WO2010143722A1 - 修飾セルロース繊維及びそのセルロース複合体 - Google Patents
修飾セルロース繊維及びそのセルロース複合体 Download PDFInfo
- Publication number
- WO2010143722A1 WO2010143722A1 PCT/JP2010/059976 JP2010059976W WO2010143722A1 WO 2010143722 A1 WO2010143722 A1 WO 2010143722A1 JP 2010059976 W JP2010059976 W JP 2010059976W WO 2010143722 A1 WO2010143722 A1 WO 2010143722A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cellulose
- cellulose fiber
- aromatic ring
- modified
- dispersion
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/04—Reinforcing macromolecular compounds with loose or coherent fibrous material
- C08J5/045—Reinforcing macromolecular compounds with loose or coherent fibrous material with vegetable or animal fibrous material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82Y—SPECIFIC USES OR APPLICATIONS OF NANOSTRUCTURES; MEASUREMENT OR ANALYSIS OF NANOSTRUCTURES; MANUFACTURE OR TREATMENT OF NANOSTRUCTURES
- B82Y30/00—Nanotechnology for materials or surface science, e.g. nanocomposites
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B11/00—Preparation of cellulose ethers
- C08B11/16—Aryl or aralkyl ethers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/005—Reinforced macromolecular compounds with nanosized materials, e.g. nanoparticles, nanofibres, nanotubes, nanowires, nanorods or nanolayered materials
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L1/00—Compositions of cellulose, modified cellulose or cellulose derivatives
- C08L1/08—Cellulose derivatives
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M13/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment
- D06M13/10—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment with compounds containing oxygen
- D06M13/11—Compounds containing epoxy groups or precursors thereof
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M13/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment
- D06M13/10—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment with compounds containing oxygen
- D06M13/152—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment with compounds containing oxygen having a hydroxy group bound to a carbon atom of a six-membered aromatic ring
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M13/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment
- D06M13/10—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment with compounds containing oxygen
- D06M13/184—Carboxylic acids; Anhydrides, halides or salts thereof
- D06M13/1845—Aromatic mono- or polycarboxylic acids
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M13/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment
- D06M13/322—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment with compounds containing nitrogen
- D06M13/395—Isocyanates
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M13/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment
- D06M13/50—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment with organometallic compounds; with organic compounds containing boron, silicon, selenium or tellurium atoms
- D06M13/503—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment with organometallic compounds; with organic compounds containing boron, silicon, selenium or tellurium atoms without bond between a carbon atom and a metal or a boron, silicon, selenium or tellurium atom
- D06M13/507—Organic silicon compounds without carbon-silicon bond
-
- D—TEXTILES; PAPER
- D21—PAPER-MAKING; PRODUCTION OF CELLULOSE
- D21H—PULP COMPOSITIONS; PREPARATION THEREOF NOT COVERED BY SUBCLASSES D21C OR D21D; IMPREGNATING OR COATING OF PAPER; TREATMENT OF FINISHED PAPER NOT COVERED BY CLASS B31 OR SUBCLASS D21G; PAPER NOT OTHERWISE PROVIDED FOR
- D21H11/00—Pulp or paper, comprising cellulose or lignocellulose fibres of natural origin only
- D21H11/16—Pulp or paper, comprising cellulose or lignocellulose fibres of natural origin only modified by a particular after-treatment
-
- D—TEXTILES; PAPER
- D21—PAPER-MAKING; PRODUCTION OF CELLULOSE
- D21H—PULP COMPOSITIONS; PREPARATION THEREOF NOT COVERED BY SUBCLASSES D21C OR D21D; IMPREGNATING OR COATING OF PAPER; TREATMENT OF FINISHED PAPER NOT COVERED BY CLASS B31 OR SUBCLASS D21G; PAPER NOT OTHERWISE PROVIDED FOR
- D21H11/00—Pulp or paper, comprising cellulose or lignocellulose fibres of natural origin only
- D21H11/16—Pulp or paper, comprising cellulose or lignocellulose fibres of natural origin only modified by a particular after-treatment
- D21H11/20—Chemically or biochemically modified fibres
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2301/00—Characterised by the use of cellulose, modified cellulose or cellulose derivatives
- C08J2301/08—Cellulose derivatives
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2351/00—Characterised by the use of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Derivatives of such polymers
- C08J2351/02—Characterised by the use of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Derivatives of such polymers grafted on to polysaccharides
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M2101/00—Chemical constitution of the fibres, threads, yarns, fabrics or fibrous goods made from such materials, to be treated
- D06M2101/02—Natural fibres, other than mineral fibres
- D06M2101/04—Vegetal fibres
- D06M2101/06—Vegetal fibres cellulosic
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/29—Coated or structually defined flake, particle, cell, strand, strand portion, rod, filament, macroscopic fiber or mass thereof
- Y10T428/2913—Rod, strand, filament or fiber
- Y10T428/298—Physical dimension
Definitions
- the present invention relates to a modified cellulose fiber and a composite using the modified cellulose fiber, and in particular, by controlling the modified substituent and the modification rate of the modified cellulose fiber, high heat resistance, easy defibration, and high production
- the present invention relates to a technology that achieves high transparency, a low linear expansion coefficient, a high elastic modulus, an improvement in strength when heated, and the like when a cellulose composite is realized.
- Patent Document 1 a composite material with improved hygroscopicity and transparency is obtained by compositing using surface-modified cellulose fine fibers.
- this surface modifier an aliphatic type is disclosed, and it is disclosed that an acetyl group or a methacryloyl group is particularly preferable.
- Patent Document 2 discloses a non-woven fabric made of modified fibers into which a compound having an aromatic ring structure is introduced as a compound for controlling optical anisotropy.
- Non-Patent Document 1 discloses benzoylated bacterial cellulose exhibiting thermotropic liquid crystal characteristics.
- the method for producing a cellulose fine fiber nonwoven fabric (cellulose sheet) specifically disclosed in Patent Document 1 described above is a method of dehydrating a mechanically defibrated cellulose fine fiber aqueous suspension by filtration through a glass filter. This is a method of forming a film.
- the present inventor manufactured a cellulose composite material according to the method disclosed in Patent Document 1, it took filtration for dehydration of an aqueous suspension of cellulose fine fibers when manufacturing a cellulose sheet. It has been found that practical productivity cannot be obtained due to the long time.
- the non-woven fabric (cellulose sheet) specifically disclosed in Patent Document 2 reacts an aromatic ring-containing compound and cellulose in the presence of N, N-dimethylformamide, ethyl cellosolve, or the like.
- N, N-dimethylformamide, ethyl cellosolve, or the like reacts an aromatic ring-containing compound and cellulose in the presence of N, N-dimethylformamide, ethyl cellosolve, or the like.
- the present inventor manufactured a cellulose sheet according to the method disclosed in Patent Document 2
- the crystal structure of cellulose was clearly destroyed and the proportion of the I-type crystal structure was reduced.
- Cellulose sheets with a low proportion of I-type crystal structure are inferior in heat resistance, so even if this cellulose sheet and a polymer material are combined, the low linear expansion coefficient and high elastic modulus are not achieved. It is thought that the strength decreases due to the effect of
- the benzoylated bacterial cellulose obtained by the method of Non-Patent Document 1 is considered to be amorphous because it has a glass transition temperature. Even if this is combined with a polymer material, a low linear expansion coefficient, a high It is not elastic modulus and high strength.
- the present invention has been made in view of the above-described conventional situation, and has high transparency when a cellulose fiber that achieves high heat resistance, easy defibration, and high productivity, and a composite thereof. It aims at realizing a low linear expansion coefficient and a high elastic modulus.
- the present inventor uses an organic acid as a solvent in the reaction for controlling or modifying the average fiber diameter, crystallinity, modification substituent and modification rate of the modified cellulose fiber. And found that high heat resistance, easy defibration, and high productivity can be achieved. That is, the gist of the present invention is as follows. [1] A method for producing a modified cellulose fiber having a modification reaction step in which cellulose is reacted with an aromatic compound in a solvent to modify the cellulose with an aromatic ring-containing substituent, and as the solvent in the modification reaction step A method for producing a modified cellulose fiber, characterized by using an organic acid.
- [2] The modified cellulose fiber according to [1], wherein the aromatic ring-containing substituent is one or more groups selected from the group consisting of an aromatic ring-containing ester group, an aromatic ring-containing ether group, and an aromatic ring-containing carbamate group. Manufacturing method. [3] The method for producing a modified cellulose fiber according to [1] or [2], wherein the modification rate of the aromatic ring-containing substituent with respect to all hydroxyl groups of cellulose is 10 mol% or more. [4] A modified cellulose fiber having a modification reaction step of modifying cellulose with an aromatic ring-containing substituent by reacting cellulose and an aromatic compound in a cellulose dispersion containing cellulose and an organic acid, and a papermaking step Sheet manufacturing method.
- the modified cellulose fiber obtained by the method according to any one of [1] to [3] or the modified cellulose fiber obtained by the method according to any one of [4] to [6] A method for producing a cellulose fiber composite, comprising a step of combining a sheet and a matrix.
- a dispersion of cellulose fibers having a cellulose I-type crystal structure in which 10 mol% or more of all hydroxyl groups of cellulose are modified with an aromatic ring-containing substituent, and “scanning” of a wide-angle X-ray diffraction image of the cellulose fibers A modified cellulose fiber dispersion in which the ratio of “average diffraction intensity at 18 to 19 degrees in the same scanning angle” to “intensity of diffraction peak derived from cellulose type I crystals at an angle of 20 to 24 degrees” is 0.8 or less.
- a modified cellulose fiber sheet comprising cellulose fibers having a cellulose I-type crystal structure having an average fiber diameter of 100 nm or less and 10 mol% or more of all hydroxyl groups of cellulose modified with an aromatic ring-containing substituent, The modified cellulose fiber sheet whose haze value by C light when it is set as the composite material shown to following (1) is 3 or less.
- a cellulose fiber sheet having a thickness adjusted to 40 g / m 2 was added to 96 parts by weight of bis (methacryloyloxymethyl) tricyclo [5.2.1.0 2,6 ] decane, pentaerythritol tetrakis ( ⁇ -Thiopropionate) After impregnating a mixed solution of 6 parts by weight, 2,4,6-trimethylbenzoyldiphenylphosphine oxide and 0.05 part by weight of benzophenone, and placing it under reduced pressure overnight.
- a cellulose fiber composite comprising the modified cellulose fiber according to any one of [11] to [13] or the modified cellulose fiber sheet according to [14] and a matrix.
- a cellulose fiber composite comprising a cellulose fiber having a cellulose I-type crystal structure in which 10 mol% or more of the total hydroxyl groups of cellulose are modified with an aromatic ring-containing substituent, and a matrix, the wide angle of the cellulose fiber Cellulose fiber having a ratio of “average diffraction intensity at 18 to 19 degrees in the same scanning angle” to “intensity of diffraction peaks derived from cellulose type I crystals at a scanning angle of 20 to 24 degrees” in the X-ray diffraction image is 0.8 or less Complex.
- the present invention by using an organic acid as a solvent in the reaction for controlling or modifying the average fiber diameter, crystallinity, modification substituent and modification rate of the modified cellulose fiber, high heat resistance and easy defibration In addition to realizing high productivity, it is possible to realize high transparency, non-coloring properties, a low linear expansion coefficient, a high elastic modulus, and improved strength during heating when a cellulose composite is formed. Furthermore, by using cellulose fibers modified with an aromatic ring-containing substituent, the effect of increasing the affinity with a resin, particularly the affinity with a resin containing an aromatic ring, is achieved.
- the method for producing the modified cellulose fiber dispersion of the present invention is a modification reaction in which cellulose is reacted with an aromatic compound in a solvent to modify the cellulose with an aromatic ring-containing substituent. And an organic acid is used as the solvent during the modification reaction.
- the cellulose in the present invention is obtained by removing impurities from a cellulose-containing material as shown below through general purification.
- Cellulose-containing materials include wood such as conifers and hardwoods, cotton such as cotton linter and cotton lint, pomace such as sugar cane and sugar radish, bast fibers such as flax, ramie, jute and kenaf, leaf veins such as sisal and pineapple Examples thereof include fiber, leaf pattern fibers such as abaca and banana, fruit fibers such as coconut palm, stem stem fibers such as bamboo, bacterial cellulose produced by bacteria, seaweeds such as valonia and falcons, and scallops. These natural celluloses are preferable because of their high crystallinity and low linear expansion and high elastic modulus.
- Bacterial cellulose is preferred in that it can be easily obtained with a fine fiber diameter.
- Cotton is also preferable in that it is easy to obtain a fine fiber diameter, and more preferable in terms of easy acquisition of raw materials.
- wood of conifers and hardwoods with fine fiber diameters can be obtained, and it is the largest amount of biological resources on the earth, and it is a sustainable resource that produces about 70 billion tons per year. Therefore, it contributes greatly to the reduction of carbon dioxide that affects global warming, which is advantageous from an economic point of view.
- Such a cellulose-containing material is subjected to general purification to obtain the cellulose raw material of the present invention.
- the fiber diameter of the cellulose used in the present invention is not particularly limited, and the number average fiber diameter is from several ⁇ m to several mm. What passed through general refining is about a few millimeters, and those obtained by defibrating cellulose are a few nanometers. For example, when a chip or the like having a size of several centimeters is refined, it is preferable to perform a mechanical treatment with a disaggregator such as a refiner or a beater to make the thickness about several mm.
- the number average fiber diameter measurement method is not particularly limited. For example, by observing with SEM, TEM, etc., draw a diagonal line in the photograph, and randomly extract 12 fibers in the vicinity, It can be obtained by averaging 10 measured values from which the finest fibers are removed.
- the cellulose used in the present invention is obtained by purifying the cellulose-containing material derived from the above by an ordinary method. For example, it can be obtained by degreasing with benzene-ethanol, followed by delignification treatment by the Wise method and dehemicellulose treatment with alkali. Or it is obtained by the manufacturing method of a general chemical pulp, for example, the manufacturing method of a kraft pulp, a salified pulp, and an alkali pulp. In general, a cellulose-containing material is heat-treated in a digester to perform delignification or the like, and further to bleaching or the like.
- cellulose is modified under conditions where an organic acid is present as a solvent.
- a hydroxyl group in cellulose is chemically modified by reacting with a chemical modifier.
- Modified substituent The functional group introduced into cellulose by chemical modification is an aromatic ring-containing substituent. Modification with an aromatic ring-containing substituent is preferable for achieving high heat resistance, easy defibration, and high productivity.
- cellulose modified with an aromatic ring-containing substituent is superior in heat resistance and easy defibration compared to unmodified cellulose or cellulose modified with an aliphatic-containing substituent.
- the aromatic ring-containing substituent is a substituent derived from a hydrocarbon aromatic compound, a heterocyclic aromatic compound, or a non-benzenoid aromatic compound.
- the hydrocarbon aromatic compound is a compound obtained by condensing 2 to 12 monocyclic benzene rings such as benzene and naphthalene or anthracene.
- the upper limit of the number of condensation is preferably 6 or less.
- the heterocyclic aromatic compound is a 5- to 10-membered heterocyclic monocyclic ring such as furan, thiophene, pyrrole or imidazole, or a compound obtained by condensing 2 to 12 thereof.
- the upper limit of the number of condensation is preferably 6 or less.
- the non-benzenoid aromatic compound include annulene, cyclopentadienyl anion, cycloheptatrienyl cation, tropone, metallocene, acebreazilene, and the like.
- the substituent derived from a hydrocarbon aromatic compound and a heterocyclic aromatic compound is preferable, and the substituent derived from a hydrocarbon aromatic compound is more preferable.
- substituents derived from benzene, naphthalene and anthracene are preferred from the viewpoint of easy availability of raw materials.
- aromatic ring-containing substituents may have hydrogen substituted with alkyl having 1 to 12 carbon atoms.
- the aromatic ring-containing substituent may be composed of two or more of the above hydrocarbon aromatic compounds, heterocyclic aromatic compounds and non-benzenoid aromatic compounds connected by a single bond or alkyl having 1 to 3 carbon atoms.
- the linking group that bonds the aromatic ring and cellulose is not particularly limited as long as it is obtained as a result of the reaction with the hydroxyl group of cellulose, but an ester group, an ether group, a carbamate group. Are preferred, and ester groups are particularly preferred.
- a benzoyl group As the aromatic ring-containing substituent of the modified substituent introduced into the modified cellulose of the present invention, a benzoyl group, a naphthoyl group, an anthroyl group, and the like are preferable, and a benzoyl group is particularly preferable.
- the modification method is not particularly limited, and there is a method of reacting cellulose with a chemical modifier as described below. In the present invention, these reactions are carried out under the condition that an organic acid is present as a solvent. However, if necessary, other solvents, catalysts, etc. can be used together, or heating, decompression, etc. can be carried out.
- the types of chemical modifiers include, for example, when an ester group is formed, an acid, an acid anhydride, a halogenating reagent, etc., and when an ether group is formed, a phenolic compound, phenoxysilane, oxirane (epoxy), etc. When the cyclic ether compound or the like forms a carbamate group, an isocyanate compound is exemplified. These chemical modifiers may be used alone or in combination of two or more.
- Examples of the acid that is a chemical modifier that forms an ester group include benzoic acid and naphthalenecarboxylic acid, and examples of the acid anhydride include benzoic anhydride and phthalic anhydride.
- Examples of the halogenating reagent include benzoyl halide and naphthoyl halide.
- phenol compound that is a chemical modifier that forms an ether group examples include phenol, naphthol, and the like, and cyclic ether compounds such as phenoxysilane, phenyloxirane (epoxy), and the like.
- benzoyl halide naphthoyl halide
- benzoic anhydride are particularly preferable.
- These chemical modifiers may be used alone or in combination of two or more.
- chemical modification rate refers to the proportion of all hydroxyl groups in cellulose that have been chemically modified, and the chemical modification rate can be measured by the following titration method.
- ⁇ Measuring method> 0.05 g of the dried modified cellulose is precisely weighed, and 6 ml of methanol and 2 ml of distilled water are added thereto. The mixture is stirred at 60 to 70 ° C. for 30 minutes, and 10 ml of 0.05N aqueous sodium hydroxide solution is added. The mixture is stirred at 60 to 70 ° C. for 15 minutes and further stirred at room temperature for one day. Titrate with 0.02N aqueous hydrochloric acid using phenolphthalein.
- the number of moles Q of the substituent introduced by chemical modification is obtained by the following formula.
- Q (mol) 0.05 (N) ⁇ 10 (ml) / 1000 ⁇ 0.02 (N) ⁇ Z (ml) / 1000
- T is the molecular weight of the substituent.
- cellulose having a predetermined modification rate is obtained by controlling a reaction method in which cellulose is modified with an aromatic ring-containing substituent and the modification rate is controlled or modified.
- the chemical modification rate is characterized by being 10 mol% or more based on the total hydroxyl groups of cellulose. Preferably it is 11 mol% or more, Most preferably, it is 15 mol% or more. Moreover, it is usually 60 mol% or less, preferably 55 mol% or less, more preferably 50 mol% or less, still more preferably 45 mol% or less, and particularly preferably 40 mol% or less.
- controlling the modification method or controlling the modification method means controlling the amount of chemical modifier added to the total hydroxyl groups of cellulose or controlling the reaction time and temperature of the modification reaction.
- Chemical modification is preferable because the thermal decomposition temperature of the cellulose fiber is increased, the heat resistance is increased, and coloring during heating is reduced.
- the I-type crystal structure of cellulose is well maintained and heat resistance is excellent. This is because the modification reaction of the present invention swells the fiber without destroying the type I crystal structure by using an organic acid as a solvent during the reaction, and enables modification more uniformly by performing modification in this state. It seems to be because.
- the thermal decomposition temperature is measured by the method shown below.
- the modified cellulose within the range of the above-mentioned chemical modification rate which is modified with an aromatic ring-containing substituent, is easy to be fibrillated and has high transparency when made into a cellulose composite. I understood. Although the detailed reason is not known, it is considered that the cellulose chains are likely to be easily separated due to the steric hindrance of the substituent.
- the thermal decomposition temperature of the modified cellulose in the present invention is preferably 300 ° C. or higher, more preferably 335 ° C. or higher.
- the upper limit is not particularly limited, but is usually 400 ° C. or lower. If the thermal decomposition temperature is too low, there is a problem that coloring due to thermal decomposition increases.
- the modified cellulose fiber of the present invention has good heat resistance because of its high thermal decomposition temperature as described above, but the reason is expected to be that the main chain cleavage is suppressed by modifying the hydroxyl group at the 6-position. Is done.
- the pyrolysis temperature is 10 ° C from room temperature to 600 ° C under nitrogen using TG-DTA (differential thermogravimetric simultaneous measurement device) after conditioning the modified cellulose to a temperature of 23 ° C and a humidity of 50% for 48 hours. It is calculated
- a cellulose dispersion having a predetermined modification rate is obtained by controlling a reaction method in which cellulose is modified with an aromatic ring-containing substituent and the modification rate is controlled or modified.
- the modified cellulose fiber dispersion obtained by the production method of the present invention is made into a cellulose composite and used for transparent applications, it is preferable to fibrillate cellulose into fine fibers.
- the modification may be performed on the cellulose obtained by purification, may be performed after the cellulose is fibrillated into fine fibers, or may be performed after the cellulose fibers are formed into, for example, a sheet.
- the surface of the fine fiber When the surface of the fine fiber is uniformly modified, it may be performed after the cellulose is defibrated into fine fibers and after the cellulose fibers are formed into, for example, a sheet or the like.
- defibration When defibration is performed after modification, defibration is easy and the filtration time for solvent removal is shortened, which is preferable in terms of productivity.
- the cellulose after purification may be modified or may be performed after defibration into fine fibers, but when performed after defibration into fine fibers, Since labor is required to remove the solvent, it is preferable to modify the undefibrated cellulose after purification.
- the chemical modification can be carried out in the usual way, but since the cellulose after purification is usually in a water-containing state, this water is replaced with a reaction solvent or the like to suppress the reaction between the chemical modifier and water as much as possible. It is important to. Moreover, since it will become difficult to advance refinement
- Chemical modification can be performed by a normal method. That is, chemical modification can be performed by reacting cellulose with a chemical modifier according to a conventional method. Under the present circumstances, in the manufacturing method of this invention, although an organic acid is used as a solvent, you may use another solvent and a catalyst, a heating, pressure reduction, etc. as needed.
- the amount of the chemical modifier is not particularly limited and varies depending on the type of the chemical modifier, but is usually 0.01 times or more, 0.05 times or more, 100 times or less, and 50 times the number of moles of hydroxyl groups of cellulose. Double or less is preferable.
- organic acid is a general term for acids of organic compounds.
- Organic acids are mainly carboxylic acids and sulfonic acids.
- carboxylic acid include aliphatic carboxylic acids such as formic acid, acetic acid, propionic acid, butyric acid and valeric acid, aliphatic unsaturated carboxylic acids such as oleic acid and linoleic acid, and aliphatic dicarboxylic acids such as oxalic acid, malonic acid and oxalic acid.
- Examples thereof include aromatic carboxylic acids such as acid, benzoic acid and phthalic acid, oxocarboxylic acids such as pyruvic acid, and carboxylic acids such as lactic acid, malic acid and citric acid.
- the sulfonic acid include aliphatic sulfonic acids such as methylsulfonic acid and ethylsulfonic acid, aliphatic unsaturated sulfonic acids, aliphatic disulfonic acids, aromatic sulfonic acids such as benzenesulfonic acid, and toluenesulfonic acid.
- organic acids those having an acid dissociation constant pKa of 5.0 or less are particularly preferred. Preferably it is 4.8 or less. Moreover, pKa is 0.1 or more, preferably 1.0 or more.
- organic acids having an acid dissociation constant pKa of 5.0 or less include formic acid, acetic acid, propionic acid, butyric acid, oleic acid, linoleic acid, succinic acid, malonic acid, succinic acid, benzoic acid, phthalic acid, pyruvic acid, lactic acid, Examples include malic acid, citric acid, methyl sulfonic acid, ethyl sulfonic acid, benzene sulfonic acid, and toluene sulfonic acid.
- aliphatic organic acids are particularly preferable. As long as it is aliphatic, it may be mono-, di- or even more. Further, the aliphatic organic acid is preferably a carboxylic acid. Furthermore, among aliphatic carboxylic acids, the number of carbon atoms in the fatty chain is preferably 1 to 7, and more preferably 1 to 5. Among these, acetic acid and succinic acid are preferable.
- the amount of the solvent is not particularly limited, but is usually preferably 0.5 times or more, 1 time or more, 200 times or less, or 100 times or less with respect to the weight of cellulose.
- an organic solvent that does not inhibit the chemical modification reaction is preferably used.
- an organic solvent compatible with an organic acid or a catalyst described later is preferable.
- examples of such an organic solvent include organic solvents such as acetone and pyridine.
- the mixing ratio of the other solvents is arbitrary as long as they are compatible with each other, and further, the catalysts described later are compatible with each other.
- the ratio of the other solvent to occupy is about 1 vol% or more, and is usually 50 vol% or less, more preferably 40 vol% or less, and particularly preferably 30 vol% or less.
- catalyst As the catalyst, it is preferable to use a basic catalyst such as pyridine, triethylamine, sodium hydroxide or sodium acetate, or an acidic catalyst such as acetic acid, sulfuric acid or perchloric acid.
- the amount of the catalyst is not particularly limited and varies depending on the type, but is usually preferably 0.01 times or more, 0.05 times or more, 100 times or less, and 50 times or less with respect to the number of moles of hydroxyl groups of cellulose.
- Temperature conditions As the temperature condition, if it is too high, yellowing of the cellulose and a decrease in the degree of polymerization are concerned, and if it is too low, the reaction rate decreases, so 10 to 130 ° C. is preferable.
- the reaction time is several minutes to several tens of hours depending on the chemical modifier and the chemical modification rate.
- Chemical modification of a molded article such as a sheet may be performed after the sheet is manufactured, after being replaced with an organic solvent such as alcohol, and after the sheet is further dried, or may be performed without drying. Is preferable because the reaction rate of chemical modification is increased. In the case of drying, it may be blown dry, dried under reduced pressure, or dried under pressure. Moreover, you may heat.
- the chemical modification of the sheet can also take a normal method as described above.
- the defibrating process is performed to obtain fine fibers, and the method will be described.
- the defibrating process may be performed on the cellulose after purification or may be performed after chemically modifying the cellulose. Since it is easy to remove and the filtration time for solvent removal becomes short, it is preferable at the point of productivity.
- a specific method of the defibration process is not particularly limited.
- ceramic beads having a diameter of about 1 mm are placed in a cellulose dispersion having a cellulose concentration of 0.1 to 10% by weight, for example, about 1% by weight.
- Examples thereof include a method of defibrating cellulose by applying vibration using a shaker or a bead mill.
- Cellulose fibers can also be obtained by blending with a blender-type disperser or slits rotating at high speed, using a cellulose dispersion such as shearing force to break up (high-speed rotating homogenizer) or by suddenly reducing the pressure from high pressure.
- Examples thereof include a method in which a shearing force is generated in the middle (high pressure homogenizer method) and a method using a counter collision type disperser (manufactured by Masuko Sangyo Co., Ltd.) such as Massomizer X.
- high-speed rotation homogenizer and high-pressure homogenizer treatment improve the efficiency of defibration.
- the solid content concentration (cellulose concentration) in the cellulose dispersion is 0.2 wt% to 10 wt%, particularly 0.3 wt% to 6 wt%. If the solid content concentration in the cellulose dispersion used in this defibrating process is too low, the amount of liquid will be too large for the amount of cellulose to be treated, and the efficiency will be poor, and if the solid content concentration is too high, the fluidity will be poor.
- the concentration of the cellulose dispersion used for the fiber treatment is adjusted by adding water as appropriate.
- the rotation speed is preferably 10,000 rpm or more, more preferably 15000 rpm or more, and particularly preferably 20000 rpm or more.
- the time is preferably 1 minute or longer, more preferably 5 minutes or longer, and particularly preferably 10 minutes or longer.
- heat is generated by shearing, it is preferable to cool the solution so that the liquid temperature does not exceed 50 ° C.
- the liquid is ejected from a high-pressure atmosphere of 100 MPa or more by a method of defibration by high-speed rotation or a high-pressure homogenizer method.
- the cellulose dispersion When using a high-pressure homogenizer, the cellulose dispersion is pressurized to 30 MPa or more, preferably 100 MPa or more, more preferably 150 MPa or more, more preferably 220 MPa or more with a pressure intensifier, and ejected from a nozzle having a pore diameter of 50 ⁇ m or more.
- the pressure is reduced to 30 MPa or more, preferably 80 MPa or more, more preferably 90 MPa or more.
- the cellulose fiber is defibrated by the cleavage phenomenon caused by this pressure difference.
- the ejection of the cellulose dispersion liquid is repeated a plurality of times as necessary, thereby increasing the degree of fineness and obtaining cellulose fibers having a desired fiber diameter.
- the number of repetitions is usually 1 or more, preferably 3 or more, and usually 20 or less, preferably 15 or less.
- the degree of miniaturization can be increased.
- an excessively large number of passes is not preferable because the cost increases.
- the high-pressure homogenizer is not particularly limited, but as a specific apparatus, a “starburst system” manufactured by Gaurin or Sugino Machine can be used.
- a “starburst system” manufactured by Gaurin or Sugino Machine can be used.
- the pressure difference from the high pressure condition to the reduced pressure is large, but generally the upper limit of the pressure difference is usually 245 MPa or less by ejecting from the pressurizing condition by the pressure intensifier to the atmospheric pressure. .
- the pore diameter is from 50 ⁇ m to 800 ⁇ m, preferably from 100 ⁇ m to 500 ⁇ m, more preferably from 150 ⁇ m to 350 ⁇ m.
- the pore diameter is from 50 ⁇ m to 800 ⁇ m, preferably from 100 ⁇ m to 500 ⁇ m, more preferably from 150 ⁇ m to 350 ⁇ m.
- the number of the ejection nozzles may be one or two, and the ejected cellulose may hit a wall, ball, or ring provided at the ejection destination. Furthermore, when there are two nozzles, celluloses may collide with each other at the ejection destination. In addition, it is possible to obtain the fine cellulose fiber dispersion of the present invention only by the treatment with such a high-pressure homogenizer. Since the efficiency is low, it is preferable to refine by performing ultrasonic treatment described later after the high-pressure homogenizer treatment about 1 to 5 times.
- the cellulose concentration of the cellulose dispersion subjected to ultrasonic treatment is preferably 0.01 to 10% by weight, particularly 0.1 to 5% by weight, and particularly preferably 0.2 to 2% by weight. If the cellulose concentration of the cellulose dispersion to be irradiated with ultrasonic waves is too low, it is inefficient, and if it is too high, the viscosity becomes high and the fibrillation process becomes uneven. Therefore, in the present invention, water and / or an organic solvent is added as necessary so that the cellulose concentration of the cellulose dispersion to be subjected to ultrasonic treatment becomes the predetermined concentration.
- the organic solvent as a dispersion medium for the cellulose dispersion liquid, alcohols such as methanol, ethanol, isopropyl alcohol, n-propyl alcohol and n-butanol, ketones such as acetone and methyl ethyl ketone, and other water-soluble organic solvents are used. Species or two or more can be used. Further, a water-insoluble organic solvent can be used in combination, but preferably, the dispersion medium is a mixed solution of water or water of an organic solvent and water, and particularly preferably water.
- the number average fiber diameter of the cellulose fibers in the cellulose dispersion subjected to ultrasonic irradiation is preferably 10 ⁇ m or less, particularly 2 ⁇ m or less by the above-described defibration. More preferably, it is 1 ⁇ m or less.
- the frequency of ultrasonic waves applied to the cellulose dispersion is 15 kHz to 1 MHz, preferably 20 kHz to 500 kHz, more preferably 20 kHz to 100 kHz. If the frequency of the ultrasonic wave to be applied is too small, cavitation described later is difficult to occur. I can't.
- an output of an ultrasonic wave it is 1 W / cm ⁇ 2 > or more as an execution output density, Preferably it is 10 W / cm ⁇ 2 > or more, More preferably, it is 20 W / cm ⁇ 2 > or more.
- the upper limit of the effective output density of the ultrasonic wave is 500 W / cm 2 or less from the viewpoint of durability of the vibrator and the horn.
- the effective output density of an ultrasonic wave can be calculated from the temperature rise of 500 mL of water. Specifically, 500 mL of water is put into a container, ultrasonic waves are irradiated to this water, the degree of temperature rise at that time is measured, and the calculation is performed according to the following formula.
- P (T / s) ⁇ 4.18 ⁇ 500 / A
- T the rising temperature (° C.)
- s time (seconds)
- A is the area (cm 2 ) of the ultrasonic vibration part
- type it is the area of the end face. In the case of a bathtub type, this corresponds to the area of the vibrator mounting surface.
- the temperature is calculated by the above formula using the temperature when the temperature rises to 10 ° C. above the room temperature and the time at that time.
- the ultrasonic irradiation method is not particularly limited, and various methods can be used. For example, by directly inserting a horn that transmits vibration of an ultrasonic vibrator into the above cellulose dispersion, a method of directly refining cellulose fibers, or ultrasonic waves on a part of the floor or wall of a container containing the cellulose dispersion By placing a vibrator to make cellulose fibers finer, or by putting a liquid such as water in a container equipped with an ultrasonic vibrator, and immersing the container containing the cellulose dispersion in it, A method in which ultrasonic vibration is indirectly applied to the cellulose dispersion liquid via a liquid to make it fine can be employed. Among them, the method of directly inserting the horn into the cellulose dispersion is efficient because it can transmit ultrasonic energy directly and increase the energy density.
- Cellulose dispersion liquid may be refined by a batch-type processing method in which the entire amount is replaced after irradiating ultrasonic waves with a predetermined frequency for a predetermined amount with a predetermined effective power density.
- the treatment may be performed by a method in which a certain amount of the cellulose-containing dispersion liquid is circulated in a processing vessel in which an ultrasonic vibrator is installed in the vicinity or on the floor or wall, and ultrasonic waves are continuously applied.
- a plurality of ultrasonic transducers may be installed in one processing container, or a plurality of processing containers in which one transducer is installed in one processing container may be used.
- the plurality of vibrators may have the same frequency, or the frequency may be changed.
- the ultrasonic waves may be irradiated continuously or intermittently at a predetermined interval.
- a method of alternately repeating ultrasonic irradiation for 0.1 to 0.9 seconds and rest operation for 0.9 to 0.1 seconds may be used.
- the applied energy is converted into heat, and the temperature of the cellulose dispersion increases. Therefore, in order to perform the micronization treatment under a certain treatment condition, it is preferable to make the temperature of the cellulose dispersion constant by cooling or heating.
- the temperature during sonication is preferably 1 to 80 ° C., more preferably 10 to 60 ° C., and still more preferably 15 to 40 ° C.
- this temperature is too low, if water is used as the dispersion medium, it will freeze and become unprocessable. In other words, it is difficult to generate cavitation with solid ice, and when water and ice are mixed, cavitation occurs on the surface of the ice and consumes energy, which reduces the efficiency of cellulose refinement. To do. On the other hand, if the treatment temperature is too high, vapor such as minute water vapor is generated on the ultrasonic transducer surface, and the energy efficiency is lowered.
- the treatment time of the ultrasonic irradiation may be a time such that the cellulose fibers in the dispersion are refined to a desired degree of refinement, and the output and frequency of the used ultrasonic waves, It is appropriately set depending on the fiber diameter and the like.
- the principle by which cellulose fibers are refined by ultrasonic treatment has not been completely elucidated, but it is presumed that the following phenomenon has occurred.
- the fiber diameter of the cellulose fiber in the cellulose dispersion defibrated by the above method can be measured and determined by observing with SEM, TEM or the like after drying and removing the dispersion medium in the dispersion.
- the number average fiber diameter of the fibrillated modified cellulose fiber obtained by the present invention is preferably 100 nm or less. It is further preferably 80 nm or less, and particularly preferably 50 nm or less. The lower limit of the average fiber diameter is usually 4 nm or more.
- the cellulose fiber used in the present invention has a cellulose type I crystal structure.
- the cellulose type I crystal structure is, for example, “The Encyclopedia of Cellulose” published by Asakura Shoten, first edition P.I. 81-P. 86, or P.I. 93-99, and most natural cellulose has a cellulose type I crystal structure.
- cellulose fibers of, for example, cellulose II, III and IV type are derived from cellulose having cellulose I type crystal structure.
- Cellulose I-type crystals have a higher crystal elastic modulus than other crystal structures, and are therefore preferable because of their high elastic modulus, high strength, and low linear expansion coefficient.
- Cellulose contained in the modified cellulose fiber dispersion of the present invention is “average diffraction intensity at 18 to 19 degrees in the scanning angle” with respect to “intensity of diffraction peak derived from cellulose type I crystal structure at 20 to 24 degrees in scanning angle” (The ratio of non-type I such as amorphous is reflected) is 0.8 or less. Preferably it is 0.7 or less, especially 0.6 or less, and especially 0.5 or less. The lower the intensity ratio, the better, but practically about 0.01 or more.
- the modified cellulose fiber dispersion of the present invention is a dispersion of cellulose fibers having a cellulose I-type crystal structure in which 10 mol% or more of the total hydroxyl groups of cellulose are modified with an aromatic ring-containing substituent.
- the ratio of the “average diffraction intensity at the same scanning angle of 18 to 19 degrees” to the “intensity of the diffraction peak derived from the cellulose I-type crystal structure at a scanning angle of 20 to 24 degrees” in the wide-angle X-ray diffraction image of the cellulose fiber. Is 0.8 or less. Preferably it is 0.7 or less, especially 0.6 or less, and especially 0.5 or less. The lower the intensity ratio, the better, but practically about 0.01 or more.
- the modified cellulose fiber dispersion of the present invention is obtained by reacting cellulose with a chemical modifier in a state where cellulose is swollen while maintaining the cellulose I-type crystal structure, and passing through a defibrating step as necessary. It is done.
- the method for swelling cellulose is not particularly limited, and it is possible to select a solvent having a fiber swelling effect as a solvent during the chemical modification reaction.
- the solvent include alcohols such as methanol and ethanol, diols such as ethylene glycol, aprotic polar solvents such as dimethylformamide, and heterocyclic compounds such as pyridine. Among them, the above-mentioned I.I. The production method is preferred. Regarding the defibrating process, I.I. Can be used.
- the chemical modification rate of cellulose contained in the modified cellulose fiber dispersion of the present invention is characterized by being 10 mol% or more based on the total hydroxyl groups of cellulose. Preferably it is 11 mol% or more, Most preferably, it is 15 mol% or more. Further, it is usually 60 mol% or less, preferably 55 mol% or less, more preferably 50 mol% or less, still more preferably 45 mol% or less, and particularly preferably 40 mol% or less.
- the method for measuring the modification rate is as described in I.S. This is the same as the manufacturing method.
- Chemical modification is preferable because the thermal decomposition temperature of the cellulose fiber is increased, the heat resistance is increased, and coloring during heating is reduced.
- the modification reaction of the present invention the I-type crystal structure of cellulose is well maintained and heat resistance is excellent.
- the modified cellulose within the range of the above-mentioned chemical modification rate which is modified with an aromatic ring-containing substituent, is easy to be fibrillated and has high transparency when made into a cellulose composite. I understood. Although the detailed reason is not known, it is considered that the cellulose chains are likely to be easily separated due to the steric hindrance of the substituent.
- the thermal decomposition temperature of cellulose contained in the modified cellulose fiber dispersion of the present invention is preferably 300 ° C. or higher, more preferably 335 ° C. or higher.
- the upper limit is not particularly limited, but is usually 400 ° C. or lower. If the thermal decomposition temperature is too low, there is a problem that coloring due to thermal decomposition increases.
- Cellulose contained in the modified cellulose fiber dispersion of the present invention has good heat resistance because of its high thermal decomposition temperature as described above. Expected to be suppressed.
- the fiber diameter of the cellulose fiber in the modified cellulose fiber dispersion of the present invention defibrated by the method is obtained by drying and removing the dispersion medium in the dispersion and then measuring and observing with SEM, TEM, etc. Can do.
- the number average fiber diameter of the cellulose fibers contained in the defibrated modified cellulose fiber dispersion obtained by the present invention is preferably 100 nm or less. More preferably, it is 80 nm or less, and it is especially preferable that it is 50 nm or less.
- the lower limit of the average fiber diameter is usually 4 nm or more.
- the modified cellulose fiber dispersion of the present invention undergoes the above-described modification step and defibration step, and then appropriately adjusts the concentration of the dispersion medium (solvent), replaces the dispersion medium, adds various additives, and the like. be able to.
- the defibrating liquid after the defibrating step basically uses water as a dispersion medium, when water is used as a dispersion medium (solvent), it can be diluted as it is or with water. Further, an organic solvent other than water can be added, and after the addition, it can be concentrated to a desired composition ratio. When using a solvent other than water, the solvent must be replaced. In this case, after adding a solvent, it is preferable to remove water by concentration.
- the solvent used as the dispersion medium is preferably water and / or a water-soluble organic solvent. Since the defibrating liquid after the defibration process is basically carried out using water and / or a water-soluble organic solvent as a dispersion medium, in order to replace it with a water-insoluble organic solvent, Time is not preferred.
- the water-soluble organic solvent here refers to a solvent that maintains a uniform appearance even after the flow has subsided when gently mixed with the same volume of pure water at 20 degrees Celsius at one atmospheric pressure.
- monohydric alcohols such as methanol, ethanol, n-propyl alcohol, isopropyl alcohol, t-butyl alcohol, allyl alcohol, ethylene glycol, butyl glycol, t-butyl glycol, methyl diglycol, ethyl diglycol
- Polyhydric alcohols (glycols) such as butyl diglycol, methyldipropylene glycol, glycerin, 1-methoxy-2-propanol, 2-ethoxyethanol, 2-n-butoxyethanol, 3-methoxybutanol, 3-methyl-3- Methoxybutanol, ethylene glycol monomethyl ether, ethylene glycol dimethyl ether, ethylene glycol monophenyl ether, propylene glycol monomethyl ether, propylene glycol
- ketones examples thereof include ketones, acetonitrile, pyridine, N, N-dimethylformamide, N, N-dimethylacetamide, N-methyl-2-pyrrolidone and the like, acetic acid, acrylic acid, methacrylic acid and butyric acid. Further, two or more of these solvents may be used in combination.
- the modified cellulose fiber of the present invention can be formed into a modified cellulose fiber sheet by forming it into a sheet shape.
- a resin can be impregnated to form a cellulose composite, or a cellulose composite can be sandwiched between resin sheets.
- the modified cellulose fiber sheet of the present invention (hereinafter also referred to as “cellulose sheet”) may be produced using the modified cellulose fiber as it is, but the one produced using the one subjected to the defibration process is more transparent. A low linear expansion coefficient and a high elastic modulus are obtained.
- the cellulose dispersion subjected to the above-described defibrating step (hereinafter also referred to as “defibrated cellulose dispersion”) is filtered, or the dispersion is applied to an appropriate substrate. It is the sheet
- the concentration of the defibrated cellulose dispersion used for filtration is 0.00. It is preferable that the content is 01% by weight or more, preferably 0.05% by weight or more, and more preferably 0.1% by weight or more. If the concentration is too low, filtration takes an enormous time, which is not preferable.
- the concentration of the defibrated cellulose dispersion is 1.5% by weight or less, preferably 1.2% by weight or less, more preferably 1.0% by weight or less. If the concentration is too high, a uniform sheet cannot be obtained.
- a filter cloth during filtration, it is important that the finely divided cellulose fibers do not pass through and the filtration speed is not too slow.
- a sheet made of an organic polymer, a woven fabric, and a porous membrane are preferable.
- the organic polymer is preferably a non-cellulose organic polymer such as polyethylene terephthalate, polyethylene, polypropylene, polytetrafluoroethylene (PTFE) or the like.
- a porous film of polytetrafluoroethylene having a pore diameter of 0.1 to 20 ⁇ m, for example, 1 ⁇ m, polyethylene terephthalate or polyethylene fabric having a pore diameter of 0.1 to 20 ⁇ m, for example, 1 ⁇ m, and the like can be given.
- the cellulose sheet can have various porosity depending on its production method.
- Examples of a method for obtaining a cellulose sheet having a large porosity include a method in which water in the cellulose sheet is finally replaced with an organic solvent such as alcohol in a film forming process by filtration. In this method, water is removed by filtration, and an organic solvent such as alcohol is added when the cellulose content reaches 5 to 99% by weight.
- the organic solvent such as alcohol can be replaced with the organic solvent such as alcohol at the end of the filtration by putting the defibrated cellulose dispersion into the filtration device and then gently introducing an organic solvent such as alcohol into the upper part of the dispersion.
- the organic solvent such as alcohol used here is not particularly limited.
- alcohols such as methanol, ethanol, 1-propanol, 2-propanol and 1-butanol, acetone, methyl ethyl ketone, tetrahydrofuran, cyclohexane 1 type, or 2 or more types of organic solvents chosen from toluene, carbon tetrachloride, etc. are mentioned.
- a water-insoluble organic solvent it is preferable to use a water-soluble organic solvent or a mixed solvent with the water-soluble organic solvent, and then replace with a water-insoluble organic solvent.
- the filtration time when obtaining a cellulose sheet having a high porosity by such a method is not particularly limited, but when filtering 40 g / m 2 by batch filtration, it is usually 120 minutes or less, preferably 110 minutes or less, Furthermore, it is 100 minutes or less. Moreover, it is 0.1 minutes or more, Preferably it is 0.3 minutes or more, More preferably, it is 0.5 minutes or more. If the filtration time is too long, the production efficiency is lowered, and there is a problem that the filtration apparatus becomes huge when continuous filtration is performed.
- the modified cellulose fiber in the present invention it is possible to filter at a higher rate compared to other unmodified cellulose and cellulose modified with an aliphatic substituent, and the obtained sheet is made of cellulose.
- the obtained sheet is made of cellulose.
- a highly transparent cellulose composite can be obtained.
- drying is performed.
- This drying may be air drying, vacuum drying, or pressure drying.
- you may heat-dry.
- the temperature is preferably 50 ° C or higher, more preferably 80 ° C or higher, 250 ° C or lower is more preferable, and 150 ° C or lower is more preferable. If the heating temperature is too low, drying may take time or drying may be insufficient. If the heating temperature is too high, the cellulose sheet may be colored or the cellulose may be decomposed.
- pressurizing 0.01 MPa or more is preferable, 0.1 MPa or more is more preferable, 5 MPa or less is preferable, and 1 MPa or less is more preferable. If the pressure is too low, drying may be insufficient, and if the pressure is too high, the planar structure of cellulose fibers may be crushed or cellulose may be decomposed.
- drying is performed after filtration or application of the defibrated cellulose dispersion. That is, as a method of obtaining a dense cellulose sheet having a low porosity, as described above, the film is formed by the filtration of the defibrated cellulose dispersion, and is not subjected to substitution with an organic solvent such as alcohol, but is directly subjected to the drying step. A method is mentioned.
- the filtration time when obtaining a cellulose sheet having a low porosity by such a method is not particularly limited, but when filtering 40 g / m 2 by batch filtration, usually 80 minutes or less, preferably 70 minutes or less, Furthermore, it is 60 minutes or less.
- it is 0.1 minutes or more, Preferably it is 0.3 minutes or more, More preferably, it is 0.5 minutes or more. If the filtration time is too long, the production efficiency is lowered, and there is a problem that the filtration apparatus becomes huge when continuous filtration is performed. Drying is as described above.
- the fiber diameter of the cellulose fiber contained in the modified cellulose fiber sheet obtained by the present invention can be measured and determined by observing with SEM, TEM or the like.
- the number average fiber diameter of cellulose fibers contained in the modified cellulose fiber sheet obtained by the present invention is preferably 100 nm or less. It is further preferably 80 nm or less, and particularly preferably 50 nm or less.
- the lower limit of the average fiber diameter is usually 4 nm or more.
- Chemical modification can also be performed after making the cellulose sheet.
- chemical modification may be performed after the sheet obtained as described above is dried, or may be performed before the sheet is dried.
- the cellulose sheet is reacted with a chemical modifier as described above, washed with water and / or an organic solvent, and then dried to obtain a modified cellulose sheet.
- the thickness of a cellulose sheet Preferably it is 1 micrometer or more, More preferably, it is 5 micrometers or more. Moreover, it is 1000 micrometers or less normally, Preferably it is 250 micrometers or less.
- the haze value by C light when it is set as the composite material shown to following (1) is 3 or less, Furthermore, 2 or less, Especially 1 or less.
- a cellulose fiber sheet having a thickness adjusted to 40 g / m 2 was added to 96 parts by weight of bis (methacryloyloxymethyl) tricyclo [5.2.1.0 2,6 ] decane, pentaerythritol tetrakis ( ⁇ -Thiopropionate) After impregnating a mixed solution of 6 parts by weight, 2,4,6-trimethylbenzoyldiphenylphosphine oxide and 0.05 part by weight of benzophenone, and placing it under reduced pressure overnight.
- the YI value is preferably 40 or less, more preferably 35 or less, and particularly preferably 25 or less even after the treatment at 190 ° C. for 4 hours under vacuum assuming heating at the time of compounding with the matrix.
- the YI value can be measured using, for example, a color computer manufactured by Suga Test Instruments.
- the modified cellulose fiber sheet obtained by the present invention has little decrease in tensile strength even after the heating step.
- the ratio of the tensile strength after heat treatment at 190 ° C. for 4 hours under vacuum to the tensile strength before treatment is 90% or more, preferably 95% or more, particularly preferably 99% or more.
- the modified cellulose fiber sheet obtained by the present invention exhibits a high tensile elastic modulus.
- the tensile modulus after heat treatment at 190 ° C. for 4 hours under vacuum is preferably 0.2 to 100 GPa, more preferably 1.0 to 50 GPa, still more preferably 4.0 to 30 GPa.
- the modified cellulose fiber of the present invention can be made into cellulose particles.
- These cellulose particles are particularly suitable for compounding with a thermoplastic resin by kneading, taking advantage of their properties such as high modulus of elasticity, low coefficient of linear expansion and surface smoothness to make various structural materials, especially surface designs. It is useful for automotive panels with excellent properties and exterior wall panels for buildings.
- the cellulose fiber obtained by the present invention is used as a dispersion, for example, by spraying it from a spray nozzle or the like using a known spray drying device, and thereby the dispersion medium is changed.
- the method of removing and granulating is mentioned.
- Specific examples of the injection method include a method using a rotating disk, a method using a pressure nozzle, and a method using a two-fluid nozzle. You may dry the particle
- cellulose particles can also be obtained by freeze-drying and then pulverizing the cellulose fibers obtained according to the present invention.
- the cellulose fiber obtained by the production method of the invention is cooled with liquid nitrogen or the like, it is pulverized with a grinder or a rotary blade.
- the cellulose fiber obtained by the present invention can be combined with a polymer other than cellulose in water without drying from the state of an aqueous dispersion, and then water can be removed or replaced with another organic solvent from water. Then, the composite can be obtained by complexing with a polymer other than cellulose in the organic solvent and then removing the organic solvent.
- Cellulose fiber composite of the present invention A cellulose fiber composite is obtained by combining the cellulose fiber or cellulose sheet or cellulose particles obtained by the method for producing a modified cellulose fiber dispersion of the present invention with a polymer as a matrix.
- the cellulose fiber composite is useful for various display substrate materials, solar cell substrates, window materials, etc., taking advantage of its high transparency, low linear expansion coefficient, and non-coloring properties, and also has a high elastic modulus. Utilizing characteristics such as low linear expansion coefficient and surface smoothness, it is useful for various structural materials, particularly for automobile panels and exterior wall panels for buildings having excellent surface design.
- the cellulose fiber composite of the present invention is a composite of the modified cellulose fiber dispersion, modified cellulose fiber sheet or cellulose particles obtained in the present invention, and a polymer other than cellulose, preferably, The cellulose sheet or cellulose particles of the present invention are combined with a polymer other than cellulose or a precursor thereof as a matrix material.
- the matrix material refers to a polymer or its precursor (for example, monomer) material that is bonded to a cellulose sheet, filled in a void, or kneaded granulated cellulose particles.
- Suitable as this matrix material is a thermoplastic resin that becomes a fluid liquid by heating, a thermosetting resin that polymerizes by heating, and a polymerized and cured by irradiation with active energy rays such as ultraviolet rays and electron beams. It is at least one resin (polymer material) obtained from an energy ray curable resin or the like or a precursor thereof.
- the precursor of the polymer material is a so-called monomer or oligomer, such as each monomer described later as a (co) polymerization component in the section of thermoplastic resin (hereinafter referred to as thermoplastic resin precursor). And the precursors described later in the section of thermosetting resin / photocurable resin.
- Examples of the method for obtaining the cellulose fiber composite of the present invention include the following methods (a) to (j).
- (b) Cellulose sheet, particles, etc., impregnating thermosetting resin precursor or active energy ray curable resin precursor (C) Cellulose sheet, particles and the like include a resin solution (one or more solutes selected from a thermoplastic resin, a thermoplastic resin precursor, a thermosetting resin precursor, and a photocurable resin precursor).
- a method of impregnating the solution) and drying, and then adhering with a hot press or the like, and polymerizing and curing if necessary (d) impregnating a cellulose sheet, particles, etc.
- thermoplastic resin melt and adhering with a hot press Method
- thermoplastic resin sheet or film
- a cellulose sheet are alternately arranged and adhered with a heating press or the like
- thermosetting resin precursor or active energy ray curable resin precursor g
- Resin solution thermoplastic resin, thermoplasticity on one or both sides of cellulose sheet
- a resin precursor, a thermosetting resin precursor, and a solution containing one or more solutes selected from a photocurable resin precursor are applied, and after removing the solvent, it is combined by polymerizing and curing as necessary.
- Method (h) Method of melt-kneading cellulose particles and thermoplastic resin and then forming into sheet or desired shape
- i Cellulose fiber dispersion and monomer solution or dispersion (thermoplastic resin precursor, thermosetting A resin precursor and a solution or dispersion containing one or more solutes or dispersoids selected from photo-curing resin precursors), and then polymerized and cured after removing the solvent. How to complex by the.
- J A method of mixing a cellulose fiber dispersion and a polymer solution or dispersion (thermoplastic resin solution or dispersion) and then removing the solvent to form a composite.
- the method (a), (b), (c), (d), (e), (f), (g) is preferable for the cellulose sheet, and the method (h) is used for the cellulose particles. Is preferred.
- a cellulose fiber composite is obtained by impregnating a polymerizable monomer or oligomer into a cellulose sheet, particles or the like and polymerizing the monomer by heat treatment or the like.
- the method of obtaining is mentioned.
- a polymerization catalyst used for polymerization of monomers can be used as a polymerization initiator.
- thermosetting resin precursor such as an epoxy resin monomer, or a photocuring property such as an acrylic resin monomer.
- a cellulose fiber composite is obtained by impregnating a cellulose sheet or particles with a mixture of a resin precursor and a curing agent, and curing the thermosetting resin precursor or the photocurable resin precursor with heat or active energy rays or the like. A method is mentioned.
- a resin solution (a solution containing one or more solutes selected from a thermoplastic resin, a thermoplastic resin precursor, a thermosetting resin precursor, and a photocurable resin precursor) is impregnated and dried, and then heated.
- a method of closely adhering with a press or the like and polymerizing and curing as necessary there is a method of dissolving a resin in a solvent, impregnating the solution into a cellulose sheet, particles, etc., and drying to obtain a cellulose fiber composite.
- a method of obtaining a higher-performance cellulose composite by adhering the voids in which the solvent is dried by a heating press after drying is mentioned.
- the solvent for dissolving the resin may be selected in consideration of the affinity with the cellulose fiber and the solubility of the resin. Specifically, from among those exemplified as the dispersion medium of the modified cellulose fiber dispersion, the resin What is necessary is just to select according to the solubility of.
- thermoplastic resin melt and adhering it with a hot press or the like As a method of impregnating a thermoplastic resin melt and adhering it with a hot press or the like, the thermoplastic resin is dissolved by heat treatment at a glass transition temperature or higher or a melting point or higher, and impregnated into a cellulose sheet, particles or the like. And a method of obtaining a polymer cellulose composite by adhering with a hot press or the like.
- the heat treatment is desirably performed under pressure, and the use of equipment having a vacuum heating press function is effective.
- thermoplastic resin sheet As a method of arranging the thermoplastic resin sheet (or film) and the cellulose sheet alternately and closely contacting with a heating press or the like, a film or sheet of the thermoplastic resin is arranged on one side or both sides of the cellulose sheet, and if necessary And a method of bonding the thermoplastic resin and the cellulose sheet by heating or pressing.
- an adhesive or a primer may be applied to the surface of the cellulose sheet and bonded.
- a method of passing between two pressurized rolls or a method of pressing in a vacuum state can be used so that bubbles are not embraced at the time of bonding.
- thermosetting resin precursor As a method of applying and curing a liquid thermoplastic resin precursor, a thermosetting resin precursor or an active energy ray curable resin precursor on one side or both sides of the cellulose sheet, on one side or both sides of the cellulose sheet
- a thermosetting resin precursor formulated with a thermal polymerization initiator is applied and cured by heating, and both are adhered, or a curable resin precursor in which a photopolymerization initiator is formulated on one or both sides of a cellulose sheet
- a resin solution (a solution containing one or more solutes selected from a thermoplastic resin, a thermoplastic resin precursor, a thermosetting resin precursor, and a photocurable resin precursor) to one side or both sides of a cellulose sheet. Then, as a method of compounding by removing the solvent, prepare a resin solution in which a solvent-soluble resin is dissolved in the solvent, apply it to one or both sides of the cellulose sheet, and remove the solvent by heating The method of doing is mentioned. In the case of a photo-curing resin, polymerization curing with active energy rays or the like is further performed as necessary.
- the solvent for dissolving the resin may be selected in consideration of the affinity with the cellulose fiber and the solubility of the resin. Specifically, from among those exemplified as the dispersion medium of the modified cellulose fiber dispersion, the resin What is necessary is just to select according to the solubility of.
- a laminate by stacking a plurality of cellulose composites of the cellulose sheet and resin thus produced. At that time, a cellulose composite containing a cellulose sheet and a resin sheet not containing the cellulose composite may be laminated. In order to adhere the cellulose composites or the resin and the cellulose composite, an adhesive may be applied or an adhesive sheet may be interposed. Moreover, it can also integrate by adding a heat press process to a laminated body.
- a method of forming into a sheet shape or a target shape includes a method of melting cellulose particles and a thermoplastic resin after dry blending, a method of melt-kneading, Etc. are preferable.
- a method of melting after dry blending both are uniformly mixed with a tumbler blender, ribbon blender, V-type blender, Henschel mixer, etc., and then an additive such as an antioxidant used as necessary is added to the mixture. It is added to obtain a cellulose composite through a molten state.
- the mixture is simply melted or melt-kneaded by a single or twin screw extruder, roll, Banbury mixer, kneader, Brabender, or the like.
- melt-kneading both are melt-mixed together with additives such as antioxidants used as necessary.
- it is melt kneaded by a single or twin screw extruder, roll, Banbury mixer, kneader, Brabender or the like. Thereafter, it is extruded from a T-die and formed into a sheet shape, or injected into a mold to form a desired shape.
- matrix materials other than cellulose (polymer materials or precursors thereof) to be complexed with cellulose sheet, cellulose particles, or cellulose fiber dispersion are exemplified below, but the matrix materials used in the present invention are as follows. It is not limited to.
- the thermoplastic resin, thermosetting resin, and light (active energy ray) curable resin in the present invention can be used in combination of two or more.
- the polymer is amorphous and has a glass transition temperature (Tg).
- Tg glass transition temperature
- the degree of amorphousness is preferably 10% or less, particularly 5% or less in terms of crystallinity
- Tg is preferably 110 ° C. or more, particularly 120 ° C. or more, and particularly preferably 130 ° C. or more. If Tg is low, there is a risk of deformation when touched with hot water, for example, which causes a practical problem.
- a polymer material having few hydrophilic functional groups such as a hydroxyl group, a carboxyl group, and an amino group.
- the polymer Tg can be determined by a general method. For example, it is obtained by measurement by the DSC method.
- the degree of crystallinity of the polymer can be calculated from the density of the amorphous part and the crystalline part, and can also be calculated from the tan ⁇ , which is the ratio of the elastic modulus to the viscosity, by dynamic viscoelasticity measurement. .
- Thermoplastic resins include styrene resins, acrylic resins, aromatic polycarbonate resins, aliphatic polycarbonate resins, aromatic polyester resins, aliphatic polyester resins, aliphatic polyolefin resins, cyclic olefin resins, polyamides. Resin, polyphenylene ether resin, thermoplastic polyimide resin, polyacetal resin, polysulfone resin, amorphous fluorine resin and the like.
- styrenic resin examples include polymers and copolymers such as styrene, chlorostyrene, divinylbenzene, and ⁇ -methylstyrene.
- acrylic resin examples include polymers and copolymers such as (meth) acrylic acid, (meth) acrylonitrile, (meth) acrylic acid ester, and (meth) acrylamide.
- (meth) acryl means “acryl and / or methacryl”.
- Aromatic polycarbonate resin is produced by reaction of one or more bisphenols that can contain polyhydric phenols having 3 or more valences as a copolymerization component with carbonate esters such as bisalkyl carbonate, bisaryl carbonate, and phosgene.
- carbonate esters such as bisalkyl carbonate, bisaryl carbonate, and phosgene.
- aromatic dicarboxylic acids such as terephthalic acid and isophthalic acid or derivatives thereof (for example, aromatic dicarboxylic acid diesters and aromatic dicarboxylic acids) Acid chloride) may be used.
- the modified cellulose of the present invention is modified with an aromatic ring-containing substituent, it is considered that compatibility with the aromatic polycarbonate resin is good, and is preferable.
- the bisphenols include bisphenol A, bisphenol C, bisphenol E, bisphenol F, bisphenol M, bisphenol P, bisphenol S, and bisphenol Z (for abbreviations, refer to the Aldrich reagent catalog). (Where the central carbon participates in the cyclohexane ring) is preferred, and bisphenol A is particularly preferred.
- copolymerizable trihydric phenols include 1,1,1- (4-hydroxyphenyl) ethane and phloroglucinol.
- the aliphatic polycarbonate resin is a copolymer produced by a reaction between an aliphatic diol component and / or an alicyclic diol component and a carbonic ester such as bisalkyl carbonate or phosgene.
- alicyclic diol include cyclohexanedimethanol and isosorbite.
- aromatic polyester resins include copolymers of diols such as ethylene glycol, propylene glycol, and 1,4-butanediol and aromatic carboxylic acids such as terephthalic acid. Moreover, the copolymer of diols, such as bisphenol A, and aromatic carboxylic acids, such as a terephthalic acid and isophthalic acid, is also mentioned like a polyarylate. Since the modified cellulose of the present invention is modified with an aromatic ring-containing substituent, it is considered preferable to be compatible with the aromatic polyester resin.
- aliphatic polyester-based resin examples include a copolymer of the diol and an aliphatic dicarboxylic acid such as succinic acid and valeric acid, and a copolymer of hydroxydicarboxylic acid such as glycolic acid and lactic acid.
- aliphatic polyolefin-based resin examples include homopolymers of ⁇ -olefins having about 2 to 8 carbon atoms, those ⁇ -olefins and other ⁇ -olefins having about 2 to 18 carbon atoms, and the like.
- a binary or ternary copolymer may be used, and two or more of these olefin polymers may be used in combination.
- the cyclic olefin-based resin is a polymer containing a cyclic olefin skeleton in a polymer chain, such as norbornene or cyclohexadiene, or a copolymer containing these.
- polyamide resins examples include aliphatic amide resins such as 6,6-nylon, 6-nylon, 11-nylon, 12-nylon, 4,6-nylon, 6,10-nylon, 6,12-nylon,
- aromatic polyamide such as an aromatic diamine such as phenylenediamine and an aromatic dicarboxylic acid such as terephthaloyl chloride or isophthaloyl chloride or a derivative thereof may be used.
- polyphenylene ether resins examples include poly (2,6-dimethyl-1,4-phenylene ether), poly (2-methyl-6-ethyl-1,4-phenylene ether), and poly (2,6-dichloro). -1,4-phenylene ether), and also copolymers of 2,6-dimethylphenol and other phenols.
- Polyimide resins include pyromellitic acid-type polyimides such as pyromellitic anhydride and 4,4'-diaminodiphenyl ether, aromatic diamines such as anhydrous chlorotrimellitic acid and p-phenylenediamine, and diisocyanate compounds.
- Benzophenone type polyimide bismaleimide, bismaleimide type polyimide made of 4,4′-diaminodiphenylmethane, and the like.
- polyacetal resin examples include a homopolymer having an oxymethylene structure as a unit structure and a copolymer containing an oxyethylene unit.
- polysulfone resin examples include copolymers such as 4,4'-dichlorodiphenylsulfone and bisphenol A.
- amorphous fluororesin examples include homopolymers or copolymers such as tetrafluoroethylene, hexafluoropropylene, chlorotrifluoroethylene, vinylidene fluoride, vinyl fluoride, and perfluoroalkyl vinyl ether.
- thermoplastic resins may be used alone or in combination of two or more.
- thermosetting resin and the light (active energy ray) curable resin mean a precursor before curing or a cured resin obtained by curing.
- the precursor means a substance that is liquid, semi-solid or solid at room temperature and exhibits fluidity at room temperature or under heating.
- These can be insoluble and infusible resins formed by forming a network-like three-dimensional structure while increasing the molecular weight by causing a polymerization reaction or a crosslinking reaction by the action of a curing agent, a catalyst, heat or light.
- the resin cured product means a resin obtained by curing the thermosetting resin precursor or the light (active energy ray) curable resin precursor.
- thermosetting resin in the present invention is not particularly limited, but epoxy resin, acrylic resin, oxetane resin, phenol resin, urea resin, melamine resin, unsaturated polyester resin, silicon resin, polyurethane resin, diallyl phthalate Examples thereof include precursors such as resins.
- the epoxy resin precursor is an organic compound having at least one epoxy group.
- the number of epoxy groups in the epoxy resin precursor is preferably 1 or more and 7 or less per molecule, and more preferably 2 or more per molecule.
- the number of epoxy groups per molecule of the precursor is obtained by dividing the total number of epoxy groups in the epoxy resin precursor by the total number of molecules in the epoxy resin. It does not specifically limit as said epoxy resin precursor, For example, the epoxy resin etc. which were shown below are mentioned. These epoxy resins may be used alone or in combination of two or more. These epoxy resins are obtained by curing a thermosetting resin precursor using a curing agent.
- aromatic epoxy resins and precursors thereof such as hydrogenated products and brominated products, alicyclic epoxy resins, aliphatic epoxy resins, glycidyl ester type epoxy resins and hydrogenated products thereof, glycidyl amine type epoxy resins and these And a copolymer of glycidyl (meth) acrylate and a radical polymerizable monomer.
- the curing agent used for the curing reaction of the epoxy resin precursor is not particularly limited.
- amine compounds compounds such as polyaminoamide compounds synthesized from amine compounds, tertiary amine compounds, imidazole compounds, hydrazide compounds
- examples include melamine compounds, acid anhydrides, phenol compounds, thermal latent cationic polymerization catalysts, photolatent cationic polymerization initiators, dicyanamide and derivatives thereof.
- curing agents may be used independently and 2 or more types may be used together.
- the acrylic resin precursor includes a monofunctional (meth) acrylate compound having one (meth) acryloyl group in the molecule, and a polyfunctional (meth) acrylate having two or three (meth) acryloyl groups in the molecule.
- Examples thereof include compounds, styrenic compounds, acrylic acid derivatives, acrylate compounds having 4 to 8 (meth) acryloyl groups in the molecule, epoxy (meth) acrylate compounds, and (meth) acrylate compounds having a urethane bond.
- Monofunctional (meth) acrylate compounds having one (meth) acryloyl group in the molecule include methyl (meth) acrylate, 2-hydroxyethyl (meth) acrylate, phenyl (meth) acrylate, benzyl (meth) acrylate, Examples thereof include cyclohexyl (meth) acrylate and alkyl (meth) acrylate having 1 to 30 carbon atoms in alkyl.
- mono (meth) acrylate having an alicyclic skeleton has high heat resistance and can be suitably used.
- Specific examples of the alicyclic skeleton mono (meth) acrylate compound include, for example, (hydroxy-acryloyloxy) tricyclo [5.2.1.0 2,6 ] decane, (hydroxy-methacryloyloxy) tricyclo [5.2.1]. .0 2,6] decane, (hydroxy - acryloyloxy) pentacyclo [6.5.1.1 3, 6. 0 2,7 . 0 9,13 ] pentadecane, (hydroxy-methacryloyloxy) pentacyclo [6.5.1.1 3,6 . 0 2,7 .
- pentadecane (hydroxymethyl-acryloyloxymethyl) tricyclo [5.2.1.0 2,6 ] decane, (hydroxymethyl-methacryloyloxymethyl) tricyclo [5.2.1.0 2,6 ] Decane, (hydroxymethyl-acryloyloxymethyl) pentacyclo [6.5.1.1 3,6 . 0 2,7 . 0 9,13 ] pentadecane, (hydroxymethyl-methacryloyloxymethyl) pentacyclo [6.5.1.1 3,6 . 0 2,7 .
- pentadecane (hydroxyethyl-acryloyloxyethyl) tricyclo [5.2.1.0 2,6 ] decane, (hydroxyethyl-methacryloyloxyethyl) tricyclo [5.2.1.0 2,6 ] Decane, (hydroxyethyl-acryloyloxyethyl) pentacyclo [6.5.1.1 3,6 . 0 2,7 . 0 9,13 ] pentadecane, (hydroxyethyl-methacryloyloxyethyl) pentacyclo [6.5.1.1 3,6 . 0 2,7 . 0 9,13 ] pentadecane and the like. Moreover, these mixtures etc. can be mentioned.
- pentadecane diacrylate, bis (hydroxy) pentacyclo [6.5.1.1 3,6 . 0 2,7 . 0 9,13 ]
- pentadecane dimethacrylate, bis (hydroxy) pentacyclo [6.5.1.1 3,6 . 0 2,7 .
- pentadecane acrylate methacrylate, 2,2-bis [4- ( ⁇ - (meth) acryloyloxyethoxy) phenyl] propane, 2,2-bis [4- ( ⁇ - (meth) acryloyloxyethoxy) Cyclohexyl] propane, 1,4-bis [(meth) acryloyloxymethyl] cyclohexane, and the like.
- styrene compound examples include styrene, chlorostyrene, divinylbenzene, ⁇ -methylstyrene, and the like.
- (meth) acrylic acid derivatives other than esters examples include acrylamide, methacrylamide, acrylonitrile, methacrylonitrile and the like.
- an alicyclic ring skeleton bis (meth) acrylate compound is preferably used.
- bis (acryloyloxy) tricyclo [5.2.1.0 2,6 ] decane bis (methacryloyloxy) tricyclo [5.2.1.0 2,6 ] decane, (acryloyloxy-methacryloyloxy) tricyclo [ 5.2.1.0 2,6 ] decane, bis (acryloyloxy) pentacyclo [6.5.1.1 3,6 . 0 2,7 . 0 9,13 ] pentadecane, bis (methacryloyloxy) pentacyclo [6.5.1.1 3,6 . 0 2,7 . 0 9,13 ] pentadecane, (acryloyloxy-methacryloyloxy) pentacyclo [6.5.1.1 3,6 .
- pentadecane bis (acryloyloxymethyl) tricyclo [5.2.1.0 2,6 ] decane, bis (methacryloyloxymethyl) tricyclo [5.2.1.0 2,6 ] decane, (Acryloyloxymethyl-methacryloyloxymethyl) tricyclo [5.2.1.0 2,6 ] decane, bis (acryloyloxymethyl) pentacyclo [6.5.1.1 3,6 . 0 2,7 . 0 9,13 ] pentadecane, bis (methacryloyloxymethyl) pentacyclo [6.5.1.1 3,6 . 0 2,7 .
- pentadecane (acryloyloxymethyl-methacryloyloxymethyl) pentacyclo [6.5.1.1 3,6 . 0 2,7 . 0 9,13 ] pentadecane, bis (acryloyloxyethyl) tricyclo [5.2.1.0 2,6 ] decane, bis (methacryloyloxyethyl) tricyclo [5.2.1.0 2,6 ] decane, ( Acryloyloxyethyl-methacryloyloxyethyl) tricyclo [5.2.1.0 2,6 ] decane, bis (acryloyloxyethyl) pentacyclo [6.5.1.1 3,6 . 0 2,7 .
- pentadecane bis (methacryloyloxyethyl) pentacyclo [6.5.1.1 3,6 . 0 2,7 . 0 9,13 ] pentadecane, (acryloyloxyethyl-methacryloyloxyethyl) pentacyclo [6.5.1.1 3,6 . 0 2,7 . 0 9,13 ] pentadecane, etc., and mixtures thereof.
- bis (acryloyloxymethyl) tricyclo [5.2.1.0 2,6 ] decane bis (methacryloyloxymethyl) tricyclo [5.2.1.0 2,6 ] decane and (acryloyloxymethyl) -Methacryloyloxymethyl) tricyclo [5.2.1.0 2,6 ] decane is preferred.
- bis (meth) acrylates can be used in combination.
- a (meth) acrylic ester of polyol or the like can be used as the (meth) acrylate having 4 to 8 (meth) acryloyl groups in the molecule.
- a (meth) acrylic ester of polyol or the like can be used.
- epoxy (meth) acrylate examples include, for example, bisphenol A type epoxy resin, bisphenol F type epoxy resin, phenol novolac type epoxy resin, compound having an alicyclic epoxy group, bisphenol A type propylene oxide addition type.
- a reaction product of terminal glycidyl ether, fluorene epoxy resin and the like and (meth) acrylic acid can be mentioned.
- the (meth) acrylate having a urethane bond in the molecule include urethane oligomers having 2 to 10 (preferably 2 to 5) (meth) acryloyl groups in one molecule.
- a (meth) acryloyl group-containing urethane oligomer produced by reacting a urethane prepolymer obtained by reacting diols and diisocyanates with a hydroxy group-containing (meth) acrylate.
- the number average molecular weight of the (meth) acrylate having a urethane bond in the molecule is preferably 1,000 to 100,000, and more preferably 2,000 to 10,000.
- urethane acrylate having methylene dicyclohexyl diisocyanate and polytetramethylene ether glycol is excellent in terms of transparency, low birefringence, flexibility, and the like, and can be suitably used.
- the oxetane resin precursor examples include a compound having at least one oxetane ring.
- the number of oxetane rings in the oxetane resin precursor is preferably 1 or more and 4 or less per molecule.
- the compound having one oxetane in the molecule include 3-ethyl-3-hydroxymethyloxetane, 3-ethyl-3- (phenoxymethyl) oxetane, and 3-ethyl-3- (2-ethylhexyloxymethyl) oxetane.
- the curing agent used for the curing reaction of the oxetane resin precursor is not particularly limited.
- amine compounds compounds such as polyaminoamide compounds synthesized from amine compounds, tertiary amine compounds, imidazole compounds, hydrazide compounds
- examples include melamine compounds, acid anhydrides, phenol compounds, thermal latent cationic polymerization catalysts, photolatent cationic polymerization initiators, dicyanamide and derivatives thereof.
- curing agents may be used independently and 2 or more types may be used together.
- the photocuring agent is preferably used from the viewpoint of effective use of energy.
- the photo-curing agent is a compound that initiates cationic polymerization by irradiation with active energy rays, and examples thereof include diaryliodonium salts and triarylsulfonium salts.
- phenol resin precursor examples include those obtained by reacting phenols such as phenol and cresol with formaldehyde and the like to synthesize novolak and the like and curing the resultant with hexamethylenetetramine or the like.
- urea resin precursor examples include polymerization reaction products such as urea and formaldehyde.
- melamine resin precursor examples include melamine and a polymerization reaction product such as formaldehyde.
- the unsaturated polyester resin examples include a resin obtained by dissolving an unsaturated polyester obtained from an unsaturated polybasic acid or the like and a polyhydric alcohol or the like in a monomer that polymerizes with the unsaturated polyester.
- Examples of the silicon resin precursor include those having an organopolysiloxane as a main skeleton.
- polyurethane resin precursor examples include polymerization reaction products composed of diols such as glycol and diisocyanate.
- diallyl phthalate resin precursor examples include reactants comprising diallyl phthalate monomers and diallyl phthalate prepolymers.
- thermosetting resins there are no particular limitations on the curing agent and curing catalyst of these thermosetting resins.
- the curing agent include polyfunctional amines, polyamides, acid anhydrides, and phenol resins
- examples of the curing catalyst include imidazole. It is done. These can be used alone or as a mixture of two or more.
- Photocurable resin Although it does not specifically limit as photocurable resin in this invention, Precursors, such as an epoxy resin illustrated in the description of the above-mentioned thermosetting resin, an acrylic resin, an oxetane resin, are mentioned. Although there is no limitation in particular as a hardening
- thermosetting resin and the photocurable resin described so far are used as a curable composition appropriately blended with a chain transfer agent, an ultraviolet absorber, a filler, a silane coupling agent and the like.
- the curable composition may contain a chain transfer agent for the purpose of causing the reaction to proceed uniformly.
- a chain transfer agent for the purpose of causing the reaction to proceed uniformly.
- a polyfunctional mercaptan compound having two or more thiol groups in the molecule can be used, thereby imparting appropriate toughness to the cured product.
- mercaptan compounds include pentaerythritol tetrakis ( ⁇ -thiopropionate), trimethylolpropane tris ( ⁇ -thiopropionate), and tris [2- ( ⁇ -thiopropionyloxyethoxy) ethyl] triisocyanurate. It is preferable to use 1 type (s) or 2 or more types.
- a mercaptan compound When a mercaptan compound is added, it is usually contained at a ratio of 30% by weight or less based on the total of radically polymerizable compounds.
- the curable composition may contain an ultraviolet absorber.
- the ultraviolet absorber is selected from a benzophenone ultraviolet absorber and a benzotriazole ultraviolet absorber, and the ultraviolet absorber may be used alone or in combination of two or more. .
- an ultraviolet absorber When an ultraviolet absorber is added, it is usually contained at a ratio of 0.01 to 1 part by weight with respect to a total of 100 parts by weight of the radically polymerizable compounds.
- filler other than cellulose
- examples of the filler include inorganic particles and organic polymers. Specific examples include inorganic particles such as silica particles, titania particles and alumina particles, transparent cycloolefin polymers such as Zeonex (Nippon Zeon) and Arton (JSR), and general-purpose thermoplastic polymers such as polycarbonate and PMMA. . Among these, the use of nano-sized silica particles is preferable because transparency can be maintained. In addition, it is preferable to use a polymer having a structure similar to that of the ultraviolet curable monomer because the polymer can be dissolved to a high concentration.
- silane coupling agent A silane coupling agent may be added.
- the silane coupling agent include ⁇ -((meth) acryloxypropyl) trimethoxysilane, ⁇ -((meth) acryloxypropyl) methyldimethoxysilane, and ⁇ -((meth) acryloxypropyl) methyldiethoxy.
- Silane, ⁇ -((meth) acryloxypropyl) triethoxysilane, ⁇ - (acryloxypropyl) trimethoxysilane, etc. have a (meth) acryl group in the molecule and must be copolymerized with other monomers. Is preferable.
- the silane coupling agent is contained in an amount of usually 0.1 to 50% by weight, preferably 1 to 20% by weight, based on the total of radically polymerizable compounds. If the blending amount is too small, the effect of containing it is not sufficiently obtained, and if it is too large, optical properties such as transparency of the cured product may be impaired.
- the curable composition for forming the cellulose fiber composite of the present invention can be polymerized and cured by a known method.
- heat curing or radiation curing can be used.
- Radiation curing is preferable.
- the radiation include infrared rays, visible rays, ultraviolet rays, electron beams, and the like, preferably light. More preferred is light having a wavelength of about 200 nm to 450 nm, and more preferred is ultraviolet light having a wavelength of 300 to 400 nm.
- thermopolymerization a method in which a thermal polymerization initiator that generates radicals upon heating is added to the curable composition in advance and polymerized by heating (hereinafter sometimes referred to as “thermal polymerization”), the curable composition in advance.
- a photopolymerization initiator that generates radicals by radiation such as ultraviolet rays is added to the polymer, and a method of polymerizing by irradiation with radiation (hereinafter sometimes referred to as “photopolymerization”), etc.
- photopolymerization a method of adding in advance using a combination of free and polymerizing by a combination of heat and light. In the present invention, photopolymerization is more preferable.
- a photo radical generator is usually used.
- the photoradical generator a known compound that can be used for this purpose can be used. Examples include benzophenone, benzoin methyl ether, benzoin propyl ether, diethoxyacetophenone, 1-hydroxycyclohexyl phenyl ketone, 2,6-dimethylbenzoyldiphenylphosphine oxide, 2,4,6-trimethylbenzoyldiphenylphosphine oxide, and the like. Among these, 2,4,6-trimethylbenzoyldiphenylphosphine oxide is preferable.
- These photopolymerization initiators may be used alone or in combination of two or more.
- the component amount of the photopolymerization initiator is 0.001 part by weight or more, preferably 0.01 part by weight or more, more preferably 0, when the total amount of radically polymerizable compounds in the curable composition is 100 parts by weight. .05 parts by weight or more.
- the upper limit is usually 1 part by weight or less, preferably 0.5 part by weight or less, more preferably 0.1 part by weight or less.
- the amount of the initiator when the amount of the initiator is 5 parts by weight, absorption of the initiator causes light to not reach the side opposite to the irradiation of the ultraviolet rays, resulting in an uncured portion. Further, it is colored yellow and the hue is markedly deteriorated. On the other hand, if the amount is too small, the polymerization may not proceed sufficiently even if ultraviolet irradiation is performed.
- a thermal polymerization initiator may be included at the same time.
- examples thereof include hydroperoxide, dialkyl peroxide, peroxy ester, diacyl peroxide, peroxy carbonate, peroxy ketal, and ketone peroxide.
- thermal polymerization initiators When thermal polymerization is initiated at the time of light irradiation, it becomes difficult to control the polymerization. Therefore, these thermal polymerization initiators preferably have a 1 minute half-life temperature of 120 ° C. or higher. These polymerization initiators may be used alone or in combination of two or more.
- the amount of radiation to be irradiated upon curing is arbitrary as long as the photopolymerization initiator generates radicals. However, when the amount is extremely small, the polymerization is incomplete, so that the cured product has sufficient heat resistance and mechanical properties. If it is not expressed and is extremely excessive, on the contrary, the cured product is deteriorated by light such as yellowing. Therefore, ultraviolet rays having a wavelength of 300 to 450 nm are used in accordance with the composition of the monomer and the type and amount of the photopolymerization initiator. preferably irradiated with 0.1 J / cm 2 or more 200 J / cm 2 or less.
- irradiation is performed in the range of 1 J / cm 2 or more and 20 J / cm 2 or less. More preferably, the radiation is divided into a plurality of times. That is, when the first irradiation is performed for about 1/20 to 1/3 of the total irradiation amount and the necessary remaining amount is irradiated for the second and subsequent times, a cured product having smaller birefringence can be obtained.
- the lamp to be used include a metal halide lamp, a high pressure mercury lamp lamp, an ultraviolet LED lamp and the like.
- Photopolymerization and thermal polymerization may be performed simultaneously for the purpose of promptly completing the polymerization.
- the curable composition is heated and cured in the range of 30 ° C. or more and 300 ° C. or less simultaneously with the radiation irradiation.
- a thermal polymerization initiator may be added to the curable composition in order to complete the polymerization, but if added in a large amount, the birefringence of the cured product is increased and the hue is deteriorated.
- the agent is usually used in an amount of 0.1 to 2% by weight, more preferably 0.3 to 1% by weight, based on the total amount of monomers.
- the cellulose fiber composite obtained by the present invention may be a laminated structure of a layer of the cellulose sheet obtained by the present invention and a planar structure layer made of a polymer other than cellulose as described above.
- the layered structure of the cellulose sheet layer obtained in the above and the layer of the polymer cellulose composite obtained in the present invention may be used, and the number of layers and the layered structure are not particularly limited.
- the cellulose fiber composite obtained in the present invention may be obtained by further laminating an inorganic film on the polymer cellulose composite layer according to its use, and further laminating the inorganic film on the above laminated structure. It may be.
- the inorganic film used here is appropriately determined according to the use of the cellulose fiber composite. For example, metals such as platinum, silver, aluminum, gold, and copper, silicon, ITO, SiO 2 , SiN, SiOxNy, ZnO, etc. A TFT etc. are mentioned, The combination and film thickness can be designed arbitrarily.
- ⁇ Characteristics and physical properties of cellulose fiber composite ⁇ Characteristics and physical properties of cellulose fiber composite ⁇
- suitable characteristics or physical properties of the cellulose fiber composite obtained in the present invention will be described.
- ⁇ Cellulose content> The cellulose content in the cellulose fiber composite of the present invention is usually 1% by weight to 99% by weight, and the content of polymers other than cellulose is 1% by weight to 99% by weight.
- the content of cellulose is 1% by weight or more and the content of polymers other than cellulose is 99% by weight or less.
- In order to express transparency it is necessary that the content of cellulose is 99% by weight or less and the content of polymers other than cellulose is 1% by weight or more.
- a preferred range is 5% to 90% by weight of cellulose, a polymer other than cellulose is 10% to 95% by weight, and a more preferred range is 10% to 80% by weight of cellulose,
- the polymer other than cellulose is 20% by weight or more and 90% by weight or less.
- the content of cellulose is preferably 30% by weight or more and 70% by weight or less, and the content of polymers other than cellulose is preferably 30% by weight or more and 70% by weight or less.
- the content of cellulose and a polymer other than cellulose in the cellulose fiber composite can be determined from, for example, the weight of cellulose before complexing and the weight of cellulose after complexing.
- the cellulose composite can be immersed in a solvent in which the polymer is soluble to remove only the polymer, and the weight can be obtained from the remaining cellulose.
- the functional group of the resin or cellulose can also be determined quantitatively using a method of obtaining from the specific gravity of the resin, NMR, or IR.
- the thickness of the cellulose fiber composite obtained by the present invention is preferably 10 ⁇ m or more and 10 cm or less, and by setting it to such a thickness, the strength as a structural material can be maintained.
- the thickness of the cellulose fiber composite is more preferably 50 ⁇ m or more and 1 cm or less, and further preferably 80 ⁇ m or more and 250 ⁇ m or less.
- the cellulose fiber composite obtained by the present invention is, for example, a film shape (film shape) or a plate shape having such a thickness, but is not limited to a flat film or a flat plate, and is a film shape or plate shape having a curved surface. It can also be. Other irregular shapes may also be used. Further, the thickness is not necessarily uniform, and may be partially different. ⁇ Coloring> The cellulose fiber composite obtained by the present invention is small in color.
- Cellulose may be yellowish by using raw materials derived from wood. This may be due to the coloring of the cellulose itself, or the substance other than the cellulose remaining depending on the degree of purification. Generally, it is not colored at the stage of cellulose alone, but it may be colored by heating when it is combined with a polymer.
- the cellulose fiber and polymer cellulose composite obtained according to the present invention are small in color even when a heating step is performed.
- YI indicating the coloration of the polymer cellulose composite obtained by the present invention is preferably 20 or less, more preferably 15 or less, and further preferably 10 or less. YI can be measured using, for example, a color computer manufactured by Suga Test Instruments.
- the cellulose fiber composite obtained by the present invention can be a high-molecular cellulose composite having high transparency, that is, low haze.
- the haze value of the polymer cellulose composite is preferably 20 or less, more preferably 10 or less, and particularly preferably 3 or less, especially 1 or less.
- the cellulose fiber composite obtained by the present invention can be made into a cellulose fiber composite having high transparency, that is, low haze.
- this cellulose fiber composite has a total light transmittance of 60% or more, further 70% or more, particularly 80% or more, particularly 90%, as measured in the thickness direction according to JIS standard K7105. % Or more is preferable. If this total light transmittance is less than 60%, it becomes translucent or opaque, and it may be difficult to use it in applications requiring transparency.
- the total light transmittance can be measured, for example, for a cellulose composite having a thickness of 10 to 100 ⁇ m using a haze meter manufactured by Suga Test Instruments, and the value of C light is used.
- the cellulose fiber composite obtained by the present invention can be made into a cellulose fiber composite having a low linear expansion coefficient by using cellulose fibers having a low linear expansion coefficient (elongation rate per 1K).
- the linear expansion coefficient of the cellulose composite is preferably 1 to 50 ppm / K, more preferably 1 to 30 ppm / K, and particularly preferably 1 to 20 ppm / K.
- the linear expansion coefficient of an inorganic thin film transistor is about 15 ppm / K
- the linear expansion coefficient of the cellulose fiber composite exceeds 50 ppm / K
- the composite film is laminated with an inorganic film.
- the difference in linear expansion coefficient between the two layers becomes large, and cracks and the like are generated.
- the linear expansion coefficient of the cellulose composite is particularly preferably 1 to 20 ppm / K.
- the tensile strength of the cellulose fiber composite obtained by the present invention is preferably 40 MPa or more, more preferably 100 MPa or more. If the tensile strength is lower than 40 MPa, sufficient strength cannot be obtained, which may affect the use of a structural material or the like to which a force is applied.
- the tensile elastic modulus of the cellulose fiber composite obtained by the present invention is preferably 0.2 to 100 GPa, more preferably 1 to 50 GPa, still more preferably 5.0 to 30 GPa.
- the tensile elastic modulus is lower than 0.2 GPa, sufficient strength cannot be obtained, which may affect the use of a structural material or the like to which a force is applied.
- the modified cellulose fiber contained in the cellulose fiber composite obtained by the present invention has a cellulose I-type crystal structure.
- Cellulose contained in the cellulose fiber composite of the present invention has an "average diffraction intensity at the same scanning angle of 18 to 19 degrees" with respect to "the intensity of the diffraction peak derived from the cellulose I-type crystal structure at a scanning angle of 20 to 24 degrees" (non- The ratio of the crystal structure and the like other than Type I is 0.8) or less. Preferably it is 0.7 or less, especially 0.6 or less, and especially 0.5 or less.
- the cellulose fiber composite obtained by the present invention has high transparency, high strength, low water absorption, high transparency, low coloration and small optical haze, and is excellent in optical properties. It is suitable as a display, substrate or panel for an emission display, a rear projection television or the like. Moreover, it is suitable for substrates for solar cells such as silicon-based solar cells and dye-sensitized solar cells.
- the substrate may be laminated with a barrier film, ITO, TFT, or the like. Further, it is suitably used for window materials for automobiles, window materials for railway vehicles, window materials for houses, window materials for offices and factories.
- window material you may laminate
- the transparent material can be used as a structure other than the transparent material utilizing the characteristics such as low linear expansion coefficient, high elasticity, and high strength.
- it is suitably used as automobile materials such as interior materials, outer plates, bumpers, etc., personal computer casings, home appliance parts, packaging materials, building materials, civil engineering materials, marine materials, and other industrial materials.
- the present invention will be described more specifically with reference to production examples, examples, and comparative examples. However, the present invention is not limited to the following examples unless it exceeds the gist.
- the measuring method of the thermal decomposition temperature of the cellulose fiber obtained by this invention, the cellulose content of a cellulose fiber composite, YI, haze, a linear expansion coefficient, and an elasticity modulus is as follows. [Chemical modification rate of modified cellulose fiber] 0.05 g of the dried modified cellulose fiber was precisely weighed, 6 ml of methanol and 2 ml of distilled water were added thereto, stirred at 60 to 70 ° C.
- the number of moles Q of the substituent introduced by chemical modification is obtained by the following formula.
- Q (mol) 0.05 (N) ⁇ 10 (ml) / 1000 ⁇ 0.02 (N) ⁇ Z (ml) / 1000
- T is the molecular weight of the substituent.
- Thermal decomposition temperature of cellulose fiber The cellulose sheet was conditioned at a temperature of 23 ° C. and a humidity of 50% for 48 hours or more, and the temperature was increased from room temperature to 600 ° C. at 10 ° C./min using TG-DTA (differential thermogravimetric simultaneous measurement device). The intersection of the tangent lines obtained from the TG at that time was defined as the thermal decomposition temperature.
- TG-DTA Differential thermogravimetric simultaneous measurement device.
- the intersection of the tangent lines obtained from the TG at that time was defined as the thermal decomposition temperature.
- Average fiber diameter The fiber diameter of the cellulose fiber was measured and determined by observing with an optical microscope, SEM, TEM or the like. The average of 10 points excluding the maximum and minimum of 12 points extracted at random was defined as the average fiber diameter.
- the cellulose sheet used for the measurement has voids, the calculation is simply performed using the thickness and width of the sample, and correction based on the void ratio is not performed.
- the cellulose content (% by weight) was determined from the weight of the cellulose sheet used for the composite and the weight of the cellulose composite.
- Linear expansion coefficient of cellulose fiber composite The cellulose fiber composite was cut into 3 mm width ⁇ 40 mm length with a laser cutter. This was stretched using a TMA6100 made by SII in a tensile mode at a temperature of 5 ° C./min. And then from 180 ° C. to 25 ° C. at 5 ° C./min. At 25 ° C. and then from 25 ° C. to 180 ° C. at 5 ° C./min. The linear expansion coefficient was determined from the measured value from 60 ° C. to 100 ° C. during the second temperature increase.
- Wood flour (Miyashita Wood Co., Ltd., Yonematsu 100) was degreased with a 2% by weight aqueous solution of sodium carbonate at 80 ° C. for 6 hours. This was washed with demineralized water and then delignified with sodium chlorite under acetic acid acidity at 80 ° C. for 5.5 hours. After washing with desalted water, it was further immersed in a 5% by weight aqueous solution of potassium hydroxide for 16 hours to dehemicellulose. Washed with demineralized water.
- Example 1 The cellulose obtained in Production Example 1 was weighed out in a solid content of 10 g, and water was replaced with acetic acid. This was added with 300 ml of acetic acid, 10 g of sodium acetate and 21 g of benzoyl chloride, and reacted at 80 ° C. for 5 hours with stirring. Thereafter, the reaction solution was filtered, washed with acetone, and further washed with a large amount of water until neutral.
- the chemical modification rate of this benzoylated cellulose was 29 mol%.
- the thermal decomposition temperature calculated from TG-DTA was 354 ° C.
- the obtained modified cellulose was made into a 0.5% by weight aqueous suspension, using a stone mill mill Supermass colloider MKCA6-2 from Masuko Sangyo Co., Ltd., using a GC6-80 stone mill, with a gap of 80 ⁇ m. Then, it was charged from the raw material charging port at a rotational speed of 1500 rpm.
- the treated cellulose dispersion that passed through the grinder was again charged into the raw material inlet, and passed through the grinder twice in total. Then, it was passed 10 times at 245 MPa through an ultra-high pressure homogenizer (Sugino Machine Ultimate).
- This cellulose fiber dispersion was diluted with water to a concentration of 0.13% by weight, and 150 g was put into a 90 mm diameter filter using PTFE having a pore diameter of 1 ⁇ m. Added and replaced. It was 38 minutes to complete the filtration. Then, it was press-dried at 120 ° C. and 0.14 MPa for 5 minutes to obtain a white cellulose sheet (weighing about 40 g / m 2 ). The average fiber diameter calculated from SEM observation of the surface of the cellulose sheet was 18 nm. Moreover, it was confirmed from the wide-angle X-ray diffraction image that the cellulose I-type crystal structure was obtained.
- the cellulose sheet was mixed with 96 parts by weight of bis (methacryloyloxymethyl) tricyclo [5.2.1.0 2,6 ] decane, 6 parts by weight of pentaerythritol tetrakis ( ⁇ -thiopropionate), 2,4,6-
- a mixed solution of 0.05 parts by weight of trimethylbenzoyldiphenylphosphine oxide (Lucirin TPO manufactured by BASF) and 0.05 parts by weight of benzophenone was impregnated and left overnight under reduced pressure. This was sandwiched between two glass plates and irradiated under an irradiance of 400 mW / cm 2 at a line speed of 7 m / min using an electrodeless mercury lamp (“D bulb” manufactured by Fusion UV Systems). The irradiation dose at this time was 0.12 J / cm 2 . This operation was performed twice by inverting the glass surface. The temperature of the glass surface after ultraviolet irradiation was 25 ° C.
- irradiation was performed under an irradiance of 1900 mW / cm 2 at a line speed of 2 m / min.
- the radiation dose at this time was 2.7 J / cm 2 .
- This operation was performed 8 times by inverting the glass surface.
- the temperature of the glass surface after ultraviolet irradiation was 44 ° C.
- the irradiation dose was 21.8 J / cm 2 .
- the glass plate was removed and heated in a vacuum oven at 190 ° C. for 4 hours to obtain a composite material.
- the irradiance of ultraviolet rays was measured at 23 ° C. by using an ultraviolet illuminance meter “UV-M02” manufactured by Oak Manufacturing Co., Ltd. and using an attachment “UV-35”.
- Example 2 The cellulose obtained in Production Example 1 was weighed out in a solid content of 10 g, and water was replaced with acetic acid. 300 ml of acetic acid, 12 g of sodium acetate, and 25 g of benzoyl chloride were added to this, and reacted at 80 ° C. for 5 hours while stirring. Thereafter, the reaction solution was filtered, washed with acetone, and further washed with a large amount of water until neutral.
- the chemical modification rate of this benzoylated cellulose was 39 mol%.
- the thermal decomposition temperature calculated from TG-DTA was 350 ° C.
- the obtained modified cellulose was made into a 0.5% by weight aqueous suspension, using a stone mill mill Supermass colloider MKCA6-2 from Masuko Sangyo Co., Ltd., using a GC6-80 stone mill, with a gap of 80 ⁇ m. Then, it was charged from the raw material charging port at a rotational speed of 1500 rpm.
- the treated cellulose dispersion that passed through the grinder was again charged into the raw material inlet, and passed through the grinder twice in total. Then, it was passed 10 times at 245 MPa through an ultra-high pressure homogenizer (Sugino Machine Ultimate).
- This cellulose fiber dispersion was diluted with water to a concentration of 0.13% by weight, and 150 g was put into a 90 mm diameter filter using PTFE having a pore diameter of 1 ⁇ m. Added and replaced. It took 20 minutes to complete the filtration. Then, it was press-dried at 120 ° C. and 0.14 MPa for 5 minutes to obtain a white cellulose sheet (weighing about 40 g / m 2 ). The average fiber diameter calculated from SEM observation of the surface of this cellulose sheet was 15 nm. Moreover, it was confirmed from the wide-angle X-ray diffraction image that the cellulose I-type crystal structure was obtained.
- a cellulose fiber composite was obtained in the same manner as in Example 1.
- the physical properties of this cellulose fiber composite are shown in Table 1 together with the physical properties of the cellulose sheet.
- Example 3 The cellulose dispersion obtained in Example 1 was further subjected to ultrasonic treatment using an ultrasonic homogenizer UH-600S (frequency 20 kHz, effective output density 22 W / cm 2 ) manufactured by SMT.
- UH-600S frequency 20 kHz, effective output density 22 W / cm 2
- a cellulose fiber composite was obtained in the same manner as in Example 1.
- the physical properties of this cellulose fiber composite are shown in Table 1 together with the physical properties of the cellulose sheet.
- Example 4 Using the cellulose dispersion obtained in Example 2, ultrasonic treatment was performed in the same manner as in Example 3. The sonicated dispersion was centrifuged at 18000 rpm (38900 G) for 10 minutes to obtain a supernatant. Using this supernatant cellulose fiber dispersion, a cellulose sheet was obtained in the same manner as in Example 1. It was 51 minutes to complete the filtration. Then, it was press-dried at 120 ° C. and 0.14 MPa for 5 minutes to obtain a white cellulose sheet (weighing about 40 g / m 2 ). The average fiber diameter calculated from SEM observation of the surface of the cellulose sheet was 12 nm. Moreover, it was confirmed from the wide-angle X-ray diffraction image that the cellulose I-type crystal structure was obtained.
- a cellulose fiber composite was obtained in the same manner as in Example 1.
- the physical properties of this cellulose fiber composite are shown in Table 1 together with the physical properties of the cellulose sheet.
- Example 5 The cellulose obtained in Production Example 1 was weighed out in a solid content of 10 g, and water was replaced with acetic acid. 300 ml of acetic acid, 5 g of sodium acetate, and 12.5 g of benzoyl chloride were added thereto, and the mixture was reacted at 80 ° C. for 5 hours while stirring. Thereafter, the reaction solution was filtered, washed with acetone, and further washed with a large amount of water until neutral.
- the chemical modification rate of this benzoylated cellulose was 11 mol%.
- the thermal decomposition temperature calculated from TG-DTA was 339 ° C.
- the obtained modified cellulose was made into a 0.5% by weight aqueous suspension, using a stone mill mill Supermass colloider MKCA6-2 from Masuko Sangyo Co., Ltd., using a GC6-80 stone mill, with a gap of 80 ⁇ m. Then, it was charged from the raw material charging port at a rotational speed of 1500 rpm.
- the treated cellulose dispersion that passed through the grinder was again charged into the raw material inlet, and passed through the grinder twice in total. Then, it was passed 10 times at 245 MPa through an ultra-high pressure homogenizer (Sugino Machine Ultimate).
- This cellulose dispersion was diluted with water to a concentration of 0.13% by weight, and 150 g was introduced into a 90 mm diameter filter using PTFE having a pore size of 1 ⁇ m. When the solid content reached about 5% by weight, 2-propanol was introduced. And replaced. It was 102 minutes to complete the filtration. Then, it was press-dried at 120 ° C. and 0.14 MPa for 5 minutes to obtain a white cellulose sheet (weighing about 40 g / m 2 ). The average fiber diameter calculated from SEM observation of the surface of the cellulose sheet was 18 nm. Moreover, it was confirmed from the wide-angle X-ray diffraction image that the cellulose I-type crystal structure was obtained.
- a cellulose fiber composite was obtained in the same manner as in Example 1.
- the physical properties of this cellulose fiber composite are shown in Table 1 together with the physical properties of the cellulose sheet.
- ⁇ Comparative Example 1> The cellulose obtained in Production Example 1 was made into a 0.5% by weight aqueous suspension, using a stone mill grinder Supermass colloider MKCA6-2 from Masuko Sangyo Co., Ltd., and using a GC6-80 stone mill, The gap was set to 80 ⁇ m, and the material was charged from the raw material charging port at a rotation speed of 1500 rpm. The treated cellulose dispersion that passed through the grinder was again charged into the raw material inlet, and passed through the grinder twice in total. Then, it was passed 10 times at 245 MPa through an ultra-high pressure homogenizer (Sugino Machine Ultimate).
- an ultra-high pressure homogenizer Sugino Machine Ultimate
- the thermal decomposition temperature calculated from TG-DTA was 296 ° C.
- This cellulose fiber dispersion was diluted with water to a concentration of 0.13% by weight, and 150 g was put into a 90 mm diameter filter using PTFE having a pore diameter of 1 ⁇ m. Added and replaced. It was 149 minutes to complete the filtration. Then, it was press-dried at 120 ° C. and 0.14 MPa for 5 minutes to obtain a white cellulose sheet (weighing about 40 g / m 2 ).
- the average fiber diameter calculated from SEM observation of the surface of this cellulose sheet was 25 nm. Moreover, it was confirmed from the wide-angle X-ray diffraction image that the cellulose I-type crystal structure was obtained.
- a cellulose fiber composite was obtained in the same manner as in Example 1.
- the physical properties of this cellulose fiber composite are shown in Table 1 together with the physical properties of the cellulose sheet.
- the cellulose sheet was taken out, washed with methanol and demineralized water in this order, finally substituted with 2-propanol, and then press-dried at 120 ° C. and 0.14 MPa for 5 minutes to obtain a white cellulose sheet.
- the average fiber diameter calculated from SEM observation of the surface of this cellulose sheet was 25 nm.
- the chemical modification rate of this acetylated cellulose was 7 mol%.
- the thermal decomposition temperature calculated from TG-DTA was 331 ° C.
- a cellulose fiber composite was obtained in the same manner as in Example 1. The physical properties of this cellulose fiber composite are shown in Table 1 together with the physical properties of the cellulose sheet.
- ⁇ Comparative Example 4> (implemented by replacing 4-biphenyl isocyanate of Example 1 with phenyl isocyanate in JP 2008-274461 A) Abaca was purified by the method of Production Example 1. Cellulose was weighed out in a solid content of 0.63 g, and water was replaced with dimethylformamide. The cellulose, 65 ml of dimethylformamide, and 11.5 g of phenyl isocyanate were mixed and reacted under stirring at 115 ° C. for 2 hours. After cooling, it was filtered and washed with water and methanol.
- the cellulose I-type crystal structure is not maintained, and the “intensity of the diffraction peak derived from the cellulose I-type crystal structure at a scanning angle of 20 to 24 degrees” is “at the same scanning angle of 18 to 19 degrees”.
- the ratio of “average diffraction intensity” (reflecting a structure other than type I such as amorphous) was 1.11, suggesting that it was amorphous.
- the thermal decomposition temperature calculated from TG-DTA was 258 ° C. The results are shown in Table 1.
- the modified cellulose fibers as shown in Examples 1 to 5 obtained by the present invention are unmodified cellulose fibers as shown in Comparative Example 1, acetylated cellulose modified with an aliphatic-containing substituent as shown in Comparative Example 2, Compared to cellulose modified with an aromatic ring-containing substituent using conventional methods as shown in Comparative Examples 3 and 4, the thermal decomposition temperature was improved.
- the sheet strength ratio is maintained with a high probability of 99% or more, and at the same time, a high tensile elastic modulus of 4 GPa or more is exhibited.
- Comparative Example 1 Although the sheet strength is maintained, the tensile elastic modulus remains at a little over 3 GPa. In Comparative Example 2, the tensile elastic modulus is improved to a little less than 4 GPa, but the tensile strength is greatly reduced. This difference also appears as a difference in tensile elastic modulus when the composite material is used, and the composite materials of Examples 1 to 5 show a clear improvement in tensile elastic modulus compared to the composite materials of Comparative Examples 1 and 2. It is done.
- the filtration time required for obtaining a cellulose sheet from the cellulose aqueous dispersion obtained in the same defibration process is compared, it is modified with an aromatic ring-containing substituent from Examples 1, 2, 5 and Comparative Example 1.
- the filtration time is faster than that of the unmodified and the haze is equal to or higher than that.
- the filtration time is more important during industrial production. Since a larger amount of the cellulose dispersion is treated by a reduced pressure or pressure filtration with a smaller differential pressure, the difference in filtration time is very large. Further, from Examples 3 and 4, a more transparent composite material was obtained by performing ultrasonic treatment.
- Comparative Example 2 which is a follow-up test of Example 6, the cellulose sheet was modified with an aliphatic acetyl group, but was modified after an unmodified cellulose sheet was prepared. Therefore, the effect of shortening the filtration time was not obtained, and the coloring after heating was not improved, and the transparency was deteriorated.
- Comparative Example 3 modification was performed with an aromatic-containing group by an esterification method different from that of the present application, but the reactivity was poor and little progressed.
- Comparative Example 4 modification with an aromatic carbamate group was performed by a method different from that of the present application, but cellulose I-type crystals were not maintained. Comparative Example 4 with a small proportion of the I-type crystal structure is expected to have a significantly lower strength than Comparative Examples 1 to 3, and various physical properties such as sheet strength retention ratio and tensile elastic modulus after heating are shown in this example. I do n’t think I ’ll reach the level. Therefore, even if the cellulose of Comparative Example 4 is made into a sheet and a cellulose fiber composite is obtained, it is unlikely that various physical properties as a composite material such as tensile elastic modulus will reach the level of the embodiment of the present application.
- Examples 1 to 5 of the present application although the hydroxyl group on the cellulose surface is modified with an aromatic ring-containing group with a high probability exceeding 10%, the type I crystal structure is maintained. It can be said that the production method of the present application is a method for efficiently introducing an aromatic-containing group while maintaining cellulose I-type crystals. It is considered that the effects of the present invention including the heat resistance of the cellulose sheet, YI when used as a composite material, tensile elastic modulus, and the like are achieved by coexistence of the chemical modification rate and the I-type crystal structure.
- Table 1 when comparing the average fiber diameters of Examples 1 and 2 and Comparative Example 1 cellulose, Examples 1 and 2 have smaller values than Comparative Example 1 despite the same defibrating process. Therefore, the present invention effectively swells cellulose fibers by using an organic acid at the time of chemical modification, achieves uniform chemical modification, and achieves both the chemical modification rate and the I-type crystal structure. Infer.
- the reaction of the modified cellulose given in the examples was carried out in acetic acid, which is an organic acid, but all of them had good defibration properties. From this, it can be said that the cellulose modified with an aromatic ring-containing substituent is easily defibrated and highly productive. Further, when the physical properties of the cellulose composite using the cellulose sheet obtained from the cellulose fiber were examined, as described above, it was highly transparent, and non-coloring property, low linear expansion coefficient, and high elastic modulus were achieved. .
- the cellulose fiber composite obtained by the production method of the present invention has high transparency, high strength, low water absorption, high transparency, low coloration and low haze, and excellent optical characteristics. Therefore, the liquid crystal display, plasma display, organic EL It is suitable as a display, a substrate or a panel of a display, a field emission display, a rear projection television or the like. Moreover, it is suitable for substrates for solar cells such as silicon-based solar cells and dye-sensitized solar cells. The substrate may be laminated with a barrier film, ITO, TFT or the like. Further, it is suitably used for window materials for automobiles, window materials for railway vehicles, window materials for houses, window materials for offices and factories, and the like. As a window material, you may laminate
- the transparent material can be used as a structure other than the transparent material utilizing the characteristics such as low linear expansion coefficient, high elasticity, and high strength.
- it is suitably used as automobile materials such as interior materials, outer plates, bumpers, etc., personal computer casings, home appliance parts, packaging materials, building materials, civil engineering materials, marine materials, and other industrial materials.
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Textile Engineering (AREA)
- Materials Engineering (AREA)
- Polymers & Plastics (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Nanotechnology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- Manufacturing & Machinery (AREA)
- Inorganic Chemistry (AREA)
- Physics & Mathematics (AREA)
- Composite Materials (AREA)
- Condensed Matter Physics & Semiconductors (AREA)
- General Physics & Mathematics (AREA)
- Crystallography & Structural Chemistry (AREA)
- Polysaccharides And Polysaccharide Derivatives (AREA)
- Treatments For Attaching Organic Compounds To Fibrous Goods (AREA)
- Nonwoven Fabrics (AREA)
Abstract
本発明は、高透明性、低線膨張係数化、高弾性率を実現するセルロース複合体を得ることができる、セルロース繊維を提供することを目的とする。本発明は、セルロースを有機酸中で芳香族化合物と反応させることにより該セルロースを芳香環含有置換基で修飾する修飾反応工程を有する修飾セルロース繊維の製造方法、芳香環含有置換基で修飾されたセルロース繊維、該セルロース繊維の分散液、及びこれを用いたセルロース繊維複合体に関する。
Description
本発明は、修飾セルロース繊維とこの修飾セルロース繊維を用いた複合体に関するものであり、特に修飾セルロース繊維の修飾置換基と修飾率を制御することで、高耐熱性と易解繊性と高生産性を実現し、セルロース複合体にした際の、高透明性、低線膨張係数化、高弾性率、加熱時強度向上等を実現する技術に関する。
近年、バクテリアセルロースをはじめとするセルロースの微細繊維を用いた複合材料が盛んに研究されている。セルロースはその伸びきり鎖結晶が故に、低線膨張係数と高弾性率と高強度とを発現することが知られている。また、微細化することにより、複合材料とした際、高透明性を示す材料として注目されている。
特許文献1においては、表面修飾したセルロースの微細繊維を用いて複合化することで、吸湿性及び透明性が向上した複合材を得ている。この表面修飾剤としては、脂肪族系のものが開示されており、特にアセチル基やメタクリロイル基が好ましいことが開示されている。
特許文献2においては、光学異方性を制御する化合物として、芳香環構造を有する化合物を導入した改質繊維からなる不織布について開示されている。
又、非特許文献1には、サーモトロピック液晶特性を示すベンゾイル化バクテリアセルロースが開示されている。
Carbohydrate Polymers,Vol.74,Issue4,p875-879(2008)
上記特許文献1において具体的に開示されているセルロースの微細繊維不織布(セルロースシート)の製造方法は、機械的に解繊したセルロース微細繊維の水懸濁液を、ガラスフィルターで濾過して脱水し、製膜する方法である。
しかしながら、本発明者が、特許文献1に開示される方法に従ってセルロース複合材料の製造を行ったところ、セルロースシートを製造する際の、セルロース微細繊維の水懸濁液の脱水のための濾過にかかる時間が長く、実用的な生産性が得られないことが判明した。
しかしながら、本発明者が、特許文献1に開示される方法に従ってセルロース複合材料の製造を行ったところ、セルロースシートを製造する際の、セルロース微細繊維の水懸濁液の脱水のための濾過にかかる時間が長く、実用的な生産性が得られないことが判明した。
上記特許文献2において具体的に開示されている不織布(セルロースシート)は、N,N-ジメチルホルムアミド、エチルセルソルブ等の存在下で芳香環含有化合物とセルロースを反応させている。
しかしながら、本発明者が、特許文献2に開示される方法に従ってセルロースシートの製造を行ったところ、セルロースの結晶構造が明らかに破壊され、I型結晶構造の割合が低くなってしまうことがわかった。I型結晶構造の割合が低いセルロースシートは耐熱性に劣ることから、このセルロースシートと高分子材料とを複合化しても、低線膨張係数、高弾性率とはならず、加熱時にはガス発生等の影響で強度が低下すると考えられる。
しかしながら、本発明者が、特許文献2に開示される方法に従ってセルロースシートの製造を行ったところ、セルロースの結晶構造が明らかに破壊され、I型結晶構造の割合が低くなってしまうことがわかった。I型結晶構造の割合が低いセルロースシートは耐熱性に劣ることから、このセルロースシートと高分子材料とを複合化しても、低線膨張係数、高弾性率とはならず、加熱時にはガス発生等の影響で強度が低下すると考えられる。
また、非特許文献1の方法で得られるベンゾイル化バクテリアセルロースは、ガラス転移温度を持つことから非晶質であると考えられ、これを高分子材料と複合化しても、低線膨張係数、高弾性率、高強度とはならない。
本発明は、上記従来の実状に鑑みてなされたものであって、高耐熱性と易解繊性と高生産性を実現するセルロース繊維と、それを、複合体にした際の、高透明性、低線膨張係数化、高弾性率を実現することを目的とする。
本発明者は、上記課題を解決すべく鋭意検討した結果、修飾セルロース繊維の平均繊維径、結晶性、修飾置換基と修飾率を制御、または修飾する反応の際に有機酸を溶媒とすることで、高耐熱性と易解繊性と高生産性を達成できることを見出した。
即ち、本発明の要旨は以下の通りである。
[1] セルロースを溶媒中で芳香族化合物と反応させることにより該セルロースを芳香環含有置換基で修飾する修飾反応工程を有する修飾セルロース繊維の製造方法であって、該修飾反応工程における該溶媒として有機酸を用いることを特徴とする修飾セルロース繊維の製造方法。
[2] 芳香環含有置換基が、芳香環含有エステル基、芳香環含有エーテル基、及び芳香環含有カルバマート基からなる群より選ばれる1種以上の基である[1]に記載の修飾セルロース繊維の製造方法。
[3] セルロースの全水酸基に対する芳香環含有置換基の修飾率が10モル%以上である[1]または[2]に記載の修飾セルロース繊維の製造方法。
[4] セルロースと有機酸とを含むセルロース分散液中でセルロースと芳香族化合物とを反応させることにより該セルロースを芳香環含有置換基で修飾する修飾反応工程と、抄紙工程とを有する修飾セルロース繊維シートの製造方法。
[5] 芳香環含有置換基が、芳香環含有エステル基、芳香環含有エーテル基、及び芳香環含有カルバマート基からなる群より選ばれる1種以上の基である[4]に記載の修飾セルロース繊維シートの製造方法。
[6] セルロースの全水酸基に対する芳香環含有置換基の修飾率が10モル%以上である[4]または[5]に記載の修飾セルロース繊維シートの製造方法。
[7] [1]~[3]のいずれか1つに記載の方法で得られた修飾セルロース繊維または[4]~[6]のいずれか1つに記載の方法で得られた修飾セルロース繊維シートと、マトリクスとを複合化する工程を有する、セルロース繊維複合体の製造方法。
[8] セルロースの全水酸基の10モル%以上が芳香環含有置換基で修飾されたセルロースI型結晶構造を有するセルロース繊維の分散液であって、該セルロース繊維の広角X線回折像の「走査角20~24度におけるセルロースI型結晶由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」の比が0.8以下である修飾セルロース繊維分散液。
[9] セルロース繊維の平均繊維径が100nm以下である[8]に記載の修飾セルロース繊維分散液。
[10] 芳香環含有置換基が、芳香環含有エステル基、芳香環含有エーテル基、及び芳香環含有カルバマート基からなる群より選ばれる1種以上の基である[8]または[9]に記載の修飾セルロース繊維分散液。
[11] セルロースの全水酸基の10モル%以上が芳香環含有置換基で修飾されたセルロースI型結晶構造を有するセルロース繊維であって、該セルロース繊維の広角X線回折像の「走査角20~24度におけるセルロースI型結晶由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」の比が0.8以下である修飾セルロース繊維。
[12] セルロース繊維の平均繊維径が100nm以下である[11]に記載の修飾セルロース繊維。
[13] 芳香環含有置換基が、芳香環含有エステル基、芳香環含有エーテル基、及び芳香環含有カルバマート基からなる群より選ばれる1種以上の基である[11]または[12]に記載の修飾セルロース繊維。
[14] 平均繊維径が100nm以下であり、セルロースの全水酸基の10モル%以上が芳香環含有置換基で修飾されたセルロースI型結晶構造を有するセルロース繊維を含む修飾セルロース繊維シートであって、下記(1)に示す複合材料としたときのC光によるヘーズ値が3以下である修飾セルロース繊維シート。
(1)秤量40g/m2となるように厚みを調整したセルロース繊維シートを、ビス(メタクリロイルオキシメチル)トリシクロ[5.2.1.02,6]デカン96重量部、ペンタエリスリトールテトラキス(β-チオプロピオネート)6重量部、2,4,6-トリメチルベンゾイルジフェニルフォスフィンオキサイド0.05重量部、ベンゾフェノン0.05重量部を混合した溶液に含浸させ、減圧下に一晩置いた後、2枚のガラス板にはさみ、無電極水銀ランプを用いて硬化後、ガラス板よりはずして190℃の真空オーブン中で4時間加熱して得た複合材料。
[15] [11]~[13]のいずれか1つに記載の修飾セルロース繊維または[14]に記載の修飾セルロース繊維シートと、マトリクスとを含むセルロース繊維複合体。
[16] セルロースの全水酸基の10モル%以上が芳香環含有置換基で修飾されたセルロースI型結晶構造を有するセルロース繊維と、マトリクスとを含むセルロース繊維複合体であって、該セルロース繊維の広角X線回折像の「走査角20~24度におけるセルロースI型結晶由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」の比が0.8以下であるセルロース繊維複合体。
即ち、本発明の要旨は以下の通りである。
[1] セルロースを溶媒中で芳香族化合物と反応させることにより該セルロースを芳香環含有置換基で修飾する修飾反応工程を有する修飾セルロース繊維の製造方法であって、該修飾反応工程における該溶媒として有機酸を用いることを特徴とする修飾セルロース繊維の製造方法。
[2] 芳香環含有置換基が、芳香環含有エステル基、芳香環含有エーテル基、及び芳香環含有カルバマート基からなる群より選ばれる1種以上の基である[1]に記載の修飾セルロース繊維の製造方法。
[3] セルロースの全水酸基に対する芳香環含有置換基の修飾率が10モル%以上である[1]または[2]に記載の修飾セルロース繊維の製造方法。
[4] セルロースと有機酸とを含むセルロース分散液中でセルロースと芳香族化合物とを反応させることにより該セルロースを芳香環含有置換基で修飾する修飾反応工程と、抄紙工程とを有する修飾セルロース繊維シートの製造方法。
[5] 芳香環含有置換基が、芳香環含有エステル基、芳香環含有エーテル基、及び芳香環含有カルバマート基からなる群より選ばれる1種以上の基である[4]に記載の修飾セルロース繊維シートの製造方法。
[6] セルロースの全水酸基に対する芳香環含有置換基の修飾率が10モル%以上である[4]または[5]に記載の修飾セルロース繊維シートの製造方法。
[7] [1]~[3]のいずれか1つに記載の方法で得られた修飾セルロース繊維または[4]~[6]のいずれか1つに記載の方法で得られた修飾セルロース繊維シートと、マトリクスとを複合化する工程を有する、セルロース繊維複合体の製造方法。
[8] セルロースの全水酸基の10モル%以上が芳香環含有置換基で修飾されたセルロースI型結晶構造を有するセルロース繊維の分散液であって、該セルロース繊維の広角X線回折像の「走査角20~24度におけるセルロースI型結晶由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」の比が0.8以下である修飾セルロース繊維分散液。
[9] セルロース繊維の平均繊維径が100nm以下である[8]に記載の修飾セルロース繊維分散液。
[10] 芳香環含有置換基が、芳香環含有エステル基、芳香環含有エーテル基、及び芳香環含有カルバマート基からなる群より選ばれる1種以上の基である[8]または[9]に記載の修飾セルロース繊維分散液。
[11] セルロースの全水酸基の10モル%以上が芳香環含有置換基で修飾されたセルロースI型結晶構造を有するセルロース繊維であって、該セルロース繊維の広角X線回折像の「走査角20~24度におけるセルロースI型結晶由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」の比が0.8以下である修飾セルロース繊維。
[12] セルロース繊維の平均繊維径が100nm以下である[11]に記載の修飾セルロース繊維。
[13] 芳香環含有置換基が、芳香環含有エステル基、芳香環含有エーテル基、及び芳香環含有カルバマート基からなる群より選ばれる1種以上の基である[11]または[12]に記載の修飾セルロース繊維。
[14] 平均繊維径が100nm以下であり、セルロースの全水酸基の10モル%以上が芳香環含有置換基で修飾されたセルロースI型結晶構造を有するセルロース繊維を含む修飾セルロース繊維シートであって、下記(1)に示す複合材料としたときのC光によるヘーズ値が3以下である修飾セルロース繊維シート。
(1)秤量40g/m2となるように厚みを調整したセルロース繊維シートを、ビス(メタクリロイルオキシメチル)トリシクロ[5.2.1.02,6]デカン96重量部、ペンタエリスリトールテトラキス(β-チオプロピオネート)6重量部、2,4,6-トリメチルベンゾイルジフェニルフォスフィンオキサイド0.05重量部、ベンゾフェノン0.05重量部を混合した溶液に含浸させ、減圧下に一晩置いた後、2枚のガラス板にはさみ、無電極水銀ランプを用いて硬化後、ガラス板よりはずして190℃の真空オーブン中で4時間加熱して得た複合材料。
[15] [11]~[13]のいずれか1つに記載の修飾セルロース繊維または[14]に記載の修飾セルロース繊維シートと、マトリクスとを含むセルロース繊維複合体。
[16] セルロースの全水酸基の10モル%以上が芳香環含有置換基で修飾されたセルロースI型結晶構造を有するセルロース繊維と、マトリクスとを含むセルロース繊維複合体であって、該セルロース繊維の広角X線回折像の「走査角20~24度におけるセルロースI型結晶由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」の比が0.8以下であるセルロース繊維複合体。
本発明によれば、修飾セルロース繊維の平均繊維径、結晶性、修飾置換基と修飾率を制御あるいは修飾する反応の際に有機酸を溶媒とすることで、高耐熱性と易解繊性と高生産性を実現するとともに、セルロース複合体にした際の、高透明性、非着色性、低線膨張係数化、高弾性率、加熱時強度向上を実現することができる。さらには、芳香環含有置換基で修飾されたセルロース繊維を用いることで樹脂との親和性、特に芳香環を含有する樹脂との親和性が高められるといった効果が奏される。
以下、本発明の実施の形態を具体的に説明するが、本発明は、以下の実施の形態に限定されるものではなく、その要旨の範囲内で種々に変更して実施することができる。
I.本発明の修飾セルロース繊維分散液の製造方法
本発明の修飾セルロース繊維分散液の製造方法は、セルロースを溶媒中で芳香族化合物と反応させることにより該セルロースを芳香環含有置換基で修飾する修飾反応工程を有し、該修飾反応時に、該溶媒として有機酸を用いることを特徴とする。
I.本発明の修飾セルロース繊維分散液の製造方法
本発明の修飾セルロース繊維分散液の製造方法は、セルロースを溶媒中で芳香族化合物と反応させることにより該セルロースを芳香環含有置換基で修飾する修飾反応工程を有し、該修飾反応時に、該溶媒として有機酸を用いることを特徴とする。
[セルロース]
本発明におけるセルロースとは、下記に示すようなセルロース含有物から一般的な精製を経て不純物を除去したものである。
本発明におけるセルロースとは、下記に示すようなセルロース含有物から一般的な精製を経て不純物を除去したものである。
<セルロース含有物>
セルロース含有物としては、針葉樹や広葉樹等の木質、コットンリンターやコットンリント等のコットン、さとうきびや砂糖大根等の絞りかす、亜麻、ラミー、ジュート、ケナフ等の靭皮繊維、サイザル、パイナップル等の葉脈繊維、アバカ、バナナ等の葉柄繊維、ココナツヤシ等の果実繊維、竹等の茎幹繊維、バクテリアが産生するバクテリアセルロース、バロニアやシオグサ等の海草やホヤの被嚢等が挙げられる。これらの天然セルロースは、結晶性が高いので低線膨張率、高弾性率になり好ましい。バクテリアセルロースは微細な繊維径のものが得やすい点で好ましい。また、コットンも微細な繊維径なものが得やすい点で好ましく、さらに原料が得やすい点で好ましい。さらには針葉樹や広葉樹等の木質も微細な繊維径のものが得られ、かつ地球上で最大量の生物資源であり、年間約700億トン以上ともいわれる量が生産されている持続型資源あることから、地球温暖化に影響する二酸化炭素削減への寄与も大きく、経済的な点から優位である。このようなセルロース含有物を一般的な精製を経て本発明のセルロース原料とする。
セルロース含有物としては、針葉樹や広葉樹等の木質、コットンリンターやコットンリント等のコットン、さとうきびや砂糖大根等の絞りかす、亜麻、ラミー、ジュート、ケナフ等の靭皮繊維、サイザル、パイナップル等の葉脈繊維、アバカ、バナナ等の葉柄繊維、ココナツヤシ等の果実繊維、竹等の茎幹繊維、バクテリアが産生するバクテリアセルロース、バロニアやシオグサ等の海草やホヤの被嚢等が挙げられる。これらの天然セルロースは、結晶性が高いので低線膨張率、高弾性率になり好ましい。バクテリアセルロースは微細な繊維径のものが得やすい点で好ましい。また、コットンも微細な繊維径なものが得やすい点で好ましく、さらに原料が得やすい点で好ましい。さらには針葉樹や広葉樹等の木質も微細な繊維径のものが得られ、かつ地球上で最大量の生物資源であり、年間約700億トン以上ともいわれる量が生産されている持続型資源あることから、地球温暖化に影響する二酸化炭素削減への寄与も大きく、経済的な点から優位である。このようなセルロース含有物を一般的な精製を経て本発明のセルロース原料とする。
<繊維径>
本発明に用いられるセルロースの繊維径は特に制限されるものではなく、数平均繊維径としては数μmから数mmである。一般的な精製を経たものは数mm程度であり、セルロースを解繊したものは数nmである。例えばチップ等の数cm大のものを精製したものである場合、リファイナーやビーター等の離解機で機械的処理を行い、数mm程度にすることが好ましい。なお、数平均繊維径の測定方法は特に限定されないが、例えばSEMやTEM等で観察して、写真の対角線に線を引き、その近傍にある繊維をランダムに12点抽出し、最も太い繊維と最も細い繊維を除去した10点の測定値を平均して求めることができる。
本発明に用いられるセルロースの繊維径は特に制限されるものではなく、数平均繊維径としては数μmから数mmである。一般的な精製を経たものは数mm程度であり、セルロースを解繊したものは数nmである。例えばチップ等の数cm大のものを精製したものである場合、リファイナーやビーター等の離解機で機械的処理を行い、数mm程度にすることが好ましい。なお、数平均繊維径の測定方法は特に限定されないが、例えばSEMやTEM等で観察して、写真の対角線に線を引き、その近傍にある繊維をランダムに12点抽出し、最も太い繊維と最も細い繊維を除去した10点の測定値を平均して求めることができる。
<セルロース含有物の精製方法>
本発明に用いられるセルロースは上記由来のセルロース含有物を通常の方法で精製して得られる。例えば、ベンゼン-エタノールで脱脂した後、ワイズ法で脱リグニン処理を行い、アルカリで脱ヘミセルロース処理をすることにより得られる。または一般的な化学パルプの製造方法、例えばクラフトパルプ、サリファイドパルプ、アルカリパルプの製造方法によって得られる。一般的に、セルロース含有物を蒸解釜で加熱処理して脱リグニン等の処理を行い、更に漂白処理等を行うものである。
本発明に用いられるセルロースは上記由来のセルロース含有物を通常の方法で精製して得られる。例えば、ベンゼン-エタノールで脱脂した後、ワイズ法で脱リグニン処理を行い、アルカリで脱ヘミセルロース処理をすることにより得られる。または一般的な化学パルプの製造方法、例えばクラフトパルプ、サリファイドパルプ、アルカリパルプの製造方法によって得られる。一般的に、セルロース含有物を蒸解釜で加熱処理して脱リグニン等の処理を行い、更に漂白処理等を行うものである。
〔セルロースの修飾〕
本発明において、セルロースは、溶媒として有機酸が存在する条件下で修飾される。修飾とは、セルロース中の水酸基が化学修飾剤と反応して化学修飾されているものである。
本発明において、セルロースは、溶媒として有機酸が存在する条件下で修飾される。修飾とは、セルロース中の水酸基が化学修飾剤と反応して化学修飾されているものである。
(修飾置換基)
化学修飾によってセルロースに導入される官能基としては、芳香環含有置換基である。芳香環含有置換基での修飾は、高耐熱性、易解繊性、高生産性を実現する上で好ましい。
化学修飾によってセルロースに導入される官能基としては、芳香環含有置換基である。芳香環含有置換基での修飾は、高耐熱性、易解繊性、高生産性を実現する上で好ましい。
本発明者の検討によれば、芳香環含有置換基で修飾されたセルロースは、未修飾のセルロースまたは脂肪族含有置換基で修飾されたセルロースと比べて、高耐熱性、易解繊性が優れており、特に高生産性である点が優れていることがわかった。
芳香環含有置換基とは、炭化水素芳香族化合物、複素環芳香族化合物や非ベンゼノイド芳香族化合物由来の置換基である。炭化水素芳香族化合物とは、ベンゼン、及びナフタレン、アントラセン等のベンゼン環の単環が2~12個縮合した化合物である。縮合数の上限は好ましくは6個以下である。複素環芳香族化合物とは、フラン、チオフェン、ピロール、イミダゾール等の5~10員環の複素環の単環又はその2~12個縮合した化合物である。縮合数の上限は好ましくは6個以下である。非ベンゼノイド芳香族化合物とは、アヌレン等、シクロペンタジエニルアニオン等、シクロヘプタトリエニルカチオン等、トロポン等、メタロセン等、アセブレイアジレン等が挙げられる。これらの中では、炭化水素芳香族化合物、複素環芳香族化合物由来の置換基が好ましく、さらには炭化水素芳香族化合物由来の置換基が好ましい。また、特にベンゼン、ナフタレン、アントラセン由来の置換基が原料の得やすさの点で好ましい。
芳香環含有置換基とは、炭化水素芳香族化合物、複素環芳香族化合物や非ベンゼノイド芳香族化合物由来の置換基である。炭化水素芳香族化合物とは、ベンゼン、及びナフタレン、アントラセン等のベンゼン環の単環が2~12個縮合した化合物である。縮合数の上限は好ましくは6個以下である。複素環芳香族化合物とは、フラン、チオフェン、ピロール、イミダゾール等の5~10員環の複素環の単環又はその2~12個縮合した化合物である。縮合数の上限は好ましくは6個以下である。非ベンゼノイド芳香族化合物とは、アヌレン等、シクロペンタジエニルアニオン等、シクロヘプタトリエニルカチオン等、トロポン等、メタロセン等、アセブレイアジレン等が挙げられる。これらの中では、炭化水素芳香族化合物、複素環芳香族化合物由来の置換基が好ましく、さらには炭化水素芳香族化合物由来の置換基が好ましい。また、特にベンゼン、ナフタレン、アントラセン由来の置換基が原料の得やすさの点で好ましい。
これらの芳香環含有置換基はその水素が炭素数1~12のアルキルで置換されていても構わない。また、芳香環含有置換基は、上記炭化水素芳香族化合物、複素環芳香族化合物や非ベンゼノイド芳香族化合物が2個以上、単結合あるいは炭素数1~3のアルキルで連結されていても構わない。
芳香環含有置換基において、芳香環とセルロースを結合する連結基としては、セルロースの水酸基と反応した結果得られたものであれば特に限定されるものではないが、エステル基、エーテル基、カルバマート基が好ましく、エステル基が特に好ましい。
芳香環含有置換基において、芳香環とセルロースを結合する連結基としては、セルロースの水酸基と反応した結果得られたものであれば特に限定されるものではないが、エステル基、エーテル基、カルバマート基が好ましく、エステル基が特に好ましい。
即ち、本発明の修飾セルロースに導入される修飾置換基の芳香環含有置換基としては、ベンゾイル基、ナフトイル基、アントロイル基等が好ましく、とりわけベンゾイル基が好ましい。
(化学修飾剤)
修飾方法としては、特に限定されるものではないが、セルロースと次に挙げるような化学修飾剤とを反応させる方法がある。本発明においては、これらの反応は溶媒として有機酸が存在する条件下で行われるが、必要に応じて他の溶媒、触媒等を併用したり、加熱、減圧等を行うこともできる。
修飾方法としては、特に限定されるものではないが、セルロースと次に挙げるような化学修飾剤とを反応させる方法がある。本発明においては、これらの反応は溶媒として有機酸が存在する条件下で行われるが、必要に応じて他の溶媒、触媒等を併用したり、加熱、減圧等を行うこともできる。
化学修飾剤の種類としては、例えば、エステル基を形成させる場合は、酸、酸無水物、ハロゲン化試薬等が、エーテル基を形成させる場合は、フェノール系化合物、フェノキシシラン、オキシラン(エポキシ)等の環状エーテル化合物等が、カルバマート基を形成させる場合はイソシアナート化合物等が挙げられる。これらの化学修飾剤は、1種又は2種以上用いても構わない。
エステル基を形成させる化学修飾剤である酸としては、例えば安息香酸、ナフタレンカルボン酸等が、酸無水物としては、例えば無水安息香酸、無水フタル酸等が挙げられる。ハロゲン化試薬としては、例えばベンゾイルハライド、ナフトイルハライド等が挙げられる。
エーテル基を形成させる化学修飾剤であるフェノール系化合物としては、フェノール、ナフトール等が、またフェノキシシラン等、フェニルオキシラン(エポキシ)等の環状エーテル化合物が挙げられる。
カルバマート基を形成させる化学修飾剤であるイソシアナート化合物としては、フェニルイソシアナート等が挙げられる。
これらの中では特にベンゾイルハライド、ナフトイルハライド、無水安息香酸が好ましい。これらの化学修飾剤は、1種又は2種以上用いても構わない。
(化学修飾率)
ここでいう化学修飾率とは、セルロース中の全水酸基のうちの化学修飾されたものの割合を示し、化学修飾率は下記の滴定法によって測定することができる。
ここでいう化学修飾率とは、セルロース中の全水酸基のうちの化学修飾されたものの割合を示し、化学修飾率は下記の滴定法によって測定することができる。
〈測定方法〉
乾燥した修飾セルロース0.05gを精秤しこれにメタノール6ml、蒸留水2mlを添加する。これを60~70℃で30分攪拌した後、0.05N水酸化ナトリウム水溶液10mlを添加する。これを60~70℃で15分攪拌しさらに室温で一日攪拌する。ここにフェノールフタレインを用いて0.02N塩酸水溶液で滴定する。
乾燥した修飾セルロース0.05gを精秤しこれにメタノール6ml、蒸留水2mlを添加する。これを60~70℃で30分攪拌した後、0.05N水酸化ナトリウム水溶液10mlを添加する。これを60~70℃で15分攪拌しさらに室温で一日攪拌する。ここにフェノールフタレインを用いて0.02N塩酸水溶液で滴定する。
ここで、滴定に要した0.02N塩酸水溶液の量Z(ml)から、化学修飾により導入された置換基のモル数Qは、下記式で求められる。
Q(mol)=0.05(N)×10(ml)/1000 -0.02(N)×Z(ml)/1000
この置換基のモル数Qと、化学修飾率X(mol%)との関係は、以下の式で算出される(セルロース=(C6O5H10)n=(162.14)n,繰り返し単位1個当たりの水酸基数=3,OHの分子量=17)。なお、以下において、Tは置換基の分子量である。
Q(mol)=0.05(N)×10(ml)/1000 -0.02(N)×Z(ml)/1000
この置換基のモル数Qと、化学修飾率X(mol%)との関係は、以下の式で算出される(セルロース=(C6O5H10)n=(162.14)n,繰り返し単位1個当たりの水酸基数=3,OHの分子量=17)。なお、以下において、Tは置換基の分子量である。

これを解いていくと、以下の通りである。

本発明の修飾セルロース繊維分散液の製造方法においては、セルロースを芳香環含有置換基で修飾し修飾率を制御あるいは修飾する反応方法を制御することで所定の修飾率を有するセルロースを得る。修飾率を制御する場合、化学修飾率は、セルロースの全水酸基に対して、10モル%以上であることを特徴とする。好ましくは11モル%以上、特に好ましくは15モル%以上である。また、通常60モル%以下、好ましくは55モル%以下、より好ましくは50モル%以下、さらに好ましくは45モル%以下、特に好ましくは40モル%以下である。
ここで修飾率を制御あるいは修飾する反応方法を制御するとは、セルロースの全水酸基に対する化学修飾剤の添加量を制御、あるいは修飾反応の反応時間と温度を制御することなどを示す。
ここで修飾率を制御あるいは修飾する反応方法を制御するとは、セルロースの全水酸基に対する化学修飾剤の添加量を制御、あるいは修飾反応の反応時間と温度を制御することなどを示す。
化学修飾することで、セルロース繊維の熱分解温度が上昇し、耐熱性が高くなり、加熱時の着色が小さくなり好ましい。特に本発明の修飾反応は、セルロースのI型結晶構造がよく維持され、耐熱性に優れる。これは、本発明の修飾反応が、反応時に溶媒として有機酸を用いることでI型結晶構造を壊さずに繊維を膨潤させ、この状態で修飾を行うことで、より均一な修飾を可能にしているためと思われる。なお、熱分解温度は下記に示す方法により測定される。
この化学修飾率が低すぎると、複合化の後処理で加熱した際に、着色が大きくなり、化学修飾率が高すぎると、セルロース繊維の結晶構造が破壊され結晶性が低下するため、得られる複合材料の線膨張係数が大きくなってしまうという問題点があり好ましくない。また、化学修飾率が低すぎると、親水性が高くなり、脱水の際の濾過時間が長くなったり、得られたセルロース繊維の含水率が高くなり好ましくない。特に、セルロース原料として木質を用いる場合、化学修飾率が低いと複合化の後処理で加熱した際に、着色してしまうため好ましくない。さらには、本発明者の検討によると、芳香環含有置換基で修飾し、上記化学修飾率の範囲の修飾セルロースは、解繊がしやすく、セルロース複合体とした際の透明性が高くなることがわかった。詳細な理由はわからないが、おそらく置換基の立体障害によりセルロース鎖同士がばらけやすくなったと考えられる。
<熱分解温度>
本発明における修飾セルロースの熱分解温度は、300℃以上であることが好ましく、さらに好ましくは335℃以上である。上限は、特に限定はないが、通常400℃以下である。熱分解温度が低すぎると、熱分解による着色が増加するといった問題点がある。本発明の修飾セルロース繊維は、上記のように熱分解温度が高いため耐熱性が良いが、その理由は、6位の水酸基が修飾されることにより、主鎖の開裂が抑制されるためと予想される。
本発明における修飾セルロースの熱分解温度は、300℃以上であることが好ましく、さらに好ましくは335℃以上である。上限は、特に限定はないが、通常400℃以下である。熱分解温度が低すぎると、熱分解による着色が増加するといった問題点がある。本発明の修飾セルロース繊維は、上記のように熱分解温度が高いため耐熱性が良いが、その理由は、6位の水酸基が修飾されることにより、主鎖の開裂が抑制されるためと予想される。
熱分解温度は、修飾セルロースを温度23℃、湿度50%に48時間以上調湿し、これをTG-DTA(示差熱熱重量同時測定装置)を用いて窒素下、室温から600℃まで10℃/分で昇温していったときのTGから求めた接線の交点を熱分解温度とすることで求められる。
〔修飾セルロース繊維分散液の製造方法〕
本発明の修飾セルロース繊維分散液の製造方法においては、セルロースを芳香環含有置換基で修飾し修飾率を制御あるいは修飾する反応方法を制御することで所定の修飾率を有するセルロースの分散液を得る。本発明の製造方法で得られた修飾セルロース繊維分散液をセルロース複合体とし、透明用途に用いる場合は、セルロースを微細繊維に解繊することが好ましい。修飾は、精製して得られたセルロースに行ってもよいし、セルロースを微細繊維に解繊してから行ってもよいし、セルロース繊維を例えばシート等に成形してから行ってもよい。微細繊維の表面を均一に修飾する場合は、セルロースを微細繊維に解繊した後及びセルロース繊維を例えばシート等に成形した後に行ってもよいが、精製して得られた未解繊のセルロースを修飾した後に解繊した場合、解繊がしやすく、脱溶媒のための濾過時間が短くなるので生産性の点で好ましい。
本発明の修飾セルロース繊維分散液の製造方法においては、セルロースを芳香環含有置換基で修飾し修飾率を制御あるいは修飾する反応方法を制御することで所定の修飾率を有するセルロースの分散液を得る。本発明の製造方法で得られた修飾セルロース繊維分散液をセルロース複合体とし、透明用途に用いる場合は、セルロースを微細繊維に解繊することが好ましい。修飾は、精製して得られたセルロースに行ってもよいし、セルロースを微細繊維に解繊してから行ってもよいし、セルロース繊維を例えばシート等に成形してから行ってもよい。微細繊維の表面を均一に修飾する場合は、セルロースを微細繊維に解繊した後及びセルロース繊維を例えばシート等に成形した後に行ってもよいが、精製して得られた未解繊のセルロースを修飾した後に解繊した場合、解繊がしやすく、脱溶媒のための濾過時間が短くなるので生産性の点で好ましい。
このような修飾セルロース繊維の製造方法について更に詳しく説明する。
シート等に成形する前のセルロースを修飾する場合、精製後のセルロースを修飾してもよいし、微細繊維に解繊してから行ってもよいが、微細繊維に解繊してから行う場合、溶媒を除去するのに労力を必要とするため、精製後未解繊のセルロースを修飾する方が好ましい。この場合、化学修飾は通常の方法をとることができるが、通常精製後のセルロースは含水状態であるので、この水を反応溶媒等と置換して、化学修飾剤と水との反応を極力抑制することが重要である。また、ここで水を除去するために乾燥すると、後の微細化が進行しにくくなるため乾燥工程を入れることは好ましくない。
シート等に成形する前のセルロースを修飾する場合、精製後のセルロースを修飾してもよいし、微細繊維に解繊してから行ってもよいが、微細繊維に解繊してから行う場合、溶媒を除去するのに労力を必要とするため、精製後未解繊のセルロースを修飾する方が好ましい。この場合、化学修飾は通常の方法をとることができるが、通常精製後のセルロースは含水状態であるので、この水を反応溶媒等と置換して、化学修飾剤と水との反応を極力抑制することが重要である。また、ここで水を除去するために乾燥すると、後の微細化が進行しにくくなるため乾燥工程を入れることは好ましくない。
化学修飾は、通常の方法をとることができる。すなわち、常法に従って、セルロースと化学修飾剤とを反応させることによって化学修飾を行うことができる。この際、本発明の製造方法では、溶媒として有機酸を用いるが、必要に応じて他の溶媒や触媒を用いたり、加熱、減圧等を行ってもよい。
化学修飾剤の量は、特に限定はなく、化学修飾剤の種類によっても異なるが、通常セルロースの水酸基のモル数に対して、0.01倍以上、0.05倍以上、100倍以下、50倍以下が好ましい。
(溶媒)
有機酸とは、有機化合物の酸の総称である。有機酸は主にカルボン酸、スルホン酸である。カルボン酸としては、例えば蟻酸、酢酸、プロピオン酸、酪酸、吉草酸等の脂肪族カルボン酸、オレイン酸、リノール酸等の脂肪族不飽和カルボン酸、蓚酸、マロン酸、琥珀酸等の脂肪族ジカルボン酸、安息香酸、フタル酸等の芳香族カルボン酸、ピルビン酸等のオキソカルボン酸、乳酸、リンゴ酸、クエン酸等のカルボン酸が挙げられる。スルホン酸としては、メチルスルホン酸、エチルスルホン酸等の脂肪族スルホン酸、脂肪族不飽和スルホン酸、脂肪族ジスルホン酸、ベンゼンスルホン酸、トルエンスルホン酸等の芳香族スルホン酸等が挙げられる。
有機酸とは、有機化合物の酸の総称である。有機酸は主にカルボン酸、スルホン酸である。カルボン酸としては、例えば蟻酸、酢酸、プロピオン酸、酪酸、吉草酸等の脂肪族カルボン酸、オレイン酸、リノール酸等の脂肪族不飽和カルボン酸、蓚酸、マロン酸、琥珀酸等の脂肪族ジカルボン酸、安息香酸、フタル酸等の芳香族カルボン酸、ピルビン酸等のオキソカルボン酸、乳酸、リンゴ酸、クエン酸等のカルボン酸が挙げられる。スルホン酸としては、メチルスルホン酸、エチルスルホン酸等の脂肪族スルホン酸、脂肪族不飽和スルホン酸、脂肪族ジスルホン酸、ベンゼンスルホン酸、トルエンスルホン酸等の芳香族スルホン酸等が挙げられる。
有機酸の中でも特に酸解離定数pKaが5.0以下であるものが好ましい。好ましくは4.8以下であるものが好ましい。また、pKaは0.1以上、好ましくは1.0以上であることが好ましい。
酸解離定数pKaが5.0以下である有機酸としては、蟻酸、酢酸、プロピオン酸、酪酸、オレイン酸、リノール酸、蓚酸、マロン酸、琥珀酸、安息香酸、フタル酸、ピルビン酸、乳酸、リンゴ酸、クエン酸、メチルスルホン酸、エチルスルホン酸、ベンゼンスルホン酸、トルエンスルホン酸が挙げられる。
上記有機酸の中でも特に脂肪族の有機酸が好ましい。脂肪族であれば、モノ体であってもジ体であってもそれ以上であっても構わない。さらにそれら脂肪族の有機酸はカルボン酸であることが好ましい。更に脂肪族カルボン酸の中でも脂肪鎖の炭素数が1から7であることが好ましく、1から5であることが更に好ましい。特にその中でも、酢酸や蓚酸が好ましい。
酢酸等の有機酸を用いることで、修飾がセルロースに均一に進行するため、後の解繊がしやすくなったり、高耐熱性、高生産性を呈するようになると考えられる。
溶媒の量は、特に限定はないが、通常セルロース重量に対して、0.5倍以上、1倍以上、200倍以下、100倍以下が好ましい。
有機酸と併用できるその他の溶媒としては、化学修飾反応を阻害しない有機溶媒を用いることが好ましい。
さらには有機酸や後述する触媒と相溶性のある有機溶媒が好ましい。かかる有機溶媒としては、例えばアセトン、ピリジン等の有機溶媒等が挙げられる。また、その他の溶媒を有機酸と併用する場合、その他の溶媒の混合比は、それぞれが相溶しており、さらには後述する触媒が相溶していれば任意であるが、通常全溶媒に占めるその他の溶媒の比率が1vol%以上程度であり、通常50vol%以下、更には40vol%以下、中でも30vol%以下であることが好ましい。
(触媒)
触媒としてはピリジンやトリエチルアミン、水酸化ナトリウム、酢酸ナトリウム等の塩基性触媒や、酢酸、硫酸、過塩素酸等の酸性触媒を用いることが好ましい。触媒の量は、特に限定はなく、種類によっても異なるが、通常セルロースの水酸基のモル数に対して、0.01倍以上、0.05倍以上、100倍以下、50倍以下が好ましい。
触媒としてはピリジンやトリエチルアミン、水酸化ナトリウム、酢酸ナトリウム等の塩基性触媒や、酢酸、硫酸、過塩素酸等の酸性触媒を用いることが好ましい。触媒の量は、特に限定はなく、種類によっても異なるが、通常セルロースの水酸基のモル数に対して、0.01倍以上、0.05倍以上、100倍以下、50倍以下が好ましい。
(温度条件)
温度条件としては、高すぎるとセルロースの黄変や重合度の低下等が懸念され、低すぎると反応速度が低下することから10~130℃が好ましい。反応時間は化学修飾剤や化学修飾率にもよるが数分から数十時間である。
温度条件としては、高すぎるとセルロースの黄変や重合度の低下等が懸念され、低すぎると反応速度が低下することから10~130℃が好ましい。反応時間は化学修飾剤や化学修飾率にもよるが数分から数十時間である。
このようにして化学修飾を行った後は、反応を終結させるために有機溶剤や水で十分に洗浄することが好ましい。未反応の化学修飾剤が残留していると、後で着色の原因になったり、樹脂と複合化する際に問題になったりするので好ましくない。
シート等の成形体の化学修飾は、シートを製造後、アルコール等の有機溶媒で置換した後、更にシートを乾燥した後に行っても、乾燥せずに行っても構わないが、乾燥した後に行った方が化学修飾の反応速度が速くなるため好ましい。乾燥する場合は送風乾燥、減圧乾燥してもよいし、加圧乾燥してもよい。また、加熱しても構わない。
シートの化学修飾も、通常の方法をとることができ、上述のとおりである。
シートの化学修飾も、通常の方法をとることができ、上述のとおりである。
(洗浄)
化学修飾を行った後は、反応を終結させるために水で十分に洗浄することが好ましい。未反応の化学修飾剤が残留していると、後に着色の原因になったり、樹脂と複合化する際に問題になったりするので好ましくない。また、水で十分に洗浄した後、さらに残留する水をアルコール等の有機溶媒で置換することが好ましい。この場合、シートをアルコール等の有機溶媒に浸漬しておくことで容易に置換することができる。
化学修飾を行った後は、反応を終結させるために水で十分に洗浄することが好ましい。未反応の化学修飾剤が残留していると、後に着色の原因になったり、樹脂と複合化する際に問題になったりするので好ましくない。また、水で十分に洗浄した後、さらに残留する水をアルコール等の有機溶媒で置換することが好ましい。この場合、シートをアルコール等の有機溶媒に浸漬しておくことで容易に置換することができる。
[解繊工程]
本発明において、透明なセルロース複合体を得るためには、セルロースを解繊し微細繊維とすることが必要である。微細繊維を得るために解繊工程を行うが、その方法について説明する。解繊工程は、精製後のセルロースに行う場合もあるし、セルロースを化学修飾した後に行う場合もあるが、精製して得られた未解繊のセルロースを修飾した後に解繊した場合、解繊がしやすく、脱溶媒のための濾過時間が短くなるので生産性の点で好ましい。
本発明において、透明なセルロース複合体を得るためには、セルロースを解繊し微細繊維とすることが必要である。微細繊維を得るために解繊工程を行うが、その方法について説明する。解繊工程は、精製後のセルロースに行う場合もあるし、セルロースを化学修飾した後に行う場合もあるが、精製して得られた未解繊のセルロースを修飾した後に解繊した場合、解繊がしやすく、脱溶媒のための濾過時間が短くなるので生産性の点で好ましい。
解繊工程の具体的な方法としては、特に制限はないが、例えば、直径1mm程度のセラミック製ビーズをセルロース濃度0.1~10重量%、例えば1重量%程度のセルロース分散液に入れ、ペイントシェーカーやビーズミル等を用いて振動を与え、セルロースを解繊する方法などが挙げられる。
また、ブレンダータイプの分散機や高速回転するスリットの間に、このようなセルロース分散液を通して剪断力を働かせて解繊する方法(高速回転ホモジナイザー)や、高圧から急に減圧することによって、セルロース繊維間に剪断力を発生させて解繊する方法(高圧ホモジナイザー法)、マスコマイザーXのような対向衝突型の分散機(増幸産業(株)社製)等を用いる方法などが挙げられる。特に、高速回転ホモジナイザーや高圧ホモジナイザー処理は、解繊の効率が向上する。
また、ブレンダータイプの分散機や高速回転するスリットの間に、このようなセルロース分散液を通して剪断力を働かせて解繊する方法(高速回転ホモジナイザー)や、高圧から急に減圧することによって、セルロース繊維間に剪断力を発生させて解繊する方法(高圧ホモジナイザー法)、マスコマイザーXのような対向衝突型の分散機(増幸産業(株)社製)等を用いる方法などが挙げられる。特に、高速回転ホモジナイザーや高圧ホモジナイザー処理は、解繊の効率が向上する。
これらの処理で解繊する場合は、セルロース分散液中の固形分濃度(セルロース濃度)が0.2重量%以上10重量%以下、特に0.3重量%以上、6重量%以下で行う。この解繊工程に供するセルロース分散液中の固形分濃度が低過ぎると処理するセルロース量に対して液量が多くなり過ぎ効率が悪く、固形分濃度が高過ぎると流動性が悪くなるため、解繊処理に供するセルロース分散液は適宜水を添加するなどして濃度調整する。
高速回転ホモジナイザーの場合、回転数が高い方が、剪断力が掛かり解繊効率が高い。回転数としては例えば10000rpm以上が好ましく、15000rpm以上が更に好ましく、20000rpm以上が特に好ましい。また、時間は1分以上が好ましく、5分以上が更に好ましく、10分以上が特に好ましい。剪断により発熱が生じる場合は液温が50℃を越えない程度に冷却することが好ましい。また、分散液に均一に剪断力がかかるように攪拌または循環することが好ましい。高速回転で解繊する方法や、高圧ホモジナイザー法で液を100MPa以上の高圧雰囲気下から噴出させる。
高圧ホモジナイザーを用いる場合、セルロース分散液を増圧機で30MPa以上、好ましくは100MPa以上、より好ましくは150MPa以上、更に好ましくは220MPa以上に加圧し、細孔直径50μm以上のノズルから噴出させ、圧力差が30MPa以上、好ましくは80MPa以上、より好ましくは90MPa以上となるように減圧する。この圧力差で生じるへき開現象により、セルロース繊維を解繊する。ここで、高圧条件の圧力が低い場合や、高圧から減圧条件への圧力差が小さい場合には、解繊効率が下がり、所望の繊維径とするための繰り返し噴出回数が多く必要となるため好ましくない。また、セルロース分散液を噴出させる細孔の細孔直径が大き過ぎる場合にも、十分な解繊効果が得られず、この場合には、噴出処理を繰り返し行っても、所望の繊維径のセルロース繊維が得られないおそれもある。
セルロース分散液の噴出は、必要に応じて複数回繰り返すことにより、微細化度を上げて所望の繊維径のセルロース繊維を得ることができる。この繰り返し回数(パス数)は、通常1回以上、好ましくは3回以上で、通常20回以下、好ましくは15回以下である。パス数が多い程、微細化の程度を上げることができるが、過度にパス数が多いとコスト高となるため好ましくない。
高圧ホモジナイザーとしては特に限定はないが、具体的装置としては、ガウリン社製やスギノマシン社製の「スターバーストシステム」を用いることができる。
噴出時の高圧条件は高い程、圧力差により大きなへき開現象でより一層の微細化を図ることができるが、装置仕様の上限として、通常245MPa以下である。
同様に、高圧条件から減圧下への圧力差も大きいことが好ましいが、一般的には、増圧機による加圧条件から大気圧下に噴出することで、圧力差の上限は通常245MPa以下である。
噴出時の高圧条件は高い程、圧力差により大きなへき開現象でより一層の微細化を図ることができるが、装置仕様の上限として、通常245MPa以下である。
同様に、高圧条件から減圧下への圧力差も大きいことが好ましいが、一般的には、増圧機による加圧条件から大気圧下に噴出することで、圧力差の上限は通常245MPa以下である。
また、セルロース分散液を噴出させる細孔の直径は小さければ容易に高圧状態を作り出せるが、過度に小さいと噴出効率が悪くなる。この細孔直径は50μm以上800μm以下、好ましくは100μm以上500μm以下、より好ましくは150μm以上350μm以下である。
噴出時の温度(分散液温度)には特に制限はないが、通常5℃以上100℃以下である。温度が高すぎると装置、具体的には送液ポンプや高圧シール部等の劣化を早める恐れがあるため好ましくない。
噴出時の温度(分散液温度)には特に制限はないが、通常5℃以上100℃以下である。温度が高すぎると装置、具体的には送液ポンプや高圧シール部等の劣化を早める恐れがあるため好ましくない。
なお、噴出ノズルは1本でも2本でもよく、噴出させたセルロースを噴出先に設けた壁やボール、リングにぶつけてもよい。更にノズルが2本の場合には噴出先でセルロース同士を衝突させてもよい。
なお、このような高圧ホモジナイザーによる処理のみでも、本発明の微細セルロース繊維分散液を得ることは可能であるが、その場合には、十分な微細化度とするための繰り返し回数が多くなり、処理効率が悪いことから、1~5回程度の高圧ホモジナイザー処理後に後述の超音波処理を行って微細化することが好ましい。
なお、このような高圧ホモジナイザーによる処理のみでも、本発明の微細セルロース繊維分散液を得ることは可能であるが、その場合には、十分な微細化度とするための繰り返し回数が多くなり、処理効率が悪いことから、1~5回程度の高圧ホモジナイザー処理後に後述の超音波処理を行って微細化することが好ましい。
本発明において、超音波処理するセルロース分散液のセルロース濃度は、0.01~10重量%、特に0.1~5重量%、とりわけ0.2~2重量%であることが好ましい。超音波を照射するセルロース分散液のセルロース濃度が低過ぎると非効率であり、高過ぎると粘度が高くなり解繊処理が不均一になる。従って、本発明においては、超音波処理に供されるセルロース分散液のセルロース濃度が上記所定濃度となるように、必要に応じて水及び/又は有機溶媒を添加する。
なお、セルロース分散液の分散媒としての有機溶媒としてはメタノール、エタノール、イソプロピルアルコール、n-プロピルアルコール、n-ブタノール等のアルコール類、アセトンやメチルエチルケトン等のケトン類、その他水溶性の有機溶剤の1種又は2種以上を用いることができる。さらには非水溶性の有機溶媒も併用することができるが、好ましくは、分散媒は有機溶媒と水との混合液又は水であり、特には水であることが好ましい。
また、超音波を照射するセルロース分散液中のセルロース繊維の数平均繊維径は、上述の解繊により10μm以下、特に2μm以下にしておくことが好ましい。さらに好ましくは1μm以下であることが好ましい。
セルロース分散液に照射する超音波の周波数は15kHz~1MHz、好ましくは20kHz~500kHz、更に好ましくは20kHz~100kHzである。照射する超音波の周波数が小さ過ぎると後述のキャビテーションが発生しにくく、大き過ぎると発生したキャビテーションが物理的な作用を発生させるまでに大きく成長することなく消滅してしまうため、微細化効果が得られない。また、超音波の出力としては、実行出力密度として1W/cm2以上であり、好ましくは10W/cm2以上、更に好ましくは20W/cm2以上である。超音波の出力が小さ過ぎると微細化効率が低下して、十分な微細化を行うために長時間の照射が必要であり、実用的ではない。なお、超音波の実行出力密度の上限は振動子やホーン等の耐久性の点から500W/cm2以下である。
なお、超音波の実効出力密度は水500mLの温度上昇から計算することができる。具体的には、容器に水500mLを投入し、この水に超音波を照射してそのときの温度上昇の程度を測定し、下記式に従って計算することにより求められる。
P=(T/s)×4.18×500/A
ここで、Pは超音波の実効出力密度(W/cm2)、Tは上昇温度(℃)、sは時間(秒)、Aは超音波の振動部の面積(cm2)であり、ホーンタイプの場合はその端面の面積である。また、浴槽式の場合は振動子取り付け面の面積に相当する。
P=(T/s)×4.18×500/A
ここで、Pは超音波の実効出力密度(W/cm2)、Tは上昇温度(℃)、sは時間(秒)、Aは超音波の振動部の面積(cm2)であり、ホーンタイプの場合はその端面の面積である。また、浴槽式の場合は振動子取り付け面の面積に相当する。
なお、温度の測定に際しては、投入した超音波のエネルギーにより生じた熱が外部に伝わらないように、水を入れる容器は十分に断熱する必要がある。また、室温よりも高い温度では熱が外部に伝わりやすいため、室温よりも10℃まで上がった時の温度とその時の時間を用いて上記式により計算する。
超音波の照射方法には特に制限はなく、各種の方法が利用できる。例えば、超音波振動子の振動を伝えるホーンを直接上記のセルロース分散液に挿入することにより、直接セルロース繊維を微細化する方法や、セルロース分散液を入れた容器の床や壁の一部に超音波振動子を設置してセルロース繊維を微細化する方法や、超音波振動子を装着した容器に水等の液体を入れ、その中にセルロース分散液を入れた容器を漬すことにより、水等の液体を介して間接的に超音波振動をセルロース分散液に与えて微細化する方法が採用できる。中でも、ホーンを直接セルロース分散液に挿入する方法は直接超音波エネルギーを伝達することができエネルギー密度を高くできるので効率がよく、好適に利用される。
セルロース分散液は一定の量に対して一定時間所定の周波数の超音波を所定の実効出力密度で照射した後、全量を入れ替えるバッチ式の処理方法で微細化処理しても良く、また、ホーンの近傍や、床や壁に超音波振動子を設置した処理容器に一定量のセルロース含有物分散液を流通させて、連続的に超音波を当てる方法で処理を行っても良い。また、一つの処理容器の中に超音波振動子を複数設置しても良いし、一つの処理容器に一つの振動子を設置した処理容器を複数個連結して用いても良い。特に、連続的にセルロース含有物分散液を流して処理する場合、振動子を有する処理容器を直列に連結して、セルロース分散液を順次流通させる方法は、効率の面から好適である。その際に、複数の振動子は同一の周波数でも良いし、周波数を変化させても良い。
また、超音波は連続的に照射しても良く、所定の間隔で間欠的に照射しても良い。例えば0.1~0.9秒間の超音波照射と0.9~0.1秒間の休止運転とを交互に繰り返し行う方法であっても良い。
超音波処理を行うと、与えたエネルギーが熱に変換されてセルロース分散液の温度が上昇する。従って、一定の処理条件で微細化処理を行うために、冷却もしくは加熱などにより、セルロース分散液の温度を一定にすることが好ましい。超音波処理時の温度は1~80℃が好ましく、より好ましくは10~60℃、更に好ましくは15~40℃である。この温度が低過ぎると水を分散媒に用いた場合、凍結してしまい、処理不能となる。即ち、固体の氷ではキャビテーションの発生が困難であり、また、水と氷が混在している場合には、氷の表面でキャビテーションが発生してエネルギーを消費するため、セルロースの微細化効率が低下する。逆に、処理温度が高過ぎると超音波振動子面に微小な水蒸気等の蒸気が発生し、エネルギー効率が低下するため、好ましくない。
超音波処理を行うと、与えたエネルギーが熱に変換されてセルロース分散液の温度が上昇する。従って、一定の処理条件で微細化処理を行うために、冷却もしくは加熱などにより、セルロース分散液の温度を一定にすることが好ましい。超音波処理時の温度は1~80℃が好ましく、より好ましくは10~60℃、更に好ましくは15~40℃である。この温度が低過ぎると水を分散媒に用いた場合、凍結してしまい、処理不能となる。即ち、固体の氷ではキャビテーションの発生が困難であり、また、水と氷が混在している場合には、氷の表面でキャビテーションが発生してエネルギーを消費するため、セルロースの微細化効率が低下する。逆に、処理温度が高過ぎると超音波振動子面に微小な水蒸気等の蒸気が発生し、エネルギー効率が低下するため、好ましくない。
超音波照射の処理時間は、分散液中のセルロース繊維が所望の微細化度に微細化されるような時間であれば良く、用いた超音波の出力や周波数、超音波照射前のセルロース繊維の繊維径等により適宜設定される。
超音波処理によりセルロース繊維が微細化される原理は完全に解明されているわけではないが、以下の現象が発生していると推測される。
超音波処理によりセルロース繊維が微細化される原理は完全に解明されているわけではないが、以下の現象が発生していると推測される。
即ち、水などの液体中にセルロース繊維が懸濁、分散している状態で、超音波を照射すると、超音波振動子から発生した超音波がセルロース繊維に当たり、セルロース繊維と水との界面にキャビテーションが発生する。発生したキャビティは急激に収縮して消滅するが、その際に、周辺に大きな剪断力を発生させる。これによりセルロース繊維の表面から微細なセルロース繊維が剥離されることにより、微細セルロース繊維が生成する。
<平均繊維径>
上記方法によって解繊されたセルロース分散液中のセルロース繊維の繊維径は、分散液中の分散媒を乾燥除去した後、SEMやTEM等で観察することにより計測して求めることができる。
本発明により得られる解繊された修飾セルロース繊維の数平均繊維径は、高透明なセルロース複合体を得るためには、好ましくは100nm以下であることを特徴とする。80nm以下であることがさらに好ましく、50nm以下であることが特に好ましい。尚、この平均繊維径の下限は通常4nm以上である。
上記方法によって解繊されたセルロース分散液中のセルロース繊維の繊維径は、分散液中の分散媒を乾燥除去した後、SEMやTEM等で観察することにより計測して求めることができる。
本発明により得られる解繊された修飾セルロース繊維の数平均繊維径は、高透明なセルロース複合体を得るためには、好ましくは100nm以下であることを特徴とする。80nm以下であることがさらに好ましく、50nm以下であることが特に好ましい。尚、この平均繊維径の下限は通常4nm以上である。
<セルロースI型結晶>
本発明に用いられるセルロース繊維は、セルロースI型結晶構造を有するものである。
セルロースI型結晶構造とは、例えば、朝倉書店発行の「セルロースの事典」新装版第一刷P.81~P.86、あるいはP.93~99に記載の通りのものであり、ほとんどの天然セルロースはセルロースI型結晶構造である。これに対して、セルロースI型結晶構造ではなく、例えばセルロースII、III、IV型構造のセルロース繊維はセルロースI型結晶構造を有するセルロースから誘導されるものである。セルロースI型結晶は、他の結晶構造より結晶弾性率が高いため、高弾性率、高強度、低線膨張係数であり好ましい。
本発明に用いられるセルロース繊維は、セルロースI型結晶構造を有するものである。
セルロースI型結晶構造とは、例えば、朝倉書店発行の「セルロースの事典」新装版第一刷P.81~P.86、あるいはP.93~99に記載の通りのものであり、ほとんどの天然セルロースはセルロースI型結晶構造である。これに対して、セルロースI型結晶構造ではなく、例えばセルロースII、III、IV型構造のセルロース繊維はセルロースI型結晶構造を有するセルロースから誘導されるものである。セルロースI型結晶は、他の結晶構造より結晶弾性率が高いため、高弾性率、高強度、低線膨張係数であり好ましい。
セルロース繊維がI型結晶構造であることは、その広角X線回折像測定により得られる回折プロファイル(広角X線回折像)において、走査角2θ=14~17°付近と2θ=22~23°付近の二つの位置に典型的なピークをもつことから同定することができる。
一方、セルロースI型結晶構造が壊れた場合、2θ=18~19°付近に非晶質に由来するハローが観測されるようになる。I型以外の結晶構造を有する場合にもこの近傍にピークを示すようになる。
本発明の修飾セルロース繊維分散液に含まれるセルロースは、「走査角20~24度におけるセルロースI型結晶構造由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」(非晶質等のI型以外の構造を反映)の比が0.8以下である。好ましくは0.7以下、特に0.6以下、中でも0.5以下である。該強度比は低いほどよいが、実用的には0.01以上程度である。
一方、セルロースI型結晶構造が壊れた場合、2θ=18~19°付近に非晶質に由来するハローが観測されるようになる。I型以外の結晶構造を有する場合にもこの近傍にピークを示すようになる。
本発明の修飾セルロース繊維分散液に含まれるセルロースは、「走査角20~24度におけるセルロースI型結晶構造由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」(非晶質等のI型以外の構造を反映)の比が0.8以下である。好ましくは0.7以下、特に0.6以下、中でも0.5以下である。該強度比は低いほどよいが、実用的には0.01以上程度である。
II.本発明の修飾セルロース繊維分散液
本発明の修飾セルロース繊維分散液は、セルロースの全水酸基の10モル%以上が芳香環含有置換基により修飾されたセルロースI型結晶構造を有するセルロース繊維の分散液であって、該セルロース繊維の広角X線回折像の「走査角20~24度におけるセルロースI型結晶構造由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」の比が0.8以下であることを特徴とする。好ましくは0.7以下、特に0.6以下、中でも0.5以下である。該強度比は低いほどよいが、実用的には0.01以上程度である。
本発明の修飾セルロース繊維分散液は、セルロースの全水酸基の10モル%以上が芳香環含有置換基により修飾されたセルロースI型結晶構造を有するセルロース繊維の分散液であって、該セルロース繊維の広角X線回折像の「走査角20~24度におけるセルロースI型結晶構造由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」の比が0.8以下であることを特徴とする。好ましくは0.7以下、特に0.6以下、中でも0.5以下である。該強度比は低いほどよいが、実用的には0.01以上程度である。
本発明の修飾セルロース繊維分散液は、セルロースI型結晶構造を保ったままセルロースを膨潤させた状態で、セルロースと化学修飾剤との反応を行い、必要に応じて解繊工程を経ることで得られる。セルロースを膨潤させる方法は特に限定されず、化学修飾反応時に溶媒として繊維膨潤効果のある溶媒を選択することが可能である。溶媒としては例えば、メタノールやエタノール等のアルコールやエチレングリコール等のジオールや、ジメチルホルムアミド等の非プロトン性極性溶媒、ピリジン等の複素環式化合物などがあげられる。中でも化学修飾反応時の溶媒として有機酸を用いる、前述I.の製造方法が好ましい。解繊工程についても、I.に記載した方法を用いることができる。
本発明の修飾セルロース繊維分散液に含まれるセルロースの化学修飾率は、セルロースの全水酸基に対して、10モル%以上であることを特徴とする。好ましくは11モル%以上、特に好ましくは15モル%以上である。また、通常60モル%以下、好ましくは55モル%以下、より好ましくは50モル%以下、さらに好ましくは45モル%以下、特に好ましくは40モル%以下である。修飾率の測定方法は前述I.の製造方法と同様である。
化学修飾することで、セルロース繊維の熱分解温度が上昇し、耐熱性が高くなり、加熱時の着色が小さくなり好ましい。特に本発明の修飾反応は、セルロースのI型結晶構造がよく維持され、耐熱性に優れる。
これは、本発明の修飾反応が、反応時にI型結晶構造を壊さずに繊維を膨潤させ、この状態で修飾を行うことで、より均一な修飾を可能にしているためと思われる。
この化学修飾率が低すぎると、複合化の後処理で加熱した際に、着色が大きくなり、化学修飾率が高すぎると、セルロース繊維の結晶構造が破壊され結晶性が低下するため、得られる複合材料の線膨張係数が大きくなってしまうという問題点があり好ましくない。また、化学修飾率が低すぎると、親水性が高くなり、脱水の際の濾過時間が長くなったり、得られたセルロース繊維の含水率が高くなり好ましくない。特に、セルロース原料として木質を用いる場合、化学修飾率が低いと複合化の後処理で加熱した際に、着色してしまうため好ましくない。さらには、本発明者の検討によると、芳香環含有置換基で修飾し、上記化学修飾率の範囲の修飾セルロースは、解繊がしやすく、セルロース複合体とした際の透明性が高くなることがわかった。詳細な理由はわからないが、おそらく置換基の立体障害によりセルロース鎖同士がばらけやすくなったと考えられる。
<熱分解温度>
本発明の修飾セルロース繊維分散液に含まれるセルロースの熱分解温度は、300℃以上であることが好ましく、さらに好ましくは335℃以上である。上限は、特に限定はないが、通常400℃以下である。熱分解温度が低すぎると、熱分解による着色が増加するといった問題点がある。本発明の修飾セルロース繊維分散液に含まれるセルロースは、上記のように熱分解温度が高いため耐熱性が良いが、その理由は、6位の水酸基が修飾されることにより、主鎖の開裂が抑制されるためと予想される。
本発明の修飾セルロース繊維分散液に含まれるセルロースの熱分解温度は、300℃以上であることが好ましく、さらに好ましくは335℃以上である。上限は、特に限定はないが、通常400℃以下である。熱分解温度が低すぎると、熱分解による着色が増加するといった問題点がある。本発明の修飾セルロース繊維分散液に含まれるセルロースは、上記のように熱分解温度が高いため耐熱性が良いが、その理由は、6位の水酸基が修飾されることにより、主鎖の開裂が抑制されるためと予想される。
<平均繊維径>
上記I.の方法によって解繊された本発明の修飾セルロース繊維分散液中のセルロース繊維の繊維径は、分散液中の分散媒を乾燥除去した後、SEMやTEM等で観察することにより計測して求めることができる。
上記I.の方法によって解繊された本発明の修飾セルロース繊維分散液中のセルロース繊維の繊維径は、分散液中の分散媒を乾燥除去した後、SEMやTEM等で観察することにより計測して求めることができる。
本発明により得られる解繊された修飾セルロース繊維分散液に含まれるセルロース繊維の数平均繊維径は、高透明なセルロース複合体を得るためには、好ましくは100nm以下であることを特徴とする。80nm以下であることがさらに好ましく、50nm以下であることが特に好ましい。尚、この平均繊維径の下限は通常4nm以上である。
本発明の修飾セルロース繊維分散液は、上記修飾工程および解繊工程を経た後、その用途に応じて、分散媒(溶媒)濃度の調整、分散媒の置換、各種添加剤の添加等を適宜行うことができる。
解繊工程を終了した解繊液は基本的に水を分散媒としているため、水を分散媒(溶媒)として用いる場合はそのまま、または水で希釈することができる。また、水以外の有機溶媒を追加することもできるし、追加したのちに所望の組成比まで濃縮することもできる。水以外の溶媒を用いる場合は、溶媒の置換が必要となる。この場合は、溶媒を追加したのち、濃縮により水を除去することが好ましい。溶媒を追加する際は、凝集がおこらないように攪拌等を行いながらゆっくりと追加することが好ましい。また、濃縮を行う際には、系内が不均一にならないよう、注意することが必要である。系内が不均一になると、セルロースの凝集が起こる場合があり、それによって、セルロース繊維複合体のヘーズが高くなるので注意が必要である。溶媒置換を行う際は、上記以外の方法も可能であるが、セルロースを乾燥させずに置換することが重要である。
解繊工程を終了した解繊液は基本的に水を分散媒としているため、水を分散媒(溶媒)として用いる場合はそのまま、または水で希釈することができる。また、水以外の有機溶媒を追加することもできるし、追加したのちに所望の組成比まで濃縮することもできる。水以外の溶媒を用いる場合は、溶媒の置換が必要となる。この場合は、溶媒を追加したのち、濃縮により水を除去することが好ましい。溶媒を追加する際は、凝集がおこらないように攪拌等を行いながらゆっくりと追加することが好ましい。また、濃縮を行う際には、系内が不均一にならないよう、注意することが必要である。系内が不均一になると、セルロースの凝集が起こる場合があり、それによって、セルロース繊維複合体のヘーズが高くなるので注意が必要である。溶媒置換を行う際は、上記以外の方法も可能であるが、セルロースを乾燥させずに置換することが重要である。
分散媒として用いる溶媒は、水及び/または水溶性有機溶媒であることが好ましい。解繊工程を終了した解繊液は基本的に水及び/または水溶性有機溶媒を分散媒として行われることが多いため、非水溶性有機溶媒に置換するには多量の置換のための溶剤と時間がかかり好ましくない。
また、ここでいう水溶性有機溶媒とは、一気圧において、摂氏20度で同容量の純水と穏やかにかき混ぜた場合、流動が収まった後もその混合液が均一な外観を維持するものをいう。具体的にはメタノール、エタノール、nープロピルアルコール、イソプロピルアルコール、t-ブチルアルコール、アリルアルコール等の一価のアルコール類、エチレングリコール、ブチルグリコール、t-ブチルグリコール、メチルジグリコール、エチルジグリコール、ブチルジグリコール、メチルジプロピレングリコール、グリセリン等の多価アルコール類(グリコール類)、1-メトキシー2-プロパノール、2-エトキシエタノール、2-n-ブトキシエタノール、3-メトキシブタノール、3―メチルー3-メトキシブタノール、エチレングリコールモノメチルエーテル、エチレングリコールジメチルエーテル、エチレングリコールモノフェニルエーテル、プロピレングリコールモノメチルエーテル、プロピレングリコールジメチルエーテル、ジエチレングリコールモノメチルエーテル、ジエチレングリコールジメチルエーテル、ジエチレングリコールエチルメチルエーテル、ジエチレングリコールモノエチルエーテル、ジエチレングリコールジエチルエーテル、ジエチレングリコールモノブチルエーテル、ジエチレングリコールブチルメチルエーテル、ジエチレングリコールジブチルエーテル、ジプロピレングリコールモノメチルエーテル、ジプロピレングリコールジメチルエーテル、トリエチレングリコールモノメチルエーテル、トリエチレングリコールジメチルエーテル、トリエチレングリコールブチルメチルエーテル、トリプロピレングリコールジメチルエーテル、テトラエチレングリコールジメチルエーテル、ポリエチレングリコールモノメチルエーテル、ポリエチレングリコールジメチルエーテル等の上記グリコール類のアルキルエーテル類、ジエチレングリコールモノブチルエーテルアセテート、エチレングリコールモノメチルエーテルアセテート、エチルラクテート等のエステル類、テトラヒドロフラン、1,4-ジオキサン等のエーテル類、アセトン、ジアセトンアルコール等のケトン類、アセトニトリル、ピリジン、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチルー2-ピロリドン等の窒素系、酢酸、アクリル酸、メタクリル酸、酪酸が挙げられる。またこれらの溶媒を2種以上組み合わせて用いても構わない。
[修飾セルロース繊維シートの製造方法]
本発明の修飾セルロース繊維はシート状に成形することで、修飾セルロース繊維シートとすることができる。セルロースシートとすることで、樹脂を含浸させてセルロース複合体としたり、樹脂シートではさんでセルロース複合体とすることができる。本発明の修飾セルロース繊維シート(以下「セルロースシート」とも称する)は修飾セルロース繊維をそのまま用いて製造してもよいが、解繊する工程を施したものを用いて製造したものの方が高透明性、低線膨張係数、高弾性率のものが得られる。具体的には、前述の解繊する工程を施したセルロース分散液(以下、「解繊セルロース分散液」とも称する。)を濾過することにより、或いは該分散液を適当な基材に塗布することにより製造されたシートである。
本発明の修飾セルロース繊維はシート状に成形することで、修飾セルロース繊維シートとすることができる。セルロースシートとすることで、樹脂を含浸させてセルロース複合体としたり、樹脂シートではさんでセルロース複合体とすることができる。本発明の修飾セルロース繊維シート(以下「セルロースシート」とも称する)は修飾セルロース繊維をそのまま用いて製造してもよいが、解繊する工程を施したものを用いて製造したものの方が高透明性、低線膨張係数、高弾性率のものが得られる。具体的には、前述の解繊する工程を施したセルロース分散液(以下、「解繊セルロース分散液」とも称する。)を濾過することにより、或いは該分散液を適当な基材に塗布することにより製造されたシートである。
<抄紙工程>
セルロースシートを、解繊セルロース分散液を濾過することによって製造する場合(以下、この工程を「抄紙」と称することがある。)、濾過に供される解繊セルロース分散液の濃度は、0.01重量%以上、好ましくは0.05重量%以上、さらに好ましくは0.1重量%以上であることが好ましい。濃度が低すぎると濾過に膨大な時間がかかるため好ましくない。また、解繊セルロース分散液の濃度は1.5重量%以下、好ましくは1.2重量%以下、さらに好ましくは1.0重量%以下であることが好ましい。濃度が高すぎると均一なシートが得られないため好ましくない。
セルロースシートを、解繊セルロース分散液を濾過することによって製造する場合(以下、この工程を「抄紙」と称することがある。)、濾過に供される解繊セルロース分散液の濃度は、0.01重量%以上、好ましくは0.05重量%以上、さらに好ましくは0.1重量%以上であることが好ましい。濃度が低すぎると濾過に膨大な時間がかかるため好ましくない。また、解繊セルロース分散液の濃度は1.5重量%以下、好ましくは1.2重量%以下、さらに好ましくは1.0重量%以下であることが好ましい。濃度が高すぎると均一なシートが得られないため好ましくない。
また、濾過時の濾布としては、微細化したセルロース繊維は通過せずかつ濾過速度が遅くなりすぎないことが重要である。このような濾布としては、有機ポリマーからなるシート、織物、多孔膜が好ましい。有機ポリマーとしてはポリエチレンテレフタレートやポリエチレン、ポリプロピレン、ポリテトラフルオロエチレン(PTFE)等のような非セルロース系の有機ポリマーが好ましい。
具体的には孔径0.1~20μm、例えば1μmのポリテトラフルオロエチレンの多孔膜、孔径0.1~20μm、例えば1μmのポリエチレンテレフタレートやポリエチレンの織物等が挙げられる。
セルロースシートはその製造方法により、様々な空隙率を有することができる。空隙率の大きなセルロースシートを得る方法としては、濾過による製膜工程において、セルロースシート中の水を最後にアルコール等の有機溶媒に置換する方法を挙げることができる。これは、濾過により水を除去し、セルロース含量が5~99重量%になったところでアルコール等の有機溶媒を加えるものである。又は、解繊セルロース分散液を濾過装置に投入した後、アルコール等の有機溶媒を分散液の上部に静かに投入することによっても濾過の最後にアルコール等の有機溶媒と置換することができる。
ここで用いるアルコール等の有機溶媒としては、特に限定されるものではないが、例えばメタノール、エタノール、1-プロパノール、2-プロパノール、1-ブタノール等のアルコール類の他、アセトン、メチルエチルケトン、テトラヒドロフラン、シクロヘキサン、トルエン、四塩化炭素等から選ばれる1種又は2種以上の有機溶媒が挙げられる。非水溶性有機溶媒を用いる場合は、水溶性有機溶媒との混合溶媒にするか水溶性有機溶媒で置換した後、非水溶性有機溶媒で置換することが好ましい。
このような方法で空隙率の大きいセルロースシートを得る場合の濾過時間は、特に限定はないが、バッチ式濾過で40g/m2量を濾過する場合、通常120分以下、好ましくは110分以下、さらには100分以下である。又、0.1分以上、好ましくは0.3分以上、さらに好ましくは0.5分以上である。濾過時間が長すぎると生産効率が落ちたり、連続式濾過を行う場合、濾過装置が巨大になってしまうといった問題点がある。
本発明における修飾セルロース繊維を用いることで、他の修飾していないセルロースや脂肪族系置換基で修飾されたセルロースと比較して、早い速度で濾過することができ、かつ得られたシートをセルロース複合体とした際、高透明なセルロース複合体を得ることができる。
その後、乾燥を行うが、この乾燥は、送風乾燥であっても良く、減圧乾燥であっても良く、また、加圧乾燥であっても良い。また、加熱乾燥しても構わない。加熱する場合、温度は50℃以上が好ましく、80℃以上がより好ましく、また、250℃以下が好ましく、150℃以下がより好ましい。加熱温度が低すぎると乾燥に時間がかかったり、乾燥が不十分になる可能性があり、加熱温度が高すぎるとセルロースシートが着色したり、セルロースが分解したりする可能性がある。また、加圧する場合は0.01MPa以上が好ましく、0.1MPa以上がより好ましく、また、5MPa以下が好ましく、1MPa以下がより好ましい。圧力が低すぎると乾燥が不十分になる可能性がり、圧力が高すぎるとセルロース繊維平面構造体がつぶれたりセルロースが分解する可能性がある。
空隙率の小さなセルロースシートを得る方法としては、解繊セルロース分散液の濾過又は塗布後、乾燥を行う。すなわち、空隙率の小さな密なセルロースシートを得る方法としては、上記のように解繊セルロース分散液の濾過による製膜工程の最後にアルコール等の有機溶媒による置換を行わず、そのまま乾燥工程に供する方法が挙げられる。このような方法で空隙率の小さいセルロースシートを得る場合の濾過時間は、特に限定はないが、バッチ式濾過で40g/m2量を濾過する場合、通常80分以下、好ましくは70分以下、さらには60分以下である。又、0.1分以上、好ましくは0.3分以上、さらに好ましくは0.5分以上である。濾過時間が長すぎると生産効率が落ちたり、連続式濾過を行う場合、濾過装置が巨大になってしまうといった問題点がある。乾燥に関しては上述の通りである。
<平均繊維径>
本発明により得られる修飾セルロース繊維シートに含まれるセルロース繊維の繊維径は、SEMやTEM等で観察することにより計測して求めることができる。
本発明により得られる修飾セルロース繊維シートに含まれるセルロース繊維の数平均繊維径は、高透明なセルロース複合体を得るためには、好ましくは100nm以下であることを特徴とする。80nm以下であることがさらに好ましく、50nm以下であることが特に好ましい。尚、この平均繊維径の下限は通常4nm以上である。
本発明により得られる修飾セルロース繊維シートに含まれるセルロース繊維の繊維径は、SEMやTEM等で観察することにより計測して求めることができる。
本発明により得られる修飾セルロース繊維シートに含まれるセルロース繊維の数平均繊維径は、高透明なセルロース複合体を得るためには、好ましくは100nm以下であることを特徴とする。80nm以下であることがさらに好ましく、50nm以下であることが特に好ましい。尚、この平均繊維径の下限は通常4nm以上である。
化学修飾はセルロースシートにした後に行うこともできる。この場合、上述のようにして得られたシートを乾燥してから化学修飾を行ってもよいし、シートを乾燥する前に行ってもよい。このセルロースシートを前述の通り化学修飾剤と反応させた後、水及び/または有機溶剤で洗浄した後、乾燥することで修飾セルロースシートが得られる。
セルロースシートの厚みには特に限定はないが、好ましくは1μm以上、さらに好ましくは5μm以上である。又、通常1000μm以下、好ましくは250μm以下である。なお、本発明で得られるセルロースシートの好ましいものは、下記(1)に示す複合材料としたときのC光によるヘーズ値が3以下、更には2以下、特には1以下である。
(1)秤量40g/m2となるように厚みを調整したセルロース繊維シートを、ビス(メタクリロイルオキシメチル)トリシクロ[5.2.1.02,6]デカン96重量部、ペンタエリスリトールテトラキス(β-チオプロピオネート)6重量部、2,4,6-トリメチルベンゾイルジフェニルフォスフィンオキサイド0.05重量部、ベンゾフェノン0.05重量部を混合した溶液に含浸させ、減圧下に一晩置いた後、2枚のガラス板にはさみ、無電極水銀ランプを用いて硬化後、ガラス板よりはずして190℃の真空オーブン中で4時間加熱して得た複合材料。
(1)秤量40g/m2となるように厚みを調整したセルロース繊維シートを、ビス(メタクリロイルオキシメチル)トリシクロ[5.2.1.02,6]デカン96重量部、ペンタエリスリトールテトラキス(β-チオプロピオネート)6重量部、2,4,6-トリメチルベンゾイルジフェニルフォスフィンオキサイド0.05重量部、ベンゾフェノン0.05重量部を混合した溶液に含浸させ、減圧下に一晩置いた後、2枚のガラス板にはさみ、無電極水銀ランプを用いて硬化後、ガラス板よりはずして190℃の真空オーブン中で4時間加熱して得た複合材料。
<着色>
本発明により得られる修飾セルロース繊維シートは、加熱工程を経ても着色が小さい。マトリクスとの複合化時の加熱を想定して真空下で190℃4時間処理を行った後にも、YI値が好ましくは40以下、更に好ましくは35以下、特に好ましくは25以下である。YI値は例えば、スガ試験機製カラーコンピューターを用いて測定することができる。
本発明により得られる修飾セルロース繊維シートは、加熱工程を経ても着色が小さい。マトリクスとの複合化時の加熱を想定して真空下で190℃4時間処理を行った後にも、YI値が好ましくは40以下、更に好ましくは35以下、特に好ましくは25以下である。YI値は例えば、スガ試験機製カラーコンピューターを用いて測定することができる。
<引張強度>
本発明により得られる修飾セルロース繊維シートは、加熱工程を経ても引張り強度の低下が少ない。処理前の引張強度に対する、真空下190℃4時間加熱処理後の引張強度の比が、90%以上、好ましくは95%以上、特に好ましくは99%以上である。
本発明により得られる修飾セルロース繊維シートは、加熱工程を経ても引張り強度の低下が少ない。処理前の引張強度に対する、真空下190℃4時間加熱処理後の引張強度の比が、90%以上、好ましくは95%以上、特に好ましくは99%以上である。
<引張弾性率>
本発明により得られる修飾セルロース繊維シートは、高い引張弾性率を示す。真空下190℃4時間加熱処理後の引張弾性率が、好ましくは0.2~100GPaであり、より好ましくは1.0~50GPa、更に好ましくは4.0~30GPaである。
本発明により得られる修飾セルロース繊維シートは、高い引張弾性率を示す。真空下190℃4時間加熱処理後の引張弾性率が、好ましくは0.2~100GPaであり、より好ましくは1.0~50GPa、更に好ましくは4.0~30GPaである。
[セルロース粒子の製造方法]
本発明の修飾セルロース繊維は、セルロース粒子とすることができる。これらのセルロース粒子は特に熱可塑性樹脂と混練によって複合化する際に好適に用いられ、その高弾性率、低線膨張率、表面平滑性といった特性を生かして、各種の構造材、特に表面の意匠性に優れた自動車用パネルや建築物の外壁パネル等に有用である。
本発明の修飾セルロース繊維は、セルロース粒子とすることができる。これらのセルロース粒子は特に熱可塑性樹脂と混練によって複合化する際に好適に用いられ、その高弾性率、低線膨張率、表面平滑性といった特性を生かして、各種の構造材、特に表面の意匠性に優れた自動車用パネルや建築物の外壁パネル等に有用である。
本発明によって得られるセルロース繊維を、粒子化する方法としては、本発明によって得られるセルロース繊維を分散液とし、例えば公知のスプレードライ装置を用いて、スプレーノズル等から噴射することにより、分散媒を除去して造粒する方法が挙げられる。この噴射方法としては、具体的には回転円盤による方法、加圧ノズルによる方法、2流体ノズルによる方法などがある。スプレードライして得られた粒子を更に他の乾燥装置を用いて乾燥させても良い。この場合の熱エネルギー源としては、赤外線やマイクロ波を用いることもできる。
また、本発明によって得られたセルロース繊維を凍結乾燥した後、粉砕することによってもセルロース粒子を得ることができる。この場合、具体的には、発明の製造方法によって得られたセルロース繊維を液体窒素などで冷却した後、グラインダーや回転刃などで粉砕する方法が挙げられる。
また、本発明によって得られたセルロース繊維は、水分散体の状態から乾燥させることなく水中でセルロース以外の高分子と複合化させた後、水を除去したり、水から他の有機溶媒に置換した後その有機溶媒中でセルロース以外の高分子と複合化させた後、その有機溶媒を除去することで複合体を得ることもできる。
III.本発明のセルロース繊維複合体
本発明の修飾セルロース繊維分散液の製造方法により得られたセルロース繊維またはセルロースシートまたはセルロース粒子はマトリクスである高分子と複合化することでセルロース繊維複合体が得られる。該セルロース繊維複合体は、その高透明性、低線膨張率、非着色性といった特性を生かして、各種ディスプレイ基板材料、太陽電池用基板、窓材等に有用であり、また、その高弾性率、低線膨張率、表面平滑性といった特性を生かして、各種の構造材、特に表面の意匠性に優れた自動車用パネルや建築物の外壁パネル等に有用である。
本発明の修飾セルロース繊維分散液の製造方法により得られたセルロース繊維またはセルロースシートまたはセルロース粒子はマトリクスである高分子と複合化することでセルロース繊維複合体が得られる。該セルロース繊維複合体は、その高透明性、低線膨張率、非着色性といった特性を生かして、各種ディスプレイ基板材料、太陽電池用基板、窓材等に有用であり、また、その高弾性率、低線膨張率、表面平滑性といった特性を生かして、各種の構造材、特に表面の意匠性に優れた自動車用パネルや建築物の外壁パネル等に有用である。
以下、セルロースを高分子と複合化するセルロース繊維複合体の製造方法について説明する。
本発明のセルロース繊維複合体は、上述の本発明で得られた修飾セルロース繊維分散液、修飾セルロース繊維シートまたはセルロース粒子と、セルロース以外の高分子とを複合化したたものであり、好ましくは、本発明のセルロースシートまたはセルロース粒子とマトリクス材料であるセルロース以外の高分子またはその前駆体とが複合化したものである。
本発明のセルロース繊維複合体は、上述の本発明で得られた修飾セルロース繊維分散液、修飾セルロース繊維シートまたはセルロース粒子と、セルロース以外の高分子とを複合化したたものであり、好ましくは、本発明のセルロースシートまたはセルロース粒子とマトリクス材料であるセルロース以外の高分子またはその前駆体とが複合化したものである。
ここでマトリクス材料とは、セルロースシートと貼り合わせたり、空隙を埋めたり、造粒したセルロース粒子を混練する高分子またはその前駆体(例えばモノマー)材料のことをいう。
このマトリクス材料として好適なのは、加熱することにより流動性のある液体になる熱可塑性樹脂、加熱により重合する熱硬化性樹脂、紫外線や電子線などの活性エネルギー線を照射することにより重合硬化する、活性エネルギー線硬化性樹脂等から得られる少なくとも1種の樹脂(高分子材料)またはその前駆体である。
このマトリクス材料として好適なのは、加熱することにより流動性のある液体になる熱可塑性樹脂、加熱により重合する熱硬化性樹脂、紫外線や電子線などの活性エネルギー線を照射することにより重合硬化する、活性エネルギー線硬化性樹脂等から得られる少なくとも1種の樹脂(高分子材料)またはその前駆体である。
なお、本発明において高分子材料の前駆体とは、いわゆるモノマー、オリゴマーであり、例えば、熱可塑性樹脂の項に(共)重合成分として後述する各単量体など(以後、熱可塑性樹脂前駆体と称することがある)、熱硬化性樹脂・光硬化性樹脂の項に後述する各前駆体などが挙げられる。
本発明のセルロース繊維複合体を得る方法としては、次の(a)~(j)の方法が挙げられる。
(a) セルロースシート、粒子等に液状の熱可塑性樹脂前駆体を含浸させて重合する方法
(b) セルロースシート、粒子等に熱硬化性樹脂前駆体又は活性エネルギー線硬化性樹脂前駆体を含浸させて重合硬化させる方法
(c) セルロースシート、粒子等に樹脂溶液(熱可塑性樹脂、熱可塑性樹脂前駆体、熱硬化性樹脂前駆体、および光硬化性樹脂前駆体から選ばれる1以上の溶質を含む溶液)を含浸させて乾燥した後、加熱プレス等で密着させ、必要に応じて重合硬化する方法
(d) セルロースシート、粒子等に熱可塑性樹脂の溶融体を含浸させ、加熱プレス等で密着させる方法
(e) 熱可塑性樹脂シート(又はフィルム)とセルロースシートを交互に配置し、加熱プレス等で密着させる方法
(f) セルロースシートの片面もしくは両面に液状の熱可塑性樹脂前駆体や熱硬化性樹脂前駆体もしくは活性エネルギー線硬化性樹脂前駆体を塗布して重合硬化させる方法
(g) セルロースシートの片面もしくは両面に樹脂溶液(熱可塑性樹脂、熱可塑性樹脂前駆体、熱硬化性樹脂前駆体、および光硬化性樹脂前駆体から選ばれる1以上の溶質を含む溶液)を塗布して、溶媒を除去後、必要に応じて重合硬化することにより複合化する方法
(h) セルロース粒子と熱可塑性樹脂を溶融混練した後、シート状や目的の形状に成形する方法
(i) セルロース繊維分散液とモノマー溶液または分散液(熱可塑性樹脂前駆体、熱硬化性樹脂前駆体、および光硬化性樹脂前駆体から選ばれる1以上の溶質または分散質を含む溶液または分散液)とを混合したのち、溶媒を除去した後重合硬化することにより複合化する方法。
(j) セルロース繊維分散液と高分子溶液または分散液(熱可塑性樹脂溶液または分散液)を混合したのち、溶媒を除去して複合化する方法。
(a) セルロースシート、粒子等に液状の熱可塑性樹脂前駆体を含浸させて重合する方法
(b) セルロースシート、粒子等に熱硬化性樹脂前駆体又は活性エネルギー線硬化性樹脂前駆体を含浸させて重合硬化させる方法
(c) セルロースシート、粒子等に樹脂溶液(熱可塑性樹脂、熱可塑性樹脂前駆体、熱硬化性樹脂前駆体、および光硬化性樹脂前駆体から選ばれる1以上の溶質を含む溶液)を含浸させて乾燥した後、加熱プレス等で密着させ、必要に応じて重合硬化する方法
(d) セルロースシート、粒子等に熱可塑性樹脂の溶融体を含浸させ、加熱プレス等で密着させる方法
(e) 熱可塑性樹脂シート(又はフィルム)とセルロースシートを交互に配置し、加熱プレス等で密着させる方法
(f) セルロースシートの片面もしくは両面に液状の熱可塑性樹脂前駆体や熱硬化性樹脂前駆体もしくは活性エネルギー線硬化性樹脂前駆体を塗布して重合硬化させる方法
(g) セルロースシートの片面もしくは両面に樹脂溶液(熱可塑性樹脂、熱可塑性樹脂前駆体、熱硬化性樹脂前駆体、および光硬化性樹脂前駆体から選ばれる1以上の溶質を含む溶液)を塗布して、溶媒を除去後、必要に応じて重合硬化することにより複合化する方法
(h) セルロース粒子と熱可塑性樹脂を溶融混練した後、シート状や目的の形状に成形する方法
(i) セルロース繊維分散液とモノマー溶液または分散液(熱可塑性樹脂前駆体、熱硬化性樹脂前駆体、および光硬化性樹脂前駆体から選ばれる1以上の溶質または分散質を含む溶液または分散液)とを混合したのち、溶媒を除去した後重合硬化することにより複合化する方法。
(j) セルロース繊維分散液と高分子溶液または分散液(熱可塑性樹脂溶液または分散液)を混合したのち、溶媒を除去して複合化する方法。
中でもセルロースシートに対しては(a)、(b)、(c)、(d)、(e)、(f)、(g)の方法が好ましく、セルロース粒子に対しては(h)の方法が好ましい。
(a)液状の熱可塑性樹脂前駆体を含浸させて重合する方法としては、重合可能なモノマーやオリゴマーをセルロースシート、粒子等に含浸させ、熱処理等により上記モノマーを重合させることによりセルロース繊維複合体を得る方法が挙げられる。一般的には、モノマーの重合に用いられる重合触媒を重合開始剤として用いることができる。
(b)熱硬化性樹脂前駆体又は活性エネルギー線硬化性樹脂前駆体を含浸させて重合硬化させる方法としては、エポキシ樹脂モノマー等の熱硬化性樹脂前駆体、又はアクリル樹脂モノマー等の光硬化性樹脂前駆体と硬化剤の混合物を、セルロースシート又は粒子に含浸させ、熱又は活性エネルギー線等により上記熱硬化性樹脂前躯体又は光硬化性樹脂前躯体を硬化させることによりセルロース繊維複合体を得る方法が挙げられる。
(c)樹脂溶液(熱可塑性樹脂、熱可塑性樹脂前駆体、熱硬化性樹脂前駆体、および光硬化性樹脂前駆体から選ばれる1以上の溶質を含む溶液)を含浸させて乾燥した後、加熱プレス等で密着させ、必要に応じて重合硬化する方法としては、樹脂が溶解する溶媒に溶解させ、その溶液をセルロースシート、粒子等に含浸させ、乾燥させることでセルロース繊維複合体を得る方法が挙げられる。この場合、乾燥後加熱プレス等で溶媒が乾燥した空隙を密着させることでより高性能なセルロース複合体を得る方法が挙げられる。光硬化性樹脂の場合には更に、必要に応じて活性エネルギー線等による重合硬化を行う。
樹脂を溶解させる溶媒としては、セルロース繊維との親和性と樹脂の溶解性を考慮して選択すればよく、具体的には修飾セルロース繊維分散液の分散媒として例示したもの等の中から、樹脂の溶解性に応じて選択すればよい。
樹脂を溶解させる溶媒としては、セルロース繊維との親和性と樹脂の溶解性を考慮して選択すればよく、具体的には修飾セルロース繊維分散液の分散媒として例示したもの等の中から、樹脂の溶解性に応じて選択すればよい。
(d)熱可塑性樹脂の溶融体を含浸させ、加熱プレス等で密着させる方法としては、熱可塑性樹脂をガラス転移温度以上又は融点以上で熱処理することにより溶解させ、セルロースシート、粒子等に含浸し、加熱プレス等で密着することにより高分子セルロース複合体を得る方法が挙げられる。熱処理は加圧下で行うことが望ましく、真空加熱プレス機能を有する設備の使用が有効である。
(e)熱可塑性樹脂シート(又はフィルム)とセルロースシートを交互に配置し、加熱プレス等で密着させる方法としては、セルロースシートの片面もしくは両面に熱可塑性樹脂のフィルムもしくはシート配置し、必要に応じて加熱やプレスすることにより、熱可塑性樹脂とセルロースシートを貼り合わせる方法が挙げられる。この場合、セルロースシートの表面に接着剤やプライマーなどを塗布して貼り合わせても良い。貼り合わせる際に気泡を抱き込まないように、加圧された2本のロールの間を通す方法や、真空状態でプレスする方法を用いることができる。
(f)セルロースシートの片面もしくは両面に液状の熱可塑性樹脂前駆体や熱硬化性樹脂前駆体もしくは活性エネルギー線硬化性樹脂前駆体を塗布して硬化させる方法としては、セルロースシートの片面もしくは両面に熱重合開始剤を処方した熱硬化性樹脂前駆体を塗布して加熱することにより硬化させて両者を密着させる方法や、セルロースシートの片面もしくは両面に光重合開始剤を処方した硬化性樹脂前駆体を塗布した後、紫外線等の活性エネルギー線を照射して硬化させる方法が挙げられる。セルロースシートに熱もしくは光硬化性樹脂前駆体を塗布した後、更にセルロースシートを重ねるなど、多層構造にしてから、硬化させても良い。
(g)セルロースシートの片面もしくは両面に樹脂溶液(熱可塑性樹脂、熱可塑性樹脂前駆体、熱硬化性樹脂前駆体、および光硬化性樹脂前駆体から選ばれる1以上の溶質を含む溶液)を塗布して、溶媒を除去することにより複合化する方法としては、溶媒に可溶な樹脂を当該溶媒に溶解させた樹脂溶液を用意し、セルロースシートの片面もしくは両面に塗布し、加熱により溶媒を除去する方法が挙げられる。光硬化性樹脂の場合には更に、必要に応じて活性エネルギー線等による重合硬化を行う。
樹脂を溶解させる溶媒としては、セルロース繊維との親和性と樹脂の溶解性を考慮して選択すればよく、具体的には修飾セルロース繊維分散液の分散媒として例示したもの等の中から、樹脂の溶解性に応じて選択すればよい。
樹脂を溶解させる溶媒としては、セルロース繊維との親和性と樹脂の溶解性を考慮して選択すればよく、具体的には修飾セルロース繊維分散液の分散媒として例示したもの等の中から、樹脂の溶解性に応じて選択すればよい。
このようにして製造したセルロースシートと樹脂のセルロース複合体を複数枚重ねて積層体を得ることもできる。その際に、セルロースシートを含むセルロース複合体と含まない樹脂シートを積層してもよい。セルロース複合体同士や樹脂とセルロース複合体を接着させるために、接着剤を塗布したり接着シートを介在させてもよい。また、積層体に加熱プレス処理を加えて一体化することもできる。
(h)セルロース粒子と熱可塑性樹脂を溶融混練した後、シート状や目的の形状に成形する方法としては、セルロース粒子と熱可塑性樹脂とを、ドライブレンドした後に溶融する方法、溶融混練する方法、等が好ましく挙げられる。ドライブレンドした後に溶融する方法は、両者を、タンブラーブレンダー、リボンブレンダー、V型ブレンダー、ヘンシェルミキサー等により均一に混合し、その後、該混合物に必要に応じて用いられる酸化防止剤などの添加剤を添加し、溶融状態を経てセルロース複合体とする。具体的には、例えば、該混合物を単に溶融するか、又は、一軸又は二軸押出機、ロール、バンバリーミキサー、ニーダー、ブラベンダー等により溶融混練する。溶融混練する場合は、両者を、必要に応じて用いられる酸化防止剤などの添加剤等と共に溶融混合する。例えば、一軸又は二軸押出機、ロール、バンバリーミキサー、ニーダー、ブラベンダー等により溶融混練する。その後、Tダイから押し出してシート状に成形したり、金型に射出するなどして、目的の形状に成形する。
(i)セルロース繊維分散液とモノマー溶液または分散液(熱可塑性樹脂前駆体、熱硬化性樹脂前駆体、および光硬化性樹脂前駆体から選ばれる1以上の溶質または分散質を含む溶液または分散液)とを混合したのち、溶媒を除去した後重合硬化することにより複合化する方法としては、溶媒に可溶なモノマーを溶解させた溶液、もしくは分散液を用意し、セルロース繊維分散液を混合する。この際、必要に応じてセルロース繊維分散液の分散媒(溶媒)溶媒は水から有機溶媒に置換することが好ましい。この混合液中でモノマーを重合もしくは、溶媒を除去した後にモノマーを重合硬化することで複合材を得ることができる。
(j)セルロース繊維分散液と高分子溶液または分散液(熱可塑性樹脂溶液または分散液)を混合したのち、溶媒を除去して複合化する方法としては、溶媒に可溶な高分子溶液または分散液を用意し、セルロース繊維分散液と混合する。この際、必要に応じてセルロース繊維分散液の分散媒(溶媒)は水から有機溶媒に置換することが好ましい。この混合液の溶媒を除去することで複合材を得ることができる。
[マトリクス材料]
本発明において、セルロースシート又はセルロース粒子又はセルロース繊維分散液に複合化させるセルロース以外のマトリクス材料(高分子材料またはその前駆体)を以下に例示するが、本発明で用いるマトリクス材料は何ら以下のものに限定されるものではない。また、本発明における熱可塑性樹脂、熱硬化性樹脂、光(活性エネルギー線)硬化性樹脂は2種以上混合して用いることができる。
本発明において、セルロースシート又はセルロース粒子又はセルロース繊維分散液に複合化させるセルロース以外のマトリクス材料(高分子材料またはその前駆体)を以下に例示するが、本発明で用いるマトリクス材料は何ら以下のものに限定されるものではない。また、本発明における熱可塑性樹脂、熱硬化性樹脂、光(活性エネルギー線)硬化性樹脂は2種以上混合して用いることができる。
本発明においては、以下のマトリクス材料(高分子材料またはその前駆体)のうち、特に、高分子材料、または前駆体の場合にはその重合体が、非晶質でガラス転移温度(Tg)の高い合成高分子であるものが、透明性に優れた高耐久性のセルロース繊維複合体を得る上で好ましい。このうち非晶質の程度としては、結晶化度で10%以下、特に5%以下であるものが好ましく、また、Tgは110℃以上、特に120℃以上、とりわけ130℃以上のものが好ましい。Tgが低いと例えば熱水等に触れた際に変形する恐れがあり、実用上問題が生じる。また、低吸水性のセルロース複合体を得るためには、ヒドロキシル基、カルボキシル基、アミノ基などの親水性の官能基が少ない高分子材料を選定することが好ましい。なお、高分子のTgは一般的な方法で求めることができる。例えば、DSC法による測定で求められる。高分子の結晶化度は、非晶質部と結晶質部の密度から算定することができ、また、動的粘弾性測定により、弾性率と粘性率の比であるtanδから算出することもできる。
<熱可塑性樹脂>
熱可塑性樹脂としては、スチレン系樹脂、アクリル系樹脂、芳香族ポリカーボネート系樹脂、脂肪族ポリカーボネート系樹脂、芳香族ポリエステル系樹脂、脂肪族ポリエステル系樹脂、脂肪族ポリオレフィン系樹脂、環状オレフィン系樹脂、ポリアミド系樹脂、ポリフェニレンエーテル系樹脂、熱可塑性ポリイミド系樹脂、ポリアセタール系樹脂、ポリスルホン系樹脂、非晶性フッ素系樹脂等が挙げられる。
熱可塑性樹脂としては、スチレン系樹脂、アクリル系樹脂、芳香族ポリカーボネート系樹脂、脂肪族ポリカーボネート系樹脂、芳香族ポリエステル系樹脂、脂肪族ポリエステル系樹脂、脂肪族ポリオレフィン系樹脂、環状オレフィン系樹脂、ポリアミド系樹脂、ポリフェニレンエーテル系樹脂、熱可塑性ポリイミド系樹脂、ポリアセタール系樹脂、ポリスルホン系樹脂、非晶性フッ素系樹脂等が挙げられる。
スチレン系樹脂としては、スチレン、クロルスチレン、ジビニルベンゼン、α-メチルスチレン等の重合体及び共重合体が挙げられる。
アクリル系樹脂としては、(メタ)アクリル酸、(メタ)アクリロニトリル、(メタ)アクリル酸エステル、(メタ)アクリルアミド等の重合体及び共重合体が挙げられる。ここで「(メタ)アクリル」とは、「アクリル及び/又はメタクリル」を意味する。
アクリル系樹脂としては、(メタ)アクリル酸、(メタ)アクリロニトリル、(メタ)アクリル酸エステル、(メタ)アクリルアミド等の重合体及び共重合体が挙げられる。ここで「(メタ)アクリル」とは、「アクリル及び/又はメタクリル」を意味する。
芳香族ポリカーボネート系樹脂とは、3価以上の多価フェノール類を共重合成分として含有できる1種以上のビスフェノール類と、ビスアルキルカーボネート、ビスアリールカーボネート、ホスゲン等の炭酸エステル類との反応により製造される共重合体であり、必要に応じて芳香族ポリエステルカーボネート類とするために共重合成分としてテレフタル酸やイソフタル酸などの芳香族ジカルボン酸又はその誘導体(例えば芳香族ジカルボン酸ジエステルや芳香族ジカルボン酸塩化物)を使用してもよいものである。
本発明の修飾セルロースは、芳香環含有置換基で修飾されたものなので、芳香族ポリカーボネネート系樹脂とは相溶性が良好であると考えられ、好ましい。
前記ビスフェノール類としては、ビスフェノールA、ビスフェノールC、ビスフェノールE、ビスフェノールF、ビスフェノールM、ビスフェノールP、ビスフェノールS、ビスフェノールZ(略号はアルドリッチ社試薬カタログを参照)等が例示され、中でもビスフェノールAとビスフェノールZ(中心炭素がシクロヘキサン環に参加しているもの)が好ましく、ビスフェノールAが特に好ましい。共重合可能な3価フェノール類としては、1,1,1-(4-ヒドロキシフェニル)エタンやフロログルシノールなどが例示できる。
前記ビスフェノール類としては、ビスフェノールA、ビスフェノールC、ビスフェノールE、ビスフェノールF、ビスフェノールM、ビスフェノールP、ビスフェノールS、ビスフェノールZ(略号はアルドリッチ社試薬カタログを参照)等が例示され、中でもビスフェノールAとビスフェノールZ(中心炭素がシクロヘキサン環に参加しているもの)が好ましく、ビスフェノールAが特に好ましい。共重合可能な3価フェノール類としては、1,1,1-(4-ヒドロキシフェニル)エタンやフロログルシノールなどが例示できる。
脂肪族ポリカーボネート系樹脂としては、脂肪族ジオール成分及び/又は脂環式ジオール成分とビスアルキルカーボネート、ホスゲン等の炭酸エステル類との反応により製造される共重合体である。脂環式ジオールとしてはシクロヘキサンジメタノールやイソソルバイト等が挙げられる。
芳香族ポリエステル系樹脂としては、エチレングリコール、プロピレングリコール、1,4-ブタンジオール等のジオール類とテレフタル酸等の芳香族カルボン酸との共重合体が挙げられる。また、ポリアリレートのように、ビスフェノールA等のジオール類とテレフタル酸やイソフタル酸等の芳香族カルボン酸との共重合体も挙げられる。
本発明の修飾セルロースは、芳香環含有置換基で修飾されたものなので、芳香族ポリエステル系樹脂とは相溶性が良好であると考えられ、好ましい。
本発明の修飾セルロースは、芳香環含有置換基で修飾されたものなので、芳香族ポリエステル系樹脂とは相溶性が良好であると考えられ、好ましい。
脂肪族ポリエステル系樹脂としては、上記ジオールとコハク酸、吉草酸等の脂肪族ジカルボン酸との共重合体やグリコール酸や乳酸等のヒドロキシジカルボン酸の共重合体等が挙げられる。
脂肪族ポリオレフィン系樹脂としては、具体的には、例えば炭素数2~8程度のα-オレフィンの単独重合体、それらのα-オレフィンと炭素数2~18程度の他のα-オレフィン等との二元或いは三元の共重合体等が挙げられ、これらのオレフィン系重合体は2種以上が併用されていてもよい。
環状オレフィン系樹脂とは、ノルボルネンやシクロヘキサジエン等、ポリマー鎖中に環状オレフィン骨格を含む重合体もしくはこれらを含む共重合体である。
ポリアミド系樹脂としては、6,6-ナイロン、6-ナイロン、11-ナイロン、12-ナイロン、4,6-ナイロン、6,10-ナイロン、6,12-ナイロン等の脂肪族アミド系樹脂や、フェニレンジアミン等の芳香族ジアミンと塩化テレフタロイルや塩化イソフタロイル等の芳香族ジカルボン酸又はその誘導体からなる芳香族ポリアミド等が挙げられる。
ポリフェニレンエーテル系樹脂としては、例えば、ポリ(2,6-ジメチル-1,4-フェニレンエーテル)、ポリ(2-メチル-6-エチル-1,4-フェニレンエーテル)、ポリ(2,6-ジクロロ-1,4-フェニレンエーテル)等が挙げられ、さらに2,6-ジメチルフェノールと他のフェノール類との共重合体も挙げられる。
ポリイミド系樹脂としては、無水ピロメリット酸や4,4’-ジアミノジフェニルエーテル等の共重合体であるピロメリット酸型ポリイミド、無水塩化トリメリット酸やp-フェニレンジアミン等の芳香族ジアミンやジイソシアネート化合物からなる共重合体であるトリメリット酸型ポリイミド、ビフェニルテトラカルボン酸、4,4’-ジアミノジフェニルエーテル、p-フェニレンジアミン等からなるビフェニル型ポリイミド、ベンゾフェノンテトラカルボン酸や4,4’-ジアミノジフェニルエーテル等からなるベンゾフェノン型ポリイミド、ビスマレイミドや4,4’-ジアミノジフェニルメタン等からなるビスマレイミド型ポリイミド等が挙げられる。
ポリアセタール系樹脂としては、オキシメチレン構造を単位構造にもつホモポリマーと、オキシエチレン単位を含む共重合体が挙げられる。
ポリスルホン系樹脂としては、4,4’-ジクロロジフェニルスルホンやビスフェノールA等の共重合体が挙げられる。
非晶性フッ素系樹脂としては、テトラフルオロエチレン、ヘキサフルオロプロピレン、クロロトリフルオロエチレン、フッ化ビニリデン、フッ化ビニル、ペルフルオロアルキルビニルエーテル等の単独重合体又は共重合体が挙げられる。
これらの熱可塑性樹脂は、1種を単独で用いても良く、2種以上を併用しても良い。
<硬化性樹脂>
熱硬化性樹脂、光(活性エネルギー線)硬化性樹脂とは、硬化する前の前駆体もしくは硬化してなる樹脂硬化物のことを意味する。ここで前駆体は、常温では液状、半固体状又は固形状等であって常温下又は加熱下で流動性を示す物質を意味する。これらは硬化剤、触媒、熱又は光の作用によって重合反応や架橋反応を起こして分子量を増大させながら網目状の三次元構造を形成してなる不溶不融の樹脂となり得る。また、樹脂硬化物とは、上記熱硬化性樹脂前駆体又は光(活性エネルギー線)硬化性樹脂前駆体が硬化してなる樹脂を意味する。
熱硬化性樹脂、光(活性エネルギー線)硬化性樹脂とは、硬化する前の前駆体もしくは硬化してなる樹脂硬化物のことを意味する。ここで前駆体は、常温では液状、半固体状又は固形状等であって常温下又は加熱下で流動性を示す物質を意味する。これらは硬化剤、触媒、熱又は光の作用によって重合反応や架橋反応を起こして分子量を増大させながら網目状の三次元構造を形成してなる不溶不融の樹脂となり得る。また、樹脂硬化物とは、上記熱硬化性樹脂前駆体又は光(活性エネルギー線)硬化性樹脂前駆体が硬化してなる樹脂を意味する。
<<熱硬化性樹脂>>
本発明における熱硬化性樹脂としては、特に限定されるものではないが、エポキシ樹脂、アクリル樹脂、オキセタン樹脂、フェノール樹脂、ユリア樹脂、メラミン樹脂、不飽和ポリエステル樹脂、珪素樹脂、ポリウレタン樹脂、ジアリルフタレート樹脂等の前駆体が挙げられる。
本発明における熱硬化性樹脂としては、特に限定されるものではないが、エポキシ樹脂、アクリル樹脂、オキセタン樹脂、フェノール樹脂、ユリア樹脂、メラミン樹脂、不飽和ポリエステル樹脂、珪素樹脂、ポリウレタン樹脂、ジアリルフタレート樹脂等の前駆体が挙げられる。
上記エポキシ樹脂前駆体としては、少なくとも1個のエポキシ基を有する有機化合物をいう。上記エポキシ樹脂前駆体中のエポキシ基の数としては、1分子あたり1個以上7個以下であることが好ましく、1分子あたり2個以上であることがより好ましい。ここで、前駆体1分子あたりのエポキシ基の数は、エポキシ樹脂前駆体中のエポキシ基の総数をエポキシ樹脂中の分子の総数で除算することにより求められる。上記エポキシ樹脂前駆体としては特に限定されず、例えば、以下に示したエポキシ樹脂等が挙げられる。これらのエポキシ樹脂は単独でも2種以上併用されてもよい。これらエポキシ樹脂は硬化剤を用いて熱硬化性樹脂前躯体を硬化することにより得られる。
例えば、芳香族エポキシ樹脂及びこれらの水添化物や臭素化物等の前駆体、脂環族エポキシ樹脂、脂肪族エポキシ樹脂、グリシジルエステル型エポキシ樹脂及びこれらの水添化物、グリシジルアミン型エポキシ樹脂及びこれらの水添化物、グリシジル(メタ)アクリレートとラジカル重合性モノマーとの共重合体等が挙げられる。上記エポキシ樹脂前駆体の硬化反応に用いられる硬化剤としては、特に限定されず、例えば、アミン化合物、アミン化合物から合成されるポリアミノアミド化合物等の化合物、3級アミン化合物、イミダゾール化合物、ヒドラジド化合物、メラミン化合物、酸無水物、フェノール化合物、熱潜在性カチオン重合触媒、光潜在性カチオン重合開始剤、ジシアンアミド及びその誘導体等が挙げられる。これらの硬化剤は、単独で用いられてもよく、2種以上が併用されてもよい。
アクリル樹脂前駆体としては、分子内に1個の(メタ)アクリロイル基を有する単官能(メタ)アクリレート化合物、分子内に2個又は3個の(メタ)アクリロイル基を有する多官能(メタ)アクリレート化合物、スチレン系化合物、アクリル酸誘導体、分子内に4~8個の(メタ)アクリロイル基を有するアクリレート化合物、エポキシ(メタ)アクリレート化合物、ウレタン結合を有する(メタ)アクリレート化合物などが挙げられる。
分子内に1個の(メタ)アクリロイル基を有する単官能(メタ)アクリレート化合物としては、メチル(メタ)アクリレート、2-ヒドロキシエチル(メタ)アクリレート、フェニル(メタ)アクリレート、ベンジル(メタ)アクリレート、シクロヘキシル(メタ)アクリレート、アルキルの炭素数が1~30であるアルキル(メタ)アクリレートなどが挙げられる。
分子内に1個の(メタ)アクリロイル基を有する単官能(メタ)アクリレート化合物としては、メチル(メタ)アクリレート、2-ヒドロキシエチル(メタ)アクリレート、フェニル(メタ)アクリレート、ベンジル(メタ)アクリレート、シクロヘキシル(メタ)アクリレート、アルキルの炭素数が1~30であるアルキル(メタ)アクリレートなどが挙げられる。
特に、脂環骨格を有するモノ(メタ)アクリレートは、耐熱性が高くなるので、好適に利用することができる。脂環骨格モノ(メタ)アクリレート化合物の具体例としては、例えば(ヒドロキシ-アクリロイルオキシ)トリシクロ[5.2.1.02,6]デカン、(ヒドロキシ-メタクリロイルオキシ)トリシクロ[5.2.1.02,6]デカン、(ヒドロキシ-アクリロイルオキシ)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、(ヒドロキシ-メタクリロイルオキシ)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、(ヒドロキシメチル-アクリロイルオキシメチル)トリシクロ[5.2.1.02,6]デカン、(ヒドロキシメチル-メタクリロイルオキシメチル)トリシクロ[5.2.1.02,6]デカン、(ヒドロキシメチル-アクリロイルオキシメチル)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、(ヒドロキシメチル-メタクリロイルオキシメチル)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、(ヒドロキシエチル-アクリロイルオキシエチル)トリシクロ[5.2.1.02,6]デカン、(ヒドロキシエチル-メタクリロイルオキシエチル)トリシクロ[5.2.1.02,6]デカン、(ヒドロキシエチル-アクリロイルオキシエチル)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、(ヒドロキシエチル-メタクリロイルオキシエチル)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン等が挙げられる。また、これらの混合物等を挙げることが出来る。
分子中に2個又は3個の(メタ)アクリロイル基を有する多官能(メタ)アクリレート化合物としては、エチレングリコールジ(メタ)アクリレート、ジエチレングリコールジ(メタ)アクリレート、トリエチレングリコールジ(メタ)アクリレート、テトラエチレングリコール以上のポリエチレングリコールのジ(メタ)アクリレート、1,3-ブチレングリコールジ(メタ)アクリレート、1,6-ヘキサンジオールジ(メタ)アクリレート、アルキルの炭素数が1~30であるアルキルジオールジ(メタ)アクリレート、ネオペンチルグリコールジ(メタ)アクリレート、2-ヒドロキシ1,3-ジ(メタ)アクリロキシプロパン、2,2-ビス[4-(メタ)アクリロイルオキシフェニル]プロパン、トリメチロールプロパントリ(メタ)アクリレート、ビス(ヒドロキシ)トリシクロ[5.2.1.02,6]デカン=ジアクリレート、ビス(ヒドロキシ)トリシクロ[5.2.1.02,6]デカン=ジメタクリレート、ビス(ヒドロキシ)トリシクロ[5.2.1.02,6]デカン=アクリレートメタクリレート、ビス(ヒドロキシ)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン=ジアクリレート、ビス(ヒドロキシ)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン=ジメタクリレート、ビス(ヒドロキシ)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン=アクリレートメタクリレート、2,2-ビス[4-(β-(メタ)アクリロイルオキシエトキシ)フェニル]プロパン、2,2-ビス[4-(β-(メタ)アクリロイルオキシエトキシ)シクロヘキシル]プロパン、1,4-ビス[(メタ)アクリロイルオキシメチル]シクロヘキサン等が挙げられる。
スチレン系化合物としては、スチレン、クロルスチレン、ジビニルベンゼン、α-メチルスチレンなどが挙げられる。
エステル以外の(メタ)アクリル酸誘導体としては、アクリルアミド、メタクリルアミド、アクリロニトリル、メタクリロニトリルなどが挙げられる。
これらの中でも、含脂環骨格ビス(メタ)アクリレート化合物が好適に用いられる。例えばビス(アクリロイルオキシ)トリシクロ[5.2.1.02,6]デカン、ビス(メタクリロイルオキシ)トリシクロ[5.2.1.02,6]デカン、(アクリロイルオキシ-メタクリロイルオキシ)トリシクロ[5.2.1.02,6]デカン、ビス(アクリロイルオキシ)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、ビス(メタクリロイルオキシ)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、(アクリロイルオキシ-メタクリロイルオキシ)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、ビス(アクリロイルオキシメチル)トリシクロ[5.2.1.02,6]デカン、ビス(メタクリロイルオキシメチル)トリシクロ[5.2.1.02,6]デカン、(アクリロイルオキシメチル-メタクリロイルオキシメチル)トリシクロ[5.2.1.02,6]デカン、ビス(アクリロイルオキシメチル)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、ビス(メタクリロイルオキシメチル)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、(アクリロイルオキシメチル-メタクリロイルオキシメチル)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、ビス(アクリロイルオキシエチル)トリシクロ[5.2.1.02,6]デカン、ビス(メタクリロイルオキシエチル)トリシクロ[5.2.1.02,6]デカン、(アクリロイルオキシエチル-メタクリロイルオキシエチル)トリシクロ[5.2.1.02,6]デカン、ビス(アクリロイルオキシエチル)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、ビス(メタクリロイルオキシエチル)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、(アクリロイルオキシエチル-メタクリロイルオキシエチル)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン等、及びこれらの混合物等を挙げることが出来る。
エステル以外の(メタ)アクリル酸誘導体としては、アクリルアミド、メタクリルアミド、アクリロニトリル、メタクリロニトリルなどが挙げられる。
これらの中でも、含脂環骨格ビス(メタ)アクリレート化合物が好適に用いられる。例えばビス(アクリロイルオキシ)トリシクロ[5.2.1.02,6]デカン、ビス(メタクリロイルオキシ)トリシクロ[5.2.1.02,6]デカン、(アクリロイルオキシ-メタクリロイルオキシ)トリシクロ[5.2.1.02,6]デカン、ビス(アクリロイルオキシ)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、ビス(メタクリロイルオキシ)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、(アクリロイルオキシ-メタクリロイルオキシ)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、ビス(アクリロイルオキシメチル)トリシクロ[5.2.1.02,6]デカン、ビス(メタクリロイルオキシメチル)トリシクロ[5.2.1.02,6]デカン、(アクリロイルオキシメチル-メタクリロイルオキシメチル)トリシクロ[5.2.1.02,6]デカン、ビス(アクリロイルオキシメチル)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、ビス(メタクリロイルオキシメチル)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、(アクリロイルオキシメチル-メタクリロイルオキシメチル)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、ビス(アクリロイルオキシエチル)トリシクロ[5.2.1.02,6]デカン、ビス(メタクリロイルオキシエチル)トリシクロ[5.2.1.02,6]デカン、(アクリロイルオキシエチル-メタクリロイルオキシエチル)トリシクロ[5.2.1.02,6]デカン、ビス(アクリロイルオキシエチル)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、ビス(メタクリロイルオキシエチル)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン、(アクリロイルオキシエチル-メタクリロイルオキシエチル)ペンタシクロ[6.5.1.13,6.02,7.09,13]ペンタデカン等、及びこれらの混合物等を挙げることが出来る。
これらのうち、ビス(アクリロイルオキシメチル)トリシクロ[5.2.1.02,6]デカン、ビス(メタクリロイルオキシメチル)トリシクロ[5.2.1.02,6]デカン及び(アクリロイルオキシメチル-メタクリロイルオキシメチル)トリシクロ[5.2.1.02,6]デカンから選ばれるものが好ましい。これらのビス(メタ)アクリレートは、いくつか併用することもできる。
分子内に4~8個の(メタ)アクリロイル基を有する(メタ)アクリレートとしては、ポリオールの(メタ)アクリル酸エステル等が利用できる。具体的には、ペンタエリスリテールテトラ(メタ)アクリレート、ペンタエリスリテールトリ(メタ)アクリレート、ジペンタエリスリトールヘキサ(メタ)アクリレート、ジペンタエリスリトールペンタ(メタ)アクリレート、ジペンタエリスリトールテトラ(メタ)アクリレート、ジペンタエリスリトールトリ(メタ)アクリレート、トリペンタエリスリトールオクタ(メタ)アクリレート、トリペンタエリスリトールセプタ(メタ)アクリレート、トリペンタエリスリトールヘキサ(メタ)アクリレート、トリペンタエリスリトールペンタ(メタ)アクリレート、トリペンタエリスリトールテトラ(メタ)アクリレート、トリペンタエリスリトールトリ(メタ)アクリレート等が挙げられる。
次にエポキシ(メタ)アクリレートの具体例としては、例えば、ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、フェノールノボラック型エポキシ樹脂、脂環式エポキシ基を有する化合物、ビスフェノールA型プロピレンオキサイド付加型の末端グリシジルエーテル、フルオレンエポキシ樹脂等と(メタ)アクリル酸との反応物を挙げることができる。
分子内にウレタン結合を有する(メタ)アクリレートとしては、1分子中に(メタ)アクリロイル基を2~10個(好ましくは2~5個)有するウレタンオリゴマー等が挙げられる。例えば、ジオール類及びジイソシアネー類を反応させて得られるウレタンプレポリマーと、ヒドロキシ基含有の(メタ)アクリレートを反応させて製造される(メタ)アクリロイル基含有ウレタンオリゴマーがある。
分子内にウレタン結合を有する(メタ)アクリレートとしては、1分子中に(メタ)アクリロイル基を2~10個(好ましくは2~5個)有するウレタンオリゴマー等が挙げられる。例えば、ジオール類及びジイソシアネー類を反応させて得られるウレタンプレポリマーと、ヒドロキシ基含有の(メタ)アクリレートを反応させて製造される(メタ)アクリロイル基含有ウレタンオリゴマーがある。
分子内にウレタン結合を有する(メタ)アクリレートの数平均分子量は1,000~100,000が好ましく、更に好ましくは2,000~10,000である。中でもメチレンジシクロヘキシルジイソシアネートとポリテトラメチレンエーテルグリコールを有するウレタンアクリレートは透明性、低複屈折性、柔軟性等の点により優れており、好適に利用することができる。
オキセタン樹脂前駆体としては、少なくとも1個のオキセタン環を有する化合物が挙げられる。上記オキセタン樹脂前駆体中のオキセタン環の数は、1分子あたり1個以上、4個以下が好ましい。分子中に1個のオキセタンを有する化合物としては、3-エチル-3-ヒドロキシメチルオキセタン、3-エチル-3-(フェノキシメチル)オキセタン、3-エチル-3-(2-エチルヘキシロキシメチル)オキセタン、3-エチル{[-3-(トリエトキシリル)プロポキシ]メチル}オキセタン、3-エチル-3-メタクリロキシメチルオキセタンなどが挙げられる。分子中に2個のオキセタンを有する化合物としては、ジ[1-エチル(3-オキセタニル)]メチルエーテル、1,4-ビス{[(3-エチル-3-オキセタニル)メトキシ]メチル}ベンゼン、4,4′-ビス[(3-エチル-3-オキセタニル)メトキシメチル]ビフェニル等が挙げられる。3~4個のオキセタン環を有する化合物としては、分枝状のポリアルキレンオキシ基やポリシロキシ基と3-アルキル-3-メチルオキセタンの反応物などが挙げられる。
上記オキセタン樹脂前駆体の硬化反応に用いられる硬化剤としては、特に限定されず、例えば、アミン化合物、アミン化合物から合成されるポリアミノアミド化合物等の化合物、3級アミン化合物、イミダゾール化合物、ヒドラジド化合物、メラミン化合物、酸無水物、フェノール化合物、熱潜在性カチオン重合触媒、光潜在性カチオン重合開始剤、ジシアンアミド及びその誘導体等が挙げられる。これらの硬化剤は、単独で用いられてもよく、2種以上が併用されてもよい。特に光硬化剤はエネルギーの有効活用の面から好適に利用される。ここで光硬化剤とは活性エネルギー線の照射によりカチオン重合を開始させる化合物であり、例えば、ジアリールヨードニウム塩、トリアリールスルホニウム塩等が挙げられる。
フェノール樹脂前駆体としては、フェノール、クレゾール等のフェノール類とホルムアルデヒド等を反応させノボラック等を合成し、これをヘキサメチレンテトラミン等で硬化させたもの等が挙げられる。
ユリア樹脂前駆体としては、尿素等とホルムアルデヒド等の重合反応物が挙げられる。
メラミン樹脂前駆体としては、メラミン等とホルムアルデヒド等の重合反応物が挙げられる。
メラミン樹脂前駆体としては、メラミン等とホルムアルデヒド等の重合反応物が挙げられる。
不飽和ポリエステル樹脂としては、不飽和多塩基酸等と多価アルコール等より得られる不飽和ポリエステルを、これと重合する単量体に溶解し硬化した樹脂等が挙げられる。
珪素樹脂前駆体としては、オルガノポリシロキサン類を主骨格とするものが挙げられる。
ポリウレタン樹脂前駆体としては、グリコール等のジオール類と、ジイソシアネートからなる重合反応物等が挙げられる。
ジアリルフタレート樹脂前駆体としては、ジアリルフタレートモノマー類とジアリルフタレートプレポリマー類からなる反応物が挙げられる。
これら熱硬化性樹脂の硬化剤、硬化触媒としては特に限定はないが、例えば、硬化剤としては多官能アミン、ポリアミド、酸無水物、フェノール樹脂等が挙げられ、硬化触媒としてはイミダゾール等が挙げられる。これらは単独又は2種以上の混合物として使用することができる。
<<光硬化性樹脂>>
本発明における光硬化性樹脂としては、特に限定されるものではないが、上述の熱硬化性樹脂の説明において例示したエポキシ樹脂、アクリル樹脂、オキセタン樹脂等の前駆体が挙げられる。
これら光硬化性樹脂の硬化剤としては特に限定はないが、例えばジアリールヨードニウム塩、トリアリールスルホニウム塩等が挙げられる。
本発明における光硬化性樹脂としては、特に限定されるものではないが、上述の熱硬化性樹脂の説明において例示したエポキシ樹脂、アクリル樹脂、オキセタン樹脂等の前駆体が挙げられる。
これら光硬化性樹脂の硬化剤としては特に限定はないが、例えばジアリールヨードニウム塩、トリアリールスルホニウム塩等が挙げられる。
<<その他の成分>>
今まで述べた熱硬化性樹脂及び光硬化性樹脂は、適宜、連鎖移動剤、紫外線吸収剤、充填剤、シランカップリング剤等と配合した硬化性組成物として用いられる。
今まで述べた熱硬化性樹脂及び光硬化性樹脂は、適宜、連鎖移動剤、紫外線吸収剤、充填剤、シランカップリング剤等と配合した硬化性組成物として用いられる。
(連鎖移動剤)
反応を均一に進行させる目的等で硬化性組成物は連鎖移動剤を含んでも良い。例えば、分子内に2個以上のチオール基を有する多官能メルカプタン化合物を用いることができ、これにより硬化物に適度な靱性を付与する事が出来る。メルカプタン化合物としては、例えばペンタエリスリトールテトラキス(β-チオプロピオネート)、トリメチロールプロパントリス(β-チオプロピオネート)、トリス[2-(β-チオプロピオニルオキシエトキシ)エチル]トリイソシアヌレートなどの1種又は2種以上を用いるのが好ましい。メルカプタン化合物を入れる場合は、ラジカル重合な可能化合物の合計に対して、通常30重量%以下の割合で含有させる。
反応を均一に進行させる目的等で硬化性組成物は連鎖移動剤を含んでも良い。例えば、分子内に2個以上のチオール基を有する多官能メルカプタン化合物を用いることができ、これにより硬化物に適度な靱性を付与する事が出来る。メルカプタン化合物としては、例えばペンタエリスリトールテトラキス(β-チオプロピオネート)、トリメチロールプロパントリス(β-チオプロピオネート)、トリス[2-(β-チオプロピオニルオキシエトキシ)エチル]トリイソシアヌレートなどの1種又は2種以上を用いるのが好ましい。メルカプタン化合物を入れる場合は、ラジカル重合な可能化合物の合計に対して、通常30重量%以下の割合で含有させる。
(紫外線吸収剤)
着色防止目的で硬化性組成物は紫外線吸収剤を含んでも良い。例えば、紫外線吸収剤としては、ベンゾフェノン系紫外線吸収剤及びベンゾトリアゾール系紫外線吸収剤から選ばれるものであり、その紫外線吸収剤は1種類を用いてもよいし、2種類以上を併用しても良い。紫外線吸収剤を入れる場合は、ラジカル重合な可能化合物の合計100重量部に対して、通常0.01~1重量部の割合で含有させる。
着色防止目的で硬化性組成物は紫外線吸収剤を含んでも良い。例えば、紫外線吸収剤としては、ベンゾフェノン系紫外線吸収剤及びベンゾトリアゾール系紫外線吸収剤から選ばれるものであり、その紫外線吸収剤は1種類を用いてもよいし、2種類以上を併用しても良い。紫外線吸収剤を入れる場合は、ラジカル重合な可能化合物の合計100重量部に対して、通常0.01~1重量部の割合で含有させる。
(セルロース以外の充填剤)
また、セルロース繊維以外の充填剤を含んでも良い。充填剤としては、例えば、無機粒子や有機高分子などが挙げられる。具体的には、シリカ粒子、チタニア粒子、アルミナ粒子などの無機粒子、ゼオネックス(日本ゼオン社)やアートン(JSR社)などの透明シクロオレフィンポリマー、ポリカーボネートやPMMAなどの汎用熱可塑性ポリマーなどが挙げられる。中でも、ナノサイズのシリカ粒子を用いると透明性を維持することができ好適である。また、紫外線硬化性モノマーと構造の似たポリマーを用いると高濃度までポリマーを溶解させることが可能であり、好適である。
また、セルロース繊維以外の充填剤を含んでも良い。充填剤としては、例えば、無機粒子や有機高分子などが挙げられる。具体的には、シリカ粒子、チタニア粒子、アルミナ粒子などの無機粒子、ゼオネックス(日本ゼオン社)やアートン(JSR社)などの透明シクロオレフィンポリマー、ポリカーボネートやPMMAなどの汎用熱可塑性ポリマーなどが挙げられる。中でも、ナノサイズのシリカ粒子を用いると透明性を維持することができ好適である。また、紫外線硬化性モノマーと構造の似たポリマーを用いると高濃度までポリマーを溶解させることが可能であり、好適である。
(シランカップリング剤)
また、シランカップリング剤を添加しても良い。シランカップリング剤としては、例えば、γ-((メタ)アクリロキシプロピル)トリメトキシシラン、γ-((メタ)アクリロキシプロピル)メチルジメトキシシラン、γ-((メタ)アクリロキシプロピル)メチルジエトキシシラン、γ-((メタ)アクリロキシプロピル)トリエトキシシラン、γ-(アクリロキシプロピル)トリメトキシシラン等は分子中に(メタ)アクリル基を有しており、他のモノマーと共重合することができるので好ましい。シランカップリング剤は、ラジカル重合な可能化合物の合計に対して通常0.1~50重量%、好ましくは1~20重量%となるように含有させる。この配合量が少な過ぎると、これを含有させる効果が十分に得られず、また、多過ぎると、硬化物の透明性などの光学特性が損なわれる恐れがある。
また、シランカップリング剤を添加しても良い。シランカップリング剤としては、例えば、γ-((メタ)アクリロキシプロピル)トリメトキシシラン、γ-((メタ)アクリロキシプロピル)メチルジメトキシシラン、γ-((メタ)アクリロキシプロピル)メチルジエトキシシラン、γ-((メタ)アクリロキシプロピル)トリエトキシシラン、γ-(アクリロキシプロピル)トリメトキシシラン等は分子中に(メタ)アクリル基を有しており、他のモノマーと共重合することができるので好ましい。シランカップリング剤は、ラジカル重合な可能化合物の合計に対して通常0.1~50重量%、好ましくは1~20重量%となるように含有させる。この配合量が少な過ぎると、これを含有させる効果が十分に得られず、また、多過ぎると、硬化物の透明性などの光学特性が損なわれる恐れがある。
<<硬化工程>>
本発明のセルロース繊維複合体を形成するための硬化性組成物は、公知の方法で重合硬化させることができる。
例えば、熱硬化、又は放射線硬化等が挙げられる。好ましくは放射線硬化である。放射線としては、赤外線、可視光線、紫外線、電子線等が挙げられるが、好ましくは光である。更に好ましくは波長が200nm~450nm程度の光であり、更に好ましくは波長が300~400nmの紫外線である。
本発明のセルロース繊維複合体を形成するための硬化性組成物は、公知の方法で重合硬化させることができる。
例えば、熱硬化、又は放射線硬化等が挙げられる。好ましくは放射線硬化である。放射線としては、赤外線、可視光線、紫外線、電子線等が挙げられるが、好ましくは光である。更に好ましくは波長が200nm~450nm程度の光であり、更に好ましくは波長が300~400nmの紫外線である。
具体的には、予め硬化性組成物に加熱によりラジカルを発生する熱重合開始剤を添加しておき、加熱して重合させる方法(以下「熱重合」という場合がある)、予め硬化性組成物に紫外線等の放射線によりラジカルを発生する光重合開始剤を添加しておき、放射線を照射して重合させる方法(以下「光重合」という場合がある)等、及び熱重合開始剤と光重合開始自在を併用して予め添加しておき、熱と光の組み合わせにより重合させる方法が挙げられ、本発明においては光重合がより好ましい。
光重合開始剤としては、通常、光ラジカル発生剤が用いられる。光ラジカル発生剤としては、この用途に用い得ることが知られている公知の化合物を用いることができる。例えば、ベンゾフェノン、ベンゾインメチルエーテル、ベンゾインプロピルエーテル、ジエトキシアセトフェノン、1-ヒドロキシシクロヘキシルフェニルケトン、2,6-ジメチルベンゾイルジフェニルホスフィンオキシド、2,4,6-トリメチルベンゾイルジフェニルホシフィンオキシド等が挙げられる。これらの中でも、2,4,6-トリメチルベンゾイルジフェニルホスフィンオキシドが好ましい。これらの光重合開始剤は単独で用いても、2種以上を併用してもよい。
光重合開始剤の成分量は、硬化性組成物中のラジカル重合可能な化合物の合計を100重量部としたとき、0.001重量部以上、好ましくは0.01重量部以上、更に好ましくは0.05重量部以上である。その上限は、通常1重量部以下、好ましくは0.5重量部以下、更に好ましくは0.1重量部以下である。光重合開始剤の添加量が多すぎると、重合が急激に進行し、得られる硬化物の複屈折を大きくするだけでなく色相も悪化する。例えば、開始剤の量を5重量部とした場合、開始剤の吸収により、紫外線の照射と反対側に光が到達できずに未硬化の部分が生ずる。また、黄色く着色し色相の劣化が著しい。一方、少なすぎると紫外線照射を行っても重合が十分に進行しないおそれがある。
また、熱重合開始剤を同時に含んでも良い。例えば、ハイドロパーオキサイド、ジアルキルパーオキサイド、パーオキシエステル、ジアシルパーオキサイド、パーオキシカーボネート、パーオキシケタール、ケトンパーオキサイド等が挙げられる。具体的にはベンゾイルパーオキシド、ジイソプロピルパーオキシカーボネート、t-ブチルパーオキシ(2-エチルヘキサノエート)ジクミルパーオキサイド、ジt-ブチルパーオキサイド、t-ブチルパーオキシベンゾエート、t-ブチルハイドロパーキサイド、ジイソプロピルベンゼンハイドロパーオキサイド、1,1,3,3-テトラメチルブチルハイドロパーオキサイド等を用いることができる。光照射時に熱重合が開始されると、重合を制御することが難しくなるので、これらの熱重合開始剤は好ましくは1分半減期温度が120℃以上であることがよい。これらの重合開始剤は単独で用いても、2種以上を併用してもよい。
硬化に際して照射する放射線の量は、光重合開始剤がラジカルを発生させる範囲であれば任意であるが、極端に少ない場合は重合が不完全となるため硬化物の耐熱性、機械特性が十分に発現されず、逆に極端に過剰な場合は硬化物の黄変等の光による劣化を生じるので、モノマーの組成及び光重合開始剤の種類、量に合わせて、波長300~450nmの紫外線を、好ましくは0.1J/cm2以上200J/cm2以下の範囲で照射する。更に好ましくは1J/cm2以上20J/cm2以下の範囲で照射する。放射線を複数回に分割して照射すると、より好ましい。すなわち1回目に全照射量の1/20~1/3程度を照射し、2回目以降に必要残量を照射すると、複屈折のより小さな硬化物が得られる。使用するランプの具体例としては、メタルハライドランプ、高圧水銀灯ランプ、紫外線LEDランプ等を挙げることができる。
重合をすみやかに完了させる目的で、光重合と熱重合を同時に行ってもよい。この場合には、放射線照射と同時に硬化性組成物を30℃以上300℃以下の範囲で加熱して硬化を行う。この場合、硬化性組成物には、重合を完結するために熱重合開始剤を添加してもよいが、大量に添加すると硬化物の複屈折の増大と色相の悪化をもたらすので、熱重合開始剤は、モノマー量の合計に対して通常0.1重量%以上2重量%以下、より好ましくは0.3重量%以上1重量%以下となるように用いる。
{積層構造体}
本発明で得られるセルロース繊維複合体は、本発明で得られるセルロースシートの層と、前述したセルロース以外の高分子よりなる平面構造体層との積層構造体であっても良く、また、本発明で得られるセルロースシートの層と、本発明で得られる高分子セルロース複合体の層との積層構造であっても良く、その積層数や積層構成には特に制限はない。
本発明で得られるセルロース繊維複合体は、本発明で得られるセルロースシートの層と、前述したセルロース以外の高分子よりなる平面構造体層との積層構造体であっても良く、また、本発明で得られるセルロースシートの層と、本発明で得られる高分子セルロース複合体の層との積層構造であっても良く、その積層数や積層構成には特に制限はない。
{無機膜}
本発明で得られるセルロース繊維複合体は、その用途に応じて、高分子セルロース複合体層に更に無機膜が積層されたものであっても良く、上述の積層構造体に更に無機膜が積層されたものであっても良い。
ここで用いられる無機膜は、セルロース繊維複合体の用途に応じて適宜決定され、例えば、白金、銀、アルミニウム、金、銅等の金属、シリコン、ITO、SiO2、SiN、SiOxNy、ZnO等、TFT等が挙げられ、その組み合わせや膜厚は任意に設計することができる。
本発明で得られるセルロース繊維複合体は、その用途に応じて、高分子セルロース複合体層に更に無機膜が積層されたものであっても良く、上述の積層構造体に更に無機膜が積層されたものであっても良い。
ここで用いられる無機膜は、セルロース繊維複合体の用途に応じて適宜決定され、例えば、白金、銀、アルミニウム、金、銅等の金属、シリコン、ITO、SiO2、SiN、SiOxNy、ZnO等、TFT等が挙げられ、その組み合わせや膜厚は任意に設計することができる。
{セルロース繊維複合体の特性ないし物性}
以下に本発明で得られるセルロース繊維複合体の好適な特性ないし物性について説明する。
<セルロース含有量>
本発明のセルロース繊維複合体中のセルロースの含有量は通常1重量%以上99重量%以下であり、セルロース以外の高分子の含有量が1重量%以上99重量%以下である。低線膨張性を発現するには、セルロースの含有量が1重量%以上、セルロース以外の高分子の含有量が99重量%以下であること必要である。透明性を発現するにはセルロースの含有量が99重量%以下、セルロース以外の高分子の含有量が1重量%以上であることが必要である。好ましい範囲はセルロースが5重量%以上90重量%以下であり、セルロース以外の高分子が10重量%以上95重量%以下であり、さらに好ましい範囲はセルロースが10重量%以上80重量%以下であり、セルロース以外の高分子が20重量%以上90重量%以下である。特に、セルロースの含有量が30重量%以上70重量%以下で、セルロース以外の高分子の含有量が30重量%以上70重量%以下であることが好ましい。
以下に本発明で得られるセルロース繊維複合体の好適な特性ないし物性について説明する。
<セルロース含有量>
本発明のセルロース繊維複合体中のセルロースの含有量は通常1重量%以上99重量%以下であり、セルロース以外の高分子の含有量が1重量%以上99重量%以下である。低線膨張性を発現するには、セルロースの含有量が1重量%以上、セルロース以外の高分子の含有量が99重量%以下であること必要である。透明性を発現するにはセルロースの含有量が99重量%以下、セルロース以外の高分子の含有量が1重量%以上であることが必要である。好ましい範囲はセルロースが5重量%以上90重量%以下であり、セルロース以外の高分子が10重量%以上95重量%以下であり、さらに好ましい範囲はセルロースが10重量%以上80重量%以下であり、セルロース以外の高分子が20重量%以上90重量%以下である。特に、セルロースの含有量が30重量%以上70重量%以下で、セルロース以外の高分子の含有量が30重量%以上70重量%以下であることが好ましい。
セルロース繊維複合体中のセルロース及びセルロース以外の高分子の含有量は、例えば、複合化前のセルロースの重量と複合化後のセルロースの重量より求めることができる。また、高分子が可溶な溶媒にセルロース複合体を浸漬して高分子のみを取り除き、残ったセルロースの重量から求めることもできる。その他、樹脂の比重から求める方法や、NMR、IRを用いて樹脂やセルロースの官能基を定量して求めることもできる。
<厚み>
本発明により得られるセルロース繊維複合体の厚みは、好ましくは10μm以上10cm以下であり、このような厚みとすることにより、構造材としての強度を保つことができる。セルロース繊維複合体の厚さはより好ましくは50μm以上1cm以下であり、さらに好ましくは80μm以上250μm以下である。
本発明により得られるセルロース繊維複合体の厚みは、好ましくは10μm以上10cm以下であり、このような厚みとすることにより、構造材としての強度を保つことができる。セルロース繊維複合体の厚さはより好ましくは50μm以上1cm以下であり、さらに好ましくは80μm以上250μm以下である。
なお、本発明により得られるセルロース繊維複合体は、例えば、このような厚さの膜状(フィルム状)又は板状であるが、平膜又は平板に限らず、曲面を有する膜状又は板状とすることもできる。また、その他の異形形状であっても良い。また、厚さは必ずしも均一である必要はなく、部分的に異なっていても良い。
<着色>
本発明により得られるセルロース繊維複合体は、着色が小さい。
<着色>
本発明により得られるセルロース繊維複合体は、着色が小さい。
セルロースは、特に木質由来の原料を用いることで黄色味がつく場合がある。これは、セルロース自身の着色の場合と、精製度合いによって残ったセルロース以外の物質が着色する場合がある。一般的に、セルロースのみの段階では着色しないが、高分子と複合化する際の加熱によって着色することがある。本発明により得られるセルロース繊維及び高分子セルロース複合体は、加熱の工程が入っても着色が小さい。本発明により得られる高分子セルロース複合体の着色を示すYIは20以下であることが好ましく、15以下であることがより好ましく、10以下であることがさらに好ましい。YIは例えば、スガ試験機製カラーコンピューターを用いて測定することができる。
<ヘーズ>
本発明により得られるセルロース繊維複合体は、透明性の高い、すなわちヘーズの小さい高分子セルロース複合体とすることができる。各種透明材料として用いる場合、この高分子セルロース複合体のヘーズ値は、好ましくは20以下、より好ましくは10以下であり、特にこの値は3以下、中でも1以下であることが好ましい。
本発明により得られるセルロース繊維複合体は、透明性の高い、すなわちヘーズの小さい高分子セルロース複合体とすることができる。各種透明材料として用いる場合、この高分子セルロース複合体のヘーズ値は、好ましくは20以下、より好ましくは10以下であり、特にこの値は3以下、中でも1以下であることが好ましい。
<全光線透過率>
本発明により得られるセルロース繊維複合体は、透明性の高い、すなわちヘーズの小さいセルロース繊維複合体とすることができる。各種透明材料として用いる場合、このセルロース繊維複合体は、JIS規格K7105に準拠してその厚み方向に測定された全光線透過率が60%以上、更には70%以上、特に80%以上、とりわけ90%以上であることが好ましい。この全光線透過率が60%未満であると半透明又は不透明となり、透明性が要求される用途への使用が困難となる場合がある。全光線透過率は例えば、厚み10~100μmのセルロース複合体について、スガ試験機製ヘーズメータを用いて測定することができ、C光の値を用いる。
本発明により得られるセルロース繊維複合体は、透明性の高い、すなわちヘーズの小さいセルロース繊維複合体とすることができる。各種透明材料として用いる場合、このセルロース繊維複合体は、JIS規格K7105に準拠してその厚み方向に測定された全光線透過率が60%以上、更には70%以上、特に80%以上、とりわけ90%以上であることが好ましい。この全光線透過率が60%未満であると半透明又は不透明となり、透明性が要求される用途への使用が困難となる場合がある。全光線透過率は例えば、厚み10~100μmのセルロース複合体について、スガ試験機製ヘーズメータを用いて測定することができ、C光の値を用いる。
<線膨張係数>
本発明により得られるセルロース繊維複合体は、線膨張係数(1Kあたりの伸び率)の低いセルロース繊維を用いることにより線膨張係数の低いセルロース繊維複合体とすることができる。このセルロース複合体の線膨張係数は1~50ppm/Kであることが好ましく、1~30ppm/Kであることがより好ましく、1~20ppm/Kであることが特に好ましい。
本発明により得られるセルロース繊維複合体は、線膨張係数(1Kあたりの伸び率)の低いセルロース繊維を用いることにより線膨張係数の低いセルロース繊維複合体とすることができる。このセルロース複合体の線膨張係数は1~50ppm/Kであることが好ましく、1~30ppm/Kであることがより好ましく、1~20ppm/Kであることが特に好ましい。
即ち、例えば、基板用途においては、無機の薄膜トランジスタの線膨張係数が15ppm/K程度であるため、セルロース繊維複合体の線膨張係数が50ppm/Kを超えると無機膜との積層複合化の際に、二層の線膨張率差が大きくなり、クラック等が発生する。従って、セルロース複合体の線膨張係数は、特に1~20ppm/Kであることが好ましい。
なお、線膨張係数は、後述の実施例の項に記載される方法により測定される。
<引張強度>
本発明により得られるセルロース繊維複合体の引張強度は、好ましくは40MPa以上であり、より好ましくは100MPa以上である。引張強度が40MPaより低いと、十分な強度が得られず、構造材料等、力の加わる用途への使用に影響を与えることがある。
<引張強度>
本発明により得られるセルロース繊維複合体の引張強度は、好ましくは40MPa以上であり、より好ましくは100MPa以上である。引張強度が40MPaより低いと、十分な強度が得られず、構造材料等、力の加わる用途への使用に影響を与えることがある。
<引張弾性率>
本発明により得られるセルロース繊維複合体の引張弾性率は、好ましくは0.2~100GPaであり、より好ましくは1~50GPa、さらに好ましくは5.0~30GPaである。引張弾性率が0.2GPaより低いと、十分な強度が得られず、構造材料等、力の加わる用途への使用に影響を与えることがある。
本発明により得られるセルロース繊維複合体の引張弾性率は、好ましくは0.2~100GPaであり、より好ましくは1~50GPa、さらに好ましくは5.0~30GPaである。引張弾性率が0.2GPaより低いと、十分な強度が得られず、構造材料等、力の加わる用途への使用に影響を与えることがある。
<セルロースの結晶性>
本発明により得られるセルロース繊維複合体に含まれる修飾セルロース繊維は、セルロースI型結晶構造を有する。
修飾セルロース繊維がI型結晶構造であることは、その広角X線回折像測定により得られる回折プロファイル(広角X線回折像)において、走査角2θ=14~17°付近と2θ=22~23°付近の二つの位置に典型的なピークをもつことから同定することができる。
本発明により得られるセルロース繊維複合体に含まれる修飾セルロース繊維は、セルロースI型結晶構造を有する。
修飾セルロース繊維がI型結晶構造であることは、その広角X線回折像測定により得られる回折プロファイル(広角X線回折像)において、走査角2θ=14~17°付近と2θ=22~23°付近の二つの位置に典型的なピークをもつことから同定することができる。
一方、セルロースI型結晶構造が壊れた場合、2θ=18~19°付近に非晶質に由来するハローが観測されるようになる。I型以外の結晶構造を有する場合にもこの近傍にピークを示すようになる。
本発明のセルロース繊維複合体に含まれるセルロースは、「走査角20~24度におけるセルロースI型結晶構造由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」(非晶質等のI型以外の構造を反映)の比が0.8以下である。好ましくは0.7以下、特に0.6以下、中でも0.5以下である。該強度比は低いほどよいが、実用的には0.01以上程度である。
セルロース繊維複合体の場合、用いるマトリクス材料によっては、マトリクス由来の信号が上記に重なることも考えられるが、マトリクス材料単体でのプロファイルによりバックグラウンド差し引きを行う、等の方法でセルロースシート同様の評価が可能である。
本発明のセルロース繊維複合体に含まれるセルロースは、「走査角20~24度におけるセルロースI型結晶構造由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」(非晶質等のI型以外の構造を反映)の比が0.8以下である。好ましくは0.7以下、特に0.6以下、中でも0.5以下である。該強度比は低いほどよいが、実用的には0.01以上程度である。
セルロース繊維複合体の場合、用いるマトリクス材料によっては、マトリクス由来の信号が上記に重なることも考えられるが、マトリクス材料単体でのプロファイルによりバックグラウンド差し引きを行う、等の方法でセルロースシート同様の評価が可能である。
[用途]
本発明により得られるセルロース繊維複合体は、透明性が高く、高強度、低吸水性、高透明性、低着色およびヘーズが小さく光学特性に優れるため、液晶ディスプレイ、プラズマディスプレイ、有機ELディスプレイ、フィールドエミッションディスプレイ、リアプロジェクションテレビ等のディスプレイや基板やパネルとして好適である。また、シリコン系太陽電池、色素増感太陽電池などの太陽電池用基板に好適である。基板としては、バリア膜、ITO、TFT等と積層してもよい。また、自動車用の窓材、鉄道車両用の窓材、住宅用の窓材、オフィスや工場などの窓材などに好適に使われる。窓材としては、必要に応じてフッ素皮膜、ハードコート膜等の膜や耐衝撃性、耐光性の素材を積層してもよい。
本発明により得られるセルロース繊維複合体は、透明性が高く、高強度、低吸水性、高透明性、低着色およびヘーズが小さく光学特性に優れるため、液晶ディスプレイ、プラズマディスプレイ、有機ELディスプレイ、フィールドエミッションディスプレイ、リアプロジェクションテレビ等のディスプレイや基板やパネルとして好適である。また、シリコン系太陽電池、色素増感太陽電池などの太陽電池用基板に好適である。基板としては、バリア膜、ITO、TFT等と積層してもよい。また、自動車用の窓材、鉄道車両用の窓材、住宅用の窓材、オフィスや工場などの窓材などに好適に使われる。窓材としては、必要に応じてフッ素皮膜、ハードコート膜等の膜や耐衝撃性、耐光性の素材を積層してもよい。
また、低線膨張係数、高弾性、高強度等の特性を生かして透明材料用途以外の構造体としても用いることができる。特に、内装材、外板、バンパー等の自動車材料やパソコンの筐体、家電部品、包装用資材、建築資材、土木資材、水産資材、その他、工業用資材等として好適に用いられる。
以下、製造例、実施例および比較例によって、本発明をさらに具体的に説明するが、本発明はその要旨を超えない限り、以下の実施例により限定されるものではない。
尚、本発明により得られるセルロース繊維の熱分解温度、セルロース繊維複合体のセルロース含量、YI、ヘーズ、線膨張係数及び弾性率の測定方法は以下の通りである。
〔修飾セルロース繊維の化学修飾率〕
乾燥した修飾セルロース繊維0.05gを精秤しこれにメタノール6ml、蒸留水2mlを添加し、60~70℃で30分攪拌した後、0.05N水酸化ナトリウム水溶液10mlを添加し、60~70℃で15分攪拌しさらに室温で一日攪拌した。これをフェノールフタレインを用いて0.02N塩酸水溶液で滴定し、下記式より化学修飾率を計算した。
尚、本発明により得られるセルロース繊維の熱分解温度、セルロース繊維複合体のセルロース含量、YI、ヘーズ、線膨張係数及び弾性率の測定方法は以下の通りである。
〔修飾セルロース繊維の化学修飾率〕
乾燥した修飾セルロース繊維0.05gを精秤しこれにメタノール6ml、蒸留水2mlを添加し、60~70℃で30分攪拌した後、0.05N水酸化ナトリウム水溶液10mlを添加し、60~70℃で15分攪拌しさらに室温で一日攪拌した。これをフェノールフタレインを用いて0.02N塩酸水溶液で滴定し、下記式より化学修飾率を計算した。
ここで、滴定に要した0.02N塩酸水溶液の量Z(ml)から、化学修飾により導入された置換基のモル数Qは、下記式で求められる。
Q(mol)=0.05(N)×10(ml)/1000 -0.02(N)×Z(ml)/1000
この置換基のモル数Qと、化学修飾率X(mol%)との関係は、以下の式で算出される(セルロース=(C6O5H10)n=(162.14)n,繰り返し単位1個当たりの水酸基数=3,OHの分子量=17)。なお、以下において、Tは置換基の分子量である。
Q(mol)=0.05(N)×10(ml)/1000 -0.02(N)×Z(ml)/1000
この置換基のモル数Qと、化学修飾率X(mol%)との関係は、以下の式で算出される(セルロース=(C6O5H10)n=(162.14)n,繰り返し単位1個当たりの水酸基数=3,OHの分子量=17)。なお、以下において、Tは置換基の分子量である。

これを解いていくと、以下の通りである。

〔セルロース繊維の熱分解温度〕
セルロースシートを温度23℃、湿度50%に48時間以上調湿し、これをTG-DTA(示差熱熱重量同時測定装置)を用いて窒素下、室温から600℃まで10℃/分で昇温していったときのTGから求めた接線の交点を熱分解温度とした。
〔平均繊維径〕
セルロース繊維の繊維径は、光学顕微鏡または、SEMやTEM等で観察することにより計測して求めた。ランダムに抽出した12点中最大と最小を除いた10点の平均を平均繊維径とした。
セルロースシートを温度23℃、湿度50%に48時間以上調湿し、これをTG-DTA(示差熱熱重量同時測定装置)を用いて窒素下、室温から600℃まで10℃/分で昇温していったときのTGから求めた接線の交点を熱分解温度とした。
〔平均繊維径〕
セルロース繊維の繊維径は、光学顕微鏡または、SEMやTEM等で観察することにより計測して求めた。ランダムに抽出した12点中最大と最小を除いた10点の平均を平均繊維径とした。
〔セルロースの広角X線回折像測定〕
セルロースシートをX線発生装置(PANanalytical社製「PW1700」)を用い、ターゲットCu/Kα線、モノクロメーター、電圧40kV、電流30mA、走査角(2θ)3.0~50.0°、ステップ角0.05°の測定条件で測定を行った。
セルロースシートをX線発生装置(PANanalytical社製「PW1700」)を用い、ターゲットCu/Kα線、モノクロメーター、電圧40kV、電流30mA、走査角(2θ)3.0~50.0°、ステップ角0.05°の測定条件で測定を行った。
(結晶性評価)
「走査角20~24度におけるセルロースI型結晶構造由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」(非晶質等のI型以外の構造を反映)の比を求めた。
「走査角20~24度におけるセルロースI型結晶構造由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」(非晶質等のI型以外の構造を反映)の比を求めた。
〔セルロースシートの加熱後のYI値〕
セルロースシートを190℃の真空オーブン中で4時間加熱した後、スガ試験機製カラーコンピュータを用いて反射モードでYI値を測定した。
セルロースシートを190℃の真空オーブン中で4時間加熱した後、スガ試験機製カラーコンピュータを用いて反射モードでYI値を測定した。
〔セルロースシートの弾性率と加熱前後の強度比〕
セルロースシートを幅8mm×長さ50mmにカットした。これを、オリエンテック社製引張試験機STA-1225を用いて引張試験を行った。チャック間距離は25mm、試験速度は 2mm/min.で行い、歪-応力曲線から最大強度を算出した。また、同様のセルロースシートを190℃の真空オーブン中で4時間加熱した後、同様に引張試験を実施し、弾性率と最大強度を算出した。引張強度の元に対する加熱後の比率を求めた。測定に用いるセルロースシートは空隙のあるものであるが、算出は単純にサンプルの厚みと幅を用いて行い、空隙率による補正等は行っていない。
〔セルロース繊維複合体中のセルロース含量〕
複合化に用いたセルロースシートの重量と、セルロース複合体の重量からセルロース含量(重量%)を求めた。
セルロースシートを幅8mm×長さ50mmにカットした。これを、オリエンテック社製引張試験機STA-1225を用いて引張試験を行った。チャック間距離は25mm、試験速度は 2mm/min.で行い、歪-応力曲線から最大強度を算出した。また、同様のセルロースシートを190℃の真空オーブン中で4時間加熱した後、同様に引張試験を実施し、弾性率と最大強度を算出した。引張強度の元に対する加熱後の比率を求めた。測定に用いるセルロースシートは空隙のあるものであるが、算出は単純にサンプルの厚みと幅を用いて行い、空隙率による補正等は行っていない。
〔セルロース繊維複合体中のセルロース含量〕
複合化に用いたセルロースシートの重量と、セルロース複合体の重量からセルロース含量(重量%)を求めた。
〔セルロース繊維複合体の厚み〕
膜厚計(PEACOK製のPDN-20)を用いて、セルロース繊維複合体の種々な位置について10点の測定を行い、その平均値を採用した。
〔セルロース繊維複合体のYI値〕
スガ試験機製カラーコンピュータを用いてYI値を測定した。
膜厚計(PEACOK製のPDN-20)を用いて、セルロース繊維複合体の種々な位置について10点の測定を行い、その平均値を採用した。
〔セルロース繊維複合体のYI値〕
スガ試験機製カラーコンピュータを用いてYI値を測定した。
〔セルロース繊維複合体のヘーズ〕
スガ試験機製ヘーズメータを用いてC光によるヘーズ値を測定した。
〔セルロース繊維複合体の全光線透過率〕
得られた複合体について、JIS規格K7105に準拠し、スガ試験機製ヘーズメータを用いてC光による全光線透過率を測定した。
スガ試験機製ヘーズメータを用いてC光によるヘーズ値を測定した。
〔セルロース繊維複合体の全光線透過率〕
得られた複合体について、JIS規格K7105に準拠し、スガ試験機製ヘーズメータを用いてC光による全光線透過率を測定した。
〔セルロース繊維複合体の線膨張係数〕
セルロース繊維複合体をレーザーカッターにより、3mm幅×40mm長にカットした。これをSII製TMA6100を用いて引張モードでチャック間20mm、荷重10g、窒素雰囲気下、室温から180℃まで5℃/min.で昇温し、次いで180℃から25℃まで5℃/min.で降温し、更に25℃から180℃まで5℃/min.で昇温した際の2度目の昇温時の60℃から100℃の測定値から線膨張係数を求めた。
セルロース繊維複合体をレーザーカッターにより、3mm幅×40mm長にカットした。これをSII製TMA6100を用いて引張モードでチャック間20mm、荷重10g、窒素雰囲気下、室温から180℃まで5℃/min.で昇温し、次いで180℃から25℃まで5℃/min.で降温し、更に25℃から180℃まで5℃/min.で昇温した際の2度目の昇温時の60℃から100℃の測定値から線膨張係数を求めた。
〔セルロース繊維複合体中の引張弾性率〕
セルロース繊維複合体をレーザーカッターにより、10mm幅×40mm長にカットした。これを、SII社製DMS6100を用いて引張モードでDMA(動的粘弾性)測定を行い、周波数10Hz、23℃における貯蔵弾性率E’(単位;GPa)を測定した。
セルロース繊維複合体をレーザーカッターにより、10mm幅×40mm長にカットした。これを、SII社製DMS6100を用いて引張モードでDMA(動的粘弾性)測定を行い、周波数10Hz、23℃における貯蔵弾性率E’(単位;GPa)を測定した。
<製造例1>
木粉((株)宮下木材、米松100)を炭酸ナトリウム2重量%水溶液で80℃にて6時間脱脂した。これを脱塩水で洗浄した後、亜塩素酸ナトリウムを用いて酢酸酸性下、80℃にて5.5時間脱リグニンした。脱塩水洗浄した後にさらに水酸化カリウム5重量%水溶液に16時間浸漬して脱ヘミセルロースした。脱塩水洗浄した。
木粉((株)宮下木材、米松100)を炭酸ナトリウム2重量%水溶液で80℃にて6時間脱脂した。これを脱塩水で洗浄した後、亜塩素酸ナトリウムを用いて酢酸酸性下、80℃にて5.5時間脱リグニンした。脱塩水洗浄した後にさらに水酸化カリウム5重量%水溶液に16時間浸漬して脱ヘミセルロースした。脱塩水洗浄した。
<実施例1>
製造例1で得られたセルロースを固形分で10g分量りとり、水分を酢酸に置換した。これを酢酸300ml、酢酸ナトリウム10g、ベンゾイルクロリド21gを添加し攪拌しながら80℃5hr反応させた。その後、反応液を濾過し、アセトンで洗浄した後、さらに大量の水で中性になるまで洗浄した。
製造例1で得られたセルロースを固形分で10g分量りとり、水分を酢酸に置換した。これを酢酸300ml、酢酸ナトリウム10g、ベンゾイルクロリド21gを添加し攪拌しながら80℃5hr反応させた。その後、反応液を濾過し、アセトンで洗浄した後、さらに大量の水で中性になるまで洗浄した。
このベンゾイル化セルロースの化学修飾率は29mol%であった。また、TG-DTAより算出した熱分解温度は354℃であった。
得られた修飾セルロースを、0.5重量%の水懸濁液とし、増幸産業株式会社の石臼式摩砕機スーパーマスコロイダーMKCA6-2を用い、GC6-80の石臼を用いて、ギャップ間を80μmにして回転数1500rpmにて、原料投入口から、投入した。摩砕機を通った処理済みセルロース分散液を再び原料投入口に投入し、合計2回摩砕機を通した。その後、超高圧ホモジナイザー(スギノマシン製アルティマイザー)に245MPaで10回通した。
得られた修飾セルロースを、0.5重量%の水懸濁液とし、増幸産業株式会社の石臼式摩砕機スーパーマスコロイダーMKCA6-2を用い、GC6-80の石臼を用いて、ギャップ間を80μmにして回転数1500rpmにて、原料投入口から、投入した。摩砕機を通った処理済みセルロース分散液を再び原料投入口に投入し、合計2回摩砕機を通した。その後、超高圧ホモジナイザー(スギノマシン製アルティマイザー)に245MPaで10回通した。
このセルロース繊維分散液を0.13重量%濃度に水で希釈し、孔径1μmのPTFEを用いた90mm径の濾過器に150g投入し、固形分が約5重量%になったところで2-プロパノールを投入して置換した。濾過終了まで38分であった。その後、120℃、0.14MPaで5分間プレス乾燥して白色のセルロースシートを得た(秤量約40g/m2)。このセルロースシートの表面のSEM観察より算出した平均繊維径は、18nmであった。また、広角X線回折像から、セルロースI型結晶構造であることが確認された。
このセルロースシートを、ビス(メタクリロイルオキシメチル)トリシクロ[5.2.1.02,6]デカン96重量部、ペンタエリスリトールテトラキス(β-チオプロピオネート)6重量部、2,4,6-トリメチルベンゾイルジフェニルフォスフィンオキサイド(BASF社製ルシリンTPO)0.05重量部、ベンゾフェノン0.05重量部を混合した溶液に含浸させ、減圧下一晩おいた。これを2枚のガラス板にはさみ、無電極水銀ランプ(フュージョンUVシステムズ社製「Dバルブ」)を用いて、放射照度400mW/cm2の下を、ライン速度7m/minで照射した。このときの放射照射量は0.12J/cm2であった。この操作をガラス面を反転して2回行った。紫外線照射後のガラス面の温度は25℃であった。
次いで、放射照度1900mW/cm2の下をライン速度2m/minで照射した。このときの放射照射量は2.7J/cm2であった。この操作をガラス面を反転して8回行った。紫外線照射後のガラス面の温度は44℃であった。放射照射量は21.8J/cm2であった。紫外線照射終了後、ガラス板よりはずし、190℃の真空オーブン中で4時間加熱して複合材料を得た。なお、紫外線の放射照度は、オーク製作所製紫外線照度計「UV-M02」で、アタッチメント「UV-35」を用いて、320~390nmの紫外線の照度を23℃で測定した。
このセルロース繊維複合体の物性をセルロースシートの物性と共に表1に示す。
<実施例2>
製造例1で得られたセルロースを固形分で10g分量りとり、水分を酢酸に置換した。これを酢酸300ml、酢酸ナトリウム12g、ベンゾイルクロリド25gを添加し攪拌しながら80℃5hr反応させた。その後、反応液を濾過し、アセトンで洗浄した後、さらに大量の水で中性になるまで洗浄した。
製造例1で得られたセルロースを固形分で10g分量りとり、水分を酢酸に置換した。これを酢酸300ml、酢酸ナトリウム12g、ベンゾイルクロリド25gを添加し攪拌しながら80℃5hr反応させた。その後、反応液を濾過し、アセトンで洗浄した後、さらに大量の水で中性になるまで洗浄した。
このベンゾイル化セルロースの化学修飾率は39mol%であった。また、TG-DTAより算出した熱分解温度は350℃であった。
得られた修飾セルロースを、0.5重量%の水懸濁液とし、増幸産業株式会社の石臼式摩砕機スーパーマスコロイダーMKCA6-2を用い、GC6-80の石臼を用いて、ギャップ間を80μmにして回転数1500rpmにて、原料投入口から、投入した。摩砕機を通った処理済みセルロース分散液を再び原料投入口に投入し、合計2回摩砕機を通した。その後、超高圧ホモジナイザー(スギノマシン製アルティマイザー)に245MPaで10回通した。
得られた修飾セルロースを、0.5重量%の水懸濁液とし、増幸産業株式会社の石臼式摩砕機スーパーマスコロイダーMKCA6-2を用い、GC6-80の石臼を用いて、ギャップ間を80μmにして回転数1500rpmにて、原料投入口から、投入した。摩砕機を通った処理済みセルロース分散液を再び原料投入口に投入し、合計2回摩砕機を通した。その後、超高圧ホモジナイザー(スギノマシン製アルティマイザー)に245MPaで10回通した。
このセルロース繊維分散液を0.13重量%濃度に水で希釈し、孔径1μmのPTFEを用いた90mm径の濾過器に150g投入し、固形分が約5重量%になったところで2-プロパノールを投入して置換した。濾過終了まで20分であった。その後、120℃、0.14MPaで5分間プレス乾燥して白色のセルロースシートを得た(秤量約40g/m2)。このセルロースシートの表面のSEM観察より算出した平均繊維径は、15nmであった。 また、広角X線回折像から、セルロースI型結晶構造であることが確認された。
このセルロースシートを用いて実施例1と同様にしてセルロース繊維複合体を得た。このセルロース繊維複合体の物性をセルロースシートの物性と共に表1に示す。
<実施例3>
実施例1で得られたセルロース分散液を、更にSMT社製超音波ホモジナイザーUH-600S(周波数20kHz、実効出力密度22W/cm2)を用いて超音波処理を行った。
実施例1で得られたセルロース分散液を、更にSMT社製超音波ホモジナイザーUH-600S(周波数20kHz、実効出力密度22W/cm2)を用いて超音波処理を行った。
36mmφのストレート型チップ(チタン合金製)を用い、アウトプットボリウム8でチューニングを行い、最適なチューニング位置で30分間超音波処理を行った。セルロース分散液は処理容器の外側から5℃の冷水で冷却し、また、マグネティックスターラーにて撹拌しながら処理を行った。
この超音波処理分散液を、18000rpm(38900G)にて10分遠心分離を行い上澄みを得た。この上澄みのセルロース繊維分散液を用いて、実施例1と同様にして、セルロースシートを得た。濾過終了まで53分であった。その後、120℃、0.14MPaで5分間プレス乾燥して白色のセルロースシートを得た(秤量約40g/m2)。このセルロースシートの表面のSEM観察より算出した平均繊維径は、14nmであった。また、広角X線回折像から、セルロースI型結晶構造であることが確認された。
この超音波処理分散液を、18000rpm(38900G)にて10分遠心分離を行い上澄みを得た。この上澄みのセルロース繊維分散液を用いて、実施例1と同様にして、セルロースシートを得た。濾過終了まで53分であった。その後、120℃、0.14MPaで5分間プレス乾燥して白色のセルロースシートを得た(秤量約40g/m2)。このセルロースシートの表面のSEM観察より算出した平均繊維径は、14nmであった。また、広角X線回折像から、セルロースI型結晶構造であることが確認された。
このセルロースシートを用いて実施例1と同様にしてセルロース繊維複合体を得た。
このセルロース繊維複合体の物性をセルロースシートの物性と共に表1に示す。
このセルロース繊維複合体の物性をセルロースシートの物性と共に表1に示す。
<実施例4>
実施例2で得られたセルロース分散液を用いて、実施例3と同様にして超音波処理を行った。この超音波処理分散液を、18000rpm(38900G)にて10分遠心分離を行い上澄みを得た。この上澄みのセルロース繊維分散液を用いて、実施例1と同様にして、セルロースシートを得た。濾過終了まで51分であった。その後、120℃、0.14MPaで5分間プレス乾燥して白色のセルロースシートを得た(秤量約40g/m2)。このセルロースシートの表面のSEM観察より算出した平均繊維径は、12nmであった。また、広角X線回折像から、セルロースI型結晶構造であることが確認された。
実施例2で得られたセルロース分散液を用いて、実施例3と同様にして超音波処理を行った。この超音波処理分散液を、18000rpm(38900G)にて10分遠心分離を行い上澄みを得た。この上澄みのセルロース繊維分散液を用いて、実施例1と同様にして、セルロースシートを得た。濾過終了まで51分であった。その後、120℃、0.14MPaで5分間プレス乾燥して白色のセルロースシートを得た(秤量約40g/m2)。このセルロースシートの表面のSEM観察より算出した平均繊維径は、12nmであった。また、広角X線回折像から、セルロースI型結晶構造であることが確認された。
このセルロースシートを用いて実施例1と同様にしてセルロース繊維複合体を得た。
このセルロース繊維複合体の物性をセルロースシートの物性と共に表1に示す。
このセルロース繊維複合体の物性をセルロースシートの物性と共に表1に示す。
<実施例5>
製造例1で得られたセルロースを固形分で10g分量りとり、水分を酢酸に置換した。これを酢酸300ml、酢酸ナトリウム5g、ベンゾイルクロリド12.5gを添加し攪拌しながら80℃5hr反応させた。その後、反応液を濾過し、アセトンで洗浄した後、さらに大量の水で中性になるまで洗浄した。
製造例1で得られたセルロースを固形分で10g分量りとり、水分を酢酸に置換した。これを酢酸300ml、酢酸ナトリウム5g、ベンゾイルクロリド12.5gを添加し攪拌しながら80℃5hr反応させた。その後、反応液を濾過し、アセトンで洗浄した後、さらに大量の水で中性になるまで洗浄した。
このベンゾイル化セルロースの化学修飾率は11mol%であった。また、TG-DTAより算出した熱分解温度は339℃であった。
得られた修飾セルロースを、0.5重量%の水懸濁液とし、増幸産業株式会社の石臼式摩砕機スーパーマスコロイダーMKCA6-2を用い、GC6-80の石臼を用いて、ギャップ間を80μmにして回転数1500rpmにて、原料投入口から、投入した。摩砕機を通った処理済みセルロース分散液を再び原料投入口に投入し、合計2回摩砕機を通した。その後、超高圧ホモジナイザー(スギノマシン製アルティマイザー)に245MPaで10回通した。
得られた修飾セルロースを、0.5重量%の水懸濁液とし、増幸産業株式会社の石臼式摩砕機スーパーマスコロイダーMKCA6-2を用い、GC6-80の石臼を用いて、ギャップ間を80μmにして回転数1500rpmにて、原料投入口から、投入した。摩砕機を通った処理済みセルロース分散液を再び原料投入口に投入し、合計2回摩砕機を通した。その後、超高圧ホモジナイザー(スギノマシン製アルティマイザー)に245MPaで10回通した。
このセルロース分散液を0.13重量%濃度に水で希釈し、孔径1μmのPTFEを用いた90mm径の濾過器に150g投入し、固形分が約5重量%になったところで2-プロパノールを投入して置換した。濾過終了まで102分であった。その後、120℃、0.14MPaで5分間プレス乾燥して白色のセルロースシートを得た(秤量約40g/m2)。このセルロースシートの表面のSEM観察より算出した平均繊維径は、18nmであった。また、広角X線回折像から、セルロースI型結晶構造であることが確認された。
このセルロースシートを用いて実施例1と同様にしてセルロース繊維複合体を得た。
このセルロース繊維複合体の物性をセルロースシートの物性と共に表1に示す。
このセルロース繊維複合体の物性をセルロースシートの物性と共に表1に示す。
<比較例1>
製造例1で得られたセルロースを、0.5重量%の水懸濁液とし、増幸産業株式会社の石臼式摩砕機スーパーマスコロイダーMKCA6-2を用い、GC6-80の石臼を用いて、ギャップ間を80μmにして回転数1500rpmにて、原料投入口から、投入した。摩砕機を通った処理済みセルロース分散液を再び原料投入口に投入し、合計2回摩砕機を通した。その後、超高圧ホモジナイザー(スギノマシン製アルティマイザー)に245MPaで10回通した。
製造例1で得られたセルロースを、0.5重量%の水懸濁液とし、増幸産業株式会社の石臼式摩砕機スーパーマスコロイダーMKCA6-2を用い、GC6-80の石臼を用いて、ギャップ間を80μmにして回転数1500rpmにて、原料投入口から、投入した。摩砕機を通った処理済みセルロース分散液を再び原料投入口に投入し、合計2回摩砕機を通した。その後、超高圧ホモジナイザー(スギノマシン製アルティマイザー)に245MPaで10回通した。
TG-DTAより算出した熱分解温度は296℃であった。
このセルロース繊維分散液を0.13重量%濃度に水で希釈し、孔径1μmのPTFEを用いた90mm径の濾過器に150g投入し、固形分が約5重量%になったところで2-プロパノールを投入して置換した。濾過終了まで149分であった。その後、120℃、0.14MPaで5分間プレス乾燥して白色のセルロースシートを得た(秤量約40g/m2)。このセルロースシートの表面のSEM観察より算出した平均繊維径は、25nmであった。また、広角X線回折像から、セルロースI型結晶構造であることが確認された。
このセルロース繊維分散液を0.13重量%濃度に水で希釈し、孔径1μmのPTFEを用いた90mm径の濾過器に150g投入し、固形分が約5重量%になったところで2-プロパノールを投入して置換した。濾過終了まで149分であった。その後、120℃、0.14MPaで5分間プレス乾燥して白色のセルロースシートを得た(秤量約40g/m2)。このセルロースシートの表面のSEM観察より算出した平均繊維径は、25nmであった。また、広角X線回折像から、セルロースI型結晶構造であることが確認された。
このセルロースシートを用いて実施例1と同様にしてセルロース繊維複合体を得た。
このセルロース繊維複合体の物性をセルロースシートの物性と共に表1に示す。
このセルロース繊維複合体の物性をセルロースシートの物性と共に表1に示す。
<比較例2>(日本国特開2007-51266号公報 実施例6の追試)
比較例1と同様にして、セルロースシートを得た。このセルロースシートを無水酢酸:酢酸=9:1(体積比)の反応液の入ったシャーレに浸し、室温下、30分、デシケーター内で減圧下、反応液をシート内部まで含浸させた。常圧に戻し、窒素雰囲気下で5日間、室温、暗所に放置した。反応後、セルロースシートを取り出し、メタノール、脱塩水の順で洗浄し、最後に2-プロパノールで置換した後、120℃、0.14MPaで5分間プレス乾燥して白色のセルロースシートを得た。このセルロースシートの表面のSEM観察より算出した平均繊維径は、25nmであった。
比較例1と同様にして、セルロースシートを得た。このセルロースシートを無水酢酸:酢酸=9:1(体積比)の反応液の入ったシャーレに浸し、室温下、30分、デシケーター内で減圧下、反応液をシート内部まで含浸させた。常圧に戻し、窒素雰囲気下で5日間、室温、暗所に放置した。反応後、セルロースシートを取り出し、メタノール、脱塩水の順で洗浄し、最後に2-プロパノールで置換した後、120℃、0.14MPaで5分間プレス乾燥して白色のセルロースシートを得た。このセルロースシートの表面のSEM観察より算出した平均繊維径は、25nmであった。
このアセチル化セルロースの化学修飾率は7mol%であった。また、TG-DTAより算出した熱分解温度は331℃であった。また、広角X線回折像から、セルロースI型結晶構造であることが確認された。
このセルロースシートを用いて実施例1と同様にしてセルロース繊維複合体を得た。
このセルロース繊維複合体の物性をセルロースシートの物性と共に表1に示す。
このセルロースシートを用いて実施例1と同様にしてセルロース繊維複合体を得た。
このセルロース繊維複合体の物性をセルロースシートの物性と共に表1に示す。
<比較例3>(日本国特開2008-274461号公報 実施例11の追試)
製造例1で得られたセルロースを固形分で1.4g分測り取り、水分をエチルセルソルブに置換した。このセルロースとβ-ナフトエ酸25gをエチルセルソルブ400mlに溶解した溶液を混合し、攪拌しながら窒素下80℃4時間反応させた。冷却後濾過し、エチルセロソルブで洗浄した後、水で洗浄した。
このナフトイル化セルロースの化学修飾率は、1.3mol%であり、ほとんどナフトイル化は進行していなかった。また、TG-DTAより算出した熱分解温度は319℃であった。結果を表1に示す。
製造例1で得られたセルロースを固形分で1.4g分測り取り、水分をエチルセルソルブに置換した。このセルロースとβ-ナフトエ酸25gをエチルセルソルブ400mlに溶解した溶液を混合し、攪拌しながら窒素下80℃4時間反応させた。冷却後濾過し、エチルセロソルブで洗浄した後、水で洗浄した。
このナフトイル化セルロースの化学修飾率は、1.3mol%であり、ほとんどナフトイル化は進行していなかった。また、TG-DTAより算出した熱分解温度は319℃であった。結果を表1に示す。
<比較例4>(日本国特開2008-274461号公報 実施例1の4-ビフェニルイソシアネートをフェニルイソシアネートに替えて実施)
アバカを製造例1の方法で精製した。セルロースを固形分で0.63g分測り取り、水分をジメチルホルムアミドに置換した。このセルロースとジメチルホルムアミド65mlとフェニルイソシアネート11.5gを混合し、攪拌しながら窒素下115℃2時間反応させた。冷却後濾過し、水とメタノールで洗浄した。広角X線回折像から、セルロースI型結晶構造は維持されておらず、「走査角20~24度におけるセルロースI型結晶構造由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」(非晶質等のI型以外の構造を反映)の比は1.11であり、非晶性になっていることが示唆された。また、TG-DTAより算出した熱分解温度は258℃であった。結果を表1に示す。
アバカを製造例1の方法で精製した。セルロースを固形分で0.63g分測り取り、水分をジメチルホルムアミドに置換した。このセルロースとジメチルホルムアミド65mlとフェニルイソシアネート11.5gを混合し、攪拌しながら窒素下115℃2時間反応させた。冷却後濾過し、水とメタノールで洗浄した。広角X線回折像から、セルロースI型結晶構造は維持されておらず、「走査角20~24度におけるセルロースI型結晶構造由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」(非晶質等のI型以外の構造を反映)の比は1.11であり、非晶性になっていることが示唆された。また、TG-DTAより算出した熱分解温度は258℃であった。結果を表1に示す。
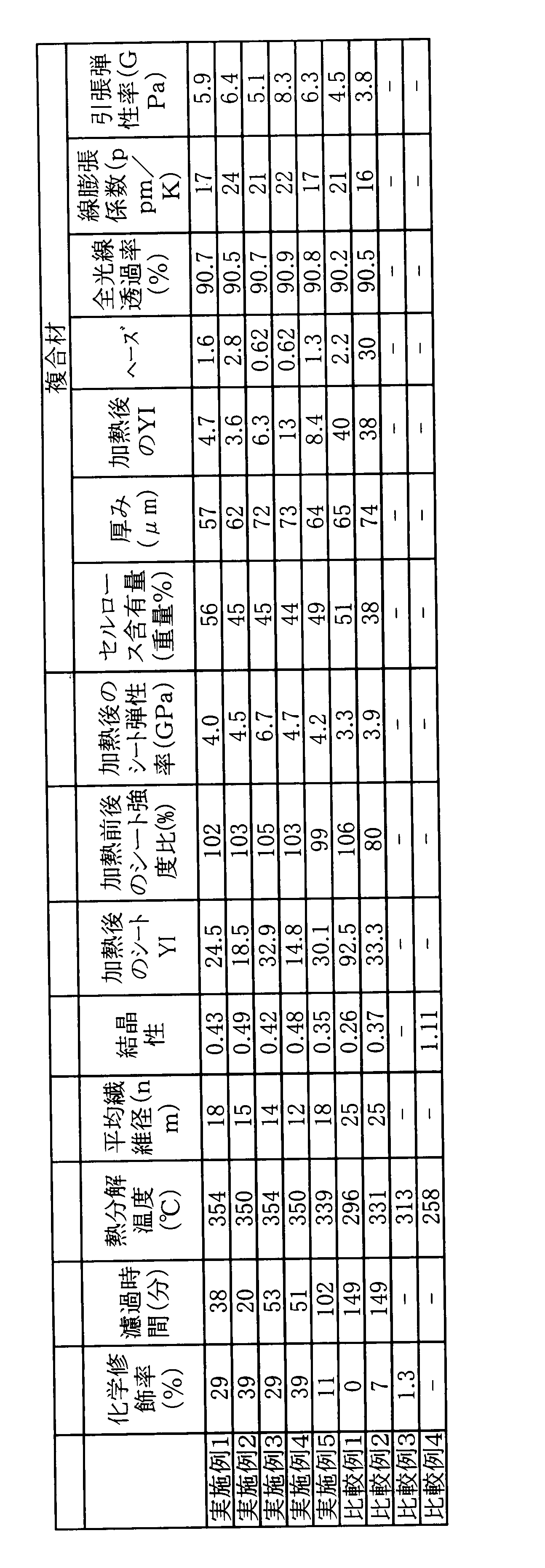
本発明により得られる実施例1~5に示すような修飾セルロース繊維は、比較例1に示すような未修飾セルロース繊維、比較例2に示すような脂肪族含有置換基で修飾したアセチル化セルロース、比較例3,4に示すような従来の方法を用いて芳香環含有置換基で修飾されたセルロースと比較して、熱分解温度が向上した。加熱後のセルロースシートの物性を対比すると、実施例1~5ではシート強度比が99%以上の高い確率で維持されると同時に、4GPa以上の高い引張弾性率を示している。一方、比較例1ではシート強度は維持されるものの引張弾性率は3GPa強に留まり、比較例2では引張弾性率は4GPa弱まで向上するものの、引張強度は大幅に低下している。この違いは複合材としたときの引張弾性率の違いとしても現れており、実施例1~5の複合材は比較例1,2の複合材に比し、明らかな引張弾性率の向上が認められる。
また、同様な解繊工程で得られたセルロース水分散液からセルロースシートを得るのに必要な濾過時間を比較すると、実施例1,2,5及び比較例1より、芳香環含有置換基で修飾した実施例は、未変性より、濾過時間が早く、ヘーズ同等以上である。尚、濾過時間については、工業的生産時にはより重要である。より大量のセルロース分散液を、より小さな差圧の減圧又は加圧濾過で処理するので、濾過時間の差は非常に大きな差となる。
また、実施例3,4より、超音波処理を行うことでより高透明な複合材料が得られた。
また、実施例3,4より、超音波処理を行うことでより高透明な複合材料が得られた。
日本国特開2007-51266号公報 実施例6の追試である比較例2は、セルロースシートを脂肪族であるアセチル基で修飾したものであるが、未変性のセルロースシートを作成してから修飾したものであるため、濾過時間の短縮効果が得られないばかりか、加熱後の着色の向上も見られず、透明性も悪化した。
比較例3では、本願とは異なるエステル化方法により、芳香族含有基で修飾したが、反応性が悪く、ほとんど進行しなかった。熱分解温度の上昇幅が比較例2にも及ばないことから、加熱後のシート強度維持率、引張弾性率、等の諸物性についても本願実施例レベルは下回ることが予想される。従って、比較例3のセルロースをシート化してセルロース繊維複合体を得たとしても、引張弾性率等、複合材としての諸物性が本願実施例レベルに到達するとは思われない。
比較例3では、本願とは異なるエステル化方法により、芳香族含有基で修飾したが、反応性が悪く、ほとんど進行しなかった。熱分解温度の上昇幅が比較例2にも及ばないことから、加熱後のシート強度維持率、引張弾性率、等の諸物性についても本願実施例レベルは下回ることが予想される。従って、比較例3のセルロースをシート化してセルロース繊維複合体を得たとしても、引張弾性率等、複合材としての諸物性が本願実施例レベルに到達するとは思われない。
また、比較例4でも、本願と異なる方法で芳香族カルバマート基で修飾したが、セルロースI型結晶が維持されなかった。I型結晶構造の比率が少ない比較例4は比較例1~3に比べても強度が大幅に下がると予想され、加熱後のシート強度維持率、引張弾性率、等の諸物性が本願実施例レベルに到達するとは思われない。従って、比較例4のセルロースをシート化してセルロース繊維複合体を得たとしても、引張弾性率等、複合材としての諸物性が本願実施例レベルに到達するとは到底思われない。
本願実施例1~5はセルロース表面の水酸基が10%を超える高い確率で芳香環含有基によって修飾されているにも関わらず、I型結晶構造が維持されている。本願の製造方法は、セルロースI型結晶が維持されつつ、芳香族含有基を効率よく導入する方法であるといえる。
セルロースシートの耐熱性、複合材としたときのYI、引張弾性率等を始めとする本発明の効果は、上記化学修飾率とI型結晶構造の両立によって達成されていると考えられる。
表1において実施例1,2および比較例1セルロースの平均繊維径を比べると、同じ解繊工程を経ているにもかかわらず実施例1,2は比較例1より値が小さい。このことから、本願発明は、化学修飾時に有機酸を用いることによりセルロース繊維を効果的に膨潤させ、均一な化学修飾を実現し、上記化学修飾率とI型結晶構造の両立を達成したものと推測する。
本願実施例1~5はセルロース表面の水酸基が10%を超える高い確率で芳香環含有基によって修飾されているにも関わらず、I型結晶構造が維持されている。本願の製造方法は、セルロースI型結晶が維持されつつ、芳香族含有基を効率よく導入する方法であるといえる。
セルロースシートの耐熱性、複合材としたときのYI、引張弾性率等を始めとする本発明の効果は、上記化学修飾率とI型結晶構造の両立によって達成されていると考えられる。
表1において実施例1,2および比較例1セルロースの平均繊維径を比べると、同じ解繊工程を経ているにもかかわらず実施例1,2は比較例1より値が小さい。このことから、本願発明は、化学修飾時に有機酸を用いることによりセルロース繊維を効果的に膨潤させ、均一な化学修飾を実現し、上記化学修飾率とI型結晶構造の両立を達成したものと推測する。
また、実施例にあげた修飾セルロースの反応は、有機酸である酢酸中で行っているが、いずれも、解繊性は良好であった。このことより、芳香環含有置換基で修飾したセルロースは易解繊性であり、かつ高生産性であるといえる。また、このセルロース繊維から得られたセルロースシートを用いたセルロース複合体の物性をみると、前述の通り、高透明性であり、かつ非着色性、低線膨張係数、高弾性率が達成された。
以上、本発明を特定の態様を用いて詳細に説明したが、本発明の意図と範囲を離れることなく様々な変更が可能であることは当業者に明らかである。
なお本出願は、2009年6月12日付で出願された日本特許出願(特願2009-140697)に基づいており、その全体が引用により援用される。
なお本出願は、2009年6月12日付で出願された日本特許出願(特願2009-140697)に基づいており、その全体が引用により援用される。
本発明の製造方法により得られるセルロース繊維複合体は、透明性が高く、高強度、低吸水性、高透明性、低着色およびヘーズが小さく光学特性に優れるため、液晶ディスプレイ、プラズマディスプレイ、有機ELディスプレイ、フィールドエミッションディスプレイ、リアプロジェクションテレビ等のディスプレイや基板やパネルとして好適である。また、シリコン系太陽電池、色素増感太陽電池などの太陽電池用基板に好適である。基板としては、バリア膜、ITO、TFT等と積層してもよい。また、自動車用の窓材、鉄道車両用の窓材、住宅用の窓材、オフィスや工場などの窓材などに好適に使われる。窓材としては、必要に応じてフッ素皮膜、ハードコート膜等の膜や耐衝撃性、耐光性の素材を積層してもよい。
また、低線膨張係数、高弾性、高強度等の特性を生かして透明材料用途以外の構造体としても用いることができる。特に、内装材、外板、バンパー等の自動車材料やパソコンの筐体、家電部品、包装用資材、建築資材、土木資材、水産資材、その他、工業用資材等として好適に用いられる。
Claims (16)
- セルロースを溶媒中で芳香族化合物と反応させることにより該セルロースを芳香環含有置換基で修飾する修飾反応工程を有する修飾セルロース繊維の製造方法であって、該修飾反応工程における該溶媒として有機酸を用いることを特徴とする修飾セルロース繊維の製造方法。
- 芳香環含有置換基が、芳香環含有エステル基、芳香環含有エーテル基、及び芳香環含有カルバマート基からなる群より選ばれる1種以上の基である請求項1に記載の修飾セルロース繊維の製造方法。
- セルロースの全水酸基に対する芳香環含有置換基の修飾率が10モル%以上である請求項1または2に記載の修飾セルロース繊維の製造方法。
- セルロースと有機酸とを含むセルロース分散液中でセルロースと芳香族化合物とを反応させることにより該セルロースを芳香環含有置換基で修飾する修飾反応工程と、抄紙工程とを有する修飾セルロース繊維シートの製造方法。
- 芳香環含有置換基が、芳香環含有エステル基、芳香環含有エーテル基、及び芳香環含有カルバマート基からなる群より選ばれる1種以上の基である請求項4に記載の修飾セルロース繊維シートの製造方法。
- セルロースの全水酸基に対する芳香環含有置換基の修飾率が10モル%以上である請求項4または5に記載の修飾セルロース繊維シートの製造方法。
- 請求項1~3のいずれか1項に記載の方法で得られた修飾セルロース繊維または請求項4~6のいずれか1項に記載の方法で得られた修飾セルロース繊維シートと、マトリクスとを複合化する工程を有する、セルロース繊維複合体の製造方法。
- セルロースの全水酸基の10モル%以上が芳香環含有置換基で修飾されたセルロースI型結晶構造を有するセルロース繊維の分散液であって、該セルロース繊維の広角X線回折像の「走査角20~24度におけるセルロースI型結晶由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」の比が0.8以下である修飾セルロース繊維分散液。
- セルロース繊維の平均繊維径が100nm以下である請求項8に記載の修飾セルロース繊維分散液。
- 芳香環含有置換基が、芳香環含有エステル基、芳香環含有エーテル基、及び芳香環含有カルバマート基からなる群より選ばれる1種以上の基である請求項8または9に記載の修飾セルロース繊維分散液。
- セルロースの全水酸基の10モル%以上が芳香環含有置換基で修飾されたセルロースI型結晶構造を有するセルロース繊維であって、該セルロース繊維の広角X線回折像の「走査角20~24度におけるセルロースI型結晶由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」の比が0.8以下である修飾セルロース繊維。
- セルロース繊維の平均繊維径が100nm以下である請求項11に記載の修飾セルロース繊維。
- 芳香環含有置換基が、芳香環含有エステル基、芳香環含有エーテル基、及び芳香環含有カルバマート基からなる群より選ばれる1種以上の基である請求項11または12に記載の修飾セルロース繊維。
- 平均繊維径が100nm以下であり、セルロースの全水酸基の10モル%以上が芳香環含有置換基で修飾されたセルロースI型結晶構造を有するセルロース繊維を含む修飾セルロース繊維シートであって、下記(1)に示す複合材料としたときのC光によるヘーズ値が3以下である修飾セルロース繊維シート。
(1)秤量40g/m2となるように厚みを調整したセルロース繊維シートを、ビス(メタクリロイルオキシメチル)トリシクロ[5.2.1.02,6]デカン96重量部、ペンタエリスリトールテトラキス(β-チオプロピオネート)6重量部、2,4,6-トリメチルベンゾイルジフェニルフォスフィンオキサイド0.05重量部、ベンゾフェノン0.05重量部を混合した溶液に含浸させ、減圧下に一晩置いた後、2枚のガラス板にはさみ、無電極水銀ランプを用いて硬化後、ガラス板よりはずして190℃の真空オーブン中で4時間加熱して得た複合材料。 - 請求項11~13のいずれか1項に記載の修飾セルロース繊維または請求項14に記載の修飾セルロース繊維シートと、マトリクスとを含むセルロース繊維複合体。
- セルロースの全水酸基の10モル%以上が芳香環含有置換基で修飾されたセルロースI型結晶構造を有するセルロース繊維と、マトリクスとを含むセルロース繊維複合体であって、該セルロース繊維の広角X線回折像の「走査角20~24度におけるセルロースI型結晶由来の回折ピークの強度」に対する、「同走査角18~19度における平均回折強度」の比が0.8以下であるセルロース繊維複合体。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10786253.4A EP2441885B1 (en) | 2009-06-12 | 2010-06-11 | Modified cellulose fiber and cellulose composite thereof |
| CN201080025432.8A CN102803600B (zh) | 2009-06-12 | 2010-06-11 | 修饰纤维素纤维及其纤维素复合体 |
| JP2011518592A JP5614402B2 (ja) | 2009-06-12 | 2010-06-11 | 修飾セルロース繊維及びそのセルロース複合体 |
| US13/323,063 US8992731B2 (en) | 2009-06-12 | 2011-12-12 | Modified cellulose fibers and cellulose composite thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009140697 | 2009-06-12 | ||
| JP2009-140697 | 2009-06-12 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/323,063 Continuation US8992731B2 (en) | 2009-06-12 | 2011-12-12 | Modified cellulose fibers and cellulose composite thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010143722A1 true WO2010143722A1 (ja) | 2010-12-16 |
Family
ID=43308977
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/059976 WO2010143722A1 (ja) | 2009-06-12 | 2010-06-11 | 修飾セルロース繊維及びそのセルロース複合体 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US8992731B2 (ja) |
| EP (1) | EP2441885B1 (ja) |
| JP (1) | JP5614402B2 (ja) |
| CN (1) | CN102803600B (ja) |
| WO (1) | WO2010143722A1 (ja) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012107199A (ja) * | 2010-10-26 | 2012-06-07 | Sumitomo Bakelite Co Ltd | 変性セルロース繊維、樹脂組成物、成形体及び樹脂組成物、成形体の製造方法 |
| JP2012222005A (ja) * | 2011-04-04 | 2012-11-12 | Mitsubishi Chemicals Corp | 発光装置、繊維複合体、及び繊維複合体の製造方法 |
| WO2013031687A1 (ja) * | 2011-08-31 | 2013-03-07 | コニカミノルタホールディングス株式会社 | ガスバリア性フィルムおよびその製造方法、ならびにこれを用いた電子素子用基板 |
| WO2013121083A3 (en) * | 2012-02-13 | 2013-10-31 | Upm-Kymmene Corporation | Method for concentrating fibril cellulose and fibril cellulose product |
| JP2016030862A (ja) * | 2014-07-28 | 2016-03-07 | 株式会社クラレ | フィブリル化繊維およびその製造方法 |
| WO2016042930A1 (ja) * | 2014-09-16 | 2016-03-24 | 富士フイルム株式会社 | 繊維強化複合材料および繊維強化複合材料の製造方法 |
| WO2016108285A1 (ja) * | 2014-12-29 | 2016-07-07 | クラレクラフレックス株式会社 | 繊維集合体およびそれを用いた液体吸収性シート状物、ならびに繊維集合体の製造方法 |
| JP2016172856A (ja) * | 2010-11-16 | 2016-09-29 | 王子ホールディングス株式会社 | セルロース繊維集合体およびセルロース繊維複合体 |
| JP2018518569A (ja) * | 2015-06-05 | 2018-07-12 | ダウ グローバル テクノロジーズ エルエルシー | セルロースエーテル粉末 |
| JP2018145153A (ja) * | 2017-03-07 | 2018-09-20 | 花王株式会社 | エーテル化セルロース繊維及び界面活性剤を含有する組成物 |
| CN112160179A (zh) * | 2020-09-04 | 2021-01-01 | 西南大学 | 一种菠萝皮渣木质纳米纤维素的制备方法 |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK2808440T3 (da) | 2009-03-30 | 2019-09-30 | Fiberlean Tech Ltd | Fremgangsmåde til fremstilling af nanofibrillære cellulosesuspensioner |
| ES2650373T3 (es) | 2009-03-30 | 2018-01-18 | Fiberlean Technologies Limited | Procedimiento para la producción de geles de celulosa nanofibrilares |
| DK2386683T3 (da) | 2010-04-27 | 2014-06-23 | Omya Int Ag | Fremgangsmåde til fremstilling af gel-baserede kompositmaterialer |
| SI2386682T1 (sl) | 2010-04-27 | 2014-07-31 | Omya International Ag | Postopek za izdelavo strukturiranih materialov z uporabo nanofibriliranih celuloznih gelov |
| CN103118804B (zh) * | 2010-09-17 | 2016-04-06 | 泰坦木业有限公司 | 木材碎块的处理 |
| JP6048365B2 (ja) | 2012-10-23 | 2016-12-21 | 三菱化学株式会社 | ゴム改質材、ゴム改質材分散液、及びゴム組成物 |
| JP6120590B2 (ja) * | 2013-02-01 | 2017-04-26 | 国立大学法人京都大学 | 変性ナノセルロース及び変性ナノセルロースを含む樹脂組成物 |
| EP3041870A4 (en) * | 2013-09-06 | 2017-04-26 | Teknologian tutkimuskeskus VTT Oy | Surface-modified cellulose nanofibres, bio composite resin composition and method for producing the same |
| TWI716892B (zh) * | 2014-05-26 | 2021-01-21 | 日商王子控股股份有限公司 | 含微細纖維素纖維片、複合片及其應用 |
| DK3328211T3 (da) * | 2015-07-30 | 2024-01-29 | Cargill Inc | Tørre citrusfibre og anvendelser deraf |
| TWI712495B (zh) * | 2015-09-18 | 2020-12-11 | 日商王子控股股份有限公司 | 積層體 |
| EP3362508B1 (en) | 2015-10-14 | 2019-06-26 | FiberLean Technologies Limited | 3d-formable sheet material |
| JP6947033B2 (ja) * | 2015-10-27 | 2021-10-13 | 王子ホールディングス株式会社 | 積層シート及び積層体 |
| KR20170130175A (ko) * | 2016-05-18 | 2017-11-28 | 삼성전자주식회사 | 셀룰로스 분리막을 제조하는 방법, 그에 의하여 제조된 셀룰로스 분리막 및 그를 포함하는 이차이온전지 |
| US20190194866A1 (en) * | 2016-06-03 | 2019-06-27 | Kri, Inc. | Method for producing cellulose fine fiber |
| WO2018123150A1 (ja) * | 2016-12-28 | 2018-07-05 | 旭化成株式会社 | セルロース含有樹脂組成物及びセルロース製剤 |
| US11597779B2 (en) | 2017-12-04 | 2023-03-07 | Nanollose Limited | Methods for producing a viscose dope from microbial cellulose |
| CN110129958B (zh) * | 2019-05-29 | 2020-11-06 | 浙江九鼎服饰有限公司 | 内衣抗菌混纺面料的制备方法 |
| CN115897274B (zh) * | 2022-11-22 | 2024-05-03 | 中国科学院广州能源研究所 | 一种木质纤维素类生物质组分分离同步制备高纯纤维素的方法 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0632801A (ja) * | 1992-05-26 | 1994-02-08 | Eastman Kodak Co | セルロースエステルの製造方法 |
| JP2007051266A (ja) | 2005-02-01 | 2007-03-01 | Kyoto Univ | 繊維強化複合材料及びその製造方法 |
| JP2008024788A (ja) * | 2006-07-19 | 2008-02-07 | Kyoto Univ | ナノファイバーシート及びその製造方法並びに繊維強化複合材料 |
| JP2008174869A (ja) * | 2007-01-19 | 2008-07-31 | Toray Ind Inc | 疎水性セルロース誘導体からなる繊維 |
| JP2008248441A (ja) * | 2007-03-30 | 2008-10-16 | Daicel Chem Ind Ltd | 疎水化された微小繊維状セルロースを含む繊維シート |
| JP2008274461A (ja) | 2007-04-26 | 2008-11-13 | Asahi Kasei Corp | 光学的に制御された不織布および複合材料 |
| JP2009140697A (ja) | 2007-12-05 | 2009-06-25 | Nec Lighting Ltd | 照明器具 |
| JP2009167397A (ja) * | 2007-12-21 | 2009-07-30 | Mitsubishi Chemicals Corp | 繊維複合体 |
| JP2009299043A (ja) * | 2008-05-13 | 2009-12-24 | Mitsubishi Chemicals Corp | 微細セルロース繊維分散液、高分子セルロース複合体及びセルロース繊維の解繊方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3443879A (en) * | 1966-04-11 | 1969-05-13 | Us Agriculture | Preparation of a weather resistant cotton product by a molecular protection process |
| JPH0617378B2 (ja) * | 1983-08-04 | 1994-03-09 | インペリアル・ケミカル・インダストリーズ・ピーエルシー | 光重合可能な組成物、その製法及び硬化製品の製法 |
| FR2794762B1 (fr) * | 1999-06-14 | 2002-06-21 | Centre Nat Rech Scient | Dispersion de microfibrilles et/ou de microcristaux, notamment de cellulose, dans un solvant organique |
| ATE295444T1 (de) * | 2000-07-05 | 2005-05-15 | Univ Bologna | Chemische oberflächenmodifizierung von naturfasern |
| FI20030490A (fi) * | 2003-04-01 | 2004-10-02 | M Real Oyj | Menetelmä kuitukoostumuksen valmistamiseksi |
| KR20120088678A (ko) * | 2003-07-31 | 2012-08-08 | 고쿠리츠 다이가쿠 호진 교토 다이가쿠 | 섬유 강화 복합 재료, 그 제조 방법 및 그 이용 |
| TWI391427B (zh) | 2005-02-01 | 2013-04-01 | Pioneer Corp | 纖維強化複合材料及其製造方法與用途,以及纖維素纖維集合體 |
| US8012573B2 (en) | 2007-12-21 | 2011-09-06 | Mitsubishi Chemical Corporation | Fiber composite |
-
2010
- 2010-06-11 EP EP10786253.4A patent/EP2441885B1/en active Active
- 2010-06-11 CN CN201080025432.8A patent/CN102803600B/zh active Active
- 2010-06-11 WO PCT/JP2010/059976 patent/WO2010143722A1/ja active Application Filing
- 2010-06-11 JP JP2011518592A patent/JP5614402B2/ja active Active
-
2011
- 2011-12-12 US US13/323,063 patent/US8992731B2/en active Active
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0632801A (ja) * | 1992-05-26 | 1994-02-08 | Eastman Kodak Co | セルロースエステルの製造方法 |
| JP2007051266A (ja) | 2005-02-01 | 2007-03-01 | Kyoto Univ | 繊維強化複合材料及びその製造方法 |
| JP2008024788A (ja) * | 2006-07-19 | 2008-02-07 | Kyoto Univ | ナノファイバーシート及びその製造方法並びに繊維強化複合材料 |
| JP2008174869A (ja) * | 2007-01-19 | 2008-07-31 | Toray Ind Inc | 疎水性セルロース誘導体からなる繊維 |
| JP2008248441A (ja) * | 2007-03-30 | 2008-10-16 | Daicel Chem Ind Ltd | 疎水化された微小繊維状セルロースを含む繊維シート |
| JP2008274461A (ja) | 2007-04-26 | 2008-11-13 | Asahi Kasei Corp | 光学的に制御された不織布および複合材料 |
| JP2009140697A (ja) | 2007-12-05 | 2009-06-25 | Nec Lighting Ltd | 照明器具 |
| JP2009167397A (ja) * | 2007-12-21 | 2009-07-30 | Mitsubishi Chemicals Corp | 繊維複合体 |
| JP2009299043A (ja) * | 2008-05-13 | 2009-12-24 | Mitsubishi Chemicals Corp | 微細セルロース繊維分散液、高分子セルロース複合体及びセルロース繊維の解繊方法 |
Non-Patent Citations (3)
| Title |
|---|
| "Serur5su No Jiten", ASAKURA PUBLISHING CO., LTD., pages: 81 - 86,93-99 |
| CARBOHYDRATE POLYMERS, vol. 74, no. 4, 2008, pages 875 - 879 |
| See also references of EP2441885A4 * |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012107199A (ja) * | 2010-10-26 | 2012-06-07 | Sumitomo Bakelite Co Ltd | 変性セルロース繊維、樹脂組成物、成形体及び樹脂組成物、成形体の製造方法 |
| JP2016172856A (ja) * | 2010-11-16 | 2016-09-29 | 王子ホールディングス株式会社 | セルロース繊維集合体およびセルロース繊維複合体 |
| JP2012222005A (ja) * | 2011-04-04 | 2012-11-12 | Mitsubishi Chemicals Corp | 発光装置、繊維複合体、及び繊維複合体の製造方法 |
| JPWO2013031687A1 (ja) * | 2011-08-31 | 2015-03-23 | コニカミノルタ株式会社 | ガスバリア性フィルムおよびその製造方法、ならびにこれを用いた電子素子用基板 |
| WO2013031687A1 (ja) * | 2011-08-31 | 2013-03-07 | コニカミノルタホールディングス株式会社 | ガスバリア性フィルムおよびその製造方法、ならびにこれを用いた電子素子用基板 |
| US9663588B2 (en) | 2012-02-13 | 2017-05-30 | Upm-Kymmene Corporation | Method for concentrating fibril cellulose and fibril cellulose product |
| WO2013121083A3 (en) * | 2012-02-13 | 2013-10-31 | Upm-Kymmene Corporation | Method for concentrating fibril cellulose and fibril cellulose product |
| JP2016030862A (ja) * | 2014-07-28 | 2016-03-07 | 株式会社クラレ | フィブリル化繊維およびその製造方法 |
| WO2016042930A1 (ja) * | 2014-09-16 | 2016-03-24 | 富士フイルム株式会社 | 繊維強化複合材料および繊維強化複合材料の製造方法 |
| JPWO2016042930A1 (ja) * | 2014-09-16 | 2017-04-27 | 富士フイルム株式会社 | 繊維強化複合材料および繊維強化複合材料の製造方法 |
| WO2016108285A1 (ja) * | 2014-12-29 | 2016-07-07 | クラレクラフレックス株式会社 | 繊維集合体およびそれを用いた液体吸収性シート状物、ならびに繊維集合体の製造方法 |
| JPWO2016108285A1 (ja) * | 2014-12-29 | 2017-11-02 | クラレクラフレックス株式会社 | 繊維集合体およびそれを用いた液体吸収性シート状物、ならびに繊維集合体の製造方法 |
| US10507142B2 (en) | 2014-12-29 | 2019-12-17 | Kuraray Kuraflex Co., Ltd. | Fiber assembly and liquid absorbent sheet-like article including the same and method of manufacturing fiber assembly |
| AU2015373036B2 (en) * | 2014-12-29 | 2021-06-24 | Kuraray Kuraflex Co., Ltd. | Fiber aggregate, liquid-absorbing sheet-shaped object including same, and process for producing fiber aggregate |
| JP2018518569A (ja) * | 2015-06-05 | 2018-07-12 | ダウ グローバル テクノロジーズ エルエルシー | セルロースエーテル粉末 |
| JP2018145153A (ja) * | 2017-03-07 | 2018-09-20 | 花王株式会社 | エーテル化セルロース繊維及び界面活性剤を含有する組成物 |
| CN112160179A (zh) * | 2020-09-04 | 2021-01-01 | 西南大学 | 一种菠萝皮渣木质纳米纤维素的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102803600B (zh) | 2015-01-14 |
| US8992731B2 (en) | 2015-03-31 |
| JP5614402B2 (ja) | 2014-10-29 |
| EP2441885A1 (en) | 2012-04-18 |
| EP2441885A4 (en) | 2012-11-21 |
| US20120125547A1 (en) | 2012-05-24 |
| EP2441885B1 (en) | 2013-11-27 |
| CN102803600A (zh) | 2012-11-28 |
| JPWO2010143722A1 (ja) | 2012-11-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5614402B2 (ja) | 修飾セルロース繊維及びそのセルロース複合体 | |
| JP6206529B2 (ja) | セルロース繊維集合体およびセルロース繊維複合体 | |
| JP5577622B2 (ja) | 微細セルロース繊維分散液、高分子セルロース複合体及びセルロース繊維の解繊方法 | |
| WO2009081881A1 (ja) | 繊維複合体 | |
| JP5510092B2 (ja) | 修飾セルロース繊維分散液の製造方法及びセルロース複合材料の製造方法 | |
| JP5708835B2 (ja) | 繊維複合体 | |
| JP2011144363A (ja) | セルロース繊維複合体及びその製造方法 | |
| JP5412912B2 (ja) | セルロース及び高分子セルロース複合体の製造方法 | |
| JP6107297B2 (ja) | 繊維樹脂複合材料 | |
| JP2012188654A (ja) | セルロース繊維およびセルロース繊維の製造方法 | |
| JP6229440B2 (ja) | 繊維樹脂複合材料の製造方法 | |
| JP5653792B2 (ja) | 光学フィルム | |
| JP2009155772A (ja) | 微細セルロース繊維の製造方法 | |
| JP2013018851A (ja) | セルロース繊維、セルロース繊維含有重合体、樹脂組成物及び成形体。 | |
| JP2015071848A (ja) | 微細セルロース繊維の製造方法 | |
| JP2012180602A (ja) | セルロース繊維分散液、セルロース繊維分散液の製造方法および微細セルロース繊維の製造方法 | |
| JP5359819B2 (ja) | セルロース繊維複合材料、およびその製造方法 | |
| JP5141516B2 (ja) | セルロース不織布およびその製造方法と複合材料 | |
| JP2010024376A (ja) | セルロース繊維複合材料 | |
| JP2015086266A (ja) | 繊維樹脂複合体の製造方法 | |
| WO2013058244A1 (ja) | 化学修飾セルロース不織布の製造方法および化学修飾セルロース不織布、並びに、これを用いたセルロース繊維樹脂複合材料およびその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080025432.8 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10786253 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011518592 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010786253 Country of ref document: EP |