ES2503551T5 - Formas pseudopolimórficas de un inhibidor de la proteasa de VIH - Google Patents
Formas pseudopolimórficas de un inhibidor de la proteasa de VIH Download PDFInfo
- Publication number
- ES2503551T5 ES2503551T5 ES10180831T ES10180831T ES2503551T5 ES 2503551 T5 ES2503551 T5 ES 2503551T5 ES 10180831 T ES10180831 T ES 10180831T ES 10180831 T ES10180831 T ES 10180831T ES 2503551 T5 ES2503551 T5 ES 2503551T5
- Authority
- ES
- Spain
- Prior art keywords
- compound
- pseudopolymorph
- formula
- water
- ethanol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 239000004030 hiv protease inhibitor Substances 0.000 title description 3
- 229940122440 HIV protease inhibitor Drugs 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims description 82
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 60
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 39
- 239000013078 crystal Substances 0.000 claims description 37
- JQUNFHFWXCXPRK-AMMMHQJVSA-N [(3as,4r,6ar)-2,3,3a,4,5,6a-hexahydrofuro[2,3-b]furan-4-yl] n-[(2s,3r)-4-[[2-[(1-cyclopentylpiperidin-4-yl)amino]-1,3-benzothiazol-6-yl]sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate Chemical group C([C@@H]([C@H](O)CN(CC(C)C)S(=O)(=O)C=1C=C2SC(NC3CCN(CC3)C3CCCC3)=NC2=CC=1)NC(=O)O[C@@H]1[C@@H]2CCO[C@@H]2OC1)C1=CC=CC=C1 JQUNFHFWXCXPRK-AMMMHQJVSA-N 0.000 claims description 8
- AHDUVPQPGGNRDS-VQTJNVASSA-N [(2s,3r)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamic acid Chemical compound C([C@@H]([C@H](O)CN(CC(C)C)S(=O)(=O)C=1C=CC(N)=CC=1)NC(O)=O)C1=CC=CC=C1 AHDUVPQPGGNRDS-VQTJNVASSA-N 0.000 claims description 7
- -1 (1S, 2R) -3 - [[(4-aminophenyl) sulfonyl] (isobutyl) amino] -1-benzyl-2-hydroxypropylcarbamate hydrate Chemical compound 0.000 claims description 5
- 206010038997 Retroviral infections Diseases 0.000 claims description 5
- 208000031886 HIV Infections Diseases 0.000 claims description 4
- 208000037357 HIV infectious disease Diseases 0.000 claims description 3
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 claims description 3
- 239000002904 solvent Substances 0.000 description 64
- 238000000034 method Methods 0.000 description 53
- 239000012453 solvate Substances 0.000 description 49
- 239000000243 solution Substances 0.000 description 37
- 239000000047 product Substances 0.000 description 25
- 239000000203 mixture Substances 0.000 description 24
- 238000002425 crystallisation Methods 0.000 description 19
- 230000008025 crystallization Effects 0.000 description 19
- 239000003814 drug Substances 0.000 description 18
- 239000000126 substance Substances 0.000 description 17
- 238000012360 testing method Methods 0.000 description 15
- 239000008194 pharmaceutical composition Substances 0.000 description 14
- 230000008569 process Effects 0.000 description 14
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 12
- 239000002245 particle Substances 0.000 description 12
- IOLCXVTUBQKXJR-UHFFFAOYSA-M potassium bromide Chemical compound [K+].[Br-] IOLCXVTUBQKXJR-UHFFFAOYSA-M 0.000 description 12
- 230000004580 weight loss Effects 0.000 description 12
- 229940079593 drug Drugs 0.000 description 11
- 238000001035 drying Methods 0.000 description 11
- 238000001704 evaporation Methods 0.000 description 11
- 230000036571 hydration Effects 0.000 description 11
- 238000006703 hydration reaction Methods 0.000 description 11
- 238000002329 infrared spectrum Methods 0.000 description 11
- 239000003960 organic solvent Substances 0.000 description 11
- 238000002360 preparation method Methods 0.000 description 11
- 238000003860 storage Methods 0.000 description 11
- 238000003795 desorption Methods 0.000 description 10
- 230000008020 evaporation Effects 0.000 description 10
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 9
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- HHFAWKCIHAUFRX-UHFFFAOYSA-N ethoxide Chemical compound CC[O-] HHFAWKCIHAUFRX-UHFFFAOYSA-N 0.000 description 9
- 238000001914 filtration Methods 0.000 description 9
- 238000004128 high performance liquid chromatography Methods 0.000 description 9
- 238000001179 sorption measurement Methods 0.000 description 9
- 238000002336 sorption--desorption measurement Methods 0.000 description 9
- 230000003595 spectral effect Effects 0.000 description 9
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 8
- 238000010521 absorption reaction Methods 0.000 description 8
- 238000010438 heat treatment Methods 0.000 description 8
- 241000124008 Mammalia Species 0.000 description 7
- 238000001237 Raman spectrum Methods 0.000 description 7
- 230000008859 change Effects 0.000 description 7
- 239000006185 dispersion Substances 0.000 description 7
- ARXJGSRGQADJSQ-UHFFFAOYSA-N 1-methoxypropan-2-ol Chemical compound COCC(C)O ARXJGSRGQADJSQ-UHFFFAOYSA-N 0.000 description 6
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- 238000001816 cooling Methods 0.000 description 6
- 238000009472 formulation Methods 0.000 description 6
- 230000007774 longterm Effects 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- 230000008018 melting Effects 0.000 description 6
- 238000002844 melting Methods 0.000 description 6
- 241000725303 Human immunodeficiency virus Species 0.000 description 5
- 238000001069 Raman spectroscopy Methods 0.000 description 5
- 229910052782 aluminium Inorganic materials 0.000 description 5
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 5
- 238000000113 differential scanning calorimetry Methods 0.000 description 5
- 239000011521 glass Substances 0.000 description 5
- 239000012535 impurity Substances 0.000 description 5
- 238000010899 nucleation Methods 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 108010010369 HIV Protease Proteins 0.000 description 4
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 4
- 239000000654 additive Substances 0.000 description 4
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 238000001938 differential scanning calorimetry curve Methods 0.000 description 4
- 238000004090 dissolution Methods 0.000 description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 4
- 150000004677 hydrates Chemical class 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N silicon dioxide Inorganic materials O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 238000002411 thermogravimetry Methods 0.000 description 4
- 238000000870 ultraviolet spectroscopy Methods 0.000 description 4
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 238000004566 IR spectroscopy Methods 0.000 description 3
- 239000004698 Polyethylene Substances 0.000 description 3
- 208000036142 Viral infection Diseases 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- 125000004429 atom Chemical group 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 238000000354 decomposition reaction Methods 0.000 description 3
- 238000006073 displacement reaction Methods 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 230000004927 fusion Effects 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 150000002576 ketones Chemical class 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 239000012299 nitrogen atmosphere Substances 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 238000001953 recrystallisation Methods 0.000 description 3
- 229930195734 saturated hydrocarbon Natural products 0.000 description 3
- 238000009738 saturating Methods 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 238000007614 solvation Methods 0.000 description 3
- 238000001228 spectrum Methods 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- 238000005292 vacuum distillation Methods 0.000 description 3
- 230000009385 viral infection Effects 0.000 description 3
- 238000011179 visual inspection Methods 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- BWLBGMIXKSTLSX-UHFFFAOYSA-N 2-hydroxyisobutyric acid Chemical compound CC(C)(O)C(O)=O BWLBGMIXKSTLSX-UHFFFAOYSA-N 0.000 description 2
- FKOZPUORKCHONH-UHFFFAOYSA-N 2-methylpropane-1-sulfonic acid Chemical compound CC(C)CS(O)(=O)=O FKOZPUORKCHONH-UHFFFAOYSA-N 0.000 description 2
- 208000030507 AIDS Diseases 0.000 description 2
- 206010001513 AIDS related complex Diseases 0.000 description 2
- 238000005079 FT-Raman Methods 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- CPLXHLVBOLITMK-UHFFFAOYSA-N Magnesium oxide Chemical compound [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 2
- 102000014961 Protein Precursors Human genes 0.000 description 2
- 238000002441 X-ray diffraction Methods 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 239000008186 active pharmaceutical agent Substances 0.000 description 2
- 230000001476 alcoholic effect Effects 0.000 description 2
- 150000001335 aliphatic alkanes Chemical class 0.000 description 2
- 229910001508 alkali metal halide Inorganic materials 0.000 description 2
- 150000008045 alkali metal halides Chemical class 0.000 description 2
- 229940124522 antiretrovirals Drugs 0.000 description 2
- 239000003903 antiretrovirus agent Substances 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- QDHFHIQKOVNCNC-UHFFFAOYSA-M butane-1-sulfonate Chemical compound CCCCS([O-])(=O)=O QDHFHIQKOVNCNC-UHFFFAOYSA-M 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 238000012937 correction Methods 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 238000011194 good manufacturing practice Methods 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 238000000227 grinding Methods 0.000 description 2
- 208000015181 infectious disease Diseases 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- NBTOZLQBSIZIKS-UHFFFAOYSA-N methoxide Chemical compound [O-]C NBTOZLQBSIZIKS-UHFFFAOYSA-N 0.000 description 2
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 2
- 239000003607 modifier Substances 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 230000006911 nucleation Effects 0.000 description 2
- 150000002894 organic compounds Chemical class 0.000 description 2
- 239000005022 packaging material Substances 0.000 description 2
- 238000004806 packaging method and process Methods 0.000 description 2
- 239000000902 placebo Substances 0.000 description 2
- 229940068196 placebo Drugs 0.000 description 2
- 238000000634 powder X-ray diffraction Methods 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- OGHBATFHNDZKSO-UHFFFAOYSA-N propan-2-olate Chemical compound CC(C)[O-] OGHBATFHNDZKSO-UHFFFAOYSA-N 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- UJJLJRQIPMGXEZ-UHFFFAOYSA-N tetrahydro-2-furoic acid Chemical compound OC(=O)C1CCCO1 UJJLJRQIPMGXEZ-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 2
- 230000003612 virological effect Effects 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 229910004261 CaF 2 Inorganic materials 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 229910000530 Gallium indium arsenide Inorganic materials 0.000 description 1
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 1
- 241000713340 Human immunodeficiency virus 2 Species 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 108010078762 Protein Precursors Proteins 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000019486 Sunflower oil Nutrition 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 238000002159 adsorption--desorption isotherm Methods 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 150000001348 alkyl chlorides Chemical class 0.000 description 1
- 238000012442 analytical experiment Methods 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000010775 animal oil Substances 0.000 description 1
- 230000000798 anti-retroviral effect Effects 0.000 description 1
- XKRFYHLGVUSROY-UHFFFAOYSA-N argon Substances [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 238000005102 attenuated total reflection Methods 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 239000003026 cod liver oil Substances 0.000 description 1
- 235000012716 cod liver oil Nutrition 0.000 description 1
- 238000005056 compaction Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 150000004292 cyclic ethers Chemical class 0.000 description 1
- 239000007857 degradation product Substances 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 239000002274 desiccant Substances 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 229940088679 drug related substance Drugs 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 238000004508 fractional distillation Methods 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 235000014380 magnesium carbonate Nutrition 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- 235000012245 magnesium oxide Nutrition 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 239000013081 microcrystal Substances 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 239000002159 nanocrystal Substances 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- WXZMFSXDPGVJKK-UHFFFAOYSA-N pentaerythritol Chemical compound OCC(CO)(CO)CO WXZMFSXDPGVJKK-UHFFFAOYSA-N 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 230000010287 polarization Effects 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000005020 polyethylene terephthalate Substances 0.000 description 1
- 229920000139 polyethylene terephthalate Polymers 0.000 description 1
- 102000054765 polymorphisms of proteins Human genes 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 239000010453 quartz Substances 0.000 description 1
- 238000005057 refrigeration Methods 0.000 description 1
- 239000013557 residual solvent Substances 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 238000009331 sowing Methods 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000001694 spray drying Methods 0.000 description 1
- 238000013112 stability test Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000002600 sunflower oil Substances 0.000 description 1
- 230000001839 systemic circulation Effects 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 238000002076 thermal analysis method Methods 0.000 description 1
- 238000001757 thermogravimetry curve Methods 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 230000029812 viral genome replication Effects 0.000 description 1
- 230000006514 viral protein processing Effects 0.000 description 1
- 210000002845 virion Anatomy 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Virology (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Tropical Medicine & Parasitology (AREA)
- Molecular Biology (AREA)
- AIDS & HIV (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Furan Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
DESCRIPCIÓN
Formas pseudopolimórficas de un inhibidor de la proteasa de VIH
Campo técnico
Esta invención se refiere a nuevas formas pseudopolimórficas de (1S,2R)-3-[[(4-aminofenil)sulfonil](isobutil)amino]-1-bencil-2-hidroxipropilcarbamato de (3R,3aS,6aR)-hexahidro-furo[2,3-b]furan-3-ilo, a un método para su preparación, así como a su uso como un medicamento.
Antecedentes de la invención
Las proteasas codificadas por virus, que son esenciales para la replicación vírica, son necesarias para el procesamiento de precursores de proteínas víricas. La interferencia con el procesamiento de precursores de proteínas inhibe la formación de viriones infecciosos. En consecuencia, los inhibidores de proteasas víricas se pueden usar para prevenir o tratar infecciones víricas crónicas y agudas.
(1 S,2R)-3-[[(4-aminofenil)sulfonil](isobutil)amino]-1 -bencil-2-hidroxipropilcarbamato de (3R,3aS,6aR)-hexahidrofuro[2,3-b]furan-3-ilo tiene actividad inhibidora de la proteasa del VIH, y es particularmente muy adecuado para inhibir los virus VIH-1 y VIH-2.
La estructura de (1S,2R)-3-[[(4-amino-fenil)sulfonil](isobutil)amino]-1-bencil-2-hidroxipropilcarbamato de (3R,3aS,6aR)-hexahidro-furo[2,3-b]furan-3-ilo se muestra a continuación:

El compuesto de fórmula (X) y los procedimientos para su preparación se describen en los documentos EP 715618, WO 99/67417, US 6.248.775, y en Bioorganic and Chemistry Letters, Vol. 8, p. 687-690, 1998, “Potent HIV protease inhibitors incorporating high-affinity P2-igands and (R)-(hydroxyethylamino)sulfonamide isostere” , todos los cuales se incorporan a la presente por referencia.
Los fármacos utilizados en la preparación de formulaciones farmacéuticas para uso comercial deben satisfacer ciertos estándar, incluyendo GMP (Buenas Prácticas de Fabricación) y las guías de ICH (Conferencia Internacional sobre Armonización). Tales estándar incluyen requisitos técnicos que engloban un intervalo heterogéneo y amplio de parámetros físicos, químicos y farmacéuticos. Es esta variedad de parámetros a considerar lo que hace a las formulaciones farmacéuticas una disciplina técnica compleja.
Por ejemplo, y como ejemplo, un fármaco utilizado para la preparación de formulaciones farmacéuticas debería satisfacer una pureza aceptable. Hay normas establecidas que definen los límites y cuantificación de impurezas en nuevas sustancias farmacéuticas producidas mediante síntesis química, es decir, las impurezas reales y potenciales que surgen muy probablemente durante la síntesis, purificación y almacenamiento de la nueva sustancia farmacéutica. Las normas están instituidas para la cantidad de productos de degradación permitidos de la sustancia farmacéutica, o productos de reacción de la sustancia farmacéutica con un excipiente y/o recipiente/sistema de cierre inmediato.
La estabilidad es también un parámetro considerado a la hora de crear formulaciones farmacéuticas. Una buena estabilidad asegurará que la integridad química deseada de la sustancia farmacéutica se mantenga durante el período de caducidad de la formulación farmacéutica, que es el marco de tiempo a lo largo del cual se puede confiar que un producto retenga sus características de calidad cuando se almacena en condiciones de almacenamiento
esperadas o dirigidas. Durante este período, el fármaco se puede administrar con poco o ningún riesgo, ya que la presencia de productos de degradación potencialmente peligrosos no plantea consecuencias perjudiciales a la salud del receptor, ni el contenido más bajo del ingrediente activo podría provocar una medicación insuficiente.
Factores diferentes, tales como radiación de luz, temperatura, oxígeno, humedad, sensibilidad al pH en disoluciones, pueden influir en la estabilidad y determinar las condiciones de almacenamiento y el período de caducidad.
La biodisponibilidad es también un parámetro a considerar en el diseño del suministro de fármacos de formulaciones farmacéuticamente aceptables. La biodisponibilidad está relacionada con la cantidad y velocidad a la que la forma intacta de un fármaco particular aparece en la circulación sistémica tras la administración del fármaco. La biodisponibilidad mostrada por un fármaco es así de importancia a la hora de determinar si se logra una concentración terapéuticamente eficaz en el sitio o sitios de acción del fármaco.
Factores fisicoquímicos y la formulación farmacotécnica pueden tener repercusiones en la biodisponibilidad del fármaco. Como tal, varias propiedades del fármaco, tales como constante de disociación, velocidad de disolución, solubilidad, forma polimórfica, tamaño de partículas, se han de considerar cuando se mejora la biodisponibilidad. También es importante establecer que la formulación farmacéutica seleccionada es capaz de fabricarse; más adecuadamente, de fabricarse a gran escala.
A la vista de los diversos y muchos requisitos técnicos, y sus parámetros influyentes, no es obvio predecir qué formulaciones farmacéuticas serán aceptables. Como tal, se encontró inesperadamente que ciertas modificaciones del estado sólido del compuesto de fórmula (X) influyeron positivamente en su aplicabilidad en formulaciones farmacéuticas.
Sumario de la invención
La presente invención se refiere a formas pseudopolimórficas del compuesto de fórmula (X) para la preparación de formulaciones farmacéuticas. Tales formas pseudopolimórficas contribuyen a formulaciones farmacéuticas en estabilidad y biodisponibilidad mejoradas. Se pueden fabricar en pureza elevada suficiente para ser aceptables para uso farmacéutico; más particularmente, en la fabricación de un medicamento para inhibir la actividad de la proteasa del VIH en mamíferos.
En un primer aspecto, la presente invención proporciona pseudopolimorfos de (1S,2R)-3-[[(4-aminofenil)sulfonil](isobutil)amino]-1-bencil-2-hidroxipropilcarbamato de (3R,3aS,6aR)-hexahidrofuro[2,3-b]furan-3-ilo.
Los pseudopolimorfos proporcionados incluyen solvatos que son alcoholes, más en particular, solvatos que son alcoholes C1-C4; solvatos que son hidratos; solvatos que son alcanos, más en particular, solvatos que son cloroalcanos C1-C4; solvatos que son cetonas, más en particular, solvatos que son cetonas C1-C5; solvatos que son éteres, más en particular, solvatos que son éteres C1-C4; solvatos que son éteres cíclicos; solvatos que son ésteres, más en particular, solvatos que son ésteres C1-C5; y solvatos sulfónicos, más en particular, solvatos sulfónicos C1-C4; del compuesto de fórmula (X). Los pseudopolimorfos preferidos son solvatos farmacéuticamente aceptables, tales como hidrato y etanolato.
Los pseudopolimorfos particulares son la Forma A (etanolato), Forma B (hidrato), Forma C (metanolato), Forma D (acetonato), Forma E (diclorometanoato), Forma F (solvato de tipo acetato de etilo), Forma G (1-metoxi-2-propanolato), Forma H (anisolato), Forma I (tetrahidrofuranato), Forma J (isopropanolato) del compuesto de fórmula (X). Otro pseudopolimorfo particular es la Forma K (mesilato) del compuesto de fórmula (X).
En un segundo aspecto, la presente invención se refiere a procedimientos para preparar pseudopolimorfos. Los pseudopolimorfos del compuesto de fórmula (X) se preparan combinando el compuesto de fórmula (X) con un disolvente orgánico, agua, o mezclas de agua y disolventes orgánicos miscibles en agua, y aplicando cualquier técnica adecuada para inducir cristalización, para obtener los pseudopolimorfos deseados.
En un tercer aspecto, la invención se refiere al uso de los pseudopolimorfos presentes, en la fabricación de formulaciones farmacéuticas para inhibir la actividad de la proteasa del VIH en mamíferos. En relación con el campo terapéutico, una realización preferida de esta invención se refiere al uso de formas pseudopolimórficas farmacéuticamente aceptables del compuesto de fórmula (X) para el tratamiento de una enfermedad vírica del VIH en un mamífero que lo necesite, método el cual comprende administrar a dicho mamífero una cantidad eficaz de una forma pseudopolimórfica farmacéuticamente aceptable del compuesto de fórmula (X).
Los siguientes dibujos proporcionan información adicional sobre las características de los pseudopolimorfos según la presente invención.
Breve descripción de los dibujos
La FIGURA 1, FIGURA 2 y FIGURA 3 son los patrones de difracción de rayos X de polvo de la Forma A (1:1).
La FIGURA 4 representa la Forma A (1:1) en tres dimensiones con los átomos identificados.
La FIGURA 5 es una comparación de los espectros de Raman de las Formas A, B, D, E, F, H, (1:1) y la forma amorfa en la región de estiramiento de carbonilo de 1800-100 cm-1 y la región 3300-2000 cm-1.
La FIGURA 6 es una comparación de los espectros de Raman expandidos de las Formas A, B, D, E, F, H, (1:1) y la forma amorfa en la región de estiramiento de carbonilo de 600-0 cm-1.
La FIGURA 7 es una comparación de los espectros de Raman expandidos de las Formas A, B, D, E, F, H, (1:1) y la forma amorfa en la región de estiramiento de carbonilo de 1400-800 cm-1.
En las Figuras 5, 6 y 7, P1 corresponde a la Forma A, P18 corresponde a la Forma B, P19 corresponde a la forma amorfa, P25 corresponde a la Forma E, P27 corresponde a la Forma F, P50 corresponde a la Forma D, P68 corresponde a la Forma H, P69 corresponde a la Forma C, P72 corresponde a la Forma I, y P81 corresponde a la Forma G.
La FIGURA 8 es el termograma calorimétrico de barrido diferencial (DSC) de la Forma A (1:1).
La FIGURA 9 es el espectro de infrarrojos (IR) que refleja los modos vibracionales de la estructura molecular de la Forma A como producto cristalino.
La FIGURA 10 es el espectro de IR que refleja los modos vibracionales de la estructura molecular de la Forma B como producto cristalino.
FIGURA 11: Espectro de IR de las formas A, B, y forma amorfa, en el intervalo espectral 4000 a 400 cm-1. FIGURA 12: Espectro de IR de las formas A, B, y forma amorfa, en el intervalo espectral 3750 a 2650 cm-1. FIGURA 13: Espectro de IR de las formas A, B, y forma amorfa, en el intervalo espectral 1760 a 1580 cm-1. FIGURA 14: Espectro de IR de las formas A, B, y forma amorfa, en el intervalo espectral 980 a 720 cm-1. En las figuras 11, 12, 13 y 14, la curva A corresponde a la Forma A, la curva B corresponde a la Forma B, y la curva C corresponde a la forma amorfa.
FIGURA 15: Curvas termográficas de DSC de la Forma A (curva D), Forma A tras la adsorción/desorción (ADS/DES) (curva E), y Forma A después de ensayos de hidratación de ADS/DES (curva F).
FIGURA 16: Curvas termogravimétricas (TG) de la Forma A (Curva D), Forma A después de ADS/DES (curva E), y Forma A después de ensayos de hidratación de ADS/DES (curva F).
FIGURA 17: Curva TG de la Forma A a 25°C en una atmósfera de nitrógeno seco en función del tiempo. FIGURA 18: Curvas de ADS/DES de la Forma A.
FIGURA 19: Curvas de ADS/DES del ensayo de hidratación de la Forma A.
FIGURA 20: Curvas de ADS/DES de la Forma B.
FIGURA 21: Espectro de IR de la Forma K.
FIGURA 22: Espectro de Raman de la Forma K.
FIGURA 23: Curva de DSC de la Forma K.
FIGURA 24: Curva TG de la Forma K.
FIGURA 25: Isoterma de ADS/DES de la Forma K, lote 1.
FIGURA 26: Isoterma de ADS/DES de la Forma K, lote 2.
Descripción detallada
El término “polimorfismo” se refiere a la capacidad de una estructura química para aparecer en diferentes formas, y se sabe que aparecen muchos compuestos orgánicos, incluyendo fármacos. Como tal, “formas polimórficas” o “polimorfismos” incluyen sustancias farmacéuticas que aparecen en forma amorfa, en forma cristalina, en forma anhidra, en diversos grados de hidratación o solvatación, con moléculas de disolvente atrapadas, así como sustancias que varían en dureza, forma y tamaño del cristal. Los diferentes polimorfos varían en propiedades físicas tales como solubilidad, disolución, estabilidad del estado sólido, así como comportamiento de procesamiento en términos de fluidez y compactación del polvo durante la formación de comprimidos.
La expresión “forma amorfa” se define como una forma en la que no existe un orden tridimensional de largo alcance. En la forma amorfa, la posición de las moléculas con respecto una de las otras es esencialmente al azar, es decir, sin disposición regular de las moléculas en una estructura de red cristalina.
El término “cristalina” se define como una forma en la que la posición de las moléculas, unas con respecto a las otras, está organizada según una estructura de red cristalina tridimensional.
La expresión “forma anhidra” se refiere a una forma particular esencialmente libre de agua. “Hidratación” se refiere al proceso de añadir moléculas de agua a una sustancia que aparece en una forma particular, y los “hidratos” son sustancias que se forman añadiendo moléculas de agua. “Solvatar” se refiere al proceso de incorporar moléculas de un disolvente en una sustancia que aparece en una forma cristalina. Por lo tanto, el término “solvato” se define como una forma cristalina que contiene cantidades estequiométricas o no estequiométricas de disolvente. Puesto que el agua es un disolvente, los solvatos también incluyen hidratos. El término “pseudopolimorfo” se aplica a formas cristalinas polimórficas que tienen moléculas de disolvente incorporadas en sus estructuras de redes cristalinas. El término pseudopolimorfismo se usa frecuentemente para designar solvatos (Byrn, Pfeiffer, Stowell, (1999) Solidstate Chemistry of Drugs, 2a Ed., publicado por SSCI, Inc).
La presente invención proporciona pseudopolimorfos de (1S,2R)-3-[[(4-aminofenil)sulfonil](isobutil)amino]-1-bencil-2-hidroxipropilcarbamato de (3R,3aS,6aR)-hexahidrofuro[2,3-b]furan-3-ilo.
En una realización, los pseudopolimorfos son solvatos de alcoholes, más en particular, solvatos de alcoholes de C1-C4 ; solvatos de hidratos; solvatos de alcanos, más en particular, solvatos de cloroalcanos de C1-C4 ; solvatos de cetonas, más en particular, solvatos de cetonas de C1-C5 ; solvatos de éteres, más en particular solvatos de éteres de C1-C4 ; solvatos de cicloéteres; solvatos de ésteres, más en particular solvatos de ésteres de C1-C5 ; o solvatos sulfónicos, más en particular solvatos sulfónicos de C1-C4, del compuesto de fórmula (X). La expresión “alcohol de C1-C4” define hidrocarburos saturados e insaturados de cadena lineal y/o ramificada que tienen de 1 a 4 átomos de carbono sustituidos con al menos un grupo hidroxilo, y opcionalmente sustituidos con un grupo alquiloxi, tales como, por ejemplo, metanol, etanol, isopropanol, butanol, 1-metoxi-2-propanol y similares. La expresión “cloroalcano de C1-C4” define hidrocarburos saturados e insaturados de cadena lineal y/o ramificada que tienen de 1 a 4 átomos de carbono sustituidos con al menos un átomo de cloro, tales como, por ejemplo, diclorometano. La expresión “cetona de C1-C5” define disolventes de la fórmula general R’-C(=O)-R, en la que R y R’ pueden ser iguales o diferentes y son metilo o etilo, tales como, acetona y similares. La expresión “éter de C1-C4” define disolventes de la fórmula general R’-O-R, en la que R y R’ pueden ser iguales o diferentes y son un grupo fenilo, metilo o etilo, tales como anisol y similares. La expresión “cicloéter” define hidrocarburos monocíclicos de 4 a 6 miembros que contienen uno o dos átomos anulares de oxígeno, tales como tetrahidrofurano y similares. La expresión “éster de C1-C5” define disolventes de la fórmula general R’-O-C(=O)-R, en la que R y R’ pueden ser iguales o diferentes y son metilo o etilo, tales como acetato de etilo y similares. La expresión “disolvente sulfónico de C1-C4” define disolventes de la fórmula general R-SO3 H, en la que R puede ser un hidrocarburo saturado de cadena lineal o ramificada que tiene de 1 a 4 átomos de carbono, tales como mesilato, etanosulfonato, butanosulfonato, 2-metil-1-propanosulfonato, y similares. Los pseudopolimorfos de la presente invención, que son farmacéuticamente aceptables, por ejemplo hidratos, solvatos de alcohol, tales como etanolato, son formas preferidas.
Varios pseudopolimorfos se ejemplifican en esta solicitud, e incluyen Forma A (etanolato), Forma B (hidrato), Forma C (metanolato), Forma D (acetonato), Forma E (diclorometanato), Forma F (solvato de acetato de etilo), Forma G (1-metoxi-2-propanolato), Forma H (anisolato), Forma I (tetrahidrofuranato), Forma J (isopropanolato), o Forma K (mesilato) del compuesto de fórmula (X).
Los solvatos pueden aparecer en diferentes relaciones de solvatación. El contenido de disolvente del cristal puede variar en diferentes relaciones, dependiendo de las condiciones aplicadas. Las formas cristalinas de solvatos del compuesto de fórmula (X) pueden comprender hasta 5 moléculas de disolvente por molécula de compuesto de fórmula (X), que aparecen en diferentes estados solvatados, incluyendo, entre otros, hemisolvato, monosolvato, disolvato, cristales de trisolvato, cristales de solvatos intermedios, y sus mezclas. Convenientemente, la relación de compuesto de fórmula (X) al disolvente puede oscilar entre (5:1) y (1:5). En particular, la relación puede oscilar desde alrededor de 0,2 hasta alrededor de 3 moléculas de disolvente por 1 molécula de compuesto de fórmula (X), más en particular, la relación puede oscilar desde alrededor de 1 hasta alrededor de 2 moléculas de disolvente por 1 molécula de compuesto de fórmula (X), preferiblemente la relación es 1 molécula de disolvente por 1 molécula de compuesto de fórmula (X).
Los solvatos también pueden aparecer en diferentes niveles de hidratación. Como tales, las formas cristalinas de solvatos de compuesto de fórmula (X) pueden comprender además, en ciertas circunstancias, moléculas de agua parcial o completamente en las estructuras cristalinas. En consecuencia, la expresión “Forma A” se usará aquí para referirse a las formas de etanolato del compuesto de fórmula (X) que comprenden hasta 5 moléculas de disolvente por 1 molécula de compuesto de fórmula (X), cristales de solvatos intermedios, y las mezclas de los mismos; y opcionalmente que comprenden moléculas de agua adicionales, parcial o completamente en las estructuras del cristal. Lo mismo se aplica para la Forma B a la Forma K. En el caso en que sea necesario señalar una “Forma A”
particular, la relación de solvatación seguirá a la “Forma A”; por ejemplo, una molécula de etanol por una molécula de compuesto (X) se señala como Forma A (1:1).
La difracción de rayos X de polvo es una técnica para caracterizar formas polimórficas que incluyen pseudopolimorfos del compuesto de fórmula (X), y para diferenciar formas de cristales de solvatos de otras formas cristalinas y no cristalinas del compuesto de fórmula (X). Como tales, se recogieron espectros de difracción de rayos
X de polvo en un difractómetro de polvo Phillips PW 1050/80, modelo Bragg-Brentano. Los polvos de Forma A (1:1), alrededor de 200 mg cada muestra, se empaquetaron en tubos capilares de vidrio de 0,5 mm y se analizaron según un método estándar en la técnica. El generador de rayos X se hizo funcionar a 45 kV y 32 mA, usando la línea de cobre Ka como la fuente de radiación. No hubo rotación de la muestra a lo largo del eje chi, y los datos se recogieron entre un tamaño de etapa 2-theta de 4 y 60°. La Forma A (1:1) tiene las posiciones de ángulo dos-theta características de los picos como se muestran en las FIG.1,2 y 3 a: 7,04° ± 0,5°, 9,24° ± 0,5°, 9,96° ± 0,5°, 10,6 0,5°, 11,30° ± 0,5°, 12,82° ± 0,5°, 13,80° ± 0,5°, 14,56° ± 0,5°, 16,66° ± 0,5°, 17,30° ± 0,5°, 18,28° ± 0,5°, 19,10° ±
0,5°, 20,00° ± 0,5°, 20,50° ± 0,5°, 21,22° ± 0,5°, 22,68° ± 0,5°, 23,08° ± 0,5°, 23,66° ± 0,5°, 25,08° ± 0,5°, 25,58° ±
0,5°, 26,28° ± 0,5°, 27,18° ± 0,5°, 28,22° ± 0,5°, 30,20° ± 0,5°, 31,34° ± 0,5°, 32,68° ± 0,5°, 33,82° ± 0,5°, 39,18° ±
0,5°, 41,20° ± 0,5°, 42,06°C ± 0,5°, y 48,74° ± 0,5°.
En otro conjunto de experimentos analíticos, se aplicó la difracción individual de rayos X a la Forma A (1:1), que dio como resultado la siguiente configuración cristalina, enunciada en la tabla a continuación.
Tabla 1

La estructura tridimensional resultante de la Forma A (1:1) se representa en la Figura 4.
La Tabla 2 muestra las coordinadas atómicas (x 104) y parámetros de los desplazamientos ¡sotrópicos equivalentes (Á2 x 103) para la Forma A (1:1). Los átomos se numeran como se muestra en la Figura 4. Las coordenadas fraccionarias x, y y z indican la posición de los átomos con respecto al origen de la celda unidad. U(eq) se define como un tercio de la traza del tensor ü¡¡ ortogonalizado.

La Tabla 3 muestra los parámetros de los desplazamientos anisotrópicos (Á2 x 103) para la Forma A (1:1). El exponente del factor del desplazamiento anisotrópico adopta la fórmula: -2p2[h2a *2ün + ... 2 h k a*b*Ü12]

La espectroscopía de Raman se ha usado ampliamente para elucidar estructuras moleculares, cristalinidad y polimorfismo. Los modos de Raman de baja frecuencia son particularmente útiles para distinguir diferentes empaquetamientos moleculares en el cristal. Como tales, se registraron espectros de Raman en un espectrómetro
de Bruker FT-Raman RFS100 equipado con un tubo fotomultiplicador y detectores de múltiples canales ópticos. Las muestras se colocaron en tubos capilares de cuarzo, y se excitaron mediante un láser de ion de argón. La potencia del láser de las muestras se ajustó a alrededor de 100 mW, y la resolución espectral fue alrededor de 2 cm-1. Se encontró que las Formas A, B, D, E, F, y H, (1:1) y la forma amorfa tienen los espectros de Raman que aparecen en las Figuras 5, 6 y 7.
Además, las Formas A y B se caracterizaron usando un accesorio pATR (Reflectancia Total Micro-Atenuada) (Harrick Split-Pea con cristal de Si). Los espectros de infrarrojos se obtuvieron con un espectrofotómetro de Nicolet Magna 560 FTIR, un separador de haces de Ge en KBr, y un detector de DTGS con una ventana de KBr. Los espectros se midieron a una resolución de 1 cm-1 y 32 barridos cada uno, en un intervalo de longitudes de onda de 4000 a 400 cm-1, y aplicación de corrección de línea base. Los números de onda para la Forma A obtenida se muestran en la siguiente Tabla 4.
Tabla 4

El espectro de IR en la Figura 9 refleja los modos vibracionales de la estructura molecular como un producto cristalino.
Los números de onda obtenidos para la Forma B se muestran en la siguiente Tabla 5.
Tabla 5

El espectro de IR en la Figura 10 refleja los modos vibracionales de la estructura molecular de la Forma B como un producto cristalino.
Siguiendo el mismo método analítico de IR, la Forma B y la forma amorfa también se caracterizaron y compararon con la Forma A, como se muestra en las Figuras 11 a 14. Los espectros de IR de las diferentes formas físicas mostraron diferencias espectrales distintas, y las más relevantes son aquellas en la Tabla 6:
Tabla 6

Las Formas A, B y forma amorfa físicas se identifican mediante interpretación espectral, centrada en las bandas de absorción específicas para cada forma. Las diferencias espectrales únicas y específicas entre las formas se observan en 3 intervalos espectrales: de 3750 a 2650 cm-1 (intervalo 1), de 1760 a 1580 cm-1 (intervalo 2) y de 980 a 720 cm-1 (intervalo 3).
Intervalo 1 (de 3750 a 2650 cm-1)
Figura 11: La Forma A muestra una banda doble con máximos de absorción a 3454 cm-1 y 3429 cm-1. La Forma B muestra una única banda de absorción a 3615 cm-1, y la forma amorfa muestra una única banda de absorción a 3362 cm-1.
Intervalo 2 (de 1760 a 1580 cm-1)
Figura 12: La Forma A muestra una única banda de absorción a 1646 cm-1, la Forma B muestra una única banda de absorción a 1630 cm-1, y la forma amorfa muestra una única banda de absorción a 1628 cm-1 con una intensidad claramente mayor en comparación con la banda de la Forma B. Adicionalmente, la forma amorfa muestra una banda ancha, menos intensa, a 1704 cm-1, en comparación con ambas bandas de las formas A y B a alrededor de 1704 cm-1.
Intervalo 3 (de 980 a 720 cm-1)
Figura 13: La Forma A muestra un conjunto distinto de 5 bandas de absorción a 911, 890, 876, 862 y 841 cm-1. La Forma B muestra un conjunto similar, pero la banda de 876 cm-1 no está. La forma amorfa muestra una única banda ancha a alrededor de 750 cm-1, ambas formas A y B muestran dos máximos a alrededor de 768 cm-1 y 743 cm-1. La termomicroscopía es otra técnica útil en el estudio de la cinética del estado sólido. La cinética de los procesos de nucleación de disoluciones o fundidos, incluyendo el análisis de la velocidad de nucleación, se puede cuantificar. El método más simple y más ampliamente usado es la determinación del punto de fusión. Como tal, se usó un controlador de Mettler modelo FP 82 con etapa de calentamiento en un microscopio Leitz. Se colocaron unas pocas partículas de la Forma A sobre un portaobjetos de vidrio y se observaron mientras se calentaban a 10°C por minuto. Se encontró que el intervalo de fusión para la Forma A (1:1) estaba entre 90° y 110°C.
En otro medio de caracterización, la solubilidad de la Forma A (1:1) fue también objeto de estudio. Su solubilidad en diferentes disolventes a aproximadamente 23°C se determinó que era la siguiente:
Tabla 7: Solubilidad aproximada para la Forma A (1:1), en mg/ml

Se llevaron a cabo otras investigaciones de solubilidad en función del pH. Como tal, se midieron las solubilidades acuosas de la Forma A (1:1) en disolventes con pH diferente. Un exceso del soluto se equilibró con el disolvente a 20°C durante al menos 24 horas. Después de eliminar el compuesto sin disolver, la concentración en disolución se determinó usando espectrometría de UV.
Tabla 8: Solubilidad para la Forma A (1:1) en función del pH
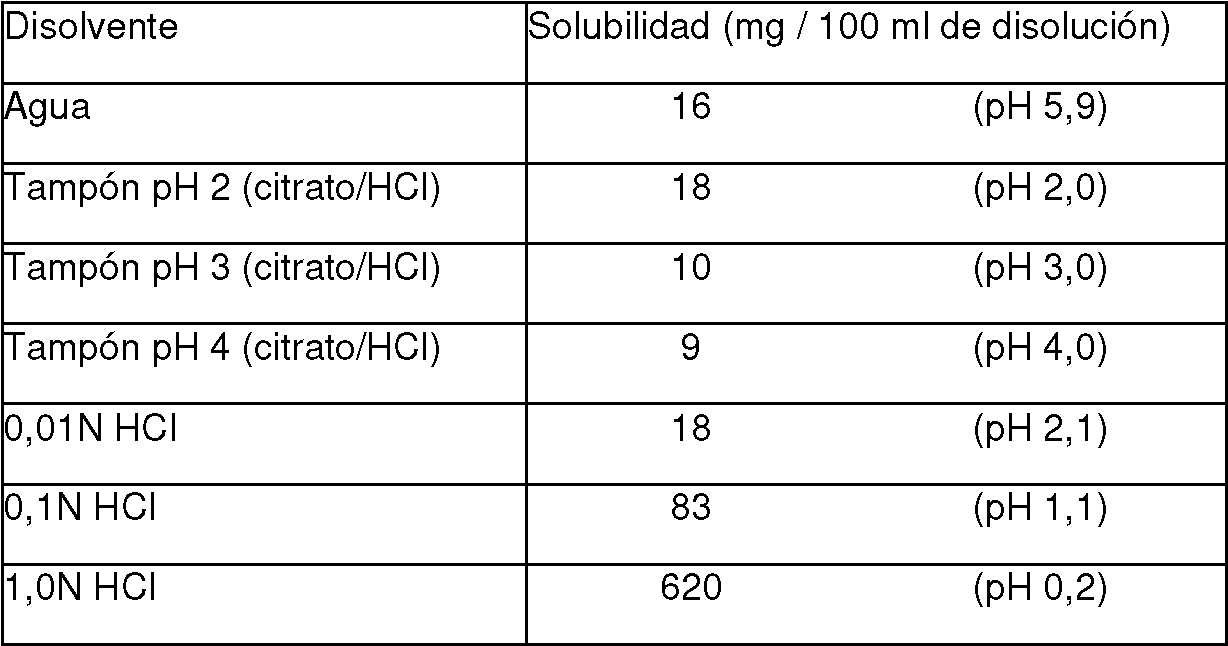
Se midió la solubilidad de la Forma A (1:1) en función de HPpCD (hidroxipropil-p-ciclodextrina). Un exceso de producto se equilibró con el disolvente durante 2 días a 20°C. Después de eliminar el compuesto sin disolver, la concentración en disolución se determinó usando espectrometría de UV.
Tabla 9: Solubilidad para la Forma A (1:1) en función de HPpCD

En un segundo aspecto, la presente invención se refiere a procedimientos para preparar pseudopolimorfos. Los pseudopolimorfos del compuesto de fórmula (X) se preparan combinando el compuesto de fórmula (X) con un disolvente orgánico, o agua, o mezclas de agua y disolventes orgánicos miscibles en agua, aplicando cualquier técnica adecuada para inducir cristalización, y aislando los pseudopolimorfos deseados.
Por técnicas para inducir cristalización se ha de entender aquellos procedimientos para la producción de cristales, que incluyen, entre otros, disolver o dispersar el compuesto de fórmula (X) en un medio disolvente, llevar la disolución o dispersión del compuesto de fórmula (X) y el disolvente o disolventes hasta una concentración deseada, llevar la mencionada disolución o dispersión hasta una temperatura deseada, efectuar cualquier presión adecuada, eliminar y/o separar cualquier material o impurezas indeseadas, secar los cristales formados para obtener los pseudopolimorfos en un estado sólido, si se desea tal estado.
Llevar la disolución o dispersión de compuesto de fórmula (X) y disolventes hasta una concentración deseada no implica necesariamente un incremento en la concentración del compuesto de fórmula (X). En ciertos casos, sería preferible una disminución o ningún cambio en la concentración. Por llevar la mencionada disolución o dispersión hasta una temperatura deseada, se entenderá los actos de calentar, enfriar o dejar a temperatura ambiente.
Las técnicas usadas para obtener una concentración deseada son aquellas habituales en la técnica, por ejemplo evaporación mediante destilación atmosférica, destilación a vacío, destilación fraccionada, destilación azeotrópica, evaporación de película, otras técnicas bien conocidas en la técnica, y sus combinaciones. Un procedimiento opcional para obtener una concentración deseada podría implicar asimismo la saturación de la disolución del compuesto de fórmula (X) y del disolvente, por ejemplo, añadiendo un volumen suficiente de un no disolvente a la disolución para alcanzar el punto de saturación. Otras técnicas adecuadas para saturar la disolución incluyen, a título de ejemplo, la introducción de compuesto de fórmula (X) adicional a la disolución y/o la evaporación de una porción del disolvente a partir de la disolución. Como se cita aquí, disolución saturada engloba disoluciones en sus puntos de saturación o que exceden sus puntos de saturación, es decir, sobresaturadas.
La eliminación y/o la separación de cualquier material o impurezas indeseadas se puede realizar mediante purificación, filtración, lavado, precipitación, o técnicas similares. La separación, por ejemplo, se podría llevar a cabo mediante técnicas de separación sólido-líquido conocidas. Los procedimientos de filtración conocidos por los expertos en la técnica se pueden usar igualmente en el presente procedimiento. Las filtraciones se pueden llevar a cabo, entre otros métodos, mediante centrifugación, o usando un filtro de tipo Buchner, un filtro de Rosenmund o placas, o una prensa de bastidor. Preferiblemente, la filtración en línea o filtración de seguridad se puede intercalar ventajosamente en los procedimientos descritos anteriormente, a fin de incrementar la pureza de la forma pseudopolimórfica resultante. Adicionalmente, también se pueden emplear agentes de filtración, tales como gel de sílice, Arbocel®, diatomita dicalita, o similar, para separar impurezas de los cristales de interés.
Los cristales obtenidos también se pueden secar, y tal procedimiento de secado se puede usar opcionalmente en las diferentes pasadas de cristalización, si se aplica más de una pasada de cristalización. Los procedimientos de secado incluyen todas las técnicas conocidas por aquellos de pericia en la técnica, tales como calentamiento, aplicación de vacío, aire o gas circulante, adición de un secante, liofilización, secado por pulverización, evaporación, o similar, o cualquier combinación de los mismos.
Los procedimientos para la cristalización de pseudopolimorfos del compuesto de fórmula (X) abarcan múltiples combinaciones de técnicas y sus variaciones. Como tales, y a título de ejemplo, la cristalización de pseudopolimorfos del compuesto de fórmula (X) se puede ejecutar disolviendo o dispersando el compuesto de fórmula (X) a una temperatura adecuada en el disolvente, con lo que se evapora una porción del mencionado disolvente, incrementando la concentración del compuesto de fórmula (X) en la mencionada disolución o dispersión, enfriando la mencionada mezcla, y opcionalmente lavando y/o filtrando y secando los cristales del solvato resultantes del compuesto de fórmula (X). Opcionalmente, los pseudopolimorfos del compuesto de fórmula (X) se pueden preparar disolviendo o dispersando el compuesto de fórmula (X) en un medio disolvente, enfriando dicha disolución de dispersión y filtrando y secando subsiguientemente el pseudopolimorfo obtenido. Otro ejemplo de preparación de solvatos del compuesto de fórmula (X) podría ser saturando el compuesto de fórmula (X) en el medio disolvente, y opcionalmente filtrando, lavando y secando los cristales obtenidos.
La formación de cristales puede implicar asimismo más de un procedimiento de cristalización. En ciertos casos, ventajosamente se pueden llevar a cabo una, dos o más etapas de cristalización extra por diferentes razones, tales como para incrementar la calidad del solvato resultante. Por ejemplo, los pseudopolimorfos de la presente invención se podrían preparar también añadiendo un disolvente a un material base de partida inicial del compuesto de fórmula (X), agitando la disolución a una temperatura fija hasta que las sustancias se disolviesen completamente, concentrando la disolución mediante destilación a vacío, y enfriando. Tendría lugar una primera cristalización, y los cristales formados se lavarían nuevamente con un disolvente, seguido de la disolución del compuesto de fórmula (X) con el disolvente para formar el pseudopolimorfo deseado. Se produciría la recristalización de la mezcla de reacción, seguido de una etapa de enfriamiento a partir del reflujo. El pseudopolimorfo formado se filtraría opcionalmente y se dejaría secar.
Disolviendo o dispersando el compuesto de fórmula (X) en el disolvente orgánico, en agua o en una mezcla de agua y disolventes orgánicos miscibles en agua, se pueden obtener diferentes grados de dispersión, tales como suspensiones, emulsiones, lechadas o mezclas; o preferiblemente se pueden obtener disoluciones homogéneas de una sola fase.
Opcionalmente, el medio disolvente puede contener aditivos, por ejemplo uno o más agentes dispersantes, tensioactivos u otros aditivos, o mezclas de los mismos del tipo normalmente usado en la preparación de suspensiones cristalinas y que están bien documentados en la bibliografía. Los aditivos se pueden usar ventajosamente para modificar la forma del cristal al incrementar la lenidad y disminuyendo el área superficial. El medio disolvente que contiene la disolución se puede agitar opcionalmente durante un cierto período de tiempo, o se puede agitar vigorosamente usando, por ejemplo, una mezcladora u homogeneizadora de alto cizallamiento, o una combinación de estas, para generar el tamaño de gotita deseado para el compuesto orgánico.
Los ejemplos de disolventes orgánicos útiles para la presente invención incluyen alcoholes de C1-C4 tales como metanol, etanol, isopropanol, butanol, 1-metoxi-2-propanol, y similares; cloroalcanos de C1-C4 , tales como diclorometano; cetonas de C1-C4 tales como acetona; éteres de C1-C4 tales como anisol, y similares; cicloéteres tales como tetrahidrofurano; ésteres de C1-C4 tales como acetato de etilo; sulfonatos de C1-C4 tales como mesilato, etanosulfonato, butanosulfonato, 2-metil-1-propanosulfonato; y similares.
Los ejemplos de mezclas de agua y disolventes orgánicos miscibles en agua incluyen mezclas de agua con todos los disolventes orgánicos enumerados anteriormente, con la condición de que sean miscibles en agua, por ejemplo etanol/agua, por ejemplo en una relación 50/50.
Los disolventes preferidos son aquellos disolventes farmacéuticamente aceptables. Sin embargo, los disolventes farmacéuticamente no aceptables también pueden encontrar su uso en la preparación de pseudopolimorfos farmacéuticamente aceptables.
En un método preferido, el disolvente es un disolvente farmacéuticamente aceptable, puesto que da como resultado un pseudopolimorfo farmacéuticamente aceptable. En un método más preferido, el disolvente es etanol.
En una realización particular, los pseudopolimorfos farmacéuticamente aceptables del compuesto de fórmula (X) se pueden preparar partiendo de formas pseudopolimórficas del compuesto de fórmula (X), que pueden no ser necesariamente farmacéuticamente aceptables. Por ejemplo, la Forma A se puede preparar partiendo de la Forma J. Los pseudopolimorfos también se pueden preparar partiendo de la forma amorfa.
En las mezclas de agua y disolventes orgánicos miscibles en agua, la cantidad de agua puede variar de alrededor de 5% en volumen a alrededor de 95% en volumen, preferiblemente de alrededor de 25% a alrededor de 75% en volumen, más preferiblemente de alrededor de 40% a alrededor de 60% en volumen.
También se debería observar que la calidad del disolvente orgánico seleccionado (absoluto, desnaturalizado, u otro) también influye en la calidad resultante del pseudopolimorfo.
El control de la temperatura de precipitación y de la siembra se puede usar adicionalmente para mejorar la reproducibilidad del procedimiento de cristalización, la distribución de tamaños de partículas, y la forma del producto. Como tal, la cristalización se puede efectuar sin sembrar con cristales del compuesto de la fórmula (X), o preferiblemente en presencia de cristales del compuesto de la fórmula (X), que se introducen en la disolución mediante siembra. La siembra se puede efectuar también varias veces a diversas temperaturas. La cantidad del material de siembra depende de la cantidad de la disolución, y se puede determinar fácilmente por una persona experta en la técnica.
El tiempo para la cristalización en cada etapa de cristalización dependerá de las condiciones aplicadas, de las técnicas empleadas y/o de los disolventes usados.
La ruptura de las partículas grandes o agregados de partículas tras la conversión del cristal se puede llevar a cabo adicionalmente a fin de obtener un tamaño de partículas deseado y homogéneo. En consecuencia, las formas de cristal de solvato del compuesto de fórmula (X) se muelen opcionalmente tras sufrir la conversión. Molienda o trituración se refiere a romper físicamente las partículas grandes o agregados de partículas usando métodos y aparatos bien conocidos en la técnica para la reducción del tamaño de partículas de los polvos. Los tamaños de partículas resultantes pueden oscilar desde milímetros hasta nanometros, produciendo por ejemplo nanocristales, microcristales.
El rendimiento del procedimiento de preparación de los pseudopolimorfos del compuesto de fórmula (X) puede ser 10% o más, y un rendimiento más preferido variaría de 40% a 100%.
De forma interesante, el rendimiento varía entre 70% y 100%.
De forma adecuada, los pseudopolimorfos de la presente invención tienen una pureza mayor que 90 por ciento. Más adecuadamente, los presentes pseudopolimorfos tienen una pureza mayor que 95 por ciento. Incluso más adecuadamente, los presentes pseudopolimorfos tienen una pureza mayor que 99 por ciento.
En un tercer aspecto, la presente invención se refiere a una formulación farmacéutica que comprende una cantidad terapéuticamente eficaz de un pseudopolimorfo del compuesto de fórmula (X), y un vehículo o diluyente farmacéuticamente aceptable del mismo.
En una realización, la presente invención se refiere al uso de formas pseudopolimórficas farmacéuticamente aceptables del compuesto de fórmula (X), preferiblemente Forma A, en la fabricación de un medicamento para tratar enfermedades provocadas por retrovirus, tales como infecciones por VIH, por ejemplo, síndrome de inmunodeficiencia adquirida (SIDA) y complejo relacionado con SIDA (ARC).
En otra realización, la presente invención proporciona un método para el tratamiento de una infección retroviral, por ejemplo, una infección por VIH, en un mamífero tal como un ser humano, que comprende administrar al mamífero que lo necesite una cantidad antirretroviral eficaz de una forma pseudopolimórfica farmacéuticamente aceptable del compuesto de fórmula (X), preferiblemente la Forma A.
La presente invención también se refiere a un método en el que el tratamiento de una infección viral por VIH comprende la reducción de la carga del VIH. La presente invención también se refiere a un método en el que el tratamiento de dicha infección vial por VIH comprende el incremento del recuento de células CD4+. La presente invención se refiere igualmente a un método en el que el tratamiento de dicha infección viral por VIH comprende inhibir la actividad de la proteasa del VIH en un mamífero.
Las formas pseudopolimórficas farmacéuticamente aceptables del compuesto de fórmula (X), preferiblemente la Forma A, también denominadas aquí como los ingredientes farmacéuticos activos, se pueden administrar mediante cualquier vía apropiada a la afección a tratar, preferiblemente oral. Sin embargo, se apreciará que la vía preferida puede variar, por ejemplo, con el estado del receptor.
Para cada una de las utilidades e indicaciones señaladas anteriormente, la cantidad requerida del ingrediente activo dependerá de un número de factores que incluyen la gravedad de la afección a tratar y la identidad del receptor, y finalmente estará a la discreción del médico o veterinario. La dosis deseada puede presentarse preferiblemente como una, dos, tres o cuatro o más subdosis, administradas a intervalos apropiados a lo largo del día.
Para una forma de administración oral, los pseudopolimorfos de la presente invención se mezclan con aditivos adecuados, tales como excipientes, estabilizantes o diluyentes inertes, y se llevan por medios de los métodos habituales a las formas de administración adecuadas, tales como comprimidos, comprimidos revestidos, cápsulas duras, disoluciones acuosas, alcohólicas u oleosas. Los ejemplos de vehículos inertes adecuados son goma arábiga, magnesia, carbonato de magnesio, fosfato de potasio, lactosa, glucosa, o almidón, en particular almidón de maíz. En este caso, la preparación se puede llevar a cabo tanto como gránulos secos y gránulos húmedos. Los excipientes o disolventes oleosos adecuados son aceites vegetales o animales, tales como aceite de girasol o aceite de hígado de bacalao. Los disolventes adecuados para disoluciones acuosas o alcohólicas son agua, etanol, disoluciones azucaradas, o sus mezclas. Los polietilenglicoles y polipropilenglicoles son también útiles como auxiliares adicionales para otras formas de administración.
Para la administración subcutánea o intravenosa, los pseudopolimorfos del compuesto de fórmula (X), si se desea con las sustancias habituales para los mismos tales como solubilizantes, emulsionantes o auxiliares adicionales, se llevan a disolución, suspensión o emulsión. Los pseudopolimorfos del compuesto de fórmula (X) también se pueden liofilizar, y los liofilizados obtenidos se pueden usar, por ejemplo, para la producción de preparaciones para inyección o infusión. Los disolventes adecuados son, por ejemplo, agua, disolución salina fisiológica, o alcoholes, por ejemplo etanol, propanol, glicerol, además también de disoluciones azucaradas tales como disoluciones de glucosa o de manitol, o como alternativa, mezclas de los diversos disolventes mencionados.
Las formulaciones farmacéuticas adecuadas para administración en forma de aerosoles o pulverizaciones son, por ejemplo, disoluciones, suspensiones o emulsiones de los pseudopolimorfos del compuesto de fórmula (X) en un disolvente farmacéuticamente aceptable, tal como etanol o agua, o una mezcla de tales disolventes. Si se requiere, la formulación también puede contener adicionalmente otros auxiliares farmacéuticos tales como tensioactivos, emulsionantes y estabilizantes, así como un propelente. Tal preparación contiene habitualmente el compuesto activo en una concentración de aproximadamente 0,1 a 50%, en particular de aproximadamente 0,3 a 3% en peso.
Los pseudopolimorfos de la presente invención también se pueden presentar en una formulación que comprende partículas del tamaño micrométrico, nanométrico o picométrico del pseudopolimorfo del compuesto de fórmula (X), formulación la cual puede contener otros agentes farmacéuticos y opcionalmente se puede convertir en forma sólida. Puede ser conveniente formular los presentes pseudopolimorfos en forma de nanopartículas que tienen un modificador de la superficie adsorbido sobre la superficie de las mismas en una cantidad suficiente para mantener un tamaño medio eficaz de partículas menor que 1000 nm. Se cree que los modificadores de la superficie útiles
incluyen aquellos que se adhieren físicamente a la superficie del agente antirretroviral pero no se unen químicamente al agente antirretroviral.
Puede ser conveniente además almacenar los pseudopolimorfos del compuesto de fórmula (X) en materiales de envasado que son protectores frente a peligros mecánicos, medioambientales, biológicos o químicos, o a la degradación. El acondicionamiento de las sustancias farmacéuticas se puede lograr empleando materiales de envasado impermeables a la humedad, tales como bolsas herméticas al vapor cerradas. El acondicionamiento de los productos farmacéuticos, tales como comprimidos, cápsulas, se puede lograr empleando, por ejemplo, blisteres de aluminio.
Se debería entender que, además de los ingredientes particularmente mencionados anteriormente, las formulaciones de esta invención incluyen otros agentes convencionales en la técnica que tienen relación con el tipo de formulación en cuestión, por ejemplo, aquellos adecuados para administración oral pueden incluir agentes saborizantes o agentes que enmascaran el sabor.
Ejemplo 1
La síntesis a escala industrial de la Forma A (1:1) se llevó a cabo usando las siguientes etapas. En primer lugar, se preparó una disolución con isopropanol y (1S,2R)-3-[[(4-aminofenil)sulfonil](isobutil)amino]-1-bencil-2-hidroxipropilcarbamato de (3R,3aS,6aR)-hexahidrofuro[2,3-b]furan-3-ilo. La disolución se concentró mediante destilación a vacío a 70°C y una presión de 200-500 mar, y se enfrió desde una T > 35° hasta una T entre 15° y 20°C durante alrededor de 10 horas. Los cristales formados se lavaron nuevamente con 13 litros de isopropanol, y se filtraron. Se llevó a cabo una recristalización subsiguiente en etanol/agua (90 litros/90 litros). Esto fue seguido de una nueva etapa de disolución, pero con 60 litros de etanol en su lugar. Se produjo la recristalización de la mezcla de reacción en etanol, seguido de una etapa de enfriamiento desde reflujo hasta -15°C aproximadamente y durante 10 horas. El etanolato formado se filtró y se dejó secar a alrededor de 50°C y alrededor de 7 mbares. El rendimiento de este procedimiento fue al menos 75%.
Ejemplo 2
En otro ejemplo, se preparó una mezcla de la Forma D y Forma B. Se usó acetona como disolvente durante el procedimiento de cristalización, para formar la Forma D. El procedimiento de cristalización comprendió entonces la etapa de agitar el compuesto de partida inicial (10 g) en 70 ml de acetona. La disolución se puso a reflujo subsiguientemente hasta que el compuesto se disolvió completamente. Se añadieron 40 ml de agua, y la disolución se enfrió subsiguientemente de forma lenta hasta la temperatura ambiente, y se agitó toda la noche. Los cristales formados se filtraron y se secaron en el horno de vacío a 50°C. Resultaron 7,6 g de producto a partir de la cristalización, siendo el rendimiento de este procedimiento de alrededor de 75%.
Ejemplo 3
En otro ejemplo, se prepararon cristales de la Forma J. Se usó isopropanol como disolvente durante el procedimiento de cristalización para formar la Forma J. El procedimiento de cristalización comprendió entonces la etapa de disolvente en material de partida inicial en el disolvente caliente. La disolución se enfrió subsiguientemente hasta la temperatura ambiente. Los cristales formados se filtraron y se secaron en el horno de vacío a 50°C. Los cristales contenían alrededor de 50% en moles de isopropanol.
Ejemplo 4
En este ejemplo, se calcularon las pérdidas de masa para diferentes pseudopolimorfos en experimentos termogravimétricos (TG). La termogravimetría es una técnica que mide el cambio de masa de una muestra a medida que se calienta, se enfría o se mantiene a temperatura constante. Se colocaron aproximadamente 2 a 5 mg de una muestra sobre una bandeja, y se insertaron en un horno de TG, modelo Netzsch Thermo-Microbalance TG 209, acoplado a un espectrómetro de Bruker FTIR vector 22. Las muestras se calentaron en una atmósfera de nitrógeno a una velocidad de 10°C/min., hasta una temperatura final de 250°C. El límite de detección de disolventes residuales estaba en el orden de 0,1% para la pérdida por etapas de disolventes distintos a lo largo de un intervalo estrecho de temperaturas (unos pocos grados Celsius).
Se obtuvieron los siguientes datos de TG:
Forma A: se observó una pérdida de peso de 4,2% en el intervalo de temperatura de 25-138°C (etanol un poco de agua) y de 6,9% (etanol CO2) en el intervalo de temperatura de 25-200°C. La velocidad de pérdida de etanol fue mínima a 120°C. La pérdida de CO2 fue debida a degradación química, y fue visible a alrededor de 190°C.
Forma B: se observó una pérdida de peso de 3,4% en el intervalo de temperatura de 25-78°C (agua) y de 5,1% en el intervalo de temperatura de 25-110°C (etanol agua para T > 78°C). A partir de 110-200°C se perdieron otros 1,1% en peso (etanol).
Forma C: se observó una pérdida de peso de 2,1% en el intervalo de temperatura de 25-832C (agua metanol) y de 4,2% en el intervalo de temperatura de 25-105°C (metanol para T > 83°C, etapa distinta). Desde 105-200°C, se perdieron otros 2,1% en peso (metanol). No se observó etanol en la fase gaseosa.
Forma D: se observó una pérdida de peso de 0,1% en el intervalo de temperatura de 25-50°C, de 4,2% en el intervalo de temperatura de 25-108°C (acetona etanol para T > 50°C), de 8,2% en el intervalo de temperatura de 25-157°C (acetona etanol para T > 108°C), y de 10,5% en el intervalo de temperatura de 25-240°C (acetona etanol para T > 157°C).
Forma E: se observó una pérdida de peso de 0,2% en el intervalo de temperatura de 25-75°C (agua), de I , 8% en el intervalo de temperatura de 25-108°C (diclorometano etanol para T > 75°C), de 6,8% en el intervalo de temperatura de 25-157°C (diclorometano etanol para T > 108°C), y de 8,8% en el intervalo de temperatura de 25-240°C (diclorometano etanol para T > 157°C).
Forma F: se observó una pérdida de peso de 0,1% en el intervalo de temperatura de 25-50°C (probablemente agua), de 1,7% en el intervalo de temperatura de 25-108°C (acetato de etilo etanol para T > 50°C), de 6,6% en el intervalo de temperatura de 25-157°C (acetato de etilo etanol para T > 108°C), y de 9% en el intervalo de temperatura de 25-240°C (acetato de etilo etanol para T > 157°C).
Forma G: se observó una pérdida de peso de 0,0% en el intervalo de temperatura de 25-50°C, de 3,7% en el intervalo de temperatura de 25-108°C (1-metoxi-2-propanol etanol para T > 50°C, etapa distinta), de 8% en el intervalo de temperatura de 25-157°C (1-metoxi-2-propanol etanol para T > 108°C), y de 12,5% en el intervalo de temperatura de 25-240°C (1-metoxi-2-propanol etanol para T > 157°C).
Forma H: se observó una pérdida de peso de 0,8% en el intervalo de temperatura de 25-100°C (anisol un poco de etanol) y de 8,8% en el intervalo de temperatura de 25-200°C (anisol etanol para T > 100°C).
Forma I: se observó una pérdida de peso de 0,3% en el intervalo de temperatura de 25-89°C (agua) y de I I , 0% en el intervalo de temperatura de 25-200°C (tetrahidrofurano para T > 89°C). No se observó etanol en la fase gaseosa.
La Tabla 10 muestra las pérdidas de masa esperadas aproximadas para diferentes Formas en experimentos termogravimétricos (TG).
Pérdida de masa en % (M x.LM = 100%)

En otro conjunto de métodos termogravimétricos, la Forma A, la Forma A después de la adsorción/desorción, y la Forma A después de ensayos de hidratación de adsorción/desorción, se transfirieron todas ellas a una bandeja de aluminio para muestras. La curva TG se registró en un termogravímetro TA Instrument Hi-Res TGA 2950 en las siguientes condiciones:
■ temperatura inicial: temperatura ambiente
■ velocidad de calentamiento: 20°C/min.
■ factor de resolución: 4
■ condición final: 300°C o <80[% (p/p)]
Las curvas TG de las muestras se recogen en la Figura 16.
La Tabla 11 muestra pérdidas de masas para las formas ensayadas:

La pérdida de peso a las temperaturas hasta 80°C es debida principalmente a la evaporación del disolvente (agua) presente en la muestra. La pérdida de peso a temperaturas por encima de 80°C es debida principalmente a la evaporación del disolvente (etanolato) presente en la muestra.
En la Figura 17 se recoge una curva TG de la forma A a 25°C en una atmósfera de nitrógeno seco en función del tiempo. La pérdida de peso a 25°C después de 10 horas fue alrededor de 0,6%. Esto fue debido a la evaporación del disolvente.
Ejemplo 5
En otro ejemplo, también se llevaron a cabo medidas de calorimetría de barrido diferencial (DSC). Para tal fin, se usó un sistema de análisis térmico Perkin Elmer DSC 204. Se pesaron exactamente de 2 a 5 mg de muestra de Forma A en una bandeja de DSC.
Los experimentos se realizaron en una bandeja abierta. La muestra se equilibró hasta aproximadamente 30°C y después se calentó a una velocidad de 10°C por minuto, hasta una temperatura final de 200°C. El dato de DSC se obtuvo siguiendo un método estándar en la técnica. La Forma A se caracterizó mediante calorimetría de barrido diferencial (DSC), en la que mostró una fuerte endotermia en el intervalo 80-119°C, mostrando un pico a alrededor de 105,6°C, con un comienzo de delta H = -98,33 J/g. En consecuencia, la Forma A de cristal de solvato de etanol del compuesto de fórmula (X) (1:1) mostró el patrón termográfico que aparece en la FIGURA 8.
En otro conjunto de medidas de DSC, se examinaron la Forma A, la Forma A tras adsorción/desorción, y la Forma A tras ensayos de hidratación de adsorción/desorción. Se transfirieron alrededor de 3 mg de las muestras a una bandeja de muestras Perkin Elmer de aluminio perforada de 30 ml. La bandeja de muestras se cerró con una cubierta apropiada, y la curva de DSC se registró en un Perkin Elmer Pyris DSC, en las siguientes condiciones:
■ temperatura inicial: 25°C
■ velocidad de calentamiento: 10°C/min.
■ temperatura final: 150C
■ caudal de nitrógeno: 30 ml/min.
La Forma A mostró una señal endotérmica a alrededor de 104,6°C y un calor de fusión de 95,8 J/g provocado por la evaporación del etanolato y la fusión del producto. La Forma A tras ADS/DES mostró una señal endotérmica ancha debido a una mezcla de Forma A de etanolato y Forma B hidratada. La Forma A tras ensayo de hidratación de ADS/DES mostró una señal endotérmica a alrededor de 73,5°C y un calor de fusión de 126 J/g provocado por la evaporación de agua y la fusión del producto. Las curvas termográficas se representan en la FIGURA 15.
Ejemplo 6
En otro ejemplo, se realizaron estudios de estabilidad de la Forma A en tres condiciones diferentes. Incluyeron condiciones de 25°C y 60% de RH, 40°C y 75% de RH, y 50°C. Estos estudios revelaron que a la estabilidad a largo plazo a 25°C y 60% de RH, la cantidad de etanol y agua es estable.
La Tabla 12 muestra el estudio de estabilidad para la Forma A. Estabilidad a largo plazo a 252C/60% de RH (humedad relativa), con botellas de vidrio marrón como recipiente de muestras

Ejemplo 7
Ensayos de adsorción-desorción
Se transfirieron alrededor de 23 mg de la Forma A en un analizador de sorción de vapor VTI modelo SGA100, y se registró el cambio de peso con respecto a la humedad atmosférica en las siguientes condiciones:
■ temperatura de secado: 40°C
■ equilibrio: < 0,05% en 5 min. o 60 min.
■ intervalo de datos: 0,05% o 2 min.
■ temperatura: 25°C
■ adsorción de RH (%) de primer ciclo: 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 95
desorción de RH (%): 95, 90, 80, 70, 60, 50, 40, 30, 20, 10, 5
■ adsorción de RH (%) de segundo ciclo: 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 95
desorción de RH (%): 95, 90, 80, 70, 60, 50, 40, 30, 20, 10, 5
En la etapa de secado, se registró una pérdida de peso de alrededor de 0,6%. El producto seco obtenido no fue higroscópico, adsorbió hasta 0,7% de agua a una humedad relativa elevada. Durante el ciclo de desorción, se registró una pérdida de peso de 1,4%, indicando esto que el producto fue perdiendo etanolato. El producto obtenido tras ADS/DES fue una mezcla de forma de etanolato y forma hidratada.
La curva de ADS/DES se recoge en la Figura 18.
Ensayos de hidratación de adsorción-desorción
Se transfirieron alrededor de 23 mg de la Forma A en un analizador de sorción de vapor VTI modelo SGA100, y se registró el cambio de peso con respecto a la humedad atmosférica en las siguientes condiciones:
■ equilibrio: < 0,0005% en 5 min. o 90 min.
■ intervalo de datos: 0,05% o 2 min.
■ temperatura: 25°C
■ adsorción/desorción de RH (%) del ciclo: 5,95
repetir el ciclo 11 veces
Al final de este ensayo, se registró una pérdida de peso de 5,2%. Esto fue comparable con el resultado de TG (TG 5,4% hasta 80°C). La forma de etanolato se transfirió a una forma de hidrato. Las curvas del ensayo de hidratación de ADS/DES se recogen en la Figura 19.
Ejemplo 8
Se estudió la estabilidad de la Forma A tras el almacenamiento del compuesto en un recipiente de muestras con una cubierta interna hecha de LD-PE solamente (sellado de cuerda), y una cubierta externa hecha de PETP/Alu/PE (Moplast) sellada por calor. Se llevó a cabo un estudio de estabilidad a largo plazo a 25°C/60% de RH, y un estudio de estabilidad acelerada a 40°C/75% de RH, durante un período de 6 meses, y las muestras se analizaron en diferentes puntos de tiempo como se muestra en las siguientes tablas.
Tabla 13: Estabilidad a largo plazo a 252C/60% de RH

Tabla 14: Estabilidad acelerada a 40°C/75% de RH

La Forma A mostró estabilidad química y cristalográfica en las condiciones mencionadas en las tablas 13 y 14.
Ejemplo 9
Se estudió la estabilidad de la Forma A tras el almacenamiento del compuesto en un recipiente de muestras con una cubierta interna hecha solamente de LD-PE (sellado de cuerda), y una cubierta externa hecha de una bolsa Vapor Loc (LPS) selladas térmicamente. Se llevó a cabo un estudio de estabilidad a largo plazo a 25°C/60% de RH, y un estudio de estabilidad acelerada a 40°C/75% de RH, durante un período de 6 meses, y las muestras se analizaron en diferentes puntos de tiempo como se muestra en las siguientes tablas.
Tabla 15: Estabilidad a largo plazo a 252C/60% de RH

Tabla 16: Estabilidad acelerada a 40°C/75% de RH

La Forma A mostró estabilidad química y cristalográfica en las condiciones mencionadas en las tablas 15 y 16.
Ejemplo 10
Para los fines del ensayo de estabilidad química, la Forma A se almacenó durante un período de 1, 4 y 8 semanas en diferentes condiciones. Estas condiciones fueron 40°C/75% de RH, 50°C, RT/<5% de RH, R t / 5 6 % de RH, RT/75% de RH y luz 0,3 da ICH. El compuesto se analizó después del almacenamiento, mediante HPLC y mediante inspección visual. El método de HPLC usado en este estudio fue el método 909 de HPLC. Los resultados de los ensayos se dan en la siguiente tabla.
Tabla 17

Se concluyó que la Forma A es químicamente estable tras el almacenamiento en todas las condiciones investigadas.
Ejemplo 11
Se caracterizaron diferentes fracciones de la Forma B con termogravimetría (TG), calorimetría de barrido diferencial (DSC) y espectroscopía de infrarrojos (IR). Los resultados de los ensayos se dan a conocer en la siguiente tabla.
Tabla 18

Ejemplo 12
Se investigó la adsorción y desorción de agua a 25°C en diferentes condiciones de humedad relativa sobre 38 mg de Forma B. Se registró el cambio de peso como función de la humedad relativa. Los resultados se presentan en la Figura 20. En la etapa de secado, se registró una pérdida de peso de alrededor de 5,6% para la Forma B. El producto seco obtenido fue higroscópico, adsorbió hasta 6,8% de agua a una humedad relativa elevada. Después del ciclo de desorción, permaneció en la muestra alrededor de 1,2% de agua. El producto obtenido tras ADS/DES fue una mezcla de producto hidratado y amorfo.
Ejemplo 13
Se midieron las solubilidades acuosas de la Forma B en disolventes con diferente pH. Un exceso del soluto se equilibró con el disolvente a 20°C durante al menos 24 horas. Tras eliminar el compuesto sin disolver, se determinó la concentración en disolución usando espectrometría UV.
Tabla 19

Ejemplo 14
Se estudió la estabilidad de la estructura de cristal de la Forma B tras almacenar el compuesto durante un período de dos semanas a temperatura ambiente (RT) a <5%, 56% y 75% de humedad relativa (RH), 50°C y 40°C/75% de RH. Las muestras se analizaron con termogravimetría (TG), calorimetría de barrido diferencial (DSC), espectroscopia infrarroja (IR) y difracción de rayos X (XRD). Los resultados de los ensayos se dan a conocer en la siguiente tabla.
Tabla 20

Ejemplo 15
En el programa del ensayo de estabilidad química, la Forma B se almacenó durante un período de 1, 4 y 9 semanas en diferentes condiciones. Estas condiciones fueron 40°C/75% de RH, 50°C, RT/<5% de RH, R t / 5 6 % de RH, RT/75% de RH y luz 0,3 da ICH. El compuesto se analizó mediante HPLC y mediante inspección visual tras el almacenamiento. El método de HPLC usado en este estudio fue el método 909 de HPLC. Los resultados de los ensayos se dan a conocer en la siguiente tabla, a partir de la cual se concluye que la Forma B es químicamente estable.
Tabla 21

Ejemplo 16
La Forma K se preparó añadiendo ácido metanosulfónico puro a una disolución de Forma A en THF a r.t. La Forma K se mezcló subsiguientemente con haluro de metal alcalino y se prensó en un pelete (Ph. Eur.) y se analizó mediante espectrometría infrarroja (IR) en las siguientes condiciones:
■ aparato: espectrofotómetro Nicolet Magna 560 FTIR
■ número de barridos: 32
■ resolución: 1 cm-1
■ intervalo de longitudes de onda: 4000 a 400 cm-1
■ corrección de la línea base: sí
■ detector: DTGS con ventanas de KBr
■ divisor de haces: Ge en KBr
■ haluro de metal alcalino: KBr (bromuro de potasio)
El espectro de IR de la Forma K, como se muestra en la Figura 21, refleja los modos vibracionales de la estructura molecular del solvato de mesilato como un producto cristalino.
Tabla 22

Ejemplo 17
La Forma K se transfirió a una celda capilar de vidrio y se analizó mediante espectrometría de Raman en las siguientes condiciones:
■ modo de Raman: Raman no dispersante
■ aparato: módulo Nicolet FT-Raman
■ número de barridos: 64
■ resolución: 4 cm-1
■ intervalo de longitudes de onda: 3700 a 100 cm-1
■ láser: Nd:YV04
■ frecuencia del láser: 1064 cm-1
■ detector: InGaAs
■ separador de haces: CaF2
■ geometría de la muestra: reflectante 180°
■ polarización: no
El espectro de Raman de la Forma K, como se muestra en la Figura 22, refleja los modos vibracionales de la estructura molecular del mesilato como un producto cristalino.
Tabla 23

Ejemplo 18
Se transfirieron alrededor de 3 mg de Forma K a una bandeja de muestras de aluminio estándar TA-Instrument. La bandeja de muestras se cerró con la cubierta apropiada, y la curva de DSC se registró en un TA-Instruments Q1000 MTDSC equipado con una unidad de refrigeración RCS, en las siguientes condiciones:
■ temperatura inicial: 25°C
■ velocidad de calentamiento: 10°C/min.
■ temperatura final: 200°C
■ caudal de nitrógeno: 50 ml/min.
La curva de DSC, como se representa en la Figura 23, muestra la fusión con descomposición de un producto cristalino. La fusión de la forma K se produce a 158,4°C. Debido a la descomposición, el cálculo del calor de fusión sólo se puede usar para indicar la propiedad cristalina del producto.
Ejemplo 19
La Forma K se transfirió a una bandeja de muestras de aluminio. La curva TG se registró en un termogravímetro TA Hi-Res TGA 2950 en las siguientes condiciones:
■ temperatura inicial: temperatura ambiente
■ velocidad de calentamiento: 20°C/min.
■ factor de resolución: 4
■ condición final: 300°C o <80[% (p/p)]
La curva TG se muestra en la Figura 24. La pérdida de peso de alrededor de 0,2% hasta 60°C fue debida a la evaporación del disolvente. La pérdida de peso a temperaturas por encima de 140°C fue debida a la evaporación y descomposición del producto.
Ejemplo 20
Adsorción-desorción
Se transfirieron alrededor de 21 mg de la Forma K a un analizador de sorción de vapor VTI modelo SGA100, y se registró el cambio de peso con respecto a la humedad atmosférica en las siguientes condiciones:
temperatura de secado: 40°C
equilibrio: < 0,05% en 5 min. o 60 min.
intervalo de datos: 0,05% o 2,0 min.
temperatura: 25°C
primer ciclo adsorción de RH (%): 5, 10, 20, 30, 40, 50, 60, 70, 80, 90,
95
desorción de RH (%): 95, 90, 80, 70, 60, 50, 40, 30, 20, 10,
5
segundo ciclo adsorción de RH (%): 5, 10, 20, 30, 40, 50, 60, 70, 80, 90,
95
desorción de RH (%): 95, 90, 80, 70, 60, 50, 40, 30, 20, 10,
5
La isoterma de adsorción-desorción se muestra en la Figura 25. La Forma K es higroscópica. En la etapa de secado inicial, se registró una pérdida de peso de 0,3%, comparable con el resultado de TG. La Forma K adsorbió hasta 1,5% de agua a una humedad relativa elevada. El producto se secó completamente durante el ciclo de desorción. Se investigó un estudio diferente de la adsorción y desorción de agua por la Forma K a 25°C a diferentes condiciones de humedad relativa sobre una cantidad de alrededor de 18 mg del solvato de mesilato. Se registró el cambio de peso en función de la humedad relativa. El resultado se presenta en la Figura 26.
En la etapa de secado, se registra una pérdida de peso de alrededor de 0,6% para la Forma K. El producto seco obtenido es ligeramente higroscópico, adsorbió hasta 1,7% de agua a humedad relativa elevada. El producto se secó completamente durante el ciclo de desorción.
Ejemplo 21
Se midieron las solubilidades acuosas de la Forma K en disolventes con diferente pH. Un exceso del soluto de libró con el disolvente a 20°C durante al menos 48 horas. Después de eliminar el compuesto sin disolver, se determinó la concentración en disolución usando espectrometría de UV.
Tabla 24

Ejemplo 22
Se estudió la estabilidad de la estructura de cristal del lote 1 de la Forma K tras el almacenamiento del compuesto durante un período de cuatro semanas a temperatura ambiente (RT) a 75% de humedad relativa (RH), 50°C y 40°C/75% de RH. Se estudió la estabilidad de la estructura de cristal del lote 2 de la Forma K tras el almacenamiento del compuesto durante un período de cuatro semanas a temperatura ambiente (RT) a <5%, 56% y 75% de humedad relativa (RH), 50°C y 40°C/75% de RH. Las muestras se analizaron con termogravimetría (TG), calorimetría de barrido diferencial (DSC) y espectroscopía infrarroja (IR). Los resultados de los ensayos se dan a conocer en la siguiente tabla.
Tabla 25

Ejemplo 23
En el programa de ensayo de estabilidad química, el lote 1 de la Forma K se almacenó durante un período de 1 y 4 semanas en diferentes condiciones. Estas condiciones fueron 40°C/75% de RH, 50°C, RT/75% de RH y luz 0,3 da ICH. El lote 2 de la Forma K también se almacenó durante un período de 1 y 4 semanas en diferentes condiciones. Estas condiciones fueron 40°C/75% de RH, 50°C, RT/<5% de R h , RT/56% de RH, RT/75% de RH y luz 0,3 da ICH. El compuesto se analizó mediante HPLC y mediante inspección visual tras del almacenamiento. El método de HPLC usado en este estudio fue el método 909 de HPLC. Los resultados de los ensayos se dan a conocer en la siguiente tabla.
Tabla 26

Ejemplo 24
Se llevó a cabo un ensayo aleatorizado, controlado con placebo, bienmascarado, de aumento de escala de múltiples dosis para examinar la seguridad, tolerabilidad y farmacocinética de la Forma A tras la administración oral dos o tres veces al día, en sujetos sanos. Se ensayaron cuatro dosis de la Forma A (400 mg b.i.d., 800 mg b.i.d., 800 mg t.i.d., y 1200 mg t.i.d.) en 4 paneles de 9 sujetos sanos. Dentro de cada panel, se trataron 6 sujetos con la Forma A y 3 sujetos con placebo durante 13 días con una sola ingesta en la mañana del día 14. (b.i.d. = dos veces al día, t.i.d. = tres veces al día).
La Forma A se absorbió fácilmente, y los perfiles de concentración frente al tiempo de la Forma A tras la dosificación repetida fueron dependientes de la dosis administrada. Las concentraciones plasmáticas de estado estacionario se alcanzaron generalmente a los 3 días, aunque Cüh (conc. en el momento de la administración) y AUC24h (área bajo la
curva o biodisponibilidad) disminuyeron ligeramente con el tiempo a todos los niveles de dosis. AUC24h y Css,av (conc. a estado estacionario medio) fueron proporcionales a la dosis (dosis diaria) a 400 mg b.i.d., 800 mg t.i.d. y 1200 mg t.i.d., pero fue más proporcional a la dosis a 800 mg b.i.d. Cmax (conc. máxima) fue proporcional a la dosis con respecto a la dosis por ingesta. Se excretó menos de 2% de Forma A sin cambiar en la orina a todos los niveles de dosis.
Claims (23)
1. Un pseudopolimorfo de etanolato de (1S,2R)-3-[[(4-aminofenil)sulfonil](isobutil)amino]-1-bencil-2-hidroxipropilcarbamato de (3R,3aS,6aR)-hexahidrofuro[2,3-b]furan-3-ilo para su uso en el tratamiento de una infección retroviral.
2. Un pseudopolimorfo según la reivindicación 1, donde la infección retroviral es una infección por VIH.
3. Un pseudopolimorfo según la reivindicación 1 o 2, donde el tratamiento comprende administrar una dosis por día al paciente.
4. Un pseudopolimorfo según la reivindicación 1 o 2, donde el tratamiento comprende administrar dos subdosis al paciente en intervalos apropiados a lo largo del día.
5. Un pseudopolimorfo según cualquiera de las reivindicaciones 1 a 4, en el que la relación de compuesto a etanol oscila entre (5:1) y (1:5).
6. Un pseudopolimorfo según cualquiera de las reivindicaciones 1 a 4, en el que la relación de etanol a compuesto oscila entre (0,2:1) y (3:1).
7. Un pseudopolimorfo según cualquiera de las reivindicaciones 1 a 4, en el que la relación de etanol a compuesto oscila entre (1:1) y (2:1).
8. Un pseudopolimorfo según cualquiera de las reivindicaciones 1 a 4, en el que la relación de compuesto a etanol es alrededor de 1:1.
9. Un pseudopolimorfo según cualquiera de las reivindicaciones 1 a 8, en el que el pseudopolimorfo tiene una pureza superior a un 90%.
10. Un pseudopolimorfo según cualquiera de las reivindicaciones 1 a 8, en el que el pseudopolimorfo tiene una pureza superior a un 95%.
11. Un pseudopolimorfo según cualquiera de las reivindicaciones 1 a 8, en el que el pseudopolimorfo tiene una pureza superior a un 99%.
12. Un pseudopolimorfo según cualquiera de las reivindicaciones 1 a 11, que comprende adicionalmente moléculas de agua en la estructura cristalina.
13. Un pseudopolimorfo de hidrato de (1S,2R)-3-[[(4-aminofenil)sulfonil](isobutil)amino]-1-bencil-2-hidroxipropilcarbamato de (3R,3aS,6aR)-hexahidrofuro[2,3-b]furan-3-ilo para su uso en el tratamiento de una infección retroviral.
14. Un pseudopolimorfo según la reivindicación 13, donde la infección retroviral es una infección por VIH.
15. Un pseudopolimorfo según la reivindicación 13 o 14, donde el tratamiento comprende administrar una dosis por día al paciente.
16. Un pseudopolimorfo según la reivindicación 13 o 14, donde el tratamiento comprende administrar dos subdosis al paciente en intervalos apropiados a lo largo del día.
17. Un pseudopolimorfo según cualquiera de las reivindicaciones 13 a 16, en el que la relación de compuesto a agua oscila entre (5:1) y (1:5).
18. Un pseudopolimorfo según cualquiera de las reivindicaciones 13 a 16, en el que la relación de agua a compuesto oscila entre (0,2:1) y (3:1).
19. Un pseudopolimorfo según cualquiera de las reivindicaciones 13 a 16, en el que la relación de agua a compuesto oscila entre (1:1) y (2:1).
20. Un pseudopolimorfo según cualquiera de las reivindicaciones 13 a 16, en el que la relación de compuesto a agua es alrededor de 1:1.
21. Un pseudopolimorfo según cualquiera de las reivindicaciones 13 a 20, en el que el pseudopolimorfo tiene una pureza superior a un 90%.
22. Un pseudopolimorfo según cualquiera de las reivindicaciones 13 a 20, en el que el pseudopolimorfo tiene una pureza superior a un 95%.
23. Un pseudopolimorfo según cualquiera de las reivindicaciones 13 a 20, en el que el pseudopolimorfo tiene una pureza superior a un 99%.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP02076929 | 2002-05-16 | ||
| EP02076929 | 2002-05-16 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| ES2503551T3 ES2503551T3 (es) | 2014-10-07 |
| ES2503551T5 true ES2503551T5 (es) | 2021-10-28 |
Family
ID=29724454
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14168686.5T Expired - Lifetime ES2638412T3 (es) | 2002-05-16 | 2003-05-16 | Formas pseudopolimórficas de un inhibidor de la proteasa del HIV |
| ES03753571T Expired - Lifetime ES2498370T5 (es) | 2002-05-16 | 2003-05-16 | Formas pseudopolimórficas de un inhibidor de la proteasa de VIH |
| ES10180831T Expired - Lifetime ES2503551T5 (es) | 2002-05-16 | 2003-05-16 | Formas pseudopolimórficas de un inhibidor de la proteasa de VIH |
| ES16158480T Expired - Lifetime ES2728735T3 (es) | 2002-05-16 | 2003-05-16 | Formas pseudopolimórficas de un inhibidor de la proteasa del HIV |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14168686.5T Expired - Lifetime ES2638412T3 (es) | 2002-05-16 | 2003-05-16 | Formas pseudopolimórficas de un inhibidor de la proteasa del HIV |
| ES03753571T Expired - Lifetime ES2498370T5 (es) | 2002-05-16 | 2003-05-16 | Formas pseudopolimórficas de un inhibidor de la proteasa de VIH |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16158480T Expired - Lifetime ES2728735T3 (es) | 2002-05-16 | 2003-05-16 | Formas pseudopolimórficas de un inhibidor de la proteasa del HIV |
Country Status (25)
| Country | Link |
|---|---|
| US (8) | US7700645B2 (es) |
| EP (4) | EP2314591B2 (es) |
| JP (1) | JP4864320B2 (es) |
| KR (2) | KR101128370B1 (es) |
| CN (1) | CN100475819C (es) |
| AP (1) | AP2052A (es) |
| AU (2) | AU2003271740B2 (es) |
| BR (1) | BRPI0311176B8 (es) |
| CA (1) | CA2485834C (es) |
| CY (3) | CY1117928T1 (es) |
| DK (3) | DK2314591T4 (es) |
| EA (1) | EA007120B8 (es) |
| ES (4) | ES2638412T3 (es) |
| HR (1) | HRP20041061B1 (es) |
| HU (1) | HUE034389T2 (es) |
| IL (1) | IL165140A0 (es) |
| LT (1) | LT2767539T (es) |
| MX (1) | MXPA04011427A (es) |
| NO (1) | NO331477B1 (es) |
| NZ (1) | NZ536497A (es) |
| PL (1) | PL215151B1 (es) |
| PT (3) | PT2767539T (es) |
| SI (3) | SI2314591T2 (es) |
| WO (1) | WO2003106461A2 (es) |
| ZA (1) | ZA200410154B (es) |
Families Citing this family (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE500823T1 (de) | 1998-06-23 | 2011-03-15 | Us Of America Represented By The Secretary Dept Of Health And Human Services | Arzneimittel zur behandlung von hiv-infizierten säugetieren |
| EA007120B8 (ru) | 2002-05-16 | 2012-03-30 | Тиботек Фармасьютикалз Лтд. | Псевдополиморфные формы ингибитора вич-протеазы |
| HRP20020614A2 (en) * | 2002-07-22 | 2004-06-30 | PLIVA-ISTRAŽIVAČKI INSTITUT d.o.o. | Rhombic pseudopolymorph of 9-deoxo-9a-aza-9a-methyl-9a-homoerythromycin a |
| JP4818124B2 (ja) | 2003-12-23 | 2011-11-16 | テイボテク・フアーマシユーチカルズ・リミテツド | (3R,3aS,6aR)−ヘキサヒドロフロ〔2,3−b〕フラン−3−イル(1S,1R)−3−〔〔(4−アミノフェニル)スルホニル〕(イソブチル)アミノ〕−1−ベンジル−2−ヒドロキシプロピルカルバマートの製造方法 |
| KR101333627B1 (ko) | 2005-02-25 | 2013-11-27 | 얀센 알 앤드 디 아일랜드 | 프로테아제 저해제 전구체 합성 |
| RU2464266C2 (ru) * | 2006-11-09 | 2012-10-20 | Тиботек Фармасьютикалз Лтд. | СПОСОБЫ ПОЛУЧЕНИЯ ГЕКСАГИДРОФУРО[2,3-b] ФУРАН-3-ОЛА |
| AR073248A1 (es) | 2008-09-01 | 2010-10-20 | Tibotec Pharm Ltd | Proceso para la preparacion de (1s, 2r)-3- ((4-aminofenil) sulfonil) ( isobutil) amino)-1- bencil-2- hidroxipropilcarbamato de (3r, 3as,6ar)- hexahidrofuro-(2,3-b) furan-3- ilo (darunavir) y compuestos intermediarios utiles en dicho proceso. |
| US8921415B2 (en) | 2009-01-29 | 2014-12-30 | Mapi Pharma Ltd. | Polymorphs of darunavir |
| MX2011007790A (es) * | 2009-01-29 | 2011-11-18 | Mapi Pharma Ltd | Polimorfos de darunavir. |
| EP2477992B1 (en) * | 2009-09-17 | 2016-12-14 | Mylan Laboratories Limited | Processes for the preparation of darunavir and the amorphous form thereof |
| EP2493891A2 (en) | 2009-10-30 | 2012-09-05 | Lupin Limited | A novel process for preparation of darunavir and darunavir ethanolate of fine particle size |
| PT2513116E (pt) * | 2009-12-16 | 2015-10-14 | Hetero Research Foundation | Polimorfos de darunavir |
| EP2521728B1 (en) * | 2010-01-05 | 2016-09-07 | Cipla Limited | Crystalline darunavir hydrate and process for preparation thereof |
| MX2012008627A (es) | 2010-01-28 | 2012-09-21 | Mapi Pharma Ltd | Proceso para la preparacion de darunavir e intermedios de darunavir. |
| US8853430B2 (en) | 2010-05-20 | 2014-10-07 | Hetero Research Foundation | Crystalline hydrochloride salt of darunavir |
| WO2012107889A1 (en) | 2011-02-10 | 2012-08-16 | Ranbaxy Laboratories Limited | Process for the preparation of amorphous darunavir |
| SI2729130T1 (en) | 2011-07-07 | 2018-03-30 | Janssen Sciences Ireland Uc | Combined formulations of darunavir |
| WO2013114382A1 (en) | 2011-12-05 | 2013-08-08 | Mylan Laboratories Ltd | Crystalline darunavir |
| WO2013108105A2 (en) * | 2012-01-18 | 2013-07-25 | Aurobindo Pharma Limited | Novel solvates of darunavir |
| CN103509031B (zh) * | 2012-06-20 | 2016-04-27 | 上海迪赛诺药业有限公司 | 制备达芦那韦无定形物的方法 |
| ES2848216T3 (es) * | 2012-07-24 | 2021-08-05 | Laurus Labs Ltd | Un proceso para la preparación de Darunavir |
| CA2889903C (en) | 2012-10-29 | 2021-03-09 | Manjinder Singh Phull | Antiviral phosphonate analogues and process for preparation thereof |
| US9346820B2 (en) * | 2013-09-11 | 2016-05-24 | Purdue Research Foundation | HIV-1 protease inhibitors having gem-di-fluoro bicyclic P2-ligands |
| WO2016092525A1 (en) * | 2014-12-12 | 2016-06-16 | Lupin Limited | Darunavir n-propanol solvate and process for preparation thereof |
| WO2016092527A1 (en) * | 2014-12-12 | 2016-06-16 | Sun Pharmaceutical Industries Limited | A process for the preparation of dolutegravir |
| DK3271347T3 (da) | 2015-03-19 | 2022-07-18 | Mycovia Pharmaceuticals Inc | Antifungale forbindelser og fremstillingsfremgangsmåder |
| WO2018029565A1 (en) | 2016-08-08 | 2018-02-15 | Hetero Labs Limited | A multi-class anti-retroviral composition |
| US11045423B2 (en) | 2016-08-08 | 2021-06-29 | Hetero Labs Limited | Anti-retroviral compositions |
| ES2881949T3 (es) | 2016-10-27 | 2021-11-30 | Gilead Sciences Inc | Forma cristalina de base libre de darunavir |
| CN108727401A (zh) * | 2017-04-20 | 2018-11-02 | 盐城迪赛诺制药有限公司 | 达鲁那韦新晶型及其制备方法和应用 |
| CN109053753A (zh) * | 2018-08-05 | 2018-12-21 | 浙江大学 | 一种制备达卢那韦二水合物晶型的方法 |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5413999A (en) * | 1991-11-08 | 1995-05-09 | Merck & Co., Inc. | HIV protease inhibitors useful for the treatment of AIDS |
| JPH05230044A (ja) | 1992-02-21 | 1993-09-07 | Hoechst Japan Ltd | ピレタニドの新規な結晶多形 |
| US5968942A (en) * | 1992-08-25 | 1999-10-19 | G. D. Searle & Co. | α- and β-amino acid hydroxyethylamino sulfonamides useful as retroviral protease inhibitors |
| ATE174587T1 (de) * | 1993-08-24 | 1999-01-15 | Searle & Co | Hydroxyaminosulfonamide verwendbar als inhibitoren retroviraler proteasen |
| ATE194607T1 (de) * | 1995-02-22 | 2000-07-15 | Hoechst Japan | Amorph piretanide, polymorphe davon, verfahren zur dessen herstellung und ihre verwendung |
| CO4940492A1 (es) * | 1997-05-29 | 2000-07-24 | Merck & Co Inc | Inhibidor de proteasa de vih |
| GB9712253D0 (en) | 1997-06-13 | 1997-08-13 | Glaxo Group Ltd | Antiviral compound |
| US6287693B1 (en) * | 1998-02-25 | 2001-09-11 | John Claude Savoir | Stable shaped particles of crystalline organic compounds |
| GB9805898D0 (en) * | 1998-03-20 | 1998-05-13 | Glaxo Group Ltd | Process for the sythesis of hiv protease inhibitors |
| GB9807354D0 (en) | 1998-04-07 | 1998-06-03 | Glaxo Group Ltd | Antiviral compound |
| WO1999067254A2 (en) * | 1998-06-23 | 1999-12-29 | The United States Of America Represented By The Secretary, Department Of Health And Human Services | Multi-drug resistant retroviral protease inhibitors and use thereof |
| ATE500823T1 (de) * | 1998-06-23 | 2011-03-15 | Us Of America Represented By The Secretary Dept Of Health And Human Services | Arzneimittel zur behandlung von hiv-infizierten säugetieren |
| US6225317B1 (en) | 1998-11-19 | 2001-05-01 | Dupont Pharmaceuticals Company | Crystalline (-)-6-chloro-4-cyclopropylethynyl-4-trifluoromethyl-3,4-dihydro-2(1H)-quinazolinone |
| WO2000047551A2 (en) | 1999-02-12 | 2000-08-17 | Vertex Pharmaceuticals Incorporated | Inhibitors of aspartyl protease |
| EA007120B8 (ru) | 2002-05-16 | 2012-03-30 | Тиботек Фармасьютикалз Лтд. | Псевдополиморфные формы ингибитора вич-протеазы |
| EP2521728B1 (en) * | 2010-01-05 | 2016-09-07 | Cipla Limited | Crystalline darunavir hydrate and process for preparation thereof |
-
2003
- 2003-05-16 EA EA200401503A patent/EA007120B8/ru unknown
- 2003-05-16 SI SI200332383T patent/SI2314591T2/sl unknown
- 2003-05-16 WO PCT/EP2003/050176 patent/WO2003106461A2/en not_active Ceased
- 2003-05-16 EP EP10180831.9A patent/EP2314591B2/en not_active Expired - Lifetime
- 2003-05-16 PT PT141686865T patent/PT2767539T/pt unknown
- 2003-05-16 HU HUE14168686A patent/HUE034389T2/en unknown
- 2003-05-16 PL PL374321A patent/PL215151B1/pl unknown
- 2003-05-16 ES ES14168686.5T patent/ES2638412T3/es not_active Expired - Lifetime
- 2003-05-16 KR KR1020047018426A patent/KR101128370B1/ko not_active Expired - Lifetime
- 2003-05-16 CA CA002485834A patent/CA2485834C/en not_active Expired - Lifetime
- 2003-05-16 DK DK10180831.9T patent/DK2314591T4/da active
- 2003-05-16 KR KR1020107024106A patent/KR20100119906A/ko not_active Ceased
- 2003-05-16 ES ES03753571T patent/ES2498370T5/es not_active Expired - Lifetime
- 2003-05-16 CN CNB038164590A patent/CN100475819C/zh not_active Expired - Lifetime
- 2003-05-16 LT LTEP14168686.5T patent/LT2767539T/lt unknown
- 2003-05-16 AP AP2004003191A patent/AP2052A/xx active
- 2003-05-16 JP JP2004513292A patent/JP4864320B2/ja not_active Expired - Lifetime
- 2003-05-16 EP EP16158480.0A patent/EP3045460B1/en not_active Expired - Lifetime
- 2003-05-16 US US10/514,352 patent/US7700645B2/en active Active
- 2003-05-16 SI SI200332389T patent/SI1567529T2/sl unknown
- 2003-05-16 HR HRP20041061AA patent/HRP20041061B1/hr not_active IP Right Cessation
- 2003-05-16 ES ES10180831T patent/ES2503551T5/es not_active Expired - Lifetime
- 2003-05-16 MX MXPA04011427A patent/MXPA04011427A/es active IP Right Grant
- 2003-05-16 DK DK14168686.5T patent/DK2767539T3/en active
- 2003-05-16 EP EP14168686.5A patent/EP2767539B1/en not_active Expired - Lifetime
- 2003-05-16 PT PT37535713T patent/PT1567529E/pt unknown
- 2003-05-16 AU AU2003271740A patent/AU2003271740B2/en not_active Revoked
- 2003-05-16 DK DK03753571.3T patent/DK1567529T4/da active
- 2003-05-16 PT PT101808319T patent/PT2314591E/pt unknown
- 2003-05-16 ES ES16158480T patent/ES2728735T3/es not_active Expired - Lifetime
- 2003-05-16 NZ NZ536497A patent/NZ536497A/en not_active IP Right Cessation
- 2003-05-16 BR BRPI0311176A patent/BRPI0311176B8/pt active IP Right Grant
- 2003-05-16 EP EP03753571.3A patent/EP1567529B2/en not_active Expired - Lifetime
- 2003-05-16 SI SI200332545T patent/SI2767539T1/sl unknown
-
2004
- 2004-11-10 IL IL16514004A patent/IL165140A0/xx unknown
- 2004-12-10 NO NO20045409A patent/NO331477B1/no not_active IP Right Cessation
- 2004-12-15 ZA ZA2004/10154A patent/ZA200410154B/en unknown
-
2009
- 2009-08-06 US US12/536,807 patent/US8518987B2/en not_active Expired - Fee Related
-
2012
- 2012-07-03 AU AU2012205289A patent/AU2012205289A1/en active Pending
-
2013
- 2013-07-11 US US13/939,494 patent/US20130303790A1/en not_active Abandoned
-
2014
- 2014-02-19 US US14/183,712 patent/US20140171499A1/en not_active Abandoned
- 2014-09-17 CY CY20141100756T patent/CY1117928T1/el unknown
- 2014-09-17 CY CY20141100755T patent/CY1115665T1/el unknown
-
2015
- 2015-08-04 US US14/817,827 patent/US20150336980A1/en not_active Abandoned
-
2017
- 2017-05-22 US US15/600,932 patent/US10000504B2/en not_active Expired - Lifetime
- 2017-08-23 CY CY20171100896T patent/CY1119311T1/el unknown
-
2018
- 2018-05-16 US US15/980,993 patent/US10858369B2/en not_active Expired - Lifetime
-
2020
- 2020-10-30 US US17/085,096 patent/US20210179631A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2503551T5 (es) | Formas pseudopolimórficas de un inhibidor de la proteasa de VIH | |
| ES2860648T5 (es) | Polimorfos cristalinos de la base libre de 2-hidroxi-6-((2-(1-isopropil-1H-pirazol-5-il)piridin-3-il)metoxi)benzaldehído | |
| ES2560955T3 (es) | Cristal de derivado de diamina y procedimiento de producción del mismo | |
| ES2553584T3 (es) | Polimorfos de Febuxostat | |
| HK1081969B (en) | Pseudopolymorphic forms of a hiv protease inhibitor |