BRPI0317176B1 - Processo para a síntese de peptídeos contendo uma subestrutura de 4-hidróxi-prolina - Google Patents
Processo para a síntese de peptídeos contendo uma subestrutura de 4-hidróxi-prolina Download PDFInfo
- Publication number
- BRPI0317176B1 BRPI0317176B1 BRPI0317176-0A BRPI0317176A BRPI0317176B1 BR PI0317176 B1 BRPI0317176 B1 BR PI0317176B1 BR PI0317176 A BRPI0317176 A BR PI0317176A BR PI0317176 B1 BRPI0317176 B1 BR PI0317176B1
- Authority
- BR
- Brazil
- Prior art keywords
- compound
- formula
- group
- removable
- reaction
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 27
- PMMYEEVYMWASQN-DMTCNVIQSA-N Hydroxyproline Chemical compound O[C@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-DMTCNVIQSA-N 0.000 title abstract description 7
- PMMYEEVYMWASQN-UHFFFAOYSA-N dl-hydroxyproline Natural products OC1C[NH2+]C(C([O-])=O)C1 PMMYEEVYMWASQN-UHFFFAOYSA-N 0.000 title abstract description 5
- 229960002591 hydroxyproline Drugs 0.000 title abstract description 5
- 238000010647 peptide synthesis reaction Methods 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 110
- 238000004519 manufacturing process Methods 0.000 claims abstract description 6
- 238000006243 chemical reaction Methods 0.000 claims description 24
- 125000006239 protecting group Chemical group 0.000 claims description 18
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 claims description 16
- 230000007062 hydrolysis Effects 0.000 claims description 14
- 238000006460 hydrolysis reaction Methods 0.000 claims description 14
- 238000007327 hydrogenolysis reaction Methods 0.000 claims description 11
- 239000001257 hydrogen Substances 0.000 claims description 8
- 229910052739 hydrogen Inorganic materials 0.000 claims description 8
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 7
- 230000000903 blocking effect Effects 0.000 claims description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims description 3
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 3
- 238000003786 synthesis reaction Methods 0.000 abstract description 11
- 102000004196 processed proteins & peptides Human genes 0.000 abstract description 10
- 108090000765 processed proteins & peptides Proteins 0.000 abstract description 10
- 230000015572 biosynthetic process Effects 0.000 abstract description 8
- 239000000543 intermediate Substances 0.000 abstract description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 36
- 125000001424 substituent group Chemical group 0.000 description 31
- 239000000243 solution Substances 0.000 description 28
- -1 hydroxy, amino Chemical group 0.000 description 23
- 229910052736 halogen Inorganic materials 0.000 description 21
- 150000002367 halogens Chemical class 0.000 description 21
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 20
- 239000007790 solid phase Chemical group 0.000 description 20
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 17
- 229910052751 metal Inorganic materials 0.000 description 17
- 239000002184 metal Substances 0.000 description 17
- 150000002902 organometallic compounds Chemical class 0.000 description 16
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- 239000000203 mixture Substances 0.000 description 15
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 14
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 14
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 13
- 230000000269 nucleophilic effect Effects 0.000 description 13
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 12
- 239000000463 material Substances 0.000 description 11
- 238000003756 stirring Methods 0.000 description 11
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 10
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 10
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 10
- 238000002360 preparation method Methods 0.000 description 10
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 9
- 239000012071 phase Substances 0.000 description 9
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 8
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 8
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 8
- 125000000217 alkyl group Chemical group 0.000 description 8
- 239000000460 chlorine Substances 0.000 description 8
- 229910052801 chlorine Inorganic materials 0.000 description 8
- 150000002736 metal compounds Chemical class 0.000 description 8
- 150000003839 salts Chemical group 0.000 description 8
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 7
- 239000012039 electrophile Substances 0.000 description 7
- 229910052744 lithium Inorganic materials 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 6
- 239000001569 carbon dioxide Substances 0.000 description 6
- 229910002092 carbon dioxide Inorganic materials 0.000 description 6
- 229910052731 fluorine Inorganic materials 0.000 description 6
- 239000011737 fluorine Substances 0.000 description 6
- 238000004128 high performance liquid chromatography Methods 0.000 description 6
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 6
- 229940011051 isopropyl acetate Drugs 0.000 description 6
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 6
- 239000011541 reaction mixture Substances 0.000 description 6
- 239000002904 solvent Substances 0.000 description 6
- BUNWPAKPJKBKIA-UHFFFAOYSA-N 3-(iodomethyl)piperidine Chemical compound ICC1CCCNC1 BUNWPAKPJKBKIA-UHFFFAOYSA-N 0.000 description 5
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 5
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 5
- 150000001266 acyl halides Chemical class 0.000 description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 5
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 4
- 239000004793 Polystyrene Substances 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 125000003277 amino group Chemical group 0.000 description 4
- 239000008346 aqueous phase Substances 0.000 description 4
- 125000003118 aryl group Chemical group 0.000 description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 4
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 4
- 229910052794 bromium Inorganic materials 0.000 description 4
- 230000008878 coupling Effects 0.000 description 4
- 238000010168 coupling process Methods 0.000 description 4
- 238000005859 coupling reaction Methods 0.000 description 4
- 239000006260 foam Substances 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 4
- 239000012074 organic phase Substances 0.000 description 4
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 4
- 229920002223 polystyrene Polymers 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 4
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 3
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 125000003545 alkoxy group Chemical group 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- 125000001188 haloalkyl group Chemical group 0.000 description 3
- 238000005984 hydrogenation reaction Methods 0.000 description 3
- 238000011065 in-situ storage Methods 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 3
- 150000002901 organomagnesium compounds Chemical class 0.000 description 3
- 229920001184 polypeptide Polymers 0.000 description 3
- 238000004886 process control Methods 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 3
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 2
- 239000004215 Carbon black (E152) Substances 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical class ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- XAKBSHICSHRJCL-UHFFFAOYSA-N [CH2]C(=O)C1=CC=CC=C1 Chemical group [CH2]C(=O)C1=CC=CC=C1 XAKBSHICSHRJCL-UHFFFAOYSA-N 0.000 description 2
- 125000004442 acylamino group Chemical group 0.000 description 2
- 125000004423 acyloxy group Chemical group 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 238000010945 base-catalyzed hydrolysis reactiony Methods 0.000 description 2
- RWCCWEUUXYIKHB-UHFFFAOYSA-N benzophenone Chemical compound C=1C=CC=CC=1C(=O)C1=CC=CC=C1 RWCCWEUUXYIKHB-UHFFFAOYSA-N 0.000 description 2
- 239000012965 benzophenone Substances 0.000 description 2
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 239000013068 control sample Substances 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 2
- 230000032050 esterification Effects 0.000 description 2
- 238000005886 esterification reaction Methods 0.000 description 2
- 150000004795 grignard reagents Chemical class 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- 229910000856 hastalloy Inorganic materials 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 2
- 125000005647 linker group Chemical group 0.000 description 2
- 239000000178 monomer Substances 0.000 description 2
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 229960005235 piperonyl butoxide Drugs 0.000 description 2
- 125000004591 piperonyl group Chemical group C(C1=CC=2OCOC2C=C1)* 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- LVTJOONKWUXEFR-FZRMHRINSA-N protoneodioscin Natural products O(C[C@@H](CC[C@]1(O)[C@H](C)[C@@H]2[C@]3(C)[C@H]([C@H]4[C@@H]([C@]5(C)C(=CC4)C[C@@H](O[C@@H]4[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@@H](O)[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@H](CO)O4)CC5)CC3)C[C@@H]2O1)C)[C@H]1[C@H](O)[C@H](O)[C@H](O)[C@@H](CO)O1 LVTJOONKWUXEFR-FZRMHRINSA-N 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 230000000087 stabilizing effect Effects 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 1
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 description 1
- SWJPEBQEEAHIGZ-UHFFFAOYSA-N 1,4-dibromobenzene Chemical compound BrC1=CC=C(Br)C=C1 SWJPEBQEEAHIGZ-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- JMTMSDXUXJISAY-UHFFFAOYSA-N 2H-benzotriazol-4-ol Chemical compound OC1=CC=CC2=C1N=NN2 JMTMSDXUXJISAY-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 102100026735 Coagulation factor VIII Human genes 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 102000002068 Glycopeptides Human genes 0.000 description 1
- 108010015899 Glycopeptides Proteins 0.000 description 1
- 239000007818 Grignard reagent Substances 0.000 description 1
- 101000911390 Homo sapiens Coagulation factor VIII Proteins 0.000 description 1
- WOBHKFSMXKNTIM-UHFFFAOYSA-N Hydroxyethyl methacrylate Chemical compound CC(=C)C(=O)OCCO WOBHKFSMXKNTIM-UHFFFAOYSA-N 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 150000001263 acyl chlorides Chemical class 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 125000005115 alkyl carbamoyl group Chemical group 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 150000001502 aryl halides Chemical class 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- OPBURCVWOKTFHZ-UHFFFAOYSA-N benzyl 1-hydroxy-2,5-dioxopyrrolidine-3-carboxylate Chemical compound O=C1N(O)C(=O)CC1C(=O)OCC1=CC=CC=C1 OPBURCVWOKTFHZ-UHFFFAOYSA-N 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 239000008366 buffered solution Substances 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 230000021523 carboxylation Effects 0.000 description 1
- 238000006473 carboxylation reaction Methods 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 125000005982 diphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 229920000592 inorganic polymer Polymers 0.000 description 1
- 238000009413 insulation Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- CETVQRFGPOGIQJ-UHFFFAOYSA-N lithium;hexane Chemical compound [Li+].CCCCC[CH2-] CETVQRFGPOGIQJ-UHFFFAOYSA-N 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 125000006502 nitrobenzyl group Chemical group 0.000 description 1
- 239000012454 non-polar solvent Substances 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 125000000962 organic group Chemical group 0.000 description 1
- 150000002900 organolithium compounds Chemical class 0.000 description 1
- 125000005740 oxycarbonyl group Chemical group [*:1]OC([*:2])=O 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 239000002861 polymer material Substances 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000011814 protection agent Substances 0.000 description 1
- 239000003223 protective agent Substances 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000002344 surface layer Substances 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- AOCSUUGBCMTKJH-UHFFFAOYSA-N tert-butyl n-(2-aminoethyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCN AOCSUUGBCMTKJH-UHFFFAOYSA-N 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 125000005424 tosyloxy group Chemical group S(=O)(=O)(C1=CC=C(C)C=C1)O* 0.000 description 1
- 230000017105 transposition Effects 0.000 description 1
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/347—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/15—Preparation of carboxylic acids or their salts, halides or anhydrides by reaction of organic compounds with carbon dioxide, e.g. Kolbe-Schmitt synthesis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C65/00—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C65/01—Compounds having carboxyl groups bound to carbon atoms of six—membered aromatic rings and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups containing hydroxy or O-metal groups
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Peptides Or Proteins (AREA)
- Pyrrole Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
"processo para a síntese de peptídeos contendo uma subestrutura de 4-hidróxi-prolina". a presente invenção refere-se a processos para preparo de peptídeos e a intermediários envolvidos em tais processos, por exemplo um processo para preparo de um composto de fórmula viii em que r~ 12~ e r~ 13~ são como definidos aqui.
Description
(54) Título: PROCESSO PARA A SÍNTESE DE PEPTÍDEOS CONTENDO UMA SUBESTRUTURA DE 4-HIDRÓXI-PROLINA (51) Int.CI.: C07D 207/16; C07C 51/347; C07C 65/01; C07K 1/04 (30) Prioridade Unionista: 12/12/2002 GB 02 29020.3, 16/12/2002 GB 02 29280.3 (73) Titular(es): NOVARTIS AG (72) Inventor(es): BERNHARD ERB; WERNER PACHINGER; BERNHARD WIETFELD; WALTER PRIKOSZOVICH • · * ··· · ··· · ··· ·· ···· · · · ··· · · • * · * * · · · ··· • · «··· · ·· · · ο°>
Relatório Descritivo da Patente de Invenção para PROCESSO PARA A SÍNTESE DE PEPTÍDEOS CONTENDO UMA SUBESTRUTURA DE 4-HIDRÓXI-PROLINA.
A presente invenção refere-se a processos para preparo de 5 peptídeos e intermediários envolvidos em tais processos.
Em um aspecto, a invenção refere-se a:
(A) um processo para preparo de um composto de fórmula I

em que Rt é um substituinte reativo ou uma ligação a uma fase sólida;
R2 é um substituinte reativo; e
R3, R4 e R5 são, cada um, independentemente hidrogênio ou um ou mais substituintes ligados a cada anel de benzeno e selecionados de hidróxi, amino, C1.10-alquila, C^^-alcóxi, C1.10-alquilamino, di-C110-alquilamino, carbamoíla, C^^-alquilcarbamoíla, di-C^^-alquilcarbamoíla, halo-C^^-alquila, halogênio e nitro;
em forma livre ou de sal; compreendendo (a) reação de um composto de fórmula VI com um eletrófilo:

em que R3, R4 e R5 são como definidos acima;
R9 é -OH, -OM ou -OMX, onde M é metal e X é um substituinte nucleofílico;
R10 é -M ou -MX, onde M é metal e X é um substituinte nucleofílico;
em forma livre ou de sal;
• · · * · • ♦ · · · • · · · • · · · · • · · e hidrólise do composto resultante para formar um composto de fórmula I em que R2 é hidróxi;
(b) opcionalmente conversão de um composto de fórmula I em que R2 é hidróxi a um composto de fórmula I em que R2 é outro que não hidróxi;
opcionalmente conversão de em um composto de fórmula I a um grupo de Rt alternativo; opcionalmente desproteção de um composto de fórmula I em forma protegida; e onde requerido, conversão de um composto de fórmula I obtido em forma livre na forma de sal desejada, ou viceversa;
um processo para o preparo de um sistema de suporte de fase sólida, compreendendo preparo de um composto de fórmula I por um processo como definido acima, e acoplamento do composto com um material de fase sólida funcionalizado ou derivatizado adequado;
um composto de fórmula V em forma livre ou de sal v FL FL (C) (d) (θ) (B) (C)
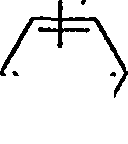
V
R
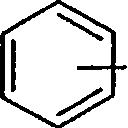
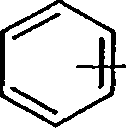
em que R3, R4, R5, e R9 são como definidos acima; e R7 é um substituinte nucleofílico;
(D) um composto de fórmula VI em forma livre ou de sal
R ^R- V'
-FL
V//
R.

FL em que R3, R4, R5 e R9 são como definidos acima; e • ·
4
444 · • 4 • ·· 4
R10 é -M ou -MX, onde M é metal e X é um substituinte nucleofílico.
A presente invenção fornece uma rotina simples para a preparação de compostos de fórmula I, os quais são úteis para síntese química de fase sólida. O processo da invenção pode diretamente produzir um composto de fórmula I ligado a uma fase sólida, ou onde Rt é um substituinte reativo, o composto de fórmula I pode facilmente ser acoplado a uma fase sólida em um último estágio. A presença do substituinte reativo R2 permite o emprego dos compostos de fórmula I como ligadores na síntese de oligôme10 ros e polímeros, tais como glicopeptídeos, nucleotídeos e proteínas, especialmente na síntese de fase sólida de peptídeos. Os compostos de fórmula I, particularmente onde R2 é halogênio, podem além disso ser empregados como agentes de proteção para grupos funcionais de proteção, por exemplo grupos de amino ou hidróxi, em síntese química.
Os compostos de fórmulas V e VI são úteis como compostos intermediários na preparação de compostos de fórmula I.
Um composto de fórmula VI pode ser preparado por reação de um composto de fórmula V com um metal ou composto organometálico:
em que R3, R4, R5 e R9 são como definidos acima; e
R7 é um substituinte nucleofílico.
Um composto de fórmula V pode ser preparado por:
(i) reação de um composto de fórmula II com um metal ou composto organometálico
II em que Re e R7, são cada um, substituinte nucleofílico e R3 é como definido • · • · • · • · acima e é protegido se necessário por um grupo de proteção removível; e (ii) reação do composto obtido em (i) com um composto de

em que R4 e R5 são como definidos acima e são protegidos se necessário por um grupo de proteção removível.
O processo da presente invenção pode adequadamente ser executado em um recipiente de reação simples sem isolamento intermediário.
Os termos empregados no relatório descritivo têm os seguintes significados:
Alquila pode ser reta ou ramificada. De preferência alquila é
CV4-alquila.
Alcóxi pode ser alcóxi reto ou ramificado. De preferência alcóxi é CM-alcóxi.
Acilamino denota um grupo de fórmula -NH-C(O)-R onde R é C^o-alquila, cicloalquila ou arila de cadeia reta ou ramificada. De preferência R é C^-alquila.
Acilóxi denota um grupo de fórmula -O-C(O)-R onde R é como definido acima.
Arila é de preferência C6.10-arila, por exemplo fenila.
Halogênio quer dizer flúor, cloro, bromo ou iodo.
Haloalquila quer dizer C^^-alquila de cadeia reta ou ramificada, substituída por um ou mais, por exemplo um, dois ou três átomos de halogênio, de preferência átomos de flúor ou cloro. De preferência haloalquila é CM-alquila substituída por um, dois ou três átomos de flúor ou cloro.
Composto organometálico denota um composto no qual um átomo de carbono de um grupo orgânico está ligado a um metal. O composto organometálico é de preferência um composto alquilmetálico, por exemplo um alquil-lítio, por exemplo um composto de C1.10 alquil-lítio de ca·· • · ·

deia reta ou ramificada ou pode alternativamente ser um composto arilmetálico, por exemplo um aril-lítio.
Mais preferivelmente o composto de alquil-lítio é um composto de C3_6 alquil-lítio, tal como butil-lítio ou hexil-lítio.
Alternativamente, o composto organometálico pode ser um composto de organomagnésio, por exemplo um composto de alquilmagnésio ou arilmagnésio de cadeia reta ou ramificada, de preferência um composto de CV6 alquilmagnésio. Os compostos de organomagnésio são geralmente conhecidos como reagentes de Grignard. O composto de organomagnésio é de preferência um haleto de organomagnésio, especialmente um iodeto ou brometo.
Em outras modalidades alternativas, o composto organometálico pode ser um composto de alquil- ou arilzinco, por exemplo um composto de C^-alquilzinco, ou um composto de C16-alquil- ou arilestanho.
M é de preferência lítio ou magnésio.
Ri pode ser um substituinte reativo adequado para ligação do composto a uma fase sólida. R, pode adequadamente ser -C(O)R’, -C(O)-OR’, -C(O)-NR’R”, -R12-NR’R, -R12-OR’, -NR’R, ou -C(O)X, em que R’e R são, cada um, independentemente hidrogênio ou C^^-alquila reta ou ramificada, por exemplo CM-alquila, R12 é Ci.10-alquila reta ou ramificada, por exemplo CM-alquila, e X é um substituinte nucleofílico, de preferência halogênio, por exemplo cloro. Ri pode adequadamente estar na posição para, orto ou meta, de preferência na posição para.
Alternativamente Ri pode ser uma ligação a um material de fase sólida, por exemplo poliestireno. De preferência a ligação é da fórmula -C(O)-P, -C(O)-OP, -C(O)-NR’-P, -R12-NR’-P, -R12-OP, -NR’-P, -C(O)-R12-P, -C(O)-OR12-P, -C(O)-NR’-R12-P, -R12-NR’-R12-P, -R12-OR12-P, -NR’-R12-P ou -Rí2-P, em que R’, R e Ri2 são como definidos acima e P é um material de fase sólida. Mais preferivelmente Ri é -C(O)-OP, -C(O)-ORí2-P, -C(O)-NH-P, -C(O)-NH-Ri2-P, -NH-Rí2-P ou -R12-P, em que Rí2 é metila, por exemplo -CH2-P.
R2 é de preferência um substituinte reativo adequado para liga• · · ‘ 14 ção do composto a um oligômero ou polímero biológico, ou uma unidade de monômero destes, por exemplo um aminoácido ou polipeptídeo. R2 pode adequadamente ser hidróxi, acilamino, acilóxi, amino, halogênio, sulfidrila, C^o-alcóxi ou C6_10-arilóxi, de preferência halogênio.
Cada anel de benzeno mostrado nas fórmulas I a VII pode ser substituído por um ou mais grupos. Por exemplo R3 pode designar um a quatro grupos de substituinte, de preferência um ou dois grupos de substituinte, ligados ao anel de benzeno mostrado em fórmulas I, II e IV a VII. R4 e R5 podem designar um a cinco grupos de substituinte, de preferência um a três grupos de substituinte, ligados a cada um dos anéis de benzeno mostrados em fórmulas I, II e IV a VII. Cada grupo de substituinte pode estar presente em qualquer posição adequada nos anéis de benzeno aos quais eles são ligados. Mais preferivelmente R4 e/ou R5 é um grupo de substituinte na posição orto ou para no anel de benzeno ao qual ele é ligado.
Cada um de R3, R4 e R5 pode ser protegido por um grupo de proteção removível se necessário, por exemplo quando ele contém um grupo de -OH ou -NH2 o qual não participa na reação. Grupos de proteção, sua introdução e remoção são descritos, por exemplo, em Protective Groups in Organic Synthesis, T. W. Greene e outros, John Wiley & Sons Inc., Segunda Edição 1991. De preferência cada um de R3, R4 ou R5 é um grupo o qual não requer proteção, por exemplo quaisquer dos grupos listados acima outros que são hidróxi, amino ou nitro.
Quando R3, R4 ou R5 é halogênio, ele é de preferência flúor ou cloro. Quando R3, R4 ou R5 é haloalquila ela é de preferência trifluorometila. De preferência R3 é Cw-alquila, halogênio, ou hidrogênio. De preferência R4 e R5 são cada um independentemente CV4-alquilcarbamoíla, di-C^-alquilcarbamoíla, carbamoíla, trifluorometila, flúor ou cloro. De preferência R4 e R5 são os mesmos.
De preferência os substituintes nucleofílicos Re e R7 são, cada um, independentemente halogênio, mais preferivelmente bromo ou iodo, e de maior preferência R6 e R7 são, cada um, bromo. R7 pode adequadamente estar na posição para, orto ou meta, de preferência na posição para.
• · · • · • ·
Ιό
Em uma modalidade da invenção, o composto de fórmula II é em primeiro lugar reagido com o metal ou composto organometálico para formar um composto de fórmula IV:
A-b, iv
R3 em que R3 e R7 são como definidos acima e R8 é -M ou -MX, onde M é metal e X é um substituinte nucleofílico, de preferência halogênio.
Onde o metal é lítio ou o composto organometálico é um composto de organolítio, R8 é -Li. Onde o metal é magnésio ou o composto organometálico é um reagente de Grignard, R8 é -MgX, e X é de preferência halogênio. O composto de fórmula IV é em seguida reagido com um composto de fórmula III para formar um composto de fórmula V.
Os compostos de fórmulas IV e V não precisam ser separados ou isolados mas podem ser preparados in situ.
Eletrófilos adequados para emprego no processo incluem dióxido de carbono, isocianatos, nitrilas, haletos de acila (tais como fosgênio), levando à formação de, por exemplo, compostos de fórmula I em que R! é carbóxi, carbamoíla, alquilcarbamoíla ou acila. Alternativamente o eletrófilo pode ser um material de fase sólida derivado, por exemplo um polímero de Merrifield, permitindo acoplamento direto do composto de fórmula VI a uma fase sólida. Em uma modalidade o eletrófilo é um composto de fórmula X’(CH2)n-P, em que X’é um substituinte nucleofílico por exemplo halogênio ou tosilóxi, n é um número inteiro entre 1 e 4, de preferência 1, e P é um material de fase sólida.
Onde o eletrófilo é dióxido de carbono, o processo de preferência compreende em primeiro lugar reação do composto de fórmula V com um metal ou composto organometálico para formar um composto de fórmula VI como definido acima e reação, de preferência in situ, o composto de fórmula VI com dióxido de carbono.
Quando o eletrófilo é dióxido de carbono, de preferência um composto de fórmula VII é formado:
• · ·

em que R3, R4, R5 e R9 são como definidos acima; e
R14 é -OH, -OM ou -OMX, onde M é metal e X é um substituinte nucleofílico, de preferência halogênio, em forma de sal ou livre.
Alternativamente, a etapa de carboxilação compreende reação de um composto de fórmula V com dióxido de carbono na presença de um metal ou composto organometálico, para formar um composto de fórmula VII.
A etapa de hidrólise de preferência compreende reação de um composto de fórmula VII em que R^ é -OM ou -OMX e/ou Rg é -OM ou -OMX com água ou um ácido produzindo um composto de fórmula I em que R1 é carbóxi e R2 é hidróxi, em forma de sal ou livre. Ácidos adequados incluem cloreto de amônio, ácido acético, ácido sulfúrico e ácido clorídrico. Uma solução tamponadà por pH pode além disso ser empregada. De preferência um ácido fraco é empregado e/ou a etapa pode ser realizada em um pH de 4 a 7. A temperatura de reação pode convenientemente ser -50 a 50°C, de preferência -10 a 10°C.
Alternativamente o composto de fórmula VII onde R^ é -OM ou OMX pode ser reagido com um nucleófilo, por exemplo uma amina ou haleto, para formar um composto de fórmula I em que R! é -C(O)-NR’R ou -C(O)-X e R’, R e X são como definidos acima.
O processo da invenção pode convenientemente ser realizado em um solvente orgânico inerte, de preferência um solvente de éter, por exemplo éter de dietila, tetrahidrofurano ou éter de metila de terc-butila. Alternativamente um solvente de hidrocarboneto pode ser empregado. A temperatura de reação para etapa (a) é convenientemente -30 a +10°C, de preferência -5 a -0°C. A reação pode, por exemplo, ser realizada empregando 0,5 a 2 equivalentes, de preferência 0,8 a 1,2 equivalentes e de maior

• · • · • · preferência em cerca de 1 equivalente do metal ou composto organometálico por equivalente do composto de fórmula II. 0,5 a 2 equivalentes, de preferência 0,8 a 1,2 equivalentes do composto de fórmula III pode ser empregado por equivalente do composto de fórmula II.
A temperatura durante a reação de um composto de fórmula V com um metal ou composto organometálico pode convenientemente ser 0 a +50°C, de preferência +20 a +30°C. A temperatura de reação para a reação com um eletrófilo (por exemplo CO2) pode convenientemente ser 0 a -30°C, de preferência -5 a -10°C. A etapa de hidrólise, por exemplo com ácido, pode convenientemente ser executada a -10°C a +10°C, por exemplo 0 a +5°C. De preferência 0,5 a 2 equivalentes, mais preferivelmente 0,8 a 1,2 equivalentes de um metal ou composto organometálico por equivalente do composto de fórmula V são empregados.
Os grupos R, e R2 podem ser convertidos a grupos de R! e R2 alternativos especificados acima por processos padrão, tais como por esterificação, amidação ou substituição nucleofílica. Por exemplo, um composto de fórmula I em que R2 é hidróxi pode ser convertido a um composto de fórmula I em que R2 é halogênio por reação com um haleto de acila, por exemplo cloreto de acila.
De preferência o composto de fórmula I está na forma livre. Os compostos em forma livre ou de sal podem ser obtidos na forma de hidratos ou solvatos contendo um solvente empregado para cristalização.
Compostos de fórmula I podem ser recuperados da mistura de reação e purificados de uma maneira convencional.
Os compostos de partida de fórmula II ou fórmula III são conhecidos ou podem ser preparados por métodos análogos àqueles conhecidos na técnica. Compostos organometálicos podem ser preparados por processos padrão, por exemplo por reação de um haleto de alquila ou arila com um metal, por exemplo lítio ou magnésio, suspensos em éter de dietila ou tetrahidrofurano. O composto organometálico é de preferência preparado e empregado em uma atmosfera anidra (livre de oxigênio) inerte, por exemplo sob nitrogênio.

• · • · • · • ύ/ϊ>
Ο processo de acordo com a invenção pode adequadamente incluir uma outra etapa de acoplamento do composto de fórmula I em que R, é um substituinte reativo a um material de fase sólida. Materiais de fase sólida adequados são descritos, por exemplo, em DE 4306839 A1, e incluem polímeros orgânicos ou inorgânicos de ocorrência naturalmente ou sintéticos em forma particulada, por exemplo como contas, ou de preferência como uma camada de superfície em um material de substrato inerte adequado. Exemplos de materiais de polímeros adequados incluem poliestireno reticulado, por exemplo pinos de poliestireno, pinos de Gly-HMD-MA / DMA e pinos de HEMA. O composto de fórmula I pode convenientemente ser acoplado a um material de fase sólida por reação de um grupo presente na fase sólida com R,. Deste modo o material de fase sólida de preferência compreende grupos reativos, tais como grupos de amino. De preferência um composto de fórmula I, em que R1 é um grupo de carbóxi ou um grupo de carbóxi ativado, por exemplo por reação com diisopropilcarbodiimida, é reagido com um polímero portando grupos de amino livre.
Um composto de fórmula I pode ser empregado como um ligador. Deste modo o processo de acordo com a invenção pode além disso adequadamente incluir uma outra etapa de acoplamento do composto de fórmula I, opcionalmente ligado a um material de fase sólida, a um oligômero ou polímero biológico, ou uma unidade de monômero destes. O composto pode convenientemente ser acoplado à molécula biológica, por exemplo um aminoácido ou polipeptídeo, por reação de um grupo presente na molécula biológica com R2. Por exemplo, onde R2 é hidróxi e a molécula biológica é um polipeptídeo ou aminoácido, o grupo de ácido carboxílico terminal da molécula biológica pode ser esterificado pelo grupo de hidróxi de R2, opcionalmente por meio de reação inicial do composto de fórmula I com um haleto de acila levando à substituição in situ de hidróxi por halogênio.
Em um outro aspecto, a presente invenção fornece:
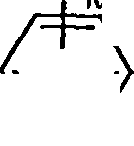
(E) um processo para preparo de um composto de fórmula VIII • · · π· Ύ YWH
VIII em que R12 e R13 são, cada um, grupo de proteção removível e R12 e R13 são diferentes: compreendendo reação de um composto de fórmula IX
ΪΜ
IX com um composto doador de R12 adequado;
(F) intermediários úteis no processo acima, definidos pela fórmula geral XIV HY°ry^°-R XIV ° Yl O I ^18 em que R16 é um grupo de proteção removível outro que não fluorenilmetoxicarbonila, e é diferente de R18;
R17 é hidrogênio ou um grupo de bloqueio removível por hidrólise ou hidrogenólise; e
R18 é hidrogênio ou um grupo de proteção removível outro que são fluorenilmetoxicarbonila.
A presente invenção fornece uma rotina simples e eficiente para a preparação de compostos de fórmula VIII, os quais são úteis na síntese de peptídeos, por exemplo como descrito em WO n° 02/10192. Os compostos de fórmula XIV são úteis como compostos intermediários na preparação de compostos de fórmula VIII.
O composto de fórmula IX pode ser preparado a partir de um composto de fórmula X kl
X
13\ γ°γ>γθ-Β'=
O Y o

• · • · • · • · em que R13 é como definido acima,
R14 é um grupo de proteção removível e R14 é diferente de R12 e
R15 é um grupo de bloqueio removível por hidrólise ou hidrogenólise
Grupos de proteção, sua introdução e remoção são descritos, por exemplo, em Protective Groups in Organic Synthesis, T. W. Greene e outros, John Wiley & Sons Inc., Segunda Edição 1991. Compostos doadores de grupo de proteção adequados, por exemplo agentes de proteção de grupo de amino, são bem-conhecidos para uma pessoa versada, por exemplo anidridos, haletos, carbamatos ou N-hidroxissuccinimidas os quais fornecem um dos grupos de proteção abaixo.
O grupo de proteção R12 é de preferência fluorenilmetoxicarboniia. R13 ou R16 é de preferência um grupo de proteção outro que não fluorenilmetoxicarbonila, e é de preferência mais resistente à remoção por hidrólise (por exemplo hidrólise catalisada por base) e/ou hidrogenólise do que R12 e/ou R14, por exemplo mais resistente do que fluorenilmetoxicarbonila e/ou benziloxicarbonila. Mais preferivelmente R13 ou R16 é terc-butoxicarbonila.
O grupo de proteção R14 ou R18 é de preferência mais resistente à remoção por hidrólise do que R12, por exemplo mais resistente do que fluorenilmetoxicarbonila. R14 ou R18 é de preferência removível por hidrogenólise. Substituintes de R14 ou R18 adequados incluem benziloxicarbonila, 1,1,dimetilpropiniloxicarbonila, viniloxicarbonila, N-hidróxipiperidiniloxicarbonila,
9-antrilmetiloxicarbonila e fenilaminotiocarbonila, alila, nitrobenzila, trifenilmetila, (p-metoxifenil)difenilmetila, difenil-4-piridilmetila ou benzilsulfonila. De preferência R14 ou R18 é um grupo de proteção contendo oxicarbonila, por exemplo benziloxicarbonila (carbobenzóxi).
R15 ou R17 pode adequadamente ser:
(i) C^w-alquila, por exemplo C14-alquila, de preferência metila, etila, propila ou butila outro que não terc-butila, mais preferivelmente metila.
(ii) C3_8-cicloalquila, opcionalmente substituída por um ou mais • · ·
Cv4 alquila, por exemplo metila. De preferência cicloalquila é C3_6-cicloalquila.
(iii) C6.10-arila, opcionalmente substituída por um ou mais substituintes de estabilização, por exemplo halogênio ou nitro. De preferência arila é fenila, opcionalmente substituída por um, dois ou três halogênios, por exemplo cloro.
(iv) (C6_10-arila)V3-C1.10-alquila, opcionalmente substituída no grupo de arila por (i) um ou mais substituintes de estabilização, por exemplo halogênio ou nitro, ou (ii) por dois substituintes os quais juntos com átomos de carbono no anel aos quais eles estão ligados formam um anel de 5 ou 6 membros, opcionalmente contendo um ou dois átomos de nitrogênio ou oxigênio. (C^Q-arilaj^^C^^-alquila é de preferência (i) (fenilh^-C^-alquila, mais preferivelmente benzila, difenilmetila ou trifenilmetila, opcionalmente substituída em cada anel de benzeno por um, dois ou três halogênios, por exemplo cloro, (ii) antrilmetila, por exemplo 9antrilmetila, ou (iii) piperonila.
(v) C6_10-aril-C14-alcóxi-CV4-alquila, de preferência benziloximetila.
(vi) C6.10-aril-carbonil-CM-alquila, de preferência fenacila.
De preferência R15 ou R17 é um grupo o qual é removível por hidrogenólise, tal como benzila, benziloximetila, fenacila, trifenilmetila, piperonila ou 9-antrilmetila, de preferência benzila.
O composto de fórmula IX pode ser preparado por (i) hidrólise do composto de éster de fórmula X para obter o correspondente ácido carboxílico e (ii) remoção do grupo de proteção R14. De preferência a etapa de hidrólise é executada antes da remoção do grupo de proteção R14. O grupo de proteção R14 pode convenientemente ser removido por hidrogenação redutiva (hidrogenólise). Esta rotina, envolvendo uma etapa de hidrólise, é adequadamente seguida quando R15 não é removível por hidrogenólise. A etapa de hidrólise é de preferência uma hidrólise catalisada por base, por

exemplo empregando hidróxido de sódio e pode adequadamente ser executada em um solvente polar, por exemplo metanol.
Alternativamente, um composto de fórmula IX pode convenientemente ser preparado por hidrogenação (hidrogenólise) de um composto de fórmula X em que R15 é um grupo o qual é removível por hidrogenólise, por exemplo benzila. A etapa de hidrogenação pode convenientemente ser executada empregando um agente catalítico adequado, por exemplo paládio em carvão vegetal.
Composto de fórmula X pode ser preparado por reação de uma composto de fórmula XI
XI ϊ nr em que X é um substituinte nucleofílico e R14 e R15 são como definidos acima, com um composto de fórmula XII R13^n/^x/NH2 X”
H em que R13 é como definido acima. Esta etapa pode ser executada em qualquer solvente orgânico adequado, de preferência em um solvente de hidro15 carboneto, mais preferencialmente tolueno.
O composto de fórmula XII é etilenodiamina protegida, na qual um grupo de amino tem sido protegido com um grupo de proteção removível. O substituinte nucleofílico X em fórmula XI é de preferência halogênio, tal como flúor, cloro, bromo ou iodo, mais preferivelmente cloro. O composto de fórmula XI na qual X é halogênio pode ser formado por reação de um composto de fórmula XIII
HO
O-R
XIII com um haleto de acila, por exemplo fosgênio, tri-fosgênio, fenilcloroformiato

Λ3 ou 4-nitrofenilcloroformiato, de preferência 4-nitrofenilcloroformato. Esta etapa pode adequadamente ser executada na presença de uma base orgânica, por exemplo dimetilaminopiridina, em um solvente não-polar, por exemplo tolueno.
O composto de fórmula XIII pode ser comercialmente disponível, por exemplo quando R15 é metila ou pode ser formado por esterificação de 4-hidróxi-prolina de acordo com métodos conhecidos na técnica, por exemplo por reação com álcool de benzila ou metanol. O éster resultante é em seguida protegido por reação com um composto doador de R14 adequado, por exemplo benziloxicarbonil-N-hidroxissuccinimida.
O composto de fórmula XI não precisa ser separado ou isolado, como o composto de fórmula XIII pode ser reagido com um haleto de acila e o produto desta reação subseqüentemente reagido com um composto de fórmula XII no mesmo recipiente.
A adição do grupo de proteção R12 ao composto de fórmula IX pode adequadamente ser executada na presença de carbonato de sódio / acetonitrila.
Compostos de fórmula VIII podem ser recuperados da mistura de reação e purificados de uma maneira convencional.
Nos compostos de fórmulas VIII a XI e XIII acima, o substituinte de óxi na prolina pode estar na posição c/s ou trans, de preferência trans. Os eis ou trans isômeros podem ser individualmente preparados, empregando a correspondente c/s ou trans hidroxiprolina como material de partida.
Na medida em que a produção dos materiais de partida não está particularmente descrita, os compostos são conhecidos ou podem ser preparados analogamente a métodos conhecidos na técnica ou como descrito daqui em diante.
Em um outro aspecto, a presente invenção refere-se a um processo para produção de um composto de fórmula VIII, em que R12 é fluorenilmetoxicarbonila e R13 é um grupo de proteção removível outro que não fluorenilmetoxicarbonila, compreendendo reação de um composto de fórmula VIII com um composto doador de fluorenilmetoxicarbonila, por exemplo ·· · · ··· · • · · • · · : : : : : :* • · · · ··· · ··· · · fluorenilmetoxicarbonil-N-hidroxissuccinimida.
A invenção no momento será descrita com referência às seguintes modalidades específicas, nas quais as seguintes abreviações são empregadas:
Fmoc = fluorenilmetoxicarbonila
Boc = terc-butoxicarbonila
Cbo = carbobenzóxi (benziloxicarbonila)
Osu = N-hidroxissuccinimida
HPTF = Fração de heptano
JT = Temperatura de jaqueta
HPLC = Cromatografia líquida de alto desempenho
THF = Tetrahidrofurano
TBME = Éter de metila de terc-butila
DMF = Dimetilformamida
Exemplo 1
Preparação de ácido 4-(difenil-hidróxi-metil)-benzóico.
1,4-dibromobenzeno (47,2 g, 0,2 M) é adicionado a THF (240 mL). A solução clara é resfriada a -65°C. Uma solução de butil-lítio (0,22 M, 94 mL de uma solução a 20% em CHX) é adicionada durante 30 minutos.
Depois de 5 minutos de agitação uma solução de benzofenona (36,4 g, 0,2 M em 180 mL de THF) é adicionada durante 30 minutos (exotérmíca). A mistura é agitada durante um adicional de 30 minutos a -65°C. Em seguida durante 30 minutos a temperatura é elevada a -10°C e a solução é agitada a esta temperatura durante uma hora.
A mistura de reação é em seguida novamente esfriada a -65°C. Durante 30 minutos uma solução de butil-lítio (0,22 M, 94 mL de uma solução a 20% em ciclohexano) é adicionada.
A suspensão resultante é diluída com 200 mL de THF. Em seguida gás de dióxido de carbono é introduzido durante 90 minutos a -65°C. A temperatura é elevada a 20°C e a mistura agitada durante a noite. A mistura é em seguida resfriada a 0°C e uma solução aquosa de cloreto de amônio (120 mL de uma solução a 10%) é adicionada durante 30 minutos.

Ácido 4-(difenil-hidróxi-metil)-benzóico é formado neste estágio.
A mistura é evaporada a 45°C sob um vácuo. O resíduo é ajustado a pH 4 com ácido acético e misturado com 400 mL de H2O. A extração é executada com 2 x 150 mL de acetato de etila. As fases orgânicas são extraídas mais uma vez com 100 mL de água. As fases de EST combinadas são agitadas com uma solução de hidróxido de potássio aquoso a 10% (2 x 120 mL). As fases aquosas combinadas são ajustadas para pH 1 a 2 com ácido clorídrico a 20°C e em seguida extraídas com 2 x 150 mL de TMBE. As fases de TBME combinadas são misturadas com 50 mL de água e 50 mL de Na2SO4 saturado, secadas com sulfato de magnésio e evaporadas a 45°C sob vácuo para obter um produto bruto.
38,3 g de produto bruto são dissolvidos em TBME (300 mL) a 40°C. A solução amarelo-clara é concentrada em um volume de 60 mL (240 mL de TBME destilado). A mistura é agitada durante uma hora a 40°C (cristalização). 50 mL de HPTF são adicionados, a mistura é resfriada a 0°C e agitada a 0°C durante 1 hora. Evaporação e lavagem com 2 x 15 mL de fração de heptano e secagem durante a noite a 45°C sob vácuo produzem cristais brancos.
Ligação de ácido 4-(difenil-hidróxi-metil)-benzóico a uma fase sólida.
g de ácido 4-(difenil-hidróxi-metil)-benzóico com 7,54 g de hidroxibenzotriazol são dissolvidos em 140 mL de DMF por agitação durante 15 minutos. 15,3 mL de diisopropilcarbodi-imida são adicionados e a solução deixada à temperatura ambiente durante 30 minutos. A solução é em seguida agitada durante a noite à temperatura ambiente na presença de poliestireno de aminometilado. Depois de lavagem com DMF, metanol e THF o suporte derivado de ligador é secado sob vácuo.
Exemplo 2
Preparação de ácido 4-(difenil-hidróxi-metil)-benzóico (método alternativo).
A 12 litros de TBME em um Reator Hastelloy de 100 litros bem seco, 3,0 kg de n-butil-lítio (20% em ciclohexano; 9,37 moles) são adicionados durante um período de 20 minutos, deixando a temperatura a -5°C (solução clara). Durante um período de 30 minutos 2,00 kg de 1,4-dibromo-

d-(p benzeno (8,48 moles) dissolvidos em 16 litros de TBME são adicionados deixando a temperatura entre -5°C e 0°C. O recipiente de adição é enxaguado com 3 litros de TBME.
Depois de 30 minutos de agitação a -5°C uma solução de 1,55 kg de benzofenona (8,50 moles) em 8 litros de TBME é adicionada durante um período de 20 minutos deixando a temperatura entre -5°C e 0°C. O recipiente de adição é enxaguado com 3 litros de TBME. Uma quantidade muito pequena de um sólido branco é formada. Depois de 15 minutos de agitação a -5°C uma amostra de controle de processo (HPLC 1) é extraída. A mistura de reação é agitada durante 25 minutos adicionais a -5°C e aquecida a +25°C.
3,2 kg de n-butil-lítio (20% em ciclohexano; 10,00 moles) são adicionados durante um período de 25 minutos deixando a temperatura entre +25 e 27°C. A adição é ligeiramente exotérmica e a cor tornou-se ligeiramente verde. Algum precipitado e espuma são formados. Depois de 20 minutos de agitação uma amostra de controle de processo é extraída (HPLC 2). Dependendo do resultado de HPLC 2 0,3 kg adicional de n-butil-lítio é adicionado depois de agitação a +25°C durante 35 minutos. Depois de 15 minutos de agitação uma amostra para controle de processo é extraída (HPLC 3). As linhas da adição de n-butil-lítio são enxaguadas com 1,5 litros de TBME e a reação é resfriada a -10°C.
1,99 kg de gelo seco (CO2 sólido) é adicionado em porções durante um período de 20 minutos deixando a temperatura entre -10 e -5°C. A reação é exotérmica e um precipitado ligeiramente amarelo é formado. Depois de 15 minutos de agitação a -10°C 11 litros de TBME são adicionados e a mistura de reação é aquecida a 0°C. 5 litros de ácido clorídrico aquoso a 18% são adicionados durante um período de 15 minutos deixando a temperatura entre 0° e +5°C. A adição é exotérmica e o precipitado é dissolvido (PH<1).
A solução clara é transferida a um tanque de separação e o reator é enxaguado com 5 litros de TBME. Depois da separação da fase aquosa a fase orgânica é lavada com 20 litros de água. Depois da separa19 ção das duas camadas a fase orgânica é extraída com 13 litros de solução de KOH aquoso a 5%. A fase de água básica é separada e a camada orgânica é extraída mais uma vez com 13 litros de solução de KOH aquoso a 5%. As camadas aquosas básicas combinadas são transferidas ao Reator Hastelloy de 100 litros. 22 litros de TBME e 6 litros de ácido clorídrico a 18% aquoso são adicionados durante um período de 20 minutos a uma temperatura entre 0° e +5°C. A adição é exotérmica e um precipitado branco é formado mas ele dissolve-se de novo em um valor de pH baixo ( pH < 1 depois de adição de HCI). A mistura é agitada durante 10 minutos e transferida a um tanque de separação. As camadas são separadas e a fase aquosa é extraída de novo com 16 litros de TBME. Depois de separação das camadas as fases orgânicas combinadas são concentradas a 500 mbar / 45°C de JT a um volume de 4 a 5 litros (32 litros de TBME são destilados) e cristais semente são adicionados. A temperatura é elevada a 50°C e 20 litros de HPTF são adicionados lentamente com boa agitação. O precipitado branco é agitado durante 2 horas a 50°C JT. A temperatura de jaqueta é regulada a 0°C e a agitação é continuada durante a noite (16 horas) permitindo esfriar-se a suspensão a 0°C. A suspensão branca é filtrada e o reator é enxaguado 5 vezes com 5 litros do Icor-mãe. O resíduo é secado a 45°C JT sob vácuo (> 10 mbar) para peso constante (durante a noite).
Exemplo 3
Preparação de Fmoc-(2S,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH partindo de Cbo-(2S,4R)-Pro(4-OH)-OMe.
1. Dimetilaminopiridina (30,5 g, 250 mmoles) e Cbo-(2S,4R)Pro(4-OH)-OMe (34,9 g, 125 mmoles) são dissolvidos em tolueno (870 mL). Uma solução de 4-nitrofenilcloroformato (31,5 g, 157 mmoles) em tolueno (206 mL) é adicionada em gotas a esta solução a 0°C até 5°C durante 20 minutos e agitada durante um adicional de 2 horas. Isto é seguido por adição de uma solução de Boc-etilenodiamina (80,1 g, 500 mmoles) em tolueno (205 mL) e agitação à temperatura ambiente durante 12 horas. Uma solução de ácido sulfúrico concentrado (43,7 g, 450 mmoles) em água (873 mL) é em seguida adicionada ao mesmo tempo que mantendo uma temperatura

de 20°C a 25°C. A suspensão branca é filtrada por sucção e lavada com tolueno (30 mL). A fase de tolueno é lavada com água (450 mL), carbonato de sódio (10% peso/peso, 450 mL) e três vezes com água (450 mL cada). A fase de tolueno é azeotropicamente secada por destilação de 300 mL, a qual é continuamente substituída por tolueno seco (2 x 300 mL). Heptano (130 mL) é adicionado à solução de tolueno seco a 50°C e resfriado a 0°C durante duas horas. O produto precipitado é filtrado, lavado duas vezes com 1 : 2 de tolueno / heptano v / v (70 mL), e secado a 50°C sob vácuo para produzir Cbo-(2S,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OMe como um sólido branco.
2. Cbo-(2S,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OMme (20,0 g, 43,0 mmoles) é dissolvido em uma mistura de 1: 1 de tetrahidrofurano e metanol (380 mL). Uma solução de 1 M de hidróxido de sódio (51,6 mL) é adicionada e a mistura resultante agitada durante 4 horas à temperatura ambiente. A mistura é ajustada a pH 3 por adição de ácido sulfúrico (50 mL, 1 M). Tetrahidrofurano e metanol são destilados a 50°C e 50 mbar até que nenhum outro solvente destile. A solução leitosa restante é diluída com acetato de isopropila (113 mL) e água (57 mL), as fases são separadas e a fase de acetato de isopropila é lavada com solução de cloreto de sódio (10%, 113 mL). O solvente é destilado (50°C, 50 mbar) para produzir uma espuma de Cbo-(2S,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH (19,8 g), a qual foi empregada sem purificação adicional na reação seguinte.
3. Paládio em carvão vegetal (10%, 1,94 g, 0,042 mmol) é adicionado a uma solução de Cbo-(2S,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH (19,4 g, 43,0 mmoles) em isopropanol (350 mL) e água (37 mL). Hidrogênio é borbulhado através desta mistura durante 4 horas, o catalisador é filtrado, e o resíduo é lavado com uma mistura de isopropanol (50 mL) e água (50 mL). A fase de isopropanol/água é azeotropicamente secada por destilação de 2/3 do volume, o qual é continuamente substituído por uma mistura de tolueno/isopropanol (1 :1 v/v). A solução seca restante é concentrada em vácuo até a secura (50°C, 200 mbar) para produzir (2S,4R)-Pro(4-OCO-NHCH2-CH2-NH-Boc)-OH como um sólido acastanhado, o qual foi empregado • · · ··· · ··· · ··· ·· ···· ··· ··· ·· • · · · · · · · ··· ······ · · ·· • · ···· ···· · sem purificação adicional.
4. (2S,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH (5,0 g, 15 mmoles) é dissolvido em uma mistura de água (25 mL) e trietilamina (1,5 g, 15 mmoles) a 40°C. Uma solução de Fmoc-Osu (4,65 g, 14 mmoles) em acetonitrila (25 mL) é adicionada à solução clara durante 30 minutos e agitada durante 2 horas. Em seguida a mistura de reação é ajustada a pH 3 com ácido clorídrico (1 m, 13 mL) e agitada durante uma hora adicional. Acetonitrila é destilado (40°C, 80 mbar) e substituído por acetato de isopropila, produzindo uma mistura de duas fases. A fase aquosa inferior é separada, enquanto a camada orgânica restante é lavada com água e destilada duas vezes com substituição com acetato de isopropil e em seguida concentrada a uma espuma acastanhada. Esta espuma é dissolvida em acetato de isopropil (25 mL) e adicionada em gotas a heptano (200 mL) por meio do qual o produto é precipitado. O sólido é filtrado, lavado com acetato de isopropil/heptano e secado em vácuo a 40°C para produzir Fmoc-(2S,4R)Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH.
Exemplo 4
Preparação de Fmoc-(2S,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH partindo de Cbo-(2S,4R)-Pro(4-OH)-OBzl.
A síntese de Cbo-(2S,4R)-Pro(4-OH)-OBzl é descrita em T. Makoto, H. Guoxia, V. J. Hruby, J. Org. Chem. 2001, 66, 1038 - 1042. O processo de exemplo 3 é repetido, mas empregando Cbo-(2S,4R)-Pro(4OH)-OBzl em vez de Cbo-(2S,4R)-Pro(4-OH)-OMe e executando etapas 1, 3 e 4 apenas (omitindo etapa 2).
Exemplo 5
Preparação de Fmoc-(2R,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH
O processo do exemplo 3 ou exemplo 4 é repetido mas empregando Cbo-(2R,4R)-Pro(4-OH)-Ome ou Cbo-(2R,4R)-Pro(4-OH)-OBzl em vez de Cbo-(2S,4R)-Pro(4-OH)-Ome ou Cbo-(2S,4R)-Pro(4-OH)-OBzl. Exemplo 6
Preparação de Fmoc-(2S,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH
O processo do exemplo 3 ou exemplo 4 é repetido mas empre30 • · · · · • ·· · ···· gando Cbo-(2S,4S)-Pro(4-OH)-Ome ou Cbo-(2S,4S)-Pro(4-OH)-OBzl em vez de Cbo-(2S,4R)-Pro(4-OH)-Ome ou Cbo-(2S,4R)-Pro(4-OH)-OBzl.
Claims (5)
- REIVINDICAÇÕES1. Processo para preparação de um composto de fórmula VIII:em que R12 e R13 são cada um grupo de proteção removível e R12 e R13 são diferentes; caracterizado pelo fato de que compreende a reação de um com- com um composto doador de R12 adequado.
- 2. Processo de acordo com reivindicação 1, caracterizado pelo fato de que o composto de fórmula IX é preparado através da (i) hidrólise de um composto de fórmula X em que R13 é como definido em reivindicação 1,R14 é um grupo de proteção removível e R14 é diferente de R12 eR13, eR15 é um grupo de bloqueio removível por hidrólise ou hidrogenólise, para obter o correspondente ácido carboxílico, e (ii) remoção do grupo de proteção R14 no ácido carboxílico resultante.
- 3. Composto, caracterizado pelo fato de que apresenta a fórmulaXIV em que R16 é um grupo de proteção removível outro que não fluorenilmetoxicarbonila, e é diferente de R18;R17 é hidrogênio ou um grupo de bloqueio removível por hidrólise ou hidrogenólise; eR18 é hidrogênio ou um grupo de proteção removível diferente de fluorenilmetoxicarbonila.
- 4. Composto de acordo com reivindicação 3, caracterizado pelo fato de que R16 é terc-butoxicarbonila.
- 5. Processo para produção de um composto de fórmula VIII, como definido em reivindicação 1, em que R12 é fluorenilmetoxicarbonila e R13 é um grupo de proteção removível diferente de fluorenilmetoxicarbonila, caracterizado pelo fato de que compreende a reação de um composto de fórmula IX com um composto doador de fluorenilmetoxicarbonila.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0229020.3 | 2002-12-12 | ||
| GB0229020A GB0229020D0 (en) | 2002-12-12 | 2002-12-12 | Organic compounds |
| GB0229280.3 | 2002-12-16 | ||
| GB0229280A GB0229280D0 (en) | 2002-12-16 | 2002-12-16 | Organic compounds |
| PCT/EP2003/014082 WO2004052817A1 (en) | 2002-12-12 | 2003-12-11 | Process for peptide synthesis of peptides containing a 4-hydroxy-proline substructure |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| BRPI0317176B1 true BRPI0317176B1 (pt) | 2018-03-13 |
Family
ID=32510405
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0317176-0A BRPI0317176B1 (pt) | 2002-12-12 | 2003-12-11 | Processo para a síntese de peptídeos contendo uma subestrutura de 4-hidróxi-prolina |
| BR0317176-0A BR0317176A (pt) | 2002-12-12 | 2003-12-11 | Processo para a sìntese de peptìdeos contendo uma subestrutura de 4-hidróxi-prolina |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BR0317176-0A BR0317176A (pt) | 2002-12-12 | 2003-12-11 | Processo para a sìntese de peptìdeos contendo uma subestrutura de 4-hidróxi-prolina |
Country Status (24)
| Country | Link |
|---|---|
| US (3) | US7294722B2 (pt) |
| EP (1) | EP1572615B8 (pt) |
| JP (2) | JP4796303B2 (pt) |
| KR (1) | KR101018491B1 (pt) |
| AT (1) | ATE461173T1 (pt) |
| AU (1) | AU2003290019B2 (pt) |
| BR (2) | BRPI0317176B1 (pt) |
| CA (1) | CA2509363C (pt) |
| CO (1) | CO5700810A2 (pt) |
| CY (1) | CY1110639T1 (pt) |
| DE (1) | DE60331770D1 (pt) |
| DK (1) | DK1572615T3 (pt) |
| EC (1) | ECSP055846A (pt) |
| ES (1) | ES2341336T3 (pt) |
| HK (1) | HK1085459A1 (pt) |
| IL (1) | IL168866A (pt) |
| MX (1) | MXPA05006267A (pt) |
| NO (1) | NO331836B1 (pt) |
| NZ (1) | NZ540441A (pt) |
| PL (1) | PL206914B1 (pt) |
| PT (1) | PT1572615E (pt) |
| RU (1) | RU2392265C2 (pt) |
| SI (1) | SI1572615T1 (pt) |
| WO (1) | WO2004052817A1 (pt) |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE4306839A1 (de) * | 1993-03-05 | 1994-09-08 | Bayer Ernst Prof Dr | Tritylgruppe enthaltendes Festphasensystem, Verfahren zu seiner Herstellung und seine Verwendung in Festphasenreaktionen |
| MY147327A (en) * | 1995-06-29 | 2012-11-30 | Novartis Ag | Somatostatin peptides |
| GB9625167D0 (en) * | 1996-12-04 | 1997-01-22 | Sandoz Ltd | Organic compounds |
| ATE321882T1 (de) * | 1997-07-01 | 2006-04-15 | Isis Pharmaceuticals Inc | Zusammensetzungen und verfahren zur verabreichung von oligonukleotiden über die speiseröhre |
| GB0018891D0 (en) * | 2000-08-01 | 2000-09-20 | Novartis Ag | Organic compounds |
-
2003
- 2003-12-11 WO PCT/EP2003/014082 patent/WO2004052817A1/en active Application Filing
- 2003-12-11 AU AU2003290019A patent/AU2003290019B2/en not_active Expired
- 2003-12-11 BR BRPI0317176-0A patent/BRPI0317176B1/pt unknown
- 2003-12-11 AT AT03782373T patent/ATE461173T1/de active
- 2003-12-11 ES ES03782373T patent/ES2341336T3/es not_active Expired - Lifetime
- 2003-12-11 EP EP03782373A patent/EP1572615B8/en not_active Expired - Lifetime
- 2003-12-11 NZ NZ540441A patent/NZ540441A/en not_active IP Right Cessation
- 2003-12-11 RU RU2005121664/04A patent/RU2392265C2/ru active
- 2003-12-11 BR BR0317176-0A patent/BR0317176A/pt not_active IP Right Cessation
- 2003-12-11 PL PL375821A patent/PL206914B1/pl unknown
- 2003-12-11 MX MXPA05006267A patent/MXPA05006267A/es active IP Right Grant
- 2003-12-11 CA CA2509363A patent/CA2509363C/en not_active Expired - Lifetime
- 2003-12-11 KR KR1020057010600A patent/KR101018491B1/ko active IP Right Grant
- 2003-12-11 SI SI200331800T patent/SI1572615T1/sl unknown
- 2003-12-11 JP JP2004558073A patent/JP4796303B2/ja not_active Expired - Lifetime
- 2003-12-11 DE DE60331770T patent/DE60331770D1/de not_active Expired - Lifetime
- 2003-12-11 PT PT03782373T patent/PT1572615E/pt unknown
- 2003-12-11 US US10/538,028 patent/US7294722B2/en not_active Expired - Lifetime
- 2003-12-11 DK DK03782373.9T patent/DK1572615T3/da active
-
2005
- 2005-05-30 IL IL168866A patent/IL168866A/en active IP Right Grant
- 2005-06-09 EC EC2005005846A patent/ECSP055846A/es unknown
- 2005-06-14 CO CO05057286A patent/CO5700810A2/es active IP Right Grant
- 2005-07-11 NO NO20053374A patent/NO331836B1/no not_active IP Right Cessation
-
2006
- 2006-03-10 HK HK06103122.9A patent/HK1085459A1/xx not_active IP Right Cessation
-
2007
- 2007-08-16 US US11/839,565 patent/US7449587B2/en not_active Expired - Lifetime
-
2008
- 2008-08-25 US US12/197,657 patent/US20080312456A1/en not_active Abandoned
-
2010
- 2010-04-05 JP JP2010086682A patent/JP5249276B2/ja not_active Expired - Lifetime
- 2010-05-14 CY CY20101100426T patent/CY1110639T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7418027B2 (ja) | ジフェニルメタン構造を含有する化合物及びその応用 | |
| EP0098865B1 (en) | Peptide synthesis and amino acid blocking agents | |
| US6239305B1 (en) | Method for producing optically active phenylpropionic acid derivative | |
| Cho et al. | Indium-mediated mild and facile method for the synthesis of amides | |
| BRPI0317176B1 (pt) | Processo para a síntese de peptídeos contendo uma subestrutura de 4-hidróxi-prolina | |
| RU2114103C1 (ru) | Способ получения (s)-энантиомеров | |
| US6492541B2 (en) | Process for the preparation of 3-amino-2-oxo-pyrrolidines, novel intermediates and their use | |
| ZA200504299B (en) | Process for peptide synthesis of peptides containing a 4-hydroxy-proline substructure | |
| JP2677927B2 (ja) | ジ炭酸ジアルキルエステルの製造方法 | |
| JP2006306784A (ja) | 高度にフッ素化されたアルコール誘導体と再利用が容易な使用法 | |
| JP2005263748A (ja) | 反応性ジアジリン基を有するホモフェニルアラニン誘導体 | |
| JPH0333149B2 (pt) | ||
| CN111606971A (zh) | 一种fapgg的制备方法 | |
| JPH04312570A (ja) | アミノ基が保護された4−ヒドロキシプロリン又は4−ヒドロキシプロリン誘導体の製造法 | |
| JPH01294690A (ja) | α−アスパルチルフェニルアラニン誘導体の製造法 | |
| JPH09194444A (ja) | 3−アミノ−2−オキソ−1−ハロゲノプロパン誘導体の製造方法 | |
| JPH10120678A (ja) | N−(1,3−ジアルキル−2−イミダゾリジニリデン)−4−ピリジンアミン | |
| JPH11158173A (ja) | 2官能性六員環カーボナート化合物 | |
| JPH09249617A (ja) | ジアルキルジカーボネートの製造方法 | |
| JPH0524145B2 (pt) |