-
BEREICH DER
ERFINDUNG
-
Die
vorliegende Erfindung betrifft ein Verfahren zur Herstellung von
Eprosartan. Diese Verbindung ist im U.S.-Patent Nr. 5,185,351 als
zur Behandlung von Hypertonie, dekompensierter Herzinsuffizienz
und Nierenversagen verwendbarer Angiotensin-II-Rezeptorantagonist beschrieben.
-
HINTERGRUND
DER ERFINDUNG
-
Die
Herstellung von 1-(Carboxybenzyl)imidazol-5-acrylsäuren als
nicht-peptidischen Angiotensin-II-Rezeptorantagonisten ist beschrieben
worden (Keenan, R.M. et al (1993) J. Med. Chem. 36 1880–1892).
-
U.S.-Patent
Nr. 5,185,351 beschreibt Verfahren zur Herstellung von Imidazol-Verbindungen.
Eines dieser in dieser Anmeldung beschriebenen Verfahren ist die
Umsetzung eines Aldehyds mit einem substituierten halb Säure-, halb
Esterderivat eines Malonats. Obwohl dieses Verfahren die darin beanspruchten
Imidazole liefert, bestand die Notwendigkeit, dieses Verfahren zu
verbessern, wenn Verbindungen wie Eprosartan im kommerziellen Maßstab hergestellt
werden.
-
Es
ist nun festgestellt worden, dass Eprosartan durch Umsetzen von
4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure oder
der Hydrogensulfit-Additionsverbindung von 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure (PCT-Anmeldung
WO 95/32189) mit (2-Thienylmethyl)propandisäure-mono-ethylester
hergestellt werden kann, um Eprosartan effizient in hoher Ausbeute
und hoher Reinheit zu liefern. Die Effizienz dieses Verfahrens und
die Qualität
und Ausbeute des Imidazol-Produkts sind besonders wichtig, wenn
das Produkt im Großmaßstab für die therapeutische
Verwendung hergestellt wird.
-
BESCHREIBUNG DER ERFINDUNG
-
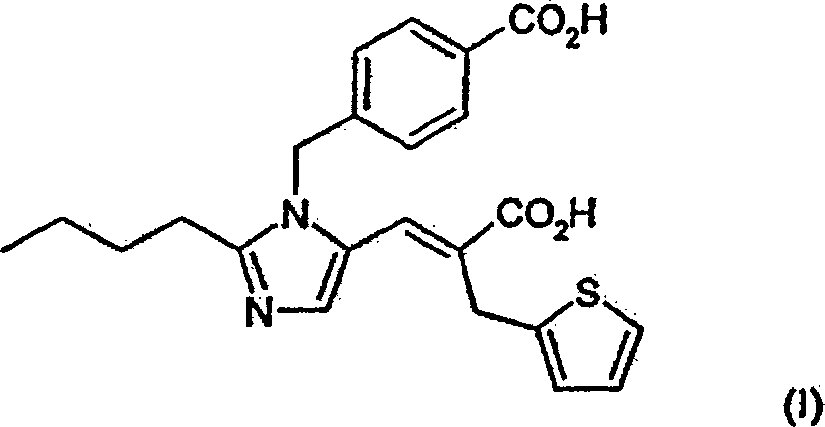
Die
vorliegende Erfindung stellt ein Verfahren zur Herstellung von Eprosartan,
das (E)-α-[[2-Butyl-1-[(4-carboxyphenyl)methyl]-1H-imidazol-5-yl]methylen]-2-thiophenpropansäure ist,
eine Verbindung der Formel (I):
bereit, wobei das Verfahren
das Umsetzen einer Verbindung der Formel (II):
oder eines Säure- oder
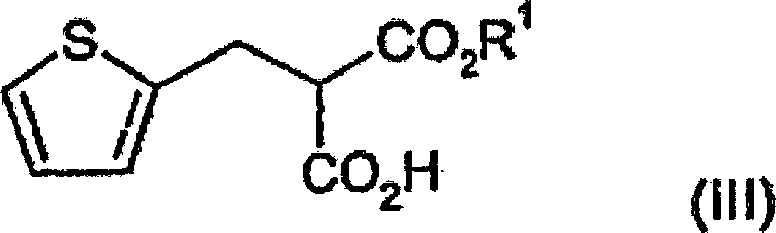
Basenadditionssalzes davon, mit einer Verbindung der Formel (III):
wobei R'C
1-4-Alkyl bedeutet,
bei
vermindertem Druck in Gegenwart eines Katalysators, insbesondere
Piperidin oder Piperidiniumpropionat in einem Überschuss an Propionsäure, und
anschließendes
Hydrolysieren des R'-Esters
und gegebenenfalls Bilden eines pharmazeutisch verträglichen
Salzes umfasst. Alternativ kann eine Formel (I)-Verbindung durch das
Umsetzen einer Verbindung der Formel (IV):
mit einer Formel (III)-Verbindung
bei vermindertem Druck in Gegenwart eines Katalysators, insbesondere
Piperidin oder Piperidiniumpropionat in einem Überschuss an Propionsäure, und
anschließendes
Hydrolysieren des R'-Esters
und gegebenenfalls Bilden eines pharmazeutisch verträglichen
Salzes hergestellt werden.
-
Säureadditionsalze
von Formel (I)- und (II)-Verbindungen werden mit den passenden anorganischen oder
organischen Säuren
mit in dem Fachgebiet bekannten Verfahren gebildet. Typische Beispiele
für geeignete
Säuren
sind Malein-, Fumar-, Essig-, Bernstein-, Salz-, Bromwasserstoff-,
Schwefel-, Phosphor- oder Methansulfonsäure. Vorzugsweise ist das pharmazeutisch
verträgliche
Säureadditionssalz
der Formel (I)-Verbindung das Methansulfonsäure-Additionssalz.
-
Basenadditionssalze
der Formel (I)- und (II)-Verbindungen werden mit den passenden anorganischen oder
organischen Basen mit in dem Fachgebiet bekannten Verfahren gebildet.
Kationensalze werden durch Behandeln der Stamm-Verbindung mit einem Überschuss
eines alkalischen Reagenz',
wie einem Hydroxid, Carbonat oder Alkoxid, das das passende Kation
enthält,
hergestellt; oder mit einem passenden organischen Amin. Typische
Beispiele für
Kationen sind Li
+, Na
+,
K
+, Ca
++, Mg
++ und NH
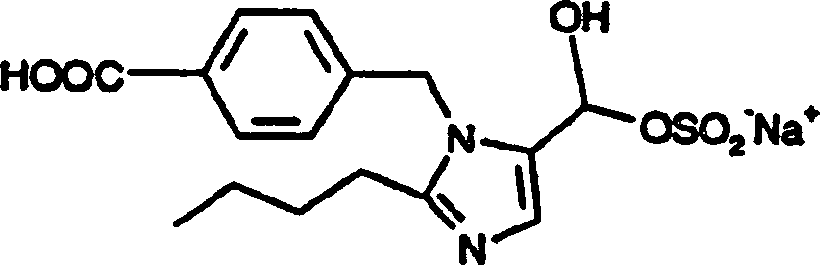
4 +. Die bevorzugte Salzform der Formel (II)-Verbindung ist
-
Wie
hier verwendet, bedeutet C1-4-Alkyl einen
Alkylrest mit 1 bis 4 Kohlenstoffatomen, verzweigt oder unverzweigt.
C1-4-Alkyl beinhaltet eine Methyl-, Ethyl-,
n-Propyl-, Isopropyl-, n-Butyl-,
Isobutyl- und t-Butylgruppe. Der bevorzugte R'C1-4-Alkylrest
ist eine Ethylgruppe.
-
Typischerweise
wird das Verfahren ausgeführt
durch Verbinden von 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure oder
der Bisulfit-Additionsverbindung von 4-[(2-n-Butyl-5-formyl-1H-imidazol-l-yl)methyl]benzoesäure mit
(2-Thienylmethyl)propandisäure-monoethylester
in einem geeigneten Lösungsmittel
wie Toluol, in Gegenwart eines Katalysators, beispielsweise in Gegenwart
von Piperidiniumpropionat in einem Überschuss an Propionsäure, bei
einer geeigneten Temperatur, wie bei einer Temperatur von etwa 75°C bis etwa
100°C, vorzugsweise
bei einer Temperatur von 80°C
bis 85°C,
bei vermindertem Druck, insbesondere bei einem auf etwa 23 bis 33
cm (9 bis 13 Zoll) Hg, vorzugsweise 28 cm (11 Zoll) Hg, verminderten
Innendruck. Die Ester-Vorstufen der Formel (I)-Verbindung werden
zu der entsprechenden Formel (I)-Carbonsäure unter Verwendung einer
Base wie wässrigem
Natrium- oder Kaliumhydroxid
hydrolisiert. Anschließend
können
pharmazeutisch verträgliche
Salze wie vorstehend beschrieben hergestellt werden.
-
Alternativ
werden 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure oder
die Bisulfit-Additionsverbindung von 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure und
(2-Thienylmethyl)propandisäure-mono-ethylester
durch Erhitzen der zwei Substrate in Toluol unter Rückfluss
unter vermindertem Druck und in Gegenwart von Piperidin als Katalysator
umgesetzt, gefolgt von Hydrolyse der Esterzwischenstufe (Ethyl-(E)-α-[[2-Butyl-1-[(4-carboxyphenyl)methyl]-1H-imidazol-5-yl]methylen]-2-thiophenpropanoat),
um (E)-α-[[2-Butyl-1-[(4-carboxyphenyl)methyl]-1H-imidazol-5-yl]methylen]-2-thiophenpropansäure zu ergeben. Bei
dieser Herstellung werden 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure oder
die Bisulfit-Additionsverbindung von 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure, (2-Thienylmethyl)propandisäure-mono-ethylester
und Toluol in einen Stahlbehälter
mit Glasfutter gefüllt
und werden anfänglich
auf 55–60°C erhitzt,
um eine homogene Lösung
zu erhalten. Der Katalysator (66 Mol-% Piperidin) wird zugegeben
und der Reaktionsansatz wird unter vermindertem Druck unter Rückfluss
erhitzt (70–75°C). Rückflussbedingungen
werden für
20 bis 35 Stunden aufrechterhalten und zusätzlicher (2-Thienylmethyl)propandisäure-mono-ethylester
wird zugegeben. Sobald die Umsetzung vollständig ist, werden Wasser und
wässrige Natriumhydroxid-Lösung zu
dem Gefäß zugegeben
und das Reaktionsgemisch unter Atmosphärenbedingungen für 1 bis
3 Stunden unter Rückfluss
erhitzt. Die Umsetzung wird als vollständig betrachtet, wenn der Anteil an
Ethyl-(E)-α-[[2-butyl-1-[(4-carboxyphenyl)methyl]-1H-imidazol-5-yl]methylen]-2-thiophenpropanoat
weniger als 2,0% beträgt.
Der Reaktionsansatz wird auf 45–50°C abgekühlt und
die wässrige
und organische Phase getrennt. Die Toluolphase wird verworfen. Ethanol
wird zu der wässrigen
Phase zugegeben und die Lösung wird
mit wässriger
Salzsäure
angesäuert,
bis ein pH-Wert von 5,0 bis 5,4 erreicht wird, wobei die Temperatur bei
50–55°C gehalten
wird. Man kühlt
die Produktaufschlämmung
ab und lässt
bei 10–15°C für 2 Stunden
rühren.
Das Produkt wird durch Zentrifugation isoliert, gewaschen und gelagert.
Anschließend
können
pharmazeutisch verträgliche
Salze wie vorstehend beschrieben hergestellt werden.
-
Die
mit Piperidin katalysierte Reaktion zwischen 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure oder
der Bisulfit-Additionsverbindung von 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure und
(2-Thienylmethyl)propandisäure-mono-ethylester
kann erfolgreich in von Toluol verschiedenen Lösungsmitteln (und/oder Lösungsmittelsystemen)
durchgeführt
werden; diese Lösungsmittel
beinhalten Cyclohexan, Cyclohexan:Dichlorethan (12:5 oder 1:1),
Cyclohexan:Pyridin (12:5) und Cyclohexan:Ethylacetat:Pyridin (8:3:1).
-
Die
Erfindung wird durch die folgenden Beispiele veranschaulicht. Die
Beispiele sollen den Umfang dieser Erfindung, wie vorstehend definiert
und wie nachstehend beansprucht, nicht begrenzen.
-
BEISPIELE
-
Beispiel 1
-
Synthese von (E)-α-[[2-Butyl-1-[4-carboxyphenyl)-methyl-1H-imidazol-5-yl]methylen]-2-thiophenpropansäure (Eprosartan)
-
Reagenzien und Lösungsmittel
-
- 1. Bisulfit-Additionsverbindung von 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure 12,03
kg 28,82 Mol
(68,57% 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure)
- 2. (2-Thienylmethyl)propandisäure-mono-ethylester 15,29 kg
(80,9% Gew./Gew.-Test) 54,18 Mol
- 3. Piperidin 2,85 l 28,82 Mol
- 4. Propionsäure
8,60 l 115,28 Mol
- 5. Toluol 56,5 l + 19,0 l = 75;5 l Gesamtmenge
- 6. Natriumhydroxid 16,7 kg (50%ige wässrige Lösung) 208,75 Mol
- 7. Wasser 65,0 l
- 8. Ethanol 41,2 kg
- 9. 6 N HCl Einstellen auf pH-Wert 5,0 bis 5,2
- 10. Wasser 75,0 l
-
Verfahren
-
- 1. Toluol(56,5 l) in den Reaktor füllen.
- 2. (2-Thienylmethyl)propandisäure-mono-ethylester (15,29
kg, 80,9% Gew./Gew.-Test) und die Bisulfit-Additionsverbindung von
4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure (12,03 kg, 68,57% 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure) in
den Reaktor füllen
und das Rühren
starten. Den Innendruck auf 28 cm (11 Zoll) Hg vermindern und unter
Rückfluss
(die Innentemperatur des Reaktionsansatzes wurde zwischen 80–85°C gehalten)
für 1 bis
2 Std erhitzen. Die Manteltemperatur auf 110°C einstellen. Das Wasser in
einer Dean-Stark-Falle auffangen.
- 3. Toluol (19,0 l), gefolgt von Propionsäure (6,45 l, 86,46 Mol), in
einen zweiten Reaktor füllen.
Die erhaltene Lösung
bei Raumtemperatur langsam mit Piperidin (2,85 l, 28,82 Mol) behandeln.
Das erhaltene Gemisch für
annähernd
30 Min rühren.
- 4. Den ersten Reaktor mit Stickstoff belüften und die Manteltemperatur
auf 80 C vermindern. Die Piperidiniumpropionat-Propionsäure-Lösung in
Toluol aus dem zweiten Reaktor in den ersten Reaktor überführen. Den
Innendruck auf 28 cm (11 Zoll) Hg vermindern und unter Rückfluss
(die Innentemperatur des Reaktionsansatzes wurde zwischen 80–85°C gehalten)
erhitzen. Die Manteltemperatur auf 140°C einstellen. Das Wasser in
einer Dean-Stark-Falle
auffangen.
- 5. Nach 7,5 Std betrug die Menge des in Lösung verbliebenen Aldehyds
(4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure) etwa
20% und die Menge des verbliebenen (2-Thienylmethyl)propandisäure-mono-ethylesters
betrug etwa 20%. Ein zusätzlicher
Anteil von (2-Thienylmethyl)propandisäure-mono-ethylester
(1,53 kg, 5,42 Mol) wurde bei der 8,5 Std-Markierung zugegeben.
- 6. Nach 13,5 Std war die Umsetzung vollständig und der Reaktionsansatz
wurde auf 70°C
abgekühlt.
(Die Menge des restlichen Aldehyds betrug etwa 5%.) Wasser (65,0
l) und Natriumhydroxid (16,7 kg; 50%ige (Gew./Gew.) wässrige Lösung) wurde
zugegeben und der Reaktionsansatz wurde zum Rückfluss gebracht.
- 7. Der Reaktionsansatz wurde für eine Stunde unter Rückfluss
erhitzt. Der Reaktionsansatz wurde auf das Vorhandensein von Ethyl-(E)-α-[[2-butyl-1-[(4-carboxyphenyl)methyl]-1H-imidazol-5-yl]methylen]-2-thiophenpropanoat
geprüft.
Falls vorhanden, eine zusätzliche
halbe Stunde unter Rückfluss
erhitzen. Den Test wiederholen.
- 8. Die Lösung
auf 60 C abkühlen.
Die Schichten trennen und Ethanol (41,2 kg) zu der Wasserschicht
geben. Den pH-Wert der Lösung
mit 6 N HCl (Temp. 60°C)
langsam auf 5,2 einstellen. Das Produkt wird anfangen zu kristallisieren
(Temp. 60°C).
Auf Raumtemperatur abkühlen
und für
zwei Stunden rühren.
Filtrieren und das Produkt mit Wasser (2 × 37,5 l) waschen.
- 9. Der Feststoff wurde vakuumgetrocknet (9,44 kg, 77,2%).
-
Analysedaten
-
HPLC
| Säule | Zorbax
SB-C18, 3,5 mm, 7,5 cm × 4,6
mm |
| Säulentemperatur | 40°C |
| Fließgeschwindigkeit | 2,0
ml/Min |
| Probenvorbereitung | 8
ml des Reaktionsansatzes werden mit einem Stickstoffstrom begast
und danach in 2 ml 50:50 Acetonitril:Wasser gelöst |
| Injektionsvolumen | 2,0
ml |
| Detektions-Wellenlänge | 235
nm |
| Mobile
Phase A | 0,1
M Ammoniumacetat (pH-Wert = 6,7) |
| Mobile
Phase B | 50:50
0,1 M Ammoniumacetat:Acetonitril |
| Gradientenprogramm | Von
0 bis 10 Minuten von 100% mobile Phase A bis 100% mobile Phase B
mit einem linearen Gradienten, 5 Minuten bei 100% |
| | mobile
Phase B, danach Re-Äquilibrieren
für 5 Minuten
mit 100% mobile Phase A |
| Entwicklungszeit | 15
Minuten |
| Äquilibrierungszeit | 5
Minuten |
| Retentionszeit | 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure: 4,81
Min
(E)-α-[[2-Butyl-1-[(4-carboxyphenyl)-methyl]-1H-imidazol-5-yl]methylen]-2-thiophenpropansäure: 4,58 Min
(2-Thienylmethyl)-propandisäure-mono-ethylester: 4,13
Min
Ethyl-(E)-α-[[2-butyl-1-[(4-carboxyphenyl)-methyl]-1H-imidazol-5-yl]methylen]-2-thiophenpropanoat:
8,43 Min |
-
Beispiel 2
-
Synthese von (E)-α-[[2-Butyl-1-[(4-carboxyphenyl)-methyl]-1H-imidazol-5-yl]methylen]-2-thiophenpropansäure (Eprosartan)
-
Ein
Stahlreaktionsgefäß mit Glasfutter
wird mit 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure, (2-Thienylmethyl)propandisäure-mono-ethylester
(etwa 1,9 Moläquivalente
bezüglich
getesteter 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure) und Toluol (etwa 6,3 g
pro Gramm getesteter 4-[(2-n-Butyl-5-formyl-1H-irnidazol-1-yl)methyl]benzoesäure) befüllt und
auf 55–60°C erhitzt.
Piperidin (annähernd
66 Mol-% bezüglich
der 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure) wird
zugegeben. Der Reaktionsansatz wird danach unter vermindertem Druck
unter Rückfluss
erhitzt, wobei Wasser azeotrop entfernt wird, so dass eine Innentemperatur
von etwa 70–75°C aufrechterhalten
wird. Die Umsetzung wird mit der IPC 1 auf das Verschwinden der Ausgangsverbindung
4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure überwacht.
Falls >10% der Ausgangsverbindung
4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure nach 12 bis 30 Stunden
verbleiben, können
zusätzliche
Anteile von (2-Thienylmethyl)propandisäure-mono-ethylester
(0,10 Äquivalente
pro Anteil bezüglich
der 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure) zugegeben
und die Umsetzung fortgesetzt werden. Wenn die IPC 1-Analyse darauf
hindeutet, dass die Umsetzung der Ausgangsverbindung 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure im Wesentlichen
vollständig
ist (< 10% Rest),
wird die Lösung
auf etwa 60–65°C abgekühlt. Die
abgekühlte
Lösung
wird mit demineralisiertem Wasser (6,8 g pro Gramm getesteter 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure) und
einer wässrigen
6,7 N Natriumhydroxid-Lösung
(etwa 2,0 ml Lösung
pro Gramm getesteter 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure) behandelt
und das Gemisch wird unter Rückfluss
für etwa
1,0 bis 3,5 Stunden erhitzt. Der Reaktionsansatz wird mit der IPC 2
getestet, um die vollständige
Umwandlung (< 2.0%)
in das Produkt zu bestätigen.
Die Lösung
wird danach auf etwa 50°C
abgekühlt
und die Schichten werden getrennt. Ethylalkohol (etwa 5,0 g pro
Gramm getesteter 4-[(2-n-Butyl-5-formyl-1H-imidazol-1-yl)methyl]benzoesäure) wird
zu der wässrigen
Phase zugegeben und der pH-Wert wird mit wässriger 6 N Salzsäure-Lösung auf
5,0 bis 5,4 eingestellt. Die so erhaltene Suspension wird bei etwa
10–15°C für etwa 2
Stunden bis zur vollständigen
Ausfällung
gerührt.
Das Produkt wird durch Zentrifugation isoliert, zweimal mit Wasser
gewaschen und der feuchte Kuchen wird direkt im nächsten Schritt
verwendet. Die korrigierte isolierte Ausbeute des Produkt auf dieser
Stufe beträgt
typischerweise etwa 70–85%. Untersuchung
einer getrockneten Produktprobe auf einer Gew./Gew.-Basis mit HPLC
gegen eine Standardprobe deutet typischerweise auf eine relative
Reinheit von etwa 97 bis 99% hin.
-
Analysedaten
-
IPC 1
-
HPLC (Gradient)
-
Apparate:
-
Die
folgende Ausstattung oder ihr Äquivalent
kann verwendet werden:
| Gerät | Hewlett
Packard, Modell 1050 |
| Pumpsystem | ternäre Niederdruck-Gradientenpumpe,
HP 1050 |
| Injektor | Autosampler,
Typ HP 1050 |
| Detektor | UV,
variable Wellenlänge,
Typ HP 1050 |
Bedingungen:
| Säule | Zorbax
SB-C18, 7,5 cm × 4,6
mm, 3,5 Mikron Teilchengröße,
hergestellt
von Rockland Technologies, Inc.
US-Distributor: MAC-MOD Analytical,
Inc. |
| Verdünnungslösungsmittel | 1:5
Acetonitril:HPLC-reines Wasser |
| Eluent
Organisch: | HPLC-reines
Acetonitril |
| Wässrig: | 0,1
M Ammoniumacetat (pH-Wert = 6,7) |
| Mobile
Phase Herstellung | Mobile
Phase A = 0,1 M Ammoniumacetat
Mobile Phase B = 50:50, 0,1
M Ammoniumacetat:Acetonitril |
| Detektions-Wellenlänge | 235
nm, 0,1 AUFS |
| Fließgeschwindigkeit | 2,0
ml/Min. |
| Temperatur | 40°C |
| Injektionsvolumen | 20
Mikroliter |
| Analysenzeit | 20
Minuten |
| Re-Äquilibrierungszeit | 6
Minuten |
| Probenvorbereitung | Annähernd 30
mg (2 Tropfen) des Reaktionsgemischs werden in einen 25 ml-Messkolben
eingewogen und unter einem Stickstoffstrom getrocknet. |
| | Der
Messkolben wird danach mit Verdünnungslösungsmittel
auf das Volumen aufgefüllt.
Man behandelt die Probe für
10 Minuten mit Ultraschall und lässt auf
Raumtemperatur abkühlen |
-
Gradienten-Programm
-
- 1.) Anfängliche
Lösungsmittelzusammensetzung-0%
mobile Phase B
- 2.) Linearer Gradient von 0% bis 100% mobile Phase B in 10 Minuten
- 3.) Halten bei 100% mobile Phase B für 5 Minuten
- 4.) Linearer Gradient von 100% bis 0% mobile Phase B in 5 Minuten
- 5.) Re-Äquilibrieren
bei 0% mobile Phase B für
sechs Minuten.
-
IPC 2
-
HPLC (Gradient)
-
Apparate:
-
Die
folgende Ausstattung oder ihr Äquivalent
kann verwendet werden:
| Gerät | Hewlett
Packard, Modell 1050 |
| Pumpsystem | ternäre Niederdruck-Gradientenpumpe,
HP 1050 |
| Injektor | Autosampler,
Typ HP 1050 |
| Detektor | UV,
variable Wellenlänge,
Typ HP 1050 |
Bedingungen:
| Säule | Spherisorb
SCX, 5μm,
250 mm × 4,6
mm |
| Verdünnungslösungsmittel1 | 1:5
Acetonitril:HPLC-reines Wasser |
| Eluent
Organisch: | HPLC-reines
Acetonitril |
| Wässriger
Puffer A: | 11,5
g Ammoniumdihydrogenphosphat, gelöst in 1000 ml Wasser,
mit
Phosphorsäure
auf pH-Wert 2,5 eingestellt |
| Mobile
Phase Herstellung | Mobile
Phase A = 200 ml Puffer A, 700 ml Wasser, 100 ml Acetonitril
Mobile
Phase B = 200 ml Puffer A, 450 ml Wasser, 350 ml Acetonitril |
| Detektions-Wellenlänge | 235
nm |
| Fließgeschwindigkeit | 2,0
ml/Min. |
| Temperatur | 60°C |
| Injektionsvolumen | 10
Mikroliter |
| Analysenzeit | 20
Minuten |
| Re-Äquilibrierungszeit | 5
Minuten |
| Probenvorbereitung | 20
mL der IPC-Probe in ein 50 ml-Becherglas überführen. Rühren und gegebenenfalls Methanol
(ein bis zwei ml) zugeben, bis die Lösung homogen ist. Mit einer
Pasteur-Pipette vier Tropfen der IPC-Probe (50 μl) in einen 25-ml-Meßkolben überführen. Mit
20 ml der mobilen Phase B verdünnen
und für
eine Minute mit Ultraschall behandeln. |
-
Gradienten-Programm
-
- 1.) Lösungsmittelzusammensetzung
von 0 bis 3 Minuten: 0% mobile Phase B
- 2.) Linearer Gradient von 0% bis 100% mobile Phase B in einer
Minute
- 3.) Halten bei 100% mobile Phase B für 16 Minuten
- 4.) Linearer Gradient von 100% bis 0% mobile Phase B in 5 Minuten
- 5.) Re-Äquilibrieren
bei 0% mobile Phase B für
5 Minuten.
-
Es
ist selbstverständlich,
dass die Erfindung nicht auf die vorstehend veranschaulichten Ausführungsformen
begrenzt ist und das Recht auf die veranschaulichten Ausführungsformen
und alle Abwandlungen, die in den Schutzbereich der folgenden Ansprüche fallen,
wird vorbehalten.
 oder eines Säure- oder Basenadditionssalzes davon, mit einer Verbindung der Formel (III):
oder eines Säure- oder Basenadditionssalzes davon, mit einer Verbindung der Formel (III):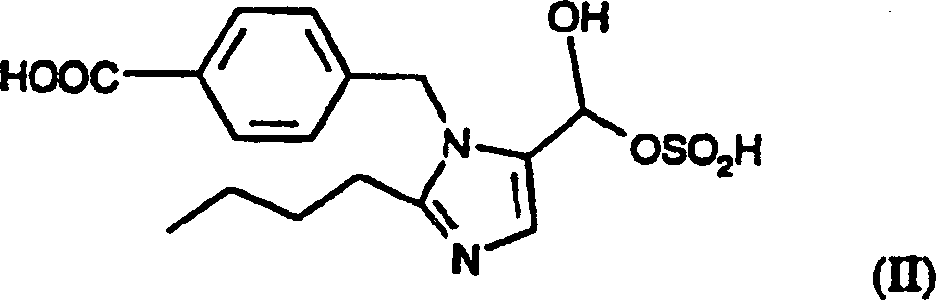 wobei R'C1-4-Alkyl bedeutet,
wobei R'C1-4-Alkyl bedeutet,
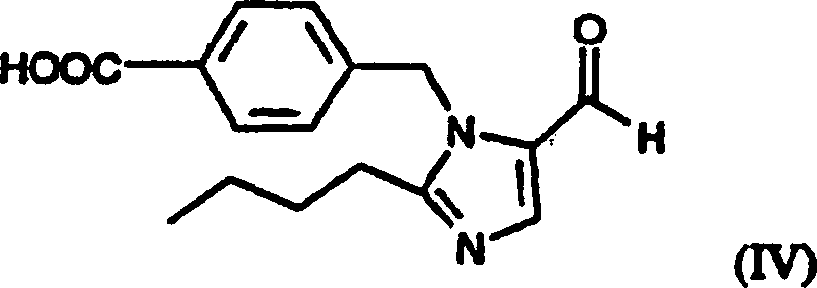
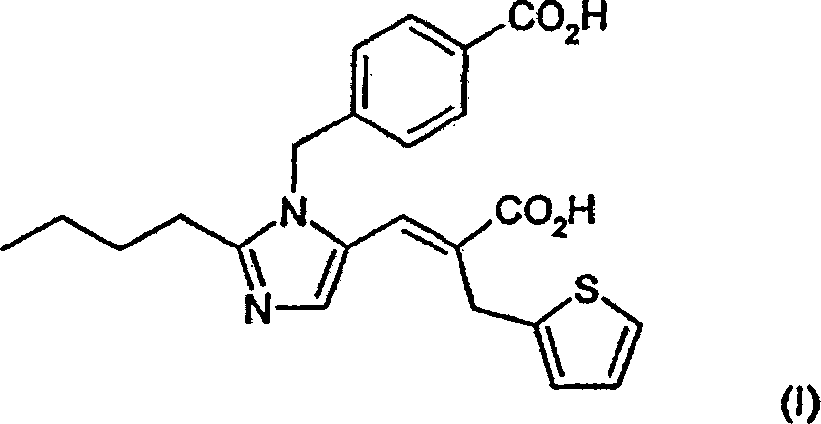 oder eines Säure- oder Basenadditionssalzes davon mit einer Verbindung der Formel (III):
oder eines Säure- oder Basenadditionssalzes davon mit einer Verbindung der Formel (III):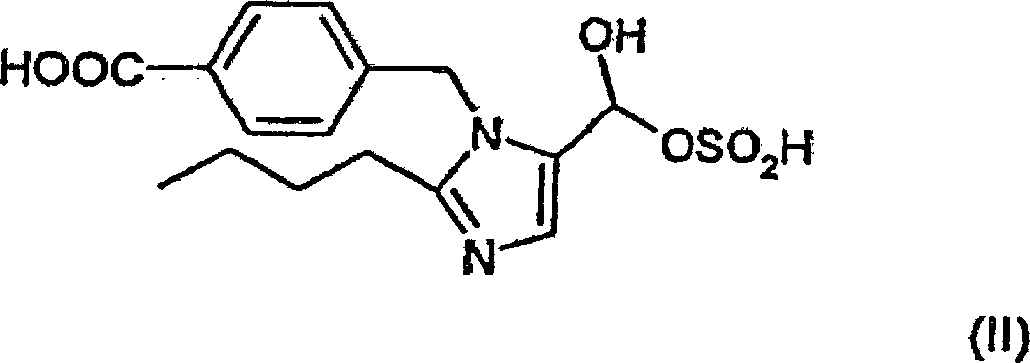 wobei R'C1-4-Alkyl bedeutet, bei vermindertem Druck von etwa 23 bis 33 cm (9 bis 13 Zoll) Hg in Gegenwart von Piperidin oder Piperidiniumpropionat und einem Überschuß an Propionsäure und anschließendes Hydrolysieren des R'-Esters unter Verwendung einer Base und gegebenenfalls Bilden eines pharmazeutsch verträglichen Salzes umfaßt.
wobei R'C1-4-Alkyl bedeutet, bei vermindertem Druck von etwa 23 bis 33 cm (9 bis 13 Zoll) Hg in Gegenwart von Piperidin oder Piperidiniumpropionat und einem Überschuß an Propionsäure und anschließendes Hydrolysieren des R'-Esters unter Verwendung einer Base und gegebenenfalls Bilden eines pharmazeutsch verträglichen Salzes umfaßt.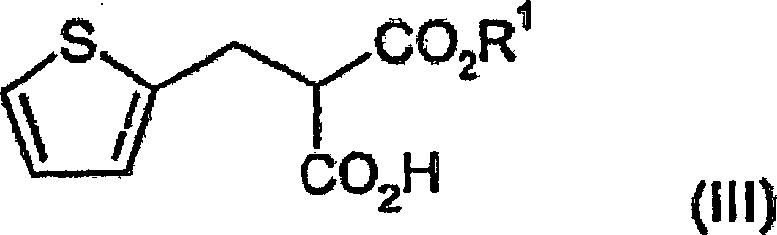

 oder eines Säure- oder Basenadditionssalzes davon mit einer Verbindung der Formel (III):
oder eines Säure- oder Basenadditionssalzes davon mit einer Verbindung der Formel (III): wobei R'C1-4-Alkyl bedeutet, bei vermindertem Druck von etwa 23 bis 33 cm (9 bis 13 Zoll) Hg in Gegenwart von Piperidin oder Piperidiniumpropionat und einem Überschuß an Propionsäure und anschließendes Hydrolysieren des R'-Esters unter Verwendung einer Base und gegebenenfalls Bilden eines pharmazeutisch verträglichen Salzes umfaßt.
wobei R'C1-4-Alkyl bedeutet, bei vermindertem Druck von etwa 23 bis 33 cm (9 bis 13 Zoll) Hg in Gegenwart von Piperidin oder Piperidiniumpropionat und einem Überschuß an Propionsäure und anschließendes Hydrolysieren des R'-Esters unter Verwendung einer Base und gegebenenfalls Bilden eines pharmazeutisch verträglichen Salzes umfaßt.