CN1169961C - 基因转变作为工具用于构建重组的工业化丝状真菌 - Google Patents
基因转变作为工具用于构建重组的工业化丝状真菌 Download PDFInfo
- Publication number
- CN1169961C CN1169961C CNB988052377A CN98805237A CN1169961C CN 1169961 C CN1169961 C CN 1169961C CN B988052377 A CNB988052377 A CN B988052377A CN 98805237 A CN98805237 A CN 98805237A CN 1169961 C CN1169961 C CN 1169961C
- Authority
- CN
- China
- Prior art keywords
- dna
- glaa
- filamentous fungus
- gene
- district
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images















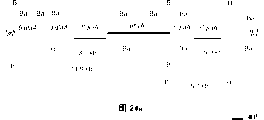











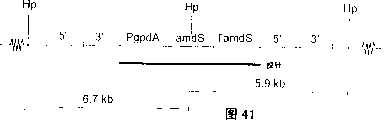




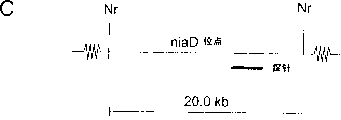
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/62—DNA sequences coding for fusion proteins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/80—Vectors or expression systems specially adapted for eukaryotic hosts for fungi
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Zoology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Wood Science & Technology (AREA)
- Organic Chemistry (AREA)
- Biomedical Technology (AREA)
- General Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Plant Pathology (AREA)
- Mycology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Enzymes And Modification Thereof (AREA)
Abstract
本发明涉及这样的丝状真菌,它们的基因组中包含适于整合一个或多个拷贝重组DNA分子的至少两个基本同源DNA区,且其中至少有两个这样的DNA区包含了重组DNA分子的整合拷贝。本发明还涉及制备这种丝状真菌的方法及通过基因转变或扩增进一步增加带有整合重组DNA分子的DNA区的方法。
Description
发明领域
本发明涉及用于工业化发酵工艺的微生物的遗传工程。
发明背景
在工业化大生产中通过微生物发酵生产的产品不断增多。这些产品的范围从初级和次级代谢产物,分别举例如柠檬酸和抗生素,到蛋白质,酶,甚至完整微生物,例如面包酵母或生物质的形式。按惯例,所用的微生物已接受了经典性菌株改良程序,这些程序包括连续多轮诱变及随后选择具有改良性能的突变体。最近,遗传工程,即重组DNA技术也已经应用于工业化微生物。该技术不仅能进一步提高由所述微生物天然产生的产品的生产水平,而且能开发全新产品和/或生产工艺,举例如异源蛋白质的生产或代谢途径的改造。
遗传工程要求将重组DNA分子导入目的生物体中并通常还要表达。该过程称为转化。微生物的稳定转化要求导入的重组DNA分子在细胞中代代相传,即稳定遗传。原则上有两种方法可保留导入的重组DNA分子。第一,重组DNA可整合在宿主细胞的基因组中,如整合在其染色体上。一旦整合的重组DNA成为该染色体的一部分,就能随该染色体一起复制而被保留。第二,重组DNA可作为能自主复制(即独立于宿主基因组的复制)的DNA分子的一部分而被导入细胞。这类自主复制型载体通常衍生自适合于容纳重组DNA插入的天然质粒或病毒。
就工业化微生物生产用生物体而言,自主的和整合型的载体系统各有其优缺点。例如,自主复制型载体系统对待导入的DNA序列的数量和/或长度有严格限制,一般被认为与整合型系统相比较不稳定并常使每个基因拷贝以较低水平表达。更重要的是,对某些十分重要的工业化微生物,主要是丝状真菌如曲霉、青霉或木霉来说,没有稳定的自主复制型载体系统可供使用。
整合型载体系统的不利之处是整合方法上的某些限制。通过同源重组向预定的基因组序列中整合难以得到高拷贝数的重组DNA分子。而多拷贝重组DNA分子的随机整合,如丝状真菌中常用的那样,将产生无法预料的转化子基因型。这不仅将导致转化后生产株的优点丧失,而且这类菌株的不可预料且不确定特点将使权威登记机构难以接受。
WO91/00920公布了其中多拷贝重组DNA分子整合在核糖体DNA重复簇中的酵母菌株。这些重组DNA分子(此例中是异源基因的表达载体)额外包含有缺陷选择性标记基因和酵母核糖体DNA序列,后者使载体通过同源重组向核糖体DNA重复簇中的整合成为可能。缺陷选择性标记基因是选择及稳定维持包含整合在核糖体DNA重复簇上的多拷贝载体的菌株所必需的。此外,WO91/000920建议,向核糖体DNA重复序列中的多拷贝整合作用可能也可应用于一般性真菌,包括丝状真菌如曲霉。然而,WO91/00920公布的多拷贝整合系统依赖于缺陷选择性标记基因的应用。而且,WO91/00920并未提供向天然适合于支持蛋白质编码基因的高水平RNA聚合酶II转录的基因组环境中整合的方法。
最近,ES2 094 088描述了在产黄青霉E-1和产黄青霉AS-P-78过量产生青霉素的菌株中扩增了的一段DNA区。扩增的DNA区大小分别被描述为75kb和106kb,并在扩增区内一段16.5kb的片段中包含基因pcbAB,pcbC和penDE。有人建议,该扩增区左端和右端的DNA序列可用于构建已导入标记基因的载体,从而启动位于它们之间的遗传物质的扩增,尤其可用于经亚硝基胍随机诱变获得青霉素产量较高的菌株,以便使载体一旦整合入微生物的基因组就可增加其拷贝数。然而,ES 2 094 088没能描述上文简述的方法是否可成功应用。事实上,ES 2 094 088所述方法有好几点缺陷。例如,复杂DNA结构的扩增频率很低。而且亚硝基胍诱变剂的应用将在微生物基因组中同时产生不希望的自发突变。更有甚者,诱变处理可导致载体中扩增区序列的缺失,并因此缺失插入其间的目的基因;也参见Fierro,美国国家科学院院报(1995),6200-6204。还有,如上所述,载体向微生物基因组的随机整合将导致不可预料的转化基因型。
因此,本发明的根本技术难题是提供普遍可用的方法以构建丝状真菌的重组生产株,该株应包含整合在其基因组中预定靶位点的多拷贝重组DNA分子,而且该系统应不依赖于特殊类型选择性标记的应用就可转化。
图的简述
图中所用缩写:
限制性酶和限制性位点:
A=ApaI;Ba=BamHI;B=BglII;Bs=BssHII;E=EcoRI;H=HindIII;K=KpnI;N=NdeI;No=NotI;Ps=PstI;P=PvuII;Sa=SalI;Sc=ScaI;S=SmaI;Sn=SnaBI;Spe=SpeI;Sp=SphI;Ss=SstII;Xb=XbaI;X=XhoI;Nr=NruI;Hp=HpaI;Sf=SfiI;Ns=NsiI;Bst=BstXI.
插图说明
图1:图示黑曲霉CBS646.97(于1997年4月11日保藏于真菌菌种保藏中心,Baarn,荷兰)的转化体(ISO-505)的glaA DNA扩增子中三个ΔglaA位点,每个均用不同的限制位点作标记(BamHI,SalI或BglII)。每个glaA位点的截短相差近20或60bp,这样可用基于PCR的DNA旗试验(DNA-flag test)显示每个截短的glaA位点。
图2:黑曲霉亲代株CBS646.97(图上半部分)的glaA位点及黑曲霉重组株ISO-505的三个截短的“X标记”glaA位点(X代表BamHI,SalI或BglII限制位点)的物理图谱。
图3:中间载体pGBGLA16的构建途径。
图4:图示融合PCR,以破坏3’-glaA DNA序列中的KpnI*,并在边界加入克隆位点以便恰当地克隆进pGBGLA16,产生中间载体pGBGLA18。
图5:图示融合PCR使5’-和3’-glaA序列之间包含一个BamHI限制位点,并在边界加入克隆位点以便恰当地克隆进pGBGLA18,产生基因置换型载体pGBDEL5。
图6:图示融合PCR使glaA的5’-和3’-序列之间包含一个SalI限制位点,并在边界加入克隆位点以便恰当地克隆进pGBGLA18中,产生基因置换型载体pGBDEL9。
图7:图示融合PCR使glaA的5’和3’序列间包含一个BglII限制位点,并在边界加入克隆位点以便恰当地克隆进入pGBGLA18中,产生基因置换型载体pGBDEL11。
图8:图示分离自黑曲霉亲代株CBS646.97的染色体DNA用KpnI或BamHI消化后,以Hind III/XhoI 5’-glaA DNA片段(a)或SalI/SalI3’-glaA DNA片段(b)为探针所观察到的杂交模式。
图9:图示用KpnI或BamHI消化分离自黑曲霉CBS646.97转化体的染色体DNA后,用探针Hind III/XhoI 5’-glaA DNA片段(a)或SalI/SalI 3’glaA DNA片段(b)所观察到的杂交模式,其中线性化pGBDEL5载体已通过一个双交换而整合到glaA靶位点。在单个转化体的基因组中,宿主的3’-glaA序列中的原始KpnI位点可能因双交换重组事件而被破坏(K*),但并非一定如此。
图10:图示分离自无amdS选择性标记基因的黑曲霉转化体GBA-201的染色体DNA用KpnI或BamHI消化后用探针HindIII/XhoI5’-glaA DNA片段(a)或SalI/SalI 3’-glaA DNA片段(b)的杂交模式,其中该转化体包含一个“ BamHI标记的”ΔglaA DNA扩增子和两个完整无损的glaA扩增子。
图11:图示分离自黑曲霉GBA-201转化体的染色体DNA用KpnI或BamHI消化后用探针HindIII/XhoI 5’-glaA DNA片段(a)或SalI/SalI 3’glaA DNA片段(b)的杂交模式,其中线性pGBDEL9载体经双交换变成整合型。在单个转化体的基因组中,宿主的3’glaA序列中的原始KpnI位点可能因双交换重组事件而被破坏(K*),但并非一定如此。
图12:图示分离自无amdS选择性标记基因的黑曲霉GBA-202转化体的染色体DNA用KpnI或BamHI消化后用探针HindIII/XhoI5’-glaA DNA片段(a)或SalI/SalI 3’-glaA DNA片段(b)的杂交模式,该转化体包含两个“BamHI和SalI标记的”ΔglaA DNA扩增子和一个仍然无损的glaA扩增子。
图13:图示分离自黑曲霉GBA-202转化体的染色体DNA用KpnI或BamHI消化后用探针HindIII/XhoI 5’-glaA DNA片段(a)或SalI/SalI 3’-glaA DNA片段(b)的杂交模式,转化体中线性pGBDEL11载体已经双交换为整合型。在单个转化体的基因组中,宿主的3’-glaA序列中的原始KpnI位点可能因双交换重组事件而被破坏(K*),但并非一定如此。
图14:图示分离自无amdS选择性标记基因的黑曲霉转化体ISO-505的染色体DNA用KpnI或BamHI消化后用探针HindIII/XhoI5’-glaA DNA片段(a)或SalI/SalI 3’-glaA DNA片段(b)的杂交模式,该转化体包括3个“BamHI,SalI和BglII标记的”ΔglaA DNA扩增子。
图15:乙酰胺选择性标记和glaA打靶载体pGBAAS-1的物理图谱。
图16:肌醇六磷酸酶表达盒和glaA打靶载体pGBTOPFYT-1的物理图谱。
图17:图例说明定向PCR检验可测定phyA表达盒(a)或amdS盒(b)是否已导入黑曲霉ISO-505转化体的一个3’-3″-glaA靶位点的附近。这些盒中之一的打靶将导致两例均扩增一个4.2Kb大小的DNA片段。
图18:图示黑曲霉ISO-505的三个ΔglaA区。所示杂交模式为BglII消化物用2.2Kb SalI/XhoI 3″-glaA DNA片段作探针杂交的。
图19:图示pGBAAS-1/pGBTOPFYT-1黑曲霉ISO-505转化体中的ΔglaA区(分别标记为B*,Sa*和Ba*),其中多个amdS盒打靶到黑曲霉ISO-505宿主的三个ΔglaA区之一。所示杂交模式为BglII消化物用探针2.2Kb SalI/XhoI 3″-glaA DNA片段杂交的。值得注意的是,7.5Kb BglII杂交片段的存在和强度取决于所整合的amdS盒数(n)。
图20:图示pGBAAS-1/pGBTOPFYT-1黑曲霉ISO-505转化体的三个ΔglaA区(分别标记为B*,Sa*和Ba*),其中多个phyA已整合在黑曲霉ISO-505宿主三个ΔglaA区之一的3’-3″-glaA靶序列邻近位点,同时还有下游的一个或多个amdS盒,所示杂交模式为BglII消化物用探针2.2Kb SalI/Xho I 3″-glaA DNA片段杂交的。值得注意的是,7.5Kb Bgl II杂交片段的强度取决于整合的phyA和amdS盒数(n)。
图21:图示pGBAAS-1/pGBTOPFYT-1黑曲霉ISO-505转化体的三个ΔglaA区(分别以B*,Sa*和Ba*标记),其中多个amdS盒已整合在黑曲霉ISO-505宿主三个ΔglaA区之一的3’-3″-glaA靶序列附近,同时还有下游的一个或多个phyA盒。所示为BglII消化物用探针2.2Kb SalI/XhoI 3″-glaA DNA片段的杂交模式。注意7.5Kb BglII杂交片段的强度取决于所整合的phyA和amdS盒数(n)。
图22:图示pGBAAS-1/pGBTOPFYT-1黑曲霉ISO-505转化体的三个ΔglaA区(分别以B*,Sa*和Ba*标记),其中多个phyA盒已整合在黑曲霉ISO-505宿主三个ΔglaA区之一的3’-3″-glaA靶序列附近,同时还有下游的一个或多个phyA和amdS盒。所示为BglII消化物用探针2.2Kb SalI/XhoI 3″-glaA DNA片段的杂交模式。注意7.5Kb BglII杂交片段的强度取决于所整合的phyA和amdS盒数目(n)。
图23:图示pGBAAS-1/pGBTOPFYT-1黑曲霉ISO-505转化体的三个ΔglaA区(分别以B*,Sa*和Ba*标记),其中多个amdS盒已整合在黑曲霉ISO-505宿主三个ΔglaA区之一的3’-3″-glaA靶序列附近,还有下游的一个或多个amdS和phyA盒。所示为BglII消化物用探针2.2Kb SalI/XhoI 3″-glaA DNA片段的杂交模式。
图24:不含amdS基因、但含有打靶到“BamHI标记的”ΔglaA位点的一个phyA拷贝(a),两个phyA拷贝(b)和三个phyA拷贝(c)的黑曲霉ISO-505转化体物理图谱。图示为BamHI和BglII消化物用探针3″-glaA的杂交模式。
图25:图示黑曲霉ISO-505-2转化体的三个glaA区,其中两个phyA盒均打靶到“BamHI标记的”ΔglaA扩增子处,表现为“DNA旗”基因型BamHI+/SalI+/BglII+(A),BamHI2+/SalI-/BglII+(B)和BamHI2+/SalI+/BglII-(C)。所示为BamHI消化物用探针2.2kbSalI/XhoI 3″-glaA DNA片段的杂交型。注意两个BamHI杂交片段的强度均取决于不同扩增子的存在和数量。
图26:图示黑曲霉ISO-505-2转化体的三个glaA区,其中两个phyA盒打靶到“BamHI标记的”ΔglaA扩增子上,显示“DNA-旗”基因型BamHI+/SalI+/BglII+(A),BamHI2+/SalI-/BglII+(B),BamHI2+/SalI+/BglII-(C)。所示为BglII消化物用探针1.5Kb HindIII/XhoI5’-glaA片段的杂交模式。注意BglII杂交片段的强度取决于不同扩增子的存在和数量。
图27:图示黑曲霉ISO-505-2转化体的glaA区,其中两个phyA盒定位到“BamHI-标记的”ΔglaA扩增子上,显示“DNA-旗”基因型BamHI3+/SalI-/BglII-(A),BamHI3+/SalI+/BglII-(B)和BamHI4+/SalI-/BglII-(C)。所示为BamHI消化物用探针1.5Kb HindIII/XhoI 5’-glaA DNA片段的杂交模式。注意两个BamHI杂交片段的强度取决于不同扩增子的存在和数量。
图28:图示黑曲霉ISO-505-2转化体的glaA区,其中两个phyA盒定位到“BamHI-标记的”ΔglaA扩增子,具有“DNA-旗”基因型BamHI3+/SalI-/BglII-(A),BamHI3+/SalI+/BglII-(B)和BamHI4+/SalI-/BglII-(C)。所示为BglII消化物用探针2.2kb SalI/XhoI 3″-glaADNA片段的杂交模式。注意两个BamHI杂交片段的强度取决于不同扩增子的存在和数量。
图29:产黄青霉的PEN扩增子。青霉素生物合成基因,HELE和HELF在单个扩增子中的相对位置(括号之间)。多个PEN扩增子以直接重复子(n)的形式存在。注意HELF的3’末端延伸进了邻近的PEN扩增子内。
图30:Southern分析定量PEN扩增子。BstXI位点的相对位置,探针及niaD位点(A)和PEN扩增子(B)的预期杂交片段的大小。与HELE探针杂交的DNA量取决于PEN扩增子的数量(n)。
图31:经CHEF定量PEN扩增子。示有NotI位点的相对位置及HELE探针。NotI位点位于PEN扩增子之外。杂交片段的预计大小取决于PEN扩增子的数量,为(30+nx57)Kb。
图32:定向整合。(A)表达载体含目的基因,该基因受适合的启动子(P)和终止子(T)调节。该盒的5″和3″区侧翼与靶位点同源(用’和”来区分基因组序列和载体序列)。将盒及侧翼克隆进一个大肠杆菌载体中以增殖表达载体。转化之前,用限制酶R消化该表达载体,产生线性片段并去除大肠杆菌序列。(B)整合发生在基因组中同源区,转化片段的游离5’和3’末端是重组热点。(C)所得转化体包含1个或多拷贝(n)的整合在靶位点的转化片段(括号之间)。
图33:表达载体pHELE-A1。包含侧翼是HELE 5’和3’区的amdS表达盒的转化片段分离成一个SfiI片段。这些位点经由用来PCR扩增相应侧翼区的寡核苷酸而导入。
图34:表达载体pHELF-A1。包含侧翼是HELF 5’和3’区的amdS表达盒的转化片段分离为一个SfiI片段。这些位点经由用来PCR相应侧翼区的寡核苷酸导入。
图35:表达载体pHELE-E1。包含侧翼是HELE 5’和3’区的cefE表达盒的转化片段分离为一个SfiI片段。
图36:表达载体pHELF-F1。包含侧翼是HELE 5’和3’区的cefF表达盒的转化片段分离为一个Sfi I片段。
图37:重组丢失amdS。(A)经表达盒的串联整合产生的5’和3’侧翼区直接重复子。(B)5’和3’侧翼配对,重组通过单交换而发生。(C)交换位点之间的区段丢失。注意任意数量的盒(包括目的基因)均可丢失,因为所有盒的侧翼均是直接重复子。
图38:cefE整合进HELE。HELE中单个cefE盒的物理图谱(A)或多个cefE盒的物理图谱。示有NruI位点的相对位置,cefE探针和杂交片段的预期大小。6.0Kb片段因盒的多重整合产生,其强度取决于盒存在的数目(n)。
图39:TAFE测定的cefE向HELE的整合。示有HpaI位点的相对位置,探针及杂交片段的预期大小。杂交片段的大小取决于所整合的盒的数量(n)(5.9+nx6.0)Kb。
图40:盒及定向PCR。特异性模板和寡核苷酸结合的图示。定向PCR必需的是一个寡核苷酸位于5’侧翼的上游。因而,尽管可出现好几个盒,只有在该寡核苷酸3’的盒产生PCR产物。图中示有有关区段,寡核苷酸和PCR产物的预期大小。(A)cefE的盒式PCR,(B)HELE内cefE的定向PCR,(C)HELE内amdS的定向PCR,(D)amdS的盒式PCR,(E)cefE-amdS结合的定向PCR,(F)amdS-cefE结合的定向PCR,(G)amdS-amdS组合的定向PCR,(H)niaD位点的盒式PCR,(I)cefF的盒式PCR,(J)整合在HELF中的cefF的定向PCR,(K)整合进HELF的amdS的定向PCR。
图41:amdS向HELE的整合。示有HpaI位点的相对位置,amdS探针和杂交片段的预期大小。
图42:探针。分离的DNA片段用于探针制备:(A)cefE探针:pHEL-E1的NdeI-NsiI片段。(B)cefF探针:pHEL-F1的NdeI-NsiI片段。(C)amdS探针:pHELE-A1的NotI片段。(D)HELE探针:pHELE-A1的SphI-NotI片段。(E)niaD探针:以产黄青霉染色体DNA为模板,寡核苷酸28和29的PCR产物。
图43:基因转换。通过以另一个PEN扩增子中的cefE替代amdS而进行的cefE基因转换和因此所致的重复。
图44:在一个扩增子中的(cefE+cefF)整合。示有NotI位点的相对位置,HELE探针和cefE和cefF向同一个扩增子上的HELE和HELF中整合的特异杂交片段的预期大小。注意NotI位点是通过cefE和cefF盒的整合而掺入的。
图45:cefF向HELF的整合。HELF中的单个cefF盒的物理图谱(A)或多个cefF盒的物理图谱(B)。示有NruI位点的相对位置,cefF探针和杂交片段的预期大小。5.7Kb片段是盒多次整合的结果,它的强度取决于盒的存在数量(n)。
图46:(cefE+cefF)基因转版本中的cefE定量。示意NruI位点的相对位置,探针及针对HELE中(A)单个或(B)多个cefE盒的杂交片段的预期大小。此6.0Kb片段是盒多次整合的结果,其强度取决于盒的数量(n)。(C)niaD位点。基因转版本将具有相同于亲代株的杂交模式,但强度相对于niaD信号更强。
图47:(cefE+cefF)基因转版本中的cefF定量。NruI位点的相对位置,探针及针对HELF中(A)单个或(B)多个cefF盒的杂交片段的预期大小。此5.7Kb片段是盒多次整合的结果,其强度取决于盒的数量(n)。(C)niaD位点。基因转版本将具有相同于亲代株的杂交模式,但强度相对于niaD信号更强。
图48:cefE基因转版本的定量。NruI位点的相对位置,探针和针对HELE中(A)单个或(B)多个cefE盒的杂交片段的预期大小。此6.0Kb片段是盒多次整合的结果,其强度取决于盒的数量(n)。(C)niaD位点。基因转版本将具有相同于亲代株的杂交模式,但强度相对于niaD信号更强。
发明详述
本发明的第一方面涉及其基因组中至少两个基本同源的DNA区内整合有一种重组DNA分子的丝状真菌,其中这些DNA区不是核糖体DNA重复序列。本发明的丝状真菌制备方法是:用一种重组DNA分子转化其基因组中至少有两个这样的DNA区的丝状真菌,并鉴定至少一个重组DNA分子整合进了至少一个这样的DNA区的转化体,随后鉴定转化体的子代中,含有该重组DNA分子的DNA区已通过与DNA区的其它形式进行基因转变或通过含该重组DNA分子的DNA区的扩增而增加了的菌株。或者说,本发明的丝状真菌在其基因组中至少一个内源DNA区内掺入了至少一个重组DNA分子。
本发明的丝状真菌是真核微生物,包括真菌界真菌门的所有丝状形式(参见如,Lasure和Bennett,1985,真菌分类学,在“真菌的基因操作”一书中,pp531-535,Academic Press公司)。本发明的丝状真菌从形态学,生理学和遗传学上区别于酵母:比较于酵母菌如酿酒酵母,丝状真菌的营养生长是通过菌丝体延伸进行,碳的代谢是专性需氧的。而且,酵母菌如酿酒酵母有明显的稳定二倍体期,而丝状真菌如构巢曲霉和粗糙脉孢菌的二倍体只是在减数分裂之前短期存在。此外,很多工业上重要的丝状真菌属于半知菌(Deuteromycotina)亚门,亦称半知菌,是一类人为划分的缺乏任何已知有性形式的真菌。本发明的优选丝状真菌属于曲霉属,木霉属,青霉属,头孢霉属,枝顶孢属,镰孢霉属,毛霉属,根霉属(Rhizophus),Phanerochaete,脉孢菌属,腐质霉属,麦角属,粪壳属,黑粉菌属,裂褶菌属,布拉霉属,被孢霉属,须霉属,弯颈霉属(Tolypocladium)各属。本发明的最优选丝状真菌是属于由Raper和Fennell所确定的黑曲酶(1965,“曲霉属”一书中,Williams & Wilkin公司,Baltimore)的真菌,如黑曲霉;由Raper和Fennell所确定的黄曲霉类(文献同上),如米曲霉;以及真菌Trichoderma reesei和产黄青霉。本发明以黑曲霉和产黄青霉示范。
本发明的丝状真菌在其基因组中包含至少两个适于整合一或多拷贝重组DNA分子的基本同源的DNA区,其中该DNA区不是核糖体DNA。本文实施例中,黑曲霉中扩增的葡糖淀粉酶(glaA)位点和产黄青霉中扩增的青霉素簇均用作整合重组DNA分子的DNA区。黑曲霉的glaA位点和产黄青霉的青霉素簇(含青霉素生物合成基因pcbAB,pcbC和penDE)在野生型黑曲霉和产黄青霉菌株的基因组中均以单拷贝出现。含多拷贝的这些DNA区的菌株,如本实施例所用的,可在经典的菌株改良程序中分别通过选择葡糖淀粉酶或青霉素产量增加了的菌株而获得。通常,这些产量的增加是所选菌株中DNA区扩增的结果。这些扩增的DNA区在后文中称扩增子。虽然本发明优选用这些扩增子作为整合重组DNA分子的DNA区,但并非仅限于此。事实上,在丝状真菌基因组中有两个或更多基本同源形式的任何DNA区均可应用,只要它们满足以下两项功能标准:1)该DNA区应适于接受重组DNA分子的整合;2)该DNA区应能与真菌基因组中其它基本同源区发生重组,以使整合了的重组DNA分子通过基因转变而增加。
为符合第一个标准,DNA区应有足够的长度以便重组DNA分子可通过同源重组打靶到该区内。为此,DNA区应包含至少100bp,优选至少1kb,更优选至少2Kb。确定DNA区是否适合于整合重组DNA分子还可看是否向该DNA区的整合符合不破坏所研究的丝状真菌有活力所必需的功能的要求。
第二个功能标准,即能与真菌基因组中该区的其它基本同源版本重组,这是为了允许在该DNA的不同版本间进行基因转变。达到这一目的的最低要求是每个版本区任一侧的DNA序列与其它版本DNA区的相应侧序列相互有足够同源性,以便在侧序列之间进行同源重组。这种同源重组的结果是发生基因转变,其中该DNA区的一个版本被含整合重组DNA分子的其它DNA区的复制体取代。允许基因转变的侧翼序列最小长度和最小同源范围尚不确知,可能因所研究的生物不同而有变化。可能最小长度为100bp且总同源性不少于60%能允许基因转变。显然,基因转变频率将随该DNA区侧序列的长度和同源性的增加而增加。优选不同区的侧序列有至少1Kb,其同源性至少80%。真菌基因组可能包含一类或不同类型的上述DNA区。不同类型的并非互为完全拷贝的这些区的实例有等位基因变异体,编码同工酶的基因家族和/或基因。最优选的区为互为确切拷贝的区,区别至多在于所整合重组DNA分子。这些相同区的实例是扩增子。故在本发明的一个优选实施方案中,上述适于整合一或多拷贝重组DNA分子的DNA区为扩增子。
DNA区的总长度并不十分重要,可在1Kb以下至几百Kb的范围内变化,如本文实施例中DNA区的长度从扩增的青霉素簇每单位约57Kb至扩增的glaA位点每单位超过80Kb的范围内。
重组DNA分子包含向本发明的丝状真菌导入所需遗传修饰所要求的遗传因子的任意组合。重组DNA分子包含任何遗传因子,其部分或其组合,如基因(编码部分或完整位点),cDNA,启动子,终止子,内含子,信号序列,DNA结合蛋白的任何调节序列或识别序列。遗传因子还可包含已被修饰,即有一个或更多核苷酸改变(如插入,缺失,替代)的DNA序列。
所需遗传修饰包括任何修饰,即在所选丝状真菌中有DNA序列的插入、缺失、和/或取代,这是因一个或更多个上述遗传因子通过转化或与重组DNA分子共转化而导入真菌的结果。可理解的是,这些遗传修饰可在互不相干的多轮转化中被导入。
根据本发明的一个实施方案,整合进丝状真菌DNA区中的重组DNA分子包含一个或多个表达盒以表达一个或多个目的基因。本文中表达盒意指包含将被表达的目的基因并可操作连接于能影响并调节该基因表达的适当表达因子的DNA片段。这样的表达因子包括启动子,信号序列,终止子等。
如果丝状真菌意图用于生产蛋白质或酶(不管异源还是同源),目的基因优选编码分泌型蛋白或酶,故应包含可引起酶分泌的信号序列。此实施方案用含黑曲霉肌醇六磷酸酶基因的表达盒向黑曲霉glaA扩增子内的整合举例说明。然而,技术人员应十分清楚本发明可应用于任何感兴趣的蛋白质或酶。
或者,如果丝状真菌意图用于生产初级或次级代谢产物,则目的基因通常编码一种或多种涉及这些代谢物的生物合成的胞内酶。根据本发明的一个优选实施方案,丝状真菌包含一个或多个整合在DNA区中的重组DNA分子,而这些重组DNA分子包含一个或多个胞内酶的表达盒,这些酶是丝状真菌天然不具有的代谢途径中的成员。这些丝状真菌的实例包括如在其青霉素扩增子中整合有脱乙酰氧基头孢菌素(deacetoxycephalosporin)C合成酶(扩展酶(expandase))和脱乙酰基头孢菌素(deacetylcephalosporin)C合成酶(羟化酶)的表达盒、能分别允许菌株合成己二酰-7-氨基脱乙酰氧基头孢菌酸(adipoyl-7-aminodeacetoxycephalosporanic acid)和己二酰氨基脱乙酰基头孢菌酸(adipoyl aminodeacetylcephalosporanic acid)的产黄青霉菌菌株。
重组DNA分子整合进DNA区的机制对本发明并不重要,可取决于本发明应用者的实际工作。重组DNA分子整合进DNA区可通过随机整合,但更优选同源重组性整合,不管是单交换重组事件的整合(即导致插入该重组DNA分子),还是双交换重组整合(即导致被重组DNA分子取代DNA区原有序列的一部分)。为促进经同源重组发生整合,重组DNA优选包含与靶序列同源以整合进DNA区的序列。这些重组DNA分子中的打靶序列优选相同于DNA区中的靶序列。
在本发明的较优选实施方案中,丝状真菌中存在的基本同源DNA区的每个版本包含一个整合拷贝的重组DNA分子。真菌中所有DNA区全部被整合重组DNA分子占据,这样可产生最大可能拷贝数的重组DNA分子,并提供更稳定状态,因为这样的真菌不含能作为与充实DNA区进行基因转变的供体的“空”DNA区,从而不会减少整合重组DNA分子的拷贝数。可以是同类型的DNA区每个版本被重组DNA分子占据,也可以是不同型的上述DNA区每个版本被占。优选丝状真菌基因组中基本同源DNA区的所有类型的每个版本均包含一个整合拷贝的重组DNA分子。
本发明的一个有利方面中,用于整合重组DNA分子的DNA区在其天然状态下包含能高水平表达的内源基因。一般已知整合的重组基因的表达水平可根据其在基因组中的整合位点不同而差异很大。用高表达区整合需表达的重组基因好处是这些区至少能支持内源基因的高水平表达。故似乎这些区也将支持整合的重组基因高水平表达。事实上我们发现,如本文实施例所述,向黑曲霉的glaA区整合和向产黄青霉的青霉素簇整合比向一些其它基因位点的整合会提供每基因拷贝的更高水平表达。在此可理解的是,能高水平表达的基因规定为最高水平表达时产生的mRNA至少占总mRNA群体的0.1%,优选至少占总mRNA的0.5%,更优选至少占总mRNA的1%的基因。这些可高度表达的内源基因(其所在区特别适于整合本发明的重组DNA分子)的实例有糖酵解酶基因,淀粉酶基因,纤维素分解酶基因和/或抗生素生物合成酶基因。更优选是包含涉及工业化生产过程、并已知高水平表达的基因如葡糖淀粉酶基因、TAKA淀粉酶基因、纤维二糖水解酶基因和青霉素生物合成基因的区。
本发明的一方面中,丝状真菌中每拷贝的DNA区内可高表达的内源基因当其表达非必需时可以被失活。此时,高表达内源基因的失活节约了能量和资源,可进一步表达目的基因。而且,若经重组DNA分子的整合产生的目的酶和内源基因编码的酶均为分泌型酶,则内源酶的失活将导致目的酶的制剂更纯。内源基因失活方法优选不可逆缺失内源基因的至少一部分,以便排除失活的回复突变。内源基因的失活更优选不可逆缺失启动子及上游激活序列的至少一部分。当经重组DNA的整合而生产的酶的编码基因表达受到该内源基因的启动子驱动时,这将特别有利,因为这将使可能限制目的基因表达所需转录因子的竞争作用减少。
本发明的另一实施方案中,丝状真菌中DNA区的每个版本通过独有的序列标记区别于其他版本。这些序列标记能用来监控不同区之间的基因转变,这将便于筛选和/或选择带所需基因型的转版本。任何形式的序列标记均可应用,只要它们允许检测DNA区的不同版本:如从可在Southern印迹中检测的限制位点到提供易于分析的表型的完整可选择标记基因。本文的一个特别有效实施方案举例说明了序列标记,允许在单PCR中用一对寡核苷酸作引物引发PCR以检测每个区。各区如此修饰以致于PCR中每个版本产生的PCR片段有各自独有的长度。所获PCR片段的长度和强度分别显示各区的存在和拷贝数。这种序列标记形式,称为“DNA旗”(DNA flag),可用来快速分析大量转化体集落的基因型,以便获得带所需基因型的转化体。
本发明进一步涉及制备本发明之丝状真菌的方法。这些方法包括用重组DNA分子转化这样的丝状真菌:其中一条或多条染色体包含至少两个基本同源的、适于一或多拷贝的重组DNA分子整合的DNA区,而且这些DNA区不是核糖体DNA重复序列。丝状真菌的转化现对于技术人员已常规化,现已有各种适用于丝状真菌的转化方案。
用重组DNA分子转化丝状真菌要求应用能区别吸收了转化DNA形式的真菌细胞与未转化细胞的可选择标记基因。多种可选择标记基因可用于丝状真菌的转化。适合的标记包括涉及氨基酸或核苷酸代谢的营养缺陷型标记基因,如编码鸟氨酸转氨甲酰酶(argB),乳清苷-5’-脱羧酶(pyrG),或谷氨酰胺酰胺转移酶,吲哚甘油磷酸合成酶,磷酸核糖基邻氨基苯甲酸异构酶(trpC)的基因,或涉及碳元素或氮代谢的营养缺陷型标记基因,如niaD或facA,以及抗生素抗性标记,如腐草霉素抗性基因,博来霉素抗性基因或新霉素(G418)抗性基因。优选应用双向选择标记,即既有阳性遗传选择又有阴性遗传选择。这些双向选择标记的实施有pyrG,facA,和amdS基因。由于其双向选择性,这些标记可从转化的丝状真菌中缺失,只剩下导入的重组DNA分子在其位点上,从而可获得不含选择标记的丝状真菌。此MARKER GENEFREETM转化技术的要素公布于EP-A-0 635 574,被本文引为文献。这些选择标记中,应用显性的双向选择标记如象构巢曲霉,黑曲霉和产黄青霉的amdS这样的乙酰胺酶基因为最优选。除了其双向性,这些标记还提供这样的优点,即作为显性选择标记,应用时无需突变型(营养缺陷型)菌株,可直接应用于野生型菌株。
因而另一个实施方案涉及本发明的丝状真菌,其中重组DNA分子缺乏选择标记基因,或更优选本发明的丝状真菌总共缺乏一个选择标记基因。
用重组DNA分子转化本发明的丝状真菌时,所用选择标记既可物理连接于被转化的重组DNA分子上,也可位于与所需重组DNA分子共转化的另一个DNA分子上。共转化经常被本领域技术人员采用,因为在丝状真菌中其发生频率相对较高。
制备本发明之丝状真菌方法的下一步包括选择有至少一个重组DNA分子整合进丝状真菌的至少一个DNA区的转化体。技术人员可采用多种常规技术检测所获转化体哪一个在其一个DNA区中整合了一个重组DNA分子。下一步对选择的转化体进行增殖,从其子代中选出其中至少两个DNA区包含整合重组DNA分子的菌株。这意味着所选择的是其中包含整合重组DNA分子的DNA区经与“空”DNA区基因转变或经扩增而增加了的菌株。这种基因转变和/或扩增事件以低频率自发发生。其确切频率取决于包括所研究真菌,DNA区的数量、长度和同源程度在内的多个变量。然而我们发现,这些频率之高足以使我们能用当今的分析技术筛选和选择发生了这些事件的菌株。其内包含整合重组DNA分子的DNA区数量已增加的菌株可通过例如简单地筛选出合成受重组DNA分子影响的产物以较高水平生产的菌株而鉴定,或者通过用例如上文简述的“DNA旗”检验分析其基因型而鉴定。
本发明的方法可包含附加的步骤,即对其中含整合重组DNA分子的DNA区有增加的菌株进行增殖,从其后代菌株中筛选到DNA区的其它拷贝含整合重组DNA分子的菌株。可将这些菌株再执行一次此程序,直至获得其中每个DNA区均包含整合重组DNA分子的菌株。如本文所概述,这些菌株优点在于重组DNA分子拷贝数高且稳定性增加。
用于制备本发明丝状真菌的方法的另一方面中,重组DNA分子包含基本同源于DNA区的序列。这些基本同源序列在重组DNA分子中的存在将明显增加重组DNA分子整合进DNA区的频率,重组DNA分子中基本同源序列的最小需求尚不清楚,但实际工作中,用至少1Kb,优选至少2Kb的同源定向序列获得了合理的定向频率。
如上所述,本发明的一个方面涉及应用双向选择标记,以便用重组DNA分子转化丝状真菌。一旦获得这样一个转化体,便可对其进行负选择,筛选双向选择标记的缺失。这些缺标记转化体可再进一步转化,或可增殖,以从子代菌株中选择含重组DNA分子的DNA区有基因转变和/或扩增的菌株。
在制备本发明之丝状真菌的另一方法中,丝状真菌用重组DNA分子转化,选择有至少一个重组DNA分子整合进预定基因组靶序列的转化体。该转化体随后经增殖,从其子代选出有至少两个DNA区包含整合重组DNA分子的菌株。此例中待用重组DNA分子转化的受体丝状真菌无需包含多拷贝的DNA区。DNA区最好是在重组DNA分子整合后扩增。
本发明能够用于制备这样的重组丝状真菌:在其一条或多条染色体中包含至少两个适于整合一或多拷贝重组DNA分子的基本同源DNA区,其中这些DNA区并不是核糖体DNA重复序列,又其中至少两个DNA区包含一个整合拷贝的重组DNA分子。这些重组真菌可应用于生产感兴趣产品的工艺中。该工艺通常包括在有助于生产感兴趣产品的培养基中培养重组细胞,从培养基和/或从真菌中回收感兴趣的产品。这些产品可以是蛋白质,如酶,和/或初级代谢产物,如CO2,乙醇或有机酸,和/或次级代谢产物,如抗生素或类胡萝卜素。感兴趣产物也可是重组真菌自身,即生产中所获生物质。本发明丝状真菌中可产生的酶和蛋白质实例有脂肪酶,磷脂酶,磷酸酶,肌醇六磷酸酶,蛋白酶,支链淀粉酶,酯酶,糖苷酶,淀粉酶,葡糖淀粉酶,过氧化氢酶,葡糖氧化酶,β-葡糖苷酶,阿拉伯呋喃糖苷酶,鼠李糖苷酶,芹菜糖苷酶,凝乳酶,乳铁蛋白,细胞壁降解酶如纤维素酶,半纤维素酶,木聚糖酶,甘露聚糖酶,果胶酶,鼠李半乳糖醛酸酶等等。
本发明的丝状真菌的一些优点及其制备方法概述如下:
-相比较于已有的向丝状真菌非随机性多拷贝整合重组DNA分子
的系统,本发明提供更广泛用途。更广泛用途是因为本发明不限
于应用缺陷选择标记基因进行转化。还因为本发明不限于只应用核
糖体DNA序列作为整合的靶序列。
-与传统的重组丝状真菌(其中重组DNA分子以串联排列方式随机整
合)相比,本发明的丝状真菌其整合的多拷贝重组DNA分子具有更
强的遗传稳定性。具体地说,有高拷贝数的随机整合重组DNA分
子的丝状真菌通常是大量串联重复的重组DNA分子只整合于极少
数基因组位点。该构型在遗传上不如本发明的构型稳定,在本发
明的构型中,只有极少数串联重复拷贝的重组DNA分子(优选不超
过5个拷贝)整合在好几个不同基因组位点,如基本同源的DNA
区。例如,本发明的丝状真菌可能有的拷贝数为15,其中在5个
基本同源DNA区中每个区整合有3个串联重复的重组DNA分子拷
贝。相比之下,有15个拷贝的传统重组丝状真菌可能有10个拷
贝整合在一个不确定的基因组位点,而另5个拷贝在另一个不确
定的基因组位点。按照本发明,我们发现,就重组DNA分子的丢
失而言,后一种构型比本发明有相同拷贝数的真菌稳定性要差得
多。
-与整合发生在核糖体DNA的丝状真菌相比,本发明的丝状真菌预期
相对于每个基因拷贝的表达水平更高,因为前者尚未进化得可支
持蛋白编码基因的高水平RNA多聚酶II转录过程。
-本发明的丝状真菌均为多拷贝菌株,至少能够完全确定就整合重组
DNA分子而言的基因型。这将有利于获得对这些真菌所涉及工艺
和产物的核准。
-基于相似的理由,对丝状真菌表型的预计将比传统型重组DNA分子
在其中随机整合的重组丝状真菌容易得多,因为随机整合能改变整
合位点之中或附近的未知基因的表达。
-制备本发明之丝状真菌的方法具有可与较早的经典菌株改良程序
协同的好处。如上简述,在本发明的优选实施方案中,用到了从
经典菌株改良程序中获得的工业化丝状真菌。应用这些真菌的好
处不仅是因为它们常包含适于整合并随后扩增本发明之重组DNA
分子的扩增子,而且还因为工业化丝状真菌带有累积的(大量)突
变,它们不仅对菌株改良程序所针对的产品(“前期”产品)的生
产有好处,而且对其它新产品的生产有好处。通过对这些改良后
的工业化丝状真菌中涉及“前期”产品生产的基因进行修饰以便
使真菌产生新产品,这些有利突变现将对新产品的生产有好处。
这样一来,本发明可在开发新生产菌株时更省事。
-本发明能够“设计和建造”出重组丝状真菌。“设计和建造”指,
在单拷贝水平上,设计由所有将被导DNA区中的所需遗传修饰
组成。例如,假如要生产所需蛋白质,则将该蛋白质的一个或多
个表达盒整合进DNA区,并使位于同一DNA区、可能对所需蛋白
质的表达有负影响的内源基因失活。假如要生产混合蛋白质,设
计过程将包括调节不同蛋白质的表达水平至所需比率。在要以预
定比率导入一系列新代谢活性、构成新代谢途径的一部分的代谢
途径改造中,后一种情况可能尤其有利。一旦在单拷贝水平上于
这些DNA区中建立了所需遗传修饰的设计,建造过程将可启动,
这意味着通过对含所需遗传修饰的区进行基因转变和/或扩增而
增加所设计的单拷贝区,直至达到所需生产水平为止。
实施例
命名
A.niger 黑曲霉
P.Chrysogenum 产黄青霉
S.clavuligerus 带小棒链霉菌
A.nidulans 构巢曲霉
phyA 黑曲霉phyA基因,编码肌醇六磷酸酶。
amdS 构巢曲霉amdS基因,编码乙酰胺酶(Corrick等,1987,基因,
53:63-71)
cefE 带小棒链霉菌cefE基因,编码脱乙酰氧基头孢霉素C合成
酶(Kovacevic等,1989,细菌学杂志,171:754-760)
cefF 带小棒链霉菌cefF基因,编码脱乙酰基头孢霉素C合成
酶(Kovacevic,S.和Miller,J.R.1991细菌学杂志,173:
398-400)
niaD 产黄青霉niaD基因,编码硝酸盐还原酶(Haas等,
1996,生物化学、生物物理学学报(Biochem.Biophys.Acta)
1309:81;-84)
glaA 黑曲霉glaA基因,编码葡糖淀粉酶
gpdA 构巢曲霉gpdA基因,编码甘油醛-3-磷酸脱氢酶(Punt
等,1988基因69:49-57)
pcbC 产黄青霉pcbC基因,编码异青霉素N合成酶(IPNS)(Carr
等,1986基因48:257-266)
PgpdA gpdA启动子
PglaA glaA启动子
PpcbC pcbC启动子
PenDE penDE终止子
TamdS amdS终止子
TglaA glaA终止子
GLA 黑曲霉葡糖淀粉酶蛋白质
缩写
CHEF 钳位均匀电场(电泳)
TAFE 横向交变电场电泳
Kb 千碱基
bp 碱基对
ADCA 氨基脱乙酰氧基头孢菌酸
ADAC 氨基脱乙酰基头孢菌酸
oligo 寡核苷酸
PCR 聚合酶链式反应
寡核苷酸
1. 5′-gta gct gcg gcc gcc tcc gtc ttc act tct tcg ccc gca
ct-3′
2. 5′-caa agg gca tgc ggc cgt atc ggc cgg tga caa aca tca
ttc aac gcc-3′
3. 5′-atg ttt aag ctt ggc cga tac ggc caa aac acc ttt gat
ttc-3′
4. 5′-caa gtt gcg gcc gct cct cac taa cga gcc agc aga tat
cga tgg-3′
5. 5′-aag ctt atg cgg ccg cga att cga gct ctg tac agt gac-
3′
6. 5′-cgg tac gtg cgg ccg ctc gta cca tgg gtt gag tgg tat
g-3′
7. 5′-ata tgt gcg gcc gct tta cat ggt caa tgc aat tag atg
gtg g-3′
8. 5′-ata act cta gag gcc cta ccg gcc ttt gca aat ata ctg
taa gaa cc-3′
9. 5′-gta tat tct gca ggg ccg gta ggg cca aca gtt tcc gca
ggt g-3′
10. 5′-gta tgg gcg gcc gct tta caa cta gaa tat ggg aac ctg
tgg g-3′
11. 5′-ctc gag tgc ggc cgc aaa gct agc ttg ata tcg aat tcc
tta tac tgg gcc tgc tgc att g-3′
12. 5′-gtc cat atg ggt gtc tag aaa aat aat ggt gaa aac ttg
aag gcg-3′
13. 5′-cat atg gcg gac acg ccc gta ccg atc ttc-3′
14. 5′-atg cat tgg ctc gtc atg aag agc cta tca tcc ggc ctg
cgg ctc gtt ctt cgc-3′
15. 5′-cag cta ccc cgc ttg agc aga cat c-3′
16. 5′-gtc agg gaa gaa cac gag ggc gca g-3′
17. 5′-ccc tct ctt cgt cgt tgt cca cgc c-3′
18. 5′-atg tcc ttg gcc gac ttc agc tcg g-3′
19. 5′-gac gag cca atg cat ctt ttg tat g-3′
20. 5′-cgg gta ctc gct cta cct act tcg g-3′
21. 5′-gcc cag tat aag gaa ttc gat atc aag-3′
22. 5′-agg gtc gac act agt tct aga gcg g-3′
23. 5′-gac gtt atc gga cgg aga ctc agt g-3′
24. 5′-gcc tac tct gtt ctg gag agc tgc-3′
25. 5′-ccc cca tcc cgg tca cgc act cgc g-3′
26. 5′-cac aga gaa tgt gcc gtt tct ttg g-3′
27. 5′-tca cat atc ccc tac tcc cga gcc g-3′
28. 5′-gtc gcg tat ccc agg-3′
29. 5′-gtc aaa gga tat gca tac-3′
30. 5′-agc tta tgc ggc cgc gaa ttc agg tac cgt atc tcg aga-
3′
31. 5′-aat ttc tcg aga tac ggt acc tga att cgc ggc cgc ata-
3′
32. 5′-gtg cga ggt acc aca atc aat cca ttt cgc-3′
33. 5′-atg gtt caa gaa ctc ggt agc ctt ttc ctt gat tct-3′
34. 5′-aga atc aag gaa aag gct acc gag ttc ttg aac cat-3′
35. 5′-atc aat cag aag ctt tct ctc gag acg ggc atc gga gtc
ccg-3′
36. 5′-gac cat gat tac gcc aag ctt-3′
37. 5′-gga tcc tta act agt taa gtg ggg gcc tgc gca aag-3′
38. 5′-tta act agt taa gga tcc aca atc aat cca ttt cgc-3′
39. 5′-gct cta gag cgg ccg cga att cat ccg gag atc c-3′
40. 5′-ctt tgc gca ggc ccc cac-3′
41. 5′-tgc agg gta aat cag gga-3′
42. 5′-tcc gct aaa ggt ggt cgc g-3′
43. 5′-ccc cag cat cat tac acc tc-3′
44. 5′-aaa gga ccc gag atc cgt ac-3′
45. 5′-tct cga tac caa ggt cac cac ggg c-3′
46. 5′-gca tcc atc ggc cac cgt cat tgg a-3′
47. 5′-atc cag acc agc aca ggc agc ttc g-3′
48. 5′-tcc gca tgc cag aaa gag tca ccg g-3′
49. 5′-gtc gac tta act agt taa ggc ttc aga cgc agc gag-3′
50. 5′-tta act agt taa gtc gac aca atc aat cca ttt cgc-3′
51. 5′-aga tct tta act agt taa gtg gcc tga aca gtg ccg-3′
52. 5′-tta act agt taa aga tct aca atc aat cca ttt cgc-3′
材料与方法
总过程
标准分子克隆技术,如DNA的分离,凝胶电泳,核酸的酶切消化修饰,Southern分析,大肠杆菌转化等,均按Sambrook等(1989)“分子克隆:实验室手册”,冷泉港实验室,冷泉港,纽约,和Innis等(1990)“PCR程序,方法及应用指南”Academic Press,圣迭哥所述进行。合成寡脱氧核苷酸得自ISOGEN Bioscience(Maarssen,荷兰)。DNA序列分析在Applied Biosystems 373A DNA测序仪上按厂家建议执行。
黑曲霉的转化
黑曲霉的转化按Tiburn,J.等(1983)基因
26,205-221和Kelly,J.& Hynes,M.(1985)EMBO J.,
4,475-479所述方法进行,其中有以下修改:
将孢子接种于曲霉基本培养基中,在转速为300rpm的旋转摇床中30℃生长16小时。曲霉基本培养基每升中含有:6g NaNO3;0.52g KCl;1.52g KH2PO4;1.12ml 4M KOH;0.52g MgSO4·7H2O;10g葡萄糖;1g酪蛋白水解物,22mg ZnSO4·7H2O;11mg H3BO3;5mg FeSO4·7H2O;1.7mgCoCl2·6H2O;1.6mg CuSO4·5H2O;5mg MnCl2·2H2O;1.5mg Na2MoO4·2H2O;50mg EDTA;2mg核黄素;2mg盐酸硫胺素;2mg烟酰胺;1mg盐酸吡哆醇;0.2mg泛酸;4μg生物素;10ml青霉素(5000IU/ml)链霉素(5000UG/ml)溶液(Gibco)。
-Novozym 234(Novo Industries)取代解旋酶用于制备原生质体;
-原生质体形成(60-90分钟)后,加入KC缓冲液(0.8M KCl,9.5mM
柠檬酸,pH6.2)至终体积为45ml,原生质体悬浮液用悬桶式转
头于4℃在3000rpm离心10分钟,再将原生质体重新悬浮于20ml
KC缓冲液,然后加入25ml STC缓冲液(1.2M山梨糖醇,10mM
Tris-HCl pH7.5,50mM CaCl2)。该原生质体悬浮液在悬桶式转
头上于4℃在3000rpm离心10分钟,在STC缓冲液中洗涤并重
新悬浮于STC缓冲液中,浓度为108个原生质体/ml;
-向200μl原生质体悬浮液中加入溶于10μl TE缓冲液(10mM
Tris-HCl pH7.5,0.1mM EDTA)的DNA片段,及100μl PEG溶
液(20%PEG4000(Merck),0.8M山梨糖醇,10mM Tris-HCl
pH7.5,50mM CaCl2);
-将DNA-原生质体悬浮液室温温育10分钟后,缓缓加入1.5ml PEG
溶液(60%PEG4000(Merck),10mM Tris-HCl pH7.5,50mM CaCl2)
并反复混合试管。室温温育20分钟后,将悬浮液用5ml 1.2M山
梨糖醇稀释,颠倒混合,室温4000rpm离心10分钟。原生质体
重新轻柔地悬浮于1ml 1.2M山梨糖醇中,铺板于选择性再生
培养基,该培养基由不含核黄素、盐酸硫胺素,烟酰胺、吡哆
醇、泛酸、生物素、酪蛋白水解物和葡萄糖的曲霉基本培养基
组成,并补加有10mM乙酰胺作为唯一氮源,1M蔗糖,并以2%
细菌学1#琼脂(Oxoid,England)固定成形。30°温育6-10天
后,将这些平板影印至选择性乙酰胺平板(曲霉基本培养基其中
2%葡萄糖取代蔗糖,1.5%琼脂糖取代琼脂)上。30℃生长5-10
天后分离出单个转化体。
产黄青霉的转化
用Ca-PEG介导的原生质体转化程序。原生质体的制备和产黄青霉的转化按Gouka等,生物技术杂志
20(1991),189-200所述方法进行,其中有以下修改:
-转化后,将原生质体铺板于选择性再生培养基平板上,该培养基
组成为曲霉基本培养基,以1.2M蔗糖渗透稳定,含0.1%乙酰胺
为唯一碳源,以1.5%细菌学1#琼脂(Oxoid,England)固定成
形。
-25℃温育5-8天后转化体出现。
TAFE
大小在20-50Kb范围的DNA片段在1%琼脂糖凝胶上经TAFE分离,所用为Beckman GenelineTMII仪器(Beckman),按厂家说明进行。电泳参数:4Sec A,4Sec B脉冲时间,于300mA电流下14℃电泳近18h。
CHEF
大于约50Kb的DNA片段在1%琼脂糖凝胶上,用CHEF-DRII模块(BioRad),配备Pulsewave 760 Switcher,200/2.0型电源及CHEF电泳室,经CHEF电泳而分离。电泳参数:启动时间50Sec,最终时间90Sec,起始比率1.0,200V电压下电泳20至24小时,14℃。
定量测定PCR产物和杂交信号
PCR产物和杂交信号的定量分别从溴乙锭染色的琼脂糖凝胶的照片或从放射自显影照片上,按ImageQuaNTTM软件(Molecular Dynamics)进行。
DNA标记和杂交
DNA标记和杂交依照ECLTM直接核酸标记和检测系统(AmershamLIFE SCIENCE,Little Chalfont,England)进行。
在黑曲霉菌落上进行的盒式和DNA-旗特异性PCR操作
将黑曲霉孢子接种于PDA平板(马铃薯葡萄糖琼脂,Oxoid;按厂家建议制备)。30℃生长48小时后,将单个菌落菌丝体的1/2-1/4转移至50μl novozym[5mg Novozym 234(Novo Industries)/每mlKC(0.8M KCl,9.5mM柠檬酸,pH6.2)],37℃温育1小时。随后,加入300μl DNA稀释缓冲液(10mM Tris-HCl pH7.5;10mM NaCl;1mMEDTA),悬浮液100℃煮沸5分钟,然后猛烈摇动破坏未受损菌丝体。以5μl这样的混合物作模板,在50μl包含5μl 10x Super Taq PCR缓冲液1(HT Biotechnology公司),8μl dNTP(每种1.25mM),20-80ng每种寡核苷酸和1U Super Taq(HT Biotechnology公司,剑桥,英国的PCR反应混合物中进行反应)。寡核苷酸的最佳用量需对每批购买的药品实验后确定。
在DNA-扩增仪(如Perkin-Elmer,Hybaid)中进行25个扩增循环(每循环为:1分钟94℃,1分钟55℃和1.5分钟72℃)和最后延伸步骤72℃ 7分钟。
至于DNA旗检验,用寡核苷酸40/41作引物对,phyA盒式PCR检验的寡核苷酸对为42/43,amdS盒检验的寡核苷酸对为15/16。随后分析PCR产物,在TBE缓冲液(0.09M Tris,0.09M H3BO3,2mM EDTA;pH8.3)中于2%的琼脂糖凝胶上经电泳分析20μl PCR混合物。
黑曲霉的定向特异性PCR操作
DNA-模板的制备和PCR反应条件与进行青霉菌的定向PCR检验所用的一样。引物对选用寡核苷酸46/48以确定amdS盒是否打靶到glaA靶位点的邻位,用寡核苷酸对46/47确定phyA盒是否打靶到glaA靶位点的邻位。用的PCR条件如青霉菌定向PCR检验所述。
产黄青霉转化体的定向PCR
将近1/3的4天龄菌落于补加5mg/ml NovozymTM 234的50μl KC缓冲液(60g/l KCl,2g/l柠檬酸,pH6.2)中37℃温育2小时。随后加入100μl 10mM Tris,50mM EDTA,150mM NaCl,1%SDS,pH8和400μl QIAquickTM PB缓冲液(Quiagen公司,Chatsworth,USA)。将提取物重新轻柔悬浮,上样于QIAquickTM旋转柱。将柱置于微量离心机中1,3000rpm离心1min,再用500μl QIAquickTMPE缓冲液洗一次。乙醇残迹经最后快速旋转去除。向柱中加入50μl水,然后于13000rpm离心1min,可从柱中洗脱出染色体DNA(PCR模板)。PCR反应物包括10μl eLONGaseTMB缓冲液(Life Technologies,Breda,荷兰),14μl dNTP(每种1.25mM),1μl eLONGaseTM酶混合物,1μl模板,每种寡核苷酸30-50ng,终体积50μl。寡核苷酸的最佳用量需对每批药实验后确定。一般用30-50ng。反应循环条件如下:1x(2min 94℃),10x(15sec 94℃,30sec 55℃,4min 68℃);20x(15sec 94℃,30sec 55℃,首循环于68℃4min,然后每循环增加20sec),1x(10min 68℃)。将8μl样本上样于琼脂糖凝胶,分析PCR产物。
产黄青霉转化体的盒式PCR
将近1/3的4天龄菌落于加有5mg/ml NovozymTM234的200μl DVB缓冲液(10mM Tris,10mM NaCl,1mM EDTA,pH7.5)中37℃温育2小时。混合物煮沸8min,随后猛烈摇动以破坏未受损菌丝体。5μl这样的混合物作为PCR模板,在50μl包含5μl 10x Super Taq PCR缓冲液(HT Biotechnology公司,剑桥,英国),8μl dNTP(每种1.25mM),1U Super Taq和20-80ng的每种寡核苷酸的PCR反应物中反应。每批寡核苷酸的最佳用量需实验确定。反应循环条件为:25x(1min 94℃,1min 55℃,1.5min 72℃),1x(8min 72℃)。将15μl样本上样于琼脂糖凝胶,分析PCR产物。
染色体DNA分离
将黑曲霉和产黄青霉的孢子接种100ml锥瓶中的20ml曲霉基本培养基(见黑曲霉的转化),30℃以300rpm的速度旋转振荡培养20-24小时。长成的培养物取5-10ml接种于100ml新鲜曲霉基本培养基中,30℃300rpm旋转振荡下再培养20-24小时。培养结束后,经Miracloth(Cal Biochem)滤膜过滤收获菌丝体,用15mlKC缓冲液(0.8M KCl,9.5mM柠檬酸,pH6.2)洗。每克洗过的菌丝体(100ml培养物中可得3-5g)中加4ml KC缓冲液和0.25ml novozym[50mgNovozym 234(Novo Industries)/ml KC缓冲液],使原生质体在30℃经轻轻振荡生成。原生质体形成后,加KC缓冲液,使之达45ml,此原生质体悬浮液用悬桶式转头于4℃3000rpm离心5分钟。再将原生质体悬浮于20ml KC缓冲液。然后加入25ml STC缓冲液(1.2M山梨糖醇,10mM Tris-HCl pH7.5,50mM CaCl2),混匀后用悬桶式转头4℃下3000rpm离心5分钟,再悬浮于50ml STC缓冲液,再用悬桶式转头4℃2500xg离心5分钟。向每0.3至0.6ml原生质体沉淀中加60μl蛋白酶K(20mg/ml)和1ml低溶点琼脂糖(0.8%溶于1M山梨糖醇/0.45MEDTA)。振荡混匀后,将悬浮液转移至预先制成的Plexiglass模具的孔中,置于冰上约15分钟。然后,将琼脂糖塞转移至2.5ml十二烷基肌氨酸钠溶液(1%十二烷基肌氨酸钠溶于0.5M EDTA)。加入60μl蛋白酶K(20mg/ml),50℃温育16-20小时后,用10ml 50mM EDTA溶液替换十二烷基肌氨酸钠溶液。50℃温育2小时后,用10ml新鲜的50mMEDTA替换原EDTA溶液,再50℃温育2小时。最后,再用10ml新鲜的50mM EDTA溶液替换原EDTA溶液,将琼脂糖塞贮于4℃。
对染色体DNA琼脂糖塞的限制酶消化
将包含1-3μg染色体DNA的部分DNA琼脂糖塞(如上述制备)于1ml TE(10mM Tris-HCl pH7.5,1mM EDTA)中37℃温育1小时。重新换1mlTE缓冲液,继续于37℃温育1小时。然后TE缓冲液由200μl适应于每种限制酶的缓冲液(按供应商的建议)替换。37℃温育1小时后,重新更换限制酶缓冲液(50μl),65℃温育10分钟以溶解琼脂糖。最后,加入20-40单位的适当限制酶,在供应商就每种限制酶推荐使用的温度下,使消化混合物温育16小时。
amdS负选择过程
在多数衍生载体中,amdS选择标记基因位于DNA重复序列之间。故,在所识别的带有这些载体的转化体中,去除amdS标记基因既可通过盒内侧翼DNA重复序列的内部重组也可经单交换事件而对因整合而产生的重复序列同源重组达到。选择已丢失amdS选择标记的细胞可通过将细胞培养于含氟乙酰胺的平板上完成。有amdS基因的细胞将氟乙酰胺代谢为铵盐和对细胞有毒性的氟乙酸。结果,只有失去了amdS基因的细胞才能在含氟乙酰胺的平板上生长。
若要去除曲霉转化体中的amdS标记,则将其孢子铺板于选择性再生培养基(上述)上,该培养基内含5mM氟乙酰胺和5mM ureum代替10mM乙酰胺,含1.1%葡萄糖代替1M蔗糖,含1.1%代替2%的细菌学1#琼脂(Oxoid,England),30℃生长7-10天后,收获单菌落,铺板于0.4%马铃薯葡萄糖琼脂(Oxoid,England)上。
若去除青霉转化体的amdS标记,则将其孢子铺板于曲霉基本培养基上,该培养基含有10mM氟乙酰胺和5%葡萄糖,以1.5%细菌学1#琼脂(Oxoid,England)固定成形。25℃生长5-10天后,耐药菌落出现。
大肠杆菌生物分析
转化体在YPD琼脂培养基上长5天。大肠杆菌ESS2231作为指示细菌用于琼脂覆盖层中,它还含有Bacto-penase,能辨别青霉素和头孢霉素的产生,方法为本领域熟知(Guttierez等,分子普通遣传学,1991 225:56-64)。菌落周围的琼脂覆盖层澄清即可鉴定为表达cefE的转化体。
产黄青霉的工业化菌株改良
利用本领域技术人员熟知的标准诱变技术和筛选过程(Rowlands,1984,酶微生物技术(Enzyme Microb.Technol.)6:3-10),从Wis54-1255(ATCC 28089)中分离产黄青霉菌株CBS 649.97(于1997年4月11日存于真菌菌种保藏中心,Baarn,荷兰)。
己二酰-7-ADCA和己二酰-7-ADAC发酵
己二酰-7-ADCA和己二酰-7-ADAC的发酵生产和定量基本如对2-(羧乙基巯基)乙酰基-和3-(羧甲基巯基)丙酰基-7-ADCA(WO95/04148)的生产的描述,除了加入培养基的是3g/l己二酸而非3’-羧甲基巯基丙酸。合成的己二酰-7-ADCA和己二酰-7-ADAC应用作为参照物质。
黑曲霉摇瓶发酵
重组及对照黑曲霉菌株的大批孢子通过将孢子或菌丝体铺板于已按供应商建议制备好的PDA平板(马铃薯葡萄糖琼脂,Oxoid)上生产。30℃生长3-7天后,向平板中加0.01%Triton x-100收集孢子。用无菌水清洗后,将所选出的转化体和对照菌株的约107个孢子接种于摇瓶中,瓶内有20ml液态预培养基质,每升内含:30g麦芽糖·H2O;5g酵母浸出膏;10g水解的酪蛋白;1g KH2PO4;0.5g MgSO4·7H2O;0.03gZnCl2;0.02g CaCl2;0.01g MnSO4·4H2O;0.3g FeSO4·7H2O;3g吐温80;10ml青霉素(5000IU/ml)/链霉素(5000UG/ml);pH5.5。这些培养物于34℃生长20-24小时。将该培养物10ml接种至100ml黑曲霉发酵基质中,该发酵基质每升含:70g麦芽糖糊精;25g水解的酪蛋白;12.5g酵母浸膏;1g KH2PO4;2g K2SO4;0.5g MgSO4·7H2O;0.03gZnCl2;0.02g CaCl2;0.01g MnSO4·4H2O;0.3g FeSO4·7H2O;10ml青霉素(5000IU/ml)/链霉素(5000UG/ml);用4N H2SO4调pH至5.6。这些培养物于34℃生长6天。对取自发酵液的样本离心(10分钟,5000rpm,用悬桶式离心机),收集上清。对这些上清进行葡糖淀粉酶或肌醇六磷酸酶活性分析(见下文)。
葡糖淀粉酶活性分析
葡糖淀粉酶活性测定如下:将在0.032M NaAC/HAC pH4.05中6倍稀释的培养物上清样本10μl与115μl于0.032M NaAc/HAc pH4.05中的0.2%(W/V)对硝基苯基α-D-葡糖吡喃糖苷(Sigma)一起温育。室温30分钟后,加入50μl 0.3M Na2CO3,在波长405nm处测吸收值。A405nm定量的是GLA产量。
肌醇六磷酸酶活性分析
取烧瓶或微滴板黑曲霉发酵产物的100μl上清(如有必要则稀释)(参照为100μl双蒸水)加入900μl混合物中,该混合物包含0.25M醋酸钠缓冲液pH5.5,1mM肌醇六磷酸(钠盐,Sigma P-3168),37℃温育30分钟。加1ml 10%TCA(三氯醋酸)终止反应。然后加入2ml试剂(3.66g FeSO4·7H2O溶于50ml钼酸铵溶液(2.5g(NH4)6MO7O24·4H2O和8ml H2SO4,用半水稀释至250ml)。测690nm处对兰色的分光光度计吸收值。将测量值对照0-1mMol/l范围内的磷酸标准曲线可指示释放的磷酸量。
微生物的培养
带小棒链霉菌株ATCC27064于27℃在大豆胰朊酶(tryptic)肉汤(Difco)中培养。
产黄青霉菌株于25℃在完全YPD培养基(1%酵母浸膏,2%蛋白胨,2%葡萄糖)中培养。固体培养基则加2%Bacto琼脂。转化体的纯化通过反复在YPD琼脂上培养而完成。利用单个稳定菌落接种琼脂斜面以产生孢子。
黑曲霉菌株于30℃在含PDA的平板中培养。通过反复培养于PDA平板上从而纯化转化体。利用单个稳定菌落再制成斜面以产生孢子。
大肠杆菌菌株的培养依照标准程序(Sambrook)。
黑曲霉实施例
1.1黑曲霉宿主的选择和鉴定
1.1.a原理说明
基因组中重复DNA区的存在是应用基因转变为工具扩增所整合的重组表达盒必需的。例如,黑曲霉葡糖淀粉酶高产菌株的glaA基因就可出现在这样的扩增后DNA区中。为了鉴定适于生产工业酶的宿主,我们筛选出具有改进的葡糖淀粉酶生产能力的黑曲霉菌株。在此我们描述如何选择和鉴定这些含有所要求的DNA扩增子的黑曲霉菌株。
1.1.b选择黑曲霉突变体
从黑曲霉CBS513.88(于1988年10月10日存放)中选择出好几个在摇瓶水平上显示有改进的葡糖淀粉酶生产能力的突变菌株。对黑曲霉CBS513.88的孢子经紫外线处理而进行诱变。检验存活孢子(近1%)在材料与方法部分中所述的摇瓶发酵中生产葡糖淀粉酶的能力。生长6天后按材料与方法中所述测定葡糖淀粉酶活性。
可选出好几株显示葡糖淀粉酶生产水平提高,(甚至高达600U葡糖淀粉酶/ml)的黑曲霉突变体。黑曲霉亲代株CBS513.88所能达到的水平近200U葡糖淀粉酶/ml。最好的黑曲霉生产突变株已作为黑曲霉CBS646.97于1997年4月11日保藏于荷兰Baarn的真菌菌种保藏中心。
1.1.c黑曲霉CBS646.97的遗传定性
为测定上述葡糖淀粉酶生产的增加是否glaA位点扩增的结果,从黑曲霉突变株CBS646.97及其亲代黑曲霉CBS513.88分离染色体DNA。对其进行Southern分析,用EcoRI和SalI消化,并以葡糖淀粉酶/肌醇六磷酸酶DNA片段(glaA/phyA融合体:来自pFYT3的EcoRI/BamHI片段,描述于EP专利申请0420358A1))为探针探查。放射自显影清楚地显示,葡糖淀粉酶改进型突变株包含多个(3-4个)glaA基因拷贝。
1.1.d黑曲霉突变株中glaA扩增子的大小
TAFE分析揭示所选黑曲霉突变株中的大DNA片段均得到扩增。按材料与方法所述从黑曲霉初始株CBS513.88及黑曲霉CBS646.97株中分离染色体DNA。用HindIII和八碱基限制酶NotI,SwaI,AscI和PacI消化,再用来自pAB6-1(见EP专利申请0357127A1)的几个glaA位点特异性探针对其进行TAFE分析。亲代株和突变株之间在由上述限制酶处理的glaA杂交性DNA片段的大小方面没有区别。
测得最大杂交DNA片段各例均有,限制酶SwaI消化片段:近80Kb。唯一所见区别在于glaA杂交性DNA带的强度。尽管很难对杂交glaA带的强度准确定量,TAFE放射自显影仍清楚地显示黑曲霉突变株CBS646.97包含额外拷贝的所见glaA扩增子。
此外,我们无法找到另一个在所检测的glaA扩增子以外切割的限制酶。这意味着glaA扩增子包含至少80Kb或更大。
1.2在黑曲霉CBS646.97株的各扩增子上修饰glaA位点
1.2.a原理说明
如前述(EP专利申请0635574A1),可用“无标记基因”法在黑曲霉中适当缺失一个基因组靶基因。在那个专利申请中,有一个实施例广泛描述了如何在黑曲霉CBS513.88的基因组中缺失glaA特异性DNA序列。所述置换型载体通过双交换同源重组整合进黑曲霉gpdA基因组序列中。缺失载体包含同源于glaA靶位点的DNA区,还包含由glaA启动子序列驱动的amdS选择标记基因。此外,此载体中amdS选择标记基因两侧均是glaA序列,它们是直接DNA重复序列,可促进随后amdS标记基因的恰当消失。
这样的载体指导amdS标记基因取代glaA基因。随后对这些转化子进行氟乙酰胺反选择,amdS标记基因可通过3’-glaA DNA直接重复序列之间的内部重组而正确缺失,从而产生最终不含任何外源DNA序列的无标记基因型ΔglaA重组黑曲霉CBS513.88菌株。
显然这种“无标记基因”法尤其适用于反复修饰和缺失宿主基因组序列。
此实施例描述了用三个一套“特别设计的”pGBDEL置换型载体所进行的三轮连续转化过程中,黑曲霉CBS646.97的所有glaA启动子及编码序列的恰当缺失。为了进行这三轮缺失,构建并应用到多于三个或少于三个的一套相同的glaA基因置换型pGBDEL载体。每个pGBDEL载体中有一个唯一的限制酶识别DNA序列,目的是经Southern分析而从衍生的ΔglaA重组黑曲霉菌株中单独识别并显示每个截短的glaA位点(述于实施例1.2.e)。每个pGBDEL载体中的唯一限制位点位于截短的glaA位点5’边缘。然而,每个pGBDEL载体中用于指导基因置换载体打靶到其中一个glaA位点的各种glaA序列稍有不同。结果黑曲霉宿主中每个截短的glaA位点也略有不同。包含这种微小差异是为了在用快速PCR检验即所谓“DNA-旗”检验(图1)分析菌落时能显示每个截短的glaA位点。尽管非必需,但这种掺入DNA特点结合本文所述发生于黑曲霉glaA扩增子之间的基因转变表型,对于监控打靶到黑曲霉宿主中三个截短glaA位点之一处的(肌醇六磷酸)表达盒的扩增这一“建造”过程将特别有用。有关利用黑曲霉中glaA扩增子之间的转变事件衍生rDNA菌株以生产酶的这一“建造”过程将详述于下文(实施例1.5和1.6)。
1.2.b glaA置换型pGBDEL载体的描述
构建的pGBDEL置换型载体包含:
-2Kb大小的5’-glaA靶序列,始于HindIII位点,直至glaA启动子序列内XhoI单切点上游200bp(pGBDEL-5)、180bp(pGBDEL-9)和140bp(pGBDEL-11);
-2Kb大小的3’-glaA靶序列,从glaA终止密码至SalI位点;
-amdS基因作为选择标记,受控于gpdA启动子;
-两个1Kb大小的3’-glaA DNA直接重复序列,用于促进后续amdS选择标记基因的去除;和
-一个额外的限制位点(pGBDEL-5中为BamHI位点,pGBDEL
-9中为SalI位点,pGBDEL-11中为BglII位点),位于glaA截短处。
通过应用这些pGBDEL置换型载体,黑曲霉宿主中包含2Kb大小的glaA启动子序列(在pGBDEL-5,pGBDEL-9,pGBDEL-11中分别为XhoI上游200bp,180bp或140bp)直至ATG起始密码子和完整glaA编码序列的4.3Kb glaA序列将缺失。图示见图2。
1.2.c pGBGLA中间载体的构建
为构建中间载体pGBGLA16,合成两个包含限制酶:NotI,EcoRI,KpnI和XhoI识别位点的寡核苷酸(30/31)。寡核苷酸5’-末端衔接上一个HindIII限制位点,3’-末端衔接上一个EcoRI限制位点。将该寡核苷酸插入pTZ18R则EcoRI位点将被破坏。
寡核苷酸30和31均克隆进质粒pTZ18R的EcoRI和HindIII位点。进行监控消化,以确定所需KpnI,XhoI,NotI和HindIII酶限制位点是否已正确掺入,EcoRI位点是否已遭破坏。衍生所得质粒命名为pTMl(见图3)。
从pTMl,经EcoRI和KpnI限制酶消化后通过凝胶电泳分离并纯化出EcoRI/KpnI DNA片段,以插入pGBDEL4L中包含PgpdA/amdS序列的EcoRI/KpnI片段,pGBDEL4L的构建详述于我们的以前专利申请(EPA0635574A1)中。pGBDEL4L用EcoRI/KpnI和XhoI消化(XhoI是为了避免克隆另一个大小相同的pGBDEL4L DNA片段)。包含PgpdA/amdS序列的正确EcoRI/KpnI DNA片段经凝胶电泳纯化,并克隆进pTMl的EcoRI/KpnI位点。此中间载体命名为pGBGLA16(见图3)。
为在pGBGLA16中正确插入大小为2.2Kb的3’-glaA序列,该片段首先需进行一些修饰。用pAB6-1为模板,两套寡核苷酸(32/33和34/35)为引物进行融合PCR,以破坏2.2Kb 3’-glaA序列内的KpnI限制位点,并在5’-边界(KpnI)和3’-边界(XhoI,HindIII)产生相应克隆位点。模板pAB6-1在pUC19中包含一个大小为16Kb的Hind III大片段,内含glaA基因组位点(其分子克隆详述于EPA 0357127A1)。
在第一轮PCR中,用寡核苷酸32和33为引物扩增3’-glaA序列的一部分(1Kb)。第二轮PCR中,用寡核苷酸34和35扩增3’-glaA侧翼中剩下的1.2Kb序列。
经凝胶电泳纯化后,两个扩增片段作为模板,用寡核苷酸32和35作引物对进行融合PCR。该PCR融合的图示见图4。
所获2.2Kb 3’-glaA PCR融合片段经凝胶电泳纯化,用KpnI和XhoI消化,并经分子克隆插入pGBGLA16的KpnI/XhoI位点,产生pGBGLA18(见图4)。
1.2.d glaA置换型pGBDEL载体的构建
为获得三个最终的pGBDEL载体,需将5’-glaA靶序列、每个载体所需的唯一限制位点及作为直接重复序列的1Kb 3’-glaA序列插入pGBGLA18的Hind III/NotI位点。为此,进行三次不同的融合PCR,以构建pGBDEL5,-9和-11。
构建pGBDEL5
用两个合成寡核苷酸进行第一轮PCR,以在3’边界修饰5’-glaA靶序列,这两个寡核苷酸其一为36,包含pUC19核苷酸序列的一部分和5’-glaA序列的HindIII位点;另一为37,与glaA启动子中XhoI位点上游约200bp处匹配,其包含一个额外的BamHI位点,所有读框均有的终止密码及1Kb 3’-glaA直接重复序列的最初18个核苷酸(5’-边界)。用两个寡核苷酸38和39进行第二轮PCR,以在5’-边界修饰3’-glaA直接重复序列,其中38为37的逆向,39相配对于3’-glaA直接重复序列中EcoRI位点附近的序列,并另附加XbaI和NotI位点(分别适用于克隆进pTZ18R和pGBGLA18中)。两轮PCR均以pAB6-1为模板。两轮PCR的扩增DNA片段均用凝胶电泳分离,并作为模板,以36和39为引物进行融合PCR。所获3Kb大小的DNA融合片段经凝胶电泳纯化,经HindIII和NotI消化,再分子克隆进入pGBGLA18的恰当位点,产生第一个基因置换型载体pGBDEL5(见图5)。
构建pGBDEL9
第一轮PCR用寡核苷酸36和49进行,49(如上述)与glaA启动子中XhoI位点上游约180bp处匹配,它包含一个额外的SalI位点,所有读框均有的终止密码及1Kb 3’-glaA直接重复序列的最初18个核苷酸(5’-边界)。用与49逆向的寡核苷酸50,以及39进行第二轮PCR,以在5’-边界修饰3’glaA直接重复序列。两轮PCR均以pAB6-1为模板。扩增片段经凝胶电泳分离,用作融合PCR(以寡核苷酸36和39为引物)的模板。所获3Kb大小的DNA融合片段经凝胶电泳纯化,用HindIII和NotI消化,再分子克隆进pGBGLA18中的恰当位点,产生第二个glaA基因置换型载体pGBDEL9(见图6)。
构建pGBDEL11
第一轮PCR用寡核苷酸36和51进行,寡核苷酸51与glaA启动子中XhoI位点上游的约140bp处配对,它包含一个额外的BglII位点,所有读框均有的终止密码及1Kb 3’-glaA直接重复序列的最初18个核苷酸(5’-边界)。用寡核苷酸为52和39进行第二轮PCR,以在5’-边界修饰3’-glaA直接重复序列,其中52为51的逆向。
两轮PCR均以pAB6-1为模板,扩增片段均用凝胶电泳分离并作为融合PCR的模板(以36和36为引物)。所获3Kb大小的DNA融合片段经凝胶电泳纯化,用HindIII和NotI消化,再分子克隆进pGBGLA18的恰当位点,产生第三个基因置换型载体pGBDEL11(见图7)。
1.2.e黑曲霉中glaA启动子和编码序列的缺失及特异性“DNA-旗”的掺入。
本实施例描述了用pGBDEL载体和“无标记基因”(MARRER-GENEFREE)技术进行三轮成功的转化,缺失黑曲霉646.97中的所有三个glaA基因。
用XhoI和HindIII使pGBDEL载体线性化,并基于事实:rDNA盒侧翼是与宿主基因组的glaA靶位点同源的DNA序列,可通过宿主某个glaA位点上的双交换事件使pGBDEL载体掺入,使glaA序列被pGBDEL载体上截短的glaA位点取代。然而,pGBDEL载体通过双交换事件打靶的频率有限,并取决于宿主的glaA基因组靶位点数。故尽管用了双侧均包含大小为2Kb的glaA同源区的线性载体,但大部分产生的转化体仍包含随机整合的载体。
然而,有所需遗传特征的转化体易于选择并检定,可用a)基于PCR的DNA旗检验,b)定向特异性PCR过程和c)详细的Southern分析(如材料与方法中所说明的)。
最后,一旦经遗传检定了正确的转化体,便用氟乙酰胺反选择过程去除amdS基因。由于掺入的3-glaA DNA重复序列为PgpdA/amdS标记基因盒的侧翼,每轮后amdS选择标记均因内部重组事件而去除。对如此获得的修饰的黑曲霉菌株进行多轮转化,以去除和修饰所剩glaA位点。
用pGBDEL5修饰黑曲霉CBS646.97中的第一个glaA扩增子
用5μg线性化(HindIII/XhoI)pGBDEL5 DNA转化黑曲霉CBS646.97。在含乙酰胺的选择性平板上选择转化体,重复生长于选择培养基后制备孢子。
选择出限制数目的转化体并分离染色体DNA,用KpnI和BamHI消化,并以5’-glaA HindIII/XhoI和2.2Kb SalI 3’-glaA片段为探针进行Southern分析。见图8,比较了宿主菌株的ΔglaA位点和转化体中两个剩余ΔglaA位点的BamHI杂交模式。
选择出示有预期杂交模式的转化体(如图9示)。
随后,对这些转化体进行反选择。将孢子接种于含氟乙酰胺的培养基(详见材料与方法)上。平均有所接种孢子的1-2%可在这些选择条件下生长。直接对长成菌落的菌丝体用amdS特异性寡核苷酸15和16作引物进行盒式PCR分析,揭示所有生长细胞均为重组细胞,丢失了amdS标记基因。
奇怪的是,对许多重组细胞进行详细的Southern分析显示,大多数杂交模式(约90%)为亲代株646.97特征型,而不是所预期的一个截短glaA位点和剩下两个无损glaA位点的杂交模式。此外,杂交DNA片段的强度显示glaA扩增子的数量在这些amdS阴性菌株中并未减少。约10%的细胞显示预期的杂交模式(如图10示)。故,那些细胞中,amdS标记基因确实经旁侧的3’-glaA直接重复序列间的内部重组而缺失,如预期的那样。
多数amdS阴性菌株中这一预料之外的重组事件只能解释为一种称为基因转变的遗传现象的发生。这一特征后来(见实施例1.5和1.6)用于对定向在黑曲霉CBS646.97宿主截短的glaA扩增子的酶编码表达盒进行扩增。
显示有针对一个截短的glaA位点和两个无损glaA位点的杂交模式的一个amdS阴性菌株(如图10),命名为黑曲霉GBA-201,用pGBDEL-9对其进行第二次转化。
用pGBDEL9修饰黑曲霉CBS646.97中的第二个glaA扩增子。
用5μg线性化(HindIII/XhoI)pGBDEL9 DNA转化黑曲霉GBA-201。同样在含乙酰胺的选择平板上选出转化体,并经重复培养于选择培养基而制备孢子。因为pGBDEL9的打靶也可发生在以前被BamHI标记的宿主glaA截短位点,只筛选出显示仍有该截短“BamHI”glaA位点的那些转化体,并经Southern分析。为此,先对这些转化体的菌丝体进行基于PCR的“DNA-旗”检验。只对那些显示“BamHI”截短glaA位点特征性的200bp DNA片段的转化体进行PCR分析,再经Southern详细分析,如上述。
随后,显将示正确杂交模式(图10)的转化体孢子接种于含氟乙酰胺的培养基(详见材料与方法)上。同样,平均1-2%的接种孢子能生长在这些选择条件下。所有这些孢子似乎均丢失了amdS基因,用盒式PCR检验分析这些菌落的菌丝体揭示了这一点。
作为第二选择标准,进行了“DNA-旗”检验。只对那些显示所预期DNA旗模式(200和220bp两条带,分别为“BamHI”截短glaA扩增子和“SalI”截短glaA扩增子的特征模式)的转化体如上述进行详细Southern分析。
显示两个截短glaA扩增子和一个无损glaA扩增子的杂交模式(图12)的一个amdS阴性菌株,命名为黑曲霉GBA-202,用pGBDEL-11对它进行第三次转化。
用pGBDEL11修饰黑曲霉646.97的第三个glaA扩增子
用5μg线性化(HindIII/XhoI)pGBDEL-11 DNA转化黑曲霉GBA-202。同样在乙酰胺选择平板上选出转化体并重复培养于选择培养基上获得孢子。同样在本例中,pGBDEL11的打靶可发生在以前的宿主glaA截短扩增子上。通过“DNA-旗”检验,只选择出那些仍显示有200和220bp片段(表示前两个截短的“BamHI”和“SalI”glaA扩增子)的转化体,对其进行Southern分析。
对显示如图13的正确杂交模式的转化体进行反选择程序,以获得无标记基因的重组体。同样,只有1-2%的接种孢子能在这些选择条件下生长。它们经应用amdS盒式PCR检验其菌丝体发现均丢失PgpdA/amdS标记基因盒。
“DNA-旗”检验作为第二选择标准也执行了。只对那些显示有所预期三条带200,220和300bp DNA旗模式的转化体进行如上述的详细Southern分析,这三条带分别指示“BamHI”,“Sal”和“BglII”截短的glaA扩增子的存在。
显示有所有三个截短glaA扩增子的杂交模式(图14)的一个amdS阴性菌株命名为黑曲霉ISO-505。
1.3描述并构建pGBAAS-1和pGBTOPFYT-1表达载体
1.3.a原理
将包含一种酶的表达盒的pGBTOP载体,与含amdS选择标记基因的命名为pGBAAS-1的载体共转化黑曲霉宿主菌株后被导入该菌株。两个载体均含有两个同源于黑曲霉宿主菌株glaA位点的DNA区,可指导线性化质粒对黑曲霉ISO-505的其中一个截短的glaA位点打靶。这些区,每个约2Kb大,均同源于glaA序列下游非编码区,并均特异为3’-和3″-glaA区。这些区之间掺入了一个唯一的限制位点(pGBAAS-1中为XhoI,pGBTOPFYT-1中为HindIII),以便在转化前经消化去除大肠杆菌pTZ18R克隆载体后获得几乎完全互补于宿主glaA靶位点的DNA限制酶末端。
pGBAAS-1中amdS基因受强gpdA启动子驱动。应用这样一个启动子,获得的大多数转化体将只有一个单独整合的选择标记盒。具有这样的转化体是进行后续步骤所严格要求的。后续步骤为应用前述“无标记基因”技术选择无标记基因型重组菌株。多个amdS基因的存在,当然会影响在“一步反选择过程”中去除它们的频率,甚至可能无法去除。
pGBTOPFYT-1中的phyA受PglaA驱动。如已述,该载体的组成可设计成使线性化表达盒整合在黑曲霉ISO-505的其中一个glaA靶位点上。为了在一次转化中获得带有多拷贝表达盒但只有单拷贝协同载体(最终获得无标记基因型重组菌株所必需)且均打靶到宿主基因组的相同glaA靶位点上的转化体,两种盒的线性化DNA片段的比例须严格控制。
3.1.b构建整合载体pGBAAS-1
有关构建pGBAAS-1的所有细节均可在我们以前的专利申请EPA0635574A1中找到。在该专利申请中,详细描述了amdS选择标记基因载体pGBGLA-50的构建。只是因命名的理由,后来这个载体重新命名为pGBAAS-1(AAS意为曲霉菌amdS穿梭型)。物理图谱见图15。
3.1.c构建整合载体pGBTOPFYT-1
1.pGBTOPFYT-1的构建也可在上述相同专利申请中找到。该专利申请中详细描述了肌醇六磷酸酶表达载体pGBGLA-53的构建。同样仅因为命名,该表达载体后重新命名为pGBTOPFYT-1(TOP意为过度表达蛋白质的工具)。物理图谱见图16。
1.4开发无标记基因型的肌醇六磷酸酶黑曲霉生产菌株,它包含一个或多个肌醇六磷酸酶表达盒,诸盒均打靶到黑曲霉ISO-505的其中一个截短glaA扩增子中。
1.4.a原则说明
本实施例的目的是显示,表达盒(一个或更多拷贝)可通过与带有选择标记基因的载体共转染而正确导向宿主细胞基因组中的预定靶位点。第二是要显示,如何通过内部重组去除选择标记基因盒而又不丢失打靶的酶表达盒。
本例中选择的靶位点为4Kb glaA序列,就位于glaA终止密码的下游。
线性化质粒pGBTOPFYT-1和pGBAAS-1的整合通过黑曲霉ISO-505菌株的其中一个截短并标记的glaA位点处发生单交换而成。
为最终选择无标记基因型重组体,只筛选出那些有两个质粒以适当方式打靶到宿主基因组的同一已标记glaA位点的转化体,对其进行随后的氟乙酰胺反选择过程。
1.4.b用线性化pGBAAS-1和pGBTOPFYT-1 DNA共转化黑曲霉ISO-505。
pGBAAS-1和pGBTOPFYT-1分别用XhoI和HindIII消化而成为线性。经凝胶电泳去除2.8Kb的大肠杆菌克隆序列后用于转化。用1μgpGBAAS-1线性化DNA比5μg pGBTOPFYT-1线性化DNA的比例,如材料与方法所述转化黑曲霉ISO-505。在乙酰胺平板上选出转化体。其孢子经将单菌落铺板于PDA平板上而分离得到。对约500个转化体进行盒式PCR检验。
1.4.c共转化体的选择
具有amdS标记基因及phyA表达盒的转化体用肌醇六磷酸酶特异性寡核苷酸42/43作引物经盒式PCR检验而鉴别出来。含有一个或多个phyA表达盒的阳性转化体应显示一条大小为482bp的特异DNA带。共转化频率变化范围在10-50%之间。
1.4.d定向的amdS+,phyA+共转化体的选择
对所鉴别的共转化体用两套寡核苷酸46/47(第1套引物)和46/48(第2套引物)进行定向PCR检验。挑选出用这两套引物中的一套有阳性结果(扩增了4.2Kb DNA片段,见图17)的共转化体。阴性结果说明所有盒都是随机整合的,用第1套引物或第2套引物有阳性结果暗示phyA或amdS分别位于glaA靶位点的邻近位点。5-50%的共转化子表现为阳性。选出有限量的所鉴定菌株进一步进行Southern分析。
1.4.e用Southern分析对amdS打靶和phyA打靶的共转化体进行遗传分析
分离染色体DNA,用BglII消化,以amdS和2.2Kb SalI/XhoI3″-glaA DNA片段为杂交探针进行Southern分析。最经常观察到的6种杂交模式图示于18-23图。对显示有单个amdS拷贝,一个或多个phyA表达盒的特征性杂交模式(图20-23)的共转化体进行氟乙酰胺反选择过程,以最终去除amdS选择标记基因。
1.4.f选择不含amdS基因(amdS-)的肌醇六磷酸酶生产菌株。
因amdS表达盒的定向整合而形成的直接重复序列之间的重组将导致amdS盒的丢失,并可在氟乙酰胺培养基上进行选择。这里我们描述经应用氟乙酰反选择过程选择仍包含相邻的(多个)整合phyA表达盒而无amdS的重组菌株。将所选出的打靶amdS+/phyA+共转化体的孢子铺板于氟乙酰胺平板。首先,通过几轮连续地基于PCR的检验分析生长的菌落。先检验所有子代的基因型是否amdS-。方法是用amdS特异性引物如材料与方法中所述对菌落进行盒式PCR检验。令人惊讶的是,在后续基于PCR的“DNA-旗”检验中,似乎大多数子代同时也丢失了完整的glaA扩增子。此外,大多数amdS-重组体中,打靶的phyA盒也失去了,这是在应用phyA特异性引物对进行的第三轮PCR检验中发现的。
这些结果只可解释为发生了基因转变这一现象。好几个例子中,极大的DNA片段或甚至完整glaA扩增子(≥80Kb)由另一个glaA扩增子所取代。然而,大多数子代(超过90%)中似乎缺失了一个完整glaA扩增子。因而,为全面说明所观察的结果,称为扩增子转变和缺失更为恰当。利用所观察到的现象确定是否共转化体中的所有amdS和phyA盒均整合在同一个截短的glaA位点以及具体是哪一个。为确定这一点,用phyA和“DNA-旗”寡核苷酸引物对进行混合PCR只检验每个共转化体的1个或两个氟乙胺抗性菌落,看它们中哪一个基因型为:amdS-/phyA-/glaA-扩增子2+。结果显示,可以获得具有整合在三个glaA扩增子之一的amdS和phyA盒的共转化体。
随后,用phyA和“DNA-旗”引物对经混合PCR检验对来自如此鉴别出的共转化体的其它子代进行检测。所需amdS-/phyA+/glaA扩增子3+基因型的出现在各个共转化体的子代之间差别很大(1至20%)。该频率似乎极大地取决于先选出的共转化体的glaA扩增子内amdS和phyA盒的遗传组成。
如果在一个共转化体中多个盒打靶到一个glaA位点,将产生许多重复序列。氟乙酰胺反选择的结果是amdS盒的丢失各不相同,从而产生的amdS-子代有不同数目的phyA盒。
因此而选择出每个先选出的共转化体的好几个子代重组体(都显示有所需基因型amdS-/phyA+/glaA扩增子3+,但包含不同数目的剩余phyA盒)。
对这些子代菌株的预选择可通过比较在上述PCR检验中扩增的phyA特异性DNA带与代表三个glaA扩增子的DNA带的强度差异而轻易完成。
最后,为确证所选amdS子代的遗传组成,染色体DNA用BamHI和BglII消化,再与2.2Kb SalI/XhoI 3’-glaA DNA片段这一特异性探针杂交进行Southern分析。选出有图24所示杂交模式、具有1,2,或3个phyA盒整合在一个BamHI标记的glaA扩增子中的菌株,分别命名为黑曲霉NP505-1,-2,和-3。
这些菌株,及在其另二个glaA扩增子之一上有肌醇六磷酸酶盒的那些菌株,均随后检验了它们产生肌醇六磷酸酶的能力。
按材料与方法所示进行摇瓶发酵。上清中肌醇六磷酸酶活性的测量揭示所有菌株的每个phyA基因拷贝产生相同量的肌醇六磷酸酶(100U/ml),并似乎与phyA基因所在的glaA扩增子无关。
1.5选择所含肌醇六磷酸酶盒由1增至2的转版本
1.5.a原理说明
打靶到某DNA扩增子的表达盒可通过基因转变增加拷贝数。结果该基因编码的酶产量增加。这里我们描述了对显示肌醇六磷酸酶产量增加的phyA转化体的选择和鉴定。
1.5.b对推定phyA转化体的选择
选黑曲霉NP505-2菌株作为实例。也可用有更少或更多phyA盒定向于另一个glaA扩增子的重组菌株。取有两个phyA盒定向于BamHI标记的glaA扩增子的黑曲霉菌株NP505-2,将其孢子接种在含PDA培养基的平板上,2-3天后用phyA DNA-旗特异性引物对单菌落进行DNA-旗检验(见材料与方法)。用这种方法可很快看到发生在三个标记glaA扩增子之间的遗传交换(转变/扩增或缺失)。尽管所得基因型的频率差异很大,但平均观察到超过95%的受检菌落显示所有三个glaA扩增子根本无任何改变。5%的子代显示或缺失了一或两个glaA扩增子,或扩增了其中一个glaA扩增子,也检测到所需转版本,是BamHI标记的glaA位点和其他两个glaA扩增之一之间的转变(每个的实施示于图25)的结果。
1.5.c phyA转版本的Southern分析
为确定这些转化体中phyA的拷贝数是否也倍增了,对好几个这样的转化体(基因型BamHI2+/SalI+/BglII-或BamHI2+/SalI-/BglII+)进行进一步Southern分析:即用BamHI和BglII消化,并分别以glaA特异性的HindIII/XhoI 5’glaA DNA片段和SalI/XhoI 3″-glaA DNA片段为探针。
亲代菌株及所选转版本的BamHI消化物的杂交模式均示于图25。如所预料,并与亲代黑曲霉菌株NP505-2比较,PCR鉴别的BamHI2+基因型显示,BamHI消化物中2.5Kb和4.2Kb杂交DNA片段的强度确实相反。在BamHI2+基因型中,2.5Kb片段表示两个BamHI标记的glaA扩增子,4.2Kb片段为所剩SalI或BglII glaA扩增子(取决于所选BamHI2+转版本的基因型)。观察结果清楚地显示,预定扩增子可通过牺牲另一相关扩增子的“基因转变”而增加。
BglII消化物的杂交模式清楚显示,所选BamHI2+转版本中,phyA基因拷贝数也增加一倍。如图26示,亲代株染色体DNA的BglII消化物用3″-glaA片段作探针显示有5个杂交DNA片段。5条带14.9,11.7,7.5,5.7和5.6kb分别代表glaA序列5’侧翼,SalI标记的扩增子,phyA-phyA转换,glaA序列3’侧翼和BglII标记的glaA扩增子。亲代菌株中所有这些带的杂交强度大致相同。亦如图26示,所选基因型为BamHI2+/SalI+/BglII-或BamHI2+/SalI-/BglII+的转版本中分别缺失了所述5.6Kb或11.7Kb的DNA片段,而14.9,7.5和5.7Kb杂交片段的强度加倍了。
这一结果说明,位于亲代株BamHI标记的glaA圹增体内的2个phyA盒也通过转变事件而在所选出的BamHI2+转版本中增加了一倍。
1.5.d分析phyA转版本的肌醇六磷酸酶生产
为确定选出的phyA转版本中肌醇六磷酸酶产量是否确实增加,按材料与方法中所述进行摇瓶发酵。
亲代株NP505-2(有2拷贝phyA基因)的平均肌醇六磷酸酶表达为约200U/ml的水平。如所预料,选出的两个转变体BamHI2+/SalI+或BamHI2+/BglII+的肌醇六磷酸酶水平可测出高达400U/ml。
这两株菌命名为黑曲霉NP505-4和-5,用于实施例1.6以进一步增加phyA基因的拷贝数。
1.6选择含多个修饰的DNA扩增子的转版本
1.6.a原理说明
对打靶到宿主菌株中其中一个DNA扩增子内的表达盒的扩增可以只需通过筛选亲代菌株的子代,并选择那些发生了所需转变的重组菌株而达到。尽管程度低,如上述,但这些DNA扩增子之间的转变是自发的。这一基因转变过程可反复应用,产生的新转版本可具有数目增多的(phyA)修饰DNA扩增子。通过应用这种方法,我们已鉴别出一个转版本,它最终有6个phyA基因拷贝,均等地分配于黑曲霉ISO-505宿主中的三个已修饰glaA扩增子内。
“酶表达盒增加”的程度取决于宿主菌株中最初具有的DNA扩增子的数目。然而,通过应用此基于PCR的DNA-旗检验,似乎可能分离到带有DNA-旗特征的子代,这种现象只能解释为三个glaA DNA扩增子之一发生了自发扩增。反复利用这一点,最终可选出比宿主菌株的glaA DNA扩增子多、每个扩增子均含2个phyA表达盒的重组菌株。
1.6.b选择推定含6拷贝phyA基因的转版本。
将述于实施例1.5.(d)、共有4个phyA表达盒(每个BamHI标记的phyA扩增子中有2个phyA盒)的黑曲霉菌株NP505-4的孢子接种在PDA平板上,2-3天后用DNA-旗特异性引物对单个子代进行DNA-旗PCR检验。如前文声明的,又有约95%的受检菌落显示其glaA扩增子的基因型未变。约5%的子代显示指示其中一个BamHI-或SalI-标记的扩增子有缺失或甚至指示其中一个BamHI-标记的glaA扩增子有自发扩增的DNA-旗模式。除这些重组菌株以外,还鉴定出有所需DNA-旗模式的转版本,其200bp大小的BamHI DNA-旗带强度略有增加。对所有显示基因型为“BamHI3+”和“BamHI3+/SalI+”的子代菌株均进行Southern分析。
为使“BamHI3+/SalI+”转版本中的phyA基因增至8个,将其孢子接种于PDA平板上,长2-3天后,用DNA-旗特异性引物对单个子代进行DNA-旗PCR检验。以一个极低频率(1/1000)检测到变显示的DNA-旗模式伴有大小为200bp的BamHI-旗DNA片段强度增加且随后丢失所剩大小为220bp的SalI-旗DNA片段的“BamHI4+”转版本(图27)。对该转版本菌株也进行Southern分析。
1.6.c经Southern分析对转版本进行遗传鉴定
上述鉴定出的转版本(基于PCR的基因型为“BamHI3+”,“BamHI3+/SalI+”和“BamHI4+”)的phyA基因拷贝数通过用BamHI和BglII消化物,并分别与HindIII/XhoI 5’-glaA和SalI/XhoI 3″-glaA特异性DNA片段这样的探针杂交,如此Southern分析测定。亲代转版本(基因型为BamHI2+/SalI+/BglII-或BamHI2+/SalI-/BglII+)及其选出的转版本BamHI消化物的预期杂交模式示于图25和27。BamHI消化物显示确有一个2.5Kb的“BamHI3+”转版本唯一有的杂交片段。对“BamHI3+/SalI+”检测到另有4.2Kb的带,说明在这个转版本中SalI标记的扩增子如DNA-旗模式所得结果预计的那样仍存在。转版本“BamHI4+”显示与“BamHI3+”转版本相同的杂交模式。原始亲代株黑曲霉NP505-2和两个亲代转版本菌株BamHI2+/SalI+/BglII-及BamHI2+/SalI-/BglII+的杂交模式的比较见前面各图。
上述菌株内DNA的BglII消化物的预期杂交模式示于图28。“BamHI3+”和“BamHI4+”转版本中均可测出三条杂交带(145,7.5和5.7),强度均相同,说明除了5’-和3’-侧翼序列以外,还有phyA基因拷贝也分别增至6和8。
对转版本“BamHI3+/SalI+”也观察到预期模式,它包含大小为11.7Kb的带(为SalI扩增子存在的特征)。此带的强度当然弱三倍。
这些结果清楚说明,可筛选得到包含3、甚至4个BamHI标记的glaA扩增子、并均包含2个phyA盒的转版本。
1.6.d摇瓶发酵分析肌醇六磷酸酶的产生
为确定所选“BamHI3+”和“BamHI4+”转版本是否显示确有肌醇六磷酸酶的产量增加,按材料与方法所述进行摇瓶发酵。亲代株NP505-2(有2个phyA基因拷贝)的肌醇六磷酸酶表达水平测得平均约200U/ml。三个选出的转版本的肌醇六磷酸酶水平测得“BamHI3+/SalI+”和“BamHI3+”转版本为约600U,“BamHI4+”转版本菌株为800U/ml,与预期一样。
后来命名这些菌株分别为黑曲霉NP505-6,-7,和-8。
1.7.直接选择肌醇六磷酸酶产量增加的重组体
1.7.a原理说明
由于glaA DNA扩增子用所谓“DNA-旗”标记了,故可只用基于PCR的DNA-旗检验选出转版本。应用这种遗传学筛选方法,我们能显示并证实phyA修饰的BamHI标记glaA扩增子和另两个扩增子之间的转变使phyA表达盒增加。
如上述,我们发现phyA基因拷贝数和肌醇六磷酸酶产量之间存在线性关系,这说明也可基于表达进行选择。本实施例中我们显示,通过在微孔板上,从最初BamHI-glaA打靶的phyA重组菌株的子代中筛选肌醇六磷酸酶表达量增加的子代,可分离到有增加的phyA基因拷贝数的转版本。
1.7.b筛选增加的肌醇六磷酸酶表达水平
将黑曲霉NP505-2的单菌落的孢子接种于含200μl黑曲霉发酵培养基(如材料与方法所示)的96孔板中。34℃,100%湿度及轻微振荡的条件下生长7天后,测每孔上清中肌醇六磷酸酶活性,检验了近千个子代的酶表达水平。收获肌醇六磷酸酶活性增强的细胞,收集孢子并进行另一轮微滴定筛选。同样发现肌醇六磷酸酶活性增强的细胞。
在随后的摇瓶发酵中,这些鉴定出的菌株显示肌醇六磷酸酶表达水平高达600U/ml。
1.7.c鉴定出的重组菌株的遗传鉴定
所观察到的上述菌株肌醇六磷酸酶产量的增加似乎是phyA基因拷贝数通过含phyA的BamHI标记glaA位点的扩增或转变而增加的结果。首先,应用DNA-旗检验,然后进行Southern分析,确定选出的这些菌株的基因型。
DNA-旗检验清楚地说明,选出的菌株囊括了所有各种“DNA-旗”基因型。这些菌株显示与以进行DNA-旗检验为选择标准而分离出的菌株相同的基因型,象:
BamHI2+/Sal+/BglII-,BamHI2+/Sal+/BglII-,
BamHI3+/Sal+/BglII-,BamHI2+/Sal-/BglII+,
BamHI3+/Sal-/BglII-.
所有这些基因型按前面实施例所述进行Southern分析确证。
青霉菌实施例
2.1选择和鉴定产黄青霉宿主
2.1.a原理说明
通过基因转变而扩增插入的基因要求基因组中有多个同源DNA区。产黄青霉的这些区已有描述(本报告中称为PEN扩增子:Fierro等,1995,美国国家科学院院报,92:6200-6204)。包含青霉素生物合成基因pcbAB,pcbC和penDE的一个15Kb青霉素簇区位于这些PEN扩增子上(图29)(Diez等1990,生物化学杂志,265:16358-16365;Smith等,1990 EMBO.J.9:741-747)。这里我们描述了对包含多拷贝PEN扩增子的产黄青霉宿主菌株CBS649.97的选择和鉴定。
2.1.b定量PEN扩增子
用经典菌株改良法,从包含单个PEN扩增子(Fierro等,1995,美国国家科学院院报,92:6200-6204)的Wis 54-1255(ATCC 28089)菌株获得产黄青霉株CBS649.97。CBS649.97中的PEN扩增子数经Southern分析确定。为此,用BstXI消化染色体DNA,并与HELE和niaD探针杂交。测得CBS649.97与HELE/niaD杂交比Wis 54-1255高7倍(图30,42)。相似地,NotI消化的DNA经CHEF分离后,与HELE探针杂交(图42)。在CBS649.97中检测到的一个主要杂交信号比在Wis 54-1544中检测到的要大约420Kb(图31)。这些结果说明CBS649.97中有约7个PEN扩增子。
2.2构建表达载体
2.2.a原理说明
rDNA分子定向插入(打靶到)基因组中通过同源重组进行。故,rDNA盒应在其侧翼有同源于基因组中靶位点的DNA片段(图32)。青霉素簇中定为HELE和HELF的两个定向区被选出,用于将表达盒定向插入PEN扩增子中(图29)。这里我们描述用于本报告的amdS,cefE和cefF表达载体的设计和构建。
2.2.b表达载体的基本设计
向基因组中定向整合要求必须是线性DNA分子,且5’和3’末端(侧翼)必须由同源于所预期整合的位点的DNA组成。故转化片段包含5’和3’侧有定向区的表达盒(目的基因受适当启动子和终止子调节)。将这些片段克隆进入用于质粒增殖的大肠杆菌载体中。所得表达载体均设计为大肠杆菌序列在转化片段的线性化和分离过程中会被去除(图32)。因此,重组菌株将不含大肠杆菌DNA(E.P.O 635 574 A1)。
2.2.c表达载体pHELE-A1的构建
用寡核苷酸1和2从产黄青霉株649.97的染色体DNA中PCR扩增5’HELE打靶区。所得产物作为NotI-SpHI片段克隆至pZErOTM-1(Invitrogen,Carlsbad,USA)中,产生质粒pHELE5’。相似地,3’HELE打靶区用寡核苷酸3和4经PCR扩增而得,并作为HindIII-NotI片段克隆进pHELE5’中,从而产生质粒pHELF53。包含受PgpdA和TamdS调节的amdS的表达盒从质粒pGBDEL4L(E.P.O 635 547 A1)中用寡核苷酸5和6经PCR进行扩增。所得产物作为NotI片段克隆进pHELE53中,产生表达载体pHELE-A1(图33)。
2.2.d表达载体pHELF-A1的构建
5’HELF打靶区用寡核苷酸7和8从产黄青霉株CBS649.97的染色体DNA中经PCR而扩增。所得产物作为NotI-XbaI片段克隆进pZEroTM-1(Invitrogen,Carlsbad,USA)中,产生质粒pHELF5’。相似地,3’HELF侧翼用寡核苷酸9和10经PCR而扩增,并作为PstI-NotI片段克隆进pHELF5’中,产生pHELF53。包含受PgpdA和TamdS调节的amdS的表达盒用寡核苷酸5和6从质粒pGBDEL4L(E.P.O 635 547 A1)中经PCR而扩增。所得产物作为NotI片段克隆至pHELF53中,产生表达载体pHELF-A1(图34)。
2.2.e表达载体pHELE-E1的构建
PpcbC用寡核苷酸11和12从产黄青霉株CBS649.97的基因组DNA中经PCR而扩增。所得产物作为XhoI-NdeI片段克隆至pGSEWA(WO95/04148)中,产生pISEWA-N。最后,受PpcbC和TpenDE调节的cefE表达盒从pISEWA-N中作为NotI片段克隆至pHELE53中,产生表达载体pHELE-E1(图35)。
2.2.f表达载体pHELF-F1的构建
cefF基因用寡核苷酸13和14,从带小棒链霉菌ATCC 27064的基因组DNA中经PCR扩增,PCR依照ExpandTM Long Template PCR System(Boehringer Mannheim)进行。循环条件为:30x(1min 98℃,5min 70℃,1x(7min 72℃)。所得产物作为NdeI-NsiI片段克隆至pISEWA-N中(取代cefE),产生pISFWA。最后,受PpcbC和TpenDE调节的cefF表达盒从pISFWA中作为NotI片段克隆至pHELF53中,产生表达载体pHELF-F1(图36)。
2.3用cefE修饰PEN扩增子
2.3.a原理说明
转化体的有效选择要求有一个可选择标记。真菌的共转化过程为人熟知,是宿主同时用两种不同的rDNA分子转化;其一包含目的基因而另一包含选择标记。双向选择性amdS标记的应用允许反复转化产黄青霉宿主(E.P.O 635 547 A1)。因打靶整合而产生的直接重复序列之间的重组将导致amdS盒的丢失,并可在氟乙酰胺培养基上选择(图37)。这里我们描述了对含有整合于其中一个PEN扩增子的cefE表达盒的无amdS重组体进行选择。
2.3.b定向的amdS,cefE共转化体的选择。
产黄青霉株CBS649.97用SfiI线性化的HELE-A1和HELE-E1片段共转染(图33,35)。含amdS的转化体从乙酰胺平板上选出,并用寡核苷酸17和18经盒式PCR检验有无cefE(图40a)。用寡核苷酸19,20和21经定向PCR鉴别有amdS或cefE整合在HELE中的共转化体(图40.b,c)。这些菌株用于分离amdS-重组体。
2.3.c amdS-(无标记)cefE重组体的选择
将2.3.b所述amdS,cefE共转染体的孢子铺板于氟乙酰胺培养基上。保留cefE的重组体经大肠杆菌生物质分析而鉴别。失去amdS但有至少一个cefE拷贝的情况用寡核苷酸15,16,17和18经盒式PCR证实(图40.a,d)。选出这些菌株作Southern分析。
2.3.d amdS-重组体的Southern分析
cefE向HELE中的整合及cefE在amdS-、cefE+重组体中的拷贝数用Southern和TAFE分析。为此,重组体的染色体DNA经NruI消化再与cefE探针杂交(图38,42)。对经TAFE分离的HpaI消化DNA进行相似的杂交过程(图39),选出有cefE整合进HELE中所预期的杂交模式的重组菌株以进行下一步实验。
2.4用amdS修饰第二扩增子
2.4.a原理说明
基因转变实质上将受体DNA链的遗传信息用供体DNA链的来取代。供体链和受体链均可被标记以显示这一事件。2.3所述的菌株已有一个扩增子被cefE标记(本例中为供体链)。这里我们描述通过向不同于cefE所在区的另一个PEN扩增子中插入一个amdS盒而修饰受体链(图43)。
2.4.b amdS定向于第二PEN扩增子。
2.3.d所述包含一个带cefE的扩增子的菌株用SfiI线性化的HELE-A1片段(图33)转化。amdS的整合及向HELE的定向分别用寡核苷酸15,16及19,20经盒式PCR及定向PCR检验(图40,c,d)。amdS整合在cefE的邻位或多个amdS拷贝整合在一个HELE位点均可用寡核苷酸20,21,22和23经定向PCR鉴别(图40e,f,g),从而选出有单一amdS盒整合在第二扩增子中的菌株,进行Southern分析。
2.4.c amdS转化体的Southern分析。
2.4.b所述转化体的染色体DNA经HpaI消化后与amdS探针杂交(图41,42)。选出有amdS正确整合在HELE中所预期的杂交模式的菌株,进行基因转变实验。
2.5通过基因转变增加产量。
2.5.a原理说明
整合基因的拷贝数可通过基因转变增加(图43)。结果该基因编码的酶的产量或酶催化活性增加。表达cefE的产黄青霉重组体在合适条件下发酵时可产生己二酰-7-ADCA(Crawford等,1995,BIO/Technol.13:58-62)。这里我们描述对cefE基因转版本的选择和鉴定,以及由此导致的己二酰-7-ADCA产量的增加。
2.5.b cefE基因转版本的选择。
将2.5.a所述在不同PEN扩增子中含有cefE和amdS的菌株的孢子铺板于氟乙酰胺培养基上,以便选择amdS-重组体。这些重组体的amdS-、cefE+基因型用寡核苷酸15,16,17,18,26和27经盒式PCR证实(图40a,d,h)。用寡核苷酸26和27可扩增唯一niaD位点的片段,该片段可作为PCR产物相对定量的内部参照。通过相比于亲代株其cefE/niaD比率增高而由此鉴定为失去了amdS标记但获得了cefE多拷贝的基因转版本菌株被选出进行Southern分析。
2.5.c cefE基因转版本的Southern分析。
2.5.b选出的菌株的染色体DNA用NruI消化,再与cefE和niaD探针杂交。具有与亲代株相同的杂交模式,但cefE/niaD杂交信号的比率增大的菌株被选出进行己二酰-7-ADCA发酵(图48)。
2.5.d cefE基因转版本的己二酰-7-ADCA生产
2.7.b选出的基因转版本的己二酰-7-ADCA产量经摇瓶发酵测定。所有选出的菌株生产的己二酰-7-ADCA比其对应的亲代株明显更多。
2.6通过基因转变使不同表达盒同时增加
2.6.a原理说明
包含大节段扩增子或甚至延伸至扩增子边界的区域可参与基因转变。从而相邻的不同基因的拷贝数可通过基因转变同时增加。表达cefE和cefF的产黄青霉重组体菌株在合适条件下发酵时可产生己二酰-7-ADAC(Crawford等,1995 BIO/Technol,13:58-62)。这里我们描述打靶到同一PEN扩增子上不同位点的cefE和cefF表达盒(cefE+cefF)的同时基因转变,及由此导致的己二酰-7-ADAC产量的增加。
2.6.b(cefE+cefF)转化体的选择
2.3.d所述有cefE整合在HELE中的菌株用SfiI线性化的HELF-A1和HELF-F1片段(图34,36)共转化。转化体在乙酰胺平板上选出并用寡核苷酸15,16,17和25经盒式PCR检验cefF和amdS(图40d,j)。amdS或cefE向HELF中的整合用寡核苷酸20,21和24经定向PCR确定(图40j,k)。这些菌株的染色体DNA用NotI消化,印迹,再用HELE探针杂交(图44)。所含杂交片段相应于cefF或amdS向已含cefE的PEN扩增子上HELF中的整合的菌株被用于选择无标记基因型(amdS-)重组体。
2.6.c无标记基因型(cefE+cefF)重组体的选择
将2.6.b所述菌株的孢子铺板于氟乙酰胺培养基上,以选择amdS-重组体。失去amdS但保留cefE和cefF的重组体用寡核苷酸15,16,17,18和25经盒式PCR鉴别(图40a,d,j)。这些菌株的染色体用NruI消化,再与cefF探针杂交(图42,45)。利用其cefF正确整合于HELF中的菌株筛选基因转版本。
2.6.d(cefE+cefF)基因转版本的选择
使2.6.c所述菌株的孢子长成单菌落,然后用寡核苷酸17,18,25,26和27经盒式PCR分析cefE,cefF和niaD的量(图40a,h,i)。基因转版本为相比于亲代株其cefE/niaD和cefF/niaD比率同时增加了的菌株。cefE和cefF增加的拷贝数用Southern分析证实。为此,将染色体DNA用NruI消化再与cefE和niaD的探针杂交(图46)。除去印迹上的探针后与cefF和niaD探针杂交(图47)。选出其cefE/niaD和cefF/niaD比率均比亲代株高的菌株进行己二酰-7-ADAC生产分析。
2.6.e(cefE+cefF)基因转版本的己二酰-7-ADAC生产
2.6.d中选出的基因转版本的己二酰-7-ADAC生产用摇瓶发酵测定。所有选出的菌株生产的己二酰-7-ADAC量均比亲代株的多。
序列表
(1)一般资料:
(i)申请人
(A)姓名:Gist-Brocades B.V.
(B)街名:Wateringseweg 1
(C)城市:Delft
(D)州名:none
(E)国家:Netherlands
(F)邮编(ZIP):2600 MA
(G)电话:+31(0)15-2799111
(H)电传:+31(0)15-2793957
(ii)发明题目:基因转变作为工具用于构建重组工业化生物体
(iii)序列数:52
(iv)计算机可读形式
(A)介质类型:软盘
(B)计算机:IBM PC可兼容机
(C)操作系统:PC-DOS/MS-DOS
(D)软件:PatentIn Release#1.0,Version#1.30(EPO)
(2)SEQ ID NO:1的资料:
(i)序列特征:
(A)长度:41个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓朴构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:1
GTAGCTGCGG CCGCCTCCGT CTTCACTTCT TCGCCCGCAC T
41
(2)SEQ ID NO:2的资料:
(i)序列特征:
(A)长度:48个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:2
CAAAGGGCAT GCGGCCGTAT CGGCCGGTGA CAAACATCAT TCAACGCC
48
(2)SEQ ID NO:3的资料:
(i)序列特征:
(A)长度:42个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQID NO:3
ATGTTTAAGC TTGGCCGATA CGGCCAAAAC ACCTTTGATT TC
42
(2)SEQ ID NO:4的资料:
(i)序列特征:
(A)长度:45个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:4
CAAGTTGCGG CCGCTCCTCA CTAACGAGCC AGCAGATATC GATGG
45
(2)SEQ ID NO:5的资料:
(i)序列特征:
(A)长度:39个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:5
AAGCTTATGC GGCCGCGAAT TCGAGCTCTG TACAGTGAC
39
(2)SEQ ID NO:6的资料:
(i)序列特征:
(A)长度:40个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:6
(2)SEQ ID NO:6的资料:
(i)序列特征:
(A)长度:40个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:6
CGGTACGTGC GGCCGCTCGT ACCATGGGTT GAGTGGTATG
40
(2)SEQ ID NO:7的资料:
(i)序列特征:
(A)长度:43个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:7
ATATGTGCGG CCGCTTTACA TGGTCAATGC AATTAGATGG TGG
43(2)SEQ ID NO:8的资料:
(i)序列特征:
(A)长度:47个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:8
ATAACTCTAG AGGCCCTACC GGCCTTTGCA AATATACTGT AAGAACC
47
(2)SEQ ID NO:9的资料:
(i)序列特征:
(A)长度:43个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:9
GTATATTCTG CAGGGCCGGT AGGGCCAACA GTTTCCGCAG GTG
43
(2)SEQ ID NO:10的资料:
(i)序列特征:
(A)长度:43个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:10
GTATGGGCGG CCGCTTTACA ACTAGAATAT GGGAACCTGT GGG
43
(2)SEQ ID NO:11的资料:
(i)序列特征:
(A)长度:61个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:11
CTCGAGTGCG GCCGCAAAGC TAGCTTGATA TCGAATTCCT TATACTGGGC CTGCTGCATT
60
G
61
(2)SEQ ID NO:12的资料:
(i)序列特征:
(A)长度:45个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:12
GTCCATATGG GTGTCTAGAA AAATAATGGT GAAAACTTGA AGGCG
45
(2)SEQ ID NO:13的资料:
(i)序列特征:
(A)长度:30个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:13
CATATGGCGG ACACGCCCGT ACCGATCTTC
30
(2)SEQ ID NO:14的资料:
(i)序列特征:
(A)长度:54个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:14
ATGCATTGGC TCGTCATGAA GAGCCTATCA TCCGGCCTGC GGCTCGTTCT TCGC
54
(2)SEQ ID NO:15的资料:
(i)序列特征:
(A)长度:25个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:15
CAGCTACCCC GCTTGAGCAG ACATC
25
(2)SEQ ID NO:16的资料:
(i)序列特征:
(A)长度:25个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:16GTCAGGGAAG AACACGAGGG CGCAG25
(2)SEQ ID NO:17的资料:
(i)序列特征:
(A)长度:25个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:17
CCCTCTCTTC GTCGTTGTCC ACGCC
25
(2)SEQ ID NO:18的资料:
(i)序列特征:
(A)长度:25个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:18
ATGTCCTTGG CCGACTTCAG CTCGG
25
(2)SEQ ID NO:19的资料:
(i)序列特征:
(A)长度:25个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:19
GACGAGCCAA TGCATCTTTT GTATG
25
(2)SEQ ID NO:20的资料:
(i)序列特征:
(A)长度:25个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:20
CGGGTACTCG CTCTACCTAC TTCGG
25
(2)SEQ ID NO:21的资料:
(i)序列特征:
(A)长度:27个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:21
GCCCAGTATA AGGAATTCGA TATCAAG
27
(2)SEQ ID NO:22的资料:
(i)序列特征:
(A)长度:25个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:22
AGGGTCGACA CTAGTTCTAG AGCGG
25
(2)SEQ ID NO:23的资料:
(i)序列特征:
(A)长度:25个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:23
GACGTTATCG GACGGAGACT CAGTG
25
(2)SEQ ID NO:24的资料:
(i)序列特征:
(A)长度:24个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:24
GCCTACTCTG TTCTGGAGAG CTGC
24
(2)SEQ ID NO:25的资料:
(i)序列特征:
(A)长度:25个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:25
CCCCCATCCC GGTCACGCAC TCGCG
25
(2)SEQ ID NO:26的资料:
(i)序列特征:
(A)长度:25个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:26
CACAGAGAAT GTGCCGTTTC TTTGG
25
(2)SEQ ID NO:27的资料:
(i)序列特征:
(A)长度:25个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:27
TCACATATCC CCTACTCCCG AGCCG
25
(2)SEQ ID NO:28的资料:
(i)序列特征:
(A)长度:15个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:28
GTCGCGTATC CCAGG
15
(2)SEQ ID NO:29的资料:
(i)序列特征:
(A)长度:18个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:29
GTCAAAGGAT ATGCATAC
18
(2)SEQ ID NO:30的资料:
(i)序列特征:
(A)长度:39个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:30
AGCTTATGCG GCCGCGAATT CAGGTACCGT ATCTCGAGA
39
(2)SEQ ID NO:31的资料:
(i)序列特征:
(A)长度:39个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:31
AATTTCTCGA GATACGGTAC CTGAATTCGC GGCCGCATA
39
(2)SEQ ID NO:32的资料:
(i)序列特征:
(A)长度:30个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:32
GTGCGAGGTA CCACAATCAA TCCATTTCGC
30
(2)SEQ ID NO:33的资料:
(i)序列特征:
(A)长度:36个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:33
ATGGTTCAAG AACTCGGTAG CCTTTTCCTT GATTCT
36
(2)SEQ ID NO:34的资料:
(i)序列特征:
(A)长度:36个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:34
AGAATCAAGG AAAGGCTAC CGAGTTCTTG AACCAT
36
(2)SEQ ID NO:35的资料:
(i)序列特征:
(A)长度:42个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:35
ATCAATCAGA AGCTTTCTCT CGAGACGGGC ATCGGAGTCC CG
42
(2)SEQ ID NO:36的资料:
(i)序列特征:
(A)长度:21个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:36
GACCATGATT ACGCCAAGCT T
21
(2)SEQ ID NO:37的资料:
(i)序列特征:
(A)长度:36个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:37
GGATCCTTAA CTAGTTAAGT GGGGGCCTGC GCAAAG
36
(2)SEQ ID NO:38的资料:
(i)序列特征:
(A)长度:36个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQID NO:38
TTAACTAGTT AAGGATCCAC AATCAATCCA TTTCGC
36
(2)SEQ ID NO:39的资料:
(i)序列特征:
(A)长度:34个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:39
GCTCTAGAGC GGCCGCGAAT TCATCCGGAG ATCC
34
(2)SEQ ID NO:40的资料:
(i)序列特征:
(A)长度:18个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:40
CTTTGCGCAG GCCCCCAC
18
(2)SEQ ID NO:41的资料:
(i)序列特征:
(A)长度:18个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:41
TGCAGGGTAA ATCAGGGA
18
(2)SEQ ID NO:42的资料:
(i)序列特征:
(A)长度:19个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:42
TCCGCTAAAG GTGGTCGCG
19
(2)SEQ ID NO:43的资料:
(i)序列特征:
(A)长度:20个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:43
CCCCAGCATC ATTACACCTC
20
(2)SEQ ID NO:44的资料:
(i)序列特征:
(A)长度:20个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:44
AAAGGACCCG AGATCCGTAC
20
(2)SEQ ID NO:45的资料:
(i)序列特征:
(A)长度:25个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:45
TCTCGATACC AAGGTCACCA CGGGC
25
(2)SEQ ID NO:46的资料:
(i)序列特征:
(A)长度:25个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:46
GCATCCATCG GCCACCGTCA TTGGA
25
(2)SEQ ID NO:47的资料:
(i)序列特征:
(A)长度:25个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:47
ATCCAGACCA GCACAGGCAG CTTCG
25
(2)SEQ ID NO:48的资料:
(i)序列特征:
(A)长度:25个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:48
TCCGCATGCC AGAAAGAGTC ACCGG
25
(2)SEQ ID NO:49的资料:
(i)序列特征:
(A)长度:36个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:49
GTCGACTTAA CTAGTTAAGG CTTCAGACGC AGCGAG
36
(2)SEQ ID NO:50的资料:
(i)序列特征:
(A)长度:36个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:50
TTAACTAGTT AAGTCGACAC AATCAATCCA TTTCGC
36
(2)SEQ ID NO:51的资料:
(i)序列特征:
(A)长度:36个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:51
AGATCTTTAA CTAGTTAAGT GGCCTGAACA GTGCCG
36
(2)SEQ ID NO:52的资料:
(i)序列特征:
(A)长度:36个碱基对
(B)类型:核酸
(C)链型:单链
(D)拓扑构型:线性
(ii)分子类型:其它核酸
(A)描述:/desc=“寡核苷酸”
(iii)假设:是
(xi)序列描述:SEQ ID NO:52
TTAACTAGTT AAAGATCTAC AATCAATCCA TTTCGC
36
Claims (29)
1.一种丝状真菌,在其染色体的两个基本同源的DNA区的至少其一之中整合有重组DNA分子,其中所述DNA区不是核糖体DNA重复序列。
2.权利要求1的丝状真菌,其中每个基本同源的DNA区中均整合有重组DNA分子。
3.权利要求1或2的丝状真菌,其中这些DNA区为扩增子。
4.权利要求1的丝状真菌,其中所述DNA区是在其天然状态下包含能高水平表达的内源基因的一个区。
5.权利要求4的丝状真菌,其中该内源基因选自以下组:糖酵解酶基因,淀粉分解酶基因,纤维素分解酶基因和抗生素生物合成酶基因。
6.权利要求5的丝状真菌,其中该内源基因选自以下组:葡糖淀粉酶基因,TAKA淀粉酶基因,纤维二糖水解酶基因和青霉素生物合成酶基因。
7.权利要求4至6中任一项的丝状真菌,其中该内源基因在所述DNA区的每个拷贝中均失活。
8.权利要求7的丝状真菌,其中该内源基因是通过使其至少一部分不可逆地缺失而失活的。
9.权利要求8的丝状真菌,其中该不可逆缺失包含启动子和上游激活序列的至少部分。
10.权利要求1的丝状真菌,其中所述DNA区的每一版本区别于其它版本的是特有的序列标记。
11.权利要求10的丝状真菌,其中该序列标记是一个限制位点。
12权利要求1的丝状真菌,其中该重组DNA分子包含可表达所需基因的表达盒。
13.权利要求12的丝状真菌,其中该所需基因编码一种分泌型酶。
14.权利要求12的丝状真菌,其中该所需基因编码一种胞内酶。
15.权利要求14的丝状真菌,其中包含一个或多个胞内酶表达盒的一个或多个重组DNA分子整合在所述DNA区内,且所述胞内酶是该丝状真菌天然不具有的代谢途径的一部分。
16.权利要求15的丝状真菌,其中所述胞内酶选自脱乙酰氧基头孢菌素C合成酶和脱乙酰基头孢菌素C合成酶。
17.权利要求1的丝状真菌,其中该重组DNA分子缺乏选择标记基因。
18.权利要求17的丝状真菌,它缺乏选择标记基因。
19.权利要求1的丝状真菌,它属于从下组中选出的属:曲霉属,木霉属,青霉属,头孢属,枝顶孢属,镰孢属,毛霉属,酒曲菌属,Phanerochaete,脉孢菌属,腐质霉属,麦角属,粪壳属,黑粉菌属,裂褶菌属,布拉霉属,被孢霉属,须霉属,和弯颈霉属。
20.权利要求19的丝状真菌,选自以下组合:黑曲霉类,米曲霉,Trichoderma reesei和产黄青霉。
21.制备权利要求1至20中任何一项的丝状真菌的方法,其中该方法包括的步骤有:
(a)用重组DNA分子转化其一条或多条染色体中有至少两个适于整合一个或多个拷贝重组DNA分子的基本同源DNA区的丝状真菌,其中这些DNA区不是核糖体DNA重复序列;
(b)选出有至少一个重组DNA分子整合在至少其中一个DNA区的转化体;
(c)增殖(b)中所获转化体,从其子代选出其中至少两个DNA区包含整合重组DNA分子的菌株。
22.权利要求21的方法,其中除了步骤(a),(b)和(c)以外,还包括以下步骤:
(d)增殖其中至少两个DNA区包含整合DNA分子的菌株,从其子代选出其中另外拷贝的DNA区也包含整合重组DNA分子的菌株;
(e)重复步骤(d)直至获得其中每个DNA区均包含整合重组DNA分子的菌株。
23.权利要求21或22的方法,其中该重组DNA分子包含基本同源于所述DNA区的序列。
24.权利要求21或22的方法,其中利用双向选择标记在步骤(a)中转化丝状真菌,且在执行步骤(c)之前,选择缺失了双向选择标记的转化子。
25.权利要求24的方法,其中所述双向选择标记为显性标记。
26.生产目的蛋白质的方法,包括的步骤有:
(a)在有利于目的蛋白质表达的条件下培养权利要求1至20中任何一项的丝状真菌,并
(b)回收目的蛋白质。
27.权利要求26的方法,其中目的蛋白质为分泌型蛋白质。
28.产生目的代谢物的方法,包括的步骤有:
(a)在有利于目的代谢物生产的条件下培养权利要求1至20中任一项的丝状真菌;并
(b)回收目的代谢物。
29.权利要求28的方法,其中目的代谢物是一种次级代谢物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP97201091 | 1997-04-11 | ||
| EP97201091.2 | 1997-04-11 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1257546A CN1257546A (zh) | 2000-06-21 |
| CN1169961C true CN1169961C (zh) | 2004-10-06 |
Family
ID=8228199
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB988052377A Expired - Fee Related CN1169961C (zh) | 1997-04-11 | 1998-04-09 | 基因转变作为工具用于构建重组的工业化丝状真菌 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US6432672B1 (zh) |
| EP (2) | EP0979294B1 (zh) |
| JP (2) | JP5303084B2 (zh) |
| CN (1) | CN1169961C (zh) |
| AU (1) | AU7642298A (zh) |
| BR (1) | BR9808859A (zh) |
| DK (1) | DK0979294T3 (zh) |
| PL (1) | PL336345A1 (zh) |
| WO (1) | WO1998046772A2 (zh) |
Families Citing this family (136)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PL336345A1 (en) * | 1997-04-11 | 2000-06-19 | Dsm Nv | Genic conversion as a tool for constructing recombined filiform fungi |
| NZ524814A (en) * | 2000-09-21 | 2004-08-27 | Basf Ag | Talaromyces xylanases |
| DK1339837T3 (da) | 2000-12-07 | 2008-06-09 | Dsm Ip Assets Bv | Prolyl-endoprotease fra Aspergillus Niger |
| US7404977B2 (en) | 2001-12-21 | 2008-07-29 | Dsm Ip Assets B.V. | Rennets |
| TWI328612B (en) | 2002-07-25 | 2010-08-11 | Dsm Ip Assets Bv | Genes and their encoded polypeptides involved in the biosynthetic pathway of vitamin b12, vectors and host cells comprising the genes, and process for producing vitamin b12 |
| WO2004033703A1 (ja) * | 2002-10-11 | 2004-04-22 | The Kitasato Institute | 新規マクロファージ泡沫化阻害物質fka−25およびその製造法 |
| US20060234333A1 (en) * | 2003-01-09 | 2006-10-19 | Basf Aktiengesellschaft Patents, Trademarks And Licenses | Method for producing carotenoids or their precursors using genetically modified organisms of the blakeslea genus, carotenoids or their precursors produced by said method and use thereof |
| KR20050092740A (ko) * | 2003-01-09 | 2005-09-22 | 바스프 악티엔게젤샤프트 | 블라케슬레아 속 유기체의 유전자 변형 방법, 해당 유기체및 그의 용도 |
| US20060127976A1 (en) * | 2003-02-05 | 2006-06-15 | Wenzel Thibaut J | Use of oxalate deficient aspergillus niger strains for producing a polypeptide |
| WO2004072225A2 (en) * | 2003-02-12 | 2004-08-26 | Ramot At Tel Aviv University Ltd. | Transgenic fungi expressing bcl-2 and methods of using bcl-2 or portions thereof for improving biomass production, survival, longevity, stress resistance and pathogenicity of fungi |
| WO2004074490A2 (en) * | 2003-02-24 | 2004-09-02 | Genoclipp Biotechnology B.V. | Method for transforming blakeslea strains |
| WO2004078919A2 (en) * | 2003-02-27 | 2004-09-16 | Midwest Research Institute | Superactive cellulase formulation using cellobiohydrolase-1 from penicillium funiculosum |
| ATE441664T1 (de) * | 2003-03-31 | 2009-09-15 | Novozymes Inc | Verfahren zur produktion biologischer substanzen in enzymmangelmutanten von aspergillus niger |
| ES2350048T3 (es) | 2004-04-02 | 2011-01-17 | Dsm Ip Assets B.V. | Mutantes fúngicos filamentosos con eficiencia mejorada de recombinación homóloga. |
| AU2005233326B2 (en) * | 2004-04-16 | 2010-09-23 | Dsm Ip Assets B.V. | Fungal promoters for expressing a gene in a fungal cell |
| WO2006040345A2 (en) * | 2004-10-15 | 2006-04-20 | Dsm Ip Assets B.V. | Amidases from aspergillus niger and their use in a food production process |
| DK2410048T3 (en) | 2005-01-24 | 2016-11-28 | Dsm Ip Assets Bv | A process for the preparation of a compound of interest in a filamentous fungal cell |
| US9410175B2 (en) * | 2005-03-24 | 2016-08-09 | Dsm Ip Assets B.V. | Process for microbial production of a valuable compound |
| DE102005056667A1 (de) * | 2005-11-28 | 2007-05-31 | Basf Ag | Fermentative Herstellung organischer Verbindungen |
| WO2007062936A2 (en) | 2005-11-29 | 2007-06-07 | Dsm Ip Assets B.V. | Dna binding site of a transcriptional activator useful in gene expression |
| CN101400694B (zh) | 2005-12-01 | 2013-07-24 | 阿德库端木营养控股有限责任公司 | 用于工业生产柠檬酸的基因 |
| EA015925B1 (ru) | 2006-06-29 | 2011-12-30 | ДСМ АйПи АССЕТС Б.В. | Способ получения полипептидов |
| EP2511372A1 (en) | 2006-11-02 | 2012-10-17 | DSM IP Assets B.V. | Improved production of secreted proteins by filamentous fungi |
| US20100112638A1 (en) | 2007-02-15 | 2010-05-06 | Cornelis Maria Jacobus Sagt | Recombinant host cell for the production of a compound of interest |
| ES2492469T3 (es) | 2007-02-20 | 2014-09-09 | Dsm Ip Assets B.V. | Nueva sialidasa |
| US20110318795A1 (en) | 2007-11-20 | 2011-12-29 | Verwaal Rene | Dicarboxylic acid production in a filamentous fungus |
| ES2522622T3 (es) | 2007-11-20 | 2014-11-17 | Dsm Ip Assets B.V. | Producción de ácido succínico en una célula eucariota |
| WO2009065779A1 (en) | 2007-11-20 | 2009-05-28 | Dsm Ip Assets B.V. | Dicarboxylic acid production in a recombinant yeast |
| EP2116136A1 (en) | 2008-05-08 | 2009-11-11 | Nederlandse Organisatie voor toegepast- natuurwetenschappelijk onderzoek TNO | Novel phytases |
| MX346700B (es) | 2009-03-10 | 2017-03-28 | Dsm Ip Assets Bv | Metodo para mejorar el rendimiento de un polipeptido. |
| US8735100B2 (en) | 2009-03-17 | 2014-05-27 | Nederlandse Organisatie Voor Toegepast -Natuurwetenschappelijk Onderzoek Tno | Cellulose and ligno-cellulose active proteins |
| EP2419518A1 (en) | 2009-04-15 | 2012-02-22 | DSM IP Assets B.V. | Dicarboxylic acid production process |
| CN102414323B (zh) | 2009-04-22 | 2015-07-08 | 帝斯曼知识产权资产管理有限公司 | 用于生产感兴趣的重组多肽的方法 |
| UA108853C2 (uk) | 2009-07-10 | 2015-06-25 | Спосіб ферментації галактози | |
| AU2010275672A1 (en) | 2009-07-22 | 2012-02-02 | Dsm Ip Assets B.V. | Improved host cell for the production of a compound of interest |
| WO2011048046A2 (de) | 2009-10-22 | 2011-04-28 | Basf Se | Synthetische phytasevarianten |
| US20130011876A1 (en) | 2010-02-04 | 2013-01-10 | Dsm Ip Assetts, B.V. | Process for preparing filamentous fungal strains having a sexual cycle and a process for preparing sexually crossed filamentous fungal strains |
| BR112012020802A2 (pt) | 2010-02-11 | 2015-09-15 | Dsm Ip Assets Bv | célula hospedeira capaz de produzir enzimas úteis para degradação de material lignocelulósico |
| WO2011131667A1 (en) | 2010-04-21 | 2011-10-27 | Dsm Ip Assets B.V. | Cell suitable for fermentation of a mixed sugar composition |
| EP2388331A1 (en) | 2010-05-21 | 2011-11-23 | Nederlandse Organisatie voor toegepast -natuurwetenschappelijk onderzoek TNO | Constitutive promoter |
| EP2588616B1 (en) | 2010-07-01 | 2018-11-14 | DSM IP Assets B.V. | A method for the production of a compound of interest |
| WO2012007445A1 (en) | 2010-07-13 | 2012-01-19 | Dsm Ip Assets B.V. | Mutarotase in crystallization |
| WO2012007446A1 (en) | 2010-07-13 | 2012-01-19 | Dsm Ip Assets B.V. | Use of a mutarotase in the production of dried powders |
| WO2012049170A2 (en) | 2010-10-13 | 2012-04-19 | Dsm Ip Assets B.V. | Pentose and glucose fermenting yeast cell |
| DK2699674T3 (da) | 2011-04-21 | 2016-03-21 | Basf Se | Syntetiske fytasevarianter |
| AR086471A1 (es) | 2011-04-22 | 2013-12-18 | Dsm Ip Assets Bv | Celula de levadura capaz de convertir azucares que incluyen arabinosa y xilosa |
| AR087423A1 (es) | 2011-08-04 | 2014-03-19 | Dsm Ip Assets Bv | Celula capaz de fermentar azucares pentosas |
| EP2554668A1 (en) | 2011-08-04 | 2013-02-06 | DSM IP Assets B.V. | A pentose sugar fermenting cell |
| EP3444338A1 (en) | 2012-01-23 | 2019-02-20 | DSM IP Assets B.V. | Diterpene production |
| BR112014031526A2 (pt) | 2012-06-19 | 2017-08-01 | Dsm Ip Assets Bv | promotores para expressar um gene em uma célula |
| MX357482B (es) | 2012-07-19 | 2018-07-11 | Dsm Ip Assets Bv | Cepa deficiente en agse. |
| US9593158B2 (en) | 2012-09-18 | 2017-03-14 | Dsm Ip Assets B.V. | Process for the preparation of gelatin |
| PL2909305T3 (pl) | 2012-10-16 | 2019-06-28 | Dsm Ip Assets B.V. | Komórki o ulepszonej konwersji pentozy |
| DK3202900T3 (en) | 2013-02-04 | 2019-04-08 | Dsm Ip Assets Bv | CARBOHYDRATE DEGRADING POLYPEPTIDE AND APPLICATIONS THEREOF |
| EP2772545A1 (en) | 2013-03-01 | 2014-09-03 | Nederlandse Organisatie voor toegepast -natuurwetenschappelijk onderzoek TNO | Starch active proteins |
| WO2014142647A1 (en) | 2013-03-14 | 2014-09-18 | Wageningen Universiteit | Fungals strains with improved citric acid and itaconic acid production |
| US20160060660A1 (en) | 2013-04-05 | 2016-03-03 | Université Du Luxembourg | Biotechnological production of itaconic acid |
| CN103952430A (zh) * | 2013-04-16 | 2014-07-30 | 中国科学院合肥物质科学研究院 | 一种产黄青霉形态突变高产菌的构建方法 |
| WO2014178717A1 (en) | 2013-05-02 | 2014-11-06 | Nederlandse Organisatie Voor Toegepast-Natuurwetenschappelijk Onderzoek Tno | Novel organic acid pathway |
| AU2014273055A1 (en) | 2013-05-31 | 2015-12-17 | Dsm Ip Assets B.V. | Microorganisms for diterpene production |
| WO2014202624A2 (en) | 2013-06-19 | 2014-12-24 | Dsm Ip Assets B.V. | Rasamsonia gene and use thereof |
| WO2014202622A2 (en) | 2013-06-19 | 2014-12-24 | Dsm Ip Assets B.V. | Rasamsonia gene and use thereof |
| WO2014202620A2 (en) | 2013-06-19 | 2014-12-24 | Dsm Ip Assets B.V. | Rasamsonia gene and use thereof |
| WO2014202621A1 (en) | 2013-06-20 | 2014-12-24 | Dsm Ip Assets B.V. | Carbohydrate degrading polypeptide and uses thereof |
| EP3021689B1 (en) | 2013-07-15 | 2021-03-24 | DSM IP Assets B.V. | Diterpene production |
| US20160185813A1 (en) | 2013-07-31 | 2016-06-30 | Dsm Ip Assets B.V. | Recovery of steviol glycosides |
| US10487125B2 (en) | 2013-12-02 | 2019-11-26 | Dsm Ip Assets B.V. | Ice structuring protein |
| WO2015177153A1 (en) | 2014-05-19 | 2015-11-26 | Dsm Ip Assets B.V. | Proline-specific endoprotease |
| EP3146043B1 (en) | 2014-05-19 | 2019-06-26 | DSM IP Assets B.V. | Proline-specific endoprotease |
| WO2015177152A1 (en) | 2014-05-19 | 2015-11-26 | Dsm Ip Assets B.V. | Proline-specific endoprotease |
| EP3242949B1 (en) | 2015-01-06 | 2021-11-03 | DSM IP Assets B.V. | A crispr-cas system for a yeast host cell |
| US10590436B2 (en) | 2015-01-06 | 2020-03-17 | Dsm Ip Assets B.V. | CRISPR-CAS system for a lipolytic yeast host cell |
| WO2016110453A1 (en) | 2015-01-06 | 2016-07-14 | Dsm Ip Assets B.V. | A crispr-cas system for a filamentous fungal host cell |
| CN107429273A (zh) | 2015-02-16 | 2017-12-01 | 帝斯曼知识产权资产管理有限公司 | 用于在厌氧条件下生产衣康酸的方法 |
| WO2016146711A1 (en) | 2015-03-16 | 2016-09-22 | Dsm Ip Assets B.V. | Udp-glycosyltransferases |
| EP3274448B1 (en) | 2015-03-23 | 2021-08-11 | DSM IP Assets B.V. | Udp-glycosyltransferases from solanum lycopersicum |
| BR112017021066B1 (pt) | 2015-04-03 | 2022-02-08 | Dsm Ip Assets B.V. | Glicosídeos de esteviol, método para a produção de um glicosídeo de esteviol, composição, usos relacionados, gênero alimentício, alimento para animais e bebida |
| AR104205A1 (es) | 2015-04-09 | 2017-07-05 | Dsm Ip Assets Bv | Fosfolipasa c |
| BR112017022488A2 (pt) | 2015-04-21 | 2018-07-17 | Dsm Ip Assets Bv | produção microbiana de terpenoides |
| MY186983A (en) | 2015-05-22 | 2021-08-26 | Dsm Ip Assets Bv | Acetate consuming yeast cell |
| AU2016273208B2 (en) | 2015-06-02 | 2021-01-21 | Dsm Ip Assets B.V. | Use of ice structuring protein AFP19 expressed in filamentous fungal strains for preparing food |
| CA2991651A1 (en) | 2015-07-10 | 2017-01-19 | Dsm Ip Assets B.V. | Steviol glycoside composition |
| US20180230505A1 (en) | 2015-08-13 | 2018-08-16 | Dsm Ip Assets B.V. | Steviol glycoside transport |
| WO2017050652A1 (en) | 2015-09-25 | 2017-03-30 | Dsm Ip Assets B.V. | Asparaginase |
| WO2017060318A2 (en) | 2015-10-05 | 2017-04-13 | Dsm Ip Assets B.V. | Kaurenoic acid hydroxylases |
| WO2017060195A1 (en) | 2015-10-06 | 2017-04-13 | Dsm Ip Assets B.V. | Eukaryotic cell with increased production of fermentation product |
| EP4361240A3 (en) | 2015-11-17 | 2024-07-31 | DSM IP Assets B.V. | Preparation of a stable beer |
| MX2018006535A (es) | 2015-12-02 | 2018-08-15 | Basf Se | Metodo para producir proteinas en hongos filamentosos con accion clr2 reducida. |
| PE20181403A1 (es) | 2015-12-30 | 2018-09-07 | Dsm Ip Assets Bv | Hidrolisis enzimatica parcial de triacilgliceroles |
| BR112018013376A2 (pt) | 2015-12-30 | 2018-12-18 | Dsm Ip Assets Bv | hidrólise enzimática parcial de triacilgliceróis |
| EP3435771A1 (en) | 2016-03-31 | 2019-02-06 | DSM IP Assets B.V. | Enzyme composition and preparation of a dairy product with improved properties |
| WO2017167849A1 (en) | 2016-03-31 | 2017-10-05 | Dsm Ip Assets B.V. | Enzyme composition and preparation of a dairy product with improved properties |
| WO2017167847A1 (en) | 2016-03-31 | 2017-10-05 | Dsm Ip Assets B.V. | Production of milk with low lactose content |
| CA3027448A1 (en) | 2016-06-14 | 2017-12-21 | Dsm Ip Assets B.V. | Recombinant yeast cell |
| EP3484994A4 (en) | 2016-07-13 | 2020-01-22 | DSM IP Assets B.V. | CRISPR-CAS-SYSTEM FOR AN ALGENE CELL |
| CN109689864B (zh) | 2016-07-13 | 2023-04-04 | 帝斯曼知识产权资产管理有限公司 | 苹果酸脱氢酶 |
| US10913938B2 (en) | 2016-07-29 | 2021-02-09 | Dsm Ip Assets B.V. | Polypeptides having cellulolytic enhancing activity and uses thereof |
| US20190169220A1 (en) | 2016-08-09 | 2019-06-06 | Dsm Ip Assets B.V. | Crystallization of steviol glycosides |
| CN109563118A (zh) | 2016-08-09 | 2019-04-02 | 帝斯曼知识产权资产管理有限公司 | 甜菊醇糖苷的结晶 |
| EP3504336A1 (en) | 2016-08-26 | 2019-07-03 | Lesaffre et Compagnie | Improved production of itaconic acid |
| KR101808192B1 (ko) * | 2016-08-26 | 2018-01-18 | 아미코젠주식회사 | 7-아미노세팔로스포란산의 고농도 생산 재조합 아크레모니움 크리소제눔 균주의 제조방법 및 이 방법으로 제조된 균주 |
| CN109715804A (zh) | 2016-09-23 | 2019-05-03 | 帝斯曼知识产权资产管理有限公司 | 用于宿主细胞的指导rna表达系统 |
| WO2018073107A1 (en) | 2016-10-19 | 2018-04-26 | Dsm Ip Assets B.V. | Eukaryotic cell comprising xylose isomerase |
| EP3998337A1 (en) | 2016-10-27 | 2022-05-18 | DSM IP Assets B.V. | Geranylgeranyl pyrophosphate synthases |
| CN110072995B (zh) | 2016-12-08 | 2024-05-10 | 帝斯曼知识产权资产管理有限公司 | 贝壳杉烯酸羟化酶 |
| US10918113B2 (en) | 2016-12-21 | 2021-02-16 | Dsm Ip Assets B.V. | Lipolytic enzyme variants |
| WO2018114912A1 (en) | 2016-12-21 | 2018-06-28 | Dsm Ip Assets B.V. | Lipolytic enzyme variants |
| CA3046153A1 (en) | 2016-12-21 | 2018-06-28 | Dsm Ip Assets B.V. | Lipolytic enzyme variants |
| WO2018114938A1 (en) | 2016-12-21 | 2018-06-28 | Dsm Ip Assets B.V. | Lipolytic enzyme variants |
| WO2018114995A1 (en) | 2016-12-22 | 2018-06-28 | Dsm Ip Assets B.V. | Fermentation process for producing steviol glycosides |
| WO2018166943A1 (en) | 2017-03-13 | 2018-09-20 | Dsm Ip Assets B.V. | Zinc binuclear cluster transcriptional regulator-deficient strain |
| CN110462044A (zh) | 2017-04-06 | 2019-11-15 | 帝斯曼知识产权资产管理有限公司 | 自引导整合构建体(sgic) |
| BR112019025777A2 (pt) | 2017-06-13 | 2020-06-30 | Dsm Ip Assets B.V. | célula de levedura recombinante |
| EP3645714B1 (en) | 2017-06-27 | 2022-08-31 | DSM IP Assets B.V. | Udp-glycosyltransferases |
| EP3688177A1 (en) | 2017-09-26 | 2020-08-05 | DSM IP Assets B.V. | Acetic acid consuming strain |
| EP3533878A1 (en) | 2018-02-28 | 2019-09-04 | Dutch DNA Biotech B.V. | Process for producing citramalic acid employing aspergillus |
| JP2021078354A (ja) * | 2018-03-08 | 2021-05-27 | 有限会社アルティザイム・インターナショナル | フラビンアデニンジヌクレオチドグルコース脱水素酵素とシトクロム分子との融合タンパク質 |
| UA126992C2 (uk) | 2018-05-07 | 2023-03-01 | Дсм Айпі Асетс Б.В. | Спосіб ферментативного дегумування олії |
| MX2020014191A (es) | 2018-06-19 | 2021-03-09 | Dsm Ip Assets Bv | Variantes de enzimas lipoliticas. |
| US20220389458A1 (en) | 2019-11-04 | 2022-12-08 | Dsm Ip Assets B.V. | Low volume transfection |
| AU2021207258A1 (en) | 2020-01-14 | 2022-08-04 | The Protein Brewery B.V. | Expression of ovalbumin and its natural variants |
| EP4117445A1 (en) | 2020-03-10 | 2023-01-18 | DSM IP Assets B.V. | Lactose reduced dairy powder |
| AU2021367159A1 (en) | 2020-10-22 | 2023-05-04 | Cargill, Incorporated | Microorganisms for diterpene production |
| AU2022297881A1 (en) | 2021-06-24 | 2024-01-18 | Fonterra Co-Operative Group Limited | Recombinant beta-lactoglobulin |
| MX2024000514A (es) | 2021-07-12 | 2024-03-11 | Danisco Us Inc | Celula de levadura recombinante. |
| CN117940570A (zh) | 2021-07-12 | 2024-04-26 | 丹尼斯科美国公司 | 重组酵母细胞 |
| EP4370690A1 (en) | 2021-07-12 | 2024-05-22 | Danisco Us Inc | Recombinant yeast cell |
| MX2024000511A (es) | 2021-07-12 | 2024-03-11 | Danisco Us Inc | Celula de levadura recombinante. |
| WO2023285297A1 (en) | 2021-07-12 | 2023-01-19 | Dsm Ip Assets B.V. | Recombinant yeast cell |
| WO2023285280A1 (en) | 2021-07-12 | 2023-01-19 | Dsm Ip Assets B.V. | Recombinant yeast cell |
| CN118176296A (zh) | 2021-11-04 | 2024-06-11 | 丹尼斯科美国公司 | 重组酵母细胞 |
| CN118215740A (zh) | 2021-11-04 | 2024-06-18 | 丹尼斯科美国公司 | 用于生产乙醇的方法和重组酵母细胞 |
| EP4426823A1 (en) | 2021-11-04 | 2024-09-11 | Danisco Us Inc | Variant polypeptide and recombinant yeast cell |
| CA3237662A1 (en) | 2021-11-09 | 2023-05-19 | Amgen Inc. | Production of therapeutic proteins |
| WO2023222614A1 (en) | 2022-05-16 | 2023-11-23 | Dsm Ip Assets B.V. | Lipolytic enzyme variants |
| WO2024133849A1 (en) | 2022-12-22 | 2024-06-27 | The Protein Brewery B.V. | Ovalbumin fermentation |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA1341226C (en) | 1988-08-16 | 2001-05-01 | Wim Van Hartingsveldt | Gene replacement as a tool for the construction of aspergillus strains |
| JP3273609B2 (ja) * | 1989-07-07 | 2002-04-08 | ユニリーバー・ナームローゼ・ベンノートシヤープ | 発現ベクターのマルチコピー組込みにより形質転換した真菌による蛋白質の製法 |
| SK466390A3 (en) | 1989-09-27 | 2000-05-16 | Gist Brocades Nv | Purified and isolated dna sequence, construct, vector, transformed cell, peptide or protein having phytase activity, process for its preparation, and its use |
| ES2198410T3 (es) * | 1993-07-23 | 2004-02-01 | Dsm Ip Assets B.V. | Cepas recombinantes de hongos filamentosos libres de gen marcador de seleccion: un metodo para obtener dichas cepas y su utilizacion. |
| HU219259B (en) | 1993-07-30 | 2001-03-28 | Gist Brocades Bv | Process for the efficient production of 7-adca via 2-(carboxyethylthio)acetyl-7-adca and 3-(carboxymethylthio)propionyl-7-adca, recombinant dna vectrors and transformed host cells |
| WO1995017513A1 (en) * | 1993-12-23 | 1995-06-29 | Novo Nordisk A/S | Retransformation of filamentous fungi |
| ES2094088B1 (es) | 1994-05-04 | 1997-09-01 | Antibioticos Sa | Identificacion, clonacion y utilizacion de una region de dna amplificada en cepas de p. chrysogenum superproductoras de penicilina. |
| JP3343567B2 (ja) * | 1995-06-29 | 2002-11-11 | 大関株式会社 | 糸状菌のエンハンサーdna塩基配列およびその用途 |
| DE69637784D1 (de) * | 1995-08-03 | 2009-02-05 | Dsm Ip Assets Bv | amdS-Gen aus Aspergillus niger für eine Acetamidase kodierend |
| PL336345A1 (en) * | 1997-04-11 | 2000-06-19 | Dsm Nv | Genic conversion as a tool for constructing recombined filiform fungi |
-
1998
- 1998-04-09 PL PL98336345A patent/PL336345A1/xx unknown
- 1998-04-09 EP EP98924103.9A patent/EP0979294B1/en not_active Expired - Lifetime
- 1998-04-09 US US09/402,631 patent/US6432672B1/en not_active Expired - Lifetime
- 1998-04-09 CN CNB988052377A patent/CN1169961C/zh not_active Expired - Fee Related
- 1998-04-09 AU AU76422/98A patent/AU7642298A/en not_active Abandoned
- 1998-04-09 DK DK98924103.9T patent/DK0979294T3/en active
- 1998-04-09 WO PCT/EP1998/002070 patent/WO1998046772A2/en active Application Filing
- 1998-04-09 BR BR9808859-9A patent/BR9808859A/pt not_active Application Discontinuation
- 1998-04-09 JP JP54345698A patent/JP5303084B2/ja not_active Expired - Fee Related
- 1998-04-09 EP EP10174795A patent/EP2298913A1/en not_active Withdrawn
-
2009
- 2009-12-01 JP JP2009273669A patent/JP2010075204A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| EP0979294B1 (en) | 2015-05-27 |
| US6432672B1 (en) | 2002-08-13 |
| JP2010075204A (ja) | 2010-04-08 |
| EP0979294A2 (en) | 2000-02-16 |
| BR9808859A (pt) | 2000-08-01 |
| DK0979294T3 (en) | 2015-08-31 |
| EP2298913A1 (en) | 2011-03-23 |
| CN1257546A (zh) | 2000-06-21 |
| AU7642298A (en) | 1998-11-11 |
| WO1998046772A3 (en) | 1999-02-25 |
| JP5303084B2 (ja) | 2013-10-02 |
| JP2001518798A (ja) | 2001-10-16 |
| WO1998046772A2 (en) | 1998-10-22 |
| PL336345A1 (en) | 2000-06-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1169961C (zh) | 基因转变作为工具用于构建重组的工业化丝状真菌 | |
| CN100347301C (zh) | 无选择标志重组基因的菌株,得到这些菌株的方法及应用 | |
| CN1261567C (zh) | 热稳定的葡糖淀粉酶 | |
| CN1262639C (zh) | 新的宿主细胞和生产蛋白质的方法 | |
| CN1170938C (zh) | 构建产生氨基酸的细菌的方法及通过发酵该经构建的产生氨基酸的细菌以制备氨基酸的方法 | |
| CN1198924C (zh) | 在丝状真菌中表达克隆 | |
| CN1128875C (zh) | 制备多肽变异体的方法 | |
| CN1197965C (zh) | 在丝状真菌细胞内构建和筛选目的dna文库 | |
| CN1253574C (zh) | 来自米曲霉的areA基因和其中areA基因已被修饰的真菌 | |
| CN1040025C (zh) | 编码可提高耐热性的脱氨基甲酰酶的dna及其用途 | |
| CN1262666C (zh) | 用人工转座子扩增基因的方法 | |
| CN1798848A (zh) | 在缺乏酶的黑曲霉突变体中生产生物物质的方法 | |
| CN1117151C (zh) | 真菌中的核黄素生物合成 | |
| CN1384190A (zh) | 通过发酵生产l-谷氨酰胺的方法和产生l-谷氨酰胺的细菌 | |
| CN100344765C (zh) | 用于调节转录的dna序列 | |
| CN1160465C (zh) | 其中areA、pepC和/或pepE基因已被灭活的真菌 | |
| CN1253575C (zh) | 用共有翻译起始区序列制备多肽的方法 | |
| CN1303434A (zh) | 用于生产核黄素的遗传方法 | |
| CN1026015C (zh) | 编码脱乙酸基头孢菌素c合成酶和脱乙酰头孢菌素c合成酶的重组dna表达载体及dna化合物 | |
| CN1839199A (zh) | 脂质生产菌的育种方法 | |
| CN1795270A (zh) | 编码具有d-乳酸脱氢酶活性蛋白质的dna及其用途 | |
| CN1268746C (zh) | 用于在真菌细胞中生产多肽的方法 | |
| CN1185174A (zh) | 核酸类的制备方法 | |
| CN1661015A (zh) | 来自产朊假丝酵母的谷胱甘肽合成酶编码基因 | |
| CN1183248C (zh) | 环状缩肽合成酶及其基因、以及环状缩肽的大规模生产系统 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20041006 Termination date: 20170409 |