WO2009113590A1 - ポリアミド、ポリアミド組成物及びポリアミドの製造方法 - Google Patents
ポリアミド、ポリアミド組成物及びポリアミドの製造方法 Download PDFInfo
- Publication number
- WO2009113590A1 WO2009113590A1 PCT/JP2009/054693 JP2009054693W WO2009113590A1 WO 2009113590 A1 WO2009113590 A1 WO 2009113590A1 JP 2009054693 W JP2009054693 W JP 2009054693W WO 2009113590 A1 WO2009113590 A1 WO 2009113590A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polyamide
- acid
- polyamide composition
- dicarboxylic acid
- mass
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/02—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids
- C08G69/26—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from polyamines and polycarboxylic acids
- C08G69/265—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from polyamines and polycarboxylic acids from at least two different diamines or at least two different dicarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/02—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids
- C08G69/26—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from polyamines and polycarboxylic acids
- C08G69/28—Preparatory processes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/02—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids
- C08G69/36—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from amino acids, polyamines and polycarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/02—Halogenated hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/09—Carboxylic acids; Metal salts thereof; Anhydrides thereof
- C08K5/098—Metal salts of carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/49—Phosphorus-containing compounds
- C08K5/51—Phosphorus bound to oxygen
- C08K5/53—Phosphorus bound to oxygen bound to oxygen and to carbon only
- C08K5/5313—Phosphinic compounds, e.g. R2=P(:O)OR'
Definitions
- the present invention relates to a polyamide, a polyamide composition, and a method for producing a polyamide.
- Polyamides represented by polyamide 6 and polyamide 66 are excellent in molding processability, mechanical properties or chemical resistance. Polyamides are widely used as various parts materials for automobiles, electric and electronic, industrial materials, industrial materials, daily use and household goods.
- PA6T terephthalic acid and hexamethylenediamine
- PA6T may be abbreviated as aliphatic polyamide such as PA6 and PA66 or amorphous aromatic polyamide (hereinafter referred to as “PA6I”) composed of isophthalic acid and hexamethylenediamine. ) And the like, and a high melting point semi-aromatic polyamide mainly composed of terephthalic acid and hexamethylenediamine (hereinafter referred to as “6T copolymer polyamide”) whose melting point is lowered to about 220 to 340 ° C. Have been proposed).
- aliphatic polyamide such as PA6 and PA66 or amorphous aromatic polyamide (hereinafter referred to as “PA6I”) composed of isophthalic acid and hexamethylenediamine. )
- PA6I amorphous aromatic polyamide
- 6T copolymer polyamide a high melting point semi-aromatic polyamide mainly composed of terephthalic acid and hexamethylenediamine
- Patent Document 1 discloses an aromatic polyamide (hereinafter referred to as “a mixture of hexamethylene diamine and 2-methylpentamethylene diamine”, which is composed of an aromatic dicarboxylic acid and an aliphatic diamine. May be abbreviated as “PA6T / 2MPDT”).
- a high melting point aliphatic polyamide composed of adipic acid and tetramethylene diamine hereinafter sometimes abbreviated as “PA46”
- PA46 tetramethylene diamine
- Patent Documents 2 and 3 disclose semialicyclic polyamides of alicyclic polyamides (hereinafter sometimes abbreviated as “PA6C”) composed of 1,4-cyclohexanedicarboxylic acid and hexamethylenediamine and other polyamides. (Hereinafter, it may be abbreviated as “PA6C copolymer polyamide”).
- Patent Document 2 discloses that an electrical and electronic member of a semi-alicyclic polyamide containing 1 to 40% 1,4-cyclohexanedicarboxylic acid as a dicarboxylic acid unit has improved solder heat resistance. Is disclosed that the automobile parts are excellent in fluidity and toughness.
- Patent Document 4 discloses that a polyamide comprising a dicarboxylic acid unit containing 1,4-cyclohexanedicarboxylic acid and a diamine unit containing 2-methyl-1,8-octanediamine is light resistance, toughness, moldability, lightness, And excellent heat resistance. Further, as a method for producing the polyamide, 1,4-cyclohexanedicarboxylic acid and 1,9-nonanediamine are reacted at 230 ° C. or less to form a prepolymer, and the prepolymer is solid-phase polymerized at 230 ° C. to obtain a melting point of 311 ° C. The production of polyamides is disclosed.
- Patent Document 5 discloses that a polyamide using 1,4-cyclohexanedicarboxylic acid having a trans / cis ratio of 50/50 to 97/3 as a raw material is excellent in heat resistance, low water absorption, light resistance, and the like. It is disclosed.
- JP-T 6-503590 Japanese National Patent Publication No. 11-512476 JP 2001-514695 A Japanese Patent Laid-Open No. 9-12868 International Publication No. 2002/048239 Pamphlet
- the 6T copolymer polyamide certainly has the characteristics of low water absorption, high heat resistance, and high chemical resistance, it has low flowability and insufficient moldability and molded product surface appearance. Inferior in light resistance. Therefore, the appearance of a molded product such as an exterior part is required, or improvement is desired in applications where it is exposed to sunlight or the like. In addition, the specific gravity is large, and improvement in lightness is also desired.
- the PA6T / 2MPDT disclosed in Patent Document 1 can partially improve the problems of the conventional PA6T copolymer polyamide, but in terms of fluidity, moldability, toughness, molded product surface appearance, and light resistance. The level of improvement is insufficient.
- PA46 has good heat resistance and moldability, but has a high water absorption rate, and has a problem that dimensional change due to water absorption and deterioration of mechanical properties are remarkably large. Dimensional change required for automotive applications, etc. There are cases where the demand cannot be met.
- the PA6C copolymer polyamides disclosed in Patent Documents 2 and 3 also have problems such as high water absorption and insufficient fluidity.
- the polyamides disclosed in Patent Documents 4 and 5 also have toughness, rigidity, and fluidity. In terms of sex, improvement is insufficient.
- the problem to be solved by the present invention is to provide a polyamide having excellent heat resistance, fluidity, toughness, low water absorption, rigidity and high melting point.
- the inventors of the present invention obtained a polyamide obtained by polymerizing alicyclic dicarboxylic acid and a diamine having a substituent branched from the main chain as main components.
- the present inventors have found that the problem can be solved and have completed the present invention.
- the present invention is as follows. (1) (a) a dicarboxylic acid containing at least 50 mol% of an alicyclic dicarboxylic acid; (B) A polyamide obtained by polymerizing at least 50 mol% of a diamine containing a diamine having a substituent branched from the main chain. (2) The polyamide according to (1), wherein the diamine having a substituent branched from the main chain is 2-methylpentamethylenediamine. (3) The polyamide according to (1) or (2), wherein the alicyclic dicarboxylic acid is 1,4-cyclohexanedicarboxylic acid.
- (9) (A) the polyamide according to any one of (1) to (8); (B) A polyamide composition containing an inorganic filler. (10) (A) the polyamide according to any one of (1) to (8); (C) A polyamide composition containing a copper compound and a metal halide. (11) (A) the polyamide according to any one of (1) to (8); (D) A polyamide composition containing a halogen-based flame retardant. (12) (A) the polyamide according to any one of (1) to (8); (E) A polyamide composition containing a phosphinate and / or a diphosphinate. (13) (A) the polyamide according to any one of (1) to (8); (F) A polyamide composition containing a stabilizer.
- An automobile part comprising the polyamide composition according to any one of (9) to (13).
- (16) (a) a dicarboxylic acid containing at least 50 mol% of an alicyclic dicarboxylic acid, and (b) at least 50 mol% of a diamine containing an aliphatic diamine having a substituent branched from the main chain.
- Including the step of A method for producing polyamide Including the step of A method for producing polyamide.
- (18) A polyamide obtained by the method according to (16) or (17).
- the present invention it is possible to provide a polyamide having excellent heat resistance, fluidity, toughness, low water absorption, rigidity, and a high melting point.
- the present embodiment the best mode for carrying out the present invention (hereinafter referred to as “the present embodiment”) will be described in detail.
- this invention is not limited to the following embodiment, It can implement by changing variously within the range of the summary.
- polyamide The polyamide of the present embodiment is a polyamide obtained by polymerizing the following (a) and (b): (A) a dicarboxylic acid containing at least 50 mol% of an alicyclic dicarboxylic acid, (B) A diamine containing at least 50 mol% of a diamine having a substituent branched from the main chain.
- polyamide means a polymer having an amide (—NHCO—) bond in the main chain.
- the (a) dicarboxylic acid used in the present embodiment contains at least 50 mol% of an alicyclic dicarboxylic acid.
- Examples of (a-1) alicyclic dicarboxylic acid include, for example, 1,4-cyclohexanedicarboxylic acid, 1,3-cyclohexanedicarboxylic acid, and 1,3-cyclopentane.
- Examples thereof include alicyclic dicarboxylic acids having 3 to 10 carbon atoms, preferably 5 to 10 carbon atoms, such as dicarboxylic acids.
- the alicyclic dicarboxylic acid may be unsubstituted or may have a substituent.
- examples of the substituent include alkyl groups having 1 to 4 carbon atoms such as a methyl group, an ethyl group, an n-propyl group, an isopropyl group, an n-butyl group, an isobutyl group, and a tert-butyl group.
- the alicyclic dicarboxylic acid is preferably 1,4-cyclohexanedicarboxylic acid from the viewpoints of heat resistance, fluidity, rigidity, and the like.
- the alicyclic dicarboxylic acid one kind may be used, or two or more kinds may be used in combination.
- An alicyclic dicarboxylic acid has a trans isomer and a cis geometric isomer.
- the alicyclic dicarboxylic acid as a raw material monomer, either a trans isomer or a cis isomer may be used, or a mixture of various proportions of a trans isomer and a cis isomer may be used. Since alicyclic dicarboxylic acids are isomerized at a high temperature to a certain ratio, and the cis isomer has higher water solubility in the equivalent salt with the diamine than the trans isomer, the trans isomer / cis cis is used as a raw material monomer.
- the body ratio in terms of molar ratio is preferably 50/50 to 0/100, more preferably 40/60 to 10/90, and further preferably 35/65 to 15/85.
- the trans isomer / cis isomer ratio (molar ratio) of the alicyclic dicarboxylic acid can be determined by liquid chromatography (HPLC) or NMR.
- Examples of (a) dicarboxylic acid other than (a-2) alicyclic dicarboxylic acid used in the present embodiment include aliphatic dicarboxylic acids and aromatic dicarboxylic acids.
- aliphatic dicarboxylic acid examples include malonic acid, dimethylmalonic acid, succinic acid, 2,2-dimethylsuccinic acid, 2,3-dimethylglutaric acid, 2,2-diethylsuccinic acid, and 2,3-diethylglutaric acid.
- Glutaric acid 2,2-dimethylglutaric acid, adipic acid, 2-methyladipic acid, trimethyladipic acid, pimelic acid, suberic acid, azelaic acid, sebacic acid, dodecanedioic acid, tetradecanedioic acid, hexadecanedioic acid, octadecane
- Examples thereof include linear or branched saturated aliphatic dicarboxylic acids having 3 to 20 carbon atoms such as diacid, eicosane diacid, and diglycolic acid.
- aromatic dicarboxylic acid examples include unsubstituted or various terephthalic acid, isophthalic acid, naphthalenedicarboxylic acid, 2-chloroterephthalic acid, 2-methylterephthalic acid, 5-methylisophthalic acid, and 5-sodium sulfoisophthalic acid. And aromatic dicarboxylic acids having 8 to 20 carbon atoms substituted with the above substituents.
- Examples of the various substituents include, for example, an alkyl group having 1 to 6 carbon atoms, an aryl group having 6 to 12 carbon atoms, an arylalkyl group having 7 to 20 carbon atoms, a halogen group such as a chloro group and a bromo group, and 3 carbon atoms. ⁇ 10 alkylsilyl groups, and sulfonic acid groups and groups that are salts thereof such as sodium salts.
- the dicarboxylic acid other than the alicyclic dicarboxylic acid is preferably an aliphatic dicarboxylic acid in terms of heat resistance, fluidity, toughness, low water absorption, rigidity, and the like, and more preferably has 6 or more carbon atoms. It is an aliphatic dicarboxylic acid. Among these, aliphatic dicarboxylic acids having 10 or more carbon atoms are preferable from the viewpoints of heat resistance and low water absorption.
- Examples of the aliphatic dicarboxylic acid having 10 or more carbon atoms include sebacic acid, dodecanedioic acid, tetradecanedioic acid, hexadecanedioic acid, octadecanedioic acid, and eicosanedioic acid. Of these, sebacic acid and dodecanedioic acid are preferable from the viewpoint of heat resistance and the like.
- the dicarboxylic acid other than the alicyclic dicarboxylic acid one kind may be used, or two or more kinds may be used in combination.
- a trivalent or higher polyvalent carboxylic acid such as trimellitic acid, trimesic acid, and pyromellitic acid may be further included within the range not impairing the object of the present embodiment.
- the polyvalent carboxylic acid one kind may be used, or two or more kinds may be used in combination.
- the proportion of (a-1) alicyclic dicarboxylic acid in (a) dicarboxylic acid is at least 50 mol%.
- the proportion of the alicyclic dicarboxylic acid is 50 to 100 mol%, and preferably 60 to 100 mol%.
- a polyamide that simultaneously satisfies heat resistance, fluidity, toughness, low water absorption, rigidity, and the like can be obtained.
- the ratio of (a-2) dicarboxylic acid other than (a-2) alicyclic dicarboxylic acid in (a) dicarboxylic acid is 0 to 50 mol%, preferably 0 to 40 mol%.
- the alicyclic dicarboxylic acid is preferably 50.0 to 99.9 mol% and (a-2) the aliphatic dicarboxylic acid having 10 or more carbon atoms is preferably 0.1 to 50.0 mol%. More preferably, (a-1) the alicyclic dicarboxylic acid is 60.0 to 90.0 mol% and (a-2) the aliphatic dicarboxylic acid having 10 or more carbon atoms is 10.0 to 40.0 mol%. (A-1) 70.0 to 85.0 mol% of alicyclic dicarboxylic acid and (a-2) 15.0 to 30.0 mol% of aliphatic dicarboxylic acid having 10 or more carbon atoms. preferable.
- the (a) dicarboxylic acid is not limited to the compounds described as the dicarboxylic acid, and may be a compound equivalent to the dicarboxylic acid.
- the compound equivalent to the dicarboxylic acid is not particularly limited as long as it can be a dicarboxylic acid structure similar to the dicarboxylic acid structure derived from the dicarboxylic acid, and examples thereof include anhydrides and halides of dicarboxylic acids. Can be mentioned.
- the diamine used in the present embodiment contains at least 50 mol% of a diamine having a substituent branched from the main chain.
- B By containing at least 50 mol% of a diamine having a substituent branched from the main chain as the diamine, a polyamide that simultaneously satisfies fluidity, toughness, rigidity, and the like can be obtained.
- Examples of the substituent branched from the main chain include alkyl groups having 1 to 4 carbon atoms such as methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, isobutyl group, and tert-butyl group. Is mentioned.
- Examples of the diamine having a substituent branched from the main chain include 2-methylpentamethylenediamine (also referred to as 2-methyl-1,5-diaminopentane), 2,2,4- And branched saturated aliphatic diamines having 3 to 20 carbon atoms such as trimethylhexamethylenediamine, 2,4,4-trimethylhexamethylenediamine, 2-methyloctamethylenediamine, and 2,4-dimethyloctamethylenediamine. .
- the diamine having a substituent branched from the main chain is preferably 2-methylpentamethylenediamine from the viewpoint of rigidity and the like.
- the diamine having a substituent branched from the main chain one kind may be used, or two or more kinds may be used in combination.
- diamines other than the (b-2) diamine having a substituent branched from the main chain of (b) diamine used in the present embodiment include aliphatic diamines, alicyclic diamines, and aromatic diamines. Can be mentioned.
- aliphatic diamine examples include ethylene diamine, propylene diamine, tetramethylene diamine, pentamethylene diamine, hexamethylene diamine, heptamethylene diamine, octamethylene diamine, nonamethylene diamine, decamethylene diamine, undecamethylene diamine, dodecamethylene diamine, And straight-chain saturated aliphatic diamines having 2 to 20 carbon atoms such as tridecamethylenediamine.
- alicyclic diamine examples include 1,4-cyclohexanediamine, 1,3-cyclohexanediamine, 1,3-cyclopentanediamine, and the like.
- aromatic diamines examples include metaxylylenediamine.
- the diamine other than the diamine having a substituent branched from the main chain is preferably an aliphatic diamine and an alicyclic diamine, more preferably in terms of heat resistance, fluidity, toughness, low water absorption, rigidity, and the like.
- the diamine other than the diamine having a substituent branched from the main chain one kind may be used, or two or more kinds may be used in combination.
- a trivalent or higher polyvalent aliphatic amine such as bishexamethylenetriamine may be further included within a range not impairing the object of the present embodiment.
- the polyvalent aliphatic amine one kind may be used, or two or more kinds may be used in combination.
- the ratio of (b-1) diamine having a substituent branched from the main chain in (b) diamine is at least 50 mol%.
- the ratio of the diamine having a substituent branched from the main chain is 50 to 100 mol%, and preferably 60 to 100 mol%.
- a polyamide having excellent fluidity, toughness, rigidity, and the like can be obtained.
- the proportion of diamine other than (b-2) diamine having a substituent branched from the main chain in (b) diamine is 0 to 50 mol%, preferably 0 to 40 mol%.
- the addition amount of dicarboxylic acid is preferably in the vicinity of the same molar amount as the addition amount of (b) diamine.
- the molar amount of (b) diamine as a whole is preferably 0. 90 to 1.20, more preferably 0.95 to 1.10, and still more preferably 0.98 to 1.05.
- the polyamide of the present embodiment is preferably further copolymerized with (c) lactam and / or aminocarboxylic acid.
- the (c) lactam and / or aminocarboxylic acid used in the present embodiment means a lactam and / or aminocarboxylic acid capable of polycondensation.
- the lactam and / or aminocarboxylic acid is preferably a lactam and / or aminocarboxylic acid having 4 to 14 carbon atoms, and more preferably a lactam and / or aminocarboxylic acid having 6 to 12 carbon atoms.
- lactam examples include butyrolactam, pivalolactam, ⁇ -caprolactam, caprilactam, enantolactam, undecanolactam, laurolactam (dodecanolactam), and the like.
- ⁇ -caprolactam, laurolactam, and the like are preferable, and ⁇ -caprolactam is more preferable.
- aminocarboxylic acids examples include ⁇ -aminocarboxylic acids and ⁇ , ⁇ -amino acids that are compounds in which the lactam is ring-opened.
- the aminocarboxylic acid is preferably a linear or branched saturated aliphatic carboxylic acid having 4 to 14 carbon atoms substituted with an amino group at the ⁇ position.
- 6-aminocaproic acid, 11-aminoundecanoic acid, And 12-aminododecanoic acid examples include paraaminomethylbenzoic acid.
- lactam and / or aminocarboxylic acid one kind may be used, or two or more kinds may be used in combination.
- the addition amount of lactam and / or aminocarboxylic acid is preferably 0 to 20 mol% with respect to the total molar amount of each monomer of (a), (b) and (c).
- a known end-capping agent can be further added for molecular weight adjustment.
- the end-capping agent include monocarboxylic acids, monoamines, acid anhydrides such as phthalic anhydride, monoisocyanates, monoacid halides, monoesters, and monoalcohols.
- monocarboxylic acid and monoamine are preferable.
- the terminal blocking agent one kind may be used, or two or more kinds may be used in combination.
- the monocarboxylic acid that can be used as the end-capping agent is not particularly limited as long as it has reactivity with an amino group.
- monocarboxylic acid you may use by 1 type and may be used in combination of 2 or more types.
- the monoamine that can be used as the end-capping agent is not particularly limited as long as it has reactivity with a carboxyl group.
- methylamine, ethylamine, propylamine, butylamine, hexylamine, octylamine Aliphatic monoamines such as decylamine, stearylamine, dimethylamine, diethylamine, dipropylamine, and dibutylamine; alicyclic monoamines such as cyclohexylamine and dicyclohexylamine; and aromatic monoamines such as aniline, toluidine, diphenylamine, and naphthylamine; Etc.
- Monoamines may be used alone or in combination of two or more.
- the combination of (a) dicarboxylic acid and (b) diamine is not limited to the following: (a-1) at least 50 mol% or more of alicyclic dicarboxylic acid and (b-1) at least 50 mol% or more
- the combination of 2-methylpentamethylenediamine is preferably (a-1) at least 50 mol% or more of 1,4-cyclohexanedicarboxylic acid and (b-1) at least 50 mol% or more of 2-methylpentamethylenediamine. preferable.
- the alicyclic dicarboxylic acid structure exists as a geometric isomer of a trans isomer and a cis isomer.
- the trans isomer ratio of the alicyclic dicarboxylic acid structure in the polyamide is This represents the ratio of trans isomers in the whole alicyclic dicarboxylic acid in the polyamide, and the trans isomer ratio is preferably 50 to 85 mol%, more preferably 50 to 80 mol%, and still more preferably. 60 to 80 mol%.
- the (a-1) alicyclic dicarboxylic acid an alicyclic dicarboxylic acid having a trans isomer / cis isomer ratio (molar ratio) of 50/50 to 0/100 is preferably used.
- the polyamide obtained by polymerization of the diamine preferably has a trans isomer ratio of 50 to 85 mol%.
- the trans isomer ratio is within the above range, in addition to the characteristics of high melting point, toughness and rigidity, polyamide has properties that are contrary to thermal rigidity due to high glass transition temperature and usually heat resistance. It has the property of simultaneously satisfying fluidity, high crystallinity and low water absorption.
- the polyamide consist of a combination of (a) at least 50 mol% or more of 1,4-cyclohexanedicarboxylic acid, (b) at least 50 mol% or more of 2-methylpentamethylenediamine, and the trans isomer ratio is This is particularly noticeable with polyamides of 50 to 85 mol%.
- the trans isomer ratio can be measured by the method described in the Examples below.
- the polyamide production method of the present embodiment includes (a) a dicarboxylic acid containing at least 50 mol% of an alicyclic dicarboxylic acid and (b) at least 50 mol% of a fatty acid having a substituent branched from the main chain. If it is a manufacturing method of polyamide including the process of superposing
- the method for producing polyamide preferably further includes a step of increasing the degree of polymerization of the polyamide.
- Examples of the production method of polyamide include various methods as exemplified below: 1) Suspension of aqueous solution or water of dicarboxylic acid and diamine, or suspension of aqueous solution or water of a mixture of dicarboxylic acid and diamine salt and other components (hereinafter abbreviated as “the mixture” in this paragraph).
- a method of polymerizing a suspended liquid while heating and maintaining a molten state hereinafter sometimes abbreviated as “hot melt polymerization method”
- a method of increasing the degree of polymerization while maintaining the solid state of the polyamide obtained by the hot melt polymerization method at a temperature below the melting point hereinafter sometimes abbreviated as “hot melt polymerization / solid phase polymerization method”.
- prepolymer / solid phase polymerization method A method in which an aqueous solution or suspension of water of dicarboxylic acid and diamine or a mixture thereof is heated, and the degree of polymerization is increased while maintaining the solid state of the precipitated prepolymer at a temperature below the melting point of the polyamide (hereinafter, “ Abbreviated as “prepolymer / solid phase polymerization method”), 5) A method of polymerizing a dicarboxylic acid and a diamine or a mixture thereof while maintaining a solid state (hereinafter sometimes abbreviated as “solid phase polymerization method”), 6) A “solution method” in which a dicarboxylic acid halide and a diamine equivalent to dicarboxylic acid are used for polymerization.
- the polyamide production method it is preferable to carry out the polymerization while maintaining the trans isomer ratio of the alicyclic dicarboxylic acid at 50 to 85%. Is more preferable.
- the trans isomer ratio within the above range, particularly 80% or less, it is possible to obtain a polyamide having an excellent color tone and tensile elongation and a high melting point.
- the production method of polyamide in order to increase the degree of polymerization and increase the melting point of the polyamide, it is necessary to increase the heating temperature and / or lengthen the heating time.
- the tensile elongation may decrease due to coloring or thermal deterioration.
- the rate at which the molecular weight increases may decrease significantly. Since it is possible to prevent a decrease in tensile elongation due to polyamide coloring or thermal deterioration, it is preferable to perform polymerization while maintaining the trans isomer ratio at 80% or less.
- hot melt polymerization method As a method for producing polyamide, since it is easy to maintain the trans isomer ratio at 80% or less and the obtained polyamide is excellent in color tone, 1) hot melt polymerization method and 2) hot melt polymerization Polyamide is preferably produced by a solid phase polymerization method.
- the polymerization form may be a batch type or a continuous type.
- the polymerization apparatus is not particularly limited, and examples thereof include known apparatuses such as an autoclave type reactor, a tumbler type reactor, and an extruder type reactor such as a kneader.
- the method for producing the polyamide is not particularly limited, and the polyamide can be produced by a batch-type hot melt polymerization method described below.
- the batch-type hot melt polymerization method include, for example, a polyamide component ((a) dicarboxylic acid, (b) diamine, and, if necessary, (c) lactam and / or aminocarboxylic acid) using water as a solvent.
- the contained solution of about 40 to 60% by mass is concentrated to about 65 to 90% by mass in a concentration tank operated at a temperature of 110 to 180 ° C. and a pressure of about 0.035 to 0.6 MPa (gauge pressure). To obtain a concentrated solution.
- the concentrated solution is then transferred to an autoclave and heating is continued until the pressure in the vessel is about 1.5-5.0 MPa (gauge pressure). Thereafter, the pressure is maintained at about 1.5 to 5.0 MPa (gauge pressure) while draining water and / or gas components, and when the temperature reaches about 250 to 350 ° C., the pressure is reduced to atmospheric pressure (the gauge pressure is , 0 MPa). By reducing the pressure to atmospheric pressure and reducing the pressure as necessary, by-product water can be effectively removed. Thereafter, pressurization is performed with an inert gas such as nitrogen to extrude the polyamide melt as a strand. The strand is cooled and cut to obtain pellets.
- an inert gas such as nitrogen
- the method for producing the polyamide is not particularly limited, and the polyamide can be produced by a continuous hot melt polymerization method described below.
- a solution of about 40 to 60% by mass containing water and a polyamide component as a solvent is preheated to about 40 to 100 ° C. in a container of a preliminary apparatus, and then concentrated layer / Concentrate to about 70-90% at a pressure of about 0.1-0.5 MPa (gauge pressure) and a temperature of about 200-270 ° C. to obtain a concentrated solution.
- the concentrated solution is discharged to a flasher maintained at a temperature of about 200 to 350 ° C., and then the pressure is reduced to atmospheric pressure (gauge pressure is 0 MPa). After reducing the pressure to atmospheric pressure, reduce the pressure as necessary.
- the polyamide melt is then extruded into strands, cooled and cut into pellets.
- a relative viscosity ⁇ r at 25 ° C. was used as an index.
- the molecular weight of the polyamide in the present embodiment is preferably 1 in terms of mechanical properties such as toughness and rigidity, moldability, etc., at a 98% sulfuric acid concentration of 1% and a relative viscosity ⁇ r at 25 ° C. measured according to JIS-K6810. It is 0.5 to 7.0, more preferably 1.7 to 6.0, and still more preferably 1.9 to 5.5.
- the relative viscosity at 25 ° C. can be measured according to JIS-K6810 as described in the following examples.
- the melting point of the polyamide in the present embodiment is preferably 270 to 350 ° C. as Tm2 from the viewpoint of heat resistance. Melting
- Tm2 By setting the polyamide melting point Tm2 to 270 ° C. or higher, a polyamide having excellent heat resistance can be obtained. By setting the melting point Tm2 of the polyamide to 350 ° C. or lower, it is possible to suppress the thermal decomposition of the polyamide in the melt processing such as extrusion and molding.
- the heat of fusion ⁇ H of the polyamide in the present embodiment is preferably 10 J / g or more, more preferably 14 J / g or more, still more preferably 18 J / g or more, and still more preferably, from the viewpoint of heat resistance. Is 20 J / g or more.
- Measurement of the melting point (Tm1 or Tm2) and heat of fusion ⁇ H of the polyamide in the present embodiment can be performed according to JIS-K7121 as described in the following examples.
- Examples of the measuring device for melting point and heat of fusion include Diamond-DSC manufactured by PERKIN-ELMER.
- the glass transition temperature Tg of the polyamide in the present embodiment is preferably 90 to 170 ° C.
- the glass transition temperature is preferably 90 ° C. or higher, more preferably 100 ° C. or higher, and further preferably 110 ° C. or higher.
- the glass transition temperature is preferably 170 ° C. or lower, more preferably 165 ° C. or lower, and further preferably 160 ° C. or lower.
- a polyamide having excellent heat resistance and chemical resistance can be obtained.
- a molded product having a good appearance can be obtained.
- the glass transition temperature can be measured according to JIS-K7121 as described in the following examples. Examples of the glass transition temperature measuring device include Diamond-DSC manufactured by PERKIN-ELMER.
- the polyamide in this embodiment has a melt shear viscosity ⁇ s of preferably 20 to 140 Pa ⁇ s, more preferably 25 to 115 Pa ⁇ s, and further preferably 30 to 90 Pa ⁇ s.
- the melt shear viscosity can be measured by the method described in the following examples. When the melt shear viscosity is within the above range, a polyamide having excellent fluidity can be obtained.
- the tensile strength of the polyamide in the present embodiment is preferably 70 MPa or more, more preferably 80 MPa or more, and further preferably 85 MPa or more.
- the tensile strength can be measured according to ASTM D638 as described in the following examples. When the tensile strength is 70 MPa or more, a polyamide having excellent rigidity can be obtained.
- the tensile elongation of the polyamide in the present embodiment is preferably 3.0% or more, more preferably 5.0% or more, and further preferably 7.0% or more.
- the tensile elongation can be measured according to ASTM D638 as described in the following examples. When the tensile elongation is 3.0% or more, a polyamide having excellent toughness can be obtained.
- the water absorption rate of the polyamide in the present embodiment is preferably 5.0% or less, more preferably 4.0% or less, and even more preferably 3.0% or less.
- the water absorption rate can be measured by the method described in the following examples. When the water absorption is 5.0% or less, a polyamide composition excellent in low water absorption can be obtained.
- the color tone b value of the polyamide in the present embodiment is preferably 0 or less, more preferably -2 or less.
- the color tone b value can be measured by the method described in Examples below. When the color tone b value is 0 or less, a polyamide composition having excellent heat discoloration resistance can be obtained.
- the polyamide composition of the present embodiment is a polyamide composition containing the (A) polyamide and (B) an inorganic filler.
- a polyamide composition by containing an inorganic filler (B), heat resistance, fluidity, toughness, low water absorption, rigidity and the like, without damaging the properties of polyamide, While satisfying fluidity, toughness, low water absorption, etc., it is possible to make a polyamide composition that is particularly excellent in rigidity.
- Polyamides such as PA6 and PA66 have a low melting point and cannot satisfy these requirements in terms of heat resistance. Even if it contains an inorganic filler, the polyamide composition is excellent in light resistance and excellent in color tone of the polyamide composition.
- the inorganic filler (B) used in the present embodiment is not particularly limited.
- the inorganic filler one kind may be used, or two or more kinds may be used in combination.
- glass fiber, carbon fiber, glass flake, talc, kaolin, mica, calcium carbonate, calcium monohydrogen phosphate, wollastonite, silica, carbon nanotube, Graphite, calcium fluoride, montmorillonite, swellable fluorine mica, apatite, and the like are preferable.
- the inorganic filler is more preferably glass fiber or carbon fiber, and among glass fiber and carbon fiber, the number average fiber diameter is 3 to 30 ⁇ m, the weight average fiber length is 100 to 750 ⁇ m, and the weight average Those having an aspect ratio (L / D) between the fiber length and the number average fiber diameter of 10 to 100 are more preferably used from the viewpoint of developing high characteristics.
- the inorganic filler (B) is more preferably wollastonite, and among the wollastonites, the number average fiber diameter is 3 to 30 ⁇ m, the weight average fiber length is 10 to 500 ⁇ m, and the aspect ratio ( Those having an L / D) of 3 to 100 are more preferably used.
- the inorganic filler (B) talc, mica, kaolin, silicon nitride, and the like are more preferable.
- the number average fiber diameter is 0.1 to 3 ⁇ m. Those are more preferably used.
- the number average fiber diameter and the weight average fiber length of the inorganic filler are measured by dissolving a polyamide composition molded article with a solvent in which polyamide is soluble, such as formic acid, and, for example, 100 of the obtained insoluble components.
- a solvent in which polyamide is soluble such as formic acid
- the above inorganic fillers can be arbitrarily selected and observed and obtained with an optical microscope or a scanning electron microscope.
- the method for producing the polyamide composition in the present embodiment is not particularly limited as long as it is a method of mixing the (A) polyamide and (B) the inorganic filler.
- a mixing method of polyamide and inorganic filler for example, polyamide and inorganic filler are mixed using a Henschel mixer, and supplied to a melt kneader and kneaded, or melted with a single screw or twin screw extruder.
- blending an inorganic filler with a polyamide from the side feeder is mentioned.
- all the components may be supplied to the same supply port at a time, or the components may be supplied from different supply ports.
- the melt kneading temperature is preferably about 250 to 375 ° C. as the resin temperature.
- the melt kneading time is preferably about 0.5 to 5 minutes.
- the apparatus for performing melt kneading is not particularly limited, and a known apparatus, for example, a melt kneader such as a single or twin screw extruder, a Banbury mixer, and a mixing roll can be used.
- the blending amount of the (B) inorganic filler is preferably 0.1 to 200 parts by mass, more preferably 1 to 180 parts by mass, and further preferably 5 to 100 parts by mass with respect to 100 parts by mass of the (A) polyamide. 150 parts by mass.
- the blending amount is preferably 0.1 to 200 parts by mass, more preferably 1 to 180 parts by mass, and further preferably 5 to 100 parts by mass with respect to 100 parts by mass of the (A) polyamide. 150 parts by mass.
- (B) Additives conventionally used for polyamides such as pigments, dyes, flame retardants, lubricants, fluorescents to the polyamide composition containing the inorganic filler, as long as the object of the present embodiment is not impaired. Bleaching agents, plasticizers, organic antioxidants, stabilizers, ultraviolet absorbers, nucleating agents, rubbers, reinforcing agents and the like can also be contained.
- the relative viscosity ⁇ r, melting point Tm2, and glass transition temperature Tg at 25 ° C. of the polyamide composition containing the inorganic filler (B) in the present embodiment can be measured by the same method as that for the polyamide.
- (B) a polyamide composition having excellent heat resistance, moldability, and chemical resistance because the measured value in the polyamide composition containing an inorganic filler is in the same range as the measured value of the polyamide. You can get things.
- the melt shear viscosity ⁇ s of the polyamide composition containing an inorganic filler is preferably 30 to 200 Pa ⁇ s, more preferably 40 to 180 Pa ⁇ s, and further preferably 50 to 150 Pa ⁇ s. .
- the melt shear viscosity can be measured by the method described in the following examples. When the melt shear viscosity is within the above range, a polyamide having excellent fluidity can be obtained.
- the tensile strength of the polyamide composition containing an inorganic filler is preferably 140 MPa or more, more preferably 150 MPa or more, and further preferably 160 MPa or more.
- the tensile strength can be measured according to ASTM D638 as described in the following examples. When the tensile strength is 140 MPa or more, a polyamide having excellent rigidity can be obtained.
- the tensile elongation of the polyamide composition containing the inorganic filler is preferably 1.0% or more, more preferably 1.5% or more, and further preferably 2.0% or more.
- the tensile elongation can be measured according to ASTM D638 as described in the following examples. When the tensile elongation is 1.0% or more, a polyamide having excellent toughness can be obtained.
- the water absorption rate of the polyamide composition containing an inorganic filler is preferably 5.0% or less, more preferably 4.0% or less, and even more preferably 3.0% or less.
- the water absorption rate can be measured by the method described in the following examples. When the water absorption is 5.0% or less, a polyamide composition excellent in low water absorption can be obtained.
- the polyamide composition of the present embodiment is a polyamide composition containing the (A) polyamide, and (C) a copper compound and a metal halide.
- a polyamide composition without impairing the properties of polyamide excellent in heat resistance, fluidity, toughness, low water absorption, rigidity, etc.
- (C) By containing a copper compound and a metal halide, heat resistance, fluidity, It can be set as the polyamide composition which is excellent in toughness, low water absorption, and rigidity, and also excellent in heat aging resistance.
- Examples of the copper compound used in the present embodiment include copper halide, copper acetate, copper propionate, copper benzoate, copper adipate, copper terephthalate, copper isophthalate, copper salicylate, copper nicotinate, and stearin.
- Examples include copper acid salts and copper complex salts coordinated to chelating agents such as ethylenediamine and ethylenediaminetetraacetic acid.
- copper compound As a copper compound, it has excellent heat aging resistance, and can suppress metal corrosion of the screw and cylinder part during extrusion (hereinafter sometimes abbreviated as “metal corrosion”). Cuprous, cupric bromide, cuprous chloride, and copper acetate are preferred, and copper iodide and / or copper acetate are more preferred. As a copper compound, you may use by 1 type and may be used in combination of 2 or more types.
- the compounding amount of the copper compound in the polyamide composition is preferably 0.01 to 0.6 parts by mass, more preferably 0.02 to 0.4 parts by mass with respect to 100 parts by mass of the (A) polyamide. is there.
- the polyamide 10 6 parts by weight preferably 50 to 2,000 parts by weight of copper, more preferably, 100-1500 parts by weight of copper, more preferably, a copper compound such that 150-1000 parts by weight of copper It is preferable to contain.
- a polyamide composition having excellent heat aging resistance can be obtained.
- the metal halide used in the present embodiment copper halide is excluded.
- the metal halide is a salt of a group 1 or 2 metal element of the periodic table of elements and a halogen, and examples thereof include potassium iodide, potassium bromide, potassium chloride, sodium iodide, and sodium chloride.
- potassium iodide and potassium bromide are examples thereof.
- the metal halide one kind may be used, or two or more kinds may be used in combination.
- potassium iodide is preferable because it is excellent in heat aging resistance and can suppress metal corrosion.
- the compounding amount of the metal halide in the polyamide composition is preferably 0.05 to 20 parts by mass, more preferably 0.2 to 10 parts by mass with respect to 100 parts by mass of the (A) polyamide.
- the ratio of the copper compound to the metal halide is preferably such that the polyamide composition contains the copper compound and the metal halide so that the molar ratio of halogen to copper (halogen / copper) is 2/1 to 50/1. .
- the molar ratio of halogen to copper (halogen / copper) is more preferably 2/1 to 40/1, and further preferably 5/1 to 30/1. It is preferable that the molar ratio of halogen to copper is 2/1 or more because copper precipitation and metal corrosion can be suppressed.
- the molar ratio of halogen and copper is 50/1 or less, the problem of corroding the screw of the molding machine can be suppressed without impairing mechanical properties such as toughness and rigidity.
- a method of adding (C) a copper compound and a metal halide compound alone or in a mixture to a polyamide hereinafter, abbreviated as “Production Method 2”. There are).
- the polyamide polymerization step in production method 1 is any step from the raw material monomer to the completion of the polyamide polymerization, and any step may be used.
- the apparatus for performing melt kneading in production method 2 is not particularly limited, and a known apparatus, for example, a melt kneader such as a single or twin screw extruder, a Banbury mixer, and a mixing roll can be used. . Of these, a twin screw extruder is preferably used.
- the temperature for melt kneading is preferably a temperature about 1 to 100 ° C. higher than the melting point of (A) polyamide, more preferably about 10 to 50 ° C.
- the shear rate in the kneader is preferably about 100 sec ⁇ 1 or more, and the average residence time during kneading is preferably about 0.5 to 5 minutes.
- other components include higher fatty acids such as lauric acid as lubricants, higher fatty acid metal salts of higher fatty acids and metals such as aluminum, higher fatty acid amides such as ethylene bisstearyl amide, and waxes such as polyethylene wax. Can be mentioned.
- the organic compound which has at least 1 amide group is also mentioned.
- a polyamide composition having excellent mechanical properties such as toughness and rigidity can be obtained by further containing (B) an inorganic filler.
- the blending amount of the inorganic filler is preferably 0.1 to 200 parts by mass, more preferably 1 to 180 parts by mass, and further preferably 5 to 150 parts by mass with respect to 100 parts by mass of the polyamide.
- the polyamide composition containing a copper compound and a metal halide has additives that are conventionally used for polyamides, such as pigments, dyes, flame retardants, and lubricants, as long as the object of the present embodiment is not impaired.
- additives such as pigments, dyes, flame retardants, and lubricants, as long as the object of the present embodiment is not impaired.
- Agents, fluorescent bleaches, plasticizers, organic antioxidants, stabilizers, ultraviolet absorbers, nucleating agents, rubbers, reinforcing agents, and the like can also be contained.
- the relative viscosity ⁇ r, melting point Tm2, and glass transition temperature Tg at 25 ° C. of the polyamide composition containing (C) the copper compound and the metal halide in the present embodiment are measured by the same method as that for the polyamide. be able to.
- the measured value in the polyamide composition containing (C) the copper compound and the metal halide is in the same range as the preferable value as the measured value of the polyamide, thereby improving heat resistance, moldability, and chemical resistance. An excellent polyamide composition can be obtained.
- the melt shear viscosity ⁇ s of the polyamide composition containing a copper compound and a metal halide is preferably 30 to 200 Pa ⁇ s, more preferably 40 to 180 Pa ⁇ s, and still more preferably 50 to 150 Pa ⁇ s. s.
- the melt shear viscosity can be measured by the method described in the following examples. When the melt shear viscosity is within the above range, a polyamide composition having excellent fluidity can be obtained.
- the tensile strength of the polyamide composition containing a copper compound and a metal halide is preferably 140 MPa or more, more preferably 150 MPa or more, and further preferably 160 MPa or more.
- the tensile strength can be measured according to ASTM D638 as described in the following examples. When the tensile strength is 140 MPa or more, a polyamide composition having excellent rigidity can be obtained.
- the tensile elongation of the polyamide composition containing a copper compound and a metal halide is preferably 1.0% or more, more preferably 1.5% or more, and further preferably 2.0% or more. It is.
- the tensile elongation can be measured according to ASTM D638 as described in the following examples. When the tensile elongation is 1.0% or more, a polyamide composition having excellent toughness can be obtained.
- the water absorption of the polyamide composition containing a copper compound and a metal halide is preferably 5.0% or less, more preferably 4.0% or less, and even more preferably 3.0% or less. is there.
- the water absorption rate can be measured by the method described in the following examples. When the water absorption is 5.0% or less, a polyamide composition excellent in low water absorption can be obtained.
- the strength half-life of the polyamide composition containing a copper compound and a metal halide is preferably 40 days or more, more preferably 45 days or more, and even more preferably 50 days or more as a molded article. .
- the strength half-life can be measured by the method described in the Examples below. When the strength half-life is 40 days or longer, a polyamide composition having excellent heat resistance, particularly heat aging resistance, can be obtained.
- the fracture stress of the polyamide composition containing a copper compound and a metal halide is preferably 45 MPa or more, more preferably 50 MPa or more, and further preferably 55 MPa or more.
- the fracture stress can be measured by the method described in the following examples. By molding a polyamide composition having a fracture stress of 45 MPa or more, a polyamide composition having excellent vibration resistance fatigue resistance can be obtained.
- the tensile strength retention after immersion of the polyamide containing a copper compound and a metal halide is preferably 60% or more, more preferably 75% or more, and further preferably 80% or more.
- the tensile strength retention after immersion can be measured by the method described in the following examples. By molding a polyamide composition having a tensile strength retention after immersion of 60% or more, a polyamide composition having excellent LLC resistance can be obtained.
- the polyamide composition in the present embodiment is a polyamide composition containing the (A) polyamide and (D) a halogen-based flame retardant.
- the polyamide composition in the present embodiment contains (D) a halogen flame retardant without impairing the properties of polyamide excellent in heat resistance, fluidity, toughness, rigidity, and low water absorption.
- it can be set as the polyamide composition which is excellent in heat resistance, fluidity
- the polyamide composition of the present embodiment is excellent in light resistance and excellent in color tone of the polyamide composition even if it contains a halogen-based flame retardant.
- the (D) halogen flame retardant used in the present embodiment is not particularly limited as long as it is a flame retardant containing a halogen element, and examples thereof include a chlorine flame retardant and a bromine flame retardant. . These flame retardants may be used alone or in combination of two or more.
- chlorinated flame retardant examples include chlorinated paraffin, chlorinated polyethylene, dodecachloropentacyclooctadeca-7,15-diene (Occidental Chemical, Dechlorane Plus 25 ⁇ registered trademark>), and heptic anhydride. Can be mentioned.
- brominated flame retardants include hexabromocyclododecane (HBCD), decabromodiphenyl oxide (DBDPO), octabromodiphenyl oxide, tetrabromobisphenol A (TBBA), bis (tribromophenoxy) ethane, and bis (pentabromo).
- HBCD hexabromocyclododecane
- DBDPO decabromodiphenyl oxide
- TBBA tetrabromobisphenol A
- bis (tribromophenoxy) ethane bis (pentabromo).
- Phenoxy) ethane (BPBPE), tetrabromobisphenol A epoxy resin (TBBA epoxy), tetrabromobisphenol A carbonate (TBBA-PC), ethylene (bistetrabromophthal) imide (EBTBPI), ethylene bispentabromodiphenyl, tris (tri Bromophenoxy) triazine (TTBPTA), bis (dibromopropyl) tetrabromobisphenol A (DBP-TBBA), bis (dibromopropyl) tetrabromo Sphenol S (DBP-TBBS), brominated polyphenylene ether (including poly (di) bromophenylene ether) (BrPPE), brominated polystyrene (including polydibromostyrene, polytribromostyrene, crosslinked brominated polystyrene, etc.) (BrPS), brominated crosslinked aromatic polymer, brominated epoxy resin, brominated phen
- brominated polyphenylene is used from the viewpoint of low generation amount of corrosive gas at the time of melt processing such as extrusion and molding, and further from mechanical properties such as flame retardancy, toughness and rigidity.
- Ether including poly (di) bromophenylene ether) and brominated polystyrene (including polydibromostyrene, polytribromostyrene, and cross-linked brominated polystyrene) are preferable, and brominated polystyrene is more preferable.
- the brominated polystyrene is not particularly limited.
- the benzene ring of the polystyrene is brominated or brominated styrene monomer (bromostyrene, dibromo). Styrene, tribromostyrene, etc.) can be produced by polymerization.
- the bromine content in the brominated polystyrene is preferably 55 to 75% by mass.
- the amount of bromine necessary for flame retardancy can be satisfied with a small blended amount of brominated polystyrene, and heat resistance and fluidity are maintained without impairing the properties of the polyamide.
- a polyamide composition having excellent toughness, low water absorption, rigidity, and excellent flame retardancy can be obtained.
- a polyamide composition that hardly causes thermal decomposition during melt processing such as extrusion and molding can suppress gas generation, and is excellent in heat discoloration. Obtainable.
- a polyamide composition further excellent in flame retardancy can be obtained by further containing (G) a flame retardant aid.
- the flame retardant aid (G) used in the present embodiment is not particularly limited.
- antimony oxides such as antimony trioxide, antimony tetroxide, antimony pentoxide, and sodium antimonate.
- Tin oxide such as tin monoxide and tin dioxide, iron oxides such as ferric oxide and gamma iron oxide, other zinc oxide, zinc borate, calcium oxide, aluminum oxide (alumina), aluminum oxide (boehmite), oxidation Metal oxides such as silicon (silica), titanium oxide, zirconium oxide, manganese oxide, molybdenum oxide, cobalt oxide, bismuth oxide, chromium oxide, tin oxide, nickel oxide, copper oxide, and tungsten oxide; magnesium hydroxide, and water Metal hydroxides such as aluminum oxide; aluminum, iron, titanium, manganese, Metal powders such as lead, molybdenum, cobalt, bismuth, chromium, tin, antimony, nickel, copper, and tungsten; metal carbonates such as zinc carbonate, calcium carbonate, magnesium carbonate, and barium carbonate; magnesium borate, calcium borate And metal borates such as aluminum borate; and silicone.
- These flame retardant aids may be used alone or in combination of
- Antimony oxides such as antimony trioxide, antimony tetroxide, antimony pentoxide, sodium antimonate and the like are used as flame retardant aids together with halogen-based flame retardants.
- Tin oxide such as tin monoxide and tin dioxide, iron oxides such as ferric oxide and gamma iron oxide, zinc oxide, and zinc borate are preferable.
- Antimony oxides such as antimony and zinc borate are more preferable, and antimony trioxide is more preferable.
- a flame retardant aid having an average particle size of 0.01 to 10 ⁇ m.
- the average particle size can be measured using a laser diffraction scattering method particle size distribution measuring device or a precision particle size distribution measuring device.
- (D) As a polyamide composition containing a halogen-based flame retardant, (H) by further containing a polymer containing an ⁇ , ⁇ unsaturated dicarboxylic acid anhydride, mechanical properties such as flame retardancy and toughness and rigidity
- a polyamide composition that is further superior to the above can be obtained.
- the polymer containing (H) ⁇ , ⁇ unsaturated dicarboxylic anhydride used in the present embodiment include a polymer containing ⁇ , ⁇ unsaturated dicarboxylic anhydride as a copolymerization component, and ⁇ , ⁇ . And polymers modified with unsaturated dicarboxylic acid anhydrides.
- Examples of the ⁇ , ⁇ unsaturated dicarboxylic acid anhydride include compounds represented by the following general formula (1).
- General formula (1) In the general formula (1), R 1 and R 2 are each independently hydrogen or an alkyl group having 1 to 3 carbon atoms.
- Examples of the ⁇ , ⁇ unsaturated dicarboxylic acid anhydride include maleic anhydride and methyl maleic anhydride, and maleic anhydride is preferable.
- Examples of the polymer containing an ⁇ , ⁇ unsaturated dicarboxylic acid anhydride as a copolymerization component include a copolymer of an aromatic vinyl compound and an ⁇ , ⁇ unsaturated dicarboxylic acid anhydride.
- Examples of the polymer modified with ⁇ , ⁇ unsaturated dicarboxylic acid anhydride include polyphenylene ether resin and polypropylene resin modified with ⁇ , ⁇ unsaturated dicarboxylic acid anhydride.
- General formula (2) As an aromatic vinyl compound used in this Embodiment, the compound represented by following General formula (2) is mentioned, for example.
- General formula (2) In the general formula (2), R 3 and R 4 are each independently hydrogen or an alkyl group having 1 to 3 carbon atoms, and k is an integer of 1 to 5.
- the aromatic vinyl compound examples include styrene, ⁇ -methylstyrene, p-methylstyrene, and the like, and styrene is preferable.
- the aromatic vinyl compound component when the polymer containing an ⁇ , ⁇ unsaturated dicarboxylic acid anhydride contains an aromatic vinyl compound component, the aromatic vinyl compound component has an affinity with a halogen-based flame retardant (such as brominated polystyrene).
- the ⁇ , ⁇ unsaturated dicarboxylic acid anhydride part has an affinity for or reacts with the polyamide, thereby helping to disperse the halogen flame retardant in the polyamide matrix and to finely disperse the halogen flame retardant. It is thought that you can.
- the ratio of aromatic vinyl compound component and ⁇ , ⁇ unsaturated dicarboxylic acid anhydride component in the copolymer of aromatic vinyl compound and ⁇ , ⁇ unsaturated dicarboxylic acid anhydride is flame retardant, fluidity, and heat decomposability.
- the aromatic vinyl compound component is preferably 50 to 99% by mass and the ⁇ , ⁇ unsaturated dicarboxylic anhydride component is preferably 1 to 50% by mass.
- the proportion of the ⁇ , ⁇ unsaturated dicarboxylic anhydride component is more preferably 5 to 20% by mass, still more preferably 8 to 15% by mass.
- a polyamide composition having excellent mechanical properties such as toughness and rigidity and flame retardancy can be obtained. Further, by setting the proportion of the ⁇ , ⁇ unsaturated dicarboxylic acid anhydride component to 50% by mass or less, the deterioration of the polyamide composition due to the ⁇ , ⁇ unsaturated dicarboxylic acid anhydride can be prevented.
- a polyamide composition containing a halogen-based flame retardant As a polyamide composition containing a halogen-based flame retardant, a polyamide composition that is further excellent in mechanical properties such as toughness and rigidity can be obtained by further containing the inorganic filler (B).
- the method for producing the polyamide composition in the present embodiment is not particularly limited as long as it is a method of mixing (A) the polyamide and (D) the halogen-based flame retardant.
- (D) a method for producing a polyamide composition containing a halogen flame retardant includes (G) a flame retardant aid, (H) a polymer containing an ⁇ , ⁇ unsaturated dicarboxylic acid anhydride, and / or ( B) A method of further mixing an inorganic filler can be mentioned.
- Examples of the mixing method of polyamide and halogen flame retardant include, for example, a polymer containing polyamide and halogen flame retardant, and optionally a flame retardant aid, ⁇ , ⁇ unsaturated dicarboxylic acid anhydride, and / or inorganic filler.
- a method of blending a polymer containing a saturated dicarboxylic acid anhydride in advance using a Henschel mixer or the like into a melt kneader and kneading the mixture and optionally mixing an inorganic filler from a side feeder is exemplified.
- all the components may be supplied to the same supply port at a time, or the components may be supplied from different supply ports. .
- the melt kneading temperature is preferably about 250 to 375 ° C. as the resin temperature.
- the melt kneading time is preferably about 0.5 to 5 minutes.
- a known apparatus for example, a melt kneader such as a single or twin screw extruder, a Banbury mixer, and a mixing roll is preferably used.
- the blending amount of is not particularly limited.
- the blending amount of the halogen-based flame retardant in the polyamide composition is preferably 30 to 60 parts by mass, more preferably 35 to 55 parts by mass, and further preferably 40 to 50 parts by mass with respect to 100 parts by mass of the polyamide. Part. By setting the blending amount of the halogen-based flame retardant to 30 parts by mass or more, a polyamide composition having excellent flame retardancy can be obtained.
- the blending amount of the halogen-based flame retardant to 60 parts by mass or less, generation of decomposition gas at the time of melt-kneading, deterioration of fluidity at the time of molding processing, and adhesion of pollutant substances to the molding die are suppressed. Can do. Furthermore, deterioration of mechanical properties such as toughness and rigidity and appearance of the molded product can be suppressed.
- the blending amount of the flame retardant aid in the polyamide composition is preferably 0 to 30 parts by mass, more preferably 1 to 30 parts by mass, and further preferably 2 to 20 parts by mass with respect to 100 parts by mass of the polyamide. More preferably, it is 4 to 15 parts by mass.
- a polyamide composition having further excellent flame retardancy can be obtained.
- the viscosity at the time of melt processing can be controlled within an appropriate range, the torque at the time of extrusion is increased, the moldability at the time of molding is reduced, and the molding Deterioration of product appearance can be suppressed.
- a polyamide composition having excellent toughness can be obtained without impairing the properties of polyamide having excellent mechanical properties such as toughness and rigidity.
- the blending amount of the polymer containing ⁇ , ⁇ unsaturated dicarboxylic acid anhydride in the polyamide composition is preferably 0 to 20 parts by mass, more preferably 0.5 to 20 parts by mass with respect to 100 parts by mass of the polyamide. Part, more preferably 1 to 15 parts by weight, and still more preferably 2 to 10 parts by weight.
- the blending amount of the inorganic filler in the polyamide composition is preferably 0 to 200 parts by mass, more preferably 0.1 to 200 parts by mass, and still more preferably 1 to 180 parts per 100 parts by mass of the polyamide. Parts by weight, more preferably 5 to 150 parts by weight.
- additives that are conventionally used for polyamides, for example, pigments, dyes, flame retardants, lubricants, as long as the object of the present embodiment is not impaired. It can also contain fluorescent bleaching agents, plasticizers, organic antioxidants, stabilizers, ultraviolet absorbers, nucleating agents, rubbers, reinforcing agents and the like.
- the relative viscosity ⁇ r, melting point Tm2, and glass transition temperature Tg at 25 ° C. of the polyamide composition containing the halogen-based flame retardant (D) in the present embodiment can be measured by the same method as that for the polyamide. .
- (D) mechanical properties such as heat resistance, moldability, toughness, and rigidity are measured in a polyamide composition containing a halogen-based flame retardant in a range similar to the preferred range as the measured value of the polyamide.
- a polyamide composition having excellent chemical resistance can be obtained.
- the tensile strength of the polyamide composition containing the halogen-based flame retardant is preferably 140 MPa or more, more preferably 150 MPa or more, and further preferably 160 MPa or more.
- the tensile strength can be measured according to ASTM D638 as described in the following examples. When the tensile strength is 140 MPa or more, a polyamide composition having excellent rigidity can be obtained.
- the tensile elongation of the polyamide composition containing the halogen-based flame retardant is preferably 1.0% or more, more preferably 1.5% or more, and further preferably 2.0% or more. .
- the tensile elongation can be measured according to ASTM D638 as described in the following examples. When the tensile elongation is 1.0% or more, a polyamide composition having excellent toughness can be obtained.
- the water absorption of the polyamide containing a halogen-based flame retardant is preferably 5.0% or less, more preferably 4.0% or less, and even more preferably 3.0% or less.
- the water absorption rate can be measured by the method described in the following examples. When the water absorption is 5.0% or less, a polyamide composition excellent in low water absorption can be obtained.
- the flame retardancy of the polyamide composition containing a halogen flame retardant was measured according to UL-94VB.
- the flame retardancy of the polyamide composition is preferably V-2 or more, more preferably V-1 or more, and further preferably V-0.
- the flow length of the polyamide composition containing the halogen-based flame retardant is preferably 15 cm or more, more preferably 17 cm or more, and further preferably 20 cm or more.
- the flow length can be measured by the method described in the examples below. When the flow length is 15 cm or more, a polyamide composition having excellent fluidity can be obtained.
- the polyamide composition of the present embodiment includes the polyamide (A) and (E) phosphinate and / or diphosphinate (hereinafter, both may be collectively referred to as “phosphinate”). And a polyamide composition containing.
- phosphinic acid include compounds represented by the following general formula (I).
- R 5 and R 6 and R 7 and R 8 are each independently an alkyl group having 1 to 6 carbon atoms, an aryl group having 6 to 12 carbon atoms, and R 9 is selected from the group consisting of arylalkyl groups having 7 to 20 carbon atoms, and R 9 is an alkylene group having 1 to 10 carbon atoms, an arylene group having 6 to 10 carbon atoms, an alkylarylene group having 7 to 20 carbon atoms, and carbon M is selected from the group consisting of calcium (ion), magnesium (ion), aluminum (ion) and zinc (ion), and m is 2 or 3 , N is 1 or 3, and x is 1 or 2.
- examples of the alkyl group include linear or branched saturated aliphatic groups.
- examples of the aryl group include an aromatic group having 6 to 20 carbon atoms which is unsubstituted or substituted with various substituents, and includes a phenyl group, a benzyl group, an o-toluyl group, 2, Examples include 3-xylyl group.
- the polyamide composition in the present embodiment by containing (E) phosphinate, the polyamide composition does not impair the properties of polyamide having excellent heat resistance, fluidity, toughness, low water absorption, and rigidity. Also, a polyamide composition having excellent heat resistance, fluidity, toughness, low water absorption, rigidity, and excellent flame retardancy can be obtained. Moreover, even if the polyamide composition of this Embodiment contains a phosphinate, it is excellent in light resistance, and is excellent also as a color tone of a polyamide composition.
- Examples of the (E) phosphinate used in the present embodiment include phosphinic acid, metal carbonate, metal water, as described in European Patent Application Publication No. 699708 and Japanese Patent Application Laid-Open No. 8-73720. It can manufacture in aqueous solution using metal components, such as an oxide or a metal oxide. Although these are essentially monomeric compounds, depending on the reaction conditions, polymeric phosphinic acid salts having a degree of condensation of 1 to 3 are also included depending on the environment.
- Examples of the phosphinic acid and diphosphinic acid in the phosphinic acid salt include dimethylphosphinic acid, ethylmethylphosphinic acid, diethylphosphinic acid, methyl-n-propylphosphinic acid, methandi (methylphosphinic acid), and benzene-1,4. -Di (methylphosphinic acid), methylphenylphosphinic acid, diphenylphosphinic acid and the like.
- Examples of the metal component in the phosphinate include calcium ions, magnesium ions, aluminum ions, and zinc ions.
- phosphinic acid salts include calcium dimethylphosphinate, magnesium dimethylphosphinate, aluminum dimethylphosphinate, zinc dimethylphosphinate, calcium ethylmethylphosphinate, magnesium ethylmethylphosphinate, aluminum ethylmethylphosphinate, ethyl Zinc methylphosphinate, calcium diethylphosphinate, magnesium diethylphosphinate, aluminum diethylphosphinate, zinc diethylphosphinate, calcium methyl-n-propylphosphinate, magnesium methyl-n-propylphosphinate, methyl-n-propylphosphinic acid
- the phosphinate from the viewpoint of flame retardancy and electrical properties of the polyamide composition, and from the viewpoint of synthesis of phosphinate, calcium dimethylphosphinate, aluminum dimethylphosphinate, zinc dimethylphosphinate, ethylmethyl Calcium phosphinate, aluminum ethylmethylphosphinate, zinc ethylmethylphosphinate, calcium diethylphosphinate, aluminum diethylphosphinate, and zinc diethylphosphinate are preferred.
- the phosphinate is used as a powder obtained by pulverizing the phosphinate to a particle size of 100 ⁇ m or less in terms of mechanical properties such as toughness and rigidity of a molded product obtained by molding a polyamide composition and appearance of the molded product. It is preferable to use a powder pulverized to 50 ⁇ m or less. The use of 0.5 to 20 ⁇ m powdery (E) phosphinate is more preferable because not only a polyamide composition exhibiting high flame retardancy can be obtained, but also the strength of the molded product is remarkably increased.
- the average particle size can be measured using a laser diffraction scattering method particle size distribution measuring device or a precision particle size distribution measuring device.
- the phosphinic acid salt is not necessarily completely pure, and some unreacted product or by-product may remain.
- the polyamide composition containing a phosphinate may further contain either (G) a flame retardant aid or (B) an inorganic filler.
- a polyamide composition containing a phosphinate a polyamide composition that is further excellent in flame retardancy can be obtained by further containing (G) a flame retardant aid.
- the flame retardant aid is not particularly limited as long as it is the above flame retardant aid.
- These (G) flame retardant aids may be used alone or in combination of two or more.
- E As a flame retardant aid used together with a phosphinate, calcium oxide, aluminum oxide (alumina), aluminum hydroxide (boehmite), magnesium hydroxide, and zinc borate are used from the viewpoint of flame retardancy. Etc. are preferable.
- the zinc borate more preferably zinc borate represented by xZnO ⁇ yB 2 O 3 ⁇ zH 2 O (x> 0, y> 0, z ⁇ 0), more preferably, 2ZnO ⁇ 3B 2 O
- Zinc borate represented by 3 ⁇ 3.5H 2 O, 4ZnO ⁇ B 2 O 3 ⁇ H 2 O, and 2ZnO ⁇ 3B 2 O 3 may be mentioned.
- These metal borate compounds may be treated with a surface treatment agent such as a silane coupling agent and a titanate coupling agent.
- a surface treatment agent such as a silane coupling agent and a titanate coupling agent.
- the average particle size of the flame retardant aid is preferably 30 ⁇ m or less, more preferably 15 ⁇ m or less, and even more preferably 7 ⁇ m or less.
- the blending amount of (E) phosphinate in the polyamide composition, and optionally (G) the flame retardant aid and / or (B) the blending amount of the inorganic filler is there is no particular limitation.
- the blending amount of the phosphinic acid salt in the polyamide composition is preferably 20 to 90 parts by mass, more preferably 25 to 80 parts by mass, and further preferably 30 to 60 parts by mass with respect to 100 parts by mass of the polyamide. It is. By setting the blending amount of the phosphinate to 20 parts by mass or more, a polyamide composition having excellent flame retardancy can be obtained.
- molding process can be suppressed by the compounding quantity of a halogenated flame retardant being 90 mass parts or less. Furthermore, deterioration of mechanical properties such as toughness and rigidity and appearance of the molded product can be suppressed.
- the blending amount of the flame retardant aid in the polyamide composition is preferably 0 to 30 parts by mass, more preferably 1 to 30 parts by mass, and further preferably 1 to 20 parts by mass with respect to 100 parts by mass of the polyamide. And more preferably 2 to 15 parts by mass.
- a polyamide composition having further excellent flame retardancy can be obtained.
- the viscosity at the time of melt processing can be controlled within an appropriate range, the torque at the time of extrusion is increased, the moldability at the time of molding is reduced, and the molding Deterioration of product appearance can be suppressed.
- a polyamide composition having excellent toughness can be obtained without impairing the properties of polyamide having excellent mechanical properties such as toughness and rigidity.
- the amount of the inorganic filler in the polyamide composition is preferably 0 to 200 parts by mass, more preferably 0.1 to 200 parts by mass, and further preferably 1 to 180 parts by mass with respect to 100 parts by mass of the polyamide. Part, more preferably 5 to 150 parts by weight.
- the relative viscosity ⁇ r at 25 ° C., the melting point Tm2, and the glass transition temperature Tg of the polyamide composition containing the phosphinate can be measured by the same method as that for the polyamide.
- the measured value in the polyamide composition containing (E) phosphinate is in the same range as the preferable value as the measured value of the polyamide, mechanical properties such as heat resistance, moldability, toughness and rigidity, and A polyamide composition having excellent chemical resistance can be obtained.
- the tensile strength of the polyamide composition containing a phosphinate is preferably 140 MPa or more, more preferably 150 MPa or more, and further preferably 160 MPa or more.
- the tensile strength can be measured according to ASTM D638 as described in the following examples. When the tensile strength is 140 MPa or more, a polyamide composition having excellent rigidity can be obtained.
- the tensile elongation of the polyamide composition containing the phosphinate is preferably 1.0% or more, more preferably 1.5% or more, and further preferably 2.0% or more.
- the tensile elongation can be measured according to ASTM D638 as described in the following examples. When the tensile elongation is 10% or more, a polyamide composition having excellent toughness can be obtained.
- the water absorption rate of the polyamide containing a phosphinate is preferably 5.0% or less, more preferably 4.0% or less, and even more preferably 3.0% or less.
- the water absorption rate can be measured by the method described in the following examples. When the water absorption is 5.0% or less, a polyamide composition excellent in low water absorption can be obtained.
- the flame retardancy of the polyamide composition containing the phosphinate was measured according to UL-94VB.
- the flame retardancy of the polyamide composition is preferably V-2 or more, more preferably V-1 or more, and further preferably V-0.
- the complete filling pressure of the polyamide composition containing the phosphinic acid salt is preferably 15 to 50%, more preferably 18 to 48%, and further preferably 20 to 45%.
- the complete filling pressure can be measured by the method described in the examples below. When the complete filling pressure is within the above range, a polyamide composition having excellent fluidity can be obtained.
- the polyamide composition of the present embodiment is a polyamide composition containing the (A) polyamide and (F) a stabilizer.
- the polyamide composition of the present embodiment by containing the stabilizer (F), the polyamide composition can be used as a polyamide composition without impairing the properties of polyamide having excellent heat resistance, fluidity, toughness, low water absorption, and rigidity. It can be set as the polyamide composition which is excellent in heat resistance, fluidity
- the (F) stabilizer used in the present embodiment includes a group consisting of a phenol stabilizer, a phosphite stabilizer, a hindered amine stabilizer, a triazine stabilizer, a sulfur stabilizer, and an inorganic phosphorus stabilizer. Is at least one selected from These stabilizers may be used alone or in combination of two or more.
- the phenol stabilizer is not particularly limited, and examples thereof include hindered phenol compounds.
- examples of the hindered phenol compound include N, N′-hexane-1,6-diylbis [3- (3,5-di-t-butyl-4-hydroxyphenylpropionamide), pentaerythrityl-tetrakis [ 3- (3,5-di-tert-butyl-4-hydroxyphenyl) propionate], N, N′-hexamethylenebis (3,5-di-tert-butyl-4-hydroxy-hydrocinnamamide), Triethylene glycol-bis [3- (3-tert-butyl-5-methyl-4-hydroxyphenyl) propionate], 3,9-bis ⁇ 2- [3- (3-tert-butyl-4-hydroxy-5] -Methylphenyl) propynyloxy] -1,1-dimethylethyl ⁇ -2,4,8,10-tetraoxapyro [5,5] unde
- the phosphite stabilizer is not particularly limited.
- phosphite stabilizer is a pentaerythritol phosphite compound.
- pentaerythritol-type phosphite compounds include 2,6-di-t-butyl-4-methylphenyl phenyl, pentaerythritol diphosphite, 2,6-di-t-butyl-4-methylphenyl, Methyl pentaerythritol diphosphite, 2,6-di-t-butyl-4-methylphenyl 2-ethylhexyl pentaerythritol diphosphite, 2,6-di-t-butyl-4-methylphenyl isodecyl Pentaerythritol diphosphite, 2,6-di-t-butyl-4-methylphenyl lauryl pentaerythritol diphosphite, 2,6-di-t-butyl-4-methyl
- Pentaerythritol phosphite compounds include bis (2,6-di-t-butyl-4-methylphenyl) pentaerythritol diphosphite, bis (2,6-di-t-butyl-4-ethylphenyl) Pentaerythritol diphosphite, bis (2,6-di-t-amyl-4-methylphenyl) pentaerythritol diphosphite, and bis (2,6-di-t-octyl-4-methylphenyl) pentaerythritol di Phosphites and the like are preferred, and bis (2,6-di-t-butyl-4-methylphenyl) pentaerythritol diphosphite is more preferred.
- the hindered amine stabilizer is not particularly limited, and examples thereof include 4-acetoxy-2,2,6,6-tetramethylpiperidine, 4-stearoyloxy-2,2,6,6-tetramethylpiperidine, 4-acryloyloxy-2,2,6,6-tetramethylpiperidine, 4- (phenylacetoxy) -2,2,6,6-tetramethylpiperidine, 4-benzoyloxy-2,2,6,6-tetra Methylpiperidine, 4-methoxy-2,2,6,6-tetramethylpiperidine, 4-stearyloxy-2,2,6,6-tetramethylpiperidine, 4-cyclohexyloxy-2,2,6,6-tetra Methylpiperidine, 4-benzyloxy-2,2,6,6-tetramethylpiperidine, 4-phenoxy-2,2,6,6-tetrame Lupiperidine, 4- (ethylcarbamoyloxy) -2,2,6,6-tetramethylpiperidine, 4- (cyclohe
- the triazine-based stabilizer is not particularly limited, and examples thereof include hydroxyphenyl triazines.
- hydroxyphenyltriazines include 2,4,6-tris (2′-hydroxy-4′-octyloxy-phenyl) -1,3,5-triazine, 2- (2′-hydroxy-4′- Hexyloxy-phenyl) -4,6-diphenyl-1,3,5-triazine, 2- (2′-hydroxy-4′-octyloxyphenyl) -4,6-bis (2 ′, 4′-dimethylphenyl) ) -1,3,5-triazine, 2- (2 ′, 4′-dihydroxyphenyl) -4,6-bis (2 ′, 4′-dimethylphenyl) -1,3,5-triazine, 2,4 -Bis (2'-hydroxy-4'-propyloxy-phenyl) -6- (2 ', 4'-dimethylphenyl) -1,3,5-
- the sulfur stabilizer is not particularly limited.
- These sulfur stabilizers may be used alone or in combination of two or more.
- the inorganic phosphorus stabilizer is not particularly limited, and examples thereof include phosphoric acids, phosphorous acids and hypophosphorous acids, and metal phosphates, metal phosphites, and metal hypophosphites. .
- Examples of phosphoric acids, phosphorous acids, and hypophosphorous acids include phosphoric acid, phosphorous acid, hypophosphorous acid, pyrophosphorous acid, and diphosphorous acid.
- metal phosphates examples include salts of the above-mentioned compounds such as phosphoric acid and Group 1 metals of the periodic table.
- the inorganic phosphorus stabilizer is preferably a soluble compound, and examples thereof include sodium phosphate, sodium phosphite, and sodium hypophosphite, and more preferably sodium phosphite and hypophosphorous acid. Sodium, more preferably sodium hypophosphite.
- the inorganic phosphorus stabilizer may be, for example, a hydrate thereof (preferably a hydrate of sodium diphosphite (NaH 2 PO 2 .nH 2 O)). These inorganic phosphorus stabilizers may be used alone or in combination of two or more.
- the blending amount of the (F) stabilizer in the polyamide composition in the present embodiment is preferably 0.01 to 5 parts by mass, more preferably 0.02 to 1 part by mass with respect to 100 parts by mass of the polyamide. More preferably, it is 0.1 to 1 part by mass.
- (F) By making the compounding quantity of a stabilizer 0.01 mass part or more, it can be set as the polyamide composition excellent in heat-resistant discoloration property and a weather resistance.
- by making the blending amount of (F) stabilizer 5 parts by mass or less it is possible to suppress the occurrence of silver on the surface of the molded product when the polyamide composition is molded, and toughness of the molded product. In addition, a molded product having excellent mechanical properties such as rigidity can be obtained.
- the method for producing the polyamide composition containing the stabilizer (F) in the present embodiment is not particularly limited as long as it is a method of mixing the polyamide (A) and the stabilizer (F).
- a method of blending a stabilizer with polyamide a method of blending a stabilizer during polymerization of polyamide, a method of blending a stabilizer when mixing polyamide and other resin, a stabilizer on the surface of polyamide powder or pellets
- Examples thereof include a method of adhering, a method of blending a stabilizer with polyamide by melt kneading, a method of blending a master batch of stabilizer with polyamide, a method of blending these methods in combination, and the like.
- the mixing method of polyamide and stabilizer is, for example, a method in which polyamide and stabilizer are mixed using a Henschel mixer and supplied to a melt kneader and kneaded, or in a polyamide melted with a single or twin screw extruder. And a method of blending a stabilizer from the side feeder.
- all the components may be supplied to the same supply port at a time, or the components may be supplied from different supply ports. .
- the melt kneading temperature is preferably about 250 to 375 ° C. as the resin temperature.
- the melt kneading time is preferably about 0.5 to 5 minutes.
- a known apparatus for example, a melt kneader such as a single or twin screw extruder, a Banbury mixer, and a mixing roll is preferably used.
- additives conventionally used for polyamide for example, inorganic fillers, pigments, dyes, flame retardants, lubrication, within a range not impairing the object of the present embodiment
- additives conventionally used for polyamide for example, inorganic fillers, pigments, dyes, flame retardants, lubrication, within a range not impairing the object of the present embodiment
- Agents, fluorescent bleaches, plasticizers, organic antioxidants, UV absorbers, nucleating agents, rubbers, reinforcing agents, and the like can also be contained.
- the relative viscosity ⁇ r, melting point Tm2 and glass transition temperature Tg at 25 ° C. of the polyamide composition containing the stabilizer (F) in the present embodiment can be measured by the same method as that for the polyamide.
- the melt shear viscosity ⁇ s of the polyamide composition containing the stabilizer (F) in the present embodiment is preferably 20 to 110, more preferably 25 to 90, and further preferably 30 to 80.
- the melt shear viscosity can be measured by the method described in the following examples. When the melt shear viscosity is within the above range, a polyamide composition having excellent fluidity can be obtained.
- the tensile strength of the polyamide composition is preferably 80 MPa or more, more preferably 85 MPa or more, and further preferably 90 MPa or more.
- the tensile strength can be measured according to ASTM D638 as described in the following examples. When the tensile strength is 80 MPa or more, a polyamide composition having excellent rigidity can be obtained.
- the tensile elongation of the polyamide composition is preferably 1.0% or more, more preferably 2.0% or more, and further preferably 3.0% or more.
- the tensile elongation can be measured according to ASTM D638 as described in the following examples. When the tensile elongation is 3.0% or more, a polyamide composition having excellent toughness can be obtained.
- the water absorption rate of the polyamide composition is preferably 5.0% or less, more preferably 4.0% or less, and even more preferably 3.0% or less.
- the water absorption rate can be measured by the method described in the following examples. When the water absorption is 5.0% or less, a polyamide composition excellent in low water absorption can be obtained.
- the change ⁇ b in color tone between before and after rework of the polyamide composition containing the stabilizer (F) in the present embodiment is preferably 9 or less, more preferably 6 or less.
- the change ⁇ b in color tone can be measured by the method described in the following examples. When the color change ⁇ b is 9 or less, a polyamide composition having excellent heat discoloration can be obtained.
- the color difference ⁇ E of the polyamide composition containing the stabilizer is preferably 9 or less, more preferably 5 or less.
- the color difference ⁇ E can be measured by the method described in the examples below. When the color difference ⁇ E is 9 or less, a polyamide composition having excellent weather resistance can be obtained.
- the polyamide or polyamide composition of the present embodiment is a known molding method such as press molding, injection molding, gas assist injection molding, welding molding, extrusion molding, blow molding, film molding, hollow molding, multilayer molding, and melting.
- Various molded articles can be obtained using spinning or the like.
- the polyamide or polyamide composition of the present embodiment can be suitably used as a raw material for automobile parts.
- automobile parts include intake system parts, cooling system parts, interior parts, exterior parts, and electrical parts.
- the automobile intake system parts are not particularly limited, and examples thereof include an air intake manifold, an intercooler inlet, an exhaust pipe cover, an inner bush, a bearing retainer, an engine mount, an engine head cover, a resonator, and a throttle body.
- the automobile cooling system component is not particularly limited, and examples thereof include a chain cover, a thermostat housing, an outlet pipe, a radiator tank, an oil netter, and a delivery pipe.
- the automobile fuel system parts are not particularly limited, and examples thereof include a fuel delivery pipe and a gasoline tank case.
- the interior part is not particularly limited, and examples thereof include an instrument panel, a console box, a glove box, a steering wheel, and a trim.
- the exterior parts are not particularly limited, and examples include a mall, a lamp housing, a front grill, a mud guard, a side bumper, a door mirror stay, and a roof rail.
- the electrical component is not particularly limited, and examples thereof include a connector, a wire harness connector, a motor component, a lamp socket, a sensor on-vehicle switch, and a combination switch.
- a molded article obtained from the polyamide composition of the present embodiment is excellent in heat resistance, rigidity, toughness, moldability, low water absorption, and the like. Since it is further excellent in vibration fatigue resistance, fluidity, and heat aging resistance, it can be suitably used as an automobile intake system component.
- the strength half-life of the molded product is preferably 40 days or longer, more preferably 45 days or longer, and even more preferably 50 days or longer. The strength half-life can be measured by the method described in the Examples below. When the strength half-life of the molded product is 40 days or more, an automobile intake system component having excellent heat resistance, particularly heat aging characteristics can be obtained.
- the fracture stress of the molded product is preferably 45 MPa or more, more preferably 50 MPa or more, and further preferably 55 MPa or more.
- the fracture stress can be measured by the method described in the following examples.
- the water absorption rate of the molded product is preferably 5.0% or less, more preferably 4.0% or less, and even more preferably 3.0% or less.
- the water absorption rate can be measured by the method described in the following examples. When the water absorption rate of the molded product is 5.0% or less, it is possible to obtain an automobile intake system component that is excellent in low water absorption.
- the molded article obtained from the polyamide composition of the present embodiment is excellent in heat resistance, rigidity, toughness, moldability, and low water absorption. Since it is further excellent in LLC property, it can be suitably used as an automotive cooling system component.
- the strength half-life of the molded product is preferably 40 days or longer, more preferably 45 days or longer, and even more preferably 50 days or longer. The strength half-life can be measured by the method described in the Examples below. When the strength half-life of the molded product is 40 days or more, it is possible to obtain an automotive cooling system component having excellent heat resistance, particularly heat aging characteristics.
- the tensile strength retention after immersion of the molded product is preferably 60% or more, more preferably 75% or more, and further preferably 80% or more.
- the tensile strength after immersion can be measured by the method described in the following examples.
- an automobile cooling system component having excellent LLC resistance can be obtained.
- the water absorption rate of the molded product is preferably 5.0% or less, more preferably 4.0% or less, and even more preferably 3.0% or less.
- the water absorption rate can be measured by the method described in the following examples. When the water absorption rate of the molded product is 5.0% or less, it is possible to obtain an automobile cooling system component that is excellent in low water absorption.
- the molded product of the polyamide or polyamide composition in the present embodiment is a known molding method, such as press molding, injection molding, gas assist injection molding, welding molding, extrusion molding, blow molding, film molding, hollow molding, multilayer molding, And a generally known plastic molding method such as melt spinning.
- the molded product obtained from the polyamide or the polyamide composition in the present embodiment is excellent in heat resistance, toughness, moldability, and low water absorption. Therefore, the polyamide and the polyamide composition of the present embodiment are not only for automobiles, but also as various parts materials for, for example, electric and electronic, industrial materials, daily use and household goods, and extrusion applications. Can be suitably used.
- the electrical and electronic use there are no particular limitations on the electrical and electronic use, and for example, it is used for connectors, switches, relays, printed wiring boards, electronic component housings, outlets, noise filters, coil bobbins, motor end caps, and the like.
- industrial equipment it is not particularly limited.
- it is used for gears, cams, insulating blocks, valves, electric tool parts, agricultural equipment parts, engine covers, and the like.
- It is not specifically limited as for daily use and household goods, for example, it is used for buttons, food containers, office furniture and the like.
- the extrusion application is not particularly limited, and for example, it is used for a film, a sheet, a filament, a tube, a rod, a hollow molded product, and the like.
- Inorganic filler (15) Glass fiber (GF) Made by Nippon Electric Glass Product name ECS03T275H Average fiber diameter 10 ⁇ m ⁇ , cut length 3 mm
- F Stabilizer
- the molar percentage of (a-1) alicyclic dicarboxylic acid is (number of moles of (a-1) alicyclic dicarboxylic acid added as raw material monomer / number of moles of all (a) dicarboxylic acid added as raw material monomer. ) ⁇ 100.
- the mol% of the diamine having a substituent branched from the main chain is the number of moles of the diamine having the substituent branched from the main chain (b-1) added as a raw material monomer, excluding the additional component. / The number of moles of (b) diamine added as a raw material monomer) ⁇ 100.
- (c) mol% of lactam and / or aminocarboxylic acid is ((c) moles of lactam and / or aminocarboxylic acid added as raw material monomer / all (a) dicarboxylic acid added as raw material monomer. (B) mole number of all diamines + (c) mole number of lactam and / or aminocarboxylic acid) ⁇ 100.
- Melting points Tm1, Tm2 (° C.) According to JIS-K7121, measurement was performed using Diamond-DSC manufactured by PERKIN-ELMER. The measurement condition is that the temperature of an endothermic peak (melting peak) that appears when about 10 mg of a sample is heated to 300 to 350 ° C. according to the melting point of the sample at a heating rate of 20 ° C./min in a nitrogen atmosphere is Tm1 (° C.). After maintaining the temperature in the molten state at the highest temperature rise for 2 minutes, the temperature was lowered to 30 ° C. at a temperature drop rate of 20 ° C./min, held at 30 ° C.
- the increment of the mass after water absorption with respect to the mass before water absorption was taken as the water absorption amount
- Copper concentration, halogen concentration, and molar ratio of halogen to copper (halogen / Cu)
- the copper concentration was determined by adding sulfuric acid to the sample, dropping nitric acid while heating to decompose the organic component, measuring the decomposition solution in pure water, and quantifying it by ICP emission analysis (high frequency plasma emission analysis).
- ICP emission analysis high frequency plasma emission analysis.
- Vista-Pro manufactured by SEIKO ELECTRONIC INDUSTRY CO., LTD. was used. Taking iodine as an example, the halogen concentration is combusted in a flask in which the sample is replaced with high-purity oxygen, and the generated gas is collected in an absorbing solution.
- the iodine in the collected solution is 1 / 100N silver nitrate solution. Quantification was performed using potentiometric titration. The molar ratio of halogen to copper (halogen / Cu) was calculated by converting the molecular weight into a mole using the above quantitative values.
- Trans isomer ratio 30-40 mg of polyamide was dissolved in 1.2 g of hexafluoroisopropanol deuteride and measured by 1 H-NMR.
- the trans isomer ratio was determined from the ratio of the peak area of 1.98 ppm derived from the trans isomer and the peak areas of 1.77 ppm and 1.86 ppm derived from the cis isomer.
- Example 1 Polymerization reaction of polyamide was carried out by “hot melt polymerization method”.
- (A) CHDA 896 g (5.20 mol) and (b) 2MPD 604 g (5.20 mol) were dissolved in 1500 g of distilled water to prepare an equimolar 50 mass% homogeneous aqueous solution of raw material monomers.
- 15 g (0.13 mol) of 2MPD was added to this homogeneous aqueous solution.
- the obtained aqueous solution was charged into an autoclave (made by Nitto High Pressure Co., Ltd.) having an internal volume of 5.4 L, kept warm until the liquid temperature (internal temperature) reached 50 ° C., and the autoclave was purged with nitrogen.
- the liquid temperature was continuously heated from about 50 ° C. until the pressure in the autoclave tank reached about 2.5 kg / cm 2 as gauge pressure (hereinafter, all pressure in the tank was expressed as gauge pressure). (The liquid temperature in this system was about 145 ° C.). While maintaining the pressure in the tank at about 2.5 Kg / cm 2 , water was removed from the system while heating was continued until the concentration of the aqueous solution was about 75% (the liquid temperature in this system was about It was 160 ° C.). The removal of water was stopped and heating was continued until the pressure in the tank reached about 30 Kg / cm 2 (the liquid temperature in this system was about 245 ° C.). Heating was continued until the final temperature reached ⁇ 50 ° C.
- the heater temperature was adjusted so that the final temperature of the resin temperature (liquid temperature) was about 350 ° C. With the resin temperature kept in this state, the inside of the tank was maintained for 30 minutes under a reduced pressure of 400 torr with a vacuum apparatus.
- Example 2 In Example 1, as the (a) dicarboxylic acid, (b) diamine, and (c) lactam and / or aminocarboxylic acid, the compounds and amounts shown in Table 1 or 2 were used, and the final temperature of the resin temperature.
- the polyamide was polymerized by the method described in Example 1 except that the temperature was set to the temperature described in Table 4 or 5 ("hot melt polymerization method"). Tables 4 and 5 show the measurement results of the obtained polyamide based on the above measurement method.
- Example 1 In Example 1, as the (a) dicarboxylic acid, (b) diamine, and (c) lactam and / or aminocarboxylic acid, the compounds and amounts shown in Table 3 were used, and the final temperature of the resin temperature was expressed. Polymerization of polyamide was carried out by the method described in Example 1 except that the temperature described in 6 was used (“hot melt polymerization method”). In Comparative Example 1, since it was solidified in the autoclave during the polymerization, it could not be taken out as a strand. Therefore, after cooling, it was taken out as a lump and pulverized with a pulverizer to a size about pellets. Since the foaming was severe, the molded product could not be obtained.
- Example 2 In Example 1, as the (a) dicarboxylic acid, (b) diamine, and (c) lactam and / or aminocarboxylic acid, the compounds and amounts shown in Table 3 were used, and the final temperature of the resin temperature was expressed. Polymerization of polyamide was carried out by the method described in Example 1 except that the temperature described in 6 was used (“hot melt polymerization method”). Table 6 shows the measurement results of the obtained polyamide based on the above measurement method.
- the polyamides of Examples 1 to 21 obtained by polymerizing specific (a) and (b) are all heat resistant, fluid, tough, low water absorption, and rigid. In this respect, it has particularly excellent characteristics.
- Comparative Example 1 which is a polyamide containing less than 50 mol% of 2-methylpentamethylenediamine, it solidifies during copolymerization and cannot be taken out as a strand, and a molded product is obtained. I could't.
- the polyamide of Comparative Example 4 produced by the method disclosed in Patent Document 1 has too low fluidity and is not sufficient in terms of moldability. Moreover, the toughness was not sufficient.
- Example 22 Polymerization reaction of polyamide was carried out by “hot melt polymerization / solid phase polymerization method”. About hot melt polymerization, the same operation as Example 1 was carried out in the same amount and procedure to obtain polyamide (polyamide (I)). Table 7 shows the measurement results of the obtained polyamide based on the above measurement method. Of this, 1300 g was charged into a ribbon stirring type heating device (ribocorn, manufactured by Okawara Seisakusho) for solid phase polymerization, and nitrogen substitution was performed at room temperature. Heating was performed for 12 hours so that the resin temperature would be 200 ° C. while nitrogen was passed.
- ribocorn manufactured by Okawara Seisakusho
- polyamide (II) polyamide (polyamide (II)).
- Table 7 shows the measurement results of the obtained polyamide based on the above measurement method.
- polyamide (I) after solid phase polymerization, the relative viscosity at 25 ° C. is increased and the tensile elongation is increased.
- the trans isomer ratio did not change before and after solid state polymerization. Also, the degree of coloring did not change.
- Table 7 also shows the measurement results obtained based on the above-described measurement method for the polyamide of Example 1 obtained by hot melt polymerization.
- Example 23 Polymerization reaction of polyamide was carried out by “prepolymer / solid phase polymerization method”. Distilled water (500 g) was added to (a) CHDA 896 g (5.20 mol) and (b) 2MPD 604 g (5.20 mol) to prepare an equimolar 33% by mass slurry solution of raw material monomers. 15 g (0.13 mol) of 2MPD was added to the slurry. The obtained slurry was charged into an autoclave (made by Nitto High Pressure Co., Ltd.) having an internal volume of 5.4 L, and the inside of the autoclave was purged with nitrogen.
- an autoclave made by Nitto High Pressure Co., Ltd.
- Table 7 shows the measurement results of the obtained prepolymer based on the above measurement method.
- the trans isomer ratio of 1,4-cyclohexanedicarboxylic acid in the prepolymer was 85%.
- coloring was seen with polyamide (I).
- polyamide (II) Using 1300 g of the obtained prepolymer, solid phase polymerization was carried out in the same manner as in Example 22 to obtain polyamide (polyamide (II)).
- Table 7 shows the measurement results of the obtained polyamide based on the above measurement method. Polyamide (II) was improved in relative viscosity as compared with the prepolymer, but colored.
- Example 24 A polyamide polymerization reaction was carried out by the “prepolymer / extrusion polymerization method”.
- the same operation as in Example 23 was carried out with the same amount and procedure, and a polyamide prepolymer was obtained (polyamide (I)).
- 1300 g of the obtained prepolymer was used for post-polymerization in an extrusion polymerization apparatus (KRC kneader manufactured by Kurimoto Iron Works Co., Ltd.).
- the prepolymer was introduced so that the jacket temperature was 350 ° C., the degree of vacuum was ⁇ 0.5 MPa (gauge pressure), and the residence time was 30 minutes.
- polyamide (II) The strand was cooled and cut to obtain polyamide as a pellet (polyamide (II)).
- Table 7 shows the measurement results of the obtained polyamide based on the above measurement method.
- Polyamide (II) was improved in relative viscosity at 25 ° C. as compared with the prepolymer, but was colored.
- Example 25 Polyamide composition containing inorganic filler
- the glass fiber (GF) is supplied from the side feed port on the downstream side of the extruder (the resin supplied from the top feed port is sufficiently melted), and the melt-kneaded product extruded from the die head is cooled in a strand shape. And pelletized to obtain polyamide composition pellets.
- the blending amount was 55 parts by mass of glass fiber (GF) with respect to 100 parts by mass of polyamide.
- Table 8 shows the measurement results of the obtained polyamide composition based on the above-described measurement method.
- Examples 26 to 45 The same procedure as in Example 25 was performed except that each polyamide of Examples 2 to 21 was used instead of the polyamide of Example 1.
- Tables 8 and 9 show the measurement results of the obtained polyamide composition based on the above measurement method.
- Example 46 In Example 29, it implemented similarly to Example 29 except having set it as 100 mass parts of glass fibers (GF) with respect to 100 mass parts of polyamides. Table 9 shows the results of measurements performed on the obtained polyamide composition based on the above-described measurement methods.
- Example 11 The same procedure as in Example 25 was performed except that the polyamide of Comparative Example 4 was used instead of the polyamide of Example 1 and the glass fiber (GF) was 100 parts by mass with respect to 100 parts by mass of the polyamide.
- Table 10 shows the measurement results of the obtained polyamide composition based on the above measurement method.
- the polyamide compositions of Examples 25 to 46 containing the polyamide obtained by polymerizing the specific (a) and (b) and the inorganic filler were found to have heat resistance, fluidity, toughness and low water absorption. It has particularly excellent characteristics in all of the properties and rigidity.
- Comparative Example 8 containing a polyamide obtained by polymerizing less than 50 mol% of 2-methylpentamethylenediamine the extruded state was unstable, and a polyamide composition could not be obtained.
- the polyamide compositions of Comparative Examples 9 and 10 containing a polyamide obtained by polymerizing less than 50 mol% of an alicyclic dicarboxylic acid were inferior in terms of heat resistance and low water absorption.
- Example 47 was carried out in the same manner as Example 47 except that each polyamide of Examples 2 to 21 was used instead of the polyamide of Example 1.
- Tables 11 and 12 show the measurement results of the obtained polyamide composition based on the above measurement method.
- Example 47 an attempt was made in the same manner as in Example 47 except that the polyamide of Comparative Example 1 was used instead of the polyamide of Example 1, but the extruded state was very unstable and a polyamide composition was obtained. I could not.
- Example 47 was carried out in the same manner as Example 47 except that each polyamide of Comparative Examples 2 to 7 was used in place of the polyamide of Example 1.
- Table 13 shows the results of measurements performed on the obtained polyamide composition based on the above-described measurement methods.
- Table 14 shows the measurement results of measurements performed on the polyamide of Example 29 based on the above measurement method.
- Example 51 it implemented like Example 51 except having used the granule (1) of the manufacture example 1 of 3.1 mass part with respect to the polyamide of Example 5 of 100 mass parts.
- Table 14 shows the results of measurements performed on the obtained polyamide composition based on the above-described measurement methods.
- Example 69 In Example 51, it implemented like Example 51 except having used the granule (1) of the manufacture example 1 of 9.2 mass parts with respect to the polyamide of Example 5 of 100 mass parts. Table 14 shows the results of measurements performed on the obtained polyamide composition based on the above-described measurement methods.
- Example 70 In Example 51, it implemented like Example 51 except having used 12.1 mass parts granule (1) of the manufacture example 1 with respect to the polyamide of Example 51 of 100 mass parts. Table 14 shows the results of measurements performed on the obtained polyamide composition based on the above-described measurement methods.
- Example 71 In Example 51, it implemented like Example 51 except having used the granule (2) of the manufacture example 2 of 3.2 mass parts with respect to the polyamide of Example 51 of 100 mass parts. Table 14 shows the results of measurements performed on the obtained polyamide composition based on the above-described measurement methods.
- Example 72 In Example 51, it implemented like Example 51 except having used 15.0 mass parts granule (3) of manufacture example 3 with respect to the polyamide of Example 51 of 100 mass parts. Table 14 shows the results of measurements performed on the obtained polyamide composition based on the above-described measurement methods.
- the polyamide compositions of Examples 51 to 72 containing the polyamide obtained by polymerizing specific (a) and (b), the copper compound and the metal halide were found to be heat resistant, fluid It has particularly excellent characteristics in terms of heat resistance, toughness, low water absorption, and rigidity, and in terms of heat aging resistance.
- Comparative Example 15 containing polyamide containing less than 50 mol% of 2-methylpentamethylenediamine the extruded state was unstable, and a polyamide composition could not be obtained.
- the polyamide compositions of Comparative Examples 16 and 17 containing a polyamide obtained by polymerizing less than 50 mol% of an alicyclic dicarboxylic acid were inferior in terms of heat resistance and low water absorption.
- the melt shear viscosity is large, the fluidity is too low, and the moldability is not sufficient. There wasn't.
- the tensile elongation was small and the toughness was not sufficient.
- Example 73 Polyamide composition containing halogenated flame retardant
- the polyamide of Example 1 was dried in a nitrogen stream and the moisture content was adjusted to about 0.2% by mass.
- top feed provided in the most upstream part of the extruder Feed and extrude a polymer containing (A) polyamide, (D) halogen flame retardant, (G) flame retardant aid, and (H) ⁇ , ⁇ unsaturated dicarboxylic anhydride in advance.
- (B) Inorganic filler is supplied from the side feed port on the machine downstream side (the resin supplied from the top feed port is sufficiently melted), and the melt-kneaded product extruded from the die head is cooled in a strand shape. Pelletization gave polyamide composition pellets.
- the blending amount is 100 parts by weight of (A) polyamide, (C) 45.0 parts by weight of halogenated flame retardant, (G) 7.0 parts by weight of flame retardant aid, (H) ⁇ , ⁇ unsaturated dicarboxylic acid. It was set as 4.0 mass parts of polymer containing an anhydride and 70.0 mass parts of (B) inorganic filler. Table 15 shows the results of measurements performed on the obtained polyamide composition based on the above-described measurement methods.
- Example 73 was carried out in the same manner as Example 73 except that the polyamides of Examples 2 to 21 were used in place of the polyamide of Example 1.
- Tables 15 and 16 show the results of measurements performed on the obtained polyamide composition based on the above measurement method.
- Example 94 In Example 77, (H) 15.0 parts by mass of a flame retardant aid and (B) an inorganic filler 75.0 without blending (H) a polymer containing an ⁇ , ⁇ unsaturated dicarboxylic acid anhydride. The same operation as in Example 77 was carried out except that the parts were by mass. Table 16 shows the results of measurements performed on the obtained polyamide composition based on the above-described measurement methods.
- Example 95 In Example 77, it implemented like Example 77 except having set magnesium hydroxide as 7.0 mass part as (G) flame retardant adjuvant. Table 16 shows the results of measurements performed on the obtained polyamide composition based on the above-described measurement methods.
- Example 73 In Example 73, an attempt was made in the same manner as in Example 73 except that the polyamide of Comparative Example 1 was used instead of the polyamide of Example 1, but the extruded state was very unstable and a polyamide composition could be obtained. could not.
- Example 73 was carried out in the same manner as Example 73 except that the polyamides of Comparative Examples 2 to 7 were used in place of the polyamide of Example 1.
- Table 17 shows the results of measurements performed on the obtained polyamide composition based on the above-described measurement methods.
- the polyamide compositions of Examples 73 to 94 containing a polyamide obtained by polymerizing specific (a) and (b) and a halogen-based flame retardant have heat resistance, fluidity, toughness, low It has particularly excellent characteristics in terms of water absorption and rigidity, and further in terms of flame retardancy.
- Comparative Example 22 containing polyamide obtained by polymerizing less than 50 mol% of 2-methylpentamethylenediamine the extruded state was unstable, and a polyamide composition could not be obtained.
- the polyamide compositions of Comparative Examples 23 and 24 containing a polyamide obtained by polymerizing less than 50 mol% of an alicyclic dicarboxylic acid were inferior in terms of heat resistance and low water absorption.
- the polyamide composition of Comparative Example 25 containing the polyamide produced by the method disclosed in Patent Document 1 the flow length is short, the fluidity is too low, and the moldability is not sufficient. It was. Moreover, the tensile elongation was small and the toughness was not sufficient. Comparative Example 28 containing PA66 was inferior in terms of heat resistance and low water absorption.
- Example 96 Polyamide composition containing phosphinate and / or diphosphinate
- the polyamide of Example 1 was dried in a nitrogen stream and the moisture content was adjusted to about 0.2% by mass.
- the blending amount is (E) 100 parts by mass of polyamide (E) 42.0 parts by mass of phosphinate, (G) 2.0 parts by mass of flame retardant aid, (B) 48.0 parts by mass of inorganic filler. It was. Table 18 shows the results of measurements performed on the obtained polyamide composition based on the above-described measurement methods.
- Example 96 was carried out in the same manner as Example 96 except that the polyamides of Examples 2 to 21 were used in place of the polyamide of Example 1. Tables 18 and 19 show the results of measurements performed on the obtained polyamide composition based on the above measurement method.
- Example 117 In Example 100, it implemented like Example 100 except not mix
- Example 118 In Example 100, it implemented similarly to Example 100 except having mix
- Example 96 In Example 96, an attempt was made in the same manner as in Example 96 except that the polyamide of Comparative Example 1 was used instead of the polyamide of Example 1, but the extruded state was very unstable and a polyamide composition could be obtained. could not.
- Example 96 was carried out in the same manner as Example 96 except that the polyamides of Comparative Examples 2 to 7 were used in place of the polyamide of Example 1.
- Table 20 shows the results of measurements performed on the obtained polyamide composition based on the above-described measurement methods.
- the polyamide compositions of Examples 96 to 118 containing the polyamides obtained by polymerizing specific (a) and (b) and phosphinic acid salts have heat resistance, fluidity, toughness and low water absorption. It has particularly excellent characteristics in terms of all properties and rigidity, and further in terms of flame retardancy.
- Comparative Example 29 containing polyamide obtained by polymerizing less than 50 mol% of 2-methylpentamethylenediamine the extruded state was unstable, and a polyamide composition could not be obtained.
- the polyamide compositions of Comparative Examples 30 and 31 containing a polyamide obtained by polymerizing less than 50 mol% of an alicyclic dicarboxylic acid were inferior in terms of heat resistance and low water absorption.
- the polyamide composition of Comparative Example 32 containing the polyamide produced by the method disclosed in Patent Document 1 the complete filling pressure is large, the fluidity is too low, and the moldability is not sufficient. There wasn't. Moreover, the tensile elongation was small and the toughness was not sufficient. Comparative Example 35 containing PA66 was inferior in terms of heat resistance and low water absorption.
- Example 119 was carried out in the same manner as Example 119, except that each polyamide of Examples 2 to 21 was used instead of the polyamide of Example 1.
- Tables 21 and 22 show the measurement results of the obtained polyamide composition based on the above measurement method.
- Example 119 In Example 119, an attempt was made in the same manner as in Example 119 except that the polyamide of Comparative Example 1 was used instead of the polyamide of Example 1, but the extruded state was very unstable, and a polyamide composition was obtained. I could not.
- Example 119 was carried out in the same manner as Example 119, except that each polyamide of Comparative Examples 2 to 7 was used instead of the polyamide of Example 1.
- Table 23 shows the results of measurements performed on the obtained polyamide composition based on the above-described measurement methods.
- Example 140 In Example 123, instead of the stabilizer (21) N, N′-hexane-1,6-diylbis [3- (3,5-di-t-butyl-4-hydroxyphenylpropionamide)], a stable Agent (22) Performed in the same manner as in Example 123 except that bis (2,6-di-t-butyl-4-methylphenyl) pentaerythritol diphosphite was used.
- Table 24 shows the measurement results of measurements performed on the obtained polyamide composition based on the above measurement methods.
- Example 141 In Example 123, instead of the stabilizer (21) N, N′-hexane-1,6-diylbis [3- (3,5-di-t-butyl-4-hydroxyphenylpropionamide)], a stable The same procedure as in Example 123 was performed except that the agent (23) bis- (2,2,6,6-tetramethyl-4-piperidyl) -sebacate was used. Table 24 shows the measurement results of measurements performed on the obtained polyamide composition based on the above measurement methods.
- Example 142 In Example 123, instead of the stabilizer (21) N, N′-hexane-1,6-diylbis [3- (3,5-di-t-butyl-4-hydroxyphenylpropionamide)], a stable Agent (24) Performed in the same manner as in Example 123 except that 2- (2′-hydroxy-4′-hexyloxyphenyl) -4,6-diphenyl-1,3,5-triazine was used. Table 24 shows the measurement results of measurements performed on the obtained polyamide composition based on the above measurement methods.
- Example 143 In Example 123, instead of the stabilizer (21) N, N′-hexane-1,6-diylbis [3- (3,5-di-t-butyl-4-hydroxyphenylpropionamide)], a stable Agent (25) Performed in the same manner as in Example 123 except that 0.1 parts by mass of sodium hypophosphite was used. Table 24 shows the measurement results of measurements performed on the obtained polyamide composition based on the above measurement methods.
- Example 144 In Example 123, 0.5 parts by weight of the stabilizer (21) N, N′-hexane-1,6-diylbis [3- (3,5-diyl) is added to 100 parts by weight of the polyamide of Example 5. -T-butyl-4-hydroxyphenylpropionamide)] was used in the same manner as in Example 123.
- Table 24 shows the measurement results of measurements performed on the obtained polyamide composition based on the above measurement methods.
- Example 145 In Example 123, 3.0 parts by weight of stabilizer (21) N, N′-hexane-1,6-diylbis [3- (3,5-diyl) in 100 parts by weight of the polyamide of Example 5 -T-butyl-4-hydroxyphenylpropionamide)] was used in the same manner as in Example 123.
- Table 24 shows the measurement results of measurements performed on the obtained polyamide composition based on the above measurement methods.
- Table 24 shows the measurement results of the polyamide of Example 5 measured based on the above measurement method.
- Table 24 shows the measurement results of measurements performed on the obtained polyamide composition based on the above measurement methods.
- Table 24 shows the measurement results of measurements performed on the obtained polyamide composition based on the above measurement methods.
- the polyamide composition was obtained by melt-kneading using a screw at 300 ° C. Table 24 shows the results of measurement performed on the obtained polyamide composition based on the above-described measurement method.
- the polyamide compositions of Examples 117 to 149 containing specific (a) polyamides obtained by polymerizing dicarboxylic acids and (b) diamines and stabilizers were found to have heat resistance and fluidity.
- the polyamide composition has particularly excellent characteristics in terms of toughness, low water absorption, and rigidity, and further in terms of heat discoloration resistance and weather resistance.
- Comparative Example 36 containing a polyamide obtained by polymerizing less than 50 mol% of 2-methylpentamethylenediamine the extruded state was unstable, and a polyamide composition could not be obtained.
- Comparative Examples 37 and 38 containing polyamide by polymerizing less than 50 mol% of alicyclic dicarboxylic acid were inferior in terms of heat resistance and low water absorption. Furthermore, in Comparative Example 39 containing a polyamide produced by the method disclosed in Patent Document 1, the melt shear viscosity is large, the fluidity is too low, and the moldability is not sufficient. Moreover, the tensile elongation was small and the toughness was not sufficient. Comparative Example 42 containing PA66 was inferior in terms of heat resistance and low water absorption.
- Japanese Patent Application No. 2008-62811 Japanese patent application filed on March 24, 2008 (Japanese Patent Application No. 2008-75926), and October 10, 2008. This is based on a Japanese patent application (Japanese Patent Application No. 2008-264182) filed in Japan, the contents of which are incorporated herein by reference.
- the present invention can provide a high-melting-point polyamide excellent in heat resistance, fluidity, toughness, low water absorption, and rigidity.
- the polyamide of the present invention can be suitably used as a molding material for various parts such as automobiles, electric and electronic, industrial materials, industrial materials, daily products and household products. Have potential.
Abstract
Description
また、家電などの電気及び電子産業において、表面実装(SMT)ハンダの鉛フリー化に対応すべく、ハンダの融点上昇に耐えることができる、ポリアミド材料に対する高耐熱化が要求されている。
PA6及びPA66などのポリアミドでは、融点が低く、耐熱性の点でこれらの要求を満たすことができない。
しかしながら、PA6Tは、融点が370℃程度という高融点ポリアミドであるため、溶融成形により成形品を得ようとしても、ポリアミドの熱分解が激しく起こり、十分な特性を有する成形品を得ることが難しい。
特許文献2には、ジカルボン酸単位として1,4-シクロヘキサンジカルボン酸を1~40%配合した半脂環族ポリアミドの電気及び電子部材はハンダ耐熱性が向上することが開示され、特許文献3には、自動車部品では、流動性及び靭性などに優れることが開示されている。
また、特許文献5には、トランス/シス比が50/50から97/3である1,4-シクロヘキサンジカルボン酸を原料として用いたポリアミドが、耐熱性、低吸水性、及び耐光性などに優れることが開示されている。
特許文献4及び5に開示されたポリアミドも、靭性、剛性、及び流動性の面で改善が不十分である。
(1)(a)少なくとも50モル%の脂環族ジカルボン酸を含むジカルボン酸と、
(b)少なくとも50モル%の、主鎖から分岐した置換基を持つジアミンを含むジアミンと、を重合させた、ポリアミド。
(2)前記主鎖から分岐した置換基を持つジアミンが、2-メチルペンタメチレンジアミンである、(1)に記載のポリアミド。
(3)前記脂環族ジカルボン酸が、1,4-シクロヘキサンジカルボン酸である、(1)又は(2)に記載のポリアミド。
(4)前記ジカルボン酸が、炭素数10以上の脂肪族ジカルボン酸をさらに含む、(1)~(3)のいずれかに記載のポリアミド。
(5)(c)ラクタム及び/又はアミノカルボン酸をさらに共重合させた、(1)~(4)のいずれかに記載のポリアミド。
(6)融点が270~350℃である、(1)~(5)のいずれかに記載のポリアミド。
(7)トランス異性体比率が50~85%である、(1)~(6)のいずれかに記載のポリアミド。
(8)b値が0以下である、(1)~(7)のいずれかに記載のポリアミド。
(9)(A)(1)~(8)のいずれかに記載のポリアミドと、
(B)無機充填材と、を含有するポリアミド組成物。
(10)(A)(1)~(8)のいずれかに記載のポリアミドと、
(C)銅化合物及び金属ハロゲン化物と、を含有するポリアミド組成物。
(11)(A)(1)~(8)のいずれかに記載のポリアミドと、
(D)ハロゲン系難燃剤と、を含有するポリアミド組成物。
(12)(A)(1)~(8)のいずれかに記載のポリアミドと、
(E)ホスフィン酸塩及び/又はジホスフィン酸塩と、を含有するポリアミド組成物。
(13)(A)(1)~(8)のいずれかに記載のポリアミドと、
(F)安定剤と、を含有するポリアミド組成物。
(14)(9)~(13)のいずれかに記載のポリアミド組成物を含む、自動車部品。
(15)自動車吸気系部品又は自動車冷却系部品である、(14)に記載の自動車部品。
(16)(a)少なくとも50モル%の脂環族ジカルボン酸を含むジカルボン酸と、 (b)少なくとも50モル%の、主鎖から分岐した置換基を持つ脂肪族ジアミンを含むジアミンと、を重合させる工程を含む、
ポリアミドの製造方法。
(17)トランス異性体比率を50~80%に維持して重合する、(16)に記載のポリアミドの製造方法。
(18)(16)又は(17)に記載の方法により得られるポリアミド。
本実施の形態のポリアミドは、下記(a)及び(b)を重合させたポリアミドである:
(a)少なくとも50モル%の脂環族ジカルボン酸を含むジカルボン酸、
(b)少なくとも50モル%の、主鎖から分岐した置換基を持つジアミンを含むジアミン。
本実施の形態において、ポリアミドとは主鎖中にアミド(-NHCO-)結合を有する重合体を意味する。
本実施の形態に用いられる(a)ジカルボン酸は、少なくとも50モル%の脂環族ジカルボン酸を含む。
(a)ジカルボン酸として、脂環族ジカルボン酸を少なくとも50モル%含むことにより、耐熱性、流動性、靭性、低吸水性、及び剛性などを同時に満足する、ポリアミドを得ることができる。
脂環族ジカルボン酸は、無置換でも置換基を有していてもよい。
脂環族ジカルボン酸としては、1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
原料モノマーとしての脂環族ジカルボン酸は、トランス体とシス体のどちらか一方を用いてもよく、トランス体とシス体の種々の比率の混合物として用いてもよい。
脂環族ジカルボン酸は、高温で異性化し一定の比率になることやシス体の方がトランス体に比べて、ジアミンとの当量塩の水溶性が高いことから、原料モノマーとして、トランス体/シス体比がモル比にして、好ましくは50/50~0/100であり、より好ましくは40/60~10/90であり、さらに好ましくは35/65~15/85である。
脂環族ジカルボン酸のトランス体/シス体比(モル比)は、液体クロマトグラフィー(HPLC)やNMRにより求めることができる。
種々の置換基としては、例えば、炭素数1~6のアルキル基、炭素数6~12のアリール基、炭素数7~20のアリールアルキル基、クロロ基及びブロモ基などのハロゲン基、炭素数3~10のアルキルシリル基、並びにスルホン酸基及びナトリウム塩などのその塩である基などが挙げられる。
中でも、耐熱性及び低吸水性などの観点で、炭素数が10以上である脂肪族ジカルボン酸が好ましい。
炭素数が10以上である脂肪族ジカルボン酸としては、例えば、セバシン酸、ドデカン二酸、テトラデカン二酸、ヘキサデカン二酸、オクタデカン二酸、及びエイコサン二酸などが挙げられる。
中でも、耐熱性などの観点で、セバシン酸及びドデカン二酸が好ましい。
脂環族ジカルボン酸以外のジカルボン酸としては、1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
多価カルボン酸としては、1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
(a)ジカルボン酸中の(a-2)脂環族ジカルボン酸以外のジカルボン酸の割合は、0~50モル%であり、0~40モル%であることが好ましい。
ジカルボン酸と等価な化合物としては、上記ジカルボン酸に由来するジカルボン酸構造と同様のジカルボン酸構造となり得る化合物であれば特に限定されるものではなく、例えば、ジカルボン酸の無水物及びハロゲン化物などが挙げられる。
本実施の形態に用いられる(b)ジアミンは、少なくとも50モル%の、主鎖から分岐した置換基を持つジアミンを含む。
(b)ジアミンとして、主鎖から分岐した置換基を持つジアミンを少なくとも50モル%含むことにより、流動性、靭性、及び剛性などを同時に満足する、ポリアミドを得ることができる。
主鎖から分岐した置換基を持つジアミンとしては、剛性などの観点で、2-メチルペンタメチレンジアミンであることが好ましい。
主鎖から分岐した置換基を持つジアミンとしては、1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
主鎖から分岐した置換基を持つジアミン以外のジアミンとしては、1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
多価脂肪族アミンとしては、1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
本実施の形態のポリアミドは、靭性の観点で、(c)ラクタム及び/又はアミノカルボン酸をさらに共重合させることが好ましい。
本実施の形態に用いられる(c)ラクタム及び/又はアミノカルボン酸とは、重縮合可能なラクタム及び/又はアミノカルボン酸を意味する。
ラクタム及び/アミノカルボン酸としては、好ましくは、炭素数4~14のラクタム及び/又はアミノカルボン酸であり、より好ましくは、炭素数6~12のラクタム及び/又はアミノカルボン酸である。
中でも、靭性の観点で、ε-カプロラクタム、ラウロラクタムなどが好ましく、ε-カプロラクタムがより好ましい。
アミノカルボン酸としては、ω位がアミノ基で置換された炭素数4~14の直鎖又は分岐状飽和脂肪族カルボン酸であることが好ましく、例えば、6-アミノカプロン酸、11-アミノウンデカン酸、及び12-アミノドデカン酸などが挙げられ、アミノカルボン酸としては、パラアミノメチル安息香酸なども挙げられる。
末端封止剤としては、例えば、モノカルボン酸、モノアミン、無水フタル酸などの酸無水物、モノイソシアネート、モノ酸ハロゲン化物、モノエステル類、及びモノアルコール類などが挙げられ、ポリアミドの熱安定性の観点で、モノカルボン酸及びモノアミンが好ましい。
末端封止剤としては、1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
モノカルボン酸としては、1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
モノアミンとしては、1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
これらの組み合わせをポリアミドの成分として重合させることにより、耐熱性、流動性、靭性、低吸水性、及び剛性に優れることを同時に満足する高融点ポリアミドとすることができる。
ポリアミド中における脂環族ジカルボン酸構造のトランス異性体比率は、
ポリアミド中の脂環族ジカルボン酸全体中のトランス異性体である比率を表し、トランス異性体比率は、好ましくは50~85モル%であり、より好ましくは50~80モル%であり、さらに好ましくは60~80モル%である。
(a-1)脂環族ジカルボン酸としては、トランス体/シス体比(モル比)が50/50~0/100である脂環族ジカルボン酸を用いることが好ましいが、(a)ジカルボン酸と(b)ジアミンの重合により得られるポリアミドとしては、トランス異性体比率が50~85モル%であることが好ましい。
トランス異性体比率が上記範囲内にあることにより、ポリアミドは、高融点、靭性及び剛性に優れるという特徴に加えて、高いガラス転移温度による熱時剛性と、通常では耐熱性と相反する性質である流動性と、高い結晶性及び低吸水性とを同時に満足するという性質を持つ。
ポリアミドのこれらの特徴は、(a)少なくとも50モル%以上の1,4-シクロヘキサンジカルボン酸、(b)少なくとも50モル%以上の2-メチルペンタメチレンジアミンの組み合わせからなり、かつトランス異性体比率が50~85モル%であるポリアミドで特に顕著である。
本実施の形態において、トランス異性体比率は、下記実施例に記載の方法により測定することができる。
ポリアミドの製造方法としては、ポリアミドの重合度を上昇させる工程を、さらに含むことが好ましい。
1)ジカルボン酸及びジアミンの水溶液又は水の懸濁液、又はジカルボン酸及びジアミン塩と他の成分との混合物(以下、本段落において、「その混合物」と略称する。)の水溶液又は水の懸濁液を、加熱し、溶融状態を維持したまま重合させる方法(以下、「熱溶融重合法」と略称する場合がある。)、
2)熱溶融重合法で得られたポリアミドを融点以下の温度で固体状態を維持したまま重合度を上昇させる方法(以下、「熱溶融重合・固相重合法」と略称する場合がある。)、
3)ジカルボン酸及びジアミン又はその混合物の水溶液又は水の懸濁液を加熱し、析出したプレポリマーをさらにニーダーなどの押出機で再び溶融して重合度を上昇させる方法(以下、「プレポリマー・押出重合法」と略称する場合がある。)、
4)ジカルボン酸及びジアミン又はその混合物の水溶液又は水の懸濁液を加熱し、析出したプレポリマーをさらにポリアミドの融点以下の温度で固体状態を維持したまま重合度を上昇させる方法(以下、「プレポリマー・固相重合法」と略称する場合がある。)、
5)ジカルボン酸及びジアミン又はその混合物を、固体状態を維持したまま重合させる方法(以下、「固相重合法」と略称する場合がある)、
6)ジカルボン酸と等価なジカルボン酸ハライド及びジアミンを用いて重合させる方法「溶液法」。
トランス異性体比率を上記範囲内に、特に、80%以下に維持することにより、色調や引張伸度に優れ、高融点のポリアミドを得ることができる。
ポリアミドの製造方法において、重合度を上昇させてポリアミドの融点を上昇させるために、加熱の温度を上昇させたり、及び/又は加熱の時間を長くする必要が生ずるが、その場合、加熱によるポリアミドの着色や熱劣化による引張伸度の低下が起こる場合がある。また、分子量の上昇する速度が著しく低下する場合がある。
ポリアミドの着色や熱劣化による引張伸度の低下を防止することができるため、トランス異性体比率を80%以下に維持して重合することが好適である。
重合装置としては、特に限定されるものではなく、公知の装置、例えば、オートクレーブ型反応器、タンブラー型反応器、及びニーダーなどの押出機型反応器などが挙げられる。
バッチ式の熱溶融重合法としては、例えば、水を溶媒として、ポリアミド成分((a)ジカルボン酸、(b)ジアミン、及び、必要に応じて、(c)ラクタム及び/又はアミノカルボン酸)を含有する約40~60質量%の溶液を、110~180℃の温度及び約0.035~0.6MPa(ゲージ圧)の圧力で操作される濃縮槽で、約65~90質量%に濃縮して濃縮溶液を得る。次いで、該濃縮溶液をオートクレーブに移し、容器における圧力が約1.5~5.0MPa(ゲージ圧)になるまで加熱を続ける。その後、水及び/又はガス成分を抜きながら圧力を約1.5~5.0MPa(ゲージ圧)に保ち、温度が約250~350℃に達した時点で、大気圧まで降圧する(ゲージ圧は、0MPa)。大気圧に降圧後、必要に応じて減圧することにより、副生する水を効果的に除くことができる。その後、窒素などの不活性ガスで加圧し、ポリアミド溶融物をストランドとして押し出す。該ストランドを、冷却、カッティングしてペレットを得る。
連続式の熱溶融重合法としては、例えば、水を溶媒としてポリアミド成分を含有する約40~60質量%の溶液を、予備装置の容器において約40~100℃まで予備加熱し、次いで、濃縮層/反応器に移し、約0.1~0.5MPa(ゲージ圧)の圧力及び約200~270℃の温度で約70~90%に濃縮して濃縮溶液を得る。該濃縮溶液を約200~350℃の温度に保ったフラッシャーに排出し、その後、大気圧まで降圧する(ゲージ圧は、0MPa)。大気圧に降圧後、必要に応じて減圧する。その後、ポリアミド溶融物は押し出されてストランドとなり、冷却、カッティングされペレットとなる。
本実施の形態におけるポリアミドの分子量は、靭性及び剛性などの機械物性並びに成形性などの観点で、JIS-K6810に従って測定した98%硫酸中濃度1%、25℃の相対粘度ηrにおいて、好ましくは1.5~7.0であり、より好ましくは1.7~6.0であり、さらに好ましくは1.9~5.5である。
25℃の相対粘度の測定は、下記実施例に記載するように、JIS-K6810に準じて行うことができる。
ポリアミドの融点Tm2を270℃以上とすることにより、耐熱性に優れるポリアミドとすることができる。ポリアミドの融点Tm2を350℃以下とすることにより、押出、成形などの溶融加工でのポリアミドの熱分解などを抑制することができる。
融点及び融解熱量の測定装置としては、例えば、PERKIN-ELMER社製Diamond-DSCなどが挙げられる。
ガラス転移温度を90℃以上とすることにより、耐熱性や耐薬品性に優れるポリアミドとすることができる。また、ガラス転移温度を170℃以下とすることにより、外観のよい成形品を得ることができる。
ガラス転移温度の測定は、下記実施例に記載するように、JIS-K7121に準じて行うことができる。
ガラス転移温度の測定装置としては、例えば、PERKIN-ELMER社製Diamond-DSCなどが挙げられる。
溶融せん断粘度は、下記実施例に記載の方法により測定することができる。
溶融せん断粘度が上記範囲内にあることにより、流動性に優れるポリアミドを得ることができる。
引張強度の測定は、下記実施例に記載するように、ASTM D638に準じて行うことができる。
引張強度が70MPa以上であることにより、剛性に優れるポリアミドを得ることができる。
引張伸度の測定は、下記実施例に記載するように、ASTM D638に準じて行うことができる。
引張伸度が3.0%以上であることにより、靭性に優れるポリアミドを得ることができる。
吸水率は、下記実施例に記載の方法により測定することができる。
吸水率が5.0%以下であることにより、低吸水性に優れるポリアミド組成物を得ることができる。
色調b値は、下記実施例に記載の方法により測定することができる。
色調b値が0以下であることにより、耐熱変色性に優れるポリアミド組成物を得ることができる。
本実施の形態のポリアミド組成物は、前記(A)ポリアミドと、(B)無機充填材と、を含有するポリアミド組成物である。
ポリアミド組成物として、(B)無機充填材を含有することにより、耐熱性、流動性、靭性、低吸水性及び剛性などに優れるポリアミドの性質を損なうことなく、ポリアミド組成物としても、耐熱性、流動性、靭性、及び低吸水性などを満足しながら、さらに、特に剛性に優れるポリアミド組成物とすることができる。
PA6及びPA66などのポリアミドでは、融点が低く、耐熱性の点でこれらの要求を満たすことができない。
ポリアミド組成物は、無機充填材を含有しても、耐光性に優れ、ポリアミド組成物の色調としても優れるものである。
無機充填材としては、1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
また、(B)無機充填材としては、ウォラストナイトがより好ましく、ウォラストナイトの中でも、数平均繊維径が3~30μmであり、重量平均繊維長が10~500μmであり、前記アスペクト比(L/D)が3~100であるものがさらに好ましく用いられる。
さらに、(B)無機充填材としては、タルク、マイカ、カオリン、及び窒化珪素などがより好ましく、タルク、マイカ、カオリン、及び窒化珪素などの中でも、数平均繊維径が0.1~3μmであるものがさらに好ましく用いられる。
ポリアミドと無機充填材の混合方法として、例えば、ポリアミドと無機充填材とをヘンシェルミキサーなどを用いて混合し溶融混練機に供給し混練する方法や、単軸又は2軸押出機で溶融状態にしたポリアミドに、サイドフィダーから無機充填材を配合する方法などが挙げられる。
溶融混練温度は、樹脂温度にして250~375℃程度であることが好ましい。
溶融混練時間は、0.5~5分程度であることが好ましい。
溶融混練を行う装置としては、特に限定されるものではなく、公知の装置、例えば、単軸又は2軸押出機、バンバリーミキサー、及びミキシングロールなどの溶融混練機を用いることができる。
配合量を0.1質量部以上とすることにより、ポリアミド組成物の靭性及び剛性などの機械物性が良好に向上し、また、配合量を200質量部以下とすることにより、成形性に優れるポリアミド組成物を得ることができる。
溶融せん断粘度は、下記実施例に記載の方法により測定することができる。
溶融せん断粘度が上記範囲内にあることにより、流動性に優れるポリアミドを得ることができる。
引張強度の測定は、下記実施例に記載するように、ASTM D638に準じて行うことができる。
引張強度が140MPa以上であることにより、剛性に優れるポリアミドを得ることができる。
引張伸度の測定は、下記実施例に記載するように、ASTM D638に準じて行うことができる。
引張伸度が1.0%以上であることにより、靭性に優れるポリアミドを得ることができる。
吸水率は、下記実施例に記載の方法により測定することができる。
吸水率が5.0%以下であることにより、低吸水性に優れるポリアミド組成物を得ることができる。
本実施の形態のポリアミド組成物は、前記(A)ポリアミドと、(C)銅化合物及び金属ハロゲン化物と、を含有するポリアミド組成物である。
ポリアミド組成物として、耐熱性、流動性、靭性、低吸水性及び剛性などに優れるポリアミドの性質を損なうことなく、(C)銅化合物及び金属ハロゲン化物を含有することにより、耐熱性、流動性、靭性、低吸水性、及び剛性に優れ、さらに、耐熱エージング性に優れるポリアミド組成物とすることができる。
銅化合物としては、1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
銅化合物の配合量を、上記範囲内にすることにより、十分な耐熱エージング性が向上し、銅析出及び金属腐食を抑制することができる。
ポリアミド組成物中に、銅として50~2000質量部含有することにより、耐熱エージング性に優れるポリアミド組成物を得ることができる。
金属ハロゲン化物としては、元素周期律表の1族又は2族金属元素とハロゲンとの塩であり、例えば、ヨウ化カリウム、臭化カリウム、塩化カリウム、ヨウ化ナトリウム、及び塩化ナトリウムなどが挙げられ、ヨウ化カリウム及び臭化カリウムであることが好ましい。
金属ハロゲン化合物としては、1種類で用いてもよく、2種類以上を組み合わせて用いてもよい。
金属ハロゲン化物としては、耐熱エージング性に優れ、金属腐食を抑制することができるので、ヨウ化カリウムが好ましい。
金属ハロゲン化物の配合量を、上記範囲内にすることにより、十分な耐熱エージング性が向上し、銅析出及び金属腐食を抑制することができる。
ハロゲンと銅のモル比が2/1以上である場合には銅析出及び金属腐食の抑制をすることができるため好ましい。また、ハロゲンと銅のモル比が50/1以下であれば靭性及び剛性などの機械物性を損なうことなく、成形機のスクリューなどを腐食するという問題を抑制することができる。
製法1におけるポリアミドの重合工程中とは、原料モノマーからポリアミドの重合完了までのいずれかの工程であって、どの段階でもよい。
製法2の溶融混練を行う装置としては、特に限定されるものではなく、公知の装置、例えば、単軸又は2軸押出機、バンバリーミキサー、及びミキシングロールなどの溶融混練機などを用いることができる。
中でも2軸押出機が好ましく用いられる。
溶融混練の温度は、好ましくは、(A)ポリアミドの融点より1~100℃程度高い温度、より好ましくは10~50℃程度高い温度である。
混練機での剪断速度は100sec-1以上程度であることが好ましく、混練時の平均滞留時間は0.5~5分程度であることが好ましい。
他の成分としては、例えば、滑剤としてラウリル酸などの高級脂肪酸、高級脂肪酸とアルミニウムなどの金属との高級脂肪酸金属塩、エチレンビスステアリルアミドなどの高級脂肪酸アミド、及びポリエチレンワックスなどのワックス類などが挙げられる。
また、少なくとも1つのアミド基を有する有機化合物も挙げられる。
無機充填材の配合量は、ポリアミド100質量部に対して、好ましくは0.1~200質量部であり、より好ましくは1~180質量部である、さらに好ましくは5~150質量部である。
無機充填材の配合量を0.1質量部以上とすることにより、ポリアミド組成物の靭性及び剛性などの機械物性が良好に向上し、また、無機充填材の配合量を200質量部以下とすることにより、成形性に優れるポリアミド組成物を得ることができる。
溶融せん断粘度は、下記実施例に記載の方法により測定することができる。
溶融せん断粘度が上記範囲内にあることにより、流動性に優れるポリアミド組成物を得ることができる。
引張強度の測定は、下記実施例に記載するように、ASTM D638に準じて行うことができる。
引張強度が140MPa以上であることにより、剛性に優れるポリアミド組成物を得ることができる。
引張伸度の測定は、下記実施例に記載するように、ASTM D638に準じて行うことができる。
引張伸度が1.0%以上であることにより、靭性に優れるポリアミド組成物を得ることができる。
吸水率は、下記実施例に記載の方法により測定することができる。
吸水率が5.0%以下であることにより、低吸水性に優れるポリアミド組成物を得ることができる。
強度半減期は、下記実施例に記載の方法により測定することができる。
強度半減期が40日以上であることにより、耐熱性、特に、耐熱エージング性に優れるポリアミド組成物を得ることができる。
破壊応力は、下記実施例に記載の方法により測定することができる。
破壊応力が45MPa以上であるポリアミド組成物を成形することにより、耐振動性疲労性に優れるポリアミド組成物を得ることができる。
浸漬後の引張強度保持率は、下記実施例に記載の方法により測定することができる。
浸漬後の引張強度保持率が60%以上であるポリアミド組成物を成形することにより、耐LLC性に優れるポリアミド組成物を得ることができる。
本実施の形態におけるポリアミド組成物は、前記(A)ポリアミドと、(D)ハロゲン系難燃剤と、を含有するポリアミド組成物である。
本実施の形態におけるポリアミド組成物として、(D)ハロゲン系難燃剤を含有することにより、耐熱性、流動性、靭性、剛性、及び低吸水性に優れるポリアミドの性質を損なうことなく、ポリアミド組成物としても、耐熱性、流動性、靭性、剛性、及び低吸水性に優れ、さらに、難燃性に優れるポリアミド組成物とすることができる。
また、本実施の形態のポリアミド組成物は、ハロゲン系難燃剤を含有しても、耐光性に優れ、ポリアミド組成物の色調としても優れるものである。
これら難燃剤を1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
臭素化ポリスチレン中の臭素含有量は55~75質量%が好ましい。臭素含有量を55質量%以上とすることにより、少ない臭素化ポリスチレンの配合量で難燃化に必要な臭素量を満足させることができ、ポリアミドの有する性質を損なうことなく、耐熱性、流動性、靭性、低吸水性、及び剛性に優れ、かつ難燃性に優れるポリアミド組成物を得ることができる。また、臭素含有量を75質量%以下とすることにより、押出や成形などの溶融加工時において熱分解を起こし難く、ガス発生などを抑制することができたり、耐熱変色性に優れるポリアミド組成物を得ることができる。
本実施の形態において用いられる(G)難燃助剤としては、特に限定されるものではなく、例えば、三酸化二アンチモン、四酸化二アンチモン、五酸化二アンチモン、アンチモン酸ナトリウムなどの酸化アンチモン類、一酸化スズ、二酸化スズなどの酸化スズ、酸化第二鉄、γ酸化鉄などの酸化鉄類、その他酸化亜鉛、ホウ酸亜鉛、酸化カルシウム、酸化アルミニウム(アルミナ)、酸化アルミニウム(ベーマイト)、酸化ケイ素(シリカ)、酸化チタン、酸化ジルコニウム、酸化マンガン、酸化モリブデン、酸化コバルト、酸化ビスマス、酸化クロム、酸化スズ、酸化ニッケル、酸化銅、及び酸化タングステンなどの金属酸化物;水酸化マグネシウム、及び水酸化アルミニウムなどの金属水酸化物;アルミニウム、鉄、チタン、マンガン、亜鉛、モリブデン、コバルト、ビスマス、クロム、スズ、アンチモン、ニッケル、銅、及びタングステンなどの金属粉末;炭酸亜鉛、炭酸カルシウム、炭酸マグネシウム、及び炭酸バリウムなどの金属炭酸塩;ホウ酸マグネシウム、ホウ酸カルシウム、及びホウ酸アルミニウムなどの金属ホウ酸塩;並びにシリコーン;などが挙げられる。
これら難燃助剤を1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
平均粒径は、レーザー回折散乱法粒度分布測定装置や精密粒度分布測定装置を用いて測定することができる。
本実施の形態において用いられる(H)α,β不飽和ジカルボン酸無水物を含む重合体としては、例えば、α,β不飽和ジカルボン酸無水物を共重合成分して含む重合体やα,β不飽和ジカルボン酸無水物で変性された重合体などが挙げられる。
一般式(1):

一般式(1)において、R1及びR2は、それぞれ独立して、水素又は炭素数1~3のアルキル基である。
α,β不飽和ジカルボン酸無水物を共重合成分して含む重合体としては、例えば、芳香族ビニル化合物とα,β不飽和ジカルボン酸無水物の共重合体などが挙げられる。
α,β不飽和ジカルボン酸無水物で変性された重合体としては、例えば、α,β不飽和ジカルボン酸無水物で変性されたポリフェニレンエーテル樹脂やポリプロピレン樹脂などが挙げられる。
る芳香族ビニル化合物としては、例えば、下記一般式(2)で表される化合物が挙げられる。
一般式(2):

一般式(2)において、R3及びR4は、それぞれ独立して、水素又は炭素数1~3のアルキル基であり、kは1~5の整数である。
本実施の形態において、α,β不飽和ジカルボン酸無水物を含む重合体が、芳香族ビニル化合物成分を含む場合には、芳香族ビニル化合物成分がハロゲン系難燃剤(臭素化ポリスチレンなど)と親和しており、また、α,β不飽和ジカルボン酸無水物部分がポリアミドと親和ないし反応することにより、ポリアミドマトリックス中にハロゲン系難燃剤が分散するのを助け、ハロゲン系難燃剤を微分散させることができると考えられる。
α,β不飽和ジカルボン酸無水物成分の割合を1質量%以上とすることにより、靭性及び剛性などの機械物性及び難燃性に優れるポリアミド組成物を得ることができる。また、α,β不飽和ジカルボン酸無水物成分の割合を50質量%以下とすることにより、α,β不飽和ジカルボン酸無水物によるポリアミド組成物の劣化を防止することができる。
ポリアミドとハロゲン系難燃剤の混合方法としては、例えば、ポリアミドとハロゲン系難燃剤と、任意に、難燃助剤、α,β不飽和ジカルボン酸無水物を含む重合体、及び/又は無機充填材とをヘンシェルミキサーなどを用いて混合し溶融混練機に供給し混練する方法や、単軸又は2軸押出機でポリアミドとハロゲン系難燃剤、任意に、難燃助剤及び/又はα,β不飽和ジカルボン酸無水物を含む重合体を予めヘンシェルミキサーなどを用いて混合したものを溶融混練機に供給し混練した後に、任意に、サイドフィダーから無機充填材を配合する方法などが挙げられる。
溶融混練時間は、0.5~5分程度であることが好ましい。
溶融混練を行う装置としては、公知の装置、例えば、単軸又は2軸押出機、バンバリーミキサー、及びミキシングロールなどの溶融混練機が好ましく用いられる。
ポリアミド組成物中のハロゲン系難燃剤の配合量は、ポリアミド100質量部に対して、好ましくは30~60質量部であり、より好ましくは35~55質量部であり、さらに好ましくは40~50質量部である。
ハロゲン系難燃剤の配合量を30質量部以上とすることにより、難燃性に優れるポリアミド組成物を得ることができる。また、ハロゲン系難燃剤の配合量を60質量部以下とすることにより、溶融混練時に分解ガスの発生、成形加工時の流動性の低下や、成形金型に汚染性物質の付着を抑制することができる。さらに、靭性及び剛性などの機械物性や成形品外観の低下も抑制することができる。
難燃助剤を配合することにより、さらに難燃性に優れるポリアミド組成物を得ることができる。また、難燃助剤の配合量を30質量部以下とすることにより、溶融加工時の粘度適切な範囲に制御することができ、押出時のトルクの上昇、成形時の成形性の低下及び成形品外観の低下を抑制することができる。また、靭性及び剛性などの機械物性に優れるポリアミドの性質を損なうことなく、靭性などに優れるポリアミド組成物を得ることができる。
α,β不飽和ジカルボン酸無水物を含む重合体を配合することにより、相溶化によるポリアミド中でのハロゲン系難燃剤の微分散効果を高めることができ、難燃性や強度の向上効果に優れるポリアミド組成物を得ることができる。また、α,β不飽和ジカルボン酸無水物を含む重合体の配合量を20質量部以下とすることにより、靭性及び剛性などの機械物性に優れるポリアミドの性質を損なうことなく、強度などに優れるポリアミド組成物を得ることできる。
無機充填材を配合することにより、ポリアミド組成物の靭性及び剛性などの機械物性が良好に向上し、また、無機充填材の配合量を200質量部以下とすることにより、成形性に優れるポリアミド組成物を得ることができる。
引張強度の測定は、下記実施例に記載するように、ASTM D638に準じて行うことができる。
引張強度が140MPa以上であることにより、剛性に優れるポリアミド組成物を得ることができる。
引張伸度の測定は、下記実施例に記載するように、ASTM D638に準じて行うことができる。
引張伸度が1.0%以上であることにより、靭性に優れるポリアミド組成物を得ることができる。
吸水率は、下記実施例に記載の方法により測定することができる。
吸水率が5.0%以下であることにより、低吸水性に優れるポリアミド組成物を得ることができる。
ポリアミド組成物の難燃性は、好ましくはV-2以上であり、より好ましくはV-1以上であり、さらに好ましくはV-0である。
流動長は、下記実施例に記載の方法により測定することができる。
流動長が15cm以上であることにより、流動性に優れるポリアミド組成物を得ることができる。
本実施の形態のポリアミド組成物は、前記(A)ポリアミドと、(E)ホスフィン酸塩及び/又はジホスフィン酸塩(以下、両者を総称して「ホスフィン酸塩」と略称する場合がある。)と、を含有するポリアミド組成物である。
ホスフィン酸としては、例えば、下記一般式(I)で表される化合物が挙げられる。
一般式(I):
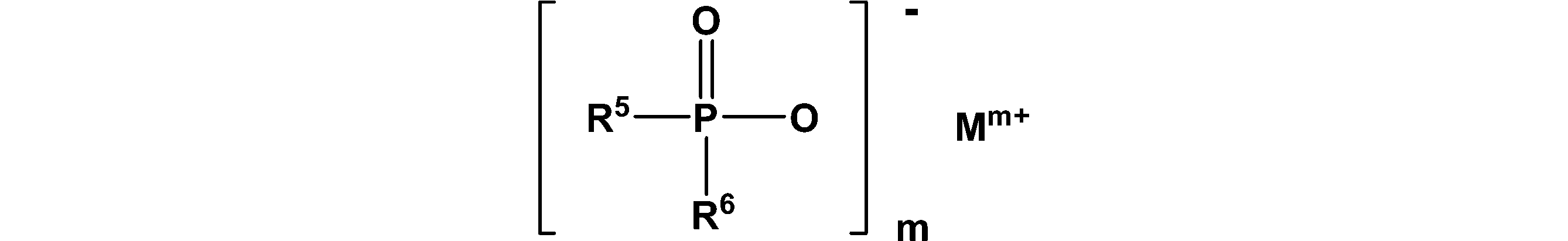
ジホスフィン酸としては、例えば、下記一般式(II)で表される化合物が挙げられる。
一般式(II):

一般式(I)及び一般式(II)中、R5及びR6並びにR7及びR8は、それぞれ独立して、炭素数1~6のアルキル基、炭素数6~12のアリール基、及び炭素数7~20のアリールアルキル基からなる群から選択され、R9は、炭素数1~10のアルキレン基、炭素数6~10のアリーレン基、炭素数7~20のアルキルアリーレン基、及び炭素数7~20のアリールアルキレン基からなる群から選択され、Mはカルシウム(イオン)、マグネシウム(イオン)、アルミニウム(イオン)及び亜鉛(イオン)からなる群から選択され、mは2又は3であり、nは1又は3であり、xは1又は2である。
本実施の形態において、アリール基としては、無置換又は種々の置換基で置換された炭素数6~20の芳香族基を挙げることができ、フェニル基、ベンジル基、o-トルイル基、2,3-キシリル基などが挙げられる。
また、本実施の形態のポリアミド組成物は、ホスフィン酸塩を含有しても、耐光性に優れ、ポリアミド組成物の色調としても優れるものである。
これらは、本質的にモノマー性化合物であるが、反応条件に依存して、環境によっては縮合度が1~3のポリマー性ホスフィン酸塩も含まれる。
これら(E)ホスフィン酸塩を1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
0.5~20μmの粉末状の(E)ホスフィン酸塩を用いると、高い難燃性を発現するポリアミド組成物を得ることができるばかりでなく、成形品の強度が著しく高くなるのでさらに好ましい。
平均粒径は、レーザー回折散乱法粒度分布測定装置や精密粒度分布測定装置を用いて測定することができる。
(E)ホスフィン酸塩を含有するポリアミド組成物として、(G)難燃助剤をさらに含有することにより、さらに難燃性に優れるポリアミド組成物を得ることができる。
(G)難燃助剤としては、上記難燃助剤であれば特に限定されるものではないが、中でも、酸化亜鉛、酸化鉄、酸化カルシウム、酸化アルミニウム(アルミナ)、酸化アルミニウム(ベーマイト)、酸化ケイ素(シリカ)、酸化チタン、酸化ジルコニウム、酸化マンガン、酸化モリブデン、酸化コバルト、酸化ビスマス、酸化クロム、酸化スズ、酸化アンチモン、酸化ニッケル、酸化銅、及び酸化タングステンなどの金属酸化物、水酸化マグネシウム、及び水酸化アルミニウムなどの金属水酸化物、アルミニウム、鉄、チタン、マンガン、亜鉛、モリブデン、コバルト、ビスマス、クロム、スズ、アンチモン、ニッケル、銅、及びタングステンなどの金属粉末、炭酸亜鉛、炭酸カルシウム、炭酸マグネシウム、及び炭酸バリウムなどの金属炭酸塩、ホウ酸亜鉛、ホウ酸マグネシウム、ホウ酸カルシウム、及びホウ酸アルミニウムなどの金属ホウ酸塩、並びにシリコーンなどが好ましい。
これら(G)難燃助剤を1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
ホウ酸亜鉛としては、より好ましくは、xZnO・yB2O3・zH2O(x>0、y>0、z≧0)で表されるホウ酸亜鉛、さらに好ましくは、2ZnO・3B2O3・3.5H2O、4ZnO・B2O3・H2O、及び2ZnO・3B2O3で表されるホウ酸亜鉛が挙げられる。
これらホウ酸金属化合物はシラン系カップリング剤及びチタネート系カップリング剤などの表面処理剤で処理されていてもよい。
難燃助剤の平均粒径は、好ましくは30μm以下であり、より好ましくは、15μm以下であり、さらに好ましくは7μm以下である。
ポリアミド組成物中のホスフィン酸塩の配合量は、ポリアミド100質量部に対して、好ましくは20~90質量部であり、より好ましくは25~80質量部であり、さらに好ましくは30~60質量部である。
ホスフィン酸塩の配合量を20質量部以上とすることにより、難燃性に優れるポリアミド組成物を得ることができる。また、ハロゲン系難燃剤の配合量を90質量部以下とすることにより、成形加工時の流動性の低下を抑制することができる。さらに靭性及び剛性などの機械物性や成形品外観の低下を抑制することができる。
難燃助剤を配合することにより、さらに難燃性に優れるポリアミド組成物を得ることができる。また、難燃助剤の配合量を30質量部以下とすることにより、溶融加工時の粘度適切な範囲に制御することができ、押出時のトルクの上昇、成形時の成形性の低下及び成形品外観の低下を抑制することができる。また、靭性及び剛性などの機械物性に優れるポリアミドの性質を損なうことなく、靭性などに優れるポリアミド組成物を得ることができる。
無機充填材をさらに配合することにより、ポリアミド組成物の靭性及び剛性などの機械物性が良好に向上し、また、無機充填材の配合量を200質量部以下とすることにより、成形性に優れるポリアミド組成物を得ることができる。
引張強度の測定は、下記実施例に記載するように、ASTM D638に準じて行うことができる。
引張強度が140MPa以上であることにより、剛性に優れるポリアミド組成物を得ることができる。
引張伸度の測定は、下記実施例に記載するように、ASTM D638に準じて行うことができる。
引張伸度が1,0%以上であることにより、靭性に優れるポリアミド組成物を得ることができる。
吸水率は、下記実施例に記載の方法により測定することができる。
吸水率が5.0%以下であることにより、低吸水性に優れるポリアミド組成物を得ることができる。
完全充填圧力は、下記実施例に記載の方法により測定することができる。
完全充填圧力が、上記範囲内にあることにより、流動性に優れるポリアミド組成物を得ることができる。
本実施の形態のポリアミド組成物は、前記(A)ポリアミドと、(F)安定剤と、を含有するポリアミド組成物である。
本実施の形態のポリアミド組成物として、(F)安定剤を含有することにより、耐熱性、流動性、靭性、低吸水性、及び剛性に優れるポリアミドの性質を損なうことなく、ポリアミド組成物としても耐熱性、流動性、靭性、低吸水性、及び剛性に優れ、さらに、耐熱変色性及び耐候性に優れるポリアミド組成物とすることができる。
これらの安定剤を1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
ヒンダードフェノール化合物としては、例えば、N,N’-へキサン-1,6-ジイルビス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニルプロピオンアミド)、ペンタエリスリチル-テトラキス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオネート]、N,N’-ヘキサメチレンビス(3,5-ジ-t-ブチル-4-ヒドロキシ-ヒドロシンナマミド)、トリエチレングリコール-ビス[3-(3-t-ブチル-5-メチル-4-ヒドロキシフェニル)プロピオネート]、3,9-ビス{2-[3-(3-t-ブチル-4-ヒドロキシ-5-メチルフェニル)プロピニロキシ]-1,1-ジメチルエチル}-2,4,8,10-テトラオキサピロ[5,5]ウンデカン、3,5-ジ-t-ブチル-4-ヒドロキシベンジルホスホネート-ジエチルエステル、1,3,5-トリメチル-2,4,6-トリス(3,5-ジ-t-ブチル-4-ヒドロキシベンジル)ベンゼン、及び1,3,5-トリス(4-t-ブチル-3-ヒドロキシ-2,6-ジメチルベンジル)イソシアヌル酸などが挙げられる。
これらフェノール系安定剤を1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
これらホスファイト系安定剤を1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
ペンタエリストール型ホスファイト化合物としては、例えば、2,6-ジ-t-ブチル-4-メチルフェニル・フェニル・ペンタエリスリトールジホスファイト、2,6-ジ-t-ブチル-4-メチルフェニル・メチル・ペンタエリスリトールジホスファイト、2,6-ジ-t-ブチル-4-メチルフェニル・2-エチルヘキシル・ペンタエリスリトールジホスファイト、2,6-ジ-t-ブチル-4-メチルフェニル・イソデシル・ペンタエリスリトールジホスファイト、2,6-ジ-t-ブチル-4-メチルフェニル・ラウリル・ペンタエリスリトールジホスファイト、2,6-ジ-t-ブチル-4-メチルフェニル・イソトリデシル・ペンタエリスリトールジホスファイト、2,6-ジ-t-ブチル-4-メチルフェニル・ステアリル・ペンタエリスリトールジホスファイト、2,6-ジ-t-ブチル-4-メチルフェニル・シクロヘキシル・ペンタエリスリトールジホスファイト、2,6-ジ-t-ブチル-4-メチルフェニル・ベンジル・ペンタエリスリトールジホスファイト、2,6-ジ-t-ブチル-4-メチルフェニル・エチルセロソルブ・ペンタエリスリトールジホスファイト、2,6-ジ-t-ブチル-4-メチルフェニル・ブチルカルビトール・ペンタエリスリトールジホスファイト、2,6-ジ-t-ブチル-4-メチルフェニル・オクチルフェニル・ペンタエリスリトールジホスファイト、2,6-ジ-t-ブチル-4-メチルフェニル・ノニルフェニル・ペンタエリスリトールジホスファイト、ビス(2,6-ジ-t-ブチル-4-メチルフェニル)ペンタエリスリトールジホスファイト、ビス(2,6-ジ-t-ブチル-4-エチルフェニル)ペンタエリスリトールジホスファイト、2,6-ジ-t-ブチル-4-メチルフェニル・2,6-ジ-t-ブチルフェニル・ペンタエリスリトールジホスファイト、2,6-ジ-t-ブチル-4-メチルフェニル・2,4-ジ-t-ブチルフェニル・ペンタエリスリトールジホスファイト、2,6-ジ-t-ブチル-4-メチルフェニル・2,4-ジ-t-オクチルフェニル・ペンタエリスリトールジホスファイト、2,6-ジ-t-ブチル-4-メチルフェニル・2-シクロヘキシルフェニル・ペンタエリスリトールジホスファイト、2,6-ジ-t-アミル-4-メチルフェニル・フェニル・ペンタエリストリトールジホスファイト、ビス(2,6-ジ-t-アミル-4-メチルフェニル)ペンタエリスリトールジホスファイト、及びビス(2,6-ジ-t-オクチル-4-メチルフェニル)ペンタエリスリトールジホスファイトなどが挙げられる。
これらペンタエリストール型ホスファイト系安定剤を1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
これらヒンダードアミン系安定剤を1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
ヒドロキシフェニルトリアジン類としては、例えば、2,4,6-トリス(2’-ヒドロキシ-4’-オクチルオキシ-フェニル)-1,3,5-トリアジン、2-(2’-ヒドロキシ-4’-ヘキシルオキシ-フェニル)-4,6-ジフェニル-1,3,5-トリアジン、2-(2’-ヒドロキシ-4’-オクチルオキシフェニル)-4,6-ビス(2’,4’-ジメチルフェニル)-1,3,5-トリアジン、2-(2’,4’-ジヒドロキシフェニル)-4,6-ビス(2’,4’-ジメチルフェニル)-1,3,5-トリアジン、2,4-ビス(2’-ヒドロキシ-4’-プロピルオキシ-フェニル)-6-(2’,4’-ジメチルフェニル)-1,3,5-トリアジン、2-(2-ヒドロキシ-4-オクチルオキシフェニル)-4,6-ビス(4’-メチルフェニル)-1,3,5-トリアジン、2-(2’-ヒドロキシ-4’-ドデシルオキシフェニル)-4,6-ビス(2’,4’-ジメチルフェニル)-1,3,5-トリアジン、2,4,6-トリス(2’-ヒドロキシ-4’-イソプロピルオキシフェニル)-1,3,5-トリアジン、2,4,6-トリス(2’-ヒドロキシ-4’-n-ヘキシルオキシフェニル)-1,3,5-トリアジン、及び2,4,6-トリス(2’-ヒドロキシ-4’-エトキシカルボニルメトキシフェニル)-1,3,5-トリアジンなどが挙げられる。
これらトリアジン系安定剤を1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
これらイオウ系安定剤を1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
無機リン系安定剤としては、例えば、その水和物(好ましくは、ジ亜リン酸ナトリウムの水和物(NaH2PO2・nH2O))であってもよい。
これら無機リン系安定剤を1種類で用いてもよいし、2種類以上を組み合わせて用いてもよい。
(F)安定剤の配合量を0.01質量部以上とすることにより、耐熱変色性や耐候性に優れるポリアミド組成物とすることができる。また、(F)安定剤の配合量を5質量部以下とすることにより、ポリアミド組成物を成形した際の成形品表面への銀状の発生を抑制することができ、また、成形品の靭性及び剛性などの機械物性に優れる成形品を得ることができる。
本実施の形態における(F)安定剤を含有するポリアミド組成物の製造方法としては、前記(A)ポリアミドと(F)安定剤とを混合する方法であれば、特に限定されるものではなく、例えば、ポリアミドに安定剤を配合する方法、ポリアミドの重合時に安定剤を配合する方法、ポリアミドと他の樹脂との混合時に安定剤を配合する方法、ポリアミドの粉体又はペレットの表面に安定剤を付着させる方法、ポリアミドに溶融混練により安定剤を配合する方法、安定剤のマスターバッチをポリアミドに配合する方法など、あるいはこれらの方法を組み合わせて配合する方法などを挙げることができる。
ポリアミド組成物を構成する成分を溶融混練機に供給する方法は、すべての構成成分を同一の供給口に一度に供給してもよいし、構成成分をそれぞれ異なる供給口から供給してもかまわない。
溶融混練時間は、0.5~5分程度であることが好ましい。
溶融混練を行う装置としては、公知の装置、例えば、単軸又は2軸押出機、バンバリーミキサー、及びミキシングロールなどの溶融混練機が好ましく用いられる。
溶融せん断粘度は、下記実施例に記載の方法により測定することができる。
溶融せん断粘度が上記範囲内にあることにより、流動性に優れるポリアミド組成物を得ることができる。
引張強度の測定は、下記実施例に記載するように、ASTM D638に準じて行うことができる。
引張強度が80MPa以上であることにより、剛性に優れるポリアミド組成物を得ることができる。
引張伸度の測定は、下記実施例に記載するように、ASTM D638に準じて行うことができる。
引張伸度が3.0%以上であることにより、靭性に優れるポリアミド組成物を得ることができる。
吸水率は、下記実施例に記載の方法により測定することができる。
吸水率が5.0%以下であることにより、低吸水性に優れるポリアミド組成物を得ることができる。
色調の変化Δbは、下記実施例に記載の方法により測定することができる。
色調の変化Δbが9以下であることにより、耐熱変色性に優れるポリアミド組成物を得ることができる。
色差ΔEの測定は、下記実施例に記載の方法により測定することができる。
色差ΔEが9以下であることにより、耐候性に優れるポリアミド組成物を得ることができる。
本実施の形態のポリアミド又はポリアミド組成物は、周知の成形方法、例えば、プレス成形、射出成形、ガスアシスト射出成形、溶着成形、押出成形、吹込成形、フィルム成形、中空成形、多層成形、及び溶融紡糸などを用いて各種成形品を得ることができる。
自動車吸気系部品としては、特に限定されるものではなく、例えば、エアインテークマニホールド、インタークーラーインレット、エキゾーストパイプカバー、インナーブッシュ、ベアリングリテーナー、エンジンマウント、エンジンヘッドカバー、リゾネーター、及びスロットルボディなどが挙げられる。
自動車冷却系部品としては、特に限定されるものではなく、例えば、チェーンカバー、サーモスタットハウジング、アウトレットパイプ、ラジエータータンク、オイルネーター、及びデリバリーパイプなどが挙げられる。
自動車燃料系部品では、特に限定されるものではなく、例えば、燃料デリバリーパイプ及びガソリンタンクケースなどが挙げられる。 内装部品としては、特に限定されるものではなく、例えば、インストルメンタルパネル、コンソールボックス、グローブボックス、ステアリングホイール、及びトリムなどが挙げられる。
外装部品としては、特に限定されるものではなく、例えば、モール、ランプハウジング、フロントグリル、マッドガード、サイドバンパー、及びドアミラーステイ、ルーフレールなどが挙げられる。
電装部品としては、特に限定されるものではなく、例えば、コネクタやワイヤーハーネスコネクタ、モーター部品、ランプソケット、センサー車載スイッチ、及びコンビネーションスイッチなどが挙げられる。
成形品の強度半減期は、好ましく40日以上であり、より好ましくは45日以上であり、さらに好ましくは50日以上である。強度半減期は、下記実施例に記載の方法により測定することができる。
成形品の強度半減期が40日以上であることにより、耐熱性、特に、耐熱エージング特性に優れる自動車吸気系部品を得ることができる。
成形品の破壊応力は、好ましくは45MPa以上であり、より好ましくは50MPa以上であり、さらに好ましくは55MPa以上である。破壊応力は、下記実施例に記載の方法により測定することができる。
成形品の破壊応力が45MPa以上であることにより、耐振動性疲労性に優れる自動車吸気系部品を得ることができる。
成形品の吸水率は、好ましくは5.0%以下であり、より好ましくは4.0%以下であり、さらに好ましくは3.0%以下である。吸水率は、下記実施例に記載の方法により測定することができる。
成形品の吸水率が5.0%以下であることにより、低吸水性に優れる自動車吸気系部品を得ることができる。
成形品の強度半減期は、好ましく40日以上であり、より好ましくは45日以上であり、さらに好ましくは50日以上である。強度半減期は、下記実施例に記載の方法により測定することができる。
成形品の強度半減期が40日以上であることにより、耐熱性、特に耐熱エージング特性に優れる自動車冷却系部品を得ることができる。
成形品の浸漬後の引張強度保持率は、好ましくは60%以上であり、より好ましくは75%以上であり、さらに好ましくは80%以上である。浸漬後の引張強度は、下記実施例に記載の方法により測定することができる。
成形品の浸漬後の引張強度保持率が60%以上であることにより、耐LLC性に優れる自動車冷却系部品を得ることができる。
成形品の吸水率は、好ましくは5.0%以下であり、より好ましくは4.0%以下であり、さらに好ましくは3.0%以下である。吸水率は、下記実施例に記載の方法により測定することができる。
成形品の吸水率が5.0%以下であることにより、低吸水性に優れる自動車冷却系部品を得ることができる。
本実施の形態におけるポリアミド又はポリアミド組成物から得られる成形品は、耐熱性、靭性、成形性に優れ、かつ低吸水性に優れる。したがって、本実施の形態のポリアミド及びポリアミド組成物は、自動車用以外にも、例えば、電気及び電子用、産業資材用、及び日用及び家庭品用などの各種部品材料として、また、押出用途などに好適に用いることができる。
産業機器用としては、特に限定されるものではなく、例えば、ギヤ、カム、絶縁ブロック、バルブ、電動工具部品、農機具部品、エンジンカバーなどに用いられる。
日用及び家庭品用としては、特に限定されるものではなく、例えば、ボタン、食品容器、及びオフィス家具などに用いられる。
押し出し用途としては、特に限定されるものではなく、例えば、フィルム、シート、フィラメント、チューブ、棒、及び中空成形品などに用いられる。
実施例及び比較例に用いた原材料及び測定方法を以下に示す。なお、本実施例において、1Kg/cm2は、0.098MPaを意味する。
本実施例において下記化合物を用いた。
(a)ジカルボン酸
(1)1,4-シクロヘキサンジカルボン酸(CHDA) イーストマンケミカル製 商品名 1,4-CHDA HPグレード(トランス体/シス体(モル比)=25/75)
(2)テレフタル酸(TPA) 和光純薬工業製 商品名 テレフタル酸
(3)アジピン酸(ADA) 和光純薬工業製 商品名 アジピン酸
(4)スベリン酸(C8DA) 和光純薬工業製 商品名 スベリン酸
(5)アゼライン酸(C9DA) 和光純薬工業製 商品名 アゼライン酸
(6)セバシン酸(C10DA) 和光純薬工業製 商品名 セバシン酸
(7)ドデカン二酸(C12DA) 和光純薬工業製 商品名 ドデカン二酸
(8)テトラデカン二酸(C14DA) 東京化成工業製 商品名 テトラデカン二酸
(9)ヘキサデカン二酸(C16DA) 東京化成工業製 商品名 ヘキサデカン二酸
(10)2-メチルペンタメチレンジアミン(2MPD) 東京化成工業製 商品名 2-メチル-1,5-ジアミノペンタン
(11)ヘキサメチレンジアミン(HMD) 和光純薬工業製 商品名 ヘキサメチレンジアミン
(12)1,9-ノナメチレンジアミン(NMD) アルドリッチ製 商品名 1,9-ノナンジアミン
(13)2-メチルオクタメチレンジアミン(2MOD) 特開平05-17413号公報に記載されている製法を参考にして製造した。
(14)2,2,4-トリメチル-1,6-ヘキサンジアミンと2,4,4-トリメチル-1,6-ヘキサンジアミンの混合物(TMHD) アルドリッチ製 商品名 C,C,C-1,6-ヘキサメチレンジアミン
(15)ガラス繊維(GF) 日本電気硝子製 商品名 ECS03T275H 平均繊維径10μmφ、カット長3mm
(16)ヨウ化銅(CuI)和光純薬工業製 商品名 ヨウ化銅(I)
(17)ヨウ化カリウム(KI)和光純薬工業製 商品名 ヨウ化カリウム
(18)エチレンビスステアリルアミド ライオン製 商品名 アーモワックス EBS
(19)臭素化ポリスチレン ALBEMARLE CORPORATION製 商品名 SAYTEX(登録商標)HP-7010G (元素分析より 臭素含有量:63質量%)
(20)特開平08-73720号公報に記載されている製法を参考にして製造した、ジエチルホスフィン酸アルミニウム(DEPAl)。
(F-1)フェノール系安定剤
(21)N,N’-へキサン-1,6-ジイルビス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニルプロピオンアミド)] チバ・ジャパン製 商品名 IRGANOX(登録商標)1098
(F-2)ホスファイト系安定剤
(22)ビス(2,6-ジ-t-ブチル-4-メチルフェニル)ペンタエリスリトールジホスファイト ADEKA製 商品名 アデカスタブ(登録商標)PEP-36
(F-3)ヒンダードアミン系安定剤
(23)ビス-(2,2,6,6-テトラメチル-4-ピペリジル)-セバケート チバ・ジャパン製 商品名 サノール(登録商標)770
(F-4)トリアジン系安定剤
(24)2-(2’-ヒドロキシ-4’-ヘキシルオキシフェニル)-4、6-ジフェニル-1,3,5-トリアジン チバ・ジャパン製 商品名 TINUBIN(登録商標)167FF
(F-5)無機リン系安定剤
(25)次亜リン酸ナトリウム 和光純薬製 商品名 ジ亜リン酸ナトリウム
(26)三酸化二アンチモン 第一エフ・アール製 商品名 三酸化アンチモン
(27)ホウ酸亜鉛 2ZnO・3B2O3・3.5H2O U.S.Borax製 商品名 Firebrake(登録商標)ZB
(28)水酸化マグネシウム 協和化学製 商品名 キスマ(登録商標)5、平均粒径:0.8μm
(29)スチレンと無水マレイン酸の共重合体 NOVA Chemicals製 商品名 DYLARK(登録商標)332(スチレン85質量%及び無水マレイン酸15質量%の共重合体)
(a-1)脂環族ジカルボン酸のモル%は、(原料モノマーとして加えた(a-1)脂環族ジカルボン酸のモル数/原料モノマーとして加えた全ての(a)ジカルボン酸のモル数)×100として、計算により求めた。
(b-1)主鎖から分岐した置換基を持つジアミンのモル%は、(追添分を除く、原料モノマーとして加えた(b-1)主鎖から分岐した置換基を持つジアミンのモル数/原料モノマーとして加えた全ての(b)ジアミンのモル数)×100として、計算により求めた。
また、(c)ラクタム及び/又はアミノカルボン酸のモル%は、(原料モノマーとして加えた(c)ラクタム及び/又はアミノカルボン酸のモル数/原料モノマーとして加えた、全ての(a)ジカルボン酸のモル数+(b)全てのジアミンのモル数+(c)ラクタム及び/又はアミノカルボン酸のモル数)×100として、計算により求めた。
(1)融点Tm1、Tm2(℃)
JIS-K7121に準じて、PERKIN-ELMER社製Diamond-DSCを用いて測定した。測定条件は、窒素雰囲気下、試料約10mgを昇温速度20℃/minでサンプルの融点に応じて300~350℃まで昇温したときに現れる吸熱ピーク(融解ピーク)の温度をTm1(℃)とし、昇温の最高温度の溶融状態で温度を2分間保った後、降温速度20℃/minで30℃まで降温し、30℃で2分間保持した後、昇温速度20℃/minで同様に昇温したときに現れる吸熱ピーク(融解ピーク)の最大ピーク温度を融点Tm2(℃)とし、その全ピーク面積を融解熱量ΔH(J/g)とした。なお、ピークが複数ある場合には、ΔHが1J/g以上のものをピークとみなした。例えば、融点295℃、ΔH=20J/gと融点325℃、ΔH=5J/gの二つのピークが存在する場合、融点は325℃とした。
JIS-K7121に準じて、PERKIN-ELMER社製Diamond-DSCを用いて測定した。測定条件は、試料をホットステージ(Mettler社製EP80)で溶融させて得られた溶融状態のサンプルを、液体窒素を用いて急冷し、固化させ、測定サンプルとした。そのサンプル10mgを用いて、昇温スピード20℃/minの条件下、30~350℃の範囲で昇温して、ガラス転移温度を測定した。
JIS-K6810に準じて実施した。具体的には、98%硫酸を用いて、1%の濃度の溶解液((ポリアミド1g)/(98%硫酸100mL)の割合)を作成し、25℃の温度条件下で測定した。
上記(1)で求めた融点+20℃の温度条件下で、せん断速度1000sec-1における溶融せん断粘度ηsで流動性を評価した。具体的な測定方法は、英国ROSAND社製ツインキャピラリーレオメーターRH7-2型を使用し、オリフィスは、ダイ径1.0mm、ダイ入口角180度のもので、L/Dが16及び0.25、の2つのオリフィスを使用した。
ASTM引張試験用のダンベル射出成形試験片(3mm厚)を用いて、ASTM D638に準じて行った。成形試験片は、射出成形機(日精樹脂(株)製PS40E)にASTM引張試験(ASTM D638)用のダンベル試験片(3mm厚)の金型(金型温度=Tg+20℃)を取り付けて、シリンダー温度=(Tm2+10)℃~(Tm2+30)℃で成形を行った。
ASTM引張試験用のダンベル射出成形試験片(3mm厚)を成形後の絶乾状態(dry as mold)で、試験前質量(吸水前質量)を測定した。80℃の純水中に24時間浸漬させた。その後、水中から試験片を取り出し、表面の付着水分をふき取り、恒温恒湿(23℃、50RH%)雰囲気下に30分放置後、試験後質量(吸水後質量)を測定した。吸水前質量に対しての吸水後質量の増分を吸水量とし、吸水前質量に対する吸水量の割合を、試行数n=3で求め、その平均値を吸水率(%)とした。
銅濃度は、試料に硫酸を加え、加熱しながら硝酸を滴下し有機分を分解し、該分解液を純水にて定容しICP発光分析(高周波プラズマ発光分析)法により定量した。ICP発光分析装置は、SEIKO電子工業社製Vista-Proを用いた。
ハロゲン濃度は、ヨウ素を例にとると、試料を高純度酸素で置換したフラスコ中で燃焼し、発生したガスを吸収液に捕集し、該捕集液中のヨウ素を1/100N硝酸銀溶液による電位差滴定法を用いて定量した。
ハロゲンと銅のモル比(ハロゲン/Cu)は、上記それぞれの定量値を用いて分子量からモルに換算し算出した。
上記(5)のASTM引張試験用のダンベル射出成形試験片(3mm厚)を熱風オーブン中で200℃、所定時間処理した後、ASTM-D638に準じて引張強度を測定した。そして熱処理前に測定した引張強度に対する熱処理後の引張強度を引張強度保持率として算出し、引張強度保持率が50%となる熱処理時間を強度半減期とした。
上記(5)のASTM引張試験用のダンベル射出成形試験片(3mm厚)を株式会社鷺宮製作所製油圧サーボ疲労試験機EHF-50-10-3を用い、120℃の雰囲気下、周波数20Hzの正弦波にて引張り荷重を負荷し、1,000,000回で破壊する応力(MPa)を求めた。
ポリマーペレットを射出成形機で、射出成形条件はシリンダ温度をTm2+30℃、金型温度Tg+20℃、成形サイクル60秒で、ASTM引張試験用のダンベル射出成形試験片(ASTMダンベル、3mm厚)を得た。日本電色社製色差計ND-300Aを用いて、初期成形品の色調b値を求めた。測定はダンベル射出成形試験片3枚を用い、反ゲート側の幅広部の中央位置について1枚ずつ3回測定し、平均値から求めた。
ポリマーペレットを射出成形機で、射出成形条件はシリンダ温度をTm2+30℃、金型温度Tg+20℃、成形サイクル60秒で、ASTM引張試験用のダンベル射出成形試験片(ASTMダンベル、3mm厚)を得た。日本電色社製色差計ND-300Aを用いて、初期成形品と1000時間後の成形品のそれぞれの色調b値を求めた。その差をΔbとした。測定はダンベル射出成形試験片3枚を用い、反ゲート側の幅広部の中央位置について1枚ずつ3回測定し、平均値から求めた。
ISO4892-2に準じ、ASTM引張試験用のダンベル射出成形試験片(3mm厚)を用いて、自然色の成形品にて1000時間後を評価した。試験機:ATLAS社製 Ci4000(キセノンランプ)雨有り。日本電色社製色差計ND-300Aを用いて、初期成形品と1000時間後の成形品の色差(ΔE)を求めた。測定はダンベル射出成形試験片3枚を用い、反ゲート側の幅広部の中央位置について1枚ずつ3回測定し、平均値から求めた。
ポリアミド30~40mgをヘキサフルオロイソプロパノール重水素化物1.2gに溶解し、1H-NMRで測定した。1,4-シクロヘキサンジカルボン酸の場合、トランス異性体に由来する1.98ppmのピーク面積とシス異性体に由来する1.77ppmと1.86ppmのピーク面積の比率からトランス異性体比率を求めた。
UL94(米国Under Writers Laboratories Incで定められた規格)の方法を用いて測定を行った。なお試験片(長さ127mm、幅12.7mm、厚みは1/32インチ)は射出成形機(日精樹脂(株)製PS40E)にUL試験片の金型(金型温度=Tg+20℃)を取り付けて、シリンダー温度=Tm2+20℃で成形を行った。射出圧力はUL試験片成形する際の完全充填圧力+2%の圧力で行った。
難燃等級には、UL94規格(垂直燃焼試験)に準じた。また、V-2不合格のものは、V-2outと記載した。
下記条件に設定した成形機で2mm厚×15mm幅を成形してその流動長(充填された長さ、cm)から流動性を評価した。
射出成形機(日精樹脂(株)製FN3000)に、流動性評価(2mm厚×15mm幅のスパイラル流路)の金型(金型温度=Tg+20℃)を取り付けて、シリンダー温度=Tm2+20℃、射出速度は20%設定、射出圧力は、34%設定で成形を行った。
上記(14)に記載のUL試験片成形する際の完全充填圧力(%)を示した。
完全充填圧力とは、射出速度(99%)は統一して、溶融させた樹脂を金型内の充填末端まで完全に充填できる最低圧力を測定し、成形機の与えることができる最大の圧力を100%としたときの比率として求めた。
上記(5)のASTM引張試験用のダンベル射出成形試験片(3mm厚)を、130℃のエチレングリコール50%水溶液に1000時間浸漬し、室温に放置した後、上記(5)の方法の引張試験を行い、引張強度を測定し、成形直後に測定した引張強度に対する割合を浸漬後の引張強度保持率として求めた。
「熱溶融重合法」によりポリアミドの重合反応を実施した。
(a)CHDA896g(5.20モル)、及び(b)2MPD604g(5.20モル)を蒸留水1500gに溶解させ、原料モノマーの等モル50質量%均一水溶液を作った。該均一水溶液に2MPD15g(0.13モル)を追添した。
得られた水溶液を内容積5.4Lのオートクレーブ(日東高圧製)に仕込み、液温(内温)が50℃になるまで保温して、オートクレーブ内を窒素置換した。オートクレーブの槽内の圧力が、ゲージ圧として(以下、槽内の圧力は全てゲージ圧として表記する。)、約2.5Kg/cm2になるまで、液温を約50℃から加熱を続けた(この系での液温は約145℃であった。)。槽内の圧力を約2.5Kg/cm2に保つため水を系外に除去しながら、加熱を続けて、水溶液の濃度が約75%になるまで濃縮した(この系での液温は約160℃であった。)。水の除去を止め、槽内の圧力が約30Kg/cm2になるまで加熱を続けた(この系での液温は約245℃であった。)。槽内の圧力を約30Kg/cm2に保つため水を系外に除去しながら、最終温度-50℃になるまで加熱を続けた。液温が最終温度-50℃(ここでは300℃)まで上昇した後に、加熱は続けながら、槽内の圧力が大気圧(ゲージ圧は0Kg/cm2)になるまで120分ほどかけながら降圧した。
その後、樹脂温度(液温)の最終温度が約350℃になるようにヒーター温度を調整した。樹脂温度はその状態のまま、槽内を真空装置で400torrの減圧下に30分維持した。その後、窒素で加圧し下部紡口(ノズル)からストランド状にし、水冷、カッティングを行いペレット状で排出して、ポリアミドを得た。得られたポリアミドの上記測定方法に基づいて行った測定結果を表4に示す。
実施例1において、(a)ジカルボン酸、(b)ジアミン、及び(c)ラクタム及び/又はアミノカルボン酸として、表1又は2に記載の化合物と量を用いたことと、樹脂温度の最終温度を表4又は5に記載の温度にしたこと以外は、実施例1に記載した方法でポリアミドの重合を行った(「熱溶融重合法」)。得られたポリアミドの上記測定方法に基づいて行った測定結果を表4及び5に示す。
実施例1において、(a)ジカルボン酸、(b)ジアミン、及び(c)ラクタム及び/又はアミノカルボン酸として、表3に記載の化合物と量を用いたことと、樹脂温度の最終温度を表6に記載の温度にしたこと以外は、実施例1に記載した方法でポリアミドの重合を行った(「熱溶融重合法」)。
比較例1においては、重合途中で、オートクレーブ内で固化したため、ストランドでの取り出しができなかったので、冷却後、塊で取り出し、粉砕機にて粉砕して、ペレットくらいの大きさにした。成形は発泡が激しかったため、成形品が得られなかった。
実施例1において、(a)ジカルボン酸、(b)ジアミン、及び(c)ラクタム及び/又はアミノカルボン酸として、表3に記載の化合物と量を用いたことと、樹脂温度の最終温度を表6に記載の温度にしたこと以外は、実施例1に記載した方法でポリアミドの重合を行った(「熱溶融重合法」)。得られたポリアミドの上記測定方法に基づいて行った測定結果を表6に示す。
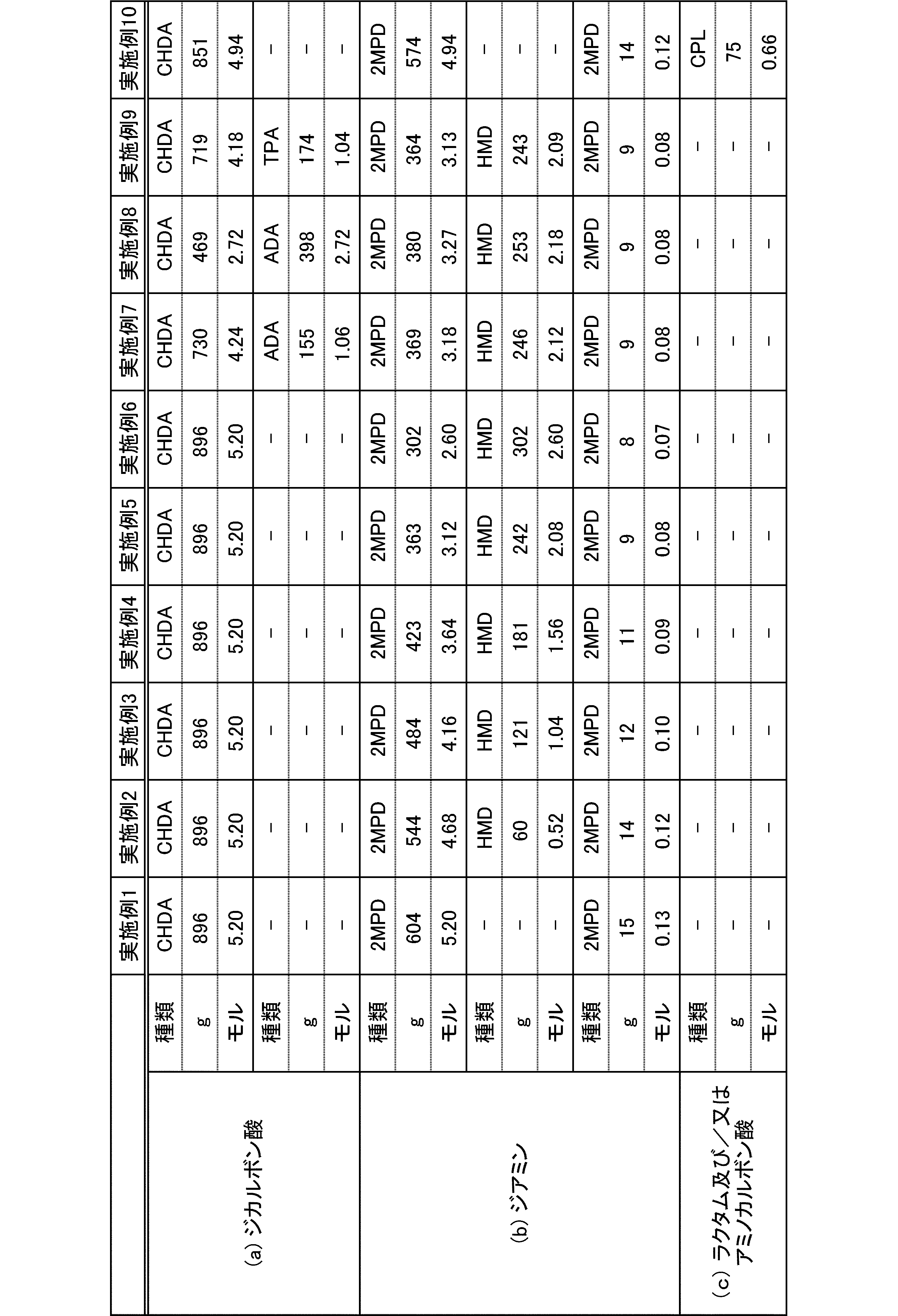


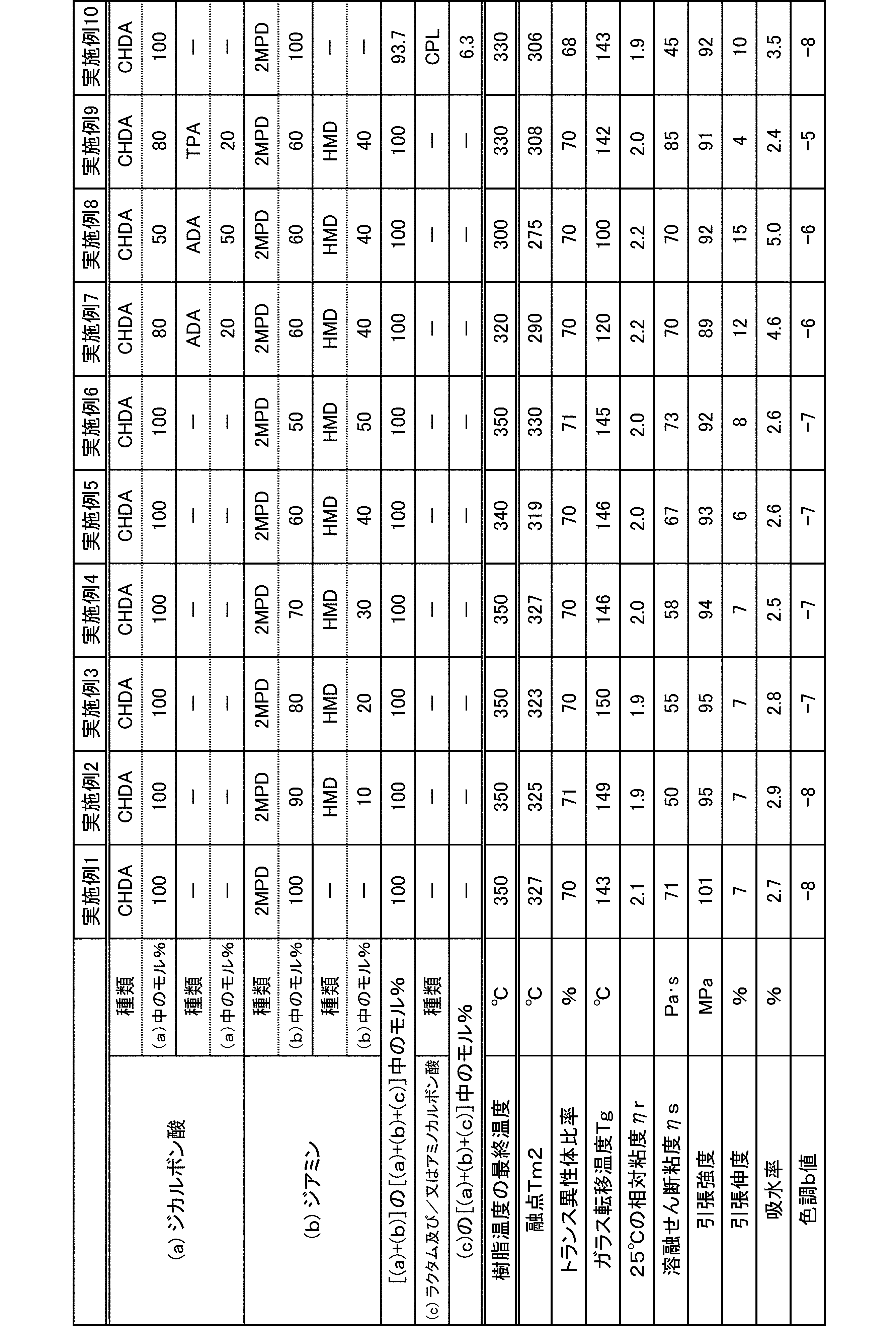
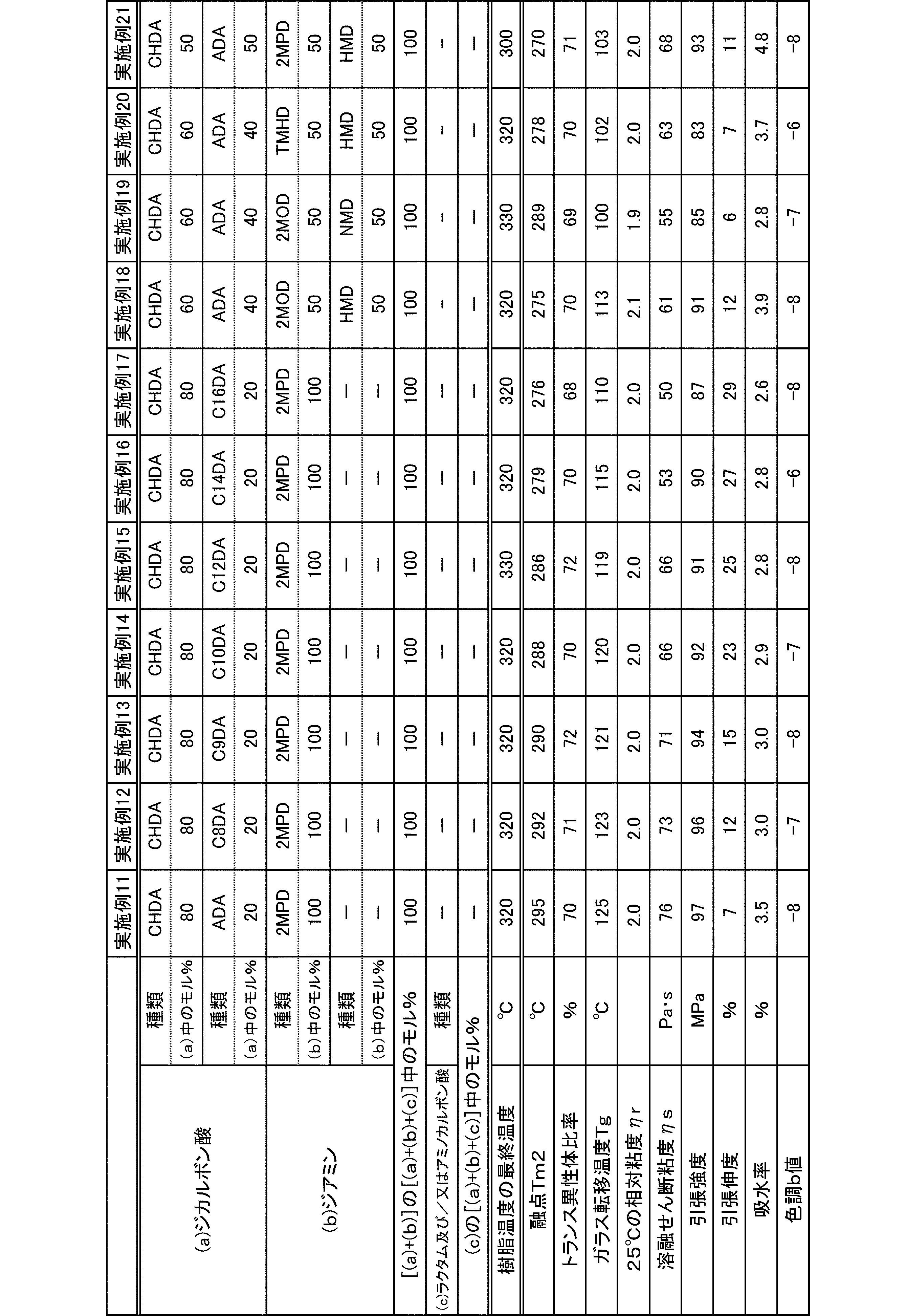

これに対して、50モル%未満の2-メチルペンタメチレンジアミンを含むポリアミドである比較例1では、共重合中に固化してしまいストランドとして取り出すことのできないものであると共に、成形品を得ることもできなかった。
さらに、特許文献1に開示された方法により製造した比較例4のポリアミドでは、流動性が低すぎて、成形性の点で十分なものではなかった。また、靭性も十分ではなかった。
「熱溶融重合・固相重合法」によりポリアミドの重合反応を実施した。
熱溶融重合については実施例1と仕込み量、手順とも同じ操作を実施し、ポリアミドを得た(ポリアミド(I))。得られたポリアミドの上記測定方法に基づいて行った測定結果を表7に示す。このうち1300gを固相重合用のリボン攪拌式加熱装置(リボコーン、大河原製作所製)に仕込み、室温で窒素置換した。窒素を流通したまま、樹脂温度が200℃になるように12時間加熱を行った。その後、窒素を流通したまま温度を下げていき約50℃になったところでペレットのまま装置から取り出し、ポリアミドを得た(ポリアミド(II))。得られたポリアミドの上記測定方法に基づいて行った測定結果を表7に示す。
ポリアミド(I)に対し、固相重合後のポリアミド(II)では、25℃における相対粘度が上昇し、引張伸度が上昇している。固相重合の前後で、トランス異性体比率は変化していない。また着色度も変化はなかった。
「プレポリマー・固相重合法」によりポリアミドの重合反応を実施した。
(a)CHDA896g(5.20モル)、及び(b)2MPD604g(5.20モル)に蒸留水500gを加え、原料モノマーの等モル33質量%スラリー液を作った。該スラリー液に2MPD15g(0.13モル)を追添した。
得られたスラリー液を内容積5.4Lのオートクレーブ(日東高圧(株)製)に仕込み、オートクレーブ内を窒素置換した。液温100℃で30分間攪拌した後、2時間かけて液温200℃になるように昇温した。この時、オートクレーブの槽内の圧力は22kg/cm2であった。220℃に昇温し、槽内の圧力を22Kg/cm2に保つため水を系外に除去しながら2時間維持した。槽内の圧力が大気圧(ゲージ圧は0Kg/cm2)になるまで、60分かけて降圧した。その後、樹脂温度(液温)を室温まで下げた後、オートクレーブの下部のフランジを外して固体状態のポリアミドのプレポリマーを得た(ポリアミド(I))。得られたプレポリマーの上記測定方法に基づいて行った測定結果を表7に示す。プレポリマーの1,4-シクロヘキサンジカルボン酸のトランス異性体比率は85%であった。また、ポリアミド(I)で着色が見られた。
得られたプレポリマーのうち1300gを用いて、実施例22と同様にして、固相重合を実施して、ポリアミドを得た(ポリアミド(II))。得られたポリアミドの上記測定方法に基づいて行った測定結果を表7に示す。ポリアミド(II)はプレポリマーに比べて相対粘度は向上しているが、着色が見られた。
「プレポリマー・押出重合法」によりポリアミドの重合反応を実施した。
プレポリマーの製造については実施例23と仕込み量、手順とも同じ操作を実施し、ポリアミドのプレポリマーを得た(ポリアミド(I))。得られたプレポリマーのうち1300gを用いて、押出重合装置(栗本鉄工所(株)製KRCニーダ)にて後重合を行った。ジャケット温度は350℃、真空度-0.5MPa(ゲージ圧)で滞留時間が30分になるようにプレポリマーを導入した。ストランドを冷却、カットして、ペレットとして、ポリアミドを得た(ポリアミド(II))。得られたポリアミドの上記測定方法に基づいて行った測定結果を表7に示す。ポリアミド(II)はプレポリマーに比べて25℃の相対粘度は向上しているが、着色が見られた。
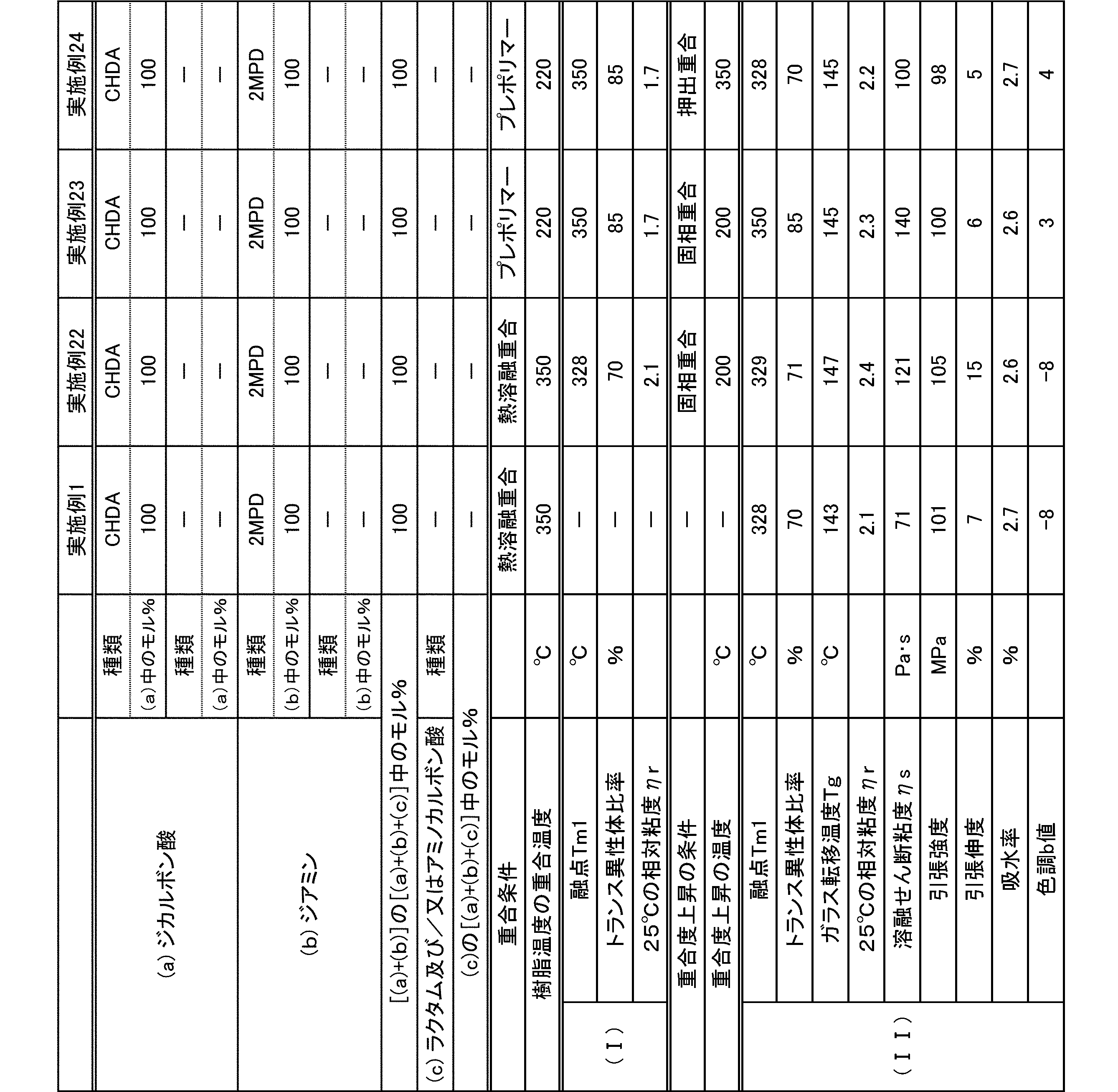
[実施例25]
実施例1のポリアミドを窒素気流中で乾燥し水分率を約0.2質量%に調整して用いた。2軸押出機(東芝機械(株)製TEM35φL/D=47.6、設定温度340℃、スクリュー回転数300rpm)を用いて、押出し機最上流部に設けられたトップフィード口よりポリアミドを供給し、押出し機下流側(トップフィード口より供給された樹脂が十分溶融している状態)のサイドフィード口よりガラス繊維(GF)を供給し、ダイヘッドより押し出された溶融混練物をストランド状で冷却し、ペレタイズしてポリアミド組成物ペレットを得た。配合量はポリアミド100質量部に対してガラス繊維(GF)55質量部とした。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表8に示す。
実施例1のポリアミドに代えて実施例2~21の各ポリアミドを用いる以外は実施例25と同様に実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表8及び9に示す。
実施例29において、ポリアミド100質量部に対してガラス繊維(GF)100質量部とした以外は、実施例29と同様に実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表9に示す。
実施例1のポリアミドに代えて比較例1のポリアミドを用いる以外は実施例25と同様に実施しようとしたが、押出状態が非常に不安定で、ポリアミド組成物を得ることができなかった。
実施例1のポリアミドに代えて比較例2、3の各ポリアミドを用いる以外は実施例25と同様に実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果結果を表10に示す。
実施例1のポリアミドに代えて比較例4のポリアミドを用い、ポリアミド100質量部に対してガラス繊維(GF)100質量部とした以外は、実施例25と同様に実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表10に示す。
実施例1のポリアミドに代えて比較例5~7の各ポリアミドを用いる以外は実施例25と同様に実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表10に示す。
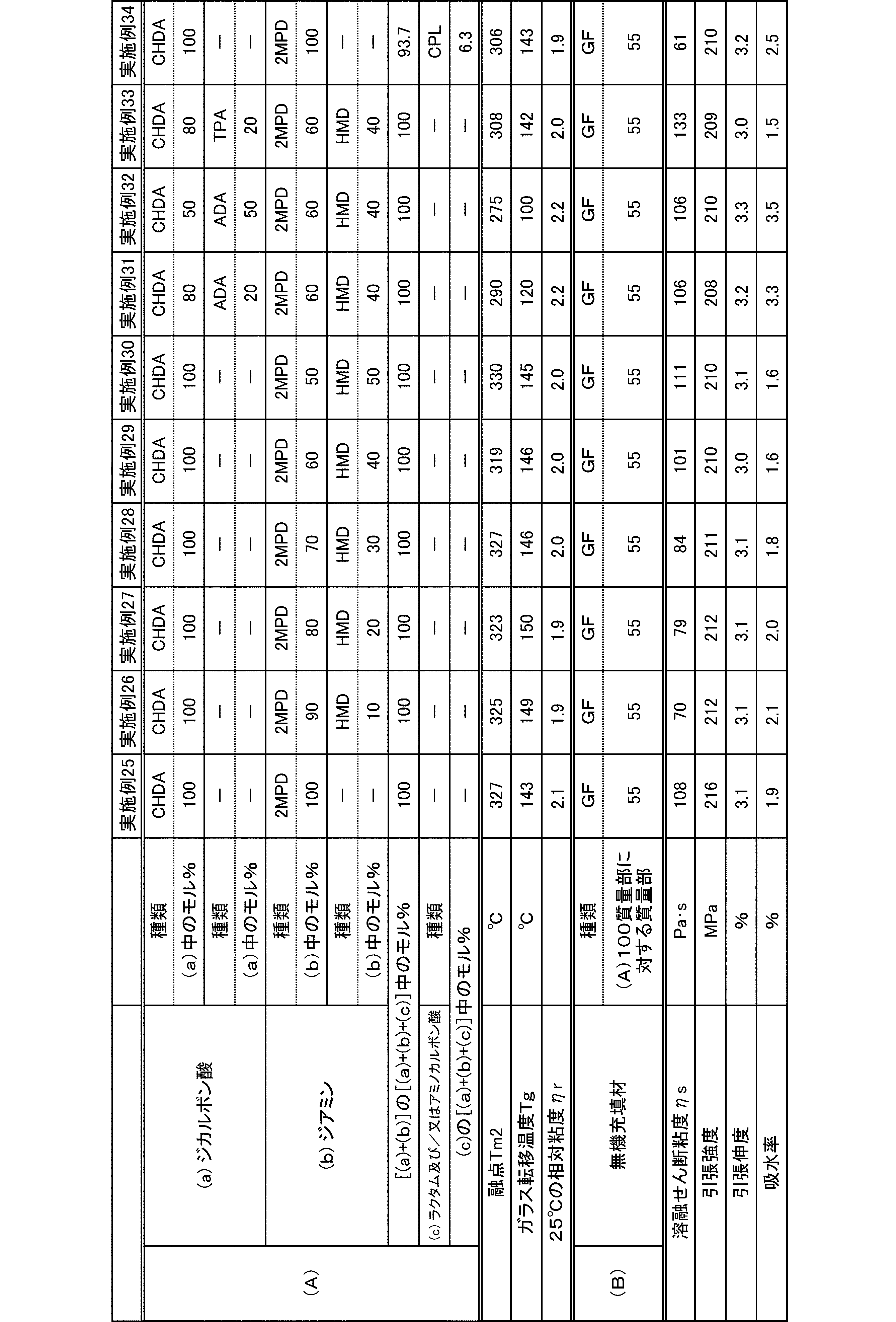
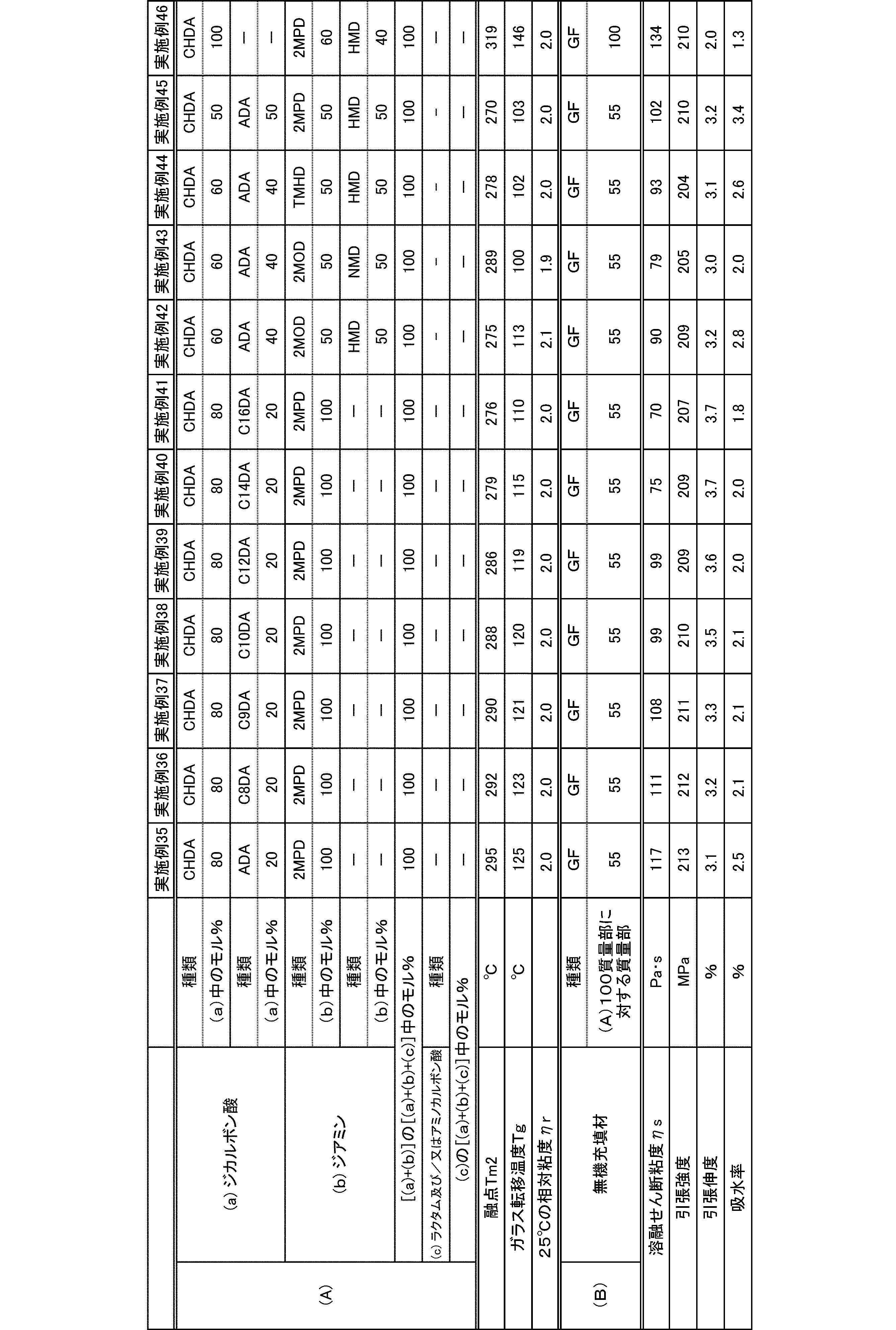

これに対して、50モル%未満の2-メチルペンタメチレンジアミンを重合させたポリアミドを含有する比較例8では、押出状態が不安定なものであり、ポリアミド組成物を得ることができなかった。
また、50モル%未満の脂環族ジカルボン酸を重合させたポリアミドを含有する比較例9及び10のポリアミド組成物では、耐熱性及び低吸水性の点で劣るものであった。
また、特許文献1に開示された方法により製造したポリアミドを含有する比較例11のポリアミド組成物では、溶融せん断粘度が大きく、流動性が低すぎるものであり、成形性の点で十分なものではなかった。また、引張伸度が小さく、靭性も十分ではなかった。
[製造例1]
KI 85.1質量部、エチレンビスステアリルアミド10質量部を混合し、KIとエチレンビスステアリルアミドの混合物を得た。該混合物にCuI 4.9質量部をよく混合し、ディスクペレッター(不二パウダル社製F5-11-175)で顆粒化し、顆粒(1)を得た。
KI 80.7質量部、エチレンビスステアリルアミド10質量部を混合し、KIとエチレンビスステアリルアミドの混合物を得た。該混合物にCuI 9.3質量部をよく混合し、ディスクペレッター(不二パウダル社製F5-11-175)で顆粒化し、顆粒(2)を得た。
KI 88.0質量部、エチレンビスステアリルアミド10質量部を混合し、KIとエチレンビスステアリルアミドの混合物を得た。該混合物にCuI 2.0質量部をよく混合し、ディスクペレッター(不二パウダル社製F5-11-175)で顆粒化し、顆粒(3)を得た。
100質量部の実施例1のポリアミド対して、6.1質量部の製造例1で製造した顆粒(1)、55質量部の無機充填材(GF)を配合し、二軸押出機(東芝機械(株)製TEM35φL/D=47.6、設定温度340℃、スクリュー回転数300rpm)で溶融混練してポリアミド組成物を得た。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表11に示す。
実施例47において、実施例1のポリアミドに代えて実施例2~21の各ポリアミドを用いる以外は実施例47と同様にして実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表11及び12に示す。
実施例47において、実施例1のポリアミドに代えて比較例1のポリアミドを用いる以外は実施例47と同様にして実施しようとしたが、押出状態が非常に不安定で、ポリアミド組成物を得ることができなかった。
実施例47において、実施例1のポリアミドに代えて比較例2~7の各ポリアミドを用いる以外は実施例47と同様にして実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表13に示す。
実施例51において、100質量部の実施例5のポリアミド対して、3.1質量部の製造例1の顆粒(1)を用いた以外は実施例51と同様にして実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表14に示す。
実施例51において、100質量部の実施例5のポリアミド対して、9.2質量部の製造例1の顆粒(1)を用いた以外は実施例51と同様にして実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表14に示す。
実施例51において、100質量部の実施例51のポリアミド対して、12.2質量部の製造例1の顆粒(1)を用いた以外は実施例51と同様にして実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表14に示す。
実施例51において、100質量部の実施例51のポリアミド対して、3.2質量部の製造例2の顆粒(2)を用いた以外は実施例51と同様にして実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表14に示す。
実施例51において、100質量部の実施例51のポリアミド対して、15.0質量部の製造例3の顆粒(3)を用いた以外は実施例51と同様にして実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表14に示す。
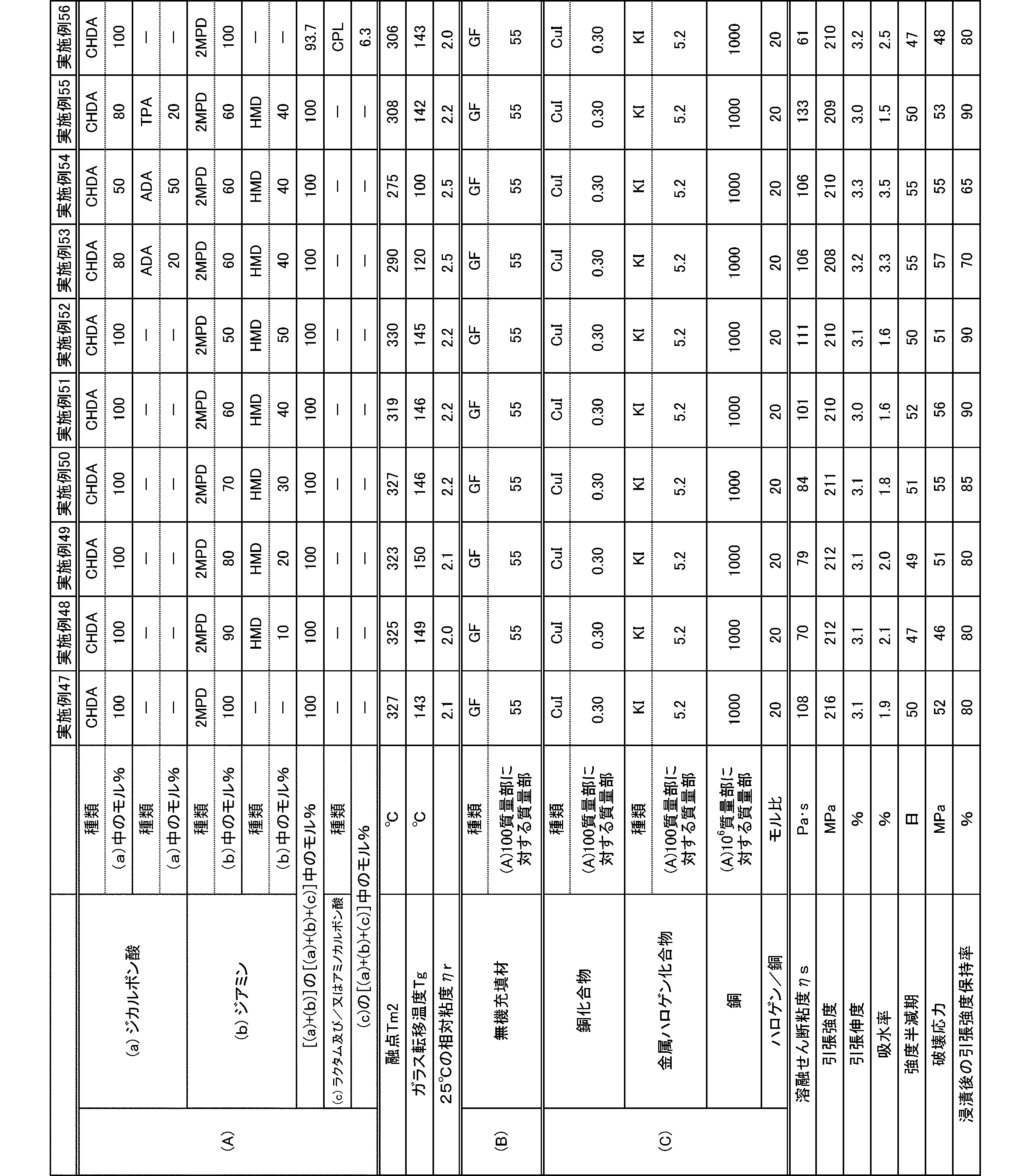



これに対して、50モル%未満の2-メチルペンタメチレンジアミンを含むポリアミドを含有する比較例15では、押出状態が不安定なものであり、ポリアミド組成物を得ることができなかった。
また、50モル%未満の脂環族ジカルボン酸を重合させたポリアミドを含有する比較例16及び17のポリアミド組成物では、耐熱性及び低吸水性の点で劣るものであった。
また、特許文献1に開示された方法により製造したポリアミドを含有する比較例18のポリアミド組成物では、溶融せん断粘度が大きく、流動性が低すぎるものであり、成形性の点で十分なものではなかった。また、引張伸度が小さく、靭性も十分ではなかった。
ミド組成物
[実施例73]
実施例1のポリアミドを窒素気流中で乾燥し水分率を約0.2質量%に調整して用いた。2軸押出機(東芝機械(株)製TEM35φL/D=47.6、設定温度340℃、スクリュー回転数300rpm、吐出量50kg/hr)を用いて、押出機最上流部に設けられたトップフィード口より(A)ポリアミド、(D)ハロゲン系難燃剤、(G)難燃助剤、及び(H)α,β不飽和ジカルボン酸無水物を含む重合体を予めブレンドしたものを供給し、押出し機下流側(トップフィード口より供給された樹脂が十分溶融している状態)のサイドフィード口より(B)無機充填材を供給し、ダイヘッドより押し出された溶融混練物をストランド状で冷却し、ペレタイズしてポリアミド組成物ペレットを得た。配合量は(A)ポリアミド100質量部に対して、(C)ハロゲン系難燃剤45.0質量部、(G)難燃助剤7.0質量部、(H)α,β不飽和ジカルボン酸無水物を含む重合体4.0質量部、及び(B)無機充填材70.0質量部とした。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表15に示す。
実施例73において、実施例1のポリアミドに代えて実施例2~21のポリアミドを用いる以外は実施例73と同様に実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表15及び16に示す。
実施例77において、(H)α,β不飽和ジカルボン酸無水物を含む重合体を配合せずに、(G)難燃助剤15.0質量部、及び(B)無機充填材75.0質量部とした以外は実施例77と同様に実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表16に示す。
実施例77において、(G)難燃助剤として水酸化マグネシウムを7.0質量部とした以外は実施例77と同様に実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表16に示す。
実施例73において、実施例1のポリアミドに代えて比較例1のポリアミドを用いる以外は実施例73と同様に実施しようとしたが、押出状態が非常に不安定で、ポリアミド組成物を得ることができなかった。
実施例73において、実施例1のポリアミドに代えて比較例2~7のポリアミドを用いる以外は実施例73と同様に実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表17に示す。
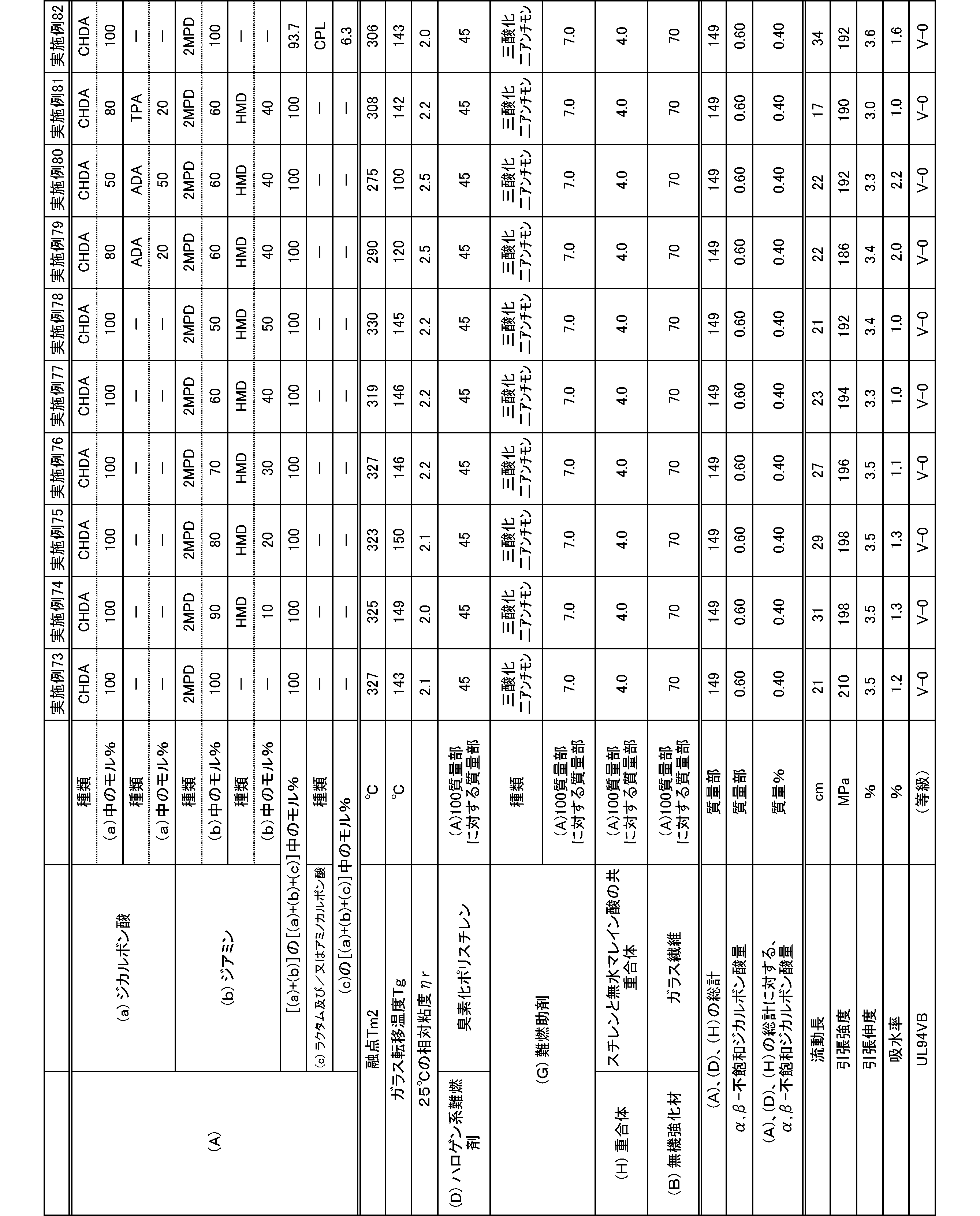

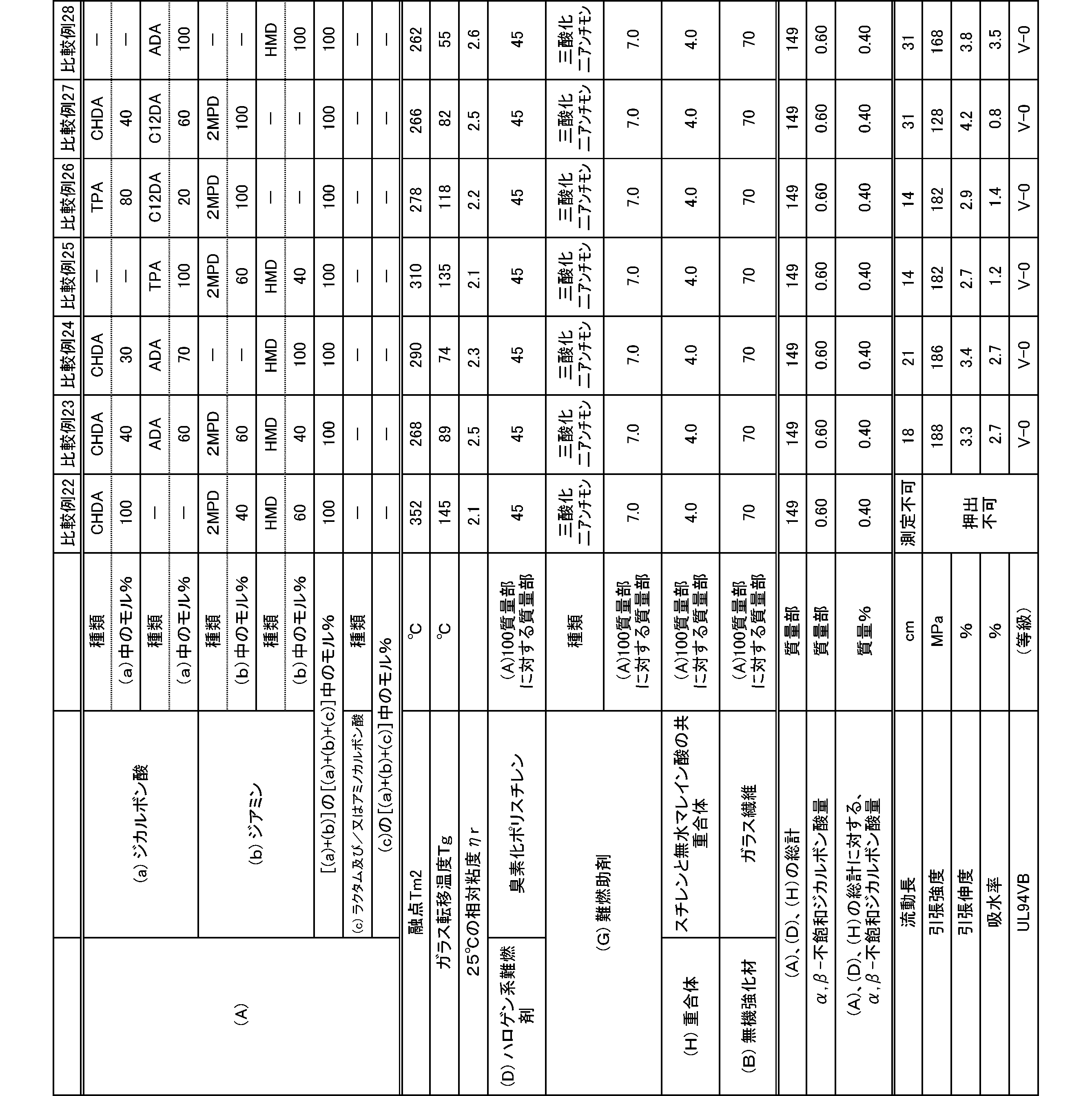
これに対して、50モル%未満の2-メチルペンタメチレンジアミンを重合させたポリアミドを含有する比較例22では、押出状態が不安定なものであり、ポリアミド組成物を得ることができなかった。
また、50モル%未満の脂環族ジカルボン酸を重合させたポリアミドを含有する比較例23及び24のポリアミド組成物では、耐熱性及び低吸水性の点で劣るものであった。
さらに、特許文献1に開示された方法により製造したポリアミドを含有する比較例25のポリアミド組成物では、流動長が短く、流動性が低すぎるものであり、成形性の点で十分なものではなかった。また、引張伸度が小さく、靭性も十分ではなかった。
PA66を含有する比較例28では、耐熱性及び低吸水性の点で劣るものであった。
[実施例96]
実施例1のポリアミドを窒素気流中で乾燥し水分率を約0.2質量%に調整して用いた。上流側に1ヶ所(トップフィード)と、押出機中央部並びにダイに近い下流側の2ヶ所に供給口を有する2軸押出機(東芝機械(株)製TEM35φL/D=47.6、設定温度340℃、スクリュー回転数100rpm、吐出量=30kg/hr)を用いて、押出機最上流部に設けられたトップフィード口より(A)ポリアミド、押出機中央部の供給口より(E)ホスフィン酸塩、(G)難燃助剤、ダイに近い下流側の供給口より(B)無機充填材を供給し、ダイヘッドより押し出された溶融混練物をストランド状で冷却し、ペレタイズしてポリアミド組成物ペレットを得た。配合量は(A)ポリアミド100質量部に対して、(E)ホスフィン酸塩42.0質量部、(G)難燃助剤2.0質量部、(B)無機充填材48.0質量部とした。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表18に示す。
実施例96において、実施例1のポリアミドに代えて実施例2~21のポリアミドを用いる以外は実施例96と同様に実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表18及び19に示す。
実施例100において、(G)難燃助剤を配合しない以外は実施例100と同様に実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表19に示す。
実施例100において、(G)難燃助剤として水酸化マグネシウム2.0質量部を配合した以外は実施例100と同様に実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表19に示す。
実施例96において、実施例1のポリアミドに代えて比較例1のポリアミドを用いる以外は実施例96と同様に実施しようとしたが、押出状態が非常に不安定で、ポリアミド組成物を得ることができなかった。
実施例96において、実施例1のポリアミドに代えて比較例2~7のポリアミドを用いる以外は実施例96と同様に実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表20に示す。

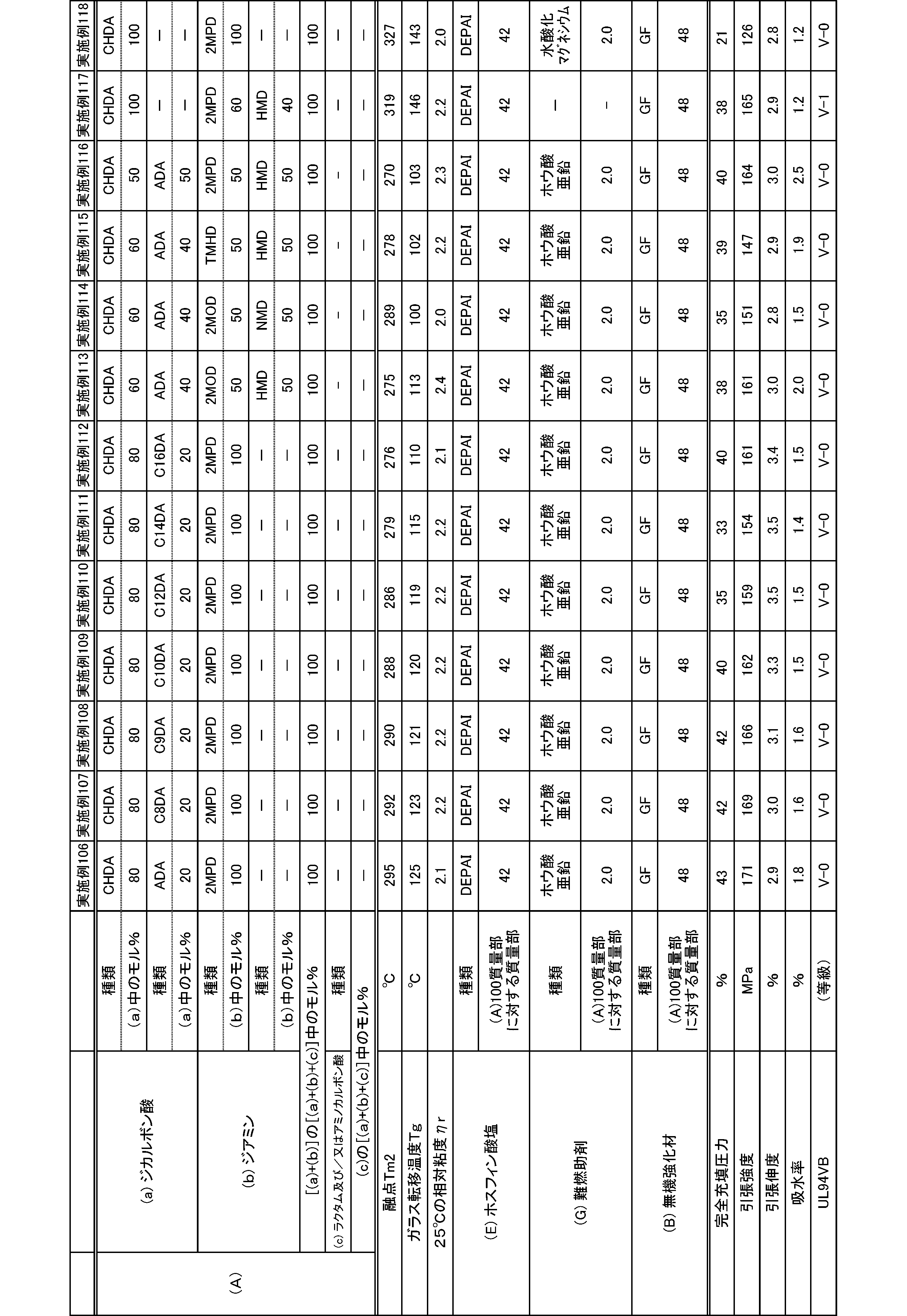
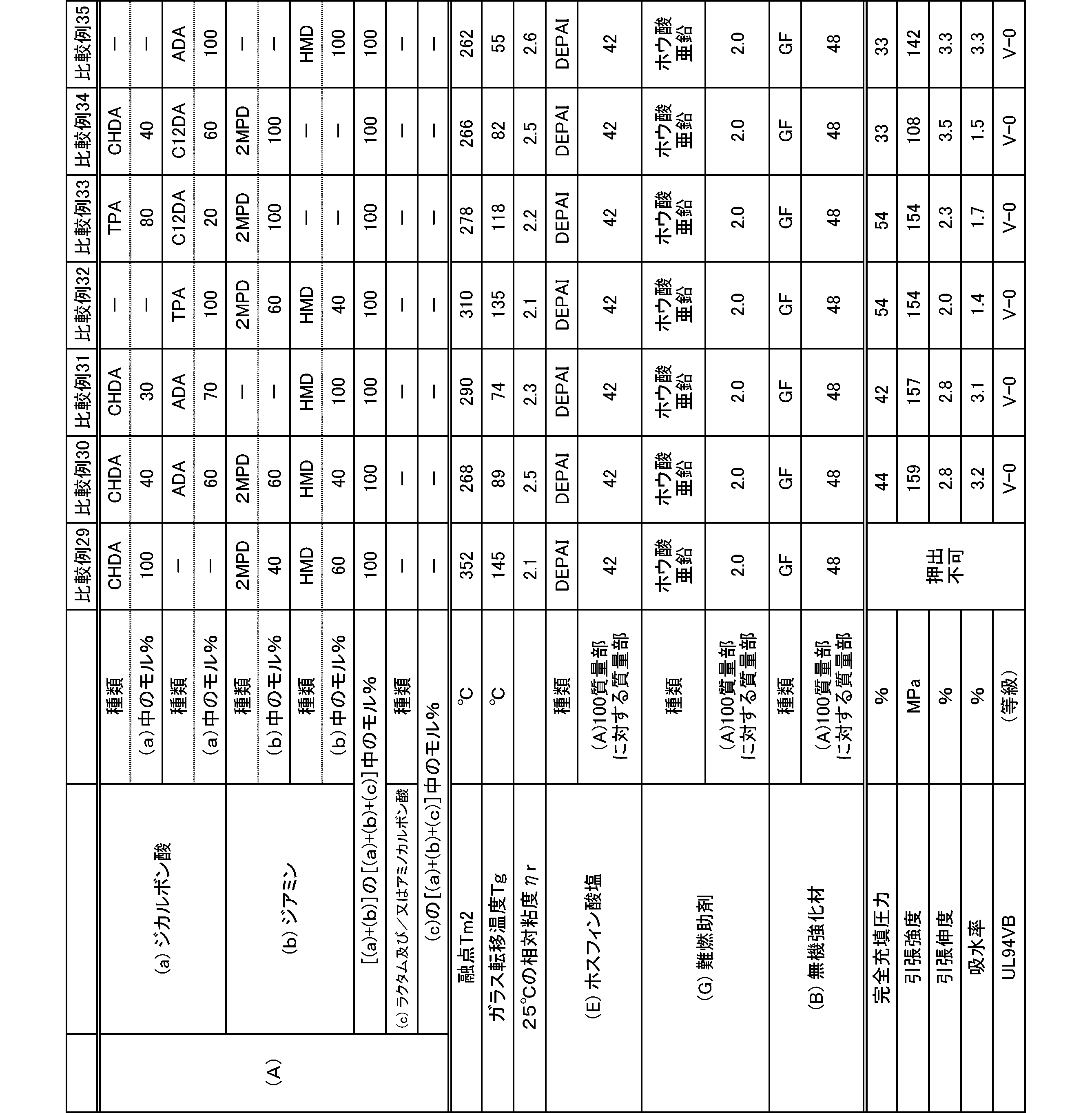
これに対して、50モル%未満の2-メチルペンタメチレンジアミンを重合させたポリアミドを含有する比較例29では、押出状態が不安定なものであり、ポリアミド組成物を得ることができなかった。
また、50モル%未満の脂環族ジカルボン酸を重合させたポリアミドを含有する比較例30及び31のポリアミド組成物では、耐熱性及び低吸水性の点で劣るものであった。
さらに、特許文献1に開示された方法により製造したポリアミドを含有する比較例32のポリアミド組成物では、完全充填圧力が大きく、流動性が低すぎるものであり、成形性の点で十分なものではなかった。また、引張伸度が小さく、靭性も十分ではなかった。
PA66を含有する比較例35では、耐熱性及び低吸水性の点で劣るものであった。
[実施例119]
100質量部の実施例1のポリアミド対して、0.3質量部の安定剤(21)N,N’-へキサン-1,6-ジイルビス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニルプロピオンアミド)]を配合し、二軸押出機(東芝機械(株)製TEM35φL/D=47.6、設定温度340℃、スクリュー回転数300rpm)を用いて溶融混練して、ポリアミド組成物を得た。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表21に示す。
実施例119において、実施例1のポリアミドに代えて実施例2~21の各ポリアミドを用いる以外は実施例119と同様にして実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表21及び22に示す。
実施例119において、実施例1のポリアミドに代えて比較例1のポリアミドを用いる以外は実施例119と同様にして実施しようとしたが、押出状態が非常に不安定で、ポリアミド組成物を得ることができなかった。
実施例119において、実施例1のポリアミドに代えて比較例2~7の各ポリアミドを用いる以外は実施例119と同様にして実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表23に示す。
実施例123において、安定剤(21)N,N’-へキサン-1,6-ジイルビス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニルプロピオンアミド)]の代わりに、安定剤(22)ビス(2,6-ジ-t-ブチル-4-メチルフェニル)ペンタエリスリトールジホスファイトを用いた以外は、実施例123と同様にして実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表24に示す。
実施例123において、安定剤(21)N,N’-へキサン-1,6-ジイルビス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニルプロピオンアミド)]の代わりに、安定剤(23)ビス-(2,2,6,6-テトラメチル-4-ピペリジル)-セバケートを用いた以外は、実施例123と同様にして実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表24に示す。
実施例123において、安定剤(21)N,N’-へキサン-1,6-ジイルビス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニルプロピオンアミド)]の代わりに、安定剤(24)2-(2’-ヒドロキシ-4’-ヘキシルオキシフェニル)-4、6-ジフェニル-1,3,5-トリアジンを用いた以外は、実施例123と同様にして実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表24に示す。
実施例123において、安定剤(21)N,N’-へキサン-1,6-ジイルビス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニルプロピオンアミド)]の代わりに、安定剤(25)次亜リン酸ナトリウム0.1質量部を用いた以外は、実施例123と同様にして実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表24に示す。
実施例123において、100質量部の実施例5のポリアミド対して、0.5質量部の安定剤(21)N,N’-へキサン-1,6-ジイルビス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニルプロピオンアミド)]を用いた以外は、実施例123と同様にして実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表24に示す。
実施例123において、100質量部の実施例5のポリアミド対して、3.0質量部の安定剤(21)N,N’-へキサン-1,6-ジイルビス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニルプロピオンアミド)]を用いた以外は、実施例123と同様にして実施した。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表24に示す。
また、実施例5のポリアミドの測定結果を、上記測定方法に基づいて行った測定結果を表24に示す。
100質量部の実施例5のポリアミド対して、それぞれが0.3質量部の安定剤(21)N,N’-へキサン-1,6-ジイルビス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニルプロピオンアミド)]及び安定剤(22)ビス(2,6-ジ-t-ブチル-4-メチルフェニル)ペンタエリスリトールジホスファイトを配合し、二軸押出機(東芝機械(株)製TEM35φL/D=47.6、設定温度340℃、スクリュー回転数300rpm)を用いて溶融混練して、ポリアミド組成物を得た。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表24に示す。
100質量部の実施例5のポリアミド対して、それぞれが0.3質量部の安定剤(22)ビス(2,6-ジ-t-ブチル-4-メチルフェニル)ペンタエリスリトールジホスファイト及び安定剤(24)2-(2’-ヒドロキシ-4’-ヘキシルオキシフェニル)-4、6-ジフェニル-1,3,5-トリアジンを配合し、二軸押出機(東芝機械(株)製TEM35φL/D=47.6、設定温度340℃、スクリュー回転数300rpm)を用いて溶融混練して、ポリアミド組成物を得た。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表24に示す。
100質量部の実施例5のポリアミド対して、それぞれが0.3質量部の安定剤(21)N,N’-へキサン-1,6-ジイルビス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニルプロピオンアミド)]、安定剤(22)ビス(2,6-ジ-t-ブチル-4-メチルフェニル)ペンタエリスリトールジホスファイト、及び安定剤(23)ビス-(2,2,6,6-テトラメチル-4-ピペリジル)-セバケートを配合し、二軸押出機(東芝機械(株)製TEM35φL/D=47.6、設定温度340℃、スクリュー回転数300rpm)を用いて溶融混練して、ポリアミド組成物を得た。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果を表24に示す。
100質量部の実施例5のポリアミド対して、それぞれが0.3質量部の安定剤(21)N,N’-へキサン-1,6-ジイルビス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニルプロピオンアミド)]、安定剤(22)ビス(2,6-ジ-t-ブチル-4-メチルフェニル)ペンタエリスリトールジホスファイト、及び安定剤(24)2-(2’-ヒドロキシ-4’-ヘキシルオキシフェニル)-4、6-ジフェニル-1,3,5-トリアジンを配合し、二軸押出機(東芝機械(株)製TEM35φL/D=47.6、設定温度340℃、スクリュー回転数300rpm)を用いて溶融混練して、ポリアミド組成物を得た。得られたポリアミド組成物の上記測定方法に基づいて行った測定結果結果を表24に示す。


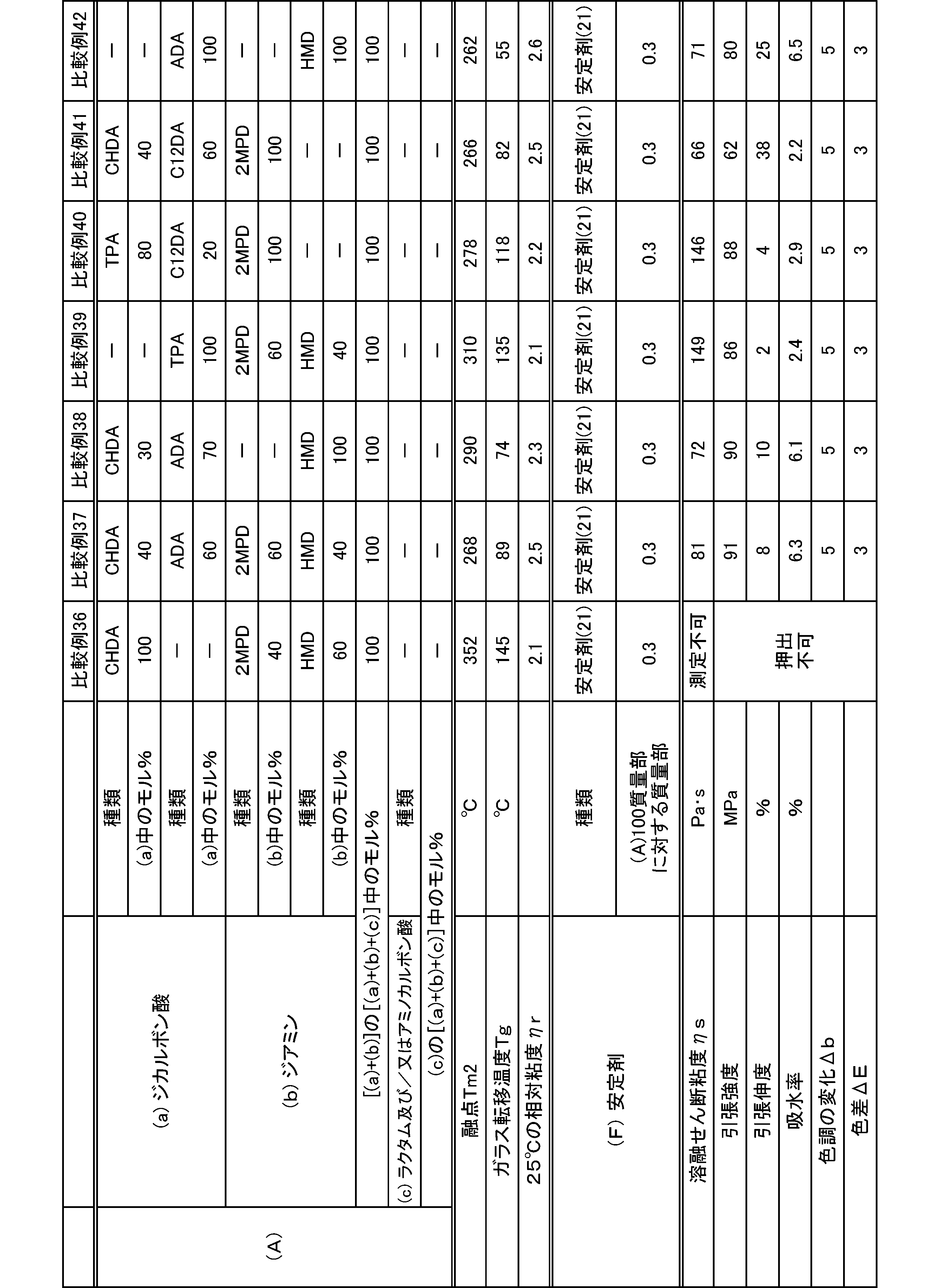

これに対して、50モル%未満の2-メチルペンタメチレンジアミンを重合させたポリアミドを含有する比較例36では、押し出し状態が不安定なものであり、ポリアミド組成物を得ることができなかった。
また、50モル%未満の脂環族ジカルボン酸を重合させポリアミドを含有する比較例37及び38では、耐熱性及び低吸水性の点で劣るものであった。
さらに、特許文献1に開示された方法により製造したポリアミドを含有する比較例39では、溶融せん断粘度が大きく、流動性が低すぎるものであり、成形性の点で十分なものではなかった。また、引張伸度が小さく、靭性も十分ではなかった。
PA66を含有する比較例42では、耐熱性及び低吸水性の点で劣るものであった。
Claims (18)
- (a)少なくとも50モル%の脂環族ジカルボン酸を含むジカルボン酸と、
(b)少なくとも50モル%の、主鎖から分岐した置換基を持つジアミンを含むジアミンと、を重合させた、ポリアミド。 - 前記主鎖から分岐した置換基を持つジアミンが、2-メチルペンタメチレンジアミンである、請求項1に記載のポリアミド。
- 前記脂環族ジカルボン酸が、1,4-シクロヘキサンジカルボン酸である、請求項1又は2に記載のポリアミド。
- 前記ジカルボン酸が、炭素数10以上の脂肪族ジカルボン酸をさらに含む、請求項1~3のいずれかに記載のポリアミド。
- (c)ラクタム及び/又はアミノカルボン酸をさらに共重合させた、請求項1~4のいずれかに記載のポリアミド。
- 融点が270~350℃である、請求項1~5のいずれかに記載のポリアミド。
- トランス異性体比率が50~85%である、請求項1~6のいずれかに記載のポリアミド。
- b値が0以下である、請求項1~7のいずれかに記載のポリアミド。
- (A)請求項1~8のいずれかに記載のポリアミドと、
(B)無機充填材と、を含有するポリアミド組成物。 - (A)請求項1~8のいずれかに記載のポリアミドと、
(C)銅化合物及び金属ハロゲン化物と、を含有するポリアミド組成物。 - (A)請求項1~8のいずれかに記載のポリアミドと、
(D)ハロゲン系難燃剤と、を含有するポリアミド組成物。 - (A)請求項1~8のいずれかに記載のポリアミドと、
(E)ホスフィン酸塩及び/又はジホスフィン酸塩と、を含有するポリアミド組成物。 - (A)請求項1~8のいずれかに記載のポリアミドと、
(F)安定剤と、を含有するポリアミド組成物。 - 請求項9~13のいずれかに記載のポリアミド組成物を含む、自動車部品。
- 自動車吸気系部品又は自動車冷却系部品である、請求項14に記載の自動車部品。
- (a)少なくとも50モル%の脂環族ジカルボン酸を含むジカルボン酸と、
(b)少なくとも50モル%の、主鎖から分岐した置換基を持つ脂肪族ジアミンを含むジアミンと、を重合させる工程を含む、
ポリアミドの製造方法。 - トランス異性体比率を50~80%に維持して重合する、請求項16に記載のポリアミドの製造方法。
- 請求項16又は17に記載の方法により得られるポリアミド。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/921,815 US8487024B2 (en) | 2008-03-12 | 2009-03-11 | Polyamide, polyamide composition, and method for producing polyamide |
| EP09720500.9A EP2270067B1 (en) | 2008-03-12 | 2009-03-11 | Polyamide, polyamide composition and method for producing polyamide |
| CN200980108831.8A CN101970535B (zh) | 2008-03-12 | 2009-03-11 | 聚酰胺、聚酰胺组合物及聚酰胺的制造方法 |
| US13/921,702 US9115247B2 (en) | 2008-03-12 | 2013-06-19 | Polyamide, polyamide composition, and method for producing polyamide |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008-062811 | 2008-03-12 | ||
| JP2008062811 | 2008-03-12 | ||
| JP2008075926 | 2008-03-24 | ||
| JP2008-075926 | 2008-03-24 | ||
| JP2008-264182 | 2008-10-10 | ||
| JP2008264182 | 2008-10-10 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/921,815 A-371-Of-International US8487024B2 (en) | 2008-03-12 | 2009-03-11 | Polyamide, polyamide composition, and method for producing polyamide |
| US13/921,702 Continuation US9115247B2 (en) | 2008-03-12 | 2013-06-19 | Polyamide, polyamide composition, and method for producing polyamide |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009113590A1 true WO2009113590A1 (ja) | 2009-09-17 |
| WO2009113590A9 WO2009113590A9 (ja) | 2010-12-09 |
Family
ID=41065251
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2009/054693 WO2009113590A1 (ja) | 2008-03-12 | 2009-03-11 | ポリアミド、ポリアミド組成物及びポリアミドの製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US8487024B2 (ja) |
| EP (1) | EP2270067B1 (ja) |
| KR (1) | KR20100115796A (ja) |
| CN (1) | CN101970535B (ja) |
| TW (2) | TWI460207B (ja) |
| WO (1) | WO2009113590A1 (ja) |
Cited By (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011042782A (ja) * | 2009-07-24 | 2011-03-03 | Asahi Kasei Chemicals Corp | ポリアミド組成物及びポリアミド組成物を成形した成形体 |
| WO2011030742A1 (ja) * | 2009-09-11 | 2011-03-17 | 旭化成ケミカルズ株式会社 | ポリアミド及びポリアミド組成物 |
| JP2011063795A (ja) * | 2009-08-21 | 2011-03-31 | Asahi Kasei Chemicals Corp | ポリアミド溶着成形品 |
| JP2012083626A (ja) * | 2010-10-13 | 2012-04-26 | Gunze Ltd | 難燃性を有する電子写真用導電性弾性部材およびその製造方法 |
| US20120170277A1 (en) * | 2009-09-07 | 2012-07-05 | Kuraray Co., Ltd. | Reflector for led and light-emitting device equipped with same |
| WO2012093722A1 (ja) | 2011-01-07 | 2012-07-12 | 旭化成ケミカルズ株式会社 | 共重合ポリアミド |
| JP2012172086A (ja) * | 2011-02-22 | 2012-09-10 | Asahi Kasei Chemicals Corp | 長繊維強化ポリアミド樹脂組成物及び成形体 |
| WO2012124740A1 (ja) * | 2011-03-15 | 2012-09-20 | 旭化成ケミカルズ株式会社 | ポリアミド及びポリアミド組成物 |
| JP2012184293A (ja) * | 2011-03-03 | 2012-09-27 | Asahi Kasei Chemicals Corp | ポリアミド組成物及び成形品 |
| US8487024B2 (en) | 2008-03-12 | 2013-07-16 | Asahi Kasei Chemicals Corporation | Polyamide, polyamide composition, and method for producing polyamide |
| US20130286654A1 (en) * | 2011-01-28 | 2013-10-31 | Kuraray Co., Ltd | Polyamide composition for reflector, reflector, light emitting device including the reflector, and lighting device and image display device each including the light emitting device |
| JP2013241506A (ja) * | 2012-05-18 | 2013-12-05 | Asahi Kasei Chemicals Corp | 耐熱変色性メタリック調ポリアミド樹脂組成物 |
| WO2014010607A1 (ja) | 2012-07-09 | 2014-01-16 | 旭化成ケミカルズ株式会社 | ポリアミド、ポリアミド組成物及び成形品 |
| JP2014005406A (ja) * | 2012-06-26 | 2014-01-16 | Asahi Kasei Chemicals Corp | 共重合ポリアミド及び共重合ポリアミド組成物 |
| JP2014005343A (ja) * | 2012-06-22 | 2014-01-16 | Asahi Kasei Chemicals Corp | 共重合ポリアミド |
| JP2014009353A (ja) * | 2012-07-03 | 2014-01-20 | Asahi Kasei Chemicals Corp | 共重合ポリアミド |
| JP2014015553A (ja) * | 2012-07-10 | 2014-01-30 | Asahi Kasei Chemicals Corp | 共重合ポリアミド組成物及び成形品 |
| JP2014015540A (ja) * | 2012-07-09 | 2014-01-30 | Asahi Kasei Chemicals Corp | ポリアミド樹脂組成物及び成形品 |
| JP2014015593A (ja) * | 2012-07-11 | 2014-01-30 | Asahi Kasei Chemicals Corp | ポリアミド樹脂組成物及び成形品 |
| JP2014015588A (ja) * | 2012-07-11 | 2014-01-30 | Asahi Kasei Chemicals Corp | 共重合ポリアミド組成物及び成形品 |
| JP2014015589A (ja) * | 2012-07-11 | 2014-01-30 | Asahi Kasei Chemicals Corp | ポリアミド樹脂組成物及び成形品 |
| JP2014084396A (ja) * | 2012-10-23 | 2014-05-12 | Cheil Industries Inc | ポリアミドの製造方法 |
| US8765861B2 (en) | 2010-05-21 | 2014-07-01 | Asahi Kasei Chemicals Corporation | Masterbatch pellet, production method therefor and polyamide resin composition containing masterbatch pellet |
| WO2021124895A1 (ja) * | 2019-12-16 | 2021-06-24 | 株式会社ブリヂストン | タイヤ |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2477243B1 (en) | 2009-09-11 | 2016-01-06 | Asahi Kasei Chemicals Corporation | Reflector for light-emitting device, and light-emitting device |
| KR20140026414A (ko) * | 2011-04-06 | 2014-03-05 | 솔베이 스페셜티 폴리머즈 유에스에이, 엘엘씨 | 폴리아미드 조성물 및 그로부터 제조되는 물품 |
| KR101437144B1 (ko) * | 2011-12-13 | 2014-09-02 | 제일모직주식회사 | 폴리아미드 수지, 이의 제조 방법 및 이를 포함하는 제품 |
| US20130168128A1 (en) * | 2011-12-29 | 2013-07-04 | Viakable, S. A. De C. V. | Sun-light resistant self-lubricated insulated conductor |
| FR2986798B1 (fr) * | 2012-02-14 | 2014-01-31 | Rhodia Operations | Nouvel agent inhibiteur de gonflement des argiles, compositions comprenant ledit agent et procedes mettant en oeuvre ledit agent |
| US9534083B2 (en) * | 2012-09-03 | 2017-01-03 | Basf Se | Production of polyamides by polycondensation |
| KR102102009B1 (ko) * | 2012-09-14 | 2020-04-17 | 주식회사 쿠라레 | 폴리아미드 수지 |
| CN104371100A (zh) * | 2013-08-15 | 2015-02-25 | 骏马化纤股份有限公司 | 一种半芳香聚酰胺树脂工业化连续生产方法 |
| CN104761886B (zh) * | 2014-01-08 | 2018-08-14 | 旭化成株式会社 | 聚酰胺树脂组合物及成形品 |
| KR101921393B1 (ko) * | 2014-01-08 | 2018-11-22 | 아사히 가세이 가부시키가이샤 | 폴리아미드 멀티필라멘트 섬유 및 이 섬유를 포함하는 타이어 코드 |
| CN103881374A (zh) * | 2014-03-13 | 2014-06-25 | 上海凯赛生物技术研发中心有限公司 | 增强聚酰胺树脂复合材料 |
| CN106164141B (zh) * | 2014-04-09 | 2019-05-28 | 旭化成株式会社 | 聚酰胺树脂组合物 |
| US20170081472A1 (en) * | 2014-05-16 | 2017-03-23 | Basf Se | Production of polyamides by hydrolytic polymerization and subsequent treatment in a kneader |
| CN106700064B (zh) * | 2016-12-28 | 2019-01-08 | 浙江新和成特种材料有限公司 | 一种半芳香族聚酰胺生产方法及半芳香族聚酰胺 |
| CN107189421A (zh) * | 2017-06-21 | 2017-09-22 | 安徽江淮汽车集团股份有限公司 | 一种抗老化耐磨pa6复合材料及其制备方法 |
| JP2019090016A (ja) * | 2017-11-10 | 2019-06-13 | 旭化成株式会社 | ポリアミド樹脂組成物及び成形体 |
| WO2019147458A1 (en) | 2018-01-23 | 2019-08-01 | Eastman Chemical Company | Novel polyesteramides, processes for the preparation thereof, and polyesteramide compositions |
| CA3123827A1 (en) * | 2018-12-18 | 2020-06-25 | Ascend Performance Materials Operations Llc | Antimicrobial polymer compositions, fibers, and yarns |
| TWI695869B (zh) * | 2019-01-24 | 2020-06-11 | 防焰塗料有限公司 | 防火塗料及其製造方法 |
| CN111487331B (zh) * | 2019-01-25 | 2022-07-12 | 南开大学 | 一种针对环境样品中微量尼龙6和尼龙66的定量检测方法 |
| EP3959254A1 (en) * | 2019-04-22 | 2022-03-02 | Solvay Specialty Polymers USA, LLC. | Copolyamides obtainable from 4-(aminomethyl)benzoic acid |
| TWI789730B (zh) * | 2020-05-11 | 2023-01-11 | 財團法人工業技術研究院 | 共聚物與其形成方法 |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0517413A (ja) | 1991-07-09 | 1993-01-26 | Koei Chem Co Ltd | ジアミンの製造法 |
| JPH05125184A (ja) * | 1991-04-10 | 1993-05-21 | Schering Ag | ポリアミド、その製造法、および接着法並びにレリーフ印刷物の製造法 |
| JPH06503590A (ja) | 1990-12-12 | 1994-04-21 | デユポン・カナダ・インコーポレーテツド | テレフタル酸コポリアミド |
| EP0699708A2 (de) | 1994-08-31 | 1996-03-06 | Hoechst Aktiengesellschaft | Flammengeschützte Polyesterformmassen |
| JPH0912868A (ja) | 1995-06-26 | 1997-01-14 | Kuraray Co Ltd | ポリアミド組成物 |
| JPH10292113A (ja) * | 1997-04-18 | 1998-11-04 | Hitachi Ltd | 液晶配向膜用組成物 |
| JPH11512476A (ja) | 1995-09-19 | 1999-10-26 | ディーエスエム エヌ.ブイ. | ポリアミド組成物から成る電気及び電子部材 |
| JP2000336167A (ja) * | 1999-05-27 | 2000-12-05 | Ube Ind Ltd | 延伸性に優れたポリアミド |
| JP2001514695A (ja) | 1997-03-13 | 2001-09-11 | ディーエスエム エヌ.ブイ. | ポリアミド組成物から作られた自動車部品 |
| JP2002097265A (ja) * | 2000-09-22 | 2002-04-02 | Mitsubishi Gas Chem Co Inc | ポリアミドの製造方法 |
| WO2002048239A1 (fr) | 2000-12-11 | 2002-06-20 | Asahi Kasei Kabushiki Kaisha | Polyamide |
| JP2003138012A (ja) * | 2001-11-08 | 2003-05-14 | Ube Ind Ltd | 延伸性に優れたポリアミド |
| JP2006522842A (ja) * | 2003-04-11 | 2006-10-05 | エムス ヒェミー アーゲー | 耐炎性ポリアミド成形組成物 |
| JP2008038125A (ja) * | 2005-11-10 | 2008-02-21 | Asahi Kasei Chemicals Corp | 難燃性に優れた樹脂組成物 |
Family Cites Families (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0073557B1 (en) | 1981-06-29 | 1985-09-25 | Toray Industries, Inc. | Copolyamide, process for producing thereof and copolyamide molding composition comprising thereof |
| JPS582327A (ja) | 1981-06-29 | 1983-01-07 | Toray Ind Inc | 共重合ポリアミドおよびその製造法 |
| JPS61174141A (ja) | 1985-01-25 | 1986-08-05 | Nitto Boseki Co Ltd | ガラス繊維及びその製造方法 |
| EP0196194B1 (en) | 1985-03-23 | 1989-12-27 | Nitto Boseki Co., Ltd. | Glass fiber strand and method of producing the same |
| JP2912964B2 (ja) | 1987-06-24 | 1999-06-28 | 清水建設株式会社 | 工程管理表作成システム |
| DE3924679A1 (de) | 1989-07-26 | 1991-01-31 | Bayer Ag | Verwendung von (meth)acryloylgruppen aufweisenden polyurethanen als bindemittel fuer pulverlacke |
| JPH0413300A (ja) | 1990-04-28 | 1992-01-17 | Fujitsu Ltd | 半導体記憶装置 |
| JP2838807B2 (ja) | 1990-05-30 | 1998-12-16 | 株式会社コパル | 加速度センサ |
| US5422420A (en) | 1990-11-20 | 1995-06-06 | E. I. Du Pont De Nemours And Company | Terpolyamides and multipolyamides containing amide units of 2-methylpentamethylenediamine and products prepared therefrom |
| JP3181697B2 (ja) | 1992-07-20 | 2001-07-03 | 旭化成株式会社 | 結晶性ポリアミド及びその組成物 |
| US5270437A (en) | 1992-11-13 | 1993-12-14 | E. I. Du Pont De Nemours And Company | Process for making partially aromatic polyamides containing 2-methylpentamethylenediamine units |
| DE19519820A1 (de) | 1995-05-31 | 1996-12-05 | Bayer Ag | Thermostabile, witterungsbeständige Polyamidformmassen |
| SG71170A1 (en) * | 1997-11-18 | 2000-03-21 | Mitsui Chemicals Inc | Process for preparing aromatic polyamides |
| US6121388A (en) * | 1998-05-12 | 2000-09-19 | Toray Industries, Inc. | Polyamide resin composition |
| WO2000058248A1 (en) | 1999-03-25 | 2000-10-05 | Eastman Chemical Company | Process for producing 1,4-cyclohexanedimethanol with enhanced cis-isomer content |
| US6297345B1 (en) * | 1999-05-27 | 2001-10-02 | Ube Industries, Ltd. | Polyamide having excellent stretching properties |
| EP1179568B1 (en) * | 2000-02-16 | 2006-05-10 | Asahi Kasei Kabushiki Kaisha | Polyamide resin composition |
| TW521082B (en) | 2000-09-12 | 2003-02-21 | Kuraray Co | Polyamide resin composition |
| JP2003119378A (ja) | 2000-09-12 | 2003-04-23 | Kuraray Co Ltd | ポリアミド樹脂組成物 |
| JP2002309083A (ja) | 2001-04-10 | 2002-10-23 | Kuraray Co Ltd | ポリアミド組成物 |
| JP2003002966A (ja) | 2001-06-22 | 2003-01-08 | Toyobo Co Ltd | ポリアミド樹脂 |
| JP4165106B2 (ja) | 2002-04-04 | 2008-10-15 | 東レ株式会社 | ポリペンタメチレンアジパミド樹脂およびその製造方法 |
| JP2003292614A (ja) | 2002-04-05 | 2003-10-15 | Toray Ind Inc | ポリアミド樹脂 |
| WO2004007614A1 (ja) * | 2002-07-10 | 2004-01-22 | Asahi Kasei Chemicals Corporation | ポリアミド組成物 |
| JP4168702B2 (ja) | 2002-08-21 | 2008-10-22 | 東レ株式会社 | ポリペンタメチレンアジパミド樹脂の製造方法 |
| JP4458231B2 (ja) | 2002-10-08 | 2010-04-28 | 三菱瓦斯化学株式会社 | ポリアミドおよび樹脂組成物 |
| JP4451130B2 (ja) | 2002-12-20 | 2010-04-14 | 旭化成ケミカルズ株式会社 | 高分子量ポリアミド樹脂組成物の製造方法 |
| CN100469838C (zh) | 2003-02-21 | 2009-03-18 | 沙伯基础创新塑料知识产权有限公司 | 半透明热塑性组合物、其制备方法及其模塑制品 |
| US20050113496A1 (en) * | 2003-10-03 | 2005-05-26 | Yuji Saga | Flame resistant polyamide resin composition containing phenolic resin and articles made therefrom |
| JP4854977B2 (ja) | 2005-03-28 | 2012-01-18 | 旭化成ケミカルズ株式会社 | ポリアミド樹脂組成物の製造方法 |
| CN101155877B (zh) | 2005-04-08 | 2011-03-23 | 三井化学株式会社 | 阻燃性聚酰胺组合物 |
| US20090275682A1 (en) | 2005-11-10 | 2009-11-05 | Asahi Kasei Chemicals Corporation | Resin Composition Excellent in Flame Retardance |
| US7960451B2 (en) | 2006-04-11 | 2011-06-14 | Asahi Kasei Chemicals Corporation | Method for producing polyamide masterbatch |
| JP5436745B2 (ja) | 2006-04-25 | 2014-03-05 | 旭化成ケミカルズ株式会社 | 難燃性ポリアミド樹脂組成物 |
| JP5236473B2 (ja) | 2006-07-18 | 2013-07-17 | 旭化成ケミカルズ株式会社 | ポリアミド組成物 |
| JP4871175B2 (ja) | 2007-03-12 | 2012-02-08 | 株式会社神戸製鋼所 | 長繊維強化熱可塑性樹脂ペレットの製造方法 |
| CN101679744B (zh) | 2007-06-04 | 2013-09-25 | 旭化成化学株式会社 | 聚酰胺-聚苯醚树脂组合物和膜 |
| JP4674827B2 (ja) | 2008-03-12 | 2011-04-20 | 旭化成ケミカルズ株式会社 | ポリアミド及びポリアミドの製造方法 |
| KR20100115796A (ko) | 2008-03-12 | 2010-10-28 | 아사히 가세이 케미칼즈 가부시키가이샤 | 폴리아미드, 폴리아미드 조성물 및 폴리아미드의 제조 방법 |
| JP5667983B2 (ja) | 2009-09-08 | 2015-02-12 | 旭化成ケミカルズ株式会社 | ポリアミド共重合体及び成形品 |
| CN102482415B (zh) | 2009-09-11 | 2013-11-06 | 旭化成化学株式会社 | 聚酰胺及聚酰胺组合物 |
| JP2011225830A (ja) | 2010-03-31 | 2011-11-10 | Toray Ind Inc | ポリアミド樹脂の製造方法 |
-
2009
- 2009-03-11 KR KR20107020021A patent/KR20100115796A/ko active Search and Examination
- 2009-03-11 CN CN200980108831.8A patent/CN101970535B/zh not_active Expired - Fee Related
- 2009-03-11 WO PCT/JP2009/054693 patent/WO2009113590A1/ja active Application Filing
- 2009-03-11 US US12/921,815 patent/US8487024B2/en active Active
- 2009-03-11 EP EP09720500.9A patent/EP2270067B1/en not_active Not-in-force
- 2009-03-12 TW TW98108003A patent/TWI460207B/zh not_active IP Right Cessation
- 2009-03-12 TW TW103105610A patent/TW201422671A/zh unknown
-
2013
- 2013-06-19 US US13/921,702 patent/US9115247B2/en not_active Expired - Fee Related
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06503590A (ja) | 1990-12-12 | 1994-04-21 | デユポン・カナダ・インコーポレーテツド | テレフタル酸コポリアミド |
| JPH05125184A (ja) * | 1991-04-10 | 1993-05-21 | Schering Ag | ポリアミド、その製造法、および接着法並びにレリーフ印刷物の製造法 |
| JPH0517413A (ja) | 1991-07-09 | 1993-01-26 | Koei Chem Co Ltd | ジアミンの製造法 |
| EP0699708A2 (de) | 1994-08-31 | 1996-03-06 | Hoechst Aktiengesellschaft | Flammengeschützte Polyesterformmassen |
| JPH0873720A (ja) | 1994-08-31 | 1996-03-19 | Hoechst Ag | 防炎性ポリエステル成形材料 |
| JPH0912868A (ja) | 1995-06-26 | 1997-01-14 | Kuraray Co Ltd | ポリアミド組成物 |
| JPH11512476A (ja) | 1995-09-19 | 1999-10-26 | ディーエスエム エヌ.ブイ. | ポリアミド組成物から成る電気及び電子部材 |
| JP2001514695A (ja) | 1997-03-13 | 2001-09-11 | ディーエスエム エヌ.ブイ. | ポリアミド組成物から作られた自動車部品 |
| JPH10292113A (ja) * | 1997-04-18 | 1998-11-04 | Hitachi Ltd | 液晶配向膜用組成物 |
| JP2000336167A (ja) * | 1999-05-27 | 2000-12-05 | Ube Ind Ltd | 延伸性に優れたポリアミド |
| JP2002097265A (ja) * | 2000-09-22 | 2002-04-02 | Mitsubishi Gas Chem Co Inc | ポリアミドの製造方法 |
| WO2002048239A1 (fr) | 2000-12-11 | 2002-06-20 | Asahi Kasei Kabushiki Kaisha | Polyamide |
| JP2003138012A (ja) * | 2001-11-08 | 2003-05-14 | Ube Ind Ltd | 延伸性に優れたポリアミド |
| JP2006522842A (ja) * | 2003-04-11 | 2006-10-05 | エムス ヒェミー アーゲー | 耐炎性ポリアミド成形組成物 |
| JP2008038125A (ja) * | 2005-11-10 | 2008-02-21 | Asahi Kasei Chemicals Corp | 難燃性に優れた樹脂組成物 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2270067A4 * |
Cited By (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9115247B2 (en) | 2008-03-12 | 2015-08-25 | Asahi Kasei Chemicals Corporation | Polyamide, polyamide composition, and method for producing polyamide |
| US8487024B2 (en) | 2008-03-12 | 2013-07-16 | Asahi Kasei Chemicals Corporation | Polyamide, polyamide composition, and method for producing polyamide |
| JP2011042782A (ja) * | 2009-07-24 | 2011-03-03 | Asahi Kasei Chemicals Corp | ポリアミド組成物及びポリアミド組成物を成形した成形体 |
| JP2011063795A (ja) * | 2009-08-21 | 2011-03-31 | Asahi Kasei Chemicals Corp | ポリアミド溶着成形品 |
| US20120170277A1 (en) * | 2009-09-07 | 2012-07-05 | Kuraray Co., Ltd. | Reflector for led and light-emitting device equipped with same |
| US10253181B2 (en) | 2009-09-07 | 2019-04-09 | Kuraray Co., Ltd. | Reflector for LED and light-emitting device equipped with same |
| EP2476731A1 (en) * | 2009-09-07 | 2012-07-18 | Kuraray Co., Ltd. | Reflector for led and light-emitting device equipped with same |
| EP2476731A4 (en) * | 2009-09-07 | 2013-06-19 | Kuraray Co | REFLECTOR FOR LEDS AND LIGHT-EMITTING DEVICE THEREFOR |
| JP5105563B2 (ja) * | 2009-09-11 | 2012-12-26 | 旭化成ケミカルズ株式会社 | ポリアミド及びポリアミド組成物 |
| WO2011030742A1 (ja) * | 2009-09-11 | 2011-03-17 | 旭化成ケミカルズ株式会社 | ポリアミド及びポリアミド組成物 |
| US9023975B2 (en) | 2009-09-11 | 2015-05-05 | Asahi Kasei Chemicals Corporation | Polyamide and polyamide composition |
| US8765861B2 (en) | 2010-05-21 | 2014-07-01 | Asahi Kasei Chemicals Corporation | Masterbatch pellet, production method therefor and polyamide resin composition containing masterbatch pellet |
| JP5667625B2 (ja) * | 2010-05-21 | 2015-02-12 | 旭化成ケミカルズ株式会社 | マスターバッチペレットおよびその製造方法ならびに該マスターバッチペレットを含むポリアミド樹脂組成物 |
| JP2012083626A (ja) * | 2010-10-13 | 2012-04-26 | Gunze Ltd | 難燃性を有する電子写真用導電性弾性部材およびその製造方法 |
| WO2012093722A1 (ja) | 2011-01-07 | 2012-07-12 | 旭化成ケミカルズ株式会社 | 共重合ポリアミド |
| US9611356B2 (en) | 2011-01-07 | 2017-04-04 | Asahi Kasei Chemicals Corporation | Copolymer polyamide |
| US10407548B2 (en) * | 2011-01-28 | 2019-09-10 | Kuraray Co., Ltd. | Polyamide composition for reflector, reflector, light emitting device including the reflector, and lighting device and image display device each including the light emitting device |
| US20130286654A1 (en) * | 2011-01-28 | 2013-10-31 | Kuraray Co., Ltd | Polyamide composition for reflector, reflector, light emitting device including the reflector, and lighting device and image display device each including the light emitting device |
| JP2012172086A (ja) * | 2011-02-22 | 2012-09-10 | Asahi Kasei Chemicals Corp | 長繊維強化ポリアミド樹脂組成物及び成形体 |
| JP2012184293A (ja) * | 2011-03-03 | 2012-09-27 | Asahi Kasei Chemicals Corp | ポリアミド組成物及び成形品 |
| WO2012124740A1 (ja) * | 2011-03-15 | 2012-09-20 | 旭化成ケミカルズ株式会社 | ポリアミド及びポリアミド組成物 |
| JP5942229B2 (ja) * | 2011-03-15 | 2016-06-29 | 旭化成株式会社 | ポリアミド及びポリアミド組成物 |
| US9090739B2 (en) | 2011-03-15 | 2015-07-28 | Asahi Kasei Chemicals Corporation | Polyamide and polyamide composition |
| JP2013241506A (ja) * | 2012-05-18 | 2013-12-05 | Asahi Kasei Chemicals Corp | 耐熱変色性メタリック調ポリアミド樹脂組成物 |
| JP2014005343A (ja) * | 2012-06-22 | 2014-01-16 | Asahi Kasei Chemicals Corp | 共重合ポリアミド |
| JP2014005406A (ja) * | 2012-06-26 | 2014-01-16 | Asahi Kasei Chemicals Corp | 共重合ポリアミド及び共重合ポリアミド組成物 |
| JP2014009353A (ja) * | 2012-07-03 | 2014-01-20 | Asahi Kasei Chemicals Corp | 共重合ポリアミド |
| KR20170019493A (ko) | 2012-07-09 | 2017-02-21 | 아사히 가세이 케미칼즈 가부시키가이샤 | 폴리아미드, 폴리아미드 조성물 및 성형품 |
| US9228057B2 (en) | 2012-07-09 | 2016-01-05 | Asahi Kasei Chemicals Corporation | Polyamide, polyamide composition, and molded article |
| JP2014015540A (ja) * | 2012-07-09 | 2014-01-30 | Asahi Kasei Chemicals Corp | ポリアミド樹脂組成物及び成形品 |
| WO2014010607A1 (ja) | 2012-07-09 | 2014-01-16 | 旭化成ケミカルズ株式会社 | ポリアミド、ポリアミド組成物及び成形品 |
| JP2014015553A (ja) * | 2012-07-10 | 2014-01-30 | Asahi Kasei Chemicals Corp | 共重合ポリアミド組成物及び成形品 |
| JP2014015589A (ja) * | 2012-07-11 | 2014-01-30 | Asahi Kasei Chemicals Corp | ポリアミド樹脂組成物及び成形品 |
| JP2014015588A (ja) * | 2012-07-11 | 2014-01-30 | Asahi Kasei Chemicals Corp | 共重合ポリアミド組成物及び成形品 |
| JP2014015593A (ja) * | 2012-07-11 | 2014-01-30 | Asahi Kasei Chemicals Corp | ポリアミド樹脂組成物及び成形品 |
| JP2014084396A (ja) * | 2012-10-23 | 2014-05-12 | Cheil Industries Inc | ポリアミドの製造方法 |
| US9840587B2 (en) | 2012-10-23 | 2017-12-12 | Lotte Advanced Materials Co., Ltd. | Polyamide production method |
| WO2021124895A1 (ja) * | 2019-12-16 | 2021-06-24 | 株式会社ブリヂストン | タイヤ |
Also Published As
| Publication number | Publication date |
|---|---|
| US8487024B2 (en) | 2013-07-16 |
| US9115247B2 (en) | 2015-08-25 |
| KR20100115796A (ko) | 2010-10-28 |
| EP2270067B1 (en) | 2016-06-15 |
| CN101970535B (zh) | 2015-05-27 |
| TWI460207B (zh) | 2014-11-11 |
| US20110028614A1 (en) | 2011-02-03 |
| EP2270067A4 (en) | 2012-12-26 |
| CN101970535A (zh) | 2011-02-09 |
| TW200938567A (en) | 2009-09-16 |
| EP2270067A1 (en) | 2011-01-05 |
| WO2009113590A9 (ja) | 2010-12-09 |
| US20130281655A1 (en) | 2013-10-24 |
| TW201422671A (zh) | 2014-06-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9115247B2 (en) | Polyamide, polyamide composition, and method for producing polyamide | |
| KR102078497B1 (ko) | 난연성 세미-방향족 폴리아마이드 조성물 및 이로부터 제조된 성형 제품 | |
| JP5497921B2 (ja) | 共重合ポリアミド | |
| EP2180018B1 (en) | Polyamide composition | |
| TWI472552B (zh) | 聚醯胺及聚醯胺組成物 | |
| JP6039945B2 (ja) | ポリアミド樹脂組成物及び成形品 | |
| JP6130400B2 (ja) | ハロゲンフリーの難燃性ポリアミド組成物の調整方法 | |
| JP5042263B2 (ja) | ポリアミド組成物 | |
| JP7107646B2 (ja) | ポリアミド組成物および成形品 | |
| JP5079733B2 (ja) | ポリアミド組成物 | |
| JP2011080046A (ja) | ポリアミド組成物、並びにポリアミド組成物を含む成形品及び電気部品 | |
| JP6937226B2 (ja) | ポリアミド組成物及び成形品 | |
| WO2022210019A1 (ja) | ポリアミド樹脂組成物およびポリアミド成形体 | |
| JP6050937B2 (ja) | ポリアミド樹脂組成物及び成形品 | |
| JP5461052B2 (ja) | ポリアミド組成物 | |
| JP2012184284A (ja) | ポリアミド樹脂組成物及び成形品 | |
| JP2020033412A (ja) | 樹脂組成物及び成形品 | |
| JP2018188533A (ja) | ポリアミド組成物及び成形品 | |
| WO2022196711A1 (ja) | ポリアミド樹脂組成物およびポリアミド成形体 | |
| JP6408637B2 (ja) | ハロゲンフリーの難燃性ポリアミド組成物の調製方法 | |
| JP2017155150A (ja) | ポリアミド組成物、ポリアミド組成物成形品およびポリアミド組成物の製造方法 | |
| JP2023028935A (ja) | ポリアミド組成物及び成形品 | |
| JP2023157584A (ja) | ポリアミド組成物及び成形品 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980108831.8 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09720500 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20107020021 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3326/KOLNP/2010 Country of ref document: IN Ref document number: 2009720500 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12921815 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |