BRPI0921097B1 - composto ou seu sal farmaceuticamente aceitável, intermediário do composto, composição farmacêutica e uso do composto - Google Patents
composto ou seu sal farmaceuticamente aceitável, intermediário do composto, composição farmacêutica e uso do composto Download PDFInfo
- Publication number
- BRPI0921097B1 BRPI0921097B1 BRPI0921097-0A BRPI0921097A BRPI0921097B1 BR PI0921097 B1 BRPI0921097 B1 BR PI0921097B1 BR PI0921097 A BRPI0921097 A BR PI0921097A BR PI0921097 B1 BRPI0921097 B1 BR PI0921097B1
- Authority
- BR
- Brazil
- Prior art keywords
- methyl
- ethyl
- carboxamide
- pyrazol
- compound
- Prior art date
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/4155—1,2-Diazoles non condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/10—Laxatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/12—Antidiarrhoeals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Pain & Pain Management (AREA)
- Pulmonology (AREA)
- Virology (AREA)
- Urology & Nephrology (AREA)
- Oncology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Dermatology (AREA)
- Communicable Diseases (AREA)
- Rheumatology (AREA)
- Psychiatry (AREA)
- Gastroenterology & Hepatology (AREA)
- Molecular Biology (AREA)
- Obesity (AREA)
- Vascular Medicine (AREA)
- Hematology (AREA)
- Diabetes (AREA)
- Hospice & Palliative Care (AREA)
- Child & Adolescent Psychology (AREA)
- Physical Education & Sports Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
Abstract
COMPOSTO DERIVADO DE PIRAZOL-3CARBOXAMIDA COM ATIVIDADE DE ANTAGONISTA DE RECEPTOR 5-HT2B, ,AGENTE PREVENTIVO OU TERAPÊUTICO, COMPOSIÇÃO FARMACÊUTICA E USO DO COMPOSTO OU SEU SAL FARMACEUTICAMENTE ACEITÁVEL E MÉTODO OU PREVENÇÃO OU TRATAMENTO É revelado um composto representado por fórmula geral (I) ou um seu sal farmaceuticamente aceitável, que é útil como um antagonista seletivo de um receptor 5-HT2B. O composto e seu sal úteis para o tratamento ou prevenção de várias doenças e condições associadas com um receptor 5-HT2B,
Description
A invenção refere-se aos derivados de pirazol-3-carboxamida novos. Os compostos desta invenção são compostos úteis como antagonistas de receptor 5-HT2B e são úteis para prevenção ou tratamento de várias doenças relacionadas a este receptor. A presente invenção também se refere a uma composição farmacêutica que compreende os derivados acima.
A serotonina (5-hidroxitriptamina), que foi descoberta a primeira vez em 1948, é um dos neurotransmissores e é um dos derivados da triptamina, que são distribuídos com alta concentração para a área do hipotálamo, gânglio basal, núcleo da rafe medula e assim por diante. A serotonina é uma substância química contida em animais, incluindo humanos e é biosintetizada a partir de triptofano. Cerca de 10 mg de serotonina são encontrados em humanos e a maior parte dela é distribuída para a célula cromafina na mucosa do intestino delgado. A serotonina sintetizada aqui atua no músculo como o intestino e refere-se altamente com a motilidade do trato gastrointestinal. A serotonina também é encontrada no sistema nervoso central e contribui para as atividades mentais no ser humano. Muita atenção está sendo prestada para o efeito da serotonina da vida diária aos transtornos mentais tais como depressão e neurose. Nos últimos anos, os medicamentos curativos contra estas doenças têm sido desenvolvidos utilizando os medicamentos que afetam a serotonina.
Por outro lado a serotonina é um dos receptores acoplados à proteina G principalmente no sistema nervoso central. A serotonina é categorizada em 7 famílias de 5-HT1 a 5-HT7 e 14 subtipos são reconhecidos. Embora as investigações farmacológicas sobre cada subtipo têm sido continuadas (literatura de não-patente 1), três subtipos, 5-HT2A, 5-HT2B e 5-HT2C, são encontrados na família 5-HT2. Além disso, sobre o receptor 5-HT2B vários efeitos farmacológicos têm sido relatados como úteis para a prevenção ou tratamento de várias doenças.
Em geral, os antagonistas dos receptores 5-HT2B são considerados úteis para a prevenção ou tratamento de uma variedade de doenças, incluindo enxaqueca, dor inflamatória, dor nociceptiva, fibromialgia, dor lombar crônica, dor visceral, doença do refluxo gastroesofágico (DRGE), constipação, diarréia, distúrbios gastrintestinal funcional, síndrome do intestino irritável (a seguir é chamado IBS para encurtar). A definição e o critério é descrito em ROMA III, (literatura de não-patente 2), asma, osteoartrite, artrite reumatóide, doença de Crohn, colite ulcerativa, glomerulonefrite, nefrite, dermatite, hepatite, vasculite, isquemia renal, derrame cerebral, infarto do miocárdio, isquemia cerebral, doença de Alzheimer, obstrução reversível das vias aéreas, síndrome da doença respiratória do adulto, doença pulmonar obstrutiva crônica (COPD), hipertensão pulmonar (PH), pneumonia intersticial idiopática, bronquite, fibrose hepática alveolite fibrosante criptogênica, esclerose múltipla, depressão, ansiedade e obesidade. (Literaturas de não-patente 3-7)
Além disso, no respeitante aos receptores 5-HT2B, a relação do referido receptor com o aparelho digestivo e artéria pulmonar é conhecida com base nos experimentos utilizando inibidores seletivos de 5-HT2B.
O que concerne ao papel do aparelho digestivo, os antagonistas do receptor 5-HT2B são úteis para IBS com base na depreciação da contração intestinal humana por estimulação elétrica (literatura de patente 1). É descrito que os antagonistas de 5-HT2B são eficazes para o tratamento de distúrbio intestinal funcional com base na contração intestinal de ratos através da estimulação de serotonina (literatura de patente 2). Além disso, a redução do limite de dor contra a distensão colônica é relatada em ratos tratados com 2,4,6-trinitrobenzeno ácido sulfônico (chamado de TNBS a seguir), que é considerado como um modelo de hipersensibilidade visceral (literatura de não-patente 8).
Além disso, os antagonistas de 5-HT2B deprimiram o aumento do peso da defecação por estresse no modelo de defecação induzida por estresse em ratos geralmente considerado como um modelo de IBS, que pode ser confirmado como útil para IBS de diarreia-predominante. Além disso, quando o estresse é dado aos ratos, a resposta de dor aumenta contra a distensão colônica, os agonistas de 5-HT2B suprimem o aumento da resposta à dor.
No que concerne ao papel na artéria pulmonar, é descrito que o receptor 5-HT2B refere-se ao apriomoramento do modelo de camundongos cronicamente hipóxicos de hipertensão pulmonar, os compostos antagônicos de 5-HT2B são eficazes para o tratamento de hipertensão pulmonar (literatura de não- patente 9). É relatado que os antagonistas seletivos de 5-HT2B mostraram redução da pressão arterial no início do estudo da fase II contra pacientes com hipertensão pulmonar com doença pulmonar obstrutiva crônica (COPD) no teste duplamente cego utilizando placebo como referência (literatura de não-patente 10) em que antagonistas seletivos de 5-HT2B tiveram confirmda a sua segurança e utilidade em humanos.
Literatura de Patente 1: Panfleto 02/056010 de publicação international
Literatura de Patente 2: Publicação japonesa de pedido de patente não examinado (Tradução do pedido PCT) No. 1997-510216
Literatura de não patente 1: Phamacol.Rev.1994, 46, 157-203
Literatura de não patente 2:. Drossman et al, Journal of Gastrointestinal and Liver Diseases (2006) Vol.15 (3), 237-241
Literatura de não patente 3: Johnson KwCephalalgia 23 (2): 117-23 (2003)
Literatura de não patente 4: Allman JM et al, TRENDS in Cognitives Sciences 9 (8): 367-373 (2005)
Literatura de não patente 5: Borman RA et al, Br J Pharmacol. 135 (5): 114, 4-51 (2002)
Literatura de não patente 6: Beattie DT et al, Br J Pharmacol. 143 (5):549-60 (2004)
Literatura de não patente 7: Kubera M et al, Psychiatry Res.30; 134 (3):251-8 (2005)
Literatura de não patente 8: The Journal of Pharmacology and Experimental Therapeutics, Vol.302, n° 3, 1013-1022 (2002), 2) Pharmacology (2008), 81 (2), 144-150))
Literatura de não patente 9: Nature Medicine, 8 (10) :1129-1135, 2002
Literatura de não patente 10: PRX-08066: EPIX Pharmaceuticals
O objetivo desta invenção é prover uma composição de medicamentos e farmacêutica que contenha compostos com atividade de antagonista de receptor 5-HT2B seletivo como ingredientes eficazes. Além disso, pela afinidade do receptor seletivo elevada e reduzindo a relação com os outros receptores, reduzindo várias ações desfavoráveis, que o antagonista de receptor 5-HT2B se relaciona, também é o objetivo da presente invenção.
Inventores da presente invenção, para resolver o problema referido acima descobriram que os derivados de pirazol-3-carboxamida novos com a estrutura química única mostram a atividade antagonista seletiva e forte contra o receptor 5-HT2B entre os subtipos de receptores de serotonina. Além disso, eles confirmaram que os derivados de pirazol-3-carboxamida novos aprimoraram efetivamente o limiar da dor visceral no modelo de IBS induzido por TNBS de rato. Portanto, derivados de 5-substituído-1H-pirazol-3-carboxamida novos são úteis para prevenção ou tratamento de condições de doença mediadas pela estimulação do receptor acima, tais como enxaqueca, dor inflamatória, dor nociceptiva, fibromialgia, dor lombar crônica, dor visceral, doença de refluxo gastroesofágico (GERD), constipação, diarréia, distúrbio gastrointestinal funcional, síndrome do intestino irritável (IBS), asma, osteoartrite, artrite reumatóide, doença de Crohn, colite ulcerativa, glomerulonefrite, nefrite, dermatite, hepatite, vasculite, isquemia renal, acidente vascular cerebral, infarto do miocárdio, isquemia cerebral, doença de Alzheimer, obstrução reversível das vias aéreas, síndrome da doença respiratória do adulto, doença pulmonar obstrutiva crônica (COPD), hipertensão pulmonar (PH), pneumonia intersticial idiopática, bronquite, fibrose hepática, alveolite fibrosante criptogênica, esclerose múltipla, depressão, ansiedade e obesidade.
Esta invenção foi concluída com base na visão acima e provê os seguintes compostos ou seus sais farmaceuticamente aceitáveis, os referidos compostos ou seus sais farmaceuticamente aceitáveis, agentes de prevenção ou tratamento de doenças relacionadas com o receptor 5-HT2B como ingrediente eficaz, composições farmacêuticas contendo os referidos compostos ou seus sais farmaceuticamente aceitáveis, ou método de tratamento dos referidos compostos ou seus sais farmaceuticamente aceitáveis.
Notadamente, a presente invenção é como se segue: [1] Um composto da seguinte fórmula geral (I0) ou seu sal farmaceuticamente aceitável,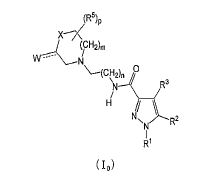 em que, R1 é grupo alquila menor de cadeia linear, cadeia ramificada ou cíclica com 1 a 6 átomos de carbono ou um grupo haloalquila de cadeia linear, cadeia ramificada ou cíclico com 1 a 6 átomos de carbono; R2 é um grupo de anel (hetero)aril da seguinte fórmula geral (Ar);
em que, R1 é grupo alquila menor de cadeia linear, cadeia ramificada ou cíclica com 1 a 6 átomos de carbono ou um grupo haloalquila de cadeia linear, cadeia ramificada ou cíclico com 1 a 6 átomos de carbono; R2 é um grupo de anel (hetero)aril da seguinte fórmula geral (Ar); 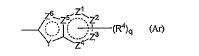 R3 é um átomo de hidrogênio ou halogênio; R4 é um grupo alquila menor de cadeia linear, cadeia ramificada ou cíclica com 1 a 6 átomos de carbono, um grupo haloalquila de cadeia linear, cadeia ramificada ou cíclica com 1 a 6 átomos de carbono, OH, OR1A, halogênio, -(CH2)aOH, CO2H, CONH2, CONHR1A, CONR1AR1A, CN, COR1A, NH2, NHR1A, NR1AR1A, NHCOR1A, SR1A, SOR1A, SO2R1A, SO2NH2, SO2NHR1A, SO2NR1AR1A ou NHSO2R1A, quando q é plural, R4 pode ser igual ou diferente; quando R4 tem dois R1A, eles podem ser iguais ou diferentes, ou R1A pode combinar com o outro R1A; R5 é um grupo alquila menor de cadeia linear, cadeia ramificada ou cíclica com 1 a 6 átomos de carbono, -(CH2)aOH, -(CH2)aOR1B, halogênio, CONH2, CONR1BR1B, COR1B, SO2R1B, -OCH2CH2NR1BR1B ou um grupo haloalquila de cadeia-linear, cadeia ramificada ou cíclica com 1 a 6 átomos de carbono; quando p é plural, R5 pode ser igual ou diferente, ou R5 pode combinar com o outro R5; R1A e R1B são cada um independentemente um grupo alquila menor de cadeia linear, cadeia ramificada ou cíclica com 1 a 6 átomos de carbono, ou um grupo haloalquila de cadeia linear, cadeia ramificada ou cíclica com 1 a 6 átomos de carbono; a é 0, 1 ou 2; m é 0, 1 ou 2; n é 1 ou 2; p é 0, 1, 2, 3, 4 ou 5; e q é 0, 1, 2 ou 3; X é CH2, NH, O, S, SO, SO2, CHR5, CR5R5 (R5 é igual conforme descrito acima, e pode ser igual ou diferente), ou NR5 (R5 é igual conforme o descrito acima); W é um átomo de oxigênio, (H, H), (H, R5) ou (R5, R5), quando X é CH2, NH, O, CHR5, CR5R5 ou NR5, ou W é (H, H), (H, R5), ou (R5, R5) quando X é S, SO ou SO2; em que (H, H), (H, R5) ou (R5, R5) significa que W representa dois grupos monovalentes, e o referido dois grupos monovalentes são H e H, H e R5, R5 e R5; Y é NH, NR1, O ou S; Z1, Z2, Z3, Z4, Z5 e Z6 e são cada um independentemente N, C, CH ou CR4 (R4 é igual conforme o descrito acima e 1, 2 ou 3 de Z1 a Z6 pode representar um átomo de nitrogênio). [2] Um composto da seguinte fórmula geral (I) ou seu sal farmaceuticamente aceitável.
R3 é um átomo de hidrogênio ou halogênio; R4 é um grupo alquila menor de cadeia linear, cadeia ramificada ou cíclica com 1 a 6 átomos de carbono, um grupo haloalquila de cadeia linear, cadeia ramificada ou cíclica com 1 a 6 átomos de carbono, OH, OR1A, halogênio, -(CH2)aOH, CO2H, CONH2, CONHR1A, CONR1AR1A, CN, COR1A, NH2, NHR1A, NR1AR1A, NHCOR1A, SR1A, SOR1A, SO2R1A, SO2NH2, SO2NHR1A, SO2NR1AR1A ou NHSO2R1A, quando q é plural, R4 pode ser igual ou diferente; quando R4 tem dois R1A, eles podem ser iguais ou diferentes, ou R1A pode combinar com o outro R1A; R5 é um grupo alquila menor de cadeia linear, cadeia ramificada ou cíclica com 1 a 6 átomos de carbono, -(CH2)aOH, -(CH2)aOR1B, halogênio, CONH2, CONR1BR1B, COR1B, SO2R1B, -OCH2CH2NR1BR1B ou um grupo haloalquila de cadeia-linear, cadeia ramificada ou cíclica com 1 a 6 átomos de carbono; quando p é plural, R5 pode ser igual ou diferente, ou R5 pode combinar com o outro R5; R1A e R1B são cada um independentemente um grupo alquila menor de cadeia linear, cadeia ramificada ou cíclica com 1 a 6 átomos de carbono, ou um grupo haloalquila de cadeia linear, cadeia ramificada ou cíclica com 1 a 6 átomos de carbono; a é 0, 1 ou 2; m é 0, 1 ou 2; n é 1 ou 2; p é 0, 1, 2, 3, 4 ou 5; e q é 0, 1, 2 ou 3; X é CH2, NH, O, S, SO, SO2, CHR5, CR5R5 (R5 é igual conforme descrito acima, e pode ser igual ou diferente), ou NR5 (R5 é igual conforme o descrito acima); W é um átomo de oxigênio, (H, H), (H, R5) ou (R5, R5), quando X é CH2, NH, O, CHR5, CR5R5 ou NR5, ou W é (H, H), (H, R5), ou (R5, R5) quando X é S, SO ou SO2; em que (H, H), (H, R5) ou (R5, R5) significa que W representa dois grupos monovalentes, e o referido dois grupos monovalentes são H e H, H e R5, R5 e R5; Y é NH, NR1, O ou S; Z1, Z2, Z3, Z4, Z5 e Z6 e são cada um independentemente N, C, CH ou CR4 (R4 é igual conforme o descrito acima e 1, 2 ou 3 de Z1 a Z6 pode representar um átomo de nitrogênio). [2] Um composto da seguinte fórmula geral (I) ou seu sal farmaceuticamente aceitável. em que, A é um anel de 3 a 8 elementos e pode conter 0 a 4 heteroátomos selecionados de O, S e N; R1 é um grupo C1-C6 alquila ou um grupo C1-C6 haloalquila; R2 é um grupo arila bicíclico ou monocíclico saturado ou parcialmente ou totalmente insaturado, que pode ser substituído por R4; R3 é um átomo de hidrogênio ou halogênio; R4 é um grupo C1-C6 alquila, um grupo C1-C6 haloalquila, OH, OR1A, halogênio, - (CH2)aOH, CO2H, CONH2, CONHR1A, CONR1AR1A, CN, COR1A, NH2, NHR1A, NR1AR1A, NHCOR1A, SR1A, SOR1A, SO2R1A, SO2NH2, SO2NHR1A, SO2NR1AR1A ou NHSO2R1A; quando q é plural, R4 pode ser igual ou diferente, quando R4 tem dois R1A, eles podem ser iguais ou diferentes ou R1A pode combinar com o outro R1A; R5 é um grupo C1-C6 alquila, -(CH2)aOH, -(CH2)aOR1B, halogênio, CONH2, CONR1BR1B, COR1B, SO2R1B, -OCH2CH2NR1BR1B ou um grupo C1-C6 haloalquil; quando p é plural, R5 pode ser igual ou diferente, ou R5 pode combinar com o outro R5; R1A, R1B são cada um independentemente um grupo C1-C6 alquila ou um grupo C1-C6 haloalquila; a é 0, 1 ou 2; n é 1 ou 2; p é 0, 1, 2, 3, 4 ou 5; e q é 0, 1, 2 ou 3. [3] O composto ou o sal farmaceuticamente aceitável, conforme descrito no [2] acima, em que R2 é o seguinte Ar1, Ar2, Ar3 ou Ar4,
em que, A é um anel de 3 a 8 elementos e pode conter 0 a 4 heteroátomos selecionados de O, S e N; R1 é um grupo C1-C6 alquila ou um grupo C1-C6 haloalquila; R2 é um grupo arila bicíclico ou monocíclico saturado ou parcialmente ou totalmente insaturado, que pode ser substituído por R4; R3 é um átomo de hidrogênio ou halogênio; R4 é um grupo C1-C6 alquila, um grupo C1-C6 haloalquila, OH, OR1A, halogênio, - (CH2)aOH, CO2H, CONH2, CONHR1A, CONR1AR1A, CN, COR1A, NH2, NHR1A, NR1AR1A, NHCOR1A, SR1A, SOR1A, SO2R1A, SO2NH2, SO2NHR1A, SO2NR1AR1A ou NHSO2R1A; quando q é plural, R4 pode ser igual ou diferente, quando R4 tem dois R1A, eles podem ser iguais ou diferentes ou R1A pode combinar com o outro R1A; R5 é um grupo C1-C6 alquila, -(CH2)aOH, -(CH2)aOR1B, halogênio, CONH2, CONR1BR1B, COR1B, SO2R1B, -OCH2CH2NR1BR1B ou um grupo C1-C6 haloalquil; quando p é plural, R5 pode ser igual ou diferente, ou R5 pode combinar com o outro R5; R1A, R1B são cada um independentemente um grupo C1-C6 alquila ou um grupo C1-C6 haloalquila; a é 0, 1 ou 2; n é 1 ou 2; p é 0, 1, 2, 3, 4 ou 5; e q é 0, 1, 2 ou 3. [3] O composto ou o sal farmaceuticamente aceitável, conforme descrito no [2] acima, em que R2 é o seguinte Ar1, Ar2, Ar3 ou Ar4, em que, R4 e q são iguais ao descrito no [2] acima; Y é NH, NR6, O ou S; Z1, Z2, Z3, Z4, Z5 e Z6 e são cada um independentemente N, C, CH, ou CR4 (1, 2 ou 3 de Z1 a Z6 pode representar um átomo de nitrogênio); e R6 é hidrogênio, um grupo C1-C6 alquila, um grupo C1-C6 haloalquila, um grupo C1-C6 alcóxi C1-C6 alquila, um grupo hidroxila C1-C6 alquila, um grupo halo C1-C6 alcóxi C1-C6 alquila, um grupo diC1-C6 alquilamino C1-C6 alquila, um grupo mono C1-C6 alquilamino C1-C6 alquila, um grupo amino C1-C6 alquila, um grupo C3-C8 ciclo C1-C6 alquila (referido grupo C3-C8 ciclo C1-C6 alquila pode ser substituído com 1 ou 2 grupos, cada um independentemente selecionado de hidróxi, C1-C6 alcóxi e C1-C6 acilóxi, e pode ter S (enxofre), O (oxigênio) ou NR1), um aminocarbonil C1-C6 alquila, um grupo mono C1-C6 alquilaminocarbonil alquila, um grupo di C1-C6 alquilaminocarbonil C1-C6 alquil, um hidróxicarbonil C1-C6 alquila ou um grupo C1-C6 alquilsulfonil,
em que, R4 e q são iguais ao descrito no [2] acima; Y é NH, NR6, O ou S; Z1, Z2, Z3, Z4, Z5 e Z6 e são cada um independentemente N, C, CH, ou CR4 (1, 2 ou 3 de Z1 a Z6 pode representar um átomo de nitrogênio); e R6 é hidrogênio, um grupo C1-C6 alquila, um grupo C1-C6 haloalquila, um grupo C1-C6 alcóxi C1-C6 alquila, um grupo hidroxila C1-C6 alquila, um grupo halo C1-C6 alcóxi C1-C6 alquila, um grupo diC1-C6 alquilamino C1-C6 alquila, um grupo mono C1-C6 alquilamino C1-C6 alquila, um grupo amino C1-C6 alquila, um grupo C3-C8 ciclo C1-C6 alquila (referido grupo C3-C8 ciclo C1-C6 alquila pode ser substituído com 1 ou 2 grupos, cada um independentemente selecionado de hidróxi, C1-C6 alcóxi e C1-C6 acilóxi, e pode ter S (enxofre), O (oxigênio) ou NR1), um aminocarbonil C1-C6 alquila, um grupo mono C1-C6 alquilaminocarbonil alquila, um grupo di C1-C6 alquilaminocarbonil C1-C6 alquil, um hidróxicarbonil C1-C6 alquila ou um grupo C1-C6 alquilsulfonil,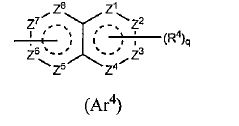 em que, R4 e q são iguais ao descrito no [2] acima, e Z1, Z2, Z3, Z4, Z5, Z6, Z7 e Z8 são cada um independentemente N, C, CH ou CR4 (1, 2 ou 3 de Z1 a Z8 pode representar um átomo de nitrogênio). [4] O composto ou o seu sal farmaceuticamente aceitável, tal como descrito no acima [3], em que Ar1, Ar2, Ar3 ou Ar4 é representado pela seguinte fórmula geral:
em que, R4 e q são iguais ao descrito no [2] acima, e Z1, Z2, Z3, Z4, Z5, Z6, Z7 e Z8 são cada um independentemente N, C, CH ou CR4 (1, 2 ou 3 de Z1 a Z8 pode representar um átomo de nitrogênio). [4] O composto ou o seu sal farmaceuticamente aceitável, tal como descrito no acima [3], em que Ar1, Ar2, Ar3 ou Ar4 é representado pela seguinte fórmula geral: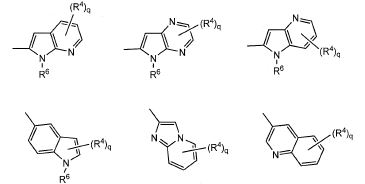 em que, R4 e q são iguais ao descrito no [2] acima; R6 é hidrogênio ou um grupo C1-C6 alquila, e (R4)q pode substituir um dos dois anéis ou ambos os anéis. [5] O composto ou o seu sal farmaceuticamente aceitável, conforme descrito no [2] acima, em que o anel A é morfolina, piperidina, pirrolidina ou azetidina que se liga em N; n é 1; p é 0, 1 ou 2; e q é 0, 1 ou 2. [6] O composto ou seu sal farmaceuticamente aceitável, conforme descrito no [2] acima, em que o composto representado pela fórmula geral (I) é selecionado do grupo constituído por 1-metil-N-[2-(morfolin-4-il)etil]-5-(quinolin-3-il)-1H-pirazol-3-carboxamida; 1-metil-5-{5-metil-1H-pirrolo[3,2-b]piridin-2-il}-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 1-metil-N-[2-(morfolin-4-il)etil]-5-{1H-pirrolo[2,3-b]piridin-2-il}-1H-pirazol-3- carboxamida; 1-metil-N-[2-(morfolin-4-il)etil]-5-{7H-pirrolo[2,3-d]pirimidin-6-il}-1H-pirazol-3- carboxamida; 1-metil-N-[2-(morfolin-4-il)etil]-5-[5-(trifluorometil)-1H-pirrolo[3,2-b]piridin-2-il]-1H- pirazol-3-carboxamida; 1-metil-5-{5-metil-1H-pirrolo[2,3-b]piridin-2-il}-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-{5-fluoro-1H-pirrolo[2,3-b]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-{5-ciano-1H-pirrolo[3,2-b]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-{6-fluoro-1H-pirrolo[3,2-b]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 1-metil-N-[2-(morfolin-4-il)etil]5-{5H-pirrolo[2,3-b]pirazin-6-il}-1H-pirazol-3- carboxamida; 5-{5-ciano-1H-pirrolo[2,3-b]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-{5-fluoro1-metil-1H-pirrolo[2,3-b]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H- pirazol-3-carboxamida; N-[2-(3,3-difluoroazetidin-1-il)etil]-5-(5-fluoro-1H-indol-2-il)-1-metil-1H-pirazol-3- carboxamida; N-[2-(azetidin-1-il)etil]-5-(5-fluoro-1H-indol-2-il)-1-metil-1H-pirazol-3-carboxamida; 1-metil-5-(2-metil-1H-indol-5-il)-N-[2-(morfolin-4-il)etil]-1H-pirazol-3-carboxamida; 5-(1,2-dimetil-1H-indol-5-il)-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-[1-(2-methoxietil)-1H-indol-3-il]-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-(4-acetamido-1H-indol-2-il)-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-{imidazo[1,2-a]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-{6-fluoroimidazo[1,2-a]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-{7-fluoroimidazo[1,2-a]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-{6-cianoimidazo[1,2-a]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; N-[2-(3,3-difluoroazetidin-1-il)etil]-1-metil-5-(quinolin-3-il)-1H-pirazol-3- carboxamida; N-[2-(3,3-difluoroazetidin-1-il)etil]-1-metil-5-{1H-pirrolo[2,3-b]piridin-2-il}-1H-pirazol- 3-carboxamida; e 5-{7-cianoimidazo[1,2-a]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida. [7] Um intermediário do composto descrito no [2] acima, que é representado pela fórmula geral (1A):
em que, R4 e q são iguais ao descrito no [2] acima; R6 é hidrogênio ou um grupo C1-C6 alquila, e (R4)q pode substituir um dos dois anéis ou ambos os anéis. [5] O composto ou o seu sal farmaceuticamente aceitável, conforme descrito no [2] acima, em que o anel A é morfolina, piperidina, pirrolidina ou azetidina que se liga em N; n é 1; p é 0, 1 ou 2; e q é 0, 1 ou 2. [6] O composto ou seu sal farmaceuticamente aceitável, conforme descrito no [2] acima, em que o composto representado pela fórmula geral (I) é selecionado do grupo constituído por 1-metil-N-[2-(morfolin-4-il)etil]-5-(quinolin-3-il)-1H-pirazol-3-carboxamida; 1-metil-5-{5-metil-1H-pirrolo[3,2-b]piridin-2-il}-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 1-metil-N-[2-(morfolin-4-il)etil]-5-{1H-pirrolo[2,3-b]piridin-2-il}-1H-pirazol-3- carboxamida; 1-metil-N-[2-(morfolin-4-il)etil]-5-{7H-pirrolo[2,3-d]pirimidin-6-il}-1H-pirazol-3- carboxamida; 1-metil-N-[2-(morfolin-4-il)etil]-5-[5-(trifluorometil)-1H-pirrolo[3,2-b]piridin-2-il]-1H- pirazol-3-carboxamida; 1-metil-5-{5-metil-1H-pirrolo[2,3-b]piridin-2-il}-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-{5-fluoro-1H-pirrolo[2,3-b]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-{5-ciano-1H-pirrolo[3,2-b]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-{6-fluoro-1H-pirrolo[3,2-b]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 1-metil-N-[2-(morfolin-4-il)etil]5-{5H-pirrolo[2,3-b]pirazin-6-il}-1H-pirazol-3- carboxamida; 5-{5-ciano-1H-pirrolo[2,3-b]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-{5-fluoro1-metil-1H-pirrolo[2,3-b]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H- pirazol-3-carboxamida; N-[2-(3,3-difluoroazetidin-1-il)etil]-5-(5-fluoro-1H-indol-2-il)-1-metil-1H-pirazol-3- carboxamida; N-[2-(azetidin-1-il)etil]-5-(5-fluoro-1H-indol-2-il)-1-metil-1H-pirazol-3-carboxamida; 1-metil-5-(2-metil-1H-indol-5-il)-N-[2-(morfolin-4-il)etil]-1H-pirazol-3-carboxamida; 5-(1,2-dimetil-1H-indol-5-il)-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-[1-(2-methoxietil)-1H-indol-3-il]-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-(4-acetamido-1H-indol-2-il)-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-{imidazo[1,2-a]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-{6-fluoroimidazo[1,2-a]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-{7-fluoroimidazo[1,2-a]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-{6-cianoimidazo[1,2-a]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; N-[2-(3,3-difluoroazetidin-1-il)etil]-1-metil-5-(quinolin-3-il)-1H-pirazol-3- carboxamida; N-[2-(3,3-difluoroazetidin-1-il)etil]-1-metil-5-{1H-pirrolo[2,3-b]piridin-2-il}-1H-pirazol- 3-carboxamida; e 5-{7-cianoimidazo[1,2-a]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida. [7] Um intermediário do composto descrito no [2] acima, que é representado pela fórmula geral (1A): em que, cada descrição é igual ao descrito no [2] acima. [8] Um intermediário do composto descrito no [2] acima, que é representado pela fórmula geral (1B):
em que, cada descrição é igual ao descrito no [2] acima. [8] Um intermediário do composto descrito no [2] acima, que é representado pela fórmula geral (1B): em que, R1, R2, R3 são os mesmos conforme definidos na fórmula (I), e OH de ácido carboxílico pode ser substituído por um substituinte removível. [9] Um agente preventivo ou terapêutico para doenças em que os receptores 5 HT2B estão envolvidos em que o composto ou seu sal farmaceuticamente aceitável, conforme descrito em qualquer um dos [2] a [6], é um ingrediente eficaz. [10] Uma composição farmacêutica compreendendo o composto ou o seu sal farmaceuticamente aceitável, conforme descrito em qualquer um dos [2] a [6], e 15 um carreador farmaceuticamente aceitável. [11] Uma composição farmacêutica para a prevenção ou tratamento de uma condição de doença mediada por receptores 5-HT2B, em um sujeito mamífero, que compreende uma quantidade eficaz do composto ou seu sal farmaceuticamente aceitável, conforme descrito em qualquer um dos [2] a [5], e um carreador farmaceuticamente aceitável. [12] Uma composição farmacêutica compreendendo o composto conforme descrito em qualquer um dos [2] a [6], compreendendo ainda outro agente farmacologicamente ativo. [13] O composto ou seu sal farmaceuticamente aceitável, conforme descrito em qualquer um dos [2] a [6], para uso na prevenção ou tratamento de uma condição de doença mediada por receptores 5-HT2B. [14] Uma utilização do composto ou seu sal farmaceuticamente aceitável, conforme descrito em qualquer um dos [2] a [6], para a fabricação de um medicamento para a prevenção ou tratamento de uma condição mediada por receptores 5-HT2B. [15] Um método de prevenção ou tratamento para enxaqueca, dor inflamatória, dor nociceptiva, dor neuropática, fibromialgia, dor lombar crônica, dor visceral, doença do refluxo gastroesofágico (GERD), constipação, diarréia, distúrbio gastrointestinal funcional, síndrome do intestino irritável, asma, osteoartrite, artrite reumatóide, doença de Crohn, colite ulcerativa, glomerulonefrite, nefrite, dermatite, hepatite, vasculite, isquemia renal, acidente vascular cerebral, infarto do miocárdio, isquemia cerebral, doença de Alzheimer, obstrução reversível das vias aéreas, síndrome da doença respiratória do adulto, doença pulmonar obstrutiva crônica (COPD), hipertensão pulmonar (PH), pneumonia intersticial idiopática, bronquite, fibrose hepática, alveolite fibrosante criptogênica, esclerose múltipla, depressão, ansiedade ou obesidade, que é caracterizada pela administração de uma quantidade eficaz de uma composição farmacêutica, compreendendo o composto ou seu sal farmaceuticamente aceitável, conforme descrito em qualquer um dos [2] a [6] e um carreador farmaceuticamente aceitável, para um sujeito humana ou um mamífero.
em que, R1, R2, R3 são os mesmos conforme definidos na fórmula (I), e OH de ácido carboxílico pode ser substituído por um substituinte removível. [9] Um agente preventivo ou terapêutico para doenças em que os receptores 5 HT2B estão envolvidos em que o composto ou seu sal farmaceuticamente aceitável, conforme descrito em qualquer um dos [2] a [6], é um ingrediente eficaz. [10] Uma composição farmacêutica compreendendo o composto ou o seu sal farmaceuticamente aceitável, conforme descrito em qualquer um dos [2] a [6], e 15 um carreador farmaceuticamente aceitável. [11] Uma composição farmacêutica para a prevenção ou tratamento de uma condição de doença mediada por receptores 5-HT2B, em um sujeito mamífero, que compreende uma quantidade eficaz do composto ou seu sal farmaceuticamente aceitável, conforme descrito em qualquer um dos [2] a [5], e um carreador farmaceuticamente aceitável. [12] Uma composição farmacêutica compreendendo o composto conforme descrito em qualquer um dos [2] a [6], compreendendo ainda outro agente farmacologicamente ativo. [13] O composto ou seu sal farmaceuticamente aceitável, conforme descrito em qualquer um dos [2] a [6], para uso na prevenção ou tratamento de uma condição de doença mediada por receptores 5-HT2B. [14] Uma utilização do composto ou seu sal farmaceuticamente aceitável, conforme descrito em qualquer um dos [2] a [6], para a fabricação de um medicamento para a prevenção ou tratamento de uma condição mediada por receptores 5-HT2B. [15] Um método de prevenção ou tratamento para enxaqueca, dor inflamatória, dor nociceptiva, dor neuropática, fibromialgia, dor lombar crônica, dor visceral, doença do refluxo gastroesofágico (GERD), constipação, diarréia, distúrbio gastrointestinal funcional, síndrome do intestino irritável, asma, osteoartrite, artrite reumatóide, doença de Crohn, colite ulcerativa, glomerulonefrite, nefrite, dermatite, hepatite, vasculite, isquemia renal, acidente vascular cerebral, infarto do miocárdio, isquemia cerebral, doença de Alzheimer, obstrução reversível das vias aéreas, síndrome da doença respiratória do adulto, doença pulmonar obstrutiva crônica (COPD), hipertensão pulmonar (PH), pneumonia intersticial idiopática, bronquite, fibrose hepática, alveolite fibrosante criptogênica, esclerose múltipla, depressão, ansiedade ou obesidade, que é caracterizada pela administração de uma quantidade eficaz de uma composição farmacêutica, compreendendo o composto ou seu sal farmaceuticamente aceitável, conforme descrito em qualquer um dos [2] a [6] e um carreador farmaceuticamente aceitável, para um sujeito humana ou um mamífero.
O ingrediente eficaz, derivados de pirazol-3-carboxamida da presente invenção tem núcleo novo e inibe fortemente e seletivamente a função de receptor 5-HT2B. A atividade antagonista do receptor 5-HT2B forte deste medicamento da invenção mostra os efeitos terapêuticos com base nos excelentes efeitos farmacêuticos. Além disso, a alta seletividade deste medicamento da invenção é útil para reduzir a ampla gama de efeitos colaterais com base nas atividades que não do receptor 5-HT2B.
Fig. 1 é um gráfico de resultados no estudo de distensão do cólon utilizando um modelo de IBS de rato induzido por TNBS sobre composto de Exemplo 24.
O composto desta invenção é caracterizado por atividades específicas de ligação ao receptor 5-HT2B. O composto desta invenção inibe selectivamente as atividades do receptor 5-HT2B pela ligação de forma antagônica ao receptor 5- HT2B, que é útil para o tratamento ou o pré tratamento em mamíferos em relação ao referido receptor.
O termo "agente antagonista" também é chamado antagonista, e significa o medicamento que age de forma antagônica contra o agonista e reduz os efeitos. A capacidade que esses antagonistas e agonistas de se ligarem parcialmente é chamada de afinidades de ligação, e a avaliação da afinidades de ligação, como os exemplos descritos a seguir, é conduzida pela comparação do valor Ki calculado no receptor de estudos de ligação in vitro, ou valores de IC50 conduzida no ensaio de ligação de receptor nas mesmas condições em alguns casos.
Nos estudos de ligação ao receptor, quando IC50 não pode ser calculado por não mostrar as atividades bastante antagônicas, o IC50 do composto pode ser considerado mais do que a referida concentração.
O composto da presente invenção tem uma afinidade de ligação, e o valor de IC50, que mostra a atividade inibindo a serotonina para o recptor 5-HT2B (atividade inibitória), é de preferência inferior a 1000 nM, mais preferivelmente menor do que 100 nM, adicionalmente de preferência inferior a 10 nM, e mais de preferência inferior a 1 nM.
O composto desta invenção ou o seu sal farmaceuticamente aceitável é favorável a ser “seletivo” na atividade inibitória para o 5-HT2B em comparação com os outros receptores. "Seletivo" significa que a atividade inibitória para o referido receptor é maior do que as atividades inibitórias para "os outros receptores”. “Selectivo” na presente invenção significa que o valor de IC50 da atividade inibitória para o referido receptor é um décimo ou menos, de preferência um centésimo ou menos, e mais preferencialmente um milésimo ou menos, comparando com o valor do IC50 "dos outros receptores”.
“Os outros receptores” aqui, significam os outros receptores relatados nos atuais antagonistas da serotonina não-seletivos. Particularmente após a avaliação da seletividade contra 5-HT2A, 5-HT2C, a avaliando os compostos representativos sobre a influência do receptore existente e enzimas é favorável.
As atividades inibitórias ou atividades antagonistas do receptor dos antagonista seletivos de 5-HT2B na presente invenção podem ser facilmente avaliadas com as tecnologias conhecidas mencionados abaixa.
Neste contexto, o termo "C1-C6", conforme definido na fórmula geral supra mencionada, salvo indicação em contrário, significa uma cadeia de carbono linear ou ramificada com 1 a 6 átomos de carbono. Assim, o "grupo C1-C6 alquila", significa um grupo alquila com 1 a 6 átomos de carbono, incluindo de preferência metila (a seguir às vezes abreviado como Me), etila (a seguir às vezes abreviado como Et), propila, isopropila, butila, isobutila, terc-butila.
O "halôgenio", significa o grupo 17 da tabela periódica, incluindo de preferência F, Cl, Br ou I.
O "grupo haloalquila" significa grupo C1-C6 alquila, que é substituído com 1 a 5 átomos de halogênio (s).
O "anel arila" significa anel mono ou bicíclico que pode ser saturado ou parcial ou totalmente insaturado. O arila, significa um substituinte que se liga na parte deixada por um átomo de hidrogênio fora do anel arila, incluindo de preferência Ar1, Ar2, Ar3 e Ar4.
Um grupo de anel monocíclico insaturado contém, por exemplo, fenila, pirazolil, furil, tienil, oxazolil, tetrazolil, tiazolil, imidazolil, thiadiazolil, piridil, pirimidinil, pirrolil, tiofenila, pirazinil, piridazinil, isoxazolil, isotiazolil, triazolil, furazanil são citados.
Um grupo de anel bicíclico insaturado contém, por exemplo, naftila, benzofuranil, isobenzofuranil, benzotiofenil, indolil, isoindolil, benzoxazolil, benzotiazolil, indazolil, benzimidazolil, quinolil, isoquinolil, cinnolinil, ftalazinil, quinazolinil, quinoxalinil são citados.
Um exemplo de grupo de anel saturado contém o anel, que é parcialmente saturado ou completamente saturado na parte insaturada do grupo de anel mono ou bicíclico descrito acima. “R1A pode combinar com o outro R1A” significa que NR1AR1A tal como NR1AR1A, CONR1AR1A e SO2NR1AR1A podem mostrar o grupo de anel contendo carbono de 3 a 13 elementos pela referida combinação (por exemplo, r é de 1 a 12 no seguinte esquema (IIa)).
Entre eles, o grupo de anel contendo carbono de 3 a 8 elementos é favorável (por exemplo, r é de 1 a 6 no esquema seguinte (IIa)). Na verdade
CONR1AR1A e NR1AR1AinR4 pode ser descrito no seguinte esquema (IIa). O estilo de ligação, porém, o estilo de ligação não se limita apenas no seguinte esquema.
"R1B pode combinar com o outro R1B” é o mesmo significado conforme descrito acima e R1A é substituído por R1B.
Os substituintes removíveis são exemplificados etóxi, fenóxi, halogênio, alcóxicarbonilóxi, arilóxicarbonilóxi, imidazol-1-il, grupo 4-nitrofenóxi, mas não limitado apenas a estes.
Os sais de um composto da fórmula (I) são sais farmaceuticamente aceitáveis e incluem a adição de ácido e adição de base (incluindo sais de diácidos e sal de dibase) dos mesmo.
Em geral, sais de adição de ácido adequados são formadas a partir dos ácidos que formam sais atóxicos. Os exemplos incluem o acetato, aspartato, benzoato, besilato, bicarbonato/carbonato, bissulfato/sulfato, borato, cansilato, citrato, edisilato, esilato, formato, fumarato, gluceptato, gluconato, hexafluorofosfato, hibenzato, cloridrato, bromidrato, hidroiodeto, isotionato, lactato, malato, maleato, malonato, mesilato, metilsulfeto, naftilato, 2-napsilato, nicotinato, nitrato, orotato, oxalato, palmitato, pamoato, fosfato/hidrogênio fosfato/dihidrogênio fosfato, sacarato, estearato, succinato, tartarato, tosilato sais de trifluoroacetato.
Sais de base adequados são formados a partir de bases que formam os sais atóxicos. Exemplos como sais de base incluem o alumínio, arginina, benzatina, cálcio, colina, dietilamina, diolamina, glicina, lisina, magnésio, meglumina, olamine, potássio, sódio, trometamina e sais de zinco. Ver Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl e Wermuth (Wiley-VCH, Weinheim, Alemanha, 2002), conforme necessário.
Sais farmaceuticamente aceitáveis dos compostos da fórmula (I) podem ser facilmente preparados misturando a solução de ácido ou base desejada. O sal resultante pode precipitar-se para fora e ser recolhido por filtração ou pode ser recuperado por evaporação do solvente. O grau de ionização do sal resultante pode variar de quase completamente ionizado para praticamente não- ionizado.
Os compostos da invenção podem existir em ambas as formas, insolvatada e solvatada. O termo "solvato" é utilizado aqui para descrever um complexo molecular compreendendo o composto da invenção e uma quantidade estequiométrica de uma ou mais moléculas de solvente farmaceuticamente aceitável, por exemplo, o etanol. O termo "hidratar" é empregado quando referido solvente é água.
Os solvatos farmaceuticamente aceitáveis, de acordo com a invenção, incluem aqueles em que o solvente de cristalização pode ser isotopicamente substituído, por exemplo, D2O, d6-acetona, d6-dimetilsulfóxido.
Estão Incluídos no escopo da invenção complexos, tais como clatratos, complexos de inclusão de hospedeiro de drogas, em que, em contraste com os solvatos acima mencionados, a droga e o hospedeiro estão presentes em quantidades estequiométricas ou não-estequiométricas. Também estão incluídos os complexos da droga contendo dois ou mais componentes orgânicos e/ou inorgânicos que podem estar em quantidades estequiométricas ou não- estequiométricas. Os complexos resultantes podem ser ionizados, parcialmente ionizados ou não-ionizados. Ver J Pharm Sci, 64 (8), 1269-1288, por Haleblian (Agosto 1975), conforme necessário.
A seguir todas as referências aos compostos da fórmula (I) incluem referências aos seus sais, solvatos e complexos e aos solvatos e complexos dos seus sais.
Os compostos da invenção incluem compostos da fórmula (I) como definidos acima, incluindo polimorfos e os seus hábitos cristalinos, pró-drogas e seus isômeros (incluindo os isômeros ópticos, geométricos e tautoméricos) a seguir definidos e rotulados isotopicamente compostos da fórmula (I).
Conforme indicado, as chamadas "pró-drogas” dos compostos da fórmula (I) ou seus sais também estão dentro do escopo da invenção. Consequentemente, certos derivados de compostos da fórmula I, que podem ter pouca ou nenhuma atividade farmacológica eles mesmos podem, quando administrados dentro ou sobre o corpo, ser convertidos em compostos da fórmula (I) com a atividade desejada, por exemplo, por clivagem hidrolítica. Tais derivados são referidos como "pró-drogas". Mais informações sobre o uso de pró-drogas podem ser encontrados no Pro-drugs as Novel Delivery Systems, vol. 14, ACS Symposium Series (T. Higuchi e W. Stella) e Bioreversible Carriers in Drug
Design, Pergamon Press, 1987 (ed. E. B. Roche, Associação Farmacêutica Americana).
Pró-drogas de acordo com a invenção podem, por exemplo, ser produzidas através da substituição de funcionalidades adequadas presentes nos compostos da fórmula (I) com certas frações conhecidas por aqueles versados na técnica como "pró-frações", conforme descrito, por exemplo, no Design of Prodrugs por H. Bundgaard (Elsevier, 1985).
Alguns exemplos de pró-drogas de acordo com a invenção incluem:quando o composto da fórmula (I) ou seu sal contém uma funcionalidade de ácido carboxílico (-COOH), um éster do mesmo e amida do mesmo, por exemplo, seu etil éster, seu fenil éster, seus carboximetil éster, seu dimetilaminometil éster, seu pivaloiloximetil éster, seu etóxicarboniloxietil éster, seu ftalidil éster, seu (5-metil-2-oxo-1,3-dioxolen-4-il)metil éster, seu 1- (ciclohexiloxicarbonilóxi)etil éster, seu metilamida e outros;quando o composto da fórmula (I) ou seu sal contém uma funcionalidade de álcool (-OH), um composto em que a funcionalidade hidróxi está sujeita a acilação, alquilação, fosforilação e boração, por exemplo, um composto de acetila, um composto de palmitoil, um composto de pronanoil, um composto de pivaloil, um composto de succinil, um composto de alanil, um composto de dimetilaminometilcarbonil e outros; Além disso, dependendo dos substituintes, a pró-droga pode formar a N-óxido. Também estão incluídos dentro do escopo da invenção tais N-óxidos;quando o composto da fórmula (I) ou seu sal contém uma funcionalidade amino, uma amida sua, por exemplo, um composto em que, conforme o caso pode ser, um ou ambos os hidrogênios da funcionalidade amina é/são sujeitos a acilação, alquilação e fosforilação, por exemplo, um composto eicosanonil, composto alanil, um composto pentilaminocarbonil, um composto (5- metil-2-oxo1,3-dioxolen-4-il)metóxicarbonil, um composto tetrahidrofuranil, um composto pirrolidinilmetil, composto terc-butil e outros.
Outros exemplos de grupos de substituição de acordo com os exemplos anteriores e exemplos de outros tipos de pró-droga podem ser encontrados nas referências mencionadas acima. Além disso, certos compostos da fórmula I podem eles mesmos agirem como pró-drogas de outros compostos da fórmula (I).
Os compostos da fórmula (I) contendo um ou mais átomos de carbono assimétricos podem existir como dois ou mais estereoisômeros. Onde um composto da fórmula geral (I) contém um grupo alquenila ou alquenileno, isômeros geométricos cis/trans (ou Z/E) são possíveis. Onde o composto contém, por exemplo, um grupo ceto ou oxima ou uma fração aromática, isomerismo tautomérico ('tautomeria) pode ocorrer. Daqui resulta que um único composto pode apresentar mais de um tipo de isomerismo.
Incluídos dentro do escopo da presente invenção estão todos os estereoisômeros, isômeros geométricos e formas tautoméricas dos compostos da fórmula geral (I), incluindo compostos exibindo mais de dois tipos iguais de isomerismo, e misturas de um ou mais dos mesmos. Também estão incluídos a adição de ácido ou sais de base, em que o contra-íon é opticamente ativo, por exemplo, D-lactato e L-lisina, ou racêmico, por exemplo, DL-tartarato ou DL- arginina.
Isômeros cis/trans podem ser separados por técnicas convencionais também conhecidas por aqueles versados na técnica, por exemplo, cromatografia e cristalização fracionada. As técnicas convencionais para a preparação/isolamento dos enantiômeros individuais incluem a síntese quiral de um precursor puro opticamente adequado ou resolução do racemato (ou o racemato de um sal ou derivado), utilizando, por exemplo, cromatografia líquida de alta pressão quiral (HPLC).
Alternativamente, o racemato (ou um precursor racêmico) pode ser reagido com um composto opticamente ativo adequado, por exemplo, um álcool, ou, no caso em que o composto da fórmula geral (I) contém uma fração acídica ou básica, um ácido ou base tal como o ácido tartárico ou 1-feniletilamina. A mistura diastereomérica resultante pode ser separada por cromatografia e/ou cristalização fracional e um ou ambos os diastereoisômeros convertidos para o(s) enantiômero(s) puro correspondente por meios bem conhecidos de uma pessoa versada.
Os compostos quirais da invenção (e seus precursores quirais) podem ser obtidos na forma enantiomericamente enriquecida utilizando cromatografia, tipicamente HPLC, em uma resina assimétrica com uma fase móvel composta de um hidrocarboneto, tipicamente heptano ou hexano, contendo de 0 a 50(w/w)% isopropanol, geralmente de 2 a 20 (w/w)% e de 0 a 5 (w/w)% de uma alquilamina, normalmente 0,1(w/w)% dietilamina. A concentração do eluato proporciona a mistura enriquecida.
Os conglomerados de estereoisômeros podem ser separados por técnicas convencionais conhecidas por aqueles versados na técnica - ver, por exemplo, Stereochemistry of Organics Compounds por E L Eliel (Wiley, Nova York, 1994).
A presente invenção inclui todos os compostos isotopicamente rotulados farmaceuticamente aceitáveis da fórmula geral (I) em que um ou mais átomos são substituídos por átomos com o mesmo número atômico, mas uma massa atômica ou número de massa diferente da massa atômica ou número de massa geralmente encontrado na natureza.
Exemplos de isótopos adequados para inclusão nos compostos da invenção incluem os isótopos de hidrogênio, tais como 2H e 3H, carbono, tais como 11C, 13C e 14C, cloro, tal como 36Cl, flúor, tal como 18F, iodo, tais como 123I e 125I, nitrogênio, tais como 13N e 15N, oxigênio, tais como 15O, 17O e 18O, fósforo, tal como 32P, e enxofre, tal como o 35S.
A substituição por isótopos mais pesados, como o deutério, 2H, ou seja, pode oferecer algumas vantagens terapêuticas decorrentes da maior estabilidade metabólica, por exemplo, meia-vida in vivo elevada ou requisitos de dosagem reduzidos e, portanto, pode ser preferencial, ao invés de composto normal de 1H em algumas circunstâncias.
A substituição com isótopos emissores de pósitrons, tais como 11C, 18F, 15O e 13N, pode ser útil nos estudos de Topografia de Emissão de Pósitron (PET) para examinar a ocupação do receptor de substrato.
Certos compostos rotulados isotopicamente da fórmula (I), por exemplo, aqueles que incorporam um isótopo radioativo, são úteis nos estudos de distribuição de tecido de droga e/ou substrato. O trítio de isótopos radioativos, ou seja, 3H, e carbono-14, ou seja, 14C, são particularmente úteis para este propósito em vista de suas facilidades de incorporação e meios rápidos de detecção.
Todos os compostos da fórmula geral (I) podem ser preparados com os procedimentos descritos nos métodos gerais apresentados a seguir ou por métodos específicos descritos na seção de Exemplos e seção de Preparações, ou por suas alterações de rotina. A presente invenção também abrange qualquer um ou mais desses processos para a preparação dos compostos da fórmula geral (I), além de quaisquer novos intermediários utilizados neles.
O composto da fórmula geral (I) na presente invenção podem ser preparados com método de preparação conhecido ou pode ser preparado de acordo com o procedimento geral ou método de preparação mostrado no esquema de reação seguinte. Salvo indicação em contrário, R1 para R5 e X, Y e Z nos métodos a seguir são conforme definidos acima. O termo "grupo de proteção", como é utilizado a seguir, significa um grupo de proteção de hidroxila ou amino que é selecionado de hidróxi típico, acetileno ou grupos de proteção amino descritos no Protective Groups in Organic Synthesis editado por T.W. Greene et al. (John Wiley & Sons, 1999). Além disso, cada composto descrito no 5 esquema de reação, a menos que iniba a reação, pode formar o sal que inclui omesmo sal conforme composto (I). A pró-droga da presente invenção pode ser preparada através da introdução de um grupo específico no estágio do intermediário ou pela reação utilizando um composto obtido, que é similar ao grupo de proteção descrito acima. A reação, tal como esterificação, amidação e 10 desidratação pode ser feita utilizando métodos padrão bem conhecidos poraqueles versados na técnica.
A preparação das formulações parenterais sob condições estéreis, por exemplo, por liofilização, pode ser facilmente conseguida utilizando técnicas farmacêuticas padrão bem conhecidas por aqueles versados na técnica.
A preparação do composto da fórmula IA a partir da fórmula II através do processo A-2 (Método 1) e a preparação do composto na fórmula IA a partir da fórmula II através do processo A-3 (Método 2) são apresentados como se segue.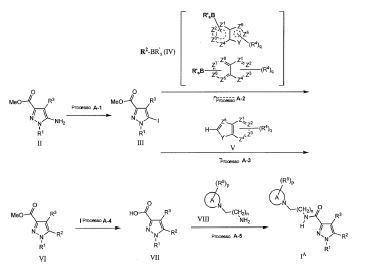
Em uma representação de R'sB, R' significa OH, O-alquila inferior, alquila inferior ou flúor, e s é 2 ou 3, B é átomo de boro. Como a representação concreta do substituinte (OH)2B, (O-alquila inferior)2B, (alquila inferior)2B, potássio trifluoroborato (BF3)(BF3K) são descritos, mas quando (O-alquila inferior)2B pode formar o anel cíclico entre os grupos alquila inferior.
Nesta etapa, os compostos de iodo da equação III podem ser preparados com uma síntese de pote na presença dos agentes de iodação adequados através de sais de diazônio, ou após a formação de sais de diazônio eles podem ser preparados pela adição de agentes iodação adequada. A formação de sais de diazônio pode ser conduzida no procedimento conhecido. No procedimento comum, a formação de diazônio é conduzida utilizando nitrito de sódio sob solução ácida. Em solução ácida, por exemplo, solução de ácido acético, ácido clorídrico, ácido fórmico ou ácido sulfúrico pode ser utilizada, em que o ácido acético é preferencial. A reação é de 10 minutos a 12 horas, mas em geral, 30 minutos a 6 horas. A temperatura de reação varia de aproximadamente - 20°C a 30°C, mas, em geral, de -10°C a 5°C. Um agente de iodação adequada, iodeto de potássio, iodeto de sódio ou iodeto, em que iodeto de potássio é preferencial. No esquema de reação, Me significa grupo metila (igual daqui para frente).
Nesta etapa, o composto (VI) pode ser preparado utilizando uma reação de acoplamento cruzado de arila com o composto (III), preparada no processo de A-1. Este pode ser preparado sob a condição de acoplamento com a presença de um catalisador de metal de transição adequado e base (ou sem base) em uma mistura de solvente orgânico em água. Como um substituinte R'sB adequado em um reagente arilmetálico, por exemplo, (OH)2B, (O-alquila inferior)2B, (alquila inferior)2B, sal de potássio(BF3K) de trifluoroborato (BF3-) são citados, mas no caso de (O-alquila inferior)2B, um anel cíclico pode ser formado entre os grupos alquila inferiores.
Como um catalisador de metal de transição, por exemplo, tetraquis (trifenilfosfina)paládio, bis(trifenilfosfina)paládio(II)cloreto, cobre(0), acetato de cobre (I), brometo de cobre(I), cloreto de cobre (I), iodeto de cobre(I), óxido de cobre(I), trifluorometano sulfonato de cobre(I), acetato de cobre (II), brometo de cobre(II), cloreto de cobre(II), iodo de cobre(II), óxido de cobre(II), trifluorometano sulfonato(II) de cobre (II), acetato de paládio(II), cloreto de paládio(II), bis(acetonitril)dicloropaládio(II), bis(dibenzilidenoacetona)paládio(0), tris(dibenzilidenoacetona)dipaládio(0), [1,1’- bis(difenilfosfino)ferroceno]paládio(II)dicloreto e assim por diante são citados. Em particular, tetraquis(trifenilfosfina)paládio, bis(trifenilfosfina)paládio(II)cloreto, acetato de paládio(II), bis(acetonitril)dicloropaládio(II), tris(dibenzilidenoacetona)dipaládio(0), [1,1’- bis(difenilfosfino)ferroceno]paládio(II)dicloreto são favoráveis. Como um reagente arilmetálico, por exemplo, reagentes de ácido borônico tal como derivado de 2- ácido indoilborônico e reagentes de ester ácido borônico tal como derivado de 2- 2-éster ácido indoilborônico são citados, mas não limitados a eles. Como um solvente orgânico adequado em solução misturada orgânica em água, por exemplo, na presença ou ausência da base solúvel em água, tal como hidróxido de potássio, hidróxido de sódio, hidróxido de lítio e solução de carbonato de potássio, tetraidrofurano, 1,4-dioxano, N,N-dimetilformamida (DMF), acetonitril, álcoois tais como metanol e etanol, hidrocarbonetos halogenados, tais como diclorometano, 1,2-dicloroetano, clorofórmio, tetracloreto de carbono ou dietiléter são citados. Essa reação pode ser conduzida na presença de fatores adicionais adequados. Como tal um fator adicional, por exemplo, trifenilfosfina, tri-terc- butilfosfina, 1,1'-bis(difenilfosfina)ferroceno, tri-2-furilfosfina, 2-(diclorohexilfosfino) bifenil, trifenilarsina, cloreto de tetrabutilamônio, fluoreto de tetrabutilamônio, ácidolítio acético, cloreto de lítio, trietilamina, metóxido de potássio (ou sódio), hidróxido de sódio, carbonato de sódio, fosfato de potássio, carbonato de césio, bicarbonato de sódio ou iodeto de sódio são citados. Esta reação é de aproximadamente 0°C a 200°C, e é geralmente de aproximadamente 20°C a 120°C. O período de reação é de aproximadamente 5 minutos a 96 horas, e é geralmente de aproximadamente 30 minutos a 24 horas. Além disso, durante a reação, um reator de microondas pode ser utilizado. Além disso, quando Y é NH, o átomo de nitrogênio pode ser protegido com um grupo alcoxicarbonila inferior (por exemplo, grupo Boc) e grupo (p-alquil)benzenosulfonila (por exemplo, grupo benzenosulfonila e p-toluenossulfonila).
À excepção de um acoplamento cruzado de Suzuki-Miyaura mostrado acima, a reação de acoplamento de Stillecross utilizando trialquiltina ao invés de substituinte R'sB, e a reação de acoplamento de Negishi zinco-halogênio, em que conforme um halogênio, cloro, bromo, iodo são citados, em vez de substituinte R'sB pode ser utilizado.
Nesta etapa, o composto heterocíclico (VI), correspondente à fórmula geral R2 pode ser preparado pela derivatização para o aril boronato éster utilizando reação de borilação de CH entre borano pinacol (HBpin) ou bis(pinacolato)diborano (B2pin2, pin.Me4C2O2) e o composto heterocíclico (V), sob um catalisador de metal de transição adequado (irídio, por exemplo) e um solvente orgânico apropriado. (borilação de CH; T. Ishiyama et al, Organic Synthesis (2005), 82, 126-133.) O composto de acoplamento (VI) pode ser preparado pela reação de Suzuki-Miyaura do aril boronato éster derivatizado com o composto (III) . Estas reações podem ser conduzidas na reação de um pote ou procedimento de reação de duas etapas.
Como um catalisador de metal de transição, por exemplo, [Ir (OMe) (COD)]2(COD significa 1,5 ciclooctadieno), Cp*Rh(n4-C6Me6)(Cp* significa C5Me5), Ir(n5-C9H7)(COD), [IrCl(COD)]2, [IrCl(COE)2]2 ou RhCl{P(i-Pr)3}(N2) são citados. Como um aditivo, por exemplo, 1,2-bis (dimetilfosfino)etano(dmpe), 2,2'-bipiridin- (dpi), 4,4'-ditercbutil-2,2'-bipiridin(dtbpi), ou dppe são citados. Como um solvente orgânico apropriado, por exemplo, hidrocarbonetos, tais como n-hexano ou ciclohexano são citados. Usando uma combinação de 1/2[IrCl(COD)]2 e 4,4'- ditercbutil-2,2'-bipiridin-(dtbpi) como um catalisador em hexano, reagindo pinacolborano ou bis(pinacolato)diborano com o composto de arila é uma preparação prática. Então, reagindo aril boronato ésteres preparado acima com o composto (III) é transferido para o composto (VI) por reação de Suzuki-Miyaura. Esta reação é substancialmente igual àquela no processo A-2. Os mesmos reagentes e condições de reação no processo A-2 podem ser utilizados, que é similar ao processo A-2 descrito acima. Ressalva-se que quando esta reação é conduzida em uma reação de pote na reação de Suzuki-Miyaura, a combinação de N,N-dimetilformamida(DMF) ou 1,4-dioxano como um solvente, fostato de potássio sólido (K3PO4) como uma base, [1,1'-bis(difenilfosfina)ferroceno]paládio (II)dicloreto(PdCl2(dppf)) como um catalisador de paládio é favorável.
A borilação de CH descrita acima, seguida pela reação de introdução do grupo heteroarila bicíclico direta (V), que é similar à reação de Suzuki-Miyaura, pode ser substituída com a reação de arilação direta mediada por paládio (literatura de não-patente 10), ródio (literatura de não-patente 11) e cobre (literatura de não-patente 12).
Literatura de não-patente 11: Aldrichimica Acta Vol.40, No.2-(2007) 35-41. Literatura de não-patente 12: Tetrahedron Letter 49 (2008) 1598-1600.
Nesta etapa, o composto de ácido carboxílico (VII) pode ser preparado pela hidrólise do composto de éster (VI) em um solvente de reação.
A hidrólise pode ser conduzida de acordo com o procedimento conhecido em público. No procedimento normal, a hidrólise pode ser conduzida sob condição básica, tal como hidróxido de sódio, hidróxido de potássio ou hidróxido de lítio. Como um solvente adequado, por exemplo, álcoois tais como metanol, etanol, propanol, butanol, 2-metóximetanol ou etileno glicol, éteres, tais como tetraidrofurano(THF), 1,2-dimetóxietano(DME) ou 1,4-dioxano; amidas, tais como N,N-dimetilformamida (DMF) ou triamida hexametilfosfórica; sulfóxidos tais como dimetilsulfóxido (DMSO) ou água são citados. O período de reação é de aproximadamente 30 minutos a 48 horas, e é geralmente de aproximadamente 60 minutos a 30 horas. A temperatura de reação é de aproximadamente -20°C a 100°C, e é geralmente de aproximadamente 20°C a 75°C.
A hidrólise pode ser conduzida sob condições ácidas, por exemplo, haleto de hidrogênio tal como cloridrato ou bromidrato; ácidos sulfônicos tais como ácido p-toluenosulfônico ou ácido benzenossulfônico; piridio ácido p- toluenosulfônico; e ácidos carboxílicos, tal como ácido acético ou ácido trifluoroacético. Como um solvente adequado, por exemplo, álcoois tais como metanol, etanol, propanol, butanol, 2-metóximetanol, ou etileno glicol, éteres, tais como tetrahidrofurano (THF), 1,2-dimetóxietano (DME) ou 1,4-dioxano; ou hidrocarboneto halogenado, tal como 1,2-dicloroetano; ou amidas, tais como N, N- dimetilformamida (DMF) ou triamida hexametilfosfórica; sulfóxidos tais como dimetilsulfóxido (DMSO), ou a água são citados. O período de reação é de aproximadamente 30 minutos a 24 horas, e é geralmente de aproximadamente 60 minutos a 10 horas. A temperatura de reação é de aproximadamente -20°C a 100°C, e é geralmente de aproximadamente 0oC a 65°C.
Nesta etapa, o composto de amida (IA) pode ser preparado na presença ou ausência de um reagente de acoplamento de um solvente inerte pela reação de acoplamento do composto de amina (VIII) com um composto de ácido carboxílico (VII) em um solvente da reação. Além disso, esta reação pode ser conduzida na presença ou ausência de aditivos tais como 1-hidróxibenzotriazol (HoBt) ou 1-hidróxiazabenzotriazol. Como um solvente adequado, por exemplo, acetona, nitrometano, N,N-dimetilformamida(DMF), sulfolano, dimetilsulfóxido (DMSO), 1-metil-2-pirrolidinona (NMP), 2-butanona, acetonitril; hidrocarbonetos halogenados, tais como diclorometano, 1,2-dicloroetano, clorofórmio; éteres, tais como tetrahidrofurano e 1,4-dioxano são citados. O período de reação é de aproximadamente 5 minutos a 1 semana, e é geralmente de aproximadamente 30 minutos a 24 horas. A temperatura de reação é de aproximadamente -20°C a 100°C, e é geralmente de aproximadamente 0oC a 60°C. Como um agente de acoplamento adequado, o agente que é utilizado na síntese de peptídeo pode ser utilizado, por exemplo, diciclohexilcarbodiimida (DCC), carbodiimida solúvel em água (WSC), hexafluorofosfato-O-benzotriazol-1-il-N,N,N',N'-tetrametiluronio (HBTU), 2-etóxi-N-etóxicarbonil-1,2-dihidroquinolina, ácido tetrafluoro bórico 2- bromo-1-etilpiridínio (BEP), 2-cloro-1,3-cloreto de dimetilimidazolínio, benzotriazol- 1-ilóxitris(dimetilamino)fosfônio hexafluorofosfato (BOP), dietil azodicarboxilato- trifenilfosfina, dietil cianofosfato, dietilfosforilazido, 2-cloro1metilpiridínio iodeto, N,N'-carbonildiimidazol, benzotriazol-1-il-dietilfosfato, etila cloroformato ou isobutila cloroformiato são citados. Além disso, é desejável para conduzir a reação na presença de bases, tais como N,N-diisopropiletilamina, N- metilmorfolina, 4-(dimetilamino)piridina ou trietilamina. O composto de amida (IA) pode ser preparado através de haleto de acila correspondente, que é obtido pela reação com o agente de halogenação tal como cloreto de oxalil, oxicloreto de fósforo ou cloreto de tionila. O haleto de acila obtido pode ser convertido para o composto de amida correspondente (IA) pelo tratamento do composto de amina (VIII), sem o uso de reagentes de condensação descritos nesta etapa.
A síntese do anel de azaindol (método 3) utilizando a reação de formação de anel no processo B-6 é mostrado como se segue.
Nesta etapa, o composto de IX pode ser preparado pela hidrólise de compostos de éster (III). Esta reação é substancialmente idêntica àquela do processo A-4 e os mesmos reagentes e condições de reação no processo A-4 podem ser utilizados na forma similar do processo A-4.
Nesta etapa, o composto de X pode ser preparado pela reação de amidação do composto de ácido carboxílico (IX) com o composto de amina (VIII). Esta reação é substancialmente idêntica àquela do processo A-5 e os mesmos reagentes e condições de reação no processo A-5 podem ser utilizados na forma similar do processo A-5.
Nesta etapa, o composto (XI) pode ser preparado utilizando reação de acoplamento de Closs do composto (X) com um composto de acetileno protegido pelo grupo trialquilsilil tal como um grupo trimetilsilil na presença de uma quantidade catalítica de reagente de paládio e um reagente de paládio ou sal de cobre (I) e um ligante de fosfina em um solvente adequado, incluindo uma base ou utilizando apenas uma base própria como um solvente. Como exemplo de reagente de paládio, tetraquis(trifenilfosfina)paládio e cloreto de bis(trifenilfosfina)paládio(II), são de preferência citados. Como um exemplo de sal de cobre (I), iodeto de cobre (I) e brometo de cobre (I) são de preferência citados. Como um ligante de fosfina, por exemplo, bis(difenilfosfino)butano (dppb) é citado. Como um exemplo de base, por exemplo dietilamina, trietilamina, diisopropiletilamina, diciclohexilamina, carbonato de potássio e carbonato de sódio são citados. Além disso, como um solvente de reação, por exemplo tetrahidrofurano, 1,4-dioxano, N,N-dimetilformamida (DMF), acetonitril, etil acetato, hidrocarbonetos, tais como n-hexano, ciclohexano, benzeno, tolueno e dietileter são citados . O período de reação é de aproximadamente 5 minutos a 96 horas e é geralmente de aproximadamente 30 minutos a 24 horas. A temperatura de reação é de aproximadamente -78°C a 200°C, e é geralmente de aproximadamente -20°C a 80°C. Além disso, durante a reação, um reator de microondas pode ser utilizado.
Nesta etapa, o composto (XII) pode ser preparado desprotegendo o grupo trialquilsilil utilizando um método usual, em geral, conhecido tal como um método descrito no John Wiley & Sons, Grupos de Proteção em Síntese Orgânica (1999). Como um método usual, a desproteção pode ser conduzida na presença de uma base tal como um carbonato de potássio e carbonato de sódio em um solvente de álcool tal como um álcool metílico e álcool etílico.
Esta reação é substancialmente idêntica àquela no processo B-3 e o composto (XIV) pode ser preparado pela reação de acoplamento de Sonogashira do compostos de acetileno (XII) e composto de arilhaleto (XIII), em que o esquema P1 é hidrogênio, grupo terc-butóxicarbonil ou grupo de proteção de amino tal como um grupo trifluoracetil, utilizando os mesmos reagentes e condições de reação no processo B-3 pode ser usado de forma similar ao do processo B-3.
Nesta etapa, o composto (IB) pode ser preparado pela reação de cicloadição intramolecular do composto de acetileno (XIV), utilizando uma base apropriada. Como uma base adequada, potássio terc-butóxido, sódio terc- butóxido, césio terc-butóxido, hidróxido de césio, 1,8-diazabiciclo[5.4.0]undeca-7- eno (DBU), 1,1,3,3-tetrametilguanidina, trietilamina e assim por diante são utilizados e a reação é conduzida em um solvente adequado. Como um solvente adequado, N,N-dimetilformamida(DMF), N-metilpirrolidinona (NMP), tolueno, 1,4- dioxano, álcoois tais como metanol e etanol. O período de reação é de aproximadamente 5 minutos a 96 horas, e é geralmente de aproximadamente 30 minutos a 24 horas. A temperatura da reação é de aproximadamente -78°C a 250°C, e geralmente é de -20°C a 150°C. De preferência, é conduzida utilizando potássio terc-butóxido em DMF na faixa da temperatura ambiente para 80°C. Em outro método de cicloadição intramolecular pode ser conduzido utilizando um catalisador de paládio em que diclorobis(trifenilfosfina)paládio(II), cobre(I)iodeto, trietilamina, DMF é citado como uma combinação representante. Além disso, um catalisador de metal ou complexos de metal, incluindo cobre, ouro, irídio, mercúrio, molibdênio, platina e ródio pode ser conduzido. Além disso, quando o substituinte de NHP1 é o grupo fenol ou tiol, a cicloadição intramolecular pode ser conduzida sob condições acima descritas, resultando na preparação de benzotiofeno correspondente e derivados de benzofurano. Além disso, após a reação de ciclização, quando o grupo de proteção (P1) permanece, a desproteção pode ser conduzida por uma condição adequada.
A síntese de anel de imidazol[1,2-a]piridina (método 4) utilizando a reação de formação de anel no processo C-3 é mostrada como se segue.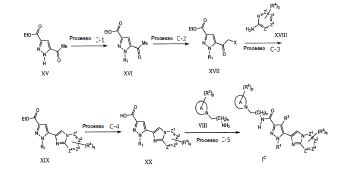
Nesta etapa, o composto de XVI pode ser preparado pela reação de alquilação de N do composto XV que pode ser facilmente preparada utilizando uma base adequada e haleto de alquila de acordo com a literatura. Como uma base adequada, por exemplo, etóxido de sódio, potássio terc-butóxido, hidreto de potássio, hidreto de sódio, sódio bis(trimetilsilil)amida, carbonato de potássio, carbonato de sódio, carbonato de césio, hidróxido de sódio são citados, mas não limitados a eles. Além disso, como um solvente orgânico apropriado, por exemplo, tetra, N,N-dimetilformamida (DMF), dietiléter, acetonitril são citados. O período de reação é de aproximadamente 5 minutos a 96 horas, e é geralmente de aproximadamente 30 minutos a 24 horas. A temperatura de reação é de aproximadamente -78°C a 250°C, e é geralmente de aproximadamente -20°C a 150°C.
Nesta etapa, o composto de XVII pode ser preparado pela reação de halogenação de alfa (X=Cl, Br, I) do composto (XVI) utilizando um reagente de halogenação apropriado. Como um reagente de halogenação adequado, por exemplo, bromo, cloro, cloreto de sulfurilo, brometo de hidrogênio, N- bromosuccinimida (NBS), 5,5-dibromo2,2-dimetil-4, 6-dioxo-1,3-dioxano, tribrometo de fenil trimetilamônio são citados. Como um solvente orgânico adequado, por exemplo, ácido acético, bissulfureto de carbono, éter, tetrahidrofurano, N,N-dimetilformamida(DMF), hidrocarboneto halogenado tal como o diclorometano, 1,2-dicloroetano, clorofórmio, tetracloreto de carbono pode ser utilizado. O período de reação é de aproximadamente 5 minutos a 96 horas, e é geralmente de aproximadamente 30 minutos a 24 horas. A temperatura de reação é de aproximadamente -78°C a 250°C, e é geralmente de aproximadamente -20°C a 150°C.
Nesta etapa, o composto de XIX pode ser preparado pela reação de condensação do anel do composto haloketone-alfa (XVII) com um composto de amina apropriada na presença de um solvente adequado com o calor. Como um solvente adequado, por exemplo tetraidrofurano, 1,4-dioxano, N,N- dimetilformamida (DMF), acetonitril, álcoois tais como metanol e etanol. O período de reação é de aproximadamente 5 minutos a 96 horas, e é geralmente de aproximadamente 30 minutos a 24 horas. A temperatura de reação é de aproximadamente 0°C a 250°C, e é geralmente de aproximadamente 30°C a 150°C.
Nesta etapa, o composto de XX pode ser preparado pela hidrólise do composto de éster (XIX). Esta reação é substancialmente idêntica àquela do processo A-4 e os mesmos reagentes e condições de reação no processo A-4 podem ser utilizados na forma similar do processo A-4.
Nesta etapa, o composto de IC pode ser preparado pela reação de amidação do composto de ácido carboxílico (XX) com um composto de amina (VIII). Esta reação é substancialmente idêntica àquela do processo A-5 e os mesmos reagentes e condições de reação no processo A-5 podem ser utilizados na forma similar do processo de A-5.
A cadeia lateral de amina de troca (método 5) utilizando o processo D3 é mostrada como se segue.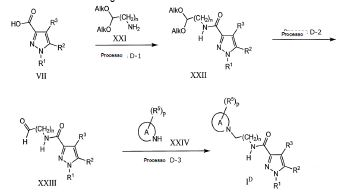
Nesta etapa, o composto de XXII pode ser preparado pela reação de amidação do composto de ácido carboxílico (VII) com um composto de amina (XXI). Esta reação é substancialmente idêntica àquela no processo A-5 e os mesmos reagentes e condições de reação no processo A-5 podem ser utilizadas na forma similar a do processo A-5.
Nesta etapa, o composto (XXIII) pode ser preparado pela desproteção do grupo acetal utilizando um método usual conhecido, em geral, tal como um método descrito no John Wiley & Sons, Grupos de Proteção em Síntese Orgânica (1999). Como um método usual, a desproteção pode ser conduzida na presença de um ácido, tal como um ácido clorídrico diluído, ácido p-toluenosulfônico ou sob uma condição ácida em um solvente orgânico geral.
Nesta etapa, o composto (ID) pode ser preparado pela reação de aminação redutiva do composto aldeído (XXIII) com um composto de amina XXIV utilizando um agente de redução adequado. Como um agente de redução adequado, por exemplo, borohidreto de sódio (NaBH4), cianoborohidreto de sódio (NaBH3CN), triacetóxiborohidreto de sódio [NaBH(OAc)3] são citados. Como um solvente adequado, por exemplo, ácido acético, tetrahidrofurano, hidrocarbonetos halogenados, tais como diclorometano, 1,2-dicloroetano, clorofórmio, tetracloreto de carbono são utilizados, e se for necessário, uma quantidade catalítica de ácido acético ou ácidos de Lewis como o tetracloreto de titânio, tetraisopropóxi titânio[Ti(O-iPr)4] pode ser utilizado. Quando cianoborohidreto (NaBH3CN) é utilizado, a reação também pode ser conduzida sob uma condição ácida. O período de reação é de aproximadamente 5 minutos a 96 horas, e é geralmente de aproximadamente 30 minutos a 24 horas. A temperatura de reação é de aproximadamente 0°C a 250°C, e é geralmente de aproximadamente 30°C a 100°C.
A cadeia lateral de amina de troca (método 6), utilizando o processo E- é mostrada como se segue.
Nesta etapa, o composto de IE pode ser preparado pela reação de acoplamento do composto halogenado (X) com um derivado de ácido arilborônico (ou éster). Esta reação é substancialmente idêntica àquela no processo A-2 e os mesmos reagentes e condições de reação no processo A-2 podem ser utilizados na forma similar à do processo A-2.
A cadeia lateral de R2 de troca (método 7) utilizando o processo de F é mostrada como se segue. No seguinte XXVI, P2 significa o grupo de proteção selecionado do grupo alcóxicarbonila inferior, grupo benzilóxicarbonil, grupo benzenosulfonil e 4-alquilbenzenosulfonil.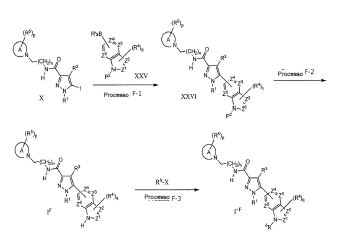
Nesta etapa, o composto de XXVI pode ser preparado pela reação de acoplamento do composto halogenado (X) com o ácido heteroarilborônico (ou éster) XXV que pode ser protegido com o grupo terc-butóxicarbonil ou grupo benzenosulfonil ou 4-alquilbenzenosulfonil. Esta reação é substancialmente idêntica àquela no processo A-2 e os mesmos reagentes e condições de reação no processo A-2 podem ser utilizados na forma similar á do processo A-2.
Nesta etapa, o composto (IF) pode ser preparado pela desproteção do grupo trialquilsilil e grupo arilsulfonil utilizando um método usual conhecido, em geral, tal como o método descrito no John Wiley & Sons, Proteção de Grupos em Síntese Orgânica (1999). Como um método usual, a desproteção do grupo terc- butóxicarbonil pode ser conduzida na presença de um catalisador ácido, como ácido clorídrico diluído, ácido p-toluenosulfônico sob condições ácidas em um solvente orgânico geral. A desproteção dos grupos benzenosulfonil ou 4- alquilbenzenosulfonil pode ser desprotegida na presença do reagente alcalino tal como o carbonato de potássio, carbonato de sódio, carbonato de césio e hidróxido de sódio na combinação de um solvente orgânico geral.
Nesta etapa, o composto (I'F) pode ser preparado convertendo a ligação de N-H sobre o anel de heteroarila em IF para a ligação de N-R6. Quando o reagente R6-X é alquilhaleto, essa reação é substancialmente idêntica àquela no processo C-1, e a reação pode ser conduzida sob a mesma condição que o processo C-1. Além disso, O-tosilato, O-mesilato e O-triflato que têm grupo de saída no grupo hidroxila (-OH) podem ser substituíveis. Além disso, quando R6 é o grupo alquilsulfonil, a reação é substancialmente idêntica áquela no processo C-1 em que o cloreto de alquilsulfonil é utilizado sob a mesma condição que o processo C-1.
O intermediário (1A) é útil para a preparação do composto da presente invenção. Por exemplo, o intermediário X mostrado no processo B na síntese geral é efetivamente utilizado para a preparação do composto da presente invenção.
O intermediário (IB) é útil para a preparação do composto da presente invenção. Por exemplo, o intermediário VI eo VII no processo A, XIX e XX, no processo sintético C, XXI no processo sintético D na síntese geral é efetivamente utilizado para a preparação do composto da presente invenção.
Os efeitos farmacológicos dos compostos da presente invenção como um antagonista de 5-HT2B podem ser estimados através da medição da pressao arterial pulmonar que aumenta em modelo de animal (rato, camundongo) exposto à hipóxia crônica. A droga existente para a hipertensão arterial pulmonar (por exemplo, preparações de sildenafil e de prostaglandinas) e RS-127445, que é conhecido como um antagonista seletivo de 5-HT2B pode ser utilizado para os compostos de referência.
Os outros efeitos farmacológicos dos compostos da presente invenção como um antagonista de 5-HT2B podem ser estimado através da medição dos efeitos antidiarréicos em modelo de animal (rato, camundongo) exposto às drogas ou estresse. As drogas antidiarréicas existentes (por exemplo, loperamida e berberina) e RS-127445, que é conhecido como um antagonista seletivo de 5- HT2B podem ser utilizados para os compostos de referência.
Assim, compostos resultantes podem ser isolados e purificados na forma livre ou como um sal por um tratamento de formação de sal convencional. Isolamento e purificação podem ser alcançados pela aplicação de um procedimento químico convencional, tais como extração, concentração, destilação, cristalização, filtração, recristalização, uma variedade de cromatografia e assim por diante.
Uma variedade de isômeros podem ser isolada por uma forma convencional utilizando a diferença das propriedades físico-químicas entre os isômeros. Por exemplo, os isômeros ópticos podem ser separados e purificados pela formação de sais diastereoméricos do racematos com um ácido orgânico opticamente ativo (por exemplo, o ácido tartárico) e recristalização fracional subseqüente, ou por cromatografia de coluna utilizando uma fase estacionária quiral. Além disso, o composto opticamente ativo pode ser produzido utilizando um composto opticamente ativo adequado como um material de partida. Neste contexto, uma mistura de diastereoisômeros pode também ser separada pela cristalização fracionada ou cromatografia para o enantiômero(s) puro correspondente.
Os compostos da invenção podem ser administrados por via oral. A administração oral pode envolver deglutição, de modo que o composto entra no trato gastrointestinal, ou a administração bucal ou sublingual pode ser empregada através da qual o composto entra na corrente sanguínea diretamente da boca.
Formulações adequadas para administração oral incluem formulações sólidas tais como, por exemplo, comprimidos, cápsulas contendo partículas, líquidos ou pós, pastilhas (incluindo cheias de líquido), gomas de mascar, multi- partículas, nano-partículas, géis, solução sólida, lipossomas, filmes (incluindo o muco-adesivo), óvulos, sprays e formulações líquidas.
As formulações líquidas incluem, por exemplo, suspensões, soluções, xaropes e elixires. Tais formulações podem ser empregadas como preenchedores em cápsulas moles ou duras e normalmente compreendem um carreador, por exemplo, água, etanol, polietileno glicol, propileno glicol, metilcelulose, ou um óleo adequado, e um ou mais agentes emulsionantes e/ou agentes de suspensão. As formulações líquidas também podem ser preparadas pela reconstituição de um sólido, por exemplo, de um saquinho em água e outros.
Os compostos da invenção também podem ser utilizados em formas de desitegração rápidas, dissolução rápida, tais como aquelas descritas no Expert Opinion in Therapeutic Patentes, 11 (6), 981-986 por Liang Chen (2001).
Para formas de dosagem em comprimidos, dependendo da dose, a droga pode fazer-se de aproximadamente 1% em peso a aproximadamente 80% em peso da forma de dosagem, mais normalmente aproximadamente de 5% em peso a aproximadamente 60% em peso da forma de dosagem.
Além da droga como um ingrediente ativo, os comprimidos contêm geralmente um desintegrante. Exemplos de desintegrantes incluem glicolato de amido de sódio, carboximetil celulose de sódio, carboximetil celulose de cálcio, croscarmelose sódica, crospovidona, polivinilpirrolidona, metil celulose, celulose microcristalina, hidroxipropilcelulose substituída por alquila inferior, amido, amido pré-gelatinizado e alginato de sódio. Geralmente, o desintegrante compreenderá de aproximadamente 1% em peso a aproximadamente 25% em peso, de preferência aproximadamente 5% em peso a aproximadamente 20% em peso da forma de dosagem.
Ligantes são geralmente utilizados para transmitir qualidades coesivas para uma formulação de comprimidos. Uma formulação de comprimidos pode conter ligantes para transmitir outras qualidades coesivas ao invés da droga como um ingrediente ativo. Os ligantes adequados incluem a celulose microcristalina, gelatina, lactose (monohidrato, monohidrato seco por spray, anidro e outros), manitol, xilitol, dextrose, sacarose, sorbitol, polietilenoglicol, gomas naturais e sintéticas, polivinilpirrolidona, amido pré-gelatinado, hidróxipropil celulose e hidróxipropil celulose, dehidrato de fosfato de cálcio dibásico e hidróxipropilmetilcelulose e outros.
Os comprimidos também podem compreender opcionalmente agentes ativos de superfície, tal como lauril sulfato de sódio e polissorbato 80 e glidantes como o dióxido de silício e talco. Quando presentes, agentes ativos de superfície podem compreender de aproximadamente 0,2% em peso a aproximadamente 5% em peso do comprimido, e glidantes podem compreender de aproximadamente 0,2% em peso a aproximadamente 1% em peso do comprimido.
Os comprimidos também contêm geralmente lubrificantes, tais como estearato de magnésio, estearato de cálcio, estearato de zinco, estearil fumarato de sódio, e as misturas de estearato de magnésio com lauril sulfato de sódio. Lubrificantes geralmente compreendem entre aproximadamente 0,25% em peso a aproximadamente 10% em peso, de preferência de aproximadamente 0,5% em peso a aproximadamente 3% em peso do comprimido.
Outros ingredientes possíveis incluem anti-oxidantes, corantes, agentes aromatizantes, conservantes, agentes mascarantes de sabor e outros.
Comprimidos exemplares contêm até aproximadamente 80% em peso de droga, de aproximadamente 10% em peso a aproximadamente 90% em peso de ligante, de aproximadamente 0% em peso a aproximadamente 85% em peso de solvente, de aproximadamente 2% em peso a aproximadamente 10% em peso de desintegrante, e de aproximadamente 0,25% em peso a aproximadamente 10% em peso de lubrificante.
Os métodos de preparação de comprimidos não são limitados, mas os métodos gerais para a preparação de comprimidos podem ser adequadamente utilizados. Por exemplo, misturas de comprimidos podem ser compactados diretamente ou através de rolo para formar comprimidos. Misturas de comprimidos ou porções de misturas podem ser, alternativamente, granuladas por umidade, secagem ou derretimento, congeladas derretidas ou extrudadas antes de se tornarem comprimidos. A formulação final pode compreender uma ou mais camadas e pode ser revestida ou sem revestimento, que pode até ser encapsulado.
Em termos de formulação de comprimidos, os conteúdos descritos em "Pharmaceutical Dosage Forms: Tablets, Vol. 1", por H. Lieberman e L. Lachman, Marcel Dekker, NY, 1980 (ISBN 0-8247-6918-X) podem ser referidos.
As formulações sólidas para a administração oral podem ser formuladas para ser imediata e/ou de libertação modificada. As formulações de liberação modificada incluem, por exemplo, liberação atrasada, sustentada, pulsada, controlada, orientada e programada.
As formulações de liberação modificada apropriadas para os fins da invenção são descritas na US N° 6,106,864. Detalhes de outras tecnologias de liberação adequadas tais como dispersões de energia elevada e partículas osmóticas e revestidas podem ser encontrados em Verma et al, Tecnologia Farmacêutica On-line, 25(2), 1-14 (2001). A utilização de goma de mascar para conseguir liberação controlada é descrita no WO 00/35298.
Os compostos da invenção também podem ser administrados diretamente na corrente sanguínea, no músculo ou em um órgão interno. Meios adequados para administração parenteral incluem intravenoso, intra-arterial, intraperitoneal, intratecal, intraventricular, intrauretral, intrastemal, intracraniano, intramuscular e subcutâneo. Os dispositivos adequados para administração parenteral incluem injetores de agulha (incluindo micro-agulha), injectores livres de agulha e técnicas de infusão.
As formulações parenterais podem conter excipientes, tais como sais, carboidratos e agentes tampão (de preferência para um pH de aproximadamente 3 a aproximadamente 9). Elas podem ser normalmente soluções aquosas, mas para algumas aplicações, elas podem ser mais adequadamente formuladas como uma solução estéril não aquosa ou como uma forma seca para ser utilizada em conjunto com um veículo adequado, tal como água apirogênica estéril.
A preparação das formulações parenterais sob condições estéreis, por exemplo, por liofilização, podem ser facilmente conseguidas utilizando técnicas farmacêuticas padrão bem conhecidas por aqueles versados na técnica.
A solubilidade dos compostos da fórmula (I) utilizados na preparação de soluções parenterais pode ser aumentada pelo uso de técnicas de formulação apropriadas, tais como a incorporação de agentes que aumentam a solubilidade.
As formulações para a administração parenteral podem ser formuladas para ser de liberação imediata e/ou modificada. As formulações de liberação modificada incluem, por exemplo, liberação atrasada, sustentada, pulsada, controlada, orientada e programada. Assim, os compostos da invenção podem ser formulados como um sólido, semi-sólido ou líquido tixotrópico para a administração como um depósito implantado providenciando a liberação modificada do composto ativo. Exemplos de tais formulações incluem stents revestidos por droga e microesferas de PGLA.
Os compostos da invenção também podem ser administrados por via tópica para a pele ou mucosa, ou seja, dérmica ou transdérmicamente. As formulações típicas para esta finalidade incluem, por exemplo, géis, hidrogéis, loções, soluções, cremes, pomadas, pós de pó, molhos, espumas, filmes, manchas na pele, bolachas, implantes, esponjas, fibras, bandagens e microemulsões. Os lipossomas também podem ser utilizados. Carreadores típicos incluem, por exemplo, álcool, água, óleo mineral, vaselina líquida, vaselina branca, glicerina, polietileno glicol e propileno glicol ou água e outros. Os promotores de penetração podem ser incorporados - ver, por exemplo, J Pharm Sci, 88 (10), 955-958 por Finnin e Morgan (outubro 1999).
Outros meios de administração tópica incluem, por exemplo, entrega por eletroporação, iontoforese, fonoforese, sonoforese e injeção de micro agulha ou livre de agulha (por exemplo, Powderject (marca registrada), Bioject (marca registada), etc.).
As formulações para administração tópica podem ser formuladas para serem de liberação imediata e/ou modificada. As formulações de liberação modificada incluem, por exemplo, liberação atrasada, sustentada, pulsada, controlada, orientada e programada.
Os compostos da invenção podem ser combinados com entidades macromoleculares solúveis, tais como ciclodextrina e seus derivados adequados ou polímeros contendo polietileno glicol, para melhorar a sua solubilidade, taxa de dissolução, mascaração de sabor, biodisponibilidade e/ou estabilidade para utilização em qualquer dos modos mencionados acima de administração.
Os complexos de ciclodextrina de droga, por exemplo, geralmente são considerados úteis para a maioria das formas de dosagem e vias de administração. Tanto complexos de inclusão quanto de não-inclusão podem ser utilizados. Como uma alternativa à complexação direta com a droga, a ciclodextrina pode ser utilizada como um aditivo auxiliar, ou seja, como um carreador, diluente, ou solubilizante. São mais comumente utilizados para esses fins ciclodextrinas de alfa, beta e gama, cujos exemplos podem ser encontrados no WO 91/11172, WO 94/02518 e WO 98/55148.
Está dentro do âmbito da presente invenção que duas ou mais composições farmacêuticas, pelo menos, uma das quais contém um composto de acordo com a invenção, podem ser convenientemente combinadas na forma de um kit adequado para administrar uma combinação, por exemplo, a coadministração das composições.
Assim, o kit da invenção compreende duas ou mais composições farmacêuticas separadas, pelo menos uma das quais contém um composto da fórmula (I), de acordo com a invenção, e os meios para manter separadamente referidas composições, tal como um recipiente, garrafa dividida ou pacote de folha dividida. Um exemplo de tal kit é o pacote de bolha familiar utilizado para a embalagem de comprimidos, cápsulas e outros.
O kit da invenção é particularmente adequado para administrar diferentes formas de dosagem, por exemplo, oral e parenteral, para administrar as composições separadas em intervalos de dosagem diferentes, ou para titulação das composições separadas umas contra as outras. Para auxiliar o cumprimento, o kit compreende normalmente direções para a administração e pode ser provido com um chamado auxiliar de memória.
Para a administração para pacientes humanos, com base em um sujeito humano médio com um peso de aproximadamente 65 kg a aproximadamente 70 kg, a dose diária total dos compostos da invenção é geralmente na faixa de aproximadamente 0,05 mg a aproximadamente 1000 mg, de preferência na faixa de aproximadamente 0,1 mg a aproximadamente100 mg e mais preferencialmente na faixa de aproximadamente 0,5 mg a aproximadamente 20 mg. Dependendo, naturalmente, do modo de administração, por exemplo, a administração oral pode necessitar de uma dose diária total de aproximadamente 1 mg a aproximadamente 500 mg, enquanto que uma dose intravenosa pode exigir apenas de aproximadamente 0,5 mg a aproximadamente 250 mg. A dose diária total pode ser administrada em dose única ou dividida. Estas doses podem ser adequadamente alteradas em função de condições de sexo, idade ou doença de seres humanos.
Conforme discutido acima, um composto da invenção apresenta atividade antagonista de 5-HT2B. Um antagonista de 5-HT2B da presente invenção pode ser útilmente combinado com outro composto farmacologicamente ativo, ou com dois ou mais outros compostos farmacologicamente ativos, particularmente no tratamento do câncer, doenças inflamatórias, doenças imunomodulatórias e desordens gastrointestinais, por exemplo, desordem motora no trato digestivo e irritação sensorial ou ajustes da pressão arterial pulmonar e reparo arterial.
Por exemplo, o antagonista de 5-HT2B, especialmente um composto da fórmula (I), ou um seu sal farmaceuticamente aceitável, conforme definido anteriormente, pode ser administrado simultâneo, seqüencial ou separadamente, em combinação com um ou mais agentes selecionados a partir de:
Laxantes: são citados, por exemplo, Regulan (marca registrada) e Celevac (marca registada);
Fator anticonvulsivante: são citados, por exemplo, mebeverina, pinaverio, brometo de otilonio e trimebutina que têm efeito relaxante do músculo liso, por exemplo dicicloverina, hiosciamina e cimetrópio que têm ações anti-muscarínicas.
Droga de ação opióide/central: são citados, por exemplo, loperamida, naltrexona, metilnaltrexona, modulon (marca registrada) e alvimopan que são MOR agonistas; por exemplo fedotozina e asimadolina que são KOR agonistas; por exemplo imipramina, amitriptilina, clomipramina, desipramina e lofepramina que são os antidepressivos tricíclicos; por exemplo sertralina, paroxetina, fluoxetina e escitalopram que são inibidores de recaptação de serotonina seletiva; por exemplo venlafaxina e duloxetina que são inibidores de recaptação de serotonina- noradrenalina seletiva; por exemplo, moclobemida que é um inibidor reversível da monoamina oxidase, por exemplo diazepam, prazepam, clonazepam e dextofisopam que são agonistas de benzodiazepina; por exemplo oximorfona ER e tramadol, que são analgésicos centrais; por exemplo, isocarboxazid, fenelzina tranicipromina e selegilina, que são inibidores de oxidase de monoamina.
Moduladores de receptor serotoninérgico: são citados, por exemplo, alosetron, ondansetron, tropisetron, palonossetrom, ramosetron, mitrazapina, indisetron, cilansetron, granisetron e dolasetron, que são antagonistas de 5-HT3; por exemplo, tegaserode e mosaprida, que são agonistas de 5-HT4; por exemplo MKC-733, que é agonista de 5-HT3, por exemplo, renzaprida, que é antagonista de 5-HT4/ antagonista de 5-HT3; por exemplo indisetron, que é antagonista de 5- HT3/5-HT4; por exemplo DR-4004, SB-269970, SB-258719 e SB-258741, que são antagonistas de 5-HT7, por exemplo, buspirona e epirona, que são agonistas ou antagonistas de 5-HT1A; por exemplo buspirona, que é agonista de 5- HT1A/1B/D; por exemplo, ergotamina, sumatriptano e rizatriptano, que são drogas de enxaqueca.
Fator de motilidade gastrintestinal: são citados, por exemplo, maropitant, aprepitant e ezlopitant, que são antagonistas de NK1, por exemplo nepadutant e saredutant, que são antagonistas de NK2; por exemplo talnetant, que é antagonista de NK3; por exemplo, CP-154526, NBI-35965 e CRA-1000, que são antagonistas do receptor de CRF1; por exemplo dexloxiglumida, que são antagonistas do receptor de CCK-A; por exemplo mitemcinal e PF-4548043, que são agonistas de motilina, por exemplo lubiprostona, que é agonista de canal de cloreto (tipo 2); por exemplo linaclotide, que é agonista de guanilato ciclase; por exemplo GTP-010, que é agonista de peptídeo-1 como glucagon; por exemplo ibutamoreno e capromorelina, que são agonistas do receptor da grelina.
Antibióticos: são citados, por exemplo, sulfacetamida, eritromicina, rifaximina, tobramicina e ciprofloxacina.
Bactérias probióticas: são citadas, por exemplo, bifidobactérias, não patogênicas, infantis 35624 e E.coli.
Fator antianalgésico: são citados, por exemplo, clonidina, medetomidina, lofexidina, dexmedetomidina e AGN-2-3818, que são drogas de alfa2-adrenérgico; por exemplo solabegron, que é a droga beta3-adrenérgico; por exemplo, GRC- 10622, GW842166 e S-777469, que são agonistas de canabinóide 1 ou 2; por exemplo celecoxib, rofecoxib, vaidecoxib, etoricoxib e lumiracoxib que são inibidores de COX-2 seletivos; por exemplo piroxicam, naproxeno, ibuprofeno, diclofenaco e indometacina, que são drogas antiinflamatórias não-esteróides (NSAIDSs); por exemplo dizocilpina, que é antagonista de NMDA, por exemplo, resiniferatoxina e capsazepina, que são moduladores de TRPs (subtipos V1, V3, V4, M8, A1); por exemplo gabapentina, pregabalina e 3 metiIgabapentina, que são ligantes de alfa-2-delta; por exemplo topiramato, cinolazepam e clonazepam, que são agonistas de GABA.
Fator anti-inflamatório: são citados, por exemplo, dexametasona, prednisolona, ciclesonida, e budesonida, que são hormônios adrenocorticais sintéticos; por exemplo anakinra, atlizumab e mepolizumab, que são terapêuticas a base de interleucina.
Fator anti-alérgico: são citados, por exemplo, montelucaste, zafirlucaste e pranlucaste, que são antagonistas de leucotrienos; por exemplo albuterol, levalbuterol, salmeterol, formotero e arformoterol, que são agoistas de beta-2; por exemplo roflumilaste, tiotrópio e israpafante, que são tratamentos de doença pulmonar obstrutiva de asma e/ou crônicas.
Outras terapêuticas: são citados por exemplo, polyful (marca registrada), Metamucil (marca registrada), crofelemer e cascas de psillium.
Hipertensão pulmonar associada: são citados, por exemplo, beraprost, que é derivado de prostaglandina; por exemplo sildenafil, que é um inibidor de PDE5; por exemplo, bosentano, que é antagonista da endotelina-1.
A seguir, a presente invenção é descrita em detalhes pelos exemplos, mas os exemplos a seguir nunca limitam a presente invenção e várias alterações podem ser feitas sem se afastar do escopo da invenção. Também estão incluídas dentro do escopo da invenção essas várias alterações feitas sem se afastar do escopo da invenção.
A invenção é ilustrada nos seguintes exemplos não limitantes em que, salvo indicação em contrário: todas as operações foram realizadas em temperatura ambiente ou sala, ou seja, na faixa de aproximadamente 18-25°C, a evaporação do solvente foi realizada utilizando um evaporador rotativo sob pressão reduzida, com uma temperatura de banho de até aproximadamente 60°C; as reações foram monitoradas por cromatografia em camada fina (CCD) e tempos de reação são dadas apenas para ilustração; pontos de fusão (m.p) dados não são corrigidos (polimorfismo pode resultar em diferentes pontos de fusão), a estrutura e a pureza de todos os compostos isolados foram assegurados por pelo menos uma das seguintes técnicas: TLC (placas de TLC pré-revestidas de Merck gel de sílica 60 F254 ou placas de HPTLC pré-revestidas de Merck Nh2 F254s), espectrometria de massa, ressonância nuclear magnética (NMR), espectros de absorção no de infravermelho vermelho (IR), ou microanálise. Os rendimentos são apresentados apenas para fins ilustrativos. Cromatografia em coluna Flash foi realizada utilizando WAKO gel de sílica 300HG (40-60 micrômetros) ou Fuji Silysia Chromatorex (marca registrada) DU3050 (Tipo Amino, 30-50 micrômetros) ou sílica Biotage (32-63 mm, KP-Sil) ou amino ligado a sílica Biotage (35-75 mm, KP-NH).
O aparelho de microondas utilizado na reação foi otimizador Emrys (química pessoal) ou Initiator (marca registrada) Sixty (Biotage). O aparelho ultra sônico utilizado na reação foi Ultra Sonic Cleaner de Frequência Única (como ONE) As abreviações de solventes de reação são as seguintes:. Tetrahidrofurano (THF), dimetilsulfóxido (DMSO) e dimetilformamida (DMF). Além disso, 2-(1H- benzotriazol-1-il)-1,1,3,3-tetraetiluronio hexafluorofosfato (HBTU).
A purificação utilizando HPLC no composto final foi realizada pelo aparelho e condições seguintes.
Aparelho: sistema MS-trigger AutoPurification (marca registrada), águas (chamado a seguir de aparelho de purificação A) Coluna: XTerra C18, 19 x 50 mm, partículas de 5 um; Método A: metanol ou acetonitrila/0.05% (v/v) de solução aquosa de ácido fórmico ou Método B: metanol ou acetonitrila/0.01% (v/v) de solução aquosa de amônia. A confirmação da pureza química no método de purificação utilizando aparelho de purificação A foi realizada pelo seguinte aparelho e condições. Aparelho: Acquity ULTRA Parformance LC no TUV Detector e espectrômetro de massa ZQ, waters, Coluna: waters ACQUITY C18, 2,1 x 50mm, partículas de 1.7 micrômetros Temperatura da coluna: 60°C, Taxa de fluxo: 10mL/min, detector de UV: 210nm, Detecção de MS: modo positivo de ESI, método: QC_neutral_full_1pt5min Eluente: solução de acetato de amónio de acetonitrila/10mM, Graduação: 5% (0-0.1min), 5-95% (0,10-8min), 95% (0,8-1 min), Tempo para análise: 1,5 min. A purificação utilizando HPLC foi realizada pelo sehuinte aparelho e condições. Aparelho: sistema de UV-trigger preparative HPLC, waters (chamado, a seguir, de aparelhos de purificação B) Coluna: XTerra MS C18, 5 micrômetros, 19 x 50 mm ou 30 x 50 mm, Detector: UV 254 nm, Taxa de fluxo: 20 ml/min (19 x 50 mm) ou 40 ml/min (30 x 50 mm) à temperatura ambiente. Dados espectrais de massa de baixa resolução (EI) foram obtidos em um espectrômetro de massa Integrity (waters) ou um espectrômetro de massa Automass 120 (JEOL) ou 6890GC/5793MSD (GC-MS Agilent Technologies). Dados espectrais de massa de baixa resolução (ESI), foram obtidos pelo seguinte aparelho e condições. Aparelho: sistema de waters Alliance HPLC no espectrômetro de massa ZQ ou ZMD e detector de UV, Quando alguns átomos de bromo estão contidos na molécula, levando a razão de abundância de isótopo em consideração, dois ou mais valores numéricos podem ser descritos dependendo do número de átomos de bromo. Coluna: waters XTerra (marca registrada) C18, 2,1 x 30 mm, partícula de 3,5 micrômetro, Graduação: 4-96% (0-2 min), 96% (2-4 min), Taxa de fluxo; 0,5 mL/min, Detecção de UV: 254 nm, Detecção de MS: modo de ESI posi/nega, Eluente: acetonitrila/0.025% (v/v) de solução aquosa de formiato de amônio (faixa completa Neutral), acetonitrila/0.05% de solução aquosa de ácido fórmico (faixa completa ácida), acetonitrila/0.01% de solução aquosa de amônia (faixa completa básica).
Os dados de NMR foram determinados em 270 MHz (espectrômetro JEOL JNM-LA 270) ou 300 MHz (JEOL JNM-LA300), utilizando clorofórmio deuterado (99,8% D) ou dimetilsulfóxido (99,9% D) como solvente, salvo indicação em contrário, em relação ao tetrametilsilano (TMS) como padrão interno em partes por milhão (ppm).
As abreviaturas convencionais utilizadas são: s = único, d = duplo, t = terceto, q = quarteto, m = multipleto, br.= amplo, etc. Espectros de IR foram medidos por um espectrômetro de infravermelho Shimadzu (IR-470). Rotações ópticas foram medidas utilizando um Polarímetro Digital JASCO DIP-370 (Japan Spectroscopic Co., Ltd.). Os símbolos químicos têm seus significados usuais; b.p. (Ponto de ebulição), m.p. (ponto de fusão), L (litro(s)), mL (mililitro(s)), g (grama (s)), mg (miligrama(s)), mol (moles), mmol (milimoles). Em seguida, grupo de proteção Ts significa ácido p-toluenosulfônico.
Um pequeno pedaço de metal de sódio (75,87 g, 2,79 mol) foi adicionado com várias porções de metanol refrigerado de gelo (1,2 L). Depois de desaparecer o metal de sódio, a mistura de ácido oxálico de dimetil (300 g, 2,541 mol) e acetonitrila (114,72 g, 2,79 mol) em metanol foi adicionada gota a gota à solução de metanol de metóxido de sódio preparada acima. Em seguida, concentrando-se a mistura sob vácuo deu o amarelo intermediário 2 (377,4 g, 99,5% de rendimento). Os intermediário 2 foi utilizados na próxima etapa sem purificação adicional.
O ácido sulfônico (23,4 g, 0,238 mol) foi adicionado gota a gota à 40% de solução de metilhidrazina (25 g, 0,217 mol). Depois de adicionado completamente, a mistura de reação foi agitada por 4 horas. A mistura resultante foi seca com gelo seco e o intermediário 3 foi obtido.
O intermediário 3 (280 g, 1,94 mol) foi adicionado à suspensão de metanol (1,5 L) do intermediário 2 (263,39 g, 1,77 mol). A mistura resultante foi agitada por 48 horas em temperatura ambiente. Depois disso, 2 M de hidróxido de sódio (200 ml) e diclorometano (1 L) foram adicionados à mistura de reação, e a camada orgânica foi separada. Depois a camada orgânica resultante foi lavada com cloreto de sódio saturado, e foi seca sobre sulfato de sódio anidroso. Depois de filtrado o agente de secagem, concentrando o filtrado resultante no vácuo deu cristal amarelo do crude intermediário 4 (82.20g, rendimento de 30%). O intermediário 4 resultante foi utilizado na etapa seguinte, sem purificação adicional.
Um solução de sulfito de sódio (17,06 g, 0,247 mol) foi adicionada cuidadosamente gota a gota à solução de água-ácido acético (3/1 (v/v), 300 ml) do intermediário 4 (32,0 g, 0,206 mol) e iodeto de potássio (342,4 g, 2,06 mol). Depois de gotejar, a mistura de reação foi agitada por 3 horas a 0oC. Após a confirmação do consumo do intermediário 4 com acetato de TLC (acetato de etila: hexano = v (1: 4 (v/v)), a mistura resultante foi ajustada para pH10 para pH11 com a adição de carbonato de hidrogênio de sódio sólido. A camada aquosa foi extraída com 1100 mL de acetato de etila três vezes. Em seguida, a camada orgânica coletada foi seca sobre sulfato de sódio anidroso. Após filtração, o filtrado concentrado sob pressao reduzida deu um intermediário 5 cru, viscoso e marrom. O itermediário 5 cru resultante foi purificado por cromatografia em coluna utilizando gel de sílica (acetato de etila / éter de petróleo, (0/1-1/3 (v / v)) e o intermediário 5 (16,4 g, 30% de rendimento) como um cristal branco.1H-NMR(270 MHz, CDCl3) δ 6.98(s, 1H), 4.01(s, 3H), 3.92(s, 3H). MS(ESI)m/z: [M+H]+267.
Uma solução de dioxiano absoluto (25 mL) de uma mistura de 1H- indol-(2,50 g, 21,3 mmol), [Ir (OMe)(COD)]2(28,4 mg, 0,042 mmol), 4,4'-diterc-butil -2,2'-bipiridil(dtbpi)(22,9 mg, 0,085 mmol) e bis(pinacolato)diborano (3,25 g, 12,8 mmol) foi agitada a 80°C durante 1 hora. Esta solução de reação foi utilizada na próxima etapa sem nenhuma purificação.
O intermediário 5 (3,73 g, 14,0 mmol), tris(dibenzilidenacetona) dipaládio (0)(128 mg, 0,14 mmol), solução de fosfato de potássio (4,88 g, 23 mmol), triciclohexilfosphino (78,5 mg, 0,28 mmol) e água (3 mL) foi adicionada à solução de mistura de reação acima. 1H-NMR(270 MHz, CDCl3) δ 8.40(br s, 1H), 7.68(d, J=7.3Hz, 1H), 7.44(d, J=6.6Hz, 1H), 7.31-7.25(m, 1H), 7.21-7.15(m, 1H), 7.04(s, 1H), 6.75(s, 1H), 4.15(s, 3H), 3.97(s, 3H).
A solução de reação de metanol(15 ml)-THF(5 mL) do intermediário 6 (742 mg, 2,91 mmol), 2M de solução de hidróxido de sódio (5 mL, 10 mmol) foram agitados a 70°C durante 1 hora. Depois de arrefecer para a temperatura ambiente, a mistura de reação foi ajustada para pH3 com 2M de solução de HCl e diluída com cloreto de sódio saturado. A mistura foi extraída com diclorometano, a camada orgânica combinada foi seca com sulfato de magnésio anidroso. Após a filtração, a camada orgânica foi concentrada sob pressão reduzida e o produto cru do intermediário 7 (673 mg, 96%) foi isolado como um sólido branco. 1H-NMR(300 MHz, DMSO-d6) δ 11.58(s, 1H), 7.61(d, J=8.0Hz, 1H), 7.42(d, J=8.0Hz, 1H), 7.22-7.10(m, 2H), 7.09-7.02(m, 1H), 6.88(s, 1H), 4.12(s, 3H). Não foi observado pico causado por NH. MS(ESI)m/z: [M+H]+242, [M-H]-240.
Os intermediário 5 (3.14g, 11,8 mmol), acetato de paládio (265 mg, 1,18 mmol) e trifenilfosfina (1.24g, 4,71 mmol) foi dissolvido em solução de dioxano/tolueno (3,5/1 (v/v), 27 ml). A solução resultante foi agitada em temperatura ambiente durante 10 minutos. Depois disso, terc-butil-2- (dihidróxiboranil)-1H-indol-1-ester ácido carboxílico (4.00g, 15,3 mmol), água (3 mL) e carbonato de sódio (3,12 g, 29,5 mmol) foi adicionado à solução de reação. A solução foi refluída durante 1,5 horas. Após o resfriamento, a solução de reação foi adicionado à água (150 ml). Então a camada aquosa foi extraída com acetato de etila (150 mLx2). Depois a camada orgânica resultante foi seca sobre sulfato de magnésio, os agentes de secagem foram filtrados. O filtrado foi concentrado sob pressão reduzida. O resíduo foi pré-tratado com cromatografia de coluna (acetato de etila) utilizando gel de sílica tratado com amina. Então, o intermediário 8 (1.72g, rendimento de 41%) foi obtido como sólido branco pela purificação utilizando cromatografia em coluna de gel de sílica (hexano-éter (1.5/1-1/1) (v/v)). 1H-NMR(300MHz, CDCl3) δ 8.29(d, J=8.1Hz, 1H), 7.61(d, J=8.0Hz, 1H), 7.42(t, J=7.3Hz, 1H), 7.31(t, J=7.3Hz, 1H), 6.88(s, 1H), 6.71(s, 1H), 3.96(s, 3H), 3.79(s, 3H), 1.39(s, 9H). MS(ESI)m/z: [M+H]+356.
Uma solução de hidróxido de sódio de 2M (12,1 mL, 24,2 mmol) foi adicionada a uma solução de metanol do intermediário 8 (1,72 mmol g, 4,84) em 50°C durante 7 horas. Após o resfriamento, a mistura resultante foi adicionada à solução de reação até pH3. Em seguida, foi adicionada água à mistura. O precipitado resultante foi filtrado e foi lavado com pequena quantidade de água. O intermediário 7 (106 g, rendimento de 90%) foi obtido como sólido branco. 1H-NMR(300MHz, DMSO-d6) δ 11.59(s, 1H), 7.61(d, J=7.3Hz, 1H), 7.42(d, J=8.1Hz, 1H), 7.18(t, J=8.0Hz, 1H), 7.14(s, 1H), 7.06(t, J=8.1Hz, 1H), 6.88(s, 1H), 4.12(s, 3H). NENHUM PICO CAUSADO POR NH FOI OBSERVADO. MS(ESI)m/z: [M+H]+242, [M-H]-240.
Carbonato de sódio (2,53 g, 23,89 mmol) e água (3 mL) foi adicionado ao intermediário 5 (2,50 g, 9,56 mmol), acetato de paládio (II) (215 mg, 0,96 mmol), trifenilfosfina (100 g, 3,82 mmol) e terc-butil2-(dihidróxiboranil)-5-fluoro-1H- indol-1- éster ácido carboxílico (3,20 g, 11,47 mmol) na mistura de dioxano (20 mL) e tolueno (10 mL). A mistura resultante foi refluída durante 1,5 horas. Após o resfriamento para a temperatura ambiente, a solução de reação foi adicionada à água (80ml) e extraída com acetato de etila (100 mL x 2). O extrato combinado foi seco sobre sulfato de magnésio anidroso. O agente de secagem é filtrado. O filtrado foi concentrado sob pressão reduzida. O resíduo obtido foi purificado com cromatografia de coluna (hexano/acetato de etila = 2:1 (v/v)) utilizando gel de sílica de amina. Em seguida, o intermediário 10 (990 mg, rendimento de 37%) foi obtida como sólido branco. MS(ESI)m/z: [M+H]+374.
Uma solução de hidróxido de sódio de 2M (6,6 mL, 13,26 mmol) foi adicionado em solução de THF (10 mL) do intermediária 9 (990 mg, 2,65 mmol). A solução resultante foi agitada a 45°C durante 2 horas e foi agitada à temperatura ambiente durante a noite. A solução de reação foi concentrada sob pressão reduzida. Então, uma solução de HCl de 2M (8 mL) foi adicionada ao resíduo obtido. A solução resultante foi extraída com acetato de etila (120 mL x 2), e o extrato combinado foi seco sobre sulfato de magnésio anidroso. Depois que os agentes de secagem foram filtrados, o filtrado foi concentrado sob pressão reduzida. Em seguida, o intermediário 10 cru (680 mg, rendimento de 100%) foi obtido como sólido amarelo marrom-claro 1H-NMR (300 MHz, CDCl3/DMSO-d6(1drop)) δ 10.49(br s, 1H), 7.40-7.25(m, 2H)7.13(s, 1H), 6.66(s, 1H), 7.00-6.94(m, 1H), 4.15(s, 3H). MS(ESI)m/z: [M+H]+260, [M-H]-258.
Uma solução de DMF (0,5 mL) de hexafluorofosfato O-benzotriazol-1- il-N,N,N',N'-tetrametiluronio (36 mg, 1,5 equivalente) foi adicionada a uma solução de DMF (0,5 mL) do composto intermediário 7 (15 mg), amino (1,1 equivalente, 14 mg como 2-(piperidina-1-il)etano-1-amina), trietilamina (0,026 mL, 3 equivalente) na temperatura ambiente. A solução resultante foi agitada a 50°C durante 2 horas. Depois a solução resultante foi concentrada sob pressão reduzida, uma solução de hidróxido de sódio de 1M (0,5 mL) foi adicionada ao resíduo, e a mistura foi extraída com acetato de etila (1 mL) duas vezes. O resíduo da “camada” orgânica combinada foi dissolvido em uma pequena quantidade de metanol. A solução foi carregada para um cartucho de SCX (cartucho de forte troca de cátions), seguido por lavagem com metanol (10 mL) e, finalmente, foi eluatado com uma solução de amônia-metanol de 1M (8 mL). O produto cru obtido pela concentração foi purificado com um HPLC preparativo (o aparelho de purificação A escrito no início dos {exemplos}). MS (ESI) m/z: [M + H]+352.
Exemplos sintetizados pela utilização de reação similar à descrita acima são indicados abaixo: Composto de exemplo 2: 5-(1H-indol-2-il)-N-[2-(4-metoxipiperidin-1-il)etil]-1-metil-1H-pirazol-3-carboxamida Composto de exemplo 3: N-[2-(4-hidroxipiperidin-1-il)etil]-5-(1H-indol-2-il)-1-metil-1H-pirazol-3-carboxamida Composto de exemplo 4: N-[2-(4-etilpiperidin-1-il)etil]-5-(1H-indol-2-il)-1-metil-1H-pirazol-3-carboxamida Composto de exemplo 5: N-[2-(3-hidroxipiperidin-1-il)etil]-5-(1H-indol-2-il)-1-metil-1H-pirazol-3-carboxamida Composto de exemplo 6: 1-(2-{[5-(1H-indol-2-il)-1-metil-1H-pirazol-3-il]formamida}etil)piperidine-3- carboxamida Composto de exemplo 7: 5-(1H-indol-2-il)-1-metil-N-{2-[4-(propan-2-il)piperidin-1-il]etil}-1H-pirazol-3- carboxamida Composto de exemplo 8: 5-(1H-indol-2-il)-1-metil-N-[2-(4-metilpiperidin-1-il)etil]-1H-pirazol-3-carboxamida Composto de exemplo 9: 5-(1H-indol-2-il)-1-metil-N-[2-(pirrolidin-1-il)etil]-1H-pirazol-3-carboxamida Composto de exemplo 10: 5-(1H-indol-2-il)-1-metil-N-[2-(2-metilpiperidin-1-il)etil]-1H-pirazol-3-carboxamida Composto de exemplo 11: N-[2-(4,4-difluoropiperidin-1-il)etil]-5-(1H-indol-2-il)-1-metil-1H-pirazol-3- carboxamida Composto de exemplo 12: N-[2-(2,6-dimetilmorpholin-4-il)etil]-5-(1H-indol-2-il)-1-metil-1H-pirazol-3-carbox- amida Composto de exemplo 13: 5-(1H-indol-2-il)-1-metil-N-[3-(morpholin-4-il)propil]-1H-pirazol-3-carboxamida Composto de exemplo 14: 5-(5-fluoro-1H-indol-2-il)-N-{2-[(2S)-2-(metoximetil)pirrolidin-1-il]etil}-1-metil-1H- pirazol-3-carboxamida Composto de exemplo 15: 5-(5-fluoro-1H-indol-2-il)-N-{2-[(2R)-2-(metoximetil)pirrolidin-1-il]etil}-1-metil-1H- pirazol-3-carboxamida Composto de exemplo 16: 5-(5-fluoro-1H-indol-2-il)-N-{2-[(3S)-3-hidroxipirrolidin-1-il]etil}-1-metil-1H-pirazol-3- carboxamida Tabela 1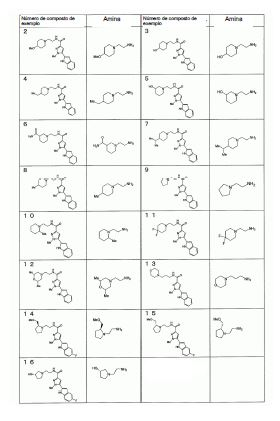 Tabela 2
Tabela 2
Tris(dibenzilidenoacetona)dipaládio(0) (540 mg, 0,59 mmol) foi adicionado à solução misturada do intermediário 5 (1,57 g, 5,90 mmol), 3-ácido quinolinaborônico (102 g, 5,90 mmol), fosfato de potássio (1,88 g , 8,85 mmol) e 1,4-dioxano (65 mL) de tri(ciclohexil)fosfina (165 mg, 0,59 mmol) e água (15 mL). A mistura de reação foi agitada a 100°C durante a noite (15 horas). Após arrefecimento à temperatura ambiente, a solução misturada foi diluído com um solvente de acetato de etila, e foi filtrada através de celite para a finalidade de eliminar o catalisador. Após a camada orgânica do filtrado ser separada, a camada aquosa foi extraída novamente com um solvente de acetato de etila. Após a camada combinada orgânica ser lavada com uma solução saturada de cloreto de sódio saturado, seca sobre sulfato de sódio. Após filtração, o resíduo foi obtido. A purificação do resíduo com cromatografia de coluna (hexano-acetato de etila = (1/1)(v/v) a (2/3)(v/v)) usando gel de sílica concedeu o intermediário 11 (873 mg, rendimento de 55%) como cristal ligeiramente amarelo. 1H-NMR (300 MHz, CDCl3) δ 8.99(d, J=2.2Hz, 1H), 8.23(d, J=2.2Hz, 1H), 8.18(d, J=8.8Hz, 1H), 7.79-7.75(m, 2H), 7.54-7.52(m, 1H), 7.04(s, 1H), 4.05(s, 3H),3.98(s, 3H). MS(ESI)m/z; [M+H]+268.
Uma solução de metanol (30 mL) do intermediário 11 (870 mg, 3,25 mmol) e 2M de solução de hidróxido de sódio (4,20 mL, 8,20 mmol) foi agitada a 75°C durante 2 horas. Após a remoção do solvente, a solução remanescente foi ajustada para PH6 a 7 com uma solução de ácido clorídrico. O sólido precipitado foi sujeito a filtração de sucção, seco na presença de pentóxido de fósforo em vácuo, e concedeu o intermediário 12 (790 mg, rendimento de 96%), como um cristal marrom claro. 1H-NMR (300 MHz, DMSO-d6) δ 12.8(br s, 1H), 9.11(d, J=2.2Hz, 1H), 8.69(d, J=2.2Hz, 1H), 8.1,3-8.04(m, 2H), 7.90-7.81(m, 1H), 7.75-7.66(m, 1H), 7.13(s, 1H), 4.05(s, 3H). MS(ESI)m/z; [M+H]+254, [M-H]-252.
De acordo com a forma similar do Composto de exemplo 1, o Composto de exemplo 17 (628 mg, 86% de rendimento) foi obtido como um cristal branco a partir do intermediário 12 (506 mg, 2,00 mmol) e 4-(2-aminoetil)morfolina (286 mg, 2,20 mmol). 1H-NMR (300 MHz, CDCl3) δ 8.99 (d, J=2.2Hz, 1H), 8.21(d, J=2.2Hz, 1H), 8.17(d, J=8.1Hz, 1H), 7.90(d, J=8.1Hz, 1H), 7.94-7.76(m, 1H), 7.70-7.60(m, 1H), 7.34- 7.22(m, 1H), 7.01(s, 1H), 4.00(s, 3H), 3.79-3.72(m, 4H), 3.63-3.54(m, 2H), 2.66- 2.48(m, 6H). MS(ESI)m/z; [M+H]+366.
Exemplos sintetizados pela utilização da reação similar descrita acima são indicados abaixo: Composto de exemplo 18: 1-metil-N-[2-(piperidina-1-il)etil]-5-(quinolin-3-il)-1H-pirazol-3-carboxamida Composto de exemplo 19: 1-metil-N-[2-(pirrolidin-1-il)etil]-5-(quinolin-3-il)-1H-pirazol-3-carboxamida Tabela 3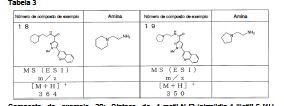
Intermediário 13: Síntese de terc-butil 2-(dihidróxiboranil)-1H-pirrolo[3,2-b]-piridina-1 -carboxilato n-Butil lítio (8,3 mL, 1,65 m, solução de hexano) foi adicionado gota a gota à solução de THF (10 ml) de diisopropilamina (1,39 g, 13,8 mmol) em condição de de frio gelado sob a atmosfera de nitrogênio durante 5 minutos. Agitando a mistura obtida em condições de frio gelado durante 20 minutos concedeu uma solução de THF de diisopropilamida de lítio.
A solução de THF de diisopropilamida de lítio preparada acima foi adicionada gota a gota à mistura de THF (20 ml) de terc-butil-1H-pirrolo-[3,2-b]piridina-1- carboxilato 12 (2,00 g, 9,16 mmol) e éster triisopropil de ácido bórico (2,76 g, 14,66 mmol) a -20°C sob a atomosphere de nitrogênio durante 1 hora. Após a mistura resultante ser agitada a -10°C durante 3 horas, uma solução de hidrogenossulfato de potássio 10% foi adicionada à solução de reação e a mistura resultante foi extraída com acetato de etila. A camada de extrato obtida foi lavada com uma solução de cloreto de sódio saturada, e foi seca sobre sulfato de magnésio anidroso. Após filtração, o filtrado foi concentrado sob pressão reduzida para isolar intermediário 13 cru. O intermediário 13 obtido foi suspenso em éter diisopropil. A precipitação obtida foi filtrada e o intermediário 13 cristalino (1,90 g, rendimento de 79%) foi obtida. 1H-NMR (300 MHz, DMSO-d6) δ 8.45(1H, d, J=4.4Hz), 8.36(2H, s), 8.33(1H, d, J=8.8Hz), 7.28(1H, dd, J=8.1, 5.1Hz), 6.72(1H, s), 1.61(9H, s). MS(ESI)m/z: [M+H]+263.
De acordo com o método de geração do intermediário 8, o intermediário 14 (0,37 g, 27% de rendimento) foi sintetizado a partir do intermediário 13 (1,3 g, 5,0 mmol) e o intermediários 5 (10 g, 3,8 mmol). 1H-NMR (300 MHz, DMSO-d6) δ 8.61(1H, d, J=5.1Hz), 8.53(1H, d, J=8.8Hz), 7.34(1H, dd, J=8.8, 5.1Hz), 6.93(2H, s), 3.96(3H, s), 3.82(3H, s), 1.41(9H, s). MS(ESI)m/z: [M+H]+357.
De acordo com o método similar (hidrólise) da síntese alternativa descrita no intermediário 7, o intermediário 15 (87mg) foi sintetizado a partir do intermediário 14 (210 mg, 0,59 mmol) com 61% de rendimento. 1H-NMR(300 MHz, DMSO-d6) δ 11.83(1H, br s), 8.38(1H, d, J=2.9Hz), 7.80(1H, d, J=8.1Hz), 7.21(1H, s), 7.18(1H, dd, J=4.4, 8.1Hz), 7.04(1H, s), 4.15(3H, s). Nenhum pico causado por NH foi observado. MS(ESI)m/z: [M+H]+243.
O intermediário 15 (120 mg, 0,5 mmol) e N-(2-amino-etil)pirrolidina (59 mg, 0,5 mmol) foi dissolvido em DMF (4 mL). Trietilamina (260 mg, 2,6 mmol) e N- (3-dimetilaminopropil)-N'-hidrocloreto de etilcarbodiimida (98 mg, 0,5 mmol) e 1- hidróxibenzotriazol monohidrato (39 mg, 0,3 mmol) foram sucessivamente adicionados à mistura resultante em temperatura ambiente. A mistura obtida foi agitada em temperatura ambiente durante 2 dias. O solvente foi removido sob pressão reduzida, e o produto resultante cru foi purificado com um sistema de HPLC preparativo (o aparelho de purificação B) para conceder o produto cristalino (15 mg, 9% de rendimento). MS(ESI)m/z: [M+H]+339.
Um sistema de HPLC preparativo (o aparelho de purificação A), que foi utilizado para a purificação no Composto de exemplo 1, foi utilizado para purificação adicional.
De acordo com o método do Composto de exemplo 20, o Composto de exemplo 21 (16 mg, 34% de rendimento) foi obtido a partir do intermediário 15 (30 mg, 0,12 mmol) e 1-(2-aminoetil)piperidin-3-ol (27 mg, 0,19 mmol). MS(ESI)m/z:[M+H]+369.
Um sistema de HPLC preparativo (o aparelho de purificação A), que foi utilizado para a purificação no Composto de exemplo 1, foi utilizado para purificação adicional.
Uma solução de hidróxido de sódio de 2M (56,4 mmol, 28,2 ml) foi adicionada a uma solução de metanol (100 mL) do intermediário 5 (6,0 g, 22,6 mmol) e a mistura foi aquecida a 50°C. Duas horas depois, o solvente de metanol foi removido sob pressão reduzida, e a solução restante foi ajustada para pH2 a 3, com uma solução de HCl de 2M sob condição de frio gelado. Após o cristal precipitado obtido ser dissolvido em acetato de etila, a camada orgânica foi separada e, em seguida a camada aquosa foi extraída com acetato de etila duas vezes. Depois a camada orgânica extraída combinada ser lavada com uma solução de cloreto de sódio saturada, e seca sobre sulfato de sódio anidroso. Após a filtração, o filtrado foi concentrado sob pressão reduzida para conceder um intermediária 16 cru (5,68 g, quantitativo) como um sólido amarelo claro. 1H-NMR (270 MHz, DMSO-d6) δ 12.81(br s, 1H), 6.89(s, 1H), 3.92(s, 3H). MS(ESI)m/z; [M+H]+253, [M-H]-251.
Intermediário 17: Síntese da Síntese de 5-iodo1-metil-N-[2-(morfolin-4-il)etil]-1H- pirazol-3-carboxamida 4-(2-amino-etil)morfolina (4,15 g, 31,9 mmol), trietilamina (12,1 mL, 86,9 mmol), 1-hidroxibenzotriazol monohidrato (8,9 g, 57,9 mmol) e N-(3- dimetilamino-propil)-N’-hidrocloreto de etilcarbodiimida (11,1 g, 57,9 mmol) foram sucessivamente adicionados a uma solução de diclorometano (150 ml) do intermediário 16 (7,3 g, 29,0 mmol) à temperatura ambiente. Após a mistura resultante ser agitada em temperatura ambiente durante 20 horas, a solução de bicarbonato de sódio saturada foi adicionada à solução de reação, e a camada aquosa foi extraída com diclorometano. A camada orgânica obtida foi seca sobre sulfato de sódio anidroso. Após a filtração, o filtrado foi concentrado sob pressão reduzida, e o intermediári 17 cru foi isolado. A purificação com cromatografia de coluna (acetato de etila/metanol = 9/1 (v/v)) utilizando gel de sílica concedeu o intermediário 15 (9,8 g, rendimento de 93%), como um cristal branco. 1H-NMR (270MHz, CDCl3) δ 6.95(s, 1H), 3.95(s, 3H), 3.80-3.64(m, 4H), 3.62- 3.40(m, 2H), 2.57(t, J=6.3Hz, 2H), 2.54-2.37(m, 4H). Nenhum pico causado por NH foi observado. MS(ESI)m/z; [M+H]+365.
Trietilamina (6,12 mL, 43,9 mmol) foi adicionado a uma solução de THF (45 mL) do intermediário 17 (4,00 g, 10,98 mmol) de iodeto de cobre (I) (209 mg, 1,10 mmol), trimetilsililacetileno (2,33 mL, 16,5 mmol) e diclorobis(acetonitrila)paládio(II) cloreto (770 mg, 1,10 mmol), sob a atmosfera de nitrogênio e a mistura de reação foi agitada à temperatura ambiente durante 2 horas. Depois disso, a mistura resultante foi filtrada através de celite e o filtrado obtido foi concentrado sob pressão reduzida. O resíduo foi purificado com cromatografia de coluna (diclorometano/metanol = 30/1 (v/v)), utilizando gel de sílica para conceder o intermediário 18 (3,67 g, 100% de rendimento) como um sólido amarelo-marrom. 1H-NMR (300 MHz, CDCl3) δ 7.22-7.14(br s, 1H), 6.90(s, 1H), 3.93(s, 3H), 3.78- 3.70(m, 4H), 3.58-3.48(m, 2H), 2.62-2.45(m, 6H), 0.28(s, 9H). MS(ESI)m/z; [M+H]+335.
Uma solução de metanol (60 mL) do intermediário 18 (3,43 g, 10,3 mmol) e carbonato de potássio (2,13 g, 15,4 mmol) foi agitada à temperatura ambiente durante 1,5 horas. Depois disso, a mistura de reação foi filtrada através de celite e o filtrado obtido foi concentrado sob pressão reduzida. O resíduo foi diluído com uma aolução/água de solução de cloreto de sódio saturada (1/1 (V/V)) (40 ml) e diclorometano (200 ml). Após o procedimento de extração, a camada orgânica foi separada. Após a camada orgânica obtida ser seca sobre sulfato de sódio anidroso, o agente de secagem foi filtrado e o filtrado foi concentrado sob pressão reduzida. O resíduo foi purificado com cromatografia de coluna (diclorometano/metanol = (30/1-20/1) (v/v)), utilizando gel de sílica para conceder o intermediário 19 (2,23 g, 83% de rendimento) como sólido marrom-claro. 1H-NMR (270MHz, CDCl3) δ 7.20(br s, 1H), 6.96(s, 1H), 3.96(s, 3H), 3.77-3.70(m, 4H), 3.58-3.48(m, 3H), 2.62-2.46(m, 6H). MS(ESI)m/z; [M+H]+263.
Acetato de paradio (19 mg, 0,027 mmol) foi adicionado à solução de acetonitrila (5,0 mL) do intermediário 19 (308 mg, 1,18 mmol), 3-amino-2-bromo- 6-metilpiridina (200 mg, 107 mmol), 1,4-bis(difenilfosfina)butano (dppb) (18,3 mg, 0,043 mmol) e carbonato de potássio (444 mg, 3,21 mmol), sob a atmosfera de nitrogênio. A mistura resultante foi agitada em 80°C durante 15 horas. Depois disso, a mistura resultante foi filtrada através de celite e o filtrado obtido foi concentrado sob pressão reduzida. O resíduo foi purificado com cromatografia de coluna (diclorometano/metanol = 25/1-10/1 (v/v)), utilizando gel de sílica para conceder o intermediário 20 (36,9 mg, 9,4% de rendimento) como um cristal amarelo escuro. 1H-NMR (300 MHz, CDCl3) δ 7.28-7.18(br s, 1H), 7.03-6.98(m, 3H), 4.14(br s, 2H), 4.04(s, 3H), 3.78-3.71(m, 4H), 3.59-3.50(m, 2H), 2.63-2.45(m, 6H), 2.47(s, 3H). MS(ESI)m/z; [M+H]+369.
Terc-butóxido de potássio (56 mg, 0,5 mmol) foi adicionado à solução de DMF (1 mL) do intermediário 20 (36,9 mg, 0,1 mmol) em temperatura ambiente em um período. A mistura resultante foi aquecida sob agitação a 35°C durante 2 horas. Após a reação completa, a mistura resultante foi tratada com água (0,5 mL) e o solvente foi removido pela concentração sob pressão reduzida. O resíduo obtido foi purificado com cromatografia de coluna (diclorometano/metanol = 25/1- 10/1(v/v)), utilizando gel de sílica para conceder o Composto de exemplo 22 (31,3 mg, 84% de rendimento) como um cristal de amarelo claro. 1H-NMR (300 MHz, CDCl3) δ 10.4(br s, 1H), 7.70(d, J=8.8Hz, 1H), 7.50-7.40(m, 1H), 7.42(s, 1H), 7.06(d, J=8.8Hz, 1H), 6.87(s, 1H), 4.15(s, 3H), 3.80-3.69(m, 4H), 3.67-3.55(m, 2H), 2.68(s, 3H), 2.66-2.57(m, 2H), 2.55-2.44(m, 4H). MS(ESI)m/z: [M+H]+369, [M-H]-367.
De acordo com o método sintético do intermediário 20, o intermediário 21 (46,8 mg, 13,3% de rendimento) foi obtido como amorfo amarelo-escuro a partir do intermediário 19 (240 mg, 0,915 mmol) e 3-amino-2-bromo-4,6- dimetilpiridina (184 mg, 0,915 mmol). 1H-NMR (300 MHz, CDCl3) δ 7.30-7.20(m, 1H), 7.02(s, 1H), 6.91(s, 1H), 4.11(br s, 2H), 4.04(s, 3H), 3.80-3.71(m, 4H), 3.60-3.50(m, 2H), 2.65-2.48(m, 6H), 2.43(s, 3H), 2.19(s, 3H). MS(ESI)m/z: [M+H]+383, [M+HCO2]-427.
De acordo com o método sintético similar do composto de exemplo 22, o composto de exemplo 23 (31,3 mg, 84% de rendimento) foi obtido como um cristal de amarelo claro a partir do intermediário 21 (46,8 mg, 0,122 mmol). 1H-NMR (300 MHz, CDCl3) δ 9.75(br s, 1H), 7.40-7.35(m, 1H), 7.18(s, 1H), 6.88(s, 1H), 6.82(s, 1H), 4.09(s, 3H), 3.80-3.67(m, 4H), 3.57-3.44(m, 2H), 2.62(s, 3H), 2.60-2.40(m, 6H), 2.54(s, 3H). MS(ESI)m/z: [M+H]+383, [M-H]-381.
Trietilamina (404 mg, 4,0 mmol) foi adicionado a uma solução de THF (6 mL) do intermediário 19 (262 mg, 1,00 mmol), 2-amino-3-iodopiridina (264 mg, 1,20 mmol), diclorobis(acetonitrila)paládio (II), (70,1 mg, 0,1 mmol) e cobre(I) (19 mg, 0,1 mmol) sob a atmosfera de nitrogênio. A solução de reação foi agitada em temperatura ambiente durante 7,5 horas. Depois disso, a mistura resultante foi filtrada através de celite e o filtrado obtido foi concentrado sob pressão reduzida. O resíduo foi purificado com cromatografia de coluna (diclorometano/metanol = 20/1 (v/v)), utilizando gel de sílica para conceder o intermediário 22 (207 mg,58,4% de rendimento) como um cristal amarelo. 1H-NMR (300 MHz, CDCl3) δ 8.14-8.08(m, 1H), 7.65-7.60(m, 1H), 7.27-7.20(m, 1H), 6.99(s, 1H), 6.73-6.65(m, 1H), 5.06(br s, 2H), 4.01(s, 3H), 3.78-3.71(m, 4H), 3.59-3.50(m, 2H), 2.63-2.47(m, 6H). MS(ESI)m/z: [M+H]+355, [M-H]-353.
De acordo com o método sintético similar do composto de exemplo 22, o composto de exemplo 24 (51,5 mg, 26% de rendimento) foi obtido como um cristal amarelo a partir do intermediário 22 (200 mg, 0,56 mmol). 1H-NMR (300 MHz, CDCl3) δ 12.2(br s, 1H), 8.45-8.38(m, 1H), 8.04-7.97(m, 1H), 7.36(s, 1H), 7.40-7.30(m, 1H), 7.23-7.14(m, 1H), 6.69(s, 1H), 4.16(s, 3H), 3.82- 3.73(m, 4H), 3.71-3.62(m, 2H), 2.70-2.61(m, 2H), 2.60-2.50(m, 4H). MS(ESI)m/z: [M+H]+355, [M-H]-353.
De acordo com o método sintético do intermediário 22, o intermediário 23 (112 mg, 41% de rendimento) foi obtido como um sólido marrom escuro por aquecimento sob agitação em 100°C durante 16 horas forma o composto 19 (200 mg, 0,762 mmol) e 4-bromopirimidin-5-amina (159 mg, 0,914 mmol). 1H-NMR (300 MHz, CDCl3) δ 8.64(s, 1H), 8.34(s, 1H), 7.09(s, 1H), 4.41(br s, 2H), 4.06(s, 3H), 3.82-3.69(m, 4H), 3.64-3.51(m, 2H), 2.62-2.46(m, 6H). No peak was observed caused by NH. MS(ESI)m/z: [M+H]+356, [M-H]-354.
De acordo com o método sintético similar do composto de exemplo 22, o composto de exemplo 25 cru (41 mg, o rendimento de 37%) foi obtido como um sólido marrom claro a partir do intermediário 23 (112 mg, 0,315 mmol). 1H-NMR (300 MHz, CDCl3) δ 11.51(br s, 1H), 9.03(s, 1H), 9.00(s, 1H), 7.65(s, 1H), 6.94(s, 1H), 4.22(s, 3H), 3.80-3.62(m, 6H), 2.67-2.42(m, 6H). No peak was observed caused by NH. MS(ESI)m/z: [M+H]+356, [M-H]-354.
Um sistema de HPLC preparativo (o aparelho de purificação A), que foi utilizado para a purificação no composto do exemplo 1, foi utilizado para purificação adicional.
De acordo com o método sintético similar do intermediário 22, o composto 19 (200 mg, 0,762 mmol) e 5-iodopirimidin-4-amina (253 mg, 1,14 mmol) foram aquecidos sob agitação em 90°C durante 16 horas para conceder o intermediário 24 cru (129 mg), como xarope amarelo claro. 1H-NMR (300 MHz, CDCl3) δ 8.56(s, 1H), 8.44(s, 1H), 7.32-7.28(m, 1H), 7.00(s, 1H), 6.08(br s, 2H), 4.00(s, 3H), 3.76-3.73(m, 4H), 3.57-3.51(m, 2H), 2.63-2.49(m, 6H). MS(ESI)m/z: [M+H]+356, [M-H]-354.
De acordo com o método sintético similar do composto de exemplo 22, o intermediário 24 (115 mg, 0,324 mmol) foi aquecido sob agitação em 70°C durante 1 hora. Em seguida, composto de exemplo cru 26 (41 mg, 37% de rendimento) foi obtido como um sólido marrom claro. 1H-NMR (300 MHz, CDCl3) δ 12.01(br s, 1H), 9.03(s, 1H), 8.90(s, 1H), 7.89(s, 1H), 7.55-7.51(m, 1H), 6.77(s, 1H), 4.22(s, 3H), 3.96-3.90(m, 2H), 3.78-3.75(m, 4H), 2.72-2.51(m, 6H). MS(ESI)m/z: [M+H]+356, [M-H]-354.
Um sistema de HPLC preparativo (o aparelho de purificação A), que foi utilizado para a purificação no composto de exemplo 1, foi utilizado para purificação adicional.
De acordo com o método sintético similar do intermediário 20, o intermediário 19 (262 mg, 1,00 mmol) e 3-amino-2-iodo-6-trifluorometilpiridina (288 mg, 1,00 mmol) foi aquecido sob agitação em 80°C durante 14 horas e o produto bruto foi obtido. O produto foi purificado com cromatografia de coluna (diclorometano/metanol=30/1-20/1(v/v)), utilizando gel de sílica para conceder o intermediário 25 (245 mg, 58% de rendimento) como um cristal branco. 1H-NMR (300 MHz, CDCl3) δ 7.48(d, J=8.8Hz, 1H), 7.28-7.20(m, 1H), 7.15(d, J=8.8Hz, 1H), 7.05(s, 1H), 4.67(br s, 2H), 4.05(s, 3H), 3.80-3.69(m, 4H), 3.59- 3.49(m, 2H), 2.63-2.45(m, 6H). MS(ESI)m/z: [M+H]+423, [M-H]-421.
De acordo com o método sintético similar do composto de exemplo 22, o produto cru foi obtido como um cristal branco a partir do intermediário 25 (240 mg, 0,578 mmol) e terc-butóxido de potássio (324 mg, 2,89 mmol). O produto foi purificado com cromatografia de coluna (diclorometano/metanol = 15/1(v/v)),utilizando gel de sílica para conceder o intermediário 27 (187 mg, 78% de rendimento) como um cristal branco. 1H-NMR (300 MHz, DMSO-d6) δ 12.3(br s, 1H), 8.17-8.08(m, 1H), 8.03(d, J=8.8Hz, 1H), 7.64(d, J=8.8Hz, 1H), 7.23(s, 1H), 7.21(s, 1H), 4.17(s, 3H), 3.62- 3.52(m, 4H), 3.4, 4-3.30(m, 2H), 2.54-2.36(m, 6H). MS(ESI)m/z: [M+H]+383, [M-H]-381.
De acordo com o método sintético similar do intermediário 20, o intermediário 19 (393 mg, 1,50 mmol) e 2-amino-3-iodo-5-metilpiridina (421 mg, 1,80 mmol) foram aquecidos sob agitação em 80°C durante 14 horas , e o produto cru foi obtido. O produto foi purificado com cromatografia de coluna (diclorometano/metanol = 30/1-20/1(v/v)), utilizando gel de sílica para conceder o intermediário 26 (245 mg, 58% de rendimento) como um cristal branco. 1H-NMR (300 MHz, CDCl3) δ 7.94(d, J=2.2Hz, 1H), 7.46(d, J=2.2Hz, 1H), 7.30- 7.18(m, 1H), 6.98(s, 1H), 4.88(br s, 2H), 4.00(s, 3H), 3.78-3.70(m, 4H), 3.60- 3.50(m, 2H), 2.64-2.46(m, 6H), 2.21(s, 3H). MS(ESI)m/z: [M+H]+369.
De acordo com o método sintético similar do composto de exemplo 22, o produto cru foi obtido a partir do intermediário 26 (460 mg, 1,25 mmol) e terc- butóxido de potássio (1200 mg, 10,7 mmol). O produto foi purificado com cromatografia de coluna (diclorometano/metanol = 50/1(v/v)), utilizando gel de sílica de amina e foi recristalizado a partir de metanol e diisopropil éter para conceder o composto de exemplo 28 (229 mg, 50% de rendimento) como um cristal branco. 1H-NMR (300 MHz, CDCl3) δ 11.8(br s, 1H), 8.25(d, J=1.5Hz, 1H), 7.78(br s, 1H), 7.38-7.27(m, 1H), 7.34(s, 1H), 6.59(s, 1H), 4.14(s, 3H), 3.80-3.62(m, 6H), 2.68- 2.50(m, 6H), 2.47(s, 3H). MS(ESI)m/z: [M+H]+369.
Trietilamina (309 mg, 3,05 mmol) foi adicionada em solução de THF (10 mL) do intermediário 19 (200 mg, 0,76 mmol), 5-fluoro3-iodopiridin-2-amina (218 mg, 0,915 mmol), diclorobis(trifenilfosfina)paládio (II) dicloreto (54 mg, 0,076 mmol), iodeto de cobre (I) (14,5 mg, 0,076 mmol) e a mistura foi agitada em temperatura ambiente durante dois dias. A solução de reação foi concentrada sob pressão reduzida, e o resíduo marrom oleoso obtido foi purificado com cromatografia de coluna (acetato de etila como um solvente), utilizando gel de sílica de amina para conceder o intermediário 27 (187 mg, 66% de rendimento) como um sólido branco. 1H-NMR (300 MHz, CDCl3) δ 8.00(d, J=2.9Hz, 1H), 7.39(dd, J=2.9, 8.0Hz, 1H), 7.01(s, 1H), 4.91(br s, 2H), 4.01(s, 3H), 3.74(t, J=4.4Hz, 4H), 3.54(dd, J=5.9, 11.7Hz, 2H), 2.59(t, J=6.6Hz, 2H), 2.50-2.20(m, 4H). Nenhum pico causado por NH de amida foi observado. MS(ESI)m/z: [M+H]+373.
De acordo com o método sintético similar do composto de exemplo 22, o produto cru foi obtido a partir do intermediário 27 (180 mg, 0,48 mmol) e terc- butóxido de potássio (271 mg, 2,41 mmol). O produto foi purificado com cromatografia de coluna (diclorometano/metanol = 10/1(v/v)), utilizando gel de sílica e foi recristalizado a partir de metanol e diisopropil éter para conceder o intermediário 29 (110 mg, 61% de rendimento) como um cristal branco. 1H-NMR (300 MHz, CDCl3) δ 11.30(br s, 1H), 8.23(d, J=2.2Hz, 1H), 7.64(dd, J=2.9, 8.8Hz, 1H), 7.58(s, 1H), 7.41(br s, 1H), 6.66(d, J=2.2Hz, 1H), 4.17(s, 3H), 3.83-3.70(m, 6H), 2.66(t, J=5.9Hz, 2H), 2.56-2.50(m, 4H). MS(ESI)m/z: [M+H]+373, [M-H]-371.
De acordo com o método similar sintético do intermediário 20, o intermediário 19 (262 mg, 1,00 mmol) e 3-amino-2-iodo-6-cianopiridina (245 mg, 1,00 mmol) foram aquecidos sob agitação em 80°C durante 14 horas , e o produto cru foi obtido. O produto foi purificado com cromatografia de coluna (diclorometano/metanol = 20/1(v/v)), utilizando gel de sílica para conceder o intermediário 28 (231 mg, 61% de rendimento) como um sólido marrom claro. 1H-NMR (300 MHz, DMSO-d6) δ 8.20-8.12(m, 1H), 7.68(d, J=8.8Hz, 1H), 7.18(d, J=8.8Hz, 1H), 7.11(s, 1H), 6.83(br s, 2H), 4.02(s, 3H), 3.60-3.53(m, 4H), 3.40- 3.30(m, 2H), 2.48-2.35(m, 6H). MS(ESI)m/z: [M+H]+380.
De acordo com o método sintético similar do composto de exemplo 22, o produto cru foi obtido a partir do intermediário 28 (227 mg, 0,60 mmol) e terc- butóxido de potássio (674 mg, 6,00 mmol). O produto foi purificado com cromatografia de coluna (diclorometano / metanol = 15/1 (v/v)), utilizando gel de sílica e foi recristalizado a partir de metanol e diisopropil éter para conceder o composto de exemplo 30 (229 mg, 50% de rendimento) como um cristal branco. 1H-NMR (300 MHz, DMSO-d6) δ 12.5(br s, 1H), 8.20-8.12(m, 1H), 8.00(d, J=8.8Hz, 1H), 7.76(d, J=8.8Hz, 1H), 7.22(total 2H due to overlapping different (s, 1H) peaks), 4.17(s, 3H), 3.62-3.52(m, 4H), 3.43-3.30(m, 2H), 2.55-2.35(m, 6H). MS(ESI)m/z: [M+H]+380, [M-H]-378.
De acordo com o método sintético similar do composto de exemplo 29, o intermediário 29 (307 mg, 50% de rendimento) foi obtido como um sólido amarelo-amarrozado a partir do intermediário 19 (400 mg, 1,53 mmol), 2-cloro-5- fluoro -3-nitropiridina (323 mg, 1,83 mmol), bis(trifenilfosfina)dicloreto de paládio(II) (176 mg, 0,15 mmol), iodeto de cobre(I) (29 mg, 0,15 mmol) e trietilamina(617 mg, 6,10 mmol). 1H-NMR (300 MHz, CDCl3) δ 8.81(d, J=2.2Hz, 1H), 8.24(dd, J=2.2, 7.3Hz, 1H), 7.18(s, 1H), 4.13(s, 3H), 3.75(t, J=5.1Hz, 4H), 3.58-3.52(m, 2H), 2.59(t, J=5.9Hz, 2H), 2.58-2.49(m, 4H). Nenhum pico caudado por NH amida foi observado.
Uma solução de metanol do intermediário 29 (690 mg, 1,72 mmol), cloreto de tin(II) (4,877 mg, 25,7 mmol), cloreto de amônio (1376 mg, 25,7 mmol) e água (927 mg, 51,4 mmol) foi agitada em 80°C durante 16 horas. Após arrefecimento à temperatura ambiente, a mistura foi ajustada para pH12 com uma solução de hidróxido de sódio de 2M. A mistura obtida foi filtrada através de celite, e foi lavada com metanol. O filtrado foi concentrado sob pressão reduzida e o resíduo foi diluído com dicloromethano e foi lavada com solução de cloreto de sódio saturada, e seca sobre sulfato de sódio anidroso. Depois que o agente de secagem foi filtrado, o resíduo obtido foi concentrado sob pressão reduzida para conceder o intermediário 30 cru (405 mg, 63%) como um sólido amarelo. MS(ESI)m/z: [M+H]+373, [M-H]-371.
De acordo com o método sintético similar do composto de exemplo 22, o produto cru foi obtido a partir do intermediário 30 (400 mg, 107 mmol) e terc- butóxido de potássio (603 mg, 5,37 mmol). O produto foi purificado com cromatografia de coluna (diclorometano/metanol = 20/1(v/v)), utilizando gel de sílica e foi recristalizado a partir de metanol para conceder o composto de exemplo 31 (71 mg, 18% de rendimento) como um sólido branco. 1H-NMR (300 MHz, CDCl3) δ 11.23(br s, 1H), 8.36(s, 1H), 7.46(d, J=8.8Hz, 1H), 7.22(s, 1H), 6.87(s, 1H), 4.14(s, 3H), 3.77-3.73(m, 4H), 3.60-3.50(m, 2H), 2.63- 2.50(m, 6H). Nenhum pico causado por NH de pirrolo[3,2-b]piridina foi observado. MS(ESI)m/z: [M+H]+373, [M-H]-371.
De acordo com o método sintético similar do intermediário 20, o intermediário 19 (393 mg, 1,50 mmol) e 2-amino-3-chloropirazina (233 mg, 1,80 mmol) foram aquecidos sob agitação em 80°C durante 14 horas e o produto cru foi obtido. O produto foi purificado com cromatografia de coluna (diclorometano/metanol = 20/1-15/1 (v/v)), utilizando gel de sílica para conceder o intermediário 31 (272 mg, 51% de rendimento) como um sólido amarelo claro. 1H-NMR (300 MHz, CDCI3) δ 8.04(d, J=2.2Hz, 1H), 7.99(d, J=2.2Hz, 1H), 7.30- 7.20(m, 1H), 7.07(s, 1H), 5.13(br s, 2H), 4.05(s, 3H), 3.80-3.70(m, 4H), 3.60- 3.50(m, 2H), 2.65-2.45(m, 6H). MS(ESI)m/z: [M+H]+380, [M-H]-378.
De acordo com o método sintético similar do composto de exemplo 22, o produto cru foi obtido a partir do intermediário 31 (267 mg, 0,75 mmol) e terc- butóxido de potássio (420 mg, 3,76 mmol). O produto foi purificado com cromatografia de coluna (diclorometano/metanol = 20/1-15/1 (v/v)) utilizando gel de sílica e foi recristalizado a partir de metanol e diisopropil éter para conceder o composto de exemplo 32 (183 mg, o rendimento de 69%) como um cristal branco. 1H-NMR (300 MHz, DMSO-d6) δ 12.6(br s, 1H), 8.46(d, J=2.9Hz, 1H), 8.31(d, J=2.9Hz, 1H), 8.18-8.09(m, 1H), 7.25(s, 1H), 7.13(s, 1H), 4.17(s, 3H), 3.62- 3.53(m, 4H), 3.43-3.30(m, 2H), 2.55-2.35(m, 6H). MS(ESI)m/z: [M+H]+356, [M-H]-354.
De acordo com o método sintético similar do intermediário 20, o intermediário 19 (393 mg, 1,50 mmol) e 2-amino-3-iodo-5-cianopiridina (441 mg, 1,80 mmol) foram aquecidos sob agitação em 80°C durante 14 horas , e o produto cru foi obtido. O produto foi purificado com cromatografia de coluna (diclorometano/metanol = 15/1 (v/v)), utilizando gel de sílica para conceder o intermediário 32 (122 mg, 21% de rendimento) como um sólido amarelo claro. 1H-NMR (300 MHz, DMSO-d6) δ 8.43(d, J=2.2Hz, 1H), 8.16(d, J=2.2Hz, 1H), 8.18- 8.08(m, 1H), 7.50(br s, 2H), 7.02(s, 1H), 4.00(s, 3H), 3.60-3.53(m, 4H), 3.40- 3.30(m, 2H), 2.48-2.35(m, 6H). MS(ESI)m/z: [M+H]+380, [M-H]-378.
De acordo com o método sintético similar do composto exemplo 22, o produto cru foi obtido a partir do intermediário 32 (122 mg, 0,32 mmol) e terc- butóxido de potássio (360 mg, 3,20 mmol). O produto foi purificado com cromatografia de coluna (diclorometano/metanol = 15/1 (v/v)), utilizando gel de sílica e foi recristalizado de metanol e diisopropil éter para conceder o composto de exemplo 33 (57 mg, 47% de rendimento) como um cristal branco. 1H-NMR (300 MHz, DMSO-d6) δ 12.9(br s, 1H), 8.68(d, J=2.2Hz, 1H), 8.60(d, J=2.2Hz, 1H), 8.16-8.08(m, 1H)7.22(s, 1H), 7.04(s, 1H)4.14(s, 3H), 3.62-3.53(m, 4H), 3.42-3.30(m, 2H), 2.55-2.35(m, 6H). MS(ESI)m/z: [M+H]+380, [M-H]-378.
Uma solução de DMF (10 ml) de etil 5-acetil1H-pirazol-3-carboxilato (1,50 g, 8,23 mmol, referência: US5470862) foi adicionada gota a gota a uma suspensão de DMF (10 mL) de 60% (w/w; em óleo ) de hidreto de sódio (442 mg, 11,5 mmol) em condição gelada. A mistura resultante foi agitada em temperatura ambiente durante 10 minutos, e iodometano (103 mL, 16,5 mmol) foi adicionado à solução de reação e foi agitada em temperatura ambiente durate 30 minutos. Depois disso, a solução de reação foi adicionada à água (20 mL) e a camada aquosa foi extraída com éter (80 ml x 2). Após a camada orgânica obtida ser seca sobre sulfato de magnésio, o agente de secagem foi filtrado, e o filtrado foi concentrado sob pressão reduzida. O resíduo foi purificado com cromatografia de coluna (hexano/acetato de etila = 10/1-6/1 (v/v)), utilizando gel de sílica para conceder o intermediário 33 (180 mg, 11% de rendimento) como um cristal branco. 1H-NMR (300 MHz, CDCl3) δ 7.36(s, 1H), 4.43(q, J=7.3Hz, 2H), 4.24(s, 3H), 2.56(s, 3H), 1.42(t, J=7.3Hz, 3H). MS(ESI)m/z: [M+H]+197.
Brometo de feniltrimetilamonio (148 mg, 0,40 mmol) foi adicionado a uma solução de THF (1 mL) do intermediário 33 (79 mg, 0,40 mmol) e a mistura foi agitada na temperatura ambiente durante 1,5 horas. A solução de reação obtida foi adicionada à água (20 mL). A camada aquosa foi extraída com éter (20 ml x 2). Após a camada orgânica obtida ser seca sobre sulfato de magnésio, o agente de secagem foi filtrado, e o filtrado foi concentrado sob pressão reduzida. O sólido residual foi lavado com uma pequena quantidade de isopropil éter/hexano = 1/2 (v/v), e o intermediários 34 (114 mg, quantitativa) foi obtido como um cristal branco. 1H-NMR (300 MHz, CDCl3) δ 7.45(s, 1H), 4.44(q, J=7.3Hz, 2H), 4.31(s, 2H), 4.26(s, 3H), 1.42(t, J=7.3Hz, 3H). MS(ESI)m/z: [M+H]+275 & 277.
Uma solução de metanol (2 mL) do intermediário 34 (114 mg, 0,41 mmol) e 2-aminopiridina (39 mg, 0,41 mmol) foi aquecida em refluxo durante 20 horas. Após o resfriamento, a solução de reação foi adicionada a uma solução de bicarbonato de sódio saturado (20 mL) e a camada aquosa foi extraída com diclorometano (20 ml x 2). A camada orgânica obtida foi seca sobre sulfato de magnésio. Depois que os agentes de secagem foram filtrados, o filtrado foi concentrado sob pressão reduzida. O resíduo foi purificado com cromatografia de coluna (hexano/acetato de etila = 1/3 (v/v)) utilizando gel de sílica para conceder o intermediário 35 (64 mg, 57% de rendimento) como um sólido amarelo claro. 1H-NMR (300 MHz, CDCl3) δ 8.16(d, J=6.6Hz, 1H), 7.81(s, 1H), 7.65(d, J=8.8Hz, 1H), 7.28-7.20(m, 1H), 7.09(s, 1H), 6.87(t, J=7.3Hz, 1H), 4.43(q, J=6.6Hz, 2H), 4.34(s, 3H), 1.43(t, J=6.6Hz, 3H). MS(ESI)m/z: [M+H]+271.
Uma solução de hidróxido de sódio de 2M (0,25 mL, 0,50 mmol) foi adicionada a uma solução de metanol (1 ml) do intermediário 35 (60 mg, 0,22 mmol) e a mistura foi agitada em 70°C durante 1 hora. Após o resfriamento, uma solução de HCl de 2M (0,25 mL, 0,50 mmol) foi adicionada à solução de reação, e a mistura resultante foi concentrada sob pressão reduzida para conceder o intermediário 36 (54 mg) de um cristal branco como uma mistura de cloreto de sódio. O intermediário foi utilizado para a reação seguinte, sem qualquer purificação adicional. 1H-NMR (300 MHz, CDCl3) δ 8.57(d, J=7.3Hz, 1H), 8.42(s, 1H), 7.64(d, J=10.2Hz, 1H), 7.33(t, 10.3Hz, 1H), 7.00(s, 1H), 6.98(t, J=7.3Hz, 1H), 4.25(s, 3H). Nenhum pico causado por COOH foi observado. MS(ESI)m/z: [M+H]+243, [M-H]-241.
Hexafluorofosfato O-benzotriazol-1-il-N,N,N',N'-tetrametiluronio (126 mg, 0,33 mmol) foi adicionado à solução de acetonitrila (1 mL) do intermediário 36 (54 mg), 4-(2-amino-etil)morfolina (32 mg, 0,24 mmol) e trietilamina (0,09 mL, 0,67 mmol) e a mistura foi agitada na temperatura ambiente. Depois disso, a solução resultante foi concentrada sob pressão reduzida. O resíduo foi purificado com cromatografia de coluna (diclorometano/metanol = 30/1 (v/v)) utilizando gel de sílica tratado com amina para conceder produtos oleosos incolores crus (79 mg). 1H-NMR (300 MHz, CDCl3) δ 8.17(d, J=7.3Hz, 1H), 7.83(s, 1H), 7.63(d, J=9.5Hz, 1H), 7.30-7.20(m, 1H), 7.04(s, 1H), 6.86(t, J=6.6Hz, 1H), 6.15(br s, 1H), 4.30(s, 3H), 3.78-3.68(m, 4H), 3.61-3.53(m, 2H), 2.65-2.45(m, 6H). MS(ESI)m/z: [M+H]+355.
Um sistema de HPLC preparativo (o aparelho de purificação A), que foi utilizado para a purificação no composto de exemplo 1, foi utilizado para purificação adicional.
Uma solução de DMSO (0,7 mL) do intermediário 17 (100 mg, 0,275 mmol), benzotiazol (31 mg, 0,229 mmol), iodeto de cobre (I)(44 mg, 0,229 mmol), trifenilfosfina (12 mg, 0,046 mmol) e fosfato de tripotássio (97 mg, 0,458 mmol) foi agitada em 160°C durante 1 hora sob a atmosfera de nitrogênio. O resíduo resultante foi purificado com cromatografia de coluna (diclorometano/metanol = 10/1 (v/v)) e foi tratado com SCX (cartucho de forte troca de cátions) em uma maneira similar ao composto de exemplo 1, para conceder o produto 35 cru (100 mg).
Um sistema de HPLC preparativo (o aparelho de purificação A), que foi usado para a purificação do composto de exemplo 1, foi utilizado para a purificação adicional. MS(ESI)m/z: [M+H]+372.
Composto de exemplo 36: Síntese de 5-{5-fluoro1-metil-1H-pirrolo[2,3-b]piridina- 2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3-carboxamida Iodeto de metila (14,2 mg, 0,10 mmol) foi adicionado a uma solução de acetonitrila (5 mL).O composto de exemplo 29 (30 mg, 0,08 mmol) e carbonato de césio (52 mg, 0,16 mmol). A solução de reação foi agitada em 60°C durante 3 horas. Após arrefecimento à temperatura ambiente, a solução de reação foi diluída com diclorometano. Depois a solução resultante foi lavada com água e uma solução de cloreto de sódio saturada, e foi seca sobre sulfato de sódio. Depois que o agente de secagem foi filtrado, o filtrado foi concentrado sob pressão reduzida e o produto cru (32 mg) foi obtido como um amorfo amarelo. Um sistema de HPLC preparativo (o aparelho de purificação A), que foi utilizado para a purificação no composto de exemplo 1, foi utilizado para purificação adicional. MS(ESI)m/z: [M+H]+387.
Hexafluorofosfato O-benzotriazol-1-il-N,N,N',N'-tetrametiluronio (505 mg, 1,33 mmol) foi adicionado a uma solução de acetonitrila (10 mL) do intermediário 10 (230 mg, 0,89 mmol), amino-acetaldeído dietil aceta (93 mg, 0,89 mmol), trietilamina (0,50 mL, 3,55 mmol) em temperatura ambiente. A solução de reação foi agitada erm temperatura ambiente durante 16 horas. A solução de reação foi diluída com acetato de etila. Depois que a solução resultante foi lavada com água, a camada orgânica foi seca sobre sulfato de magnésio. Depois que o agente de secagem foi filtrado, o filtrado foi concentrado sob pressão reduzida. O resíduo foi purificado com cromatografia de coluna (hexano/acetato de etila = 1/2 (v/v)), utilizando gel de sílica para conceder o intermediário 37 (307 mg,quantitativo) como um sólido branco. 1H-NMR (300 MHz, CDCl3) δ 9.28(s, 1H), 7.42-7.39(m, 1H), 7.32-7.26(m, 1H), 7.20-7.12(m, 2H), 7.05-6.98(m, 1H), 6.68(d, J=1.5Hz, 1H), 4.50(t, J=5.9Hz, 1H), 4.11(s, 3H), 3.64(t, J=5.9Hz, 2H), 3.42(s, 6H). MS(ESI)m/z: [M-H]-345.
Uma solução de HCl de 2M (5 mL) foi adicionada a uma solução de THF (5 mL) do intermediário 37 (294 mg, 0,85 mmol) e a solução resultante foi agitada em 50°C durante 2 horas. Após o resfriamento, a solução de reação foi neutralizada com a adição de uma solução de hidróxido de sódio de 2M (5 mL), e foi extraída com acetato de etila. A camada orgânica obtida foi lavada com uma solução de cloreto de sódio saturada, e foi seca sobre sulfato de magnésio. Depois que os agentes de secagem foram filtrados, o filtrado foi concentrado sob pressão reduzida para conceder o intermediário 38 (300 mg, quantitativo) como um sólido branco. O intermediário foi utilizado para a reação seguinte, sem qualquer purificação adicional. MS(ESI)m/z: [M+H]+301
Uma solução de THF (4,2 mL) do intermediário 38 (30 mg, 0,10 mmol), (R)-(-)-2-pirrolidinametanol (15 mg, 0,15 mmol) e ácido acético (0,15 mL) foi agitada na temperatura ambiente durante 10 minutos. Depois disso, uma solução de THF (0,2 mL) do triacetóxiborohidreto de sódio (63 mg, 0,30 mmol) foi adicionado à solução, e a solução resultante foi agitada durante a noite em temperatura ambiente. Após a solução de reação ter sido concentrada, o resíduo foi completamente dissolvido com a adição de acetato de etila (0,7 mL) e uma solução de hidróxido de sódio de 2M (0,5 mL). Depois a camada orgânica obtida foi carregada para o SCX (cartucho de forte troca de cátions), lavada com metanol (5 mL), e finalmente foi eluída com amônia-metanol (1M, 4 mL). O produto cru obtido pela concentração foi purificado com um HPLC preparativp (o aparelho de purificação A escrito no início dos EXEMPLOS). MS (ESI) m/z: [M + H]+386.
Exemplos sintetizados pela utilização de reação similar às descritos acima são indicados abaixo: Composto de exemplo 38: 5-(5-fluoro-1H-indol-2-il)-N-{2-[(3R)-3-(hidróxipirrolidin- 1-il]etil}-1-metil-1H-pirazol-3-carboxamida Composto de exemplo 39: 5-(5-fluoro-1H-indol-2-il)-N-{2-[(3S)-3-(hidróxipiperidin- 1-il]etil}-1-metil-1H-pirazol-3-carboxamida Composto de exemplo 40: 5-(5-fluoro-1H-indol-2-il)-1-metil-N-{2-[(1S,4S)-2-oxa-5- azabiciclo[2.2.1]heptan-5-il]etil}-1H-pirazol-3-carboxamida Composto de exemplo 41: N-[2-(3, 3-difluoroazetidin-1-il)etil]-5-(5-fluoro-1H-indol- 2-il)-1-metil-1H-pirazol-3-carboxamida Composto de exemplo 42: 5-(5-fluoro-1H-indol-2-il)-N-{2-[(2S)-2- (hidróximetil)pirrolidin-1-il]etil}-1-metil-1H-pirazol-3-carboxamida Composto de exemplo 43: 5-(5-fluoro-1H-indol-2-il)-N-{2-[(3R)-3-fluoropirrolidin-1- il]etil}-1-metil-1H-pirazol-3-carboxamida Composto de exemplo 44: 5-(5-fluoro-1H-indol-2-il)-N-{2-[(3S)-3-fluoropirrolidin-1- il]etil}-1-metil-1H-pirazol-3-carboxamida Composto de exemplo 45: 5-(5-fluoro-1H-indol-2-il)-N-[2-(4-fluoropiperidin-1-il]etil}- 1-metil-1H-pirazol-3-carboxamida Composto de exemplo 46: 5-(5-fluoro-1H-indol-2-il)-1-metil-N-[2-(1,4-oxazepam-4- il]etil}-1H-pirazol-3-carboxamida Composto de exemplo 47: N-[2-(azetidin1-il)etil]-5-(5-fluoro-1H-indol-2-il)-1-metil- 1H-pirazol-3-carboxamida Tabela 4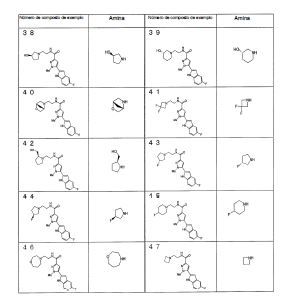 Tabela 5
Tabela 5 
De acordo com o método sintético similar do intermediário 11 acima, tris(dibenzilidenoacetona)dipaládio(0) (66 mg, 0,0723 mmol) foi adicionado ao 1,4- dioxano (20 mL)- solução misturada de água (3 mL) do intermediário 17 (526 mg, 1,44 mmol), 3-ácido quinolinoborônico (250 mg, 1,44 mmol), fosfato de potássio (458 mg, 2,16 mmol) e da triciclohexilfosfina (40,4 mg, 0,14 mmol). A solução de reação foi agitada em 100°C durante a noite (15 horas). O resíduo obtido foi purificado com cromatografia de coluna (diclorometano/metanol = 20/1 (v/v)), utilizando gel de sílica para conceder um cristal branco. Então o composto de exemplo 17 (204 mg, 39% de rendimento) foi obtido como um cristal branco por recristalização a partir de hexano-acetato de etila.
Exemplos sintetizados pela utilização da reação similar às descritas acima, ou utilizando a condição de reação no intermediário 8 são mostradas abaixo: Composto de exemplo 48 1-metil-N-[2-(morfolin-4-il)etil]-5-(quinolin-6-il)-1H-pirazole-3-carboxamida Composto de exemplo 49: 1-metil-N-[2-(morfolin-4-il)etil]-5-(quinolin-7-il)-1H-pirazole-3-carboxamida Composto de exemplo 50: 1-metil-N-[2-(morfolin-4-il)etil]-5-(naftalen-2-il)-1H-pirazole-3-carboxamida Composto de exemplo 51: 5-(6-metoxinaftalen-2-il)-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazole-3-carboxamida Composto de exemplo 52: 5-(7-metoxinaftalen-2-il)-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazole-3-carboxamida Composto de exemplo 53: 5-(1-benzotiofen-3-il)-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazole-3-carboxamida Composto de exemplo 54: 1-metil-N-[2-(morfolin-4-il)etil]-5-(quinoxalin -6-il)-1H-pirazole-3-carboxamida Tabela 6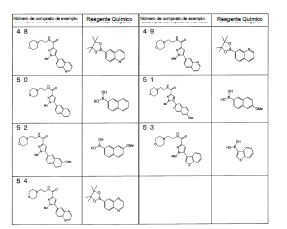 Tabela 7
Tabela 7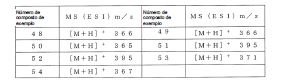 Síntese de compostos de exemplo 55: Síntese de 5-(1H-indol-3-il)-1-metil-N-[2- (morfolin-4-il)etil)-1H-pirazol-3-carboxamida
Síntese de compostos de exemplo 55: Síntese de 5-(1H-indol-3-il)-1-metil-N-[2- (morfolin-4-il)etil)-1H-pirazol-3-carboxamida
O intermediário 17 (200 mg, 0,55 mmol), acetato de paládio (25 mg, 0,11 mmol) e trifenilfosfina (58 mg, 0,22 mmol) foi dissolvido em solução de dioxano/tolueno (3,3/1 (v/v), 5,2 mL) e a mistura resultante foi agitada em temperatura ambiente durante 10 minutos. Depois disso, 1-(fenilsulfonil)-3-ácido indoleboronico (215 mg, 0,71 mmol), água (1,2 mL), e carbonato de sódio (233 mg, 2,20 mmol) foi adicionado à solução de reação, e a mistura resultante foi aquecida em refluxo durante 16 horas. Após o resfriamento, a solução de reação foi diluída com acetato de etila. O sulfato de sódio foi adicionado à solução resultante para remover a água e a mistura resultante foi filtrada. Depois o filtrado foi concentrado sob pressão reduzida, o resíduo foi pré tratado com cromatografia de coluna (acetato de etila) utilizando gel de sílica. Depois disso, a camada orgânica obtida foi carregada para SCX (cartucho de forte troca de cátions), lavada com metanol e foi finalmente eluída com amônia-metanol (1M). O eluato foi concentrado sob pressão reduzida para conceder o intermediário 39 (271 mg, quantitativo) como produto cru. O intermediário foi utilizado para a reação seguinte, sem qualquer purificação adicional. MS(ESI)m/z: [M+H]+494.
O intermediário 39 (271 mg) foi dissolvido em metanol (5 mL), e uma solução de hidróxido de sódio de 2M (5 mL) foi adicionada à solução, a solução resultante foi agitada em 70°C durante 30 minutos. Após o resfriamento, a água (10 mL) foi adicionada à solução de reação, e a mistura resultante foi extraída com diclorometano (50 mL) três vezes. Depois a camada orgânica combinada foi seca sobre sulfato de magnésio, o agente de secagem foi filtrado e o filtrado foi concentrado sob pressão reduzida. O resíduo foi purificado com cromatografia de coluna (diclorometano/metanol = 10/1 (v/v)), utilizando gel de sílica para dar o produto cru (169 mg) como um sólido marrom claro. Um sistema de HPLC preparativo (o aparelho de purificação A), que foi usado para a purificação do composto de exemplo 1, foi utilizado para purificação adicional. MS(ESI)m/z: [M+H]+354.
O composto de exemplo 55 (50 mg) foi dissolvido em acetonitrila (1 mL) e carbonato de césio (138 mg, 0,42 mmol) e iodeto de metila (0,013 mL, 0,21 mmol) foi adicionado à solução resultante e a mistura foi aquecida em refluxo durante 1 hora. Após o resfriamento, as substâncias insolúveis foram retiradas por filtração, e o filtrado foi concentrado sob pressão reduzida. O resíduo foi purificado com cromatografia de coluna (diclorometano/metanol = 10/1 (v/v)), utilizando gel de sílica para conceder o produto cru (45 mg), como xarope incolor. Um sistema de HPLC preparativo (o aparelho de purificação A), que foi utilizado para a purificação no composto de exemplo 1, foi utilizado para purificação adicional. MS(ESI)m/z: [M+H]+368. Intermediários e exemplos sintetizados pela utilização de reação 5 similar às descritas acima são os indicados abaixo:Tabela 8 Composto de exemplo 57: 1-metil-5-(3-metil-1H-indol-5-il)-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 58: 1-metil-5-(2-metil-1H-indol-5-il)-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 59: 1-metil-N-[2-(morfolin-4-il)etil]-5-{1H-pirrolo[2,3- b]piridin-3-il}-1H-pirazol-3-carboxamida Composto de exemplo 60: 5-(5-metóxi1H-indol-3-il)-1-metil-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 61: 5-(5-fluoro-1H-indol-3-il)-1-metil-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 62: 5-(1, 2-dimetil-1H-indol-5-il)-1-metil-N-[2-(morfolin-4- il)etil]-1H-pirazol-3-carboxamida Composto de exemplo 63: 5-(1,3-dimetil-1H-indol-5-il)-1-metil-N-[2-(morfolin-4- il)etil]-1H-pirazol-3-carboxamida Composto de exemplo 64: 1-metil-5-{1-metil-1H-pirrolo[2,3-b]piridin-3-il}-N-[2- (morfolin-4-il)etil]-1H-pirazol-3-carboxamida Composto de exemplo 65: 5-(5-metóxi1-metil-1H-indol-3-il)-1-metil-N-[2-(morfolin- 4-il)etil]-1H-pirazol-3-carboxamida Composto de exemplo 66: 5-(5-fluoro1-metil-1H-indol-3-il)-1-metil-N-[2-(morfolin- 4-il)etil]-1H-pirazol-3- carboxamida Composto de exemplo 67: 1-metil-5-(1-metil-1H-indol-4-il)-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 68: 5-[1-(2-metóxietil)-1H-indol-3-il] -1-metil-N-[2-(morfolin- 4-il)etil]-1H-pirazol-3-carboxamida Composto de exemplo 69: 5-(1-etil1H-indol-3-il)-1-metil-N-[2-(morfolin-4-il)etil]-1H- pirazol-3-carboxamida Composto de exemplo 70: 5-(4-fluoro-1H-indol-3-il)-1-metil-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 71: 5-(6-fluoro-1H-indol-3-il)-1-metil-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 72: 5-(6-metóxi1H-indol-3-il)-1-metil-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 73: 5-(7-metóxi1H-indol-3-il)-1-metil-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 74: 5-[1-(2-metóxietil)-2-metil-1H-indol-5-il]-1-metil-N-[2- (morfolin-4-il)etil]-1H-pirazol-3-carboxamida Composto de exemplo 75: 5-[1-(2-hidróxietil)-2-metil-1H-indol-5-il]-1-metil-N-[2- (morfolin-4-il)etil]-1H-pirazol-3-carboxamida
Composto de exemplo 57: 1-metil-5-(3-metil-1H-indol-5-il)-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 58: 1-metil-5-(2-metil-1H-indol-5-il)-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 59: 1-metil-N-[2-(morfolin-4-il)etil]-5-{1H-pirrolo[2,3- b]piridin-3-il}-1H-pirazol-3-carboxamida Composto de exemplo 60: 5-(5-metóxi1H-indol-3-il)-1-metil-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 61: 5-(5-fluoro-1H-indol-3-il)-1-metil-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 62: 5-(1, 2-dimetil-1H-indol-5-il)-1-metil-N-[2-(morfolin-4- il)etil]-1H-pirazol-3-carboxamida Composto de exemplo 63: 5-(1,3-dimetil-1H-indol-5-il)-1-metil-N-[2-(morfolin-4- il)etil]-1H-pirazol-3-carboxamida Composto de exemplo 64: 1-metil-5-{1-metil-1H-pirrolo[2,3-b]piridin-3-il}-N-[2- (morfolin-4-il)etil]-1H-pirazol-3-carboxamida Composto de exemplo 65: 5-(5-metóxi1-metil-1H-indol-3-il)-1-metil-N-[2-(morfolin- 4-il)etil]-1H-pirazol-3-carboxamida Composto de exemplo 66: 5-(5-fluoro1-metil-1H-indol-3-il)-1-metil-N-[2-(morfolin- 4-il)etil]-1H-pirazol-3- carboxamida Composto de exemplo 67: 1-metil-5-(1-metil-1H-indol-4-il)-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 68: 5-[1-(2-metóxietil)-1H-indol-3-il] -1-metil-N-[2-(morfolin- 4-il)etil]-1H-pirazol-3-carboxamida Composto de exemplo 69: 5-(1-etil1H-indol-3-il)-1-metil-N-[2-(morfolin-4-il)etil]-1H- pirazol-3-carboxamida Composto de exemplo 70: 5-(4-fluoro-1H-indol-3-il)-1-metil-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 71: 5-(6-fluoro-1H-indol-3-il)-1-metil-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 72: 5-(6-metóxi1H-indol-3-il)-1-metil-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 73: 5-(7-metóxi1H-indol-3-il)-1-metil-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida Composto de exemplo 74: 5-[1-(2-metóxietil)-2-metil-1H-indol-5-il]-1-metil-N-[2- (morfolin-4-il)etil]-1H-pirazol-3-carboxamida Composto de exemplo 75: 5-[1-(2-hidróxietil)-2-metil-1H-indol-5-il]-1-metil-N-[2- (morfolin-4-il)etil]-1H-pirazol-3-carboxamida
Exemplos sintetizados pela uitilização de reação similar às descritas acima são indicados abaixo: em que, facilmente reconhecido por aqueles 5 versados na técnica, haletos de alquila substituídos correspondendo aos exemplos podem ser utilizado ao invés de iodeto de metila. Tabela 9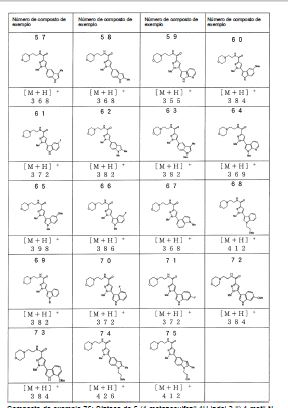
Metanotiolato de sódio (1300 mg, 18,16 mmol) e uma solução de um 1N HCl-dietil éter (20 mL) foi adicionado em um banho de gelo para uma solução de THF (50 mL) de 1-(benzenosulfonil)-4,5,6,7-tetrahidro-1H-indol-4-ona (1250 mg, 4,54 mmol), que foi preparado a partir de 4, 5, 6, 7-tetrahidro-1H-4-ona, em referência à literatura de patente (JP-7-247263A), e a mistura foi agitada à temperatura ambiente durante a noite. Depois o dietil éter (30 mL) foi adicionado à mistura. A mistura resultante foi lavada com uma solução de bicarbonato de sódio saturada (50 mL), e foi seca sobre sulfato de magnésio anidroso. Depois o agente de secagem foi filtrado, o filtrado obtido foi concentrado sob pressão reduzida para conceder um resíduo oleoso. O resíduo oleoso foi dissolvido em tolueno (15 mL), e DDQ (1500 mg, 6,81 mmol) foi adicionado à mistura. A mistura foi aquecida em refluxo durante 2 horas. Após o resfriamento, a mistura resultante foi concentrada sob pressão reduzida para conceder o intermediário 49 oleoso bruto. O intermediário 49 cru obtido foi purificado com cromatografia de coluna (hexano- etilacetato = 10:1 (v/v)), utilizando gel de sílica para conceder o intermediário 49 (690 mg, 50% de rendimento) como um semi-sólido branco. 1H-NMR (300 MHz, CDCl3) δ 7.88-7.79(m, 3H), 7.58(d, J=4.0Hz, 1H), 7.57- 7.50(m, 1H), 7.46-7.41(m, 2H), 7.43-7.24(m, 1H), 7.08(d, J=7.3Hz, 1H), 6.78(d, J=3.7Hz, 1H), 3.58(s, 3H). MS(ESI)m/z: [M+H]+304.
n-butillítio (2,5 mL, 4,12 mmol, solução de ciclohexano de 1.65M) foi adicionado gota a gota a uma solução de THF (5 mL) de diisopropilamina (417 mg, 0,58 mmol) em um banho de gelo, e a mistura foi agitada durante 20 minutos, o que concedeu diisopropilamida de lítio.
O diisopropilamida de lítio preparado acima foi adicionado gota a gota em um banho de gelo a uma solução de THF (25 mL) do intermediário 49 (833 mg, 2,75 mmol) e o éster de ácido triisopropilborônico (620 mg, 3,29 mmol), que foi preparado separadamente. Duas horas depois, a mesma quantidade de diisopropilamida de lítio também foi adicionada gota a gota em um banho de gelo. Depois a mistura foi agitada em um banho de gelo durante 1 hora, uma solução de HCl de 1M foi adicionada à mistura, e a solução foi ajustada para o pH3. A solução resultante foi extraída com acetato de etila (25 mL x 2), e a mistura combinada foi seca sobre sulfato de magnésio anidroso. Depois que o agente de secagem foi filtrado, o filtrado obtido foi concentrado sob pressão reduzida. O resíduo obtido foi purificado com cromatografia de coluna (hexano/acetato de etila) utilizando gel de sílica para conceder o intermediário 50 cru (953 mg, 100% de rendimento) como um semi-sólido amarelo.
De acordo com a forma similar à síntese do intermediário 8 no composto do exemplo 1, o intermediário 51 (390 mg, 29% de rendimento) foi obtido como um semi-sólido branco a partir do intermediário 17 (909 mg, 2,50 mmol), os intermediário 50 (953 mg, 2,74 mmol), acetato de paládio (II) (56 mg, 0,25 mmol), trifenilfosfina (262 mg, 1,00 mmol) e carbonato de sódio (661 mg, 6,24 mmol). MS(ESI)m/z: [M+H]+539.
O composto de oxona-persulfato (683 mg, 1,11 mmol) foi adicionado a uma solução de dioxano/água (8 ml / 3 ml) do intermediário 51 (200 mg, 0,37 mmol) em temperatura ambiente. A solução de reação foi agitada em temperatura ambiente durante 2,5 horas. A solução de reação foi seca sobre sulfato de magnésio anidroso. Depois o agente de secagem foi filtrado, o filtrado obtido foi concentrado sob pressão reduzida para conceder o intermediário 52 cru oleoso amarelo (61 mg, 29% de rendimento). MS(ESI)m/z: [M+H]+572.
Tetrabutilamoniofluorado monohidreto (140 mg, 0,53 mmol) foi adicionado a uma solução de TF (5 mL) do intermediário 52 (61 mg, 0,11 mmol) e a mistura foi aquecida em refluxo durante 3 horas. Após arrefecimento à temperatura ambiente, o resíduo obtido sob pressão reduzida foi purificado com SCX (cartucho de forte troca de cátions) em forma similar ao composto de 1 para conceder o produto cru 76 como um sólido amarelo. Purificação adicional com cromatografia de coluna (diclorometano/metanol = 10/1 (v/v)), utilizando gel de sílica para conceder o composto de exemplo 76 (17 mg, 37% de rendimento) como um sólido amarelo. MS(ESI)m/z: [M+H]+432.
n-Butillítio (0,63 mL, 1,65 M, solução de hexano) foi adicionado gota a gota a uma solução de TF resfriada (5 mL) de diisopropilamina (105 mg, 104 mmol) em condição gelada durante 5 minutos, sob atmosfera de nitrogênio. A mistura resultante foi agitada durante 20 minutos, o que concedeu uma solução de TF de diisopropilamida de lítio.
Uma solução de TF diisopropilamida de lítio preparada acima foi adicionada gota a gota em um banho de gelo a uma solução de TF (5 mL) de 1- (terc-butil)-4-(di-terc-butil)aminoindol (300 mg, 0,694 mmol) e borato de triisopropil (157 mg, 0,832 mmol), que foi preparado separadamente. Depois a solução de reação foi agitada em um banho de gelo durante 1 hora, a solução foi ajustada ao pH3 com a adição de uma solução de HCl de 1M. A solução resultante foi extraída com acetato de etila (15 mL x 2), e a solução combinada foi seca sobre sulfato de magnésio anidroso para conceder o produto 53 cru de 1-(terc-butil)-4- (diterc-butil)amino-2-ácido indolborônico. O produto cru foi utilizado para a reação seguinte, sem qualquer purificação adicional.
Um intermediário 17 (178 mg, 0,49 mmol), acetato de paládio (14 mg, 0,063 mmol) e trifenilfosfina (52 mg, 0,20 mmol) foram dissolvidos em solução de dioxano/tolueno (3,3 / 1 (v / v), 5,2 mL ), e a mistura foi agitada em temperatura ambiente durante 10 minutos. Depois disso, 1-(terc-butil)-4-(diterc-butil)amino-2- ácido indolborônico 53 (256 mg, 0,54 mmol), água (5 mL), e carbonato de sódio (129 mg, 1,22 mmol) foram adicionados à solução de reação, e a mistura foi aquecida em refluxo durante 1,5 horas. Após o resfriamento, a solução de reação foi diluída com acetato de etila e sulfato de sódio foi adicionado à solução para a remoção da água. Então, a solução resultante foi filtrada. Depois o filtrado foi concentrado sob pressão reduzida, o produto foi purificado com cromatografia de coluna (acetato de etila), utilizando gel de sílica revestido de amina para conceder 5-{1-(terc-butil)-4-(diterc-butil)amino-indol-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H- pirazol-3-carboxamida 54 e 5-{-4-(diterc-butil)aminoindol-2-il}-1-metil-N-[2- (morfolin-4-il)etil]-1H-pirazol-3-carboxamida foi obtido como a mistura (127 mg, sólido incolor). Essa mistura não foi sujeita a qualquer isolamento adicional e foi utilizada para a reação seguinte.
A mistura (127 mg) que contém o intermediário 54 acima e uma solução de HCl-metanol 10% (15 mL) em 100 mL do frasco foi agitada sob a atmosfera de nitrogênio em temperatura ambiente durante 16 horas. A solução resultante foi concentrada sob pressão reduzida, e 5-(1H-4-aminoindol-2-il)-1- metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3-carboxamida sal de dicloridrato 55 foi obtido (99 mg, quantitativa). 1H-NMR (300 MHz, CD3OD) δ 7.50-7.47(m, 1H), 7.23-7.08(m, 4H), 4.10(s, 3H), 4.03-3.99(m, 2H), 3.87-3.76(m, 4H), 3.67-3.63(m, 2H), 3.43-3.40(m, 2H), 3.29- 3.19(m, 2H). Não foram observados mais prótons devido à sobreposição de picos de solvente com base em CD3OD. MS(ESI)m/z: [M+H]+369.
Os compostos de exemplos abaixo estão substancialmente na mesma forma como o processo de F-3 descrito acima, e foi preparado sob a condição selecionada do processo F-3.
O composto de exemplo 77 e compostos 78 na tabela seguinte foram preparados na presença de base (trietilamina) em solução de 1,2-dicloroetano utilizando o reagente de cada um (cloreto de acetila ou anidrido acético, cloreto de metanosulfonil) e o intermediário 55. Um sistema de HPLC preparativo (o aparelho de purificação A), que foi utilizado para a purificação no composto do exemplo 1, foi utilizado para purificação adicional.
O composto de exemplo 79 e o composto 80 na tabela seguinte foram preparados na presença de base (hidreto de sódio) em uma solução de DMF utilizando o reagente de cada um (cloreto de metanosulfonil) e o intermediário 55. Um sistema de HPLC preparativo (o aparelho de purificação A), que foi utilizado para a purificação no composto de exemplo 1, foi utilizado para purificação adicional.
Os reagentes e exemplos são mostrados abaixo.Tabela 10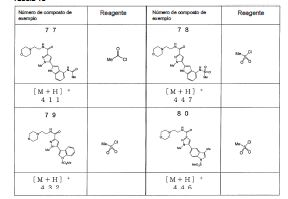
Composto de exemplo 81: Síntese do 1-metil-5-(3-metil-1H-indol-1-il)-N-[2- (morfolin-4-il)etil]-1H-pirazol-3-carboxamida 3-metilindol (27 mg, 0,21 mmol), carbonato de césio (179 mg, 0,55 mmol) e iodeto de cobre (I) (52 mg, 0,28 mmol) foram adicionados a uma solução de intermediário 17 (50 mg, 0,14 mmol) dissolvida em DMF (1 mL), e a mistura foi agitada em 110°C durante 20 horas. Após o resfriamento, as substâncias insolúveis foram retiradas por filtração, o filtrado foi concentrado sob pressão reduzida. O resíduo foi pré-tratado com cromatografia de coluna (diclorometano/etanol (10/1) (v/v)) utilizando gel de sílica tratada com amina e foi então carregado para o SCX (cartucho de forte troca de cátions), lavado com metanol (10 mL) e foi finalmente eluído com amônia-metanol (1M, 8 mL). O produto cru obtido pela concentração foi purificado com um HPLC preparativo (o aparelho de purificação A escrito no início dos EXEMPLOS). MS(ESI)m/z: [M+H]+368.
De acordo com o método sintético similar do composto do exemplo 1, trietilamina (3,32 mL, 23,80 mmol) foi adicionada a uma solução de mistura de DMF anidrosa (50 mL) do intermediário 16 (2,00 g, 7,94 mmol), aminoacetaldeído dietil acetal (1,27 g , 9,52 mmol) e HBTU (4,52 g, 11,9 mmol) e a mistura foi agitada em temperatura ambiente. O resíduo resultante foi purificado com cromatografia de coluna (hexano/acetato de etilacetatoetil = 2: 1 (v/v)), utilizando gel de sílica para conceder o intermediário 56 (3.84g) como um óleo amarelo claro. 1H-NMR (300 MHz, CDCl3) δ 6.99(br s, 1H), 6.94(s, 1H), 4.61-4.55(m, 1H), 3.94(s, 3H), 3.80-3.68(m, 2H), 3.64-3.50(m, 4H), 1.23(t, J=7.0Hz, 6H).
A mistura de reação de uma solução de TF (50 mL) do intermediário 56 (3,84 g, 10,45 mmol) e uma solução de HCl de 2M (25 mL) foi agitada em 50°C durante 2 horas. Após esfriar até a temperatura ambiente, a mistura de reação foi ajustada para mais do que pH10 com uma solução de bicarbonato de sódio saturada e foi extraída com acetato de etila duas vezes. A camada orgânica obtida foi lavada com uma solução de cloreto de sódio saturada, e foi seca sobre hidrocloreto de sódio. Depois o agente de secagem foi filtrado, o filtrado foi concentrado sob pressão reduzida para conceder uma sólido amarelo claro. Triacetóxiborohidrida de sódio (6,64 g, 31,35 mmol) foi adicionado em várias porções à mistura de 1,2-dicloroetan (50 mL) do intermediário de aldeído cru e 3, 3-monocloridrato de difluoroazetidina (1,35 g, 10,45 mmol) e acetato de etila (10 mL). Depois a mistura resultante foi agitada em temperatura ambiente durante 15 horas, a mistura de reação foi ajustada para mais do que pH10 com solução de bicarbonato de sódio saturada, e a solução foi extraída com diclorometano três vezes. A camada orgânica resultante foi lavada com solução de cloreto de sódio saturada, e seca sobre sulfato de sódio. Depois o agente de secagem foi filtrado, o filtrado foi concentrado sob pressão reduzida. O resíduo obtido foi purificado com cromatografia de coluna (diclorometano/metanol= 40/1-30/1 (v/v)), utilizando gel de sílica para conceder o intermediário 57 (2,93 g, 76% de rendimento) como um óleo incolor. 1H-NMR (300 MHz, CDCl3) δ 7.05(br s, 1H), 6.94(s, 1H), 3.95(s, 3H), 3.70-3.58(m, 4H), 3.47-3.39(m, 2H), 2.80-2.74(m, 2H). MS(ESI)m/z: [M+H]+371.
De acordo com o mesmo método sintético do intermediário 18 descrito no composto de exemplo 22, trietilamina (3,46 mL, 24,86 mmol) foi adicionada a uma solução de TF anidrosa (30 mL) do intermediário 57 (2. 30 g, 6,21 mmol), trimetilsililacetileno (1,32 mL, 9,32 mmol), iodeto de cobre(I) (118 mg, 0,621 mmol) e diclorobis(acetonitrila) cloreto de paládio(II) (436 mg, 0,621 mmol) e a mistura resultante foi agitada na temperatura ambiente durante 1,5 horas. Depois do tratamento regular, o resíduo foi purificado com cromatografia de coluna (hexano/acetato de etila = 3: 2-1:1 (v/v)), utilizando gel de sílica para conceder o intermediário 58 (1,76 g, 83% de rendimento) como um óleo marrom amarelado. E o intermediário 58 resultado foi utilizado para a próxima etapa sem obter qualquer dado físico.
Uma mistura de metanol (30 mL) do intermediário 58 (1,76 g, 5,17 mmol) e carbonato de potássio (107 g, 7,75 mmol) foi agitada em temperatura ambiente durante 2 horas. Depois que a reação foi concluída, carbonato de potássio foi filtrado. O filtrado foi concentrado sob pressão reduzida. O resíduo resultante foi diluído com diclorometano novamente, e foi lavado com água, seguida por uma solução de cloreto de sódio saturada. Então, a solução foi seca sobre sulfato de sódio. Depois o agente de secagem foi filtrado, o filtrado foi concentrado sob pressão reduzida. O resíduo resultante foi purificado com cromatografia de coluna (hexano / acetato de etila = 1: 2 (v/v)), utilizando gel de sílica para conceder o intermediário 59 (1,32 g, 95% de rendimento) como um sólido amarelo claro. 1H-NMR (300 MHz, CDCl3) δ 7.12(br s, 1H), 6.95(s, 1H), 3.96(s, 3H), 3.70- 3.54(m, 5H), 3.48-3.40(m, 2H), 2.82-2.74(m, 2H). MS(ESI)m/z: [M+H]+269.
De acordo com o método sintético do intermediário 20, o intermediário 59 (300 mg, 1,12 mmol) e 2-amino-3-iodo-5-cianopiridina (329 mg, 1,34 mmol) foram aquecidos sob agitação em 85°C durante 14 horas, e o intermediário 60 (29,0 mg, 6,7% de rendimento) foi obtido como um sólido amarelo claro. MS(ESI)m/z: [M+H]+386, [M-H]-384.
De acordo com o método similar para o processo de síntese do composto de exemplo 22, o composto de exemplo 82 (21 mg, 72% de rendimento) foi obtido como um sólido amarelo claro a partir do intermediário 60 (29 mg, 0,075 mmol). 1H-NMR (300 MHz, DMSO-d6) δ 12.85(br s, 1H), 8.68(d, J=2.2Hz, 1H), 8.59(d, J=2.2Hz, 1H), 8.20-8.12(m, 1H), 7.23(s, 1H), 7.04(s, 1H), 4.14(s, 3H), 3.68- 3.53(m, 4H), 3.31-3.20(m, 2H), 2.73-2.63(m, 2H) MS(ESI)m/z: [M+H]+355, [M-H]-353.
De acordo com o método sintético do intermediário 18, o intermediário 59 (250 mg, 1,12 mmol) e 3-iodo-2-aminopiridina (226 mg, 1,03 mmol) foram agitados na temperatura ambiente durante 3 horas para conceder o intermediário 61 ( 50,3 mg, 15% de rendimento) como um sólido amarelo claro. MS(ESI)m/z: [M+H]+361.
De acordo com o método similar para o processo de síntese do composto de exemplo 22, o composto de exemplo 83 (37,9 mg, 75% de rendimento) foi obtido como um sólido amarelo claro a partir do intermediário 61 (50 mg, 0,075 mmol). 1H-NMR (300 MHz, DMSO-d6) δ 12.19(br s, 1H), 8.31-8.26(m, 1H), 8.15-8.08(m, 1H), 8.05-7.98(m, 1H), 7.18-7.08(m, 2H), 6.89(s, 1H), 4.12(s, 3H), 3.66-3.54(m, 4H), 3.30-3.20(m, 2H), 2.72-2.63(m, 2H). MS(ESI)m/z: [M+H]+361, [M-H]-359.
De acordo com o método de síntese similar do intermediário 35 no composto de exemplo 34, uma solução de metanol (2 mL) do intermediário 34 (114 mg, 0,41 mmol) e 2-amino-5-fluoropiridina (39 mg, 0,41 mmol) foi aquecido sob agitação durante 20 horas. O resíduo foi purificado com cromatografia de coluna (hexano/acetato de etila = 1/3 (v/v)), utilizando gel de sílica para conceder o intermediário 62 (101 mg, 48% de rendimento) como um sólido amarelo claro. 1H-NMR (300 MHz, CDCl3) δ 8.11-8.09(m, 1H), 7.81(s, 1H), 7.65-7.60(m, 1H), 7.21-7.14(m, 1H), 7.07(s, 1H), 4.43(t, J=7.3Hz, 2H), 4.33(s, 3H), 1.42(t, J=7.3Hz, 3H). MS(ESI)m/z: [M+H]+289.
De acordo com o método sintético similar do intermediário 36 no composto de exemplo 34, uma solução de hidróxido de sódio de 2M (0,175 mL, 0,350 mmol) foi adicionada a uma solução de metanol (5 mL) do intermediário 62 (101 mg, 0,350 mmol), e a mistura resultante foi agitada em 70°C durante 1 hora. Após a neutralização com uma solução de HCl de 2M, o intermediário 63 (67 mg, 73% de rendimento) foi obtido como um cristal branco. O intermediário foi utilizado para a reação seguinte, sem qualquer purificação adicional. 1H-NMR (300 MHz, DMSO-d6) δ 8.75(br s, 1H), 8.38(s, 1H), 8.23(s, 1H), 7.70- 7.65(m, 1H), 7.39-7.33(m, 1H), 7.06(s, 1H), 4.24(s, 3H). MS(ESI)m/z: [M+H]+261.
De acordo com o método sintético similar do composto do exemplo 1, o composto de exemplo 84 cru (52,3 mg, 54% de rendimento) foi obtido como um cristal branco a partir do intermediário 63 (67 mg, 0,257 mmol) e 4-(2-aminoetil) morfolina (36,9 mg, 0,283 mmol). Um sistema de HPLC preparativo (o aparelho de purificação A), que foi utilizado para a purificação no composto do exemplo 1, foi utilizado para purificação adicional. MS(ESI)m/z: [M+H]+373.
De acordo com o método sintético similar do intermediário 35 no composto de exemplo 34, uma solução de metanol (2 mL) do intermediário 34 (114 mg, 0,41 mmol) e 2-amino-4-fluoropiridina (39 mg, 0,41 mmol) foi aquecida sob agitação durante 20 horas. Em seguida, o resíduo foi purificado com cromatografia de coluna (hexano/acetato de etila = 1/3 (v/v)), utilizando gel de sílica para conceder o intermediário 64 (120 mg, 57% de rendimento) como um sólido amarelo claro. 1H-NMR (300 MHz, CDCl3) δ 8.10(br s, 1H), 7.81(s, 1H), 7.65-7.60(m, 1H), 7.22- 7.15(m, 1H), 7.07(s, 1H), 4.46-4.40(t, J=7.3Hz, 2H), 4.33(s, 3H), 1.42(t, J=7.3Hz, 3H). MS(ESI)m/z: [M+H]+289.
De acordo com o método sintético similar do intermediário 36 no composto de exemplo 34, uma solução de hidróxido de sódio de 2M (0,21 mL, 0,416 mmol) foi adicionado a uma solução de metanol (5 mL) do intermediário 64 (120 mg, 0,416 mmol), e a mistura resultante foi agitada em 70°C durante 1 hora. Após a neutralização com uma solução de HCl de 2M, o intermediário 65 (67 mg) foi obtido como um cristal branco. O intermediário foi utilizado para a reação seguinte, sem qualquer purificação adicional. 1H-NMR (300 MHz, DMSO-d6) δ 8.81-8.79(m, 1H), 8.41(s, 1H), 7.75-7.70(m, 1H), 7.46-7.39(m, 1H), 7.07(s, 1H), 4.23(s, 3H). MS(ESI)m/z: [M+H]+261.
De acordo com o método sintético similar do composto do exemplo 1, o composto de exemplo 85 cru (102 mg) foi obtido como um cristal branco a partir do intermediário 65 (67 mg, 0,257 mmol) e 4-(2-aminoetil)morfolina (36,9 mg , mmol 0,283). Um sistema de HPLC preparativo (o aparelho de purificação A), que foi utilizado para a purificação no composto do exemplo 1, foi utilizado para purificação adicional. MS(ESI)m/z: [M+H]+373.
De acordo com o método sintético similar do intermediário 35 no composto 34, uma solução de etanol (10 mL) do intermediário 34 (235mg, 0,854 mmol) e 2-amino-5-bromopiridina (148 mg, 0,854 mmol) foi aquecida em refluxo durante 20 horas e, em seguida o resíduo foi purificado com cromatografia de coluna (hexano/acetato de etila 1/4 (v/v) diclorometano/metanol = 30/1 (v/v)),utilizando gel de sílica para conceder o intermediário 66 (106 mg, o rendimento de 36%) foi obtido como um sólido amarelo claro. 1H-NMR (300 MHz, CDCl3) δ 8.32(s, 1H), 7.77(s, 1H), 7.56-7.53(m, 1H), 7.32- 7.27(m, 1H), 7.09-7.08(m, 1H), 4.4, 4-4.39(m, 2H), 4.34-4.33(m, 3H), 1.42(t, J=7.3Hz, 3H) MS(ESI)m/z: [M+H]+349 & 351.
De acordo com o método sintético similar do intermediário 36 no composto de exemplo 34, uma solução de hidróxido de sódio de 2M (0,304 mL, 0,608 mmol) foi adicionada a uma solução de metanol (15 mL) do intermediário 66 (106 mg, 0,304 mmol), e a mistura resultante foi agitada em 70°C durante 1 hora. Após a neutralização com solução de HCl de 2M, o intermediário 67 (66,7 mg) foi obtido como sólido branco. O intermediário foi utilizado para a reação seguinte, sem qualquer purificação adicional. 1H-NMR (300 MHz, DMSO-d6) δ 8.93(s, 1H), 8.38(s, 1H), 7.65(d, J=10.4Hz, 1H), 7.50(d, J=10.4Hz, 1H), 7.10(s, 1H), 4.23(s, 3H) MS(ESI)m/z: [M+H]+321 & 323.
De acordo com o método sintético similar do composto de exemplo 1, o intermediário 68 cru (77 mg, 0,078 mmol) foi obtido a partir do intermediário 67 (67 mg, 0,209 mmol) e 4-(2-aminoetil)morfolina (29,9 mg, 0,23 mmol). Em seguida, o resíduo foi purificado com SCX, e intermediário 68 (74,9 mg, 83% de rendimento) foi obtido como sólido branco. MS(ESI)m/z: [M+H]+433 & 435.
Uma mistura de DMF anidrosa (4 mL) do intermediário 68 (74,9 mg, 0,173 mmol) e cianeto de zinco (12,5 mg, 0,107 mmol) e tetraquis(trifenilfosfina) paládio (20,0 mg, 0,017 mmol) foi agitada em 100°C durante 20 horas. Após arrefecimento à temperatura ambiente, a água foi adicionada à solução de reação, e a mistura resultante foi extraída com solução de acetato de etila/tolueno (01/09). Como a maioria dos compostos foram transferidos para a camada aquosa, a camada aquosa foi concentrada sob pressão reduzida novamente. O resíduo sólido resultante foi fracionado com uma pequena quantidade de metanol. O sólido obtido foi seco sob pressão reduzida para conceder o composto de exemplo 86 cru (58,0 mg, 88% de rendimento) como um sólido branco. Um sistema de HPLC preparativo (o aparelho de purificação A), que foi utilizado para a purificação do composto do exemplo 1, foi utilizado para purificação adicional. MS(ESI)m/z: [M+H]+380, [M-H]-378.
De acordo com o método sintético similar ao intermediário 11 no composto de exemplo 17, a solução de 1,4-dioxano do (10 mL) intermediário 57 (224 mg, 0,605 mmol) e ácido 3-quinolinaborônico (122 mg, 0,666 mmol) foi aquecido sob agitaçãor em 100°C durante 15 horas, o resíduo resultante foi purificado com cromatografia de coluna (diclorometano/metanol = 30/1-20/1 (v/v)), utilizando gel de sílica para conceder o composto de exemplo 81 (181 mg, 80% produção) como o sólido marrom claro. O composto de exemplo (181 mg) foi purificado por recristalização a partir de acetato de etila e solução de hexano para conceder o composto de exemplo 87 (121 mg). 1H-NMR (300 MHz, CDCl3) δ 9.00-8.97(m, 1H), 8.23-8.14(m, 2H), 7.93-7.77(m, 2H), 7.69-7.60(m, 1H), 7.19(br s, 1H), 7.01(s, 1H), 4.00(s, 3H), 3.73-3.61(m, 4H), 3.54-3.45(m, 2H), 2.86-2.78(m, 2H). MS(ESI)m/z: [M+H]+372.
De acordo com o método sintético similar para o intermediário 16 no composto de exemplo 22, uma solução de hidróxido de sódio de 2M (2,55 mL, 5,10 mmol) foi adicionada a uma solução de metanol (12 mL) do intermediário 33 (500 mg, 2,55 mmol), e a mistura resultante foi agitada na temperatura ambiente durante 16 horas. Após neutralização com uma solução de HCl de 2M, o resíduo foi lavado com água fria para conceder o intermediário 69 (220 mg, 51% de rendimento) como um sólido branco. MS(ESI)m/z: [M+H]+169, [M-H]-167.
De acordo com o método sintético similar para o intermediário 56 no composto de exemplo 82, trietilamina (397 mg, 3,93 mmol) foi adicionada à solução mista de DMF anidrosa (5 mL) do intermediário 69 (220 mg, 1,31 mmol) e aminoacetaldehida dietil acetal (192 mg, 1,44 mmol), HBTU (595 mg, 1,57 mmol). A mistura resultante foi agitada em temperatura ambiente durante 3 horas. Depois do tratamento regular, o resíduo resultante foi purificado com cromatografia de coluna (hexano/acetato de etila = 2/1 (v/v)), utilizando gel de sílica para conceder o intermediário 70 (351 mg, 95% de rendimento) como um sólido branco. MS(ESI)m/z: [M-H]-282.27.
De acordo com o método sintético similar do intermediário 34 no Composto de Exemplo 34, brometo de feniltrimetilamonia (466 mg, 1,24 mmol) foi adicionado a uma solução de THF (5 mL) do intermediário 70 (351 mg, 1,24 mmol), a mistura resultante foi agitada na temperatura ambiente. Após o tratamento regular, o resíduo obtido foi purificado com cromatografia de coluna (hexano/acetato de etila = 3/1 (v/v)), utilizando gel de sílica para conceder ointermediário 71 (223 mg, 50% de rendimento) como um sólido branco. MS(ESI)m/z: [M+H]+362 & 364, [M-H]-360 & 362.
De acordo com o método sintético similar para o intermediário 35 no composto de exemplo 34, uma solução de etanol (10 mL) do intermediário 71 (222,9 mg, 0,615 mmol) e 2-amino-4-cianopiridina (73,3 mg, 0,615 mmol) foi aquecida sob agitação durante 15 horas. O resíduo foi purificado com cromatografia de coluna (diclorometano/metanol = 30/1 (v/v)), utilizando gel de sílica para conceder o intermediário 72 (174,1 mg, 74% de rendimento) como um sólido amarelo claro. MS(ESI)m/z: [M+H]+337.
De acordo com o método sintético similar o intermediário 57 no composto de exemplo 82, a mistura de reação de uma solução de THF (5 mL) do intermediário 72 (174,1 mg, 0,455 mmol) e uma solução de HCl de 2M (2,5 mL) foi agitada em 50°C durante 1 hora. Após arrefecimento até a temperatura ambiente, a solução de reação foi ajustada para mais do que pH10 com uma solução de hidróxido de sódio de 2M (2,5 mL), e a solução resultante foi extraída com acetato de etila. A camada orgânica obtida foi lavada com uma solução de cloreto de sódio saturada, e a solução foi seca sobre sulfato de sódio. Depois o agente de secagem foi filtrado, o filtrado foi concentrado sob pressão reduzida, e um sólido amarelo claro foi obtido. Triacetóxiborohidreto de sódio (289,3 mg, 1,365 mmol) foi adicionado à mistura do intermediário de aldeído cru e 1, 2 dicloroetano (5 mL) de morfolina (39,2 mg, 0,455 mmol) e ácido acético (1 mL) na temperatura ambiente. Depois a mistura resultante foi agitada na temperatura ambiente durante 15 horas, a mistura de reação foi ajustada para mais do que pH10 com uma solução de bicarbonato de sódio, e a solução foi extraída com diclorometano, três vezes, a camada orgânica obtida foi lavada com uma solução de cloreto de sódio saturada e foi seca sobre sulfato de sódio. Depois que o agente de secagem foi filtrada, o filtrado foi concentrado sob pressão reduzida. O resíduo foi purificado com cromatografia de coluna (diclorometano/metanol = 30/1-10/1 (v/v)) utilizando gel de sílica. Então, o composto de exemplo 88 (7 mg, 4% de rendimento) foi obtido como cólido laranja claro. MS(ESI)m/z: [M+H]+380.
As listas dos intermediários para a sintetização do composto de exemplo são mostradas na seguinte tabela 11-1 a 11-4-.Tabela 11-1

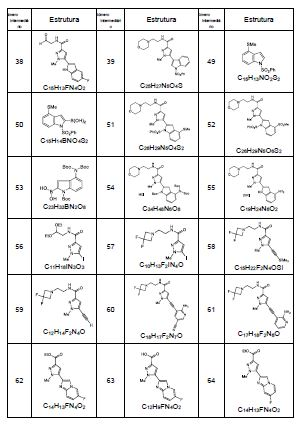

As afinidade de ligação do receptor de 5-HT2B dos compostos desta invenção são determinadas pelos procedimentos seguintes.
Células de CHO-K1 transfectadas de 5-HT2B humano foram obtidas de Euroscreen (N° de cat: ES-314-F) e cultivadas em casa. As células coletadas foram suspensas em 50 mM de HEPES (pH 7,4) suplementados com coquetel inibidor da protease (SIGMA, diluição 1:100) e 1 mM de EDTA, homogeneizado utilizando um disruptor Polytron PT1200 definido na potência máxima durante 30 segundos no gelo. Os homogeneizados foram centrifugados em 1.000 rpm em 4°C durante 5 min e os sobrenadantes foram congelados em -80°C durante 10 min. Os sobrenadantes foram congelados, em seguida, re-suspensos em 50 mM de HEPES (pH 7,4), homogeneizados e centrifugados mais uma vez da mesma maneira. Os sobrenadantes foram centrifugados em 25.000 rpm em 4°C durante 60 min. Os pellets foram então ressuspensos em 50 mM de HEPES (pH 7,4), homogeneizados, divididos e armazenados em -80°C até o uso. Uma alíquota de frações de membrana foi utilizada para determinação da concentração de proteína utilizando o kit de análise protéica BCA (PIERCE) e o leitor de placa ARVOsx (Wallac).
Para os experimentos de ligação ao receptor, 20 microL de compostos de teste foram incubados com 100 microL de [3H]-mesulergina (GE Healthcare, 10 nM) e 80 microL de homogenato de membrana (20 mcg de proteína) durante 120 minutos em temperatura ambiente. A ligação não específica foi determinada em 10 microM de mianserin (SIGMA) na concentração final. Todas as incubações foram encerradas por filtração a vácuo rápida superior a 0,2(v/v)% de PEI papéis filtro de vidro embebidos utiilizando colheitadeira de Filtermate (PerkinElmer), seguido de cinco lavagens com 50 mM de HEPES (pH 7,4). A radioactividade de ligação de receptor foi quantificada por contagem de cintilação líquida utilizando TopCount (PerkinElmer).
Como resultado do experimento, todos os compostos de exemplos mostraram afinidade de receptor 5-HT2B humano.
As afinidades de ligação ao receptor de 5-HT2B dos compostos desta invenção são determinadas pelos procedimentos seguintes.
Células CHO-K1 transfectadas de 5-HT2B humano foram obtidas de Euroscreen e cultivadas. As células foram cultivadas em 37°C e 5% de CO2 em meio de UltraCHO (Cambrex) suplementado com 400 mcrog/mL G418, 250 mcrog/mL de zeocina, 100 U/mL de penicilina, 100 mcrog/ml de estreptomicina e 1 (v/v)% de FBS dialisado (soro fetal bovino). Após o crescimento para 60-80% de confluência, o meio de cultura das células foi substituído por tampão de KRH (1,8 mM CaCl2, 1 mM de MgSO4, 115 mM de NaCl, 5,4 mM de KCl, 11 mM de D- glicose, 0,96 mM de NaH2PO4, 25 mM de HEPES, ajustado para pH7,4 com NaOH), incluindo 5 microM Fura-2 AM. As células foram incubadas durante 120 minutos em temperatura ambiente. Após a incubação, as células foram desacopladas com 0,05 (w/w)% de Tripsina/1 mM de EDTA e lavadas com PBS. Em seguida, essas células foram suspensas em tampão de KRH para dar 1,0 x 106 células/mL.
Os compostos desta invenção foram preparados em placas de 384 well (50 microL/well). Os 34 microL de suspensão de células (3,4 x 104 células) foi distribuído em cada well da placa de ensaio preta 384 well com fundo transparente. As placas de ensaio foram ajustadas sobre o FDSS6000 (Hamamatsu Photonics), e o monitoramento do sinal foi iniciado. Trinta segundos depois, 6 microL das várias diluições dos compostos foram adicionadas a cada well automaticamente e o FDSS6000 continuou a monitorar em mais 4,5 minutos para o exame da atividade antagonista. Em seguida, as células foram incubadas durante 10 minutos em temperatura ambiente sib a escuridão. As placas de 5 ensaio foram re-ajustadas no FDSS6000, e o monitoramento do sinal foi iniciado.
Trinta segundos depois, 20 microL de 9 nM 5-HT foi adicionado a cada well automaticamente e o FDSS6000 continuou a monitorar em mais 4,5 minutos para o exame dos valores de IC50 dos compostos de teste. Este experimento foi referido à Br. J. Pharmacol, Setembro de 1999, 128(1):. 13-20.
As atividades antagonistas do receptor de 5-HT2B (IC50, nM) de todos os 88 compostos de exemplos mostrados pela Tabela 12 a Tabela 15 seguintes foram a partir de 0,1 nM a 100 nM.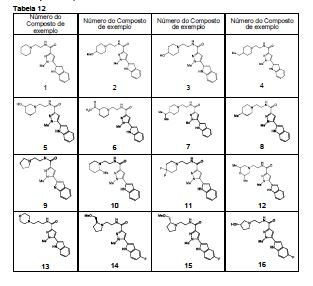
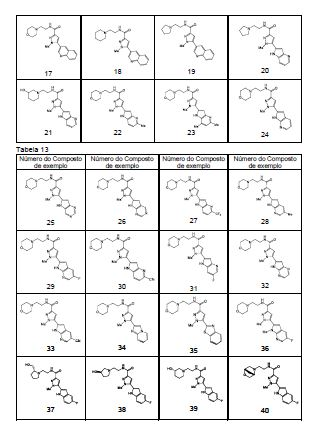


As afinidades de ligação ao receptor 5-HT2B dos compostos desta invenção são determinadas pelos procedimentos seguintes.
As células 3T3 transfectas de 5-HT2B humano foram preparadas em casa. As células foram cultivadas em 37°C e 5% de CO2 em meio de DMEM (Invitrogen) suplementado com 400 mcrog/mL G418, 100 U/mL de penicilina, 100 mcrog/ml de estreptomicina e 10(v/v)% de FBS. Após o crescimento para 60-80% de confluência, o meio de cultura das células foi substituído com tampão de KRH (1,8 mM de CaCl2, 1 mM de MgSO4, 115 mM de NaCl, 5,4 mM de KCl, 11 mM de D-glicose, 0,96 mM de NaH2PO4, 25 mM de HEPES, ajustado para pH 7,4 com NaOH), incluindo 5 microM de Fura-2 AM. As células foram incubadas durante 120 minutos em temperatura ambiente. Após a incubação, as células foram desacopladas com 0,05% de tripsina/1 mM de EDTA e lavadas com PBS. Essas células foram suspensas em tampão de KRH para dar 0,3 x 106 células/mL.
Os compostos desta invenção foram preparados em placas de 384- well (50 microL/well). Os 34 microL de suspensão de células (1,0 x 104 células) foi distribuído em cada well da placa de ensaio preto de 384-well com fundo transparente. As placas de ensaio foram ajustadas no FDSS6000 (Hamamatsu Photonics), e o monitoramento do sinal foi iniciado. Trinta segundos depois, 6 microL das várias diluições dos compostos foram adicionados a cada well automaticamente e o FDSS6000 continuou a monitorar em mais 4,5 minutos para o exame da atividade antagonista. Em seguida, as células foram incubadas durante 10 minutos em temperatura ambiente sob a escuridão. As placas de ensaio foram re-ajustadas no FDSS6000, e o monitoramento do sinal foi iniciado. Trinta segundos depois, 20 microL de 90 nm de 5-HT foi adicionado a cada well automaticamente e o FDSS6000 continuou a monitorar em mais 4,5 minutos para o exame dos valores de IC50 dos compostos de teste. Este experimento foi referido para Br. J. Pharmacol, em Setembro de 1999, 128 (1):. 13-20.
As afinidades de ligação ao receptor 5-HT2B dos compostos desta invenção são determinadas pelos procedimentos seguintes.
As células 3T3 transfectas de 5-HT2B humano foram preparadas em casa. As células foram cultivadas em 37°C e 5% de CO2 em meio de DMEM (Invitrogen) suplementado com 20 mcrog/mL de G418, 100 U/mL de penicilina, 100 mcrog/ml de estreptomicina e 10(v/v)% de FBS. Após o crescimento para 6080% de confluência, o meio de cultura das células foi substituído por tampão de KRH (1,8 mM de CaCl2, 1 mM de MgSO4, 115 mM de NaCl, 5,4 mM de KCl, 11 mM de D-glicose, 0,96 mM de NaH2PO4, 25 mM de HEPES, ajustado ao pH 7,4 com NaOH), incluindo 5 microM de Fura-2 AM. As células foram incubadas durante 120 minutos em temperatura ambiente. Após a incubação, as células foram isoladas, com 0,05% de tripsina/1 mM de EDTA e lavadas com PBS. Essas células foram suspensas em tampão de KRH para dar 0,45 x 106 células/mL.
Os compostos desta invenção foram preparados em placas de 384- well (50 microL /well). Os 34 microL de suspensão de células (1,5 x 104 células) foi distribuído em cada well da placa de ensaio preta de 384-well com fundo transparente. As placas de ensaio foram ajustadas no FDSS6000 (Hamamatsu Photonics), e o monitoramento do sinal foi iniciado. Trinta segundos depois, 6 microL das várias diluições dos compostos foram adicionados a cada well automaticamente e o FDSS6000 continuou a monitorar em mais 4,5 minutos para o exame da atividade antagonista. Em seguida, as células foram incubadas durante 10 minutos em temperatura ambiente sob a escuridão. As placas de ensaio foram re-ajustadas no FDSS6000, e o monitoramento do sinal foi iniciado. Trinta segundos depois, 20 microL de 3 nM de 5-HT foi adicionado a cada well automaticamente e o FDSS6000 continuou a monitorar em mais 4,5 minutos para o exame dos valores de IC50 dos compostos de teste. Este experimento foi referido para Br. J. Pharmacol, em Setembro de 1999, 128(1):. 13-20.
Os efeitos farmacológicos dos compostos de teste nesta invenção foi avaliado através da medição do efeito de melhora contra a inferiorização do limite de dor em estimulação de extensão do cólon em modelo deIBS induzido por TNBS.
Para detalhes, consultar a literatura, Katsuyo Ohashi et al, Pharmacology, 81 (2):. 144-150(2008).
A incisão mediana foi realizada sob anestesia em animais, ratos SD machos, 240-270 g. A solução de TNBS (50mg/kg, 30% metanol) foi tratada no início do cólon nos ratos. Após o tratamento, o ceco é colocado de volta na cavidade abdominal. A parede muscular é então suturada. Após a operação os animais foram alojados no ambiente normal, e foram utilizados para a avaliação farmacológica após 7 dias a partir da cirurgia. A estimulação de extensão do cólon foi utilizada para a avaliação dos compostos, Diop L. et al, J Pharmacol Exp Ther.302(3): 1013-1022 (2002). O balão (5cm de comprimento) é introduzido através do ânus e mantido em posição (ponta do balão é de 5 cm a partir do ânus). Em seguida, o balão foi insuflado progressivamente até o passo de 5 mm de Hg, de 0 a 70 mm de Hg utilizando Barostat (Barostat DISTENDER II R, G & J, 5 Canadá). O limite de dor foi avaliada pela pressão que correspondeu à a primeira contração abdominal produzida (cólica abdominal:. Wesselmann U et al, (1998) Neurosci Lett 246: 73-76).
O resultado do composto do Exemplo 24 foi mostrado na figura 1. O número de animais é de 8 em cada grupo. Os dados no gráfico apresentaram 10 uma media. A barra mostrou 25% e 75% destes valores. A análise estatística foi realizada com teste fechado utilizando teste de Mann-Whitney.
O eixo vertical revelou uma pressão de limite de dor. Neste caso, 10mg/kg deu os efeitos aprimorados contra a inferiorização do limiter de dor em TNBS. Portanto, novos derivados de pirazol-3-carboxamida podem ser úteis para 15 o tratamento de IBS.
Um composto desta invenção é útil como um antagonista seletivo de um receptor de 5-HT2B e é útil para o pré-tratamento ou prevenção de várias doenças associadas a um receptor de 5-HT2B.
Claims (6)
1. Composto da seguinte fórmula geral (I) ou seu sal farmaceuticamente aceitável: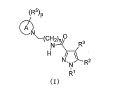 caracterizado pelo fato de que A é uma morfolina, piperidina, pirrolidina ou azetidina que se liga em N; R1 é um C1-C6 grupo alquil, ou um C1-C6 grupo haloalquil; R2 é representado pela seguinte fórmula geral:
caracterizado pelo fato de que A é uma morfolina, piperidina, pirrolidina ou azetidina que se liga em N; R1 é um C1-C6 grupo alquil, ou um C1-C6 grupo haloalquil; R2 é representado pela seguinte fórmula geral: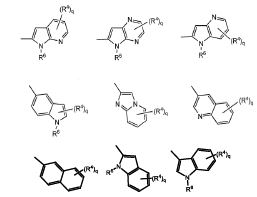 R3 é um átomo de halogênio ou hidrogênio; R4 é um C1-C6 grupo alquil, um C1-C6 grupo haloalquil, OH, OMe, halogênio, -(CH2)aOH, CONH2, CN, NH2, NHCOMe, SO2Me ou NHSO2Me; q é 0, 1, 2 ou 3; (R4)q pode substituir um dos dois anéis ou ambos os anéis; R5 é um C1-C6 grupo alquil, -(CH2)aOH, -(CH2)aOMe, halogênio ou CONH2, quando p é plural, R5 pode ser igual ou diferente; ou R5 pode combinar com o outro R5; R6 é hidrogênio ou um C1-C6 grupo alquil; a é independentemente 0, 1 ou 2; n é 1 ou 2; e p é 0, 1, ou 2 ou 3.
R3 é um átomo de halogênio ou hidrogênio; R4 é um C1-C6 grupo alquil, um C1-C6 grupo haloalquil, OH, OMe, halogênio, -(CH2)aOH, CONH2, CN, NH2, NHCOMe, SO2Me ou NHSO2Me; q é 0, 1, 2 ou 3; (R4)q pode substituir um dos dois anéis ou ambos os anéis; R5 é um C1-C6 grupo alquil, -(CH2)aOH, -(CH2)aOMe, halogênio ou CONH2, quando p é plural, R5 pode ser igual ou diferente; ou R5 pode combinar com o outro R5; R6 é hidrogênio ou um C1-C6 grupo alquil; a é independentemente 0, 1 ou 2; n é 1 ou 2; e p é 0, 1, ou 2 ou 3.
2. Composto ou seu sal farmaceuticamente aceitável, de acordo com a reivindicação 1, caracterizado pelo fato de que n é 1; p é 0, 1 ou 2; e q é 0, 1 ou 2.
3. Composto ou seu sal farmaceuticamente aceitável, de acordo com a reivindicação 1, caracterizado pelo fato de que é selecionado do grupo constituído por: 1-metil-N-[2-(morfolin-4-il)etil]-5-(quinolin-3-il)-1H-pirazol-3- carboxamida; 1-metil-5-{5-metil-1H-pirrolo[3,2-b]piridin-2-il}-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida; 1-metil-N-[2-(morfolin-4-il)etil]-5-{1H-pirrolo[2,3-b]piridin-2-il}-1H- pirazol-3-carboxamida; 1-metil-N-[2-(morfolin-4-il)etil]-5-{7H-pirrolo[2,3-d]pirimidin-6-il}-1H- pirazol-3-carboxamida; 1-metil-N-[2-(morfolin-4-il)etil]-5-[5-(trifluorometil)-1H-pirrolo[3,2- b]piridin-2-il]-1H-pirazol-3-carboxamida; 1-metil-5-{5-metil-1H-pirrolo[2,3-b]piridin-2-il}-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida; 5-{5-fluoro-1H-pirrolo[2,3-b]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida; 5-{5-ciano-1H-pirrolo[3,2-b]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida; 5-{6-fluoro-1H-pirrolo[3,2-b]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida; 1-metil-N-[2-(morfolin-4-il)etil]5-{5H-pirrolo[2,3-b]pirazin-6-il}-1H-pirazol- 3-carboxamida; 5-{5-ciano-1H-pirrolo[2,3-b]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]- 1H-pirazol-3-carboxamida; 5-{5-fluoro1-metil-1H-pirrolo[2,3-b]piridin-2-il}-1-metil-N-[2-(morfolin-4- il)etil]-1H-pirazol-3-carboxamida; N-[2-(3,3-difluoroazetidin-1-il)etil]-5-(5-fluoro-1H-indol-2-il)-1-metil-1H- pirazol-3-carboxamida; N-[2-(azetidin-1-il)etil]-5-(5-fluoro-1H-indol-2-il)-1-metil-1H-pirazol-3- carboxamida; 1-metil-5-(2-metil-1H-indol-5-il)-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-(1,2-dimetil-1H-indol-5-il)-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-(4-acetamido-1H-indol-2-il)-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol- 3-carboxamida; 5-{imidazo[1,2-a]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H-pirazol-3- carboxamida; 5-{6-fluoroimidazo[1,2-a]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H- pirazol-3-carboxamida; 5-{7-fluoroimidazo[1,2-a]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H- pirazol-3-carboxamida; 5-{6-cianoimidazo[1,2-a]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H- pirazol-3-carboxamida; N-[2-(3,3-difluoroazetidin-1-il)etil]-1-metil-5-(quinolin-3-il)-1H-pirazol-3- carboxamida; N-[2-(3,3-difluoroazetidin-1-il)etil]-1-metil-5-{1H-pirrolo[2,3-b]piridin-2- il}-1H-pirazol-3-carboxamida; e 5-{7-cianoimidazo[1,2-a]piridin-2-il}-1-metil-N-[2-(morfolin-4-il)etil]-1H- pirazol-3-carboxamida.
4. Intermediário do composto definido na reivindicação 1, caracterizado pelo fato de que é representado pela fórmula geral (1A): em que cada descrição é definida na reivindicação 1.
em que cada descrição é definida na reivindicação 1.
5. Composição farmacêutica, caracterizada pelo fato de que compreende o composto ou seu sal farmaceuticamente aceitável conforme definido em qualquer uma das reivindicações 1 a 3, e um carreador farmaceuticamente aceitável.
6. Uso do composto ou seu sal farmaceuticamente aceitável conforme definido em qualquer uma das reivindicações 1 a 3, caracterizado pelo fato de ser para a fabricação de um medicamento para a prevenção ou tratamento de enxaqueca, dor inflamatória, dor nociceptiva, dor neuropática, fibromialgia, dor lombar crônica, dor visceral, doença do refluxo gastroesofágico (DRGE), constipação, diarreia, desordem gastrointestinal funcional, síndrome do intestino irritável, asma, osteoartrite, artrite reumatoide, doença de Crohn, colite ulcerativa, glomerulonefrite, nefrite, dermatite, hepatite, vasculite, isquemia renal, acidente vascular cerebral, infarto do miocárdio, isquemia cerebral, doença de Alzheimer, obstrução reversível das vias aéreas, síndrome da doença respiratória do adulto, doença pulmonar obstrutiva crônica (DPOC), hipertensão pulmonar (HP), pneumonia intersticial idiopática, bronquite, fibrose hepática, alveolite fibrosante criptogênica, esclerose múltipla, depressão, ansiedade ou obesidade.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008-298821 | 2008-11-21 | ||
| JP2008298821 | 2008-11-21 | ||
| JP2009112344 | 2009-05-01 | ||
| JP2009-112344 | 2009-05-01 | ||
| PCT/JP2009/069816 WO2010058858A1 (ja) | 2008-11-21 | 2009-11-24 | 5-ht2b受容体拮抗活性を有する新規ピラゾール-3-カルボキサミド誘導体 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| BRPI0921097A2 BRPI0921097A2 (pt) | 2016-09-06 |
| BRPI0921097B1 true BRPI0921097B1 (pt) | 2021-03-02 |
| BRPI0921097B8 BRPI0921097B8 (pt) | 2021-05-25 |
Family
ID=42198297
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0921097A BRPI0921097B8 (pt) | 2008-11-21 | 2009-11-24 | composto ou seu sal farmaceuticamente aceitável, intermediário do composto, composição farmacêutica e uso do composto |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US8252790B2 (pt) |
| EP (1) | EP2386546B1 (pt) |
| JP (1) | JP5621148B2 (pt) |
| KR (1) | KR101637337B1 (pt) |
| CN (1) | CN102224142B (pt) |
| BR (1) | BRPI0921097B8 (pt) |
| CA (1) | CA2741511C (pt) |
| ES (1) | ES2549005T3 (pt) |
| HK (1) | HK1163063A1 (pt) |
| MX (1) | MX2011005221A (pt) |
| RU (1) | RU2528406C2 (pt) |
| WO (1) | WO2010058858A1 (pt) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101582335B1 (ko) * | 2008-10-29 | 2016-01-04 | 삼성전자주식회사 | 휴대용 단말기에서 전압 강하 보상 장치 및 방법 |
| US8889730B2 (en) | 2012-04-10 | 2014-11-18 | Pfizer Inc. | Indole and indazole compounds that activate AMPK |
| JP6064062B2 (ja) | 2013-03-15 | 2017-01-18 | ファイザー・インク | Ampkを活性化させるインダゾール化合物 |
| EP3089972B1 (de) | 2014-01-03 | 2018-05-16 | Bayer Animal Health GmbH | Neue pyrazolyl-heteroarylamide als schädlingsbekämpfungsmittel |
| WO2015116460A1 (en) * | 2014-01-28 | 2015-08-06 | Virginia Commonwealth University | 2-substituted-5-hydroxy-4h-chromen-4-ones as novel ligands for the serotonin receptor 2b (5-ht2b) |
| WO2017070320A1 (en) | 2015-10-21 | 2017-04-27 | University Of Pittsburgh - Of The Commonwealth System Of Higher Education | Phenyl indole allosteric inhibitors of p97 atpase |
| AU2016366310C1 (en) * | 2015-12-09 | 2021-09-09 | Research Triangle Institute | Improved apelin receptor (APJ) agonists and uses thereof |
| EP3753924A1 (en) | 2019-06-18 | 2020-12-23 | AnaMar AB | New tricyclic 5-ht2 antagonists |
| KR20220097434A (ko) * | 2019-11-01 | 2022-07-07 | 브리스톨-마이어스 스큅 컴퍼니 | 톨 수용체 억제제로서의 치환된 피라졸 화합물 |
| US11807644B2 (en) * | 2020-05-12 | 2023-11-07 | Pmv Pharmaceuticals, Inc. | Methods and compounds for restoring mutant p53 function |
| KR20220014854A (ko) * | 2020-07-29 | 2022-02-07 | 주식회사 비보존 | mGluR5 및 5-HT2A 수용체의 이중 조절제 및 이의 용도 |
| CN116270635A (zh) * | 2021-12-13 | 2023-06-23 | 清华大学 | 杂环化合物用于减轻化疗药物引起的不良反应的用途 |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2745553B2 (ja) | 1988-08-22 | 1998-04-28 | ミツミ電機株式会社 | 低電圧基準電源回路 |
| US5376645A (en) | 1990-01-23 | 1994-12-27 | University Of Kansas | Derivatives of cyclodextrins exhibiting enhanced aqueous solubility and the use thereof |
| KR0166088B1 (ko) | 1990-01-23 | 1999-01-15 | . | 수용해도가 증가된 시클로덱스트린 유도체 및 이의 용도 |
| JP3534816B2 (ja) | 1994-03-10 | 2004-06-07 | 日本曹達株式会社 | メチルチオベンゼン類の製造方法 |
| AU679635B2 (en) | 1994-03-11 | 1997-07-03 | Eli Lilly And Company | Method for treating 5HT-2B receptor related conditions |
| US5470862A (en) | 1995-02-03 | 1995-11-28 | Ohmeda Pharmaceutical Products Division Inc. | Substituted pyrazolyl compounds and methods employing such compounds |
| GB9518953D0 (en) | 1995-09-15 | 1995-11-15 | Pfizer Ltd | Pharmaceutical formulations |
| TW438796B (en) * | 1996-05-15 | 2001-06-07 | Hoffmann La Roche | 2,4-diaminopyrimidine derivatives, the manufacture process thereof, and the antibiotically-active pharmaceutical composition containing the same |
| WO2000035296A1 (en) | 1996-11-27 | 2000-06-22 | Wm. Wrigley Jr. Company | Improved release of medicament active agents from a chewing gum coating |
| EA003925B1 (ru) * | 1997-05-22 | 2003-10-30 | Дж.Д.Сирл Энд Ко | ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ, КАК ИНГИБИТОРЫ p 38 КИНАЗЫ |
| GB9711643D0 (en) | 1997-06-05 | 1997-07-30 | Janssen Pharmaceutica Nv | Glass thermoplastic systems |
| IL136588A0 (en) * | 1998-02-27 | 2001-06-14 | Pfizer Prod Inc | N-[(substituted five-membered di-or triaza diunsaturated ring) carbonyl] guanidine derivatives for the treatment of ischemia |
| TWI262185B (en) * | 1999-10-01 | 2006-09-21 | Eisai Co Ltd | Carboxylic acid derivatives having anti-hyperglycemia and anti-hyperlipemia action, and pharmaceutical composition containing the derivatives |
| US6439387B1 (en) | 2000-07-20 | 2002-08-27 | Air Fresh Inc. | Liquid detergent container and dispensing |
| WO2002080899A1 (fr) * | 2001-03-30 | 2002-10-17 | Eisai Co., Ltd. | Agent de traitement de maladie digestive |
| CN1832741A (zh) * | 2003-08-07 | 2006-09-13 | 默克公司 | 作为11-β-羟甾类脱氢酶-1抑制剂的吡唑甲酰胺类 |
| TW200526588A (en) | 2003-11-17 | 2005-08-16 | Smithkline Beecham Corp | Chemical compounds |
| RU2375357C2 (ru) * | 2004-03-09 | 2009-12-10 | Ф.Хоффманн-Ля Рош Аг | Производные пиразолилиндолила в качестве активаторов ppar |
| CN101277932A (zh) * | 2005-09-30 | 2008-10-01 | 万有制药株式会社 | 芳基取代含氮杂环化合物 |
| CN101384549B (zh) * | 2006-02-20 | 2011-04-13 | 安斯泰来制药株式会社 | 吡咯衍生物或其盐 |
| EP2016068A1 (en) * | 2006-05-03 | 2009-01-21 | AstraZeneca AB | Pyrazole derivatives and their use as pi3k inhibitors |
| TW200831498A (en) | 2006-10-13 | 2008-08-01 | Otsuka Pharma Co Ltd | Heterocyclic compound |
-
2009
- 2009-11-24 EP EP09827639.7A patent/EP2386546B1/en active Active
- 2009-11-24 KR KR1020117014181A patent/KR101637337B1/ko active IP Right Grant
- 2009-11-24 MX MX2011005221A patent/MX2011005221A/es active IP Right Grant
- 2009-11-24 WO PCT/JP2009/069816 patent/WO2010058858A1/ja active Application Filing
- 2009-11-24 RU RU2011125314/04A patent/RU2528406C2/ru active
- 2009-11-24 ES ES09827639.7T patent/ES2549005T3/es active Active
- 2009-11-24 US US13/129,916 patent/US8252790B2/en active Active
- 2009-11-24 CA CA2741511A patent/CA2741511C/en active Active
- 2009-11-24 BR BRPI0921097A patent/BRPI0921097B8/pt active IP Right Grant
- 2009-11-24 CN CN200980146558.8A patent/CN102224142B/zh active Active
- 2009-11-24 JP JP2010539267A patent/JP5621148B2/ja active Active
-
2012
- 2012-03-28 HK HK12103083.8A patent/HK1163063A1/xx unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CA2741511A1 (en) | 2010-05-27 |
| ES2549005T3 (es) | 2015-10-22 |
| WO2010058858A1 (ja) | 2010-05-27 |
| RU2011125314A (ru) | 2012-12-27 |
| KR101637337B1 (ko) | 2016-07-07 |
| HK1163063A1 (en) | 2012-09-07 |
| US8252790B2 (en) | 2012-08-28 |
| US20110275628A1 (en) | 2011-11-10 |
| EP2386546A4 (en) | 2012-08-15 |
| EP2386546B1 (en) | 2015-08-19 |
| JPWO2010058858A1 (ja) | 2012-04-19 |
| MX2011005221A (es) | 2011-08-03 |
| EP2386546A1 (en) | 2011-11-16 |
| JP5621148B2 (ja) | 2014-11-05 |
| BRPI0921097A2 (pt) | 2016-09-06 |
| CN102224142B (zh) | 2015-07-22 |
| RU2528406C2 (ru) | 2014-09-20 |
| KR20110091550A (ko) | 2011-08-11 |
| CN102224142A (zh) | 2011-10-19 |
| BRPI0921097B8 (pt) | 2021-05-25 |
| CA2741511C (en) | 2017-01-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2386546B1 (en) | Novel pyrazole-3-carboxamide derivate having 5-ht2b receptor antagonist activity | |
| JP5301469B2 (ja) | Ep4受容体アンタゴニストとしてのインドール及びインドリンシクロプロピルアミド誘導体 | |
| JP5685203B2 (ja) | カルシウムチャネル遮断薬またはナトリウムチャネル遮断薬としてのアリール置換カルボキサミド誘導体 | |
| JP7426398B2 (ja) | Ras阻害剤としての新規イソインドリノン置換インドールおよび誘導体 | |
| CN116041346A (zh) | 作为ehmt2抑制剂的胺取代的杂环化合物及其使用方法 | |
| WO2020210828A1 (en) | (aza)indazolyl-aryl sulfonamide and related compounds and their use in treating medical conditions | |
| US20110312992A1 (en) | Inhibitors of C-Kit and Uses Thereof | |
| JP5140080B2 (ja) | 選択的アシッドポンプ阻害剤としてのベンゾイミダゾール誘導体 | |
| EP3562809B1 (en) | Pyrazolopyrimidine compounds and methods of use thereof | |
| JP2012521428A (ja) | 疼痛治療用のp2x3受容体アンタゴニスト | |
| KR20080114711A (ko) | 17β HSD 타입 5 저해제 | |
| WO2019205983A1 (zh) | 氧杂螺环类化合物及其制备方法和用途 | |
| KR20200013718A (ko) | 바닌 억제제로서의 헤테로방향족 화합물 | |
| KR20230026418A (ko) | 아릴설포닐 유도체 및 무스카린성 아세틸콜린 수용체 m5 억제제로서의 이의 용도 | |
| JP2016540811A (ja) | N−アシルピペリジンエーテルトロポミオシン関連キナーゼ阻害剤 | |
| BR112016005606B1 (pt) | Composto, composição farmacêutica, e usos de um composto | |
| KR20130122531A (ko) | Mglur5 양성 알로스테릭 조절물로서 치환된 6메틸니코틴아미드 | |
| JP6762300B2 (ja) | アミノピラゾール誘導体 | |
| JP2015537018A (ja) | 置換ピラゾロ[3,4−d]ピリミジン化合物、その製造及びシグマ受容体リガンドとしての使用 | |
| WO2012022120A1 (zh) | 二氢吡唑类化合物 | |
| TW200924765A (en) | Piperidine derivative | |
| CN113631548B (zh) | 血清素5-ht2b抑制化合物 | |
| EP4066895A1 (en) | Compound having lysophosphatidic acid receptor agonistic activity and pharmaceutical use of said compound | |
| JP6401156B2 (ja) | イミダゾ[2,1−b]チアゾール誘導体、その製造方法、および医薬品としての使用 | |
| JP2010155802A (ja) | 5−ht2b受容体拮抗活性を有する新規5−エチニルピラゾール−3−カルボキサミド誘導体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] | ||
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B07E | Notification of approval relating to section 229 industrial property law [chapter 7.5 patent gazette] | ||
| B06U | Preliminary requirement: requests with searches performed by other patent offices: procedure suspended [chapter 6.21 patent gazette] | ||
| B06A | Patent application procedure suspended [chapter 6.1 patent gazette] | ||
| B06A | Patent application procedure suspended [chapter 6.1 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 10 (DEZ) ANOS CONTADOS A PARTIR DE 02/03/2021, OBSERVADAS AS CONDICOES LEGAIS. |
|
| B16C | Correction of notification of the grant [chapter 16.3 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 24/11/2009 OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF |
|
| B25G | Requested change of headquarter approved |
Owner name: RAQUALIA PHARMA INC. (JP) |