CN103796830A - 气体阻隔性膜及其制造方法、以及使用了其的电子元件用基板 - Google Patents
气体阻隔性膜及其制造方法、以及使用了其的电子元件用基板 Download PDFInfo
- Publication number
- CN103796830A CN103796830A CN201280042683.6A CN201280042683A CN103796830A CN 103796830 A CN103796830 A CN 103796830A CN 201280042683 A CN201280042683 A CN 201280042683A CN 103796830 A CN103796830 A CN 103796830A
- Authority
- CN
- China
- Prior art keywords
- fibrous
- film
- gas barrier
- cellulose nano
- ester
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images

Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C41/00—Shaping by coating a mould, core or other substrate, i.e. by depositing material and stripping-off the shaped article; Apparatus therefor
- B29C41/24—Shaping by coating a mould, core or other substrate, i.e. by depositing material and stripping-off the shaped article; Apparatus therefor for making articles of indefinite length
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C55/00—Shaping by stretching, e.g. drawing through a die; Apparatus therefor
- B29C55/02—Shaping by stretching, e.g. drawing through a die; Apparatus therefor of plates or sheets
- B29C55/10—Shaping by stretching, e.g. drawing through a die; Apparatus therefor of plates or sheets multiaxial
- B29C55/12—Shaping by stretching, e.g. drawing through a die; Apparatus therefor of plates or sheets multiaxial biaxial
-
- G—PHYSICS
- G02—OPTICS
- G02B—OPTICAL ELEMENTS, SYSTEMS OR APPARATUS
- G02B1/00—Optical elements characterised by the material of which they are made; Optical coatings for optical elements
- G02B1/10—Optical coatings produced by application to, or surface treatment of, optical elements
- G02B1/14—Protective coatings, e.g. hard coatings
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/03—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor characterised by the shape of the extruded material at extrusion
- B29C48/07—Flat, e.g. panels
- B29C48/08—Flat, e.g. panels flexible, e.g. films
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/16—Articles comprising two or more components, e.g. co-extruded layers
- B29C48/18—Articles comprising two or more components, e.g. co-extruded layers the components being layers
- B29C48/21—Articles comprising two or more components, e.g. co-extruded layers the components being layers the layers being joined at their surfaces
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2001/00—Use of cellulose, modified cellulose or cellulose derivatives, e.g. viscose, as moulding material
- B29K2001/08—Cellulose derivatives
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2995/00—Properties of moulding materials, reinforcements, fillers, preformed parts or moulds
- B29K2995/0037—Other properties
- B29K2995/0065—Permeability to gases
- B29K2995/0067—Permeability to gases non-permeable
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2995/00—Properties of moulding materials, reinforcements, fillers, preformed parts or moulds
- B29K2995/0037—Other properties
- B29K2995/0068—Permeability to liquids; Adsorption
- B29K2995/0069—Permeability to liquids; Adsorption non-permeable
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/31504—Composite [nonstructural laminate]
- Y10T428/31971—Of carbohydrate
- Y10T428/31975—Of cellulosic next to another carbohydrate
Landscapes
- Engineering & Computer Science (AREA)
- Mechanical Engineering (AREA)
- Physics & Mathematics (AREA)
- General Physics & Mathematics (AREA)
- Optics & Photonics (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Laminated Bodies (AREA)
- Liquid Crystal (AREA)
Abstract
本发明提供气体阻隔性膜及其制造方法、以及使用了其的电子元件用基板。本发明的气体阻隔性膜具有片状基材和形成于上述片状基材的至少单面的气体阻隔层,所述片状基材含有纤维素纳米纤维的羟基的氢原子的至少一部分被碳数1~8的酰基取代了的表面改性纤维素纳米纤维,基体树脂的含量相对于上述纤维素纳米纤维和所述基体树脂的总量为10质量%以下。另外,本发明的气体阻隔性膜的制造方法,具有将纤维素纳米纤维的羟基的氢原子的至少一部分用碳数1~8的酰基进行取代而得到表面改性纤维素纳米纤维、将上述表面改性纤维素纳米纤维用熔融挤出法或溶液浇铸法进行制膜而得到片状基材的工序A和在上述片状基材上形成气体阻隔层的工序B。
Description
技术领域
本发明涉及气体阻隔性膜及其制造方法、以及使用了其的电子元件用基板。
背景技术
一般而言,作为液晶、有机EL等的显示元件基板、滤色器基板、太阳能电池用基板等,广泛使用玻璃板。然而,玻璃板,由于易碎、不弯曲、比重大、不适合轻量化等的理由,近年来作为玻璃板的替代而正在研究塑料原材料。
例如,已知有将玻璃布无纺布含浸于环氧树脂而热固化了的树脂基材(专利文献1)、由含有纤维素和纤维素以外的树脂的复合体构成的液晶显示元件用塑料基板(专利文献2)。
然而,上述的玻璃代替用塑料材料,由于与玻璃板相比在透明性、线膨胀率方面较差,因此,存在因制造工序中的热处理等而产生透明性的劣化、卷曲等导致的翘曲·断线等这样的问题。另外,由于无纺布的空隙率不均匀,因此,在将树脂含浸于无纺布片时,存在树脂的浸透变得不均匀、产生泡而产生缺陷等的问题。因此,将上述的代替材料应用于显示元件等的基板用途是困难的。
作为改善这些问题的方法,公开有改性纤维素纳米纤维而使基体树脂(基体材料)的浸透提高的技术、将纤维素纳米纤维和基体树脂用熔融混合法、溶液浇铸法而进行膜化的技术(专利文献3及4)。
另一方面,各种显示元件用的基板,除上述的性能以外,要求高的气体阻隔性。因此,近年来,进行了许多在基材的单面或两面设置各种的硬涂层、气体阻隔层、从基板固有的水平进一步使气体阻隔特性提高的尝试。
作为不伴随液晶显示元件、有机EL元件等的性能劣化地赋予气体阻隔性的方法,有将由SiO2等构成的气体阻隔层进行蒸镀的方法;通过涂布烷氧基硅烷的有机溶剂溶液这样的涂布系二氧化硅材料、进行加热而使其进行三维反应来形成阻隔层的方法;通过涂布含聚硅氮烷液体、实施改性处理(等离子体处理、紫外线照射等)而形成气体阻隔层的方法(例如专利文献5)等。
现有技术文献
专利文献
专利文献1:美国专利申请公开第2004/132867号说明书
专利文献2:特开2006-316253号公报
专利文献3:特开2008-208231号公报
专利文献4:特开2008-209595号公报
专利文献5:特开2007-237588号公报
发明内容
发明所要解决的课题
在如上述专利文献3及4中所公开那样的纤维素纳米纤维基材中,在纤维素纤维的周围存在纤维素树脂等的基体树脂。这些技术由于伴随纤维素纳米纤维和基体树脂的混合而表面平滑性及透明性不充分。
另外,如专利文献5中所公开那样的气体阻隔层,存在可应用的基材受限这样的问题。例如,在如上述专利文献3、4中所记载那样的在具有基体树脂的纤维素纳米纤维基材表面形成了专利文献5中记载的气体阻隔层的情况下,不仅因形成气体阻隔层时的改性处理引起基体树脂和纤维素纳米纤维的界面的层分离、微小的表面性状的不均匀,气体阻隔性得不到提高,而且存在基材和气体阻隔层的粘接性、表面的平滑性受损这样的问题。
这样,即使通过专利文献3~5中所记载的技术,也难以得到满足显示元件基板所要求的透明性、平滑性、粘接性及气体阻隔性的塑料基板。
本发明,是鉴于上述课题完成的,其目的在于:提供透明性、表面平滑性、气体阻隔性及粘接性优异的气体阻隔性膜及其制造方法、以及使用了其的电子元件用基板。
用于解决课题的手段
本发明人等,为了改善上述课题进行了潜心研究,结果发现:通过相对于实质上不含有基体树脂、由纤维素纳米纤维的表面的纤维素的羟基的氢原子的至少一部分被碳数1~8的酰基取代了的表面改性纤维素纳米纤维构成的基材来形成气体阻隔层,可解决上述课题,以至完成了本发明。
即,本发明的上述目的由以下的构成来达到。
(1)一种气体阻隔性膜,其具有片状基材和在上述片状基材的至少单面形成了的气体阻隔层,所述片状基材为:含有纤维素纳米纤维的羟基的氢原子的至少一部分被碳数1~8的酰基取代了的表面改性纤维素纳米纤维、且基体树脂的含量相对于上述纤维素纳米纤维和上述基体树脂的总量为10质量%以下。
(2)如(1)所述的气体阻隔性膜,其中,上述酰基包含丙酰基。
(3)如(1)或(2)所述的气体阻隔性膜,其中,上述气体阻隔层含有硅氧化物或氮化硅氧化物。
(4)一种气体阻隔性膜的制造方法,其具有以下工序:将纤维素纳米纤维的羟基的氢原子的至少一部分用碳数1~8的酰基进行取代而得到表面改性纤维素纳米纤维、将上述表面改性纤维素纳米纤维用熔融挤出法或溶液浇铸法进行制膜而得到片状基材的工序A;和在上述片状基材上形成气体阻隔层的工序B。
(5)如(4)所述的制造方法,其中,在上述工序A中,在制膜后进行拉伸处理或/及加热压延处理。
(6)如(4)或(5)所述的制造方法,其中,上述工序B包含:在上述片状基材上涂布含有聚硅氮烷化合物的涂布液后、进行准分子照射处理。
(7)一种电子元件用基板,其使用了(1)~(3)的任一项所述的气体阻隔性膜或通过(4)~(6)的任一项所述的制造方法而制造的气体阻隔性膜。
发明的效果
由于构成本发明的气体阻隔性膜的片状基材实质上不含基体树脂,因此,可以形成多种气体阻隔层,可谋求高水平的透明性、表面平滑性、气体阻隔性、及粘接性的实现。特别是即使在电子元件的制造工序中被热处理了的情况下也可维持良好的粘接性。
附图说明
图1是表示作为本发明的一实施方式的气体阻隔性膜的基本构成的示意剖面图。
具体实施方式
以下,一边参照附图一边对本发明的实施方式进行说明。需要说明的是,本发明不只限于以下的实施方式。附图的尺寸比率为了便于说明而夸张,有时与实际的比率不同。
根据本发明的一方式,提供具有具有片状基材和形成于上述片状基材的至少单面的气体阻隔层的气体阻隔性膜,所述片状基材为含有纤维素纳米纤维的羟基的氢原子的至少一部分被碳数1~8的酰基取代了的表面改性纤维素纳米纤维,且基体树脂的含量相对于上述纤维素纳米纤维和上述基体树脂的总量为10质量%以下。
本发明的特征在于,由特定的表面改性纤维素纳米纤维构成,在基体树脂的含量小(实质上不含基体树脂)的基材上形成气体阻隔层。即,发现:通过使用实质上不含有基体树脂地对表面改性纤维素纳米纤维进行制膜了的膜基材,与使用了以往的基体树脂的树脂含浸膜相比,能够实现高水平的透明性、表面平滑性、气体阻隔性及粘接性,以至完成了本发明。
本发明的详细的机制尚未明确,但通过实质上不含基体树脂地使用纤维素纳米纤维的表面被酰基取代了的纤维素纳米纤维,维持纤维素纳米纤维成分的互相缠绕,同时表层的非晶性的树脂成分(酰基成分)熔融而均匀地扩展,因此,与将基体树脂混合的体系相比,折射率差小,膜内的纳米纤维的均匀性也良好。因此,在后面的电子元件的制造工序中的被热加工了时也能够维持透明性、粘接性。
以下,详细地说明本发明。
图1是示出作为本发明的一实施方式的气体阻隔性膜的基本构成的示意剖面图。如图1中所示,气体阻隔性膜10由片状基材1、夹持其的1对中间层(中间层2a及中间层2b)、夹持片状基材1及中间层(2a及2b)的层叠体的1对气体阻隔层(气体阻隔层3a及气体阻隔层3b)构成。具体而言,在片状基材1的两面设有中间层(2a、2b),在该中间层(2a、2b)的上部层叠有气体阻隔层3。
在图1种所示的方式中,中间层(2a及2b)介于片状基材1和气体阻隔层3之间。在中间层(2a、2b)介于片状基材1和气体阻隔层(3a、3b)之间的情况下,其部分的膜厚增加,且均匀地进行气体阻隔层的形成,因此气体阻隔性可提高。需要说明的是,由中间层引起的气体阻隔特性的提高效果是限定性的,仅仅中间层不能发挥充分的气体阻隔特性。但是,本发明只要在片状基材上形成气体阻隔层即可,也可以不配置中间层(2a、2b)地直接在片状基材1的上面层叠气体阻隔层(3a、3b)。
另外,在图1中所示的方式中,气体阻隔层(3a、3b)形成于片状基材1的两面,但气体阻隔层(3a或3b)也可以仅形成于片状基材1的单面。
进而,当然也可以形成在片状基材1的一面设置中间层(2a或2b)、不在另一面设置中间层的构成。
以下,对构成气体阻隔性膜10的部件进行说明。
(片状基材)
片状基材1含有纤维素纳米纤维的羟基的氢原子的至少一部分被碳数1~8的酰基取代了的表面改性纤维素纳米纤维(以下也简称为“表面改性纤维素纳米纤维”)、以及根据需要的微量的基体树脂及碳自由基捕捉剂、一次抗氧化剂、二次抗氧化剂、酸捕捉剂、紫外线吸收剂、增塑剂、消光剂、光学各向异性控制剂、交联剂等的添加剂而构成。
(a)纤维素纳米纤维
所谓本发明中所使用的纤维素纳米纤维,是指平均纤维直径为1~1000nm的纤维素纤维。优选为4~400nm的纤维直径的纤维。若纤维的平均纤维直径为400nm以下,则由于比可见光的波长小,因此可抑制透明性的降低。若平均纤维直径为4nm以上,则制造容易。更优选的是,为了使片状基材的强度提高,为4~200nm,更优选为4~100nm,进一步优选为4~50nm的纤维直径的纤维。
所谓“纤维素纤维”,是指构成植物细胞壁的基本骨架等的纤维素的微原纤维或该构成纤维,通常为由纤维直径4nm左右的单纤维(纤维素分子链以几十个氢键键合了的结晶性的纤维)构成的集合体。纤维素纤维,从得到高的强度和低的热膨胀的方面考虑,优选含有40%以上的结晶结构的纤维。
纤维素纳米纤维,可以由单纤维不合丝、而以在相互间进入地充分隔离而存在的状态构成。此时,纤维直径为单纤维的直径。或者,也可以为多根单纤维集合成束状而构成1根线条的纤维,此时,纤维直径定义为1根线条的直径。
需要说明的是,本发明中使用的纤维素纳米纤维的平均纤维直径,可为上述范围内,也可以含有上述范围以外的纤维直径的纤维。但是,上述范围以外的纤维直径的纤维相对于纤维素纳米纤维全部的比例,优选为20质量%以下,更优选全部的纤维素纳米纤维的纤维直径在上述范围内。
对于纳米纤维的长度没有特别限定,但以平均纤维长计优选50nm以上,进一步优选100nm以上。若为这样的范围,则纤维的互相缠绕良好,增强效果高,可抑制热膨胀的增大。
在本发明中,“平均纤维直径”、“平均纤维长”,可从用透射型电子显微镜(TEM)(例如H-1700FA型(日立制作所公司制))或扫描型电子显微镜(SEM)以10000倍的倍率观察纤维素纳米纤维而得到的图像随机地选择100根纤维,使用图像处理软件(例如WINROOF)分析每一根的纤维直径(直径)及纤维长、以这些的简单数均值的形式算出。
纤维素纳米纤维,通过对原料纤维素纤维进行解纤处理而得到。作为原料纤维素纤维,可以举出从来自植物的纸浆、木材、棉花、麻、竹、棉、洋麻、大麻、黄麻、香蕉、椰子、海草等的植物纤维分离的纤维、从作为海洋动物的海鞘类产生的动物纤维分离的纤维、或由乙酸菌产生的细菌纤维素等。其中,优选从植物纤维分离的纤维,更优选由纸浆、棉花得到的纤维。
作为原料纤维素纤维的解纤处理的方法,只要纤维素纤维保持纤维状态就没有任何限制,可以举出使用了均质器、研磨机等的机械解纤处理、使用了2,2,6,6-四甲基哌啶-1-氧自由基(TEMPO)等的氧化催化剂的化学解纤处理。进而,为了促进这些解纤处理,可以利用酶等来微细化为微原纤维状。
作为机械解纤处理的具体方法,例如,首先将纸浆等的原料纤维素纤维投入到放入了水的分散容器中以使得到达到0.1~3质量%,将其用高压均质器进行解纤处理,得到解纤为平均纤维直径0.1~10μm左右的微原纤维的纤维素纤维的水分散液。接着,通过用研磨机等重复进行磨碎处理,可以得到平均纤维直径2~几百nm左右的纤维素纳米纤维。作为上述磨碎处理中所使用的研磨机,例如可以举出ピュァファィンミル(栗田机械制作所公司制)等。
另外,作为其它的方法,已知有使用通过在250MPa左右的高压下从一对喷嘴分别喷射原料纤维素纤维的分散液、使其喷射流互相以高速碰撞来粉碎纤维素纤维的高压均质器的方法。作为所使用的装置,例如可以举出:三和机械公司制的“ホモジナィザ一”、スギノマシン(株)制的“ァルテマィザ一システム”等。
作为化学解纤处理的具体方法,例如可以举出使用氧化催化剂及根据需要的共氧化剂对原料纤维素纤维进行氧化处理的方法。由此,在吡喃糖单元的C6位存在的伯羟基被氧化为羧基,通过原纤维彼此的静电排斥而被化学解纤。需要说明的是,通过经过氧化反应处理,在原料纤维素纤维的分子中导入羧基,但也有时部分地根据氧化处理的进行程度而导入醛基。因此,氧化处理后的解纤纤维的羟基被醛基及羧基的至少一方取代。
作为氧化催化剂,可使用N-氧化合物。例如从常温下的反应速度良好的方面考虑,优选选自由2,6,6-四甲基哌啶-N-氧(TEMPO)、4-乙酰胺-TEMPO、4-羧基-TEMPO、4-磷酰氧-TEMPO、2-氮杂金刚烷-N-氧、1-甲基-2-氮杂金刚烷-N-氧、及1,3-二甲基-2-氮杂金刚烷-N-氧(DMAO)组成的组中的至少1个。其中,为了实现膜的高的透明性和耐热性,优选使用2,2,6,6-四甲基哌啶-1-氧自由基(TEMPO)作为氧化催化剂、对纤维素非晶区域的伯羟基进行氧化而导入羧基、利用原纤维相互的静电排斥来进行化学解纤的方法。
作为共氧化剂,可以举出选自由次卤酸或其盐、亚卤酸或其盐、高卤酸或其盐、过氧化氢、及过有机酸组成的组中的至少1种。对于上述的共氧化剂中作为盐的物质,优选选自由碱金属、镁及碱土金属组成的组中的至少1种的盐,其中,更优选碱金属次卤酸盐,例如次氯酸钠、次溴酸钠。在使用次氯酸钠这样的次卤酸盐的情况下,从提高反应速度的方面考虑,特别优选在溴化碱金属、例如溴化钠的存在下进行反应。在使共氧化剂与氧化催化剂一起作用来进行氧化反应的情况下,由吡喃糖单元构成的高分子链为分子链水平、而且只是C6位的伯羟基被选择性地氧化、经由醛而被氧化至羧基,故优选。
上述氧化反应,优选使原料纤维素纤维分散于溶剂中来进行。作为溶剂,需要为与原料纤维素纤维、氧化催化剂及共氧化剂在氧化反应、操作的条件下不显示显著的反应性且解纤纤维和羧基导入后的纤维良好地分散的溶剂。其中,从廉价且容易操作等的方面考虑,最优选水。此时,优选使原料纤维素纤维相对于作为溶剂的水的浓度为0.1质量%以上且3质量%以下。
使上述氧化催化剂、及根据需要的共氧化剂与解纤纤维作用、得到导入了羧基的改性解纤纤维时的具体方法、条件,可优选使用特开2008-1728号公报中所公开的方法、条件。
基于这样的C6位的羧基的静电排斥的化学解纤,与机械解纤相比,可以得到均匀的更小的纤维直径。
纤维素纤维,一般而言是聚合度为1,000~3,000(以重均分子量计为几万~几百万)的范围的不溶性的天然纤维。在本发明中,解纤后的结晶性原纤维的纤维直径是重要的,只要使用聚合度(重均分子量)在该范围的不溶性的天然纤维即可。
在本发明中,“重均分子量”采用使用高速液相色谱在下述的测定条件下测得的值。
溶剂:二氯甲烷
柱:Shodex K806、K805、K803G(昭和电工(株)制,将3根连接来使用)
柱温:25℃
试样浓度:0.1重量%
检测器:RI Model504(GLサィェンス公司制)
泵:L6000(日立制作所(株)制)
流量:1.0m1/min
校正曲线:使用利用标准聚苯乙烯STK标准聚苯乙烯(东ン(株))制)重均分子量1000000~500的13个样品的校正曲线
(b)表面改性纤维素纳米纤维
本发明中的表面改性纤维素纳米纤维,为构成纤维素纳米纤维的纤维素的葡萄糖单元的2位、3位及/或6位的羟基(-OH)的氢原子的至少一部分通过化学改性而被碳数1~8的酰基取代了的纤维。
所谓纤维素,是多个的β-葡萄糖分子通过糖苷键直链状地聚合而成的,在C2位、C3位、及C6位具有羟基。因此,一般而言未被化学改性的纤维素纳米纤维,含有下述化学式(A)作为重复单元。
[化学式1]

对本方式涉及的表面改性纤维素纳米纤维而言,上述纤维素纳米纤维的C2位、C3位、及C6位的至少一个的羟基被酯化。即,本方式涉及的纤维素纳米纤维在C2位、C3位、及C6位的至少一个上具有碳数1~8的酰基。
更具体而言,本发明的表面改性纤维素纳米纤维,可推定为纤维素纳米纤维的表面的羟基的氢原子被取代为酰基,认为其成为结晶性的纳米纤维成分为核、非晶性的改性了的纤维素酯成分(酰基成分)为壳的具有核壳形的剖面的纤维。
表面改性纤维素纳米纤维的平均纤维直径及平均纤维长,与上述了的纤维素纳米纤维的平均纤维直径及平均纤维长的规定相同。
碳数1~8的酰基没有特别限制,可以举出甲酰基、乙酰基、丙酰基(丙酰基)、异丙酰基、丁酰基(丁酰基)、异丁酰基(异丁酰基)、戊酰基、异戊酰基、2-甲基戊酰基、3-甲基戊酰基、4-甲基戊酰基、叔丁基乙酰基、特戊酰基、己酰基、2-乙基己酰基、2-甲基己酰基、庚酰基、辛酰基、苯甲酰基等。其中,优选碳数2~4的酰基,更优选乙酰基、丙酰基、丁酰基,特别优选丙酰基。即,在特别优选的方式中,酰基包含丙酰基。由于丙酸酯成分与其它的酰基成分相比流动性等良好,因此,透明性及平滑性可提高。需要说明的是,纤维素纳米纤维的羟基的氢原子可以被单一种类的酰基取代,也可以被多种酰基取代。
通过用酰基取代纤维素纳米纤维的羟基的氢原子的至少一部分,可以对纤维的表层进行非晶化(树脂化),可以一边维持纤维素纳米纤维成分的互相缠绕一边对结晶性的纤维素纳米纤维赋予柔软性。由此,即使在未与基体树脂混合的情况下,成形加工性也优异,可形成均匀的制膜。进而,通过对纤维的表层进行非晶化(树脂化),可提高透明性及表面平滑性。
纤维素纳米纤维的酰基的取代度,优选为0.5~2.5。若取代度为0.5以上,则纤维表面的树脂成分(酰基成分)变多,制膜性及透明性提高,进而可减少缺陷,故优选。若取代度为2.5以下,则结晶性纳米纤维部分(核部)变多,纳米纤维的互相缠绕增大,热线膨胀性优异,故优选。更优选取代度为0.5~2.0。
如上述化学式(A)中所示,构成纤维素的β-1,4进行键合的葡萄糖单元,在2位、3位及6位具有游离的羟基(-OH)。所谓“纤维素纳米纤维的酰基的取代度”,表示每1葡萄糖单元的酰基的平均数,表示1葡萄糖单元的2位、3位及6位的羟基的氢原子的任意被取代为酰基。即,2位、3位及6位的羟基的氢原子全部被酰基取代了时的取代度(最大的取代度)为3.0。酰基可以在葡萄糖单元的2位、3位、6位平均地取代,也可以具有分布地取代。取代度可利用ASTM-D817-96中规定的方法求出。
表面改性纤维素纳米纤维的结晶度,优选为30~90%。若结晶度为30%以上,则可抑制纳米纤维的热线膨胀特性的劣化及伴随其的膜的热线膨胀特性的劣化。另一方面,若为90%以下,则可抑制制膜性、透明性及表面平滑性的降低。更优选结晶度为50~90%,进一步优选为40~80%。
结晶度可用以下记载的方法算出。
[结晶度的算出方法]
测定X射线衍射强度,基于下述数学式(1)算出结晶度CrI。需要说明的是,I8表示2θ=8°衍射峰强度,I18表示2θ=18°的衍射峰强度。
衍射峰强度根据树脂而不同,但可以通过从各光谱的峰的强度中减去基线的强度来算出。
[数学式1]
CrI=(I8-I18)/I8
(混合取代度和结晶度不同的纤维素纳米纤维)
在本发明中,表面改性纤维素纳米纤维,优选混合了酰基的取代度及结晶度不同的表面改性纤维素纳米纤维的纤维。通过混合取代度和结晶度不同的纳米纤维,性能(透明性、生产率)的稳定性提高,因此是有效的。具体而言,优选将酰基的取代度小且结晶度高的表面改性纤维素纳米纤维和酰基的取代度大且结晶度小的表面改性纤维素纳米纤维混合来使用。前者为对热膨胀性的降低有利的纤维,后者为对透明性、生产率有利的纤维。通过混合它们,作为本发明的效果的性能的稳定性更稳定,故优选。
本发明中的表面改性纤维素纳米纤维,可以在不损害本发明的效果的范围用酰基以外的官能团进行取代、改性。改性方法,可使用将纤维素纳米纤维的羟基用酸、醇类、卤化试剂、酸酐、异氰酸酯类、硅烷偶联剂等的改性剂进行化学改性等的公知的方法。
(c)基体树脂
在本发明中,片状基材1的特征之一在于:基体树脂的含量相对于纤维素纳米纤维和上述基体树脂的总量为10质量%以下。该基体树脂的含量优选为5质量%以下,更优选为3质量%以下,进一步优选为1质量%以下,特别优选为0质量%、即不含基体树脂。
在本发明中,所谓“基体树脂”,是指分子量为10,000以上的无机高分子或有机高分子。具体而言,作为无机高分子,可以举出玻璃、硅酸盐(酯)材料、钛酸盐(酯)材料等的陶瓷等,作为有机高分子,可以举出纤维素树脂、纤维素酯树脂等的纤维素系树脂、乙烯基系树脂、缩聚系树脂、加聚系树脂、加成缩合系树脂、开环聚合系树脂等。
(d)其它的添加剂
片状基材,以使使用气体阻隔性膜及气体阻隔性膜而制作了的电子元件用基板的性能进一步地提高的目的,优选添加以下(1)碳自由基捕捉剂、(2)一次抗氧化剂、(3)二次抗氧化剂、(4)酸捕捉剂、(5)紫外线吸收剂、(6)增塑剂、(7)消光剂、(8)光学各向异性控制剂、(9)交联剂等的添加剂。其中,在使用后述的熔融挤出法的情况下,优选添加(2)一次抗氧化剂、(3)二次抗氧化剂、(6)增塑剂的添加剂中的至少1种以上,特别优选添加(2)、(3)、(6)的全部。另一方面,在使用熔融浇铸法的情况下,优选添加(6)增塑剂、(9)交联剂中的至少1种以上,特别优选添加(6)及(9)的全部2种。
(1)碳自由基捕捉剂
片状基材,优选含有至少1种以上的碳自由基捕捉剂。所谓“碳自由基捕捉剂”,是指具有碳自由基能够迅速地进行加成反应的基团(例如双键、三键等的不饱和基团)、且在碳自由基加成后产生不引起聚合等的后续反应的稳定的产物的化合物。
作为上述碳自由基捕捉剂,在分子内迅速地与碳自由基反应的基团((甲基)丙烯酰基、芳基等的不饱和基团)及酚系、内酯系化合物等的具有阻止自由基聚合能力的化合物是有用的,特别优选由下述通式(1)或通式(2)所示的化合物。
[化学式2]
通式(1)
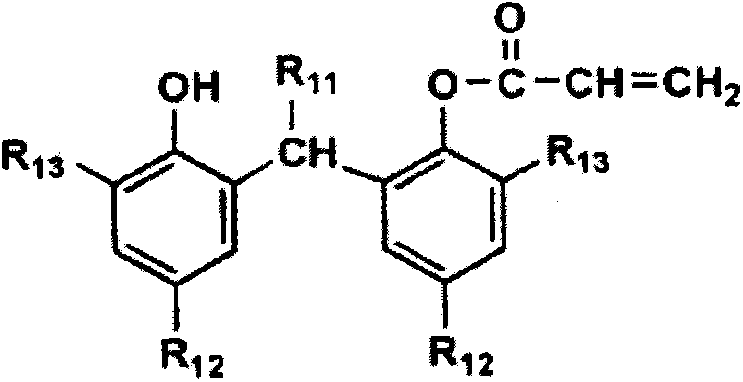
通式(2)
在通式(1)中,R11表示氢原子或碳原子数1~10的烷基,优选为氢原子或碳原子数1~4的烷基,特别优选为氢原子或甲基。
R12及R13分别独立地表示碳数1~8的烷基,可以具有直链,也可以具有支链结构或环结构。
R12及R13优选为含有季碳的“*-C(CH3)2-R”所示的结构(*表示在芳香环中的连接部位,R’表示碳数1~5的烷基。)。
R12更优选为叔丁基、叔戊基或叔辛基。R13更优选为叔丁基、叔戊基。作为由上述通式(1)所示的化合物,在市售的物质中,可以举出“SumilizerGM、SumilizerGS”(均为商品名、住友化学(株)制)等。
在以下例示由上述通式(1)所示的化合物的具体例(I-1~I-18),但本发明并不限定于这些化合物。
[化学式3-1]

[化学式3-2]

[化学式3-3]

在上述通式(2)中,R22~R25分别独立地表示氢原子或取代基,作为由R22~R25表示的取代基没有特别限制,例如可以举出烷基(例如甲基、乙基、丙基、异丙基、叔丁基、戊基、己基、辛基、十二烷基、三氟甲基等)、环烷基(例如环戊基、环己基等)、芳基(例如苯基、萘基等)、酰氨基(例如乙酰氨基、苯甲酰氨基等)、烷硫基(例如甲硫基、乙硫基等)、芳硫基(例如苯硫基、萘硫基等)、烯基(例如乙烯基、2-丙烯基、3-丁烯基、1-甲基-3-丙烯基、3-戊烯基、1-甲基-3-丁烯基、4-己烯基、环己烯基等)、卤素原子(例如氟原子、氯原子、溴原子、碘原子等)、炔基(例如炔丙基等)、杂环基(例如吡啶基、噻唑基、噁唑基、咪唑基等)、烷基磺酰基(例如甲基磺酰基、乙基磺酰基等)、芳基磺酰基(例如苯基磺酰基、萘基磺酰基等)、烷基亚磺酰基(例如甲基亚磺酰基等)、芳基亚磺酰基(例如苯基亚磺酰基等)、膦酰基、酰基(例如乙酰基、特戊酰基、苯甲酰基等)、氨基甲酰基(例如氨基羰基、甲基氨基羰基、二甲基氨基羰基、丁基氨基羰基、环己基氨基羰基、苯基氨基羰基、2-吡啶基氨基羰基等)、氨磺酰基(例如氨基磺酰基、甲基氨基磺酰基、二甲基氨基磺酰基、丁基氨基磺酰基、己基氨基磺酰基、环己基氨基磺酰基、辛基氨基磺酰基、十二烷基氨基磺酰基、苯基氨基磺酰基、萘基氨基磺酰基、2-吡啶基氨基磺酰基等)、磺酰胺基(例如甲磺酰胺基、苯磺酰胺基等)、氰基、烷氧基(例如甲氧基、乙氧基、丙氧基等)、芳氧基(例如苯氧基、萘氧基等)、杂环氧基、甲硅烷氧基、酰氧基(例如乙酰氧基、苯甲酰氧基等)、磺酸基、磺酸的盐、氨基羰氧基、氨基(例如氨基、乙基氨基、二甲基氨基、丁基氨基、环戊基氨基、2-乙基己基氨基、十二烷基氨基等)、苯胺基(例如苯基氨基、氯苯基氨基、甲苯胺基、甲氧苯胺基、萘基氨基、2-吡啶基氨基等)、酰亚胺基、脲基(例如甲基脲基、乙基脲基、戊基脲基、环己基脲基、辛基脲基、十二烷基脲基、苯基脲基、萘基脲基、2-吡啶基氨基脲基等)、烷氧基羰基氨基(例如甲氧基羰基氨基、苯氧基羰基氨基等)、烷氧基羰基(例如甲氧基羰基、乙氧基羰基、苯氧基羰基等)、芳氧基羰基(例如苯氧基羰基等)、杂环硫基、硫脲基、羧基、羧酸的盐、羟基、巯基、硝基等的各基团。这些取代基可以进一步被同样的取代基取代。
在上述通式(2)中,R26表示氢原子或取代基,由R26表示的取代基可以举出与上述由R22~R25表示的取代基同样的基团。
在上述通式(2)中,n表示1或2。
在上述通式(2)中,n为1时,R21表示取代基,n为2时,R21表示2价的连接基团。R21表示取代基时,作为取代基,可以举出与上述由R22~R25表示的取代基同样的基团。
R21表示2价的连接基团时,作为2价的连接基团,例如可以举出可以具有取代基的亚烷基、可以具有取代基的亚芳基、氧原子、氮原子、硫原子、或者这些连接基团的组合。
在上述通式(2)中,n优选1。
接着,示出本发明中的由上述通式(2)所示的化合物的具体例,但本发明并不受以下的具体例限定。
[化学式4-1]

[化学式4-2]
[化学式4-3]

上述碳自由基捕捉剂,可以单独使用1种或组合使用2种以上,其配合量可在不损害本发明的目的的范围内适宜选择,但相对于表面改性纤维素纳米纤维的总质量(100质量份),通常优选添加0.001~10.0质量份,进一步优选为0.01~5.0质量份,特别优选为0.1~1.0质量份。
(2)一次抗氧化剂
片状基材优选含有至少1种具有相对于过氧自由基的氢自由基供给能力的一次抗氧化剂。
所谓“具有相对于过氧自由基的氢自由基供给能力的一次抗氧化剂”,为在分子内至少具有1个以上通过过氧自由基而迅速地脱去的氢原子的化合物,优选为被羟基或者伯氨基或仲氨基取代了的芳香族化合物或具有空间位阻性基团的杂环化合物,更优选为在邻位具有烷基的酚系化合物或者受阻胺系化合物。
(酚系化合物)
本发明中所优选使用的酚化合物,例如包含美国专利第4,839,405号说明书的第12~14栏中所记载的化合物等的、2,6-二烷基酚衍生物化合物。这样的化合物中包含由下述通式(3)所示的化合物。
[化学式5]
通式(3)

式中,R31~R36表示氢原子或取代基。作为取代基,可以举出卤素原子(例如氟原子、氯原子等)、烷基(例如甲基、乙基、异丙基、羟基乙基、甲氧基甲基、三氟甲基、叔丁基等)、环烷基(例如环戊基、环己基等)、芳烷基(例如苄基、2-苯乙基等)、芳基(例如苯基、萘基、对甲苯基、对氯苯基等)、烷氧基(例如甲氧基、乙氧基、异丙氧基、丁氧基等)、芳氧基(例如苯氧基等)、氰基、酰氨基(例如乙酰氨基、丙酰氨基等)、烷硫基(例如甲硫基、乙硫基、丁硫基等)、芳硫基(例如苯硫基等)、磺酰氨基(例如甲烷磺酰氨基、苯磺酰氨基等)、脲基(例如3-甲基脲基、3,3-二甲基脲基、1,3-二甲基脲基等)、氨基磺酰氨基(二甲基氨基磺酰氨基等)、氨基甲酰基(例如甲基氨基甲酰基、乙基氨基甲酰基、二甲基氨基甲酰基等)、氨基磺酰基(例如乙基氨基磺酰基、二甲基氨基磺酰基等)、烷氧基羰基(例如甲氧基羰基、乙氧基羰基等)、芳氧基羰基(例如苯氧基羰基等)、磺酰基(例如甲基磺酰基、丁基磺酰基、苯基磺酰基等)、酰基(例如乙酰基、丙酰基、丁酰基等)、氨基(甲基氨基、乙基氨基、二甲基氨基等)、氰基、羟基、硝基、亚硝基、氧化胺基(例如氧化吡啶基)、酰亚胺基(例如邻苯二甲酰亚胺基等)、二硫基(例如苯二硫基、苯并噻唑-2-二硫基等)、羧基、磺基、杂环基(例如吡咯基、吡咯烷基、吡唑基、咪唑基、吡啶基、苯并咪唑基、苯并噻唑基、苯并噁唑基等)等。这些取代基可以进一步被取代。
另外,优选R21为氢原子,R32、R36为叔丁基的化合物。作为酚系化合物的具体例,包含3-(3,5-二叔丁基-4-羟基苯基)丙酸正十八烷基酯、3-(3,5-二叔丁基-4-羟基苯基)乙酸正十八烷基酯、3,5-二叔丁基-4-羟基苯甲酸正十八烷基酯、3,5-二叔丁基-4-羟基苯基苯甲酸正己酯、3,5-二叔丁基-4-羟基苯基苯甲酸正十二烷基酯、3-(3,5-二叔丁基-4-羟基苯基)丙酸新十二烷基酯、β-(3,5-二叔丁基-4-羟基苯基)丙酸十二烷基酯、α-(4-羟基-3,5-二叔丁基苯基)异丁酸乙酯、α-(4-羟基-3,5-二叔丁基苯基)异丁酸十八烷基酯、α-(4-羟基-3,5-二叔丁基-4-羟基苯基)丙酸十八烷基酯、3,5-二叔丁基-4-羟基-苯甲酸2-(正辛基硫代)乙酯、3,5-二叔丁基-4-羟基苯乙酸2-(正辛基硫代)乙酯、3,5-二叔丁基-4-羟基苯乙酸2-(正十八烷基硫代)乙酯、3,5-二叔丁基-4-羟基苯甲酸2-(正十八烷基硫代)乙酯、3,5-二叔丁基-4-羟基苯甲酸2-(2-羟基乙基硫代)乙酯、二乙二醇双(3,5-二叔丁基-4-羟基苯基)丙酸酯、3-(3,5-二叔丁基-4-羟基苯基)丙酸2-(正十八烷基硫代)乙酯、硬脂酰胺-N,N-双[亚乙基-3-(3,5-二叔丁基-4-羟基苯基)丙酸酯]、正丁基亚氨基-N,N-双[亚乙基-3-(3,5-二叔丁基-4-羟基苯基)丙酸酯]、3,5-二叔丁基-4-羟基苯甲酸2-(2-硬脂酰氧基乙基硫代)乙酯、7-(3-甲基-5-叔丁基-4-羟基苯基)庚酸2-(2-硬脂酰氧基乙基硫代)乙酯、1,2-丙二醇双[3-(3,5-二叔丁基-4-羟基苯基)丙酸酯]、乙二醇双[3-(3,5-二叔丁基-4-羟基苯基)丙酸酯]、新戊二醇双[3-(3,5-二叔丁基-4-羟基苯基)丙酸酯]、乙二醇双(3,5-二叔丁基-4-羟基苯基乙酸酯)、甘油-1-正硬脂酸酯-2,3-双(3,5-二叔丁基-4-羟基苯基乙酸酯)、季戊四醇四[3-(3’,5’-二叔丁基-4’-羟基苯基)丙酸酯]、3,9-双{2-[3-(3-叔丁基-4-羟基-5-甲基苯基)丙酰氧基]-1,1-二甲基乙基}-2,4,8,10-四氧杂螺[5.5]十一烷、1,1,1-三羟甲基乙烷三[3-(3,5-二叔丁基-4-羟基苯基)丙酸酯]、山梨糖醇六[3-(3,5-二叔丁基-4-羟基苯基)丙酸酯]、2-羟基乙基7-(3-甲基-5-叔丁基-4-羟基苯基)丙酸酯、2-硬脂酰氧基乙基7-(3-甲基-5-叔丁基-4-羟基苯基)庚酸酯、1,6-正己二醇双[(3’,5’-二叔丁基-4-羟基苯基)丙酸酯]、季戊四醇四(3,5-二叔丁基-4-羟基氢化肉桂酸酯)。上述类型的酚化合物,例如可由BASFジヤパン公司以“Irganox1076”及“Irganox1010”这样的商品名而被市售。
上述酚化合物,可以单独使用1种或组合使用2种以上,其配合量可以在不损害本发明的目的的范围内适宜选择,但相对于表面改性纤维素纳米纤维的总质量(100质量份),通常优选添加0.001~10.0质量份,进一步优选为0.05~5.0质量份,特别优选为0.1~2.0质量份。
(受阻胺系化合物)
作为受阻胺系化合物,优选由下述通式(4)所示的化合物。
[化学式6]
通式(4)

式中,R41~R47表示取代基。作为取代基与上述通式(3)的R31~R36表示的取代基意义相同。R44优选氢原子、甲基,R47优选氢原子,R42、R43、R45、R46优选甲基。作为受阻胺系化合物的具体例,可以举出双(2,2,6,6-四甲基-4-哌啶基)癸二酸酯、双(2,2,6,6-四甲基-4-哌啶基)琥珀酸酯、双(1,2,2,6,6-五甲基-4-哌啶基)癸二酸酯、双(N-辛氧基-2,2,6,6-四甲基-4-哌啶基)癸二酸酯、双(N-苄氧基-2,2,6,6-四甲基-4-哌啶基)癸二酸酯、双(N-环己氧基-2,2,6,6-四甲基-4-哌啶基)癸二酸酯、双(1,2,2,6,6-五甲基-4-哌啶基)-2-(3,5-二叔丁基-4-羟基苄基)-2-丁基丙二酸酯、双(1-丙烯酰基-2,2,6,6-四甲基-4-哌啶基)-2,2-双(3,5-二叔丁基-4-羟基苄基)-2-丁基丙二酸酯、双(1,2,2,6,6-五甲基-4-哌啶基)癸二酸酯、2,2,6,6-四甲基-4-哌啶基甲基丙烯酸酯、4-[3-(3,5-二叔丁基-4-羟基苯基)丙酰氧基]-1-[2-(3-(3,5-二叔丁基-4-羟基苯基)丙酰氧基)乙基]-2,2,6,6-四甲基哌啶、2-甲基-2-(2,2,6,6-四甲基-4-哌啶基)-氨基-N-(2,2,6,6-四甲基-4-哌啶基)丙酰胺、四(2,2,6,6-四甲基-4-哌啶基)-1,2,3,4-丁烷四羧酸酯、四(1,2,2,6,6-五甲基-4-哌啶基)-1,2,3,4-丁烷四羧酸酯等。
另外,也可以为高分子类型的化合物,作为具体例,可以举出N,N’,N”,N”’-四-[4,6-双[丁基-(N-甲基-2,2,6,6-四甲基哌啶-4-基)氨基]三嗪-2-基]-4,7-二氮杂癸烷-1,10-二胺、二丁胺和1,3,5-三嗪-N,N’-双(2,2,6,6-四甲基-4-哌啶基)-1,6-六亚甲基二胺和N-(2,2,6,6-四甲基-4-哌啶基)丁胺的缩聚物、二丁胺和1,3,5-三嗪和N,N’-双(2,2,6,6-四甲基-4-哌啶基)丁胺的缩聚物、聚[{(1,1,3,3-四甲基丁基)氨基-1,3,5-三嗪-2,4-二基}{(2,2,6,6-四甲基-4-哌啶基)亚氨基}六亚甲基{(2,2,6,6-四甲基-4-哌啶基)亚氨基}]、1,6-己二胺-N,N’-双(2,2,6,6-四甲基-4-哌啶基)和吗啉-2,4,6-三氯-1,3,5-三嗪的缩聚物、聚[(6-吗啉基-均三嗪-2,4-二基)[(2,2,6,6-四甲基-4-哌啶基)亚氨基]-六亚甲基[(2,2,6,6-四甲基-4-哌啶基)亚氨基]]等的多个哌啶环经由三嗪骨架而键合了的高分子量HALS;琥珀酸二甲酯和4-羟基-2,2,6,6-四甲基-1-哌啶乙醇的聚合物、1,2,3,4-丁烷四羧酸和1,2,2,6,6-五甲基-4-哌啶醇和3,9-双(2-羟基-1,1-二甲基乙基)-2,4,8,10-四氧杂螺[5,5]十-烷的混合酯化物等的哌啶环经由酯键而键合了的化合物等,但并不限定于这些。需要说明的是,高分子类型的受阻胺系化合物的数均分子量(Mn)为500~10,000。
其中,优选二丁胺和1,3,5-三嗪和N,N’-双(2,2,6,6-四甲基-4-哌啶基)丁胺的缩聚物、聚[{(1,1,3,3-四甲基丁基)氨基-1,3,5-三嗪-2,4-二基}{(2,2,6,6-四甲基-4-哌啶基)亚氨基}六亚甲基{(2,2,6,6-四甲基-4-哌啶基)亚氨基}]、琥珀酸二甲酯和4-羟基-2,2,6,6-四甲基-1-哌啶乙醇的聚合物等、数均分子量(Mn)为2,000~5,000的物质。
上述类型的受阻胺化合物,例如可由BASFジヤパン公司以“Tinuvin144”及“Tinuvin770”、由株式会社ADEKA以“ADK STAB LA-52”这样的商品名而被市售。
上述受阻胺化合物可以单独使用1种或组合使用2种以上,其配合量可在不损害本发明的目的的范围内适宜选择,但相对于表面改性纤维素纳米纤维的总质量(100质量份),通常优选添加0.001~10.0质量份,进-步优选为0.05~5.0质量份,特别优选为0.1~2.0质量份。
(3)二次抗氧化剂
片状基材,优选含有至少1种以上具有对于过氧化物的还原作用的二次抗氧化剂。
所谓“具有对于过氧化物的还原作用的二次抗氧化剂”,是指迅速地将过氧化物还原而转化为羟基的还原剂。
作为具有对于过氧化物的还原能力的二次抗氧化剂,优选磷系化合物或硫系化合物。
(磷系化合物)
作为磷系化合物,优选选自由亚磷酸酯(phosphite)、亚膦酸酯(phosphonite)、次亚膦酸酯(phosphinite)、或叔磷烷(phosphane)组成的组中的磷系化合物,具体而言,优选分子内具有由下述通式(5-1)、(5-2)、(5-3)、(5-4)、(C-5)所示的部分结构的化合物。
[化学式7]
通式(5-1)

式中,Ph1及Ph1’表示取代基。作为取代基,与上述通式(3)的R31~R36表示的取代基意义相同。更优选Ph1及Ph1’表示亚苯基,该亚苯基的氢原子也可以被苯基、碳数1~8的烷基、碳数5~8的环烷基、碳数6~12的烷基环烷基或碳数7~12的芳烷基取代。Ph1及Ph1’互相可以相同或不同。X表示单键、硫原子或-CHR-基。R表示氢原子、碳数1~8的烷基或碳数5~8的环烷基。另外,这些可以通过与上述通式(3)的R31~R36表示的取代基意义相同的取代基而被取代。
[化学式8]
通式(5-2)

式中,Ph2及Ph2’表示取代基。作为取代基,与上述通式(3)的R31~R36表示的取代基意义相同。更优选Ph2及Ph2’表示苯基或联苯基,该苯基或联苯基的氢原子可以被碳数1~8的烷基、碳数5~8的环烷基、碳数6~12的烷基环烷基或碳数7~12的芳烷基取代。Ph2及Ph2’互相可以相同或不同。另外,这些可以通过与上述通式(3)的R31~R36表示的取代基意义相同的取代基而被取代。
[化学式9]
通式(5-3)

式中,Ph3表示取代基。作为取代基,与上述通式(3)的R31~R36表示的取代基意义相同。更优选Ph3表示苯基或联苯基,该苯基或联苯基的氢原子可以被碳数1~8的烷基、碳数5~8的环烷基、碳数6~12的烷基环烷基或碳数7~12的芳烷基取代。另外,这些可以通过与上述通式(3)的R31~R36表示的取代基意义相同的取代基而被取代。
[化学式10]
通式(5-4)
式中,Ph4表示取代基。作为取代基,与上述通式(3)的R31~R36表示的取代基意义相同。更优选Ph4表示碳数1~20的烷基或苯基,该烷基或苯基可以通过与上述通式(3)的R31~R36表示的取代基意义相同的取代基而被取代。
[化学式11]
通式(5-5)

式中,Ph5、Ph5’及Ph5”表示取代基。作为取代基,与上述通式(3)的R31~R36表示的取代基含义相同。更优选Ph5、Ph5’及Ph5”表示碳数1~20的烷基或苯基,该烷基或苯基可以通过与上述通式(3)的R31~R36表示的取代基含义相同的取代基而被取代。
作为磷系化合物的具体例,可以举出亚磷酸三苯酯、亚磷酸二苯基异癸酯、亚磷酸苯基二异癸酯、亚磷酸三(壬基苯基)酯、亚磷酸三(二壬基苯基)酯、亚磷酸三(2,4-二叔丁基苯基)酯、10-(3,5-二叔丁基-4-羟基苄基)-9,10-二氢-9-氧杂-10-磷杂菲-10-氧化物、6-[3-(3-叔丁基-4-羟基-5-甲基苯基)丙氧基]-2,4,8,10-四叔丁基二苯并[d,f][1,3,2]二噁磷环庚烷、亚磷酸三癸酯等单亚磷酸酯系化合物;4,4’-亚丁基-双(3-甲基-6-叔丁基苯基-二-十三烷基亚磷酸酯)、4,4’-亚异丙基-双(苯基-二-烷基(C12~C15)亚磷酸酯)等的二亚磷酸酯系化合物;亚膦酸三苯酯、四(2,4-二-叔丁基苯基)[1,1-联苯]-4,4’-二基双亚膦酸酯、四(2,4-二-叔丁基-5-甲基苯基)[1,1-联苯]-4,4’-二基双亚膦酸酯等的亚膦酸酯系化合物;次亚膦酸三苯酯、2,6-二甲基苯基二苯基次亚膦酸酯等的次亚膦酸酯系化合物;三苯基膦、三(2,6-二甲氧基苯基)膦等的膦系化合物等。
上述类型的磷系化合物,例如可由住友化学株式会社以“SumilizerGP”、由株式会社ADEKA以“ADK STAB PEP-24G”、“ADKSTAB PEP-36”及“ADK STAB3010”、由BASFジヤパン公司以“I RGAFOSP-EP Q”、由堺化学工业株式会社以“GSY-P101”这样的商品名被市售。
上述的磷系化合物,可以单独使用1种或组合使用2种以上,其配合量可在不损害本发明的目的的范围内适宜选择,但相对于表面改性纤维素纳米纤维的总质量(100质量份),通常优选添加0.001~10.0质量份,进一步优选为0.05~5.0质量份,特别优选为0.05~2.0质量份。
(硫系化合物)
作为硫系化合物,优选由下述通式(6)所示的硫系化合物。
[化学式12]
通式(6)
R61-S-R62
式中,R61及R62表示取代基。作为取代基,与上述通式(3)的R31~R36表示的取代基意义相同。
作为硫系化合物的具体例,可以举出3,3-硫代二丙酸二月桂酯、3,3’-硫代二丙酸二肉豆蔻酯、3,3-硫代二丙酸二(十八烷基)酯、3,3-硫代二丙酸月桂基十八烷基酯、季戊四醇-四(硫代丙酸β-月桂酯)、3,9-双(2-十二烷基硫代乙基)-2,4,8,10-四氧杂螺[5,5]十一烷等。
上述类型的硫系化合物,例如可由住友化学株式会社以“SumilizerTPL-R”及“Sumilizer TP-D”这样的商品名而被市售。
上述硫系化合物,可以单独使用1种或组合使用2种以上,其配合量可在不损害本发明的目的的范围内适宜选择,但相对于表面改性纤维素纳米纤维的总质量(100质量份),通常优选添加0.001~10.0质量份,进一步优选为0.05~5.0质量份,特别优选为0.05~2.0质量份。
(4)酸捕捉剂
由于在进行熔融制膜这样的高温环境下分解因酸而被促进,因此片用基材优选含有酸捕捉剂作为稳定剂。
作为酸捕捉剂,只要为与酸反应而使酸钝化的化合物就可以没有限制地使用,其中,优选美国专利第4,137,201号说明书中所记载那样的具有环氧基的化合物。作为这样的酸捕捉剂的环氧化合物,在该技术领域中是已知的,包含各种聚乙二醇的二缩水甘油醚,特别是由每聚乙二醇1摩尔约8~40摩尔的环氧乙烷等的缩合而衍生的聚乙二醇、甘油的二缩水甘油醚等、金属环氧化合物(例如在氯乙烯聚合物组合物中,及与氯乙烯聚合物组合物一同一直以来被利用的物质)、环氧化醚缩合产物、双酚A的二缩水甘油醚(即4,4’-二羟基二苯基二甲基甲烷)、可由环氧化不饱和脂肪酸酯(特别是2~22个碳原子的脂肪酸的4~2个左右的碳原子的烷基的酯(例如环氧硬脂酸丁酯)等)、及各种环氧化长链脂肪酸三甘油酯等(例如环氧化大豆油、环氧化亚麻仁油等)的组合物所代表、例示的环氧化植物油及其它的不饱和天然油(这些有时称为环氧化天然甘油酯或不饱和脂肪酸,这些脂肪酸通常含有12~22个碳原子)。另外,作为市售的含有环氧基的环氧化物树脂化合物,也可优选使用EPON815C、及下述通式(7)的其它的环氧化醚低聚物缩合产物。
[化学式13]
通式(7)

式中,n为0~12的整数。作为可以使用的其它的酸捕捉剂,包含特开平5-194788号公报的段落87~105中所记载的物质。
酸捕捉剂可以单独使用1种或组合使用2种以上,其配合量可在不损害本发明的目的的范围内适宜选择,但相对于表面改性纤维素纳米纤维的总质量(100质量份),通常优选添加0.001~10.0质量份,进一步优选为0.05~5.0质量份,特别优选为0.05~2.0质量份。
需要说明的是,酸捕捉剂也有时相对于树脂被称为酸清除剂、酸捕获剂、缚酸剂等,但在本发明中可以不存在由这些称呼所引起的差异地使用。
(5)紫外线吸收剂
片状基材,可含有紫外线吸收剂。紫外线吸收剂以通过吸收400nm以下的紫外线而使耐久性提高为目的,特别优选波长370nm处的透过率为10%以下,更优选为5%以下,进一步优选为2%以下。进而,在液晶显示装置用途中,从液晶显示性的观点考虑,优选波长400nm以上的可见光的吸收少。
上述紫外线吸收剂没有特别限定,可以举出例如羟基二苯甲酮系化合物、苯并三唑系化合物、水杨酸酯系化合物、二苯甲酮系化合物、氰基丙烯酸酯系化合物、三嗪系化合物、镍络合盐系化合物、无机粉体等。优选苯并三唑系化合物、二苯甲酮系化合物、三嗪系化合物,特别优选苯并三唑系化合物、二苯甲酮系化合物。
作为苯并三唑系化合物的具体例,可以举出2-(2’-羟基-5’-甲基苯基)苯并三唑、2-(2’-羟基-3’,5’-二-叔丁基苯基)苯并三唑、2-(2’-羟基-3’-叔丁基-5’-甲基苯基)苯并三唑、2-(2’-羟基-3’,5’-二-叔丁基苯基)-5-氯苯并三唑、2-(2’-羟基-3’-(3”,4”,5”,6”-四氢邻苯二甲酰亚胺甲基)-5’-甲基苯基)苯并三唑、2,2-亚甲基双(4-(1,1,3,3-四甲基丁基)-6-(2H-苯并三唑-2-基)苯酚)、2-(2’-羟基-3,-叔丁基-5’-甲基苯基)-5-氯苯并三唑、2-(2’-羟基-3’-叔丁基-5’-(2-辛氧基羰基乙基)-苯基)-5-氯苯并三唑、2-(2’-羟基-3’-(1-甲基-1-苯基乙基)-5’(1,1,3,3-四甲基丁基)-苯基)苯并三唑、2-(2H-苯并三唑-2-基)-6-(直链及侧链十二烷基)-4-甲基苯酚、辛基-3-[3-叔丁基-4-羟基-5-(氯-2H-苯并三唑-2-基)苯基]丙酸酯和2-乙基己基-3-[3-叔丁基-4-羟基-5-(5-氯-2H-苯并三唑-2-基)苯基]丙酸酯的混合物等,但不限定于这些化合物。
另外,作为市售品,可以举出チヌビン(TINUVIN)171、チヌビン(TINUVIN)900、チヌビン(TINUVIN)928、チヌビン(TINUVIN)360(均为BASFジヤパン公司制)、LA31(株式会社ADEKA公司制)、RUVA-100(大塜化学制)。
作为二苯甲酮系化合物的具体例,可以举出2,4-二羟基二苯甲酮、2,2’-二羟基-4-甲氧基二苯甲酮、2-羟基-4-甲氧基-5-磺基二苯甲酮、双(2-甲氧基-4-羟基-5-苯甲酰基苯基甲烷)等,但并不限定于这些化合物。
需要说明的是,通过使苯并三唑结构、三嗪结构导入到增塑剂、抗氧化剂、酸清除剂等的其它的添加剂的分子结构的一部分,可以赋予作为紫外线吸收剂的功能。
上述紫外线吸收剂可以单独使用1种或组合使用2种以上。
紫外线吸收剂的配合量可在不损害本发明的目的的范围内适宜选择,但相对于表面改性纤维素纳米纤维的总质量(100质量份),通常优选添加0.1~5质量份,进一步优选为0.2~3质量份,特别优选为0.5~2质量份。
(6)增塑剂
片状基材可含有增塑剂。在本发明中,所谓增塑剂,是指分子量为500~10,000的可以改善脆弱性或赋予柔软性的化合物。在本发明中,增塑剂可以改善表面改性纤维素纳米纤维的亲水性、改善气体阻隔性膜的透湿度、具有作为防透湿剂的功能。
另外,在本发明的优选的实施方式中,为了使熔融挤出时的膜构成材料的熔融温度、熔融粘度降低,可添加增塑剂。在此,所谓熔融温度,是指材料被加热、呈现出了流动性的状态的温度。为了使高分子材料熔融流动,需要至少加热到比玻璃化转变温度高的温度。在玻璃化转变温度以上时,弹性模量、粘度因热量的吸收而降低,呈现流动性。但是,在高温下时,在熔融的同时因热分解而产生表面改性纤维素纳米纤维的分子量的降低,有时对得到的膜的力学特性等造成不良影响,需要在低的温度下使树脂熔融。因此,为了使膜构成材料的熔融温度降低,可添加具有比表面改性纤维素纳米纤维的玻璃化转变温度低的熔点或玻璃化转变温度的增塑剂。
作为增塑剂没有特别限定,优选由多元醇和一元羧酸构成的酯系增塑剂、由多元羧酸和一元醇构成的酯系增塑剂。
(多元醇酯系增塑剂)
作为酯系增塑剂的原料即多元醇的例子,可以举出例如如以下的物质,但本发明并不限定于这些。可以举出核糖醇、阿拉伯糖醇、乙二醇、甘油、二甘油、二乙二醇、三乙二醇、四乙二醇、1,2-丙二醇、1,3-丙二醇、二丙二醇、三丙二醇、1,2-丁二醇、1,3-丁二醇、1,4-丁二醇、二丁二醇、1,2,4-丁三醇、1,5-戊二醇、1,6-己二醇、己三醇、半乳糖醇、甘露醇、3-甲基戊烷-1,3,5-三醇、频哪醇、山梨糖醇、三羟甲基丙烷、二(三羟甲基)丙烷、三羟甲基乙烷、季戊四醇、二季戊四醇、木糖醇等。特别优选乙二醇、甘油、三羟甲基丙烷。
作为多元醇酯系之一即乙二醇酯系的增塑剂而言,具体可以举出乙二醇二乙酸酯、乙二醇二丁酸酯等的乙二醇烷基酯系的增塑剂;乙二醇二环丙基羧酸酯、乙二醇二环己基羧酸酯等的乙二醇环烷基酯系的增塑剂;乙二醇二苯甲酸酯、乙二醇二4-甲基苯甲酸酯等的乙二醇芳基酯系的增塑剂。这些烷基化物基、环烷基化物基、芳基化物基可以相同或不同,也可以进一步被取代。另外也可以为烷基化物基、环烷基化物基、芳基化物的混合,另外这些取代基彼此也可以以共价键键合。进而乙二醇部也可以被取代,乙二醇酯的部分结构可以在聚合物的一部分、或者规则地形成侧链,另外可以导入到抗氧化剂、酸清除剂、紫外线吸收剂等的添加剂的分子结构的一部分中。
作为多元醇酯系之一即甘油酯系的增塑剂,具体而言可以举出三乙酸甘油酯、三丁酸甘油酯、甘油二乙酸酯辛酸酯、甘油油酸酯丙酸酯等的甘油烷基酯;甘油三环丙基羧酸酯、甘油三环己基羧酸酯等的甘油环烷基酯;甘油三苯甲酸酯、甘油4-甲基苯甲酸酯等的甘油芳基酯;二甘油四乙酸酯、二甘油四丙酸酯、二甘油乙酸酯三辛酸酯、二甘油四月桂酯等的二甘油烷基酯;二甘油四环丁基羧酸酯、二甘油四环戊基羧酸酯等的二甘油环烷基酯;二甘油四苯甲酸酯、二甘油3-甲基苯甲酸酯等的二甘油芳基酯等。这些烷基化物基、环烷基羧酸酯基、芳基化物基可以相同或不同,也可以被取代。另外可以为烷基化物基、环烷基羧酸酯基、芳基化物基的混合,另外这些取代基彼此也可以以共价键键合。进而甘油、二甘油部也可以被取代,甘油酯、二甘油酯的部分结构可以在聚合物的一部分、或者规则地形成侧链,另外可以导入到抗氧化剂、酸清除剂、紫外线吸收剂等的添加剂的分子结构的一部分中。
作为其它的多元醇酯系的增塑剂,具体而言可以举出特开2003-12823号公报的段落30~33记载的多元醇酯系增塑剂、特开2006-188663号公报的段落64~74记载的多元醇酯系增塑剂。
这些烷基化物基、环烷基羧酸酯基、芳基化物基,可以相同或不同,也可以被取代。另外,可以为烷基化物基、环烷基羧酸酯基、芳基化物基的混合,另外,这些取代基彼此也可以以共价键而键合。进而,多元醇部也可以被取代,多元醇的部分结构可以在聚合物的一部分、或者规则地形成侧链,另外可以导入到抗氧化剂、酸清除剂、紫外线吸收剂等的添加剂的分子结构的一部分。
在上述由多元醇和一元的羧酸构成的酯系增塑剂中,优选烷基多元醇芳基酯,具体而言可以举出上述的乙二醇二苯甲酸酯、甘油三苯甲酸酯、二甘油四苯甲酸酯、季戊四醇四苯甲酸酯、三羟甲基丙烷三苯甲酸酯、特开2003-12823号公报的段落31记载例示化合物16、特开2006-188663号公报的段落71记载例示化合物48。
(多元羧酸酯系增塑剂)
作为多元羧酸酯系之一即二羧酸酯系的增塑剂,具体可以举出丙二酸二(十二烷基)酯、己二酸二辛酯、癸二酸二丁酯等的烷基二羧酸烷基酯系的增塑剂;琥珀酸二环戊基酯、己二酸二环己基酯等的烷基二羧酸环烷基酯系的增塑剂;琥珀酸二苯酯、二4-甲基苯基戊二酸酯等的烷基二羧酸芳基酯系的增塑剂;二己基-1,4-环己烷二羧酸酯、二癸基双环[2.2.1]庚烷-2,3-二羧酸酯等的环烷基二羧酸烷基酯系的增塑剂;二环己基-1,2-环丁烷二羧酸酯、二环丙基-1,2-环己烷二羧酸酯等的环烷基二羧酸环烷基酯系的增塑剂;二苯基-1,1-环丙基二羧酸酯、二2-萘基-1,4-环己烷二羧酸酯等的环烷基二羧酸芳基酯系的增塑剂;邻苯二甲酸二乙酯、邻苯二甲酸二甲酯、邻苯二甲酸二辛酯、邻苯二甲酸二丁酯、邻苯二甲酸二-2-乙基己酯等的芳基二羧酸烷基酯系的增塑剂;邻苯二甲酸二环丙酯、邻苯二甲酸二环己酯等的芳基二羧酸环烷基酯系的增塑剂;邻苯二甲酸二苯酯、邻苯二甲酸二4-甲基苯酯等的芳基二羧酸芳基酯系的增塑剂。这些烷氧基、环烷氧基可以相同或不同,还可以为一取代,这些取代基也可以进一步被取代。烷基、环烷基可以为混合,另外,这些取代基彼此也可以以共价键键合。进而邻苯二甲酸的芳香环也可以被取代,可以为二聚物、三聚物、四聚物等的多聚物。
另外邻苯二甲酸酯的部分结构,可以在聚合物的一部分、或规则地在聚合物形成侧基,另外可以导入到抗氧化剂、酸清除剂、紫外线吸收剂等的添加剂的分子结构的一部分。
另外,来自一元的醇的烷基、环烷基、芳基的氢原子,可以被烷氧基羰基取代。作为这样的增塑剂,例如可以举出乙基邻苯二甲酰基乙基乙醇酸酯。
作为其它的多元羧酸酯系的增塑剂,具体而言,可以举出三(十二烷基)三苯氨甲酸酯(トリドデシルトリカルバレ一ト)、三丁基-内消旋丁烷-1,2,3,4-四羧酸酯等的烷基多元羧酸烷基酯系的增塑剂;三环己基三苯氨甲酸酯(トリシクロヘキシルトリカルバレ一ト)、三环丙基-2-羟基-1,2,3-丙烷三羧酸酯等的烷基多元羧酸环烷基酯系的增塑剂;三苯基2-羟基-1,2,3-丙烷三羧酸酯、四3-甲基苯基四氢呋喃-2,3,4,5-四羧酸酯等的烷基多元羧酸芳基酯系的增塑剂;四己基-1,2,3,4-环丁烷四羧酸酯、四丁基-1,2,3,4-环戊烷四羧酸酯等的环烷基多元羧酸烷基酯系的增塑剂;四环丙基-1,2,3,4-环丁烷四羧酸酯、三环己基-1,3,5-环己基三羧酸酯等的环烷基多元羧酸环烷基酯系的增塑剂;三苯基-1,3,5-环己基三羧酸酯、六4-甲基苯基-1,2,3,4,5,6-环己基六羧酸酯等的环烷基多元羧酸芳基酯系的增塑剂;三(十二烷基)苯-1,2,4-三羧酸酯、四辛基苯-1,2,4,5-四羧酸酯等的芳基多元羧酸烷基酯系的增塑剂;三环戊基苯-1,3,5-三羧酸酯、四环己基苯-1,2,3,5-四羧酸酯等的芳基多元羧酸环烷基酯系的增塑剂;三苯基苯-1,3,5-四羧酸酯、六4-甲基苯基苯-1,2,3,4,5,6-六羧酸酯等的芳基多元羧酸芳基酯系的增塑剂。这些烷氧基、环烷氧基可以相同或不同,还可以为1取代,这些取代基也可以进一步进行取代。烷基、环烷基可以为混合,另外这些取代基彼此也可以以共价键键合。进而邻苯二甲酸的芳香环也可以被取代,还可以为二聚物、三聚物、四聚物等的多聚物。另外邻苯二甲酸酯的部分结构可以在聚合物的一部分、或者规则地在聚合物上形成侧链,也可以导入到抗氧化剂、酸清除剂、紫外线吸收剂等的添加剂的分子结构的一部分中。
在上述由多元羧酸和一元的醇构成的酯系增塑剂中,优选烷基二羧酸烷基酯,具体而言,可以举出上述的己二酸二辛酯。
(其它的增塑剂)
作为本发明中所使用的其它的增塑剂,可以举出磷酸酯系增塑剂、碳水化物酯系增塑剂、聚合物增塑剂等。
(磷酸酯系增塑剂)
作为磷酸酯系增塑剂,具体而言可以举出磷酸三乙酰酯、磷酸三丁酯等的磷酸烷基酯;磷酸三环戊酯、磷酸环己酯等的磷酸环烷基酯;磷酸三苯酯、磷酸三甲苯酯、磷酸甲苯基苯基酯、磷酸辛基二苯基酯、磷酸二苯基联苯基酯、磷酸三辛酯、磷酸三丁酯、磷酸三萘酯、磷酸三(二甲苯基)酯、磷酸三邻联苯基酯等的磷酸芳基酯。这些取代基可以相同或不同,也可以被取代。另外,可以为烷基、环烷基、芳基的混合,另外取代基彼此也可以以共价键键合。
另外,可以举出亚乙基双(二甲基磷酸酯)、亚丁基双(二乙基磷酸酯)等的亚烷基双(二烷基磷酸酯)、亚乙基双(二苯基磷酸酯)、亚丙基双(二萘基磷酸酯)等的亚烷基双(二芳基磷酸酯)、亚苯基双(二丁基磷酸酯)、联亚苯基双(二辛基磷酸酯)等的亚芳基双(二烷基磷酸酯)、亚苯基双(二苯基磷酸酯)、亚萘基双(二甲苯基磷酸酯)等的亚芳基双(二芳基磷酸酯)等的磷酸酯。这些取代基可以相同或不同,也可以进一步进行取代。另外,可以为烷基、环烷基、芳基的混合,另外取代基彼此也可以以共价键键合。
进而,磷酸酯的部分结构,可以在聚合物的一部分、或者规则地形成侧链,另外,可以导入到抗氧化剂、酸清除剂、紫外线吸收剂等添加剂的分子结构的一部分。在上述化合物中,优选磷酸芳基酯、亚芳基双(二芳基磷酸酯),具体而言,优选磷酸三苯酯、亚苯基双(二苯基磷酸酯)。
(碳水化物酯系增塑剂)
所谓碳水化物,是指糖类以吡喃糖或呋喃糖(六元环或五元环)的方式存在的单糖类、双糖类或三糖类。作为碳水化物的非限定的例子,可以举出葡萄糖、蔗糖、乳糖、纤维二糖、甘露糖、木糖、核糖、半乳糖、阿拉伯糖、果糖、山梨糖、纤维三糖及棉子糖等。所谓碳水化物酯是指碳水化物的羟基与羧酸进行脱水缩合而形成了酯化合物的物质,详细而言,是指碳水化物的脂肪族羧酸酯、或者芳香族羧酸酯。作为脂肪族羧酸,可以举出例如乙酸、丙酸等,作为芳香族羧酸,可以举出例如苯甲酸、甲基苯甲酸、茴香酸等。碳水化物具有对应其种类的羟基数,但可以为羟基的一部分与羧酸反应而形成酯化合物,也可以为羟基的全部与羧酸反应而形成酯化合物。在本发明中,优选羟基的全部与羧酸反应而形成酯化合物。
作为碳水化物酯系增塑剂,具体而言,可优选举出葡萄糖五乙酸酯、葡萄糖五丙酸酯、葡萄糖五丁酸酯、蔗糖八乙酸酯、蔗糖八苯甲酸酯等,其中,更优选蔗糖八乙酸酯、蔗糖八苯甲酸酯,特别优选蔗糖八苯甲酸酯。
下述中举出这些化合物的一个例子,但本发明并不限定于这些化合物。
モノペツトSB:第一工业制药公司制;
モノペットSOA:第一工业制药公司制。
(聚合物增塑剂)
作为聚合物增塑剂,具体而言,可以举出脂肪族烃系聚合物;脂环式烃系聚合物;聚丙烯酸乙酯、聚甲基丙烯酸甲酯、甲基丙烯酸甲酯和甲基丙烯酸-2-羟基乙酯的共聚物(例如共聚比1∶99~99∶1之间的任意的比率)等的丙烯酸系聚合物;聚乙烯基异丁基醚、聚N-乙烯基吡咯烷酮的等乙烯基系聚合物;聚苯乙烯、聚4-羟基苯乙烯等的苯乙烯系聚合物;聚丁二酸丁二醇酯、聚对苯二甲酸乙二醇酯、聚萘二甲酸乙二醇酯等的聚酯;聚环氧乙烷、聚环氧丙烷等的聚醚;聚酰胺;聚氨酯、聚脲等。数均分子量优选为1,000~10,000左右,特别优选为5,000~10,000。若为1,000以上,则可抑制挥发性的问题,若为10,000以下,则可发挥增塑剂的功能,可提高光学膜的机械性质。这些聚合物增塑剂可以为由1种重复单元构成的均聚物,也可以为具有多个重复结构体的共聚物。另外,可以并用2种以上的上述聚合物。
上述增塑剂可以单独使用1种或组合使用2种以上,但在使用2种以上的增塑剂的情况下,优选至少1种为多元醇酯系增塑剂。
增塑剂的配合量可在不损害本发明的目的的范围内适宜选择,但相对于表面改性纳米纤维的总质量(100质量份),优选添加0.1~20质量%,进一步优选为0.2~10质量份。
(7)消光剂
对于片状基材而言,为了赋予平滑性、光学功能、机械功能,可含有消光剂。
作为消光剂,可以举出无机化合物的微粒或有机化合物的微粒。消光剂的形状,可优选使用球状、棒状、针状、层状、平板状等的形状。
作为消光剂,例如可以举出二氧化硅、二氧化钛、氧化铝、氧化锆、碳酸钙、高岭土、滑石、煅烧硅酸钙、水合硅酸钙、硅酸铝、硅酸镁、磷酸钙等的金属的氧化物;磷酸盐、硅酸盐、碳酸盐等的无机微粒、交联高分子微粒。其中,二氧化硅可降低膜的雾度,因此优选。
这些微粒通过有机物进行表面处理,可降低膜的雾度,因此优选。表面处理优选用卤代硅烷类、烷氧基硅烷类、硅氮烷、硅氧烷等进行。
平均粒径大的微粒的平滑性效果大,相反平均粒径小的微粒的透明性优异。通常,微粒的一次粒子的平均粒径为0.01~1.0μm的范围。优选的微粒的一次粒子的平均粒径优选为5~50nm,进一步优选为7~14nm。这些微粒,为了在基材表面生成0.01~1.0μm的凹凸,因此优选使用。
这样的二氧化硅的微粒,以日本ァェロジル(株)制的ァェロジル(AEROSIL)200、200V、300、R972、R972V、R974、R202、R812、OX50、TT600、NAX50等、日本催化剂(株)制的KE-P10、KE-P30、KE-P100、KE-P150等的商品名被市售,可使用。
其中,由于一边低地保持膜的浊度、一边降低摩擦系数的效果大,因此优选为ァェロジル200V、R972V、NAX50、KE-P30、KE-P100。
这些微粒可以并用2种以上。在并用2种以上的情况下,可以以任意的比例混合来使用。可以将平均粒径、材质不同的微粒、例如ァェロジル200V和R972V以质量比计在0.1∶99.9~99.9∶0.1的范围使用。
越添加消光剂,得到的膜的平滑性越提高,但由于越添加雾度越增加,因此,其配合量可在不损害本发明的目的的范围内适宜选择。若举出一个例子,则相对于表面改性纳米纤维的总质量(100质量份),优选添加0.001~5质量份,更优选为0.005~1质量份,进一步优选为0.01~0.5质量份。
(8)光学各向异性控制剂
可将用于控制光学各向异性的延迟增加剂根据情况而添加。这些为了调整膜的延迟,优选将具有至少二个芳香族环的芳香族化合物作为延迟增加剂来使用。芳香族化合物,在相对于表面改性纤维素纳米纤维的总质量(100质量份)为0.01~20质量份的范围使用。进而,优选在0.05~15质量份的范围使用,进一步优选在0.1~10质量份的范围使用。也可以并用二种以上的芳香族化合物。芳香族化合物的芳香族环中,除芳香族烃环以外,含有芳香族性杂环。芳香族烃环,特别优选为六元环(即苯环)。芳香族性杂环,一般而言为不饱和杂环。芳香族性杂环,优选为5元环、6元环或7元环,进一步优选为5元环或6元环。芳香族性杂环,一般而言具有最多的双键。作为杂原子,优选氮原子、氧原子及硫原子,特别优选氮原子。芳香族性杂环的例子中,包括呋喃环、噻吩环、吡咯环、噁唑环、异噁唑环、噻唑环、异噻唑环、咪唑环、吡唑环、呋咱环、三唑环、吡喃环、吡啶环、哒嗪环、嘧啶环、吡嗪环及1,3,5-三嗪环。对于这些,详细记载在特开2004-109410号、特开2003-344655号、特开2000-275434号、特开2000-111914号、特开平12-275434号公报等中。
(9)交联剂
片状基材,可以含有交联剂。通过添加交联剂,可以使纤维素纳米纤维间的互相缠绕紧密,透明性提高,且热膨胀性降低,因此优选。
作为交联剂,优选金属氧化物,例如氧化铝、硼酸、氧化钴等。另外,可以使用选自由以下组成的组中的至少1种:间二甲苯乙烯基磺酸等的具有乙烯基砜基的化合物;双酚缩水甘油基醚等的具有环氧基的化合物;具有异氰酸酯基的化合物;具有封端异氰酸酯基的化合物;2-甲氧基-4,6-二氯三嗪、2-钠氧基-4,6-二氯三嗪等的具有活性卤素基团的化合物、甲醛、乙二醛等的具有醛基的化合物;糠氯酸、四亚甲基-1,4-双(亚乙基脲)、六亚甲基-1,6-双(亚乙基脲)等的具有亚乙基亚胺基的化合物及具有活性酯生成基的化合物。这些交联剂可以组合使用2种以上。其中,特别优选金属氧化物、具有乙烯基砜基的化合物、具有亚乙基亚胺基的化合物、具有环氧基的化合物。
在本发明中,所谓具有乙烯基砜基的化合物,为具有与磺酰基键合了的乙烯基或者可形成乙烯基的基团的化合物,优选具有至少2个与磺酰基键合了的乙烯基或者可形成乙烯基的基团,优选由下述通式(8)所示的物质。
[化学式14]
通式(8)
(CH2=CHSO2)nA
式中,A为n价的连接基团,例如亚烷基、取代亚烷基、亚苯基、取代亚苯基,在中间也可以具有酰胺连接部分、氨基连接部分、醚连接部分或者硫醚连接部分。作为取代基,可以举出卤素原子、羟基、羟基烷基、氨基、磺酸基、硫酸酯基等。n为1、2、3或4。
以下举出乙烯基砜系交联剂的代表性具体例。
[化学式15-1]
[化学式15-2]
作为具有环氧基的化合物,特别优选具有2个以上环氧基、每1个官能团的分子量为300以下的物质。以下举出具有环氧基的交联剂的具体例。
[化学式16-1]

[化学式16-2]

作为具有亚乙基亚胺基的化合物,可特别优选使用2官能、3官能且分子量为700以下的物质。以下举出具有亚乙基亚胺基的交联剂的具体例。
[化学式17]
交联剂的使用量,可在不损害本发明的目的的范围适宜选择,但相对于表面改性纤维素纳米纤维的总质量(100质量份),优选为0.1~10质量%,更优选为1~8质量%。
片状基材的厚度没有特别限制,但优选为10~200μm,进一步优选为50~150μm,特别优选为50~125μm。
(气体阻隔层)
气体阻隔层,形成于片状基材1的至少单面,是指主要是对于水蒸气和氧的气体阻隔性高的层。气体阻隔层,为特别是用于防止对于高湿度的基材及用该基材保护的各种电子元件的劣化的层。
气体阻隔层只要为具有上述功能的透明性良好的无机膜就没有特别限制。从透明性、气体阻隔性的观点考虑,可使用硅氧化物、硅氮化物、硅氧化氮化物、氧化铝、氧化钽、氧化氮化铝、SiAlON等。
进而从耐酸性、耐碱性的观点考虑,优选以硅氧化物、硅氮化物及/或硅氧化氮化务为主要成分(相对于气体阻隔层的构成材料100质量%为30质量%以上),相对于气体阻隔层的构成材料100质量%更优选为40质量%以上,进一步优选为50质量%以上。气体阻隔层,可以具有单层结构,也可以从使气体阻隔性进一步提高的方面考虑具有由多个层形成的层叠结构。
气体阻隔层的表面的表面粗糙度(Ra),优选为2nm以下,进一步优选为1nm以下。通过表面粗糙度在上述范围而作为有机电子元件用基板来使用时,可得到凹凸少的平滑的膜面带来的光透过效率的提高效果和电极间泄露电流的减少带来的能量转化效率的提高效果。需要说明的是,气体阻隔层的表面粗糙度(Ra),可使用AFM(原子力显微镜)而通过实施例中记载的方法来算出。
气体阻隔层的厚度没有特别限制,为0.01~5μm,更优选为0.05~3μm,最优选为0.1~1μm。
(中间层)
对本发明的气体阻隔性膜而言,可以使中间层介于片状基材和气体阻隔层之间。作为这样的中间层,例如可以举出平滑层、防渗层、锚涂层等。通过形成这样的中间层,可谋求气体阻隔层和基材的密合性、气体阻隔特性的提高。
(气体阻隔性膜的物性)
气体阻隔性,可以通过依据了JIS-K7129:1992的方法来测定。氧透过度,可以通过依据了JIS-K7126:1987的方法来测定。在本发明中,水蒸气透过度(60±0.5℃、相对湿度(90±2)%RH)只要为1×10-3g/(m2·24h)以下即可。一般而言,由于氧透过度比水蒸气透过度小,因此若为满足上述水蒸气透过度的膜,则作为有机元件很少成为问题。
对于透明性而言,优选总透光率为85%以上,特别优选具有90%以上的高的透明性。在低于85%的情况下,应用用途的范围狭窄,特别是有可能图像紊乱或清晰性劣化。另外在制造工序中的热加工后也需要上述的高的透明性。透光率,可以通过分光光度计来测定。
雾度值优选低于1.5%,更优选低于1%,进一步优选低于0.5%。雾度可以使用浊度计来测定。
作为着色性的指标可以使用黄色度(黄色指数、Y I),优选为3.0以下,更优选为1.0以下。黄色度可以基于JI S-K7103:1994来测定。
20~200℃下的线热膨胀系数,优选为15ppm/K以下,更优选为10ppm/K以下,进一步优选为5ppm/K以下。若大于15ppm/K,则因为形成元件器件的导电膜、阻隔膜等的无机膜、进而和与玻璃的线热膨胀系数的不同,由于制造工序中的热加工等,有时膜破裂而不能发挥功能,或在膜上产生弯曲、变形,或作为元件用零件产生成像性能、折射率失常等的问题。
气体阻隔性膜的膜厚,没有特别限定,可优选使用10~200μm。特别是膜厚特别优选为50~150μm。进一步优选为75~125μm。
(气体阻隔性膜的制造方法)
上述气体阻隔性膜的制造方法没有特别限制,可以适宜参照现有公知的方法来制作。
根据本发明的其他的一方式,提供气体阻隔性膜的制造方法。本方式的制造方法,具有(1)将纤维素纳米纤维的羟基的氢原子的至少一部分用碳数1~8的酰基进行取代而得到表面改性纤维素纳米纤维,将上述表面改性纤维素纳米纤维用熔融挤出法或溶液浇铸法进行制膜而得到片状基材的工序A;(2)在上述片状基材上形成气体阻隔层的工序B。
(1)工序A
(1-1)表面改性纤维素纳米纤维的制造
首先,将纤维素纳米纤维的羟基的氢原子的至少一部分用酰基进行取代而得到表面改性纤维素纳米纤维。
作为纤维素纳米纤维,如上所述,可以使用通过原料纤维素纤维的解纤处理而得到的纤维。
将纤维素纳米纤维的羟基的氢原子用酰基取代的方法没有特别限制,可以根据公知的方法进行。例如,只要将通过解纤处理得到的纤维素纳米纤维添加到水或适当的溶剂中而使其分散后,在其中添加酰卤、羧酸酐、羧酸、或醛而使其在适当的反应条件下反应即可。
此时,可以根据需要添加反应催化剂,例如可以使用吡啶、N,N-二甲基氨基吡啶、三乙胺、甲醇钠、乙醇钠、氢氧化钠等的碱性催化剂、乙酸、硫酸、高氯酸等的酸性催化剂,但为了防止反应速度、聚合度的降低,优选使用吡啶等的碱性催化剂。作为反应温度,从抑制纤维素纤维的黄变、聚合度的降低等的变质、确保反应速度的观点考虑,优选40~100℃左右。对于反应时间可根据使用的酰基化剂、处理条件适宜选定。
(1-2)制膜
接着,将上述中得到了的表面改性纤维素纳米纤维用熔融挤出法或溶液浇铸法进行制膜而得到片状基材。
(a)熔融挤出法
在使用熔融挤出法(熔融流延法)的情况下,可以用以下的方法来制造片状基材:将在高温下熔融含有表面改性纤维素纳米纤维及根据需要的微量的基体树脂、添加剂的纤维素纳米纤维组合物而得到的熔融物由加压模头等挤出,例如流延到无限地进行移送的环形的金属带或进行旋转的金属鼓的流延用支撑体上、进行制膜。
(a-1)纤维素纳米纤维组合物的调制
首先,调制含有纤维素纳米纤维及根据需要所添加的基体树脂、添加剂的纤维素纳米纤维组合物。该组合物的调制,可以在纤维素纳米纤维的解纤处理后至熔融前的任意的工序中进行。优选的是,该组合物在熔融前混合,进一步优选在加热前混合。或者,也可以在树脂熔融物的制造过程中添加添加剂。此时,在使用多种添加剂的情况下,可以预先在溶剂中使它们混合分散后,得到使溶剂挥发或沉淀了的固体物,将其在树脂熔融物的制造过程中添加。
混合手段没有特别限制,例如可以使用V型混合机、圆锥螺杆型混合机、水平圆筒型混合机等、亨舍尔混合机、螺带混合机、拉伸流动分散机等的一般的混合机。
进而,纤维素纳米纤维组合物优选在熔融前进行热风干燥或真空干燥。
(a-2)熔融挤出
将上述中得到的纤维素纳米纤维组合物使用挤出机进行熔融而制膜。此时,可以在调制了纤维素纳米纤维组合物后,将该组合物使用挤出机直接熔融而进行制膜,或也可以在对纤维素纳米纤维组合物进行粒料化后,将该粒料用挤出机进行熔融而制膜。
另外,在纤维素纳米纤维组合物含有熔点不同的多种材料的情况下,也可在仅熔点低的材料进行熔融的温度下暂时制作所谓的江米糖(ぉこし)状的半熔融物,将半熔融物投入到挤出机中而制膜。
在纤维素纳米纤维组合物中含有容易热分解的材料情况下,以减少熔融次数的目的,优选在未制作粒料地直接制膜的方法、制作如上所述的江米糖状的半熔融物后进行制膜的方法。
挤出机,可使用能够在市场中获得的各种挤出机,但优选熔融混炼挤出机,可以为单轴挤出机,也可以为双轴挤出机。在未由纤维素纳米纤维组合物制作粒料地直接进行制膜的情况下,由于需要适当的混炼度,因此优选使用双轴挤出机,但即使为单双轴挤出机,通过将螺杆的形状变更为Maddock(マドック)型、Un ime lt(ュニメルト)、Du1madage(ダルメ一ジ)等的混炼型的螺杆,可得到适度的混炼,因此可使用。在暂时使用粒料、江米糖状的半熔融物的情况下,单轴挤出机或双轴挤出机均可使用。
对于熔融温度而言,根据纤维素纳米纤维组合物(膜构成材料)的粘度、喷出量、进行制造的片的厚度等而优选条件不同,但一般而言,相对于膜的玻璃化转变温度Tg,为Tg以上且Tg+100℃以下,优选为Tg+10℃以上且Tg+90℃以下。
在本发明中,纤维素纳米纤维的用酰基所改性了的部分的Tg成为标准。但是,也担心在高温下纤维素纳米纤维的热分解,因此,具体而言,熔融挤出时的温度优选为150~300℃,更优选为180~270℃的范围,进一步优选为200~250℃的范围。
挤出时的熔融粘度,优选为10~100000P(1~10000Pa·s),更优选为100~10000P(10~1000Pa·s)。
纤维素纳米纤维组合物在挤出机内的滞留时间优选短,优选为5分钟以内,更优选为3分钟以内,进一步优选为2分钟以内。滞留时间也受挤出机1的种类、挤出条件影响,但可通过对组合物的供给量、L/D、螺杆转数、螺杆的槽的深度等进行调整来缩短。
(a-3)冷却
熔融挤出,优选由T型模头挤出为膜状。进而,优选在挤出后通过静电施加法等使膜状的挤出物与冷却鼓密合,使其冷却固化,得到未拉伸膜。此时,冷却鼓的温度优选维持为90~150℃。
挤出机内及挤出后的冷却工序,优选通过在氮气等的不活泼气体下进行替换或者进行减压来降低氧的浓度。
利用上述工序,得到未拉伸膜(片状基材)。
(b)溶液浇铸法
在使用溶液浇铸法的情况下,工序A包括以下工序:使表面改性纤维素纳米纤维及根据需要的微量的基体树脂、添加剂溶解于溶剂中而调制胶浆的工序、将胶浆流延到无限地进行移动的环状的金属支撑体上的工序、将流延了的胶浆制成网状物而进行干燥的工序、将上述网状物从金属支撑体进行剥离的工序、将完成的膜进行卷绕的工序。
(b-1)胶浆调制工序
首先,使表面改性纤维素纳米纤维及根据需要的微量的基体树脂、添加剂溶解于溶剂中,得到胶浆。
胶浆中所使用的溶剂,可以单独使用,也可以并用2种以上,但从生产效率的方面考虑,优选混合使用表面改性纤维素纳米纤维的良溶剂和不良溶剂,从表面改性纤维素纳米纤维的溶解性的方面考虑,优选良溶剂多。对良溶剂和不良溶剂的混合比率的优选的范围而言,良溶剂为2~30质量%,不良溶剂为70~98质量%。所谓良溶剂、不良溶剂,将单独溶解使用的纤维素纳米纤维的溶剂定义为良溶剂,将单独溶胀或不溶解使用的纤维素纳米纤维的溶剂定义为不良溶剂。这些根据表面改性纤维素纳米纤维的酰基的取代度、结晶度而发生变化,因此可以适宜选择。
上述良溶剂没有特别限定,可以举出二氯甲烷等的有机卤素化合物及二氧戊烷类、丙酮、乙酸甲酯、乙酰乙酸甲酯等。可特别优选可举出二氯甲烷或乙酸甲酯。
上述不良溶剂没有特别限定,例如可优选使用甲醇、乙醇、正丁醇、环己烷、环己酮等。另外,胶浆中优选含有0.01~2质量%的水。
对胶浆中的表面改性纤维素纳米纤维浓度而言,浓者可减少流延到金属支撑体后的干燥负荷,优选,但表面改性纤维素纳米纤维的浓度过浓时过滤时的负荷增加,过滤精度变差。作为兼具这些的浓度,优选为10~35质量%,进一步优选为15~25质量%。
作为调制上述记载的胶浆时的表面改性纤维素纳米纤维的溶解方法,可以使用一般的方法。若组合加热和加压,则可加热到常压下的沸点以上,因此优选。即,若在溶剂的常压下的沸点以上且在加压下溶剂不沸腾的范围的温度下一边加热一边搅拌溶解时,则防止被称为凝胶或面团的块状未溶解物的产生,因此优选。
另外,也可优选使用在将表面改性纤维素纳米纤维与不良溶剂混合而使其湿润或溶胀后、进一步添加良溶剂来进行溶解的方法。加压可通过压入氮气等的不活泼气体的方法或通过加热而使溶剂的蒸汽压呈现的方法来进行。加热优选从外部进行,例如夹套型的装置的温度控制容易,优选。
对于添加溶剂后的加热温度而言,从纤维素纳米纤维的溶解性的观点考虑,优选高者,但如果加热温度过高,则所需要的压力变大,生产率变差。优选的加热温度为45~120℃,更优选为60~110℃,进一步优选为70℃~105℃。另外,压力调整为使得在设定温度下溶剂不沸腾。或者也可优选使用冷却溶解法。
各种添加剂可以分批添加到制膜前的胶浆中,也可以另行准备将添加剂于甲醇、乙醇、丁醇等的醇、二氯甲烷、乙酸甲酯、丙酮、二氧戊烷等的有机溶剂或者这些的混合溶剂中的溶液而在线添加。特别是为了减少微粒对过滤材料的负荷,优选在线添加一部分或总量。为了进行在线添加、混合,例如,可优选使用静态混合器(东レェンジニァリング制)、SWJ(东レ静止型管内混合器Hi-Mixer)等的在线混合器等。
使表面改性纤维素纳米纤维溶解了的胶浆,优选通过过滤将原料的纤维素纳米纤维中所含的杂质、特别是辉点异物除去、减少。所谓辉点异物,为将2张偏振片以正交尼科尔状态配置、在其间设置光学膜等、从一方的偏振片侧照射光而从另一方的偏振片侧观察时来自相反侧的光泄漏而可看到的点(异物),优选直径在0.01mm以上的辉点数为200个/cm2以下。更优选为100个/cm2以下,进一步优选为50个/m2以下,进一步优选为0~10个/cm2以下。另外,优选0.01mm以下的辉点也少。
过滤的方法没有特别限制,可以用通常的方法进行,优选使用滤纸等的适当的过滤材料来过滤。
作为过滤材料,为了除去不溶物等而优选绝对过滤精度小,但若绝对过滤精度过小,则存在容易产生过滤材料堵塞这样的问题。因此优选绝对过滤精度0.008mm以下的过滤材料,更优选0.001~0.008mm的过滤材料,进一步优选0.003~0.006mm的过滤材料。
过滤材料的材质没有特别限制,可以使用通常的过滤材料,但聚丙烯、特氟龙(注册商标)等的塑料制的过滤材料、不锈钢等的金属制的过滤材料没有纤维的脱落等,优选。
作为过滤条件没有特别限制,但在溶剂的常压下的沸点以上且在加压下溶剂不会沸腾的范围的温度下一边加热一边过滤的方法,过滤前后的滤压的差(称为差压)的呈现小,优选。优选的温度为45~120℃,更优选为45~70℃,进一步优选为45~55℃。滤压优选小者。滤压优选为1.6MPa以下,更优选为1.2MPa以下,进一步优选为1.0MPa以下。
(b-2)胶浆流延工序
接着,将胶浆流延(浇铸)到金属支撑体上。
金属支撑体,优选对表面进行了镜面精加工的支撑体,作为金属支撑体,可优选使用用不锈钢带或者铸件对表面进行了镀敷精加工了的鼓。浇铸的宽度可以设为1~4m。
(b-3)干燥工序
接着,使流延了的胶浆干燥成网状物。
金属支撑体的表面温度为-50℃~低于溶剂的沸点的温度。由于温度高的一方可加快网状物的干燥速度,故优选,但若过高,则有时网状物发泡或平面性劣化。优选的支撑体温度为0~40℃,进一步优选5~30℃。
控制金属支撑体的温度的方法没有特别限制,有吹暖风或冷风的方法、使温水与金属支撑体的背侧接触的方法。由于使用温水的一方可有效地进行热传递,因此,金属支撑体的温度达到一定的时间短,优选。在使用暖风的情况下,有时使用温度比目标温度高的风。
需要说明的是,可以回收在干燥工序中所除去的溶剂、作为上述(b-1)胶浆制备工序中的上述表面改性纤维素纳米纤维的溶解中所使用的溶剂进行再利用来使用。需要说明的是,在回收溶剂中也有时含有微量的添加剂(例如增塑剂、紫外线吸收剂、聚合物、单体成分等),但即使含有这些添加剂也可优选进行再利用,也可以根据需要精制来进行再利用。
(b-4)剥离工序
接着,将网状物从金属支撑体剥离。
为了制膜后的膜显示良好的平面性,从金属支撑体剥离网状物时的残留溶剂量优选10~150质量%,进一步优选为20~40质量%或60~130质量%,特别优选为20~30质量%或70~120质量%。
在本发明中,残留溶剂量以下述数学式(2)定义。
[数学式2]
残留溶剂量(质量%)={(M-N)/N}×100
式中,M为在制造中或制造后的任意的时刻采集网状物或膜的试样的质量,N为在115℃下将上述采集的试样(质量M的试样)加热1小时后的质量。
但是,在通过进行冷却而使网状物凝胶化来多地含有残留溶剂的状态下从鼓进行剥离也为优选的方法。
需要说明的是,剥离了的网状物优选进一步进行干燥,优选使残留溶剂量为1质量%以下,更优选为0.1质量%以下,特别优选为0~0.01质量%以下。
该干燥,一般而言采用以辊干燥方式(在上下配置了的多个辊上交替通过网状物、使其干燥的方式)、拉幅机方式将网状物一边输送一边干燥的方式。
(b-5)膜卷绕工序
最后,通过卷绕得到的网状物(完成的膜)来得到片状基材。
(1-3)拉伸处理
上述得到的片状基材,可以在制膜后至少在一方向上拉伸。通过进行拉伸处理,可以调整膜的延迟,光学特性可提高。
作为拉伸方法,优选将从上述的冷却鼓上剥离、得到的未拉伸膜经由多个辊组及/或红外线加热器等的加热装置而从纤维素纳米纤维的用酰基所改性了的部分的玻璃化转变温度(Tg)-50℃加热至Tg+100℃的范围内、在膜输送方向(也称为长度方向)进行一段或多段纵向拉伸。接着,也优选将如上所述得到的被拉伸了的表面改性纤维素膜在与膜输送方向正交的方向(也称为横向方向)进行拉伸。为了在宽度方向将膜拉伸,优选使用拉幅机装置。
在膜输送方向或与膜输送方向正交的方向上进行拉伸的情况下,优选以2.5倍以下的倍率进行拉伸,更优选为1.1~2.0倍的范围。若为2.5倍以下,则可防止纳米纤维周边的空隙产生,可抑制透明性的劣化。
另外,也可以接着拉伸进行热加工。热加工,优选在Tg-100℃~Tg+50℃的范围内通常一边进行输送0.5~300秒钟一边进行。
热加工手段没有特别限制,一般而言可以用热风、红外线、加热辊、微波等进行,但从简便性的方面考虑,优选用热风进行。膜的加热优选阶段性地提高。
被热加工了的膜通常被冷却至Tg以下,切割膜两端的夹具把持部分、进行卷绕。另外冷却优选以每秒100℃以下的冷却速度从最终热加工温度缓慢冷却至Tg。
进行冷却的手段没有特别限定,可以用以往公知的方法进行,但从膜的尺寸稳定性提高的方面考虑,特别优选一边在多个温度区域下依次冷却一边进行这些处理。需要说明的是,对于冷却速度而言,为在将最终热加工温度设为T1、将膜从最终热加工温度直到达到Tg的时间设为t时,以(T1-Tg)/t而求出的值。
(c)多层化
另外,也可以通过共流延法来得到形成了多层构成的膜。通过形成为多层构成,可以调整制造工序的热加工中的翘曲、变形等,或调整透明性、热膨胀性,因此是有效的。例如,通过形成将酰基的取代度小、结晶度高的纤维配置于中心、将酰基的取代度大、结晶度小的纤维配置于两面而成的构成,可改善热加工中的翘曲、变形等。通过共流延法而形成为多层构成时的膜厚构成,可以适宜调整。
(1-4)压延处理
上述得到的片状基材,可在制膜后通过加热压延处理进行透明、平滑化。需要说明的是,也可以除了加热压延处理外进行拉伸处理,在制膜后进行拉伸处理及压延处理这两者的情况下,其顺序没有特别限制,先进行哪一个均可。
通过加热压延处理,可以使纤维素纳米纤维的改性了的树脂成分(酰基成分)在膜中扩散,由此,透明性、生产率、热膨胀、平滑性提高。
作为加热压延处理,除利用单压辊的通常的压延装置以外,也可以使用具有多段式地设置了这些的结构的超级压延装置。这些装置及压延处理时的辊两侧各自的材质(材质硬度)或线压力可以根据目的来选择。
(2)工序B
接着,在上述片状基材上形成气体阻隔层。
气体阻隔层的形成方法,没有特别限制,可以使用涂布、溶胶凝胶法、蒸镀法、CVD(化学气相沉积法)、溅射法等的公知方法。
但是,与如CVD法那样以膜材料为气体而进行供给相比,涂布在基材表面能够形成更均匀、平滑的气体阻隔层。特别是在使用了CVD的情况下,在气相中反应性增加了的原料物质堆积在基材表面的工序的同时,恐怕生成气相中不需要的被称为颗粒的异物。从这样的观点考虑,优选使用在上述片状基材上涂布了气体阻隔层的前体材料后、对涂布膜进行改性的方法。在涂布法中,通过不在气相反应空间存在原料,可抑制这些颗粒的产生。
该前体材料,可根据气体阻隔层的材料来选择,可以举出聚硅氮烷化合物、溶胶状的有机金属化合物等。作为有机金属化合物,只要为可水解的物质即可,没有特别限定,但作为优选的有机金属化合物,可以举出金属醇盐。
优选使用聚硅氮烷化合物作为气体阻隔层的前体材料。即,工序B优选包含在上述片状基材上涂布含有聚硅氮烷化合物的涂布液(涂布工序)后、进行改性处理(改性工序)。
在使基体树脂存在于纤维素纳米纤维的周围的以往的纤维素纳米纤维基材表面使用聚硅氮烷化合物而形成了气体阻隔层的情况下,存在如下问题:由于含聚硅氮烷液体的涂布后的紫外线照射等的改性处理,基体树脂受到影响,不仅引起在基材表面附近的层分离、微小的表面性状的不均匀、无法使气体阻隔性提高,而且基材与气体阻隔层的粘接性、表面的平滑性受损。进而,为了解决平滑性、粘接性的问题,即使在基材和气体阻隔层之间设置了中间层的情况下,也形成长期保存时的粘接性受损、保存性恶化的结果。
本发明的详细机制尚未明确,但由于本发明的片状基材实质上不含基体树脂,因此,可提高片状基材与气体阻隔层的粘接性,特别是长期保存时的粘接性(保存性)。
以下,对该优选的方式进行说明。
(2-1)含有聚硅氮烷化合物涂布液的涂布工序
首先,使聚硅氮烷化合物溶解于有机溶剂中、调制含有聚硅氮烷化合物的涂布液。
所谓“聚硅氮烷化合物”,为具有硅-氮键的聚合物,为由Si-N、Si-H、N-H等构成的SiO2、Si3N4及两者的中间固溶体SiOxNy等的陶瓷前体无机聚合物。
为了在片状基材上形成均匀的涂装层,改性后形成具有良好的气体阻隔性的气体阻隔层,并且不损害基材的特性,可使用在比较低温下进行陶瓷化而改性为二氧化硅的具有下述通式(9)所示的构成单元的聚硅氮烷化合物。
[化学式18]
通式(9)

式中,R91、R92、及R93分别独立地为氢原子、碳数1~3的烷基、碳数2~3的烯基、碳数1~3的烷基甲硅烷基、碳数1~3的烷基氨基、碳数1~3的烷氧基。
从得到的气体阻隔性膜的致密性的观点考虑,特别优选R91、R92及R93的全部为氢原子的全氢聚硅氮烷。
全氢聚硅氮烷推定为存在直链结构和以六元环及八元环为中心的环结构的结构。其分子量以数均分子量(Mn)计为600~2000左右(聚苯乙烯换算),在常温下为液体或固体的物质,根据分子量而不同。这些能够以溶解在有机溶剂中的溶液状态而被市售,可以将市售品直接作为含聚硅氮烷涂布液来使用。
另一方面,与Si键合的氢部分的一部分用烷基等取代了的有机聚硅氮烷(R91、R92及/或R93具有烷基的化合物)通过具有甲基等的烷基,与基底基材的粘接性得到改善,且可以使硬且脆的由聚硅氮烷形成的陶瓷膜具有韧性,具有即使在进一步加厚(平均)膜厚的情况下也可抑制破裂的产生的优点。
因此,可以根据用途适宜选择全氢聚硅氮烷和有机聚硅氮烷,也可以混合使用。
作为在低温下进行陶瓷化的聚硅氮烷化合物的另一例,可以举出使硅醇盐与上述通式(9)的聚硅氮烷反应而得到的硅醇盐加成聚硅氮烷(特开平5-238827号公报)、使缩水甘油与上述通式(9)的聚硅氮烷反应而得到的缩水甘油加成聚硅氮烷(特开平6-122852号公报)、使醇与上述通式(9)的聚硅氮烷反应而得到的醇加成聚硅氮烷(特开平6-240208号公报)、使金属羧酸盐与上述通式(9)的聚硅氮烷反应而得到的金属羧酸盐加成聚硅氮烷(特开平6-299118号公报)、使含有金属的乙酰丙酮络合物与上述通式(9)的聚硅氮烷反应而得到的乙酰丙酮络合物加成聚硅氮烷(特开平6-306329号公报)、向上述通式(9)的聚硅氮烷添加金属微粒而得到的添加有金属微粒的聚硅氮烷(特开平7-196986号公报)等。
作为有机溶剂,只要为不含有容易与聚硅氮烷化合物反应的醇系、水分的物质就没有特别限制。具体而言,可使用脂肪族烃、脂环式烃、芳香族烃等的烃溶剂;卤代烃溶剂;脂肪族醚、脂环式醚等发醚类。具体而言,有戊烷、己烷、环己烷、甲苯、二甲苯、So1Vesso、松节油等的烃;二氯甲烷、三氯乙烷等的卤代烃;二丁醚、二噁烷、四氢呋喃等的醚类等。这些溶剂可考虑聚硅氮烷的溶解度、溶剂的蒸发速度等根据目的来选择,也可以混合多种溶剂。
含有聚硅氮烷化合物涂布液中的聚硅氮烷浓度也根据目标气体阻隔层的膜厚、涂布液的适用期而不同,但相对于涂布液的总质量为0.2~35质量%左右。
在含有聚硅氮烷化合物的涂布液中,为了促进向氧化硅化合物的转化,也可以添加胺、金属的催化剂。具体可以举出AZェレクトロニツクマテリァルズ(株)制クァミカNAX120-20、NN110、NN310、NN320、NL110A、NL120A、NL150A、NP110、NP140、SP140等。
接着,将至少1层的含有聚硅氮烷化合物的涂布液涂布在片状基材上。
作为涂布方法,可采用任意的适当的方法。作为具体例,可以举出旋涂法、辊涂法、流涂法、喷墨法、喷涂法、印刷法、浸涂法、流延成膜法、棒涂法、凹版印刷法等。
涂布厚度,可根据目的适当地设定。例如涂布厚度可设定为使得干燥后的厚度优选为1nm~100μm左右、进一步优选为10nm~10μm左右、最优选为10nm~1μm左右。
(2-2)除湿工序
优选在上述涂布工序后、接着进行改性工序之前或改性工序中包含从含聚硅氮烷液体的涂布膜除去水分的工序(除湿工序)。通过在改性处理前或改性中除去水分,可以促进转化为硅烷醇的聚硅氮烷膜的脱水反应。因此,优选聚硅氮烷膜在利用除湿工序去除水分后、维持其状态来进行改性处理。
<聚硅氮烷膜的含水量>
聚硅氮烷膜中的含水率,定义为由利用下述的分析方法得到的含水量除以聚硅氮烷膜的体积的值。通过除湿工序而去除了水分的状态的聚硅氮烷膜中的含水率,优选为0.1%以下,更优选为0.01%以下(检测限以下)。
聚硅氮烷膜的含水率可以用以下的分析方法来检测。
顶空-气相色谱/质量分析法
装置:HP6890GC/HP5973MSD
烘箱:40℃(2min),然后,以10℃/min的速度升温至150℃
柱:DB-624(0.25mmid×30m)
注入口:230℃
检测器:SIM m/z=18
HS条件:190℃·30min。
更优选的是,除湿工序包含去除聚硅氮烷膜中的溶剂的第一除湿工序和接着其的去除聚硅氮烷膜中的水分的第二除湿工序。
在第一除湿工序中,可以用热处理等的方法适宜设定主要用于去除溶剂的干燥条件。但是,也可以根据此时的条件来除去水分。
从迅速处理的观点考虑,热处理温度优选高的温度,但可以考虑对树脂基材的热损伤、设定温度和处理时间。若举出一个例子,则在片状基材(表面改性纤维素纳米纤维)的玻璃化转变温度(Tg)为70℃的情况下,热处理温度可以设定为200℃以下。
处理时间优选以可除去溶剂、且对基材的热损伤变少地设定在短时间,例如在热处理温度为200℃以下的情况下,优选设为30分钟以内。
第二除湿工序为用于去除聚硅氮烷膜中的水分的工序。
作为优选的方法,为维持在低湿度环境的方式。低湿度环境中的湿度,根据温度发生变化,因此温度和湿度的关系可通过露点的规定而示出优选的方式。优选的露点为4度以下(温度25度/湿度25%),更优选的露点为-8度(温度25度/湿度10%)以下,维持的时间根据聚硅氮烷膜的膜厚而适宜改变。例如在聚硅氮烷膜厚1μm以下的条件下,优选的露点为-8度以下,所维持的时间为5分钟以上。另外,为了容易去除水分,也可以进行减压干燥。减压干燥中的压力可以选择常压~0.1MPa。
作为第一除湿工序及第二除湿工序的优选的条件的组合,例如有在第一除湿工序中以温度60~150℃、处理时间1分钟~30分钟来除去溶剂,通过第二除湿工序的露点为4度以下、处理时间为5分钟~120分钟来除去水分的条件。设置第一除湿工序及第二除湿工序时的这些的区分,可以在露点的变化、即工序环境的露点之差变化10度以上的时刻进行区别。
(2-3)改性工序
在本发明中所谓改性处理,是指将作为气体阻隔层的前体材料的聚硅氮烷化合物通过活性能量射线的照射或热处理等而添加到硅氧化物或氮化硅氧化物中的处理。
改性处理的方法,可以选择基于聚硅氮烷化合物的转化反应的公知的方法。但是,由于利用热处理的硅氮烷化合物的转化反应需要450℃以上的高温,因此,恐怕因改性处理而使基材的性能劣化。从这样的观点考虑,在本发明中优选在更低温下可进行转化反应的使用了等离子体、紫外线的照射的转化反应,更优选紫外线的照射,特别是更优选利用准分子照射的添加反应。
(a)等离子体处理
作为等离子体处理,可以使用公知的方法,但优选大气压等离子体处理。在大气压等离子体处理的情况下,作为放电气体,可使用氮气及/或稀有气体(具体而言为氦、氖、氩、氪、氙、氡等)。其中,可优选使用氮、氦、氩,特别是氮的成本也便宜,故优选。
《形成了二个以上不同频率的电场的大气压等离子体》
接着,对于上述大气压等离子体,说明优选的方式。大气压等离子体,具体而言,如国际公开第2007-026545号中所记载那样,在放电空间形成2个以上不同频率的电场,优选形成重叠了第1高频电场和第2高频电场的电场。
上述第2高频电场的频率ω2比上述第1高频电场的频率ω1高,且上述第1高频电场的强度V1、上述第2高频电场的强度V2和放电开始电场的强度IV的关系,满足以下的数学式(3),上述第2高频电场的输出功率密度为1W/cm2以上。
[数学式3]
V1≥IV>V2或V1>IV≥V2
通过采用这样的放电条件,即使为如例如氮气那样放电开始电场强度高的放电气体,也可以开始放电、维持高密度、稳定的等离子体状态,可以进行高性能的薄膜形成。
在通过上述的测定而以放电气体为氮气的情况下,其放电开始电场强度IV(1/2Vp-p)为3.7kV/mm左右,因此,在上述的关系中,通过使第1施加电场强度为V1≥3.7kV/mm来进行施加,可以激发氮气、形成等离子体状态。
在此,作为第1电源的频率,可优选使用200kHz以下。另外,作为其电场波形,可以为连续波,也可以为脉冲波。下限优选为1kHz左右。
另一方面,作为第2电源的频率,可优选使用800kHz以上。该第2电源的频率越高,等离子体密度变得越高,可得到致优质的薄膜。上限优选为200MHz左右。
对这样地由2个电源形成高频电场而言,需要利用第1高频电场来开始具有高的放电开始电场强度的放电气体的放电,另外,可以利用第2高频电场的高的频率及高的输出功率密度而提高等离子体密度来形成致密优质的薄膜。
(b)紫外线照射处理
作为改性处理的方法,也优选利用紫外线照射的处理。在本发明中,所谓“紫外线”,一般而言是指具有10~400nm的波长的电磁波,但在后述的真空紫外线(10~200nm)处理以外的紫外线照射处理的情况下,优选使用210~350nm的紫外线。
通过紫外线(与紫外光意义相同)而生成的臭氧、活性氧原子具有高的氧化能力,可制作在低温下具有高的致密性和绝缘性的氧化硅膜或氧化氮化硅膜。
由于基材通过该紫外线照射而被加热,有助于陶瓷化(二氧化硅转化)的O2和H2O、紫外线吸收剂、聚硅氮烷化合物自身被激发、被活化,因此聚硅氮烷化合物的陶瓷化(转化反应)得到促进,另外得到的气体阻隔层变得更加致密。紫外线照射若在涂膜形成后,则在任意的时刻实施均是有效的。
作为紫外线照射装置,常用的任意的紫外线产生装置均可使用。
紫外线的照射,应在被照射的担载涂膜的基材不受损伤的范围设定照射强度及/或照射时间。若举出一个例子,则可以使用2kW(80W/cm×25cm)的灯、以基材表面的强度为20~300mW/cm2、优选为50~200mW/cm2地设定基材-灯间距离,进行0.1秒钟~10分钟的照射。
一般而言,若紫外线照射处理时的基材温度为150℃以上,则在塑料膜等的情况下基材发生变形或其强度劣化等,基材受损害。因此,该紫外线照射时的基材温度优选低于150℃。需要说明的是,紫外线照射气氛没有特别限制,可在空气中实施。
作为这样的紫外线的产生方法,例如可以举出金属卤化物灯、高压水银灯、低压水银灯、氙弧灯、碳弧灯、准分子灯(172nm、222nm、308nm的单一波长,例如ウシオ电机(株)制)、UV光激光等,没有特别限定。另外,在对聚硅氮烷涂膜照射产生了的紫外线时,为了效率的提高也为了达到均匀的照射,优选将来自产生源的紫外线用反射板反射之后向涂膜进行照射。
对紫外线照射而言,分批处理、连续处理均可适合,可根据被涂布基材的形状来适宜选择。例如在分批处理的情况下,可以用具备如上所述的紫外线产生源的紫外线烧成炉将在表面具有聚硅氮烷涂膜的基材(例如硅晶片)进行处理。紫外线烧成炉自身一般而言是已知的,例如可以使用ァィグラフィクス(株)制。另外,在表面具有聚硅氮烷涂膜的基材为长条膜状的情况下,可以通过将其一边输送一边在具备如上所述的紫外线产生源的干燥区连续地照射紫外线来陶瓷化。
紫外线照射所需的时间,也取决于被涂布的基材、涂布膜的组成、浓度,但一般而言为0.1秒钟~10分钟,优选为0.5秒钟~3分钟。
在本发明中,特别优选的是,利用真空紫外线(准分子)照射的改性。即,在本发明的特别优选的实施方式中,工序B包含在上述片状基材上涂布含有聚硅氮烷化合物的涂布液后、进行准分子照射处理。
(准分子照射处理)
所谓准分子光,为以稀有气体准分子或异核准分子为工作介质的激光。Xe、Kr、Ar、Ne等的稀有气体的原子通过放电等得到能量而被激发,与其它的原子结合而形成分子。例如在稀有气体为氙的情况下,成为
[化学式19]
e+Xe→e+×e*
Xe*+Xe+Xe→Xe2 *+Xe,
作为被激发了的准分子分子的Xe2*跃迁到基底状态时发出172nm的准分子光。
利用真空紫外线(准分子)照射的处理为如下方法:使用比硅氮烷化合物内的原子间结合力大的100~200nm(优选100~180nm)的光能,一边通过被称为光量子工艺的仅由光子引起的作用来直接切断原子的结合一边进行利用活性氧、臭氧的氧化反应,由此可以在较低温下进行氧化硅膜的形成。
作为准分子照射所需要的真空紫外光源,可优选使用稀有气体准分子灯。作为准分子灯的特征,可以举出:放射集中于一个波长,几乎未放射需要的光以外,因此效率高。另外,由于未放射多余的光,因此,可以低地保持对象物的温度。进而,起动·再起动不需要时间,因此可以进行瞬时的点亮点灭。因此,适于容易受到热的影响的柔性膜材料。
更优选的是,从以单一波长放射波长短的172nm的紫外线的方面考虑,更优选为发光效率优异的Xe准分子灯。该光,由于氧的吸收系数大而可以用微量的氧以高浓度产生自由基的氧原子种、臭氧。另外,已知使有机物的结合解离的波长短的172nm的光的能量的能力高。通过该活性氧、臭氧和紫外线放射所具有的高的能量,可在短时间实现聚硅氮烷膜的改性。因此,与发出波长185nm、254nm的低压水银灯、等离子体清洗相比,可以缩短伴随高通过量的工艺时间、缩小设备面积,可以进行向容易受到热引起的损伤的有机材料、塑料基板等的照射。
准分子灯的种类没有特别限制,可使用双重圆筒型灯、细管准分子灯。双重圆筒型灯与细管灯相比,在操作、输送中容易破损。细管准分子灯的结构简单,可提供非常廉价的光源。但是,若细管灯的管的外径太粗,则启动需要高的电压。
放电的方式,可以为电介质阻挡放电,也可以为无电极电场放电。所谓电介质阻挡放电为在两电极间经由电介质(在准分子灯的情况下为透明石英)配置气体空间、对电极施加数10kHz的高频高电压,由此在气体空间产生的与雷相似的非常细小的被称为micro discharge的放电,另一方面,无电极电场放电别名也称为RF放电。灯和电极及其配置基本上可以与电介质阻挡放电相同,但施加于两极间的高频以数MHz被点亮。无电极电场放电可得到如上所述空间上或时间上一样的放电,因此,与电介质阻挡放电相比,可得到没有闪烁的长寿命的灯。
对电极的形状而言,与灯相接的面可以为平面,但若为符合灯的曲面的形状,则可以牢固地固定灯,并且通过电极密合于灯,放电更稳定。另外,若用铝将曲面形成镜面,则也可以成为光的反射板。
需要说明的是,在片状基材和气体阻隔层之间配置中间层的情况下,可以片状基材制膜后、在该片状基材上形成中间层、在上述中间层上形成气体阻隔层。中间层的形成方法没有特别限制,可以参照专利文献5中记载的方法,或将其适宜改变来应用。
[电子元件用基板]
上述气体阻隔性膜,从透明性、表面平滑性、气体阻隔性、及粘接性优异的方面考虑,可以作为电子元件用的透明基板(电子元件用基板)来使用。特别是可应用于液晶、有机元件用基板,作为有机元件,可以举出有机电致发光元件、有机光电转换元件等。
在将本发明的气体阻隔性膜作为电子元件用的透明基板来使用的情况下,可以根据需要在气体阻隔性膜上设置透明导电膜、硬涂层。
(透明导电膜)
可以在本发明的电子元件用基板中使用的透明导电膜没有特别限定,可以根据元件构成来选择。例如,在作为透明电极来使用的情况下,优选为透过380~800nm的光的电极。作为材料,例如可以使用氧化铟锡(ITO)、SnO2、ZnO等的透明导电性金属氧化物;金、银、铂等的金属薄膜;金属纳米线、碳纳米管。另外,也可以使用选自聚吡咯、聚苯胺、聚噻吩、聚噻吩乙炔、聚甘菊环、聚异硫茚、聚咔唑、聚乙炔、聚亚苯基、聚亚苯基亚乙烯基、聚并苯、聚苯基乙炔、聚二乙炔及聚萘的各衍生物组成的组中的导电性高分子等。另外,也可以组合使用多种这些导电性化合物。
(硬涂层)
可以在本发明的电子元件用基板中使用的硬涂层没有特别限定,可以根据元件构成来选择。通过设置硬涂层,可以对基材赋予硬度、平滑性、透明性、耐热性。
作为可应用的硬涂层树脂,只要为通过固化而形成透明的树脂组合物的物质就可以没有特别限制地使用,例如可以举出硅树脂、环氧树脂、乙烯基酯树脂、丙烯酸系树脂、烯丙基酯系树脂等。特别优选的是,从可能的方面考虑,可使用丙烯酸系树脂。对固化方法而言,光、热均可,但从生产率的方面考虑,优选利用光、特别是UV光的固化。
实施例
以下,举出实施例对本发明具体地进行说明,但本发明并不限定于这些实施例。
需要说明的是,在实施例中,使用“%”、“份”的表示,但只要没有特别说明就表示“质量%”、“质量份”。
另外,在实施例中,取代度通过ASTM-D817-96中规定的方法算出,结晶度使用下述装置、由通过X射线衍射法而测得的衍射峰强度来算出。
X射线产生装置:理学电机制RINT TTR2
X射线源:CuKα
输出功率:50kV/300mA
第一狭缝:0.04mm
第二狭缝:0.03mm
光接收狭缝:0.1mm
<计数记录装置>
2θ/θ:连续扫描
测定范围:2θ=2~45°
取样:0.02°
累积时间:1.2秒钟。
[纤维素纳米纤维的制作]
(制造例1.纤维素纳米纤维A)
将由针叶树得到的亚硫酸漂白纸浆(纤维素纤维)添加到纯水以使得成为0.1质量%,使用石臼式粉碎机(ピュァファィンミルKMG1-10;栗田机械制作所公司制)进行50次磨碎处理(转数:1500转/分钟)而将纤维素纤维进行解纤。过滤该水分散液后,用纯水清洗,在70℃下使其干燥而得到纤维素纳米纤维A。
通过扫描型电子显微镜(SEM)观察,确认为得到的纤维素纳米纤维A被解纤为平均纤维直径32nm,进行了微原纤维化。
(制造例2.纤维素纳米纤维B)
在丙酸酐/吡啶(摩尔比1/1)溶液500质量份中添加上述制造例1中得到的纤维素纳米纤维A10质量份而使其分散,在室温下搅拌1小时。接着,过滤分散了的纤维素纳米纤维,用500质量份的水水洗3次后,用200质量份的乙醇清洗2次。进而,用500质量份的水进行2次水洗后,在70℃下使其干燥,得到用丙酰基取代了纤维素纳米纤维的羟基的氢原子的纤维素纳米纤维B。
通过扫描型电子显微镜(SEM)观察,确认为得到的纤维素纳米纤维B的平均纤维直径保持在32nm。
丙酰基的取代度为0.5,结晶度为89%。
(制造例3.纤维素纳米纤维C)
将使纤维素纳米纤维A溶解于丙酸酐/吡啶(摩尔比1/1)溶液中的溶液的搅拌时间变更为6小时,除此以外与制造例2同样地得到用丙酰基取代了纤维素纳米纤维的羟基的氢原子的纤维素纳米纤维C。
通过扫描型电子显微镜(SEM)观察,确认为得到的纤维素纳米纤维C的平均纤维直径保持在32nm。
丙酰基的取代度为2.0,结晶度为56%。
(制造例4.纤维素纳米纤维D)
使以干燥质量计相当于1g分量的纤维素纳米纤维A、0.0125g的TEMPO(2,2,6,6-四甲基哌啶-N-氧)及0.125g的溴化钠分散于水100ml中后,添加13质量%次氯酸钠水溶液(次氯酸钠的量达到2.5mmo1的量)来开始反应。反应中,滴加0.5M的氢氧化钠水溶液而将pH保持在10.5。将无法确认为pH变化的时刻看作反应结束。将反应物用玻璃过滤器过滤后,重复5次利用充分的量的水的水洗及过滤,进而,用超声波分散机进行1小时处理。在70℃下使其干燥而得到纤维素纳米纤维D。
扫描型电子显微镜(SEM)观察的结果,为纤维素纳米纤维D的平均纤维直径4nm。
(制造例5.纤维素纳米纤维E)
将纤维素纳米纤维A变更为纤维素纳米纤维D,除此以外与制造例2同样地得到用丙酰基取代了纤维素纳米纤维的羟基的氢原子的纤维素纳米纤维E。
通过扫描型电子显微镜(SEM)观察,确认为得到的纤维素纳米纤维E的平均纤维直径保持在4nm。
丙酰基的取代度为0.6,结晶度为88%。
(制造例6.纤维素纳米纤维F)
将纤维素纳米纤维A变更为纤维素纳米纤维D,除此以外与制造例3同样地得到用丙酰基取代了纤维素纳米纤维的羟基的氢原子的纤维素纳米纤维F。
通过扫描型电子显微镜(SEM)观察,确认为得到的纤维素纳米纤维F的平均纤维直径保持在4nm。
丙酰基的取代度为2.2,结晶度为52%。
(制造例7.纤维素纳米纤维G)
将丙酸酐变更为乙酸酐,除此以外与制造例2同样地得到用乙酰基取代了纤维素纳米纤维的羟基的氢原子的纤维素纳米纤维G。
通过扫描型电子显微镜(SEM)观察,确认为得到的纤维素纳米纤维G的平均纤维直径保持在32nm。
乙酰基的取代度为1.0,结晶度为82%。
(制造例8.纤维素纳米纤维H)
将丙酸酐变更为丁酸酐,除此以外与制造例2同样地得到用丁酰基取代了纤维素纳米纤维的羟基的氢原子的纤维素纳米纤维H。
通过扫描型电子显微镜(SEM)观察,确认为得到的纤维素纳米纤维H的平均纤维直径保持在32nm。
丁酰基的取代度为0.9,结晶度为84%。
对于上述制造例1~8中制作了的纤维素纳米纤维A、B、C、D、E、F、G、H,将制造方法、取代度、结晶度及平均纤维直径示于表1。

[膜基材的制作]
(熔融制膜方法)
(制膜例1.膜基材1)
1.熔融挤出
将上述制造例1中得到的纤维素纳米纤维A:100质量份通过(株)松井制作所制除湿热风式干燥机在热风温度150℃、露点-36℃下干燥后,与增塑剂P-1:8质量份、抗氧化剂A-1:1质量份、抗氧化剂A-2:0.5质量份一起在V型滚筒(タンブラ一)中混合30分钟。需要说明的是,作为增塑剂P-1、抗氧化剂A-1、A-2,使用下述物质。
增塑剂P-1:三羟甲基丙烷三苯甲酸酯
一次抗氧化剂A-1:IRGANOX-1010(BASFジャパン公司制)
二次抗氧化剂A-2:スミラィザ一GP(住友化学株式会社)
接着,将混合物以120kg/hr供给到双轴挤出机(テクノベル株式会社制)中。对于螺杆设计而言,减少捏合盘来抑制混炼发热。使筒的温度设定为200℃~250℃,在前端附近设置通风口,除去挥发成分。在挤出机下游配置过滤器、齿轮泵、过滤器,从衣架型T模头挤出,落在温度调节为20℃的2根镀铬镜面辊之间而回收,在3根辊间通过,切边后,卷绕于卷绕机。在挤出机内的纤维素纳米纤维组合物的滞留时间为1分30秒钟。调整挤出量和牵引辊的旋转速度以使得卷绕了的膜的厚度达到125μm。
2.压延处理
对得到的膜使用由利ロ一ル公司制辊压制装置实施压延处理。对压延处理而言,上部下部均使用金属辊,作为辊温度,设定为200℃,以线压力0.5吨、2m/min的运行速度进行。
3.拉伸处理
接着,将通过压延处理而得到了的膜预热后,通过辊速度差而在膜输送方向上拉伸(纵向拉伸),接着,导入拉幅机式拉伸机,在与膜输送方向正交的方向上拉伸(横向拉伸)。拉伸倍率设为纵向拉伸1.5倍、横向拉伸1.5倍。
通过上述工序,得到膜基材1。
(制膜例2~7.膜基材2~7)
将纤维素纳米纤维A变更为纤维素纳米纤维D、G、H、B、C或E,除此以外与制膜例1同样地得到膜基材2~7。
(制膜例8.膜基材8)
将纤维素纳米纤维A变更为纤维素纳米纤维E及纤维素纳米纤维F的混合物(E∶F的质量比=70∶30),除此以外与制膜例1同样地得到膜基材8。
(制膜例9.膜基材9)
通过从模头将熔融了的聚合物使用了供料头的同时挤出法,得到膜基材。即,以纤维素纳米纤维C/纤维素纳米纤维B/纤维素纳米纤维C地进行层叠,以符合各层的质量比的流量比而以与制膜例1~8相同的总送液量的在模头展开来实施挤出,由此制作从下层向上层具有纤维素纳米纤维C、纤维素纳米纤维B、及纤维素纳米纤维C的3层结构的由纤维素纳米纤维C/B/C形成的膜基材(各层的质量比=15∶70∶15)。
将纤维素纳米纤维A变更为上述纤维素纳米纤维C/B/C,除此以外与制膜例1同样地得到膜基材9。
(制膜例10.膜基材10)
将纤维素纳米纤维A:95质量份通过(株)松井制作所制除湿热风式干燥机在热风温度150℃、露点-36℃下干燥后,与作为基体树脂的纤维素乙酸酯丙酸酯(CAP)(乙酰基取代度=1.5、丙酰基取代度=1.2、数均分子量Mn=70000、重均分子量Mw=220000、Mw/Mn=3):5质量份、增塑剂P-1:8质量份、抗氧化剂A-1:1质量份、抗氧化剂A-2:0.5质量份一起在V型滚筒中混合30分钟。需要说明的是,增塑剂P-1、抗氧化剂A-1、A-2与上述比较例1中使用的相同。
使用上述混合物来进行熔融挤出、压延处理及拉伸处理,除此以外与制膜例1同样地得到膜基材10。
(制膜例11.膜基材11)
混合纤维素纳米纤维A:90质量份、作为基体树脂的纤维素乙酸酯丙酸酯(CAP):10质量份、增塑剂P-1:8质量份、抗氧化剂A-1:1质量份、抗氧化剂A-2:0.5质量份,除此以外与制膜例10同样地得到膜基材11。
(制膜例12.膜基材12)
混合纤维素纳米纤维A:85质量份、作为基体树脂的纤维素乙酸酯丙酸酯(CAP):15质量份、增塑剂P-1:8质量份、抗氧化剂A-1:1质量份、抗氧化剂A-2:0.5质量份,除此以外与制膜例10同样地得到膜基材12。
(制膜例13.膜基材13)
混合纤维素纳米纤维C:95质量份、作为基体树脂的纤维素乙酸酯丙酸酯(CAP):5质量份、增塑剂P-1:8质量份、抗氧化剂A-1:1质量份、抗氧化剂A-2:0.5质量份,除此以外与制膜例10同样地得到膜基材13。
(制膜例14.膜基材14)
混合纤维素纳米纤维C:90质量份、作为基体树脂的纤维素乙酸酯丙酸酯(CAP):10质量份、增塑剂P-1:8质量份、抗氧化剂A-1:1质量份、抗氧化剂A-2:0.5质量份,除此以外与制膜例10同样地得到膜基材14。
(制膜例15.膜基材15)
混合纤维素纳米纤维C:85质量份、作为基体树脂的纤维素乙酸酯丙酸酯(CAP):15质量份、增塑剂P-1:8质量份、抗氧化剂A-1:1质量份、抗氧化剂A-2:0.5质量份,除此以外与制膜例10同样地得到膜基材15。
(溶液浇铸制膜方法)
(制膜例16.膜基材16)
1.溶液浇铸
将纤维素纳米纤维A的乙醇溶液(固体成分10质量%)一边搅拌一边投入到密闭容器中,一边加热、搅拌一边混合30分钟,制备胶浆液。
接着,在胶浆液:840质量份中添加作为增塑剂的三苯基磷酸酯:10质量份、作为增塑剂的乙基邻苯二甲酰乙基乙醇酸酯:5质量份、作为良溶剂的二氯甲烷:140质量份及交联剂E-5:5质量份,在70℃下完全混合,冷却至进行流延的温度而静置一晚,实施脱泡操作,然后使用安积滤纸(株)制的安积滤纸No.244进行过滤,得到胶浆A。
将上述中调制了的胶浆A(温度:35℃)使用带流延装置在30℃的不锈钢带支撑体上均匀地流延。然后,使其干燥至可剥离的范围后,从不锈钢带支撑体上剥离网状物。此时的网状物的残留溶剂量为80质量%。
将上述得到的网状物在85℃的干燥区一边进行辊输送一边使其干燥,得到膜(膜厚:125μm)。卷绕时的残留溶剂量低于0.1质量%。
2.拉伸处理
将得到的膜在残留溶剂量低于35质量%时预热,然后通过辊速度差在膜输送方向上拉伸(纵向拉伸),接着,导入拉幅机式拉伸机,在与膜输送方向正交的方向上拉伸(横向拉伸)。拉伸倍率设为纵向拉伸1.5倍、横向拉伸1.5倍。
3.压延处理
对得到的膜使用由利ロ一ル公司制辊压制装置而实施压延处理。对压延处理而言,上部下部均使用金属辊,作为辊温度,设定为200℃,以线压力0.5吨、2m/min的运行速度进行。
通过上述工序,得到膜基材16。
(制膜例17~22.膜基材17~22)
将纤维素纳米纤维A变更为纤维素纳米纤维D、G、H、B、C或E,除此以外与制膜例16同样地得到膜基材17~22。
(制膜例23.膜基材23)
将纤维素纳米纤维A变更为纤维素纳米纤维E及纤维素纳米纤维F的混合物(E∶F的质量比=70∶30),除此以外与制膜例16同样地得到膜基材23。
(制膜例24.膜基材24)
由3系列的供给线以符合各层的质量比的流量比而以与制膜例16~23相同的总送液量进行送液,由此通过分割浇铸来制作了从下层向上层具有纤维素纳米纤维C、纤维素纳米纤维B、及纤维素纳米纤维C这3层结构的纤维素纳米纤维C/B/C的膜基材24(各层的质量比=15∶70∶15)。需要说明的是,对于分割浇铸而言,通过在金属支撑体上配置3处模涂机,进行制膜以使得达到表2的层构成的组成、膜厚比来实施。需要说明的是,上述以外的制膜条件与制膜例16相同。
(制膜例25.膜基材25)
使用纤维素纳米纤维A:95质量份及作为基体树脂的纤维素乙酸酯丙酸酯(CAP)(乙酰基取代度=1.5、丙酰基取代度1.2、数均分子量Mn=70000、重均分子量Mw=220000、Mw/Mn=3):5质量份的乙醇溶液(固体成分10质量%)代替纤维素纳米纤维A的乙醇溶液(固体成分10质量%),除此以外与制膜例16同样地得到膜基材25。
(制膜例26.膜基材26)
使用纤维素纳米纤维A:90质量份及作为基体树脂的纤维素乙酸酯丙酸酯(CAP)(乙酰基取代度=1.5、丙酰基取代度1.2、数均分子量Mn=70000、重均分子量Mw=220000、Mw/Mn=3):10质量份的乙醇溶液(固体成分10质量%)代替纤维素纳米纤维A的乙醇溶液(固体成分10质量%),除此以外与制膜例16同样地得到膜基材26。
(制膜例27.膜基材27)
使用纤维素纳米纤维A:80质量份及作为基体树脂的纤维素乙酸酯丙酸酯(CAP)(乙酰基取代度=1.5、丙酰基取代度1.2、数均分子量Mn=70000、重均分子量Mw=220000、Mw/Mn=3):20质量份的乙醇溶液(固体成分10质量%)代替纤维素纳米纤维A的乙醇溶液(固体成分10质量%),除此以外,与制膜例16同样地得到膜基材27。
(制膜例28.膜基材28)
使用纤维素纳米纤维C:95质量份及作为基体树脂的纤维素乙酸酯丙酸酯(CAP)(乙酰基取代度=1.5、丙酰基取代度1.2、数均分子量Mn=70000、重均分子量Mw=220000、Mw/Mn=3):5质量份的乙醇溶液(固体成分10质量%)代替纤维素纳米纤维A的乙醇溶液(固体成分10质量%),除此以外,与制膜例16同样地得到膜基材28。
(制膜例29.膜基材29)
使用纤维素纳米纤维C:90质量份及作为基体树脂的纤维素乙酸酯丙酸酯(CAP)(乙酰基取代度=1.5、丙酰基取代度1.2、数均分子量Mn=70000、重均分子量Mw=220000、Mw/Mn=3):10质量份的乙醇溶液(固体成分10质量%)代替纤维素纳米纤维A的乙醇溶液(固体成分10质量%),除此以外与制膜例16同样地得到膜基材29。
(制膜例30.膜基材30)
使用纤维素纳米纤维C:85质量份及作为基体树脂的纤维素乙酸酯丙酸酯(CAP)(乙酰基取代度=1.5、丙酰基取代度1.2、数均分子量Mn=70000、重均分子量Mw=220000、Mw/Mn=3):15质量份的乙醇溶液(固体成分10质量%)代替纤维素纳米纤维A的乙醇溶液(固体成分10质量%),除此以外与制膜例16同样地得到膜基材30。
将上述制膜例1~30中制作了的膜基材1~30的构成及制造方法示于表2。
[表2]

1)C/B/C:从中心向外侧具有纤维素纳米纤维C、纤维素纳米纤维B及纤维素纳米纤维C的3层结构。
2)表示CAP和纤维素纳米纤维A或C的含有比率(质量比)。
[气体阻隔性膜的制作]
(中间层的形成)
将膜基材1~30一边以30m/分钟的速度进行输送一边通过以下的形成方法在表面侧形成中间层1、在背面侧形成中间层2,得到膜层叠体1~30。
(中间层1)
在膜基材的单面用线棒涂布JSR株式会社制UV固化型有机/无机混合硬涂层材料OPSTAR Z7535以使得干燥后的平均膜厚达到4μm。然后,在干燥条件(80℃、3分钟)下使其干燥后,在1.0J/cm2的固化条件下、在空气气氛下使用高压水银灯进行固化,形成中间层1。
(中间层2)
在膜基材的相反面用线棒涂布JSR株式会社制UV固化型有机/无机混合硬涂层材料OPSTAR Z7501以使得干燥后的平均膜厚达到4μm。然后,在干燥条件(80℃、3分钟)下使其干燥后,在1.0J/cm2的固化条件下、在空气气氛下使用高压水银灯进行固化,形成中间层2。
中间层2的最大剖面高度Rt(p)为8nm。
(气体阻隔层的形成)
A.熔融挤出膜
(聚硅氮烷膜的准分子照射)
(比较例1.气体阻隔性膜1)
1.涂布工序
调制全氢聚硅氮烷(PHPS;AZェレクトロニックマテリァルズ(株)制ァクァミカNN320)的20质量%二丁醚溶液作为含聚硅氮烷涂布液。
在设有上述中间层1及中间层2的膜层叠体1的两面用无线棒进行涂布以使得干燥后的平均膜厚达到0.30μm。
2.除湿工序
使得到的涂膜在温度85℃、湿度55%RH的气氛(露点:70℃)下干燥1分钟,得到干燥试样(第一除湿工序)。
将上述干燥试样进一步在温度25℃、湿度10%RH(露点:-8℃)的气氛下保持10分钟,进行除湿处理(第二除湿工序)。
3.改性工序
将进行了除湿处理的试样固定在下述改性处理装置的操作台上,在以下的条件下进行改性处理,得到气体阻隔性膜1。改性处理时的露点为-8℃。
(改性处理装置)
株式会社ェム·ディ·コム制准分子照射装置MODEL:MECL-M-1-200、波长172nm、灯封入气体Xe
(改性处理条件)
准分子光强度:130mW/cm2(172nm)
试样与光源的距离:1mm
台加热温度:70℃
照射装置内的氧浓度:1%
准分子照射时间:3秒钟。
(比较例2、实施例1~7、比较例3~5、实施例8~9、比较例6.气体阻隔性膜2~15)
将设有中间层1及中间层2的膜层叠体1变更为设有中间层1及中间层2的膜层叠体2~15,除此以外与比较例1同样地得到气体阻隔性膜2~15。
(比较例7、实施例10.气体阻隔性膜16~17)
将设有中间层1及中间层2的膜层叠体1变更为未设有中间层1及中间层2的膜基材1或膜基材6,除此以外与比较例1同样地得到气体阻隔性膜16~17。
(比较例8.气体阻隔性膜18)
将改性工序中的改性处理条件的准分子光强度130mW/cm2(172nm)变更为180mW/cm2(172nm),除此以外与比较例1同样地得到气体阻隔性膜18。
(实施例11.气体阻隔性膜19)
将设有中间层1及中间层2的膜层叠体1变更为设有中间层1及中间层2的膜层叠体6,除此以外与比较例8同样地得到气体阻隔性膜19。
(比较例9.气体阻隔性膜20)
将改性工序中的改性处理条件的准分子光强度130mW/cm2(172nm)变更为80mW/cm2(172nm),除此以外与比较例1同样地得到气体阻隔性膜20。
(实施例12.气体阻隔性膜21)
将设有中间层1及中间层2的膜层叠体1变更为设有中间层1及中间层2的膜层叠体6,除此以外与比较例9同样地得到气体阻隔性膜21。
(比较例10、实施例13.气体阻隔性膜22~23)
将设有中间层1及中间层2的膜层叠体1变更为未设有中间层1及中间层2的膜基材1或膜基材6,除此以外与比较例9同样地得到气体阻隔性膜22~23。
(SiOx的等离子体溅射)
(比较例11.气体阻隔性膜24)
在未设有中间层1及中间层2的膜基材1的两面,使用等离子体产生溅射辊涂装置,通过DC磁电管溅射,使用Si作为靶,在成膜温度180℃下通过导入了氩气及氧气作为工艺气体的反应性溅射,形成膜厚70nm的SiOx(x=1.8,通过XPS)的气体阻隔层,得到气体阻隔性膜24。此时,气体阻隔层的膜厚,通过反应时间来调整。
(实施例14.气体阻隔性膜25)
将未设有中间层1及中间层2的膜基材1变更为未设有中间层1及中间层2的膜基材6,除此以外与比较例11同样地得到气体阻隔性膜25。
B.溶液浇铸膜
(聚硅氮烷膜的准分子照射)
(比较例11.气体阻隔性膜26)
1.涂布工序
调制全氢聚硅氮烷(PHPS;AZェレクトロニツクマテリァルズ(株)制ァクァミカNN320)的20质量%二丁醚溶液作为含聚硅氮烷涂布液。
在设有上述中间层1及中间层2的膜层叠体16的两面用无线棒进行涂布以使得干燥后的平均膜厚达到0.30μm。
2.干燥工序
使得到的涂膜在温度85℃、湿度55%RH的气氛下干燥1分钟,得到干燥试样。
3.除湿工序
将上述干燥试样进一步在温度25℃、湿度10%RH(露点:-8℃)的气氛下保持10分钟,进行除湿处理。
4.改性工序
将进行了除湿处理的试样固定在下述改性处理装置的操作台上,在以下的条件下进行改性处理,得到气体阻隔性膜26。改性处理时的露点为-8℃。
(改性处理装置)
株式会社ェム.ディ.コム制准分子照射装置MODEL:MECL-M-1-200、波长172nm、灯封入气体Xe
(改性处理条件)
准分子光强度:130mW/cm2(172nm)
试样与光源的距离:1mm
台加热温度:70℃
照射装置内的氧浓度:1%
准分子照射时间:3秒钟。
(比较例13、实施例15~21、比较例14~16、实施例22~23、比较例17.气体阻隔性膜27~40)
将设有中间层1及中间层2的膜层叠体16变更为设有中间层1及中间层2的膜层叠体17~30,除此以外与比较例12同样地得到气体阻隔性膜27~40。
(比较例18、实施例24.气体阻隔性膜41~42)
将设有中间层1及中间层2的膜层叠体16变更为未设有中间层1及中间层2的膜基材16或膜基材21,除此以外与比较例12同样地得到气体阻隔性膜41~42。
(比较例19.气体阻隔性膜43)
将改性工序中的改性处理条件的准分子光强度130mW/cm2(172nm)变更为180mW/cm2(172nm),除此以外与比较例12同样地得到气体阻隔性膜43。
(实施例25.气体阻隔性膜44)
将设有中间层1及中间层2的膜层叠体16变更为设有中间层1及中间层2的膜层叠体21,除此以外与比较例19同样地得到气体阻隔性膜44。
(比较例20.气体阻隔性膜45)
将改性工序中的改性处理条件的准分子光强度130mW/cm2(172nm)变更为80mW/cm2(172nm),除此以外与比较例12同样地得到气体阻隔性膜45。
(实施例26.气体阻隔性膜46)
将设有中间层1及中间层2的膜层叠体16变更为设有中间层1及中间层2的膜层叠体21,除此以外与比较例20同样地得到气体阻隔性膜46。
(比较例21、实施例27.气体阻隔性膜47~48)
将设有中间层1及中间层2的膜层叠体16变更为未设有中间层1及中间层2的膜基材16或膜基材21,除此以外与比较例20同样地得到气体阻隔性膜47~48。
(SiOx的等离子体溅射)
(比较例22.气体阻隔性膜49)
在未设有中间层1及中间层2的膜基材16的两面,使用等离子体产生溅射辊涂装置,通过DC磁电管溅射,使用Si作为靶,在成膜温度180℃下通过导入了氩气及氧气作为工艺气体的反应性溅射,形成膜厚70nm的SiOx(x=1.8,通过XPS)的气体阻隔层,得到气体阻隔性膜49。此时,气体阻隔层的膜厚,根据反应时间调整。
(实施例28.气体阻隔性膜50)
将未设有中间层1及中间层2的膜基材16变更为未设有中间层1及中间层2的膜基材21,除此以外与比较例22同样地得到气体阻隔性膜50。
将上述比较例1~22、实施例1~28中制作了的气体阻隔性膜1~50的构成及制造方法示于表3及表4。
[评价]
对气体阻隔性膜1~50的水蒸气透过性(水蒸气阻隔评价)、表面粗糙度(表面平滑性评价)、透明性、弯曲特性、切断加工性、保存性用以下的方法来评价。
(水蒸气透过性)
1.水蒸气阻隔性评价用单元的制作
在气体阻隔性膜1~50的气体阻隔层的单面使用真空蒸镀装置(日本电子(株)制真空蒸镀装置JEE-400)而蒸镀作为透明导电膜的金属钙(粒状)。此时,将使透明导电膜蒸镀的部分(9处12mm×12mm)以外的部分进行掩模而蒸镀。需要说明的是,钙为与水分反应而发生腐蚀的金属。
然后,在真空状态下解除掩模,由另一个金属蒸镀源在气体阻隔性膜1~44的另一单面的整个面使作为水蒸气不透过性的金属的铝(φ3~5mm、粒状)蒸镀。
铝密封后,解除真空状态,在干燥氮气气氛下迅速地使厚度0.2mm的石英玻璃经由密封用紫外线固化树脂(ナガセケムテックス制)与铝密封侧相对,照射紫外线,由此制作评价用单元。
2.透过水分量的测定
将得到的密封了两面的评价用单元使用恒温恒湿度烘箱(Yama toHumidic Chamber IG47M)在60℃、90%RH的高温高湿下保存,基于特开2005-283561号公报中记载的方法由金属钙的腐蚀量计算透过到单元内的水分量。
需要说明的是,为了确认没有从阻隔膜面透过以外的水蒸气,作为比较试样,将在厚度0.2mm的石英玻璃板上蒸镀了金属钙的试样代替气体阻隔性膜而与上述同样地使用恒温恒湿度烘箱(Yamato HumidicChamber IG47M)在60℃、90%RH的高温高湿下保存,确认为即使经过1000小时后也不产生金属钙腐蚀。
将得到的透过水分量分类为以下的5个等级。
5:低于1×10-4g/m2/day
4:1×10-4g/m2/day以上、低于1×10-3g/m2/day
3:1×10-3g/m2/day以上、低于1×10-2g/m2/day
2:1×10-2g/m2/day以上、低于1×10-1g/m2/day
1:1×10-1g/m2/day以上
将结果示于表3及表4。
(表面粗糙度Ra:表面平滑性)
表面粗糙度Ra,可使用原子力显微镜(AFM;Digita1 Instruments公司制DI3100)由用具有极小的前端半径的触针的检测器而连续测得的凹凸的剖面曲线算出,通过极小的前端半径的触针在测定方向为30μm的区间内进行多次测定,由涉及微细的凹凸的振幅的平均的粗糙度而求出。
将结果示于表3及表4。
(透明性:雾度值)
作为透明性的尺度,使用雾度计(日本电色工业公司制、NDH2000)来测定雾度值(%)。
将结果示于表3及表4。
(弯曲特性)
对于气体阻隔性膜1~50,在180度的角度下重复弯曲100次以使得达到半径10mm的曲率。
使用弯曲后的气体阻隔性膜1~50,用与上述同样的方法制作水蒸气阻隔性评价用单元,进行水蒸气透过率的评价。
算出弯曲后的气体阻隔性膜的水蒸气透过度相对于弯曲前的气体阻隔性膜的水蒸气透过度的比例(弯曲后的水蒸气透过度/弯曲前的水蒸气透过度×100(%)),评价由弯曲导致的劣化程度。
弯曲后的水蒸气透过度/弯曲前的水蒸气透过度×100(%)
○:85%以上
△:低于60%
×:低于30%
将结果示于表3及表4。
(切断加工性)
在将气体阻隔性膜1~50使用圆盘式切刀DC-230(CADL公司)而切断为B5尺寸时,评价在切断了的端部产生的破裂。
○:没有产生破裂
△:产生5条以下的破裂
×:产生5条以上的破裂。
(粘接性)
对于气体阻隔性膜1~50,在100℃的烘箱中实施5小时加热处理。
在该加热处理前后,依据基于JIS K5400的棋盘格试验,使用间隙间隔2mm的刀具导承(カッタ一ガィド)棋盘格状地切成刀伤,使用胶带进行180°剥离,测定膜的残留率(%),将其作为粘接性来评价。
将结果示于表3及表4。

由表3及表4所示的结果可确认:在含有本发明涉及的纤维素纳米纤维的表面的纤维素的羟基的氢原子的至少一部分被酰基取代了的表面改性纤维素纳米纤维、实质上不含基体树脂的片状基材上形成了气体阻隔层的实施例的气体阻隔性膜的透明性、平滑性(表面粗糙度Ra)、气体阻隔性(水蒸气透过性)、粘接性、弯曲特性、切断加工性优异。特别是实施例的气体阻隔性膜,即使在被热处理了的情况下也可维持良好的粘接性。
通过聚硅氮烷化合物的涂布膜的准分子照射而形成了气体阻隔层的实施例的气体阻隔性膜,与通过利用等离子体的反应性溅射而形成了气体阻隔层的实施例14及28的气体阻隔性膜(No.25、50)相比,气体阻隔性及切断加工性显著提高。
将纤维素纳米纤维用丙酰基取代了的气体阻隔性膜,与用乙酰基或丁酰基取代了的情况(实施例1、2、15、16)相比,平滑性及透明性有意地提高。
配置了中间层的情况(实施例4、12、18、26)与未配置中间层的情况(实施例10、13、24、27)相比,可知气体阻隔性提高。
与此相对,使用了非取代的纤维素纳米纤维的比较例的气体阻隔性膜与实施例的气体阻隔性膜相比,在透明性、平滑性(表面粗糙度Ra)、气体阻隔性(水蒸气透过性)、保存性(粘接性)的方面差。特别是基体树脂的含量多的比较例5及比较例16的气体阻隔性膜(No.12、37)的平滑性、保存性有意地恶化。
符号的说明
1 片状基材、
2a、2b 中间层
3a、3b 气体阻隔层
10 气体阻隔性膜。
Claims (7)
1.一种气体阻隔性膜,其具有片状基材和在所述片状基材的至少单面形成了的气体阻隔层,所述片状基材为:含有纤维素纳米纤维的羟基的氢原子的至少一部分被碳数1~8的酰基取代了的表面改性纤维素纳米纤维、基体树脂的含量相对于所述纤维素纳米纤维和所述基体树脂的总量为10质量%以下。
2.如权利要求1所述的气体阻隔性膜,其中,所述酰基包含丙酰基。
3.如权利要求1或2所述的气体阻隔性膜,其中,所述气体阻隔层含有硅氧化物、氮化硅氧化物、及硅氧化氮化物的至少一种。
4.一种气体阻隔性膜的制造方法,其具有以下工序:
将纤维素纳米纤维的羟基的氢原子的至少一部分用碳数1~8的酰基进行取代而得到表面改性纤维素纳米纤维、将所述表面改性纤维素纳米纤维用熔融挤出法或溶液浇铸法进行制膜而得到片状基材的工序A;和
在所述片状基材上形成气体阻隔层的工序B。
5.如权利要求4所述的制造方法,其中,在所述工序A中,在制膜后进行拉伸处理或/及加热压延处理。
6.如权利要求4或5所述的制造方法,其中,所述工序B包含:在所述片状基材上涂布含有聚硅氮烷化合物的涂布液后、进行准分子照射处理。
7.一种电子元件用基板,其使用了权利要求1~3的任一项所述的气体阻隔性膜或通过权利要求4~6的任一项所述的制造方法而制造的气体阻隔性膜。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011189749 | 2011-08-31 | ||
| JP2011-189749 | 2011-08-31 | ||
| PCT/JP2012/071454 WO2013031687A1 (ja) | 2011-08-31 | 2012-08-24 | ガスバリア性フィルムおよびその製造方法、ならびにこれを用いた電子素子用基板 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN103796830A true CN103796830A (zh) | 2014-05-14 |
| CN103796830B CN103796830B (zh) | 2016-04-27 |
Family
ID=47756177
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201280042683.6A Expired - Fee Related CN103796830B (zh) | 2011-08-31 | 2012-08-24 | 气体阻隔性膜及其制造方法、以及使用了其的电子元件用基板 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20140234640A1 (zh) |
| JP (1) | JP5942995B2 (zh) |
| CN (1) | CN103796830B (zh) |
| WO (1) | WO2013031687A1 (zh) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107662385A (zh) * | 2017-10-12 | 2018-02-06 | 丹阳市维尼光学有限公司 | 双层阻隔膜 |
| CN108350198A (zh) * | 2015-12-31 | 2018-07-31 | 芬兰国家技术研究中心股份公司 | 由高稠度酶原纤化纳米纤维生产薄膜的方法 |
| CN109071063A (zh) * | 2016-04-14 | 2018-12-21 | 凸版印刷株式会社 | 纸杯、酸性食品用纸杯 |
| CN109195793A (zh) * | 2016-05-13 | 2019-01-11 | 株式会社雷尼阿斯 | 树脂玻璃板及其制造方法 |
| CN109844047A (zh) * | 2016-11-10 | 2019-06-04 | 琳得科株式会社 | 阻气性层合片、阻气性层合片的制造方法和电子部件或光学部件 |
| CN110446722A (zh) * | 2017-01-16 | 2019-11-12 | 株式会社Kri | 硫酸酯化修饰纤维素纳米纤维和纤维素纳米纤维的制造方法 |
| CN111093974A (zh) * | 2017-09-13 | 2020-05-01 | 住友化学株式会社 | 阻气性膜和柔性电子设备 |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FI123630B (fi) | 2011-10-24 | 2013-08-30 | Teknologian Tutkimuskeskus Vtt | Menetelmä NFC-kalvojen valmistamiseksi alustoille |
| WO2014098275A1 (ko) * | 2012-12-17 | 2014-06-26 | 코오롱글로텍주식회사 | 플렉서블 디스플레이를 위한 평탄화 섬유기판의 제조방법 |
| EP3025857B1 (en) * | 2013-07-25 | 2018-06-06 | Toppan Printing Co., Ltd. | Sheet material and barrier packaging container using same, and process for manufacturing sheet material |
| KR20160145608A (ko) * | 2014-04-22 | 2016-12-20 | 오지 홀딩스 가부시키가이샤 | 복합체 및 그 제조 방법 |
| JP6691750B2 (ja) * | 2014-07-23 | 2020-05-13 | 日信工業株式会社 | 熱可塑性樹脂組成物の製造方法及び熱可塑性樹脂組成物 |
| FI126761B (en) | 2014-11-28 | 2017-05-15 | Teknologian Tutkimuskeskus Vtt Oy | A method for improving the water resistance of bio-based CNF films |
| JP6117828B2 (ja) * | 2015-01-08 | 2017-04-19 | デクセリアルズ株式会社 | 無機偏光板 |
| CN108603343A (zh) * | 2015-11-30 | 2018-09-28 | 王子控股株式会社 | 片材及片材的制造方法 |
| JP6896997B2 (ja) * | 2016-02-02 | 2021-06-30 | 富士フイルムビジネスイノベーション株式会社 | 樹脂組成物、樹脂成形体、及び樹脂組成物の製造方法 |
| PL3449056T3 (pl) * | 2016-04-29 | 2022-04-04 | Stora Enso Oyj | Folia zawierająca mikrofibrylowaną celulozę i wykonane z niej produkty |
| JP6984649B2 (ja) * | 2017-03-17 | 2021-12-22 | コニカミノルタ株式会社 | 電子デバイス作製用組成物、電子デバイス作製用組成物の製造方法、有機薄膜及び有機薄膜の製造方法 |
| JP2019178216A (ja) * | 2018-03-30 | 2019-10-17 | 第一工業製薬株式会社 | 微細繊維状セルロースおよびその樹脂組成物 |
| WO2022030014A1 (ja) * | 2020-08-07 | 2022-02-10 | 株式会社ダイセル | セルロースアセテート樹脂組成物 |
| KR20220041323A (ko) * | 2020-09-25 | 2022-04-01 | 현대자동차주식회사 | 셀룰로오스 나노섬유 필름 및 이의 제조방법 |
| JP7040592B2 (ja) * | 2020-12-25 | 2022-03-23 | 王子ホールディングス株式会社 | 微細繊維を含有する基材シート層と無機層とを含む、耐湿性複合シート |
| KR20230146284A (ko) * | 2022-04-12 | 2023-10-19 | 현대자동차주식회사 | 투명도 및 강성이 뛰어난 복합 시트의 제조방법 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008010462A1 (fr) * | 2006-07-19 | 2008-01-24 | Pioneer Corporation | feuille de nanofibre, son processus de fabrication, et matériau composite renforcé de fibre |
| CN101772516A (zh) * | 2007-08-07 | 2010-07-07 | 花王株式会社 | 阻气用材料 |
| JP2011068709A (ja) * | 2009-09-24 | 2011-04-07 | Konica Minolta Opto Inc | 光学フィルム、それを用いた透明ガスバリアフィルム、および光学フィルムの製造方法 |
| JP2011148914A (ja) * | 2010-01-22 | 2011-08-04 | Konica Minolta Holdings Inc | 繊維複合材料、光学フィルム、光学フィルムの製造方法、それを用いた偏光板および液晶表示装置 |
| JP2011152693A (ja) * | 2010-01-27 | 2011-08-11 | Konica Minolta Opto Inc | 樹脂フィルム基板とその製造方法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3907650B2 (ja) * | 2004-09-16 | 2007-04-18 | 大王製紙株式会社 | クラフト包装紙 |
| JP5386866B2 (ja) * | 2008-06-30 | 2014-01-15 | 国立大学法人京都大学 | ナノファイバーシート |
| EP2441885B1 (en) * | 2009-06-12 | 2013-11-27 | Mitsubishi Chemical Corporation | Modified cellulose fiber and cellulose composite thereof |
| JP5589354B2 (ja) * | 2009-11-09 | 2014-09-17 | 住友ベークライト株式会社 | セルロース繊維、成形体および表示素子用基板 |
| JP2011143551A (ja) * | 2010-01-12 | 2011-07-28 | Konica Minolta Holdings Inc | ガスバリア性フィルム、ガスバリア性フィルムの製造方法及び有機光電変換素子 |
| JP5691175B2 (ja) * | 2010-01-13 | 2015-04-01 | コニカミノルタ株式会社 | ガスバリアフィルムの製造方法、ガスバリアフィルム及び有機光電変換素子 |
-
2012
- 2012-08-24 WO PCT/JP2012/071454 patent/WO2013031687A1/ja active Application Filing
- 2012-08-24 JP JP2013531282A patent/JP5942995B2/ja active Active
- 2012-08-24 US US14/241,201 patent/US20140234640A1/en not_active Abandoned
- 2012-08-24 CN CN201280042683.6A patent/CN103796830B/zh not_active Expired - Fee Related
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008010462A1 (fr) * | 2006-07-19 | 2008-01-24 | Pioneer Corporation | feuille de nanofibre, son processus de fabrication, et matériau composite renforcé de fibre |
| CN101772516A (zh) * | 2007-08-07 | 2010-07-07 | 花王株式会社 | 阻气用材料 |
| JP2011068709A (ja) * | 2009-09-24 | 2011-04-07 | Konica Minolta Opto Inc | 光学フィルム、それを用いた透明ガスバリアフィルム、および光学フィルムの製造方法 |
| JP2011148914A (ja) * | 2010-01-22 | 2011-08-04 | Konica Minolta Holdings Inc | 繊維複合材料、光学フィルム、光学フィルムの製造方法、それを用いた偏光板および液晶表示装置 |
| JP2011152693A (ja) * | 2010-01-27 | 2011-08-11 | Konica Minolta Opto Inc | 樹脂フィルム基板とその製造方法 |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108350198A (zh) * | 2015-12-31 | 2018-07-31 | 芬兰国家技术研究中心股份公司 | 由高稠度酶原纤化纳米纤维生产薄膜的方法 |
| CN109071063A (zh) * | 2016-04-14 | 2018-12-21 | 凸版印刷株式会社 | 纸杯、酸性食品用纸杯 |
| CN109195793A (zh) * | 2016-05-13 | 2019-01-11 | 株式会社雷尼阿斯 | 树脂玻璃板及其制造方法 |
| CN109195793B (zh) * | 2016-05-13 | 2020-11-27 | 株式会社雷尼阿斯 | 树脂玻璃板及其制造方法 |
| CN109844047A (zh) * | 2016-11-10 | 2019-06-04 | 琳得科株式会社 | 阻气性层合片、阻气性层合片的制造方法和电子部件或光学部件 |
| CN109844047B (zh) * | 2016-11-10 | 2021-11-23 | 琳得科株式会社 | 阻气性层合片、阻气性层合片的制造方法和电子部件或光学部件 |
| US11535682B2 (en) | 2017-01-16 | 2022-12-27 | Yokogawa Electric Corporation | Sulfate ester modified cellulose nanofibers and method for producing cellulose nanofibers |
| CN110446722A (zh) * | 2017-01-16 | 2019-11-12 | 株式会社Kri | 硫酸酯化修饰纤维素纳米纤维和纤维素纳米纤维的制造方法 |
| US12084525B2 (en) | 2017-01-16 | 2024-09-10 | Yokogawa Electric Corporation | Sulfate ester modified cellulose nanofibers and method for producing cellulose nanofibers |
| CN110446722B (zh) * | 2017-01-16 | 2024-06-11 | 横河电机株式会社 | 硫酸酯化修饰纤维素纳米纤维和纤维素纳米纤维的制造方法 |
| CN111093974A (zh) * | 2017-09-13 | 2020-05-01 | 住友化学株式会社 | 阻气性膜和柔性电子设备 |
| CN111093974B (zh) * | 2017-09-13 | 2022-09-13 | 住友化学株式会社 | 阻气性膜和柔性电子设备 |
| CN107662385A (zh) * | 2017-10-12 | 2018-02-06 | 丹阳市维尼光学有限公司 | 双层阻隔膜 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2013031687A1 (ja) | 2013-03-07 |
| JPWO2013031687A1 (ja) | 2015-03-23 |
| JP5942995B2 (ja) | 2016-06-29 |
| US20140234640A1 (en) | 2014-08-21 |
| CN103796830B (zh) | 2016-04-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN103796830B (zh) | 气体阻隔性膜及其制造方法、以及使用了其的电子元件用基板 | |
| CN101680989B (zh) | 纤维素酯光学膜、使用该纤维素酯光学膜的偏振片及液晶显示装置、纤维素酯光学膜的制造方法 | |
| CN101300121B (zh) | 纤维素类树脂膜、纤维素类树脂膜的制造方法、防反射膜、偏振片及液晶显示装置 | |
| JP4906308B2 (ja) | ポリビニルアルコール系フィルム、およびその製造方法 | |
| CN104508544B (zh) | 液晶显示装置 | |
| CN101277997B (zh) | 纤维素酯膜的制造方法、纤维素酯膜、偏振片和液晶显示装置 | |
| KR101471691B1 (ko) | 셀룰로오스 에스테르 필름, 셀룰로오스 에스테르 필름의 제조 방법, 그것을 사용한 편광판 보호 필름, 편광판 및 액정 표시 장치 | |
| JP5699862B2 (ja) | 光学フィルムの製造方法及び該光学フィルムを用いた素子用基板 | |
| CN106537310B (zh) | 导电性薄膜、偏振片及带触摸面板的显示装置 | |
| US20070009674A1 (en) | Transparent film for display substrate, display substrate using the film and method of manufacturing the same, liquid crystal display, organic electroluminescence display, and touch panel | |
| CN105672023A (zh) | 纤维素纤维集合体及其制造方法、原纤化纤维素纤维及其制造方法、以及纤维素纤维复合体 | |
| JP5929916B2 (ja) | 光学フィルム、その製造方法及び該光学フィルムを用いた素子用基板 | |
| KR20080009309A (ko) | 셀룰로오스 아실레이트막의 제조 방법 및 장치, 그리고셀룰로오스 아실레이트막 | |
| CN104220965A (zh) | 带触摸面板的液晶显示装置 | |
| WO2006068221A1 (ja) | セルロースアシレートフィルムおよびその製造方法 | |
| WO2007138910A1 (ja) | 偏光板保護フィルム及びその製造方法、偏光板及びその製造方法、液晶表示装置 | |
| JP2011144363A (ja) | セルロース繊維複合体及びその製造方法 | |
| CN109804282A (zh) | 偏振片和液晶显示装置 | |
| CN102190806A (zh) | 纤维素乙酸酯膜及其制备方法、偏振片和液晶显示装置 | |
| JP2013064029A (ja) | ナノファイバーフィルム、およびこれを用いた電子素子用樹脂基板 | |
| CN109476119A (zh) | 光学层叠体的制造方法、以及光学层叠体中间体 | |
| CN101490585A (zh) | 光学薄膜、其制造方法、偏光板及液晶显示装置 | |
| KR20030094104A (ko) | 용액제막방법, 셀룰로오스에스테르필름, 보호필름 및 편광판 | |
| JP2011068709A (ja) | 光学フィルム、それを用いた透明ガスバリアフィルム、および光学フィルムの製造方法 | |
| JP2010194790A (ja) | フィルム基板 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20160427 |