WO2010150835A1 - 塩素化炭化水素の製造方法 - Google Patents
塩素化炭化水素の製造方法 Download PDFInfo
- Publication number
- WO2010150835A1 WO2010150835A1 PCT/JP2010/060695 JP2010060695W WO2010150835A1 WO 2010150835 A1 WO2010150835 A1 WO 2010150835A1 JP 2010060695 W JP2010060695 W JP 2010060695W WO 2010150835 A1 WO2010150835 A1 WO 2010150835A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- reaction
- ccl
- general formula
- compound represented
- iron
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/013—Preparation of halogenated hydrocarbons by addition of halogens
- C07C17/04—Preparation of halogenated hydrocarbons by addition of halogens to unsaturated halogenated hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B7/00—Halogens; Halogen acids
- C01B7/01—Chlorine; Hydrogen chloride
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B7/00—Halogens; Halogen acids
- C01B7/01—Chlorine; Hydrogen chloride
- C01B7/03—Preparation from chlorides
- C01B7/04—Preparation of chlorine from hydrogen chloride
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B7/00—Halogens; Halogen acids
- C01B7/01—Chlorine; Hydrogen chloride
- C01B7/07—Purification ; Separation
- C01B7/0706—Purification ; Separation of hydrogen chloride
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/25—Preparation of halogenated hydrocarbons by splitting-off hydrogen halides from halogenated hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/26—Preparation of halogenated hydrocarbons by reactions involving an increase in the number of carbon atoms in the skeleton
- C07C17/272—Preparation of halogenated hydrocarbons by reactions involving an increase in the number of carbon atoms in the skeleton by addition reactions
- C07C17/275—Preparation of halogenated hydrocarbons by reactions involving an increase in the number of carbon atoms in the skeleton by addition reactions of hydrocarbons and halogenated hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/26—Preparation of halogenated hydrocarbons by reactions involving an increase in the number of carbon atoms in the skeleton
- C07C17/272—Preparation of halogenated hydrocarbons by reactions involving an increase in the number of carbon atoms in the skeleton by addition reactions
- C07C17/278—Preparation of halogenated hydrocarbons by reactions involving an increase in the number of carbon atoms in the skeleton by addition reactions of only halogenated hydrocarbons
Definitions
- the present invention relates to a method of producing chlorinated hydrocarbons. More particularly, the present invention relates to a method for producing high purity chlorinated hydrocarbons by a simple method.
- Chlorinated hydrocarbons are important as raw materials or intermediates for producing various products such as agricultural chemicals, medicines, fluorocarbon substitutes and the like.
- trichloroallyldiisopropylthiocarbamates useful as herbicides can be prepared starting from 1,1,1,2,3-pentachloropropane via 1,1,2,3-tetrachloropropene.
- Chlorinated hydrocarbons, which are raw materials or intermediates for such agricultural and pharmaceutical products, are required to have extremely high purity.
- a method for producing such a chlorinated hydrocarbon for example, a first reaction in which carbon tetrachloride is added to an unsaturated compound having 2 carbon atoms to obtain a chlorinated saturated hydrocarbon having 3 carbon atoms, A second reaction for dehydrochlorinating the chlorinated saturated hydrocarbon to obtain a chlorinated unsaturated hydrocarbon having 3 carbon atoms; A three-step reaction is known which comprises a third reaction in which chlorine is further added to the chlorinated unsaturated hydrocarbon to obtain a chlorinated saturated hydrocarbon having 3 carbon atoms.
- the known chlorinated hydrocarbon production methods as described above have the following problems to be improved at each step.
- the iron-phosphoryl compound catalyst used exhibits high activity immediately after preparation, there is a problem that the subsequent activity falls rapidly with time, so to obtain the desired high conversion rate It is necessary to use a large amount of catalyst.
- the start of the reaction at the initial stage of the reaction may be so rapid that control of the reaction may be difficult and the reaction yield of the desired product may be impaired.
- the use of a large amount of catalyst requires labor and cost for the treatment of the spent catalyst, which also hinders the reduction of the production cost.
- metallic iron used to prepare the iron-phosphoryl compound catalyst has a problem that the surface is gradually oxidized during storage, and the initial state of the reaction largely changes depending on the oxidation state.
- the second reaction in addition to the problem of the cost of sodium hydroxide consumed in large amounts, it requires a great deal of labor to treat the organic chlorine compounds dissolved in the aqueous phase to be discarded.
- it is necessary to prolong the reaction time (residence time).
- the first reaction there is a method of efficiently carrying out the addition reaction at a stable reaction rate of the desired product at a high conversion rate while reducing the amount of catalyst used;
- the second reaction there is a method of carrying out a dehydrochlorination reaction without using an expensive alkali source and suppressing the formation of by-products to be treated;
- the third reaction methods for carrying out more efficient chlorination reactions are respectively desired.
- a chlorinated hydrocarbon is characterized in that it is subjected to a thermal dehydrochlorination step to obtain an unsaturated compound represented by the following general formula (2) by thermally decomposing a saturated compound represented by the following general formula (1) Achieved by a method for manufacturing.
- a chlorination step of obtaining a saturated compound represented by the following general formula (3) by further reacting an unsaturated compound represented by the following general formula (2) with chlorine is carried out it can.
- the saturated compound represented by the above general formula (1) is preferably an iron-phosphate ester catalyst in the reaction system of liquid phase, preferably carbon tetrachloride and the unsaturated compound represented by the following general formula (0) It is a saturated compound represented by following General formula (1 ') obtained by the addition reaction process made to add in presence.
- the first reaction in the present invention is an addition reaction in which a saturated compound represented by the above general formula (1 ′) is obtained by the addition reaction of carbon tetrachloride with the unsaturated compound represented by the above general formula (0). is there. This reaction is preferably carried out in the presence of a catalyst in a liquid phase reaction system.
- Examples of the unsaturated compound represented by the above general formula (0) include ethylene, vinyl chloride, 1,1-dichloroethylene, 1,2-dichloroethylene, 1,1,2-trichloroethylene, etc.
- ethylene or vinyl chloride is used.
- the kind of chloropropane which is a saturated compound represented by the said General formula (1 ') obtained by this reaction depends on the kind of unsaturated compound represented by the said General formula (0) used as a raw material. For example, when ethylene is used as a raw material, 1,1,1,3-tetrachloropropane is obtained as a saturated compound represented by the above general formula (1 ').
- the iron-phosphate ester catalyst is prepared by contacting a predetermined amount of iron and a predetermined amount of phosphoric acid ester in a liquid phase reaction system (i.e., in liquid carbon tetrachloride).
- the contact of iron with the phosphate ester may be carried out by introducing all the amounts of iron and phosphate ester into the reaction system at once before the start of the reaction, or Alternatively, the total amount of iron and a part of the phosphate ester can be added before the start of the reaction, and the remaining phosphate ester can be added by addition during the progress of the addition reaction.
- iron used herein include metallic iron, pure iron, soft iron, carbon steel, ferrosilicon steel, alloys containing iron (for example, stainless steel and the like), and the like.
- the shape of iron may be, for example, any shape such as powdery, granular, massive, rod-like, spherical, plate-like, and fibrous, and further, metal pieces further optionally processed using these, distillation filling It may be a thing etc.
- Examples of the processed metal piece include coils, nets, steel wool, and other irregularly shaped pieces; and examples of the distillation packing include Raschig rings, helices and the like.
- the specific surface area of iron measured by BET method using nitrogen as an adsorbate is 0.001 to 5 m. 2 It is preferable that it is / g.
- the amount of iron used in the case of adding phosphate esters all at once before the start of the reaction is 0 with respect to 1 mol of carbon tetrachloride used, from the viewpoint of achieving both high reaction conversion rate and high selectivity. It is preferable to use .001 mol or more, more preferably 0.005 mol or more, still more preferably 0.01 mol or more, and particularly preferably 0.05 mol or more.
- the upper limit of the amount of iron used is not particularly limited. Increasing the amount of iron used has little effect on activity and selectivity, but is economically disadvantageous in that more iron is wasted without taking part in the reaction.
- the amount of iron used is preferably 10 moles or less, more preferably 5 moles or less, and still more preferably 1 mole or less, with respect to 1 mole of carbon tetrachloride used. In particular, the amount is preferably 0.1 mol or less.
- phosphoric acid ester for example, following General formula (4) (In the formula (4), R 1 Is a phenyl group or an alkyl group having 1 to 4 carbon atoms, and R 2 And R 3 Each independently represents a hydrogen atom, a phenyl group or an alkyl group having 1 to 4 carbon atoms.
- Specific examples thereof include trimethyl phosphate, triethyl phosphate, tripropyl phosphate, tributyl phosphate, diethyl phosphate, dibutyl phosphate, monophenyl phosphate, monobutyl phosphate, and the like.
- Examples include dimethylphenyl phosphate, diethylphenyl phosphate, dimethylethyl phosphate, phenylethyl methyl phosphate and the like.
- R 1 , R 2 And R 3 Preferred is a trialkyl phosphate which is an alkyl group having 1 to 4 carbon atoms, and particularly preferred is trimethyl phosphate, triethyl phosphate, tripropyl phosphate or tributyl phosphate.
- the amount of phosphoric acid ester used is preferably 0.001 mol or more, particularly preferably 0.002 or more, with respect to 1 mol of carbon tetrachloride used, from the viewpoint of securing high conversion and high selectivity. It is preferable to The upper limit of the amount of phosphoric acid ester used is not particularly limited.
- the amount of phosphoric acid ester used is preferably 5 moles or less, more preferably 1 mole or less, per 1 mole of carbon tetrachloride, even if it is 0.5 moles or less Good.
- the reaction temperature of the first reaction is preferably 70 to 180 ° C., and more preferably 90 to 150 ° C., in order to achieve both a high conversion rate and a high selectivity.
- the reaction pressure may be a pressure at which the reaction system can maintain the liquid phase at the reaction temperature, and can generally be 0.05 to 3 MPaG, preferably 0.1 to 2 MPaG.
- the concentration of the raw material (the unsaturated compound represented by the above formula (0)) in the liquid phase may be too low, and the reaction addition rate may be insufficient. If the pressure is exceeded, the rate of formation of the multimer may be increased to impair the selectivity, which is not preferable.
- the reaction time of the first reaction is preferably 2 to 24 hours, and more preferably 2 to 10 hours. Here, if the reaction time is shorter than 2 hours, the reaction conversion may be insufficient. On the other hand, there is no real benefit to perform the reaction longer than 24 hours.
- the whole amount of iron and a part of the phosphate ester are added before the reaction starts, and the remaining phosphate ester is additionally added during the progress of the addition reaction. It is preferable in that it has good controllability, high selectivity, and can reduce the amount of iron used.
- the amount of iron to be added all at once before the start of the reaction can be smaller than the above-mentioned value as the lower limit of the amount of iron used in the case of adding phosphate esters all at once before the start of the reaction.
- the amount of iron used in this case is preferably 0.0001 mol or more, more preferably 0.0005 mol or more, and more preferably 0.001 mol or more per 1 mol of carbon tetrachloride used. It is more preferable to carry out, and it is particularly preferable to use 0.005 mol or more.
- the upper limit of iron usage is set from the economic point of view.
- the amount of iron used is preferably 1 mol or less, more preferably 0.5 mol or less, and more preferably 0.1 mol or less, per 1 mol of carbon tetrachloride used. Is more preferred.
- the addition of the phosphoric acid ester may be performed only once, may be divided into several times, or may be performed continuously.
- the number of times of additional addition in the case of dividing into several times is preferably 2 to 10 times, and more preferably 2 to 6 times.
- the total amount of phosphoric acid ester used (total amount of all the components added before reaction start and additional components) is preferably 0.001 mol or more per 1 mol of carbon tetrachloride used, particularly 0 It is preferable to set it as .002 mol or more.
- the total addition amount of phosphoric acid ester in the case of additional addition is not particularly limited. However, also in this case, if the total addition amount of the phosphate ester is excessively increased, there is an economic disadvantage in that the amount of phosphate ester which is wasted without participating in the reaction increases. From this point of view, the total addition amount of phosphoric acid ester in the case of additional addition is preferably 5 mol or less, more preferably 1 mol or less, with respect to 1 mol of carbon tetrachloride. It may be less than the mole. In the method of additionally adding a phosphate, the amount of the phosphate used is higher than that of the prior art, for example, the method described in the above-mentioned Japanese Examined Patent Publication No.
- a portion of the phosphoric acid ester is added prior to the start of the reaction.
- the addition amount of the phosphoric acid ester before the start of the reaction is preferably 0.0005 mol or more, more preferably 0.001 mol or more, with respect to 1 mol of carbon tetrachloride to be used.
- the upper limit value of the phosphate ester added before the start of the reaction does not depend on the mode of the additional addition (whether the additional addition is performed only once, divided into several times, or continuously performed), and In the case of dividing and adding several times, the addition amount is preferably 80% or less, and more preferably 70% or less, of the total amount of phosphate ester used, regardless of the addition frequency.
- the addition amount of the phosphate ester in the range as described above before the start of the reaction, the reaction can be stably started, the control of the reaction becomes easy, and as a result, a high conversion can be achieved. Become.
- the addition reaction thus initiated is preferably carried out while continuously monitoring the consumption rate of the unsaturated compound represented by the above general formula (0).
- the continuous monitoring of the rate of consumption of unsaturated compounds is, for example, to maintain the amount of unsaturated compounds continuously supplied in the gaseous state and an appropriate reaction pressure in liquid phase batch reactions under gas phase flow. It can carry out by comparing the amount of unsaturated compounds discharged from the gas phase at any time. Then, when the consumption rate decreases from the initial value to a predetermined degree, additional addition of phosphoric acid ester is performed or started. When the additional addition of the phosphate ester is performed only once, the consumption rate of the unsaturated compound is preferably 5 to 50%, more preferably 10 to 40% of the average consumption rate in 60 minutes after the start of the reaction. , The entire remaining amount of phosphate ester is additionally added.
- the consumption rate of the unsaturated compound once reduced is recovered, and thereafter, the remaining addition reaction proceeds while the consumption rate gradually decreases again.
- the consumption rate is preferably 5 to 50%, more preferably 10 to 40% of the average consumption rate in 60 minutes after the start of the reaction.
- the first additional addition of phosphate ester is performed.
- the first additional addition recovers the consumption rate of the unsaturated compound once decreased, and thereafter the consumption rate gradually decreases again.
- the consumption rate of the unsaturated compound becomes preferably 5 to 50%, more preferably 10 to 40% of the average consumption rate in 60 minutes after the start of the reaction again, addition of the second and subsequent phosphate esters Addition takes place.
- This additional addition restores the rate of consumption of unsaturated compounds again. Thereafter, the consumption rate of the unsaturated compound represented by the above general formula (0) can be monitored continuously, and the phosphate ester can be additionally added a predetermined number of times. In the case where the phosphate ester is additionally added several times in divided portions, it is preferable to set equal amounts of addition each time equal to each other or to gradually reduce the amount added each time the number is repeated.
- the phosphoric acid is preferably used when the consumption rate is 5 to 50%, more preferably 10 to 40% of the average consumption rate in 60 minutes after the start of the reaction. Additional addition of ester is initiated.
- the continuous addition of this phosphoric acid is carried out continuously over preferably 1 to 400 minutes, more preferably 2 to 360 minutes, of the entire remaining amount of phosphoric acid ester from the above-mentioned time point.
- additional addition of phosphoric acid ester it is preferable to carry out only once or continuously.
- the total reaction time is preferably 2 to 12 hours, and more preferably 2 to 10 hours.
- the reaction mixture thus obtained contains the desired product converted to the desired product with high conversion and high selectivity.
- the reaction mixture obtained by the method of additionally adding phosphoric acid ester contains unreacted carbon tetrachloride (the content of which is small) contained therein, iron-phosphate ester catalyst residue and excess If the unsaturated compound represented by the above general formula (0) is separated, it can be used as a product in many cases as it is or can be used as it is in the reaction of the next step. If desired, purification can be carried out after the first reaction, but the purification method may be very simple, and for example, a simple purification with about 2 to 10 theoretical plates may provide a high purity product.
- the second reaction in the present invention is a thermal dehydrochlorination step in which the saturated compound represented by the general formula (1) is thermally decomposed to obtain the unsaturated compound represented by the general formula (2).
- the present inventors have found that the saturated compound represented by the above general formula (1) has the property of easily causing a dehydrochlorination reaction by heating. Such characteristics are determined by the fact that CCl in the saturated compound represented by the above general formula (1) is 3 It is presumed to be due to That is, the present inventors, the above CCl 3 It is extremely easy to find that Cl in the base is easily released by heat, so that it is possible to easily cause the dehydrochlorination reaction by thermal decomposition in a short heating time of about 1 to 10 seconds.
- saturated compound represented by the above formula (1) examples include, for example, 1,1,1-trichloropropane, 1,1,1,2-tetrachloropropane, 1,1,1,3-tetrachloropropane, 1 1,1,1,2,3- heptachloropropane, 1,1,1,3,3-pentachloropropane, 1,1,1,2,3,3-hexachloropropane, 1,1,1,3,3,3 3-hexachloropropane, 1,1,1,2,3,3,3-heptachloropropane etc. can be mentioned.
- the saturated compound represented by the said General formula (1) used here is a saturated compound represented by the said General formula (1 ') manufactured by 1st reaction as mentioned above.
- the specific example of the unsaturated compound represented by the above general formula (2) obtained by the second reaction corresponds to the order of the compounds listed above as the specific example of the saturated compound represented by the above formula (1)
- 1,1-dichloropropene, 1,1,2-trichloropropene, 1,1,3-trichloropropene, 1,1,2,3-tetrachloropropene, 1,1,3,3-tetrachloro Propene, 1,1,2,3,3-pentachloropropene, 1,1,3,3,3-pentachloropropene and 1,1,2,3,3,3-hexachloropropene are exemplified.
- the second reaction in the present invention can be performed by a method of heating the saturated compound represented by the above general formula (1) at a temperature above its thermal decomposition temperature.
- the thermal decomposition reaction is preferably carried out in the gas phase, and the reaction system may be either a flow system or a batch system. However, since the thermal decomposition reaction of the second reaction in the present invention can achieve high reaction conversion rate in a short reaction time, it is preferable from the aspect of reaction efficiency that it depends on the flow system.
- the heating temperature is preferably 300 to 600 ° C., more preferably 350 to 550 ° C.
- the heating time (staying time) is, for example, preferably 1 to 10 seconds, more preferably 1 to 5 seconds, and still more preferably 1 to 3 seconds.
- the heating in the second reaction of the present invention can be carried out by a known method.
- a reaction tube (cracking furnace) provided with a heating device on the outer wall is suitable.
- a material which comprises a reactor quartz, a ceramic, a metal etc. can be illustrated, for example.
- a heating apparatus a burner, an electric heater, a high frequency heating apparatus etc. can be used, for example.
- a method of supplying the saturated compound represented by the above formula (1) to the reactor for example, a method of vaporizing the compound by a vaporizer and introducing it as a gas into the reactor, or spraying a liquid compound A method such as introducing into a reactor can be adopted.
- a method of vaporizing the compound by a vaporizer and introducing it as a gas into the reactor, or spraying a liquid compound A method such as introducing into a reactor can be adopted.
- only the saturated compound represented by the above formula (1) may be supplied to the reactor, or a mixture of the saturated compound represented by the above formula (1) and a suitable dilution gas may be introduced. Also good.
- a dilution gas used here it is preferable to use an inert gas, for example, nitrogen, argon, helium etc.
- the dilution gas can be preferably used, but nitrogen is particularly preferably used from the viewpoint of the cost required for the dilution gas preferable.
- the oxygen concentration in the dilution gas is preferably adjusted to 1% by weight or less.
- the product obtained by the dehydrochlorination step of the second reaction contains water, an extra purification step is required for its removal, which is an economic disadvantage.
- the reaction pressure in the second reaction is not particularly limited, and may be carried out under reduced pressure or increased pressure. Although it is possible to make the reactor smaller under pressure, the higher the reaction pressure, the more likely the deposition of carbon on the pipe. In the present invention, by carrying out the second reaction under normal pressure, it is possible to carry out dehydrochlorination by thermal decomposition extremely efficiently, so there is little merit in selecting the reaction under pressurized conditions.
- the thermal decomposition in the second reaction proceeds sufficiently rapidly under noncatalytic conditions, but a fixed bed or fluidized bed catalyst bed is provided in the reactor, and the thermal decomposition reaction is heated in the presence of a catalyst. You may go by. This is preferable because high conversion can be achieved even at relatively low temperatures within the above preferred temperature range.
- oxides such as a silica, an alumina, a titania, etc., activated carbon etc. can be mentioned, for example. It is preferable that the gas after the second reaction be rapidly cooled from the viewpoint of reducing by-products.
- the gas discharged from the pyrolysis reactor at the above pyrolysis temperature is preferably cooled to a temperature of less than 300 ° C., more preferably less than 200 ° C., particularly preferably less than 100 ° C., preferably within 2 seconds It is advantageous to do.
- This cooling can be performed by a known method such as a method of heat exchange with a supplied gas, a method of spraying droplets of a dehydrochlorinated hydrolyzate, and cooling by the latent heat of vaporization thereof.
- the reaction mixture of the second reaction (exhaust gas from the reactor) obtained as described above may be used as it is for the third reaction, or hydrogen chloride produced (by-produced) in the second reaction may be used.
- the third reaction in the present invention is a chlorination step in which the unsaturated compound represented by the above general formula (2) and chlorine are reacted to obtain the saturated compound represented by the above general formula (3).
- the third reaction rapidly proceeds only by contacting the unsaturated compound represented by the above general formula (2) with chlorine.
- Specific examples of the saturated compound represented by the above general formula (3) include 1,1,1,2,3-pentachloropropane, 1,1,1,2,3,3-hexachloropropane, 1,1,1,3.
- 1,2,3,3,3-heptachloropropane 1,1,1,2,2,3,3-heptachloropropane, 1,1,1,2,2,2,3,3,3-octachloropropane, etc. It can be mentioned. Among these, 1,1,1,2,3-tetrachloropropane is useful as an intermediate of various compounds and is preferable. It will be obvious to those skilled in the art which product can be obtained as the saturated compound represented by the general formula (3) depending on the unsaturated compound represented by the general formula (2) to be used. By performing the third reaction in the present invention following the second reaction, the following merits can be obtained. First, there is the merit that the product of the second reaction in the present invention contains almost no water.
- the second reaction since the second reaction, the dehydrochlorination step, is carried out preferably by thermal decomposition reaction in the gas phase, the exhaust gas of the second reaction contains almost no water.
- the present invention has the advantage of not requiring extensive dehydration equipment before or after the third reaction.
- the product of the second reaction contains almost no iron.
- the second reaction since the second reaction is preferably carried out in the gas phase, it is one of the merits that iron is hardly mixed into the product of the second reaction.
- the third reaction in the present invention may be carried out in the gas phase without condensing the unsaturated compound represented by the above general formula (2), or may be carried out in the liquid phase by condensing the same.
- the third reaction in the present invention is carried out in the gas phase, either of the flow system and the batch system may be used, but since the method of the present invention can achieve high reaction conversion in short reaction time, the flow system is used. Is preferred from the viewpoint of the reaction efficiency.
- the third reaction in the gas phase in the method of the present invention, it can be carried out by heating the mixed gas of the exhaust gas of the second reaction and chlorine gas at a predetermined temperature for a predetermined time.
- the use ratio of chlorine gas is preferably 0.9 to 1 mole of the dehydrochlorinated substance (the unsaturated compound represented by the above general formula (2)) contained in the reaction mixture of the second reaction. It is 2.0 moles, more preferably 1.0 to 1.5 moles. If this value is less than 0.9 mol, the reaction conversion rate of the third reaction may be impaired, while if it exceeds 2.0 mol, there may be a disadvantage that the formation of by-products increases, Not desirable.
- the reaction temperature of the chlorination reaction in the gas phase is from the boiling point of the unsaturated compound represented by the above formula (2) to 300 ° C. or less from the viewpoint of maintaining the gas phase and suppressing the formation of by-products.
- the boiling point is at or above 280 ° C.
- the reaction time is preferably 1 to 60 seconds, more preferably 1 to 40 seconds.
- the reaction temperature is not particularly limited as long as the unsaturated compound represented by the above general formula (2) used as a raw material does not vaporize.
- the temperature is 120 ° C. or less to reduce by-products.
- the amount of chlorine to be supplied is preferably 0.9 to 2.0 with respect to 1 mole of the unsaturated compound represented by the above general formula (2), from the viewpoint of increasing the conversion rate and selectivity.
- the reaction time is preferably 1 to 10 hours.
- a method of using chlorine as a fine bubble blowing method, irradiating ultraviolet light, and the like are also preferable embodiments.
- the exhaust gas or the discharge liquid of the third reaction obtained as described above is a crude product containing the saturated compound represented by the general formula (3), which is the target product, at a high concentration.
- the crude product is, if necessary, purified by a known method, and made into a product.
- distillation purification can be mentioned.
- phenol such as p-methoxyphenol, o-t-butylphenol, eugenol, o-allylphenol etc. It is preferable to carry out distillation after adding a derivative, particularly a phenol derivative having an allyl group.
- the mixture of the unsaturated compound represented by the above formula (2) and hydrogen chloride was supplied to the third reaction for chlorination without separating hydrogen chloride by-produced from the exhaust gas obtained in the second reaction
- the crude product obtained by the third reaction contains hydrogen chloride and optionally unreacted chlorine.
- the above-mentioned (A) hydrogen chloride separation step can be carried out by a known method capable of separating hydrogen chloride, and optionally hydrogen chloride and chlorine, from the crude product.
- a method of condensing a saturated compound represented by the above formula (3) in a crude product to separate hydrogen chloride or hydrogen chloride and chlorine as a gas there can be mentioned, for example, a method of condensing a saturated compound represented by the above formula (3) in a crude product to separate hydrogen chloride or hydrogen chloride and chlorine as a gas.
- known methods are adopted without particular limitation.
- a catalytic oxidation method and the like can be mentioned.
- a method of passing hydrogen chloride or hydrogen chloride and chlorine in a gaseous state, for example, through a chromia or ruthenium-supported titania catalyst layer can be mentioned.
- the reaction temperature at this time can be, for example, 200 to 450 ° C.
- 1,1,1,2,3-pentachloropropane uses ethylene as an unsaturated compound represented by the above general formula (0) and adds carbon tetrachloride to it to produce 1,1,1,3-tetra
- the first reaction to obtain chloropropane The 1,1,1,3-tetrachloropropane is dehydrochlorinated to form at least one chlorinated saturated hydrocarbon selected from the group consisting of 3,3,3-trichloropropene and 1,1,3-trichloropropene
- a second reaction to obtain A third reaction adding chlorine to the trichloropropene to give 1,1,1,2,3-pentachloropropane
- the reaction mixture obtained by the first reaction contains 1,1,1,3-tetrachloropropane as a main component, but in addition to this, carbon tetrachloride, hexachloroethane, tetrachloroethylene, 1,1,1,3 as impurities.
- Trichloropropene, 1,1,1,3,3-pentachloropropane and the like in the purification after the first reaction, it is preferable to reduce the concentration of hexachloroethane, tetrachloroethylene and 1,1,1,3,3-pentachloropropane, among the above-mentioned impurities, as much as possible.
- the total weight of hexachloroethane, tetrachloroethylene and 1,1,1,3,3-pentachloropropane is 1 weight based on the total weight of these and 1,1,1,3-tetrachloropropane. % Or less is preferable, and 0.9 wt% or less is more preferable.
- 1,1,1,2,3-pentachloropropane obtained as a target product after the third reaction is And impurities substantially difficult to separate.
- 1,1,1,2,3-pentachloropropane obtained by the method of the present invention can be used as a product as it is, or it can be extremely purified by performing a simple purification method thereafter.
- 1,1,1,2,3-pentachloropropane In order to achieve the object of the present invention, the present inventors examined in detail the trends of impurities generated in each of the first, second and third reactions.
- the boiling points of the target product and impurities in the first reaction under normal pressure are shown in Table 1
- the boiling points of the target product and impurities in the third reaction under normal pressure are shown in Table 2.
- the distillation column to be used may be any column known in the art as being used for the rectification of a liquid substance under normal temperature and pressure. And column or packed columns may be mentioned as preferred. It is preferable that the number of stages of the column or the equivalent number of columns of the packed column converted into the column is 2 to 20.
- the distillation column may be performed in one column or several columns. Among these, an embodiment in which several distillation columns are used is a preferable embodiment because the removal efficiency of iron chloride and the like dissolved in the reaction mixture is improved.
- a cross flow tray, a shower tray, etc. can be used as the above-mentioned column.
- a cross flow tray for example, perforated plate tray, bubble bell tray, valve tray, turbo grid tray, etc .;
- a turbo grid tray, a ripple tray, etc. can be mentioned, for example.
- a distillation column packed with ordered packing or irregular packing can be used. Examples of the irregular packing include Raschig ring, Bell saddle, McMahon, Nuttering, Polling, Cascade miniring, Helipak and the like.
- the distillation temperature is preferably set to 60 to 140 ° C., from the viewpoint that the decomposition of 1,1,1,3-tetrachloropropane can be suppressed, and the control of the distillation pressure is easy, and the like. It is more preferable to set it as ° C.
- the distillation pressure is preferably set to a pressure that can maintain the distillation operation at the above preferable temperature, but can be, for example, 1 to 20 kPa.
- the reaction mixture of the first reaction (crude 1,1,1,3-tetrachloropropane, hereinafter referred to as "crude TCP") is introduced into the distillation column from an appropriate position in the lower part of the distillation column, preferably from the middle stage. .
- the target substance and acid content in the crude TCP rise in the distillation column and the target substance be withdrawn as a column side stream of the distillation column in a stage where the concentration of impurities is reduced to a desired level.
- “extracted as a column side stream” refers to a liquid at least one lower stage, preferably 3 to 10 lower stages than the uppermost stage of the distillation column and in a stage sufficient to remove impurities. It refers to removing the purified product from the phase.
- the overhead stream is separated into the condensed liquid and the non-condensed gas through the condenser, and the non-condensed gas (containing the acid content at a high concentration) It is preferable to bleed off and all or part of the condensate is refluxed to a stage higher than the stage for drawing out the purified product.
- the amount of withdrawal of the condensate is the amount of distillate and the amount of reflux is the amount of reflux
- the reflux ratio defined by the amount of reflux / the amount of distillate is preferably 0.1 to 50. And 1 to 40 are more preferable.
- introducing an inert gas from the lower stage of the distillation column rather than the stage for introducing crude TCP can be suitably adopted to promote the stripping effect of the acid in the liquid in the column.
- an inert gas used here nitrogen, helium etc. can be mentioned, for example.
- the amount of inert gas introduced is 1 ⁇ 10 6 as the volume ratio to the entire rising gas volume in the column.
- purified TCP 1,1,1,3-tetrachloropropane
- purified TCP substantially contains hexachloroethane, tetrachloroethylene and 1,1,1,3,3-pentachloropropane as impurities.
- 1,1,1,2,3-pentachloropropane after the third reaction can be used as it is as a product, or it can be made a product of extremely high purity only by performing simple purification.
- a simple purification to be carried out for example, distillation purification, deacidification treatment, etc. can be mentioned.
- distillative purification can be carried out according to the above-mentioned as distillative purification preferably carried out after the above first reaction
- 1,1,1,2,3-pentachloropropane obtained by the method of the present invention is Since it does not contain impurities close to the boiling point, the number of stages or the equivalent number of stages of the distillation column used for purification may be about 2 to 20.
- This distillation is preferably carried out after adding the above-mentioned phenol derivative, particularly a phenol derivative having an allyl group, to crude 1,1,1,2,3-pentachloropropane.
- the above deacidification treatment can be carried out by contacting 1,1,1,2,3-pentachloropropane with a suitable deacidification agent.
- a deoxidizer used here a silica, an alumina, a silicate etc. can be mentioned, for example.
- silica, an amorphous silica, crystalline silica, a silica hydrate etc. can be mentioned, for example, A silica gel, a diatomaceous earth etc. can be mentioned as the specific example.
- alumina an amorphous alumina, a crystalline alumina, an alumina hydrate etc.
- the said silicate can be used without a restriction
- alkali silicates such as sodium silicate
- alkaline earth metal silicates such as calcium silicate and magnesium silicate
- aluminum silicates such as silica-alumina gel.
- zeolite, synthetic zeolite, kaolinite, activated clay, halloysite, montmorillonite, allophane, bentonite and the like can be used.
- the deoxidizer for example, a commercially available product such as Secard KW (manufactured by Shinagawa Kasei Co., Ltd.) may be used.
- a shape of a deoxidizer appropriate things, such as a powdery form, a granular form, and a granular form, can be used without particular restriction.
- the particle diameter of the deoxidizer can be preferably small from the viewpoint of contact efficiency, and the average particle diameter is preferably 9 mm or less, and more preferably 0.3 to 5 mm.
- the space velocity when contacting 1,1,1,2,3-pentachloropropane with a deoxidizer is 0.001 to 20 hours. -1 Is preferably 0.005 to 10 hours. -1 It is more preferable to The contact temperature is preferably 5 to 80 ° C., and more preferably 10 to 70 ° C.
- Example 1-1 (1) First reaction 769 g of carbon tetrachloride, 4.5 g of triethyl phosphate and 14 g of iron powder (manufactured by Wako Pure Chemical Industries, Ltd., reduced iron) were charged into a SUS-made autoclave equipped with a stirrer, and the temperature was adjusted The reaction was started by setting ethylene at 110 ° C. and introducing ethylene so as to maintain a reaction pressure of 0.4 MPaG.
- Example 1-2 (1) First reaction 769 g of carbon tetrachloride, 4.5 g of triethyl phosphate and 14 g of iron powder (reduced iron, manufactured by Wako Pure Chemical Industries, Ltd.) were charged in a SUS-made autoclave equipped with a stirrer, and the temperature was 110 The reaction was initiated by introducing vinyl chloride at a reaction pressure of 0.4 MPaG set at ° C. After 6 hours, the autoclave was cooled and the reaction solution after reaction was recovered and analyzed by gas chromatography to determine the conversion and selectivity. As a result, the conversion on a vinyl chloride basis was 81%, and the selectivity to 1,1,1,3,3-pentachloropropane was 91%.
- a mixed gas (1,1,3,3-tetrachloropropene: chlorine 100: 110 (molar ratio)) in which chlorine gas is joined to the above product gas supply line, a reaction tube with an inner diameter of 4.35 mm and a length of 300 mm
- the crude chloropropane obtained from the outlet of the reaction tube is liquefied by cooling to 0 ° C. to remove hydrogen chloride and unreacted chlorine.
- the target 1,1,1,2,3,3-hexachloropropane was obtained.
- a part of the above-mentioned liquefied crude chloropropane was taken out and analyzed by gas chromatography to determine the conversion and selectivity.
- Example 1-3 The same procedure as in Example 1-1 is carried out except that the third reaction (chlorination step) is carried out as follows, to obtain 1,1,1,2,3-pentachloropropane. The That is, after trichloropropene produced by the second reaction (dehydrochlorination step) is liquefied to separate hydrogen chloride, it is supplied to the third reaction and chlorination reaction is carried out at a temperature of 80 ° C. while being supplied at 360 NmL / min of chlorine.
- Example 2-1 Charge 1,560 g of carbon tetrachloride, 8 g of triethyl phosphate and 4 g of pure iron powder for chemical reaction in an autoclave, set the temperature to 110 ° C., introduce ethylene so that the reaction pressure is 0.4 MPaG, and perform addition reaction Started. Some time after the start of ethylene introduction, the consumption of ethylene increased sharply. The average ethylene consumption rate for 60 minutes from the start of ethylene introduction was 950 NmL / min.
- Example 2-2 Charge 1,560 g of carbon tetrachloride, 8 g of triethyl phosphate and 4 g of pure iron powder for chemical reaction in an autoclave, set the temperature to 110 ° C., introduce ethylene so that the reaction pressure is 0.4 MPaG, and perform addition reaction Started. Some time after the start of ethylene introduction, the consumption of ethylene increased sharply. The average ethylene consumption rate for 60 minutes from the start of ethylene introduction was 950 NmL / min. Sixty minutes after the start of ethylene introduction, the ethylene consumption rate fell below 400 NmL / min.
- triethyl phosphate was continuously added continuously for 250 minutes at an addition rate of 0.016 g / min from 60 minutes after the start of ethylene introduction (addition amount of triethyl phosphate is 4 g in total). After 6 hours from the start of ethylene introduction, the autoclave is cooled, and the reaction mixture after completion of the reaction is recovered and analyzed by gas chromatography. The reaction conversion rate based on carbon tetrachloride used is 93%, 1, 1 The selectivity of 1,3-tetrachloropropane was 90%.
- Reference Example 2-1 Charge 1,560 g of carbon tetrachloride, 12 g of triethyl phosphate and 60 g of pure iron powder for chemical reaction in an autoclave, set the temperature to 110 ° C., introduce ethylene so that the reaction pressure is 0.4 MPaG, and perform addition reaction Started. After 6 hours from the start of ethylene introduction, the autoclave is cooled, and the reaction mixture after completion of the reaction is recovered and analyzed by gas chromatography. The reaction conversion rate based on carbon tetrachloride used is 85%, 1, 1 The selectivity of 1,3-tetrachloropropane was 86%.
- Reference Example 2-2 Add 1,560 g of carbon tetrachloride, 14 g of dibutyl phosphate and 60 g of pure iron powder for chemical reaction in an autoclave, set the temperature to 100 ° C, introduce ethylene so that the reaction pressure is 0.4 MPaG, and add the reaction Started. After 6 hours from the start of ethylene introduction, the autoclave is cooled, and the reaction mixture after completion of the reaction is recovered and analyzed by gas chromatography. The reaction conversion based on carbon tetrachloride used is 21%, 1, 1 The selectivity of 1,3-tetrachloropropane was 85%.
- Example 3-1 ⁇ Example of three-step reaction in which distillation purification of the product is performed after the first reaction and before the second reaction>
- Example 3-1 (1) First reaction 1,560 g of carbon tetrachloride, 8 g of triethyl phosphate and 4 g of pure iron powder for chemical reaction are charged in an autoclave, the temperature is set to 110 ° C., and the reaction pressure is 0.4 MPaG ethylene. Was introduced to initiate the first reaction (addition reaction). One hour after the start of ethylene introduction, 4 g of triethyl phosphate was additionally added.
- the autoclave was cooled after 6 hours from the start of ethylene introduction, and the reaction mixture after completion of the reaction was recovered and analyzed by gas chromatography to find that the reaction conversion rate based on carbon tetrachloride used was 85%, 1, 1, The selectivity of 1,3-tetrachloropropane was 90%.
- (2) Purification after the First Reaction The reaction mixture obtained by the first reaction was subjected to two-stage distillation purification as follows. As a first distillation, batch distillation is performed at a distillation pressure of 10 kPa and a distillation temperature of 95 ° C without refluxing in a distillation column with a column diameter of 30 mm ⁇ packed with glass packing made by Shibata Scientific Co., Ltd. at a height of 500 mm.
- the purity of 1,1,1,3-tetrachloropropane in the purified fraction obtained here was 98.5%.
- a reflux ratio of 3 and a distillation pressure of 10 kPa in which the purified fraction obtained above is filled with glass packing made by Shibata Scientific Co., Ltd. at a height of 1,000 mm as the second distillation.
- Batch distillation was performed at a distillation temperature of 95 ° C.
- the purity of purified 1,1,1,3-tetrachloropropane which is a distillate component obtained by removing 4 wt% of the low boiling fraction and 6 wt% of the bottom high boiling residue was almost 100%. .
- Second reaction The purified 1,1,1,3-tetrachloropropane obtained above is subjected to a second reaction (dehydrochlorination) under the conditions of a residence time of 2.5 seconds and a reaction temperature of 500 ° C.
- the conversion ratio of the reaction in this dehydrochlorination reaction was 99.7%, and the selectivity of the desired product 1,1,3-trichloropropene was 99.0%.
- the above conversion and selectivity were calculated based on 1,1,1,3-tetrachloropropane, respectively.
- Third reaction In the third reaction (chlorination reaction), the exhaust gas of the second reaction is cooled to 150 ° C., mixed with chlorine gas whose flow rate is adjusted, and then supplied to the reactor for a residence time of 1.
- the chlorination reaction was carried out under the conditions of 24 seconds to obtain crude 1,1,1,2,3-pentachloropropane.
- the flow rate of chlorine gas was 1.1 m 3 (0 ° C., 98 kPa) with respect to 1 m 3 (0 ° C., 98 kPa) of the exhaust gas of the second reaction.
- the reaction conversion rate in this chlorination reaction was 99.0%, and the selectivity of the target product 1,1,1,2,3-pentachloropropane was 95.0%.
- the above conversion and selectivity were calculated based on 1,1,3-trichloropropene, respectively.
- Example 3-2 In the above Example 3-1, a column in which a crude outlet of the distillation column of crude 1,1,1,2,3-pentachloropropane (top of column) was packed with SEKAD KW (trade name, manufactured by Shinagawa Kasei Co., Ltd.) was used.
- Example 3-1 In Example 3-1, the distillation pressure of the distillation column used for purification after the third reaction is 5 kPa, and the distillation temperature is 110 ° C.
- Example 3-1 An effluent component of purified 1,1,1,2,3-pentachloropropane was obtained.
- the trend of each component in the present example was the same as in Example 3-1.
- the acid content of the purified 1,1,1,2,3-pentachloropropane was 90 ppm by weight.
- Comparative Example 3-1 The same procedure as in Example 1 is carried out except that distillation purification after the first reaction is not carried out in the above-mentioned Example 1, and the outflow component of the purified 1,1,1,2,3-pentachloropropane from the distillation outlet at the top of the column I got The acid content of this purified 1,1,1,2,3-pentachloropropane was 210 ppm by weight.
- Table 4 shows the trend of each component in this comparative example.
- Example 4-1 The flow rate of 1,1,1,3-tetrachloropropane vaporized with a preheater at 200 ° C is adjusted to 500 ° C with an electric furnace so that the residence time is 2.5 seconds based on the inlet gas flow rate. Gas was introduced into a heated reaction tube (material: SUS316, inner diameter: 4.35 mm, length: 300 mm), and a thermal decomposition reaction was performed in the gas phase at normal pressure.
- Example 4-2 Thermal decomposition was performed in the same manner as in Example 4-1 except that the temperature of the reaction tube was changed to 450 ° C. The analysis results of conversion and selectivity are shown in Table 5.
- Example 4-3 Thermal decomposition was performed in the same manner as in Example 4-1 except that the temperature of the reaction tube was changed to 550 ° C. The analysis results of conversion and selectivity are shown in Table 5.
- Example 4-4 Thermal decomposition was performed in the same manner as in Example 4-1 except that the staying time was set to 1.25 seconds. The analysis results of conversion and selectivity are shown in Table 5.
- Example 4-5 In the above Example 4-1, it was carried out except using 1,1,1,2-tetrachloropropane as a raw material instead of 1,1,1,3-tetrachloropropane and setting the temperature of the reaction tube to 450 ° C. The thermal decomposition was carried out in the same manner as in Example 4-1. The analysis results of the conversion of 1,1,1,2-tetrachloropropane and the selectivity to 1,1,2-trichloropropene are shown in Table 5.
- Example 4-6 Example 4-1 except that in the above Example 4-1 the raw material 1,1,1,3-tetrachloropropane was diluted with nitrogen gas to a concentration of 75% and supplied into the reaction tube, Thermal decomposition was performed in the same manner as in The analysis results of conversion and selectivity are shown in Table 5.
- Example 4-7 The flow rate of 1,1,1,3-tetrachloropropane vaporized with a preheater at 200 ° C is adjusted to 450 ° C with an electric furnace so that the residence time is 9.0 seconds based on the inlet gas flow rate. Gas was introduced into a heated reaction tube (material: SUS316, inner diameter: 7.3 mm, length: 600 mm), and a thermal decomposition reaction was performed in the gas phase at normal pressure.
Abstract
Description
このような塩素化炭化水素の製造方法としては、例えば炭素数2の不飽和化合物に四塩化炭素を付加して炭素数3の塩素化飽和炭化水素を得る第一反応と、
該塩素化飽和炭化水素を脱塩化水素して炭素数3の塩素化不飽和炭化水素を得る第二反応と、
該塩素化不飽和炭化水素にさらに塩素を付加して炭素数3の塩素化飽和炭化水素を得る第三反応と
からなる三段階反応が知られている。例えば特公平2−47969号公報には、エチレンと四塩化炭素との付加反応を、金属鉄とホスホリル化合物とからなる相間移動触媒の存在下で行って1,1,1,3−テトラクロロプロパンとし(第一反応)、
次いでこれを第4級アンモニウム塩又は第4級ホスホニウム塩の存在下に水酸化ナトリウム水溶液中、40~80℃の温度で処理して脱塩化水素することによって1,1,3−トリクロロプロペン及び3,3,3−トリクロロプロペンからなるトリクロロプロペン混合物を得て(第二反応)、
さらに該トリクロロプロペン混合物に、塩素の存在下で紫外光を照射して塩素化することによって1,1,1,2,3−ペンタクロロプロパンとする(第三反応)方法が記載されている。
当業者は、適当な原料化合物を選択したうえで、上記三段階反応の一部又は全部を行うことにより、所望の炭素数及び塩素数を有する塩素化された飽和又は不飽和の炭化水素を得ることができる。
しかしながら上記の如き公知の塩素化炭化水素製造方法は、その各段階においてそれぞれ以下のような改善すべき問題点を有している。
第一反応においては、使用される鉄−ホスホリル化合物触媒が、調製直後こそ高い活性を示すものの、その後の活性は経時的に急激に落ちる問題があるため、所望の高転化率を得るためには多量の触媒を使用する必要がある。しかし、触媒を多量に使用すると、反応初期における反応の立ち上がりが急激となって反応の制御が困難となり、目的物の反応収率が損なわれる場合がある。また、多量の触媒を使用すると、廃触媒の処理に労力及びコストがかかり、製造コストの削減の障害ともなっている。さらに、鉄−ホスホリル化合物触媒の調製に使用される金属鉄は、保存中に表面が徐々に酸化され、その酸化状態によって反応の初期速度が大きく変わるとの問題がある。
次に第二反応においては、多量に消費される水酸化ナトリウムのコストの問題があるほか、廃棄されることとなる水相中に溶解している有機塩素化合物の処理に多大の労力を要する。
第三反応の光塩素化反応において十分に高い反応転化率を実現するためには、反応時間(滞留時間)を長くすることを要する。
さらに、塩素化炭化水素として例えば1,1,1,2,3−ペンタクロロプロパンを高純度で得たいときには、第三反応の後に生成物の蒸留精製を行う方法によることが一般的である。しかしながらこのような塩素数の多い塩素化炭化水素を蒸留精製する場合、不純物との分離性が極めて悪い。そのため、高純度品を得るための蒸留塔としては極めて高性能のものが必要となるほか、精密蒸留を行うには長時間を要することとなり、コスト上の問題がある。
上記のように、塩素化炭化水素を製造するための三段階反応において、
第一反応においては、触媒の使用量を削減しつつ、高い転化率で目的生成物を安定した反応速度で効率的に付加反応を行う方法が;
第二反応においては、高価なアルカリ源を使用せず、また処理すべき副生成物の生成が抑制された脱塩化水素反応を行う方法が;
第三反応においては、より効率性の高い塩素化反応を行う方法が、それぞれ熱望されている。
本発明によれば、上記目的は、
下記一般式(1)で表される飽和化合物を熱分解して下記一般式(2)で表される不飽和化合物を得る熱脱塩化水素工程を経ることを特徴とする、塩素化炭化水素を製造するための方法によって達成される。この脱塩化水素工程の後、さらに下記一般式(2)で表される不飽和化合物と塩素とを反応させて下記一般式(3)で表される飽和化合物を得る塩素化工程を行うことができる。
CCl3−CCl2−mHm−CCl3−nHn (1)
CCl2=CCl2−mHm−1−CCl3−nHn (2)
CCl3−CCl3−mHm−1−CCl3−nHn (3)
(式中、mは1又は2であり、nは0~3の整数である。)
上記一般式(1)で表される飽和化合物は、好ましくは四塩化炭素と下記一般式(0)で表される不飽和化合物とを、液相の反応系中で鉄−リン酸エステル触媒の存在下に付加させる付加反応工程によって得られた下記一般式(1’)で表される飽和化合物である。
CCl2−mHm=CCl2−pHP (0)
CCl3−CCl2−mHm−CCl3−pHp (1’)
(式中、mは1又は2であり、pは0~2の整数であり、ただしm≧pである。)
以下、本発明について、反応の各段階ごとに分けて順次に詳説する。
<第一反応>
本発明における第一反応は、四塩化炭素と上記一般式(0)で表される不飽和化合物との付加反応によって、上記一般式(1’)で表される飽和化合物を得る、付加反応である。本反応は、好ましくは液相の反応系中で、触媒の存在下に行われる。
上記一般式(0)で表される不飽和化合物としては、エチレン、塩化ビニル、1,1−ジクロロエチレン、1,2−ジクロロエチレン、1,1,2−トリクロロエチレン等を挙げることができ、これらのうち、エチレン又は塩化ビニルを用いることが好ましい。
本反応により得られる上記一般式(1’)で表される飽和化合物であるクロロプロパンの種類は、原料として使用する上記一般式(0)で表される不飽和化合物の種類に依存する。例えば原料としてエチレンを使用した場合には上記一般式(1’)で表される飽和化合物として1,1,1,3−テトラクロロプロパンが得られる。また原料として塩化ビニルを使用した場合には上記一般式(1’)で表される飽和化合物として1,1,1,3,3−ペンタクロロプロパンが得られる。上記一般式(0)で表される不飽和化合物としてその他の化合物を用いた場合に、上記一般式(1’)で表される飽和化合物としていかなる生成物が得られるかは、当業者には自明であろう。
使用される触媒としては、例えば鉄−リン酸エステル触媒、鉄−非プロトン極性溶媒触媒、銅−アミン触媒等を挙げることができるが、これらのうち鉄−リン酸エステル触媒が好ましい。
本反応は、液相に鉄−リン酸エステル触媒が存在する状態で行われる。この鉄−リン酸エステル触媒は、液相の反応系中(すなわち液体状の四塩化炭素中)で、所定量の鉄及び所定量のリン酸エステルを接触させることにより調製される。鉄とリン酸エステルとの接触は、反応開始前に鉄及びリン酸エステルの各全量を反応系中に一度に投入して行う方法によるか、
あるいは鉄の全量及びリン酸エステルの一部を反応開始前に添加し、残余のリン酸エステルは付加反応の進行中に追加添加することにより行うことができる。ここで、「反応開始前」とは、四塩化炭素中に上記一般式(0)で表される不飽和化合物を導入する前の時点をいう。
ここで使用される鉄としては、例えば金属鉄、純鉄、軟鉄、炭素鋼、フェロシリコン鋼、鉄を含む合金(例えばステンレス鋼等)等を挙げることができる。鉄の形状としては、例えば粉末状、粒状、塊状、棒状、球状、板状、繊維状等の任意の形状であることができるほか、これらを用いてさらに任意の加工をした金属片、蒸留充填物等であってもよい。前記加工金属片としては、例えばコイル、網、スチールウール、その他の不定形片状を;前記蒸留充填物としては、例えばラシヒリング、ヘリックス等を、それぞれ挙げることができる。これらのいずれの形態であっても使用することができるが、リン酸エステル及び反応物との接触面積を十分に確保する観点から、粉末状又は繊維状であることが好ましい。同様の観点から、窒素を吸着質としてBET法により測定した鉄の比表面積は0.001~5m2/gであることが好ましい。
反応開始前にリン酸エステルを一括して添加する場合における鉄の使用量としては、高い反応転化率及び高い選択率を両立するとの観点から、使用する四塩化炭素の1モルに対して、0.001モル以上とすることが好ましく、0.005モル以上とすることがより好ましく、0.01モル以上とすることがさらに好ましく、特に0.05モル以上とすることが好ましい。鉄の使用量の上限は特に限定されない。鉄の使用量を多くしても、活性及び選択性にはほとんど影響しないが、反応に関与せずに無駄となる鉄が多くなる点で、経済上不利益となる。かかる観点から、鉄の使用量は使用する四塩化炭素の1モルに対して、10モル以下とすることが好ましく、5モル以下とすることがより好ましく、1モル以下とすることがさらに好ましく、特に0.1モル以下とすることが好ましい。
上記リン酸エステルとしては、例えば下記一般式(4)
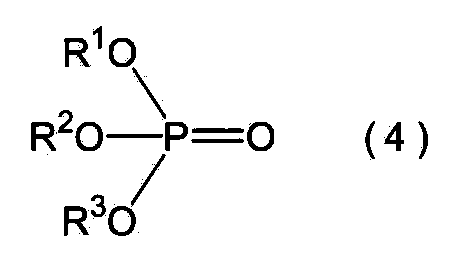
で表される化合物を挙げることができ、その具体例として例えばリン酸トリメチル、リン酸トリエチル、リン酸トリプロピル、リン酸トリブチル、リン酸ジエチル、リン酸ジブチル、リン酸モノフェニル、リン酸モノブチル、リン酸ジメチルフェニル、リン酸ジエチルフェニル、リン酸ジメチルエチル、リン酸フェニルエチルメチル等を挙げることができる。これらのうち、上記一般式(4)において、R1,R2及びR3のすべてが炭素数1~4のアルキル基であるリン酸トリアルキルエステルが好ましく、特にリン酸トリメチル、リン酸トリエチル、リン酸トリプロピル又はリン酸トリブチルが好ましい。
リン酸エステルの使用量は、高い転化率及び高い選択率を担保するとの観点から、使用する四塩化炭素の1モルに対して、0.001モル以上とすることが好ましく、特に0.002以上とすることが好ましい。リン酸エステルの使用量の上限は特に限定されないが、使用量を過度に多くすると、反応に関与せずに無駄となるリン酸エステルが多くなる点で、経済上不利益となる。かかる観点から、リン酸エステルの使用量は、四塩化炭素の1モルに対して、5モル以下とすることが好ましく、1モル以下とすることがより好ましく、0.5モル以下であってもよい。
第一反応の反応温度は、高い転化率と高い選択率とを両立するために、70~180℃とすることが好ましく、90~150℃とすることが更に好ましい。反応圧力は、上記反応温度において反応系が液相を維持し得る圧力であればよく、一般には0.05~3MPaGとすることができ、好ましくは0.1~2MPaGである。反応圧力を0.05MPa未満とすると、液相中における原料(上記式(0)で表される不飽和化合物)の濃度が過小となって反応添加率が不足する場合があり、一方、3MPaを超える圧力では多量体が生成する割合が高くなって選択率が損なわれる場合があり、いずれも好ましくない。
第一反応の反応時間は、2~24時間とすることが好ましく、2~10時間とすることがより好ましい。ここで、反応時間を2時間よりも短くすると反応転化率が不十分となる場合があり、一方、24時間を超えて長く反応を行う実益はない。
本発明においては、上記の如き第一反応において、鉄の全量及びリン酸エステルの一部を反応開始前に添加し、残余のリン酸エステルは付加反応の進行中に追加添加することが、反応の制御性を良好とし、選択率を高くし、そして使用する鉄の量を低減しうる点で好ましい。
反応開始前に一括添加される鉄の量は、反応開始前にリン酸エステルを一括して添加する場合における鉄の使用量の下限として上記した値よりも少なくすることができる。この場合における鉄の使用量は、使用する四塩化炭素の1モルに対して、0.0001モル以上とすることが好ましく、0.0005モル以上とすることがより好ましく、0.001モル以上とすることがさらに好ましく、特に0.005モル以上とすることが好ましい。鉄の使用量の上限は、経済上の観点から設定される。この場合における鉄の使用量は、使用する四塩化炭素の1モルに対して、1モル以下とすることが好ましく、0.5モル以下とすることがより好ましく、0.1モル以下とすることがさらに好ましい。
本反応においては、リン酸エステルは反応開始前にその一部を添加し、残余のリン酸エステルは付加反応進行中に追加添加することが好ましい。リン酸エステルの追加添加は、1回だけ行ってもよく、数回に分割して行ってもよく、あるいは連続的に行ってもよい。数回に分割して行う場合における追加添加の回数としては、2~10回とすることが好ましく、2~6回とすることが好ましい。
リン酸エステルの全使用量(反応開始前添加分及び追加添加分の全部の合計量)は、使用する四塩化炭素の1モルに対して、0.001モル以上とすることが好ましく、特に0.002モル以上とすることが好ましい。追加添加する場合のリン酸エステルの総添加量は特に限定されない。しかしながらこの場合もリン酸エステルの総添加量を過度に多くすると、反応に関与せずに無駄となるリン酸エステルが多くなる点で、経済上不利益となる。かかる観点から、追加添加する場合のリン酸エステルの総添加量は、四塩化炭素の1モルに対して、5モル以下とすることが好ましく、1モル以下とすることがより好ましく、0.5モル以下であってもよい。
リン酸エステルを追加添加する方法においては、リン酸エステルの使用量を、従来技術、例えば上記特公平2−47969号公報に記載された方法よりも少ない量としても、目的の化合物を、より高い転化率及び安定した反応速度にて効率的に製造することができる利点を有する。
本反応の好ましい態様においては、リン酸エステルは、その一部が反応開始前に添加される。反応開始前におけるリン酸エステルの添加量としては、使用する四塩化炭素の1モルに対して0.0005モル以上とすることが好ましく、0.001モル以上とすることがより好ましい。反応開始前に添加されるリン酸エステルの上限値は、追加添加の態様(追加添加を1回だけ行うか、数回に分割して行うか、あるいは連続的に行うか)によらず、また数回に分割して追加添加する場合にはその添加回数によらず、リン酸エステルの全使用量の80%以下とすることが好ましく、70%以下とすることがより好ましい。反応開始前におけるリン酸エステルの添加量を上記の如き範囲とすることにより、反応を安定して立ち上げることができ、反応のコントロールが容易となり、結果として高い転化率を達成することができることとなる。
かくして開始された付加反応は、上記一般式(0)で表される不飽和化合物の消費速度を連続的にモニターしながら行うことが好ましい。この不飽和化合物の消費速度の連続的モニターは、例えば気相流通下における液相バッチ反応において、気体状で連続的に供給される不飽和化合物の量と、適当な反応圧力を維持するために気相から排出される不飽和化合物の量とを随時比較することにより行うことができる。そして、該消費速度が初期値から所定の程度に低下したときに、リン酸エステルの追加添加が行われ、あるいは開始される。
リン酸エステルの追加添加を1回だけ行う場合には、不飽和化合物の消費速度が反応開始後60分間における平均消費速度の好ましくは5~50%、より好ましくは10~40%となったときに、リン酸エステルの残りの全量が追加添加される。この追加添加により、一旦減少した不飽和化合物の消費速度が回復し、以後、該消費速度が再び漸減しながら残余の付加反応が進行していくこととなる。
リン酸エステルの追加添加を数回に分割して行う場合には、消費速度が反応開始後60分間における平均消費速度の好ましくは5~50%、より好ましくは10~40%となったときに、第1回目のリン酸エステルの追加添加が行われる。この第1回目の追加添加により、一旦減少した不飽和化合物の消費速度が回復し、以後、該消費速度が再び漸減して行く。そして、不飽和化合物の消費速度が再度反応開始後60分間における平均消費速度の好ましくは5~50%、より好ましくは10~40%となったときに、第2回目以降のリン酸エステルの追加添加が行われる。この追加添加により、不飽和化合物の消費速度は再度回復する。以降、さらに継続して上記一般式(0)で表される不飽和化合物の消費速度をモニターし、所定の回数だけリン酸エステルの追加添加を行うことができる。
リン酸エステルの追加添加を数回に分割して行う場合の各分割添加量は、各回毎の添加量を等しく設定するか、あるいは回数を重ねるごとに徐々に少ない添加量とすることが好ましい。
リン酸エステルの追加添加を連続的に行う場合には、消費速度が反応開始後60分間における平均消費速度の好ましくは5~50%、より好ましくは10~40%となったときに、リン酸エステルの追加添加が開始される。このリン酸の連続的な追加添加は、リン酸エステルの残りの全量を前記の時点から好ましくは1~400分、より好ましくは2~360分かけて連続的に行われる。
リン酸エステルの追加添加の態様としては、1回のみ又は連続的に行うことが好ましい。ここで、リン酸エステルの追加添加を1回のみ行う場合には操作が簡便となる利点があり、これを連続的に行う場合には反応のコントロールが容易になる利点がある。
上記のようにして行われる付加反応は、その合計の反応時間を2~12時間とすることが好ましく、2~10時間とすることがより好ましい。
かくして得られる反応混合物は、高い転化率及び高い選択率で目的物に転化した目的物を含むものである。従って、リン酸エステルを追加添加する方法によって得られた反応混合物は、これに含有される未反応の四塩化炭素(その含有率はわずかである。)、鉄−リン酸エステル触媒残滓及び過剰の上記一般式(0)で表される不飽和化合物を分離すれば、多くの場合においてこれをそのまま製品として用いることができ、あるいはそのまま次工程の反応に供することができる。所望により第一反応後に精製を行うことができるが、該精製方法はごく簡単なものでよく、例えば理論段数2~10段程度の簡易な精製により高純度の製品とすることができる。
<第二反応>
本発明における第二反応は、上記一般式(1)で表される飽和化合物を熱分解して上記一般式(2)で表される不飽和化合物を得る熱脱塩化水素工程である。
本発明者らは、上記一般式(1)で表される飽和化合物が加熱によって容易に脱塩化水素化反応を起こす特性を有するものであることを見出した。かかる特性は、上記一般式(1)で表される飽和化合物中のCCl3基によるものと推察される。即ち本発明者らは、上記CCl3基中のClが熱による脱離を起こし易く、従って1~10秒程度の短い加熱時間で容易に熱分解による脱塩化水素反応を起こすことが可能であることを見出すことにより、極めて簡易な第二反応を実現したのである。
一方、上記一般式(1)で表される飽和化合物以外の、CCl3基を持たない塩素化飽和炭化水素に対して熱分解による脱塩化水素化反応を行おうすると、長い滞在時間が必要となり、その結果として反応中に生成する副生成物の割合が増え、また過度に熱分解して生成した炭素が配管中に析出するという問題も発生する。
上記式(1)で表される飽和化合物の具体例としては、例えば1,1,1−トリクロロプロパン、1,1,1,2−テトラクロロプロパン、1,1,1,3−テトラクロロプロパン、1,1,1,2,3−ヘプタクロロプロパン、1,1,1,3,3−ペンタクロロプロパン、1,1,1,2,3,3−ヘキサクロロプロパン、1,1,1,3,3,3−ヘキサクロロプロパン、1,1,1,2,3,3,3−ヘプタクロロプロパン等を挙げることができる。これらのうち、1,1,1,3−テトラクロロプロパンを使用すると、第二反応によって上記一般式(2)で表される不飽和化合物として種々の化合物の中間体として有用である1,1,3−トリクロロプロペンが得られる点で好ましい。
また、ここで使用される上記一般式(1)で表される飽和化合物は、上記の如き第一反応によって製造された上記一般式(1’)で表される飽和化合物であることが好ましい。
第二反応によって得られる上記一般式(2)で表される不飽和化合物の具体例を、上記式(1)で表される飽和化合物の具体例として上記に列記した化合物の順番に対応させて列記すると、1,1−ジクロロプロペン、1,1,2−トリクロロプロペン、1,1,3−トリクロロプロペン、1,1,2,3−テトラクロロプロペン、1,1,3,3−テトラクロロプロペン、1,1,2,3,3−ペンタクロロプロペン、1,1,3,3,3−ペンタクロロプロペン及び1,1,2,3,3,3−ヘキサクロロプロペンが例示される。
本発明における第二反応は、上記一般式(1)で表される飽和化合物を、その熱分解温度以上の温度において加熱する方法によることができる。
この熱分解反応は気相で行われることが好ましく、その反応方式は、流通方式及びバッチ方式のいずれによってもよい。しかしながら、本発明における第二反応の熱分解反応は短い反応時間で高い反応転化率を達成することができるので、流通方式によることが、反応の効率性の面から好ましい。
上記加熱温度としては好ましくは300~600℃であり、より好ましくは350~550℃である。加熱時間(滞在時間)は、例えば1~10秒とすることが好ましく、1~5秒とすることがより好ましく、さらに1~3秒とすることが好ましい。ここで、加熱温度が300℃未満であるかあるいは加熱時間が1秒未満であると熱分解が困難となり、一方、加熱温度600℃を超えるかあるいは加熱時間が10秒を超えると反応の選択率が低くなる場合があり、いずれも好ましくない。
本発明の第二反応における加熱は、公知の方法により行うことができる。反応器としては、外壁に加熱装置を備えた反応管(分解炉)が好適である。反応器を構成する材質としては、例えば石英、セラミック、金属等を例示することができる。加熱装置としては、例えばバーナー、電気ヒーター、高周波加熱装置等を使用することができる。
第二反応において、上記式(1)で表される飽和化合物を反応器に供給する方法としては、例えば気化器によって化合物を気化し、ガスとして反応器に導入する方法、あるいは液状の化合物を噴霧して反応器に導入する方法等を採用することができる。このとき反応器には、上記式(1)で表される飽和化合物のみを供給してもよく、あるいは上記式(1)で表される飽和化合物と適当な希釈ガスとの混合物を導入してもよい。ここで使用される希釈ガスとしては、不活性ガスを使用することが好ましく、例えば窒素、アルゴン、ヘリウム等を好ましく使用することができるが、希釈ガスに要するコストの観点から特に窒素を用いることが好ましい。希釈ガス中の酸素濃度は1重量%以下に調整することが好ましい。また、第二反応の脱塩化水素工程によって得られる生成物が水を含んでいると、その除去のために余分の精製工程が必要となり、経済上の不利益となる。これを避けるため、希釈ガス中の水分濃度は1,000重量ppm以下に調整することが好ましい。
上記式(1)で表される飽和化合物を希釈することによって、急激な反応の発生を抑えることが可能となるが、希釈しすぎると反応効率が低下する。これらのバランスをとる観点から、希釈する場合の希釈倍率は1.1~2倍程度とすることが好ましい。
第二反応における反応圧力は特に限定されず、減圧下でも加圧下でも実施することが可能である。加圧下の方が反応器を小さくすることが可能であるが、反応圧を高くするほど配管への炭素の析出が起こりやすい傾向にある。本発明においては、第二反応を常圧で行うことにより、熱分解による脱塩化水素化を極めて効率的に行うことが可能であるから、加圧条件下の反応をあえて選択する実益は少ない。
第二反応における熱分解は、無触媒条件下で十分迅速に進行するが、前記反応器内に固定床方式又は流動床方式の触媒床を設け、熱分解反応を触媒の存在下に加熱する態様によって行ってもよい。このことにより、前記の好ましい温度範囲のうちの比較的に低い温度においても高い転化率を達成することができ、好ましい。ここで使用される触媒としては、例えばシリカ、アルミナ、チタニア等の酸化物、活性炭等を挙げることができる。
第二反応後のガスは、速やかに冷却することが、副生成物を減らす観点から好ましい。ここで、熱分解反応器から上記の熱分解温度で排出されるガスを、好ましくは300℃未満、より好ましくは200℃未満、特に好ましくは100℃未満の温度に、好ましくは2秒以内に冷却することが有利である。この冷却は公知の方法、例えば供給ガスとの熱交換法、脱塩化水素体の液滴を噴霧してその気化潜熱により冷却する方法等、により行うことができる。
上記の如くして得られた第二反応の反応混合物(反応器からの排出ガス)は、これをそのまま第三反応に供してもよく、あるいは第二反応で生成(副生)した塩化水素を何らかの方法(例えば蒸留)によって除去した後に第三反応に供してもよい。ここで、反応混合物である排出ガスから特に塩化水素(HCl)を除く必要のないことが本発明の方法の特徴の一つである。このことにより、脱塩化水素反応の反応器と塩素化反応の反応器との間に蒸留塔や中間タンクの如き、塩化水素を分離するための設備を設ける必要がなくなり、塩素化炭化水素の製造設備をコンパクトに設計することが可能となる。
なお上記において、「反応混合物(排出ガス)をそのまま第三反応に供する」とは、第二反応の反応器からの排出ガスについて特段の精製(特に塩化水素の除去)を行わない出口組成のまま第三反応の反応器に移送することをいうのであって、決して排出後のガス温度の調整や第三反応に必要な塩素ガスの追加までを除外するものではない。
<第三反応>
本発明における第三反応は、上記一般式(2)で表される不飽和化合物と塩素とを反応させて上記一般式(3)で表される飽和化合物を得る、塩素化工程である。本第三反応は、上記一般式(2)で表される不飽和化合物を塩素を接触させるだけで迅速に進行する。
上記一般式(3)で示される飽和化合物を具体的に例示すると、1,1,1,2,3−ペンタクロロプロパン、1,1,1,2,3,3−ヘキサクロロプロパン,1,1,1,2,3,3,3−ヘプタクロロプロパン、1,1,1,2,2,3,3−ヘプタクロロプロパン、1,1,1,2,2,3,3,3−オクタクロロプロパン等を挙げることができる。これらのうち1,1,1,2,3−テトラクロロプロパンは、種々の化合物の中間体として有用であり、好ましい。使用する上記一般式(2)で表される不飽和化合物に応じて、上記一般式(3)で表される飽和化合物としていかなる生成物が得られるかは、当業者には自明であろう。
本発明における第三反応を、上記第二反応に引き続いて行うことにより、以下のようなメリットを享受することができる。
第一に、本発明における第二反応の生成物が水をほとんど含有しないことによるメリットである。本発明では、上記第二反応である脱塩化水素工程を、好ましくは気相における熱分解反応によって行うため、第二反応の排出ガスはほとんど水を含まない。従って本発明は、第三反応の前又は後において大規模な脱水設備を必要としないメリットがある。
第二に、第二反応の生成物が鉄をほとんど含有しないことによるメリットである。一般に、塩素化不飽和炭化水素の塩素化工程においては、鉄が存在しない方が好ましいとされる。これは、塩素化反応の生成物である塩素化飽和炭化水素の分解ないし逆反応が、鉄の存在によって促進されるからである。本発明においては、第二反応を好ましくは気相中で行うため、好都合なことに第二反応の生成物中に鉄がほとんど混入しないことも、メリットの一つである。
本発明における第三反応は、上記一般式(2)で表される不飽和化合物を凝縮せずに気相の状態で行ってもよく、あるいはこれを凝縮して液相において行ってもよい。
本発明における第三反応を気相で行う場合、流通方式及びバッチ方式のいずれによってもよいが、本発明における方法は短い反応時間で高い反応転化率を達成することができるので、流通方式によることが、反応の効率性の面から好ましい。
本発明の方法における第三反応を気相で行うには、第二反応の排出ガスと塩素ガスとの混合ガスを、所定温度で所定時間加熱することによって行うことができる。塩素ガスの使用割合としては、第二反応の反応混合物中に含まれる脱塩化水素体(上記一般式(2)で表される不飽和化合物)の1モルに対して、好ましくは0.9~2.0モルであり、より好ましくは1.0~1.5モルである。この値が0.9モル未満であると第三反応の反応転化率が損なわれる場合があり、一方2.0モルを超えると副生物の生成が増大するとの不都合が生じる場合があり、いずれも好ましくない。
気相における塩素化反応の反応温度としては、気相状態を維持し、且つ副生物の生成を抑制するとの観点から、上記式(2)で表される不飽和化合物の沸点以上且つ300℃以下とすることが好ましく、沸点~280℃とすることがより好ましい。反応時間は、好ましくは1~60秒であり、より好ましくは1~40秒である。
一方、本発明における第三反応を液相で行う場合、その反応温度としては、原料として使用する上記一般式(2)で表される不飽和化合物が気化しない温度領域であれば特に限定されないが、副生物を少なくするために120℃以下とすることが好ましい。供給する塩素量は,転化率及び選択率を高くするとの観点から、上記一般式(2)で表される不飽和化合物の1モルに対して0.9~2.0とすることが好ましい。第三反応を液相で行う場合の反応時間は、好ましくは1~10時間である。液相反応を促進するために、塩素の吹き込み方法を微細気泡とする方法、紫外光を照射すること等も好適な態様である。
上記のようにして得られる第三反応の排出ガス又は排出液は、目的物である上記一般式(3)で表される飽和化合物を高濃度で含有する粗生成物である。この粗生成物は、必要に応じて公知の方法によって精製した後に、製品とされる。任意的に行われる精製方法としては、例えば蒸留精製を挙げることができる。蒸留を行う際には、上記一般式(3)で表される化合物の熱分解を防止するため、粗生成物にp−メトキシフェノール、o−t−ブチルフェノール、オイゲノール、o−アリルフェノール等のフェノール誘導体、特にアリル基を有するフェノール誘導体を添加した後、蒸留を行うことが好ましい。
第二反応で得られた排出ガスから副生した塩化水素を分離せずに、上記式(2)で表される不飽和化合物と塩化水素との混合物を第三反応に供給して塩素化した場合、該第三反応によって得られる粗生成物は、塩化水素、及び場合によって未反応の塩素を含有している。このような場合は、粗生成物について、下記の工程(A)~(C)を実施し、粗生成物中の塩化水素を回収して再利用することが好ましい。
(A)上記第三反応から得られた粗生成物から塩化水素(未反応の塩素を含む場合もある)を分離する、塩化水素分離工程、
(B)上記塩化水素分離工程から得られた塩化水素を酸化処理する酸化工程、及び
(C)上記酸化工程より得られた塩素を、前記塩素化工程の塩素源として循環使用する塩素循環工程。
上記(A)塩化水素分離工程は、粗生成物から塩化水素、場合によって塩化水素及び塩素を分離し得る公知の方法によって行うことができる。かかる方法としては、例えば粗生成物中の上記式(3)で表される飽和化合物を凝縮して、塩化水素、又は塩化水素と塩素とを、ガスとして分離する方法を挙げることができる。
上記(B)酸化工程における塩化水素の酸化条件としては、公知の方法が特に制限なく採用される。かかる方法としては、例えば触媒酸化法等を挙げることができる。具体的には、塩化水素又は塩化水素と塩素とを、ガス状態で、例えばクロミア又はルテニウムを担持したチタニア触媒層を通過させる方法等を挙げることができる。このときの反応温度は例えば200~450℃とすることができる。
<高純度の1,1,1,2,3−ペンタクロロプロパンの製造>
以上述べてきたように、本発明の方法に従って三段階反応又はそのうちの任意の段階の反応を行えば、純度の高い塩素化炭化水素を効率的に製造することができる。
しかしながら、上記の如き三段階反応によって特に高純度の1,1,1,2,3−ペンタクロロプロパンを迅速、安価に製造する場合には、上記第一反応後且つ第二反応前に1,1,1,3−テトラクロロプロパンの蒸留精製を行うことが有利である。
1,1,1,2,3−ペンタクロロプロパンは、上記一般式(0)で表される不飽和化合物としてエチレンを用い、これに四塩化炭素を付加して1,1,1,3−テトラクロロプロパンを得る第一反応と、
該1,1,1,3−テトラクロロプロパンを脱塩化水素して3,3,3−トリクロロプロペン及び1,1,3−トリクロロプロペンよりなる群から選択される少なくとも1種の塩素化飽和炭化水素を得る第二反応と、
該トリクロロプロペンに塩素を付加して1,1,1,2,3−ペンタクロロプロパンを得る第三反応と
からなる三段階反応によって製造することができる。
ここで、第一反応によって得られる反応混合物は、主成分として1,1,1,3−テトラクロロプロパンを含有するが、これ以外に不純物として四塩化炭素、ヘキサクロロエタン、テトラクロロエチレン、1,1,3−トリクロロプロペン、1,1,1,3,3−ペンタクロロプロパン等を含有する。本発明においては、第一反応後の精製において、上記不純物のうち特にヘキサクロロエタン、テトラクロロエチレン及び1,1,1,3,3−ペンタクロロプロパンの濃度を可及的に低くしておくことが好ましい。具体的には、ヘキサクロロエタン、テトラクロロエチレン及び1,1,1,3,3−ペンタクロロプロパンの合計の重量を、これらと1,1,1,3−テトラクロロプロパンとの合計重量に対して、1重量%以下とすることが好ましく、0.9重量%以下とすることがより好ましい。第一反応の反応混合物について、第一反応後かつ第二反応前に、上記の程度に精製することによって、第三反応後に目的物として得られる1,1,1,2,3−ペンタクロロプロパンは、分離困難な不純物を実質的に含まないこととなる。このことにより、本発明の方法で得られた1,1,1,2,3−ペンタクロロプロパンは、これをそのまま製品とすることができ、あるいはその後に簡易な精製方法を行うことによって極めて高純度の1,1,1,2,3−ペンタクロロプロパンとすることができるのである。
本願発明者らは、本願発明の目的を達成するために、第一、第二及び第三反応の各反応において生成する不純物の動向を詳細に検討した。その結果、第一反応において不純物として生成するヘキサクロロエタン、テトラクロロエチレン及び1,1,1,3,3−ペンタクロロプロパンが、第三反応後の精製において問題となることを突き止めた。
即ち、ヘキサクロロエタンは第二反応及び第三反応を経由しても反応せずにそのまま第三反応後まで残存し;
テトラクロロエチレンは第二反応においては反応しないが第三反応の塩素化反応においてヘキサクロロエタンに変換し;
1,1,1,3,3−ペンタクロロプロパンは、そのうちの一部が第二反応及び第三反応を経由してもそのまま反応せずに第三反応後まで残存することが分かった。
ここで説明のために、第一反応における目的物及び不純物の常圧における沸点を第1表に、第三反応における目的物及び不純物の常圧における沸点を第2表に、それぞれ示す。
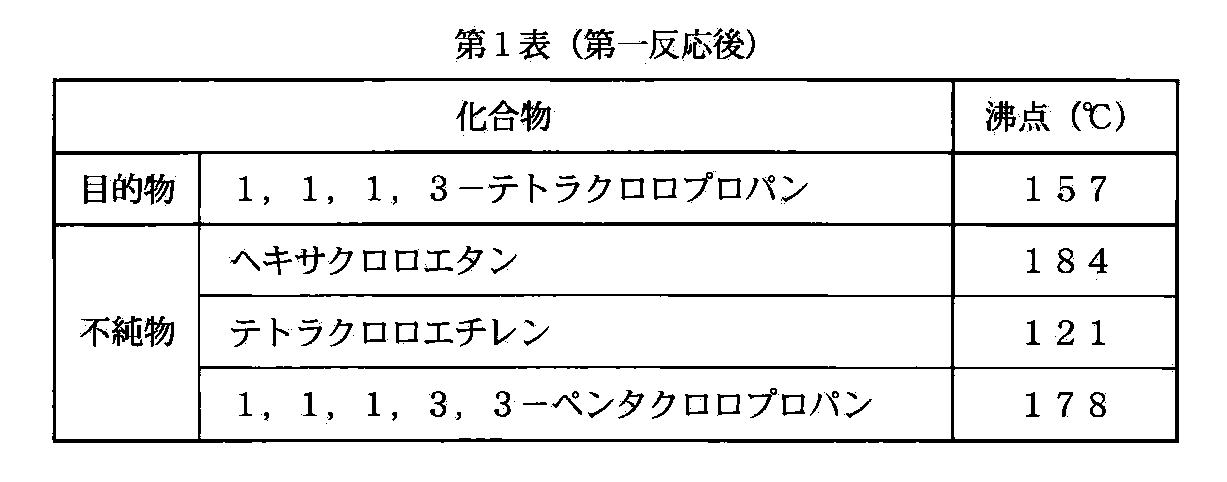
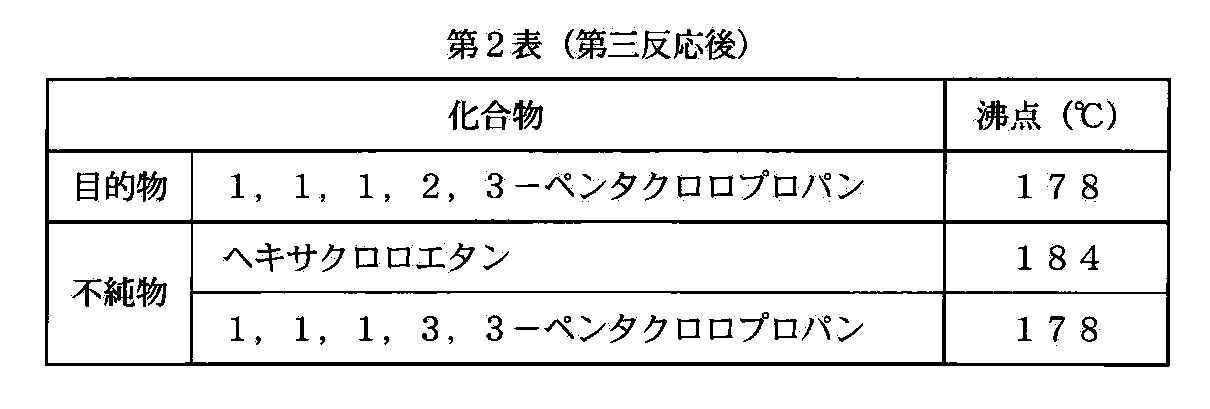
第一反応後では目的物と不純物との沸点差が大きく(第1表)、従って簡易な蒸留装置で精製の目的を達することができる。
そして、第一反応後に不純物のうちのヘキサクロロエタン、テトラクロロエチレン及び1,1,1,3,3−ペンタクロロプロパンを除去すれば、第三反応後の反応混合物中に、ヘキサクロロエタン及び1,1,1,3,3−ペンタクロロプロパンは存在しえないこととなるから、第一反応後に精製を行った場合には、第三反応後に精製を行わずとも目的物を得ることができ、あるいは簡易な精製によって極めて高純度の目的物を得ることができるのである。
このような発想に基づいて高純度のペンタクロロプロパンを製造しようとする試みは、今まで知られていない。
第一反応後の精製を蒸留精製による場合、使用する蒸留塔としては、常温常圧下で液体状の物質の精留に使用されるものとして当業界において知られているものを制限なく使用することができ、段塔又は充填塔を好ましいものとして挙げることができる。段塔の段数又は段塔に換算した充填塔の相当段数は、2~20段であることが好ましい。
蒸留塔は一塔で行ってもよいし、数塔で行ってもよい。これらのうち、蒸留塔を数塔で行う態様は、反応混合物中に溶解した塩化鉄等の除去効率がよくなるため、好ましい態様である。
上記段塔としては、十字流トレイ、シャワートレイ等を用いることができる。これらの具体例としては、十字流トレイとして、例えば多孔板トレイ、泡鐘トレイ、バルブトレイ、ターボグリッドトレイ等を;
シャワートレイとして、例えばターボグリッドトレイ、リップルトレイ等を、それぞれ挙げることができる。これらのうち、十字流トレイを用いることが好ましい。
上記充填塔としては、規則充填物又は不規則充填物を充填した蒸留塔を使用することができる。上記不規則充填物としては、例えばラシヒリング、ベルサドル、マクマホン、ナッターリング、ポーリング、カスケードミニリング、ヘリパック等を挙げることができる。これらのうち、規則充填物又はカスケードミニリングを充填した蒸留塔を使用することが、蒸留効率を高くすることができる点で好ましい。
蒸留温度は、1,1,1,3−テトラクロロプロパンの分解を抑制することができ、且つ蒸留圧力の制御が容易である等の観点から、60~140℃とすることが好ましく、80~130℃とすることがより好ましい。蒸留圧力は、前記の好ましい温度における蒸留操作を維持しうる圧力に設定することが好ましいが、例えば1~20kPaとすることができる。
第一反応の反応混合物(粗1,1,1,3−テトラクロロプロパン、以下、「粗TCP」という。)は、蒸留塔の下部の適当な位置、好ましくは中間段から蒸留塔に導入される。粗TCP中の重質分は導入位置から下降して缶出液として除去される。他方、粗TCP中の目的物及び酸分は蒸留塔内を上昇し、不純物の濃度が所望の程度まで減じた段において、目的物を蒸留塔の塔側流として抜き出すことが好ましい。ここで、「塔側流として抜き出す」とは、蒸留塔の最上段よりも少なくとも1段下の段、好ましくは3~10段下であって、かつ不純物を除去するのに十分な段における液相から精製物を抜き出すことをいう。
抜き出し段よりも上の段においては酸分が濃縮されるから、塔頂流出物は、凝縮器を通して凝縮液と非凝縮気体とに分離したうえ、非凝縮気体(酸分を高濃度で含む)は抽気排除し、凝縮液の全部又は一部は精製物の抜き出し段よりも上の段に還流させることが好ましい。ここで、凝縮液の抜き出し量を留出液量、還流液を還流液量とした場合、還流液量/留出液量で定義される還流比は、0.1~50とすることが好ましく、1~40とすることがより好ましい。
さらに、蒸留塔の粗TCPを導入する段よりも下の段から不活性ガスを導入して塔内液中の酸分のストリッピング効果を促進することも好適に採用することができる。ここで使用される不活性ガスとしては、例えば窒素、ヘリウム等を挙げることができる。不活性ガスの導入量としては、塔内の上昇気体容量全体に対する体積比として、1×10−5~1×10−3とすることが好ましく、1×10−5~1×10−4とすることがより好ましい。
かくして得られた精製1,1,1,3−テトラクロロプロパン(以下、「精製TCP」という。)は、不純物としてヘキサクロロエタン、テトラクロロエチレン及び1,1,1,3,3−ペンタクロロプロパンを実質的に含有せず、従ってかかる精製TCPを使用して第二反応及び第三反応を経由して得られる1,1,1,2,3−ペンタクロロプロパンは、不純物としてヘキサクロロエタン及び1,1,1,3,3−ペンタクロロプロパンを含有しないことになる。従って、第三反応後の1,1,1,2,3−ペンタクロロプロパンは、これをそのまま製品とすることができ、あるいは簡易な精製を行うことのみで極めて高純度の製品とすることができる。
ここで、実施される簡易な精製としては、例えば蒸留精製、脱酸剤処理等を挙げることができる。
蒸留精製は、上記第一反応後に好ましく行われる蒸留精製として上記したところに準じて実施することができるが、本発明の方法によって得られる1,1,1,2,3−ペンタクロロプロパンは、これと沸点の近い不純物を含有しないため、精製に使用する蒸留塔の段数又は相当段数は、2~20程度で足りる。この蒸留は、粗1,1,1,2,3−ペンタクロロプロパンに上述のフェノール誘導体、特にアリル基を有するフェノール誘導体を添加した後に行うことが好ましい。
上記脱酸剤処理は、1,1,1,2,3−ペンタクロロプロパンを適当な脱酸剤と接触させることにより行うことができる。ここで使用される脱酸剤としては、例えばシリカ、アルミナ、ケイ酸塩等を挙げることができる。
上記シリカとしては、例えば無定形シリカ、結晶性シリカ、シリカ水和物等を挙げることができ、その具体例として、例えばシリカゲル、ケイソウ土等を挙げることができる。
上記アルミナとしては、例えば無定形アルミナ、結晶性アルミナ、アルミナ水和物等を挙げることができる。
上記ケイ酸塩は、二酸化ケイ素と金属酸化物との塩であればその種類に特に制限なく用いることができる。具体的には、ケイ酸ナトリウム等のケイ酸アルカリ;ケイ酸カルシウム、ケイ酸マグネシウム等のケイ酸アルカリ土類金属塩;シリカアルミナゲル等のケイ酸アルミニウム等を挙げることができる。このようなケイ酸塩を含む鉱物としては、ゼオライト、合成ゼオライト、カオリナイト、活性白土、ハロイサイト、モンモリロナイト、アロフェン、ベントナイト等を用いることができる。
上記の如き脱酸剤としては、例えばセカードKW(品川化成(株)製)等の市販品を使用してもよい。
脱酸剤の形状としては、粉末状、顆粒状、粒状等の適宜のものを特に制限なく用いることができる。脱酸剤の粒径は、接触効率の点から小さいものを好ましく使用することができ、その平均粒径が9mm以下であることが好ましく、0.3~5mmであることがより好ましい。
1,1,1,2,3−ペンタクロロプロパンを脱酸剤と接触させる際の空間速度としては、0.001~20hr−1とすることが好ましく、0.005~10hr−1とすることがより好ましい。接触温度は、5~80℃とすることが好ましく、10~70℃とすることがより好ましい。
<三段階反応の実施例>
実施例1−1
(1)第一反応
攪拌器を取り付けたSUS製のオートクレーブに、四塩化炭素769g、リン酸トリエチル4.5g及び鉄粉(和光純薬工業(株)製、還元鉄)14gを仕込み、温度を110℃に設定し、0.4MPaGの反応圧力を保つようにエチレンを導入して反応を開始した。6時間後、オートクレーブを冷却して、反応後の反応液を回収してガスクロマトグラフィーにより分析し、転化率及び選択率を求めた。その結果、四塩化炭素基準での転化率は80%、1,1,1,3−テトラクロロプロパンへの選択率は90%であった。
(2)第二反応(脱塩化水素工程)
上記工程で生成した1,1,1,3−テトラクロロプロパンを200℃の予熱器で気化し、入口ガス流量を基準として滞在時間が2.5秒になるように流量を調整し、電気炉によって500℃に加熱した反応管(材質SUS316、内径4.35mm、長さ300mm)中にガス状で導入し、常圧にて気相で熱分解反応を行った。
このとき、上記第二反応において得られた生成ガスの一部を0℃に冷却し、液化してガスクロマトグラフィーにより分析し、転化率及び選択率を求めた。その結果、転化率は99.7%、1,1,3−トリクロロプロペンへの選択率は99.0%であった。
(3)第三反応(塩素化工程)
上記第二反応で生成した生成ガスを150℃に冷却したうえで、そのまま(第二反応において副生した塩化水素を除去せずに)塩素化工程に供給し、気相で塩素化を以下の条件で行った。
上記生成ガスの供給ラインに塩素ガスを合流した混合ガス(1,1,3−トリクロロプロペン:塩素=100:110(モル比))を、内径4.35mm、長さ300mmの反応管中に通過させ、滞在時間1.24秒で塩素化反応を行なった後、反応管出口より得られる粗クロロプロパンを0℃に冷却することにより液化して、塩化水素と未反応塩素を除去した後、蒸留精製して、目的とする1,1,1,2,3−ペンタクロロプロパンを得た。
このとき、上述の液化した粗クロロプロパンの一部を取出し、ガスクロマトグラフィーで分析し、転化率及び選択率を求めた。その結果、1,1,3−トリクロロプロペン基準の転化率は99%、1,1,1,2,3−ペンタクロロプロパンへの選択率は95%であった。
実施例1−2
(1)第一反応
攪拌器を取り付けたSUS製のオートクレーブに四塩化炭素769g、リン酸トリエチル4.5g及び鉄粉(和光純薬工業(株)製、還元鉄)14gを仕込み、温度を110℃に設定し、0.4MPaGの反応圧力を保つように塩化ビニルを導入して反応を開始した。6時間後、オートクレーブを冷却して、反応後の反応液を回収してガスクロマトグラフィーにより分析し、転化率及び選択率を求めた。その結果、塩化ビニル基準での転化率は81%、1,1,1,3,3−ペンタクロロプロパンへの選択率は91%であった。
(2)第二反応(脱塩化水素工程)
上記工程で生成した1,1,1,3,3−ペンタクロロプロパンを200℃の予熱器で気化し、入口ガス流量を基準として滞在時間が2.5秒になるように流量を調整し、電気炉によって500℃に加熱した反応管(材質SUS316、内径4.35mm、長さ300mm)中にガス状で導入し、常圧にて気相で熱分解反応を行った。
このとき、上記第二反応において得られた生成ガスの一部を0℃に冷却し、液化してガスクロマトグラフィーにより分析し、転化率及び選択率を求めた。その結果、転化率は99.1%、1,1,3,3−テトラクロロプロペンへの選択率は98.0%であった。
(3)第三反応(塩素化工程)
上記第二反応で生成した生成ガスを180℃に冷却したうえで、そのまま(第二反応において副生した塩化水素を除去せずに)塩素化工程に供給し、気相で塩素化を以下の条件で行った。
上記生成ガスの供給ラインに塩素ガスを合流した混合ガス(1,1,3,3−テトラクロロプロペン:塩素=100:110(モル比))を、内径4.35mm、長さ300mmの反応管中に通過させ、滞在時間1.24秒で塩素化反応を行なった後、反応管出口より得られる粗クロロプロパンを0℃に冷却することにより液化して、塩化水素と未反応塩素を除去した後、蒸留精製して、目的とする1,1,1,2,3,3−ヘキサクロロプロパンを得た。
このとき、上述の液化した粗クロロプロパンの一部を取出し、ガスクロマトグラフィーで分析し、転化率及び選択率を求めた。その結果、1,1,3,3−テトラクロロプロペン基準の転化率は99%、1,1,1,2,3,3−ヘキサクロロプロパンへの選択率は90%であった。
実施例1−3
上記実施例1−1において、第三反応(塩素化工程)を以下の通り実施した以外は実施例1−1と同様に実施して、1,1,1,2,3−ペンタクロロプロパンを得た。
即ち、第二反応(脱塩化水素工程)により生成したトリクロロプロペンを液化して塩化水素を分離した後、第三反応に供給し、塩素360NmL/minで供給しつつ、温度80℃で塩素化反応を行った(1,1,3−トリクロロプロペン:塩素=100:120(モル比))。なお、上記塩素供給量は標準状態(SATP)換算値である。
反応開始から270分後、反応液をガスクロマトグラフィーで分析して転化率及び選択率を求めた。その結果、1,1,3−トリクロロプロペン基準の転化率は99%、1,1,1,2,3−ペンタクロロプロパンへの選択率は96%であった。
本発明の方法における三段階反応の効果を考察するに際しては、後述の実施例4−3、4−4及び4−6の結果も参照されたい。
<第一反応においてリン酸エステルを追加点添加する態様の実施例>
以下の比較例、実施例及び参考例では、エチレンと四塩化炭素との付加反応を、気相流通下における液相バッチ反応により試験した。
反応装置としては、撹拌機、エチレン用ガス導入口及びガス排出口並びにリン酸エステルの追加添加口を有するSUS製のオートクレーブ(内容積1,500mL)を用いた。エチレンは、上記ガス導入口を介して気相に導入した。反応中は、ガス導入口から導入されるエチレン量とガス排出口から排出されるエチレン量との差分からエチレンの消費速度を連続的にモニターした。反応中、反応圧力は0.4MPaG±0.02MPa(5%)となるように制御した。
反応終了後の反応混合物はガスクロマトグラフィーにより分析し、使用した四塩化炭素基準の反応転化率及び目的生成物である1,1,1,3−テトラクロロプロパンの選択率を求めた。
なお、以下におけるエチレン消費速度の単位は、すべて標準状態(SATP)換算値である。
比較例2−1
オートクレーブ中に四塩化炭素1,560g、リン酸トリエチル12g及び化学反応用純鉄粉(JFEスチール(株)製、K−100T)4gを仕込み、温度を110℃に設定し、反応圧力が0.4MPaGとなるようにエチレンを導入し、付加反応を開始した。
エチレン導入開始後しばらくするとエチレンの消費量が急激に上昇した。エチレン導入開始から60分間のエチレン平均消費速度は1,000NmL/分であり、エチレン導入開始から60分後のエチレン消費速度は400NmL/分を下回った。
エチレン導入開始から6時間後、オートクレーブを冷却し、反応終了後の反応混合物を回収してガスクロマトグラフィーにより分析したところ、使用した四塩化炭素基準の反応転化率は27%であり、1,1,1,3−テトラクロロプロパンの選択率は86%であった。
実施例2−1
オートクレーブ中に四塩化炭素1,560g、リン酸トリエチル8g及び化学反応用純鉄粉4gを仕込み、温度を110℃に設定し、反応圧力が0.4MPaGとなるようにエチレンを導入し、付加反応を開始した。
エチレン導入開始後しばらくするとエチレンの消費量が急激に上昇した。エチレン導入開始から60分間のエチレン平均消費速度は950NmL/分であった。エチレン導入開始から60分後にエチレン消費速度は400NmL/分を下回ったので、リン酸トリエチル4gを1回で追加添加した。
エチレン導入開始から6時間後、オートクレーブを冷却し、反応終了後の反応混合物を回収してガスクロマトグラフィーにより分析したところ、使用した四塩化炭素基準の反応転化率は85%であり、1,1,1,3−テトラクロロプロパンの選択率は90%であった。
実施例2−2
オートクレーブ中に四塩化炭素1,560g、リン酸トリエチル8g及び化学反応用純鉄粉4gを仕込み、温度を110℃に設定し、反応圧力が0.4MPaGとなるようにエチレンを導入し、付加反応を開始した。
エチレン導入開始後しばらくするとエチレンの消費量が急激に上昇した。エチレン導入開始から60分間のエチレン平均消費速度は950NmL/分であった。
エチレン導入開始から60分後にエチレン消費速度は400NmL/分を下回った。そこでリン酸トリエチルを、エチレン導入開始から60分後から0.016g/分の添加速度で250分間連続的に追加添加した(リン酸トリエチルの追加添加量は合計4gである。)。
エチレン導入開始から6時間後、オートクレーブを冷却し、反応終了後の反応混合物を回収してガスクロマトグラフィーにより分析したところ、使用した四塩化炭素基準の反応転化率は93%であり、1,1,1,3−テトラクロロプロパンの選択率は90%であった。
参考例2−1
オートクレーブ中に四塩化炭素1,560g、リン酸トリエチル12g及び化学反応用純鉄粉60gを仕込み、温度を110℃に設定し、反応圧力が0.4MPaGとなるようにエチレンを導入し、付加反応を開始した。
エチレン導入開始から6時間後、オートクレーブを冷却し、反応終了後の反応混合物を回収してガスクロマトグラフィーにより分析したところ、使用した四塩化炭素基準の反応転化率は85%であり、1,1,1,3−テトラクロロプロパンの選択率は86%であった。
参考例2−2
オートクレーブ中に四塩化炭素1,560g、リン酸ジブチル14g及び化学反応用純鉄粉60gを仕込み、温度を100℃に設定し、反応圧力が0.4MPaGとなるようにエチレンを導入し、付加反応を開始した。
エチレン導入開始から6時間後、オートクレーブを冷却し、反応終了後の反応混合物を回収してガスクロマトグラフィーにより分析したところ、使用した四塩化炭素基準の反応転化率は21%であり、1,1,1,3−テトラクロロプロパンの選択率は85%であった。
以上の結果から、鉄及びリン酸エステルを同じ量だけ用いた場合、これらの全量を反応開始前に一括添加する従来技術の方法(比較例2−1)よりも、反応開始前に鉄の全量及びリン酸エステルの一部を添加し、残余のリン酸エステルは上記付加反応進行中に追加添加する本発明の方法(実施例2−1及び2−2)の方が、反応転化率及び選択率の双方において優れていることが理解される。
<第一反応後且つ第二反応前に生成物の蒸留精製を行う、三段階反応の実施例>
実施例3−1
(1)第一反応
オートクレーブ中に四塩化炭素1,560g、リン酸トリエチル8g及び化学反応用純鉄粉4gを仕込み、温度を110℃に設定し、反応圧力が0.4MPaGとなるようにエチレンを導入し、第一反応(付加反応)を開始した。エチレン導入開始から1時間後、リン酸トリエチル4gを追加添加した。
エチレン導入開始から6時間後にオートクレーブを冷却し、反応終了後の反応混合物を回収してガスクロマトグラフィーにより分析したところ、使用した四塩化炭素基準の反応転化率は85%であり、1,1,1,3−テトラクロロプロパンの選択率は90%であった。
(2)第一反応後の精製
上記第一反応によって得られた反応混合物につき、以下のようにして二段階の蒸留精製を行った。
一回目蒸留として、内部に柴田科学(株)製ガラスパッキンを高さ500mmに充填した塔径30mmφの蒸留塔において、還流をかけずに、蒸留圧力10kPa、蒸留温度95℃にてバッチ蒸留を行った。ここで得られた精製留分中の1,1,1,3−テトラクロロプロパンの純度は、98.5%であった。
上記で得られた精製留分について、二回目の蒸留として、内部に柴田科学(株)製ガラスパッキンを高さ1,000mmに充填した塔径30mmφの蒸留塔において、還流比3、蒸留圧力10kPa、蒸留温度95℃にてバッチ蒸留を行った。このとき、低沸留分を4重量%、ボトム高沸残渣を6重量%除いて得られた留出成分である精製1,1,1,3−テトラクロロプロパンの純度はほぼ100%であった。
(3)第二反応
上記で得た精製1,1,1,3−テトラクロロプロパンを用いて、滞在時間2.5秒、反応温度500℃の条件で第二反応(脱塩化水素反応)を行った。この脱塩化水素反応における反応の転化率は99.7%、目的物である1,1,3−トリクロロプロペンの選択率は99.0%であった。なお、上記転化率及び選択率は、それぞれ、1,1,1,3−テトラクロロプロパンを基準として算出した。
(4)第三反応
第三反応(塩素化反応)では、上記第二反応の排出ガスを150℃に冷却し、流量調整した塩素ガスと混合したうえで反応器に供給し、滞在時間1.24秒の条件で塩素化反応を行い、粗1,1,1,2,3−ペンタクロロプロパンを得た。ここで、塩素ガスの流量は、第二反応の排出ガスの1m3(0℃、98kPa)に対して1.1m3(0℃、98kPa)とした。この塩素化反応における反応転化率は99.0%、目的物である1,1,1,2,3−ペンタクロロプロパンの選択率は95.0%であった。なお、上記転化率及び選択率は、それぞれ、1,1,3−トリクロロプロペンを基準として算出した。
(5)第三反応後の精製
上記第三反応で得られた粗1,1,1,2,3−ペンタクロロプロパンを、内部に柴田科学(株)製ガラスパッキンを高さ1,000mm充填した塔径30mmφの蒸留塔において、還流比3、蒸留圧力10kPa、蒸留温度135℃にてバッチ蒸留を行った。このとき、低沸留分を3重量%、ボトム高沸残渣を7重量%除いて得られた留出成分である精製1,1,1,2,3−ペンタクロロプロパンの純度はほぼ100%であった。また、この時点における酸分は210重量ppmであった。別途、この蒸留塔の塔頂より100mm下部(塔頂から0.2段下に相当する。)から、留出成分をサンプリングしたところ、精製1,1,1,2,3−ペンタクロロプロパンの純度はほぼ100%であり、酸分は40重量ppmであった。
(6)各成分の動向
上記の各反応及び各精製における各成分の動向を第3表に示した。
実施例3−2
上記実施例3−1において、粗1,1,1,2,3−ペンタクロロプロパンの蒸留塔の留出口(塔頂)にセカードKW(商品名、品川化成(株)製)を充填したカラムを設置したほかは、実施例1と同様に行い、精製1,1,1,2,3−ペンタクロロプロパンの流出成分を得た。ここで、1,1,1,2,3−ペンタクロロプロパン留分とセカードKWとの接触における空間速度は1hr−1とした。
本実施例における各成分の動向は、実施例3−1におけるのと同様であった。上記精製1,1,1,2,3−ペンタクロロプロパンにおける酸分は20重量ppm以下であった。
実施例3−3
上記実施例3−1において、第三反応後の精製に使用した蒸留塔の蒸留圧力5kPaとし、蒸留温度を110℃としたほかは、実施例1と同様に行い、留出口(塔頂)から精製1,1,1,2,3−ペンタクロロプロパンの流出成分を得た。
本実施例における各成分の動向は、実施例3−1におけるのと同様であった。上記精製1,1,1,2,3−ペンタクロロプロパンにおける酸分は90重量ppmであった。
比較例3−1
上記実施例1において、第一反応後の蒸留精製を行わなかったほかは、実施例1と同様に行い、塔頂の留出口から精製1,1,1,2,3−ペンタクロロプロパンの流出成分を得た。この精製1,1,1,2,3−ペンタクロロプロパンにおける酸分は210重量ppmであった。
本比較例における各成分の動向を第4表に示した。

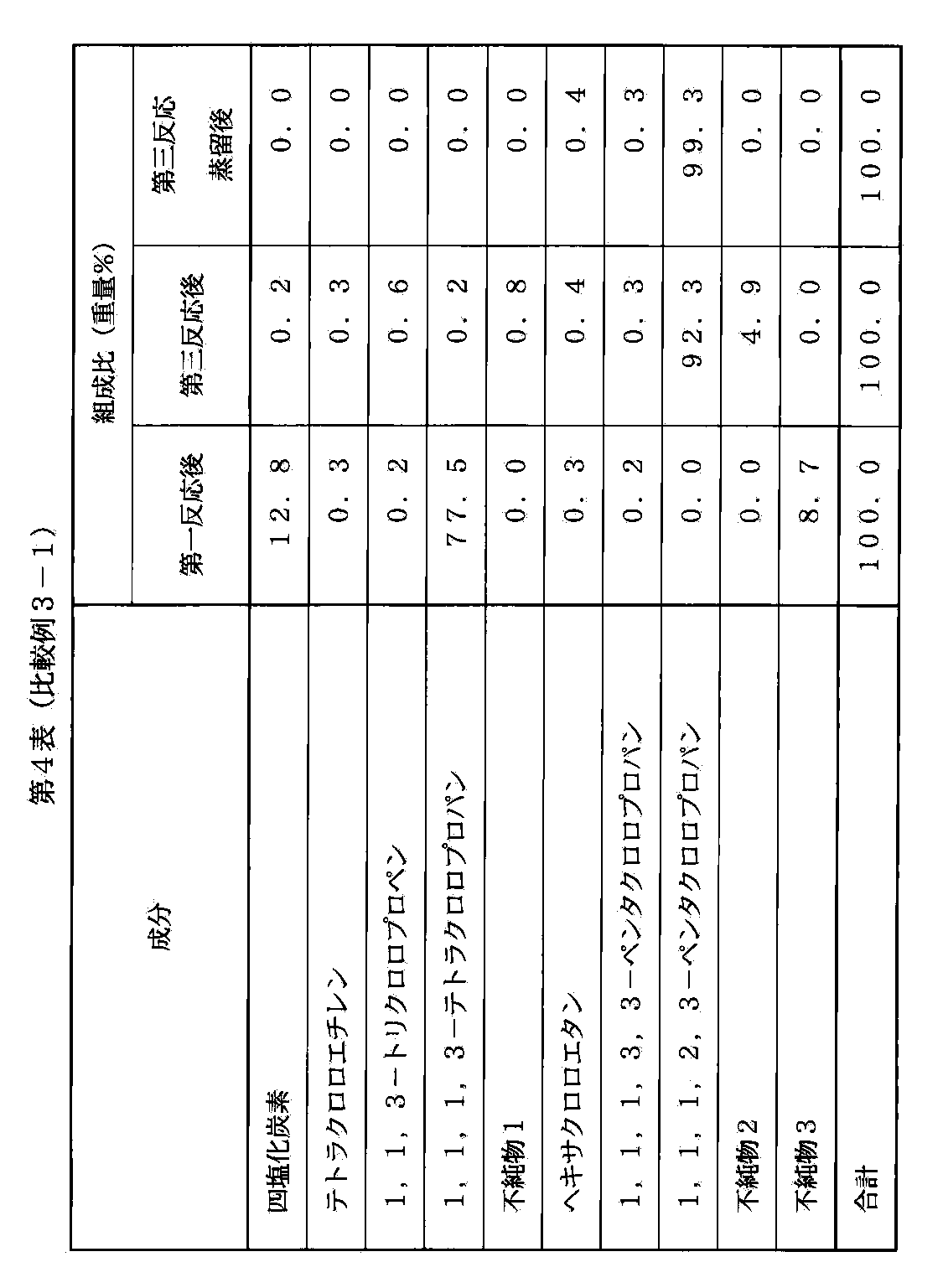
<第二反応を熱分解で行う態様の実施例>
実施例4−1
200℃の予熱器で気化させた1,1,1,3−テトラクロロプロパンを、入口ガス流量を基準として滞在時間が2.5秒になるように流量を調整して、電気炉により500℃に加熱した反応管(材質SUS316、内径4.35mm、長さ300mm)中にガス状で導入し、常圧にて気相で熱分解反応を行った。
出口ガスを0℃に冷却し、液化してガスクロマトグラフィーにより分析し、転化率及び1,1,3−トリクロロプロペンへの選択率を求めた。
結果は第5表に示した。
実施例4−2
反応管の温度を450℃とした以外は、実施例4−1と同様に熱分解を行った。
転化率及び選択率の分析結果を第5表に示した。
実施例4−3
反応管の温度を550℃とした以外は、実施例4−1と同様に熱分解を行った。
転化率及び選択率の分析結果を第5表に示した。
実施例4−4
滞在時間を1.25秒とした以外は、実施例4−1と同様に熱分解を行った。
転化率及び選択率の分析結果を第5表に示した。
実施例4−5
上記実施例4−1において、1,1,1,3−テトラクロロプロパンの代わりに1,1,1,2−テトラクロロプロパンを原料として使用し、反応管の温度を450℃とした以外は、実施例4−1と同様に熱分解を行った。
1,1,1,2−テトラクロロプロパンの転化率及び1,1,2−トリクロロプロペンへの選択率の分析結果を第5表に示した。
実施例4−6
上記実施例4−1において、原料である1,1,1,3−テトラクロロプロパンを75%の濃度となるように窒素ガスで希釈して反応管中に供給した以外は、実施例4−1と同様に熱分解を行った。
転化率及び選択率の分析結果を第5表に示した。
実施例4−7
200℃の予熱器で気化させた1,1,1,3−テトラクロロプロパンを、入口ガス流量を基準として滞在時間が9.0秒になるように流量を調整して、電気炉により450℃に加熱した反応管(材質SUS316、内径7.3mm、長さ600mm)中にガス状で導入し、常圧にて気相で熱分解反応を行った。
転化率及び選択率の分析結果を第5表に示した。

本発明によると、塩素化炭化水素を製造するための三段階工程のそれぞれにおいて、以下のような利点を伴う方法が提供される。
第一反応においては、触媒の使用量を削減しつつ、高い転化率で目的生成物を安定した反応速度で効率的に付加反応を行う方法が;
第二反応においては、高価なアルカリ源を使用せず、また処理すべき副生成物の発生が抑制された脱塩化水素反応を行う方法が;
第三反応においては、より効率性の高い塩素化反応を行う方法が、それぞれ提供される。
本発明によると、簡易な操作によって効率的に高純度の塩素化炭化水素を製造することができる。
Claims (10)
- 下記一般式(1)で表される飽和化合物を熱分解して下記一般式(2)で表される不飽和化合物を得る熱脱塩化水素工程を経ることを特徴とする、塩素化炭化水素を製造するための方法。
CCl3−CCl2−mHm−CCl3−nHn (1)
CCl2=CCl2−mHm−1−CCl3−nHn (2)
(式中、mは1又は2であり、nは0~3の整数である。) - 熱分解温度が350~550℃である、請求項1に記載の方法。
- 上記熱脱塩化水素工程の後、さらに上記一般式(2)で表される不飽和化合物と塩素とを反応させて下記一般式(3)で表される飽和化合物を得る塩素化工程を行う、請求項1又は2に記載の方法。
CCl3−CCl3−mHm−1−CCl3−nHn (3)
(式中、mは1又は2であり、nは0~3の整数である。) - 上記熱脱塩化水素工程の後、得られた反応混合物から塩化水素を分離せずに該反応混合物を上記塩素化工程に供する、請求項3に記載の方法。
- 上記熱脱塩化水素工程の後、生成した反応混合物から塩化水素を分離し、該分離した塩化水素を酸化して塩素へ変換し、該塩素を上記塩素化工程の塩素源として用いる、請求項3に記載の方法。
- 上記一般式(1)で表される飽和化合物が、四塩化炭素と下記一般式(0)で表される不飽和化合物とを、液相の反応系中で鉄−リン酸エステル触媒の存在下に付加させる付加反応工程によって得られた下記一般式(1’)で表される飽和化合物である、請求項3に記載の方法。
CCl2−mHm=CCl2−pHP (0)
CCl3−CCl2−mHm−CCl3−pHp (1’)
(式中、mは1又は2であり、pは0~2の整数であり、ただしm≧pである。) - 上記付加反応工程における鉄−リン酸エステル触媒が、反応系に鉄とリン酸エステルとを添加して反応系中で調製されるものであり、
反応開始前に鉄の全量及びリン酸エステルの一部を添加し、残余のリン酸エステルを上記付加反応進行中に追加添加する、請求項6に記載の方法。 - 上記付加反応工程後、且つ上記熱脱塩化水素工程前に、上記一般式(1)で表される飽和化合物の蒸留精製を行う、請求項6に記載の方法。
- 四塩化炭素と下記一般式(0)で表される不飽和化合物とを、液相の反応系中で鉄−リン酸エステル触媒の存在下に付加させる付加反応工程によって下記一般式(1’)で表される飽和化合物を得る、塩素化炭化水素を製造するための方法であって、
上記付加反応工程における鉄−リン酸エステル触媒が、反応系に鉄とリン酸エステルとを添加して反応系中で調製されるものであり、
反応開始前に鉄の全量及びリン酸エステルの一部を添加し、残余のリン酸エステルを上記付加反応進行中に追加添加することを特徴とする、前記方法。
CCl2−mHm=CCl2−PHP (0)
CCl3−CCl2−mHm−CCl3−PHP (1’)
(式中、mは1又は2であり、pは0~2の整数であり、ただしm≧pである。) - 四塩化炭素と下記一般式(0)で表される不飽和化合物とを、液相の反応系中で鉄−リン酸エステル触媒の存在下に付加させて下記一般式(1’)で表される飽和化合物を得る付加反応工程を行い、次いで
下記一般式(1’)で表される飽和化合物を熱分解して下記一般式(2’)で表される不飽和化合物を得る熱脱塩化水素工程を行い、そして
下記一般式(2’)で表される不飽和化合物と塩素とを反応させて下記一般式(3’)で表される飽和化合物を得る塩素化工程を行う、塩素化炭化水素を製造するための方法であって、
前記付加反応工程後、且つ上記熱脱塩化水素工程前に、下記一般式(1’)で表される飽和化合物の蒸留精製を行うことを特徴とする、前記方法。
CCl2−mHm=CCl2−PHP (0)
CCl3−CCl2−mHm−CCl3−PHP (1’)
CCl2=CCl2−mHm−1−CCl3−PHP (2’)
CCl3−CCl3−mHm−1−CCl3−PHP (3’)
(式中、m1又は2であり、pは0~2の整数であり、ただしm≧pである。)
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011502583A JP4855550B2 (ja) | 2009-06-24 | 2010-06-17 | 塩素化炭化水素の製造方法 |
| US13/319,912 US8877990B2 (en) | 2009-06-24 | 2010-06-17 | Process of making a chlorinated hydrocarbon |
| EP10792153A EP2447238A4 (en) | 2009-06-24 | 2010-06-17 | METHOD FOR PRODUCING CHLORINE-CARBONATED HYDROGEN |
| CN201080002886.3A CN102177116B (zh) | 2009-06-24 | 2010-06-17 | 氯化烃的制备方法 |
| US13/406,238 US8304589B2 (en) | 2009-06-24 | 2012-02-27 | Process of making a chlorinated hydrocarbon |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009-150096 | 2009-06-24 | ||
| JP2009150096 | 2009-06-24 | ||
| JP2009-164149 | 2009-07-10 | ||
| JP2009164149 | 2009-07-10 | ||
| JP2009208370 | 2009-09-09 | ||
| JP2009-208370 | 2009-09-09 | ||
| JP2009210812 | 2009-09-11 | ||
| JP2009-210812 | 2009-09-11 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/319,912 A-371-Of-International US8877990B2 (en) | 2009-06-24 | 2010-06-17 | Process of making a chlorinated hydrocarbon |
| US13/406,238 Division US8304589B2 (en) | 2009-06-24 | 2012-02-27 | Process of making a chlorinated hydrocarbon |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010150835A1 true WO2010150835A1 (ja) | 2010-12-29 |
Family
ID=43386604
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/060695 WO2010150835A1 (ja) | 2009-06-24 | 2010-06-17 | 塩素化炭化水素の製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US8877990B2 (ja) |
| EP (2) | EP2447238A4 (ja) |
| JP (2) | JP4855550B2 (ja) |
| CN (1) | CN102177116B (ja) |
| MY (1) | MY152994A (ja) |
| WO (1) | WO2010150835A1 (ja) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012081482A1 (ja) * | 2010-12-16 | 2012-06-21 | 株式会社トクヤマ | 炭素数3の塩素化炭化水素の製造方法 |
| JP2013189402A (ja) * | 2012-03-14 | 2013-09-26 | Tokuyama Corp | ポリクロロプロパンの製造方法 |
| EP2814796A4 (en) * | 2012-02-14 | 2015-10-28 | Honeywell Int Inc | METHOD FOR PRODUCING TETRAFLUORPROPES |
| JP2016509072A (ja) * | 2013-03-09 | 2016-03-24 | ブルー キューブ アイピー エルエルシー | クロロアルカンの製造方法 |
| WO2016178433A1 (ja) * | 2015-05-07 | 2016-11-10 | 株式会社トクヤマ | 脱塩化水素反応を伴うクロロアルケンの製造方法 |
| JP2017149689A (ja) * | 2016-02-25 | 2017-08-31 | 株式会社トクヤマ | クロロプロパン類の製造方法 |
| JP2017178897A (ja) * | 2016-03-31 | 2017-10-05 | 株式会社トクヤマ | クロロプロパン類の製造方法 |
| JP2017531014A (ja) * | 2014-10-16 | 2017-10-19 | アルケマ フランス | 1,1,1,2,3−ペンタクロロプロパンに基づく組成物 |
| JP2017532379A (ja) * | 2014-10-16 | 2017-11-02 | スポレク プロ ヘミコウ アー フツニ ブイロブ,アクツィオバ スポレチェノスト | 方法 |
| JP2017532380A (ja) * | 2014-10-16 | 2017-11-02 | スポレク プロ ヘミコウ アー フツニ ブイロブ,アクツィオバ スポレチェノスト | 方法 |
| JP2017534679A (ja) * | 2014-10-16 | 2017-11-24 | スポレク プロ ヘミコウ アー フツニ ブイロブ,アクツィオバ スポレチェノスト | 高純度塩素化アルカンの製造方法 |
| JP2018527393A (ja) * | 2015-09-21 | 2018-09-20 | アーケマ・インコーポレイテッド | ペンタクロロプロパンの触媒気相脱塩化水素化によるテトラクロロプロペンの製造方法 |
| JP7348993B2 (ja) | 2015-08-19 | 2023-09-21 | スポルケミ ゼブラ,アクツィオバ スポレチェノスト | C3塩素化アルカン及びアルケン化合物の製造方法 |
Families Citing this family (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5495676B2 (ja) * | 2009-08-27 | 2014-05-21 | 株式会社トクヤマ | クロロアルカンの製造方法 |
| CN102630221B (zh) | 2009-11-27 | 2015-03-25 | 大金工业株式会社 | 制备1,1,2,3-四氯丙烯的方法 |
| EP2433922A1 (en) * | 2010-08-25 | 2012-03-28 | Syngenta Participations AG | Process for the preparation of dichlorofulvene |
| JP6158032B2 (ja) * | 2012-10-17 | 2017-07-05 | 株式会社トクヤマ | 反応ガスをリサイクルした塩素化プロパンの製造方法 |
| US9289758B2 (en) | 2013-01-22 | 2016-03-22 | Axiall Ohio, Inc. | Processes for producing chlorinated hydrocarbons and methods for recovering polyvalent antimony catalysts therefrom |
| US8889930B2 (en) | 2013-01-22 | 2014-11-18 | Axiall Ohio, Inc. | Process for producing chlorinated hydrocarbons |
| US20140215970A1 (en) * | 2013-02-04 | 2014-08-07 | Honeywell International Inc. | METHODS OF HANDLING CHLORINATED COMPOUNDS USED FOR MANUFACTURING HFO-1234yf |
| CN104058925B (zh) * | 2013-03-22 | 2017-07-07 | 中化蓝天集团有限公司 | 1,1,1,2,3‑五氯丙烷的连续制备方法 |
| CN103524291B (zh) * | 2013-09-18 | 2015-09-23 | 巨化集团技术中心 | 一种氯代烷烃的连续合成方法 |
| US9139497B2 (en) | 2013-10-23 | 2015-09-22 | Axiall Ohio, Inc. | Process for producing chlorinated hydrocarbons in the presence of a polyvalent bismuth compound |
| US9255045B2 (en) | 2014-01-13 | 2016-02-09 | Arkema France | E-1-chloro-3,3,3-trifluoropropene production process from 1,1,3,3-tetrachloropropene |
| CA3226491A1 (en) | 2014-05-16 | 2015-11-19 | Occidental Chemical Corporation | Method for making 1,1,3,3-tetrachloropropene |
| JP6791860B2 (ja) | 2014-10-16 | 2020-11-25 | スポレク プロ ヘミコウ アー フツニ ブイロブ,アクツィオバ スポレチェノスト | プロセス |
| FR3036398B1 (fr) * | 2015-05-22 | 2019-05-03 | Arkema France | Compositions a base de 1,1,3,3-tetrachloropropene |
| CN109071382B (zh) | 2016-04-13 | 2022-04-15 | 斯波齐梅斑马公司 | 方法 |
| US20190127303A1 (en) * | 2016-04-13 | 2019-05-02 | Arkema France | Process for the manufacture of 2,3,3,3-tetrafluoropropene |
| JP6751239B2 (ja) | 2016-04-19 | 2020-09-02 | セントラル硝子株式会社 | 1,2−ジクロロ−3,3,3−トリフルオロプロペンの製造方法 |
| FR3081158B1 (fr) | 2018-05-16 | 2020-07-31 | Arkema France | Procede de production du 1-chloro-3,3,3-trifluoropropene. |
| CN108658722A (zh) * | 2018-06-21 | 2018-10-16 | 岳阳景嘉化工有限公司 | 一种1,2,3-三氯丙烷的合成方法 |
| CN108640812A (zh) * | 2018-06-21 | 2018-10-12 | 岳阳景嘉化工有限公司 | 一种2,3-二氯丙烯的合成方法 |
| FR3083232B1 (fr) | 2018-06-27 | 2021-11-12 | Arkema France | Procede de production du 1-chloro-3,3,3-trifluoropropene |
| FR3086287B1 (fr) | 2018-09-26 | 2020-09-18 | Arkema France | Stabilisation du 1-chloro-3,3,3-trifluoropropene |
| CN109796300B (zh) * | 2018-12-29 | 2020-12-18 | 浙江巨化技术中心有限公司 | 一种2,3,3,3-四氟丙烯的连续制备方法 |
| CN110903161A (zh) * | 2019-12-03 | 2020-03-24 | 湖南莱万特化工有限公司 | 一种1,2,3-三氯丙烷化合物制备方法及其制备装置 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS504006A (ja) * | 1971-12-17 | 1975-01-16 | ||
| JPS6036429A (ja) * | 1983-07-06 | 1985-02-25 | モンサント・カンパニー | 1,1,2,3‐テトラクロロプロペンの製造方法 |
| JP2007023050A (ja) * | 1994-05-16 | 2007-02-01 | Allied Signal Inc | フッ素化オレフィンの製造方法およびフッ素化オレフィンを用いるヒドロフルオロカーボンの製造方法 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE261689C (ja) * | ||||
| US2725411A (en) * | 1948-04-30 | 1955-11-29 | Us Rubber Co | Vapor phase dehydrohalogenation of polyhalogenomethyl compounds |
| US3823195A (en) | 1971-12-27 | 1974-07-09 | Monsanto Co | Preparation of 1,1,2,3-tetrachloropropene from 1,2,3-trichloropropane |
| US3926758A (en) | 1971-12-27 | 1975-12-16 | Monsanto Co | Preparation of 1,1,2,3-tetrachloropropene from 2,3-trichloropropane |
| US4650914A (en) * | 1983-07-06 | 1987-03-17 | Monsanto Company | Process for producing 1,1,2,3-tetrachloropropene |
| DE3460965D1 (en) * | 1983-07-06 | 1986-11-20 | Monsanto Co | Process for producing monoadducts of olefins and telogens reactive therewith |
| US20050177012A1 (en) * | 2001-07-20 | 2005-08-11 | Pcbu Services, Inc. | Halocarbon production processes, halocarbon separation processes, and halocarbon separation systems |
| US7335806B2 (en) * | 2006-02-14 | 2008-02-26 | Ppg Industries Ohio, Inc. | Integrated process for producing 1,2-dichloroethylene |
| JP5360230B2 (ja) * | 2009-04-23 | 2013-12-04 | ダイキン工業株式会社 | 2−クロロ−3,3,3−トリフルオロプロペンの製造方法 |
| US8779219B2 (en) * | 2009-05-13 | 2014-07-15 | Daikin Industries, Ltd. | Process for preparing chlorine-containing fluorocarbon compound |
| CN102630221B (zh) * | 2009-11-27 | 2015-03-25 | 大金工业株式会社 | 制备1,1,2,3-四氯丙烯的方法 |
-
2010
- 2010-06-17 EP EP10792153A patent/EP2447238A4/en not_active Withdrawn
- 2010-06-17 US US13/319,912 patent/US8877990B2/en not_active Expired - Fee Related
- 2010-06-17 EP EP12157901A patent/EP2463261A3/en not_active Withdrawn
- 2010-06-17 JP JP2011502583A patent/JP4855550B2/ja not_active Expired - Fee Related
- 2010-06-17 WO PCT/JP2010/060695 patent/WO2010150835A1/ja active Application Filing
- 2010-06-17 CN CN201080002886.3A patent/CN102177116B/zh not_active Expired - Fee Related
- 2010-06-17 MY MYPI2011005311 patent/MY152994A/en unknown
-
2011
- 2011-09-02 JP JP2011191859A patent/JP5501313B2/ja not_active Expired - Fee Related
-
2012
- 2012-02-27 US US13/406,238 patent/US8304589B2/en not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS504006A (ja) * | 1971-12-17 | 1975-01-16 | ||
| JPS6036429A (ja) * | 1983-07-06 | 1985-02-25 | モンサント・カンパニー | 1,1,2,3‐テトラクロロプロペンの製造方法 |
| JPH0247969B2 (ja) | 1983-07-06 | 1990-10-23 | Monsanto Co | |
| JP2007023050A (ja) * | 1994-05-16 | 2007-02-01 | Allied Signal Inc | フッ素化オレフィンの製造方法およびフッ素化オレフィンを用いるヒドロフルオロカーボンの製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2447238A4 |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012081482A1 (ja) * | 2010-12-16 | 2012-06-21 | 株式会社トクヤマ | 炭素数3の塩素化炭化水素の製造方法 |
| US8912371B2 (en) | 2010-12-16 | 2014-12-16 | Tokuyama Corporation | Method of producing a chlorinated hydrocarbon having 3 carbon atoms |
| EP2814796A4 (en) * | 2012-02-14 | 2015-10-28 | Honeywell Int Inc | METHOD FOR PRODUCING TETRAFLUORPROPES |
| EP3339280A1 (en) * | 2012-02-14 | 2018-06-27 | Honeywell International Inc. | Process for making tetrafluoropropene |
| JP2017061485A (ja) * | 2012-02-14 | 2017-03-30 | ハネウェル・インターナショナル・インコーポレーテッド | テトラフルオロプロペンの作製方法 |
| JP2013189402A (ja) * | 2012-03-14 | 2013-09-26 | Tokuyama Corp | ポリクロロプロパンの製造方法 |
| JP2016509072A (ja) * | 2013-03-09 | 2016-03-24 | ブルー キューブ アイピー エルエルシー | クロロアルカンの製造方法 |
| JP2017534679A (ja) * | 2014-10-16 | 2017-11-24 | スポレク プロ ヘミコウ アー フツニ ブイロブ,アクツィオバ スポレチェノスト | 高純度塩素化アルカンの製造方法 |
| JP2017531014A (ja) * | 2014-10-16 | 2017-10-19 | アルケマ フランス | 1,1,1,2,3−ペンタクロロプロパンに基づく組成物 |
| JP2017532379A (ja) * | 2014-10-16 | 2017-11-02 | スポレク プロ ヘミコウ アー フツニ ブイロブ,アクツィオバ スポレチェノスト | 方法 |
| JP2017532380A (ja) * | 2014-10-16 | 2017-11-02 | スポレク プロ ヘミコウ アー フツニ ブイロブ,アクツィオバ スポレチェノスト | 方法 |
| US10494321B2 (en) | 2014-10-16 | 2019-12-03 | Arkema France | Compositions comprising 1,1,1,2,3 pentachloropropane |
| JP2020203898A (ja) * | 2014-10-16 | 2020-12-24 | アルケマ フランス | 1,1,1,2,3−ペンタクロロプロパンに基づく組成物 |
| JP7168321B2 (ja) | 2014-10-16 | 2022-11-09 | スポレク プロ ヘミコウ アー フツニ ブイロブ,アクツィオバ スポレチェノスト | 方法 |
| WO2016178433A1 (ja) * | 2015-05-07 | 2016-11-10 | 株式会社トクヤマ | 脱塩化水素反応を伴うクロロアルケンの製造方法 |
| JP7348993B2 (ja) | 2015-08-19 | 2023-09-21 | スポルケミ ゼブラ,アクツィオバ スポレチェノスト | C3塩素化アルカン及びアルケン化合物の製造方法 |
| JP2018527393A (ja) * | 2015-09-21 | 2018-09-20 | アーケマ・インコーポレイテッド | ペンタクロロプロパンの触媒気相脱塩化水素化によるテトラクロロプロペンの製造方法 |
| JP2017149689A (ja) * | 2016-02-25 | 2017-08-31 | 株式会社トクヤマ | クロロプロパン類の製造方法 |
| JP2017178897A (ja) * | 2016-03-31 | 2017-10-05 | 株式会社トクヤマ | クロロプロパン類の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP4855550B2 (ja) | 2012-01-18 |
| EP2463261A2 (en) | 2012-06-13 |
| EP2463261A3 (en) | 2012-12-19 |
| MY152994A (en) | 2014-12-31 |
| JPWO2010150835A1 (ja) | 2012-12-10 |
| JP5501313B2 (ja) | 2014-05-21 |
| CN102177116A (zh) | 2011-09-07 |
| US8877990B2 (en) | 2014-11-04 |
| EP2447238A4 (en) | 2012-12-19 |
| US20120157723A1 (en) | 2012-06-21 |
| US20120053374A1 (en) | 2012-03-01 |
| US8304589B2 (en) | 2012-11-06 |
| EP2447238A1 (en) | 2012-05-02 |
| CN102177116B (zh) | 2014-07-16 |
| JP2012036190A (ja) | 2012-02-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4855550B2 (ja) | 塩素化炭化水素の製造方法 | |
| JP6527575B2 (ja) | 高純度e−1−クロロ−3,3,3−トリフルオロプロペン及びその製造方法 | |
| ES2728780T3 (es) | Síntesis de 1,1,2,3-tetracloropropeno | |
| WO2012081482A1 (ja) | 炭素数3の塩素化炭化水素の製造方法 | |
| JP6258973B2 (ja) | 塩素化炭化水素の生成プロセス | |
| JP5532131B2 (ja) | 1,1,2,3−テトラクロロプロペンの製造方法 | |
| JP5360230B2 (ja) | 2−クロロ−3,3,3−トリフルオロプロペンの製造方法 | |
| JP2018109010A (ja) | 2,3,3,3−テトラフルオロプロぺンの製造方法 | |
| TW200936538A (en) | Methods of making chlorinated hydrocarbons | |
| JP2015529247A (ja) | 塩素化プロペンの生成のためのプロセス | |
| JP2011057650A (ja) | クロロプロペンの製造方法 | |
| JP5653833B2 (ja) | ポリクロロプロパンの製造方法 | |
| JP5495676B2 (ja) | クロロアルカンの製造方法 | |
| JP5610917B2 (ja) | クロロプロパンの製造方法 | |
| JP5709656B2 (ja) | ポリクロロプロパンの製造方法 | |
| JP6173908B2 (ja) | クロロ高次アルケンの製造方法 | |
| JP5858830B2 (ja) | ポリクロロプロパンの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080002886.3 Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: 2011502583 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10792153 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13319912 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010792153 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |