivanib alaninato (BMS-582664, (S)-((R)-1-(4-(4-Fluoro-2-metil-1 H-indol-5-iloxi)-5-metilpirrolo[2,1-f][1,2,4]triazin-6-iloxi)propan-2-il)2-aminopropanoato); Vargatef (BIBF1120, CAS 928326-83-4); ácido dovitinib diláctico (TKI258, CAS 852433-84-2); 3-(2,6-Dicloro-3,5-dimetoxi-fenil)-1-{6-[4-(4-etil-piperazin-1-il)-fenilamino]-pirimidin-4-il}-1-metil-urea (BGJ398, CAS 872511-34-7); Danusertib (pHa - 739358); y (PD173074, CAS 219580-11-7). En un aspecto específico, la presente divulgación provee un método de tratamiento del cáncer administrándole a un sujeto en necesidad del mismo un conjugado anticuerpo-fármaco en combinación con un inhibidor de FGFR2, tal como 3-(2,6-dicloro-3,5-dimetoxifenil)-1-(6((4-(4-etilpiperazin-1-il)fenil)amino)pirimidin-4-il)-1 - metilurea (conocida también como BGJ-398); o 4-amino-5-fluoro-3-(5-(4-metilpiperazin1-il)-1 H-benzo[d]imidazol-2 - il)quinolin-2(1H)-ona (conocido también como dovitinib o TKI-258). AZD4547 (Gavine et al., 2012, Cáncer Research 72, 2045-56, N-[5-[2-(3,5-Dimetoxifenil)etil]-2H-pirazol-3-il]-4-(3R,5S)-diemetilpiperazin-1-il)benzamida), Ponatinib (AP24534; Gozgit et al., 2012, Mol Cáncer Ther., 11; 690-99; 3-[2-(imidazo[1,2-b]piridazin-3-il)etinil]-4-metil-N-{4-[(4-metilpiperazin-1-il)metil]-3-(trifluorometil)fenil}benzamida, CAS 943319-70-8).
Composiciones farmacéuticas
Para preparar composiciones farmacéuticas o estériles que incluyen inmunoconjugados, los inmunoconjugados de la presente divulgación se mezclan con un vehículo o excipiente farmacéuticamente aceptable. Adicionalmente, las composiciones pueden contener uno o más de otros agentes terapéuticos que son adecuados para el tratamiento o prevención de tumores estromales gastrointestinales (GIST), cáncer de pulmón de célula pequeña (SCLC), leucemia mieloide aguda (AML), melanoma, leucemia de mastocitos (MCL), mastocitosis, neurofibromatosis, cáncer de mama, cáncer de pulmón de célula no pequeña (NSCLC), y cáncer pancreático.
Las formulaciones de agentes terapéuticos y diagnósticos se pueden preparar mezclando con vehículos, excipientes o estabilizadores fisiológicamente aceptables en forma, por ejemplo, de polvos liofilizados, suspensiones, soluciones acuosas, lociones o suspensiones (véase por ejemplo Hardman et al., “Goodman and Gilman's The Pharmacological Basis of Therapeutics”, McGraw-Hill, Nueva York, N.Y., 2001; Gennaro, “Remington: The Science and Practice of Pharmacy”, Lippincott, Williams and Wilkins, Nueva York, N.Y., 2000; Avis, et al. (eds.), “Pharmaceutical Dosage Forms: Parenteral Medications”, Marcel Dekker, NY, 1993; Lieberman, et al. (eds.), “Pharmaceutical Dosage Forms: Tablets”, Marcel Dekker, NY, 1990; Lieberman, et al. (eds.) “Pharmaceutical Dosage Forms: Disperse Systems”, Marcel Dekker, NY, 1990; Weiner y Kotkoskie, “Excipient Toxicity and Safety”, Marcel Dekker, Inc., Nueva York, N.Y., 2000).
En un aspecto específico, la forma de servicio clínico (CSF) de los conjugados anticuerpo-fármaco de la presente divulgación es un liofilizado en un vial que contiene el ADC, succinato de sodio y polisorbato 20. El liofilizado se puede reconstituir con agua para inyección; la solución comprende el ADC, succionato de sodio, sacarosa y polisorbato 20 a un pH de aproximadamente de 5,0. Para administración intravenosa subsecuente la solución obtenida usualmente se diluirá adicionalmente en una solución vehículo.
La selección de un régimen de administración para un agente terapéutico depende de varios factores que incluyen la velocidad de recambio de la entidad en el suero o tejido, la severidad de los síntomas, la inmunogenicidad de la entidad, y la accesibilidad de las células objetivo en la matriz biológica. En ciertos aspectos, un régimen de administración maximiza la cantidad de agente terapéutico suministrado al paciente consistentemente con un grado aceptable de efectos secundarios. Por consiguiente, la cantidad de agente biológico suministrado depende en parte de la entidad particular y la severidad de la afección tratada. Se tienen disponibles guías para seleccionar dosis apropiadas de anticuerpos, citocinas y moléculas pequeñas (véase por ejemplo Wawrzynczak, “Antibody Therapy”, Bios Scientific Pub. Ltd, Oxfordshire, Reino Unido, 1996; Kresina (ed.), “Monoclonal Antibodies, Cytokines and Arthritis”, Marcel Dekker, Nueva York, N.Y., 1991; Bach (ed.), “Monoclonal Antibodies and Peptide Therapy in Autoimmune Diseases”, Marcel Dekker, Nueva York, N.Y., 1993; Baert et al., New Engl. J. Med. 348:601-608, 2003; Milgrom et al., New Engl. J. Med. 341:1966-1973, 1999; Slamon et al., New Engl. J. Med. 344:783-792, 2001; Beniaminovitz et al., New Engl. J. Med. 342:613-619, 2000; Ghosh et al., New Engl. J. Med. 348:24-32, 2003; Lipsky et al., New Engl. J. Med. 343:1594-1602, 2000).
La determinación de la dosis apropiada la realiza el médico, por ejemplo, usando parámetros o factores conocidos o sospechosos de afectar el tratamiento, o que se espera afecten el tratamiento. En general, la dosis empieza con una cantidad un poco menor que la dosis óptima y se aumenta en incrementos pequeños posteriormente hasta lograr el efecto deseado u óptimo con respecto a cualquier efecto secundario negativo. Las mediciones diagnósticas importantes incluyen
las de los síntomas, por ejemplo la inflamación o cantidad de citocinas inflamatorias producidas.
Las dosis reales de los ingredientes activos en los conjugados anticuerpo-fármaco de las composiciones farmacéuticas se pueden variar a fin de obtener una cantidad de ingrediente activo que sea eficaz para obtener la respuesta terapéutica deseada para un paciente particular, composición y modo de administración, sin ser tóxica para el paciente. La dosis seleccionada dependerá de una variedad de factores farmacocinéticos que incluyen la actividad de las composiciones particulares de la presente divulgación utilizadas, o el éster, sal o amida del mismo, la vía de administración, el tiempo de administración, la velocidad de excreción del compuesto particular utilizado, la duración del tratamiento, otros fármacos, compuestos o materiales usados en combinación con las composiciones particulares utilizadas, la edad, sexo, peso, condición, salud general, e historia médica previa del paciente tratado, y factores similares conocidos en las ciencias médicas.
Las composiciones que comprenden los anticuerpos o fragmentos de los mismos pueden ser provistas por infusión continua, o por dosis a intervalos de por ejemplo 1 día, una semana, o 1-7 veces a la semana. Las dosis se pueden proveer por vía intravenosa, subcutánea, tópica, oral, nasal, rectal, intramuscular, intracerebral, o por inhalación. Un protocolo de dosis específico es el que incluye la dosis máxima o frecuencia de dosis que evita efectos secundarios indeseables significativos.
Para los inmunoconjugados de la presente divulgación, la dosis administrada a un paciente puede ser de 0,0001 mg/kg a 100 mg/kg del peso corporal del paciente. La dosificacion puede ser entre 0,0001 mg/kg y 20 mg/kg, 0,0001 mg/kg y 10 mg/kg, 0,0001 mg/kg y 5 mg/kg, 0,0001 y 2 mg/kg, 0,0001 y 1 mg/kg, 0,0001 mg/kg y 0,75 mg/kg, 0,0001 mg/kg y 0,5 mg/kg, 0,0001 mg/kg a 0,25 mg/kg, 0,0001 a 0,15 mg/kg, 0,0001 a 0,10 mg/kg, 0,001 a 0,5 mg/kg, 0,01 a 0,25 mg/kg o 0,01 a 0,10 mg/kg del peso corporal del paciente. La dosis de los anticuerpos o fragmentos de los mismos se puede calcular usando el peso del paciente en kilogramos (kg) multiplicado por la dosis por administrar en mg/kg.
Las dosis de los inmunoconjugados se pueden repetir y las administraciones se pueden separar por al menos 1 día, 2 días, 3 días, 5 días, 10 días, 15 días, 30 días, 45 días, 2 meses, 75 días, 3 meses, o por lo menos 6 meses. En un aspecto específico, las dosis de los inmunoconjugados de la presente divulgación se repiten cada 3 semanas.
Una cantidad eficaz para un paciente particular puede variar dependiendo de factores tales como la afección tratada, la salud general del paciente, el método, vía y dosis de administración, y la severidad de los efectos secundarios (véase por ejemplo Maynard etal., “A Handbook of SOPs for Good Clinical Practice”, Interpharm Press, Boca Raton, Fla., 1996; Dent, “Good Laboratory and Good Clinical Practice”, Urch Publ., Londres, Reino Unido, 2001).
La vía de administración puede ser por ejemplo, por aplicación tópica o cutánea, inyección o infusión por vía intravenosa, intraperitoneal, intracerebral, intramuscular, intraocular, intraarterial, intracerebroespinal, intralesional, o mediante sistemas de liberación sostenida o un implante (véase por ejemplo Sidman et al., Biopolymers 22:547-556, 1983; Langer et al., J. Biomed. Mater. Res. 15:167-277, 1981; Langer, Chem. Tech. 12:98-105, 1982; Epstein et al., Proc. Natl. Acad. Sci. USA 82:3688-3692, 1985; Hwang et al., Proc. Natl. Acad. Sci. USA 77:4030-4034, 1980; patentes de EE. UU. Nos.
6.350.466 y 6.316.024). Cuando sea necesario, la composición también puede incluir un agente de solubilizacion o un anestésico local tal como lidocaína para mitigar el dolor en el sitio de la inyección, o ambas cosas. Además también se puede utilizar administración pulmonar, por ejemplo usando un inhalador o nebulizador, y formulación con un agente de atomización. Véase por ejemplo las patentes de EE. UU. Nos. 6.019.968, 5.985.320, 5.985.309, 5.934.272, 5.874.064, 5.855.913, 5.290.540 y 4.880.078; y las publicaciones de PCT Nos. WO 92/19244, WO 97/32572, WO 97/44013, WO 98/31346, y WO 99/66903, cada una de las cuales se incorpora aquí como referencia en su totalidad.
Una composición de la presente divulgación también se puede administrar por medio de una o más vías de administración usando uno o más de una variedad de métodos conocidos. Como apreciará el experto en la materia, la vía y/o modo de administración variarán dependiendo de los resultados deseados. Las vías de administración seleccionadas para los inmunoconjugados incluyen la vía de administración intravenosa, intramuscular, intradérmica, intraperitoneal, subcutánea, espinal u otras vías de administración parenterales, por ejemplo por inyección o infusión. La administración parenteral puede representar modos de administración diferentes de la administración enteral y tópica, usualmente por inyección, e incluye, sin limitación, inyección e infusión intravenosa, intramuscular, intraarterial, intratecal, intracapsular, intraorbital, intracardiaca, intradérmica, intraperinoteal, transtraqueal, subcutánea, subcuticular, intraarticular, subcapsular, subaracnoide, intraespinal, epidural e intraesternal. Alternativamente, una composición de la presente divulgación se puede administrar por medio de una vía no parenteral, tal como una vía de administración tópica, epidérmica o mucosal, por ejemplo por vía intranasal, oral, vaginal, rectal, sublingual o tópica. En un aspecto, los inmunoconjugados de la presente divulgación se administran por infusión. En otro aspecto, los inmunoconjugados se administran por vía subcutánea.
Si los inmunoconjugados de la presente divulgación se administran en un sistema de liberación controlada o liberación sostenida, se puede usar una bomba para lograr la liberación controlada o sostenida (véase Langer, supra; Sefton, CRC Crit. Ref Biomed. Eng. 14:20, 1987; Buchwald etal., Surgery88:507, 1980; Saudek etal., N. Engl. J. Med.321:574, 1989). Se pueden usar materiales poliméricos para obtener la liberación controlada o sostenida de las terapias de los inmunoconjugados (véase, por ejemplo, “Medical Applications of Controlled Release”, Langer y Wise (eds.), CRC Pres., Boca Raton, Florida, 1974; “Controlled Drug Bioavailability, Drug Product Design and Performance”, Smolen y Ball (eds.), Wiley, Nueva York, 1984; Ranger y Peppas, J. Macromol. Sc¡. Rev. Macromol. Chem. 23:61, 1983; véase también Levy et al., Science 228:190, 1985; During et al., Ann. Neurol. 25:351, 1989; Howard et al., J. Neurosurg. 71:105, 1989; patente de EE. UU. No. 5.679.377; patente de EE. UU. No. 5.916.597; patente de EE. UU. No. 5.912.015; patente de EE. Uu . No.
5.989.463; patente de EE. UU. No. 5.128.326; la publicación de PCT No. WO 99/15154; y la publicación de PCT No. WO 99/20253. Los ejemplos de polímeros usados en las formulaciones de liberación sostenida incluyen, sin limitación, poli(2-hidroxietilmetacrilato), poli(metilmetacrilato), poli(ácido acrílico), poli(etilen-co-acetato de vinilo), poli(ácido metacrílico), poliglicólidos (PLG), polianhídridos, poli(N-vinilpirrolidona), poli(alcohol vinílico), poliacrilamida, poli(etilenglicol), poliláctidas (pLa ), poli(láctida-co-glicólidos) (PLGa ), y poliortoésteres. En un aspecto, el polímero usado en una formulación de liberación sostenida es inerte, libre de impurezas lixiviables, estable en almacenamiento, estéril y biodegradable. Un sistema de liberación controlada o sostenida se puede colocar en la proximidad del agente profiláctico o terapéutico, requiriéndose así sólo una fracción de la dosis sistémica (véase, por ejemplo, Goodson, en “Medical Applications of Controlled Release”, supra, volumen 2, p. 115-138, 1984).
Los sistemas de liberación controlada se exponen en la revisión de Langer, Science 249:1527-1533, 1990. Cualquier técnica conocida para el experto en la materia se puede usar para producir formulaciones de liberación sostenida que comprenden uno o más inmunoconjugados de la presente divulgación; véase, por ejemplo, la patente de EE. UU. No.
4.526.938; la publicación de PCT WO 91/05548; la publicación de PCT w O 96/20698; Ning et al., Radiotherapy & Oncology 39:179-189, 1996; Song et al., PDA Journal of Pharmaceutical Science & Technology 50:372-397, 1995; Cleek et al., Pro. Int'l. Symp. Control. Rel. Bioact. Mater. 24:853-854, 1997; y Lam et al., Proc. Int'l. Symp. Control Rel. Bioact. Mater. 24:759-760, 1997, cada una de las cuales se incorpora aquí como referencia en su totalidad.
Si los inmunoconjugados de la divulgación se administran por vía tópica, se pueden formular en forma de un ungüento, crema, parche transdérmico, loción, gel, champú, espray, aerosol, solución, emulsión, u otra forma muy conocida para el experto en la materia; véase, por ejemplo, “Remington's Pharmaceutical Sciences and Introduction to Pharmaceutical Dosage Forms”, 19a edición, Mack Pub. Co., Easton, Pa. (1995). Para las formas farmacéuticas tópicas no atomizables, típicamente se emplean formas viscosas a semisólidas o sólidas, que comprenden un vehículo o uno o más excipientes compatibles con la aplicación tópica, y que tienen una viscosidad dinámica, en algunos casos, mayor que la del agua. Las formulaciones adecuadas incluyen, sin limitación, soluciones, suspensiones, emulsiones, cremas, ungüentos, polvos, linimentos, pomadas y similares que, si se desea, se esterilizan o se mezclan con agentes auxiliares (por ejemplo conservadores, estabilizadores, agentes de mojado, amortiguadores o sales) para alterar varias propiedades tales como por ejemplo la presión osmótica. Otras formas farmacéuticas tópicas adecuadas incluyen las preparaciones de aerosol atomizables, en donde el ingrediente activo, en algunos casos, en combinación con un vehículo sólido o líquido inerte, se envasa en una mezcla con un material volátil presurizado (por ejemplo, un propulsor gaseoso como freón) o en una botella exprimible. Si así se desea también se le pueden añadir agentes de mojado o humectantes a las composiciones farmacéuticas y formas farmacéuticas. Los ejemplos de tales ingredientes adicionales son muy conocidos.
Si las composiciones que comprenden los inmunoconjugados se administran por vía intranasal, se puede formular en forma de aerosol, espray, niebla o en forma de gotas. En particular, los agentes profilácticos o terapéuticos para usarse de acuerdo con la presente divulgación se pueden suministrar convenientemente en forma de una presentación de espray de aerosol desde envases presurizados o un nebulizador, utilizando un propulsor adecuado (por ejemplo, diclorodifluorometano, triclorofluorometano, diclorotetrafluoroetano, dióxido de carbono u otro gas adecuado). En el caso de un aerosol presurizado, la dosis unitaria puede ser determinada suministrando una válvula para suministrar una cantidad medida. Se pueden formular cápsulas y cartuchos (compuestos, por ejemplo, de gelatina) para usarse en un inhalador o insuflador, que contiene una mezcla en polvo del compuesto y una base de polvo adecuada como lactosa o almidón.
Los métodos de coadministración o tratamiento con un segundo agente terapéutico, por ejemplo una citocina, esteroide, agente quimioterapéutico, antibiótico o radiación, son conocidos (véase, por ejemplo, Hardman et al. (eds.) (2001) Goodman y Gilman's The Pharmacological Basis of Therapeutics, 10a ed., McGraw-Hill, Nueva York, N.Y.; Poole y Peterson (eds.) (2001) “Pharmacotherapeutics for Advanced Practice: A Practical Approach”, Lippincott, Williams & Wilkins, Phila., Pa.; Chabner y Longo (eds.) (2001) “Cancer Chemotherapy and Biotherapy”, Lippincott, Williams & Wilkins, Phila., Pa.). Una cantidad eficaz del agente terapéutico puede reducir los síntomas en por lo menos 10%; por lo menos 20%; por lo menos aproximadamente 30%; por lo menos 40%, o por lo menos 50%.
Las terapias adicionales (por ejemplo, agentes profilácticos o terapéuticos) que se pueden administrar en combinación con los inmunoconjugados, se pueden administrar con menos de 5 minutos de separación, menos de 30 minutos de separación, 1 hora de separación, con aproximadamente 1 hora de separación, con aproximadamente 1 a aproximadamente 2 horas de separación, con aproximadamente 2 horas a aproximadamente 3 horas de separación, con aproximadamente 3 horas a aproximadamente 4 horas de separación, con aproximadamente 4 horas a aproximadamente 5 horas de separación, con aproximadamente 5 horas a aproximadamente 6 horas de separación, con aproximadamente 6 horas a aproximadamente 7 horas de separación, con aproximadamente 7 horas a aproximadamente 8 horas de separación, con aproximadamente 8 horas a aproximadamente 9 horas de separación, con aproximadamente 9 horas a aproximadamente 10 horas de separación, con aproximadamente 10 horas a aproximadamente 11 horas de separación, con aproximadamente 11 horas a aproximadamente 12 horas de separación, con aproximadamente 12 horas a 18 horas de separación, 18 horas a 24 horas de separación, 24 horas a 36 horas de separación, 36 horas a 48 horas de separación, 48 horas a 52 horas de separación, 52 horas a 60 horas de separación, 60 horas a 72 horas de separación, 72 horas a 84 horas de separación, 84 horas a 96 horas de separación, o 96 horas a 120 horas de separación de los inmunoconjugados de la presente divulgación. Las dos o más terapias se pueden administrar en la misma visita del paciente.
En ciertos aspectos, los inmunoconjugados se pueden formular para asegurar una distribución adecuada in vivo. Por ejemplo, la barrera hematoencefálica (BBB) excluye muchos compuestos muy hidrófilos. Para asegurar que los compuestos terapéuticos de la divulgación atraviesen la BBB (si así se desea), se pueden formular, por ejemplo, en liposomas. Para métodos de fabricación de liposomas véanse por ejemplo las patentes de EE. UU. Nos. 4.522.811; 5.374.548; y 5.399.331. Los liposomas pueden comprender una o más porciones que son transportadas selectivamente a células u órganos específicos, de esta manera incrementan el suministro dirigido del fármaco (véase por ejemplo, Ranade (1989) J. Clin. Pharmacol. 29:685). Porciones de direccionamiento ejemplares incluyen folato o biotina (véase por ejemplo la patente de EE. UU. No. 5.416.016 de Low etal); manósidos (Umezawa etal, (1988) Biochem. Biophys. Res. Commun. 153: 1038); anticuerpos (Bloeman et al. (1995) FEBS Lett. 357:140; Owais et al. (1995) Antimicrob. Agents Chemother. 39:180); receptor de proteína A tensoactivo (Briscoe et al. (1995) Am. J. Physiol 1233:134); p120 (Schreier et al. (1994) J. Biol. Chem. 269:9090); véase también K. Keinanen; M.L. Laukkanen (1994) FEBS Lett. 346:123; JJ. Killion; I.J. Fidler (1994; Immunomethods 4:273).
La presente divulgación provee protocolos para la administración de una composición farmacéutica que comprende inmunoconjugados solos o en combinación con otras terapias a un sujeto en necesidad de los mismos. Las terapias de combinaciónn (por ejemplo, agentes profilácticos o terapéuticos) se pueden administrar de forma concomitante o secuencial a un sujeto. La terapia (por ejemplo, agentes profilácticos o terapéuticos) de las terapias de combinación también se puede administrar cíclicamente. La terapia cíclica incluye la administración de una primera terapia (por ejemplo, un primer agente profiláctico o terapéutico) durante un periodo, seguido por la administración de una segunda terapia (por ejemplo, un segundo agente profiláctico o terapéutico) durante un periodo, y repetir esta administración secuencial, esto es, el ciclo, para reducir el desarrollo de resistencia a una de las terapias (por ejemplo, agentes) para evitar o reducir los efectos secundarios de una de las terapias (por ejemplo, agentes) o para mejorar la eficacia de las terapias.
Las terapias (por ejemplo, agentes profilácticos o terapéuticos) de las terapias de combinación de la divulgación se le pueden administrar a un sujeto de forma concurrente.
El término “de forma concurrente” no se limita a la administración de terapias (por ejemplo, agentes profilácticos o terapéuticos) exactamente al mismo tiempo, más bien significa que una composición farmacéutica que comprende anticuerpos o fragmentos de los mismos se administra a un sujeto en una secuencia y dentro de un intervalo de tiempo, de tal manera que los inmunoconjugados pueden actuar juntos con las otras terapias para proveer un mayor beneficio que si se administraran de otra manera. Por ejemplo, cada terapia se le puede administrar a un sujeto al mismo tiempo o secuencialmente en cualquier orden en diferentes puntos de tiempo; sin embargo, si no se administran al mismo tiempo, deben ser administradas suficientemente próximas en el tiempo a fin de proveer el efecto terapéutico o profiláctico deseado. Cada terapia se puede administrar a un sujeto por separado, en cualquier forma apropiada y por cualquier vía adecuada. En diversos aspectos, las terapias (por ejemplo, agentes profilácticos o terapéuticos) se administran a un sujeto con menos de 15 minutos, menos de 30 minutos, menos de 1 hora de separación, con aproximadamente 1 hora de separación, con aproximadamente 1 hora a aproximadamente 2 horas de separación, con aproximadamente 2 horas a aproximadamente 3 horas de separación, con aproximadamente 3 horas a aproximadamente 4 horas de separación, con aproximadamente 4 horas a aproximadamente 5 horas de separación, con aproximadamente 5 horas a aproximadamente 6 horas de separación, con aproximadamente 6 horas a aproximadamente 7 horas de separación, con aproximadamente 7 horas a aproximadamente 8 horas de separación, con aproximadamente 8 horas a aproximadamente 9 horas de separación, con aproximadamente 9 horas a aproximadamente 10 horas de separación, con aproximadamente 10 horas a aproximadamente 11 horas de separación, con aproximadamente 11 horas a aproximadamente 12 horas de separación,
24 horas de separación, 48 horas de separación, 72 horas de separación, o 1 semana de separación. En otros aspectos, dos o más terapias (por ejemplo agentes profilácticos o terapéuticos) se administran en la misma visita del paciente.
Los agentes profilácticos o terapéuticos de las terapias de combinación se pueden administrar a un sujeto en la misma composición farmacéutica. Alternativamente, los agentes profilácticos o terapéuticos de las terapias de combinación se le pueden administrar simultáneamente a un sujeto en composiciones farmacéuticas separadas. Los agentes profilácticos o terapéuticos pueden administrarse a un sujeto por la misma vía o diferentes vías de administración.
Ejemplos
Ejemplo 1: Generación de Abs de cKIT por tecnología de hibridoma
Antígeno y otras proteínas
Se generó una línea celular de expresión transitoria que secreta proteína cKIT humana por transfección de células 293 Freestyle™ (Invitrogen, Carlsbad, Ca). Brevemente, las células cultivadas en medio Freestyle™ (Invitrogen) se transfectaron usando reactivo de transfección 293Fectin™ y un plásmido recombinante que contiene el ECD del ADNc de cKIT humano y una etiqueta His6 en el extremo C de la secuencia, o un Fc murino (pFUSE, Invivogen, San Diego, CA). De 48 a 72 horas después el medio se centrifuga para quitar las células, se esterilizan por filtración y el lisado aclarado se usa para la purificación de proteína.
Para la cKIT etiquetada con His6: El concentrado resultante se aplicó a una columna NiNTA His-Bind Superflow a 0,5 ml/min. Después del lavado de línea de base con PBS, el material enlazado se eluyó con PBS con un gradiente escalonado de imidazol (10-500 mM). El eluato resultante se dializó contra PBS, pH 7,3, se esterilizó por filtración y se distribuyó en alícuotas. Para la fusión Fc-cKIT, se usaron columnas Protein G fastFlow (en lugar de NiNTA) como se describe arriba, y se eluyó con un amortiguador de glicina de pH 3, que se neutralizó con Tris, pH 8.
Generación de hibridoma
Inmunización de ratones y producción de hibridomas
La cKIT purificada se diluyó 1:1 con adyuvante completo de Freund antes de inmunización de ratones transgénicos Bcl-2 (raza C57BL/6-Tgn (bcl-2) 22 Wehi). Los ratones se inmunizaron usando un procedimiento que se denomina inmunización repetitiva en sitios múltiples (RIMMS) (McIntyre GD., Hybridoma, 1997). Brevemente, los ratones se inyectaron con 1-3 pg de antígeno en 8 sitios específicos próximos a los nódulos linfáticos periféricos (PLN). Este procedimiento se repitió 8 veces durante un periodo de 12 días. El día 12 se recolectó un sangrado de prueba y el título de anticuerpo en el suero se analizó por ELISA. Los PLN reunidos se removieron de los ratones de alto título el día 15. Para cosechar los linfocitos, los PLN se lavaron dos veces con DMEM simple y después se disociaron por pasaje a través de un tamiz de 0,22 micrómetros (Falcon #352350, BD Bioscience, San Jose, CA). Los linfocitos resultantes se lavaron dos veces más antes de la fusión. Células de mieloma F0 se mezclaron con linfocitos a una proporción de 2,5 linfocitos a 1 célula F0. La mezcla de células se centrifugó, y subsiguientemente se agregó 1 mL de PEG 1500 gota a gota a la pella celular durante 1 minuto. Después de 30 segundos se agregó lentamente 1 mL de DMEM, y 1 minuto después se agregaron 19 mL de DMEM durante 5 minutos. Las células fusionadas se hicieron pella, se suspendieron a una densidad de 2 x 105 células/mL en medio HAT (DMEM 20 % FBS, Pen/Strep/Glu, 1x NEAA, 1x HAT, 0,5x HFCS), y se pusieron a 37 °C durante 1 hora. Después las células se sembraron en placas de 384 pocillos a razón de 60 pL/pocillo.
Tamizado de hibridomas que secretan anticuerpos para cKIT
Diez días después de la fusión las placas de hibridomas se tamizaron para determinar la presencia de anticuerpos específicos de cKIT. Para el tamizado de ELISA, placas de 384 pocillos Maxisorp (Nunc #464718) se recubrieron con 50 pL de cKIT (diluido a 15 ng/pocillo en PBS) y se incubaron durante la noche a 4 °C. La proteína remanente se aspiró y los pocillos se bloquearon con BSA al 1 % en PBs . Después de 30 minutos de incubación a temperatura ambiente, los pocillos se lavaron 4 veces con PBS Tween al 0.05 % (PBST). Se transfirieron 15 pL del sobrenadante de hibridoma a las placas de ELISA. Se diluyeron 15 pL de suero de ratón, tomado en el momento de la remoción de PLN, 1:1000 en PBS, y se agregaron como control positivo. Se agregaron 50 pL de anticuerpo secundario (anti-IgG de ratón de cabra - HRP (Jackson Immuno Research #115-035-071, West Grove, PA), diluido 1:5000 en PBS) a todos los pocillos sobre las placas de ELISA. Después de incubación a temperatura ambiente durante 1 hora, las placas se lavaron 8 veces con PBSt . Se agregaron 25 pL de TMB (KPL #50-76-05) y después de 30 minutos de incubación a temperatura ambiente; las placas se leyeron a una absorbancia de 605 nm. Las células de los pocillos positivos se expandieron en placas de 24 pocillos en medio HT (DMEM 20 % FBS, Pen/Strep/Glu, 1x NEAA, 1x HT, 0,5x HFCS).
Purificación del anticuerpo
El sobrenadante que contenía los anticuerpos de cKIT se purificaron usando proteína G (Upstate # 16-266 (Billerica, MA)). Antes de cargar el sobrenadante, la resina se equilibró con 10 volúmenes de columna de PBS. Después de la unión de la muestra, la columna se lavó con 10 volúmenes de columna de PBS, y después el anticuerpo se eluyó con 5 volúmenes de columna de glicina 0,1 M, pH 2,0. Las fracciones de la columna se neutralizaron inmediatamente con 1/10 de volumen de T ris HCl, pH 9,0. Se midió la OD280 de las fracciones, y las fracciones positivas se reunieron y se dializaron durante la noche contra PBS, pH 7,2.
Ejemplo 2: Humanización y maduración de afinidad de anticuerpos anti-cKIT
Diseño de la humanización
Las secuencias de VH y VL del anticuerpo anti-cKIT derivado de hibridoma, 9P3, son SEQ ID NO.9 y SEQ ID NO.18, respectivamente. Las secuencias de aminoácidos de los dominios constantes de IgG 1 de humano usadas para generar IgG completa son SEQ ID NO.10 para la cadena pesada y SEQ ID NO.19 para la cadena ligera. La humanización de la cadena pesada se logró injertando las tres regiones CDR (GFTFSDYYMA (SeQ ID NO. 148)) (NINYDGSSTYYLDS (SEQ ID NO.149)) Y (GDYYGTTYWYFDV (SEQ ID NO.150) del anticuerpo anti-cKIT 9P3, sobre el armazón aceptor de línea germinal humana VH3_3-07 (base de datos vBASE). La humanización de la cadena ligera se efectuó injertando las 3 regiones CDR (RASQDISNYLN (SEQ ID NO.151)), (YTSRLQS (SEQ ID NO.152)) y (QQGKKLWS (SEQ ID NO.153)) del anticuerpo anti-cKIT 9P3, sobre el armazón de aceptor de línea germinal humana VK3-L25 (base de datos vBASE) o injertando 2 regiones CDR (SEQ ID NO.152 y SEQ iD NO.153) sobre el armazón aceptor de línea germinal humana VK1-O12 (base de datos vBASE). Además de las regiones CDR, un residuo de armazón del dominio variable de cadena ligera, esto es, VL #71, y en el caso de VK3-L25 VL #79 (numeración de residuos basada en SEQ ID NO. 21) fue retenida de la secuencia 9P3. Además, los elementos J humanos JH4 y JK4 se usaron para la cadena pesada y ligera respectivamente. Las secuencias de aminoácidos resultantes de la cadena pesada de anticuerpo humanizado son SEQ ID NO.11, y para las dos cadenas ligeras SEQ ID NO.20 (VK1-O12) y SEQ ID NO. 21 (VK3-L6).
Creemos que el motivo de aminoácido aspartato seguido por glicina (DG) puede ser susceptible de modificación posterior a la traducción (formación de iso-aspartato) y que las lisinas dentro de las CDR pueden disminuir la fracción de anticuerpo activo después de la conjugación anticuerpo-fármaco. Se aplicó una combinación de mutagénesis aleatoria (esto es, PCR susceptible de error) y mutagénesis dirigida para optimizar los anticuerpos humanizados.
Generación de secuencias humanizadas
Las secuencias de ADN que codifican dominios VL y VH humanizados se compraron en GeneArt (Life Technologies Inc. Regensburg, Alemania), incluyendo optimización de codón para Homo sapiens. Las secuencias que codifican los dominios VL y VH se subclonaron por corte y pegado de los vectores derivados en GeneArt en vectores de expresión adecuados para secreción en células de mamífero. Las cadenas pesada y ligera se clonaron en vectores de expresión individuales para permitir la cotransfección. Los elementos del vector de expresión incluyen un promotor (intensificador-promotor de citomegalovirus (CMV)), una secuencia señal para facilitar la secreción, una señal de poliadenilación y un terminador de transcripción (gen de hormona de crecimiento bovino (BGH)), un elemento que permite la replicación episómica y replicación en procariotas (por ejemplo origen de SV40 y ColE1 u otros conocidos) y elementos que permiten la selección (gen de resistencia a ampicilina y marcador de zeocina).
Expresión y purificación de anticuerpos humanizados
Las células de riñón embrionario humanas que expresan constitutivamente el gen T grande de SV40 (HEK293-T ATCC11268) son una de las líneas celulares hospederas preferidas para la expresión transitoria de proteínas IgG humanizadas u optimizadas. La transfección se realiza usando PEI (Polietilenimina, MW 25,000 lineal, Polysciences, USA Cat.No. 23966) como reactivo de transfección. La solución de reserva de PEI se prepara disolviendo cuidadosamente 1 g de PEI en 900 ml de agua grado cultivo celular a temperatura ambiente (t.a.). Para facilitar la disolución de la PEI, la solución se acidifica agregando HCl a pH 3-5, seguido por neutralización con NaOH a un pH final de 7,05. Finalmente, el volumen se ajusta a 1 L y la solución se filtra a través de un filtro de 0,22 pm, se distribuye en alícuotas y se congela a -80 °C hasta su uso ulterior. Una vez descongelada, una alícuota puede ser congelada nuevamente hasta 3 veces a -20 °C pero no se debe almacenar mucho tiempo a -20 °C. Las células HEK 293T se cultivan usando un medio de cultivo libre de suero patentado de Novartis para transfección y propagación de las células, y medio de cultivo libre de suero ExCell VPRO (SAFC Biosciences, USA, Cat.No. 24561C) como medio de producción/alimentación. Las células preparadas para transfecciones transitorias son cultivadas en cultivo en suspensión. Para transfecciones a escala
pequeña (<5 L), las células se desarrollan en matraces agitados Corning (Corning, Tewksbury, MA) sobre un agitador orbital (100-120 rpm) en un incubador humidificado a 5% de CO2 (matraces de semilla). Las células en los cultivos semilla deben mantenerse en la fase de crecimiento exponencial (densidades celulares entre 5x105 y 3x106/mL) y exhiben una viabilidad de >90% para la transfección. Las densidades celulares fuera de esta escala resultarán en una fase de retraso después de la dilución o eficiencia de transfección reducida. Para transfección a escala pequeña (<5 L) se toma una alícuota de las células de los cultivos semilla y se ajusta a 1,4x106 células/mL en 36 % del volumen final con medio de cultivo libre de suero de Novartis. La solución de ADN (solución 1: 0,5 mg de plásmido de expresión de cadena pesada y 0,5 mg de cadena ligera para una transfección de 1 L) se prepara diluyendo el ADN a 1 mg/L (volumen final) en 7% del volumen de cultivo final, seguido por agitación suave. Para prevenir la contaminación bacteriana, esta solución se filtra usando un filtro de 0,22gm (por ejemplo Millipore Stericup). Después 3 mg/L (volumen final) de solución de PEI también se diluye en 7% del volumen de cultivo final y se mezcla suavemente (solución 2). Ambas soluciones se incuban durante 5-10 minutos a temperatura ambiente (t.a.). Posteriormente la solución 2 se añade a la solución 1 con agitación suave y se incuba otros 5-15 minutos a temperatura ambiente. Después se agrega a las células la mezcla de transfección y el cultivo de las células se continúa durante 4 a 6 horas. Finalmente, el restante 50% de volumen de producción total se obtiene con la adición de medio de cultivo libre de suero ExCell® VPRO. El cultivo celular se continúa durante 11 días después de la transfección. El cultivo se cosecha por centrifugación a 4500 rpm durante 20 minutos a 4 °C (Heraeus ®, Multifuge 3 S-R, Thermo Scientific, Rockford, IL). El sobrenadante celular recuperado se esteriliza por filtración a través de un filtro stericup (0,22 gm) y se guarda a 4 °C hasta su ulterior procesamiento.
La purificación se realizó en un sistema de cromatografía “AKTA 100 explorer Air” a 4 °C en una cabina de enfriamiento, usando una columna recién sanitizada (0,25 M NaOH) HiT rap ProtA MabSelect®SuRe, 5 ml. La columna se equilibró con 5 volúmenes de columna (CV) de PBS (Gibco, Life Technologies, Carlsbad, CA) y después el sobrenadante filtrado estéril (2 L) se cargó a 4,0 ml/min. La columna se lavó con 8 CV de PBS para eluir la muestra no enlazada y nuevamente se lavó con 5 CV de PBS. El anticuerpo se eluyó con 5 CV de citrato 50 mM, 70 mM NaCl pH 3,2. El eluato se recolectó en fracciones de 3 mL; las fracciones se reunieron y se ajustaron a pH 7 con Tris HCl 1 M, pH 10. Las fracciones reunidas se juntaron y se esterilizaron por filtración (Millipore Steriflip, 0,22 um), se midió la OD 280 nm en un espectrofotómetro ND-1000 (NanoDrop), y la concentración de proteína se calculó basándose en los datos de la secuencia. El eluato se analizó para determinar la agregación (SEC-MALS) y la pureza (SDS-PAGE, LAL y MS). Para el segundo paso de purificación, en caso necesario, las fracciones reunidas de la primera purificación se cargaron en una SPX recién sanitizada (0,5 M NaOH) (Hi Load 16/60 Superdex 200 grade 120 mL (GE-Healthcare). La columna se equilibró con PBS y la corrida se hizo con amortiguador PBS a 1 ml/min, el eluato se recogió en fracciones de 1,2 ml y se analizó como se describe en el primer paso de purificación.
Ejemplo 3: Tamizado de anticuerpos anti-cKIT
Inmunopurificaciones HuCAL PLATINUM®
Para la selección de anticuerpos que reconocen cKIT humano se utilizaron muchas estrategias de inmunopurificación. Los anticuerpos terapéuticos contra las proteínas cKIT humanas se generaron por selección de clones que tienen altas afinidades de unión, usando como fuente de proteínas variantes de anticuerpo una colección de presentación de fago disponible comercialmente, la colección Morphosys HuCAL PLATINUM® (Morphosys, Munich DE). La colección de fagémido se basa en el concepto HuCAL® (Knappik et al., (2000) J Mol Biol 296: 57-86) y utiliza la tecnología CysDisplay® para presentar el Fab sobre la superficie del fago (Lohning, WO 01/05950). Para el aislamiento de los anticuerpos anticKIT, se realizaron estrategias de inmunopurificación estándares usando enfoques de inmunopurificación en fase sólida, solución, célula entera, y célula entera diferencial.
Inmunopurificación en fase solida contra cKIT
Una placa Maxisorp™ de 96 pocillos se recubrió con proteína de fusión cKIT-Fc de humano o ratón durante la noche a 4 °C. Para cada inmunopurificación, se agregaron aproximadamente 4x1013 fago-anticuerpos HuCAL PLATINUM® a cada antígeno recubierto y se incubó 2 horas a t.a. sobre una agitadora de placa de microtitulación. Posteriormente, los fagos enlazados no específicos se lavaron por medio de varios pasos de lavado y los fagos enlazados específicamente se eluyeron usando DTT 25 mM en Tris/HCl 10 mM, pH 8.
El eluato se transfirió a 14 ml de bacterias E. coli y se incubó para la infección del fago. Las bacterias infectadas se resuspendieron en medio 2xYT, se sembró sobre placas de agar LB/Cam y se incubó durante la noche. Las colonias se rasparon de las placas y se usaron para rescate de fago, amplificación policlonal de clones seleccionados y producción de fago. Con el fago purificado se inició la siguiente ronda de inmunopurificación.
La segunda y tercera ronda de inmunopurificación en fase solida se realizó de acuerdo con el protocolo de la primera
ronda, excepto por cantidades disminuidas de antígeno y condiciones de lavado más severas.
Inmunopurificación de captura contra cKIT
Para inmunopurificación de captura, las proteínas de fusión antígeno cKIT/Fc murino se inmovilizaron sobre una placa Maxisorp™ de 96 pocillos por medio de un anticuerpo de captura de cabra anti-Fc de ratón. Durante el bloqueo del fago, se agregó y globulina de humano y ratón al amortiguador de bloqueo para evitar la selección de anticuerpos contra el anticuerpo de captura y la parte Fc de ratón del antígeno. Los procedimientos de recubrimiento de antígeno y bloqueo de fago en la inmunopurificación de captura se realizaron como se describe en el protocolo de inmunopurificación de fase sólida (véase arriba).
Protocolo de inmunopurificación en solución con perlas magnéticas acopladas con estreptavidina.
Se hicieron inmunopurificaciones en solución en 2 modos diferentes (“clásico” y “alternativo”). Para cada inmunopurificación, aproximadamente 4x1013 fago-anticuerpos HuCAL PLATINUM® se bloquearon con un volumen igual de 2x Chemiblocker/ 0,1% Tween20. Para la remoción de fago enlazado a estreptavidina o a perla, se hizo una preadsorción de partículas de fago bloqueadas 2 veces usando cada vez 1 mg de perlas de estraptividina bloqueadas.
a) Modo “clásico”: mAb 16P23 biotinilado se incubó con proteína cKIT ECD His humana y se agregó a las partículas de fago bloqueadas. El anticuerpo 16P23 es un hibridoma generado internamente y se usó en varios protocolos de tamizado como anticuerpo de captura para exponer diferentes dominios sobre el ECD de cKIT. El anticuerpo de 16P23 también se usó para fines de agrupación de anticuerpo. Después de incubación, los complejos fago-antígeno se capturaron usando perlas de estreptavidina y las partículas de fago enlazadas a las perlas de estraptividina se recolectaron con un separador magnético.
b) Modo “alternativo”: Se agregó mAb 16P23 biotinilado a perlas de estraptividina y la mezcla anticuerpo-perlas se incubó en un aparato giratorio a t.a. por 30 minutos. Las perlas se lavaron y se resuspendieron en PBS que contenía la proteína cKIT ECD-His humana. Subsiguientemente los fagos se agregaron y el complejo anticuerpo-perla-antígenofago se giró durante 1 hora más a t.a. en un aparato giratorio. Después de este último paso de incubación las perlas se capturaron con un separador magnético y los sobrenadantes se desecharon.
Usando ambos métodos de presentación el fago enlazado no específico se deslavó por medio de varios pasos de lavado usando PBS/0,05% Tween20 y PBS. Los fagos enlazados específicamente se eluyeron de perlas de estreptavidina usando DTT 25 mM en Tris/HCl 10 mM, pH 8. Se hizo infección de fago y producción de fago subsecuentes de acuerdo con el protocolo de inmunopurificación en fase sólida.
La segunda y tercera ronda de la inmunopurificación en solución se realizó de acuerdo con el protocolo de la primera ronda, excepto por cantidades disminuidas de antígeno y condiciones de lavado más severas.
Inmunopurificación de célula entera contra cKIT
Células objetivo que expresan el antígeno cKIT de humano, ratón o rata, se usaron como antígenos y se pusieron en contacto con fago-anticuerpos HuCAL PLATINUM® para inmunopurificaciones. Los complejos fago-célula se lavaron 3 veces con PBS/5% FCS. La elución de fago enlazado específicamente de las células objetivo se realizó con 0,1 M glicina-HCl/0,5 M NaCl, pH 2,2. Se hizo una infección de fago subsiguiente y producción de fago de acuerdo con el protocolo de inmunopurificación en fase sólida. La segunda y tercera ronda de la inmunopurificación de célula entera se realizó de acuerdo al protocolo de la primera ronda.
Inmunopurificación de célula entera diferencial contra cKIT
En la inmunopurificación de célula entera diferencial, la selección se hizo alternando células y proteína purificada. Las rondas de selección sobre el antígeno purificado se hicieron como se describe en el protocolo de inmunpurificación en fase sólida. Para las rondas de selección sobre las células, consulte el procedimiento en la sección de inmunopurificación de célula entera contra cKIT.
Inmunopurificaciones de maduración
Para obtener anticuerpos específicos con mayor afinidad, se hicieron inmunopurificaciones de maduración (Prassler et al., Future Med. Immuno. 2009 1(4):571-583). Para este propósito, clones secuenciados ya probados para la unión especifica de cKIT se usaron para intercambio de casete LCDR3 o HCDR2. Posteriormente se hicieron 2 rondas de inmunopurificaciones en fase solida con proteína de fusión cKIT-Fc de humano o ratón como se describe en el protocolo
de inmunopurificación en fase sólida.
a) Para LCDR3 RapMAT®: Fragmentos que codifican Fab de ADN del vector pMORPH30® derivado de fago (Morphosys, Munich DE) se digirieron enzimáticamente y los insertos se reemplazaron con casetes de maduración TRIM™ LCDR3 (Virnekaes et al., NAR 1994 22(25):5600-5607). Subsecuentemente se ligaron 1,25 gg de vector de presentación pMORPH30® con el fragmento de inserto que lleva las LCDR3 diversificadas.
b) Para HCDR2 RapMAT®: Después de la segunda ronda de inmunopurificación, fragmentos que codifican Fab de ADN del vector pMORPH30® derivado de fago se digirieron enzimáticamente y los insertos se reemplazaron con casetes de maduración TRIM™ HCDR2 (Virnekas et al., supra). Subsecuentemente se ligaron 1,25 gg del vector de presentación pMORPH30® con el fragmento de inserto que lleva las HCDR2 diversificadas.
Las colecciones generadas se amplificaron y sometieron a 2 rondas de inmunopurificación con severidad incrementada y concentración de antígeno reducida o alternación de antígeno cKIT humano y de ratón para identificar los clones de afinidad mejorada.
Preparación de lisados bacterianos que contienen Fab para inmunopurificación por ELISA
Para el tamizado y caracterización inicial en un cultivo nocturno de clones de E. coli que expresan Fab individuales se lisaron usando lisozima, 4 mM EDTA y 10 U/gl Benzonase. Los lisados de E. coli que contenían Fab se usaron para tamizado de ELISA, FACS y SET.
Tamizado de lisados bacterianos crudos que contienen Fab
Tamizado por ELISA
Usando tamizado de ELISA se identificaron clones de Fab individuales del resultado de inmunopurificación para la unión de antígeno objetivo. Los Fab se probaron usando lisados de E. coli crudos que contienen Fab.
ELISA de la verificación de expresión de Fab
Para la verificación de la expresión de Fab en los lisados de E. coli preparados, placas de 384 pocillos Maxisorp™ (Nunc, Sigma-Aldrich, St. Louis MO) se recubrieron con anti-IgG humana de oveja especifica de fragmento Fd diluido 1:1000 en PBS. Después de bloquear con leche descremada en polvo al 5% en PBS que contenía Tween20 al 0,05%, se agregaron los lisados de E. coli que contenían Fab. Subsecuentemente los fragmentos HuCAL®- Fab enlazados se detectaron por incubación con anti-IgG humana de cabra específico de F(ab)2 , conjugado con fosfatasa alcalina (diluido 1:5000), seguido por adición del sustrato de fluorescencia AttoPhos® (Roche, #11681982001, Mannheim, DE). La emisión de fluorescencia a 535 nm se registró con excitación a 430 nm.
Tamizado de ELISA sobre antígeno recubierto directamente
Placas de 384 pocillos Maxisorp™ se recubrieron con proteína ECD de cKIT humana con etiqueta de mFc a una concentración de 10 gg/ml en pBs . Después de bloquear las placas con leche descremada en polvo al 5% en PBS, se agregaron los lisados de E. coli que contenían Fab. La unión de los Fab fue detectada por anti-IgG humana de cabra específico de F(ab)2, conjugado con fosfatasa alcalina (diluido 1:5000), usando el sustrato de fluorescencia Attophos® (Roche, #11681982001, Mannheim, DE). La emisión de fluorescencia a 535 nm se registró con excitación a 430 nm. Agrupación en bins de epítopos con lisados Fab BEL
Para identificar posibles competidores de unión de ligando antes de la maduración de afinidad, se hizo un tamizado de ELISA de competencia con lisados de E. coli de Fab y anticuerpo 16P23, un competidor de unión de ligando conocido. Para este propósito, placas de 384 pocillos Maxisorp™ se recubrieron con proteína cKIT ECD humana etiquetada con mFc y se bloquearon como se describe arriba (tamizado de ELISA sobre antígeno recubierto directamente).
El mAb 16P23 se agregó a una concentración final de 5 gg/ml, seguido por incubación con lisados de E. coli que contienen Fab. Finalmente, la unión de los Fab fue detectada con un anticuerpo anti-FLAG conjugado con fosfatasa alcalina (Sigma A-9469, diluido a 1:10000) usando el substrato de fluorescencia Attophos ® (Roche, #11681982001). La emisión de fluorescencia a 535 nm se registró con excitación a 430 nm.
Tamizado de FACS
En el tamizado de FACS, se identifican clones de Fab individuales que se unen al antígeno expresado en la superficie celular a partir del resultado de inmunopurificación. Los Fab se prueban usando lisados de E. co licrudos que contienen Fab.
El tamizado de FACS se realizó en formato de placa de 96 o 384 pocillos:
a) En un formato de placa de 96 pocillos usando un dispositivo de matriz BD FACS, 100 gl de suspensión celular se transfirieron a una placa nueva de 96 pocillos (dando por resultado 1x105 células/pocillo). La placa que contenía la suspensión celular objetivo se centrifugó y el sobrenadante se desechó. La pella celular remanente se resuspendió y 50 gl de los extractos de BEL que contiene Fab se agregaron a los pocillos correspondientes. La placa se incubó sobre hielo durante 1 hora. Después de la incubación, las células se centrifugaron y se lavaron 3 veces con 200 gl de amortiguador FACS (PBS, 3% FCS). Después de cada paso de lavado, las células se centrifugaron y se resuspendieron cuidadosamente. Se agregó anticuerpo de detección secundario (anti-IgG humana de cabra conjugado con PE; Dianova, Hamburg, DE) y las muestras se incubaron sobre hielo y se lavaron subsecuentemente de acuerdo con la incubación de Fab. Finalmente, las pellas celulares se resuspendieron en 150 gl de amortiguador FACS por pocillo y las muestras se analizaron en una matriz BD FACS.
b) En un formato de placa de 384 pocillos usando un dispositivo BD Calibur ® HTS (BD Biosciences, San Jose, CA), 20 gl de suspensión celular se transfirieron a una placa nueva de 384 pocillos redondos (dando como resultado 4x104 células/pocillo). La placa que contenía la suspensión celular objetivo se centrifugó y el sobrenadante se desechó. La pella celular remanente se resuspendió y 20 gl de extractos que contenían Fab se agregaron a los pocillos correspondientes. La placa se incubó 1 hora con agitación a 4 °C. Después de la incubación las células se centrifugaron y se lavaron 3 veces con 40 gl de amortiguador FACS (PBS, 3% FCS). Después de cada paso de lavado, las células se centrifugaron y se resuspendieron cuidadosamente. Se agregaron 40 gl de anticuerpo de detección anti-humano de cabra conjugado con PE, y las muestras se incubaron sobre hielo y subsecuentemente se lavaron de acuerdo con la incubación de Fab. Finalmente, las pellas celulares se resuspendieron en 35 gl de amortiguador FACS por pocillo y las muestras se midieron con el dispositivo BD FACS Calibur/HTS.
Determinación de la afinidad
Para las determinaciones de Kd se usaron fracciones de monómero de la proteína anticuerpo (por lo menos 90% de contenido de monómero, analizado por SEC analítica; Superdex es 75 PC3.2/30 (GE Healthcare, Pittsburgh, PA) para Fab, o Tosoh TSKgel G3000 SWx l (7,8 mm / 30,0 cm) (Tosoh Bioscience GmbH, Stuttgart, Alemania) para IgG, respectivamente).
Método de titulación de solución en equilibrio (SET) para determinación de KD usando Sector Imager 6000 (MSD)
La determinación de afinidad en solución fue realizada básicamente como se describe en la literatura (Friquet et al., J. Immuno. Meth. 1985; 77:305-319). Para mejorar la sensibilidad y precisión del método SET, se transfirió del ELISA clásico a la tecnología basada en ECL (Haenel et al., Anal. Biochem. 2005 339(1):182-4). Se marcó 1 mg/ml de anticuerpos específicos anti-fragmento (Fab)2 humano de cabra (Dianova) con MSD Sulfo-TAGTM NHS-Ester (Meso Scale Discovery, Gaithersburg, MD, EE.UU), de acuerdo con las instrucciones del fabricante. Las placas MSD se recubrieron con antígeno y las muestras equilibradas se transfirieron a las placas. Después de lavar, 30 gl por pocillo del anticuerpo de detección marcado con la etiqueta MSD-sulfo (anti-(Fab)2humano) se agregaron a la placa MSD y se incubó sobre un agitador. Después de lavar la placa MSD y agregar 30 gl/pocillo de amortiguador MSD Read Buffer T con agente tensoactivo, las señales electroquimioluminiscentes se detectaron usando un aparato Sector Imager 6000 (Meso Scale Discovery, Gaithersburg, MD, EE.UU.).
Los datos se evaluaron con software XLfit (IDBS) aplicando modelos de ajuste personalizado. Para la determinación de Kd de moléculas de Fab se usó el siguiente modelo de ajuste (de acuerdo con (Haenel et al., Anal. Biochem 2005;339(1):182-184), modificado de acuerdo con (Abraham et al., J. Mol. Recogn 1996; 9:456-461)):
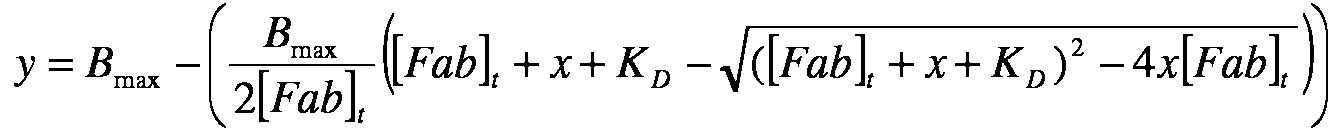
[Fab]t: concentración total aplicada de Fab
x: concentración total aplicada de antígeno soluble (sitios de unión)
Bmax: señal máxima de Fab sin antígeno
Kd : afinidad
Para la determinación de KD de moléculas de IgG se usó el siguiente modelo de ajuste para IgG (modificado de acuerdo con (Piehler et al., 1997)):

[IgG]: concentración total aplicada de IgG
x: concentración total aplicada de antígeno soluble (sitios de unión)
Bmax: señal máxima de IgG sin antígeno
Kd : afinidad
Ajustes experimentales:
La determinación de Kd de anti-IgG de cKIT HuCAL® se realizó básicamente como sigue: cKIT-Fc humano se recubrió a 0,1 pg/ml en PBS durante la noche a 4 °C sobre placas MSD estándares/amortiguador de ensayo durante 1 hora a t.a. sobre placas MSD de estreptavidina. Subsecuentemente las placas MSD se bloquearon con PBS con 3% BSA durante 1 hora a t.a. Las placas de estreptavidina se bloquearon durante la noche a 4 °C con PBS con BSA al 5% antes del recubrimiento con el antígeno. Para la titulación de antígeno se aplicó cKIT-His de humano.
Subsecuentemente la concentración de Fab no enlazado se cuantificó por medio de detección de ECL usando el Sector Imager 6000 (Meso Scale Discovery, Gaithersburg, MD, EE.UU.). Los resultados se procesaron usando software XLfit (IDBS), aplicando el modelo de ajuste correspondiente para calcular las afinidades e identificar así los clones más mejorados por la maduración.
Ensayos bioquímicos in vltro (reactividad cruzada y análisis de unión de dominio)
Las IgG purificadas se probaron en ELISA para la unión a proteínas ECD de longitud completa de cKit de humano, mono cynomolgus y ratón, y también construcciones del dominio ECD cKIT de humano D1-3 y D4-5. Para este propósito unas placas se recubrieron con antígeno a una concentración de 5 pg/ml en PBS durante la noche a 4 °C. La unión de IgG se detectó por medio de anti-F(ab)2 de humano o anti-F(ab)2 de ratón conjugado con fosfatasa alcalina (diluida 1:5000 en MPBS al 1%) usando Attophos ® como sustrato. La emisión de fluorescencia se midió a una excitación de 430 nm y una emisión de 535 nm.
Agrupación en Bins de epítopo de IgG purificadas
Los candidatos de IgG purificados se analizaron para determinar su competencia con anticuerpos herramienta generados internamente, que previamente mostraron definir bins individuales sobre el dominio extracelular de cKIT. Para este propósito, las IgG se recubrieron en cantidades constantes sobre placas Maxisorp™ y se analizaron para determinar su competencia con cantidades crecientes de IgG competidora en solución. Como control positivo la IgG recubierta se analizó para competencia consigo misma en solución. Todas las IgG probadas se incubaron previamente en un exceso 50x con la fusión cKIT-fc de humano glicobiotinilado durante 1 hora a t.a. en solución. Después los complejos antígeno/anticuerpo se agregaron a los anticuerpos recubiertos y la detección de los complejos enlazados ocurrió a través del antígeno biotinilado. En general, solo se pudieron obtener señales a concentración alta de IgG cuando la IgG recubierta fue capaz de unirse a epítopos accesibles sobre el antígeno diferentes de la IgG probada en solución (eso es, un anticuerpo no
competitivo). En contraste, para anticuerpos competitivos, los anticuerpos con epítopos parcialmente traslapados o anticuerpos que bloquean el epítopo por impedimento estérico, señales de unión a concentración alta de IgG disminuyeron significativamente en contraste con los controles.
Los pocillos respectivos de las placas Maxisorp™ se recubrieron con 20 pl/pocillo de dilución de IgG a una concentración de 1,2 pg/ml en PBS, se incubó durante la noche a 4 °C y después se lavó 3x con PBST. Las placas se bloquearon bien con 90 pl de BSA 3%/PBS durante 1 hora a t.a. y se lavaron 3x con PBST.
Determinación de EC50 sobre las células por medio de FACS
Las IgG purificadas se probaron a una sola concentración o se titularon en FACS para determinar los valores de EC50 para la unión a cKIT de humano, ratón o rata expresado en la superficie celular. Para este propósito se cosecharon células Mo7e, P815 o RBL-2H3 con Accutase ® (Life Technologies, Carlsbad, CA) y se diluyeron 1x106/ml en amortiguador FACS. Todos los pasos subsecuentes se hicieron sobre hielo para prevenir el internamiento del receptor. La suspensión celular se llenó con 100pl/pocillo en una placa de fondo en forma de U de 96 pocillos. Después de centrifugación a 210g durante 5 minutos a 4 °C, el amortiguador se desechó. Después se agregaron 100 pl de los mAb específicos diluidos en amortiguador FACS por pocillo, a una concentración de 15 pg/ml en experimentos de titulación a una dilución seriada de concentraciones de anticuerpo (pasos de dilución 1:3, concentración inicial de 15 pg/ml). Después de 1 hora de incubación sobre hielo las células se lavaron 3 veces con 150 pl de amortiguador FACS. Se agregó a las células un anticuerpo secundario de detección anti-humano de cabra conjugado con PE (diluido 1:200 en amortiguador FACS) con 100 pl/pocillo y se incubó sobre hielo durante 1 hora. Las células se lavaron 3 veces con 150 pl de amortiguador FACS. Finalmente, las pellas celulares se resuspendieron en 250 pl de amortiguador FACS por pocillo y las muestran se analizaron en la matriz BD FACS.
Bioensayos in vltro
Ensayo de proliferación dependiente de SCF
Se hicieron ensayos de proliferación sobre la línea celular Mo7e (leucemia megacarioblástica aguda humana, DSMZ no.: ACC 104) cultivada en RPMI1640 con glutamina estable (PAN #P04-18500), FCS al 10% y SCF a 10 ng/ml (R&D CAT. #255-SC; Lote #CM2810061, R&D Corp, Berkeley CA).
En el ensayo de proliferación dependiente de SCF, IgG purificadas o sobrenadantes de cultivo celular que contienen IgG se analizaron. En ambas situaciones experimentales las células se cosecharon y se resuspendieron en 50 ml de medio de privación (medio de cultivo sin SCF) de una concentración de 0,5x106 células/ml y se incubaron a 37 °C por 18 horas. Después las células se resuspendieron a una concentración de 1x106 células/ml en medio de privación con SCF a 60 ng/ml (concentrado 2X, concentración final después de la adición de anticuerpo es de 30 ng/ml). Se agregaron 50 pl de células (5x104 células/pocillo) y 50 pl de anticuerpos purificados concentrados 2x o sobrenadantes de cultivo celular no diluido por pocillo de un plano blanco de 96 pocillos con placas de fondo transparente. Para controles negativo y positivo, se incluyeron células sin SCF y sin anticuerpo o células con SCF y sin anticuerpo. Las placas se incubaron 48 horas a 37 °C y finalmente se determinaron los números de células usando CellTiter-Glo ® (Promega #G7571, Promega, Madison, WI) de acuerdo con las instrucciones del fabricante.
Ensayo Fab-ZAP ADC Piggyback
Para probar la capacidad de los anticuerpos para internarse después de la unión del receptor, se hizo un ensayo de ADC mezclando el reactivo Fab-ZAP (anti- hu-mAb de cabra conjugado con saporina; ATS Biotechnology, Cat. # IT-51-250, ATS Bio, San Diego, CA) con IgG purificadas o con sobrenadantes de cultivo celular que contienen IgG. El potencial citotóxico se probó sobre la línea de células de cáncer CMK-11-5 (células de leucemia megacarioblástica aguda, cultivadas en RPMI1640 FCS al 10%) ya que estas células muestran alta expresión de cKIT.
Las células en cultivo se contaron y se diluyeron en un medio a una concentración de 1x105 células/ml. Se transfirieron 50 pl de suspensión celular (5000 células/pocillo) a placas de 96 pocillos (Placa blanca de fondo transparente plano TC-tratada, Corning Cat. # 3903, Corning, Tewksbury, MA). En una placa separada (fondo en forma de V de 96 pocillos, Nunc, Cat. # 249946, Nunc Sigma-Aldrich, St. Louis, MO), se diluyeron IgG en el medio. Los sobrenadantes de cultivo celular que contenían IgG se diluyeron 1:125 y las IgG purificadas a una concentración de 0,4 nm, dando como resultado un volumen total de 60 pl/pocillo. Un volumen igual de solución FabZAP a una concentración de 5 nm se agregó y la placa se incubó durante 60 minutos a 37 °C. Se transfirieron 50 pl de conjugados anticuerpo/Fab-ZAP a células CMK-11-5 (volumen total 100 pl). Para control se prepararon pocillos con células únicamente (=100% de control de viabilidad) y células incubadas solo con Fab-ZAP (para verificar la destrucción no especifica del reactivo secundario). La concentración
final de Fab-ZAP fue de 1,25 nM. Las placas se incubaron 72 horas a 37 °C y 5% de CO2. El número de células se determinó usando CellTiter-Glo ® (Promega #G7571) de acuerdo con las instrucciones del fabricante. La viabilidad se normalizó para el control de solo células.
Síntesis
En el tamizado de anticuerpos de cKIT, se efectuaron dos estrategias diferentes:
Estrategia 1:
Los candidatos con reactividad cruzada humano/cynomolgus (217 familias de HCDR3) se seleccionaron sobre alta afinidad y después de conversión de IgG los clones se tamizaron para funcionalidad en el ensayo CMK-11-5 FabZAP ADC y el ensayo de proliferación de Mo7e. Basándose en la actividad y diversidad funcional los candidatos se seleccionaron para expresión a escala exploratoria.
Estrategia 2:
Los candidatos con reactividad cruzada humano/cynomolgus/ratón (5 familias de HCDR3) se maduraron de afinidad y después de conversión de IgG los candidatos se seleccionaron para expresión.
En resumen, 82 IgG candidatos purificados de la estrategia 1 y 2 se sometieron a caracterización en profundidad. De este conjunto de 82, se seleccionaron 26 candidatos de IgG para producción a escala elevada, conjugación de toxina y análisis subsecuente como conjugados anticuerpo-fármaco en experimentos in vitro e in vivo.
Después de la caracterización a profundidad, los 26 anticuerpos (14 candidatos de la estrategia 1 y 12 candidatos de la estrategia 2) que pertenecen a 16 familias de HCDR3 diferentes, se seleccionaron para producción a escala elevada y se analizaron como conjugado anticuerpo-DM1. Los candidatos se seleccionaron de acuerdo con los siguientes criterios: 1) destrucción potente de células que expresan cKIT de tipo silvestre y mutante en un ensayo Fab-DM1 piggyback con EC50 en la escala sub-nanomolar o más baja, 2) los valores KD de 24/26 IgG para cKIT de cynomolgus están dentro de la escala de 3 veces la determinada para cKIT humano. Además, 12/26 IgG reaccionaron cruzadamente con cKIT de ratón y rata expresado en las células.
Los candidatos seleccionados de este tamizado pudieron asignarse a diferentes bins de epítopos:
1) 19/26 IgG pertenecen al bin 1 o bin 6 (unión a dominios de unión de ligando D1-3 de cKIT)
2) 6/26 IgG pertenecen al bin 8 (unión a dominios de dimerización D4-5 de cKIT).
3) 1/26 IgG pertenecen al bin 2, que tiene alta afinidad por cKIT humano pero tiene solo débil afinidad para cKIT de cynomolgus. Un ejemplo de un anticuerpo que viene de este tipo de protocolo de tamizado es el anticuerpo 20376.
Ejemplo 4: Construcciones para proteínas ECD de cKIT de humano, cvnomolaus. ratón v rata
Dominios extracelulares de cKIT de genes de humano, ratón y rata se sintetizaron basándose en las secuencias de aminoácidos de las bases de datos GenBank o Uniprot (véase la tabla 2 más abajo). ADNc molde de cKIT de genes de Cynomolgus y 1 ECD se sintetizaron sobre la información de secuencia de aminoácidos generados usando ARNm de varios tejidos de Cynomolgus (por ejemplo, Zyagen Laboratories; tabla 2 abajo). Todos los fragmentos de ADN sintetizados se clonaron en vectores de expresión apropiados, por ejemplo el vector a base de hEF1-HTLV (pFUSE-mIgG2A-Fc2) con etiquetas C-terminales para permitir la purificación.
Tabla 2


Tabla 3: S ecuenc ias de p ro te ína cK IT de cynom o lgus

Expresión de proteínas cKIT recombinantes
Las proteínas recombinantes cKIT deseadas se expresaron en líneas celulares derivadas de HEK293 (293FS) previamente adaptadas a cultivo en suspensión y se desarrollaron en medio libre de suero FreeStyle-293 (Gibco, cat. # 12338018). La producción de proteína tanto a escala pequeña como escala grande fue por medio de transfección transitoria y se realizó en muchos matraces agitados (Nalgene), hasta 1 L cada uno con 293 Fectin (Life Technologies, cat. #12347019) como un plásmido portador. El ADN total y 293 Fectin se usó a una relación de 1:1,5 (p:v). La relación de ADN a cultivo fue de 1 mg/L. Los sobrenadantes de cultivo celular se cosecharon 3-4 días después de la transfección, se centrifugaron y se esterilizaron por filtración antes de la purificación.
Ejemplo 5: Purificación de proteína ECD de cKIT de humano, cynomolgus. ratón y rata, y de los subdominios 1 3 y 4-5 de cKIT
Purificación de proteína etiquetada
Las proteínas del dominio extracelular de cKIT recombinantes etiquetadas con Fc (por ejemplo ECD-Fc de cKIT de humano, cKIT humano (subdominios ECD 1-3, 4-5)-Fc, cKIT-mFc, de cynomolgus, cKIT-mFc de rata, cKIT-mFc de ratón) se purificaron del sobrenadante de cultivo celular. El sobrenadante clarificado se pasó sobre una columna de proteína A-Sepharose que se había equilibrado con PBS. Después de lavar a la línea de base, el material enlazado se eluyó con amortiguador de elución de pH bajo Pierce Immunopure, o glicina 100 mM (pH 2,7) e inmediatamente se neutralizó con 1/8 del volumen de elución de Tris 1M pH 9. La proteína reunida se concentró en caso necesario usando concentradores centrífugos de 15 ml Amicon Ultra con cortes de peso molecular nominales de 10kD o 30kD. Después los conjuntos se purificaron por SEC usando una columna Superdex 200 26/60 para remover los agregados. Después la proteína purificada se caracterizó por SDS-PAGE y SEC-MALLS (dispersión de luz láser de ángulo múltiple). La concentración se determinó por absorbancia a 280 nm, usando los coeficientes de absorción teóricos calculados de la secuencia por vector NTI.
Ejemplo 6: Unión de Abs de cKIT a subdominios de ECD de cKIT
Para ayudar a definir los sitios de unión de los Abs de cKIT, el ECD de cKIT de humano se dividió en los subdominios 1 3 (dominio de unión de ligando) y los subdominios 4-5 (dominios de dimerización). Para determinar que subdominios se enlazaron se utilizó un ensayo ELISA de sándwich. Se diluyó 1 pg/ml de ECD en solución salina amortiguadora de fosfato 1X que corresponde a los subdominios de cKIT 1-3, subdominios 4-5 o ECD de cKIT de longitud completa se recubrieron sobre placas Immulon 4-HBX de 96 pocillos (Thermo Scientific cat. # 3855, Rockford, IL) y se incubaron durante la noche a 4 °C. Las placas se lavaron 3 veces con amortiguador de lavado (solución salina amortiguadora de fosfato (PBS) 1X con Tween-20 al 0,01% (Bio-Rad 101-0781)). Las placas se bloquearon con 280 pl/pocillo de albumina de suero bovino
al 3% diluida en PBS 1X durante 2 horas a temperatura ambiente. Las placas se lavaron 3 veces con amortiguador de lavado. Los anticuerpos se prepararon a 2 pg/ml en amortiguador de lavado con diluciones de 5 veces para 8 puntos y se agregaron a placas de ELISA a 100 pl/pocillo por triplicado. Las placas se incubaron en un agitador orbital y se agitaron a 200 rpm 1 hora a temperatura ambiente. Las placas del ensayo se lavaron 3 veces con amortiguador de lavado. Se preparó el anticuerpo secundario anti-IgG humana de cabra (H+L) del fragmento F(ab’)2 (Jackson Immunoresearch Cat. # 109-036-088, West Grove, PA) a 1:10000 en amortiguador de lavado y se agregó a las placas de ELISA a 100 pl/pocillo. Las placas se incubaron con anticuerpo secundario durante 1 hora a temperatura ambiente agitando a 200 rpm en un agitador orbital. Las placas del ensayo se lavaron 3 veces con amortiguador de lavado. Para desarrollar la señal de ELISA se agregaron a las placas 100 pl/pocillo de sustrato de Sure blue ® TMB (KPL Cat. # 52-00-03, Gaithersburg, MD), y se dejó incubar 10 minutos a temperatura ambiente. Para detener la reacción se agregaron a cada pocillo 50 pl de ácido clorhídrico 1N. La absorbancia se midió a 450 nM usando una lectora de placas Molecular Devices SpectraMax M5. Para determinar la respuesta de unión de cada anticuerpo las mediciones de densidad ópticas se promediaron, se generaron los valores de desviación estándar y se graficaron usando Excel. Los dominios de unión de cada anticuerpo anti-cKIT individual se encuentran en la tabla 5 más abajo.
Ejemplo 7: Mediciones de afinidad de Abs de cKIT
La afinidad de los anticuerpos para ortólogos de especie de cKIT y también para cKIT se determinó usando tecnología SPR usando un instrumento Biacore® 2000 (GE Healthcare, Pittsburgh, PA) y con chips sensores CM5.
Brevemente, se usó HBS-P (0,01 M HEPES, pH 7,4, 0,15 M NaCl, 0,005% Surfactant P20) suplementado con amortiguador de bloqueo Odyssey® al 2% (Li-Cor Biosciences, Lincoln, NE) como el amortiguador de desarrollo para todos los experimentos. El grado de inmovilización y las interacciones de analitos se midieron por unidad de respuesta (RU). Se hicieron experimentos pilotos para probar y confirmar la factibilidad de la inmovilización del anticuerpo anti-Fc humano (Catálogo # BR100839, GE Healthcare, Pittsburgh, PA) y la captura de los anticuerpos de prueba.
Para mediciones de cinética, se realizaron experimentos en los que los anticuerpos se capturaron en la superficie del chip sensor por medio del anticuerpo anti-Fc humano inmovilizado y se determinó la capacidad de las proteínas cKIT para unirse en solución libre. Brevemente, se inmovilizaron 25 pg/ml de anticuerpo anti-Fc humano a pH 5 sobre un chip sensor CM5 por medio de acoplamiento de amina a una velocidad de flujo de 5 pl/minuto en las dos celdas de flujo para alcanzar 10500 RU. Después se inyectaron 0,1-1 pg/ml de anticuerpos de prueba a 10 pl/minuto durante 1 minuto. Los niveles capturados de anticuerpos generalmente se mantuvieron por debajo de 200 RU. Subsecuentemente se diluyeron 3,125 -50 nM de dominios extracelulares (ECD) del receptor cKIT en una serie de dos veces y se inyectaron a una velocidad de flujo de 40 p/minuto por 3 minutos sobre las celdas de flujo tanto de referencia como de prueba. Más abajo se muestra la tabla de eCd probados. La disociación de la unión fue seguida durante 10 minutos. Después de cada ciclo de inyección la superficie del chip fue regenerada con MgCl23 M a 10 p/minuto por 30 segundos. Todos los experimentos se realizaron a 25 °C y los datos de respuesta se ajustaron globalmente con un modelo de interacción simple 1:1 (usando el software Scrubber 2 ® versión 2.0b (BioLogic Software) para obtener estimados de la constante de asociación (ka),constante de disociación (kd) y afinidad (Kd).
Tabla 4: Isotipo y fuente de ECD

La tabla 5 lista la unión y afinidad del dominio. Como se muestra en la tabla, los anticuerpos 9p3, NEG024, NEG027, NEG085, NEG086, NEG087 y 20376 reaccionaron todos con cKIT humano a nivel nanomolar, y tienen afinidades similares para los probados contra ECD de mono cynomolgus. Sin embargo, únicamente 20376 reaccionó cruzadamente con ratón. Ninguno de los anticuerpos probados reaccionó cruzadamente con cKIT de rata.
Tabla 5

Ejemplo 8: Preparación de ADC
Preparación de los conjugados DM1 por un proceso de un paso
Anticuerpos de cKIT individuales se sometieron a diafiltración en un amortiguador de reacción (fosfato de potasio 15 mM, EDTA 2 mM, pH 7,6) mediante filtración de flujo tangencial (TFF#1) antes de iniciar la reacción de conjugación. Subsecuentemente un anticuerpo de cKIT (aproximadamente 5.0 mg/mL) se mezcló con DM1 (exceso molar de 5,6 veces con respecto a la cantidad de anticuerpo) y después con SMCC (aproximadamente un exceso 5,0 veces con respecto a la cantidad de anticuerpo). La reacción se hizo a 20 °C en amortiguador de fosfato de potasio 15 mM (pH 7,6) que contenía EDTA 2 mM y DMA al 10%, durante aproximadamente 16 horas. La reacción se apagó agregando ácido acético 1 M para ajustar el pH a 5,0. Después de ajuste del pH la mezcla de reacción se filtró a través de un filtro PVDF de multicapa (0,45/0,22 pm) y se purifico y sometió a diafiltración en un amortiguador de succinato 20 mM (pH 5,0) que contenía sacarosa al 8,22% usando filtración de flujo Tangencial (TFF#2). Un ejemplo de los parámetros instrumentales para la filtración de flujo Tangencial se muestra abajo en la tabla 6.
Tabla 6: Parámetros instrumentales para la filtración de flujo tangencial

Los conjugados obtenidos del proceso anteriormente descrito se analizaron por: espectroscopia de UV para carga de agentes citotóxicos (relación de maitansinoide a anticuerpo, MAR); SEC-HPLC para determinación de monómero conjugado; y HPLC de fase inversa o HPLC de fase protegida hidrofóbica (Hisep) para porcentaje de maitansinoide libre.
Preparación de conjugados de DM1 mediante proceso in situ
Los anticuerpos anti-cKIT también se pueden conjugar por medio de un proceso in situ de acuerdo con el siguiente procedimiento. Los anticuerpos de cKIT se conjugaron con DM1 usando el enlazador sulfosuccinimidil 4 -(N -maleimidometil) ciclohexan-1-carboxilato (sulfo-sMcC). Se prepararon soluciones de reserva de DM1 y enlazador heterobifuncional Sulfo-SMCC en DMA. El Sulfo-SMCC y DM1 tiol se mezclaron entre sí para reaccionar por 10 minutos a 25 °C en DMA que contenía 40% v/v de amortiguador de succinato acuoso 50 mM, EDTA 2 mM, pH 5,0, a la relación de DM1 a enlazador de 1,3:1 equivalentes molares y una concentración final de DM1 de 1,95 mM. Después el anticuerpo se hizo reaccionar con una alícuota de la reacción para dar una relación equivalente molar de SMCC a Ab de alrededor de 6,5:1 bajo condiciones de conjugación final de 2,5 mg/mL de Ab en EPPS 50 mM, pH 8,0 y DMA al 10% (v/v). Después de aproximadamente 18 horas a 25 °C, la mezcla de reacción de conjugación se purificó usando una columna SEpHa DEX™ G25, equilibrada con succinato 10 mM, glicina 250 mM, sacarosa al 0,5%, Tween 20 al 0,01%, pH 5.5.
Cualquier método es útil en la conjugación de anticuerpos. La tabla siguiente provee un ejemplo de ADC de cKIT.
Tabla 7: P rop iedades de los an ticue rpos con ju gad os con DM1
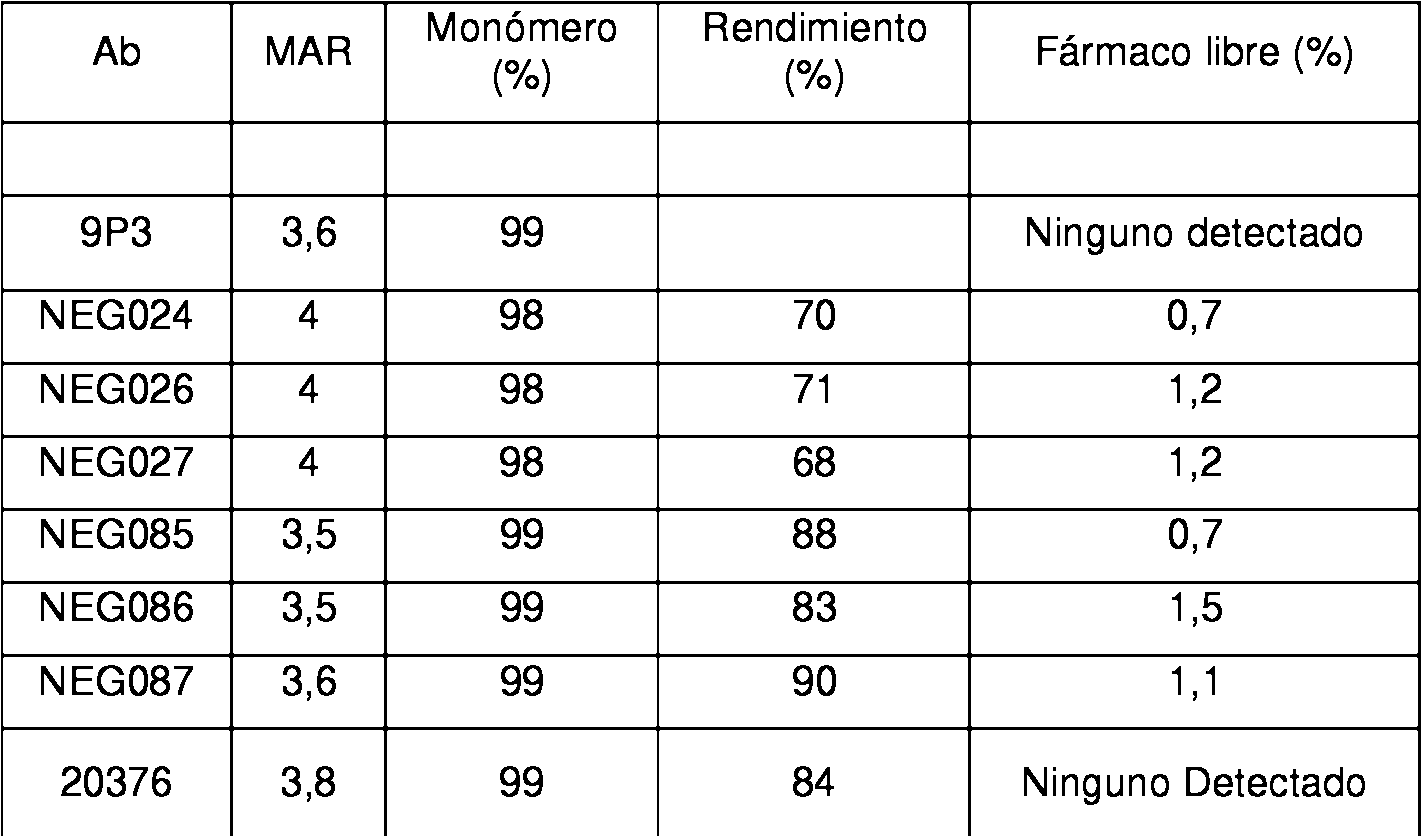
Preparación de ADC con el enlazador SPDB
Los anticuerpos anti-cKIT, por ejemplo el anticuerpo 9P3, (8 mg/ml) se modificaron con N-succinimidil-4-(2-piridilditio)butanoato (SPDB, exceso molar de 5,0, 5,5 y 4,.9 veces, respectivamente), por 120 minutos a 25 °C en amortiguador de fosfato de potasio 50 mM (pH 7,5) que contenía NaCl 50 mM, EDTA 2 mM y DMA al 5%. El Ab modificado sin purificación se conjugó subsecuentemente con DM4 (exceso molar de 1,7 veces sobre el enlazador no enlazado) a una concentración de anticuerpo modificado final de 4 mg/mL en amortiguador de fosfato de potasio 50 mM (pH 7,5) que contenía NaCl 50 mM, EDTA 2 mM y DMA al 5% por 18 horas a 25 °C. La mezcla de reacción de conjugación se purificó usando una columna SEPHADEX™ G25, equilibrada y eluida con succinato 10 mM, glicina 250 mM, sacarosa al 0,5%, Tween 20 al 0,01%, pH 5,5.
Preparación de ADC con el enlazador CX1-1
Anticuerpos anti-cKIT, por ejemplo el anticuerpo 9P3, (5.0 mg/ml) se mezclaron con DM1 (exceso molar de 7,15 veces con respecto a la cantidad de anticuerpo) y después con CX1-1 (exceso de 5,5 veces con respecto a la cantidad de anticuerpo). La reacción se realizó a 25°C en amortiguador EEPS 60 mM [ácido 4-(2-hidroxietil)-1-piperazinpropanosulfónico] (pH 8,5) que contenía EDTA 2 mM y DMA al 5% por aproximadamente 16 horas. Después la mezcla de reacción se purificó usando una columna SEPHADEX™ G25, equilibrada y eluida en succinato 10 mM, glicina 250 mM, sacarosa al 0,5%, Tween 20 al 0,.01%, pH 5,5.
En la figura 2 se muestra un ejemplo que compara las eficacias in vitro de anticuerpo-MCC-DM1, anticuerpo-SPDB-DM4 y anticuerpo-CX1-1-DM1.
Ejemplo 9: Afinidad de ADC con respecto a los anticuerpos de origen
La afinidad de los anticuerpos a cKIT después de conjugación con SMCC-DM1 se determinó usando tecnología Biacore usando un instrumento Biacore® T100 (GE Healthcare, Pittsburgh, PA) y chips sensores CM5, usando una metodología similar a la que se describe en el ejemplo 7 anterior.
Para los anticuerpos evaluados, se obtuvieron estimados de afinidad similares para la unión a cKIT humano para anticuerpos conjugados con SMCC-DM1 con respecto a los anticuerpos de origen no conjugados, lo que sugiere que la conjugación no tiene un impacto apreciable en la unión del anticuerpo (tabla 8).
Tabla 8: A fin ida des de an ticue rpos con jugados y no con ju gad os con M C C -D M 1

Ejemplo 10: Actividad de 9P3-MCC-DM1, 9P3-SPDB-DM4 y 9P3-CX1-1-DM1 sobre un panel de líneas celulares
Después de conjugación con la carga útil de enlazador MCC-DM1 se determinó la capacidad de los conjugados anticuerpo-fármaco (ADC) para inhibir la proliferación de AML, SCLC, GIST y líneas de células de melanoma. La línea celular GlST-T1 fue provista generosamente por el Dr. Takahiro Taguchi, Kochi U., Japón. Las líneas celulares GIST430 y GIST882 fueron provistas amablemente por el Dr. Jonathan Fletcher, Brigham and Women’s Hospital, Boston, MA.
Para cáncer de pulmón de célula pequeña (SCLC), se usaron las líneas celulares NCI-H526 y NCI-H1048. La NCI-H526 es un expresador superior de cKIT y se obtuvo de ATCC (CRL-5811, ATCC Manassas, VA). La NCI-H1048 expresa cKIT a un nivel más bajo y también fue obtenida de la ATCC (CRL-5853). La CMK-11-5 es una línea de AML que expresa altos niveles de cKIT ((JCRB Cat. # IFO50430, Japón) véase también Nagano et al., Int. J. Hematol. 1992; 56:67-78)). La UKE-1 también es una línea celular de AML y expresa cantidades bajas de cKIT. La línea celular UKE-1 fue provista amablemente por el Profesor Walter Fiedler, University Hospital Eppendorf, Hamburgo, Alemania. La Kasumi 1 fue obtenida de la ATCC (CRL-2724). La Kasumi 6 fue obtenida de la ATCC (CRL-2775). La MDA-MB-453 fue obtenida de la ATCC (HTB-131). Las líneas NCI-H889 y NCI-H1930 se compraron a ATCC (CRL-5817 y CRL-5906, respectivamente). Las células Hel92.1.7 se obtuvieron de Sigma-Aldrich (Cat. # 92111706-1VL, Sigma Aldrich, St. Louis, MO). Las células M-07e y SKNO1 se le compraron a DSMZ, ACC-104 y ACC-690 respectivamente (DSMZ, Braunschweig, DE). La línea celular OCI-M1 también es de DSMZ (ACC-529).
Brevemente, las células se cultivaron en un incubador de cultivo de tejido a 37 °C con 5% de CO2 en medio de cultivo como lo recomienda el proveedor. El día del ensayo las células se lavaron 2 veces con PBS (Cellgro, Corning, Tewksbury MA (# catálogo 21-031-CV)), antes de tratarse con tripsina 0,1%-EDTA (servicios técnicos dentro del laboratorio) por 5 minutos y se volvieron a suspender en el medio de cultivo recomendado. Después las células se contaron y se sembraron en placas de 96 pocillos (Costar, # catálogo 3603, Corning, Tewksbury, MA) a densidades de 2000-10000 células/pocillo en 100 gl de medio de cultivo celular. Se generó una placa duplicada para una medición del día 0 y todas las placas se incubaron en un incubador de cultivo de tejido a 37 °C con 5% CO2 durante la noche. También se generaron pocillos de medio solo para actuar como controles negativos. Después de esta incubación, se agregaron 100 gl/pocillo del reactivo Cell titer Glo® (Promega, # catálogo G7573, Madison, WI) a las placas del día 0, que entonces se agitaron suavemente durante 2 minutos, se incubaron por 10 minutos y la intensidad de luminiscencia resultante se midió usando una lectora de placa Perkin Elmer Wallac Microbeta Trilux® (Perkin Elmer, Waltham, MA). Los ADC de prueba se diluyeron en serie a una solución de reserva 3X en el medio de cultivo celular apropiado y 50 gl de los ADC diluidos seriadamente 3X se agregaron (concentración final del ensayo de 0,0002-68 nM de equivalentes de DM1) antes de incubación en una incubadora de cultivo de tejido a 37 °C con 5% CO2 por 5 días. Después de este periodo de incubación, la viabilidad celular relativa se determinó mediante la adición del reactivo Cell titer Glo® descrito anteriormente. El efecto de los ADC sobre la proliferación celular se calculó usando el promedio de los duplicados como sigue: (% Inhibición = (tratado con ADC -no tratado) / (no tratado - Día 0)*100). Los datos de % de inhibición se ajustaron a una ecuación logística de 4 parámetros y se determinaron los valores de GI50.
Como se muestra en la figura 1, los ADC de cKIT se probaron en un ensayo de proliferación en un panel de líneas celulares GIST (GIST T-1, GIST882, GIST430), SCLC (NCI-H526, NCI-H1048) y AML (Kasumi-6, Kasumi-1). Los valores de IC50 y destrucción máxima se indican en la tabla. MDA-MB453 (línea celular de cáncer de mama) no expresa
cKIT. IgG-MCC-DM1 es el control de isotipo. Como se muestra en la figura 1, todos los ADC de cKIT tuvieron IC50 nanomolares a sub-nanomolares en las 7 líneas utilizadas. Esto indica que los ADC de cKIT tienen un amplio espectro de indicaciones, y se podrían usar siempre que un tumor exprese niveles apropiados de cKIT.
La capacidad de un anticuerpo anti-cKIT (9P3) conjugado con la carga útil de enlazador SPDB-DM4 and CX1-1-DM1 también se evaluó y se muestra en la figura 2. Estos estudios, que se realizaron como se describe arriba, revelaron que el ADC anti-cKIT evaluado también fue un potente inhibidor de la proliferación celular usando SPDB-DM4 o CX1-1-DM1, lo que sugiere que su capacidad para suministrar exitosamente toxina para destruir células no se limita a MCC-DM1. Tanto la figura 1 como la figura 2 proveen ACD de cKIT que son eficaces en la escala nanomolar a sub-nanomolar.
Además, la figura 3 es una gráfica de GI50 de ADC anti-cKIT contra el nivel del receptor cKIT, y qué indicaciones (AML, GIST, melanoma y SCLC). Como se muestra en la figura 3, el ADC anti-cKIT es eficaz en todas las indicaciones listadas.
Ejemplo 11: Actividad in v itro de ADC cKIT-MCC-DM1 sobre líneas celulares GIST, SCLC y AML
Después de la conjugación a la carga útil de enlazador MCC-DM1, la capacidad de los conjugados de anticuerpo-fármaco (ADC) para inhibir la proliferación de las líneas celulares AML, SCLC y GIST fue determinada. Para un listado de las células usadas en estos experimentos por proveedor véase el ejemplo 10 anterior.
Brevemente, las células se cultivaron en un incubador de cultivo de tejido a 37 °C con 5% de CO2 en medio de cultivo como lo recomienda el proveedor. El día del ensayo, las células se lavaron dos veces con PBS (Cellgro, Cat #21-031-CV, Corning Tewksbury, MA), antes de ser tratadas con tripsina al 0,1% -EDTA (servicios técnicos en el laboratorio) durante 5 minutos y se resuspendieron en el medio de cultivo recomendado. Después se contaron las células y se sembraron en placas de 96 pocillos (Costar, # catálogo 3603, Corning, Tewksbury, MA) a densidades de 5000 células/pocillo para células AML y SCLC, y 10000 células/pocillo para células GIST en 100 gl de medio de cultivo celular. Se generó una placa duplicada para una medición del día 0 y todas las placas se incubaron en un incubador de cultivo de tejido a 37 °C con 5% CO2 durante la noche. Después de esta incubación se agregaron 100 gl/pocillo del reactivo Cell titer Glo® (Promega , # catálogo G7573, Promega, Madison, WI) a las placas del día 0 que entonces se agitaron suavemente por 2 minutos, se incubaron 10 minutos y la intensidad de luminiscencia resultante se midió usando una lectora de placa Perkin Elmer Wallac Microbeta ® Trilux (Perkin Elmer, Waltham, MA). Los ADC de prueba se diluyeron en serie a una solución de reserva 3X en el medio de cultivo celular apropiado y se agregaron los 50 gl de ADC diluidos en serie 3X (concentración final del ensayo de 0,0002-68 nM equivalentes de DM1) antes de incubación en un incubador de cultivo de tejido a 37 °C con 5% CO2 durante 5 a 8 días. Después de este periodo de incubación se determinó la viabilidad celular relativa mediante la adición del reactivo Cell titer Glo ® como se describe arriba. El efecto de los ADC sobre la proliferación celular se calculó usando el promedio de los duplicados como sigue: (% de efecto máximo (Em a x) = (no tratado - tratado con la concentración de ADC más alta)*100).
Los datos de porcentaje de inhibición se ajustaron a una ecuación logística de 4 parámetros y se determinaron los valores de GI50. Estos datos se muestran en las figuras 4 a 9. Como se muestra en las gráficas, se usa como control un conjugado IgG-MCC-DM1. Todos los ADC probados tienen mayor actividad que el anticuerpo de control. Como lo muestran las curvas de las figuras 4 a 9, los ADC anti-cKIT, por ejemplo los anticuerpos NEG085, NEG024 y 20376 fueron muy eficaces para reducir la proliferación celular y por lo tanto serian eficaces en el tratamiento de GIST, AML y SCLC.
Ejemplo 12: Cuantificación de la densidad del receptor de superficie de cKIT sobre líneas celulares por medio de FACS (clasificación celular activada por fluorescencia)
Se usaron como estándares perlas celulares Quantum Simply (Bangs Laboratories, Inc. Catálogo #815, Fishers, IN). La capacidad de unión de anticuerpo de los estándares de perla varía de 0 a aproximadamente 310000. Las perlas o 500000 células se centrifugaron y se lavaron 2 veces con 100 gl/muestra de amortiguador FACS (PBS, 0,2% BSA, 0,1% NaAz). Después de cada paso de lavado, las perlas o células fueron centrifugadas o cuidadosamente resuspendidas. Después de los lavados se agregó amortiguador FACS y se agregaron a los pocillos correspondientes 10 gg/ml de APC-Mouse Anti-Human CD117 (BD Pharmigen Catálogo #550412, BD Biosciences, San Jose, CA) o 10 gg/ml of APC-Mouse IgG k Isotype Control (BD Pharmigen Catálogo #554681), para un volumen final de 100 g/muestra.
Después las suspensiones de célula-anticuerpos se incubaron sobre hielo durante 1 hora. Después de la incubación las células se centrifugaron y se lavaron 2 veces con 100 gl de amortiguador FACS. Después de cada paso de lavado las perlas o células se centrifugaron y se resuspendieron cuidadosamente.
Las células no viables se excluyeron por resuspensión en 100 gl/muestra de amortiguador FACS que contenía 7-AAD (BD Pharmigen Catálogo #559925). Las muestras se incubaron sobre hielo por 10 minutos y se analizaron en BD FACS
Canto II ® (BD Biosciences, San Jose, CA). Se determinó la media geométrica de señal por muestra usando software FlowJo ® y la densidad de antígeno se determinó como se describe en el manual de Quantum Simply Cellular. Se hicieron análisis de la sensibilidad de la línea celular in vitro a los ADC y la densidad del receptor de línea celular en TIBCO Spotfire 4.0.
Esta densidad de receptor se muestra en el eje Y de la Figura 3. Un análisis de densidad de receptor es útil en este aspecto como un biomarcador inicial para la estratificación de paciente. Por ejemplo, en la figura 3, una densidad de receptor alta se correlaciona con la eficacia de la GI50 del ADC anti-cKIT mostrada en el eje X. El análisis de densidad de receptor es útil en una situación clínica para determinar qué pacientes deben recibir una terapia de ADC anti-cKIT.
Ejemplo 13: Mapeo de epítopo de cKIT al anticuerpo 9P3 por medio de espectrometría de masa de intercambio de deuterio (HDx-MS)
La espectrometría de masa de intercambio de deuterio (HDx-MS) mide la captación de deuterio sobre el esqueleto de amida de una proteína. Estas mediciones son sensibles a la accesibilidad de disolvente de la amida y a cambios en la red de unión de hidrogeno de las amidas del esqueleto. La HDx-MS se usa frecuentemente para comparar proteínas en dos estados diferentes, tal como apo y enlazada al ligando, y acoplada con digestión rápida con pepsina. En estos experimentos se pueden localizar regiones, típicamente de 10 a 15 aminoácidos que muestran captación de deuterio diferencial entre dos estados diferentes. Las regiones que son protegidas están implicadas directamente en la unión de ligando o afectadas alostéricamente por la unión del anticuerpo al ligando.
En estos experimentos, la captación de deuterio del dominio extracelular cKIT (SEQ ID NO:160, véase abajo) se midió en ausencia y presencia de un mAb terapéutico, 9P3. Las regiones en cKIT que muestran una disminución de la captación de deuterio después de la unión del anticuerpo probablemente estén implicadas en el epítopo; sin embargo, debido a la naturaleza de la medición también es posible detectar cambios remotos del sitio de unión directo (efectos alostéricos). Usualmente, las regiones que tienen la mayor cantidad de protección están implicadas en la unión directa, aunque este puede no ser siempre el caso. Para delinear los eventos de unión directa de los efectos alostéricos son necesarias mediciones ortogonales (por ejemplo cristalografía de rayos X, mutagénesis de alanina).
Tabla 9: Construcción del dominio extracelular de cKIT

Los experimentos de mapeo de epítopo de cKIT se realizan en una plataforma Waters Synapt ® G2 HDx-MS, que incluye sistema robótico LEAP ®, sistema nanoACQUITY® UPLC, y espectrómetro de masa Synapt ® G2. En este método se efectúan experimentos de control por triplicado de la siguiente manera. Se diluyen 300 pmol (1,4 mg/ml) del antígeno cKIT en 110 gl de amortiguador PBS deuterado al 95% (pH 7,4) y se incuba a temperatura ambiente en un aparato rotativo de laboratorio durante 25 minutos (%D = 85,5%). El intercambio de deuterio es apagado por dilución 1:1 con amortiguador de apagado frio (6M Urea y 1M TCEP pH = 2,5) sobre hielo durante 5 minutos. Después del apagado el tubo se transfiere a un sistema LEAP (el Thermo box se pone a 2 °C) y la muestra apagada es inyectada por el sistema LEAP sobre el sistema UPLC para su análisis. El sistema UPLC incorpora una columna de pepsina inmovilizada de 2,1 mm x 30 mm (Life Technologies 2-3131-00) que es mantenida a 12 °C. Se usa un gradiente de acetonitrilo de 2 a 35% en 8 minutos y una columna Waters UPLC CSH C181,0 x 100 mm para la separación. En seguida se efectúan experimentos por triplicado usando el anticuerpo. Se inmovilizan 300 pmol de anticuerpo 9P3 sobre las perlas de agarosa de proteína G (Thermo Scientific Cat. #22851) usando las técnicas estándares. Brevemente, el anticuerpo se centrifuga para remover amortiguador de almacenamiento. Después se agregan 200 gl de amortiguador PBS (pH 7,4) y 300 pmol de cKIT al Ab inmovilizado y se incuba 30 minutos a temperatura ambiente. Después de la incubación el complejo se centrifuga y se
lava con 200 gl de amortiguador PBS y se centrifuga nuevamente. Para intercambio de deuterio, se agregan 200 gl de PBS deuterado al complejo antígeno-anticuerpo para incubación a temperatura ambiente por 25 minutos (%D = 85,5%). Después se remueve el amortiguador de deuterio e inmediatamente se agregan 125 gl de amortiguador apagado enfriado con hielo. Después de apagar durante 5 minutos la columna se centrifuga y el flujo pasante se transfiere a un vial de HPLC previamente enfriado. La muestra se analiza usando la misma preparación en línea de digestión de pepsina/ LC-MS que el experimento de control.
Los resultados de estas mediciones se resumen en la figura 10 y en la figura 11. La figura 10 muestra las diferencias corregidas de la línea de base entre el control y la muestra enlazada de anticuerpo 9P3 dividida entre el error estándar en la medición. En esta gráfica el valor más negativo indica una mayor cantidad de protección en una región dada después de la unión del anticuerpo 9P3 al antígeno cKIT. Después de la unión de 9P3 a cKIT se observan las cantidades más significativas de protección en las siguientes 2 regiones de cKIT VFVRDPAKLFL ((Región 1, 109-119 (SEQ ID NO: 161)) y HCSVDQEGKSVLSE ((Región 2, 185-198 (SEQ ID NO:162)). La región 1 comprende los residuos 109-119 y es parte de los dominios de D1 y D2. La región 2 comprende los residuos 185-198 y es parte del dominio D2. En la figura 11, hemos mapeado las dos regiones más protegidas (véase la figura 10) sobre la estructura de cristal del dominio extracelular de cKIT (PDB ID 2e9w). Además, también hemos marcado los sitios de unión de SCF sobre cKIT como sitio I, II y III usando los valores de la literatura (Yuzawa et al., Cell 2007;130: 323-334). Hay dos hallazgos clave en la figura 11. En primer lugar, las regiones 1 y 2 están muy cercanas en la estructura de cristal aunque están apartadas en el espacio de la secuencia primaria. Esta observación sugiere que ambas podrían ser parte potencialmente del epítopo, y si es así el epítopo para 9P3 es discontinuo. En segundo lugar, las regiones 1 y 2 son remotas de los sitios de unión de SCF reportados en la literatura. Esta es una observación importante porque sugiere que el anticuerpo 9P3 no altera directamente la unión del ligando. En cambio, el anticuerpo puede interferir estéricamente con la unión de ligando o con la dimerización del receptor después de la unión de ligando. En ensayos de competencia separados, usando ELISA y FACS, observamos bloqueo parcial de la unión de SCF a cKIT por 9P3, de modo que parece ser una interferencia estérica parcial. En conclusión, los datos de HDx-MS indican que el epítopo para el anticuerpo 9P3 consiste en un epítopo discontinuo que es remoto de los sitios de unión de SCF. Es de esperar que NEG024, NEG085, NEG086, NEG027 y NEG087 tengan el mismo mecanismo de acción.
Ejemplo 14: La capacidad de los ADC de cKIT para actuar como agonistas se evaluó usando una línea celular de cKIT de tipo silvestre Mo7e, y una línea celular mutante de cKIT GIST T-1
Para evaluar las posibles propiedades agonistas de ADC de cKIT, 2x106 células GIST T-1 (provistas amablemente por el Dr. Takahiro Taguchi, Kochi U., Japón) o Mo7e (DSMZ, ACC-104) se privaron de suero durante la noche a 37 °C con 5% CO2 (DMEM para GIST T-1 y RPMI para Mo7e suplementado con FBS al 0,1%) en una placa de 6 pocillos (NUNC, # catálogo 14067). Las células se trataron con rh-SCF a 10 ng/ml (R&D, Cat. # 255-SC, R&D, Berkeley, CA), 5 gg/ml de NEG085-MCC-DM1, NEG024-MCC-DM1, y 20376-MCC-DM1 por 15 minutos a 37 °C. Un pocillo fue designado como no tratado (UT). Las células se cosecharon en 1 ml de PBS. Las pellas celulares se lisaron sobre hielo durante 60 minutos en 30 gl de amortiguador de lisis: 20 mM Tris-HCl; pH 7,5, 137 mM NaCl, 1% Triton X-100, 15% glicerol, proteasa e inhibidores de fosfatasa. Después los lisados se centrifugaron 40 minutos a 12000 rpm a 4 °C. Se hirvieron 20 gg de cada nuestra durante 10 minutos a 75 °C y se cargaron en un gel de 12 pocillos NuPAGE® 4-12% Bis-Tris (Life Technologies, NP0322BOX, Carlsbad, CA). Después de la transferencia de proteína a los blots de membrana, las membranas se bloquearon con TBST-5 % leche a temperatura ambiente durante 1 hora y después se sondearon con anticuerpos primarios durante la noche a 4 °C. Los blots se lavaron con TBST (4x5 mins) el día siguiente. Los blots se incubaron en el anticuerpo secundario (anti-HRP de conejo de cabra 1:30000, Santa Cruz) durante 1 hora a temperatura ambiente. Los blots se lavaron en TBST (4x5 minutos) y se desarrollaron.
Los anticuerpos primarios usados para Western blotting fueron a-cKIT, Tyr703 (Cell Signaling Technology Cat. # 3073, Beverly, MA), a-cKIT Tyr721 (NOVUS, Cat. # NBP1-51412, Novus, Littleton, CO), AKT Ser473 (Cell Signaling Technology Cat. # 9271), AKT (Cell Signaling Technology Cat. # 4691), ERK Thr202/Tyr204 (Cell Signaling Technology Cat. # 9101), ERK (Cell Signaling Technology Cat. # 9102), y GAPDH (Cell Signaling Technology Cat. #3683).
Como se muestra en la figura 12, los anticuerpos de cKIT NEG085, NEG024 20376 pueden mediar la fosforilación de cKIT en ausencia de ligando (SCF). Sin embargo, las rutas de señalización secuencia abajo no son afectadas, ya que la señal no se transduce a fosfo-ERK o fosfo-AKT.
Ejemplo 15: Internamiento mediado por Ab de cKIT de cKIT de superficies sobre células GIST-T1, determinado por citometría de flujo
La cinética del internamiento mediado por anticuerpo de cKIT se evaluó tratando con anticuerpo en una monocapa celular usando un método de cambio de temperatura y lectura de citometría de flujo. Células GIST-T1 (provistas amablemente
por el Dr. Takahiro Taguchi, Kochi U., Japón) se sembraron a 2,5x105 células/pocillo en 5 placas tratadas de cultivo de tejido de 12 pocillos (BD Falcon 353043). Las células se incubaron en un incubador de cultivo de tejido a 37 °C con 5% CO2 durante la noche. El día siguiente el medio se removió y se remplazó con 450 gl de medio nuevo. Los anticuerpos de cKIT NEG085, 20376 y un control de isotipo se preparó a una concentración 10X 10 gg/ml en medio de cultivo de células apropiado, y 50 gl de anticuerpo de cKIT de prueba o isotipo se agregaron por pocillo con una concentración final de 10 gg/ml. Todas las células se encubaron 1 hora sobre hielo seguido de por 2 lavados con 1 ml de solución salina amortiguadora de fosfato (PBS) 1X y se resuspendieron en 500 gl de medio de cultivo celular. Las placas # 2-5 se transfirieron a 37 °C y se cosecharon en los puntos de tiempo: 30 minutos, 2 horas, 4 horas y 24 horas a 37 °C con 5% CO2. Se agregaron a la placa 1, 100 gl de amortiguador de disociación celular (Gibco Cat. #13150-016, Life Technologies, Carlsbad, CA) (control de unión 4 °C) y se incubó a 37 °C hasta que las células se desprendieron. Las células se neutralizaron con 100 gl de medio y se transfirieron a una placa tratada de cultivo de tejido con fondo de forma de V con 96 pocillos (Costar 3894). Las células se centrifugaron y se lavaron 2 veces con amortiguador FACS (solución salina amortiguadora de fosfato 1X, suero bovino fetal al 2%, azida de sodio al 0,1%). El Ab secundario anti-IgG humana de cabra conjugado con ficoeritrina (Invitrogen H10104, Life Technologies, Carlsbad, CA) se preparó a una proporción de 1 a 100 en un amortiguador FACS. Se agregó a las células anticuerpo secundario a 100 gl/pocillo y se incubó con las células sobre hielo por 45 minutos. Al final del periodo de incubación las células se centrifugaron y se lavaron con amortiguador FACS 3 veces. Las células se fijaron con 100 gl/pocillo de paraformadehído al 1% y se guardaron a 4 °C en la oscuridad. Se repitieron los pasos de disociación celular, incubación con anticuerpo secundario y fijación para las células incubadas a 37 °C para los diversos puntos de tiempo. Al día siguiente todas las muestras se analizaron usando el equipo BD FACSCanto II® usando un sistema HTS (BD Biosciences, San Jose, CA). Las muestras se analizaron con software FlowJo para obtener los valores de media geométrica de fluorescencia para el canal de ficoeritrina. La figura 13A es una gráfica del porcentaje de unión de superficie celular inicial contra la media geométrica-PE unión a 4 °C/media geométrica-punto de tiempo PE a 37 °C x 100. Como lo muestra la figura 13A, los dos anticuerpos NEG085 y 20376 se unen a cKIT sobre la superficie celular y son internados rápidamente en la célula. Esto indica que los ADC de cKIT descritos serían internados rápidamente, suministrando así la toxina a la célula eficientemente.
En otro experimento de internamiento se evaluó el impacto de NEG085 sobre los niveles del receptor cKIT sobre células de medula ósea humana. Células de medula ósea normales CD34+ de humano (All Cells, Cat #ABM022F, Emeryville, CA) se descongelaron y se lavaron con 10 mL de medio StemPro®-34 SFM (Gibco, Life Technologies, Carlsbad, CA). Las células se resuspendieron en 1,25 mL de medio StemPro-34 SFM a 4 x 105 células/mL y se dividieron equitativamente en 2 tubos. Un tubo se dejó sin tratar y el otro se trató con 10 gg/ml de NEG085, y ambos se incubaron a 37 °C, 5% de CO2. Se recolectaron 100 gL de suspensión celular en cada punto de tiempo (0, 15, 30, 60, 120, y 240 minutos) de cada condición, y se pusieron en un tubo de recolección enfriado con hielo para cesar el internamiento. Las células se lavaron con 3 mL de amortiguador de marcación FBS enfriado con hielo y se resuspendieron en 100 glde amortiguador de marcación FBS. Se agregaron a cada tubo 5 ml de 104D2-BV421 (anti-IgG 1 k humana de ratón, Biolegend, San Diego CA), y se incubó sobre hielo durante 1 hora. Después de otro lavado con amortiguador de marcación FBS, el total de receptores cKIT se midió por citometría de flujo evaluando la intensidad de fluorescencia media de BV421 en un FACS Canto II® (BD Biosciences, San José, CA).
Como se muestra en la figura 13B, cKIT es internado rápidamente después de la unión de NEG085, con la mayor parte del internamiento sucediendo rápidamente (15 minutos) y después continuando hasta declinar constantemente la cantidad de cKIT sobre la superficie hasta el punto final de 4 horas.
Ejemplo 16: Evaluación de la capacidad de NEG085-MCC-DM1 para modular la degradación de cKIT en una línea celular de cKIT de tipo silvestre (NCI-H526) o una línea celular de cKIT mutante (GIST-T1).
Se sembraron 5x106 células GIST-T1 (provistas amablemente por Dr. Takahiro Taguchi, Kochi U., Japón) o NCI-H526 (ATCC CRL-5811) en medio de crecimiento (DMEM, 10% FBS para GIST T-1 y RPMI, 10% FBS para NCI-H526) la noche anterior a 37 °C 5% de CO2. Después las células se trataron con cicloheximida 100 mM (CHX) (Cat. # 090M4009, Sigma-Aldrich, St. Louis, MO) en medio libre de metionina (GIBCO: DMEM, 21013-024; RPMI, A14517-01, Life Technologies, Carlsbad, CA). Las células se trataron con ADC con 5 gg/ml (NEG085-MCC-DM1), 10 ng/ml rh-SCF (R&D, 255-SC), o tanto ADC como rh-SCF durante 1,4 o 6 horas a 37 °C con 5% de CO2. Las células se cosecharon a 1 hora, 4 horas y 6 horas después del tratamiento en 1 mL de PBS. Las pellas celulares se lisaron sobre hielo por 60 minutos en 50 gl de amortiguador de lisis (20 mM Tris-HCl; pH 7,5, 137 mM NaCl, 1% Triton X-100, 15% Glicerol, proteasa e inhibidores de fosfatasa. Después los lisados se centrifugaron por 40 minutos a 12000 rpm a 4 °C. Se hirvieron 5 gg de cada muestra por 10 minutos a 75 °C y se cargaron en un gel NuPAGE® 4-12% Bis-Tris (NP0323BOX Life Technologies, Carlsbad, CA). Después de transferencia de proteína a los blots de membrana, las membranas se bloquearon en TBST 5% de leche a temperatura ambiente por 1 hora, y después se sondearon con un anticuerpo anti-cKIT (Cell Signaling Technology Cat. # 3074, Beverly, MA) durante la noche a 4 °C. Los blots se lavaron en TBST (4x5 minutos) el día siguiente. El blot se incubó en el anticuerpo secundario (anti-conejo de cabra-HRP 1:30000, Santa Cruz Biotechnologies, Dallas,
TX) durante 1 hora a temperatura ambiente. El blot se lavó en TBST (4x5 minutos) y se desarrolló. Los anticuerpos primarios usados para el Western blot fueron anti-cKIT (Cell Signaling Technology Cat. # 3074) and GAPDH (Cell Signaling Technology Cat. # 3683). Las figuras 14A/B muestran el curso de tiempo de degradación de receptor cKIT mediada por NEG085-MCC-DM1. La degradación fue rápida con los niveles haciéndose muy bajos/indetectables después de 6 horas. Es de observar que la degradación del receptor cKIT sucede más rápido que SCF con NEG085-MCC-DM1 en las células GIST T1 que expresan un receptor cKIT mutante (panel 14A). También, el NEG085-MCC-DM1 no bloquea el receptor cKIT de la unión de SCF, ya que la adición de NEG085-MCC-DM1 y SCF provee una degradación más rápida como se observa en la figura 14B. Si el NEG085-MCC-DM1 fuera un bloqueador de ligando, no habría diferencia entre NEG085-MCC-DM1 y NEG085-MCC-DM1 con SCF.
Ejemplo 17: NEG085 y 20376 no conjugados no inhiben la proliferación de Mo7e, una línea celular dependiente de SCF
Para evaluar las propiedades antagónicas potenciales de los anticuerpos desnudos y la capacidad de los conjugados anticuerpo-fármaco (ADC) para inhibir la proliferación de una línea celular que expresa cKIT, se desarrollaron MO7e (DSMZ, Catálogo # ACC-104, Braunschweig, DE) en presencia o ausencia de ligando de cKIT, factor de células madre (SCF), para supervivencia. Las células MO7e se desarrollaron en factor estimulador de colonia de granulocitosmacrófagos de humano a 10 ng/ml GM-CSF (R&D Systems Cat. # 215-GM, Minneapolis, MN) o SCF factor de células madre de humano a 10 ng/ml (R&D Systems Cat. # 255-SC) antes de sembrar en placas de 96 pocillos (Costar Cat # 3904, Corning, Tewksbury, MA) a 5000 células/pocillo en 100 pl de medio de dilución. Se generó una placa duplicada para una medición del día 0 y todas las placas se incubaron en un incubador de cultivo de tejido a 37 °C con 5% de CO2 durante la noche. Después de esta incubación se agregaron 50 pl adicionales de medio de dilución, seguido por 90 pl/pocillo del reactivo Cell titer Glo ® (Promega Cat. # G7573, Madison, WI) a cada pocillo de la placa designada “día 0”. Las placas del ensayo se agitaron suavemente por 20 minutos y la intensidad de luminiscencia resultante se midió usando una lectora de placa Perkin Elmer 1450 Microbeta TriLux ® (Perkin Elmer, Waltham, MA). Se prepararon Ab desnudos de prueba y ADC a concentración 3X; 30 pg/ml en el medio de cultivo celular apropiado y se diluyó en series 5 veces para 8 puntos. También se generaron pocillos de medio solo para actuar como controles negativos. Se agregaron 50 pl de anticuerpos diluidos en serie 3X o ADC (concentración del ensayo final 0,0009-68 nM) antes de incubación en un incubador de cultivo de tejido a 37 °C con 5% de CO2 por 5 días. Después de este periodo de incubación se determinó la viabilidad celular relativa por medio de la adición del reactivo Cell titer Glo como se describe arriba. El efecto de los ADC sobre la proliferación celular se calculó usando el promedio de los duplicados de la siguiente manera: (% Inhibición = (tratado con ADC o Ab) / (no tratado)*100). Los datos de porcentaje de inhibición se ajustaron a una ecuación logística de 4 parámetros y se determinaron los valores de IC50.
Como se muestra en la figura 15 y la figura 16, los anticuerpos anti-cKIt desnudos no inhiben la proliferación celular. En la figura 15 se compara NEG085-MCC-DM1 con NEG085, NEG024 y 20376 no conjugados. Como se muestra claramente en la gráfica, NEG085-MCC-DM1 inhibe la proliferación celular de las células M07e a una concentración baja, mientras que los anticuerpos no conjugados no tienen este efecto. El control IgG-MCC-DM1 tiene un mayor efecto antiproliferativo que los NEG085, NEG024 o 20376 no conjugados.
Esto también se observa en la figura 16, en donde el experimento usa GM-CSF en lugar de SCF para anular el efecto de internamiento del receptor cKIT que tiene el ligando SCF. El resultado de la figura 16 es consistente con la figura 15, ya que un anticuerpo NEG085 no conjugado no tiene efecto nocivo sobre la proliferación celular, de forma similar a una IgG no conjugada de control. En resumen, los resultados mostrados en las figuras 15 y 16 indican que la reducción en la proliferación celular se debe a la conjugación de los anticuerpos anti-cKIT con la toxina.
Ejemplo 18: Evaluación de la actividad ADCC in vitro
La capacidad de los anticuerpos anti-cKIT no conjugados (NEG085, 20376) para mediar la citotoxicidad celular dependiente de anticuerpo se determinó contra células Uke-1 (células objetivo; provistas generosamente por el Profesor Walter Fiedler, University Hospital Eppendorf, Hamburg, Alemania) en coincubación con células NK3.3 (células asesinas o células efectoras; provistas amablemente por Jacky Kornbluth de Saint Louis University). Brevemente, células Uke-1 se marcaron con calceín acetoxi-metíl éster (Calcein-AM; Sigma-Aldrich , # catálogo 17783-5MG, St. Louis, MO), se lavaron 2 veces, se tomaron con pipeta a una placa de microtitulación de 96 pocillos (96 pocillos, fondo en forma de U, plástico transparente; Corning Costar, # catálogo 650 160, Tewksbury, MA) a una concentración de 5000 células por pocillo y se pre-incubaron 10 minutos con una dilución en serie de los anticuerpos y proteínas anteriormente mencionados (de 50000 a 0,003 pg por ml) antes de agregar las células NK3.3 efectoras durante 1 hora a una relación de efector a objetivo de 20 a 1. Para calcular la lisis especifica de anticuerpo de las células objetivo, una incubación paralela de células objetivo solas sin anticuerpo o células efectoras sirvió como una línea de base y control negativo, mientras que el control positivo o lisis máxima o 100% de lisis especifica se determinó por lisis de células objetivo solas con una solución de
Tritón X 100 al 1%. Como un control positivo adicional se utilizó MabCampath® (Sanofi, Paris, FR), que reconoce CD52 sobre las células Uke-1. Después de una coincubación de las células objetivo y efectoras, la placa de microtitulación se centrifugó y una alícuota del fluido sobrenadante se transfirió a otra placa de microtitulación (96 pocillos, fondo plano, negra con fondo transparente (Corning Costar, # catálogo 3904, Tewksbury, MA) y la concentración de calceín libre en solución se determinó con un contador de fluorescencia (Victor 3 ® multilabel counter, Perkin Elmer, Waltham, MA).
Los resultados se presentan en la figura 17. La citotoxicidad mediada por célula dependiente de anticuerpo (ADCC) es un mecanismo de inmunidad mediada por célula. Por medio del cual una célula efectora lisa una célula objetivo que ha sido enlazada por anticuerpos específicos. En este experimento el MabCampath® y también los anticuerpos anti-cKIT 20376 y NEG085 son anticuerpos IgG1 humanos no conjugados. Como se muestra en la figura 17, solo el anticuerpo MabCampath media la destrucción por ADCC de las células objetivo. Tanto 20376 como NEG085 no fueron capaces de inducir ADCC incluso a concentraciones más altas. Por lo tanto, cualquier muerte celular observada cuando se usó uno de los ADCC, por ejemplo, NEG085-MCC-DM1, no se debe a un mecanismo de acción ADCC.
Ejemplo 19: La capacidad de NEG085 y 20376 para ocasionar apoptosis de mastocitos fue investigada usando mastocitos humanos primarios
Mastocitos humanos primarios se cultivaron de sangre periférica humana de acuerdo con los métodos descritos por Saito et al., Nature Protocols 2006;1(4):2178-2183. Los mastocitos, que habían estado en cultivo líquido durante un mínimo de una semana, se incubaron con concentraciones crecientes (0,05 - 100 nM) de los Ab anti-cKIT humano, NEG085 y 20376, o una IgG de control de isotipo, en presencia de rhSCF 1,6 nM (Genscript, Cat # Z00400, Piscataway, NJ), durante 48 horas a 37 °C antes de la adición del reactivo Caspase-Glo® 3/7 (Promega, Cat. # G8093, Madison, W i) para medir la apoptosis. Después de 30 minutos de incubación a t.a., la luminiscencia se registró en la lectora de placa BioTek® Synergy (BioTek, Winooski, VT).
Como cKIT es expresada sobre mastocitos, cualquier anticuerpo anti-cKIT terapéutico no debe causar agotamiento de mastocitos. La figura 18 muestra un ensayo de apoptosis con mastocitos primarios humanos después de tratamiento con Abs anti-cKIT humano o un Ab de control de isotipo, en presencia de rhSCF 1,6 nM. Los mastocitos humanos primarios se incubaron con concentraciones crecientes de los anticuerpos anti-cKIT, NEG085 y 20376, o una IgG de control de isotipo. Como se muestra en la figura 18, tanto el anticuerpo NEG085 como el 20376 no conjugados no producen apoptosis de mastocitos primarios humanos ex vivo.
Ejemplo 20: La capacidad de NEG085 y 20376 para mediar la desgranulación de mastocitos fue determinada usando mastocitos humanos primarios
Se cultivaron mastocitos humanos primarios de sangre periférica humana de acuerdo con los métodos descritos por Saito et al., (supra). Los mastocitos, que habían estado en cultivo líquido durante un mínimo de una semana, se pre-trataron con IgE JW8 especifica de Ag al 5% (lote del laboratorio ACE 27283), 95% de IgE humana monoclonal no específica (Abbiotec, Cat # 12030635, San Diego, CA) y rhIL-4 a 10 ng/mL (R&D Systems Cat # 204-IL, Minneapolis, MN) durante 5 días a 37 °C. Después las células se incubaron con concentraciones crecientes (0,05 - 100 nM) de una IgG de control de isotipo, los Ab anti-cKIT humano 20376 y NEG085, el Ab anti-IgE, LE27, o el antígeno NIP(5)BsA, en presencia de 1 Ab específico de Fc anti-IgG humana (H+L) de cabra (Jackson ImmunoResearch, Cat # 109-005-008-JlR, West Grove, PA) por 90 minutos a 37 °C. Después las células se centrifugaron y los sobrenadantes se transfirieron a placas de paredes negras de 96 pocillos antes de la adición del sustrato de p-hexosaminidasa. Después de 90 minutos de incubación a 37 °C, la reacción se detuvo agregando tris-base (Sigma, Cat # T1503-500G, pH 12, St. Louis, MO) y la intensidad de fluorescencia se registró en la lectora de placa Envision®.
Como en el experimento anterior del ejemplo 19, es importante evaluar cualquier efecto nocivo de los anticuerpos anticKIT sobre los mastocitos. En donde el experimento previo examinó la apoptosis de mastocitos, aquí los experimentos están dirigidos a la desgranulación de mastocitos. Como se muestra en la figura 19, los controles positivos NlP(5) y LE27 muestran niveles altos de desgranulación de mastocitos. En contraste, los anticuerpos anti-cKIT NEG085 y 20376 no inducen desgranulación de mastocitos de mastocitos primarios humanos ex vivo.
Ejemplo 21: Modulación de marcador farmacodinámico sobre el objetivo in v ivo por ADC de cKIT
Se hicieron estudios para evaluar la capacidad del ACD de cKIT NEG027-MCC-DM1 para modular marcadores farmacodinámicos in vivo, incluso un examen de la colocalización de anticuerpo NEG027 hacia el evento farmacodinámico (PD) de detención mitótica en el xenoinjerto de tumor GIST T1 que expresa cKIT mutante. La meta de estos estudios fue evaluar el grado y duración de la detención del ciclo celular G2/M.
La presencia de ACD fue calculada indirectamente detectando anticuerpo IgG humano (que es NEG027 en el ratón) en el tumor usando un enfoque inmunohistoquímico. Un anti-IgG humana de conejo (H+L) purificado por afinidad se obtuvo de Jackson ImmunoResearch Laboratories (Cat. # 309-005-082, West Grove, PA). El anticuerpo reacciona con la IgG humana de molécula completa y las cadenas ligeras de otras inmunoglobulinas humanas con mínima reacción cruzada a las proteínas del suero del ratón. Brevemente, el protocolo de IHC incluyó calentamiento y exposición estándar al reactivo de recuperación de antígeno Ventana Cell Conditioning #1 (Ventana, Tucson, AZ). El anticuerpo primario se diluyó a una concentración de trabajo de 2 pg/ml y se incubó 32 minutos a temperatura ambiente. Subsecuentemente se hizo una incubación con anticuerpo anti-conejo conjugado con HRP prediluido Ventana UltraMap (Cat # 760-4315, Ventana, Tucson, AZ) durante 32 minutos.
La acumulación de núcleos pHH3 positivos, evaluada por inmunohistoquímica, se usó como un marcador de la detención de G2/M. Un anticuerpo policlonal de conejo producido por animales en inmunización con un fosfopéptido sintético que corresponde a los residuos circundantes a Ser10 de histona humana H3 (pHH3) se obtuvo de Cell Signaling Technology (Danvers, MA, Cat. # 9701). Brevemente, el protocolo de IHC incluyó calor y exposición estándar al reactivo de recuperación de antígeno Ventana Cell Conditioning #1. El anticuerpo primario se diluyó 1:50 y se incubó 60 minutos a temperatura ambiente. Subsecuentemente, se incubó con el anticuerpo secundario anti-conejo de cabra biotinilado de Jackson ImmunoResearch Laboratorios (Cat. # 111-065-144, West Grove, PA) durante 32 minutos.
Para evaluar los cambios de marcador PD inducidos por ADC anti-cKIT en el modelo de xenoinjerto del tumor subcutáneo GIST T1, ratones SCID-beige hembras fueron implantados por vía subcutánea con 10x106 células en una suspensión que contenía Matrigel™ al 50% (BD Biosciences) en solución salina equilibrada de Hank. El volumen de inyección total que contenía las células en suspensión era de 200 pl. Los ratones se asignaron aleatoriamente para recibir una sola dosis i.v. de NEG027-MCC-DM1 (2,5 mg/kg), control de isotipo no específico IgG1-MCC-DM1 (2,5 mg/kg) o solución salina amortiguadora tris (TBS; 5 ml/kg) una vez que los tumores alcanzaron entre 300 y 500 mm3 (n=3/grupo). La inmunotinción para IgG humana muestra en donde está localizado NEG027 y esto se correlaciona con áreas de una mayor densidad de inmunotinción de pHH3 (imágenes representativas mostradas en la figura 20, suministrando soporte para la colocalización de anticuerpo con el efecto farmacodinámico. Consistentemente con el mecanismo de acción esperado de la carga útil de maitansinoide, NEG027-MCC-DM1 produjo un aumento marcado, dependiente del tiempo, en el porcentaje de células positivas para positividad de pHH3, alcanzando un pico a 33 h y 48 h después de la dosis con respecto a los controles de isotipo no específico IgG1-MCC-DM1 o tratados con PBS, con una señal de vuelta a la línea de base a alrededor de una semana (imágenes representativas en la figura 21, la gráfica mostrada en la figura 22). También se evaluaron los cambios dependientes del tiempo en caspasa 3 escindida. En estos estudios, un anticuerpo policlonal de conejo producido inmunizando animales con un péptido sintético que corresponde a residuos amino-terminales adyacentes a (Asp175) en caspasa 3 humana se obtuvieron de EMD Millipore (Cat. #PC679). El protocolo de IHC incluyó calor y exposición estándar al reactivo de recuperación de antígeno Ventana Cell Conditioning #1. El anticuerpo primario se diluyó a 20 pg/ml y se incubó durante 32 minutos a temperatura ambiente. Subsecuentemente, se incubó con anticuerpo secundario anti-conejo de cabra biotinilidado de Jackson ImmunoResearch Laboratorios (Cat. # 111-065-144, West Grove, PA) durante 32 minutos.
De forma similar a pHH3, los cambios dependientes del tiempo en la caspasa 3 escindida también se observaron (imágenes representativas mostradas en la figura 21, gráfica mostrada en la figura 22). Estos datos demuestran que el ADC de cKIT NEG027-MCC-DM1 es capaz de provocar efectos PD celulares in vivo robustos consistentes con el mecanismo de acción de la carga útil de maitansinoide.
Una fotografía representativa de la inmunotinción de cKIT sobre el tumor GIST T1 se muestra para visualizar el patrón de tinción en este modelo de xenoinjerto (figura 21). Un anticuerpo policlonal de conejo producido inmunizando animales con un péptido sintético que corresponde a los aminoácidos 963 a 976 en la parte c-terminal citoplásmica de cKIT fue obtenida de Dako (Cat. # A4502). Brevemente, el protocolo de IHC incluyó calor y exposición estándar al reactivo de recuperación de antígeno Ventana Cell Conditioning #1. El anticuerpo primario se diluyó a una concentración de trabajo de 14 pg/m y se incubó por 60 minutos a temperatura ambiente. Subsecuentemente se incubó con el anticuerpo anti-conejo conjugado con HRP prediluido de Ventana UltraMap (Cat # 760-4315) durante 16 minutos.
Ejemplo 22: Eficacia in vivo de ADC anti-cKIT contra el tumor estromal gastrointestinal (GIST) en ratones
La actividad antitumoral de ADC anti-cKIT se evaluó en varios modelos de xenoinjerto de tumor. La actividad antitumoral dependiente de la dosis y la farmacocinética (PK) de un ADC anti-cKIT humano reactivo cruzadamente con cKIT diferente de ratón NEG027-MCC-DM1 se evaluó en el modelo de xenoinjerto de tumor subcutáneo GIST T1 que expresa cKIT mutante. Ratones SCID-beige hembras recibieron implantes por vía subcutánea de 10x106 células que contenían Matrigel™ al 50% (BD Biosciences) en solución salina balanceada de Hank. El volumen de inyección total que contenía las células en suspensión era de 200 pl.
Los ratones se enrolaron en el estudio 10 días después de la implantación con un volumen de tumor promedio de 207 mm3. Después de ser asignados aleatoriamente a uno de cinco grupos (n = 9/grupo), los ratones se les administró una sola dosis i.v. de TBS, el vehículo de ADC (5 ml/kg), un control de isotipo no específico IgG1-MCC-DM1 (2,5 mg/kg), o NEG027-MCC-DM1 (0,625, 1,25 o 2,5 mg/kg). Los volúmenes de tumor y los pesos corporales se midieron 2 veces a la semana. La IgG1-MCC-DM1 de control no fue activa significativamente a 2,5 mg/kg. NEG027-MCC-DM1 a 0,625 mostró eficacia estadísticamente significativa en comparación con el grupo tratado con TBS, sin embargo, 1,25 y 2,5 mg/kg indujeron una eficacia aún mayor, induciendo estasis de volumen de tumor similar según las mediciones del calibrador, aunque una evaluación histológica no mostrara la presencia de células de tumor. En su lugar, una mezcla de tejido conectivo, tejido adiposo y segmentos de nervios periféricos y músculo estriado fueron los principales componentes de tejido en estas secciones. Esto apoya una regresión histológica en el tumor (figuras 23-26).
De este estudio, también se recolectó el suero a 1 hora, 24 horas y 4, 7, 11, y 21 días después de la dosis para medir la concentración de anticuerpo/ADC con el tiempo usando un ELISA de anti—IgG 1 humana y un ELISA de anti-DM, respectivamente. Para evaluar los parámetros PK el suero se recolectó por medio de sangrados retroorbitales y se analizó por medio de ELISA. El ensayo PK de anticuerpo total mide la concentración total de anticuerpo con/sin DM1 mediante ELISA colorimétrico. Las placas se recubren con anti—IgG humana (específico de Fc), y la detección es con un anti—IgG humana-HRP antes de ser leída en una lectora de placa apropiada. El ensayo PK del conjugado mide el anticuerpo que está enlazado a por lo menos una (1) molécula de DM1 por medio de ELISA colorimétrico. En este formato, las placas se recubren con anticuerpo anti—maitansina y se detecta con anti—IgG humana-HRP. La PK es proporcional a la dosis con una vida media en el suero aproximada de 7 días (figuras 23-24).
Puesto que una sola dosis de 0,625 mg/kg de NEG027—MCC—DM1 solo causó retraso del crecimiento del tumor GIST T1, suministrando así una escala dinámica para evaluar diversas actividades de ADC, este nivel de dosis se seleccionó para evaluar la eficacia de un grupo de ADC estrechamente relacionados, también derivados del 9P3—MCC—DM1 ADC murino original. Ratones SCID—beige hembras recibieron implantes por vía subcutánea de 10x106 células que contenían Matrigel™ al 50% (BD Biosciences, San Jose, CA) en solución salina balanceada de Hank. El volumen total de inyección que contenía las células en suspensión fue de 200 pl. Los ratones se enrolaron en el estudio 10 días después de la implantación con un volumen de tumor promedio de 195 mm3. Después de ser asignados aleatoriamente a los grupos (n = 8/grupo), a los ratones se les administró una sola dosis i.v. de t Bs (8 ml/kg), un control de isotipo no específico IgG1 — MCC—DM1 (10 mg/kg), NEG085—MCC—DM1 (0,625 mg/kg), NEG086—MCC—DM1 (0,625 mg/kg), NEG087—MCC—DM1 (0,625 mg/kg), NEG024—MCC—DM1 (0,625 mg/kg), o NEG026—MCC—DM1 (0,625 mg/kg). Los volúmenes de tumor y pesos corporales se midieron 2 veces a la semana (figuras 27-29). El control IgG1—MCC—DM1 incluso a la dosis alta de 10 mg/kg no fue activo. Los ADC anti—cKIT a 0,625 mg/kg no fueron estadísticamente diferentes entre sí. Los grupos tratados con NEG085—MCC—DM1 y NEG024—MCC—DM1 tuvieron los volúmenes de tumor más pequeños en la escala más estrecha.
De este estudio, también se recolectó el suero a 1 hora, 24 horas y 3, 7, 10, 14 y 21 días después de la dosis para medir la concentración de anticuerpo/ADC con el tiempo usando un ELISA de anti—IgG1 humana y un ELISA de anti—DM, respectivamente. Para evaluar los parámetros PK el suero se recolectó por medio de sangrados retrorbitales y se analizó por medio de ELISA. El ensayo PK de anticuerpo total mide la concentración total de anticuerpo, con/sin DM1 por ELISA colorimétrico. Las placas se recubren con anti—IgG humana (específico de Fc) y la detección es con anti—IgG humana— HRP, antes de ser leída en una lectora de placas apropiada. El ensayo PK del conjugado mide el anticuerpo que está enlazado a por lo menos una (1) molécula de DM1 por medio de ELISA colorimétrico. En este formato, las placas se recubren con anticuerpo anti—maitansina y se detectan con anti—IgG humana—HRP. Estos ADC mostraron exposiciones al suero similares (figura 30).
Ejemplo 23: Eficacia in vivo de ADC anti-cKIT contra cáncer de pulmón de célula pequeña en ratones
La actividad antitumoral de un grupo de ADC se evaluó en el modelo de xenoinjerto de cáncer de pulmón de célula pequeña NCI—H1048 con inmunotinción de cKIT moderada que exhibe mayor heterogeneidad en comparación con xenoinjertos de tumor GIST T1 (Figura 21-Figura 30). El NEG085—MCC—DM1 se comparó con un grupo de ADC de cKIT que son fuertes antagonistas de la señalización de cKIT, ninguno de los cuales se une a cKIT de ratón. Ratones SCID— beige hembras recibieron implantes por vía subcutánea de 10x106 células que contenían Matrigel™ al 50% (BD Biosciences) en solución salina equilibrada de Hank. El volumen de inyección total que contenía las células en suspensión era de 200 pl. Los ratones se enrolaron en el estudio 15 días después de la implantación con un volumen de tumor promedio de aproximadamente 120 mm3. Todos los grupos tratados recibieron una sola dosis intravenosa de 2 mg/kg. Después de asignarse aleatoriamente a los grupos (n = 8/grupo), se administró a los ratones una sola dosis i.v. de TBS (5 ml/kg), un control de isotipo no específico IgG1—MCC—DM1 (2 mg/kg), NEG024—MCC—DM1, NEG085—MCC—DM1, y NEG086—MCC—DM1. Los volúmenes de tumor y pesos corporales se midieron 2 veces a la semana (figuras 31 y 32). El
IgG1-MCC-DM1 de control no fue activo. El NEG085-MCC-DM1 tiende a ser eficaz con una AT/AC baja de 9%, pero no fue estadísticamente diferente del vehículo a esta dosis de 2 mg/kg. El NEG024-MCC-DM1 y NEG026-MCC-DM1 fueron significativamente eficaces.
También se evaluó la eficacia antitumoral de ADC de cKIT en el modelo de xenoinjerto de cáncer de pulmón de célula pequeña NCI-H1048 de actividad antitumoral dependiente de la dosis de NEG085-MCC-DM1. A ratones SCID-beige hembras se les implantó por vía subcutánea Matrigel™ al 50% que contenía 10x106 células (BD Biosciences) en solución salina equilibrada de Hank. El volumen total de inyección que contenía las células en suspensión era de 200 gl. Los ratones fueron enrolados en el estudio 11 días después de la implantación con un volumen de tumor promedio de aproximadamente 150-200 mm3. Después de ser asignados aleatoriamente a los grupos (n = 8/grupo), se administró a los ratones una sola dosis i.v. de TBS (5 ml/kg), un control de isotipo no específico IgG1-MCC-DM1 (10 mg/kg), o NEG085-MCC-DM1 (2,5, 5 y 10 mg/kg). Se midieron los volúmenes de tumor y los pesos corporales 2 veces a la semana (figura 33 y figura 34). El IgG1-MCC-DM1 control no fue activo, tampoco la dosis de 2,5 mg/kg de NEG085-MCC-DM1. Sin embargo, las dosis de 5 y 10 mg/kg fueron significativamente eficaces.
La actividad antitumoral de dos ADC anti-cKIT se evaluó en un segundo modelo de xenoinjerto de cáncer de pulmón de célula pequeña con niveles más altos de cKIT de forma similar a los xenoinjertos de tumor GIST T1 (fotografías representativas en la figura 21 y gráficas en la figura 35). Ratones SCID-beige hembras se les implantó por vía subcutánea con 10x106 células que contenía Matrigel™ al 50% (BD Biosciences) en solución salina equilibrada de Hank. El volumen total de inyección que contenía las células en suspensión era de 200 gl. Los ratones fueron enrolados en el estudio 6 días después de la implantación con un volumen de tumor promedio de aproximadamente 150 mm3. Después de ser asignados aleatoriamente a los grupos (n = 9/grupo), se administró a los ratones una sola dosis i.v. de TBS (8 ml/kg), un control de isotipo no específico IgG1-MCC-DM1 (10 mg/kg), NEG024-MCC-DM1 (2,5, 5 y 10 mg/kg) y ADC 20376-MCC-DM1 (10 mg/kg) cruzadamente reactivo de ratón. Se midieron los volúmenes de tumor y los pesos corporales 2 veces a la semana (figuras 35 y 36). El IgG1-MCC-DM1 de control no fue activo. El 20376-MCC-DM1 a 10 mg/kg inicialmente regresó los tumores, sin embargo después de la regresión inicial se observó recurrencia de tumor. Se observó una eficacia significativa dependiente de la dosis con las tres dosis de NEG024-MCC-DM1, con regresión sostenida a largo plazo a 10 mg/kg, empezando los tumores a volver a crecer después de 60 días, sugiriendo que 20376-MCC-DM1 puede requerir más de la sola dosis administrada en este estudio. La exposición sérica de una dosis de 10 mg/kg de 20376-MCC-DM1 y NEG024-MCC-DM1 fue aproximadamente equivalente.
Ejemplo 24: Eficacia in vivo de ADC anti-cKIT contra leucemia mielógena aguda en ratones
La actividad antitumoral dependiente de la dosis de ADC anti-cKIT 9P3-MCC-DM1 y 9P3-SPDB-DM4 de murino se evaluó en el modelo de xenoinjerto de tumor subcutáneo Kasumi-1 de leucemia mielógena aguda que expresa cKIT mutante. Ratones SCID-beige hembras recibieron trasplantes por vía subcutánea de 2-3 piezas de 1 mm3 de tejidos de tumor de Kasumi-1 fragmentados en el costado derecho con Matrigel™ (BD Biosciences). Los ratones con tumores de Kasumi-1 fueron enrolados en el estudio 21 días después de la implantación con un volumen promedio de tumor de 150 mm3. Después de ser asignados aleatoriamente a 1 de 8 grupos (n = 8/grupo), se administró a los ratones una sola dosis i.v. de PBS (200 gl), un control de isotipo no específico IgG1-SPDB-DM4 (10 mg/kg), 9P3-MCC-DM1 (10 mg/kg) y 9P3-SPDB-DM4 (1 o 5 mg/kg). Se midieron los volúmenes de tumor y pesos corporales 3 veces a la semana (figura 37). El IgG1-SPDB-DM4 de control no fue significativamente activo a 10 mg/kg. Se observó regresión del crecimiento del tumor con 9P3-SPDB-DM4 a dosis de 5 mg/kg y 10 mg/kg.
Tabla 10: Eficacia en Kasumi-1
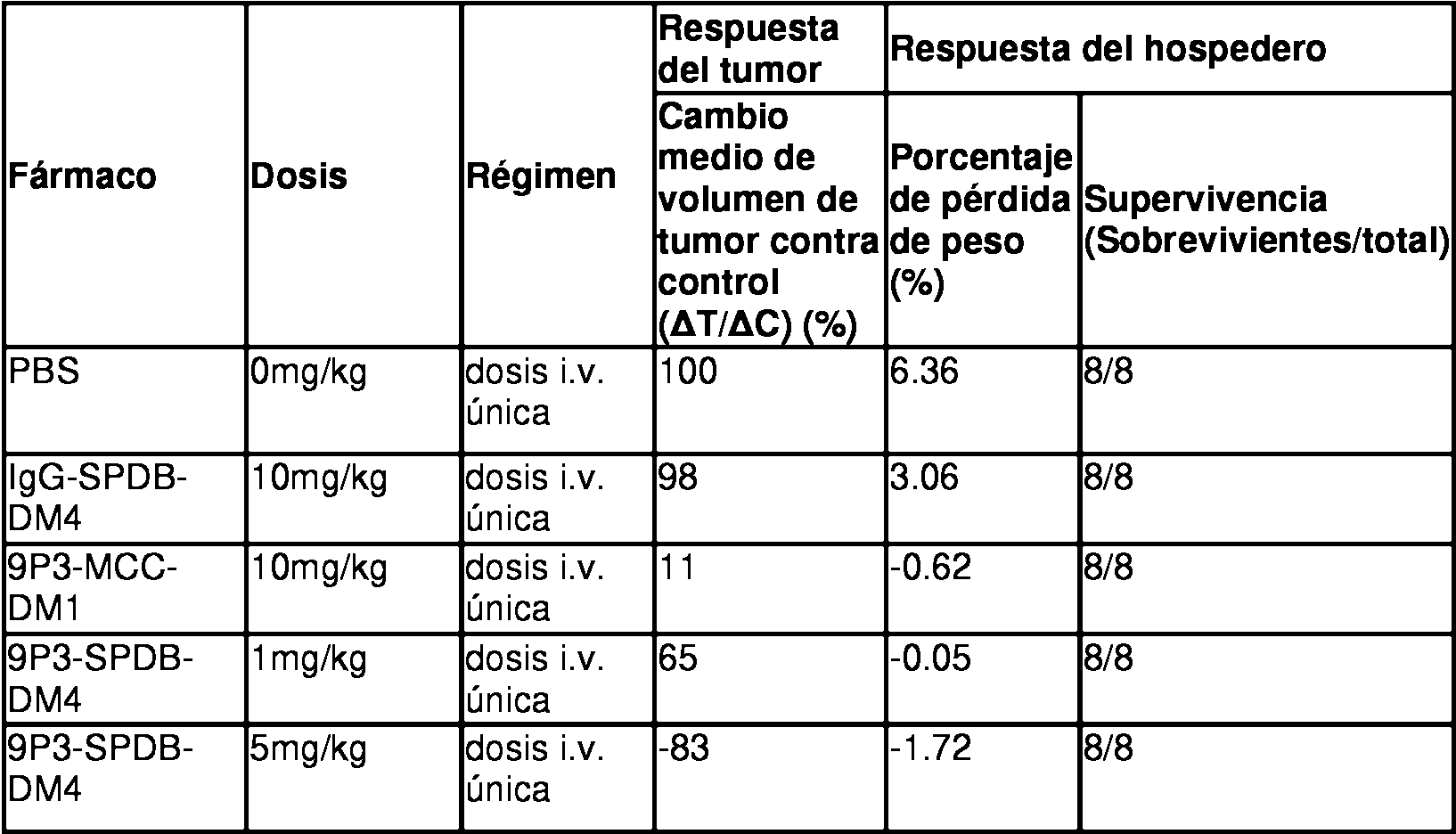
Ejemplo 25: Eficacia in vivo de ADC anti-cKIT contra mastocitocis en ratones
La actividad antitumoral de ADC anti-cKIT 9P3-MCC-DM1 y 9P3-SPDB-DM4 de murino se evaluó en el modelo de xenoinjerto de tumor subcutáneo HMC-1.2 que expresa cKIT mutante. La línea celular HMC-1.2 fue provista amablemente por el Dr. Joseph Butterfield, Mayo Clinic, Rochester, MN. Ratones sin pelo Foxn-1 hembras recibieron implantes por vía subcutánea de 3, 5, y 10x106 células que contenían Matrigel™ al 50% (Bd Biosciences) en medio DMEM libre de FBS. El volumen total de inyección que contenía las células en suspensión era de 100 gl.
Los ratones que llevan el tumor HMC-1.2 en este estudio se enrolaron 33 días después de la implantación con un volumen de tumor promedio 100 mm3. Después de ser asignados aleatoriamente a 1 de 3 grupos (n = 4/grupo), se administró a los ratones una sola dosis i.v. de pBS (200 gl), 9P3-MCC-DM1 (10 mg/kg) o 9P3-SPDB-DM4 (10 mg/kg). Se midieron los volúmenes de tumor y pesos corporales 3 veces a la semana (figura 38). Se observó regresión de tumor con 9P3-SPDB-DM4 y 9P3-MCC-DM1 a 10 mg/kg.
Tabla 11: Estudio de HMC-1

Ejemplo 26: Eficacia in vivo de un ADC de cKIT cruzadamente reactivo con ratón 20376-MCC-DM1
La actividad antitumoral dependiente de la dosis y farmacocinética (PK) del ADC anti-cKIT humano 20376-MCC-DM1 reactivo cruzadamente con cKIT de ratón se evaluó en el modelo de xenoinjerto de tumor subcutáneo GIST T1 que expresa cKIT mutante. Ratones SCID-beige hembras fueron enrolados en el estudio 10 días después de la implantación con un volumen de tumor promedio de aproximadamente 200 mm3. Después de ser asignados aleatoriamente a 1 de 5 grupos (n = 9/grupo), se administró a los ratones una sola dosis i.v. de TBS (5 ml/kg), un control de isotipo no específico IgG1-MCC-DM1 (10 mg/kg), NEG085-MCC-DM1 (0,625 mg/kg) o 20376-MCC-DM1 (0,625, 2,5, 5 o 10 mg/kg). Se midieron los volúmenes de tumor y pesos corporales 2 veces a la semana (figuras 39-41). El IgG1-MCC-DM1 de control no fue significativamente activo a 10 mg/kg. El 203786-MCC-DM1 también fue ineficaz, mientras que NEG085-MCC-DM1 a 0,625 mostró baja eficacia, aunque tampoco fue estadísticamente significativo. El 20376-MCC-DM1 a 2,5, 5 y 10 mg/kg fue significativamente eficaz.
De este estudio también fue recolectado el suero a 1 hora, 24 horas y 4, 7, 11 y 21 días después de la dosificación para medir la concentración de anticuerpo/ADC con el tiempo usando un ELISA de anti-IgG1 humana, y un ELISA de anti-DM respectivamente. Para evaluar los parámetros PK el suero se recolectó por medio de sangrados retrorbitales y se analizó por medio de ELISA. El ensayo PK de anticuerpo total mide la concentración total de anticuerpo con/sin DM1 mediante ELISA colorimétrico. Las placas se recubren con anti-IgG humana (específico de Fc), y la detección es con un anti-IgG humana-HRP antes de ser leída en una lectora de placa apropiada. El ensayo PK del conjugado mide el anticuerpo que se enlaza a por lo menos una (1) molécula de DM1 por medio de ELISA colorimétrico. En este formato, las placas se recubren con anticuerpo anti-maitansina y se detecta con anti-IgG humana-HRP. Con el ADC 20376-MCC-DM1 reactivo cruzadamente con cKIT de ratón, la PK no fue proporcional a la dosis debido a que el ADC que se une a cKIT de ratón en tejidos normales afecta la exposición (disposición de fármaco mediada por tejido), y por lo tanto hay una clara diferencia en concentraciones séricas entre 20376-MCC-DM1 y el ADC NEG085-MCC-DM1 no reactivo cruzadamente con cKIT de ratón (figura 40). Esto representa la diferencia de eficacia entre los dos ADC a la dosis baja de 0,625 mg/kg. A las dosis más altas, el efecto de disposición de fármaco mediada por tejido es menos pronunciado y la eficacia se hace evidente en el modelo de xenoinjerto de tumor GIST T1 en ratones.
Ejemplo 28: Eficacia in vivo de ADC de cKIT con la carga útil SPDB-enlazador DM4 contra tumores estromales gastrointestinales
La actividad antitumoral dependiente de la dosis de los ADC 9P3 murinos (de los cuales derivan NEG024 y NEG085) con los enlazadores/cargas útiles MCC-DM1 (no escindible) y SPDB-DM4 (escindible) se comparó en el modelo de xenoinjerto de tumor subcutáneo GIST T1 que expresa cKIT mutante. Ratones SCID-beige hembras fueron enrolados en el estudio 18 días después de la implantación con un volumen de tumor promedio de aproximadamente 170 mm3. Después
de ser asignados aleatoriamente a los grupos (n = 8/grupo), se administró a los ratones una sola dosis i.v. de TBS (5 ml/kg), anticuerpo 9P3 murino no conjugado (10 mg/kg), un control de isotipo no específico IgG1-MCC-DM1 (5 mg/kg), control de isotipo no específico IgG1-McC-DMl (5 mg/kg), control de isotipo no específico IgG1-SPDB-DM4 (10 mg/kg), 9P3-M C C -d M i (5 y 10 mg/kg) o 9P3-SPDB-DM4 (2,5 y 5 mg/kg). Se midieron los volúmenes de tumor y pesos corporales 2 veces a la semana (figuras 42, 43). Ni los ADC de IgG 1 de control no específicos ni el p93 no conjugado fueron eficaces. Todos los ADC 9P3 fueron eficaces a las dosis probadas; sin embargo, en los tumores del grupo tratado con 2,5 mg/kg de 9P3-SPDB-DM4 se observó eficacia ligeramente menor que los otros grupos.
La actividad antitumoral dependiente de la dosis de los ADC 9P3 murinos (de los que derivan NEG024 y NEG085) con los enlazadores/cargas útiles MCC-DC1 (no escindible) y SPDB-DM4 (escindible) se comparó en un segundo modelo de xenoinjerto de tumor estromal gastrointestial que expresa cKIT mutante, GIST430. Ratones SCID-beige hembras fueron enrolados en el estudio 11 días después de la implantación con un volumen de tumor promedio de aproximadamente 200 mm3. Después de ser asignados aleatoriamente a los grupos (n = 9/grupo), se administró a los ratones una sola dosis de i.v. de TBS (5 ml/kg), anticuerpo 9P3 no conjugado de murino (10 mg/kg), un control de isotipo no específico IgG1-MCC-DM1 (5 mg/kg), control de isotipo no específico IgG1-MCC-DM1 (10 mg/kg), control de isotipo no específico IgG1-SPDB-DM4 (5 mg/kg), 9P3-MCC-DM1 (10 mg/kg) o 9P3-SPDB-DM4 (5 mg/kg). Se midieron los volúmenes de tumor y pesos corporales 2 veces a la semana (figuras 44, 45). Ninguno de los ADC de IgG 1 de control no específicos fue eficaz. Sin embargo, ambos ADC 9P3 fueron similarmente eficaces a las dosis probadas.
Ejemplo 29: Formulación
La forma de servicio clínico (CSF) del ADC es un liofilizado en un vial que contiene 50 mg de anti-cKIT-MCC-DM1, 16,2 mg de succinato de sodio, 410,8 mg de sacarosa y 1 mg de polisorbato 20 (sin considerar el sobrerelleno de 10% para permitir el retiro del contenido declarado). Después de la reconstitución del liofilizado con 5 mL de agua para inyección, se obtiene una solución que contiene 10 mg/mL de anti-cKIT-MCC-DM1, succinato de sodio 20 mM, sacarosa 240 mM y polisorbato 20 al 0,02 % a un pH de 5,0.
Para administración intravenosa subsecuente, la solución obtenida usualmente se diluirá adicionalmente en una solución vehículo para obtener una solución de ADC lista para usarse para infusión.
Para la CSF se escogió una concentración de ADC de 10 mg/mL basándose en las pruebas preliminares de estabilidad. Se seleccionó una concentración de sacarosa de 240 mM para crear una formulación isotónica, para mantener una estructura de torta de liofilizado amorfo y producir estabilización de proteína.
Los métodos analíticos importantes que indican estabilidad para seleccionar la formulación más estable abarcan, entre otros, cromatografía de exclusión de tamaño para determinar el grado de agregación, prueba de materia en partículas sub-visibles, determinación de toxina libre y pruebas de potencia.
El estudio de pretamizado mostró que el polisorbato 20 a una concentración de 0,02% provee estabilización suficiente contra el esfuerzo mecánico. Los estudios de estabilidad en líquido y liofilizado en tiempo real y condiciones de estabilidad acelerada (25 °C y 40 °C) mostraron que una formulación de succinato pH 5,0 provee mejor estabilidad general en almacenamiento. Mas notablemente en esta formulación el mejor equilibrio de todas las formulaciones probadas entre agregación y liberación de la toxina libre se pudieron cumplir. Después de 3 meses a 40 °C no pudo determinarse un aumento notable en productos de degradación.
Ejemplo: 30 Modulación de marcador farmacodinámico in vivo en el objetivo por ADC de cKIT
Se hicieron estudios para evaluar la capacidad del ADC de cKIT NEG085-MCC-DM1 para modular marcadores farmacodinámicos in vivo, incluso un examen de la colocalización del anticuerpo NEG085 para el evento farmacodinámico (PD) de detención mitótica en el xenoinjerto de tumor GIST T1 que expresa cKIT mutante. La meta de estos estudios fue evaluar el grado y duración de detención del ciclo celular G2/M.
La acumulación de núcleos fosfo-Histona H3 (pHH3) positivos, evaluada por inmunohistoquímica, se usó como un marcador de la detención G2/M. Un anticuerpo policlonal de conejo producido inmunizando animales con un fosfopéptido sintético que corresponde a los residuos que rodean a Ser10 de histona H3 humana (pHH3) se obtuvo de Cell Signaling Technology (Danvers, MA, Cat. # 9701). Brevemente, el protocolo de IHC incluyó calor y exposición estándar al reactivo de recuperación de antígeno Ventana Cell Conditioning #1 (Ventana, Tucson, AZ). El anticuerpo primario se diluyó a 1:50 y se incubó 60 minutos a temperatura ambiente. Subsecuentemente se incubó con anticuerpo secundario anti-conejo de cabra biotinilado de Jackson ImmunoResearch Laboratories (Cat. # 111-065-144, West Grove, PA) durante 32 minutos.
Para evaluar los cambios de marcador PD inducidos por ADC anti-cKIT en el modelo de xenoinjerto de tumor subcutáneo GIST T1, a ratones SCID-beige hembras se les implantó por vía subcutánea 10x106 células en una suspensión que contenía Matrigel™ al 50% (BD Biosciences) en solución salina equilibrada de Hank. El volumen total de la inyección que contenía las células en suspensión era de 200 pl. Los ratones se asignaron al azar para recibir una dosis i.v. única de NEG085-MCC-DM1 (5 mg/kg), control de isotipo no específico IgG1-MCC-DM1 (5 mg/kg) o amortiguador tris (10 mM Tris-HCl, 80 mM NaCl, 3,5% sacarosa, 0,01% Tween 20, pH 7,5) una vez que los tumores alcanzaron entre 200 y 300 mm3 (n=3/grupo).
Consistentemente con el mecanismo de acción esperado de la carga útil de maitansinoides, NEG085-MCC-DM1 produjo un aumento notable dependiente del tiempo en el porcentaje de células positivas para positividad de pHH3, y por lo tanto detención del ciclo celular. La positividad de pHH3 alcanzó un pico 1-2 días posdosis con respecto a los controles tratados con isotipo no específico IgG1-MCC-DM1 o amortiguador tris, con una caída de señal cuatro días después del tratamiento (imágenes representativas mostradas en la figura 46, y gráfica mostrada en la figura 47).
Ejemplo 31: Eficacia in vivo de ADC anti-cKIT contra tumor estromal gastrointestinal (GIST) en ratones
La actividad antitumoral del ADC anti-cKIT NEG085-MCC-DM1 se evaluó en dos modelos de xenoinjerto de tumor GIST. Ratones SCID-beige hembras recibieron implantes por vía subcutánea de 10x106 células que contenían Matrigel™ al 50% (BD Biosciences) en solución salina balanceada de Hank. El volumen de inyección total que contenía las células en suspensión era de 200 pl.
Se muestra una fotografía representativa de la inmunotinción de cKIT en los tumores GIST T1 y GIST430 para visualizar el patrón de tinción en estos modelos de xenoinjerto (Figuras 48A y 49A, respectivamente). Un anticuerpo policlonal de conejo producido inmunizando animales con un péptido sintético que corresponde a los aminoácidos 963 a 976 en la parte c-terminal citoplásmica de cKIT se obtuvo de Dako (Cat. # A4502). Brevemente, el protocolo de IHC incluyó calor y exposición estándar al reactivo de recuperación de antígeno Ventana Cell Conditioning #1 (Ventana, Tucson, AZ). El anticuerpo primario se diluyó a una concentración de trabajo de 14 pg/ml y se incubó 60 minutos a temperatura ambiente. Subsecuentemente se incubó con anticuerpo anti-conejo conjugado con HRP previamente diluido con Ventana UltraMap (Cat # 760-4315) durante 16 minutos.
Para el estudio de eficacia en GIST T1, los ratones se enrolaron en el estudio 18 días después de la implantación con un volumen de tumor promedio de ~118 mm3 - 234 mm3. Después de ser asignados aleatoriamente a uno de cinco grupos (n = 9/grupo), se administró a los ratones una sola dosis i.v. de amortiguador tris (el vehículo de ADC), un control de isotipo no específico IgG1-MCC-DM1 (5 mg/kg), o NEG085-MCC-DM1 (1,25, 2,5 o 5 mg/kg). Se midieron los volúmenes de tumor y pesos corporales 2 veces a la semana (figuras 48B y 48C). El IgG1-MCC-DM1 de control no fue significativamente activo a 5 mg/kg. Los ratones tratados con NEG085-MCC-DM1 a 1,25, 2,5 y 5 mg/kg tuvieron tumores que mostraron un porcentaje de cambio promedio del volumen de tumor en comparación con el control tratado con amortiguador tris (AT/AC) de 63, 11 y 12 %, respectivamente, y en la tabla 12 se da un resumen de estos datos. Los tratamientos con NEG085-MCC-DM1 fueron bien tolerados a todas las dosis.
Tabla 12: Respuesta a la dosis de NEG085-MCC-DM1 en un modelo de xenoinjerto GIST T1 de ratón el día 38

Para el estudio de eficacia de GIST 430, los ratones se enrolaron en el estudio 12 días después de la implantación con un volumen de tumor promedio de 125 mm3 - 200 mm3. Después de ser asignados aleatoriamente a los grupos (n = 8/grupo), se administró a los ratones una sola dosis i.v. de amortiguador tris (el vehículo de ADC), un control de isotipo no específico IgG1-MCC-DM1 (10 mg/kg), anticuerpo NEG085 no conjugado o NEG085-MCC-DM1 (2,5, 5 o 10 mg/kg). Se midieron los volúmenes de tumor y pesos corporales 2 veces a la semana (figuras 49B y 49C). El IgG1-MCC-DM1 de control y NEG085 no conjugado no fueron activos a 10 mg/kg. Los ratones tratados con NEG085-MCC-DM1 a 2,5 y 5 mg/kg no fueron significativamente activos (AT/AC de 78% y 56%, respectivamente), ni lo fue el comparador Imatinib (AT/AC de 47%), administrado inicialmente a 100 mg/kg con una reducción de dosis a 80 mg/kg debido a la baja tolerabilidad a la dosis de 100 mg/kg en los ratones SCID-beige. La dosis de 10 mg/kg fue significativamente eficaz (AT/AC de 19%) como se muestra gráficamente en la figura 49B y se resume en la tabla 13. Los tratamientos con NEG085-MCC-DM1 fueron bien tolerados a todas las dosis.
Tabla 13: R espuesta a la dosis de N E G 085 -M C C -D M 1 en un m ode lo de xeno in je rto G IS T430 de ratón e l dí

Ejemplo 32: Eficacia in vivo de ADC anti-cKIT contra cáncer de pulmón de célula pequeña en ratones
La actividad antitumoral del ADC anti-cKIT_NEG085-MCC-DM1 se evaluó en el modelo de xenoinjerto de cáncer de pulmón de célula pequeña NCI-H526. A ratones SCID-beige hembras se les implantó por vía subcutánea 10x1056 células que contenían Matrigel™ al 50% (BD Biosciences) en solución salina equilibrada de Hank. El volumen total de la inyección que contenía las células en suspensión era de 200 gl.
Una fotografía representativa de la inmunotinción de cKIT en el tumor NCI-H526 se muestra para visualizar el patrón de tinción en este modelo de xenoinjerto (figura 50A). Un anticuerpo policlonal de conejo producido inmunizando a los animales con un péptido sintético que corresponde a los aminoácidos 963 a 976 en la parte C-terminal citoplásmica de cKIT, se obtuvo de Dako (No. de catálogo A4502). En resumen, el protocolo de IHC incluyó calor y exposición estándar al reactivo de recuperación de antígenos Cell Conditioning #1 de Ventana (Ventana, Tucson, AZ). El anticuerpo primario se diluyó hasta una concentración útil de 14 gg/ml, y se incubó por 60 minutos a temperatura ambiente. Posteriormente, la incubación con el anticuerpo anti-conejo conjugado con HRP pre-diluido UltraMap de Ventana (No. de catálogo 760 4315) se llevó a cabo por 16 minutos.
Se enroló a los ratones en el estudio seis días postimplante con el volumen de tumores promedio de aproximadamente 180 mm3. Después de que son asignados aleatoriamente a los grupos (n = 9/grupo), se administró a los ratones una dosis i.v. individual de amortiguador Tris (el vehículo de ADC), un control de isotipos no específico IgG1-MCC-DM1 (5 mg/kg) o NEG085-MCC-DM1 (1,25, 2,5 y 5 mg/kg). Se midieron dos veces por semana los volúmenes de los tumores y los pesos corporales (figura 50B y figura 50C). IgG1-MCC-DM1 control no fue activo. NEG085-MCC-DM1 a 1,25 mg/kg indujo inicialmente estasis en el volumen del tumor seguida por el rebrote del tumor. Los tratamientos de 2,5 y 5 mg/kg fueron significativamente eficaces, induciendo regresiones de los tumores (74% y 96% de regresiones, respectivamente), y esto se muestra en la figura 50B y se resume en la tabla 14. Los tratamientos a estas dosis mostraron rebrote de los tumores aproximadamente 3 semanas después del tratamiento individual. Todos los tratamientos de NEG085-MCC-DM1
fueron bien tolerados.
Tabla 14: R espuesta a la dosis de N E G 085 -M C C -D M 1 en un m ode lo de xeno in je rto de ratón N C I-H 526 e l d ía 19

A partir de este estudio, se recolectó también suero a 1 hora, 24 horas y 5, 7, 9, 14, 21 y 28 días posdosis para medir la concentración de anticuerpo/ADC con el tiempo usando un ELISA de anti-IgG 1 humana y un ELISA de anti-DM, respectivamente. Para evaluar los parámetros PK, se colectó suero por medio de sangrías retro-orbitales, y se analizó por medio de ELISA. La prueba PK de los anticuerpos totales mide la concentración total de anticuerpos, con/sin DM1 por ELISA colorimétrico. Las placas se recubren con anti-IgG humana (específico del Fc), y la detección es con anti-IgG humana-HRP antes de que sea leída en un lector de placas apropiado. La prueba PK del conjugado mide el anticuerpo que es unido a por lo menos una (1) molécula de DM1 por ELISA colorimétrico. En este formato, las placas son recubiertas con anticuerpo anti-maitansina, y detectadas con anti-IgG humana-HRP. La eficacia dependiente de la dosis en este estudio con NEG085-MCC-DM1 se correlacionó con una exposición al suero, dependiente de la dosis, del anticuerpo total y ADC, como es medido por ELISA del anticuerpo anti-total y anti-maitansina (figura 51A y figura 51B, respectivamente).
Ejemplo 33: Eficacia in vivo de ADC anti-cKIT contra la leucemia mielógena aguda en ratones
La actividad antitumoral de ADC anti-cKIT NEG085-MCC-DM1 se evaluó en el modelo de xenoinjerto de AML (leucemia mielógena aguda) primaria sistémica HAMLX5343 establecido en Novartis. Se implantó sistémicamente a ratones NSG hembras (por medio de inyección en la vena de la cola) con 5x106 células en solución salina amortiguadora de fosfato. El volumen de inyección total que contenía células en suspensión fue de 200 pl.
Se enroló a los ratones en el estudio 43 días posimplante con la carga leucémica promedio de aproximadamente 14,8% células mononucleares de sangre periférica (PBMC) positivas para CD45. Después de que son asignados aleatoriamente a los grupos (n = 6/grupo), se dejó a los ratones sin tratar o se les administró intraperitonealmente ARA-C (citarabina) diariamente por 6 días, o NEG085-MCC-DM1 (10 mg/kg) vía intravenosa una vez cada dos semanas. La carga leucémica se midió por citometría de flujo. Semanalmente, se colectó sangre de todos los animales de estudio por medio de la cola. Los eritrocitos fueron lisados, y las PMBC restantes se tiñeron con un anticuerpo anti-hCD45 (eBioscience, San Diego CA, No. de catálogo P/N 17-9459-42). Las células teñidas se analizaron en un citómetro de flujo FACS Canto (BD
Biosciences) (figura 52A). Se midieron los pesos corporales dos veces por semana (figura 52B). La ARA-C, mientras que es eficaz en tres de los animales, fue altamente tóxica, causando >20% de pérdida de peso corporal en los otros tres animales. El tratamiento de NEG085-MCC-DM1 a 10 mg/kg resultó en progresión retardada de los tumores, incluyendo una breve regresión después de la segunda dosis (figura 52A). El tratamiento de NEG085-MCC-DM1 fue significativamente eficaz en comparación con el control no tratado como se muestra en la figura 52 y en la tabla 15. Los tratamientos de NEG085-MCC-DM1 fueron bien tolerados (figura 52B).
Tabla 15: NEG085-MCC-DM1 en el modelo de xenoinjerto de AML primaria sistémica HAMLX5343 el día 71

Ejemplo 34. NEG085-MCC-DM1 en la terapia de combinación
Se puso a prueba a NEG085-MCC-DM1 en combinación con inhibidores de pequeña molécula usando diferentes matrices de dosis. La inhibición relativa de la viabilidad de las células se calculó para cada combinación de dosis. Mediante el uso del software Chalice (Zalicus, Cambridge MA) o la aplicación ComboExplorer (Novartis, Basel CH), la respuesta de la combinación se comparó con sus agentes individuales, contra el modelo de Loewe ampliamente usado para la aditividad de la dosis de fármaco consigo misma (Lehar etal. Nat. Biotechnol. (2009) 27: 659-666; Zimmermann etal., Drug Discov. Today (2007) 12: 34-42). El exceso de inhibición comparado con la aditividad puede graficarse como un diagrama de matriz de dosis completa para visualizar las concentraciones de fármaco en donde ocurren sinergias. Las combinaciones sinérgicas produjeron regiones de exceso de inhibición dentro de la matriz de dosis. La tabla 16 muestra los datos de varias combinaciones de NEG085-MCC-DM1. Se encontró “aditividad” cuando la combinación generó la misma inhibición de la respuesta de la viabilidad de las células cuando se comparó con la respuesta con el agente individual por sí mismo. Se indicó “sinergia” cuando la inhibición de la viabilidad de las células fue mayor que la respuesta del agente individual comparado consigo mismo. En forma alternativa, se indicó “aditividad” con una puntuación de Loewe de menos de 5, y se indicó “sinergia” con una puntuación de Loewe de más de 5.
La viabilidad de las células se determinó midiendo el contenido de ATP celular usando la prueba de luminiscencia CellTiter Glo (Promega, Madison WI). Un día antes de la adición del fármaco, se sembraron 250-500 células GIST de 2 diferentes líneas de células en placas de 384 cavidades (Greiner, Monroe, NC) en 20 pl de medio de crecimiento. Las células GIST430 contienen una doble mutación en cKIT, que hace que sean parcialmente resistentes a Glivec® (Imatinib). Las células GIST882 tienen una mutación individual en cKIT, y son sensibles a Glivec® (Imatinib). Las células se incubaron entonces por 120 h con varias concentraciones de NEG085-MCC-DM1 como un agente individual, compuestos de agente individual o combinaciones de NEG085-MCC-DM1/compuesto antes de que se añadiera el reactivo CellTiter Glo a cada cavidad, y se registró la luminiscencia en un lector de placas Envision (Perkin Elmer, Waltham MA). Se usaron los valores de luminiscencia para calcular la inhibición de la viabilidad de las células respecto de las células tratadas con DMSO (0%
de inhibición).
Tabla 16


En resumen, los anticuerpos anti-cKIT descritos en la presente tienen efectos sinérgicos cuando se usan en combinación con otras moléculas, lo cual lleva a más opciones de tratamiento. Por ejemplo, NEG085-MCC-DM1 puede co administrarse con un inhibitor de IAP (por ejemplo, NVP-LCL161) como una terapia para obtener un efecto sinérgico. E jem p lo 35. M ap eo de e p íto p o s de N E G 085
Proteólisis limitada in situ
Se obtuvo la proteína de dominio extracelular (ECD) cKIT humana (código de registro NM_000222, dominio 1 (D1)-dominio 3 (D3), con los residuos Q26-G311 (N130S, N145S - estos cambios resultan en deficiencia de glucosilación, para expresar la proteína en una forma libre de glucanos). Se mezclaron relaciones molares iguales de cKIT humana D1-D3 ECD y NEG085 Fab, y se sometieron a un paso final de filtración en gel equilibradas con Tris-HCl 20 mM pH 7,5 y NaCl 100 mM, y se concentraron hasta 20 mg/ml. Se añadió tripsina a la muestra de cristalización del complejo de proteínas para crear una dilución de 1:100 p/p. La mezcla de proteasa/muestra se incubó a temperatura ambiente por 30 minutos antes de preparar el experimento de cristalización.
Cristalización
Se obtuvieron cristales de difracción del complejo de cKIT ECD/NEG085 Fab directamente de Protein Complex Suite F5 (NaCl 0,1M, Tris 0,1M pH 8,0, PEG 8% 20 k; Qiagen) a 4 °C y, después de optimización menor, llevó a cristales que difractan internamente a 5 Á. Los cristales usados para la recolección de datos se desarrollaron a 4 °C equilibrando volúmenes iguales de proteína (20 mg/mL) y solución de depósito (NaCl 0,1M, Tris 0,1M pH 8,0, PEG 10% 20k) por el método de difusión de vapor por asentamiento de una gota. Antes de la recolección de los datos, los cristales fueron crioprotegidos en solución de depósito que contenía glicerol 30% y enfriados instantáneamente en nitrógeno líquido. Recolección de datos
Se recolectaron los datos de un cristal individual enfriado hasta 100°K usando un detector ADSC QUANTUM 315 (ADSC, Poway CA) y radiación sincrotrónica (A=1.0000Á) en la línea del haz 5.0.2 de la fuente de luz avanzada. Los cristales de cKIT ECD/NEG085 Fab difractaron hasta resolución de 3.1 Á y pertenecieron al grupo espacial C2 con los parámetros de células individuales a=213.76 Á, b=117.48 Á, c=171.92 Á, a=90°, |3=118.47°, y=90°. El cristal contiene cuatro copias del complejo cKIT ECD/NEG085 Fab en la unidad asimétrica con un contenido de disolvente calculado de 66%. Los datos se procesaron usando autoPROC (Global Phasing Ltd, Cambridge Reino Unido).
Determinación y refinamiento de la estructura
La estructura del complejo cKIT ECD/NEG085 Fab se resolvió a resolución de 3.1 Á por reemplazo molecular con PHASER (McCoy etal., J. Appl. Crystallogr. 2007; 40(4): 658-74) usando la estructura de cristal publicada de cKIT ECD (código de ID del PDB: 2EC8) (Yuzawa et al., Cell 2007; 130 (2):323-34) e integrina Fab anti-a1|31I (código de ID del Pd B: 1MHP) (Karpusas et al., J. Mol. Biol. 2003; 327 (5): 1031-41) como modelos de partida. Los bucles de CDR del fragmento NEG085 Fab se reconstruyeron manualmente en COOT (Emsley y Cowtan, Acta Crystallogr. D. Biol. Crystallogr. 2004; 60 (12): 2126-32) usando material mixto de union simulada omit map implementado en Phenix (Adams et al., Acta Crystallogr. D. Biol. Crystallogr. 2010; 66(2): 213-21). Rondas subsiguientes de construcción y refinamiento del modelo con el programa Phenix.refine, se llevaron a cabo hasta convergencia.
Resultados: Estructura del cKIT D1-D2 humano en complejo con el fragmento Fab de NEG085
La coestructura de cKIT D1-D3/NEG085 Fab demuestra que NEG085 reconoce y se une al dominio distal extracelular de la membrana de cKIT interactuando específicamente con un gran número de residuos contenidos dentro de los dominios 1, 2 y los residuos del enlazador entre ellos. El epítopo de cKIT reconocido por NEG085, puede definirse por lo tanto como:
Residuos del dominio 1 de cKIT: R49, V50
Residuos del dominio 2 de cKIT: Q152, G153, H185 y bucle Q190-E198
Enlazador entre el dominio 1 y 2: D113-L117
NEG085 se une a cKIT D1-D2 usando todas las CDR (H1-H3 y L1-L3) (figura 53). La figura 53 es una representación
de la estructura de cristal de 3.1 Á del complejo de kit D1-D2/NEG085 Fab, que muestra los dominios de las cadenas pesadas de Fab (gris oscuro), cadenas ligeras de Fab (blanco) y Kit D1-D2 (gris claro). Los epítopos y parátopos se colorearon de negro. La interfaz NEG085/cKIT entierra un total de ~ 1890 Á2 de área de superficie accesible al disolvente (1211 Á2 y 679 Á2 de la cadena H y la cadena L, respectivamente). El epítopo está centrado en la región del enlazador de cKIT D1-D2 D113-L117 (SEQ ID NO. 163) y el bucle Q190-E198 (SEQ ID NO. 164), y estos residuos están en negritas y subrayados en la tabla 2, SEQ ID NO. 155. Se observa que estos epítopos son discontinuos en la secuencia primaria, pero están muy cerca juntos en la estructura de cristal. Estas interacciones de epítopos son suplementadas por interacciones periféricas con R49, V50, Q152, G153 y H185 (figura 53). Las interacciones intermoleculares entre cKIT D1-D2 y NEG085 Fab se examinaron usando PISA (Interfaces, Superficies y Ensambles de Proteínas) (Krissinel y Henrick, J. Mol. Biol. 2007, 372(3): 774-97). Estos datos se muestran en la tabla 17.
La superposición de cKIT en el complejo de señalización dimérico cKIT/SCF (Yuzawa etal., Cell 2007; 130 (2); 323-34) y el complejo cKIT/NEG085, muestra que NEG085 y SCF parecen no competir entre sí por la unión a cKIT. NEG085 se une a un epítopo que es distinto de los epítopos de unión responsables de la unión a SCF. Por lo tanto, la unión de NEG085 a cKIT no competiría directamente por la asociación para SCF.
Tabla 17


Los residuos de VH y VL de NEG085 Fab se enumeran con base en su secuencia de aminoácidos lineal. Los residuos de cKIT se enumeran con base en el código de registro NM_000222. Las interacciones intermoleculares se examinaron usando PISA (Interfaces, Superficies y Ensambles de Proteínas) (Krissinel y Henrick, J. Mol. Biol. 2007, 372(3): 774-97).
Claims (10)
1. Un conjugado de anticuerpo-fármaco de la fórmula:
Ab-(L-(D)m)n
o una sal farmacéuticamente aceptable del mismo; en donde:
Ab es un anticuerpo o fragmento de unión a antígeno del mismo que comprende:
(i) una región variable de cadena pesada que comprende (a) una HCDR1 (CDR- Región Determinante de Complementariedad) de SEQ ID NO: 76, (b) una HCDR2 de SEQ ID NO: 77, (c) una HCDR3 de SEQ ID NO: 78; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 85, (e) una LCDR2 de SEQ ID n O: 86, y (f) una LCDR3 de SEQ ID NO: 87;
(ii) una región variable de cadena pesada que comprende (a) una HCDR1 de SEQ ID NO: 22, (b) una HCDR2 de SEQ ID NO: 23, (c) una HCDR3 de SEQ ID NO: 24; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 31, (e) una LCDR2 de SEQ ID NO: 32, y (f) una LCDR3 de SEQ ID NO: 33;
(iii) una región variable de cadena pesada que comprende (a) una HCDR1 de SEQ ID NO: 130, (b) una HCDR2 de SEQ ID NO: 131, (c) una HCDR3 de SEQ ID NO: 132; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 139, (e) una LCDR2 de SEQ ID NO: 140, y (f) una LCDR3 de SEQ ID NO: 141;
(iv) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 58, (b) una HCDR2 de SEQ ID NO: 59, (c) una HCDR3 de SEQ ID NO: 60; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 67, (e) una LCDR2 de SEQ ID NO: 68, y (f) una LCDR3 de SEQ ID NO: 69;
(v) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 40, (b) una HCDR2 de SEQ ID NO: 41, (c) una HCDR3 de SEQ ID NO: 42; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 49, (e) una LCDR2 de SEQ ID NO: 50, y (f) una LCDR3 de SEQ ID NO: 51;
(vi) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 94, (b) una HCDR2 de SEQ ID NO: 95, (c) una HCDR3 de SEQ ID NO: 96; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 103, (d) una LCDR2 de SEQ ID NO: 104, y (f) una LCDR3 de SEQ ID NO: 105;
(vii) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 112, (b) una HCDR2 de SEQ ID NO: 113, (c) una HCDR3 de SEQ ID NO: 114; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 121, (e) una LCDR2 de SEQ ID NO: 122, y (f) una LCDR3 de SEQ ID NO: 123; o
(viii) una región variable de cadena pesada que comprende: (a) una HCDR1 de SEQ ID NO: 3, (b) una HCDR2 de SEQ ID NO: 4, (c) una HCDR3 de SEQ ID NO: 5; y una región variable de cadena ligera que comprende: (d) una LCDR1 de SEQ ID NO: 12, (e) una LCDR2 de SEQ ID NO: 13, y (f) una LCDR3 de SEQ ID NO: 14.
L es un enlazador;
D es un maitansinoide;
m es un entero de 1 a 8; y
n es un entero de 1 a 10.
2. El conjugado anticuerpo-fármaco de acuerdo con la reivindicación 1, en donde dicho n es 3 ó 4.
3. El conjugado anticuerpo-fármaco de acuerdo con cualquiera de las reivindicaciones precedentes, en donde el anticuerpo es un anticuerpo monoclonal, un anticuerpo quimérico, un anticuerpo humanizado, un anticuerpo humano modificado por ingeniería, un anticuerpo humano, un anticuerpo de cadena simple (scFv) o un fragmento de anticuerpo.
4. El conjugado anticuerpo-fármaco de acuerdo con cualquiera de las reivindicaciones 1-3, en donde dicho enlazador (L) se selecciona del grupo que consiste en un enlazador escindible, un enlazador no escindible, un enlazador hidrófilo, un
enlazador procargado y un enlazador a base de ácido dicarboxílico.
5. El conjugado anticuerpo-fármaco de acuerdo con la reivindicación 4, en donde el enlazador se deriva de un reactivo de entrelazamiento seleccionado del grupo que consiste en N-succinimidil-3-(2-piridilditio)propionato (SPDP), N-succinimidil-4-(2-piridilditio)pentanoato (SPP), N-succinimidil-4-(2-piridilditio)butanoato (SPDB), N-succinimidil-4-(2-piridilditio)2-sulfo-butanoato (sulfo-SPDB), N-succinimidil-yodoacetato (SIA), N-succinimidil(4-yodoacetil)aminobenzoato (SIAB), maleimida-PEG-NHS, N-succinimidil-4-(maleimidometil)-ciclohexancarboxilato (SMCC), N-sulfosuccinimidil-4-(maleimidometil)ciclohexancarboxilato (sulfo-SMCC), o 2,5-dioxopirrolidin -1 -il-17-(2,5-dioxo-2,5-dihidro-1 H-pirrol-1-il)-5,8,11,14-tetraoxo-4,7,10,13-tetraazaheptadecan-1-oato (CX1-1).
6. El conjugado anticuerpo-fármaco de acuerdo con la reivindicación 5, en donde dicho enlazador se deriva del reactivo de entrelazamiento N-succinimidil-4-(maleimidometil) ciclohexancarboxilato (SMCC).
7. El conjugado anticuerpo-fármaco de acuerdo con cualquiera de las reivindicaciones precedentes, en donde el maitansinoide es N(2')-desacetil-N(2')-(3-mercapto-1-oxopropil)-maitansina (DM1) o N(2')-desacetil-N2-(4-mercapto-4-metil-1-oxopentil)-maitansina (DM4).
8. El conjugado anticuerpo-fármaco de cualquiera de las reivindicaciones precedentes, en combinación con otro agente terapéutico.
9. El conjugado anticuerpo-fármaco de cualquiera de las reivindicaciones precedentes, en combinación con un agente terapéutico indicado en la tabla 16.
10. Una composición farmacéutica que comprende el conjugado anticuerpo-fármaco de acuerdo con cualquiera de las reivindicaciones 1 a 9 y un portador farmacéuticamente aceptable.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361793641P | 2013-03-15 | 2013-03-15 | |
| PCT/US2014/024597 WO2014150937A1 (en) | 2013-03-15 | 2014-03-12 | Antibody drug conjugates |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2701051T3 true ES2701051T3 (es) | 2019-02-20 |
Family
ID=50721874
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14723915T Active ES2701051T3 (es) | 2013-03-15 | 2014-03-12 | Conjugados de anticuerpo-fármaco |
Country Status (26)
| Country | Link |
|---|---|
| US (6) | US9498543B2 (es) |
| EP (2) | EP3514178A1 (es) |
| JP (2) | JP6333943B2 (es) |
| KR (1) | KR20150131173A (es) |
| CN (1) | CN105007950B (es) |
| AP (1) | AP2015008656A0 (es) |
| AR (1) | AR095666A1 (es) |
| AU (1) | AU2014235474B2 (es) |
| BR (1) | BR112015021134A2 (es) |
| CA (1) | CA2903772A1 (es) |
| CL (1) | CL2015002246A1 (es) |
| CR (1) | CR20150488A (es) |
| CU (1) | CU20150128A7 (es) |
| EA (1) | EA035253B1 (es) |
| ES (1) | ES2701051T3 (es) |
| IL (1) | IL241277A0 (es) |
| MA (1) | MA38396B1 (es) |
| MX (1) | MX366978B (es) |
| NZ (1) | NZ710929A (es) |
| PH (1) | PH12015501902A1 (es) |
| SG (1) | SG11201506395RA (es) |
| TN (1) | TN2015000396A1 (es) |
| TW (1) | TW201446806A (es) |
| UY (1) | UY35490A (es) |
| WO (1) | WO2014150937A1 (es) |
| ZA (1) | ZA201505656B (es) |
Families Citing this family (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MY166083A (en) | 2011-06-17 | 2018-05-24 | Invista Tech Sarl | Use of hydrolases to increase monomer content in waste stream |
| CA2903772A1 (en) * | 2013-03-15 | 2014-09-25 | Novartis Ag | Antibody drug conjugates |
| WO2015109391A1 (en) | 2014-01-24 | 2015-07-30 | Children's Hospital Of Eastern Ontario Research Institute Inc. | Smc combination therapy for the treatment of cancer |
| WO2016020791A1 (en) * | 2014-08-05 | 2016-02-11 | Novartis Ag | Ckit antibody drug conjugates |
| US10238748B2 (en) | 2014-08-12 | 2019-03-26 | Novartis Ag | Anti-CDH6 antibody drug conjugates |
| MX2017011380A (es) | 2015-03-09 | 2018-04-26 | Agensys Inc | Conjugados anticuerpo-farmaco (adc) que se ligan a proteinas flt3. |
| WO2017060322A2 (en) * | 2015-10-10 | 2017-04-13 | Bayer Pharma Aktiengesellschaft | Ptefb-inhibitor-adc |
| MA44334A (fr) * | 2015-10-29 | 2018-09-05 | Novartis Ag | Conjugués d'anticorps comprenant un agoniste du récepteur de type toll |
| US11052111B2 (en) * | 2015-12-08 | 2021-07-06 | Chimera Bioengineering, Inc. | Smart CAR devices and DE CAR polypeptides for treating disease and methods for enhancing immune responses |
| WO2017125897A1 (en) | 2016-01-21 | 2017-07-27 | Novartis Ag | Multispecific molecules targeting cll-1 |
| CN109843919A (zh) | 2016-03-25 | 2019-06-04 | 西雅图基因公司 | 用于制备聚乙二醇化的药物-接头及其中间体的方法 |
| EP3448874A4 (en) | 2016-04-29 | 2020-04-22 | Voyager Therapeutics, Inc. | COMPOSITIONS FOR TREATING A DISEASE |
| US11299751B2 (en) | 2016-04-29 | 2022-04-12 | Voyager Therapeutics, Inc. | Compositions for the treatment of disease |
| CN109661400A (zh) | 2016-06-17 | 2019-04-19 | 美真达治疗公司 | 用于耗尽cd117+细胞的组合物和方法 |
| CN107913410B (zh) * | 2016-10-08 | 2021-02-02 | 浙江大学 | 两亲性共聚物-美登素类共价偶联药物、制备方法及应用 |
| JOP20190155A1 (ar) * | 2016-12-21 | 2019-06-23 | Novartis Ag | مترافقات عقار جسم مضاد لإزالة خلايا جذعية مكونة للدم |
| MX2019008205A (es) | 2017-01-20 | 2020-01-23 | Magenta Therapeutics Inc | Composiciones y metodos para el agotamiento de células cd137+. |
| JOP20190187A1 (ar) * | 2017-02-03 | 2019-08-01 | Novartis Ag | مترافقات عقار جسم مضاد لـ ccr7 |
| CN108456213B (zh) * | 2017-02-22 | 2021-01-15 | 浙江九洲药业股份有限公司 | 一种3-氟-4-羟基环己烷羧酸酯的制备方法 |
| MX2019010769A (es) | 2017-03-24 | 2019-12-11 | Seattle Genetics Inc | Proceso para la preparacion de enlazadores de farmacos de glucuronidos y compuestos intermediarios de los mismos. |
| JOP20190245A1 (ar) | 2017-04-20 | 2019-10-15 | Novartis Ag | أنظمة توصيل إطلاق مستدام تتضمن روابط بلا أثر لنقطة الربط |
| UY37758A (es) | 2017-06-12 | 2019-01-31 | Novartis Ag | Método de fabricación de anticuerpos biespecíficos, anticuerpos biespecíficos y uso terapéutico de dichos anticuerpos |
| TW201945026A (zh) * | 2018-01-29 | 2019-12-01 | 美商希拉諾斯醫療公司 | 結合erfe之抗體及使用方法 |
| GB201802201D0 (en) | 2018-02-09 | 2018-03-28 | Ultrahuman Five Ltd | Binding agents |
| EP3552631A1 (en) * | 2018-04-10 | 2019-10-16 | Inatherys | Antibody-drug conjugates and their uses for the treatment of cancer |
| UY38265A (es) * | 2018-06-20 | 2020-01-31 | Novartis Ag | Conjugados anticuerpo droga para ablación de células madre hematopoyéticas |
| AR116566A1 (es) | 2018-10-03 | 2021-05-19 | Novartis Ag | Administración sostenida de polipéptidos similares a la angiopoyetina 3 |
| KR20200040407A (ko) * | 2018-10-10 | 2020-04-20 | 주식회사 노벨티노빌리티 | 신규 항-c-KIT 항체 |
| KR20210094610A (ko) * | 2018-11-26 | 2021-07-29 | 포티 세븐, 인코포레이티드 | c-Kit에 대한 인간화 항체 |
| JP2022522777A (ja) | 2019-03-01 | 2022-04-20 | レボリューション メディシンズ インコーポレイテッド | 二環式ヘテロアリール化合物及びその使用 |
| CA3130083A1 (en) | 2019-03-01 | 2020-09-10 | Revolution Medicines, Inc. | Bicyclic heterocyclyl compounds and uses thereof |
| SG11202110287QA (en) * | 2019-04-24 | 2021-10-28 | Heidelberg Pharma Res Gmbh | Amatoxin antibody-drug conjugates and uses thereof |
| WO2020219748A2 (en) * | 2019-04-24 | 2020-10-29 | Magenta Therapeutics, Inc. | Anti-cd117 antibodies and uses thereof |
| CN111620927B (zh) * | 2019-05-20 | 2022-11-11 | 烟台迈百瑞国际生物医药股份有限公司 | 一种抗体药物偶联物中间体的一锅法制备工艺 |
| US20230092679A1 (en) * | 2019-05-20 | 2023-03-23 | Novartis Ag | Mcl-1 inhibitor antibody-drug conjugates and methods of use |
| CN112206327A (zh) * | 2019-07-12 | 2021-01-12 | 上海药明生物技术有限公司 | 一种抗体偶联药物的制备及其高通量筛选方法 |
| US11608346B2 (en) | 2019-11-04 | 2023-03-21 | Revolution Medicines, Inc. | Ras inhibitors |
| CA3159559A1 (en) | 2019-11-04 | 2021-05-14 | Revolution Medicines, Inc. | Ras inhibitors |
| JP2022553858A (ja) | 2019-11-04 | 2022-12-26 | レボリューション メディシンズ インコーポレイテッド | Ras阻害剤 |
| JP2023500328A (ja) | 2019-11-08 | 2023-01-05 | レボリューション メディシンズ インコーポレイテッド | 二環式ヘテロアリール化合物及びその使用 |
| KR20210063782A (ko) | 2019-11-25 | 2021-06-02 | 주식회사 노벨티노빌리티 | c-kit에 대한 항체 및 이의 용도 |
| JP2023505100A (ja) | 2019-11-27 | 2023-02-08 | レボリューション メディシンズ インコーポレイテッド | 共有ras阻害剤及びその使用 |
| EP4267250A1 (en) | 2020-12-22 | 2023-11-01 | Qilu Regor Therapeutics Inc. | Sos1 inhibitors and uses thereof |
| IL308134A (en) | 2021-06-22 | 2023-12-01 | Novartis Ag | BISPECIFIC ANTIBODIES FOR USE IN THE TREATMENT OF HIDRADENITIS SUPPURATIVA |
| KR102544135B1 (ko) * | 2022-01-26 | 2023-06-19 | 주식회사 노벨티노빌리티 | c-Kit을 표적으로 하는 면역접합체 |
| KR20230125129A (ko) * | 2022-02-17 | 2023-08-29 | 주식회사 노벨티노빌리티 | 항체-약물 접합체 |
| US20230357381A1 (en) | 2022-04-26 | 2023-11-09 | Novartis Ag | Multispecific antibodies targeting il-13 and il-18 |
| WO2024040195A1 (en) | 2022-08-17 | 2024-02-22 | Capstan Therapeutics, Inc. | Conditioning for in vivo immune cell engineering |
Family Cites Families (128)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US633410A (en) | 1898-09-22 | 1899-09-19 | George A Ames | Ice-cutter. |
| US2429001A (en) | 1946-12-28 | 1947-10-14 | Axel H Stone | Artificial hand |
| US2779780A (en) | 1955-03-01 | 1957-01-29 | Du Pont | 1, 4-diamino-2, 3-dicyano-1, 4-bis (substituted mercapto) butadienes and their preparation |
| US3720760A (en) | 1968-09-06 | 1973-03-13 | Pharmacia Ab | Method for determining the presence of reagin-immunoglobulins(reagin-ig)directed against certain allergens,in aqueous samples |
| US3896111A (en) | 1973-02-20 | 1975-07-22 | Research Corp | Ansa macrolides |
| US4151042A (en) | 1977-03-31 | 1979-04-24 | Takeda Chemical Industries, Ltd. | Method for producing maytansinol and its derivatives |
| US4137230A (en) | 1977-11-14 | 1979-01-30 | Takeda Chemical Industries, Ltd. | Method for the production of maytansinoids |
| US4307016A (en) | 1978-03-24 | 1981-12-22 | Takeda Chemical Industries, Ltd. | Demethyl maytansinoids |
| US4265814A (en) | 1978-03-24 | 1981-05-05 | Takeda Chemical Industries | Matansinol 3-n-hexadecanoate |
| JPS5562090A (en) | 1978-10-27 | 1980-05-10 | Takeda Chem Ind Ltd | Novel maytansinoid compound and its preparation |
| US4256746A (en) | 1978-11-14 | 1981-03-17 | Takeda Chemical Industries | Dechloromaytansinoids, their pharmaceutical compositions and method of use |
| JPS5566585A (en) | 1978-11-14 | 1980-05-20 | Takeda Chem Ind Ltd | Novel maytansinoid compound and its preparation |
| JPS55164687A (en) | 1979-06-11 | 1980-12-22 | Takeda Chem Ind Ltd | Novel maytansinoid compound and its preparation |
| JPS55102583A (en) | 1979-01-31 | 1980-08-05 | Takeda Chem Ind Ltd | 20-acyloxy-20-demethylmaytansinoid compound |
| JPS55162791A (en) | 1979-06-05 | 1980-12-18 | Takeda Chem Ind Ltd | Antibiotic c-15003pnd and its preparation |
| JPS55164685A (en) | 1979-06-08 | 1980-12-22 | Takeda Chem Ind Ltd | Novel maytansinoid compound and its preparation |
| JPS55164686A (en) | 1979-06-11 | 1980-12-22 | Takeda Chem Ind Ltd | Novel maytansinoid compound and its preparation |
| US4309428A (en) | 1979-07-30 | 1982-01-05 | Takeda Chemical Industries, Ltd. | Maytansinoids |
| JPS5645483A (en) | 1979-09-19 | 1981-04-25 | Takeda Chem Ind Ltd | C-15003phm and its preparation |
| EP0028683A1 (en) | 1979-09-21 | 1981-05-20 | Takeda Chemical Industries, Ltd. | Antibiotic C-15003 PHO and production thereof |
| JPS5645485A (en) | 1979-09-21 | 1981-04-25 | Takeda Chem Ind Ltd | Production of c-15003pnd |
| US4458066A (en) | 1980-02-29 | 1984-07-03 | University Patents, Inc. | Process for preparing polynucleotides |
| WO1982001188A1 (en) | 1980-10-08 | 1982-04-15 | Takeda Chemical Industries Ltd | 4,5-deoxymaytansinoide compounds and process for preparing same |
| US4450254A (en) | 1980-11-03 | 1984-05-22 | Standard Oil Company | Impact improvement of high nitrile resins |
| US4313946A (en) | 1981-01-27 | 1982-02-02 | The United States Of America As Represented By The Secretary Of Agriculture | Chemotherapeutically active maytansinoids from Trewia nudiflora |
| US4315929A (en) | 1981-01-27 | 1982-02-16 | The United States Of America As Represented By The Secretary Of Agriculture | Method of controlling the European corn borer with trewiasine |
| JPS57192389A (en) | 1981-05-20 | 1982-11-26 | Takeda Chem Ind Ltd | Novel maytansinoid |
| ATE37983T1 (de) | 1982-04-22 | 1988-11-15 | Ici Plc | Mittel mit verzoegerter freigabe. |
| US4522811A (en) | 1982-07-08 | 1985-06-11 | Syntex (U.S.A.) Inc. | Serial injection of muramyldipeptides and liposomes enhances the anti-infective activity of muramyldipeptides |
| US5128326A (en) | 1984-12-06 | 1992-07-07 | Biomatrix, Inc. | Drug delivery systems based on hyaluronans derivatives thereof and their salts and methods of producing same |
| US5374548A (en) | 1986-05-02 | 1994-12-20 | Genentech, Inc. | Methods and compositions for the attachment of proteins to liposomes using a glycophospholipid anchor |
| MX9203291A (es) | 1985-06-26 | 1992-08-01 | Liposome Co Inc | Metodo para acoplamiento de liposomas. |
| AU600575B2 (en) | 1987-03-18 | 1990-08-16 | Sb2, Inc. | Altered antibodies |
| US4880078A (en) | 1987-06-29 | 1989-11-14 | Honda Giken Kogyo Kabushiki Kaisha | Exhaust muffler |
| US5677425A (en) | 1987-09-04 | 1997-10-14 | Celltech Therapeutics Limited | Recombinant antibody |
| US5108921A (en) | 1989-04-03 | 1992-04-28 | Purdue Research Foundation | Method for enhanced transmembrane transport of exogenous molecules |
| AU6430190A (en) | 1989-10-10 | 1991-05-16 | Pitman-Moore, Inc. | Sustained release composition for macromolecular proteins |
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| WO1991006287A1 (en) | 1989-11-06 | 1991-05-16 | Enzytech, Inc. | Protein microspheres and methods of using them |
| ATE169031T1 (de) | 1991-04-05 | 1998-08-15 | Univ Washington | Zellrezeptor spezifische monoklonale antikörper gegen stammzell-faktor-rezeptor |
| ES2181673T3 (es) | 1991-05-01 | 2003-03-01 | Jackson H M Found Military Med | Procedimiento de tratamiento de las enfermedades respiratorias infecciosas. |
| US5545533A (en) | 1991-05-25 | 1996-08-13 | Boehringer Mannheim Gmbh | Monoclonal antibodies against c-kit and method of detecting a malignancy using c-kit specific antibodies |
| US5714350A (en) | 1992-03-09 | 1998-02-03 | Protein Design Labs, Inc. | Increasing antibody affinity by altering glycosylation in the immunoglobulin variable region |
| US5912015A (en) | 1992-03-12 | 1999-06-15 | Alkermes Controlled Therapeutics, Inc. | Modulated release from biocompatible polymers |
| ES2149768T3 (es) | 1992-03-25 | 2000-11-16 | Immunogen Inc | Conjugados de agentes enlazantes de celulas derivados de cc-1065. |
| EP0640094A1 (en) | 1992-04-24 | 1995-03-01 | The Board Of Regents, The University Of Texas System | Recombinant production of immunoglobulin-like domains in prokaryotic cells |
| US5934272A (en) | 1993-01-29 | 1999-08-10 | Aradigm Corporation | Device and method of creating aerosolized mist of respiratory drug |
| EP0714409A1 (en) | 1993-06-16 | 1996-06-05 | Celltech Therapeutics Limited | Antibodies |
| US5837458A (en) | 1994-02-17 | 1998-11-17 | Maxygen, Inc. | Methods and compositions for cellular and metabolic engineering |
| US5605793A (en) | 1994-02-17 | 1997-02-25 | Affymax Technologies N.V. | Methods for in vitro recombination |
| US5834252A (en) | 1995-04-18 | 1998-11-10 | Glaxo Group Limited | End-complementary polymerase reaction |
| US6132764A (en) | 1994-08-05 | 2000-10-17 | Targesome, Inc. | Targeted polymerized liposome diagnostic and treatment agents |
| EP0805678B1 (en) | 1995-01-05 | 2003-10-29 | THE BOARD OF REGENTS acting for and on behalf of THE UNIVERSITY OF MICHIGAN | Surface-modified nanoparticles and method of making and using same |
| US5641870A (en) | 1995-04-20 | 1997-06-24 | Genentech, Inc. | Low pH hydrophobic interaction chromatography for antibody purification |
| US6121022A (en) | 1995-04-14 | 2000-09-19 | Genentech, Inc. | Altered polypeptides with increased half-life |
| US6019968A (en) | 1995-04-14 | 2000-02-01 | Inhale Therapeutic Systems, Inc. | Dispersible antibody compositions and methods for their preparation and use |
| US5869046A (en) | 1995-04-14 | 1999-02-09 | Genentech, Inc. | Altered polypeptides with increased half-life |
| US6685940B2 (en) | 1995-07-27 | 2004-02-03 | Genentech, Inc. | Protein formulation |
| AU710347B2 (en) | 1995-08-31 | 1999-09-16 | Alkermes Controlled Therapeutics, Inc. | Composition for sustained release of an agent |
| DE19600589C1 (de) | 1996-01-10 | 1997-01-16 | Univ Eberhard Karls | Antikörper A3C6E2 |
| JP2000506165A (ja) | 1996-03-04 | 2000-05-23 | ザ ペン ステイト リサーチ ファウンデーション | 細胞インターナリゼーションを増強するための物質および方法 |
| US5985309A (en) | 1996-05-24 | 1999-11-16 | Massachusetts Institute Of Technology | Preparation of particles for inhalation |
| US5874064A (en) | 1996-05-24 | 1999-02-23 | Massachusetts Institute Of Technology | Aerodynamically light particles for pulmonary drug delivery |
| US5855913A (en) | 1997-01-16 | 1999-01-05 | Massachusetts Instite Of Technology | Particles incorporating surfactants for pulmonary drug delivery |
| US6056973A (en) | 1996-10-11 | 2000-05-02 | Sequus Pharmaceuticals, Inc. | Therapeutic liposome composition and method of preparation |
| EP0954282B1 (en) | 1997-01-16 | 2005-01-19 | Massachusetts Institute Of Technology | Preparation of particles for inhalation |
| US6277375B1 (en) | 1997-03-03 | 2001-08-21 | Board Of Regents, The University Of Texas System | Immunoglobulin-like domains with increased half-lives |
| GB2339430A (en) | 1997-05-21 | 2000-01-26 | Biovation Ltd | Method for the production of non-immunogenic proteins |
| ATE386802T1 (de) | 1997-06-12 | 2008-03-15 | Novartis Int Pharm Ltd | Künstliche antikörperpolypeptide |
| US5989463A (en) | 1997-09-24 | 1999-11-23 | Alkermes Controlled Therapeutics, Inc. | Methods for fabricating polymer-based controlled release devices |
| SE512663C2 (sv) | 1997-10-23 | 2000-04-17 | Biogram Ab | Inkapslingsförfarande för aktiv substans i en bionedbrytbar polymer |
| US6194551B1 (en) | 1998-04-02 | 2001-02-27 | Genentech, Inc. | Polypeptide variants |
| ES2340112T3 (es) | 1998-04-20 | 2010-05-28 | Glycart Biotechnology Ag | Ingenieria de glicosilacion de anticuerpos para la mejora de la citotoxicidad celular dependiente de anticuerpos. |
| ES2198922T3 (es) | 1998-06-24 | 2004-02-01 | Advanced Inhalation Research, Inc. | Particulas porosas grandes emitadas por un inhalador. |
| WO2000035436A2 (en) | 1998-12-16 | 2000-06-22 | Warner-Lambert Company | Treatment of arthritis with mek inhibitors |
| WO2000042072A2 (en) | 1999-01-15 | 2000-07-20 | Genentech, Inc. | Polypeptide variants with altered effector function |
| US6737056B1 (en) | 1999-01-15 | 2004-05-18 | Genentech, Inc. | Polypeptide variants with altered effector function |
| EP1176195B1 (en) | 1999-04-09 | 2013-05-22 | Kyowa Hakko Kirin Co., Ltd. | Method for controlling the activity of immunologically functional molecule |
| EP1144607B1 (en) | 1999-07-20 | 2008-12-17 | MorphoSys AG | Methods for displaying (poly)peptides/proteins on bacteriophage particles via disulfide bonds |
| DK1242401T3 (da) | 1999-11-24 | 2007-05-07 | Immunogen Inc | Cytotoksiske midler, der omfatter taxaner, og den terapeutiske anvendelse deraf |
| DK1242438T3 (da) | 1999-12-29 | 2007-02-12 | Immunogen Inc | Cytotoksiske midler omfattende modificerede doxorubiciner og daunorubiciner og deres terapeutiske anvendelse |
| BRPI0112584B8 (pt) | 2000-07-19 | 2021-05-25 | Warner Lambert Co | ésteres oxigenados de derivados de ácido benzidroxâmico de 4-iodofenilamino, composição farmacêutica e combinação |
| US6411163B1 (en) | 2000-08-14 | 2002-06-25 | Intersil Americas Inc. | Transconductance amplifier circuit |
| AU2002348477A1 (en) | 2001-05-01 | 2002-12-23 | The General Hospital Corporation | Photoimmunotherapies for cancer using photosensitizer immunoconjugates and combination therapies |
| US6441163B1 (en) | 2001-05-31 | 2002-08-27 | Immunogen, Inc. | Methods for preparation of cytotoxic conjugates of maytansinoids and cell binding agents |
| WO2003035835A2 (en) | 2001-10-25 | 2003-05-01 | Genentech, Inc. | Glycoprotein compositions |
| IL162734A0 (en) | 2002-02-01 | 2005-11-20 | Ariad Gene Therapeutics Inc | Phosphorus-containing compounds & uses thereof |
| ES2555307T3 (es) | 2002-03-08 | 2015-12-30 | Eisai R&D Management Co., Ltd. | Compuestos macrocíclicos útiles como agentes farmacéuticos |
| US20050069551A1 (en) * | 2002-03-08 | 2005-03-31 | Emory University | Cytotoxic compound-protein conjugates as suppressors of tumor growth and angiogenesis |
| TWI275390B (en) | 2002-04-30 | 2007-03-11 | Wyeth Corp | Process for the preparation of 7-substituted-3- quinolinecarbonitriles |
| GB0215823D0 (en) | 2002-07-09 | 2002-08-14 | Astrazeneca Ab | Quinazoline derivatives |
| ES2318167T3 (es) | 2002-07-15 | 2009-05-01 | The Trustees Of Princeton University | Compuestos de union a iap. |
| US8088387B2 (en) * | 2003-10-10 | 2012-01-03 | Immunogen Inc. | Method of targeting specific cell populations using cell-binding agent maytansinoid conjugates linked via a non-cleavable linker, said conjugates, and methods of making said conjugates |
| US7399865B2 (en) | 2003-09-15 | 2008-07-15 | Wyeth | Protein tyrosine kinase enzyme inhibitors |
| CN101087611B (zh) * | 2003-10-10 | 2019-04-23 | 伊缪诺金公司 | 用经不可切割接头连接的细胞结合剂美登木素生物碱偶联物靶向特定细胞群的方法、所述偶联物和制备所述偶联物的方法 |
| CA2553871A1 (en) | 2004-01-16 | 2005-08-04 | The Regents Of The University Of Michigan | Smac peptidomimetics and the uses thereof |
| JP2007522116A (ja) | 2004-01-16 | 2007-08-09 | ザ リージェンツ オブ ザ ユニバーシティ オブ ミシガン | コンホメーションが制約されたSmac模倣物およびその使用 |
| US7345081B2 (en) | 2004-03-23 | 2008-03-18 | Genentech, Inc. | Azabicyclo-octane inhibitors of IAP |
| RU2425838C2 (ru) | 2004-04-07 | 2011-08-10 | Новартис Аг | Ингибиторы iap |
| PL1778718T3 (pl) | 2004-07-02 | 2015-03-31 | Genentech Inc | Inhibitory IAP |
| US7674787B2 (en) | 2004-07-09 | 2010-03-09 | The Regents Of The University Of Michigan | Conformationally constrained Smac mimetics and the uses thereof |
| US20100190688A1 (en) | 2004-07-12 | 2010-07-29 | Bin Chao | Tetrapeptide analogs |
| ES2475207T3 (es) | 2004-07-15 | 2014-07-10 | Tetralogic Pharmaceuticals Corporation | Compuestos de unión a IAP |
| NZ589670A (en) | 2004-12-20 | 2013-01-25 | Genentech Inc | Pyrrolidine inhibitors of iap |
| US20110166319A1 (en) | 2005-02-11 | 2011-07-07 | Immunogen, Inc. | Process for preparing purified drug conjugates |
| CA2597407C (en) | 2005-02-11 | 2013-09-10 | Immunogen, Inc. | Process for preparing stable drug conjugates |
| GB0510390D0 (en) | 2005-05-20 | 2005-06-29 | Novartis Ag | Organic compounds |
| MX2008002114A (es) | 2005-07-21 | 2008-04-17 | Ardea Biosciences Inc | Inhibidores de n-(arilamino)-sulfonamida de mek. |
| RS52470B (en) | 2005-08-24 | 2013-02-28 | Immunogen Inc. | PROCEDURE FOR PREPARATION OF PURE CONJUGATED MEDICINES |
| EP1928912A4 (en) | 2005-09-07 | 2010-02-24 | Medimmune Inc | EPH ANTI-RECEPTOR ANTIBODIES CONJUGATED TO TOXINS |
| JO2660B1 (en) | 2006-01-20 | 2012-06-17 | نوفارتيس ايه جي | Pi-3 inhibitors and methods of use |
| TWI395754B (zh) | 2006-04-24 | 2013-05-11 | Amgen Inc | 人類化之c-kit抗體 |
| TWI432212B (zh) | 2007-04-30 | 2014-04-01 | Genentech Inc | Iap抑制劑 |
| CN103694351A (zh) * | 2007-06-05 | 2014-04-02 | 耶鲁大学 | 受体酪氨酸激酶抑制剂及其使用方法 |
| RU2523890C2 (ru) | 2007-09-12 | 2014-07-27 | Дженентек, Инк. | Комбинации ингибиторов фосфоинозитид 3-киназы и химиотерапевтических агентов и способы применения |
| JP5348725B2 (ja) | 2007-10-25 | 2013-11-20 | ジェネンテック, インコーポレイテッド | チエノピリミジン化合物の製造方法 |
| JP5769616B2 (ja) | 2008-04-30 | 2015-08-26 | イミュノジェン・インコーポレーテッド | クロスリンカーおよびそれらの使用 |
| US8168784B2 (en) | 2008-06-20 | 2012-05-01 | Abbott Laboratories | Processes to make apoptosis promoters |
| SG10201810743WA (en) | 2009-06-03 | 2018-12-28 | Immunogen Inc | Conjugation methods |
| WO2011119948A1 (en) | 2010-03-26 | 2011-09-29 | Kolltan Pharmaceuticals, Inc. | Anti-kit antibodies and uses thereof |
| AR083847A1 (es) | 2010-11-15 | 2013-03-27 | Novartis Ag | Variantes de fc (fragmento constante) silenciosas de los anticuerpos anti-cd40 |
| US9540443B2 (en) * | 2011-01-26 | 2017-01-10 | Kolltan Pharmaceuticals, Inc. | Anti-kit antibodies |
| US20120259100A1 (en) | 2011-03-29 | 2012-10-11 | Immunogen, Inc. | Process for manufacturing conjugates of improved homogeneity |
| EA201991268A3 (ru) | 2011-03-29 | 2020-01-31 | Иммуноджен, Инк. | Получение конъюгатов "майтансиноид-антитело" одностадийным способом |
| AR086044A1 (es) * | 2011-05-12 | 2013-11-13 | Imclone Llc | Anticuerpos que se unen especificamente a un dominio extracelular de c-kit y usos de los mismos |
| BR112015001459B1 (pt) * | 2012-07-25 | 2023-02-14 | Celldex Therapeutics, Inc | Anticorpo isolado ou fragmento do mesmo, conjugado, usos dos mesmos, composição farmacêutica, polinucleotídeo, vetor, célula hospedeira, célula isolada, kit, método in vitro para inibir atividade da kit, método para produzir um anticorpo |
| CA2903772A1 (en) * | 2013-03-15 | 2014-09-25 | Novartis Ag | Antibody drug conjugates |
| US11605302B2 (en) | 2020-11-10 | 2023-03-14 | Rockwell Collins, Inc. | Time-critical obstacle avoidance path planning in uncertain environments |
-
2014
- 2014-03-12 CA CA2903772A patent/CA2903772A1/en not_active Abandoned
- 2014-03-12 MX MX2015013203A patent/MX366978B/es active IP Right Grant
- 2014-03-12 CN CN201480013720.XA patent/CN105007950B/zh not_active Expired - Fee Related
- 2014-03-12 US US14/205,915 patent/US9498543B2/en active Active
- 2014-03-12 MA MA38396A patent/MA38396B1/fr unknown
- 2014-03-12 AP AP2015008656A patent/AP2015008656A0/xx unknown
- 2014-03-12 EA EA201591801A patent/EA035253B1/ru not_active IP Right Cessation
- 2014-03-12 WO PCT/US2014/024597 patent/WO2014150937A1/en active Application Filing
- 2014-03-12 SG SG11201506395RA patent/SG11201506395RA/en unknown
- 2014-03-12 EP EP18188339.8A patent/EP3514178A1/en not_active Withdrawn
- 2014-03-12 US US14/774,512 patent/US9789203B2/en active Active
- 2014-03-12 EP EP14723915.6A patent/EP2968590B1/en active Active
- 2014-03-12 BR BR112015021134A patent/BR112015021134A2/pt not_active Application Discontinuation
- 2014-03-12 AU AU2014235474A patent/AU2014235474B2/en not_active Ceased
- 2014-03-12 ES ES14723915T patent/ES2701051T3/es active Active
- 2014-03-12 JP JP2016501584A patent/JP6333943B2/ja not_active Expired - Fee Related
- 2014-03-12 KR KR1020157028519A patent/KR20150131173A/ko not_active Application Discontinuation
- 2014-03-12 NZ NZ710929A patent/NZ710929A/en not_active IP Right Cessation
- 2014-03-14 TW TW103109292A patent/TW201446806A/zh unknown
- 2014-03-17 UY UY0001035490A patent/UY35490A/es not_active Application Discontinuation
- 2014-03-18 AR ARP140101272A patent/AR095666A1/es unknown
-
2015
- 2015-05-20 US US14/717,578 patent/US20150320880A1/en not_active Abandoned
- 2015-08-05 ZA ZA2015/05656A patent/ZA201505656B/en unknown
- 2015-08-12 CL CL2015002246A patent/CL2015002246A1/es unknown
- 2015-08-27 PH PH12015501902A patent/PH12015501902A1/en unknown
- 2015-09-04 TN TN2015000396A patent/TN2015000396A1/en unknown
- 2015-09-07 IL IL241277A patent/IL241277A0/en unknown
- 2015-09-14 CR CR20150488A patent/CR20150488A/es unknown
- 2015-09-15 CU CUP2015000128A patent/CU20150128A7/es unknown
-
2016
- 2016-10-03 US US15/284,444 patent/US10117953B2/en not_active Expired - Fee Related
-
2017
- 2017-08-30 US US15/690,737 patent/US20180125996A1/en not_active Abandoned
-
2018
- 2018-03-16 JP JP2018049209A patent/JP6578400B2/ja not_active Expired - Fee Related
-
2019
- 2019-12-09 US US16/708,196 patent/US20200129632A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2701051T3 (es) | Conjugados de anticuerpo-fármaco | |
| US11833215B2 (en) | CKIT antibody drug conjugates | |
| US10626172B2 (en) | Antibody drug conjugates | |
| US9498532B2 (en) | Antibody drug conjugates | |
| US20190345250A1 (en) | Antibody drug conjugates | |
| JP2017524364A (ja) | 抗cdh6抗体薬物コンジュゲート | |
| BR112019015950A2 (pt) | conjugados de anticorpo anti-ccr7 e fármaco | |
| CN116096426A (zh) | 用于治疗癌症的ccr7抗体药物缀合物 |