DE60009911T2 - Aminothiazolderivate und ihre verwendung als crf-rezeptor-liganden - Google Patents
Aminothiazolderivate und ihre verwendung als crf-rezeptor-liganden Download PDFInfo
- Publication number
- DE60009911T2 DE60009911T2 DE60009911T DE60009911T DE60009911T2 DE 60009911 T2 DE60009911 T2 DE 60009911T2 DE 60009911 T DE60009911 T DE 60009911T DE 60009911 T DE60009911 T DE 60009911T DE 60009911 T2 DE60009911 T2 DE 60009911T2
- Authority
- DE
- Germany
- Prior art keywords
- methylphenyl
- group
- methoxy
- prop
- methylthiazol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- RAIPHJJURHTUIC-UHFFFAOYSA-N 1,3-thiazol-2-amine Chemical class NC1=NC=CS1 RAIPHJJURHTUIC-UHFFFAOYSA-N 0.000 title description 8
- 108010056643 Corticotropin-Releasing Hormone Receptors Proteins 0.000 title description 4
- 239000003446 ligand Substances 0.000 title description 2
- -1 2-chloro-4-methoxy-5-methylphenyl Chemical group 0.000 claims description 124
- 150000001875 compounds Chemical class 0.000 claims description 104
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 37
- 238000000034 method Methods 0.000 claims description 35
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 24
- 238000002360 preparation method Methods 0.000 claims description 21
- 208000035475 disorder Diseases 0.000 claims description 20
- 125000006527 (C1-C5) alkyl group Chemical group 0.000 claims description 18
- 239000004480 active ingredient Substances 0.000 claims description 14
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 13
- 150000003839 salts Chemical class 0.000 claims description 13
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 10
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 9
- 229910052739 hydrogen Inorganic materials 0.000 claims description 9
- 239000001257 hydrogen Substances 0.000 claims description 9
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 9
- 239000012453 solvate Substances 0.000 claims description 9
- 125000000217 alkyl group Chemical group 0.000 claims description 8
- 229940079593 drug Drugs 0.000 claims description 8
- 239000003814 drug Substances 0.000 claims description 8
- 125000005843 halogen group Chemical group 0.000 claims description 8
- 125000006555 (C3-C5) cycloalkyl group Chemical group 0.000 claims description 7
- CIUQDSCDWFSTQR-UHFFFAOYSA-N [C]1=CC=CC=C1 Chemical compound [C]1=CC=CC=C1 CIUQDSCDWFSTQR-UHFFFAOYSA-N 0.000 claims description 7
- 230000001575 pathological effect Effects 0.000 claims description 7
- 239000000825 pharmaceutical preparation Substances 0.000 claims description 7
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 6
- 208000024827 Alzheimer disease Diseases 0.000 claims description 6
- 229910003827 NRaRb Inorganic materials 0.000 claims description 6
- 125000003545 alkoxy group Chemical group 0.000 claims description 6
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 6
- 125000003118 aryl group Chemical group 0.000 claims description 6
- 229910052736 halogen Inorganic materials 0.000 claims description 6
- 150000002367 halogens Chemical class 0.000 claims description 6
- 150000004677 hydrates Chemical class 0.000 claims description 6
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 claims description 5
- 208000019901 Anxiety disease Diseases 0.000 claims description 5
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 claims description 5
- 208000022531 anorexia Diseases 0.000 claims description 5
- 230000036506 anxiety Effects 0.000 claims description 5
- 206010061428 decreased appetite Diseases 0.000 claims description 5
- 230000004054 inflammatory process Effects 0.000 claims description 5
- 230000008569 process Effects 0.000 claims description 5
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 4
- 208000023105 Huntington disease Diseases 0.000 claims description 4
- 230000003542 behavioural effect Effects 0.000 claims description 4
- 230000001419 dependent effect Effects 0.000 claims description 4
- 201000010099 disease Diseases 0.000 claims description 4
- 125000000623 heterocyclic group Chemical group 0.000 claims description 4
- 230000001568 sexual effect Effects 0.000 claims description 4
- 208000019116 sleep disease Diseases 0.000 claims description 4
- 208000018522 Gastrointestinal disease Diseases 0.000 claims description 3
- 206010062016 Immunosuppression Diseases 0.000 claims description 3
- 208000019695 Migraine disease Diseases 0.000 claims description 3
- 206010028980 Neoplasm Diseases 0.000 claims description 3
- 208000018737 Parkinson disease Diseases 0.000 claims description 3
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 claims description 3
- 201000011510 cancer Diseases 0.000 claims description 3
- 125000004432 carbon atom Chemical group C* 0.000 claims description 3
- 206010012601 diabetes mellitus Diseases 0.000 claims description 3
- 206010015037 epilepsy Diseases 0.000 claims description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 3
- 206010016256 fatigue Diseases 0.000 claims description 3
- 201000001421 hyperglycemia Diseases 0.000 claims description 3
- 230000001506 immunosuppresive effect Effects 0.000 claims description 3
- 208000015181 infectious disease Diseases 0.000 claims description 3
- 230000004968 inflammatory condition Effects 0.000 claims description 3
- 208000002551 irritable bowel syndrome Diseases 0.000 claims description 3
- 206010027599 migraine Diseases 0.000 claims description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 3
- 229910052757 nitrogen Inorganic materials 0.000 claims description 3
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 claims description 3
- 125000004076 pyridyl group Chemical group 0.000 claims description 3
- 229910052717 sulfur Inorganic materials 0.000 claims description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 3
- 125000006700 (C1-C6) alkylthio group Chemical group 0.000 claims description 2
- 206010001488 Aggression Diseases 0.000 claims description 2
- 208000032841 Bulimia Diseases 0.000 claims description 2
- 206010006550 Bulimia nervosa Diseases 0.000 claims description 2
- 206010012735 Diarrhoea Diseases 0.000 claims description 2
- 208000001640 Fibromyalgia Diseases 0.000 claims description 2
- 206010061218 Inflammation Diseases 0.000 claims description 2
- 208000019022 Mood disease Diseases 0.000 claims description 2
- 208000010067 Pituitary ACTH Hypersecretion Diseases 0.000 claims description 2
- 208000020627 Pituitary-dependent Cushing syndrome Diseases 0.000 claims description 2
- 208000005107 Premature Birth Diseases 0.000 claims description 2
- 201000004681 Psoriasis Diseases 0.000 claims description 2
- 206010046851 Uveitis Diseases 0.000 claims description 2
- 238000009109 curative therapy Methods 0.000 claims description 2
- 230000035558 fertility Effects 0.000 claims description 2
- 150000002431 hydrogen Chemical group 0.000 claims description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 2
- 125000002883 imidazolyl group Chemical group 0.000 claims description 2
- 125000002560 nitrile group Chemical group 0.000 claims description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 2
- 201000008482 osteoarthritis Diseases 0.000 claims description 2
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 2
- 230000003449 preventive effect Effects 0.000 claims description 2
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 2
- 125000004434 sulfur atom Chemical group 0.000 claims description 2
- 208000024891 symptom Diseases 0.000 claims description 2
- 229930192474 thiophene Natural products 0.000 claims description 2
- 230000001960 triggered effect Effects 0.000 claims description 2
- GNAYLSUBLKWESD-RSAXXLAASA-N Cl.Cc1ccc(cc1F)[C@H](CC1CC1)NCC#C Chemical compound Cl.Cc1ccc(cc1F)[C@H](CC1CC1)NCC#C GNAYLSUBLKWESD-RSAXXLAASA-N 0.000 claims 2
- IKXNIQJDNKPPCH-UHFFFAOYSA-N hydron;prop-2-yn-1-amine;chloride Chemical compound [Cl-].[NH3+]CC#C IKXNIQJDNKPPCH-UHFFFAOYSA-N 0.000 claims 2
- 208000007848 Alcoholism Diseases 0.000 claims 1
- 208000014644 Brain disease Diseases 0.000 claims 1
- 208000020446 Cardiac disease Diseases 0.000 claims 1
- 206010053759 Growth retardation Diseases 0.000 claims 1
- 208000036818 High risk pregnancy Diseases 0.000 claims 1
- 208000021384 Obsessive-Compulsive disease Diseases 0.000 claims 1
- 208000007271 Substance Withdrawal Syndrome Diseases 0.000 claims 1
- 201000007930 alcohol dependence Diseases 0.000 claims 1
- 125000005332 alkyl sulfoxy group Chemical group 0.000 claims 1
- 230000002152 alkylating effect Effects 0.000 claims 1
- 231100000001 growth retardation Toxicity 0.000 claims 1
- 208000019622 heart disease Diseases 0.000 claims 1
- 230000000968 intestinal effect Effects 0.000 claims 1
- 230000037324 pain perception Effects 0.000 claims 1
- 230000002265 prevention Effects 0.000 claims 1
- 125000003944 tolyl group Chemical group 0.000 claims 1
- 208000019553 vascular disease Diseases 0.000 claims 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 93
- 239000000243 solution Substances 0.000 description 79
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 78
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 49
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 44
- 239000000055 Corticotropin-Releasing Hormone Substances 0.000 description 43
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 43
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 43
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 42
- 102100021752 Corticoliberin Human genes 0.000 description 40
- 108010022152 Corticotropin-Releasing Hormone Proteins 0.000 description 40
- 239000011541 reaction mixture Substances 0.000 description 38
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 36
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 34
- 239000000203 mixture Substances 0.000 description 34
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 33
- FKLJPTJMIBLJAV-UHFFFAOYSA-N Compound IV Chemical compound O1N=C(C)C=C1CCCCCCCOC1=CC=C(C=2OCCN=2)C=C1 FKLJPTJMIBLJAV-UHFFFAOYSA-N 0.000 description 31
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 30
- 239000000460 chlorine Substances 0.000 description 27
- 239000012074 organic phase Substances 0.000 description 27
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 24
- 238000003756 stirring Methods 0.000 description 21
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 21
- 229910052938 sodium sulfate Inorganic materials 0.000 description 20
- 235000011152 sodium sulphate Nutrition 0.000 description 20
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 15
- 230000000694 effects Effects 0.000 description 14
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- 150000001412 amines Chemical class 0.000 description 12
- 230000015572 biosynthetic process Effects 0.000 description 12
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 12
- 150000003585 thioureas Chemical class 0.000 description 12
- 102100027467 Pro-opiomelanocortin Human genes 0.000 description 11
- 229940126214 compound 3 Drugs 0.000 description 11
- 239000000047 product Substances 0.000 description 11
- 238000010992 reflux Methods 0.000 description 11
- 239000000741 silica gel Substances 0.000 description 11
- 229910002027 silica gel Inorganic materials 0.000 description 11
- 238000003786 synthesis reaction Methods 0.000 description 11
- 101800000414 Corticotropin Proteins 0.000 description 10
- 229910052786 argon Inorganic materials 0.000 description 10
- IDLFZVILOHSSID-OVLDLUHVSA-N corticotropin Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)NC(=O)[C@@H](N)CO)C1=CC=C(O)C=C1 IDLFZVILOHSSID-OVLDLUHVSA-N 0.000 description 10
- 229960000258 corticotropin Drugs 0.000 description 10
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 10
- 239000012071 phase Substances 0.000 description 10
- 108090000765 processed proteins & peptides Proteins 0.000 description 10
- 239000011780 sodium chloride Substances 0.000 description 9
- 239000000275 Adrenocorticotropic Hormone Substances 0.000 description 8
- 238000004587 chromatography analysis Methods 0.000 description 8
- 239000003480 eluent Substances 0.000 description 8
- 229920006395 saturated elastomer Polymers 0.000 description 8
- 239000000725 suspension Substances 0.000 description 8
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 7
- NLFBCYMMUAKCPC-KQQUZDAGSA-N ethyl (e)-3-[3-amino-2-cyano-1-[(e)-3-ethoxy-3-oxoprop-1-enyl]sulfanyl-3-oxoprop-1-enyl]sulfanylprop-2-enoate Chemical compound CCOC(=O)\C=C\SC(=C(C#N)C(N)=O)S\C=C\C(=O)OCC NLFBCYMMUAKCPC-KQQUZDAGSA-N 0.000 description 7
- 238000001704 evaporation Methods 0.000 description 7
- 230000008020 evaporation Effects 0.000 description 7
- AFABGHUZZDYHJO-UHFFFAOYSA-N 2-Methylpentane Chemical compound CCCC(C)C AFABGHUZZDYHJO-UHFFFAOYSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 6
- 230000009471 action Effects 0.000 description 6
- 239000000284 extract Substances 0.000 description 6
- 150000002576 ketones Chemical class 0.000 description 6
- 230000028327 secretion Effects 0.000 description 6
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 5
- 241000282412 Homo Species 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 241000700159 Rattus Species 0.000 description 5
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 5
- 239000008346 aqueous phase Substances 0.000 description 5
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 239000000543 intermediate Substances 0.000 description 5
- 238000004519 manufacturing process Methods 0.000 description 5
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 5
- 102000004196 processed proteins & peptides Human genes 0.000 description 5
- 108020003175 receptors Proteins 0.000 description 5
- 102000005962 receptors Human genes 0.000 description 5
- 239000000377 silicon dioxide Substances 0.000 description 5
- 239000012312 sodium hydride Substances 0.000 description 5
- 229910000104 sodium hydride Inorganic materials 0.000 description 5
- 230000035882 stress Effects 0.000 description 5
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- DRSHXJFUUPIBHX-UHFFFAOYSA-N COc1ccc(cc1)N1N=CC2C=NC(Nc3cc(OC)c(OC)c(OCCCN4CCN(C)CC4)c3)=NC12 Chemical compound COc1ccc(cc1)N1N=CC2C=NC(Nc3cc(OC)c(OC)c(OCCCN4CCN(C)CC4)c3)=NC12 DRSHXJFUUPIBHX-UHFFFAOYSA-N 0.000 description 4
- 229920000858 Cyclodextrin Polymers 0.000 description 4
- 241000282414 Homo sapiens Species 0.000 description 4
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 4
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 4
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 4
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- CZHLJFDJWWMBCF-NSHDSACASA-N [(1s)-2-cyclopropyl-1-phenylethyl]thiourea Chemical compound C([C@H](NC(=S)N)C=1C=CC=CC=1)C1CC1 CZHLJFDJWWMBCF-NSHDSACASA-N 0.000 description 4
- 239000005557 antagonist Substances 0.000 description 4
- 230000006399 behavior Effects 0.000 description 4
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 4
- 210000004556 brain Anatomy 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 239000000499 gel Substances 0.000 description 4
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine hydrate Chemical compound O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 4
- 229910052740 iodine Inorganic materials 0.000 description 4
- 239000012280 lithium aluminium hydride Substances 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 150000002923 oximes Chemical class 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 239000002904 solvent Substances 0.000 description 4
- NTHHQWJMKRQMNJ-UHFFFAOYSA-N 1-(4-chlorophenyl)-2-methoxy-n-phenylmethoxyethanimine Chemical compound C=1C=C(Cl)C=CC=1C(COC)=NOCC1=CC=CC=C1 NTHHQWJMKRQMNJ-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 3
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 3
- WXNXCEHXYPACJF-ZETCQYMHSA-M N-acetyl-L-leucinate Chemical compound CC(C)C[C@@H](C([O-])=O)NC(C)=O WXNXCEHXYPACJF-ZETCQYMHSA-M 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 230000003042 antagnostic effect Effects 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 238000004296 chiral HPLC Methods 0.000 description 3
- 229940125782 compound 2 Drugs 0.000 description 3
- 239000000287 crude extract Substances 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 238000010828 elution Methods 0.000 description 3
- 239000011630 iodine Substances 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- 206010025482 malaise Diseases 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- 238000004808 supercritical fluid chromatography Methods 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- CDNKJSYTIWINBS-UHFFFAOYSA-N 1-(1,3-benzodioxol-5-yl)-2-cyclopropyl-n-phenylmethoxyethanimine Chemical compound C=1C=CC=CC=1CON=C(C=1C=C2OCOC2=CC=1)CC1CC1 CDNKJSYTIWINBS-UHFFFAOYSA-N 0.000 description 2
- FMMVUADITNBALS-UHFFFAOYSA-N 1-(1,3-benzodioxol-5-yl)-2-cyclopropylethanone Chemical compound C=1C=C2OCOC2=CC=1C(=O)CC1CC1 FMMVUADITNBALS-UHFFFAOYSA-N 0.000 description 2
- QBZXPYPUARDCKX-UHFFFAOYSA-N 1-(1,3-benzodioxol-5-yl)-2-methoxyethanone Chemical compound COCC(=O)C1=CC=C2OCOC2=C1 QBZXPYPUARDCKX-UHFFFAOYSA-N 0.000 description 2
- CIJVBOFREGVENE-UHFFFAOYSA-N 1-(1,3-benzodioxol-5-yl)-n-phenylmethoxybutan-1-imine Chemical compound C=1C=C2OCOC2=CC=1C(CCC)=NOCC1=CC=CC=C1 CIJVBOFREGVENE-UHFFFAOYSA-N 0.000 description 2
- VHOZZIUFGVJOEL-UHFFFAOYSA-N 1-(1,3-benzodioxol-5-yl)butan-1-one Chemical compound CCCC(=O)C1=CC=C2OCOC2=C1 VHOZZIUFGVJOEL-UHFFFAOYSA-N 0.000 description 2
- QGAFDQLNBYTCCS-UHFFFAOYSA-N 1-(3-fluoro-4-methylphenyl)-n-phenylmethoxybutan-1-imine Chemical compound C=1C=C(C)C(F)=CC=1C(CCC)=NOCC1=CC=CC=C1 QGAFDQLNBYTCCS-UHFFFAOYSA-N 0.000 description 2
- ZTDXGIKSKMTVPX-UHFFFAOYSA-N 1-(3-fluoro-4-methylphenyl)-n-phenylmethoxypentan-1-imine Chemical compound C=1C=C(C)C(F)=CC=1C(CCCC)=NOCC1=CC=CC=C1 ZTDXGIKSKMTVPX-UHFFFAOYSA-N 0.000 description 2
- CCQQJZYSNOZDOS-UHFFFAOYSA-N 1-(3-fluoro-4-methylphenyl)butan-1-one Chemical compound CCCC(=O)C1=CC=C(C)C(F)=C1 CCQQJZYSNOZDOS-UHFFFAOYSA-N 0.000 description 2
- GVJLXQFPBYBNQF-UHFFFAOYSA-N 1-(3-fluoro-4-methylphenyl)pentan-1-one Chemical compound CCCCC(=O)C1=CC=C(C)C(F)=C1 GVJLXQFPBYBNQF-UHFFFAOYSA-N 0.000 description 2
- SGFDPBCDFTXBIH-UHFFFAOYSA-N 1-(4-bromophenyl)-2-cyclopropyl-n-phenylmethoxyethanimine Chemical compound C1=CC(Br)=CC=C1C(CC1CC1)=NOCC1=CC=CC=C1 SGFDPBCDFTXBIH-UHFFFAOYSA-N 0.000 description 2
- WXYYSMFEPWSPKJ-UHFFFAOYSA-N 1-(4-chlorophenyl)-2-cyclopropyl-n-phenylmethoxyethanimine Chemical compound C1=CC(Cl)=CC=C1C(CC1CC1)=NOCC1=CC=CC=C1 WXYYSMFEPWSPKJ-UHFFFAOYSA-N 0.000 description 2
- ZLUGEAOUDPQUSA-UHFFFAOYSA-N 1-(4-ethylphenyl)-2-methoxy-n-phenylmethoxyethanimine Chemical compound C1=CC(CC)=CC=C1C(COC)=NOCC1=CC=CC=C1 ZLUGEAOUDPQUSA-UHFFFAOYSA-N 0.000 description 2
- FZJFYVNQYIEBRP-UHFFFAOYSA-N 1-(4-ethylphenyl)-2-methoxyethanone Chemical compound CCC1=CC=C(C(=O)COC)C=C1 FZJFYVNQYIEBRP-UHFFFAOYSA-N 0.000 description 2
- MFKCRZCWDMFWLE-UHFFFAOYSA-N 1-(4-fluorophenyl)-n-phenylmethoxybutan-1-imine Chemical compound C=1C=C(F)C=CC=1C(CCC)=NOCC1=CC=CC=C1 MFKCRZCWDMFWLE-UHFFFAOYSA-N 0.000 description 2
- HXMIHKLBYDRDBU-UHFFFAOYSA-N 1-(4-fluorophenyl)-n-phenylmethoxypentan-1-imine Chemical compound C=1C=C(F)C=CC=1C(CCCC)=NOCC1=CC=CC=C1 HXMIHKLBYDRDBU-UHFFFAOYSA-N 0.000 description 2
- SZWBZWDCIJIWOA-UHFFFAOYSA-N 1-[3-fluoro-4-(methoxymethyl)phenyl]-n-phenylmethoxybutan-1-imine Chemical compound C=1C=C(COC)C(F)=CC=1C(CCC)=NOCC1=CC=CC=C1 SZWBZWDCIJIWOA-UHFFFAOYSA-N 0.000 description 2
- ICZZOQNXXHAGBD-UHFFFAOYSA-N 1-[3-fluoro-4-(methoxymethyl)phenyl]butan-1-one Chemical compound CCCC(=O)C1=CC=C(COC)C(F)=C1 ICZZOQNXXHAGBD-UHFFFAOYSA-N 0.000 description 2
- YGYNQXJTTFDLST-UHFFFAOYSA-N 1-[4-(methoxymethyl)phenyl]-n-phenylmethoxybutan-1-imine Chemical compound C=1C=C(COC)C=CC=1C(CCC)=NOCC1=CC=CC=C1 YGYNQXJTTFDLST-UHFFFAOYSA-N 0.000 description 2
- RNKSAAVSCPNQGZ-UHFFFAOYSA-N 1-[4-(methoxymethyl)phenyl]-n-phenylmethoxypentan-1-imine Chemical compound C=1C=C(COC)C=CC=1C(CCCC)=NOCC1=CC=CC=C1 RNKSAAVSCPNQGZ-UHFFFAOYSA-N 0.000 description 2
- KCSYDCGLIMJDJQ-UHFFFAOYSA-N 1-[4-(methoxymethyl)phenyl]pentan-1-one Chemical compound CCCCC(=O)C1=CC=C(COC)C=C1 KCSYDCGLIMJDJQ-UHFFFAOYSA-N 0.000 description 2
- KXVHSXZNHMAWJI-UHFFFAOYSA-N 1-cyclopropyl-1-phenyl-n-phenylmethoxymethanimine Chemical compound C=1C=CC=CC=1CON=C(C=1C=CC=CC=1)C1CC1 KXVHSXZNHMAWJI-UHFFFAOYSA-N 0.000 description 2
- ZNAXTORDSHSVNX-UHFFFAOYSA-N 1-phenyl-n-phenylmethoxybutan-1-imine Chemical compound C=1C=CC=CC=1C(CCC)=NOCC1=CC=CC=C1 ZNAXTORDSHSVNX-UHFFFAOYSA-N 0.000 description 2
- PAYDWBSNMUTRQV-UHFFFAOYSA-N 1-phenylpentylthiourea Chemical compound CCCCC(NC(N)=S)C1=CC=CC=C1 PAYDWBSNMUTRQV-UHFFFAOYSA-N 0.000 description 2
- LNQVZZGGOZBOQS-UHFFFAOYSA-N 2-amino-3-methyl-1,1-diphenylbutan-1-ol Chemical compound C=1C=CC=CC=1C(O)(C(N)C(C)C)C1=CC=CC=C1 LNQVZZGGOZBOQS-UHFFFAOYSA-N 0.000 description 2
- QMQSVQZHACYLON-UHFFFAOYSA-N 2-bromo-1-(2,4-dichloro-5-methylphenyl)propan-1-one Chemical compound CC(Br)C(=O)C1=CC(C)=C(Cl)C=C1Cl QMQSVQZHACYLON-UHFFFAOYSA-N 0.000 description 2
- WCOSWDUCCDUPIM-UHFFFAOYSA-N 2-bromo-1-(2,4-dimethoxy-5-methylphenyl)propan-1-one Chemical compound COC1=CC(OC)=C(C(=O)C(C)Br)C=C1C WCOSWDUCCDUPIM-UHFFFAOYSA-N 0.000 description 2
- JDUMYIRHFTZPHA-UHFFFAOYSA-N 2-bromo-1-(2-chloro-4-methoxyphenyl)propan-1-one Chemical compound COC1=CC=C(C(=O)C(C)Br)C(Cl)=C1 JDUMYIRHFTZPHA-UHFFFAOYSA-N 0.000 description 2
- OFWUBMOUIVHXFE-UHFFFAOYSA-N 2-bromo-1-(4-methoxy-2,5-dimethylphenyl)propan-1-one Chemical compound COC1=CC(C)=C(C(=O)C(C)Br)C=C1C OFWUBMOUIVHXFE-UHFFFAOYSA-N 0.000 description 2
- ILLHORFDXDLILE-UHFFFAOYSA-N 2-bromopropanoyl bromide Chemical compound CC(Br)C(Br)=O ILLHORFDXDLILE-UHFFFAOYSA-N 0.000 description 2
- DIGANWFLFCXCSM-UHFFFAOYSA-N 2-cyclobutyl-1-(4-fluorophenyl)-n-phenylmethoxyethanimine Chemical compound C1=CC(F)=CC=C1C(CC1CCC1)=NOCC1=CC=CC=C1 DIGANWFLFCXCSM-UHFFFAOYSA-N 0.000 description 2
- NQQOLUFXFBTYIW-UHFFFAOYSA-N 2-cyclobutyl-1-(4-fluorophenyl)ethanone Chemical compound C1=CC(F)=CC=C1C(=O)CC1CCC1 NQQOLUFXFBTYIW-UHFFFAOYSA-N 0.000 description 2
- KZTBTEGBDNDYSX-UHFFFAOYSA-N 2-cyclopropyl-1-(3-fluoro-4-methylphenyl)-n-phenylmethoxyethanimine Chemical compound C1=C(F)C(C)=CC=C1C(CC1CC1)=NOCC1=CC=CC=C1 KZTBTEGBDNDYSX-UHFFFAOYSA-N 0.000 description 2
- JEXGCIGZLRHGRJ-UHFFFAOYSA-N 2-cyclopropyl-1-(4-fluorophenyl)-n-phenylmethoxyethanimine Chemical compound C1=CC(F)=CC=C1C(CC1CC1)=NOCC1=CC=CC=C1 JEXGCIGZLRHGRJ-UHFFFAOYSA-N 0.000 description 2
- XVOYCKUASIHFRJ-UHFFFAOYSA-N 2-cyclopropyl-1-(4-methylphenyl)-n-phenylmethoxyethanimine Chemical compound C1=CC(C)=CC=C1C(CC1CC1)=NOCC1=CC=CC=C1 XVOYCKUASIHFRJ-UHFFFAOYSA-N 0.000 description 2
- JBGVKJVTILEZNJ-UHFFFAOYSA-N 2-cyclopropyl-1-(4-methylphenyl)ethanone Chemical compound C1=CC(C)=CC=C1C(=O)CC1CC1 JBGVKJVTILEZNJ-UHFFFAOYSA-N 0.000 description 2
- NMEBUSJSLZDTEO-UHFFFAOYSA-N 2-cyclopropyl-1-phenyl-n-phenylmethoxyethanimine Chemical compound C=1C=CC=CC=1CON=C(C=1C=CC=CC=1)CC1CC1 NMEBUSJSLZDTEO-UHFFFAOYSA-N 0.000 description 2
- USSJCFQCQHTMGR-UHFFFAOYSA-N 2-methoxy-1-[4-(methoxymethyl)phenyl]-n-phenylmethoxyethanimine Chemical compound C=1C=C(COC)C=CC=1C(COC)=NOCC1=CC=CC=C1 USSJCFQCQHTMGR-UHFFFAOYSA-N 0.000 description 2
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 2
- WUNUPSGQEDDRBD-UHFFFAOYSA-N 3-cyclopropyl-1-(4-fluorophenyl)-n-phenylmethoxypropan-1-imine Chemical compound C1=CC(F)=CC=C1C(CCC1CC1)=NOCC1=CC=CC=C1 WUNUPSGQEDDRBD-UHFFFAOYSA-N 0.000 description 2
- APSCIIYYEZZAFL-FTBISJDPSA-N 4-(2-chloro-4-methoxy-5-methylphenyl)-n-[(1r)-1-(3-fluoro-4-methylphenyl)-2-methoxyethyl]-5-methyl-n-prop-2-ynyl-1,3-thiazol-2-amine;hydrochloride Chemical compound Cl.C#CCN([C@@H](COC)C=1C=C(F)C(C)=CC=1)C(SC=1C)=NC=1C1=CC(C)=C(OC)C=C1Cl APSCIIYYEZZAFL-FTBISJDPSA-N 0.000 description 2
- YZFVUQSAJMLFOZ-UHFFFAOYSA-N 4-bromo-2-fluoro-1-methylbenzene Chemical compound CC1=CC=C(Br)C=C1F YZFVUQSAJMLFOZ-UHFFFAOYSA-N 0.000 description 2
- ILCVKEBXPLEGOY-ZLFMSJRASA-N 74434-59-6 Chemical compound CC[C@H](C)[C@@H](C(N)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H](CO)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@@H]1CCCN1C(=O)[C@H]1N(C(=O)CNC(=O)[C@H]2NC(=O)CC2)CCC1 ILCVKEBXPLEGOY-ZLFMSJRASA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 208000024172 Cardiovascular disease Diseases 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 206010012289 Dementia Diseases 0.000 description 2
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 2
- 238000005727 Friedel-Crafts reaction Methods 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 206010020772 Hypertension Diseases 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 2
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 2
- BYEZPXBHFNAKNM-QMMMGPOBSA-N [(1r)-1-(1,3-benzodioxol-5-yl)-2-methoxyethyl]thiourea Chemical compound COC[C@H](NC(N)=S)C1=CC=C2OCOC2=C1 BYEZPXBHFNAKNM-QMMMGPOBSA-N 0.000 description 2
- FBAYZHYUJHAFHR-VIFPVBQESA-N [(1r)-1-(4-chlorophenyl)-2-methoxyethyl]thiourea Chemical compound COC[C@H](NC(N)=S)C1=CC=C(Cl)C=C1 FBAYZHYUJHAFHR-VIFPVBQESA-N 0.000 description 2
- UOFDJSBLKIOPLK-NSHDSACASA-N [(1r)-1-(4-ethylphenyl)-2-methoxyethyl]thiourea Chemical compound CCC1=CC=C([C@H](COC)NC(N)=S)C=C1 UOFDJSBLKIOPLK-NSHDSACASA-N 0.000 description 2
- FYSVDXYACDSUGR-NSHDSACASA-N [(1r)-2-methoxy-1-[4-(methoxymethyl)phenyl]ethyl]thiourea Chemical compound COC[C@H](NC(N)=S)C1=CC=C(COC)C=C1 FYSVDXYACDSUGR-NSHDSACASA-N 0.000 description 2
- ZWBDIGRAYDJSKJ-VIFPVBQESA-N [(1r)-2-methoxy-1-phenylethyl]thiourea Chemical compound COC[C@H](NC(N)=S)C1=CC=CC=C1 ZWBDIGRAYDJSKJ-VIFPVBQESA-N 0.000 description 2
- GGGGDQZLIYHRHY-NSHDSACASA-N [(1s)-1-(3-fluoro-4-methylphenyl)butyl]thiourea Chemical compound CCC[C@H](NC(N)=S)C1=CC=C(C)C(F)=C1 GGGGDQZLIYHRHY-NSHDSACASA-N 0.000 description 2
- UBESSRMKTPYBSK-LBPRGKRZSA-N [(1s)-1-(3-fluoro-4-methylphenyl)pentyl]thiourea Chemical compound CCCC[C@H](NC(N)=S)C1=CC=C(C)C(F)=C1 UBESSRMKTPYBSK-LBPRGKRZSA-N 0.000 description 2
- BAXHUMXGIWAYJL-NSHDSACASA-N [(1s)-1-(4-bromophenyl)-2-cyclopropylethyl]thiourea Chemical compound C([C@H](NC(=S)N)C=1C=CC(Br)=CC=1)C1CC1 BAXHUMXGIWAYJL-NSHDSACASA-N 0.000 description 2
- SECFEVWALRUEDZ-NSHDSACASA-N [(1s)-1-(4-chlorophenyl)-2-cyclopropylethyl]thiourea Chemical compound C([C@H](NC(=S)N)C=1C=CC(Cl)=CC=1)C1CC1 SECFEVWALRUEDZ-NSHDSACASA-N 0.000 description 2
- DXAOTJKTXCEFBZ-JTQLQIEISA-N [(1s)-1-(4-fluorophenyl)butyl]thiourea Chemical compound CCC[C@H](NC(N)=S)C1=CC=C(F)C=C1 DXAOTJKTXCEFBZ-JTQLQIEISA-N 0.000 description 2
- TUUOEZMWKKGYRP-NSHDSACASA-N [(1s)-1-(4-fluorophenyl)pentyl]thiourea Chemical compound CCCC[C@H](NC(N)=S)C1=CC=C(F)C=C1 TUUOEZMWKKGYRP-NSHDSACASA-N 0.000 description 2
- QZKVGBHGQMRXGQ-LBPRGKRZSA-N [(1s)-1-[3-fluoro-4-(methoxymethyl)phenyl]butyl]thiourea Chemical compound CCC[C@H](NC(N)=S)C1=CC=C(COC)C(F)=C1 QZKVGBHGQMRXGQ-LBPRGKRZSA-N 0.000 description 2
- QXINUPXURAZBSD-JTQLQIEISA-N [(1s)-1-phenylbutyl]thiourea Chemical compound CCC[C@H](NC(N)=S)C1=CC=CC=C1 QXINUPXURAZBSD-JTQLQIEISA-N 0.000 description 2
- PAYDWBSNMUTRQV-NSHDSACASA-N [(1s)-1-phenylpentyl]thiourea Chemical compound CCCC[C@H](NC(N)=S)C1=CC=CC=C1 PAYDWBSNMUTRQV-NSHDSACASA-N 0.000 description 2
- NZIVMBWCIGNVDS-LBPRGKRZSA-N [(1s)-2-cyclobutyl-1-(4-fluorophenyl)ethyl]thiourea Chemical compound C([C@H](NC(=S)N)C=1C=CC(F)=CC=1)C1CCC1 NZIVMBWCIGNVDS-LBPRGKRZSA-N 0.000 description 2
- AWQGFODYZLOESK-LBPRGKRZSA-N [(1s)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]thiourea Chemical compound C1=C(F)C(C)=CC=C1[C@@H](NC(N)=S)CC1CC1 AWQGFODYZLOESK-LBPRGKRZSA-N 0.000 description 2
- UXWRDBUXDKDHLM-NSHDSACASA-N [(1s)-2-cyclopropyl-1-(4-fluorophenyl)ethyl]thiourea Chemical compound C([C@H](NC(=S)N)C=1C=CC(F)=CC=1)C1CC1 UXWRDBUXDKDHLM-NSHDSACASA-N 0.000 description 2
- OEQQMIGIQGCMQD-LBPRGKRZSA-N [(1s)-2-cyclopropyl-1-(4-methylphenyl)ethyl]thiourea Chemical compound C1=CC(C)=CC=C1[C@@H](NC(N)=S)CC1CC1 OEQQMIGIQGCMQD-LBPRGKRZSA-N 0.000 description 2
- GCFVKDMLYRRHRA-SNVBAGLBSA-N [(s)-cyclopropyl(phenyl)methyl]thiourea Chemical compound C1([C@H](NC(=S)N)C=2C=CC=CC=2)CC1 GCFVKDMLYRRHRA-SNVBAGLBSA-N 0.000 description 2
- 230000009858 acid secretion Effects 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 238000005804 alkylation reaction Methods 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 229950003476 aminothiazole Drugs 0.000 description 2
- SOIFLUNRINLCBN-UHFFFAOYSA-N ammonium thiocyanate Chemical compound [NH4+].[S-]C#N SOIFLUNRINLCBN-UHFFFAOYSA-N 0.000 description 2
- 230000003110 anti-inflammatory effect Effects 0.000 description 2
- 239000002249 anxiolytic agent Substances 0.000 description 2
- 230000000949 anxiolytic effect Effects 0.000 description 2
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 2
- MDKCFLQDBWCQCV-UHFFFAOYSA-N benzyl isothiocyanate Chemical compound S=C=NCC1=CC=CC=C1 MDKCFLQDBWCQCV-UHFFFAOYSA-N 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- 229910000085 borane Inorganic materials 0.000 description 2
- 239000012876 carrier material Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 238000013375 chromatographic separation Methods 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- QTMDXZNDVAMKGV-UHFFFAOYSA-L copper(ii) bromide Chemical compound [Cu+2].[Br-].[Br-] QTMDXZNDVAMKGV-UHFFFAOYSA-L 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- 125000004093 cyano group Chemical group *C#N 0.000 description 2
- 230000000994 depressogenic effect Effects 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- IIEWJVIFRVWJOD-UHFFFAOYSA-N ethyl cyclohexane Natural products CCC1CCCCC1 IIEWJVIFRVWJOD-UHFFFAOYSA-N 0.000 description 2
- 230000029142 excretion Effects 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 230000002550 fecal effect Effects 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 230000030136 gastric emptying Effects 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 239000011491 glass wool Substances 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 230000004179 hypothalamic–pituitary–adrenal axis Effects 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 208000021267 infertility disease Diseases 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000000185 intracerebroventricular administration Methods 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- UHOVQNZJYSORNB-UHFFFAOYSA-N monobenzene Natural products C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 2
- GJUGETZWYKVYDW-UHFFFAOYSA-N n,2-dimethoxy-1-[4-(methoxymethyl)phenyl]ethanimine Chemical compound COCC(=NOC)C1=CC=C(COC)C=C1 GJUGETZWYKVYDW-UHFFFAOYSA-N 0.000 description 2
- JDFNNPHDXMUFHJ-UHFFFAOYSA-N n-(1-phenylbutylidene)hydroxylamine Chemical compound CCCC(=NO)C1=CC=CC=C1 JDFNNPHDXMUFHJ-UHFFFAOYSA-N 0.000 description 2
- MEGCHFZCFPXPDK-JIDHJSLPSA-N n-[(1s)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-4-(2,4-dimethoxy-5-methylphenyl)-5-methyl-n-prop-2-ynyl-1,3-thiazol-2-amine;hydrochloride Chemical compound Cl.C1=C(C)C(OC)=CC(OC)=C1C1=C(C)SC(N(CC#C)[C@@H](CC2CC2)C=2C=C(F)C(C)=CC=2)=N1 MEGCHFZCFPXPDK-JIDHJSLPSA-N 0.000 description 2
- MVDKSBBYWLMXJQ-UQIIZPHYSA-N n-[(1s)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-4-(4-methoxy-2,5-dimethylphenyl)-5-methyl-n-prop-2-ynyl-1,3-thiazol-2-amine;hydrochloride Chemical compound Cl.C1=C(C)C(OC)=CC(C)=C1C1=C(C)SC(N(CC#C)[C@@H](CC2CC2)C=2C=C(F)C(C)=CC=2)=N1 MVDKSBBYWLMXJQ-UQIIZPHYSA-N 0.000 description 2
- SLXLFTIZFIAPFE-UHFFFAOYSA-N n-[1-(3-fluoro-4-methylphenyl)-2-methoxyethylidene]hydroxylamine Chemical compound COCC(=NO)C1=CC=C(C)C(F)=C1 SLXLFTIZFIAPFE-UHFFFAOYSA-N 0.000 description 2
- GREYVYREOPQVGM-UHFFFAOYSA-N n-methoxy-1-phenylbutan-1-imine Chemical compound CCCC(=NOC)C1=CC=CC=C1 GREYVYREOPQVGM-UHFFFAOYSA-N 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 210000002569 neuron Anatomy 0.000 description 2
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 2
- 229910000105 potassium hydride Inorganic materials 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 230000035935 pregnancy Effects 0.000 description 2
- YORCIIVHUBAYBQ-UHFFFAOYSA-N propargyl bromide Chemical compound BrCC#C YORCIIVHUBAYBQ-UHFFFAOYSA-N 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 108010086511 sauvagine Proteins 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 150000007979 thiazole derivatives Chemical class 0.000 description 2
- 229920002554 vinyl polymer Polymers 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 1
- YZOUYRAONFXZSI-SBHWVFSVSA-N (1S,3R,5R,6R,8R,10R,11R,13R,15R,16R,18R,20R,21R,23R,25R,26R,28R,30R,31S,33R,35R,36R,37S,38R,39S,40R,41S,42R,43S,44R,45S,46R,47S,48R,49S)-5,10,15,20,25,30,35-heptakis(hydroxymethyl)-37,39,40,41,42,43,44,45,46,47,48,49-dodecamethoxy-2,4,7,9,12,14,17,19,22,24,27,29,32,34-tetradecaoxaoctacyclo[31.2.2.23,6.28,11.213,16.218,21.223,26.228,31]nonatetracontane-36,38-diol Chemical compound O([C@@H]([C@H]([C@@H]1OC)OC)O[C@H]2[C@@H](O)[C@@H]([C@@H](O[C@@H]3[C@@H](CO)O[C@@H]([C@H]([C@@H]3O)OC)O[C@@H]3[C@@H](CO)O[C@@H]([C@H]([C@@H]3OC)OC)O[C@@H]3[C@@H](CO)O[C@@H]([C@H]([C@@H]3OC)OC)O[C@@H]3[C@@H](CO)O[C@@H]([C@H]([C@@H]3OC)OC)O3)O[C@@H]2CO)OC)[C@H](CO)[C@H]1O[C@@H]1[C@@H](OC)[C@H](OC)[C@H]3[C@@H](CO)O1 YZOUYRAONFXZSI-SBHWVFSVSA-N 0.000 description 1
- DSMMLIWKSJQDAG-VIFPVBQESA-N (1r)-1-(4-chloro-3-fluorophenyl)-2-methoxyethanamine Chemical compound COC[C@H](N)C1=CC=C(Cl)C(F)=C1 DSMMLIWKSJQDAG-VIFPVBQESA-N 0.000 description 1
- WPXVXJBFCKQSST-VIFPVBQESA-N (1r)-1-(4-chlorophenyl)-2-methoxyethanamine Chemical compound COC[C@H](N)C1=CC=C(Cl)C=C1 WPXVXJBFCKQSST-VIFPVBQESA-N 0.000 description 1
- BAASAJGPRXQZQJ-NSHDSACASA-N (1r)-1-(4-ethylphenyl)-2-methoxyethanamine Chemical compound CCC1=CC=C([C@@H](N)COC)C=C1 BAASAJGPRXQZQJ-NSHDSACASA-N 0.000 description 1
- OJIDQUDUQKAHIG-NSHDSACASA-N (1r)-2-methoxy-1-[4-(methoxymethyl)phenyl]ethanamine Chemical compound COC[C@H](N)C1=CC=C(COC)C=C1 OJIDQUDUQKAHIG-NSHDSACASA-N 0.000 description 1
- CMTDMIYJXVBUDX-VIFPVBQESA-N (1r)-2-methoxy-1-phenylethanamine Chemical compound COC[C@H](N)C1=CC=CC=C1 CMTDMIYJXVBUDX-VIFPVBQESA-N 0.000 description 1
- PWDQSUMPGQHCIB-JTQLQIEISA-N (1s)-1-(1,3-benzodioxol-5-yl)-2-cyclopropylethanamine Chemical compound C([C@H](N)C=1C=C2OCOC2=CC=1)C1CC1 PWDQSUMPGQHCIB-JTQLQIEISA-N 0.000 description 1
- DFTBFIMTDRVUQC-VIFPVBQESA-N (1s)-1-(1,3-benzodioxol-5-yl)butan-1-amine Chemical compound CCC[C@H](N)C1=CC=C2OCOC2=C1 DFTBFIMTDRVUQC-VIFPVBQESA-N 0.000 description 1
- BPNTUXMMEQLTDU-NSHDSACASA-N (1s)-1-(3-fluoro-4-methylphenyl)butan-1-amine Chemical compound CCC[C@H](N)C1=CC=C(C)C(F)=C1 BPNTUXMMEQLTDU-NSHDSACASA-N 0.000 description 1
- OCYXRXJLROJZSU-LBPRGKRZSA-N (1s)-1-(3-fluoro-4-methylphenyl)pentan-1-amine Chemical compound CCCC[C@H](N)C1=CC=C(C)C(F)=C1 OCYXRXJLROJZSU-LBPRGKRZSA-N 0.000 description 1
- XHCMWCBPIBFIBN-NSHDSACASA-N (1s)-1-(4-bromophenyl)-2-cyclopropylethanamine Chemical compound C([C@H](N)C=1C=CC(Br)=CC=1)C1CC1 XHCMWCBPIBFIBN-NSHDSACASA-N 0.000 description 1
- XXBWILFWEPHBDY-NSHDSACASA-N (1s)-1-(4-chlorophenyl)-2-cyclopropylethanamine Chemical compound C([C@H](N)C=1C=CC(Cl)=CC=1)C1CC1 XXBWILFWEPHBDY-NSHDSACASA-N 0.000 description 1
- ZIQUQHBBALXDFL-JTQLQIEISA-N (1s)-1-(4-fluorophenyl)butan-1-amine Chemical compound CCC[C@H](N)C1=CC=C(F)C=C1 ZIQUQHBBALXDFL-JTQLQIEISA-N 0.000 description 1
- HCJGKNUUHOOUJS-NSHDSACASA-N (1s)-1-(4-fluorophenyl)pentan-1-amine Chemical compound CCCC[C@H](N)C1=CC=C(F)C=C1 HCJGKNUUHOOUJS-NSHDSACASA-N 0.000 description 1
- XKIZOGWEUAYTSW-LBPRGKRZSA-N (1s)-1-[3-fluoro-4-(methoxymethyl)phenyl]butan-1-amine Chemical compound CCC[C@H](N)C1=CC=C(COC)C(F)=C1 XKIZOGWEUAYTSW-LBPRGKRZSA-N 0.000 description 1
- LAZACPUCELIAQA-LBPRGKRZSA-N (1s)-1-[4-(methoxymethyl)phenyl]butan-1-amine Chemical compound CCC[C@H](N)C1=CC=C(COC)C=C1 LAZACPUCELIAQA-LBPRGKRZSA-N 0.000 description 1
- UYUCMDMVWMBYSE-ZDUSSCGKSA-N (1s)-1-[4-(methoxymethyl)phenyl]pentan-1-amine Chemical compound CCCC[C@H](N)C1=CC=C(COC)C=C1 UYUCMDMVWMBYSE-ZDUSSCGKSA-N 0.000 description 1
- VQDUMXPOSJPPGZ-LBPRGKRZSA-N (1s)-2-cyclobutyl-1-(4-fluorophenyl)ethanamine Chemical compound C([C@H](N)C=1C=CC(F)=CC=1)C1CCC1 VQDUMXPOSJPPGZ-LBPRGKRZSA-N 0.000 description 1
- NQCCWXKFKYSSBV-LBPRGKRZSA-N (1s)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethanamine Chemical compound C1=C(F)C(C)=CC=C1[C@@H](N)CC1CC1 NQCCWXKFKYSSBV-LBPRGKRZSA-N 0.000 description 1
- VPJZNTCBXCDHFF-NSHDSACASA-N (1s)-2-cyclopropyl-1-(4-fluorophenyl)ethanamine Chemical compound C([C@H](N)C=1C=CC(F)=CC=1)C1CC1 VPJZNTCBXCDHFF-NSHDSACASA-N 0.000 description 1
- YIAJNUVBVQPDIN-LBPRGKRZSA-N (1s)-2-cyclopropyl-1-(4-methylphenyl)ethanamine Chemical compound C1=CC(C)=CC=C1[C@@H](N)CC1CC1 YIAJNUVBVQPDIN-LBPRGKRZSA-N 0.000 description 1
- CZNFPPQWCGEPGX-NSHDSACASA-N (1s)-2-cyclopropyl-1-phenylethanamine Chemical compound C([C@H](N)C=1C=CC=CC=1)C1CC1 CZNFPPQWCGEPGX-NSHDSACASA-N 0.000 description 1
- SRQPEYLZIUEVIA-QMMMGPOBSA-N (2r)-2-amino-2-(4-fluorophenyl)ethanol Chemical compound OC[C@H](N)C1=CC=C(F)C=C1 SRQPEYLZIUEVIA-QMMMGPOBSA-N 0.000 description 1
- LNQVZZGGOZBOQS-INIZCTEOSA-N (2s)-2-amino-3-methyl-1,1-diphenylbutan-1-ol Chemical compound C=1C=CC=CC=1C(O)([C@@H](N)C(C)C)C1=CC=CC=C1 LNQVZZGGOZBOQS-INIZCTEOSA-N 0.000 description 1
- HOFUGGFJBSCHEW-UHFFFAOYSA-N (4-prop-2-enoxyphenyl)boronic acid Chemical compound OB(O)C1=CC=C(OCC=C)C=C1 HOFUGGFJBSCHEW-UHFFFAOYSA-N 0.000 description 1
- MLXCGHFYQVFSLZ-UHFFFAOYSA-N 1-(3-fluoro-4-methylphenyl)-2-methoxy-n-phenylmethoxyethanimine Chemical compound C=1C=C(C)C(F)=CC=1C(COC)=NOCC1=CC=CC=C1 MLXCGHFYQVFSLZ-UHFFFAOYSA-N 0.000 description 1
- DCMSBJXQUAUMQM-UHFFFAOYSA-N 1-(3-fluoro-4-methylphenyl)-2-methoxyethanone Chemical compound COCC(=O)C1=CC=C(C)C(F)=C1 DCMSBJXQUAUMQM-UHFFFAOYSA-N 0.000 description 1
- OCYXRXJLROJZSU-UHFFFAOYSA-N 1-(3-fluoro-4-methylphenyl)pentan-1-amine Chemical compound CCCCC(N)C1=CC=C(C)C(F)=C1 OCYXRXJLROJZSU-UHFFFAOYSA-N 0.000 description 1
- VHKLGKYMXNRCFN-UHFFFAOYSA-N 1-(4-chloro-3-fluorophenyl)-2-methoxy-n-phenylmethoxyethanimine Chemical compound C=1C=C(Cl)C(F)=CC=1C(COC)=NOCC1=CC=CC=C1 VHKLGKYMXNRCFN-UHFFFAOYSA-N 0.000 description 1
- DSMMLIWKSJQDAG-UHFFFAOYSA-N 1-(4-chloro-3-fluorophenyl)-2-methoxyethanamine Chemical compound COCC(N)C1=CC=C(Cl)C(F)=C1 DSMMLIWKSJQDAG-UHFFFAOYSA-N 0.000 description 1
- KXNVDQPVFIMODI-UHFFFAOYSA-N 1-(4-chloro-3-fluorophenyl)-2-methoxyethanone Chemical compound COCC(=O)C1=CC=C(Cl)C(F)=C1 KXNVDQPVFIMODI-UHFFFAOYSA-N 0.000 description 1
- WPXVXJBFCKQSST-UHFFFAOYSA-N 1-(4-chlorophenyl)-2-methoxyethanamine Chemical compound COCC(N)C1=CC=C(Cl)C=C1 WPXVXJBFCKQSST-UHFFFAOYSA-N 0.000 description 1
- DFVRECUTZCVSSA-UHFFFAOYSA-N 1-(4-chlorophenyl)-2-methoxyethanone Chemical compound COCC(=O)C1=CC=C(Cl)C=C1 DFVRECUTZCVSSA-UHFFFAOYSA-N 0.000 description 1
- IZTWRIBUKHAEDP-UHFFFAOYSA-N 1-[4-(methoxymethyl)phenyl]butylthiourea Chemical compound CCCC(NC(N)=S)C1=CC=C(COC)C=C1 IZTWRIBUKHAEDP-UHFFFAOYSA-N 0.000 description 1
- AITNMTXHTIIIBB-UHFFFAOYSA-N 1-bromo-4-fluorobenzene Chemical compound FC1=CC=C(Br)C=C1 AITNMTXHTIIIBB-UHFFFAOYSA-N 0.000 description 1
- KGWPEAIPPHCGKM-UHFFFAOYSA-N 1-cyclobutyl-1-(4-fluorophenyl)-n-phenylmethoxymethanimine Chemical compound C1=CC(F)=CC=C1C(C1CCC1)=NOCC1=CC=CC=C1 KGWPEAIPPHCGKM-UHFFFAOYSA-N 0.000 description 1
- LQUVVWBTBOYJAU-UHFFFAOYSA-N 1-phenylpentan-1-amine Chemical compound CCCCC(N)C1=CC=CC=C1 LQUVVWBTBOYJAU-UHFFFAOYSA-N 0.000 description 1
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 1
- FJZRSVQOVMQGBD-UHFFFAOYSA-N 2,2-dimethylpropanoyl isothiocyanate Chemical compound CC(C)(C)C(=O)N=C=S FJZRSVQOVMQGBD-UHFFFAOYSA-N 0.000 description 1
- HVAUUPRFYPCOCA-AREMUKBSSA-N 2-O-acetyl-1-O-hexadecyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCOC[C@@H](OC(C)=O)COP([O-])(=O)OCC[N+](C)(C)C HVAUUPRFYPCOCA-AREMUKBSSA-N 0.000 description 1
- SRQPEYLZIUEVIA-UHFFFAOYSA-N 2-amino-2-(4-fluorophenyl)ethanol Chemical compound OCC(N)C1=CC=C(F)C=C1 SRQPEYLZIUEVIA-UHFFFAOYSA-N 0.000 description 1
- JKFYKCYQEWQPTM-UHFFFAOYSA-N 2-azaniumyl-2-(4-fluorophenyl)acetate Chemical compound OC(=O)C(N)C1=CC=C(F)C=C1 JKFYKCYQEWQPTM-UHFFFAOYSA-N 0.000 description 1
- GCPRCXZISRDEDG-UHFFFAOYSA-N 2-bromo-1-(2-chloro-4-methoxy-5-methylphenyl)propan-1-one Chemical compound COC1=CC(Cl)=C(C(=O)C(C)Br)C=C1C GCPRCXZISRDEDG-UHFFFAOYSA-N 0.000 description 1
- VYWUMKDOVBOZFU-UHFFFAOYSA-N 2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethanone Chemical compound C1=C(F)C(C)=CC=C1C(=O)CC1CC1 VYWUMKDOVBOZFU-UHFFFAOYSA-N 0.000 description 1
- FAUQRRGKJKMEIW-UHFFFAOYSA-N 2-cyclopropylacetonitrile Chemical compound N#CCC1CC1 FAUQRRGKJKMEIW-UHFFFAOYSA-N 0.000 description 1
- OJIDQUDUQKAHIG-UHFFFAOYSA-N 2-methoxy-1-[4-(methoxymethyl)phenyl]ethanamine Chemical compound COCC(N)C1=CC=C(COC)C=C1 OJIDQUDUQKAHIG-UHFFFAOYSA-N 0.000 description 1
- IIXMFSVEDLMWOQ-UHFFFAOYSA-N 2-methoxy-1-[4-(methoxymethyl)phenyl]ethanone Chemical compound COCC(=O)C1=CC=C(COC)C=C1 IIXMFSVEDLMWOQ-UHFFFAOYSA-N 0.000 description 1
- QKPVEISEHYYHRH-UHFFFAOYSA-N 2-methoxyacetonitrile Chemical compound COCC#N QKPVEISEHYYHRH-UHFFFAOYSA-N 0.000 description 1
- LOCHGJFCGIWSBZ-UHFFFAOYSA-N 3-cyclopropyl-1-(4-fluorophenyl)propan-1-amine Chemical compound C=1C=C(F)C=CC=1C(N)CCC1CC1 LOCHGJFCGIWSBZ-UHFFFAOYSA-N 0.000 description 1
- LXHJZWZQICEKID-UHFFFAOYSA-N 3-cyclopropyl-1-(4-fluorophenyl)propan-1-one Chemical compound C1=CC(F)=CC=C1C(=O)CCC1CC1 LXHJZWZQICEKID-UHFFFAOYSA-N 0.000 description 1
- XDWGEGZDMFHZBL-UHFFFAOYSA-N 4,5-diphenyl-1,3-thiazol-2-amine Chemical class S1C(N)=NC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 XDWGEGZDMFHZBL-UHFFFAOYSA-N 0.000 description 1
- QGSJYYIRAFRPIT-UHFFFAOYSA-N 4-(2-amino-1,3-thiazol-4-yl)phenol Chemical class S1C(N)=NC(C=2C=CC(O)=CC=2)=C1 QGSJYYIRAFRPIT-UHFFFAOYSA-N 0.000 description 1
- NWGPIGBBTPBTBU-SFHVURJKSA-N 4-(2-chloro-4-methoxy-5-methylphenyl)-n-[(1r)-1-(4-fluorophenyl)-2-methoxyethyl]-5-methyl-1,3-thiazol-2-amine Chemical compound N([C@@H](COC)C=1C=CC(F)=CC=1)C(SC=1C)=NC=1C1=CC(C)=C(OC)C=C1Cl NWGPIGBBTPBTBU-SFHVURJKSA-N 0.000 description 1
- RFZOGPABZLMDQW-UHFFFAOYSA-N 4-chloro-2-methoxy-1-methylbenzene Chemical compound COC1=CC(Cl)=CC=C1C RFZOGPABZLMDQW-UHFFFAOYSA-N 0.000 description 1
- AEKVBBNGWBBYLL-UHFFFAOYSA-N 4-fluorobenzonitrile Chemical compound FC1=CC=C(C#N)C=C1 AEKVBBNGWBBYLL-UHFFFAOYSA-N 0.000 description 1
- JMHFFDIMOUKDCZ-NTXHZHDSSA-N 61214-51-5 Chemical compound C([C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CCSC)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)CNC(=O)CNC(=O)[C@@H](N)CC=1C=CC(O)=CC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)[C@@H](C)O)C1=CC=CC=C1 JMHFFDIMOUKDCZ-NTXHZHDSSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 229920001450 Alpha-Cyclodextrin Polymers 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 101800005049 Beta-endorphin Proteins 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 0 CC1C(C)=C*(*)=CC1 Chemical compound CC1C(C)=C*(*)=CC1 0.000 description 1
- 125000006519 CCH3 Chemical group 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- 101800001982 Cholecystokinin Proteins 0.000 description 1
- 102100025841 Cholecystokinin Human genes 0.000 description 1
- 206010010144 Completed suicide Diseases 0.000 description 1
- 229910021590 Copper(II) bromide Inorganic materials 0.000 description 1
- OMFXVFTZEKFJBZ-UHFFFAOYSA-N Corticosterone Natural products O=C1CCC2(C)C3C(O)CC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 OMFXVFTZEKFJBZ-UHFFFAOYSA-N 0.000 description 1
- 102400000739 Corticotropin Human genes 0.000 description 1
- 229940122010 Corticotropin releasing factor antagonist Drugs 0.000 description 1
- ZGUNAGUHMKGQNY-SSDOTTSWSA-N D-alpha-phenylglycine Chemical compound OC(=O)[C@H](N)C1=CC=CC=C1 ZGUNAGUHMKGQNY-SSDOTTSWSA-N 0.000 description 1
- 208000020401 Depressive disease Diseases 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 102400000921 Gastrin Human genes 0.000 description 1
- 108010052343 Gastrins Proteins 0.000 description 1
- 206010017969 Gastrointestinal inflammatory conditions Diseases 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 102000014415 Muscarinic acetylcholine receptor Human genes 0.000 description 1
- 108050003473 Muscarinic acetylcholine receptor Proteins 0.000 description 1
- QAADZYUXQLUXFX-UHFFFAOYSA-N N-phenylmethylthioformamide Natural products S=CNCC1=CC=CC=C1 QAADZYUXQLUXFX-UHFFFAOYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 102000003797 Neuropeptides Human genes 0.000 description 1
- 108090000189 Neuropeptides Proteins 0.000 description 1
- 206010029897 Obsessive thoughts Diseases 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 229940083963 Peptide antagonist Drugs 0.000 description 1
- 108010003541 Platelet Activating Factor Proteins 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 206010039966 Senile dementia Diseases 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229940123445 Tricyclic antidepressant Drugs 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 108010059705 Urocortins Proteins 0.000 description 1
- 102000005630 Urocortins Human genes 0.000 description 1
- 108010011107 Urotensins Proteins 0.000 description 1
- 102000026557 Urotensins Human genes 0.000 description 1
- FZNCGRZWXLXZSZ-CIQUZCHMSA-N Voglibose Chemical compound OCC(CO)N[C@H]1C[C@](O)(CO)[C@@H](O)[C@H](O)[C@H]1O FZNCGRZWXLXZSZ-CIQUZCHMSA-N 0.000 description 1
- IOVMDNVQMAKRNZ-VIFPVBQESA-N [(1r)-1-(4-fluorophenyl)-2-methoxyethyl]thiourea Chemical compound COC[C@H](NC(N)=S)C1=CC=C(F)C=C1 IOVMDNVQMAKRNZ-VIFPVBQESA-N 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 150000001253 acrylic acids Chemical class 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 230000001919 adrenal effect Effects 0.000 description 1
- 210000004100 adrenal gland Anatomy 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 239000012670 alkaline solution Substances 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- HFHDHCJBZVLPGP-RWMJIURBSA-N alpha-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO HFHDHCJBZVLPGP-RWMJIURBSA-N 0.000 description 1
- 229940043377 alpha-cyclodextrin Drugs 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 229940124332 anorexigenic agent Drugs 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000002421 anti-septic effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 239000000935 antidepressant agent Substances 0.000 description 1
- 229940005513 antidepressants Drugs 0.000 description 1
- 230000000338 anxiogenic effect Effects 0.000 description 1
- 229940005530 anxiolytics Drugs 0.000 description 1
- 239000002830 appetite depressant Substances 0.000 description 1
- 239000011260 aqueous acid Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 210000003403 autonomic nervous system Anatomy 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 150000001555 benzenes Chemical class 0.000 description 1
- 150000001557 benzodiazepines Chemical class 0.000 description 1
- RWCCWEUUXYIKHB-UHFFFAOYSA-N benzophenone Chemical class C=1C=CC=CC=1C(=O)C1=CC=CC=C1 RWCCWEUUXYIKHB-UHFFFAOYSA-N 0.000 description 1
- 150000008366 benzophenones Chemical class 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 238000005574 benzylation reaction Methods 0.000 description 1
- 108010042362 beta-Lipotropin Proteins 0.000 description 1
- WHGYBXFWUBPSRW-FOUAGVGXSA-N beta-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO WHGYBXFWUBPSRW-FOUAGVGXSA-N 0.000 description 1
- 229960004853 betadex Drugs 0.000 description 1
- 239000003833 bile salt Substances 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- KVNRLNFWIYMESJ-UHFFFAOYSA-N butyronitrile Chemical compound CCCC#N KVNRLNFWIYMESJ-UHFFFAOYSA-N 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- AOXOCDRNSPFDPE-UKEONUMOSA-N chembl413654 Chemical compound C([C@H](C(=O)NCC(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](C)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)CNC(=O)[C@@H](N)CCC(O)=O)C1=CC=C(O)C=C1 AOXOCDRNSPFDPE-UKEONUMOSA-N 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 229940107137 cholecystokinin Drugs 0.000 description 1
- 230000037326 chronic stress Effects 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 206010009887 colitis Diseases 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- JNGZXGGOCLZBFB-IVCQMTBJSA-N compound E Chemical compound N([C@@H](C)C(=O)N[C@@H]1C(N(C)C2=CC=CC=C2C(C=2C=CC=CC=2)=N1)=O)C(=O)CC1=CC(F)=CC(F)=C1 JNGZXGGOCLZBFB-IVCQMTBJSA-N 0.000 description 1
- ONCCWDRMOZMNSM-FBCQKBJTSA-N compound Z Chemical compound N1=C2C(=O)NC(N)=NC2=NC=C1C(=O)[C@H]1OP(O)(=O)OC[C@H]1O ONCCWDRMOZMNSM-FBCQKBJTSA-N 0.000 description 1
- 229940124558 contraceptive agent Drugs 0.000 description 1
- 239000003433 contraceptive agent Substances 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- OMFXVFTZEKFJBZ-HJTSIMOOSA-N corticosterone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@H](CC4)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OMFXVFTZEKFJBZ-HJTSIMOOSA-N 0.000 description 1
- 230000003131 corticotrophic effect Effects 0.000 description 1
- 239000002769 corticotropin releasing factor antagonist Substances 0.000 description 1
- 108010050742 corticotropin releasing hormone (9-41) Proteins 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- KMWTUCKAZFNWNG-UHFFFAOYSA-N cyclobutyl-(4-fluorophenyl)methanone Chemical compound C1=CC(F)=CC=C1C(=O)C1CCC1 KMWTUCKAZFNWNG-UHFFFAOYSA-N 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- VWHSNGZQBUUEBK-UHFFFAOYSA-N di(cyclobutyl)methanamine Chemical compound C1CCC1C(N)C1CCC1 VWHSNGZQBUUEBK-UHFFFAOYSA-N 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 230000002996 emotional effect Effects 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 230000003826 endocrine responses Effects 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 235000012631 food intake Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- GDSRMADSINPKSL-HSEONFRVSA-N gamma-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO GDSRMADSINPKSL-HSEONFRVSA-N 0.000 description 1
- 229940080345 gamma-cyclodextrin Drugs 0.000 description 1
- 239000007897 gelcap Substances 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 125000001475 halogen functional group Chemical group 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 210000003016 hypothalamus Anatomy 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 230000008799 immune stress Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 239000002917 insecticide Substances 0.000 description 1
- 210000002011 intestinal secretion Anatomy 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 150000002540 isothiocyanates Chemical class 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 210000000627 locus coeruleus Anatomy 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 150000002681 magnesium compounds Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 108020004084 membrane receptors Proteins 0.000 description 1
- 102000006240 membrane receptors Human genes 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- KUGLDBMQKZTXPW-JEDNCBNOSA-N methyl (2s)-2-amino-3-methylbutanoate;hydrochloride Chemical compound Cl.COC(=O)[C@@H](N)C(C)C KUGLDBMQKZTXPW-JEDNCBNOSA-N 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- QUSNBJAOOMFDIB-UHFFFAOYSA-N monoethyl amine Natural products CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 1
- 230000036651 mood Effects 0.000 description 1
- 210000000754 myometrium Anatomy 0.000 description 1
- YDABARPWSZPQDA-UHFFFAOYSA-N n-(1-phenylpentylidene)hydroxylamine Chemical compound CCCCC(=NO)C1=CC=CC=C1 YDABARPWSZPQDA-UHFFFAOYSA-N 0.000 description 1
- KVHMQBYTRDFQBL-UHFFFAOYSA-N n-[1-(4-chloro-3-fluorophenyl)-2-methoxyethylidene]hydroxylamine Chemical compound COCC(=NO)C1=CC=C(Cl)C(F)=C1 KVHMQBYTRDFQBL-UHFFFAOYSA-N 0.000 description 1
- QUOJIVHMMXOHCJ-UHFFFAOYSA-N n-[1-(4-chlorophenyl)-2-methoxyethylidene]hydroxylamine Chemical compound COCC(=NO)C1=CC=C(Cl)C=C1 QUOJIVHMMXOHCJ-UHFFFAOYSA-N 0.000 description 1
- BDFCTCHHUWGPLE-UHFFFAOYSA-N n-[1-[4-(methoxymethyl)phenyl]butylidene]hydroxylamine Chemical compound CCCC(=NO)C1=CC=C(COC)C=C1 BDFCTCHHUWGPLE-UHFFFAOYSA-N 0.000 description 1
- YAGHLPPCRRCMOY-UHFFFAOYSA-N n-[2-methoxy-1-[4-(methoxymethyl)phenyl]ethylidene]hydroxylamine Chemical compound COCC(=NO)C1=CC=C(COC)C=C1 YAGHLPPCRRCMOY-UHFFFAOYSA-N 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- DKJVLGJSHVEHHG-UHFFFAOYSA-N n-[di(cyclobutyl)methylidene]hydroxylamine Chemical compound C1CCC1C(=NO)C1CCC1 DKJVLGJSHVEHHG-UHFFFAOYSA-N 0.000 description 1
- PWTLHDIOHIPRPR-UHFFFAOYSA-N n-benzyl-n-methyl-1,3-thiazol-2-amine Chemical class N=1C=CSC=1N(C)CC1=CC=CC=C1 PWTLHDIOHIPRPR-UHFFFAOYSA-N 0.000 description 1
- UBSMCXXKGLHECF-UHFFFAOYSA-N n-cyclobutyl-4-fluoro-n-methylaniline Chemical compound C=1C=C(F)C=CC=1N(C)C1CCC1 UBSMCXXKGLHECF-UHFFFAOYSA-N 0.000 description 1
- HJOUHLJNIJTZRR-UHFFFAOYSA-N n-methoxy-1-[4-(methoxymethyl)phenyl]butan-1-imine Chemical compound CCCC(=NOC)C1=CC=C(COC)C=C1 HJOUHLJNIJTZRR-UHFFFAOYSA-N 0.000 description 1
- RNDVXOSSNFWIMI-UHFFFAOYSA-N n-methoxy-1-phenylpentan-1-imine Chemical compound CCCCC(=NOC)C1=CC=CC=C1 RNDVXOSSNFWIMI-UHFFFAOYSA-N 0.000 description 1
- 230000000955 neuroendocrine Effects 0.000 description 1
- 235000013615 non-nutritive sweetener Nutrition 0.000 description 1
- 230000002474 noradrenergic effect Effects 0.000 description 1
- 231100000926 not very toxic Toxicity 0.000 description 1
- HYDZPXNVHXJHBG-UHFFFAOYSA-N o-benzylhydroxylamine;hydron;chloride Chemical compound Cl.NOCC1=CC=CC=C1 HYDZPXNVHXJHBG-UHFFFAOYSA-N 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 230000008447 perception Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- NHKJPPKXDNZFBJ-UHFFFAOYSA-N phenyllithium Chemical compound [Li]C1=CC=CC=C1 NHKJPPKXDNZFBJ-UHFFFAOYSA-N 0.000 description 1
- ANRQGKOBLBYXFM-UHFFFAOYSA-M phenylmagnesium bromide Chemical compound Br[Mg]C1=CC=CC=C1 ANRQGKOBLBYXFM-UHFFFAOYSA-M 0.000 description 1
- 230000001817 pituitary effect Effects 0.000 description 1
- 210000002826 placenta Anatomy 0.000 description 1
- 230000010118 platelet activation Effects 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 208000020016 psychiatric disease Diseases 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 239000012429 reaction media Substances 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000001850 reproductive effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000003938 response to stress Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 125000000467 secondary amino group Chemical group [H]N([*:1])[*:2] 0.000 description 1
- IZTQOLKUZKXIRV-YRVFCXMDSA-N sincalide Chemical compound C([C@@H](C(=O)N[C@@H](CCSC)C(=O)NCC(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](N)CC(O)=O)C1=CC=C(OS(O)(=O)=O)C=C1 IZTQOLKUZKXIRV-YRVFCXMDSA-N 0.000 description 1
- 210000002027 skeletal muscle Anatomy 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000016978 synaptic transmission, cholinergic Effects 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 125000001302 tertiary amino group Chemical group 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- ODLHGICHYURWBS-LKONHMLTSA-N trappsol cyclo Chemical compound CC(O)COC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)COCC(O)C)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1COCC(C)O ODLHGICHYURWBS-LKONHMLTSA-N 0.000 description 1
- 239000003029 tricyclic antidepressant agent Substances 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 239000000777 urocortin Substances 0.000 description 1
- 239000000780 urotensin Substances 0.000 description 1
- 230000008728 vascular permeability Effects 0.000 description 1
- 229920006163 vinyl copolymer Polymers 0.000 description 1
- 230000009278 visceral effect Effects 0.000 description 1
- MMSLSKDMIZOHRX-WYDAWZGFSA-N α-helical crf(9-41) Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(N)=O MMSLSKDMIZOHRX-WYDAWZGFSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/12—Antidiarrhoeals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/06—Antiabortive agents; Labour repressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/32—Alcohol-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/02—Drugs for disorders of the endocrine system of the hypothalamic hormones, e.g. TRH, GnRH, CRH, GRH, somatostatin
- A61P5/04—Drugs for disorders of the endocrine system of the hypothalamic hormones, e.g. TRH, GnRH, CRH, GRH, somatostatin for decreasing, blocking or antagonising the activity of the hypothalamic hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/38—Drugs for disorders of the endocrine system of the suprarenal hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/14—Vasoprotectives; Antihaemorrhoidals; Drugs for varicose therapy; Capillary stabilisers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/42—Amino or imino radicals substituted by hydrocarbon or substituted hydrocarbon radicals
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Diabetes (AREA)
- Endocrinology (AREA)
- Immunology (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Addiction (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Reproductive Health (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Rheumatology (AREA)
- Psychology (AREA)
- Physical Education & Sports Medicine (AREA)
- Child & Adolescent Psychology (AREA)
- Emergency Medicine (AREA)
- Anesthesiology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Ophthalmology & Optometry (AREA)
- Transplantation (AREA)
Description
- Die vorliegende Erfindung betrifft neue verzweigte Aminoderivate des Thiazols, ein Verfahren zu ihrer Herstellung und sie enthaltende pharmazeutische Zubereitungen. Diese neuen Thiazolderivate besitzen CRF-(Corticotropin Releasing Factor)-antagonistische Wirkung und können demzufolge Wirkstoffe für pharmazeutische Zubereitungen darstellen.
- Der Faktor der Freisetzung des corticotropen Hormons (CRF) ist ein Peptid , dessen Sequenz von 41 Aminosäuren von W. Vale et al. im Jahr 1981 aufgeklärt worden ist (Science, 213 (1981), 1394–1397). CRF ist der wesentliche endogene Faktor, der bei der Regulierung der Hypothalamus-Hypophysen-Nebennieren-Achse (Freisetzung des adrenocorticotropen Hormons: ACTH) und ihrer pathologischen Zustände beteiligt ist sowie bei depressiven Syndromen, die dadurch verursacht werden. CRF verursacht weiterhin die Sekretion von β-Endorphin, β-Lipotropin und Corticosteron. CRF stellt damit einen physiologischen Regulator der Sekretion des adrenocorticotropen Hormons (ACTH) und insbesondere von Peptiden dar, die von Propiomelanocortin (POMC) abgeleitet sind. Neben seiner Lokalisierung im Hypothalamus ist CRF in starkem Maße in dem Zentralnervensystem verteilt, jedoch auch in extraneuronalen Geweben, wie den Nebennierendrüsen und den Testikeln. Die Anwesenheit von CRF wurde auch im Verlaufe von Entzündungsprozessen nachgewiesen.
- Eine Vielzahl von Tierexperimenten hat gezeigt, daß die zentrale Verabreichtung von CRF verschiedenartige anxiogene Wirkungen hervorruft, wie die Modifizierung des Allgemeinverhaltens: beispielsweise Neophobie, Verminderung der sexuellen Empfänglichkeit, Verringerung der Nahrungsmittelaufnahme und des langsamen Schlafes bei der Ratte. Die intrazerebroventrikuläre Injektion von CRF erhöht weiterhin die Reizung der noradrenergischen Neuronen im Locus coeruleus, was beim Tier häufig mit einem Angstzustand verknüpft ist. Bei der Ratte induziert die zentrale oder periphere Verabreichung von CRF oder von verwandten Peptiden (beispielsweise Urocortin oder Sauvagin) neben zentralen Effekten, wie der Erhöhung der Aufmerksamkeit und der emotionellen Reaktivität gegenüber der Umgebung, Veränderungen der Magenentleerung, der Säuresekretion, des Darmdurchgangs und der Fäzesausscheidung sowie Spannungseffekte. CRF ist weiterhin bei der komplexen Steuerung von Entzündungsantworten beteiligt, einerseits durch eine pro-inflammatorische Wirkung bei bestimmten Tiermodellen und andererseits als Inhibitor von Effekten, die durch eine Erhöhung der Gefäßpermeabilität als Folge einer Entzündung hervorgerufen sind.
- Die Verwendung eines peptidischen Antagonisten, nämlich alpha-helikales CRF (9-41) (α-CRF) oder von spezifischen Antikörpern (J. Rivier et al., Science, 224 (1984), 889–891) hat es ermöglicht, die Rolle dieses Peptids bei der Gesamtheit dieser Effekte zu bestätigen. Diese Unersuchungen haben weiterhin die wichtige Rolle des CRF beim Menschen bei der Integration von komplexen Antworten bestätigt, die man bei einem physiologischen, psychologischen oder immunologischen Stress auf den neuroendokrinen, viszeralen oder verhaltensmäßigen Bereich beobachtet (J.E. Morley et al., Endocrine Review 8, 3 (1987), 256-287; M.A. Smith et al., Horm. Res., 31 (1989), 66–71). Weiterhin stützen klinische Ergebnisse die wirksame Beteiligung von CRF bei einer Vielzahl von Störungen, die von einem Stresszustand ausgelöst werden (L.R. Gulley et al., J. Clin. Psychiatry, 54, 1 (Suppl.) (1993), 16–19), beispielsweise:
- – So hat die Existenz des CRF-Tests (i.v.-Verabreichung) beim Menschen es ermöglicht, die Modifizierung der ACTH-Antwort bei depressiven Patienten zu beeinflussen (A. Breier et al., Am. J. Psychiatry, 144 (1987), 1419–1425).
- – Die Entdeckung einer endogenen CRF-Hypersekretion bei bestimmten pathologischen Zuständen, beispielsweise einem erhöhten CRF-Spiegel in der Zephalorachidialflüssigkeit bei nicht behandelten, deprimierten Patienten oder Patienten, die von einer Demenz vom Typ der Alzheimerschen Krankheit befallen sind (C.B. Nemeroff et al., Science, 226, 4680 (1984), 1342–1343; Regul. Pept., 25 (1989), 123–130), oder einer verringerten Dichte der CRF-Rezeptoren im Kortex von Selbstmordopfern (C.B. Nemeroff et al.., Arch. Gen. Psychiatry, 45 (1988), 577–579).
- – Die Dysfunktion von CRF-abhängigen Neuronen wird bei schweren pathologischen Zuständen angenommen, wie der Alzheimerschen Krankheit, der Parkinsonschen Krankheit, der Huntington-Chorea und der amyotrophischen Lateralsklerose (E.B. De Souza, Hospital Practice, 23 (1988), 59).
- Die zentrale Verabreichung von CRF verursacht bei einer Vielzahl von Tierarten verhaltensmäßige Wirkungen, die vergleichbar sind mit jenen, die man beim Menschen bei Stress-Situationen vorfindet. Wenn sie im Laufe der Zeit wiederholt werden, können diese Effekte zu verschiedenartigen pathologischen Zuständen führen: Müdigkeit, Hypertension, Herz- und Spannungsstörungen, Modifizierung der Magenentleerung, der Fäzesexkretion (Colitis, Reizdarm), Veränderung der Säuresekretion, Hyperglykämie, verlangsamtes Wachstum, Anorexie, Neophobie, Migräne, Reproduktionsstörungen, Immunosuppression (Entzün dungsprozesse, multiple Infektionen und Krebs) und verschiedenartige neuropsychiatrische Störungen (Depressionen, nervöse Anorexie und Angst).
- Die intrazerebroventrikulare Injektion des Referenzpeptid-Antagonisten, α-CRF, beugt den Wirkungen vor, die man entweder durch die Verabreichung von exogenem CRF erzielt oder durch die Anwendung von Stress (Ether, Zwang, laute Geräusche, Elektroschock, Ethanolentzug, Operationen), die ihrerseits dazu geeignet sind, eine Erhöhung des endogenen CRF-Spiegels zu induzieren. Diese Ergebnisse wurden durch die Untersuchung einer Vielzahl von antagonistischen Peptidmolekülen mit einer dem CRF verwandten Struktur und die eine längere Wirkungsdauer im Vergleich zu α-CRF aufweisen, bestätigt (J. Rivier, et al., J. Med. Chem., 36 (1993), 2851–2859; F. Menzaghi et al., J. Pharmacol. Exp. Ther., 269, 2 (1994), 564–572; J.F. Hernandez et al., J. Med. Chem., 36 (1993), 2860–2867).
- Solche CRF-antagonistischen Peptidverbindungen sind beispielsweise in den Patenten
US 5,109,111 ,US 5,132,111 ,US 5,245,009 und in den Patentanmeldungen WO 92/22576 und WO 96/19499 beschrieben. - Weiterhin haben vorläufige Untersuchungen gezeigt, daß tricyclische Antidepressiva den CRF-Spiegel sowie die Anzahl der CRF-Rezeptoren im Gehirn modulieren können (D.E. Grigoriadis et al., Neuropsychopharmacology, 2 (1989), 53–60). In gleicher Weise sind anxiolytische Benzodiazepin-Verbindungen dazu in der Lage, die Wirkung des CRF umzukehren (K.T. Britton et al., Psychopharmacology, 94 (1988), 306), ohne daß der Wirkungsmechanismus dieser Substanzen vollständig aufgeklärt wäre. Diese Ergebnisse bestätigen das wachsende Bedürfnis für antagonistische Nichtpeptid-Moleküle bezüglich der CRF-Rezeptoren.
- Es ist wichtig, auf drei mögliche Konsequenzen von chronischen Stresszuständen hinzuweisen, nämlich die Immunodepression, Fertilitätsstörungen sowie die Verursachung des Diabetes.
- CRF übt solche Wirkungen aus, indem es mit spezifischen Membranrezeptoren in Wechselwirkung tritt, die in der Hypophyse und dem Gehirn einer Vielzahl von Tieren (Maus, Ratte und Menschen) sowie im Herzen, der Skelettmuskulatur (Ratte, Maus) und im Myometrium und der Plazenta im Verlaufe der Schwangerschaft gefunden wurden.
- Eine große Vielzahl von 2-Aminothiazol-Derivaten ist bereits bekannt. So beschreibt die Patentanmeldung
EP 462 264 - Die Anmeldung GB 2 022 285 beschreibt Verbindungen, die eine regulierende Wirkung auf die Immunantwort ausüben und antiinflammatorische Wirkungen besitzen. Es handelt sich um Thiazolderivate, die in der 2-Stellung durch sekundäre Aminogruppen substituiert sind.
- In der Patentanmeldung
EP 432 040 - Es sind auch 2-Amino-4,5-diphenylthiazol-Derivate mit antiinflammatorischen Wirkungen bekannt (Patentanmeldung JP-01 75 475).
- Man kennt auch 2-Amino-4-(4-hydroxyphenyl)-thiazol-Derivate, die als Zwischenprodukte der Synthese von 2,2-Diarylchromenothiazol-Derivaten nützlich sind (Patentanmeldung
EP 205 069 - Weiterhin sind 2-(N-Methyl-N-benzylamino)-thiazol-Derivate in J. Chem, Soc. Perkin, Trans 1, 2 (1984), 147–153 und in J. Chem. Soc. Perkin, Trans 1, 2 (1983), 341–347 beschrieben.
- Die Patentanmeldung WO 94/01423 beschreibt 2-Aminothiazol-Derivate. Diese Verbindungen werden als Insektizide verwendet und besitzen keinerlei Substitution in der 5-Stellung des Heterocyclus.
- In gleicher Weise beschreibt die Patentanmeldung WO 96/ 16650 von 2-Aminothiazol abgeleitete Verbindungen, die als Antibiotika nützlich sind.
- Die Patentanmeldung
EP 283 390 - Diese Verbindungen besitzen insbesondere eine stimulierende Wirkung auf die zentrale cholinergische Transmission. Sie können daher als Agonisten von Muscarin-Rezeptoren verwendet werden und finden Anwendung bei der Behandlung von Störungen des Gedächtnisses und von senilen Dementien.
- Die 2-Aminothiazol-Derivate, deren Aminogruppe in der 2-Stellung eine tertiäre Aminogruppe ist, die einen verzweigten Alkyl- oder Aralkyl-Substituenten aufweist, sind in der
EP 576 350 EP 659 747 - Das US-Patent 5,063,245 offenbart CRF-Antagonisten, welche es ermöglichen, in vitro die Bindung von CRF an seine spezifischen Rezeptoren bei einer Konzentration im Bereich eines Mikromols zu verdrängen. Seitdem wurde eine Vielzahl von Patentanmeldungen veröffentlicht, welche nicht-peptidische Moleküle betreffen, beispielsweise die Anmeldungen WO 94/13643, WO 94/ 13644, WO 94/13661, WO 94/13676, WO 94/13677, WO 94/10333, WO 95/00640, WO 95/10506, WO 95/13372, WO 95/33727, WO 95/33750, WO 95/34563,
EP 691 128 EP 729 758 - Es wurde im Rahmen der vorliegenden Erfindung nunmehr gefunden, daß bestimmte verzweigte Aminoderivate des Thiazols, die Gegenstand der vorliegenden Erfindung sind, eine ausgezeichnete Affinität für die Rezeptoren des CRF besitzen. Weiterhin besitzen diese Moleküle aufgrund ihrer Struktur eine gute Dispergierbarkeit und/oder Löslichkeit in üblicherweise in der Therapie verwendeten Lösungsmitteln oder Lösungen, was ihnen eine pharmakologische Wirkung verleiht und auch ihre leichte Verarbeitung zu oral und parenteral zu verabreichenden galenischen Formen ermöglicht.
- Dies ist überraschend und unerwartet, da die erfindungsgemäßen Verbindungen in vivo wirksamer sind als Verbindungen mit ähnlicher Struktur, insbesondere durch eine signifikantere Inhibierung der durch CRF im Bereich der Hypothalamus-Hypophysen-Nebennieren-Achse induzierten Antwort.
- Die vorliegende Erfindung betrifft daher die Verbindungen der folgenden Formel in racemischer oder enantiomerenreiner Form:in der

- – R1 und R2, die gleichartig oder verschieden sind, jeweils unabhängig voneinander ein Halogenatom; eine Hydroxy-(C1-C5)-alkylgruppe; eine (C1-C5)-Alkylgruppe; eine Aralkylgruppe, in der der Arylteil (C6-C8) und der Alkylteil (C1-C4) ist; eine (C1-C5)-Alkoxygruppe; eine Trifluormethylgruppe; eine Nitrogruppe; eine Nitrilgruppe; eine Gruppe -SR, in der R Wasserstoff, eine (C1-C5)-Alkylgruppe oder eine Aralkylgruppe, in der der Arylteil (C6-C8) und der Alkylteil (C1-C4) ist, bedeutet; eine Gruppe -S-CO-R, in der R eine (C1-C5)-Alkylgruppe oder eine Aralkylgruppe, in der der Arylteil (C6-C8) und der Alkylteil (C1-C4) ist, bedeutet; eine Gruppe -COORa, worin Ra Wasserstoff oder eine (C1-C5)-Alkylgruppe darstellt; eine Gruppe -CONRaRb, worin Ra und Rb die oben für Ra angegebenen Bedeutungen besitzen; eine Gruppe -NRaRb, worin Ra und Rb die oben für Ra angegebenen Bedeutungen besitzen; eine Gruppe -CONRcRd oder -NRcRd, worin Rc und Rd zusammen mit dem Stickstoffatom, an das sie gebunden sind, einen Heterocyclus mit 5 bis 7 Kettengliedern bilden; oder eine Gruppe -NHCO-NRaRb, worin Ra und Rb die oben für Ra angegebenen Bedeutungen besitzen; bedeuten;
- – R3 Wasserstoff bedeutet oder die oben für R1 und R2 angegebenen Bedeutungen besitzt;
- – oder R2 mit R3, wenn dieser Rest den Phenylrest in der 5-Stellung substituiert, eine Gruppe -X-CH2-X- bildet, worin X unabhängig voneinander eine CH2-Gruppe, ein Sauerstoffatom oder ein Schwefelatom bedeutet;
- – R6 eine (C1-C6)-Alkylgruppe; eine (C1-C6)-Alkoxy-(C1-C3)-alkylgruppe; eine (C3-C5)-Cycloalkylgruppe; eine (C3-C6)-Cycloalkyl-(C1-C6)-alkylgruppe; eine (C1-C6)-Alkylthio-(C1-C3)-alkylgruppe; eine (C1-C6)-Alkylsulfoxy-(C1-C3)-alkylgruppe; oder eine (C1-C6)-Alkylsulfodioxy-(C1-C3)-alkylgruppe bedeutet;
- – R7 eine nichtsubstituierte Phenylgruppe bedeutet oder eine Phenylgruppe, die in den Stellungen 3, 4 oder 5 einfach, zweifach oder dreifach substituiert ist durch ein Halogen, eine (C1-C5)-Alkylgruppe, eine Gruppe -O-CH2-O- an zwei benachbarten Kohlenstoffatomen des Phenylrests, durch eine Gruppe -CF3, -NO2, -CN, durch eine Gruppe -COOR8, -CONR8R9 oder durch eine Gruppe -CH2OR8, worin R8 und R9 eine (C1-C3)-Alkylgruppe bedeuten, OR10, worin R10 eine (C1-C5)-Alkylgruppe darstellt; oder R7 eine Pyridyl-, Thiophen-, Pyrazolyl-, Imidazolyl-, (C3-C5)-Cycloalkyl- oder (C3-C6)-Cycloalkyl-(C1-C6)-alkylgruppe bedeutet;
- In der vorliegenden Beschreibung sind die Alkylgruppen oder Alkoxygruppen geradkettig oder verzweigt.
- Unter einem Halogenatom versteht man ein Fluor-, Chlor-, Brom- oder Iodatom.
- Die für R7 definierten Heterocyclen können durch die gleichen Substituenten substituiert sein wie der Phenylrest.
- Gemäß einer weiteren Ausführungsform betrifft die Erfindung die Verbindungen der Formel (I.2) in racemischer oder enantiomerenreiner Form, in der:
- – R1 und R2, die gleichartig oder verschieden sind, jeweils unabhängig voneinander ein Halogenatom; eine (C1-C5)-Alkylgruppe; oder eine (C1-C5)-Alkoxygruppe bedeuten;
- – R3 Wasserstoff bedeutet oder die oben für R1 und R2 angegebenen Bedeutungen besitzt;
- – R6 eine (C1-C6)-Alkylgruppe; eine (C1-C6)-Alkoxy-(C1-C3)-alkylgruppe; eine (C3-C5)-Cycloalkylgruppe; oder eine (C3-C6)-Cycloalkyl-(C1-C6)-alkylgruppe bedeu tet;
- – R7 eine nichtsubstituierte Phenylgruppe oder eine Phenylgruppe, die in der 3- oder 4-Stellung mono- oder disubstituiert ist durch ein Halogen, eine (C1-C5)-Alkylgruppe, eine Gruppe -CH2OR8, in der R8 eine (C1-C3)-Alkylgruppe bedeutet, oder durch eine Gruppe -O-CH2-O- in der Stellung 3, 4; oder R7 eine (C3-C5)-Cycloalkylgruppe bedeutet, deren Additionssalze, deren Hydrate und/oder deren Solvate.
- Die Erfindung betrifft weiterhin die Verbindungen der Formel (I.2) in racemischer oder enantiomerenreiner Form, bei denen R3 in der 5-Stellung des Phenylrests steht, sowie deren Additionssalze, deren Hydrate und/oder deren Solvate.
- Gemäß einer weiteren Ausführungsform betrifft die Erfindung die folgenden Verbindungen:
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1R)-(1-(3-fluor-4-methylphenyl)-2-methoxyethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 31)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(1-phenylbutyl)-prop-2-inylamin-Hydrochlorid (BEISPIEL 33)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-phenylethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 34)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-phenylethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 35)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(4-fluorhenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 36)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(1-phenylpentyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 37)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1R)-(2-methoxy-1-(4-methoxymethylphenyl)-ethyl]-prop-2-inylamin-Hydrochlorid (BEISPIEL 40)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(1-(4-methoxymethylphenyl)-pentyl]-prop-2-inylamin-Hydrochlorid (BEISPIEL 42)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(1-(4-fluorphenyl)-pentyl]-prop-2-inylamin-Hydrochlorid (BEISPIEL 45)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(cyclopropylphenyl-methyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 47)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(1-(3-fluor-4-methylphenyl)-pentyl]-prop-2-inylamin-Hydrochlorid (BEISPIEL 49)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(3-fluor-4-methylphenyl)-ethyl]-prop-2-inylamin-Hydrochlorid (BEISPIEL 50)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(1-(4-fluorphenyl)-butyl]-prop-2-inylamin-Hydrochlorid (BEISPIEL 51)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(1-(3-fluor-4-methoxymethylphenyl)-butyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 52)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(4-chlorphenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 53)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(4-methylphenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 54)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclobutyl-1-(4-fluorphenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 55)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(4-bromphenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 56)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(3,4-methylendioxyphenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 57)
- – [4-(2-Chlor-4-methoxyphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(3-fluor-4-methylphenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 58)
- – [4-(2,4-Dimethoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(3-fluor-4-methylphenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 59)
- – [4-(4-Methoxy-2,5-dimethylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(3-fluor-4-methylphenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 60)
- – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(1-(3,4-methylendioxyphenyl)-butyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 61)
- Die erfindungsgemäßen Verbindungen besitzen in freier Form im allgemeinen die Wirkungen einer schwachen Base. In Abhängigkeit von der Art bestimmter Substituenten können sie jedoch auch saure Eigenschaften besitzen.
- Die Salze der Verbindungen der Formel (I) mit pharmazeutisch annehmbaren Säuren oder Basen (wenn dies möglich ist) sind die bevorzugten Salze, jedoch sind auch jene, die es ermöglichen, die Verbindungen der Formel (I) zu isolieren, und insbesondere sie zu reinigen und die reinen Isomeren zu erhalten, ebenfalls Gegenstand der Erfindung.
- Als pharmazeutisch annehmbare Säuren für die Herstellung von Additionssalzen der Verbindungen der Formel (I) kann man nennen: Chlorwasserstoffsäure, Bromwasserstoffsäure, Phosphorsäure, Fumarsäure, Citronensäure, Oxalsäure, Schwefelsäure, Ascorbinsäure, Weinsäure, Maleinsäure, Mandelsäure, Methansulfonsäure, Lactobionsäure, Gluconsäure, Glucarsäure, Bernsteinsäure, Sulfonsäure und Hydroxypropansulfonsäure.
- Als pharmazeutisch annehmbare Basen für die Herstellung von Additionssalzen der Verbindungen der Formel (I), wenn diese saure Wirkungen besitzen, kann man Natriumhydroxid, Kaliumhydroxid oder Ammoniumhydroxid nennen.
- Die erfindungsgemäßen Verbindungen sowie die Zwischenprodukte für ihre Herstellung werden mit Hilfe von dem Fachmann bekannten Methoden hergestellt, insbesondere gemäß
EP 576 350 EP 659 747 - Das nachfolgende Reaktionsschema verdeutlicht ein Herstellungsverfahrer. für die Synthese der Verbindungen (I), bei denen die Gruppe R5 eine Propargylgruppe ist. SCHEMA 1
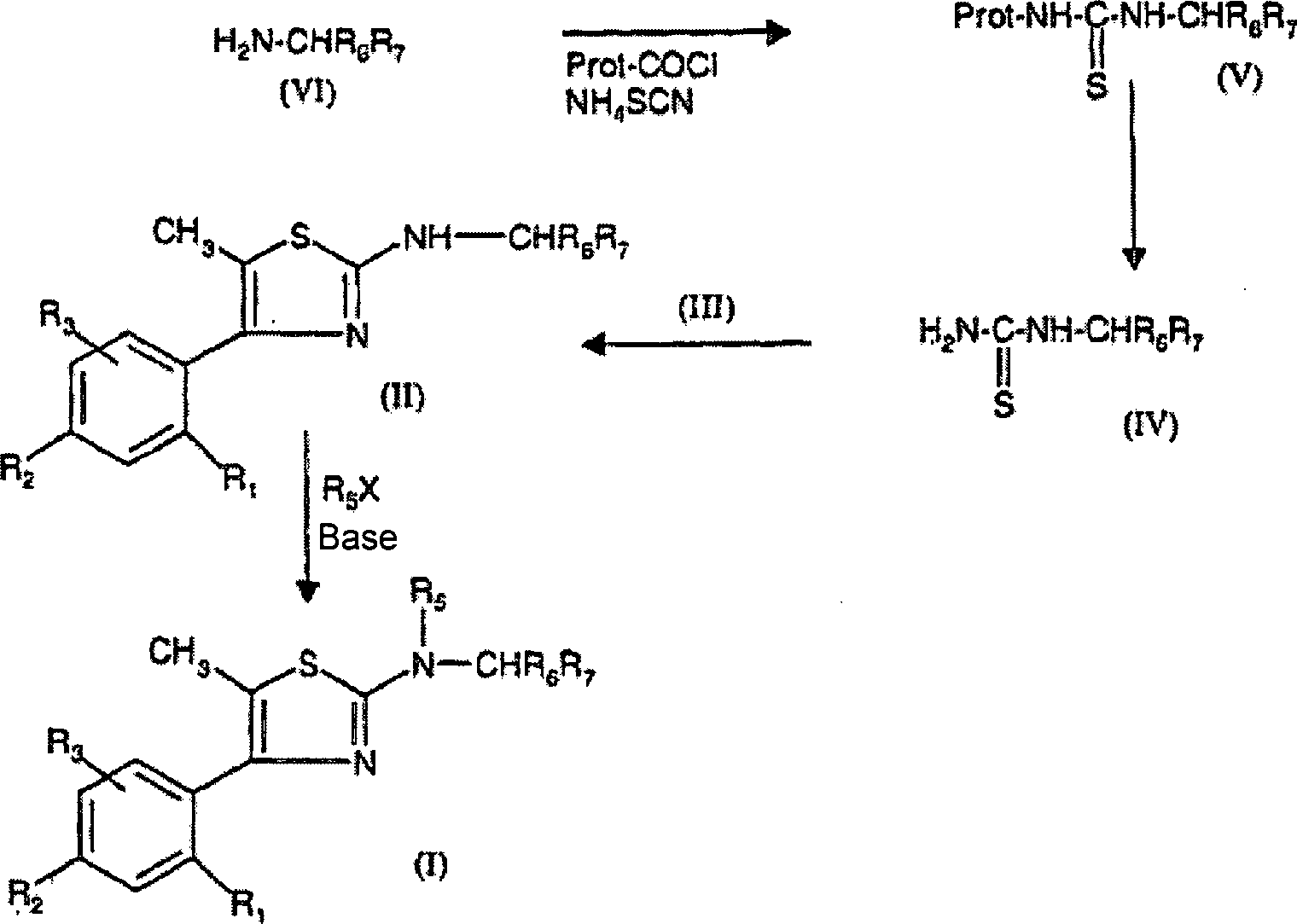
- Gemäß einer weiteren Ausführungsform betrifft die Erfindung ein Verfahren zur Herstellung der Verbindungen der Formel (I), welches dadurch gekennzeichnet ist, daß man ein alpha-Halogenderivat, vorzugsweise ein alpha-Brom- oder alpha-Chlorderivat der Formel (III)in der R1, R2 und R3 die bezüglich (I.2) angegebenen Bedeutungen besitzen und Hal ein Halogenatom, vorzugsweise ein Brom- oder Chloratom darstellt, mit einem Thioharnstoff der Formel:
 in der R6 und R7 die bezüglich (I) angegebenen Bedeutungen besitzen, umsetzt zur Bildung einer Verbindung der Formel (II)
in der R6 und R7 die bezüglich (I) angegebenen Bedeutungen besitzen, umsetzt zur Bildung einer Verbindung der Formel (II) in der R1, R2, R3, R6 und R7 die bezüglich (I) angegebenen Bedeutungen besitzen, welche man anschließend einer Alkylierungsreaktion unterwirft zur Bildung der Verbindung (I).
in der R1, R2, R3, R6 und R7 die bezüglich (I) angegebenen Bedeutungen besitzen, welche man anschließend einer Alkylierungsreaktion unterwirft zur Bildung der Verbindung (I).
- Die bei dem oben beschriebenen Verfahren angewandten Alkylierungsreaktionen werden unter den üblichen, dem Fachmann bekannten Bedingungen durchgeführt durch Einwirkung eines geeigneten Alkylierungsmittels, wie beispielsweise einem Alkenylhalogenids oder Alkinylhalogenids in Gegenwart einer Base, vorzugsweise von Natriumhydrid.
- Die Derivate der Formel (III) kann man ausgehend von den entsprechenden nichthalogenierten Ketonen der Formelin der R1, R2 und R3 die bezüglich (I) angegebenen Bedeutungen besitzen, entweder (i) durch Einwirkung von Brom in einem geeigneten organischen Lösungsmittel, wie Essigsäure, Tetrachlorkohlenstoff oder Diethylether oder (ii) durch Einwirkung von quaternären Ammoniumtribromiden gemäß der in Bull. Chem. Soc. Japan, 60 (1987), 1159–1160 und 2667–2668 beschriebenen Methode, oder (iii) durch Einwirkung von Kupfer(II)-bromid in einem organischen Lösungsmittel, wie einer Mischung aus Chloroform und Ethylacetat gemäß J. Org. Chem, 29 (1964), 3451–3461 erhalten. Gemäß einer Variante können die Verbindungen der Formel (III) erhalten werden durch Einwirkung von 2-Brompropionylbromid auf ein sub stituiertes Benzol der Formel:
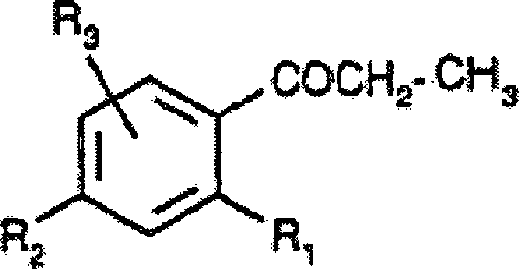 in der R1, R2 und R3 die bezüglich (I) angegebenen Bedeutungen besitzen, gemäß einer Friedel-Crafts-Reaktion.
in der R1, R2 und R3 die bezüglich (I) angegebenen Bedeutungen besitzen, gemäß einer Friedel-Crafts-Reaktion.
- Die oben erwähnten Ketone sind im allgemeinen bekannte Produkte oder im Handel erhältlich. Diese Verbindungen können durch Friedel-Crafts-Reaktion in Gegenwart einer Lewis-Säure gemäß dem Fachmann gut bekannter Methoden hergestellt werden.
- Man erhält die Thioharnstoff-Derivate (IV) ausgehend von den geschützen Thioharnstoffen (V)in deren Prot eine Schutzgruppe bedeutet, beispielsweise eine Benzoyl- oder Pivaloylgruppe, und worin R6 und R7 die bezüglich (I) angegebenen Bedeutungen besitzen, entweder durch basische Behandlung, wobei man vorzugsweise Ammoniak, Natriumhydroxid oder Hydrazin bei einer Temperatur von Raumtemperatur bis zu r Rückflußtemperatur der Reaktionsmischung verwendet, oder durch Behandeln mit Säure unter Verwendung von vorzugsweise Chlorwasserstoffsäure.
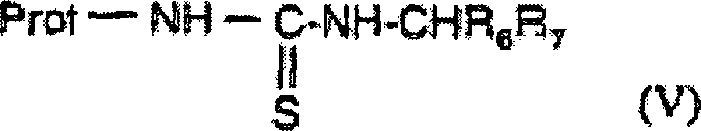
- Man bereitet die Verbindungen der Formel (V) durch Umsetzen eines Isothiocyanats, beispielsweise eines Benzylisothiocyanats oder eines Pivaloylisothiocyanats, mit entsprechenden Aminen der Formel (VI):in der R6 und R7 die bezüglich (I) angegebenen Bedeutungen besitzen, unter Anwendung bekannter Methoden.
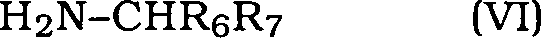
- Die Herstellung der optisch aktiven Aminothiazole, das heißt, in Form der reinen Enantiomeren, erfolgt ausgehend von optisch aktiven primären Aminen gemäß dem SCHEMA 2 mit Hilfe eines Verfahrens, das identisch dem oben beschriebenen ist. SCHEMA 2

- Die obigen Verbindungen der Formel (I) umfassen auch jene, bei denen eines oder mehrere Wasserstoff- oder Kohlenstoffatome durch ihr radioaktives Isotop, beispielsweise Tritium oder Kohlenstoff-14, ersetzt sind. Solche markierten Verbindungen sind nützlich bei Arbeiten in der Forschung, bezüglich des Stoffwechsels oder der Pharmakokinetik oder auch bei biochemischen Untersuchungen als Rezeptor-Liganden.
- Die Verbindungen der vorliegenden Erfindung waren Gegenstand biochemischer und pharmakologischer Untersuchungen. Sie besitzen sehr interessante pharmakologische Wirkungen.
- Die erfindungsgemäßen Verbindungen verdrängen bei der von E.B. De Souza (J. Neurosci., 7, 1 (1987), 88–100) beschriebenen Methode in Konzentrationen von weniger als 10 μM die Bindung von CRF oder von verwandten iodierten Peptiden (Urotensin, Sauvagin), beispielsweise 125I-Tyrosin-CRF, von den Rezeptoren, die auf Membranen des Gehirns oder auf Kulturzellen vorliegen.
- Die antagonistische Wirkung der erfindungsgemäßen Verbindungen wurde durch ihre Fähigkeit, bestimmte mit CRF verknüpfte Wirkungen zu inhibieren , nachgewiesen. Insbesondere sind die Verbindungen der Formel (I) dazu in der Lage, die durch CRF induzierte Adrenocorticotropin-Sekretion (ACTH) zu inhibieren. Die Untersuchung an der durch CRF induzierten ACTH-Sekretion er folgte in vivo an wachen Ratten nach einer von C. Rivier et al. (Endocrinology, 110(1), (1982), 272–278) angepaßten Methode.
- CRF ist ein Neuropeptid, welches die Aktivität der Hypothalamo-Hypophyso Nebennieren-Achse steuert. Dieser Faktor ist verantwortlich für endokrine und verhaltensmäßige Antworten, die mit Stress verknüpft sind.
- In der Tat wurde gezeigt, daß CRF das Verhalten sowie bestimmte Funktionen des autonomen Nervensystems modulieren kann (G.F. Koob, F.E. Bloom, Fed. Proc. 44, (1985), 259; M.R. Brown, L.A. Fisher, Fed. Proc., 44 (1985), 243). Insbesondere induziert CRF die Sekretion von Corticotropin (ACTH), von (3-Endorphinen und anderen von Pro-opiomelanocortin abgeleiteten Peptidderivaten (A. Tazi et al., Regul. Peptides, 18 (1987), 37; M.R. Brown et al., Regul. Peptides, 16 (1986), 321; C.L. Williams et al., Am. J. Physiol., G 582 (1987), 253).
- Die erfindungsgemäßen Verbindungen können daher nützlich sein bei der Steuerung der Sekretion dieser endogenen Substanzen. Sie finden insbesondere Anwendung als Wirkstoffe in Arzneimitteln zur Verringerung der Antwort auf Stress (Verhalten, Gemütszustände, Magen-Darm-Störungen und kardiovaskuläre Störungen, Störungen des Immunsystems) und allgemeiner bei pathologischen Zuständen, bei denen CRF eine Rolle spielt, beispielsweise psychiatrischen Störungen, der Angst, Depressionen, nervöser Anorexie, Epilepsie, Störungen der Sexualaktivität und der Fertilität, der Alzheimerschen Krankheit oder dergleichen.
- Die erfindungsgemäßen Verbindungen sind sehr stabil und daher besonders gut geeignet als Wirkstoffe von Arzneimitteln.
- Die Erfindung erstreckt sich daher auch auf pharmazeutische Zubereitungen, die als Wirkstoff eine Verbindung der Formel (I) oder eines ihrer pharmazeutisch annehmbaren Salze gegebenenfalls in Kombination mit einem oder mehreren inerten und geeigneten Trägermaterialien enthalten.
- In jeder Einheitsdosierung ist der Wirkstoff der Formel (I) in Mengen enthalten, die auf die angestrebten täglichen Dosierungen eingestellt sind. Jede Dosiseinheit wird in geeigneter Weise an die Dosierung und die vorgesehene Verabreichungsform angepaßt, beispielsweise in Form von Tabletten, Gelkapseln oder dergleichen, Sachets, Ampullen, Sirupe oder dergleichen, Tropfen, transdermale oder transmucosale Pflaster, und zwar in der Weise, daß eine Einheitsdosis 0,5 bis 800 mg des Wirkstoffs, vorzugsweise 0,5 bis 200 mg enthält.
- Die erfindungsgemäßen Verbindungen können auch in Kombination mit einem anderen Wirkstoff, der für die angestrebte Therapie nützlich ist, verwendet werden, beispielsweise Anxiolytika, Antidepressiva oder anorexigene Mittel.
- Die Verbindungen der Formel (I) sind wenig toxisch; ihre Toxizität ist verträglich mit ihrer Anwendung als Arzneimittel bei der Behandlung der oben angesprochenen Störungen und Krankheiten.
- Die Verbindungen der Formel (I) können zur Behandlung der oben genannten Krankheiten zu pharmazeutischen Zubereitungen für die Verabreichung an Säuger, einschließlich Menschen, formuliert werden.
- Die in dieser Weise erhaltenen pharmazeutischen Zubereitungen liegen im Vorteil in unterschiedlichen Formen vor, beispielsweise injizierbaren oder trinkbaren Lösungen, Dragees, Tabletten oder Gelkapseln. Die pharmazeutischen Zubereitungen, die als Wirkstoff mindestens eine Verbindung der Formel (I) oder eines ihrer Salze enthalten, sind besonders nützlich für die vorbeugende oder heilende Behandlung von Erkrankungen, die mit dem Stress verknüpft sind, und insbesondere bei der Behandlung jeglicher pathologischer Zustände, bei denen CRF beteiligt ist, wie beispielsweise: die Cushing-Krankheit, neuropsychiatrische Störungen, wie Depressionen, Angst, Panik, krampfartige Obsessionsstörungen, Gemütstörungen, posttraumatischem Stress, Verhaltensstörungen, der Aggressivität, der Anorexie, der Bulimie, der Hyperglykämie, Frühgeburten, riskanten Schwangerschaften, verlangsamtem Wachstum, Schlafstörungen, der Epilepsie und Depressionen jeglicher Art; der Alzheimerschen Krankheit, der Parkinsonschen Krankheit; der Huntington-Chorea; der amyotrophischen Lateralsklerose; von Störungen der Gefäße, des Herzens und des Gehirns; von Störungen der sexuellen Aktivität und der Fertilität; der Immunodepression, der Immunosuppression, Entzündungszuständen, multiplen Infekten, rheumatoider Arthritis, Osteoarthritis, Uveitis, Psoriasis sowie Diabetes; Krebs; funktionelle Magen-Darm-Störungen und Entzündungszustände, die sich daraus ergeben (Reizdarm und entzündeter Darm, Diarrhoe); Störungen der Wahrnehmung von Schmerz, Fibromyalgien, die gegebenenfalls mit Schlafstörungen verknüpft sind, Müdigkeit, Migräne; Symptome, die mit der Abhängigkeit (Alkohol) und dem Entzug von Drogen verknüpft sind.
- Die Dosierung kann in Abhängigkeit von dem Alter, dem Gewicht und dem Gesundheitszustand des Patienten, der Art und der Schwere der Erkrankung sowie dem Verabreichungsweg variieren. Diese Dosierung umfaßt die Verabreichung von einer oder mehreren Dosen von etwa 0,5 mg bis 800 mg, vorzugsweise von etwa 0,5 bis 200 mg täglich.
- In den erfindungsgemäßen pharmazeutischen Zubereitungen für die Verabreichung auf oralem, sublingualem, subkutanem, intramuskulärem, intravenösem, transdermalem, transmucosalem, lokalem oder rektalem Wege kann der Wirkstoff in Mischung mit klassischen pharmazeutischen Trägermaterialien in Einheitsverabreichungsformen an Tiere und Menschen gegeben werden. Die geeigneten Einheitsverabreichungsformen umfassen Formen, die auf oralem Wege gegeben werden, wie Tabletten, Gelkapseln, Pulver, Granulate und oral zu nehmende Lösungen oder Suspensionen, sublinguale und bukkale Verabreichungsformen, Verabreichungsformen für die subkutane, intramuskuläre, intravenöse, intranasale oder intraokulare Verabreichung und Formen für die rektale Verabreichung.
- Wenn man eine feste Zubereitung in Form von Tabletten bereitet, vermischt man den Hauptwirkstoff mit einem pharmazeutischen Trägermaterial, wie Gelatine, Stärke, Lactose, Magnesiumstearat, Talkum, Gummi arabicum oder dergleichen. Man kann die Tabletten mit Saccharose oder anderen geeigneten Materialien umhüllen oder kann sie in der Weise behandeln, daß sie eine verlängerte oder verzögerte Wirkung entfalten und daß sie eine vorbestimmte Menge des Wirkstoffs in kontinuierlicher Weise freisetzen.
- Man erhält ein Präparat in Form von Gelkapseln durch Vermischen des Wirkstoffs mit einem Verdünnungsmittel und Einbringen der erhaltenen Mischung in weiche oder harte Gelkapseln.
- Ein Präparat in Form eines Sirups oder Elixiers kann den Wirkstoff zusammen mit einem vorzugsweise kalorienfreien Süßungsmittel, Methylparaben und Propylparaben als Antiseptikum sowie einem geeigneten Geschmacksbildner und Farbstoff enthalten.
- In Wasser dispergierbare Pulver oder Granulate können den Wirkstoff in Mischung mit Dispergiermitteln oder Netzmitteln oder Suspendiermitteln, wie Polyvinylpyrrolidon, und zusammen mit Süßungsmitteln oder Geschmacksmodifizierungsmitteln enthalten.
- Für die rektale Verabreichung greift man auf Suppositorien zurück, die mit bei der Rektaltemperatur schmelzenden Bindemitteln, wie beispielsweise Kakaobutter oder Polyethylenglykolen, hergestellt werden.
- Für die parenterale, intranasale oder intraokulare Verabreichung verwendet man wäßrige Suspensionen, isotonische Salzlösungen oder sterile und injizierbare Lösungen, welche pharmakologisch verträgliche Dispergiermittel und/oder Netzmittel enthalten, beispielsweise Propylenglykol oder Butylenglykol.
- Für die transmucosale Verabreichung kann der Wirkstoff in Gegenwart eines Promotors, wie ein Gallensalz, ein hydrophiles Polymer, wie beispielsweise Hydroxypropoylcellulose, Hydroxypropylmethylcellulose, Hydroxyethylcellulose, Ethylcellulose, Carboxymethylcellulose, Dextran, Polyvinylpyrrolidon, Pektine, Stärken, Gelatine, Casein, Acrylsäuren, Acrylester und deren Copolymere, Vinylpolymere oder -copolymere, Vinylalkohole, Alkoxypolymere, Polyethylenoxid-poly mere, Polyether oder Mischungen davon formuliert werden.
- Der Wirkstoff kann auch in Form von Mikrokapseln, gegebenenfalls zusammen mit einem oder mehreren Trägermaterialien oder Hilfsstoffen formuliert werden.
- Der Wirkstoff kann auch in Komplexform mit einem Cyclodextrin vorliegen, beispielsweise α-, β- oder γ-Cyclodextrin, 2-Hydroxypropyl-β-cyclodextrin oder Methyl-β-cyclodextrin.
- Die nachfolgenden BEISPIELE dienen der weiteren Erläuterung der Erfindung, ohne sie jedoch einzuschränken.
- In den HERSTELLUNGSBEISPIELEN sind Synthesemethoden für die verschiedenen Zwischenprodukte beschrieben, welche die Herstellung der erfindungsgemäßen Verbindungen ermöglichen. Diese Zwischenprodukte werden sämtlich mit Hilfe dem Fachmann gut bekannter Verfahrensweisen erhalten.
- Die Schmelzpunkte wurden nach der Mikro-Kofler-Technik gemessen und sind in Grad Celsius angegeben.
- Die protonenkernmagnetischen Resonanzspektren (1H-NMR) wurden, wenn nichts anderes angegeben ist, in CDCl3 bei 200 MHz oder 300 MHz aufgezeichnet. Die chemischen Verschiebungen sind in ppm und die Kupplungskonstanten in Hertz angegeben.
- Die Enantiomeren-Überschüsse (EÜ) sind mit Hilfe von Chromatogrammen ermittelt worden, die entweder durch HPLC-Chromatographie auf chiraler Phase oder durch superkritische chirale Flüssigchromatographie (SFC) erhalten worden sind.
- Die Drehwerte der optisch aktiven Produkte sind durch ihre [α]t°D-Werte angegeben (wobei die Konzentrationen c der analysierten Lösungen in Gramm pro 100 ml angegeben sind).
- Die nachfolgend verwendeten Abkürzungen sind die folgenden: s = Singulett; m = Multiplett; d = Doublett; t = Triplett; q = Quadruplett.
- Die erfindungsgemäßen Verbindungen besitzen eine der Theorie entsprechende Elementaranalyse.
- Die in den TABELLEN 3 und 5 beschriebenen erfindungsgemäßen Verbindungen besitzen ebenfalls NMR-Spektren, die mit ihrer Struktur im Einklang stehen.
- HERSTELLUNG DER α-BROMKETONE DER FORMEL (III)
- 2-Brom-1-(2-chlor-4-methoxy-5-methylphenyl)-propan-1-on
- Verbindung III.1
- Man rührt eine Lösung von 46 g (280 mMol) 4-Chlor-2-methoxy-toluol in 150 ml Dichlormethan bei 0°C mit 29,4 g (280 mMol) 2-Brompropionylbromid. Man gibt dann portionsweise 39,2 g (294 mMol) Aluminiumtrichlorid zu der Mischung. Man rührt und läßt die Temperatur nach und nach auf Raumtemperatur ansteigen. Nach dem Rühren während 4 Stunden gießt man die Reaktionsmischung langsam auf Eis. Man versetzt diese Mischung unter Rühren mit 50 ml 1 N Chlorwasserstoffsäure und 1 Liter Wasser und extrahiert dann mit 1,2 Liter tert.-Butylmethylether. Man wäscht die organische Phase mit Wasser, einer gesättigten wäßrigen Natriumhydrogencarbonatlösung, mit Wasser und dann mit einer gesättigten Natriumchloridlösung. Man trocknet über wasserfreiem Natriumsulfat und dampft zur Trockne ein. der rohe Rückstand wird durch Chromatographie über Kieselgel (Lösungsmittel: Cyclohexan/Ethylacetat 50/1) gereinigt. Man erhält 67 g der Verbindung III-1. Ausbeute = 82 %. 1H-NMR-Spektrum: 7,44 (s, Ar, 1H); 6,86 (s, Ar, 1H); 5,41 (q, J = 5,35 Hz, CH, 1H); 3,90 (s, OCH3, 3H); 2,23 (s, CH3, 3H); 1,91 (d, J = 5,35 Hz, CH3, 3H).
- In gleicher Weise synthetisiert man:
- 2-Brom-1-(2-chlor-4-methoxyphenyl)-propan-1-on Verbindung III.2
- 2-Brom-1-(2,4-dichlor-5-methylphenyl)-propan-1-on Verbindung III.3
- 2-Brom-1-(2,4-dimethoxy-5-methylphenyl)-propan-1-on Verbindung III.4
- 2-Brom-1-(4-methoxy-2,5-dimethylphenyl)-propan-1-on Verbindung III.5
- HERSTELLUNG DER RACEMISCHEN AMINE DER FORMEL (VI)
- Erste Methode
- a) 2-Amino-2-(4-fluorphenyl)-ethanol Verbindung 1.1
- Man rührt 60 ml (60 mMol) einer 1 M Lösung von Lithiumaluminiumhydrid in Tetrahydrofuran am Rückfluß und gibt portionsweise 5 g (29 mMol) 4-Fluor-DL-α-phenylglycin (FLUKA) zu. Nach dem Rühren während 6 Stunden am Rückfluß rührt man die Reaktionsmischung bei 0°C und gibt langsam 2,5 ml Wasser, 2,5 ml ener wäßrigen 15 %-igen Natriumhydroxidlösung und dann 7,5 ml Weisser zu. Man filtriert die erhaltene Suspension über Celit, engt das Filtrat ein und nimmt mit 300 ml Dichlormethan auf. Man wäscht die Lösung mit einer gesättigten Natriumchloridlösung, trocknet über wasserfreiem Natriumsulfat und dampft zur Trockne ein. Man erhält 3,3 g eines gelben öligen Produkts. Ausbeute = 73 %. MS (MH+ = 156) 1H-NMR-Spektrum: 7,23 – 7,33 (m, Ar, 2H); 6,95 – 7,07 (m, Ar, 2H); 4,08 (m, CH, 1H); 3,45 – 3,86 (m, CH2O, 2H); 2,03 (s, NH2 und OH, 3H).
- b) 1-(4-Fluorphenyl)-2-methoxyethylamin Verbindung VI.1
- Man suspendiert 0,94 g (23 mMol) Kaliumhydrid, welches man durch Waschen von 2,2 g einer öligen Suspension in Pentan erhalten hat, in 18 ml Tetrahydrofuran und rührt bei 10°C. Dann gibt man langsam eine Lösung von 3,3 g (21 mMol) der Verbindung 1.1 in 43 ml Tetrahydrofuran zu. Nach dem Rühren während 16 Stunden bei Raumtemperatur gibt man im Verlaufe von einer Stunde und dreißig Minuten eine Lösung von 1,3 ml (20,8 mMol) Iodmethan in 25 ml Tetrahydrofuran zu. Man rührt die Reaktionsmischung während drei Stunden bei Raumtemperatur und gießt dann auf 300 ml einer eisgekühlten wäßrigen Salzlösung. Man extrahiert die Mischung mit 500 ml tert.-Butylmethyloxid, wäscht die organische Phase mit Wasser und dann mit einer gesättigten Natriumchloridlösung, trocknet über wasserfreiem Natriumsulfat und dampft zur Trockne ein. Man erhält 3,2 g des öligen Amins. Ausbeute = 88 %. 1H-NMR-Spektrum: 7,24 – 7,38 (m, Ar, 2H); 6,93 – 7,05 (m, Ar, 2H); 4,16 (m, CH, 1H); 3 45 (dd, CH2, 1H); 3,36 (s, OCH3, 3H); 3,29 (d, CH2, 1H); 1,70 (s, NH2, 2H). In gleicher Weise erhält man 2-Methoxy-1-phenylethylamin Verbindung VI.2
- Zweite Methode
- a) Synthese der substituierten Phenylketone. Verbindungen 3
- Verfahrensweise A
- 1-(3-Fluor-4-methylphenyl)-2-methoxyethan-1-on Verbindung 3.1
- Zur Herstellung der Magnesiumverbindung rührt man 14 g (583 mMol, 1 Äq.) Magnesiumspäne in Gegenwart von zerstoßenem Glas über Nacht unter Argon. Man isoliert sie mit 400 ml Diethylether und gibt dann eine Spatelspitze Iod zu. Man gibt langsam 110 g (582 mMol) 4-Brom-2-fluortoluol in Lösung in 700 ml Diethylether in der Weise zu, daß sich ein schwaches Sieden am Rückfluß ergibt, wonach man die Reaktionsmischung während drei Stunden zum Sieden am Rückfluß erhitzt. Man gibt dann 39 ml Methoxyacetonitril (610 mMol, 1,1 Äq.) zu und rührt während zwei Stunden. Nach Beendigung der Reaktion gießt man die Reaktionsmischung auf 1, 5 kg Eis und gibt unter Rühren 300 ml konzentrierte Chlorwasserstoffsäure zu. Man extrahiert mit Diethylether, trocknet über Natriumsulfat und dampft ein. Man gewinnt 77 g der Verbindung 3.1, die man direkt ohne weitere Reinigung in der nächsten Stufe verwendet.
- In gleicher Weise erhält man die folgenden Verbindungen:
- 1-(4-Chlor-3-fluorphenyl)-2-methoxyethan-1-on Verbindung 3.2
- 1-(4-Chlorphenyl)-2-methoxyethan-1-on Verbindung 3.3
- 3-Cyclopropyl-1-(4-fluorphenyl)-propan-1-on Verbindung 3.4
- Verfahrensweise B
- 2-Methoxy-1-(4-methoxymethylphenyl)-ethan-1-on Verbindung 3.5
- Man rührt eine Lösung von 62 g (308 mMol) 1-Brom-4-methoxymethylphenyl in 600 ml Tetrahydrofuran bei –70°C und gibt langsam 200 ml (320 mMol) einer 1,6 M Lösung von Butyllithium zu. Man rührt die Reaktionsmischung während 30 Minuten bei –70°C und gibt dann langsam eine Lösung von 50 g (380 mMol) 2-N-Dimethoxy-N-methyl-acetamid zu. Man rührt die Reaktionsmischung, wobei man die Temperatur langsam auf Raumtemperatur ansteigen läßt. Nach dem Rühren während 4 Stunden kühlt man auf 0°C ab und gibt langsam eine wäßrige gesättigte Ammoniumchloridlösung zu. Man extrahiert die Mischung mit Ethylacetat und wäscht die organische Phase mit Wasser und dann mit einer gesättigten Natriumchloridlösung, trocknet über wasserfreiem Natriumsulfat und dampft zur Trockne ein. Man reinigt den erhaltenen Rückstand durch Chromatographie über Kieselgel (Lösungsmittel: Cyclohexan/Ethylacetat 9/1, dann 3/1) und erhält 32 g des Ketons. Ausbeute = 53 %. 1H-NMR-Spektrum:7,89 (d, J = 8,1 Hz, Ar, 2H); 7,40 (d, J = 8,1 Hz, Ar, 2H); 4,66 (s, OCH2, 2H); 4,48 (s, OCH2, 2H), 3,47 (s, CH3, 3H); 3,38 (s, CH3, 3H).
- b) Synthese der Oxime. Verbindungen 4
- 1-(3-Fluor-4-methylphenyl)-2-methoxyethan-1-on-oxim Verbindung 4.1
- Verfahrensweise A
- Man vermischt 33 g Hydroxylamin-Hydrochlorid (475 mMol, 1,6 Äq.) mit 30 ml Wasser und 100 ml Ethanol. Dann gibt man bei 0°C 54 g (296 mMol) der Vrbindung 3.1 in Verdünnung mit 30 ml Ethanol zu. Nach Beendigung der Zugabe gibt man 60 g Natriumhydroxidplätzchen (1,5 Mol, 5 Äq.), die man zuvor zerstoßen hat, zu, wobei man die Temperatur unterhalb 30°C hält. Man hält die Reaktionsmischung über Nacht bei Raumtemperatur und kühlt sie dann auf 0°C ab zur Neutralisation der Reaktionsmischung mit konzentrierter Chlorwasserstoffsäure (pH < 7). Anschließend extrahiert man mit Ethylacetat, wäscht die organische Phase mit Wasser und mit einer gesättigten Natriumchloridlösung. Man trocknet die organische Phase über Natriumsulfat, dampft ein und chromatographiert das in dieser Weise erhaltene Öl über Kieselgel, wobei man als Elutionsmittel eine Ethylacetat/Cyclohexan-Mischung (1/9, V/V) verwendet. Man erhält 26 g des Isomeren (Z) und 9 g des Isomeren (E), was einer Ausbeute von 45 % an (Z) und 16 % an (E) entspricht.
- Verfahrensweise B
- Man vermischt 47 g Hydroxylamin-Hydrochlorid (676 mMol, 1,6 Äq.) mit 275 ml Pyridin. Dann gibt man bei 0°C 77 g (423 mMol) der Verbindung 3.1 zu und läßt die Reaktionsmischung während fünf Stunden bei Raumtemperatur. Nach Beendigung der Reaktion verdampft man das Pyridin und extrahiert anschließend mit Dichlormethan. Man wäscht die organische Phase mit Wasser und dann mit einer gesättigten Natriumchloridlösung. Man trocknet die organische Phase über Natriumsulfat, dampft ein und chromatographiert das in dieser Weise erhaltene Öl über Kieselgel, wobei man mit einer Ethylacetat/Cyclohexan-Mischung (1/9, V/V) eluiert und erhält 42,5 g der Verbindung (Z) und 14 g der Ver bindung (E), was einer Ausbeute von 51 % (Z) und 17 % (E) entspricht. 1H-NMR-Spektrum der Verbindung Z: 11,59 (N-OH, s, 1H); 7,20 – 7,40 (Ar, m, 3H); 4,51 (-O-CH2-, s, 2H); 3,18 (OCH3, s, 3H); 2,20 (CH3-Ph, s, 3H). 1H-NMR-Spektrum der Verbindung E: 11,30 (N-OH, s, 1H); 7,20 – 7,50 (Ar, m, 3H); 4, 21 (-O-CH2-, s, 2H); 3,17 (OCH3, s, 3H); 2,22 (CH3-Ph, s, 3H).
- In gleicher Weise erhält man durch eine der beiden oben genannten Verfahrensweisen die nachfolgenden Produkte:
- 1-(4-Chlor-3-fluorphenyl)-2-methoxyethan-1-on-oxim Verbindung 4.2
- 1-(4-Chlorphenyl)-2-methoxyethan-1-on-oxim Verbindung 4.3
- 1-Phenylbutan-1-on-oxim Verbindung 4.4
- 1-(4-Methoxymethylphenyl)-2-methoxyethan-1-on-oxim Verbindung 4.5
- 1-(4-Methoxymethylphenyl)-butan-1-on-oxim Verbindung 4.6
- Dicyclobutylketon-oxim Verbindung 4.7
- 1-Phenyl-pentan-1-on-oxim Verbindung 4.8
- c) Synthese der Amine. Verbindungen VI
- 1-(3-Fluor-4-methylphenyl)-2-methoxyethylamin Verbindung VI.3 Man gibt zu 10 ml einer 1 M Lithiumaluminiumhydrid-Lösung in Tetrahydrofuran (10 mMol, 8 Äq.) langsam bei 0°C eine Lösung von 1 g der Verbindung 4.1 (5 mMol) in Lösung in 15 ml Tetrahydrofuran. Man läßt die Temperatur auf Raumtemperatur ansteigen und läßt die Mischung dann während zwei Stunden reagieren, wonach man während einer Stunde zum Sieden am Rückfluß erhitzt. Man kühlt die Reaktionsmischung auf Null Grad ab und gibt 10 ml Wasser zu. Man extrahiert die wäßrige Phase mit Diethylether und extrahiert die vereinigten organischen Phasen mit einer 2N Chlorwasserstoffsäurelösung.
- Man rührt die erhaltene wäßrige Phase bei 0°C und gibt eine 35 %-ige Natriumhydroxidlösung zu. Man extrahiert die erhaltene alkalische Lösung mit Dichlormethan, wäscht die organische Phase mit mit Natriumchlorid gesättigtem Wasser, trocknet über Natriumsulfat und dampft zur Trockne ein. Nach der Filtration über Siliciumdioxid unter Verwendung einer Dichlormethan/Methanol-Mischung (95/5, V/V als Elutionsmittel erhält man 0,6 g der Verbindung VI.3. Ausbeute = 65 %. 1H-NMR-Spektrum: 6,90 – 7,20 (Ar, m, 3H); 4,14 (-CH-N, dd, J = 4 und 8,5, 1H); 3,47 (-CH2-O, dd, J = 4 und 9, 1H); 3,37 (OCH3, s, 3H); 3,32 (-CH2-O, dd, 1H, J = 8,5 und 9); 2,24 (CH3-Ph, d, J = 1,8, 3H); 1,68 (-NH2, s, 2H).
- In gleicher Weise erhält man die folgenden Verbindungen:
- 1-(4-Chlor-3-fluorphenyl)-2-methoxyethylamin Verbindung VI.4
- Dicyclobutylmethylamin Verbindung VI.5
- Dritte Methode
- a) Synthese des O-Alkyl-oxims. Verbindungen 6
- Verfahrensweise A
- 1-Phenylbutan-1-on-O-methyloxim Verbindung 6.1
- Man gibt zu 66 g (0,40 Mol) 1-Phenylbutan-1-on-oxim (Verbindung 4.4) in einer Mischung aus 400 ml Dimethylformamid und Tetrahydrofuran (1:1) 18 g (0,45 Mol) 55 %-iges Natriumhydrid in Öl in kleinen Portionen bei 0°C im Verlaufe einer Stunde. Nach der Zugabe von 31 ml (0,5 Mol) Methyliodid verdickt sich die Reaktionsmischung nach und nach. Nach der Zugabe von 50 ml Ethanol und dann von Wasser extrahiert man die Reaktionsmischung mit 4 × 250 ml Ethylacetat. Man wäscht die organische Phase mit mit Natriumchlorid gesättigtem Wasser, trocknet über Natriumsulfat und dampft im Vakuum ein. Man erhält 75 g eines hellgelben Öls in Form einer Mischung der geometrischen Isomeren ((Z) 7 % und (E) 93 %). Ausbeute = 94 % (Z + E).
- Diese beiden Isomeren können durch Chromatographie über Siliciumdioxid unter Elution mit einer Cyclohexan/Ethylacetat-Mischung getrennt werden. 1H-NMR-Spektrum: 7,56 – 7,93 (m, Ar, 2H); 7,24 – 7,40 (m, Ar, 3H); 3,95 [s, OCH3, (E)]; 3,82 [s, OCH3, (Z)]; 2,71 [m, CH2 (E)]; 2,50 [m, CH2, (Z)]; 1,41 – 1,64 (m, CH2, 2H); 0,84 – 1,03 (m, CH3), 3H).
- In gleicher Weise erhält man die folgenden alkylierten Oxime:
- 1-Phenylpentan-1-on-O-methyloxim Verbindung 6.2
- 1-(4-Chlorphenyl)-2-methoxyethan-1-on-O-benzyloxim Verbindung 6.3
- 2-Methoxy-1-(4-methoxymethylphenyl)-ethan-1-on-O-methyloxim Verbindung 6.4
- 1-(4-Methoxymethylphenyl)-butan-1-on-O-methyloxim Verbindung 6.5
- Verfahrensweise B
- Cyclobutyl-(4-fluorphenyl)-keton-O-benzyloxim Verbindung 6.6
- Man rührt eine Lösung von 15 g (84 mMol) Cyclobutyl-(4-fluorphenyl)-keton in 80 ml Ethanol bei Raumtemperatur und gibt 20,2 g (126 mMol) O-Benzylhydroxylamin-Hydrochlorid zu. Anschließend versetzt man die Mischung portionsweise mit 8,4 g (210 mMol) Natriumhydroxid und rührt während 4 Stunden bei Raumtemperatur Man versetzt die Mischung mit Wasser und extrahiert mit Ethylacetat. Man wäscht die organische Phase bis zur Neutralität mit Wasser und dann mit einer gesättigten Natriumchloridlösung. Man trocknet über Natriumsulfat, dampft zur Trockne ein und erhält 28,4 g einer Mischung der Isomeren (E 58 %, Z 42 %). 1H-NMR-Spektrum (DMSO D6): 7,15 – 7,48 (m, Ar, 9H); 5,07 [s, OCH2, (E)]; 5,01 [s, OCH2 (Z)]; 3,68 – 3,82 [m, CH Cyclobutyl, (E)]; 3,36 – 3,52 [m, CH Cyclobutyl, (Z)]; 1,58 – 2,30 (m, CH2 Cyclobutyl, 6H).
- Man verwendet die gleiche Methode zur Herstellung der folgenden Verbindung:
- 3-Cyclopropyl-1-(4-fluorphenyl)-propan-1-on-O-benzyloxim Verbindung 6.7
- b) Synthese der Amine Verbindungen VI
- 1-Phenylbutylamin Verbindung VI.6
- Man gibt zu 85 ml einer 1 M Lösung von Lithiumaluminiumhydrid in Tetrahydrofuran unter Argon tropfenweise eine Lösung von 14,2 g (0,085 Mol) 1-Phenylbutan-1-on-O-methyloxim (Verbindung 6.1) in 85 ml Tetrahydrofuran. Nach Beendigung der Zugabe erhitzt man die Reaktionsmischung während einer Stunde und dreißig Minuten zum Sieden am Rückfluß. Nach dem Aufbewahren über Nacht bei Raumtemperatur gibt man nacheinander 3,5 ml H2O, dann 3,5 ml 15 %-iges Natriumhydroxid und dann 10,5 ml H2O zu. Man filtriert den Niederschlag ab, wäscht ihn mit Diethylether, wäscht das Tetrahydrofuran/Diethylether-Filtrat mit Wasser und extrahiert dreimal mit einer 1 N Chlorwasserstoffsäurelösung. Man vereinigt die wäßrigen sauren Phasen und stellt sie mit 35 %-igem Natriumhydroxid bei 0°C alkalisch. Nach der Extraktion mit Dichlormethan, dem Waschen mit Wasser, dem Trocknen über Natriumsulfat und dem Eindampfen im Vakuum erhält man 0,3 g eines Öls. Ausbeute = 73 %. 1H-NMR-Spektrum: 7,11 – 7,36 (m, Ar, 5H); 3,81 – 3,95 (m, CH, 1H); 1,73 (s, NH2, 2H); 1,60 – 1,70 (m, CH2, 2H); 1,15 – 1,36 (m, CH2, 2H); 0,95 – 0,98 (m, CH3, 3H).
- In geicher Weise erhält man die folgenden Amine:
- 1-Phenylpentylamin Verbindung VI.7
- 1-(4-Chlorphenyl)-2-methoxyethylamin Verbindung VI.8
- 2-Methoxy-1-(4-methoxymethylphenyl)-ethylamin Verbindung VI.9
- 1-(4-Methosymethylphenyl)-butylamin Verbindung VI.10
- Cyclobutyl-(4-fluorphenyl)-methylamin Verbindung VI.11
- 3-Cyclopropyl-1-(4-fluorphenyl)-propylamin Verbindung VI.12
- Vierte Methode
- 1-(4-Fluorphenyl)-pentylamin Verbindung VI.13
- Man gibt zu einer Lösung von 1,21 g (10 mMol) 4-Fluorbenzonitril in 10 ml Tetrahydrofuran bei 0°C unter Rühren tropfenweise 10 ml (10 mMol) einer 1M Lösung von Boran in Tetrahydrofuran. Man rührt die Reaktionsmischung während einer Stunde und dreißig Minuten bei Raumtemperatur und gibt sie dann langsam zu 18,8 ml einer 1,6 M Lösung von Butyllithium in Hexan, die man zuvor auf –78°C abgekühlt hat. Man rührt die Reaktionsmischung während zwei Stunden bei –78°C und hydrolysiert dann bei dieser Temperatur mit 10 ml 2N Chlorwasserstoffsäure. Man extrahiert die organische Phase mit 2N Chlorwasserstoffsäure und neutralisiert die erhaltene wäßrige Phase bei 0°C durch langsame Zugabe von 35 %-igem Natriumhydroxid und extrahiert dann mit Ethylacetat. Man wäscht die organische Phase mit Wasser und dann mit einer gesättigten Natriumchloridlösung, trocknet über wasserfreiem Natriumsulfat und dampft zur Trockne ein. Man erhält 0,95 g des öligen Amins. Ausbeute = 53 %. 1H-NMR-Spektrum: 7,21 – 7,31 (m, Ar, 2H); 6,93 – 7,05 (m, Ar, 2H); 4,13 (t, CH, 1H); 1,59 – 1,75 (m, CH2, 2H); 1,49 (s, NH2, 2H); 1,24 – 1,33 (m, CH2-CH2, 4H); 0,85 (1, CH3, 3H).
- In gleicher Weise erhält man:
- 1-(3-Fluor-4-methylphenyl)-pentylamin Verbindung VI.14
- Fünfte Methode
- 1.(4-Fluorphenyl)-butylamin Verbindung VI.15
- Man gibt zu einer Suspension von 2,4 g (100 mMol) Magnesium in 30 ml Diethylether einen Iodkristall und dann 17,4 g (100 mMol) 4-Bromfluorbenzol (verdünnt in 70 ml Diethylether) in der Weise, daß sich ein schwaches Sieden am Rückfluß ergibt. Man erhitzt die Reaktionsmischung während einer Stunde zum Sieden am Rückflug und kühlt dann auf Raumtemperatur ab und gibt 5,75 g (85 mMol) in 30 ml Diethylether verdünntes Butyronitril zu. Man erhitzt die Reaktionsmischung während zwei Stunden zum Sieden am Rückflug, kühlt ab und filtriert über Glaswolle. Man rührt das Filtrat bei Raumtemperatur und gibt langsam 100 ml (100 mMol) einer 1M Lithiumaluminiumhydridlösung in Tetrahydrofuran zu. Man rührt die Reaktionsmischung während achtzehn Stunden am Rückfluß, kühlt dann auf 0°C ab und gibt nacheinander 3,8 ml Wasser, 3,8 ml 15 %-iges Natriumhydroxid und dann 11,4 ml Wasser zu. Man filtriert die Mischung über Celit, dampft das Filtrat zur Trockne ein, filtriert den erhaltenen Rückstand über Siliciumdioxid, wobei man mit einer Dichlormethan/Methanol-Mischung (98/2, V/V) eluiert und erhält 6,3 g des öligen Produkts. Ausbeute = 37 %. 1H-NMR-Spektrum: 7,22 – 7,36 (m, Ar, 2H); 6,92 – 7,05 (m, Ar, 2H); 3,87 (t, CH, 1H); 1,45 – 1,65 (m, CH2, 2H); 1,12 – 1,40 (m, CH2, 2H); 0,88 (t, CH3, 3H).
- HERSTELLUNG DER RACEMISCHEN THIOHARNSTOFFE, Verbindungen IV
- N-[1.-(4-Fluorphenyl)-2-methosyethyl]-thioharnstoff Verbindung IV.1
- Man gibt zu einer Lösung von 4,5 g (58 mMol) Ammoniumisothiocyanat in 115 ml Aceton unter Rühren bei 0°C 6,5 ml (56,6 mMol) Benzoylchlorid. Nach dreißig Minuten gibt man langsam 8,6 g (56 mMol) der Verbindung VI.1 in Lösung in 100 ml Aceton zu. Man rührt die Reaktionsmischung während zwei Stunden bei Raumtemperatur und engt dann unter vermindertem Druck ein. Man nimmt die Suspension mit 200 ml tert.-Butylmethyloxid und 200 ml Wasser auf, wäscht die organische Phase mit Wasser und dann mit einer gesättigten Natriumchloridlösung , trocknet über wasserfreiem Natriumsulfat und dampft zur Trockne ein. Man löst den Verdampfungsrückstand in 180 ml Ethanol und versetzt die erhaltene Lösung mit 5,85 ml (116 mMol) Hydrazin-Monohydrat. Nach dem Rühren während sechszehn Stunden bei Raumtemperatur ist die Reaktion nicht vollständig. M an gibt daher 1, 7 ml Hydazin zu. Nach dem Rühren während vierundzwanzig Stunden bei Raumtemperatur dampft man die Reaktionsmischung ein, löst den Verdampfungsrückstand in 500 ml Ethylacetat und wäscht die organische Phase mit Wasser und dann mit einer gesättigten Natriumchloridlösung, trocknet über wasserfreiem Natriumsulfat und dampft zur Trockne ein. Man chromatographiert den Verdampfungsrückstand über einer mit Siliciumdioxid beschickten Säule (Elutionsmittel: Cyclohexan/Ethylacetat 1/1, V/V) und erhält 8,5 g (40 mMol) des festen weißen Produkts. Ausbeute = 69 %, F = 154°C. 1H-NMR-Spektrum (DMSO-D6): 8,10 (d, NH, 1H); 7,28 – 7,32 (m, Ar, 2H); 7,08-7,17 (m, Ar und NH2, 4H); 5,45 (m, CH, 1H); 3,54 – 3,62 (m, CH-CH2, 2H); 3,24 (s, OCH3, 3H).
- In gleicher Weise erhält man die in der nachfolgenden TABELLE 1 beschriebenen Thioharnstoffe:TABELLE 1 (Fortsetzung)
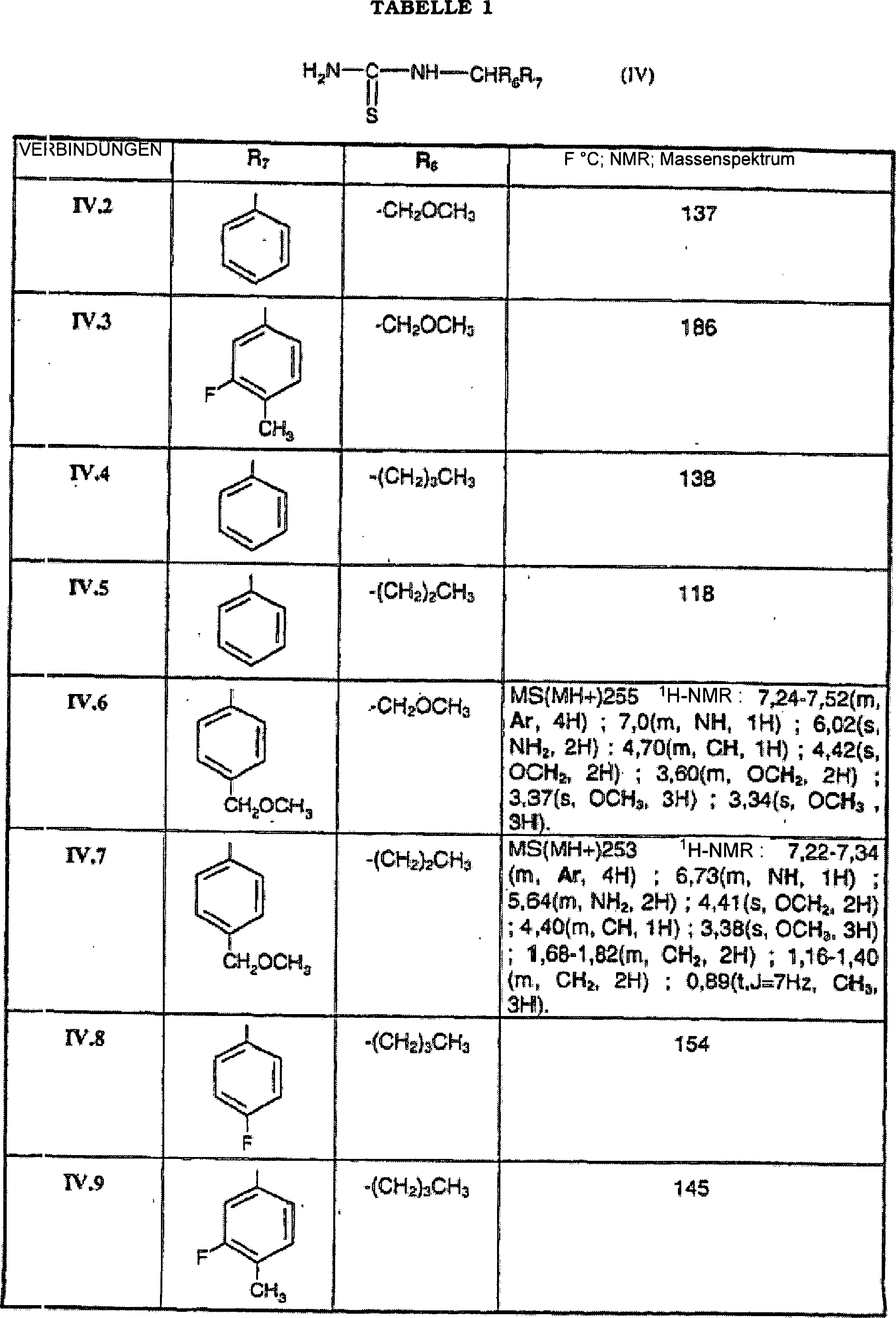
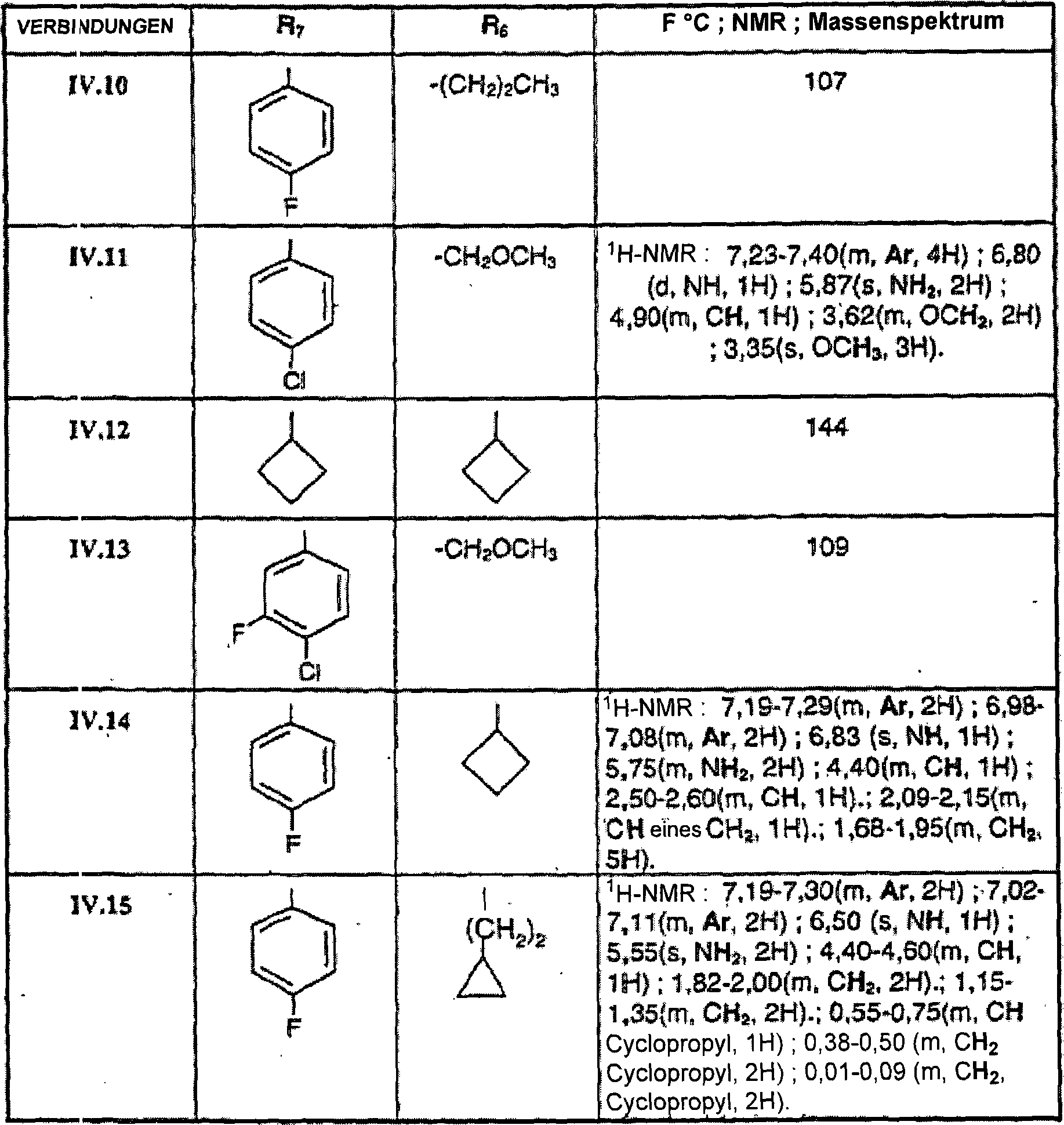
- HERSTELLUNG DER NH-THIAZOLE Verbindungen II
- [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[1-(4-methoxymethylphenyl)-butyl]-amin Verbindung II.1
- Man gibt zu 1,4 g (5,54 mMol) 1-(4-Methoxymethylphenyl)-butylthioharnstoff (Verbindung IV.7 in 60 ml Ethanol 1,92 g (6 mMol) 2-Brom-1-(2-chlor-4-methoxy-5-methylphenyl)-propan-1-on (Verbindung III.1) und 1,5 ml Triethylamin. Man rührt die Reaktionsmischung während drei Stunden bei 85°C und engt dann unter vermindertem Druck ein. Man nimmt den Rückstand mit 100 ml Dichlormethan und 50 ml Wasser auf. Man wäscht die organische Phase mit einer gesättigten wäßrigen Natriumchloridlösung, trocknet über Natriumsulfat und dampft im Vakuum zur Trockne ein. Man reinigt den rohen Extrakt durch Säulenchromatographie über Kieselgel unter Elution mit einer Cyclohexan/Ethylacetat-Mischung (9/1, V/V) und erhält 2,35 g des Aminothiazols. Ausbeute = 96 %. MS(MH+) = 445 7,26 – 7,36 (m, Ar, 4H); 7,10 (s, Ar, 1H); 6,83 (s, Ar, 1H); 5,44 – 5,47 (m, NH, 1H); 4,43 (s, OCH2, 2H); 4,17 – 4,33 (m, CH, 1H); 3,81 (s, OCH3, 3H); 3,39 (s, OCH3, 3H); 2,14 (s, CH3, 3H); 2,05 (s, CH3, 3H); 1,63 – 1,88 (m, CH2, 2H); 1,23 – 1,48 (m, CH2, 2H); 0,90 (t, CH3, 3H).
- In gleicher Weise bereitet man die in der nachfolgenden TABELLE 2 beschriebenen Produkte.
-

-
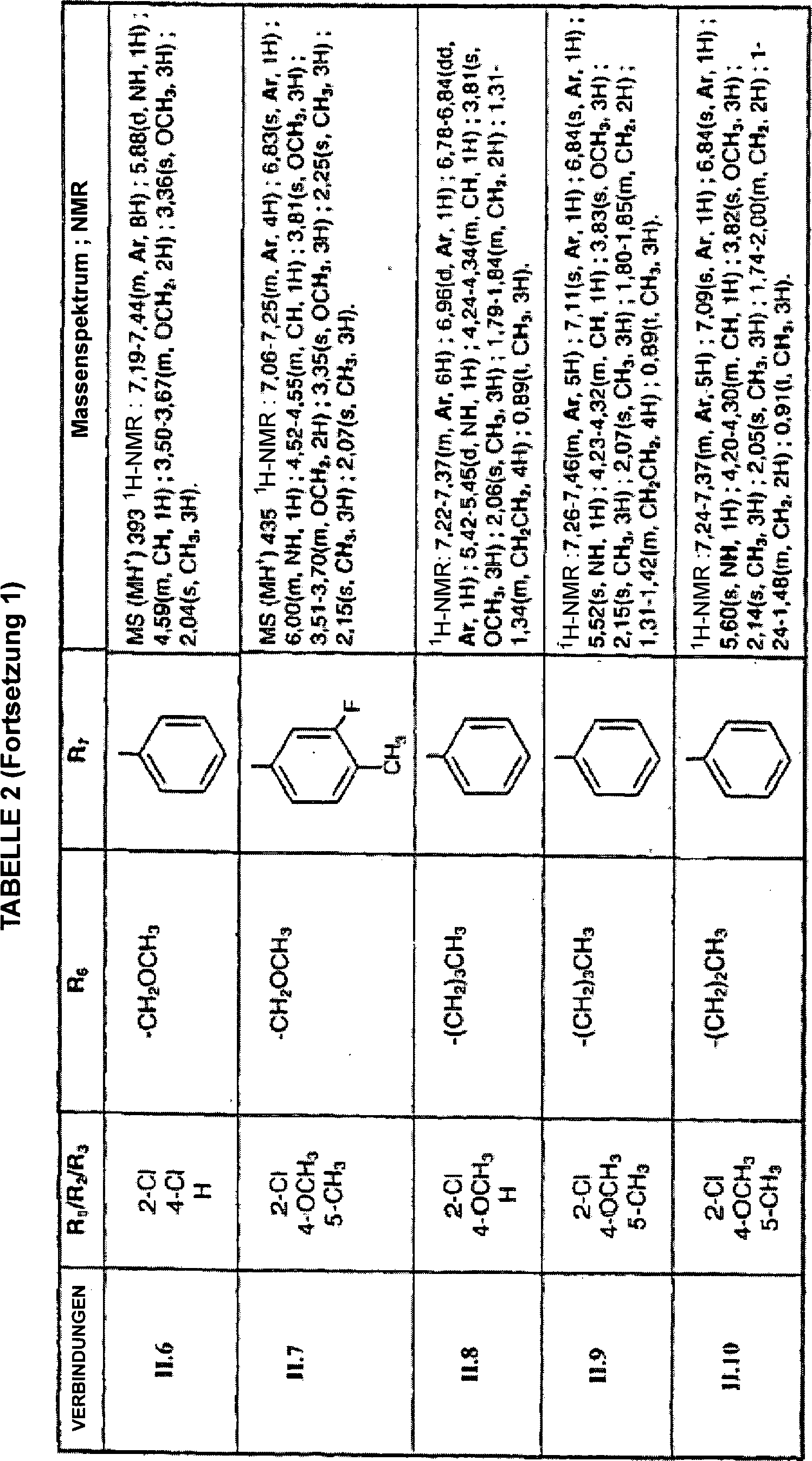
-
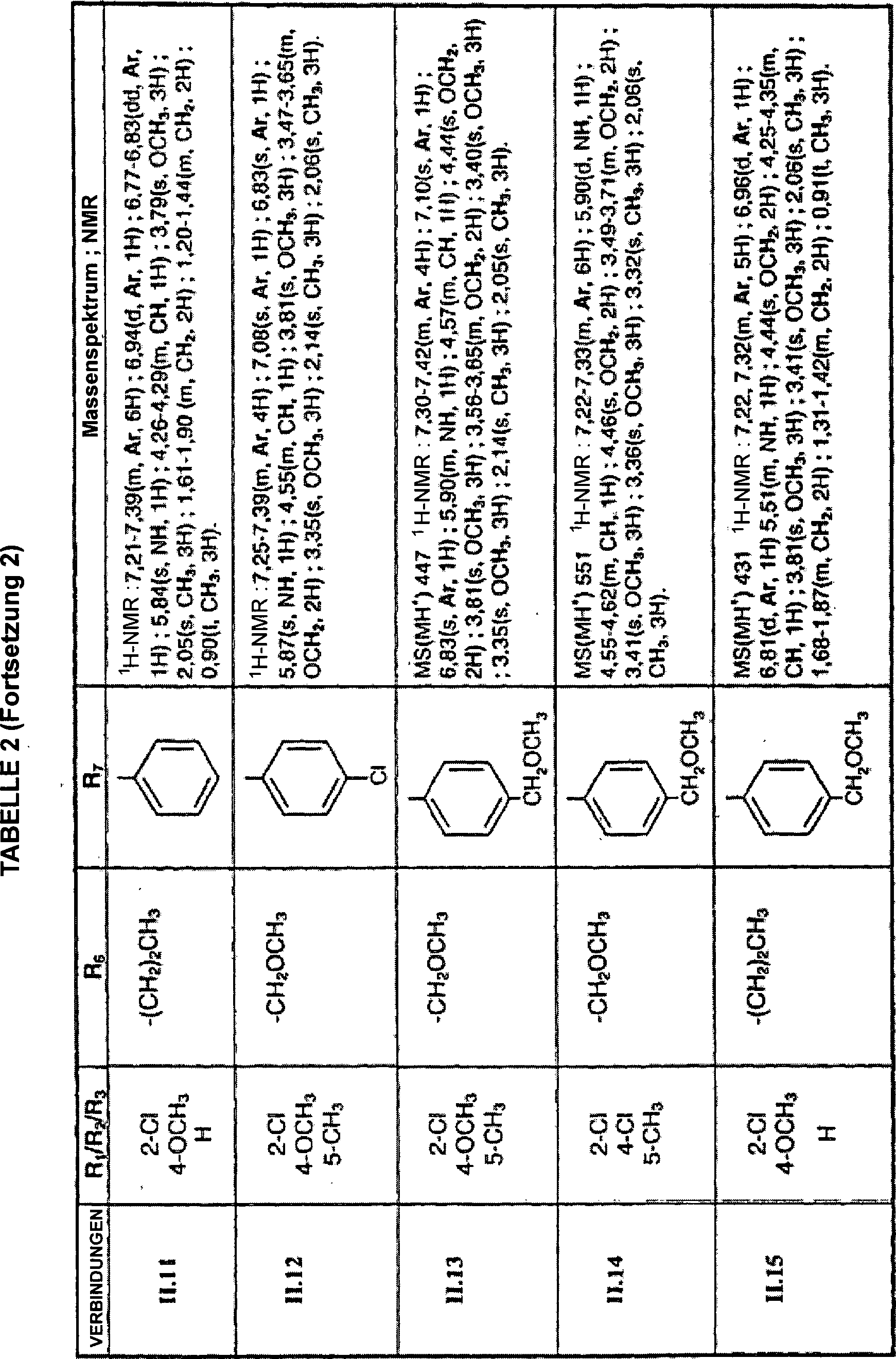
-
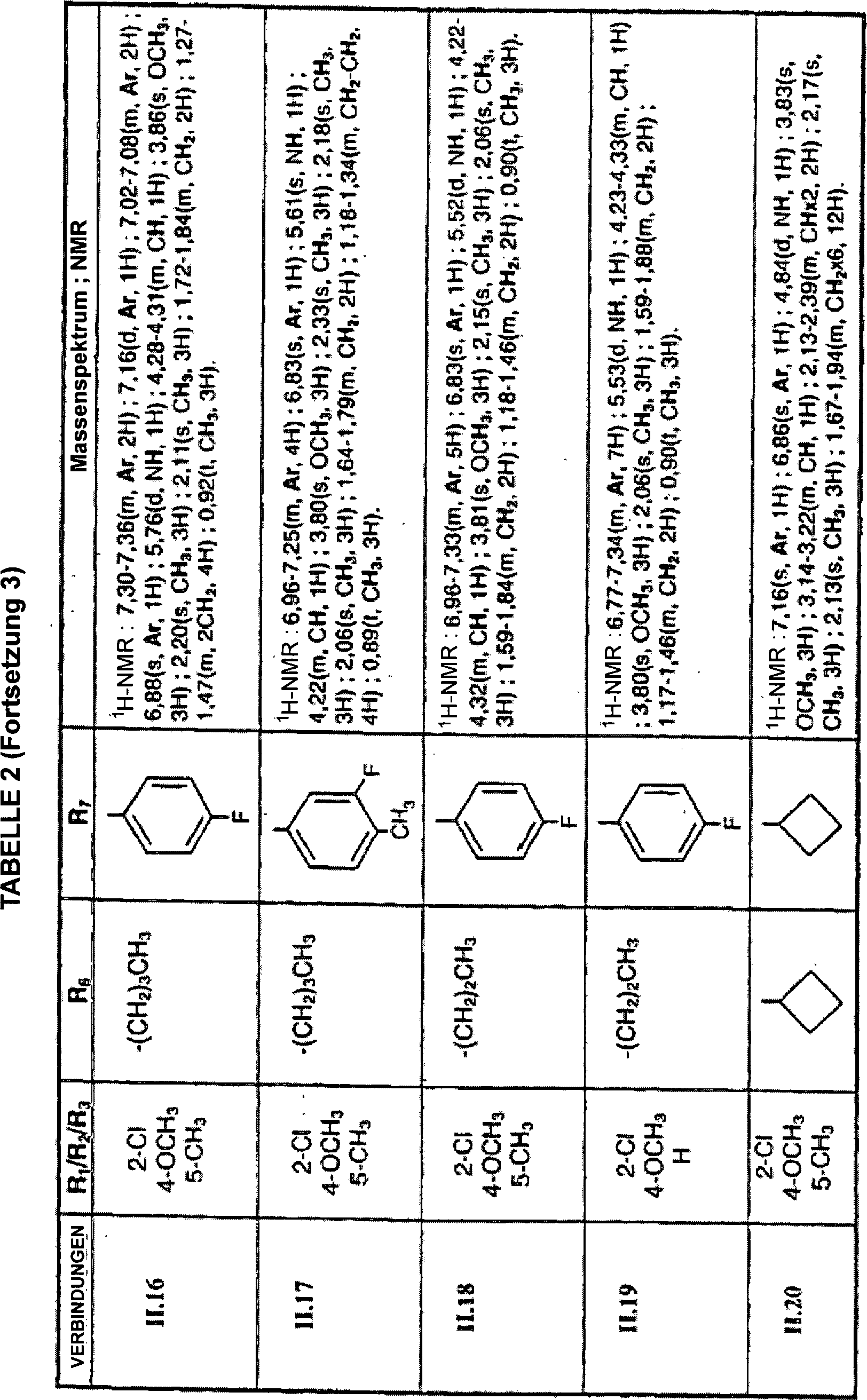
-
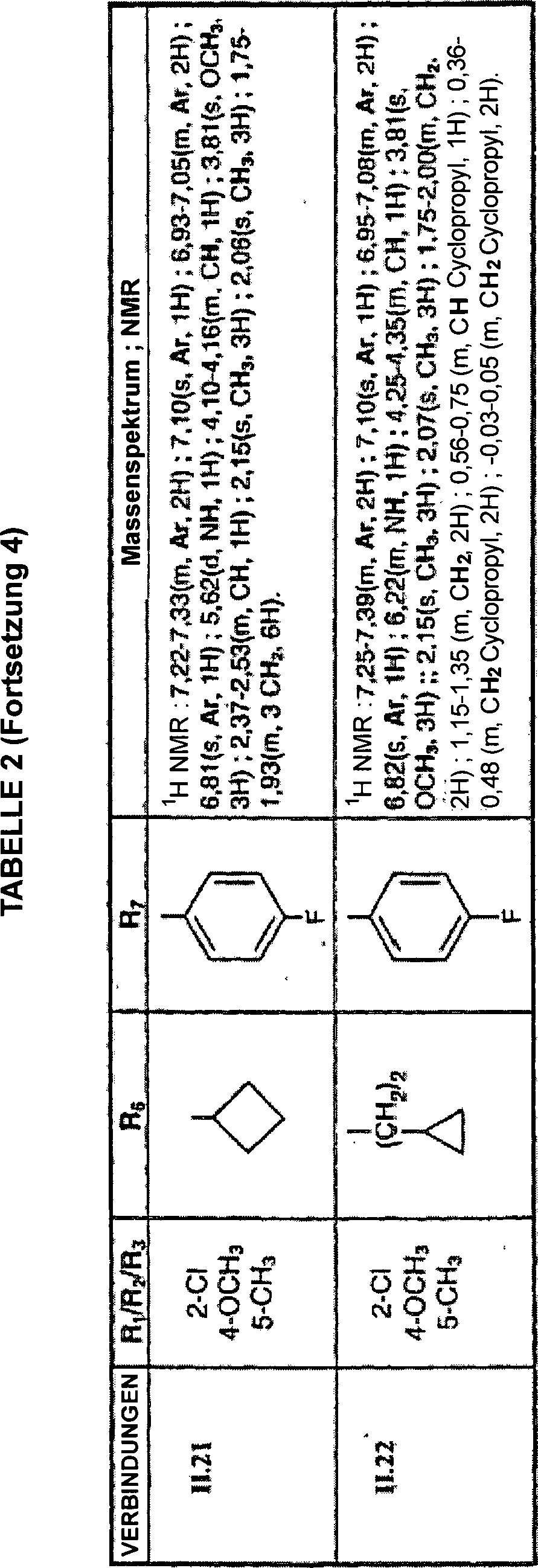
- HERSTELLUNG DER N-SUBSTITUIERTEN THIAZOLE Verbindungen 1
- BEISPIEL 1
- [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[1-(4-fluorphenyl)-2-methoxyethyl]-prop-2-inyl-amin Verbindung I.1
- Man gibt zu 500 mg (1,2 mMol) der Verbindung 77.2 in 6 ml wasserfreiem Dimethylformamid unter Rühren und bei 0°C 50 mg 60 %-iges Natriumhydrid in Öl. Man rührt die Reaktionsmischung während zwanzig Minuten bei 0°C und gibt dann 0,22 ml (2 mMol) einer 80 %-igen Lösung von Propargylbromid in Toluol zu. Man rührt die Reaktionsmischung während einer Stunde bei 10°C und gibt dann 0,5 ml Ethanol und dann 10 ml Wasser zu. Man extrahiert die Mischung zweimal mit 50 ml Ethylacetat, wäscht die organische Phase mit Wasser und dann mit einer gesättigten wäßrigen Natriumchloridlösung, trocknet über wasserfreiem Natriumsulfat und dampft zur Trockne ein. Man chromatographiert den rohen Rückstand über einer mit Kieselgel beschickten Säule [Elutionsmittel: Cyclohexan/Ethylacetat 9/1 (V/V)] und erhält 400 mg der erwarteten reinen Verbindung. Ausbeute = 73 %; Hydrochlorid, Hemihydrat: F = 94°C
- In gleicher Weise bereitet man die in der nachfolgenden TABELLE 3 beschriebenen Produkte.
 TABELLE 3 (Fortsetzung 2)
TABELLE 3 (Fortsetzung 2)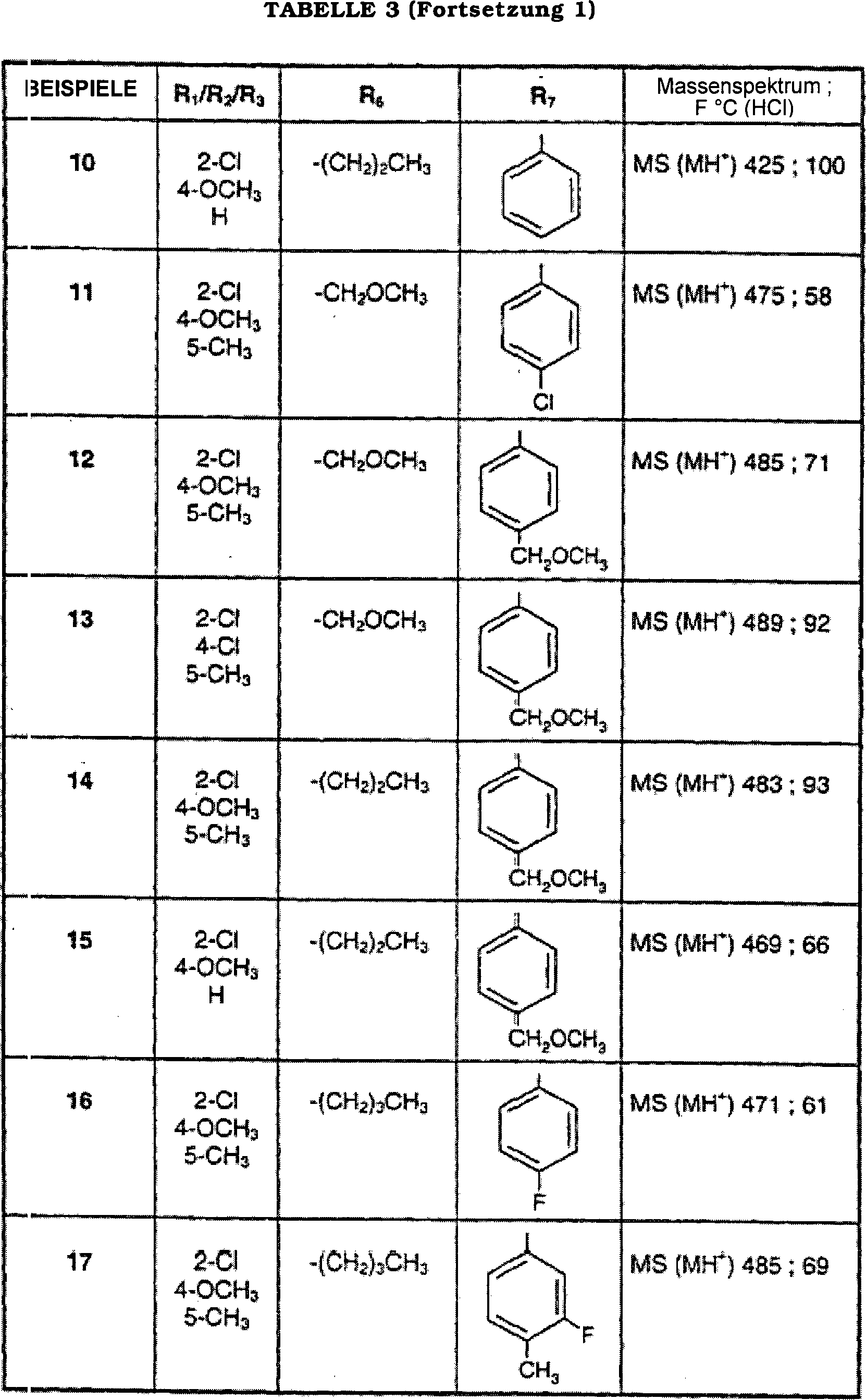

- HERSTELLUNG DER AMINE IN FORM EINES ENANTIOMEREN Verbindung VI'
- Erste Methode
- a) (R)-2-Amino-2-(4-fluorphenyl)-ethanol Verbindung 1'.1
- Man rührt 240 ml (240 mMol) einer 1 M Lösung von Lithiumaluminiumhydrid in Tetrahydrofuran am Rückfluß und gibt dann portionsweise 20 g (118 mMol) (R)-(4-Fluorphenyl)-glycin zu. Nach dem Rühren während sechs Stunden und dreißig Minuten am Rückfluß rührt man die Reaktionsmischung bei 0°C und gibt dann langsam 9,5 ml Wasser, 9,5 ml einer 15 %-igen Natriumhydroxidlösung und dann 28,5 ml Wasser zu. Man filtriert die erhaltene Suspension über Celit, engt das Filtrat ein und nimmt es mit 1 Liter Dichlormethan auf. Man wäscht die Lösung mit einer gesättigten Natriumchloridlösung, trocknet über wasserfreiem Natriumsulfat und dampft zur Trockne ein. Eine Kristallisation aus Isopropylether liefert 13,22 g (85,2 mMol) des kristallinen Produkts. Ausbeute = 72 %; F = 95°C ; MS (MH+) : 156. 1H-NMR-Spektrum (DMSO-D6): 7,30 – 7,41 (m, Ar, AH); 7,01 – 7,13 (m, Ar, 2H); 4,73 (s, OH, 1H); 3,84 (m, CH, 1H); 3,35 – 3,45 (m, CH2O, 2H); 1,82 (s, NH2, 2H).
- b) (R)-1-(4-Fluorphenyl)-2-methoxyethylamin Verbindung VI'.1
- Man suspendiert 3,64 g (91 mMol) Kaliumhydrid, welches man durch Waschen von 8,1 g einer öligen Suspension in Pentan erhalten hat, in 70 ml Tetrahydrofuran und rührt bei 10°C. Dann gibt man langsam eine Lösung von 13,22 g (85 mMol) der Verbindung 1'.1 in 175 ml Tetrahydrofuran zu. Nach dem Rühren während sechzehn Stunden bei Raumtemperatur gibt man im Verlaufe von zwei Stunden eine Lösung von 5,2 ml (83,5 mMol) Iodmethan in 105 ml Tetrahydrofuran zu. Man rührt die Reaktionsmischung während drei Stunden bei Raumtemperatur und gießt sie dann auf 1 Liter eisgekühlten salzhaltigen Wassers. Man extrahiert die Mischung mit 1 Liter tert.-Butyl-methyloxid, wäscht die organische Phase mit Wasser und dann mit einer gesättigten Natriumchloridlösung, trocknet über wasserfreiem Natriumsulfat und dampft zur Trockne ein. Man erhält 11,87 g (70 mMol) des öligen Amins. Ausbeute = 82 %. 1H-NMR-Spektrum: 7,24 – 7,38 (m, Ar, 2H); 6,93 – 7,05 (m, Ar, 2H); 4,16 (m, CH, 1H); 3,45 (dd, CH2, 1H); 3,36 (s, OCH3, 3H); 3,29 (d, CH2, 1H); 1,66 (s, NH2, 2H).
- In gleicher Weise erhält man ausgehend von (R)-Phenylglycin:
- (R)-2-Methoxy-1-phenylethylamin Verbindung VI'.2
- Zweite Methode
- a) (S)-2-Amino-3-methyl-1,1-diphenylbutan-1-ol Verbindung 2'.1
- Man rührt 600 ml einer 3,0 M Lösung von Phenylmagnesiumbromid (1790 mMol) in Diethylether bei 0°C, verdünnt mit 300 ml THF und gibt dann portionsweise 50 g (298 mMol) L-Valin-methylester-Hydrochlorid zu, wobei man die Temperatur unterhalb 10°C hält. Nach dem Rühren während 3 Stunden bei Raumtemperatur gießt man die Reaktionsmischung langsam auf eine eisgekühlte Lösung von Ammoniumchlorid. Man rührt die mit 500 ml Diethylether und 500 ml Ethylacetat versetzte Mischung über Nacht bei Raumtemperatur. Nach dem Dekantieren und dem Trennen der Phasen extrahiert man die wäßrige Phase erneut mit 1 Liter TBME (tert.-Butylmethylether), rührt die vereinigten organischen Phasen bei 0°C und säuert sie langsam mit etwa 40 ml 35 %-iger wäßriger Chlorwasserstoffsäure an. Der in dieser Weise gebildete Niederschlag des Hydrochlorids wird filtriert und mit TMBE gespült. Anschließend nimmt man ihn mit 1 Liter Dichlormethan und 1 Liter Wasser auf und stellt die Mischung bei 0°C mit etwa 50 ml 35 %-iger Natronlauge alkalisch. Nach dem Dekantieren und dem Trennen der Phasen extrahiert man die wäßrige Phase erneut mit 1 Liter Dichlormethan. Man wäscht die vereinigten organischen Phasen mit Wasser und dann mit Salzwasser, trocknet über Natriumsulfat und engt ein. Nach der Kristallisation aus Isopropylether erhält man 61 g der 'Verbindung 2'.1. (Ausbeute = 87 %). [αD]20D = –127,8° (CHCl3 c = 0,639) 1H-NMR-Spektrum: 7,00 – 7,60 (Ar, m, 10H); 5,24 (-OH, s, 1H); 3,66 (-CH-N, d, J = 1,5, 1H); 1,53 (-CH-, Hept d, J = 1,5 und 7, 1H); 1,16 (-NH2, s, 2H); 0,81 (-CH3, 2d, J = 7, 6H).
- In gleicher Weise erhält man ausgehend von D-Valin-methylester-Hydrochlorid:
- (R)-'2-Amino-3-methyl-1,1-diphenylbutan-1-ol Verbindung 2'.2
- Diese Verbindungen werden als chirale Hilfsstoffe bei der enantioselektiven Reduktion der O-Benzyl-oxime 6' verwendet.
- b) Synthese der substituierten Phenylketone Verbindungen 3'
- Verfahrensweise A
- 2-Cyclopropyl-1-(3-fluor-4-methylphenyl)-ethan-1-on Verbindung 3'.1
- Man versetzt 10,2 g (418 mMol) Magnesiumspäne mit 50 ml Diethylether und einem Iodkristall und rührt bei Raumtemperatur. Man gibt dann im Verlaufe von drei Stunden eine Lösung von 75,35 g (398 mMol) 4-Brom-2-fluortoluol in 370 ml Diethylether in der Weise zu, daß sich ein schwaches Sieden am Rückfluß ergibt. Anschließend erhitzt man die Reaktionsmischung während einer Stunde und dreißig Minuten zum Sieden am Rückfluß, kühlt ab und filtriert über Glaswolle. Man gibt die erhaltene Lösung langsam zu einer Lösung von 32,3 g (398 mMol) Cyclopropylacetonitril in 230 ml bei 0°C gerührtem Diethylether. Man rührt die Reaktionsmischung über Nacht bei Raumtemperatur und gibt dann unter Rühren bei 0°C langsam 200 ml 2N Chlorwasserstoffsäure zu. Nach dem Abtrennen der Etherphase extrahiert man die wäßrige saure Phase mit Ethylacetat, wäscht die vereinigten organischen Phasen mit Wasser und dann mit einer gesättigten Natriumchloridlösung, trocknet über wasserfreiem Natriumsulfat und dampft zur Trockne ein. Man reinigt den rohen Extrakt chromatographisch über Kieselgel (Elutionslösungsmittel Cyclohexan/Ethylacetat 20/1) und erhält 53,3 g des Ketons 3'.1 (Ausbeute = 70 %). 1H-NMR-Spektrum: 7,54 – 7,64 (m, 2H, Ar); 7,22 – 7,30 (m, 1H, Ar); 2,82 (d, J = 6,7 Hz, 2H, CH2); 2,31 (s, 3H, CH3); 1,07 – 1,20 (m, 1H, CH Cyclopropyl); 0,55-0,65 (m, 2H, CH2 Cyclopropyl); 0,15 – 0,21 (m, 2H, CH2 Cyclopropyl).
- Mit Hilfe der gleichen Verfahrensweise wurden die folgenden Ketone synthetisiert:
- 1-(4-Ethylphenyl)-2-methoxyethan-1-on Verbindung 3'.2
- 2-Cyclopropyl-l-(4-methylphenyl)-ethan-1-on Verbindung 3'.3
- 2-Cyclobutyl-1-(4-fluorphenyl)-ethan-1-on Verbindung 3'.4
- Verfahrensweise B
- Man wendet die für die Verbindung 3.5 beschriebene Verfahrensweise an (Reduktion einer Phenyl-lithium-Verbindung mit dem WEINREB-Amid):
- 2-Methoxy-1-(3,4-methylendioxyphenyl)-ethan-1-on Verbindung 3'.5
- 1-(4-Methoxymethylphenyl)-pentan-1-on Verbindung 3'.6
- 1-(3-Fluor-4-methylphenyl)-butan-1-on Verbindung 3'.7
- 1-(3-Fluor-4-methylphenyl)-pentan-1-on Verbindung 3'.8
- 1-(3-Fluor-4-methoxymethylphenyl)-butan-1-on Verbindung 3'.9
- 2-Cyclopropyl-1-(3,4-methylendioxyphenyl)-ethan-1-on Verbindung 3'.10
- 1-(3,4-Methylendioxyphenyl)-butan-1-on Verbindung 3'.11
- c) Synthese der O-Benzyl-oxime. Verbindungen 6'
- Man bereitet die O-Benzyl-oxime durch O-Benzylierung der entsprechenden Oxime nach der folgenden Verfahrensweise (man erhält die als Ausgangsmaterial eingesetzten Oxime ausgehend von den Ketonen durch eine der beiden oben beschriebenen Methoden der Synthese der Verbindung 4.1).
- 1-(3-Fluor-4-methylphenyl)-2-methoxyethan-1-on-O-benzyloxim Isomeres Z Verbindung 6'.1
- Man gibt zu einer bei 0°C gerührten Lösung von 42,5 g (217 mMol) 1-(3-Fluor-4-methyl-phenyl)-2-methoxyethan-1-on-oxim Z (Verbindung 4.1) in 100 ml Dimethylformamid in kleinen Portionen 15,6 g (325 mMol, 1,5 Äq.) 50 %-iges Natriumhydrid in Öl. Man rührt die Reaktionsmischung während fünfzehn Minuten und gibt dann langsam eine Lösung zu, die 30 ml (280 mMol, 1,3 Äq.) Benzylbromid in 100 ml Dimethylformamid enthält. Man rührt die Reaktionsmischung während zwei Stunden bei Raumtemperatur, kühlt dann auf 0°C ab und gibt 5 ml Ethanol und dann 50 ml Wasser zu. Man extrahiert mit Ethylacetat, wäscht die organische Phase mit Wasser und dann mit einer gesättigten Natriumchloridlösung, trocknet anschließend über Natriumsulfat und dampft zur Trockne ein. Man reinigt das in dieser Weise erhaltene Öl durch Chromatographie über Kieselgel (Elutionsmittel: Cyclohexan/Dichlormethan 7/3, V/V) und erhält 39 g der Verbindung 6'.1 (Z); Ausbeute = 63 %. 1H-NMR-Spektrum: 7,10 – 7,50 (Ar, m, 8H); 5,22 (-O-CH2-Ph, s, 2H); 4,58 (-CH2-O, s, 2H); 3,28 (OCH3, s, 3H); 2,26 (CH3-Ph, d, J = 1,8, 3H).
- In gleicher Weise bereitet man die folgenden Verbindungen:
- 1-(4-Chlor-3-fluor-phenyl)-2-methoxyethan-1-on-O-benzyloxim (Z) Verbindung 6'.2
- 1-(4-Chlorphenyl)-2-methoxyethan-1-on-O-benzyloxim (Z) Verbindung 6'.3
- 2-M'ethoxy-1-(3,4-methylendioxyphenyl)-ethan-1-on-O-benzyloxim (Z) Verbindung 6'.4
- 1-(4-Ethylphenyl)-2-methoxyethan-1-on-O-benzyloxim (Z) Verbindung 6'.5
- 2-Methoxy-1-(4-methoxymethylphenyl)-ethan-1-on-O-benzyloxim (Z) Verbindung 6'.6
- 1-Phenylbutan-1-on-O-benzyloxim (E) Verbindung 6'.7
- 1-(4-Methoxymethylphenyl)-butan-1-on-O-benzyloxim (E) Verbindung 6'.8
- 1-(4-Methoxymethylphenyl)-pentan-1-on-O-benzyloxim (E) Verbindung 6'.9
- 2-Cyclopropyl-1-phenylethan-1-on-O-benzyloxim (E) Verbindung 6'.10
- 2-Cyclopropyl-1-(4-fluorphenyl)-ethan-1-on-O-benzyloxim (E) Verbindung 6'.11
- 1-(4-Fluorphenyl)-pentan-1-on-O-benzyloxim (E) Verbindung 6'.12
- Cyclopropyl-phenyl-keton-O-benzyloxim (E) Verbindung 6'.13
- 1-(3-Fluor-4-methylphenyl)-butan-1-on-O-benzyloxim (E) Verbindung 6'.14
- 1-(3-Fluor-4-methylphenyl)-pentan-1-on-O-benzyloxim (E) Verbindung 6'.15
- 2-Cyclopropyl-1-(3-fluor-4-methylphenyl)-ethan-1-on-O-benzyloxim (E) Verbindung 6'.16
- 1-(4-Fluorphenyl)-butan-1-on-O-benzyloxim (E) Verbindung 6'.17
- 1-(3-Fluor-4-methoxymethylphenyl)-butan-1-on-O-benzyloxim (E) Verbindung 6'.18
- 2-Cyclopropyl-1-(4-chlorphenyl)-ethan-1-on-O-benzyloxim (E) Verbindung 6'.19
- 2-Cyclopropyl-1-(4-methylphenyl)-ethan-1-on-O-benzyloxim (E) Verbindung 6'.20
- 2-Cyclobutyl-1-(4-fluorphenyl)-ethan-1-on-O-benzyloxim (E) Verbindung 6'.21
- 2-Cyclopropyl-1-(4-bromphenyl)-ethan-1-on-O-benzyloxim (E) Verbindung 6'.22
- 2-Cyclopropyl-1-(3,4-methylendioxyphenyl)-ethan-1-on-O-benzyloxim (E) Verbindung 6'.23 1-(3,4-Methylendioxyphenyl)-butan-1-on-O-benzyloxim (E) Verbindung 6'.24
- d) Synthese der enantiomeren Amine
- (R)-1-(3-Fluor-4-methylphenyl)-2-methoxyethylamin Verbindung VI'.3
- Man rührt eine Lösung von 86,5 g der Verbindung 2'.1 (330 mMol) in 600 ml Tetrahydrofuran bei einer Temperatur von unterhalb 30°C und gibt langsam 670 ml einer 1 M Lösung von Boran in Tetrahydrofuran (670 mMol) zu. Man läßt die Temperatur im Verlaufe von zwei Stunden auf Raumtemperatur ansteigen. Dann rührt man das Reaktionsmedium bei 0°C und gibt 39 g (132 mMol) der zuvor in 100 ml Tetrahydrofuran gelösten Verbindung 6'.1 zu. Nach dem Rühren während zwanzig Stunden bei Raumtemperatur kühlt man die Reaktionsmischung; auf 0°C ab und gibt 1 Liter 1N Chlorwasserstoffsäure zu. Man rührt während sechszehn Stunden, stellt die Mischung bei 0°C durch Zugabe von 35 %-igem Natriumhydroxid alkalisch und extrahiert mit Ethylacetat. Man wäscht diesen Extrakt mit Wasser und mit Natriumchlorid gesättigtem Wasser, trocknet über Natriumsulfat und dampft zur Trockne ein. Man chromatographiert den erhaltenen Rückstand über einer mit Kieselgel beschickten Säule (Elutionsmittel: Dichlormethan/Methan 95/5, V/V) und erhält 17 g der Verbindung VI'.3; Ausbeute = 79 %. 1H-NMR-Spektrum: 6,90 – 7,20 (m, Ar, 3H; 4,14 (dd, J1 = 4 Hz, J2 = 8,5 Hz, CHN, 1H); 3,47 (dd, J1 = 4 Hz, J2 = 9 Hz, -CH2-O, 1H); 3,37 (s, OCH3, 3H); 3,32 (dd, J1 = 8,5 Hz, J2 = 9 Hz, -CH2-O, 1H); 2,24 (d, J = 1,8 Hz, CH3-Ph, 3H); 1,68 (s, -NH2, 2H). Chirale HPLC: %-Satz der Enantiomeren : R = 99,5 %, S = 0,5 %, EÜ = 99,0 %.
- Allgemeine Bemerkung Die Enantiomeren-Überschüsse (EÜ) wurden über die Chromatogramme (chirale HPLC oder SFC) dieser Amine oder der entsprechenden Thioharnstoffe IV' ermittelt.
- In gleicher Weise erhält man:
- (R)-1-(4-Chlor-3-fluorphenyl)-2-methoxyethylamin Verbindung VI'.4 EÜ = 98,2 %
- (R)-1-(4-Chlorphenyl)-2-methoxyethylamin Verbindung VI'.5 EÜ = 98,6 %
- (R)-2-Methosy-1-(3,4-methylendioayphenyl)-ethylamin Verbindung VI'.6 EÜ>99%
- (R)-1-(4-Ethylphenyl)-2-methoxyethylamin Verbindung VI'.7 EÜ > 99 %
- (R)-2-Methoxy-1-(4-methoxymethylphenyl)-ethylamin Verbindung VI'.8 EÜ>99%
- (S)-(1-Phenyl)-butylamin Verbindung VI'.9 EÜ = 97,1 %
- (S)-1-(4-Methoxymethylphenyl)-butylamin Verbindung VI'.10 EÜ = 97,1 %
- (S)-1-(4-Methoxymethylphenyl)-pentylamin Verbindung VI'.11 EÜ = 96,8 %
- (S)-2-Cyclopropyl-1-phenylethylamin Verbindung VI'.12 EÜ = 95,8 %
- (S)-2-Cyclopropyl-1-(4-fluorphenyl)-ethylamin Verbindung VI'.13 EÜ = 95,4 %
- (S)-1-(4-Fluorphenyl)-pentylamin Verbindung VI'.14
- (S)-Cyclopropyl-phenyl-methylamin Verbindung VI'.15 EÜ = 90 %
- (S)-1-(3-Fluor-4-methylphenyl)-butylamin Verbindung VI'.16 EÜ > 99 %
- (S)-1-(3-Fluor-4-methylphenyl)-pentylamin Verbindung VI'.17 EÜ = 97 %
- (S)-2-Cyclopropyl-1-(3-fluor-4-methylphenyl)-ethylamin Verbindung VI'.18 EÜ>99%
- (S)-1-(4-Fluorphenyl)-butylamin Verbindung VI'.19 EÜ = 98,4 %
- (S)-1-(3-Fluor-4-methoxymethylphenyl)-butylamin Verbindung VI'.20 EÜ = 90,5 %
- (S)-2-Cyclopropyl-1-(4-chlorphenyl)-ethylamin Verbindung VI'.21 EÜ > 99 %
- (S)-2-Cyclopropyl-1-(4-methylphenyl)-ethylamin Verbindung VI.22 EÜ = 85,6 %
- (S)-2-Cyclobutyl-1-(4-fluorphenyl)-ethylamin Verbindung VI'.23 EÜ = 98,5 %
- (S)-2-Cyclopropyl-1-(4-bromphenyl)-ethylamin Verbindung VI'.24 EÜ = 98,3 %
- (S)-2-Cyclopropyl-1-(3,4-methylendioxyphenyl)-ethylamin Verbindung VI'.25 EÜ = 96,7 %
- (S)-1-(3,4-Methylendioxyphenyl)-butylamin Verbindung VI'.26 EÜ = 84 %
- Dritte Methode
- Zur Verbesserung des Enantiomeren-Überschusses kann man die obigen Amine mit organischen Säuren in Form der reinen Enantiomeren (beispielsweise N-Acetyl-L-leucin) behandeln und umkrisetallisieren:
- (S)-(1-Phenyl)-butylamin Verbindung VI'.9
- Salzbildung mit N-Acetyl-L-leucin:
- Man rührt eine Lösung von 10,4 g (60 mMol) N-Acetyl-L-leucin in 70 ml wasserfreiem Methanol bei 60°C und gibt tropfenweise eine Lösung von 9,0 g (60 mMol) (S)-(1-Phenyl)-butylamin Verbindung VI'.9 (EÜ 97,1 %) in 30 ml wasserfreiem Methanol zu. Nach Beendigung der Zugabe erhitzt man die methanolische Lösung zum Sieden (zur vollständigen Auflösung) und läßt dann über Nacht stehen. Nach dem Filtrieren und dem Spülen mit 20 ml wasserfreiem kaltem Methanol gewinnt man 7,7 g Kristalle, die man in der minimalen Menge Wasser löst. Nach dem Alkalischstellen mit 1N Natriumhydroxid unter Extraktion mit Dichlormethan wäscht man die organische Phase mit einer gesättigten Natriumchloridlösung, trocknet über Natriumsulfat und dampft im Vakuum ein. Man erhält 3,4 g des Amins in Form eines Öls. 1H-NMR-Spektrum: 7,16 – 7,36 (m, Ar, 5H); 3,87 (m, -CH-N, 1H); 1,57 – 1,69 (m, -CH-CH2, 2H); 1,47 (s, NH2, 2H); 1,15 – 1,40 (m, -CH2CH3, 2H); 0,88 (t, -CH2CH3, 3H). Chirale HPLC: %-Satz der Enantiomeren: S = 100 % R = 0 % EÜ = 100 %. [α]D 25 = – 22,0° (c = 1,05 CHCl3).
- HERSTELLUNG DER THIOHARNSTOFFE IN FORM EINES ENANTIOMEREN Verbindung IV'
- Erste Methode
- N-[(R)-1-(4-Fluorphenyl)-2-methoxyethyl]-thioharnstoff Verbindung IV'.1
- Man gibt zu einer Lösung von 2,83 g (37,2 mMol) Ammoniumisothiocyanat in 75 ml Aceton unter Rühren bei 0°C 4,23 ml (36,6 mMol) Benzoylchlorid. Nach dreißig Minuten gibt man langsam 6 g (35,5 mMol) der Verbindung VI'.1 in Lösung; in 75 ml Aceton zu. Man rührt die Reaktionsmischung während zwei Stunden bei Raumtemperatur und engt dann unter vermindertem Druck ein. Man nimmt die Suspension mit 200 ml tert.-Butylmethyloxid und 200 ml Wasser auf. Man wäscht die organische Phase mit Wasser und dann mit einer gesättigten Natriumchloridlösung, trocknet über wasserfreiem Natriumsulfat und dampft zur Trockne ein. Man löst den Verdampfungsrückstand in 180 ml Ethanol und versetzt die erhaltene Lösung mit 3,75 ml (75 mMol) Hydrazin-Monohydrat. Nach dem Rühren während vierundzwanzig Stunden bei Raumtemperatur dampft man die Reaktionsmischung ein, löst den Verdampfungsrückstand in 200 ml Ethylacetat und wäscht die organische Phase mit Wasser und dann mit einer gesättigten Natriumchloridlösung, trocknet über wasserfreiem Natriumsulfat und dampft zur Trockne ein. Man chromatographiert den Verdampfungsrückstand über einer mit Siliciumdioxid beschickten Säule (Elutionsmittel: Cyclohexan/Ethylacetat 1 / 1, V/V) und erhält 5 g (23 mMol) des weißen festen Produkts; Ausbeute = 63 %; F = 119 'C. 1H-NMR-Spektrum (DMSO D6): 8,10 (d, NH, 1H); 7,28 – 7,32 (m, Ar, 2H); 7,08-7,17 (m, Ar und NH2, 4H); 5,45 (m, CH-N, 1H); 3,54 – 3,62 (m, CH-CH2, 2H); 3,24 (s, OCH3, 3H). [α]19D = + 32,0° (c = 0,87 CH2Cl2)
- Chirale superkritische Chromatographie: EÜ = 100 %
- In gleicher Weise erhält man die folgenden Produkte:
- N-[(R)-2-Methoxy-1-phenylethyl]-thioharnstoff Verbindung IV'.2 F = 140°C [α]19D = + 4,6° (c = 1,0 CH2Cl2).
- N-[(R)-1-(4-Chlorphenyl)-2-methoxyethyl]-thioharnstoff Verbindung IV'.3 F = 133°C [α]19D = + 25,7° (c = 1,04 CH2Cl2).
- N-[(R)-2-Methoxy-1-(3,4-methylendioxyphenyl)-ethyl]-thioharnstoff Verbindung IV'.4 F = 160°C [α]19D = + 19,4° (c = 0,68 CH2Cl2).
- N-[(R)-1-(4-Ethylphenyl)-2-methoxyethyl]-thioharnstoff Verbindung IV'.5 F = 116°C [α]19 D = + 20,0° (c = 0,93 CH2Cl2).
- N-[(S)-1-Phenylbutyl]-thioharnstoff Verbindung IV'.6 F = 140°C [α]19D = + 48,7° (c = 0,82 CH2Cl2).
- N-[(R)-2-Methoxy-1-(4-methoxymethylphenyl)-ethyl]-thioharnstoff Verbindung IV'.7 1H-NMR-Spektrum: 7,25 – 7,36 (m, Ar, 4H); 6,85 (m, NH, 1H); 5,93 (m, NH2, 2H); 4,78 (m, CH-N, 1H); 4,43 (s, O-CH2, 2H); 3,58 – 3,65 (m, O-CH2, 2H); 3,38 (s, OCH3, 3H); 3,35 (s, O-CH3, 3H). [α]19p = + 20,5° (c = 0,95 CH2Cl2).
- N-[(S)-1-(4-Methosymethylphenyl)-pentyl]-thioharnstoff Verbindung IV '.8 1H-NMR-Spektrum: 7,22 – 7,35 (m, Ar, 4H); 6,71 (m, NH, 1H); 5,63 (m, NH2, 2H); 4,42 (s, O-CH2, 2H); 4,40 (m, CH, 1H); 3,39 (s, OCH3, 3H); 1,68 – 1,79 (m, CH2, 2H); 1,14 – 1,30 (m, CH2-CH2, 4H); 0,81 – 0,87 (m, CH3, 3H). [α]19 D = + 49,8° (c = 1,04 CH2Cl2).
- N-[(S)-1-(4-Methosymethylphenyl)-butyl]-thioharnstoff Verbindung IV'.9 1H-NMR-Spektrum: 7,20 – 7,40 (m, Ar, 4H); 6,69 (m, NH, 1H); 5,63 (m, NH2, 2H); 4,41 (s, O-CH2, 2H); 4,40 (m, CH, 1H); 3,39 (s, OCH3, 3H); 1,59 – 1,88 (m, CH-CH2-CH2, 2H); 1,15 – 1,44 (m, CH2-CH2-CH3, 2H); 0,85 – 0,92 (m, CH2-CH3, 3H). [α]19 D= + 43,9° (c = 1,17 CH2Cl2).
- N-[(S)-2-Cyclopropyl-1-phenylethyl]-thioharnstoff Verbindung IV'.10 F = 80°C [α]19 D = + 55,0° (e = 0,97 CH2Cl2); EÜ = 95,8 %
- N-((R)-1-(3-Fluor-4-methylphenyl)-2-methosyethyl]-thioharnstoff Verbindung IV'.11 F = 149°C [α]20 D = + 30,3° (c = 0,97 CH2Cl2).
- N-[(R)-1-(3-Fluor-4-chlorphenyl)-2-methogyethyl]-thiohasnstoff Verbindung IV'.12 F = 110°C [α]20 D = + 29,1° (c = 1,04 CH2Cl2).
- N-[(S)-1-(4-Fluorphenyl)-pentyl]-thioharnstoff Verbindung IV'.13 F = 118°C [α]20 D = – 19,2° (c = 0,78 Methanol)
- N-[(S)-Cyclopropyl-phenyl-methyl]-thioharnstoff Verbindung IV'.14 1H-NMR-Spektrum: 7,25 – 7,41 (m, Ar, 5H); 6,92 (m, NH, 1H); 5,58 (m, NH2, 2H) 3,92 (m, CH, 1H); 1,08 – 1,25 (m, CH Cyclopropyl, 1H); 0,35 – 0,69 (m, 2CH2 Cyclopropyl, 4H). [α]20 D = + 33,5° (c = 0,48 Methanol); EÜ = 90 %.
- N-[(S)-1-(3-Fluor-4-methylphenyl)-butyl]-thioharnstoff Verbindung IV'.15 F = 129°C [α]20 D = 44,4° (c = 0,81 CH2Cl2); EÜ = 99 %.
- N-[(S)-1-(3-Fluor-4-methylphenyl)-pentyl]-thioharnstoff Verbindung IV'.16 F = 124°C [α]20 D = + 4,6° (c = 1,4 CH2Cl2); EÜ = 97 %.
- N-[(S)-2-Cyclopropyl-1-(3-fluor-4-methylphenyl)-ethyl]-thioharnstoff Ver bindung IV'.17 F = 91°C [α]22 D = + 55,4° (c = 0,9 CH2Cl2); EÜ = 99 %.
- N-[(S)-1-(4-Fluorphenyl)-butyl]-thioharnstoff Verbindung IV'.18 1H-NMR-Spektrum: 7,21 – 7,28 (m, Ar, 2H); 6,99 – 7,09 (m, Ar, 2H); 6,75 (s, NH, 1H); 5,71 (s, NH2, 2H); 4,35 – 4,60 (m, CH, 1H); 1,65 – 1,85 (m, CH2, 2H); 1,18 – 1,45 (m, CH2, 2H); 0,86 – 0,93 (m, CH3, 3H). [α]22 D = + 49° (c = 0,95 CH2Cl2); EÜ = 98,4 %.
- N-[(S)-2-Cyclopropyl-1-(4-chlorphenyl)-ethyl]-thioharnstoff Verbindung IV'.19 F = 93,7°C [α]23 D = + 53° (c = 0,5 CH2Cl2); EÜ = 99,1 %.
- N-[(S)-2-Cyclobutyl-1-(4-fluorphenyl)-ethyl]-thioharnstoff Verbindung IV'.20 F = 104°C [α]20 D = – 21° (c = 1 Methanol); EÜ = 98,5 %.
- N-[(S)-2-Cyclopropyl-1-(4-bromphenyl)-ethyl]-thioharnstoff Verbindung IV'.21 F = 130°C [α]20 D = + 57° (c = 0,67 CH2Cl2); EÜ = 98,3 %.
- N-((S)-2-Cyclopropyl-1-(3,4-methylendioxyphenyl)-ethyl]-thioharnstoff Verbindung IV'.22 F = 125°C [α]19 D = + 63° (c = 0,75 CH2Cl2); EÜ = 96,7 %.
- Zweite Methode
- a) Man erhält die folgenden Verbindungen durch Chromatographie der Thioharnstoffe in Form der Enantiomeren (EÜ > 99 %) ausgehend von den an ei nem Enantiomeren angereicherten Thioharnstoffen:
- N-[(S)-2-Cyclopropyl-1-phenylethyl]-thioharnstoff Verbindung IV'.10 Ausgehend von einer Mischung, die überwiegend das Enantiomere S enthält (EÜ 95,8 %) erhält man nach der chromatographischen Trennung über der Phase CHIRACEL OJ unter Elution mit einer Isohexan/Ethanol-Mischung (97/3) das reine Enantiomere S (EÜ 100 %). F = 84°C [α]19 D = + 59,3° (c = 1,06 CH2Cl2).
- N-[(S)-2-Cyclopropyl-1-(4-fluorphenyl)-ethyl]-thioharnstoff Verbindung IV'.23 F = 105° [α]22 D = + 61,0° (e = 0,53 CH2Cl2); EÜ = 100 %.
- N-[(S)-1-(3-Fluor-4-methoxymethylphenyl)-butyl]-thioharnstoff Verbindung IV'.24 1H-NMR-Spektrum: 7,39 – 7,46 (m, Ar, 1H); 6,93 – 7,07 (m, Ar, 2H und NH, 1H); 5,85 (m, NH2, 2H); 4,45 (s, O-CH2, 2H); 4,35 (m, CH, 1H); 3,38 (s, CCH3, 3H); 1,59 – 1,88 (m, CH-CH2-CH2, 2H); 1,18 – 1,40 (m, CH2-CH2-CH3, 2H); 0,85 – 0,92 (m, CH2-CH3, 3H). [α]19 D = + 30,5° (c = 0,77 CH2Cl2); EÜ = 100 %.
- N-[(S)-2-Cyclopropyl-1-(4-methylphenyl)-ethyl]-thioharnstoff Verbindung IV'.25 1H-NMR-Spektrum: 7,10 – 7,20 (m, Ar, 4H); 6,93 (m, NH, 1H); 5,75 (m, NH2, 2H); 4,43 (m, CH, 1H); 2,30 (s, CH3, 3H); 1,62 – 1,73 (m, CH2, 2H); 0,40-0,59 (m, CH und CH2, Cyclopropyl, 3H); 0,04 – 0,13 (m, CH2, Cyclopropyl, 2H). [α]19 D = + 75,5° (c = 0,42 CH2Cl2); EÜ = 100 %.
- N-[(S)-1-(3,4-Methylendiosyphenyl)-butyl]-thioharnstoff Verbindung IV'.26 F = 140°C [α]20 D = + 40,3° (c = 1,18 CH2Cl2); EÜ = 100 %.
- b) Man erhält die folgenden Verbindungen durch Chromatographie der optisch aktiven Thioharnstoffe (EÜ > 99 %) ausgehend von den racemischen Thioharnstoffen:
- N-[(S)-1-Phenylpentyl]-thioharnstoff Verbindung IV'.27 Ausgehend von racemischem N-(1-Phenylpentyl)-thioharnstoff erhält man nach der chromatographischen Trennung über der Phase CHIRACEL OJ unter Elution mit einer Isohexan/Ethanol-Mischung (95/5) das Enantiomere S, dessen Enantiomerenreinheit 99,8 % beträgt. F = 147°C [α]19 D = + 46,0° (c = 1,00 CH2Cl2).
- HERSTELLUNG DER NH-AMINOTHIAZOLE IN FORM EINES ENANTIOMEREN Verbindung II'
- [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1R)-1-(4-fluorphenyl)-2-methoxyethyl]-amin Verbindung II'.1
- Man gibt zu 3,28 g (14,3 mMol) des Thioharnstoffs (Verbindung IV'.1) in Lösung in 70 ml Ethanol 4,23 g (14,5 mMol) 2-Brom-1-(2'-chlor-4'-methoxy-5'-methylphenyl)-propan-1-on (Verbindung III.1) und 4,2 ml (30 mMol) Triethylamin. Man rührt die Reaktionsmischung während 3 Stunden bei 90°C und engt dann unter vermindertem Druck ein. Man nimmt den Rückstand mit 200 ml Dichlormethan und 100 ml Wasser auf. Man wäscht die organische Phase mit einer gesättigten Natriumchloridlösung, trocknet über wasserfreiem Natriumsulfat und dampft zur Trockne ein. Man reinigt den rohen Extrakt säulenchromatographisch über Kieselgel (Elutionsmittel: Cyclohexan/Ethylacetat 4/1 (V/V)) und erhält 5,27 g (12,5 mMol) der Verbindung II'.1; Ausbeute = 87 % ; MS (MH+) 421. 1H-NMR-Spektrum: 7,34 – 7,44 (m, Ar, 2H); 7,0 – 7,09 (m, Ar, 3H); 6,83 (s, Ar, 1H); 5,87 (d, NH, 1H); 4,57 (m, CH, 1H); 3,81 (s, OCH3, 3H); 3,46 – 3,62 (m, OCH2 2H); 3,35 (s, OCH3, 3H); 2,14 (s, CH3, 3H); 2,04 (s, CH3, 3H).
- In gleicher Weise erhält man die folgenden Zwischenprodukte:
-

-
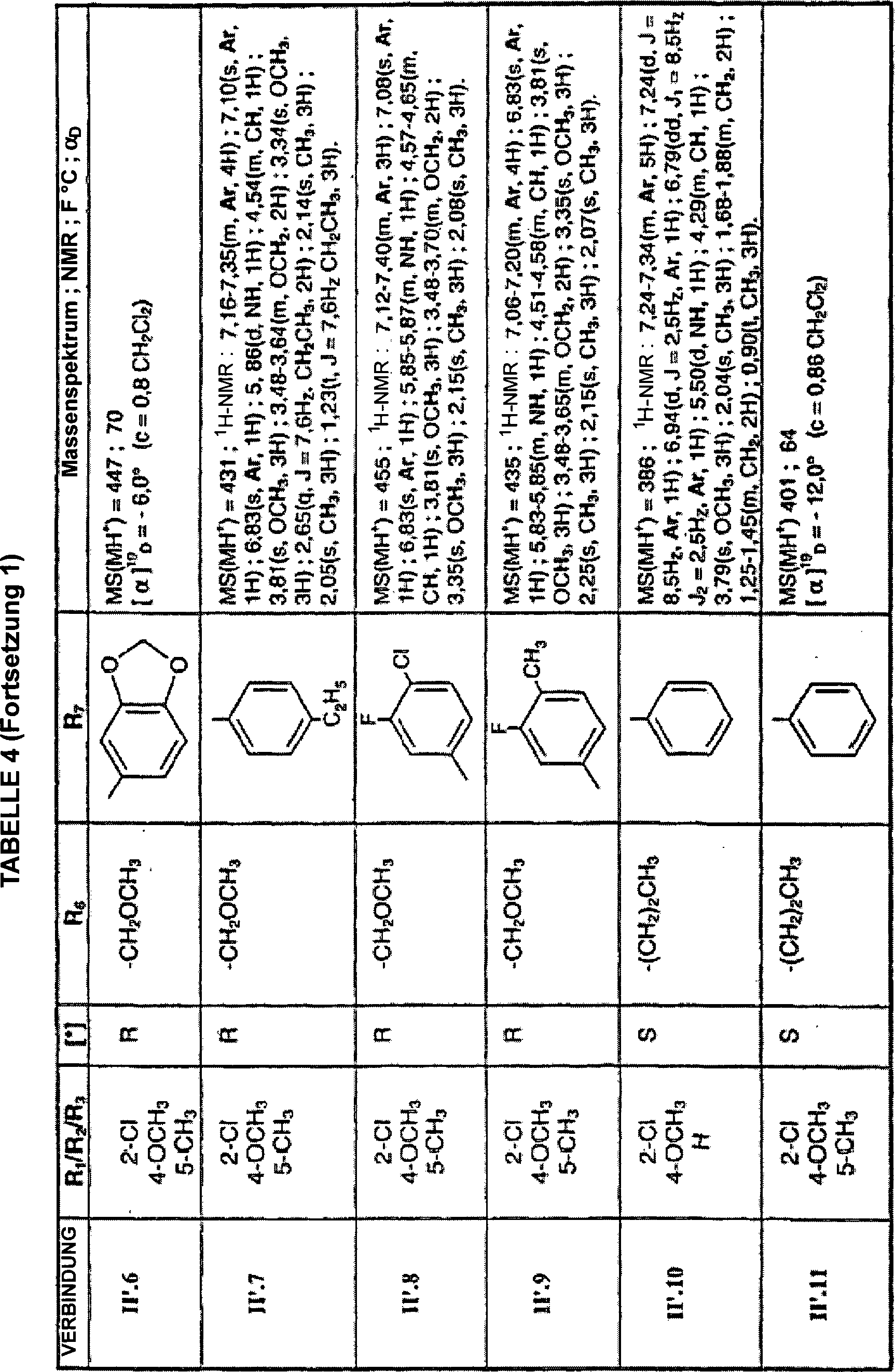
-
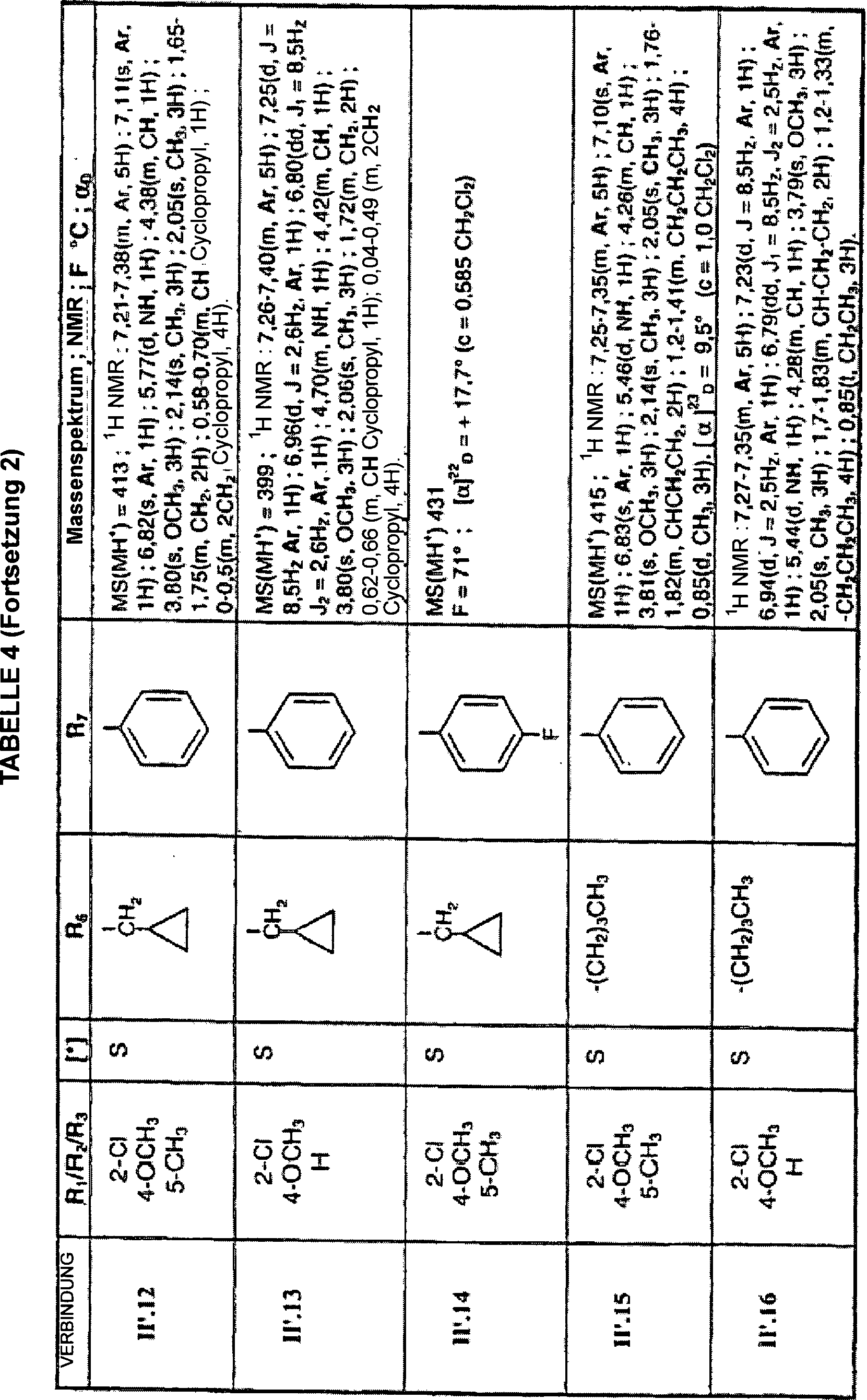
-
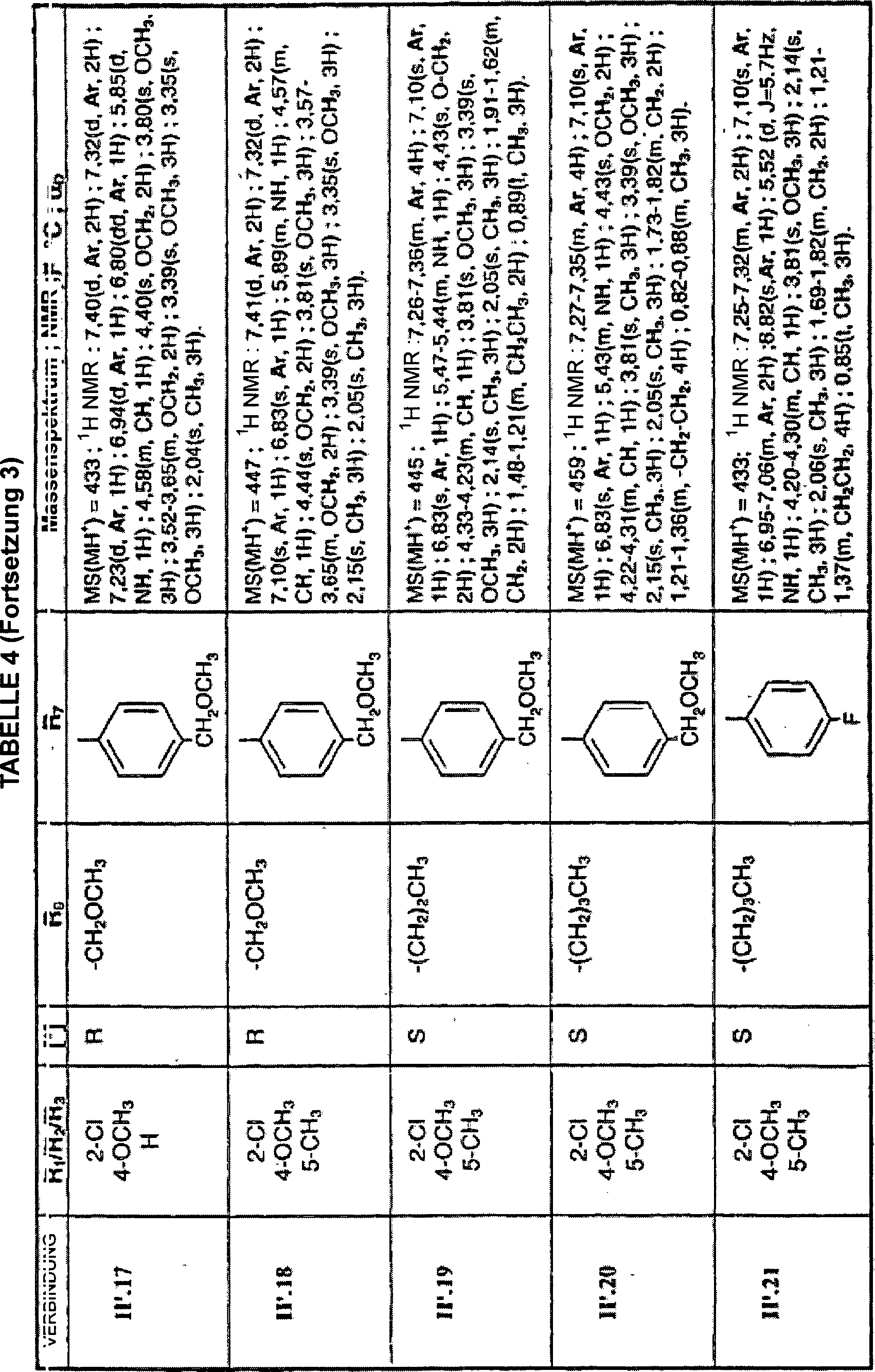
-
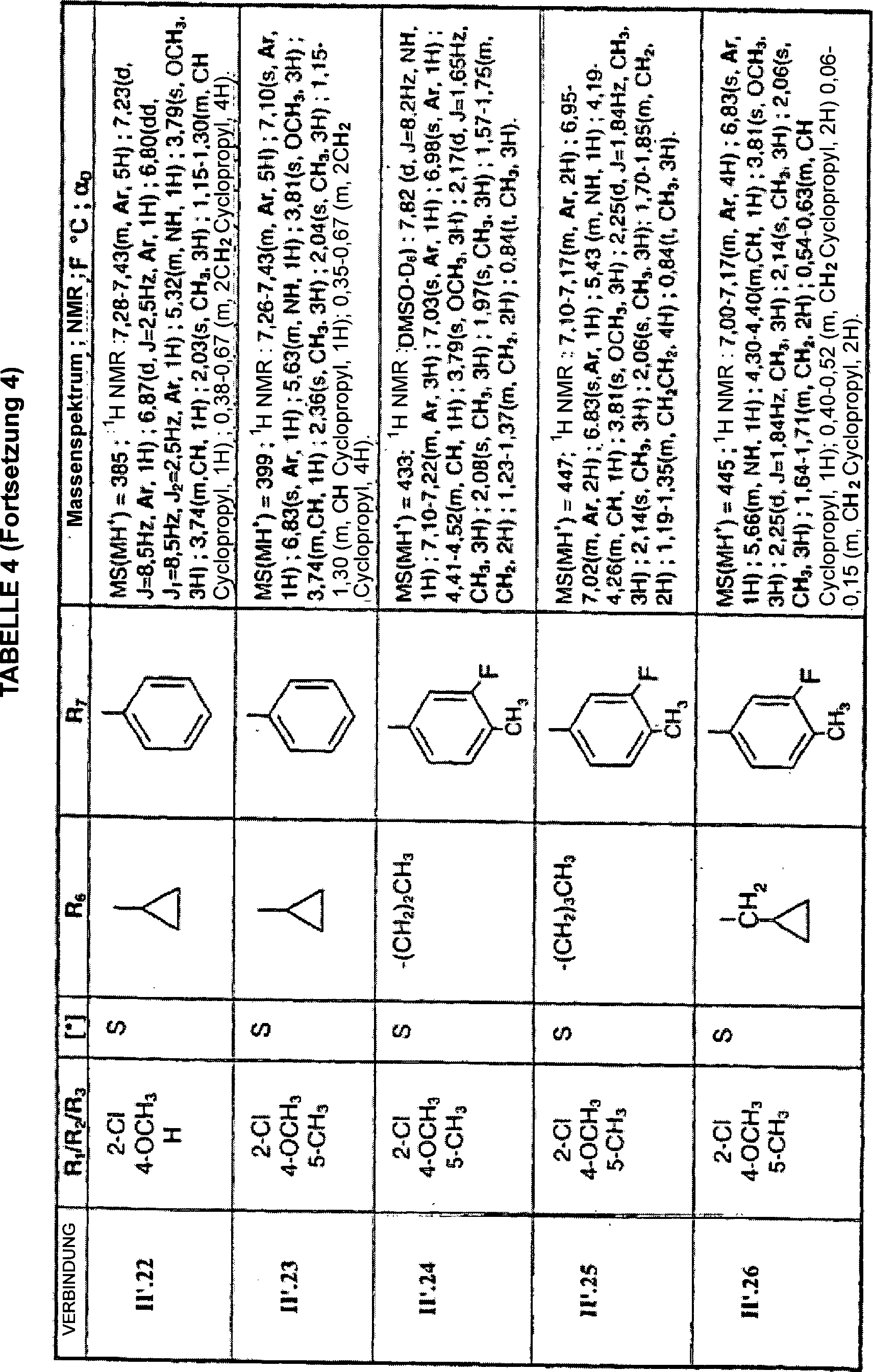
-
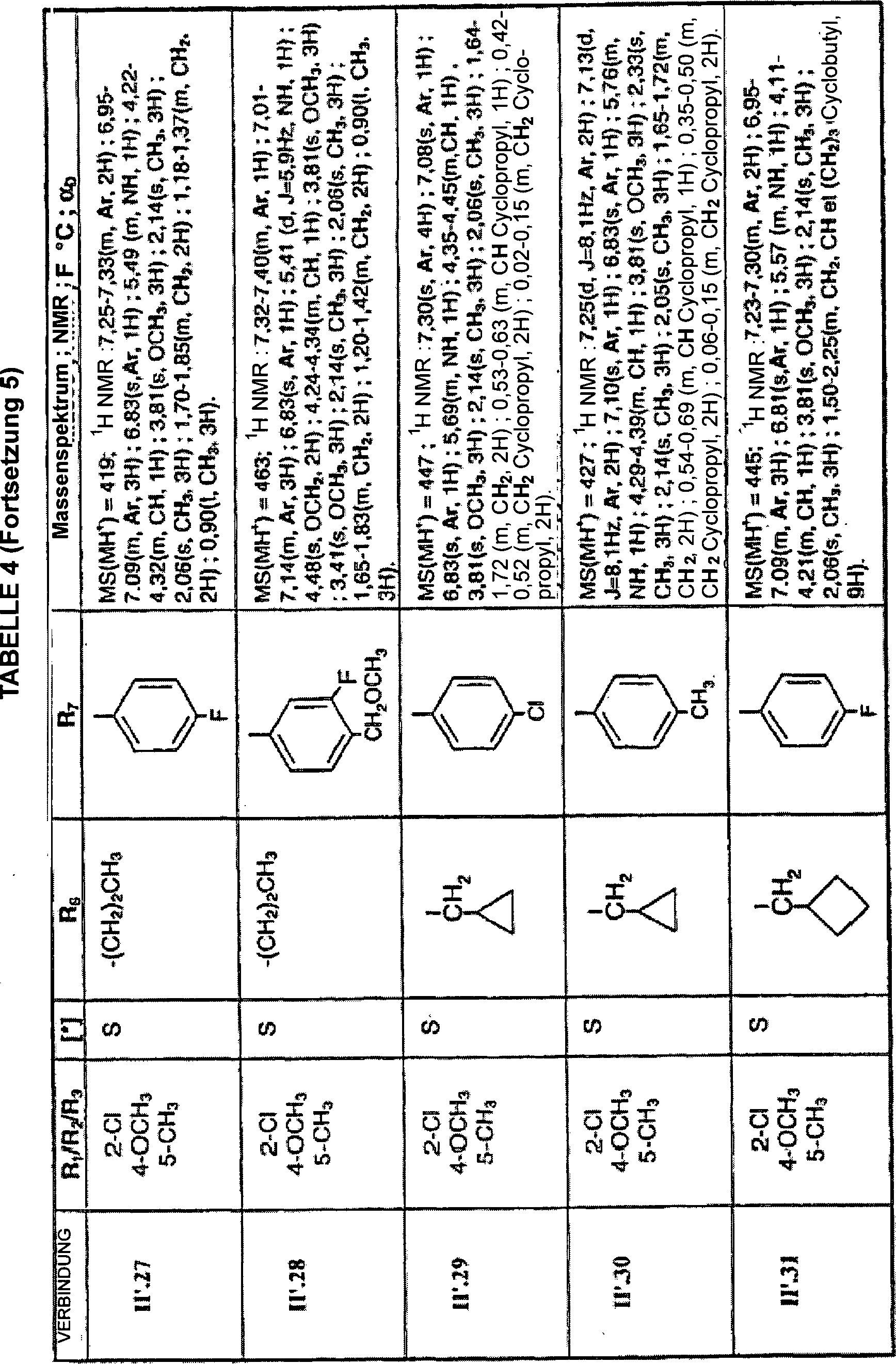
-
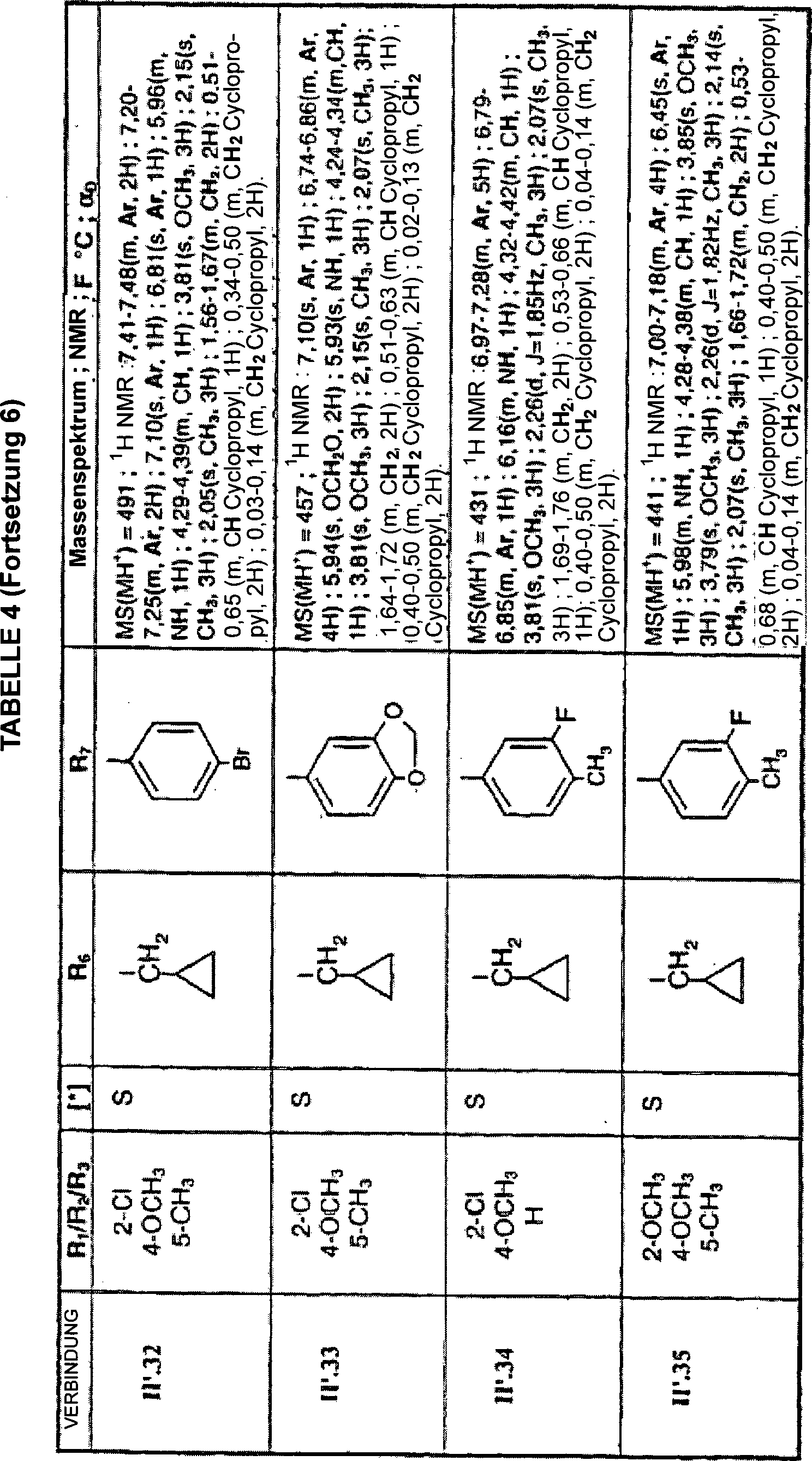
-

- HERSTELLUNG DER N-SUBSTITUIERTEN AMINOTHIAZOLE IN FORM EINES ENANTIOMEREN
- BEISPIEL 25
- [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1R)-1-(4-fluorphenyl)-2-methoxyethyl]-prop-2-inyl-amin
- Man gibt zu einer Lösung von 2,5 g (5,9 mMol) der Verbindung II'.1 in 30 ml Dimethylformamid unter Rühren bei 0°C 260 mg (6,5 mMol) 60 %-iges Natriumhydrid in Öl. Man rührt die Reaktionsmischung während 20 Minuten bei 0°C und gibt dann 0,83 ml (7,5 mMol) einer 80 %-igen Lösung von Propargylbromid in Toluol zu. Man rührt die Reaktionsmischung während einer Stunde bei 10°C und gibt dann bei 0°C 2 ml Ethanol und dann 50 ml Wasser zu. Man extrahiert die Mischung mit 200 ml Ethylacetat, wäscht die organische Phase mit Wasser und dann mit einer gesättigten Natriumchloridlösung, trocknet über wasserfreiem Natriumsulfat und dampft zur Trockne ein. Man chromatographiert den rohen Rückstand über einer mit Kieselgel beschickten Säule (Elutionsmittel: Cyclohexan/Ethylacetat 9/1, V/V) und erhält 2,14 g eines reinen gummiartigen Produkts. Ausbeute = 80 % ; MS (MH+) 459. 1H-NMR-Spektrum: 7,37 – 7,46 (m, Ar, 2H); 7,14 (s, Ar, 1H); 6,95 – 7,07 (m, Ar, 2H); 6,81 (s, Ar, 1H); 5,54 (m, CH, 1H) ; 4,15 (dd, J1 = 18 HZ, J2 = 2,4 HZ, CH2-N, 1H); 4,00 – 4,08 (m, OCH2, 2H); 3,98 (dd, J1 = 18 HZ, J2 = 2,4 HZ, CH2-N, 1H); 3,82 (s, OCH3, 3H); 3,40 (s, OCH3, 3H); 2,18 (t, J = 2,4 HZ, CH Propargyl, 1H); 2,17 (s, CH3, 3H); 2,16 (s, CH3, 3H). [α]19 D = – 127° (c = 0,99 CH2Cl2). Chirale superkritische HPLC : EÜ = 99,4 %.
-

-
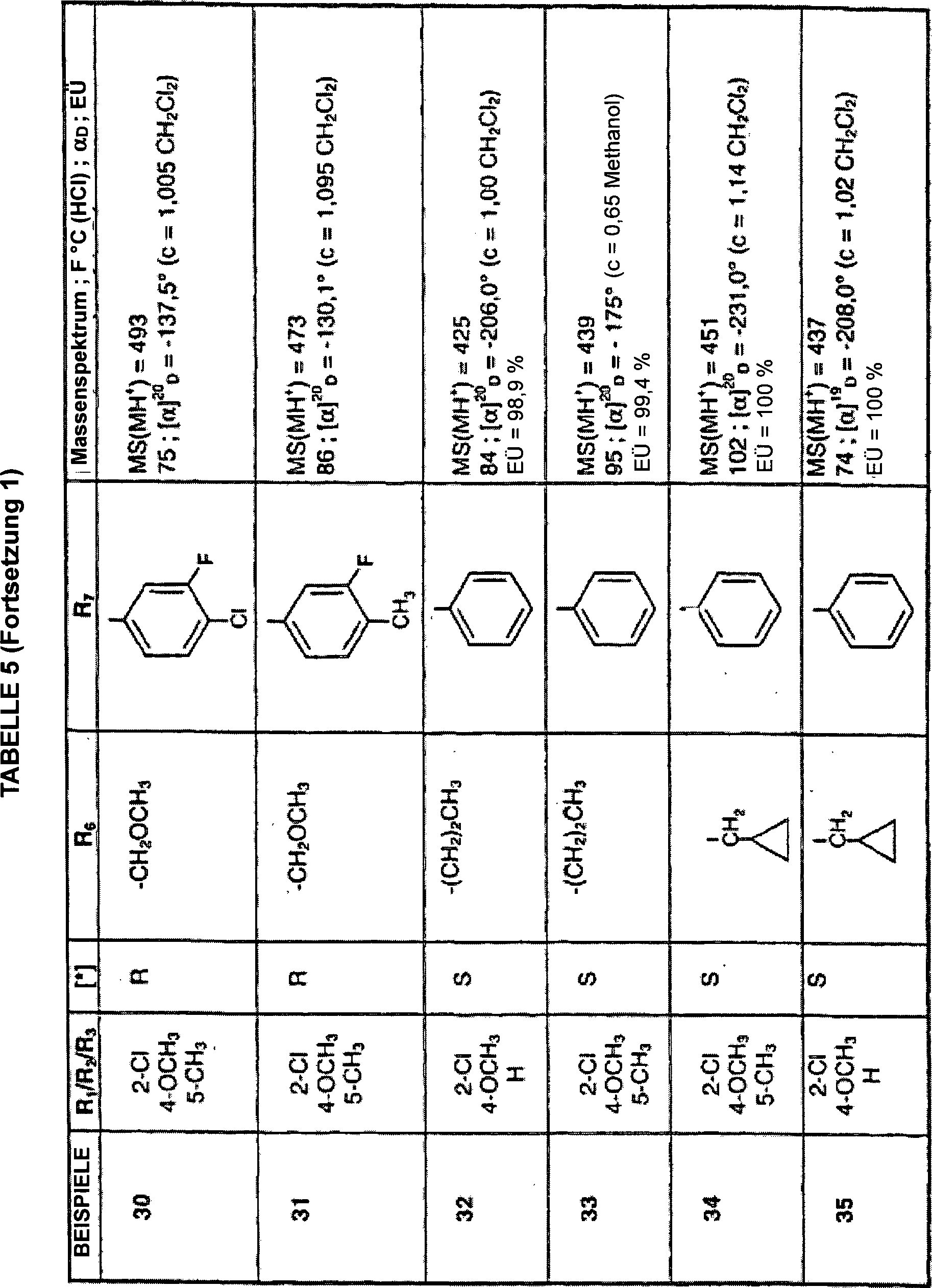
-
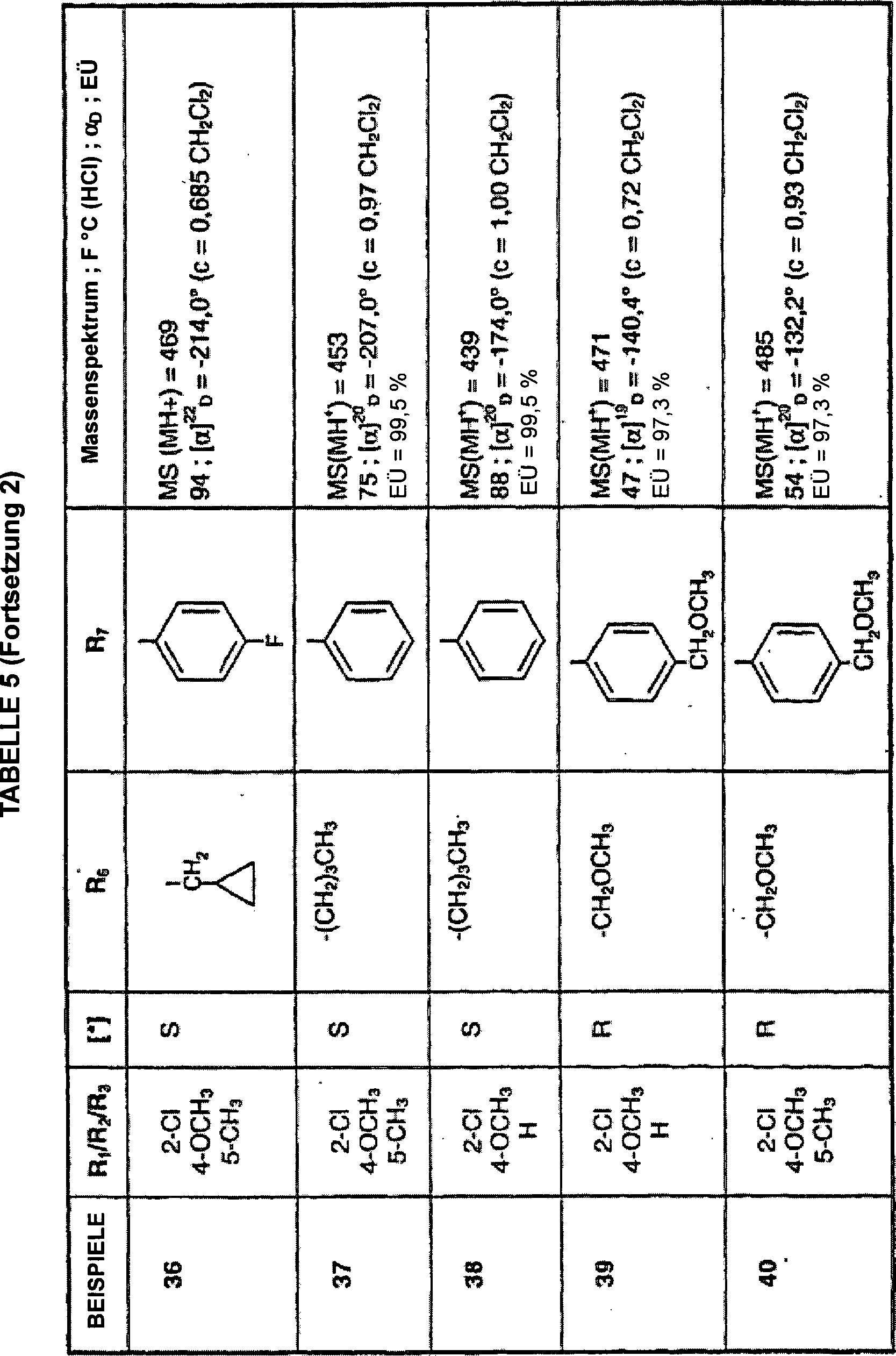
-

-
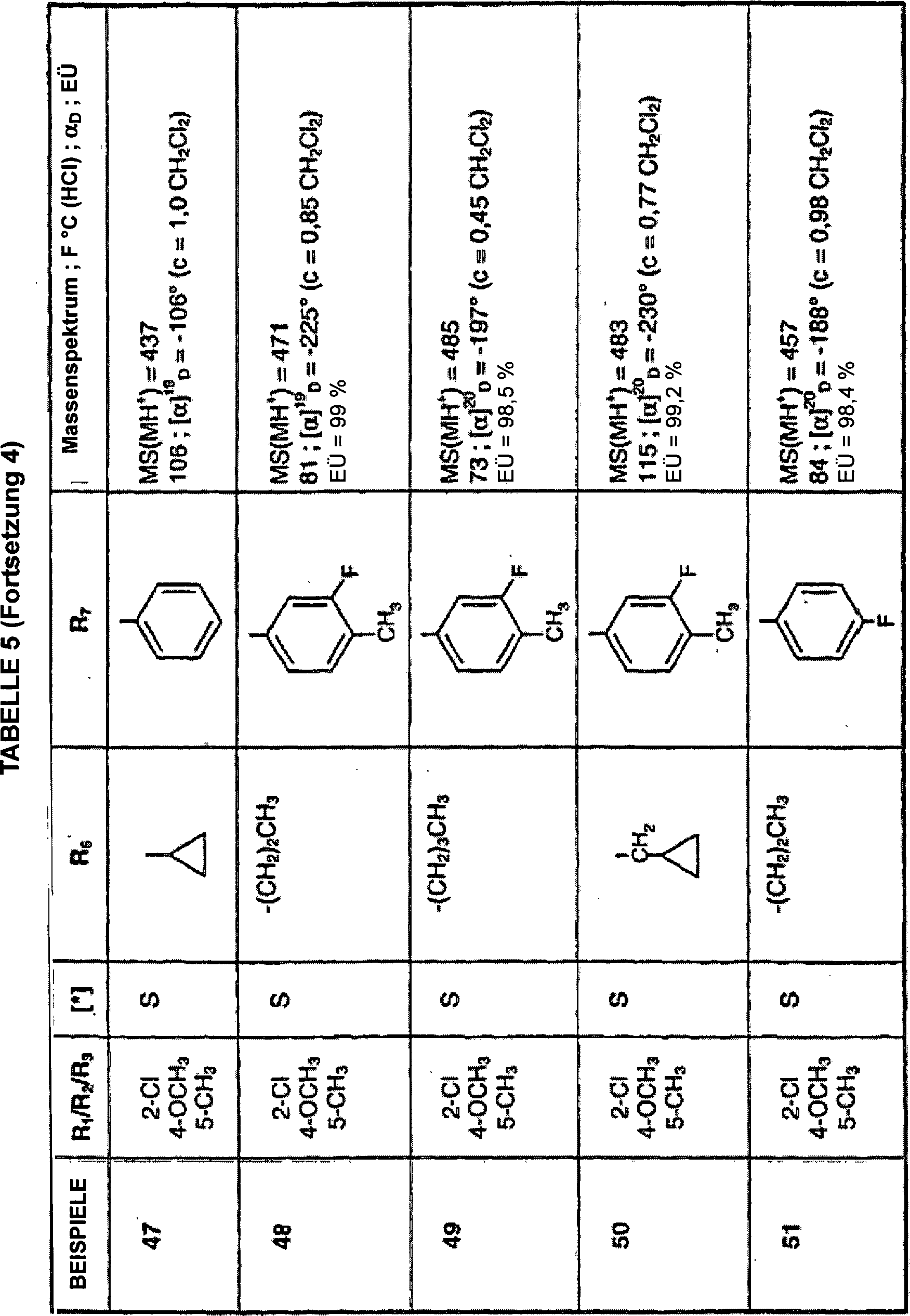
-
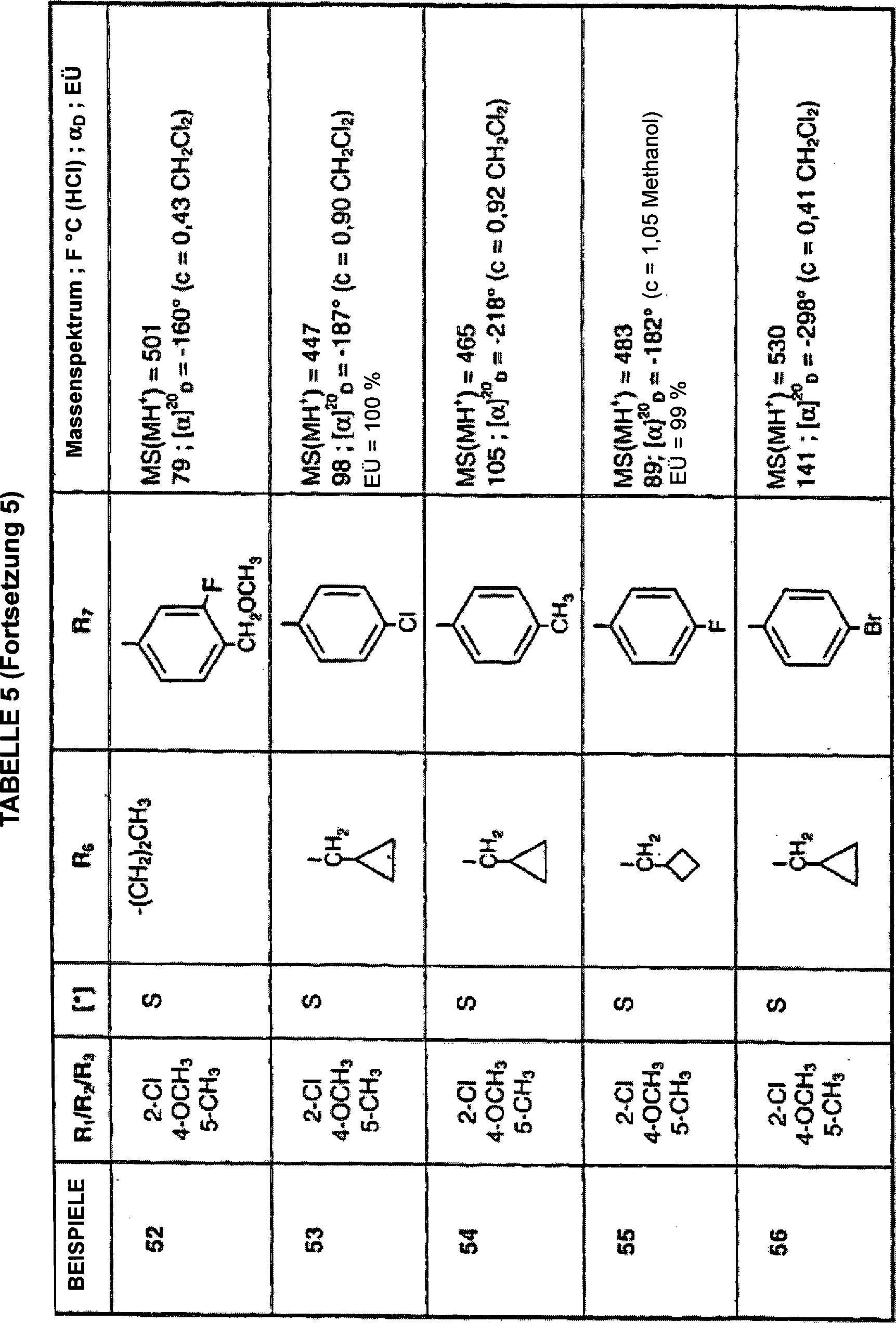
-
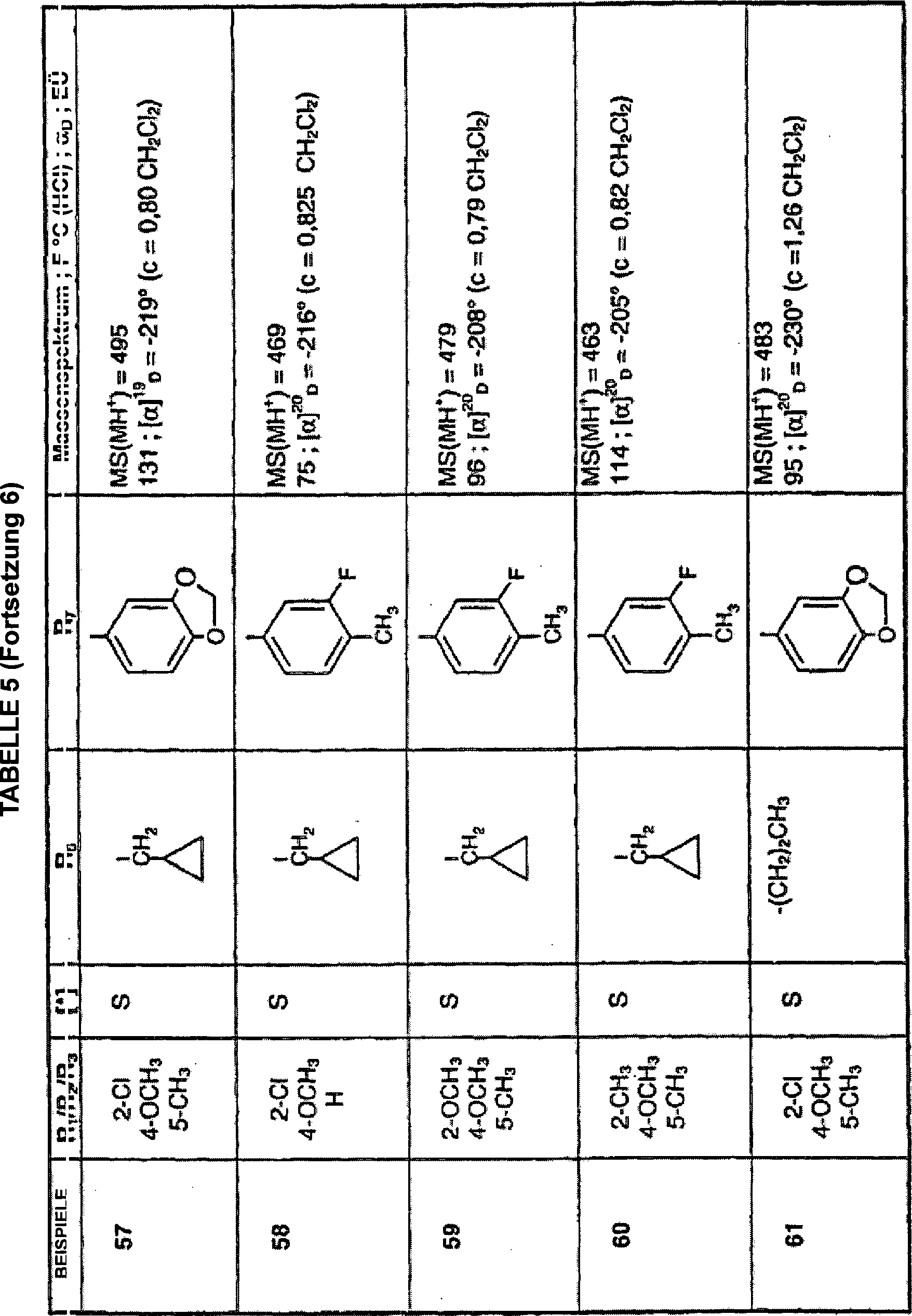
Claims (8)
- Verbindungen in racemischer oder enantiomerenreiner Form der Formel:in der – R1 und R2, die gleichartig oder verschieden sind, jeweils unabhängig voneinander ein Halogenatom; eine Hydroxy-(C1-C5)-alkylgruppe; eine (C1-C5)-Alkylgruppe; eine Aralkylgruppe, in der der Arylteil (C6-C8) und der Alkylteil (C1-C4) ist; eine (C1-C5)-Alkoxygruppe; eine Trifluormethylgruppe; eine Nitrogruppe; eine Nitrilgruppe; eine Gruppe -SR, in der R Wasserstoff, eine (C1-C5)-Alkylgruppe oder eine Aralkylgruppe, in der der Arylteil (C6-C8) und der Alkylteil (C1-C4) ist, bedeutet; eine Gruppe -S-CO-R, in der R eine (C1-C5)-Alkylgruppe oder eine Aralkylgruppe, in der der Arylteil (C6-C8) und der Alkylteil (C1-C4) ist, bedeutet; eine Gruppe -COORa, worin Ra Wasserstoff oder eine (C1-C5)-Alkylgruppe darstellt; eine Gruppe -CONRaRb, worin Ra und Rb die oben für Ra angegebenen Bedeutungen besitzen; eine Gruppe -NRaRb, worin Ra und Rb die oben für Ra angegebenen Bedeutungen besitzen; eine Gruppe -CONRcRd oder -NRcRd, worin Rc und Rd zusammen mit dem Stickstoffatom, an das sie gebunden sind, einen Heterocyclus mit 5 bis 7 Kettengliedern bilden; oder eine Gruppe -NHCO-NRaRb, worin Ra und Rb die oben für Ra angegebenen Bedeutungen besitzen; bedeuten; – R3 Wasserstoff bedeutet oder die oben für R1 und R2 angegebenen Bedeutungen besitzt; – oder R2 mit R3, wenn dieser Rest den Phenylrest in der 5-Stellung substituiert, eine Gruppe -X-CH2-X- bildet, worin X unabhängig voneinander eine CH2-Gruppe, ein Sauerstoffatom oder ein Schwefelatom bedeutet; – R6 eine (C1-C6)-Alkylgruppe; eine (C1-C6)-Alkoxy-(C1-C3)-alkylgruppe; eine (C3-C5)-Cycloalkylgruppe; eine (C3-C6)-Cycloalkyl-(C1-C6)-alkylgruppe; eine (C1-C6)-Alkylthio-(C1-C3)-alkylgruppe; eine (C1-C6)-Alkylsulfoxy-(C1-C3)-alkylgruppe; oder eine (C1-C6)-Alkylsulfodioxy-(C1-C3)-alkylgruppe bedeutet; – R7 eine nichtsubstituierte Phenylgruppe bedeutet oder Phenylgruppe, die einfach, zweifach oder dreifach in den Stellungen 3, 4 oder 5 substituiert ist durch ein Halogen, eine (C1-C5)-Alkylgruppe, eine Gruppe -O-CH2-O- an zwei benachbarten Kohlenstoffatomen des Phenylrests, durch eine Gruppe -CF3, -NO2, -CN, durch eine Gruppe -COOR8, -CONR8R9 oder durch eine Gruppe -CH2OR8, worin R8 und R9 eine (C1-C3)-Alkylgruppe bedeuten, OR10, worin R10 eine (C1-C5)-Alkylgruppe darstellt; oder R7 eine Pyridyl-, Thiophen-, Pyrazolyl-, Imidazolyl-, (C3-C5)-Cycloalkyl- oder (C3-C6)-Cycloalkyl-(C1-C6)-alkylgruppe bedeutet; deren Additionssalze, deren Hydrate und/oder deren Solvate.
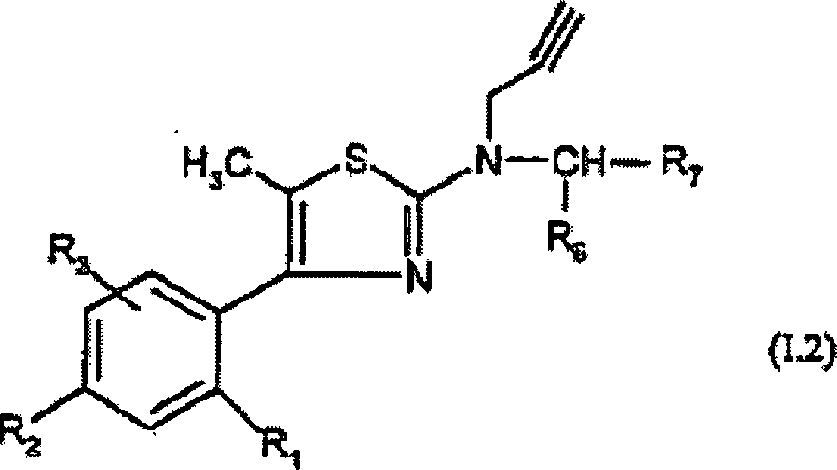
- Verbindungen nach Anspruch 1, dadurch gekennzeichnet, daß: – R1 und R2, die gleichartig oder verschieden sind, jeweils unabhängig voneinander ein Halogenatom; eine (C1-C5)-Alkylgruppe; oder eine (C1-C-)-Alkoxygruppe bedeuten; – R3 Wasserstoff bedeutet oder die oben für R1 und R2 angegebenen Bedeutungen. besitzt; – R6 eine (C1-C6)-Alkylgruppe; eine (C1-C6)-Alkoxy-(C1-C3)-alkylgruppe; eine (C3-C5) -Cycloalkylgruppe; oder eine (C3-C6)-Cycloalkyl-(C1-C6)-alkylgruppe bedeutet; – R7 eine nichtsubstituierte Phenylgruppe oder eine Phenylgruppe, die mono- oder disubstituiert ist in der 3- oder 4-Stellung durch ein Halogen, eine (C1-C5)Alkylgruppe, eine Gruppe -CH2OR8, in der R8 eine (C1-C3)-Alkylgruppe bedeutet, oder durch eine Gruppe -O-CH2-O- in der Stellung 3, 4; oder R7 eine (C3..C5)-Cycloalkylgruppe bedeutet.
- Verbindungen nach einem der Ansprüche 1 oder 2, dadurch gekennzeichnet, daß R3 in der 5-Stellung des Phenylrests steht.
- Verbindungen nach Anspruch 1, ausgewählt aus: – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1R)-(1-(3-fluor-4-methylphenyl)-2-methoxyethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 31) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(1-phenylbutyl)-prop-2-inylamin-Hydrochlorid (BEISPIEL 33) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-phenylethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 34) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-phenylethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 35) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(4-fluorhenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 36) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(1-phenylpentyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 37) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1R)-(2-methoxy-1-(4-methoxymethylphenyl)-ethyl]-prop-2-inylamin-Hydrochlorid (BEISPIEL 40) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(1-(4-methoxymethylphenyl)-pentyl]-prop-2-inylamin-Hydrochlorid (BEISPIEL 42) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(1-(4-fluorphenyl)-pentyl]-prop-2-inylamin-Hydrochlorid (BEISPIEL 45) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(cyclopropylphenyl-methyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 47) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(1-(3-fluor-4-methylphenyl)-pentyl]-prop-2-inylamin-Hydrochlorid (BEISPIEL 49) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(3-fluor-4-methylphenyl)-ethyl]-prop-2-inylamin-Hydrochlorid (BEISPIEL 50) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(1-(4-fluorphenyl)-butyl]-prop-2-inylamin-Hydrochlorid (BEISPIEL 51) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(1-(3-fluor-4-methoxymethylphenyl)-butyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 52) – (4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(4-chlorphenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 53) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(4-methylphenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 54) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclobutyl-1-(4-fluorphenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 55) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(4-bromphenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 56) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(3,4-methylendioxyphenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 57) – [4-(2-Chlor-4-methoxyphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(3-fluor-4-methylphenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 58) – [4-(2,4-Dimethoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(3-fluor-4-methylphenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 59) – [4-(4-Methoxy-2,5-dimethylphenyl)-5-methylthiazol-2-yl]-[(1S)-(2-cyclopropyl-1-(3-fluor-4-methylphenyl)-ethyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 60) – [4-(2-Chlor-4-methoxy-5-methylphenyl)-5-methylthiazol-2-yl]-[(1S)-(1-(3,4-methylendioxyphenyl)-butyl)]-prop-2-inylamin-Hydrochlorid (BEISPIEL 61) sowie die entsprechenden Basen, die weiteren Additionssalze, ihre Solvate und/oder Hydrate.
- Verfahren zur Herstellung der Verbindungen der Formel (I.2) nach Anspruch 1, dadurch gekennzeichnet, daß man sie durch Alkylierung von Verbindungen der Formel:in der R1, R2, R3, R6 und R7 die für (I.2) angegebenen Bedeutungen besitzen, erhält.

- Pharmazeutische Zubereitung, dadurch gekennzeichnet, daß sie als Wirkstoff eine Verbindung nach einem der Ansprüche 1 bis 4 enthält.
- Verwendung einer Verbindung nach einem der Ansprüche 1 bis 4 zur Herstellung von Arzneimitteln, die für die Vorbeugung und/oder Behandlung von CRF-abhängigen Erkrankungen bestimmt sind.
- Verwendung einer Verbindung nach einem der Ansprüche 1 bis 4 für die Herstellung eines Arzneimittels zur vorbeugenden oder heilenden Behandlung von Erkrankungen, die mit dem Stress verbunden sind, und insbesondere für die Behandlung sämtlicher pathologischer Zustände, bei denen CRF beteiligt ist, ausgewählt aus der Cushing-Krankheit, neuropsychiatrischen Störungen, wie der Depression, der Angst, der Panik, krampfartigen Besessenheitsstörungen, Stimmungstörungen, posttraumatischen Stress, Verhaltensstörungen, Aggressivität, Anorexie, Bulimie, Hyperglykämie, Frühgeburten, Risikoschwangerschaften, Wachstumsverzögerungen, Schlafstörungen, Epilepsie, Depressionen jeglicher Art; der Alzheimerschen Krankheit, der Parkinsonschen Krankheit; der Huntington Chorea; der amyotrophischen Lateralsklerose; Gefäßstörungen, Herzstörungen und Gehirnstörungen; Störungen der sexuellen Aktivität und der Fertilität; der Immunodepression, der Immunosuppression, von Entzündungsprozessen, multiplen Infektionen, der rheumatoiden Arthritis, der Osteoarthritis, der Uveitis, Psoriasis sowie Diabetes; Krebs; funktionellen Magen-Darm-Störungen und die dadurch ausgelösten Entzündungszustände, wie Reizdarm und Darmentzündung, Diarrhoen; Störungen der Schmerzwahrnehmung; Fibromyalgien, die mit Schlafstörungen, Müdigkeit oder Migräne verknüpft sind oder auch nicht; und Symptome, die mit der Alkoholabhängigkeit und dem Drogenentzug verknüpft sind.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9909144A FR2796380B3 (fr) | 1999-07-15 | 1999-07-15 | Nouveaux derives d'aminothiazoles, leur preparation et les compositions pharmaceutiques les contenant |
| FR9909144 | 1999-07-15 | ||
| PCT/FR2000/001995 WO2001005776A1 (fr) | 1999-07-15 | 2000-07-11 | Derives d'aminothiazole et leur utilisation comme ligands des recepteurs crf |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE60009911D1 DE60009911D1 (de) | 2004-05-19 |
| DE60009911T2 true DE60009911T2 (de) | 2005-03-10 |
Family
ID=9548107
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE60009911T Expired - Lifetime DE60009911T2 (de) | 1999-07-15 | 2000-07-11 | Aminothiazolderivate und ihre verwendung als crf-rezeptor-liganden |
Country Status (36)
| Country | Link |
|---|---|
| US (2) | US6586456B1 (de) |
| EP (1) | EP1200419B1 (de) |
| JP (1) | JP4949582B2 (de) |
| KR (1) | KR100707320B1 (de) |
| CN (1) | CN1167692C (de) |
| AR (1) | AR024747A1 (de) |
| AT (1) | ATE264315T1 (de) |
| AU (1) | AU772683B2 (de) |
| BR (1) | BR0012478B1 (de) |
| CA (1) | CA2378792C (de) |
| CO (1) | CO5200792A1 (de) |
| CZ (1) | CZ300937B6 (de) |
| DE (1) | DE60009911T2 (de) |
| DK (1) | DK1200419T3 (de) |
| EA (1) | EA005159B1 (de) |
| EE (1) | EE05004B1 (de) |
| ES (1) | ES2220504T3 (de) |
| FR (1) | FR2796380B3 (de) |
| HK (1) | HK1045692B (de) |
| HU (1) | HU229069B1 (de) |
| IL (2) | IL147282A0 (de) |
| IS (1) | IS2314B (de) |
| ME (1) | MEP26608A (de) |
| MX (1) | MXPA02000545A (de) |
| NO (1) | NO324352B1 (de) |
| NZ (1) | NZ516397A (de) |
| PL (1) | PL206515B1 (de) |
| PT (1) | PT1200419E (de) |
| RS (1) | RS50443B (de) |
| SI (1) | SI1200419T1 (de) |
| SK (1) | SK287069B6 (de) |
| TR (1) | TR200200125T2 (de) |
| TW (1) | TWI278453B (de) |
| UA (1) | UA73125C2 (de) |
| WO (1) | WO2001005776A1 (de) |
| ZA (1) | ZA200200221B (de) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002024200A1 (en) * | 2000-09-21 | 2002-03-28 | Bristol-Myers Squibb Company | Substituted azole derivatives as inhibitors of corticotropin releasing factor |
| WO2003015777A1 (en) * | 2001-08-13 | 2003-02-27 | Lion Bioscience Ag | Nr1h4 nuclear receptor binding compounds |
| AU2002334772A1 (en) | 2001-10-12 | 2003-04-28 | Duke University | Methods for image analysis of high-density synthetic dna microarrays |
| WO2005118810A1 (en) | 2004-06-03 | 2005-12-15 | Athlomics Pty Ltd | Agents and methods for diagnosing stress |
| ES2401138T3 (es) * | 2004-08-13 | 2013-04-17 | Genentech, Inc. | Inhibidores basados en tiazol de enzimas que usan ATP |
| EP1834641A1 (de) * | 2006-03-16 | 2007-09-19 | Sanofi-Aventis | CRF1-Rezeptorantagonisten zur Herstellung von Medikamenten zur Behandlung des Metabolischen Syndromes, und/oder Übergewicht und/oder Dyslipoproteinemie |
| HUP0900267A2 (en) * | 2009-04-30 | 2011-03-28 | Sanofi Aventis | Process for preparing of thiazole amines and intermediates thereof |
| JP2015516979A (ja) * | 2012-04-23 | 2015-06-18 | ホルスボールマッシュマイヤー ニューロケミー ゲーエムベーハーHolsboermaschmeyer Neurochemie Gmbh | Crh過剰活性を有する患者の治療に用いるためのcrhr1アンタゴニスト |
| GB201210686D0 (en) | 2012-06-15 | 2012-08-01 | Holsboermaschmeyer Neurochemie Gmbh | V1B receptor antagonist for use in the treatment of patients having an elevated AVP level and/or an elevated copeptin level |
| GB201310782D0 (en) | 2013-06-17 | 2013-07-31 | Max Planck Innovation Gmbh | Method for predicting a treatment response to a CRHR1 antagonist and/or V1B antagonist in a patient with depressive and/or anxiety symptoms |
| LT3096756T (lt) | 2014-01-21 | 2024-08-26 | Neurocrine Biosciences, Inc. | Crf1 receptoriaus antagonistai, skirti įgimtos antinksčių hiperplazijos gydymui |
| JP7532328B2 (ja) * | 2018-12-07 | 2024-08-13 | ニューロクライン バイオサイエンシーズ,インコーポレイテッド | 先天性副腎過形成を処置するためのcrf1受容体アンタゴニスト、その医薬製剤および固体形態 |
| FI3984523T3 (fi) | 2018-12-07 | 2025-10-21 | Neurocrine Biosciences Inc | Crf1-reseptorin antagonisti, lääkeformulaatioita ja niiden kiinteitä muotoja synnynnäisen lisämunuaisten liikakasvun hoitoon |
| CA3152590A1 (en) * | 2019-09-27 | 2021-04-01 | Evan Smith | Crf receptor antagonists and methods of use |
| CA3181084A1 (en) * | 2020-06-10 | 2021-12-16 | Robert H. FARBER | Crf1 receptor antagonist for the treatment of congenital adrenal hyperplasia |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2692893B1 (fr) * | 1992-06-24 | 1994-09-02 | Sanofi Elf | Dérivés alkylamino ramifiés du thiazole, leurs procédés de préparation et les compositions pharmaceutiques qui les contiennent. |
| FR2714059B1 (fr) | 1993-12-21 | 1996-03-08 | Sanofi Elf | Dérivés amino ramifiés du thiazole, leurs procédés de préparation et les compositions pharmaceutiques qui les contiennent. |
| FR2735777B1 (fr) | 1995-06-21 | 1997-09-12 | Sanofi Sa | Derives de 4-phenylaminothiazole, leur procede de preparation et les compositions pharmaceutiques les contenant |
| FR2754258B1 (fr) | 1996-10-08 | 1998-12-31 | Sanofi Sa | Derives d'aminothiazole, leur procede de preparation et les compositions pharmaceutiques les contenant |
-
1999
- 1999-07-15 FR FR9909144A patent/FR2796380B3/fr not_active Expired - Lifetime
-
2000
- 2000-07-11 CZ CZ20020137A patent/CZ300937B6/cs not_active IP Right Cessation
- 2000-07-11 HU HU0202387A patent/HU229069B1/hu not_active IP Right Cessation
- 2000-07-11 EP EP00951647A patent/EP1200419B1/de not_active Expired - Lifetime
- 2000-07-11 ME MEP-266/08A patent/MEP26608A/xx unknown
- 2000-07-11 AT AT00951647T patent/ATE264315T1/de active
- 2000-07-11 RS YUP-13/02A patent/RS50443B/sr unknown
- 2000-07-11 EA EA200101234A patent/EA005159B1/ru not_active IP Right Cessation
- 2000-07-11 SK SK38-2002A patent/SK287069B6/sk not_active IP Right Cessation
- 2000-07-11 BR BRPI0012478-8B1A patent/BR0012478B1/pt not_active IP Right Cessation
- 2000-07-11 CN CNB008116911A patent/CN1167692C/zh not_active Expired - Fee Related
- 2000-07-11 JP JP2001511437A patent/JP4949582B2/ja not_active Expired - Lifetime
- 2000-07-11 KR KR1020027000549A patent/KR100707320B1/ko not_active Expired - Fee Related
- 2000-07-11 NZ NZ516397A patent/NZ516397A/xx not_active IP Right Cessation
- 2000-07-11 US US10/031,038 patent/US6586456B1/en not_active Expired - Lifetime
- 2000-07-11 IL IL14728200A patent/IL147282A0/xx active IP Right Grant
- 2000-07-11 DK DK00951647T patent/DK1200419T3/da active
- 2000-07-11 SI SI200030377T patent/SI1200419T1/xx unknown
- 2000-07-11 DE DE60009911T patent/DE60009911T2/de not_active Expired - Lifetime
- 2000-07-11 EE EEP200200022A patent/EE05004B1/xx not_active IP Right Cessation
- 2000-07-11 HK HK02107310.6A patent/HK1045692B/zh not_active IP Right Cessation
- 2000-07-11 PT PT00951647T patent/PT1200419E/pt unknown
- 2000-07-11 ES ES00951647T patent/ES2220504T3/es not_active Expired - Lifetime
- 2000-07-11 PL PL352901A patent/PL206515B1/pl unknown
- 2000-07-11 MX MXPA02000545A patent/MXPA02000545A/es active IP Right Grant
- 2000-07-11 TR TR2002/00125T patent/TR200200125T2/xx unknown
- 2000-07-11 WO PCT/FR2000/001995 patent/WO2001005776A1/fr not_active Ceased
- 2000-07-11 CA CA002378792A patent/CA2378792C/en not_active Expired - Fee Related
- 2000-07-11 AU AU64521/00A patent/AU772683B2/en not_active Ceased
- 2000-07-13 CO CO00052852A patent/CO5200792A1/es active IP Right Grant
- 2000-07-14 AR ARP000103626A patent/AR024747A1/es not_active Application Discontinuation
- 2000-07-14 TW TW089114114A patent/TWI278453B/zh not_active IP Right Cessation
- 2000-11-07 UA UA2001129084A patent/UA73125C2/uk unknown
-
2001
- 2001-12-24 IL IL147282A patent/IL147282A/en not_active IP Right Cessation
-
2002
- 2002-01-03 IS IS6219A patent/IS2314B/is unknown
- 2002-01-10 ZA ZA200200221A patent/ZA200200221B/en unknown
- 2002-01-11 NO NO20020155A patent/NO324352B1/no not_active IP Right Cessation
-
2007
- 2007-04-25 US US11/740,001 patent/US8420679B2/en not_active Expired - Fee Related
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69314855T2 (de) | Verzweigte Alkylaminothiazolederivate, Verfahren zu ihrer Herstellung und pharmazeutische Zusammenstellungen die sie enthalten | |
| DE69712084T2 (de) | Aminothiazole derivate, ihre herstellung und sie enthaltenden pharmazeutischen zusammensetzungen | |
| DE60009911T2 (de) | Aminothiazolderivate und ihre verwendung als crf-rezeptor-liganden | |
| US5880135A (en) | Substituted 4-phenylaminothiazoles, their process of preparation and the pharmaceutical compositions containing them | |
| DE69409337T2 (de) | Verzweigte Aminothiazole-Derivate, Verfahren zu ihrer Herstellung und pharmazeutische Zusammenstellungen die sie enthalten | |
| DE69006907T2 (de) | Arylthiazolylimidazole als 5HT3-Antagonisten. | |
| DE60010126T2 (de) | Verzweigte substituierte aminoderivate vom 3-amino-1-phenyl-1h(1,2,4)triazol, verfahren zu deren herstellung und sie enthaltende pharmazeutische zusammensetzungen | |
| WO1994013642A1 (de) | Substituierte benzazepinone | |
| DE69406678T2 (de) | Heterozyclische amine mit zns-wirksamkeit | |
| DE3923675A1 (de) | Amonoalkylsubstituierte 2-aminothiazole und diese enthaltende therapeutische mittel | |
| EP0673371A1 (de) | Benzazepinon-derivate | |
| EP0477141B1 (de) | Heteroarylmethylbenzole-Aromatasehemmer | |
| DE69616357T2 (de) | Benzocycloalken-verbindungen mit bindungsaffinität an den melatonin-rezeptor, ihre herstellung und verwendung | |
| DE69203445T2 (de) | Neuro-schützende Mittel. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8327 | Change in the person/name/address of the patent owner |
Owner name: SANOFI-AVENTIS, PARIS, FR |
|
| 8364 | No opposition during term of opposition | ||
| R082 | Change of representative |
Ref document number: 1200419 Country of ref document: EP Representative=s name: TER MEER STEINMEISTER & PARTNER GBR PATENTANWA, DE |