WO2012133124A1 - 太陽電池用ポリマーシート及びその製造方法、太陽電池用バックシート、並びに太陽電池モジュール - Google Patents
太陽電池用ポリマーシート及びその製造方法、太陽電池用バックシート、並びに太陽電池モジュール Download PDFInfo
- Publication number
- WO2012133124A1 WO2012133124A1 PCT/JP2012/057390 JP2012057390W WO2012133124A1 WO 2012133124 A1 WO2012133124 A1 WO 2012133124A1 JP 2012057390 W JP2012057390 W JP 2012057390W WO 2012133124 A1 WO2012133124 A1 WO 2012133124A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polymer
- solar cell
- layer
- polyester
- polymer sheet
- Prior art date
Links
- 229920000642 polymer Polymers 0.000 title claims abstract description 447
- 238000000034 method Methods 0.000 title claims description 91
- 238000004519 manufacturing process Methods 0.000 title claims description 41
- 230000008569 process Effects 0.000 title description 2
- 229920000728 polyester Polymers 0.000 claims abstract description 228
- 239000000758 substrate Substances 0.000 claims abstract description 91
- 239000002131 composite material Substances 0.000 claims abstract description 68
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims abstract description 44
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical class [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 claims abstract description 34
- 230000014759 maintenance of location Effects 0.000 claims abstract description 26
- 125000000962 organic group Chemical group 0.000 claims abstract description 12
- 125000005843 halogen group Chemical group 0.000 claims abstract description 9
- 239000000463 material Substances 0.000 claims description 107
- 238000000576 coating method Methods 0.000 claims description 84
- -1 polysiloxane Polymers 0.000 claims description 84
- 239000011248 coating agent Substances 0.000 claims description 78
- 239000003431 cross linking reagent Substances 0.000 claims description 45
- 239000003566 sealing material Substances 0.000 claims description 31
- 239000000853 adhesive Substances 0.000 claims description 29
- 230000001070 adhesive effect Effects 0.000 claims description 29
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 28
- 239000007788 liquid Substances 0.000 claims description 24
- 230000004888 barrier function Effects 0.000 claims description 20
- 239000007789 gas Substances 0.000 claims description 20
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 claims description 18
- 238000003851 corona treatment Methods 0.000 claims description 16
- 239000002904 solvent Substances 0.000 claims description 16
- 229920001296 polysiloxane Polymers 0.000 claims description 15
- 238000000113 differential scanning calorimetry Methods 0.000 claims description 11
- 238000007789 sealing Methods 0.000 claims description 11
- 238000011282 treatment Methods 0.000 claims description 11
- 239000012463 white pigment Substances 0.000 claims description 10
- 239000004593 Epoxy Substances 0.000 claims description 9
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 9
- 125000003118 aryl group Chemical group 0.000 claims description 6
- 238000001579 optical reflectometry Methods 0.000 claims description 6
- 238000009832 plasma treatment Methods 0.000 claims description 6
- 238000004381 surface treatment Methods 0.000 claims description 6
- 125000003277 amino group Chemical group 0.000 claims description 5
- 125000003545 alkoxy group Chemical group 0.000 claims description 4
- 125000000217 alkyl group Chemical group 0.000 claims description 4
- 125000003368 amide group Chemical group 0.000 claims description 4
- 229920000307 polymer substrate Polymers 0.000 claims description 4
- 125000003396 thiol group Chemical group [H]S* 0.000 claims description 4
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 3
- 125000004104 aryloxy group Chemical group 0.000 claims description 3
- 239000000565 sealant Substances 0.000 claims description 2
- 238000004737 colorimetric analysis Methods 0.000 abstract 1
- 239000010410 layer Substances 0.000 description 335
- 210000004027 cell Anatomy 0.000 description 157
- 239000002585 base Substances 0.000 description 102
- 239000010408 film Substances 0.000 description 57
- 229920006267 polyester film Polymers 0.000 description 57
- 239000000243 solution Substances 0.000 description 46
- 239000011230 binding agent Substances 0.000 description 45
- 238000009826 distribution Methods 0.000 description 44
- 238000006116 polymerization reaction Methods 0.000 description 32
- 230000015572 biosynthetic process Effects 0.000 description 27
- 239000003795 chemical substances by application Substances 0.000 description 27
- 150000001875 compounds Chemical class 0.000 description 27
- 238000006460 hydrolysis reaction Methods 0.000 description 27
- 230000007062 hydrolysis Effects 0.000 description 26
- 239000005038 ethylene vinyl acetate Substances 0.000 description 25
- DQXBYHZEEUGOBF-UHFFFAOYSA-N but-3-enoic acid;ethene Chemical compound C=C.OC(=O)CC=C DQXBYHZEEUGOBF-UHFFFAOYSA-N 0.000 description 24
- 239000000470 constituent Substances 0.000 description 24
- 239000000203 mixture Substances 0.000 description 24
- 239000000049 pigment Substances 0.000 description 24
- 229920001200 poly(ethylene-vinyl acetate) Polymers 0.000 description 24
- 230000032683 aging Effects 0.000 description 23
- 239000007787 solid Substances 0.000 description 22
- 238000010248 power generation Methods 0.000 description 21
- SOGAXMICEFXMKE-UHFFFAOYSA-N Butylmethacrylate Chemical compound CCCCOC(=O)C(C)=C SOGAXMICEFXMKE-UHFFFAOYSA-N 0.000 description 20
- 239000012790 adhesive layer Substances 0.000 description 20
- 230000000694 effects Effects 0.000 description 20
- 238000010438 heat treatment Methods 0.000 description 20
- 229920005989 resin Polymers 0.000 description 19
- 239000011347 resin Substances 0.000 description 19
- 229910052698 phosphorus Inorganic materials 0.000 description 18
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 16
- 229910052783 alkali metal Inorganic materials 0.000 description 16
- 230000007423 decrease Effects 0.000 description 16
- 239000010419 fine particle Substances 0.000 description 16
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 16
- 238000002844 melting Methods 0.000 description 16
- 230000008018 melting Effects 0.000 description 16
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 15
- 239000002245 particle Substances 0.000 description 15
- 230000035882 stress Effects 0.000 description 15
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 14
- 239000000654 additive Substances 0.000 description 14
- 238000011156 evaluation Methods 0.000 description 14
- 229920000139 polyethylene terephthalate Polymers 0.000 description 14
- 239000005020 polyethylene terephthalate Substances 0.000 description 14
- 239000000126 substance Substances 0.000 description 14
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 13
- 230000006870 function Effects 0.000 description 13
- 239000011574 phosphorus Substances 0.000 description 13
- 239000007790 solid phase Substances 0.000 description 13
- 150000001718 carbodiimides Chemical class 0.000 description 12
- 238000003786 synthesis reaction Methods 0.000 description 12
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 11
- IMSODMZESSGVBE-UHFFFAOYSA-N 2-Oxazoline Chemical compound C1CN=CO1 IMSODMZESSGVBE-UHFFFAOYSA-N 0.000 description 11
- 150000001340 alkali metals Chemical class 0.000 description 11
- 238000001816 cooling Methods 0.000 description 11
- 238000002360 preparation method Methods 0.000 description 11
- 239000004094 surface-active agent Substances 0.000 description 11
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 10
- 239000012298 atmosphere Substances 0.000 description 10
- 239000000872 buffer Substances 0.000 description 10
- 239000006172 buffering agent Substances 0.000 description 10
- CQEYYJKEWSMYFG-UHFFFAOYSA-N butyl acrylate Chemical compound CCCCOC(=O)C=C CQEYYJKEWSMYFG-UHFFFAOYSA-N 0.000 description 10
- 230000000052 comparative effect Effects 0.000 description 10
- JJQZDUKDJDQPMQ-UHFFFAOYSA-N dimethoxy(dimethyl)silane Chemical compound CO[Si](C)(C)OC JJQZDUKDJDQPMQ-UHFFFAOYSA-N 0.000 description 10
- 230000001965 increasing effect Effects 0.000 description 10
- 239000008188 pellet Substances 0.000 description 10
- ZNOCGWVLWPVKAO-UHFFFAOYSA-N trimethoxy(phenyl)silane Chemical compound CO[Si](OC)(OC)C1=CC=CC=C1 ZNOCGWVLWPVKAO-UHFFFAOYSA-N 0.000 description 10
- 229920002554 vinyl polymer Polymers 0.000 description 10
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 239000000155 melt Substances 0.000 description 9
- 239000002243 precursor Substances 0.000 description 9
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 8
- 239000002253 acid Substances 0.000 description 8
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 8
- 239000003054 catalyst Substances 0.000 description 8
- 239000006185 dispersion Substances 0.000 description 8
- 229920000058 polyacrylate Polymers 0.000 description 8
- 239000010936 titanium Substances 0.000 description 8
- ARCGXLSVLAOJQL-UHFFFAOYSA-N trimellitic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C(C(O)=O)=C1 ARCGXLSVLAOJQL-UHFFFAOYSA-N 0.000 description 8
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 7
- 239000013078 crystal Substances 0.000 description 7
- 229910001873 dinitrogen Inorganic materials 0.000 description 7
- 238000001125 extrusion Methods 0.000 description 7
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- TZCXTZWJZNENPQ-UHFFFAOYSA-L barium sulfate Chemical compound [Ba+2].[O-]S([O-])(=O)=O TZCXTZWJZNENPQ-UHFFFAOYSA-L 0.000 description 6
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 6
- 230000006866 deterioration Effects 0.000 description 6
- 229910001882 dioxygen Inorganic materials 0.000 description 6
- 230000007613 environmental effect Effects 0.000 description 6
- 239000000945 filler Substances 0.000 description 6
- 125000001905 inorganic group Chemical group 0.000 description 6
- 239000000178 monomer Substances 0.000 description 6
- 229920001225 polyester resin Polymers 0.000 description 6
- 239000004645 polyester resin Substances 0.000 description 6
- 230000009257 reactivity Effects 0.000 description 6
- XDLMVUHYZWKMMD-UHFFFAOYSA-N 3-trimethoxysilylpropyl 2-methylprop-2-enoate Chemical compound CO[Si](OC)(OC)CCCOC(=O)C(C)=C XDLMVUHYZWKMMD-UHFFFAOYSA-N 0.000 description 5
- 239000004925 Acrylic resin Substances 0.000 description 5
- 229920000178 Acrylic resin Polymers 0.000 description 5
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 5
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 5
- 229910052782 aluminium Inorganic materials 0.000 description 5
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 5
- 238000006243 chemical reaction Methods 0.000 description 5
- 238000004132 cross linking Methods 0.000 description 5
- 150000002009 diols Chemical class 0.000 description 5
- 239000012153 distilled water Substances 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 239000004744 fabric Substances 0.000 description 5
- 239000011521 glass Substances 0.000 description 5
- 230000009477 glass transition Effects 0.000 description 5
- 229910052809 inorganic oxide Inorganic materials 0.000 description 5
- 230000007774 longterm Effects 0.000 description 5
- 125000004437 phosphorous atom Chemical group 0.000 description 5
- 239000000377 silicon dioxide Substances 0.000 description 5
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 5
- 238000005979 thermal decomposition reaction Methods 0.000 description 5
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 4
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 4
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 4
- 229910000019 calcium carbonate Inorganic materials 0.000 description 4
- 235000010216 calcium carbonate Nutrition 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- WOZVHXUHUFLZGK-UHFFFAOYSA-N dimethyl terephthalate Chemical compound COC(=O)C1=CC=C(C(=O)OC)C=C1 WOZVHXUHUFLZGK-UHFFFAOYSA-N 0.000 description 4
- 238000001035 drying Methods 0.000 description 4
- 239000011888 foil Substances 0.000 description 4
- 239000001257 hydrogen Substances 0.000 description 4
- 229910052739 hydrogen Inorganic materials 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- 229910001502 inorganic halide Inorganic materials 0.000 description 4
- 229910052945 inorganic sulfide Inorganic materials 0.000 description 4
- 239000012948 isocyanate Substances 0.000 description 4
- 150000002513 isocyanates Chemical class 0.000 description 4
- 238000004898 kneading Methods 0.000 description 4
- 239000002346 layers by function Substances 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- 150000004767 nitrides Chemical class 0.000 description 4
- 239000003960 organic solvent Substances 0.000 description 4
- 229920000098 polyolefin Polymers 0.000 description 4
- 229920002635 polyurethane Polymers 0.000 description 4
- 239000004814 polyurethane Substances 0.000 description 4
- CYIDZMCFTVVTJO-UHFFFAOYSA-N pyromellitic acid Chemical compound OC(=O)C1=CC(C(O)=O)=C(C(O)=O)C=C1C(O)=O CYIDZMCFTVVTJO-UHFFFAOYSA-N 0.000 description 4
- 239000002356 single layer Substances 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 238000003860 storage Methods 0.000 description 4
- 239000004408 titanium dioxide Substances 0.000 description 4
- KKKKCPPTESQGQH-UHFFFAOYSA-N 2-(4,5-dihydro-1,3-oxazol-2-yl)-4,5-dihydro-1,3-oxazole Chemical compound O1CCN=C1C1=NCCO1 KKKKCPPTESQGQH-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 229920000877 Melamine resin Polymers 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 230000000996 additive effect Effects 0.000 description 3
- 125000000129 anionic group Chemical group 0.000 description 3
- 239000002216 antistatic agent Substances 0.000 description 3
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical compound C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000009833 condensation Methods 0.000 description 3
- 230000005494 condensation Effects 0.000 description 3
- 230000008602 contraction Effects 0.000 description 3
- 229920001577 copolymer Polymers 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- 229910052736 halogen Inorganic materials 0.000 description 3
- 150000002367 halogens Chemical class 0.000 description 3
- 239000000395 magnesium oxide Substances 0.000 description 3
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 3
- 235000012245 magnesium oxide Nutrition 0.000 description 3
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 3
- JDSHMPZPIAZGSV-UHFFFAOYSA-N melamine Chemical compound NC1=NC(N)=NC(N)=N1 JDSHMPZPIAZGSV-UHFFFAOYSA-N 0.000 description 3
- 229910052751 metal Inorganic materials 0.000 description 3
- 239000002184 metal Substances 0.000 description 3
- 238000000465 moulding Methods 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 150000002918 oxazolines Chemical class 0.000 description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 3
- 230000000704 physical effect Effects 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 239000002994 raw material Substances 0.000 description 3
- 230000000630 rising effect Effects 0.000 description 3
- 125000005372 silanol group Chemical group 0.000 description 3
- 229920002050 silicone resin Polymers 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 239000011593 sulfur Substances 0.000 description 3
- 239000000454 talc Substances 0.000 description 3
- 229910052623 talc Inorganic materials 0.000 description 3
- 235000012222 talc Nutrition 0.000 description 3
- ISIJQEHRDSCQIU-UHFFFAOYSA-N tert-butyl 2,7-diazaspiro[4.5]decane-7-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCCC11CNCC1 ISIJQEHRDSCQIU-UHFFFAOYSA-N 0.000 description 3
- 238000005809 transesterification reaction Methods 0.000 description 3
- 230000000007 visual effect Effects 0.000 description 3
- JJTUDXZGHPGLLC-QWWZWVQMSA-N (3r,6r)-3,6-dimethyl-1,4-dioxane-2,5-dione Chemical compound C[C@H]1OC(=O)[C@@H](C)OC1=O JJTUDXZGHPGLLC-QWWZWVQMSA-N 0.000 description 2
- DNIAPMSPPWPWGF-GSVOUGTGSA-N (R)-(-)-Propylene glycol Chemical compound C[C@@H](O)CO DNIAPMSPPWPWGF-GSVOUGTGSA-N 0.000 description 2
- QPFMBZIOSGYJDE-UHFFFAOYSA-N 1,1,2,2-tetrachloroethane Chemical compound ClC(Cl)C(Cl)Cl QPFMBZIOSGYJDE-UHFFFAOYSA-N 0.000 description 2
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 2
- OOSZCNKVJAVHJI-UHFFFAOYSA-N 1-[(4-fluorophenyl)methyl]piperazine Chemical compound C1=CC(F)=CC=C1CN1CCNCC1 OOSZCNKVJAVHJI-UHFFFAOYSA-N 0.000 description 2
- OHLSHRJUBRUKAN-UHFFFAOYSA-N 2,3-dihydroxyterephthalic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C(O)=C1O OHLSHRJUBRUKAN-UHFFFAOYSA-N 0.000 description 2
- KFNAHVKJFHDCSK-UHFFFAOYSA-N 2-[2-(4,5-dihydro-1,3-oxazol-2-yl)ethyl]-4,5-dihydro-1,3-oxazole Chemical compound N=1CCOC=1CCC1=NCCO1 KFNAHVKJFHDCSK-UHFFFAOYSA-N 0.000 description 2
- GZQKJQLFIGBEIE-UHFFFAOYSA-N 2-[4-(4,5-dihydro-1,3-oxazol-2-yl)butyl]-4,5-dihydro-1,3-oxazole Chemical compound N=1CCOC=1CCCCC1=NCCO1 GZQKJQLFIGBEIE-UHFFFAOYSA-N 0.000 description 2
- ZDNUPMSZKVCETJ-UHFFFAOYSA-N 2-[4-(4,5-dihydro-1,3-oxazol-2-yl)phenyl]-4,5-dihydro-1,3-oxazole Chemical compound O1CCN=C1C1=CC=C(C=2OCCN=2)C=C1 ZDNUPMSZKVCETJ-UHFFFAOYSA-N 0.000 description 2
- LDXQWLJXDIZULP-UHFFFAOYSA-N 2-[6-(4,5-dihydro-1,3-oxazol-2-yl)hexyl]-4,5-dihydro-1,3-oxazole Chemical compound N=1CCOC=1CCCCCCC1=NCCO1 LDXQWLJXDIZULP-UHFFFAOYSA-N 0.000 description 2
- MPPNPBNSYXFIBF-UHFFFAOYSA-N 2-[8-(4,5-dihydro-1,3-oxazol-2-yl)octyl]-4,5-dihydro-1,3-oxazole Chemical compound N=1CCOC=1CCCCCCCCC1=NCCO1 MPPNPBNSYXFIBF-UHFFFAOYSA-N 0.000 description 2
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 2
- 239000005995 Aluminium silicate Substances 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 239000004372 Polyvinyl alcohol Substances 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 2
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 2
- KKEYFWRCBNTPAC-UHFFFAOYSA-N Terephthalic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-N 0.000 description 2
- MCMNRKCIXSYSNV-UHFFFAOYSA-N Zirconium dioxide Chemical compound O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 description 2
- 239000006096 absorbing agent Substances 0.000 description 2
- 150000008065 acid anhydrides Chemical class 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 150000007933 aliphatic carboxylic acids Chemical class 0.000 description 2
- 235000012211 aluminium silicate Nutrition 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000003945 anionic surfactant Substances 0.000 description 2
- ADCOVFLJGNWWNZ-UHFFFAOYSA-N antimony trioxide Chemical compound O=[Sb]O[Sb]=O ADCOVFLJGNWWNZ-UHFFFAOYSA-N 0.000 description 2
- GCAIEATUVJFSMC-UHFFFAOYSA-N benzene-1,2,3,4-tetracarboxylic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C(C(O)=O)=C1C(O)=O GCAIEATUVJFSMC-UHFFFAOYSA-N 0.000 description 2
- QMKYBPDZANOJGF-UHFFFAOYSA-N benzene-1,3,5-tricarboxylic acid Chemical compound OC(=O)C1=CC(C(O)=O)=CC(C(O)=O)=C1 QMKYBPDZANOJGF-UHFFFAOYSA-N 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- 239000002981 blocking agent Substances 0.000 description 2
- 210000004899 c-terminal region Anatomy 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 238000005266 casting Methods 0.000 description 2
- 238000005229 chemical vapour deposition Methods 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 238000004040 coloring Methods 0.000 description 2
- 239000000805 composite resin Substances 0.000 description 2
- XATSGKXHVKZFKL-UHFFFAOYSA-N cyclohexylmethylcyclohexane;methanediimine Chemical compound N=C=N.C1CCCCC1CC1CCCCC1 XATSGKXHVKZFKL-UHFFFAOYSA-N 0.000 description 2
- 238000000354 decomposition reaction Methods 0.000 description 2
- 239000008367 deionised water Substances 0.000 description 2
- 229910021641 deionized water Inorganic materials 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 2
- SNRUBQQJIBEYMU-UHFFFAOYSA-N dodecane Chemical compound CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 2
- 239000000975 dye Substances 0.000 description 2
- 239000003822 epoxy resin Substances 0.000 description 2
- 125000004494 ethyl ester group Chemical group 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 239000003063 flame retardant Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 125000003055 glycidyl group Chemical group C(C1CO1)* 0.000 description 2
- 230000020169 heat generation Effects 0.000 description 2
- 238000009998 heat setting Methods 0.000 description 2
- 229920001519 homopolymer Polymers 0.000 description 2
- 238000007731 hot pressing Methods 0.000 description 2
- 239000010954 inorganic particle Substances 0.000 description 2
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 238000010030 laminating Methods 0.000 description 2
- 238000003475 lamination Methods 0.000 description 2
- YDSWCNNOKPMOTP-UHFFFAOYSA-N mellitic acid Chemical compound OC(=O)C1=C(C(O)=O)C(C(O)=O)=C(C(O)=O)C(C(O)=O)=C1C(O)=O YDSWCNNOKPMOTP-UHFFFAOYSA-N 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- FRVOOSTVCDXSRG-UHFFFAOYSA-N methanediimine;3,5,5-trimethylcyclohex-2-en-1-one Chemical compound N=C=N.CC1=CC(=O)CC(C)(C)C1 FRVOOSTVCDXSRG-UHFFFAOYSA-N 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 239000011259 mixed solution Substances 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- 239000002736 nonionic surfactant Substances 0.000 description 2
- XULSCZPZVQIMFM-IPZQJPLYSA-N odevixibat Chemical compound C12=CC(SC)=C(OCC(=O)N[C@@H](C(=O)N[C@@H](CC)C(O)=O)C=3C=CC(O)=CC=3)C=C2S(=O)(=O)NC(CCCC)(CCCC)CN1C1=CC=CC=C1 XULSCZPZVQIMFM-IPZQJPLYSA-N 0.000 description 2
- 239000011146 organic particle Substances 0.000 description 2
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 description 2
- 230000035699 permeability Effects 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 238000005240 physical vapour deposition Methods 0.000 description 2
- 229920002037 poly(vinyl butyral) polymer Polymers 0.000 description 2
- 229920000647 polyepoxide Polymers 0.000 description 2
- 229920002451 polyvinyl alcohol Polymers 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- WQGWDDDVZFFDIG-UHFFFAOYSA-N pyrogallol Chemical compound OC1=CC=CC(O)=C1O WQGWDDDVZFFDIG-UHFFFAOYSA-N 0.000 description 2
- 229910000077 silane Inorganic materials 0.000 description 2
- SCPYDCQAZCOKTP-UHFFFAOYSA-N silanol Chemical compound [SiH3]O SCPYDCQAZCOKTP-UHFFFAOYSA-N 0.000 description 2
- 229910052814 silicon oxide Inorganic materials 0.000 description 2
- 229940074545 sodium dihydrogen phosphate dihydrate Drugs 0.000 description 2
- 238000003892 spreading Methods 0.000 description 2
- 230000007480 spreading Effects 0.000 description 2
- 238000004544 sputter deposition Methods 0.000 description 2
- 125000005415 substituted alkoxy group Chemical group 0.000 description 2
- WYKYCHHWIJXDAO-UHFFFAOYSA-N tert-butyl 2-ethylhexaneperoxoate Chemical compound CCCCC(CC)C(=O)OOC(C)(C)C WYKYCHHWIJXDAO-UHFFFAOYSA-N 0.000 description 2
- 239000010409 thin film Substances 0.000 description 2
- XOLBLPGZBRYERU-UHFFFAOYSA-N tin dioxide Chemical compound O=[Sn]=O XOLBLPGZBRYERU-UHFFFAOYSA-N 0.000 description 2
- 229910001887 tin oxide Inorganic materials 0.000 description 2
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 2
- 238000007740 vapor deposition Methods 0.000 description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 2
- IQVLXQGNLCPZCL-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2,6-bis[(2-methylpropan-2-yl)oxycarbonylamino]hexanoate Chemical compound CC(C)(C)OC(=O)NCCCCC(NC(=O)OC(C)(C)C)C(=O)ON1C(=O)CCC1=O IQVLXQGNLCPZCL-UHFFFAOYSA-N 0.000 description 1
- GGAUUQHSCNMCAU-ZXZARUISSA-N (2s,3r)-butane-1,2,3,4-tetracarboxylic acid Chemical compound OC(=O)C[C@H](C(O)=O)[C@H](C(O)=O)CC(O)=O GGAUUQHSCNMCAU-ZXZARUISSA-N 0.000 description 1
- CEGRHPCDLKAHJD-UHFFFAOYSA-N 1,1,1-propanetricarboxylic acid Chemical compound CCC(C(O)=O)(C(O)=O)C(O)=O CEGRHPCDLKAHJD-UHFFFAOYSA-N 0.000 description 1
- 125000001140 1,4-phenylene group Chemical group [H]C1=C([H])C([*:2])=C([H])C([H])=C1[*:1] 0.000 description 1
- UYLNEXRLQPVSOZ-UHFFFAOYSA-N 1-[3-(hydroxymethyl)-2,2,5,5-tetramethylpyrrol-1-yl]ethanone Chemical compound CC(=O)N1C(C)(C)C=C(CO)C1(C)C UYLNEXRLQPVSOZ-UHFFFAOYSA-N 0.000 description 1
- FENFUOGYJVOCRY-UHFFFAOYSA-N 1-propoxypropan-2-ol Chemical compound CCCOCC(C)O FENFUOGYJVOCRY-UHFFFAOYSA-N 0.000 description 1
- RNFJDJUURJAICM-UHFFFAOYSA-N 2,2,4,4,6,6-hexaphenoxy-1,3,5-triaza-2$l^{5},4$l^{5},6$l^{5}-triphosphacyclohexa-1,3,5-triene Chemical compound N=1P(OC=2C=CC=CC=2)(OC=2C=CC=CC=2)=NP(OC=2C=CC=CC=2)(OC=2C=CC=CC=2)=NP=1(OC=1C=CC=CC=1)OC1=CC=CC=C1 RNFJDJUURJAICM-UHFFFAOYSA-N 0.000 description 1
- BEQDKWKSUMQVMX-UHFFFAOYSA-N 2,4-dimethyl-4,5-dihydro-1,3-oxazole Chemical compound CC1COC(C)=N1 BEQDKWKSUMQVMX-UHFFFAOYSA-N 0.000 description 1
- QEEZSWGDNCHFKC-UHFFFAOYSA-N 2-(4,5-dihydro-1,3-oxazol-2-ylmethyl)-4,5-dihydro-1,3-oxazole Chemical compound N=1CCOC=1CC1=NCCO1 QEEZSWGDNCHFKC-UHFFFAOYSA-N 0.000 description 1
- VJIADJVPBWWZEC-UHFFFAOYSA-N 2-(4-bicyclo[2.2.1]heptanyl)-4,5-dihydro-1,3-oxazole Chemical compound O1CCN=C1C1(C2)CCC2CC1 VJIADJVPBWWZEC-UHFFFAOYSA-N 0.000 description 1
- KMJHEUHUGMDAIY-UHFFFAOYSA-N 2-[10-(4,5-dihydro-1,3-oxazol-2-yl)decyl]-4,5-dihydro-1,3-oxazole Chemical compound N=1CCOC=1CCCCCCCCCCC1=NCCO1 KMJHEUHUGMDAIY-UHFFFAOYSA-N 0.000 description 1
- VOGDKZZTBPDRBD-UHFFFAOYSA-N 2-[2-(4,5-dihydro-1,3-oxazol-2-yl)phenyl]-4,5-dihydro-1,3-oxazole Chemical compound O1CCN=C1C1=CC=CC=C1C1=NCCO1 VOGDKZZTBPDRBD-UHFFFAOYSA-N 0.000 description 1
- MUBZACKCHQIRSY-UHFFFAOYSA-N 2-[3-(4,4-Dimethyl-5H-1,3-oxazol-2-yl)phenyl]-4,4-dimethyl-5H-1,3-oxazole Chemical compound CC1(C)COC(C=2C=C(C=CC=2)C=2OCC(C)(C)N=2)=N1 MUBZACKCHQIRSY-UHFFFAOYSA-N 0.000 description 1
- HMOZDINWBHMBSQ-UHFFFAOYSA-N 2-[3-(4,5-dihydro-1,3-oxazol-2-yl)phenyl]-4,5-dihydro-1,3-oxazole Chemical compound O1CCN=C1C1=CC=CC(C=2OCCN=2)=C1 HMOZDINWBHMBSQ-UHFFFAOYSA-N 0.000 description 1
- XRMPKRMBDKCXII-UHFFFAOYSA-N 2-[3-(4,5-dihydro-1,3-oxazol-2-yl)propyl]-4,5-dihydro-1,3-oxazole Chemical compound N=1CCOC=1CCCC1=NCCO1 XRMPKRMBDKCXII-UHFFFAOYSA-N 0.000 description 1
- GATDZUUWVARTOQ-UHFFFAOYSA-N 2-[4-(4,4-dimethyl-5h-1,3-oxazol-2-yl)phenyl]-4,4-dimethyl-5h-1,3-oxazole Chemical compound CC1(C)COC(C=2C=CC(=CC=2)C=2OCC(C)(C)N=2)=N1 GATDZUUWVARTOQ-UHFFFAOYSA-N 0.000 description 1
- ZKTPGXSTOJXHIG-UHFFFAOYSA-N 2-cyclohexyl-4,5-dihydro-1,3-oxazole Chemical compound O1CCN=C1C1CCCCC1 ZKTPGXSTOJXHIG-UHFFFAOYSA-N 0.000 description 1
- BQBSIHIZDSHADD-UHFFFAOYSA-N 2-ethenyl-4,5-dihydro-1,3-oxazole Chemical compound C=CC1=NCCO1 BQBSIHIZDSHADD-UHFFFAOYSA-N 0.000 description 1
- PBYIFPWEHGSUEY-UHFFFAOYSA-N 2-ethenyl-4-methyl-4,5-dihydro-1,3-oxazole Chemical compound CC1COC(C=C)=N1 PBYIFPWEHGSUEY-UHFFFAOYSA-N 0.000 description 1
- HMEVYZZCEGUONQ-UHFFFAOYSA-N 2-ethenyl-5-methyl-4,5-dihydro-1,3-oxazole Chemical compound CC1CN=C(C=C)O1 HMEVYZZCEGUONQ-UHFFFAOYSA-N 0.000 description 1
- WVDGHGISNBRCAO-UHFFFAOYSA-N 2-hydroxyisophthalic acid Chemical compound OC(=O)C1=CC=CC(C(O)=O)=C1O WVDGHGISNBRCAO-UHFFFAOYSA-N 0.000 description 1
- GUXJXWKCUUWCLX-UHFFFAOYSA-N 2-methyl-2-oxazoline Chemical compound CC1=NCCO1 GUXJXWKCUUWCLX-UHFFFAOYSA-N 0.000 description 1
- 125000003504 2-oxazolinyl group Chemical group O1C(=NCC1)* 0.000 description 1
- LPIQIQPLUVLISR-UHFFFAOYSA-N 2-prop-1-en-2-yl-4,5-dihydro-1,3-oxazole Chemical compound CC(=C)C1=NCCO1 LPIQIQPLUVLISR-UHFFFAOYSA-N 0.000 description 1
- RKFNMGWLDGIONW-UHFFFAOYSA-N 3,4,5-trihydroxychromen-2-one Chemical compound O1C(=O)C(O)=C(O)C2=C1C=CC=C2O RKFNMGWLDGIONW-UHFFFAOYSA-N 0.000 description 1
- QLHSZVGLSUHUOI-UHFFFAOYSA-N 3,5,6-trihydroxy-2-phenylchromen-4-one Chemical compound OC=1C(=O)C2=C(O)C(O)=CC=C2OC=1C1=CC=CC=C1 QLHSZVGLSUHUOI-UHFFFAOYSA-N 0.000 description 1
- KOAMXHRRVFDWRQ-UHFFFAOYSA-N 4,4-dimethyl-5h-1,3-oxazole Chemical compound CC1(C)COC=N1 KOAMXHRRVFDWRQ-UHFFFAOYSA-N 0.000 description 1
- UITKHKNFVCYWNG-UHFFFAOYSA-N 4-(3,4-dicarboxybenzoyl)phthalic acid Chemical compound C1=C(C(O)=O)C(C(=O)O)=CC=C1C(=O)C1=CC=C(C(O)=O)C(C(O)=O)=C1 UITKHKNFVCYWNG-UHFFFAOYSA-N 0.000 description 1
- CYDQOEWLBCCFJZ-UHFFFAOYSA-N 4-(4-fluorophenyl)oxane-4-carboxylic acid Chemical compound C=1C=C(F)C=CC=1C1(C(=O)O)CCOCC1 CYDQOEWLBCCFJZ-UHFFFAOYSA-N 0.000 description 1
- RWGLROKEYRSHME-UHFFFAOYSA-N 4-benzyl-4,5-dihydro-1,3-oxazole Chemical compound C=1C=CC=CC=1CC1COC=N1 RWGLROKEYRSHME-UHFFFAOYSA-N 0.000 description 1
- VITTZDWCUGTYIB-UHFFFAOYSA-N 4-butyl-4,5-dihydro-1,3-oxazole Chemical compound CCCCC1COC=N1 VITTZDWCUGTYIB-UHFFFAOYSA-N 0.000 description 1
- CJFNLGVLNYZLEA-UHFFFAOYSA-N 4-cyclohexyl-4,5-dihydro-1,3-oxazole Chemical compound C1OC=NC1C1CCCCC1 CJFNLGVLNYZLEA-UHFFFAOYSA-N 0.000 description 1
- RWMKXFCUXJWKBU-UHFFFAOYSA-N 4-ethyl-4,5-dihydro-1,3-oxazole Chemical compound CCC1COC=N1 RWMKXFCUXJWKBU-UHFFFAOYSA-N 0.000 description 1
- YTDWINDMGUQTBS-UHFFFAOYSA-N 4-hexyl-4,5-dihydro-1,3-oxazole Chemical compound CCCCCCC1COC=N1 YTDWINDMGUQTBS-UHFFFAOYSA-N 0.000 description 1
- 125000004203 4-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- WSGMRMBWRVIQRG-UHFFFAOYSA-N 4-methyl-2-[2-(4-methyl-4,5-dihydro-1,3-oxazol-2-yl)ethyl]-4,5-dihydro-1,3-oxazole Chemical compound CC1COC(CCC=2OCC(C)N=2)=N1 WSGMRMBWRVIQRG-UHFFFAOYSA-N 0.000 description 1
- RSCVPGQKACSLBP-UHFFFAOYSA-N 4-methyl-2-[3-(4-methyl-4,5-dihydro-1,3-oxazol-2-yl)phenyl]-4,5-dihydro-1,3-oxazole Chemical compound CC1COC(C=2C=C(C=CC=2)C=2OCC(C)N=2)=N1 RSCVPGQKACSLBP-UHFFFAOYSA-N 0.000 description 1
- FYQUELMPDYVBFY-UHFFFAOYSA-N 4-methyl-2-[4-(4-methyl-4,5-dihydro-1,3-oxazol-2-yl)phenyl]-4,5-dihydro-1,3-oxazole Chemical compound CC1COC(C=2C=CC(=CC=2)C=2OCC(C)N=2)=N1 FYQUELMPDYVBFY-UHFFFAOYSA-N 0.000 description 1
- MBLQIMSKMPEILU-UHFFFAOYSA-N 4-methyl-2-prop-1-en-2-yl-4,5-dihydro-1,3-oxazole Chemical compound CC1COC(C(C)=C)=N1 MBLQIMSKMPEILU-UHFFFAOYSA-N 0.000 description 1
- IFIUFEBEPGGBIJ-UHFFFAOYSA-N 4-methyl-4,5-dihydro-1,3-oxazole Chemical compound CC1COC=N1 IFIUFEBEPGGBIJ-UHFFFAOYSA-N 0.000 description 1
- DBTPMQIQJZFVAB-UHFFFAOYSA-N 4-phenyl-4,5-dihydro-1,3-oxazole Chemical compound C1OC=NC1C1=CC=CC=C1 DBTPMQIQJZFVAB-UHFFFAOYSA-N 0.000 description 1
- HLIYUPUYSLFMEB-UHFFFAOYSA-N 4-propyl-4,5-dihydro-1,3-oxazole Chemical compound CCCC1COC=N1 HLIYUPUYSLFMEB-UHFFFAOYSA-N 0.000 description 1
- KWSLGOVYXMQPPX-UHFFFAOYSA-N 5-[3-(trifluoromethyl)phenyl]-2h-tetrazole Chemical compound FC(F)(F)C1=CC=CC(C2=NNN=N2)=C1 KWSLGOVYXMQPPX-UHFFFAOYSA-N 0.000 description 1
- IRHWINGBSHBXAD-UHFFFAOYSA-N 5-ethyl-2-prop-1-en-2-yl-4,5-dihydro-1,3-oxazole Chemical compound CCC1CN=C(C(C)=C)O1 IRHWINGBSHBXAD-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- JBRZTFJDHDCESZ-UHFFFAOYSA-N AsGa Chemical compound [As]#[Ga] JBRZTFJDHDCESZ-UHFFFAOYSA-N 0.000 description 1
- 229930185605 Bisphenol Natural products 0.000 description 1
- 229920002799 BoPET Polymers 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- QCMYYKRYFNMIEC-UHFFFAOYSA-N COP(O)=O Chemical compound COP(O)=O QCMYYKRYFNMIEC-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- BRLQWZUYTZBJKN-UHFFFAOYSA-N Epichlorohydrin Chemical compound ClCC1CO1 BRLQWZUYTZBJKN-UHFFFAOYSA-N 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- VWYIWOYBERNXLX-KTKRTIGZSA-N Glycidyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC1CO1 VWYIWOYBERNXLX-KTKRTIGZSA-N 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- BELBBZDIHDAJOR-UHFFFAOYSA-N Phenolsulfonephthalein Chemical compound C1=CC(O)=CC=C1C1(C=2C=CC(O)=CC=2)C2=CC=CC=C2S(=O)(=O)O1 BELBBZDIHDAJOR-UHFFFAOYSA-N 0.000 description 1
- FQYUMYWMJTYZTK-UHFFFAOYSA-N Phenyl glycidyl ether Chemical compound C1OC1COC1=CC=CC=C1 FQYUMYWMJTYZTK-UHFFFAOYSA-N 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical compound OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- 239000004642 Polyimide Substances 0.000 description 1
- 239000004721 Polyphenylene oxide Substances 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- ZJCCRDAZUWHFQH-UHFFFAOYSA-N Trimethylolpropane Chemical compound CCC(CO)(CO)CO ZJCCRDAZUWHFQH-UHFFFAOYSA-N 0.000 description 1
- 238000005299 abrasion Methods 0.000 description 1
- 230000001133 acceleration Effects 0.000 description 1
- 125000004018 acid anhydride group Chemical group 0.000 description 1
- 239000003377 acid catalyst Substances 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- JISINGGEJRZDCJ-UHFFFAOYSA-N adamantane-1,2,2,3,4,4-hexacarboxylic acid Chemical compound C1C(C2)CC3CC1(C(=O)O)C(C(O)=O)(C(O)=O)C2(C(O)=O)C3(C(O)=O)C(O)=O JISINGGEJRZDCJ-UHFFFAOYSA-N 0.000 description 1
- JXRZUHPTUFAFSQ-UHFFFAOYSA-N adamantane-1,2,2,3,4-pentacarboxylic acid Chemical compound C1C(C2)CC3(C(O)=O)CC1C(C(=O)O)C2(C(O)=O)C3(C(O)=O)C(O)=O JXRZUHPTUFAFSQ-UHFFFAOYSA-N 0.000 description 1
- JZKNMIIVCWHHGX-UHFFFAOYSA-N adamantane-1,2,2,3-tetracarboxylic acid Chemical compound C1C(C2)CC3CC1(C(=O)O)C(C(O)=O)(C(O)=O)C2(C(O)=O)C3 JZKNMIIVCWHHGX-UHFFFAOYSA-N 0.000 description 1
- 239000002313 adhesive film Substances 0.000 description 1
- 150000007824 aliphatic compounds Chemical class 0.000 description 1
- 150000005215 alkyl ethers Chemical class 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 229910021417 amorphous silicon Inorganic materials 0.000 description 1
- MLVUJAOMAFIVBM-UHFFFAOYSA-N anthracene-1,2,3,4,5,10-hexacarboxylic acid Chemical compound OC(=O)C1=C(C(O)=O)C(C(O)=O)=C2C(C(O)=O)=C3C(C(=O)O)=CC=CC3=CC2=C1C(O)=O MLVUJAOMAFIVBM-UHFFFAOYSA-N 0.000 description 1
- STTZXRSDHVPZRO-UHFFFAOYSA-N anthracene-1,2,3,4,5,6,10-heptacarboxylic acid Chemical compound OC(=O)C1=C(C(O)=O)C(C(O)=O)=C(C(O)=O)C2=C(C(O)=O)C3=C(C(O)=O)C(C(=O)O)=CC=C3C=C21 STTZXRSDHVPZRO-UHFFFAOYSA-N 0.000 description 1
- YUZBGMHFHKMDLM-UHFFFAOYSA-N anthracene-1,2,3,4,5,6,7,10-octacarboxylic acid Chemical compound OC(=O)C1=C(C(O)=O)C(C(O)=O)=C2C(C(O)=O)=C(C(C(O)=O)=C(C(C(=O)O)=C3)C(O)=O)C3=CC2=C1C(O)=O YUZBGMHFHKMDLM-UHFFFAOYSA-N 0.000 description 1
- VBSLSHMYAWTHQM-UHFFFAOYSA-N anthracene-1,2,3,4,9-pentacarboxylic acid Chemical compound C1=CC=CC2=CC3=C(C(O)=O)C(C(=O)O)=C(C(O)=O)C(C(O)=O)=C3C(C(O)=O)=C21 VBSLSHMYAWTHQM-UHFFFAOYSA-N 0.000 description 1
- RSHYBLDYLNAJJV-UHFFFAOYSA-N anthracene-1,2,3,4-tetracarboxylic acid Chemical compound C1=CC=CC2=CC3=C(C(O)=O)C(C(=O)O)=C(C(O)=O)C(C(O)=O)=C3C=C21 RSHYBLDYLNAJJV-UHFFFAOYSA-N 0.000 description 1
- HGMQPWXYDSTESW-UHFFFAOYSA-N anthracene-1,2,3-tricarboxylic acid Chemical compound C1=CC=C2C=C(C(C(O)=O)=C(C(C(=O)O)=C3)C(O)=O)C3=CC2=C1 HGMQPWXYDSTESW-UHFFFAOYSA-N 0.000 description 1
- SLOLMTWBBAFOKJ-UHFFFAOYSA-N anthracene-1,2,3-triol Chemical compound C1=CC=C2C=C(C(O)=C(C(O)=C3)O)C3=CC2=C1 SLOLMTWBBAFOKJ-UHFFFAOYSA-N 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 150000001491 aromatic compounds Chemical class 0.000 description 1
- 125000000732 arylene group Chemical group 0.000 description 1
- 239000010426 asphalt Substances 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- IRERQBUNZFJFGC-UHFFFAOYSA-L azure blue Chemical compound [Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Al+3].[Al+3].[Al+3].[Al+3].[Al+3].[Al+3].[S-]S[S-].[O-][Si]([O-])([O-])[O-].[O-][Si]([O-])([O-])[O-].[O-][Si]([O-])([O-])[O-].[O-][Si]([O-])([O-])[O-].[O-][Si]([O-])([O-])[O-].[O-][Si]([O-])([O-])[O-] IRERQBUNZFJFGC-UHFFFAOYSA-L 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- QNSOHXTZPUMONC-UHFFFAOYSA-N benzene pentacarboxylic acid Natural products OC(=O)C1=CC(C(O)=O)=C(C(O)=O)C(C(O)=O)=C1C(O)=O QNSOHXTZPUMONC-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- MRDNMQDAPCZRNE-UHFFFAOYSA-N bis(oxiran-2-ylmethyl) 2-heptadecylpropanedioate Chemical compound C1OC1COC(=O)C(CCCCCCCCCCCCCCCCC)C(=O)OCC1CO1 MRDNMQDAPCZRNE-UHFFFAOYSA-N 0.000 description 1
- VKEFGOTVPWVWCH-UHFFFAOYSA-N bis(oxiran-2-ylmethyl) 2-methylbenzene-1,4-dicarboxylate Chemical compound CC1=CC(C(=O)OCC2OC2)=CC=C1C(=O)OCC1CO1 VKEFGOTVPWVWCH-UHFFFAOYSA-N 0.000 description 1
- JRPRCOLKIYRSNH-UHFFFAOYSA-N bis(oxiran-2-ylmethyl) benzene-1,2-dicarboxylate Chemical compound C=1C=CC=C(C(=O)OCC2OC2)C=1C(=O)OCC1CO1 JRPRCOLKIYRSNH-UHFFFAOYSA-N 0.000 description 1
- ZXOATMQSUNJNNG-UHFFFAOYSA-N bis(oxiran-2-ylmethyl) benzene-1,3-dicarboxylate Chemical compound C=1C=CC(C(=O)OCC2OC2)=CC=1C(=O)OCC1CO1 ZXOATMQSUNJNNG-UHFFFAOYSA-N 0.000 description 1
- NEPKLUNSRVEBIX-UHFFFAOYSA-N bis(oxiran-2-ylmethyl) benzene-1,4-dicarboxylate Chemical compound C=1C=C(C(=O)OCC2OC2)C=CC=1C(=O)OCC1CO1 NEPKLUNSRVEBIX-UHFFFAOYSA-N 0.000 description 1
- JQDCYGOHLMJDNA-UHFFFAOYSA-N bis(oxiran-2-ylmethyl) butanedioate Chemical compound C1OC1COC(=O)CCC(=O)OCC1CO1 JQDCYGOHLMJDNA-UHFFFAOYSA-N 0.000 description 1
- KTPIWUHKYIJBCR-UHFFFAOYSA-N bis(oxiran-2-ylmethyl) cyclohex-4-ene-1,2-dicarboxylate Chemical compound C1C=CCC(C(=O)OCC2OC2)C1C(=O)OCC1CO1 KTPIWUHKYIJBCR-UHFFFAOYSA-N 0.000 description 1
- UTDPRMMIFFJALK-UHFFFAOYSA-N bis(oxiran-2-ylmethyl) cyclohexane-1,1-dicarboxylate Chemical compound C1CCCCC1(C(=O)OCC1OC1)C(=O)OCC1CO1 UTDPRMMIFFJALK-UHFFFAOYSA-N 0.000 description 1
- XFUOBHWPTSIEOV-UHFFFAOYSA-N bis(oxiran-2-ylmethyl) cyclohexane-1,2-dicarboxylate Chemical compound C1CCCC(C(=O)OCC2OC2)C1C(=O)OCC1CO1 XFUOBHWPTSIEOV-UHFFFAOYSA-N 0.000 description 1
- NFVGWOSADNLNHZ-UHFFFAOYSA-N bis(oxiran-2-ylmethyl) decanedioate Chemical compound C1OC1COC(=O)CCCCCCCCC(=O)OCC1CO1 NFVGWOSADNLNHZ-UHFFFAOYSA-N 0.000 description 1
- KBWLNCUTNDKMPN-UHFFFAOYSA-N bis(oxiran-2-ylmethyl) hexanedioate Chemical compound C1OC1COC(=O)CCCCC(=O)OCC1CO1 KBWLNCUTNDKMPN-UHFFFAOYSA-N 0.000 description 1
- AUNDMQBAPCPNJA-UHFFFAOYSA-N bis(oxiran-2-ylmethyl) naphthalene-1,2-dicarboxylate Chemical compound C=1C=C2C=CC=CC2=C(C(=O)OCC2OC2)C=1C(=O)OCC1CO1 AUNDMQBAPCPNJA-UHFFFAOYSA-N 0.000 description 1
- 239000007844 bleaching agent Substances 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- ULCATBZEAGEBAW-UHFFFAOYSA-N butane-1,1,1,2,2,3,3-heptacarboxylic acid Chemical compound OC(=O)C(C)(C(O)=O)C(C(O)=O)(C(O)=O)C(C(O)=O)(C(O)=O)C(O)=O ULCATBZEAGEBAW-UHFFFAOYSA-N 0.000 description 1
- VSPPGHYPJUBMQG-UHFFFAOYSA-N butane-1,1,1,2,2-pentacarboxylic acid Chemical compound CCC(C(O)=O)(C(O)=O)C(C(O)=O)(C(O)=O)C(O)=O VSPPGHYPJUBMQG-UHFFFAOYSA-N 0.000 description 1
- ZNFNDZCXTPWRLQ-UHFFFAOYSA-N butane-1,1,1-tricarboxylic acid Chemical compound CCCC(C(O)=O)(C(O)=O)C(O)=O ZNFNDZCXTPWRLQ-UHFFFAOYSA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- RPPBZEBXAAZZJH-UHFFFAOYSA-N cadmium telluride Chemical compound [Te]=[Cd] RPPBZEBXAAZZJH-UHFFFAOYSA-N 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 239000006229 carbon black Substances 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 229920006317 cationic polymer Polymers 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 229910052570 clay Inorganic materials 0.000 description 1
- 239000011247 coating layer Substances 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 239000006258 conductive agent Substances 0.000 description 1
- 238000011437 continuous method Methods 0.000 description 1
- XCJYREBRNVKWGJ-UHFFFAOYSA-N copper(II) phthalocyanine Chemical compound [Cu+2].C12=CC=CC=C2C(N=C2[N-]C(C3=CC=CC=C32)=N2)=NC1=NC([C]1C=CC=CC1=1)=NC=1N=C1[C]3C=CC=CC3=C2[N-]1 XCJYREBRNVKWGJ-UHFFFAOYSA-N 0.000 description 1
- UIPVMGDJUWUZEI-UHFFFAOYSA-N copper;selanylideneindium Chemical compound [Cu].[In]=[Se] UIPVMGDJUWUZEI-UHFFFAOYSA-N 0.000 description 1
- 229920006038 crystalline resin Polymers 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- WLWKIJKUDWYINL-UHFFFAOYSA-N cyclohexane-1,1,2,2,3,3-hexacarboxylic acid Chemical compound OC(=O)C1(C(O)=O)CCCC(C(O)=O)(C(O)=O)C1(C(O)=O)C(O)=O WLWKIJKUDWYINL-UHFFFAOYSA-N 0.000 description 1
- DTLWQZBOAYMHTO-UHFFFAOYSA-N cyclohexane-1,1,2,2,3-pentacarboxylic acid Chemical compound OC(=O)C1CCCC(C(O)=O)(C(O)=O)C1(C(O)=O)C(O)=O DTLWQZBOAYMHTO-UHFFFAOYSA-N 0.000 description 1
- RZIPTXDCNDIINL-UHFFFAOYSA-N cyclohexane-1,1,2,2-tetracarboxylic acid Chemical compound OC(=O)C1(C(O)=O)CCCCC1(C(O)=O)C(O)=O RZIPTXDCNDIINL-UHFFFAOYSA-N 0.000 description 1
- MTGGGUZRAGKYEM-UHFFFAOYSA-N cyclopentane-1,1,2,2,3-pentacarboxylic acid Chemical compound OC(=O)C1CCC(C(O)=O)(C(O)=O)C1(C(O)=O)C(O)=O MTGGGUZRAGKYEM-UHFFFAOYSA-N 0.000 description 1
- STZIXLPVKZUAMV-UHFFFAOYSA-N cyclopentane-1,1,2,2-tetracarboxylic acid Chemical compound OC(=O)C1(C(O)=O)CCCC1(C(O)=O)C(O)=O STZIXLPVKZUAMV-UHFFFAOYSA-N 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 238000005034 decoration Methods 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000032798 delamination Effects 0.000 description 1
- 230000002542 deteriorative effect Effects 0.000 description 1
- 125000001664 diethylamino group Chemical group [H]C([H])([H])C([H])([H])N(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- GYZLOYUZLJXAJU-UHFFFAOYSA-N diglycidyl ether Chemical class C1OC1COCC1CO1 GYZLOYUZLJXAJU-UHFFFAOYSA-N 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- UZLGHNUASUZUOR-UHFFFAOYSA-L dipotassium;3-carboxy-3-hydroxypentanedioate Chemical compound [K+].[K+].OC(=O)CC(O)(C([O-])=O)CC([O-])=O UZLGHNUASUZUOR-UHFFFAOYSA-L 0.000 description 1
- XBMOWLAOINHDLR-UHFFFAOYSA-N dipotassium;hydrogen phosphite Chemical compound [K+].[K+].OP([O-])[O-] XBMOWLAOINHDLR-UHFFFAOYSA-N 0.000 description 1
- 208000028659 discharge Diseases 0.000 description 1
- 235000019262 disodium citrate Nutrition 0.000 description 1
- 239000002526 disodium citrate Substances 0.000 description 1
- 229940079896 disodium hydrogen citrate Drugs 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- CEYULKASIQJZGP-UHFFFAOYSA-L disodium;2-(carboxymethyl)-2-hydroxybutanedioate Chemical compound [Na+].[Na+].[O-]C(=O)CC(O)(C(=O)O)CC([O-])=O CEYULKASIQJZGP-UHFFFAOYSA-L 0.000 description 1
- ZRRLFMPOAYZELW-UHFFFAOYSA-N disodium;hydrogen phosphite Chemical compound [Na+].[Na+].OP([O-])[O-] ZRRLFMPOAYZELW-UHFFFAOYSA-N 0.000 description 1
- BXKDSDJJOVIHMX-UHFFFAOYSA-N edrophonium chloride Chemical compound [Cl-].CC[N+](C)(C)C1=CC=CC(O)=C1 BXKDSDJJOVIHMX-UHFFFAOYSA-N 0.000 description 1
- 238000005566 electron beam evaporation Methods 0.000 description 1
- 238000004945 emulsification Methods 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- CXPTYTWMNYNURO-UHFFFAOYSA-N ethane-1,1,1,2,2,2-hexacarboxylic acid Chemical compound OC(=O)C(C(O)=O)(C(O)=O)C(C(O)=O)(C(O)=O)C(O)=O CXPTYTWMNYNURO-UHFFFAOYSA-N 0.000 description 1
- DIZMSOQXELWOLG-UHFFFAOYSA-N ethane-1,1,1,2,2-pentacarboxylic acid Chemical compound OC(=O)C(C(O)=O)C(C(O)=O)(C(O)=O)C(O)=O DIZMSOQXELWOLG-UHFFFAOYSA-N 0.000 description 1
- HFKBPAKZRASAGX-UHFFFAOYSA-N ethane-1,1,1,2-tetracarboxylic acid Chemical compound OC(=O)CC(C(O)=O)(C(O)=O)C(O)=O HFKBPAKZRASAGX-UHFFFAOYSA-N 0.000 description 1
- MCOFCVVDZHTYIX-UHFFFAOYSA-N ethane-1,1,1-tricarboxylic acid Chemical compound OC(=O)C(C)(C(O)=O)C(O)=O MCOFCVVDZHTYIX-UHFFFAOYSA-N 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- AIJZIRPGCQPZSL-UHFFFAOYSA-N ethylenetetracarboxylic acid Chemical compound OC(=O)C(C(O)=O)=C(C(O)=O)C(O)=O AIJZIRPGCQPZSL-UHFFFAOYSA-N 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- NBVXSUQYWXRMNV-UHFFFAOYSA-N fluoromethane Chemical compound FC NBVXSUQYWXRMNV-UHFFFAOYSA-N 0.000 description 1
- 238000005338 heat storage Methods 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 229910010272 inorganic material Inorganic materials 0.000 description 1
- 239000011147 inorganic material Substances 0.000 description 1
- 239000001023 inorganic pigment Substances 0.000 description 1
- 239000011229 interlayer Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 238000007733 ion plating Methods 0.000 description 1
- 238000001659 ion-beam spectroscopy Methods 0.000 description 1
- 238000010884 ion-beam technique Methods 0.000 description 1
- 230000001788 irregular Effects 0.000 description 1
- DXDRHHKMWQZJHT-FPYGCLRLSA-N isoliquiritigenin Chemical compound C1=CC(O)=CC=C1\C=C\C(=O)C1=CC=C(O)C=C1O DXDRHHKMWQZJHT-FPYGCLRLSA-N 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 125000005647 linker group Chemical group 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- UEGPKNKPLBYCNK-UHFFFAOYSA-L magnesium acetate Chemical compound [Mg+2].CC([O-])=O.CC([O-])=O UEGPKNKPLBYCNK-UHFFFAOYSA-L 0.000 description 1
- 239000011654 magnesium acetate Substances 0.000 description 1
- 235000011285 magnesium acetate Nutrition 0.000 description 1
- 229940069446 magnesium acetate Drugs 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 239000006224 matting agent Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- RKGQUTNLMXNUME-UHFFFAOYSA-N methanetricarboxylic acid Chemical compound OC(=O)C(C(O)=O)C(O)=O RKGQUTNLMXNUME-UHFFFAOYSA-N 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 239000010445 mica Substances 0.000 description 1
- 229910052618 mica group Inorganic materials 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- XONPDZSGENTBNJ-UHFFFAOYSA-N molecular hydrogen;sodium Chemical compound [Na].[H][H] XONPDZSGENTBNJ-UHFFFAOYSA-N 0.000 description 1
- 229910021421 monocrystalline silicon Inorganic materials 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- HWPKGOGLCKPRLZ-UHFFFAOYSA-M monosodium citrate Chemical compound [Na+].OC(=O)CC(O)(C([O-])=O)CC(O)=O HWPKGOGLCKPRLZ-UHFFFAOYSA-M 0.000 description 1
- 235000018342 monosodium citrate Nutrition 0.000 description 1
- 239000002524 monosodium citrate Substances 0.000 description 1
- 229910000403 monosodium phosphate Inorganic materials 0.000 description 1
- 235000019799 monosodium phosphate Nutrition 0.000 description 1
- SQEHYBILZVXINP-UHFFFAOYSA-N n'-tert-butyl-n-propan-2-ylmethanediimine Chemical compound CC(C)N=C=NC(C)(C)C SQEHYBILZVXINP-UHFFFAOYSA-N 0.000 description 1
- JEQPWXGHMRFTRF-UHFFFAOYSA-N n,n'-bis(2-methylpropyl)methanediimine Chemical compound CC(C)CN=C=NCC(C)C JEQPWXGHMRFTRF-UHFFFAOYSA-N 0.000 description 1
- NASVTBDJHWPMOO-UHFFFAOYSA-N n,n'-dimethylmethanediimine Chemical compound CN=C=NC NASVTBDJHWPMOO-UHFFFAOYSA-N 0.000 description 1
- ATYQZACNIHLXIS-UHFFFAOYSA-N n,n'-dinaphthalen-2-ylmethanediimine Chemical compound C1=CC=CC2=CC(N=C=NC3=CC4=CC=CC=C4C=C3)=CC=C21 ATYQZACNIHLXIS-UHFFFAOYSA-N 0.000 description 1
- NWBVGPKHJHHPTA-UHFFFAOYSA-N n,n'-dioctylmethanediimine Chemical compound CCCCCCCCN=C=NCCCCCCCC NWBVGPKHJHHPTA-UHFFFAOYSA-N 0.000 description 1
- CMESPBFFDMPSIY-UHFFFAOYSA-N n,n'-diphenylmethanediimine Chemical compound C1=CC=CC=C1N=C=NC1=CC=CC=C1 CMESPBFFDMPSIY-UHFFFAOYSA-N 0.000 description 1
- IDVWLLCLTVBSCS-UHFFFAOYSA-N n,n'-ditert-butylmethanediimine Chemical compound CC(C)(C)N=C=NC(C)(C)C IDVWLLCLTVBSCS-UHFFFAOYSA-N 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 1
- GHHOKORHSWHMCC-UHFFFAOYSA-N naphthalene-1,2,3,4,5,6,7,8-octacarboxylic acid Chemical compound OC(=O)C1=C(C(O)=O)C(C(O)=O)=C(C(O)=O)C2=C(C(O)=O)C(C(=O)O)=C(C(O)=O)C(C(O)=O)=C21 GHHOKORHSWHMCC-UHFFFAOYSA-N 0.000 description 1
- YSMULDKSBPCDFH-UHFFFAOYSA-N naphthalene-1,2,3,4,5,6,7-heptacarboxylic acid Chemical compound OC(=O)C1=C(C(O)=O)C(C(O)=O)=C2C(C(O)=O)=C(C(O)=O)C(C(=O)O)=CC2=C1C(O)=O YSMULDKSBPCDFH-UHFFFAOYSA-N 0.000 description 1
- NMIOQQFCVOLKMH-UHFFFAOYSA-N naphthalene-1,2,3,4,5,6-hexacarboxylic acid Chemical compound OC(=O)C1=C(C(O)=O)C(C(O)=O)=C(C(O)=O)C2=C(C(O)=O)C(C(=O)O)=CC=C21 NMIOQQFCVOLKMH-UHFFFAOYSA-N 0.000 description 1
- UAKKJHRECUCKMN-UHFFFAOYSA-N naphthalene-1,2,3,4,5-pentacarboxylic acid Chemical compound OC(=O)C1=C(C(O)=O)C(C(O)=O)=C2C(C(=O)O)=CC=CC2=C1C(O)=O UAKKJHRECUCKMN-UHFFFAOYSA-N 0.000 description 1
- KVQQRFDIKYXJTJ-UHFFFAOYSA-N naphthalene-1,2,3-tricarboxylic acid Chemical compound C1=CC=C2C(C(O)=O)=C(C(O)=O)C(C(=O)O)=CC2=C1 KVQQRFDIKYXJTJ-UHFFFAOYSA-N 0.000 description 1
- NCIAGQNZQHYKGR-UHFFFAOYSA-N naphthalene-1,2,3-triol Chemical compound C1=CC=C2C(O)=C(O)C(O)=CC2=C1 NCIAGQNZQHYKGR-UHFFFAOYSA-N 0.000 description 1
- 239000012860 organic pigment Substances 0.000 description 1
- LOGTZDQTPQYKEN-HZJYTTRNSA-N oxiran-2-ylmethyl (9z,12z)-octadeca-9,12-dienoate Chemical compound CCCCC\C=C/C\C=C/CCCCCCCC(=O)OCC1CO1 LOGTZDQTPQYKEN-HZJYTTRNSA-N 0.000 description 1
- XRQKARZTFMEBBY-UHFFFAOYSA-N oxiran-2-ylmethyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1CO1 XRQKARZTFMEBBY-UHFFFAOYSA-N 0.000 description 1
- BEJZOPYRAQVYHS-UHFFFAOYSA-N oxiran-2-ylmethyl cyclohexanecarboxylate Chemical compound C1CCCCC1C(=O)OCC1CO1 BEJZOPYRAQVYHS-UHFFFAOYSA-N 0.000 description 1
- PGXFPHPLDLNGQY-UHFFFAOYSA-N oxiran-2-ylmethyl docosanoate Chemical compound CCCCCCCCCCCCCCCCCCCCCC(=O)OCC1CO1 PGXFPHPLDLNGQY-UHFFFAOYSA-N 0.000 description 1
- PTLZMJYQEBOHHM-UHFFFAOYSA-N oxiran-2-ylmethyl dodecanoate Chemical compound CCCCCCCCCCCC(=O)OCC1CO1 PTLZMJYQEBOHHM-UHFFFAOYSA-N 0.000 description 1
- KYVUJPJYTYQNGJ-UHFFFAOYSA-N oxiran-2-ylmethyl hexadecanoate Chemical compound CCCCCCCCCCCCCCCC(=O)OCC1CO1 KYVUJPJYTYQNGJ-UHFFFAOYSA-N 0.000 description 1
- DJTYNOVDSWHTJM-UHFFFAOYSA-N oxiran-2-ylmethyl nonanoate Chemical compound CCCCCCCCC(=O)OCC1CO1 DJTYNOVDSWHTJM-UHFFFAOYSA-N 0.000 description 1
- LPNBBFKOUUSUDB-UHFFFAOYSA-N p-toluenecarboxylic acid Natural products CC1=CC=C(C(O)=O)C=C1 LPNBBFKOUUSUDB-UHFFFAOYSA-N 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- WXZMFSXDPGVJKK-UHFFFAOYSA-N pentaerythritol Chemical compound OCC(CO)(CO)CO WXZMFSXDPGVJKK-UHFFFAOYSA-N 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 125000002080 perylenyl group Chemical group C1(=CC=C2C=CC=C3C4=CC=CC5=CC=CC(C1=C23)=C45)* 0.000 description 1
- CSHWQDPOILHKBI-UHFFFAOYSA-N peryrene Natural products C1=CC(C2=CC=CC=3C2=C2C=CC=3)=C3C2=CC=CC3=C1 CSHWQDPOILHKBI-UHFFFAOYSA-N 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 239000005011 phenolic resin Substances 0.000 description 1
- 229960003531 phenolsulfonphthalein Drugs 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- 125000000286 phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- ACVYVLVWPXVTIT-UHFFFAOYSA-N phosphinic acid Chemical compound O[PH2]=O ACVYVLVWPXVTIT-UHFFFAOYSA-N 0.000 description 1
- 235000011007 phosphoric acid Nutrition 0.000 description 1
- PJNZPQUBCPKICU-UHFFFAOYSA-N phosphoric acid;potassium Chemical compound [K].OP(O)(O)=O PJNZPQUBCPKICU-UHFFFAOYSA-N 0.000 description 1
- 150000003018 phosphorus compounds Chemical class 0.000 description 1
- IEQIEDJGQAUEQZ-UHFFFAOYSA-N phthalocyanine Chemical compound N1C(N=C2C3=CC=CC=C3C(N=C3C4=CC=CC=C4C(=N4)N3)=N2)=C(C=CC=C2)C2=C1N=C1C2=CC=CC=C2C4=N1 IEQIEDJGQAUEQZ-UHFFFAOYSA-N 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 1
- 229920001495 poly(sodium acrylate) polymer Polymers 0.000 description 1
- 239000004584 polyacrylic acid Substances 0.000 description 1
- 229920001707 polybutylene terephthalate Polymers 0.000 description 1
- 238000006068 polycondensation reaction Methods 0.000 description 1
- 229910021420 polycrystalline silicon Inorganic materials 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 229920001721 polyimide Polymers 0.000 description 1
- 239000002685 polymerization catalyst Substances 0.000 description 1
- 230000000379 polymerizing effect Effects 0.000 description 1
- 239000004926 polymethyl methacrylate Substances 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229920005749 polyurethane resin Polymers 0.000 description 1
- 229960003975 potassium Drugs 0.000 description 1
- 235000007686 potassium Nutrition 0.000 description 1
- KYKNRZGSIGMXFH-ZVGUSBNCSA-M potassium bitartrate Chemical compound [K+].OC(=O)[C@H](O)[C@@H](O)C([O-])=O KYKNRZGSIGMXFH-ZVGUSBNCSA-M 0.000 description 1
- IWZKICVEHNUQTL-UHFFFAOYSA-M potassium hydrogen phthalate Chemical compound [K+].OC(=O)C1=CC=CC=C1C([O-])=O IWZKICVEHNUQTL-UHFFFAOYSA-M 0.000 description 1
- 229910001380 potassium hypophosphite Inorganic materials 0.000 description 1
- PHZLMBHDXVLRIX-UHFFFAOYSA-M potassium lactate Chemical compound [K+].CC(O)C([O-])=O PHZLMBHDXVLRIX-UHFFFAOYSA-M 0.000 description 1
- 239000001521 potassium lactate Substances 0.000 description 1
- 235000011085 potassium lactate Nutrition 0.000 description 1
- 229960001304 potassium lactate Drugs 0.000 description 1
- CRGPNLUFHHUKCM-UHFFFAOYSA-M potassium phosphinate Chemical compound [K+].[O-]P=O CRGPNLUFHHUKCM-UHFFFAOYSA-M 0.000 description 1
- 239000001472 potassium tartrate Substances 0.000 description 1
- 229940111695 potassium tartrate Drugs 0.000 description 1
- 235000011005 potassium tartrates Nutrition 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 239000011241 protective layer Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000005546 reactive sputtering Methods 0.000 description 1
- 239000012088 reference solution Substances 0.000 description 1
- 230000002040 relaxant effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000004576 sand Substances 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 239000004065 semiconductor Substances 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 238000010008 shearing Methods 0.000 description 1
- 150000004756 silanes Chemical class 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000002210 silicon-based material Substances 0.000 description 1
- HELHAJAZNSDZJO-OLXYHTOASA-L sodium L-tartrate Chemical compound [Na+].[Na+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O HELHAJAZNSDZJO-OLXYHTOASA-L 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 229940001593 sodium carbonate Drugs 0.000 description 1
- 235000017550 sodium carbonate Nutrition 0.000 description 1
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 1
- 229910001379 sodium hypophosphite Inorganic materials 0.000 description 1
- 239000001540 sodium lactate Substances 0.000 description 1
- 235000011088 sodium lactate Nutrition 0.000 description 1
- 229940005581 sodium lactate Drugs 0.000 description 1
- NNMHYFLPFNGQFZ-UHFFFAOYSA-M sodium polyacrylate Chemical compound [Na+].[O-]C(=O)C=C NNMHYFLPFNGQFZ-UHFFFAOYSA-M 0.000 description 1
- 239000001433 sodium tartrate Substances 0.000 description 1
- 229960002167 sodium tartrate Drugs 0.000 description 1
- 235000011004 sodium tartrates Nutrition 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 238000009864 tensile test Methods 0.000 description 1
- KKEYFWRCBNTPAC-UHFFFAOYSA-L terephthalate(2-) Chemical compound [O-]C(=O)C1=CC=C(C([O-])=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-L 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 150000000000 tetracarboxylic acids Chemical class 0.000 description 1
- 239000003017 thermal stabilizer Substances 0.000 description 1
- 229920001187 thermosetting polymer Polymers 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 239000005341 toughened glass Substances 0.000 description 1
- 238000002834 transmittance Methods 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 235000013799 ultramarine blue Nutrition 0.000 description 1
- 238000001771 vacuum deposition Methods 0.000 description 1
- 229920006163 vinyl copolymer Polymers 0.000 description 1
- 229920001567 vinyl ester resin Polymers 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J7/00—Chemical treatment or coating of shaped articles made of macromolecular substances
- C08J7/04—Coating
- C08J7/048—Forming gas barrier coatings
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/42—Block-or graft-polymers containing polysiloxane sequences
- C08G77/442—Block-or graft-polymers containing polysiloxane sequences containing vinyl polymer sequences
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J7/00—Chemical treatment or coating of shaped articles made of macromolecular substances
- C08J7/04—Coating
- C08J7/042—Coating with two or more layers, where at least one layer of a composition contains a polymer binder
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J7/00—Chemical treatment or coating of shaped articles made of macromolecular substances
- C08J7/04—Coating
- C08J7/0427—Coating with only one layer of a composition containing a polymer binder
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J7/00—Chemical treatment or coating of shaped articles made of macromolecular substances
- C08J7/04—Coating
- C08J7/043—Improving the adhesiveness of the coatings per se, e.g. forming primers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J7/00—Chemical treatment or coating of shaped articles made of macromolecular substances
- C08J7/04—Coating
- C08J7/046—Forming abrasion-resistant coatings; Forming surface-hardening coatings
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L83/00—Compositions of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon only; Compositions of derivatives of such polymers
- C08L83/10—Block- or graft-copolymers containing polysiloxane sequences
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D183/00—Coating compositions based on macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing silicon, with or without sulfur, nitrogen, oxygen, or carbon only; Coating compositions based on derivatives of such polymers
- C09D183/10—Block or graft copolymers containing polysiloxane sequences
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L31/00—Semiconductor devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation and specially adapted either for the conversion of the energy of such radiation into electrical energy or for the control of electrical energy by such radiation; Processes or apparatus specially adapted for the manufacture or treatment thereof or of parts thereof; Details thereof
- H01L31/02—Details
- H01L31/0203—Containers; Encapsulations, e.g. encapsulation of photodiodes
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L31/00—Semiconductor devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation and specially adapted either for the conversion of the energy of such radiation into electrical energy or for the control of electrical energy by such radiation; Processes or apparatus specially adapted for the manufacture or treatment thereof or of parts thereof; Details thereof
- H01L31/04—Semiconductor devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation and specially adapted either for the conversion of the energy of such radiation into electrical energy or for the control of electrical energy by such radiation; Processes or apparatus specially adapted for the manufacture or treatment thereof or of parts thereof; Details thereof adapted as photovoltaic [PV] conversion devices
- H01L31/042—PV modules or arrays of single PV cells
- H01L31/048—Encapsulation of modules
- H01L31/0481—Encapsulation of modules characterised by the composition of the encapsulation material
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01L—SEMICONDUCTOR DEVICES NOT COVERED BY CLASS H10
- H01L31/00—Semiconductor devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation and specially adapted either for the conversion of the energy of such radiation into electrical energy or for the control of electrical energy by such radiation; Processes or apparatus specially adapted for the manufacture or treatment thereof or of parts thereof; Details thereof
- H01L31/04—Semiconductor devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation and specially adapted either for the conversion of the energy of such radiation into electrical energy or for the control of electrical energy by such radiation; Processes or apparatus specially adapted for the manufacture or treatment thereof or of parts thereof; Details thereof adapted as photovoltaic [PV] conversion devices
- H01L31/042—PV modules or arrays of single PV cells
- H01L31/048—Encapsulation of modules
- H01L31/049—Protective back sheets
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2367/00—Characterised by the use of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Derivatives of such polymers
- C08J2367/02—Polyesters derived from dicarboxylic acids and dihydroxy compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2483/00—Characterised by the use of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing silicon with or without sulfur, nitrogen, oxygen, or carbon only; Derivatives of such polymers
- C08J2483/10—Block- or graft-copolymers containing polysiloxane sequences
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E10/00—Energy generation through renewable energy sources
- Y02E10/50—Photovoltaic [PV] energy
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E10/00—Energy generation through renewable energy sources
- Y02E10/50—Photovoltaic [PV] energy
- Y02E10/52—PV systems with concentrators
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/26—Web or sheet containing structurally defined element or component, the element or component having a specified physical dimension
- Y10T428/263—Coating layer not in excess of 5 mils thick or equivalent
- Y10T428/264—Up to 3 mils
- Y10T428/265—1 mil or less
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/31504—Composite [nonstructural laminate]
- Y10T428/31652—Of asbestos
- Y10T428/31663—As siloxane, silicone or silane
Definitions
- the present invention relates to a polymer sheet for solar cells and a method for producing the same, a back sheet for solar cells, and a solar cell module.
- Solar cells are a power generation system that emits no carbon dioxide during power generation and has a low environmental load, and has been rapidly spreading in recent years.
- the polymer sheet used for solar cells has various properties such as durability corresponding to the usage environment of solar cells that are placed on the roof etc. and exposed to rain, and transparency so as not to disturb the power generation efficiency of solar cells. It has been demanded.
- a solar cell sealing material also simply referred to as “sealing material” for sealing solar cell elements (cells), or a solar cell for protecting the sealing material from the outside.
- a back sheet is known as a polymer sheet for solar cells.
- a solar cell module usually has a solar cell between a front side glass on which sunlight is incident and a so-called back sheet disposed on the side opposite to the side on which sunlight is incident (back side). It has a sandwiched structure and is sealed with EVA (ethylene-vinyl acetate) resin or the like between the front side glass and the solar battery cell and between the solar battery cell and the back sheet.
- EVA ethylene-vinyl acetate
- the back sheet has a function of preventing moisture from entering from the back surface of the solar cell module, and conventionally glass or fluororesin has been used, but in recent years, polyester has been used from the viewpoint of cost. It has become to. And a back sheet is not a mere polymer sheet, but may have various functions as shown below.
- a back sheet is added with white inorganic fine particles such as titanium oxide to have a reflection performance. This is to increase power generation efficiency by irregularly reflecting light that has passed through the cell out of sunlight incident from the front side of the module and returning it to the cell.
- white polyethylene terephthalate films added with white inorganic fine particles have been disclosed (see, for example, Japanese Patent Application Laid-Open Nos. 2003-060218 and 2006-210557), and white containing a white pigment.
- An example of a back surface protection sheet having an ink layer is also disclosed (see, for example, JP-A-2006-210557).
- a back sheet for a solar cell to which a perylene pigment as a black pigment is added in order to improve decorativeness is disclosed (see, for example, JP-A-2007-128943).
- a polymer layer may be provided on the outermost layer of the back sheet.
- a technique for providing a thermal adhesive layer on a white polyethylene terephthalate film has been described (see, for example, JP-A-2003-060218).
- the back sheet has a structure in which layers having other functions are laminated on a support.
- a means of lamination there is a method of bonding sheets having various functions to a support.
- a method of forming a back sheet by bonding a plurality of resin films is disclosed (see, for example, JP-A-2002-1000078).
- methods for applying layers having various functions to a support have been disclosed (for example, JP-A-2006-210557, JP-A-2007-128943). Issue gazette).
- a white polyester film for a reflector having a white polyester film provided with a coating layer containing an antistatic agent and a silicone compound, and an adhesive film containing an epoxy resin, a phenol resin, a vinyl copolymer, and a siloxane compound are organic films.
- a disclosure relating to a solar cell backsheet laminated thereon see, for example, Japanese Patent Application Laid-Open Nos. 2008-189828 and 2008-282873).
- the cost is high, and the adhesion between layers for a long period of time is inferior, and the durability is insufficient. That is, since the back sheet is directly exposed to moisture, heat, and light, performance that can withstand these over a long period of time is required.
- the back sheet generally has a structure bonded to an EVA-based sealing material, and in this case, the durability of bonding between the back sheet and EVA over time is extremely important.
- durability of adhesion between the support and each layer is indispensable.
- the former has poor durability of the cationic polymer contained as an antistatic agent, and the latter has a resin or copolymer other than the siloxane compound. Since the durability is poor, it is difficult to maintain long-term adhesion in an environment where the temperature and humidity are relatively high.
- a fluororesin also referred to as a fluorocarbon resin
- a polymer layer using a fluororesin as a binder has a problem of poor adhesion to a substrate. It was. Therefore, the durability of the base material and the adhesiveness of the polymer layer are not compatible.
- a solar cell that has adhesiveness with an EVA-based sealing material that can withstand long-term aging and, in some cases, other functions (for example, reflection performance and decoration), can be manufactured at low cost, and can satisfy wet heat durability.
- polymer sheets for solar cells such as backsheets for use have not yet been provided.
- the present invention has been made in view of the above, and the adhesion durability between each layer in a humid heat environment, the constituent substrate of the polymer sheet and the constituent substrate of the battery side substrate (for example, a sealing material such as EVA)
- An object of the present invention is to provide a solar cell polymer sheet and method for manufacturing the solar cell polymer sheet, a solar cell back sheet, and an inexpensive solar cell module having stable power generation efficiency, which have excellent adhesion durability between the solar cell and the manufacturing method thereof.
- the objective is to achieve the objective.
- R 1 and R 2 each independently represent a hydrogen atom, a halogen atom, or a monovalent organic group, and R 1 and R 2 may be the same or different.
- n represents an integer of 1 or more.
- the plurality of R 1 and R 2 may be the same as or different from each other.
- ⁇ 2> The polymer layer according to ⁇ 1>, wherein the polymer layer includes a structural portion derived from a crosslinking agent that crosslinks the composite polymer.
- ⁇ 3> The solar cell polymer sheet according to ⁇ 1> or ⁇ 2>, wherein the non-polysiloxane structural unit is an acrylic structural unit.
- the crosslinking agent is at least one selected from a carbodiimide compound, an oxazoline compound, and an epoxy crosslinking agent.
- ⁇ 5> The solar cell according to any one of ⁇ 2> to ⁇ 4>, wherein a mass ratio of the structural part derived from the crosslinking agent to the composite polymer in the polymer layer is 1 to 30% by mass.
- ⁇ 6> The polyester substrate according to ⁇ 1> to ⁇ 5>, wherein the polyester base material is subjected to at least one surface treatment selected from corona treatment, flame treatment, low pressure plasma treatment, atmospheric pressure plasma treatment, and ultraviolet treatment. It is a polymer sheet for solar cells as described in any one.
- the monovalent organic group represented by R 1 and R 2 is an alkyl group, an aryl group, an aralkyl group, an alkoxy group, an aryloxy group, a mercapto group, an amino group
- the polymer sheet for solar cells according to any one of ⁇ 1> to ⁇ 6>, wherein the polymer sheet is at least one selected from amide groups.
- ⁇ 9> The solar cell polymer sheet according to any one of ⁇ 1> to ⁇ 8>, wherein at least one of the polymer layers has a thickness of 0.8 ⁇ m to 12 ⁇ m.
- ⁇ 10> The solar cell polymer sheet according to any one of ⁇ 1> to ⁇ 9>, wherein at least one of the polymer layers is provided in contact with a surface of the polyester substrate.
- ⁇ 11> The polymer sheet for solar cells according to any one of ⁇ 1> to ⁇ 10>, wherein at least one of the polymer layers is an outermost layer disposed at a position farthest from the polyester base material. It is.
- ⁇ 12> The polymer sheet for solar cells according to any one of ⁇ 1> to ⁇ 11>, wherein at least one layer of the polymer layer further includes a white pigment, and is a reflective layer having light reflectivity. is there.
- ⁇ 13> The solar cell according to ⁇ 12>, including at least two polymer layers, having the reflective layer as one of the two layers, and having the other between the reflective layer and the polyester substrate. It is a polymer sheet.
- ⁇ 14> The above ⁇ 1> to ⁇ 11, further comprising a reflective layer containing a white pigment and having light reflectivity, and having at least one polymer layer between the reflective layer and the polyester base material. > It is a polymer sheet for solar cells as described in any one of>. An embodiment in which the reflective layer and the polyester base material are bonded by the polymer layer is preferable.
- the content of the carboxyl group is 15 equivalent / t or less, the minute endothermic peak temperature Tmeta (° C.) obtained by differential scanning calorimetry is 220 ° C. or less, the temperature is 125 ° C., and the relative humidity is 100% RH.
- the siloxane structural unit represented by the general formula (1) having a mass ratio of 15 to 85 mass% in the molecule and the mass
- a method for producing a polymer sheet for a solar cell comprising a step of applying a coating solution containing a composite polymer containing a non-siloxane structural unit having a ratio of 85 to 15% by mass to form at least one polymer layer.
- the said coating liquid is a manufacturing method of the polymer sheet for solar cells as described in said ⁇ 15> containing the crosslinking agent further chosen from a carbodiimide type compound, an oxazoline type compound, and an epoxy-type crosslinking agent.
- ⁇ 20> The polymer sheet for solar cells according to any one of ⁇ 1> to ⁇ 14>, and the polymer sheet for solar cells according to any one of ⁇ 15> to ⁇ 17>.
- a solar cell module comprising the solar cell backsheet according to any one of ⁇ 18> to ⁇ 21>.
- a transparent front substrate on which sunlight is incident a cell structure portion provided on the front substrate and having a solar cell element and a sealing material for sealing the solar cell device, and the cell structure portion
- the solar cell backsheet according to any one of ⁇ 18> to ⁇ 21>, provided on the side opposite to the side on which the front substrate is positioned and disposed adjacent to the sealing material And a solar cell module.
- the present invention it is excellent in adhesion durability between each layer in a wet heat environment, and adhesion durability between a constituent substrate of a polymer sheet and a constituent substrate of a battery side substrate (for example, a sealing material such as EVA). It is possible to provide a polymer sheet for solar cell that can be manufactured at low cost, a method for manufacturing the polymer sheet, and a back sheet for solar cell. Also, According to the present invention, an inexpensive solar cell module having stable power generation efficiency can be provided.
- the polymer sheet for solar cells of the present invention has a carboxyl group content of 15 equivalent / t or less, a minute endothermic peak temperature Tmeta (° C.) determined by differential scanning calorimetry is 220 ° C. or less, and a temperature of 125 ° C.
- R 1 and R 2 each independently represent a hydrogen atom, a halogen atom, or a monovalent organic group, and R 1 and R 2 may be the same or different.
- N represents an integer.
- R 1 and R 2 each independently represent a hydrogen atom, a halogen atom, or a monovalent organic group, and R 1 and R 2 may be the same or different.
- n represents an integer of 1 or more.
- the plurality of R 1 and R 2 may be the same as or different from each other.
- the polymer layer which is a constituent layer of the polymer sheet, is constituted by using a specific composite polymer containing a non-siloxane structural unit and a (poly) siloxane structural unit copolymerized therewith in the molecule, Adhesive strength between the layers and the adhesive strength between the polyester base material and the constituent base material of the battery side substrate (for example, a sealing material such as EVA) are improved, and deterioration due to heat and moisture is suppressed. Therefore, the adhesive strength can be kept high over a long period of time under environmental conditions exposed to heat and moisture for a long time, and long-term durability can be ensured. Thereby, when a solar cell module is configured, good power generation performance can be obtained, and power generation efficiency can be kept stable over a long period of time.
- a specific composite polymer containing a non-siloxane structural unit and a (poly) siloxane structural unit copolymerized therewith in the molecule Adhesive strength between the layers and the
- the polymer layer in the present invention can be applied to any layer constituting the polymer sheet.
- the polymer layer can be applied as an adhesive layer that bonds a functional layer such as a reflective layer, a back layer, or a reflective layer, which will be described later, and a polyester substrate.
- a functional layer such as a reflective layer, a back layer, or a reflective layer, which will be described later
- a polyester substrate a polyester substrate.
- a polymer layer disposed between a reflective layer containing a white pigment or the like and a polyester base material, or a solar cell module because of its excellent durability under humid heat environments such as heat and moisture
- polyester base material The polyester base material in the present invention has a carboxyl group content of 15 equivalent / t or less, a minute endothermic peak temperature Tmeta (° C.) determined by differential scanning calorimetry is 220 ° C. or less, a temperature of 125 ° C., and a relative humidity. The average elongation retention after standing for 72 hours under the condition of 100% RH is 10% or more.
- a base material can have high durability because it is such a configuration. Conventionally, when such a highly durable polyester base material is used, the adhesiveness between the base material and the polymer layer in a moist heat environment is lowered.
- the polymer sheet for solar cells of the present invention has a siloxane structure represented by the following general formula (1) having a mass ratio of 15 to 85% by mass in the molecule on the above highly durable polyester base material.
- a polymer layer containing a composite polymer containing units and a non-siloxane structural unit having a mass ratio of 85 to 15% by mass is provided.
- the carboxyl group content (Acid Value; AV) in the polyester used for the polyester substrate is 15 equivalents / t (tons; the same shall apply hereinafter) or less, preferably 12 equivalents / t or less, more preferably 8 equivalents / t or less. is there.
- the carboxyl group functions to form a hydrogen bond with a hydroxyl group present on the surface of the member or layer adjacent to the polyester base material to improve adhesion.
- the lower limit of the carboxyl group content is desirably 1 equivalent / t.
- “equivalent / ton (eq / t)” represents a molar equivalent per ton.
- H + in the carboxyl group acts as an acid catalyst and has an action of hydrolyzing the polyester molecule. Therefore, at an AV exceeding 15 equivalents / t, the molecular weight of the polyester base material surface decreases due to hydrolysis and the mechanical strength decreases due to hydrolysis, and as a result, the polyester base material surface is destroyed. As a result, peeling (adhesion failure) of the polymer layer from the polyester base material occurs.
- Specific AV adjustment methods include adjustment of the “plane orientation coefficient” of the polyester base material, adjustment of the type and content of “components” constituting the polyester, “buffering agent”, “end-capping agent”, etc.
- AV is within the scope of the present invention, depending on the amount of additives such as “buffering agent” and “end-capping agent” and / or “phosphorus atomic weight”. It is necessary to increase the content of these in the polyester.
- an excessive amount of additive or phosphorus atom in the polyester causes the additive to be deposited on the surface of the base material when the base material is subjected to heat and humidity, or an increase in thermal shrinkage due to excessive orientation.
- an increase in thermal shrinkage due to excessive orientation causes a problem of adhesion, which in turn causes poor adhesion.
- the polyester substrate in the present invention has a minute endothermic peak temperature Tmeta (° C.) determined by differential scanning calorimetry (hereinafter also referred to as “DSC”) of 220 ° C. or less, more preferably 150 ° C. or more and 215 ° C. or less. More preferably, it is 160 ° C. or higher and 210 ° C. or lower.
- DSC differential scanning calorimetry
- the minute endothermic peak temperature Tmeta (° C.) relates to the present invention by controlling the “plane orientation coefficient” in the polyester base material and the “temperature of heat setting performed after stretching” in forming the polyester base material into a film. It can be a temperature range.
- the temperature of heat setting carried out after stretching is preferably 150 ° C. or higher and 220 ° C. or lower, more preferably 160 ° C. or higher and 210 ° C. or lower, and further preferably 170 ° C. or higher and 200 ° C. or lower.
- the backsheet of the present invention is characterized by having high adhesion even after wet heat aging. For that purpose, it is preferable that the fall of adhesive force is suppressed by suppressing the hydrolysis in the polyester base-material surface. From such a viewpoint, as an index of hydrolysis on the surface of the polyester base material, “average elongation retention after standing for 72 hours under conditions of a temperature of 125 ° C. and a relative humidity of 100% RH” is employed. The average elongation retention is required to be 10% or more.
- the “average elongation retention rate” in the present invention is a value obtained by measuring the elongation retention rate in the longitudinal direction (MD) and the orthogonal direction (TD) of the polyester base material and expressing the average value.
- Examples of methods for adjusting the elongation retention rate include adjustment of the “plane orientation coefficient” of the polyester base material, adjustment of the “inherent viscosity” of the polyester, adjustment of the type and content of the “component” constituting the polyester polymer, Examples include addition of additives such as “buffering agent” and “end-capping agent”, and adjustment of “phosphorus atomic weight” present in the polyester.
- the polyester base material in the present invention needs to have an average elongation retention of 10% or more, more preferably 20% or more and 95% or less, and further preferably 30% or more and 90% or less. It is. By setting the average elongation retention to 10% or more, it is possible to effectively suppress peeling (adhesion failure) of the back sheet due to hydrolysis of the polyester.
- One of the preferred embodiments of the polyester substrate in the present invention is that the thermal shrinkage rate at 150 ° C. for 30 minutes in the longitudinal direction (MD) and the orthogonal direction (TD) of the polyester substrate is 1.0% or less, respectively. And the heat shrinkage variation ratio is 1% or more and 20% or less, respectively.
- the poor adhesion of the polyester base material and the polymer layer due to aging with heat and heat may be due to the occurrence of thermal shrinkage due to residual strain in the polyester base material. That is, when thermal shrinkage due to residual strain occurs in a polyester substrate that has been subjected to wet heat, shrinkage stress is generated between the polymer layer and the polyester substrate due to the thermal shrinkage, which causes poor adhesion of the polymer layer. That's it.
- the preferable thermal shrinkage variation ratio of the polyester base material of a preferred embodiment in the present invention is 1% or more and 20% or less, more preferably 2% or more and 15% or less, and further preferably 3% or more and 12% or less.
- the thermal shrinkage variation ratio of the polyester base material is measured at 10 points at intervals of 10 cm in the longitudinal direction (MD) and the orthogonal direction (TD), and the thermal shrinkage variation ratio (Bts) (%) from the following formula. And point to the larger value.
- (Bts) (%) 100 ⁇ (Bmax) ⁇ (Bmin)) / (Bav)
- Bts represents the heat shrinkage variation ratio
- Bmax represents the maximum value of heat shrinkage
- Bmin represents the minimum value of heat shrinkage
- Bav represents the average value of heat shrinkage.
- thermal shrinkage variation ratio exceeds 20%, the dimensional change between the large and small areas of thermal shrinkage becomes too large, and a crater-shaped shrinkage tends to occur, and stress concentration occurs along the edge of this crater. , Peeling (adhesion failure) is likely to occur.
- the thermal shrinkage variation ratio is less than 1%, it is difficult to achieve the effect of suppressing shrinkage as described above.
- control of the heat shrinkage rate and the heat shrinkage variation ratio is particularly useful for revealing the effect of improving the adhesion after aging with wet heat. That is, in the case where heat shrinkage occurs with the passage of moisture and heat under high humidity and the humidity is high, water is present at the interface between the polyester base material and the adjacent member or the adjacent layer capable of forming a hydrogen bond with the polyester base material. Penetration and breakage of hydrogen bonds, and adhesion tends to decrease, but even in such a situation, the shrinkage stress due to residual strain can be controlled by setting the heat shrinkage rate and the heat shrinkage variation ratio within the above ranges. Therefore, it is easy to ensure adhesion.
- the thermal contraction rate of the polyester base material in the present invention is measured at 150 ° C. for 30 minutes.
- the preferable range is preferably 1% or less in both the longitudinal direction (MD) and the orthogonal direction (TD), more preferably from ⁇ 0.5% to 0.8%, and still more preferably ⁇ 0.3. % Or more and 0.6% or less. (Here, “-” means “extension”).
- the effect of setting the heat shrinkage variation ratio in the specific range can be effectively expressed.
- the heat shrinkage rate exceeds 1%, the dimensional change of the polyester base material cannot be completely suppressed, and the effect of setting the heat shrinkage variation ratio in a specific range tends to be not obtained.
- the elongation of the polyester base becomes too large, the effect of suppressing the dimensional change of the polyester base by controlling the thermal shrinkage variation ratio tends to be not obtained.
- the heat shrinkage rate can be adjusted by performing a heat treatment after stretching when forming a polyester substrate.
- a preferable temperature for the heat treatment is 150 ° C. or higher and 220 ° C. or lower, more preferably 160 ° C. or higher and 210 ° C. or lower, more preferably 170 ° C. or higher and 200 ° C. or lower, and 10 seconds or longer and 120 seconds or shorter, more preferably 15 seconds or longer and 90 seconds or shorter. More preferably, it is 20 seconds or more and 60 seconds or less.
- the preferable relaxation amount is 0.5% or more and 10% or less, more preferably 1.5% or more and 9% or less. More preferably, it is 3% or more and 8% or less.
- the heat shrinkage variation ratio is adjusted by forming a temperature distribution when solidifying on a cooling roll and producing an unstretched film (raw fabric) after melt extrusion when forming a polyester substrate.
- a temperature distribution when solidifying on a cooling roll and producing an unstretched film (raw fabric) after melt extrusion when forming a polyester substrate.
- spherulites are formed when the melt cools, and this spherulite distribution is formed by changing the cooling rate. This causes an orientation distribution during longitudinal and lateral stretching, which appears as a distribution of shrinkage.
- Such distribution of the cooling rate of the melt can be achieved by giving a temperature distribution to the cooling roll.
- Such temperature distribution is achieved by disturbing the baffle plate with the flow of the heat medium flowing for temperature control in the cooling roll.
- a preferable temperature distribution is 0.2 ° C. or more and 10 ° C. or less, more preferably 0.4 ° C. or more and 5 ° C. or less, and further preferably 0.6 ° C. or more and
- end-capping agent is contained in the polyester, and “trifunctional or higher functional component” is contained as a component of the polyester.
- the end-capping agent can make the ends bulky by reacting with the polyester, which becomes a trap and decreases the mobility between the polyester molecules. Moreover, since the molecule
- the polyester base material in the present invention preferably has a plane orientation coefficient of 0.165 or more, more preferably 0.168 or more and 0.18 or less, still more preferably 0.170 or more and 0.175 or less.
- the plane orientation coefficient By setting the plane orientation coefficient to 0.165 or more, the molecules can be oriented, the formation of the “half-crystal” can be promoted, and the hydrolysis resistance can be further improved.
- the plane orientation coefficient fPO referred to in the present invention is obtained from the following formula (A) using an Abbe refractometer.
- f PO (nMD + nTD) / 2 ⁇ nZD (A)
- nMD represents the refractive index in the longitudinal direction (MD) of the film
- nTD represents the refractive index in the perpendicular direction (TD) of the film
- nZD represents the refractive index in the film thickness direction.
- the refractive index of each said direction of a film can be measured based on A method etc. of JISK7142.
- the plane orientation coefficient of the polyester base material can be adjusted by increasing the draw ratio during film formation.
- the draw ratio may be adjusted to 2.5 to 6.0 times in both the longitudinal direction (MD) of the film and the orthogonal direction (TD) of the film.
- MD longitudinal direction
- TD orthogonal direction
- the plane orientation coefficient can be improved by “preheating” and “multistage stretching” (described later) during longitudinal stretching.
- the plane orientation coefficient 0.165 or more, hydrolysis resistance can be suppressed and adhesion failure due to a decrease in molecular weight on the surface of the polyester film can be suppressed.
- the upper limit of the plane orientation coefficient of the film is that delamination (layer peeling) occurs because the film-forming stability deteriorates when the stretching ratio is increased to increase the plane orientation coefficient. ) Can be suppressed and the adhesion can be increased, so that it is preferably 0.180 or less, more preferably 0.175 or less.
- the distribution of the plane orientation coefficient is preferably 1% or more and 20% or less, more preferably 2% or more and 15% or less, and further preferably 3% or more and 12% or less.
- the adhesion can be further improved. That is, since the polyester base material shrinks after aging with wet heat, a shrinkage stress is generated between the film and a sealing agent such as EVA, thereby causing poor adhesion. This heat shrinkage stress is proportional to the elastic modulus of the film, which is proportional to the plane orientation coefficient. Accordingly, if there is a distribution in the polyester substrate surface orientation coefficient, a distribution is also generated in the elastic modulus, thereby forming a portion having a high elastic modulus (hard) and a portion having a low elastic modulus (soft).
- the portion having a low elastic modulus has a function of absorbing the generated heat shrinkage stress, and this acts as a buffer portion and exhibits the effect of suppressing the decrease in adhesion.
- the distribution of the plane orientation coefficient is less than 1%, the heat shrinkage stress cannot be relaxed and the adhesion tends to decrease.
- the distribution of the plane orientation coefficient exceeds 20%, the shrinkage stress is excessively concentrated on the small plane orientation, and the adhesion failure tends to occur.
- the distribution of the plane orientation coefficient in the polyester substrate can be formed by adjusting the preheating temperature distribution in the longitudinal stretching when forming the polyester substrate. That is, the orientation distribution in longitudinal stretching and the accompanying crystal distribution are formed by the preheating temperature distribution, thereby forming the orientation distribution in transverse stretching.
- the temperature distribution here refers to a temperature distribution in the width direction. That is, a crystal and orientation distribution occurs in the width direction after longitudinal stretching due to the temperature distribution formed in the width direction. When the film is stretched in the lateral direction thereafter, uneven distribution is formed over the entire surface of the film, so that a distribution of the plane orientation coefficient is formed.
- the distribution of preheating temperature can be adjusted by giving a temperature distribution to the preheating roll. Specifically, the preheating temperature distribution may be adjusted by disturbing the baffle plate with the flow of the heat medium flowing for temperature control in the preheating roll.
- a preferable temperature distribution of the preheating temperature is 0.2 ° C. or more and 10 ° C. or less, more preferably 0.4 ° C. or more and 5 ° C. or less, and further preferably 0.6 ° C. or more and 3 ° C. or less.
- the “end-capping agent” is contained in the polyester, and as a component of the polyester, “a trifunctional or higher functional component” is contained. Adhesion after wet heat aging can be improved more effectively.
- the end-capping agent can make the ends bulky by reacting with the polyester, which becomes a trap and decreases the mobility between the polyester molecules.
- the trifunctional or higher functional component (C) has a molecule branched via a trifunctional group, the mobility of the polyester molecule is lowered.
- it can be made easy to form a plane orientation distribution by reducing the mobility. That is, molecules flow (creep) due to a difference in stress generated between a portion with a large plane orientation and a portion with a small plane orientation, and try to eliminate this. At this time, if the mobility of the molecule is lowered as described above, such a plane orientation distribution is hardly eliminated and a plane orientation coefficient distribution is easily formed.
- the polyester in the polyester substrate of the present invention preferably has an intrinsic viscosity (hereinafter referred to as “IV” as appropriate) in the range of 0.6 to 1.2 dl / g.
- IV intrinsic viscosity
- a more preferable intrinsic viscosity is 0.65 to 1.0 dl / g, and even more preferably 0.70 to 0.95 dl / g.
- the intrinsic viscosity of the polyester in the polyester base material is less than 0.6 dl / g, the mobility of the molecules is large, and the above-described thermal shrinkage and the distribution of plane orientation tend to be easily relaxed (resolved).
- the IV of the polyester in the polyester substrate can be adjusted by the temperature and reaction time in the solid phase polymerization.
- the polyester pellets are 180 ° C. or higher and 250 ° C. or lower, more preferably 190 ° C. or higher and 240 ° C. or lower, and further preferably 195 ° C. or higher and 230 ° C. or lower.
- the heat treatment be performed in a nitrogen stream or in a vacuum for 10 hours to 40 hours, more preferably 15 hours to 30 hours.
- the solid phase polymerization may be performed at a constant temperature or may be performed while being varied.
- the polyester raw material (pellet) used for forming the polyester base film preferably has an intrinsic viscosity in the range of 0.6 to 1.2 dl / g in order to satisfy hydrolysis resistance. More preferably, it is 0.65 to 1.0 dl / g, and still more preferably 0.70 to 0.95 dl / g. In order to improve the hydrolysis resistance, it is preferable to increase the intrinsic viscosity. However, when the intrinsic viscosity exceeds 1.2 dl / g, it is necessary to lengthen the solid phase polymerization time when producing the polyester resin, and the cost is remarkably high. Therefore, it may not be preferable.
- the degree of polymerization is low, and the heat resistance and hydrolysis resistance are remarkably lowered, which is not preferable.
- the intrinsic viscosity of the pellet can be adjusted to the above preferred range by adjusting the polymerization conditions and the solid phase polymerization conditions during the production of the polyester resin.
- the polyester substrate is not particularly limited as long as it has the above physical properties.
- Specific examples of such polyester include polyethylene terephthalate, polyethylene isophthalate, polybutylene terephthalate, poly (1,4-cyclohexylenedimethylene terephthalate), polyethylene-2,6-naphthalate and the like. Of these, polyethylene terephthalate or polyethylene-2,6-naphthalate is particularly preferable from the viewpoint of balance between mechanical properties and cost.
- the polyester may be a homopolymer or a copolymer. Further, the polyester may be blended with a small amount of another type of resin such as polyimide.
- an Sb-based, Ge-based, or Ti-based compound is used as a catalyst. It is preferable to use, and Ti compound is particularly preferable among them.
- Ti compound is particularly preferable among them.
- the amount of the Ti compound used is within the above range in terms of Ti element, the terminal carboxyl group in the polyester after polymerization can be adjusted to the following range, and the hydrolysis resistance of the polyester base is kept low. be able to.
- Examples of the synthesis of polyester using a Ti compound include Japanese Patent Publication No. 8-301198, Japanese Patent No. 2543624, Japanese Patent No. 3335683, Japanese Patent No. 3717380, Japanese Patent No. 3897756, Japanese Patent No. 396226, and Japanese Patent No. 39786666.
- No. 3, Patent No. 3,996,871, Patent No. 40000867, Patent No. 4053837, Patent No. 4,127,119, Patent No. 4,134,710, Patent No. 4,159,154, Patent No. 4,269,704, Patent No. 4,313,538 and the like can be applied.
- the polyester in the present invention is preferably solid-phase polymerized after polymerization.
- Solid-phase polymerization may be a continuous method (a method in which a tower is filled with a resin, which is slowly heated for a predetermined time and then sent out), or a batch method (a resin is charged into a container). , A method of heating for a predetermined time).
- Japanese Patent No. 2621563, Japanese Patent No. 3121876, Japanese Patent No. 3136774, Japanese Patent No. 3603585, Japanese Patent No. 3616522, Japanese Patent No. 3617340, Japanese Patent No. 3680523, Japanese Patent No. 3717392 are disclosed.
- the method described in Japanese Patent No. 4167159 can be applied.
- the temperature of the solid phase polymerization is preferably 170 ° C. or higher and 240 ° C. or lower, more preferably 180 ° C. or higher and 230 ° C. or lower, and further preferably 190 ° C. or higher and 220 ° C. or lower.
- the solid phase polymerization time is preferably 5 hours to 100 hours, more preferably 10 hours to 75 hours, and still more preferably 15 hours to 50 hours.
- the solid phase polymerization is preferably performed in a vacuum or in a nitrogen atmosphere.
- One preferred embodiment of the polyester in the present invention is a dicarboxylic acid component, a diol component, and a component (p) in which the sum (a + b) of the number of carboxyl groups (a) and the number of hydroxyl groups (b) is 3 or more (p ), And the content of the component (p) is 0.005 mol% or more and 2.5 mol% or less with respect to all the components included in the polyester.
- component (p) examples include carboxylic acid components having 3 or more carboxyl groups (a), components having 3 or more hydroxyl groups (b), and oxyacids having both hydroxyl groups and carboxyl groups in one molecule. And the total component (a + b) of the number of carboxyl groups (a) and the number of hydroxyl groups (b) is 3 or more.
- carboxylic acid constituents having a carboxyl group number (a) of 3 or more include trifunctional aromatic carboxylic acid constituents such as trimesic acid, trimellitic acid, naphthalenetricarboxylic acid, anthracentricarboxylic acid, etc.
- aromatic carboxylic acid components include methanetricarboxylic acid, ethanetricarboxylic acid, propanetricarboxylic acid, and butanetricarboxylic acid.
- Tetrafunctional aromatic carboxylic acid components include benzenetetracarboxylic acid, pyromellitic acid, benzophenonetetracarboxylic acid, and naphthalene.
- Tetracarboxylic acid, anthracenetetracarboxylic acid, berylenetetracarboxylic acid, etc. are tetrafunctional aliphatic carboxylic acid constituents, such as ethanetetracarboxylic acid, ethylenetetracarboxylic acid, butanetetracarboxylic acid, cyclopentanetetracarboxylic acid. Acid, cyclohexanetetracarboxylic acid, adamantanetetracarboxylic acid, etc.
- aromatic carboxylic acid constituents such as benzenepentacarboxylic acid, benzenehexacarboxylic acid, naphthalenepentacarboxylic acid, naphthalenehexacarboxylic acid, naphthaleneheptacarboxylic acid , Naphthalene octacarboxylic acid, anthracene pentacarboxylic acid, anthracene hexacarboxylic acid, anthracene heptacarboxylic acid, anthracene octacarboxylic acid, etc.
- esters and acid anhydrides thereof are pentafunctional or higher aliphatic carboxylic acid components, ethane pentacarboxylic acid, ethane hexacarboxylic acid, Butanepentacarboxylic acid, butaneheptacarboxylic acid, cyclopentanepentacarboxylic acid, cyclohexanepentacarboxylic acid, cyclohexanehexacarboxylic acid, adamantanepentacarboxylic acid , Etc. adamantane hexacarboxylic acid, and the like These ester derivatives and acid anhydrides thereof as examples without limitation.
- oxyacids such as l-lactide, d-lactide, hydroxybenzoic acid and the like, or a combination of a plurality of the oxyacids to the carboxy terminus of the carboxylic acid component. Used for. Moreover, these may be used independently or may be used in multiple types as needed.
- components having a hydroxyl number (b) of 3 or more include trifunctional aromatic components such as trihydroxybenzene, trihydroxynaphthalene, trihydroxyanthracene, trihydroxychalcone, trihydroxyflavone, trihydroxycoumarin,
- trifunctional aromatic components such as trihydroxybenzene, trihydroxynaphthalene, trihydroxyanthracene, trihydroxychalcone, trihydroxyflavone, trihydroxycoumarin
- trifunctional aliphatic alcohol constituents glycerin, trimethylolpropane, propanetriol, tetrafunctional aliphatic alcohol constituents, compounds such as pentaerythritol, and diols were added to the hydroxyl terminal of the above-mentioned compounds
- Component (p) is also preferably used. Moreover, these may be used independently or may be used in multiple types as needed.
- oxyacids having both a hydroxyl group and a carboxyl group number in one molecule and the total component (a + b) of the carboxyl group number (a) and the hydroxyl group number (b) is 3 or more
- hydroxyisophthalic acid examples include terephthalic acid, dihydroxyterephthalic acid, and dihydroxyterephthalic acid.
- a product obtained by adding oxyacids such as l-lactide, d-lactide, hydroxybenzoic acid and the like, or a combination of a plurality of the oxyacids to the carboxy terminus of the above-described constituents is also preferably used. It is done. Moreover, these may be used independently or may be used in multiple types as needed.
- the content of the constituent component (p) is preferably 0.005 mol% or more and 2.5 mol% with respect to all the constituent components in the polyester.
- the content of component (p) is more preferably 0.020 or more and 1 or less, further preferably 0.025 or more and 1 or less, further preferably 0.035 or more and 0.5 or less, and further preferably 0.05 or more and 0. 0.5 or less, particularly preferably 0.1 or more and 0.25 or less.
- the content of the constituent component (p) in the polyester is 0.005 mol% or less with respect to all the constituent components in the polyester, the effect of improving the heat and moisture resistance may not be confirmed, and 2.5 mol%. If it exceeds, it is difficult to realize because the resin is gelled and melt extrusion is difficult, even if it is present, the gel exists as a foreign substance, and the biaxial stretchability when it is made into a film decreases, A film obtained by stretching may have many foreign matter defects.
- the content of the component (p) in the polyester is 0.005 mol% or more and 2.5 mol% with respect to all the components in the polyester, so that heat and moisture resistance is maintained while maintaining melt extrudability. It is possible to enhance the stretching property and maintain the stretchability during biaxial stretching and the quality of the obtained film.
- the component (p) is a compound having a carboxyl group number (a) of 3 or more and a compound having a carboxylic acid is an aromatic compound, or a compound having a hydroxyl group number (b) of 3 or more and a hydroxyl group. It is preferably an aliphatic compound.
- a crosslinked structure can be formed without deteriorating the orientation characteristics of the polyester film, the molecular mobility can be further reduced, and the heat and moisture resistance can be further increased.
- polyester contains a structural component (p) it is also preferable to add the below-mentioned buffering agent and terminal blocker at the time of shaping
- the polyester containing the component (p) is preferably a highly crystalline resin. Specifically, the resin is heated from 25 ° C. to 300 ° C. at a temperature rising rate of 20 ° C./min according to JIS K7122 (1999). Heat at a rate of 20 ° C / min (1stRUN), hold in that state for 5 minutes, then rapidly cool to 25 ° C or less, and then increase again from room temperature to 300 ° C at a rate of 20 ° C / min. In the differential scanning calorimetry chart of 2ndRUN obtained and obtained, it is preferable that the crystal melting heat amount ⁇ Hm obtained from the peak area of the melting peak is 15 J / g or more.
- orientation crystallization can be achieved by stretching and heat treatment, and as a result, a polyester base material having superior mechanical strength and heat-and-moisture resistance can be obtained.
- the melting point Tm of the polyester containing the component (p) is preferably 245 ° C. to 290 ° C.
- the melting point Tm here is the melting point Tm in the temperature rising process (temperature rising rate: 20 ° C./min) obtained by DSC, and at 25 ° C. by the method based on JIS K-7121 (1999) as described above. Heat from 1 to 300 ° C. at a heating rate of 20 ° C./min (1st RUN), hold in that state for 5 minutes, then rapidly cool to 25 ° C. or less, and again from room temperature at a heating rate of 20 ° C./min to 300 ° C.
- the melting point Tm1 of the polyester is determined by the temperature at the peak top in the 2ndRun crystal melting peak obtained by raising the temperature to 2 m. More preferably, the melting point Tm is 247 to 275 ° C, still more preferably 250 to 265 ° C. If the melting point Tm is less than 245 ° C, the heat resistance of the film may be inferior, and it is not preferable. If the melting point Tm exceeds 290 ° C, extrusion may be difficult. By setting the melting point Tm of the polyester to 245 to 290 ° C., a polyester base material having both heat resistance and processability can be obtained.
- the polyester base material in this invention contains a buffering agent.
- the inclusion of the buffering agent is particularly preferable when the polyester contains the component (p) as a component.
- the buffer is an alkali metal salt from the viewpoint of polymerization reactivity and heat and humidity resistance, for example, phthalic acid, citric acid, carbonic acid, lactic acid, tartaric acid, phosphoric acid, phosphorous acid.
- alkali metal salts with compounds such as hypophosphorous acid and polyacrylic acid potassium and sodium are preferable as alkali metal elements from the viewpoint of hardly generating precipitates due to catalyst residues.
- sodium dihydrogen, sodium hydrogen phosphite, potassium hydrogen phosphite, sodium hypophosphite, potassium hypophosphite, and sodium polyacrylate examples thereof include sodium dihydrogen, sodium hydrogen phosphite, potassium hydrogen phosphite, sodium hypophosphite, potassium hypophosphite, and sodium polyacrylate.
- the buffer is preferably an alkali metal salt represented by the following formula (I) from the viewpoint of polymerization reactivity of polyester and heat resistance during melt molding, and the alkali metal is sodium and / or potassium. It is preferable from the viewpoints of polymerization reactivity, heat resistance, and heat and humidity resistance, and phosphoric acid and sodium and / or potassium metal salts are particularly preferable from the viewpoint of polymerization reactivity and heat and humidity resistance.
- the content of the buffer is preferably 0.1 mol / ton or more and 5.0 mol / ton or less, more preferably 0.3 mol / ton or more and 3.0 mol / ton with respect to the total mass of the polyester. It is. When the content of the buffering agent is within the above range, the heat and moisture resistance and mechanical properties can be further improved.
- the alkali metal salt represented by the formula (I) When the alkali metal salt represented by the formula (I) is used as the buffer, it is preferable to use phosphoric acid in combination. Thereby, it becomes possible to further raise the hydrolysis inhibitory effect by a buffer, and to improve the heat-and-moisture resistance of the obtained polyester base material.
- the alkali metal element content W1 in the polyester base material is 2.5 ppm or more and 125 ppm or less
- the ratio W1 / W2 of the alkali metal element content W1 and the phosphorus element content W2 is 0.01 or more and 1 or less. It is preferable to be in the range. By setting it as this range, it becomes possible to raise the hydrolysis inhibitory effect more.
- the alkali metal element W1 is 15 ppm or more and 75 ppm or less, and the ratio W1 / W2 of the alkali metal element content W1 and the phosphorus element content W2 is 0.1 or more and 0.5 or less. If the alkali metal element content W1 is less than 2.5 ppm, the effect of inhibiting hydrolysis is insufficient, and the obtained polyester base material may not have sufficient wet heat resistance. On the other hand, if it exceeds 125 ppm, the excessively present alkali metal promotes the thermal decomposition reaction at the time of melt-extrusion and the molecular weight is lowered, which may cause the moist heat resistance and mechanical properties to be lowered.
- the hydrolysis inhibiting effect is insufficient, and if it exceeds 125 ppm, excess phosphoric acid is converted into polyester during the polymerization reaction. Reaction, the phosphate ester skeleton is formed in the molecular chain, and the portion promotes the hydrolysis reaction, which may reduce the hydrolysis resistance.
- the alkali metal element W1 in the polyester base material is 15 ppm or more and 75 ppm or less, and the ratio W1 / W2 of the alkali metal element content W1 and W2 is 0.1 or more and 0.5 or less, thereby suppressing hydrolysis resistance. As a result, it becomes possible to obtain high heat and humidity resistance.
- the buffer may be added at the time of polymerization of the polyester or may be added at the time of melt molding, but is preferably added at the time of polymerization from the viewpoint of uniform dispersion of the buffer in the polyester.
- the addition time may be any time as long as it is from the end of the esterification reaction or the transesterification reaction at the time of polymerization of the polyester to the beginning of the polycondensation reaction (inherent viscosity is less than 0.3). Can be added.
- any method such as a method of directly adding powder or a method of preparing and adding a solution dissolved in a diol component such as ethylene glycol may be used, but a diol component such as ethylene glycol It is preferable to add as a dissolved solution.
- the solution concentration is preferably 10% by mass or less after diluting, so that the buffering agent hardly adheres to the vicinity of the addition port, and the addition error is reduced, and the reactivity is preferable.
- polyester containing the structural component (p) it is preferable from the point of heat resistance and heat-and-moisture resistance that content of the diethylene glycol which is a by-product at the time of superposition
- polymerization is less than 2.0 mass%, and also 1 It is preferable that it is less than 0.0 mass%.
- the polyester base material in this invention contains terminal blocker.
- the end-capping agent is an additive that reacts with the carboxyl group at the end of the polyester to reduce the terminal amount of the carboxyl group of the polyester.
- Examples of the end capping agent include a carbodiimide compound, an epoxy compound, and an oxazoline compound.
- the end-capping agent is more effective when added together with the polyester during the formation of the polyester substrate.
- An end-capping agent may be used in the solid phase polymerization.
- the end-capping agent may be used in combination with a polyester containing the constituent component (p) in which the total (a + b) of the number of carboxyl groups (a) and the number of hydroxyl groups (b) is 3 or more.
- the content of the terminal blocking agent in the polyester base material is preferably 0.1 to 5% by mass.
- the terminal blocking agent is less than 0.1% by mass, the effect of sealing the carboxyl group is small and the hydrolysis resistance may be deteriorated.
- the end-capping agent is larger than 5% by mass, a lot of foreign matters may be generated during film formation or decomposition gas may be generated, which may affect productivity.
- a more preferable upper limit of the content of the end-capping agent is 4% by mass, and a more preferable upper limit is 2% by mass.
- a more preferable lower limit of the content of the end-capping agent is 0.3% by mass, and a more preferable lower limit is 0.5% by mass.
- a more preferable range of the content of the end-capping agent is 0.3 to 4% by weight, and a further preferable range is 0.5 to 2% by weight.
- the carbodiimide compound includes a monofunctional carbodiimide and a polyfunctional carbodiimide.
- monofunctional carbodiimides include dicyclohexylcarbodiimide, diisopropylcarbodiimide, dimethylcarbodiimide, diisobutylcarbodiimide, dioctylcarbodiimide, t-butylisopropylcarbodiimide, diphenylcarbodiimide, di-t-butylcarbodiimide and di- ⁇ -naphthylcarbodiimide. Particularly preferred are dicyclohexylcarbodiimide and diisopropylcarbodiimide.
- polycarbodiimide having a polymerization degree of 3 to 15 is preferably used.
- the polycarbodiimide generally has a repeating unit represented by “—R—N ⁇ C ⁇ N—” or the like, and R represents a divalent linking group such as alkylene or arylene. Specific examples of such a repeating unit include 1,5-naphthalenecarbodiimide, 4,4′-diphenylmethanecarbodiimide, 4,4′-diphenyldimethylmethanecarbodiimide, 1,3-phenylenecarbodiimide, 1,4-phenylene.
- alkylsulfonyl carbodiimide and 1,3,5-triisopropylbenzene-2,4-carbodiimide can be exemplified. These can use 1 type (s) or 2 or more types.
- the carbodiimide compound is preferably a carbodiimide compound having high heat resistance because an isocyanate gas is generated by thermal decomposition.
- the molecular weight degree of polymerization
- the terminal of the carbodiimide compound has a structure with high heat resistance. Further, once thermal decomposition occurs, further thermal decomposition is likely to occur. Therefore, it is necessary to devise measures such as setting the extrusion temperature of the polyester as low as possible.
- epoxy compound Preferable examples of the epoxy compound include glycidyl ester compounds and glycidyl ether compounds.
- glycidyl ester compounds include benzoic acid glycidyl ester, t-Bu-benzoic acid glycidyl ester, P-toluic acid glycidyl ester, cyclohexanecarboxylic acid glycidyl ester, pelargonic acid glycidyl ester, stearic acid glycidyl ester, lauric acid glycidyl ester , Glycidyl palmitate, glycidyl behenate, glycidyl versatate, glycidyl oleate, glycidyl linoleate, glycidyl linolein, glycidyl behenol, glycidyl stearol, diglycidyl terephthalate, isophthalic acid Diglycidyl ester, diglycidyl phthalate, diglycidyl naphthalene dicar
- the glycidyl ether compound examples include phenyl glycidyl ether, O-phenyl glycidyl ether, 1,4-bis ( ⁇ , ⁇ -epoxypropoxy) butane, 1,6-bis ( ⁇ , ⁇ - Epoxypropoxy) hexane, 1,4-bis ( ⁇ , ⁇ -epoxypropoxy) benzene, 1- ( ⁇ , ⁇ -epoxypropoxy) -2-ethoxyethane, 1- ( ⁇ , ⁇ -epoxypropoxy) -2-benzyl Oxyethane, 2,2-bis- [ politician- ( ⁇ , ⁇ -epoxypropoxy) phenyl] propane and 2,2-bis- (4-hydroxyphenyl) propane and 2,2-bis- (4-hydroxyphenyl) Examples thereof include bisglycidyl polyether obtained by reaction of bisphenol such as methane and epichlorohydrin, and these use one kind or two or more kinds. Door can be.
- a bisoxazoline compound is preferable, and specifically, 2,2′-bis (2-oxazoline), 2,2′-bis (4-methyl-2-oxazoline), 2,2′-bis (4,4-dimethyl-2-oxazoline), 2,2′-bis (4-ethyl-2-oxazoline), 2,2′-bis (4,4′-diethyl-2-oxazoline), 2,2 '-Bis (4-propyl-2-oxazoline), 2,2'-bis (4-butyl-2-oxazoline), 2,2'-bis (4-hexyl-2-oxazoline), 2,2'- Bis (4-phenyl-2-oxazoline), 2,2′-bis (4-cyclohexyl-2-oxazoline), 2,2′-bis (4-benzyl-2-oxazoline), 2,2′-p- Phenylenebis (2-oxazoline), 2,2'-m- Enylene bis (2-
- polyester base material in this invention it is also preferable to contain a phosphorus compound from a viewpoint of suppressing hydrolysis decomposition.
- the phosphorus compound When the phosphorus compound is contained, it is preferable that the phosphorus atom amount obtained by fluorescent X-ray measurement on the polyester base material is 200 ppm or more.
- the phosphorus atomic weight is more preferably 300 ppm or more, and still more preferably 400 ppm or more.
- the phosphorus compound it is preferable to use one or more phosphorus compounds selected from the group consisting of phosphoric acid, phosphorous acid, phosphonic acid, their methyl ester, ethyl ester, phenyl ester, half ester and other derivatives.
- phosphoric acid, phosphorous acid, phosphonic acid methyl ester, ethyl ester, and phenyl ester are particularly preferable.
- a method of containing the phosphorus compound it is preferable to add the phosphorus compound when manufacturing the polyester raw material chip.
- the polyester base material in the present invention is a constituent element of the polymer sheet, it is preferable that the polyester base material is less susceptible to deterioration by sunlight. Therefore, a UV (ultraviolet) absorber or a material that reflects UV may be added to the polyester. It is also a preferred embodiment that the average reflectance at a wavelength of 400 to 700 nm on at least one substrate surface is 80% or more. More preferably, it is 85% or more, and particularly preferably 90% or more. By setting the average reflectance at a wavelength of 400 to 700 nm to 80% or more, even when a solar cell using the polymer sheet of the present invention is used in direct sunlight, the deterioration of the polymer sheet is reduced.
- the biaxially-oriented polyester film which used polyethylene terephthalate (PET) as polyester is demonstrated as a representative example.
- PET polyethylene terephthalate
- the present invention is not limited to a biaxially oriented polyester film using a PET film, and may be one using another polymer.
- a polyester film is formed using polyethylene-2,6-naphthalenedicarboxylate having a high glass transition temperature or a high melting point, extrusion or stretching may be performed at a temperature higher than the following temperature.
- the polyester base material in this invention is manufactured as follows, for example. First, the raw fabric (unstretched) polyester sheet which comprises a polyester base material is manufactured. In order to produce the raw polyester sheet, for example, the polyester pellets obtained above are melted using an extruder, discharged from a die, and then cooled and solidified to form a sheet. At this time, in order to remove the unmelted material in the polymer, it is preferable to filter the polymer with a fiber sintered stainless metal filter.
- inorganic and organic particles such as clay, mica, titanium oxide, calcium carbonate, carion, talc, wet silica are used to impart easy slipping, abrasion resistance, scratch resistance, etc. to the surface of the polyester substrate.
- Dry silica, colloidal silica, inorganic particles such as calcium phosphate, barium sulfate, alumina and zirconia, organic particles containing acrylic acid, styrene resin, thermosetting resin, silicone and imide compound as constituents, and polyester polymerization reaction It is also a preferred aspect to add particles (so-called internal particles) that are precipitated by a catalyst that is sometimes added.
- additives such as compatibilizers, plasticizers, weathering agents, antioxidants, thermal stabilizers, lubricants, antistatic agents, whitening agents, coloring, as long as the effects of the present invention are not impaired.
- Agents, conductive agents, ultraviolet absorbers, flame retardants, flame retardant aids, pigments and dyes may be added.
- a vent type twin-screw kneading extruder in which the end-capping agent is directly mixed with PET pellets and heated to a temperature of 270 to 275 ° C. is used.
- a method of kneading into PET to form a high-concentration master pellet is effective.
- the obtained PET pellets are dried under reduced pressure for 3 hours or more at a temperature of 180 ° C., and then in a nitrogen stream or under reduced pressure so that the intrinsic viscosity does not decrease, a temperature of 265 to 280 ° C., more preferably 270 to 280 ° C. It supplies to the extruder heated to the temperature of 275 degreeC, it extrudes from a slit-shaped die
- melt lamination a plurality of different polymers are melt laminated using two or more extruders and manifolds or merge blocks.
- the melt lamination is preferably used, for example, when the above-described reflective layer (white layer) is coextruded.
- the melt (melt) extruded from the extruder in this way is solidified on the casting (cooling) roll given the temperature distribution as described above to obtain an original fabric (unstretched film).
- the temperature of the cooling roll is preferably 10 ° C. or more and 60 ° C. or less, more preferably 15 ° C. or more and 55 ° C. or less, and further preferably 20 ° C. or more and 50 ° C. or less.
- an electrostatic application method, an air knife method, a method of forming a water film on the cooling roll, or the like can be preferably used.
- the linear velocity of the cast roll is preferably 10 m / min or more, more preferably 15 / min to 50 m / min, and even more preferably 18 m / min or more. 40 m / min or less. Below this range, the residence time of the melt on the cast roll becomes longer, and the temperature difference applied by the above method is equalized and the effect is reduced. On the other hand, if this range is exceeded, uneven thickness of the melt is likely to occur, and the resulting uneven temperature of the melt exceeds the above range, which is not preferable.
- the present invention is characterized by adding fine resin particles to the extruder. That is, the shearing heat is most easily generated at the start of melting at the initial stage of kneading.
- the pellet and the screw are strongly rubbed to generate heat.
- the fine particles preferably have a size of 200 mesh or more and 10 mesh or less, and can be obtained by crushing the pellet and then passing it through a sieve.
- the addition amount of the fine particles is preferably 0.1% or more and 5% or less, more preferably 0.3% or more and 4% or less, and further preferably 0.5% or more and 3% or less. If it is less than this range, the above effect is insufficient, and if this range is exceeded, friction with the screw becomes too large, slip occurs, uneven melt thickness occurs due to discharge fluctuation, and the temperature distribution on the cast roll is in accordance with the present invention. Exceeding range is not preferable.
- the original fabric (unstretched film) obtained as described above is stretched biaxially in the longitudinal direction and the width direction, and then heat-treated.
- a sequential biaxial stretching method such as stretching in the width direction after stretching in the longitudinal direction, a simultaneous biaxial stretching method in which the longitudinal direction and the width direction are simultaneously stretched using a simultaneous biaxial tenter, etc.
- a method combining a sequential biaxial stretching method and a simultaneous biaxial stretching method is included.
- the unstretched film is stretched in the machine direction using the difference in peripheral speed of the rolls (MD stretching) using a longitudinal stretching machine in which several rolls are arranged, and then stretched laterally by a tenter.
- MD stretching difference in peripheral speed of the rolls
- TD stretching biaxial stretching method
- the unstretched film is MD-stretched.
- the preheating temperature is preferably 40 ° C. or higher and 90 ° C. or lower, more preferably 50 ° C. or higher and 85 ° C. or lower, and further preferably 60 ° C. or higher and 80 ° C. or lower.
- Such preheating is performed by passing the raw material over a heating (temperature control) roll. At this time, it is preferable to impart a temperature distribution in the width direction to the roll as described above.
- the preheating time is preferably 1 second to 120 seconds, more preferably 5 seconds to 60 seconds, and still more preferably 10 seconds to 40 seconds.
- MD stretching may be performed in one step or in multiple steps.
- the glass transition temperature is Tg to Tg + 15 ° C. (more preferably Tg + 10 ° C.), and the preferred draw ratio is 2.0 to 6.0 times, more preferably 3.0 to 5. 5 times, more preferably 3.5 to 5.0 times.
- a cooling roll group having a temperature of 20 to 50 ° C.
- the first low-temperature stretching (MD1 stretching) is carried out with a heating roll group in the range of (Tg ⁇ 20) to (Tg + 10) ° C., more preferably in the range of (Tg ⁇ 10) to (Tg + 5) ° C.
- the film is stretched 1.1 to 3.0 times, more preferably 1.2 to 2.5 times, and still more preferably 1.5 to 2.0 times, and then higher than the MD stretching 1 temperature (Tg + 10) MD stretching 2 is performed at ⁇ (Tg + 50). A more preferable temperature is (Tg + 15) ( ⁇ Tg + 30).
- the preferred draw ratio of MD stretch 2 is 1.2 to 4.0 times, more preferably 1.5 to 3.0 times.
- the MD stretching ratio of MD stretching 1 and MD stretching 2 is preferably 2.0 to 6.0 times, more preferably 3.0 to 5.5 times, still more preferably 3.5 to 5 times. .0 times.
- the ratio between the draw ratios of the first stage and the second stage is preferably 1.1 to 3, more preferably 1.15 to 2 times, Preferably they are 1.2 times or more and 1.8 times or less. After stretching, it is preferably cooled by a cooling roll group having a temperature of 20 to 50 ° C.
- stretching in the width direction is performed using a tenter (sometimes referred to as a stenter).
- the draw ratio is preferably 2.0 to 6.0 times, more preferably 3.0 to 5.5 times, and still more preferably 3.5 to 5.0 times.
- the temperature is preferably in the range of (Tg) to (Tg + 50) ° C., more preferably in the range of (Tg) to (Tg + 30) ° C. (TD stretching).
- Tg represents a glass transition temperature and can be measured based on JIS K7121 or ASTM D3418-82. For example. In the present invention, measurement is performed using a differential scanning calorimeter (DSC) manufactured by Shimadzu Corporation.
- DSC differential scanning calorimeter
- a polymer such as polyester is weighed as a sample, set in an aluminum pan, and heated at a rate of temperature increase of 10 ° C./min from room temperature to a final temperature of 300 ° C., with a DSC apparatus, the amount of heat with respect to temperature. was measured as the glass transition temperature.
- the thickness of the polyester base material is preferably about 25 to 300 ⁇ m. If the thickness is 25 ⁇ m or more, the mechanical strength is good, and if it is 300 ⁇ m or less, it is advantageous in terms of cost. In particular, the polyester base material has a tendency that the hydrolysis resistance deteriorates as the thickness increases, and the durability during long-term use tends to decrease. In the present invention, the thickness of the polyester base material is 120 ⁇ m or more and 300 ⁇ m or less. In addition, when the content of the carboxyl group in the polyester is 2 to 15 equivalent / t, the effect of improving the wet heat durability is further exhibited.
- the polyester base material preferably has a surface treated by corona treatment (also referred to as corona discharge treatment), flame treatment, low-pressure plasma treatment, atmospheric pressure plasma treatment, or ultraviolet treatment.
- corona treatment also referred to as corona discharge treatment
- flame treatment low-pressure plasma treatment
- atmospheric pressure plasma treatment or ultraviolet treatment.
- ultraviolet treatment ultraviolet treatment
- these surface treatments improve adhesion by increasing carboxyl groups and hydroxyl groups on the surface of the polyester substrate.
- a cross-linking agent especially an oxazoline-based or carbodiimide-based cross-linking agent highly reactive with the carboxyl group
- strong adhesiveness can be obtained. This is more remarkable in the case of corona treatment.
- the polymer layer of the present invention is a layer disposed in contact with the surface of the polyester substrate or via another layer.
- the polymer layer is composed of at least a specific composite polymer including a non-siloxane structural unit and a (poly) siloxane structural unit represented by the following general formula (1) in the molecule. Since the polymer layer in the present invention includes a composite polymer, the adhesion to the polyester base material and the adhesion between the layers (particularly the adhesion between the sealing material provided on the battery side substrate) are improved. It is preferably formed directly on the polyester substrate. Moreover, since a polymer layer having heat and heat storage resistance is formed, the polymer layer is preferably used as the outermost layer exposed to the external environment, that is, the back layer.
- This polymer layer can be constituted by using other components depending on the case, and the components differ depending on the application.
- the polymer layer can constitute a colored layer that bears the function of reflecting sunlight, imparting appearance design, and the like, a back layer disposed on the side opposite to the side on which sunlight is incident, and the like.
- the polymer layer when configured as a reflective layer that reflects sunlight toward the incident side, the polymer layer can be configured by further using a colorant such as a white pigment.
- the reflective layer is formed as a polymer layer containing a composite polymer.
- it may be configured as a white layer (polymer layer) / polymer layer / polyester base material laminated structure.
- the white layer can be configured as a reflective layer. It is possible to further improve the adhesion and adhesion within the polymer sheet of the reflective layer.
- the polymer layer in the present invention is composed of a (poly) siloxane structural unit represented by the following general formula (1) having a mass ratio of 15 to 85 mass% and a non-siloxane structural unit having a mass ratio of 85 to 15 mass% in the molecule. And at least one kind of composite polymer.
- a (poly) siloxane structural unit represented by the following general formula (1) having a mass ratio of 15 to 85 mass% and a non-siloxane structural unit having a mass ratio of 85 to 15 mass% in the molecule.
- at least one kind of composite polymer By containing this composite polymer, adhesion between the polyester base material that is the support and the constituent base material of the interlayer substrate or battery side substrate (for example, a sealing material such as EVA), that is, heat and moisture are given.
- the peel resistance and shape stability that are easily deteriorated can be dramatically improved as compared with the conventional case.
- the composite polymer in the present invention is a block copolymer obtained by copolymerizing (poly) siloxane and at least one polymer.
- the (poly) siloxane and the copolymerized polymer may be one kind alone, or two or more kinds.
- R 1 and R 2 each independently represent a hydrogen atom, a halogen atom, or a monovalent organic group.
- R 1 and R 2 may be the same or different, and the plurality of R 1 and R 2 may be the same or different from each other.
- n represents an integer of 1 or more.
- R 1 and R 2 may be the same or different and each represents a hydrogen atom, a halogen atom, or a monovalent organic group.
- — (Si (R 1 ) (R 2 ) —O) n —” is a (poly) siloxane segment derived from various (poly) siloxanes having a linear, branched or cyclic structure.
- Examples of the halogen atom represented by R 1 and R 2 include a fluorine atom, a chlorine atom, and an iodine atom.
- the “monovalent organic group” represented by R 1 and R 2 is a group that can be covalently bonded to an Si atom, and may be unsubstituted or may have a substituent.
- Examples of the monovalent organic group include an alkyl group (e.g., methyl group, ethyl group), an aryl group (e.g., phenyl group), an aralkyl group (e.g., benzyl group, phenylethyl), and an alkoxy group (e.g.
- Methoxy group, ethoxy group, propoxy group, etc. Methoxy group, ethoxy group, propoxy group, etc.), aryloxy group (eg, phenoxy group etc.), mercapto group, amino group (eg: amino group, diethylamino group etc.), amide group and the like.
- R 1 and R 2 are each independently a hydrogen atom, a chlorine atom, a bromine atom, an unsubstituted or substituted, in terms of adhesion to adjacent materials such as a polyester base material and durability in a humid heat environment.
- Alkyl groups having 1 to 4 carbon atoms particularly methyl group, ethyl group
- unsubstituted or substituted phenyl group unsubstituted or substituted alkoxy group
- mercapto group unsubstituted amino group
- amide group More preferably an unsubstituted or substituted alkoxy group (preferably an alkoxy group having 1 to 4 carbon atoms) from the viewpoint of durability in a moist heat environment.
- the n is preferably 1 to 5000, and more preferably 1 to 1000.
- the ratio of “— (Si (R 1 ) (R 2 ) —O) n —” in the composite polymer is the total mass of the composite polymer.
- the content is preferably from 15 to 85% by mass, more preferably from 20 to 80% by mass in terms of adhesion to the polyester base material and durability in a moist heat environment. If the ratio of the polysiloxane moiety is less than 15% by mass, the adhesion to the polyester substrate and the adhesion durability when exposed to a moist heat environment are poor, and if it exceeds 85% by mass, the liquid becomes unstable.
- non-siloxane structural unit copolymerized with the siloxane structural unit is not particularly limited except that it does not have a siloxane structure, and is derived from an arbitrary polymer. Any of the polymer segments may be used. Examples of the polymer (precursor polymer) that is a precursor of the polymer segment include various polymers such as a vinyl polymer, a polyester polymer, and a polyurethane polymer. From the viewpoint of easy preparation and excellent hydrolysis resistance, vinyl polymers and polyurethane polymers are preferred, and vinyl polymers are particularly preferred.
- Typical examples of the vinyl polymer include various polymers such as an acrylic polymer, a carboxylic acid vinyl ester polymer, an aromatic vinyl polymer, and a fluoroolefin polymer.
- acrylic polymers that is, acrylic structural units as non-siloxane structural units
- the polymer which comprises a non-siloxane type structural unit may be single 1 type, and 2 or more types of combined use may be sufficient as it.
- the precursor polymer forming the non-siloxane structural unit is preferably one containing at least one of an acid group and a neutralized acid group and / or a hydrolyzable silyl group.
- the vinyl polymer includes, for example, (a) a vinyl monomer containing an acid group and a vinyl monomer containing a hydrolyzable silyl group and / or a silanol group.
- Such a precursor polymer can be produced and obtained by using, for example, the method described in paragraph Nos. 0021 to 0078 of JP-A-2009-52011.
- the composite polymer may be used alone as a binder, or may be used in combination with other polymers.
- the ratio of the composite polymer in the present invention is preferably 30% by mass or more, more preferably 60% by mass or more of the total binder.
- the ratio of the composite polymer is 30% by mass or more, the adhesiveness with the polyester base material and the durability in a wet heat environment are more excellent.
- the molecular weight of the composite polymer is preferably 5,000 to 100,000, and more preferably 10,000 to 50,000.
- the temperature is about 20 to 150 ° C. for about 30 minutes to 30 hours ( (Preferably, the reaction can be performed at 50 to 130 ° C. for 1 to 20 hours).
- various silanol condensation catalysts such as an acidic compound, a basic compound, and a metal containing compound, can be added.
- water and a silanol condensation catalyst are added to a mixture of a precursor polymer and an alkoxysilane compound, and a temperature of about 20 to 150 ° C. for 30 minutes to 30 minutes. It can be prepared by performing hydrolysis condensation for about an hour (preferably at 50 to 130 ° C. for 1 to 20 hours).
- the polymer layer has a structural portion derived from a cross-linking agent that cross-links between the composite polymers. That is, the polymer layer can be formed using a crosslinking agent capable of crosslinking between the composite polymers.
- a crosslinking agent capable of crosslinking between the composite polymers.
- crosslinking agent examples include epoxy-based, isocyanate-based, melamine-based, carbodiimide-based, and oxazoline-based crosslinking agents.
- crosslinking agents such as carbodiimide compounds and oxazoline compounds are preferable.
- oxazoline-based crosslinking agent examples include 2-vinyl-2-oxazoline, 2-vinyl-4-methyl-2-oxazoline, 2-vinyl-5-methyl-2-oxazoline, 2-isopropenyl-2- Oxazoline, 2-isopropenyl-4-methyl-2-oxazoline, 2-isopropenyl-5-ethyl-2-oxazoline, 2,2'-bis- (2-oxazoline), 2,2'-methylene-bis- (2-oxazoline), 2,2′-ethylene-bis- (2-oxazoline), 2,2′-trimethylene-bis- (2-oxazoline), 2,2′-tetramethylene-bis- (2-oxazoline) ), 2,2′-hexamethylene-bis- (2-oxazoline), 2,2′-octamethylene-bis- (2-oxazoline), 2,2′-ethylene-bis- (4,4 ′) Dimethyl-2-oxazoline), 2,2'-p-pheny
- (co) polymers of these compounds are also preferably used.
- a compound having an oxazoline group Epocros K2010E, K2020E, K2030E, WS-500, WS-700 (all manufactured by Nippon Shokubai Chemical Co., Ltd.) and the like can be used.
- carbodiimide-based crosslinking agent examples include dicyclohexylmethane carbodiimide, tetramethylxylylene carbodiimide, dicyclohexylmethane carbodiimide, and the like.
- a carbodiimide compound described in JP-A-2009-235278 is also preferable.
- carbodiimide-based crosslinking agents such as Carbodilite SV-02, Carbodilite V-02, Carbodilite V-02-L2, Carbodilite V-04, Carbodilite E-01, Carbodilite E-02 (all Nisshinbo Chemical Co., Ltd.) (Commercially available) can also be used.
- the mass ratio of the structural portion derived from the crosslinking agent to the composite polymer is preferably 1 to 30% by mass, more preferably 5 to 20% by mass.
- the content of the cross-linking agent is 1% by mass or more, the strength of the polymer layer and the adhesion after wet heat aging are excellent, and when it is 30% by mass or less, the pot life of the coating solution can be kept long.
- the polymer layer includes the composite polymer as described above, so that the adhesion to the polyester base material is improved, and the adhesion between the layers (the polymer sheet of the present invention is used as a back sheet).
- adhesion between the sealing material provided on the battery side substrate is improved.
- it is excellent in deterioration resistance (adhesion durability) in a humid heat environment.
- the outermost layer disposed at the position farthest from the polyester base material is provided.
- a back layer disposed on the opposite side (back side) to the side facing the battery side substrate including the solar cell element (front side), and the sealing for sealing the solar cell element on the battery side substrate.
- a light-reflective reflective layer disposed in contact with a stopper.
- the thickness of one layer of the polymer layer is usually preferably from 0.3 ⁇ m to 22 ⁇ m, more preferably from 0.5 ⁇ m to 15 ⁇ m, still more preferably from 0.8 ⁇ m to 12 ⁇ m, and particularly preferably from 1.0 ⁇ m to 8 ⁇ m. The range of 2 to 6 ⁇ m is most preferable.
- the thickness of the polymer layer is 0.3 ⁇ m, further 0.8 ⁇ m or more, it is difficult for moisture to penetrate from the surface of the polymer layer to the inside when exposed to a humid heat environment, and the interface between the polymer layer and the polyester base material Adhesiveness is remarkably improved by making it difficult for moisture to reach the surface.
- the thickness of the polymer layer is 22 ⁇ m or less, and further 12 ⁇ m or less, the polymer layer itself is not easily fragile, and the adhesion of the polymer layer is improved by being less likely to break when exposed to a humid heat environment.
- the polymer layer in the present invention has a cross-linked structure in which the composite polymer and the polymer molecules of the composite polymer are cross-linked with a cross-linking agent, and the ratio of the structural portion derived from the cross-linking agent to the composite polymer is 1 to 30% by mass In the case where the thickness of the polymer layer is 0.8 ⁇ m to 12 ⁇ m, the effect of improving the adhesiveness after wet heat aging is particularly excellent.
- the polymer layer in the present invention may be configured as a back layer.
- other components such as various additives may be further included as necessary.
- the back layer is supported. It is a back surface protective layer disposed on the opposite side of the polyester substrate that is the body and facing the battery side substrate, and may have a single-layer structure or a structure in which two or more layers are laminated.
- the back layer which is the polymer layer in the present invention is disposed as the outermost layer farthest from the polyester base material is preferable.
- both back layers may be the composite polymer or a polymer layer containing both the composite polymer and the cross-linking agent, and only one back layer is the composite polymer, Or the polymer layer containing both the said composite polymer and the said crosslinking agent may be sufficient.
- at least the back layer in contact with the polyester base material includes the composite polymer or the composite polymer and the cross-linking agent. It is preferable that it is composed of layers.
- the second back layer provided further above the first back layer on the polyester base is composed of a (poly) siloxane structural unit represented by the general formula (1) and a non-polysiloxane structure. It is not necessary to include a composite polymer containing units, but in that case, a uniform film without voids of the resin alone is formed to prevent moisture intrusion from the voids between the polymer and the pigment, and adhesion in a humid heat environment From the viewpoint of enhancing the properties, it is preferable not to include a polysiloxane homopolymer.
- surfactants As other components that can be contained in the back layer, surfactants, fillers and the like can be mentioned as described later. Moreover, you may include the pigment used for a colored layer. Details of these other components and pigments and preferred embodiments will be described later.
- the polymer layer in the present invention is configured as a colored layer (preferably a reflective layer), it further contains a pigment in addition to the composite polymer.
- the colored layer may further include other components such as various additives as necessary.
- the polymer sheet for solar cells of the present invention is used as a back sheet for solar cells
- the incident light passes through the solar cells and is not used for power generation.
- Increasing the power generation efficiency of the solar cell module by reflecting the light that reaches the sheet and returning it to the solar cell.
- a back sheet can be seen around the solar cell, and by providing a colored layer on the back sheet, the decorativeness can be improved and the appearance can be improved.
- the colored layer in the present invention can contain at least one pigment.
- the pigment include inorganic pigments such as titanium dioxide, barium sulfate, silicon oxide, aluminum oxide, magnesium oxide, calcium carbonate, kaolin, talc, ultramarine blue, bitumen, and carbon black, and organic pigments such as phthalocyanine blue and phthalocyanine green. It can be appropriately selected and contained.
- a white pigment is preferable when the polymer layer is configured as a reflective layer that reflects light that has entered the solar cell and passed through the solar cell and returns it to the solar cell.
- the white pigment titanium dioxide, barium sulfate, silicon oxide, aluminum oxide, magnesium oxide, calcium carbonate, kaolin, talc and the like are preferable.
- the content of the pigment in the colored layer is preferably in the range of 2.5 to 8.5 g / m 2 .
- the pigment content is 2.5 g / m 2 or more, necessary coloring can be obtained, and reflectance and decorative properties can be effectively provided.
- the content of the pigment in the colored layer is 8.5 g / m 2 or less, the planar shape of the colored layer is easily maintained and the film strength is excellent.
- the pigment content is more preferably in the range of 4.5 to 8.0 g / m 2 .
- the average particle diameter of the pigment is preferably 0.03 to 0.8 ⁇ m, more preferably about 0.15 to 0.5 ⁇ m in volume average particle diameter. When the average particle size is within the above range, the light reflection efficiency is high.
- the average particle diameter is a value measured by a laser analysis / scattering particle size distribution measuring apparatus LA950 (manufactured by Horiba, Ltd.).
- the content of the binder component is preferably in the range of 15 to 200% by mass, more preferably in the range of 17 to 100% by mass with respect to the pigment.
- the content of the binder is 15% by mass or more, the strength of the colored layer is sufficiently obtained, and when it is 200% by mass or less, the reflectance and the decorativeness can be kept good.
- surfactant a filler, etc.
- a known surfactant such as an anionic or nonionic surfactant can be used.
- the range of the amount added in the polymer layer is preferably 0.1 to 15 mg / m 2 , more preferably 0.5 to 5 mg / m 2 .
- the addition amount of the surfactant is 0.1 mg / m 2 or more, generation of repellency can be suppressed and good layer formation can be obtained, and when it is 15 mg / m 2 or less, adhesion can be performed satisfactorily. .
- a filler may further be added to the polymer layer in the present invention.
- the addition amount of the filler is preferably 20% by mass or less, more preferably 15% by mass or less per binder of the polymer layer. When the addition amount of the filler is 20% by mass or less, the planar shape of the polymer layer can be kept better.
- the light reflectance at 550 nm on the surface on which the colored layer and the easily adhesive layer are provided is preferably 75% or more.
- the light reflectivity is the ratio of the amount of light incident from the surface of the easy-adhesion layer to the amount of incident light reflected from the reflection layer and emitted again from the easy-adhesion layer.
- light having a wavelength of 550 nm is used as the representative wavelength light.
- the light reflectance can be adjusted to 75% or more by controlling the content of the colorant in the range of 2.5 to 30 g / m 2 .
- the polymer sheet for solar cells of the present invention may have other functional layers in addition to the polyester substrate and the polymer layer.
- an undercoat layer and an easy adhesion layer can be provided.
- an undercoat layer may be provided between the polyester substrate (support) and the polymer layer.
- the thickness of the undercoat layer is preferably in the range of 2 ⁇ m or less, more preferably 0.05 ⁇ m to 2 ⁇ m, and still more preferably 0.1 ⁇ m to 1.5 ⁇ m. When the thickness is 2 ⁇ m or less, the planar shape can be kept good. Moreover, it is easy to ensure required adhesiveness because thickness is 0.05 micrometer or more.
- the undercoat layer can contain a binder.
- a binder for example, polyester, polyurethane, acrylic resin, polyolefin, or the like can be used.
- epoxy, isocyanate, melamine, carbodiimide, oxazoline and other crosslinking agents, anionic and nonionic surfactants, silica and other fillers are added to the undercoat layer. Also good.
- the method for applying the undercoat layer and the solvent of the coating solution used there is no particular limitation on the method for applying the undercoat layer and the solvent of the coating solution used.
- a coating method for example, a gravure coater or a bar coater can be used.
- the solvent used for the coating solution may be water or an organic solvent such as toluene or methyl ethyl ketone.
- a solvent may be used individually by 1 type and may be used in mixture of 2 or more types.
- the coating may be applied to the polyester substrate after biaxial stretching, or may be a method of stretching in a direction different from the initial stretching after coating to the polyester substrate after uniaxial stretching. Furthermore, you may extend
- the solar cell backsheet When using the solar cell polymer sheet of the present invention as a solar cell backsheet, the solar cell backsheet is brought into contact with the sealing agent of the battery-side substrate in which the solar cell element is sealed with a sealing material, If it is a method which can arrange
- the polymer sheet of the present invention may be provided with a colored layer (preferably a reflective layer) substantially free of the composite polymer.
- a colored layer preferably a reflective layer
- it can be suitably configured by providing a polymer layer containing a composite polymer between the colored layer (particularly the reflective layer) and the polyester substrate.
- the colored layer in this case includes at least a polymer component other than the composite polymer and a pigment, and can be configured using other components such as various additives as necessary.
- a polymer component other than the composite polymer is not particularly limited and can be appropriately selected according to the purpose.
- substantially free means that the composite layer is not actively contained in the colored layer. Specifically, the content of the composite polymer in the colored layer is 15% by mass or less. Preferably, the case where the composite polymer is not contained (the content is 0 (zero) mass%) is preferred.
- the reflective layer is not limited to an embodiment containing a composite polymer, and one layer or a layer between the reflective layer substantially free of the composite polymer and the polyester substrate. You may be comprised in the aspect in which the polymer layer of two or more layers was provided. In this case, by providing a polymer layer containing a composite polymer between the polyester substrate and the colored layer, the adhesion and adhesion between the reflective layer and the polyester substrate can be improved, and the water resistance can be further increased. it can. Thereby, deterioration of the weather resistance resulting from poor adhesion is prevented.
- the polymer sheet for solar cells of the present invention is a method capable of forming the polymer layer in the present invention and, if necessary, a colored layer, an undercoat layer, etc. on the polyester substrate described above. Any method may be used.
- the carboxyl group content is 15 equivalent / t or less
- the minute endothermic peak temperature Tmeta (° C.) determined by differential scanning calorimetry is 220 ° C. or less
- the temperature is 125 ° C.
- the relative humidity is 100% RH.
- the siloxane structural unit represented by the above general formula (1) having a mass ratio of 15 to 85% by mass in the molecule on a polyester substrate having an average elongation retention rate of 10% or more after standing for 72 hours under the above conditions
- a coating solution containing a composite polymer containing 85 to 15% by mass of a non-siloxane structural unit and preferably a crosslinking agent (and, if necessary, a coating solution for an easy-adhesive layer) It can be suitably produced by a method of providing a step of forming a single polymer layer (a method for producing a polymer sheet for solar cells of the present invention).
- the polymer layer coating solution is a coating solution containing at least a composite polymer as described above. The details of the polyester base material, the composite polymer constituting each coating solution, and other components are as described above.
- Suitable coating methods are also as described above, and for example, a gravure coater or a bar coater can be used.
- the polymer layer coating solution is applied directly to the surface of the polyester substrate or through an undercoat layer having a thickness of 2 ⁇ m or less, and the polymer layer (for example, a colored layer (preferably a colored layer (preferably Can form a reflective layer) and a back layer).
- the polymer layer can be formed by a method in which a sheet-like polymer member is bonded to a polyester substrate, a method in which a polymer layer is coextruded when forming a polyester substrate, a method by coating, or the like.
- coating is preferable at the point which is easy and can form in a thin film with uniformity.
- a coating method for example, a known coating method such as a gravure coater or a bar coater can be used.
- the coating solution may be an aqueous system using water as an application solvent, or a solvent system using an organic solvent such as toluene or methyl ethyl ketone. Among these, from the viewpoint of environmental burden, it is preferable to use water as a solvent.
- a coating solvent may be used individually by 1 type, and may mix and use 2 or more types.
- the polymer layer coating liquid is preferably an aqueous coating liquid in which 50% by mass or more, preferably 60% by mass or more, of the solvent contained therein is water.
- the aqueous coating solution is preferable in terms of environmental load, and is advantageous in that the environmental load is particularly reduced when the ratio of water is 50% by mass or more.
- the proportion of water in the coating liquid for the polymer layer is preferably larger from the viewpoint of environmental load, and more preferably 90% by mass or more of water is contained in the total solvent.
- a drying process for drying under desired conditions may be provided.
- the solar cell backsheet of the present invention is a solar cell backsheet that is disposed in contact with the sealing agent of the battery-side substrate in which the solar cell elements are sealed with a sealing material. It is comprised using the polymer sheet for solar cells of the invention, or the polymer sheet for solar cells manufactured by the manufacturing method of the polymer sheet for solar cells of the present invention described above.
- the polymer sheet for solar cells of the present invention may be used as it is as the polymer sheet for solar cells, or the polymer sheet for solar cells of the present invention is provided with an easy-adhesive layer and a barrier layer described later. Also good.
- the solar cell backsheet of the present invention may be configured to include two or more solar cell polymer sheets of the present invention.
- the solar cell polymer sheet and the solar cell polymer sheet include It is also a preferred embodiment that the layers are bonded together with an adhesive.
- the adhesive for example, LX660 (K) [DIC Co., Ltd. adhesive] mixed with 10 parts of a curing agent KW75 [DIC Co., Ltd.] is used.
- the laminated polymer sheet laminate for solar cells may be further subjected to hot press bonding using a press apparatus such as a vacuum laminator (vacuum laminator manufactured by Nisshinbo Co., Ltd.).
- an easy-adhesive layer is further provided on the surface of the polyester base opposite to the surface on which the polymer layer is provided, or on the polymer layer (particularly the reflective layer). It may be done.
- the easy-adhesion layer is a layer for firmly bonding the back sheet to a sealing material that seals a solar cell element (hereinafter also referred to as a power generation element) of the battery side substrate (battery body).
- the easy-adhesion layer can be formed using a binder and inorganic fine particles, and may further include other components such as additives as necessary.
- the easy-adhesion layer is for the sealing material that seals the power generating element on the battery side substrate ⁇ eg, ethylene-vinyl acetate (EVA); polyvinyl butyral (PVB), epoxy resin, etc.).
- EVA ethylene-vinyl acetate
- PVB polyvinyl butyral
- epoxy resin etc.
- the adhesive strength is 5 N / cm or more. When the adhesive force is 5 N / cm or more, wet heat resistance capable of maintaining adhesiveness is easily obtained.
- the adhesive force is preferably 10 N / cm or more, and more preferably 20 N / cm or more.
- the adhesive strength can be adjusted by adjusting the amount of the binder and inorganic fine particles in the easy-adhesive layer, or applying a corona treatment to the surface of the back sheet that is bonded to the sealing material.
- the easy-adhesion layer can contain at least one binder.
- the binder suitable for the easy-adhesive layer include polyester, polyurethane, acrylic resin, polyolefin, and the like. Among these, acrylic resin and polyolefin are preferable from the viewpoint of durability. As the acrylic resin, a composite resin of acrylic and silicone is also preferable.
- binders examples include Chemipearl S-120 and S-75N (both manufactured by Mitsui Chemicals) as specific examples of polyolefins, and Jurimer ET-410 and SEK-301 (both Nippon Pure Chemical (both manufactured by Mitsui Chemicals, Inc.)).
- Jurimer ET-410 and SEK-301 both Nippon Pure Chemical (both manufactured by Mitsui Chemicals, Inc.)
- Ceranate WSA1060, WSA1070 both manufactured by DIC Corporation
- H7620, H7630, H7650 both manufactured by Asahi Kasei Chemicals Corporation
- the content of the binder in the easy-adhesive layer is preferably in the range of 0.05 to 5 g / m 2 . In particular, the range of 0.08 to 3 g / m 2 is more preferable.
- the content of the binder, 0.05 g / m 2 or more is desired as easy adhesion obtained to that, better surface state is obtained when the is 5 g / m 2 or less.
- the easily adhesive layer can contain at least one kind of inorganic fine particles.
- the inorganic fine particles include silica, calcium carbonate, magnesium oxide, magnesium carbonate, and tin oxide.
- fine particles of tin oxide and silica are preferable in that the decrease in adhesiveness when exposed to a humid heat atmosphere is small.
- the particle size of the inorganic fine particles is preferably about 10 to 700 nm, more preferably about 20 to 300 nm in terms of volume average particle size. When the particle size is within this range, better easy adhesion can be obtained.
- the particle size is a value measured by a laser analysis / scattering particle size distribution measuring apparatus LA950 (manufactured by Horiba, Ltd.).
- the shape of the inorganic fine particles is not particularly limited, and any shape such as a spherical shape, an irregular shape, or a needle shape can be used.
- the content of the inorganic fine particles is in the range of 5 to 400% by mass with respect to the binder in the easy-adhesive layer.
- the content of the inorganic fine particles is less than 5% by mass, good adhesiveness cannot be maintained when exposed to a moist heat atmosphere, and when it exceeds 400% by mass, the surface state of the easily adhesive layer is deteriorated.
- the content of inorganic fine particles is preferably in the range of 50 to 300% by mass.
- the easily adhesive layer can contain at least one crosslinking agent.
- the crosslinking agent suitable for the easily adhesive layer include epoxy-based, isocyanate-based, melamine-based, carbodiimide-based, and oxazoline-based crosslinking agents.
- an oxazoline-based cross-linking agent is particularly preferable from the viewpoint of ensuring adhesiveness after wet heat aging.
- Specific examples of the oxazoline-based crosslinking agent include the same specific examples as described in the above-mentioned polymer layer section.
- the content of the crosslinking agent in the easy-adhesive layer is preferably 5 to 50% by mass, more preferably 20 to 40% by mass, based on the binder in the easy-adhesive layer.
- the content of the crosslinking agent is 5% by mass or more, a good crosslinking effect can be obtained, and the strength and adhesiveness of the colored layer can be maintained.
- the content is 50% by mass or less, the pot life of the coating liquid Can be kept long.
- the easily adhesive layer in the present invention may further contain a known matting agent such as polystyrene, polymethylmethacrylate, or silica, or a known surfactant such as anionic or nonionic. .
- the easy-adhesive layer can be formed by a method in which a sheet-like polymer member having easy adhesive properties is bonded to a base material or a method by coating. Especially, the method by application
- a coating method for example, a known coating method such as a gravure coater or a bar coater can be used.
- the coating solvent used for preparing the coating solution may be water or an organic solvent such as toluene or methyl ethyl ketone.
- a coating solvent may be used individually by 1 type, and may mix and use 2 or more types.
- the thickness of the easy-adhesion layer is not particularly limited, but is usually preferably 0.05 to 8 ⁇ m, more preferably 0.1 to 5 ⁇ m.
- the thickness of the easy-adhesion layer is 0.05 ⁇ m or more, necessary easy adhesion can be suitably obtained, and when it is 8 ⁇ m or less, the surface shape becomes better.
- the easily adhesive layer of the present invention needs to be transparent so as not to reduce the effect of the colored layer.
- the solar cell backsheet of the present invention has an adhesive force with the sealing material after storage for 48 hours in an atmosphere of 120 ° C. and 100% RH, with respect to the adhesive strength with the sealing material before storage, It is preferable that it is 75% or more.
- the solar cell backsheet of the present invention includes a predetermined amount of binder and a predetermined amount of inorganic fine particles with respect to the binder, and has an adhesive strength of 10 N / cm or more with respect to the EVA-based sealing material.
- the solar cell backsheet of the present invention has a barrier layer.
- the water vapor transmission rate (moisture permeability) of the barrier layer is preferably 10 0 g / m 2 ⁇ d to 10 ⁇ 6 g / m 2 ⁇ d, more preferably 10 ⁇ 1 g / m 2 ⁇ d to 10 ⁇ 5 g. / M 2 ⁇ d, more preferably 10 ⁇ 2 g / m 2 ⁇ d to 10 ⁇ 4 g / m 2 ⁇ d.
- the moisture permeability can be measured based on JIS Z0208.
- the following dry method is preferably used for the formation of the barrier layer.
- -Barrier layer formation method As a method of forming a gas barrier layer by a dry method, resistance heating evaporation, electron beam evaporation, induction heating evaporation, and vacuum evaporation methods such as an assist method using plasma or ion beam, reactive sputtering method, ion beam sputtering method, Sputtering methods such as ECR (electron cyclotron) sputtering, physical vapor deposition methods such as ion plating (PVD method), chemical vapor deposition methods using heat, light, plasma, etc. (CVD method), etc. Can be mentioned. Among these, a vacuum vapor deposition method in which a film is formed by a vapor deposition method under vacuum is preferable.
- the gas barrier layer is an inorganic layer formed of a material mainly composed of inorganic oxide, inorganic nitride, inorganic oxynitride, inorganic halide, inorganic sulfide, etc.
- the gas barrier layer to be formed It is also possible to directly volatilize the same material as the composition of the material and deposit it on the substrate or the like. However, when this method is used, the composition changes during volatilization, and as a result, the formed film is uniform. May not exhibit characteristics.
- a material having the same composition as the barrier layer formed as a volatilization source is used, oxygen gas in the case of inorganic oxide, nitrogen gas in the case of inorganic nitride, oxygen gas and nitrogen in the case of inorganic oxynitride
- a method of volatilizing a mixed gas of gases by introducing a halogen-based gas in the case of an inorganic halide, and a sulfur-based gas in the case of an inorganic sulfide while introducing them into the system, and 2) selecting an inorganic group as a volatilization source.
- an oxygen gas atmosphere in the case of an inorganic oxide is an oxygen gas atmosphere in the case of an inorganic oxide, a nitrogen gas atmosphere in the case of an inorganic nitride, and an oxygen gas and a nitrogen gas in the case of an inorganic oxynitride.
- examples thereof include a method of reacting an introduced gas with an inorganic layer by holding in a mixed gas atmosphere, in the case of an inorganic halide, in a halogen-based gas atmosphere, and in the case of an inorganic sulfide, in a sulfur-based gas atmosphere.
- the barrier layer is an inorganic oxide
- the inorganic group is used as a volatilization source, volatilized to form an inorganic group layer, and then left in the air to naturally oxidize the inorganic group.
- the method is also preferable because it is easy to form.
- the thickness is preferably 1 ⁇ m or more and 30 ⁇ m or less.
- the thickness of the barrier layer is 1 ⁇ m or more, water hardly penetrates into the polyester film during the lapse of time (thermo) and hardly causes hydrolysis, and when it is 30 ⁇ m or less, the thickness of the barrier layer does not become too thick. In addition, the film does not bend due to the stress of the barrier layer.
- the solar cell module of the present invention is configured by providing the solar cell backsheet of the present invention described above or the solar cell backsheet manufactured by the method of manufacturing the solar cell backsheet described above.
- a solar cell element that converts light energy of sunlight into electrical energy is disposed between a transparent front substrate on which sunlight is incident and the above-described solar cell backsheet of the present invention.
- the solar cell element is sealed and bonded with a sealing material such as ethylene-vinyl acetate between the front substrate and the back sheet. That is, a cell structure portion having a solar cell element and a sealing material for sealing the solar cell element is provided between the front substrate and the back sheet.
- the transparent substrate only needs to have a light-transmitting property through which sunlight can be transmitted, and can be appropriately selected from base materials that transmit light. From the viewpoint of power generation efficiency, the higher the light transmittance, the better.
- a transparent resin such as an acrylic resin, or the like can be suitably used.
- Solar cell elements include silicon-based materials such as single crystal silicon, polycrystalline silicon, and amorphous silicon, III-V groups such as copper-indium-gallium-selenium, copper-indium-selenium, cadmium-tellurium, gallium-arsenic, and II Various known solar cell elements such as a group VI compound semiconductor can be applied.
- Step 2 After completion of the transesterification reaction, 0.019 part of phosphoric acid (equivalent to 1.9 mol / t) and 0.027 part of sodium dihydrogen phosphate dihydrate (equivalent to 1.5 mol / t) were mixed with 0.5% ethylene glycol. An ethylene glycol solution (PH 5.0) dissolved in the part was added.
- PH 5.0 ethylene glycol solution
- Step 3 The polymerization reaction was carried out at a final temperature of 285 ° C. and a vacuum of 0.1 Torr to obtain a polyester having an intrinsic viscosity of 0.54 and a carboxyl group terminal group number of 13 eq / ton.
- Step 4 The obtained polyethylene terephthalate was dried and crystallized at 160 ° C.
- the component (p) was 0.15 mol%, inherent A polyester having a viscosity of 0.90, a carboxyl group terminal group number of 12 eq / ton, a melting point of 255 ° C., and a glass transition temperature of Tg 83 ° C. was obtained.
- Step 5 To 99 parts of the polyester obtained in Step 4, 1 part of Rhein Chemis' Starbuxol P100 ”(polycarbodiimide) was added and compounded.
- Step 6 The compound product obtained above was dried under reduced pressure for 2 hours under conditions of a temperature of 180 ° C. and a vacuum degree of 0.5 mmHg, supplied to an extruder heated to 295 ° C., and filtered with a 50 ⁇ m cut filter. Introduced into T die die. Next, from the inside of the T die die, it is extruded into a sheet shape to form a molten single layer sheet. The molten single layer sheet is closely cooled and solidified by electrostatic application on a drum maintained at a surface temperature of 20 ° C.
- a layer film was obtained.
- Step 7 Subsequently, after preheating the obtained unstretched monolayer film with a heated roll group, 1.8 times MD stretching 1 is performed at a temperature of 80 ° C., and 2.3 times MD stretching 2 is further performed at a temperature of 95 ° C. went.
- the film was stretched 4.1 times in the longitudinal direction (MD direction) in total, and then cooled with a roll group having a temperature of 25 ° C. to obtain a uniaxially stretched film. While holding both ends of the obtained uniaxially stretched film with clips, it is led to a preheating zone at a temperature of 95 ° C. in the tenter, and then continuously in the heating zone at a temperature of 100 ° C.
- Step 8 Subsequently, a heat treatment for 20 seconds was performed at a temperature of 205 ° C. (first heat treatment temperature) in a heat treatment zone in the tenter. Subsequently, at a temperature of 180 ° C., the film was relaxed in the width direction (TD) with a relaxation rate of 3%, and the clip interval of the tenter was shortened, thereby reducing the longitudinal direction (1.5% relaxation rate in the MD). Next, the film was uniformly cooled to 25 ° C. and wound up to obtain a biaxially stretched polyester film (PET-1) having a thickness of 250 ⁇ m.
- PET-1 biaxially stretched polyester film
- the relaxation rate is calculated by the following formula (c), where La is the length of the polyester film before relaxation and Lb is the length of the polyester film after relaxation.
- La and Lb of the width direction of a polyester film and La and Lb of the longitudinal direction of a polyester film are defined as follows. [Width direction] The maximum width of the polyester film at the time of stretching when the polyester film is stretched with a tenter is defined as the length La of the polyester film before relaxation.
- tensile_strength (relaxing) and taking out a polyester film from a tenter be length Lb of the polyester film after relaxation
- PET-2-O-Preparation of PET-2- Regarding [Step 8] of the PET-1 production method a biaxially stretched polyester film (PET-2) was produced in the same manner as PET-1, except that the first heat treatment temperature was changed to 230 ° C.
- Tmeta was changed to 225 ° C. and the average elongation retention was changed to 7% as compared with PET-1.
- PET-3 A biaxially stretched polyester film (PET-3) was produced in the same manner as PET-1, except that sodium dihydrogen phosphate dihydrate was not added in [Step 2] of the method for producing PET-1. Produced. When the properties of PET-3 were evaluated, the average elongation retention was changed to 40% and the phosphorus atom content was changed to 150 ppm as compared with PET-1.
- PET-1 The properties of PET-1 were measured by the following method.
- -Carboxyl group content (AV)- The carboxyl group content (AV) was determined by dissolving polyester completely in a mixed solution of benzyl alcohol / chloroform ( 2/3; volume ratio), using phenol red as an indicator, and using this as a reference solution (0.01 N KOH- Benzyl alcohol mixed solution) and calculated from the titration amount.
- Tmeta (° C.) was defined as the minute endothermic peak temperature before the crystal melting peak.
- the graph reading method of a micro endothermic peak is not described in JIS, it implemented based on the following methods. First, a straight line was drawn at a value of 135 ° C. and a value of 155 ° C., and the area on the endothermic side with respect to the curve of the graph was obtained.
- the area was also obtained for 17 points at 240 ° C.
- the endothermic amount of the minute peak is usually 0.2 to 5.0 J / g
- data having an area of 0.2 J / g or more and 5.0 J / g or less was handled as effective data.
- the peak temperature of the endothermic peak in the temperature region of the data indicating the effective area and the largest area was defined as Tmeta (° C.). In the absence of valid data, Tmeta (° C.) was assumed to be none.
- the elongation at break was measured in accordance with ASTM-D882-97 (referred to 1999 ANNUAL BOOK OF ASTM STANDARDS), cut into a size of 1 cm ⁇ 20 cm, and pulled at a chucking speed of 5 cm and a pulling speed of 300 mm / min.
- the elongation at break (initial) was measured.
- the measurement was performed about 5 samples, and it was set as breaking elongation (initial) A2 by the average value.
- the sample was cut into a size of 1 cm ⁇ 20 cm and treated for 72 hours under the conditions of 125 ° C. and 100% humidity using Hirayama Seisakusho Co., Ltd., High Acceleration Life Test Device (HAST device), PC-304R8D.
- HAST device High Acceleration Life Test Device
- PC-304R8D High Acceleration Life Test Device
- ASTM-D882 (1999) -97 referred to 1999 edition ANNUAL BOOK OF ASTM STANDARDS
- the elongation at break (after treatment) was measured.
- the measurement was performed on five samples, and the elongation at break (after treatment) was A3 based on the average value.
- the average elongation retention (Lave) was calculated by the following (4).
- (Lave) (%) (LrMD + LrTD) / 2 (4)
- LrMD represents the elongation retention in the MD direction
- LrTD represents the elongation retention in the TD direction.
- nMD represents the refractive index in the longitudinal direction (MD) of the film
- nTD represents the refractive index in the perpendicular direction (TD) of the film
- nZD represents the refractive index in the film thickness direction.
- IV -Intrinsic viscosity-
- Table 1 shows the carboxyl group content (AV), Tmeta, average elongation retention, and surface treatment species of the obtained PET-1 to PET-3.
- the surface on which the polymer layer (polymer layer 1 or polymer layer 1 and polymer layer 2) is formed is referred to as a front surface of the polymer sheet.
- the surface opposite to the front side of the polymer sheet is the back side of the polymer sheet (PET-1 to PET-3), and the polymer layer 1 or the polymer layer 1 and the polymer layer 2 are formed.
- This surface is called the front side of the support.
- the surface opposite to the front side of the support is called the back side of the support.
- P-1 to P-5 binders were used.
- P-1 Ceranate WSA-1070 (acrylic / silicone binder) [Dic silicone resin, solid content: 40% by mass]
- P-2 Ceranate WSA-1060 (acrylic / silicone binder) [Dic silicone resin, solid content: 40% by mass]
- P-3 Obligato SW0011F [Fluorine resin manufactured by AGC Co-Tech, solid content: 40% by mass]
- P-4 Finetex Es650 [DIC polyester resin, solid content: 29% by mass]
- P-5 Olester UD350 [Mitsui Chemicals polyurethane resin, solid content: 38% by mass]
- P-1 and P-2 which are silicone resins (composite polymers)
- P-1 has a polysiloxane moiety of about 30% by mass and an acrylic polymer part of about 70% by mass.
- P-2 has a polysiloxane moiety of about 75% by mass and an acrylic polymer portion of about 25% by mass.
- Example 1 Preparation of pigment dispersion- Each component in the following composition was mixed, and the mixture was subjected to dispersion treatment for 1 hour using a dynomill type disperser.
- Binder (P-1) 362.3 parts (Ceranate WSA-1070, manufactured by DIC Corporation, solid content: 40% by mass)
- Carbodiimide compound (crosslinking agent) 36.2 parts (Carbodilite V-02-L2, manufactured by Nisshinbo Co., Ltd., solid content: 40% by mass)
- Surfactant 9.7 parts (Naroacty CL95, manufactured by Sanyo Chemical Industries, Ltd., solid content: 1% by mass)
- -Pigment dispersion 157.0% by mass ⁇ Distilled water: 434.8 parts
- the obtained coating solution 1-P1 for forming the polymer layer 1 was applied to the surface of the support subjected to the corona treatment so that the binder amount was 3.0 g / m 2 and dried at 180 ° C. for 1 minute. Thus, a polymer layer 1 having a dry thickness of about 5 ⁇ m was formed.
- the polymer sheet 1 of Example 1 was produced.
- Adhesiveness- [A] Adhesiveness before wet heat aging
- the polymer sheet prepared as described above was cut into a width of 20 mm and 150 mm to prepare two sample pieces. These two sample pieces are arranged so that the polymer layer side is inside, and an EVA sheet (EV sheet manufactured by Mitsui Chemicals Fabro Co., Ltd .: SC50B) is sandwiched between them and vacuumed. It was made to adhere to EVA by hot pressing using a laminator (vacuum laminator manufactured by Nisshinbo Co., Ltd.). The bonding conditions at this time were as follows. Using a vacuum laminator, evacuation was performed at 128 ° C.
- the EVA non-adhered portion of the obtained adhesion evaluation sample was sandwiched between upper and lower clips with Tensilon (RTENT-1210A manufactured by ORIENTEC), a tensile test was performed at a peeling angle of 180 °, and a tensile speed of 300 mm / min, and the adhesive strength was measured. . Based on the measured adhesive strength, ranking was performed according to the following evaluation criteria. Among these, ranks 4 and 5 are practically acceptable ranges.
- ⁇ Evaluation criteria> 5 No change is observed on the surface even when observed with an optical microscope. 4: When observed with an optical microscope, slight cracks and deformations are observed on the surface. 3: Visual observation reveals that surface gloss has disappeared. 2: A slight crack is seen by visual observation. 1: Cracks are observed on the entire surface even by visual observation.
- Example 2 to 4 In the formation of the polymer layer 1 of Example 1, the coating solution 1-P1 for forming the polymer layer 1 was applied in the same manner as in Example 1 except that the thickness of the polymer layer 1 was as shown in Table 2. Polymer sheets 2 to 4 of Examples 2 to 4 were produced. The obtained polymer sheets 2 to 4 were evaluated in the same manner as the polymer sheet 1. The results are shown in Table 2.
- Example 5 In the production of the polymer sheet 1 of Example 1, the coating liquid 1-P1 for forming the polymer layer 1 was changed to the coating liquid 1-P2 for forming the polymer layer 1 in the same manner as in Example 1 except that A polymer sheet 5 was produced. About the obtained polymer sheet 5, the same evaluation as the polymer sheet 1 was performed. The results are shown in Table 2.
- the coating solution 1-P2 for forming the polymer layer 1 is the same as the coating solution 1-P1 for forming the polymer layer 1 except that the binder (P-1) is changed to the binder (P-2). Prepared.
- Example 6 On the polymer layer 1 of the polymer sheet 3 of Example 3, the following coating solution for forming the polymer layer 2 was further applied to provide the polymer layer 2 to prepare the polymer sheet 6 of Example 6. About the obtained polymer sheet 6, the same evaluation as the polymer sheet 1 was performed. The results are shown in Table 2.
- Binder (P-1): 362.3 parts (Ceranate WSA-1070, manufactured by DIC Corporation, solid content: 40% by mass) Carbodiimide compound (crosslinking agent) 24.2 parts (Carbodilite V-02-L2, manufactured by Nisshinbo Industries, Inc., solid content: 40% by mass)
- Surfactant 24.2 parts (Naroacty CL95, manufactured by Sanyo Chemical Industries, Ltd., solid content: 1% by mass)
- Distilled water 703.8 parts
- the obtained coating liquid 2-P1 for forming the polymer layer 2 was applied onto the polymer layer 1 of the polymer sheet 3 so that the binder amount was 2.0 g / m 2 in terms of the coating amount, and at 180 ° C. for 1 minute.
- the polymer layer 2 having a dry thickness of about 2 ⁇ m was formed by drying.
- Example 7 to 8 In the formation of the polymer layer 2 of Example 6, the coating liquid 2-P1 for forming the polymer layer 2 was applied in the same manner as in Example 6 except that the thickness of the polymer layer 2 was applied as shown in Table 2. Polymer sheets 7 to 8 of Examples 7 to 8 were produced. The obtained polymer sheets 7 to 8 were evaluated in the same manner as the polymer sheet 1. The results are shown in Table 2.
- Example 9 In the production of the polymer sheet 6 of Example 6, the coating liquid 2-P1 for forming the polymer layer 2 was changed to the coating liquid 1-P2 for forming the polymer layer 2 in the same manner as in Example 6, except that A polymer sheet 9 was produced. About the obtained polymer sheet 9, the same evaluation as the polymer sheet 1 was performed. The results are shown in Table 2.
- the coating liquid 2-P2 for forming the polymer layer 2 was the same as the coating liquid 2-P1 for forming the polymer layer 2, except that the binder (P-1) was changed to the binder (P-2). Prepared.
- Example 10 In the production of the polymer sheet 6 of Example 6, the coating solution 2-P1 for forming the polymer layer 2 was changed to the coating solution 1-P3 for forming the polymer layer 2, and the same procedure as in Example 6 was repeated. A polymer sheet 10 was produced. About the obtained polymer sheet 10, the same evaluation as the polymer sheet 1 was performed. The results are shown in Table 2.
- the coating solution 2-P3 for forming the polymer layer 2 is the same as the coating solution 2-P1 for forming the polymer layer 2, except that the binder (P-1) is changed to the binder (P-3). Prepared.
- Example 11 to 12 In the formation of the polymer layer 2 in Example 10, the coating solution 2-P3 for forming the polymer layer 2 was applied in the same manner as in Example 10 except that the thickness of the polymer layer 2 was applied as shown in Table 2. Polymer sheets 11 to 12 of Examples 11 to 12 were produced. The obtained polymer sheets 11 to 12 were evaluated in the same manner as the polymer sheet 1. The results are shown in Table 2.
- Example 1 In the production of the polymer sheet 1 of Example 1, the same procedure as in Example 1 was carried out except that the coating liquid 1-P1 for forming the polymer layer 1 was changed to the coating liquid 1-P4 for forming the polymer layer 1. A polymer sheet 101 was produced. About the obtained polymer sheet 101, the same evaluation as the polymer sheet 1 was performed. The results are shown in Table 2.
- the coating liquid 1-P4 for forming the polymer layer 1 was the same as the coating liquid 1-P1 for forming the polymer layer 1 except that the binder (P-1) was changed to the binder (P-4). Prepared.
- Example 2 In the production of the polymer sheet 1 of Example 1, the same as in Example 1 except that the coating liquid 1-P1 for forming the polymer layer 1 was changed to the coating liquid 1-P5 for forming the polymer layer 1. A polymer sheet 102 was produced. The obtained polymer sheet 102 was evaluated in the same manner as the polymer sheet 1. The results are shown in Table 2.
- the coating solution 1-P5 for forming the polymer layer 1 was the same as the coating solution 1-P1 for forming the polymer layer 1 except that the binder (P-1) was changed to the binder (P-5). Prepared.
- Comparative Example 3 In the production of the polymer sheet 1 of Example 1, the polymer sheet 103 of Comparative Example 3 was produced in the same manner as in Example 1 except that the support PET-1 was changed to PET-2. The obtained polymer sheet 103 was evaluated in the same manner as the polymer sheet 1. The results are shown in Table 2.
- Example 13 to 15 and Comparative Examples 4 to 5 In the production of the polymer sheet 7 of Example 7, Example 13 to Example were similarly performed except that the binder type of the polymer layer 1 was changed to any of PS-1 to PS-5 as shown in Table 2. 15 polymer sheets 13 to 15 and polymer sheets 104 to 105 of Comparative Examples 4 to 5 were produced. The obtained polymer sheets 13 to 15 and 104 to 105 were evaluated in the same manner as the polymer sheet 1. However, the system using PS-5 as a binder (Comparative Example 5) was poor in stability and could not be evaluated. The results are shown in Table 2.
- Binders PS-1 to PS-5 used in Examples 13 to 15 and Comparative Examples 4 to 5 were synthesized as follows. In Synthesis Example-1 to Synthesis Example-5, “%” is based on mass unless otherwise specified.
- MMA methyl methacrylate
- BMA n-butyl methacrylate
- BA n-butyl acrylate
- AA acrylic acid
- TBPO tert-butylperoxy-2-ethylhexanoate
- an aqueous dispersion (PS-1) of a composite polymer having a carboxyl group-containing acrylic polymer portion and a polysiloxane portion having a solid content concentration of 42% and an average particle size of 110 nm was obtained.
- the polysiloxane moiety of PS-1 is about 25%.
- PS-2 was synthesized in the same manner as in Synthesis Example 1, except that the amount of monomer used was changed as follows. 210 parts of phenyltrimethoxysilane (PTMS), 166 parts of dimethyldimethoxysilane (DMDMS), 24 parts of 3-methacryloyloxypropyltrimethoxysilane (MPTMS), 200 parts of methyl methacrylate (MMA), 100 parts of n-butyl methacrylate (BMA) N-butyl acrylate (BA) 70 parts, acrylic acid (AA) 30 parts.
- the polysiloxane moiety of PS-2 is about 50%.
- PS-3 was synthesized in the same manner as in Synthesis Example 1 except that the amount of monomer used was changed as follows. 320 parts of phenyltrimethoxysilane (PTMS), 244 parts of dimethyldimethoxysilane (DMDMS), 36 parts of 3-methacryloyloxypropyltrimethoxysilane (MPTMS), 90 parts of methyl methacrylate (MMA), 60 parts of n-butyl methacrylate (BMA) N-butyl acrylate (BA) 20 parts, acrylic acid (AA) 30 parts.
- the polysiloxane moiety of PS-3 is about 75%.
- PS-4 was synthesized in the same manner as in Synthesis Example 1 except that the amount of monomer used was changed as follows. 60 parts of phenyltrimethoxysilane (PTMS), 25 parts of dimethyldimethoxysilane (DMDMS), 15 parts of 3-methacryloyloxypropyltrimethoxysilane (MPTMS), 300 parts of methyl methacrylate (MMA), 220 parts of n-butyl methacrylate (BMA) N-butyl acrylate (BA) 150 parts, acrylic acid (AA) 30 parts.
- PTMS phenyltrimethoxysilane
- DDMS dimethyldimethoxysilane
- MPTMS 3-methacryloyloxypropyltrimethoxysilane
- MMA methyl methacrylate
- BMA n-butyl methacrylate
- BA N-butyl acrylate
- acrylic acid AA
- PS-5 was synthesized in the same manner as in Synthesis Example 1, except that the amount of monomer used was changed as follows. 360 parts of phenyltrimethoxysilane (PTMS), 320 parts of dimethyldimethoxysilane (DMDMS), 40 parts of 3-methacryloyloxypropyltrimethoxysilane (MPTMS), 20 parts of methyl methacrylate (MMA), 20 parts of n-butyl methacrylate (BMA) N-butyl acrylate (BA) 10 parts, acrylic acid (AA) 30 parts.
- PTMS phenyltrimethoxysilane
- DDMS dimethyldimethoxysilane
- MPTMS 3-methacryloyloxypropyltrimethoxysilane
- MMA methyl methacrylate
- BMA n-butyl methacrylate
- BA N-butyl acrylate
- PS-5 is a polymer not classified as a composite polymer of the present invention in which the
- Example 1 to Example 15 Polymer Sheet 1 to Polymer Sheet 15
- the corona treatment conditions were the same as the corona treatment performed on the front side of PET-1 to PET-3.
- Back sheets 1 to 15 were prepared by providing the following undercoat layer and colored layer on the back surface of the support subjected to corona treatment.
- Polyester binder 48.0 parts (Vaironal DM1245 (manufactured by Toyobo Co., Ltd., solid content 30% by mass)) Carbodiimide compound (crosslinking agent) 10.0 parts (Carbodilite V-02-L2, manufactured by Nisshinbo Co., Ltd., solid content: 10% by mass) ⁇ Oxazoline compound (crosslinking agent): 3.0 parts (Epocross WS700, manufactured by Nippon Shokubai Co., Ltd., solid content: 25% by mass) ⁇ Surfactant: 15.0 parts (Naroacty CL95, manufactured by Sanyo Chemical Industries, Ltd., solid content: 1% by mass) ⁇ Distilled water: 907.0 parts
- undercoat layer The obtained coating solution for forming the undercoat layer was applied to the back surfaces (back surfaces of the support) of the polymer sheets 1 to 15 so that the binder amount was 0.1 g / m 2 in terms of the coating amount.
- An undercoat layer having a dry thickness of about 0.1 ⁇ m was formed by drying at 180 ° C. for 1 minute.
- Pigment dispersion 1 (pigment dispersion prepared in Example 1) 80.0 parts Silanol-modified polyvinyl alcohol binder 11.4 parts R1130, manufactured by Kuraray Co., Ltd., solid content: 7 mass %] ⁇ Polyoxyalkylene alkyl ether: 1.0 part [Naroacty CL95, manufactured by Sanyo Chemical Industries, Ltd., solid content: 1% by mass] ⁇ Oxazoline compound: 2.0 parts [Epocross WS-700, manufactured by Nippon Shokubai Co., Ltd., solid content: 25%; cross-linking agent] ⁇ Distilled water: 5.6 parts
- the obtained coating solution for the colored layer was applied to the surface of the polymer sheet 1 to polymer sheet 15 on which the undercoat layer was formed and dried at 180 ° C. for 1 minute, so that the amount of titanium dioxide was 7.0 g / m2, thereby forming a colored layer of binder 1.2 g / m 2.
- back sheets 1 to 15 of Examples 16 to 30 were produced.
- the obtained backsheet 1 to backsheet 15 were evaluated in the same manner as the polymer sheet 1 of Example 1. As a result, all the ratings were 5 and all the backsheets were excellent in adhesion and durability. I found out.
- Examples 31 to 45 A tempered glass having a thickness of 3.2 mm, an EVA sheet (SC50B manufactured by Mitsui Chemicals Fabro), a crystalline solar cell, an EVA sheet (SC50B manufactured by Mitsui Chemicals Fabro), and Examples 16 to 30 Any one of the backsheets 1 to 15 in this order is superposed in this order, and hot pressing is performed using a vacuum laminator (Nisshinbo Co., Ltd., vacuum laminating machine) to bond each member and the EVA sheet. It was. However, the back sheet was disposed so that the colored layer of the back sheet was in contact with the EVA sheet. Moreover, the adhesion conditions of the EVA sheet are as follows.
- Example 46 In the production of the polymer sheet 1 of Example 1, a polymer sheet 16 was produced in the same manner as in Example 1 except that PET-3 was used instead of PET-1 as a support.
- the obtained polymer sheet 16 an aluminum foil (barrier layer) having a thickness of 20 ⁇ m, a PET support (PET-4) having a thickness of 188 ⁇ m, and a white PET support (PET-5) having a thickness of 50 ⁇ m,
- PET-4 PET support
- PET-5 white PET support
- the back sheet 16 was produced by bonding in this order. During bonding, the surfaces of PET-4 and PET-5 were previously subjected to corona treatment similar to PET-1 to PET-3.
- Adhesion conditions As an adhesive, 10 parts of a curing agent KW75 [adhesive manufactured by DIC Corporation] mixed with LX660 (K) [adhesive manufactured by DIC Corporation] was used, and polymer sheet 16, aluminum foil, PET- 4 and PET-5 were hot-press bonded with a vacuum laminator (a vacuum laminator manufactured by Nisshinbo Co., Ltd.). Adhesion was performed by evacuating at 80 ° C. for 3 minutes and then applying pressure for 2 minutes, and then maintained at 40 ° C. for 4 days.
- a vacuum laminator a vacuum laminator manufactured by Nisshinbo Co., Ltd.
- a solar cell module 16 was produced in the same manner except that the obtained back sheet 16 was used instead of the back sheet 1.
- the produced solar cell module 16 was subjected to a power generation operation, it showed good power generation performance as a solar cell.
- Example 47 A back sheet 17 was produced in the same manner as in the production of the back sheet 16 in Example 46, except that a 12 ⁇ m thick PET sheet with a barrier layer was used instead of the aluminum foil. Further, in the production of the solar cell module 16 in Example 46, a solar cell module 17 was produced in the same manner except that a PET sheet with a barrier layer having a thickness of 12 ⁇ m was used instead of the back sheet 16. When the produced solar cell module 17 was subjected to a power generation operation, it showed good power generation performance as a solar cell.
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Engineering & Computer Science (AREA)
- Microelectronics & Electronic Packaging (AREA)
- Power Engineering (AREA)
- General Physics & Mathematics (AREA)
- Physics & Mathematics (AREA)
- Computer Hardware Design (AREA)
- Electromagnetism (AREA)
- Condensed Matter Physics & Semiconductors (AREA)
- Life Sciences & Earth Sciences (AREA)
- Materials Engineering (AREA)
- Wood Science & Technology (AREA)
- Laminated Bodies (AREA)
- Photovoltaic Devices (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
本発明は、カルボキシル基の含有量が15当量/t以下であり、示差走査熱量測定により求められる微少吸熱ピーク温度Tmeta(℃)が220℃以下であり、温度125℃、相対湿度100%RHの条件下72時間放置後の平均伸度保持率が10%以上であるポリエステル基材と、ポリエステル基材上に設けられ、分子中に質量割合が15~85質量%の下記一般式(1)〔R1、R2:H、ハロゲン原子、1価の有機基(R1とR2とは同一でも異なってもよい。複数のR1及びR2は各々、互いに同一でも異なってもよい。)、n:1以上の整数〕で表されるシロキサン構造単位と質量割合が85~15質量%の非シロキサン系構造単位とを含む複合ポリマーを含有するポリマー層とを有する、湿熱環境下における接着耐久性に優れ、安価に製造可能な太陽電池用ポリマーシートを提供する。
Description
本発明は、太陽電池用ポリマーシート及びその製造方法、太陽電池用バックシート、並びに太陽電池モジュールに関する。
太陽電池は、発電時に二酸化炭素の排出がなく環境負荷が小さい発電方式であり、近年急速に普及が進んでいる。太陽電池に用いられるポリマーシートは、屋根の上などに置かれ雨曝しになる太陽電池の使用環境に対応した耐久性や、太陽電池の発電効率を妨げないための透明性等、種々の性質が求められている。また、太陽電池用のポリマーシートとしては、太陽電池素子(セル)を封止する太陽電池用封止材(単に「封止材」ともいう)や、前記封止材を外部から保護する太陽電池用バックシートなどが知られている。
太陽電池モジュールは、通常、太陽光が入射する側のオモテ面ガラスと、太陽光が入射する側とは反対側(裏面側)に配置される、いわゆるバックシートとの間に、太陽電池セルが挟まれた構造を有しており、オモテ面ガラスと太陽電池セルとの間、及び太陽電池セルとバックシートとの間は、それぞれEVA(エチレン-ビニルアセテート)樹脂などで封止されている。
バックシートは、太陽電池モジュールの裏面からの水分の浸入を防止する働きを有するもので、従来はガラスやフッ素樹脂等が用いられていたが、近年では、コストの観点からポリエステルが用いられるようになってきている。そして、バックシートは、単なるポリマーシートではなく、以下に示すような種々の機能が付与される場合がある。
前記機能として、例えば、バックシートに酸化チタン等の白色無機微粒子を添加し、反射性能を持たせたものが要求される場合がある。これは、モジュールのオモテ面から入射した太陽光のうち、セルを素通りした光を乱反射して、セルに戻すことで発電効率を上げるためである。この点について、白色無機微粒子が添加された白色ポリエチレンテレフタレートフィルムの例が開示されており(例えば、特開2003-060218号公報、特開2006-210557号公報参照)、また白色顔料を含有する白色インキ層を有する裏面保護シートの例も開示されている(例えば、特開2006-210557号公報参照)。
また、バックシートに装飾性が要求される場合がある。この点、装飾性を改良するため、黒色顔料であるペリレン系顔料を添加した太陽電池用バックシートの例が開示されている(例えば、特開2007-128943号公報参照)。
更に、バックシートとEVA封止材との間の強固な接着を得るために、バックシートの最表層にポリマー層を設ける場合がある。この点について、白色のポリエチレンテレフタレートフィルムの上に熱接着層を設ける技術が記載されている(例えば、特開2003-060218号公報参照)。
更に、バックシートとEVA封止材との間の強固な接着を得るために、バックシートの最表層にポリマー層を設ける場合がある。この点について、白色のポリエチレンテレフタレートフィルムの上に熱接着層を設ける技術が記載されている(例えば、特開2003-060218号公報参照)。
以上のような機能を付与するためには、バックシートは、支持体上に他の機能を有する層が積層された構造になる。積層の手段としては、色々な機能を持つシートを支持体に貼合する方法がある。例えば、複数の樹脂フィルムの貼合によりバックシートを形成する方法が開示されている(例えば、特開2002-100788号公報参照)。更に、貼合より低コストでバックシートを形成する方法として、支持体に色々な機能を持つ層を塗布する方法が開示されている(例えば、特開2006-210557号公報、特開2007-128943号公報参照)。
また、白色ポリエステルフィルムに帯電防止剤およびシリコーン化合物を含有する塗布層が設けられた反射板用白色ポリエステルフィルムや、エポキシ樹脂、フェノール樹脂、ビニル共重合体、シロキサン化合物を含有する接着層が有機フィルム上に積層された太陽電池用バックシートに関する開示がある(例えば、特開2008-189828号公報、特開2008-282873号公報参照)。
しかしながら、貼合によるバックシートの形成方法については開示された技術はあるが、コストが高く、しかも層間の長期使用での接着性に劣り、耐久性の点で不充分であった。すなわち、バックシートは直接、水分や熱、光に曝されるため、これらに対して長期に亘って耐え得る性能が要求される。例えば、バックシートは一般にEVA系封止材に接着された構造を有しているが、この場合はバックシート/EVA間の経時での接着耐久性は極めて重要である。また、支持体と各層の間の接着耐久性も不可欠である。
塗布による方法に関する開示もあるものの、温湿度が比較的高い環境などでも長期にわたり接着性を保つことは難しく、低廉での製造が可能で、光反射性等の機能とEVA系封止材との間の接着性とを両立させた太陽電池用のポリマーシートが提供されるには至っていない。
上記のようにシリコーン化合物やシロキサン化合物を含有するポリエステルフィルムやバックシートにおいては、前者では帯電防止剤として含有されるカチオンポリマーの耐久性が悪く、また後者ではシロキサン化合物以外の樹脂や共重合体の耐久性が悪いため、温湿度が比較的高い環境などでは長期に亘る接着性を保つことは難しい。
また、ポリマーシートに耐久性を持たせるために、ポリマーシートの基材の耐久性、特に湿熱耐久性を上げると、基材とポリマー層との接着性が低下した。一方、ポリマー層の耐久性を上げるためには、一般にフッ素樹脂(フッ化炭素系樹脂とも言う)をバインダーとして用いるが、フッ素樹脂をバインダーとしたポリマー層は基板への密着が悪いという問題があった。従って、基材の耐久性とポリマー層の接着性は両立できていなかった。
以上のように、長期経時に耐えるEVA系封止材との接着性及び場合により他の機能(例えば反射性能や装飾)を兼ね備えるとともに、安価に製造可能で、しかも湿熱耐久性を満足できる太陽電池用のバックシート等の太陽電池用ポリマーシートは、未だ提供されるに至っていないのが実情である。
本発明は、上記に鑑みなされたものであり、湿熱環境下における、各層間の接着耐久性、ポリマーシートの構成基材や電池側基板の構成基材(例えばEVA等の封止材)との間の接着耐久性に優れ、安価に製造可能な太陽電池用ポリマーシート及びその製造方法、太陽電池用バックシート、並びに安定した発電効率を有する安価な太陽電池モジュールを提供することを目的とし、該目的を達成することを課題とする。
前記課題を達成するための具体的手段は以下の通りである。
<1> カルボキシル基の含有量が15当量/t以下であり、示差走査熱量測定により求められる微少吸熱ピーク温度Tmeta(℃)が220℃以下であり、温度125℃、相対湿度100%RHの条件下72時間放置後の平均伸度保持率が10%以上であるポリエステル基材と、前記ポリエステル基材上に設けられ、分子中に質量割合が15~85質量%の下記一般式(1)で表されるシロキサン構造単位と質量割合が85~15質量%の非シロキサン系構造単位とを含む複合ポリマーを含有するポリマー層と、
を有する太陽電池用ポリマーシートである。
<1> カルボキシル基の含有量が15当量/t以下であり、示差走査熱量測定により求められる微少吸熱ピーク温度Tmeta(℃)が220℃以下であり、温度125℃、相対湿度100%RHの条件下72時間放置後の平均伸度保持率が10%以上であるポリエステル基材と、前記ポリエステル基材上に設けられ、分子中に質量割合が15~85質量%の下記一般式(1)で表されるシロキサン構造単位と質量割合が85~15質量%の非シロキサン系構造単位とを含む複合ポリマーを含有するポリマー層と、
を有する太陽電池用ポリマーシートである。

前記一般式(1)において、R1及びR2は、各々独立に、水素原子、ハロゲン原子、又は1価の有機基を表し、R1とR2とは同一でも異なってもよい。nは、1以上の整数を表す。複数のR1及びR2は各々、互いに同一でも異なってもよい。
<2> 前記ポリマー層は、前記複合ポリマーを架橋する架橋剤由来の構造部分を含む前記<1>に記載の太陽電池用ポリマーシートである。
<3> 前記非ポリシロキサン系構造単位が、アクリル系構造単位である前記<1>又は前記<2>に記載の太陽電池用ポリマーシートである。
<4> 前記架橋剤は、カルボジイミド系化合物、オキサゾリン系化合物、及びエポキシ系架橋剤から選ばれる少なくとも1種である前記<2>又は前記<3>に記載の太陽電池用ポリマーシートである。
<5> 前記ポリマー層中における、前記複合ポリマーに対する前記架橋剤由来の構造部分の質量割合が1~30質量%である前記<2>~前記<4>のいずれか1つに記載の太陽電池用ポリマーシートである。
<6> 前記ポリエステル基材は、コロナ処理、火炎処理、低圧プラズマ処理、大気圧プラズマ処理、及び紫外線処理から選ばれる少なくとも1つの表面処理が施されている前記<1>~前記<5>のいずれか1つに記載の太陽電池用ポリマーシートである。
<3> 前記非ポリシロキサン系構造単位が、アクリル系構造単位である前記<1>又は前記<2>に記載の太陽電池用ポリマーシートである。
<4> 前記架橋剤は、カルボジイミド系化合物、オキサゾリン系化合物、及びエポキシ系架橋剤から選ばれる少なくとも1種である前記<2>又は前記<3>に記載の太陽電池用ポリマーシートである。
<5> 前記ポリマー層中における、前記複合ポリマーに対する前記架橋剤由来の構造部分の質量割合が1~30質量%である前記<2>~前記<4>のいずれか1つに記載の太陽電池用ポリマーシートである。
<6> 前記ポリエステル基材は、コロナ処理、火炎処理、低圧プラズマ処理、大気圧プラズマ処理、及び紫外線処理から選ばれる少なくとも1つの表面処理が施されている前記<1>~前記<5>のいずれか1つに記載の太陽電池用ポリマーシートである。
<7> 前記一般式(1)において、前記R1及びR2で表される1価の有機基が、アルキル基、アリール基、アラルキル基、アルコキシ基、アリールオキシ基、メルカプト基、アミノ基、及びアミド基から選択される少なくとも一種である前記<1>~前記<6>のいずれか1つに記載の太陽電池用ポリマーシートである。
<8> 前記ポリエステル基材は、ポリエステル系樹脂を含み、前記ポリエステル系樹脂のカルボキシル基の含有量が1~15当量/tの範囲である前記<1>~前記<7>のいずれか1つに記載の太陽電池用ポリマーシートである。
<9> 前記ポリマー層の少なくとも1層は、厚みが0.8μm~12μmである前記<1>~前記<8>のいずれか1つに記載の太陽電池用ポリマーシートである。
<10> 前記ポリマー層の少なくとも一層は、前記ポリエステル基材の表面に接触させて設けられている前記<1>~前記<9>のいずれか1つに記載の太陽電池用ポリマーシートである。
<11> 前記ポリマー層の少なくとも一層は、前記ポリエステル基材から最も離れた位置に配置された最外層である前記<1>~前記<10>のいずれか1つに記載の太陽電池用ポリマーシートである。
<12> 前記ポリマー層の少なくとも一層は、更に白色系顔料を含み、光反射性を有する反射層である前記<1>~前記<11>のいずれか1つに記載の太陽電池用ポリマーシートである。
<13> 前記ポリマー層を少なくとも2層含み、前記2層の一方として前記反射層を有し、他方を前記反射層と前記ポリエステル基材との間に有する前記<12>に記載の太陽電池用ポリマーシートである。
<14> 白色系顔料を含み、光反射性を有する反射層を更に有し、該反射層と前記ポリエステル基材との間に、少なくとも一層の前記ポリマー層を有する前記<1>~前記<11>のいずれか1つに記載の太陽電池用ポリマーシートである。前記反射層と前記ポリエステル基材との間が、前記ポリマー層によって接着された態様が好ましい。
<8> 前記ポリエステル基材は、ポリエステル系樹脂を含み、前記ポリエステル系樹脂のカルボキシル基の含有量が1~15当量/tの範囲である前記<1>~前記<7>のいずれか1つに記載の太陽電池用ポリマーシートである。
<9> 前記ポリマー層の少なくとも1層は、厚みが0.8μm~12μmである前記<1>~前記<8>のいずれか1つに記載の太陽電池用ポリマーシートである。
<10> 前記ポリマー層の少なくとも一層は、前記ポリエステル基材の表面に接触させて設けられている前記<1>~前記<9>のいずれか1つに記載の太陽電池用ポリマーシートである。
<11> 前記ポリマー層の少なくとも一層は、前記ポリエステル基材から最も離れた位置に配置された最外層である前記<1>~前記<10>のいずれか1つに記載の太陽電池用ポリマーシートである。
<12> 前記ポリマー層の少なくとも一層は、更に白色系顔料を含み、光反射性を有する反射層である前記<1>~前記<11>のいずれか1つに記載の太陽電池用ポリマーシートである。
<13> 前記ポリマー層を少なくとも2層含み、前記2層の一方として前記反射層を有し、他方を前記反射層と前記ポリエステル基材との間に有する前記<12>に記載の太陽電池用ポリマーシートである。
<14> 白色系顔料を含み、光反射性を有する反射層を更に有し、該反射層と前記ポリエステル基材との間に、少なくとも一層の前記ポリマー層を有する前記<1>~前記<11>のいずれか1つに記載の太陽電池用ポリマーシートである。前記反射層と前記ポリエステル基材との間が、前記ポリマー層によって接着された態様が好ましい。
<15> カルボキシル基の含有量が15当量/t以下であり、示差走査熱量測定により求められる微少吸熱ピーク温度Tmeta(℃)が220℃以下であり、温度125℃、相対湿度100%RHの条件下72時間放置後の平均伸度保持率が10%以上であるポリエステル基材上に、分子中に質量割合が15~85質量%の前記一般式(1)で表されるシロキサン構造単位と質量割合が85~15質量%の非シロキサン系構造単位とを含む複合ポリマーを含有する塗布液を塗布して少なくとも1層のポリマー層を形成する工程を有する太陽電池用ポリマーシートの製造方法である。
<16> 前記塗布液は、更に、カルボジイミド系化合物、オキサゾリン系化合物、及びエポキシ系架橋剤から選ばれる架橋剤を含有する前記<15>に記載の太陽電池用ポリマーシートの製造方法である。
<17> 前記塗布液は、更に溶媒を含有し、該溶媒の50質量%以上が水である水系塗布液である前記<15>又は前記<16>に記載の太陽電池用ポリマーシートの製造方法である。
<16> 前記塗布液は、更に、カルボジイミド系化合物、オキサゾリン系化合物、及びエポキシ系架橋剤から選ばれる架橋剤を含有する前記<15>に記載の太陽電池用ポリマーシートの製造方法である。
<17> 前記塗布液は、更に溶媒を含有し、該溶媒の50質量%以上が水である水系塗布液である前記<15>又は前記<16>に記載の太陽電池用ポリマーシートの製造方法である。
<18> 太陽電池素子が封止材で封止された電池側基板の前記封止剤と接触させて配置される太陽電池用バックシートであって、前記<1>~前記<14>のいずれか1つに記載の太陽電池用ポリマーシート、または、前記<15>~前記<17>のいずれか1つに記載の太陽電池用ポリマーシートの製造方法により製造された太陽電池用ポリマーシートを用いた太陽電池用バックシートである。
<19> 前記ポリエステル基材の前記ポリマー層が設けられている面とは反対側の表面に、前記封止材に対して5N/cm以上の接着力を持つ易接着層を有する前記<18>に記載の太陽電池用バックシートである。
<20> 前記<1>~前記<14>のいずれか1つに記載の太陽電池用ポリマーシート、および、前記<15>~前記<17>のいずれか1つに記載の太陽電池用ポリマーシートの製造方法により製造された太陽電池用ポリマーシートから選択される2つ以上の太陽電池用ポリマーシートが粘着剤により貼り合わせられている前記<18>または前記<19>に記載の太陽電池用バックシートである。
<21> 水および気体の少なくとも一方の浸入を防止するバリア層を有する前記<18>~前記<20>のいずれか1つに記載の太陽電池用バックシートである。
<19> 前記ポリエステル基材の前記ポリマー層が設けられている面とは反対側の表面に、前記封止材に対して5N/cm以上の接着力を持つ易接着層を有する前記<18>に記載の太陽電池用バックシートである。
<20> 前記<1>~前記<14>のいずれか1つに記載の太陽電池用ポリマーシート、および、前記<15>~前記<17>のいずれか1つに記載の太陽電池用ポリマーシートの製造方法により製造された太陽電池用ポリマーシートから選択される2つ以上の太陽電池用ポリマーシートが粘着剤により貼り合わせられている前記<18>または前記<19>に記載の太陽電池用バックシートである。
<21> 水および気体の少なくとも一方の浸入を防止するバリア層を有する前記<18>~前記<20>のいずれか1つに記載の太陽電池用バックシートである。
<22> 前記<18>~前記<21>のいずれか1つに記載の太陽電池用バックシートを備えた太陽電池モジュールである。
<23> 太陽光が入射する透明性のフロント基板と、前記フロント基板の上に設けられ、太陽電池素子及び前記太陽電池素子を封止する封止材を有するセル構造部分と、前記セル構造部分の前記フロント基板が位置する側と反対側に設けられ、前記封止材と隣接して配置された、前記<18>~前記<21>のいずれか1つに記載の太陽電池用バックシートと、を備えた太陽電池モジュールである。
本発明によれば、湿熱環境下における、各層間の接着耐久性、ポリマーシートの構成基材や電池側基板の構成基材(例えばEVA等の封止材)との間の接着耐久性に優れ、安価に製造可能な太陽電池用ポリマーシート及びその製造方法、並びに太陽電池用バックシートを提供することができる。また、
本発明によれば、安定した発電効率を有する安価な太陽電池モジュールを提供することができる。
本発明によれば、安定した発電効率を有する安価な太陽電池モジュールを提供することができる。
以下、本発明の太陽電池用ポリマーシート及びその製造方法、並びに太陽電池モジュールについて詳細に説明する。
<太陽電池用ポリマーシート及びその製造方法>
本発明の太陽電池用ポリマーシートは、カルボキシル基の含有量が15当量/t以下であり、示差走査熱量測定により求められる微少吸熱ピーク温度Tmeta(℃)が220℃以下であり、温度125℃、相対湿度100%RHの条件下72時間放置後の平均伸度保持率が10%以上であるポリエステル基材と、ポリエステル基材上に設けられ、分子中に質量割合が15~85質量%の下記一般式(1)で表されるシロキサン構造単位と質量割合が85~15質量%の非シロキサン系構造単位とを含む複合ポリマーを含有するポリマー層と、を設けて構成されたものである。
なお、前記R1及び前記R2は、各々独立に、水素原子、ハロゲン原子、又は1価の有機基を表し、R1とR2とは同一でも異なってもよい。また、nは、整数を表す。
本発明の太陽電池用ポリマーシートは、カルボキシル基の含有量が15当量/t以下であり、示差走査熱量測定により求められる微少吸熱ピーク温度Tmeta(℃)が220℃以下であり、温度125℃、相対湿度100%RHの条件下72時間放置後の平均伸度保持率が10%以上であるポリエステル基材と、ポリエステル基材上に設けられ、分子中に質量割合が15~85質量%の下記一般式(1)で表されるシロキサン構造単位と質量割合が85~15質量%の非シロキサン系構造単位とを含む複合ポリマーを含有するポリマー層と、を設けて構成されたものである。
なお、前記R1及び前記R2は、各々独立に、水素原子、ハロゲン原子、又は1価の有機基を表し、R1とR2とは同一でも異なってもよい。また、nは、整数を表す。

前記一般式(1)において、R1及びR2は、各々独立に、水素原子、ハロゲン原子、又は1価の有機基を表し、R1とR2とは同一でも異なってもよい。nは、1以上の整数を表す。複数のR1及びR2は各々、互いに同一でも異なってもよい。
本発明においては、ポリマーシートの構成層であるポリマー層を、分子内に非シロキサン系構造単位とこれに共重合する(ポリ)シロキサン構造単位を含む特定の複合ポリマーを用いて構成することで、各層間の接着力、ポリエステル基材や電池側基板の構成基材(例えばEVA等の封止材)との間の接着力が改善され、熱や水分による劣化が抑えられる。したがって、熱や水分に長時間曝される環境条件下において、長期に亘って接着強度を高く保つことができ、長期耐久性を確保することができる。これにより、太陽電池モジュールを構成したときには、良好な発電性能が得られると共に、長期に亘って発電効率を安定に保つことができる。
本発明におけるポリマー層は、ポリマーシートを構成する任意の層に適用することができる。ポリマー層は、例えば、後述する反射層やバック層あるいは反射層等の機能性層とポリエステル基材との間を接着する接着層として適用することができる。熱や水分等の湿熱環境下での耐久性に優れる点から、ポリマーシートの構成層のうち、白色顔料等を含む反射層とポリエステル基材との間に配されるポリマー層、あるいは太陽電池モジュールとした場合に、外部環境に暴露される最外層、つまりバック層として用いることも特に好ましい。
(ポリエステル基材)
本発明におけるポリエステル基材は、カルボキシル基の含有量が15当量/t以下であり、示差走査熱量測定により求められる微少吸熱ピーク温度Tmeta(℃)が220℃以下であり、温度125℃、相対湿度100%RHの条件下72時間放置後の平均伸度保持率が10%以上である。
基材がかかる構成であることで、基材は高い耐久性を有することができる。
従来は、このような高耐久性のポリエステル基材を用いると、湿熱環境下における基材とポリマー層との接着性が低下した。かかる機構については明らかではないが、基材としてポリエステル基材を用いた場合、高耐久性のポリエステル基材は、ポリエステルの分子配向が進むため、基材表面が結晶状態に近い構造になり、基材の分子とポリマー層の分子との混合が起こりにくくなることによるものと考えられる。
しかし、本発明の太陽電池用ポリマーシートは、上記のような高耐久性のポリエステル基材上に、分子中に質量割合が15~85質量%の下記一般式(1)で表されるシロキサン構造単位と質量割合が85~15質量%の非シロキサン系構造単位とを含む複合ポリマーを含有するポリマー層が設けられている。ポリマーシートがかかる構成であることで、理由は明らかではないが、基材が高耐久性を有するにも関わらず、湿熱環境下における接着耐久性に優れる。
以下、本発明におけるポリエステル基材の詳細を説明する。
本発明におけるポリエステル基材は、カルボキシル基の含有量が15当量/t以下であり、示差走査熱量測定により求められる微少吸熱ピーク温度Tmeta(℃)が220℃以下であり、温度125℃、相対湿度100%RHの条件下72時間放置後の平均伸度保持率が10%以上である。
基材がかかる構成であることで、基材は高い耐久性を有することができる。
従来は、このような高耐久性のポリエステル基材を用いると、湿熱環境下における基材とポリマー層との接着性が低下した。かかる機構については明らかではないが、基材としてポリエステル基材を用いた場合、高耐久性のポリエステル基材は、ポリエステルの分子配向が進むため、基材表面が結晶状態に近い構造になり、基材の分子とポリマー層の分子との混合が起こりにくくなることによるものと考えられる。
しかし、本発明の太陽電池用ポリマーシートは、上記のような高耐久性のポリエステル基材上に、分子中に質量割合が15~85質量%の下記一般式(1)で表されるシロキサン構造単位と質量割合が85~15質量%の非シロキサン系構造単位とを含む複合ポリマーを含有するポリマー層が設けられている。ポリマーシートがかかる構成であることで、理由は明らかではないが、基材が高耐久性を有するにも関わらず、湿熱環境下における接着耐久性に優れる。
以下、本発明におけるポリエステル基材の詳細を説明する。
-カルボキシル基の含有量(AV)-
ポリエステル基材に用いられるポリエステル中のカルボキシル基含量(Acid Value;AV)は15当量/t(トン;以下同様)以下であり、好ましくは12当量/t以下、より好ましくは8当量/t以下である。
カルボキシル基含量が15当量/t以下であると、耐加水分解性を保持し、湿熱経時したときの強度低下を小さく抑制することができる。
カルボキシル基は、ポリエステル基材に隣接する部材又は層の表面に存在する水酸基と水素結合を形成し密着力を向上させる働きがある。このため、カルボキシル基含量の下限は、1当量/tが望ましい。なお、本明細書中において、「当量/トン(eq/t)」は1トンあたりのモル当量を表す。
ポリエステル基材に用いられるポリエステル中のカルボキシル基含量(Acid Value;AV)は15当量/t(トン;以下同様)以下であり、好ましくは12当量/t以下、より好ましくは8当量/t以下である。
カルボキシル基含量が15当量/t以下であると、耐加水分解性を保持し、湿熱経時したときの強度低下を小さく抑制することができる。
カルボキシル基は、ポリエステル基材に隣接する部材又は層の表面に存在する水酸基と水素結合を形成し密着力を向上させる働きがある。このため、カルボキシル基含量の下限は、1当量/tが望ましい。なお、本明細書中において、「当量/トン(eq/t)」は1トンあたりのモル当量を表す。
カルボキシル基におけるH+は、酸触媒として働きポリエステル分子を加水分解する作用を有する。従って、15当量/tを超えるAVでは、高湿度下で経時させた場合において、加水分解によりポリエステル基材表面の分子量が低下し力学強度が低下し、その結果、ポリエステル基材表面が破壊されることにより、ポリマー層のポリエステル基材からの剥離(密着不良)が発生してしまう。
AVの具体的な調整方法としては、ポリエステル基材の「面配向係数」の調整、ポリエステルを構成する「構成成分」の種類及び含有量の調整、「緩衝剤」や「末端封止剤」等の添加剤の添加、ポリエステル中に存在する「リン原子量」の調整などのほか、重合触媒種、製膜条件(製膜温度や時間)により調整することが可能である。
ここで、上記具体的な調整方法のうち、「緩衝剤」及び「末端封止剤」等の添加剤の添加量、及び/又は、「リン原子量」により、AVを本発明の範囲内とするには、ポリエステルにおけるこれらの含有量をより多くすることが必要となる。しかしながら、ポリエステル中における過剰量の添加剤やリン原子の含有は、当該基材を湿熱経時させた際において基材表面に添加剤等が析出したり、配向が強すぎることによる熱収縮の増大などの問題を招来し、延いては密着不良を発生させる。
ここで、上記具体的な調整方法のうち、「緩衝剤」及び「末端封止剤」等の添加剤の添加量、及び/又は、「リン原子量」により、AVを本発明の範囲内とするには、ポリエステルにおけるこれらの含有量をより多くすることが必要となる。しかしながら、ポリエステル中における過剰量の添加剤やリン原子の含有は、当該基材を湿熱経時させた際において基材表面に添加剤等が析出したり、配向が強すぎることによる熱収縮の増大などの問題を招来し、延いては密着不良を発生させる。
-示差走査熱量測定により求められる微小吸熱ピーク温度Tmeta(℃)-
本発明におけるポリエステル基材は、示差走査熱量測定(以下、「DSC」とも称する。)により求められる微小吸熱ピーク温度Tmeta(℃)が、220℃以下であり、より好ましくは150℃以上215℃以下、さらに好ましくは160℃以上210℃以下である。
本発明におけるポリエステル基材は、示差走査熱量測定(以下、「DSC」とも称する。)により求められる微小吸熱ピーク温度Tmeta(℃)が、220℃以下であり、より好ましくは150℃以上215℃以下、さらに好ましくは160℃以上210℃以下である。
微小吸熱ピーク温度Tmeta(℃)は、ポリエステル基材における「面配向係数」と、ポリエステル基材を製膜する際における「延伸後に実施する熱固定の温度」を制御することにより、本発明に係る温度範囲とすることができる。延伸後に実施する熱固定の温度としては、150℃以上220℃以下が好ましく、より好ましくは160℃以上210℃以下、さらに好ましくは170℃以上200℃以下である。
Tmeta(℃)の具体的な測定方法については後述する。
-平均伸度保持率-
本発明のバックシートは、湿熱経時後においても高い密着力を有することが特徴である。そのためには、ポリエステル基材表面における加水分解を抑制することで、密着力の低下が抑制されることが好ましい。かかる観点から、ポリエステル基材表面における加水分解の目安として、「温度125℃、相対湿度100%RHの条件下で72時間放置した後の平均伸度保持率」が採用され、本発明においては、該平均伸度保持率が10%以上であることを要する。
本発明のバックシートは、湿熱経時後においても高い密着力を有することが特徴である。そのためには、ポリエステル基材表面における加水分解を抑制することで、密着力の低下が抑制されることが好ましい。かかる観点から、ポリエステル基材表面における加水分解の目安として、「温度125℃、相対湿度100%RHの条件下で72時間放置した後の平均伸度保持率」が採用され、本発明においては、該平均伸度保持率が10%以上であることを要する。
ここで「伸度保持率(Lr)」とは、湿熱経時前の破断伸度(Li)と、湿熱経時後の破断伸度(Lt)の比率(%)を指し、下記式で求められた値である。
Lr(%)=100×(Lt)/(Li)
Lr(%)=100×(Lt)/(Li)
本発明における「平均伸度保持率」は、伸度保持率の測定を、ポリエステル基材の長手方向(MD)及びその直行方向(TD)で行い、その平均値で表したものである。
伸度保持率の調整方法としては、例えば、ポリエステル基材の「面配向係数」の調整、ポリエステルの「固有粘度」の調整、ポリエステルポリマーを構成する「構成成分」の種類及び含有量の調整、「緩衝剤」や「末端封止剤」等の添加剤の添加、ポリエステル中に存在する「リン原子量」の調整などが挙げられる。
加水分解し易いほど分子量が低下することから、ポリエステル基材が示す平均伸度保持率の値が低下し易い。かかる観点から、本発明におけるポリエステル基材は、平均伸度保持率が10%以上であることが必要であり、より好ましくは20%以上95%以下であり、さらに好ましくは30%以上90%以下である。
平均伸度保持率を10%以上にすることで、ポリエステルの加水分解に起因するバックシートの剥がれ(密着不良)を効果的に抑制できる。
平均伸度保持率を10%以上にすることで、ポリエステルの加水分解に起因するバックシートの剥がれ(密着不良)を効果的に抑制できる。
平均伸度保持率の具体的な測定方法については後述する。
-熱収縮率及び分布-
本発明におけるポリエステル基材の好適な態様の一つは、該ポリエステル基材の長手方向(MD)とその直行方向(TD)とにおける150℃30分の熱収縮率が、それぞれ1.0%以下であり、且つ、熱収縮バラツキ割合が、それぞれ1%以上20%以下である態様である。
本発明におけるポリエステル基材の好適な態様の一つは、該ポリエステル基材の長手方向(MD)とその直行方向(TD)とにおける150℃30分の熱収縮率が、それぞれ1.0%以下であり、且つ、熱収縮バラツキ割合が、それぞれ1%以上20%以下である態様である。
本発明者らは、ポリエステル基材とポリマー層との湿熱経時による密着不良は、ポリエステル基材中の残留歪による熱収縮の発生による場合があるとの知見を得た。即ち、湿熱経時されたポリエステル基材中に残留歪による熱収縮が発生した場合、該熱収縮によりポリマー層とポリエステル基材との間に収縮応力が発生し、これがポリマー層の密着不良を引起すというものである。
その作用は明確ではないが、次のように考えている。即ち、ポリエステル基材における熱収縮が、基材面内に均一であれば、応力も均一に発生することから、ポリエステル層は剥離し易い。これに対し、本発明における好適な態様のポリエステル基材のように、熱収縮に分布が存在すると、基材面内に熱収縮の大きなところが存在しても、同一面内に熱収縮の小さな箇所が存在することにより、そこで熱収縮が停止し(即ち、収縮が伝播しない。)、基材全体に波及する大きな収縮力にはならず、延いてはポリマー層の剥離が抑制される。
本発明における好適な態様のポリエステル基材の好ましい熱収縮バラツキ割合は1%以上20%以下であり、より好ましくは2%以上15%以下、さらに好ましくは3%以上12%以下である。
ここで、ポリエステル基材の熱収縮バラツキ割合は、その長手方向(MD)とその直行方向(TD)とに、10cm間隔で5点測定し、下記式から熱収縮バラツキ割合(Bts)(%)を求め、大きいほうの値を指す。
(Bts)(%)=100×(Bmax)-(Bmin))/(Bav)
ここで、Btsは熱収縮バラツキ割合を、Bmaxは熱収縮の最大値を、Bminは熱収縮の最小値を、Bavは熱収縮の平均値を表す。
(Bts)(%)=100×(Bmax)-(Bmin))/(Bav)
ここで、Btsは熱収縮バラツキ割合を、Bmaxは熱収縮の最大値を、Bminは熱収縮の最小値を、Bavは熱収縮の平均値を表す。
熱収縮バラツキ割合が20%を超えると、熱収縮の大きい箇所と小さい箇所の寸法変化が大きくなりすぎ、クレーター状の収縮部分が発生する傾向となり、このクレーターの縁にそって応力集中が発生し、剥離(密着不良)が発生し易い。一方、熱収縮バラツキ割合が1%を下回ると、上記のような収縮抑止の効果が発現し難くなり好ましくない。
このようなポリエステル基材における収縮応力の発生は、小さな面積であれば発現し難い。このため、熱収縮バラツキ割合を上記範囲とすることは、ポリマー層を0.5m2以上(より好ましくは0.75m2以上、さらに好ましくは1m2以上)の如き大面積のパネルに貼り付けた際に、その効果が特に顕在化する。これは即ち、小面積であれば、収縮量の大きな部分と小さな部分とが共存する確率が低いためである。
さらに、このような熱収縮率及び熱収縮バラツキ割合の制御は、湿熱経時後の密着性の向上効果の顕在化に特に有用である。即ち、高湿下での湿熱経時中に熱収縮が発生し、しかも高湿度である場合では、ポリエステル基材及び該ポリエステル基材と水素結合を形成しうる隣接部材又は隣接層の界面に水が浸透し、水素結合を切断するため、密着が低下し易くなるが、このような状況に於いても、熱収縮率及び熱収縮バラツキ割合の制御を上記範囲とすることで、残留歪による収縮応力を低減できるため、密着力を確保し易い。
本発明におけるポリエステル基材の熱収縮率は、150℃30分で測定される。
その好ましい範囲は、長手方向(MD)及びその直行方向(TD)ともに、1%以下であることが好ましく、より好ましくは-0.5%以上0.8%以下、さらに好ましくは-0.3%以上0.6%以下である。(なお、ここで云う「-」とは「伸張」を意味する)。
その好ましい範囲は、長手方向(MD)及びその直行方向(TD)ともに、1%以下であることが好ましく、より好ましくは-0.5%以上0.8%以下、さらに好ましくは-0.3%以上0.6%以下である。(なお、ここで云う「-」とは「伸張」を意味する)。
熱収縮率が1%以下であれば、熱収縮バラツキ割合を前記特定範囲とした効果を有効に発現しうる。熱収縮率が1%を超えると、ポリエステル基材の寸法変化を抑制し切れなくなり、熱収縮バラツキ割合を特定範囲とする効果が得られなくなる傾向となる。一方、ポリエステル基材の伸張が大きくなりすぎる場合には、熱収縮バラツキ割合の制御によるポリエステル基材の寸法変化の抑制効果が得られない傾向となる。
熱収縮率は、ポリエステル基材を製膜する際の延伸後に熱処理を行うことで調整することができる。熱処理の好ましい温度は150℃以上220℃以下、より好ましくは160℃以上210℃以下、さらに好ましくは170℃以上200℃以下であり、10秒以上120秒以下、より好ましくは15秒以上90秒以下、さらに好ましくは20秒以上60秒以下である。
さらに、延伸後に熱処理に合せて、縦方向及び横方向の少なくとも一方に緩和することが好ましく、好ましい緩和量は0.5%以上10%以下であり、より好ましくは1.5%以上9%以下、さらに好ましくは3%以上8%以下である。
また、熱収縮バラツキ割合は、ポリエステル基材を製膜する際の溶融押出し後に、冷却ロール上で固化し未延伸フィルム(原反)を作製する際において、温度分布を形成することで調整することができる。即ち、溶融体が冷却する際に球晶を形成するが、冷却速度を変えることでこの球晶分布を形成する。これが縦、横延伸中に配向分布を引起し、これが収縮量の分布となって現れる。このような溶融体の冷却速度の分布は、冷却ロールに温度分布を与えることで達成できる。このような温度分布は、冷却ロール中に温調のために流している熱媒の流れを、邪魔板を儲け乱すことで達成される。好ましい温度分布は0.2℃以上10℃以下であり、より好ましくは0.4℃以上5℃以下、さらに好ましくは0.6℃以上3℃以下である。これらの温度分布は、長手方向、幅方向、いずれの方向でも構わない。
このような熱収縮率及び熱収縮バラツキ割合の制御とともに、後述するように、「末端封止剤」をポリエステル中に含有させること、ポリエステルの構成成分としては「3官能以上の構成成分」含有させることで、より効果的に湿熱経時後の密着性を向上させることができる。
末端封止剤は、ポリエステルと反応させることで末端を嵩高くでき、これが引っ掛りとなりポリエステル分子間の易動性を低下させる。また、3官能以上の構成成分は、3官能基を介して分子が枝分かれするため、ポリエステル分子の易動性を低下させる。このように運動性が低下することで熱収縮バラツキ割合を形成し易くできる。即ち、熱収縮の大きな箇所と小さな箇所には応力が発生するが、この応力によってポリエステル分子が移動し応力(熱収縮の分布による歪)を解消しようとする。このとき、上記のように運動性が低下すると。このような熱収縮バラツキ割合の解消が起きづらく、本発明における熱収縮バラツキ割合を形成し易い。
熱収縮率の具体的な測定方法については後述する。
-面配向係数及びその分布-
本発明におけるポリエステル基材は、面配向係数が0.165以上であることが好ましく、より好ましくは0.168以上0.18以下、さらに好ましくは0.170以上、0.175以下である。面配向係数を0.165以上とすることにより、分子を配向させ、上記の「半結晶」の形成を促し、耐加水分解性をさらに向上させることができる。
本発明におけるポリエステル基材は、面配向係数が0.165以上であることが好ましく、より好ましくは0.168以上0.18以下、さらに好ましくは0.170以上、0.175以下である。面配向係数を0.165以上とすることにより、分子を配向させ、上記の「半結晶」の形成を促し、耐加水分解性をさらに向上させることができる。
ここで、本発明で言う面配向係数fPOとは、アッベ屈折計を用い、下記(A)式より求められるものである。
fPO= (nMD+nTD)/2-nZD・・・(A)
fPO= (nMD+nTD)/2-nZD・・・(A)
式(A)中、nMDは、フィルムの長手方向(MD)の屈折率を表し、nTDはフィルムの直行方向(TD)の屈折率を表し、nZDはフィルム厚み方向の屈折率を表す。
なお、フィルムの上記各方向の屈折率は、JIS K7142のA法等に基づいて測定することができる。
なお、フィルムの上記各方向の屈折率は、JIS K7142のA法等に基づいて測定することができる。
ポリエステル基材の面配向係数は、製膜時の延伸倍率を大きくすることで調整することができる。好ましくはフィルムの長手方向(MD)、フィルムの直行方向(TD)ともに延伸倍率を2.5~6.0倍に調整すればよい。フィルムの面配向係数を0.165以上とするためには、MD方向及びTD方向の延伸倍率を、それぞれ3.0~5.0倍に調整することが好ましい。さらに、面配向係数は、縦延伸中での「予熱」「多段延伸」(後述)によっても向上させることができる。
さらに、面配向係数を0.165以上とすることで、耐加水分解性を抑制しポリエステルフィルム表面の分子量低下による密着不良を抑制できる。
また、フィルムの面配向係数の上限は、面配向係数を上げるために延伸倍率を大きくしていくと製膜安定性が悪化するため、また、面配向が進みすぎることで発生するデラミ(層状剥離)を抑制し密着力を高めることができるため、0.180以下であることが好ましく、より好ましくは0.175以下である。
また、フィルムの面配向係数の上限は、面配向係数を上げるために延伸倍率を大きくしていくと製膜安定性が悪化するため、また、面配向が進みすぎることで発生するデラミ(層状剥離)を抑制し密着力を高めることができるため、0.180以下であることが好ましく、より好ましくは0.175以下である。
さらに、本発明においては、面配向係数に分布を設けることが好ましい。該面配向係数の分布としては、1%以上20%以下であることが好ましく、より好ましくは2%以上15%以下であり、さらに好ましくは3%以上12%以下である。
面配向係数に分布を設けることにより、密着力をより一層向上させることができる。即ち、湿熱経時後にポリエステル基材が収縮するため、該フィルムとEVA等の封止剤との間に収縮応力が発生、これにより密着不良が発生する。この熱収縮応力は、フィルムの弾性率に比例し、これは面配向係数に比例する。従って、ポリエステル基材面配向係数に分布が存在すると、弾性率にも分布が発生し、これにより弾性率の高い(硬い)箇所と、弾性率の低い(柔らかい)箇所が形成される。弾性率の低い箇所は、発生した熱収縮応力を吸収する働きがあり、これが緩衝部となり密着低下を抑制する効果を発揮する。
面配向係数の分布が1%未満となる場合には、熱収縮応力を緩和できずに密着力が低下する傾向がある。一方、面配向係数の分布が20%を超える場合には、面配向の小さいところに収縮応力が集中しすぎ、密着不良が発生し易い傾向がある。
面配向係数の分布が1%未満となる場合には、熱収縮応力を緩和できずに密着力が低下する傾向がある。一方、面配向係数の分布が20%を超える場合には、面配向の小さいところに収縮応力が集中しすぎ、密着不良が発生し易い傾向がある。
ポリエステル基材における面配向係数の分布は、ポリエステル基材を製膜する際の縦延伸において、予熱温度分布を調整することにより形成できる。即ち、予熱温度分布により、縦延伸での配向分布、及びこれに伴う結晶分布を形成し、これにより横延伸の配向分布を形成する。ここで云う温度分布とは、幅方向の温度分布を指す。即ち幅方向に形成された温度分布により縦延伸後に幅方向に結晶、配向分布が発生する。これがこの後に横方向に延伸される際、フィルム全面に渡り配向むらを形成することで、面配向係数の分布が形成される。
予熱温度の分布は、予熱ロールに温度分布を与えることで調整できる。具体的には、予熱温度分布は、予熱ロール中に温調のために流している熱媒の流れを、邪魔板を儲け乱すことで調整すればよい。予熱温度の好ましい温度分布は0.2℃以上10℃以下であり、より好ましくは0.4℃以上5℃以下、さらに好ましくは0.6℃以上3℃以下である。
このような面配向係数の分布の制御とともに、後述するように、「末端封止剤」をポリエステル中に含有させること、ポリエステルの構成成分としては「3官能以上の構成成分」含有させることで、より効果的に湿熱経時後の密着性を向上させることができる。
末端封止剤は、ポリエステルと反応させることで末端を嵩高くでき、これが引っ掛りとなりポリエステル分子間の易動性を低下させる。また、3官能以上の構成成分(C)は3官能基を介して分子が枝分かれするため、ポリエステル分子の易動性を低下させる。このように運動性が低下することで面配向分布を形成し易くできる。即ち、面配向の大きな箇所と小さな箇所の間で発生する応力差により分子が流動(クリープ)し、これを解消しようとする。このとき、上記のように分子の運動性が低下すると、このような面配向分布の解消が起きづらく、面配向係数の分布を形成し易い。
面配向係数の具体的な測定方法については後述する。
-固有粘度(Interisic Viscosity;IV)-
本発明におけるポリエステル基材中のポリエステルは、固有粘度(以下、適宜「IV」と称する。)が0.6~1.2dl/gの範囲にあることが好ましい。より好ましい固有粘度は0.65~1.0dl/gであり、さらに好ましくは0.70~0.95dl/gである。
ポリエステル基材中のポリエステルの固有粘度が、0.6dl/g未満であると、分子の易動性が大きく、上述した熱収縮や面配向の分布が緩和(解消)され易くなる傾向がある。一方、固有粘度が1.2dl/gを超えると、溶融押出しの際に剪断発熱し易く、これがポリエステル樹脂の熱分解を促し、この結果、ポリエステル中のカルボン酸量(AV)が増加し易い。これが湿熱経時中のポリエステルの加水分解を促し密着不良を発現し易くなる傾向がある。
本発明におけるポリエステル基材中のポリエステルは、固有粘度(以下、適宜「IV」と称する。)が0.6~1.2dl/gの範囲にあることが好ましい。より好ましい固有粘度は0.65~1.0dl/gであり、さらに好ましくは0.70~0.95dl/gである。
ポリエステル基材中のポリエステルの固有粘度が、0.6dl/g未満であると、分子の易動性が大きく、上述した熱収縮や面配向の分布が緩和(解消)され易くなる傾向がある。一方、固有粘度が1.2dl/gを超えると、溶融押出しの際に剪断発熱し易く、これがポリエステル樹脂の熱分解を促し、この結果、ポリエステル中のカルボン酸量(AV)が増加し易い。これが湿熱経時中のポリエステルの加水分解を促し密着不良を発現し易くなる傾向がある。
ポリエステル基材中のポリエステルのIVは、固相重合における温度及び反応時間により調整することができる。固相重合の好適な態様としては、ポリエステルペレットを、180℃以上250℃以下、より好ましくは190℃以上240℃以下、さらに好ましくは195℃以上230℃以下の温度条件において、5時間以上50時間以下、より好ましくは10時間以上40時間以下、さらに好ましくは15時間以上30時間以下、窒素気流中あるいは真空中で熱処理することである。固相重合は、一定温度で実施してもよく、変動させながら実施してもよい。
また、ポリエステル基材の製膜に供するポリエステル原料(ペレット)については、耐加水分解性を満たすために、固有粘度が0.6~1.2dl/gの範囲にあることが好ましい。より好ましくは0.65~1.0dl/gであり、さらに好ましくは0.70~0.95dl/gである。耐加水分解性を向上させるためには、固有粘度を高めることが好ましいが、固有粘度が1.2dl/gを越える場合はポリエステル樹脂製造時に固相重合時間を長くする必要がありコストが著しく高くなるため好ましくないことがある。また0.6dl/gより小さい場合は、重合度が低いため耐熱性・耐加水分解性が著しく落ちるため好ましくない。ペレットの固有粘度は、ポリエステル樹脂の製造時の重合条件、固相重合条件を調整することで、上記好ましい範囲にすることができる。
ポリエステル基材としては、上記物性を有するものであれば、特に制限されず、例えば、芳香族二塩基酸又はそのエステル形成性誘導体とジオール又はそのエステル形成性誘導体とから合成される線状飽和ポリエステルである。かかるポリエステルの具体例としては、ポリエチレンテレフタレート、ポリエチレンイソフタレート、ポリブチレンテレフタレート、ポリ(1,4-シクロヘキシレンジメチレンテレフタレート)、ポリエチレン-2,6-ナフタレートなどを挙げることができる。このうち、力学的物性やコストのバランスの点で、ポリエチレンテレフタレート又はポリエチレン-2,6-ナフタレートが特に好ましい。
前記ポリエステルは、単独重合体であってもよいし、共重合体であってもよい。更に、前記ポリエステルに他の種類の樹脂、例えばポリイミド等を少量ブレンドしたものであってもよい。
本発明におけるポリエステルを重合する際には、重合後のポリエステル中のカルボキシル基含量(カルボキシル基の含有量)を所定の範囲以下に抑える観点から、Sb系、Ge系、Ti系の化合物を触媒として用いることが好ましく、中でも特にTi系化合物が好ましい。Ti系化合物を用いる場合、Ti系化合物を重合後のポリエステル中のTi元素換算値が1ppm以上30ppm以下、より好ましくは3ppm以上15ppm以下の範囲となるように触媒として用いることにより重合する態様が好ましい。Ti系化合物の使用量がTi元素換算で前記範囲内であると、重合後のポリエステル中の末端カルボキシル基を下記範囲に調整することが可能であり、ポリエステル基材の耐加水分解性を低く保つことができる。
Ti系化合物を用いたポリエステルの合成には、例えば、特公平8-301198号公報、特許第2543624号、特許第3335683号、特許第3717380号、特許第3897756号、特許第3962226号、特許第3979866号、特許第3996871号、特許第4000867号、特許第4053837号、特許第4127119号、特許第4134710号、特許第4159154号、特許第4269704号、特許第4313538号等に記載の方法を適用できる。
本発明におけるポリエステルは、重合後に固相重合されていることが好ましい。これにより、好ましいカルボキシル基含量を達成することができる。固相重合は、連続法(タワーの中に樹脂を充満させ、これを加熱しながらゆっくり所定の時間滞流させた後、送り出す方法)でもよいし、バッチ法(容器の中に樹脂を投入し、所定の時間加熱する方法)でもよい。具体的には、固相重合には、特許第2621563号、特許第3121876号、特許第3136774号、特許第3603585号、特許第3616522号、特許第3617340号、特許第3680523号、特許第3717392号、特許第4167159号等に記載の方法を適用することができる。
固相重合の温度は、170℃以上240℃以下が好ましく、より好ましくは180℃以上230℃以下であり、さらに好ましくは190℃以上220℃以下である。また、固相重合時間は、5時間以上100時間以下が好ましく、より好ましくは10時間以上75時間以下であり、さらに好ましくは15時間以上50時間以下である。固相重合は、真空中あるいは窒素雰囲気下で行なうことが好ましい。
本発明におけるポリエステルの好適な態様の一つは、ジカルボン酸構成成分、ジオール構成成分、及び、カルボキシル基数(a)と水酸基数(b)との合計(a+b)が3以上である構成成分(p)を有するポリエステルを含み、前記構成成分(p)の含有量が、ポリエステルに含まれる全構成成分に対して0.005モル%以上2.5モル%以下であるポリエステルである。
~構成成分(p)~
カルボキシル基数(a)と水酸基数(b)との合計(a+b)が3以上である構成成分(p)について説明する。
カルボキシル基数(a)と水酸基数(b)との合計(a+b)が3以上である構成成分(p)について説明する。
構成成分(p)の例としては、カルボキシル基数(a)が3以上のカルボン酸構成成分、水酸基数(b)が3以上の構成成分、一分子中に水酸基とカルボキシル基数の両方を有するオキシ酸類であり、かつカルボキシル基数(a)と水酸基数(b)との合計(a+b)が3以上である構成成分が挙げられる。
カルボキシル基数(a)が3以上のカルボン酸構成成分の例としては、三官能の芳香族カルボン酸構成成分として、トリメシン酸、トリメリット酸、ナフタレントリカルボン酸、アントラセントリカルボン酸等が、三官能の脂肪族カルボン酸構成成分として、メタントリカルボン酸、エタントリカルボン酸、プロパントリカルボン酸、ブタントリカルボン酸等が、四官能の芳香族カルボン酸構成成分としてベンゼンテトラカルボン酸、ピロメリット酸、ベンゾフェノンテトラカルボン酸、ナフタレンテトラカルボン酸、アントラセンテトラカルボン酸、ベリレンテトラカルボン酸等が、四官能の脂肪族カルボン酸構成成分として、エタンテトラカルボン酸、エチレンテトラカルボン酸、ブタンテトラカルボン酸、シクロペンタンテトラカルボン酸、シクロヘキサンテトラカルボン酸、アダマンタンテトラカルボン酸等が、五官能以上の芳香族カルボン酸構成成分として、ベンゼンペンタカルボン酸、ベンゼンヘキサカルボン酸、ナフタレンペンタカルボン酸、ナフタレンヘキサカルボン酸、ナフタレンヘプタカルボン酸、ナフタレンオクタカルボン酸、アントラセンペンタカルボン酸、アントラセンヘキサカルボン酸、アントラセンヘプタカルボン酸、アントラセンオクタカルボン酸等が、五官能以上の脂肪族カルボン酸構成成分として、エタンペンタカルボン酸、エタンヘキサカルボン酸、ブタンペンタカルボン酸、ブタンヘプタカルボン酸、シクロペンタンペンタカルボン酸、シクロヘキサンペンタカルボン酸、シクロヘキサンヘキサカルボン酸、アダマンタンペンタカルボン酸、アダマンタンヘキサカルボン酸等が、及びこれらエステル誘導体や酸無水物等が例として挙げられるがこれらに限定されない。
また、上述のカルボン酸構成成分のカルボキシ末端に、l-ラクチド、d-ラクチド、ヒドロキシ安息香酸などのオキシ酸類、及びその誘導体、そのオキシ酸類が複数個連なったもの等を付加させたものも好適に用いられる。
また、これらは単独で用いても、必要に応じて、複数種類用いても構わない。
また、上述のカルボン酸構成成分のカルボキシ末端に、l-ラクチド、d-ラクチド、ヒドロキシ安息香酸などのオキシ酸類、及びその誘導体、そのオキシ酸類が複数個連なったもの等を付加させたものも好適に用いられる。
また、これらは単独で用いても、必要に応じて、複数種類用いても構わない。
水酸基数(b)が3以上の構成成分の例としては、三官能の芳香族構成成分としては、トリヒドロキシベンゼン、トリヒドロキシナフタレン、トリヒドロキシアントラセン、トリヒドロキシカルコン、トリヒドロキシフラボン、トリヒドロキシクマリン、三官能の脂肪族アルコール構成成分として、グリセリン、トリメチロールプロパン、プロパントリオール、四官能の脂肪族アルコール構成成分として、ペンタエリスリトール等の化合物、また、上述の化合物の水酸基末端にジオール類を付加させた構成成分(p)も好ましく用いられる。また、これらは単独で用いても、必要に応じて、複数種類用いても構わない。
一分子中に水酸基とカルボキシル基数の両方を有するオキシ酸類のうち、かつカルボキシル基数(a)と水酸基数(b)との合計(a+b)が3以上である構成成分としては、ヒドロキシイソフタル酸、ヒドロキシテレフタル酸、ジヒドロキシテレフタル酸、ジヒドロキシテレフタル酸などが挙げられる。
また、上述の構成成分のカルボキシ末端に、l-ラクチド、d-ラクチド、ヒドロキシ安息香酸などのオキシ酸類、及びその誘導体、そのオキシ酸類が複数個連なったもの等を付加させたものも好適に用いられる。
また、これらは単独で用いても、必要に応じて、複数種類用いても構わない。
また、上述の構成成分のカルボキシ末端に、l-ラクチド、d-ラクチド、ヒドロキシ安息香酸などのオキシ酸類、及びその誘導体、そのオキシ酸類が複数個連なったもの等を付加させたものも好適に用いられる。
また、これらは単独で用いても、必要に応じて、複数種類用いても構わない。
ポリエステルが構成成分(p)を含む場合、該構成成分(p)の含有量は、ポリエステル中の全構成成分に対して0.005モル%以上2.5モル%であることが好ましい。構成成分(p)の含有量は、より好ましくは0.020以上1以下、更好ましくは0.025以上1以下、更に好ましくは0.035以上0.5以下、更に好ましくは0.05以上0.5以下、特に好ましくは0.1以上0.25以下である。
ポリエステル中における構成成分(p)の含有量が、ポリエステル中の全構成成分に対して0.005モル%以下であると、耐湿熱性の向上効果が確認されない場合があり、また2.5モル%を越えると、樹脂がゲル化して溶融押出が困難となる等の理由で現実化困難であり、できたとしてもゲルが異物として存在し、フィルムにした場合の二軸延伸性が低下したり、延伸して得たフィルムが異物欠点を多数有する場合がある。
ポリエステル中の構成成分(p)の含有量が、該ポリエステル中の全構成成分に対して0.005モル%以上2.5モル%とすることで、溶融押出性を維持しながら、耐湿熱性を高めることが可能となり、また、二軸延伸時の延伸性や、得られたフィルムの品質を維持することができる。
ポリエステル中の構成成分(p)の含有量が、該ポリエステル中の全構成成分に対して0.005モル%以上2.5モル%とすることで、溶融押出性を維持しながら、耐湿熱性を高めることが可能となり、また、二軸延伸時の延伸性や、得られたフィルムの品質を維持することができる。
構成成分(p)は、カルボキシル基数(a)が3以上でありかつカルボン酸を有する化合物が芳香族系化合物であるか、または、水酸基数(b)が3以上でありかつ水酸基を有する化合物が脂肪族系化合物であることが好ましい。ポリエステルフィルムの配向特性を落とすことなく、架橋構造を形成することが可能となり、分子運動性を更に低下させることが可能となり、耐湿熱性を更に高めることが可能となる。
また、ポリエステルが構成成分(p)を含む場合には、後述の緩衝剤や末端封止剤を成形時に添加することも好ましい。
また、ポリエステルが構成成分(p)を含む場合には、後述の緩衝剤や末端封止剤を成形時に添加することも好ましい。
構成成分(p)を含むポリエステルは、高結晶性樹脂であることが好ましく、具体的には、JIS K7122(1999)に準じて、昇温速度20℃/minで樹脂を25℃から300℃まで20℃/分の昇温速度で加熱(1stRUN)、その状態で5分間保持後、次いで25℃以下となるよう急冷し、再度室温から20℃/minの昇温速度で300℃まで昇温を行って得られた2ndRUNの示差走査熱量測定チャートにおいて、融解ピークのピーク面積から求められる結晶融解熱量ΔHmが、15J/g以上であるのが好ましい。好ましくは結晶融解熱量が20J/g以上、より好ましくは25J/g以上、更に好ましくは30J/g以上の樹脂を用いるのがよい。このように高結晶化することによって、延伸、熱処理により、配向結晶化させることが可能となり、その結果、機械的強度、耐湿熱性により優れるポリエステル基材とすることができる。
構成成分(p)を含むポリエステルの融点Tmは、245℃~290℃であることが好ましい。ここでいう融点TmとはDSCにより得られる、昇温過程(昇温速度:20℃/min)における融点Tmであり、上述と同様にJIS K-7121(1999)に基づいた方法により、25℃から300℃まで20℃/分の昇温速度で加熱(1stRUN)、その状態で5分間保持し、次いで25℃以下となるよう急冷し、再度室温から20℃/分の昇温速度で300℃まで昇温を行って得られた2ndRunの結晶融解ピークにおけるピークトップの温度でもってポリエステルの融点Tm1とする。より好ましくは融点Tmが247~275℃、更に好ましくは250~265℃である。融点Tmが245℃に満たないと、フィルムの耐熱性に劣ったりすることがあり好ましくなく、また、融点Tmが290℃を越えると、押出加工が困難となる場合があるため好ましくない。ポリエステルの融点Tmを245~290℃とすることにより、耐熱性と加工性を両立したポリエステル基材とすることができる。
(緩衝剤)
本発明におけるポリエステル基材は、緩衝剤を含むことが好ましい。緩衝剤の含有は、ポリエステルがその構成成分として、構成成分(p)を含む場合に特に好ましい。
本発明におけるポリエステル基材は、緩衝剤を含むことが好ましい。緩衝剤の含有は、ポリエステルがその構成成分として、構成成分(p)を含む場合に特に好ましい。
緩衝剤の具体例としては、重合反応性、耐湿熱性の点から、緩衝剤がアルカリ金属塩であることが好ましく、例えば、フタル酸、クエン酸、炭酸、乳酸、酒石酸、リン酸、亜リン酸、次亜リン酸、ポリアクリル酸などの化合物とのアルカリ金属塩を挙げることができる。中でも、アルカリ金属元素として、カリウム、ナトリウムであることが触媒残渣による析出物を生成しにくい点から好ましく、具体的には、フタル酸水素カリウム、クエン酸二水素ナトリウム、クエン酸水素二ナトリウム、クエン酸二水素カリウム、クエン酸水素二カリウム、炭酸ナトリウム、酒石酸ナトリウム、酒石酸カリウム、乳酸ナトリウム、乳酸カリウム、炭酸水素ナトリウム、リン酸水素二ナトリウム、リン酸水素二カリウム、リン酸二水素カリウム、リン酸二水素ナトリウム、亜リン酸水素ナトリウム、亜リン酸水素カリウム、次亜リン酸ナトリウム、次亜リン酸カリウム、ポリアクリル酸ナトリウムなどを挙げることができる。
また、緩衝剤は、下記式(I)で示されるアルカリ金属塩であることがポリエステルの重合反応性や溶融成形時の耐熱性の点で好ましく、さらにはアルカリ金属がナトリウム、及び/又はカリウムであることが重合反応性、耐熱性、耐湿熱性の点で好ましく、特にリン酸とナトリウム及び/又はカリウムの金属塩であることが重合反応性、耐湿熱性の点で好ましい。
POxHyMz ・・・(I)
(ここで、xは2~4の整数、yは1又は2、zは11又は2であり、Mはアルカリ金属である。)
(ここで、xは2~4の整数、yは1又は2、zは11又は2であり、Mはアルカリ金属である。)
緩衝剤の含有量はポリエステルの全質量に対して、0.1モル/ton以上5.0モル/ton以下であることが好ましく、更に好ましくは0.3モル/ton以上3.0モル/tonである。緩衝剤の含有量が、上記範囲内であることで、耐湿熱性や機械特性をより向上させることができる。
緩衝剤として式(I)で表されるアルカリ金属塩を用いる場合には、リン酸を併用することが好ましい。これにより、緩衝剤による加水分解抑制効果をさらに高めることが可能となり、得られたポリエステル基材の耐湿熱性をより高めることができる。
その場合、ポリエステル基材中のアルカリ金属元素含有量W1が2.5ppm以上125ppm以下であり、かつアルカリ金属元素含有量W1とリン元素含有量W2の比W1/W2が0.01以上1以下の範囲とすることが好ましい。この範囲とすることによって、加水分解抑制効果をより高めることが可能となる。より好ましくは、アルカリ金属元素W1が15ppm以上75ppm以下であり、アルカリ金属元素含有量W1とリン元素含有量W2の比W1/W2が0.1以上0.5以下である。アルカリ金属元素含有量W1が2.5ppmに満たないと加水分解抑制効果が不足し、得られたポリエステル基材が十分な耐湿熱性が得られない場合がある。また、125ppmを越えると、過剰に存在するアルカリ金属が溶融押出時に熱分解反応を促進して分子量が低下し、耐湿熱性や機械特性低下の原因となる場合がある。また、アルカリ金属元素含有量W1とリン元素含有量W2の比W1/W2が0.1に満たないと加水分解抑制効果が不足し、125ppmを越えると、過剰なリン酸が重合反応中にポリエステルと反応し、リン酸エステル骨格が分子鎖に形成されその部分が加水分解反応を促進してしまうため、耐加水分解性が低下することがある。
ポリエステル基材におけるアルカリ金属元素W1が、15ppm以上75ppm以下であり、アルカリ金属元素含有量W1とW2の比W1/W2が0.1以上0.5以下とすることで、耐加水分解抑制効果をより高めることが可能となる結果、高い耐湿熱性を得ることが可能となる。
その場合、ポリエステル基材中のアルカリ金属元素含有量W1が2.5ppm以上125ppm以下であり、かつアルカリ金属元素含有量W1とリン元素含有量W2の比W1/W2が0.01以上1以下の範囲とすることが好ましい。この範囲とすることによって、加水分解抑制効果をより高めることが可能となる。より好ましくは、アルカリ金属元素W1が15ppm以上75ppm以下であり、アルカリ金属元素含有量W1とリン元素含有量W2の比W1/W2が0.1以上0.5以下である。アルカリ金属元素含有量W1が2.5ppmに満たないと加水分解抑制効果が不足し、得られたポリエステル基材が十分な耐湿熱性が得られない場合がある。また、125ppmを越えると、過剰に存在するアルカリ金属が溶融押出時に熱分解反応を促進して分子量が低下し、耐湿熱性や機械特性低下の原因となる場合がある。また、アルカリ金属元素含有量W1とリン元素含有量W2の比W1/W2が0.1に満たないと加水分解抑制効果が不足し、125ppmを越えると、過剰なリン酸が重合反応中にポリエステルと反応し、リン酸エステル骨格が分子鎖に形成されその部分が加水分解反応を促進してしまうため、耐加水分解性が低下することがある。
ポリエステル基材におけるアルカリ金属元素W1が、15ppm以上75ppm以下であり、アルカリ金属元素含有量W1とW2の比W1/W2が0.1以上0.5以下とすることで、耐加水分解抑制効果をより高めることが可能となる結果、高い耐湿熱性を得ることが可能となる。
緩衝剤は、ポリエステルの重合時に添加しても、溶融成形時に添加してもいずれも構わないが、緩衝剤のポリエステル中への均一分散の点から、重合時に添加することが好ましい。重合時に添加する場合、添加時期は、ポリエステルの重合時のエステル化反応、またはエステル交換反応終了後から、重縮合反応初期(固有粘度が0.3未満)までの間であれば任意の時期に添加することができる。緩衝剤の添加方法としては、粉体を直接添加する方法、エチレングリコール等のジオール構成成分へ溶解させた溶液を調製して添加する方法等、いずれも構わないが、エチレングリコール等のジオール構成成分へ溶解させた溶液として添加することが好ましい。その場合の溶液濃度は場合も10質量%以下に希釈して添加すると、添加口付近への緩衝剤の付着が少なく、添加量の誤差が小さくなる点、及び反応性の点で好ましい。
また、構成成分(p)を含むポリエステルである場合は、重合時の副生物であるジエチレングリコールの含有量が2.0質量%未満であることが耐熱性、耐湿熱性の点から好ましく、さらには1.0質量%未満であることが好ましい。
また、構成成分(p)を含むポリエステルである場合は、重合時の副生物であるジエチレングリコールの含有量が2.0質量%未満であることが耐熱性、耐湿熱性の点から好ましく、さらには1.0質量%未満であることが好ましい。
(末端封止剤)
本発明におけるポリエステル基材は、末端封止剤を含むことも好ましい態様の一つである。末端封止剤とは、ポリエステルの末端のカルボキシル基と反応し、ポリエステルのカルボキシル基末端量を減少させる添加剤である。
末端封止剤としては、カルボジイミド化合物、エポキシ化合物、オキサゾリン化合物などが挙げられる。
本発明におけるポリエステル基材は、末端封止剤を含むことも好ましい態様の一つである。末端封止剤とは、ポリエステルの末端のカルボキシル基と反応し、ポリエステルのカルボキシル基末端量を減少させる添加剤である。
末端封止剤としては、カルボジイミド化合物、エポキシ化合物、オキサゾリン化合物などが挙げられる。
末端封止剤は、ポリエステル基材の製膜時にポリエステルと一緒に添加するとより効果が高い。固相重合の際に末端封止剤を利用してもよい。
さらに、末端封止剤は、前記カルボキシル基数(a)と水酸基数(b)との合計(a+b)が3以上である構成成分(p)を含有するポリエステルと併用してもよい。
ポリエステル基材における末端封止剤の含有量は、0.1~5質量%であることが好ましい。末端封止剤が0.1質量%より少ないと、カルボキシル基を封止する効果が小さく耐加水分解性が悪化する場合がある。また、末端封止剤が5質量%よりも大きいと製膜時に異物が多く発生したり、分解ガスが発生したりして生産性に影響がある場合がある。より好ましい末端封止剤の含有量の上限値は4質量%であり、さらに好ましい上限値は2質量%である。より好ましい末端封止剤の含有量の下限値は0.3質量%、さらに好ましい下限値は0.5質量%である。末端封止剤の含有量のより好ましい範囲は0.3~4重量%であり、さらに好ましい範囲は0.5~2重量%である。
~カルボジイミド化合物~
カルボジイミド化合物には、一官能性カルボジイミドと多官能性カルボジイミドとがある。-
一官能性カルボジイミドとしては、ジシクロヘキシルカルボジイミド、ジイソプロピルカルボジイミド、ジメチルカルボジイミド、ジイソブチルカルボジイミド、ジオクチルカルボジイミド、t-ブチルイソプロピルカルボジイミド、ジフェニルカルボジイミド、ジ-t-ブチルカルボジイミド及びジ-β-ナフチルカルボジイミドなどが挙げられる。特に好ましくは、ジシクロヘキシルカルボジイミドやジイソプロピルカルボジイミドである。
カルボジイミド化合物には、一官能性カルボジイミドと多官能性カルボジイミドとがある。-
一官能性カルボジイミドとしては、ジシクロヘキシルカルボジイミド、ジイソプロピルカルボジイミド、ジメチルカルボジイミド、ジイソブチルカルボジイミド、ジオクチルカルボジイミド、t-ブチルイソプロピルカルボジイミド、ジフェニルカルボジイミド、ジ-t-ブチルカルボジイミド及びジ-β-ナフチルカルボジイミドなどが挙げられる。特に好ましくは、ジシクロヘキシルカルボジイミドやジイソプロピルカルボジイミドである。
また、多官能性カルボジイミドとしては、重合度3~15のポリカルボジイミドが好ましく用いられる。ポリカルボジイミドは、一般に、「-R-N=C=N-」等で表される繰り返し単位を有し、前記Rは、アルキレン、アリーレン等の2価の連結基を表す。このような繰り返し単位としては、具体的には、1,5-ナフタレンカルボジイミド、4,4’-ジフェニルメタンカルボジイミド、4,4’-ジフェニルジメチルメタンカルボジイミド、1,3-フェニレンカルボジイミド、1,4-フェニレンカルボジイミド、2,4-トリレンカルボジイミド、2,6-トリレンカルボジイミド、2,4-トリレンカルボジイミドと2,6-トリレンカルボジイミドの混合物、ヘキサメチレンカルボジイミド、シクロヘキサン-1,4-カルボジイミド、キシリレンカルボジイミド、イソホロンカルボジイミド、イソホロンカルボジイミド、ジシクロヘキシルメタン-4,4’-カルボジイミド、メチルシクロヘキサンカルボジイミド、テトラメチルキシリレンカルボジイミド、2,6-ジイソプロピルフェニルカルボジイミド及び1,3,5-トリイソプロピルベンゼン-2,4-カルボジイミドなどを例示することができる。
これらは1種または2種以上を用いることができる。
これらは1種または2種以上を用いることができる。
カルボジイミド化合物は、熱分解によりイソシアネート系ガスが発生するため、耐熱性の高いカルボジイミド化合物が好ましい。耐熱性を高めるためには、分子量(重合度)が高いほど好ましく、より好ましくはカルボジイミド化合物の末端を耐熱性の高い構造にすることが好ましい。また、一度熱分解を起こすとさらなる熱分解を起こし易くなるため、ポリエステルの押出温度をなるべく低温下にするなどの工夫が必要である。
~エポキシ化合物~
エポキシ化合物の好ましい例としては、グリシジルエステル化合物やグリシジルエーテル化合物などが挙げられる。
エポキシ化合物の好ましい例としては、グリシジルエステル化合物やグリシジルエーテル化合物などが挙げられる。
グリシジルエステル化合物の具体例としては、安息香酸グリシジルエステル、t-Bu-安息香酸グリシジルエステル、P-トルイル酸グリシジルエステル、シクロヘキサンカルボン酸グリシジルエステル、ペラルゴン酸グリシジルエステル、ステアリン酸グリシジルエステル、ラウリン酸グリシジルエステル、パルミチン酸グリシジルエステル、ベヘン酸グリシジルエステル、バーサティク酸グリシジルエステル、オレイン酸グリシジルエステル、リノール酸グリシジルエステル、リノレイン酸グリシジルエステル、ベヘノール酸グリシジルエステル、ステアロール酸グリシジルエステル、テレフタル酸ジグリシジルエステル、イソフタル酸ジグリシジルエステル、フタル酸ジグリシジルエステル、ナフタレンジカルボン酸ジグリシジルエステル、メチルテレフタル酸ジグリシジルエステル、ヘキサヒドロフタル酸ジグリシジルエステル、テトラヒドロフタル酸ジグリシジルエステル、シクロヘキサンジカルボン酸ジグリシジルエステル、アジピン酸ジグリシジルエステル、コハク酸ジグリシジルエステル、セバシン酸ジグリシジルエステル、ドデカンジオン酸ジグリシジルエステル、オクタデカンジカルボン酸ジグリシジルエステル、トリメリット酸トリグリシジルエステル及びピロメリット酸テトラグリシジルエステルなどを挙げられ、これらは1種または2種以上を用いることができる。
また、グリシジルエーテル化合物の具体例としては、フェニルグリシジルエ-テル、O-フェニルグリシジルエ-テル、1,4-ビス(β,γ-エポキシプロポキシ)ブタン、1,6-ビス(β,γ-エポキシプロポキシ)ヘキサン、1,4-ビス(β,γ-エポキシプロポキシ)ベンゼン、1-(β,γ-エポキシプロポキシ)-2-エトキシエタン、1-(β,γ-エポキシプロポキシ)-2-ベンジルオキシエタン、2,2-ビス-[р-(β,γ-エポキシプロポキシ)フェニル]プロパン及び2,2-ビス-(4-ヒドロキシフェニル)プロパンや2,2-ビス-(4-ヒドロキシフェニル)メタンなどのビスフェノールとエピクロルヒドリンの反応で得られるビスグリシジルポリエーテルなどが挙げられ、これらは1種または2種以上を用いることができる。
~オキサゾリン化合物~
オキサゾリン化合物としては、ビスオキサゾリン化合物が好ましく、具体的には、2,2’-ビス(2-オキサゾリン)、2,2’-ビス(4-メチル-2-オキサゾリン)、2,2’-ビス(4,4-ジメチル-2-オキサゾリン)、2,2’-ビス(4-エチル-2-オキサゾリン)、2,2’-ビス(4,4’-ジエチル-2-オキサゾリン)、2,2’-ビス(4-プロピル-2-オキサゾリン)、2,2’-ビス(4-ブチル-2-オキサゾリン)、2,2’-ビス(4-ヘキシル-2-オキサゾリン)、2,2’-ビス(4-フェニル-2-オキサゾリン)、2,2’-ビス(4-シクロヘキシル-2-オキサゾリン)、2,2’-ビス(4-ベンジル-2-オキサゾリン)、2,2’-p-フェニレンビス(2-オキサゾリン)、2,2’-m-フェニレンビス(2-オキサゾリン)、2,2’-o-フェニレンビス(2-オキサゾリン)、2,2’-p-フェニレンビス(4-メチル-2-オキサゾリン)、2,2’-p-フェニレンビス(4,4-ジメチル-2-オキサゾリン)、2,2’-m-フェニレンビス(4-メチル-2-オキサゾリン)、2,2’-m-フェニレンビス(4,4-ジメチル-2-オキサゾリン)、2,2’-エチレンビス(2-オキサゾリン)、2,2’-テトラメチレンビス(2-オキサゾリン)、2,2’-ヘキサメチレンビス(2-オキサゾリン)、2,2’-オクタメチレンビス(2-オキサゾリン)、2,2’-デカメチレンビス(2-オキサゾリン)、2,2’-エチレンビス(4-メチル-2-オキサゾリン)、2,2’-テトラメチレンビス(4,4-ジメチル-2-オキサゾリン)、2,2’-9,9’-ジフェノキシエタンビス(2-オキサゾリン)、2,2’-シクロヘキシレンビス(2-オキサゾリン)及び2,2’-ジフェニレンビス(2-オキサゾリン)等を例示することができる。これらの中では、ポリエステルとの反応性の観点から、2,2’-ビス(2-オキサゾリン)が最も好ましく用いられる。
ビスオキサゾリン化合物は、一種を単独で用いても、二種以上を併用してもどちらでも良い。
オキサゾリン化合物としては、ビスオキサゾリン化合物が好ましく、具体的には、2,2’-ビス(2-オキサゾリン)、2,2’-ビス(4-メチル-2-オキサゾリン)、2,2’-ビス(4,4-ジメチル-2-オキサゾリン)、2,2’-ビス(4-エチル-2-オキサゾリン)、2,2’-ビス(4,4’-ジエチル-2-オキサゾリン)、2,2’-ビス(4-プロピル-2-オキサゾリン)、2,2’-ビス(4-ブチル-2-オキサゾリン)、2,2’-ビス(4-ヘキシル-2-オキサゾリン)、2,2’-ビス(4-フェニル-2-オキサゾリン)、2,2’-ビス(4-シクロヘキシル-2-オキサゾリン)、2,2’-ビス(4-ベンジル-2-オキサゾリン)、2,2’-p-フェニレンビス(2-オキサゾリン)、2,2’-m-フェニレンビス(2-オキサゾリン)、2,2’-o-フェニレンビス(2-オキサゾリン)、2,2’-p-フェニレンビス(4-メチル-2-オキサゾリン)、2,2’-p-フェニレンビス(4,4-ジメチル-2-オキサゾリン)、2,2’-m-フェニレンビス(4-メチル-2-オキサゾリン)、2,2’-m-フェニレンビス(4,4-ジメチル-2-オキサゾリン)、2,2’-エチレンビス(2-オキサゾリン)、2,2’-テトラメチレンビス(2-オキサゾリン)、2,2’-ヘキサメチレンビス(2-オキサゾリン)、2,2’-オクタメチレンビス(2-オキサゾリン)、2,2’-デカメチレンビス(2-オキサゾリン)、2,2’-エチレンビス(4-メチル-2-オキサゾリン)、2,2’-テトラメチレンビス(4,4-ジメチル-2-オキサゾリン)、2,2’-9,9’-ジフェノキシエタンビス(2-オキサゾリン)、2,2’-シクロヘキシレンビス(2-オキサゾリン)及び2,2’-ジフェニレンビス(2-オキサゾリン)等を例示することができる。これらの中では、ポリエステルとの反応性の観点から、2,2’-ビス(2-オキサゾリン)が最も好ましく用いられる。
ビスオキサゾリン化合物は、一種を単独で用いても、二種以上を併用してもどちらでも良い。
(リン化合物)
本発明におけるポリエステル基材においては、加水分解の分解を抑制の観点から、リン化合物を含有させることも好ましい。
本発明におけるポリエステル基材においては、加水分解の分解を抑制の観点から、リン化合物を含有させることも好ましい。
リン化合物を含有させる場合、ポリエステル基材における蛍光X線測定により求められるリン原子量が200ppm以上であることが好ましい。リン原子量は、より好ましくは300ppm以上、さらに好ましくは400ppm以上である。
リン化合物としては、リン酸、亜リン酸、ホスホン酸、これらのメチルエステル、エチルエステル、フェニルエステル、ハーフエステル及びその他誘導体からなる群から選ばれた一種以上のリン化合物を用いることが好ましい。本発明では、特にリン酸、亜リン酸、ホスホン酸のメチルエステル、エチルエステル、フェニルエステルが好ましい。また、リン化合物の含有方法としては、ポリエステル原料チップを製造するときにリン化合物を添加することが好ましい。
(その他の添加剤)
本発明におけるポリエステル基材は、ポリマーシートの構成要素であることから、太陽光による劣化の影響を受けにくい方が好ましい。そのため、UV(紫外線)吸収剤やUVを反射する特性のものをポリエステル中に添加してもよい。また、少なくとも一方の基材表面における波長400~700nmの平均反射率を80%以上とすることも好ましい態様の一つである。さらに好ましくは85%以上であり、特に好ましくは90%以上である。波長400~700nmの平均反射率を80%以上とすることにより、本発明のポリマーシートを用いた太陽電池を太陽光が直接当たるところにて使用してもポリマーシートの劣化が少なくなる。
本発明におけるポリエステル基材は、ポリマーシートの構成要素であることから、太陽光による劣化の影響を受けにくい方が好ましい。そのため、UV(紫外線)吸収剤やUVを反射する特性のものをポリエステル中に添加してもよい。また、少なくとも一方の基材表面における波長400~700nmの平均反射率を80%以上とすることも好ましい態様の一つである。さらに好ましくは85%以上であり、特に好ましくは90%以上である。波長400~700nmの平均反射率を80%以上とすることにより、本発明のポリマーシートを用いた太陽電池を太陽光が直接当たるところにて使用してもポリマーシートの劣化が少なくなる。
(ポリエステル基材の製造方法)
次に、本発明におけるポリエステル基材の製造方法について、ポリエチレンテレフタレート(PET)をポリエステルとして用いた二軸配向ポリエステルフィルムを、代表例として説明する。
もちろん、本発明は、PETフィルムを用いた二軸配向ポリエステルフィルムに限定されるものではなく、他のポリマーを用いたものものでもよい。例えば、ガラス転移温度や融点の高いポリエチレン-2,6-ナフタレンジカルボキシレートなどを用いてポリエステルフィルムを構成する場合は、以下に示す温度よりも高温で押出や延伸を行えばよい。
次に、本発明におけるポリエステル基材の製造方法について、ポリエチレンテレフタレート(PET)をポリエステルとして用いた二軸配向ポリエステルフィルムを、代表例として説明する。
もちろん、本発明は、PETフィルムを用いた二軸配向ポリエステルフィルムに限定されるものではなく、他のポリマーを用いたものものでもよい。例えば、ガラス転移温度や融点の高いポリエチレン-2,6-ナフタレンジカルボキシレートなどを用いてポリエステルフィルムを構成する場合は、以下に示す温度よりも高温で押出や延伸を行えばよい。
-製膜/押出し-
本発明におけるポリエステル基材は、例えば、次のようにして製造される。
まず、ポリエステル基材を構成する原反(未延伸)ポリエステルシートを製造する。原反ポリエステルシートを製造するには、例えば、上記で得たポリエステルのペレットを、押出機を用いて溶融し、口金(ダイ)から吐出した後、冷却固化してシート状に成形する。このとき、ポリマー中の未溶融物を除去するために、繊維焼結ステンレス金属フィルターによりポリマーを濾過することが好ましい。
本発明におけるポリエステル基材は、例えば、次のようにして製造される。
まず、ポリエステル基材を構成する原反(未延伸)ポリエステルシートを製造する。原反ポリエステルシートを製造するには、例えば、上記で得たポリエステルのペレットを、押出機を用いて溶融し、口金(ダイ)から吐出した後、冷却固化してシート状に成形する。このとき、ポリマー中の未溶融物を除去するために、繊維焼結ステンレス金属フィルターによりポリマーを濾過することが好ましい。
また、ポリエステル基材の表面に、易滑性、耐摩耗性及び耐スクラッチ性などを付与するため、無機粒子や有機粒子、例えば、クレー、マイカ、酸化チタン、炭酸カルシウム、カリオン、タルク、湿式シリカ、乾式シリカ、コロイド状シリカ、リン酸カルシウム、硫酸バリウム、アルミナ及びジルコニア等の無機粒子、アクリル酸類、スチレン系樹脂、熱硬化樹脂、シリコーン及びイミド系化合物等を構成成分とする有機粒子、及びポリエステル重合反応時に添加する触媒等によって析出する粒子(いわゆる内部粒子)などを添加することも好ましい態様である。
さらに、本発明の効果を阻害しない範囲内であれば、各種添加剤、例えば、相溶化剤、可塑剤、耐候剤、酸化防止剤、熱安定剤、滑剤、帯電防止剤、増白剤、着色剤、導電剤、紫外線吸収剤、難燃剤、難燃助剤、顔料及び染料などが添加されてもよい。
これらの添加剤や、末端封止剤をポリエステル中に含有させる場合には、末端封止剤を直接PETペレットと混合し、270~275℃の温度に加熱したベント式二軸混練押出機を用いて、PETに練り込み高濃度マスターペレット化する方法が有効である。
次に、得られたPETのペレットを、180℃の温度で3時間以上減圧乾燥した後、固有粘度が低下しないように窒素気流下あるいは減圧下で、265~280℃の温度より好ましくは270~275℃の温度に加熱された押出機に供給し、スリット状のダイから押出し、キャスティングロール上で冷却して未延伸フィルムを得る。この際、異物や変質ポリマーを除去するために各種のフィルター、例えば、焼結金属、多孔性セラミック、サンド及び金網などの素材からなるフィルターを用いることが好ましい。また、必要に応じて、定量供給性を向上させるためにギアポンプを設けてもよい。フィルムを積層する場合には、2台以上の押出機及びマニホールドまたは合流ブロックを用いて、複数の異なるポリマーを溶融積層する。溶融積層は、例えば、前記した反射層(白色層)共押出しする際、好ましく用いられる。
このようにして押出し機から押出された融体(メルト)は、上記のように温度分布を付与されたキャスティング(冷却)ロール上で固化し原反(未延伸フィルム)を得る。好ましい冷却ロールの温度は10℃以上60℃以下が好ましく、より好ましくは15℃以上55℃以下、さらに好ましくは20℃以上50℃以下である。このとき、メルトと冷却ロールとの密着力を向上するため、静電印加法や、エアナイフ法、冷却ロール上に水膜を形成する方法等を好ましく用いることができる。
さらに、本発明では、メルトをキャストロールに押出す際、キャストロールの線速度を10m/分以上にするのが好ましく、より好ましくは15/分以上50m/分以下、さらに好ましくは18m/分以上40m/分以下である。この範囲以下では、キャストロール上でのメルトの滞留時間が長くなり、せっかく上記方法でつけた温度差が均等化し効果が減少する。一方、この範囲を超えると、メルトの厚みむらが発生し易く、これによるメルトの温度むらが上記範囲を超え好ましくない。このようなキャストロールの速度を達成するには、押出し機での混練速度を大きくする必要があり、通常の方法ではスクリューの回転速度増加に伴う樹脂の剪断発熱によりAVが上昇し易い。このような現象は高IVの樹脂を用いる本発明において特に顕著に発現し易い。このため、本発明では押出し機に樹脂の微粒子を添加することを特徴としている。即ち最も剪断発熱し易いのが混練初期の溶融開始時であり、ここではペレットとスクリューが強く擦れあい発熱する。ここに樹脂の微粒子を添加することでペレット間の摩擦を低減しAVの上昇を抑え、本発明の範囲にすることができる。この微粒子のサイズは200メッシュ以上10メッシュ以下のものが好ましく、ペレットを破砕した後、篩にかけることで得られる。また、この微粒子の添加量は0.1%以上5%以下が好ましく、より好ましくは0.3%以上4%以下、さらに好ましくは0.5%以上3%以下である。この範囲未満では上記効果が不十分であり、この範囲を超えるとスクリューとの摩擦が大きくなりすぎスリップが発生、吐出変動によるメルトの厚みむらが発生し、キャストロール上の温度分布が本発明の範囲を超え好ましくない。
-製膜/縦延伸-
続いて、上記のようにして得られた原反(未延伸フィルム)を、長手方向と幅方向の二軸に延伸した後、熱処理する。延伸形式としては、長手方向に延伸した後に幅方向に延伸を行うなどの逐次二軸延伸法、同時二軸テンター等を用いて長手方向と幅方向を同時に延伸する同時二軸延伸法、さらに、逐次二軸延伸法と同時二軸延伸法を組み合わせた方法などが包含される。
続いて、上記のようにして得られた原反(未延伸フィルム)を、長手方向と幅方向の二軸に延伸した後、熱処理する。延伸形式としては、長手方向に延伸した後に幅方向に延伸を行うなどの逐次二軸延伸法、同時二軸テンター等を用いて長手方向と幅方向を同時に延伸する同時二軸延伸法、さらに、逐次二軸延伸法と同時二軸延伸法を組み合わせた方法などが包含される。
ここでは、未延伸フィルムを、数本のロールの配置された縦延伸機を用いて、ロールの周速差を利用して縦方向に延伸し(MD延伸)、続いてテンターにより横延伸を行う(TD延伸)という二軸延伸方法について説明する。
まず、未延伸フィルムをMD延伸するが、本発明ではMD延伸に先立って原反を十分に予熱するのが好ましい。好ましい予熱温度は40℃以上90℃以下であり、より好ましくは50℃以上85℃以下、さらに好ましくは60℃以上80℃以下である。このような予熱は原反を加熱(調温)ロール上に通して行うが、この際上述のようにロールに幅方向に温度分布を付与するのが好ましい。また、好ましい予熱時間は1秒以上120秒以下、より好ましくは5秒以上60秒以下、さらに好ましくは10秒以上40秒以下である。
MD延伸は1段でおこなってもよく、多段で行ってもよい。
MD延伸は1段でおこなってもよく、多段で行ってもよい。
1段で行う場合、ガラス転移温度Tg以上Tg+15℃以下(より好ましくはTg+10℃以下)の温度とし、好ましい延伸倍率は2.0~6.0倍であり、より好ましくは3.0~5.5倍であり、さらに好ましくは3.5~5.0倍である。延伸後、20~50℃の温度の冷却ロール群で冷却することが好ましい。
ポリエステルのIVが大きく分子量が大きいほど、分子の運動性が低下し配向結晶化が起こりにくい。そこで、多段延伸を行うことがより好ましい。即ち、最初に低温で延伸を行い、その後温度を上げて2段階延伸すると配向結晶化が起こり配向を高めることができる。最初の低温での延伸(MD1延伸)は(Tg-20)~(Tg+10)℃の範囲、さらに好ましくは(Tg-10)~(Tg+5)℃の範囲にある加熱ロール群で加熱し、長手方向に好ましくは1.1~3.0倍、より好ましくは1.2~2.5倍、さらに好ましくは1.5~2.0倍に延伸し、次にMD延伸1温度より高温(Tg+10)~(Tg+50)でMD延伸2を行う。より好ましい温度は(Tg+15)(~Tg+30)である。MD延伸2の好ましい延伸倍率は1.2~4.0倍であり、より好ましくは1.5~3.0倍である。MD延伸1とMD延伸2の合わせたMD延伸倍率は、好ましくは2.0~6.0倍であり、より好ましくは3.0~5.5倍であり、さらに好ましくは3.5~5.0倍である。第1段と第2段の延伸倍率の比(第2段/第1段=多段倍率比と称する)は1.1以上3以下が好ましく、より好ましくは1.15倍以上2倍以下、さらに好ましくは1.2倍以上1.8倍以下である。
延伸後、20~50℃の温度の冷却ロール群で冷却することが好ましい。
延伸後、20~50℃の温度の冷却ロール群で冷却することが好ましい。
-製膜/横延伸-
次に、テンター(ステンターと称することもある)を用いて、幅方向の延伸を行う。その延伸倍率は、好ましくは2.0~6.0倍であり、より好ましくは3.0~5.5倍であり、さらに好ましくは3.5~5.0倍である。また、温度は好ましくは(Tg)~(Tg+50)℃の範囲であり、さらに好ましくは(Tg)~(Tg+30)℃の範囲で行う(TD延伸)。 なお、Tgはガラス転移温度を表し、JIS K7121或いはASTM D3418-82等に基づいて測定することができる。例えば。本発明では、島津製作所社製の示差走査熱量測定装置(DSC)を用いて測定する。
具体的には、試料としてポリエステル等のポリマーを10mg秤量し、アルミパンにセットし、昇温速度10℃/minで、室温から最終温度300℃まで昇温しながら、DSC装置で、温度に対する熱量を測定したとき、DSC曲線が屈曲する温度をガラス転移温度とした。
次に、テンター(ステンターと称することもある)を用いて、幅方向の延伸を行う。その延伸倍率は、好ましくは2.0~6.0倍であり、より好ましくは3.0~5.5倍であり、さらに好ましくは3.5~5.0倍である。また、温度は好ましくは(Tg)~(Tg+50)℃の範囲であり、さらに好ましくは(Tg)~(Tg+30)℃の範囲で行う(TD延伸)。 なお、Tgはガラス転移温度を表し、JIS K7121或いはASTM D3418-82等に基づいて測定することができる。例えば。本発明では、島津製作所社製の示差走査熱量測定装置(DSC)を用いて測定する。
具体的には、試料としてポリエステル等のポリマーを10mg秤量し、アルミパンにセットし、昇温速度10℃/minで、室温から最終温度300℃まで昇温しながら、DSC装置で、温度に対する熱量を測定したとき、DSC曲線が屈曲する温度をガラス転移温度とした。
ポリエステル基材の厚みは、25~300μm程度が好ましい。厚みは、25μm以上であると力学強度が良好であり、300μm以下であるとコスト的に有利である。
特にポリエステル基材は、厚みが増すに伴なって耐加水分解性が悪化し、長期使用時の耐久性が低下する傾向にあり、本発明においては、ポリエステル基材の厚みが120μm以上300μm以下であって、かつポリエステル中のカルボキシル基含量が2~15当量/tである場合に、より湿熱耐久性の向上効果が奏される。
特にポリエステル基材は、厚みが増すに伴なって耐加水分解性が悪化し、長期使用時の耐久性が低下する傾向にあり、本発明においては、ポリエステル基材の厚みが120μm以上300μm以下であって、かつポリエステル中のカルボキシル基含量が2~15当量/tである場合に、より湿熱耐久性の向上効果が奏される。
ポリエステル基材は、コロナ処理(コロナ放電処理ともいう)、火炎処理、低圧プラズマ処理、大気圧プラズマ処理、又は紫外線処理により表面処理が施された態様が好ましい。これらの表面処理を施すことで、湿熱環境下に曝された場合の接着性をさらに高めることができる。中でも特に、コロナ処理を行なうことで、より優れた接着性の向上効果が得られる。
これらの表面処理は、ポリエステル基材表面にカルボキシル基や水酸基が増加することにより接着性が高められるが、架橋剤(特にカルボキシル基と反応性の高いオキサゾリン系もしくはカルボジイミド系の架橋剤)を併用した場合により強力な接着性が得られる。これは、コロナ処理による場合により顕著である。
これらの表面処理は、ポリエステル基材表面にカルボキシル基や水酸基が増加することにより接着性が高められるが、架橋剤(特にカルボキシル基と反応性の高いオキサゾリン系もしくはカルボジイミド系の架橋剤)を併用した場合により強力な接着性が得られる。これは、コロナ処理による場合により顕著である。
(ポリマー層)
本発明のポリマー層は、前記ポリエステル基材の表面に接触させてあるいは他の層を介して配置される層である。ポリマー層は、少なくとも、分子内に非シロキサン系構造単位と下記一般式(1)で表される(ポリ)シロキサン構造単位を含む特定の複合ポリマーを用いて構成されている。本発明におけるポリマー層は、複合ポリマーを含む構成により、ポリエステル基材との接着、及び層間の接着性(特に電池側基板に設けられた封止材との間の接着性)が改善されるので、ポリエステル基材に直に形成されることが好ましい。また、耐湿熱保存性を有するポリマー層が形成されるため、外部環境に暴露される最外層、つまりバック層として用いることも好ましい。
本発明のポリマー層は、前記ポリエステル基材の表面に接触させてあるいは他の層を介して配置される層である。ポリマー層は、少なくとも、分子内に非シロキサン系構造単位と下記一般式(1)で表される(ポリ)シロキサン構造単位を含む特定の複合ポリマーを用いて構成されている。本発明におけるポリマー層は、複合ポリマーを含む構成により、ポリエステル基材との接着、及び層間の接着性(特に電池側基板に設けられた封止材との間の接着性)が改善されるので、ポリエステル基材に直に形成されることが好ましい。また、耐湿熱保存性を有するポリマー層が形成されるため、外部環境に暴露される最外層、つまりバック層として用いることも好ましい。
このポリマー層は、場合に応じて更に他の成分を用いて構成することができ、適用する用途によりその構成成分が異なる。ポリマー層は、太陽光の反射機能や外観意匠性の付与などを担う着色層や、太陽光が入射する側と反対側に配されるバック層などを構成することができる。
ポリマー層を例えば、太陽光をその入射側に反射させる反射層として構成する場合、白色顔料等の着色剤を更に用いて構成することができる。この場合、反射層は複合ポリマーを含ませてポリマー層として形成される。ポリエステル基材上に2層以上のポリマー層を有する場合には、白色層(ポリマー層)/ポリマー層/ポリエステル基材の積層構造に構成されてもよい。白色層は、反射層として構成することができる。反射層のポリマーシート内での接着性、密着性をより向上させることが可能である。
-複合ポリマー-
本発明におけるポリマー層は、分子中に質量割合が15~85質量%の下記一般式(1)で表される(ポリ)シロキサン構造単位と質量割合が85~15質量%の非シロキサン系構造単位とを含む複合ポリマーの少なくとも一種を含有する。この複合ポリマーを含有することにより、支持体であるポリエステル基材や層間あるいは電池側基板の構成基材(例えばEVA等の封止材)との間の接着性、すなわち熱や水分が与えられて劣化しやすい剥離耐性、形状安定性を従来に比べて飛躍的に向上させることができる。
本発明におけるポリマー層は、分子中に質量割合が15~85質量%の下記一般式(1)で表される(ポリ)シロキサン構造単位と質量割合が85~15質量%の非シロキサン系構造単位とを含む複合ポリマーの少なくとも一種を含有する。この複合ポリマーを含有することにより、支持体であるポリエステル基材や層間あるいは電池側基板の構成基材(例えばEVA等の封止材)との間の接着性、すなわち熱や水分が与えられて劣化しやすい剥離耐性、形状安定性を従来に比べて飛躍的に向上させることができる。
本発明における複合ポリマーは、(ポリ)シロキサンと少なくとも一種のポリマーとが共重合したブロック共重合体である。(ポリ)シロキサン、及び共重合されるポリマーは、一種単独でもよく、二種以上であってもよい。

前記一般式(1)において、R1及びR2は、各々独立に、水素原子、ハロゲン原子、又は1価の有機基を表す。ここで、R1とR2とは同一でも異なってもよく、複数のR1及びR2は各々、互いに同一でも異なってもよい。nは、1以上の整数を表す。
複合ポリマー中の(ポリ)シロキサンセグメントである「-(Si(R1) (R2)-O)n-」の部分(一般式(1)で表される(ポリ)シロキサン構造単位)において、R1及びR2は同一でも異なってもよく、水素原子、ハロゲン原子、又は1価の有機基を表す。
「-(Si(R1) (R2)-O)n-」は、線状、分岐状あるいは環状の構造を有する各種の(ポリ)シロキサンに由来する(ポリ)シロキサンセグメントである。
R1及びR2で表されるハロゲン原子としては、フッ素原子、塩素原子、ヨウ素原子等を挙げることができる。
R1及びR2で表される「1価の有機基」は、Si原子と共有結合可能な基であり、無置換でも置換基を有してもよい。前記1価の有機基は、例えば、アルキル基(例:メチル基、エチル基など)、アリール基(例:フェニル基など)、アラルキル基(例:ベンジル基、フェニルエチルなど)、アルコキシ基(例:メトキシ基、エトキシ基、プロポキシ基など)、アリールオキシ基(例;フェノキシ基など)、メルカプト基、アミノ基(例:アミノ基、ジエチルアミノ基など)、アミド基等が挙げられる。
中でも、ポリエステル基材などの隣接材料との接着性及び湿熱環境下での耐久性の点で、R1、R2としては各々独立に、水素原子、塩素原子、臭素原子、無置換の又は置換された炭素数1~4のアルキル基(特にメチル基、エチル基)、無置換の又は置換されたフェニル基、無置換の又は置換されたアルコキシ基、メルカプト基、無置換のアミノ基、アミド基が好ましく、より好ましくは、湿熱環境下での耐久性の点で、無置換の又は置換されたアルコキシ基(好ましくは炭素数1~4のアルコキシ基)である。
前記nは、1~5000であることが好ましく、1~1000であることがより好ましい。
複合ポリマー中における「-(Si(R1) (R2)-O)n-」の部分(一般式(1)で表される(ポリ)シロキサン構造単位)の比率は、複合ポリマーの全質量に対して15~85質量%であり、その中でもポリエステル基材との接着性及び湿熱環境下での耐久性の点で、20~80質量%の範囲が好ましい。
ポリシロキサン部位の比率は、15質量%未満であるとポリエステル基材との接着性及び湿熱環境下に曝された際の接着耐久性が劣り、85質量%を超えると液が不安定になる。
ポリシロキサン部位の比率は、15質量%未満であるとポリエステル基材との接着性及び湿熱環境下に曝された際の接着耐久性が劣り、85質量%を超えると液が不安定になる。
また、前記シロキサン構造単位と共重合している非シロキサン系構造単位(ポリマーに由来の構造部分)は、シロキサン構造を有していないこと以外は特に制限されるものではなく、任意のポリマーに由来のポリマーセグメントのいずれであってもよい。ポリマーセグメントの前駆体である重合体(前駆ポリマー)としては、例えば、ビニル系重合体、ポリエステル系重合体、ポリウレタン系重合体等の各種の重合体等が挙げられる。調製が容易なこと及び耐加水分解性に優れる点から、ビニル系重合体及びポリウレタン系重合体が好ましく、ビニル系重合体が特に好ましい。
前記ビニル系重合体の代表的な例としては、アクリル系重合体、カルボン酸ビニルエステル系重合体、芳香族ビニル系重合体、フルオロオレフィン系重合体等の各種の重合体が挙げられる。中でも、設計の自由度の観点から、アクリル系重合体(すなわち非シロキサン系構造単位としてアクリル系構造単位)が特に好ましい。
なお、非シロキサン系構造単位を構成する重合体は、一種単独でもよいし、2種以上の併用であってもよい。
なお、非シロキサン系構造単位を構成する重合体は、一種単独でもよいし、2種以上の併用であってもよい。
また、非シロキサン系構造単位をなす前駆ポリマーは、酸基及び中和された酸基の少なくとも1つ並びに/又は加水分解性シリル基を含有するものが好ましい。このような前駆ポリマーのうち、ビニル系重合体は、例えば、(a)酸基を含むビニル系単量体と加水分解性シリル基及び/又はシラノール基を含むビニル系単量体とを、これらと共重合可能な単量体と共重合させる方法、(2)予め調製した水酸基並びに加水分解性シリル基及び/又はシラノール基を含むビニル系重合体にポリカルボン酸無水物を反応させる方法、(3)予め調製した酸無水基並びに加水分解性シリル基及び/又はシラノール基を含むビニル系重合体を、活性水素を有する化合物(水、アルコール、アミン等)と反応させる方法などの各種方法を利用して調製することができる。
このような前駆ポリマーは、例えば、特開2009-52011号公報の段落番号0021~0078に記載の方法を利用して製造、入手することができる。
本発明におけるポリマー層は、バインダーとして、前記複合ポリマーを単独で用いてもよいし、他のポリマーと併用してもよい。他のポリマーを併用する場合、本発明における複合ポリマーの比率は、全バインダーの30質量%以上が好ましく、より好ましくは60質量%以上である。複合ポリマーの比率が30質量%以上であることにより、ポリエステル基材との接着性及び湿熱環境下での耐久性により優れる。
前記複合ポリマーの分子量は、5,000~100,000であることが好ましく、10,000~50,000であることがより好ましい。
複合ポリマーの調製には、(i)前駆ポリマーと、前記一般式(1)〔-(Si(R1) (R2)-O)n-〕の構造を有するポリシロキサンとを反応させる方法、(ii)前駆ポリマーの存在下に、R1及び/又はR2が加水分解性基である「-(Si(R1) (R2)-O)n-」の構造を有するシラン化合物を加水分解縮合させる方法、等の方法を利用することができる。
前記(ii)の方法で用いられるシラン化合物としては、各種シラン化合物が挙げられるが、アルコキシシラン化合物が特に好ましい。
前記(ii)の方法で用いられるシラン化合物としては、各種シラン化合物が挙げられるが、アルコキシシラン化合物が特に好ましい。
前記(i)の方法により複合ポリマーを調製する場合、例えば、前駆ポリマーとポリシロキサンの混合物に、必要に応じて水と触媒を加え、20~150℃程度の温度で30分~30時間程度(好ましくは50~130℃で1~20時間)反応させることにより調製することができる。触媒としては、酸性化合物、塩基性化合物、金属含有化合物等の各種のシラノール縮合触媒を添加することができる。
また、前記(ii)の方法により複合ポリマーを調製する場合、例えば、前駆ポリマーとアルコキシシラン化合物の混合物に、水とシラノール縮合触媒を添加して、20~150℃程度の温度で30分~30時間程度(好ましくは50~130℃で1~20時間)加水分解縮合を行なうことにより調製することができる。
また、前記(ii)の方法により複合ポリマーを調製する場合、例えば、前駆ポリマーとアルコキシシラン化合物の混合物に、水とシラノール縮合触媒を添加して、20~150℃程度の温度で30分~30時間程度(好ましくは50~130℃で1~20時間)加水分解縮合を行なうことにより調製することができる。
-架橋剤-
本発明においては、ポリマー層が、前記複合ポリマー間を架橋する架橋剤由来の構造部分を有していることが好ましい。つまり、ポリマー層は、複合ポリマー間を架橋しうる架橋剤を用いて構成することができる。架橋剤で架橋されることにより、湿熱経時後の接着性、具体的には湿熱環境下に曝された場合のポリエステル基材に対する接着、及び層間の接着をより向上させることができる。
本発明においては、ポリマー層が、前記複合ポリマー間を架橋する架橋剤由来の構造部分を有していることが好ましい。つまり、ポリマー層は、複合ポリマー間を架橋しうる架橋剤を用いて構成することができる。架橋剤で架橋されることにより、湿熱経時後の接着性、具体的には湿熱環境下に曝された場合のポリエステル基材に対する接着、及び層間の接着をより向上させることができる。
前記架橋剤としては、エポキシ系、イソシアネート系、メラミン系、カルボジイミド系、オキサゾリン系等の架橋剤を挙げることができる。架橋剤の中でも、カルボジイミド系化合物やオキサゾリン系化合物などの架橋剤が好ましい。
前記オキサゾリン系架橋剤の具体例としては、2-ビニル-2-オキサゾリン、2-ビニル-4-メチル-2-オキサゾリン、2-ビニル-5-メチル-2-オキサゾリン、2-イソプロペニル-2-オキサゾリン、2-イソプロペニル-4-メチル-2-オキサゾリン、2-イソプロペニル-5-エチル-2-オキサゾリン、2,2’-ビス-(2-オキサゾリン)、2,2’-メチレン-ビス-(2-オキサゾリン)、2,2’-エチレン-ビス-(2-オキサゾリン)、2,2’-トリメチレン-ビス-(2-オキサゾリン)、2,2’-テトラメチレン-ビス-(2-オキサゾリン)、2、2’-ヘキサメチレン-ビス-(2-オキサゾリン)、2,2’-オクタメチレン-ビス-(2-オキサゾリン)、2,2’-エチレン-ビス-(4,4’-ジメチル-2-オキサゾリン)、2,2’-p-フェニレン-ビス-(2-オキサゾリン)、2,2’-m-フェニレン-ビス-(2-オキサゾリン)、2,2’-m-フェニレン-ビス-(4,4’-ジメチル-2-オキサゾリン)、ビス-(2-オキサゾリニルシクロヘキサン)スルフィド、ビス-(2-オキサゾリニルノルボルナン)スルフィド等が挙げられる。さらに、これらの化合物の(共)重合体も好ましく用いられる。
また、オキサゾリン基を有する化合物として、エポクロスK2010E、同K2020E、同K2030E、同WS-500、同WS-700(いずれも日本触媒化学工業(株)製)等も利用できる。
また、オキサゾリン基を有する化合物として、エポクロスK2010E、同K2020E、同K2030E、同WS-500、同WS-700(いずれも日本触媒化学工業(株)製)等も利用できる。
前記カルボジイミド系架橋剤の具体例としては、ジシクロヘキシルメタンカルボジイミド、テトラメチルキシリレンカルボジイミド、ジシクロヘキシルメタンカルボジイミド等を挙げることができる。また、特開2009-235278号公報に記載のカルボジイミド化合物も好ましい。具体的には、カルボジイミド系架橋剤として、カルボジライトSV-02、カルボジライトV-02、カルボジライトV-02-L2、カルボジライトV-04、カルボジライトE-01、カルボジライトE-02(いずれも日清紡ケミカル(株)製)等の市販品も利用できる。
また、ポリマー層中における、架橋剤由来の構造部分の複合ポリマーに対する質量割合としては、1~30質量%が好ましく、より好ましくは5~20質量%である。架橋剤の含有割合は、1質量%以上であると、ポリマー層の強度、及び湿熱経時後の接着性に優れ、30質量%以下であると、塗布液のポットライフを長く保てる。
本発明のポリマーシートにおいては、ポリマー層は、該層が上記のように複合ポリマーを含むことで、ポリエステル基材に対する接着が良化し、層間の接着性(本発明のポリマーシートをバックシートとして用いるときは、特に、電池側基板に設けられた封止材との間の接着性)が良化する。さらに、湿熱環境下での劣化耐性(接着耐久性)に優れている。このことから、ポリエステル基材から最も離れた位置に配された最外層として設けられることも好ましい。具体的には、例えば、太陽電池素子を備えた電池側基板と対向する側(オモテ側)とは反対側(裏側)に配置されるバック層、電池側基板の太陽電池素子を封止する封止剤と接触させて配される光反射性の反射層などである。
ポリマー層は、1層のみ設けられてもよいし、複数のポリマー層が形成されてもよい。
ポリマー層の1層の厚みとしては、通常は0.3μm~22μmが好ましく、0.5μm~15μmがより好ましく、0.8μm~12μmの範囲が更に好ましく、1.0μm~8μmの範囲が特に好ましく、2~6μmの範囲が最も好ましい。ポリマー層の厚みが0.3μm、更には0.8μm以上であることで、湿熱環境下に曝されたときにポリマー層表面から内部に水分が浸透し難く、ポリマー層とポリエステル基材との界面に水分が到達し難くなることで接着性が顕著に改善される。また、ポリマー層の厚みが22μm以下、更には12μm以下であると、ポリマー層自身が脆弱になり難く、湿熱環境下に暴露したときにポリマー層の破壊が生じにくくなることで接着性が改善される。
ポリマー層の1層の厚みとしては、通常は0.3μm~22μmが好ましく、0.5μm~15μmがより好ましく、0.8μm~12μmの範囲が更に好ましく、1.0μm~8μmの範囲が特に好ましく、2~6μmの範囲が最も好ましい。ポリマー層の厚みが0.3μm、更には0.8μm以上であることで、湿熱環境下に曝されたときにポリマー層表面から内部に水分が浸透し難く、ポリマー層とポリエステル基材との界面に水分が到達し難くなることで接着性が顕著に改善される。また、ポリマー層の厚みが22μm以下、更には12μm以下であると、ポリマー層自身が脆弱になり難く、湿熱環境下に暴露したときにポリマー層の破壊が生じにくくなることで接着性が改善される。
本発明におけるポリマー層は、前記複合ポリマーと、前記複合ポリマーのポリマー分子間が架橋剤で架橋された架橋構造を有し、該架橋剤由来の構造部分の複合ポリマーに対する比率が1~30質量%であって、ポリマー層の厚みが0.8μm~12μmである場合が特に湿熱経時後の接着性に対する向上効果に優れる。
~バック層~
本発明の太陽電池用ポリマーシートを太陽電池用バックシートとして用いるときは、本発明におけるポリマー層をバック層として構成すればよい。この場合、前記複合ポリマーに加え、必要に応じて、さらに各種添加剤などの他の成分を含んで構成されてもよい。電池側基板(=太陽光が入射する側の透明性の基板(ガラス基板等)/太陽電池素子を含む素子構造部分)/太陽電池用バックシートの積層構造を有する太陽電池において、バック層は支持体であるポリエステル基材の前記電池側基板と対向する側と反対側に配される裏面保護層であり、1層構造でもよいし、2層以上を積層した構造であってもよい。複合ポリマーを含むことで、ポリエステル基材に対する接着や、バック層が2層以上からなる場合の層間における接着が良化するとともに、更には湿熱環境下での劣化耐性が得られる。そのため、本発明におけるポリマー層であるバック層が、ポリエステル基材から最も離れた最外層として配置された形態が好ましい。
本発明の太陽電池用ポリマーシートを太陽電池用バックシートとして用いるときは、本発明におけるポリマー層をバック層として構成すればよい。この場合、前記複合ポリマーに加え、必要に応じて、さらに各種添加剤などの他の成分を含んで構成されてもよい。電池側基板(=太陽光が入射する側の透明性の基板(ガラス基板等)/太陽電池素子を含む素子構造部分)/太陽電池用バックシートの積層構造を有する太陽電池において、バック層は支持体であるポリエステル基材の前記電池側基板と対向する側と反対側に配される裏面保護層であり、1層構造でもよいし、2層以上を積層した構造であってもよい。複合ポリマーを含むことで、ポリエステル基材に対する接着や、バック層が2層以上からなる場合の層間における接着が良化するとともに、更には湿熱環境下での劣化耐性が得られる。そのため、本発明におけるポリマー層であるバック層が、ポリエステル基材から最も離れた最外層として配置された形態が好ましい。
バック層を2層以上設ける場合は、両方のバック層が前記複合ポリマー、又は前記複合ポリマーと前記架橋剤との双方を含むポリマー層であってもよく、一方のみのバック層が前記複合ポリマー、又は前記複合ポリマーと前記架橋剤との双方を含むポリマー層であってもよい。
中でも、湿熱環境下における接着耐久性を改善する観点から、少なくとも、ポリエステル基材と接するバック層(第1のバック層)が前記複合ポリマー、又は前記複合ポリマーと前記架橋剤との双方を含むポリマー層で構成されていることが好ましい。なお、この場合、ポリエステル基材上の前記第1のバック層の更に上に設けられる第2のバック層は、前記一般式(1)で表される(ポリ)シロキサン構造単位と非ポリシロキサン構造単位を含有する複合ポリマーを含まなくてもよいが、その場合は樹脂単独の空隙のない均一膜を形成してポリマーと顔料との間の空隙からの水分侵入を防ぎ、湿熱環境下での接着性を高める観点から、ポリシロキサンの単独重合体をも含まないことが好ましい。
中でも、湿熱環境下における接着耐久性を改善する観点から、少なくとも、ポリエステル基材と接するバック層(第1のバック層)が前記複合ポリマー、又は前記複合ポリマーと前記架橋剤との双方を含むポリマー層で構成されていることが好ましい。なお、この場合、ポリエステル基材上の前記第1のバック層の更に上に設けられる第2のバック層は、前記一般式(1)で表される(ポリ)シロキサン構造単位と非ポリシロキサン構造単位を含有する複合ポリマーを含まなくてもよいが、その場合は樹脂単独の空隙のない均一膜を形成してポリマーと顔料との間の空隙からの水分侵入を防ぎ、湿熱環境下での接着性を高める観点から、ポリシロキサンの単独重合体をも含まないことが好ましい。
バック層中に含むことができる他の成分については、後述するように、界面活性剤、フィラー等が挙げられる。また、着色層に用いられる顔料を含んでもよい。これらの他の成分及び顔料の詳細、好ましい態様については、後述する。
~着色層~
本発明におけるポリマー層を着色層(好ましくは反射層)として構成する場合、前記複合ポリマーに加え、さらに顔料を含有する。着色層は、必要に応じて、さらに各種添加剤などの他の成分を含んで構成されてもよい。
本発明におけるポリマー層を着色層(好ましくは反射層)として構成する場合、前記複合ポリマーに加え、さらに顔料を含有する。着色層は、必要に応じて、さらに各種添加剤などの他の成分を含んで構成されてもよい。
着色層の機能としては、例えば、本発明の太陽電池用ポリマーシートを太陽電池用バックシートとして用いたとき、第1に、入射光のうち太陽電池セルを通過して発電に使用されずにバックシートに到達した光を反射させて太陽電池セルに戻すことにより、太陽電池モジュールの発電効率を上げること、第2に、太陽電池モジュールを太陽光が入射する側(オモテ面側)から見た場合の外観の装飾性を向上すること、等が挙げられる。一般に太陽電池モジュールをオモテ面側から見ると、太陽電池セルの周囲にバックシートが見えており、バックシートに着色層を設けることにより装飾性を向上させて見栄えを改善することができる。
-顔料-
本発明における着色層は、顔料の少なくとも一種を含有することができる。
顔料としては、例えば、二酸化チタン、硫酸バリウム、酸化珪素、酸化アルミニウム、酸化マグネシウム、炭酸カルシウム、カオリン、タルク、群青、紺青、カーボンブラック等の無機顔料、フタロシアニンブルー、フタロシアニングリーン等の有機顔料を、適宜選択して含有することができる。
本発明における着色層は、顔料の少なくとも一種を含有することができる。
顔料としては、例えば、二酸化チタン、硫酸バリウム、酸化珪素、酸化アルミニウム、酸化マグネシウム、炭酸カルシウム、カオリン、タルク、群青、紺青、カーボンブラック等の無機顔料、フタロシアニンブルー、フタロシアニングリーン等の有機顔料を、適宜選択して含有することができる。
顔料のうち、ポリマー層を、太陽電池に入射して太陽電池セルを通過した光を反射して太陽電池セルに戻す反射層として構成する場合、白色顔料が好ましい。白色顔料としては、二酸化チタン、硫酸バリウム、酸化珪素、酸化アルミニウム、酸化マグネシウム、炭酸カルシウム、カオリン、タルク等が好ましい。
顔料の着色層中における含有量は、2.5~8.5g/m2の範囲が好ましい。顔料の含有量が2.5g/m2以上であると、必要な着色が得られ、反射率や装飾性を効果的に与えることができる。また、顔料の着色層中における含量が8.5g/m2以下であると、着色層の面状を良好に維持しやすく、膜強度により優れる。中でも、顔料の含有量は、4.5~8.0g/m2の範囲がより好ましい。
顔料の平均粒径としては、体積平均粒径で0.03~0.8μmが好ましく、より好ましくは0.15~0.5μm程度である。平均粒径が前記範囲内であると、光の反射効率が高い。平均粒径は、レーザー解析/散乱式粒子径分布測定装置LA950〔(株)堀場製作所製〕により測定される値である。
ポリマー層を着色層として構成する場合、バインダー成分(前記複合ポリマーを含む)の含有量は、顔料に対して、15~200質量%の範囲が好ましく、17~100質量%の範囲がより好ましい。バインダーの含有量は、15質量%以上であると、着色層の強度が充分に得られ、また200質量%以下であると、反射率や装飾性を良好に保つことができる。
-添加剤-
本発明のポリマー層には、必要に応じて、界面活性剤、フィラー等を添加してもよい。
前記界面活性剤としては、アニオン系やノニオン系等の公知の界面活性剤を用いることができる。界面活性剤を添加する場合、ポリマー層中のその添加量の範囲は0.1~15mg/m2が好ましく、より好ましくは0.5~5mg/m2である。界面活性剤の添加量は、0.1mg/m2以上であると、ハジキの発生を抑えて良好な層形成が得られ、15mg/m2以下であると、接着を良好に行なうことができる。
本発明のポリマー層には、必要に応じて、界面活性剤、フィラー等を添加してもよい。
前記界面活性剤としては、アニオン系やノニオン系等の公知の界面活性剤を用いることができる。界面活性剤を添加する場合、ポリマー層中のその添加量の範囲は0.1~15mg/m2が好ましく、より好ましくは0.5~5mg/m2である。界面活性剤の添加量は、0.1mg/m2以上であると、ハジキの発生を抑えて良好な層形成が得られ、15mg/m2以下であると、接着を良好に行なうことができる。
本発明におけるポリマー層には、更に、フィラーを添加してもよい。フィラーの添加量は、ポリマー層のバインダー当たり20質量%以下が好ましく、より好ましくは15質量%以下である。フィラーの添加量が20質量%以下であると、ポリマー層の面状がより良好に保てる。
~物性~
着色層に顔料として白色顔料を添加して反射層とする場合、着色層及び易接着性層が設けられている側の表面における550nmの光反射率は、75%以上であることが好ましい。なお、光反射率とは、易接着性層の表面から入射した光が反射層で反射して再び易接着性層から出射した光量の入射光量に対する比率である。ここでは、代表波長光として、波長550nmの光が用いられる。
光反射率が75%以上であると、セルを素通りして内部に入射した光を効果的にセルに戻すことができ、発電効率の向上効果が大きい。着色剤の含有量を2.5~30g/m2の範囲で制御することにより、光反射率を75%以上に調整することができる。
着色層に顔料として白色顔料を添加して反射層とする場合、着色層及び易接着性層が設けられている側の表面における550nmの光反射率は、75%以上であることが好ましい。なお、光反射率とは、易接着性層の表面から入射した光が反射層で反射して再び易接着性層から出射した光量の入射光量に対する比率である。ここでは、代表波長光として、波長550nmの光が用いられる。
光反射率が75%以上であると、セルを素通りして内部に入射した光を効果的にセルに戻すことができ、発電効率の向上効果が大きい。着色剤の含有量を2.5~30g/m2の範囲で制御することにより、光反射率を75%以上に調整することができる。
(他の機能層)
本発明の太陽電池用ポリマーシートは、ポリエステル基材とポリマー層以外に他の機能層を有していてもよい。他の機能層として、下塗り層、易接着層を設けることができる。
本発明の太陽電池用ポリマーシートは、ポリエステル基材とポリマー層以外に他の機能層を有していてもよい。他の機能層として、下塗り層、易接着層を設けることができる。
[下塗り層]
本発明の太陽電池用ポリマーシートには、前記ポリエステル基材(支持体)と前記ポリマー層との間に下塗り層を設けてもよい。下塗り層の厚みは、厚み2μm以下の範囲が好ましく、より好ましくは0.05μm~2μmであり、更に好ましくは0.1μm~1.5μmである。厚みが2μm以下であると、面状を良好に保つことができる。また、厚みが0.05μm以上であることにより、必要な接着性を確保しやすい。
本発明の太陽電池用ポリマーシートには、前記ポリエステル基材(支持体)と前記ポリマー層との間に下塗り層を設けてもよい。下塗り層の厚みは、厚み2μm以下の範囲が好ましく、より好ましくは0.05μm~2μmであり、更に好ましくは0.1μm~1.5μmである。厚みが2μm以下であると、面状を良好に保つことができる。また、厚みが0.05μm以上であることにより、必要な接着性を確保しやすい。
下塗り層は、バインダーを含有することができる。バインダーとしては、例えば、ポリエステル、ポリウレタン、アクリル樹脂、ポリオレフィン等を用いることができる。また、下塗り層には、バインダー以外に、エポキシ系、イソシアネート系、メラミン系、カルボジイミド系、オキサゾリン系等の架橋剤、アニオン系やノニオン系等の界面活性剤、シリカ等のフィラーなどを添加してもよい。
下塗り層を塗布するための方法や用いる塗布液の溶媒には、特に制限はない。
塗布方法としては、例えばグラビアコーターやバーコーターを利用することができる。
塗布液に用いる溶媒は、水でもよいし、トルエンやメチルエチルケトン等の有機溶媒でもよい。溶媒は1種類を単独で用いてもよいし、2種類以上を混合して用いてもよい。
また、塗布は、2軸延伸した後のポリエステル基材に塗布してもよいし、1軸延伸後のポリエステル基材に塗布した後に初めの延伸と異なる方向に延伸する方法でもよい。さらに、延伸前の基材に塗布した後に2方向に延伸してもよい。
塗布方法としては、例えばグラビアコーターやバーコーターを利用することができる。
塗布液に用いる溶媒は、水でもよいし、トルエンやメチルエチルケトン等の有機溶媒でもよい。溶媒は1種類を単独で用いてもよいし、2種類以上を混合して用いてもよい。
また、塗布は、2軸延伸した後のポリエステル基材に塗布してもよいし、1軸延伸後のポリエステル基材に塗布した後に初めの延伸と異なる方向に延伸する方法でもよい。さらに、延伸前の基材に塗布した後に2方向に延伸してもよい。
本発明の太陽電池用ポリマーシートを太陽電池用バックシートとして用いるとき、太陽電池用バックシートは、太陽電池素子が封止材で封止された電池側基板の前記封止剤と接触させて、既述の特定の複合ポリマー(非シロキサン系構造単位と一般式(1)で表されるシロキサン構造単位とを含む複合ポリマー)を含有するポリマー層を配設できる方法であれば適宜選択して製造することができる。中でも、ポリマー層の形成は、以下に示す本発明の太陽電池用ポリマーシートの製造方法により最も好適に行なえる。
[着色層]
本発明のポリマーシートには、前記複合ポリマーを実質的に含まない着色層(好ましくは反射層)が設けられてもよい。この場合、着色層(特に反射層)とポリエステル基材との間に複合ポリマーを含むポリマー層を設けることにより好適に構成することができる。この場合の着色層は、前記複合ポリマー以外のポリマー成分と顔料とを少なくとも含み、必要に応じて、さらに各種添加剤などの他の成分を用いて構成することができる。
なお、顔料及び各種添加剤の詳細については、ポリマー層が着色層として形成される場合について既述した通りである。複合ポリマー以外のポリマー成分については、特に制限はなく適宜目的等に応じて選択することができる。
本発明のポリマーシートには、前記複合ポリマーを実質的に含まない着色層(好ましくは反射層)が設けられてもよい。この場合、着色層(特に反射層)とポリエステル基材との間に複合ポリマーを含むポリマー層を設けることにより好適に構成することができる。この場合の着色層は、前記複合ポリマー以外のポリマー成分と顔料とを少なくとも含み、必要に応じて、さらに各種添加剤などの他の成分を用いて構成することができる。
なお、顔料及び各種添加剤の詳細については、ポリマー層が着色層として形成される場合について既述した通りである。複合ポリマー以外のポリマー成分については、特に制限はなく適宜目的等に応じて選択することができる。
前記「実質的に含まない」とは、着色層中に複合ポリマーを積極的に含有しないことを意味し、具体的には、着色層中における複合ポリマーの含有量が15質量%以下であることをいい、好ましくは複合ポリマーを含有しない(含有量が0(ゼロ)質量%である)場合が好ましい。
ポリエステル基材上に反射層を設けるときには、上記のように、反射層が複合ポリマーを含有する態様に限らず、複合ポリマーを実質的に含まない反射層とポリエステル基材との間に1層又は2層以上のポリマー層が設けられた態様に構成されてもよい。この場合、ポリエステル基材と着色層との間に複合ポリマーを含むポリマー層を設けることにより、反射層とポリエステル基材との間の接着性、密着性を向上させ、耐水性をより高めることができる。これにより、密着不良に起因する耐候性の悪化が防止される。
<太陽電池用ポリマーシートの製造>
本発明の太陽電池用ポリマーシートは、上記のように、既述のポリエステル基材の上に本発明におけるポリマー層と、必要に応じて着色層、下塗層等を形成することができる方法であればいずれの方法により作製されてもよい。本発明においては、カルボキシル基の含有量が15当量/t以下であり、示差走査熱量測定により求められる微少吸熱ピーク温度Tmeta(℃)が220℃以下であり、温度125℃、相対湿度100%RHの条件下72時間放置後の平均伸度保持率が10%以上であるポリエステル基材上に、分子中に質量割合が15~85質量%の前記一般式(1)で表されるシロキサン構造単位と質量割合が85~15質量%の非シロキサン系構造単位とを含む複合ポリマー及び好ましくは架橋剤を含有する塗布液(及び必要に応じて易接着性層用塗布液等)を塗布し、少なくとも1層のポリマー層を形成する工程を設けて作製する方法(本発明の太陽電池用ポリマーシートの製造方法)により好適に作製することができる。
なお、ポリマー層用塗布液は、既述のように少なくとも複合ポリマーを含有する塗布液である。ポリエステル基材、及び各塗布液を構成する複合ポリマー及び他の成分などの詳細については、既述の通りである。
本発明の太陽電池用ポリマーシートは、上記のように、既述のポリエステル基材の上に本発明におけるポリマー層と、必要に応じて着色層、下塗層等を形成することができる方法であればいずれの方法により作製されてもよい。本発明においては、カルボキシル基の含有量が15当量/t以下であり、示差走査熱量測定により求められる微少吸熱ピーク温度Tmeta(℃)が220℃以下であり、温度125℃、相対湿度100%RHの条件下72時間放置後の平均伸度保持率が10%以上であるポリエステル基材上に、分子中に質量割合が15~85質量%の前記一般式(1)で表されるシロキサン構造単位と質量割合が85~15質量%の非シロキサン系構造単位とを含む複合ポリマー及び好ましくは架橋剤を含有する塗布液(及び必要に応じて易接着性層用塗布液等)を塗布し、少なくとも1層のポリマー層を形成する工程を設けて作製する方法(本発明の太陽電池用ポリマーシートの製造方法)により好適に作製することができる。
なお、ポリマー層用塗布液は、既述のように少なくとも複合ポリマーを含有する塗布液である。ポリエステル基材、及び各塗布液を構成する複合ポリマー及び他の成分などの詳細については、既述の通りである。
好適な塗布法も既述の通りであり、例えばグラビアコーターやバーコーターを利用することができる。また、本発明における塗布工程では、ポリエステル基材の表面に直にあるいは厚み2μm以下の下塗り層を介して、ポリマー層用塗布液を塗布し、ポリエステル基材上にポリマー層(例えば着色層(好ましくは反射層)やバック層)を形成することができる。
ポリマー層の形成は、シート状のポリマー部材をポリエステル基材に貼合する方法、ポリエステル基材形成時にポリマー層を共押出しする方法、塗布による方法等により行なえる。中でも、塗布による方法は、簡便であると共に、均一性で薄膜での形成が可能である点で好ましい。塗布による場合、塗布方法としては、例えば、グラビアコーター、バーコーターなどの公知の塗布方法を利用することができる。
塗布液は、塗布溶媒として水を用いた水系でもよいし、トルエンやメチルエチルケトン等の有機溶媒を用いた溶剤系でもよい。中でも、環境負荷の観点から、水を溶媒とすることが好ましい。塗布溶媒は、1種類を単独で用いてもよいし、2種類以上を混合して用いてもよい。
ポリマー層用塗布液としては、これに含まれる溶媒中の50質量%以上、好ましくは60質量%以上が水である水系塗布液であることが好ましい。水系塗布液は、環境負荷の点で好ましく、また水の割合が50質量%以上であることにより、環境負荷が特に小さくなる点で有利である。ポリマー層用塗布液中の水の割合は、環境負荷の観点からは、さらに多い方が望ましく、水が全溶媒の90質量%以上含まれる場合がより好ましい。
塗布後は、所望の条件で乾燥を行なう乾燥工程が設けられてもよい。
<太陽電池用バックシート>
本発明の太陽電池用バックシートは、太陽電池素子が封止材で封止された電池側基板の前記封止剤と接触させて配置される太陽電池用バックシートであって、既述の本発明の太陽電池用ポリマーシート、または、既述の本発明の太陽電池用ポリマーシートの製造方法により製造された太陽電池用ポリマーシートを用いて構成される。
例えば、本発明の太陽電池用ポリマーシートをそのまま太陽電池用ポリマーシートとして用いてもよいし、本発明の太陽電池用ポリマーシートに、後述する易接着性層やバリア層を備えて構成されていてもよい。
本発明の太陽電池用バックシートは、太陽電池素子が封止材で封止された電池側基板の前記封止剤と接触させて配置される太陽電池用バックシートであって、既述の本発明の太陽電池用ポリマーシート、または、既述の本発明の太陽電池用ポリマーシートの製造方法により製造された太陽電池用ポリマーシートを用いて構成される。
例えば、本発明の太陽電池用ポリマーシートをそのまま太陽電池用ポリマーシートとして用いてもよいし、本発明の太陽電池用ポリマーシートに、後述する易接着性層やバリア層を備えて構成されていてもよい。
また、本発明の太陽電池用バックシートは、2つ以上の本発明の太陽電池用ポリマーシートを備えて構成されていてもよく、この場合、太陽電池用ポリマーシートと太陽電池用ポリマーシートとが粘着剤により貼り合わせられて構成されていることも好ましい態様である。接着剤としては、例えば、LX660(K)〔DIC(株)製接着剤〕に、硬化剤KW75〔DIC(株)製接着剤〕を10部混合したものが用いられる。貼り合わせた太陽電池用ポリマーシート積層体は、さらに、例えば、真空ラミネータ〔日清紡(株)製真空ラミネート機〕等のプレス装置を用いてホットプレス接着してもよい。
[易接着性層]
本発明のバックシートには、前記ポリエステル基材の前記ポリマー層が設けられている面とは反対側の表面、または、前記ポリマー層(特に反射層)の上に、さらに易接着性層が設けられていてもよい。易接着性層は、バックシートを電池側基板(電池本体)の太陽電池素子(以下、発電素子ともいう)を封止する封止材と強固に接着するための層である。
本発明のバックシートには、前記ポリエステル基材の前記ポリマー層が設けられている面とは反対側の表面、または、前記ポリマー層(特に反射層)の上に、さらに易接着性層が設けられていてもよい。易接着性層は、バックシートを電池側基板(電池本体)の太陽電池素子(以下、発電素子ともいう)を封止する封止材と強固に接着するための層である。
易接着性層は、バインダー、無機微粒子を用いて構成することができ、必要に応じて、さらに添加剤などの他の成分を含んで構成されてもよい。易接着性層は、電池側基板の発電素子を封止する封止材{例えばエチレン-ビニルアセテート(EVA;エチレン-酢酸ビニル)共重合体}、ポリビニルブチラール(PVB)、エポキシ樹脂等)に対して、5N/cm以上の接着力を有するように構成されていることが好ましい。接着力が5N/cm以上であると、接着性を維持し得る湿熱耐性が得られやすい。接着力は、10N/cm以上であることが好ましく、さらに20N/cm以上であることが好ましい。
なお、接着力は、易接着性層中のバインダー及び無機微粒子の量を調節する方法、バックシートの封止材と接着する面にコロナ処理を施す方法などにより調整が可能である。
-バインダー-
易接着性層は、バインダーの少なくとも一種を含有することができる。
易接着性層に好適なバインダーとしては、例えば、ポリエステル、ポリウレタン、アクリル樹脂、ポリオレフィン等が挙げられ、中でも耐久性の観点から、アクリル樹脂、ポリオレフィンが好ましい。また、アクリル樹脂として、アクリルとシリコーンとの複合樹脂も好ましい。
易接着性層は、バインダーの少なくとも一種を含有することができる。
易接着性層に好適なバインダーとしては、例えば、ポリエステル、ポリウレタン、アクリル樹脂、ポリオレフィン等が挙げられ、中でも耐久性の観点から、アクリル樹脂、ポリオレフィンが好ましい。また、アクリル樹脂として、アクリルとシリコーンとの複合樹脂も好ましい。
好ましいバインダーの例としては、ポリオレフィンの具体例としてケミパールS-120、S-75N(ともに三井化学(株)製)、アクリル樹脂の具体例としてジュリマーET-410、SEK-301(ともに日本純薬(株)製)、アクリルとシリコーンとの複合樹脂の具体例としてセラネートWSA1060、WSA1070(ともにDIC(株)製)とH7620、H7630、H7650(ともに旭化成ケミカルズ(株)製)などを挙げることができる。
バインダーの易接着性層中における含有量は、0.05~5g/m2の範囲とすることが好ましい。中でも、0.08~3g/m2の範囲がより好ましい。バインダーの含有量は、0.05g/m2以上であると所望とする接着力が得られやすく、5g/m2以下であるとより良好な面状が得られる。
-微粒子-
易接着性層は、無機微粒子の少なくとも一種を含有することができる。
無機微粒子としては、例えば、シリカ、炭酸カルシウム、酸化マグネシウム、炭酸マグネシウム、酸化錫等が挙げられる。中でも、湿熱雰囲気に曝されたときの接着性の低下が小さい点で、酸化錫、シリカの微粒子が好ましい。
易接着性層は、無機微粒子の少なくとも一種を含有することができる。
無機微粒子としては、例えば、シリカ、炭酸カルシウム、酸化マグネシウム、炭酸マグネシウム、酸化錫等が挙げられる。中でも、湿熱雰囲気に曝されたときの接着性の低下が小さい点で、酸化錫、シリカの微粒子が好ましい。
無機微粒子の粒径は、体積平均粒径で10~700nm程度が好ましく、より好ましくは20~300nm程度である。粒径がこの範囲内であると、より良好な易接着性を得ることができる。粒径は、レーザー解析/散乱式粒子径分布測定装置LA950〔(株)堀場製作所製〕により測定される値である。
無機微粒子の形状には、特に制限はなく、球形、不定形、針状形等のいずれのものを用いることができる。
無機微粒子の含有量は、易接着性層中のバインダーに対して、5~400質量%の範囲とする。無機微粒子の含有量は、5質量%未満であると、湿熱雰囲気に曝されたときに良好な接着性が保持できず、400質量%を超えると、易接着性層の面状が悪化する。
中でも、無機微粒子の含有量は、50~300質量%の範囲が好ましい。
中でも、無機微粒子の含有量は、50~300質量%の範囲が好ましい。
-架橋剤-
易接着性層には、架橋剤の少なくとも一種を含有することができる。
易接着性層に好適な架橋剤としては、エポキシ系、イソシアネート系、メラミン系、カルボジイミド系、オキサゾリン系等の架橋剤を挙げることができる。中でも、湿熱経時後の接着性を確保する観点から、オキサゾリン系架橋剤が特に好ましい。オキサゾリン系架橋剤の具体例については、既述のポリマー層の項で説明した具体例と同様のものが挙げられる。
易接着性層には、架橋剤の少なくとも一種を含有することができる。
易接着性層に好適な架橋剤としては、エポキシ系、イソシアネート系、メラミン系、カルボジイミド系、オキサゾリン系等の架橋剤を挙げることができる。中でも、湿熱経時後の接着性を確保する観点から、オキサゾリン系架橋剤が特に好ましい。オキサゾリン系架橋剤の具体例については、既述のポリマー層の項で説明した具体例と同様のものが挙げられる。
架橋剤の易接着性層中における含有量としては、易接着性層中のバインダーに対して、5~50質量%が好ましく、中でもより好ましくは20~40質量%である。架橋剤の含有量は、5質量%以上であると、良好な架橋効果が得られ、着色層の強度や接着性を保持することができ、50質量%以下であると、塗布液のポットライフを長く保つことができる。
-添加剤-
本発明における易接着性層には、必要に応じて、更に、ポリスチレン、ポリメチルメタクリレート、シリカ等の公知のマット剤、アニオン系やノニオン系などの公知の界面活性剤などを添加してもよい。
本発明における易接着性層には、必要に応じて、更に、ポリスチレン、ポリメチルメタクリレート、シリカ等の公知のマット剤、アニオン系やノニオン系などの公知の界面活性剤などを添加してもよい。
~易接着性層の形成方法~
易接着性層の形成は、易接着性を有するシート状のポリマー部材を基材に貼合する方法や、塗布による方法が挙げられる。中でも、塗布による方法は、簡便であると共に、均一性で薄膜での形成が可能である点で好ましい。塗布方法としては、例えば、グラビアコーターやバーコーターなどの公知の塗布法を利用することができる。塗布液の調製に用いる塗布溶媒は、水でもよいし、トルエンやメチルエチルケトン等の有機溶媒でもよい。塗布溶媒は、1種類を単独で用いてもよいし、2種類以上を混合して用いてもよい。
易接着性層の形成は、易接着性を有するシート状のポリマー部材を基材に貼合する方法や、塗布による方法が挙げられる。中でも、塗布による方法は、簡便であると共に、均一性で薄膜での形成が可能である点で好ましい。塗布方法としては、例えば、グラビアコーターやバーコーターなどの公知の塗布法を利用することができる。塗布液の調製に用いる塗布溶媒は、水でもよいし、トルエンやメチルエチルケトン等の有機溶媒でもよい。塗布溶媒は、1種類を単独で用いてもよいし、2種類以上を混合して用いてもよい。
易接着性層の厚みには、特に制限はないが、通常は0.05~8μmが好ましく、より好ましくは0.1~5μmの範囲である。易接着性層の厚みは、0.05μm以上であると必要な易接着性を好適に得ることができ、8μm以下であると面状がより良好になる。
また、本発明の易接着性層は、着色層の効果を低減させないために、透明であることが必要である。
また、本発明の易接着性層は、着色層の効果を低減させないために、透明であることが必要である。
~物性~
また、本発明の太陽電池用バックシートは、120℃、100%RHの雰囲気下に48時間保存した後の封止材との接着力が、保存前の封止材との接着力に対し、75%以上であることが好ましい。本発明の太陽電池用バックシートは、既述の通り、所定量のバインダーと該バインダーに対して所定量の無機微粒子とを含み、EVA系封止材に対して10N/cm以上の接着力を持つ易接着層を有することにより、前記保存後にも保存前の75%以上の接着力が得られる。これにより、作製された太陽電池モジュールは、バックシートの剥がれやそれに伴う発電性能の低下が抑制され、長期耐久性がより向上する。
また、本発明の太陽電池用バックシートは、120℃、100%RHの雰囲気下に48時間保存した後の封止材との接着力が、保存前の封止材との接着力に対し、75%以上であることが好ましい。本発明の太陽電池用バックシートは、既述の通り、所定量のバインダーと該バインダーに対して所定量の無機微粒子とを含み、EVA系封止材に対して10N/cm以上の接着力を持つ易接着層を有することにより、前記保存後にも保存前の75%以上の接着力が得られる。これにより、作製された太陽電池モジュールは、バックシートの剥がれやそれに伴う発電性能の低下が抑制され、長期耐久性がより向上する。
[バリア層]
本発明の太陽電池用バックシートは、バリア層を有していることも好ましい。バリア層を設けることで、太陽電池用バックシートへの水や気体の浸入を防止することができる。バリア層の水蒸気透過量(透湿度)は、100g/m2・d~10-6g/m2・dが好ましく、より好ましくは10-1g/m2・d~10-5g/m2・dであり、さらに好ましくは10-2g/m2・d~10-4g/m2・dである。 尚、透湿度はJIS Z0208等に基づいて測定することができる。
バリア層の形成には、以下のような乾式法が好ましく用いられる。
本発明の太陽電池用バックシートは、バリア層を有していることも好ましい。バリア層を設けることで、太陽電池用バックシートへの水や気体の浸入を防止することができる。バリア層の水蒸気透過量(透湿度)は、100g/m2・d~10-6g/m2・dが好ましく、より好ましくは10-1g/m2・d~10-5g/m2・dであり、さらに好ましくは10-2g/m2・d~10-4g/m2・dである。 尚、透湿度はJIS Z0208等に基づいて測定することができる。
バリア層の形成には、以下のような乾式法が好ましく用いられる。
~バリア層の形成方法~
乾式法によりガスバリア層を形成する方法としては、抵抗加熱蒸着、電子ビーム蒸着、誘導加熱蒸着、及びこれらにプラズマやイオンビームによるアシスト法などの真空蒸着法、反応性スパッタリング法、イオンビームスパッタリング法、ECR(電子サイクロトロン)スパッタリング法などのスパッタリング法、イオンプレーティング法などの物理的気相成長法(PVD法)、熱や光、プラズマなどを利用した化学的気相成長法(CVD法)などが挙げられる。中でも、真空下で蒸着法により膜形成する真空蒸着法が好ましい。
乾式法によりガスバリア層を形成する方法としては、抵抗加熱蒸着、電子ビーム蒸着、誘導加熱蒸着、及びこれらにプラズマやイオンビームによるアシスト法などの真空蒸着法、反応性スパッタリング法、イオンビームスパッタリング法、ECR(電子サイクロトロン)スパッタリング法などのスパッタリング法、イオンプレーティング法などの物理的気相成長法(PVD法)、熱や光、プラズマなどを利用した化学的気相成長法(CVD法)などが挙げられる。中でも、真空下で蒸着法により膜形成する真空蒸着法が好ましい。
ここで、ガスバリア層が、無機酸化物、無機窒化物、無機酸窒化物、無機ハロゲン化物、無機硫化物などを主たる構成成分とする材料で形成される無機層である場合は、形成するガスバリア層の組成と同一の材料を直接揮発させて基材などに堆積させることも可能であるが、この方法で行なう場合には、揮発中に組成が変化し、その結果、形成された膜が均一な特性を呈さない場合がある。そのため、1)揮発源として形成するバリア層と同一組成の材料を用い、無機酸化物の場合は酸素ガスを、無機窒化物の場合は窒素ガスを、無機酸窒化物の場合は酸素ガスと窒素ガスの混合ガスを、無機ハロゲン化物の場合はハロゲン系ガスを、無機硫化物の場合は硫黄系ガスを、それぞれ系内に補助的に導入しながら揮発させる方法、2)揮発源として無機物群を用い、これを揮発させながら、無機酸化物の場合は酸素ガスを、無機窒化物の場合は窒素ガスを、無機酸窒化物の場合は酸素ガスと窒素ガスの混合ガスを、無機ハロゲン化物の場合はハロゲン系ガスを、無機硫化物の場合は硫黄系ガスを、それぞれ系内に導入し、無機物と導入したガスを反応させながら基材表面に堆積させる方法、3)揮発源として無機物群を用い、これを揮発させて、無機物群の層を形成させた後、それを無機酸化物の場合は酸素ガス雰囲気下、無機窒化物の場合は窒素ガス雰囲気下、無機酸窒化物の場合は酸素ガスと窒素ガスの混合ガス雰囲気下、無機ハロゲン化物の場合はハロゲン系ガス雰囲気下、無機硫化物の場合は硫黄系ガス雰囲気下で保持することにより無機物層と導入したガスを反応させる方法、等が挙げられる。
これらのうち、揮発源から揮発させることが容易であるという点で、2)又は3)がより好ましく用いられる。さらには、膜質の制御が容易である点で2)の方法が更に好ましく用いられる。また、バリア層が無機酸化物の場合は、揮発源として無機物群を用い、これを揮発させて、無機物群の層を形成させた後、空気中で放置することで、無機物群を自然酸化させる方法も、形成が容易であるという点で好ましい。
また、アルミ箔を貼り合わせてバリア層として使用することも好ましい。厚みは、1μm以上30μm以下が好ましい。バリア層の厚みは、1μm以上であると、経時(サーモ)中にポリエステルフィルム中に水が浸透し難くなって加水分解を生じ難く、30μm以下であると、バリア層の厚みが厚くなり過ぎず、バリア層の応力でフィルムにベコが発生することもない。
<太陽電池モジュール>
本発明の太陽電池モジュールは、既述の本発明の太陽電池用バックシート、又は既述の太陽電池用バックシートの製造方法により製造された太陽電池用バックシートを設けて構成されている。本発明の好ましい形態として、太陽光の光エネルギーを電気エネルギーに変換する太陽電池素子を、太陽光が入射する透明性のフロント基板と既述の本発明の太陽電池用バックシートとの間に配置し、該フロント基板とバックシートとの間で太陽電池素子をエチレン-ビニルアセテート系等の封止材で封止、接着して構成されている。すなわち、フロント基板とバックシートとの間に、太陽電池素子及び前記太陽電池素子を封止する封止材を有するセル構造部分が設けられている。
本発明の太陽電池モジュールは、既述の本発明の太陽電池用バックシート、又は既述の太陽電池用バックシートの製造方法により製造された太陽電池用バックシートを設けて構成されている。本発明の好ましい形態として、太陽光の光エネルギーを電気エネルギーに変換する太陽電池素子を、太陽光が入射する透明性のフロント基板と既述の本発明の太陽電池用バックシートとの間に配置し、該フロント基板とバックシートとの間で太陽電池素子をエチレン-ビニルアセテート系等の封止材で封止、接着して構成されている。すなわち、フロント基板とバックシートとの間に、太陽電池素子及び前記太陽電池素子を封止する封止材を有するセル構造部分が設けられている。
太陽電池モジュール、太陽電池セル、バックシート以外の部材については、例えば、「太陽光発電システム構成材料」(杉本栄一監修、(株)工業調査会、2008年発行)に詳細に記載されている。
透明性の基板は、太陽光が透過し得る光透過性を有していればよく、光を透過する基材から適宜選択することができる。発電効率の観点からは、光の透過率が高いものほど好ましく、このような基板として、例えば、ガラス基板、アクリル樹脂などの透明樹脂などを好適に用いることができる。
太陽電池素子としては、単結晶シリコン、多結晶シリコン、アモルファスシリコンなどのシリコン系、銅-インジウム-ガリウム-セレン、銅-インジウム-セレン、カドミウム-テルル、ガリウム-砒素などのIII-V族やII-VI族化合物半導体系など、各種公知の太陽電池素子を適用することができる。
以下、本発明を実施例により更に具体的に説明するが、本発明はその主旨を越えない限り、以下の実施例に限定されるものではない。なお、実施例中の「部」は、質量基準である。
<ポリマー基材の作製>
~PET-1の作製~
[工程1]
テレフタル酸ジメチル100部、トリメリット酸トリメチル(テレフタル酸ジメチル/トリメリット酸トリメチル=99.7/0.3のモル比となるように添加)、エチレングリコール57.5部、酢酸マグネシウム0.06部、三酸化アンチモン0.03部を150℃、窒素雰囲気下で溶融後、攪拌しながら230℃まで3時間かけて昇温し、メタノールを留出させ、エステル交換反応を終了した。
[工程2]
エステル交換反応終了後、リン酸0.019部(1.9モル/t相当)とリン酸二水素ナトリウム2水和物0.027部(1.5モル/t相当)をエチレングリコール0.5部に溶解したエチレングリコール溶液(PH5.0)を添加した。
[工程3]
重合反応を最終到達温度285℃、真空度0.1Torrで行い、固有粘度0.54、カルボキシル基末端基数13eq/tonのポリエステルを得た。
[工程4]
得られたポリエチレンテレフタレートを160℃で6時間乾燥、結晶化させたのち、220℃、真空度0.3Torr、9時間の固相重合を行い、構成成分(p)が0.15モル%、固有粘度0.90、カルボキシル基末端基数12eq/ton、融点255℃、ガラス転移温度Tg83℃のポリエステルを得た。
~PET-1の作製~
[工程1]
テレフタル酸ジメチル100部、トリメリット酸トリメチル(テレフタル酸ジメチル/トリメリット酸トリメチル=99.7/0.3のモル比となるように添加)、エチレングリコール57.5部、酢酸マグネシウム0.06部、三酸化アンチモン0.03部を150℃、窒素雰囲気下で溶融後、攪拌しながら230℃まで3時間かけて昇温し、メタノールを留出させ、エステル交換反応を終了した。
[工程2]
エステル交換反応終了後、リン酸0.019部(1.9モル/t相当)とリン酸二水素ナトリウム2水和物0.027部(1.5モル/t相当)をエチレングリコール0.5部に溶解したエチレングリコール溶液(PH5.0)を添加した。
[工程3]
重合反応を最終到達温度285℃、真空度0.1Torrで行い、固有粘度0.54、カルボキシル基末端基数13eq/tonのポリエステルを得た。
[工程4]
得られたポリエチレンテレフタレートを160℃で6時間乾燥、結晶化させたのち、220℃、真空度0.3Torr、9時間の固相重合を行い、構成成分(p)が0.15モル%、固有粘度0.90、カルボキシル基末端基数12eq/ton、融点255℃、ガラス転移温度Tg83℃のポリエステルを得た。
[工程5]
工程4にて得られたポリエステル 99部に対して、ラインケミー社製スタバクゾールP100」(ポリカルボジイミド)を1部加えてコンパウンドした。
[工程6]
上記で得られたコンパウンド品を温度180℃、真空度0.5mmHgの条件下、2時間の減圧乾燥を行い、295℃に加熱した押出機に供給し、50μmカットフィルターにより異物濾過を行ったのちにTダイ口金に導入した。次いで、Tダイ口金内より、シート状に押出して溶融単層シートとし、該溶融単層シートを、表面温度20℃に保たれたドラム上に静電印加法で密着冷却固化させて未延伸単層フィルムを得た。
[工程7]
続いて、得られた未延伸単層フィルムを加熱したロール群で予熱した後、80℃の温度で1.8倍MD延伸1を行い、さらに95℃の温度で2.3倍MD延伸2を行った。トータルで長手方向(MD方向)に4.1倍延伸を行った後、25℃の温度のロール群で冷却して一軸延伸フィルムを得た。得られた一軸延伸フィルムの両端をクリップで把持しながらテンター内の95℃の温度の予熱ゾーンに導き、引き続き連続的に100℃の温度の加熱ゾーンで長手方向に直角な幅方向(TD方向)に4.0倍延伸した。
[工程8]
さらに引き続いて、テンター内の熱処理ゾーンで205℃の温度(第1熱処理温度)で20秒間の熱処理を施した。引き続き、180℃の温度下において、フィルムを幅方向(TD)に3%の弛緩率にて弛緩させ、また、テンターのクリップ間隔を縮めることによって、長手方向(MDに1.5%の弛緩率にて弛緩させた。次いで、25℃まで均一に冷却後巻取り、厚さ250μmの二軸延伸ポリエステルフィルム(PET-1)を得た。
尚、弛緩率は、弛緩前のポリエステルフィルムの長さをLa、弛緩後のポリエステルフィルムの長さをLbとしたとき、下記式(c)により算出される。
式(c) 100×(La-Lb)/La
なお、ポリエステルフィルムの幅方向のLaおよびLb、並びに、ポリエステルフィルムの長手方向のLaおよびLbは、次のように定義する。
[幅方向]
テンターでポリエステルフィルムに緊張を与えて延伸したときの、延伸時におけるポリエステルフィルムの最大の幅を、弛緩前のポリエステルフィルムの長さLaとする。また、緊張を解いて(弛緩して)ポリエステルフィルムをテンターから取り出すときのポリエステルフィルムの幅の長さを、弛緩後のポリエステルフィルムの長さLbとする。
[長手方向]
テンターでポリエステルフィルムに緊張を与えて延伸したときの、延伸時におけるポリエステルフィルムに、長手方向に2点の印をつけ、その2点間の距離を弛緩前のポリエステルフィルムの長さLaとする。また、緊張を解いて(弛緩して)テンターから取り出した後の前記2点間の距離を弛緩後のポリエステルフィルムの長さLbとする。
工程4にて得られたポリエステル 99部に対して、ラインケミー社製スタバクゾールP100」(ポリカルボジイミド)を1部加えてコンパウンドした。
[工程6]
上記で得られたコンパウンド品を温度180℃、真空度0.5mmHgの条件下、2時間の減圧乾燥を行い、295℃に加熱した押出機に供給し、50μmカットフィルターにより異物濾過を行ったのちにTダイ口金に導入した。次いで、Tダイ口金内より、シート状に押出して溶融単層シートとし、該溶融単層シートを、表面温度20℃に保たれたドラム上に静電印加法で密着冷却固化させて未延伸単層フィルムを得た。
[工程7]
続いて、得られた未延伸単層フィルムを加熱したロール群で予熱した後、80℃の温度で1.8倍MD延伸1を行い、さらに95℃の温度で2.3倍MD延伸2を行った。トータルで長手方向(MD方向)に4.1倍延伸を行った後、25℃の温度のロール群で冷却して一軸延伸フィルムを得た。得られた一軸延伸フィルムの両端をクリップで把持しながらテンター内の95℃の温度の予熱ゾーンに導き、引き続き連続的に100℃の温度の加熱ゾーンで長手方向に直角な幅方向(TD方向)に4.0倍延伸した。
[工程8]
さらに引き続いて、テンター内の熱処理ゾーンで205℃の温度(第1熱処理温度)で20秒間の熱処理を施した。引き続き、180℃の温度下において、フィルムを幅方向(TD)に3%の弛緩率にて弛緩させ、また、テンターのクリップ間隔を縮めることによって、長手方向(MDに1.5%の弛緩率にて弛緩させた。次いで、25℃まで均一に冷却後巻取り、厚さ250μmの二軸延伸ポリエステルフィルム(PET-1)を得た。
尚、弛緩率は、弛緩前のポリエステルフィルムの長さをLa、弛緩後のポリエステルフィルムの長さをLbとしたとき、下記式(c)により算出される。
式(c) 100×(La-Lb)/La
なお、ポリエステルフィルムの幅方向のLaおよびLb、並びに、ポリエステルフィルムの長手方向のLaおよびLbは、次のように定義する。
[幅方向]
テンターでポリエステルフィルムに緊張を与えて延伸したときの、延伸時におけるポリエステルフィルムの最大の幅を、弛緩前のポリエステルフィルムの長さLaとする。また、緊張を解いて(弛緩して)ポリエステルフィルムをテンターから取り出すときのポリエステルフィルムの幅の長さを、弛緩後のポリエステルフィルムの長さLbとする。
[長手方向]
テンターでポリエステルフィルムに緊張を与えて延伸したときの、延伸時におけるポリエステルフィルムに、長手方向に2点の印をつけ、その2点間の距離を弛緩前のポリエステルフィルムの長さLaとする。また、緊張を解いて(弛緩して)テンターから取り出した後の前記2点間の距離を弛緩後のポリエステルフィルムの長さLbとする。
PET-1の特性を評価した結果を以下に示す。
・末端カルボキシル基含有量:5eq/t
・Tmeta :190℃
・平均伸度保持率 :50%
・面配向係数 :0.170
・固有粘度 :0.75dl/g
・熱収縮率(MD/TD) :0.4%/0.2%
・構成成分(p)含有量 :0.15モル%
・緩衝剤:リン酸二水素ナトリウム 1.5モル/t
・末端封止剤:ポリカルボジイミド 1重量%
・リン原子の含有量 :230ppm
・末端カルボキシル基含有量:5eq/t
・Tmeta :190℃
・平均伸度保持率 :50%
・面配向係数 :0.170
・固有粘度 :0.75dl/g
・熱収縮率(MD/TD) :0.4%/0.2%
・構成成分(p)含有量 :0.15モル%
・緩衝剤:リン酸二水素ナトリウム 1.5モル/t
・末端封止剤:ポリカルボジイミド 1重量%
・リン原子の含有量 :230ppm
~PET-2の作製~
PET-1の製造方法の[工程8]について、第1熱処理温度を230℃に変更した以外は、PET-1と同様の方法で二軸延伸ポリエステルフィルム(PET-2)を作製した。
PET-2の特性を評価したところ、PET-1と比較して、Tmetaが225℃に、平均伸度保持率が7%に変化した。
PET-1の製造方法の[工程8]について、第1熱処理温度を230℃に変更した以外は、PET-1と同様の方法で二軸延伸ポリエステルフィルム(PET-2)を作製した。
PET-2の特性を評価したところ、PET-1と比較して、Tmetaが225℃に、平均伸度保持率が7%に変化した。
~PET-3の作製~
PET-1の製造方法の[工程2]において、リン酸二水素ナトリウム2水和物を添加しなかったこと以外は、PET-1と同様の方法で二軸延伸ポリエステルフィルム(PET-3)を作製した。
PET-3の特性を評価したところ、PET-1と比較して平均伸度保持率が40%に、リン原子の含有量が150ppmに変化した。
PET-1の製造方法の[工程2]において、リン酸二水素ナトリウム2水和物を添加しなかったこと以外は、PET-1と同様の方法で二軸延伸ポリエステルフィルム(PET-3)を作製した。
PET-3の特性を評価したところ、PET-1と比較して平均伸度保持率が40%に、リン原子の含有量が150ppmに変化した。
なお、PET-1の特性は、次の方法により測定した。
-カルボキシル基の含有量(AV)-
カルボキシル基の含有量(AV)は、ポリエステルをベンジルアルコール/クロロホルム(=2/3;体積比)の混合溶液に完全溶解させ、指示薬としてフェノールレッドを用い、これを基準液(0.01N KOH-ベンジルアルコール混合溶液)で滴定し、その滴定量から算出した。
-カルボキシル基の含有量(AV)-
カルボキシル基の含有量(AV)は、ポリエステルをベンジルアルコール/クロロホルム(=2/3;体積比)の混合溶液に完全溶解させ、指示薬としてフェノールレッドを用い、これを基準液(0.01N KOH-ベンジルアルコール混合溶液)で滴定し、その滴定量から算出した。
-示差走査熱量測定(DSC)により求められる微少吸熱ピーク温度Tmeta(℃)-
微少吸熱ピーク温度Tmeta(℃)は、JIS K7122-1987(JISハンドブック1999年版を参照した)に準じて、セイコー電子工業(株)製示差走査熱量測定装置”ロボットDSC-RDC220”を、データ解析にはディスクセッション”SSC/5200”を用いて測定した。具体的には、サンプルパンにフィルムを5mg秤量し、25℃から300℃まで20℃/分の昇温速度で昇温を行って測定した。
得られた示差走査熱量測定チャートにおける結晶融解ピーク前の微少吸熱ピーク温度でもってTmeta(℃)とした。微小な吸熱のピークが観測しにくい場合は、データ解析部にてピーク付近を拡大して、ピークを読みとった。
微少吸熱ピーク温度Tmeta(℃)は、JIS K7122-1987(JISハンドブック1999年版を参照した)に準じて、セイコー電子工業(株)製示差走査熱量測定装置”ロボットDSC-RDC220”を、データ解析にはディスクセッション”SSC/5200”を用いて測定した。具体的には、サンプルパンにフィルムを5mg秤量し、25℃から300℃まで20℃/分の昇温速度で昇温を行って測定した。
得られた示差走査熱量測定チャートにおける結晶融解ピーク前の微少吸熱ピーク温度でもってTmeta(℃)とした。微小な吸熱のピークが観測しにくい場合は、データ解析部にてピーク付近を拡大して、ピークを読みとった。
なお、微小吸熱ピークのグラフ読み取り方法は、JISに記載されていないが、以下の方法に基づいて実施した。
まず、135℃の値と155℃の値で直線を引き、グラフの曲線との吸熱側の面積を求めた。同様に140℃と160℃、145℃と165℃、150℃と170℃、155℃と175℃、160℃と180℃、165℃と185℃、170℃と190℃、175℃と195℃、180℃と200℃、185℃と205℃、190℃と210℃、195℃と215℃、200℃と220℃、205℃と225℃、210℃と230℃、215℃と235℃、220℃と240℃の17点についても面積を求めた。微小ピークの吸熱量は、通常、0.2~5.0J/gであることから、面積が0.2J/g以上5.0J/g以下であるデータのみを有効データとして取り扱った。合計18個の面積データの中から、有効データでありかつ最も大きい面積を示すデータの温度領域おける吸熱ピークのピーク温度をもってTmeta(℃)とした。有効データがない場合、Tmeta(℃)はなしとした。
まず、135℃の値と155℃の値で直線を引き、グラフの曲線との吸熱側の面積を求めた。同様に140℃と160℃、145℃と165℃、150℃と170℃、155℃と175℃、160℃と180℃、165℃と185℃、170℃と190℃、175℃と195℃、180℃と200℃、185℃と205℃、190℃と210℃、195℃と215℃、200℃と220℃、205℃と225℃、210℃と230℃、215℃と235℃、220℃と240℃の17点についても面積を求めた。微小ピークの吸熱量は、通常、0.2~5.0J/gであることから、面積が0.2J/g以上5.0J/g以下であるデータのみを有効データとして取り扱った。合計18個の面積データの中から、有効データでありかつ最も大きい面積を示すデータの温度領域おける吸熱ピークのピーク温度をもってTmeta(℃)とした。有効データがない場合、Tmeta(℃)はなしとした。
-平均伸度保持率-
破断伸度の測定はASTM-D882-97(1999年版ANNUAL BOOK OF ASTM STANDARDSを参照した)に準じて、サンプルを1cm×20cmの大きさに切り出し、チャック間5cm、引っ張り速度300mm/minにて引っ張ったときの破断伸度(初期)を測定した。なお、測定は5サンプルについて測定を実施し、その平均値でもって破断伸度(初期)A2とした。
破断伸度の測定はASTM-D882-97(1999年版ANNUAL BOOK OF ASTM STANDARDSを参照した)に準じて、サンプルを1cm×20cmの大きさに切り出し、チャック間5cm、引っ張り速度300mm/minにて引っ張ったときの破断伸度(初期)を測定した。なお、測定は5サンプルについて測定を実施し、その平均値でもって破断伸度(初期)A2とした。
次いで、サンプルを1cm×20cmの大きさに切り出し、(株)平山製作所 高加速寿命試験装置(HAST装置)、PC-304R8Dを用いて、125℃、湿度100%の条件下72時間処理を行った後、処理後のサンプルの破断伸度をASTM-D882(1999)-97(1999年版ANNUAL BOOK OF ASTM STANDARDSを参照した)に準じて、チャック間5cm、引っ張り速度300mm/minにて引っ張ったときの破断伸度(処理後)を測定した。なお、測定は5サンプルについて測定を実施しその平均値でもって破断伸度(処理後)A3とした。
得られた破断伸度A2とA3を用いて、下記式(3)により伸度保持率(Lr)を算出した。
Lr(%)=A3/A2×100 (3)
Lr(%)=A3/A2×100 (3)
また、下記(4)により平均伸度保持率(Lave)を算出した。
(Lave)(%)=(LrMD+LrTD)/2 (4)
ここでLrMDはMD方向の伸度保持率を、LrTDはTD方向の伸度保持率を表す。
(Lave)(%)=(LrMD+LrTD)/2 (4)
ここでLrMDはMD方向の伸度保持率を、LrTDはTD方向の伸度保持率を表す。
-面配向係数(fPO)-
アタゴ社(株)製アッベ屈折率計Type 4Tを用い、光源をナトリウムランプとして、フィルム屈折率の測定を行った。
fPO= (nMD+nTD)/2 - nZD ・・・ (A)
上記式(A)におけるnMDはフィルムの長手方向(MD)の屈折率を表し、nTDはフィルムの直行方向(TD)の屈折率を表し、nZDはフィルム厚み方向の屈折率を表している。
アタゴ社(株)製アッベ屈折率計Type 4Tを用い、光源をナトリウムランプとして、フィルム屈折率の測定を行った。
fPO= (nMD+nTD)/2 - nZD ・・・ (A)
上記式(A)におけるnMDはフィルムの長手方向(MD)の屈折率を表し、nTDはフィルムの直行方向(TD)の屈折率を表し、nZDはフィルム厚み方向の屈折率を表している。
-固有粘度(IV)-
固有粘度(IV)は、溶液粘度(η)と溶媒粘度(η0)の比ηr(=η/η0;相対粘度)から1を引いた比粘度(ηsp=ηr-1)を濃度で割った値を濃度がゼロの状態に外挿した値である。IVは、ウベローデ型粘度計を用い、ポリエステルを1,1,2,2-テトラクロルエタン/フェノール(=2/3[質量比])混合溶媒に溶解させ、25℃の溶液粘度から求めた。
固有粘度(IV)は、溶液粘度(η)と溶媒粘度(η0)の比ηr(=η/η0;相対粘度)から1を引いた比粘度(ηsp=ηr-1)を濃度で割った値を濃度がゼロの状態に外挿した値である。IVは、ウベローデ型粘度計を用い、ポリエステルを1,1,2,2-テトラクロルエタン/フェノール(=2/3[質量比])混合溶媒に溶解させ、25℃の溶液粘度から求めた。
-熱収縮率(MD/TD)-
JIS-C2318(2007)に準じて、幅10mm、標線間隙約100mmのサンプルを、温度150℃、荷重0.5gで30分間熱処理した。その熱処理前後の標線間隙を(株)テクノニーズ製熱収縮率測定器(AMM-1号機)を用いて測定し、次式より熱収縮率を算出した。
Rts(%)={(L0-L)/L0}×100
Rts:熱収縮率
L0:加熱処理前の標線間隙
L :加熱処理後の標線間隙
JIS-C2318(2007)に準じて、幅10mm、標線間隙約100mmのサンプルを、温度150℃、荷重0.5gで30分間熱処理した。その熱処理前後の標線間隙を(株)テクノニーズ製熱収縮率測定器(AMM-1号機)を用いて測定し、次式より熱収縮率を算出した。
Rts(%)={(L0-L)/L0}×100
Rts:熱収縮率
L0:加熱処理前の標線間隙
L :加熱処理後の標線間隙
-リン原子の含有量-
蛍光X線法(リガク製ZSX100e)により、リン原子の含有量を測定した。
蛍光X線法(リガク製ZSX100e)により、リン原子の含有量を測定した。
[表面処理]
得られたPET-1~PET-3の各支持体の一方の面を、下記の条件でコロナ処理した。
装置 :ピラー社製ソリッドステートコロナ処理機6KVAモデル
電極と誘電体ロールギャップクリアランス:1.6mm
処理周波数:9.6kHz
処理速度 :20m/分
処理強度 :0.375kV・A・分/m2
得られたPET-1~PET-3の各支持体の一方の面を、下記の条件でコロナ処理した。
装置 :ピラー社製ソリッドステートコロナ処理機6KVAモデル
電極と誘電体ロールギャップクリアランス:1.6mm
処理周波数:9.6kHz
処理速度 :20m/分
処理強度 :0.375kV・A・分/m2
得られたPET-1~PET-3のカルボキシル基の含有量(AV)、Tmeta、平均伸度保持率、及び表面処理種を、表1に示す。

<ポリマーシートの作成>
支持体として表1に示すPET-1またはPET-2を用い、コロナ処理を施した面に、表1に示す構成のポリマー層(ポリマー層1、または、ポリマー層1及びポリマー層2)を有するポリマーシートを作成した。ポリマー層2を形成する場合は、支持体の上にポリマー層1を形成し、さらにポリマー層1の上にポリマー層2を形成した。
支持体として表1に示すPET-1またはPET-2を用い、コロナ処理を施した面に、表1に示す構成のポリマー層(ポリマー層1、または、ポリマー層1及びポリマー層2)を有するポリマーシートを作成した。ポリマー層2を形成する場合は、支持体の上にポリマー層1を形成し、さらにポリマー層1の上にポリマー層2を形成した。
なお、ポリマーシートにおいて、ポリマー層(ポリマー層1、または、ポリマー層1及びポリマー層2)が形成されている面を、ポリマーシートのオモテ面という。また、ポリマーシートのオモテ面とは反対の面を、ポリマーシートのウラ面という
支持体(PET-1~PET-3)において、ポリマー層1、または、ポリマー層1及びポリマー層2が形成されている面を、支持体のオモテ面という。また、支持体のオモテ面とは反対の面を、支持体のウラ面という
支持体(PET-1~PET-3)において、ポリマー層1、または、ポリマー層1及びポリマー層2が形成されている面を、支持体のオモテ面という。また、支持体のオモテ面とは反対の面を、支持体のウラ面という
ポリマー層1およびポリマー層2の形成には、次のP-1~P-5のバインダーを用いた。
P-1:セラネートWSA-1070(アクリル/シリコーン系バインダー)
〔DIC社製シリコーン系樹脂、固形分:40質量%〕
P-2:セラネートWSA-1060(アクリル/シリコーン系バインダー)
〔DIC社製シリコーン系樹脂、固形分:40質量%〕
P-3:オブリガートSW0011F
〔AGCコーテック社製フッ素樹脂、固形分:40質量%〕
P-4:ファインテックスEs650
〔DIC社製ポリエステル樹脂、固形分:29質量%〕
P-5:オレスターUD350
〔三井化学社製ポリウレタン樹脂、固形分:38質量%〕
P-1:セラネートWSA-1070(アクリル/シリコーン系バインダー)
〔DIC社製シリコーン系樹脂、固形分:40質量%〕
P-2:セラネートWSA-1060(アクリル/シリコーン系バインダー)
〔DIC社製シリコーン系樹脂、固形分:40質量%〕
P-3:オブリガートSW0011F
〔AGCコーテック社製フッ素樹脂、固形分:40質量%〕
P-4:ファインテックスEs650
〔DIC社製ポリエステル樹脂、固形分:29質量%〕
P-5:オレスターUD350
〔三井化学社製ポリウレタン樹脂、固形分:38質量%〕
ここで、シリコーン系樹脂(複合ポリマー)であるP-1およびP-2の分子構成は次のとおりである。
P-1は、ポリシロキサン部位が約30質量%であり、アクリル系ポリマー部分が約70質量%である。
P-2は、ポリシロキサン部位が約75質量%であり、アクリル系ポリマー部分が約25質量%である。
P-1は、ポリシロキサン部位が約30質量%であり、アクリル系ポリマー部分が約70質量%である。
P-2は、ポリシロキサン部位が約75質量%であり、アクリル系ポリマー部分が約25質量%である。
〔実施例1〕
-顔料分散物の調製-
下記組成中の各成分を混合し、その混合物をダイノミル型分散機により1時間、分散処理を施した。
(顔料分散物の組成)
・二酸化チタン(体積平均粒子径=0.42μm)・・・40質量%
(タイペークR-780-2、石原産業(株)製、固形分100質量%)
・ポリビニルアルコール水溶液(10質量%)・・・20.0質量%
(PVA-105、(株)クラレ製)
・界面活性剤(デモールEP、花王(株)製、固形分:25質量%)・・・0.5質量%
・蒸留水・・・39.5質量%
-顔料分散物の調製-
下記組成中の各成分を混合し、その混合物をダイノミル型分散機により1時間、分散処理を施した。
(顔料分散物の組成)
・二酸化チタン(体積平均粒子径=0.42μm)・・・40質量%
(タイペークR-780-2、石原産業(株)製、固形分100質量%)
・ポリビニルアルコール水溶液(10質量%)・・・20.0質量%
(PVA-105、(株)クラレ製)
・界面活性剤(デモールEP、花王(株)製、固形分:25質量%)・・・0.5質量%
・蒸留水・・・39.5質量%
-ポリマー層1形成用塗布液の調製-
下記組成中の各成分を混合し、ポリマー層1形成用塗布液1-P1を調製した。
(塗布液の組成)
・バインダー(P-1)・・・362.3部
(セラネートWSA-1070、DIC(株)製、固形分:40質量%)
・カルボジイミド化合物(架橋剤)・・・36.2部
(カルボジライトV-02-L2、日清紡績(株)製、固形分:40質量%)
・界面活性剤・・・9.7部
(ナロアクティーCL95、三洋化成工業(株)製、固形分:1質量%)
・上記顔料分散物・・・157.0質量%
・蒸留水・・・434.8部
下記組成中の各成分を混合し、ポリマー層1形成用塗布液1-P1を調製した。
(塗布液の組成)
・バインダー(P-1)・・・362.3部
(セラネートWSA-1070、DIC(株)製、固形分:40質量%)
・カルボジイミド化合物(架橋剤)・・・36.2部
(カルボジライトV-02-L2、日清紡績(株)製、固形分:40質量%)
・界面活性剤・・・9.7部
(ナロアクティーCL95、三洋化成工業(株)製、固形分:1質量%)
・上記顔料分散物・・・157.0質量%
・蒸留水・・・434.8部
-ポリマー層1の形成-
得られたポリマー層1形成用塗布液1-P1を、支持体のコロナ処理を施した面に、バインダー量が塗布量で3.0g/m2になるように塗布し、180℃で1分間乾燥させて、乾燥厚みが約5μmのポリマー層1を形成した。
得られたポリマー層1形成用塗布液1-P1を、支持体のコロナ処理を施した面に、バインダー量が塗布量で3.0g/m2になるように塗布し、180℃で1分間乾燥させて、乾燥厚みが約5μmのポリマー層1を形成した。
以上のようにして、実施例1のポリマーシート1を作製した。
<評価>
-1.接着性-
[A]湿熱経時前の接着性
上記のようにして作製したポリマーシートを20mm幅×150mmにカットして、サンプル片を2枚準備した。この2枚のサンプル片を、互いにポリマー層側が内側になるように配置し、この間に20mm幅×100mm長にカットしたEVAシート(三井化学ファブロ(株)製のEVAシート:SC50B)を挟み、真空ラミネータ(日清紡(株)製の真空ラミネート機)を用いてホットプレスすることにより、EVAと接着させた。このときの接着条件は、以下の通りとした。
真空ラミネータを用いて、128℃で3分間の真空引き後、2分間加圧して仮接着した。その後、ドライオーブンを用いて、150℃で30分間、本接着処理を施した。このようにして、互いに接着した2枚のサンプル片の一端から20mmの部分はEVAと未接着で、残りの100mmの部分にEVAシートが接着された接着評価用試料を得た。
-1.接着性-
[A]湿熱経時前の接着性
上記のようにして作製したポリマーシートを20mm幅×150mmにカットして、サンプル片を2枚準備した。この2枚のサンプル片を、互いにポリマー層側が内側になるように配置し、この間に20mm幅×100mm長にカットしたEVAシート(三井化学ファブロ(株)製のEVAシート:SC50B)を挟み、真空ラミネータ(日清紡(株)製の真空ラミネート機)を用いてホットプレスすることにより、EVAと接着させた。このときの接着条件は、以下の通りとした。
真空ラミネータを用いて、128℃で3分間の真空引き後、2分間加圧して仮接着した。その後、ドライオーブンを用いて、150℃で30分間、本接着処理を施した。このようにして、互いに接着した2枚のサンプル片の一端から20mmの部分はEVAと未接着で、残りの100mmの部分にEVAシートが接着された接着評価用試料を得た。
得られた接着評価用試料のEVA未接着部分を、テンシロン(ORIENTEC製 RTC-1210A)にて上下クリップに挟み、剥離角度180°、引っ張り速度300mm/分で引っ張り試験を行ない、接着力を測定した。
測定された接着力をもとに以下の評価基準にしたがってランク付けした。このうち、ランク4、5が実用上許容可能な範囲である。
測定された接着力をもとに以下の評価基準にしたがってランク付けした。このうち、ランク4、5が実用上許容可能な範囲である。
<評価基準>
5:密着が非常に良好であった(60N/20mm以上)
4:密着は良好であった (30N/20mm以上60N/20mm未満)
3:密着がやや不良であった (20N/20mm以上30N/20mm未満)
2:密着不良が生じた (10N/20mm以上20N/20mm未満)
1:密着不良が顕著であった (10N/20mm未満)
5:密着が非常に良好であった(60N/20mm以上)
4:密着は良好であった (30N/20mm以上60N/20mm未満)
3:密着がやや不良であった (20N/20mm以上30N/20mm未満)
2:密着不良が生じた (10N/20mm以上20N/20mm未満)
1:密着不良が顕著であった (10N/20mm未満)
[B]湿熱経時後の接着性
得られた接着評価用試料を、120℃、100%RHの環境条件下で48時間保持(湿熱経時)した後、前記[A]と同様の方法にて接着力を測定した。測定された保持後の接着力について、同じ接着評価用試料の前記[A]湿熱経時前の接着力に対する比率〔%;=湿熱経時後の接着力/[A]湿熱経時前の接着力×100〕を算出した。また、測定された湿熱経時後の接着力をもとに、前記[A]と同様の方法にて接着力を評価した。
得られた接着評価用試料を、120℃、100%RHの環境条件下で48時間保持(湿熱経時)した後、前記[A]と同様の方法にて接着力を測定した。測定された保持後の接着力について、同じ接着評価用試料の前記[A]湿熱経時前の接着力に対する比率〔%;=湿熱経時後の接着力/[A]湿熱経時前の接着力×100〕を算出した。また、測定された湿熱経時後の接着力をもとに、前記[A]と同様の方法にて接着力を評価した。
-2.耐久性-
作製したポリマーシートを120℃、100%RHの雰囲気下で50時間保持した後、ポリマーシートのオモテ面(ポリマー層が設けられている側の面)の表面を、目視と光学顕微鏡(ME-600、(株)ニコン製、倍率100倍)で観察して以下のようにランク分けした。
評価ランク4、5が、実用上許容可能な範囲である。
作製したポリマーシートを120℃、100%RHの雰囲気下で50時間保持した後、ポリマーシートのオモテ面(ポリマー層が設けられている側の面)の表面を、目視と光学顕微鏡(ME-600、(株)ニコン製、倍率100倍)で観察して以下のようにランク分けした。
評価ランク4、5が、実用上許容可能な範囲である。
<評価基準>
5:光学顕微鏡観察しても表面に変化が認められない。
4:光学顕微鏡観察すると表面にわずかな亀裂や変形が見られる。
3:目視観察すると、表面のツヤが消失しているのがわかる。
2:目視観察でわずかな亀裂が見られる。
1:目視観察でも全面に亀裂が見られる。
5:光学顕微鏡観察しても表面に変化が認められない。
4:光学顕微鏡観察すると表面にわずかな亀裂や変形が見られる。
3:目視観察すると、表面のツヤが消失しているのがわかる。
2:目視観察でわずかな亀裂が見られる。
1:目視観察でも全面に亀裂が見られる。
〔実施例2~実施例4〕
実施例1のポリマー層1の形成において、ポリマー層1形成用塗布液1-P1を、ポリマー層1の厚みが表2に示す厚みとなるように塗布した以外は実施例1と同様にして、実施例2~実施例4のポリマーシート2~ポリマーシート4を作製した。得られたポリマーシート2~ポリマーシート4について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
実施例1のポリマー層1の形成において、ポリマー層1形成用塗布液1-P1を、ポリマー層1の厚みが表2に示す厚みとなるように塗布した以外は実施例1と同様にして、実施例2~実施例4のポリマーシート2~ポリマーシート4を作製した。得られたポリマーシート2~ポリマーシート4について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
〔実施例5〕
実施例1のポリマーシート1の作製において、ポリマー層1形成用塗布液1-P1を、ポリマー層1形成用塗布液1-P2に変更した以外は実施例1と同様にして、実施例5のポリマーシート5を作製した。得られたポリマーシート5について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
実施例1のポリマーシート1の作製において、ポリマー層1形成用塗布液1-P1を、ポリマー層1形成用塗布液1-P2に変更した以外は実施例1と同様にして、実施例5のポリマーシート5を作製した。得られたポリマーシート5について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
なお、ポリマー層1形成用塗布液1-P2は、ポリマー層1形成用塗布液1-P1の調製において、バインダー(P-1)を、バインダー(P-2)に変更したほかは、同様にして調製した。
〔実施例6〕
実施例3のポリマーシート3のポリマー層1の上に、更に下記のポリマー層2形成用塗布液を塗布して、ポリマー層2を設け、実施例6のポリマーシート6を作製した。得られたポリマーシート6について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
実施例3のポリマーシート3のポリマー層1の上に、更に下記のポリマー層2形成用塗布液を塗布して、ポリマー層2を設け、実施例6のポリマーシート6を作製した。得られたポリマーシート6について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
-ポリマー層2形成用塗布液の調製-
下記組成中の各成分を混合し、ポリマー層2形成用塗布液2-P1を調製した。
(塗布液の組成)
・バインダー(P-1)・・・362.3部
(セラネートWSA-1070、DIC(株)製、固形分:40質量%)
・カルボジイミド化合物(架橋剤)・・・24.2部
(カルボジライトV-02-L2、日清紡績(株)製、固形分:40質量%)
・界面活性剤・・・24.2部
(ナロアクティーCL95、三洋化成工業(株)製、固形分:1質量%)
・蒸留水・・・703.8部
下記組成中の各成分を混合し、ポリマー層2形成用塗布液2-P1を調製した。
(塗布液の組成)
・バインダー(P-1)・・・362.3部
(セラネートWSA-1070、DIC(株)製、固形分:40質量%)
・カルボジイミド化合物(架橋剤)・・・24.2部
(カルボジライトV-02-L2、日清紡績(株)製、固形分:40質量%)
・界面活性剤・・・24.2部
(ナロアクティーCL95、三洋化成工業(株)製、固形分:1質量%)
・蒸留水・・・703.8部
-ポリマー層2の形成-
得られたポリマー層2形成用塗布液2-P1を、ポリマーシート3のポリマー層1の上に、バインダー量が塗布量で2.0g/m2になるように塗布し、180℃で1分間乾燥させて、乾燥厚み約2μmのポリマー層2を形成した。
得られたポリマー層2形成用塗布液2-P1を、ポリマーシート3のポリマー層1の上に、バインダー量が塗布量で2.0g/m2になるように塗布し、180℃で1分間乾燥させて、乾燥厚み約2μmのポリマー層2を形成した。
〔実施例7~実施例8〕
実施例6のポリマー層2の形成において、ポリマー層2形成用塗布液2-P1を、ポリマー層2の厚みが表2に示す厚みとなるように塗布した以外は実施例6と同様にして、実施例7~実施例8のポリマーシート7~ポリマーシート8を作製した。得られたポリマーシート7~ポリマーシート8について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
実施例6のポリマー層2の形成において、ポリマー層2形成用塗布液2-P1を、ポリマー層2の厚みが表2に示す厚みとなるように塗布した以外は実施例6と同様にして、実施例7~実施例8のポリマーシート7~ポリマーシート8を作製した。得られたポリマーシート7~ポリマーシート8について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
〔実施例9〕
実施例6のポリマーシート6の作製において、ポリマー層2形成用塗布液2-P1を、ポリマー層2形成用塗布液1-P2に変更した以外は実施例6と同様にして、実施例9のポリマーシート9を作製した。得られたポリマーシート9について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
実施例6のポリマーシート6の作製において、ポリマー層2形成用塗布液2-P1を、ポリマー層2形成用塗布液1-P2に変更した以外は実施例6と同様にして、実施例9のポリマーシート9を作製した。得られたポリマーシート9について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
なお、ポリマー層2形成用塗布液2-P2は、ポリマー層2形成用塗布液2-P1の調製において、バインダー(P-1)を、バインダー(P-2)に変更したほかは、同様にして調製した。
〔実施例10〕
実施例6のポリマーシート6の作製において、ポリマー層2形成用塗布液2-P1を、ポリマー層2形成用塗布液1-P3に変更した以外は実施例6と同様にして、実施例10のポリマーシート10を作製した。得られたポリマーシート10について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
実施例6のポリマーシート6の作製において、ポリマー層2形成用塗布液2-P1を、ポリマー層2形成用塗布液1-P3に変更した以外は実施例6と同様にして、実施例10のポリマーシート10を作製した。得られたポリマーシート10について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
なお、ポリマー層2形成用塗布液2-P3は、ポリマー層2形成用塗布液2-P1の調製において、バインダー(P-1)を、バインダー(P-3)に変更したほかは、同様にして調製した。
〔実施例11~実施例12〕
実施例10のポリマー層2の形成において、ポリマー層2形成用塗布液2-P3を、ポリマー層2の厚みが表2に示す厚みとなるように塗布した以外は実施例10と同様にして、実施例11~実施例12のポリマーシート11~ポリマーシート12を作製した。得られたポリマーシート11~ポリマーシート12について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
実施例10のポリマー層2の形成において、ポリマー層2形成用塗布液2-P3を、ポリマー層2の厚みが表2に示す厚みとなるように塗布した以外は実施例10と同様にして、実施例11~実施例12のポリマーシート11~ポリマーシート12を作製した。得られたポリマーシート11~ポリマーシート12について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
〔比較例1〕
実施例1のポリマーシート1の作製において、ポリマー層1形成用塗布液1-P1を、ポリマー層1形成用塗布液1-P4に変更した以外は実施例1と同様にして、比較例1のポリマーシート101を作製した。得られたポリマーシート101について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
実施例1のポリマーシート1の作製において、ポリマー層1形成用塗布液1-P1を、ポリマー層1形成用塗布液1-P4に変更した以外は実施例1と同様にして、比較例1のポリマーシート101を作製した。得られたポリマーシート101について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
なお、ポリマー層1形成用塗布液1-P4は、ポリマー層1形成用塗布液1-P1の調製において、バインダー(P-1)を、バインダー(P-4)に変更したほかは、同様にして調製した。
〔比較例2〕
実施例1のポリマーシート1の作製において、ポリマー層1形成用塗布液1-P1を、ポリマー層1形成用塗布液1-P5に変更した以外は実施例1と同様にして、比較例2のポリマーシート102を作製した。得られたポリマーシート102について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
実施例1のポリマーシート1の作製において、ポリマー層1形成用塗布液1-P1を、ポリマー層1形成用塗布液1-P5に変更した以外は実施例1と同様にして、比較例2のポリマーシート102を作製した。得られたポリマーシート102について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
なお、ポリマー層1形成用塗布液1-P5は、ポリマー層1形成用塗布液1-P1の調製において、バインダー(P-1)を、バインダー(P-5)に変更したほかは、同様にして調製した。
〔比較例3〕
実施例1のポリマーシート1の作製において、支持体PET-1を、PET-2に変更した以外は実施例1と同様にして、比較例3のポリマーシート103を作製した。得られたポリマーシート103について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
実施例1のポリマーシート1の作製において、支持体PET-1を、PET-2に変更した以外は実施例1と同様にして、比較例3のポリマーシート103を作製した。得られたポリマーシート103について、ポリマーシート1と同様の評価を行った。結果を表2に示す。
〔実施例13~実施例15、および比較例4~比較例5〕
実施例7のポリマーシート7の作製において、ポリマー層1のバインダー種を表2に示すように、PS-1~PS-5のいずれかに変更した以外は同様にして、実施例13~実施例15のポリマーシート13~15、および比較例4~比較例5のポリマーシート104~105を作製した。
得られたポリマーシート13~15および104~105について、ポリマーシート1と同様の評価を行った。ただし、バインダーとしてPS-5を用いた系(比較例5)は、安定性が不良で評価を行えなかった。結果を表2に示す。
実施例7のポリマーシート7の作製において、ポリマー層1のバインダー種を表2に示すように、PS-1~PS-5のいずれかに変更した以外は同様にして、実施例13~実施例15のポリマーシート13~15、および比較例4~比較例5のポリマーシート104~105を作製した。
得られたポリマーシート13~15および104~105について、ポリマーシート1と同様の評価を行った。ただし、バインダーとしてPS-5を用いた系(比較例5)は、安定性が不良で評価を行えなかった。結果を表2に示す。
実施例13~実施例15、および比較例4~比較例5に用いたバインダーPS-1~PS-5は、次のように合成した。なお、合成例-1~合成例-5において、「%」は、特に記載のない限り、質量基準である。
(合成例-1)
撹拌装置、滴下ロートを備え、窒素ガス置換した反応容器に、プロピレングリコールモノ-n-プロピルエーテル(PNP)81部、イソプロピルアルコール(IPA)360部、フェニルトリメトキシシラン(PTMS)110部およびジメチルジメトキシシラン(DMDMS)71部を仕込み、窒素ガス雰囲気下に撹拌しながら60℃に昇温した。
次いで、同温度で、メチルメタクリレート(MMA)260部、n-ブチルメタクリレート(BMA)200部、n-ブチルアクリレート(BA)110部、アクリル酸(AA)30部、3-メタクリロイルオキシプロピルトリメトキシシラン(MPTMS)19部、tert-ブチルパーオキシ-2-エチルヘキサノエート31.5部(TBPO)およびPNP部31.5部からなる混合物を4時間かけて滴下した。
撹拌装置、滴下ロートを備え、窒素ガス置換した反応容器に、プロピレングリコールモノ-n-プロピルエーテル(PNP)81部、イソプロピルアルコール(IPA)360部、フェニルトリメトキシシラン(PTMS)110部およびジメチルジメトキシシラン(DMDMS)71部を仕込み、窒素ガス雰囲気下に撹拌しながら60℃に昇温した。
次いで、同温度で、メチルメタクリレート(MMA)260部、n-ブチルメタクリレート(BMA)200部、n-ブチルアクリレート(BA)110部、アクリル酸(AA)30部、3-メタクリロイルオキシプロピルトリメトキシシラン(MPTMS)19部、tert-ブチルパーオキシ-2-エチルヘキサノエート31.5部(TBPO)およびPNP部31.5部からなる混合物を4時間かけて滴下した。
その後、同温度で2.5時間加熱撹拌を行って重量平均分子量が約30000であるカルボキシル基と加水分解性シリル基を含有するアクリル系ポリマーの溶液を得た。
次いで、脱イオン水54.8部の混合物を加えて16時間加熱撹拌を継続してアルコキシシランを加水分解し、さらにアクリル系重合体と縮合させ、不揮発分(NV)が56%、溶液酸価が22mgKOH/gである、カルボキシル基含有アクリル系重合体部位とポリシロキサン部位を有する複合ポリマーの溶液を得た。
次いで、同温度で撹拌しながらトリエチルアミン42部を添加して10分間撹拌を行った。これにより、含有されるカルボキシル基の100%が中和された。
この後、同温度で脱イオン水1250.0部を1.5間かけて滴下して転相乳化させた後、50℃に昇温して30分間撹拌を行った。次いで内温40℃で3.5時間をかけて、有機溶剤とともに水の一部分を減圧下除去した。こうして固形分濃度が42%、平均粒子径が110nmのカルボキシル基含有アクリル系重合体部位とポリシロキサン部位を有する複合ポリマーの水分散物(PS-1)を得た。PS-1のポリシロキサン部位は約25%である。
次いで、脱イオン水54.8部の混合物を加えて16時間加熱撹拌を継続してアルコキシシランを加水分解し、さらにアクリル系重合体と縮合させ、不揮発分(NV)が56%、溶液酸価が22mgKOH/gである、カルボキシル基含有アクリル系重合体部位とポリシロキサン部位を有する複合ポリマーの溶液を得た。
次いで、同温度で撹拌しながらトリエチルアミン42部を添加して10分間撹拌を行った。これにより、含有されるカルボキシル基の100%が中和された。
この後、同温度で脱イオン水1250.0部を1.5間かけて滴下して転相乳化させた後、50℃に昇温して30分間撹拌を行った。次いで内温40℃で3.5時間をかけて、有機溶剤とともに水の一部分を減圧下除去した。こうして固形分濃度が42%、平均粒子径が110nmのカルボキシル基含有アクリル系重合体部位とポリシロキサン部位を有する複合ポリマーの水分散物(PS-1)を得た。PS-1のポリシロキサン部位は約25%である。
(合成例-2)
用いるモノマー量を下記のように変更する以外、合成例-1と同様にしてPS-2を合成した。
フェニルトリメトキシシラン(PTMS)210部、ジメチルジメトキシシラン(DMDMS)166部、3-メタクリロイルオキシプロピルトリメトキシシラン(MPTMS)24部、メチルメタクリレート(MMA)200部、n-ブチルメタクリレート(BMA)100部、n-ブチルアクリレート(BA)70部、アクリル酸(AA)30部。PS-2のポリシロキサン部位は約50%である。
用いるモノマー量を下記のように変更する以外、合成例-1と同様にしてPS-2を合成した。
フェニルトリメトキシシラン(PTMS)210部、ジメチルジメトキシシラン(DMDMS)166部、3-メタクリロイルオキシプロピルトリメトキシシラン(MPTMS)24部、メチルメタクリレート(MMA)200部、n-ブチルメタクリレート(BMA)100部、n-ブチルアクリレート(BA)70部、アクリル酸(AA)30部。PS-2のポリシロキサン部位は約50%である。
(合成例-3)
用いるモノマー量を下記のように変更する以外、合成例-1と同様にしてPS-3を合成した。
フェニルトリメトキシシラン(PTMS)320部、ジメチルジメトキシシラン(DMDMS)244部、3-メタクリロイルオキシプロピルトリメトキシシラン(MPTMS)36部、メチルメタクリレート(MMA)90部、n-ブチルメタクリレート(BMA)60部、n-ブチルアクリレート(BA)20部、アクリル酸(AA)30部。PS-3のポリシロキサン部位は約75%である。
用いるモノマー量を下記のように変更する以外、合成例-1と同様にしてPS-3を合成した。
フェニルトリメトキシシラン(PTMS)320部、ジメチルジメトキシシラン(DMDMS)244部、3-メタクリロイルオキシプロピルトリメトキシシラン(MPTMS)36部、メチルメタクリレート(MMA)90部、n-ブチルメタクリレート(BMA)60部、n-ブチルアクリレート(BA)20部、アクリル酸(AA)30部。PS-3のポリシロキサン部位は約75%である。
(合成例-4)
用いるモノマー量を下記のように変更する以外、合成例-1と同様にしてPS-4を合成した。
フェニルトリメトキシシラン(PTMS)60部、ジメチルジメトキシシラン(DMDMS)25部、3-メタクリロイルオキシプロピルトリメトキシシラン(MPTMS)15部、メチルメタクリレート(MMA)300部、n-ブチルメタクリレート(BMA)220部、n-ブチルアクリレート(BA)150部、アクリル酸(AA)30部。PS-4はポリシロキサン部位が約13%である本発明の複合ポリマーには分類されないポリマーである。
用いるモノマー量を下記のように変更する以外、合成例-1と同様にしてPS-4を合成した。
フェニルトリメトキシシラン(PTMS)60部、ジメチルジメトキシシラン(DMDMS)25部、3-メタクリロイルオキシプロピルトリメトキシシラン(MPTMS)15部、メチルメタクリレート(MMA)300部、n-ブチルメタクリレート(BMA)220部、n-ブチルアクリレート(BA)150部、アクリル酸(AA)30部。PS-4はポリシロキサン部位が約13%である本発明の複合ポリマーには分類されないポリマーである。
(合成例-5)
用いるモノマー量を下記のように変更する以外、合成例-1と同様にしてPS-5を合成した。
フェニルトリメトキシシラン(PTMS)360部、ジメチルジメトキシシラン(DMDMS)320部、3-メタクリロイルオキシプロピルトリメトキシシラン(MPTMS)40部、メチルメタクリレート(MMA)20部、n-ブチルメタクリレート(BMA)20部、n-ブチルアクリレート(BA)10部、アクリル酸(AA)30部。PS-5のポリシロキサン部位は約90%である本発明の複合ポリマーには分類されないポリマーである。なお、このポリマー水分散物は凝集が発生しており、安定性が不良であった。
用いるモノマー量を下記のように変更する以外、合成例-1と同様にしてPS-5を合成した。
フェニルトリメトキシシラン(PTMS)360部、ジメチルジメトキシシラン(DMDMS)320部、3-メタクリロイルオキシプロピルトリメトキシシラン(MPTMS)40部、メチルメタクリレート(MMA)20部、n-ブチルメタクリレート(BMA)20部、n-ブチルアクリレート(BA)10部、アクリル酸(AA)30部。PS-5のポリシロキサン部位は約90%である本発明の複合ポリマーには分類されないポリマーである。なお、このポリマー水分散物は凝集が発生しており、安定性が不良であった。
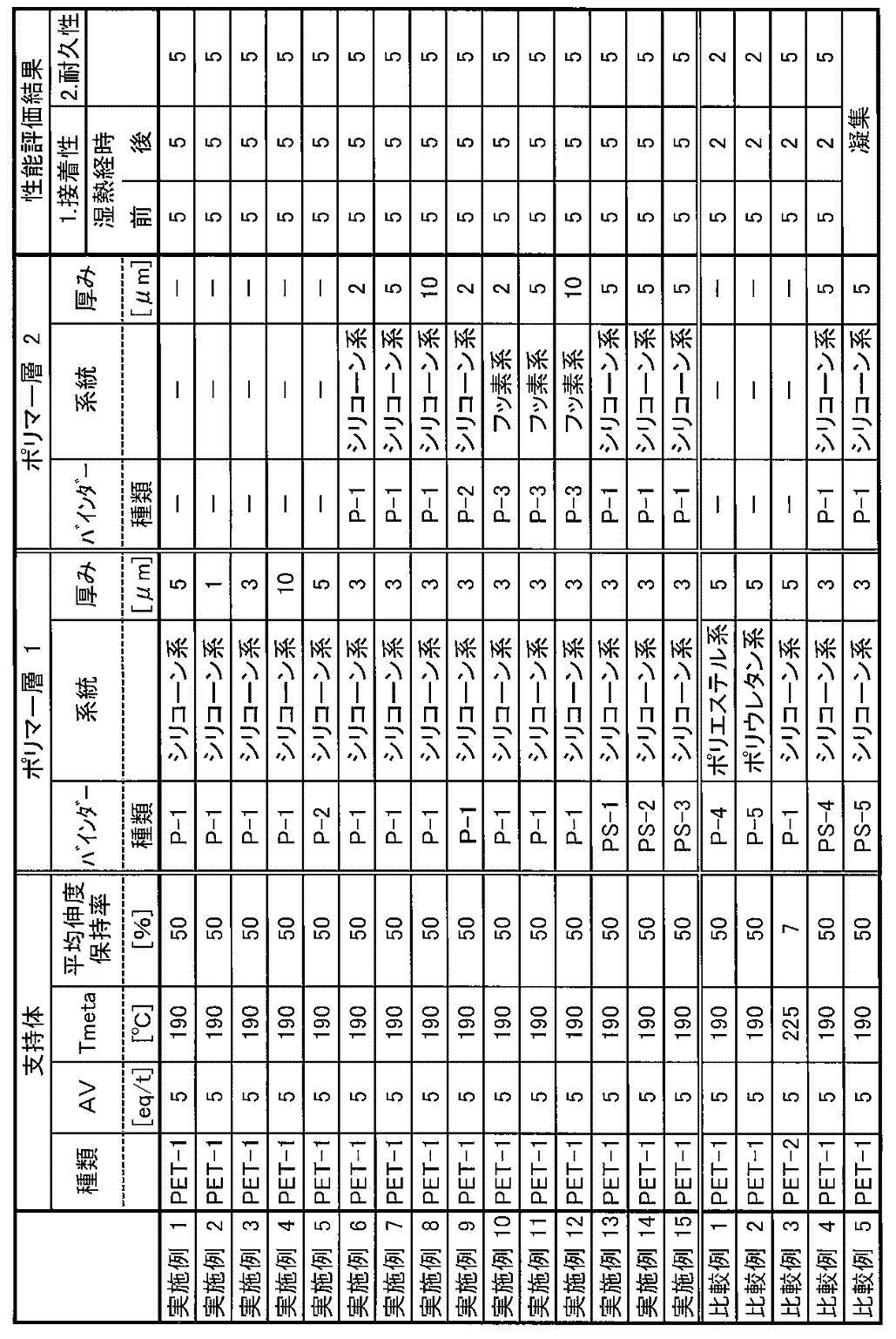
表2からわかるように、実施例のポリマーシートは、いずれも、比較例のポリマーシートに比べ、湿熱経時後の接着性に優れた。さらに、耐久性に関しては、ポリマー層に、本発明における複合ポリマーを用いていない比較例1及び2のポリマーシート101および102よりも、実施例のポリマーシートの方が優れていた。
<バックシート(太陽電池用バックシート)の作製>
〔実施例16~実施例30〕
実施例1~実施例15のポリマーシート1~ポリマーシート15のポリマー層が設けられている面(オモテ面)とは反対側の面、すなわち、支持体(PET-1またはPET-2)のウラ面表面に対してコロナ処理を施した。コロナ処理の条件は、PET-1~PET-3のオモテ面に対して行なったコロナ処理と同じ条件とした。コロナ処理を施した支持体のウラ面に、以下の下塗り層、及び着色層を設けてバックシート1~バックシート15を作成した。
〔実施例16~実施例30〕
実施例1~実施例15のポリマーシート1~ポリマーシート15のポリマー層が設けられている面(オモテ面)とは反対側の面、すなわち、支持体(PET-1またはPET-2)のウラ面表面に対してコロナ処理を施した。コロナ処理の条件は、PET-1~PET-3のオモテ面に対して行なったコロナ処理と同じ条件とした。コロナ処理を施した支持体のウラ面に、以下の下塗り層、及び着色層を設けてバックシート1~バックシート15を作成した。
[下塗り層]
-下塗り層形成用塗布液の調製-
下記組成中の各成分を混合し、下塗り層形成用塗布液を調製した。
-下塗り層形成用塗布液の調製-
下記組成中の各成分を混合し、下塗り層形成用塗布液を調製した。
(塗布液の組成)
・ポリエステル系バインダー ・・・48.0部
(バイロナールDM1245(東洋紡(株)製、固形分30質量%))
・カルボジイミド化合物(架橋剤)・・・10.0部
(カルボジライトV-02-L2、日清紡績(株)製、固形分:10質量%)
・オキサゾリン化合物(架橋剤)・・・3.0部
(エポクロスWS700、(株)日本触媒製、固形分:25質量%)
・界面活性剤・・・15.0部
(ナロアクティーCL95、三洋化成工業(株)製、固形分:1質量%)
・蒸留水・・・907.0部
・ポリエステル系バインダー ・・・48.0部
(バイロナールDM1245(東洋紡(株)製、固形分30質量%))
・カルボジイミド化合物(架橋剤)・・・10.0部
(カルボジライトV-02-L2、日清紡績(株)製、固形分:10質量%)
・オキサゾリン化合物(架橋剤)・・・3.0部
(エポクロスWS700、(株)日本触媒製、固形分:25質量%)
・界面活性剤・・・15.0部
(ナロアクティーCL95、三洋化成工業(株)製、固形分:1質量%)
・蒸留水・・・907.0部
-下塗り層の形成-
得られた下塗り層形成用塗布液を、ポリマーシート1~ポリマーシート15のウラ面(支持体のウラ面)表面に、バインダー量が塗布量で0.1g/m2になるように塗布し、180℃で1分間乾燥させて、乾燥厚みが約0.1μmの下塗り層を形成した。
得られた下塗り層形成用塗布液を、ポリマーシート1~ポリマーシート15のウラ面(支持体のウラ面)表面に、バインダー量が塗布量で0.1g/m2になるように塗布し、180℃で1分間乾燥させて、乾燥厚みが約0.1μmの下塗り層を形成した。
[着色層]
-着色層用塗布液の調製-
下記組成の成分を混合し、着色層用塗布液を調製した。
-着色層用塗布液の調製-
下記組成の成分を混合し、着色層用塗布液を調製した。
(塗布液の組成)
・顔料分散物1(実施例1で調製した顔料分散物)・・・80.0部
・シラノール変性ポリビニルアルコールバインダー・・・11.4部
〔R1130、(株)クラレ製、固形分:7質量%〕
・ポリオキシアルキレンアルキルエーテル ・・・1.0部
〔ナロアクティーCL95、三洋化成工業(株)製、固形分:1質量%〕
・オキサゾリン化合物・・・2.0部
〔エポクロスWS-700、日本触媒(株)製、固形分:25%;架橋剤〕
・蒸留水・・・5.6部
・顔料分散物1(実施例1で調製した顔料分散物)・・・80.0部
・シラノール変性ポリビニルアルコールバインダー・・・11.4部
〔R1130、(株)クラレ製、固形分:7質量%〕
・ポリオキシアルキレンアルキルエーテル ・・・1.0部
〔ナロアクティーCL95、三洋化成工業(株)製、固形分:1質量%〕
・オキサゾリン化合物・・・2.0部
〔エポクロスWS-700、日本触媒(株)製、固形分:25%;架橋剤〕
・蒸留水・・・5.6部
-着色層の形成-
得られた着色層用塗布液を、ポリマーシート1~ポリマーシート15のウラ面の下塗り層が形成されている面に塗布し、180℃で1分間乾燥させて、二酸化チタン量が7.0g/m2、バインダー1.2g/m2の着色層を形成した。
得られた着色層用塗布液を、ポリマーシート1~ポリマーシート15のウラ面の下塗り層が形成されている面に塗布し、180℃で1分間乾燥させて、二酸化チタン量が7.0g/m2、バインダー1.2g/m2の着色層を形成した。
以上のようにして、実施例16~実施例30のバックシート1~バックシート15を作製した。得られたバックシート1~バックシート15について、実施例1のポリマーシート1と同様の評価を行なったところ、いずれの評価もランクが5であり、いずれのバックシートも接着性および耐久性に優れていることがわかった。
<太陽電池モジュールの作製>
〔実施例31~実施例45〕
厚さ3.2mmの強化ガラスと、EVAシート〔三井化学ファブロ社製のSC50B〕と、結晶系太陽電池セルと、EVAシート〔三井化学ファブロ社製のSC50B〕と、実施例16~実施例30のバックシート1~バックシート15のいずれか1枚とを、この順に重ね合わせ、真空ラミネータ〔日清紡社製、真空ラミネート機〕を用いてホットプレスすることにより、各部材とEVAシートとを接着させた。ただし、バックシートは、バックシートの着色層がEVAシートと接触するように配置した。また、EVAシートの接着条件は、以下の通りである。
〔実施例31~実施例45〕
厚さ3.2mmの強化ガラスと、EVAシート〔三井化学ファブロ社製のSC50B〕と、結晶系太陽電池セルと、EVAシート〔三井化学ファブロ社製のSC50B〕と、実施例16~実施例30のバックシート1~バックシート15のいずれか1枚とを、この順に重ね合わせ、真空ラミネータ〔日清紡社製、真空ラミネート機〕を用いてホットプレスすることにより、各部材とEVAシートとを接着させた。ただし、バックシートは、バックシートの着色層がEVAシートと接触するように配置した。また、EVAシートの接着条件は、以下の通りである。
真空ラミネータを用いて、128℃で3分間の真空引き後、2分間加圧して仮接着した。その後、ドライオーブンにて150℃で30分間、本接着処理を施した。
このようにして、結晶系の太陽電池モジュール1~太陽電池モジュール15を作製した。
作製した太陽電池モジュール1~太陽電池モジュール15について、発電運転をしたところ、いずれも太陽電池として良好な発電性能を示した。
このようにして、結晶系の太陽電池モジュール1~太陽電池モジュール15を作製した。
作製した太陽電池モジュール1~太陽電池モジュール15について、発電運転をしたところ、いずれも太陽電池として良好な発電性能を示した。
〔実施例46〕
実施例1のポリマーシート1の作製において、支持体としてPET-1を用いる代わりに、PET-3を用いた以外は実施例1と同様にして、ポリマーシート16を作成した。得られたポリマーシート16と、厚さ20μmのアルミ箔(バリア層)と、厚さ188μmのPET支持体(PET-4)と、厚さ50μmの白色PET支持体(PET-5)とを、この順に接着してバックシート16を作製した。
なお接着に際して、PET-4およびPET-5の各表面は、予めPET-1~PET-3と同様のコロナ処理を施した。
実施例1のポリマーシート1の作製において、支持体としてPET-1を用いる代わりに、PET-3を用いた以外は実施例1と同様にして、ポリマーシート16を作成した。得られたポリマーシート16と、厚さ20μmのアルミ箔(バリア層)と、厚さ188μmのPET支持体(PET-4)と、厚さ50μmの白色PET支持体(PET-5)とを、この順に接着してバックシート16を作製した。
なお接着に際して、PET-4およびPET-5の各表面は、予めPET-1~PET-3と同様のコロナ処理を施した。
(接着条件)
接着剤としてLX660(K)〔DIC(株)製接着剤〕に、硬化剤KW75〔DIC(株)製接着剤〕を10部混合したものを用い、ポリマーシート16と、アルミ箔と、PET-4と、PET-5とを、真空ラミネータ〔日清紡(株)製 真空ラミネート機〕でホットプレス接着した。
接着は80℃で3分の真空引き後、2分間加圧することで行い、その後40℃で4日間保持した。
接着剤としてLX660(K)〔DIC(株)製接着剤〕に、硬化剤KW75〔DIC(株)製接着剤〕を10部混合したものを用い、ポリマーシート16と、アルミ箔と、PET-4と、PET-5とを、真空ラミネータ〔日清紡(株)製 真空ラミネート機〕でホットプレス接着した。
接着は80℃で3分の真空引き後、2分間加圧することで行い、その後40℃で4日間保持した。
実施例31の太陽電池モジュール1の作製において、バックシート1の代わりに、得られたバックシート16を用いた他は同様の方法で、太陽電池モジュール16を作製した。
作製した太陽電池モジュール16について、発電運転をしたところ、太陽電池として良好な発電性能を示した。
作製した太陽電池モジュール16について、発電運転をしたところ、太陽電池として良好な発電性能を示した。
〔実施例47〕
実施例46におけるバックシート16の作製において、アルミ箔の代わりに、厚さ12μmのバリア層付PETシートを用いたほかは同様にして、バックシート17を作製した。
さらに、実施例46における太陽電池モジュール16の作製において、バックシート16の代わりに、厚さ12μmのバリア層付PETシートを用いたほかは同様にして、太陽電池モジュール17を作製した。
作製した太陽電池モジュール17について、発電運転をしたところ、太陽電池として良好な発電性能を示した。
実施例46におけるバックシート16の作製において、アルミ箔の代わりに、厚さ12μmのバリア層付PETシートを用いたほかは同様にして、バックシート17を作製した。
さらに、実施例46における太陽電池モジュール16の作製において、バックシート16の代わりに、厚さ12μmのバリア層付PETシートを用いたほかは同様にして、太陽電池モジュール17を作製した。
作製した太陽電池モジュール17について、発電運転をしたところ、太陽電池として良好な発電性能を示した。
本発明の具体的態様の前記記述は、記述と説明の目的で提供するものである。開示された、まさにその形態に本発明を限定することを企図するものでもなく、或いは網羅的なものを企図するものでもない。明らかに、当業者が多くの修飾や変形をすることができることは自明である。該態様は、本発明の概念やその実際の応用を最もよく説明するために選定されたものであって、それによって、当業者の他者が企図する特定の用途に適合させるべく種々の態様や種々の変形をなすことができるように、当業者の他者に本発明を理解せしめるためのものである。
2011年3月25日出願の日本特許出願第2011-068658号公報は、その開示全体がここに参照文献として組み込まれるものである。
本明細書に記述された全ての刊行物や特許出願、並びに技術標準は、それら個々の刊行物や特許出願、並びに技術標準が引用文献として特別に、そして個々に組み込むことが指定されている場合には、該引用文献と同じ限定範囲においてここに組み込まれるものである。本発明の範囲は下記特許請求の範囲及びその等価物に拠って決定されることを企図するものである。
本明細書に記述された全ての刊行物や特許出願、並びに技術標準は、それら個々の刊行物や特許出願、並びに技術標準が引用文献として特別に、そして個々に組み込むことが指定されている場合には、該引用文献と同じ限定範囲においてここに組み込まれるものである。本発明の範囲は下記特許請求の範囲及びその等価物に拠って決定されることを企図するものである。
Claims (23)
- カルボキシル基の含有量が15当量/t以下であり、示差走査熱量測定により求められる微少吸熱ピーク温度Tmeta(℃)が220℃以下であり、温度125℃、相対湿度100%RHの条件下72時間放置後の平均伸度保持率が10%以上であるポリエステル基材と、
前記ポリエステル基材上に設けられ、分子中に質量割合が15~85質量%の下記一般式(1)で表されるシロキサン構造単位と質量割合が85~15質量%の非シロキサン系構造単位とを含む複合ポリマーを含有するポリマー層と、
を有する太陽電池用ポリマーシート。

〔式中、R1及びR2は、各々独立に、水素原子、ハロゲン原子、又は1価の有機基を表し、R1とR2とは同一でも異なってもよい。nは、1以上の整数を表す。複数のR1及びR2は各々、互いに同一でも異なってもよい。〕 - 前記ポリマー層は、前記複合ポリマーを架橋する架橋剤由来の構造部分を含む請求項1に記載の太陽電池用ポリマーシート。
- 前記非ポリシロキサン系構造単位が、アクリル系構造単位である請求項1に記載の太陽電池用ポリマーシート。
- 前記架橋剤は、カルボジイミド系化合物、オキサゾリン系化合物、及びエポキシ系架橋剤から選ばれる少なくとも1種である請求項2に記載の太陽電池用ポリマーシート。
- 前記ポリマー層中における、前記複合ポリマーに対する前記架橋剤由来の構造部分の質量割合が1~30質量%である請求項2に記載の太陽電池用ポリマーシート。
- 前記ポリエステル基材は、コロナ処理、火炎処理、低圧プラズマ処理、大気圧プラズマ処理、及び紫外線処理から選ばれる少なくとも1つの表面処理が施されている請求項1に記載の太陽電池用ポリマーシート。
- 前記一般式(1)中の前記R1及びR2で表される1価の有機基が、アルキル基、アリール基、アラルキル基、アルコキシ基、アリールオキシ基、メルカプト基、アミノ基、及びアミド基から選択される少なくとも一種である請求項1に記載の太陽電池用ポリマーシート。
- 前記ポリエステル基材のカルボキシル基の含有量が1~15当量/tの範囲である請求項1~請求項7のいずれか1項に記載の太陽電池用ポリマーシート。
- 前記ポリマー層の少なくとも1層は、厚みが0.8μm~12μmである請求項1~請求項7のいずれか1項に記載の太陽電池用ポリマーシート。
- 前記ポリマー層の少なくとも1層は、前記ポリマー基材の表面に接触させて設けられている請求項1~請求項7のいずれか1項に記載の太陽電池用ポリマーシート。
- 前記ポリマー層の少なくとも1層は、前記ポリマー基材から最も離れた位置に配置された最外層である請求項1~請求項7のいずれか1項に記載の太陽電池用ポリマーシート。
- 前記ポリマー層の少なくとも1層は、更に白色系顔料を含み、光反射性を有する反射層である請求項1~請求項7のいずれか1項に記載の太陽電池用ポリマーシート。
- 前記ポリマー層を少なくとも2層含み、前記2層の一方として前記反射層を有し、他方を前記反射層と前記ポリエステル基材との間に有する請求項12に記載の太陽電池用ポリマーシート。
- 白色系顔料を含み、光反射性を有する反射層を更に有し、該反射層と前記ポリマー基材との間に、少なくとも一層の前記ポリマー層を有する請求項1~請求項7のいずれか1項に記載の太陽電池用ポリマーシート。
- カルボキシル基の含有量が15当量/t以下であり、示差走査熱量測定により求められる微少吸熱ピーク温度Tmeta(℃)が220℃以下であり、温度125℃、相対湿度100%RHの条件下72時間放置後の平均伸度保持率が10%以上であるポリエステル基材上に、分子中に質量割合が15~85質量%の下記一般式(1)で表されるシロキサン構造単位と質量割合が85~15質量%の非シロキサン系構造単位とを含む複合ポリマーを含有する塗布液を塗布して少なくとも1層のポリマー層を形成する工程を有する太陽電池用ポリマーシートの製造方法。

〔式中、R1及びR2は、各々独立に、水素原子、ハロゲン原子、又は1価の有機基を表し、R1とR2とは同一でも異なってもよい。nは、1以上の整数を表す。複数のR1及びR2は各々、互いに同一でも異なってもよい。〕 - 前記塗布液は、更に、カルボジイミド系化合物、オキサゾリン系化合物、及びエポキシ系架橋剤から選ばれる架橋剤を含有する請求項15に記載の太陽電池用ポリマーシートの製造方法。
- 前記塗布液は、更に溶媒を含有し、該溶媒の50質量%以上が水である請求項15に記載の太陽電池用ポリマーシートの製造方法。
- 太陽電池素子が封止材で封止された電池側基板の前記封止剤と接触させて配置される太陽電池用バックシートであって、
請求項1~請求項7のいずれか1項に記載の太陽電池用ポリマーシート、または、請求項15~請求項17のいずれか1項に記載の太陽電池用ポリマーシートの製造方法により製造された太陽電池用ポリマーシートを用いた太陽電池用バックシート。 - 前記ポリエステル基材の前記ポリマー層が設けられている面とは反対側の表面に、前記封止材に対して5N/cm以上の接着力を持つ易接着層を有する請求項18に記載の太陽電池用バックシート。
- 前記太陽電池用ポリマーシートから選択される2つ以上の太陽電池用ポリマーシートが粘着剤により貼り合わせられている請求項18に記載の太陽電池用バックシート。
- 水および気体の少なくとも一方の浸入を防止するバリア層を有する請求項18に記載の太陽電池用バックシート。
- 請求項18に記載の太陽電池用バックシートを備えた太陽電池モジュール。
- 太陽光が入射する透明性のフロント基板と、
前記フロント基板の上に設けられ、太陽電池素子及び前記太陽電池素子を封止する封止材を有するセル構造部分と、
前記セル構造部分の前記フロント基板が位置する側と反対側に設けられ、前記封止材と隣接して配置された、請求項18に記載の太陽電池用バックシートと、
を備えた太陽電池モジュール。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201280011843.0A CN103430321B (zh) | 2011-03-25 | 2012-03-22 | 太阳能电池用聚合物板及其制造方法、太阳能电池用背板、以及太阳能电池模块 |
| KR1020137023639A KR101663554B1 (ko) | 2011-03-25 | 2012-03-22 | 태양전지용 폴리머 시트 및 그 제조 방법, 태양전지용 백 시트, 및 태양전지 모듈 |
| US14/013,899 US20130340829A1 (en) | 2011-03-25 | 2013-08-29 | Polymer sheet for solar cell, process for production thereof, solar cell backsheet and solar cell module |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011-068658 | 2011-03-25 | ||
| JP2011068658A JP5623952B2 (ja) | 2011-03-25 | 2011-03-25 | 太陽電池用ポリマーシート及びその製造方法、太陽電池用バックシート、並びに太陽電池モジュール |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/013,899 Continuation US20130340829A1 (en) | 2011-03-25 | 2013-08-29 | Polymer sheet for solar cell, process for production thereof, solar cell backsheet and solar cell module |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012133124A1 true WO2012133124A1 (ja) | 2012-10-04 |
Family
ID=46930851
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/057390 WO2012133124A1 (ja) | 2011-03-25 | 2012-03-22 | 太陽電池用ポリマーシート及びその製造方法、太陽電池用バックシート、並びに太陽電池モジュール |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20130340829A1 (ja) |
| JP (1) | JP5623952B2 (ja) |
| KR (1) | KR101663554B1 (ja) |
| CN (1) | CN103430321B (ja) |
| WO (1) | WO2012133124A1 (ja) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5587230B2 (ja) * | 2011-03-25 | 2014-09-10 | 富士フイルム株式会社 | 太陽電池用バックシート及びその製造方法、並びに太陽電池モジュール |
| JP6655785B2 (ja) * | 2014-04-17 | 2020-02-26 | パナソニックIpマネジメント株式会社 | 樹脂組成物およびその製造方法並びに半導体装置 |
| CN105489685B (zh) * | 2014-10-03 | 2017-05-10 | 财团法人工业技术研究院 | 板材与模组结构体 |
| GB201504291D0 (en) * | 2015-03-13 | 2015-04-29 | Dupont Teijin Films Us Ltd | PV cells |
| US10074626B2 (en) * | 2016-06-06 | 2018-09-11 | Shin-Etsu Chemical Co., Ltd. | Wafer laminate and making method |
| KR102346652B1 (ko) * | 2020-06-18 | 2022-01-03 | 도레이첨단소재 주식회사 | 태양광 배리어필름, 및 이를 포함하는 전기화학소자 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002083988A (ja) * | 2000-09-08 | 2002-03-22 | Dainippon Printing Co Ltd | 太陽電池モジュ−ル用裏面保護シ−トおよびそれを使用した太陽電池モジュ−ル |
| WO2006067984A1 (ja) * | 2004-12-21 | 2006-06-29 | Toyo Aluminium Kabushiki Kaisha | 太陽電池用シート部材 |
| WO2009133827A1 (ja) * | 2008-04-28 | 2009-11-05 | 旭化成ケミカルズ株式会社 | 太陽電池バックシート用積層体およびそれを有するバックシート |
| WO2010110119A1 (ja) * | 2009-03-26 | 2010-09-30 | 東レ株式会社 | 太陽電池用ポリエステルフィルム、それを用いた太陽電池バックシート、および太陽電池 |
| JP2012049331A (ja) * | 2010-08-26 | 2012-03-08 | Fujifilm Corp | 太陽電池保護シート及びその製造方法、太陽電池用バックシート並びに太陽電池モジュール |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3239523B2 (ja) * | 1993-03-19 | 2001-12-17 | ジェイエスアール株式会社 | ポリシロキサン複合重合体ラテックス |
| JP2006255927A (ja) * | 2005-03-15 | 2006-09-28 | Teijin Dupont Films Japan Ltd | 太陽電池用表面保護フィルムおよびそれを用いた太陽電池積層体 |
| EP1862490B1 (en) * | 2005-03-24 | 2011-12-28 | Sakata Inx Corporation | Liquid developer comprising carbodiimide compound |
| JP2007150084A (ja) * | 2005-11-29 | 2007-06-14 | Dainippon Printing Co Ltd | 太陽電池モジュール用裏面保護シート、太陽電池モジュール用裏面積層体、および、太陽電池モジュール |
| US8143342B2 (en) * | 2005-12-27 | 2012-03-27 | Sk Kaken Co., Ltd. | Coating agent |
| JP5142603B2 (ja) * | 2007-06-27 | 2013-02-13 | 株式会社巴川製紙所 | 太陽電池用保護シート |
| JP2009071236A (ja) * | 2007-09-18 | 2009-04-02 | Tomoegawa Paper Co Ltd | 太陽電池用バックシート |
| DE102009000450A1 (de) * | 2009-01-28 | 2010-07-29 | Evonik Degussa Gmbh | Transparente, witterungsbeständige Barrierefolie, Herstellung durch Lamination, Extrusionslamination oder Extrusionbeschichtung |
| CN101823355B (zh) * | 2009-03-03 | 2013-09-25 | E.I.内穆尔杜邦公司 | 聚合物叠层膜和使用该叠层膜的太阳能电池板 |
| JP5565020B2 (ja) * | 2009-03-27 | 2014-08-06 | 東レ株式会社 | ポリエステルフィルム、およびそれを用いた太陽電池 |
| JP2010232513A (ja) * | 2009-03-27 | 2010-10-14 | Lintec Corp | 太陽電池モジュール用裏面保護シート及び太陽電池モジュール |
| US20120198780A1 (en) * | 2009-10-14 | 2012-08-09 | Adco Products, Inc. | Method for attaching a solar module to a substrate using an adhesive |
| JP5606956B2 (ja) * | 2010-02-23 | 2014-10-15 | 富士フイルム株式会社 | 太陽電池用バックシート及びその製造方法、並びに太陽電池モジュール |
-
2011
- 2011-03-25 JP JP2011068658A patent/JP5623952B2/ja not_active Expired - Fee Related
-
2012
- 2012-03-22 KR KR1020137023639A patent/KR101663554B1/ko active IP Right Grant
- 2012-03-22 CN CN201280011843.0A patent/CN103430321B/zh active Active
- 2012-03-22 WO PCT/JP2012/057390 patent/WO2012133124A1/ja active Application Filing
-
2013
- 2013-08-29 US US14/013,899 patent/US20130340829A1/en not_active Abandoned
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002083988A (ja) * | 2000-09-08 | 2002-03-22 | Dainippon Printing Co Ltd | 太陽電池モジュ−ル用裏面保護シ−トおよびそれを使用した太陽電池モジュ−ル |
| WO2006067984A1 (ja) * | 2004-12-21 | 2006-06-29 | Toyo Aluminium Kabushiki Kaisha | 太陽電池用シート部材 |
| WO2009133827A1 (ja) * | 2008-04-28 | 2009-11-05 | 旭化成ケミカルズ株式会社 | 太陽電池バックシート用積層体およびそれを有するバックシート |
| WO2010110119A1 (ja) * | 2009-03-26 | 2010-09-30 | 東レ株式会社 | 太陽電池用ポリエステルフィルム、それを用いた太陽電池バックシート、および太陽電池 |
| JP2012049331A (ja) * | 2010-08-26 | 2012-03-08 | Fujifilm Corp | 太陽電池保護シート及びその製造方法、太陽電池用バックシート並びに太陽電池モジュール |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5623952B2 (ja) | 2014-11-12 |
| CN103430321A (zh) | 2013-12-04 |
| CN103430321B (zh) | 2016-11-16 |
| JP2012204664A (ja) | 2012-10-22 |
| US20130340829A1 (en) | 2013-12-26 |
| KR101663554B1 (ko) | 2016-10-07 |
| KR20140071954A (ko) | 2014-06-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5815276B2 (ja) | 太陽電池用バックシート用ポリマーシート及びその製造方法並びに太陽電池モジュール | |
| JP5587230B2 (ja) | 太陽電池用バックシート及びその製造方法、並びに太陽電池モジュール | |
| JP5623952B2 (ja) | 太陽電池用ポリマーシート及びその製造方法、太陽電池用バックシート、並びに太陽電池モジュール | |
| KR101649054B1 (ko) | 태양 전지용 보호 시트와 그 제조 방법, 태양 전지용 백시트, 태양 전지 모듈 | |
| JP5752733B2 (ja) | ポリエステルフィルム、太陽電池用バックシート、および、太陽電池モジュール | |
| JP5722287B2 (ja) | 太陽電池用バックシート及びその製造方法並びに太陽電池モジュール | |
| JP5606956B2 (ja) | 太陽電池用バックシート及びその製造方法、並びに太陽電池モジュール | |
| WO2013024892A1 (ja) | 太陽電池用バックシート及びその製造方法、並びに太陽電池モジュール | |
| JP2013042007A (ja) | 太陽電池モジュール用バックシートおよび太陽電池モジュール | |
| JP5763021B2 (ja) | 太陽電池用ポリマーシート及びその製造方法、並びに太陽電池モジュール | |
| WO2013008945A1 (ja) | 太陽電池用ポリマーシート及び太陽電池モジュール | |
| JP5653853B2 (ja) | 太陽電池用バックシート及びその製造方法並びに太陽電池モジュール | |
| JP5562889B2 (ja) | 積層フィルム、太陽電池用バックシートおよび積層フィルムの製造方法 | |
| JP6215273B2 (ja) | 太陽電池用保護シート及びその製造方法、並びに太陽電池モジュール | |
| JP2013042016A (ja) | 太陽電池用ポリマーシート、太陽電池用バックシート、及び太陽電池モジュール | |
| JP2015188015A (ja) | 太陽電池裏面保護用積層ポリエステルフィルム、太陽電池裏面保護シート、及び太陽電池モジュール | |
| JP2012049331A (ja) | 太陽電池保護シート及びその製造方法、太陽電池用バックシート並びに太陽電池モジュール | |
| JP2011060791A (ja) | 太陽電池モジュール用裏面保護シートおよびそれを用いた太陽電池モジュール | |
| WO2013146180A1 (ja) | 太陽電池用裏面保護シートおよび太陽電池モジュール | |
| WO2013115119A1 (ja) | 2軸延伸ポリエステルフィルム、太陽電池モジュール用バックシートおよび太陽電池モジュール | |
| JP5694881B2 (ja) | 太陽電池用ポリマーシート、太陽電池用バックシート、及び太陽電池モジュール |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12765124 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20137023639 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 12765124 Country of ref document: EP Kind code of ref document: A1 |