WO2011118647A1 - オキシムの製造方法 - Google Patents
オキシムの製造方法 Download PDFInfo
- Publication number
- WO2011118647A1 WO2011118647A1 PCT/JP2011/057018 JP2011057018W WO2011118647A1 WO 2011118647 A1 WO2011118647 A1 WO 2011118647A1 JP 2011057018 W JP2011057018 W JP 2011057018W WO 2011118647 A1 WO2011118647 A1 WO 2011118647A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- reaction
- group
- hydroxylamine
- tank
- cyclododecanone
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C249/00—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C249/04—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton of oximes
- C07C249/08—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton of oximes by reaction of hydroxylamines with carbonyl compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/18—Systems containing only non-condensed rings with a ring being at least seven-membered
- C07C2601/20—Systems containing only non-condensed rings with a ring being at least seven-membered the ring being twelve-membered
Definitions
- the present invention relates to a method for producing a corresponding oxime from a ketone and hydroxylamine.
- An oxime can be derived into an amide compound by Beckmann rearrangement reaction, and in particular, an oxime derived from a cyclic ketone can be derived into a lactam.
- cyclohexanone oxime provides ⁇ -caprolactam, which is a raw material for nylon 6
- cyclododecanone oxime provides laurolactam, which is a raw material for nylon 12.
- the following methods are known as oxime production methods.
- a protecting group for example, an acyl group such as an acetyl group
- the N-hydroxyimide compound is derived from an aliphatic polycarboxylic anhydride (cyclic anhydride) or an aromatic polycarboxylic anhydride (cyclic anhydride) such as N-hydroxysuccinimide. .
- the present invention solves the above problem of reaction time in a method for industrially producing a corresponding oxime from a ketone and hydroxylamine in a two-phase system of a hydrophobic solvent and water, thereby reducing the size of the apparatus. For the purpose.
- the present invention relates to the following matters.
- a method for producing an oxime comprising reacting a ketone and hydroxylamine in a system comprising an aqueous phase and a hydrophobic solvent phase in the presence of a carboxylic acid and / or a salt thereof.
- hydrophobic solvent is an aromatic hydrocarbon or an aliphatic hydrocarbon.
- a method for accelerating the reaction speed and miniaturizing the reaction apparatus in a method for producing an oxime from a ketone and hydroxylamine in a two-phase system of a hydrophobic solvent and water, a method for accelerating the reaction speed and miniaturizing the reaction apparatus is provided. can do.
- the present invention relates to a method for producing an oxime from a ketone and hydroxylamine in the presence of a carboxylic acid and / or a salt thereof in a two-phase system of a hydrophobic solvent and water.
- a carboxylic acid and / or a salt thereof in a two-phase system of a hydrophobic solvent and water.
- the present invention is characterized in that a carboxylic acid and / or a salt thereof is used in the method for producing an oxime.
- a carboxylic acid and / or a salt thereof is used in the method for producing an oxime.
- the reaction rate is accelerated.
- a ketone that is more easily distributed to the hydrophobic solvent phase than the aqueous phase hereinafter sometimes referred to as “highly hydrophobic ketone”
- the reaction field for oximation is in the oil phase, so the carboxylic acid or salt thereof used together may be a carboxylic acid or salt thereof that is easily distributed to the hydrophobic solvent phase. preferable.
- carboxylic acid or a salt thereof By using carboxylic acid or a salt thereof, hydroxylamine present in the aqueous phase is easily transferred to the oil phase as a reaction field, and the reaction rate of oximation is promoted.
- the carboxylic acid or a salt thereof used in the present invention is not particularly limited, but is preferably a saturated or unsaturated linear aliphatic carboxylic acid having 5 or more carbon atoms, an aromatic carboxylic acid, or a salt thereof.
- Carboxylic acids having 4 or less carbon atoms such as acetic acid and propionic acid, are highly soluble in water and are easily distributed to the aqueous phase in the oximation reaction, so that a remarkable acceleration effect of the oximation reaction is hardly exhibited.
- the upper limit of the carbon number of the carboxylic acid is not particularly limited, but if it exceeds 28, it will be difficult to dissolve in the reaction solvent.
- carboxylic acids include saturated aliphatic monocarboxylic acids such as caproic acid, capric acid, undecanoic acid, lauric acid, palmitic acid, stearic acid, and arachidic acid; saturated aliphatic dicarboxylic acids such as sebacic acid and dodecanedioic acid. Acid; unsaturated aliphatic carboxylic acids such as oleic acid, linoleic acid, and linolenic acid, and aromatic carboxylic acids such as benzoic acid, phthalic acid, and naphthoic acid.
- saturated aliphatic monocarboxylic acids such as caproic acid, capric acid, undecanoic acid, lauric acid, palmitic acid, stearic acid, and arachidic acid
- saturated aliphatic dicarboxylic acids such as sebacic acid and dodecanedioic acid. Acid
- unsaturated aliphatic carboxylic acids such as oleic acid, lino
- carboxylic acid salts include salts of alkali metals (sodium, potassium, etc.), alkaline earth metals (magnesium, calcium, etc.) and the above carboxylic acids. Specific examples include sodium laurate and calcium laurate.
- the above carboxylic acids or salts thereof may be used in combination of two or more.
- the oximation reaction is carried out in a two-phase system comprising a hydrophobic solvent phase and an aqueous phase.
- a ketone that is easily distributed to the aqueous phase is used, the reaction rate is relatively fast because oximation proceeds in the aqueous phase, and the reaction acceleration effect by addition of carboxylic acid or a salt thereof is relatively small.
- a highly hydrophobic ketone is used, since the reaction field is limited to the oil phase, the mass transfer rate of hydroxylamine to the oil phase becomes rate-limiting.
- the effect of the present invention (the acceleration effect of the oximation rate by the addition of carboxylic acid or a salt thereof) is more remarkable when a highly hydrophobic ketone is used. Appear in That is, the present invention is particularly important when a highly hydrophobic ketone is desired.
- examples of the ketone include a compound represented by the following formula (1).
- R 1 and R 2 each represents an organic group.
- R 1 and R 2 together represent a divalent organic group, and carbon atoms to which R 1 and R 2 are bonded
- And may form a ring together.
- ketone having a total number of carbon atoms of R 1 and R 2 of preferably 8 or more, more preferably 8 or more and 30 or less is used, it is easily distributed to the phase of the hydrophobic solvent in the oximation step. preferable.
- Examples of the organic group in R 1 and R 2 include an alkyl group, an alkenyl group, an alkynyl group, a cycloalkyl group, a cycloalkenyl group, an aryl group, an aralkyl group, an aromatic or non-aromatic heterocyclic group, and the like. Is mentioned.
- examples of the alkyl group include an alkyl group having 1 to 20 carbon atoms, preferably an alkyl group having 1 to 12 carbon atoms, and more preferably an alkyl group having 2 to 8 carbon atoms. It is. Specifically, methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, s-butyl group, t-butyl group, pentyl group, isopentyl group, hexyl group, isohexyl group, heptyl group, octyl group , Nonyl group, decyl group, dodecyl group, pentadecyl group and the like.
- alkenyl group examples include an alkenyl group having 2 to 20 carbon atoms, preferably an alkenyl group having 2 to 12 carbon atoms, and more preferably an alkenyl group having 2 to 8 carbon atoms.
- Specific examples include a vinyl group, an allyl group, a 1-propenyl group, a 1-butenyl group, a 1-pentenyl group, and a 1-octenyl group.
- alkynyl group examples include an alkynyl group having 2 to 20 carbon atoms, preferably an alkynyl group having 2 to 12 carbon atoms, and more preferably an alkynyl group having 2 to 8 carbon atoms. Specific examples include ethynyl group and 1-propynyl group.
- cycloalkyl group examples include a cycloalkyl group having 3 to 20 carbon atoms, and a cycloalkyl group having 3 to 15 carbon atoms is preferable. Specific examples include a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, a cycloheptyl group, a cyclooctyl group, and a cyclododecyl group.
- Examples of the cycloalkenyl group include a cycloalkenyl group having 3 to 20 carbon atoms, and a cycloalkenyl group having 3 to 15 carbon atoms is preferable. Specific examples include a cyclopentenyl group, a cyclohexenyl group, and a cyclooctenyl group.
- aryl group examples include a phenyl group and a naphthyl group.
- Examples of the aralkyl group include a benzyl group, a 2-phenylethyl group, and a 3-phenylpropyl group.
- aromatic or non-aromatic heterocyclic group examples include a 2-pyridyl group, a 2-quinolyl group, a 2-furyl group, a 2-thienyl group, and a 4-piperidinyl group.
- R 1 and R 2 together represent a divalent organic group, they form a ring with the carbon atom to which they are attached.
- the divalent organic group include a linear or branched alkylene group, preferably a linear alkylene group.
- the effect of the present invention is remarkable when it is an 8-membered ring or more that is easily distributed to the phase of the hydrophobic solvent in the oximation step, and the formed ring is, for example, an 8-30 membered ring, preferably In the case of an 8- to 20-membered ring, more preferably an 8- to 14-membered ring, the present invention is particularly effective.
- organic groups may have various substituents without particular limitation as long as they do not inhibit the reaction regardless of the presence or absence of ring formation.
- substituents a halogen atom, an oxo group, a mercapto group, a substituted oxy group (alkoxy group, aryloxy group, acyloxy group, etc.), a substituted thio group, a substituted oxycarbonyl group, a substituted or unsubstituted carbamoyl group, a cyano group Nitro group, substituted aminoalkyl group, alkenyl group, alkynyl group, cycloalkyl group, cycloalkenyl group, aryl group (phenyl group, naphthyl group, etc.), aralkyl group, heterocyclic group and the like.
- examples of the ketone in which the effects of the present invention are remarkably exhibited include 1-cyclohexyl-1-propanone, acetophenone, benzophenone, 4-hydroxyacetophenone, and the like that form a ring.
- examples of the ketone in which the effects of the present invention are remarkably exhibited include 1-cyclohexyl-1-propanone, acetophenone, benzophenone, 4-hydroxyacetophenone, and the like that form a ring.
- cyclododecanone is extremely important industrially.
- hydroxylamine Since hydroxylamine is unstable, it is produced and sold as an aqueous solution of an inorganic salt of hydroxylamine such as hydroxylamine sulfate or hydroxylamine carbonate. Usually, a base such as aqueous ammonia is added during reaction to liberate hydroxylamine. In the present invention, an aqueous hydroxylamine solution from which hydroxylamine has been liberated in advance may be used. Usually, however, an aqueous solution of an inorganic salt of hydroxylamine (preferably sulfate) and a base (preferably aqueous ammonia) in the reaction apparatus. ) Is used to liberate hydroxylamine in the reactor.
- a hydrophobic solvent is used. For this reason, the manufacturing method of the oxime of this invention becomes a two-phase system of the water derived from the above-mentioned hydroxylamine aqueous solution, and a hydrophobic solvent.
- the hydrophobic solvent is not particularly limited, but is preferably a solvent having high solubility of the raw material ketone and the produced oxime.
- aliphatic hydrocarbons such as hexane, heptane, octane, cyclododecane and isopropylcyclohexane; aromatic hydrocarbons such as benzene, toluene and xylene; chloroform, dichloromethane, dichloroethane, carbon tetrachloride, chlorobenzene, tri Halogenated hydrocarbons such as fluoromethylbenzene; nitro compounds such as nitrobenzene, nitromethane, and nitroethane; fluorine-based alcohols such as hexafluoroisopropyl alcohol and trifluoroethanol; or a mixed solvent thereof.
- the oxime obtained by the production method of the present invention can be used for the Beckmann rearrangement reaction for obtaining an amide compound, particularly a lactam, but if the solvent used for the production of the oxime is directly used for the Beckmann rearrangement reaction, the solvent This is preferable because it is not necessary to perform exchange.
- the solvent used for the production of oxime is the same as the solvent used for the Beckmann rearrangement reaction and thionyl chloride is used as a catalyst in the Beckmann rearrangement reaction, the use of alcohols and esters as the solvent adversely affects the Beckmann rearrangement reaction. Therefore, it is preferable not to use them.
- the amount ratio of ketone to hydroxylamine is not particularly limited, but it is preferable to react equimolarly.
- ketone and hydroxylamine are fed to each other, that is, ketone is fed to the first reaction tank.
- the light liquid phase (oil phase) containing ketone and oxime produced by the reaction is sequentially sent to the subsequent reaction tank, hydroxylamine is fed to the final reaction tank, and the heavy liquid phase containing hydroxylamine is sequentially fed to the previous reaction tank.
- the feed reaction method is a preferred embodiment for reducing unreacted ketone and / or hydroxylamine.
- the amount of carboxylic acid or its salt used is not particularly limited, but is preferably 0.001 mol% to 5 mol%, more preferably 0.01 mol% to 1 mol%, relative to the ketone.
- the addition amount is too small, the effect of accelerating the reaction rate of oximation is poor.
- the reaction temperature is preferably 115 ° C. or lower, more preferably 100 ° C. or lower, and further preferably 75 ° C. or higher and 100 ° C. or lower.
- the reaction temperature is high.
- hydroxylamine is decomposed and dangerous.
- a pressurized container is required to perform the reaction at a high temperature. Therefore, when the temperature is 100 ° C. or lower, the reaction can be performed at normal pressure, which is simpler.
- the hydrogen ion concentration (pH) of the aqueous phase in the oximation reaction is preferably from 5 to 6.
- the higher the pH the faster the reaction rate because the free hydroxylamine concentration increases. Accordingly, a higher pH is preferable in order to maintain an industrially suitable reaction rate. If the pH is too low, an industrially sufficient reaction rate cannot be obtained, which is not preferable.
- hydroxylamine when hydroxylamine is liberated, it is unstable and causes autolysis, and from the viewpoint of safety, retention of hydroxylamine at a high concentration must be avoided.
- the hydroxylamine liberated in the aqueous phase moves to the oil phase and is consumed by the oximation reaction.
- the addition of carboxylic acid or a salt thereof promotes mass transfer of hydroxylamine into the oil phase and increases the consumption rate of hydroxylamine, so that the pH of the aqueous phase is allowed up to 6.
- PH adjustment is performed by adding a base (preferably ammonia water).
- a base preferably ammonia water.
- Examples of the pH adjustment method include a method of constantly monitoring the pH of the aqueous phase with a pH controller and adding an appropriate amount of base so as not to exceed a desired value.
- the reaction apparatus used for the oximation reaction can be a commonly used reaction apparatus such as a batch reaction apparatus, a tube-type continuous reaction apparatus, a stirred tank type continuous reaction apparatus, etc., but maintains a reaction rate that can be produced industrially.
- a batch reaction apparatus or a stirred tank type continuous reaction apparatus having a mixing apparatus capable of sufficiently mixing the two phases of the hydrophobic solvent and the aqueous phase is preferable.
- the reaction time varies depending on the ketone, solvent type, ketone concentration, molar ratio of ketone to hydroxylamine, reaction temperature, pH, etc., but various conditions are set to be 15 hours or less from the viewpoint of preventing the apparatus from becoming too long. It is preferable to do. In the present invention, the reaction time may exceed 15 hours, but the use of the above-described carboxylic acid and / or salt thereof shortens the reaction time compared to the case where these are not used.
- the oximation reaction may be performed in the air, but the reaction system may be filled with an inert gas such as nitrogen gas or argon gas.
- the pressurized reactor is economically disadvantageous because it is more expensive than the atmospheric reactor, so it is carried out at atmospheric pressure. It is preferable.
- the oxime produced by the above reaction can be obtained by separating the organic phase from the reaction mixture and distilling off all or part of the solvent. .
- the kind of oxime produced by the above method corresponds to the kind of ketone used.
- cyclododecanone is used, cyclododecanone oxime is produced.
- Example 1 1162.0 g of a 15 wt% aqueous solution of hydroxylamine sulfate was added to a 1 L vertical reactor, and 25 wt% aqueous ammonia was added dropwise so that the pH was 4 while maintaining the temperature at 40 ° C. or lower. .1 g was prepared. (First tank, cyclododecanone excess tank) After adding 339.7 g of the above hydroxylamine aqueous solution, 167.3 g of cyclododecanone, 0.104 g of caproic acid and 71.9 g of toluene to a 1 L vertical reactor, 25 wt% aqueous ammonia was added at 90 ° C.
- Example 2 The same operation as in Example 1 was performed except that caproic acid was changed to undecanoic acid.
- First tank, cyclododecanone excess tank The oximation reaction was performed until the hydroxylamine sulfate concentration in the aqueous phase was 0.1 wt% or less. The reaction time was 4 hours.
- Hydroxylamine conversion 99.2% (Second tank, hydroxylamine excess tank) The reaction was carried out until the cyclododecanone concentration in the organic phase was 0.1% by weight or less. The reaction time was 6 hours.
- Cyclododecanone conversion 99.8% The same operation as in Example 1 was performed except that caproic acid was changed to undecanoic acid.
- Example 3 The same operation as in Example 1 was performed except that caproic acid was changed to lauric acid. (First tank, cyclododecanone excess tank) The oximation reaction was performed until the hydroxylamine sulfate concentration in the aqueous phase was 0.1 wt% or less. The reaction time was 4 hours. (Hydroxylamine conversion 99.2%) (Second tank, hydroxylamine excess tank) The reaction was carried out until the cyclododecanone concentration in the organic phase was 0.1% by weight or less. The reaction time was 6 hours. (Cyclododecanone conversion 99.8%)
- Example 4 The same operation as in Example 1 was performed except that caproic acid was changed to dodecanedioic acid.
- First tank, cyclododecanone excess tank The oximation reaction was performed until the hydroxylamine sulfate concentration in the aqueous phase was 0.1 wt% or less. The reaction time was 4 hours.
- Hydroxylamine conversion 99.2% (Second tank, hydroxylamine excess tank) The reaction was carried out until the cyclododecanone concentration in the organic phase was 0.1% by weight or less. The reaction time was 6 hours.
- Cyclododecanone conversion 99.8% The same operation as in Example 1 was performed except that caproic acid was changed to dodecanedioic acid.
- Example 5 The same operation as in Example 1 was performed except that caproic acid was changed to stearic acid. (First tank, cyclododecanone excess tank) The oximation reaction was performed until the hydroxylamine sulfate concentration in the aqueous phase was 0.1 wt% or less. The reaction time was 4 hours. (Hydroxylamine conversion 99.2%) (Second tank, hydroxylamine excess tank) The reaction was carried out until the cyclododecanone concentration in the organic phase was 0.1% by weight or less. The reaction time was 6 hours. (Cyclododecanone conversion 99.8%)
- Example 6 The same operation as in Example 5 was performed except that the amount of stearic acid added was changed from 0.261 g to 1.305 g. (First tank, cyclododecanone excess tank) The oximation reaction was performed until the hydroxylamine sulfate concentration in the aqueous phase was 0.1 wt% or less. The reaction time was 2 hours. (Hydroxylamine conversion 99.2%) (Second tank, hydroxylamine excess tank) The reaction was carried out until the cyclododecanone concentration in the organic phase was 0.1% by weight or less. The reaction time was 3 hours. (Cyclododecanone conversion 99.8%)
- Example 7 The same operation as in Example 1 was performed except that caproic acid was changed to sodium laurate. (First tank, cyclododecanone excess tank) The oximation reaction was performed until the hydroxylamine sulfate concentration in the aqueous phase was 0.1 wt% or less. The reaction time was 4 hours. (Hydroxylamine conversion 99.2%) (Second tank, hydroxylamine excess tank) The reaction was carried out until the cyclododecanone concentration in the organic phase was 0.1% by weight or less. The reaction time was 6 hours. (Cyclododecanone conversion 99.8%)
- Example 8> The same operation as in Example 1 was performed except that caproic acid was changed to propionic acid. (First tank, cyclododecanone excess tank) The reaction was carried out until the hydroxylamine sulfate concentration in the aqueous phase was 0.1% by weight or less. The reaction time was 6 hours. (Hydroxylamine conversion 99.2%) (Second tank, hydroxylamine excess tank) The reaction was carried out until the cyclododecanone concentration in the organic phase was 0.1% by weight or less. The reaction time was 9 hours. (Cyclododecanone conversion 99.8%)
- Example 1 The same operation as in Example 1 was performed except that caproic acid was not added. (First tank, cyclododecanone excess tank) The reaction was carried out until the hydroxylamine sulfate concentration in the aqueous phase was 0.1% by weight or less. The reaction time was 8 hours. (Hydroxylamine conversion 99.2%) (Second tank, hydroxylamine excess tank) The reaction was carried out until the cyclododecanone concentration in the organic phase was 0.1% by weight or less. The reaction time was 9 hours. (Cyclododecanone conversion 99.8%)
- Example 2 The same operation as in Example 1 was performed except that caproic acid was changed to tetrabutylammonium hydrogen sulfate. (First tank, cyclododecanone excess tank) The reaction was carried out until the hydroxylamine sulfate concentration in the aqueous phase was 0.1% by weight or less. The reaction time was 10 hours. (Hydroxylamine conversion 99.2%) (Second tank, hydroxylamine excess tank) The reaction was performed until the concentration of cyclododecanone in the organic phase was 0.5% by weight or less. The reaction time was 7 hours. (Cyclododecanone conversion 99.0%)
- Example 9 The same operation as in Example 5 was performed except that the solvent was changed to isopropylcyclohexane. (First tank, cyclododecanone excess tank) The reaction was carried out until the hydroxylamine sulfate concentration in the aqueous phase was 0.1% by weight or less. The reaction time was 7 hours. (Hydroxylamine conversion 99.2%) (Second tank, hydroxylamine excess tank) The reaction was carried out until the cyclododecanone concentration in the organic phase was 0.3% by weight or less. The reaction time was 10 hours. (Cyclododecanone conversion 98.9%)
- Example 3 The same operation as in Example 9 was performed except that stearic acid was not added. (First tank, cyclododecanone excess tank) The reaction was carried out until the hydroxylamine sulfate concentration in the aqueous phase was 0.1% by weight or less. The reaction time was 12 hours. (Hydroxylamine conversion 99.2%) (Second tank, hydroxylamine excess tank) The reaction was carried out until the cyclododecanone concentration in the organic phase was 0.2% by weight or less. The reaction time was 14 hours. (Cyclododecanone conversion 99.3%)
- the table below shows the type and amount of carboxylic acid added and its salt used in each example and comparative example, the solvent used for oximation, and the reaction time in each reaction tank.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description
(i)N-ヒドロキシイミド化合物及び該N-ヒドロキシイミド化合物のヒドロキシル基に保護基(例えば、アセチル基等のアシル基など)を導入することにより得られる化合物の存在下、メチル基又はメチレン基を有する化合物と、亜硝酸エステル又は亜硝酸塩とを反応させる製造方法(特許文献1)。ここで、前記N-ヒドロキシイミド化合物は、N-ヒドロキシコハク酸イミドなどの脂肪族多価カルボン酸無水物(環状無水物)又は芳香族多価カルボン酸無水物(環状無水物)から誘導される。
(ii)シクロアルカン等を光ニトロソ化する方法(特許文献2)。
(iii)チタノシリケートのような触媒の存在下にケトンと、アンモニア及び過酸化水素とを反応させる方法(特許文献3)。
(iv)対応するケトンに硫酸ヒドロキシルアミン鉱酸塩の複分解によって生成したヒドロキシルアミンを縮合反応させる方法。
これらのうち、(iv)の方法が汎用的かつ一般的である。例えば、ラウロラクタムの原料であるシクロドデカノンオキシムは、シクロドデカノンと、ヒドロキシルアミン鉱酸塩との反応により製造される(特許文献4)。
本発明は、オキシムの製造方法において、カルボン酸および/またはその塩を用いることを特徴とする。カルボン酸および/またはその塩の存在下でオキシム化反応を行うことにより、反応速度が加速される。後述するように、本発明によるオキシムの製造においては、水相より疎水性溶媒の相に分配されやすいケトン(以下、「疎水性の高いケトン」と表記することもある。)を用いる場合、反応速度の加速効果がより顕著にあらわれる。疎水性の高いケトンが用いられる場合、オキシム化の反応場は油相中となるため、共に用いるカルボン酸またはその塩も、疎水性溶媒の相に分配されやすいカルボン酸またはその塩であることが好ましい。カルボン酸またはその塩を使用することにより、水相中に存在するヒドロキシルアミンが、反応場である油相中に物質移動しやすくなり、オキシム化の反応速度が促進される。
本発明においては、オキシム化反応は疎水性溶媒の相と水相との2相系中で行われる。水相に分配されやすいケトンを用いる場合、水相においてもオキシム化が進行するため比較的反応速度が速く、カルボン酸またはその塩の添加による反応加速効果は比較的小さい。一方、疎水性の高いケトンを用いた場合、反応場が油相に限られるため、ヒドロキシルアミンの油相への物質移動速度が律速となる。カルボン酸またはその塩の添加は当該物質移動速度を加速するため、疎水性の高いケトンを用いた場合、本発明の効果(カルボン酸またはその塩の添加によるオキシム化速度の加速効果)はより顕著に現れる。すなわち、疎水性の高いケトンが所望される場合において、本発明は特に重要である。
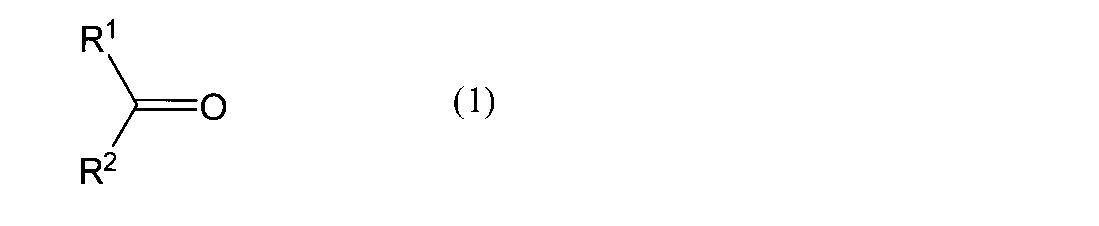
ヒドロキシルアミンは不安定なため、ヒドロキシルアミン硫酸塩又はヒドロキシルアミン炭酸塩等のヒドロキシルアミンの無機塩の水溶液として製造、販売されている。通常、反応時にアンモニア水等の塩基を加えて、ヒドロキシルアミンを遊離させて使用される。本発明においては、予めヒドロキシルアミンを遊離させたヒドロキシルアミン水溶液を使用してもよいが、通常は反応装置中にヒドロキシルアミンの無機塩(好ましくは硫酸塩)の水溶液と、塩基(好ましくはアンモニア水)を供給して、反応装置内でヒドロキシルアミンを遊離させて使用する。
本発明のオキシムの製造方法においては、疎水性溶媒が使用される。このため、本発明のオキシムの製造方法は、上述のヒドロキシルアミン水溶液由来の水と、疎水性溶媒との2相系となる。
次に、上記化合物を用いたオキシムの製造方法について説明する。本発明のオキシムの製造方法においては、水相と疎水性溶媒との2相系中、カルボン酸および/またはその塩の存在下、ケトンとヒドロキシルアミンとを反応させる。
1L竪型反応器にヒドロキシルアミン硫酸塩の15重量%水溶液1162.0gを添加し、40℃以下を保持しながらpHが4になるように25重量%アンモニア水を滴下し、ヒドロキシルアミンの水溶液1313.1gを調製した。
(第一槽、シクロドデカノン過剰槽)
1L竪型反応器に、上記ヒドロキシルアミン水溶液339.7g、シクロドデカノン167.3g、カプロン酸0.104gおよびトルエン71.9gを添加後、90℃で25重量%アンモニア水を水相のpHを5.8に保持するように滴下し、水相中のヒドロキシルアミン硫酸塩濃度が0.1重量%以下になるまでオキシム化反応を行った。反応時間は4時間であった。(ヒドロキシルアミン転化率99.2%)
反応終了後、水相を抜き出し、有機相は下記第二槽での反応にそのまま使用した。
(第二槽、ヒドロキシルアミン過剰槽)
上記有機相へ上記ヒドロキシルアミン水溶液567.4gを更に添加後、90℃で25重量%アンモニア水を水相のpHを5.8に保持するように滴下しながら、有機相中のシクロドデカノン濃度が0.1重量%以下になるまで反応をおこなった。反応時間は7時間であった。(シクロドデカノン転化率99.8%)
カプロン酸をウンデカン酸に変えた以外は実施例1と同様の操作を行った。
(第一槽、シクロドデカノン過剰槽)
水相中のヒドロキシルアミン硫酸塩濃度が0.1重量%以下になるまでオキシム化反応を行った。反応時間は4時間であった。(ヒドロキシルアミン転化率99.2%)
(第二槽、ヒドロキシルアミン過剰槽)
有機相中のシクロドデカノン濃度が0.1重量%以下になるまで反応を行った。反応時間は6時間であった。(シクロドデカノン転化率99.8%)
カプロン酸をラウリン酸に変えた以外は実施例1と同様の操作を行った。
(第一槽、シクロドデカノン過剰槽)
水相中のヒドロキシルアミン硫酸塩濃度が0.1重量%以下になるまでオキシム化反応を行った。反応時間は4時間であった。(ヒドロキシルアミン転化率99.2%)
(第二槽、ヒドロキシルアミン過剰槽)
有機相中のシクロドデカノン濃度が0.1重量%以下になるまで反応を行った。反応時間は6時間であった。(シクロドデカノン転化率99.8%)
カプロン酸をドデカン二酸に変えた以外は実施例1と同様の操作を行った。
(第一槽、シクロドデカノン過剰槽)
水相中のヒドロキシルアミン硫酸塩濃度が0.1重量%以下になるまでオキシム化反応を行った。反応時間は4時間であった。(ヒドロキシルアミン転化率99.2%)
(第二槽、ヒドロキシルアミン過剰槽)
有機相中のシクロドデカノン濃度が0.1重量%以下になるまで反応を行った。反応時間は6時間であった。(シクロドデカノン転化率99.8%)
カプロン酸をステアリン酸に変えた以外は実施例1と同様の操作を行った。
(第一槽、シクロドデカノン過剰槽)
水相中のヒドロキシルアミン硫酸塩濃度が0.1重量%以下になるまでオキシム化反応を行った。反応時間は4時間であった。(ヒドロキシルアミン転化率99.2%)
(第二槽、ヒドロキシルアミン過剰槽)
有機相中のシクロドデカノン濃度が0.1重量%以下になるまで反応を行った。反応時間は6時間であった。(シクロドデカノン転化率99.8%)
ステアリン酸添加量を0.261gから1.305gに変えた以外は実施例5と同様の操作を行った。
(第一槽、シクロドデカノン過剰槽)
水相中のヒドロキシルアミン硫酸塩濃度が0.1重量%以下になるまでオキシム化反応を行った。反応時間は2時間であった。(ヒドロキシルアミン転化率99.2%)
(第二槽、ヒドロキシルアミン過剰槽)
有機相中のシクロドデカノン濃度が0.1重量%以下になるまで反応を行った。反応時間は3時間であった。(シクロドデカノン転化率99.8%)
カプロン酸をラウリン酸ナトリウムに変えた以外は実施例1と同様の操作を行った。
(第一槽、シクロドデカノン過剰槽)
水相中のヒドロキシルアミン硫酸塩濃度が0.1重量%以下になるまでオキシム化反応を行った。反応時間は4時間であった。(ヒドロキシルアミン転化率99.2%)
(第二槽、ヒドロキシルアミン過剰槽)
有機相中のシクロドデカノン濃度が0.1重量%以下になるまで反応を行った。反応時間は6時間であった。(シクロドデカノン転化率99.8%)
カプロン酸をプロピオン酸に変えた以外は、実施例1と同様の操作を行った。
(第一槽、シクロドデカノン過剰槽)
水相中のヒドロキシルアミン硫酸塩濃度が0.1重量%以下になるまで反応を行った。反応時間は6時間であった。(ヒドロキシルアミン転化率99.2%)
(第二槽、ヒドロキシルアミン過剰槽)
有機相中のシクロドデカノン濃度が0.1重量%以下になるまで反応を行った。反応時間は9時間であった。(シクロドデカノン転化率99.8%)
カプロン酸を添加しなかった以外は、実施例1と同様の操作を行った。
(第一槽、シクロドデカノン過剰槽)
水相中のヒドロキシルアミン硫酸塩濃度が0.1重量%以下になるまで反応を行った。反応時間は8時間であった。(ヒドロキシルアミン転化率99.2%)
(第二槽、ヒドロキシルアミン過剰槽)
有機相中のシクロドデカノン濃度が0.1重量%以下になるまで反応を行った。反応時間は9時間であった。(シクロドデカノン転化率99.8%)
カプロン酸をテトラブチルアンモニウム硫酸水素塩に変えた以外は、実施例1と同様の操作を行った。
(第一槽、シクロドデカノン過剰槽)
水相中のヒドロキシルアミン硫酸塩濃度が0.1重量%以下になるまで反応を行った。反応時間は10時間であった。(ヒドロキシルアミン転化率99.2%)
(第二槽、ヒドロキシルアミン過剰槽)
有機相中のシクロドデカノン濃度が0.5重量%以下になるまで反応を行った。反応時間は7時間であった。(シクロドデカノン転化率99.0%)
溶媒をイソプロピルシクロヘキサンに変えた以外は、実施例5と同様の操作を行った。
(第一槽、シクロドデカノン過剰槽)
水相中のヒドロキシルアミン硫酸塩濃度が0.1重量%以下になるまで反応を行った。反応時間は7時間であった。(ヒドロキシルアミン転化率99.2%)
(第二槽、ヒドロキシルアミン過剰槽)
有機相中のシクロドデカノン濃度が0.3重量%以下になるまで反応を行った。反応時間は10時間であった。(シクロドデカノン転化率98.9%)
ステアリン酸を添加しなかったこと以外は、実施例9と同様の操作を行った。
(第一槽、シクロドデカノン過剰槽)
水相中のヒドロキシルアミン硫酸塩濃度が0.1重量%以下になるまで反応を行った。反応時間は12時間であった。(ヒドロキシルアミン転化率99.2%)
(第二槽、ヒドロキシルアミン過剰槽)
有機相中のシクロドデカノン濃度が0.2重量%以下になるまで反応を行った。反応時間は14時間であった。(シクロドデカノン転化率99.3%)

Claims (6)
- 水相と、疎水性溶媒の相とからなる系中、カルボン酸および/またはその塩の存在下、ケトンとヒドロキシルアミンとを反応させることを特徴とする、オキシムの製造方法。
- ケトンの炭素原子数が8以上30以下である、請求項1記載の方法。
- ケトンがシクロドデカノンである、請求項1記載の方法。
- カルボン酸および/またはその塩の炭素原子数が5以上である、請求項1記載の方法。
- 反応時における水相の水素イオン濃度(pH)がpH5からpH6の範囲である、請求項1記載の方法。
- 疎水性溶媒が芳香族炭化水素または脂肪族炭化水素である、請求項1記載の方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11759452.3A EP2551261B1 (en) | 2010-03-24 | 2011-03-23 | Method for producing oxime |
| CN201180025500.5A CN102906065B (zh) | 2010-03-24 | 2011-03-23 | 肟的制备方法 |
| US13/636,433 US9376375B2 (en) | 2010-03-24 | 2011-03-23 | Method for producing oxime |
| ES11759452.3T ES2614085T3 (es) | 2010-03-24 | 2011-03-23 | Método para producir oxima |
| JP2012507039A JP5794227B2 (ja) | 2010-03-24 | 2011-03-23 | オキシムの製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010067479 | 2010-03-24 | ||
| JP2010-067479 | 2010-03-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011118647A1 true WO2011118647A1 (ja) | 2011-09-29 |
Family
ID=44673197
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/057018 WO2011118647A1 (ja) | 2010-03-24 | 2011-03-23 | オキシムの製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9376375B2 (ja) |
| EP (1) | EP2551261B1 (ja) |
| JP (1) | JP5794227B2 (ja) |
| CN (1) | CN102906065B (ja) |
| ES (1) | ES2614085T3 (ja) |
| WO (1) | WO2011118647A1 (ja) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3498759A1 (de) | 2017-12-13 | 2019-06-19 | Evonik Degussa GmbH | Verfahren zur herstellung von polymeren aus monomeren umfassend laurinlactam |
| EP3498758A1 (de) | 2017-12-13 | 2019-06-19 | Evonik Degussa GmbH | Verfahren zur herstellung von polymeren aus monomeren umfassend laurinlactam |
| CN110272356B (zh) * | 2019-07-25 | 2022-07-12 | 万华化学集团股份有限公司 | 一种环十二酮肟化的方法 |
| CN110498748B (zh) * | 2019-09-17 | 2022-07-12 | 万华化学集团股份有限公司 | L-精氨酸及其衍生物用于制备环十二酮肟的用途及一种制备环十二酮肟的方法 |
| CN113548980B (zh) * | 2020-04-23 | 2023-08-11 | 万华化学集团股份有限公司 | 一种制备环十二酮肟的方法 |
| CN114989041B (zh) * | 2022-05-30 | 2024-02-02 | 万华化学集团股份有限公司 | 一种环十二酮肟化的方法 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0225457A (ja) * | 1988-07-12 | 1990-01-26 | Mitsubishi Kasei Corp | オキシム類の製法 |
| JPH07504912A (ja) * | 1992-03-20 | 1995-06-01 | ヘンケル・コーポレーション | オキシム製造法 |
| JP2004059553A (ja) | 2002-07-31 | 2004-02-26 | Mitsubishi Chemicals Corp | アミド化合物の製造方法 |
| JP2006206476A (ja) | 2005-01-27 | 2006-08-10 | Sumitomo Chemical Co Ltd | シクロアルカノンオキシムの製造方法 |
| WO2009069522A1 (ja) | 2007-11-29 | 2009-06-04 | Ube Industries, Ltd. | ラウロラクタムの製造方法 |
| JP2009298706A (ja) | 2008-06-11 | 2009-12-24 | Daicel Chem Ind Ltd | アミド又はラクタムの製造法 |
| JP2010006775A (ja) | 2008-06-30 | 2010-01-14 | Toray Ind Inc | シクロアルカノンオキシムの製造方法 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL301053A (ja) * | 1963-11-27 | |||
| US3808275A (en) * | 1970-03-25 | 1974-04-30 | Toa Gosei Chem Ind | Process for producing oximes |
| US4128580A (en) * | 1976-01-30 | 1978-12-05 | Stauffer Chemical Company | Process for the production of 1,3-dichloroacetone oxime and its acetate derivative |
| US5488161A (en) | 1992-03-20 | 1996-01-30 | Henkel Corporation | Oximation process |
| US5434307A (en) * | 1993-05-21 | 1995-07-18 | Howard University | Synthesis of 12-oxododecanoic acid oxime from vernolic acid |
| US6235935B1 (en) * | 1998-11-24 | 2001-05-22 | Silicon Valley Chemlabs, Inc. | Method of manufacturing high purity oximes from aqueous hydroxylamine and ketones |
| JP2003535841A (ja) * | 2000-06-05 | 2003-12-02 | ディーエスエム エヌ.ブイ. | シクロヘキサノンオキシムの製造法 |
| DE10158352A1 (de) | 2001-11-28 | 2003-06-12 | Degussa | Zweiphasige Ammoximierung |
| CN102770409B (zh) * | 2010-02-26 | 2015-07-15 | 住友化学株式会社 | 环丙甲酸酯化合物的制造方法 |
-
2011
- 2011-03-23 EP EP11759452.3A patent/EP2551261B1/en not_active Not-in-force
- 2011-03-23 US US13/636,433 patent/US9376375B2/en active Active
- 2011-03-23 CN CN201180025500.5A patent/CN102906065B/zh not_active Expired - Fee Related
- 2011-03-23 ES ES11759452.3T patent/ES2614085T3/es active Active
- 2011-03-23 WO PCT/JP2011/057018 patent/WO2011118647A1/ja active Application Filing
- 2011-03-23 JP JP2012507039A patent/JP5794227B2/ja not_active Expired - Fee Related
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0225457A (ja) * | 1988-07-12 | 1990-01-26 | Mitsubishi Kasei Corp | オキシム類の製法 |
| JPH07504912A (ja) * | 1992-03-20 | 1995-06-01 | ヘンケル・コーポレーション | オキシム製造法 |
| JP2004059553A (ja) | 2002-07-31 | 2004-02-26 | Mitsubishi Chemicals Corp | アミド化合物の製造方法 |
| JP2006206476A (ja) | 2005-01-27 | 2006-08-10 | Sumitomo Chemical Co Ltd | シクロアルカノンオキシムの製造方法 |
| WO2009069522A1 (ja) | 2007-11-29 | 2009-06-04 | Ube Industries, Ltd. | ラウロラクタムの製造方法 |
| JP2009298706A (ja) | 2008-06-11 | 2009-12-24 | Daicel Chem Ind Ltd | アミド又はラクタムの製造法 |
| JP2010006775A (ja) | 2008-06-30 | 2010-01-14 | Toray Ind Inc | シクロアルカノンオキシムの製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2551261A4 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2551261B1 (en) | 2016-11-02 |
| CN102906065B (zh) | 2016-03-02 |
| EP2551261A4 (en) | 2014-01-15 |
| CN102906065A (zh) | 2013-01-30 |
| EP2551261A1 (en) | 2013-01-30 |
| ES2614085T3 (es) | 2017-05-29 |
| JP5794227B2 (ja) | 2015-10-14 |
| JPWO2011118647A1 (ja) | 2013-07-04 |
| US20130023697A1 (en) | 2013-01-24 |
| US9376375B2 (en) | 2016-06-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5794227B2 (ja) | オキシムの製造方法 | |
| JP5446872B2 (ja) | ラウロラクタムの製造方法 | |
| US7608738B2 (en) | Coammoxidation of ketones | |
| EP2275407B1 (en) | Process for production of amide or lactam | |
| US9593073B2 (en) | Method for the preparation of diazoalkanes | |
| JP2012505828A (ja) | N,n−ジアルキルラクタミドを製造する方法 | |
| JP5447502B2 (ja) | アミド化合物の製造方法 | |
| KR101904568B1 (ko) | 하이드록실아민의 제조 방법 | |
| EP1471047B1 (en) | Process for producing dicarboxylic acid | |
| TW201008903A (en) | Method for producing oxime | |
| JP2010229042A (ja) | ラウロラクタムの製造方法 | |
| JP5369653B2 (ja) | アミド又はラクタムの製造方法 | |
| JP5574327B2 (ja) | アミド化合物の製造方法 | |
| JP5640985B2 (ja) | 新規化合物およびそれを用いたアミド化合物の製造方法 | |
| JP5572839B2 (ja) | アミド化合物の製造方法 | |
| JP2011173814A (ja) | アミド化合物の製造方法 | |
| JP2011201816A (ja) | ラクタムの製造方法 | |
| US8865892B2 (en) | Catalyst composition and method for preparing amide using the same | |
| JP2016175870A (ja) | アミド化合物の製造方法 | |
| TW201134550A (en) | Process for preparing a deperoxidation catalyst | |
| JP2010222272A (ja) | ラウロラクタムの製造方法 | |
| JP5593095B2 (ja) | アミド化合物の製造方法 | |
| US5292945A (en) | Process for preparing α-chloro-α-oximino-4-hydroxyacetophenone | |
| JP2004043406A (ja) | ヒドロキシラクトンの製造方法 | |
| WO2004000827A1 (fr) | Procede de fabrication de composes carbonyles |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180025500.5 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11759452 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13636433 Country of ref document: US Ref document number: 2012507039 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1201004898 Country of ref document: TH |
|
| REEP | Request for entry into the european phase |
Ref document number: 2011759452 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011759452 Country of ref document: EP |