ES2728735T3 - Formas pseudopolimórficas de un inhibidor de la proteasa del HIV - Google Patents
Formas pseudopolimórficas de un inhibidor de la proteasa del HIV Download PDFInfo
- Publication number
- ES2728735T3 ES2728735T3 ES16158480T ES16158480T ES2728735T3 ES 2728735 T3 ES2728735 T3 ES 2728735T3 ES 16158480 T ES16158480 T ES 16158480T ES 16158480 T ES16158480 T ES 16158480T ES 2728735 T3 ES2728735 T3 ES 2728735T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- formula
- solvent
- pseudopolymorphs
- water
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Virology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Tropical Medicine & Parasitology (AREA)
- Molecular Biology (AREA)
- AIDS & HIV (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Furan Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Un pseudopolimorfo de (3R,3aS,6aR)-hexahidrofuro [2,3-b] furan-3-il (1S,2R)-3-[[(4-aminofenil) sulfonil] (isobutil) amino]-1-bencil-2-hidroxipropilcarbamato, en donde el pseudopolimorfo es un solvato de isopropanol, que contiene 50% en moles de isopropanol.
Description
DESCRIPCIÓN
Formas pseudopolimórficas de un inhibidor de la proteasa del HIV
Campo técnico
Esta invención se refiere a una novedosa forma pseudopolimórfica de (3R,3aS,6aR)-hexahidro-furo [2,3-b] furan-3-il (1S,2R)-3-[[(4-aminofenil) sulfonil] (isobutil) amino]-1-bencil-2-hidroxipropilcarbamato, y su uso médico.
Antecedentes de la invención
Las proteasas codificadas por virus, que son esenciales para la replicación vírica, son necesarias para el procesamiento de precursores de proteínas víricas. La interferencia con el procesamiento de precursores proteicos inhibe la formación de viriones infecciosos. De acuerdo con lo anterior, se pueden usar inhibidores de proteasas virales para prevenir o tratar infecciones virales crónicas y agudas. (3R,3aS,6aR)-hexahidrofuro [2,3-b] furan-3-il (1S,2R)-3-[[(4-aminofenil) sulfonil] (isobutil) amino]-1-bencil-2-hidroxipropilcarbamato tiene actividad inhibidora de la proteasa del HIV y es particularmente adecuado para inhibir los virus HIV-1 y HIV-2.
La estructura del (3R,3aS,6aR)-hexahidrofuro [2,3-b] furan-3-il (1S,2R)-3-[[(4-aminofenil) sulfonil] (isobutil) amino]-1-bencil-2-hidroxipropilcarbamato, se muestra a continuación:

El compuesto de fórmula (X) y los procedimientos para su preparación se describen en los documentos EP 715618, WO 99/67417, US 6,248,775, y en Bioorganic and Chemistry Letters, Vol. 8, pp. 687-690, 1998, "Potent HIV protease inhibitors incorporating high-affinity P2-igands and (R)-(hydroxyethylamino)sulfonamide isostere", todos los cuales se incorporan en este documento como referencia.
Los fármacos utilizados en la preparación de formulaciones farmacéuticas para uso comercial deben cumplir con ciertas normas, incluidas las directrices GMP (Good Manufacturing Practices) e ICH (International Conference on Harmonization). Dichas normas incluyen requisitos técnicos que abarcan una heterogénea y amplia gama de parámetros físicos, químicos y farmacéuticos. Es esta variedad de parámetros a considerar, que hacen de las formulaciones farmacéuticas una disciplina técnica compleja.
Por ejemplo, y como ejemplo, un fármaco utilizado para la preparación de formulaciones farmacéuticas debe cumplir una pureza aceptable. Existen pautas establecidas que definen los límites y calificación de impurezas en nuevas sustancias farmacéuticas producidas por síntesis química, esto es, impurezas reales y potenciales más probables de surgir durante la síntesis, purificación y almacenamiento de la nueva sustancia farmacéutica. Se establecen directrices para la cantidad de productos de degradación permitidos de la sustancia farmacéutica, o productos de reacción de la sustancia farmacéutica con un excipiente y/o un sistema de recipiente/cierre inmediato.
La estabilidad es también un parámetro considerado en la creación de formulaciones farmacéuticas. Una buena estabilidad asegurará que la integridad química deseada de las sustancias farmacéuticas se mantenga durante la vida útil de la formulación farmacéutica, que es el marco de tiempo sobre el cual se puede confiar en que un producto conserve sus características de calidad cuando se almacena bajo condiciones de almacenamiento esperado o dirigido. Durante este período, el fármaco se puede administrar con poco o ningún riesgo, ya que la presencia de productos de degradación potencialmente peligrosos no tiene consecuencias perjudiciales para la salud del receptor, ni el menor contenido del ingrediente activo puede causar submedicación.
Diferentes factores, tales como radiación luminosa, temperatura, oxígeno, humedad, sensibilidad al pH en soluciones, pueden influir en la estabilidad y pueden determinar la vida útil y las condiciones de almacenamiento. La biodisponibilidad es también un parámetro a considerar en el diseño de administración de fármacos de formulaciones farmacéuticamente aceptables. La biodisponibilidad se refiere a la cantidad y velocidad a la que la
forma intacta de un fármaco particular aparece en la circulación sistémica después de la administración del fármaco. La biodisponibilidad exhibida por un fármaco es, de este modo, de relevancia para determinar si se alcanza una concentración terapéuticamente eficaz en el(los) sitio(s) de acción del fármaco.
Los factores fisicoquímicos y la formulación farmacotécnica pueden tener repercusiones en la biodisponibilidad del fármaco. Como tales, se deben considerar varias propiedades del fármaco tales como la constante de disociación, la velocidad de disolución, la solubilidad, la forma polimórfica, el tamaño de partícula, al mejorar la biodisponibilidad. También es relevante establecer que la formulación farmacéutica seleccionada es capaz de fabricar, más adecuadamente, de fabricar a gran escala.
En vista de los diversos y muchos requisitos técnicos, y sus parámetros de influencia, no es obvio prever qué formulaciones farmacéuticas serán aceptables. Como tal, se descubrió inesperadamente que ciertas modificaciones del estado sólido del compuesto de fórmula (X) influyeron positivamente en su aplicabilidad en formulaciones farmacéuticas.
Resumen de la invención
La presente descripción se refiere a formas pseudopolimórficas del compuesto de fórmula (X) para la preparación de formulaciones farmacéuticas. Tales formas pseudopolimórficas contribuyen a formulaciones farmacéuticas en una estabilidad y biodisponibilidad mejoradas. Se pueden fabricar con una pureza suficientemente alta para ser aceptables para uso farmacéutico, más particularmente en la fabricación de un medicamento para inhibir la actividad de la proteasa del HIV en mamíferos.
En un primer aspecto, la presente invención proporciona un pseudopolimorfo de (3R,3aS,6aR)-hexahidrofuro [2,3-b] furan-3-il (1S,2R)-3-[[(4-aminofenil) sulfonil] (isobutil) amino]-1-bencil-2-hidroxipropilcarbamato, que es la Forma J (isopropanolato), como se define en la reivindicación 1.
Los pseudopolimorfos proporcionados como referencia incluyen solvatos de alcohol, más en particular, solvatos de alcohol C1-C4; solvatos hidratados; solvatos de alcano, más en particular, solvatos de cloroalcano C1-C4; solvatos cetónicos, más en particular, solvatos cetona C1-C5; solvatos de éter, más en particular, solvatos de éter C1-C4; solvatos de cicloéter; solvatos de éster, más en particular, solvatos de ésteres C1-C5; y solvatos sulfónicos, más en particular, solvatos sulfónicos C1-4 del compuesto de fórmula (X). Los pseudopolimorfos preferidos son solvatos farmacéuticamente aceptables, tales como hidrato y etanolato.
Los pseudopolimorfos particulares son la forma A (etanolato), la forma B (hidrato), la forma C (metanolato), la forma D (acetonato), la forma E (diclorometano), la forma F (solvato de acetato de etilo), la forma G (1-metoxi-2-propanolato), la forma H (anisolato), la forma I (tetrahidrofuranato) del compuesto de fórmula (X). Otro pseudopolimorfo particular es la forma K (mesilato) del compuesto de fórmula (X).
Los pseudopolimorfos del compuesto de fórmula (X) se preparan combinando el compuesto de fórmula (X) con un solvente orgánico, agua, o mezclas de agua y solventes orgánicos miscibles en agua, y aplicando cualquier técnica apropiada para inducir la cristalización, para obtener los pseudopolimorfos deseados.
La descripción se refiere al uso de los pseudopolimorfos presentes, en la fabricación de formulaciones farmacéuticas para inhibir la actividad de la proteasa del HIV en mamíferos. En relación con el campo terapéutico, una realización preferida de esta invención se refiere a la forma pseudopolimórfica del compuesto de fórmula (X) como se define en la reivindicación 1, para uso en el tratamiento de una enfermedad viral de HIV en un mamífero que lo necesite, método que comprende administrar a dicho mamífero una cantidad eficaz de la forma pseudopolimórfica del compuesto de fórmula (X).
Los siguientes dibujos proporcionan información adicional sobre las características de los pseudopolimorfos del compuesto de fórmula (X).
Breve descripción de los dibujos
La figura 1, la figura 2 y la figura 3 son los patrones de difracción de rayos X en polvo de la forma A (1:1).
La figura 4 representa la forma A (1:1) en tres dimensiones con los átomos identificados.
La figura 5 es una comparación de los espectros Raman de las formas A, B, D, E, F, H, (1:1) y la forma amorfa en la región de estiramiento de carbonilo de 1800-100 cm-1 y la región 3300-2000 cm-1.
La figura 6 es una comparación de los espectros Raman expandidos de las formas A, B, D, E, F, H (1:1) y la forma amorfa en la región de estiramiento de carbonilo de 600-0 cirr1.
La figura 7 es una comparación de los espectros Raman expandidos de las formas A, B, D, E, F, H, (1:1) y la forma amorfa en la región de estiramiento de carbonilo de 1400-800 cm-1.
En las Figuras 5, 6 y 7, P1 corresponde a la forma A, P18 corresponde a la forma B, P19 corresponde a la forma amorfa, P25 corresponde a la forma E, P27 corresponde a la forma F, P50 corresponde a la forma D, P68 corresponde a la forma H, P69 corresponde a la forma C, P72 corresponde a la forma I, y P81 corresponde a la forma G.
La figura 8 es el termógrafo calorimétrico de barrido diferencial (DSC) de la forma A (1:1).
La figura 9 es el espectro infrarrojo (IR) que refleja los modos vibracionales de la estructura molecular de la forma A como un producto cristalino
La figura 10 es el espectro IR que refleja los modos vibratorios de la estructura molecular de la forma B como un producto cristalino
Figura 11: Espectro IR de las formas A, B y forma amorfa, a un intervalo espectral de 4000 a 400 cm-1.
Figura 12: Espectro  las formas
las formas  B y forma amorfa, a un intervalo espectral de 3750 a 2650 Figura 13: Espectro
B y forma amorfa, a un intervalo espectral de 3750 a 2650 Figura 13: Espectro  las formas
las formas  B y forma amorfa, a un intervalo espectral de 1760 a 1580 Figura 14: Espectro
B y forma amorfa, a un intervalo espectral de 1760 a 1580 Figura 14: Espectro  las formas
las formas  B y forma amorfa, un intervalo espectral de 980 a 720 cm-1 En las figuras 11, 12, 13 y 14, la curva A corresponde a la forma A, la curva B corresponde a la forma B, y la curva C corresponde a la forma amorfa.
B y forma amorfa, un intervalo espectral de 980 a 720 cm-1 En las figuras 11, 12, 13 y 14, la curva A corresponde a la forma A, la curva B corresponde a la forma B, y la curva C corresponde a la forma amorfa.
Figura 15: Curvas del termógrafo DSC de la forma A (curva D), la forma A después de adsorción/desorción (ADS/DES) (curva E), y la forma A después de los ensayos de hidratación ADS/DES (curva F)
Figura 16: Curvas termogravimétricas (TG) de la forma A (curva D), la forma A después de ADS/DES (curva E), y la forma A después de los ensayos de hidratación ADS/DES (curva F)
Figura 17: Curva TG de la forma A a 25°C en atmósfera de nitrógeno seco en función del tiempo
Figura 18: Curvas ADS/DES de la forma A.
Figura 19: Curvas ADS/DES de la prueba de hidratación de la forma A
Figura 20: Curvas ADS/DES de la forma B
Figura 21: Espectro IR de la forma K
Figura 22: Espectro Raman de la forma K
Figura 23: Curva DSC de la forma K
Figura 24: Curva TG de la forma K
Figura 25: Isoterma ADS/DES de la forma K, lote 1
Figura 26: Isoterma ADS/DES de la forma K, lote 2
Descripción detallada
El término “polimorfismo” se refiere a la capacidad de una estructura química para que se produzca en diferentes formas y se sabe que se produce en muchos compuestos orgánicos, incluyendo fármacos. Como tales, las "formas polimórficas" o "polimorfos" incluyen sustancias farmacéuticas que aparecen en forma amorfa, en forma cristalina, en forma anhidra, a diversos grados de hidratación o solvatación, con moléculas de solvente atrapadas, así como sustancias que varían en dureza cristalina, forma y tamaño. Los diferentes polimorfos varían en propiedades físicas tales como solubilidad, disolución, estabilidad en estado sólido, así como comportamiento de procesamiento en términos de flujo de polvo y compactación durante la formación de comprimidos.
El término "forma amorfa" se define como una forma en la que no existe un orden tridimensional de largo alcance.
En la forma amorfa, la posición de las moléculas entre sí es esencialmente aleatoria, esto es, sin disposición regular de las moléculas sobre una estructura de celosía.
El término "cristalino" se define como una forma en la que la posición de las moléculas entre sí se organiza de acuerdo con una estructura de celosía tridimensional.
El término "forma anhidra" se refiere a una forma particular esencialmente exenta de agua. "Hidratación" se refiere al procedimiento de adicionar moléculas de agua a una sustancia que se produce en una forma particular e "hidratos" son sustancias que se forman mediante la adición de moléculas de agua. "Solvatación" se refiere al procedimiento
de incorporación de moléculas de un solvente a una sustancia que se presenta en forma cristalina. Por lo tanto, el término "solvato" se define como una forma cristalina que contiene cantidades ya sea estequiométricas o no estequiométricas de solvente. Dado que el agua es un solvente, los solvatos también incluyen hidratos. El término "pseudopolimorfo" se aplica a formas cristalinas polimórficas que tienen moléculas de solvente incorporadas en sus estructuras de celosía. El término pseudopolimorfismo se usa frecuentemente para designar solvatos (Byrn, Pfeiffer, Stowell, (1999) Solid-state Chemistry of Drugs, 2nd Ed., published by SSCI, Inc).
La presente descripción proporciona pseudopolimorfos de (3R,3aS,6aR)-hexahidrofuro [2,3-b] furan-3-il (1S,2R)-3-[[(4-aminofenil) sulfonil] (isobutil) amino]-1-bencil-2-hidroxipropilcarbamato.
En esta solicitud se dan ejemplos de varios pseudopolimorfos e incluyen la Forma J (isopropanolato), así como las formas de referencia, a saber, la Forma A (etanolato), la Forma B (hidrato), la Forma C (metanolato), la Forma D (acetonato), la Forma E (diclorometano), la Forma F (solvato de acetato de etilo), la Forma G (1-metoxi-2-propanolato), la Forma H (anisolato), la Forma I (tetrahidrofuranato), o la Forma K (mesilato) del compuesto de fórmula (X).
Los solvatos pueden ocurrir en diferentes proporciones de solvatación. El contenido de solvente del cristal puede variar en diferentes proporciones dependiendo de las condiciones aplicadas. Las formas cristalinas de solvato del compuesto de fórmula (X) pueden comprender hasta 5 moléculas de solvente por molécula del compuesto de fórmula (X), que aparecen en diferentes estados solvatados incluyendo, entre otros, los cristales de hemisolvato, monosolvato, disolvato, trisolvato, cristales de solvatos intermedios, y mezclas de los mismos. Convenientemente, la proporción del compuesto de fórmula (X) con el solvente puede variar entre (5:1) y (1:5). En particular, la proporción puede variar de aproximadamente 0.2 a aproximadamente 3 moléculas de solvente por 1 molécula del compuesto de fórmula (X), más en particular, la proporción puede variar de aproximadamente 1 a aproximadamente 2 moléculas de solvente por 1 molécula del compuesto de fórmula (X), preferiblemente la proporción es 1 molécula de solvente por 1 molécula del compuesto de fórmula (X). El pseudopolimorfo de la reivindicación 1 contiene 50% en moles de isopropanol.
La difracción de rayos X en polvo es una técnica para caracterizar formas polimórficas incluyendo pseudopolimorfos del compuesto de fórmula (X) y diferenciar formas cristalinas de solvato de otras formas cristalinas y no cristalinas del compuesto de fórmula (X). Como tal, los espectros de difracción de rayos X en polvo se recogieron en un difractómetro de polvo Phillips PW 1050/80, modelo Bragg-Brentano. Los polvos de la forma A (1:1), alrededor de 200 mg de cada muestra, se empaquetaron en tubos capilares de vidrio de 0.5 mm y se analizaron de acuerdo con un método estándar en la técnica. El generador de rayos X se hizo funcionar a 45 Kv y 32 mA, utilizando la línea de cobre Ka como fuente de radiación. No hubo rotación de la muestra a lo largo del eje chi y los datos se recogieron entre 4 y 60° 2-theta de tamaño del paso. La forma A (1:1) tiene las posiciones de ángulo de dos-theta característicos de los picos como se muestra en la figura 1,2 y 3 a: 7,04° ± 0,5°, 9,24° ± 0,5°, 9,96° ± 0,5°, 10,66° ± 0,5°, 11,30° ± 0,5°, 12,82° ± 0,5°, 13,80° ± 0,5°, 14,56° ± 0,5°, 16,66° ± 0,5°, 17,30° ± 0,5°, 18,28° ± 0,5°, 19,10° ± 0,5-, 20,00- ± 0,5°, 20,50° ± 0,5°, 21,22° ± 0,5°, 22,68° ± 0,5°, 23,08° ± 0,5°, 23,66° ± 0,5°, 25,08° ± 0,5°, 25,58° ± 0,5°, 26,28° ± 0,5°, 27,18° ± 0,5°, 28,22° ± 0,5°, 30,20° ± 0,5°, 31,34° ± 0,5°, 32,68° ± 0,5°, 33,82° ± 0,5°, 39,18° ± 0,5°, 41,20° ± 0,5°, 42,06° ± 0,5°, y 48,74° ± 0,5°.
En otro conjunto de experimentos analíticos, se aplicó difracción única de rayos X a la forma A (1:1), que dio como resultado la siguiente configuración de los cristales, que se enumeran en la siguiente tabla.
Tabla 1


La estructura tridimensional resultante de la forma A (1:1) se representa en la figura 4.
La tabla 2 muestra las coordenadas atómicas (x 104) y los parámetros de desplazamiento isotrópico equivalente (A2 x 103) para la forma A (1:1). Los átomos se numeran como se muestra en la figura 4. Las coordenadas fraccionales x, y y z indican la posición de los átomos en relación con el origen de la celda unitaria. U(eq) se define como un tercio de la traza del tensor Uy ortogonalizado.


La tabla 3 muestra los parámetros de desplazamiento anisótropo (A2 x 103) para la forma A (1:1). El exponente del factor de desplazamiento anisotrópico toma la fórmula:
■2ji2[/?2a*2í7|i ... 2
h k a*
b*J7i2]



La espectroscopia Raman se ha utilizado ampliamente para elucidar estructuras moleculares, cristalinidad y polimorfismo. Los modos Raman de baja frecuencia son particularmente útiles para distinguir diferentes envases moleculares en cristal. Como tal, los espectros Raman se registraron en un espectrómetro Bruker FT-Raman RFS100 equipado con un tubo fotomultiplicador y detectores ópticos multicanal. Las muestras colocadas en tubos capilares de cuarzo fueron excitadas por un láser de iones de argón. La potencia del láser en las muestras se ajustó a aproximadamente 100 mW y la resolución espectral fue de aproximadamente 2 cm-1. Se encontró que las formas A, B, D, E, F y H, (1:1) y la forma amorfa tienen los espectros Raman que aparecen en las figuras 5, 6 y 7.
Además, las formas A y B se caracterizaron usando un accesorio |oATR (reflectancia total microatenuada) (Harrick Split-Pea con cristal Si). Los espectros infrarrojos se obtuvieron con un espectrofotómetro Nicolet Magna 560 FTIR, un separador de haz Ge en KBr y un detector de ventanas DTGS con KBr. Los espectros se midieron a una resolución de 1 cm-1 y 32 exploraciones cada uno, en un intervalo de longitud de onda desde 4000 a 400 cm-1 y aplicación de corrección de línea de base. Los números de onda para la forma A obtenidos se muestran en la siguiente tabla 4.
Tabla 4

El espectro IR de la figura 9 refleja los modos vibracionales de la estructura molecular como un producto cristalino. Los números de onda obtenidos para la forma B se muestran en la siguiente tabla 5.
Tabla 5


El espectro IR de la figura 10 refleja los modos vibracionales de la estructura molecular de la forma B como un producto cristalino.
Siguiendo el mismo método IR analítico, la forma B y la forma amorfa también se caracterizaron y compararon con la forma A, como se muestra en las figuras 11 a 14. Los espectros IR de las diferentes formas físicas mostraron diferencias espectrales distintas, las más relevantes son las de la tabla 6:
Tabla 6

Las formas físicas A, B y la forma amorfa se identifican mediante la interpretación espectral, centrándose en bandas de absorción específicas para cada forma. Las diferencias espectrales únicas y específicas entre formas se observan en 3 intervalos espectrales: desde 3750 a 2650 cm-1 (intervalo 1), desde 1760 a 1580 cm-1 (intervalo 2) y desde 980 a 720 cm-1 (intervalo 3).
Intervalo 1 (desde 3750 a 2650 cm-1)
Figura 11: La forma A muestra una banda doble con máximos de absorción a 3454 cm-1 y 3429 cm-1. La forma B muestra una sola banda de absorción a 3615 cm -1 y la forma amorfa muestra una banda de absorción única a 3362 cm -1.
Intervalo 2 (desde 1760 a 1580 cm-1)
Figura 12: La forma A muestra una banda de absorción única a 1646 cm-1, la forma B muestra una banda de absorción única a 1630 cm-1 y la forma amorfa muestra una banda de absorción única a 1628 cm-1 con una intensidad claramente superior comparada con la banda de la forma B. Además, la forma amorfa muestra una banda ancha menos intensa a 1704 cm-1 comparada con ambas bandas de las formas A y B a aproximadamente 1704 cm-1.
Intervalo 3 (desde 980 a 720 cm-1)
Figura 13: La forma A muestra un conjunto distinto de 5 bandas de absorción a 911, 890, 876, 862 y 841 cm-1. La forma B muestra un conjunto similar, pero falta la banda de 876 cm-1. La forma amorfa muestra una única banda ancha a aproximadamente 750 cm-1, ambas formas A y B muestran dos máximos a aproximadamente 768 cm-1 y 743 cm-1.
La termomicroscopia es otra técnica útil en el estudio de la cinética de estado sólido. Se puede cuantificar la cinética de los procedimientos de nucleación a partir de soluciones o fusión, incluyendo el análisis de la velocidad de nucleación. El método más simple y más utilizado es la determinación del punto de fusión. Como tal, se utilizó un controlador Mettler modelo FP 82 con etapa de calentamiento en un microscopio Leitz. Se colocaron unas pocas partículas de la forma A en un portaobjetos de vidrio y se observaron mientras se calentaba a 10°C por minuto. Se encontró que el intervalo de fusión para la forma A (1:1) estaba entre 90° y 110°C.
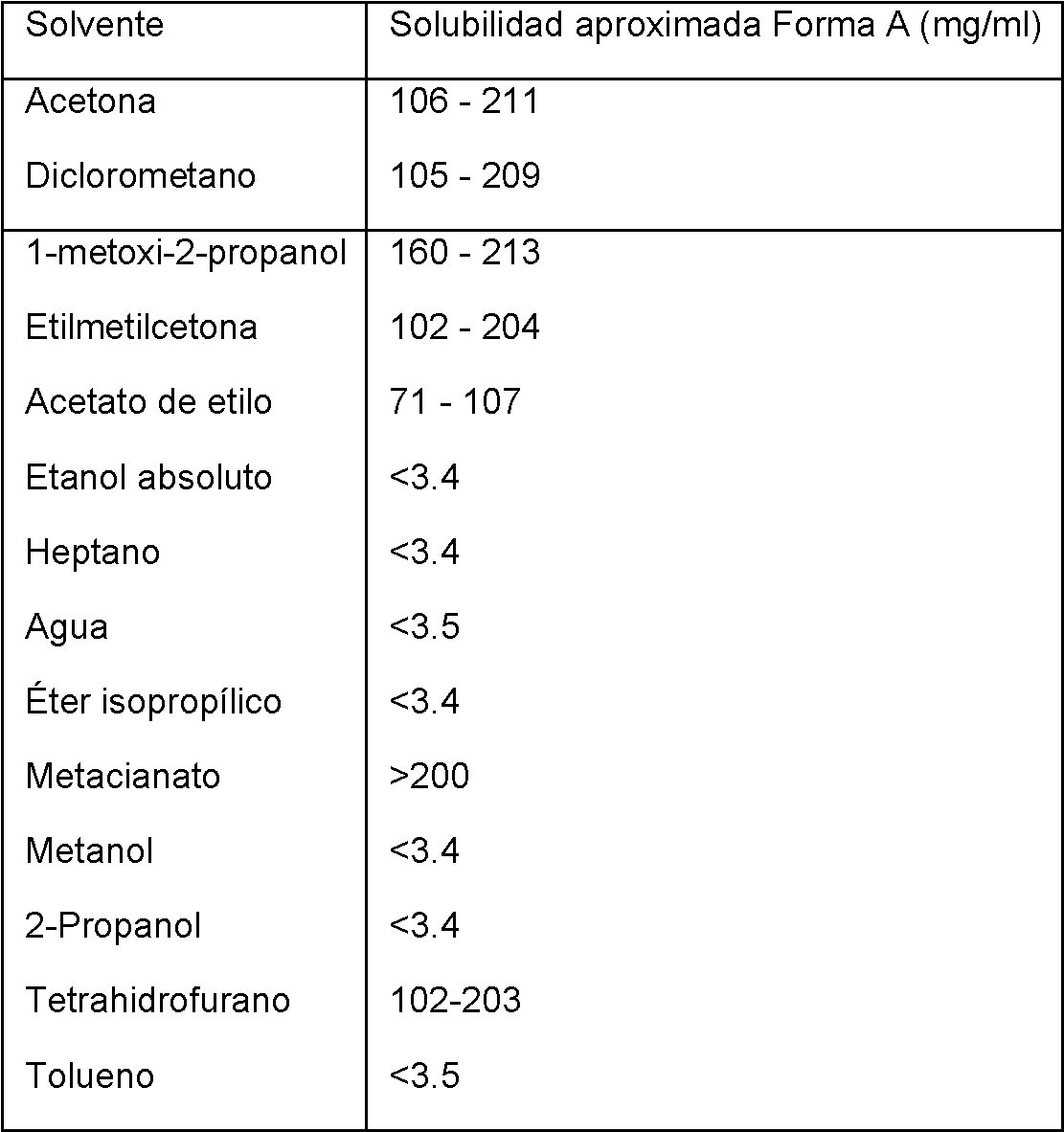
En otro medio de caracterización, la solubilidad de la forma A (1:1) era también una materia objeto de estudio. Se determinó que su solubilidad en diferentes solventes a aproximadamente 23°C era la siguiente:
Tabla 7: Solubilidad aproximada para la forma A (1:1), en mg/ml
Se llevaron a cabo investigaciones de solubilidad adicionales en función del pH. Como tal, las solubilidades acuosas de la forma A (1:1) se midieron en solventes con diferentes pH. Se equilibró un exceso del soluto con el solvente a 20°C durante al menos 24 horas. Después de retirar el compuesto no disuelto, se determinó la concentración en solución usando espectrometría UV.
Tabla 8: Solubilidad para la forma A (1:1) en función del pH

Se midió la solubilidad de la forma A (1:1) en función de HPpCD (hidroxipropil-p-ciclodextrina). Se equilibró un exceso de producto con el solvente durante 2 días a 20°C. Después de retirar el compuesto no disuelto, se determinó la concentración en solución usando espectrometría UV.
Tabla 9: Solubilidad para la forma A (1:1) en función de HPpCD

En un segundo aspecto, la presente descripción se refiere a procedimientos de preparación de pseudopolimorfos. Los pseudopolimorfos del compuesto de fórmula (X) se preparan combinando el compuesto de fórmula (X) con un solvente orgánico, o agua, o mezclas de agua y solventes orgánicos miscibles en agua, aplicando cualquier técnica apropiada para inducir la cristalización, y aislando los pseudopolimorfos deseados.
Por técnicas para inducir la cristalización se entienden los procedimientos para la producción de los cristales que incluyen entre otros disolver o dispersar el compuesto de fórmula (X) en un medio solvente, llevando la solución o dispersión del compuesto de fórmula (X) y el(los) solvente(s) a una concentración deseada, llevando dicha solución o dispersión a una temperatura deseada, efectuando cualquier presión apropiada, eliminando y/o separando cualquier material o impurezas no deseados, secando los cristales formados para obtener los pseudopolimorfos en un estado sólido, si tal estado es deseado.
Llevar la solución o dispersión del compuesto de fórmula (X) y solventes a una concentración deseada no implica necesariamente un aumento en la concentración del compuesto de fórmula (X). En ciertos casos, podría ser preferible una disminución o ningún cambio en la concentración. Al llevar dicha solución o dispersión a una temperatura deseada, se comprenderán los actos de calentamiento, enfriamiento o salida a temperatura ambiente. Las técnicas utilizadas para obtener una concentración deseada son las comunes en la técnica, por ejemplo, evaporación por destilación atmosférica, destilación al vacío, destilación fraccionada, destilación azeotrópica, evaporación de película, otras técnicas bien conocidas en la técnica y combinaciones de las mismas. Un procedimiento opcional para obtener una concentración deseada podría implicar también la saturación de la solución del compuesto de fórmula (X) y solvente, por ejemplo, adicionando un volumen suficiente de un no solvente a la solución para alcanzar el punto de saturación. Otras técnicas apropiadas para saturar la solución incluyen, a modo de ejemplo, la introducción de compuesto adicional de fórmula (X) a la solución y/o evaporación de una porción del solvente de la solución. Como se hace referencia en este documento, la solución saturada comprende soluciones en sus puntos de saturación o que exceden sus puntos de saturación, esto es, sobresaturada.
La eliminación y/o separación de cualquier material no deseado o impurezas se puede realizar por purificación, filtración, lavado, precipitación o técnicas similares. La separación, por ejemplo, se puede llevar a cabo mediante técnicas conocidas de separación sólido-líquido. Los procedimientos de filtración conocidos para los expertos en el arte también se pueden usar en el presente procedimiento. Las filtraciones se pueden realizar, entre otros métodos, mediante centrifugación, o utilizando un filtro de estilo Buchner, un filtro o placas Rosenmund o una prensa de marco. Preferiblemente, la filtración en línea o la filtración de seguridad se pueden intercalar ventajosamente en los procedimientos descritos anteriormente, con el fin de aumentar la pureza de la forma pseudopolimórfica resultante. Además, también se pueden emplear agentes de filtración tales como gel de sílice, Arbocel®, diatomita de dicalita, o similares, para separar impurezas de los cristales de interés.
Los cristales obtenidos se pueden secar también, y dicho procedimiento de secado se puede usar opcionalmente en los diferentes pasos de cristalización, si se aplica más de un paso de cristalización. Los procedimientos de secado incluyen todas las técnicas conocidas para los expertos en el arte, tales como calentamiento, aplicación de vacío, circulación de aire o gas, adición de un desecante, liofilización, secado por pulverización, evaporación o similares, o cualquier combinación de los mismos.
Los procedimientos para la cristalización de pseudopolimorfos del compuesto de fórmula (X) abarcan combinaciones múltiples de técnicas y variaciones de las mismas. Como tal, y a modo de ejemplo, la cristalización de pseudopolimorfos del compuesto de fórmula (X) se puede ejecutar disolviendo o dispersando el compuesto de fórmula (X) a una temperatura apropiada en el solvente, por lo que una parte de dicho solvente se evapora aumentando la concentración del compuesto de fórmula (X) en la dicha solución o dispersión, enfriando la dicha
mezcla, y opcionalmente lavando y/o filtrando y secando los cristales de solvato resultantes del compuesto de fórmula (X). Opcionalmente, se pueden preparar pseudopolimorfos del compuesto de fórmula (X) disolviendo o dispersando el compuesto de fórmula (X) en un medio solvente, enfriando dicha solución o dispersión y posteriormente filtrando y secando el pseudopolimorfo obtenido. Otro ejemplo de preparación de solvatos del compuesto de fórmula (X) podría ser saturando el compuesto de fórmula (X) en el medio solvente, y opcionalmente filtrando, lavando y secando cristales obtenidos.
La formación de los cristales también puede implicar más de un procedimiento de cristalización. En ciertos casos, se pueden realizar ventajosamente una, dos o más etapas de cristalización adicionales por diferentes razones, tales como, para aumentar la calidad del solvato resultante. Por ejemplo, los pseudopolimorfos de la presente invención también se podrían preparar adicionando un solvente a un material base de partida inicial del compuesto de fórmula (X), agitando la solución a una temperatura fija hasta que las sustancias se resuelvan completamente, concentrando la solución por destilación a vacío, y enfriamiento. Se llevaría a cabo una primera cristalización y los cristales formados se volverían a lavar con un solvente, y seguido de la disolución del compuesto de fórmula (X) con el solvente para formar el pseudopolimorfo deseado. La recristalización de la mezcla de reacción ocurriría, seguida de una etapa de enfriamiento del reflujo. El pseudopolimorfo formado se filtraría opcionalmente y se dejaría secar. Al disolver o dispersar el compuesto de fórmula (X) en el solvente orgánico, agua o una mezcla de agua y solventes orgánicos miscibles en agua, se pueden obtener diferentes grados de dispersión, tales como suspensiones, emulsiones, suspensiones o mezclas; o preferiblemente obtener soluciones homogéneas de una fase.
Opcionalmente, el medio de solvente puede contener aditivos, por ejemplo, uno o más agentes dispersantes, surfactantes u otros aditivos, o mezclas de los mismos del tipo normalmente utilizado en la preparación de suspensiones cristalinas y que están bien documentados en la bibliografía. Los aditivos se pueden utilizar ventajosamente para modificar la forma del cristal aumentando la indulgencia y disminuyendo el área superficial. El medio de solvente que contiene la solución se puede agitar opcionalmente durante un cierto periodo de tiempo, o agitar vigorosamente usando, por ejemplo, un mezclador u homogeneizador de alto cizallamiento o una combinación de estos, para generar el tamaño de gotita deseado para el compuesto orgánico.
Ejemplos de solventes orgánicos útiles para la presente invención incluyen alcoholes C1-C4 tales como metanol, etanol, isopropanol, butanol, 1-metoxi-2-propanol, y similares; cloroalcanos C1-C4 tales como diclorometano; cetonas C1-C4 tales como acetona; éteres C1-C4 tales como anisol, y similares; cicloéteres tales como tetrahidrofurano; ésteres C1-C4 tales como acetato de etilo; sulfonatos C1-C4 tales como mesilato, etanosulfonato, butanosulfonato, 2-metil-1-propanosulfonato; y similares.
Ejemplos de mezclas de agua y solventes orgánicos miscibles en agua incluyen, mezclas de agua con todos los solventes orgánicos enumerados anteriormente, siempre que sean miscibles en agua, por ejemplo, etanol/agua, por ejemplo, en una proporción 50/50.
Los solventes preferidos son aquellos solventes farmacéuticamente aceptables. Sin embargo, los solventes farmacéuticamente no aceptables también pueden encontrar su uso en la preparación de pseudopolimorfos farmacéuticamente aceptables.
En un método preferido, el solvente es un solvente farmacéuticamente aceptable ya que da como resultado un pseudopolimorfo farmacéuticamente aceptable. En un método más preferido, el solvente es etanol.
En una realización particular, los pseudopolimorfos farmacéuticamente aceptables del compuesto de fórmula (X) se pueden preparar a partir de formas pseudopolimórficas del compuesto de fórmula (X), que puede no ser necesariamente farmacéuticamente aceptable. Por ejemplo, la forma A se puede preparar a partir de la forma J. Los pseudopolimorfos también se pueden preparar a partir de la forma amorfa.
En las mezclas de agua y solventes orgánicos miscibles en agua, la cantidad de agua puede variar de aproximadamente 5% en volumen a aproximadamente 95% en volumen, preferiblemente de aproximadamente 25% a aproximadamente 75% en volumen, más preferiblemente de aproximadamente 40% a aproximadamente 60% en volumen.
También se debe observar que la calidad del solvente orgánico seleccionado (absoluto, desnaturalizado u otro) también influye en la calidad resultante del pseudopolimorfo.
El control de la temperatura de precipitación y la siembra se puede utilizar adicionalmente para mejorar la reproducibilidad del procedimiento de cristalización, la distribución del tamaño de partícula y la forma del producto. Como tal, la cristalización se puede efectuar sin siembra con cristales del compuesto de fórmula (X) o preferiblemente en presencia de los cristales del compuesto de fórmula (X), que se introducen en la solución por siembra. La siembra también se puede efectuar varias veces a diversas temperaturas. La cantidad de material de siembra depende de la cantidad de la solución y se puede determinar fácilmente por un experto en el arte.
El tiempo de cristalización en cada etapa de cristalización dependerá de las condiciones aplicadas, de las técnicas empleadas y/o de los solventes utilizados.
La ruptura de las partículas o agregados grandes de partículas después de la conversión del cristal se puede realizar adicionalmente con el fin de obtener un tamaño de partícula deseado y homogéneo. De acuerdo con lo anterior, las formas cristalinas de solvato del compuesto de fórmula (X) se trituran opcionalmente después de experimentar la conversión. La trituración o molienda se refiere a romper físicamente las partículas grandes o agregados de partículas usando métodos y aparatos bien conocidos en la técnica para la reducción del tamaño de partícula de polvos. Los tamaños de partícula resultantes pueden variar desde milímetros hasta nanómetros, produciendo, por ejemplo, nanocristales, microcristales.
El rendimiento del procedimiento de preparación de los pseudopolimorfos del compuesto de fórmula (X) puede ser de 10% o más, un rendimiento más preferido variaría desde 40% a 100%.
Curiosamente, el rendimiento varía entre 70% y 100%.
De manera apropiada, los pseudopolimorfos de la presente invención tienen una pureza superior al 90 por ciento. Más adecuadamente, los presentes pseudopolimorfos tienen una pureza superior al 95 por ciento. Aún más adecuadamente, los pseudopolimorfos presentes tienen una pureza superior al 99 por ciento.
En un tercer aspecto, la presente descripción se refiere a una formulación farmacéutica que comprende una cantidad terapéuticamente eficaz de un pseudopolimorfo del compuesto de fórmula (X), y un portador o diluyente farmacéuticamente aceptable del mismo.
En una realización, la presente descripción se refiere al uso de formas pseudopolimórficas farmacéuticamente aceptables del compuesto de fórmula (X), preferiblemente la forma A, en la fabricación de un medicamento para tratar enfermedades causadas por retrovirus, tales como infecciones por HIV, por ejemplo, síndrome de inmunodeficiencia adquirida (AIDS) y complejo relacionado con el AIDS (ARC).
En otra realización, la presente descripción proporciona un método para el tratamiento de una infección retroviral, por ejemplo, una infección por HIV, en un mamífero tal como un ser humano, que comprende administrar al mamífero que lo necesite una cantidad antirretroviral eficaz de una forma pseudopolimórfica farmacéuticamente aceptable del compuesto de fórmula (X), preferiblemente la forma A.
La presente descripción también se refiere a un método en el que el tratamiento de una infección vírica de HIV comprende la reducción de la carga de HIV. La presente invención también se refiere a un método en el que el tratamiento de dicha infección vírica por HIV comprende el aumento del recuento de células CD4+. La presente invención se refiere también a un método en el que el tratamiento de dicha infección vírica de HIV comprende inhibir la actividad de la proteasa del HIV en un mamífero.
Las formas pseudopolimórficas farmacéuticamente aceptables del compuesto de fórmula (X), preferiblemente la forma A, también denominadas en este documento como los ingredientes farmacéuticos activos, se pueden administrar por cualquier ruta apropiada a la afección que se va a tratar, preferiblemente oralmente. Sin embargo, se apreciará que la ruta preferida puede variar con, por ejemplo, la condición del receptor.
Para cada una de las utilidades e indicaciones indicadas anteriormente, la cantidad requerida del ingrediente activo dependerá de un número de factores que incluyen la gravedad de la afección que se va a tratar y la identidad del receptor y, en última instancia, estará a la discreción del médico o veterinario. La dosis deseada preferiblemente se puede presentar como una, dos, tres o cuatro o más subdosis administradas a intervalos apropiados a lo largo del día.
Para una forma de administración oral, los pseudopolimorfos de la presente divulgación se mezclan con aditivos apropiados, tales como excipientes, estabilizadores o diluyentes inertes, y se llevan mediante los métodos habituales a las formas de administración apropiadas, tales como comprimidos, comprimidos recubiertos, cápsulas duras, soluciones acuosas, alcohólicas u oleosas. Ejemplos de portadores inertes apropiados son goma arábiga, magnesia, carbonato de magnesio, fosfato de potasio, lactosa, glucosa o almidón, en particular almidón de maíz. En este caso, la preparación se puede llevar a cabo tanto en gránulos secos como en gránulos húmedos. Los excipientes o solventes aceitosos apropiados son aceites vegetales o animales, tales como aceite de girasol o aceite de hígado de bacalao. Los solventes apropiados para soluciones acuosas o alcohólicas son agua, etanol, soluciones de azúcar, o mezclas de las mismas. Los polietilenglicoles y polipropilenglicoles son también útiles como otros auxiliares para otras formas de administración.
Para la administración subcutánea o intravenosa, los pseudopolimorfos del compuesto de fórmula (X), si se desea con las sustancias habituales para ello, tales como solubilizantes, emulsionantes u otros auxiliares, se ponen en solución, suspensión o emulsión. Los pseudopolimorfos del compuesto de fórmula (X) también se pueden liofilizar y los liofilizados obtenidos se utilizan, por ejemplo, para la producción de preparaciones para inyección o infusión. Los solventes apropiados son, por ejemplo, agua, solución salina fisiológica o alcoholes, por ejemplo, etanol, propanol,
glicerol, además también de soluciones de azúcares tales como soluciones de glucosa o manitol, o alternativamente mezclas de los diversos solventes mencionados.
Las formulaciones farmacéuticas apropiadas para la administración en forma de aerosoles o pulverizaciones son, por ejemplo, soluciones, suspensiones o emulsiones de los pseudopolimorfos del compuesto de fórmula (X) en un solvente farmacéuticamente aceptable, tal como etanol o agua, o una mezcla de tales solventes. Si se requiere, la formulación también puede contener adicionalmente otros auxiliares farmacéuticos tales como surfactantes, emulsionantes y estabilizantes, así como un propulsor. Dicha preparación contiene habitualmente el compuesto activo en una concentración de aproximadamente 0.1 a 50%, en particular de aproximadamente 0.3 a 3% en peso. Los pseudopolimorfos de la presente divulgación también se pueden presentar en una formulación que comprende partículas de tamaño micrómetro, nanómetro o picómetro del pseudopolimorfo del compuesto de fórmula (X), cuya formulación puede contener otros agentes farmacéuticos y puede convertirse opcionalmente en forma sólida.
Puede ser conveniente formular los presentes pseudopolimorfos en forma de nanopartículas que tienen un modificador de superficie adsorbido sobre su superficie en una cantidad suficiente para mantener un tamaño de partícula medio eficaz de menos de 1000 nm. Se cree que los modificadores de superficie útiles incluyen aquellos que se adhieren físicamente a la superficie del agente antirretroviral pero no se unen químicamente al agente antirretroviral.
Puede ser además conveniente almacenar los pseudopolimorfos del compuesto de fórmula (X) en materiales de envase que sean protectores frente a riesgos mecánicos, ambientales, biológicos o químicos, o degradación. Las sustancias farmacéuticas de acondicionamiento se pueden conseguir empleando materiales de embalaje impermeables a la humedad, tales como bolsas selladas de cierre de vapor. Los productos farmacéuticos de acondicionamiento, tales como comprimidos, cápsulas, se pueden conseguir empleando, por ejemplo, blísteres de aluminio.
Se debe entender que, además de los ingredientes particularmente mencionados anteriormente, las formulaciones de esta invención incluyen otros agentes convencionales en la técnica teniendo en cuenta el tipo de formulación en cuestión, por ejemplo, los apropiados para administración oral pueden incluir agentes aromatizantes o agentes enmascaradores del sabor.
Los siguientes ejemplos están destinados sólo a la ilustración y no pretenden limitar el alcance de la invención de ninguna manera.
Ejemplo 1
La síntesis a escala industrial de la forma A (1:1) se llevó a cabo utilizando las siguientes etapas. Primero se preparó una solución con isopropanol y (3R,3aS,6aR)-hexahidrofuro [2,3-b] furan-3-il (1S,2R)-3-[[(4-aminofenil) sulfonil] (isobutil) amino]-1-bencil-2-hidroxipropilcarbamato. La solución se concentró por destilación a vacío a 70°C y 200 500 mbar de presión y se enfrió de una T >35° a una T entre 15° y 20°C durante aproximadamente 10 horas. Los cristales formados se lavaron nuevamente con 13 litros de isopropanol y se filtraron. Se realizó una recristalización posterior a partir de etanol/agua (90 litros/90 litros). Esto fue seguido por una nueva etapa de disolución, pero con 60 litros de etanol en su lugar. Se produjo la recristalización de la mezcla de reacción a partir de etanol, seguido de una etapa de enfriamiento desde reflujo a -15°C aproximadamente y durante 10 horas. El etanolato formado se filtró y se dejó secar a aproximadamente 50°C y aproximadamente 7 mbar. El rendimiento de este procedimiento fue de al menos el 75%.
Ejemplo 2 (Referencia)
En otro ejemplo se preparó una mezcla de la forma D y la forma B. Se usó acetona como solvente durante el procedimiento de cristalización para formar la forma D. El procedimiento de cristalización comprendió entonces la etapa de agitar el compuesto inicial de partida (10 g) en 70 ml de acetona. La solución se sometió a reflujo posteriormente hasta que el compuesto se resolvió por completo. Se adicionaron 40 ml de agua y la solución se enfrió posteriormente lentamente hasta temperatura ambiente y se agitó durante la noche. Los cristales formados se filtraron y se secaron en el horno de vacío a 50°C. 7.6 g de producto resultaron de la cristalización, siendo el rendimiento de este procedimiento de aproximadamente el 75%.
Ejemplo 3
En otro ejemplo se prepararon cristales de la forma J. Se usó isopropanol como solvente durante el procedimiento de cristalización para formar la forma J. El procedimiento de cristalización comprendió entonces la etapa de resolver el material inicial de partida en el solvente caliente. La solución se enfrió posteriormente hasta temperatura ambiente. Los cristales formados se filtraron y se secaron en el horno de vacío a 50°C. Los cristales contenían aproximadamente 50% molar de isopropanol.
Ejemplo 4 (Referencia)
En este ejemplo, se calcularon las pérdidas de masa para diferentes pseudopolimorfos en experimentos termogravimétricos (TG). La termogravimetría es una técnica que mide el cambio de masa de una muestra a medida que se calienta, enfría o se mantiene a temperatura constante. Se colocaron aproximadamente 2 a 5 mg de muestra en una bandeja y se insertaron en el horno TG, modelo Netzsch Thermo-Microbalance TG 209 acoplado a un vector 22 de Espectrómetro FTIR Bruker. Las muestras se calentaron en una atmósfera de nitrógeno a una velocidad de 10°C/min, hasta una temperatura final de 250°C. El límite de detección de los solventes residuales fue del orden del 0.1% para una pérdida de solvente escalonada distinta en un intervalo de temperatura de estrecho (pocos grados Celsius).
Se obtuvieron los siguientes datos de TG:
Forma A: se observó una pérdida de peso del 4.2% en el intervalo de temperatura de 25-138°C (etanol poca agua) y de 6.9% (etanol CO2) en el intervalo de temperatura de 25-200°C. La tasa de pérdida de etanol fue máxima a 120°C. La pérdida de CO2 se debió a la degradación química y fue visible a alrededor de 190°C.
Forma B: se observó una pérdida de peso de 3.4% en el intervalo de temperatura de 25-78°C (agua) y de 5.1% en el intervalo de temperatura de 25-110°C (etanol agua para T>78°C). De l1o-200°C, se perdió 1.1% de peso (etanol). Forma C: se observó una pérdida de peso del 2.1% en el intervalo de temperatura de 25-83°C (agua metanol) y de 4.2% en el intervalo de temperatura de 25-105°C (metanol para T>83°C, etapa distinta). De 105-200°C, además se perdió un 2.1% de peso (metanol). No se observó etanol en la fase gaseosa.
Forma D: se observó una pérdida de peso del 0.1% en el intervalo de temperatura de 25-50°C, del 4.2% en el intervalo de temperatura de 25-108°C (acetona etanol para T>50°C), del 8.2% en el intervalo de temperatura de 25-157°C (acetona etanol para T>108°C) y del 10.5% en el intervalo de temperatura de 25-240°C (acetona etanol para T>157°C).
Forma E: se observó una pérdida de peso del 0.2% en el intervalo de temperatura de 25-75°C (agua), del 1.8% en el intervalo de temperatura de 25-108°C (diclorometano etanol para T>75°C), del 6.8 % en el intervalo de temperatura de 25-157°C (diclorometano etanol para T>108°C) y del 8.8% en el intervalo de temperatura de 25-240°C (diclorometano etanol para T>157°C).
Forma F: se observó una pérdida de peso de 0.1% en el intervalo de temperatura de 25-50°C (probablemente agua), del 1.7% en el intervalo de temperatura de 25-108°C (acetato de etilo etanol para T>50°C), del 6.6% en el intervalo de temperatura de 25-157°C (acetato de etilo etanol para T>108°C) y del 9% en el intervalo de temperatura de 25-240°C (acetato de etilo etanol para T>157°C).
Forma G: se observó una pérdida de peso del 0.0% en el intervalo de temperatura de 25-50°C, del 3.7% en el intervalo de temperatura de 25-108°C (1-metoxi-2-propanol etanol para T>50°C, , etapa distinta), del 8% en el intervalo de temperatura de 25-157°C (1-metoxi-2-propanol etanol para T>108°C) y del 12.5% en el intervalo de temperatura de 25-240°C (1-metoxi) -2-propanol etanol para T>157°C).
Forma H: se observó una pérdida de peso del 0.8% en el intervalo de temperatura de 25-100°C (anisol poco etanol) y del 8.8% en el intervalo de temperatura de 25-200°C (anisol etanol para T>100°C).
Forma I: se observó una pérdida de peso del 0.3% en el intervalo de temperatura de 25-89°C (agua) y de 11.0% en el intervalo de temperatura de 25-200°C (tetrahidrofurano para T>89°C). No se observó etanol en la fase gaseosa. La tabla 10 muestra las pérdidas de masa esperadas aproximadas para diferentes formas en experimentos termogravimétricos (TG).


En otro conjunto de métodos termogravimétricos, la forma A, la forma A después de la adsorción/desorción, y Forma A después de los ensayos de hidratación de adsorción/desorción, se transfirieron todos a una bandeja de muestra de aluminio. La curva TG se registró en un termogravímetro TA Instrument Hi-Res TGA 2950 en las siguientes condiciones:
temperatura inicial: temperatura ambiente
velocidad de calentamiento: 20°C/min
factor de resolución: 4
condición final: 300°C o <80 [% (p/p)]
Las curvas TG de las muestras se recogen en la figura 16.
Tabla 11 muestra las pérdidas de masa para las formas ensayadas:

La pérdida de peso a temperaturas de hasta 80°C se debe principalmente a la evaporación del solvente (agua) presente en la muestra. La pérdida de peso a temperaturas superiores a 80°C se debe principalmente a la evaporación del solvente (etanolato) presente en la muestra.
Una curva de TG de la forma A a 25°C en atmósfera de nitrógeno seco en función del tiempo se recoge en la figura 17. La pérdida de peso a 25°C después de 10 horas fue alrededor del 0.6%. Esto se debió a la evaporación del solvente.
Ejemplo 5 (Referencia)
En otro ejemplo, se realizaron también mediciones de calorimetría diferencial de barrido (DSC). Para ello, se utilizó un sistema de análisis térmico Perkin Elmer DSC 204. De 2 a 5 mg de muestra de la forma A se pesaron con precisión en una bandeja DSC. Los experimentos se realizaron en una bandeja abierta. La muestra se equilibró a aproximadamente 30°C y luego se calentó a una velocidad de 10°C por minuto, hasta una temperatura final de 200°C. Los datos de DSC se obtuvieron siguiendo un método estándar en la técnica. La forma A se caracterizó por calorimetría diferencial de barrido (DSC) en la que mostró una endotermia aguda en el intervalo de 80-119°C, mostrando un pico a aproximadamente 105.6°C, con un inicio delta H=-98.33 J/g. De acuerdo con lo anterior, la forma A cristalina de solvato de etanol A del compuesto de fórmula (X) (1:1) mostró el patrón termográfico, que aparece en la figura 8.
En otro conjunto de mediciones de DSC, se examinó la forma A, la forma A después de la adsorción/desorción, y la forma A después de los ensayos de hidratación de adsorción/desorción. Se transfirieron aproximadamente 3 mg de las muestras a una bandeja de muestra Perkin Elmer de aluminio perforada de 30 pl. La bandeja de muestra se cerró con la cubierta apropiada y se registró la curva de DSC en un Perkin Elmer Pyris DSC, en las siguientes condiciones:
temperatura inicial: 25°C
velocidad de calentamiento: 10°C/min
temperatura final: 150°C
flujo de nitrógeno: 30 ml/min
La forma A mostró una señal endotérmica a aproximadamente 104.6°C y un calor de fusión de 95.8 J/g provocado por la evaporación del etanolato y la fusión del producto. La forma A después de ADS/DES mostró una señal endotérmica amplia debido a una mezcla de la forma A etanolato y la forma B hidratada. La forma A después del ensayo de hidratación ADS/DES mostró una señal endotérmica a aproximadamente 73.5°C y un calor de fusión de 126 J/g causada por la evaporación del agua y la fusión del producto. Las curvas termográficas se representan en la figura 15.
Ejemplo 6 (Referencia)
En otro ejemplo se ensayaron estudios de estabilidad de la forma A en tres condiciones diferentes. Se incluyeron condiciones de 25°C y 60% de RH, 40°C y 75% de RH, y 50°C. Estos estudios revelaron que a 25°C y 60% de RH, la estabilidad a largo plazo, la cantidad de etanol y agua es estable.
Tabla 12 muestra el estudio de estabilidad para la forma A. Estabilidad a largo plazo a 25°C/60% de RH (humedad relativa), con botellas de vidrio marrón como recipiente de muestra.

Ejemplo 7 (Referencia)
Pruebas de adsorción-desorción
Se transfirieron aproximadamente 23 mg de la forma A en un analizador de sorción de vapor VTI modelo SGA100 y se registró el cambio de peso con respecto a la humedad atmosférica en las siguientes condiciones:
temperatura de secado: 40°C
equilibrio: < 0.05% en 5 minutos o 60 minutos.
intervalo de datos: 0.05% o 2 min.
temperatura: 25°C
primer ciclo RH (%) adsorción: 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 95
RH (%) desorción: 95, 90, 80, 70, 60, 50, 40, 30, 20, 10, 5
segundo ciclo RH (%) adsorción: 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 95
RH (%) desorción: 95, 90, 80, 70, 60, 50, 40, 30, 20, 10, 5
En la etapa de secado se registró aproximadamente 0.6% de pérdida de peso. El producto seco obtenido no era higroscópico, adsorbe hasta 0.7% de agua a alta humedad relativa. Durante el ciclo de desorción se registró una pérdida de peso del 1.4%, lo que indicó que el producto estaba perdiendo etanolato. El producto obtenido después de ADS/DES fue una mezcla de la forma etanolato y la forma hidratada.
La curva ADS/DES se recoge en la figura 18.
Ensayos de hidratación de adsorción-desorción
Se transfirieron aproximadamente 23 mg de la forma A en un analizador de sorción de vapor VTI modelo SGA100 y se registró el cambio de peso con respecto a la humedad atmosférica en las siguientes condiciones:
equilibrio: <0.0005% en 5min. o 90min.
intervalo de datos: 0.05% o 2min
temperatura: 25°C
ciclo RH (%) adsorción/desorción: 5.95 repetir el ciclo 11 veces
Al final de este ensayo se registró una pérdida de peso del 5.2%. Esto fue comparable con el resultado TG (TG 5.4% hasta 80°C). La forma de etanolato se transfirió a una forma hidratada. Las curvas de prueba de hidratación ADS/DES se recogen en la figura 19.
Ejemplo 8 (Referencia)
La estabilidad de la forma A se estudió después del almacenamiento del compuesto en un recipiente de muestra con una cubierta interior hecha de un solo LD-PE (sellado en cadena) y una cubierta externa hecha de PETP/Alu/PE (Moplast) termosellada. Se realizó un estudio de estabilidad a largo plazo a 25°C/60% de RH, y un estudio de estabilidad acelerado a 40°C/75% de RH durante un período de 6 meses, y las muestras se analizaron en diferentes momentos como se muestra en las siguientes tablas.
Tabla 13 Estabilidad a largo plazo a 25°C/60% de RH

Tabla 14 Estabilidad acelerada a 40°C/75% de RH

La forma A mostró estabilidad química y cristalina en las condiciones mencionadas en las Tablas 13 y 14.
Ejemplo 9 (Referencia)
La estabilidad de la forma A se estudió después de almacenar el compuesto en un recipiente de muestra con una cubierta interior hecha de LD-PE individual (sellado en cadena) y una cubierta externa hecha de bolsa de vapor loc (LPS) sellada térmicamente. Se realizó un estudio de estabilidad a largo plazo a 25°C/60% de RH, y un estudio de estabilidad acelerado a 40°C/75% de RH durante un período de 6 meses y las muestras se analizaron en diferentes momentos como se muestra en las siguientes tablas.
Tabla 15 Estabilidad a largo plazo a 25°C/60% de RH

Tabla 16: Estabilidad acelerada a 40°C/75% de RH

La forma A mostró estabilidad química y cristalográfica en las condiciones mencionadas en las tablas 15 y 16.
Ejemplo 10 (Referencia)
Para los ensayos de estabilidad química, la forma A se almacenó durante un período de 1, 4 y 8 semanas bajo diferentes condiciones. Estas condiciones fueron 40°C/75% de RH, 50°C, RT/<5% de RH, RT/56% de RH, RT/75% de RH y luz 0.3da ICH. El compuesto se analizó después de almacenamiento por HPLC y por inspección visual. El método de HPLC utilizado en este estudio fue el método de HPLC 909. Los resultados de los ensayos se presentan en la siguiente tabla.
Tabla 17

Se concluyó que la forma A es químicamente estable después del almacenamiento en todas las condiciones investigadas.
Ejemplo 11 (Referencia)
Se caracterizaron diferentes fracciones de la forma B con termogravimetría (TG), calorimetría diferencial de barrido (DSC) y espectroscopia infrarroja (IR). Los resultados de los ensayos se presentan en la siguiente tabla.
Tabla 18

Ejemplo 12 (Referencia)
Se investigó la adsorción y desorción de agua a 25°C en diferentes condiciones de humedad relativa en 38 mg de la forma B. Se registró el cambio de peso en función de la humedad relativa. Los resultados se muestran en la figura 20. En la etapa de secado se registró aproximadamente un 5.6% de pérdida de peso para la forma B. El producto seco obtenido era higroscópico, adsorbe hasta 6.8% de agua a alta humedad relativa. Después del ciclo de desorción, permaneció en la muestra aproximadamente 1.2% de agua. El producto obtenido después de ADS/DES fue una mezcla de hidrato y producto amorfo.
Ejemplo 13 (Referencia)
Las solubilidades acuosas de la forma B se midieron en solventes con diferentes pH. Se equilibró un exceso del soluto con el solvente a 20°C, durante al menos 24 horas. Después de retirar el compuesto no disuelto, se determinó la concentración en solución usando espectrometría UV.
Tabla 19

Ejemplo 14 (Referencia)
La estabilidad de la estructura cristalina de la forma B se estudió después del almacenamiento del compuesto durante un período de dos semanas a temperatura ambiente (RT) bajo <5%, 56% y 75% de humedad relativa (RH), 50°C y 40°C/75% de RH. Las muestras fueron analizadas mediante termogravimetría (TG), calorimetría diferencial de barrido (DSC), espectroscopia infrarroja (IR) y difracción de rayos X (DRX). Los resultados de los ensayos se presentan en la siguiente tabla.
Tabla 20

Ejemplo 15 (Referencia)
En el programa de ensayo de estabilidad química, la forma B se almacenó durante un periodo de 1,4 y 9 semanas bajo diferentes condiciones. Estas condiciones fueron 40°C/75% de RH, 50°C, RT/<5% de RH, RT/56% de RH, rT/75% de RH y luz 0.3da ICH. El compuesto se analizó después de almacenamiento por HPLC y por inspección visual. El método de HPLC utilizado en este estudio fue el método de HPLC 909. Los resultados de los ensayos se presentan en la siguiente tabla, a partir de la cual se concluyó que la forma B es químicamente estable.
Tabla 21

Ejemplo 16 (Referencia)
La forma K se preparó adicionando ácido metanosulfónico puro a una solución de la forma A en THF a r.t. La forma K se mezcló posteriormente con haluro de álcali y se prensó en un pella (Ph. Eur.) y se analizó por espectrometría de infrarrojos (IR) en las siguientes condiciones:
Aparato Espectrofotómetro Nicolet Magna 560 FTIR
número de barridos: 32
resolución: 1 cm-1
intervalo de longitud de onda: 4000 a 400 cm-1
corrección de línea base: sí
detector: DTGS con ventanas de KBr
divisor de haz: Ge en KBr
haluro de álcali: KBr (bromuro de potasio)
El espectro IR de la forma K, como se muestra en la figura 21, refleja los modos vibratorios de la estructura molecular del solvato mesilato como un producto cristalino.
Tabla 22


Ejemplo 17 (Referencia)
La forma K se transfirió a una celda capilar de vidrio y se analizó mediante espectrometría Raman en las siguientes condiciones:
Modo Raman: Raman No dispersivo
Aparato Módulo Nicolet FT-Raman
número de barridos: 64
resolución: 4 cm-1
intervalo de longitud de onda: 3700 a 100 cm-1
láser: Nd: YVO4
frecuencia de láser: 1064 cm-1
detector: InGaAs
divisor de haz: CaF2
geometría de la muestra: 180° reflectante
polarización: no
El espectro de Raman de la forma K, como se muestra en la figura 22, refleja los modos de vibración de la estructura molecular del mesilato como un producto cristalino.
Tabla 23


Ejemplo 18 (Referencia)
Se transfirieron aproximadamente 3 mg de la forma K a una bandeja de muestra de aluminio TA-Instrument estándar. La bandeja de muestras se cerró con la tapa apropiada y la curva DSC se registró en un TA-Instruments Q1000 MTDSC equipado con una unidad de refrigeración RCS, en las siguientes condiciones:
temperatura inicial: 25°C
velocidad de calentamiento: 10°C/min
temperatura final: 200°C
flujo de nitrógeno: 50 ml/min
La curva DSC como se representa en la figura 23, muestra la fusión con descomposición de un producto cristalino. La fusión de la forma K tiene lugar a 158.4°C. Debido a la descomposición, el cálculo del calor de fusión sólo se puede usar para indicar la propiedad cristalina del producto.
Ejemplo 19 (Referencia)
La forma K se transfirió a una bandeja de muestra de aluminio. La curva TG se registró en un termogravímetro TA Instruments H-Res TGA 2950 en las siguientes condiciones:
temperatura inicial: temperatura ambiente
velocidad de calentamiento: 20°C/min
factor de resolución: 4
condición final: 300°C o <80 [% (p/p)]
La curva TG se muestra en la figura 24. La pérdida de peso de aproximadamente 0.2% hasta 60°C se debió a la evaporación del solvente. La pérdida de peso a temperaturas superiores a 140°C se debió a la evaporación y descomposición del producto.
Ejemplo 20 (Referencia)
Adsorción-Desorción
Se transfirieron aproximadamente 21 mg de la forma K a un analizador de sorción de vapor VTI modelo SGA100 y se registró el cambio de peso con respecto a la humedad atmosférica en las siguientes condiciones:
temperatura de secado: 40°C
equilibrio: < 0.05% en 5 min. o 60 minutos.
intervalo de datos: 0.05% o 2.0 min.
temperatura: 25°C
primer ciclo RH (%) adsorción: 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 95
RH (%) desorción: 95, 90, 80, 70, 60, 50, 40, 30, 20, 10, 5
segundo ciclo RH (%) adsorción: 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 95
RH (%) desorción: 95, 90, 80, 70, 60, 50, 40, 30, 20, 10, 5
La isoterma de adsorción-desorción se muestra en la figura 25. La forma K es higroscópica. En la etapa de secado inicial se registró una pérdida de peso del 0.3%, comparable al resultado TG. La forma K adsorbió hasta 1.5% de agua a alta humedad relativa. El producto se secó completamente durante el ciclo de desorción.
Se investigó un estudio diferente de la adsorción y desorción de agua por la forma K a 25°C en diferentes condiciones de humedad relativa en una cantidad de aproximadamente 18 mg del solvato de mesilato. Se registró el cambio de peso en función de la humedad relativa. El resultado se muestra en la figura 26.
En la etapa de secado se registra aproximadamente 0.6% de pérdida de peso para la forma K. El producto seco obtenido es ligeramente higroscópico, adsorbe hasta 1.7% de agua a humedad relativa alta. El producto se secó completamente durante el ciclo de desorción.
Ejemplo 21 (Referencia)
Las solubilidades acuosas de la forma K se midieron en solventes con diferentes pH. Se equilibró un exceso del soluto con el solvente a 20°C durante al menos 48 horas. Después de retirar el compuesto no disuelto, se determinó la concentración en solución usando espectrometría UV.
Tabla 24

Ejemplo 22 (Referencia)
La estabilidad de la estructura cristalina de la forma K lote 1 se estudió después del almacenamiento del compuesto durante un período de cuatro semanas a temperatura ambiente (RT) bajo una humedad relativa (RH) del 75%, 50°C y 40°C/75% de RH. La estabilidad de la estructura cristalina de la forma K lote 2 se estudió después del almacenamiento del compuesto durante un período de cuatro semanas a temperatura ambiente (RT) bajo <5%, 56% y 75% de humedad relativa (RH), 50°C y 40°C/75% de RH. Las muestras se analizaron con termogravimetría (TG), calorimetría diferencial de barrido (DSC) y espectroscopia infrarroja (IR). Los resultados de los ensayos se presentan en la siguiente tabla.
Tabla 25


Ejemplo 23 (Referencia)
En el programa de ensayo de estabilidad química, la forma K lote 1 se almacenó durante un período de 1 y 4 semanas bajo diferentes condiciones. Estas condiciones fueron 40°C/75% de RH, 50°C, RT/75% de RH y luz 0.3da ICH. La forma K lote 2 también se almacenó durante un período de 1 y 4 semanas bajo diferentes condiciones. Estas condiciones fueron 40°C/75% de RH, 50°C, RT/<5% de RH, RT/56% de RH, RT/75% de RH y luz 0.3da ICH. El compuesto se analizó después de almacenamiento por HPLC y por inspección visual. El método de HPLC utilizado en este estudio fue el método de HPLC 909. Los resultados de los ensayos se presentan en la siguiente tabla.
Tabla 26

Ejemplo 24 (Referencia)
Se realizó un ensayo aleatorizado, doble ciego, controlado con placebo, de dosis múltiples, para examinar la seguridad, tolerabilidad y farmacocinética de la forma A después de la administración oral dos o tres veces al día, en sujetos sanos. Se ensayaron cuatro dosificaciones de la forma A (400 mg b.i.d., 800 mg b.i.d., 800 mg t.i.d. y 1200 mg t.i.d.) en 4 paneles de 9 sujetos sanos. Dentro de cada panel, 6 sujetos fueron tratados con la forma A y 3 sujetos con placebo durante 13 días con una sola ingesta en la mañana del día 14. (b.i.d. = dos veces al día, t.i.d. = tres veces al día).
La forma A se absorbió fácilmente y los perfiles de tiempo de concentración de la forma A después de la dosificación repetida dependieron de la dosis administrada. Las concentraciones plasmáticas en estado estacionario se alcanzaron generalmente en 3 días, aunque C0h (conc. en el momento de la administración) y AUC24h (área bajo la curva o biodisponibilidad) disminuyeron ligeramente con el tiempo en todos los niveles de dosis. AUC24h y Css,av (conc. en estado estacionario medio) fueron proporcionales a la dosis (dosis diaria) a 400 mg b.i.d., 800 mg t.i.d. y 1200 mg t.i.d., pero era más que proporcional a la dosis a 800 mg b.i.d. La Cmax (concentración máxima) fue proporcional a la dosis con respecto a la dosis por toma. Menos del 2% de la forma A inalterada se excretó en la orina en todos los niveles de dosis.
Claims (3)
1. Un pseudopolimorfo de (3R,3aS,6aR)-hexahidrofuro [2,3-b] furan-3-il (1S,2R)-3-[[(4-aminofenil) sulfonil] (isobutil) amino]-1-bencil-2-hidroxipropilcarbamato, en donde el pseudopolimorfo es un solvato de isopropanol, que contiene 50% en moles de isopropanol.
2. Un pseudopolimorfo de acuerdo con la reivindicación 1, para usar como una medicina.
3. Un pseudopolimorfo de acuerdo con la reivindicación 1, para uso en el tratamiento de una infección retroviral.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP02076929 | 2002-05-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2728735T3 true ES2728735T3 (es) | 2019-10-28 |
Family
ID=29724454
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14168686.5T Expired - Lifetime ES2638412T3 (es) | 2002-05-16 | 2003-05-16 | Formas pseudopolimórficas de un inhibidor de la proteasa del HIV |
| ES16158480T Expired - Lifetime ES2728735T3 (es) | 2002-05-16 | 2003-05-16 | Formas pseudopolimórficas de un inhibidor de la proteasa del HIV |
| ES03753571T Expired - Lifetime ES2498370T5 (es) | 2002-05-16 | 2003-05-16 | Formas pseudopolimórficas de un inhibidor de la proteasa de VIH |
| ES10180831T Expired - Lifetime ES2503551T5 (es) | 2002-05-16 | 2003-05-16 | Formas pseudopolimórficas de un inhibidor de la proteasa de VIH |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14168686.5T Expired - Lifetime ES2638412T3 (es) | 2002-05-16 | 2003-05-16 | Formas pseudopolimórficas de un inhibidor de la proteasa del HIV |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES03753571T Expired - Lifetime ES2498370T5 (es) | 2002-05-16 | 2003-05-16 | Formas pseudopolimórficas de un inhibidor de la proteasa de VIH |
| ES10180831T Expired - Lifetime ES2503551T5 (es) | 2002-05-16 | 2003-05-16 | Formas pseudopolimórficas de un inhibidor de la proteasa de VIH |
Country Status (26)
| Country | Link |
|---|---|
| US (8) | US7700645B2 (es) |
| EP (4) | EP3045460B1 (es) |
| JP (1) | JP4864320B2 (es) |
| KR (2) | KR101128370B1 (es) |
| CN (1) | CN100475819C (es) |
| AP (1) | AP2052A (es) |
| AU (2) | AU2003271740B2 (es) |
| BR (1) | BRPI0311176B8 (es) |
| CA (1) | CA2485834C (es) |
| CY (3) | CY1117928T1 (es) |
| DK (3) | DK2314591T4 (es) |
| EA (1) | EA007120B8 (es) |
| ES (4) | ES2638412T3 (es) |
| HK (1) | HK1081969A1 (es) |
| HR (1) | HRP20041061B1 (es) |
| HU (1) | HUE034389T2 (es) |
| IL (1) | IL165140A0 (es) |
| LT (1) | LT2767539T (es) |
| MX (1) | MXPA04011427A (es) |
| NO (1) | NO331477B1 (es) |
| NZ (1) | NZ536497A (es) |
| PL (1) | PL215151B1 (es) |
| PT (3) | PT1567529E (es) |
| SI (3) | SI1567529T2 (es) |
| WO (1) | WO2003106461A2 (es) |
| ZA (1) | ZA200410154B (es) |
Families Citing this family (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK1088098T4 (en) | 1998-06-23 | 2015-09-14 | Us Of America Represented By The Secretary Dept Of Health And Human Services | Fitnessassay and methods for reducing HIV resistance to therapy |
| LT2767539T (lt) * | 2002-05-16 | 2017-09-25 | Janssen Sciences Ireland Uc | Pseudopolimorfinės živ proteazės inhibitorių formos |
| HRP20020614A2 (en) * | 2002-07-22 | 2004-06-30 | PLIVA-ISTRAŽIVAČKI INSTITUT d.o.o. | Rhombic pseudopolymorph of 9-deoxo-9a-aza-9a-methyl-9a-homoerythromycin a |
| UA85567C2 (en) | 2003-12-23 | 2009-02-10 | Тиботек Фармасьютикелз Лтд. | Process for the preparation of (3r,3as,6ar)-hexahydrofuro [2,3-b] furan-3-yl (1s,2r)-3-[[(4-aminophenyl) sulfonyl] (isobutyl) amino]-1-benzyl-2-hydroxypropylcarbamate |
| BRPI0607827B8 (pt) | 2005-02-25 | 2021-05-25 | Janssen R & D Ireland | composto precursor de inibidor de protease, processo para a produção do referido composto e seu uso |
| PL2089371T3 (pl) * | 2006-11-09 | 2011-06-30 | Janssen Sciences Ireland Uc | Sposoby wytwarzania heksahydrofuro[2,3-b]furan-3-olu |
| TWI482775B (zh) | 2008-09-01 | 2015-05-01 | Tibotec Pharm Ltd | 用於製備(3r,3as,6ar)-六氫呋喃并〔2,3-b〕呋喃-3-基(1s,2r)-3-〔〔(4-胺基苯基)磺醯基〕(異丁基)胺基〕-1-苯甲基-2-羥基丙基胺基甲酸酯之方法 |
| US20120035142A1 (en) * | 2009-01-29 | 2012-02-09 | Mapi Pharma Limited | Polymorphs of darunavir |
| US8921415B2 (en) | 2009-01-29 | 2014-12-30 | Mapi Pharma Ltd. | Polymorphs of darunavir |
| ES2619562T3 (es) * | 2009-09-17 | 2017-06-26 | Mylan Laboratories Limited | Procesos para la preparación de darunavir y su forma amorfa |
| JP2013509400A (ja) * | 2009-10-30 | 2013-03-14 | ルパン リミテッド | ダルナビルの新規調製方法および微粒子径のダルナビルエタノール付加物 |
| CN102686594A (zh) * | 2009-12-16 | 2012-09-19 | 熙德隆研究基金会 | 地瑞那韦的多晶型物 |
| AU2010340799B2 (en) * | 2010-01-05 | 2015-06-11 | Cipla Limited | Darunavir polymorph and process for preparation thereof |
| CA2784398A1 (en) | 2010-01-28 | 2011-08-04 | Mapi Pharma Limited | Process for the preparation of darunavir and darunavir intermediates |
| PT2571355T (pt) * | 2010-05-20 | 2016-10-17 | Hetero Research Foundation | Sal de cloridrato cristalino de darunavir |
| WO2012107889A1 (en) | 2011-02-10 | 2012-08-16 | Ranbaxy Laboratories Limited | Process for the preparation of amorphous darunavir |
| SI2729130T1 (en) | 2011-07-07 | 2018-03-30 | Janssen Sciences Ireland Uc | Combined formulations of darunavir |
| US9216990B2 (en) | 2011-12-05 | 2015-12-22 | Mylan Labs Limited | Crystalline Darunavir |
| WO2013108105A2 (en) * | 2012-01-18 | 2013-07-25 | Aurobindo Pharma Limited | Novel solvates of darunavir |
| CN103509031B (zh) * | 2012-06-20 | 2016-04-27 | 上海迪赛诺药业有限公司 | 制备达芦那韦无定形物的方法 |
| ES2848216T3 (es) * | 2012-07-24 | 2021-08-05 | Laurus Labs Ltd | Un proceso para la preparación de Darunavir |
| WO2014068265A1 (en) | 2012-10-29 | 2014-05-08 | Cipla Limited | Antiviral phosphonate analogues and process for preparation thereof |
| US9346820B2 (en) * | 2013-09-11 | 2016-05-24 | Purdue Research Foundation | HIV-1 protease inhibitors having gem-di-fluoro bicyclic P2-ligands |
| WO2016092525A1 (en) * | 2014-12-12 | 2016-06-16 | Lupin Limited | Darunavir n-propanol solvate and process for preparation thereof |
| WO2016092527A1 (en) * | 2014-12-12 | 2016-06-16 | Sun Pharmaceutical Industries Limited | A process for the preparation of dolutegravir |
| HUE059771T2 (hu) | 2015-03-19 | 2022-12-28 | Mycovia Pharmaceuticals Inc | Gombaellenes vegyületek és eljárások azok elõállítására |
| BR112019002120A2 (pt) | 2016-08-08 | 2019-05-14 | Hetero Labs Limited | composição anti-retroviral de várias classes |
| EP3496718A4 (en) | 2016-08-08 | 2020-01-22 | Hetero Labs Limited | ANTIRETROVIRAL COMPOSITIONS |
| US10407438B2 (en) | 2016-10-27 | 2019-09-10 | Gilead Sciences, Inc. | Crystalline forms of darunavir |
| CN108727401A (zh) * | 2017-04-20 | 2018-11-02 | 盐城迪赛诺制药有限公司 | 达鲁那韦新晶型及其制备方法和应用 |
| CN109053753A (zh) * | 2018-08-05 | 2018-12-21 | 浙江大学 | 一种制备达卢那韦二水合物晶型的方法 |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5413999A (en) * | 1991-11-08 | 1995-05-09 | Merck & Co., Inc. | HIV protease inhibitors useful for the treatment of AIDS |
| JPH05230044A (ja) | 1992-02-21 | 1993-09-07 | Hoechst Japan Ltd | ピレタニドの新規な結晶多形 |
| US5968942A (en) | 1992-08-25 | 1999-10-19 | G. D. Searle & Co. | α- and β-amino acid hydroxyethylamino sulfonamides useful as retroviral protease inhibitors |
| ES2127938T3 (es) | 1993-08-24 | 1999-05-01 | Searle & Co | Hidroxietilamino sulfonamidas utiles como inhibidores de proteasas retroviricas. |
| EP0812320B1 (en) * | 1995-02-22 | 2000-07-12 | Hoechst Pharmaceuticals & Chemicals K.K. | Amorphous piretanide, piretanide polymorphs, process for their preparation and their use |
| CO4940492A1 (es) * | 1997-05-29 | 2000-07-24 | Merck & Co Inc | Inhibidor de proteasa de vih |
| GB9712253D0 (en) | 1997-06-13 | 1997-08-13 | Glaxo Group Ltd | Antiviral compound |
| US6287693B1 (en) * | 1998-02-25 | 2001-09-11 | John Claude Savoir | Stable shaped particles of crystalline organic compounds |
| GB9805898D0 (en) * | 1998-03-20 | 1998-05-13 | Glaxo Group Ltd | Process for the sythesis of hiv protease inhibitors |
| GB9807354D0 (en) | 1998-04-07 | 1998-06-03 | Glaxo Group Ltd | Antiviral compound |
| DK1088098T4 (en) | 1998-06-23 | 2015-09-14 | Us Of America Represented By The Secretary Dept Of Health And Human Services | Fitnessassay and methods for reducing HIV resistance to therapy |
| AU4828199A (en) * | 1998-06-23 | 2000-01-10 | Board Of Trustees Of The University Of Illinois, The | Multi-drug resistant retroviral protease inhibitors and associated methods |
| AU1735000A (en) | 1998-11-19 | 2000-06-05 | Du Pont Pharmaceuticals Company | Crystalline (-)-6- chloro-4- cyclopropylethynyl- 4-trifluoromethyl- 3,4-dihydro-2(1h)-quinazolinone |
| PT1159278E (pt) | 1999-02-12 | 2006-06-30 | Vertex Pharma | Inibidores de aspartil-protease |
| LT2767539T (lt) * | 2002-05-16 | 2017-09-25 | Janssen Sciences Ireland Uc | Pseudopolimorfinės živ proteazės inhibitorių formos |
| AU2010340799B2 (en) * | 2010-01-05 | 2015-06-11 | Cipla Limited | Darunavir polymorph and process for preparation thereof |
-
2003
- 2003-05-16 LT LTEP14168686.5T patent/LT2767539T/lt unknown
- 2003-05-16 US US10/514,352 patent/US7700645B2/en active Active
- 2003-05-16 NZ NZ536497A patent/NZ536497A/en not_active IP Right Cessation
- 2003-05-16 DK DK10180831.9T patent/DK2314591T4/da active
- 2003-05-16 CN CNB038164590A patent/CN100475819C/zh not_active Expired - Lifetime
- 2003-05-16 BR BRPI0311176A patent/BRPI0311176B8/pt active IP Right Grant
- 2003-05-16 EP EP16158480.0A patent/EP3045460B1/en not_active Expired - Lifetime
- 2003-05-16 JP JP2004513292A patent/JP4864320B2/ja not_active Expired - Lifetime
- 2003-05-16 WO PCT/EP2003/050176 patent/WO2003106461A2/en active Application Filing
- 2003-05-16 MX MXPA04011427A patent/MXPA04011427A/es active IP Right Grant
- 2003-05-16 ES ES14168686.5T patent/ES2638412T3/es not_active Expired - Lifetime
- 2003-05-16 ES ES16158480T patent/ES2728735T3/es not_active Expired - Lifetime
- 2003-05-16 DK DK14168686.5T patent/DK2767539T3/en active
- 2003-05-16 PT PT37535713T patent/PT1567529E/pt unknown
- 2003-05-16 SI SI200332389T patent/SI1567529T2/sl unknown
- 2003-05-16 HU HUE14168686A patent/HUE034389T2/en unknown
- 2003-05-16 SI SI200332545T patent/SI2767539T1/sl unknown
- 2003-05-16 CA CA002485834A patent/CA2485834C/en not_active Expired - Lifetime
- 2003-05-16 EP EP10180831.9A patent/EP2314591B2/en not_active Expired - Lifetime
- 2003-05-16 KR KR1020047018426A patent/KR101128370B1/ko active IP Right Grant
- 2003-05-16 DK DK03753571.3T patent/DK1567529T4/da active
- 2003-05-16 ES ES03753571T patent/ES2498370T5/es not_active Expired - Lifetime
- 2003-05-16 SI SI200332383T patent/SI2314591T2/sl unknown
- 2003-05-16 PL PL374321A patent/PL215151B1/pl unknown
- 2003-05-16 AP AP2004003191A patent/AP2052A/xx active
- 2003-05-16 EA EA200401503A patent/EA007120B8/ru unknown
- 2003-05-16 AU AU2003271740A patent/AU2003271740B2/en not_active Revoked
- 2003-05-16 EP EP14168686.5A patent/EP2767539B1/en not_active Expired - Lifetime
- 2003-05-16 PT PT101808319T patent/PT2314591E/pt unknown
- 2003-05-16 ES ES10180831T patent/ES2503551T5/es not_active Expired - Lifetime
- 2003-05-16 PT PT141686865T patent/PT2767539T/pt unknown
- 2003-05-16 KR KR1020107024106A patent/KR20100119906A/ko not_active Application Discontinuation
- 2003-05-16 EP EP03753571.3A patent/EP1567529B2/en not_active Expired - Lifetime
-
2004
- 2004-11-10 IL IL16514004A patent/IL165140A0/xx unknown
- 2004-11-12 HR HRP20041061AA patent/HRP20041061B1/hr not_active IP Right Cessation
- 2004-12-10 NO NO20045409A patent/NO331477B1/no not_active IP Right Cessation
- 2004-12-15 ZA ZA2004/10154A patent/ZA200410154B/en unknown
-
2006
- 2006-02-23 HK HK06102443.3A patent/HK1081969A1/xx not_active IP Right Cessation
-
2009
- 2009-08-06 US US12/536,807 patent/US8518987B2/en not_active Expired - Lifetime
-
2012
- 2012-07-03 AU AU2012205289A patent/AU2012205289A1/en active Pending
-
2013
- 2013-07-11 US US13/939,494 patent/US20130303790A1/en not_active Abandoned
-
2014
- 2014-02-19 US US14/183,712 patent/US20140171499A1/en not_active Abandoned
- 2014-09-17 CY CY20141100756T patent/CY1117928T1/el unknown
- 2014-09-17 CY CY20141100755T patent/CY1115665T1/el unknown
-
2015
- 2015-08-04 US US14/817,827 patent/US20150336980A1/en not_active Abandoned
-
2017
- 2017-05-22 US US15/600,932 patent/US10000504B2/en not_active Expired - Lifetime
- 2017-08-23 CY CY20171100896T patent/CY1119311T1/el unknown
-
2018
- 2018-05-16 US US15/980,993 patent/US10858369B2/en not_active Expired - Lifetime
-
2020
- 2020-10-30 US US17/085,096 patent/US20210179631A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2728735T3 (es) | Formas pseudopolimórficas de un inhibidor de la proteasa del HIV | |
| ES2560955T3 (es) | Cristal de derivado de diamina y procedimiento de producción del mismo | |
| ES2806604T3 (es) | Formas cristalinas de monofumarato de tenofovir alafenamida | |
| ES2553574T3 (es) | Polimorfos de un principio activo farmacéutico | |
| ES2875402T3 (es) | Tosilato de palbociclib | |
| ES2327265T3 (es) | Una nueva forma cristalina estable de andolast. |