WO2014115799A1 - 多能性幹細胞の継代培養方法 - Google Patents
多能性幹細胞の継代培養方法 Download PDFInfo
- Publication number
- WO2014115799A1 WO2014115799A1 PCT/JP2014/051362 JP2014051362W WO2014115799A1 WO 2014115799 A1 WO2014115799 A1 WO 2014115799A1 JP 2014051362 W JP2014051362 W JP 2014051362W WO 2014115799 A1 WO2014115799 A1 WO 2014115799A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cells
- cell
- microwell
- pluripotent stem
- culture
- Prior art date
Links
Images






















Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0607—Non-embryonic pluripotent stem cells, e.g. MASC
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12M—APPARATUS FOR ENZYMOLOGY OR MICROBIOLOGY; APPARATUS FOR CULTURING MICROORGANISMS FOR PRODUCING BIOMASS, FOR GROWING CELLS OR FOR OBTAINING FERMENTATION OR METABOLIC PRODUCTS, i.e. BIOREACTORS OR FERMENTERS
- C12M23/00—Constructional details, e.g. recesses, hinges
- C12M23/02—Form or structure of the vessel
- C12M23/12—Well or multiwell plates
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/0081—Purging biological preparations of unwanted cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0603—Embryonic cells ; Embryoid bodies
- C12N5/0606—Pluripotent embryonic cells, e.g. embryonic stem cells [ES]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0696—Artificially induced pluripotent stem cells, e.g. iPS
Definitions
- the present invention relates to a method for subculturing pluripotent stem cells. More specifically, the present invention relates to a simple and homogeneous method for subculturing pluripotent stem cells.
- the cells are detached from the adhesion surface of the culture container using an enzyme treatment or a cell scraper, and subcultured to a culture container containing a fresh medium. It is known that pluripotent stem cells are killed when dissociated into single cells, and it is indispensable to pass in the state of a cell cluster during passage. Therefore, in the passage of pluripotent stem cells, when detaching the cells from the culture vessel, the pluripotent stem cells are detached as colonies, crushed into a cell mass of an appropriate size by pipetting, etc., and then new Sowing in a dish.
- a method of seeding pluripotent stem cells into a single cell is known, but this method is a method for cloning pluripotent stem cells having a single property, called cell proliferation. From the viewpoint, the method is very inefficient, and is not a suitable method for subculture of pluripotent stem cells (Non-patent Document 1). Further, a method is known in which a single-cell pluripotent stem cell is aggregated to form a uniform-sized embryoid body (Embryonic Body) to improve differentiation induction efficiency (Non-patent Document 2). In the first place, such an embryoid body is produced in order to promote differentiation induction of pluripotent stem cells (Non-patent Documents 2 and 3). It is considered that pluripotent stem cells lose pluripotency once they start differentiation, and it is impossible to make pluripotent stem cells form embryoid bodies for the purpose of maintaining an undifferentiated state.
- An object of the present invention is to provide an efficient and homogeneous subculture method for maintenance culture of pluripotent stem cells.
- the present inventors subcultured pluripotent stem cells, even if the cell mass obtained by culturing is dispersed to a single cell level, the cell mass is rapidly reformed thereafter. (Ie, by forming a cell clump), cell death due to dispersion into a single cell can be prevented, and a certain size and shape can be achieved by devising a method for forming a cell clump. It has been found that a homogeneous cell clump having the same can be obtained.
- the present inventors also obtained a cell agglomeration in which cells were three-dimensionally aggregated, but when seeded in a dish, the cell agglomerates rapidly spread on the cell adhesion surface of the dish and proliferate well and uniformly.
- the present inventors have further found that the cell culture efficiency can be easily improved by adjusting the size of the cell clump. Furthermore, the present inventors have also found that the proportion of undifferentiated cells in the subcultured cells can be improved by classifying the cells dispersed to the single cell level based on the size.
- the inventors of the present invention inoculate the cell conglomerate formed in the well at a specific position in the culture container by dropping the container provided with the well onto the culture surface of the culture container by turning the container upside down. It was found that (precise sowing) can be performed.
- the present invention is based on such knowledge.
- a method for subculturing pluripotent stem cells (A) dispersing the cell mass of pluripotent stem cells during passage; (B) seeding cells obtained by dispersion in microwells; (C) forming a cell clump from cells seeded in a microwell; (D) seeding the obtained cell conglomerate on the culture surface of the culture vessel.
- the step (d) comprises the step (d ′) of reversing the container provided with the microwells so that the cell clump is dropped on the culture surface of the culture container.
- Method (3) The method according to (1) or (2) above, wherein the cell mass is dissociated into a cell mass composed of 1 to 100 cells in the step (a).
- step (9) The method according to (8) above, wherein the number of cells seeded in each microwell in step (b) is 55 to 220.
- step (10) The method according to any one of (1) to (9) above, wherein the step (c) comprises allowing the cells to stand in the microwell for a time sufficient to form a cell clump. .
- step (c) comprises allowing the cells to stand for 8 to 24 hours in a microwell.
- step (c) comprises allowing the cells to stand in a microwell for 8 to 12 hours.
- step (c) is performed without using centrifugation.
- the pluripotent stem cell is a human pluripotent stem cell.
- the human pluripotent stem cells are human ES cells or human iPS cells.
- the step (a ′) of removing differentiated cells comprises removing differentiated cells by classification.
- a closed culture vessel comprising a surface with a microwell and a culture surface, and arranged so that these two surfaces face each other.
- (28) A fully automated subculture system for pluripotent stem cells for carrying out the method according to any one of (1) to (22) and (27) above.
- the method of the present invention is advantageous in that homogeneous and pluripotent stem cell clumps can be obtained simply and rapidly, thereby enabling stable passage of pluripotent stem cells.
- the method of the present invention can also control the inoculation position of the cell agglomeration by dropping the cell agglomeration into the culture container by reversing the container equipped with the microwells, for example, allowing the cells to be uniformly inoculated. This is advantageous.
- the method of the present invention is further advantageous in that the proportion of undifferentiated cells can be increased because differentiated cells can be removed during passage.
- the pluripotent stem cell subculture method of the present invention is suitable for full automation, and can automate the whole process.
- FIG. 1 shows the state of cells (FIG. 1A) immediately after and 1 day after seeding human iPS cells dispersed to the single cell level in AggreWell 800 (FIG. 1A) and the resulting cell agglomeration (FIG. 1B). is there.
- FIG. 2 is a phase contrast microscopic image of cells after passage of cell clumps obtained with or without centrifugation. The dark part of the cell cluster or cell colony is the part where the cells are multi-layered.
- FIG. 3 is a diagram showing a growth curve after passage of the obtained cell conglomerate. Cell growth was determined by measuring the area (mm 2 ) occupied by cells on the dish.
- FIG. 4 is a diagram showing a fluorescent immunostaining image of colonies after passage.
- FIG. 5 is a phase-contrast microscope image obtained by observing the formation process of cell clumps on the AggreWell over time.
- FIG. 6 is a diagram showing a phase contrast microscopic image of the standing time on AggreWell and subsequent cell proliferation. In FIG. 6, the phase-contrast microscope image was acquired 48 hours after seed
- FIG. 7 is a diagram showing a phase contrast microscopic image of the standing time on AggreWell and subsequent cell growth. In FIG. 7, the phase-contrast microscope image was acquired 168 hours after seed
- FIG. 8 is a phase-contrast microscope image showing the state of cells 7 days after seeding when the cell aggregate formation conditions are changed.
- FIG. 8 is a phase-contrast microscope image showing the state of cells 7 days after seeding when the cell aggregate formation conditions are changed.
- FIG. 9 is a phase-contrast microscope image showing the state of cells 7 days after seeding when the conditions for forming cell clumps are changed.
- FIG. 10 is a phase-contrast microscope image showing the state of cells at the fifth passage when centrifugation is performed to form cell clumps.
- FIG. 10 shows the state of cell clump development and the state of proliferation from 2 to 7 days after seeding.
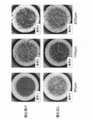
- FIG. 11 is a diagram showing colony survival rates when the conditions for forming cell clumps are changed.
- P1 to P5 indicate passage numbers. Specifically, P1 indicates data after the first passage, and P2 to P5 indicate data after the second to fifth passages, respectively.
- FIG. 12 is a diagram showing the ratio of multi-layered colonies when the conditions for forming cell clumps are changed.
- FIG. 13 is a graph showing the cell recovery rate when the cell aggregate formation conditions are changed.
- P1 to P5 indicate passage numbers.
- FIG. 14 is a diagram showing cell mortality when the conditions for forming cell clumps are changed.
- P1 to P5 indicate passage numbers.
- FIG. 15 is a diagram showing the relationship between the number of cells contained in one cell clump and the diameter of the cell clump.
- FIG. 16 is a diagram showing the relationship between the number of cells contained in one cell clump and the cell expansion rate after cell clump seeding.
- FIG. 12 is a diagram showing the ratio of multi-layered colonies when the conditions for forming cell clumps are changed.
- FIG. 13 is a graph showing the cell recovery rate when the cell aggregate formation conditions are changed.
- P1 to P5 indicate passage numbers.
- FIG. 14 is a diagram showing cell mortality when the conditions for forming
- FIG. 17 is a diagram showing the relationship between the number of cells contained per cell clump and the cell adhesion rate after cell clump seeding.
- FIG. 18 is a diagram showing the relationship between the number of cells contained in one cell aggregate and the number of cell aggregates obtained when cell aggregates of equal size are obtained using 10,000 cells. It is.
- FIG. 19 is a diagram showing the relationship between the number of cells contained in one cell clump and the amplification factor of cells per passage.
- FIG. 20 is a diagram showing the relationship between the number of cells contained in one cell clump and the number of days required for a colony derived from each cell clump to reach a diameter of 2 mm.
- FIG. 21 is a diagram showing the relationship between the number of cells contained in one cell clump and the amplification factor of cells per day.
- FIG. 22 is a diagram showing the relationship between the number of cells contained per cell cluster and the amplification factor of cells after 45.42 days.
- FIG. 23 is a diagram showing the difference between the diameter of cells obtained from good iPS cell colonies and the diameter of cells obtained from bad iPS cell colonies.
- FIG. 24 is a diagram showing the arrangement on the culture surface of the cell conglomerate dropped on the culture surface of the culture container.
- pluripotent stem cells used in the present invention include embryonic stem cells (ES cells), inducible pluripotent stem cells (iPS cells or induced pluripotent stem cells), Muse cells (Multilineage-differentiating Stress Enduring Cell), embryonic Examples include pluripotent stem cells such as tumor cells (EC cells) or embryonic germ stem cells (EG cells), preferably ES cells or iPS cells.
- the pluripotent stem cells used in the present invention are also preferably pluripotent stem cells of mammals such as primates or rodents, and more preferably human pluripotent stem cells.
- the pluripotent stem cell used in the present invention is most preferably a human ES cell or a human iPS cell.
- the method for subculturing pluripotent stem cells of the present invention comprises: (A) dispersing the cell mass of pluripotent stem cells during passage; (B) seeding cells obtained by dispersion in microwells; (C) forming a cell clump from cells seeded in microwells with or without centrifugation techniques; (D) seeding the obtained cell conglomerate on the culture surface of the culture vessel.
- the subculture of the pluripotent stem cells of the present invention is performed in an adhesion culture system.
- pluripotent stem cells can be maintained and cultured while maintaining a good undifferentiated state.
- the cell clump obtained by the subculture method of the pluripotent stem cell of the present invention has a loose bond between cells. Therefore, although cell death due to unicellularization can be avoided, the cell clump obtained by the method of the present invention rapidly develops on the cell adhesion surface of the culture vessel after passage because of the loose binding between the cells. Can do.
- the cell conglomerate obtained by the method of the present invention is also seeded at a controlled position in the culture container (hereinafter referred to as “ Sometimes referred to as precision sowing).
- a step of dispersing a cell mass of pluripotent stem cells at the time of passage detachment of cells from the cell adhesion surface
- pluripotent stem cells that are physiologically or physically detached from the cell adhesion surface can be used in the adhesion culture.
- an enzyme for detaching pluripotent stem cells from the cell adhesion surface an enzyme used in a conventional method can be used, and for example, enzymes such as trypsin, dispase, actase and collagenase can be used.
- the detachment of pluripotent stem cells from the cell adhesion surface may also be performed using a chemical substance having a cell detaching action, such as a chelating agent of divalent ions (particularly Mg 2+ ) such as ethylenediaminetetraacetic acid (EDTA), These may be performed in combination with the above enzyme.
- a chemical substance having a cell detaching action such as a chelating agent of divalent ions (particularly Mg 2+ ) such as ethylenediaminetetraacetic acid (EDTA), These may be performed in combination with the above enzyme.
- vibration such as high-frequency vibration may be applied to the cell adhesion surface, and / or a cell scraper may be used.
- the cells may be detached by combining the above physiological detachment method and physical detachment method. Those skilled in the art will be able to appropriately remove the pluripotent stem cells from the cell adhesion surface using the well-known peeling method as described above.
- the cell mass can be dissociated to the single cell level.
- the dissociation of the cell mass may be performed simultaneously with the detachment of the cell from the culture surface, or may be performed after the cell mass is detached.
- the exfoliated cell mass can be dispersed after being dissociated to a single cell level by, for example, pipetting water flow.
- “dispersed to a single cell level” means a cell mass such that the average number of cells contained in one cell mass is 1 to 100, preferably 1 to 10. Is dissociated and then dispersed, and includes completely dissociating into single cells and then dispersing. Therefore, in the present invention, the cell mass may be dissociated after being completely dissociated into single cells, or may be dispersed after being dissociated so that most of the cells become single cells.
- cell mass after dispersion is large, when the cell mass is seeded in the microwell, the number of cells seeded in each microwell tends to vary, and thus the cell mass after dispersion is preferably small.
- the cell mass separated from the culture surface may be dispersed to the single cell level by further processing using an enzyme.
- Enzymes that can be used to dissociate cells to the single cell level include those that can break cell-cell bonds and cell-extracellular matrix (ECM) bonds. Can be mentioned and these enzymes are well known to those skilled in the art. Dissociation of the cell mass using an enzyme or a water flow can be automated, and the process (a) can be automated.
- a compound that suppresses adverse effects eg, cell death
- Y-27632 and other ROCK inhibitors can be added.
- step (B) Step of seeding cells obtained by dispersion in microwells
- the cells or cell mass obtained by dispersion in step (a) (hereinafter sometimes simply referred to as “cells”) ) Can be successfully grown after passage after forming a cell clump. Formation of cell clumps can be performed by seeding the obtained cells in microwells.
- the cells obtained by dispersing in the step (a) naturally sink by gravity in the culture solution. Therefore, when cells are seeded in a well having a slope, the cells gather in the well by utilizing the slope of the well. Thereafter, the cells form cell clumps with adjacent cells to form cell clumps.
- the well has a shape that gathers when the cells sink, for example, a shape that becomes smaller as the inner circumference approaches the bottom surface. That is, the well preferably has a shape with a concave bottom, for example, a conical shape, a round bottom, a V bottom, a U bottom, and a chamfered flat bottom (having a bottom surface but with a corner and a concave bottom. It is preferable to have a shape of a shape.
- the shape of the upper opening of each well can be appropriately selected in consideration of ease of processing and the shape in which a large number of wells can be arranged. For example, the shape of a polygon such as a triangle, a quadrangle, or a hexagon Or it can have a circular shape.
- the cell agglomerates to be prepared have a certain size.
- the size of the cell clump to be prepared is determined by the number of cells seeded in each microwell, in step (b), the number of cells seeded in each microwell is made uniform. Preferably it is.
- the fixed amount means that the average number of cells seeded in each microwell in step (b) is, for example, 10 to 3,500 (that is, the average diameter of the formed cell clump is 35 to 350 ⁇ m).
- the average diameter of the formed cell clumps is from 50 to 200 ⁇ m), more preferably from 40 to 500 (ie the average of the formed cell clumps) It means that the diameter is 60 to 160 ⁇ m), more preferably 55 to 220 (that is, the average diameter of the formed cell clumps is 69 to 115 ⁇ m).
- the cells are sufficiently suspended in the suspension. Sowing.
- a well for forming a cell agglomeration is called a “microwell”.
- the “microwell” is an upper opening having a side or a diameter of 1 mm or more. It is not a term intended to exclude wells having a section, and “microwell” includes a well having a top opening with a side or diameter of 1 mm or more.
- the size of the upper opening of the microwell can be determined by the size of the cell clump to be formed.
- the area of the microwell is equivalent to a circle having a diameter of 100 ⁇ m to 3 mm, a diameter of 200 ⁇ m to 800 ⁇ m, or a diameter of 400 ⁇ m to 600 ⁇ m. It can be a well with an upper opening.
- a large number of microwells are arranged on the bottom surface of the container, and there are no gaps (no flat portion between adjacent wells). Or the like so that the gap between wells is minimized.
- the arrangement of the microwells on the bottom surface of the container will be described in more detail in the item of step (d ′) relating to precision seeding.
- the shape of the microwell of the container is uniform from the viewpoint of uniforming the size of the cell clump to be formed.
- the cells obtained by dispersing in the step (a) are preferably prepared using a multiwell plate in which a large number of uniform-shaped microwells are arranged on the bottom surface.
- a multiwell plate is not particularly limited.
- AggreWell trademark
- the step (b) can be performed on the surface provided with the microwell using the closed culture vessel of the present invention described later.
- the surface of the microwell can be coated with a non-adhesive coating for cells.
- the number of cells seeded in the microwell can be adjusted as appropriate.
- seeding of the cells in the microwell can be performed uniformly by sufficiently suspending the cells. Since these operations can be automated, the step of seeding a certain amount of cells in a microwell in step (b) can also be fully automated.
- Step of forming a cell clump from cells seeded in the microwell the cells seeded in the microwell are allowed to stand as they are, so that they are placed on the bottom of the microwell by gravity. The cells gather and adhere to each other to form a cell clump. The cells may be collected at the bottom of the microwell by centrifuging a container with the microwell using a centrifugation technique.
- centrifugation at 400 g to 3000 g for 1 minute to 10 minutes can be performed. In this way, cells can be effectively collected on the bottom surface of the microwell.
- cells can be densely aggregated when a cell clump is formed using a centrifugation technique, but in the present invention, it is not always necessary to use a centrifugation technique. That is, after seeding cells dispersed to the level of a single cell in a microwell, without using a centrifugation method, for example, by simply allowing the cells to stand in a microwell, the cells become pluripotent stem cells within a few hours. Cell clumps can be formed. Thus, in the present invention, cell clumps can be formed without using a centrifugation method.
- cell clumps can be formed in pluripotent stem cells by allowing them to stand in microwells.
- the standing time may be longer than the time necessary for the pluripotent stem cells to form a cell clump, for example, 8 hours or longer.
- the standing time can be, for example, 8 hours to 24 hours, preferably 8 hours to 16 hours, and more preferably 8 hours to 12 hours.
- the shorter the standing time the looser the bonds between the cells in the cell clumps, so that the cell clumps are more fragile, while the cell clumps develop quickly after seeding in the culture vessel. is there.
- the cell agglomerates prepared in the step (c) of the present invention have a uniform size.
- the average number of cells contained in each cell clump is, for example, 10 to 3,500 (that is, the average diameter of the cell clump is 35 to 350 ⁇ m), preferably Is 25 to 870 (that is, the average diameter of the cell clump is 50 to 200 ⁇ m), more preferably 40 to 500 (that is, the average diameter of the cell clump is 60 to 160 ⁇ m), and even more preferably Is 55 to 220 (that is, the average diameter of the cell clump is 69 to 115 ⁇ m), and the average diameter of the cell clump can be appropriately adjusted according to the size of the microwell and the number of cells to be seeded.
- a microwell having a size larger than the size of the cell clump to be produced can be selected.
- a cell clump having an average diameter of 50 ⁇ m using human iPS cells 24 wells in which 1200 microwells per well (for example, the microwell size is 400 ⁇ m ⁇ 400 ⁇ m) are engraved. Plates (2 cm 2 / well) can be seeded with 2.8 ⁇ 10 4 cells / well.
- 1.8 ⁇ 10 5 cells per 24-well plate (2 cm 2 / well) engraved with 1200 microwells per well. / Well cells can be seeded.
- the cell aggregates of human iPS cells with diameters of 50 ⁇ m, 100 ⁇ m and 200 ⁇ m are calculated to include about 23, about 151 and about 981 cells, respectively.
- a person skilled in the art will be able to calculate the required number of cells according to the diameter of the cell clump to be produced and obtain a cell clump having a desired size.
- step (c) can be fully automated.
- the cell agglomerate formed in the container provided with the microwell by the step (c) of seeding the obtained cell agglomeration on the culture surface of the culture container is then transferred to the culture container (that is, Culture surface).
- This step (d) can be performed by collecting the cell clumps, suspending them in a medium, and seeding them on the culture surface of the culture vessel, and this step (d) can be fully automated.
- the seeded cell cluster can be rapidly developed after seeding, and then cultured well in a state where pluripotency is maintained. From the viewpoint of uniformly seeding the cell clumps on the culture surface of the culture vessel, it is preferable to seed the cell suspension containing the cell clumps after sufficient suspension.
- step (d) cell seeding can be precisely seeded in step (d).
- the step (d ′) is carried out by reversing the container provided with the microwells so that the cell agglomeration is dropped on the culture surface of the culture vessel. Precise seeding can be performed on the culture surface of the culture vessel.
- the container equipped with microwells in the step (d ′) is turned upside down, the cell conglomerate falls from the microwells almost vertically onto the culture surface of the culture vessel. So that it is seeded on the culture surface.
- the arrangement of the microwells is designed based on how the cell clumps are to be dropped (similarly, the arrangement of the microwells in the container having the microwells used in the step (b))
- Cell clumps can be seeded (precision seeding) in a controlled arrangement.
- the cell clump can be precisely seeded on the culture surface of the culture vessel. Precision seeding can be used, for example, to uniformly seed cell clumps on the culture surface of a culture vessel, whereby the cells can be uniformly grown on the culture surface.
- the microwells are preferably arranged in a honeycomb shape, for example (the arrangement of the microwells in the vessel equipped with the microwells used in the step (b) is also possible. The same). By doing in this way, the useless space between colonies formed by pluripotent stem cells can be reduced, and the culture surface can be used efficiently.
- the microwells are arranged in a honeycomb shape means that a plurality of microwell rows extending in one predetermined direction are formed, and each microwell row is a plurality of microwells continuously arranged in the one direction. It means that microwells including wells and included in a certain microwell array are alternately arranged with respect to microwells included in an adjacent microwell array.
- the step (d) includes the step (d ′) including the step of dropping the cell conglomerate onto the culture surface of the culture vessel by reversing the vessel equipped with the microwells upside down.
- the sedimentation rate of the cell agglomeration due to gravity is not so fast, so even if the microwell and the cell agglomeration are not adhered, if the container equipped with the microwell is inverted upside down, the cells will be relatively good.
- Agglomerates can be aligned on the culture surface of the culture vessel.
- the adhesion between the microwell and the cell clump can be dissociated using impact, liquid flow and vibration (for example, low frequency vibration or high frequency vibration). it can.
- the top-bottom reversal of the culture vessel comprises a closed-system culture vessel, for example a closed-system culture comprising a surface with a microwell and a culture surface, the two surfaces being arranged facing each other. This can be done easily using a container.
- the present invention provides a closed culture vessel comprising a surface having a microwell (preferably a plurality of aligned microwells) and a culture surface, and these two surfaces are arranged to face each other.
- seeding of the cells dispersed in step (a) into the microwell is performed by, for example, using a sufficiently suspended cell suspension. It can be performed by pouring into a container and then leaving the surface with the microwells on the ground side. Moreover, what is necessary is just to reverse the whole container upside down for the upside down reversal of the container.
- the process (d ') can also be fully automated. From the viewpoint of facilitating full automation of the process, as described above, it is preferable to use the closed culture vessel of the present invention in the process (d ′).
- the method for subculturing pluripotent stem cells of the present invention can be performed by steps (a) to (d). As mentioned above, all of these steps can be automated.
- the method for subculturing pluripotent stem cells of the present invention may include (a ′) a step of removing differentiated cells between steps (a) and (b).
- Step (A ′) Step of removing differentiated cells
- Step (a ′) is a step relating to the separation and removal of differentiated cells, and is introduced into the subculture of pluripotent stem cells for the first time in the present invention that can disperse pluripotent stem cells into single cells. This is a process that has become possible. That is, the step (a ′) is a step based on the premise that the pluripotent stem cells are dispersed into single cells in the step (a).
- the inventor of the present invention revealed that cells separated into single cells can be classified into undifferentiated cells and cells that have started differentiation based on the size, according to the examples described later. Specifically, in human iPS cells, undifferentiated cells were distributed in a size of about 14 to 20 ⁇ m centering on a diameter of 17 ⁇ m, but cells that started differentiation were mainly distributed in a diameter of 23 ⁇ m or more. Based on this finding, if the step (a ′) of classifying the cells based on the size is performed after the step (a), cells that have started differentiation are removed, and the proportion of undifferentiated cells is increased and passaged. Is possible.
- step (a ′) cells having a diameter exceeding a threshold (threshold is a value of 20 ⁇ m or more, preferably 23 ⁇ m or more) are removed by classification to remove undifferentiated cells.
- the ratio can be increased.
- the classification threshold value in the step (a ′) is preferably a value of 25 ⁇ m or less, for example, 20 ⁇ m, 21 ⁇ m, 22 ⁇ m, 23 ⁇ m, 24 ⁇ m or 25 ⁇ m, more preferably 23 ⁇ m, 24 ⁇ m or 25 ⁇ m, More preferably, it is 23 ⁇ m.
- the threshold is set low, the ratio of mixed differentiated cells decreases, but at the same time, the recovery rate of undifferentiated cells decreases.
- the threshold value is set high, the recovery rate of undifferentiated cells is improved, but the contamination rate of differentiated cells is also increased.
- a person skilled in the art can appropriately set the threshold based on the cell recovery rate and the contamination rate.
- the classification of cells in the step (a ′) is not particularly limited, and can be performed using, for example, a cell fractionation filter or a cell sorter.
- Cell fractionation filters are often used to classify cells in suspension cell systems and can classify cells of various sizes.
- a cell fractionation filter for example, a filter of Filcon S, size 20 ⁇ m (manufactured by ASONE, product number: 2-7210-01) is commercially available and can be used.
- methods for producing a cell separation filter for example, JP-A-2001-178) are known, and those skilled in the art can produce a cell separation filter to classify cells.
- Cell classification can also be performed using a cell sorter, and those skilled in the art can classify cells based on, for example, a manufacturer's manual.
- the removal of differentiated cells can be performed based on the presence or absence of the expression of a marker (surface marker) that is expressed on the surface of the cell.
- Surface markers that can be used to remove differentiated cells include undifferentiated markers expressed by pluripotent stem cells. Examples of such undifferentiated markers include alkaline phosphatase, SSEA-3, SSEA-4, Undifferentiated markers such as TRA-1-60 and TRA-1-81 are known. Methods for separating cells based on the presence or absence of surface marker expression are well known, and those skilled in the art can appropriately separate and remove differentiated cells. Separation of cells based on the surface marker can be performed using a technique such as flow cytometry.
- the fully automated subculture system of pluripotent stem cells for carrying out the method of the present invention is a means (1) for culturing pluripotent stem cells; pluripotent stem cells are detached from the culture surface and brought to a single cell level.
- Means for dispersing (2); means for seeding the dispersed cells on the multi-microwell plate (where the multi-microwell plate may be centrifuged after seeding) (3); and the formed cell clumps One or more means selected from the group consisting of means (4) for seeding on the culture surface of the culture vessel, preferably all means.
- a fully automated subculture system for pluripotent stem cells for carrying out the method of the present invention comprises the closed culture vessel of the present invention, and means (4) comprises the closed culture of the present invention. This is achieved by means (5) for reversing the container upside down.
- the cells subcultured by the method of the present invention can be cryopreserved according to a normal cryopreservation protocol. After thawing the frozen cells, for example, steps (a) to (d) subsequent to step (a) and step (a) can be performed.
- the cell conglomerate can be stored frozen after step (c).
- the cell aggregate can be cryopreserved, for example, after the cell aggregate has been pelletized using a centrifugation technique, and then can be performed according to a normal cryopreservation protocol. After the frozen cells are thawed, the step following step (a) and step (a) may be performed, or the step following step (c) and step (c) (for example, step (d)) is performed. May be.
- Freezing of the dispersed cells or cell clumps can be performed by using a freezing method corresponding to the cell freezing solution, and those skilled in the art can appropriately select a freezing method.
- Example 1 Examination of human iPS cells into single cells and subsequent passage methods When pluripotent stem cells such as ES cells and iPS cells are separated into single cells during passage, cell death is induced. Is done. In addition, there is a concern that cells may start to differentiate when embryoid bodies are formed. In this example, it was examined whether cell death or differentiation problems would occur when cell clumps were rapidly formed after being dissociated into single cells and then passaged.
- human iPS cells established strain by Kawasada Laboratory, Cell Evaluation Group, Advanced Medical Promotion Foundation
- the culture was performed under feeder-less conditions.
- a medium obtained by adding bFGF (manufactured by Wako Pure Chemical Industries, product number: 064-04541) final concentration of 5 ng / mL to ReproFF2 medium (manufactured by ReproCell, product number: RCHEMD006) was used.
- a culture vessel a 10 mm cell culture dish (manufactured by BD, product number: REF353003) is used, and in order to ensure adhesion of iPS cells to the culture vessel, according to the method described in the manufacturer's instruction manual, Before culturing, the inside of the culture vessel was coated with ECM.
- ECM BD Matrigel (manufactured by BD, product number: 356234) was used.
- the cells were cultured until they became confluent by a conventional method. Thereafter, the medium was removed by aspiration for passage, and the cells were washed once with 10 mL of phosphate buffered saline (Life Technologies, product number: 14190). Thereafter, the cells were treated with 1 mL of accutase solution (Innovative cell technologies, product number: AT104) at 37 ° C. for 5 minutes, and the cells were collected in a tube.
- phosphate buffered saline Life Technologies, product number: 14190
- the cells were collected by centrifugation (440 g, 5 minutes), the supernatant was removed by aspiration, the cells were resuspended in 1 mL of medium, and ROCK inhibitor Y-27632 (manufactured by STEMGENT, product number: 04-0012) was added to the medium. ) was added to a final concentration of 10 ⁇ M to obtain a cell suspension. The cells were then crushed by pipetting water flow until single cell level. The cell concentration in the cell suspension was adjusted, and the cell suspension was injected into the well of AggreWell 800 (manufactured by STEMCELL Technologies). Thereafter, the AggreWell 800 was centrifuged (2000 g, 5 minutes) or not, and cultured at 37 ° C.
- AggreWell 800 manufactured by STEMCELL Technologies
- FIG. 1A The state of the cells immediately after seeding and after 1 day with and without centrifugation was as shown in FIG. 1A. That is, when centrifugation was performed, cells gathered at the bottom of the inverted pyramid-shaped well immediately after seeding (FIG. 1A—upper left), but when centrifugation was not performed, no cells gathered ( FIG. 1A—upper right). However, after one day, the cells gathered at the bottom of the well to form a cell conglomerate whether centrifugation was performed (FIG. 1A-lower left) or not (FIG. 1A-lower right). Moreover, the obtained cell clump had a substantially uniform size (FIG. 1B).
- the obtained cell clumps were collected with a pipette so as not to break as much as possible, and seeded on a 6-well plate (300 cell clumps / well).
- a pipette so as not to break as much as possible
- a 6-well plate 300 cell clumps / well.
- the cell conglomerate obtained in this example has a three-dimensional structure artificially accumulated, the situation is greatly different from that of normal subculture, but in this example, it was unexpectedly produced in this way. Even when a multi-layered cell agglomeration was cultured, it naturally developed, and after that, it was possible to form a colony of pluripotent stem cells found in normal culture and to carry out the culture suitably.
- the cells are seeded on a 6-well plate, collected 5 days later, fixed with paraformaldehyde, and then subjected to fluorescent immunostaining. It was. Nuclear staining was performed by incubating the cells for 15 minutes in phosphate buffered saline containing 1 ⁇ g / mL DAPI.
- Fluorescent immunostaining uses an anti-Nanog antibody (manufactured by ReproCell, product number: RAB0003P) as an antibody to confirm the expression of Nanog, Oct3 / 4 is an anti-Oct3 / 4 antibody (manufactured by Santa Cruz Biotechnology, product number) : Sc-5279) according to a conventional method.
- the bright field image was acquired using a phase contrast microscope (Olympus, product number: IX-81).
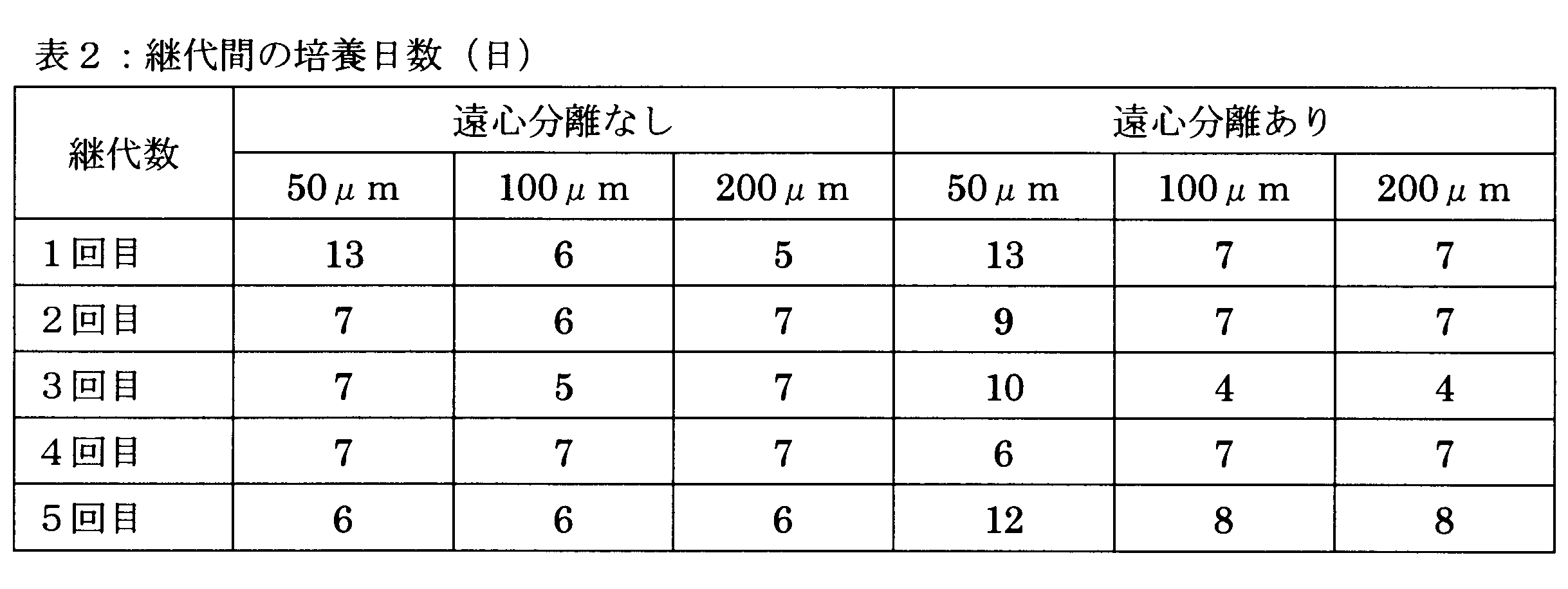
- Example 2 Examination of formation time of cell clumps
- cell clumps were formed using AggreWell, but the formation time was 24 hours. In this example, the optimum time for forming a cell clump was examined.
- Example 2 a cell suspension was obtained in the same manner as in Example 1. The obtained cell suspension was injected into the wells of AggreWell, and the subsequent formation of cell clumps was monitored over time (FIG. 5). As a result, no change in the shape of the cells was observed in about 10 hours, whether or not centrifugation was performed after cell suspension injection. Probably, the reason why the change in shape is no longer observed is that a bond is formed between cells.
- the number of cells 168 hours (7 days) after the injection of the cell suspension into the wells of AggreWell was counted. Then, the number of cells was particularly large when the standing time was 8 hours, but when the standing time was 10 hours or more, the number of cells did not change so much (FIG. 7).
- Example 3-1 Examination of appropriate cell clump size
- the optimum cell clump size in subculture was examined.
- Example 2 a cell suspension was obtained in the same manner as in Example 1.
- the cell clump formation conditions were set as shown in Table 1, and the cell suspension was injected into the wells of AggreWell.
- the cell mass is passaged in a substantially monolayer state.
- a multilayer cell cluster was formed using AggreWell and seeded. According to Examples 1 to 3, the cell conglomerate developed during culture under various conditions and was monolayered.
- the survival rate of the colonies after subculture under each condition was examined.
- the survival rate (%) is calculated by the following formula: The number of colonies on the dish was counted 3 days after passage, and divided by the number of cell clumps seeded.
- the cell engraftment rate tended to be good under the condition of a large cell clump size (FIG. 11).
- the survival rate also tended to be good under conditions where no centrifugation with AggreWell was performed (FIG. 11).
- the ratio of colonies with multiple cells was compared under each condition. Then, in conditions 3 and 6 where the cell clump size was large, the ratio of multilayer colonies was large, and in conditions 1 and 4 where the cell agglomerate size was small, the ratio of multilayer colonies was small (FIG. 12).
- pluripotent stem cells could be cultured well under various conditions as described above, it was found that differences in the subsequent culture occurred due to the size of the cell clumps to be formed and the conditions for forming the cell clumps. It was. That is, if the cell clumps are large, it takes time for the cell clumps to expand, which means that the cell clumps cannot be fully expanded by the next passage, and centrifugation is performed when forming the cell clumps. If the above method is used, the cell agglomeration becomes strong and the cell agglomeration is difficult to break, but it takes time to expand.If the cell agglomeration is small, the cell recovery rate increases, but the colony growth takes time. When the method of centrifugation was not used for the formation of the clumps, it became clear that shortening the cell clump formation time softens the cell clumps, and the cell clumps tend to collapse, but the development is accelerated.
- the size of the growing colonies was relatively uniform even when subculture was performed under any condition (FIG. 8). This means that the method of the present invention is excellent in terms of stability of the quality of cells to be cultured and culture efficiency.
- Example 3-2 Examination of an appropriate cell clump size In Example 3-1, a result suggesting that the subsequent culture is affected by the size of the cell clump to be formed was obtained. In this example, an appropriate cell clump size was examined in more detail.
- a cell suspension was obtained according to the method described in Example 1. Thereafter, the cell concentration in the cell suspension was appropriately adjusted to form a cell clump, and the relationship between the cell clump diameter (y) and the average number of cells per cell clump ( ⁇ ) was examined. .
- the diameter of the cell clump was measured immediately after the cells were allowed to stand in the well for 24 hours to form a cell clump and seeded on the culture surface of the culture vessel.
- the measurement is performed using an optical microscope, and the diameter of the cell clump is obtained by first obtaining the area of the cell clump calculated from the microscopic image of the cell clump and assuming that the cell clump is spherical. Calculated.
- the cell concentration was adjusted so that the average number of cells per cell clump was 11 to 3,415 (FIG. 15). In any case, cell clumps were formed well. I was able to.
- Example 3-1 if the size of the cell clump is large, it takes a long time to develop on the culture surface. For example, depending on the size of the cell clump, it may not be able to expand even after 8 days of passage. It was done. Therefore, the relationship between the development of the cell clump after passage and the size of the cell clump was examined in detail.
- the average number of cells per cell cluster is preferably 1,209 (corresponding to a diameter of 216 ⁇ m) or less, preferably 867 (corresponding to a diameter of 195 ⁇ m) or less from the viewpoint of developability. It became clear that it was more preferable.
- the adhesion rate to the culture surface and the cell agglomeration after seeding the cell agglomerate on the culture surface The relationship with size was examined in detail. Then, it became clear that the adhesion rate to the culture surface increased as the number of cells per cell cluster increased (the size of the cell cluster increased) (FIG. 17).
- the adhesion rate (y) of the cell clumps to the culture surface has a relationship approximated by the following formula with the average number of cells per cell clump ( ⁇ ). It was found (Fig. 17).
- the average number of cells per cell cluster is preferably adjusted to be about 28 (corresponding to a diameter of about 51.6 ⁇ m) or more.
- the approximation error is considered to be relatively large particularly in a region where the engraftment rate is low (for example, the average number of cells is around 28 (that is, equivalent to a diameter of around 50 ⁇ m)). This does not mean that culturing is not possible with cell clumps having an average number of cells of 28 or less (ie, 50 ⁇ m in diameter).
- FIG. 18 shows the relationship between the average number of cells per cell clump and the number of cell clumps obtained when the number of cells used for passage is constant. Then, from the result of the adhesion rate of the obtained cell clump (FIG. 17) and the result of FIG. 18, the amplification factor of the cells up to the next passage (cell amplification factor per passage) was calculated. Specifically, the passage timing is when the size of the colony formed by the development of the cell clumps reaches a diameter of 2 mm, and the number of cells per unit area of the colony is 4000 cells / mm 2 . .
- passage is performed when the number of cells per colony reaches 12,566. Then, in consideration of the relationship between the size of the cell clump, the number of cell clumps, and the adhesion rate (FIGS. 17 and 18), the amplification factor of the cells up to the next passage after the passage was calculated. Then, when the number of cells per cell clump was 76, a convex graph with the maximum cell amplification factor was drawn (FIG. 19). Further, when the size of the cell clump and the number of days until the next passage were examined, it was found that there was a relationship as shown in FIG. From this, the amplification factor of the cells per day was calculated.
- the size of the cell clump when seeded is not uniform.
- the size of the cell clump can be easily adjusted to a certain size by adjusting the number of cells to be seeded in each microwell. Each size can be made uniform. Therefore, it is clear that the proliferation efficiency of pluripotent stem cells can be easily increased by the method of the present invention.
- Example 4 Examination of seeding method of cell agglomeration on culture surface of culture vessel
- the cell agglomerate formed in the microwell was passaged to a new culture vessel by collecting with a pipette. Careful pipetting operation was required, taking time to avoid breaking the cell conglomerate. Therefore, in this example, a simpler cell agglomeration method was examined.
- the inventors examined the use of a closed culture vessel having a surface having a microwell and a culture surface, and these two surfaces are arranged to face each other.
- the surface of the closed culture vessel having the microwells was formed by arranging square-shaped microwells having a square bottom shape and a top opening of 1000 ⁇ m ⁇ 1000 ⁇ m in a grid shape.
- the culture surface was coated with BD Matrigel (trademark).
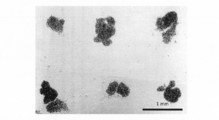
- FIG. 23 is a diagram showing the arrangement of cell clumps dropped on the culture surface. As shown in FIG. 23, the cell clumps were regularly aligned on the culture surface. The arrangement of these cell clumps reflects the arrangement pattern of the microwells used, and the cell clump spacing in FIG. 23 is the pitch of the microwells (1000 ⁇ m) of the closed culture vessel used in this example. Matched.
- the cell agglomeration in the microwell can be dropped onto the culture surface of the culture vessel while maintaining the arrangement pattern of the microwell. It is considered that the seeding position of the cell cluster can be freely controlled by changing the arrangement of the microwells. Moreover, it became clear that control of sowing and sowing position can be performed by a very easy operation of turning the container upside down.
- pluripotent stem cells can be well cultured while maintaining an undifferentiated state by forming cell clusters immediately after being dispersed into single cells. It was possible. Further, by seeding the cell suspension in a culture vessel having a surface on which a plurality of microwells are arranged, it was possible to easily form a cell aggregate having a uniform size. Furthermore, the cell clumps developed rapidly after seeding in the culture vessel, and the cells proliferated well. Since the size of the formed cell clump was uniform, the cell clump development rate and the subsequent growth rate were also uniform. In addition, the uniform size of the cell clumps increased the efficiency of subculture and facilitated cell quality control.
- the cell clumps in the microwells could be seeded on the culture surface of the culture vessel by a simple operation.
- the arrangement pattern of the seeded cell agglomeration reflects the arrangement pattern of the microwells, and it was shown that the cell agglomeration can be precisely seeded by a simple operation.
- formation of a uniform cell clump, uniform seeding of the cell clump, and the like can be performed by a very simple mechanical operation.
- the method of the present invention not only facilitates maintaining the quality of pluripotent stem cells, but also opens the way to full automation of subculture of pluripotent stem cells.
- Example 5 Classification of cells based on size According to the above example, even when pluripotent stem cells are dissociated to the level of a single cell, they can be cultured well by rapidly forming cell clusters. I understood. In this example, taking advantage of the present invention that can be made into a single cell, the possibility of classifying cells based on size was evaluated.
- iPS cell colony judged visually undifferentiated (i.e., good) and iPS cell colony judged to have started differentiation (i.e., poor) in a part of the colony were isolated using a pipette, The cells were dispersed into single cells using an enzyme, and then observed under a microscope to confirm the distribution of the size of the single cells.
- the cells obtained from the iPS cell colonies determined to be good had a size of about 14 to 20 ⁇ m centering on 17 ⁇ m, and showed a tendency that the sizes of the respective cells were uniform, but poor From the cells obtained from the iPS cell colonies determined to be, the presence of cells having a size of 22 ⁇ m or more or 23 ⁇ m or more was confirmed (FIG.
- the cells having a size of 22 ⁇ m or more or 23 ⁇ m or more are expected to be cells that have started to differentiate within the colony in comparison with cells obtained from a good colony.
- the cells were evaluated as having lost their ability. From these results, it was shown that the classification operation could remove cells (differentiated cells) that started differentiation and lost pluripotency.
- the removal of differentiated cells can also be performed by flow cytometry.
- pluripotent stem cells Maintaining the quality of pluripotent stem cells is an important issue in subculture of pluripotent stem cells.
- differentiated cells in pluripotent stem cells dispersed into single cells can be removed by a simple mechanical operation. Therefore, it can be said that the present invention opens up the path of automating the removal of differentiated cells in subculture.
Abstract
Description
(1)多能性幹細胞の継代培養方法であって、
(a)継代時に多能性幹細胞の細胞塊を分散させる工程と、
(b)分散させて得られた細胞をマイクロウェル中に播種する工程と、
(c)マイクロウェル中に播種された細胞から細胞集塊を形成させる工程と、
(d)得られた細胞集塊を培養容器の培養面に播種する工程
とを含んでなる、方法。
(2)工程(d)が、(d’)マイクロウェルを備えた容器を天地逆転させて細胞集塊を培養容器の培養面上に落下させる工程を含んでなる、上記(1)に記載の方法。
(3)工程(a)において細胞塊を1~100個の細胞からなる細胞塊に解離させる、上記(1)または(2)に記載の方法。
(4)工程(a)において細胞塊を1~10個の細胞からなる細胞塊に解離させる、上記(3)に記載の方法。
(5)工程(a)において細胞塊を単一細胞にまで解離させる、上記(4)に記載の方法。
(6)工程(b)において各マイクロウェルに播種される細胞の平均細胞数が、10~3,500個である、上記(1)~(5)のいずれかに記載の方法。
(7)工程(b)において各マイクロウェルに播種される細胞の平均細胞数が、25~870個である、上記(6)に記載の方法。
(8)工程(b)において各マイクロウェルに播種される細胞の平均細胞数が、40~500個である、上記(7)に記載の方法。
(9)工程(b)において各マイクロウェルに播種される細胞数が、55~220個である、上記(8)に記載の方法。
(10)工程(c)がマイクロウェル中で細胞を、細胞集塊を形成するために十分な時間静置することを含んでなる、上記(1)~(9)のいずれかに記載の方法。
(11)工程(c)がマイクロウェル中で細胞を8~24時間静置することを含んでなる、上記(10)に記載の方法。
(12)工程(c)がマイクロウェル中で細胞を8~12時間静置することを含んでなる、上記(11)に記載の方法。
(13)工程(c)が遠心分離を用いることなく行われる、上記(1)~(12)のいずれかに記載の方法。
(14)多能性幹細胞が、ヒト多能性幹細胞である、上記(1)~(13)のいずれかに記載の方法。
(15)ヒト多能性幹細胞が、ヒトES細胞またはヒトiPS細胞である、上記(14)に記載の方法。
(16)工程(a)の後に、分化細胞を除去する工程(a’)をさらに含んでなる、上記(1)~(15)のいずれかに記載の方法。
(17)分化細胞を除去する工程(a’)が、分化細胞を分級することにより除去することを含んでなる、上記(16)に記載の方法。
(18)閾値(閾値は、20μm以上の値である)を超えた直径を有する細胞を分級により除去する、上記(17)に記載の方法。
(19)閾値(閾値は、23μm以上の値である)を超えた直径を有する細胞を分級により除去する、上記(18)に記載の方法。
(20)分化細胞を除去する工程(a’)が、細胞の表面マーカーの発現の有無に基づいて行われる、上記(16)に記載の方法。
(21)細胞の表面マーカーが、多能性幹細胞が細胞表面に発現する未分化マーカーである、上記(20)に記載の方法。
(22)未分化マーカーが、アルカリフォスファターゼ、SSEA-3、SSEA-4、TRA-1-60およびTRA-1-81からなる群から選択される1以上の未分化マーカーである、上記(21)に記載の方法。
(23)マイクロウェルを備えた面と培養面とを備えてなり、これら2つの面が向かい合うように配置された、閉鎖系培養容器。
(24)マイクロウェルが、その内周が底面に近づくほど小さくなる形状を有する、上記(23)に記載の閉鎖系培養容器。
(25)マイクロウェルが、丸底、V底、U底または角取平面底を有する、上記(24)に記載の閉鎖系培養容器。
(26)整列した複数のマイクロウェルを備えた、上記(23)~(25)のいずれかに記載の閉鎖系培養容器。
(27)工程(d’)が、上記(23)~(26)のいずれかに記載の閉鎖系培養容器を天地逆転させることにより行われる、上記(2)に記載の方法。
(28)上記(1)~(22)および(27)のいずれかに記載の方法を実施するための多能性幹細胞の全自動継代培養システム。
(a)継代時に多能性幹細胞の細胞塊を分散させる工程と、
(b)分散させて得られた細胞をマイクロウェル中に播種する工程と、
(c)遠心分離の手法を用いてまたは用いずにマイクロウェル中に播種された細胞から細胞集塊を形成させる工程と、
(d)得られた細胞集塊を培養容器の培養面に播種する工程
とを含んでなる。本発明の多能性幹細胞の継代培養は、接着培養系で行われる。本発明では、多能性幹細胞は良好な未分化状態を維持したまま維持培養することができる。
(細胞接着面からの細胞の剥離)
本発明では、接着培養において、生理学的にまたは物理的に細胞接着面から剥離させた多能性幹細胞を用いることができる。本発明では、多能性幹細胞を細胞接着面から剥離させる酵素としては、常法で用いられる酵素を用いることができ、例えば、トリプシン、ディスパーゼ、アキュターゼおよびコラゲナーゼなどの酵素を用いることができる。多能性幹細胞の細胞接着面からの剥離はまた、エチレンジアミン四酢酸(EDTA)などの二価イオン(特にMg2+)のキレート剤など、細胞剥離作用を有する化学物質を用いて行ってもよく、これらを上記酵素と組み合わせて用いて行ってもよい。本発明ではまた、多能性幹細胞を細胞接着面から剥離させるために、細胞の接着表面に高周波振動などの振動を与えてもよく、および/または、セルスクレイパーを用いてもよい。本発明では更に、上記の生理学的な剥離法と物理的な剥離法を組み合わせて細胞を剥離してもよい。当業者であれば、細胞接着面からの多能性幹細胞の剥離は上記のような周知の剥離法を用いて適宜行うことができるであろう。
接着面から剥離させた細胞が細胞塊の形状を保っている場合には、剥離させた細胞塊は、例えば、ピペッティングの水流により単一細胞レベルにまで解離させてから分散させることができる。本明細書では、「単一細胞レベルにまで分散させる」とは、細胞塊1つ当りに含まれる細胞数の平均が、1~100個、好ましくは、1~10個となるように細胞塊を解離させてから分散させることを意味し、完全に単一細胞にまで解離させてから分散させることを含むものとする。従って、本発明では、細胞塊は完全に単一細胞にまで解離させてから分散させてもよいし、大部分が単一細胞となるように解離させてから分散させてもよいし、大部分が1~100個、好ましくは、1~10個の細胞からなる細胞塊となるように解離させてから分散させてもよい。分散後の細胞塊が大きい場合は、細胞塊をマイクロウェルに播種する際に、それぞれのマイクロウェルに播種される細胞数にばらつきが生じやすくなるため、分散後の細胞塊は小さい方が好ましい。
本発明によれば、工程(a)で分散させて得られた細胞または細胞塊(以下、単に「細胞」ということがある)は、細胞集塊を形成させた後に継代するとその後良好に増殖させることができる。細胞集塊の形成は、得られた細胞をマイクロウェルに播種することにより行うことができる。工程(a)で分散させて得られた細胞は、培養液中では重力により自然と沈む。従って、傾斜を有するウェル(くぼみ)中に細胞を播種すると、ウェル中では細胞がウェルの傾斜を利用して集まる。その後、細胞は、隣り合う細胞と細胞接着を形成して細胞集塊を形成する。従って、本発明では、ウェルは、細胞が沈んだときに集まる形状、例えば、その内周が底面に近づくほど小さくなる形状を有していることが好ましい。すなわち、ウェルは、底がすぼんだ形状を有していることが好ましく、例えば、錐形状、丸底、V底、U底および角取平面底(底面を有するが角が取れて底がすぼんだ形状となったもの)の形状を有していることが好ましい。また、各ウェルの上部開口部の形状は、加工の容易性やウェルを大量に配置できる形状を考慮して適宜選択することができるが、例えば、三角形、四角形若しくは六角形などの多角形の形状または円の形状を有することができる。
本発明によれば、マイクロウェル中に播種された細胞は、そのまま静置しておくことで重力によりマイクロウェルの底に集まり、細胞同士が接着し、細胞集塊を形成する。細胞は、マイクロウェルを有する容器を遠心分離の手法を用いて遠心することによってマイクロウェルの底に集めてもよい。
工程(c)によりマイクロウェルを備えた容器中で形成された細胞集塊は、その後、培養容器(すなわち、培養容器の培養面)に播種することができる。この工程(d)は、細胞集塊を回収し、培地に懸濁し、培養容器の培養面に播種することにより行うことができ、この工程(d)は、全自動化が可能である。播種された細胞集塊は、播種後速やかに展開し、その後、多能性を維持した状態で良好に培養することができる。細胞集塊を培養容器の培養面上に均一に播種する観点では、細胞集塊を含む細胞懸濁液は、十分に懸濁してから播種することが好ましい。
本発明の方法によれば、多能性幹細胞を単一細胞にまで分散させた場合であっても、細胞に細胞死等の有害事象の発生を抑制することができる。また、その後細胞集塊を形成させることにより効率的に細胞の継代が可能である。本発明では、多能性幹細胞を単一細胞にまで分散させる利点として、細胞1つ1つの性状に基づく分化細胞の分離除去が可能となる。工程(a’)は、分化細胞の分離除去に関する工程であり、多能性幹細胞を単一細胞にまで分散させることができるようになった本発明で初めて多能性幹細胞の継代培養に導入可能となった工程である。すなわち、工程(a’)は、工程(a)において多能性幹細胞を単一細胞にまで分散させることを前提とする工程である。
ES細胞やiPS細胞などの多能性幹細胞は、継代の際に単一細胞にまでばらばらにすると細胞死が誘導される。また、胚様体様にすると細胞が分化を開始することが懸念される。本実施例では、単一細胞にまでばらばらにした後に、速やかに細胞集塊を形成させてから、継代を行った場合に細胞死や分化の問題が生じるか否かを検討した。
実施例1では、AggreWellを用いて細胞集塊を形成させたが、形成させる時間は24時間としていた。本実施例では、細胞集塊を形成させるための最適な時間の検討を行った。
本実施例では、継代培養における最適な細胞集塊サイズを検討した。

※2 AggreWell 400では、ウェルの底面に、400μm×400μmの逆ピラミッド状のマイクロウェルが1ウェル当り1,200個刻まれている。
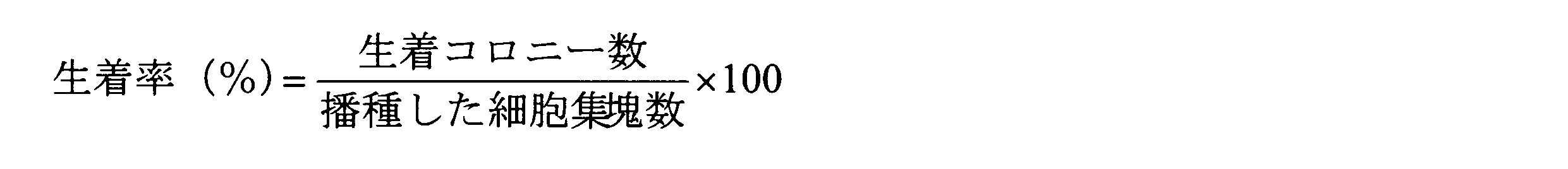


実施例3-1では、形成させる細胞集塊のサイズにより、その後の培養が影響を受けることを示唆する結果が得られた。本実施例では、適切な細胞集塊サイズをより詳細に検討した。


上記実施例では、マイクロウェル中で形成された細胞集塊は、ピペットで回収することにより新しい培養容器に継代したが、細胞集塊を壊さないために時間をかけた慎重なピペッティング操作が求められた。そこで、本実施例では、より簡便な細胞集塊の播種法を検討した。
上記実施例により、多能性幹細胞が、単一細胞レベルに解離させた場合でも迅速に細胞集塊を形成させることにより、その後良好に培養することができることが分かった。本実施例では、単一細胞化できる本発明の利点を活かして、サイズに基づく細胞の分級の可能性を評価した。
Claims (28)
- 多能性幹細胞の継代培養方法であって、
(a)継代時に多能性幹細胞の細胞塊を分散させる工程と、
(b)分散させて得られた細胞をマイクロウェル中に播種する工程と、
(c)マイクロウェル中に播種された細胞から細胞集塊を形成させる工程と、
(d)得られた細胞集塊を培養容器の培養面に播種する工程
とを含んでなる、方法。 - 工程(d)が、(d’)マイクロウェルを備えた容器を天地逆転させて細胞集塊を培養容器の培養面上に落下させる工程を含んでなる、請求項1に記載の方法。
- 工程(a)において細胞塊を1~100個の細胞からなる細胞塊に解離させる、請求項1または2に記載の方法。
- 工程(a)において細胞塊を1~10個の細胞からなる細胞塊に解離させる、請求項3に記載の方法。
- 工程(a)において細胞塊を単一細胞にまで解離させる、請求項4に記載の方法。
- 工程(b)において各マイクロウェルに播種される細胞の平均細胞数が、10~3,500個である、請求項1~5のいずれか一項に記載の方法。
- 工程(b)において各マイクロウェルに播種される細胞の平均細胞数が、25~870個である、請求項6に記載の方法。
- 工程(b)において各マイクロウェルに播種される細胞の平均細胞数が、40~500個である、請求項7に記載の方法。
- 工程(b)において各マイクロウェルに播種される細胞数が、55~220個である、請求項8に記載の方法。
- 工程(c)がマイクロウェル中で細胞を、細胞集塊を形成するために十分な時間静置することを含んでなる、請求項1~9のいずれか一項に記載の方法。
- 工程(c)がマイクロウェル中で細胞を8~24時間静置することを含んでなる、請求項10に記載の方法。
- 工程(c)がマイクロウェル中で細胞を8~12時間静置することを含んでなる、請求項11に記載の方法。
- 工程(c)が遠心分離を用いることなく行われる、請求項1~12のいずれか一項に記載の方法。
- 多能性幹細胞が、ヒト多能性幹細胞である、請求項1~13のいずれか一項に記載の方法。
- ヒト多能性幹細胞が、ヒトES細胞またはヒトiPS細胞である、請求項14に記載の方法。
- 工程(a)の後に、分化細胞を除去する工程(a’)をさらに含んでなる、請求項1~15のいずれか一項に記載の方法。
- 分化細胞を除去する工程(a’)が、分化細胞を分級することにより除去することを含んでなる、請求項16に記載の方法。
- 閾値(閾値は、20μm以上の値である)を超えた直径を有する細胞を分級により除去する、請求項17に記載の方法。
- 閾値(閾値は、23μm以上の値である)を超えた直径を有する細胞を分級により除去する、請求項18に記載の方法。
- 分化細胞を除去する工程(a’)が、細胞の表面マーカーの発現の有無に基づいて行われる、請求項16に記載の方法。
- 細胞の表面マーカーが、多能性幹細胞が細胞表面に発現する未分化マーカーである、請求項20に記載の方法。
- 未分化マーカーが、アルカリフォスファターゼ、SSEA-3、SSEA-4、TRA-1-60およびTRA-1-81からなる群から選択される1以上の未分化マーカーである、請求項21に記載の方法。
- マイクロウェルを備えた面と培養面とを備えてなり、これら2つの面が向かい合うように配置された、閉鎖系培養容器。
- マイクロウェルが、その内周が底面に近づくほど小さくなる形状を有する、請求項23に記載の閉鎖系培養容器。
- マイクロウェルが、丸底、V底、U底または角取平面底を有する、請求項24に記載の閉鎖系培養容器。
- 整列した複数のマイクロウェルを備えた、請求項23~25のいずれか一項に記載の閉鎖系培養容器。
- 工程(d’)が、請求項23~26のいずれか一項に記載の閉鎖系培養容器を天地逆転させることにより行われる、請求項2に記載の方法。
- 請求項1~22および27のいずれか一項に記載の方法を実施するための多能性幹細胞の全自動継代培養システム。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/762,921 US20150353884A1 (en) | 2013-01-23 | 2014-01-23 | Method of subculturing pluripotent stem cells |
| EP14743585.3A EP2949746A4 (en) | 2013-01-23 | 2014-01-23 | METHOD OF SUB-CULTURE OF PLURIPOTENT STEM CELLS |
| JP2014558609A JPWO2014115799A1 (ja) | 2013-01-23 | 2014-01-23 | 多能性幹細胞の継代培養方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013010161 | 2013-01-23 | ||
| JP2013-010161 | 2013-01-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014115799A1 true WO2014115799A1 (ja) | 2014-07-31 |
Family
ID=51227588
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/051362 WO2014115799A1 (ja) | 2013-01-23 | 2014-01-23 | 多能性幹細胞の継代培養方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20150353884A1 (ja) |
| EP (1) | EP2949746A4 (ja) |
| JP (1) | JPWO2014115799A1 (ja) |
| WO (1) | WO2014115799A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015159950A1 (ja) * | 2014-04-17 | 2015-10-22 | 東京エレクトロン株式会社 | 多能性幹細胞の細胞集塊製造方法および細胞集塊製造システム |
| WO2016052657A1 (ja) * | 2014-09-30 | 2016-04-07 | 国立研究開発法人産業技術総合研究所 | 万能性幹細胞の培養方法 |
| WO2016147239A1 (ja) * | 2015-03-16 | 2016-09-22 | パナソニック株式会社 | ピペットチップ及びピペッティング方法 |
| WO2017199737A1 (ja) * | 2016-05-16 | 2017-11-23 | 富士フイルム株式会社 | 培養細胞の回収方法および培養細胞分散液 |
| JPWO2016140327A1 (ja) * | 2015-03-04 | 2017-12-14 | 国立研究開発法人産業技術総合研究所 | マイクロチャンバーアレイプレート |
| WO2020218579A1 (ja) * | 2019-04-26 | 2020-10-29 | 国立大学法人京都大学 | 分化誘導のために馴化された多能性幹細胞の作製方法 |
| JPWO2019069378A1 (ja) * | 2017-10-03 | 2020-11-05 | オリンパス株式会社 | 培養情報処理装置 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102136478B1 (ko) * | 2016-05-06 | 2020-07-21 | 후지필름 가부시키가이샤 | 다능성 줄기 세포의 계대 방법 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002000178A (ja) | 2000-06-19 | 2002-01-08 | Miyamura Tekkosho:Kk | トラブル解消機構を備えた回転胴形茶葉蒸機 |
| WO2007114351A1 (ja) * | 2006-03-31 | 2007-10-11 | Asubio Pharma Co., Ltd. | 新規細胞培養方法、およびその方法を用いた細胞塊の生産および回収方法 |
| JP2010233456A (ja) * | 2009-03-30 | 2010-10-21 | Kitakyushu Foundation For The Advancement Of Industry Science & Technology | 細胞集合体形成器具、細胞集合体培養器具、細胞集合体転写キット及び細胞集合体の培養方法 |
| WO2011022507A1 (en) * | 2009-08-21 | 2011-02-24 | The Board Of Trustees Of The Leland Stanford Junior University | Enhanced efficiency of induced pluripotent stem cell generation from human somatic cells |
| WO2011068879A2 (en) * | 2009-12-02 | 2011-06-09 | Research Development Foundation | Selection of stem cell clones with defined differentiation capabilities |
| WO2011161962A1 (ja) * | 2010-06-25 | 2011-12-29 | 川崎重工業株式会社 | 多能性幹細胞コロニーの識別方法及び装置並びに多能性幹細胞の自動培養方法及び装置 |
| WO2012087965A2 (en) * | 2010-12-22 | 2012-06-28 | Fate Therapauetics, Inc. | Cell culture platform for single cell sorting and enhanced reprogramming of ipscs |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8956867B2 (en) * | 2008-11-07 | 2015-02-17 | Wisconsin Alumni Research Foundation | Method for culturing stem cells |
| KR101330327B1 (ko) * | 2010-05-07 | 2013-11-14 | 한국생명공학연구원 | 줄기세포로부터 생성된 배아체를 대량 증식 및 유지하는 방법 |
-
2014
- 2014-01-23 JP JP2014558609A patent/JPWO2014115799A1/ja active Pending
- 2014-01-23 WO PCT/JP2014/051362 patent/WO2014115799A1/ja active Application Filing
- 2014-01-23 US US14/762,921 patent/US20150353884A1/en not_active Abandoned
- 2014-01-23 EP EP14743585.3A patent/EP2949746A4/en not_active Withdrawn
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002000178A (ja) | 2000-06-19 | 2002-01-08 | Miyamura Tekkosho:Kk | トラブル解消機構を備えた回転胴形茶葉蒸機 |
| WO2007114351A1 (ja) * | 2006-03-31 | 2007-10-11 | Asubio Pharma Co., Ltd. | 新規細胞培養方法、およびその方法を用いた細胞塊の生産および回収方法 |
| JP2010233456A (ja) * | 2009-03-30 | 2010-10-21 | Kitakyushu Foundation For The Advancement Of Industry Science & Technology | 細胞集合体形成器具、細胞集合体培養器具、細胞集合体転写キット及び細胞集合体の培養方法 |
| WO2011022507A1 (en) * | 2009-08-21 | 2011-02-24 | The Board Of Trustees Of The Leland Stanford Junior University | Enhanced efficiency of induced pluripotent stem cell generation from human somatic cells |
| WO2011068879A2 (en) * | 2009-12-02 | 2011-06-09 | Research Development Foundation | Selection of stem cell clones with defined differentiation capabilities |
| WO2011161962A1 (ja) * | 2010-06-25 | 2011-12-29 | 川崎重工業株式会社 | 多能性幹細胞コロニーの識別方法及び装置並びに多能性幹細胞の自動培養方法及び装置 |
| WO2012087965A2 (en) * | 2010-12-22 | 2012-06-28 | Fate Therapauetics, Inc. | Cell culture platform for single cell sorting and enhanced reprogramming of ipscs |
Non-Patent Citations (8)
| Title |
|---|
| "AGGREWELL: ARE YOU MAKING EMBRYOID BODIES? (STEMCELL TECHNOLOGIES)", 25 September 2009 (2009-09-25), XP054976485, Retrieved from the Internet <URL:HTTP://WWW.VERITASTK.CO.JP/NEWS.PHP?ID=345, HTTPS://WWW.YOUTUBE.COM/WATCH?V=2P-KGB9WT9I> [retrieved on 20140409] * |
| HIROMICH YOSHIOKA ET AL.: "iPS Saibo Yurai Bunka Saibo to Mibunka iPS Saibo no Isosa ni yoru Hi Shinshuteki Shikibetsu", DAI 64 KAI ABSTRACTS OF THE ANNUAL MEETING OF THE SOCIETY FOR BIOTECHNOLOGY, 25 September 2012 (2012-09-25), JAPAN, pages 112, XP008179912 * |
| KANJI YAHIRO ET AL.: "Introduction of stem cell culture, differentiation and assay tools", THE CELL, vol. 44, no. 9, 20 August 2012 (2012-08-20), pages 393 - 397, XP008179927 * |
| See also references of EP2949746A4 |
| SHIMAZAKI TAKUYA; OKADA YOHEI; YOSIJAKI DHAKAHITO; OKANO HIDEYUKI: "Protein, Nucleic acid and Enzyme", KIORITZ PUBLICATION, vol. 51, no. 13, 2006, pages 1854 - 1861 |
| SPELKE D.P.: "Methods for embryoid body formation: the microwell approach", METHODS IN MOLECULAR BIOLOGY, vol. 690, 2011, pages 151 - 162 |
| STOVER, A.E. ET AL.: "The Generation of Embryoid Bodies from Feeder-Based or Feeder- Free Human Pluripotent Stem Cell Cultures", METHODS IN MOLECULAR BIOLOGY, vol. 767, 2011, pages 391 - 398, XP055265964 * |
| WATANABE, K.: "A ROCK inhibitor permits survival of dissociated human embryonic stem cells", NATURE BBOTECHNOLOGY, vol. 25, 2007, pages 681, XP002478043, DOI: doi:10.1038/nbt1310 |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015159950A1 (ja) * | 2014-04-17 | 2015-10-22 | 東京エレクトロン株式会社 | 多能性幹細胞の細胞集塊製造方法および細胞集塊製造システム |
| EP3202896A4 (en) * | 2014-09-30 | 2018-07-04 | JTEC Corporation | Method for culturing pluripotent stem cells |
| JPWO2016052657A1 (ja) * | 2014-09-30 | 2017-07-20 | 国立研究開発法人産業技術総合研究所 | 万能性幹細胞の培養方法 |
| WO2016052657A1 (ja) * | 2014-09-30 | 2016-04-07 | 国立研究開発法人産業技術総合研究所 | 万能性幹細胞の培養方法 |
| US10696951B2 (en) | 2014-09-30 | 2020-06-30 | Jtec Corporation | Method for culturing pluripotent stem cells |
| JPWO2016140327A1 (ja) * | 2015-03-04 | 2017-12-14 | 国立研究開発法人産業技術総合研究所 | マイクロチャンバーアレイプレート |
| US10456779B2 (en) | 2015-03-16 | 2019-10-29 | Panasonic Corporation | Pipette tip having a straight pipe section with inner protrusion and pipetting method for a liquid including cells |
| JP6004149B1 (ja) * | 2015-03-16 | 2016-10-05 | パナソニック株式会社 | ピペットチップ及びピペッティング方法 |
| WO2016147239A1 (ja) * | 2015-03-16 | 2016-09-22 | パナソニック株式会社 | ピペットチップ及びピペッティング方法 |
| EP3272851A4 (en) * | 2015-03-16 | 2018-01-24 | Panasonic Corporation | Pipette tip and pipetting method |
| WO2017199737A1 (ja) * | 2016-05-16 | 2017-11-23 | 富士フイルム株式会社 | 培養細胞の回収方法および培養細胞分散液 |
| JPWO2017199737A1 (ja) * | 2016-05-16 | 2019-03-07 | 富士フイルム株式会社 | 培養細胞の回収方法および培養細胞分散液 |
| JPWO2019069378A1 (ja) * | 2017-10-03 | 2020-11-05 | オリンパス株式会社 | 培養情報処理装置 |
| JP7018955B2 (ja) | 2017-10-03 | 2022-02-14 | オリンパス株式会社 | 培養情報処理装置 |
| US11256898B2 (en) | 2017-10-03 | 2022-02-22 | Olympus Corporation | Culture information processing device |
| WO2020218579A1 (ja) * | 2019-04-26 | 2020-10-29 | 国立大学法人京都大学 | 分化誘導のために馴化された多能性幹細胞の作製方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20150353884A1 (en) | 2015-12-10 |
| JPWO2014115799A1 (ja) | 2017-01-26 |
| EP2949746A4 (en) | 2016-07-27 |
| EP2949746A1 (en) | 2015-12-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2014115799A1 (ja) | 多能性幹細胞の継代培養方法 | |
| Bajpai et al. | Efficient propagation of single cells accutase‐dissociated human embryonic stem cells | |
| Otsuji et al. | A 3D sphere culture system containing functional polymers for large-scale human pluripotent stem cell production | |
| JP6979687B2 (ja) | 幹細胞の凝集塊の集団の調製方法 | |
| US20080026460A1 (en) | Method for culturing stem cells | |
| US10487312B2 (en) | Passaging and harvesting formulation and method for human pluripotent stem cells | |
| JP6935101B2 (ja) | 幹細胞を再樹立する方法 | |
| US20090130754A1 (en) | Method for culturing human embryonic stem cells | |
| EP3202896B1 (en) | Method for culturing pluripotent stem cells | |
| CN106381282B (zh) | 一种诱导多能干细胞传代方法 | |
| JP6983907B2 (ja) | 奇形腫の形成が抑制された多分化能性幹細胞由来の神経前駆体球の製造方法 | |
| US20190359946A1 (en) | Passaging and harvesting formulation for single-cell human pluripotent stem cells | |
| Matsushita et al. | Expansion and differentiation of human iPS cells in a three-dimensional culture using hollow fibers and separation of the specific population by magnetic-activated cell sorting | |
| WO2022188395A1 (zh) | 一种多能干细胞的培养方法 | |
| Li et al. | Simple autogeneic feeder cell preparation for pluripotent stem cells | |
| JP2017046592A (ja) | 細胞培養容器および細胞継代培養システム並びに細胞継代培養方法 | |
| JP2009529859A (ja) | ヒト胚盤胞からの幹細胞を増殖ための培養系および方法 | |
| WO2021149823A1 (ja) | 因子を導入された細胞の培養方法 | |
| JP2022104813A (ja) | リプログラミング因子を導入された細胞の培養方法 | |
| Wang et al. | P-1018: Induction of hES cells to endothelial cells | |
| JP2022001027A (ja) | 細胞分離方法及び多能性幹細胞塊の細胞懸濁液の製造方法 | |
| Choo et al. | Expansion of undifferentiated human embryonic stem cells | |
| CA2659133A1 (en) | Device and methods for production of cell aggregates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14743585 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014558609 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014743585 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14762921 Country of ref document: US Ref document number: 2014743585 Country of ref document: EP |