WO2017010390A1 - 延伸フィルム、延伸フィルムの製造方法、および、ポリアミド樹脂組成物 - Google Patents
延伸フィルム、延伸フィルムの製造方法、および、ポリアミド樹脂組成物 Download PDFInfo
- Publication number
- WO2017010390A1 WO2017010390A1 PCT/JP2016/070081 JP2016070081W WO2017010390A1 WO 2017010390 A1 WO2017010390 A1 WO 2017010390A1 JP 2016070081 W JP2016070081 W JP 2016070081W WO 2017010390 A1 WO2017010390 A1 WO 2017010390A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polyamide resin
- stretched film
- derived
- general formula
- dicarboxylic acid
- Prior art date
Links
- 229920006122 polyamide resin Polymers 0.000 title claims abstract description 176
- 239000011342 resin composition Substances 0.000 title claims abstract description 45
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 30
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 claims abstract description 63
- 150000001875 compounds Chemical class 0.000 claims abstract description 54
- 150000004985 diamines Chemical class 0.000 claims abstract description 43
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 39
- GKXVJHDEWHKBFH-UHFFFAOYSA-N [2-(aminomethyl)phenyl]methanamine Chemical compound NCC1=CC=CC=C1CN GKXVJHDEWHKBFH-UHFFFAOYSA-N 0.000 claims abstract description 20
- 125000001931 aliphatic group Chemical group 0.000 claims abstract description 19
- 125000004432 carbon atom Chemical group C* 0.000 claims description 62
- 238000002844 melting Methods 0.000 claims description 37
- 230000008018 melting Effects 0.000 claims description 37
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 claims description 22
- CXMXRPHRNRROMY-UHFFFAOYSA-N sebacic acid Chemical compound OC(=O)CCCCCCCCC(O)=O CXMXRPHRNRROMY-UHFFFAOYSA-N 0.000 claims description 18
- 239000001361 adipic acid Substances 0.000 claims description 11
- 235000011037 adipic acid Nutrition 0.000 claims description 11
- FDLQZKYLHJJBHD-UHFFFAOYSA-N [3-(aminomethyl)phenyl]methanamine Chemical compound NCC1=CC=CC(CN)=C1 FDLQZKYLHJJBHD-UHFFFAOYSA-N 0.000 claims description 9
- 125000004437 phosphorous atom Chemical group 0.000 claims description 7
- ISKQADXMHQSTHK-UHFFFAOYSA-N [4-(aminomethyl)phenyl]methanamine Chemical compound NCC1=CC=C(CN)C=C1 ISKQADXMHQSTHK-UHFFFAOYSA-N 0.000 claims description 6
- 229910052698 phosphorus Inorganic materials 0.000 claims description 6
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 abstract 1
- -1 aliphatic diamine Chemical class 0.000 description 36
- 229920005989 resin Polymers 0.000 description 36
- 239000011347 resin Substances 0.000 description 36
- 238000002425 crystallisation Methods 0.000 description 24
- 230000008025 crystallization Effects 0.000 description 24
- 239000004743 Polypropylene Substances 0.000 description 23
- 229920001155 polypropylene Polymers 0.000 description 19
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 16
- 239000001301 oxygen Substances 0.000 description 16
- 229910052760 oxygen Inorganic materials 0.000 description 16
- 238000000034 method Methods 0.000 description 15
- 239000004014 plasticizer Substances 0.000 description 15
- 230000004888 barrier function Effects 0.000 description 12
- 229920002647 polyamide Polymers 0.000 description 12
- 230000000052 comparative effect Effects 0.000 description 10
- 238000010438 heat treatment Methods 0.000 description 10
- 238000005259 measurement Methods 0.000 description 9
- 239000000203 mixture Substances 0.000 description 9
- 239000004952 Polyamide Substances 0.000 description 7
- 238000009998 heat setting Methods 0.000 description 7
- QQVIHTHCMHWDBS-UHFFFAOYSA-N isophthalic acid Chemical group OC(=O)C1=CC=CC(C(O)=O)=C1 QQVIHTHCMHWDBS-UHFFFAOYSA-N 0.000 description 7
- 238000002156 mixing Methods 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- KKEYFWRCBNTPAC-UHFFFAOYSA-N Terephthalic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-N 0.000 description 6
- 230000008569 process Effects 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 101000576320 Homo sapiens Max-binding protein MNT Proteins 0.000 description 5
- 229920006121 Polyxylylene adipamide Polymers 0.000 description 5
- 239000004760 aramid Substances 0.000 description 5
- 229920003235 aromatic polyamide Polymers 0.000 description 5
- 238000006243 chemical reaction Methods 0.000 description 5
- 238000009826 distribution Methods 0.000 description 5
- 238000001125 extrusion Methods 0.000 description 5
- 230000009477 glass transition Effects 0.000 description 5
- 239000008188 pellet Substances 0.000 description 5
- 230000035699 permeability Effects 0.000 description 5
- 238000006116 polymerization reaction Methods 0.000 description 5
- 239000002994 raw material Substances 0.000 description 5
- 230000000630 rising effect Effects 0.000 description 5
- 229920005992 thermoplastic resin Polymers 0.000 description 5
- 239000000853 adhesive Substances 0.000 description 4
- 230000001070 adhesive effect Effects 0.000 description 4
- 125000003277 amino group Chemical group 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 239000007789 gas Substances 0.000 description 4
- 239000000178 monomer Substances 0.000 description 4
- BYEAHWXPCBROCE-UHFFFAOYSA-N 1,1,1,3,3,3-hexafluoropropan-2-ol Chemical compound FC(F)(F)C(O)C(F)(F)F BYEAHWXPCBROCE-UHFFFAOYSA-N 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 229920002292 Nylon 6 Polymers 0.000 description 3
- 229920002302 Nylon 6,6 Polymers 0.000 description 3
- 125000003118 aryl group Chemical group 0.000 description 3
- 230000005540 biological transmission Effects 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- KYTZHLUVELPASH-UHFFFAOYSA-N naphthalene-1,2-dicarboxylic acid Chemical compound C1=CC=CC2=C(C(O)=O)C(C(=O)O)=CC=C21 KYTZHLUVELPASH-UHFFFAOYSA-N 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 229920006111 poly(hexamethylene terephthalamide) Polymers 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- 229920000049 Carbon (fiber) Polymers 0.000 description 2
- 229920000219 Ethylene vinyl alcohol Polymers 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- IPRJXAGUEGOFGG-UHFFFAOYSA-N N-butylbenzenesulfonamide Chemical compound CCCCNS(=O)(=O)C1=CC=CC=C1 IPRJXAGUEGOFGG-UHFFFAOYSA-N 0.000 description 2
- 229920000571 Nylon 11 Polymers 0.000 description 2
- 229920000299 Nylon 12 Polymers 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- VHRGRCVQAFMJIZ-UHFFFAOYSA-N cadaverine Chemical compound NCCCCCN VHRGRCVQAFMJIZ-UHFFFAOYSA-N 0.000 description 2
- 239000004917 carbon fiber Substances 0.000 description 2
- 238000005266 casting Methods 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 238000013329 compounding Methods 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 238000000113 differential scanning calorimetry Methods 0.000 description 2
- TVIDDXQYHWJXFK-UHFFFAOYSA-N dodecanedioic acid Chemical compound OC(=O)CCCCCCCCCCC(O)=O TVIDDXQYHWJXFK-UHFFFAOYSA-N 0.000 description 2
- JBKVHLHDHHXQEQ-UHFFFAOYSA-N epsilon-caprolactam Chemical compound O=C1CCCCCN1 JBKVHLHDHHXQEQ-UHFFFAOYSA-N 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- NAQMVNRVTILPCV-UHFFFAOYSA-N hexane-1,6-diamine Chemical compound NCCCCCCN NAQMVNRVTILPCV-UHFFFAOYSA-N 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 230000001771 impaired effect Effects 0.000 description 2
- 150000003951 lactams Chemical class 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 238000000465 moulding Methods 0.000 description 2
- BDJRBEYXGGNYIS-UHFFFAOYSA-N nonanedioic acid Chemical compound OC(=O)CCCCCCCC(O)=O BDJRBEYXGGNYIS-UHFFFAOYSA-N 0.000 description 2
- 230000002093 peripheral effect Effects 0.000 description 2
- WLJVNTCWHIRURA-UHFFFAOYSA-N pimelic acid Chemical compound OC(=O)CCCCCC(O)=O WLJVNTCWHIRURA-UHFFFAOYSA-N 0.000 description 2
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 239000004926 polymethyl methacrylate Substances 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- KIDHWZJUCRJVML-UHFFFAOYSA-N putrescine Chemical compound NCCCCN KIDHWZJUCRJVML-UHFFFAOYSA-N 0.000 description 2
- 239000001632 sodium acetate Substances 0.000 description 2
- 235000017281 sodium acetate Nutrition 0.000 description 2
- KOUDKOMXLMXFKX-UHFFFAOYSA-N sodium oxido(oxo)phosphanium hydrate Chemical compound O.[Na+].[O-][PH+]=O KOUDKOMXLMXFKX-UHFFFAOYSA-N 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- TYFQFVWCELRYAO-UHFFFAOYSA-N suberic acid Chemical compound OC(=O)CCCCCCC(O)=O TYFQFVWCELRYAO-UHFFFAOYSA-N 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- LWBHHRRTOZQPDM-UHFFFAOYSA-N undecanedioic acid Chemical compound OC(=O)CCCCCCCCCC(O)=O LWBHHRRTOZQPDM-UHFFFAOYSA-N 0.000 description 2
- 238000004383 yellowing Methods 0.000 description 2
- CBCKQZAAMUWICA-UHFFFAOYSA-N 1,4-phenylenediamine Chemical group NC1=CC=C(N)C=C1 CBCKQZAAMUWICA-UHFFFAOYSA-N 0.000 description 1
- PWGJDPKCLMLPJW-UHFFFAOYSA-N 1,8-diaminooctane Chemical compound NCCCCCCCCN PWGJDPKCLMLPJW-UHFFFAOYSA-N 0.000 description 1
- RTBFRGCFXZNCOE-UHFFFAOYSA-N 1-methylsulfonylpiperidin-4-one Chemical compound CS(=O)(=O)N1CCC(=O)CC1 RTBFRGCFXZNCOE-UHFFFAOYSA-N 0.000 description 1
- JCUZDQXWVYNXHD-UHFFFAOYSA-N 2,2,4-trimethylhexane-1,6-diamine Chemical compound NCCC(C)CC(C)(C)CN JCUZDQXWVYNXHD-UHFFFAOYSA-N 0.000 description 1
- DPQHRXRAZHNGRU-UHFFFAOYSA-N 2,4,4-trimethylhexane-1,6-diamine Chemical compound NCC(C)CC(C)(C)CCN DPQHRXRAZHNGRU-UHFFFAOYSA-N 0.000 description 1
- HASUJDLTAYUWCO-UHFFFAOYSA-N 2-aminoundecanoic acid Chemical class CCCCCCCCCC(N)C(O)=O HASUJDLTAYUWCO-UHFFFAOYSA-N 0.000 description 1
- JZUHIOJYCPIVLQ-UHFFFAOYSA-N 2-methylpentane-1,5-diamine Chemical compound NCC(C)CCCN JZUHIOJYCPIVLQ-UHFFFAOYSA-N 0.000 description 1
- HLBLWEWZXPIGSM-UHFFFAOYSA-N 4-Aminophenyl ether Chemical group C1=CC(N)=CC=C1OC1=CC=C(N)C=C1 HLBLWEWZXPIGSM-UHFFFAOYSA-N 0.000 description 1
- DZIHTWJGPDVSGE-UHFFFAOYSA-N 4-[(4-aminocyclohexyl)methyl]cyclohexan-1-amine Chemical compound C1CC(N)CCC1CC1CCC(N)CC1 DZIHTWJGPDVSGE-UHFFFAOYSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical compound NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- 206010065042 Immune reconstitution inflammatory syndrome Diseases 0.000 description 1
- JHWNWJKBPDFINM-UHFFFAOYSA-N Laurolactam Chemical compound O=C1CCCCCCCCCCCN1 JHWNWJKBPDFINM-UHFFFAOYSA-N 0.000 description 1
- IIHKGAZZIDXOAT-UHFFFAOYSA-N NCC12CCCCC2(CCCC1)CN Chemical compound NCC12CCCCC2(CCCC1)CN IIHKGAZZIDXOAT-UHFFFAOYSA-N 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- 229920001007 Nylon 4 Polymers 0.000 description 1
- 229920003189 Nylon 4,6 Polymers 0.000 description 1
- 229920000305 Nylon 6,10 Polymers 0.000 description 1
- 229920000572 Nylon 6/12 Polymers 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- WPWNSTTVSOUHRP-UHFFFAOYSA-N [1-(aminomethyl)naphthalen-2-yl]methanamine Chemical group C1=CC=CC2=C(CN)C(CN)=CC=C21 WPWNSTTVSOUHRP-UHFFFAOYSA-N 0.000 description 1
- QLBRROYTTDFLDX-UHFFFAOYSA-N [3-(aminomethyl)cyclohexyl]methanamine Chemical compound NCC1CCCC(CN)C1 QLBRROYTTDFLDX-UHFFFAOYSA-N 0.000 description 1
- OXIKYYJDTWKERT-UHFFFAOYSA-N [4-(aminomethyl)cyclohexyl]methanamine Chemical compound NCC1CCC(CN)CC1 OXIKYYJDTWKERT-UHFFFAOYSA-N 0.000 description 1
- 239000006096 absorbing agent Substances 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 150000001412 amines Chemical group 0.000 description 1
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- JFCQEDHGNNZCLN-UHFFFAOYSA-N anhydrous glutaric acid Natural products OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 239000002216 antistatic agent Substances 0.000 description 1
- 150000004984 aromatic diamines Chemical class 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 235000008429 bread Nutrition 0.000 description 1
- 239000004566 building material Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 238000011088 calibration curve Methods 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000010924 continuous production Methods 0.000 description 1
- 238000007334 copolymerization reaction Methods 0.000 description 1
- GEQHKFFSPGPGLN-UHFFFAOYSA-N cyclohexane-1,3-diamine Chemical compound NC1CCCC(N)C1 GEQHKFFSPGPGLN-UHFFFAOYSA-N 0.000 description 1
- VKIRRGRTJUUZHS-UHFFFAOYSA-N cyclohexane-1,4-diamine Chemical compound NC1CCC(N)CC1 VKIRRGRTJUUZHS-UHFFFAOYSA-N 0.000 description 1
- YQLZOAVZWJBZSY-UHFFFAOYSA-N decane-1,10-diamine Chemical compound NCCCCCCCCCCN YQLZOAVZWJBZSY-UHFFFAOYSA-N 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000007123 defense Effects 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- QFTYSVGGYOXFRQ-UHFFFAOYSA-N dodecane-1,12-diamine Chemical compound NCCCCCCCCCCCCN QFTYSVGGYOXFRQ-UHFFFAOYSA-N 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 239000004715 ethylene vinyl alcohol Substances 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 239000003733 fiber-reinforced composite Substances 0.000 description 1
- 239000003063 flame retardant Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 239000003349 gelling agent Substances 0.000 description 1
- 239000003365 glass fiber Substances 0.000 description 1
- 239000012760 heat stabilizer Substances 0.000 description 1
- PWSKHLMYTZNYKO-UHFFFAOYSA-N heptane-1,7-diamine Chemical compound NCCCCCCCN PWSKHLMYTZNYKO-UHFFFAOYSA-N 0.000 description 1
- LYQZSWJVUFQXLF-UHFFFAOYSA-N hexadecan-7-yl 4-hydroxybenzoate Chemical compound CCCCCCCCCC(CCCCCC)OC(=O)C1=CC=C(O)C=C1 LYQZSWJVUFQXLF-UHFFFAOYSA-N 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 125000004464 hydroxyphenyl group Chemical group 0.000 description 1
- 150000003949 imides Chemical class 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000009616 inductively coupled plasma Methods 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 238000004898 kneading Methods 0.000 description 1
- 125000005647 linker group Chemical group 0.000 description 1
- 239000006224 matting agent Substances 0.000 description 1
- 238000000691 measurement method Methods 0.000 description 1
- 230000028161 membrane depolarization Effects 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- ABMFBCRYHDZLRD-UHFFFAOYSA-N naphthalene-1,4-dicarboxylic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=C(C(O)=O)C2=C1 ABMFBCRYHDZLRD-UHFFFAOYSA-N 0.000 description 1
- DFFZOPXDTCDZDP-UHFFFAOYSA-N naphthalene-1,5-dicarboxylic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=CC2=C1C(O)=O DFFZOPXDTCDZDP-UHFFFAOYSA-N 0.000 description 1
- VAWFFNJAPKXVPH-UHFFFAOYSA-N naphthalene-1,6-dicarboxylic acid Chemical compound OC(=O)C1=CC=CC2=CC(C(=O)O)=CC=C21 VAWFFNJAPKXVPH-UHFFFAOYSA-N 0.000 description 1
- JSKSILUXAHIKNP-UHFFFAOYSA-N naphthalene-1,7-dicarboxylic acid Chemical compound C1=CC=C(C(O)=O)C2=CC(C(=O)O)=CC=C21 JSKSILUXAHIKNP-UHFFFAOYSA-N 0.000 description 1
- HRRDCWDFRIJIQZ-UHFFFAOYSA-N naphthalene-1,8-dicarboxylic acid Chemical compound C1=CC(C(O)=O)=C2C(C(=O)O)=CC=CC2=C1 HRRDCWDFRIJIQZ-UHFFFAOYSA-N 0.000 description 1
- RXOHFPCZGPKIRD-UHFFFAOYSA-N naphthalene-2,6-dicarboxylic acid Chemical compound C1=C(C(O)=O)C=CC2=CC(C(=O)O)=CC=C21 RXOHFPCZGPKIRD-UHFFFAOYSA-N 0.000 description 1
- WPUMVKJOWWJPRK-UHFFFAOYSA-N naphthalene-2,7-dicarboxylic acid Chemical compound C1=CC(C(O)=O)=CC2=CC(C(=O)O)=CC=C21 WPUMVKJOWWJPRK-UHFFFAOYSA-N 0.000 description 1
- 239000002667 nucleating agent Substances 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- 229920006280 packaging film Polymers 0.000 description 1
- 239000012785 packaging film Substances 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 1
- 150000003022 phthalic acids Chemical class 0.000 description 1
- 229920001643 poly(ether ketone) Polymers 0.000 description 1
- 229920006131 poly(hexamethylene isophthalamide-co-terephthalamide) Polymers 0.000 description 1
- 229920006128 poly(nonamethylene terephthalamide) Polymers 0.000 description 1
- 229920001707 polybutylene terephthalate Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920005668 polycarbonate resin Polymers 0.000 description 1
- 239000004431 polycarbonate resin Substances 0.000 description 1
- 229920001225 polyester resin Polymers 0.000 description 1
- 239000004645 polyester resin Substances 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 229920006393 polyether sulfone Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920000139 polyethylene terephthalate Polymers 0.000 description 1
- 239000005020 polyethylene terephthalate Substances 0.000 description 1
- 230000000379 polymerizing effect Effects 0.000 description 1
- 229920005672 polyolefin resin Polymers 0.000 description 1
- 229920006324 polyoxymethylene Polymers 0.000 description 1
- 229920001955 polyphenylene ether Polymers 0.000 description 1
- 230000001376 precipitating effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 239000012783 reinforcing fiber Substances 0.000 description 1
- 229920006024 semi-aromatic copolyamide Polymers 0.000 description 1
- UYCAUPASBSROMS-AWQJXPNKSA-M sodium;2,2,2-trifluoroacetate Chemical compound [Na+].[O-][13C](=O)[13C](F)(F)F UYCAUPASBSROMS-AWQJXPNKSA-M 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 229920006302 stretch film Polymers 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid Substances OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 238000009864 tensile test Methods 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 229920001169 thermoplastic Polymers 0.000 description 1
- 239000004416 thermosoftening plastic Substances 0.000 description 1
- 238000004804 winding Methods 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L77/00—Compositions of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Compositions of derivatives of such polymers
- C08L77/06—Polyamides derived from polyamines and polycarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/18—Manufacture of films or sheets
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C55/00—Shaping by stretching, e.g. drawing through a die; Apparatus therefor
- B29C55/02—Shaping by stretching, e.g. drawing through a die; Apparatus therefor of plates or sheets
- B29C55/10—Shaping by stretching, e.g. drawing through a die; Apparatus therefor of plates or sheets multiaxial
- B29C55/12—Shaping by stretching, e.g. drawing through a die; Apparatus therefor of plates or sheets multiaxial biaxial
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/06—Layered products comprising a layer of synthetic resin as the main or only constituent of a layer, which is next to another layer of the same or of a different material
- B32B27/08—Layered products comprising a layer of synthetic resin as the main or only constituent of a layer, which is next to another layer of the same or of a different material of synthetic resin
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/18—Layered products comprising a layer of synthetic resin characterised by the use of special additives
- B32B27/22—Layered products comprising a layer of synthetic resin characterised by the use of special additives using plasticisers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/32—Layered products comprising a layer of synthetic resin comprising polyolefins
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/34—Layered products comprising a layer of synthetic resin comprising polyamides
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B7/00—Layered products characterised by the relation between layers; Layered products characterised by the relative orientation of features between layers, or by the relative values of a measurable parameter between layers, i.e. products comprising layers having different physical, chemical or physicochemical properties; Layered products characterised by the interconnection of layers
- B32B7/04—Interconnection of layers
- B32B7/12—Interconnection of layers using interposed adhesives or interposed materials with bonding properties
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/02—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids
- C08G69/26—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from polyamines and polycarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/13—Phenols; Phenolates
- C08K5/134—Phenols containing ester groups
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2250/00—Layers arrangement
- B32B2250/24—All layers being polymeric
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2250/00—Layers arrangement
- B32B2250/40—Symmetrical or sandwich layers, e.g. ABA, ABCBA, ABCCBA
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/30—Properties of the layers or laminate having particular thermal properties
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/50—Properties of the layers or laminate having particular mechanical properties
- B32B2307/514—Oriented
- B32B2307/518—Oriented bi-axially
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/50—Properties of the layers or laminate having particular mechanical properties
- B32B2307/54—Yield strength; Tensile strength
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/70—Other properties
- B32B2307/724—Permeability to gases, adsorption
- B32B2307/7242—Non-permeable
- B32B2307/7244—Oxygen barrier
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/70—Other properties
- B32B2307/732—Dimensional properties
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2419/00—Buildings or parts thereof
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2457/00—Electrical equipment
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2535/00—Medical equipment, e.g. bandage, prostheses or catheter
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2553/00—Packaging equipment or accessories not otherwise provided for
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2571/00—Protective equipment
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2605/00—Vehicles
- B32B2605/08—Cars
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2377/00—Characterised by the use of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Derivatives of such polymers
- C08J2377/06—Polyamides derived from polyamines and polycarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2203/00—Applications
- C08L2203/16—Applications used for films
Definitions
- the present invention relates to a stretched film, a method for producing the stretched film, and a polyamide resin composition.
- Polyamide resins having a diamine structural unit derived from xylylenediamine are frequently used because of their low water absorption and excellent chemical resistance.
- Patent Document 1 (A) 5 to 90 parts by weight of a polyphenylene ether-based resin and (B) 95 to 10 parts by weight of a polyamide resin are combined with 100 parts by weight of (C) a compatibilizing agent 0.01
- a thermoplastic resin composition comprising: 30 parts by weight, (D) 0.1 to 100 parts by weight of a plasticizer that gives flexibility to polyamide, and (E) 0 to 100 parts by weight of a rubber-like substance. It is disclosed. Further, Patent Document 1 describes that such a thermoplastic resin composition has flexibility and high tensile strength at the same time.
- Patent Document 2 discloses a stretched aromatic polyamide film obtained by stretching an aromatic polyamide resin in the MD direction and / or TD direction at a magnification exceeding 4 times, and the aromatic polyamide resin is metaxylylene range.
- Dicarboxylic acid containing diamine constituent unit containing 70 mol% or more of amine unit, 80 to 97 mol% of ⁇ , ⁇ -linear aliphatic dicarboxylic acid unit having 4 to 20 carbon atoms and 3 to 20 mol% of isophthalic acid unit The shortest half crystallization time of the aromatic polyamide resin in the measurement temperature range from the glass transition point to the melting point is 40 to 2000 seconds when it is measured by constant temperature crystallization by the depolarization intensity method including the structural unit.
- a stretched aromatic polyamide film is described.
- Patent Document 3 discloses a multilayer structure comprising A) a polyamide layer produced from a polyamide composition and B) a barrier layer produced from an ethylene vinyl alcohol copolymer (EVOH), wherein the polyamide layer is a barrier.
- the polyamide composition is a) i) an aromatic dicarboxylic acid having 8-20 carbon atoms and an aliphatic diamine having 4-20 carbon atoms, or ii) 6-20 A group A monomer selected from an aliphatic dicarboxylic acid having 5 to 20 carbon atoms and an aromatic diamine having 6 to 20 carbon atoms, or iii) an aromatic aminocarboxylic acid having 7 to 20 carbon atoms; b) iv) an aliphatic dicarboxylic acid having 6-20 carbon atoms and an aliphatic diamine having 4-20 carbon atoms, or v) 4 One or more semi-aromatic copolyamides selected from copolyamides made from lactams having 20 carbon atom
- Patent Document 4 (A) 2 to 60 parts by weight of a compound represented by the general formula (1) with respect to 100 parts by weight of a polyamide resin selected from polyamide 11, polyamide 12 or a mixture thereof, (In the formula, m is an integer of 7 or more and 10 or less.) (B) A polyamide resin composition comprising 0.05 to 5 parts by weight of a monohydric alcohol having 16 to 24 carbon atoms having a branched chain is disclosed.
- JP-A-7-157651 International Publication WO2006 / 049281 Pamphlet Special table 2013-514212 gazette JP 7-11131 A
- the polyamide resin film having a diamine structural unit derived from xylylenediamine may not be stretched at a high stretch ratio. Therefore, it may be difficult to stretch in a state where it is laminated with another resin film. For example, when laminated with a resin film stretched at a high stretch ratio such as a polypropylene resin film and stretched, the polyamide resin film may be broken.
- the present invention aims to solve such a problem, and an object of the present invention is to provide a stretched film having a high stretch ratio. Moreover, it aims at providing the polyamide resin composition for manufacturing the manufacturing method of a stretched film, and the said stretched film.
- the polyamide resin is composed of a structural unit derived from a diamine and a structural unit derived from a dicarboxylic acid, 50 mol% or more of the structural unit derived from diamine is derived from xylylenediamine, and 50 mol% or more of the structural unit derived from dicarboxylic acid is an ⁇ , ⁇ -linear aliphatic dicarboxylic acid having 4 to 20 carbon atoms.
- R 1 is an alkyl group having 1 to 10 carbon atoms
- R 2 is an alkyl group having 2 to 12 carbon atoms
- n is an integer of 1 to 3.
- ⁇ 4> The stretched film according to any one of ⁇ 1> to ⁇ 3>, wherein 50 mol% or more of the structural units derived from the dicarboxylic acid are derived from adipic acid.
- ⁇ 5> The stretched film according to any one of ⁇ 1> to ⁇ 4>, wherein a phosphorus atom concentration in the stretched film is 0.1 to 10 ppm.
- R 1 is an alkyl group having 3 to 7 carbon atoms
- R 2 is an alkyl group having 5 to 9 carbon atoms.
- ⁇ 7> The stretched film according to any one of ⁇ 1> to ⁇ 6>, wherein the final stretch ratio is 20.0 times or more.
- ⁇ 8> The stretched film according to any one of ⁇ 1> to ⁇ 7>, wherein the tensile modulus measured according to JIS K 7127 is 2.0 GPa or more.
- ⁇ 9> The stretched film according to any one of ⁇ 1> to ⁇ 8>, wherein the stretched film has a thickness of 1 to 100 ⁇ m.
- ⁇ 10> 0.5 to 15 parts by weight of the compound represented by the general formula (1) with respect to 100 parts by weight of the polyamide resin, and the polyamide resin is composed of a structural unit derived from a diamine and a structural unit derived from a dicarboxylic acid.
- a method for producing a stretched film comprising stretching a film comprising a polyamide resin composition derived from dicarboxylic acid;
- R 1 is an alkyl group having 1 to 10 carbon atoms
- R 2 is an alkyl group having 2 to 12 carbon atoms
- n is an integer of 1 to 3.
- ⁇ 12> The method for producing a stretched film according to ⁇ 10>, wherein the stretching is performed so as to be 4.2 times or more in two orthogonal directions.
- ⁇ 13> The method for producing a stretched film according to any one of ⁇ 10> to ⁇ 12>, wherein the stretching temperature is (melting point ⁇ 200 ° C.) or more and less than the melting point of the polyamide resin.
- ⁇ 14> The method for producing a stretched film according to any one of ⁇ 10> to ⁇ 13>, wherein the stretching is performed so that a total stretching ratio is 18.0 times or more.
- the polyamide resin is composed of a structural unit derived from a diamine and a structural unit derived from a dicarboxylic acid.
- 50 mol% or more of the structural unit derived from the diamine is derived from xylylenediamine
- 50 mol% or more of the structural unit derived from the dicarboxylic acid is an ⁇ , ⁇ -linear aliphatic having 4 to 20 carbon atoms.
- a polyamide resin composition for producing a stretched film derived from dicarboxylic acid General formula (1)
- R 1 is an alkyl group having 1 to 10 carbon atoms
- R 2 is an alkyl group having 2 to 12 carbon atoms
- n is an integer of 1 to 3.
- the polyamide resin is composed of a structural unit derived from a diamine and a structural unit derived from a dicarboxylic acid.
- 50 mol% or more of the structural unit derived from the diamine is derived from xylylenediamine
- 50 mol% or more of the structural unit derived from the dicarboxylic acid is an ⁇ , ⁇ -linear aliphatic having 4 to 20 carbon atoms.
- a method for producing a laminated film comprising simultaneously stretching a polyamide resin film and a polypropylene resin film derived from dicarboxylic acid;
- R 1 is an alkyl group having 1 to 10 carbon atoms
- R 2 is an alkyl group having 2 to 12 carbon atoms
- n is an integer of 1 to 3.
- a stretched film having a high stretch ratio a method for producing a stretched film, and a polyamide resin composition can be provided.
- the stretched film of the present invention contains 0.5 to 15 parts by weight of the compound represented by the general formula (1) with respect to 100 parts by weight of the polyamide resin, and the polyamide resin contains a diamine-derived structural unit and a dicarboxylic acid.
- 50 mol% or more of the structural unit derived from the diamine is derived from xylylenediamine
- 50 mol% or more of the structural unit derived from the dicarboxylic acid is composed of ⁇ , 4 to 20 carbon atoms, It is derived from an ⁇ -linear aliphatic dicarboxylic acid (hereinafter sometimes referred to as “XD polyamide resin”).
- R 1 is an alkyl group having 1 to 10 carbon atoms
- R 2 is an alkyl group having 2 to 12 carbon atoms
- n is an integer of 1 to 3.
- a plasticizer such as a compound represented by the general formula (1) with a polyamide resin
- the polyamide resin film can be stretched at a high stretch ratio by blending the compound represented by the general formula (1) at a predetermined ratio into the XD-based polyamide resin. is there. That is, when a plasticizer such as a compound represented by the general formula (1) is added, the crystallization temperature of the polyamide resin at the time of temperature rise tends to decrease. And if crystallization temperature falls, it will become easy to advance crystallization, As a result of extending
- the polyamide resin composed of the combination of the XD-based polyamide resin and the compound represented by the general formula (1) despite the increase in stretching stress
- the film could be easily stretched and the stretch ratio could be increased.
- the strain of the molecular chain that becomes the starting point of the breaking at the time of stretching is eliminated. It is considered that the draw ratio can be increased more than in the absence. Details of the present invention will be described below.
- the stretched film of the present invention stretches a film (polyamide resin film) comprising a polyamide resin composition containing 0.5 to 15 parts by weight of the compound represented by the general formula (1) with respect to 100 parts by weight of the XD-based polyamide resin. Do it.
- a film polyamide resin film
- the details of the polyamide resin composition of the present invention will be described.
- the XD-based polyamide resin used as an essential component in the present invention is composed of a structural unit derived from a diamine and a structural unit derived from a dicarboxylic acid, and 50 mol% or more of the structural unit derived from the diamine is derived from xylylenediamine, More than 50 mol% of the structural unit derived from the acid is derived from the ⁇ , ⁇ -linear aliphatic dicarboxylic acid having 4 to 20 carbon atoms.
- the XD-based polyamide resin is preferably derived from at least one mol of xylylenediamine, more preferably at least 70 mol%, more preferably at least 80 mol%, more preferably at least 90 mol% of the structural unit derived from diamine.
- the diamine component that is the raw material of the XD-based polyamide resin contains 70% by mole or more of metaxylylenediamine, more preferably 80% by mole or more, and still more preferably 90% by mole or more.
- the metaxylylenediamine in the diamine component is 70 mol% or more
- the polyamide resin obtained therefrom can exhibit excellent gas barrier properties.
- the xylylenediamine is preferably composed of 30 to 100 mol% metaxylylenediamine and 0 to 70 mol% paraxylylenediamine, and preferably 70 to 100 mol% metaxylylenediamine and 0. More preferably, it consists of ⁇ 30 mol% paraxylylenediamine.
- diamines other than xylylenediamine that can be used as the raw material diamine component of the XD polyamide resin include tetramethylenediamine, pentamethylenediamine, 2-methylpentanediamine, hexamethylenediamine, heptamethylenediamine, octamethylenediamine, and nonamethylene.
- Aliphatic diamines such as diamine, decamethylenediamine, dodecamethylenediamine, 2,2,4-trimethyl-hexamethylenediamine, 2,4,4-trimethylhexamethylenediamine, 1,3-bis (aminomethyl) cyclohexane, 1 , 4-bis (aminomethyl) cyclohexane, 1,3-diaminocyclohexane, 1,4-diaminocyclohexane, bis (4-aminocyclohexyl) methane, 2,2-bis (4-aminocyclohexyl) propyl Aliphatic diamines such as bread, bis (aminomethyl) decalin, bis (aminomethyl) tricyclodecane, diamines having aromatic rings such as bis (4-aminophenyl) ether, paraphenylenediamine, bis (aminomethyl) naphthalene Etc., and one kind or a mixture of two or more kinds can be used.
- a diamine other than xylylenediamine is used as the diamine component, it is used in an amount of 30 mol% or less, preferably 1 to 25 mol%, particularly preferably 5 to 20 mol% of the structural unit derived from the diamine.
- the ⁇ , ⁇ -linear aliphatic dicarboxylic acid having 4 to 20 carbon atoms that is preferably used as the raw material dicarboxylic acid component of the XD-based polyamide resin includes ⁇ , ⁇ -linear aliphatic dicarboxylic acid having 6 to 16 carbon atoms. ⁇ , ⁇ -linear aliphatic dicarboxylic acid having 6 to 10 carbon atoms is more preferable.
- ⁇ , ⁇ -linear aliphatic dicarboxylic acid having 4 to 20 carbon atoms examples include succinic acid, glutaric acid, pimelic acid, suberic acid, azelaic acid, adipic acid, sebacic acid, undecanedioic acid, and dodecanedioic acid.
- Aliphatic dicarboxylic acids can be exemplified, and one or more can be mixed and used, but among these, the melting point of the polyamide resin is in an appropriate range for molding, so at least one of adipic acid and sebacic acid One is preferred, and adipic acid is particularly preferred.
- dicarboxylic acid component other than the ⁇ , ⁇ -linear aliphatic dicarboxylic acid having 4 to 20 carbon atoms examples include phthalic acid compounds such as isophthalic acid, terephthalic acid and orthophthalic acid, 1,2-naphthalenedicarboxylic acid, 3-naphthalenedicarboxylic acid, 1,4-naphthalenedicarboxylic acid, 1,5-naphthalenedicarboxylic acid, 1,6-naphthalenedicarboxylic acid, 1,7-naphthalenedicarboxylic acid, 1,8-naphthalenedicarboxylic acid, 2,3- Examples thereof include naphthalenedicarboxylic acids such as isomers such as naphthalenedicarboxylic acid, 2,6-naphthalenedicarboxylic acid, and 2,7-naphthalenedicarboxylic acid, and one kind or a mixture of two or more kinds can be used.
- phthalic acid compounds such as isophthalic acid,
- terephthalic acid or isophthalic acid may be used from the viewpoint of molding processability and barrier properties. preferable.
- the proportion of terephthalic acid and isophthalic acid is preferably 30 mol% or less, more preferably 1 to 30 mol%, particularly preferably 5 to 20 mol% of the structural unit derived from dicarboxylic acid.
- a structural unit derived from a diamine and a structural unit derived from a dicarboxylic acid means that the amide bond constituting the XD-based polyamide resin is formed by a bond between the dicarboxylic acid and the diamine.
- XD type polyamide resin contains other site
- a repeating unit having an amide bond that is not derived from a bond between a dicarboxylic acid and a diamine, a trace amount of impurities, and the like are included.
- the XD polyamide resin is a component constituting the polyamide resin in addition to the diamine component and the dicarboxylic acid component, and lactams such as ⁇ -caprolactam and laurolactam, aminocapron as long as the effects of the present invention are not impaired.
- Aliphatic aminocarboxylic acids such as acids and aminoundecanoic acids can also be used as copolymerization components.
- 90% by weight or more of the XD polyamide resin is preferably a structural unit derived from diamine or a structural unit derived from dicarboxylic acid.
- the XD polyamide resin used in the present invention preferably has a phosphorus atom concentration of 0.1 to 10 ppm, more preferably 1 to 8 ppm. By setting it as such a range, the improvement of continuous productivity by prevention of yellowing of a film and suppression of clogging of a polymer filter is compatible, and the effect of the present invention is more effectively exhibited.
- the stretched film used in the present invention preferably has a phosphorus atom concentration within a predetermined range, but such phosphorus atoms are usually derived from a polyamide resin.
- the XD polyamide resin used in the present invention preferably has a number average molecular weight (Mn) of 6,000 to 30,000, more preferably 8,000 to 28,000, still more preferably 9,000 to 26,000. When it is in such a range, the moldability becomes better.
- the XD polyamide resin used in the present invention preferably has a molecular weight distribution (weight average molecular weight / number average molecular weight (Mw / Mn)) of 1.8 to 3.1.
- the molecular weight distribution is more preferably 1.9 to 3.0, still more preferably 2.0 to 2.9.
- the molecular weight distribution of the XD polyamide resin can be adjusted, for example, by appropriately selecting the polymerization reaction conditions such as the type and amount of the initiator and catalyst used during the polymerization, and the reaction temperature, pressure, and time. It can also be adjusted by mixing a plurality of types of XD polyamide resins having different average molecular weights obtained under different polymerization conditions or by separately precipitating the polymerized XD polyamide resins.
- the molecular weight distribution can be obtained by GPC measurement. Specifically, using “HLC-8320GPC” manufactured by Tosoh Corporation as an apparatus and two “TSK gel Super HM-H” manufactured by Tosoh Corporation as a column, eluent Measured under conditions of hexafluoroisopropanol (HFIP) having a sodium trifluoroacetate concentration of 10 mmol / l, a resin concentration of 0.02% by weight, a column temperature of 40 ° C., a flow rate of 0.3 ml / min, and a refractive index detector (RI). It can obtain
- a calibration curve is prepared by dissolving 6 levels of PMMA in HFIP.
- the XD polyamide resin has a terminal amino group concentration ([NH 2 ]) of preferably less than 100 ⁇ equivalent / g, more preferably 5 to 75 ⁇ equivalent / g, and still more preferably 10 to 60 ⁇ equivalent / g.
- the carboxyl group concentration ([COOH]) is preferably less than 150 ⁇ eq / g, more preferably 10 to 120 ⁇ eq / g, and still more preferably 10 to 100 ⁇ eq / g.
- the viscosity is easily stabilized when the XD-based polyamide resin is processed into a film or film, and the reactivity with a carbodiimide compound described later is good. Tend to be.
- the ratio of the terminal amino group concentration to the terminal carboxyl group concentration is preferably 0.7 or less, more preferably 0.6 or less, particularly preferably 0. .5 or less. When this ratio is larger than 0.7, it may be difficult to control the molecular weight when polymerizing the XD polyamide resin.
- the terminal amino group concentration can be measured by dissolving 0.5 g of XD polyamide resin in 30 ml of a phenol / methanol (4: 1) mixed solution with stirring at 20-30 ° C. and titrating with 0.01 N hydrochloric acid. .
- the terminal carboxyl group concentration was determined by dissolving 0.1 g of XD polyamide resin in 30 ml of benzyl alcohol at 190 ° C., cooling in a 40 ° C. water bath, and titrating 0.01 N (normative) KOH solution. Can be measured.
- the description of paragraphs 0052 to 0053 of JP-A-2014-173196 can be referred to for the production method of the XD polyamide resin, and the contents thereof are incorporated in the present specification.
- the melting point of the XD polyamide resin is preferably 150 to 350 ° C., more preferably 180 to 300 ° C., and still more preferably 180 to 250 ° C. Further, the glass transition point of the XD polyamide resin is preferably 50 to 100 ° C., more preferably 55 to 100 ° C., and particularly preferably 60 to 100 ° C. Within this range, the heat resistance tends to be good.
- the melting point in the present invention is the temperature at the peak top of the endothermic peak at the time of temperature rise observed by the DSC (Differential Scanning Calorimetry) method. Specifically, it is measured by the method described in the examples described later. Value. When the equipment used in the examples is not available due to reasons such as out of print, it can be measured using other equipment having equivalent performance. Hereinafter, the same applies to other measurement methods.
- the glass transition point refers to a glass transition point that is measured by once heating and melting a sample to eliminate the influence of the thermal history on crystallinity and then raising the temperature again.
- DSC-60 manufactured by Shimadzu Corporation
- the sample amount is about 5 mg
- nitrogen is flowed at 30 ml / min as the atmospheric gas
- the temperature rising rate is 10 ° C./min.
- the polyamide resin heated and melted from room temperature to a temperature higher than the expected melting point under dry conditions is rapidly cooled with dry ice, and the temperature is raised again to a temperature higher than the melting point at a rate of 10 ° C./min to obtain the glass transition point.
- the lower limit of the crystallization temperature at the time of temperature rise of the XD polyamide resin is preferably 50 ° C. or higher, more preferably 80 ° C. or higher, further preferably 100 ° C. or higher, particularly preferably 120 ° C. or higher, and 140 ° C. or higher. Even more preferred.
- the upper limit of the crystallization temperature of the XD polyamide resin at the time of temperature rise is preferably 180 ° C. or less, more preferably 170 ° C. or less, further preferably 162 ° C. or less, particularly preferably 155 ° C. or less, and preferably 148 ° C. or less. Even more preferred.
- the XD-based polyamide resin used in the present invention is a compound in which the crystallization temperature at the time of temperature rise when the compound represented by 5% by weight of the general formula (1) is blended is represented by the general formula (1) Is preferably lower than the crystallization temperature at the time of temperature rise when not blended, the difference is more preferably 3 ° C. or more, further preferably 5 ° C. or more, and preferably 10 ° C. or more. Particularly preferred.
- the upper limit of the difference in crystallization temperature at the time of temperature rise is not particularly defined, but can be, for example, 40 ° C. or lower, further 35 ° C. or lower, and particularly 30 ° C. or lower. it can.
- the measuring method of the crystallization temperature at the time of temperature rising in this invention follows the method as described in the Example mentioned later.
- the proportion of the XD polyamide resin in the polyamide resin composition of the present invention is 50% by weight or more, preferably 60% by weight or more, more preferably 70% by weight or more, and 80% by weight or more. You can also.
- the polyamide resin composition of the present invention may contain a polyamide resin other than the XD polyamide resin.
- examples of such other polyamide resins include polyamide 4, polyamide 6, polyamide 11, polyamide 12, polyamide 46, polyamide 66, polyamide 610, polyamide 612, polyhexamethylene terephthalamide (polyamide 6T), polyhexamethylene isophthalate.
- Examples include ramid (polyamide 6I), polyamide 66 / 6T, polyamide 9T, polyamide 9MT, polyamide 6I / 6T, and the like.
- the content of the other polyamide resin in the polyamide resin composition of the present invention is preferably 1 to 50 parts by weight, more preferably 5 to 40 parts by weight, based on 100 parts by weight of the XD polyamide resin.
- the polyamide resin composition of this invention contains the compound represented by General formula (1).
- R 1 is an alkyl group having 1 to 10 carbon atoms
- R 2 is an alkyl group having 2 to 12 carbon atoms
- n is an integer of 1 to 3.
- the OH group part improves the familiarity with the XD polyamide resin.
- the OH group moiety may be substituted at any of the ortho, para and meta positions, but the para position is more preferred.
- the group represented by — (CH 2 ) n — is presumed to play the role of a linking group that connects the hydroxyphenyl ester group and —CR 1 R 2. . n is preferably 1 or 2, and more preferably 1.
- R 1 is preferably an alkyl group having 1 to 9 carbon atoms, more preferably an alkyl group having 2 to 9 carbon atoms, and 2 to 8 carbon atoms.
- the alkyl group is more preferably an alkyl group having 3 to 7 carbon atoms, particularly preferably an alkyl group having 4 to 6 carbon atoms.
- the alkyl group as R 1 is preferably a linear or branched alkyl group, and more preferably a linear alkyl group.
- R 2 is preferably an alkyl group having 2 to 10 carbon atoms, more preferably an alkyl group having 3 to 9 carbon atoms, and 5 to 9 carbon atoms. Are more preferable, and an alkyl group having 6 to 8 carbon atoms is particularly preferable.
- the alkyl group as R 2 is preferably a linear or branched alkyl group, and more preferably a linear alkyl group.
- the number of carbon atoms constituting R 2 is preferably 2 or more, and preferably 2 to 4 larger than the number of carbon atoms constituting R 1. More preferred. By adopting such a configuration, the effect of the present invention is more effectively exhibited.
- the compound represented by the general formula (1) contains 0.5 to 15 parts by weight of the compound represented by the general formula (1) with respect to 100 parts by weight of the polyamide resin in the polyamide resin composition.
- the lower limit of the compound represented by the general formula (1) is preferably 0.6 parts by weight or more, more preferably 0.7 parts by weight or more, further preferably 0.8 parts by weight or more, and 0.9 parts by weight. The above is particularly preferable.
- the upper limit is preferably 15 parts by weight or less, more preferably 11 parts by weight or less, still more preferably 10 parts by weight or less, still more preferably 8 parts by weight or less, still more preferably 6 parts by weight or less, and 4 parts by weight.
- the total amount is preferably within the above range.
- the oxygen barrier property can be further improved.
- the polyamide resin composition of the present invention may contain one or more plasticizers other than the compound represented by the general formula (1).
- plasticizers include the plasticizers described in paragraph 0039 of JP-A No. 7-11131, the contents of which are incorporated herein.
- the polyamide resin composition of the present invention preferably has a configuration that does not substantially contain a plasticizer other than the compound represented by the general formula (1). “Substantially free” means that, for example, in the polyamide resin composition of the present invention, the content of the other plasticizer is 0.1% by weight or less of the weight of the compound represented by the general formula (1). Say.
- the polyamide resin composition of the present invention may contain a resin component other than the polyamide resin.
- resin components other than polyamide resins include polyolefin resins such as polyethylene and polypropylene, polyester resins such as polyethylene terephthalate and polybutylene terephthalate, polycarbonate resins, polyoxymethylene resins, polyether ketones, polyether sulfones, and thermoplastic polyethers.
- a thermoplastic resin such as imide is exemplified.
- the polyamide resin composition of the present invention can be configured to contain substantially no thermoplastic resin other than the polyamide resin. “Substantially not contained” means that, for example, in the polyamide resin composition of the present invention, the content of the thermoplastic resin other than the polyamide resin is 5% by weight or less of the weight of the polyamide resin.
- the polyamide resin composition of the present invention includes an antioxidant, a stabilizer such as a heat stabilizer, a hydrolysis resistance improver, a weather resistance stabilizer, and a matting agent as long as the objects and effects of the present invention are not impaired.
- Additives such as ultraviolet absorbers, nucleating agents, plasticizers, dispersants, flame retardants, antistatic agents, anti-coloring agents, anti-gelling agents, coloring agents, release agents, and the like can be added. Details of these can be referred to the description of paragraphs 0130 to 0155 of Japanese Patent No. 4894982, the contents of which are incorporated herein.
- the polyamide resin composition is an XD polyamide resin and the compound represented by the general formula (1), and preferably accounts for 80% by weight or more, more preferably 90% by weight or more.
- the polyamide resin composition of this invention may contain fillers, such as carbon fiber, it is preferable not to contain substantially. “Substantially free” means, for example, that the blending amount of the filler is 3% by weight or less of the polyamide resin composition of the present invention.
- the polyamide resin composition of the present invention is preferably used for producing a stretched film as described above.
- the polyamide resin composition of the present invention in particular, a film composed of the polyamide resin composition before stretching (film before stretching) satisfies the following characteristics.
- the melting point of the polyamide resin composition of the present invention is preferably 150 to 350 ° C., more preferably 180 to 300 ° C., and further preferably 180 to 250 ° C.
- the lower limit of the crystallization temperature at the time of temperature rise of the polyamide resin composition of the present invention is preferably 50 ° C. or higher, more preferably 80 ° C. or higher, further preferably 100 ° C. or higher, and particularly preferably 120 ° C. or higher.
- the upper limit of the crystallization temperature at the time of temperature rising of the polyamide resin composition of the present invention is preferably 180 ° C. or less, more preferably 170 ° C. or less, further preferably 162 ° C. or less, particularly preferably 155 ° C. or less. It is even more preferable that the temperature is not higher than ° C.
- the crystallization temperature at the time of temperature rise is a compound represented by the general formula (1). It is preferably lower than the crystallization temperature at the time of temperature rise when not blended, the difference is more preferably 3 ° C. or more, and further preferably 5 ° C.
- the upper limit of the difference in crystallization temperature at the time of temperature rise is not particularly defined, but can be, for example, 40 ° C. or lower, further 35 ° C. or lower, and particularly 30 ° C. or lower. In particular, it can be set to 10 ° C. or lower.
- the stretched film of the present invention can be a stretched film with a high magnification.
- the final MD stretch ratio and final TD stretch ratio of the stretched film of the present invention are each independently preferably 4.2 times or more, more preferably 4.4 times or more, and more preferably 4.6 times or more. It is more preferable to use a polyamide resin in which 50 mol% or more of the structural unit derived from dicarboxylic acid is derived from sebacic acid.
- the upper limit of the final MD draw ratio and the final TD draw ratio is not particularly defined. For example, when a polyamide resin in which 50 mol% or more of the structural units derived from dicarboxylic acid are derived from adipic acid is used, 5.8 Even below double is practical level.
- the final stretch ratio of the stretched film of the present invention is preferably 20.0 times or more, more preferably 21.0 times or more, more preferably 23.0 times or more, further preferably 24.0 times or more, and 24.5 times.
- the above is particularly preferable, and in particular, by using a polyamide resin in which 50 mol% or more of the structural unit derived from dicarboxylic acid is derived from sebacic acid, it can be 55 times or more.
- the upper limit of the final draw ratio is not particularly defined.
- the final stretch ratio means the stretch ratio of the stretched film finally obtained based on the overall stretch ratio and the relaxation rate, compared to before stretching, and in the case of biaxial stretching, the final MD stretch ratio, The product of the final TD stretch ratio.
- the final MD stretch ratio means the stretch ratio in the MD direction of the stretched film finally obtained based on the MD stretch ratio and the MD relaxation rate compared to before stretching.
- the final TD stretch ratio is the TD stretch ratio and TD. Based on the relaxation rate, it means the draw ratio in the TD direction of the stretched film finally obtained compared to before stretching, and is a value calculated from the following formula.
- the stretched film of the present invention can have a tensile modulus measured according to JIS K 7127 of 2 GPa or more, further 2.5 GPa or more, and particularly 3.0 GPa or more. it can.
- the upper limit value of the tensile modulus of elasticity is not particularly defined, but can be, for example, 6 GPa or less. The detailed conditions of the tensile modulus here are as described in the examples described later.
- the thickness of the stretched film of the present invention is preferably 1 ⁇ m or more, more preferably 3 ⁇ m or more, and can be 4 ⁇ m or more, and in particular, 20 ⁇ m or more. Moreover, the thickness of the stretched film of the present invention is preferably 100 ⁇ m or less, more preferably 90 ⁇ m or less, can be 80 ⁇ m or less, and can be particularly 40 ⁇ m or less. Although the length of the stretched film of the present invention is not particularly defined, for example, as described later, in the case of continuous production by roll-to-roll, it will usually be 10 m or more.
- the phosphorus atom concentration of the stretched film of the present invention is preferably 0.1 to 10 ppm, and more preferably 1 to 8 ppm. By setting it as such a range, yellowing of a film can be suppressed effectively, clogging of a polymer filter can be suppressed effectively, and continuous productivity can be improved more.
- the oxygen transmission coefficient of the stretched film of the present invention at 23 ° C. and relative humidity (RH) 60% RH can be 1.1 cc ⁇ mm / (m 2 ⁇ day ⁇ atm) or less, and further 0.08 cc ⁇ mm / (m 2 ⁇ day ⁇ atm) or less, and more preferably 0.05 cc ⁇ mm / (m 2 ⁇ day ⁇ atm).
- the lower limit value, 0cc ⁇ mm / (m 2 ⁇ day ⁇ atm) is preferred, a 0.002cc ⁇ mm / (m 2 ⁇ day ⁇ atm) sufficiently practical level in extent.
- the method for measuring the oxygen permeability coefficient in the present invention follows the method described in the examples described later.
- the process for producing a stretched film of the present invention comprises 0.5 to 15 parts by weight of the compound represented by the general formula (1) with respect to 100 parts by weight of the polyamide resin, and the polyamide resin is derived from diamine. And 50 mol% or more of the structural unit derived from the diamine is derived from xylylenediamine, and 50 mol% or more of the structural unit derived from the dicarboxylic acid is composed of carbon atoms.
- a film comprising a polyamide resin composition derived from 4 to 20 ⁇ , ⁇ -linear aliphatic dicarboxylic acid is stretched.
- R 1 is an alkyl group having 1 to 10 carbon atoms
- R 2 is an alkyl group having 2 to 12 carbon atoms
- n is an integer of 1 to 3.
- the said polyamide resin (XD type polyamide resin) and the compound represented by General formula (1) are synonymous with the above-mentioned, and a preferable range is also the same.
- the manufacturing method of the stretched film of this invention is demonstrated. Needless to say, the present invention is not limited to these examples.
- a polyamide resin composition containing 0.5 to 15 parts by weight of a compound represented by the general formula (1) is melt-kneaded with 100 parts by weight of an XD polyamide resin Then, it is pushed out from the T die 11 to the casting roll 12.
- the extrusion temperature at the time of extrusion is not particularly determined as long as the polyamide resin composition is melted.
- the thickness of the polyamide resin film made of the melt-extruded polyamide resin composition is preferably 5 to 60 times that of the stretched film after stretching, although it depends on the application and the stretch ratio.
- the thickness is more preferably ⁇ 40 times, more preferably 10 to 30 times, and particularly preferably 15 to 28 times.
- the stretching stress of the polyamide resin film before stretching in the present invention can be, for example, 0.24 MPa or more, further 0.26 MPa or more, and even 0.30 MPa or more. .
- the upper limit of the stretching stress is not particularly defined, but an example is 5 MPa or less, further 4 MPa or less, and particularly 1 MPa or less is a practical level. . Since the polyamide resin film used in the present invention can increase the stretching stress, it can be stretched at a high stretching ratio.
- the polyamide resin film is stretched.
- stretching is performed in the stretching / relaxation zone 13. Stretching may be performed only in one direction (uniaxial stretching), or may be performed in two orthogonal directions (biaxial stretching), and biaxial stretching is preferred.
- MD stretching in two directions may be performed simultaneously or sequentially.
- MD stretching can be performed by passing a polyamide resin film between rolls having different peripheral speeds.
- the polyamide resin film is set so that the peripheral speed is higher when it passes later. Moreover, it can also extend
- the stretching ratio is preferably 4.2 times or more, preferably 4.4 times or more, more preferably 4.8 times or more. It is particularly preferable to stretch the film by 0 times or more.
- the stretching ratio is preferably 4.2 times or more, preferably 4.4 times or more, and preferably 4.8 times or more in each direction. More preferably, it is particularly preferable to stretch 5.0 times or more.
- the upper limit value of each stretching ratio in the case of uniaxial or biaxial stretching is not particularly defined, but is, for example, 20.0 times or less, further 10.0 times or less, particularly 8.0 times. It can be as follows.
- the overall draw ratio in the present invention is preferably 18.0 times or more, more preferably 19.0 times or more, further preferably 20.0 times or more, and more preferably 22.0 times or more. More preferably, it is more preferably 24.0 times or more, and it can also be 50 times or more.
- the upper limit of the overall draw ratio is not particularly defined, but can be, for example, 100.0 times or less, further 40.0 times or less, and particularly 30.0 times or less.
- Stretching may be performed at room temperature, but is preferably performed under heating conditions.
- the heating is preferably performed while the polyamide resin film is passed through the heating zone.
- the stretching is preferably performed at a melting point of the XD polyamide resin of ⁇ 200 ° C. to less than the melting point, more preferably at a melting point of XD polyamide resin of ⁇ 150 ° C. to a melting point of ⁇ 100 ° C., and the melting point of the XD polyamide resin. More preferably, it is carried out at -145 ° C to a melting point of -110 ° C.
- the temperature of the XD polyamide resin at the time of extrusion may be set based on the melting point of the XD polyamide resin having the lowest melting point.
- the temperature may be set based on the lowest melting point.
- the relaxation is preferably performed during the heat setting step.
- the heat setting time is preferably 5 seconds to 5 minutes, more preferably 10 seconds to 1 minute.
- the relaxation can be started from 15 to 16 seconds after the start of heat setting.
- the heat setting is preferably performed at a melting point of the XD polyamide resin of ⁇ 70 ° C. to less than the melting point, more preferably a melting point of the XD polyamide resin of ⁇ 50 ° C. to a melting point of ⁇ 5 ° C., and a melting point of the XD polyamide resin.
- the relaxation rate is preferably 0.5 to 10% in the stretching direction, more preferably 1 to 8%, and more preferably 1.5 to 6%. Further preferred.
- the relaxation rate is preferably 0.5 to 10% in each stretching direction, more preferably 1 to 8%, and more preferably 1.5 to 6%. More preferably.
- the relaxation rate is calculated as follows in the case of uniaxial stretching.
- the stretched film obtained by obtaining the above steps is usually wound and stored on a roll or the like (step 14 in FIG. 1). Furthermore, the stretched film is cut and used for various applications. It is preferable to adjust the amount of stretching and the amount of relaxation so that the final stretch ratio of the stretched film obtained by the production method of the present invention becomes the final stretch ratio of the stretched film described above.
- the stretched film of the present invention may be used as it is, but is usefully used as a laminated film having a stretched polypropylene resin film. That is, when a polypropylene resin film is stretched, it is usually stretched at a high magnification.
- FIG. 2 it can be considered that the stretched film of the present invention and the polypropylene resin film are stretched simultaneously.
- a polyamide resin film 23 is laminated between two polypropylene resin films 22 (a ′) and stretched (b ′). In some cases, the polyamide resin film 23 was broken, and stretching at a high stretching ratio was difficult (c ′).
- FIGS. 2 (a ′) to (c ′) a polyamide resin film 23 is laminated between two polypropylene resin films 22 (a ′) and stretched (b ′). In some cases, the polyamide resin film 23 was broken, and stretching at a high stretching ratio was difficult (c ′).
- the stretched film 21 and the polypropylene resin film 22 can be easily stretched simultaneously. That is, as a preferred embodiment of the present invention, a laminated film production method including simultaneously stretching the stretched film of the present invention and a polypropylene resin film is exemplified.
- the polypropylene resin film has two layers, it may be only one layer or a laminated film having three or more layers. Further, the stretched film of the present invention and the stretched polypropylene resin film may be separately prepared and bonded together with an adhesive or the like.
- the stretched polypropylene resin film may be uniaxially stretched or biaxially stretched, but is more preferably biaxially stretched.
- the stretch ratio of the stretched polypropylene resin is preferably in the same range as the stretch ratio described in the method for producing a stretched film of the present invention.
- the stretched film of the present invention may be used as a fiber-reinforced composite material by impregnating reinforcing fibers in addition to the above laminated film.
- the fibers in this case include carbon fibers and glass fibers.
- the stretched film of the present invention is used for transportation parts such as automobiles, general machine parts, precision machine parts, electronic / electric equipment parts, OA equipment parts, building materials / residential equipment parts, medical equipment, leisure sports equipment, play equipment, medical goods. Widely used in daily necessities such as food packaging films, defense and aerospace products.
- the obtained pellets were charged into a tumbler (rotary vacuum tank) having a heating medium heating mantle, and heated at 200 ° C. for 1 hour in a reduced pressure state (0.5 to 10 Torr), thereby solidifying the obtained pellets.
- Phase polymerization was performed to obtain a polyamide resin (MXD6, melting point: 237 ° C., relative viscosity: 2.65, moisture content: 0.05%).
- the contents were taken out in a strand shape and pelletized with a pelletizer to obtain 15 kg pellets.
- the obtained pellets were placed in a tumbler (rotary vacuum tank) having a heating medium heating mantle, and heated at 195 ° C. for 1 hour in a reduced pressure state (0.5 to 10 Torr). Phase polymerization was performed to obtain a polyamide resin (MP10, melting point: 213 ° C., relative viscosity: 2.60, moisture content: 0.03%).
- Example 1 Method for producing stretched film> A polyamide resin composition in which the plasticizer shown in Table 1 was blended in the proportion (unit: parts by weight) shown in Table 1 with respect to 100 parts by weight of the polyamide resin shown in Table 1 was melt extruded from a die. Specifically, the polyamide resin composition obtained by melt-kneading each component was extruded to a width of 130 mm and cut into 90 mm squares with the thicknesses shown in Table 1. Then, after extending
- a biaxial stretching apparatus Teenter method, EX105S, Toyo Seiki Seisakusho Co., Ltd.
- ICP inductively coupled plasma
- ⁇ Oxygen permeability coefficient of stretched film The oxygen permeability coefficient of the stretched film was measured in an atmosphere of 23 ° C. and 60% relative humidity according to ASTM D3985, using an oxygen permeability measuring device (manufactured by MOCON, model: OX-TRAN 2/21). . The lower the measured value, the better the oxygen barrier property. In Reference Examples 1 to 3, the oxygen transmission coefficient was measured in the same manner for the unstretched polyamide resin film.
- Example 1 As shown in Table 1 or 2, the raw materials and production conditions were changed, and the others were performed in the same manner.
- the reference example is an example in which stretching is not performed.
- the stretched film of the present invention had a high stretching stress and could be stretched at a high stretch ratio (Examples 1 to 7).
- the compound represented by the general formula (1) is not blended (Comparative Examples 1, 2, 6, 7)
- the film is derived from the same resin, and the compound represented by the general formula (1) Compared with the case where it mix
- Example 8 Stretching of Multilayer Sheet Polypropylene (Novatech PP FL203D, manufactured by Nippon Polypro Co., Ltd.) melted at 240 ° C. with a single screw extruder, Admer QF580 (made by Mitsui Chemicals, Inc.) melted at 240 ° C. with a single screw extruder , PP-based adhesive), MXD6 melted at 260 ° C. in a single screw extruder is supplied to a co-extrusion multilayer film manufacturing apparatus (feed block and T-die temperature: 260 ° C.), and melt extrusion is performed.
- a co-extrusion multilayer film manufacturing apparatus feed block and T-die temperature: 260 ° C.
- the resulting film was stretched according to the conditions described in Table 3. As a result, it was found that the MXD6 film of the center layer can be stretched following the PP film. In particular, it is significant in that it can be stretched 5.0 times in the MD direction and TD direction.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Engineering & Computer Science (AREA)
- Manufacturing & Machinery (AREA)
- Materials Engineering (AREA)
- Mechanical Engineering (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Polyamides (AREA)
- Manufacture Of Macromolecular Shaped Articles (AREA)
- Shaping By String And By Release Of Stress In Plastics And The Like (AREA)
Abstract
Description
例えば、特許文献1には、(A)ポリフェニレンエーテル系樹脂 5~90重量部、および(B)ポリアミド樹脂 95~10重量部の合計100重量部に対し、(C)相容化剤 0.01~30重量部、(D)ポリアミドに柔軟性を与える可塑剤 0.1重量部~100重量部、(E)ゴム様物質 0~100重量部からなることを特徴とする熱可塑性樹脂組成物が開示されている。さらに、特許文献1には、このような熱可塑性樹脂組成物が柔軟性と高い引っ張り強度を同時に有することが記載されている。
例えば、特許文献2には、芳香族ポリアミド樹脂をMD方向および/またはTD方向に4倍を超える倍率で延伸して得られた芳香族ポリアミド延伸フィルムであり、前記芳香族ポリアミド樹脂がメタキシリレンジアミン単位を70モル%以上含むジアミン構成単位と、炭素数4~20のα,ω-直鎖脂肪族ジカルボン酸単位を80~97モル%およびイソフタル酸単位を3~20モル%を含むジカルボン酸構成単位を含み、かつ、脱偏光強度法による定温結晶化によって測定した場合、ガラス転移点以上~融点未満の測定温度範囲内での前記芳香族ポリアミド樹脂の最短半結晶化時間が40~2000秒の範囲である芳香族ポリアミド延伸フィルムが記載されている。
また、特許文献3には、A)ポリアミド組成物から製造されたポリアミド層と、 B)エチレンビニルアルコールコポリマー(EVOH)から製造されたバリア層とを含む多層構造物であって、ポリアミド層がバリア層に直接に付着され、ポリアミド組成物が、a)i)8~20個の炭素原子を有する芳香族ジカルボン酸および4~20個の炭素原子を有する脂肪族ジアミン、またはii)6~20個の炭素原子を有する脂肪族ジカルボン酸および6~20個の炭素原子を有する芳香族ジアミン、またはiii)7~20個の炭素原子を有する芳香族アミノカルボン酸から選択されたグループAのモノマーと、b)iv)6~20個の炭素原子を有する脂肪族ジカルボン酸および4~20個の炭素原子を有する脂肪族ジアミン、またはv)4~20個の炭素原子を有するラクタムおよび/または脂肪族アミノカルボン酸から選択されたグループBのモノマーとから製造されたコポリアミドから選択された1つまたは複数の半芳香族コポリアミドを含み、グループAの前記モノマーが、前記コポリアミドに基づいて大体10モルパーセント~大体40モルパーセントの量において存在し、グループBの前記モノマーが、前記コポリアミドに基づいて大体60モルパーセント~大体90モルパーセントの量において存在する、多層構造物が開示されている。

(B)分岐鎖を有する炭素数16~24の1価のアルコール0.05~5重量部を配合してなるポリアミド樹脂組成物が開示されている。
<1>ポリアミド樹脂100重量部に対し、一般式(1)で表される化合物を0.5~15重量部含み、
前記ポリアミド樹脂が、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、
前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来する、延伸フィルム;
一般式(1)

<2>前記キシリレンジアミンが、30~100モル%のメタキシリレンジアミンと0~70モル%のパラキシリレンジアミンからなる、<1>に記載の延伸フィルム。
<3>前記ジカルボン酸由来の構成単位の50モル%以上が、セバシン酸およびアジピン酸の少なくとも1種に由来する、<1>または<2>に記載の延伸フィルム。
<4>前記ジカルボン酸由来の構成単位の50モル%以上が、アジピン酸に由来する、<1>~<3>のいずれかに記載の延伸フィルム。
<5>前記延伸フィルム中のリン原子濃度が0.1~10ppmである、<1>~<4>のいずれかに記載の延伸フィルム。
<6>前記一般式(1)で表される化合物において、R1が、炭素数3~7のアルキル基であり、R2が、炭素数5~9のアルキル基である、<1>~<5>のいずれかに記載の延伸フィルム。
<7>最終延伸倍率が、20.0倍以上である、<1>~<6>のいずれかに記載の延伸フィルム。
<8>JIS K 7127に従って測定した引張弾性率が、2.0GPa以上である、<1>~<7>のいずれかに記載の延伸フィルム。
<9>前記延伸フィルムの厚さが、1~100μmである、<1>~<8>のいずれかに記載の延伸フィルム。
<10>ポリアミド樹脂100重量部に対し、一般式(1)で表される化合物を0.5~15重量部含み、前記ポリアミド樹脂が、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来するポリアミド樹脂組成物からなるフィルムを、延伸することを含む、延伸フィルムの製造方法;
一般式(1)
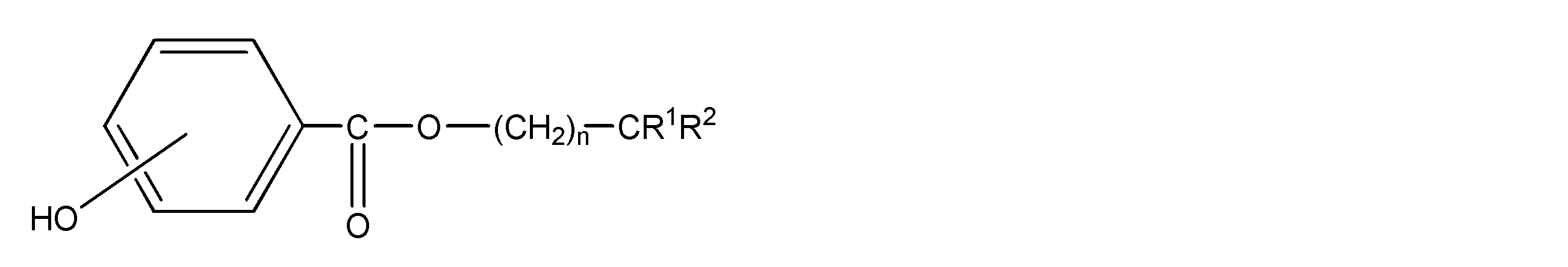
<11>前記延伸を、直交する2方向に行う、<10>に記載の延伸フィルムの製造方法。
<12>前記延伸を、直交する2方向に、それぞれ、4.2倍以上となるように行う、<10>に記載の延伸フィルムの製造方法。
<13>前記延伸温度が、前記ポリアミド樹脂の(融点-200℃)以上融点未満である、<10>~<12>のいずれかに記載の延伸フィルムの製造方法。
<14>前記延伸を、総合延伸倍率が、18.0倍以上となるように行う、<10>~<13>のいずれかに記載の延伸フィルムの製造方法。
<15>ポリアミド樹脂100重量部に対し、一般式(1)で表される化合物を0.5~15重量部含み、前記ポリアミド樹脂が、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来する、延伸フィルム製造用のポリアミド樹脂組成物;
一般式(1)

<16><1>~<9>のいずれかに記載の延伸フィルムと、延伸されたポリプロピレン樹脂フィルムを有する積層フィルム。
<17>2軸延伸フィルムである、<16>に記載の積層フィルム。
<18>ポリアミド樹脂100重量部に対し、一般式(1)で表される化合物を0.5~15重量部含み、前記ポリアミド樹脂が、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来する、ポリアミド樹脂フィルムと、ポリプロピレン樹脂フィルムを同時に延伸することを含む、積層フィルムの製造方法;
一般式(1)

<19>前記延伸を、総合延伸倍率が、20.0~30.0倍となるように行う、<18>に記載の積層フィルムの製造方法。
本発明の延伸フィルムは、ポリアミド樹脂100重量部に対し、一般式(1)で表される化合物を0.5~15重量部含み、前記ポリアミド樹脂が、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来する(以下、「XD系ポリアミド樹脂」ということがある)ことを特徴とする。このような構成とすることにより、高い延伸倍率の延伸フィルムとすることができる。そのため、高い延伸倍率で延伸された樹脂フィルムとも積層可能な延伸フィルムが得られる。
一般式(1)

ポリアミド樹脂に一般式(1)で表される化合物のような可塑剤を配合することは、例えば、上記特許文献1および特許文献4に記載されている。しかしながら、本発明者が検討したところ、ポリアミド樹脂の種類によって、一般式(1)で表される化合物の挙動が異なることが分かった。そして、驚くべきことに、XD系ポリアミド樹脂に、一般式(1)で表される化合物を所定の割合で配合することによって、ポリアミド樹脂フィルムを高い延伸倍率で延伸することを可能にしたものである。
すなわち、一般式(1)で表される化合物のような可塑剤を添加すると、通常、ポリアミド樹脂の昇温時の結晶化温度は、低下する傾向にある。そして、結晶化温度が低下すると結晶化が進行しやすくなり、延伸応力が増大した結果、通常は、延伸が困難になる。ここで、XD系ポリアミド樹脂に、一般式(1)で表される化合物を配合した場合も、昇温時の結晶化温度が低下し、延伸応力が増大した。しかしながら、一般式(1)で表される化合物を配合した場合、延伸応力が増加したにもかかわらず、XD系ポリアミド樹脂と一般式(1)で表される化合物の組み合わせから構成されるポリアミド樹脂フィルムは、容易に延伸が可能であり、延伸倍率を高めることが可能であった。推測の域を出ないが、この原理として、一般式(1)で表される化合物の存在下で適度な延伸応力がかかることで、延伸時の破断の起点となる分子鎖の歪が解消され、非存在下よりも延伸倍率を高めることが可能になったと考える。
以下、本発明の詳細について説明する。
本発明で必須成分として用いるXD系ポリアミド樹脂は、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来する。
より具体的には、キシリレンジアミンが、30~100モル%のメタキシリレンジアミンと0~70モル%のパラキシリレンジアミンからなることが好ましく、70~100モル%のメタキシリレンジアミンと0~30モル%のパラキシリレンジアミンからなることがさらに好ましい。
ジアミン成分として、キシリレンジアミン以外のジアミンを用いる場合は、ジアミン由来の構成単位の30モル%以下であり、好ましくは1~25モル%、特に好ましくは5~20モル%の割合で用いる。
尚、後述するとおり、本発明で用いる延伸フィルムは、リン原子濃度が所定の範囲であることが好ましいが、かかるリン原子は、通常、ポリアミド樹脂に由来する。
数平均分子量(Mn)=2,000,000/([COOH]+[NH2])
XD系ポリアミド樹脂の分子量分布は、例えば、重合時に使用する開始剤や触媒の種類、量および反応温度、圧力、時間等の重合反応条件などを適宜選択することにより調整できる。また、異なる重合条件によって得られた平均分子量の異なる複数種のXD系ポリアミド樹脂を混合したり、重合後のXD系ポリアミド樹脂を分別沈殿させることにより調整することもできる。
XD系ポリアミド樹脂の製造方法は、特開2014-173196号公報の段落0052~0053の記載を参酌でき、これらの内容は本明細書に組み込まれる。
また、XD系ポリアミド樹脂のガラス転移点は、50~100℃が好ましく、55~100℃がより好ましく、特に好ましくは60~100℃である。この範囲であると、耐熱性が良好となる傾向にある。
ガラス転移点とは、試料を一度加熱溶融させ熱履歴による結晶性への影響をなくした後、再度昇温して測定されるガラス転移点をいう。測定には、例えば、島津製作所社(SHIMADZU CORPORATION)製「DSC-60」を用い、試料量は約5mgとし、雰囲気ガスとしては窒素を30ml/分で流し、昇温速度は10℃/分の条件で室温から予想される融点以上の温度まで加熱し溶融させたポリアミド樹脂を、ドライアイスで急冷し、10℃/分の速度で融点以上の温度まで再度昇温し、ガラス転移点を求めることができる。
さらに、本発明で用いるXD系ポリアミド樹脂は、5重量%の一般式(1)で表される化合物を配合したときの昇温時の結晶化温度が、一般式(1)で表される化合物を配合しない場合の昇温時の結晶化温度よりも低いことが好ましく、その差が、3℃以上であることがより好ましく、5℃以上であることがさらに好ましく、10℃以上であることが特に好ましい。昇温時の結晶化温度の差の上限値については、特に定めるものでは無いが、例えば、40℃以下とでき、さらには35℃以下とすることもでき、特には30℃以下とすることもできる。
本発明における昇温時の結晶化温度の測定方法は、後述する実施例に記載の方法に従う。
本発明のポリアミド樹脂組成物は、上記XD系ポリアミド樹脂以外のポリアミド樹脂を含んでいても良い。このような他のポリアミド樹脂としては、ポリアミド4、ポリアミド6、ポリアミド11、ポリアミド12、ポリアミド46、ポリアミド66、ポリアミド610、ポリアミド612、ポリヘキサメチレンテレフタラミド(ポリアミド6T)、ポリヘキサメチレンイソフタラミド(ポリアミド6I)、ポリアミド66/6T、ポリアミド9T、ポリアミド9MT、ポリアミド6I/6T等が挙げられる。この中でも、他のポリアミド樹脂を含む場合、ポリアミド6およびポリアミド66の少なくとも1種が好ましい。
本発明のポリアミド樹脂組成物における、他のポリアミド樹脂の含有量は、配合する場合、XD系ポリアミド樹脂100重量部に対し、1~50重量部が好ましく、5~40重量部がより好ましい。
本発明のポリアミド樹脂組成物は、一般式(1)で表される化合物を含む。

一般式(1)で表される化合物において、-(CH2)n-で表される基は、上記ヒドロキシフェニルエステル基と、-CR1R2をつなぐ連結基の役割を果たすと推定される。nは1または2が好ましく、1がさらに好ましい。
一般式(1)で表される化合物において、R1は、炭素数1~9のアルキル基であることが好ましく、炭素数2~9のアルキル基であることがより好ましく、炭素数2~8のアルキル基であることがさらに好ましく、炭素数3~7のアルキル基であることが特に好ましく、炭素数4~6のアルキル基であることが一層好ましい。R1としてのアルキル基は、直鎖または分岐アルキル基が好ましく、直鎖アルキル基がより好ましい。
一般式(1)で表される化合物において、R2は、炭素数2~10のアルキル基であることが好ましく、炭素数3~9のアルキル基であることがより好ましく、炭素数5~9のアルキル基であることがさらに好ましく、炭素数6~8のアルキル基であることが特に好ましい。R2としてのアルキル基は、直鎖または分岐アルキル基が好ましく、直鎖アルキル基がより好ましい。
また、本発明では、一般式(1)で表される化合物において、R2を構成する炭素数が、R1を構成する炭素数よりも、2以上大きいことが好ましく、2~4大きいことがより好ましい。このような構成とすることにより、本発明の効果がより効果的に発揮される。

一般式(1)で表される化合物は、1種のみであってもよいし、2種以上であってもよい。2種以上含む場合、合計量が上記範囲であることが好ましい。
特に、本発明では、ジカルボン酸由来の構成単位の50モル%以上がアジピン酸に由来する、ポリアミド樹脂100重量部に対し、0.5~4重量部、特には、0.8~2.0重量部配合した場合に、酸素バリア性をより向上させることができる。
本発明のポリアミド樹脂組成物は、ポリアミド樹脂以外の他の樹脂成分を含んでいても良い。ポリアミド樹脂以外の他の樹脂成分としては、ポリエチレン、ポリプロピレン等のポリオレフィン樹脂、ポリエチレンテレフタレート、ポリブチレンテレフタレート等のポリエステル樹脂、ポリカーボネート樹脂、ポリオキシメチレン樹脂、ポリエーテルケトン、ポリエーテルスルフォン、熱可塑性ポリエーテルイミド等の熱可塑性樹脂が例示される。
また、本発明のポリアミド樹脂組成物では、ポリアミド樹脂以外の熱可塑性樹脂を実質的に含まない構成とすることができる。実質的に含まないとは、例えば、本発明のポリアミド樹脂組成物において、ポリアミド樹脂以外の熱可塑性樹脂の含有量が、ポリアミド樹脂の重量の5重量%以下であることをいう。
さらに、本発明の目的・効果を損なわない範囲で、本発明のポリアミド樹脂組成物には、酸化防止剤、熱安定剤等の安定剤、耐加水分解性改良剤、耐候安定剤、艶消剤、紫外線吸収剤、核剤、可塑剤、分散剤、難燃剤、帯電防止剤、着色防止剤、ゲル化防止剤、着色剤、離型剤等の添加剤等を加えることができる。これらの詳細は、特許第4894982号公報の段落0130~0155の記載を参酌でき、これらの内容は本明細書に組み込まれる。
しかしながら、ポリアミド樹脂組成物は、XD系ポリアミド樹脂と一般式(1)で表される化合物で、全体の80重量%以上を占めることが好ましく、90重量%以上を占めることがより好ましい。
また、本発明のポリアミド樹脂組成物は、炭素繊維等の充填剤を含んでいてもよいが、実質的に含まないことが好ましい。実質的に含まないとは、例えば、充填剤の配合量が、本発明のポリアミド樹脂組成物の3重量%以下であることをいう。
本発明のポリアミド樹脂組成物は、上述のとおり、好ましくは延伸フィルムの製造に用いられる。
本発明のポリアミド樹脂組成物、特に、延伸前のポリアミド樹脂組成物からなるフィルム(延伸前フィルム)は、以下の特性を満たすことが好ましい。
本発明のポリアミド樹脂組成物の融点は、150~350℃であることが好ましく、180~300℃であることがより好ましく、180~250℃であることがさらに好ましい。
また、本発明のポリアミド樹脂組成物の昇温時の結晶化温度の下限値は、50℃以上が好ましく、80℃以上がより好ましく、100℃以上がさらに好ましく、120℃以上が特に好ましい。また、本発明のポリアミド樹脂組成物の昇温時の結晶化温度の上限値は、180℃以下が好ましく、170℃以下がより好ましく、162℃以下がさらに好ましく、155℃以下が特に好ましく、148℃以下がより一層好ましい。
さらに、本発明のポリアミド樹脂組成物は、5重量%の一般式(1)で表される化合物を配合したときの昇温時の結晶化温度が、一般式(1)で表される化合物を配合しない場合の昇温時の結晶化温度よりも低いことが好ましく、その差が、3℃以上であることがより好ましく、5℃以上であることがさらに好ましい。昇温時の結晶化温度の差の上限値については、特に定めるものでは無いが、例えば、40℃以下とでき、さらには35℃以下とすることもでき、特には30℃以下とすることもでき、特には、10℃以下とすることもできる。
本発明の延伸フィルムは、高倍率の延伸フィルムとすることができる。本発明の延伸フィルムの最終MD延伸倍率および最終TD延伸倍率は、それぞれ独立に、4.2倍以上が好ましく、4.4倍以上がより好ましく、4.6倍以上がより好ましく、4.8倍以上がさらに好ましく、特に、ジカルボン酸由来の構成単位の50モル%以上がセバシン酸に由来するポリアミド樹脂を用いることにより、7.0倍以上とすることもできる。最終MD延伸倍率および最終TD延伸倍率の上限については、特に定めるものではないが、例えば、ジカルボン酸由来の構成単位の50モル%以上がアジピン酸に由来するポリアミド樹脂を用いた場合、5.8倍以下でも実用レベルである。また、ジカルボン酸由来の構成単位の50モル%以上がセバシン酸に由来するポリアミド樹脂を用いる場合、8.0倍以下でも実用レベルである。
本発明の延伸フィルムの最終延伸倍率は、20.0倍以上が好ましく、21.0倍以上がより好ましく、23.0倍以上がより好ましく、24.0倍以上がさらに好ましく、24.5倍以上が特に好ましく、特に、ジカルボン酸由来の構成単位の50モル%以上がセバシン酸に由来するポリアミド樹脂を用いることにより、55倍以上とすることもできる。最終延伸倍率の上限については、特に定めるものではないが、例えば、ジカルボン酸由来の構成単位の50モル%以上がアジピン酸に由来するポリアミド樹脂を用いた場合、30倍以下でも実用レベルである。また、ジカルボン酸由来の構成単位の50モル%以上がセバシン酸に由来するポリアミド樹脂を用いる場合、80倍以下でも実用レベルである。
ここで、最終延伸倍率とは、総合延伸倍率と緩和率に基づき、最終的に得られる延伸フィルムの、延伸前と比較した延伸倍率を意味し、2軸延伸の場合、最終MD延伸倍率と、最終TD延伸倍率の積をいう。最終MD延伸倍率は、MD延伸倍率とMD緩和率に基づき、最終的に得られる延伸フィルムの、延伸前と比較したMD方向の延伸倍率を意味し、最終TD延伸倍率は、TD延伸倍率とTD緩和率に基づき、最終的に得られる延伸フィルムの、延伸前と比較したTD方向の延伸倍率を意味し、以下式から算出される値である。
最終MD延伸倍率=(MD延伸倍率-1)x{(100-MD緩和率)/100}+1
最終TD延伸倍率=(TD延伸倍率-1)x{(100-TD緩和率)/100}+1
本発明の延伸フィルムは、JIS K 7127に従って測定した引張弾性率を、2GPa以上とすることができ、さらには、2.5GPa以上とすることもでき、特には、3.0GPa以上とすることもできる。引張弾性率の上限値については、特に定めるものではないが、例えば、6GPa以下とすることができる。ここでの引張弾性率の詳細な条件は、後述する実施例の記載に従う。
本発明の延伸フィルムの長さは、特に定めるものではないが、例えば、後述するとおり、ロールトゥロールで連続生産する場合は、通常、10m以上であろう。
本発明の延伸フィルムの23℃、相対湿度(RH)60%RHにおける酸素透過係数は、1.1cc・mm/(m2・day・atm)以下とすることができ、さらには0.08cc・mm/(m2・day・atm)以下とすることができ、よりさらには、0.05cc・mm/(m2・day・atm)とすることもできる。下限値については、0cc・mm/(m2・day・atm)が好ましいが、0.002cc・mm/(m2・day・atm)程度でも十分実用レベルである。
本発明における酸素透過係数の測定方法は、後述する実施例に記載の方法に従う。
本発明の延伸フィルムの製造方法は、ポリアミド樹脂100重量部に対し、一般式(1)で表される化合物を0.5~15重量部含み、前記ポリアミド樹脂が、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来するポリアミド樹脂組成物からなるフィルムを、延伸することを特徴とする。
一般式(1)

このように、一般式(1)で表される化合物を配合することにより、高い延伸倍率で延伸可能になる。
ここで、上記ポリアミド樹脂(XD系ポリアミド樹脂)および一般式(1)で表される化合物は、上述と同義であり、好ましい範囲も同様である。
本発明の延伸フィルムの製造方法では、まず、XD系ポリアミド樹脂100重量部に対し、一般式(1)で表される化合物を0.5~15重量部含むポリアミド樹脂組成物が溶融混練した状態で、Tダイ11からキャスティングロール12に押し出される。押出時の押出温度は、ポリアミド樹脂組成物が溶融している限り特に定めるものではない。溶融押出されたポリアミド樹脂組成物からなるポリアミド樹脂フィルムの厚さは、用途や延伸倍率にもよるが、一例として、延伸後の延伸フィルムの5~60倍の厚さであることが好ましく、5~40倍の厚さであることがより好ましく、10~30倍の厚さであることがさらに好ましく、15~28倍の厚さであることが特に好ましい。
本発明における延伸前のポリアミド樹脂フィルムの延伸応力は、例えば、0.24MPa以上とすることができ、さらには0.26MPa以上とすることもでき、よりさらには0.30MPa以上とすることもできる。延伸応力の上限については、特に定めるものではないが、一例として、5MPa以下とすることが挙げられ、さらには、4MPa以下とすることが挙げられ、特には1MPa以下としても十分に実用レベルである。本発明で用いるポリアミド樹脂フィルムは延伸応力を大きくできるため、高い延伸倍率で延伸可能になる。
延伸は、1方向のみに行ってもよいし(1軸延伸)、直交する2方向に行ってもよく(2軸延伸)、2軸延伸が好ましい。ポリアミド樹脂フィルムの搬送方向(Machine direction、「MD」ということがある)またはポリアミド樹脂フィルムの幅方向(Transverse Direction、「TD」ということがある)の1方向(より好ましくは、MD)、または、MDおよびTDの2方向に延伸することが好ましい。2軸延伸の場合、2方向の延伸は同時に行ってもよいし、逐次に行ってもよい。
MD延伸は、周速速度の異なるロール間を、ポリアミド樹脂フィルムを通過させて、延伸することができる。この場合、ポリアミド樹脂フィルムが後に通過する方が、周速速度が速くなるように設定される。また、テンターを用いて延伸することもできる。一方、TD延伸は、テンターを用いて延伸することができる。また、バッチ式の二軸延伸機を用いてもよい。
本発明における総合延伸倍率は、18.0倍以上であることが好ましく、19.0倍以上であることがより好ましく、20.0倍以上であることがさらに好ましく、22.0倍以上であることが一層好ましく、24.0倍以上であることがより一層好ましく、50倍以上とすることもできる。総合延伸倍率の上限については、特に定めるものではないが、例えば、100.0倍以下とでき、さらには40.0倍以下とすることができ、特には30.0倍以下とすることもできる。ここで、総合延伸倍率とは、延伸前のフィルムに対する、延伸した量の割合であり、下記式で表される値である。
総合延伸倍率=MD延伸倍率xTD延伸倍率
尚、XD系ポリアミド樹脂を2種以上含む場合、最も融点の低いXD系ポリアミド樹脂の融点を基準に、押出時のXD系ポリアミド樹脂の温度を設定するとよい。また、XD系ポリアミド樹脂が融点を2つ以上有する場合についても、最も低い融点を基準に温度を設定すると良い。
また、詳細を後述するとおり、本発明の延伸フィルムを、延伸フィルムと他の樹脂フィルムを含む積層フィルムとして用いる場合、他の樹脂フィルムと同時に延伸してもよい。
熱固定は、XD系ポリアミド樹脂の融点-70℃~融点未満で行うことが好ましく、XD系ポリアミド樹脂の融点-50℃~融点-5℃で行うことがより好ましく、XD系ポリアミド樹脂の融点-40℃~融点-10℃で行うことがさらに好ましい。
緩和は、例えば、延伸方向と逆方向にチャック間距離を戻すことによって行うことが好ましい。
緩和率は、ポリアミド樹脂フィルムを1軸延伸した場合、延伸方向に0.5~10%であることが好ましく、1~8%であることがより好ましく、1.5~6%であることがさらに好ましい。
緩和率は、ポリアミド樹脂フィルムを2軸延伸した場合、それぞれの延伸方向に0.5~10%であることが好ましく、1~8%であることがより好ましく、1.5~6%であることがさらに好ましい。
ここで、緩和率は、1軸延伸の場合、以下の通り計算される。
緩和率(%)=緩和量/延伸量x100
また、2軸延伸の場合、以下の通り計算される。
MD緩和率(%)=MD緩和量/MD延伸量x100
TD緩和率(%)=TD緩和量/TD延伸量x100
本発明の製造方法で得られる延伸フィルムの最終延伸倍率は、上述の延伸フィルムの最終延伸倍率となるように延伸量および緩和量を調整することが好ましい。
本発明の延伸フィルムは、そのまま用いてもよいが、延伸されたポリプロピレン樹脂フィルムを有する積層フィルムとして有益に用いられる。すなわち、ポリプロピレン樹脂フィルムは、延伸される場合、通常、高倍率に延伸される。ここで、図2に示すように、本発明の延伸フィルムと、ポリプロピレン樹脂フィルムを同時に延伸することが考えられる。ここで、従来、図2の(a’)~(c’)に示すように、ポリアミド樹脂フィルム23を2枚のポリプロピレン樹脂フィルム22で挟んで積層し(a’)、延伸すると(b’)、ポリアミド樹脂フィルム23が破断してしまうことがあり、高い延伸倍率での延伸が困難であった(c’)。これに対し、図2の(a)~(c)に示すように、本発明の延伸フィルムを用いると、延伸フィルム21とポリプロピレン樹脂フィルム22は、容易に同時に延伸できる。すなわち、本発明の好ましい実施形態として、本発明の延伸フィルムと、ポリプロピレン樹脂フィルムを同時に延伸することを含む、積層フィルムの製造方法が例示される。また、図2では、ポリプロピレン樹脂フィルムは、2層となっているが、1層のみであってもよいし、3層以上有する積層フィルムであってもよい。
また、本発明の延伸フィルムと延伸されたポリプロピレン樹脂フィルムを別々に準備し接着剤等で貼り合わせてもよい。このような場合でも、延伸フィルムと、延伸されたポリプロピレン樹脂フィルムを有する積層フィルムをさらに加工したりすると、延伸フィルムが追随せず、破断したりしていたが、本発明ではこのような問題を回避できる。
本発明では、延伸されたポリプロピレン樹脂フィルムは、1軸延伸されていても、2軸延伸されていてもよいが、2軸延伸されていることがより好ましい。
延伸されたポリプロピレン樹脂の延伸倍率は、上記本発明の延伸フィルムの製造方法のところで述べた延伸倍率と同様の範囲が好ましい。
本発明の延伸フィルムは、自動車等輸送機部品、一般機械部品、精密機械部品、電子・電気機器部品、OA機器部品、建材・住設関連部品、医療装置、レジャースポーツ用品、遊戯具、医療品、食品包装用フィルム等の日用品、防衛および航空宇宙製品等に広く用いられる。
<<ポリアミド樹脂MXD6の合成>>
アジピン酸8.9kgに次亜リン酸ナトリウム一水和物0.3gおよび酢酸ナトリウム0.1gを加え、反応缶内で0.1MPaAにおいて170℃にて加熱し溶融した後、内容物を撹拌しながら、メタキシリレンジアミン8.3kgを2時間かけて徐々に滴下し、温度を250℃まで上昇させた。温度上昇後、1時間かけて圧力を0.08MPaAまで緩やかに低下させ、0.5時間保持した。反応終了後、内容物をストランド状に取り出し、ペレタイザーにてペレット化して、15kgのペレットを得た。得られたペレットを熱媒加熱の外套を有するタンブラー(回転式の真空槽)に仕込み、減圧状態(0.5~10Torr)において200℃で1時間加熱を続けることで、得られたペレットの固相重合を行い、ポリアミド樹脂(MXD6、融点:237℃、相対粘度:2.65、水分率:0.05%)を得た。
セバシン酸10.1kgに次亜リン酸ナトリウム一水和物0.3gおよび酢酸ナトリウム0.1gを加え、反応缶内で0.1MPaAにおいて170℃にて加熱し溶融した後、内容物を撹拌しながら、メタキシリレンジアミンとパラキシリレンジアミンの混合ジアミン(メタキシリレンジアミン/パラキシリレンジアミン=70/30 (重量%))6.7kgを2時間かけて徐々に滴下し、温度を250℃まで上昇させた。温度上昇後、1時間かけて圧力を0.08MPaAまで緩やかに低下させ、0.5時間保持した。反応終了後、内容物をストランド状に取り出し、ペレタイザーにてペレット化して、15kgのペレットを得た。得られたペレットを熱媒加熱の外套を有するタンブラー(回転式の真空槽)に仕込み、減圧状態(0.5~10Torr)において195℃で1時間加熱を続けることで、得られたペレットの固相重合を行い、ポリアミド樹脂(MP10、融点:213℃、相対粘度:2.60、水分率:0.03%)を得た。
PA6:UBEナイロン 1024B(宇部興産株式会社製)
<<可塑剤>>
HD-PB:p-ヒドロキシ安息香酸ヘキシルデシル、花王株式会社製、エキセパール HD-PB
EH-PB:p-ヒドロキシ安息香酸エチルヘキシル、東京化成工業株式会社より入手
EH-OB:o-ヒドロキシ安息香酸エチルヘキシル、東京化成工業株式会社より入手
BBSA:N-ブチルベンゼンスルホンアミド、大八化学工業株式会社製、BM-4
<延伸フィルムの製造方法>
表1に示すポリアミド樹脂100重量部に対し、表1に示す可塑剤を、表1に示す割合(単位:重量部)で配合したポリアミド樹脂組成物を、ダイから溶融押出した。具体的には、各成分を溶融混練したポリアミド樹脂組成物を、表1に示す厚みで、130mm幅に押し出し、90mm角にカットした。その後、二軸延伸装置(テンター法、EX105S、株式会社東洋精機製作所製)を用いて、表1に示す延伸倍率となるように、MD方向およびTD方向にそれぞれ延伸を行った後、表1に示す熱工程温度で熱固定しながら、表1に示す緩和率となるように緩和して延伸フィルムを得た。
延伸前のポリアミド樹脂組成物からなるフィルム(延伸前フィルム)および延伸フィルムについて、以下の通り、各種特性の評価を行った。結果を下記表1に示す。
PCリンク型高機能レコーダ(THERMO PRO GR3000、株式会社キーエンス製)に延伸時に掛かる応力を0.1秒間隔で取り込み、最大応力を延伸応力とした。
島津製作所(SHIMADZU CORPORATION)製、DSC-60を用い、試料(延伸前フィルム)は5mgとし、雰囲気ガスとしては窒素を30ml/分で流し、昇温速度は10℃/分の条件で室温(25℃)から予想される融点以上の温度まで加熱した際に観測される発熱ピークのピークトップの温度を延伸前フィルムの昇温時の結晶化温度とした。
各実施例および比較例の延伸前フィルムの昇温時の結晶化温度と、各実施例、比較例において、それぞれ、可塑剤を除いて他は同様に行った延伸前フィルムの昇温時の結晶化温度の差を算出した。
島津製作所(SHIMADZU CORPORATION)製、DSC-60を用い、試料(延伸前フィルム)は5mgとし、雰囲気ガスとしては窒素を30ml/分で流し、昇温速度は10℃/分の条件で室温(25℃)から予想される融点以上の温度まで加熱し溶融させ次いで、溶融した試料を、ドライアイスで急冷し、10℃/分の速度で融点以上の温度まで再度昇温した際に観測される吸熱ピークのピークトップの温度を延伸前フィルムの融点とした。
延伸フィルム0.5gを秤量し、濃硫酸を20ml加え、ヒーター上で湿式分解した。冷却後、過酸化水素5mlを加え、ヒーター上で加熱し、全量が2~3mlになるまで濃縮した。再び冷却し、純水で500mlとした。Thermo Jarrell Ash社製 IRIS/IPを用いて、高周波誘導結合プラズマ(ICP)発光分析により、波長213.618nmにて定量した。
JIS K 7127に従い、10mm幅の短冊を用い、50mm/minの試験速度で測定した。測定に際し、延伸フィルムのMD方向に引張り試験を行い、チャック間距離を50mmとした。
延伸フィルムの酸素透過係数は、酸素透過率測定装置(MOCON社製、型式:OX-TRAN2/21)を使用し、ASTM D3985に準じて、23℃、相対湿度60%の雰囲気下にて測定した。測定値が低いほど酸素バリア性が良好であることを示す。なお、参考例1~3については、未延伸のポリアミド樹脂フィルムについて、同様に酸素透過係数を測定した。
実施例1において、表1または2に示す通り、原料および製造条件を変更し、他は同様に行った。参考例は、延伸を行っていない例である。


一方、参考例1~5に示すように、ポリアミド樹脂フィルムを延伸しない場合、一般式(1)で表される化合物を配合すると、配合量に準じて、酸素透過係数が上昇してしまった。すなわち、酸素バリア性が劣ってしまった。一般に、可塑剤を配合した場合には、酸素バリア性は低下する傾向にあり、参考例1~5の酸素バリア性も一般的な傾向の通りであった。これに対し、実施例3と比較例1の比較から顕著な通り、一般式(1)で表される化合物を特定の配合量で配合したポリアミド樹脂フィルムを延伸した場合、酸素透過係数が低下した。すなわち、酸素バリア性が向上することが分かった。一般式(1)で表される化合物を添加することによる酸素バリア性低下を上回るほどの、延伸倍率の向上による酸素バリア性の向上が見られたことは、極めて驚くべきことである。
単軸押出機にて240℃で溶融したポリプロピレン(ノバテックPP FL203D、日本ポリプロ株式会社製)、単軸押出機にて240℃で溶融したアドマーQF580(三井化学株式会社製、PPベースの接着剤)、単軸押出機にて260℃で溶融したMXD6を共押出多層フィルム製造装置(フィードブロックおよびTダイ温度:260℃)に供給して溶融押出を行って、多層フィルムを得た。フィルムの層構成は、PP/接着剤/MXD6/接着剤/PP = 390/10/100/10/390 (μm)とした。
得られたフィルムを表3に記載の条件に従って延伸した。その結果、中心層のMXD6フィルムがPPフィルムに追随して延伸可能であることが分かった。特に、MD方向およびTD方向に5.0倍ずつ延伸できた点で意義がある。

12 キャスティングロール
13 延伸・緩和ゾーン
14 巻き取り工程
21 延伸フィルム
22 ポリプロピレン樹脂フィルム
23 従来のポリアミド樹脂フィルム
Claims (15)
- ポリアミド樹脂100重量部に対し、一般式(1)で表される化合物を0.5~15重量部含み、
前記ポリアミド樹脂が、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、
前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来する、延伸フィルム;
一般式(1)

- 前記キシリレンジアミンが、30~100モル%のメタキシリレンジアミンと0~70モル%のパラキシリレンジアミンからなる、請求項1に記載の延伸フィルム。
- 前記ジカルボン酸由来の構成単位の50モル%以上が、セバシン酸およびアジピン酸の少なくとも1種に由来する、請求項1または2に記載の延伸フィルム。
- 前記ジカルボン酸由来の構成単位の50モル%以上が、アジピン酸に由来する、請求項1~3のいずれか1項に記載の延伸フィルム。
- 前記延伸フィルム中のリン原子濃度が0.1~10ppmである、請求項1~4のいずれか1項に記載の延伸フィルム。
- 前記一般式(1)で表される化合物において、R1が、炭素数3~7のアルキル基であり、R2が、炭素数5~9のアルキル基である、請求項1~5のいずれか1項に記載の延伸フィルム。
- 最終延伸倍率が、20.0倍以上である、請求項1~6のいずれか1項に記載の延伸フィルム。
- JIS K 7127に従って測定した引張弾性率が、2.0GPa以上である、請求項1~7のいずれか1項に記載の延伸フィルム。
- 前記延伸フィルムの厚さが、1~100μmである、請求項1~8のいずれか1項に記載の延伸フィルム。
- ポリアミド樹脂100重量部に対し、一般式(1)で表される化合物を0.5~15重量部含み、
前記ポリアミド樹脂が、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、
前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来するポリアミド樹脂組成物からなるフィルムを、延伸することを含む、延伸フィルムの製造方法;
一般式(1)

- 前記延伸を、直交する2方向に行う、請求項10に記載の延伸フィルムの製造方法。
- 前記延伸を、直交する2方向に、それぞれ、4.2倍以上となるように行う、請求項10に記載の延伸フィルムの製造方法。
- 前記延伸温度が、前記ポリアミド樹脂の(融点-200℃)以上融点未満である、請求項10~12のいずれか1項に記載の延伸フィルムの製造方法。
- 前記延伸を、総合延伸倍率が、18.0倍以上となるように行う、請求項10~13のいずれか1項に記載の延伸フィルムの製造方法。
- ポリアミド樹脂100重量部に対し、一般式(1)で表される化合物を0.5~15重量部含み、
前記ポリアミド樹脂が、ジアミン由来の構成単位とジカルボン酸由来の構成単位から構成され、
前記ジアミン由来の構成単位の50モル%以上がキシリレンジアミンに由来し、前記ジカルボン酸由来の構成単位の50モル%以上が、炭素原子数4~20のα,ω-直鎖脂肪族ジカルボン酸に由来する、延伸フィルム製造用のポリアミド樹脂組成物;
一般式(1)

Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2018105596A RU2018105596A (ru) | 2015-07-16 | 2016-07-07 | Растянутая пленка, способ изготовления растянутой пленки и композиция полиамидной смолы |
| EP16824370.7A EP3323856B1 (en) | 2015-07-16 | 2016-07-07 | Stretched film, method for manufacturing stretched film, and polyamide resin composition |
| US15/743,973 US10662303B2 (en) | 2015-07-16 | 2016-07-07 | Stretched film, method for manufacturing stretched film, and, polyamide resin composition |
| CA2969826A CA2969826C (en) | 2015-07-16 | 2016-07-07 | Polyamide composition for manufacturing stretched films |
| KR1020177018152A KR101775422B1 (ko) | 2015-07-16 | 2016-07-07 | 연신필름, 연신필름의 제조방법, 및, 폴리아미드 수지 조성물 |
| JP2016569867A JP6108055B1 (ja) | 2015-07-16 | 2016-07-07 | 延伸フィルム、延伸フィルムの製造方法、および、ポリアミド樹脂組成物 |
| CN201680040902.5A CN107849350B (zh) | 2015-07-16 | 2016-07-07 | 拉伸薄膜、拉伸薄膜的制造方法和聚酰胺树脂组合物 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015141872 | 2015-07-16 | ||
| JP2015-141872 | 2015-07-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017010390A1 true WO2017010390A1 (ja) | 2017-01-19 |
Family
ID=57756963
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/070081 WO2017010390A1 (ja) | 2015-07-16 | 2016-07-07 | 延伸フィルム、延伸フィルムの製造方法、および、ポリアミド樹脂組成物 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US10662303B2 (ja) |
| EP (1) | EP3323856B1 (ja) |
| JP (1) | JP6108055B1 (ja) |
| KR (1) | KR101775422B1 (ja) |
| CN (1) | CN107849350B (ja) |
| CA (1) | CA2969826C (ja) |
| RU (1) | RU2018105596A (ja) |
| TW (1) | TWI593752B (ja) |
| WO (1) | WO2017010390A1 (ja) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018092623A1 (ja) * | 2016-11-18 | 2018-05-24 | 三菱瓦斯化学株式会社 | 樹脂組成物、成形品および成形品の製造方法 |
| JP2018087319A (ja) * | 2016-11-18 | 2018-06-07 | 三菱瓦斯化学株式会社 | 樹脂組成物、成形品および成形品の製造方法 |
| WO2019026499A1 (ja) | 2017-07-31 | 2019-02-07 | 三菱瓦斯化学株式会社 | 易裂性フィルム、多層フィルム、包装材料および容器 |
| WO2019026500A1 (ja) | 2017-07-31 | 2019-02-07 | 三菱瓦斯化学株式会社 | 易裂性フィルム、多層フィルム、包装材料および容器 |
| WO2019208687A1 (ja) | 2018-04-26 | 2019-10-31 | 三菱瓦斯化学株式会社 | 延伸フィルム、包装材料および延伸フィルムの製造方法 |
| WO2023013253A1 (ja) | 2021-08-02 | 2023-02-09 | 三菱瓦斯化学株式会社 | 多層フィルムおよび多層フィルムの製造方法 |
| WO2023021807A1 (ja) | 2021-08-17 | 2023-02-23 | 三菱瓦斯化学株式会社 | 多層体 |
| WO2023026593A1 (ja) | 2021-08-23 | 2023-03-02 | 三菱瓦斯化学株式会社 | 延伸フィルム、多層フィルムおよび包装材料 |
| WO2023032408A1 (ja) * | 2021-09-03 | 2023-03-09 | 三菱瓦斯化学株式会社 | 樹脂組成物および成形品 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7342439B2 (ja) | 2019-06-11 | 2023-09-12 | 三菱瓦斯化学株式会社 | 延伸フィルムおよび多層体 |
| JP7133528B2 (ja) * | 2019-10-03 | 2022-09-08 | 芝浦機械株式会社 | 延伸装置のクリップ移動速度計測装置及びクリップ移動速度計測方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0711131A (ja) * | 1993-06-25 | 1995-01-13 | Kao Corp | ポリアミド樹脂組成物 |
| JP2002309082A (ja) * | 2001-04-12 | 2002-10-23 | Mitsubishi Engineering Plastics Corp | 難燃ポリアミド樹脂組成物及び押出成形品 |
| JP2003160728A (ja) * | 2001-11-28 | 2003-06-06 | Mitsubishi Engineering Plastics Corp | ポリアミド樹脂組成物及びシート状押出成形品 |
| WO2006049281A1 (ja) * | 2004-11-08 | 2006-05-11 | Mitsubishi Gas Chemical Company, Inc. | 芳香族ポリアミド延伸フィルム |
| JP2006321193A (ja) * | 2005-05-20 | 2006-11-30 | Toyobo Co Ltd | ガスバリア性ポリアミドフィルムおよびその製造方法 |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2450788A1 (de) | 1974-10-25 | 1976-05-06 | Basf Ag | Verfahren zur herstellung von polyamid-folien |
| JPH07157651A (ja) | 1993-12-08 | 1995-06-20 | Sumitomo Chem Co Ltd | 熱可塑性樹脂組成物 |
| JP4432174B2 (ja) * | 1999-11-29 | 2010-03-17 | 三菱化学株式会社 | ポリアミド系樹脂及びその製造方法、並びに、その樹脂組成物及びそれからなる包装用フィルム |
| JP2001279091A (ja) * | 2000-03-31 | 2001-10-10 | Asahi Kasei Corp | 難燃性強化ポリアミド組成物 |
| US6740275B2 (en) | 2000-09-04 | 2004-05-25 | Mitsubishi Engineering-Plastics Corporation | Flame-retardant polyamide-based protective sheet |
| DE60308581T2 (de) * | 2002-04-03 | 2007-01-18 | Mitsubishi Gas Chemical Co., Inc. | Biaxial gereckte Folie und Verfahren zu deren Herstellung |
| CN1169880C (zh) * | 2002-09-19 | 2004-10-06 | 上海交通大学 | 高阻隔性尼龙复合材料的制备方法 |
| RU2301816C2 (ru) | 2003-03-06 | 2007-06-27 | Эксонмобил Кемикал Пэйтентс, Инк. | Ориентированная термопластичная эластомерная пленка и способ ее получения |
| FR2858626B1 (fr) | 2003-08-05 | 2005-10-07 | Atofina | Polyamides semi aromatiques souple a faible reprise en humidite |
| BRPI0510314B1 (pt) | 2004-04-27 | 2016-10-18 | Kuraray Co | estrutura em multicamadas |
| DE102004045775B4 (de) * | 2004-09-21 | 2009-01-08 | Ems-Chemie Ag | Verwendung von stabilisierten, thermoplastischen Polyamid-Formmassen als Beschichtung von Lichtwellenleitern |
| JP5024550B2 (ja) | 2008-03-28 | 2012-09-12 | 宇部興産株式会社 | 積層体 |
| CA2718675C (en) | 2008-04-16 | 2016-03-22 | Unitika Ltd. | Biaxially stretched polyamide resin film, and process for production thereof |
| US20110139258A1 (en) | 2009-12-16 | 2011-06-16 | E.I. Du Pont De Nemours And Company | Multilayer structures comprising a barrier layer and their use to convey fluids |
| WO2012077473A1 (ja) * | 2010-12-07 | 2012-06-14 | 三菱瓦斯化学株式会社 | ポリアミド樹脂フィルム及びその製造方法 |
| KR101953421B1 (ko) | 2011-10-21 | 2019-02-28 | 우베 고산 가부시키가이샤 | 폴리아미드 수지 조성물 및 그것을 포함하는 중공 성형체 |
-
2016
- 2016-07-07 JP JP2016569867A patent/JP6108055B1/ja active Active
- 2016-07-07 KR KR1020177018152A patent/KR101775422B1/ko active IP Right Grant
- 2016-07-07 EP EP16824370.7A patent/EP3323856B1/en active Active
- 2016-07-07 US US15/743,973 patent/US10662303B2/en active Active
- 2016-07-07 WO PCT/JP2016/070081 patent/WO2017010390A1/ja active Application Filing
- 2016-07-07 RU RU2018105596A patent/RU2018105596A/ru not_active Application Discontinuation
- 2016-07-07 CN CN201680040902.5A patent/CN107849350B/zh active Active
- 2016-07-07 CA CA2969826A patent/CA2969826C/en active Active
- 2016-07-15 TW TW105122327A patent/TWI593752B/zh active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0711131A (ja) * | 1993-06-25 | 1995-01-13 | Kao Corp | ポリアミド樹脂組成物 |
| JP2002309082A (ja) * | 2001-04-12 | 2002-10-23 | Mitsubishi Engineering Plastics Corp | 難燃ポリアミド樹脂組成物及び押出成形品 |
| JP2003160728A (ja) * | 2001-11-28 | 2003-06-06 | Mitsubishi Engineering Plastics Corp | ポリアミド樹脂組成物及びシート状押出成形品 |
| WO2006049281A1 (ja) * | 2004-11-08 | 2006-05-11 | Mitsubishi Gas Chemical Company, Inc. | 芳香族ポリアミド延伸フィルム |
| JP2006321193A (ja) * | 2005-05-20 | 2006-11-30 | Toyobo Co Ltd | ガスバリア性ポリアミドフィルムおよびその製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3323856A4 * |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018092623A1 (ja) * | 2016-11-18 | 2018-05-24 | 三菱瓦斯化学株式会社 | 樹脂組成物、成形品および成形品の製造方法 |
| JP2018087319A (ja) * | 2016-11-18 | 2018-06-07 | 三菱瓦斯化学株式会社 | 樹脂組成物、成形品および成形品の製造方法 |
| US11034836B2 (en) | 2016-11-18 | 2021-06-15 | Mitsubishi Gas Chemical Company, Inc. | Resin composition, molded article and method for manufacturing molded article |
| WO2019026499A1 (ja) | 2017-07-31 | 2019-02-07 | 三菱瓦斯化学株式会社 | 易裂性フィルム、多層フィルム、包装材料および容器 |
| WO2019026500A1 (ja) | 2017-07-31 | 2019-02-07 | 三菱瓦斯化学株式会社 | 易裂性フィルム、多層フィルム、包装材料および容器 |
| WO2019208687A1 (ja) | 2018-04-26 | 2019-10-31 | 三菱瓦斯化学株式会社 | 延伸フィルム、包装材料および延伸フィルムの製造方法 |
| EP3786217A4 (en) * | 2018-04-26 | 2021-06-02 | Mitsubishi Gas Chemical Company, Inc. | STRETCHED FILM, PACKAGING MATERIAL AND PROCESS FOR THE PRODUCTION OF STRETCHED FILM |
| WO2023013253A1 (ja) | 2021-08-02 | 2023-02-09 | 三菱瓦斯化学株式会社 | 多層フィルムおよび多層フィルムの製造方法 |
| WO2023021807A1 (ja) | 2021-08-17 | 2023-02-23 | 三菱瓦斯化学株式会社 | 多層体 |
| WO2023026593A1 (ja) | 2021-08-23 | 2023-03-02 | 三菱瓦斯化学株式会社 | 延伸フィルム、多層フィルムおよび包装材料 |
| WO2023032408A1 (ja) * | 2021-09-03 | 2023-03-09 | 三菱瓦斯化学株式会社 | 樹脂組成物および成形品 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20180201744A1 (en) | 2018-07-19 |
| RU2018105596A (ru) | 2019-08-16 |
| CN107849350A (zh) | 2018-03-27 |
| US10662303B2 (en) | 2020-05-26 |
| CA2969826C (en) | 2018-07-24 |
| EP3323856A4 (en) | 2019-03-20 |
| TW201708392A (zh) | 2017-03-01 |
| TWI593752B (zh) | 2017-08-01 |
| CN107849350B (zh) | 2020-11-20 |
| JPWO2017010390A1 (ja) | 2017-07-13 |
| RU2018105596A3 (ja) | 2019-08-16 |
| KR20170081726A (ko) | 2017-07-12 |
| CA2969826A1 (en) | 2017-01-19 |
| KR101775422B1 (ko) | 2017-09-06 |
| EP3323856A1 (en) | 2018-05-23 |
| EP3323856B1 (en) | 2020-11-04 |
| JP6108055B1 (ja) | 2017-04-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6108055B1 (ja) | 延伸フィルム、延伸フィルムの製造方法、および、ポリアミド樹脂組成物 | |
| KR101315921B1 (ko) | 폴리아미드 수지계 복합재 및 그 제조 방법 | |
| KR102186748B1 (ko) | Me-bht를 포함하는 폴리아미드, 이와 같은 폴리아미드를 포함하는 조성물, 이와 같은 폴리아미드 또는 이와 같은 조성물을 포함하는 성형품 | |
| KR20130140541A (ko) | 폴리아미드 수지 조성물 | |
| JP5729189B2 (ja) | ポリアミド樹脂組成物 | |
| JP2011102360A (ja) | 繊維強化ポリアミド樹脂組成物 | |
| JP6468030B2 (ja) | 複合フィルムの製造方法および複合材料 | |
| JP2016216661A (ja) | 複合シートの製造方法 | |
| JP5853446B2 (ja) | ポリアミド樹脂組成物 | |
| JP2018168383A (ja) | ポリアミド樹脂及びそれを含む成形品 | |
| JP5652025B2 (ja) | 複合材の製造方法および成形品 | |
| JP6922519B2 (ja) | 樹脂組成物、成形品、繊維およびフィルム | |
| WO2019208687A1 (ja) | 延伸フィルム、包装材料および延伸フィルムの製造方法 | |
| JP7342439B2 (ja) | 延伸フィルムおよび多層体 | |
| JP6597131B2 (ja) | 長繊維強化複合材料の製造方法および長繊維強化複合材料 | |
| JP2018145291A (ja) | 延伸成形体および延伸成形体の製造方法 | |
| TW202000730A (zh) | 聚醯胺樹脂、成形品、以及聚醯胺樹脂之製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2016569867 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16824370 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2969826 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 20177018152 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15743973 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016824370 Country of ref document: EP Ref document number: 2018105596 Country of ref document: RU |