WO2012132855A1 - 光電変換素子及び光電気化学電池 - Google Patents
光電変換素子及び光電気化学電池 Download PDFInfo
- Publication number
- WO2012132855A1 WO2012132855A1 PCT/JP2012/056259 JP2012056259W WO2012132855A1 WO 2012132855 A1 WO2012132855 A1 WO 2012132855A1 JP 2012056259 W JP2012056259 W JP 2012056259W WO 2012132855 A1 WO2012132855 A1 WO 2012132855A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- metal
- semiconductor fine
- fine particles
- photoelectric conversion
- Prior art date
Links
- 239000004065 semiconductor Substances 0.000 claims abstract description 212
- 229910052751 metal Inorganic materials 0.000 claims abstract description 128
- 239000002184 metal Substances 0.000 claims abstract description 128
- 239000003446 ligand Substances 0.000 claims abstract description 65
- 150000001875 compounds Chemical class 0.000 claims abstract description 48
- 150000002736 metal compounds Chemical class 0.000 claims abstract description 30
- 150000002739 metals Chemical class 0.000 claims abstract description 27
- 238000012546 transfer Methods 0.000 claims abstract description 14
- 239000010419 fine particle Substances 0.000 claims description 216
- -1 thiocarbonate group Chemical group 0.000 claims description 157
- 238000006243 chemical reaction Methods 0.000 claims description 97
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 claims description 61
- 125000004429 atom Chemical group 0.000 claims description 40
- 125000000217 alkyl group Chemical group 0.000 claims description 38
- 150000004770 chalcogenides Chemical class 0.000 claims description 36
- 125000001424 substituent group Chemical group 0.000 claims description 36
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 claims description 34
- 239000002245 particle Substances 0.000 claims description 33
- 125000003118 aryl group Chemical group 0.000 claims description 29
- 125000000623 heterocyclic group Chemical group 0.000 claims description 29
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 claims description 26
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 26
- 125000003545 alkoxy group Chemical group 0.000 claims description 23
- 239000010936 titanium Substances 0.000 claims description 23
- 239000011258 core-shell material Substances 0.000 claims description 21
- 150000002500 ions Chemical class 0.000 claims description 21
- 125000003342 alkenyl group Chemical group 0.000 claims description 20
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 20
- 229910052719 titanium Inorganic materials 0.000 claims description 19
- 229910001887 tin oxide Inorganic materials 0.000 claims description 18
- 229910052782 aluminium Inorganic materials 0.000 claims description 16
- 239000004020 conductor Substances 0.000 claims description 16
- 108091008695 photoreceptors Proteins 0.000 claims description 16
- 239000011135 tin Substances 0.000 claims description 16
- XOLBLPGZBRYERU-UHFFFAOYSA-N tin dioxide Chemical compound O=[Sn]=O XOLBLPGZBRYERU-UHFFFAOYSA-N 0.000 claims description 16
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 claims description 15
- 125000004104 aryloxy group Chemical group 0.000 claims description 15
- 239000002253 acid Substances 0.000 claims description 14
- 125000005843 halogen group Chemical group 0.000 claims description 14
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 claims description 14
- 229910052718 tin Inorganic materials 0.000 claims description 14
- 229910000019 calcium carbonate Inorganic materials 0.000 claims description 13
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 13
- 239000000395 magnesium oxide Substances 0.000 claims description 13
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 claims description 13
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 claims description 13
- 229910001960 metal nitrate Inorganic materials 0.000 claims description 13
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 12
- ZBKFYXZXZJPWNQ-UHFFFAOYSA-N isothiocyanate group Chemical group [N-]=C=S ZBKFYXZXZJPWNQ-UHFFFAOYSA-N 0.000 claims description 12
- 229910052715 tantalum Inorganic materials 0.000 claims description 12
- 125000004423 acyloxy group Chemical group 0.000 claims description 11
- 229910021389 graphene Inorganic materials 0.000 claims description 11
- 229910052758 niobium Inorganic materials 0.000 claims description 11
- 229910052720 vanadium Inorganic materials 0.000 claims description 11
- 239000011701 zinc Substances 0.000 claims description 11
- 125000004414 alkyl thio group Chemical group 0.000 claims description 10
- 125000005110 aryl thio group Chemical group 0.000 claims description 10
- 229910044991 metal oxide Inorganic materials 0.000 claims description 10
- 150000004706 metal oxides Chemical class 0.000 claims description 10
- 125000005499 phosphonyl group Chemical group 0.000 claims description 10
- NEAQRZUHTPSBBM-UHFFFAOYSA-N 2-hydroxy-3,3-dimethyl-7-nitro-4h-isoquinolin-1-one Chemical compound C1=C([N+]([O-])=O)C=C2C(=O)N(O)C(C)(C)CC2=C1 NEAQRZUHTPSBBM-UHFFFAOYSA-N 0.000 claims description 9
- 230000002378 acidificating effect Effects 0.000 claims description 9
- 239000000654 additive Substances 0.000 claims description 9
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 9
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M thiocyanate group Chemical group [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 claims description 9
- 229910052725 zinc Inorganic materials 0.000 claims description 9
- 125000002252 acyl group Chemical group 0.000 claims description 8
- 229910052802 copper Inorganic materials 0.000 claims description 8
- 239000011777 magnesium Substances 0.000 claims description 8
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 claims description 7
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 7
- XLJMAIOERFSOGZ-UHFFFAOYSA-M cyanate group Chemical group [O-]C#N XLJMAIOERFSOGZ-UHFFFAOYSA-M 0.000 claims description 7
- 125000005678 ethenylene group Chemical group [H]C([*:1])=C([H])[*:2] 0.000 claims description 7
- IQPQWNKOIGAROB-UHFFFAOYSA-N isocyanate group Chemical group [N-]=C=O IQPQWNKOIGAROB-UHFFFAOYSA-N 0.000 claims description 7
- 229910052749 magnesium Inorganic materials 0.000 claims description 7
- 125000000542 sulfonic acid group Chemical group 0.000 claims description 7
- 229910052726 zirconium Inorganic materials 0.000 claims description 7
- AYJRCSIUFZENHW-UHFFFAOYSA-L barium carbonate Chemical compound [Ba+2].[O-]C([O-])=O AYJRCSIUFZENHW-UHFFFAOYSA-L 0.000 claims description 6
- 150000002576 ketones Chemical class 0.000 claims description 6
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 claims description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 6
- HIZCIEIDIFGZSS-UHFFFAOYSA-L trithiocarbonate Chemical group [S-]C([S-])=S HIZCIEIDIFGZSS-UHFFFAOYSA-L 0.000 claims description 6
- 125000005035 acylthio group Chemical group 0.000 claims description 5
- 230000000996 additive effect Effects 0.000 claims description 5
- GNVMUORYQLCPJZ-UHFFFAOYSA-N carbamothioic s-acid Chemical group NC(S)=O GNVMUORYQLCPJZ-UHFFFAOYSA-N 0.000 claims description 5
- 125000005521 carbonamide group Chemical group 0.000 claims description 5
- 150000004649 carbonic acid derivatives Chemical class 0.000 claims description 5
- 229910052709 silver Inorganic materials 0.000 claims description 5
- 125000005677 ethinylene group Chemical group [*:2]C#C[*:1] 0.000 claims description 4
- 229910052737 gold Inorganic materials 0.000 claims description 4
- 229910052738 indium Inorganic materials 0.000 claims description 4
- FYDKNKUEBJQCCN-UHFFFAOYSA-N lanthanum(3+);trinitrate Chemical group [La+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O FYDKNKUEBJQCCN-UHFFFAOYSA-N 0.000 claims description 4
- 229910052712 strontium Inorganic materials 0.000 claims description 4
- 229910052721 tungsten Inorganic materials 0.000 claims description 4
- BGPJLYIFDLICMR-UHFFFAOYSA-N 1,4,2,3-dioxadithiolan-5-one Chemical group O=C1OSSO1 BGPJLYIFDLICMR-UHFFFAOYSA-N 0.000 claims description 3
- WUPHOULIZUERAE-UHFFFAOYSA-N 3-(oxolan-2-yl)propanoic acid Chemical compound OC(=O)CCC1CCCO1 WUPHOULIZUERAE-UHFFFAOYSA-N 0.000 claims description 3
- 229910052684 Cerium Inorganic materials 0.000 claims description 3
- 229910052980 cadmium sulfide Inorganic materials 0.000 claims description 3
- UHYPYGJEEGLRJD-UHFFFAOYSA-N cadmium(2+);selenium(2-) Chemical compound [Se-2].[Cd+2] UHYPYGJEEGLRJD-UHFFFAOYSA-N 0.000 claims description 3
- 229910052735 hafnium Inorganic materials 0.000 claims description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 3
- 229910052727 yttrium Inorganic materials 0.000 claims description 3
- DKVNPHBNOWQYFE-UHFFFAOYSA-N carbamodithioic acid Chemical group NC(S)=S DKVNPHBNOWQYFE-UHFFFAOYSA-N 0.000 claims 1
- 239000000049 pigment Substances 0.000 abstract description 9
- 239000000758 substrate Substances 0.000 abstract description 9
- 239000011859 microparticle Substances 0.000 abstract 2
- 239000000975 dye Substances 0.000 description 138
- 238000000034 method Methods 0.000 description 100
- 125000004432 carbon atom Chemical group C* 0.000 description 87
- 239000010410 layer Substances 0.000 description 78
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 60
- 239000006185 dispersion Substances 0.000 description 34
- 239000000243 solution Substances 0.000 description 31
- 239000010408 film Substances 0.000 description 30
- 238000002360 preparation method Methods 0.000 description 27
- 239000000203 mixture Substances 0.000 description 26
- 239000000463 material Substances 0.000 description 25
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 24
- 239000002904 solvent Substances 0.000 description 21
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 20
- 239000003792 electrolyte Substances 0.000 description 19
- 238000010521 absorption reaction Methods 0.000 description 18
- 125000003277 amino group Chemical group 0.000 description 16
- 239000000178 monomer Substances 0.000 description 14
- 239000010955 niobium Substances 0.000 description 14
- 125000004442 acylamino group Chemical group 0.000 description 13
- 229910052799 carbon Inorganic materials 0.000 description 13
- 239000011248 coating agent Substances 0.000 description 13
- 238000000576 coating method Methods 0.000 description 13
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 12
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 12
- 230000015572 biosynthetic process Effects 0.000 description 12
- 239000011521 glass Substances 0.000 description 12
- 238000010438 heat treatment Methods 0.000 description 11
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Substances [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 11
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 10
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 10
- 125000000304 alkynyl group Chemical group 0.000 description 10
- 239000007864 aqueous solution Substances 0.000 description 10
- 238000005259 measurement Methods 0.000 description 10
- 238000001179 sorption measurement Methods 0.000 description 10
- 238000011282 treatment Methods 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 125000000753 cycloalkyl group Chemical group 0.000 description 9
- 239000012153 distilled water Substances 0.000 description 9
- 230000006870 function Effects 0.000 description 9
- 239000007788 liquid Substances 0.000 description 9
- 238000003756 stirring Methods 0.000 description 9
- 238000003786 synthesis reaction Methods 0.000 description 9
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 8
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 8
- DKVNPHBNOWQYFE-UHFFFAOYSA-M carbamodithioate Chemical group NC([S-])=S DKVNPHBNOWQYFE-UHFFFAOYSA-M 0.000 description 8
- 239000010949 copper Substances 0.000 description 8
- 230000005525 hole transport Effects 0.000 description 8
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 8
- 229920000642 polymer Polymers 0.000 description 8
- 230000001235 sensitizing effect Effects 0.000 description 8
- 230000003595 spectral effect Effects 0.000 description 8
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 7
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 7
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 7
- 229910052740 iodine Inorganic materials 0.000 description 7
- DNIAPMSPPWPWGF-UHFFFAOYSA-N monopropylene glycol Natural products CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 7
- 229920005989 resin Polymers 0.000 description 7
- 239000011347 resin Substances 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical group C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 6
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 6
- 230000005540 biological transmission Effects 0.000 description 6
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 6
- 150000001721 carbon Chemical group 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 229910002804 graphite Inorganic materials 0.000 description 6
- 239000010439 graphite Substances 0.000 description 6
- 229910052697 platinum Inorganic materials 0.000 description 6
- 239000002243 precursor Substances 0.000 description 6
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 5
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 5
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 5
- 239000013078 crystal Substances 0.000 description 5
- 238000009826 distribution Methods 0.000 description 5
- 239000008151 electrolyte solution Substances 0.000 description 5
- 239000011630 iodine Substances 0.000 description 5
- 239000002861 polymer material Substances 0.000 description 5
- 238000006116 polymerization reaction Methods 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 4
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 4
- 239000002202 Polyethylene glycol Substances 0.000 description 4
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 4
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 229910010413 TiO 2 Inorganic materials 0.000 description 4
- 239000011230 binding agent Substances 0.000 description 4
- 239000001913 cellulose Substances 0.000 description 4
- 229920002678 cellulose Polymers 0.000 description 4
- 235000010980 cellulose Nutrition 0.000 description 4
- 229910052801 chlorine Inorganic materials 0.000 description 4
- 229920001940 conductive polymer Polymers 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 238000011156 evaluation Methods 0.000 description 4
- 229910052731 fluorine Inorganic materials 0.000 description 4
- 239000010931 gold Substances 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- 125000002883 imidazolyl group Chemical group 0.000 description 4
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 238000002156 mixing Methods 0.000 description 4
- 125000002950 monocyclic group Chemical group 0.000 description 4
- 229910017604 nitric acid Inorganic materials 0.000 description 4
- 125000003386 piperidinyl group Chemical group 0.000 description 4
- 229920001223 polyethylene glycol Polymers 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 239000002002 slurry Substances 0.000 description 4
- 239000008279 sol Substances 0.000 description 4
- 238000003980 solgel method Methods 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- VXUYXOFXAQZZMF-UHFFFAOYSA-N titanium(IV) isopropoxide Chemical compound CC(C)O[Ti](OC(C)C)(OC(C)C)OC(C)C VXUYXOFXAQZZMF-UHFFFAOYSA-N 0.000 description 4
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- 0 C*c1ccc(C#*)[s]1 Chemical compound C*c1ccc(C#*)[s]1 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical group C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 3
- 229910006404 SnO 2 Inorganic materials 0.000 description 3
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical group C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 125000001931 aliphatic group Chemical group 0.000 description 3
- 125000000751 azo group Chemical group [*]N=N[*] 0.000 description 3
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzonitrile Chemical compound N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 description 3
- VSGNNIFQASZAOI-UHFFFAOYSA-L calcium acetate Chemical compound [Ca+2].CC([O-])=O.CC([O-])=O VSGNNIFQASZAOI-UHFFFAOYSA-L 0.000 description 3
- 239000001639 calcium acetate Substances 0.000 description 3
- 229960005147 calcium acetate Drugs 0.000 description 3
- 235000011092 calcium acetate Nutrition 0.000 description 3
- 229910021393 carbon nanotube Inorganic materials 0.000 description 3
- 239000002041 carbon nanotube Substances 0.000 description 3
- 125000001309 chloro group Chemical group Cl* 0.000 description 3
- 230000000052 comparative effect Effects 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 3
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 238000010304 firing Methods 0.000 description 3
- 239000011245 gel electrolyte Substances 0.000 description 3
- 239000012535 impurity Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 150000002496 iodine Chemical class 0.000 description 3
- XEEYBQQBJWHFJM-UHFFFAOYSA-N iron Substances [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 125000005647 linker group Chemical group 0.000 description 3
- HSZCZNFXUDYRKD-UHFFFAOYSA-M lithium iodide Chemical compound [Li+].[I-] HSZCZNFXUDYRKD-UHFFFAOYSA-M 0.000 description 3
- 238000002844 melting Methods 0.000 description 3
- 230000008018 melting Effects 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 125000001624 naphthyl group Chemical group 0.000 description 3
- PXHVJJICTQNCMI-UHFFFAOYSA-N nickel Substances [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 239000012044 organic layer Substances 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 125000002971 oxazolyl group Chemical group 0.000 description 3
- 125000005328 phosphinyl group Chemical group [PH2](=O)* 0.000 description 3
- 239000003505 polymerization initiator Substances 0.000 description 3
- 229920001451 polypropylene glycol Polymers 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 238000010526 radical polymerization reaction Methods 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 229910052710 silicon Inorganic materials 0.000 description 3
- 239000010703 silicon Substances 0.000 description 3
- 239000004332 silver Substances 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- 125000001425 triazolyl group Chemical group 0.000 description 3
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- WUOACPNHFRMFPN-SECBINFHSA-N (S)-(-)-alpha-terpineol Chemical compound CC1=CC[C@@H](C(C)(C)O)CC1 WUOACPNHFRMFPN-SECBINFHSA-N 0.000 description 2
- MYRTYDVEIRVNKP-UHFFFAOYSA-N 1,2-Divinylbenzene Chemical compound C=CC1=CC=CC=C1C=C MYRTYDVEIRVNKP-UHFFFAOYSA-N 0.000 description 2
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 2
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 2
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 2
- LEJBBGNFPAFPKQ-UHFFFAOYSA-N 2-(2-prop-2-enoyloxyethoxy)ethyl prop-2-enoate Chemical compound C=CC(=O)OCCOCCOC(=O)C=C LEJBBGNFPAFPKQ-UHFFFAOYSA-N 0.000 description 2
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 2
- XFCMNSHQOZQILR-UHFFFAOYSA-N 2-[2-(2-methylprop-2-enoyloxy)ethoxy]ethyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCCOCCOC(=O)C(C)=C XFCMNSHQOZQILR-UHFFFAOYSA-N 0.000 description 2
- INQDDHNZXOAFFD-UHFFFAOYSA-N 2-[2-(2-prop-2-enoyloxyethoxy)ethoxy]ethyl prop-2-enoate Chemical compound C=CC(=O)OCCOCCOCCOC(=O)C=C INQDDHNZXOAFFD-UHFFFAOYSA-N 0.000 description 2
- BSKHPKMHTQYZBB-UHFFFAOYSA-N 2-methylpyridine Chemical compound CC1=CC=CC=N1 BSKHPKMHTQYZBB-UHFFFAOYSA-N 0.000 description 2
- KUDUQBURMYMBIJ-UHFFFAOYSA-N 2-prop-2-enoyloxyethyl prop-2-enoate Chemical compound C=CC(=O)OCCOC(=O)C=C KUDUQBURMYMBIJ-UHFFFAOYSA-N 0.000 description 2
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 2
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 2
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 2
- YSHMQTRICHYLGF-UHFFFAOYSA-N 4-tert-butylpyridine Chemical compound CC(C)(C)C1=CC=NC=C1 YSHMQTRICHYLGF-UHFFFAOYSA-N 0.000 description 2
- UJOBWOGCFQCDNV-UHFFFAOYSA-N 9H-carbazole Chemical compound C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 2
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 2
- 239000004925 Acrylic resin Substances 0.000 description 2
- 229910018072 Al 2 O 3 Inorganic materials 0.000 description 2
- KAKZBPTYRLMSJV-UHFFFAOYSA-N Butadiene Chemical compound C=CC=C KAKZBPTYRLMSJV-UHFFFAOYSA-N 0.000 description 2
- SOGAXMICEFXMKE-UHFFFAOYSA-N Butylmethacrylate Chemical compound CCCCOC(=O)C(C)=C SOGAXMICEFXMKE-UHFFFAOYSA-N 0.000 description 2
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 2
- 239000001856 Ethyl cellulose Substances 0.000 description 2
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 2
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical compound CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 2
- RRHGJUQNOFWUDK-UHFFFAOYSA-N Isoprene Chemical compound CC(=C)C=C RRHGJUQNOFWUDK-UHFFFAOYSA-N 0.000 description 2
- BAPJBEWLBFYGME-UHFFFAOYSA-N Methyl acrylate Chemical compound COC(=O)C=C BAPJBEWLBFYGME-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical group C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 239000004697 Polyetherimide Substances 0.000 description 2
- 239000004734 Polyphenylene sulfide Substances 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 2
- 239000012327 Ruthenium complex Substances 0.000 description 2
- 206010070834 Sensitisation Diseases 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 229920010524 Syndiotactic polystyrene Polymers 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical group C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 2
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 2
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 description 2
- 238000000862 absorption spectrum Methods 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- YRKCREAYFQTBPV-UHFFFAOYSA-N acetylacetone Chemical compound CC(=O)CC(C)=O YRKCREAYFQTBPV-UHFFFAOYSA-N 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 150000004703 alkoxides Chemical class 0.000 description 2
- 125000004466 alkoxycarbonylamino group Chemical group 0.000 description 2
- 125000005194 alkoxycarbonyloxy group Chemical group 0.000 description 2
- 150000001346 alkyl aryl ethers Chemical class 0.000 description 2
- OVKDFILSBMEKLT-UHFFFAOYSA-N alpha-Terpineol Natural products CC(=C)C1(O)CCC(C)=CC1 OVKDFILSBMEKLT-UHFFFAOYSA-N 0.000 description 2
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 2
- 229940088601 alpha-terpineol Drugs 0.000 description 2
- 150000001408 amides Chemical group 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 125000006598 aminocarbonylamino group Chemical group 0.000 description 2
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 2
- 125000005162 aryl oxy carbonyl amino group Chemical group 0.000 description 2
- 125000005161 aryl oxy carbonyl group Chemical group 0.000 description 2
- 125000005135 aryl sulfinyl group Chemical group 0.000 description 2
- 125000004657 aryl sulfonyl amino group Chemical group 0.000 description 2
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 2
- 125000005200 aryloxy carbonyloxy group Chemical group 0.000 description 2
- 230000004888 barrier function Effects 0.000 description 2
- 150000007514 bases Chemical class 0.000 description 2
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 2
- 150000001602 bicycloalkyls Chemical group 0.000 description 2
- LYQFWZFBNBDLEO-UHFFFAOYSA-M caesium bromide Chemical compound [Br-].[Cs+] LYQFWZFBNBDLEO-UHFFFAOYSA-M 0.000 description 2
- 239000003575 carbonaceous material Substances 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 2
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 2
- 150000001768 cations Chemical class 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- QMVPMAAFGQKVCJ-UHFFFAOYSA-N citronellol Chemical compound OCCC(C)CCC=C(C)C QMVPMAAFGQKVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000002131 composite material Substances 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 238000004132 cross linking Methods 0.000 description 2
- 239000003431 cross linking reagent Substances 0.000 description 2
- 125000000392 cycloalkenyl group Chemical group 0.000 description 2
- ZSWFCLXCOIISFI-UHFFFAOYSA-N cyclopentadiene Chemical compound C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 2
- 238000000151 deposition Methods 0.000 description 2
- 150000001983 dialkylethers Chemical class 0.000 description 2
- 238000009792 diffusion process Methods 0.000 description 2
- 229920001971 elastomer Polymers 0.000 description 2
- 238000004070 electrodeposition Methods 0.000 description 2
- 238000001962 electrophoresis Methods 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 2
- DQYBDCGIPTYXML-UHFFFAOYSA-N ethoxyethane;hydrate Chemical compound O.CCOCC DQYBDCGIPTYXML-UHFFFAOYSA-N 0.000 description 2
- 229920001249 ethyl cellulose Polymers 0.000 description 2
- 235000019325 ethyl cellulose Nutrition 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 239000003349 gelling agent Substances 0.000 description 2
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 230000003301 hydrolyzing effect Effects 0.000 description 2
- 150000004693 imidazolium salts Chemical class 0.000 description 2
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 2
- 239000003999 initiator Substances 0.000 description 2
- 229910010272 inorganic material Inorganic materials 0.000 description 2
- 229910052742 iron Inorganic materials 0.000 description 2
- 230000001678 irradiating effect Effects 0.000 description 2
- 229910052746 lanthanum Inorganic materials 0.000 description 2
- AMXOYNBUYSYVKV-UHFFFAOYSA-M lithium bromide Chemical compound [Li+].[Br-] AMXOYNBUYSYVKV-UHFFFAOYSA-M 0.000 description 2
- UEGPKNKPLBYCNK-UHFFFAOYSA-L magnesium acetate Chemical compound [Mg+2].CC([O-])=O.CC([O-])=O UEGPKNKPLBYCNK-UHFFFAOYSA-L 0.000 description 2
- 239000011654 magnesium acetate Substances 0.000 description 2
- 235000011285 magnesium acetate Nutrition 0.000 description 2
- 229940069446 magnesium acetate Drugs 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 239000011976 maleic acid Substances 0.000 description 2
- 239000011259 mixed solution Substances 0.000 description 2
- 229910052759 nickel Inorganic materials 0.000 description 2
- GUCVJGMIXFAOAE-UHFFFAOYSA-N niobium atom Chemical compound [Nb] GUCVJGMIXFAOAE-UHFFFAOYSA-N 0.000 description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 125000001117 oleyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])/C([H])=C([H])\C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000007800 oxidant agent Substances 0.000 description 2
- 230000001590 oxidative effect Effects 0.000 description 2
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- 150000002978 peroxides Chemical class 0.000 description 2
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- 229920000548 poly(silane) polymer Polymers 0.000 description 2
- 229920002492 poly(sulfone) Polymers 0.000 description 2
- 229920001230 polyarylate Polymers 0.000 description 2
- 229920001601 polyetherimide Polymers 0.000 description 2
- 229920000139 polyethylene terephthalate Polymers 0.000 description 2
- 239000005020 polyethylene terephthalate Substances 0.000 description 2
- 229920000069 polyphenylene sulfide Polymers 0.000 description 2
- IOLCXVTUBQKXJR-UHFFFAOYSA-M potassium bromide Chemical compound [K+].[Br-] IOLCXVTUBQKXJR-UHFFFAOYSA-M 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 238000007639 printing Methods 0.000 description 2
- 125000003373 pyrazinyl group Chemical group 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 229910052703 rhodium Inorganic materials 0.000 description 2
- 239000010948 rhodium Substances 0.000 description 2
- 229910052707 ruthenium Inorganic materials 0.000 description 2
- 238000007789 sealing Methods 0.000 description 2
- 230000008313 sensitization Effects 0.000 description 2
- 125000004469 siloxy group Chemical group [SiH3]O* 0.000 description 2
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 2
- JHJLBTNAGRQEKS-UHFFFAOYSA-M sodium bromide Chemical compound [Na+].[Br-] JHJLBTNAGRQEKS-UHFFFAOYSA-M 0.000 description 2
- 238000004544 sputter deposition Methods 0.000 description 2
- 125000000565 sulfonamide group Chemical group 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- GUVRBAGPIYLISA-UHFFFAOYSA-N tantalum atom Chemical compound [Ta] GUVRBAGPIYLISA-UHFFFAOYSA-N 0.000 description 2
- 125000005207 tetraalkylammonium group Chemical group 0.000 description 2
- VDZOOKBUILJEDG-UHFFFAOYSA-M tetrabutylammonium hydroxide Chemical compound [OH-].CCCC[N+](CCCC)(CCCC)CCCC VDZOOKBUILJEDG-UHFFFAOYSA-M 0.000 description 2
- 239000010409 thin film Substances 0.000 description 2
- 125000004149 thio group Chemical group *S* 0.000 description 2
- XJDNKRIXUMDJCW-UHFFFAOYSA-J titanium tetrachloride Chemical compound Cl[Ti](Cl)(Cl)Cl XJDNKRIXUMDJCW-UHFFFAOYSA-J 0.000 description 2
- 230000032258 transport Effects 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 239000011882 ultra-fine particle Substances 0.000 description 2
- GPPXJZIENCGNKB-UHFFFAOYSA-N vanadium Chemical compound [V]#[V] GPPXJZIENCGNKB-UHFFFAOYSA-N 0.000 description 2
- 229920002554 vinyl polymer Polymers 0.000 description 2
- FDPPXZRLXIPXJB-UHFFFAOYSA-N (2-methyl-2-nitropropyl) prop-2-enoate Chemical compound [O-][N+](=O)C(C)(C)COC(=O)C=C FDPPXZRLXIPXJB-UHFFFAOYSA-N 0.000 description 1
- GNYGBUNVQVEWRG-UHFFFAOYSA-N (2-methyl-3-bicyclo[2.2.1]heptanyl)methyl 2-methylprop-2-enoate Chemical compound C1CC2C(COC(=O)C(C)=C)C(C)C1C2 GNYGBUNVQVEWRG-UHFFFAOYSA-N 0.000 description 1
- BHQCQFFYRZLCQQ-UHFFFAOYSA-N (3alpha,5alpha,7alpha,12alpha)-3,7,12-trihydroxy-cholan-24-oic acid Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 BHQCQFFYRZLCQQ-UHFFFAOYSA-N 0.000 description 1
- 229920002818 (Hydroxyethyl)methacrylate Polymers 0.000 description 1
- QMVPMAAFGQKVCJ-SNVBAGLBSA-N (R)-(+)-citronellol Natural products OCC[C@H](C)CCC=C(C)C QMVPMAAFGQKVCJ-SNVBAGLBSA-N 0.000 description 1
- DYLIWHYUXAJDOJ-OWOJBTEDSA-N (e)-4-(6-aminopurin-9-yl)but-2-en-1-ol Chemical compound NC1=NC=NC2=C1N=CN2C\C=C\CO DYLIWHYUXAJDOJ-OWOJBTEDSA-N 0.000 description 1
- SHXHPUAKLCCLDV-UHFFFAOYSA-N 1,1,1-trifluoropentane-2,4-dione Chemical compound CC(=O)CC(=O)C(F)(F)F SHXHPUAKLCCLDV-UHFFFAOYSA-N 0.000 description 1
- UIQCRIFSBWGDTQ-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5,5,6,6,7,7,10,10,10-heptadecafluorodecyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)CCC(F)(F)F UIQCRIFSBWGDTQ-UHFFFAOYSA-N 0.000 description 1
- OVQQQQUJAGEBHH-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5,5,6,6,7,7,10,10,10-heptadecafluorodecyl prop-2-enoate Chemical compound FC(F)(F)CCC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)OC(=O)C=C OVQQQQUJAGEBHH-UHFFFAOYSA-N 0.000 description 1
- GWYSWOQRJGLJPA-UHFFFAOYSA-N 1,1,2,2-tetrafluoropropyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OC(F)(F)C(C)(F)F GWYSWOQRJGLJPA-UHFFFAOYSA-N 0.000 description 1
- CEXMTZSYTLNAOG-UHFFFAOYSA-N 1,1,2,3,3,3-hexafluoropropyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OC(F)(F)C(F)C(F)(F)F CEXMTZSYTLNAOG-UHFFFAOYSA-N 0.000 description 1
- BQCIDUSAKPWEOX-UHFFFAOYSA-N 1,1-Difluoroethene Chemical compound FC(F)=C BQCIDUSAKPWEOX-UHFFFAOYSA-N 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 description 1
- FRZPYEHDSAQGAS-UHFFFAOYSA-M 1-butyl-3-methylimidazol-3-ium;trifluoromethanesulfonate Chemical compound [O-]S(=O)(=O)C(F)(F)F.CCCC[N+]=1C=CN(C)C=1 FRZPYEHDSAQGAS-UHFFFAOYSA-M 0.000 description 1
- KTZVZZJJVJQZHV-UHFFFAOYSA-N 1-chloro-4-ethenylbenzene Chemical compound ClC1=CC=C(C=C)C=C1 KTZVZZJJVJQZHV-UHFFFAOYSA-N 0.000 description 1
- LGJCFVYMIJLQJO-UHFFFAOYSA-N 1-dodecylperoxydodecane Chemical compound CCCCCCCCCCCCOOCCCCCCCCCCCC LGJCFVYMIJLQJO-UHFFFAOYSA-N 0.000 description 1
- ZPTRYWVRCNOTAS-UHFFFAOYSA-M 1-ethyl-3-methylimidazol-3-ium;trifluoromethanesulfonate Chemical compound CC[N+]=1C=CN(C)C=1.[O-]S(=O)(=O)C(F)(F)F ZPTRYWVRCNOTAS-UHFFFAOYSA-M 0.000 description 1
- 125000006219 1-ethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- IOWQHNLDXBGENT-UHFFFAOYSA-N 1-hexyl-4-(1-hexylpyridin-1-ium-4-yl)pyridin-1-ium Chemical compound C1=C[N+](CCCCCC)=CC=C1C1=CC=[N+](CCCCCC)C=C1 IOWQHNLDXBGENT-UHFFFAOYSA-N 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- HIDBROSJWZYGSZ-UHFFFAOYSA-N 1-phenylpyrrole-2,5-dione Chemical compound O=C1C=CC(=O)N1C1=CC=CC=C1 HIDBROSJWZYGSZ-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- QTKPMCIBUROOGY-UHFFFAOYSA-N 2,2,2-trifluoroethyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCC(F)(F)F QTKPMCIBUROOGY-UHFFFAOYSA-N 0.000 description 1
- VBHXIMACZBQHPX-UHFFFAOYSA-N 2,2,2-trifluoroethyl prop-2-enoate Chemical compound FC(F)(F)COC(=O)C=C VBHXIMACZBQHPX-UHFFFAOYSA-N 0.000 description 1
- YRAJNWYBUCUFBD-UHFFFAOYSA-N 2,2,6,6-tetramethylheptane-3,5-dione Chemical compound CC(C)(C)C(=O)CC(=O)C(C)(C)C YRAJNWYBUCUFBD-UHFFFAOYSA-N 0.000 description 1
- DEJYALGDDUTZFR-UHFFFAOYSA-N 2,2-dimethylbutyl prop-2-enoate Chemical compound CCC(C)(C)COC(=O)C=C DEJYALGDDUTZFR-UHFFFAOYSA-N 0.000 description 1
- LWHDQPLUIFIFFT-UHFFFAOYSA-N 2,3,5,6-tetrabromocyclohexa-2,5-diene-1,4-dione Chemical group BrC1=C(Br)C(=O)C(Br)=C(Br)C1=O LWHDQPLUIFIFFT-UHFFFAOYSA-N 0.000 description 1
- HQJLEFDAYKUXSA-UHFFFAOYSA-N 2,3-dihydroxycyclohexa-2,5-diene-1,4-dione Chemical compound OC1=C(O)C(=O)C=CC1=O HQJLEFDAYKUXSA-UHFFFAOYSA-N 0.000 description 1
- SZTSOGYCXBVMMT-UHFFFAOYSA-N 2,4-dimethyl-1-propylimidazole;hydroiodide Chemical compound [I-].CCC[NH+]1C=C(C)N=C1C SZTSOGYCXBVMMT-UHFFFAOYSA-N 0.000 description 1
- NTFJXDRAVMOYBG-UHFFFAOYSA-N 2-(2,2-dicyanoethoxymethyl)propanedinitrile Chemical compound N#CC(C#N)COCC(C#N)C#N NTFJXDRAVMOYBG-UHFFFAOYSA-N 0.000 description 1
- FTALTLPZDVFJSS-UHFFFAOYSA-N 2-(2-ethoxyethoxy)ethyl prop-2-enoate Chemical compound CCOCCOCCOC(=O)C=C FTALTLPZDVFJSS-UHFFFAOYSA-N 0.000 description 1
- HZMXJTJBSWOCQB-UHFFFAOYSA-N 2-(2-methoxyethoxy)ethyl prop-2-enoate Chemical compound COCCOCCOC(=O)C=C HZMXJTJBSWOCQB-UHFFFAOYSA-N 0.000 description 1
- JAHNSTQSQJOJLO-UHFFFAOYSA-N 2-(3-fluorophenyl)-1h-imidazole Chemical compound FC1=CC=CC(C=2NC=CN=2)=C1 JAHNSTQSQJOJLO-UHFFFAOYSA-N 0.000 description 1
- OEPOKWHJYJXUGD-UHFFFAOYSA-N 2-(3-phenylmethoxyphenyl)-1,3-thiazole-4-carbaldehyde Chemical compound O=CC1=CSC(C=2C=C(OCC=3C=CC=CC=3)C=CC=2)=N1 OEPOKWHJYJXUGD-UHFFFAOYSA-N 0.000 description 1
- JKNCOURZONDCGV-UHFFFAOYSA-N 2-(dimethylamino)ethyl 2-methylprop-2-enoate Chemical compound CN(C)CCOC(=O)C(C)=C JKNCOURZONDCGV-UHFFFAOYSA-N 0.000 description 1
- 229920000536 2-Acrylamido-2-methylpropane sulfonic acid Polymers 0.000 description 1
- XHZPRMZZQOIPDS-UHFFFAOYSA-N 2-Methyl-2-[(1-oxo-2-propenyl)amino]-1-propanesulfonic acid Chemical compound OS(=O)(=O)CC(C)(C)NC(=O)C=C XHZPRMZZQOIPDS-UHFFFAOYSA-N 0.000 description 1
- WYGWHHGCAGTUCH-UHFFFAOYSA-N 2-[(2-cyano-4-methylpentan-2-yl)diazenyl]-2,4-dimethylpentanenitrile Chemical compound CC(C)CC(C)(C#N)N=NC(C)(C#N)CC(C)C WYGWHHGCAGTUCH-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- SFBPBKYJRXFWKV-UHFFFAOYSA-N 2-[2-(2-hydroxyethoxy)ethoxy]ethanol;2-(2-methylprop-2-enoyloxy)ethyl 2-methylprop-2-enoate Chemical compound OCCOCCOCCO.CC(=C)C(=O)OCCOC(=O)C(C)=C SFBPBKYJRXFWKV-UHFFFAOYSA-N 0.000 description 1
- 125000004174 2-benzimidazolyl group Chemical group [H]N1C(*)=NC2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- 125000001340 2-chloroethyl group Chemical group [H]C([H])(Cl)C([H])([H])* 0.000 description 1
- 125000004182 2-chlorophenyl group Chemical group [H]C1=C([H])C(Cl)=C(*)C([H])=C1[H] 0.000 description 1
- 125000001731 2-cyanoethyl group Chemical group [H]C([H])(*)C([H])([H])C#N 0.000 description 1
- WBIQQQGBSDOWNP-UHFFFAOYSA-N 2-dodecylbenzenesulfonic acid Chemical compound CCCCCCCCCCCCC1=CC=CC=C1S(O)(=O)=O WBIQQQGBSDOWNP-UHFFFAOYSA-N 0.000 description 1
- AFNPEQUYBAVCOU-UHFFFAOYSA-N 2-ethoxycarbonyloxyethyl 2-methylprop-2-enoate Chemical compound CCOC(=O)OCCOC(=O)C(C)=C AFNPEQUYBAVCOU-UHFFFAOYSA-N 0.000 description 1
- FWWXYLGCHHIKNY-UHFFFAOYSA-N 2-ethoxyethyl prop-2-enoate Chemical compound CCOCCOC(=O)C=C FWWXYLGCHHIKNY-UHFFFAOYSA-N 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- OMIGHNLMNHATMP-UHFFFAOYSA-N 2-hydroxyethyl prop-2-enoate Chemical compound OCCOC(=O)C=C OMIGHNLMNHATMP-UHFFFAOYSA-N 0.000 description 1
- VHSHLMUCYSAUQU-UHFFFAOYSA-N 2-hydroxypropyl methacrylate Chemical compound CC(O)COC(=O)C(C)=C VHSHLMUCYSAUQU-UHFFFAOYSA-N 0.000 description 1
- QKPVEISEHYYHRH-UHFFFAOYSA-N 2-methoxyacetonitrile Chemical compound COCC#N QKPVEISEHYYHRH-UHFFFAOYSA-N 0.000 description 1
- YXYJVFYWCLAXHO-UHFFFAOYSA-N 2-methoxyethyl 2-methylprop-2-enoate Chemical compound COCCOC(=O)C(C)=C YXYJVFYWCLAXHO-UHFFFAOYSA-N 0.000 description 1
- HFCUBKYHMMPGBY-UHFFFAOYSA-N 2-methoxyethyl prop-2-enoate Chemical compound COCCOC(=O)C=C HFCUBKYHMMPGBY-UHFFFAOYSA-N 0.000 description 1
- SFPQDYSOPQHZAQ-UHFFFAOYSA-N 2-methoxypropanenitrile Chemical compound COC(C)C#N SFPQDYSOPQHZAQ-UHFFFAOYSA-N 0.000 description 1
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 1
- DLAXSFQUZITJGP-UHFFFAOYSA-N 2-methylbutan-2-yl 2-methylprop-2-enoate Chemical compound CCC(C)(C)OC(=O)C(C)=C DLAXSFQUZITJGP-UHFFFAOYSA-N 0.000 description 1
- FSVQAZDYQRQQKH-UHFFFAOYSA-N 2-methylbutan-2-yl prop-2-enoate Chemical compound CCC(C)(C)OC(=O)C=C FSVQAZDYQRQQKH-UHFFFAOYSA-N 0.000 description 1
- RUMACXVDVNRZJZ-UHFFFAOYSA-N 2-methylpropyl 2-methylprop-2-enoate Chemical compound CC(C)COC(=O)C(C)=C RUMACXVDVNRZJZ-UHFFFAOYSA-N 0.000 description 1
- CFVWNXQPGQOHRJ-UHFFFAOYSA-N 2-methylpropyl prop-2-enoate Chemical compound CC(C)COC(=O)C=C CFVWNXQPGQOHRJ-UHFFFAOYSA-N 0.000 description 1
- RZVINYQDSSQUKO-UHFFFAOYSA-N 2-phenoxyethyl prop-2-enoate Chemical compound C=CC(=O)OCCOC1=CC=CC=C1 RZVINYQDSSQUKO-UHFFFAOYSA-N 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- UUIMDJFBHNDZOW-UHFFFAOYSA-N 2-tert-butylpyridine Chemical compound CC(C)(C)C1=CC=CC=N1 UUIMDJFBHNDZOW-UHFFFAOYSA-N 0.000 description 1
- YEBFFEVZMLHDGY-UHFFFAOYSA-N 3-acetyloxy-2-benzoylperoxybenzoic acid Chemical compound C(C)(=O)OC1=C(C(C(=O)O)=CC=C1)OOC(C1=CC=CC=C1)=O YEBFFEVZMLHDGY-UHFFFAOYSA-N 0.000 description 1
- LTZJEGMQCRHIKJ-UHFFFAOYSA-N 3-bicyclo[2.2.1]heptanylmethyl 2-methylprop-2-enoate Chemical compound C1CC2C(COC(=O)C(=C)C)CC1C2 LTZJEGMQCRHIKJ-UHFFFAOYSA-N 0.000 description 1
- VLRGXXKFHVJQOL-UHFFFAOYSA-N 3-chloropentane-2,4-dione Chemical compound CC(=O)C(Cl)C(C)=O VLRGXXKFHVJQOL-UHFFFAOYSA-N 0.000 description 1
- 125000004179 3-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(Cl)=C1[H] 0.000 description 1
- NPYMXLXNEYZTMQ-UHFFFAOYSA-N 3-methoxybutyl prop-2-enoate Chemical compound COC(C)CCOC(=O)C=C NPYMXLXNEYZTMQ-UHFFFAOYSA-N 0.000 description 1
- VWIIJDNADIEEDB-UHFFFAOYSA-N 3-methyl-1,3-oxazolidin-2-one Chemical compound CN1CCOC1=O VWIIJDNADIEEDB-UHFFFAOYSA-N 0.000 description 1
- DBCAQXHNJOFNGC-UHFFFAOYSA-N 4-bromo-1,1,1-trifluorobutane Chemical compound FC(F)(F)CCCBr DBCAQXHNJOFNGC-UHFFFAOYSA-N 0.000 description 1
- MAGFQRLKWCCTQJ-UHFFFAOYSA-N 4-ethenylbenzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=C(C=C)C=C1 MAGFQRLKWCCTQJ-UHFFFAOYSA-N 0.000 description 1
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 1
- NYOZFCHGWPIBJO-UHFFFAOYSA-N 5-bicyclo[2.2.1]hept-2-enylmethyl 2-methylprop-2-enoate Chemical compound C1C2C(COC(=O)C(=C)C)CC1C=C2 NYOZFCHGWPIBJO-UHFFFAOYSA-N 0.000 description 1
- NUXLDNTZFXDNBA-UHFFFAOYSA-N 6-bromo-2-methyl-4h-1,4-benzoxazin-3-one Chemical compound C1=C(Br)C=C2NC(=O)C(C)OC2=C1 NUXLDNTZFXDNBA-UHFFFAOYSA-N 0.000 description 1
- SNFCXVRWFNAHQX-UHFFFAOYSA-N 9,9'-spirobi[fluorene] Chemical class C12=CC=CC=C2C2=CC=CC=C2C21C1=CC=CC=C1C1=CC=CC=C21 SNFCXVRWFNAHQX-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 1
- 229920000178 Acrylic resin Polymers 0.000 description 1
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 1
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 1
- 229910000952 Be alloy Inorganic materials 0.000 description 1
- 239000004342 Benzoyl peroxide Substances 0.000 description 1
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 1
- LUAZZOXZPVVGSO-UHFFFAOYSA-N Benzyl viologen Chemical compound C=1C=C(C=2C=C[N+](CC=3C=CC=CC=3)=CC=2)C=C[N+]=1CC1=CC=CC=C1 LUAZZOXZPVVGSO-UHFFFAOYSA-N 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- UCCUXODGPMAHRL-UHFFFAOYSA-N Brc(cc1)ccc1I Chemical compound Brc(cc1)ccc1I UCCUXODGPMAHRL-UHFFFAOYSA-N 0.000 description 1
- CWUWVFMUSWUDTO-UHFFFAOYSA-N C(C=C)(=O)OCC(CC(C)C)CCC.C(C=C)(=O)OCC(CCCC)CC Chemical compound C(C=C)(=O)OCC(CC(C)C)CCC.C(C=C)(=O)OCC(CCCC)CC CWUWVFMUSWUDTO-UHFFFAOYSA-N 0.000 description 1
- XMWRBQBLMFGWIX-UHFFFAOYSA-N C60 fullerene Chemical class C12=C3C(C4=C56)=C7C8=C5C5=C9C%10=C6C6=C4C1=C1C4=C6C6=C%10C%10=C9C9=C%11C5=C8C5=C8C7=C3C3=C7C2=C1C1=C2C4=C6C4=C%10C6=C9C9=C%11C5=C5C8=C3C3=C7C1=C1C2=C4C6=C2C9=C5C3=C12 XMWRBQBLMFGWIX-UHFFFAOYSA-N 0.000 description 1
- HLKNISNQQJTYCI-UHFFFAOYSA-N CC1C(Br)=CC=NC1c1nccc(Br)c1 Chemical compound CC1C(Br)=CC=NC1c1nccc(Br)c1 HLKNISNQQJTYCI-UHFFFAOYSA-N 0.000 description 1
- LVRCEUVOXCJYSV-UHFFFAOYSA-N CN(C)S(=O)=O Chemical compound CN(C)S(=O)=O LVRCEUVOXCJYSV-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 239000004380 Cholic acid Substances 0.000 description 1
- 239000004593 Epoxy Substances 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical class C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- JIGUQPWFLRLWPJ-UHFFFAOYSA-N Ethyl acrylate Chemical compound CCOC(=O)C=C JIGUQPWFLRLWPJ-UHFFFAOYSA-N 0.000 description 1
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- WOBHKFSMXKNTIM-UHFFFAOYSA-N Hydroxyethyl methacrylate Chemical compound CC(=C)C(=O)OCCO WOBHKFSMXKNTIM-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 1
- GYCMBHHDWRMZGG-UHFFFAOYSA-N Methylacrylonitrile Chemical compound CC(=C)C#N GYCMBHHDWRMZGG-UHFFFAOYSA-N 0.000 description 1
- 241001024304 Mino Species 0.000 description 1
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 1
- CNCOEDDPFOAUMB-UHFFFAOYSA-N N-Methylolacrylamide Chemical compound OCNC(=O)C=C CNCOEDDPFOAUMB-UHFFFAOYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 229930192627 Naphthoquinone Natural products 0.000 description 1
- JVRHADZWRWBICE-UHFFFAOYSA-N O=S(=O)NC1=CC=CC=C1 Chemical compound O=S(=O)NC1=CC=CC=C1 JVRHADZWRWBICE-UHFFFAOYSA-N 0.000 description 1
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 1
- 239000002033 PVDF binder Substances 0.000 description 1
- 101100272976 Panax ginseng CYP716A53v2 gene Proteins 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical class OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- 206010034972 Photosensitivity reaction Diseases 0.000 description 1
- 229920001609 Poly(3,4-ethylenedioxythiophene) Polymers 0.000 description 1
- 229920002367 Polyisobutene Polymers 0.000 description 1
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 description 1
- 239000002042 Silver nanowire Substances 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- DAKWPKUUDNSNPN-UHFFFAOYSA-N Trimethylolpropane triacrylate Chemical compound C=CC(=O)OCC(CC)(COC(=O)C=C)COC(=O)C=C DAKWPKUUDNSNPN-UHFFFAOYSA-N 0.000 description 1
- XTXRWKRVRITETP-UHFFFAOYSA-N Vinyl acetate Chemical compound CC(=O)OC=C XTXRWKRVRITETP-UHFFFAOYSA-N 0.000 description 1
- 238000004833 X-ray photoelectron spectroscopy Methods 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- HVVWZTWDBSEWIH-UHFFFAOYSA-N [2-(hydroxymethyl)-3-prop-2-enoyloxy-2-(prop-2-enoyloxymethyl)propyl] prop-2-enoate Chemical compound C=CC(=O)OCC(CO)(COC(=O)C=C)COC(=O)C=C HVVWZTWDBSEWIH-UHFFFAOYSA-N 0.000 description 1
- MCEWYIDBDVPMES-UHFFFAOYSA-N [60]pcbm Chemical compound C123C(C4=C5C6=C7C8=C9C%10=C%11C%12=C%13C%14=C%15C%16=C%17C%18=C(C=%19C=%20C%18=C%18C%16=C%13C%13=C%11C9=C9C7=C(C=%20C9=C%13%18)C(C7=%19)=C96)C6=C%11C%17=C%15C%13=C%15C%14=C%12C%12=C%10C%10=C85)=C9C7=C6C2=C%11C%13=C2C%15=C%12C%10=C4C23C1(CCCC(=O)OC)C1=CC=CC=C1 MCEWYIDBDVPMES-UHFFFAOYSA-N 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910001413 alkali metal ion Inorganic materials 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000003806 alkyl carbonyl amino group Chemical group 0.000 description 1
- 125000004448 alkyl carbonyl group Chemical group 0.000 description 1
- 125000005196 alkyl carbonyloxy group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 150000008051 alkyl sulfates Chemical class 0.000 description 1
- 125000004644 alkyl sulfinyl group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 125000004656 alkyl sulfonylamino group Chemical group 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- XYLMUPLGERFSHI-UHFFFAOYSA-N alpha-Methylstyrene Chemical compound CC(=C)C1=CC=CC=C1 XYLMUPLGERFSHI-UHFFFAOYSA-N 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- SOIFLUNRINLCBN-UHFFFAOYSA-N ammonium thiocyanate Chemical compound [NH4+].[S-]C#N SOIFLUNRINLCBN-UHFFFAOYSA-N 0.000 description 1
- 229910021417 amorphous silicon Inorganic materials 0.000 description 1
- 125000002490 anilino group Chemical group [H]N(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 150000001449 anionic compounds Chemical class 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 125000001204 arachidyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000000149 argon plasma sintering Methods 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 125000004658 aryl carbonyl amino group Chemical group 0.000 description 1
- 125000005129 aryl carbonyl group Chemical group 0.000 description 1
- 125000005199 aryl carbonyloxy group Chemical group 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- AYJRCSIUFZENHW-DEQYMQKBSA-L barium(2+);oxomethanediolate Chemical compound [Ba+2].[O-][14C]([O-])=O AYJRCSIUFZENHW-DEQYMQKBSA-L 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 235000019400 benzoyl peroxide Nutrition 0.000 description 1
- AOJOEFVRHOZDFN-UHFFFAOYSA-N benzyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCC1=CC=CC=C1 AOJOEFVRHOZDFN-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- GCTPMLUUWLLESL-UHFFFAOYSA-N benzyl prop-2-enoate Chemical compound C=CC(=O)OCC1=CC=CC=C1 GCTPMLUUWLLESL-UHFFFAOYSA-N 0.000 description 1
- JGQFVRIQXUFPAH-UHFFFAOYSA-N beta-citronellol Natural products OCCC(C)CCCC(C)=C JGQFVRIQXUFPAH-UHFFFAOYSA-N 0.000 description 1
- 230000001588 bifunctional effect Effects 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- GRADOOOISCPIDG-UHFFFAOYSA-N buta-1,3-diyne Chemical group [C]#CC#C GRADOOOISCPIDG-UHFFFAOYSA-N 0.000 description 1
- YHWCPXVTRSHPNY-UHFFFAOYSA-N butan-1-olate;titanium(4+) Chemical compound [Ti+4].CCCC[O-].CCCC[O-].CCCC[O-].CCCC[O-] YHWCPXVTRSHPNY-UHFFFAOYSA-N 0.000 description 1
- CQEYYJKEWSMYFG-UHFFFAOYSA-N butyl acrylate Chemical compound CCCCOC(=O)C=C CQEYYJKEWSMYFG-UHFFFAOYSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000013877 carbamide Nutrition 0.000 description 1
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 1
- 125000005587 carbonate group Chemical group 0.000 description 1
- 150000001733 carboxylic acid esters Chemical class 0.000 description 1
- 238000005266 casting Methods 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- ZMIGMASIKSOYAM-UHFFFAOYSA-N cerium Chemical compound [Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce] ZMIGMASIKSOYAM-UHFFFAOYSA-N 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- BHQCQFFYRZLCQQ-OELDTZBJSA-N cholic acid Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 BHQCQFFYRZLCQQ-OELDTZBJSA-N 0.000 description 1
- 235000019416 cholic acid Nutrition 0.000 description 1
- 229960002471 cholic acid Drugs 0.000 description 1
- 229910052804 chromium Inorganic materials 0.000 description 1
- 235000000484 citronellol Nutrition 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000009841 combustion method Methods 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 229920000547 conjugated polymer Polymers 0.000 description 1
- 150000004696 coordination complex Chemical class 0.000 description 1
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 1
- 150000001925 cycloalkenes Chemical class 0.000 description 1
- KBLWLMPSVYBVDK-UHFFFAOYSA-N cyclohexyl prop-2-enoate Chemical compound C=CC(=O)OC1CCCCC1 KBLWLMPSVYBVDK-UHFFFAOYSA-N 0.000 description 1
- BTQLDZMOTPTCGG-UHFFFAOYSA-N cyclopentyl prop-2-enoate Chemical compound C=CC(=O)OC1CCCC1 BTQLDZMOTPTCGG-UHFFFAOYSA-N 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 238000006356 dehydrogenation reaction Methods 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 239000000412 dendrimer Substances 0.000 description 1
- 229920000736 dendritic polymer Polymers 0.000 description 1
- KXGVEGMKQFWNSR-UHFFFAOYSA-N deoxycholic acid Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 KXGVEGMKQFWNSR-UHFFFAOYSA-N 0.000 description 1
- 230000000779 depleting effect Effects 0.000 description 1
- 238000003795 desorption Methods 0.000 description 1
- NZZIMKJIVMHWJC-UHFFFAOYSA-N dibenzoylmethane Chemical compound C=1C=CC=CC=1C(=O)CC(=O)C1=CC=CC=C1 NZZIMKJIVMHWJC-UHFFFAOYSA-N 0.000 description 1
- 150000001993 dienes Chemical class 0.000 description 1
- IEPRKVQEAMIZSS-AATRIKPKSA-N diethyl fumarate Chemical compound CCOC(=O)\C=C\C(=O)OCC IEPRKVQEAMIZSS-AATRIKPKSA-N 0.000 description 1
- LMBWSYZSUOEYSN-UHFFFAOYSA-N diethyldithiocarbamic acid Chemical compound CCN(CC)C(S)=S LMBWSYZSUOEYSN-UHFFFAOYSA-N 0.000 description 1
- LDCRTTXIJACKKU-ARJAWSKDSA-N dimethyl maleate Chemical compound COC(=O)\C=C/C(=O)OC LDCRTTXIJACKKU-ARJAWSKDSA-N 0.000 description 1
- MZGNSEAPZQGJRB-UHFFFAOYSA-N dimethyldithiocarbamic acid Chemical compound CN(C)C(S)=S MZGNSEAPZQGJRB-UHFFFAOYSA-N 0.000 description 1
- 238000007598 dipping method Methods 0.000 description 1
- 125000004119 disulfanediyl group Chemical group *SS* 0.000 description 1
- 229950004394 ditiocarb Drugs 0.000 description 1
- 229940060296 dodecylbenzenesulfonic acid Drugs 0.000 description 1
- 239000002019 doping agent Substances 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 230000005611 electricity Effects 0.000 description 1
- 230000005518 electrochemistry Effects 0.000 description 1
- 238000010894 electron beam technology Methods 0.000 description 1
- 239000012039 electrophile Substances 0.000 description 1
- 239000003822 epoxy resin Substances 0.000 description 1
- 238000005530 etching Methods 0.000 description 1
- 125000005670 ethenylalkyl group Chemical group 0.000 description 1
- 125000001033 ether group Chemical group 0.000 description 1
- ZOOODBUHSVUZEM-UHFFFAOYSA-N ethoxymethanedithioic acid Chemical compound CCOC(S)=S ZOOODBUHSVUZEM-UHFFFAOYSA-N 0.000 description 1
- MYXNWWDJRWCURH-UHFFFAOYSA-N ethoxymethanethioic s-acid Chemical compound CCOC(S)=O MYXNWWDJRWCURH-UHFFFAOYSA-N 0.000 description 1
- STVZJERGLQHEKB-UHFFFAOYSA-N ethylene glycol dimethacrylate Substances CC(=C)C(=O)OCCOC(=O)C(C)=C STVZJERGLQHEKB-UHFFFAOYSA-N 0.000 description 1
- XSIQSWHENMREAF-UHFFFAOYSA-N ethylsulfanylmethanedithioic acid Chemical compound CCSC(S)=S XSIQSWHENMREAF-UHFFFAOYSA-N 0.000 description 1
- 125000006125 ethylsulfonyl group Chemical group 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 238000001125 extrusion Methods 0.000 description 1
- 239000002657 fibrous material Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229920002313 fluoropolymer Polymers 0.000 description 1
- 239000004811 fluoropolymer Substances 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 239000000446 fuel Substances 0.000 description 1
- 229910003472 fullerene Inorganic materials 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 125000002350 geranyl group Chemical group [H]C([*])([H])/C([H])=C(C([H])([H])[H])/C([H])([H])C([H])([H])C([H])=C(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- ZTOMUSMDRMJOTH-UHFFFAOYSA-N glutaronitrile Chemical compound N#CCCCC#N ZTOMUSMDRMJOTH-UHFFFAOYSA-N 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 230000005283 ground state Effects 0.000 description 1
- VBJZVLUMGGDVMO-UHFFFAOYSA-N hafnium atom Chemical compound [Hf] VBJZVLUMGGDVMO-UHFFFAOYSA-N 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- PZDUWXKXFAIFOR-UHFFFAOYSA-N hexadecyl prop-2-enoate Chemical compound CCCCCCCCCCCCCCCCOC(=O)C=C PZDUWXKXFAIFOR-UHFFFAOYSA-N 0.000 description 1
- LNMQRPPRQDGUDR-UHFFFAOYSA-N hexyl prop-2-enoate Chemical compound CCCCCCOC(=O)C=C LNMQRPPRQDGUDR-UHFFFAOYSA-N 0.000 description 1
- 150000007857 hydrazones Chemical class 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 238000001027 hydrothermal synthesis Methods 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 125000005462 imide group Chemical group 0.000 description 1
- 238000005470 impregnation Methods 0.000 description 1
- RHZWSUVWRRXEJF-UHFFFAOYSA-N indium tin Chemical compound [In].[Sn] RHZWSUVWRRXEJF-UHFFFAOYSA-N 0.000 description 1
- 229910001412 inorganic anion Inorganic materials 0.000 description 1
- 150000002484 inorganic compounds Chemical class 0.000 description 1
- 239000011147 inorganic material Substances 0.000 description 1
- 229910017053 inorganic salt Inorganic materials 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 239000011229 interlayer Substances 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 229920000831 ionic polymer Polymers 0.000 description 1
- 229920000554 ionomer Polymers 0.000 description 1
- 229910052741 iridium Inorganic materials 0.000 description 1
- 150000002505 iron Chemical class 0.000 description 1
- 229940119545 isobornyl methacrylate Drugs 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical group C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- FZLIPJUXYLNCLC-UHFFFAOYSA-N lanthanum atom Chemical compound [La] FZLIPJUXYLNCLC-UHFFFAOYSA-N 0.000 description 1
- 230000031700 light absorption Effects 0.000 description 1
- 125000000040 m-tolyl group Chemical group [H]C1=C([H])C(*)=C([H])C(=C1[H])C([H])([H])[H] 0.000 description 1
- 229910052748 manganese Inorganic materials 0.000 description 1
- 238000010299 mechanically pulverizing process Methods 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 229910001509 metal bromide Inorganic materials 0.000 description 1
- 239000000434 metal complex dye Substances 0.000 description 1
- 229910001511 metal iodide Inorganic materials 0.000 description 1
- FQPSGWSUVKBHSU-UHFFFAOYSA-N methacrylamide Chemical compound CC(=C)C(N)=O FQPSGWSUVKBHSU-UHFFFAOYSA-N 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- XELZGAJCZANUQH-UHFFFAOYSA-N methyl 1-acetylthieno[3,2-c]pyrazole-5-carboxylate Chemical compound CC(=O)N1N=CC2=C1C=C(C(=O)OC)S2 XELZGAJCZANUQH-UHFFFAOYSA-N 0.000 description 1
- ZQMHJBXHRFJKOT-UHFFFAOYSA-N methyl 2-[(1-methoxy-2-methyl-1-oxopropan-2-yl)diazenyl]-2-methylpropanoate Chemical compound COC(=O)C(C)(C)N=NC(C)(C)C(=O)OC ZQMHJBXHRFJKOT-UHFFFAOYSA-N 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- XJRBAMWJDBPFIM-UHFFFAOYSA-N methyl vinyl ether Chemical compound COC=C XJRBAMWJDBPFIM-UHFFFAOYSA-N 0.000 description 1
- LVHBHZANLOWSRM-UHFFFAOYSA-N methylenebutanedioic acid Natural products OC(=O)CC(=C)C(O)=O LVHBHZANLOWSRM-UHFFFAOYSA-N 0.000 description 1
- 125000006216 methylsulfinyl group Chemical group [H]C([H])([H])S(*)=O 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 229910052750 molybdenum Inorganic materials 0.000 description 1
- 239000004570 mortar (masonry) Substances 0.000 description 1
- WFKDPJRCBCBQNT-UHFFFAOYSA-N n,2-dimethylprop-2-enamide Chemical compound CNC(=O)C(C)=C WFKDPJRCBCBQNT-UHFFFAOYSA-N 0.000 description 1
- PCERBVBQNKZCFS-UHFFFAOYSA-M n,n-dibenzylcarbamodithioate Chemical compound C=1C=CC=CC=1CN(C(=S)[S-])CC1=CC=CC=C1 PCERBVBQNKZCFS-UHFFFAOYSA-M 0.000 description 1
- UAGGVDVXSRGPRP-UHFFFAOYSA-M n,n-diethylcarbamothioate Chemical compound CCN(CC)C([O-])=S UAGGVDVXSRGPRP-UHFFFAOYSA-M 0.000 description 1
- 229940088644 n,n-dimethylacrylamide Drugs 0.000 description 1
- YLGYACDQVQQZSW-UHFFFAOYSA-N n,n-dimethylprop-2-enamide Chemical compound CN(C)C(=O)C=C YLGYACDQVQQZSW-UHFFFAOYSA-N 0.000 description 1
- OMNKZBIFPJNNIO-UHFFFAOYSA-N n-(2-methyl-4-oxopentan-2-yl)prop-2-enamide Chemical compound CC(=O)CC(C)(C)NC(=O)C=C OMNKZBIFPJNNIO-UHFFFAOYSA-N 0.000 description 1
- DNTMQTKDNSEIFO-UHFFFAOYSA-N n-(hydroxymethyl)-2-methylprop-2-enamide Chemical compound CC(=C)C(=O)NCO DNTMQTKDNSEIFO-UHFFFAOYSA-N 0.000 description 1
- PNLUGRYDUHRLOF-UHFFFAOYSA-N n-ethenyl-n-methylacetamide Chemical compound C=CN(C)C(C)=O PNLUGRYDUHRLOF-UHFFFAOYSA-N 0.000 description 1
- OFESGEKAXKKFQT-UHFFFAOYSA-N n-ethenyl-n-methylformamide Chemical compound C=CN(C)C=O OFESGEKAXKKFQT-UHFFFAOYSA-N 0.000 description 1
- RQAKESSLMFZVMC-UHFFFAOYSA-N n-ethenylacetamide Chemical compound CC(=O)NC=C RQAKESSLMFZVMC-UHFFFAOYSA-N 0.000 description 1
- ZQXSMRAEXCEDJD-UHFFFAOYSA-N n-ethenylformamide Chemical compound C=CNC=O ZQXSMRAEXCEDJD-UHFFFAOYSA-N 0.000 description 1
- SUSXWKRWORPGPG-UHFFFAOYSA-M n-phenylcarbamodithioate Chemical compound [S-]C(=S)NC1=CC=CC=C1 SUSXWKRWORPGPG-UHFFFAOYSA-M 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 239000002073 nanorod Substances 0.000 description 1
- 239000002071 nanotube Substances 0.000 description 1
- 239000002070 nanowire Substances 0.000 description 1
- PCILLCXFKWDRMK-UHFFFAOYSA-N naphthalene-1,4-diol Chemical compound C1=CC=C2C(O)=CC=C(O)C2=C1 PCILLCXFKWDRMK-UHFFFAOYSA-N 0.000 description 1
- 150000002791 naphthoquinones Chemical class 0.000 description 1
- 229920003986 novolac Polymers 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- HMZGPNHSPWNGEP-UHFFFAOYSA-N octadecyl 2-methylprop-2-enoate Chemical compound CCCCCCCCCCCCCCCCCCOC(=O)C(C)=C HMZGPNHSPWNGEP-UHFFFAOYSA-N 0.000 description 1
- NOPZJEGEHWRZSE-UHFFFAOYSA-N octadecyl formate Chemical group CCCCCCCCCCCCCCCCCCOC=O NOPZJEGEHWRZSE-UHFFFAOYSA-N 0.000 description 1
- FSAJWMJJORKPKS-UHFFFAOYSA-N octadecyl prop-2-enoate Chemical compound CCCCCCCCCCCCCCCCCCOC(=O)C=C FSAJWMJJORKPKS-UHFFFAOYSA-N 0.000 description 1
- ANISOHQJBAQUQP-UHFFFAOYSA-N octyl prop-2-enoate Chemical compound CCCCCCCCOC(=O)C=C ANISOHQJBAQUQP-UHFFFAOYSA-N 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000002891 organic anions Chemical class 0.000 description 1
- 239000012860 organic pigment Substances 0.000 description 1
- 229910052762 osmium Inorganic materials 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- SOQBVABWOPYFQZ-UHFFFAOYSA-N oxygen(2-);titanium(4+) Chemical compound [O-2].[O-2].[Ti+4] SOQBVABWOPYFQZ-UHFFFAOYSA-N 0.000 description 1
- 125000001037 p-tolyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- FIKAKWIAUPDISJ-UHFFFAOYSA-L paraquat dichloride Chemical compound [Cl-].[Cl-].C1=C[N+](C)=CC=C1C1=CC=[N+](C)C=C1 FIKAKWIAUPDISJ-UHFFFAOYSA-L 0.000 description 1
- QONHNMFEHWGACQ-UHFFFAOYSA-N pentan-3-yl prop-2-enoate Chemical compound CCC(CC)OC(=O)C=C QONHNMFEHWGACQ-UHFFFAOYSA-N 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- ULDDEWDFUNBUCM-UHFFFAOYSA-N pentyl prop-2-enoate Chemical compound CCCCCOC(=O)C=C ULDDEWDFUNBUCM-UHFFFAOYSA-N 0.000 description 1
- PNJWIWWMYCMZRO-UHFFFAOYSA-N pent‐4‐en‐2‐one Natural products CC(=O)CC=C PNJWIWWMYCMZRO-UHFFFAOYSA-N 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-M perchlorate Chemical compound [O-]Cl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-M 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- LQJARUQXWJSDFL-UHFFFAOYSA-N phenamine Chemical class CCOC1=CC=C(NC(=O)CN)C=C1 LQJARUQXWJSDFL-UHFFFAOYSA-N 0.000 description 1
- 125000006678 phenoxycarbonyl group Chemical group 0.000 description 1
- 125000003170 phenylsulfonyl group Chemical group C1(=CC=CC=C1)S(=O)(=O)* 0.000 description 1
- 150000003014 phosphoric acid esters Chemical class 0.000 description 1
- 125000004437 phosphorous atom Chemical group 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011941 photocatalyst Substances 0.000 description 1
- 238000000016 photochemical curing Methods 0.000 description 1
- 238000006303 photolysis reaction Methods 0.000 description 1
- 230000015843 photosynthesis, light reaction Effects 0.000 description 1
- 238000000053 physical method Methods 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003227 poly(N-vinyl carbazole) Polymers 0.000 description 1
- 229920000172 poly(styrenesulfonic acid) Polymers 0.000 description 1
- 229920002239 polyacrylonitrile Polymers 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229920000767 polyaniline Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 229920000647 polyepoxide Polymers 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 239000011112 polyethylene naphthalate Substances 0.000 description 1
- 239000005518 polymer electrolyte Substances 0.000 description 1
- 238000001955 polymer synthesis method Methods 0.000 description 1
- 229920000098 polyolefin Polymers 0.000 description 1
- 229940005642 polystyrene sulfonic acid Drugs 0.000 description 1
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 1
- 229920002717 polyvinylpyridine Polymers 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 125000001844 prenyl group Chemical group [H]C([*])([H])C([H])=C(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- FVSKHRXBFJPNKK-UHFFFAOYSA-N propionitrile Chemical compound CCC#N FVSKHRXBFJPNKK-UHFFFAOYSA-N 0.000 description 1
- PNXMTCDJUBJHQJ-UHFFFAOYSA-N propyl prop-2-enoate Chemical compound CCCOC(=O)C=C PNXMTCDJUBJHQJ-UHFFFAOYSA-N 0.000 description 1
- RUOJZAUFBMNUDX-UHFFFAOYSA-N propylene carbonate Chemical compound CC1COC(=O)O1 RUOJZAUFBMNUDX-UHFFFAOYSA-N 0.000 description 1
- 239000003586 protic polar solvent Substances 0.000 description 1
- 239000012264 purified product Substances 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical group C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- BBFCIBZLAVOLCF-UHFFFAOYSA-N pyridin-1-ium;bromide Chemical compound Br.C1=CC=NC=C1 BBFCIBZLAVOLCF-UHFFFAOYSA-N 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 1
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 description 1
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 150000004053 quinones Chemical class 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000027756 respiratory electron transport chain Effects 0.000 description 1
- 229910052702 rhenium Inorganic materials 0.000 description 1
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000007650 screen-printing Methods 0.000 description 1
- 229910052711 selenium Inorganic materials 0.000 description 1
- 239000011669 selenium Substances 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 150000004756 silanes Chemical class 0.000 description 1
- 239000010944 silver (metal) Substances 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000012247 sodium ferrocyanide Nutrition 0.000 description 1
- HYHCSLBZRBJJCH-UHFFFAOYSA-N sodium polysulfide Chemical compound [Na+].S HYHCSLBZRBJJCH-UHFFFAOYSA-N 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- SONHXMAHPHADTF-UHFFFAOYSA-M sodium;2-methylprop-2-enoate Chemical compound [Na+].CC(=C)C([O-])=O SONHXMAHPHADTF-UHFFFAOYSA-M 0.000 description 1
- MNCGMVDMOKPCSQ-UHFFFAOYSA-M sodium;2-phenylethenesulfonate Chemical compound [Na+].[O-]S(=O)(=O)C=CC1=CC=CC=C1 MNCGMVDMOKPCSQ-UHFFFAOYSA-M 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- BWYYYTVSBPRQCN-UHFFFAOYSA-M sodium;ethenesulfonate Chemical compound [Na+].[O-]S(=O)(=O)C=C BWYYYTVSBPRQCN-UHFFFAOYSA-M 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000003892 spreading Methods 0.000 description 1
- 230000007480 spreading Effects 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 125000003696 stearoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- PJANXHGTPQOBST-UHFFFAOYSA-N stilbene Chemical class C=1C=CC=CC=1C=CC1=CC=CC=C1 PJANXHGTPQOBST-UHFFFAOYSA-N 0.000 description 1
- CIOAGBVUUVVLOB-UHFFFAOYSA-N strontium atom Chemical compound [Sr] CIOAGBVUUVVLOB-UHFFFAOYSA-N 0.000 description 1
- 125000003107 substituted aryl group Chemical group 0.000 description 1
- 229960002317 succinimide Drugs 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 description 1
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 1
- 125000005420 sulfonamido group Chemical group S(=O)(=O)(N*)* 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 150000003459 sulfonic acid esters Chemical class 0.000 description 1
- 150000003464 sulfur compounds Chemical class 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- SJMYWORNLPSJQO-UHFFFAOYSA-N tert-butyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OC(C)(C)C SJMYWORNLPSJQO-UHFFFAOYSA-N 0.000 description 1
- ISXSCDLOGDJUNJ-UHFFFAOYSA-N tert-butyl prop-2-enoate Chemical compound CC(C)(C)OC(=O)C=C ISXSCDLOGDJUNJ-UHFFFAOYSA-N 0.000 description 1
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical group C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 1
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 1
- UGNWTBMOAKPKBL-UHFFFAOYSA-N tetrachloro-1,4-benzoquinone Chemical class ClC1=C(Cl)C(=O)C(Cl)=C(Cl)C1=O UGNWTBMOAKPKBL-UHFFFAOYSA-N 0.000 description 1
- PCCVSPMFGIFTHU-UHFFFAOYSA-N tetracyanoquinodimethane Chemical class N#CC(C#N)=C1C=CC(=C(C#N)C#N)C=C1 PCCVSPMFGIFTHU-UHFFFAOYSA-N 0.000 description 1
- GKXDJYKZFZVASJ-UHFFFAOYSA-M tetrapropylazanium;iodide Chemical compound [I-].CCC[N+](CCC)(CCC)CCC GKXDJYKZFZVASJ-UHFFFAOYSA-M 0.000 description 1
- FHCPAXDKURNIOZ-UHFFFAOYSA-N tetrathiafulvalene Chemical compound S1C=CSC1=C1SC=CS1 FHCPAXDKURNIOZ-UHFFFAOYSA-N 0.000 description 1
- 229920002397 thermoplastic olefin Polymers 0.000 description 1
- 229920001187 thermosetting polymer Polymers 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- LLZRNZOLAXHGLL-UHFFFAOYSA-J titanic acid Chemical compound O[Ti](O)(O)O LLZRNZOLAXHGLL-UHFFFAOYSA-J 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 238000002834 transmittance Methods 0.000 description 1
- JLTRXTDYQLMHGR-UHFFFAOYSA-N trimethylaluminium Chemical compound C[Al](C)C JLTRXTDYQLMHGR-UHFFFAOYSA-N 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000006617 triphenylamine group Chemical group 0.000 description 1
- AAAQKTZKLRYKHR-UHFFFAOYSA-N triphenylmethane Chemical compound C1=CC=CC=C1C(C=1C=CC=CC=1)C1=CC=CC=C1 AAAQKTZKLRYKHR-UHFFFAOYSA-N 0.000 description 1
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 1
- 239000010937 tungsten Substances 0.000 description 1
- 239000006097 ultraviolet radiation absorber Substances 0.000 description 1
- 150000003672 ureas Chemical class 0.000 description 1
- 238000007740 vapor deposition Methods 0.000 description 1
- 229920001567 vinyl ester resin Polymers 0.000 description 1
- NLVXSWCKKBEXTG-UHFFFAOYSA-N vinylsulfonic acid Chemical compound OS(=O)(=O)C=C NLVXSWCKKBEXTG-UHFFFAOYSA-N 0.000 description 1
- 229910052724 xenon Inorganic materials 0.000 description 1
- FHNFHKCVQCLJFQ-UHFFFAOYSA-N xenon atom Chemical compound [Xe] FHNFHKCVQCLJFQ-UHFFFAOYSA-N 0.000 description 1
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical compound [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 description 1
- 239000011787 zinc oxide Substances 0.000 description 1
Images

Classifications
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/30—Coordination compounds
- H10K85/341—Transition metal complexes, e.g. Ru(II)polypyridine complexes
- H10K85/344—Transition metal complexes, e.g. Ru(II)polypyridine complexes comprising ruthenium
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F15/00—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic Table
- C07F15/0006—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic Table compounds of the platinum group
- C07F15/0046—Ruthenium compounds
- C07F15/0053—Ruthenium compounds without a metal-carbon linkage
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B23/00—Methine or polymethine dyes, e.g. cyanine dyes
- C09B23/10—The polymethine chain containing an even number of >CH- groups
- C09B23/105—The polymethine chain containing an even number of >CH- groups two >CH- groups
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B57/00—Other synthetic dyes of known constitution
- C09B57/10—Metal complexes of organic compounds not being dyes in uncomplexed form
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES, LIGHT-SENSITIVE OR TEMPERATURE-SENSITIVE DEVICES OF THE ELECTROLYTIC TYPE
- H01G9/00—Electrolytic capacitors, rectifiers, detectors, switching devices, light-sensitive or temperature-sensitive devices; Processes of their manufacture
- H01G9/20—Light-sensitive devices
- H01G9/2059—Light-sensitive devices comprising an organic dye as the active light absorbing material, e.g. adsorbed on an electrode or dissolved in solution
-
- H01L31/04—
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M14/00—Electrochemical current or voltage generators not provided for in groups H01M6/00 - H01M12/00; Manufacture thereof
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES, LIGHT-SENSITIVE OR TEMPERATURE-SENSITIVE DEVICES OF THE ELECTROLYTIC TYPE
- H01G9/00—Electrolytic capacitors, rectifiers, detectors, switching devices, light-sensitive or temperature-sensitive devices; Processes of their manufacture
- H01G9/20—Light-sensitive devices
- H01G9/2027—Light-sensitive devices comprising an oxide semiconductor electrode
- H01G9/2031—Light-sensitive devices comprising an oxide semiconductor electrode comprising titanium oxide, e.g. TiO2
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E10/00—Energy generation through renewable energy sources
- Y02E10/50—Photovoltaic [PV] energy
- Y02E10/542—Dye sensitized solar cells
Definitions
- the present invention relates to a photoelectric conversion element and a photoelectrochemical cell having high conversion efficiency and excellent durability.
- the photoelectric conversion element is used in various optical sensors, copying machines, photoelectrochemical cells (for example, solar cells) and the like.
- Various types of photoelectric conversion elements have been put to practical use, such as those using metals, semiconductors, organic pigments and dyes, or combinations thereof.
- a solar cell using non-depleting solar energy does not require fuel, and its full-scale practical use is expected greatly as it uses inexhaustible clean energy.
- silicon solar cells have been researched and developed for a long time. It is spreading due to the policy considerations of each country. However, silicon is an inorganic material, and its throughput and molecular modification are naturally limited.
- Patent Document 1 describes a dye-sensitized photoelectric conversion element using semiconductor fine particles sensitized with a ruthenium complex dye by applying this technique.
- Patent Document 2 proposes a photosensitized solar cell that effectively absorbs sunlight and improves photoelectric conversion efficiency by using a dye having a specific structure.
- a photovoltaic cell in which photoelectric conversion efficiency is improved by incorporating a dopant into at least one of the core and shell of multi-layered titanium oxide fine particles having a core as a central part and a shell as an outer shell. (For example, refer to Patent Document 3).
- An object of the present invention is to provide a photoelectric conversion element and a photoelectrochemical cell having high photoelectric conversion efficiency and excellent durability.
- the inventors of the present invention made photoelectric conversion elements using various semiconductor fine particles and dyes, and intensively studied the photoelectric conversion efficiency and durability. As a result, the durability cannot be improved by simply mixing different semiconductor fine particles, and the durability can be improved simply by using tin oxide coated with aluminum oxide or magnesium oxide as the semiconductor fine particles. I found it difficult to do. Therefore, the present inventors conducted extensive studies on semiconductor fine particles and pigments. As a result, the photoelectric conversion element and the photoelectrochemical cell using the metal complex dye in which the semiconductor fine particles have two or more kinds of metals or metal compounds and the ligand has a specific substituent are the initial photoelectric It was found that not only conversion efficiency but also durability was excellent. The present invention has been made based on these findings.
- a photoelectric conversion element comprising a conductive support, a photoreceptor layer composed of a semiconductor fine particle layer containing a dye, a charge transfer layer, and a counter electrode, wherein the semiconductor fine particles are two or more kinds of metals.
- dye is a compound represented by following General formula (1).
- X is acyloxy group, acylthio group, thioacyloxy group, thioacylthio group, acylaminooxy group, thiocarbamate group, dithiocarbamate group, thiocarbonate group, dithiocarbonate group, trithiocarbonate group, acyl group, thiocyanate group , Isothiocyanate group, cyanate group, isocyanate group, cyano group, alkylthio group, arylthio group, monodentate or bidentate ligand selected from the group consisting of alkoxy group and aryloxy group, or halogen atom, carbonyl, dialkyl ketone Represents a monodentate or bidentate ligand selected from the group consisting of 1,3-diketone, carbonamide, thiocarbonamide and thiourea.
- CI represents a counter ion that neutralizes the charge of the compound represented by the general formula (1).
- m1 represents an integer of 1 to 3, and when m1 is 2 or more, LL 1 may be the same or different.
- m2 represents an integer of 0 to 2, and when m2 is 2, LL 2 may be the same or different.
- m3 represents an integer of 0 to 3, and when m3 is 2 or more, Xs may be the same or different.
- m4 represents an integer of 0 to 3, and when m4 is 2 or more, CIs may be the same or different.
- R 101 and R 102 each independently represents a heterocyclic group, a carboxyl group, a sulfonic acid group, a hydroxyl group, a hydroxamic acid group, a phosphoryl group, or a phosphonyl group.
- R 103 and R 104 each independently represent a substituent, and R 105 and R 106 each independently represent a group consisting of at least one selected from the group consisting of an alkyl group, an aryl group and a heterocyclic group.
- L 1 and L 2 each independently represents a conjugated chain composed of an ethenylene group and / or an ethynylene group.
- a1 and a2 each independently represent an integer of 0 to 3, and when a1 is 2 or more, R 101 may be the same or different, and when a2 is 2 or more, R 102 may be the same or different.
- b1 and b2 each independently represents an integer of 0 to 3.
- R 103 may be the same or different and may be connected to each other to form a ring.
- R 104 may be the same or different and may be connected to each other to form a ring.
- both b1 and b2 are 1 or more, R 103 and R 104 may be linked to form a ring.
- d1 and d2 each independently represents an integer of 0 to 5.
- d3 represents 0 or 1.
- Za, Zb and Zc each independently represent a nonmetallic atom group capable of forming a 5- or 6-membered ring, and c represents 0 or 1.
- the metal atom is at least one selected from the group consisting of Ti, Sn, Au, Ag, Cu, Al, Zr, Nb, V, and Ta.
- the metal chalcogenide is selected from the group consisting of cadmium sulfide, cadmium selenide, or Ti, Sn, Zn, Mg, Al, W, Zr, Hf, Sr, In, Ce, Y, La, V, and Ta.
- the metal carbonate is at least one selected from the group consisting of calcium carbonate, potassium carbonate, and barium carbonate.
- ⁇ 6> The photoelectric conversion device according to any one of ⁇ 2> to ⁇ 5>, wherein the metal nitrate is lanthanum nitrate.
- the semiconductor fine particle has the metal atom, the metal chalcogenide, the metal carbonate and / or the metal nitrate by a core-shell structure Item 1.
- ⁇ 8> The photoelectric conversion element according to ⁇ 7>, wherein the semiconductor fine particles have the metal chalcogenide as a core portion and the metal chalcogenide or the metal carbonate as a shell portion.
- the semiconductor fine particle has, as a core part, a metal chalcogenide selected from the group consisting of titanium oxide and tin oxide, and from the group consisting of aluminum oxide, magnesium oxide, calcium carbonate, titanium oxide, and titanium oxide / magnesium oxide.
- the semiconductor fine particles are semiconductor fine particles obtained by doping the metal chalcogenide with the metal atom.
- ⁇ 12> Semiconductor fine particles obtained by doping a metal chalcogenide selected from the group consisting of titanium oxide and tin oxide with at least one metal atom selected from the group consisting of Nb, V and Ta
- the photoelectric conversion element according to ⁇ 11>, wherein ⁇ 13> The photoelectric conversion element according to any one of ⁇ 1> to ⁇ 12>, wherein the semiconductor fine particles have a particle diameter of 1 to 1000 nm.
- the semiconductor fine particles include an additive made of a conductive material.
- the conductive material is graphene.
- ⁇ 16> In the general formula (1), ⁇ 1> to ⁇ 15>, wherein Mz is Ru, m1 is 1, m2 is 1, X is an isothiocyanate group, and m3 is 2.
- ⁇ 17> The photoelectric device according to any one of ⁇ 1> to ⁇ 16>, wherein LL 1 is represented by any one of general formulas (4-1) to (4-3) in the general formula (1): Conversion element.
- R 101 to R 104 , a 1, a 2, b 1, b 2, and d 3 have the same meaning as in general formula (2).
- R107 represents an acidic group.
- a3 represents an integer of 0 to 3.
- R108 represents a substituent.
- b3 represents an integer of 0 to 3.
- R 121 to R 124 each independently represents a hydrogen atom, an alkyl group, an alkenyl group or an aryl group.
- R 125 , R 126 , R 127 and R 128 each independently represent a substituent.
- d4 and d5 each independently represents an integer of 0 to 4.
- a photoelectrochemical cell comprising the photoelectric conversion element according to any one of ⁇ 1> to ⁇ 17>.
- a photoelectric conversion element and a photoelectrochemical cell having high conversion efficiency and excellent durability can be provided.
- a photoelectric conversion comprising a conductive support, a photosensitive layer composed of a semiconductor fine particle layer containing a dye of a specific compound, a charge transfer layer, and a counter electrode. It was discovered that a photoelectric conversion element and a photoelectrochemical cell, each of which is an element and the semiconductor fine particles locally have two or more kinds of metals or metal compounds, have high conversion efficiency and excellent durability. .
- the present invention has been made based on these findings.
- the photoelectric conversion element 10 includes a conductive support 1, a photosensitive layer 2, a charge transfer layer 3, and a counter electrode 4 arranged in that order on the conductive support 1.
- the conductive support 1 and the photoreceptor layer 2 constitute a light receiving electrode 5.
- the photoreceptor layer 2 has semiconductor fine particles 22 and a sensitizing dye (hereinafter also simply referred to as a dye) 21. At least a part of the sensitizing dye 21 is adsorbed on the semiconductor fine particles 22 (the sensitizing dye 21 is in an adsorption equilibrium state and may be partially present in the charge transfer layer 3).
- the charge transfer body layer 3 functions as, for example, a hole transport layer that transports holes.
- the conductive support 1 on which the photoreceptor layer 2 is formed functions as a working electrode in the photoelectric conversion element 10.
- the photoelectric conversion element 10 can be operated as the photoelectrochemical cell 100 by causing the external circuit 6 to work.
- the light receiving electrode 5 is an electrode composed of a conductive support 1 and a photosensitive layer 2 (semiconductor film) of semiconductor fine particles 22 adsorbed by a sensitizing dye 21 coated on the conductive support 1.
- a photosensitive layer 2 semiconductor film
- the excited dye has high energy electrons. Therefore, the electrons are transferred from the sensitizing dye 21 to the conduction band of the semiconductor fine particles 22 and reach the conductive support 1 by diffusion.
- the molecule of the sensitizing dye 21 is an oxidant.
- the electrons on the electrode return to the oxidant while working in the external circuit 6, thereby acting as the photoelectrochemical cell 100.
- the light receiving electrode 5 functions as a negative electrode of the battery.
- the photoreceptor layer 2 is composed of a porous semiconductor layer composed of a layer of semiconductor fine particles 22 to which a dye described later is adsorbed. This dye may be partially dissociated in the electrolyte.
- the photoreceptor layer 2 is designed according to the purpose and has a multilayer structure.
- the photosensitive layer 2 includes the semiconductor fine particles 22 on which a specific dye is adsorbed, the light receiving sensitivity is high, and when used as the photoelectrochemical cell 100, high photoelectric conversion efficiency can be obtained. Furthermore, it has high durability.
- the porous semiconductor layer is sensitized with at least one dye 21 represented by the following general formula (1).
- Metal atom Mz Mz represents a metal atom.
- Mz is preferably a metal capable of tetracoordinate or hexacoordinate, and more preferably Ru, Fe, Os, Cu, W, Cr, Mo, Ni, Pd, Pt, Co, Ir, Rh, Re, Mn or Zn. Ru, Os, Zn or Cu is particularly preferable, and Ru is most preferable.
- the ligand LL 1 is a bidentate ligand represented by the general formula (2).
- M1 representing the number of ligands LL 1 is an integer of 1 to 3.
- m1 is 2 or more, the ligands LL 1 may be the same or different.
- m1 is preferably 1.
- R 101 and R 102 are each independently a heterocyclic group, a carboxyl group, a sulfonic acid group, a hydroxyl group, a hydroxamic acid group (preferably a hydroxamic acid group having 1 to 20 carbon atoms, for example, —CONHOH, —CONCH 3 OH, etc.), phosphoryl group (eg, —OP (O) (OH) 2 etc.) or phosphonyl group (eg, —P (O) (OH) 2 etc.).
- the heterocyclic group may be unsubstituted or substituted with a substituent described later.
- R 101 and R 102 are preferably a carboxyl group or a phosphonyl group, and more preferably a carboxyl group.
- R 101 and R 102 may be substituted on any carbon atom on the pyridine ring.
- a1 and a2 each independently represents an integer of 0 to 3.
- a1 is R 101 when 2 or more may be the same or different, a2 is 2 or more when R 102 may be the same or different.
- a1 is preferably 0 or 1
- a2 is preferably an integer of 0-2.
- the sum of a1 and a2 is preferably an integer of 0-2.
- R 103 and R 104 each independently represents a substituent, preferably an alkyl group (preferably an alkyl group having 1 to 20 carbon atoms such as methyl, ethyl, isopropyl, t-butyl, pentyl).
- alkenyl groups preferably alkenyl groups having 2 to 20 carbon atoms, such as vinyl, allyl, oleyl, etc.
- alkynyl groups Preferably an alkynyl group having 2 to 20 carbon atoms such as ethynyl, butadiynyl, phenylethynyl, etc., a cycloalkyl group (preferably a cycloalkyl group having 3 to 20 carbon atoms such as cyclopropyl, cyclopentyl, cyclohexyl, 4 -Methylcyclohexyl, etc.), an aryl group (preferably an ant having 6 to 26 carbon atoms) Group such as phenyl, 1-naphthyl, 4-methoxyphenyl, 2-chlorophenyl,
- b1 and b2 each independently represents an integer of 0 to 3, preferably an integer of 0 to 2.
- R 103 may be the same or different and may be connected to each other to form a ring.
- R 104 may be the same or different and may be connected to each other to form a ring. Further, when b1 and b2 are both 1 or more, they may form a ring R 103 and R 104 are.
- a benzene ring a pyridine ring, a thiophene ring, a pyrrole ring, a cyclohexane ring, a cyclopentane ring etc. are mentioned.
- R 105 and R 106 each independently represent a group consisting of at least one selected from the group consisting of an alkyl group, an aryl group, and a heterocyclic group.
- R 105 and R 106 are each independently an aromatic group (preferably an aromatic group having 6 to 30 carbon atoms such as phenyl, substituted phenyl, naphthyl, substituted naphthyl, etc.) or a heterocyclic group (preferably a carbon atom Preferred is a heterocyclic group having a number of 1 to 30, for example, 2-thienyl group, 2-pyrrolyl group, 2-imidazolyl group, 1-imidazolyl group, 4-pyridyl group, 3-indolyl group), and preferably 1 to 3 electrons
- a heterocyclic group having a donor group is more preferred, and a thienyl group is more preferred.
- Electric child Azukamoto alkyl group, an alkenyl group, an alkynyl group, a cycloalkyl group, an alkoxy group, an aryloxy group, an amino group, an acylamino group (preferred examples above if the same R 101 and R 102) are or hydroxyl groups preferably An alkyl group, an alkoxy group, an amino group or a hydroxyl group is more preferable, and an alkyl group is particularly preferable.
- R 105 and R 106 may be the same or different, but are preferably the same.
- L 1 and L 2 each independently represent a conjugated chain composed of an ethenylene group and / or an ethynylene group.
- substituent include those shown as specific examples of the substituent for R 103 and R 104 .
- the substituent is preferably an alkyl group, and more preferably methyl.
- L 1 and L 2 are each independently preferably a conjugated chain having 2 to 6 carbon atoms, more preferably ethenylene, butadienylene, ethynylene, butadienylene, methylethenylene or dimethylethenylene, particularly ethenylene or butadienylene.
- L 1 and L 2 may be the same or different, but are preferably the same.
- each double bond may be a trans isomer, a cis isomer, or a mixture thereof.
- d1 and d2 each independently represents an integer of 0 to 5. If d1 and d2 is 0, R 105 and R 106 are directly bonded to the benzene ring. When d1 and d2 are integers of 1 or more, R 105 and R 106 are bonded to the benzene ring via L 1 or L 2 . d1 and d2 are each preferably 0 or 1.
- D3 is 0 or 1, and when d3 is 0, a2 is preferably 1 or 2, and when d3 is 1, a2 is preferably 0 or 1.
- the ligand LL 1 When the ligand LL 1 contains an alkyl group, an alkenyl group or the like, these may be linear or branched and may be substituted or unsubstituted. Further, when the ligand LL 1 includes an aromatic group such as an aryl group, a heterocyclic group, or the like, they may be monocyclic or condensed, and may be substituted or unsubstituted. Examples of the substituent include those shown as specific examples of the substituent for R 103 and R 104 .
- the ligand LL 1 in the general formula (1) is preferably represented by the following general formula (4-1), (4-2) or (4-3).
- R 101 to R 104 , a1, a2, b1, b2 and d3 have the same meanings as those in the general formula (2), and preferred ranges are also the same. .
- R 107 represents an acidic group, preferably a carboxyl group, a sulfonic acid group, a hydroxyl group, a hydroxamic acid group, a phosphoryl group or a phosphonyl group, more preferably a carboxyl group or a phosphoryl group.
- a carboxyl group is more preferable.
- a3 represents an integer of 0 to 3, preferably an integer of 0 to 2.
- a3 is preferably 1 or 2
- a3 is preferably 0 or 1.
- a3 is the R 107 when two or more may be the same or different.
- R 108 represents a substituent, preferably an alkyl group, alkenyl group, alkynyl group, cycloalkyl group, alkoxy group, aryloxy group, amino group or acylamino group (above preferred examples are R 103 and R 104 in the general formula (2)), and more preferably an alkyl group, an alkoxy group, an amino group, or an acylamino group.
- b3 represents an integer of 0 to 3, preferably an integer of 0 to 2.
- R 108 may be the same or different.
- R 121 to R 124 each independently represents a hydrogen atom, an alkyl group, an alkenyl group, or an aryl group.
- Preferred examples of R 121 to R 124 are the same as the preferred examples of R 103 and R 104 in the general formula (2).
- R 121 to R 124 are more preferably an alkyl group or an aryl group, and even more preferably an alkyl group.
- R 121 to R 124 are alkyl groups, they may further have a substituent, and the substituent is preferably an alkoxy group, a cyano group, an alkoxycarbonyl group or a carbonamido group, particularly preferably an alkoxy group.
- R 121 and R 122 and R 123 and R 124 may be connected to each other to form a ring.
- a pyrrolidine ring, a piperidine ring, a piperazine ring, a morpholine ring or the like is preferable.
- R 125 , R 126 , R 127 and R 128 each independently represent a substituent, preferably an alkyl group, an alkenyl group, an alkynyl group, a cycloalkyl group, An alkoxy group, an aryloxy group, an amino group, an acylamino group (preferred examples are the same as those for R 103 and R 104 in the general formula (2)) or a hydroxyl group, more preferably an alkyl group, An alkenyl group, an alkynyl group, an alkoxy group, an amino group or an acylamino group, particularly preferably an alkyl group or an alkynyl group.
- d4 and d5 each independently represents an integer of 0 to 4.
- R 125 may be linked to one or both of R 121 and R 122 to form a ring.
- the ring formed is preferably a piperidine ring or a pyrrolidine ring.
- R 125 may be the same or different and may be linked to each other to form a ring.
- R 126 may be linked to one or both of R 123 and R 124 to form a ring.
- the ring formed is preferably a piperidine ring or a pyrrolidine ring.
- R 126 may be the same or different and may be linked to each other to form a ring.
- the ligand LL 2 is a bidentate or tridentate ligand represented by the general formula (3), and is preferably a bidentate ligand.
- M2 representing the number of the ligand LL 2 is an integer of 0 to 2, preferably 0 or 1, and more preferably 1.
- the ligands LL 2 may be the same or different.
- the ligand LL 2 preferably has an acidic group such as a carboxyl group, a sulfonic acid group, a hydroxyl group, a hydroxamic acid group, a phosphoryl group, or a phosphonyl group.
- Za, Zb and Zc each independently represent a nonmetallic atom group capable of forming a 5-membered ring or a 6-membered ring.
- the formed 5-membered ring or 6-membered ring may be substituted or unsubstituted, and may be monocyclic or condensed. Examples of the substituent include the substituent W described later.
- Za, Zb and Zc are preferably a 5-membered ring or 6-membered ring having a carbon atom, a nitrogen atom, an oxygen atom, a sulfur atom and / or a phosphorus atom, and the 5-membered ring or 6-membered ring is a hydrogen atom. And may have a halogen atom.
- Za, Zb or Zc is preferably an aromatic ring. In the case of a 5-membered ring, an imidazole ring, an oxazole ring, a thiazole ring or a triazole ring is preferably formed.
- a 6-membered ring a pyridine ring, a pyrimidine ring, a pyridazine ring or a pyrazine ring is preferably formed.
- an imidazole ring or a pyridine ring is more preferable.
- c 0 or 1.
- the ligand LL 2 is preferably represented by any of the following general formulas (5-1) to (5-8), and the general formulas (5-1), (5-2), (5-4) ) Or (5-6) is more preferred, represented by general formula (5-1) or (5-2) is particularly preferred, and represented by general formula (5-1) Most preferably.
- R 151 to R 166 are depicted as substituted on one ring for the sake of illustration, but even on the ring, Alternatively, it may be substituted with a different ring from that shown.
- R 151 to R 158 each independently represent an acidic group.
- R 151 to R 158 are, for example, a carboxyl group, a sulfonic acid group, a hydroxyl group, a hydroxamic acid group (preferably a hydroxamic acid group having 1 to 20 carbon atoms, such as —CONHOH, —CONCH 3 OH, etc.), a phosphoryl group ( For example, -OP (O) (OH) 2 or the like) or a phosphonyl group (eg -P (O) (OH) 2 or the like) is represented.
- R 151 to R 158 are preferably a carboxyl group, a phosphoryl group or a phosphonyl group, more preferably a carboxyl group or a phosphonyl group, and still more preferably a carboxyl group.
- R 159 to R 166 each independently represent a substituent, preferably an alkyl group, alkenyl group, cycloalkyl group, aryl group, heterocyclic group, alkoxy group , Aryloxy group, alkoxycarbonyl group, amino group, acyl group, sulfonamido group, acyloxy group, carbamoyl group, acylamino group, cyano group or halogen atom (above preferred examples are R 103 and R 104 in formula (2)) More preferably an alkyl group, an alkenyl group, an aryl group, a heterocyclic group, an alkoxy group, an alkoxycarbonyl group, an amino group, an acylamino group or a halogen atom, particularly preferably an alkyl group or an alkenyl group. , An alkoxy group, an alkoxycarbonyl group, an amino group, or an acylamino group.
- R 167 to R 171 each independently represents a hydrogen atom, an aliphatic group, an aromatic group, or a heterocyclic group bonded with a carbon atom.
- An aliphatic group and an aromatic group are preferable, and an aliphatic group having a carboxyl group is more preferable.
- R 151 to R 166 may be bonded to any position on the ring.
- e1 to e6 each independently represents an integer of 0 to 4, preferably an integer of 1 to 2.
- e7 and e8 each independently represents an integer of 0 to 4, preferably an integer of 0 to 3, more preferably an integer of 1 to 3.
- e9 to e12 and e15 each independently represents an integer of 0 to 6, and e13, e14 and e16 each independently represents an integer of 0 to 4. It is preferable that e9 to e16 are each independently an integer of 0 to 3.
- R 151 to R 158 may be the same or different from each other, and may be connected to each other to form a ring.
- R 159 to R 166 may be the same or different from each other, and may be connected to each other to form a ring.
- the ligand LL 2 When the ligand LL 2 contains an alkyl group, an alkenyl group or the like, they may be linear or branched and may be unsubstituted substituted. Further, when the ligand LL 2 contains an aromatic group such as an aryl group, a heterocyclic group or the like, these may be monocyclic or condensed, and may be substituted or unsubstituted.
- the ligand X represents a monodentate or bidentate ligand shown below.
- M3 representing the number of ligands X represents an integer of 0 to 3, preferably an integer of 0 to 2, and more preferably 1 or 2.
- m3 is preferably 2.
- m3 is preferably 1.
- m3 is an integer of 2 or more, the ligands X may be the same or different, and the ligands X may be linked to each other.
- Ligand X is an acyloxy group (preferably an acyloxy group having 1 to 20 carbon atoms such as acetyloxy, benzoyloxy, salicylic acid, glycyloxy, N, N-dimethylglycyloxy, oxalylene (—OC (O) C (O) O—), etc.), an acylthio group (preferably an acylthio group having 1 to 20 carbon atoms, such as acetylthio, benzoylthio, etc.), a thioacyloxy group (preferably a thioacyloxy group having 1 to 20 carbon atoms, For example, a thioacetyloxy group (CH 3 C (S) O—) and the like)), a thioacylthio group (preferably a thioacylthio group having 1 to 20 carbon atoms, such as thioacetylthio (CH 3 C (S) S—) , Thiobenzoyl
- dialkyl ketone preferably a dialkyl ketone having 3 to 20 carbon atoms such as acetone ((CH 3 ) 2 CO...)
- the ligand X is preferably an acyloxy group, a thioacylthio group, an acylaminooxy group, a dithiocarbamate group, a dithiocarbonate group, a trithiocarbonate group, a thiocyanate group, an isothiocyanate group, a cyanate group, an isocyanate group, a cyano group, A ligand selected from the group consisting of an alkylthio group, an arylthio group, an alkoxy group and an aryloxy group, or a ligand selected from the group consisting of a halogen atom, carbonyl, 1,3-diketone and thiourea, more preferably Is a monodentate or bidentate ligand selected from the group consisting of acyloxy group, acylaminooxy group, dithiocarbamate group, thiocyanate group, isothiocyanate group, cyanate group, isocyanate group,
- the ligand X contains an alkyl group, an alkenyl group, an alkynyl group, an alkylene group or the like, these may be linear or branched, and may be substituted or unsubstituted.
- an aromatic group such as an aryl group, a heterocyclic group, a cycloalkyl group, and the like are included, they may be substituted or unsubstituted, and may be monocyclic or condensed.
- the ligand X is an acyloxy group, acylthio group, thioacyloxy group, thioacylthio group, acylaminooxy group, thiocarbamate group, dithiocarbamate group, thiocarbonate group, dithio group.
- Counter ion CI CI in the general formula (1) represents a counter ion when a counter ion is necessary to neutralize the charge.
- a dye is a cation or an anion, or has a net ionic charge, depends on the metal, ligand and substituent in the dye.
- the dye of the general formula (1) may be dissociated and have a negative charge because the substituent has a dissociable group.
- the entire charge of the dye of the general formula (1) is electrically neutralized by the counter ion CI.
- M4 which is the number of CIs, is an integer from 0 to 3.
- examples of the counter ion CI include inorganic or organic ammonium ions (for example, tetraalkylammonium ions, pyridinium ions, etc.), alkali metal ions, and protons.
- the counter ion CI may be an inorganic anion or an organic anion.
- halogen anions eg, fluoride ions, chloride ions, bromide ions, iodide ions, etc.
- substituted aryl sulfonate ions eg, p-toluene sulfonate ions, p-chlorobenzene sulfonate ions, etc.
- aryl disulfones Acid ions for example, 1,3-benzenedisulfonate ion, 1,5-naphthalenedisulfonate ion, 2,6-naphthalenedisulfonate ion, etc.
- alkyl sulfate ions for example, methyl sulfate ion
- sulfate ions thiocyanate ions
- an ionic polymer or another dye having a charge opposite to that of the dye may be used as the charge balance counter ion, and a metal complex ion (for example, bisbenzene-1,2-dithiolatonickel (III)) can also be used. is there.
- the dye having the structure represented by the general formula (1) has one or more suitable acidic groups (bonding groups) to the surface of the semiconductor fine particles.
- the group preferably has 1 to 6 groups, and particularly preferably has 1 to 4 groups. Carboxyl group, sulfonic acid group, hydroxyl group, hydroxamic acid group (for example, —CONHOH), phosphoryl group (for example, —OP (O) (OH) 2, etc.), phosphonyl group (for example, —P (O) (OH) 2, etc.) It is preferable that the dye has an acidic group (substituent having a dissociable proton).
- an acidic group refers to a substituent that releases a proton.
- the functional substituent when “having a specific functional substituent” such as “having an acidic group”, the functional substituent is directly bonded to the mother nucleus within a range not impairing the effects of the present invention.
- it is meant to include those linked (linked) via a predetermined linking group.
- the substituent in this specification can represent the substituent W shown below, for example except what was described especially.
- Halogen atoms for example, fluorine atom, chlorine atom, bromine atom, iodine atom
- -Alkyl group [Represents a linear, branched, or cyclic substituted or unsubstituted alkyl group.
- alkyl groups preferably alkyl groups having 1 to 30 carbon atoms such as methyl, ethyl, n-propyl, isopropyl, t-butyl, n-octyl, eicosyl, 2-chloroethyl, 2-cyanoethyl, 2-ethylhexyl
- a cycloalkyl group preferably a substituted or unsubstituted cycloalkyl group having 3 to 30 carbon atoms, such as cyclohexyl, cyclopentyl, 4-n-dodecylcyclohexyl
- a bicycloalkyl group preferably having 5 to 30 carbon atoms.
- a substituted or unsubstituted bicycloalkyl group that is, a monovalent group obtained by removing one hydrogen atom from a bicycloalkane having 5 to 30 carbon atoms, for example, bicyclo [1,2,2] heptan-2-yl, bicyclo [2,2,2] octan-3-yl), and tricyclo structures having more ring structures are also included.
- An alkyl group (for example, an alkyl group of an alkylthio group) in the substituents described below also represents such an alkyl group.
- -Alkenyl group [Represents a linear, branched, or cyclic substituted or unsubstituted alkenyl group.
- alkenyl groups preferably substituted or unsubstituted alkenyl groups having 2 to 30 carbon atoms, such as vinyl, allyl, prenyl, geranyl, oleyl
- cycloalkenyl groups preferably substituted or substituted groups having 3 to 30 carbon atoms
- An unsubstituted cycloalkenyl group that is, a monovalent group obtained by removing one hydrogen atom of a cycloalkene having 3 to 30 carbon atoms (for example, 2-cyclopenten-1-yl, 2-cyclohexen-1-yl), Bicycloalkenyl group (a substituted or unsubstituted bicycloalkenyl group, preferably a substituted or unsubstituted bicycloalkenyl group having 5 to 30 carbon atoms, that is, a monovalent group obtained by removing one hydrogen atom of a bicycloalkene having one double bond.
- An alkynyl group preferably a substituted or unsubstituted alkynyl group having 2 to 30 carbon atoms, such as ethynyl, propargyl, trimethylsilylethynyl group, aryl group (preferably a substituted or unsubstituted aryl group having 6 to 30 carbon atoms, Such as phenyl, p-tolyl, naphthyl, m-chlorophenyl, o-hexadecanoylaminophenyl), Aromatic groups (for example, benzene ring, furan ring, pyrrole ring, pyridine ring, thiophene ring, imidazole ring, oxazole ring, thiazole ring, pyrazole
- the dye having the structure represented by the general formula (1) used in the present invention is shown below, but the present invention is not limited thereto.
- dye in the following specific example contains the ligand which has a proton dissociable group, this ligand may dissociate as needed and may discharge
- the method for synthesizing the dye represented by the general formula (1) can be referred to the method described in Examples below, and can be synthesized by appropriately applying a conventional method based thereon.
- Information described in JP 2001-291534 A and WO 2007/091525 can also be referred to, and the dyes and methods described therein are cited in this specification.
- the maximum absorption wavelength in the dye solution represented by the general formula (1) is preferably in the range of 300 to 1000 nm, more preferably in the range of 350 to 950 nm, and particularly preferably in the range of 370 to 900 nm.
- the content of the dye represented by the general formula (1) is not particularly limited, but is preferably 0.001 to 1 mmol, and preferably 0.1 to 0.5 mmol, with respect to 1 g of semiconductor fine particles. It is more preferable that By setting it as the said lower limit or more, the sensitization effect in a semiconductor can fully be acquired, and the reduction of the sensitization effect by desorption of a pigment
- two or more dyes represented by the general formula (1) may be used.
- the layer which consists of electrolyte composition is applicable to the charge transfer body layer used for the photoelectric conversion element of this embodiment.
- the redox pair for example, a combination of iodine and iodide (eg, lithium iodide, tetrabutylammonium iodide, tetrapropylammonium iodide, etc.), alkyl viologen (eg, methyl viologen chloride, hexyl viologen bromide, benzyl viologen tetrafluoro) Borate) and its reduced form, a combination of polyhydroxybenzenes (for example, hydroquinone, naphthohydroquinone, etc.) and its oxidized form, a combination of divalent and trivalent iron complexes (for example, red blood salt and yellow blood salt) Etc.
- iodine and iodide eg, lithium iodide, tetrabutylammoni
- the cation of the iodine salt is preferably a 5-membered or 6-membered nitrogen-containing aromatic cation.
- the compound represented by the general formula (1) is not an iodine salt, it is described in WO95 / 18456, JP-A-8-259543, Electrochemistry, Vol. 65, No. 11, page 923 (1997), etc. It is preferable to use iodine salts such as pyridinium salts, imidazolium salts, and triazolium salts that are used in combination.
- the electrolyte composition used for the photoelectric conversion element preferably contains iodine together with the heterocyclic quaternary salt compound.
- the iodine content is preferably from 0.1 to 20% by mass, more preferably from 0.5 to 5% by mass, based on the entire electrolyte composition.
- the electrolyte composition may contain a solvent.
- the solvent content in the electrolyte composition is preferably 50% by mass or less, more preferably 30% by mass or less, and particularly preferably 10% by mass or less of the entire composition.
- a solvent having a low viscosity and high ion mobility, a high dielectric constant and capable of increasing the effective carrier concentration, or both is preferable because it exhibits excellent ion conductivity.
- Such solvents include carbonate compounds (ethylene carbonate, propylene carbonate, etc.), heterocyclic compounds (3-methyl-2-oxazolidinone, etc.), ether compounds (dioxane, diethyl ether, etc.), chain ethers (ethylene glycol dialkyl ether, Propylene glycol dialkyl ether, polyethylene glycol dialkyl ether, polypropylene glycol dialkyl ether, etc.), alcohols (methanol, ethanol, ethylene glycol monoalkyl ether, propylene glycol monoalkyl ether, polyethylene glycol monoalkyl ether, polypropylene glycol monoalkyl ether, etc.), Polyhydric alcohols (ethylene glycol, propylene glycol, polyethylene glycol Polypropylene glycol, glycerin, etc.), nitrile compounds (acetonitrile, glutarodinitrile, methoxyacetonitrile, propionitrile, benzonitrile, biscyanoethyl ether
- an electrochemically inert salt that is in a liquid state at room temperature and / or has a melting point lower than room temperature may be used as the electrolyte solvent.
- the electrolyte solvent For example, 1-ethyl-3-methylimidazolium trifluoromethanesulfonate, 1-butyl-3-methylimidazolium trifluoromethanesulfonate, etc., nitrogen-containing heterocyclic quaternary salt compounds such as imidazolium salt and pyridinium salt, or tetraalkylammonium Examples include salt.
- a polymer or an oil gelling agent may be added, or gelled (solidified) by a technique such as polymerization of a polyfunctional monomer or a crosslinking reaction of the polymer.
- the polyfunctional monomers are preferably compounds having two or more ethylenically unsaturated groups, such as divinylbenzene, ethylene glycol diacrylate, ethylene glycol dimethacrylate, diethylene glycol diacrylate, diethylene glycol dimethacrylate, triethylene glycol diacrylate, triethylene glycol Ethylene glycol dimethacrylate, pentaerythritol triacrylate, trimethylolpropane triacrylate and the like are preferable.
- divinylbenzene ethylene glycol diacrylate, ethylene glycol dimethacrylate, diethylene glycol diacrylate, diethylene glycol dimethacrylate, triethylene glycol diacrylate, triethylene glycol Ethylene glycol dimethacrylate, pentaerythritol triacrylate, trimethylolpropane triacrylate and the like are preferable.
- the gel electrolyte may be formed by polymerization of a mixture containing a monofunctional monomer in addition to the above polyfunctional monomers.
- Monofunctional monomers include acrylic acid or ⁇ -alkylacrylic acid (acrylic acid, methacrylic acid, itaconic acid, etc.) or esters or amides thereof (methyl acrylate, ethyl acrylate, n-propyl acrylate, i-propyl acrylate, n- Butyl acrylate, i-butyl acrylate, t-butyl acrylate, n-pentyl acrylate, 3-pentyl acrylate, t-pentyl acrylate, n-hexyl acrylate, 2,2-dimethylbutyl acrylate, n-octyl acrylate, 2-ethylhexyl acrylate 4-methyl-2-propylpentyl acrylate, cetyl acrylate, n-octa
- the blending amount of the polyfunctional monomer is preferably 0.5 to 70% by mass, and more preferably 1.0 to 50% by mass with respect to the whole monomer.
- the above-mentioned monomers are commonly used in Takayuki Otsu and Masato Kinoshita “Experimental Methods for Polymer Synthesis” (Chemical Doujin) and Takatsu Otsu “Lecture Polymerization Reaction Theory 1 Radical Polymerization (I)” (Chemical Doujin).
- Polymerization can be performed by radical polymerization which is a polymer synthesis method.
- the monomer for gel electrolyte used in the present invention can be radically polymerized by heating, light or electron beam, or electrochemically, and is particularly preferably radically polymerized by heating.
- polymerization initiators are 2,2′-azobisisobutyronitrile, 2,2′-azobis (2,4-dimethylvaleronitrile), dimethyl 2,2′-azobis (2-methylpropyl). Pionate), azo initiators such as dimethyl 2,2′-azobisisobutyrate, peroxide initiators such as lauryl peroxide, benzoyl peroxide, and t-butyl peroctoate.
- a preferable addition amount of the polymerization initiator is 0.01 to 20% by mass, and more preferably 0.1 to 10% by mass with respect to the total amount of monomers.
- the weight composition range of the monomer in the gel electrolyte is preferably 0.5 to 70% by mass.
- the content is 1.0 to 50% by mass.
- a polymer having a crosslinkable reactive group and a crosslinking agent is added to the composition.
- Preferred reactive groups are nitrogen-containing heterocycles such as pyridine ring, imidazole ring, thiazole ring, oxazole ring, triazole ring, morpholine ring, piperidine ring, piperazine ring, and the preferred crosslinking agent is a functional group capable of nucleophilic attack by the nitrogen atom.
- the electrolyte composition the metal iodide (LiI, NaI, KI, CsI , CaI 2 , etc.), a metal bromide (LiBr, NaBr, KBr, CsBr , CaBr 2 , etc.), quaternary ammonium bromine salt (tetraalkylammonium bromide, Pyridinium bromide, etc.), metal complexes (ferrocyanate-ferricyanate, ferrocene-ferricinium ion, etc.), sulfur compounds (sodium polysulfide, alkylthiol-alkyl disulfides, etc.), viologen dye, hydroquinone-quinone, etc. You can do it. These may be used as a mixture.
- (C) Conductive support As the conductive support, glass or a polymer material having a conductive film layer on the surface, such as a metal having a conductive property as the support itself, can be used. It is preferable that the conductive support is substantially transparent. Substantially transparent means that the light transmittance is 10% or more, preferably 50% or more, particularly preferably 80% or more.
- a glass or polymer material coated with a conductive metal oxide can be used as the conductive support. The coating amount of the conductive metal oxide at this time is preferably 0.1 to 100 g per 1 m 2 of the support of glass or polymer material. When a transparent conductive support is used, light is preferably incident from the support side.
- polymer materials examples include tetraacetyl cellulose (TAC), polyethylene terephthalate (PET), polyethylene naphthalate (PEN), syndiotactic polystyrene (SPS), polyphenylene sulfide (PPS), polycarbonate (PC), Examples include polyarylate (PAR), polysulfone (PSF), polyester sulfone (PES), polyetherimide (PEI), cyclic polyolefin, and brominated phenoxy.
- TAC tetraacetyl cellulose
- PET polyethylene terephthalate
- PEN polyethylene naphthalate
- SPS syndiotactic polystyrene
- PPS polyphenylene sulfide
- PC polycarbonate
- Examples include polyarylate (PAR), polysulfone (PSF), polyester sulfone (PES), polyetherimide (PEI), cyclic polyolefin, and brominated phenoxy
- an antireflection film in which high refractive films and low refractive index oxide films described in JP-A-2003-123859 are alternately laminated The light guide function described in Open 2002-260746 is raised.
- a metal support can also be preferably used. Examples thereof include titanium, aluminum, copper, nickel, iron, stainless steel, and copper. These metals may be alloys. More preferably, titanium, aluminum, and copper are preferable, and titanium and aluminum are particularly preferable.
- the conductive support has a function of blocking ultraviolet light.
- a method of allowing a fluorescent material capable of changing ultraviolet light to visible light in the transparent support or on the surface of the transparent support, or a method using an ultraviolet absorber is also included.
- a function described in JP-A-11-250944 may be further provided on the conductive support.
- Preferred conductive films include metals (eg, platinum, gold, silver, copper, aluminum, rhodium, indium, etc.), carbon, or conductive metal oxides (indium-tin composite oxide, tin oxide doped with fluorine, etc.) ).
- the thickness of the conductive film layer is preferably 0.01 to 30 ⁇ m, more preferably 0.03 to 25 ⁇ m, and particularly preferably 0.05 to 20 ⁇ m.
- the range of the surface resistance is preferably 50 ⁇ / cm 2 or less, more preferably 10 ⁇ / cm 2 or less. This lower limit is not particularly limited, but is usually about 0.1 ⁇ / cm 2 .
- a collecting electrode may be disposed.
- a gas barrier film and / or an ion diffusion prevention film may be disposed between the support and the transparent conductive film.
- the gas barrier layer a resin film or an inorganic film can be used.
- the transparent conductive layer may have a laminated structure, and as a preferable method, for example, FTO can be laminated on ITO.
- the semiconductor fine particles locally contain two or more kinds of metals or metal compounds.
- the metal compound represents an inorganic compound containing a metal and one or more atoms other than the metal in the molecule, and examples thereof include metal chalcogenides, metal carbonates, and metal nitrates.
- the semiconductor fine particles locally having two or more kinds of metals or metal compounds are treated with two or more kinds of metals or metal compounds in advance to thereby obtain two or more kinds of metals or fine particles in the fine particles.
- a metal compound is present locally.
- the semiconductor fine particles locally having two or more kinds of metals or metal compounds include those having a core-shell structure formed of two or more kinds of metals or metal compounds as described later, The part and other parts are formed of different metals or metal compounds. Therefore, a simple mixture of two or more kinds of semiconductor fine particles is not included.
- the dye By using a dye having a specific substituent on the semiconductor fine particles locally having two or more kinds of metals or metal compounds, the dye can be efficiently adsorbed onto the semiconductor fine particles and has high durability. A photoelectric conversion element can be realized.
- the semiconductor fine particles preferably contain a metal atom, a metal chalcogenide, a metal carbonate and / or a metal nitrate.
- Metal atoms include Ti (titanium), Sn (tin), Au (gold), Ag (silver), Cu (copper), Al (aluminum), Zr (zirconium), Nb (niobium), V (vanadium) and Ta At least one selected from the group consisting of (tantalum) is preferred. More preferred are Ti, Sn, Zr, Nb, V, and Ta, and particularly preferred are Nb, V, and Ta.
- Metal chalcogenides are cadmium sulfide, cadmium selenide or Ti (titanium), Sn (tin), Zn (zinc), Mg (magnesium), Al (aluminum), W (tungsten), Zr (zirconium), Hf (hafnium) At least one metal oxide selected from the group consisting of Sr (strontium), In (indium), Ce (cerium), Y (yttrium), La (lanthanum), V (vanadium) and Ta (tantalum). preferable.
- the metal oxide selected from the group consisting of Ti, Sn, Zn, Mg, Al, and more preferably at least one selected from the group consisting of Ti, Sn, Mg, Al. It is a kind of metal oxide, and particularly preferably at least one kind of metal oxide selected from the group consisting of Ti, Sn, and Al.
- the metal carbonate is preferably at least one selected from the group consisting of calcium carbonate, potassium carbonate and barium carbonate. More preferred are calcium carbonate and barium carbonate, and particularly preferred is calcium carbonate.
- the metal nitrate is preferably lanthanum nitrate.
- the semiconductor fine particles having two or more kinds of metals or metal compounds locally have a core-shell structure, and the metal atom, metal chalcogenide, metal carbonate and / or metal nitrate are preferably contained in the core-shell structure. More preferably.
- the “core-shell structure” means a structure having a shell portion so as to cover a core portion serving as a nucleus. It is not necessary for the core part to be entirely covered with the shell part, but preferably 50% or more, more preferably 80% or more, particularly 90% of the surface area of the core part is preferably covered with the shell part. .
- the semiconductor fine particles having a core-shell structure can exhibit an effect of improving the open circuit voltage by suppressing the electrons injected from the excited dye from returning to I 3 ⁇ in the electrolytic solution.
- Semiconductor fine particles having a core-shell structure can be obtained by adding the semiconductor fine particles to be the core to a solution of metal atoms or metal compounds to be the shell and reacting them appropriately.
- One kind or two or more kinds of semiconductor fine particles to be used as the core may be used, and one kind or two or more kinds of metal atoms or metal compounds to be used as the shell may be used.
- the core-shell semiconductor fine particles for example, the core-shell semiconductor fine particles having titanium oxide as the core and calcium carbonate as the shell can be prepared by the following method, but the method and conditions are limited. There is nothing.
- the dispersion liquid of a core is obtained by stirring with an ultrasonic homogenizer.
- titanium oxide particles as a core are added to a 1 to 3% by mass calcium acetate aqueous solution and stirred for 30 minutes to 3 hours.
- the core-shell with titanium oxide as the core and calcium carbonate as the shell Semiconductor fine particles having a structure can be obtained. Whether the obtained semiconductor fine particles have a core-shell structure can be determined by observation with a transmission electron microscope (TEM).
- TEM transmission electron microscope
- the volume ratio of the core portion to the shell portion is not particularly limited, but the core: shell volume ratio is preferably 50:50 to 98: 2, and more preferably 70:30 to 95: 5. This volume ratio can be obtained by observing with TEM.
- the core portion preferably has a metal atom, a metal chalcogenide, or a metal nitrate. More preferably, the core portion is a metal atom or a metal chalcogenide, and particularly preferably, the core portion is a metal chalcogenide. It is preferred that the shell portion comprises a metal chalcogenide or metal carbonate.
- a metal atom is used as the core portion, at least one metal atom selected from the group consisting of Ti, Nb, Sn, Zn and La is preferable. More preferably, they are Ti, Sn, and Zn. Particularly preferred are Ti and Sn.
- metal chalcogenide is used as the core part, it is preferably at least one metal oxide selected from oxides of Ti, Sn, Zn, Mg and Al. More preferred are an oxide of Ti, an oxide of Sn, and an oxide of Zn. Particularly preferred are an oxide of Ti and an oxide of Sn.
- metal nitrate is used as the core portion, lanthanum nitrate is preferred.
- a metal chalcogenide is used as the shell portion, it is preferable to use oxides of Ti, Mg, and Al.
- a metal carbonate it is preferable to use calcium carbonate.
- the particle diameter of the semiconductor fine particles is preferably 1 nm or more and 1000 nm or less, more preferably 2 nm or more and 100 nm or less, in order to keep the viscosity of the semiconductor fine particle dispersion high.
- the particle size refers to that measured by a laser diffraction type particle size distribution measuring device, for example, a master sizer (trade name) manufactured by MALVERN. Two or more kinds of fine particles having different particle size distributions may be mixed. In this case, the average size of the small particles is preferably 5 nm or less.
- large particles having an average particle size exceeding 50 nm can be added to the ultrafine particles at a low content, or another layer can be applied.
- the content of the large particles is preferably 50% or less, more preferably 20% or less of the mass of particles having an average particle size of 50 nm or less.
- the average particle size of the large particles added and mixed for the above purpose is preferably 100 nm or more, and more preferably 250 nm or more.
- the gel-sol method described in Sakuo Sakuo's “Science of Sol-Gel Method”, Agne Jofu Co., Ltd. (1998) and the like is preferable. Also preferred is a method of producing an oxide by high-temperature hydrolysis of a chloride developed by Degussa in an oxyhydrogen salt.
- the semiconductor fine particles are titanium oxide
- the above sol-gel method, gel-sol method, and high-temperature hydrolysis method in oxyhydrogen salt of chloride are preferred, but Kiyoshi Manabu's “Titanium oxide properties and applied technology”
- the sulfuric acid method and the chlorine method described in Gihodo Publishing (1997) can also be used.
- the sol-gel method the method described in Journal of American Ceramic Society, Vol. 80, No. 12, 3157-3171 (1997), or the chemistry of Burnside et al. -The method described in Materials, Vol. 10, No. 9, pages 2419-2425 is also preferable.
- the fine semiconductor particles to be the core portion can be produced by the conventional method.
- the production method of titania nanoparticles is preferably a method by flame hydrolysis of titanium tetrachloride, a combustion method of titanium tetrachloride, hydrolysis of a stable chalcogenide complex, orthotitanic acid
- the semiconductor fine particles that become the core portion are produced by hydrolyzing, dissolving and removing the soluble portions after forming the semiconductor fine particles from the soluble portion and the insoluble portion, and hydrothermal synthesis of an aqueous peroxide solution.
- the semiconductor fine particle which should be a core in the solution of the metal atom or metal compound which should be a shell, and making it react suitably.
- the crystal structure of titania as a core part include anatase type, brookite type, and rutile type, and anatase type and brookite type are preferable. Titania nanotubes, nanowires, and nanorods may be mixed with titania fine particles.
- the semiconductor fine particles locally having two or more kinds of metals or metal compounds used in the present invention are semiconductor fine particles having two or more kinds of metal atoms by doping the semiconductor fine particles with metal atoms. There may be. By doping metal atoms, the flat band potential is positively shifted, and the effect of increasing the charge injection efficiency can increase the short-circuit current.
- the metal atom to be doped include Nb, V, and Ta. More preferably, they are Nb and V.
- the obtained solution is transferred to an autoclave made of Teflon (registered trademark) and stirred at 180 ° C. for 20 hours.
- the semiconductor fine particles may contain additives other than metal atoms, metal chalcogenides, metal carbonates and metal nitrates.
- a conductive material is preferable.
- An example of the conductive material is a coating type conductive material. Examples thereof include carbon materials such as carbon nanotubes, graphene, and graphite, ⁇ -conjugated polymers that are conductive polymers, and silver nanowires. These materials can form a thin film exhibiting conductivity by coating, and can be manufactured at low cost. Among these, carbon materials such as graphite, graphene, and carbon nanotube are preferable, and graphene is more preferable.
- a conductive material By adding a conductive material to the semiconductor fine particles, the aforementioned dye excited by light irradiation can be held as it is, the reaction of returning the dye to the ground state can be suppressed, and battery performance, particularly photoelectric conversion efficiency can be improved. Can do. More preferred is graphite or graphene having a planar structure.
- An additive such as a conductive material can be added to the semiconductor fine particles by a method in which the additive is added to the semiconductor fine particle paste and dispersed with an ultrasonic homogenizer.
- the conductive material preferably has an electric resistance value of 10 7 ⁇ ⁇ cm or less, more preferably 10 5 ⁇ ⁇ cm or less.
- additives for improving the necking between the semiconductor fine particles and additives on the surface for preventing reverse electron transfer may be used.
- preferred additives include ITO, SnO particles, whiskers, fibrous graphite / carbon nanotubes, zinc oxide necking binders, fibrous materials such as cellulose, metals, organic silicon, dodecylbenzenesulfonic acid, silane compounds, etc. Examples thereof include a mobile binding molecule and a potential gradient dendrimer.
- the semiconductor fine particles may be subjected to acid-base or redox treatment before dye adsorption. Etching, oxidation treatment, hydrogen peroxide treatment, dehydrogenation treatment, UV-ozone, oxygen plasma, or the like may be used.
- the core has a metal chalcogenide as a core portion and a metal chalcogenide or a metal carbonate as a shell portion.
- a semiconductor fine particle having a shell structure and a semiconductor fine particle obtained by doping a metal chalcogenide with a metal atom are preferable, and a metal chalcogenide selected from the group consisting of titanium oxide (TiO 2 ) and tin oxide (SnO 2 ) is used as a core.
- a semiconductor fine particle layer can be obtained by applying the semiconductor fine particle dispersion to the conductive support and heating it appropriately.
- the solid content other than the semiconductor fine particles is preferably 10% by mass or less of the entire semiconductor fine particle dispersion.
- a method of preparing a semiconductor fine particle dispersion is a method of depositing fine particles in a solvent and using them as they are when synthesizing a semiconductor. Ultrafine particles are irradiated with ultrasonic waves. Or a method of mechanically pulverizing and grinding using a mill or a mortar.
- the dispersion solvent water and / or various organic solvents can be used.
- the organic solvent include alcohols such as methanol, ethanol, isopropyl alcohol, citronellol and ⁇ -terpineol, ketones such as acetone, esters such as ethyl acetate, dichloromethane, acetonitrile and the like.
- a polymer such as polyethylene glycol, butyl cellulose, ethyl cellulose, hydroxyethyl cellulose, carboxymethyl cellulose, a surfactant, an acid, a chelating agent or the like may be used in a small amount as a dispersion aid.
- the solid content other than the semiconductor fine particles can be 10% by mass or less of the total dispersion. This concentration is preferably 5% or less, more preferably 3% or less, and particularly preferably 1% or less. More preferably, it is 0.5% or less, and particularly preferably 0.2%. That is, in the semiconductor fine particle dispersion, the solid content other than the solvent and the semiconductor fine particles can be 10% by mass or less of the entire semiconductor fine dispersion.
- the viscosity of the semiconductor fine particle dispersion is preferably 10 to 300 N ⁇ s / m 2 at 25 ° C. More preferably, it is 50 to 200 N ⁇ s / m 2 at 25 ° C.
- a roller method, a dip method, or the like can be used as an application method.
- an air knife method, a blade method, etc. can be used as a metering method.
- the application method and the metering method can be made the same part.
- the wire bar method disclosed in Japanese Patent Publication No. 58-4589, the slide hopper method described in US Pat. No. 2,681,294, etc., the extrusion The method and the curtain method are preferable. It is also preferable to apply by a spin method or a spray method using a general-purpose machine.
- the wet printing method intaglio, rubber plate, screen printing and the like are preferred, including the three major printing methods of letterpress, offset and gravure. From these, a preferred film forming method is selected according to the liquid viscosity and the wet thickness. Further, since the semiconductor fine particle dispersion has a high viscosity and has a viscous property, it may have a strong cohesive force and may not be well adapted to the support during coating. In such a case, by performing cleaning and hydrophilization of the surface by UV ozone treatment, the binding force between the applied semiconductor fine particle dispersion and the surface of the conductive support increases, and the semiconductor fine particle dispersion can be easily applied. The preferred thickness of the entire semiconductor fine particle layer is 0.1 to 100 ⁇ m.
- the thickness of the semiconductor fine particle layer is further preferably 1 to 30 ⁇ m, and more preferably 2 to 25 ⁇ m.
- the amount of the semiconductor fine particles supported per 1 m 2 of the support is preferably 0.5 to 400 g, more preferably 5 to 100 g.
- the applied semiconductor fine particle layer is subjected to heat treatment to enhance the electronic contact between the semiconductor fine particles and to improve the adhesion to the support, and to dry the applied semiconductor fine particle dispersion. .
- heat treatment to enhance the electronic contact between the semiconductor fine particles and to improve the adhesion to the support, and to dry the applied semiconductor fine particle dispersion.
- a porous semiconductor fine particle layer can be formed.
- the semiconductor fine particle layer may be appropriately formed by a known method according to the characteristics and use of the member.
- the materials, preparation methods, and manufacturing methods described in Japanese Patent Application Laid-Open No. 2001-291534 can be referred to and are cited in this specification.
- light energy can also be used.
- the surface when titanium oxide is used as the semiconductor fine particles, the surface may be activated by applying light absorbed by the semiconductor fine particles such as ultraviolet light, or only the surface of the semiconductor fine particles may be activated by laser light or the like. Can do.
- the impurities adsorbed on the particle surface are decomposed by the activation of the particle surface, and can be brought into a preferable state for the above purpose.
- heat treatment and ultraviolet light it is preferable that heating be performed at 100 ° C. or higher and 250 ° C. or lower, or preferably 100 ° C. or higher and 150 ° C. or lower, while irradiating the semiconductor fine particles with light absorbed by the fine particles.
- impurities mixed in the fine particle layer can be washed by photolysis, and physical bonding between the fine particles can be strengthened.
- the semiconductor fine particle dispersion may be applied to the conductive support, and other treatments may be performed in addition to heating and light irradiation.
- Examples of preferred methods include energization and chemical treatment.
- a pressure may be applied after the application, and a method for applying the pressure includes Japanese Patent Publication No. 2003-500857.
- Examples of light irradiation include JP-A No. 2001-357896.
- Examples of plasma, microwave, and energization include JP-A No. 2002-353453.
- Examples of the chemical treatment include JP-A-2001-357896.
- the method for coating the above-mentioned semiconductor fine particles on the conductive support is not only the method for applying the above-mentioned semiconductor fine particle dispersion on the conductive support, but also the semiconductor fine particle precursor described in Japanese Patent No. 2664194.
- a method such as a method of obtaining a semiconductor fine particle film by applying on a conductive support and hydrolyzing with moisture in the air can be used.
- the precursor include (NH 4 ) 2 TiF 6 , titanium peroxide, metal alkoxide / metal complex / metal organic acid salt, and the like.
- a method of forming a semiconductor film by applying a slurry in which a metal organic oxide (alkoxide, etc.) coexists, and heat treatment, light treatment, etc., a slurry in which an inorganic precursor coexists, titania dispersed in the pH of the slurry The method which specified the property of particle
- a binder may be added in a small amount, and examples of the binder include cellulose, fluoropolymer, crosslinked rubber, polybutyl titanate, carboxymethyl cellulose and the like.
- Techniques related to the formation of semiconductor fine particles or precursor layers thereof include corona discharge, plasma, a method of hydrophilizing by a physical method such as UV, a chemical treatment with alkali, polyethylenedioxythiophene and polystyrenesulfonic acid, polyaniline, etc. For example, formation of an interlayer film for bonding may be mentioned.
- Examples of the dry method include vapor deposition, sputtering, and aerosol deposition method. Further, electrophoresis or electrodeposition may be used. Moreover, after producing a coating film once on a heat-resistant board
- the semiconductor fine particles preferably have a large surface area so that many dyes can be adsorbed.
- the surface area is preferably 10 times or more, more preferably 100 times or more the projected area.
- limiting in particular in this upper limit Usually, it is about 5000 times. JP-A-2001-93591 and the like are preferable as the structure of semiconductor fine particles.
- the thickness of the semiconductor fine particle layer increases, the amount of dye that can be supported per unit area increases, so that the light absorption efficiency increases.
- the preferred thickness of the semiconductor fine particle layer varies depending on the use of the device, but is typically 0.1 to 100 ⁇ m. When used as a photoelectrochemical cell, the thickness is preferably 1 to 50 ⁇ m, more preferably 3 to 30 ⁇ m.
- the semiconductor fine particles may be heated at a temperature of 100 to 800 ° C. for 10 minutes to 10 hours in order to adhere the particles to each other after being applied to the support.
- the film forming temperature is preferably 400 to 600 ° C.
- a polymer material is used as the support, it is preferably heated after film formation at 250 ° C.
- the film forming method may be any of (1) a wet method, (2) a dry method, and (3) an electrophoresis method (including an electrodeposition method), and preferably (1) a wet method or ( 2) Dry method, more preferably (1) Wet method.
- the coating amount of semiconductor fine particles per 1 m 2 of support is preferably 0.5 to 500 g, more preferably 5 to 100 g.
- a photoreceptor layer can be formed by adsorbing a dye to the semiconductor fine particle layer produced as described above.
- a dye adsorbing dye solution composed of a solution and a dye for a long time.
- the solution used for the dye solution for dye adsorption can be used without particular limitation as long as the solution can dissolve the dye.
- ethanol, methanol, isopropanol, toluene, t-butanol, acetonitrile, acetone, n-butanol and the like can be used.
- a dye solution for dye adsorption composed of a solution and a dye may be heated to 50 ° C. to 100 ° C. as necessary. The adsorption of the dye may be performed before or after application of the semiconductor fine particles. Further, the semiconductor fine particles and the dye may be applied and adsorbed simultaneously. Unadsorbed dye is removed by washing.
- baking a coating film it is preferable to adsorb
- the dye to be mixed is selected so as to make the wavelength range of photoelectric conversion as wide as possible. When mixing the dye, it is preferable to prepare a dye solution for dye adsorption by dissolving all the dyes.
- the total amount of the dye used is preferably 0.01 to 100 mmol, more preferably 0.1 to 50 mmol, and particularly preferably 0.1 to 10 mmol per m 2 of the support. In this case, it is preferable that the usage-amount of a pigment
- a colorless compound may be co-adsorbed for the purpose of reducing the interaction between dyes such as association.
- the hydrophobic compound to be co-adsorbed include steroid compounds having a carboxyl group (for example, cholic acid and pivaloyl acid).
- the surface of the semiconductor fine particles may be treated with amines.
- Preferred amines include 4-tert-butylpyridine and polyvinylpyridine. These may be used as they are in the case of a liquid, or may be used by dissolving in an organic solvent.
- the counter electrode serves as the positive electrode of the photoelectrochemical cell.
- the counter electrode is usually synonymous with the conductive support described above, but the support is not necessarily required in a configuration in which the strength is sufficiently maintained. However, having a support is advantageous in terms of hermeticity.
- the material for the counter electrode include platinum, carbon, conductive polymer, and the like. Preferable examples include platinum, carbon, and conductive polymer.
- a structure having a high current collecting effect is preferable.
- Preferred examples include JP-A-10-505192.
- a composite electrode such as titanium oxide and tin oxide (TiO 2 / SnO 2 ) may be used as the light receiving electrode, and as a mixed electrode of titania, for example, Japanese Patent Application Laid-Open No. 2000-119393 may be cited.
- Examples of mixed electrodes other than titania include Japanese Patent Application Laid-Open Nos. 2001-185243 and 2003-282164.
- a conductive support electrode layer
- a photoelectric conversion layer photosensitive layer and charge transfer layer
- a hole transport layer a conductive layer
- a counter electrode layer a hole transport layer
- a hole transport material that functions as a p-type semiconductor can be used as a hole transport layer.
- an inorganic or organic hole transport material can be used as a hole transport layer.
- the inorganic hole transport material include CuI, CuO, and NiO.
- the organic hole transport material include high molecular weight materials and low molecular weight materials.
- the high molecular weight material examples include polyvinyl carbazole, polyamine, and organic polysilane.
- a triphenylamine derivative, a stilbene derivative, a hydrazone derivative, a phenamine derivative etc. are mentioned, for example.
- organic polysilanes are preferable because, unlike conventional carbon polymers, ⁇ electrons delocalized along the main chain Si contribute to photoconductivity and have high hole mobility (Phys. Rev. B, 1987, vol. 35, p.
- the conductive layer is not particularly limited as long as it has good conductivity, and examples thereof include inorganic conductive materials, organic conductive materials, conductive polymers, and intermolecular charge transfer complexes. Among them, an intermolecular charge transfer complex formed from a donor material and an acceptor material is preferable. Among these, what was formed from the organic donor and the organic acceptor can be used preferably.
- the donor material is preferably a material rich in electrons in the molecular structure.
- organic donor materials include those having a substituted or unsubstituted amine group, hydroxyl group, ether group, selenium or sulfur atom in the ⁇ -electron system of the molecule, specifically, phenylamine-based, triphenylmethane , Carbazole, phenol, and tetrathiafulvalene materials.
- acceptor material those lacking electrons in the molecular structure are preferable.
- organic acceptor materials include fullerenes, those having a substituent such as a nitro group, a cyano group, a carboxyl group or a halogen group in the ⁇ -electron system of the molecule, specifically, PCBM, benzoquinone, naphthoquinone, etc.
- the thickness of the conductive layer is not particularly limited, but is preferably such that the porous layer can be completely filled.
- the structure of the element may have a structure in which a first electrode layer, a first photoreceptor layer, a conductive layer, a second photoreceptor layer, and a second electrode layer are sequentially laminated.
- the dyes used in the first photoreceptor layer and the second photoreceptor layer may be the same or different, and if they are different, it is preferable that the absorption spectra are different.
- structures and members that are applied to this type of electrochemical element can be applied as appropriate.
- the light receiving electrode may be a tandem type in order to increase the utilization rate of incident light.
- Examples of preferred tandem type configurations include those described in JP-A Nos. 2000-90989 and 2002-90989.
- a light management function for efficiently performing light scattering and reflection inside the light receiving electrode layer may be provided.
- Preferable examples include those described in JP-A-2002-93476.
- a short-circuit prevention layer between the conductive support and the photoreceptor layer.
- Preferable examples include Japanese Patent Application Laid-Open No. 06-507999.
- a spacer or a separator In order to prevent contact between the light receiving electrode and the counter electrode, it is preferable to use a spacer or a separator. A preferable example is JP-A-2001-283941.
- Cell and module sealing methods include polyisobutylene thermosetting resin, novolak resin, photo-curing (meth) acrylate resin, epoxy resin, ionomer resin, glass frit, method using aluminum alkoxide for alumina, low melting point glass paste It is preferable to use a laser melting method. When glass frit is used, powder glass mixed with acrylic resin as a binder may be used.
- the crude product was dissolved in a methanol solution together with TBAOH (tetrabutylammonium hydroxide) and purified with a Sephadex LH-20 column.
- the main layer fraction was collected and concentrated, and then 0.2 M nitric acid was added.
- the precipitate was filtered and washed with water and diethyl ether to obtain 600 mg of a crude product.
- the crude product was dissolved in a methanol solution, 1M nitric acid was added, the precipitate was filtered, and washed with water and diethyl ether to obtain 570 mg of Exemplified dye (X-26).
- the obtained exemplary dye (X-26) was prepared in an ethanol solvent so that the dye concentration was 8.5 ⁇ mol / L and subjected to spectral absorption measurement. As a result, the absorption maximum wavelength was 568 nm.
- the obtained exemplary dye (X-30) was prepared in an ethanol solvent so that the dye concentration was 8.5 ⁇ mol / L and subjected to spectral absorption measurement. As a result, the absorption maximum wavelength was 570 nm.
- the obtained exemplary dye (X-32) was prepared in an ethanol solvent so that the dye concentration was 8.5 ⁇ mol / L and subjected to spectral absorption measurement. As a result, the absorption maximum wavelength was 574 nm.
- the obtained exemplary dye (X-31) was prepared in an ethanol solvent so that the dye concentration was 8.5 ⁇ mol / L and subjected to spectral absorption measurement. As a result, the absorption maximum wavelength was 588 nm.
- the obtained exemplary dye (X-33) was prepared in an ethanol solvent so that the dye concentration was 8.5 ⁇ mol / L and subjected to spectral absorption measurement. As a result, the absorption maximum wavelength was 570 nm.
- the obtained exemplary dye (X-34) was prepared in an ethanol solvent so that the dye concentration was 8.5 ⁇ mol / L and subjected to spectral absorption measurement. As a result, the absorption maximum wavelength was 571 nm.
- the obtained exemplary dye (X-35) was prepared with an ethanol solvent so that the dye concentration was 8.5 ⁇ mol / L and subjected to spectral absorption measurement. As a result, the absorption maximum wavelength was 574 nm.
- the obtained exemplary dye (X-36) was prepared with an ethanol solvent so that the dye concentration was 8.5 ⁇ mol / L and subjected to spectral absorption measurement. As a result, the absorption maximum wavelength was 580 nm.
- Example Dye (X-22), Example Dye (X-23), Example Dye (X-24), Example Dye (X-25), Example Dye (X-27), and Example Dye (X-28) It was prepared by the method.
- comparative dyes the following dyes (X-19), (X-20) and (X-21) Am. Chem. Soc., 2001, vol. 123, p. It was prepared with reference to the method described in 1613-1624.
- Titanium oxide (TiO 2 ) Acetic acid (0.2 mol) was added dropwise to Titanium isopropoxide (0.2 mol) at room temperature and stirred for 15 minutes. Thereafter, 290 mL of distilled water was added and stirred for 1 hour. After 1 hour, 65% aqueous HNO 3 solution was added, heated to 78 ° C. over 40 minutes, and stirred for 75 minutes. After stirring, 290 mL of distilled water was added to prepare a titanium oxide sol solution (crystal system: amorphous). This titanium oxide sol solution was stirred at 250 ° C. for 12 hours using an autoclave to obtain a titanium oxide particle-dispersed aqueous solution. This aqueous solution was filtered to obtain titanium oxide. The crystal system of the obtained titanium oxide was anatase type. The particle size of the titanium oxide was measured by a laser diffraction type particle size distribution analyzer (Mastersizer (trade name) manufactured by MALVERN), and it was 10 to 30 nm.
- the particle size of the semiconductor fine particles was measured with a laser diffraction particle size distribution analyzer (Mastersizer (trade name) manufactured by MALVERN), and it was 20 to 30 nm.
- Graphene was used as the conductive material.
- Graphene was prepared from Qingdao (trade name) manufactured by Qingdao Tianhe Graphite (People's Republic of China) using flaky kraftite (average particle size: 4 ⁇ m, purity 99.95%). 5 g of the above graphite and 3.75 g of NaNO 3 were added to the flask, and 375 mL of H 2 SO 4 was added and stirred under ice cooling. Thereafter, 22.5 g of KMnO 4 was added over about 1 hour. After stirring for 2 hours under ice cooling, the mixture was stirred at room temperature for 5 days.
- aqueous solution is purified by passing through an ion exchange resin (D301T, Nankai University Chemical Plant). Purification was performed by the above method, and graphene was obtained by removing distilled water. It was confirmed by X-ray photoelectron spectroscopy and a scanning electron microscope that the purified product was graphene. About what added graphene to the semiconductor fine particle, 1 mass% was mix
- tin oxide doped with fluorine was formed as a transparent conductive film by sputtering.
- the semiconductor fine particle dispersion was applied to a transparent conductive film and heated at 500 ° C. to form a semiconductor fine particle layer.
- the thickness of the semiconductor fine particle layer thus obtained was 10 ⁇ m, and the coating amount of the semiconductor fine particles was 20 g / m 2 .
- the semiconductor fine particle layer formed on the glass substrate was immersed in a 10% ethanol solution of the dyes shown in the following table at 40 ° C. for 3 hours in the dark.
- the light-receiving electrode obtained by adsorbing the dye was desorbed using a 10% TBAOH methanol solution, and the initial adsorption amount of each dye was quantified by measuring the absorption spectrum.
- B what adsorption amount is less than 2.0 ⁇ 10 -4 mM / cm 2 , the 2.0 ⁇ 10 -4 mM / cm 2 or more of the the A.
- the thickness of the photosensitive layer thus obtained was 10 ⁇ m, and the coating amount of semiconductor fine particles was 20 g / m 2 .
- the semiconductor fine particle electrode was disposed opposite to the platinum sputtered FTO substrate through a 50 ⁇ m thick thermoplastic polyolefin resin sheet, and the resin sheet portion was melted by heat to fix the bipolar plate.
- the electrolyte solution was injected from the injection port of the electrolyte solution previously opened in the platinum sputter
- As the electrolytic solution a methoxypropionitrile solution of dimethylpropylimidazolium iodide (0.5 mol / L) and iodine (0.1 mol / L) was used.
- conversion efficiency is 4% or more and less than 5%
- a value of 8% or more and less than 9% was evaluated as A, and A, B, and C were regarded as acceptable.
- durability the conversion efficiency after 500 hours with respect to the initial value of the conversion efficiency is evaluated as A with a conversion efficiency of 90% or more, B with 80% or more and less than 90%, and C with a conversion efficiency of less than 80%. , A and B were accepted.
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Materials Engineering (AREA)
- Power Engineering (AREA)
- Crystallography & Structural Chemistry (AREA)
- Microelectronics & Electronic Packaging (AREA)
- Inorganic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Hybrid Cells (AREA)
- Photovoltaic Devices (AREA)
Abstract
Description
特許文献2には、特定の構造の色素を用いることにより、太陽光を有効に吸収して光電変換効率を向上させた光増感太陽電池が提案されている。
また、中心部であるコアと外殻部であるシェルとを有する多重構造酸化チタン微粒子のうち、コアとシェルの少なくとも一方にドーパントを含有させることにより、光電変換効率を向上させた光電池が提案されている(例えば、特許文献3参照)。
本発明者らがこれらの特許文献に記載された色素と半導体微粒子を用いて光電変換素子を作製し、評価したところ、耐久性の点では十分でない場合があることがわかった。光電変換素子には、初期の変換効率が高く、使用後も変換効率の低下が少なく耐久性に優れることが必要とされるため、これらの特許文献記載の光電変換素子では十分とはいえない。
そこで本発明者等は半導体微粒子と色素について鋭意検討を行った。その結果、半導体微粒子が2種以上の金属又は金属化合物を有してなり、かつ配位子に特定の置換基を有する金属錯体色素を用いた光電変換素子及び光電気化学電池が、初期の光電変換効率だけでなく、耐久性に優れることを見出した。
本発明はこれらの知見に基づきなされたものである。
<1>導電性支持体と、色素を含む半導体微粒子層で構成された感光体層と、電荷移動体層と、対極とからなる光電変換素子であって、前記半導体微粒子が2種以上の金属又は金属化合物を局部的に有してなり、前記色素が下記一般式(1)で表される化合物である、光電変換素子。
Mz(LL1)m1(LL2)m2(X)m3・(CI)m4 一般式(1)
[Mzは金属原子を表し、LL1は下記一般式(2)で表される2座の配位子であり、LL2は下記一般式(3)で表される2座又は3座の配位子である。
Xは、アシルオキシ基、アシルチオ基、チオアシルオキシ基、チオアシルチオ基、アシルアミノオキシ基、チオカルバメート基、ジチオカルバメート基、チオカルボネート基、ジチオカルボネート基、トリチオカルボネート基、アシル基、チオシアネート基、イソチオシアネート基、シアネート基、イソシアネート基、シアノ基、アルキルチオ基、アリールチオ基、アルコキシ基及びアリールオキシ基からなる群から選ばれる1座若しくは2座の配位子、又はハロゲン原子、カルボニル、ジアルキルケトン、1,3-ジケトン、カルボンアミド、チオカルボンアミド及びチオ尿素からなる群から選ばれる1座若しくは2座の配位子を表す。
CIは、一般式(1)で表される化合物の電荷を中和させる対イオンを表す。
m1は1~3の整数を表し、m1が2以上のときLL1は同じでも異なっていてもよい。m2は0~2の整数を表し、m2が2のときLL2は同じでも異なっていてもよい。m3は0~3の整数を表し、m3が2以上のときXは同じでも異なっていてもよい。m4は0~3の整数を表し、m4が2以上のときCIは同じでも異なっていてもよい。

L1及びL2はそれぞれ独立に、エテニレン基及び/又はエチニレン基からなる共役鎖を表す。
a1及びa2はそれぞれ独立に0~3の整数を表し、a1が2以上のときR101は同じでも異なっていてもよく、a2が2以上のときR102は同じでも異なっていてもよい。b1及びb2はそれぞれ独立に0~3の整数を表す。b1が2以上のときR103は同じでも異なっていてもよく、互いに連結して環を形成してもよい。b2が2以上のときR104は同じでも異なっていてもよく、互いに連結して環を形成してもよい。b1及びb2が共に1以上のときR103とR104とが連結して環を形成してもよい。d1及びd2はそれぞれ独立に0~5の整数を表す。d3は0又は1を表す。

<2>前記半導体微粒子における2種以上の金属又は金属化合物が、金属原子、金属のカルコゲニド、金属炭酸塩又は金属硝酸塩である、前記<1>項記載の光電変換素子。
<3>前記金属原子がTi、Sn、Au、Ag、Cu、Al、Zr、Nb、V及びTaからなる群から選ばれた少なくとも1種である、前記<2>項記載の光電変換素子。
<4>前記金属カルコゲニドが硫化カドミウム、セレン化カドミウム又はTi、Sn、Zn、Mg、Al、W、Zr、Hf、Sr、In、Ce、Y、La、V及びTaからなる群から選ばれた少なくとも1種の金属酸化物である、前記<2>又は<3>項記載の光電変換素子。
<5>前記金属炭酸塩が炭酸カルシウム、炭酸カリウム及び炭酸バリウムからなる群から選ばれた少なくとも1種である、前記<2>~<4>のいずれか1項記載の光電変換素子。
<6>前記金属硝酸塩が硝酸ランタンである、前記<2>~<5>のいずれか1項記載の光電変換素子。
<7>前記半導体微粒子が、コア-シェル構造により、前記金属原子、前記金属のカルコゲニド、前記金属炭酸塩及び/又は前記金属硝酸塩を有してなる、前記<2>~<6>のいずれか1項記載の光電変換素子。
<8>前記半導体微粒子が、前記金属のカルコゲニドをコア部分として有し、前記金属のカルコゲニド又は前記金属炭酸塩をシェル部分として有する、前記<7>項記載の光電変換素子。
<9>前記半導体微粒子が、酸化チタン及び酸化スズからなる群より選ばれる金属のカルコゲニドをコア部分として有し、酸化アルミニウム、酸化マグネシウム、炭酸カルシウム、酸化チタン及び酸化チタン/酸化マグネシウムからなる群より選ばれる金属のカルコゲニド又は金属炭酸塩をシェル部分として有する、前記<8>項記載の光電変換素子。
<10>前記半導体微粒子が、金属原子をドープすることにより、2種以上の金属原子を有してなる、前記<1>~<6>のいずれか1項記載の光電変換素子。
<11>前記半導体微粒子が、前記金属のカルコゲニドに前記金属原子をドープして得られた半導体微粒子である、前記<10>項記載の光電変換素子。
<12>前記半導体微粒子が、酸化チタン及び酸化スズからなる群より選ばれる金属のカルコゲニドに、Nb、V及びTaからなる群より選ばれる少なくとも1種の金属原子をドープして得られた半導体微粒子である、前記<11>項記載の光電変換素子。
<13>前記半導体微粒子の粒径が、1~1000nmである、前記<1>~<12>のいずれか1項記載の光電変換素子。
<14>前記半導体微粒子が、導電性材料からなる添加剤を含む、前記<1>~<13>のいずれか1項記載の光電変換素子。
<15>前記導電性材料がグラフェンである、前記<14>項記載の光電変換素子。
<16>一般式(1)において、MzがRuであり、m1が1であり、m2が1であり、Xがイソチオシアネート基であり、m3が2である、前記<1>~<15>のいずれか1項記載の光電変換素子。
<17>一般式(1)において、LL1が一般式(4-1)~(4-3)のいずれかで表される、前記<1>~<16>のいずれか1項記載の光電変換素子。

<18>前記<1>~<17>のいずれか1項に記載の光電変換素子を備える、光電気化学電池。
その感光体層2は半導体微粒子22と増感色素(以下、単に、色素ともいう。)21とを有している。増感色素21はその少なくとも一部において半導体微粒子22に吸着している(増感色素21は吸着平衡状態になっており、一部電荷移動体層3に存在していてもよい。)。電荷移動体層3は、例えば正孔(ホール)を輸送する正孔輸送層として機能する。感光体層2が形成された導電性支持体1は、光電変換素子10において作用電極として機能する。この光電変換素子10を外部回路6で仕事をさせるようにして、光電気化学電池100として作動させることができる。
上記感光体層2では、多孔質半導体層が下記一般式(1)で表される少なくとも1種の色素21で増感されている。
Mz(LL1)m1(LL2)m2(X)m3・CI 一般式(1)
Mzは金属原子を表す。Mzは4配位又は6配位が可能な金属が好ましく、Ru、Fe、Os、Cu、W、Cr、Mo、Ni、Pd、Pt、Co、Ir、Rh、Re、Mn又はZnがより好ましく、Ru、Os、Zn又はCuが特に好ましく、Ruが最も好ましい。
配位子LL1は、一般式(2)で表される2座の配位子である。
配位子LL1の数を表すm1は1~3の整数である。m1が2以上のとき、配位子LL1は同じでも異なっていてもよい。m1は1であることが好ましい。


R121とR122並びにR123とR124はそれぞれ互いに連結して環を形成していてもよい。形成する環としてはピロリジン環、ピペリジン環、ピペラジン環、又はモルホリン環等が好ましい。
配位子LL2は、一般式(3)で表される2座又は3座の配位子であり、2座配位子であるのが好ましい。
配位子LL2の数を表すm2は0~2の整数であり、0又は1であるのが好ましく、1がより好ましい。m2が2のとき配位子LL2は同じでも異なっていてもよい。
本発明において、配位子LL2は、カルボキシル基、スルホン酸基、ヒドロキシル基、ヒドロキサム酸基、ホスホリル基、ホスホニル基等の酸性基を有することが好ましい。


一般式(1)中、配位子Xは下記に示す1座又は2座の配位子を表す。配位子Xの数を表すm3は0~3の整数を表し、0~2の整数が好ましく、1又は2がより好ましい。配位子Xが1座の配位子のとき、m3は2であるのが好ましく、配位子Xが2座配位子のとき、m3は1であるのが好ましい。m3が2以上の整数であるとき、配位子Xは同じでも異なっていてもよく、配位子X同士が連結していてもよい。
一般式(1)中のCIは電荷を中和させるのに対イオンが必要な場合の対イオンを表す。一般に、色素が陽イオン又は陰イオンであるか、あるいは正味のイオン電荷を有するかどうかは、色素中の金属、配位子及び置換基に依存する。
一般式(1)で表される構造を有する色素は、半導体微粒子の表面に対する適当な酸性基(結合基、interlocking group)を1つ以上有する。この基を色素中に1~6個有するのがより好ましく、1~4個有するのが特に好ましい。カルボキシル基、スルホン酸基、ヒドロキシル基、ヒドロキサム酸基(例えば-CONHOH等)、ホスホリル基(例えば-OP(O)(OH)2等)、ホスホニル基(例えば-P(O)(OH)2等)等の酸性基(解離性のプロトンを有する置換基)を色素中に有することが好ましい。なかでも、カルボキシル基(COOH基)を配位子上に有することが好ましい。本明細書において酸性基とはプロトンを放出する置換基を指す。また、「酸性基を有する」など、「特定の機能性の置換基を有する」というとき、本発明の効果を損ねない範囲で、当該機能性の置換基が母核に直接結合されていることのほか、所定の連結基を介して結合(連結)されたものを含む意味である。
・ハロゲン原子(例えば、フッ素原子、塩素原子、臭素原子、ヨウ素原子)、
・アルキル基〔直鎖、分岐、環状の置換もしくは無置換のアルキル基を表す。それらは、アルキル基(好ましくは炭素数1から30のアルキル基、例えばメチル、エチル、n-プロピル、イソプロピル、t-ブチル、n-オクチル、エイコシル、2-クロロエチル、2-シアノエチル、2-エチルヘキシル)、シクロアルキル基(好ましくは、炭素数3から30の置換または無置換のシクロアルキル基、例えば、シクロヘキシル、シクロペンチル、4-n-ドデシルシクロヘキシル)、ビシクロアルキル基(好ましくは、炭素数5から30の置換もしくは無置換のビシクロアルキル基、つまり、炭素数5から30のビシクロアルカンから水素原子を一個取り去った一価の基である。例えば、ビシクロ[1,2,2]ヘプタン-2-イル、ビシクロ[2,2,2]オクタン-3-イル)、更に環構造が多いトリシクロ構造なども包含するものである。以下に説明する置換基の中のアルキル基(例えばアルキルチオ基のアルキル基)もこのような概念のアルキル基を表す。]、
・アルケニル基[直鎖、分岐、環状の置換もしくは無置換のアルケニル基を表す。それらは、アルケニル基(好ましくは炭素数2から30の置換または無置換のアルケニル基、例えば、ビニル、アリル、プレニル、ゲラニル、オレイル)、シクロアルケニル基(好ましくは、炭素数3から30の置換もしくは無置換のシクロアルケニル基、つまり、炭素数3から30のシクロアルケンの水素原子を一個取り去った一価の基である。例えば、2-シクロペンテン-1-イル、2-シクロヘキセン-1-イル)、ビシクロアルケニル基(置換もしくは無置換のビシクロアルケニル基、好ましくは、炭素数5から30の置換もしくは無置換のビシクロアルケニル基、つまり二重結合を一個持つビシクロアルケンの水素原子を一個取り去った一価の基である。例えば、ビシクロ[2,2,1]ヘプト-2-エン-1-イル、ビシクロ[2,2,2]オクト-2-エン-4-イル)を包含するものである。]、
・アルキニル基(好ましくは、炭素数2から30の置換または無置換のアルキニル基、例えば、エチニル、プロパルギル、トリメチルシリルエチニル基、アリール基(好ましくは炭素数6から30の置換もしくは無置換のアリール基、例えばフェニル、p-トリル、ナフチル、m-クロロフェニル、o-ヘキサデカノイルアミノフェニル)、
・芳香族基(例えば、ベンゼン環、フラン環、ピロール環、ピリジン環、チオフェン環、イミダゾール環、オキサゾール環、、チアゾール環、ピラゾール環、イソオキサゾール環、イソチアゾール環、ピリミジン環、ピラジン環もしくはこれらが縮環した環)
・ヘテロ環基(好ましくは5または6員の置換もしくは無置換の、芳香族もしくは非芳香族のヘテロ環化合物から一個の水素原子を取り除いた一価の基であり、更に好ましくは、炭素数3から30の5もしくは6員の芳香族のヘテロ環基である。例えば、2-フリル、2-チエニル、2-ピリミジニル、2-ベンゾチアゾリル)、
・シアノ基、
・ヒドロキシル基、
・ニトロ基、
・カルボキシル基、
・アルコキシ基(好ましくは、炭素数1から30の置換もしくは無置換のアルコキシ基、例えば、メトキシ、エトキシ、イソプロポキシ、t-ブトキシ、n-オクチルオキシ、2-メトキシエトキシ)、
・アリールオキシ基(好ましくは、炭素数6から30の置換もしくは無置換のアリールオキシ基、例えば、フェノキシ、2-メチルフェノキシ、4-t-ブチルフェノキシ、3-ニトロフェノキシ、2-テトラデカノイルアミノフェノキシ)、
・シリルオキシ基(好ましくは、炭素数3から20のシリルオキシ基、例えば、トリメチルシリルオキシ、t-ブチルジメチルシリルオキシ)、
・ヘテロ環オキシ基(好ましくは、炭素数2から30の置換もしくは無置換のヘテロ環オキシ基、1-フェニルテトラゾール-5-オキシ、2-テトラヒドロピラニルオキシ)、
・アシルオキシ基(好ましくはホルミルオキシ基、炭素数2から30の置換もしくは無置換のアルキルカルボニルオキシ基、炭素数6から30の置換もしくは無置換のアリールカルボニルオキシ基、例えば、ホルミルオキシ、アセチルオキシ、ピバロイルオキシ、ステアロイルオキシ、ベンゾイルオキシ、p-メトキシフェニルカルボニルオキシ)、
・カルバモイルオキシ基(好ましくは、炭素数1から30の置換もしくは無置換のカルバモイルオキシ基、例えば、N,N-ジメチルカルバモイルオキシ、N,N-ジエチルカルバモイルオキシ、モルホリノカルボニルオキシ、N,N-ジ-n-オクチルアミノカルボニルオキシ、N-n-オクチルカルバモイルオキシ)、
・アルコキシカルボニルオキシ基(好ましくは、炭素数2から30の置換もしくは無置換アルコキシカルボニルオキシ基、例えばメトキシカルボニルオキシ、エトキシカルボニルオキシ、t-ブトキシカルボニルオキシ、n-オクチルカルボニルオキシ)、
・アリールオキシカルボニルオキシ基(好ましくは、炭素数7から30の置換もしくは無置換のアリールオキシカルボニルオキシ基、例えば、フェノキシカルボニルオキシ、p-メトキシフェノキシカルボニルオキシ、p-n-ヘキサデシルオキシフェノキシカルボニルオキシ)、
・アミノ基(好ましくは、アミノ基、炭素数1から30の置換もしくは無置換のアルキルアミノ基、炭素数6から30の置換もしくは無置換のアニリノ基、例えば、アミノ、メチルアミノ、ジメチルアミノ、アニリノ、N-メチル-アニリノ、ジフェニルアミノ)、
・アシルアミノ基(好ましくは、ホルミルアミノ基、炭素数1から30の置換もしくは無置換のアルキルカルボニルアミノ基、炭素数6から30の置換もしくは無置換のアリールカルボニルアミノ基、例えば、ホルミルアミノ、アセチルアミノ、ピバロイルアミノ、ラウロイルアミノ、ベンゾイルアミノ、3,4,5-トリ-n-オクチルオキシフェニルカルボニルアミノ)、
・アミノカルボニルアミノ基(好ましくは、炭素数1から30の置換もしくは無置換のアミノカルボニルアミノ、例えば、カルバモイルアミノ、N,N-ジメチルアミノカルボニルアミノ、N,N-ジエチルアミノカルボニルアミノ、モルホリノカルボニルアミノ)、
・アルコキシカルボニルアミノ基(好ましくは炭素数2から30の置換もしくは無置換アルコキシカルボニルアミノ基、例えば、メトキシカルボニルアミノ、エトキシカルボニルアミノ、t-ブトキシカルボニルアミノ、n-オクタデシルオキシカルボニルアミノ、N-メチル-メトキシカルボニルアミノ)、
・アリールオキシカルボニルアミノ基(好ましくは、炭素数7から30の置換もしくは無置換のアリールオキシカルボニルアミノ基、例えば、フェノキシカルボニルアミノ、p-クロロフェノキシカルボニルアミノ、m-n-オクチルオキシフェノキシカルボニルアミノ)、
・スルファモイルアミノ基(好ましくは、炭素数0から30の置換もしくは無置換のスルファモイルアミノ基、例えば、スルファモイルアミノ、N,N-ジメチルアミノスルホニルアミノ、N-n-オクチルアミノスルホニルアミノ)、
・アルキル及びアリールスルホニルアミノ基(好ましくは炭素数1から30の置換もしくは無置換のアルキルスルホニルアミノ、炭素数6から30の置換もしくは無置換のアリールスルホニルアミノ、例えば、メチルスルホニルアミノ、ブチルスルホニルアミノ、フェニルスルホニルアミノ、2,3,5-トリクロロフェニルスルホニルアミノ、p-メチルフェニルスルホニルアミノ)、
・メルカプト基、
・アルキルチオ基(好ましくは、炭素数1から30の置換もしくは無置換のアルキルチオ基、例えばメチルチオ、エチルチオ、n-ヘキサデシルチオ)、
・アリールチオ基(好ましくは炭素数6から30の置換もしくは無置換のアリールチオ、例えば、フェニルチオ、p-クロロフェニルチオ、m-メトキシフェニルチオ)、
・ヘテロ環チオ基(好ましくは炭素数2から30の置換または無置換のヘテロ環チオ基、例えば、2-ベンゾチアゾリルチオ、1-フェニルテトラゾール-5-イルチオ)、
・スルファモイル基(好ましくは炭素数0から30の置換もしくは無置換のスルファモイル基、例えば、N-エチルスルファモイル、N-(3-ドデシルオキシプロピル)スルファモイル、N,N-ジメチルスルファモイル、N-アセチルスルファモイル、N-ベンゾイルスルファモイル、N-(N’-フェニルカルバモイル)スルファモイル)、
・スルホ基、
・アルキル及びアリールスルフィニル基(好ましくは、炭素数1から30の置換または無置換のアルキルスルフィニル基、6から30の置換または無置換のアリールスルフィニル基、例えば、メチルスルフィニル、エチルスルフィニル、フェニルスルフィニル、p-メチルフェニルスルフィニル)、
・アルキル及びアリールスルホニル基(好ましくは炭素数1から30の置換または無置換のアルキルスルホニル基、6から30の置換または無置換のアリールスルホニル基、例えば、メチルスルホニル、エチルスルホニル、フェニルスルホニル、p-メチルフェニルスルホニル)、
・アシル基(好ましくはホルミル基、炭素数2から30の置換または無置換のアルキルカルボニル基、炭素数7から30の置換もしくは無置換のアリールカルボニル基、炭素数4から30の置換もしくは無置換の炭素原子でカルボニル基と結合しているヘテロ環カルボニル基、例えば、アセチル、ピバロイル、2-クロロアセチル、ステアロイル、ベンゾイル、p-n-オクチルオキシフェニルカルボニル、2-ピリジルカルボニル、2-フリルカルボニル)、
・アリールオキシカルボニル基(好ましくは、炭素数7から30の置換もしくは無置換のアリールオキシカルボニル基、例えば、フェノキシカルボニル、o-クロロフェノキシカルボニル、m-ニトロフェノキシカルボニル、p-t-ブチルフェノキシカルボニル)、
・アルコキシカルボニル基(好ましくは、炭素数2から30の置換もしくは無置換アルコキシカルボニル基、例えば、メトキシカルボニル、エトキシカルボニル、t-ブトキシカルボニル、n-オクタデシルオキシカルボニル)、
・カルバモイル基(好ましくは、炭素数1から30の置換もしくは無置換のカルバモイル、例えば、カルバモイル、N-メチルカルバモイル、N,N-ジメチルカルバモイル、N,N-ジ-n-オクチルカルバモイル、N-(メチルスルホニル)カルバモイル)、
・アリール及びヘテロ環アゾ基(好ましくは炭素数6から30の置換もしくは無置換のアリールアゾ基、炭素数3から30の置換もしくは無置換のヘテロ環アゾ基、例えば、フェニルアゾ、p-クロロフェニルアゾ、5-エチルチオ-1,3,4-チアジアゾール-2-イルアゾ)、
・イミド基(好ましくは、N-スクシンイミド、N-フタルイミド)、
・ホスフィノ基(好ましくは、炭素数2から30の置換もしくは無置換のホスフィノ基、例えば、ジメチルホスフィノ、ジフェニルホスフィノ、メチルフェノキシホスフィノ)、
・ホスフィニル基(好ましくは、炭素数2から30の置換もしくは無置換のホスフィニル基、例えば、ホスフィニル、ジオクチルオキシホスフィニル、ジエトキシホスフィニル)、
・ホスフィニルオキシ基(好ましくは炭素数2から30の置換もしくは無置換のホスフィニルオキシ基、例えば、ジフェノキシホスフィニルオキシ、ジオクチルオキシホスフィニルオキシ)、
・ホスフィニルアミノ基(好ましくは、炭素数2から30の置換もしくは無置換のホスフィニルアミノ基、例えば、ジメトキシホスフィニルアミノ、ジメチルアミノホスフィニルアミノ)、
・シリル基(好ましくは、炭素数3から30の置換もしくは無置換のシリル基、例えば、トリメチルシリル、t-ブチルジメチルシリル、フェニルジメチルシリル)。
また、置換基は更に置換されていても良い。その際、置換基の例としては、上述の置換基を挙げることができる。




本実施形態の光電変換素子に用いられる電荷移動体層には、電解質組成物からなる層が適用できる。その酸化還元対として、例えばヨウ素とヨウ化物(例えばヨウ化リチウム、ヨウ化テトラブチルアンモニウム、ヨウ化テトラプロピルアンモニウム等)との組み合わせ、アルキルビオローゲン(例えばメチルビオローゲンクロリド、ヘキシルビオローゲンブロミド、ベンジルビオローゲンテトラフルオロボレート)とその還元体との組み合わせ、ポリヒドロキシベンゼン類(例えばハイドロキノン、ナフトハイドロキノン等)とその酸化体との組み合わせ、2価と3価の鉄錯体(例えば赤血塩と黄血塩)の組み合わせ等が挙げられる。これらのうちヨウ素とヨウ化物との組み合わせが好ましい。
ヨウ素塩のカチオンは5員環又は6員環の含窒素芳香族カチオンであるのが好ましい。特に、一般式(1)により表される化合物がヨウ素塩でない場合は、WO95/18456号、特開平8-259543号、電気化学,第65巻,11号,923頁(1997年)等に記載されているピリジニウム塩、イミダゾリウム塩、トリアゾリウム塩等のヨウ素塩を併用するのが好ましい。
光電変換素子に使用される電解質組成物中には、ヘテロ環4級塩化合物と共にヨウ素を含有するのが好ましい。ヨウ素の含有量は電解質組成物全体に対して0.1~20質量%であるのが好ましく、0.5~5質量%であるのがより好ましい。
溶媒としては低粘度でイオン移動度が高いか、高誘電率で有効キャリアー濃度を高めることができるか、あるいはその両方であるために優れたイオン伝導性を発現できるものが好ましい。このような溶媒としてカーボネート化合物(エチレンカーボネート、プロピレンカーボネート等)、複素環化合物(3-メチル-2-オキサゾリジノン等)、エーテル化合物(ジオキサン、ジエチルエーテル等)、鎖状エーテル類(エチレングリコールジアルキルエーテル、プロピレングリコールジアルキルエーテル、ポリエチレングリコールジアルキルエーテル、ポリプロピレングリコールジアルキルエーテル等)、アルコール類(メタノール、エタノール、エチレングリコールモノアルキルエーテル、プロピレングリコールモノアルキルエーテル、ポリエチレングリコールモノアルキルエーテル、ポリプロピレングリコールモノアルキルエーテル等)、多価アルコール類(エチレングリコール、プロピレングリコール、ポリエチレングリコール、ポリプロピレングリコール、グリセリン等)、ニトリル化合物(アセトニトリル、グルタロジニトリル、メトキシアセトニトリル、プロピオニトリル、ベンゾニトリル、ビスシアノエチルエーテル等)、エステル類(カルボン酸エステル、リン酸エステル、ホスホン酸エステル等)、非プロトン性極性溶媒(ジメチルスルホキシド(DMSO)、スルフォラン等)、水、特開2002-110262記載の含水電解液、特開2000-36332号公報、特開2000-243134号公報、及び再公表WO/00-54361号公報記載の電解質溶媒などが挙げられる。これらの溶媒は二種以上を混合して用いてもよい。
ゲル電解質に占めるモノマーの重量組成範囲は0.5~70質量%であるのが好ましい。より好ましくは1.0~50質量%である。ポリマーの架橋反応により電解質組成物をゲル化させる場合は、組成物に架橋可能な反応性基を有するポリマー及び架橋剤を添加するのが好ましい。好ましい反応性基はピリジン環、イミダゾール環、チアゾール環、オキサゾール環、トリアゾール環、モルホリン環、ピペリジン環、ピペラジン環等の含窒素複素環であり、好ましい架橋剤は窒素原子が求核攻撃できる官能基を2つ以上有する化合物(求電子剤)であり、例えば2官能以上のハロゲン化アルキル、ハロゲン化アラルキル、スルホン酸エステル、酸無水物、酸クロライド、イソシアネート等である。
導電性支持体としては、金属のように支持体そのものに導電性があるものか、または表面に導電膜層を有するガラスや高分子材料を使用することができる。導電性支持体は実質的に透明であることが好ましい。実質的に透明であるとは光の透過率が10%以上であることを意味し、50%以上であることが好ましく、80%以上が特に好ましい。導電性支持体としては、ガラスや高分子材料に導電性の金属酸化物を塗設したものを使用することができる。このときの導電性の金属酸化物の塗布量は、ガラスや高分子材料の支持体1m2当たり、0.1~100gが好ましい。透明導電性支持体を用いる場合、光は支持体側から入射させることが好ましい。好ましく使用される高分子材料の一例として、テトラアセチルセルロース(TAC)、ポリエチレンテレフタレート(PET)、ポリエチレンナフタレート(PEN)、シンジオタクチックポリスチレン(SPS)、ポリフェニレンスルフィド(PPS)、ポリカーボネート(PC)、ポリアリレート(PAR)、ポリスルフォン(PSF)、ポリエステルスルフォン(PES)、ポリエーテルイミド(PEI)、環状ポリオレフィン、ブロム化フェノキシ等を挙げることができる。導電性支持体上には、表面に光マネージメント機能を施してもよく、例えば、特開2003-123859記載の高屈折膜及び低屈性率の酸化物膜を交互に積層した反射防止膜、特開2002-260746記載のライトガイド機能が上げられる。
この他にも、金属支持体も好ましく使用することができる。その一例としては、チタン、アルミニウム、銅、ニッケル、鉄、ステンレス、銅を挙げることができる。これらの金属は合金であってもよい。さらに好ましくは、チタン、アルミニウム、銅が好ましく、特に好ましくは、チタンやアルミニウムである。
導電性支持体上には、さらに特開平11-250944号公報等に記載の機能を付与してもよい。
導電膜層の厚さは0.01~30μmであることが好ましく、0.03~25μmであることが更に好ましく、特に好ましくは0.05~20μmである。
導電性支持体は表面抵抗が低い程よい。好ましい表面抵抗の範囲としては50Ω/cm2以下であり、さらに好ましくは10Ω/cm2以下である。この下限に特に制限はないが、通常0.1Ω/cm2程度である。
また、透明電極と多孔質半導体電極光触媒含有層を設けてもよい。透明導電層は積層構造でも良く、好ましい方法としてたとえば、ITO上にFTOを積層することができる。
半導体微粒子は2種以上の金属又は金属化合物を局部的に有してなる。本発明において、金属化合物とは、分子内に金属と金属以外の1種以上の原子とを含む無機化合物をあらわし、たとえば金属のカルコゲニド、金属炭酸塩又は金属硝酸塩が挙げられる。本発明において、2種以上の金属又は金属化合物を局部的に有してなる半導体微粒子とは、予め、2種以上の金属又は金属化合物で処理することにより、微粒子中に2種以上の金属又は金属化合物が局部的に存在しているものをいう。2種以上の金属又は金属化合物を局部的に有してなる半導体微粒子としては、後述のように、2種以上の金属又は金属化合物によりコア-シェル構造を形成しているものや、表面の一部とそれ以外の部分とが異なる金属又は金属化合物で形成されているものをいう。したがって、2種以上の半導体微粒子を単に混合したものは含まれない。2種以上の金属又は金属化合物を局部的に有してなる半導体微粒子に特定の置換基を有する色素を用いることにより、色素の半導体微粒子への吸着を効率よく行うことができ、耐久性の高い光電変換素子を実現することができる。
金属原子は、Ti(チタン)、Sn(スズ)、Au(金)、Ag(銀)、Cu(銅)、Al(アルミニウム)、Zr(ジルコニウム)、Nb(ニオブ)、V(バナジウム)及びTa(タンタル)からなる群から選ばれた少なくとも1種が好ましい。さらに好ましくは、Ti、Sn、Zr、Nb、V、Taであり、特に好ましくは、Nb、V、Taである。
金属カルコゲニドは、硫化カドミウム、セレン化カドミウム又はTi(チタン)、Sn(スズ)、Zn(亜鉛)、Mg(マグネシウム)、Al(アルミニウム)、W(タングステン)、Zr(ジルコニウム)、Hf(ハフニウム)、Sr(ストロンチウム)、In(インジウム)、Ce(セリウム)、Y(イットリウム)、La(ランタン)、V(バナジウム)及びTa(タンタル)からなる群から選ばれた少なくとも1種の金属酸化物が好ましい。さらに好ましくは、Ti、Sn、Zn、Mg、Alからなる群から選ばれた少なくとも1種の金属酸化物であり、さらに好ましくは、Ti、Sn、Mg、Alからなる群から選ばれた少なくとも1種の金属酸化物であり、特に好ましくは、Ti、Sn、Alからなる群から選ばれた少なくとも1種の金属酸化物である。
金属炭酸塩は、炭酸カルシウム、炭酸カリウム及び炭酸バリウムからなる群から選ばれた少なくとも1種が好ましい。さらに好ましくは、炭酸カルシウム、炭酸バリウムであり、特に好ましくは、炭酸カルシウムである。
金属硝酸塩は硝酸ランタンが好ましい。
コア-シェル構造の半導体微粒子として、例えば、酸化チタンをコアとし、炭酸カルシウムをシェルとするコア-シェル構造の半導体微粒子は以下の方法で調製することができるが、この方法及び条件に限定されることはない。まず、12g(0.2mol)の酢酸を58.6g(0.2mol)のチタン酸テトライソプロピルにスターラーで攪拌しながら滴下する。得られた混合物を15分間攪拌し、290mLの蒸留水に添加する。1時間攪拌後、4mlの65%硝酸を加え、40分間かけて78℃まで加熱し、75分間温度を一定に保つ。反応容器をヒーターから外し、370mLの水を加える。得られた液体をチタン製のオートクレーブに移し、250℃で12時間加熱する。その後、2.4mLの65%硝酸を加え、超音波ホモジナイザーで攪拌してから,酸化チタン量が13~15%になるまで分散液を濃縮する。濃縮液を遠心分離により分離し上澄みの蒸留水を捨て、蒸留水と同量のエタノールを加える。その後、超音波ホモジナイザーにより攪拌することでコアの酸化チタン分散液を得る。次に、1~3質量%の酢酸カルシウム水溶液にコアである酸化チタン粒子を添加して30分~3時間攪拌する。攪拌後、遠心分離により酢酸カルシウム水溶液を除去し、蒸留水で洗浄、遠心分離を行った後、525℃で1時間焼成する方法により、酸化チタンをコアとし、炭酸カルシウムをシェルとするコア-シェル構造の半導体微粒子を得ることができる。
得られた半導体微粒子がコア-シェル構造を有することは、透過電子顕微鏡(TEM)で観察して判断することができる。コア部分とシェル部分の体積比は特に制限されないが、コア:シェルの体積比が、50:50~98:2が好ましく、70:30~95:5がより好ましい。この体積比は、TEMで観察して求めることができる。
コア部分として金属原子を用いた場合は、Ti、Nb、Sn、Zn及びLaからなる群から選ばれた少なくとも1種の金属原子が好ましい。さらに好ましくは、Ti、Sn、Znである。特に好ましくは、Ti、Snである。コア部分として金属カルコゲニドを用いた場合は、Ti、Sn、Zn、Mg及びAlの酸化物から選ばれた少なくとも1種の金属酸化物であることが好ましい。さらに好ましくは、Tiの酸化物、Snの酸化物、Znの酸化物である。特に好ましくは、Tiの酸化物、Snの酸化物である。コア部分として金属硝酸塩を用いた場合は、硝酸ランタンが好ましい。
シェル部分として、金属カルコゲニドを用いる場合は、Ti、Mg及びAlの酸化物を用いることが好ましい。シェル部分として、金属炭酸塩を用いる場合は、炭酸カルシウムを用いることが好ましい。
コア-シェル構造の半導体微粒子を製造するに際し、上記のように、従来の方法で、まず、コア部分となる半導体微粒子を製造することができる。例えば、コアとして酸化チタン(チタニア)を用いる場合、チタニアナノ粒子の製造方法として好ましくは、四塩化チタンの火炎加水分解による方法、四塩化チタンの燃焼法、安定なカルコゲナイド錯体の加水分解、オルトチタン酸の加水分解、可溶部と不溶部から半導体微粒子を形成後可溶部を溶解除去する方法、過酸化物水溶液の水熱合成でコア部分となる半導体微粒子を製造する。その後、前述の方法により、シェルとすべき金属原子又は金属化合物の溶液中に、コアとすべき半導体微粒子を加えて、適宜反応させることにより、得ることができる。
コア部分となるチタニアの結晶構造としては、アナターゼ型、ブルッカイト型、または、ルチル型があげられ、アナターゼ型、ブルッカイト型が好ましい。チタニアナノチューブ・ナノワイヤー・ナノロッドをチタニア微粒子に混合してもよい。
ドープされる金属原子は、例えば、Nb、V、Taを挙げることができる。さらに好ましくは、Nb、Vである。例えば、Nbパウダーとtetrabutyl titanateを過酸化水素とアンモニア(v/v=5/1)を含む水溶液中に添加し攪拌する。攪拌後、過剰の過酸化水素とアンモニアを80℃に熱することにより除去する。得られた溶液をテフロン(登録商標)製のオートクレーブに移し、180℃で20時間攪拌する。得られた沈殿物をpH=7の蒸留水で洗浄し、100℃で6時間乾燥させる方法により、金属原子をドープすることができる。
半導体微粒子には、このほか、半導体微粒子同士のネッキングを改善する為のバインダーや逆電子移動防止のために表面へ添加剤を用いてもよい。好ましい添加剤の例としては、ITO、SnO粒子、ウイスカー、繊維状グラファイト・カーボンナノチューブ、酸化亜鉛ネッキング結合子、セルロース等の繊維状物質、金属、有機シリコン、ドデシルベンゼンスルホン酸、シラン化合物等の電荷移動結合分子、及び電位傾斜型デンドリマーなどが挙げられる。半導体微粒子上の表面欠陥を除去するなどの目的で、色素吸着前に半導体微粒子を酸塩基又は酸化還元処理しても良い。エッチング、酸化処理、過酸化水素処理、脱水素処理、UV-オゾン、酸素プラズマなどで処理してもよい。
半導体微粒子分散液を前記の導電性支持体に塗布し、適度に加熱することにより、半導体微粒子層を得ることができる。半導体微粒子分散液には、半導体微粒子以外の固形分の含量が、半導体微粒子分散液全体の10質量%以下とすることが好ましい。
半導体微粒子分散液を作製する方法としては、前述のゾル・ゲル法の他に、半導体を合成する際に溶媒中で微粒子として析出させそのまま使用する方法、微粒子に超音波などを照射して超微粒子に粉砕する方法、あるいはミルや乳鉢などを使って機械的に粉砕しすり潰す方法、等が挙げられる。分散溶媒としては、水及び/又は各種の有機溶媒を用いることができる。有機溶媒としては、メタノール、エタノール、イソプロピルアルコール、シトロネロール、α-テルピネオールなどのアルコール類、アセトンなどのケトン類、酢酸エチルなどのエステル類、ジクロロメタン、アセトニトリル等が挙げられる。
分散の際、必要に応じて例えばポリエチレングリコール、ブチルセルロース、エチルセルロース、ヒドロキシエチルセルロース、カルボキシメチルセルロースのようなポリマー、界面活性剤、酸、またはキレート剤等を分散助剤として少量用いてもよい。しかし、これらの分散助剤は、導電性支持体上へ製膜する工程の前に、ろ過法や分離膜を用いる方法、あるいは遠心分離法などによって大部分を除去しておくことが好ましい。半導体微粒子分散液は、半導体微粒子以外の固形分の含量が分散液全体の10質量%以下とすることができる。この濃度は好ましくは5%以下であり、さらに好ましくは3%以下であり、特に好ましくは1%以下である。さらに好ましくは0.5%以下であり、特に好ましくは0.2%である。すなわち、半導体微粒子分散液中に、溶媒と半導体微粒子以外の固形分を半導体微分散液全体の10質量%以下とすることができる。実質的に半導体微粒子と分散溶媒のみからなることが好ましい。
半導体微粒子分散液の粘度が高すぎると分散液が凝集してしまい製膜することができず、逆に半導体微粒子分散液の粘度が低すぎると液が流れてしまい製膜することができないことがある。したがって分散液の粘度は、25℃で10~300N・s/m2が好ましい。さらに好ましくは、25℃で50~200N・s/m2である。
半導体微粒子層全体の好ましい厚さは0.1~100μmである。半導体微粒子層の厚さはさらに1~30μmが好ましく、2~25μmがより好ましい。半導体微粒子の支持体1m2当りの担持量は0.5g~400gが好ましく、5~100gがより好ましい。
また、加熱処理に加えて光のエネルギーを用いることもできる。例えば、半導体微粒子として酸化チタンを用いた場合に、紫外光のような半導体微粒子が吸収する光を与えることで表面を活性化してもよいし、レーザー光などで半導体微粒子表面のみを活性化することができる。半導体微粒子に対して該微粒子が吸収する光を照射することで、粒子表面に吸着した不純物が粒子表面の活性化によって分解され、上記の目的のために好ましい状態とすることができる。加熱処理と紫外光を組み合わせる場合は、半導体微粒子に対して該微粒子が吸収する光を照射しながら、加熱が100℃以上250℃以下あるいは好ましくは100℃以上150℃以下で行われることが好ましい。このように、半導体微粒子を光励起することによって、微粒子層内に混入した不純物を光分解により洗浄するとともに、微粒子の間の物理的接合を強めることができる。
塗布後に圧力をかけても良く、圧力をかける方法としては、特表2003-500857号公報等が挙げられる。光照射の例としては、特開2001-357896号公報等が挙げられる。プラズマ・マイクロ波・通電の例としては、特開2002-353453号公報等が挙げられる。化学的処理としては、例えば特開2001-357896号公報が挙げられる。
前駆体として例えば、(NH4)2TiF6、過酸化チタン、金属アルコキシド・金属錯体・金属有機酸塩等が挙げられる。
また、金属有機酸化物(アルコキシドなど)を共存させたスラリーを塗布し加熱処理、光処理などで半導体膜を形成する方法、無機系前駆体を共存させたスラリー、スラリーのpHと分散させたチタニア粒子の性状を特定した方法が挙げられる。これらスラリーには、少量であればバインダーを添加しても良く、バインダーとしては、セルロース、フッ素ポリマー、架橋ゴム、ポリブチルチタネート、カルボキシメチルセルロースなどが挙げられる。
半導体微粒子又はその前駆体層の形成に関する技術としては、コロナ放電、プラズマ、UVなどの物理的な方法で親水化する方法、アルカリやポリエチレンジオキシチオフェンとポリスチレンスルホン酸などによる化学処理、ポリアニリンなどの接合用中間膜の形成などが挙げられる。
また、耐熱基板上でいったん塗膜を作製した後、プラスチック等のフィルムに転写する方法を用いても良い。好ましくは、特開2002-184475号公報記載のEVAを介して転写する方法、特開2003-98977号公報記載の紫外線、水系溶媒で除去可能な無機塩を含む犠牲基盤上に半導体層・導電層を形成後、有機基板に転写後、犠牲基板を除去する方法などが挙げられる。
支持体として高分子材料を用いる場合、250℃以下で製膜後加熱することが好ましい。その場合の製膜方法としては、(1)湿式法、(2)乾式法、(3)電気泳動法(電析法を含む)の何れでも良く、好ましくは、(1)湿式法、又は(2)乾式であり、更に好ましくは、(1)湿式法である。
なお、半導体微粒子の支持体1m2当たりの塗布量は0.5~500g、さらには5~100gが好ましい。
上記のように作製された半導体微粒子層に色素を吸着させることにより、感光体層を形成することができる。
半導体微粒子に色素を吸着させるには、溶液と色素よりなる色素吸着用色素溶液の中に、よく乾燥した半導体微粒子を長時間浸漬するのが好ましい。色素吸着用色素溶液に使用される溶液は、色素が溶解できる溶液なら特に制限なく使用することができる。例えば、エタノール、メタノール、イソプロパノール、トルエン、t-ブタノール、アセトニトリル、アセトン、n-ブタノールなどを使用することができる。その中でも、エタノール、トルエンを好ましく使用することができる。
溶液と色素よりなる色素吸着用色素溶液は必要に応じて50℃ないし100℃に加熱してもよい。色素の吸着は半導体微粒子の塗布前に行っても塗布後に行ってもよい。また、半導体微粒子と色素を同時に塗布して吸着させてもよい。未吸着の色素は洗浄によって除去する。塗布膜の焼成を行う場合は色素の吸着は焼成後に行うことが好ましい。焼成後、塗布膜表面に水が吸着する前にすばやく色素を吸着させるのが特に好ましい。光電変換の波長域をできるだけ広くするように、混合する色素が選ばれる。色素を混合する場合は、すべての色素が溶解するようにして、色素吸着用色素溶液とすることが好ましい。
また、色素の半導体微粒子に対する吸着量は半導体微粒子1gに対して0.001~1ミリモルが好ましく、より好ましくは0.1~0.5ミリモルである。このような色素量とすることによって、半導体における増感効果が十分に得られる。
色素を吸着した後に、アミン類を用いて半導体微粒子の表面を処理してもよい。好ましいアミン類としては4-tert-ブチルピリジン、ポリビニルピリジン等が挙げられる。これらは液体の場合はそのまま用いてもよいし有機溶媒に溶解して用いてもよい。
対極(対向電極)は、光電気化学電池の正極として働くものである。対向電極は、通常前述の導電性支持体と同義であるが、強度が十分に保たれるような構成では支持体は必ずしも必要でない。ただし、支持体を有する方が密閉性の点で有利である。対向電極の材料としては、白金、カーボン、導電性ポリマー、などがあげられる。好ましい例としては、白金、カーボン、導電性ポリマーが挙げられる。
受光電極は酸化チタンと酸化スズ(TiO2/SnO2)などの複合電極を用いても良く、チタニアの混合電極として例えば、特開2000-113913号公報等が挙げられる。チタニア以外の混合電極として例えば、特開2001-185243号公報、特開2003-282164号公報等が挙げられる。
光電変換素子の構成として、導電性支持体(電極層)、光電変換層(感光体層及び電荷移動体層)、ホール輸送層、伝導層、対極層を順次に積層することができる。p型半導体として機能するホール輸送材料をホール輸送層としてもちいることができる。好ましいホール輸送層としては、例えば無機系又は有機系のホール輸送材料を用いることができる。無機系ホール輸送材料としては、CuI、CuO,NiO等が挙げられる。また、有機系ホール輸送材料としては、高分子系と低分子系のものが挙げられ、高分子系のものとしては、例えばポリビニルカルバゾール、ポリアミン、有機ポリシラン等が挙げられる。また、低分子系のものとしては、例えばトリフェニルアミン誘導体、スチルベン誘導体、ヒドラゾン誘導体、フェナミン誘導体等が挙げられる。この中でも有機ポリシランは、従来の炭素系高分子と異なり、主鎖のSiに沿って非局化されたσ電子が光伝導に寄与し、高いホール移動度を有するため、好ましい(Phys.Rev.B,1987,vol.35,p.2818)。
伝導層の厚みは、特に限定されないが、多孔質を完全に埋めることができる程度が好ましい。
受光電極層内部で光散乱、反射を効率的に行う光マネージメント機能を設けてもよい。好ましくは、特開2002-93476号公報に記載のものが挙げられる。
受光電極と対極の接触を防ぐ為に、スペーサーやセパレータを用いることが好ましい。好ましい例としては、特開2001-283941号公報が挙げられる。
下記のスキームの方法に従って例示色素(X-26)を調製した。

化合物(d-1-1)25g、Pd2(dba)3 33.8g、トリフェニルホスフィン8.6g、ヨウ化銅2.5g、1-へプチン25.2gをトリエチルアミン70mL及びテトラヒドロフラン(THF)50mLの混合溶液に室温で攪拌し、80℃で4.5時間攪拌した。濃縮後カラムクロマトグラフィーで精製することで化合物(d-1-2)26.4gを得た。
化合物(d-1-3)6.7gを窒素雰囲気下、-15℃でテトラヒドロフラン200mLに溶解し、別途調製したLDA(リチウムジイソプロピルアミド)を化合物(d-1-3)の2.5等量を滴下し、75分攪拌した。その後化合物(d-1-2)15gをテトラヒドロフラン30mLに溶解した溶液を滴下し0℃で1時間攪拌し、室温で17時間攪拌した。濃縮後、水150mLを加え、塩化メチレン150mLで分液・抽出し、塩水で有機層を洗浄し、有機層を濃縮した。得られた結晶をメタノールで再結晶し、化合物(d-1-4)18.9gを得た。
化合物(d-1-4)13.2g、PPTS(ピリジニウムパラトルエンスルホン酸)1.7gを、トルエン1000mLに加え、窒素雰囲気下で5時間加熱還流を行った。濃縮後、飽和重曹水及び塩化メチレンで分液を行い、有機層を濃縮した。得られた結晶をメタノール及び塩化メチレンで再結晶し、化合物(d-1-5)11.7gを得た。
化合物(d-1-5)4.0g、化合物(d-1-6)2.2g、をDMF(ジメチルホルムアミド)60mLに加え70℃で4時間攪拌した。その後化合物(d-1-7)2.1gを加え160℃で3.5時間加熱攪拌した。その後チオシアン酸アンモニウム19.0gを加え130℃で5時間攪拌した。濃縮後、水1.3mL加えろかし、ジエチルエーテルで洗った。粗精製物をTBAOH(水酸化テトラブチルアンモニウム)と共にメタノール溶液に溶解し、Sephadex LH-20カラムで精製した。主層の分画を回収し濃縮後硝酸0.2Mを添加して、沈殿物をろ過後、水及びジエチルエーテルで洗い、粗精製物600mgを得た。粗精製物をメタノール溶液に溶解し、硝酸1Mを添加して沈殿物をろ過後、水及びジエチルエーテルで洗い、例示色素(X-26)570mg得た。
1H-NMR(DMSO-d6、400MHz):δ(ppm)in aromatic regions:9.37(1H,d),9.11(1H,d),9.04(1H,s)、8.89(2H),8.74(1H,s),8.26(1H,d),8.10-7.98(2H),7.85-7.73(2H),7.60(1H,d),7.45-7.33(2H),7.33-7.12(5H,m),6.92(1H,d)
下記のスキームの方法に従って化合物(d-2-4)を調製し、化合物(d-1-2)を化合物(d-2-4)に代えた以外は例示色素(X-26)と同様にして例示色素(X-30)を調製した。

下記のスキームの方法に従って化合物(d-3-2)を調製し、化合物(d-1-2)を化合物(d-3-2)に代えた以外は例示色素(X-26)と同様にして例示色素(X-32)を調製した。

下記のスキームの方法に従って化合物(d-4-2)を調製し、化合物(d-1-2)を化合物(d-4-2)に代えた以外は例示色素(X-26)と同様にして例示色素(X-31)を調製した。

下記のスキームの方法に従って化合物(d-5-6)を調製し、化合物(d-1-5)を化合物(d-5-6)に代えた以外は例示色素(X-26)と同様にして、例示色素(X-33)を調製した。

下記のスキームの方法に従って化合物(d-6-3)を調製し、化合物(d-1-5)を化合物(d-6-3)に代えた以外は例示色素(X-26)と同様にして例示色素(X-34)を調製した。

前記例示色素(X-30)の調製において、化合物(d-2-2)の代わりに下記化合物(d-7-1)を用いた以外は同様にして、例示色素(X-35)を調製した。

下記のスキームの方法に従って、以下例示色素(X-26)と同様にして、例示色素(X-36)を調製した。

また、比較色素として、以下の色素(X-19)、(X-20)及び(X-21)を、J.Am.Chem.Soc.,2001,vol.123,p.1613-1624に記載の方法を参考に調製した。


色素(X-19)~(X-36)の最大吸収波長を測定した。その結果を表1に示す。測定は、分光光度計(U-4100(商品名)、日立ハイテク社製)によって行い、溶液はTHF:エタノール=1:1を用い、濃度が2μMになるように調整した。

半導体微粒子(I)を調製し、それを用いて、2種以上の金属又は金属化合物を局部的に有してなる半導体微粒子(II)を調製した。
1.半導体微粒子(I)の調製
(1)酸化スズ(SnO2)
Alfa Aesar社製のPuratronic(商品名)を、精製せずに使用した。この酸化スズの粒径を、レーザー回折式粒度分布測定装置(MALVERN社製のマスターサイザー(商品名))により測定したところ、20~30nmであった。
(2)酸化チタン(TiO2)
Titanium isopropoxide(0.2mol)に室温で酢酸(0.2mol)を滴下して15分間攪拌した。その後、290mLの蒸留水を添加して1時間攪拌した。1時間後、65%のHNO3水溶液を添加して、40分かけて78℃まで加熱し、75分攪拌した。攪拌後、蒸留水を290mL添加して酸化チタンゾル溶液(結晶系:アモルファス)を作製した。この酸化チタンゾル溶液を、オートクレーブを用いて250℃、12時間攪拌することにより酸化チタン粒子分散水溶液を得た。
この水溶液をろ過して、酸化チタンを得た。得られた酸化チタンの結晶系はアナターゼ型であった。この酸化チタンの粒径をレーザー回折式粒度分布測定装置(MALVERN社製のマスターサイザー(商品名))により測定したところ、10~30nmであった。
(1)コア-シェル型半導体微粒子の調製
(a)酸化アルミニウム(Al2O3)をシェル部分とする半導体微粒子(II)の調製
前記半導体微粒子(I)を、2~150mMのトリメチルアルミニウム水溶液に分散させ、200℃雰囲気で8秒間反応させ、半導体微粒子(II)を得た。得られた半導体微粒子を透過電子顕微鏡(TEM)で観察したところ、酸化スズ又は酸化チタンをコア部分とし、酸化アルミニウムをシェル部分とする、コア-シェル構造である半導体微粒子(II)であることがわかった。コア:シェルの体積比をTEMで観察したところ、90:10~98:2であった。この半導体微粒子の粒径をレーザー回折式粒度分布測定装置(MALVERN社製のマスターサイザー(商品名))で測定したところ、20~30nmであった。
前記半導体微粒子(I)を、2~150mMの酢酸マグネシウムが溶解したエタノール溶液(60~70℃)中に1分間浸漬し、洗浄後、500℃で焼成することにより、半導体微粒子(II)を得た。得られた半導体微粒子を透過電子顕微鏡(TEM)で観察したところ、酸化スズ又は酸化チタンをコア部分とし、酸化マグネシウムをシェル部分とする、コア-シェル構造である半導体微粒子(II)であることがわかった。上記と同様の方法でコア:シェルの体積比をTEMで観察したところ、90:10~98:2であった。上記と同様の方法で半導体微粒子の粒径を測定したところ、20~30nmであった。
前記半導体微粒子(I)を、2~20mMのTiCl4水溶液(70℃)に1時間浸漬し、洗浄後、500℃で焼成することにより、半導体微粒子(II)を得た。得られた半導体微粒子を透過電子顕微鏡(TEM)で観察したところ、酸化スズ又は酸化チタンをコア部分とし、酸化チタンをシェル部分とする、コア-シェル構造である半導体微粒子(II)であることがわかった。上記と同様の方法でコア:シェルの体積比をTEMで観察したところ、90:10~98:2であった。上記と同様の方法で半導体微粒子の粒径を測定したところ、20~30nmであった。
前記半導体微粒子(I)を、1~3質量%の酢酸カルシウム水溶液に所定時間浸漬して、525℃で焼成することにより、半導体微粒子(II)を得た。得られた半導体微粒子を透過電子顕微鏡(TEM)で観察したところ、酸化スズ又は酸化チタンをコア部分とし、炭酸カルシウムをシェル部分とする、コア-シェル構造である半導体微粒子(II)であることがわかった。上記と同様の方法でコア:シェルの体積比をTEMで観察したところ、90:10~98:2であった。上記と同様の方法で半導体微粒子の粒径を測定したところ、20~30nmであった。
2種類以上の材料をシェル部分とする半導体微粒子は、前記(a)~(d)に記載された方法を繰り返し行うことにより調製した。得られた半導体微粒子を透過電子顕微鏡(TEM)で観察したところ、コア部分に対して、使用した2種類以上の材料をシェル部分とする、コア-シェル構造である半導体微粒子(II)であることがわかった。上記と同様の方法でコア:シェルの体積比をTEMで観察したところ、90:10~98:2であった。上記と同様の方法で半導体微粒子の粒径を測定したところ、20~40nmであった。
導電性材料として、グラフェンを用いた。グラフェンは、フレーク状のクラファイト(平均粒径:4μm、純度99.95%、Qingdao Tianhe Graphite社製(中華人民共和国)のQingdao(商品名)から、以下の方法で調製した。
5gの上記グラファイトと3.75gのNaNO3をフラスコに加え、375mLのH2SO4を加え氷冷下で攪拌した。その後、22.5gのKMnO4を約1時間かけて添加した。氷冷下で2時間攪拌後、混合物を室温で5日間攪拌した。その後、5wt%の硫酸水溶液を700mL加え1時間攪拌し、温度を98℃に維持した。得られた混合物を98℃で更に2時間攪拌した。温度を60℃に下げた後、15mLの過酸化水素水を添加し、室温で2時間攪拌した。不純物イオンを除去するために、得られた混合物は下記の操作を15回行うことにより精製した。
(精製方法)
遠心分離を行い、上澄みを除去する。3wt%H2SO4/0.5wt%H2O2の混合水溶液を2L加え、強く攪拌しながら30分間超音波処理を行う。その後、3wt%のHCl水溶液2Lで3回洗浄し、蒸留水で1回洗浄する。得られた水溶液をイオン交換樹脂(D301T、Nankai University Chemical Plant)に通すことにより精製する。
上記の方法で精製を行い、蒸留水を除去することによりグラフェンを得た。精製物がグラフェンであることは、X線光電子分光法、走査型電子顕微鏡により確認した。半導体微粒子にグラフェンを添加したものについては、半導体微粒子に対して1質量%配合した。
Nbをドープした酸化チタンの半導体微粒子を次の方法により作製した。
ニオブ粉末(0.002mol)とTetrabutyl Titanete(0.018mol)をH2O2/NH3混合溶液(v/v=5/1)に添加して攪拌し前駆体を作製した。前駆体を80℃に加熱して過剰なH2O2とNH3を除去し、その後、オートクレーブを用いて180℃で20時間加熱した。得られた分散物をpH=7以下の脱イオン水で洗浄して100℃で6時間乾燥させることによりNbがドープされた酸化チタンの半導体微粒子を得た。この半導体微粒子にNbがドープされていることは、XRDまたはSTEMにより、確認した。上記と同様の方法で半導体微粒子の粒径を測定したところ、10~30nmであった。
前記半導体微粒子(II)をそれぞれ、エチルセルロースを5質量%含むα-テルピネオール溶液に分散させて、半導体微粒子(II)15質量%の分散液を得た。この分散液を、自転/公転併用式のミキシングコンディショナーを使用して均一に分散、混合した。
下記表に示す比較例50~105においては、下記表に示す2種の微粒子1及び微粒子2を質量比で1:1で混合したものを使用した。なお、下記表に示す微粒子2のうち、「TiO2/MgO」とは、TiO2微粒子を、2~150mMの酢酸マグネシウムが溶解したエタノール溶液(60~70℃)中に1分間浸漬し、洗浄後、500℃で焼成して調製した微粒子である。
ガラス基板上に、透明導電膜としてフッ素をドープした酸化スズをスパッタリングにより形成した。次に、上記の半導体微粒子分散液を透明導電膜に塗布し、500℃で加熱して半導体微粒子層を形成した。このようにして得られた半導体微粒子層の厚さは10μmであり、半導体微粒子の塗布量は20g/m2であった。
上記のガラス基板上に半導体微粒子層が形成されたものを、下記表に示す色素の10%エタノール溶液に暗所で40℃で3時間浸漬した。色素が吸着されて得られた受光電極を、10%TBAOHメタノール溶液を用いて色素を脱着し、吸収スペクトル測定により各色素の初期吸着量を定量した。吸着量が2.0×10-4mM/cm2未満のものをB、2.0×10-4mM/cm2以上のものをAとした。
(光電変換素子の作製)
上記の吸着性評価と同様の方法で作製した受光電極を作製した。その後、同じ半導体微粒子分散液をこの受光電極に塗布し、500℃で加熱して絶縁性多孔体を形成した。次に、表2~9に示す色素の10%エタノール溶液に、上記の絶縁性多孔体が形成されたガラス基板を12時間浸漬した。色素の染着したガラスを4-tert-ブチルピリジンの10%エタノール溶液に30分間浸漬した後、エタノールで洗浄し自然乾燥させた。このようにして得られる感光層の厚さは10μmであり、半導体微粒子の塗布量は20g/m2であった。
その後、半導体微粒子電極を50μm厚の熱可塑性ポリオレフィン樹脂シートを介して白金スパッタFTO基板と対向して配置し、樹脂シート部を熱溶融させて両極板を固定した。
なおあらかじめ白金スパッタ極側に開けておいた電解液の注液口から、電解液を注液し、電極間に満たした。さらに周辺部及び電解液注液口をエポキシ系封止樹脂を用いて本封止し、集電端子部に銀ペーストを塗布して光電変換素子とした。
電解液は、ヨウ化ジメチルプロピルイミダゾリウム(0.5モル/L)、ヨウ素(0.1モル/L)のメトキシプロピオニトリル溶液を用いた。
500Wのキセノンランプ(ウシオ製)の光をAM1.5Gフィルター(Oriel社製)およびシャープカットフィルター(KenkoL-42、商品名)を通すことにより紫外線を含まない模擬太陽光を発生させた。この光の強度は89mW/cm2であった。作製した光電変換素子にこの光を照射し、発生した電気を電流電圧測定装置(ケースレー238型、商品名)にて測定した。これにより求められた光電気化学電池の変換効率の初期値を測定した結果を表2~9に示した。結果は、変換効率が4%以上5%未満のものをE、5%以上6%未満のものをD、6%以上7%未満のものをC、7%以上8%未満のものをB、8%以上9%未満のものをAとして評価し、A、B、Cを合格とした。また、耐久性としては、変換効率の初期値に対し500時間後の変換効率が90%以上のものをA、80%以上90%未満のものをB、80%未満のものをCとして評価し、AとBを合格とした。








これに対して、半導体微粒子が2種以上の金属又は金属化合物を局部的に有してなるものを用い、一般式(1)で示される色素を用いた場合は、光電変換効率の初期値と耐久性に優れていることがわかった。
2 感光体層
21 増感色素
22 半導体微粒子
3 電荷移動体層
4 対極
5 受光電極
6 外部回路
10 光電変換素子
100 光電気化学電池
Claims (18)
- 導電性支持体と、色素を含む半導体微粒子層で構成された感光体層と、電荷移動体層と、対極とからなる光電変換素子であって、前記半導体微粒子が2種以上の金属又は金属化合物を局部的に有してなり、前記色素が下記一般式(1)で表される化合物である、光電変換素子。
Mz(LL1)m1(LL2)m2(X)m3・(CI)m4 一般式(1)
[Mzは金属原子を表し、LL1は下記一般式(2)で表される2座の配位子であり、LL2は下記一般式(3)で表される2座又は3座の配位子である。
Xは、アシルオキシ基、アシルチオ基、チオアシルオキシ基、チオアシルチオ基、アシルアミノオキシ基、チオカルバメート基、ジチオカルバメート基、チオカルボネート基、ジチオカルボネート基、トリチオカルボネート基、アシル基、チオシアネート基、イソチオシアネート基、シアネート基、イソシアネート基、シアノ基、アルキルチオ基、アリールチオ基、アルコキシ基及びアリールオキシ基からなる群から選ばれる1座若しくは2座の配位子、又はハロゲン原子、カルボニル、ジアルキルケトン、1,3-ジケトン、カルボンアミド、チオカルボンアミド及びチオ尿素からなる群から選ばれる1座若しくは2座の配位子を表す。
CIは、一般式(1)で表される化合物の電荷を中和させる対イオンを表す。
m1は1~3の整数を表し、m1が2以上のときLL1は同じでも異なっていてもよい。m2は0~2の整数を表し、m2が2のときLL2は同じでも異なっていてもよい。m3は0~3の整数を表し、m3が2以上のときXは同じでも異なっていてもよい。m4は0~3の整数を表し、m4が2以上のときCIは同じでも異なっていてもよい。
一般式(2)において、R101及びR102はそれぞれ独立に、ヘテロ環基、カルボキシル基、スルホン酸基、ヒドロキシル基、ヒドロキサム酸基、ホスホリル基又はホスホニル基を表す。R103及びR104はそれぞれ独立に置換基を表し、R105及びR106はそれぞれ独立にアルキル基、アリール基及びヘテロ環基からなる群より選ばれる少なくとも1種からなる基を表す。
L1及びL2はそれぞれ独立に、エテニレン基及び/又はエチニレン基からなる共役鎖を表す。
a1及びa2はそれぞれ独立に0~3の整数を表し、a1が2以上のときR101は同じでも異なっていてもよく、a2が2以上のときR102は同じでも異なっていてもよい。b1及びb2はそれぞれ独立に0~3の整数を表す。b1が2以上のときR103は同じでも異なっていてもよく、互いに連結して環を形成してもよい。b2が2以上のときR104は同じでも異なっていてもよく、互いに連結して環を形成してもよい。b1及びb2が共に1以上のときR103とR104とが連結して環を形成してもよい。d1及びd2はそれぞれ独立に0~5の整数を表す。d3は0又は1を表す。
一般式(3)において、Za、Zb及びZcはそれぞれ独立に、5又は6員環を形成しうる非金属原子群を表し、cは0又は1を表す。]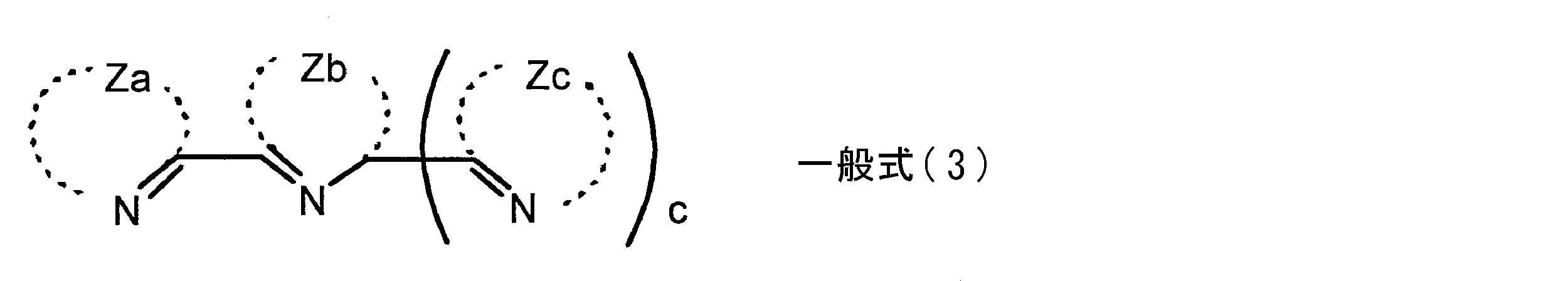
- 前記半導体微粒子における2種以上の金属又は金属化合物が、金属原子、金属のカルコゲニド、金属炭酸塩又は金属硝酸塩である、請求項1記載の光電変換素子。
- 前記金属原子がTi、Sn、Au、Ag、Cu、Al、Zr、Nb、V及びTaからなる群から選ばれた少なくとも1種である、請求項2記載の光電変換素子。
- 前記金属カルコゲニドが硫化カドミウム、セレン化カドミウム又はTi、Sn、Zn、Mg、Al、W、Zr、Hf、Sr、In、Ce、Y、La、V及びTaからなる群から選ばれた少なくとも1種の金属酸化物である、請求項2又は3記載の光電変換素子。
- 前記金属炭酸塩が炭酸カルシウム、炭酸カリウム及び炭酸バリウムからなる群から選ばれた少なくとも1種である、請求項2~4のいずれか1項記載の光電変換素子。
- 前記金属硝酸塩が硝酸ランタンである、請求項2~5のいずれか1項記載の光電変換素子。
- 前記半導体微粒子が、コア-シェル構造により、前記金属原子、前記金属のカルコゲニド、前記金属炭酸塩及び/又は前記金属硝酸塩を有してなる、請求項2~6のいずれか1項記載の光電変換素子。
- 前記半導体微粒子が、前記金属のカルコゲニドをコア部分として有し、前記金属のカルコゲニド又は前記金属炭酸塩をシェル部分として有する、請求項7記載の光電変換素子。
- 前記半導体微粒子が、酸化チタン及び酸化スズからなる群より選ばれる金属のカルコゲニドをコア部分として有し、酸化アルミニウム、酸化マグネシウム、炭酸カルシウム、酸化チタン及び酸化チタン/酸化マグネシウムからなる群より選ばれる金属のカルコゲニド又は金属炭酸塩をシェル部分として有する、請求項8記載の光電変換素子。
- 前記半導体微粒子が、金属原子をドープすることにより、2種以上の金属原子を有してなる、請求項1~6のいずれか1項記載の光電変換素子。
- 前記半導体微粒子が、前記金属のカルコゲニドに前記金属原子をドープして得られた半導体微粒子である、請求項10記載の光電変換素子。
- 前記半導体微粒子が、酸化チタン及び酸化スズからなる群より選ばれる金属のカルコゲニドに、Nb、V及びTaからなる群より選ばれる少なくとも1種の金属原子をドープして得られた半導体微粒子である、請求項11記載の光電変換素子。
- 前記半導体微粒子の粒径が、1~1000nmである、請求項1~12のいずれか1項記載の光電変換素子。
- 前記半導体微粒子が、導電性材料からなる添加剤を含む、請求項1~13のいずれか1項記載の光電変換素子。
- 前記導電性材料がグラフェンである、請求項14記載の光電変換素子。
- 一般式(1)において、MzがRuであり、m1が1であり、m2が1であり、Xがイソチオシアネート基であり、m3が2である、請求項1~15のいずれか1項記載の光電変換素子。
- 一般式(1)において、LL1が一般式(4-1)~(4-3)のいずれかで表される、請求項1~16のいずれか1項記載の光電変換素子。
[R101~R104、a1、a2、b1、b2及びd3は一般式(2)におけるものと同義である。R107は酸性基を表す。a3は0~3の整数を表す。R108は置換基を表す。b3は0~3の整数を表す。R121~R124はそれぞれ独立に、水素原子、アルキル基、アルケニル基又はアリール基を表す。R125、R126、R127及びR128はそれぞれ独立に置換基を表す。d4及びd5はそれぞれ独立に0~4の整数を表す。]
- 請求項1~17のいずれか1項に記載の光電変換素子を備える、光電気化学電池。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020137027663A KR101505767B1 (ko) | 2011-03-30 | 2012-03-12 | 광전 변환 소자 및 광전기화학 전지 |
| GB1317100.4A GB2505093B (en) | 2011-03-30 | 2012-03-12 | Photoelectric converter and photoelectrochemical cell |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011076724 | 2011-03-30 | ||
| JP2011-076724 | 2011-03-30 | ||
| JP2012-052699 | 2012-03-09 | ||
| JP2012052699A JP5756772B2 (ja) | 2011-03-30 | 2012-03-09 | 光電変換素子及び光電気化学電池 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012132855A1 true WO2012132855A1 (ja) | 2012-10-04 |
Family
ID=46930592
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/056259 WO2012132855A1 (ja) | 2011-03-30 | 2012-03-12 | 光電変換素子及び光電気化学電池 |
Country Status (4)
| Country | Link |
|---|---|
| JP (1) | JP5756772B2 (ja) |
| KR (1) | KR101505767B1 (ja) |
| GB (1) | GB2505093B (ja) |
| WO (1) | WO2012132855A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103012491A (zh) * | 2012-12-27 | 2013-04-03 | 中国科学院上海硅酸盐研究所 | 利用一锅法合成钌络合物的方法 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014177695A (ja) * | 2013-02-15 | 2014-09-25 | Sekisui Chem Co Ltd | 複合膜の製造方法、複合膜、光電極および色素増感太陽電池 |
| JP6033143B2 (ja) * | 2013-03-25 | 2016-11-30 | 富士フイルム株式会社 | 光電変換素子、色素増感太陽電池、金属錯体、金属錯体色素、色素溶液、色素吸着電極の製造方法および色素増感太陽電池の製造方法 |
| JP6033144B2 (ja) * | 2013-03-25 | 2016-11-30 | 富士フイルム株式会社 | 光電変換素子、色素増感太陽電池、金属錯体、金属錯体色素、色素溶液、色素吸着電極の製造方法および色素増感太陽電池の製造方法 |
Citations (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2681294A (en) | 1951-08-23 | 1954-06-15 | Eastman Kodak Co | Method of coating strip material |
| JPS584589B2 (ja) | 1976-08-12 | 1983-01-27 | 富士写真フイルム株式会社 | 塗布方法 |
| JPH06507999A (ja) | 1992-03-26 | 1994-09-08 | アスラブ・エス アー | 透明再生光電化学電池 |
| WO1995018456A1 (fr) | 1993-12-29 | 1995-07-06 | Ecole Polytechnique Federale De Lausanne | Pile photo-electrochimique et electrolyte pour cette pile |
| US5463057A (en) | 1992-08-21 | 1995-10-31 | Ecole Polytechnique Federale De Lausanne, (Epfl) | Bi-pyridyl-rumetal complexes |
| JPH08259543A (ja) | 1994-12-21 | 1996-10-08 | Asulab Sa | 疎水性液状塩とその生成法ならびに電気化学への応用 |
| JP2664194B2 (ja) | 1988-02-12 | 1997-10-15 | エコル ポリテクニク フェデラル ドゥ ローザンヌ | 光電気化学電池・その製法及び使用法 |
| JPH10505192A (ja) | 1994-09-02 | 1998-05-19 | キータ ホールデイング ソシエテ アノニム | 電気化学式太陽電池セル |
| JPH11250944A (ja) | 1998-02-26 | 1999-09-17 | Nikon Corp | 湿式太陽電池 |
| JP2000036332A (ja) | 1998-07-17 | 2000-02-02 | Fuji Photo Film Co Ltd | 電解液、光電変換素子および光再生型光電気化学電池 |
| JP2000090989A (ja) | 1998-09-16 | 2000-03-31 | Toshiba Corp | 色素増感型光化学電池 |
| JP2000113913A (ja) | 1998-10-02 | 2000-04-21 | Sumitomo Osaka Cement Co Ltd | 色素増感型太陽電池 |
| JP2000231943A (ja) | 1998-12-08 | 2000-08-22 | Toyota Central Res & Dev Lab Inc | 半導体電極およびその製造方法 |
| JP2000243134A (ja) | 1999-02-22 | 2000-09-08 | Fuji Photo Film Co Ltd | 電解質、光電変換素子および光電気化学電池 |
| WO2000054361A1 (fr) | 1999-03-10 | 2000-09-14 | Daiso Co., Ltd. | Dispositifs de conversion photoelectrique fabriques au moyen d'electrolytes etheres |
| JP2001093591A (ja) | 1999-09-28 | 2001-04-06 | Toshiba Corp | 光電変換素子 |
| JP2001185243A (ja) | 1999-12-27 | 2001-07-06 | Tdk Corp | 酸化物半導体色素結合電極および色素増感型太陽電池 |
| JP2001283941A (ja) | 2000-03-29 | 2001-10-12 | Hitachi Maxell Ltd | 光電変換素子 |
| JP2001291534A (ja) | 2000-01-31 | 2001-10-19 | Fuji Photo Film Co Ltd | 光電変換素子および光電池ならびに金属錯体色素 |
| JP2001357896A (ja) | 2000-06-13 | 2001-12-26 | Fuji Photo Film Co Ltd | 光電変換素子および光電池 |
| JP2002090989A (ja) | 1999-08-23 | 2002-03-27 | Mitsubishi Chemicals Corp | 光重合性組成物及び光重合性平版印刷版 |
| JP2002093476A (ja) | 2000-09-20 | 2002-03-29 | Dainippon Printing Co Ltd | 色素増感型太陽電池セルおよびそれを用いた色素増感型太陽電池モジュール、およびそれらの製造方法 |
| JP2002110262A (ja) | 2000-10-03 | 2002-04-12 | Nippon Kayaku Co Ltd | 含水電解液を用いた光電変換素子 |
| JP2002134435A (ja) | 2000-10-20 | 2002-05-10 | Fuji Photo Film Co Ltd | 半導体電極の製造方法、半導体電極、およびその用途 |
| JP2002184475A (ja) | 2000-12-12 | 2002-06-28 | Lintec Corp | 半導体電極の製造方法及び光化学電池 |
| JP2002260746A (ja) | 2001-02-28 | 2002-09-13 | Toyota Central Res & Dev Lab Inc | 色素増感型太陽電池及び色素増感型太陽電池モジュール |
| JP2002353453A (ja) | 2002-03-29 | 2002-12-06 | Mitsubishi Electric Corp | 絶縁ゲート型半導体装置 |
| JP2003500857A (ja) | 1999-05-25 | 2003-01-07 | フォッシュカルパテント・イー・ウプサラ・アクチボラゲット | ナノ構造型薄膜電極の製造方法 |
| JP2003098977A (ja) | 2001-09-19 | 2003-04-04 | Sony Corp | 素子の転写方法、素子の配列方法、及び画像表示装置の製造方法 |
| JP2003123859A (ja) | 2001-10-19 | 2003-04-25 | Bridgestone Corp | 有機色素増感型金属酸化物半導体電極及びこの半導体電極を有する太陽電池 |
| JP2003282164A (ja) | 2002-03-26 | 2003-10-03 | Canon Inc | 光電変換装置及びその製造方法 |
| JP2004010403A (ja) | 2002-06-05 | 2004-01-15 | Fuji Photo Film Co Ltd | 多重構造酸化チタン微粒子、及びその作製方法、及びそれを含有する光電変換素子並びに光電池 |
| WO2007091525A1 (ja) | 2006-02-08 | 2007-08-16 | Shimane Prefectural Government | 光増感色素 |
| JP2008174734A (ja) * | 2006-12-18 | 2008-07-31 | Sumitomo Chemical Co Ltd | 化合物、光電変換素子及び光電気化学電池 |
| JP2009200028A (ja) | 2008-02-19 | 2009-09-03 | National Central Univ | 光増感色素 |
| JP2009215539A (ja) * | 2008-02-19 | 2009-09-24 | National Central Univ | 感光剤染料 |
| JP2010108855A (ja) * | 2008-10-31 | 2010-05-13 | Jgc Catalysts & Chemicals Ltd | 光電気セルおよび該光電気セル用多孔質金属酸化物半導体膜形成用塗料 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009174734A (ja) | 2008-01-22 | 2009-08-06 | Sanyo Electric Co Ltd | 空調制御装置、空調制御方法、空調機器、および空調システム |
| JP2011014356A (ja) * | 2009-07-01 | 2011-01-20 | Sony Corp | 光電変換素子およびその製造方法ならびに電子機器 |
-
2012
- 2012-03-09 JP JP2012052699A patent/JP5756772B2/ja active Active
- 2012-03-12 GB GB1317100.4A patent/GB2505093B/en not_active Expired - Fee Related
- 2012-03-12 KR KR1020137027663A patent/KR101505767B1/ko active IP Right Grant
- 2012-03-12 WO PCT/JP2012/056259 patent/WO2012132855A1/ja active Application Filing
Patent Citations (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2681294A (en) | 1951-08-23 | 1954-06-15 | Eastman Kodak Co | Method of coating strip material |
| JPS584589B2 (ja) | 1976-08-12 | 1983-01-27 | 富士写真フイルム株式会社 | 塗布方法 |
| JP2664194B2 (ja) | 1988-02-12 | 1997-10-15 | エコル ポリテクニク フェデラル ドゥ ローザンヌ | 光電気化学電池・その製法及び使用法 |
| JPH06507999A (ja) | 1992-03-26 | 1994-09-08 | アスラブ・エス アー | 透明再生光電化学電池 |
| US5463057A (en) | 1992-08-21 | 1995-10-31 | Ecole Polytechnique Federale De Lausanne, (Epfl) | Bi-pyridyl-rumetal complexes |
| WO1995018456A1 (fr) | 1993-12-29 | 1995-07-06 | Ecole Polytechnique Federale De Lausanne | Pile photo-electrochimique et electrolyte pour cette pile |
| JPH10505192A (ja) | 1994-09-02 | 1998-05-19 | キータ ホールデイング ソシエテ アノニム | 電気化学式太陽電池セル |
| JPH08259543A (ja) | 1994-12-21 | 1996-10-08 | Asulab Sa | 疎水性液状塩とその生成法ならびに電気化学への応用 |
| JPH11250944A (ja) | 1998-02-26 | 1999-09-17 | Nikon Corp | 湿式太陽電池 |
| JP2000036332A (ja) | 1998-07-17 | 2000-02-02 | Fuji Photo Film Co Ltd | 電解液、光電変換素子および光再生型光電気化学電池 |
| JP2000090989A (ja) | 1998-09-16 | 2000-03-31 | Toshiba Corp | 色素増感型光化学電池 |
| JP2000113913A (ja) | 1998-10-02 | 2000-04-21 | Sumitomo Osaka Cement Co Ltd | 色素増感型太陽電池 |
| JP2000231943A (ja) | 1998-12-08 | 2000-08-22 | Toyota Central Res & Dev Lab Inc | 半導体電極およびその製造方法 |
| JP2000243134A (ja) | 1999-02-22 | 2000-09-08 | Fuji Photo Film Co Ltd | 電解質、光電変換素子および光電気化学電池 |
| WO2000054361A1 (fr) | 1999-03-10 | 2000-09-14 | Daiso Co., Ltd. | Dispositifs de conversion photoelectrique fabriques au moyen d'electrolytes etheres |
| JP2003500857A (ja) | 1999-05-25 | 2003-01-07 | フォッシュカルパテント・イー・ウプサラ・アクチボラゲット | ナノ構造型薄膜電極の製造方法 |
| JP2002090989A (ja) | 1999-08-23 | 2002-03-27 | Mitsubishi Chemicals Corp | 光重合性組成物及び光重合性平版印刷版 |
| JP2001093591A (ja) | 1999-09-28 | 2001-04-06 | Toshiba Corp | 光電変換素子 |
| JP2001185243A (ja) | 1999-12-27 | 2001-07-06 | Tdk Corp | 酸化物半導体色素結合電極および色素増感型太陽電池 |
| JP2001291534A (ja) | 2000-01-31 | 2001-10-19 | Fuji Photo Film Co Ltd | 光電変換素子および光電池ならびに金属錯体色素 |
| JP2001283941A (ja) | 2000-03-29 | 2001-10-12 | Hitachi Maxell Ltd | 光電変換素子 |
| JP2001357896A (ja) | 2000-06-13 | 2001-12-26 | Fuji Photo Film Co Ltd | 光電変換素子および光電池 |
| JP2002093476A (ja) | 2000-09-20 | 2002-03-29 | Dainippon Printing Co Ltd | 色素増感型太陽電池セルおよびそれを用いた色素増感型太陽電池モジュール、およびそれらの製造方法 |
| JP2002110262A (ja) | 2000-10-03 | 2002-04-12 | Nippon Kayaku Co Ltd | 含水電解液を用いた光電変換素子 |
| JP2002134435A (ja) | 2000-10-20 | 2002-05-10 | Fuji Photo Film Co Ltd | 半導体電極の製造方法、半導体電極、およびその用途 |
| JP2002184475A (ja) | 2000-12-12 | 2002-06-28 | Lintec Corp | 半導体電極の製造方法及び光化学電池 |
| JP2002260746A (ja) | 2001-02-28 | 2002-09-13 | Toyota Central Res & Dev Lab Inc | 色素増感型太陽電池及び色素増感型太陽電池モジュール |
| JP2003098977A (ja) | 2001-09-19 | 2003-04-04 | Sony Corp | 素子の転写方法、素子の配列方法、及び画像表示装置の製造方法 |
| JP2003123859A (ja) | 2001-10-19 | 2003-04-25 | Bridgestone Corp | 有機色素増感型金属酸化物半導体電極及びこの半導体電極を有する太陽電池 |
| JP2003282164A (ja) | 2002-03-26 | 2003-10-03 | Canon Inc | 光電変換装置及びその製造方法 |
| JP2002353453A (ja) | 2002-03-29 | 2002-12-06 | Mitsubishi Electric Corp | 絶縁ゲート型半導体装置 |
| JP2004010403A (ja) | 2002-06-05 | 2004-01-15 | Fuji Photo Film Co Ltd | 多重構造酸化チタン微粒子、及びその作製方法、及びそれを含有する光電変換素子並びに光電池 |
| WO2007091525A1 (ja) | 2006-02-08 | 2007-08-16 | Shimane Prefectural Government | 光増感色素 |
| JP2008174734A (ja) * | 2006-12-18 | 2008-07-31 | Sumitomo Chemical Co Ltd | 化合物、光電変換素子及び光電気化学電池 |
| JP2009200028A (ja) | 2008-02-19 | 2009-09-03 | National Central Univ | 光増感色素 |
| JP2009215539A (ja) * | 2008-02-19 | 2009-09-24 | National Central Univ | 感光剤染料 |
| JP2010108855A (ja) * | 2008-10-31 | 2010-05-13 | Jgc Catalysts & Chemicals Ltd | 光電気セルおよび該光電気セル用多孔質金属酸化物半導体膜形成用塗料 |
Non-Patent Citations (20)
| Title |
|---|
| "Kobunshi Gosei no Jikken Hou", KAGAKU-DOJIN PUBLISHING COMPANY, INC. |
| ANGEW. CHEM. INT. ED. ENGL., vol. 35, 1996, pages 1949 |
| BARBE ET AL., JOURNAL OF AMERICAN CERAMIC SOCIETY, vol. 80, no. 12, 1997, pages 3157 - 3171 |
| BURNSIDE ET AL., CHEMISTRY OF MATERIALS, vol. 10, no. 9, pages 2419 - 2425 |
| CAN. J. CHEM., vol. 75, 1997, pages 318 |
| CHEM. LETT., 1996, pages 885 |
| ELECTROCHEMISTRY, vol. 65, no. 11, 1997, pages 923 |
| INORG. CHEM., vol. 27, 1988, pages 4007 |
| J. AM. CERAM. SOC., vol. 80, no. 12, 1997, pages 3157 - 3171 |
| J. AM. CHEM. SOC., vol. 121, 1997, pages 4047 |
| J. AM. CHEM. SOC., vol. 123, 2001, pages 1613 - 1624 |
| J. AM. CHEM. SOC., vol. ILL, 1989, pages 5542 |
| J. CHEM. SOC. JAPAN, IND. CHEM. SOC., 1943, pages 46779 |
| J. CHEM. SOC., CHEM. COMMUN., 1993, pages 390 |
| J. CHEM. SOC., CHEM. COMMUN., 1997, pages 545 |
| J. R. MACCALLUM AND C. A. VINCENT: "Polymer Electrolyte Reviews 1 and 2", ELSEVIER APPLIED SCIENCE |
| PHYS. REV. B, vol. 35, 1987, pages 2818 |
| SAKKA, SUMIO: "Science of Sol-Gel Processes", 1998, AGNE SHOFU PUBLISHING, INC. |
| SEINO MANABU: "Titanium Oxide: Material Properties and Application Technologies", 1997, GIHODO SHUPPAN CO., LTD. |
| TAKAYUKI OTSU: "Koza Jugo Hannou Ron 1 (Polymerization reaction theory course 1), Radical polymerization (I", KAGAKU-DOJIN PUBLISHING COMPANY, INC. |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103012491A (zh) * | 2012-12-27 | 2013-04-03 | 中国科学院上海硅酸盐研究所 | 利用一锅法合成钌络合物的方法 |
| CN103012491B (zh) * | 2012-12-27 | 2015-06-17 | 中国科学院上海硅酸盐研究所 | 利用一锅法合成钌络合物的方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| GB2505093A (en) | 2014-02-19 |
| KR20140027168A (ko) | 2014-03-06 |
| KR101505767B1 (ko) | 2015-03-24 |
| JP2012216506A (ja) | 2012-11-08 |
| JP5756772B2 (ja) | 2015-07-29 |
| GB201317100D0 (en) | 2013-11-06 |
| GB2505093B (en) | 2019-10-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5620316B2 (ja) | 光電変換素子、光電気化学電池及び色素 | |
| WO2010050575A1 (ja) | 色素、これを用いた光電変換素子、光電気化学電池、および色素の製造方法 | |
| JP2011026376A (ja) | 色素、これを用いた光電変換素子及び光電気化学電池 | |
| JP5721717B2 (ja) | 金属錯体色素、光電変換素子及び光電気化学電池 | |
| JP5689351B2 (ja) | 光電変換素子及び光電気化学電池 | |
| JP5771092B2 (ja) | 色素、光電変換素子及び光電気化学電池 | |
| JP5649368B2 (ja) | 光電変換素子及び光電気化学電池 | |
| JP5756772B2 (ja) | 光電変換素子及び光電気化学電池 | |
| WO2012017868A1 (ja) | 金属錯体色素、光電変換素子及び光電気化学電池 | |
| JP5816111B2 (ja) | 金属錯体色素組成物、光電変換素子及び光電気化学電池 | |
| WO2012017874A1 (ja) | 金属錯体色素、光電変換素子及び光電気化学電池 | |
| JP5662728B2 (ja) | 色素、これを用いた光電変換素子及び光電気化学電池 | |
| WO2011108611A1 (ja) | 光電変換素子及び光電気化学電池 | |
| JP5607338B2 (ja) | 色素、これを用いた光電変換素子、光電気化学電池、および色素の製造方法 | |
| JP5636317B2 (ja) | 金属錯体色素、金属錯体色素組成物、光電変換素子及び光電気化学電池 | |
| JP2012033443A (ja) | 光電変換素子及び光電気化学電池、それに用いられる色素 | |
| JP2012038435A (ja) | 光電変換素子、光電気化学電池及び光電変換素子用色素溶液 | |
| JP2012038436A (ja) | 光電変換素子及びこれを用いた光電気化学電池、光電変換素子用組成物 | |
| JP5572028B2 (ja) | 光電変換素子及びこれを用いた光電気化学電池、光電変換素子用組成物 | |
| WO2012017873A1 (ja) | 金属錯体色素、光電変換素子及び光電気化学電池 | |
| WO2011108612A1 (ja) | 光電変換素子及び光電気化学電池 | |
| JP5572027B2 (ja) | 光電変換素子及びこれに用いられる光電変換素子用組成物 | |
| JP5756766B2 (ja) | 光電変換素子、光電気化学電池及び色素 | |
| WO2011108613A1 (ja) | 光電変換素子及び光電気化学電池 | |
| JP2012038437A (ja) | 光電変換素子及びこれを用いた光電気化学電池、光電変換素子用組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12763293 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 1317100 Country of ref document: GB Kind code of ref document: A Free format text: PCT FILING DATE = 20120312 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1317100.4 Country of ref document: GB |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20137027663 Country of ref document: KR Kind code of ref document: A |