WO2003015777A1 - Nr1h4 nuclear receptor binding compounds - Google Patents
Nr1h4 nuclear receptor binding compounds Download PDFInfo
- Publication number
- WO2003015777A1 WO2003015777A1 PCT/US2002/025438 US0225438W WO03015777A1 WO 2003015777 A1 WO2003015777 A1 WO 2003015777A1 US 0225438 W US0225438 W US 0225438W WO 03015777 A1 WO03015777 A1 WO 03015777A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- compound
- alkyl
- alkylphenyl
- phenyl
- Prior art date
Links
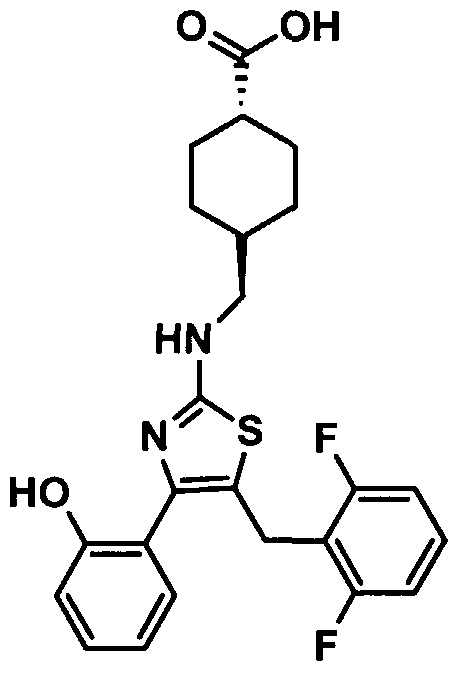
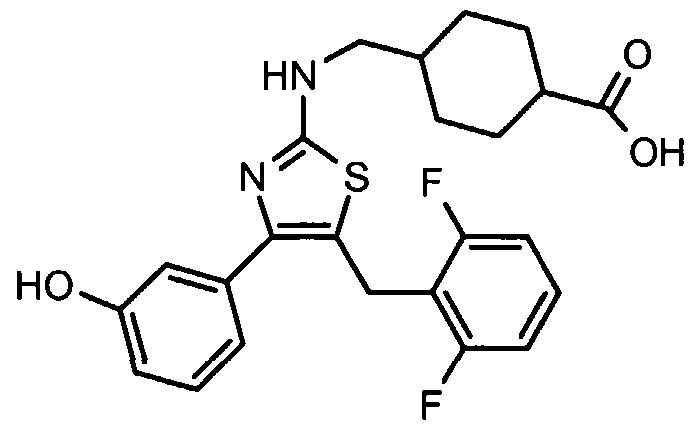
- MOJFYAFFYOWVQX-UHFFFAOYSA-N OC(C1CCC(CNc2nc(-c(cccc3)c3O)c(Cc(c(F)ccc3)c3F)[s]2)CC1)=O Chemical compound OC(C1CCC(CNc2nc(-c(cccc3)c3O)c(Cc(c(F)ccc3)c3F)[s]2)CC1)=O MOJFYAFFYOWVQX-UHFFFAOYSA-N 0.000 description 2
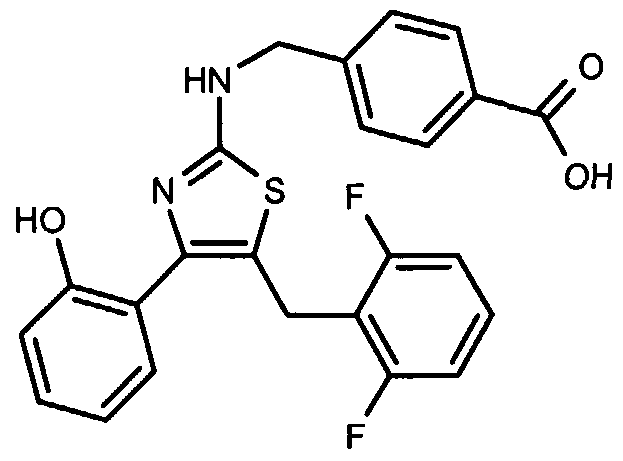
- SCQQRZGNOYUJMM-UHFFFAOYSA-N OC(c1ccc(CNc2nc(-c(cccc3)c3O)c(Cc(c(F)ccc3)c3F)[s]2)cc1)=O Chemical compound OC(c1ccc(CNc2nc(-c(cccc3)c3O)c(Cc(c(F)ccc3)c3F)[s]2)cc1)=O SCQQRZGNOYUJMM-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/42—Amino or imino radicals substituted by hydrocarbon or substituted hydrocarbon radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/50—Nitrogen atoms bound to hetero atoms
- C07D277/52—Nitrogen atoms bound to hetero atoms to sulfur atoms, e.g. sulfonamides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/11—Compounds covalently bound to a solid support
Definitions
- the present invention relates to compounds that bind to the NR1 H4 nuclear receptor and methods of treating diseases or pathological conditions influenced by NR1 H4.
- Multicellular organisms are dependent on advanced mechanisms of information transfer between cells and body compartments.
- the information that is transmitted can be highly complex and can result in the alteration of genetic programs involved in cellular differentiation, proliferation, or reproduction.
- the signals, or hormones are often simple molecules, such as peptides, fatty acid, or cholesterol derivatives.
- NR nuclear receptors
- Orphan receptors may be indicative of unknown signaling pathways in the cell or may be nuclear receptors that function without ligand activation. The activation of transcription by some of these orphan receptors may occur in the absence of an exogenous ligand and/or through signal transduction pathways originating from the cell surface (Mangelsdorf, D. J. et al., The nuclear receptor superfamily: the second decade, Cell 83, 835-839, 1995).
- Mangelsdorf, D. J. et al. The nuclear receptor superfamily: the second decade, Cell 83, 835-839, 1995.
- three functional domains have been defined in NRs. An amino terminal domain is believed to have some regulatory function.
- a DNA-binding domain hereinafter referred to as “DBD” usually comprises two zinc finger elements and recognizes a specific Hormone Responsive Element (hereinafter referred to as "HRE") within the promoters of responsive genes. Specific amino acid residues in the “DBD” have been shown to confer DNA sequence binding specificity (Schena, M. & Yamamoto, K.R., Mammalian Glucocorticoid Receptor Derivatives Enhance Transcription in Yeast, Science, 241 :965-967, 1988).
- a ligand-binding-domain (hereinafter referred to as "LBD”) is at the carboxy-terminal region of known NRs.
- Coactivators or transcriptional activators are proposed to bridge between sequence specific transcription factors, the basal transcription machinery, and in addition, to influence the chromatin structure of a target cell.
- proteins like SRC-1 , ACTR, and Gripl interact with NRs in a ligand enhanced manner (Heery et al., A signature motif in transcriptional coactivators mediates binding to nuclear receptors, Nature, 387, 733-736; Heinzel et al., A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression, Nature 387, 43-47, 1997).
- the physical interaction with negative receptor-interacting proteins or corepressors has been demonstrated (Xu et al., Coactivator and Corepressor complexes in nuclear receptor function, Curr Opin Genet Dev, 9 (2), 140-147, 1999).
- Nuclear receptor modulators like steroid hormones affect the growth and function of specific cells by binding to intracellular receptors and forming nuclear receptor-ligand complexes. Nuclear receptor-hormone complexes then interact with a HRE in the control region of specific genes and alter specific gene expression.
- the Farnesoid X Receptor alpha (hereinafter to as "FXR” and also often referred to as "NR1 H4" when referring to the human receptor) is a prototypical type 2 nuclear receptor which activates genes upon binding to promoter region of target genes in a heterodimeric fashion with Retinoid X Receptor (hereinafter referred to as "RXR”) (Forman et al., Cell, 81 , 687-93, 1995).
- NR1 H4 The relevant physiological ligands of NR1 H4 seem to be bile acids (Makishima et al., Science, 284, 1362-65, 1999; Parks et al., Science, 284, 1365-68, 1999). The most potent is chenodeoxycholic acid, which regulates the expression of several genes that participate in bile acid homeostasis. Farnesoid, originally described to activate the rat ortholog at high concentration does not activate the human or mouse receptor. FXR is expressed in the liver, small intestine, colon, ovary, adrenal gland and kidney. Like FXR ⁇ , NR1 H4 is involved in intrachne signaling.
- FXR is proposed to be a nuclear bile acid sensor. As a result, it modulates both, the synthetic output of bile acids in the liver and their recycling in the intestine (by regulating bile acid binding proteins). Upon activation (e.g. binding of chenodeoxycholic acid) it influences the conversion of dietary cholesterol into bile acids by inhibiting the transcription of key genes which are involved in bile acid synthesis such as CYP7A1. This seems to be a major mechanism of feedback regulation onto bile acid synthesis.
- the synthetic compounds, 1 ,1-bisphosphonate esters appear to display a number of similar activities to the two identified prototypes of natural FXR agonists, farnesol, and chenodeoxycholc acid.
- the 1 ,1- bisphosphonate esters increase the rate of 3-Hydroxy-3-methylglutaryl-CoA (HMG- CoA) reductase degradation and like bile acids they induce the expression of the intestinal bile acid binding protein (hereinafter referred to as "l-BABP”) and repress the cholesterol 7 ⁇ -hydroxylase gene.
- HMG- CoA 3-Hydroxy-3-methylglutaryl-CoA reductase degradation
- l-BABP intestinal bile acid binding protein
- Certain 1 ,1-bisphosphonate esters also bind to FXR (Niesor et al., Curr Pharm Des,7(4):231-59, 2001). That means that activation of FXR could lead to opposing effects (lowering the rate of cholesterol synthesis by increasing degradation of HMG-CoA reductase and increasing the cholesterol pool by inhibition of cholesterol degradation into bile acids).
- the FXR agonist does not change cholesterol and lipoprotein levels significantly in patients, although a repression of bile acid synthesis as well as a decreased HMG-CoA reductase activity was observed (Einarsson et al., Hepatology, 33(5), 1189-93, 2001 ) confirming that cellular cholesterol synthesis and degradation are controlled by numerous regulatory loops including the coordinate regulation of HMGCoA reductase and cholesterol 7 ⁇ -hydroxylase and that compounds modulating FXR acitvity might have different effects on blood lipid parameters.
- NR1 H4 receptor shows utility for treating diseases or conditions which are due to or influenced by this nuclear receptor (Maloney at al., J Med Chem, 10; 43(16): 2971-2974, 2000).
- the present invention provides, inter alia, novel NR1 H4 nuclear receptor protein binding compounds according to the general formula (I) shown below. These compounds are also binders of mammalian homologues of the receptor. Further the object of the invention is solved by providing for, amongst the NR1 H4 nuclear receptor protein binding, compounds according to the general formula (I) which act as agonists and compounds which act as antagonists of the human FXR receptor or a mammalian homologue thereof.
- the invention provides for FXR agonists which may be used for the treatment of cholesterol associated conditions or diseases.
- cholesterol lowering or cholestatic compounds are disclosed.
- the compounds according to the invention may be used for manufacture of antitumor medicaments and/or for the treatment of diseases such as cancer.
- FIGs. 1A and 1 B show the synthesis of the compounds of the invention described in Example 2.
- Fig. 2 shows the synthesis of the compounds of the invention described in Example 3.
- Fig. 3 shows the synthesis of the compounds of the invention described in Example 4.
- Fig. 4A shows SEQ ID No. 1 which is a protein sequence of the FRX protein, a portion of which can be used for cloning.
- Fig. 4B shows SEQ ID NO. 2 which is the mRNA sequence encoding the FRX protein.
- Fig. 4C shows SEQ ID NO. 3 which shows the protein sequence of
- TIF2 (Ace. No.: XM 011633 RefSeq DB).
- Fig. 4D shows SEQ ID NO. 4 which is respective mRNA sequence corresponding to the TIF2 protein.
- Fig 5 A shows a dose response with LN6348 in the HEK293-FXR reporter assay
- Fig 5B shows a dose response with LN6316 in the HEK293-FXR reporter assay
- Fig 5C shows a dose response with LN6365 in the HEK293-FXR reporter assay
- Fig 5D shows a dose response with LN6322 in the HEK293-FXR reporter assay
- the invention provides for a compound including resolved diastereoisomers and enantiomers, and tautomers, pharmaceutical acceptable salts or solvates thereof (hereinafter also referred to as the "compounds according to the invention"), having the following formula (I):
- R T and R 2 are independently selected from the group consisting of Ci to C 8 alkyl, d to C 8 substituted alkyl, phenyl, substituted phenyl, C 7 to C 12 phenylalkyl, C 7 to C 12 substituted phenylalkyl, heterocyclic ring, substituted heterocyclic ring, heteroaryl, and substituted heteroaryl; and
- R 3 and R 4 are independently selected from the group consisting of hydrogen, d to C 8 alkyl, d to C 8 substituted alkyl, phenyl, substituted phenyl, C 7 to C 12 phenylalkyl, C 7 to C 12 substituted phenylalkyl, naphthyl, substituted naphthyl, d to C 8 alkanesulfonyl, to C 8 substituted alkanesulfonyl, benzenesulfonyl, substituted benzenesulfonyl, d to C 8 acyl, and d to C 8 substituted acyl; where R 3 and R 4 may be taken together with nitrogen to form a heterocycle or substituted heterocycle or a heteroaryl or substituted heteroaryl ring.
- R ⁇ and R 2 in formula (I) are independently selected from the group consisting of phenyl, substituted phenyl, C 7 to C 12 phenylalkyl, C 7 to C 12 substituted phenylalkyl, heteroaryl, and substituted heteroaryl;
- R 3 and R 4 are independently selected from the group consisting of hydrogen, d to C 8 alkyl, d to C 8 substituted alkyl, substituted phenyl, C 7 to C 12 phenylalkyl, C 7 to C 12 substituted phenylalkyl, substituted naphthyl, d to C 8 substituted alkanesulfonyl, and substituted benzenesulfonyl; where R 3 and R 4 may be taken together with nitrogen to form a heterocycle or a substituted heterocycle or a heteroaryl or a substituted heteroaryl ring.
- R T and R 2 are independently selected from the group consisting of phenyl, substituted phenyl, C 7 to C 12 phenylalkyl, C 7 to C 12 substituted phenylalkyl, heteroaryl, and substituted heteroaryl;
- R 3 and R 4 are independently selected from the group consisting of hydrogen, Ci to C 8 substituted alkyl, substituted phenyl, C 7 to C 12 substituted phenylalkyl, substituted naphthyl, d to C 8 substituted alkanesulfonyl, and substituted benzenesulfonyl, wherein at least one of the mentioned above groups is substituted with carboxylic acid functionality as shown in formula (II) below:
- R 3 and R t may be taken together with nitrogen to form a heterocycle or a substituted heterocycle or a heteroaryl or substituted heteroaryl ring, also substituted with carboxylic acid functionality.
- Ri and R 2 are independently selected from the group consisting of phenyl, substituted phenyl, C 7 to C 12 phenylalkyl, C 7 to C 12 substituted phenylalkyl, heteroaryl, and substituted heteroaryl;
- R 3 is hydrogen, d to C 8 alkyl, d to C 8 substituted alkyl, to C 8 acyl, d to C 8 substituted acyl, Ci to C 8 alkanesulfonyl, d to C 8 substituted alkanesulfonyl, benzenesulfonyl, and substituted benzenesulfonyl;
- R 4 is one of the following structural formulas:
- n is an integer from 0 to 3.
- R 1 and R 2 are independently selected from the group consisting of substituted phenyl, C 7 to C i2 substituted phenylalkyl, and substituted heteroaryl, where preferred substituents are taken from hydrogen, halogen, hydroxy or alkoxy groups;
- R 3 is hydrogen, Ci to C 8 alkyl, Ci to C 8 substituted alkyl, Ci to C 8 acyl, and Ci to C 8 substituted acyl; and
- R 4 is one of the structures set forth above.
- a particularly preferred compound which may act as agonist of NR1 H4 is shown in formula (III) below.
- the inventors have been able to demonstrate that the compound according to formula (III) has a low effective binding concentration at FXR with an EC 50 of 0.2 ⁇ M wherein the EC 50 reflects the half-maximal effective concentration, and which is higher than the EC 50 of 0.015 ⁇ M for the published FXR agonist GW4064 (B.Goodwin et al., Molecular Cell 6, 517-526, 2000).
- the compounds of the invention can also exist as solvates and hydrates. Thus, these compounds may crystallize with, for example, waters of hydration, or one, a number of, or any fraction thereof of molecules of the mother liquor solvent.
- the solvates and hydrates of such compounds are included within the scope of this invention.
- a solid carrier can be one or more substances which can also act as diluents, flavoring agents, solubilizers, lubricants, suspending agents, binders, or tablet disintegrating agents; it can also be an encapsulating material.
- the carrier is generally a finely divided solid, which is in a mixture with the finely divided active component.
- the active compound or combination of active compounds is mixed with the carrier having the necessary binding properties in suitable proportions and compacted in the shape and size desired.
- a low-melting wax such as a mixture of fatty acid glycerides and cocoa butter is first melted and the active ingredient or combination of active ingredients is dispersed therein by, for example, stirring. The molten homogeneous mixture is then poured into convenient-sized molds and allowed to cool and solidify.
- Powders and tablets preferably contain between about 5% to about
- Suitable carriers include, for example, magnesium carbonate, magnesium stearate, talc, lactose, sugar, pectin, dextrin, starch, tragacanth, methyl cellulose, sodium carboxymethyl cellulose, a low-melting wax, cocoa butter and the like.
- the pharmaceutical compositions can include the formulation of the active compound(s) with encapsulating material as a carrier providing a capsule in which the active component (with or without other carriers) is surrounded by a carrier, which is thus in association with it.
- a carrier which is thus in association with it.
- cachets are also included. Tablets, powders, cachets, and capsules can be used as solid dosage forms suitable for oral administration.
- Liquid pharmaceutical compositions include, for example, solutions suitable for oral or parenteral administration, or suspensions, and emulsions suitable for oral administration.
- Sterile water solutions of the active component or sterile solutions of the active component in solvents comprising water, ethanol, or propylene glycol are examples of liquid compositions suitable for parenteral administration.
- Sterile solutions can be prepared by dissolving the active component(s) in the desired solvent system, and then passing the resulting solution through a membrane filter to sterilize it or, alternatively, by dissolving the sterile compound in a previously sterilized solvent under sterile conditions.
- halogen refers to the fluoro, chloro, bromo or iodo atoms.
- halogen there can be one or more halogen, which are the same or different.
- Preferred halogens are chloro and fluoro.
- Ci to C 8 alkyl denotes such radicals as methyl, ethyl, n- propyl, isopropyl, n-butyl, iso-butyl, sec-butyl, tert-butyl, amyl, tert-amyl, hexyl , n- heptyl, 2-heptyl, 3-heptyl, 4-heptyl, 2-methyl-1 -hexyl, 2-methyl-2-hexyl, 2-methyl-3- hexyl, n-octyl ,and the like.
- Ci to C 8 substituted alkyl denotes that the above d to C 8 alkyl groups are substituted by one or more, and preferably one or two, halogen, hydroxy, protected hydroxy, oxo, protected oxo, C 3 to C 7 cycloalkyl, phenyl, naphthyl, amino, protected amino, monosubstituted amino, protected monosubstituted amino, disubstituted amino, guanidino, protected guanidino, heterocyclic ring, substituted heterocyclic ring, Ci to C 8 alkoxy, Ci to C 8 acyl, Ci to C 8 acyloxy, nitro, carboxy, protected carboxy, carbamoyl, carboxamide, protected carboxamide, N-(C ⁇ to C 6 alkyl)carboxamide, protected N-(C ⁇ to C 6 alkyl)carboxamide, N,N-di(C ⁇ to C 6 alkyl)carboxamide, cyan
- Examples of the above substituted alkyl groups include the 2-oxo- prop-1-yl, 3-oxo-but-1-yl, cyanomethyl, nitromethyl, chloromethyl, hydroxymethyl, tetrahydropyranyloxymethyl, trityloxymethyl, propionyloxymethyl, amino, methylamino, aminomethyl, dimethylamino, carboxymethyl, allyloxycarbonylmethyl, allyloxycarbonylaminomethyl, methoxymethyl, ethoxymethyl, t-butoxymethyl, acetoxymethyl, 4-carboxybutyl, 5-carboxypentyl, 6-carboxyhexyl, chloromethyl, bromomethyl, iodomethyl, trifluoromethyl, 6-hydroxyhexyl, 2,4-dichloro(n-butyl), 2- aminopropyl, 1-chloroethyl, 2-chloroethyl, 1- bromoethyl, 2-chloroethyl,
- substituted phenyl specifies a phenyl group substituted with one or more, and preferably one or two, moieties chosen from the groups consisting of halogen, hydroxy, protected hydroxy, cyano, nitro, d to C 8 alkyl, Ci to C 8 substituted alkyl, Ci to C 8 alkoxy, d to C 8 substituted alkoxy, Ci to C 8 acyl, Ci to C 8 substituted acyl, Ci to C 8 acyloxy, carboxy, protected carboxy, carboxymethyl, protected carboxymethyl, hydroxymethyl, protected hydroxymethyl, amino, protected amino, monosubstituted amino, protected monosubstituted amino, disubstituted amino, carboxamide, protected carboxamide, N-(C ⁇ to C 6 alkyl)carboxamide, protected N-(C ⁇ to C 6 alkyl)carboxamide, N, N-di(C ⁇ to C 6 alkyl)carboxamide, trifluoromethyl
- substituted phenyl includes a mono- or di(halo)phenyl group such as 2, 3 or 4-chlorophenyl, 2,6-difluorophenyl, 2,3- difluorophenyl, 2,6-dichlorophenyl, 2,5-dichlorophenyl, 3,4-dichlorophenyl, 2, 3 or 4- bromophenyl, 3,4-dibromophenyl, 3-chloro-4-fluorophenyl, 2, 3 or 4-fluorophenyl and the like; a mono or di(hydroxy)phenyl group such as 2, 3 or 4-hydroxyphenyl, 2,4-dihydroxyphenyl, the protected-hydroxy derivatives thereof and the like; a nitrophenyl group such as 2, 3 or 4-nitrophenyl; a cyanophenyl group, for example, 2, 3 or 4-cyanophenyl; a mono- or di(alkyl)phenyl group such as 2, 3 or 4-chlorophenyl,
- substituted phenyl represents disubstituted phenyl groups wherein the substituents are different, for example, 3- methyl-4-hydroxyphenyl, 3-chloro-4-hydroxyphenyl, 2-methoxy-4-bromophenyl, 4-ethyl-2-hydroxyphenyl, 3-hydroxy-4-nitrophenyl, 2-hydroxy 4-chlorophenyl, and the like.
- C 7 to d 2 phenylalkyl denotes a d to C 6 alkyl group substituted at any position by a phenyl, substituted phenyl, heteroaryl or substituted heteroaryl. Examples of such a group include benzyl, 2-phenylethyl, 3-phenyl(n- propyl), 4-phenylhexyl, 3-phenyl(n-amyl), 3-phenyl(sec-butyl), and the like.
- Preferred C 7 to C 12 phenylalkyl groups are the benzyl and the phenylethyl groups.
- C 7 to C ⁇ 2 substituted phenylalkyl denotes a C 7 to C ⁇ 2 phenylalkyl group substituted on the Ci to C 6 alkyl portion with one or more, and preferably one or two, groups chosen from halogen, hydroxy, protected hydroxy, oxo, protected oxo, amino, protected amino, monosubstituted amino, protected monosubstituted amino, disubstituted amino, guanidino, protected guanidino, heterocyclic ring, substituted heterocyclic ring, d to C 8 alkyl, d to C 8 substituted alkyl, Ci to C 8 alkoxy, Ci to C 8 substituted alkoxy, C 1 to C 8 acyl, Ci to C 8 substituted acyl, Ci to C 8 acyloxy, nitro, carboxy, protected carboxy, carbamoyl, carboxamide, protected carboxamide, N-(C ⁇ to C 6 alkyl)carboxamide,
- C 7 to C ⁇ 2 substituted phenylalkyl examples include groups such as 2-hydroxyphenylmethyl, 3-hydroxyphenylmethyl, 2- methoxyphenylmethyl, 3-methoxyphenylmethyl, 2,6-difluorophenylmethyl, 2,3- difluorophenylmethyl, 2,6-dichlorophenylmethyl, 2,3-dichlorophenylmethyl, 3,5- dichlorophenylmethyl, 2-hydroxyphenylethyl, 3-hydroxyphenylethyl, 2- methoxyphenylethyl, 3-methoxyphenylethyl, 2,6-difluorophenylethyl, 2,3- difluorophenylethyl, 2,6-dichlorophenylethyl, 2,3-dichlorophenylethyl, 3,5- dichlorophenylmethyl 2-phenyl-1-chloroethyl, 2-(4-methoxyphenyl)ethyl
- heterocycle or “heterocyclic ring” denotes optionally substituted five-membered to eight-membered rings that have 1 to 4 heteroatoms, such as oxygen, sulfur and/or nitrogen, in particular nitrogen, either alone or in conjunction with sulfur or oxygen ring atoms.
- heteroatoms such as oxygen, sulfur and/or nitrogen, in particular nitrogen, either alone or in conjunction with sulfur or oxygen ring atoms.
- These five-membered to eight- membered rings may be saturated, fully unsaturated or partially unsaturated, with fully saturated rings being preferred.
- Preferred heterocyclic rings include morpholino, piperidinyl, piperazinyl, 2-amino-imidazoyl, tetrahydrofurano, pyrrolo, tetrahydrothiophen-yl, hexamethyleneimino and heptamethyleneimino.
- substituted heterocycle or "substituted heterocyclic ring” means the above-described heterocyclic ring is substituted with, for example, one or more, and preferably one or two, substituents which are the same or different which substituents can be halogen, hydroxy, protected hydroxy, cyano, nitro, Ci to C 8 alkyl, Ci to C 8 alkoxy, Ci to C 8 substituted alkoxy, Ci to C 8 acyl, d to C 8 acyloxy, carboxy, protected carboxy, carboxymethyl, protected carboxymethyl, hydroxymethyl, protected hydroxymethyl, amino, protected amino, (monosubstituted)amino, protected (monosubstituted)amino, (disubstituted)amino carboxamide, protected carboxamide, N-(d to C 12 alkyl)carboxamide, protected N-(C ⁇ to C 6 alkyl)carboxamide, N, N-di(C ⁇ to
- heteroaryl means a heterocyclic aromatic derivative which is a five-membered or six-membered ring system having from 1 to 4 heteroatoms, such as oxygen, sulfur and/or nitrogen, in particular nitrogen, either alone or in conjunction with sulfur or oxygen ring atoms.
- heteroaryls include pyridinyl, pyrimidinyl, and pyrazinyl, pyridazinyl, pyrrolo, furano, thiopheno, oxazolo, isoxazolo, phthalimido, thiazolo, and the like.
- substituted heteroaryl means the above-described heteroaryl is substituted with, for example, one or more, and preferably one or two, substituents which are the same or different which substituents can be halogen, hydroxy, protected hydroxy, cyano, nitro, Ci to C 8 alkyl, Ci to C 8 alkoxy, Ci to C 8 substituted alkoxy, Ci to C 8 acyl, Ci to C 8 substituted acyl, C 1 to C 8 acyloxy, carboxy, protected carboxy, carboxymethyl, protected carboxymethyl, hydroxymethyl, protected hydroxymethyl, amino, protected amino, (monosubstituted)amino, protected (monosubstituted)amino, (disubstituted)amino, carboxamide, protected carboxamide, N-(d to C 6 alkyl)carboxamide, protected N-(C 1 to C 6 alkyl)carboxamide, N, N-di(C ⁇ to C 6 alkyl
- substituted naphthyl specifies a naphthyl group substituted with one or more, and preferably one or two, moieties either on the same ring or on different rings chosen from the groups consisting of halogen, hydroxy, protected hydroxy, cyano, nitro, Ci to C 8 alkyl, Ci to C 8 alkoxy, Ci to C 8 acyl, Ci to C 8 acyloxy, carboxy, protected carboxy, carboxymethyl, protected carboxymethyl, hydroxymethyl, protected hydroxymethyl, amino, protected amino, monosubstituted amino, protected monosubstituted amino, disubstituted amino, carboxamide, protected carboxamide, N-(C ⁇ to C 6 alkyl)carboxamide, protected N-(C ⁇ to C 6 alkyl)carboxamide, N, N-di(C ⁇ to C 6 alkyl)carboxamide, trifluoromethyl, N-((C ⁇ to C 6 alkyl)sulfonyl
- substituted naphthyl includes a mono or di(halo)naphthyl group such as 1 , 2, 3, 4, 5, 6, 7 or 8-chloronaphthyl, 2, 6- dichloronaphthyl, 2, 5-dichloronaphthyl, 3, 4-dichloronaphthyl, 1 , 2, 3, 4, 5, 6, 7 or 8- bromonaphthyl, 3, 4-dibromonaphthyl, 3-chloro-4-fluoronaphthyl, 1 , 2, 3, 4, 5, 6, 7 or 8-fluoronaphthyl and the like; a mono or di(hydroxy)naphthyl group such as 1 , 2, 3, 4,
- a nitronaphthyl group such as 3- or 4-nitronaphthyl
- a cyanonaphthyl group for example, 1 , 2, 3, 4, 5, 6, 7 or 8-cyanonaphthyl
- a mono- or di(alkyl)naphthyl group such as 2, 3, 4, 5, 6, 7 or 8-methylnaphthyl, I, 2, 4-dimethylnaphthyl, I, 2, 3, 4, 5, 6, 7 or 8-(isopropyl)naphthyl, I, 2, 3, 4, 5, 6, 7 or 8-ethylnaphthyl, I, 2, 3, 4, 5, 6, 7 or 8-(n-propyl)naphthyl and the like
- a mono or di(alkoxy)naphthyl group for example
- substituted naphthyl represents disubstituted naphthyl groups wherein the substituents are different, for example, 3- methyl-4-hydroxynaphth-1 -yl, 3-chloro-4-hydroxynaphth-2-yl, 2-methoxy-4- bromonaphth-1-yl, 4-ethyl-2-hydroxynaphth-1-yl, 3-hydroxy-4-nitronaphth-2-yl, 2- hydroxy-4-chloronaphth-1-yl, 2-methoxy-7-bromonaphth-1-yl, 4-ethyl-5- hydroxynaphth-2-yl, 3-hydroxy-8-nitronaphth-2-yl, 2-hydroxy-5-chloronaphth-1-yl and the like.
- R 3 and R 4 may be taken together with nitrogen to form a heterocycle or substituted heterocycle of the following kind aziridine, azetidine, pyrrolidine, 3-methylpyrrolidine, 3-aminopyrrolidine, 3-hydroxypyrrolidine, pyrazolidine, imidazolidine, piperidine, 2-methylpiperidine, 4-carboxypiperidine, 4- (carboxymethyl)piperidine, piperazine, morpholine, azepine, and tetrahydroisoquinoline.
- C to C 8 acyl encompasses groups such as formyl, acetyl, propionyl, butyryl, pentanoyl, pivaloyl, hexanoyl, heptanoyl, benzoyl and the like. Preferred acyl groups are acetyl and benzoyl.
- Ci to C 8 substituted acyl denotes the acyl group substituted by one or more, and preferably one or two, halogen, hydroxy, protected hydroxy, oxo, protected oxo, cyclohexyl, naphthyl, amino, protected amino, monosubstituted amino, protected monosubstituted amino, disubstituted amino, guanidino, heterocyclic ring, substituted heterocyclic ring, imidazolyl, indolyl, pyrrolidinyl, Ci to C 8 alkoxy, Ci to C 8 acyl, Ci to C 8 acyloxy, nitro, Ci to C 8 alkyl ester, carboxy, protected carboxy, carbamoyl, carboxamide, protected carboxamide, N-(C ⁇ to C 6 alkyl)carboxamide, protected N-(C ⁇ to C ⁇ alkyl)carboxamide, N,N-di(C ⁇ to C 6 alkyl
- Ci to C 8 substituted acyl groups include 4-phenylbutyroyl
- Ci to C 8 alkoxy denotes groups such as methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, t-butoxy and like groups. A preferred alkoxy is methoxy.
- Ci to C 8 substituted alkoxy means the alkyl portion of the alkoxy can be substituted in the same manner as in relation to Ci to C 8 substituted alkyl.
- Ci to C 8 substituted aminoacyl denotes the acyl group substituted by one or more, and preferably one or two, halogen, hydroxy, protected hydroxy, oxo, protected oxo, cyclohexyl, naphthyl, amino, protected amino, monosubstituted amino, protected monosubstituted amino, disubstituted amino, guanidino, heterocyclic ring, substituted heterocyclic ring, imidazolyl, indolyl, pyrrolidinyl, Ci to C 8 alkoxy, Ci to C 8 acyl, Ci to C 8 acyloxy, nitro, Ci to C 8 alkyl ester, carboxy, protected carboxy, carbamoyl, carboxamide, protected carboxamide, N-(C ⁇ to C 6 alkyl)carboxamide, protected N-(C ⁇ to C 6 alkyl)carboxamide, N,N-di(C ⁇ to C 6 alkyl)
- compositions comprising an effective amount of a compound according to the invention.
- Such compositions can be administered by various routes, for example oral, rectal, subcutaneous, intramuscular, intravenous or intracerebral.
- the preferred route of administration would be oral at daily doses of the compound for adult human treatment of about 0.01-5000 mg, preferably 1-1500 mg per day.
- the appropriate dose may be administered in a single dose or as divided doses presented at appropriate intervals for example as two, three four or more subdoses per day.
- inert, pharmaceutically acceptable carriers are used.
- the pharmaceutical carrier can be either solid or liquid. Solid form preparations include, for example, powders, tablets, dispersible granules, capsules, cachets, and suppositories.
- the invention relates to compounds as described above wherein the compounds are capable of binding the NR1 H4 receptor protein or a portion thereof according to SEQ ID NOS 1-4 shown in Figs. 4A-4D, respectively, or a mammalian homologue thereof.
- the claimed compound can bind to the NR1 H4 receptor protein or a portion thereof in a mixture comprising 10-200 ng of NR1 H4 receptor protein or a portion thereof, preferably the ligand binding domain, 20 mM Tris /HCI at pH 7.9; 60 mM KCl; 5 mM MgCI 2 ; 160ng/ ⁇ l BSA in a total volume of preferably about 25 ⁇ l.
- SEQ ID NO. 1 shown in Fig. 4A as used herein is a protein that performs substantially the same function as NR1 H4 does in humans and shares at least 40% sequence identity at the amino acid level, preferably 50% sequence identity at the amino acid level more preferably 65% sequence identity at the amino acid level, even more preferably 75% sequence identity at the amino acid level and most preferably over 85% sequence identity at the amino acid level.
- the invention in particular concerns a method for prevention or treatment of a NR1 H4 receptor protein- or NR1 H4 receptor protein homologue- mediated disease or condition in a mammal comprising administration of a therapeutically effective amount of a compound according to the invention wherein the prevention or treatment is directly or indirectly accomplished through the binding of a compound according to the invention to the NR1 H4 receptor protein or to the NR1 H4 receptor protein homologue.
- the term mediated herein means that the physiological pathway in which the NR1 H4 receptor protein acts is either directly or indirectly involved in the disease or condition to be treated or prevented. In the case where it is indirectly involved it could be that, e.g.
- modulating the activity of NR1 H4 by a compound according to the invention influences a parameter which has a beneficial effect on a disease or a condition.
- modulation of NR1 H4 activity leads to decreased levels of serum cholesterol or certain lipoproteins, which, in turn, have a beneficial effect on the prevention and treatment of artherosclerosis.
- a condition is a physiological or phenotypic state which is desirably altered.
- One such example would be obesity which is not necessarily medically harmful but nonetheless a non desirable phenotypic condition.
- the method for prevention or treatment of a NR1 H4 receptor protein mediated disease or condition is applied to a human. This may be male or female.
- compositions generally are administered in an amount effective for treatment or prophylaxis of a specific condition or conditions. Initial dosing in a human is accompanied by clinical monitoring of symptoms, such symptoms being determined for the selected condition.
- the compositions are administered in an amount of active agent of at least about 100 ⁇ g/kg body weight. In most cases they will be administered in one or more doses in an amount not in excess of about 20 mg/kg body weight per day. Preferably, in most cases, the dose is from about 100 ⁇ g/kg to about 5 mg/kg body weight, daily.
- the daily dosage level of active agent will be 0.1 mg/kg to 10 mg/kg and typically around 1 mg/kg.
- terapéuticaally effective amount is meant a symptom- alleviating or symptom -reducing amount, a cholesterol-reducing amount, a fatty acid absorption blocking amount, a protein and/or carbohydrate digestion-blocking amount and/or a de novo cholesterol biosynthesis-blocking amount of a compound according to the invention.
- blocking means either total blockage or partial blockage.
- FXR is proposed to be a bile acid sensor. As a result, it modulates both the synthetic output of bile acids in the liver and their recycling in the intestine, by regulating bile acid binding proteins.
- the invention concerns a method for regulating bile transport in a mammal, in a preferred embodiment a human, which comprises activating the NR1 H4 receptor with a therapeutically effective amount of a compound according to the invention.
- the invention concerns a method of treating in mammal a disease which is affected by cholesterol, triglyceride, or bile acid levels comprising administering to a mammal in need of such treatment a therapeutically effective amount of a compound according to the invention.
- the compounds according to the invention may also be used as a method of prevention or treatment of mammalian atherosclerosis, gallstone disease, lipid disorders, obesity or cardiovascular disorders such as coronary heart disease or stroke.
- the invention further concerns a method of blocking fatty acid absorption in the intestine of a mammal comprising administering to the mammal a therapeutically effective amount of a compound according to the invention.
- the invention may also be used to treat obesity in a mammal, particularly in humans.
- FXR alpha is a prototypical type 2 nuclear receptor which activates genes upon binding to the promoter region of target genes in a heterodimeric fashion with RXR.
- the relevant physiological ligands of NR1 H4 are bile acids.
- the present compounds according to the invention have been demonstrated to have high binding efficacy binding coefficients measured as IC50 in the range 400 nM to 1000 nM as well as agonistic and/or antagonistic properties. Consequently they may be applied to regulate genes that participate in bile acid homeostasis as well as other downstream regulated genes.
- genes are, but are not limited to, genes encoding proteins or factors involved directly or indirectly in lipid absorption, cholesterol biosynthesis, cholesterol transport or binding, bile acid transport or binding, proteolysis, amino acid metabolism, glucose biosynthesis, protein translation, electron transport, and hepatic fatty acid metabolism.
- FXR often functions in vivo as a heterodimer with the RXR.
- Published FXR agonists such as the Glaxo SmithKline compound "GW 4064" are known to influence the regulation of various liver genes. Examples of known agonists are showin in Table 1 below.
- Genes found to be regulated by GW 4064 are genes down-regulated in the liver, genes up-regulated in the liver and genes having altered expression in the intestine.
- Genes down-regulated in the liver include apolipoprotein B; plasma proteinase inhibitor alpha-1-inhibitor III group 3(m22360); L-glucono-gamma-lactone oxidase (d 12754); peroxisomal enoyl-CoA:hydrotase-3-hydroxyacyl-CoA bifunctional enzyme (k03249); liver fatty acid binding protein (L-FABP, m13501), CYP4A2 (m57719), CYP3A23 (x96721 ) and CYP3A1 (x64401 ); Cholesterol-7-alpha- hydroxylase, CYP7A1 (RefSeq NM000780, XM 005022, XM 044651 , and XM 044652); and sodium-taurocholate cotransport protein, ntcp (RefSeq NM003049, XM007466).
- Genes that up-regulate in the liver include small heterodimer partner homolog (d86580); bile salt export pump, bsep (RefSeq NM 003742, XM 003644, and XM 033122); phospholipid transfer protein, PLTP (RefSeq NM 006227, XM 009490, XM 029929, and XM 029930); amithine palmitoyltransferase II, CPTII (RefSeq NM 000098, XM 001758, XM 038866, and XM 038867); phenylethanolamine-N-methyltransferase, PNMT (RefSeq NM 002686, XM 008597, andXM 049837); insulin-induced growth-response protein CL-6 (113619); elongation factor 2, EF-2 (y07504); mouse cornichon;
- pancreatic lipase (d88534); colipase (m58370); pancreatic phospholipase A-2 (d00036); pancreatic amylase (m24962); carboxypeptidase A1 (m23986); carboxypeptidase A2 (m23721 ): carboxypeptidase B (m23959): pancreatic trypsin I Q00778): pancreatic cationic trypsinogen (m16624); pancreatic trypsinogen II (v01274); elastase I (v01234, 100112); elastase II (100118, 100124); l-BABP (I22788); intestinal fatty acid binding protein (FABP, k01180); hepatic squalenesynthetase (m95591 ); protein kinase C receptor (u003390): longation factor 2, EF-2 (y07504) and small heterodimer
- the invention also concerns a method of modulating a gene whose expression is regulated by the NR1 H4 receptor in a mammal comprising administration of a therapeutically effective amount of a compound according to the invention to the mammal.
- the orphan receptor FXR can bind the response element of the SHP gene as a heterodimer with RXR (9-cis retinoic acid receptor) and the SHP-protein, in turn, prevents efficient transcription from the cyp7a1 promoter (Lu et al., Mol Cell, 6(3):505-17; Goodwin et al. Mol Cell, 6(3), 717-26, 2000).
- NTCP sodium/bile acid cotransporter gene
- NTCP a membrane transport protein which is required for the import of conjugated bile acids in the hepatocyte
- the gene for the bile salt export pump, a membrane transporter responsible for the secretion of bile acids into the gall is directly activated by FXR (Ananthanarayanan et al., J Biol Chem, 3;276(31):28857- 28865, 2001).
- the invention likewise concerns a method for lowering the expression of cholesterol 7-alpha-hydroxylase and NTCP and increasing expression of BSEP in parallel by use of the compounds according to the invention.
- the invention concerns a method for enhancing the expression of the Intestinal Bile Acid Binding Protein (l-BABP) (Grober et al., J Biol Chem, 15;274(42):29749-54, 1999 and/or the activity of the canicular bile salt excretion pump.
- l-BABP Intestinal Bile Acid Binding Protein
- the compounds according to the invention may be used as medicaments, in particular for the prevention or treatment of a NR1 H4 receptor protein- or NR1 H4 receptor protein homologue- mediated disease or condition in a mammal wherein the prevention or treatment is directly or indirectly accomplished through the binding of the compound according to the invention to the NR1 H4 receptor protein or NR1H4 receptor protein homologue.
- These pharmaceutical compositions contain 0.1% to 99.5% of the compound according to the invention, more particularly 0.5% to 90% of the compound according to the invention in combination with a pharmaceutically acceptable carrier.
- the invention also concerns the use of a compound or combination of compounds according to the invention for the prevention or treatment of a NR1 H4 receptor protein-mediated disease or condition wherein the mammal described above is a human.
- the medicament may be used for regulating the bile transport system in a mammal, preferentially a human, by activating the NR1 H4 receptor, for regulating levels of cholesterol, triglyceride, and/or bile acid.
- the medicament may be used for the treatment of atherosclerosis, gallstone disease, lipid disorders, obesity or a cardiovascular disorder.
- the invention further concerns the use of a compound or combination of compounds according to the invention for blocking in a mammal, preferentially a human, fatty acid absorption in the intestine. Further the compounds of the invention may be used for treating obesity in humans and for modulating a gene whose expression is regulated by the NR1 H4 receptor (see details above and figures).
- the invention also further concerns the use of a compound or combination of compounds as antitumor medicaments. The antitumor effects of such medicaments could be exerted by selective inhibition of cell proliferation and induction of apotptosis of tumor cells in a way similar to described activities for certain bisphosphonates (Alberts DS, et al., Clin Cancer Res 2001 May;7(5): 1246-50).
- This construct was used to express a recombinant GST-FXR fusion protein in E.coli (BL21 strain).
- a pDEST 17 derivative clone harboring an additional sequence encoding amino acids 548-878 of human TIF2 (Ace. No: XM_011633 RefSeq) was constructed using GatewayTM recombination technology (Invitrogen, USA) in order to obtain a construct which was used to express recombinant His-tagged TIF2 fragment in E. coli.
- GatewayTM recombination technology Invitrogen, USA
- coli BL21 (Invitrogen, USA) and cells were grown to an OD600 of 0.4-0.7 before expression was induced by addition of 0.5 mM IPTG according instructions of the manufacturer (Invitrogen). After induction for 8 hours at 30°C, the cells were harvested by centrifugation for 10 minutes at 5000 x g. Fusion proteins were affinity purified using Glutathion sepharose (Pharmacia) or Ni-NTA Agarose (QIAGEN) according to the instructions of the respective manufacturer. Recombinant proteins were dialyzed against 20 mM Tris/HCL pH 7.9; 60 mM KCl; 5 mM MgCI 2 ; 1 mM DTT, 0.2 mM PMSF; 10% glycerol.
- the TIF2 fragment was subsequently biotinylated by addition of 40-120 ⁇ l of a biotinamidocaproate N-hydroxysuccinimide-ester (Sigma) solution (20 mg/ml in DMSO). Overhead rotating samples were incubated for 2 hours at room temperature. Unincorporated label was then separated using G25 Gel filtration chromatography (Pharmacia Biotech, Sweden). Protein containing fractions from the column were pooled and tested for activity in the assay as described below.
- a biotinamidocaproate N-hydroxysuccinimide-ester Sigma
- Perkin Elmer LANCE technology For screening of the compound libraries as provided for by the methods described in Examples 2, 3 and 4 below for substances which influence the FXR/Tif 2 interaction, Perkin Elmer LANCE technology was used. This technoligy relies on the binding dependent energy transfer from a donor to an acceptor fluorophore attached to the binding partners of interest. For ease of handling and reduction of background from compound fluorescence, LANCE technology makes use of generic fluorophore labels and time resoved detection.
- the LANCE signal was detected by a Perkin Elmer VICTOR2VTM
- Argogel-MB-CHO resin (Argonaut Technologies Inc.) (100 mg each tea-bag [Houghten, U.S. Patent No. 4,631 ,211], 0.41 mmol/g substitution) (compound 4) was swollen in 1% acetic acid (AcOH) in DMF (by volume). The amine (10 eq.) was added and the bottle(s) placed on a shaker for 30 min. Solid NaBH 3 CN (20 eq) was added and the reaction bottle(s) placed on a shaker at room temperature for 18 hrs to form resin aminated Argogel-MB-HCO resin (5). The resin was washed as follows: DMF (4x), MeOH (4x), CH 2 CI 2 (2x) and then allowed to air dry. For the amines that were hydrochloride salts, 1 eq of Et 3 N was added.
- Step 5 Preparation of a resin-bound thiourea (compound 7) [0099] A 0.2 M solution of Fmoc-NCS (5 eq) in anhydrous CH 2 CI 2 was added to a bottle containing the aminated Argogel-MB-HCO resin (resin compound 5). The bottle was placed on a shaker for one hour to form resin compound 6. The resin was washed with CH 2 CI 2 (3x) and DMF (3x) and subsequently reacted with 20 % piperidine in DMF (5 eq) for one hour to produce a resin-bound thiourea (resin compound 7). The resin was then washed with DMF (3x) and MeOH (3x) and used directly in the next step.
- the resin was placed a reaction bottle and the resin was swollen in MeOH and NaHC0 3 (10 eq) was added to the solution.
- R 3 in compound 8 in Fig. 1 B is -(CH 2 -CH 2 ) n -CH 2 -C0 2 CH 3 where n is an integer from 0 to 8, preferably 1 to 6 and more preferably 1 to 4.
- the bottle was placed on a shaker for 24 hrs.
- Step 1 Immobilization of a carboxylic acid (compound 11 ) on Bromo-Wang resin
- Resin compound 12 was placed in a bottle and the resin compound was swollen in 20 % piperidine in DMF (5 eq). Tea-bags were placed in the bottle and the bottle was placed on a shaker at room temperature for one hour. Then the resin was washed with DMF (3x), MeOH (3x), CH 2 CI 2 (3x) to form resin compound 13. Then a 0.1 M solution of Fmoc-NCS (5 eq) in anhydrous CH 2 CI 2 was applied to resin compound 13 in the bottle and the bottle was placed on a shaker for one hour.
- the resin was then washed with CH 2 CI 2 (3x) and DMF (3x) and subsequently reacted again with 20 % piperidine in DMF (5 eq) for one hour to produce the resin-bound thiourea (compound 15).
- the resin was then washed with DMF (3x) and MeOH (3x) and used directly in the next step.
- Step 4 Cleavage of 2-aminothiazole to form products 10 from resin.
- Tables 3, 4, 5 and 6 illustrate the preferred compounds according to the invention that can mediate transactivation of FXR mediated transcription in a HEK293 reporter cell line.
- the Table provides the EC 50 values (EC50 AGV) as established as well as their respective average efficacy (% activity relative to CDCA control agonist).
- Stable HEK293FXR reporter cell lines were generated by stably transfecting with the pTRexDest30 (Invitrogen) derivatives pTRexDest30-hFXR, pTRexDest30-hRXR and the pGL2promoter (Promega) derivative pGL2promoter- FXRRE.
- the full length human FXR (accession U68233) and the full length human RXR ⁇ (accession P19793) were cloned into the pTRexDest30 applying the manufacturer protocols for the GatewayTM system (Invitrogen).
- the FXR response elements were cloned (upper case and undrelined). 5' - cccaGGGTGAaTAACCTcq ⁇ qctct ⁇ tccctccaatcccaGGGTGAaTAACCTcqqg 3' (SEQ ID NO. 5) was created from the human IBAB-P promoter (Grober et al 1999, JBC 274, pp. 29749-29754). A stable clone was selected and seeded at a density of 1x10 4 cells per well in 96 well plates.
- Luciferase reporter activity was measured in triplicates from extracts of cells after incubating cells in culture medium (DMEM [Gibco-BRL] + 10% FCS [PAA laboratories]) for 16 hours (5% C0 2 , 37°C) containing 0.5% DMSO (control) or 0.5% DMSO with increasing concentrations of compounds.
- Examples of such dose response assays in the HEK293-FXR cell line are shown in Fig. 5A for LN6348, in Fig 5B for LN6316, in Fig.5C for LN6365 and in Fig. 5D for LN6322.
Abstract
Description
















Claims












Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP02750474A EP1423113A4 (en) | 2001-08-13 | 2002-08-13 | Nr1h4 nuclear receptor binding compounds |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP01119473.5 | 2001-08-13 | ||
| EP01119473A EP1285914B1 (en) | 2001-08-13 | 2001-08-13 | Nr1h4 nuclear receptor binding compounds |
| US10/185,731 US6974830B2 (en) | 2001-08-13 | 2002-07-01 | NR1H4 nuclear receptor binding compounds |
| US10/185,731 | 2002-07-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2003015777A1 true WO2003015777A1 (en) | 2003-02-27 |
Family
ID=26076685
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2002/025438 WO2003015777A1 (en) | 2001-08-13 | 2002-08-13 | Nr1h4 nuclear receptor binding compounds |
Country Status (2)
| Country | Link |
|---|---|
| EP (1) | EP1423113A4 (en) |
| WO (1) | WO2003015777A1 (en) |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005000300A1 (en) * | 2003-06-27 | 2005-01-06 | Vernalis (Cambridge) Limited | Substituted 5-membered ring compounds and their use |
| WO2005089316A2 (en) | 2004-03-12 | 2005-09-29 | Intercept Pharmaceuticals, Inc. | Treatment of fibrosis using fxr ligands |
| US6987121B2 (en) | 2002-04-25 | 2006-01-17 | Smithkline Beecham Corporation | Compositions and methods for hepatoprotection and treatment of cholestasis |
| WO2006029719A2 (en) * | 2004-09-14 | 2006-03-23 | Bayer Healthcare Ag | Diagnostics and therapeutics for diseases associated with nuclear receptor subfamily 1, group h, member 4 (nr1h4) |
| EP1773768A2 (en) * | 2004-07-30 | 2007-04-18 | Exelixis, Inc. | Pyrrole derivatives as pharmaceutical agents |
| US7718808B2 (en) | 2003-12-26 | 2010-05-18 | Kyowa Hakko Kirin Co., Ltd. | Thiazole derivatives |
| WO2010069604A1 (en) | 2008-12-19 | 2010-06-24 | Royal College Of Surgeons In Ireland | Treatment of diarrhoea |
| US7994352B2 (en) | 2005-05-19 | 2011-08-09 | Intercept Pharmaceuticals, Inc. | Process for preparing 3a(β)-7a(β)-dihydroxy-6a(β)-alkyl-5β-cholanic acid |
| US8058267B2 (en) | 2001-03-12 | 2011-11-15 | Intercept Pharmaceuticals, Inc. | Steroids as agonists for FXR |
| US8114862B2 (en) | 2008-11-19 | 2012-02-14 | Intercept Pharmaceuticals, Inc. | TGR5 modulators and methods of use thereof |
| US8410083B2 (en) | 2007-01-19 | 2013-04-02 | Intercept Pharmaceuticals, Inc. | 23-substituted bile acids as TGR5 modulators and methods of use thereof |
| US8796249B2 (en) | 2008-07-30 | 2014-08-05 | Intercept Pharmaceuticals, Inc. | TGR5 modulators and methods of use thereof |
| US9238673B2 (en) | 2012-06-19 | 2016-01-19 | Intercept Pharmaceuticals, Inc. | Preparation and uses of obeticholic acid |
| US9982008B2 (en) | 2012-06-19 | 2018-05-29 | Intercept Pharmaceuticals, Inc. | Preparation and uses of obeticholic acid |
| US10220027B2 (en) | 2011-07-13 | 2019-03-05 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| US10329286B2 (en) | 2016-06-13 | 2019-06-25 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US10421730B2 (en) | 2016-06-13 | 2019-09-24 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| WO2020249064A1 (en) * | 2019-06-14 | 2020-12-17 | Nanjing Ruijie Pharma Co., Ltd. | Compounds for modulating fxr |
| WO2021009332A1 (en) | 2019-07-18 | 2021-01-21 | Enyo Pharma | Method for decreasing adverse-effects of interferon |
| US10987362B2 (en) | 2004-03-12 | 2021-04-27 | Intercept Pharmaceuticals, Inc. | Treatment of fibrosis using FXR ligands |
| WO2021144330A1 (en) | 2020-01-15 | 2021-07-22 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of fxr agonists for treating an infection by hepatitis d virus |
| US11225473B2 (en) | 2019-01-15 | 2022-01-18 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| WO2022152770A1 (en) | 2021-01-14 | 2022-07-21 | Enyo Pharma | Synergistic effect of a fxr agonist and ifn for the treatment of hbv infection |
| US11478533B2 (en) | 2020-04-27 | 2022-10-25 | Novo Nordisk A/S | Semaglutide for use in medicine |
| WO2022229302A1 (en) | 2021-04-28 | 2022-11-03 | Enyo Pharma | Strong potentiation of tlr3 agonists effects using fxr agonists as a combined treatment |
| US11524005B2 (en) | 2019-02-19 | 2022-12-13 | Gilead Sciences, Inc. | Solid forms of FXR agonists |
| US11833150B2 (en) | 2017-03-28 | 2023-12-05 | Gilead Sciences, Inc. | Methods of treating liver disease |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5464847A (en) * | 1992-06-24 | 1995-11-07 | Elf Sanofi | Branched alkylamino derivatives of thiazole, processes for preparing them and pharmaceutical compositions containing them |
| US5602132A (en) * | 1993-12-21 | 1997-02-11 | Sanofi | Branched-amino-substituted thiazoles, processes for their preparation and the pharmaceutical compositions which contain them |
| JPH09235278A (en) * | 1995-12-29 | 1997-09-09 | Toyama Chem Co Ltd | Derivatives of 2-aminothiazole or the salts of the same |
| US5856347A (en) * | 1994-11-29 | 1999-01-05 | Hisamitsu Pharmaceutical Co., Inc. | Antibacterial preparation or bactericide comprising 2-aminothiazole derivative and/or salt thereof |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK150068C (en) * | 1978-06-02 | 1987-06-29 | Pfizer | METHOD OF ANALOGUE FOR THE PREPARATION OF AMINOTHIAZOLES |
| JPH04173782A (en) * | 1990-11-08 | 1992-06-22 | Taisho Pharmaceut Co Ltd | 2-sulfonamide-4,5-diphenylthiazole derivative |
| WO1992015570A1 (en) * | 1991-03-07 | 1992-09-17 | Hisamitsu Pharmaceutical Co., Inc. | Novel diphenylthyazole derivative |
| JPH07149745A (en) * | 1993-11-30 | 1995-06-13 | Hisamitsu Pharmaceut Co Inc | New 2-aminothiazole derivative |
| FR2735777B1 (en) * | 1995-06-21 | 1997-09-12 | Sanofi Sa | 4-PHENYLAMINOTHIAZOLE DERIVATIVES, PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| FR2796380B3 (en) * | 1999-07-15 | 2001-08-17 | Sanofi Synthelabo | NOVEL AMINOTHIAZOL DERIVATIVES, THEIR PREPARATION AND THE PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
-
2002
- 2002-08-13 EP EP02750474A patent/EP1423113A4/en not_active Ceased
- 2002-08-13 WO PCT/US2002/025438 patent/WO2003015777A1/en not_active Application Discontinuation
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5464847A (en) * | 1992-06-24 | 1995-11-07 | Elf Sanofi | Branched alkylamino derivatives of thiazole, processes for preparing them and pharmaceutical compositions containing them |
| US5602132A (en) * | 1993-12-21 | 1997-02-11 | Sanofi | Branched-amino-substituted thiazoles, processes for their preparation and the pharmaceutical compositions which contain them |
| US5856347A (en) * | 1994-11-29 | 1999-01-05 | Hisamitsu Pharmaceutical Co., Inc. | Antibacterial preparation or bactericide comprising 2-aminothiazole derivative and/or salt thereof |
| JPH09235278A (en) * | 1995-12-29 | 1997-09-09 | Toyama Chem Co Ltd | Derivatives of 2-aminothiazole or the salts of the same |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1423113A4 * |
Cited By (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8058267B2 (en) | 2001-03-12 | 2011-11-15 | Intercept Pharmaceuticals, Inc. | Steroids as agonists for FXR |
| US9732117B2 (en) | 2001-03-12 | 2017-08-15 | Intercept Pharmaceuticals, Inc. | Steroids as agonists for FXR |
| US8969330B2 (en) | 2001-03-12 | 2015-03-03 | Intercept Pharmaceuticals, Inc. | Steroids as agonists for FXR |
| US10421772B2 (en) | 2001-03-12 | 2019-09-24 | Intercept Pharmaceuticals, Inc. | Steroids as agonists for FXR |
| US8377916B2 (en) | 2001-03-12 | 2013-02-19 | Intercept Pharmaceuticals, Inc. | Steroids as agonists for FXR |
| USRE48286E1 (en) | 2001-03-12 | 2020-10-27 | Intercept Pharmaceuticals, Inc. | Steroids as agonists for FXR |
| US6987121B2 (en) | 2002-04-25 | 2006-01-17 | Smithkline Beecham Corporation | Compositions and methods for hepatoprotection and treatment of cholestasis |
| WO2005000300A1 (en) * | 2003-06-27 | 2005-01-06 | Vernalis (Cambridge) Limited | Substituted 5-membered ring compounds and their use |
| US7728016B2 (en) | 2003-06-27 | 2010-06-01 | Vernalis (Cambridge) Limited | Substituted 5-membered ring compounds and their use |
| US7718808B2 (en) | 2003-12-26 | 2010-05-18 | Kyowa Hakko Kirin Co., Ltd. | Thiazole derivatives |
| US8889718B2 (en) | 2003-12-26 | 2014-11-18 | Kyowa Hakko Kirin Co., Ltd. | Thiazole derivatives |
| US7880013B2 (en) | 2003-12-26 | 2011-02-01 | Kyowa Hakko Kirin Co., Ltd. | Thiazole derivatives |
| US8420827B2 (en) | 2003-12-26 | 2013-04-16 | Kyowa Hakko Kirin Co., Ltd. | Thiazole derivatives |
| US10987362B2 (en) | 2004-03-12 | 2021-04-27 | Intercept Pharmaceuticals, Inc. | Treatment of fibrosis using FXR ligands |
| EP3175855A1 (en) | 2004-03-12 | 2017-06-07 | Intercept Pharmaceuticals, Inc. | Treatment of fibrosis using fxr ligands |
| WO2005089316A2 (en) | 2004-03-12 | 2005-09-29 | Intercept Pharmaceuticals, Inc. | Treatment of fibrosis using fxr ligands |
| US9498484B2 (en) | 2004-03-12 | 2016-11-22 | Intercept Pharmaceuticals, Inc. | Treatment of fibrosis using FXR ligands |
| US10258633B2 (en) | 2004-03-12 | 2019-04-16 | Intercept Pharmaceuticals, Inc. | Treatment of fibrosis using FXR ligands |
| EP2712617A1 (en) | 2004-03-12 | 2014-04-02 | Intercept Pharmaceuticals, Inc. | Treatment of fibrosis using Fxr ligands |
| EP1773768A2 (en) * | 2004-07-30 | 2007-04-18 | Exelixis, Inc. | Pyrrole derivatives as pharmaceutical agents |
| US8026237B2 (en) | 2004-07-30 | 2011-09-27 | Exelixis, Inc. | Pyrrole derivatives as pharmaceutical agents |
| EP1773768A4 (en) * | 2004-07-30 | 2008-08-06 | Exelixis Inc | Pyrrole derivatives as pharmaceutical agents |
| US8367667B2 (en) | 2004-07-30 | 2013-02-05 | Exelixis, Inc. | Pyrrole derivatives as pharmaceutical agents |
| WO2006029719A3 (en) * | 2004-09-14 | 2006-06-22 | Bayer Healthcare Ag | Diagnostics and therapeutics for diseases associated with nuclear receptor subfamily 1, group h, member 4 (nr1h4) |
| WO2006029719A2 (en) * | 2004-09-14 | 2006-03-23 | Bayer Healthcare Ag | Diagnostics and therapeutics for diseases associated with nuclear receptor subfamily 1, group h, member 4 (nr1h4) |
| US7994352B2 (en) | 2005-05-19 | 2011-08-09 | Intercept Pharmaceuticals, Inc. | Process for preparing 3a(β)-7a(β)-dihydroxy-6a(β)-alkyl-5β-cholanic acid |
| US8410083B2 (en) | 2007-01-19 | 2013-04-02 | Intercept Pharmaceuticals, Inc. | 23-substituted bile acids as TGR5 modulators and methods of use thereof |
| US9243027B2 (en) | 2007-01-19 | 2016-01-26 | Intercept Pharmaceuticals, Inc. | TGR5 modulators and methods of use thereof |
| US9540414B2 (en) | 2008-07-30 | 2017-01-10 | Intercept Pharmaceuticals, Inc. | TGR5 modulators and methods of use thereof |
| US8796249B2 (en) | 2008-07-30 | 2014-08-05 | Intercept Pharmaceuticals, Inc. | TGR5 modulators and methods of use thereof |
| US9650409B2 (en) | 2008-11-19 | 2017-05-16 | Intercept Pharmaceuticals, Inc. | TGR5 modulators and methods of use thereof |
| US8114862B2 (en) | 2008-11-19 | 2012-02-14 | Intercept Pharmaceuticals, Inc. | TGR5 modulators and methods of use thereof |
| US8445472B2 (en) | 2008-11-19 | 2013-05-21 | Intercept Pharmaceuticals, Inc. | TGR5 modulators and methods of use thereof |
| WO2010069604A1 (en) | 2008-12-19 | 2010-06-24 | Royal College Of Surgeons In Ireland | Treatment of diarrhoea |
| US10220027B2 (en) | 2011-07-13 | 2019-03-05 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| US10485795B2 (en) | 2011-07-13 | 2019-11-26 | Gilead Sciences, Inc. | FXR (NR1H4) binding and activity modulating compounds |
| US9238673B2 (en) | 2012-06-19 | 2016-01-19 | Intercept Pharmaceuticals, Inc. | Preparation and uses of obeticholic acid |
| US10047117B2 (en) | 2012-06-19 | 2018-08-14 | Intercept Pharmaceuticals, Inc. | Preparation and uses of obeticholic acid |
| US10174073B2 (en) | 2012-06-19 | 2019-01-08 | Intercept Pharmaceuticals, Inc. | Preparation and uses of obeticholic acid |
| US9732116B2 (en) | 2012-06-19 | 2017-08-15 | Intercept Pharmaceuticals, Inc. | Preparation and uses of obeticholic acid |
| US9982008B2 (en) | 2012-06-19 | 2018-05-29 | Intercept Pharmaceuticals, Inc. | Preparation and uses of obeticholic acid |
| US10155787B2 (en) | 2012-06-19 | 2018-12-18 | Intercept Pharmaceuticals, Inc. | Preparation and uses of obeticholic acid |
| US10981881B2 (en) | 2016-06-13 | 2021-04-20 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US10774054B2 (en) | 2016-06-13 | 2020-09-15 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US11739065B2 (en) | 2016-06-13 | 2023-08-29 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US10421730B2 (en) | 2016-06-13 | 2019-09-24 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US11247986B2 (en) | 2016-06-13 | 2022-02-15 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US10329286B2 (en) | 2016-06-13 | 2019-06-25 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US11833150B2 (en) | 2017-03-28 | 2023-12-05 | Gilead Sciences, Inc. | Methods of treating liver disease |
| US11225473B2 (en) | 2019-01-15 | 2022-01-18 | Gilead Sciences, Inc. | FXR (NR1H4) modulating compounds |
| US11524005B2 (en) | 2019-02-19 | 2022-12-13 | Gilead Sciences, Inc. | Solid forms of FXR agonists |
| CN114008040A (en) * | 2019-06-14 | 2022-02-01 | 南京瑞捷医药科技有限公司 | Compounds for modulating FXR |
| WO2020249064A1 (en) * | 2019-06-14 | 2020-12-17 | Nanjing Ruijie Pharma Co., Ltd. | Compounds for modulating fxr |
| WO2021009332A1 (en) | 2019-07-18 | 2021-01-21 | Enyo Pharma | Method for decreasing adverse-effects of interferon |
| WO2021144330A1 (en) | 2020-01-15 | 2021-07-22 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Use of fxr agonists for treating an infection by hepatitis d virus |
| US11478533B2 (en) | 2020-04-27 | 2022-10-25 | Novo Nordisk A/S | Semaglutide for use in medicine |
| WO2022152770A1 (en) | 2021-01-14 | 2022-07-21 | Enyo Pharma | Synergistic effect of a fxr agonist and ifn for the treatment of hbv infection |
| WO2022229302A1 (en) | 2021-04-28 | 2022-11-03 | Enyo Pharma | Strong potentiation of tlr3 agonists effects using fxr agonists as a combined treatment |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1423113A4 (en) | 2007-04-18 |
| EP1423113A1 (en) | 2004-06-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6974830B2 (en) | NR1H4 nuclear receptor binding compounds | |
| EP1423113A1 (en) | Nr1h4 nuclear receptor binding compounds | |
| US20070010562A1 (en) | Nr1h4 nuclear receptor binding compounds | |
| EP1407774A1 (en) | 2-Amino-4-quinazolinones as LXR nuclear receptor binding compounds | |
| Abd El-Karim et al. | New thiazol-hydrazono-coumarin hybrids targeting human cervical cancer cells: Synthesis, CDK2 inhibition, QSAR and molecular docking studies | |
| US6699879B1 (en) | Phenyl urea and phenyl thiourea derivatives as orexin receptor antagonists | |
| Ibrahim et al. | Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of novel quinoxaline derivatives as potential PPARγ and SUR agonists | |
| US6545009B1 (en) | Retinoid-related receptor function regulating agent | |
| KR101120916B1 (en) | Phenylacetamides and their use as glucokinase modulators | |
| US20060148876A1 (en) | Nr3b1 nuclear receptor binding 3-substituted pyrazoles | |
| EP1398032A1 (en) | 4-Oxo-quinazolines as LXR nuclear receptor binding compounds | |
| US20050203151A1 (en) | Novel compounds, compositions and uses thereof for treatment of metabolic disorders and related conditions | |
| CZ20021903A3 (en) | Substituted oxazole and thiazole derivatives functioning as HPPAR-alpha activators | |
| US20040063775A1 (en) | Five-membered heterocyclic alkanoic acid derivative | |
| SK288236B6 (en) | Quinolinyl phenyl sulfonamide compound, its use as medicament and pharmaceutical preparation comprising it | |
| Crocetti et al. | Synthesis and pharmacological evaluation of new pyridazin‐based thioderivatives as formyl peptide receptor (FPR) agonists | |
| EP1940815B1 (en) | Salts of modulators of ppar and methods of treating metabolic disorders | |
| CA2382581C (en) | Substituted benzylthiazolidine-2,4-dione derivatives | |
| WO2003053976A1 (en) | PIPAZOLO [1,5-a] PYRIMIDINE DERIVATIVES AS MODULATORS OF PPAR | |
| KR102101235B1 (en) | Novel SIRT 1 activator and medical use thereof | |
| JP2002348281A (en) | Five-membered heterocyclic alkane acid derivative | |
| JP2008519085A (en) | Amide derivatives, their production and use as pharmaceuticals |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SD SE SG SI SK SL TJ TM TN TR TT TZ UA UG US UZ VN YU ZA ZM ZW Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BY BZ CA CH CN CO CR CU CZ DE DM DZ EC EE ES FI GB GD GE GH HR HU ID IL IN IS JP KE KG KP KR LC LK LR LS LT LU LV MA MD MG MN MW MX MZ NO NZ OM PH PL PT RU SD SE SG SI SK SL TJ TM TN TR TZ UA UG US UZ VN YU ZA ZM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ UG ZM ZW AM AZ BY KG KZ RU TJ TM AT BE BG CH CY CZ DK EE ES FI FR GB GR IE IT LU MC PT SE SK TR BF BJ CF CG CI GA GN GQ GW ML MR NE SN TD TG Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR IE IT LU MC NL PT SE SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2002750474 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2002750474 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: JP |