JP7250737B2 - Rbポジティブ異常細胞増殖に対するhspc温存治療 - Google Patents
Rbポジティブ異常細胞増殖に対するhspc温存治療 Download PDFInfo
- Publication number
- JP7250737B2 JP7250737B2 JP2020123189A JP2020123189A JP7250737B2 JP 7250737 B2 JP7250737 B2 JP 7250737B2 JP 2020123189 A JP2020123189 A JP 2020123189A JP 2020123189 A JP2020123189 A JP 2020123189A JP 7250737 B2 JP7250737 B2 JP 7250737B2
- Authority
- JP
- Japan
- Prior art keywords
- compound
- positive
- cancer
- pharmaceutical composition
- cdk4
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/527—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim spiro-condensed
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/14—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/555—Heterocyclic compounds containing heavy metals, e.g. hemin, hematin, melarsoprol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7048—Compounds having saccharide radicals and heterocyclic rings having oxygen as a ring hetero atom, e.g. leucoglucosan, hesperidin, erythromycin, nystatin, digitoxin or digoxin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Molecular Biology (AREA)
- Oncology (AREA)
- Physiology (AREA)
- Nutrition Science (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Physics & Mathematics (AREA)
- Optics & Photonics (AREA)
Description
本出願は、2013年3月15日に出願の米国仮特許出願番号第61/798,772号、2013年8月1日に出願の米国仮特許出願番号第61/861,374号、2013年12月3日に出願の仮米国特許出願番号第61/911,354号、及び、仮2014年3月7日に出願の米国特許出願番号第61/949,786号に関連し、かつ、これらの利益を主張するものである。これらの出願の全部が、全ての目的のために参照事項としてここに包含される。
米国政府は、本発明に対して、アレルギー及び感染症学会により与えられる承認番号5R44AI084284号の支持に基づく権利を有する。
初期から中Gl段階まで、細胞がマイトジェン刺激に感応すれば、CDK4-サイクリンD及びCDK6-サイクリンDの活性化が、網膜芽細胞腫タンパク質(pRb)のリン酸化を誘発する。
Vander Welらは、強力かつ選択的CDK4阻害物質としてヨウ化含有ピリド[2、3-d]ピリミジン-7-オン(CKIA)を開示する(VanderWel et al., J. Med. Chem. 48 (2005) 2371-2387参照)。グラクソグループ社により出願された国際出願公報WO99/15500号は、プロテインキナーゼ及びセリン/トレオニンキナーゼ阻害物質を記載する。ノバルティスAGによって出願された国際出願公報WO2010/020675号は、CDK阻害物質としてピロロピリミジン化合物を記載する。ノバルティス社によって出願された国際出願公報WO2011/101409号においても、CDK4/6阻害力を有するピロロピリミジンを記載する。ノバルティス社によって出願された国際出願公報WO2005/052147号及びジャンセンファルマ社によって出願された国際出願公報WO2006/074985号は、添加CDK4阻害物質を開示する。Tavaresによって出願されG1 Therapeutics社に譲渡された国際出願公報WO2012/061156号は、CDK阻害物質を記載する。Francis Tavaresにより出願されGl Therapeuticsに譲渡された国際出願公報WO2013/148748号は、ラクタムキナーゼ阻害剤を記載する。
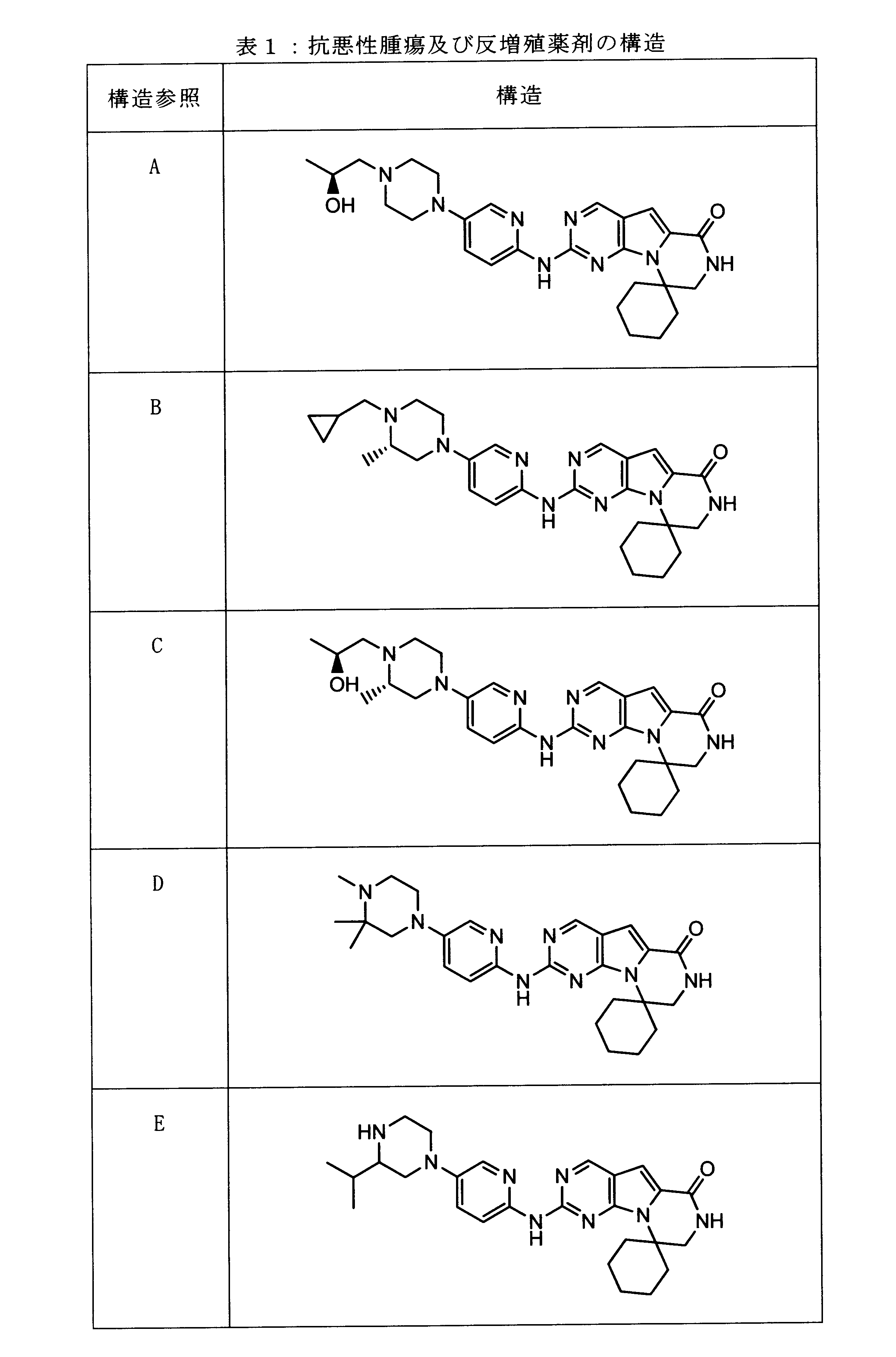
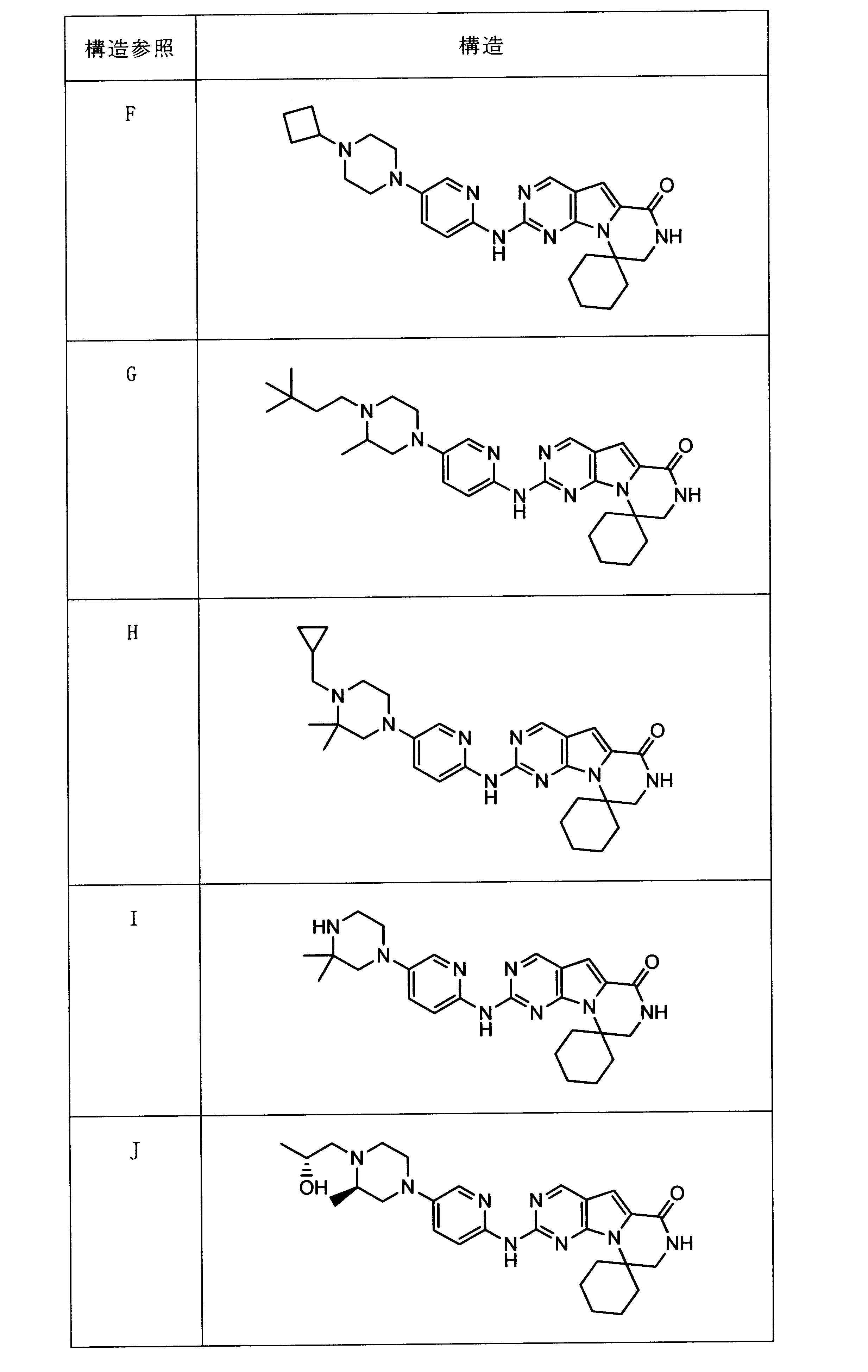
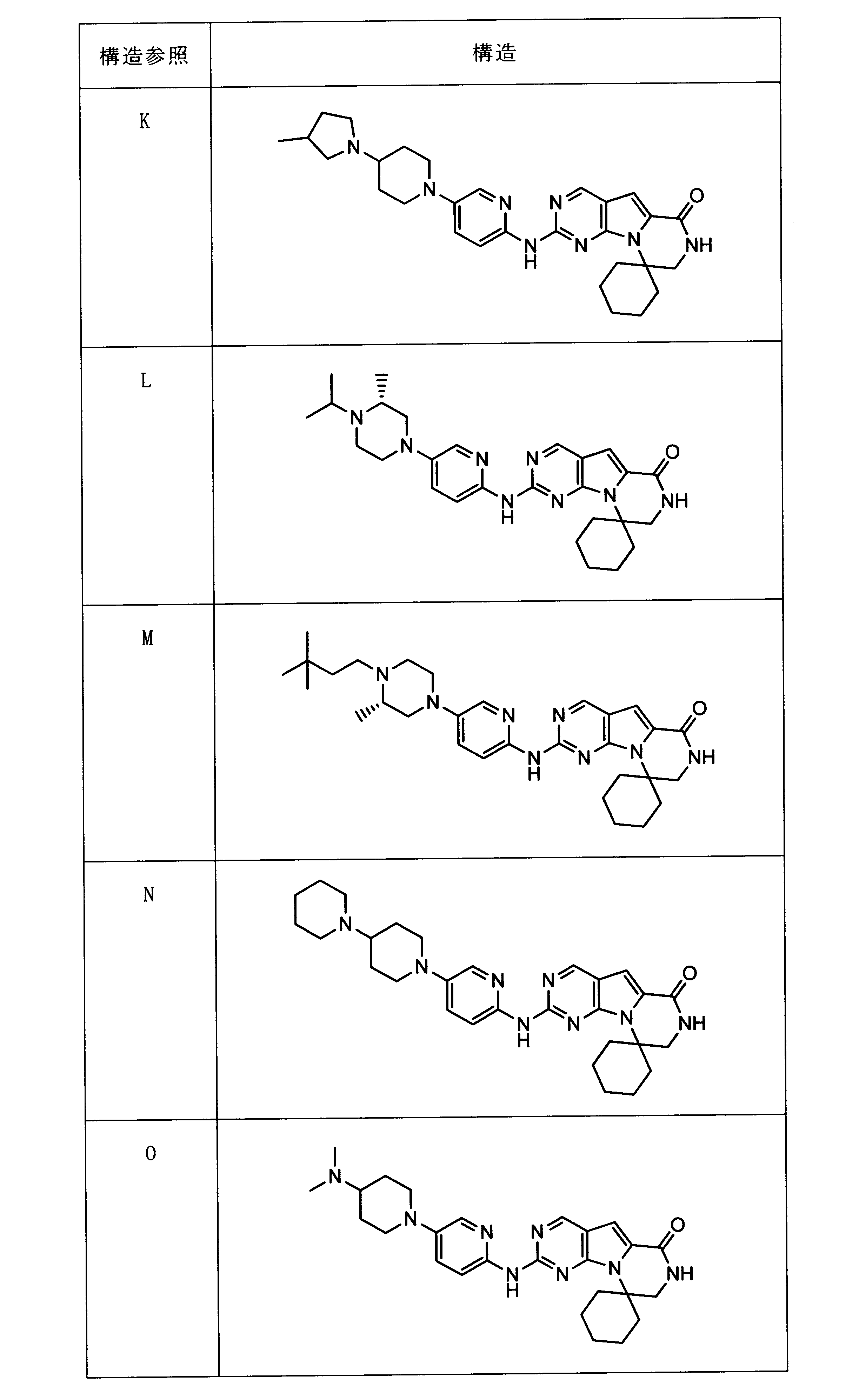
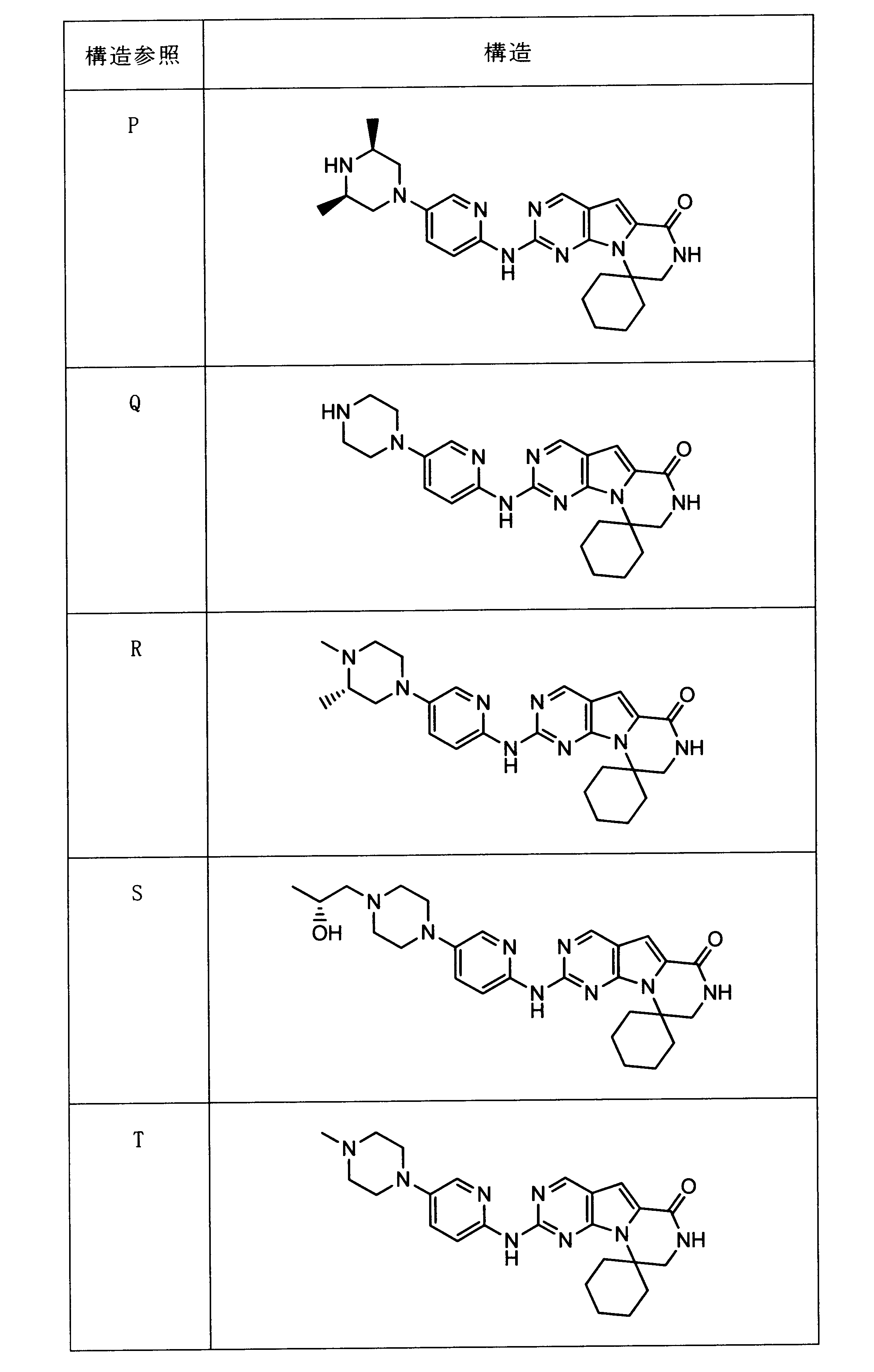
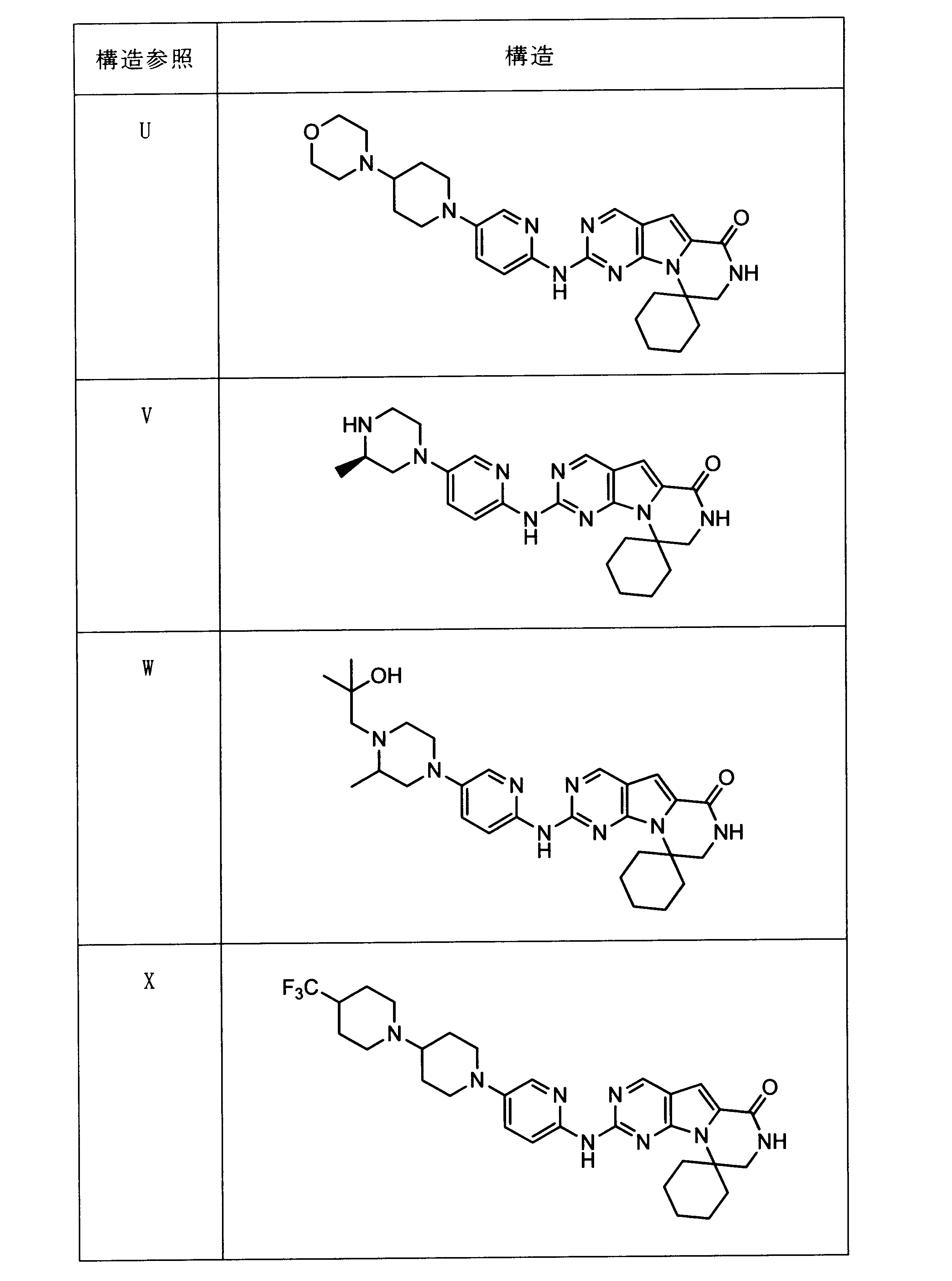
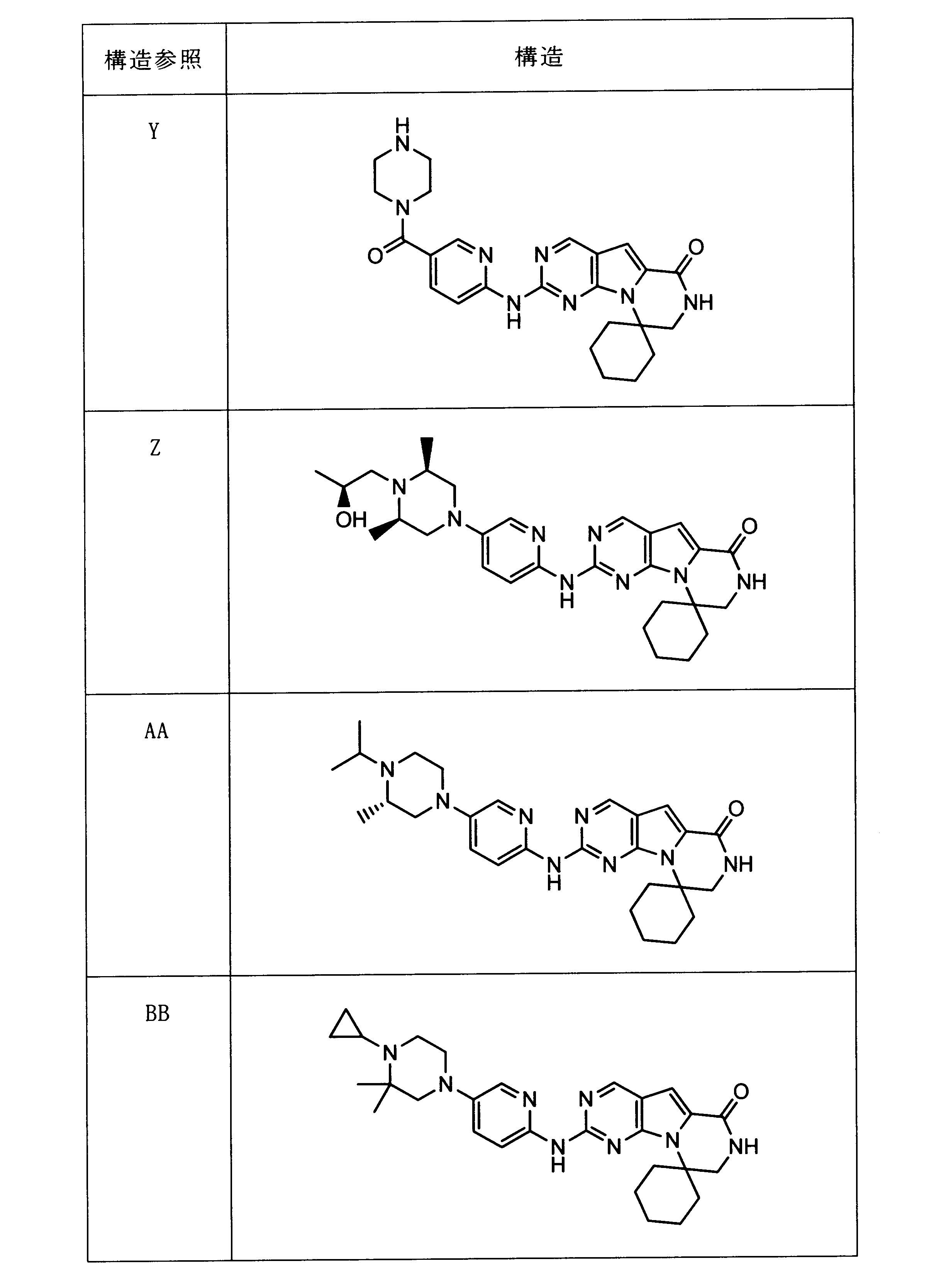
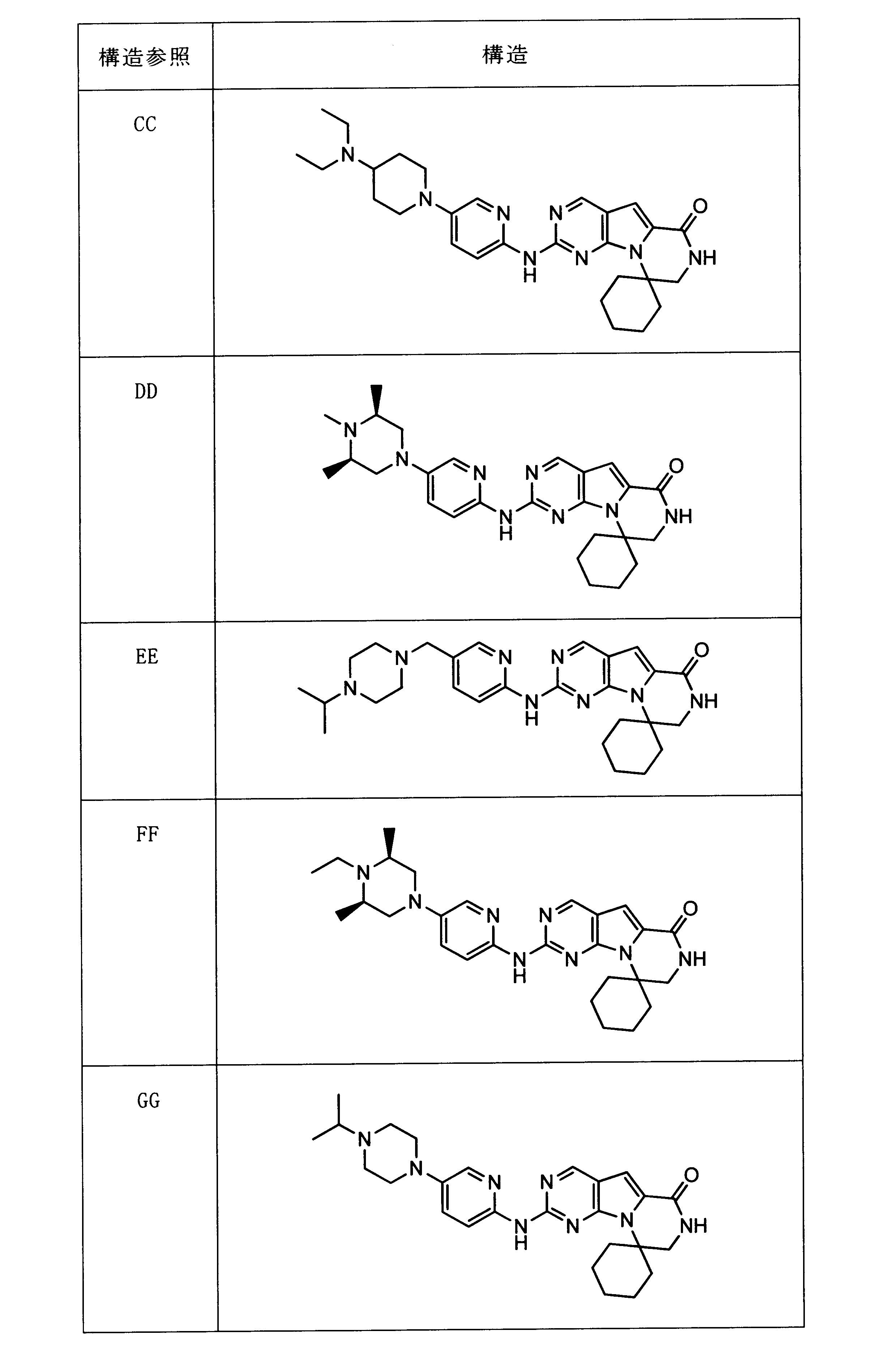
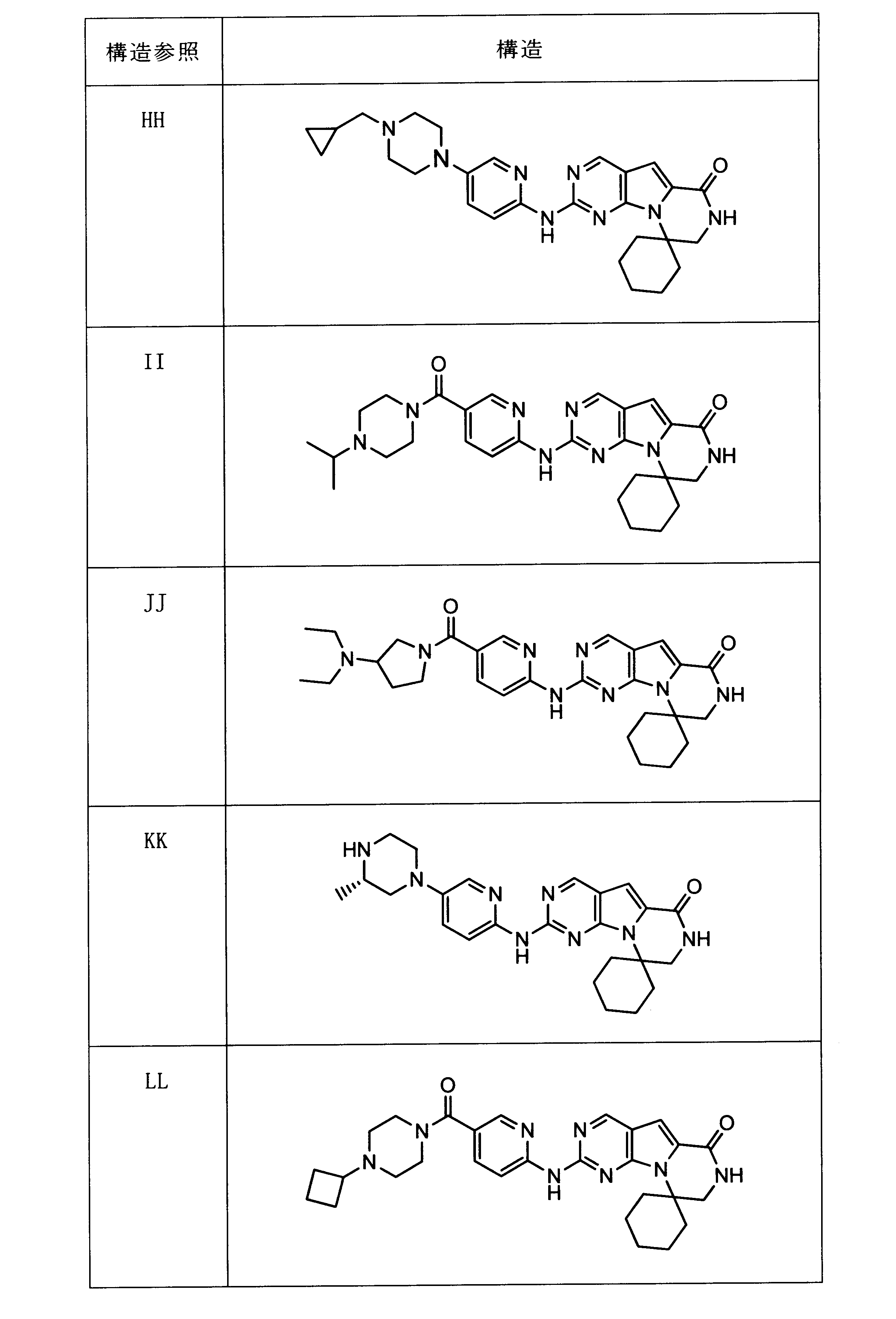
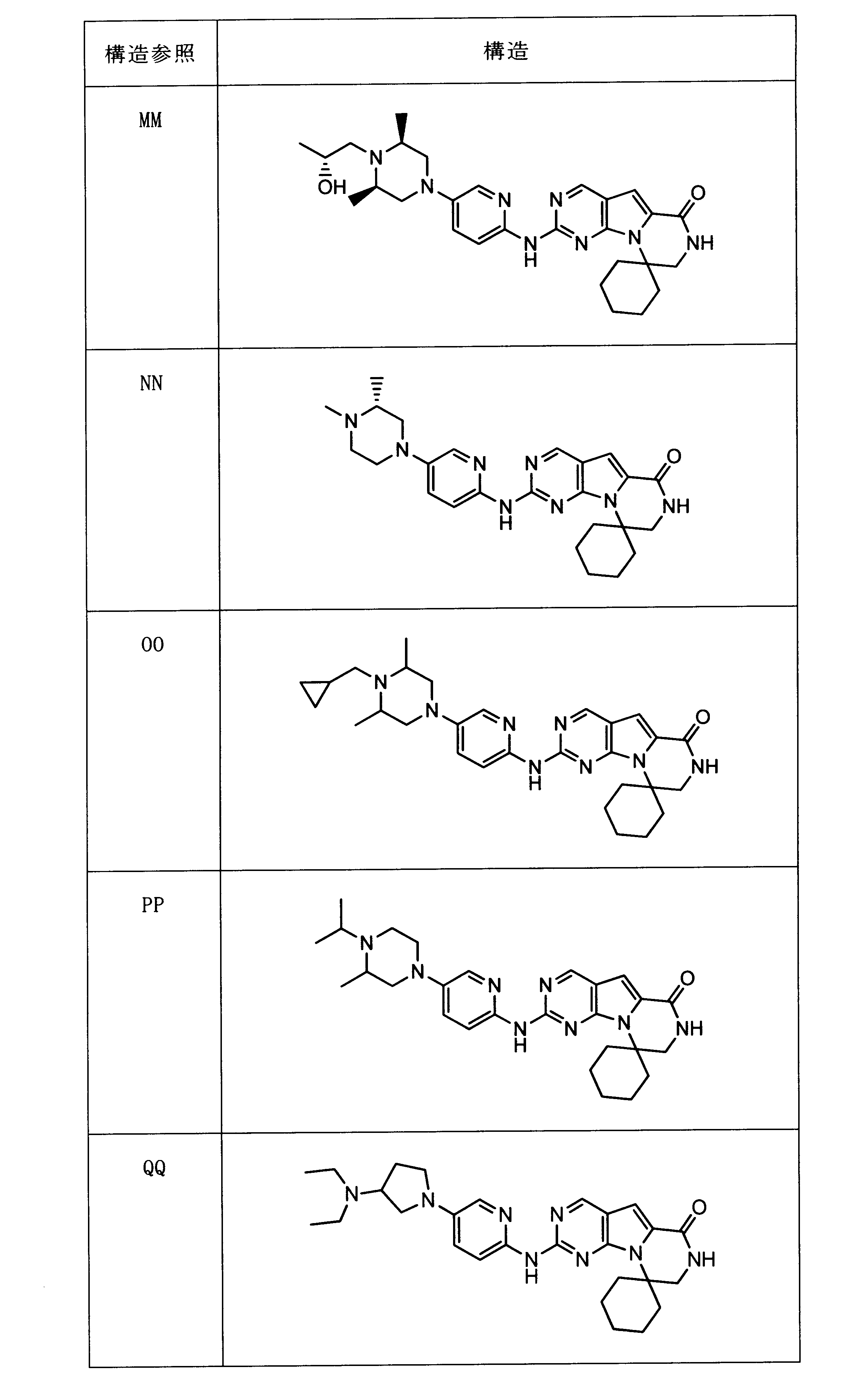
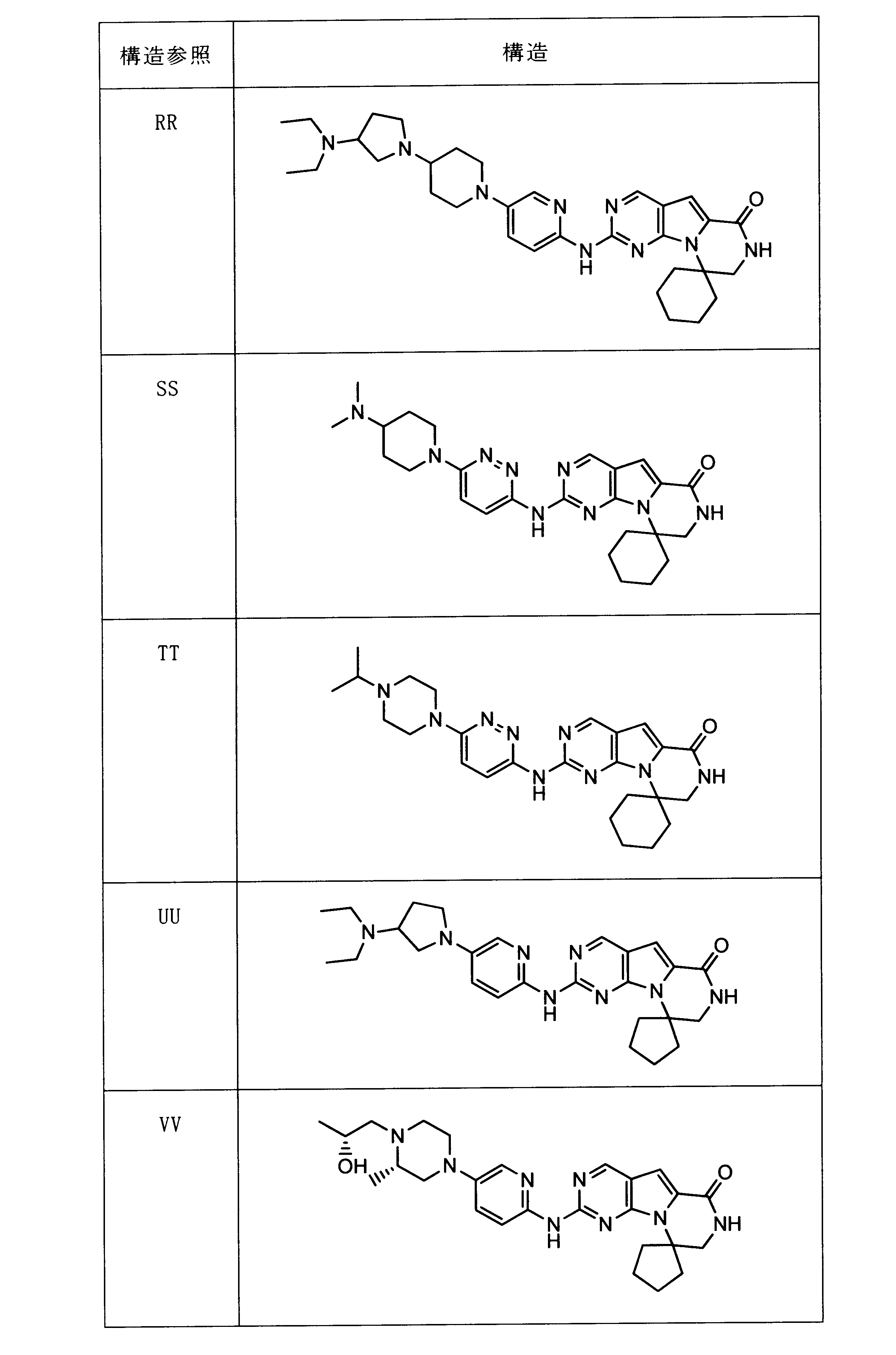
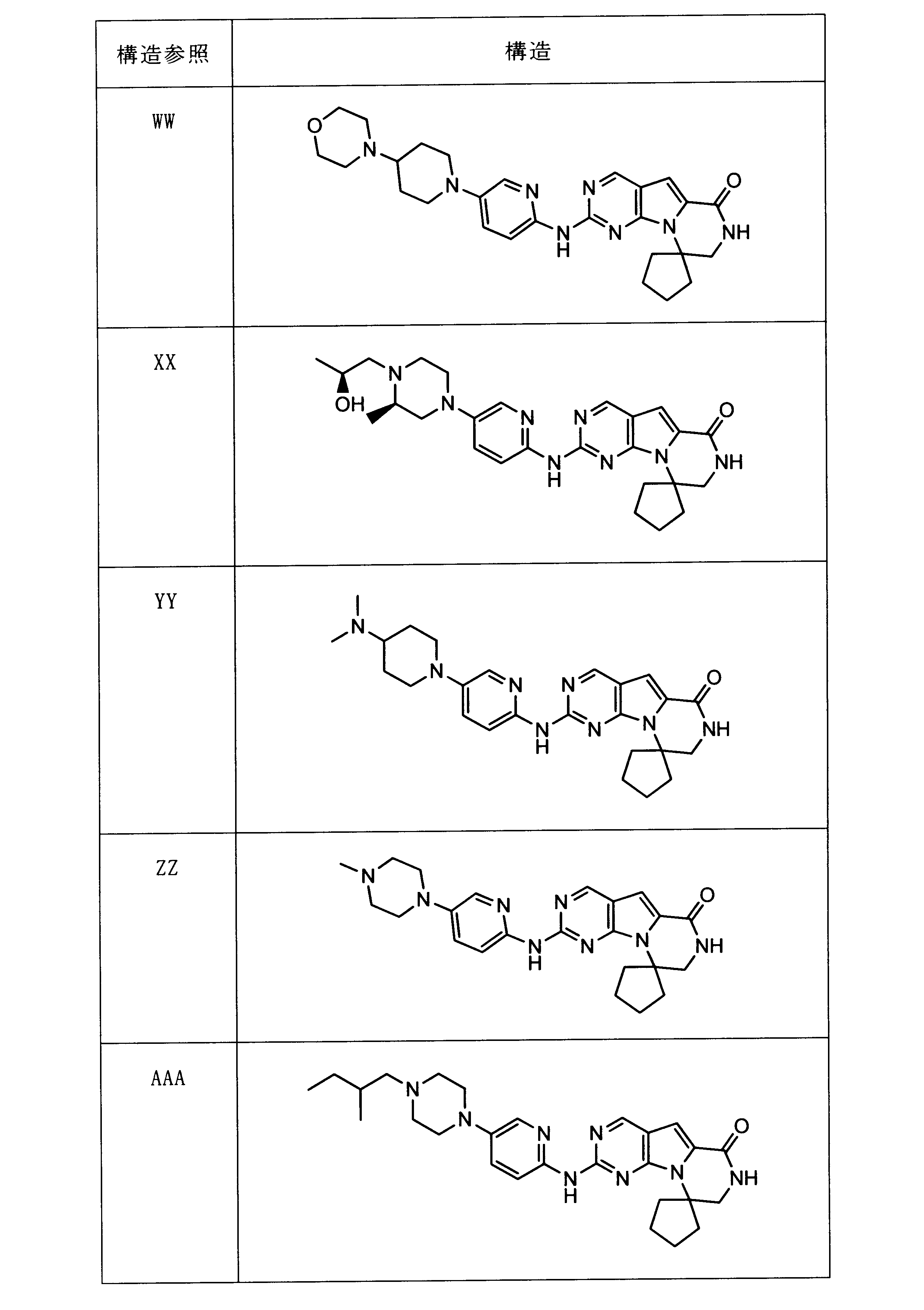
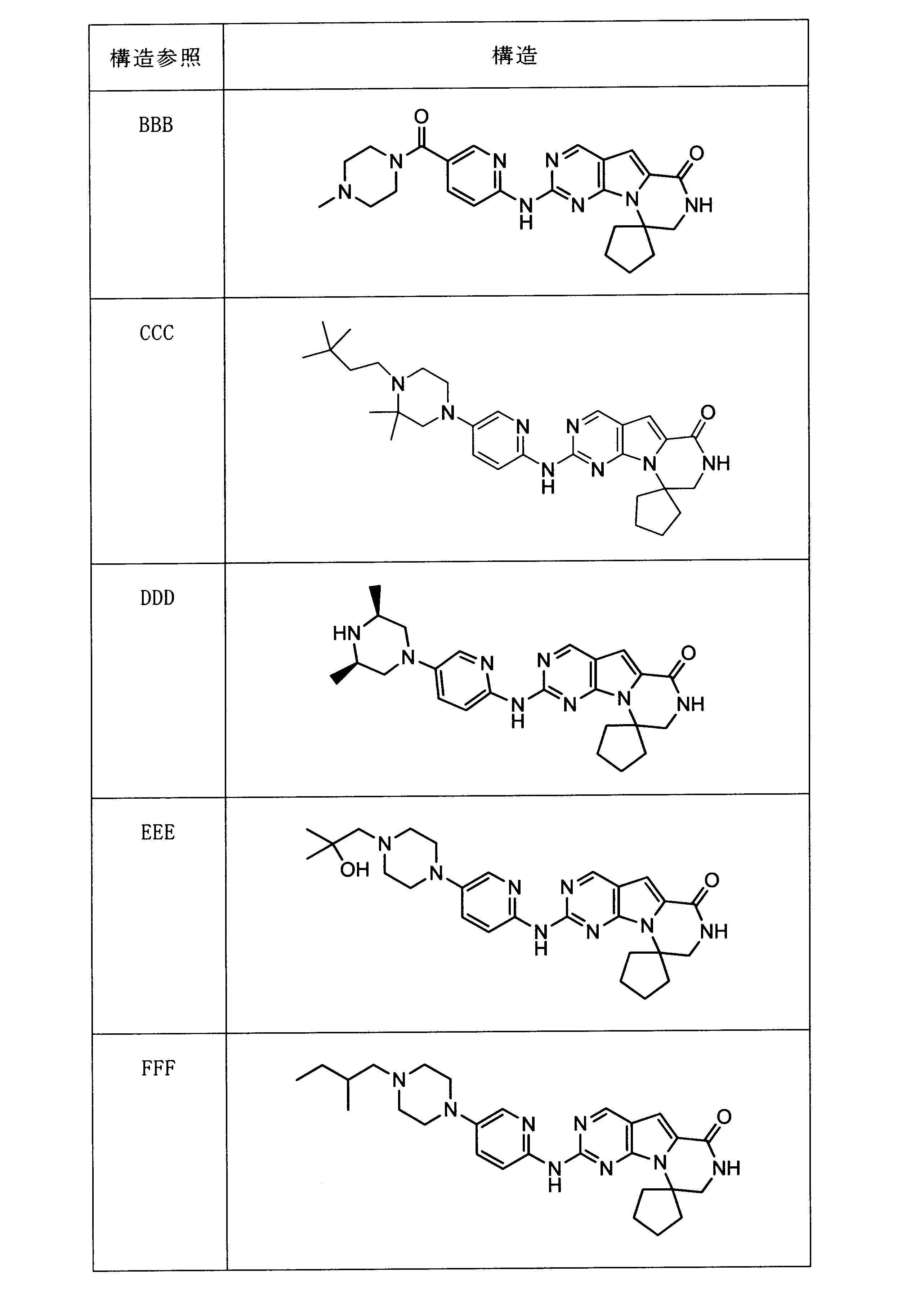
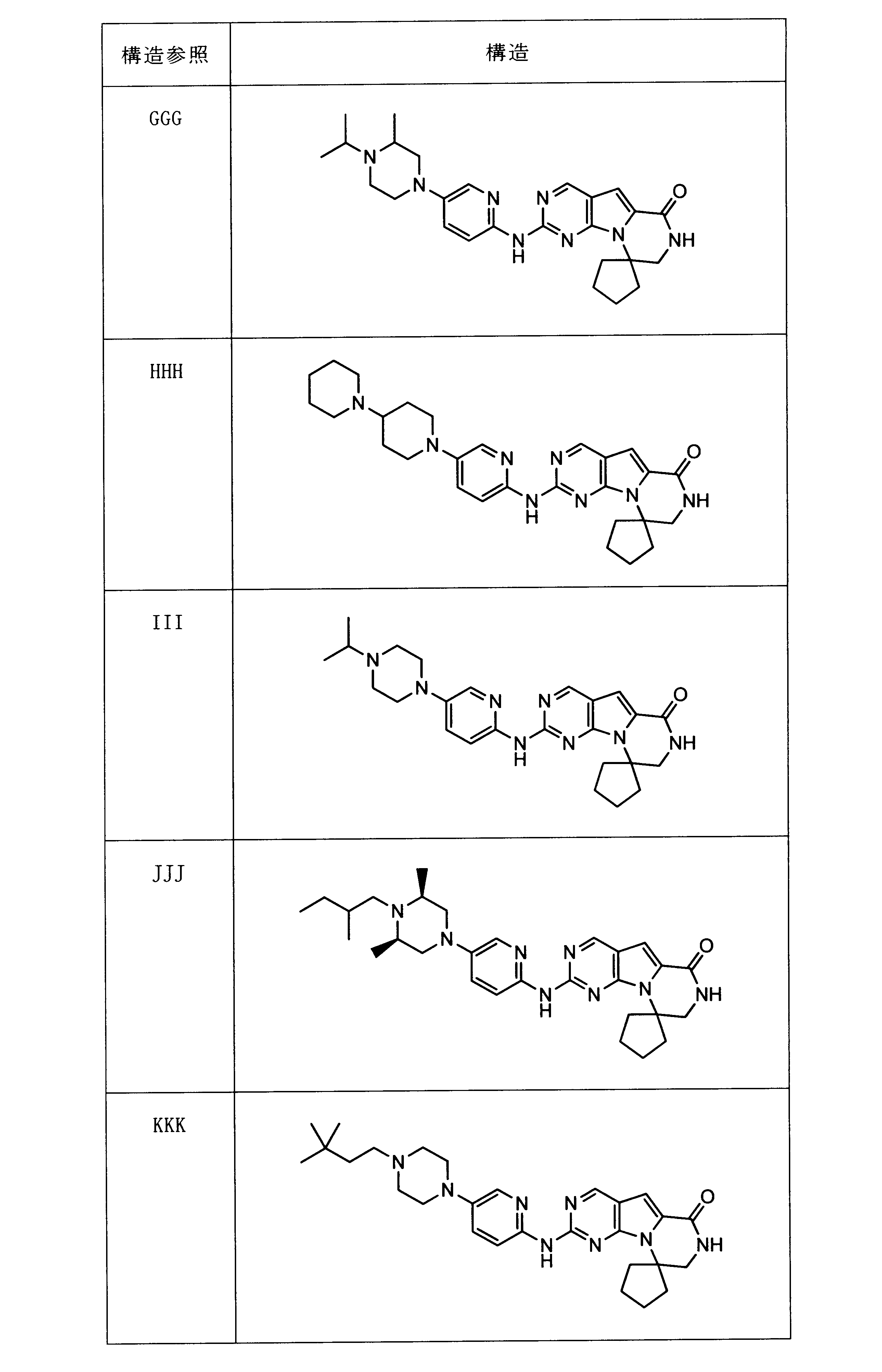
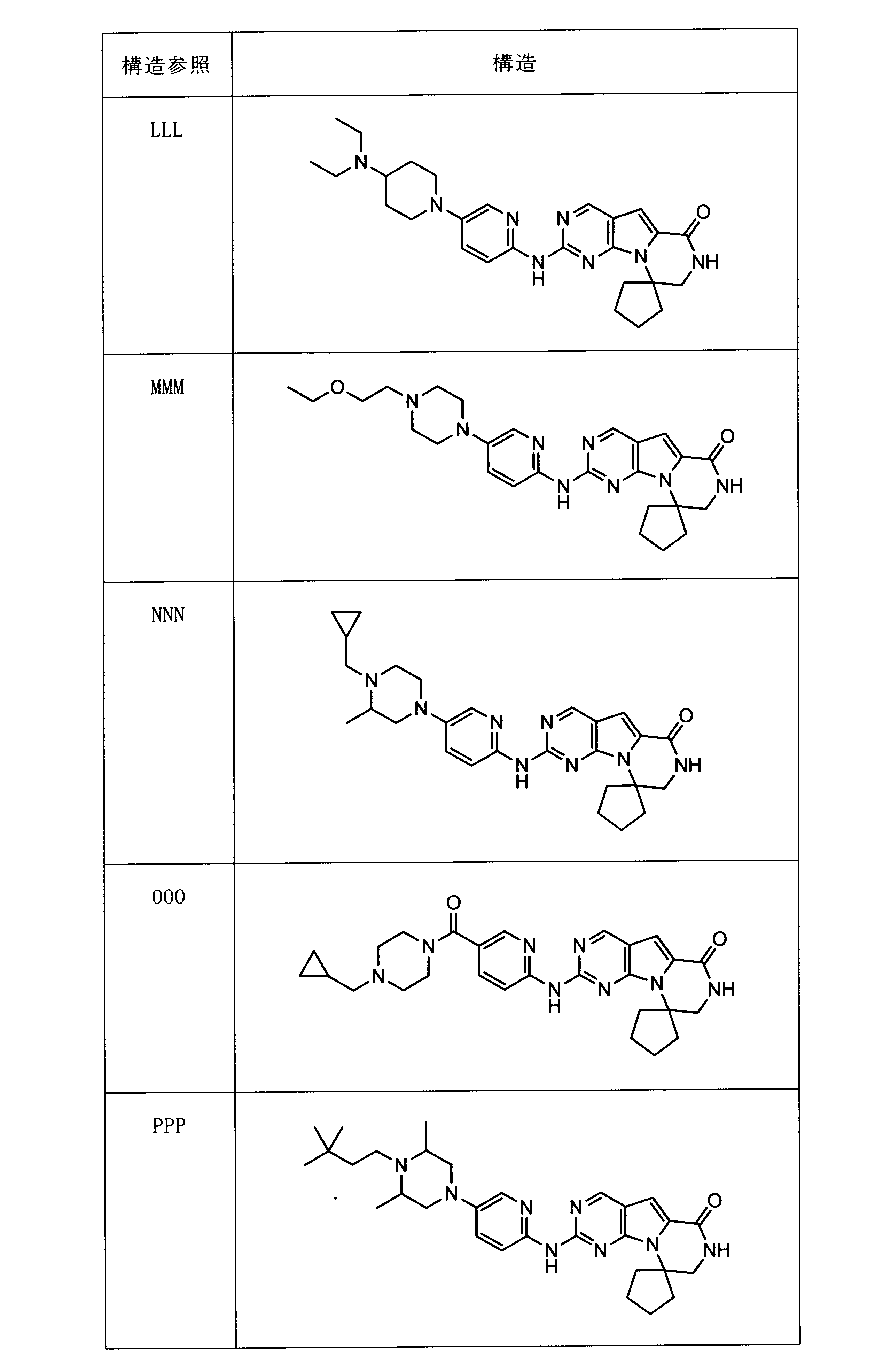
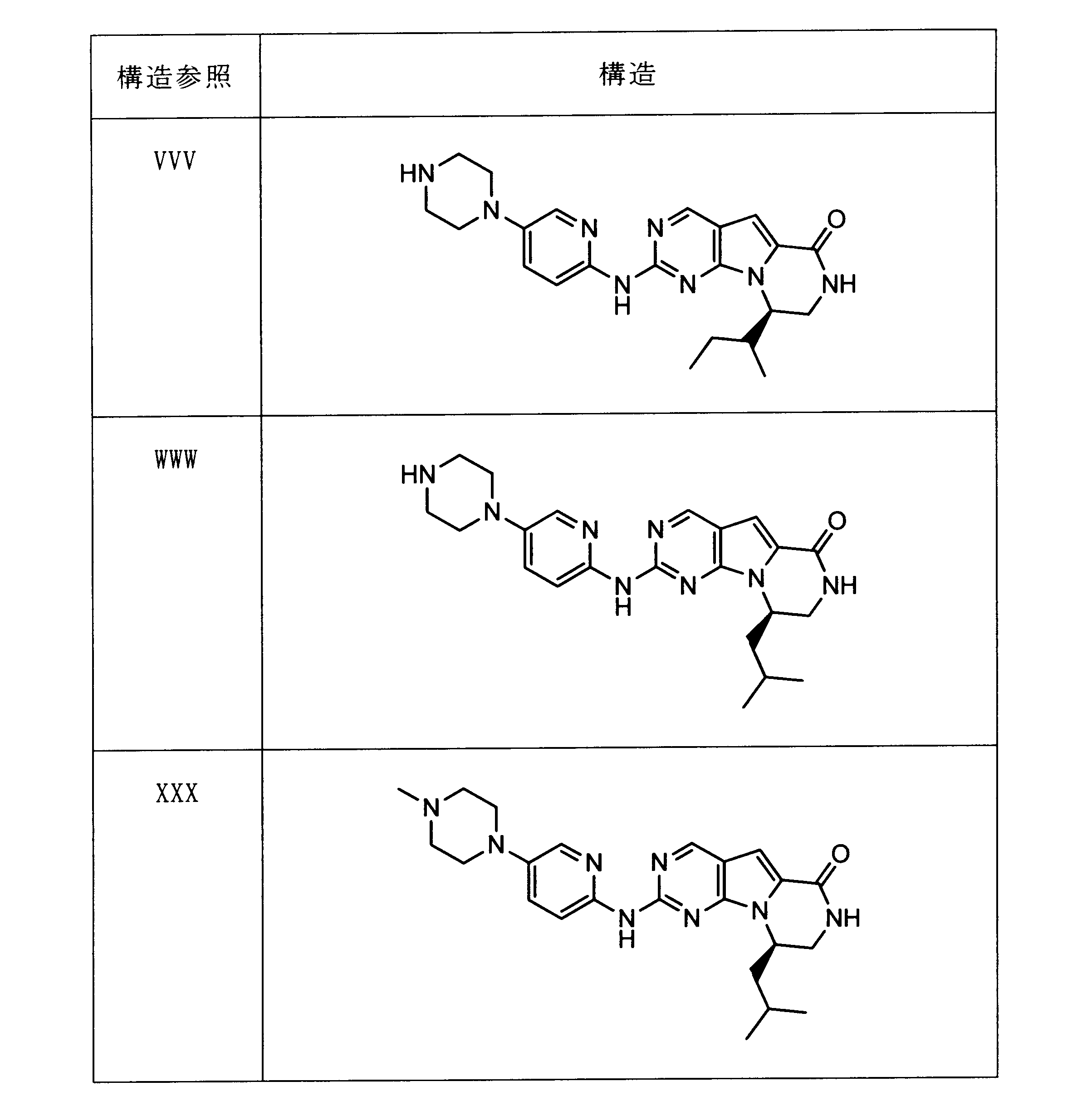
一つの非限定的な例において、化合物は、下記の表1の化合物、又はそれらの薬理学的に許容される組成物、塩、アイソトープ類似体又はプロドラッグから選択することができる。
Rbポジティブ癌は、コロンのRbポジティブ腺癌であってもよい。
Rbポジティブ癌は、直腸のRbポジティブ腺癌であってもよい。
代替的には、Rbポジティブ癌は、Rbポジティブ退形成星細胞腫であってもよい。
Rbポジティブ癌は、Rbポジティブ乳癌であってもよい。
代替的には、Rbポジティブ癌は、Rbポジティブエストロゲン受容体ネガティブ乳癌であってもよい。
Rbポジティブ癌は、Rbポジティブエストロゲン受容体に対して陽性の乳癌であってもよい。
Rbポジティブ癌は、Rbポジティブ遅発性系統転移性乳癌であってもよい。
Rbポジティブ癌は、Rbポジティブ管腔A乳癌であってもよい。
Rbポジティブ癌は、Rbポジティブ管腔B乳癌であってもよい。
Rbポジティブ癌は、RbポジティブHer2-ネガティブ乳癌又はRbポジティブHER2-ポジティブ乳癌であってもよい。
Rbポジティブ癌は、Rbポジティブ雄性乳癌である。
Rbポジティブ癌は、Rbポジティブプロゲステロンレセプター-ポジティブ乳癌であってもよい。
Rbポジティブ癌は、Rbポジティブ再発性乳癌であってもよい。
Rbポジティブ癌は、Rbポジティブ気管支癌であってもよい。
Rbポジティブ癌は、Rbポジティブ大腸癌であってもよい。
Rbポジティブ癌は、Rbポジティブ再発性大腸癌であってもよい。
Rbポジティブ癌は、RbポジティブステージIV大腸癌であってもよい。
Rbポジティブ癌は、Rbポジティブ性腺外精上皮腫であってもよい。
Rbポジティブ癌は、RbポジティブステージIII性腺外精上皮腫であってもよい。 Rbポジティブ癌は、RbポジティブステージIV性腺外精上皮腫であってもよい。
Rbポジティブ癌は、Rbポジティブ生殖細胞癌であってもよい。
Rbポジティブ癌は、Rbポジティブ中枢神経系胚細胞腫瘍であってもよい。
Rbポジティブ癌は、Rbポジティブ家族性精巣胚細胞腫瘍であってもよい。
Rbポジティブ癌は、Rbポジティブ再発性生殖腺の胚細胞腫瘍であってもよい。
Rbポジティブ癌は、Rbポジティブ再発性性腺外非精上皮腫性胚細胞腫瘍であってもよい。
Rbポジティブ癌は、Rbポジティブ性腺外精上皮腫性胚細胞腫瘍であってもよい。
Rbポジティブ癌は、Rbポジティブ再発性悪性精巣胚細胞腫瘍であってもよい。
Rbポジティブ癌は、Rbポジティブ再発性卵巣の胚細胞腫瘍であってもよい。
Rbポジティブ癌は、RbポジティブステージIII悪性精巣胚細胞腫瘍であってもよい。
Rbポジティブ癌は、RbポジティブステージIII卵巣の胚細胞腫瘍であってもよい。
Rbポジティブ癌は、RbポジティブステージIV卵巣の胚細胞腫瘍であってもよい。 Rbポジティブ癌は、RbポジティブステージIII性腺外非精上皮腫性胚細胞腫瘍であってもよい。
Rbポジティブ癌は、RbポジティブステージIV性腺外非精上皮腫性胚細胞腫瘍であってもよい。
Rbポジティブ癌は、Rbポジティブ肝細胞性癌であってもよい。
Rbポジティブ癌は、Rbポジティブ肺癌であってもよい。
Rbポジティブ癌は、Rbポジティブメラノーマであってもよい。
Rbポジティブ癌は、Rbポジティブ卵巣癌であってもよい。
Rbポジティブ癌は、Rbポジティブ膵癌であってもよい。
Rbポジティブ癌は、Rbポジティブ前立腺癌であってもよい。
Rbポジティブ癌は、Rbポジティブ再発性直腸癌であってもよい。
Rbポジティブ癌は、RbポジティブステージIV直腸癌であってもよい。
Rbポジティブ癌は、Rbポジティブ肉腫であってもよい。
Rbポジティブ癌は、Rbポジティブ神経膠肉腫であってもよい。
Rbポジティブ癌は、Rbポジティブ脂肪肉腫であってもよい。
Rbポジティブ癌は、Rbポジティブ繊維肉腫であってもよい。
Rbポジティブ癌は、Rbポジティブ粘液肉腫であってもよい。
Rbポジティブ癌は、Rbポジティブ骨肉腫であってもよい。
Rbポジティブ癌は、Rbポジティブ悪性線維性組織球腫であってもよい。
Rbポジティブ癌は、Rbポジティブ血管肉腫であってもよい。
Rbポジティブ癌は、Rbポジティブ血管肉腫であってもよい。
Rbポジティブ癌は、Rbポジティブリンパ管肉腫であってもよい。
Rbポジティブ癌は、Rbポジティブ中皮腫であってもよい。
Rbポジティブ癌は、Rbポジティブ平滑筋肉腫であってもよい。
Rbポジティブ癌は、Rbポジティブ横紋筋肉腫であってもよい。
Rbポジティブ癌は、Rbポジティブ髄膜腫であってもよい。
Rbポジティブ癌は、Rbポジティブシュワン細胞腫であってもよい。
Rbポジティブ癌は、Rbポジティブ島細胞癌であってもよい。
Rbポジティブ癌は、Rbポジティブカルチノイドであってもよい。
Rbポジティブ癌は、Rbポジティブパラガングリオーマであってもよい。
Rbポジティブ癌は、Rbポジティブ腺癌であってもよい。
Rbポジティブ癌は、Rbポジティブ肝細胞癌であってもよい。
Rbポジティブ癌は、Rbポジティブ腎細胞癌であってもよい。
Rbポジティブ癌は、Rbポジティブ胆管癌であってもよい。
Rbポジティブ癌は、Rbポジティブ難治性固形腫瘍であってもよい。
Rbポジティブ癌は、Rbポジティブ神経芽細胞腫であってもよい。
Rbポジティブ癌は、Rbポジティブ髄芽細胞腫であってもよい。
Rbポジティブ癌は、Rbポジティブ卵巣未熟奇形腫であってもよい。
Rbポジティブ癌は、Rbポジティブ卵巣成熟奇形腫であってもよい。
Rbポジティブ癌は、Rbポジティブ卵巣専門テラトーマであってもよい。
Rbポジティブ癌は、Rbポジティブ精巣未熟奇形腫であってもよい。
Rbポジティブ癌は、Rbポジティブ精巣成熟奇形腫であってもよい。
Rbポジティブ癌は、Rbポジティブテラトーマであってもよい。
Rbポジティブ癌は、Rbポジティブ卵巣単胚葉性テラトーマであってもよい。
Rbポジティブ癌は、Rbポジティブ精巣癌であってもよい。
特定の実施形態では、ここに記載される化合物は、選択されたRbポジティブ細胞増殖異常症(例えば癌)を治療するために用いられる場合に、正常細胞が常の細胞サイクルへ迅速にリエントリーすることを可能にし、かつ、血液細胞等の損害を受けた組織及び後代細胞の急速な再結合を可能にする。
この態様では、ここに記載される化合物を、Rbポジティブ癌を治療するために用いれば、CDK4/6阻害物質の現行の抗悪性腫瘍使用に関係したドラッグホリデイ及びドーズ遅れを、排除、低減、及び/又は最小にすることにより、前駆細胞及び親細胞の複製及び分化を通してダメージを受けた血球の迅速な回復が可能となる。
具体的に、本発明は、Rbポジティブ癌等の癌を有している患者に、ここに記載される化合物の有効量を投与することを含み、ここで、合物は、CDK4/6-複製依存的細胞の過渡的、可逆的Gl停止を提供する、薬物動態及び酵素の半減期を有する。
化合物は、本出願の中に記載されるもののいずれかであってもよい。
活性化合物の非限定的な例は、下記に提示される表1に記載されるもの、又は、それらの薬理学的に許容される組成物、塩、同位体的類似体、又はプロドラッグである。
この方法は、CDK4/6-複製依存的正常細胞に対する影響を低減又は最小にするものであり、何故なら、それらは、
(i)CDK4/6-複製依存的正常細胞の上への比較的短期的、過渡的かつ可逆的なGl停止効果を提供する薬物動態及び酵素の半減期を示す化合物を利用するからであり、また、
(ii)投与の中断の後、又は被験体中の治療的に有効なレベルの消失の後、正常細胞に対して迅速な細胞サイクルへのリエントリーを可能にするからである。
特定の実施形態では、ここに記載される化合物の使用は、オフサイクル又はドラッグホリデイの必要なしに、より長い期間、被験体の持続性治療を可能にする。
CDK4/6-複製依存的正常細胞増殖のタイムリーな再開は、組織修復のために必要であり、そして、正常細胞細胞サイクル進行、たとえばHSPC細胞サイクル進行の停止があまりに長い期間停止することは、好ましくない。
選択的CDK4/6阻害物質PD0332991がRbポジティブ乳癌の有効な阻害剤であることを示している報告があるにもかかわらず、この化合物は過剰な骨髄抑制作用があるため、この阻害剤は、化学療法剤として最も理想的な化合物ではないということが見出されている。
たとえば、そのような長時間作用型効果は、CDK4/6活性を抑制し得る治療のために、成長抑制された血液細胞系統を再構成するのに必要なHSPC細胞系統の増殖を遅らせ、すなわち、すなわちRb受容細胞内のRbリン酸化を遅れさせる。
選択されたRbポジティブ癌の治療のための抗腫瘍薬としてここに記載される化合物の使用は、必要なオフサイクル期間又はドラッグホリデイの長さを排除、低減又は、最小にすることができるので、抗悪性腫瘍計画において、より長く有効な癌に対するCDK4/6抑制性期間を与えることができる。
i)たとえばHSPC等のCDK4/6-複製依存的正常細胞(例えば少なくとも80%以上)の相当な部分が、ここに記載される化合物のヒトへの最後の投与から24時間未満、30時間未満又は36時間未満で、プレ治療ベースライン細胞サイクル活性に戻る又は接近する(すなわち、細胞サイクルにリエントリーする)。
ii)たとえばHSPC等のCDK4/6-複製依存的正常細胞の相当な部分が、ここに記載される化合物のヒトへの最後の投与から24時間未満、30時間未満又は36時間未満で、同時に細胞サイクルにリエントリーする。
(iii)たとえばHSPC等のCDK4/6-複製依存的正常細胞上の化合物の抑制性効果の消失が、化合物の投与から24時間未満、30時間未満又は36時間未満で発生する。
(iv)化合物のCDK4/6禁止効果の消失から24時間未満、30時間未満又は36時間未満で、たとえばHSPC等のCDK4/6-複製依存的正常細胞の相当な部分が、プレ治療ベースライン細胞サイクル活性に戻る又は接近する(すなわち、細胞サイクルにリエントリーする)。
又は(vi)たとえばHSPC等のCDK4/6-複製依存的正常細胞の相当な部分が、投与された化合物の被験体の血液中の濃度レベルが治療有効濃度を下回った時点から約24時間未満、約30時間未満又は約36時間未満以内で、プレ治療ベースライン細胞サイクル活性に戻る又は接近する(すなわち、細胞サイクルにリエントリーする)。
別の実施形態では、ここに記載される化合物は、局所、経皮又は他の所望された投与方法を介して投与される。
本発明に有用な化合物は、CDK4やCDK6の阻害に対して、他のCDK、たとえばCDK2と比べて、際立った選択性を示す。
たとえば、本発明に有用な化合物は、被験体のRbポジティブ癌細胞へのドーズ依存性Gl-停止効果を提供し、そして、ここに提供される方法は、CDK4/6-複製非依存的細胞に影響を及ぼさずに、Rbポジティブ癌細胞の化学療法剤治療及び成長阻害を提供するのに十分である。
被検者の血液の中にCDK4/6阻害物質の濃度レベルが治療有効濃度未満に下がった時点から、約24時間、30時間、36時間、40時間未満以内に、又は、約48時間以内に、G1期及びS期の通常細胞の一部が、迅速かつ効率的に復旧されるように、細胞-サイクルにリエントリーしたCDK4/6-複製依存的正常細胞は振る舞う。
化合物の迅速のオフ効果に関連した迅速の細胞サイクルリエントリーにより、有利なことに、PD0332991等の他のCDK4/6阻害物質と比較して、さらに多くのCDK4/6-複製依存的正常細胞がGl停止の消失に際して複製を開始することが可能となる。
従って、オフサイクル期間の間又は投与期間の間に、HSPC等のCDK4/6-複製依存的正常細胞が、複製を迅速に開始することが可能となる。
CDK4/6-複製依存的癌をターゲットにする治療計画における、ここに記載される化合物の使用により、現在利用できる抗悪性腫瘍化学療法剤による治療後、治療の通常後、又はこの治療と関連して、典型的に予想される状態と比較して、貧血の軽減、リンパ球減少の軽減、血小板減少の軽減、又は好中球減少症の軽減をもたらす。
ここに記載されるCDK4/6阻害物質の使用により、CDK4/6阻害物質の長期の使用に関係した骨髄抑制、例えばCDK4/6阻害物質の使用の中断に続いた骨髄抑制、貧血、リンパ球減少、血小板減少、又は好中球減少症等、からのより急速な回復がもたらされ得る。
ある実施形態では、被検者又はホストは、ヒトを含む哺乳類である。
化合物は、静脈、舌下、頬側、経口、門脈内、局所、鼻腔内、経静脈、経皮、浸透性、筋肉内、又は吸入を含むあらゆる所望されたルートで、被験体に投与することが可能である。
A)ここに記載される表1から選択される化合物を含む、式I、II、III、IV又はVの化合物の有効量を投与することを含むことを有する、選択されたRbポジティブ癌の治療を受けている被験体において、造血幹細胞及び前駆細胞(HSPC)等CDK4/6複製依存的正常細胞への有害作用を最小にする、化学療法剤として最適な化合物、方法及び組成物;
B)ここに記載される化合物を効果的量で投与することを含む、Rbポジティブ癌の治療を受けている被験体において、造血幹細胞及び前駆細胞(HSPC)等CDK4/6複製依存的正常細胞への有害作用を最小にする、化学療法剤として最適な化合物、方法及び組成物であって、
CDK4/6阻害物質の最後の投与から約24時間未満、30時間、36時間又は約40時間以内に、正常細胞の相当な部分は、プレ治療ベースライン細胞サイクル活性に戻る又は接近(すなわち、細胞サイクルにリエントリー)し、また、CDK4/6阻害物質は、CDK2阻害に対するIC50濃度の約1500倍以上小さな、CDK4阻害に対するIC50濃度を有している。
C)ここに記載される化合物を効果的量で投与することを含む、Rbポジティブ癌の治療を受けている被験体における、CDK4/6複製依存的正常細胞への有害作用を最小にする、化学療法剤として最適な化合物、方法及び組成物であって、
化合物のCDK4/6禁止効果の消失後、約24時間未満、30時間、36時間又は約40時間以内に、CDK-複製依存的正常細胞の相当な部分が、細胞サイクルに同時にリエントリーし、また、化合物は、CDK2阻害に対するIC50濃度の約1500倍以上小さな、CDK4阻害に対するIC50濃度を有している。
D)ここに記載される化合物から成る群より選択される選択的CDK4/6阻害物質の有効量を、Rbポジティブ異常細胞増殖異常症を有する被験体に投与することを含む、被験体におけるCDK4/6複製依存的正常細胞への有害作用を最小にする化学療法剤として最適な化合物、方法及び組成物。
E)Rbポジティブ癌を含むRbポジティブ異常細胞増殖異常症の治療における化学療法剤としての、ここに記載される化合物、又はそれらの薬理学的に許容される組成物、塩、アイソトープ類似体又はプロドラッグ。
F)たとえばHSPC又は腎細胞等のCDK4/6-複製依存的正常細胞への有害作用を最小にする、Rbポジティブ癌を含むRbポジティブ異常細胞増殖異常症の治療のための化学療法剤療法としての、ここに記載される化合物、又はそれらの薬理学的に許容される組成物、塩、アイソトープ類似体又はプロドラッグ。
G)Rbポジティブ癌を含むRbポジティブ異常細胞増殖異常症を治療するための治療計画を受けている被験体に造血成長因子と組み合わせて使用するための、ここに記載される化合物、又はそれらの薬理学的に許容される組成物、塩、アイソトープ類似体又はプロドラッグ。
H)Rbポジティブ癌を含むRbポジティブ異常細胞増殖異常症を治療するための治療計画を受けている被験体に対して、第2の化学療法剤と組み合わせて使用するための、ここに記載される化合物、又はそれらの薬理学的に許容される組成物、塩、アイソトープ類似体又はプロドラッグ。
I)Rbポジティブ癌を含Rbポジティブ異常細胞増殖異常症の被験体を治療する化学療法剤としての薬剤の生産における、ここに記載される化合物又は薬理学的に許容される組成物、塩、同位体的類似体又はプロドラッグの使用。
J)CDK4/6阻害物質に曝露されるときに成長停止又は成長抑制されるRbポジティブ癌を含むRbポジティブ細胞増殖異常症を有する被験体を治療する化学療法剤として薬剤の製造における、ここに記載される化合物又は薬理学的に許容される組成物、塩、同位体的類似体又はプロドラッグの使用。
K)癌等のRbポジティブ異常細胞増殖異常症を有する被験体の治療に使用するための、ここに記載される化合物の有効量を含む治療製品の製造のための方法。及び
L)CDK4/6阻害物質に感応する癌等のRbポジティブ異常細胞増殖異常症の治療における、化学療法剤としての治療使用が意図される、ここに記載される化合物から選択される薬剤を製造する方法。
特に明記しない限り、明細書及び特許請求の範囲を含む本出願に用いられる以下の用語は、以下の定義を有する。
明細書及び添付の特許請求の範囲にて用いられているように、文脈が明らかに指図しない限り、単数形「an」及び「the」は複数指示物を含む。
「低級アルキル」ラジカルは、1~約6個の炭素原子を有する。
このラジカルの例としては、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、イソブチル、第二級ブチル、第三級ブチル、ペンチル、イソアミル、ヘキシル等が挙げられる。
その例としては、プロピレンで、メチレン、エチレン、プロピレン、イソプロピレン等を含む。
「低級アルケニル」ラジカルは、2~約6個の炭素原子を有している。
アルケニルラジカルの例としては、エテニル、プロペニル、アリル、プロペニル、ブテニル及び4-メチルブテニルが挙げられる。
このラジカルの例としては、プロパルギル、ブチニル等が挙げられる。
アルキル、アルケニル及びアルキニルラジカルは、ハロ、ヒドロキシ、ニトロ、アミノ、シアノ、ハロアルキル、アリール、ヘテロアリール、ヘテロシクロ等。の1つ以上の官能基で任意に置換されてもよい。
適切なアルキルアミノラジカルとしては、N-メチルアミノ等のモノ又はジアルキルアミノ、N-エチルアミノ、N.N-ジメチルアミノ、N,N-ジエチルアミノ等が挙げられる。
「ハロ」という用語は、フッ素、塩素、臭素またはヨウ素原子のようなハロゲンを意味する。
その例としては、モノハロアルキル、ジハロアルキル)及びパーハロアルキルを含むポリハロアルキルラジカルを含む。
ジハロ及びポリハロアルキルラジカルは、同じハロ原子の二つ以上又は異なるハロラジカルの組合せを有していてもよい。
その例としては、トリフロロメチル及びペンタフルオロエチルが挙げられる。
さらなる好ましいアリールは、フェニルである。
アリール基は、ハロ、ヒドロキシ、ニトロ、アミノ、シアノ、ハロアルキル、アリール、ヘテロアリール、ヘテロシクロ等の官能基1つ以上で任意に置換されてもよい。
ヘテロシクロ環は、単環6~8員環ならびに5~16員二環式環構造を含む(架橋融合及びスピロ融合二環式環構造を含んでもよい)。
それは、-O-O-、-O-S-又は-S-S-部分を有する環を含まない。
部分的に飽和及び飽和ヘテロシクロ基の特定の例としては、ピロリジニル、イミダゾリジニル、ピペリジニル、ピロリニル、ピラゾリジニル、ピペラジニル、モルホリニル、テトラヒドロピラニル、チアゾリジニル、ジヒドロチエニル、2、3-ジヒドロベンゾ[1、4]ジオキサニル、インドリニル、イソインドリニル、ジヒドロベンゾチエニル、ジヒドロベンゾフリル、イソクロマニル、クロマニル、1、2-ジヒドロキノリル、1,2,3,4-テトラヒドロ-イソキノリル、1,2,3,4-テトラヒドロ-キノリル、2,3,4,4a,9,9a-ヘキサヒドロ-1H-3-アザ-フルオレニル、5,6,7-トリヒドロ-、2、4--トリアゾロ[3,4-a]イソキノリル、3,4-ジヒドロ-2H-ベンゾ[1、4]オキサゾリル、ベンゾ[1,4]ジオキサニル、2,3-ジヒドロ-1H-1λ-ベンゾ[d]イソチアゾール-6-イル、ジヒドロピラニル、ジヒドロフリル及びジヒドロチアゾリル等が挙げられる。
用語「ヘテロアリールアルキル」は、ヘテロアリール基で置換されるアルキルラジカルのことを意味する。その例としては、ピリジルメチル及びチエニルエチルが挙げられる。
「カルボニル」という用語は、単独で用いられるか、又は、たとえば「アミノカルボニル」など、他の用語に関連づけられるかどうかにかかわらず、-C(O)-を意味する。
用語「アミノカルボニル」は、式-C(O)-NH2のアミド基を意味する。
その例としては、ピペリジルメチル及びモルホリニルエチルが挙げられる。
その例としては、ベンジル、ジフェニルメチル及びフェニルエチルが挙げられる。
前記アラルキルにおけるアリールは、ハロ、アルキル、アルコキシ、ハロアルキル及びハロアルコキシでさらに置換されてもよい。
低級シクロアルキル基は、C3-C6環を含む。
その例としては、シクロペンチル、シクロプロピル及びシクロヘキシルが挙げられる。 シクロアルキル基は、たとえばハロ、ヒドロキシ、ニトロ、アミノ、シアノ、ハロアルキル、アリール、ヘテロアリール、ヘテロシクロ等の1つ以上の官能基で任意に置換されてもよい。
「低級シクロアルキルアルキル」ラジカルは、1~6つの炭素原子を有するアルキルラジカルに結合されるシクロアルキルラジカルである。
その例としては、シクロヘキシルメチルが挙げられる。
前記ラジカルにおけるシクロアルキルは、ハロ、アルキル、アルコキシ及びヒドロキシでさらに置換されてもよい。
その例としては、シクロペンテニル、シクロペンタジエニル、シクロヘキセニル及びシクロヘプタジエニルが挙げられる。
ここで用いられる用語「オキソ」は、二重結合で結合される酸素原子を想定している。 ここで用いられる用語「ニトロ」は、-NO2を想定する。
ここで用いられる用語「シアノ」は、-CNを想定する。
ここで用いられる例では、「プロドラッグ」という用語は、生体内でホストに投与された際に、ペアレントドラッグに変換される化合物を意味する。
全てここに含まれるとみなされるペアレントドラッグの、インヴィヴォ生成の条件を調節することに関しての選択を提供するような、プロドラッグストラテジーが存在する。
プロドラッグストラテジーの非限定的な例は、除去可能な基又は基の除去可能な部分の共有結合性結合を含み、たとえば、アシル化、リン酸化、ホスホニル化、アミド亜リン酸エステル誘導体、アミド化、低下、酸化処理、エステル化、アルキル化、他のカルボキシ誘導体、スルホキシ又はスルホン誘導体、カルボニル化又は無水物等である。
特に明記しない限り、明細書及び特許請求の範囲を通して、所与の化学式又は名前は、全ての光学異性体及び立体異性体、ならびにラセミ混合体を、これらの異性体及び混合物が存在する場合に、包摂するものとする。
従って、ここで用いられる例では、用語「HSPC」は、健康な造血幹細胞及び/又は造血前駆細胞を記載することを意味し、これは、関連した血液由来の病気に罹患したHSPC又は細胞と対照的である。
HSPCは、たとえば長期造血幹細胞(LT-HSC)及び短期造血幹細胞(ST-HSC)等の造血幹細胞と、多能性前駆細胞(MPP)一般の骨髄性前駆細胞(CMP)、一般のリンパ様前駆細胞(CLP)、顆粒球-単核細胞前駆細胞(GMP)及び巨核球赤血球前駆細胞(MEP)等を含む造血前駆細胞と、を含んでいる。
ある実施形態では、CDK4/6-複製依存的正常細胞は、非造血組織、例えば非限定的な例としては、肝臓、腎臓、すい臓、脳、肺、副腎、腸、腸、胃、皮膚、聴覚系、骨、膀胱、卵巣、子宮、睾丸、胆嚢、甲状腺、心臓、膵島、血液血管、等の中の細胞であってもよい。
ここで用いられる例では、用語「化学療法剤」又は「化学療法用剤」とは、細胞増殖抑制性又は細胞毒性剤(すなわち化合物)により処理をして、好ましくない細胞、たとえば癌細胞等の成長又は増殖を低減する又は排除することをいう。
したがって、ここで用いられる例では、「化学療法剤」又は「化学療法用剤」は、増殖異常症、たとえば癌を治療するために用いられる細胞毒又は細胞増殖抑制性剤のことをいう。
「血液欠乏」とは、血液細胞系統計数の低下、又は、血球(すなわち、脊髄形成異常)及び/又はリンパ球(すなわち、リンパ球減少、循環リンパ球数、たとえばB細胞及びT細胞、の減少)の不十分な生成のことを意味する。
たとえば、貧血の形態での骨髄抑制、血小板数の低下(すなわち、血小板減少)、白血球数の低下(すなわち、白血球減少症)、又は顆粒球の低下(例えば、好中球減少症)として、血液欠乏が観察され得る。
これに対して、「細胞サイクルへの非同時性のリエントリー」とは、CDK4/6阻害物質化合物の影響のためにG1期停止にある正常細胞、たとえばHSPCが、PD0332991等の化合物の影響の消失に対して、比較的異なる総体的な時間枠内に又は比較的異なる速度でリエントリーすることを意味する。
たとえば、被検者が21日間連続で化学療法剤を投与されそして7日間化学療法剤を投与されないという治療計画において、この計画が何度も繰り返される場合、非投与の期間である7日は、「オフサイクル」又は「ドラッグホリデイ」と考えられる。
オフターゲット及びドラッグホリデイとは、たとえば骨髄抑制等の有害な副作用のために、被検者にしばらく化学療法剤を投与しない治療計画中の中断のことを指してもよい。 治療される被検者は、典型的にはヒト被検者であるが、ここに記載される方法は、他の動物、例えば哺乳類及び脊椎動物種、に関して有効であることが理解されよう。
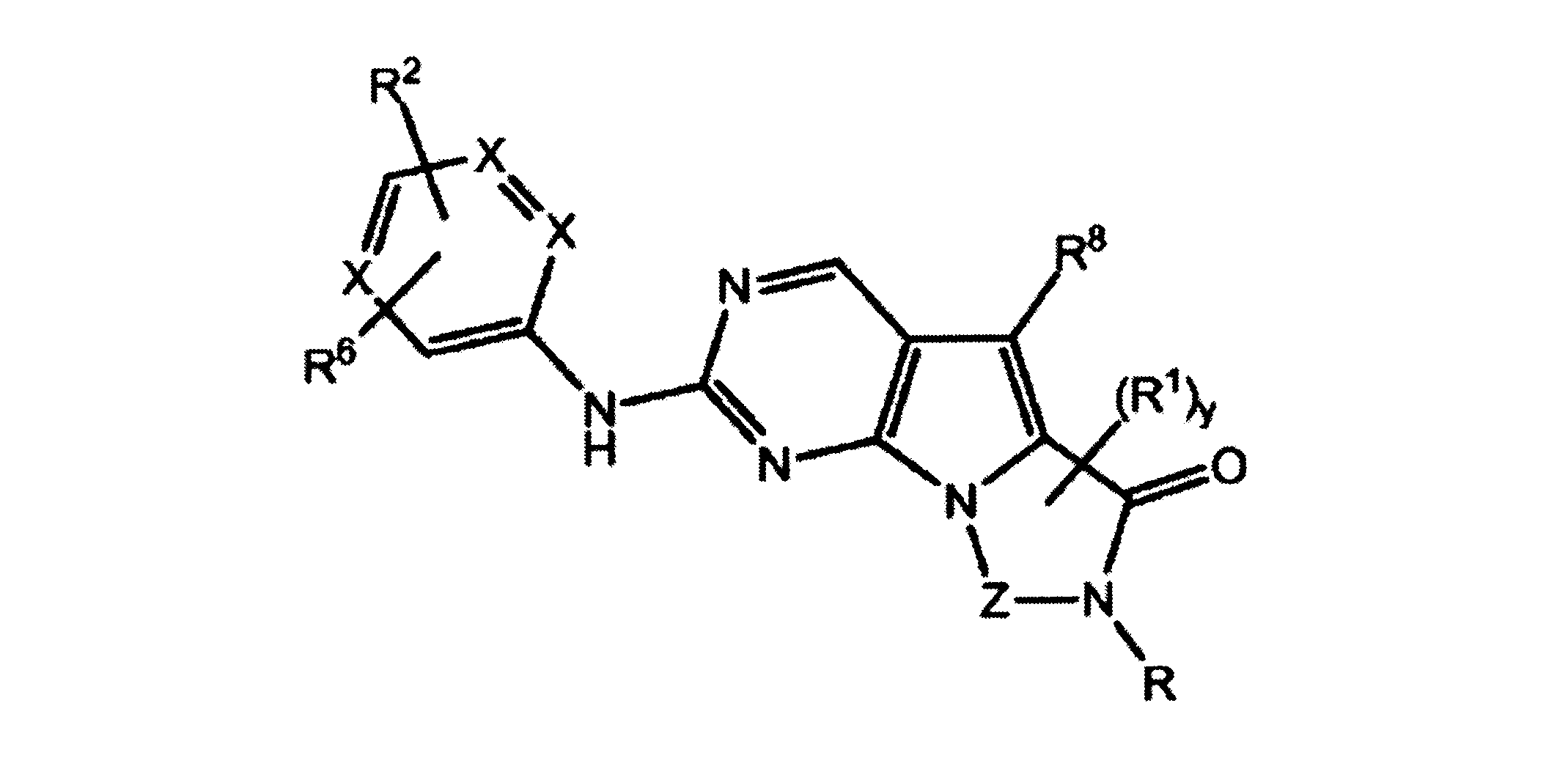
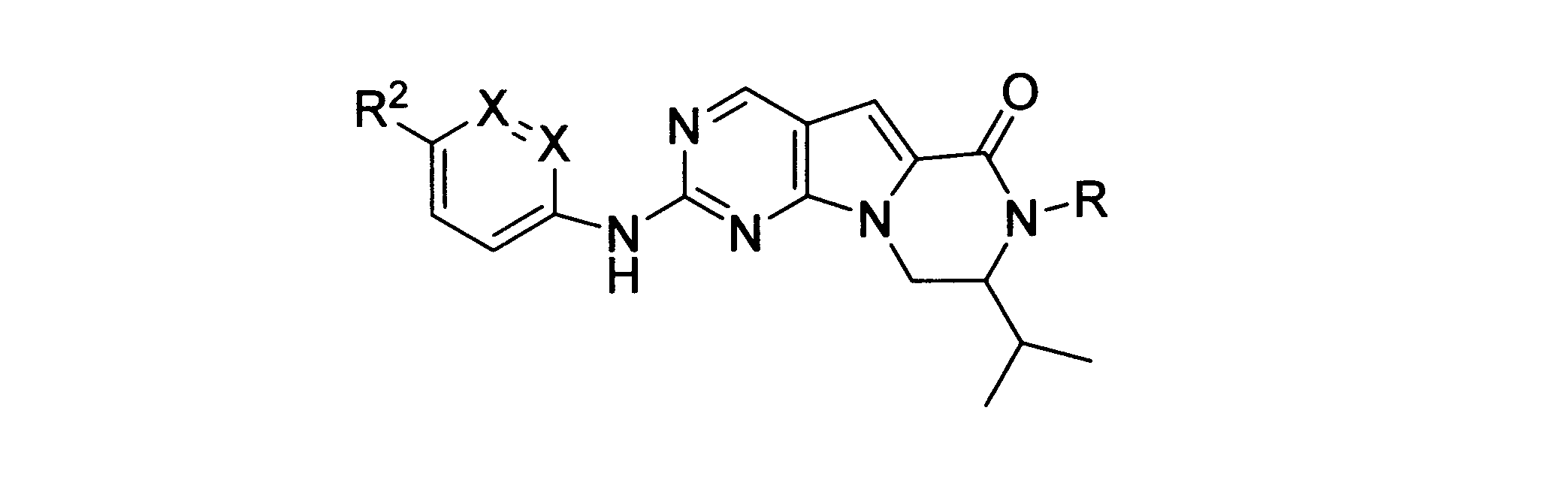
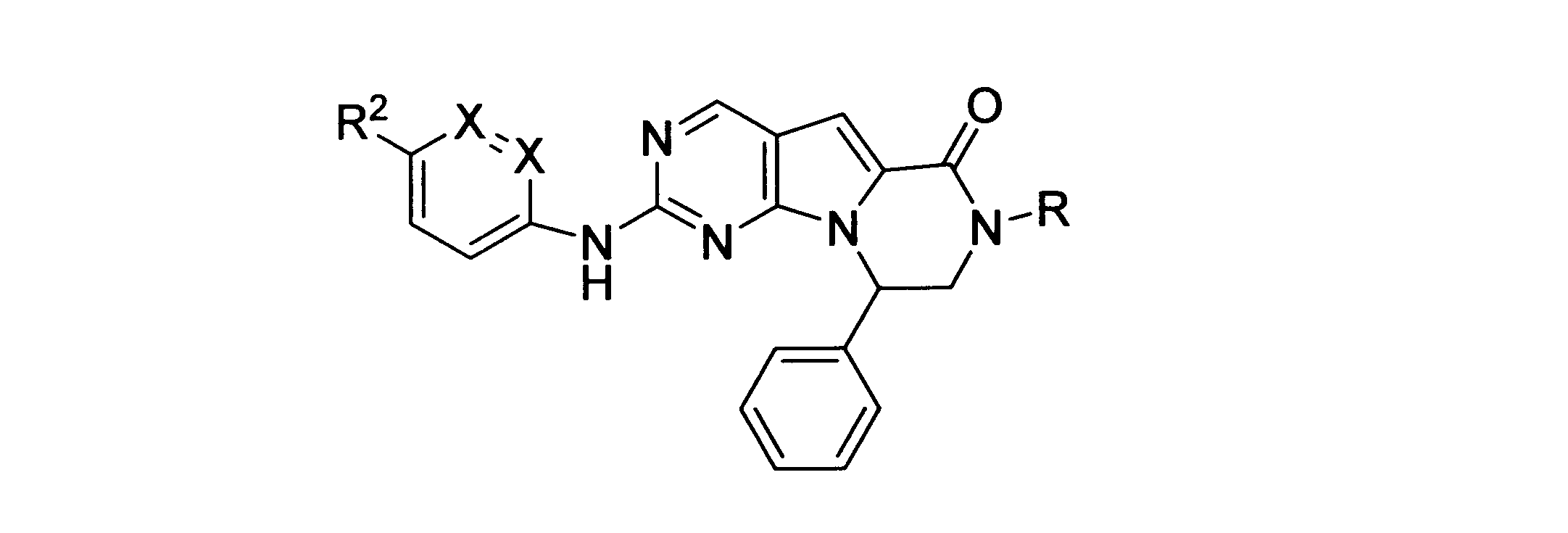
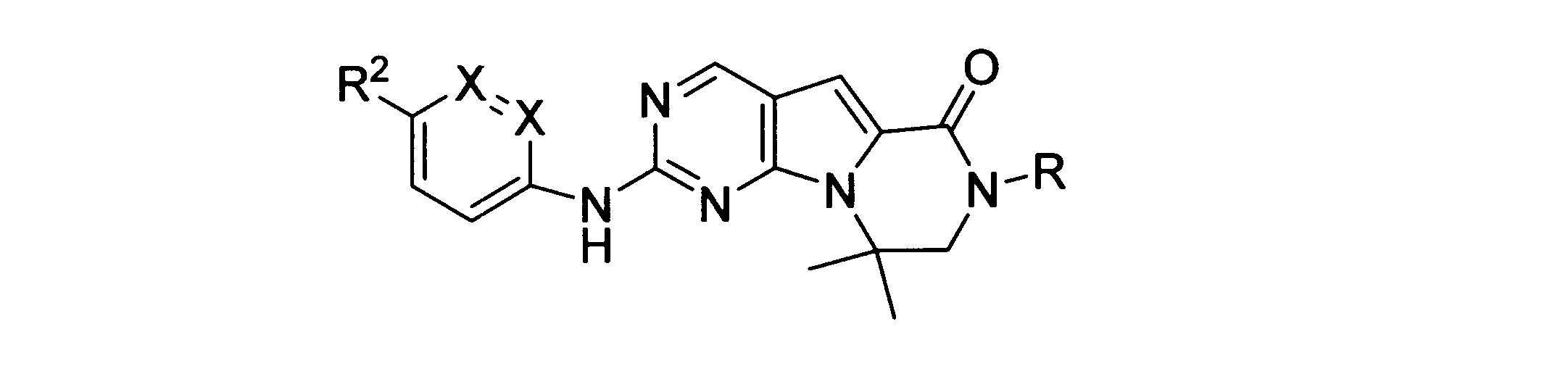
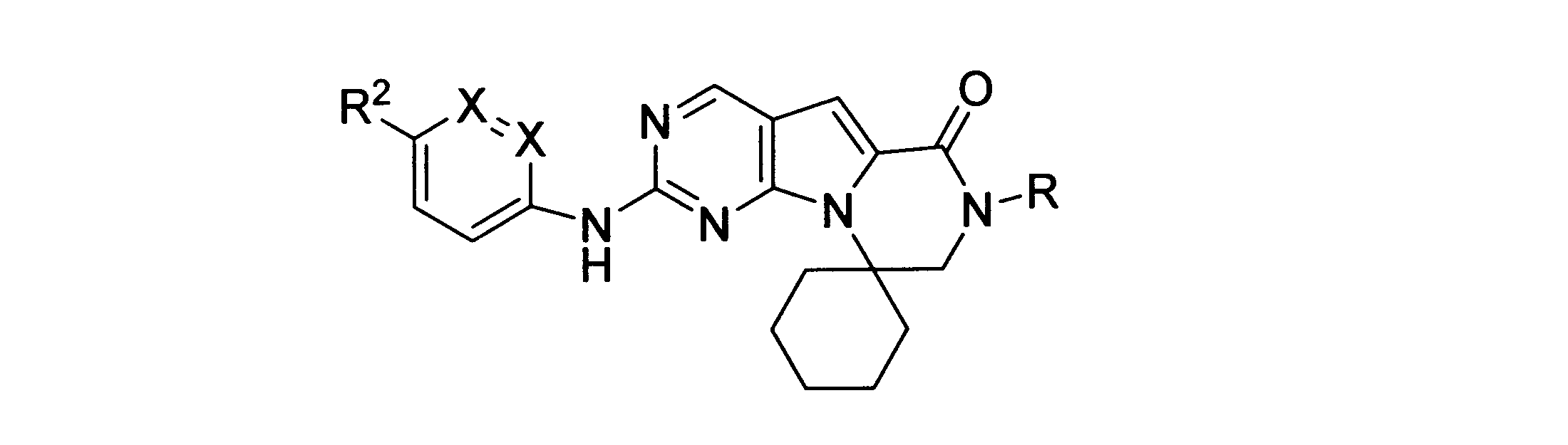
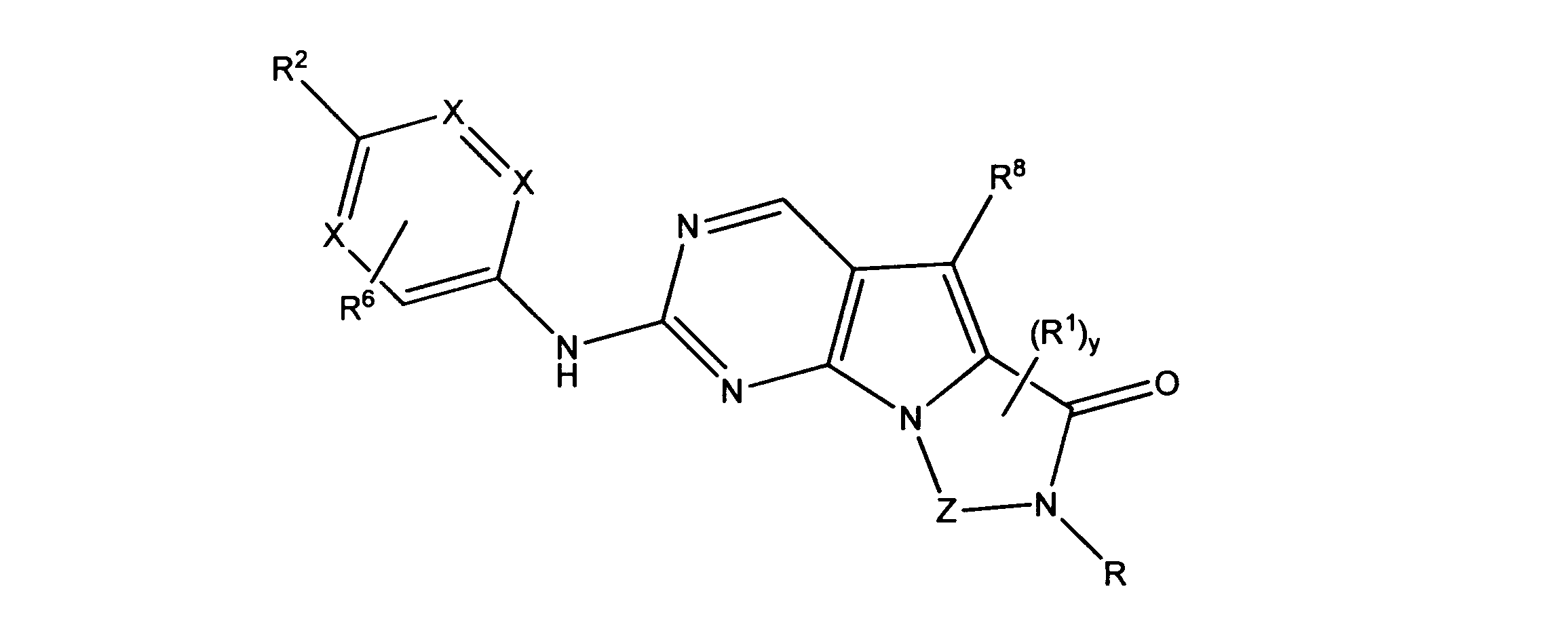
一実施形態では、本発明は、式I、II、III、IV又はVの化合物、又は、その薬理学的に許容される塩、又は、これらの化合物の使用に関し:
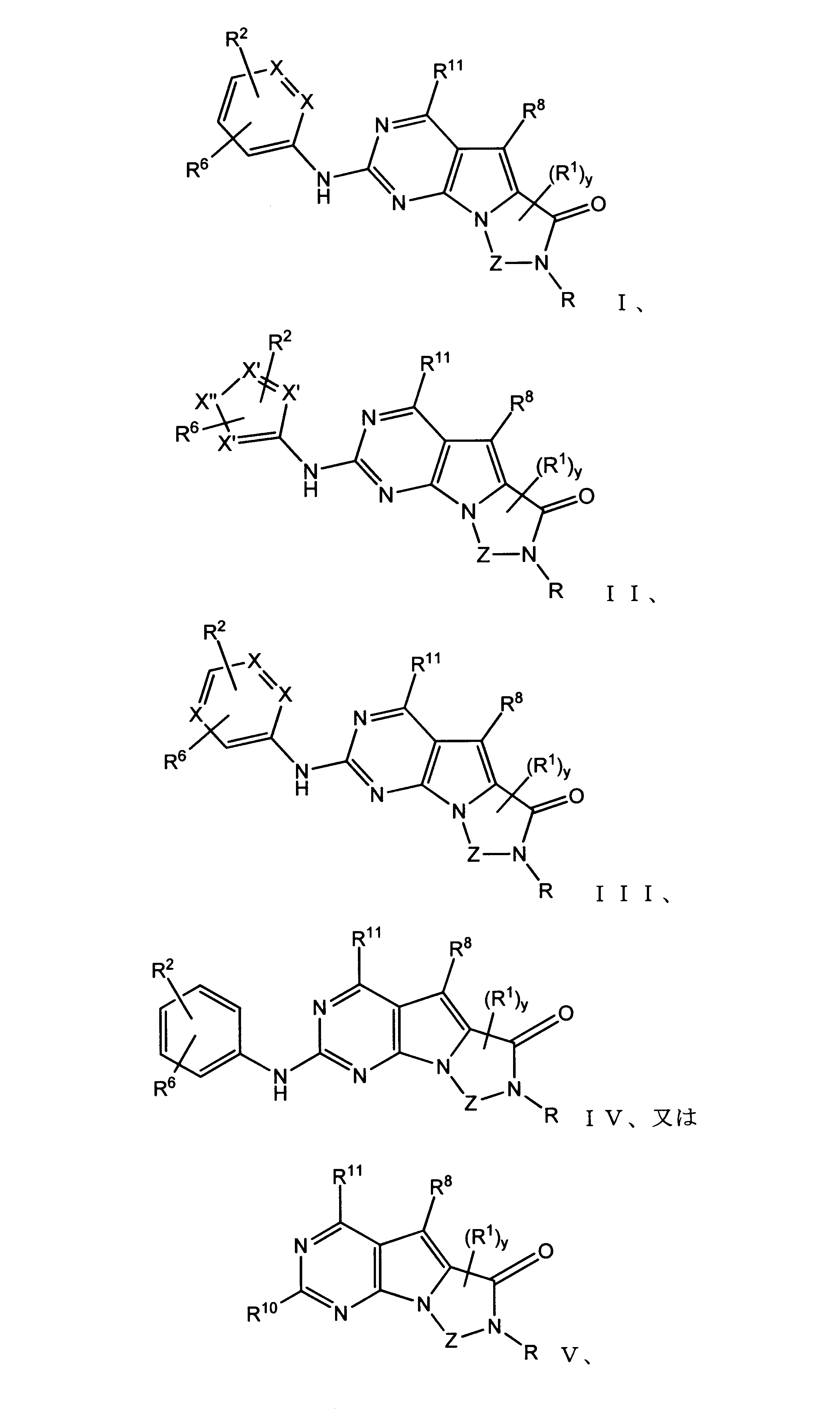
各Xは、独立してCH又はNであり;
各X’は、独立してCH又はNであり;
X’’は、独立してCH2、S又はNHであり、構成部分が安定5員環となるよう配置され;
R、R8及びR11は、独立してH、C1-C3のアルキル又はハロアルキル、シクロアルキル、又は、N、O又はSから選択される1つ以上のヘテロ原子を含んだシクロアルキル;-(アルキレン)m-C3-C8シクロアルキル、-(アルキレン)m-アリール、-(アルキレン)m、-ヘテロシクロ、-(アルキレン)m-ヘテロアリール、-(アルキレン)m-NR3R4、-(アルキレン)m-C(O)-NR3R4;-(アルキレン)m-O-R5、-(アルキレン)m-S(O)n-R5、又は-(アルキレン)m-S(O)n-NR3R4であり、これらのいずれも、電子価的に許される1つ以上のR基で任意に独立に置換されてもよく、同じ又は隣接した原子に結合した2つのRx基は任意に組み合わされて環を形成してもよく;
各R1は独立して、アリール、アルキル、シクロアルキル又はハロアルキルであり、前記アルキル、シクロアルキル及びハロアルキル基の各々は、鎖中の炭素の代わりに任意にO又はNヘテロ原子を含んでもよく、隣接した環原子又は同じ環原子の2つのR1は、それらが結合する環原子と共に、任意に3-8員環を形成してもよく;
yは、0、1、2,3又は4であり;
R2は、-(アルキレン)m-ヘテロシクロ、-(アルキレン)m-ヘテロアリール、-(アルキレン)m-NR3R4、-(アルキレン)m-C(O)-NR3R4;-(アルキレン)m-C(O)-O-アルキル;-(アルキレン)m-O-R5、-(アルキレン)m-S(O)n-R5、又は-(アルキレン)m-S(O)n-NR3R4であり、これらのいずれも、電子価的に許される1つ以上のRx基で任意に独立して置換されてもよく、同じ又は隣接した原子に結合した2つのRx基は、任意に組み合わされて環を形成してもよく、
mは0又は1、nは0、1又は2であり;
R3及びR4はその発生毎に、独立して、
(i)水素、又は
(ii)アルキル、シクロアルキル、ヘテロシクロ、アリール、ヘテロアリール、シクロアルキルアルキル、ヘテロシクロアルキル、アリールアルキル又はヘテロアリールアルキルであり、これらのいずれも、電子価的に許される1つ以上のRx基で任意に独立して置換されてもよく、同じ又は隣接した原子に結合した2つのRx基は、任意に組み合わされて環を形成してもよく、又は、
R3及びR4は、それらが結合する窒素原子と共に組み合わされて、電子価的に許される1つ以上のRx基で任意に独立して置換されてもよいヘテロシクロ環を形成してもよく、同じ又は隣接した原子に結合した2つのRx基は、任意に組み合わされて環を形成してもよく、
R5及びR5*はその発生毎に、独立して、
(i)水素、又は
(ii)アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロ、アリール、ヘテロアリール、シクロアルキルアルキル、ヘテロシクロアルキル、アリールアルキル又はヘテロアリールアルキルであり、これらのいずれも、電子価的に許される1つ以上のRx基で任意に独立して置換されてもよく;
Rxは、その発生毎に独立して、ハロ、シアノ、ニトロ、オキソ、アルキル、ハロアルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、ヘテロシクロ、アリール、ヘテロアリール、アリールアルキル、ヘテロアリールアルキル、シクロアルキルアルキル、ヘテロシクロアルキル、-(アルキレン)m-OR5、-(アルキレン)m-O-アルキレン-OR5、-(アルキレン)m-S(O)n-R5、-(アルキレン)m-NR3R4、-(アルキレン)m-CN、-(アルキレン)m-C(O)-R5、-(アルキレン)m-C(S)-R5、-(アルキレン)m-C(O)-OR5、-(アルキレン)m-O-C(O)-R5、-(アルキレン)m-C(S)-OR5、-(アルキレン)m-C(O)-(アルキレン)m-NR3R4、-(アルキレン)m-C(S)-NR3R4、-(アルキレン)m-N(R3)-C(O)-NR3R4、-(アルキレン)m-N(R3)-C(S)-NR3R4、-(アルキレン)m-N(R3)-C(O)-R5、-(アルキレン)m-N(R3)-C(S)-R5、-(アルキレン)m-O-C(O)-NR3R4、-(アルキレン)m-O-C(S)-NR3R4、-(アルキレン)m-SO2-NR3R4、-(アルキレン)m-N(R3)-SO2-R5、-(アルキレン)m-N(R3)-SO2-NR3R4、-(アルキレン)m-N(R3)-C(O)-OR5、-(アルキレン)m-N(R3)-C(S)-OR5、又は-(アルキレン)m-N(R3)-SO2-R5であり; ここで、前記アルキル、ハロアルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、ヘテロシクロ、アリール、ヘテロアリール、アリールアルキル、ヘテロアリールアルキル、シクロアルキルアルキル及びヘテロシクロアルキル基は、-(アルキレン)m-CN、-(アルキレン)m-OR5*、-(アルキレン)m-S(O)n-R5*、-(アルキレン)m-NR3*R4*、-(アルキレン)m-C(O)-R5*、-(アルキレン)m-C(=S)R5*、-(アルキレン)m-C(=O)OR5*、-(アルキレン)m-OC(=O)R5*、-(アルキレン)m-C(S)-OR5*、-(アルキレン)m-C(O)-NR3*R4*、-(アルキレン)m-C(S)-NR3*R4*、-(アルキレン)m-N(R3*)-C(O)-NR3*R4*、-(アルキレン)m-N(R3*)-C(S)-NR3*R4*、-(アルキレン)m-N(R3*)-C(O)-R5*、-(アルキレン)m-N(R3*)-C(S)-R5*、-(アルキレン)m-O-C(O)-NR3*R4*、-(アルキレン)m-O-C(S)-NR3*R4*、-(アルキレン)m-SO2-NR3*R4*、-(アルキレン)m-N(R3*)-SO2R5*、-(アルキレン)m-N(R3*)-SO2NR3*R4*、-(アルキレン)m-N(R3*)-C(O)-OR5*、-(アルキレン)m-N(R3*)-C(S)-OR5*、又は-(アルキレン)m-N(R3*)-SO2R5*、の1つ以上でさらに独立して置換されてもよく;
nは、0、1又は2であり、mは0又は1であり;
R3*及びR4*は、その発生毎に、独立して、
(i)水素、又は
(ii)アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロ、アリール、ヘテロアリール、シクロアルキルアルキル、ヘテロシクロアルキル、アリールアルキル又はヘテロアリールアルキルであって、これらのいずれも、電子価的に許される1つ以上のRx基で任意に独立して置換されてもよく、又は
R3*及びR4*は、それらが結合する窒素原子と共に組み合わされて、電子価的に許される1つ以上のRx基で任意に独立して置換されてもよいヘテロシクロ環を形成してもよく;
R6はH又は低級アルキル、-(アルキレン)m-ヘテロシクロ、-(アルキレン)m-ヘテロアリール、-(アルキレン)m-NR3R4、-(アルキレン)m-C(O)-NR3R4、-(アルキレン)m-O-R5、-(アルキレン)m-S(O)n-R5、又は-(アルキレン)m-S(O)n-NR3R4であり、これらのいずれも、電子価的に許される1つ以上のRx基で任意に独立して置換されてもよく、同じ又は隣接した原子に結合した2つのRx基は任意に組み合わされて環を形成してもよく、
R10は
(i)NHRAであり、
ここで、RAは非置換又は置換C1-C8アルキル、シクロアルキルアルキル、又は、-TT-RR、C1-C8シクロアルキル、又は、N、O及びSから選択されるヘテロ原子を1つ以上含むシクロアルキルであり;
TTは、非置換又は置換C1-C8アルキル又はC3-C8シクロアルキルリンカーであり;そして、
RRはヒドロキシル、非置換又は置換C1-C6アルコキシ、アミノ、非置換又は置換C1-C6アルキルアミノ、非置換又は置換ジC1-C6アルキルアミノ、非置換又は置換C6-C10アリール、1又は2個の5員又は6員環及びN、O及びSから選択される1-4個のヘテロ原子を含む非置換又は置換ヘテロアリール、非置換又は置換C3-C10炭素環、又は、1又は2個の5員又は6員環及びN、O及びSから選択される1-4個のヘテロ原子を含む非置換又は置換ヘテロ環であり;又は
(ii)-C(O)-R12又は-C(O)O-R13、ここで、R12はNHRA又はRAであり、R13はRAであり;又はその薬理学的に許容される塩、プロドラッグ又はアイソトープ変種、たとえば、一部又は全部に重水素を導入した形態である。
ある態様では、化合物は、式IIIであり:
ある態様では、Rxは、さらに置換されない。
ある態様では、R2は、-(アルキレン)m-ヘテロシクロ、-(アルキレン)m-ヘテロアリール、-(アルキレン)m-NR3R4、-(アルキレン)m-C(O)-NR3R4であり、;-(アルキレン)m-O-R5-(アルキレン)m-S(O)n-R5又は-(アルキレン)m-S(O)n-NR3R4のいずれも、電子価的に許される1つ以上のRx基で任意に独立して置換されてもよく、同じ又は隣接した原子に結合した2つのRx基は任意に組み合わされて環を形成してもよく、
mは0又は1、nは0、1又は2である。
ある態様では、R8は、水素又はC1-C3アルキルである。 ある態様では、Rは、水素又はC1-C3アルキルである。
ある態様では、R2は、-(アルキレン)m-ヘテロシクロ、-(アルキレン)m-NR3R4、-(アルキレン)m-C(O)-NR3R4、-(アルキレン)m-C(O)-O-アルキル又は-(アルキレン)m-OR5であり、これらのいずれも、電子価的に許される1つ以上のRx基で任意に独立して置換されてもよく、同じ又は隣接した原子に結合した2つのRx基は任意に組み合わされて環を形成してもよい。
ある態様では、R2は、-(アルキレン)m-ヘテロシクロ、-(アルキレン)m-NR3R4、-(アルキレン)m-C(O)-NR3R4、-(アルキレン)m-C(O)-O-アルキル又は-(アルキレン)m-OR5であり、さらなる置換はない。 ある態様では、R2におけるmは、1である。
更なる態様では、R2のアルキレンは、メチレン基である。
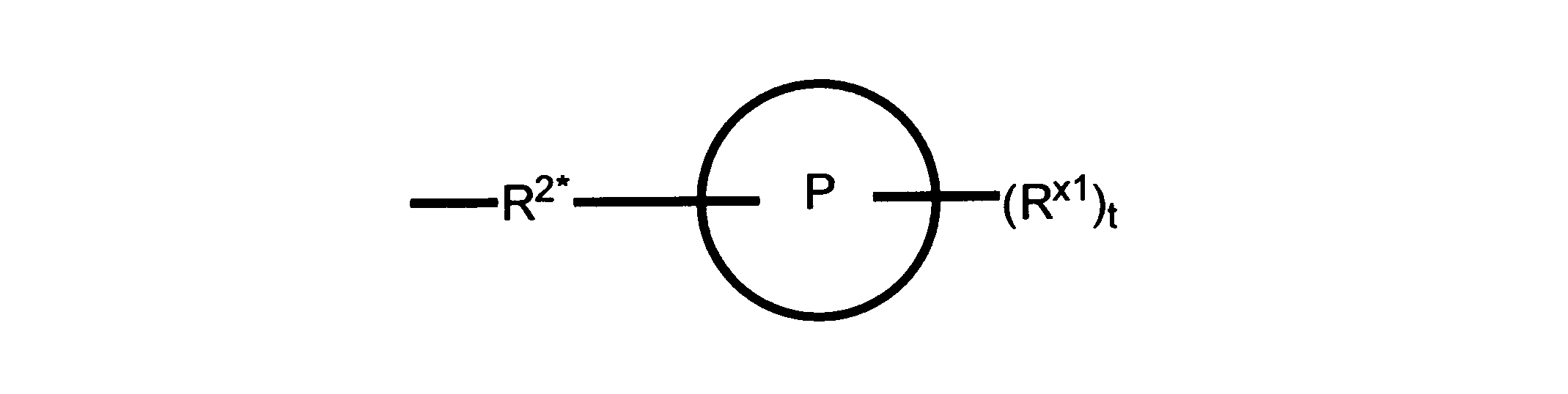
Pは、4員~8員の単環又は二環式飽和ヘテロシクリル基であり;
各Rx1は、独立して、-(アルキレン)m-(C(O))m-(アルキレン)m-(N(Rn))m-(アルキル)m、ここで各mは、独立して0又は1であるが、少なくとも一つのmは1、-(C(O))-O-アルキル、-(アルキレン)m-シクロアルキル、ここでmは0又は1、-N(Rn)-シクロアルキル、-C(O)-シクロアルキル、-(アルキレン)m-ヘテロシクリル、ここでmは0又は1、又は、-N(Rn)-ヘテロシクリル、-C(O)-ヘテロシクリル、-S(O)2-(アルキレン)m、ここでmは、1又は2であり、 ここで、Rnは、H、C1~C4アルキル又はC1~C6ヘテロアルキルであり、そして、
2個のRx1は、Pに結合する原子(これは同じ原子でもよい)と共に、環を形成することができ;tは、0、1又は2である。
ある態様では、各Rx1は、非置換のアルキル、ハロゲン又はヒドロキシによって任意に置換されるのみである。
ある態様では、Rx1は、水素又は非置換のC1~C4アルキルである。 ある態様では、少なくとも1つのRx1は、-(アルキレン)m-ヘテロシクリルであり、ここで、mは0又は1である。
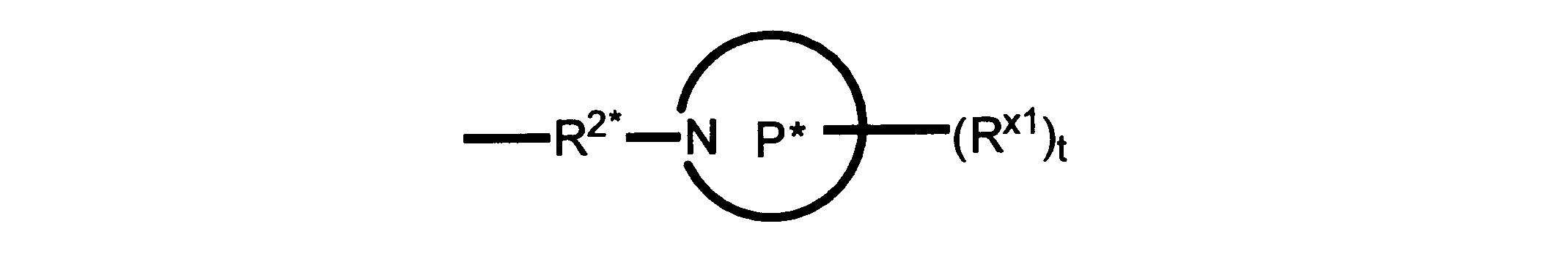
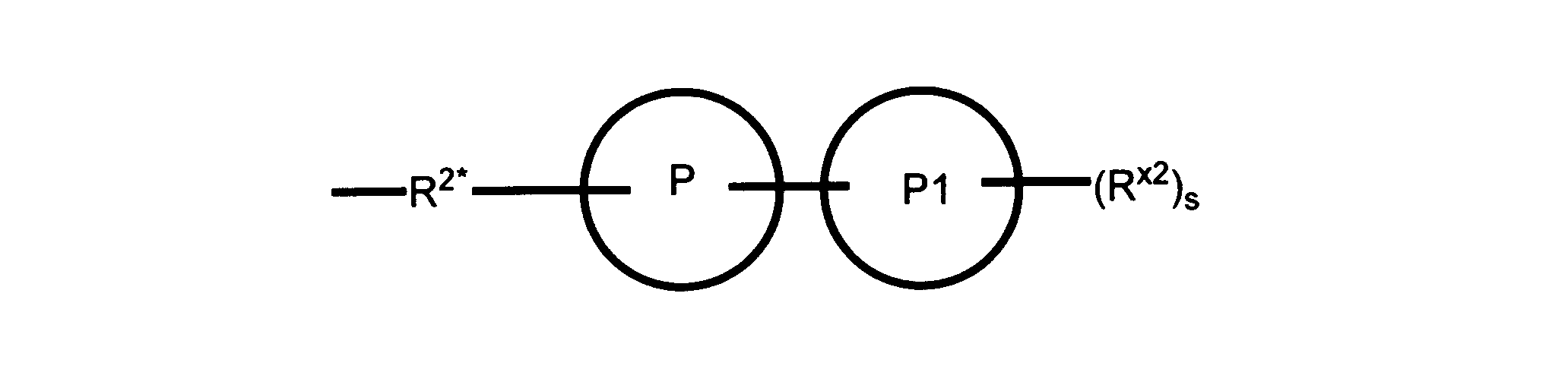
R2*は、結合、アルキレン、-(アルキレン)m-O-(アルキレン)m-、-(アルキレン)m-C(O)-(アルキレン)m-、-(アルキレン)m-S(O)2-(アルキレン)m-及び-(アルキレン)m-NH-(アルキレン)m-であり、 ここで、各mは、独立して0又は1であり;
Pは、4員~8員の単環又は二環式飽和ヘテロシクリル基であり;
P1は、4員又は6員の単環式飽和ヘテロシクリル基であり;
各Rx2は、独立して水素又はアルキルであり;
sは0、1又は2である。
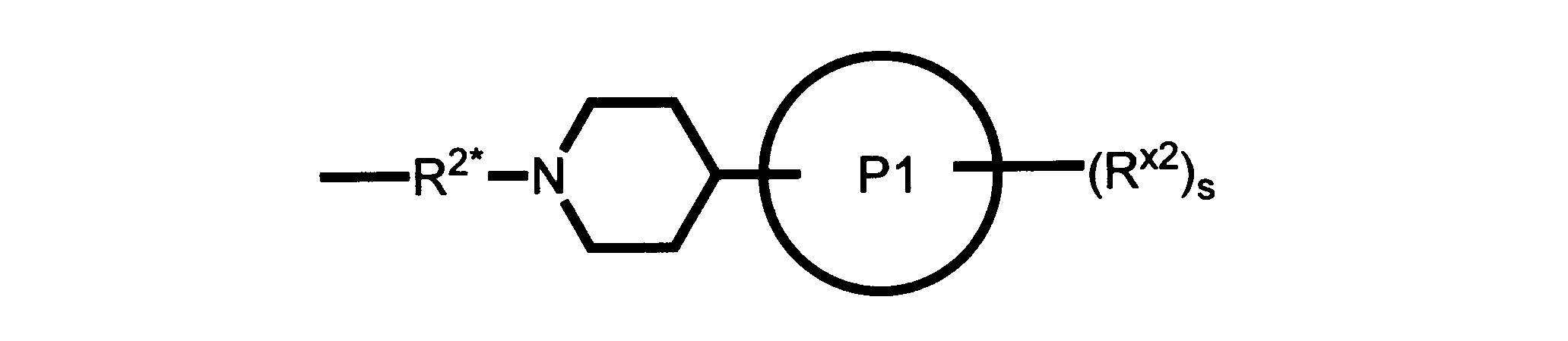
ある態様では、前述の全ての態様におけるR2*のあらゆるアルキレンは、さらに置換されない。
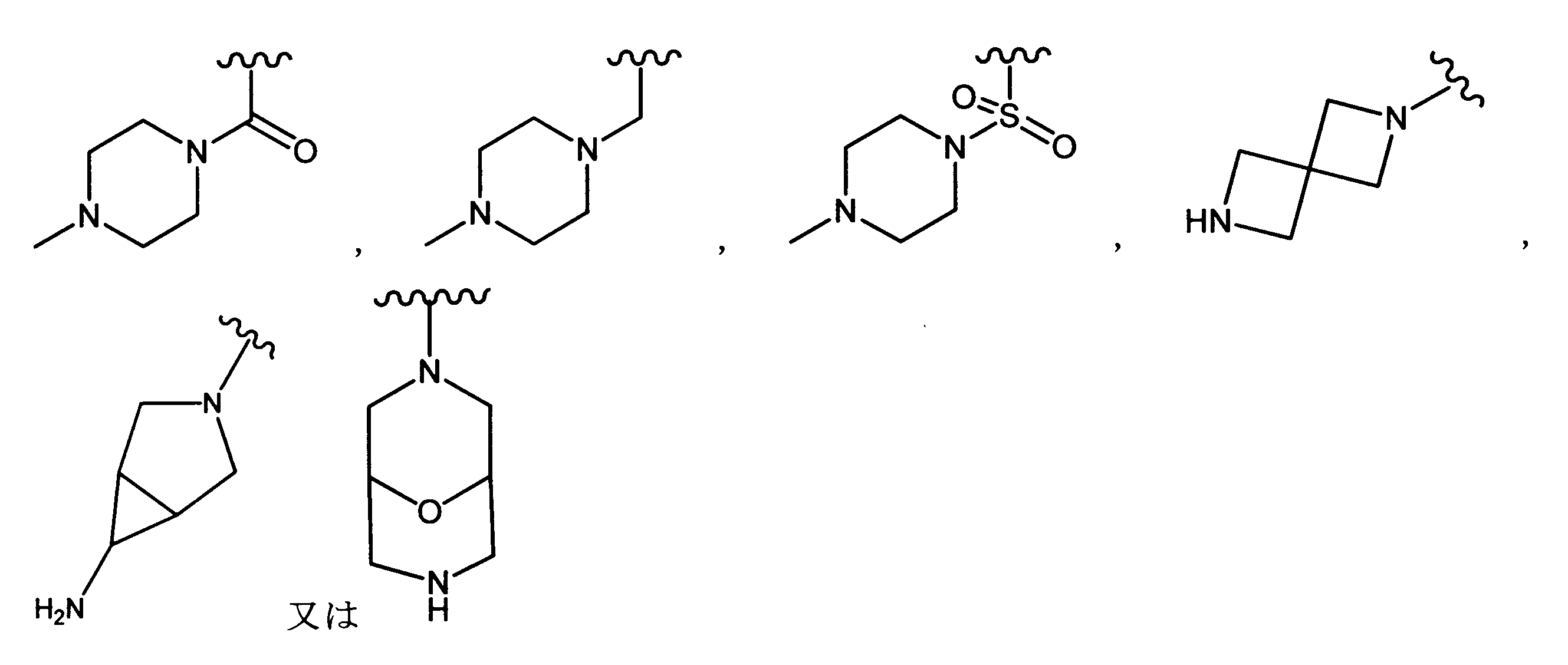
ある態様では、R2は、図13-15に示される構造から選択される。
ある態様では、化合物は、一般式Iaを有する:
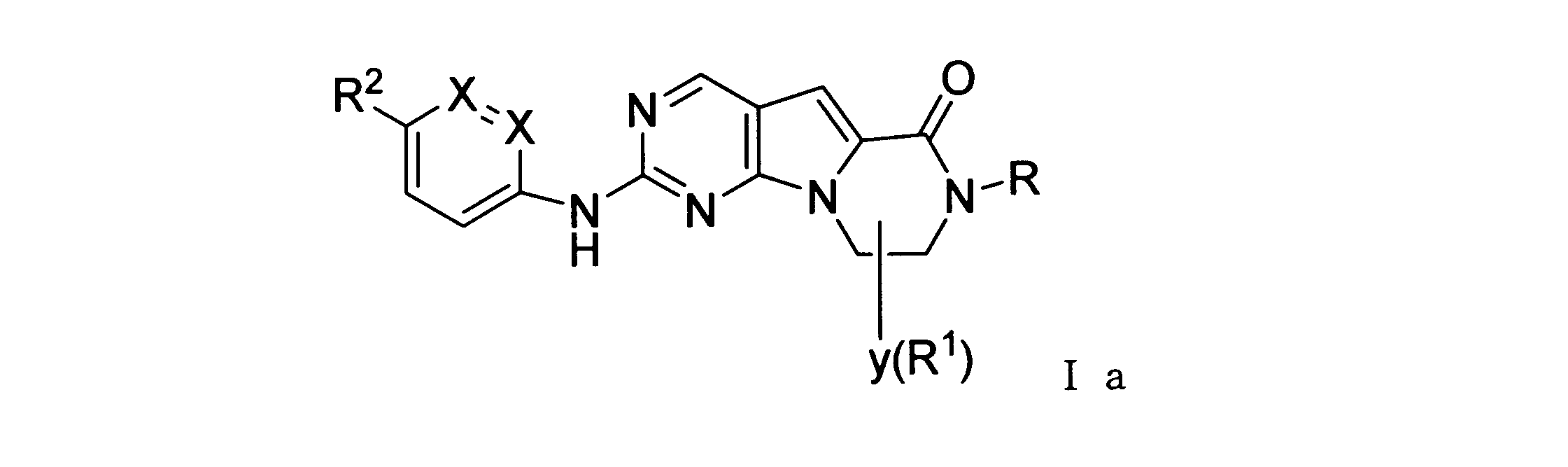
ある実施形態では、化合物は式Iaを有し、RはHである。
ある実施形態では、化合物は、式Iaを有し、R2は:
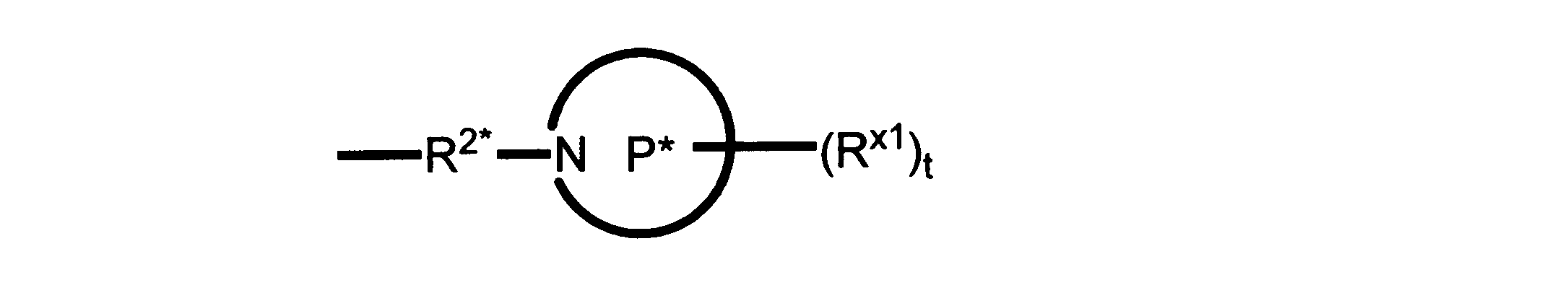
ある実施形態では、化合物は式Iaを有し、R2は:
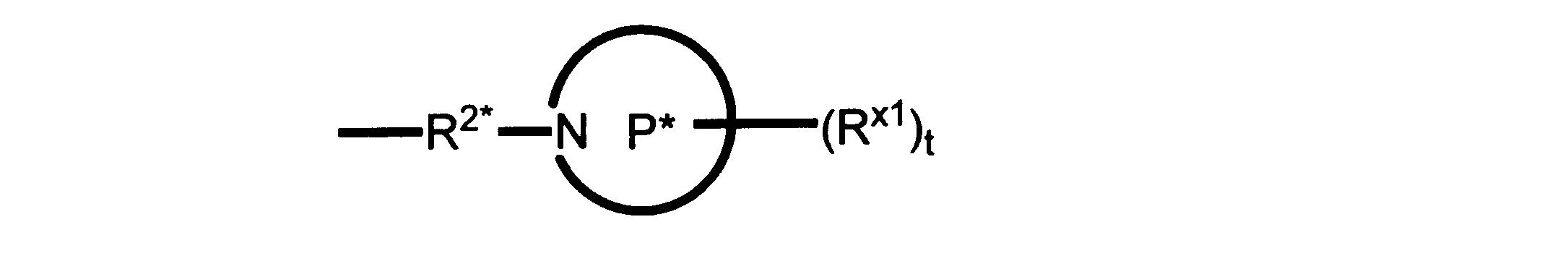
Rx1は、水素又は非置換のC1~C4アルキルであり、及び R2*は、あらかじめ定義されている通りである。
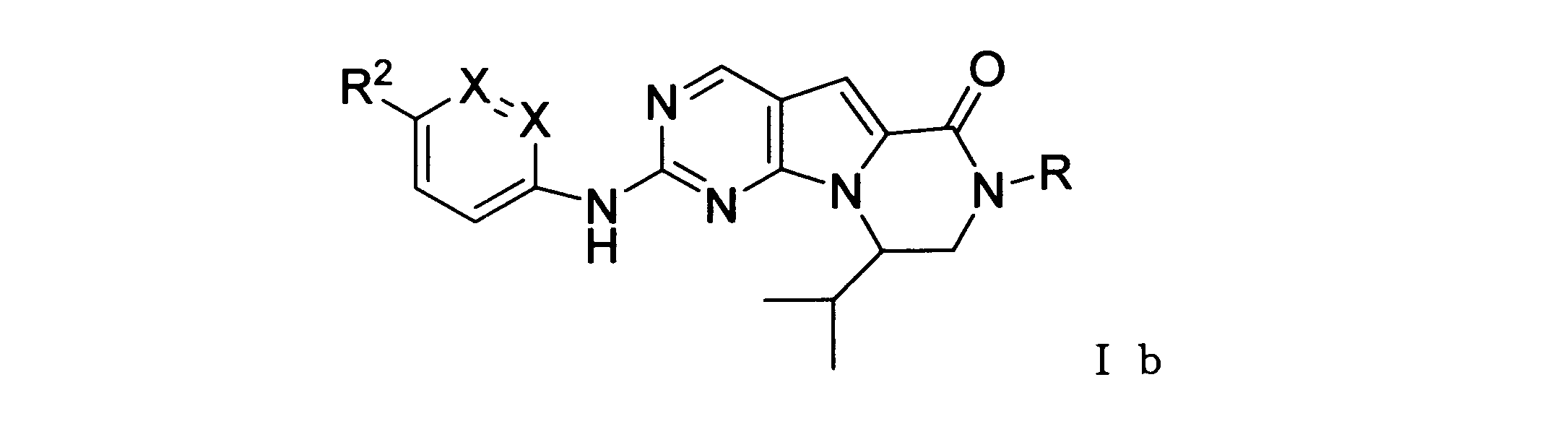
ある実施形態では、化合物は式Ibを有し、Rはアルキルである。
ある実施形態では、化合物は式Ibを有し、RはHである。
ある実施形態では、化合物は、式Ibを有し、R2は:
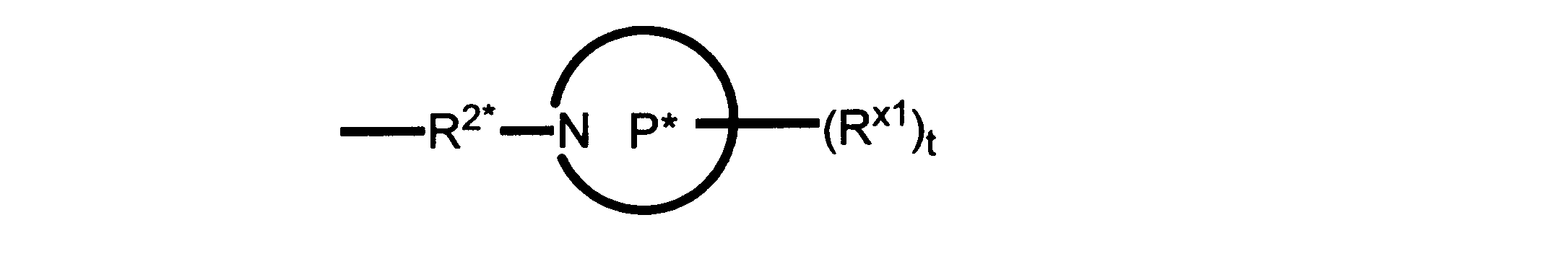
ある実施形態では、化合物は、式Ibを有し、R2は:
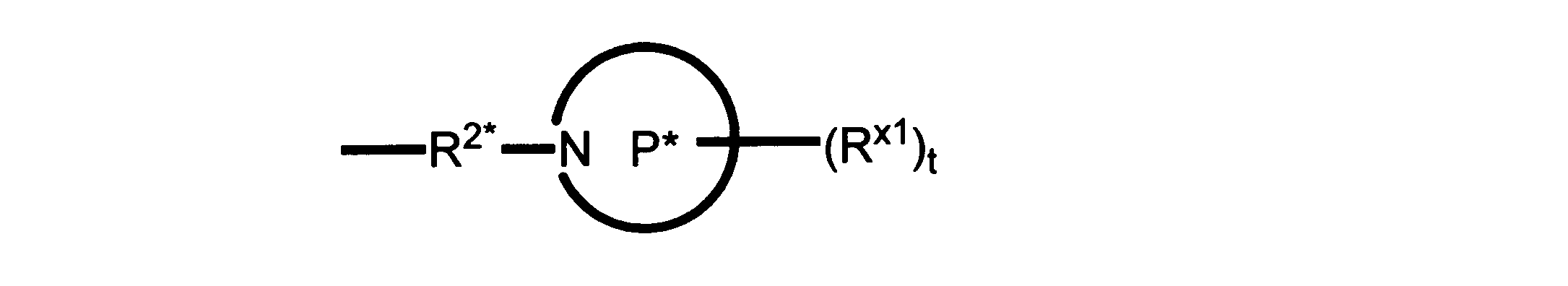
ある実施形態では、化合物は式Icを有し、Rはアルキルである。
ある実施形態では、化合物は式Icを有し、RはHである。
ある実施形態では、化合物は式Icを有し、R2は:
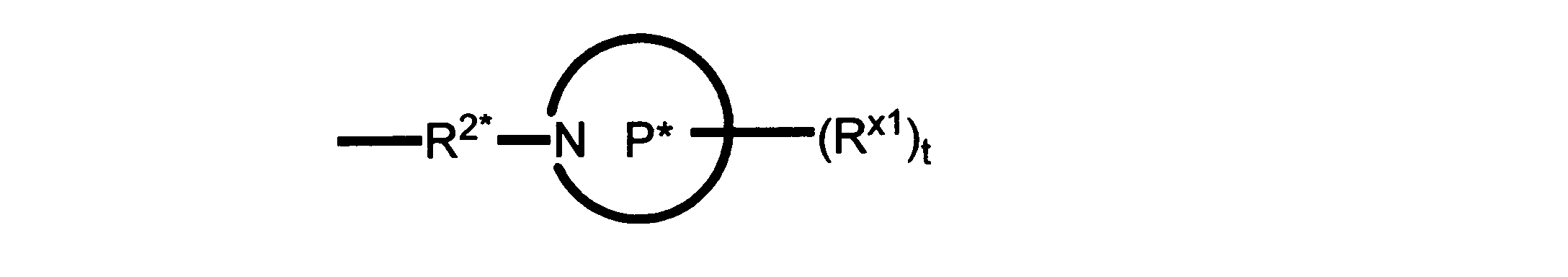
ある実施形態では、化合物は式Icを有し、R2は:
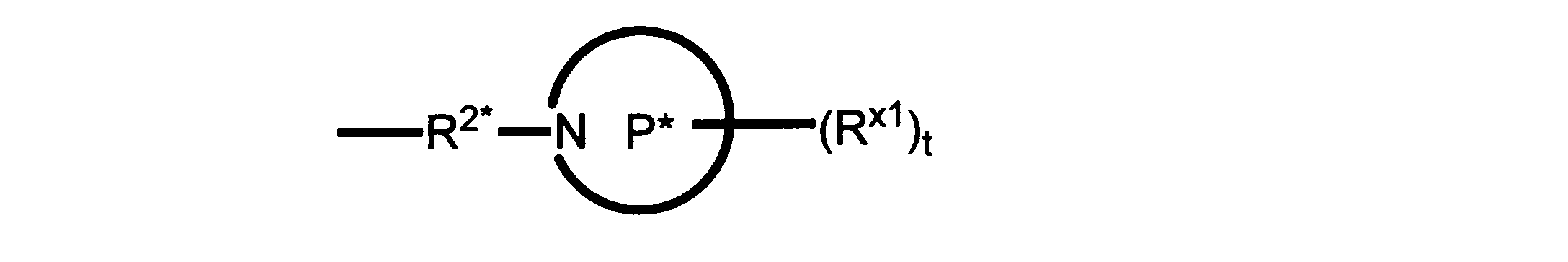
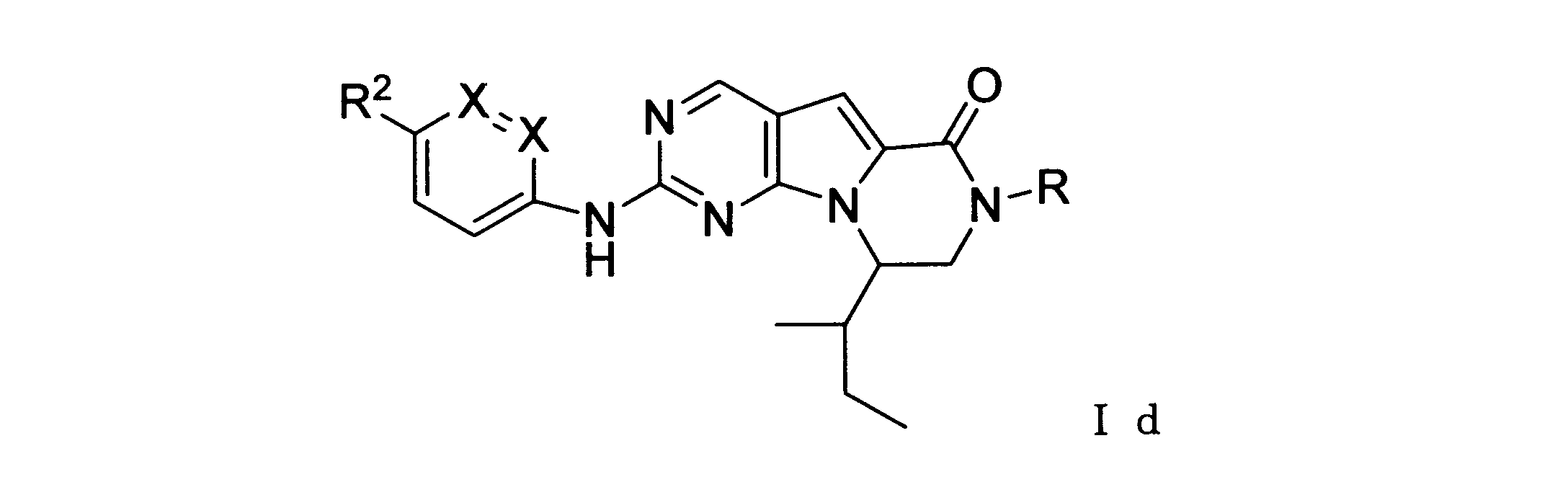
ある実施形態では、化合物は式Idを有し、Rはアルキルである。
ある実施形態では、化合物は式Idを有し、RはHである。
ある実施形態では、化合物は、式Idを有し、R2は:

ある実施形態では、化合物は、式Idを有し、R2は:
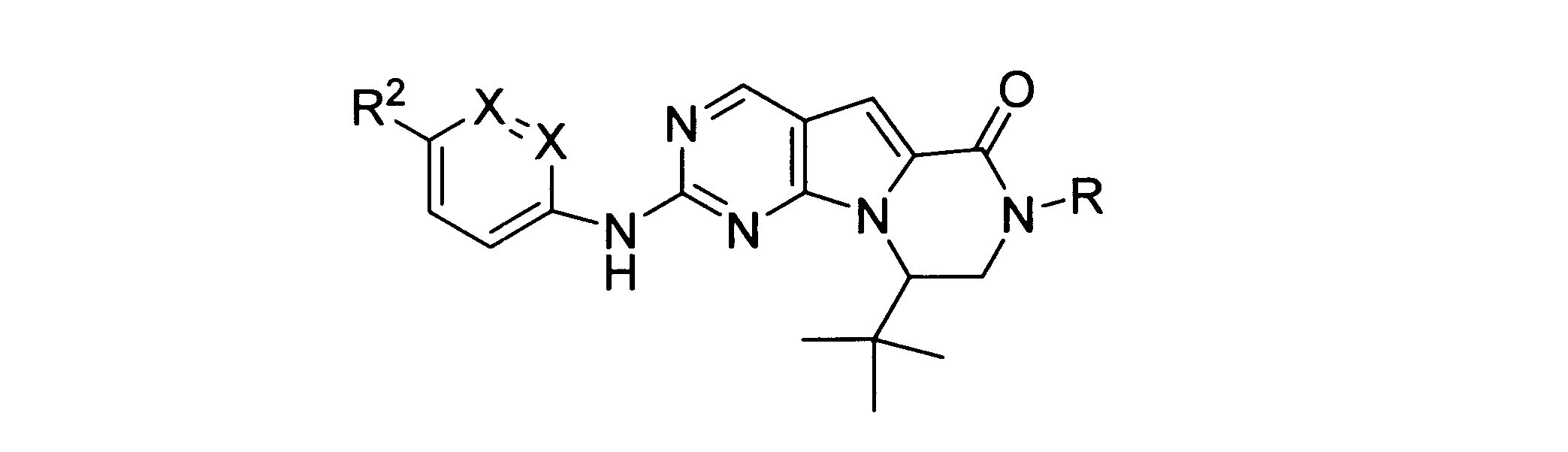
ある実施形態では、化合物は式Ieを有し、RはHである。
ある実施形態では、化合物は式Ieを有し、R2は:
ある実施形態では、化合物は式Ieを有し、R2は:
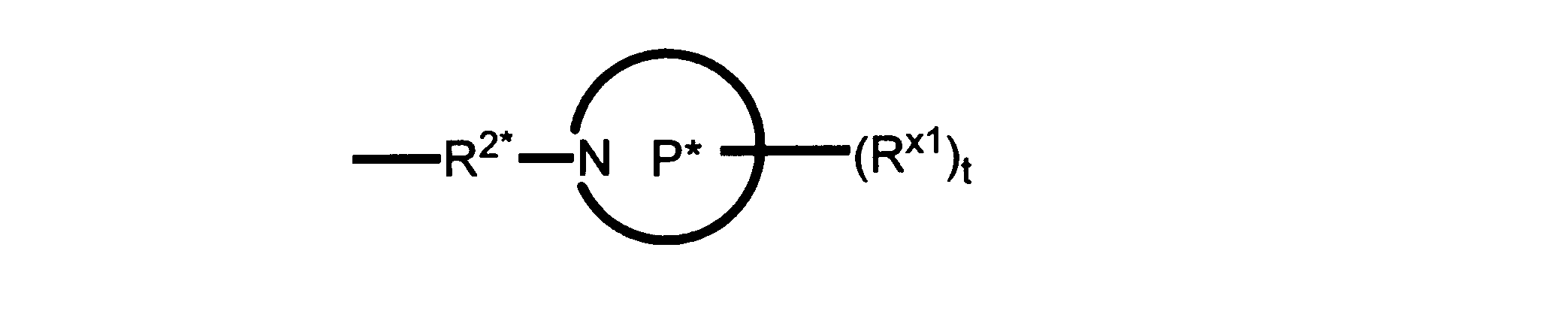
ある実施形態では、化合物は式Ifを有し、RはHである。
ある実施形態では、化合物は式Ifを有し、R2は:
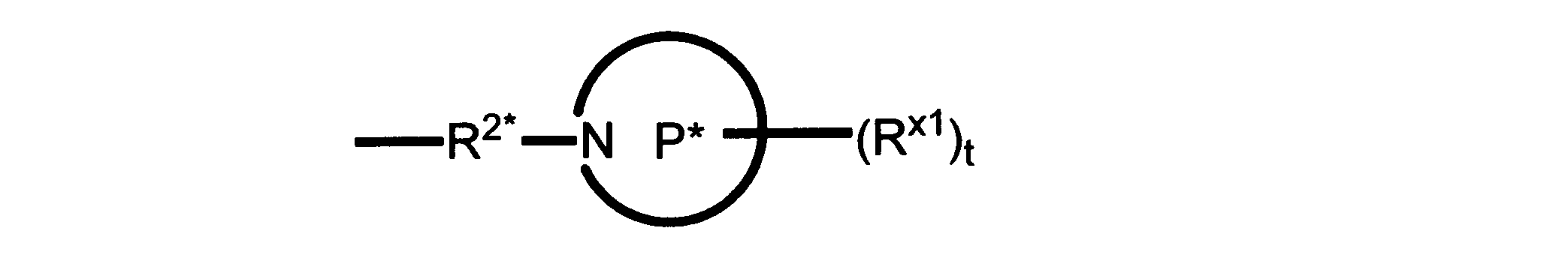
ある実施形態では、化合物は式Ifを有し、R2は:
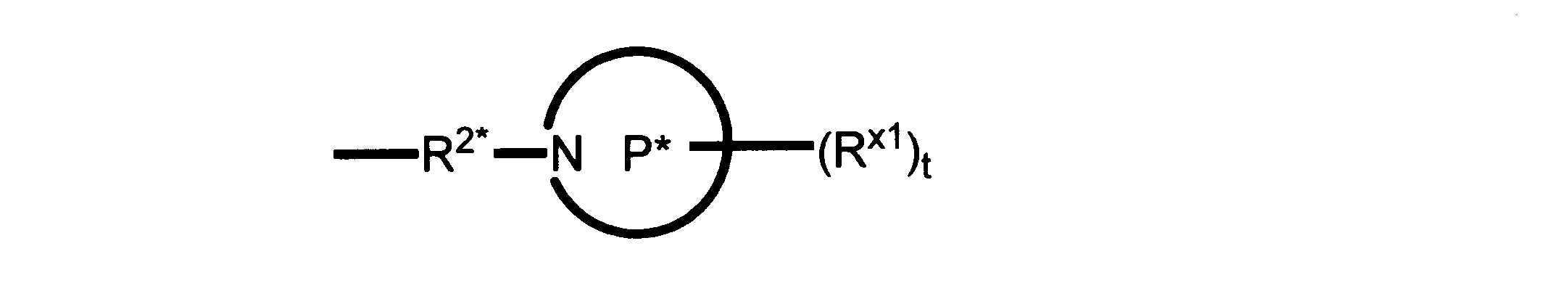
ある実施形態では、化合物は式Igを有し、RはHである。
ある実施形態では、化合物は式Igを有し、R2は:
ある実施形態では、化合物は式Igを有し、R2は:
ある実施形態では、化合物は式Ihを有し、RはHである。
ある実施形態では、化合物は、式Ihを有し、R2は:
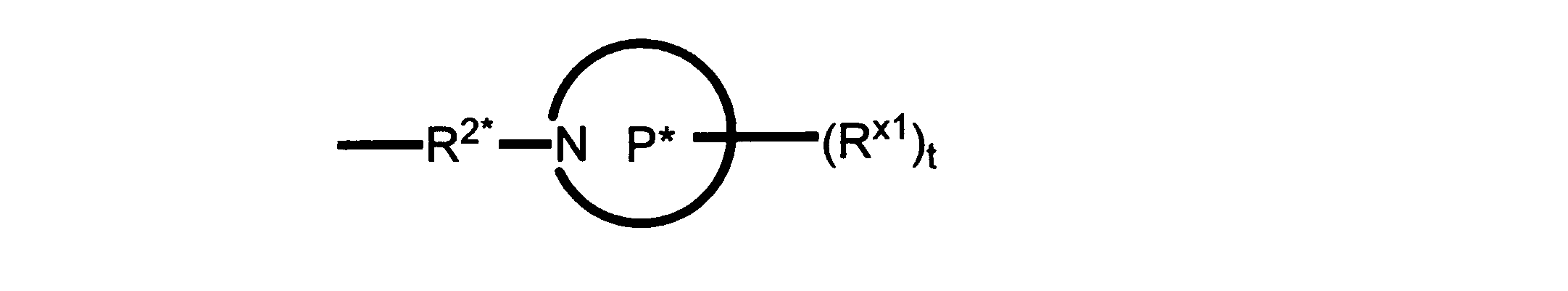
ある実施形態では、化合物は、式Ihを有し、R2は:
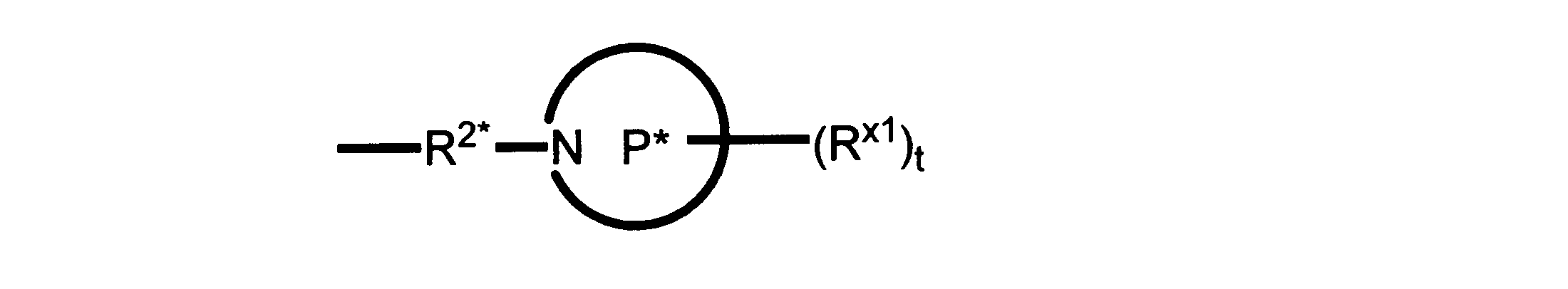
ある実施形態では、化合物は式Iiを有し、RはHである。
ある実施形態では、化合物は式Iiを有し、R2は:
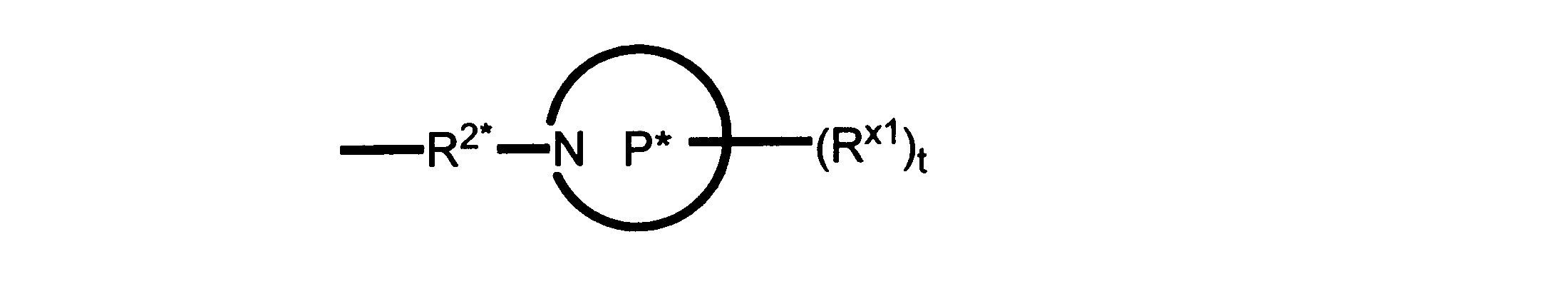
ある実施形態では、化合物は式Iiを有し、R2は:
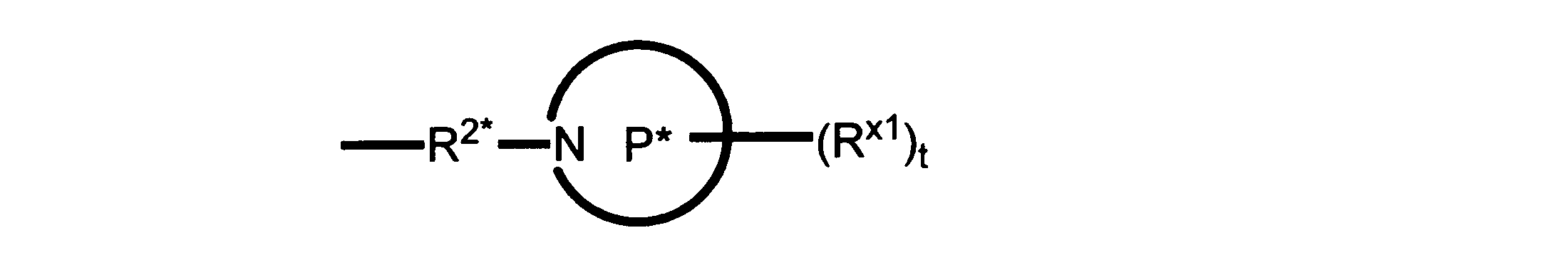
ある実施形態では、化合物は式Ijを有し、RはHである。
ある実施形態では、化合物は式Ijを有し、R2は:
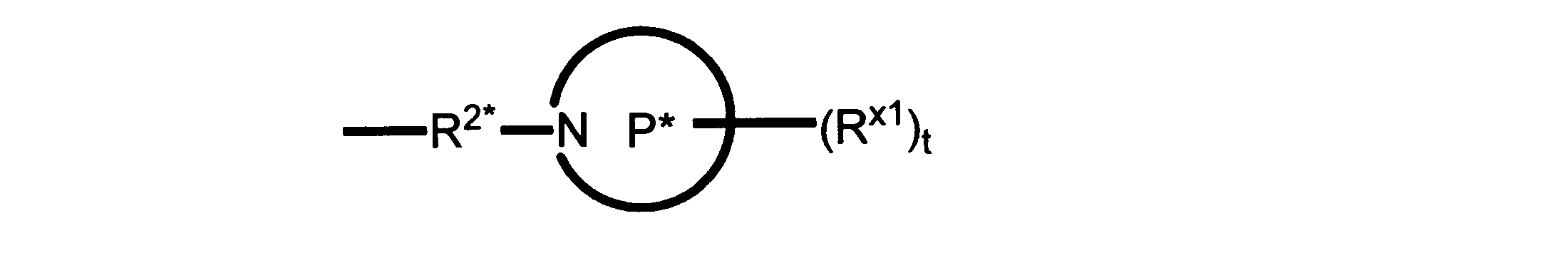
ある実施形態では、化合物は式Ijを有し、R2は:
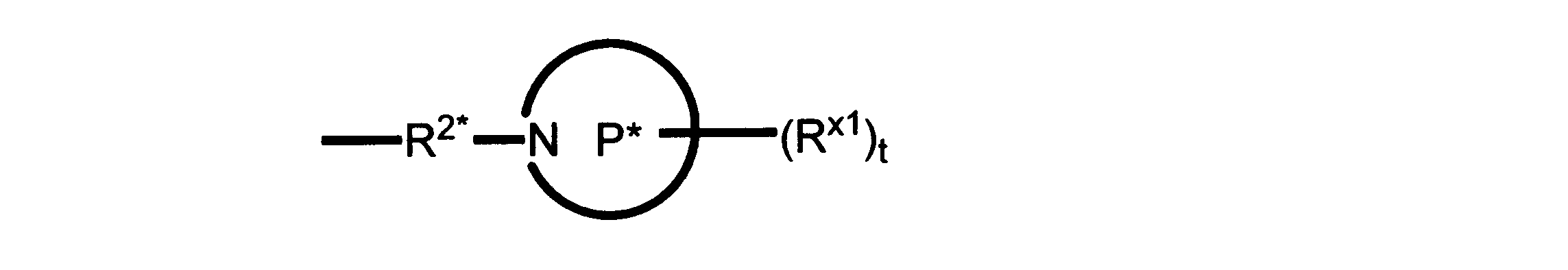
ある実施形態では、化合物は式Ijを有し、RはHであり、両方のXはNである。
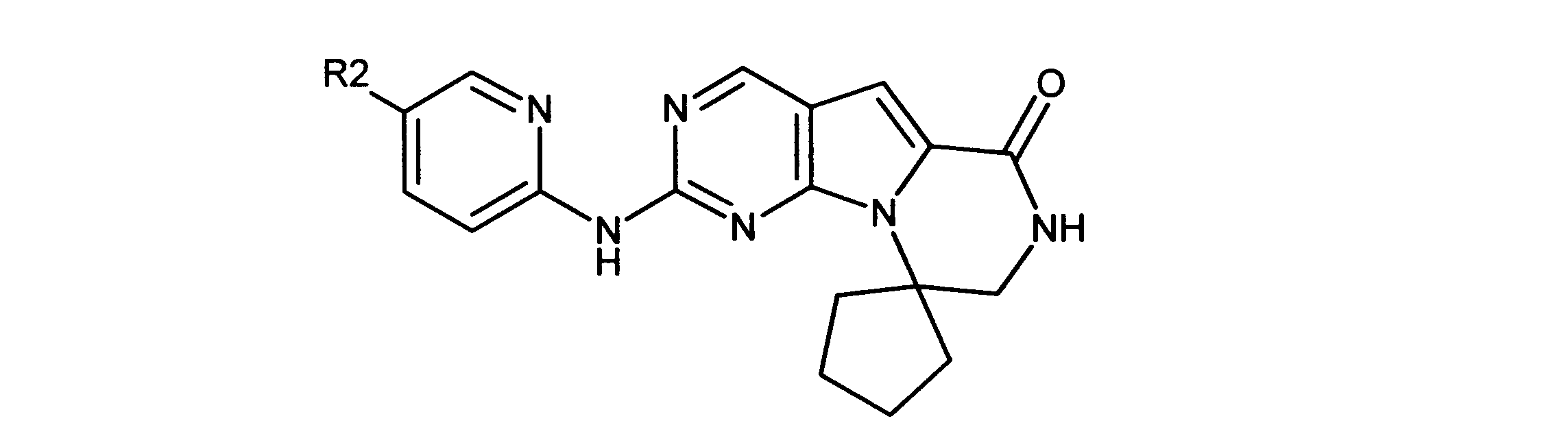
ある実施形態では、化合物は式Ikを有し、R2は:
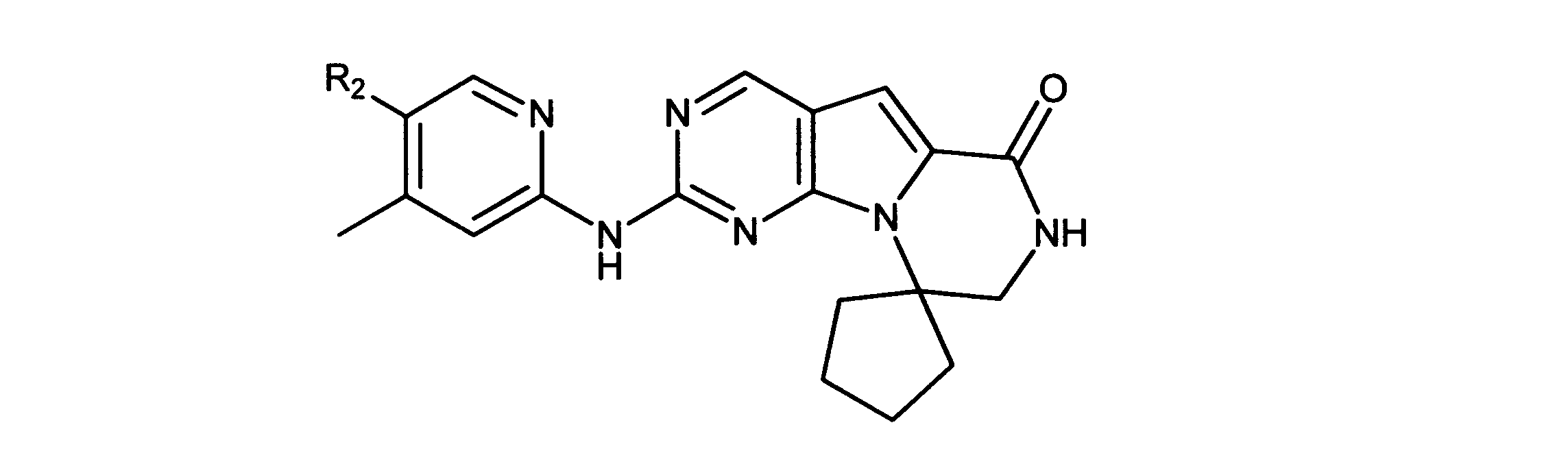
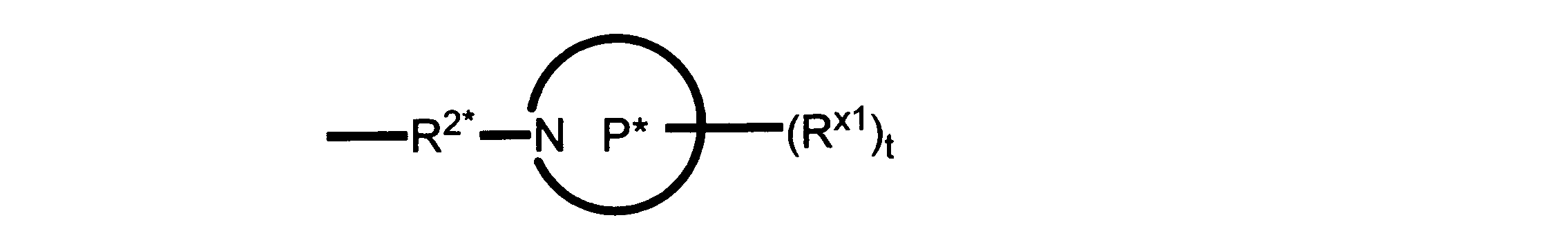
ある実施形態では、化合物は式Ilを有し、R2は:
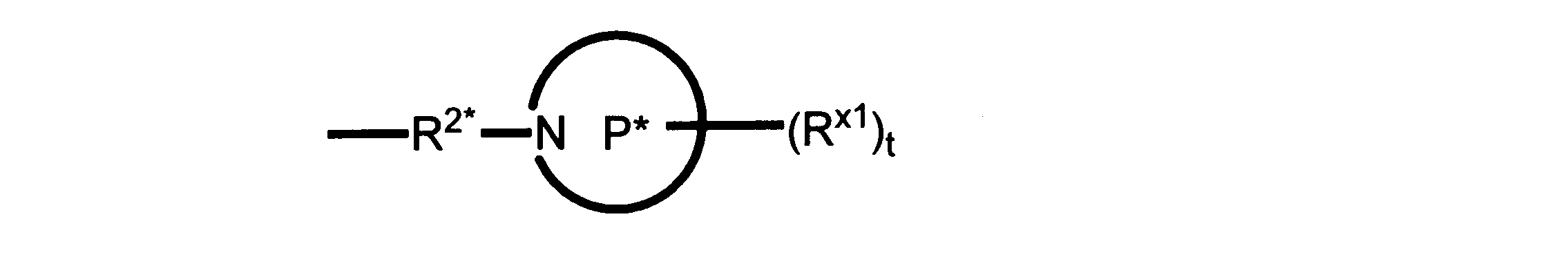
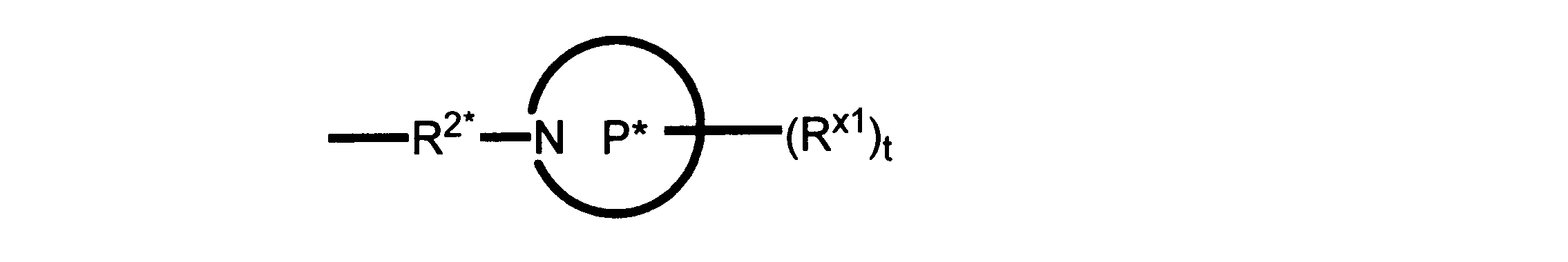
ある実施形態では、化合物は式Imを有し、R2は:
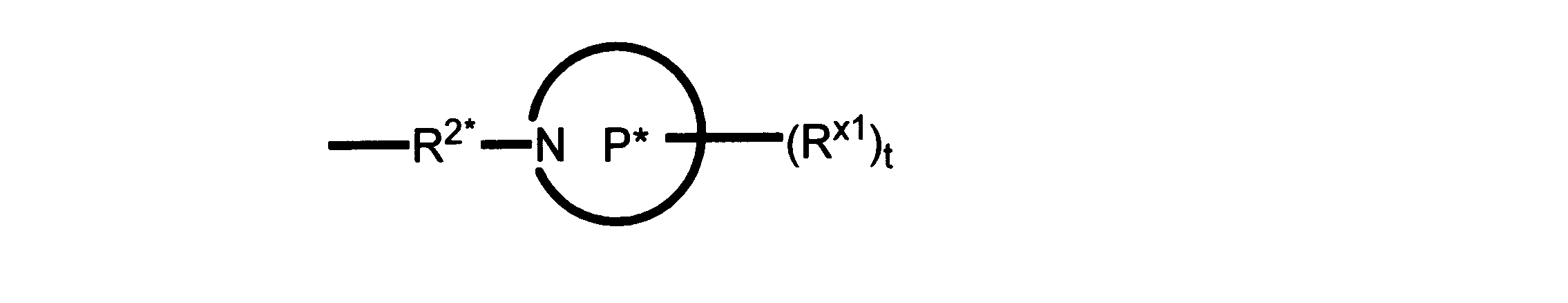

ある実施形態では、化合物は、式IIaを有し、R2は:
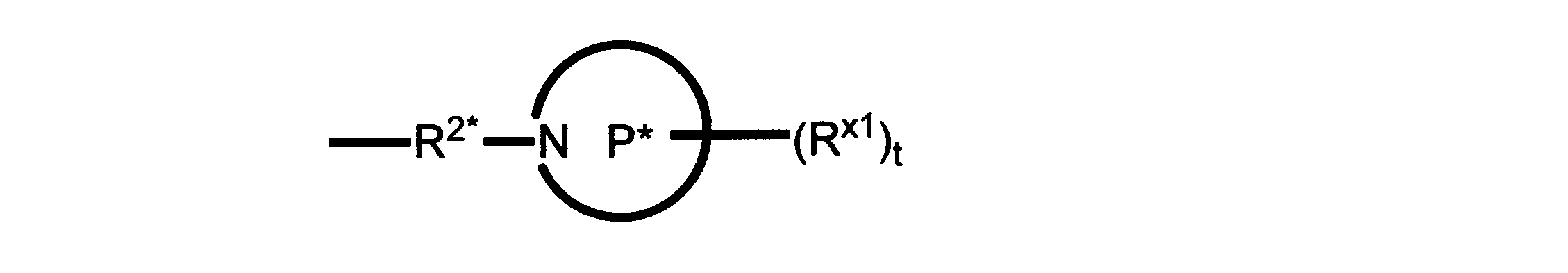
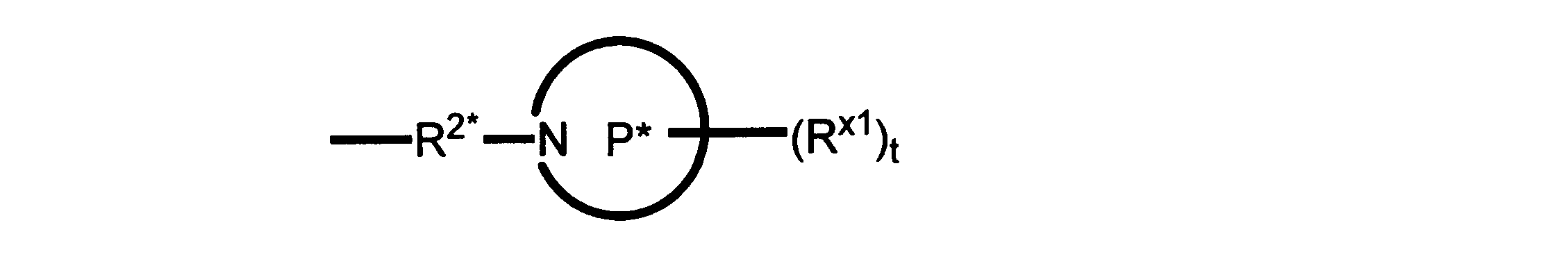
ある実施形態では、化合物は式Imを有し、R2は:

本発明は、同位体の量が自然の存在率より多くなるよう、すなわち濃縮により、望ましい原子の同位体で置換する、化合物及び化合物の使用を含む。同位元素は、同じ原子番号であるが異なる質量数を有している、すなわち、中性子の数が異なるが陽子の数は同じである原子である。一般例として、及び、限定なしで、水素の同位体、たとえば、重水素(2H)及び三重水素(3H)が、ここに記載されたいずれの構造においても、用いられてよい。代替的に又は付加的に、炭素の同位体、例えば、13C及び14Cを用いてもよい。好ましい同位体的置換は、水素に対して重水素を分子の1つ以上の位置で用いて、薬剤の性能を向上させることである。
特に、ここに記載される活性化合物は、Rbポジティブ癌又は他のRbポジティブ異常細胞増殖異常症に罹患した被験体を治療するために用いられてもよい。ある実施形態では、癌又は細胞増殖異常症は、CDK4/6-複製依存的癌又は細胞増殖異常症であり、れは癌又は細胞増殖異常症と呼ばれ、複製又は増殖のためにCDK4/6の活性を必要とする、又は選択的CDK4/6阻害物質の活性を通して成長抑制され得る。このタイプの癌及び異常症は、官能性網膜芽細胞腫タンパク質の存在(例えば、その存在を示す細胞を有する)によって特徴付けられてもよい。この癌及び異常症は、Rb-ポジティブであると分類される。
一実施形態では、Rbポジティブ癌は、肺癌、骨製癌、膵癌、皮膚癌、頭部又は頸部の癌、皮膚又は眼内メラノーマ、子宮癌、卵巣癌、直腸癌、肛門部の癌、胃癌、結腸癌、乳癌、子宮癌、卵管の癌腫、子宮内膜の癌腫、頸部の癌腫、膣の癌腫、陰門の癌腫、食道の癌、小腸の癌、内分泌系の癌、甲状腺の癌、上皮小体の癌、副腎の癌、軟組織の肉腫、尿道の癌、ペニスの癌、前立腺癌、膀胱の癌、腎臓又は尿管の癌、腎細胞癌、腎盂の癌腫、中枢神経系(CNS)の腫瘍、原発性中枢神経系リンパ腫、脊髄軸腫瘍、脳幹神経膠腫、下垂体腺腫、又は前記の癌の1つ以上の組合せを非限定的に含む、Rbポジティブ癌腫・肉腫から選択される。
繊維肉腫、粘液肉腫、軟骨肉腫、骨肉腫、脊索腫、悪性線維性組織球腫、血管肉腫、血管肉腫、リンパ管肉腫。中皮腫、平滑筋肉腫、横紋筋肉腫、鱗状細胞癌腫;類表皮癌、悪性皮膚付属器腫瘍、腺癌、肝癌、肝細胞癌、腎細胞癌、副腎腫、胆管癌、移行上皮癌、絨毛膜癌、精上皮腫、胚的細胞癌腫、未分化神経膠腫;多形神経膠芽腫、神経芽細胞腫、髄芽細胞腫、悪性髄膜腫、悪性シュワン細胞腫、神経線維肉腫、副甲状腺癌腫、甲状腺髄様癌、気管支カルチノイド、褐色細胞腫、島細胞癌、悪性カルチノイド、悪性傍神経節腫、メラノーマ、メルケル細胞腫瘍、嚢肉腫仮葉、唾液癌、胸腺癌、膀胱癌及びウィルムス腫瘍。
たとえば、米国特許公開公報US20070212736号、標題"Functional Immunohistochemical Cell Cycle Analysis as a Prognostic Indicator for Cancer”を参照。
代替的には、分子遺伝子の試験は、網膜芽細胞腫遺伝子の状態の決定に用いられてもよい。
網膜芽細胞腫のための分子遺伝子の試験は、Lohmann and Gallie,"Retinoblastoma Gene Reviews"(2010), http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=retinoblastoma, 又は、Parsamら、"A comprehensive, sensitive and economical approach for the detection of mutations in the RB1 gene in retinoblastoma" Journal of Genetics, 88(4), 517-527 (2009) に記載される以下を含む。
CDK-複製依存的細胞及びサイクリン依存的キナーゼ阻害剤
組織特異幹細胞及びその他のレジデント増殖細胞のサブセットは自己複製ができ、そのことは、成熟した哺乳類の寿命全体を通じて、管理された複製を通してそれ自身を置換することができることをを意味する。
たとえば、細網内皮系では、造血幹細胞は前駆細胞を生み出し、そしてそれは次いで、血液の全ての分化した要素(例えば、白血球、赤血球及び血小板)を生み出す(図1を参照のこと)。
ある特定の増殖細胞(例えばHSPC)は、増殖キナーゼサイクリン-依存的キナーゼ4(CDK4)及び/又は細胞複製のためのサイクリン依存的キナーゼ6(CDK6)の酵素活性を必要とすることが見出された。
一実施形態では、化学療法剤としてここに記載される化合物を使用することにより、PD0332991等の他のCDK4/6阻害物質の使用と比較してHSPC複製が遅いことによる血液回復の促進及び血液欠乏リスクの低減が、可能になる。
一実施形態では、化学療法剤としてここに記載される化合物を使用することにより、PD0332991等他のCDK4/6阻害物質を用いた現行の治療方法と比較して、治療中におけるオフサイクル又はドラッグホリデイの減少又は最小化が可能となる。
一実施形態では、化学療法剤としてここに記載される化合物を使用することにより、オフサイクル又はドラッグホリデイの除去が可能となる。
一実施形態では、化学療法剤としてここに記載される化合物を使用することにより、PD0332991等他のCDK4/6阻害物質を用いた現行の治療方法の使用と比較して、より少ないオフサイクル日又はドラッグホリデイで、長期間の投与が可能になる。
一実施形態では、化学療法剤としてここに記載される化合物を使用することにより、オフサイクル又はドラッグホリデイの間、PD0332991等他のCDK4/6阻害物質を用いた現行の方法の使用よりもより急速な、血球値の回復が可能になる。
特定の態様では、Rbポジティブ癌のCDK4/6抑制性治療を受けている被験体におけるCDK4/6-複製依存的正常細胞に対するCDK4/6阻害の有害作用を低減又は限定する化合物、方法及び組成物が、化学療法剤として提供され、この方法は、ここに記載される化合物を効果的量で投与することを含み、ここで、CDK4/6-複製依存的正常細胞の相当な部分が、化合物の投与から約24、30、36又は40時間未満以内に、プレ治療ベースライン細胞サイクル活性へリエントリーする(すなわち、細胞サイクルにリエントリーする)。
特定の実施形態では、投与される化合物は、式I、式II、式III、式IV若しくは式V、又はそれらの薬理学的に許容される組成物、塩、アイソトープ類似体又はプロドラッグを含む化合物又は組成物から成る群より選択される。
特定の実施形態では、投与される化合物は、表1中に含まれる化合物、又はそれらの薬理学的に許容される組成物、塩、アイソトープ類似体又はプロドラッグから選択される。 一実施形態では、CDK4/6-複製依存的細胞は、造血幹細胞や前駆細胞(HSPC)である。
特定の態様では、化合物、方法及び組成物は、Rbポジティブ癌の治療を受けている被験体におけるCDK4/6-複製依存的正常細胞へのCDK4/6阻害の有害作用を制限する化学療法剤として提供され、この方法は、ここに記載される化合物を効果的量で投与することを含み、CDK4/6-複製依存的正常細胞の相当な部分は、化合物の抑制性効果の消失後、約24、30、36又は40時間未満内に、細胞サイクルに同時にリエントリーする。
特定の実施形態では、化合物のIC50 CDK4抑制濃度が、CDK2のIC50抑制濃度の少なくとも1500倍小さい。
特定の実施形態では、投与される化合物は、式I、式II、式III、式IV若しくは式V、又はそれらの薬理学的に許容される組成物、塩、アイソトープ類似体又はプロドラッグを含む化合物又は組成物から成る群より選択される。
特定の実施形態では、投与される化合物は、表1又は薬理学的に許容される組成物、又はそれらの塩、同位体的類似体又はそのプロドラッグを含む化合物から選択される。
一実施形態では、CDK4/6-複製依存的細胞は、造血幹細胞及び/又は前駆細胞(HSPC)である。
一実施形態では、投与された化合物は、IC50 CDK4抑制濃度が、CDK2のIC50抑制濃度の500倍以上小さい。
特定の実施形態では、CDK4/6-複製依存的正常細胞の相当な部分は、被験体の血液中の化合物の濃度レベルが治療有効濃度以下に下がった時点から約24、30、36又は40時間未満内に、同時に、細胞サイクルにリエントリーする。
特定の実施形態では、投与される化合物は、式I、式II、式III、式IV若しくは式V、又はそれらの薬理学的に許容される組成物、塩、アイソトープ類似体又はプロドラッグを含む化合物又は組成物から成る群より選択される。
特定の実施形態では、投与される化合物は、表1中に含まれる化合物、又はそれらの薬理学的に許容される組成物、塩、アイソトープ類似体又はプロドラッグから選択される。 一実施形態では、CDK4/6-複製依存的細胞は、造血幹細胞及び/又は前駆細胞(HSPC)である。
一実施形態では、CDK4/6複製依存的正常細胞は、腎臓上皮細胞である。
1)被験体が隙間の無い間隔の治療に曝露される状況において、より長時間作用するCDK4/6阻害物質の使用が、曝露中にCDK4/6-複製依存的正常細胞のサイクリングを禁止する状況。
2)連続的又は長期の治療計画において、CDK4/6-複製依存的正常細胞の長期のG1期停止は、ターゲットの癌の成長阻害の副作用であり、被験体が、複製遅れを制限するため、連続的計画における阻害剤ドーズと阻害剤ドーズの間、又は治療中のブレークとブレークの間で、CDK4/6-複製依存的正常細胞が治療計画の中断の後迅速に細胞サイクルにリエントリーするという利益を得て、治療の中断に際して、たとえば骨髄抑制等のさらなる正常細胞障害を、低下、限定又は改善する状況。
一実施形態では、ここに記載される化合物は、被験体は、Rbポジティブ癌の定期的な反復化学療法剤治療に曝露されるCDK4/6-複製依存的正常細胞サイクリングストラテジーに用いられる。
このサイクリングは、定期的な反復治療と定期的な反復治療との間で損害を受けた血液細胞系統を再生させるためのCDK4/6-複製依存的細胞を与え、長期CDK4/6阻害に関連するリスクを低減する。
G1-停止の状態と複製の状態との間のこのサイクリングは、PD0332991等のより長時間作用するCDK4/6阻害物質を用いた、限定的な、時間を空けた反復的な薬剤曝露においては実行可能ではなく、何故なら、CDK4/6阻害物質への次の曝露の前のCDK4/6-複製依存的細胞により、又は正常細胞が細胞サイクルへ進入すること及び治療中断の後損傷した組織又は細胞を再構成することへの遅れにより、長引いた化合物のG1期停止効果が、細胞サイクルへの顕著かつ有意義なリエントリーを禁止するからである。
一実施形態では、ここに記載される化合物の使用は、たとえばHSPC等のCDK4/6-複製依存的正常細胞によって細胞サイクルへの迅速のリエントリーを提供するため、細胞は、約40時間未満、36時間、30時間、28時間、24時間、18時間、16時間、14時間、12時間又はより少ない時間以内に、プレ治療ベースライン細胞サイクル活性に接近する。
一実施形態では、ここに記載される化合物の使用は、CDK4/6-複製依存的細胞によって細胞サイクルへの迅速のリエントリーを提供するため、細胞は、ここに記載される化合物の最後の投与から約40時間未満、36時間、30時間、28時間、24時間、18時間、16時間、14時間、12時間又はより少ない時間以内に、プレ治療ベースライン細胞サイクル活性に戻る。
一実施形態では、ここに記載される化合物の使用は、CDK4/6-複製依存的正常細胞によって細胞サイクルへの迅速のリエントリーを提供するため、細胞は、化合物の最後の投与から約40時間未満、36時間、30時間、28時間、24時間、18時間、16時間、14時間、12時間又はより少ない時間以内にプレ治療ベースライン細胞サイクル活性に接近する。
一実施形態では、ここに記載される化合物の使用は、CDK4/6-複製依存的正常細胞によって細胞サイクルへの迅速のリエントリーを提供するため、細胞は、被験体の血液の中に化合物の濃度レベルが治療有効濃度以下に下がった時点から約40時間未満、36時間、30時間、28時間、24時間、18時間、16時間、14時間、12時間又はより少ない時間以内にプレ治療ベースライン細胞サイクル活性に接近する。
一実施形態では、CDK4/6複製依存的正常細胞は、腎臓上皮細胞である。
一実施形態では、細胞サイクルへの迅速リエントリーは、同時発生的である。
一実施形態では、ここに記載される化合物の使用は、たとえばHSPC等のCDK4/6-複製依存的正常細胞によって細胞サイクルへの迅速のリエントリーを提供するため、一部の細胞は、細胞サイクル活性のレベルを示し、又は細胞サイクルに進入することができ、たとえば、化合物が、長期間、例えば、連続5日、連続7日、連続10日、連続14日、連続18日、連続21日、連続24日、連続28日、連続35日又はそれ以上の間、投与される治療計画等の、連続治療計画の間に増殖する。
一実施形態では、記載された方法に有用な化合物は、オフサイクル期間又はドラッグホリデイの必要無しに、たとえば、21、28、35日又はそれ以上等の連続期間、投与される。
一実施形態では、ここに記載される化合物の使用により、オフサイクル期間、ドラッグホリデイの必要が排除され、又は治療中の共同投与抗新生物薬化合物濃度が低減される。 本発明に従い、ここに記載される化合物は、Rbポジティブ増殖異常症を有する被験体への化学療法剤として、あらゆる治療スケジュール及び所定の治療過程と調和するあらゆるドーズで、投与することができる。
たとえば化合物は、1日に1回、1日に2回又は1日に3回投与されてもよい。
化合物は、1日おきに、又は3日目ごと、又は4日目ごと、又は5日目ごと、又は6日目ごと又は週に一度、投与されてもよい。
化合物は、一週おき又は毎月投与されてもよい。
本発明の一つの態様では、ここに開示される化合物は、有益的、追加的又は協力的効果のため、他の治療療法と組み合わせて有益に投与することができる。
一実施形態では、本発明の化合物/方法は、Rbポジティブ癌を治療するために、他の治療と組み合わせて用いられる。
第2の治療は、免疫療法であってもよい。
下記の更に詳細に論じられるように、化合物は、抗体、放射性薬剤、又は罹病又は異常増殖細胞に化合物を向けるその他のターゲッティング薬剤に接合されてもよい。
別の実施形態では、化合物は、他の製薬又は生物学的薬剤(たとえば抗体)と組み合わせて用いられることにより、複合又は共働アプローチで治療の有効性を増加させる。
一実施形態では、化合物を、典型的に不活化自動反応性T細胞で免疫化に関与するT細胞ワクチン投与で用いることにより、ここに記載されるRbポジティブ癌細胞集団を排除することができる。
別の実施形態では、化合物は、ここに記載される内因性T細胞上及びRbポジティブ癌細胞上の特定の抗原に同時に結合するようにデザインされる抗体である二重特異性T 細胞誘導(BiTE)と組み合わせて用いられ、2つのタイプの細胞を結合する。
MAbの一部は、癌細胞を破壊する免疫応答を刺激する。
B細胞によって自然に生成される抗体と同様に、これらのMAbは、癌細胞表面を「コーティング」し、免疫系によるその破壊を誘発する。
例えば、ベバシズマブは、腫瘍血管の発現を促進する腫瘍の微小環境において、腫瘍細胞及び他の細胞によって分泌されるタンパク質である血管内皮増殖因子(VEGF)をターゲットにする。
ベバシズマブに結合されれば、VEGFは、その細胞レセプタと相互作用することができず、新しい血液血管の成長に至るシグナリングを防止する。
同様に、セツキシマブ及びパニツムマブは上皮生長因子レセプター(EGFR)をターゲットにし、トラスツズマブはヒト上皮生長因子レセプタ2(HER-2)をターゲットにする。
細胞表面に生長因子レセプタを結合するMAbは、ターゲットのレセプタが通常の生育促進信号を送ることを防止する。それらは、アポトーシスを誘発し、免疫系を活性化して、腫瘍細胞を破壊する。
抗体は、癌細胞の表面上のその特定の抗原に接着し、細胞死滅物質は細胞に吸収される。
このように作用するFDA認可の複合MAbは、転移乳癌細胞を発現するHER-2に、細胞増殖を抑制する薬剤DM1を供給する、HER-2分子をターゲットにするアドトラスツズマブエムタンシンを含む。
二重特異性抗体(bsAb)又はキメラ抗原リセプター(CAR)を介して癌細胞を認識するために設計されるT細胞による免疫療法は、癌細胞の割出し及び非/遅い割出し亜細胞を除去すること可能性のあるアプローチである。
二重特異性抗体は、免疫エフェクター細胞の表面上のターゲット抗原及び活性化レセプタを同時に認識することにより、癌細胞を死滅させるために免疫エフェクター細胞を向け直す機会を提供する。
キメラ抗原リセプター設計T細胞は、MHC-非依存的方法で、特異的に腫瘍細胞を死滅させることができる。
ある実施形態では、化合物は、他の化学療法剤と組み合わせて被験体に投与されてもよい。
容易な場合は、ここに記載される化合物は、治療計画を単純化するため、他の化学療法剤として同時に投与されてもよい。
ある実施形態では、化合物及び他の化学療法剤は、単一の製剤で提供されてもよい。
一実施形態では、ここに記載される化合物の使用は、他の薬剤で治療計画に併用される。
この薬剤は、タモキシフェン、ミダゾラム、レトロゾール、ボルテゾミブ、アナストロゾール、ゴセレリン、mTOR阻害剤、PI3キナーゼ阻害剤、デュアルmTOR-PI3K阻害剤、MEK阻害剤、RAS阻害剤、ALK阻害剤、HSP阻害剤、(たとえば、HSP70及びHSP90阻害剤又はその組合せ)、BCL-2阻害剤から非限定的に選択される化学療法剤、アポトーシス誘発化合物、GSK690693(ペリホシン)(KRX-0401)GDC-0068、トリシリビン、AZD5363、ホオノキオール、PF-04691502及びミルテフォシン、MK-2206、を非限定的に含むAKT阻害剤、ニボルマブ、CT-011、MK-3475、BMS936558を非限定的に含むPD-1阻害剤、及びP406、ドビチニブ、クイザルチニブ(AC220)、アムバチニブ(MP-470)、タンデュチニブ(MLN518)(ENMD-2076)及びKW-2449、またはこれらの組合せを非限定的に含むAMP-514又はFLT-3阻害剤、と混合することができる。
P13キナーゼ阻害剤の非限定的な例としては、ワートマニン、デメトキシビリジン、ペリホシン、イデラリシブ、PX-866、IPI-145、BAY 80-6946、BEZ235、RP6503、TGR1202(RP5264)、MLN1117(INK1117)、ピクチリシブ、ブパルリシブ、SAR245408(XL147)、SAR245409(XL765)、パロミド529、ZSTK474、PWT33597、RP6530、CUDC-907及びAEZS-136.が挙げられる。
RAS阻害剤の非限定的な例としては、レオリシン及びsiG12Dローダーが挙げられる。
ALK阻害剤の非限定的な例としては、クリゾチニブ、AP26113及びLDK378が挙げられる。
HSP阻害剤の非限定的な例としては、ゲルダナマイシン又は17-N-アリルアミノ-17-デメトキシゲルダナマイシン(17AAG)及びラジシコールが挙げられる。
特定の実施形態では、ここに記載される化合物は、レトロゾールやタモキシフェンと組み合わせて投与される。
一実施形態では、ここに記載されるCDK4/6阻害物質は、(イマチニブメシル酸塩(Gleevac(登録商標)))イマチニブメシル酸塩(Gleevac(登録商標))、イマチニブメシル酸塩(Gleevac(登録商標);)ダサチニブ(Sprycel(登録商標))、ニロチニブ(Tasigna(登録商標))、ボスチニブ(Bosulif(登録商標))、トラスツズマブ(ハーセプチン(登録商標))、ペルツズマブ(PerjetaTM)、ラパチニブ(Tykerb(登録商標))、ゲフィチニブ(イレッサ(登録商標))、エルロチニブ(タルセバ(登録商標))、セツキシマブ(アービタックス(登録商標))、パニツムマブ(ベクティビックス(登録商標))、バンデタニブ(Caprelsa(登録商標))、ベムラフェニブ(Zelboraf(登録商標))、ボリノスタット(ゾリンザ(登録商標))、ロミデプシン(Istodax(登録商標))、ベキサロテン(Tagretin(登録商標))、アリトレチノイン(Panretin(登録商標))、トレチノイン(Vesanoid(登録商標))、カーフィルゾミブ(KyprolisTM)、プララトレキサート(Folotyn(登録商標))、ベバシズマブ(Avastin(登録商標))、ジフ-アフリバーセプト(Zaltrap(登録商標))、ソラフェニブ(Nexavar(登録商標))、スニチニブ(Sutent(登録商標))、パゾパニブ(Votrient(登録商標))、レゴラフェニブ(Stivarga(登録商標))及びカボザンチニブ(Cometriq(登録商標))から、非限定的な例として選択される化学療法剤と併用してもよい。
適切な化学療法剤の非限定的な例としては、放射性分子、細胞障害抗体又は細胞毒性薬剤と呼ばれる毒物が挙げられ、これには細胞、薬剤及びリポソームの生存度に有害なあらゆる薬剤又は化学療法剤化合物を含む他のベシクルを含む。
ビンクリスチン(Oncovin(登録商標))又はリポソーム型ビンクリスチン(Marqibo(登録商標))、ダウノルビシン(ダウノマイシン又はCerubidine(登録商標))又はドキソルビシン(アドリアマイシン(登録商標))、シタラビン(cytosine arabinoside、ara-C又はCytosar(登録商標))、エルアスパラギナーゼ(Elspar(登録商標))又はPEG-L-アスパラギナーゼ(pegaspargase又はOncaspar(登録商標))、エトポシド(VP-16)、テニポシド(Vumon(登録商標))、6‐メルカプトプリン(6-MP又はPurinethol(登録商標))、メトトレキセート、シクロホスファミド(Cytoxan(登録商標))、プレドニソン、デキサメタゾン(Decadron)、イマチニブ(G1eevec(登録商標))、ダサチニブ(Sprycel(登録商標))、ニロチニブ(Tasigna(登録商標))、ボスチニブ(Bosulif(登録商標))及びポナチニブ(Iclusig(登録商標))。
ある実施形態では、より高用量(化学療法剤ドーズ強度を上昇)又はより高頻度で(化学療法剤ドーズ密度を上昇)他の化学療法剤を投与できるよう、化合物を選択して被験体に投与してもよい。
高ドーズ密度の化学療法剤は、標準的な化学療法剤治療計画において、薬剤を、治療間の時間を短くして与える化学療法剤治療計画である。
化学療法剤用量強度ないしドーズ強度とは、単位時間当たり投与される化学療法剤の単位ドーズを示す。
投与されるドーズ、投与の時間的間隔、又はその両方を変えることにより、ドーズ強度を増加又は減少させることができる。
ここに記載した半減期の短い選択的化合物の投与と、例えば影響を受けた細胞が成長停止状態にはない時点で造血成長因子のタイムリーな投与を行う本発明の方法とを組み合わせることにより、成長因子の量を低減して望ましい治療利益を達成しつつ不必要な副作用を最小にすることが可能となる。
したがって、この実施形態では、抗新生物性治療計画において、ここに記載される選択的化合物を使用することにより、被験体が受ける成長因子の量を減らすことが可能になり、何故なら、ターゲットの造血細胞は、他のCDK4/6阻害物質(たとえばPD0332991)の場合よりも迅速に細胞サイクルにリエントリーするだろうからである。
さらに、ここに記載される化合物を用いてG1期停止後に迅速な細胞サイクルにリエントリーすることにより、造血細胞系の再結合を援助し成長因子効果を最大にする、すなわち、成長因子が最も有効である時に、造血成長因子を投与する時間を定める能力を与える。
一実施形態では、HSPCに対する化合物の効果が消失するように、造血成長因子投与時間が定められる。
一実施形態では、成長因子は、ここに記載される化合物の投与の少なくとも20時間後に投与される。
所望の場合、ここに記載される化合物の複数のドーズが被験体に投与されてもよい。
代替的には、被験体は、ここに記載される化合物の単一ドーズを与えられてもよい。
例えば、CDK4/6-複製依存的正常細胞がG1期停止となるように、化合物は投与されてもよく、ここでは化合物のG1-停止効果が迅速に消失するため、大多数の正常細胞は、細胞サイクルにリエントリーし、曝露の直後に、例えば約24-48時間以下以内に、複製が可能になり、化合物の次の投与まで複製が継続する。
一実施形態では、化合物の投与により、G1期停止間のCDK4/6-複製依存的正常細胞のサイクリング及び細胞サイクルへのリエントリーが可能となり、反復的なドーズ治療計画、たとえば長期反復ドーズ治療計画に適合するようになる。
ある実施形態では、CDK4/6-複製依存的正常細胞は、ここに記載される化合物の複数回の限定的に時間を空けた投与により、より長い期間、たとえば数時間、数日、数週および/または数月にわたって、停止することができる。
化合物抑制性菌体内効果の消失に際して、CDK4/6-複製依存的正常細胞、たとえばHSPC等が細胞サイクルへ迅速にリエントリーするため、細胞は、たとえばPD0332991等のG1期停止プロファイルが長いCDK4/6阻害物質の場合よりも迅速に、細胞系統を再構成することができる。
適切な場合、経口、局所、鼻腔内、吸入、静脈、又はあらゆる他の望ましい投与形態のために、小分子を調製することができる。
ここに記載される方法の中に有用な化合物は、CDK4及びCDK6の少なくとも一つを選択的に抑制し又はRbポジティブ癌の細胞複製の阻害を通して抑制する選択的CDK4/6阻害物質化合物である。
一実施形態では、CDK4/6阻害物質は、PD0332991よりも、少なくとも約10倍以上有効である(すなわち、少なくとも10倍以上低いCDK4/CycD1リン酸化アッセイにおける、IC50を有する)。
ここに記載される化合物の使用は、CDK4/6依存的細胞に選択的G1期停止を誘発することができる(例えば、細胞ベースのインビトロアッセイで測定した場合に)。
一実施形態では、CDK4/6阻害物質は、G2/M期及びS期でCDK4/6依存的細胞のパーセンテージを低減させつつ、G1期にCDK4/6依存的細胞のパーセンテージを上昇させることができる。
細胞の集団の細胞段階を評価する方法は、当該技術分野で知られており(たとえば、米国特許出願公開番号第2002/0224522号を参照)、これは、血球計算分析顕微鏡分析、勾配遠心、湿式粉砕、免疫蛍光法を含む蛍光技術及びこれらの組合せを含む。
血球計算技術は、DNA結合染料等の、標識剤又は染色法に細胞を露出することを含み、例えば、PI、及びフローサイトメトリーによって細胞DNA含有率を分析することが含まれる。
免疫蛍光法は、たとえばチミジン類似体(例えば、5-ブロモ-2-デオキシウリジン(BrdU)又はヨウ化デオキシウリジン)等の特定の細胞サイクル指標を、蛍光抗体で検出することを含む。
ある実施形態では、ここに記載される化合物の使用により、特に、CDK2等、CDK4及び又はCDK6以外のキナーゼの阻害に関連したオフターゲット効果が低減され又は実質的に無くなるが、それは、ここに記載される化合物は、CDK2に対して弱い阻害剤(例えば>1μM IC50)であるからである。
さらに、CDK4/6に対する高い選択性のため、ここに記載される化合物の使用は、CDK4/6-非依存的細胞の細胞サイクル進行の停止を誘発してはならない。
さらに、G1期停止効果の短期の過渡的性質のため、CDK4/6-複製依存的正常細胞は、PD0332991の使用が提供する場合に比べて比較的より迅速に細胞サイクルにリエントリーし、その結果、化学療法剤の治療と治療の間でのHSPCの複製能力のため、一実施形態では、長期治療計画の間の血液欠乏のリスクが減少する。
付加的な実施形態では、ここに開示される化合物は、自己免疫異常症をターゲットにする治療薬剤と組み合わせて、有益に投与されてもよい。
一実施形態では、ここに記載される目的のための活性化合物の活性は、罹病又は異常増殖している細胞をターゲットにする又は活性、デリバリー、薬物動態学又は他の有益な特性を改良する薬剤への接合を通して、増大させてもよい。
たとえば、化合物は抗体-薬剤接合体(ADC)として投与されてもよい。
特定の実施形態では、ここに記載される選択化合物は、抗体又は抗体フラグメントとの接合又は組合せで投与されてもよい。
抗体のフラグメントが、化学又は遺伝子のメカニズムによって生成されてもよい。
抗体フラグメントは、フラグメントに結合した抗原であってもよい。
例えば、フラグメントを結合している抗原は、Fab、Fab’、(Fab’)2又はFvから選択されてもよい。
抗体フラグメントは、Fabであってもよい。
一価のF(ab)フラグメントは、1つの抗原結合部位を有する。
抗体は二価の(Fab’)2フラグメントでもよく、これはS‐S結合によって結合される領域を結合する2つの抗原を有する。
F(ab’)2フラグメントへの還元により、他の分子への接合のために有用なフリーなスルフヒドリル基を有する2つの一価のFab’フラグメントを生成する。
ここに記載される選択化合物は、Fvフラグメントとの接合又は組合せで投与されてもよい。
Fvフラグメントは、IgG及びIgMクラス抗体の酵素の分割から製造される最も小さなフラグメントである。
Fvフラグメントは、VH及びVC領域から製造される抗原結合部位を有するが、それらはCH1及びCL領域を有しない。
VH及びVL鎖は、非共有結合的相互作用によって、Fvフラグメント内に一緒に保持される。
一実施形態では、ここに記載される選択化合物は、ScFv、ドメイン抗体、二重特異性抗体、三重特異性抗体、四重特異性抗体、ビス-scFv、低分子化抗体、Fab2又はFab3抗体フラグメントから成る群より選択される抗体フラグメントと組み合わせて投与されてもよい。
一実施形態では、抗体フラグメントはScFvである。
遺伝子工学方法は、柔軟なペプチドに結合されるVH及びVLドメインを含むFvタイプフラグメントである単一鎖状可変フラグメント(ScFv)の生産を可能にする。
リンカーが少なくとも12残留基の長さの場合、ScFvフラグメントは一次単一遺伝子性である。
V-ドメイン及びリンカー長の配向を操作することにより、生成するFv分子リンカー形態を変えることができ、これは3~11残留基の長さに生成したscFv分子であり、官能性Fvドメインに折り畳むことができない。
二価二重特異性抗体を生成するために、これらの分子は、第2のscFv分子と結合されてもよい。
リンカー長が3残留基より短い場合、scFv分子は三重特異性抗体又は四重特異性抗体に関連づけられる。
一実施形態では、抗体フラグメントは三重特異性抗体である。
一実施形態では、抗体フラグメントは四重特異性抗体である。
多価scFvsは、抗体フラグメントのオフ率を低減する2つのさらなるターゲット抗原と結合することにより、それらの標的抗原に対して、その一価の対応物よりも大きな官能性結合親和力を有する。
一実施形態では、抗体フラグメントは低分子化抗体である。
低分子化抗体は、集合して二価ダイマーになるscFv-CH3融合タンパクである。 一実施形態では、抗体フラグメントはビス-scFvフラグメントである。
ビス-scFvフラグメントは二重特異性である。
2つの異なる可変ドメインを有する小型化されたScFvフラグメントを生成することにより、これらのビス-scFv分子が、2つの異なるエピトープに同時に結合することが可能になる。
一実施形態では、ここに記載される選択化合物は、二重特異性ダイマー(Fab2)又は三重特異性ダイマー(Fab3)との接合又は組合せで投与される。
また、遺伝学的方法を用いて、二重特異性Fabダイマー(Fab2)及び三重特異性Fabトリマー(Fab3)を生成する。
これらの抗体フラグメントは、一度に2つ(Fab2)又は3つ(Fab3)の異なる抗原を結合することができる。
rIgG抗体フラグメントとは、還元IgG(75,000ダルトン)又は半IgGのことをいう。
それは、ヒンジ領域S‐S結合だけを選択的に還元した生成物である。
いくつかのS‐S結合がIgGの中に発生するが、ヒンジ領域中S‐S結合は、最もアクセス容易で、特に2-メルカプトエチルアミン(2-MEA)等のマイルドな還元剤で最も容易に還元可能である。
半IgGは、抗体固定化又は酵素標識の接合のためのターゲット可能な曝露ヒンジ領域スルフヒドリル基をターゲットにする目的で、頻繁に調製される。
Rbポジティブ癌細胞に対して有用であるあらゆる放射性同位体は、接合体に取り込まれてもよく、たとえば、非限定的に、131I、123I、192Ir、32P、90Sr、198Au、226Ra、90Y、241Am、252Cf、60Co又は137Csが挙げられる。
チオエーテルにリンクされたT-DM1は、二硫化物リンカーバージョンに対する血清安定性を上昇させ、エンドソームの分解を経ると考えられ、細胞毒薬剤の細胞内発散をもたらし、それにより、有効性及び許容度を改良する。Barginear,M.F. and Budman,D.R.,TrastuzumabDM1、A review of the novel immune-conjugate for HER2-overexpressing breast cancer, The Open Breast Cancer Journal,1 :25-30,2009を参照。
Casi, G. and Neri, D., Antibody-drug conjugates: basic concepts, examples and future perspectives, J. Control Release 161(2):422-428, 2012, Chari, R.V., Targeted cancer therapy: conferring specificity to cytotoxic drugs, Acc. Chem. Rev., 41(1):98-107, 2008, Sapra, P. and Shor, B., Monoclonal antibody-based therapies in cancer: advances and challenges, Pharmacol. Ther., 138(3):452-69, 2013, Schliemann, C. and Neri, D., Antibody-based targeting of the tumor vasculature, Biochim. Biophys. Acta., 1776(2): 175-92, 2007, Sun, Y., Yu, F., and Sun, B.W., Antibody-drug conjugates as targeted cancer therapeutics, Yao Xue Xue Bao, 44(9):943-52, 2009, Teicher, B.A., and Chari, R.V., Antibody conjugate therapeutics: challenges and potential, Clin. Cancer Res., 17(20):6389-97, 2011, Firer, M.A., and Gellerman, G.J., Targeted drug delivery for cancer therapy: the other side of antibodies, J. Hematol. Oncol, 5:70, 2012, Vlachakis, D. and Kossida, S., Antibody Drug Conjugate bioinformatics: drug delivery through the letterbox, Comput. Math. Methods Med., 2013; 2013:282398, Epub 2013 Jun 19, Lambert, J.M., Drug-conjugated antibodies for the treatment of cancer, Br. J. Clin. Pharmacol, 76(2):248-62, 2013, Concalves, A., Tredan, O., Villanueva, C. and Dumontet, C, Antibody-drug conjugates in oncology: from the concept to trastuzumab emtansine (T-DM1), Bull. Cancer, 99(12): 1183- 1191 , 2012, Newland, AM, Brentuximab vedotin: a CD-30-directed antibody-cytotoxic drug conjugate, Pharmacotherapy, 33(1):93-104, 2013, Lopus, M., Antibody-DMl conjugates as cancer therapeutics, Cancer Lett., 307(2): 113-118, 2011, Chu, Y.W. and Poison, A., Antibody-drug conjugates for the treatment of B-cell non-Hodgkin's lymphoma and leukemia, Future Oncol, 9(3):355-368, 2013, Bertholjotti, I., Antibody-drug conjugate--a new age for personalized cancer treatment, Chimia, 65(9): 746-748, 2011, Vincent, K.J., and Zurini, M., Current strategies in antibody engineering: Fc engineering and pH-dependent antigen binding, bispecific antibodies and antibody drug conjugates, Biotechnol. J., 7(12): 1444-1450, 2012, Haeuw, J.F., Caussanel, V., and Beck, A., Immunoconjugates, drug-armed antibodies to fight against cancer, Med. Sci., 25(12): 1046-1052, 2009 及び Govindan, S.V., and Goldenberg, D.M., Designing immunoconjugates for cancer therapy, Expert Opin. Biol. Ther., 12(7):873-890, 2012。
ここに記載される活性化合物、または、その塩、同位体的類似体、又は、プロドラッグは、所望された治療結果を達成するあらゆる適切なアプローチを用いているホストに、有効量で投与することができる。
投与される活性化合物の量及びタイミングは、無論、治療されているホスト、監督する医学専門家の指導、曝露の時間経過、投与の方法、特定の活性化合物の薬物動態特性、及び処方している医師の判断によって定められる。
したがって、ホストの多様性のため、以下の用量はガイドラインであり、そして、医師は、ホストに適切であると考える治療を達成するため、化合物のドーズを漸増することができる。
要求される治療の程度の考慮において、医師は、ホストの年齢及び体重、既存の疾病の存在、ならびに他の疾病の存在等、様々な因子のバランスをとることができる。
ここに記載されるあらゆる活性化合物の治療的に有効な用量は、患者のコンディション、大きさ及び年齢ならびにデリバリーのルートに応じて、健康管理開業医によって決定される。
1つの非限定された実施形態では、約0.1~約200mg/kgの用量が、治療有効性を有しており、この全ての重量が、活性化合物の重量に基づいて計算され、それは塩が用いられる場合を含んでいる。
ある実施形態では、約10mg/kg~約50mg/kgの用量を、経口薬投与のために用いることができる。
典型的には、約0.5mg/kg~5mg/kgの用量を、筋肉内投与のために用いることができる。
経口薬用量の形態は、活性材料のあらゆる適切な量を含むことができ、たとえば錠剤当たり5mgから50、100、200又は500mgまでを含み、又はその他の固体投与形態であってもよい。
また、ある実施形態では、化合物又は塩は、リポソーム型懸濁液として、吸入、静注、又は筋内に投与することができる。
本発明の中に開示される化合物は、たとえば経口薬又は静脈ルートで投与されるときに、良好な薬物動態及び薬力学特性を実証した。
溶液が要求される場合、水溶性化合物又は塩のために選択すべきキャリアは、水であってもよい。水溶性化合物又は塩に関して、有機ビヒクル、例えばグリセリン、プロピレングリコール、ポリエチレングリコール又はそれらの混合物が、適切となり得る。後者の例では、有機ビヒクルは、相当量の水を含むことができる。溶液のいずれの場合でも、当該技術分野で人々に知られている適切な方法で、その後に殺菌ができ、例示としては、0.22ミクロンフィルタによる濾過が挙げられる。殺菌に引き続き、溶液を、適切な容器、例えば発熱物質を除去されたガラスバイアルに分配することができる。その分配は、任意に無菌方法によりなされる。殺菌されたクロージャを、その後バイアル上に置くことができ、望ましい場合は、バイアルの内容物を凍結乾燥することができる。
さらに、ステアリン酸マグネシウム、ラウリル硫酸ナトリウム及びタルカムパウダー等の平滑剤は、目的物を錠剤にするために、しばしば非常に有用である。類似したタイプの固体の組成物は、柔らかい及び堅い充填ゼラチンカプセル内で、フィラーとして用いられてもよい。これに関連して、この材料は、ラクトース又はラクトースならびに高分子量ポリエチレングリコールも含んでいる。
特に有用な乳化剤には、ホスファチジルコリン類及びレシチンが含まれる。ここに提供されている付加的な実施形態は、ここに開示される活性化合物のリポソーム型製剤を含む。
合成
開示された化合物は、以下の汎用のスキームによって製造されてもよい。
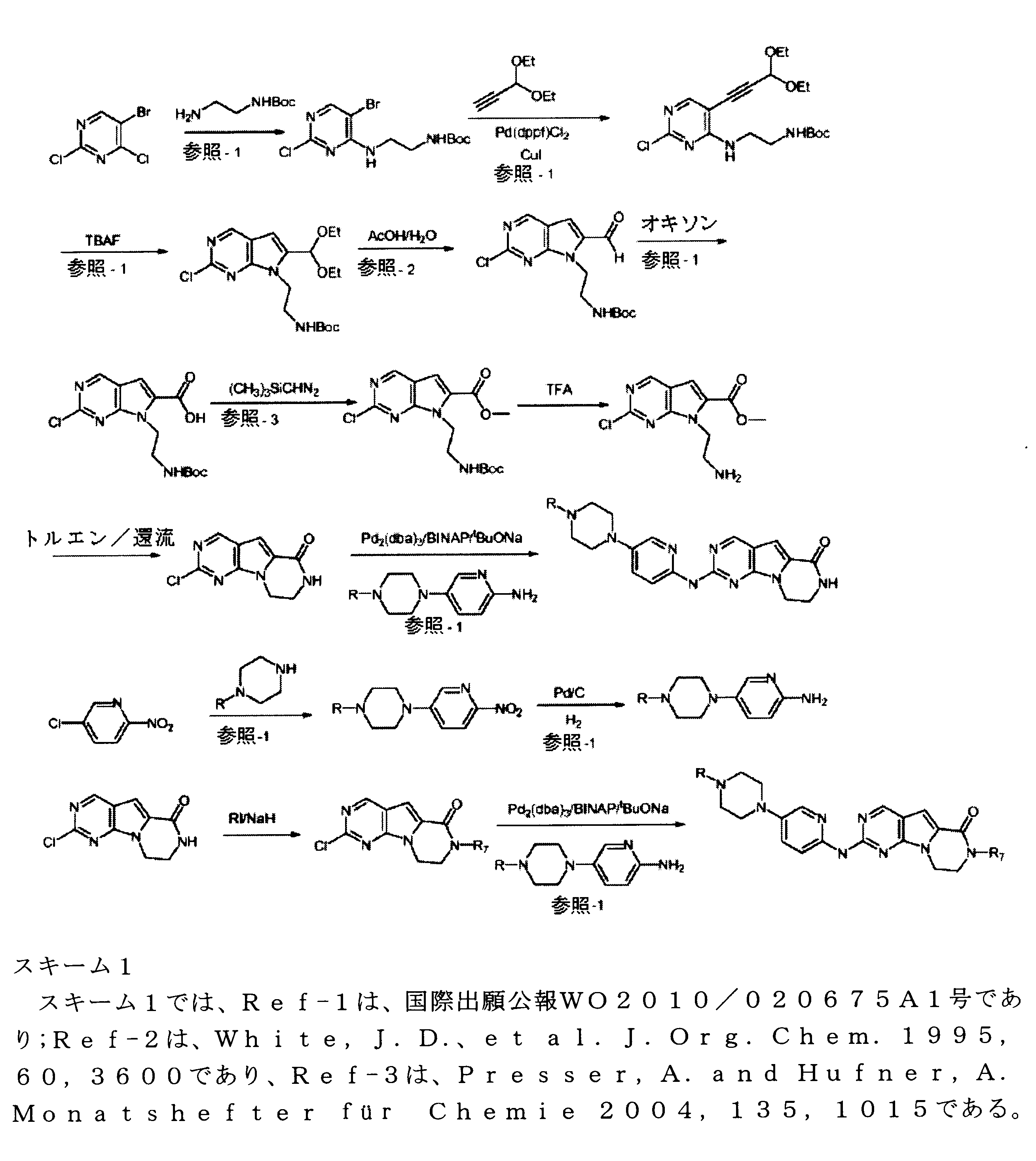
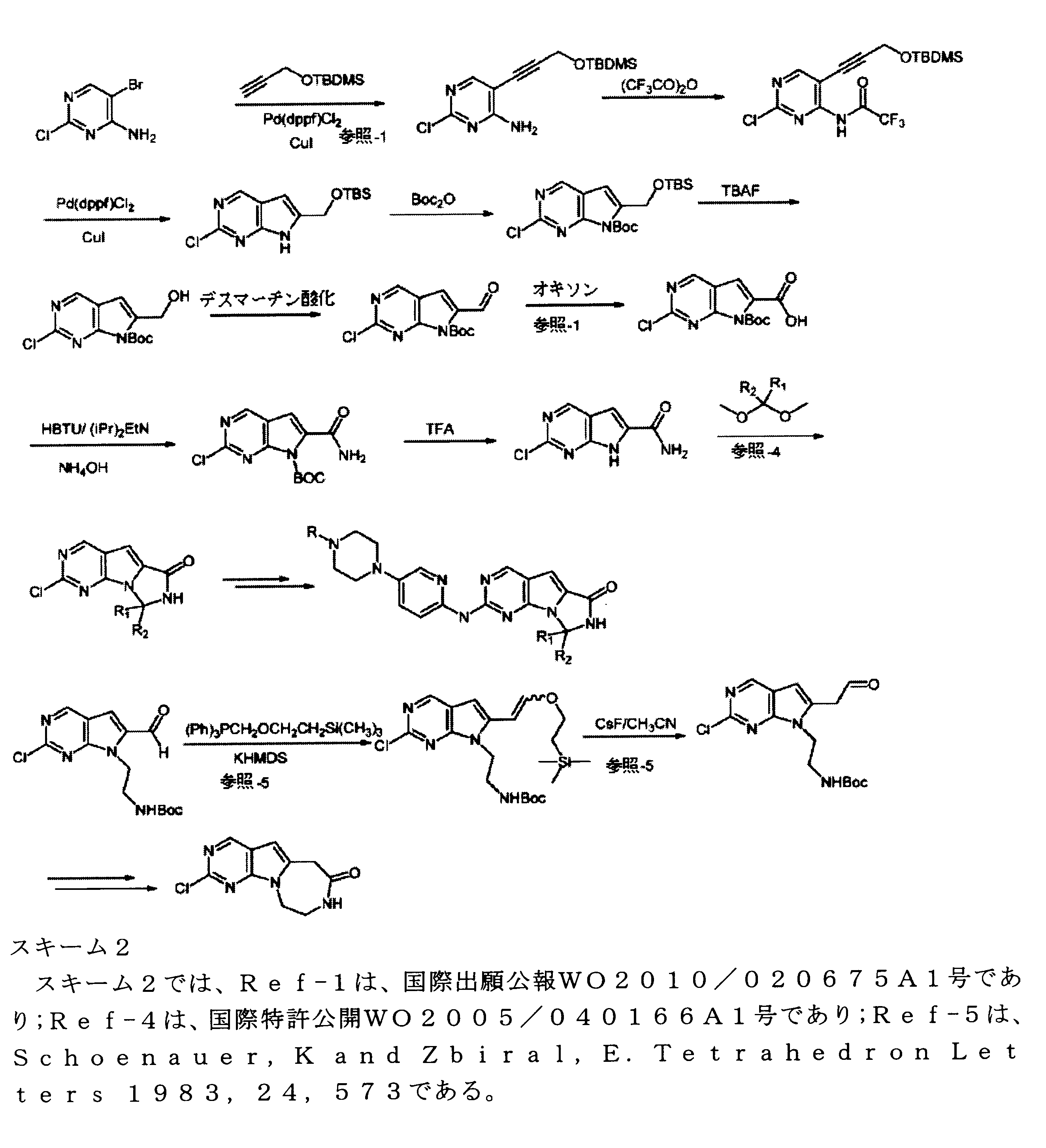
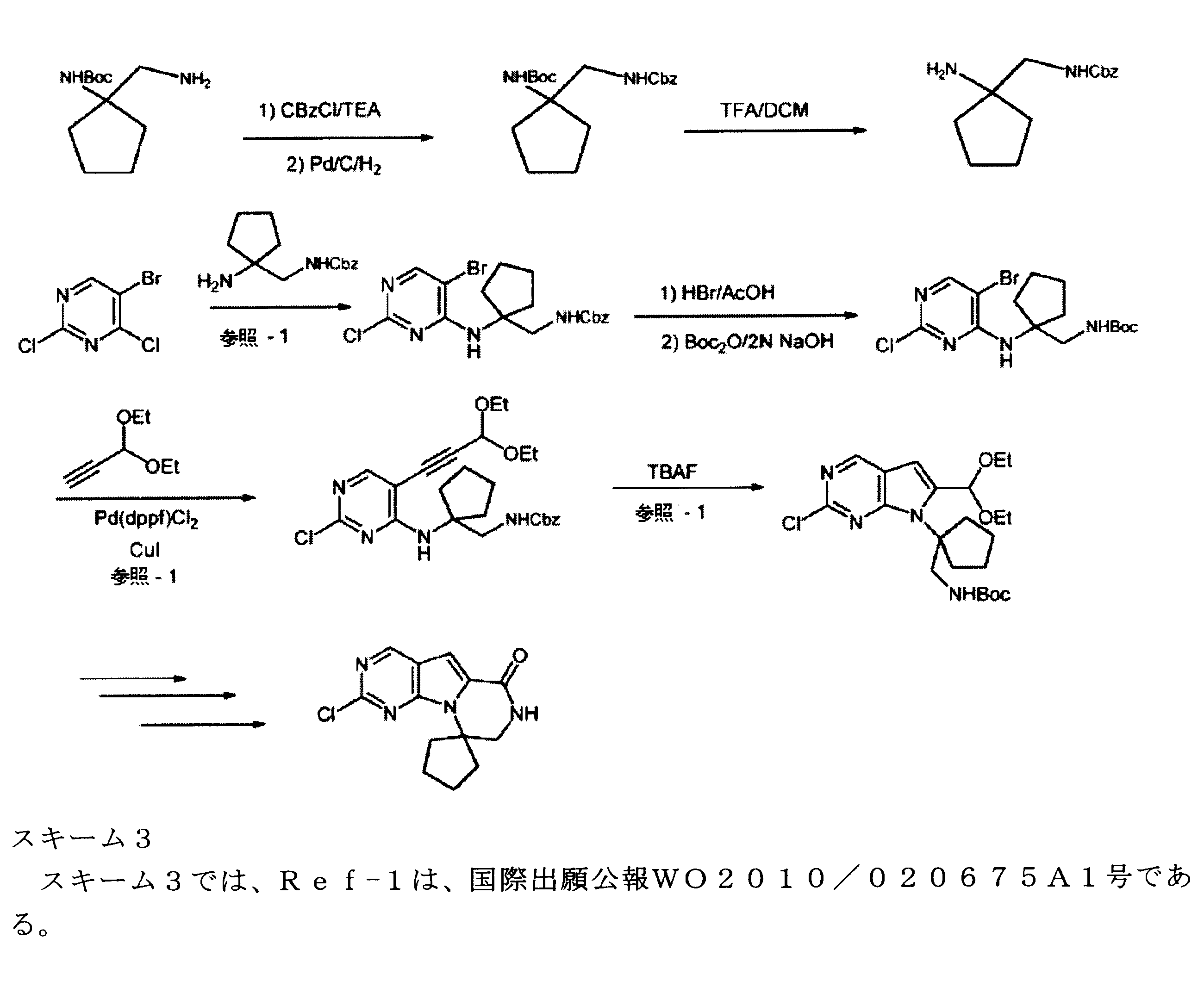
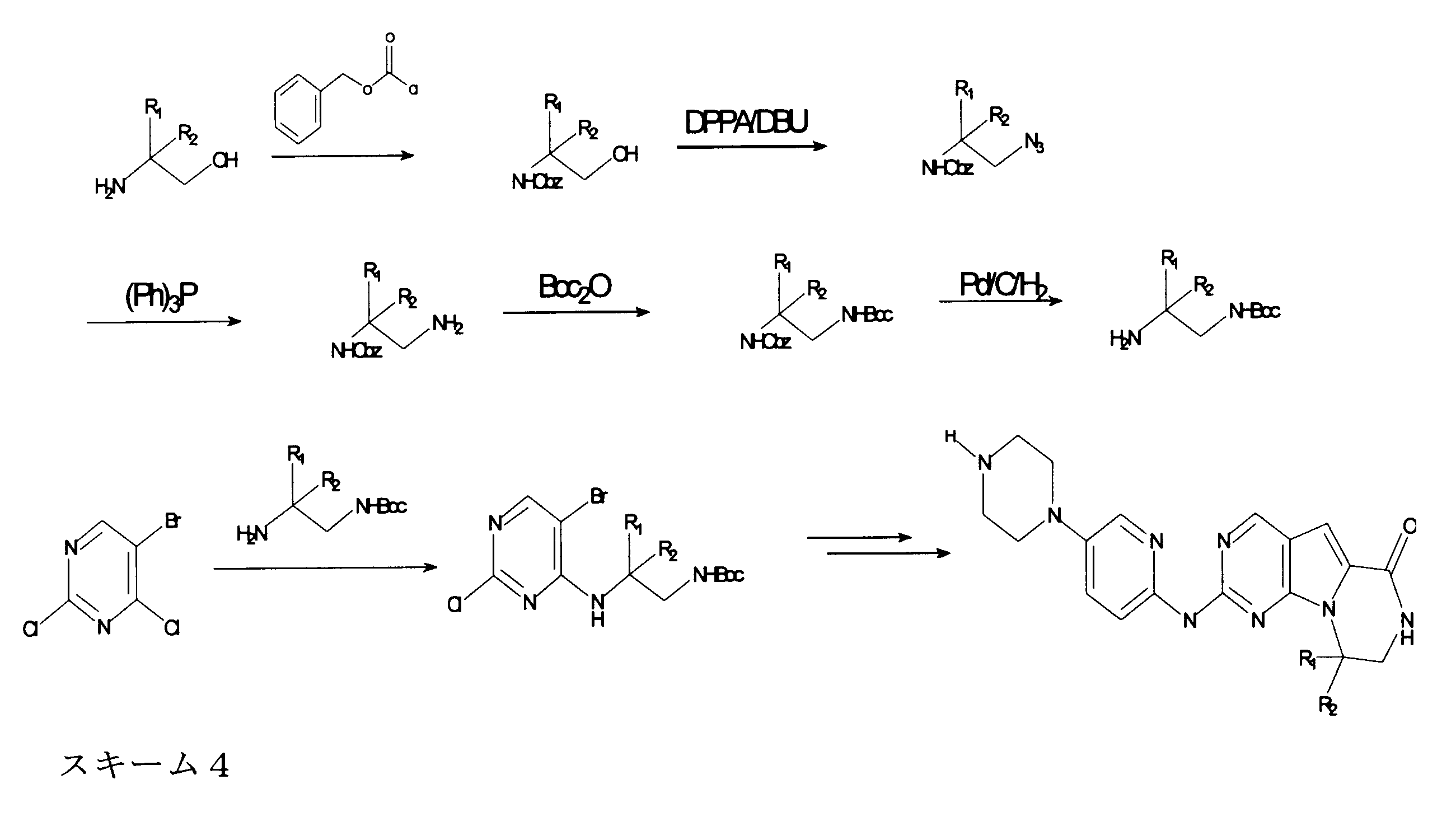
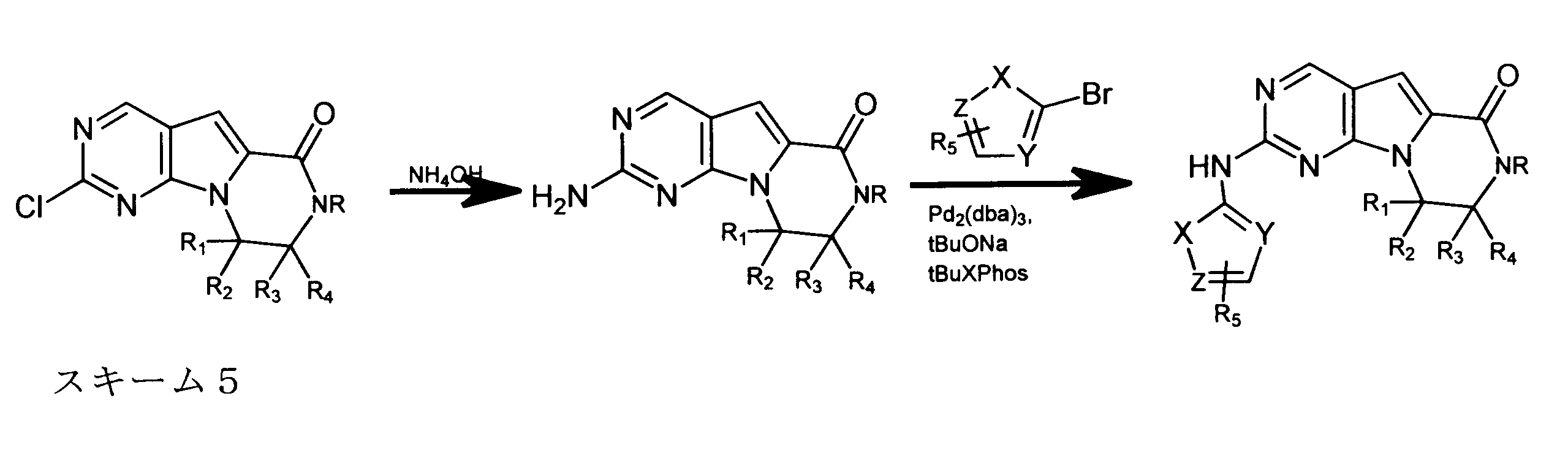
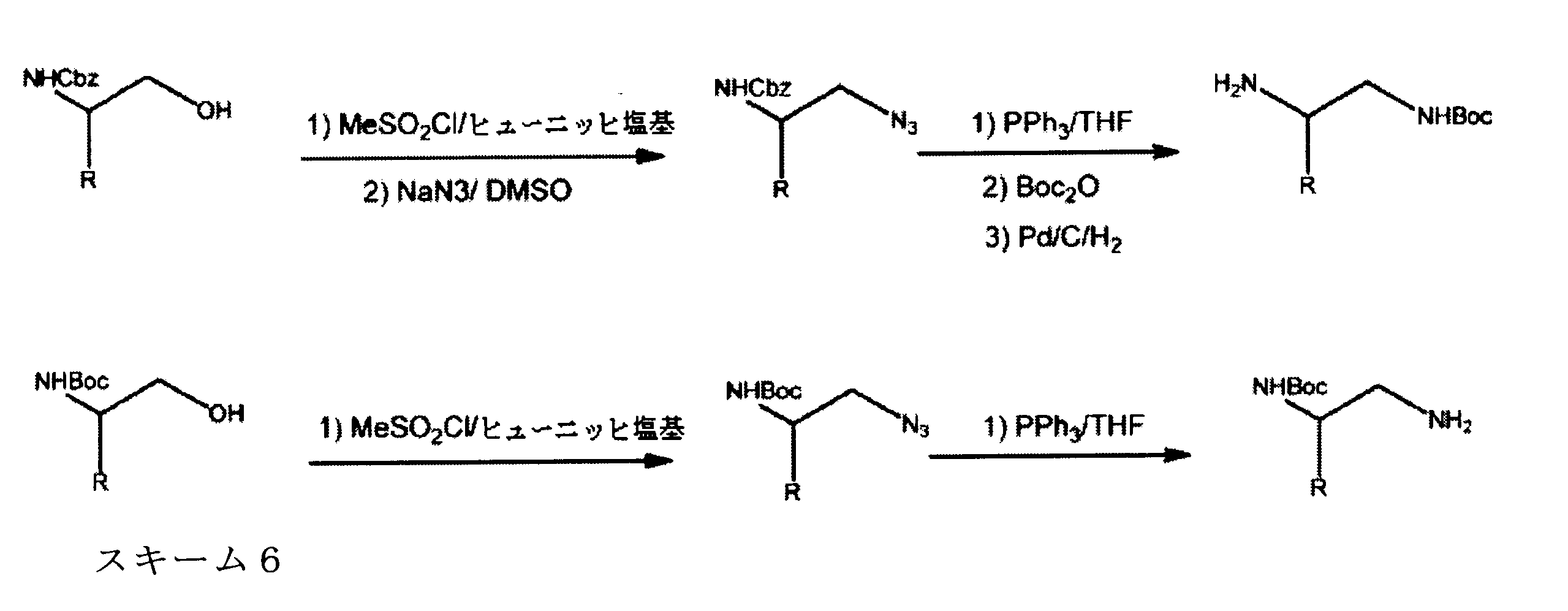
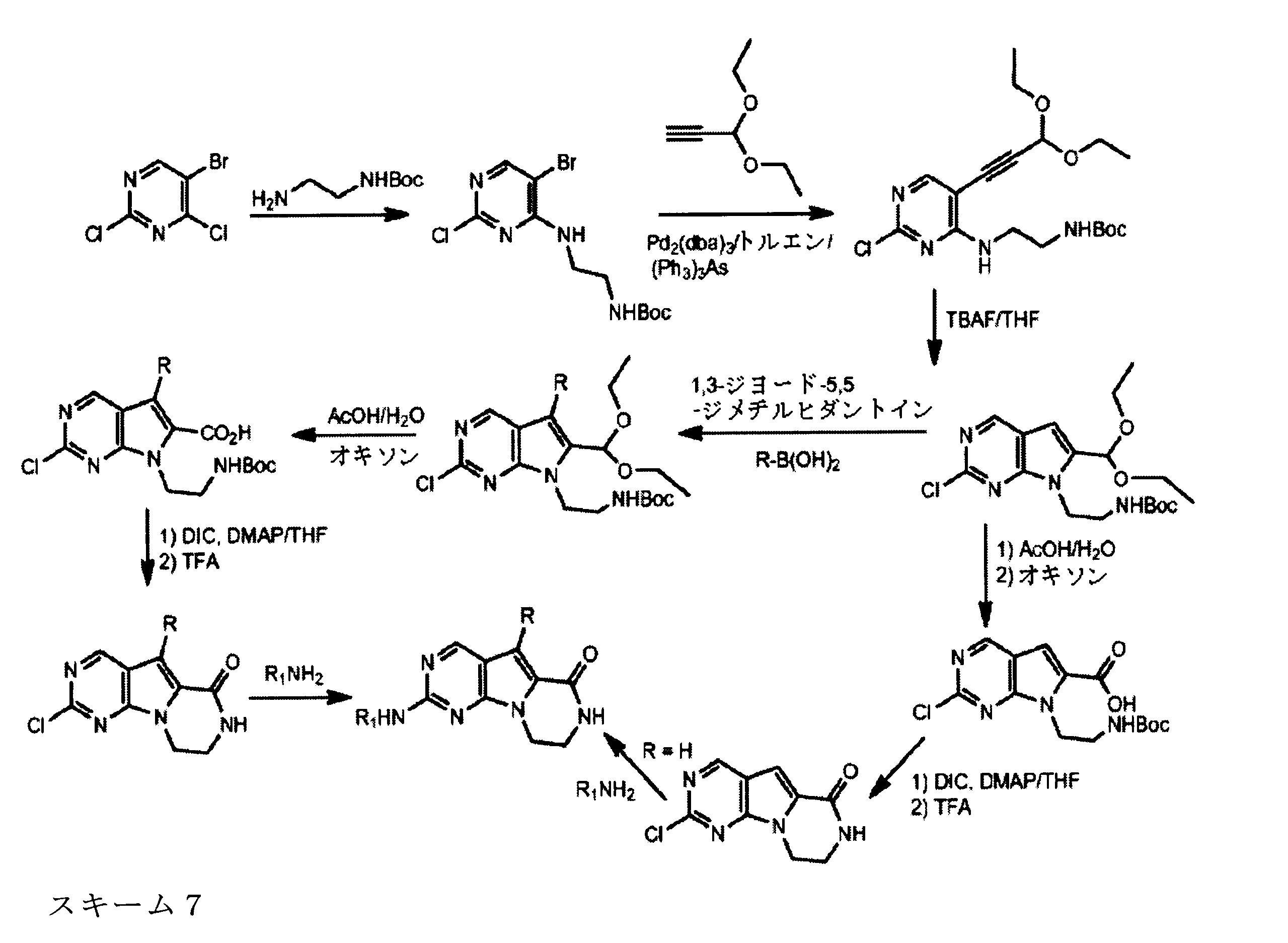
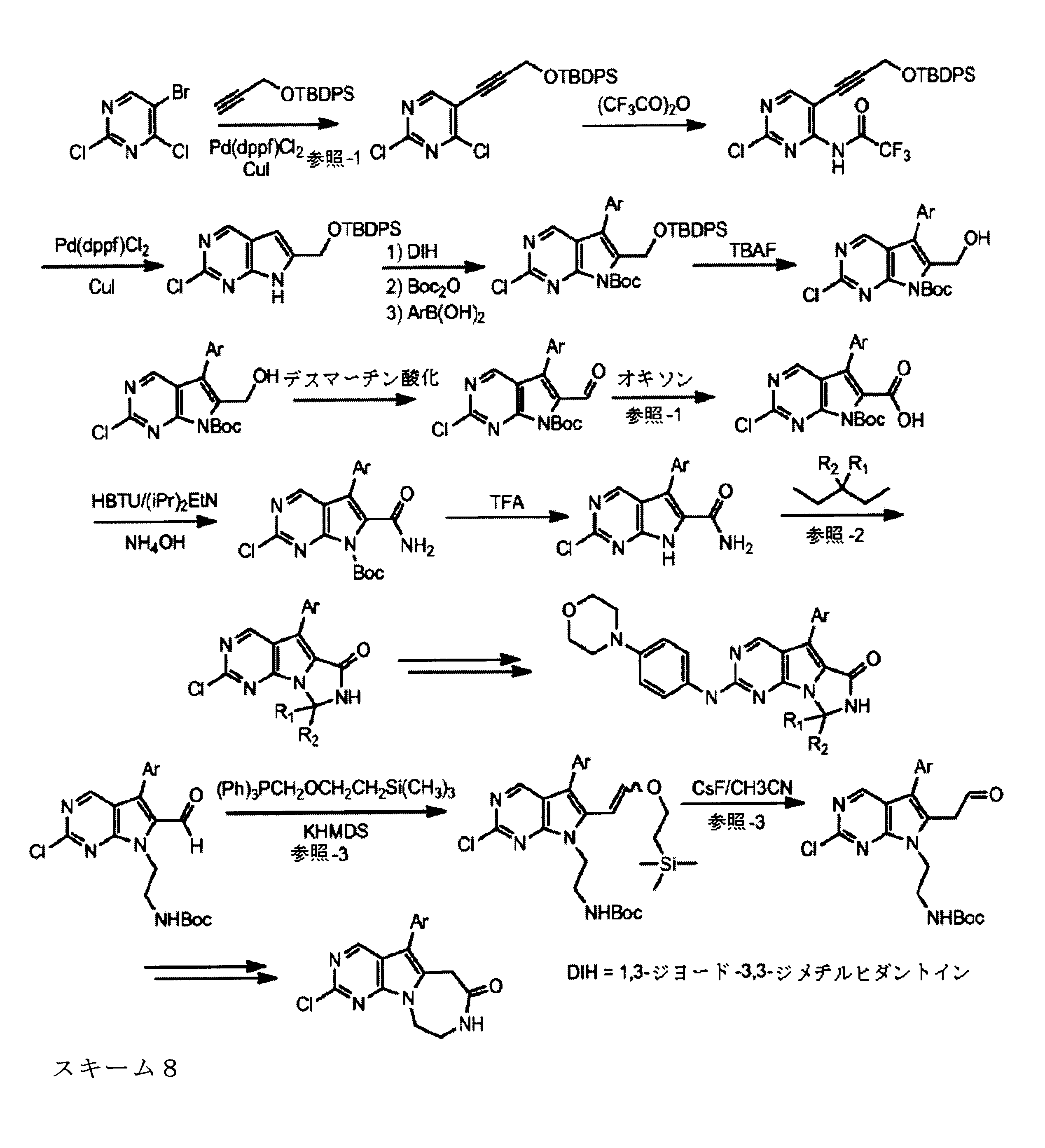
強力な酸無水物の非限定的な例としては、トリフルオロ酢酸無水物、トリブロモ酢酸無水物、トリクロロ酢酸酸無水物又は混合無水物が挙げられる。
脱水剤は、カルボジイミドベースの化合物であってもよく、この非限定的な例としては、DCC(N,N-ジシクロヘキシルカルボジイミド)、EDC(1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド又はDIC(N,N-ジイソプロピルカルボジイミド)が挙げられる。
N-保護基を除去するために、付加的なステップが必要となる場合があり、その方法は当業者に知られている。
代替的には、ピリミジン環に結合されるハロゲン部分は、あらゆる脱離基で置換することができ、この脱離基は、一次アミンによって置換可能であり、これにより、最終生産物のための中間体、たとえばBr、I、F、SMe、SO2Me、SOアルキル、SO2アルキル等をを生成する。
たとえば、Tavaresの国際特許出願PCT/US2013/037878号を参照のこと。
他のアミン中間体及び最終的なアミン化合物は、当業者により合成することができる。 この化学反応は、保護及び脱保護が可能な反応性官能性を含む試薬を用いることができ、本発明の時に当業者に知られていることが、理解されよう。
Greene, T.W. and Wuts, P.G.M., Greene's Protective Groups in Organic Synthesis, 4th edition, John Wiley and Sonsを参照のこと。

例えば、置換2-アミノピリミジンの特定の合成及びキャラクタリゼーションは、国際特許公報WO2012/061156に見出すことができる。
上記で調製された化合物T、Q、GG及びUは、以下に示すようにマススペクトル分析及びNMRによって特徴づけられた:
1HNMR (600 MHz, DMSO-d6) δ ppm 1.47 (br. s., 6 H) 1.72 (br. s., 2 H) 1.92 (br. s., 2 H) 2.77 (br. s., 3 H) 3.18 (br. s., 2 H) 3.46 (br. s., 2 H) 3.63 (br. s., 2 H) 3.66 (d, J=6.15 Hz, 2 H) 3.80 (br. s., 2 H) 7.25 (s, 1 H) 7.63 (br. s., 2 H) 7.94 (br. s., 1 H) 8.10 (br. s., 1 H) 8.39 (br. s., 1 H) 9.08 (br. s., 1 H) 11.59 (br. s., 1 H). LCMS (ESI) 447 (M + H).
1H NMR (600 MHz, DMSO-d6) dδ ppm 0.82 (d, J=7.32 Hz, 2 H) 1.08 - 1.37 (m, 3 H) 1.38 - 1.64 (m, 2 H) 1.71 (br. s., 1 H) 1.91 (br. s., 1 H) 2.80 (br. s., 1 H) 3.12 (s, 1 H) 3.41 (br. s., 4 H) 3.65 (br. s., 4 H) 4.09 (br. s., 1 H) 7.26 (s, 1 H) 7.52 - 7.74 (m, 2 H) 7.94 (br. s., 1 H) 8.13 (br. s., 1 H) 8.40 (br. s., 1 H) 9.09 (br. s., 1 H) 9.62 (br. s., 1 H) 11.71 (br. s., 1 H). LCMS ESI (M + H) 433.
1H NMR (600 MHz, DMSO-d6) dδ ppm 0.85 (br. s., 1 H) 1.17 - 1.39 (m, 7 H) 1.42 - 1.58 (m, 2 H) 1.67 - 1.84 (m,3 H) 1.88 - 2.02 (m, 1 H) 2.76 - 2.93 (m, 1 H) 3.07 - 3.22 (m, 1 H) 3.29 - 3.39 (m, 1 H) 3.41 - 3.61 (m, 4 H) 3.62 - 3.76 (m, 4 H) 3.78 - 3.88 (m, 1 H) 4.12 (br. s., 1H) 7.28 (s, 1 H) 7.60 - 7.76 (m, 2 H) 7.98 (s, 1 H) 8.13 (br. s., 1 H) 8.41 (s, 1 H) 9.10 (br. s., 1 H) 11.21 (br. s., 1 H) 11.54 (s, 1 H). LCMS ESI (M + H) 475.
1H NMR (600 MHz, DMSO-d6) dδ ppm 0.84 (t, J=7.61 Hz, 2 H) 1.13 - 1.39 (m, 4 H) 1.46 (d, J=14.05 Hz, 2 H) 1.64 - 1.99 (m, 6 H) 2.21 (br. s., 1 H) 2.66 - 2.89 (m, 2 H) 3.06 (br. s., 1 H) 3.24 - 3.36 (m, 1 H) 3.37 - 3.50 (m, 2 H) 3.56 - 3.72 (m, 2 H) 3.77 - 4.00 (m, 4 H) 4.02 - 4.19 (m, 2 H) 7.25 (s, 1 H) 7.50 - 7.75 (m, 2 H) 7.89 (d, J=2.93 Hz, 1 H) 8.14 (d, J=7.32 Hz, 1 H) 8.38 (br. s., 1 H) 9.06 (s, 1 H) 11.53 (br. s., 1 H). LCMS ESI (M +H) 517.
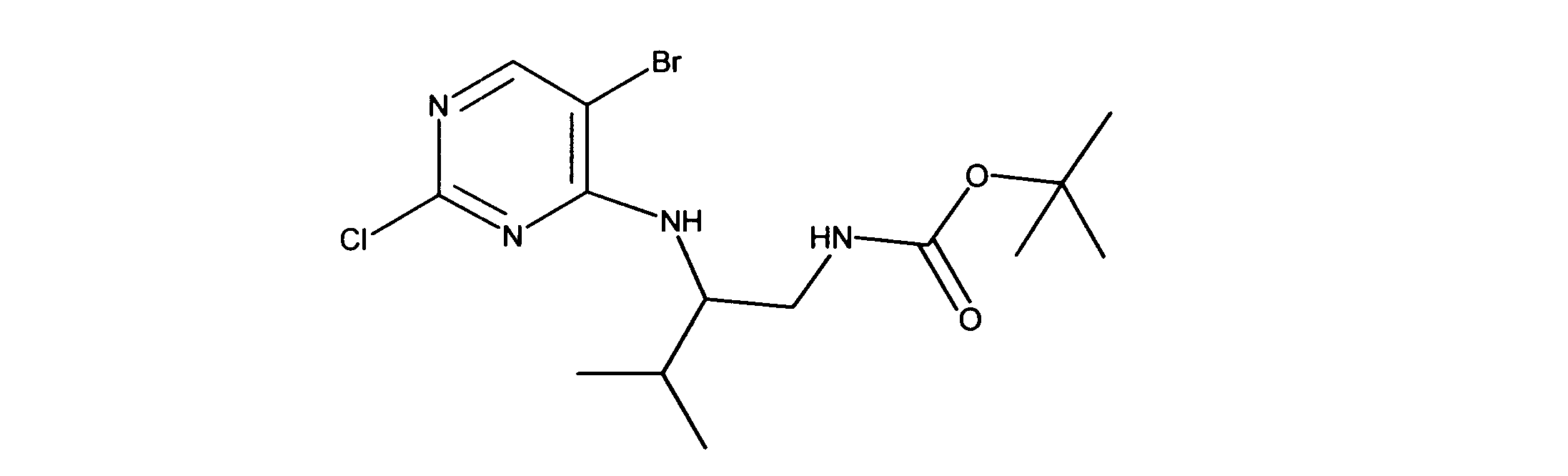
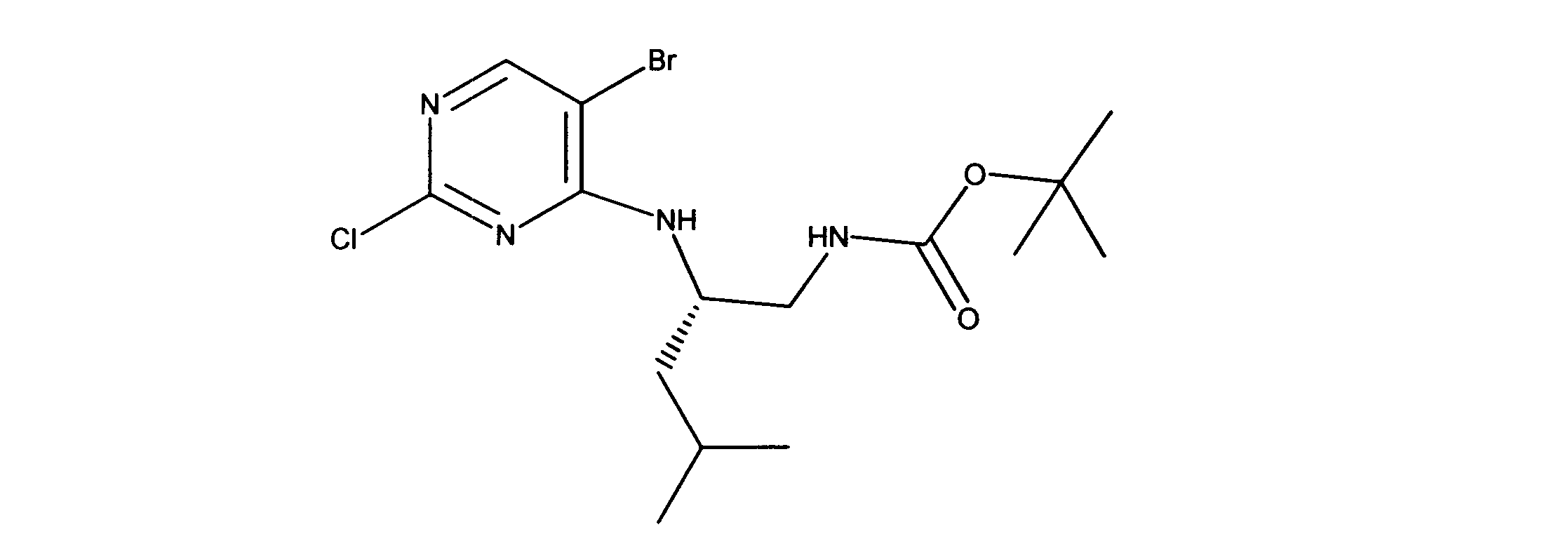
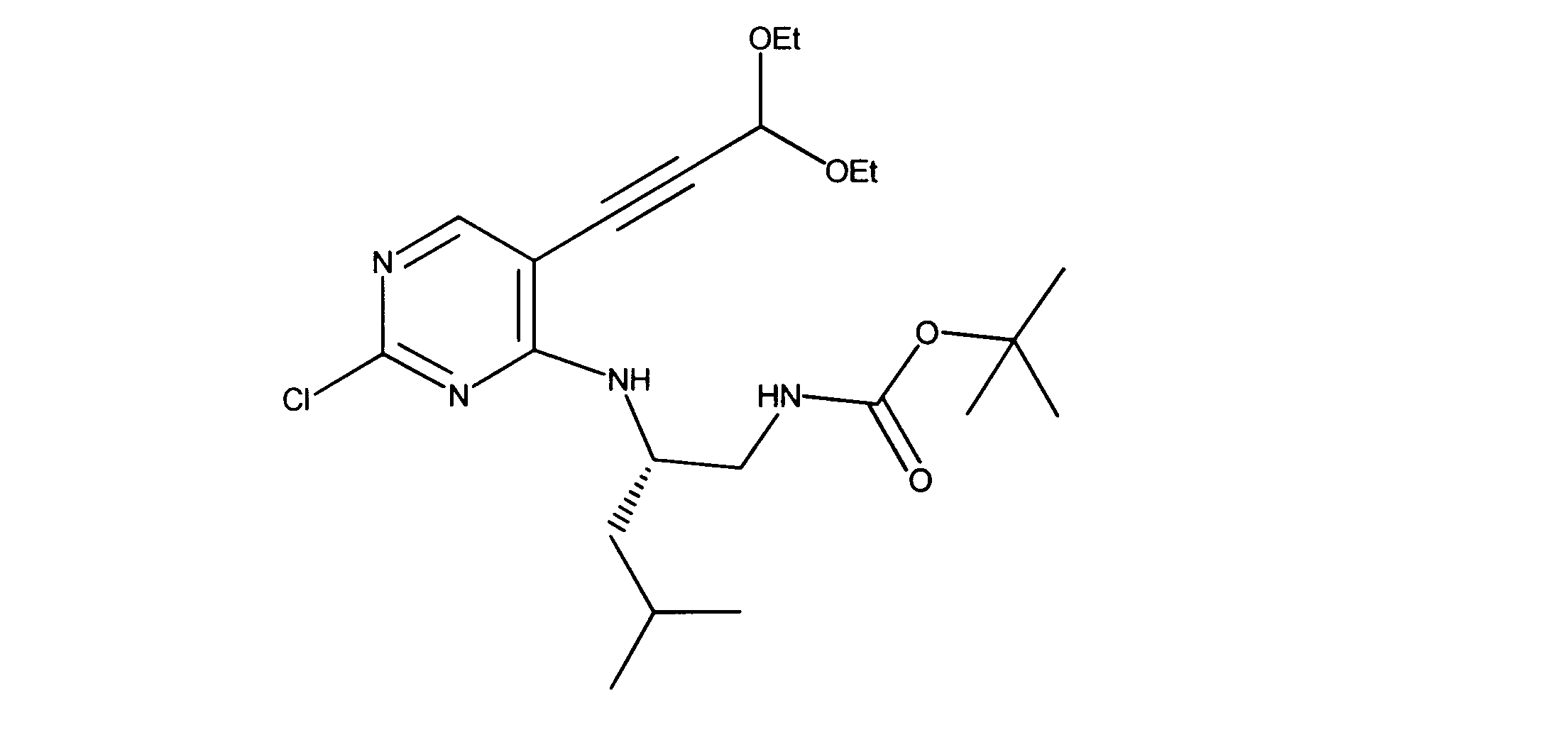
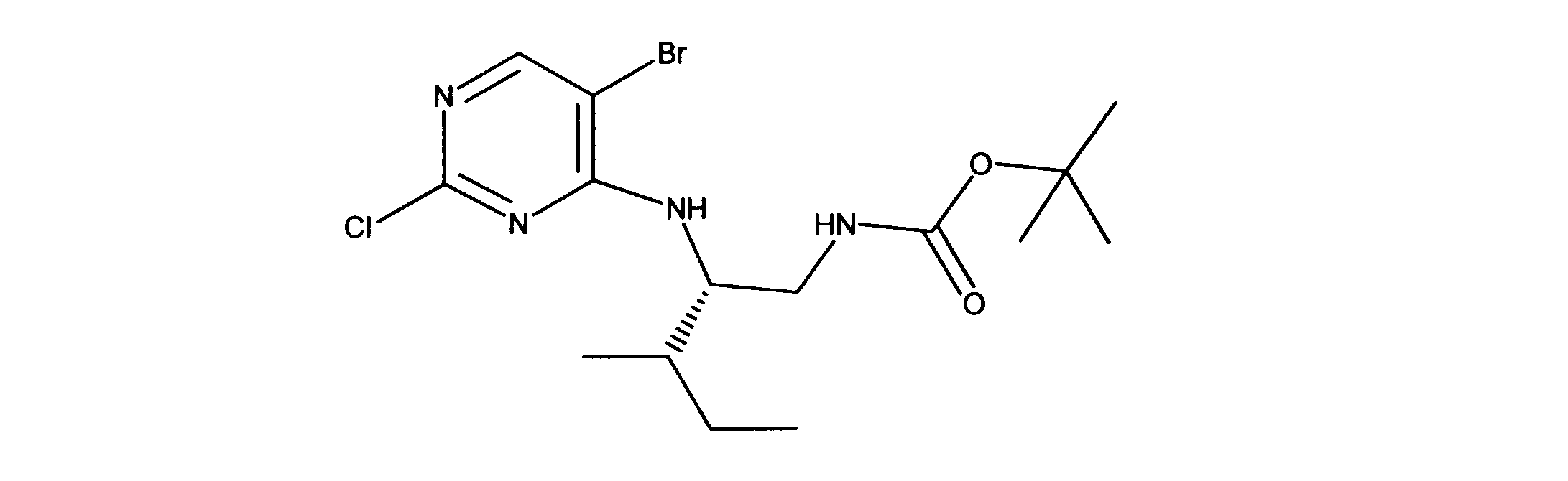
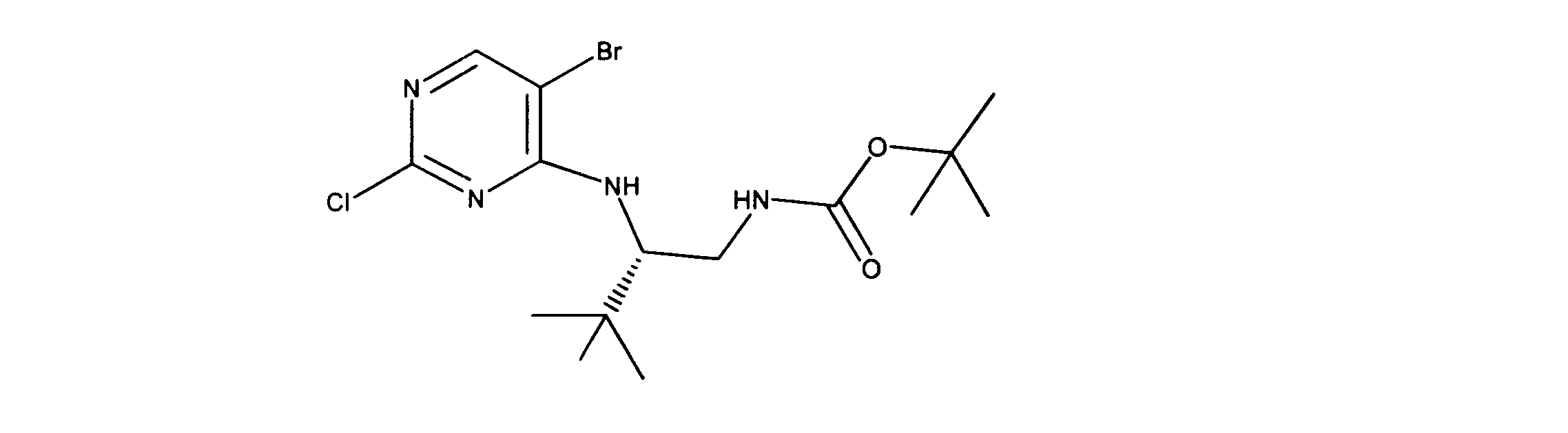
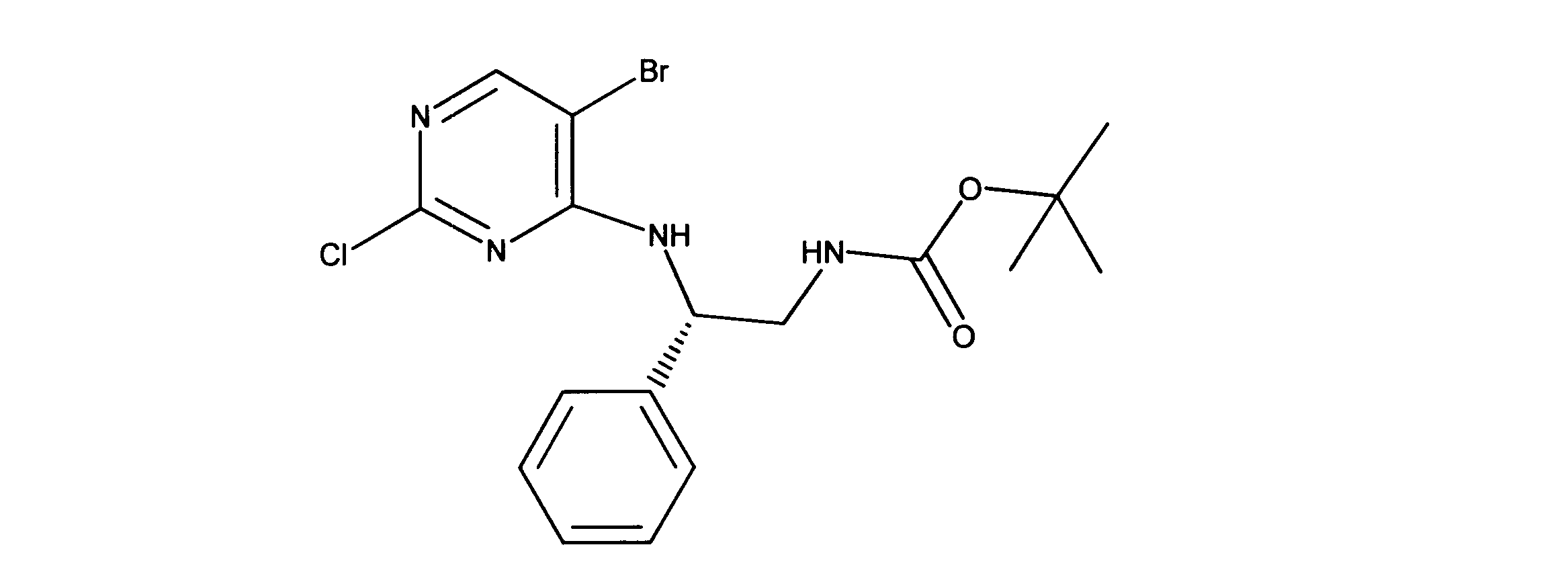
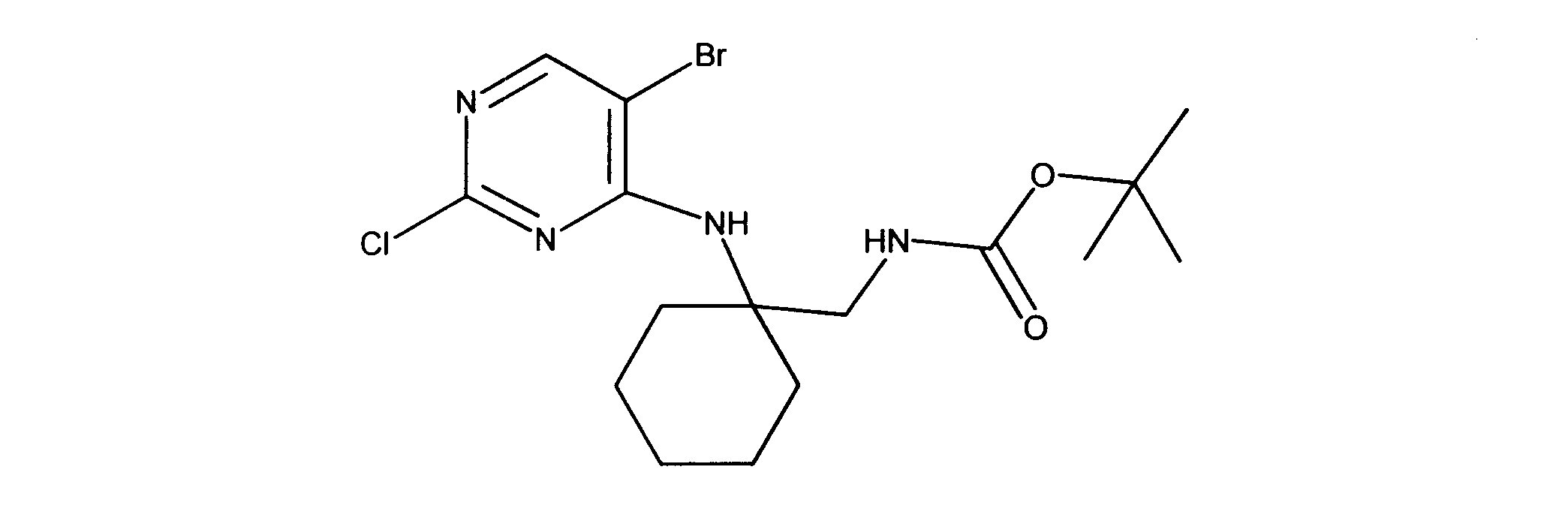
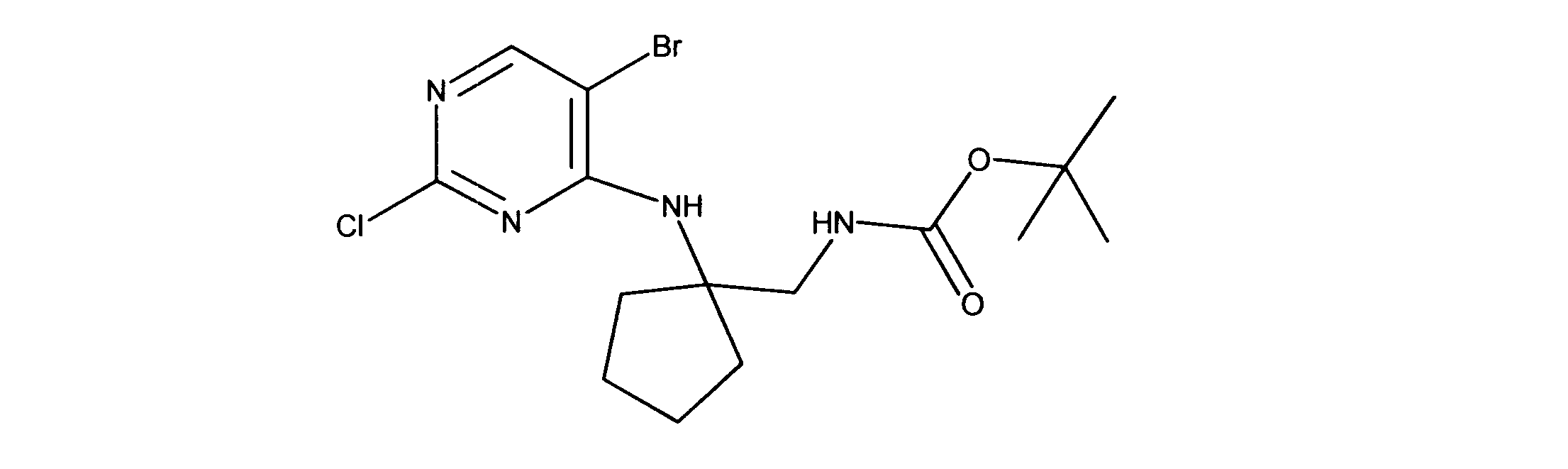
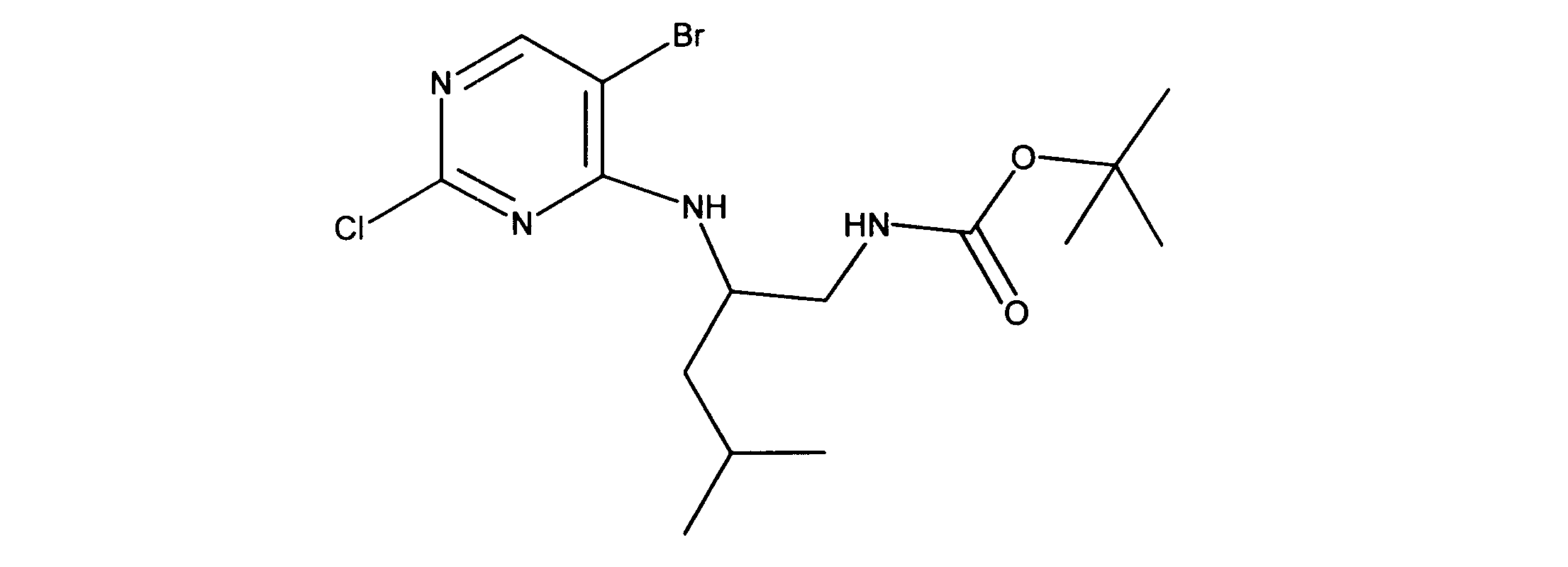
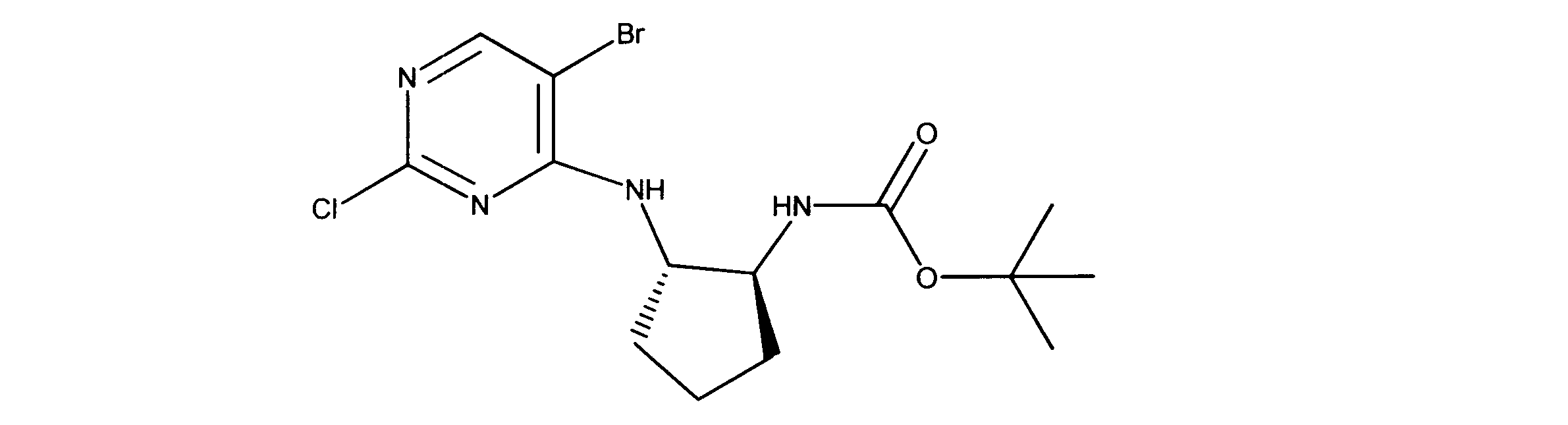
tert-ブチルN-[2-[(5-ブロモ2クロロピリミジン-4イル)アミノ]エチル]カルバメートの合成、化合物1
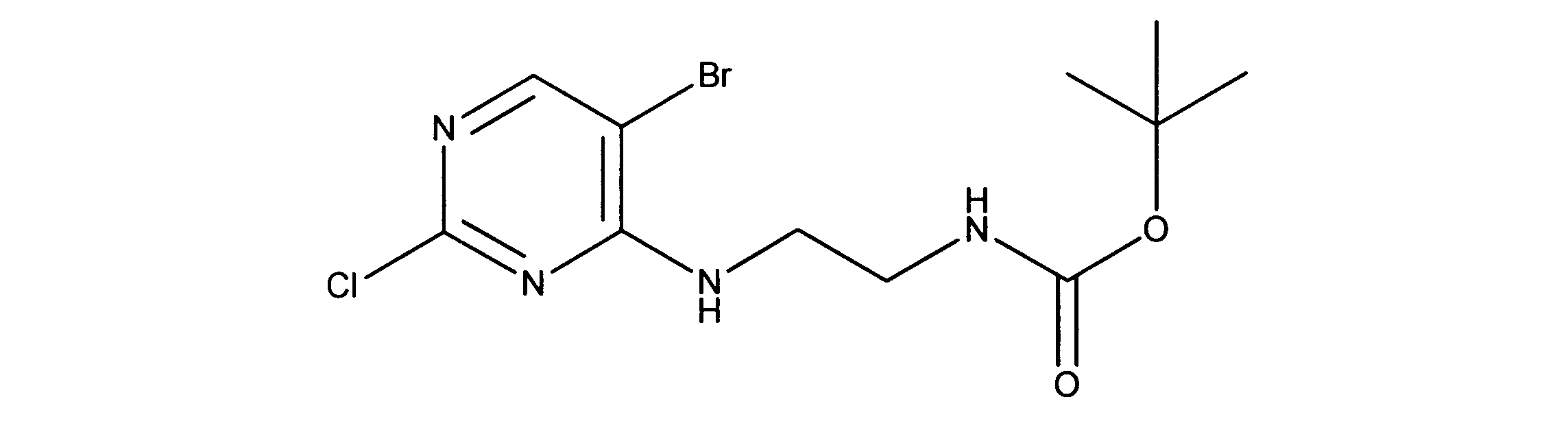
1HNMR (d6-DMSO) δ ppm 8.21 (s, 1H), 7.62 (brs, 1H), 7.27 (brs, 1H), 3.39 (m, 2H), 3.12 (m, 2H), 1.34 (s, 9H). LCMS (ESI) 351 (M + H).
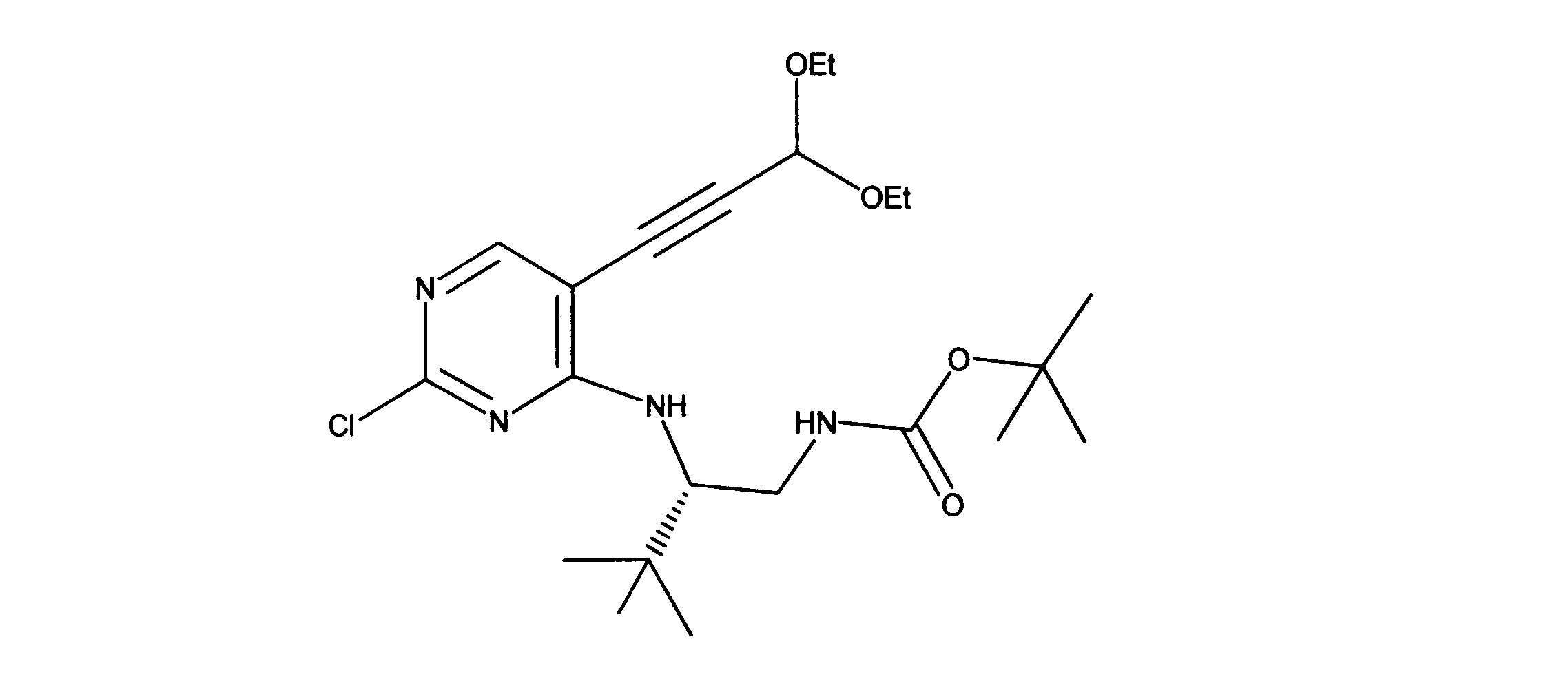
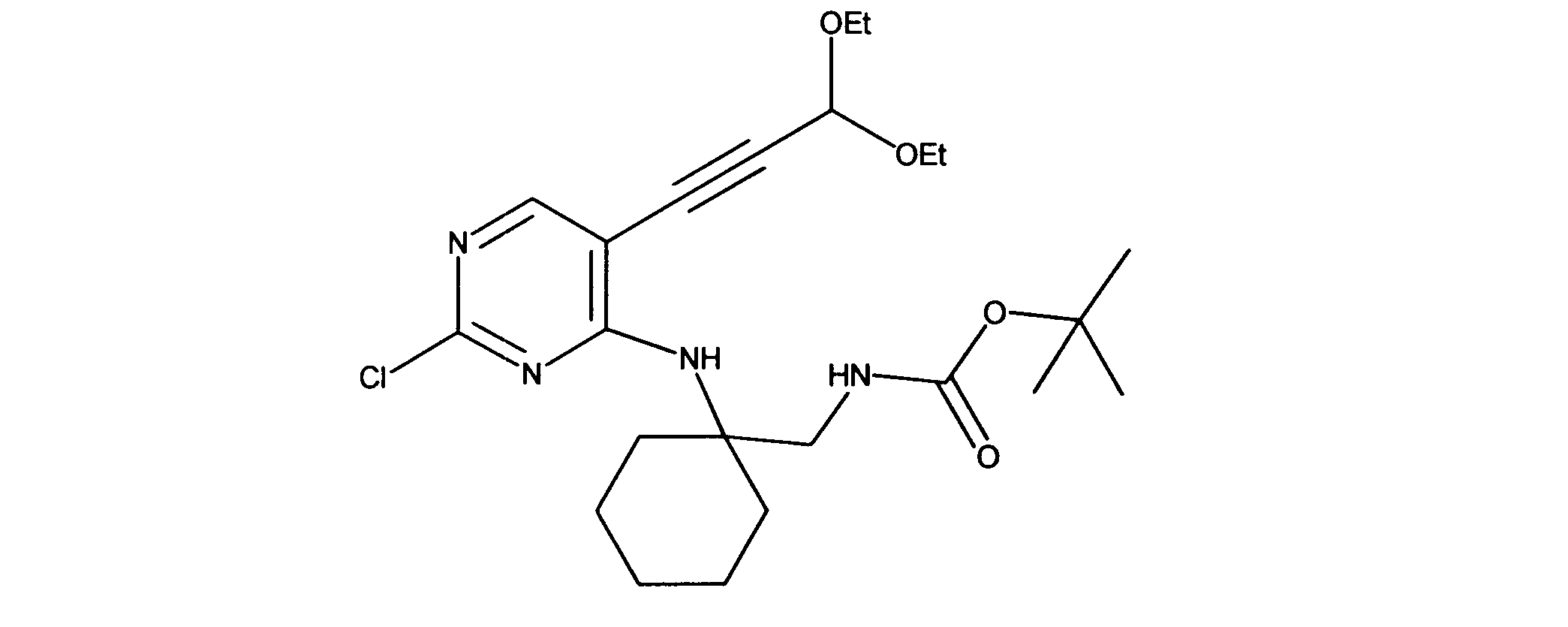
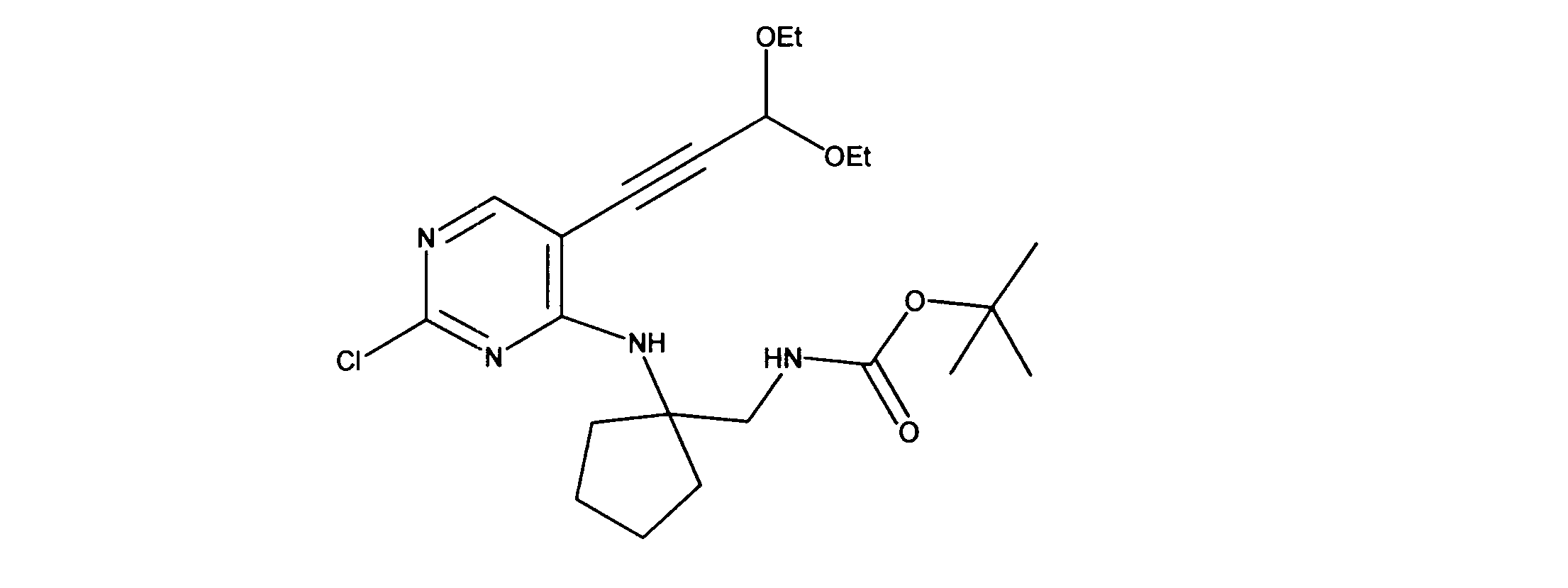
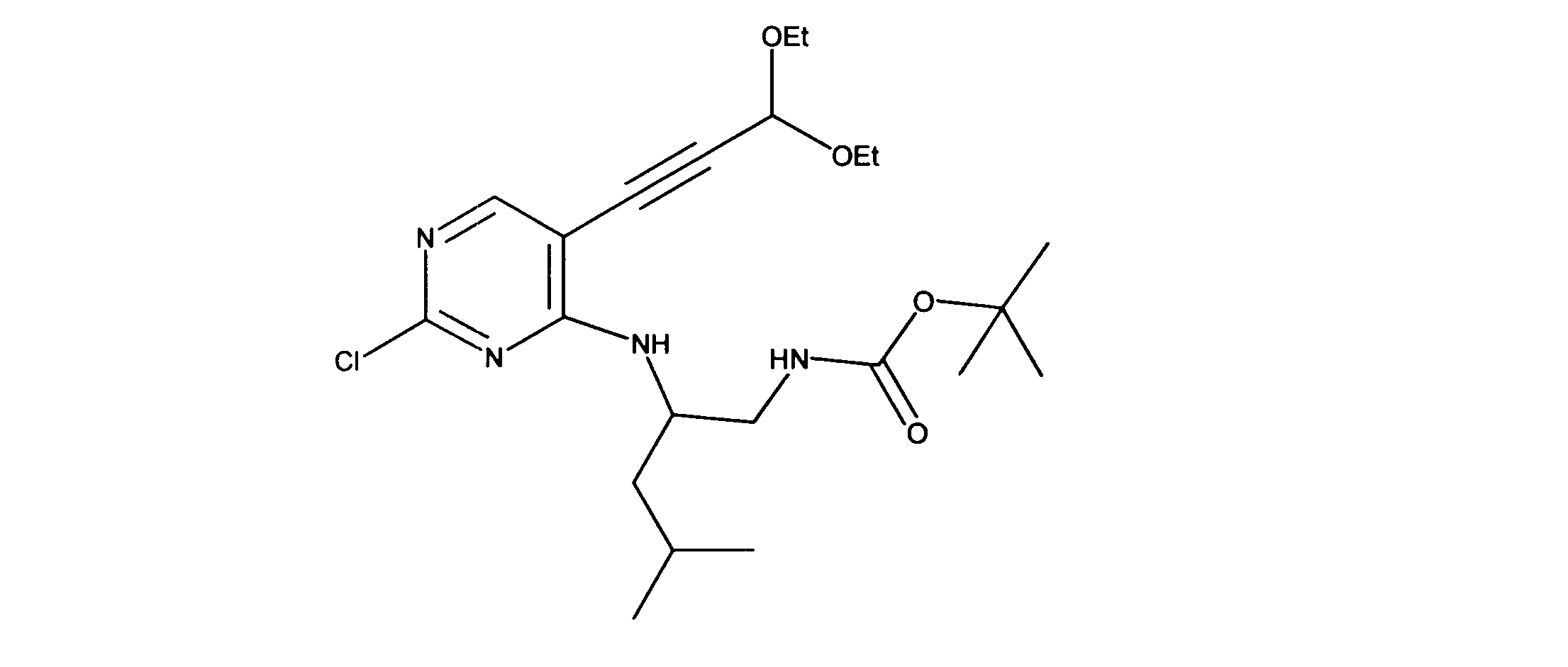
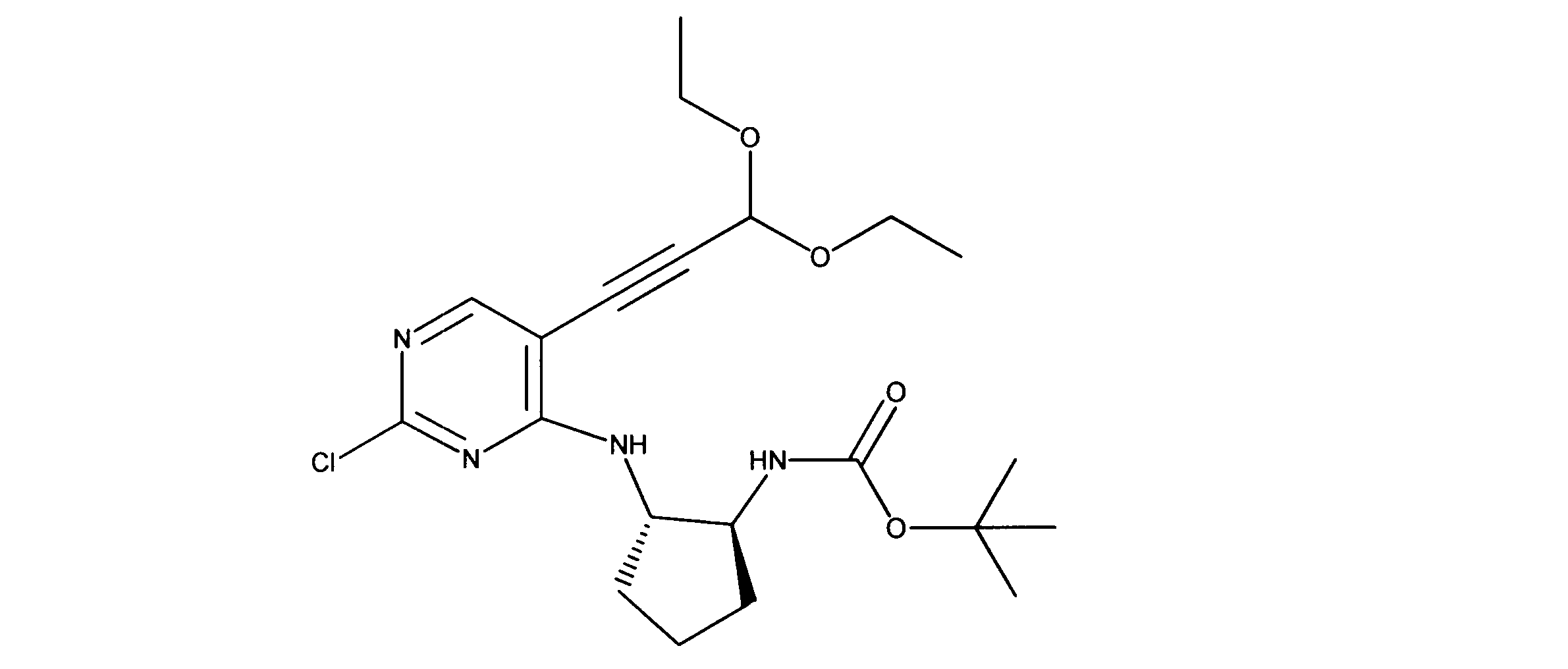
tert-ブチルN-[2-[[2-クロロ-5-(3、3-ジエトキシプロプ-1-イニル)ピリミジン-4-イル]アミノ]エチル]カルバメートの合成、化合物2
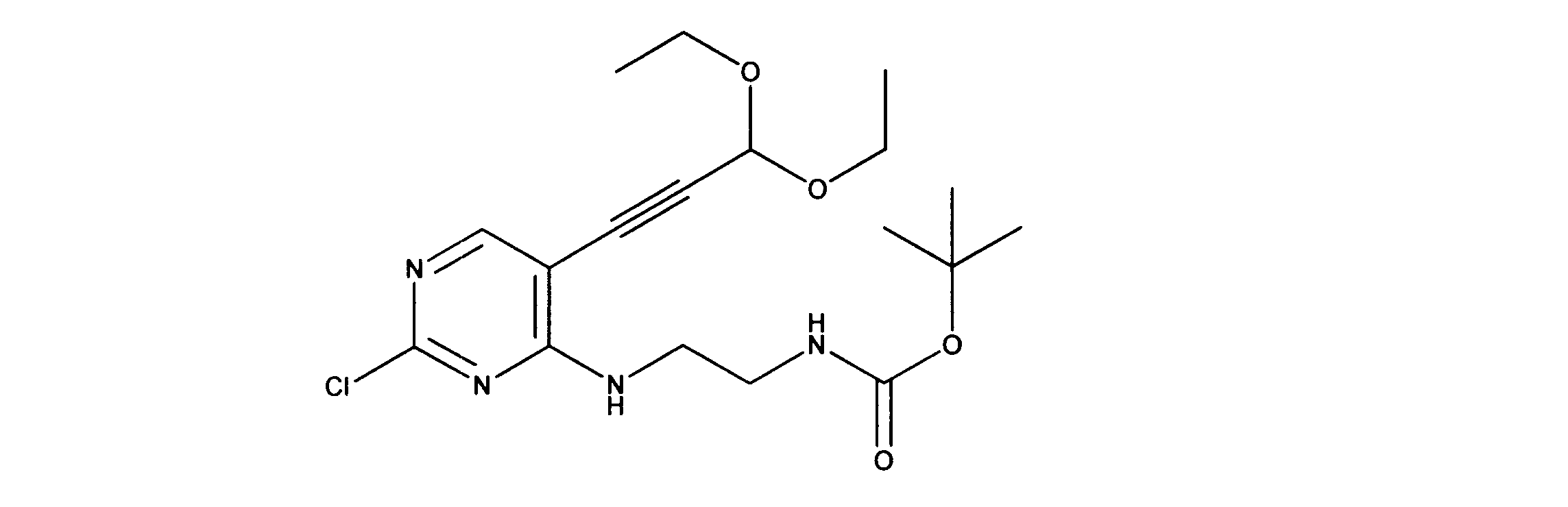
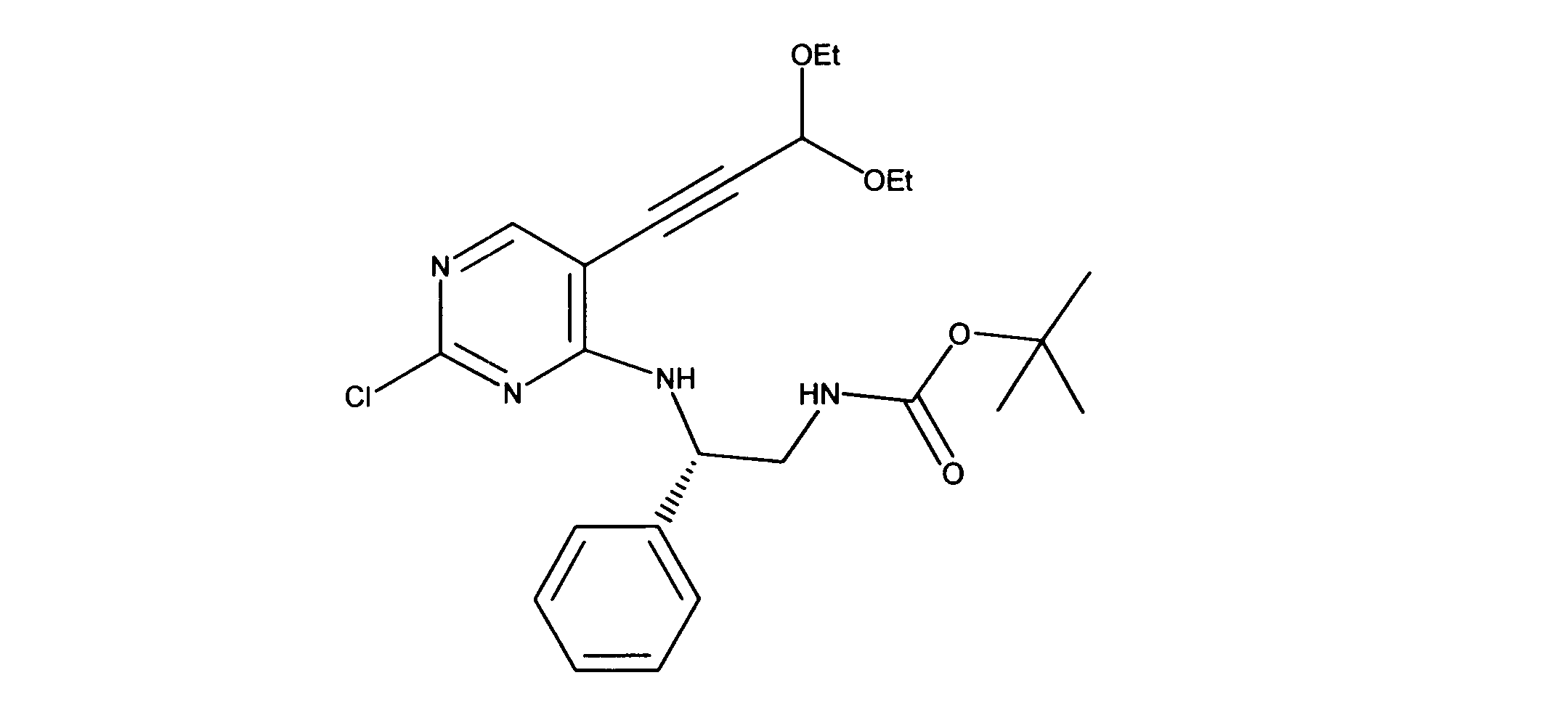
1HNMR (d6-DMSO) δ ppm 8.18 (s, 1H), 7.63 (brs, 1H), 7.40 (brs, 1H), 5.55 (s, 1H), 3.70 (m, 2H), 3.60 (m, 2H), 3.42 (m, 2H), 3.15 (m, 2H), 1.19 - 1.16 (m, 15H). LCMS (ESI) 399 (M + H).
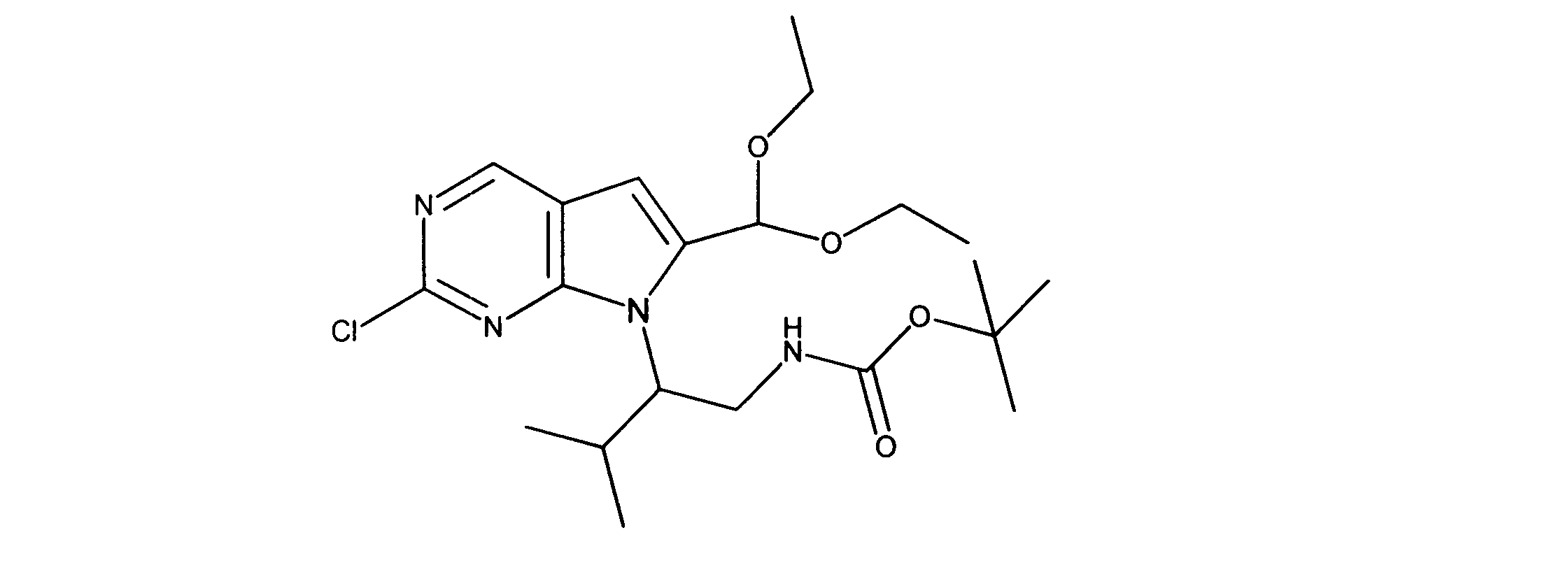
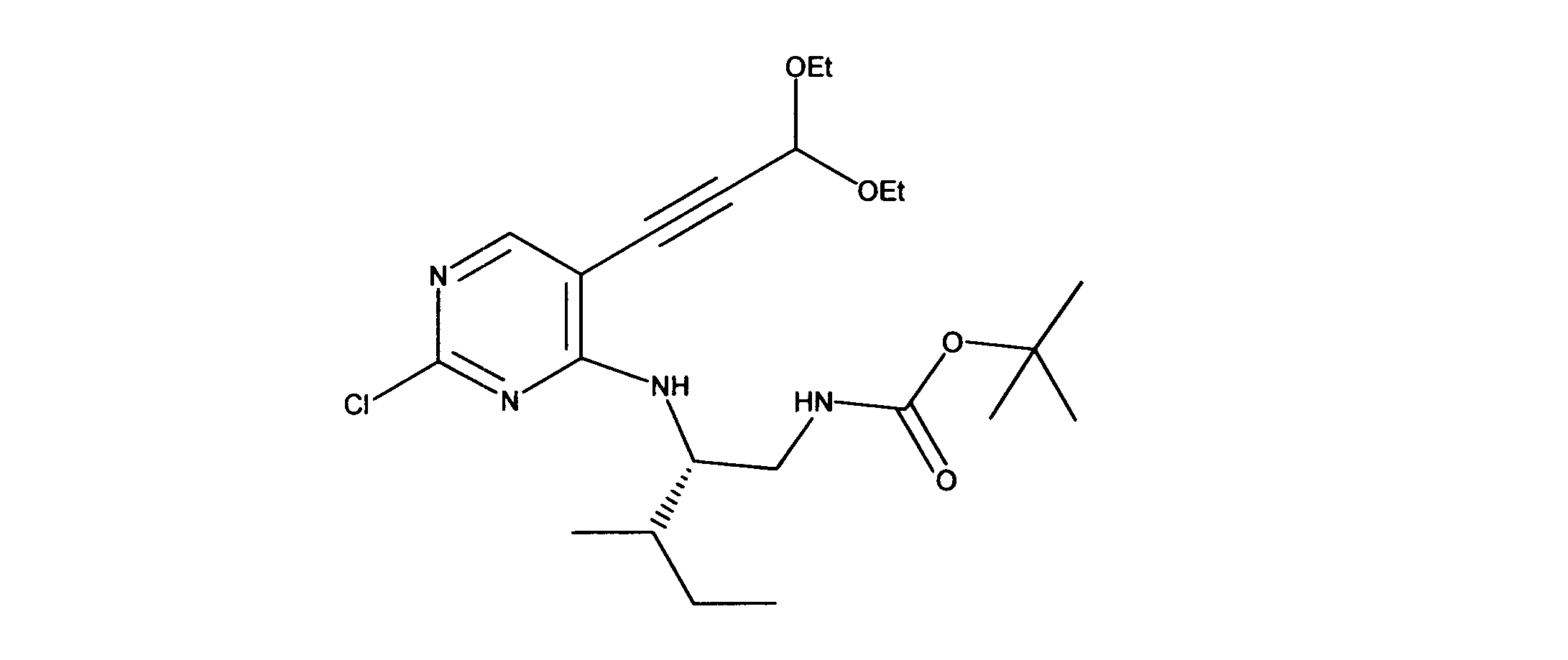
tert-ブチルN-[2-[2-クロロ-6-(ジエトキシメチル)ピロロ[2,3-d]ピリミジン-7-イル]エチル]カルバメートの合成、化合物3
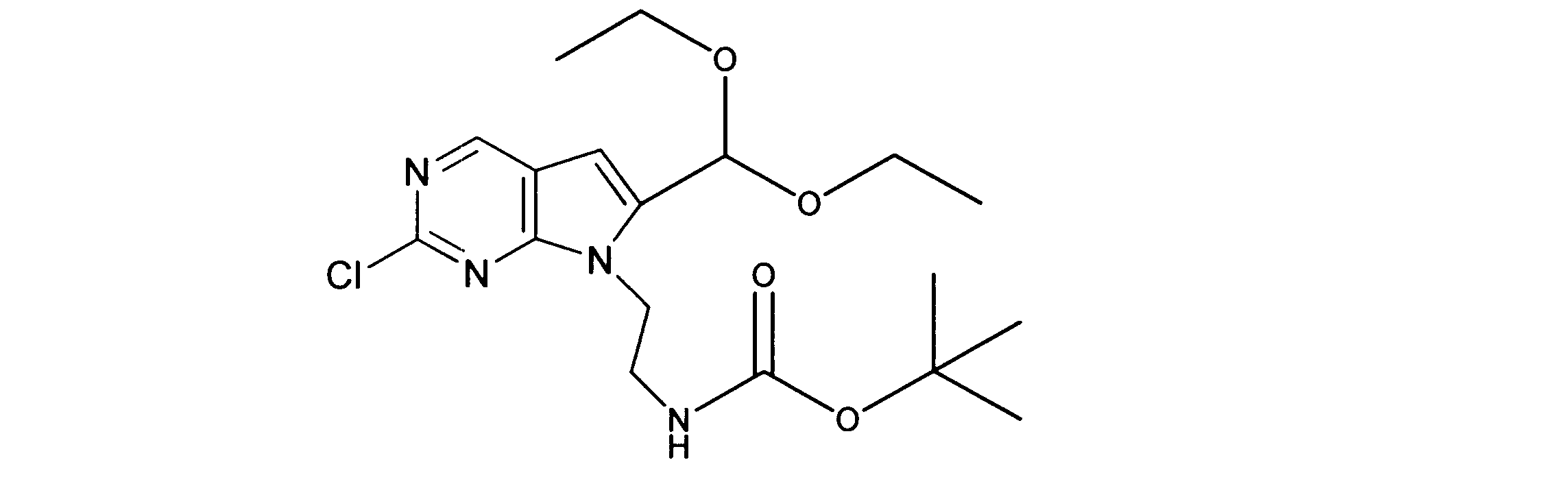
1HNMR (d6-DMSO) δ ppm 8.88 (s, 1H), 6.95 (brs, 1H), 6.69 (s, 1H), 5.79 (s, 1H), 4.29 (m, 2H), 3.59 (m, 4H), 3.34 (m, 1H), 3.18 (m, 1H), 1.19 (m, 9H), 1.17 (m, 6H). LCMS (ESI) 399 (M + H).
tert-ブチルN-[2-(2-クロロ-6-ホルミル-ピロロ[2,3-d]ピリミジン-7-イル)エチル]カルバメートの合成、化合物4
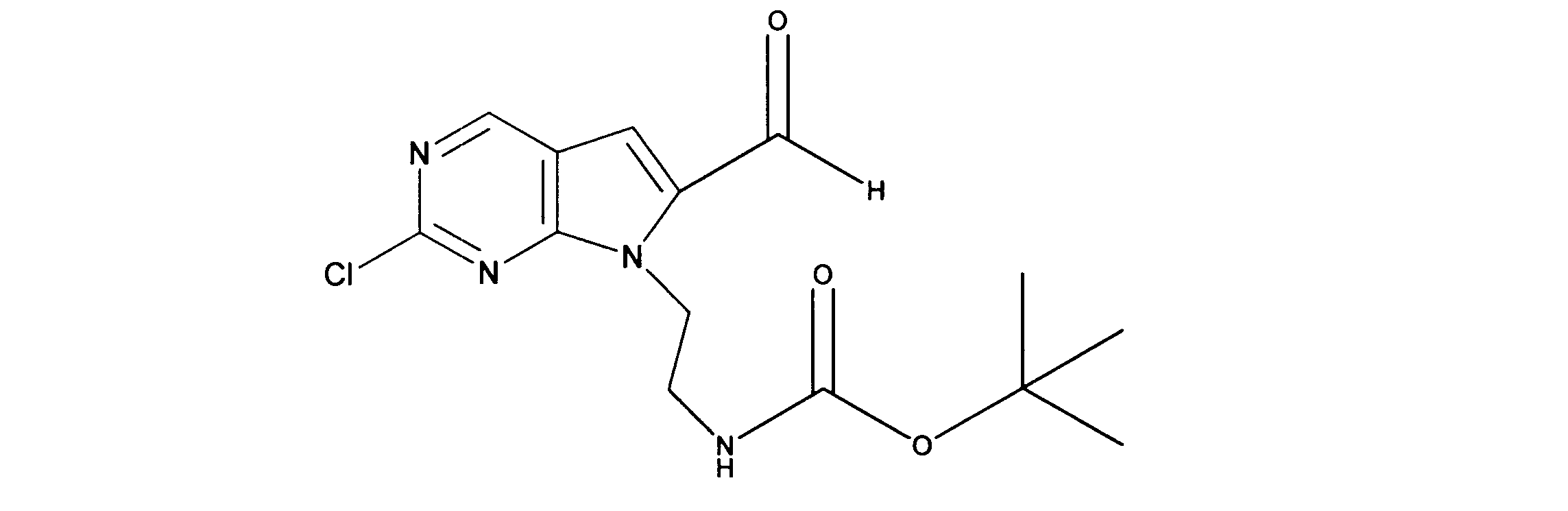
1HNMR (d6-DMSO) δ ppm 9.98 (s, 1H), 9.18 (s, 1H), 7.66 (s, 1H), 6.80 (brs, 1H), 4.52 (m, 2H), 4.36 (m, 2H), 1.14 (s, 9H). LCMS (ESI) 325 (M + H).
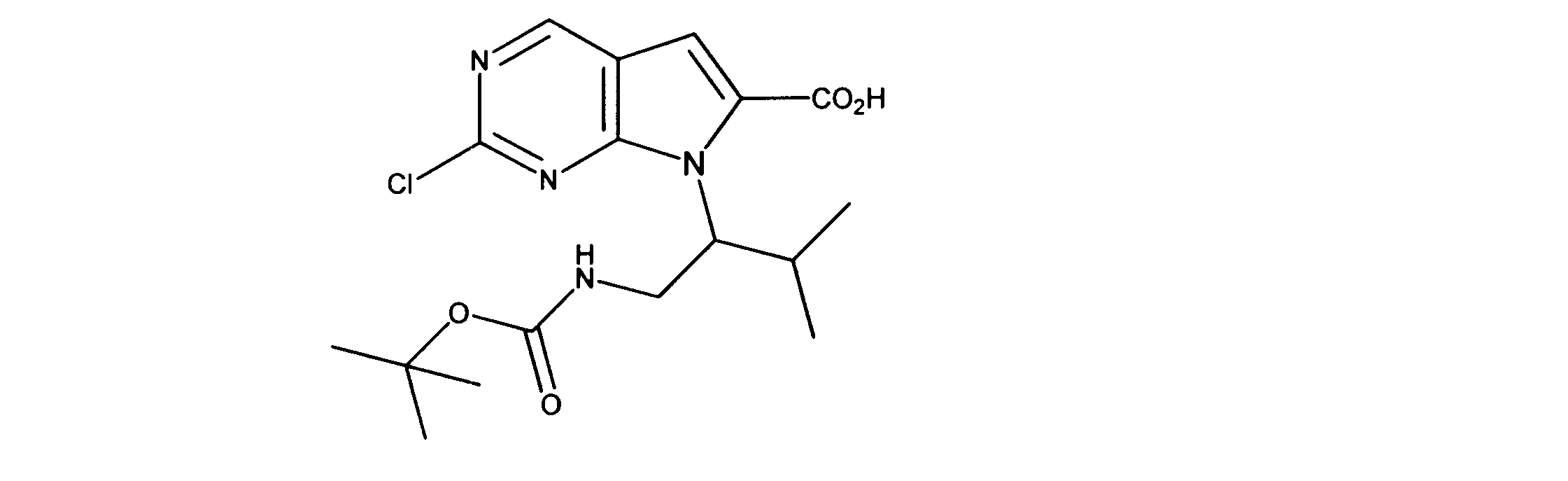
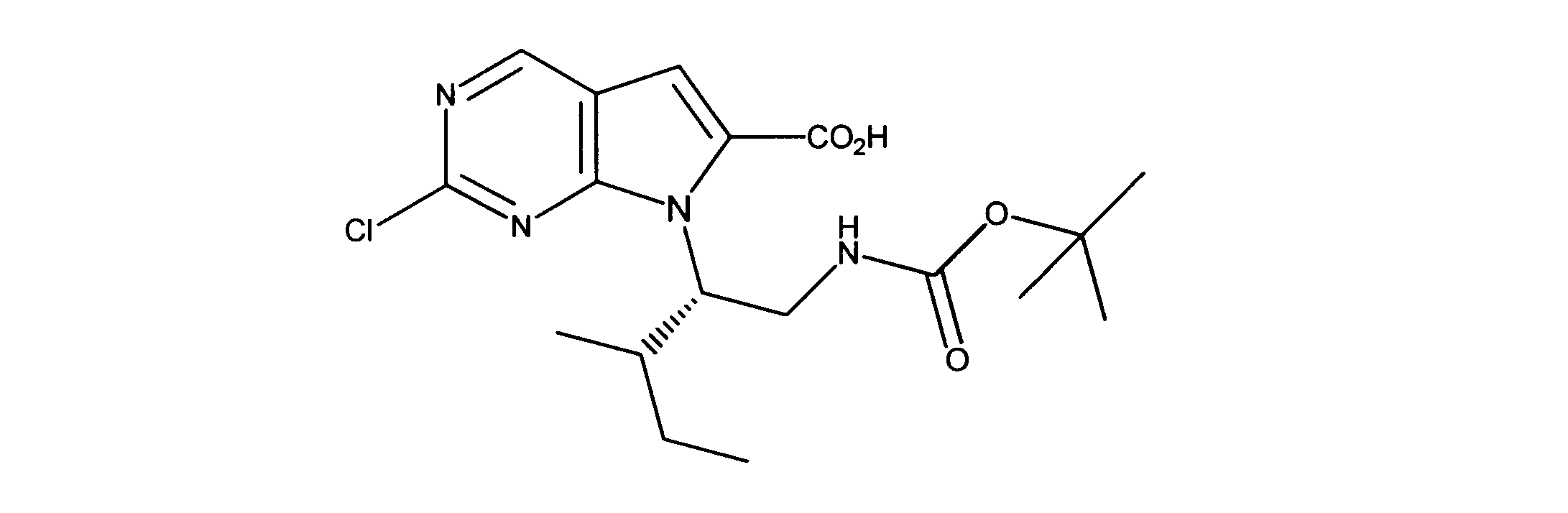
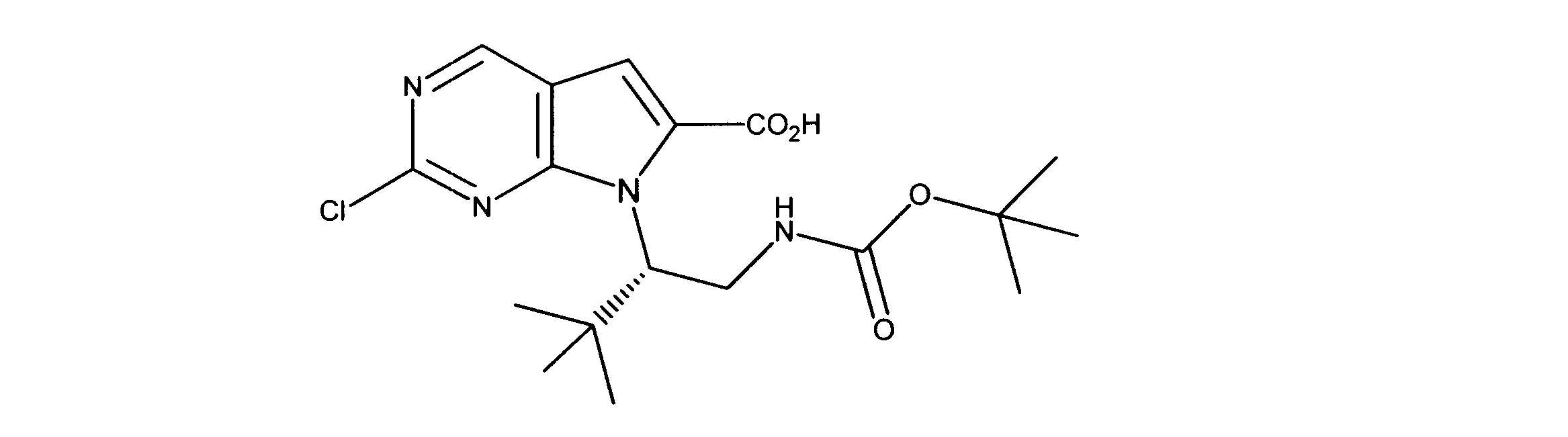
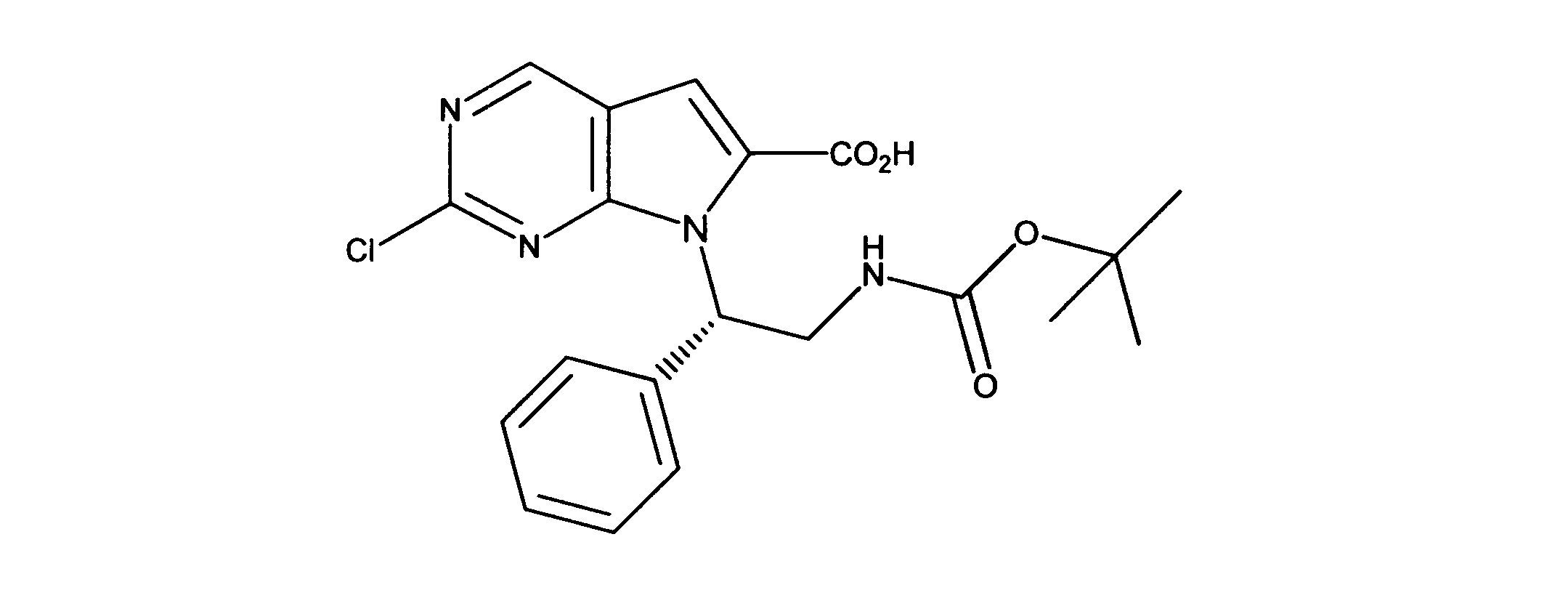
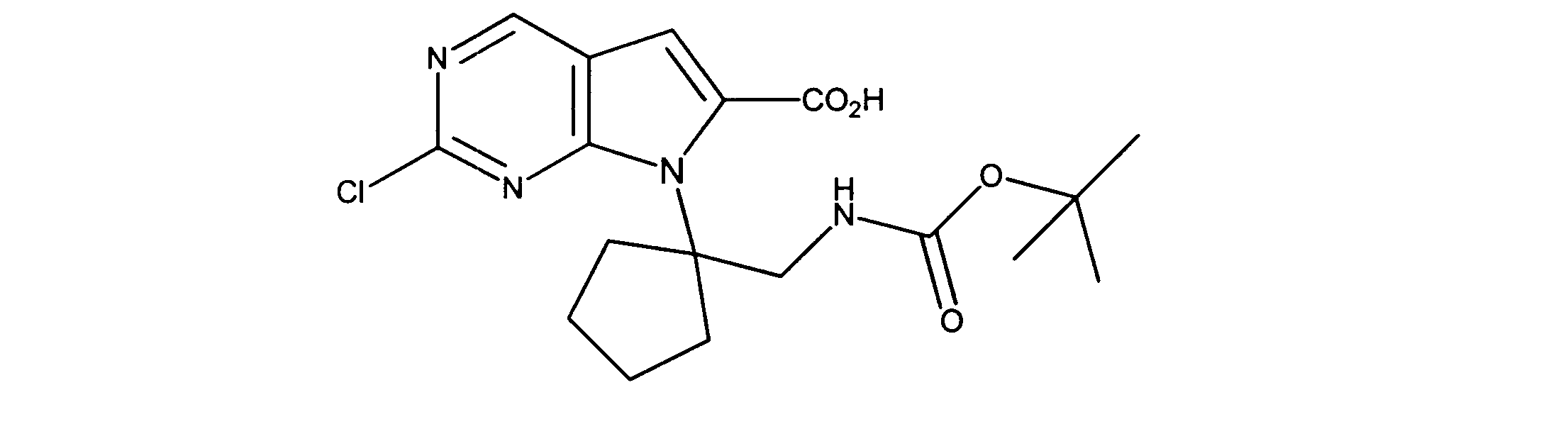
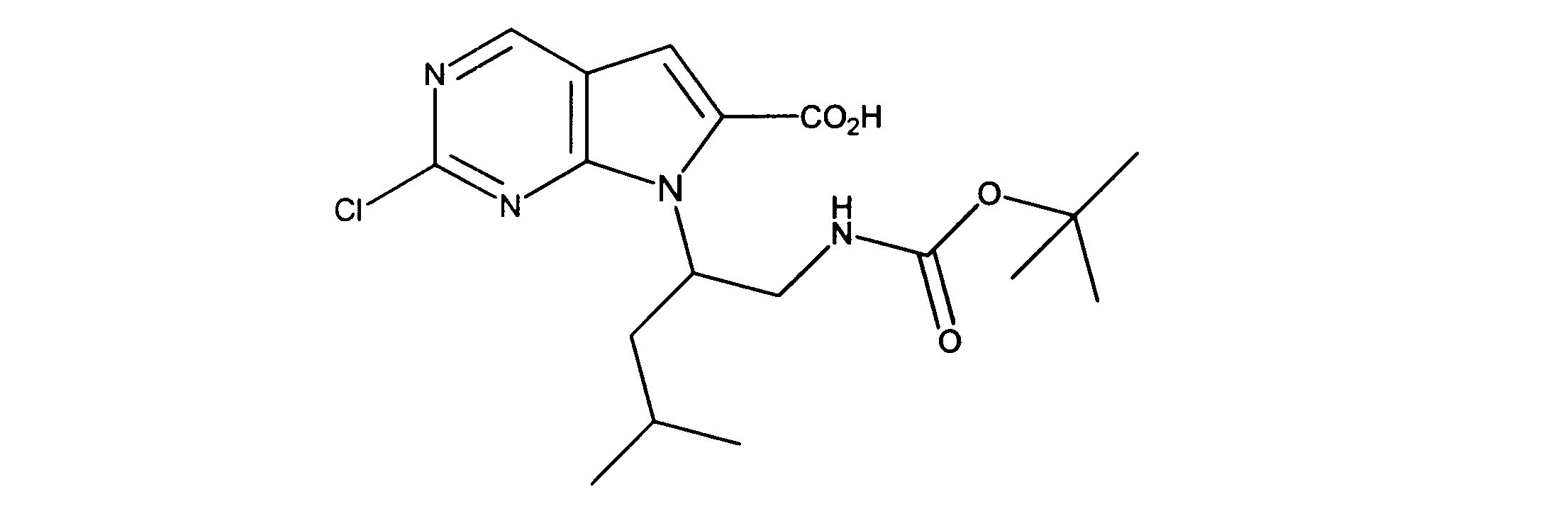
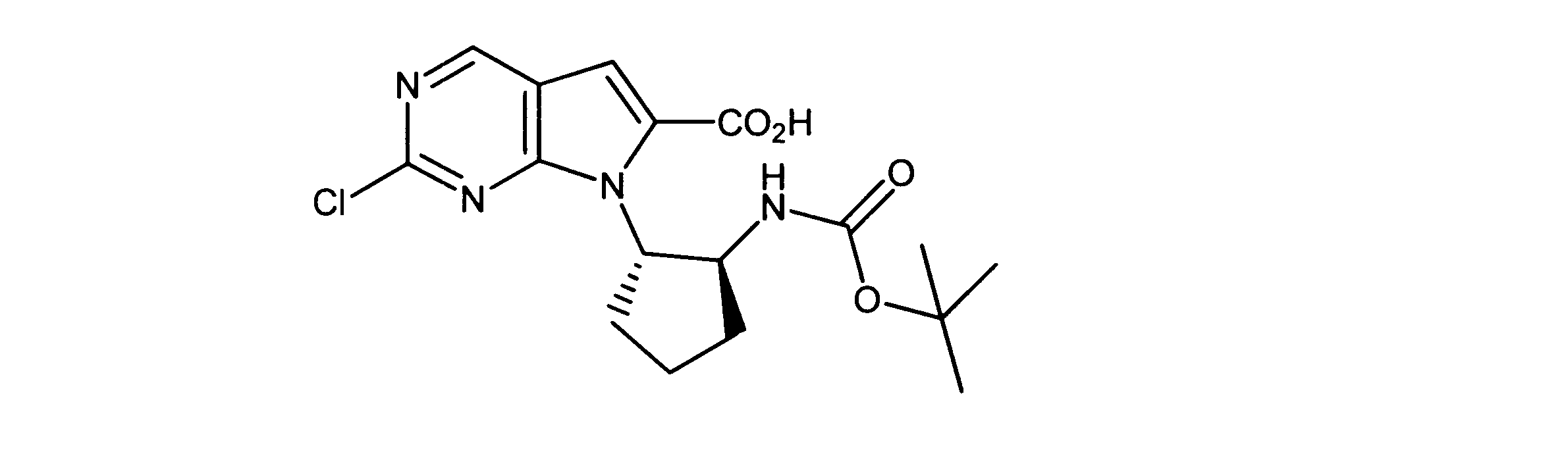
7-[2-(第三級ブトキシカルボニルアミノ)エチル]-2-クロロ-ピロロ[2,3-d]ピリミジン-6-カルボン酸の合成、化合物5
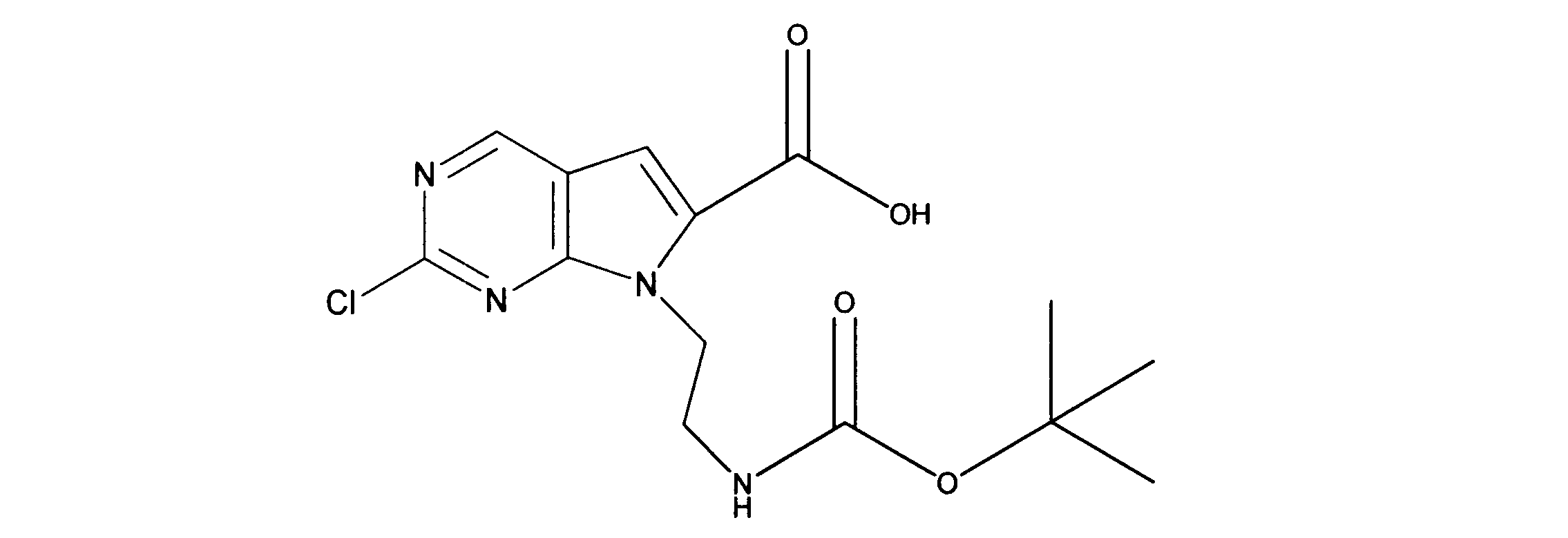
1HNMR (d6-DMSO) δ ppm 9.11 (s, 1H), 7.39 (s, 1H), 4.38 (m, 2H), 4.15 (m, 2H), 1.48 (m, 9H). LCMS (ESI) 341(M + H).
メチル7-[2-(第三級ブトキシカルボニルアミノ)エチル]-2-クロロ-ピロロ[2,3-d]ピリミジン-6-カルボン酸塩の合成、化合物6
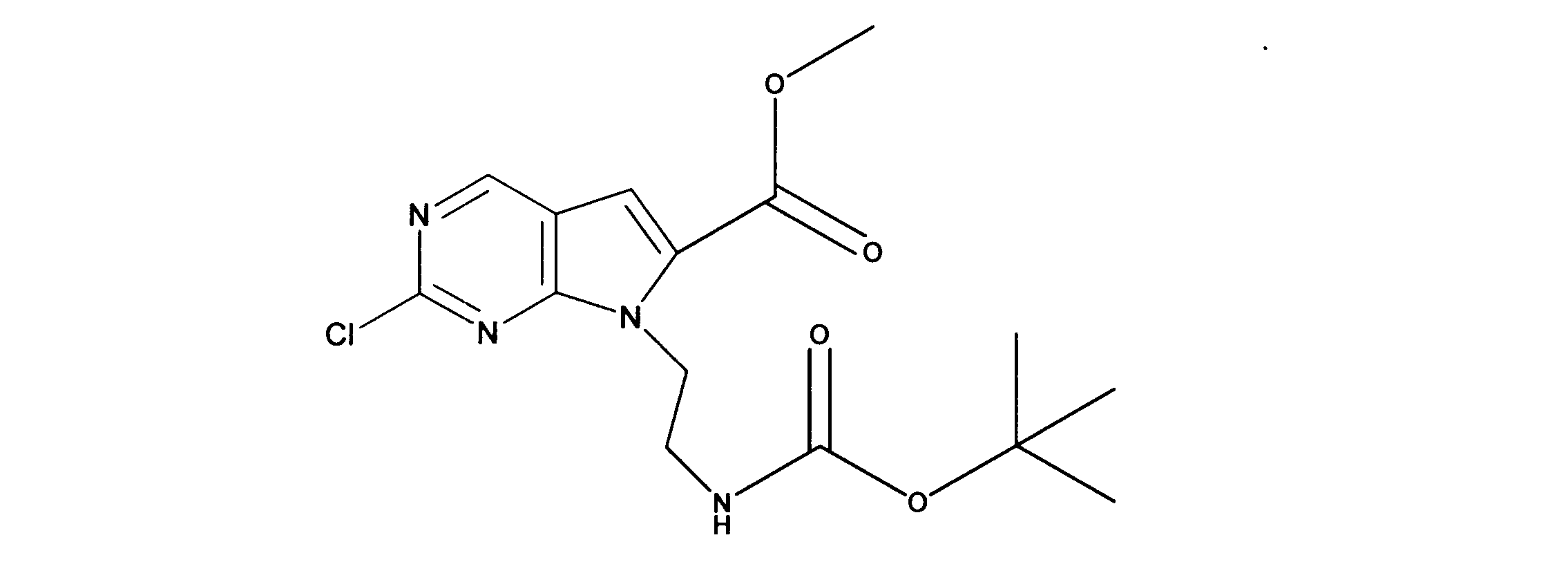
1HNMR (d6-DMSO) δ ppm 9.10 (s, 1H), 7.45 (s, 1H), 6.81 (brs, 1H) 4.60 (m, 2H), 3.91 (s, 3H), 3.29 (m, 2H), 1.18 (m, 9H) LCMS (ESI) 355 (M + H).
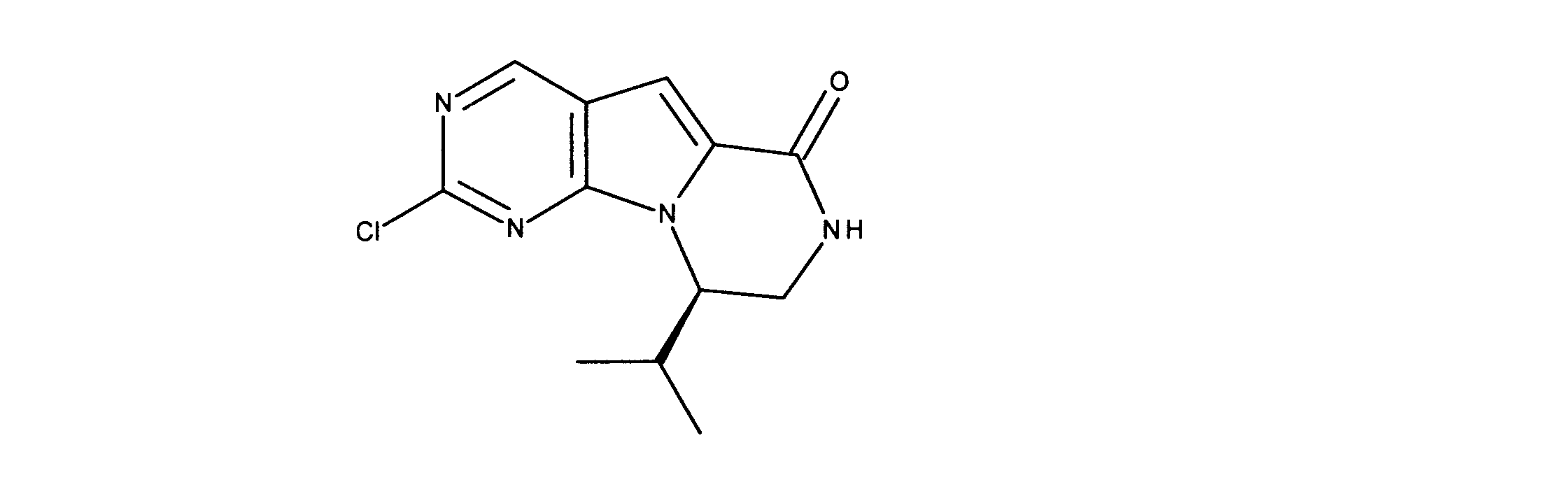
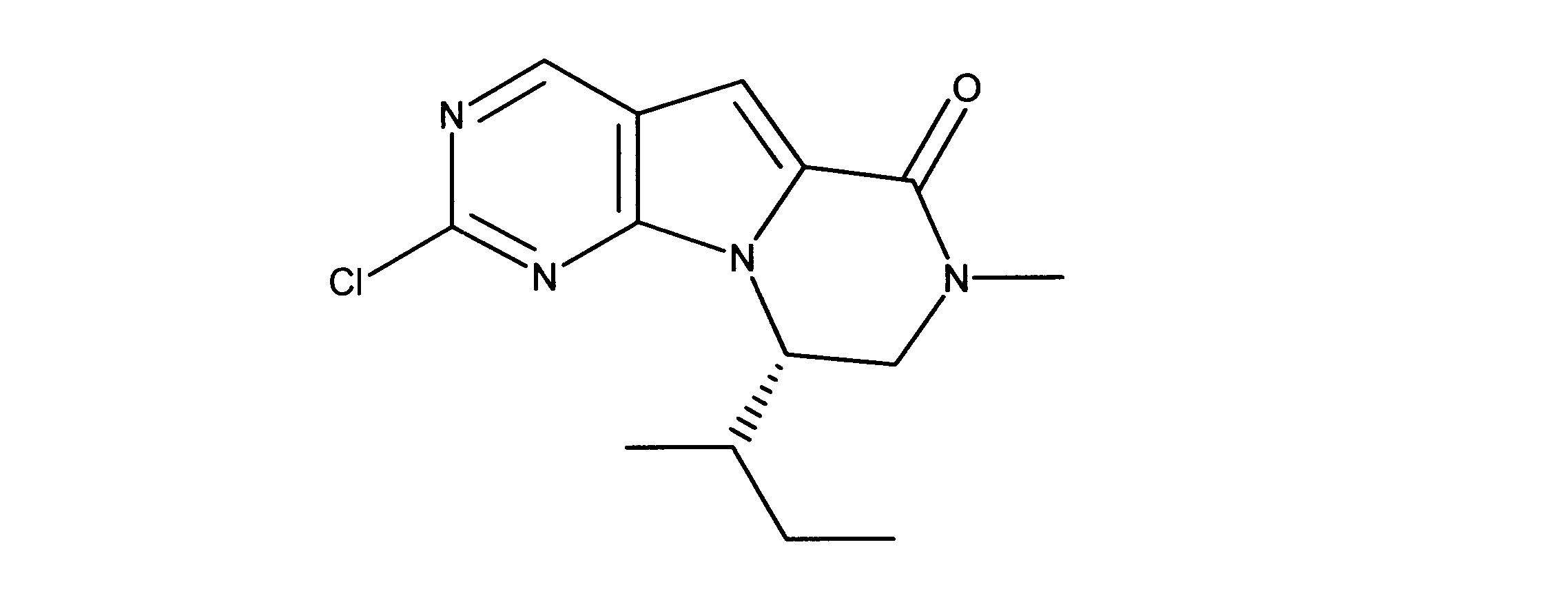
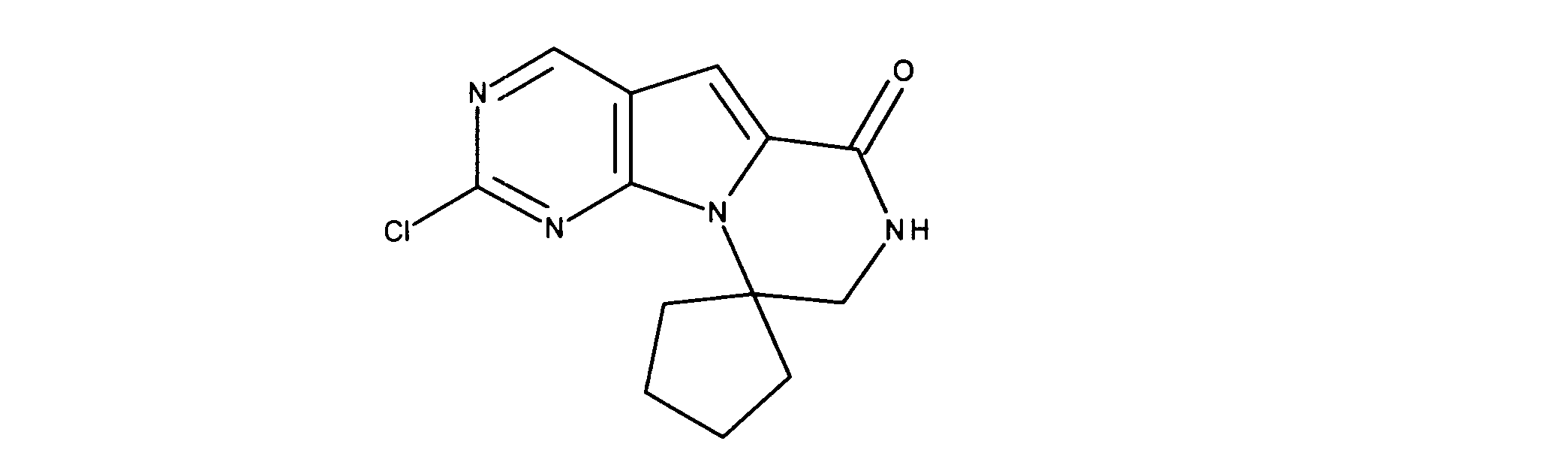
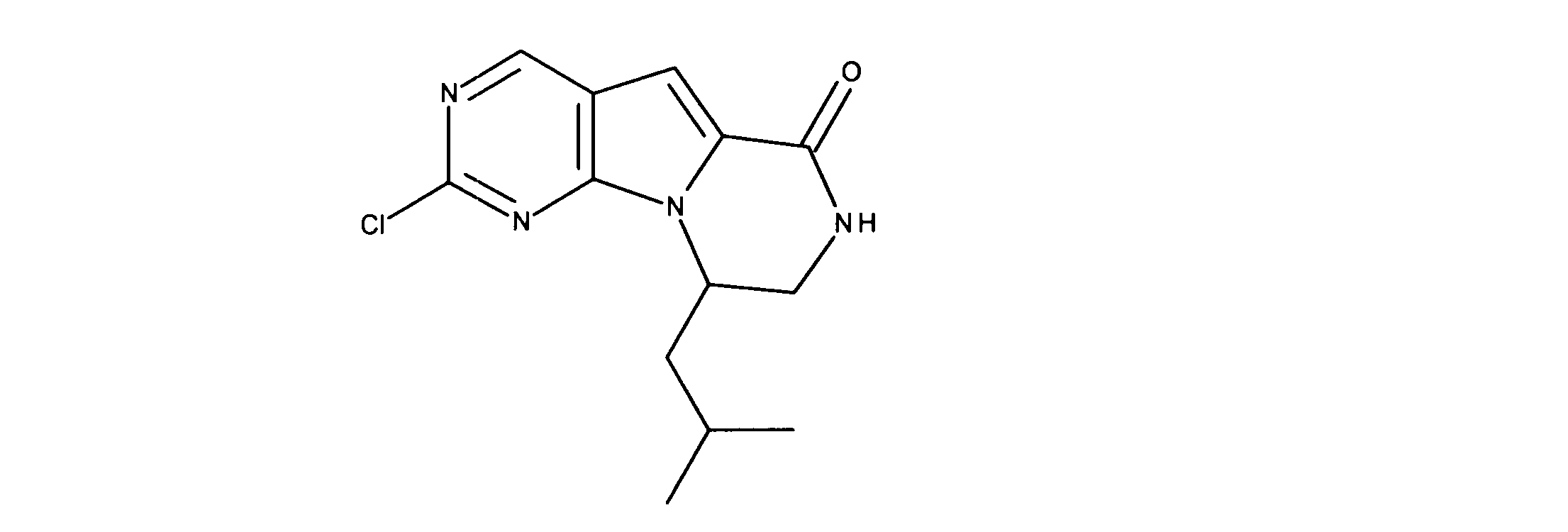
クロロ三環系アミドの合成、化合物7
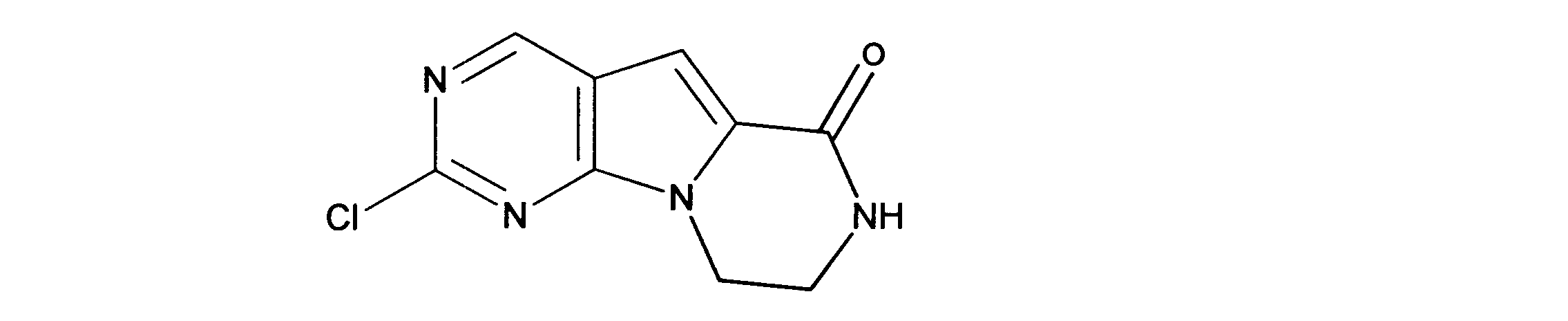
1HNMR (d6-DMSO) δ ppm 9.08 (s, 1H), 8.48 (brs, 1H), 7.21 (s, 1H) 4.33 (m, 2H), 3.64 (m, 2H). LCMS (ESI) 223 (M + H).
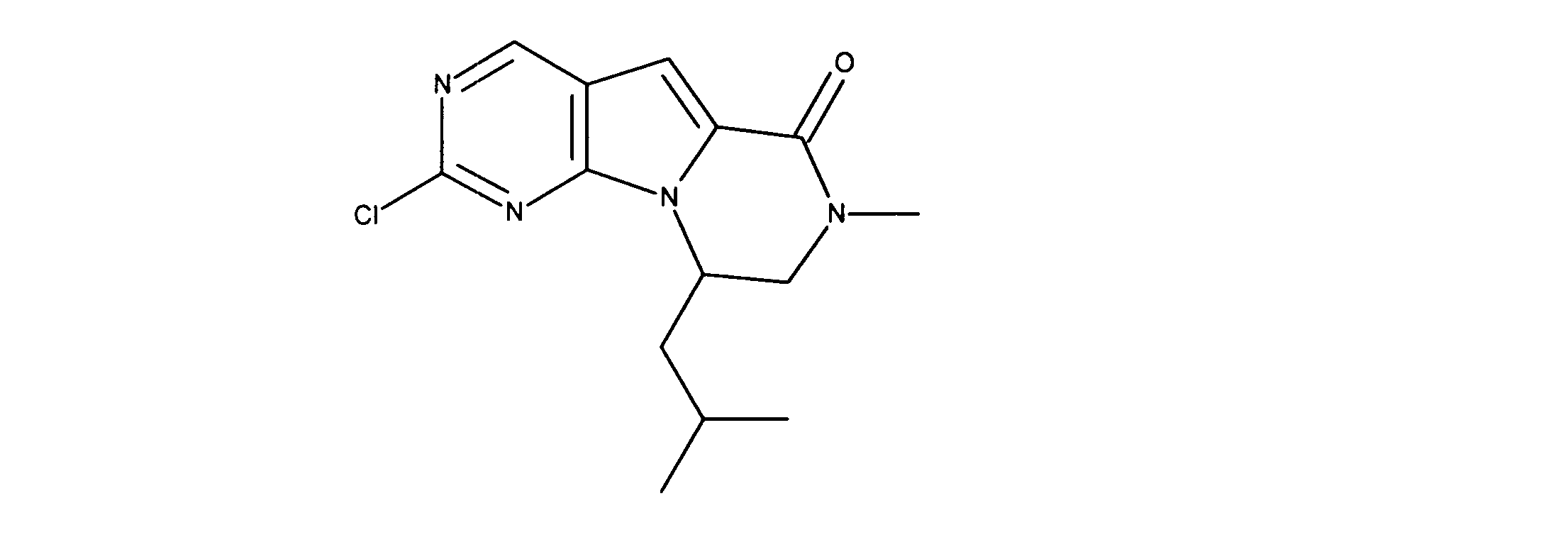
クロロ-N-メチル三環アミドの合成、化合物8
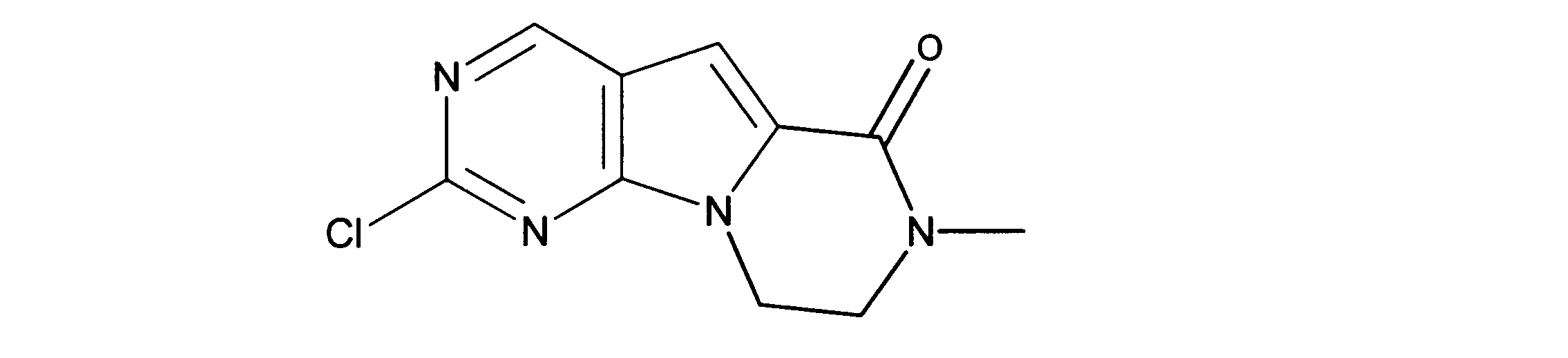
1HNMR (d6-DMSO) δ ppm 9.05 (s, 1H), 7.17 (s, 1H) 4.38 (m, 2H), 3.80 (m, 2H), 3.05 (s, 3H). LCMS (ESI) 237 (M + H).
1-メチル-4-(6-ニトロ-3-ピリジル)ピペラジンの合成、化合物9
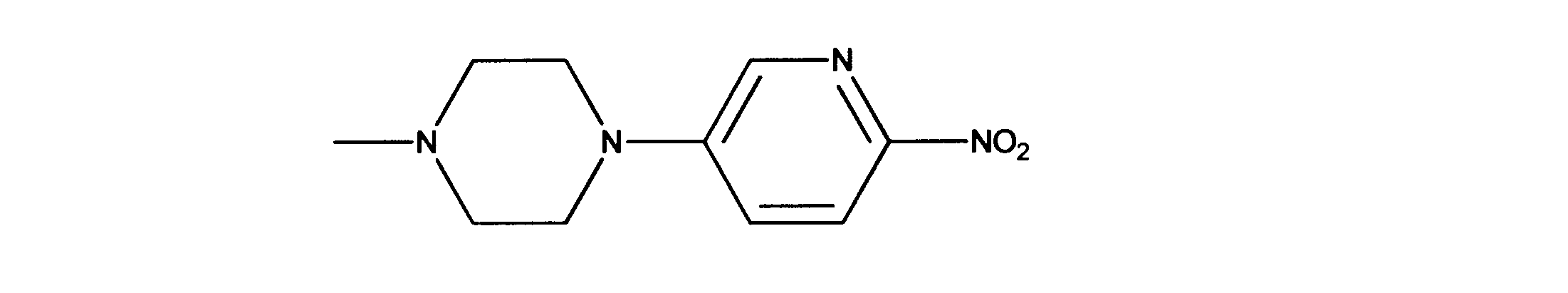
1HNMR (d6-DMSO) δ ppm 8.26 (s, 1H), 8.15 (1H, d, J = 9.3 Hz), 7.49 (1H, d, J = 9.4 Hz), 3.50 (m, 4H), 2.49 (m, 4H), 2.22 (s, 3H).
5-(4-メチルピペラジン-1-イル)ピリジン-2-アミンの合成、化合物10
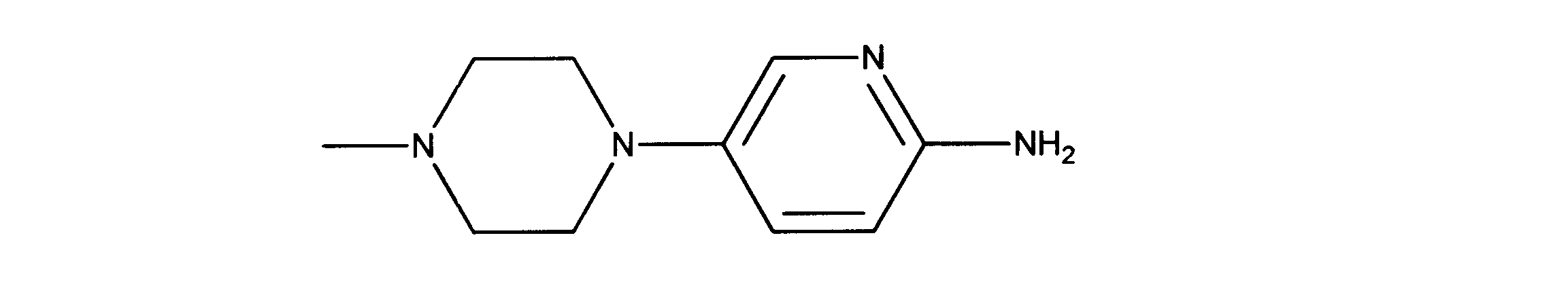
1HNMR (d6-DMSO) δ ppm 7.56 (1H, d, J = 3 Hz), 7.13 (1H, m), 6.36 (1H, d, J = 8.8 Hz), 5.33 (brs, 2H), 2.88 (m, 4H), 2.47 (m, 4H), 2.16 (s, 3H).
tert-ブチル4-(6アミノ3-ピリジル)ピペラジン-1-カルボン酸塩の合成、化合物11
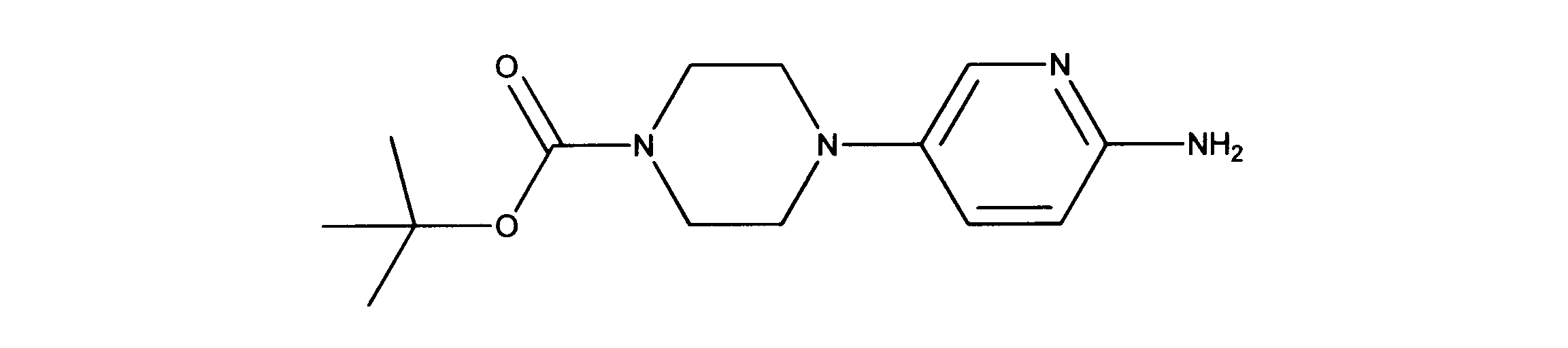
tert-ブチルN-[2-(ベンジルオキシカルボニルアミノ)-3-メチルブチル]カルバメートの合成、化合物12
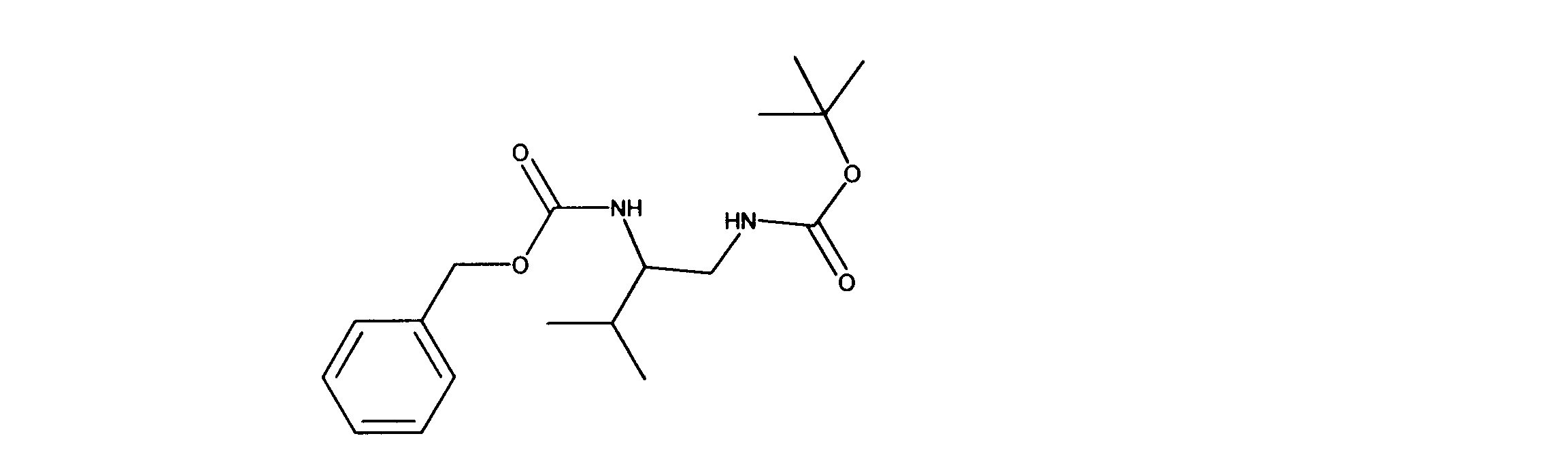
1HNMR (600 MHz, CHLOROFORM-d) δ ppm 0.89 (d, J=6.73 Hz, 3 H) 0.92 (d, J=6.73 Hz, 3 H) 1.38 (s, 9 H) 1.70 - 1.81 (m, 1 H) 3.18 (d, J=5.56 Hz, 2 H) 3.47 - 3.60 (m, 1 H) 4.76 (s, 1 H) 4.89 (d, J=7.90 Hz, 1 H) 5.07 (s, 2 H) 7.25 - 7.36 (m, 5 H). LCMS (ESI) 337 (M + H).
tert-ブチルN-[2-(ベンジルオキシカルボニルアミノ)-4-メチル-ペンチル]カルバメートの合成、化合物13
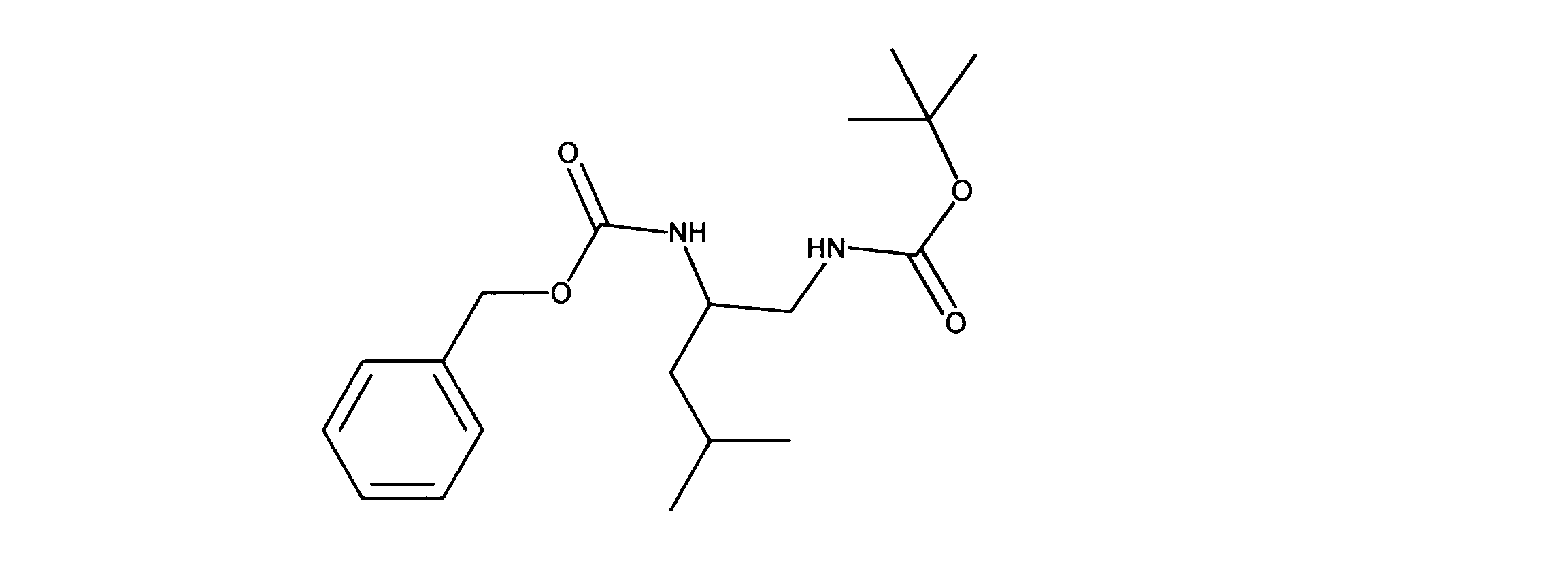
1HNMR (600 MHz, CHLOROFORM-d) δ ppm 0.89 (d, J=6.73 Hz, 6 H) 1.25 - 1.34 (m, 1 H) 1.39 (s, 9 H) 1.57 - 1.71 (m, 2 H) 3.04 - 3.26 (m, 2 H) 3.68 - 3.80 (m, 1 H) 4.72 - 4.89 (m, 2 H) 5.06 (s, 2 H) 7.25 - 7.38 (m, 5 H). LCMS (ESI) 351 (M + H).
tert-ブチルN-[(2R)-2-(ベンジルオキシカルボニルアミノ)-3-メチルブチル]カルバメートの合成、化合物14
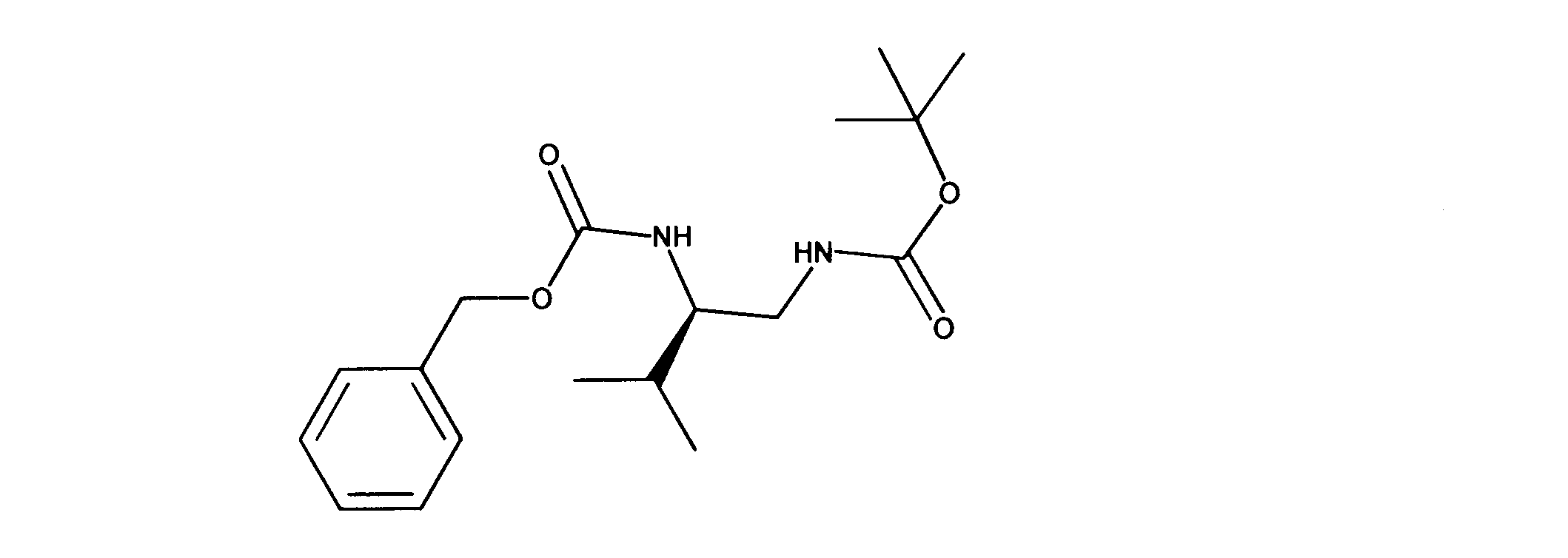
tert-ブチルN-[(2S)-2-(ベンジルオキシカルボニルアミノ)-3-メチルブチル]カルバメートの合成、化合物15
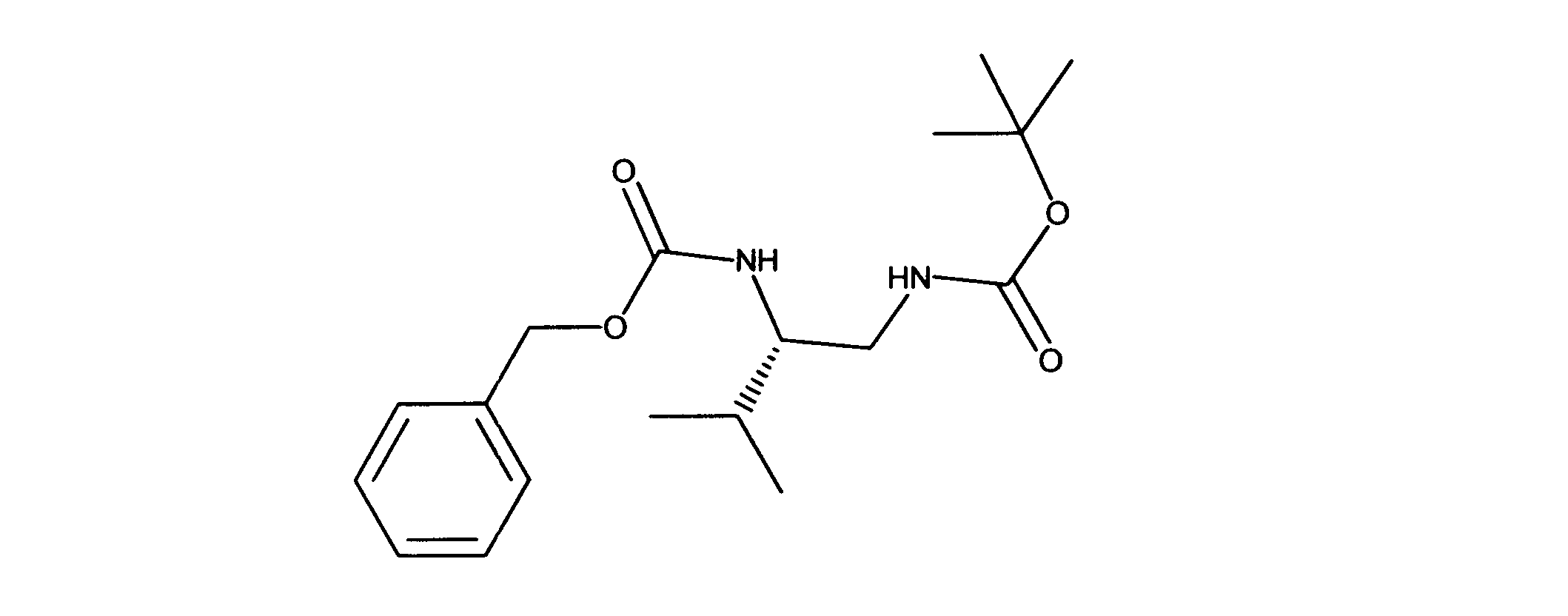
tert-ブチルN-[(1S)-1-(アミノメチル)-2メチル-プロピル]カルバメートの合成、化合物16
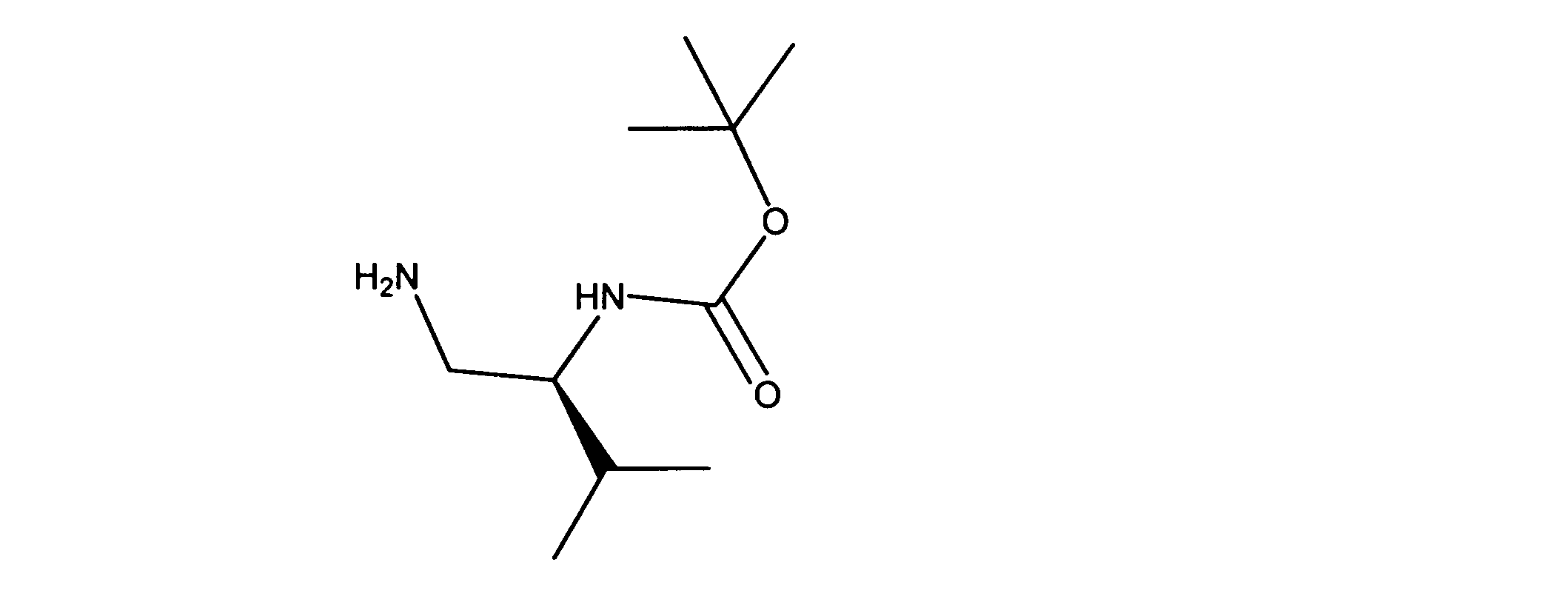
LCMS (ESI) 203 (M + H).
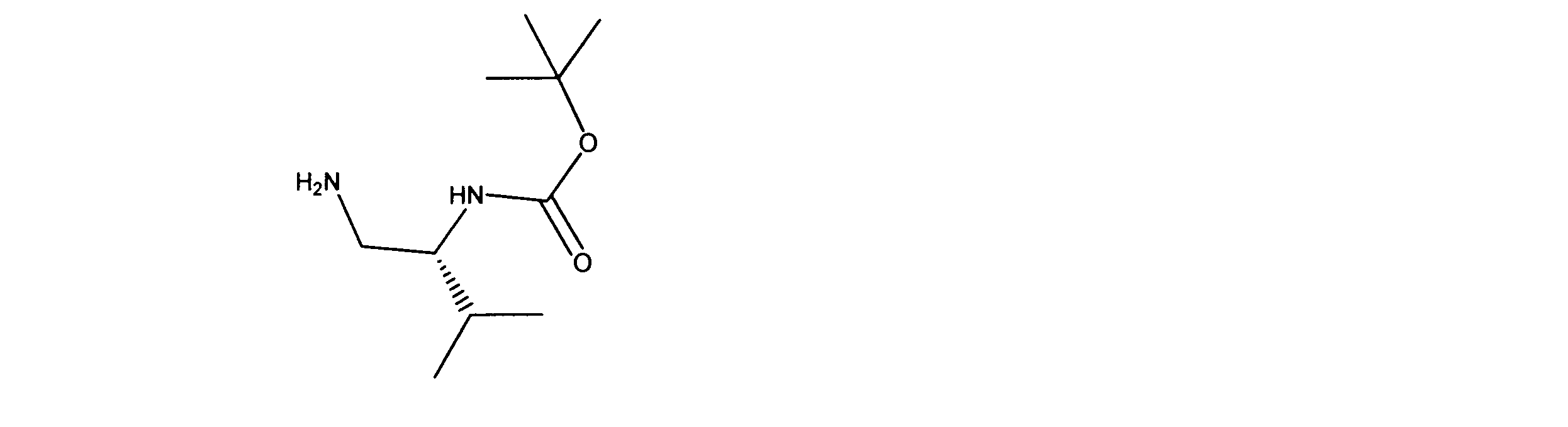
tert-ブチルN-[(1R)-1-(アミノメチル)-2メチル-プロピル]カルバメートの合成、化合物17
tert-ブチルN-[(2S)-2-(ベンジルオキシカルボニルアミノ)-4-メチル-ペンチル]カルバメートの合成、化合物18
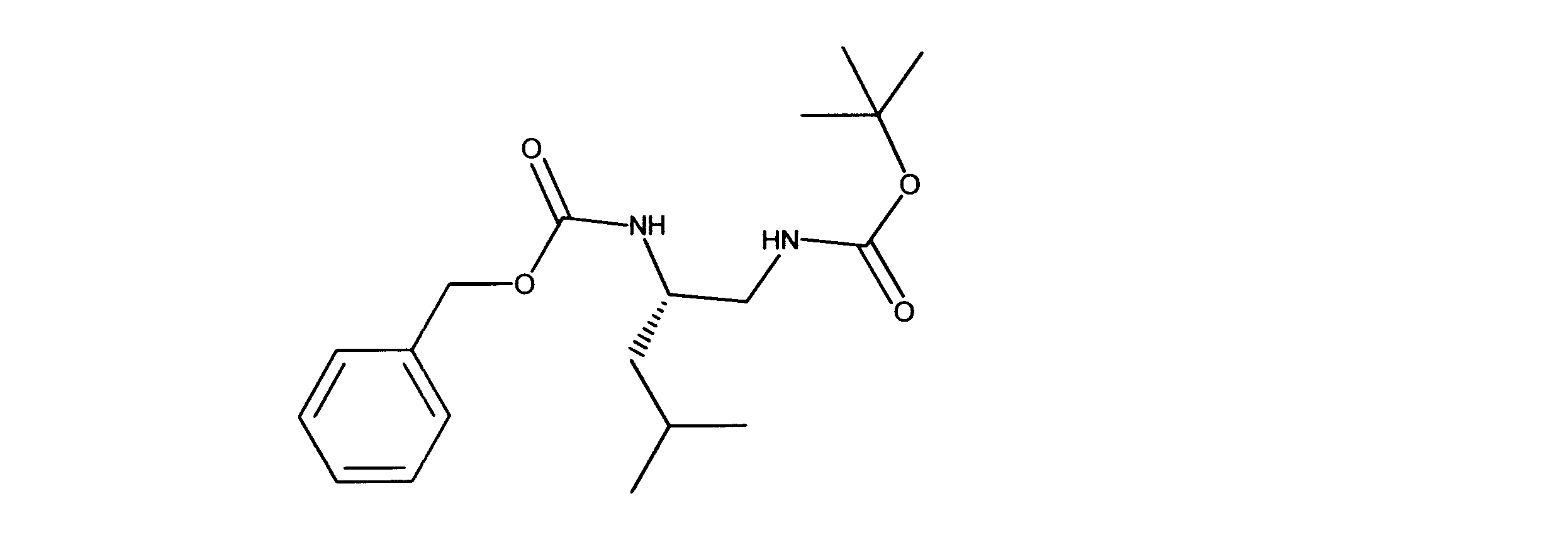
tert-ブチルN-[(2S)-2-(ベンジルオキシカルボニルアミノ)-2-フェニルエチル]カルバメートの合成、化合物19
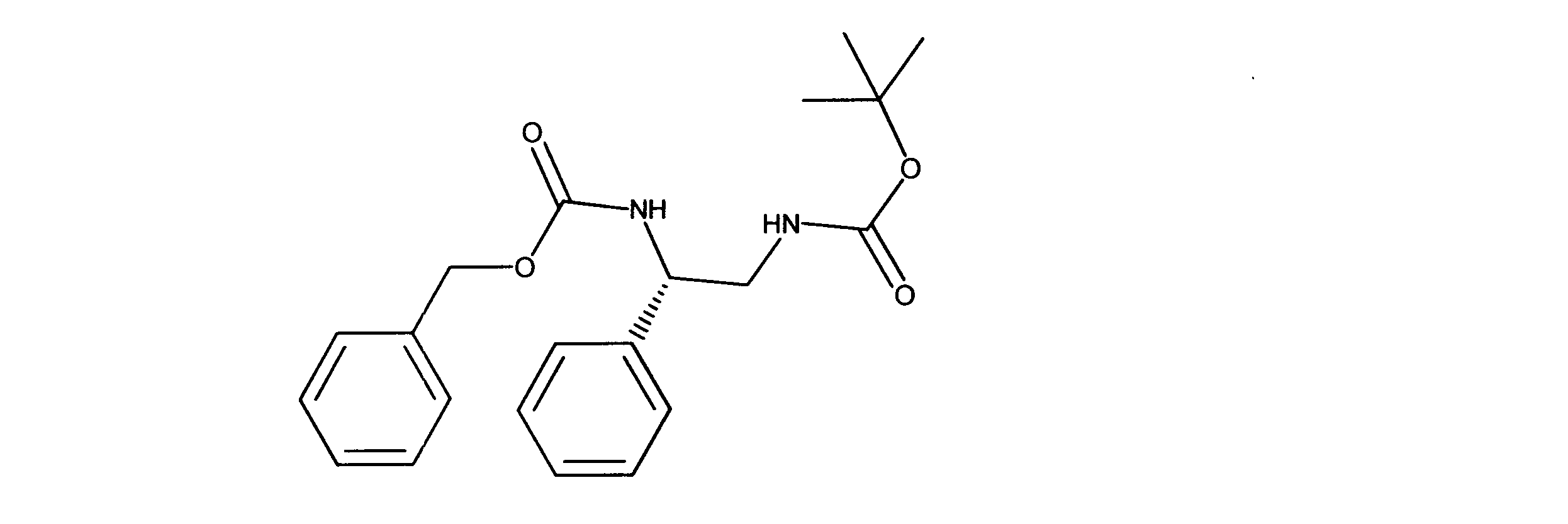
1HNMR (600 MHz, DMSO-d6) δ ppm 1.20 - 1.33 (m, 9 H) 3.11 (t, J=6.29 Hz, 2 H) 4.59 - 4.68 (m, 1 H) 4.88 - 5.01 (m, 2 H) 6.81 (t, J=5.42 Hz, 1 H) 7.14 - 7.35 (m, 10 H) 7.69 (d, J=8.49 Hz, 1 H). LCMS (ESI) 371 (M + H).
tert-ブチルN-[(2S)-2-(ベンジルオキシカルボニルアミノ)-3-メチル-ペンチル]カルバメートの合成、化合物20
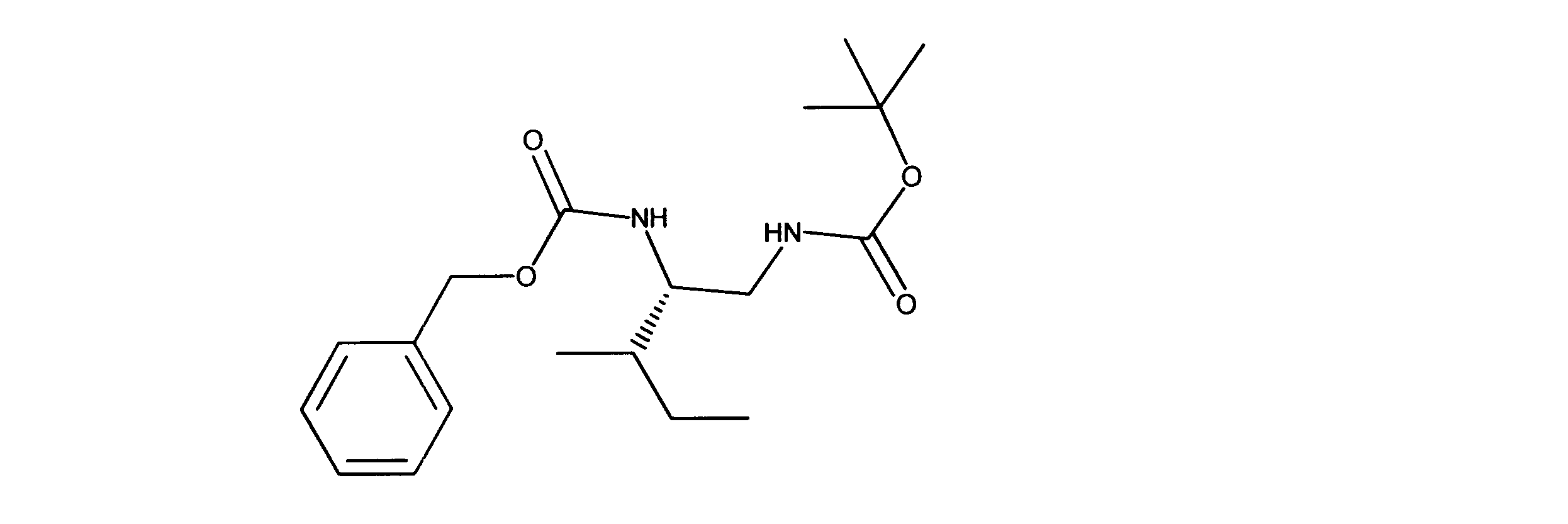
1HNMR (600 MHz, CHLOROFORM-d) δ ppm 0.85 - 0.92 (m, 6 H) 1.05 - 1.15 (m, 1 H) 1.35 - 1.41 (m, 9 H) 1.45 - 1.56 (m, 2 H) 3.14 - 3.24 (m, 2 H) 3.54 - 3.64 (m, 1 H) 4.78 (s, 1 H) 4.96 (d, J=7.91 Hz, 1 H) 5.06 (s, 2 H) 7.27 - 7.37 (m, 5 H). LCMS (ESI) 351 (M + H).
tert-ブチルN-[(2S)-2-(ベンジルオキシカルボニルアミノ)-3、3-ジメチルブチル]カルバメートの合成、化合物21
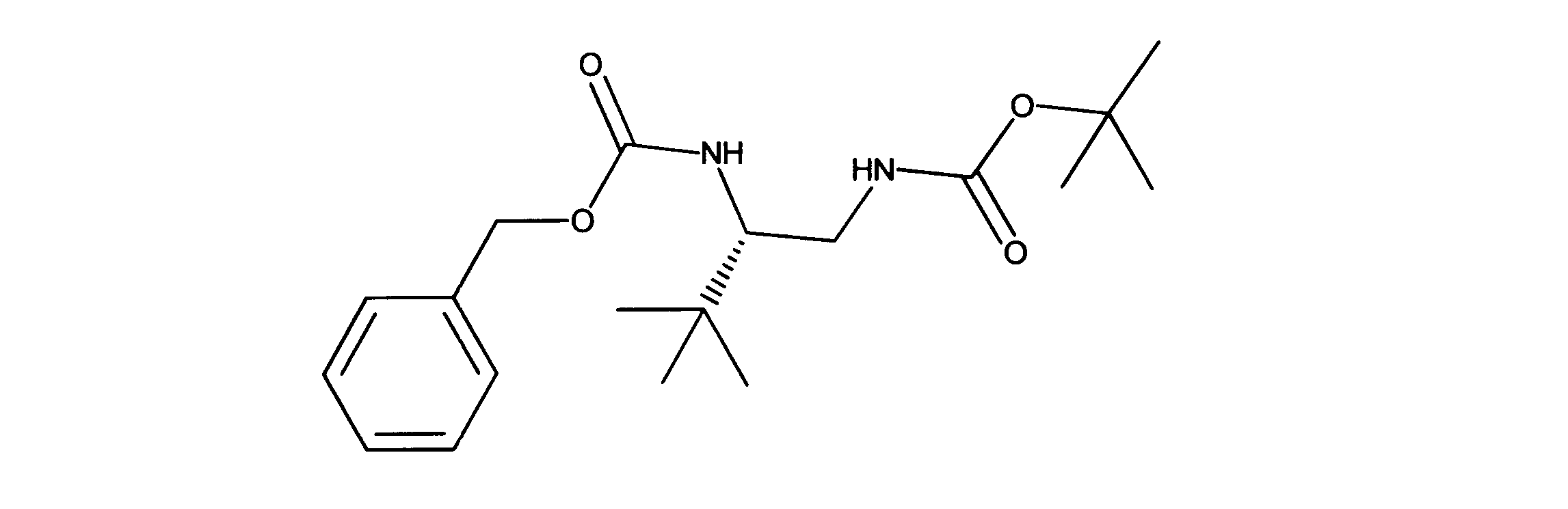
LCMS (ESI) 351.
tert-ブチルN-[[1-(ベンジルオキシカルボニルアミノ)シクロヘキシル]メチル]カルバメートの合成、化合物22
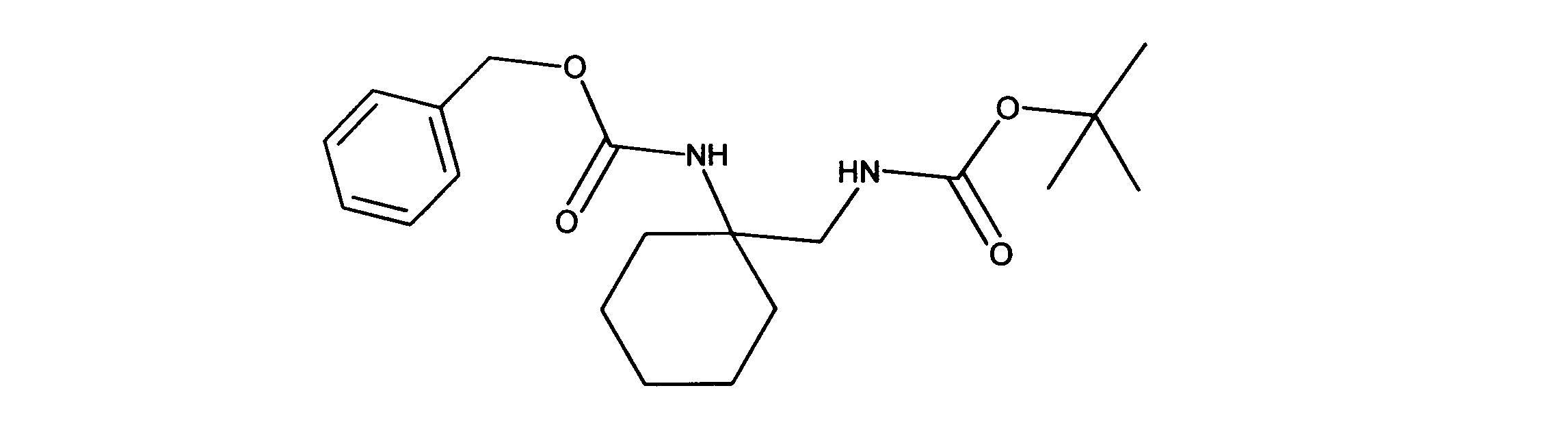
1HNMR (600 MHz, DMSO-d6) δ ppm 0.92 - 1.54 (m, 17 H) 1.76 - 2.06 (m, 2 H) 3.09 (d, J=6.15 Hz, 2 H) 4.92 (s, 2 H) 6.63 (d, J=17.27 Hz, 1 H) 7.16 - 7.49 (m, 6 H). LCMS (ESI) 363 (M + H).
tert-ブチルN-[[1-(ベンジルオキシカルボニルアミノ)シクロペンチル]メチル]カルバメートの合成、化合物23
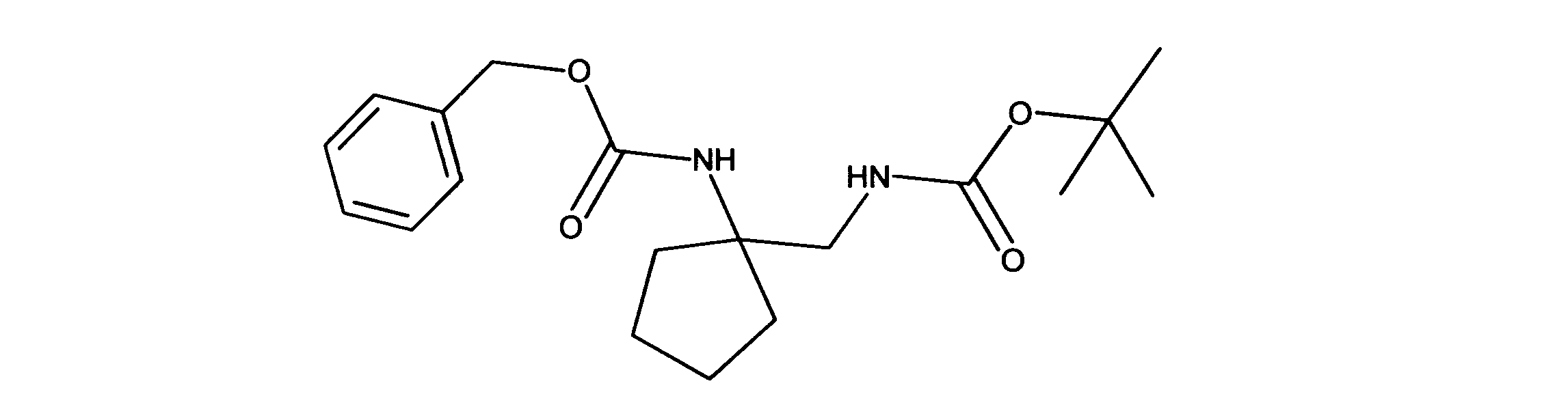
LCMS (ESI) 349 (M + H).
2-ニトロ-5-[4-(1-ピペリジル)-1-ピペリジル]ピリジンの合成、化合物24
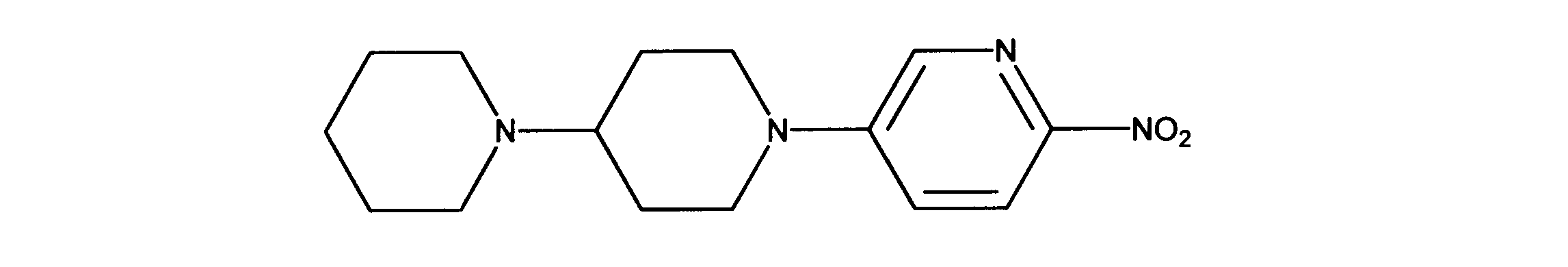
1HNMR (600 MHz, DMSO-d6) δ ppm 1.26 - 1.36 (m, 2 H) 1.43 (m, 6 H) 1.76 (m, 2 H) 2.37 (m, 5 H) 2.94 (t, J=12.74 Hz, 2 H) 4.06 (d, J=13.47 Hz, 2 H) 7.41 (dd, J=9.37, 2.64 Hz, 1 H) 8.08 (d, J=9.37 Hz, 1 H) 8.20 (d, J=2.64 Hz, 1 H)
5-[4-(1-ピペリジル)-1-ピペリジル]ピリジン-2-アミンの合成、化合物25
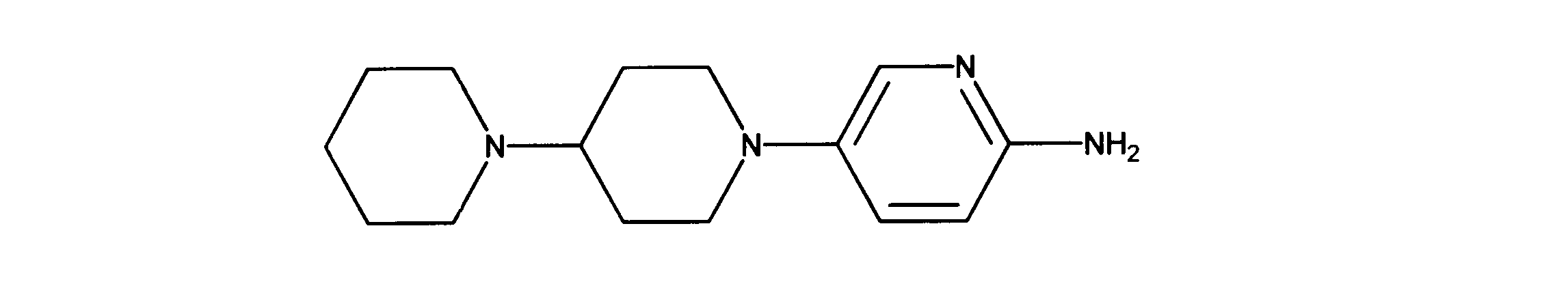
4-[1-(6-ニトロ-3-ピリジル)-4-ピペリジル]モルホリンの合成、化合物26
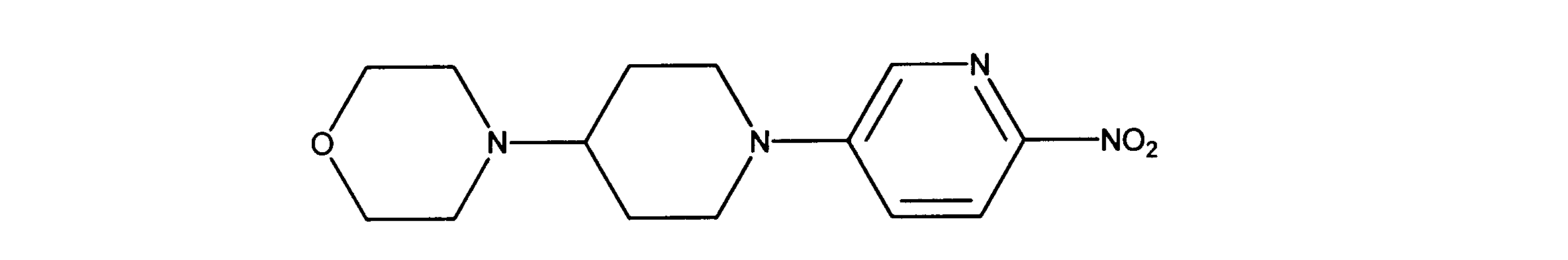
1HNMR (600 MHz, DMSO-d6) δ ppm 1.41 (m, 2 H) 1.82 (m, 2 H) 2.42 (m, 5 H) 2.98 (t, J=12.44 Hz, 2 H) 3.52 (s, 4 H) 4.04 (d, J=12.88 Hz, 2 H) 7.42 (d, J=9.37 Hz, 1 H) 8.08 (d, J=9.08 Hz, 1 H) 8.21 (s, 1 H).
5-(4-モルホリノ-1-ピペリジル)ピリジン-2-アミンの合成、化合物27

1HNMR (600 MHz, DMSO-d6) δ ppm 1.34 - 1.52 (m, 2 H) 1.78 (m, 2 H) 2.14 (m, 1 H) 2.43 (m, 4 H) 3.32 (d, J=12.30 Hz, 4 H) 3.47 - 3.59 (m, 4 H) 5.32 (s, 2 H) 6.34 (d, J=8.78 Hz, 1 H) 7.11 (dd, J=8.93, 2.78 Hz, 1 H) 7.47 - 7.62 (m, 1 H). LCMS (ESI) 263 (M + H).
4-[1-(6-ニトロ-3-ピリジル)-4-ピペリジル]チオモルホリンの合成、化合物28

1HNMR (600 MHz, DMSO-d6) δ ppm 1.40 - 1.52 (m, 2 H) 1.71 (m, 2 H) 2.49 - 2.55 (m, 4 H) 2.56 - 2.63 (m, 1 H) 2.68 - 2.75 (m, 4 H) 2.88 - 2.98 (m, 2 H) 4.09 (d, J=13.18 Hz, 2 H) 7.42 (dd, J=9.22, 3.07 Hz, 1 H) 8.08 (d, J=9.37 Hz, 1 H) 8.20 (d, J=3.22 Hz, 1 H) .
5-(4-チオモルホリノ-1-ピペリジル)ピリジン-2-アミンの合成、化合物29
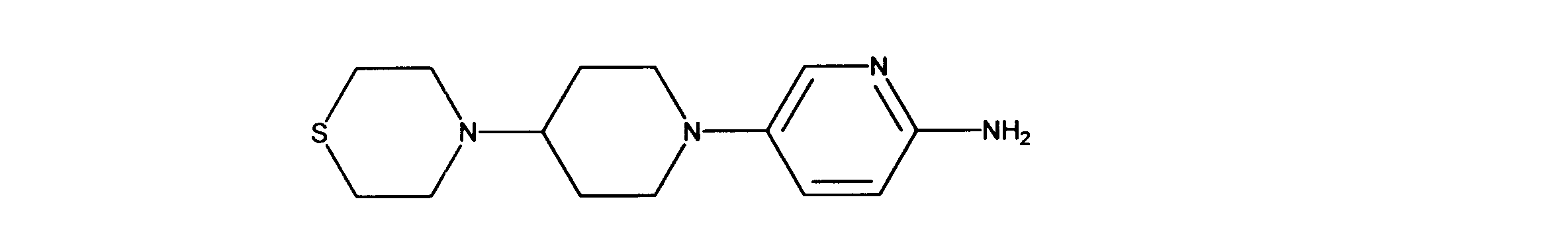
1HNMR (600 MHz, DMSO-d6) δ ppm 1.47 - 1.59 (m, 2 H) 1.65 (m, 2 H) 2.22 - 2.38 (m, 1 H) 2.50 - 2.59 (m, 6 H) 2.68 - 2.82 (m, 4 H) 3.33 (d, J=12.00 Hz, 2 H) 5.31 (s, 2 H) 6.33 (d, J=9.08 Hz, 1 H) 7.10 (dd, J=8.78, 2.93 Hz, 1 H) 7.55 (d, J=2.64 Hz, 1 H). LCMS (ESI) 279 (M + H) .
2-ニトロ-5-(1-ピペリジル)ピリジンの合成、化合物30
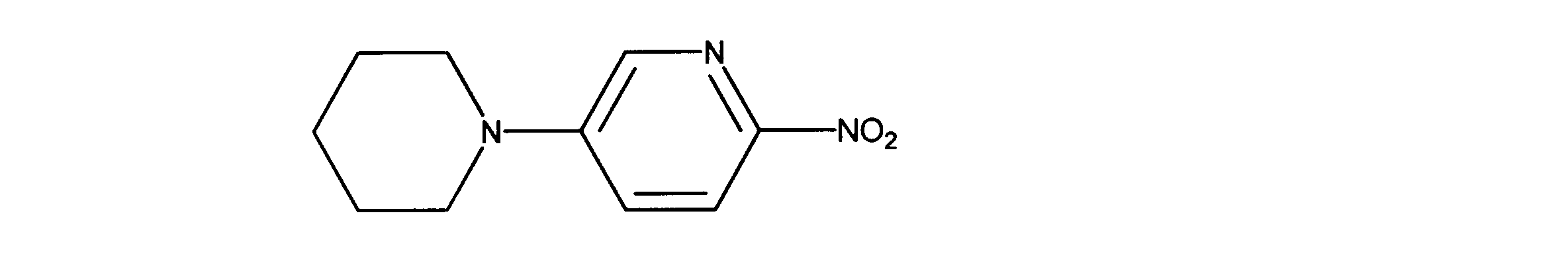
1HNMR (600 MHz, DMSO-d6) δ ppm 1.56 (m, 6 H) 3.49 (d, J=4.39 Hz, 4 H) 7.30 - 7.47 (m, 1 H) 8.02 - 8.12 (m, 1 H) 8.15 - 8.26 (m, 1 H).
5-(1-ピペリジル)ピリジン-2-アミンの合成、化合物31
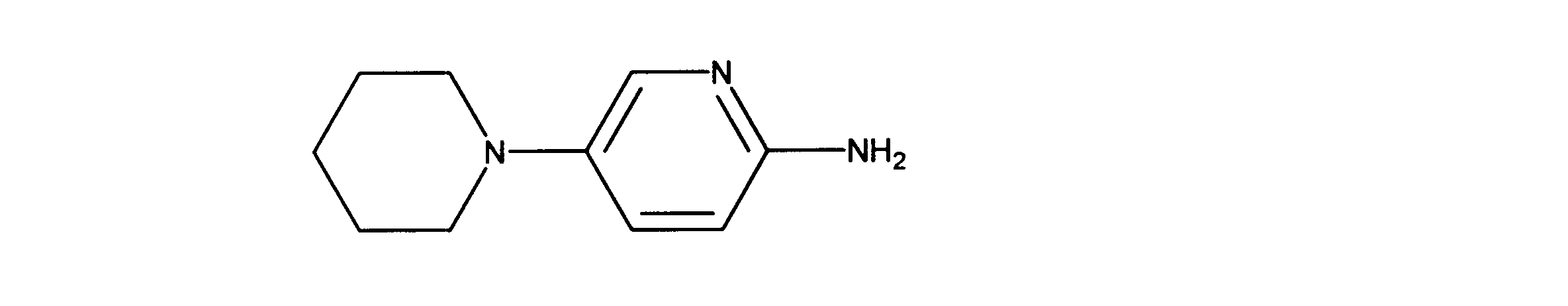
1HNMR (600 MHz, DMSO-d6) δ ppm 1.39 - 1.46 (m, 2 H) 1.51 - 1.62 (m, 4 H) 2.75 - 2.92 (m, 4 H) 5.30 (s, 2 H) 6.34 (d, J=8.78 Hz, 1 H) 7.09 (dd, J=8.78, 2.93 Hz, 1 H) 7.54 (d, J=2.93 Hz, 1 H). LCMS (ESI) 178 (M + H).
4-(6-ニトロ-3-ピリジル)チオモルホリンの合成、化合物32
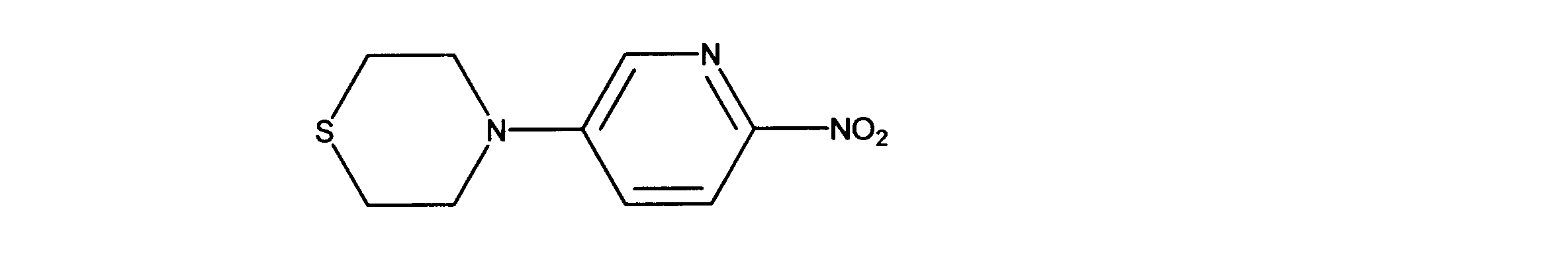
1HNMR (600 MHz, DMSO-d6) δ ppm 2.56 - 2.69 (m, 4 H) 3.79 - 3.92 (m, 4 H) 7.43 (dd, J=9.22, 3.07 Hz, 1 H) 8.10 (d, J=9.37 Hz, 1 H) 8.20 (d, J=2.93 Hz, 1 H).
5-チオモルホリノピリジン-2-アミンの合成、化合物33
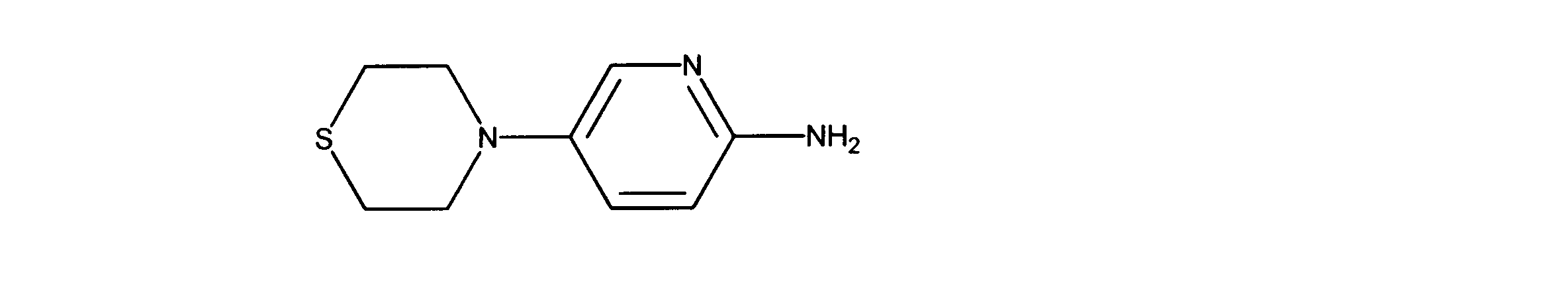
1HNMR (600 MHz, DMSO-d6) δ ppm 2.59 - 2.73 (m, 4 H) 3.04 - 3.20 (m, 4 H) 5.41 (s, 2 H) 6.35 (d, J=8.78 Hz, 1 H) 7.10 (dd, J=8.78, 2.93 Hz, 1 H) 7.57 (d, J=2.64 Hz, 1 H). LCMS (ESI) 196 (M + H).
tert-ブチル(4R)-5-(6-ニトロ-3-ピリジル)-2、5-ジアザビシクロ[2.2.1]ヘプタン-2-カルボン酸塩の合成、化合物34
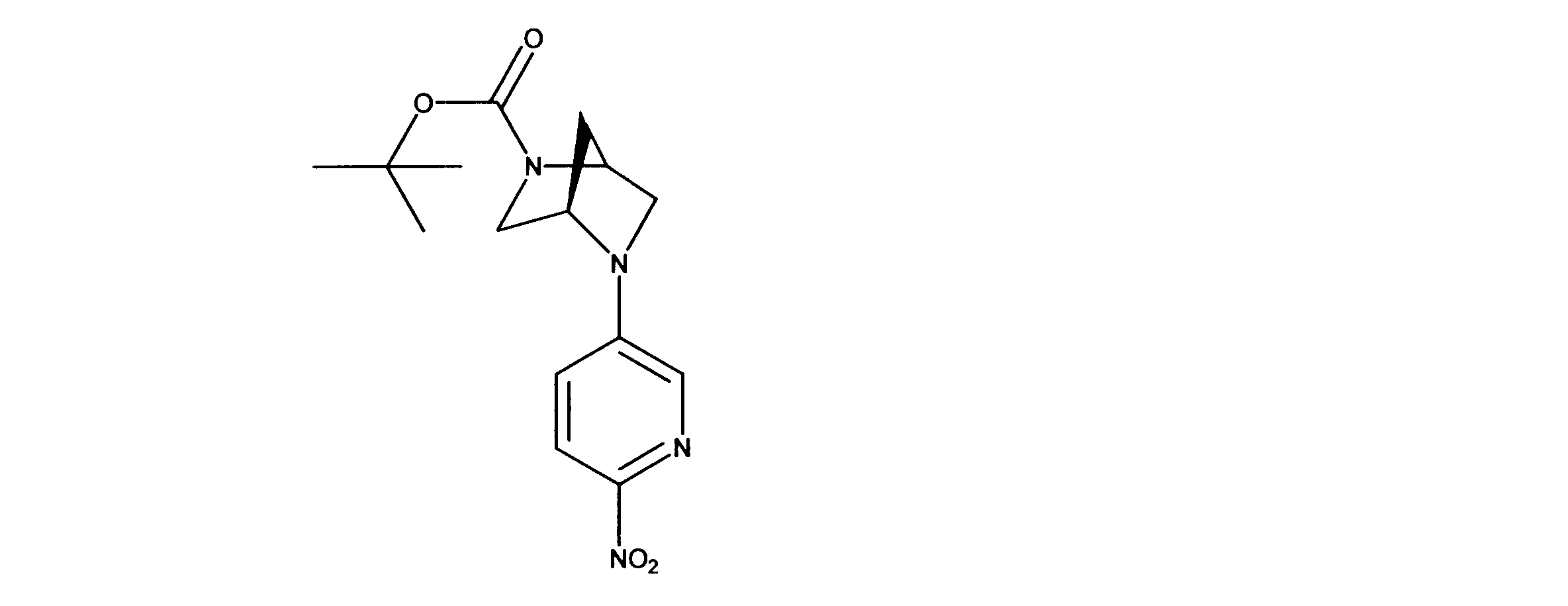
1HNMR (600 MHz, DMSO-d6) δ ppm 1.33 (d, J=32.21 Hz, 11 H) 1.91 (m, 2 H) 3.15 (d, J=10.25 Hz, 1 H) 3.58 (m, 1 H) 4.46 (m, 1 H) 4.83 (s, 1 H) 7.16 (s, 1 H) 7.94 (s, 1 H) 8.05 - 8.16 (m, 1 H).
tert-ブチル(4R)-5-(6アミノ3-ピリジル)-2、5-ジアザビシクロ[2.2.1]ヘプタン-2-カルボン酸塩の合成、化合物35
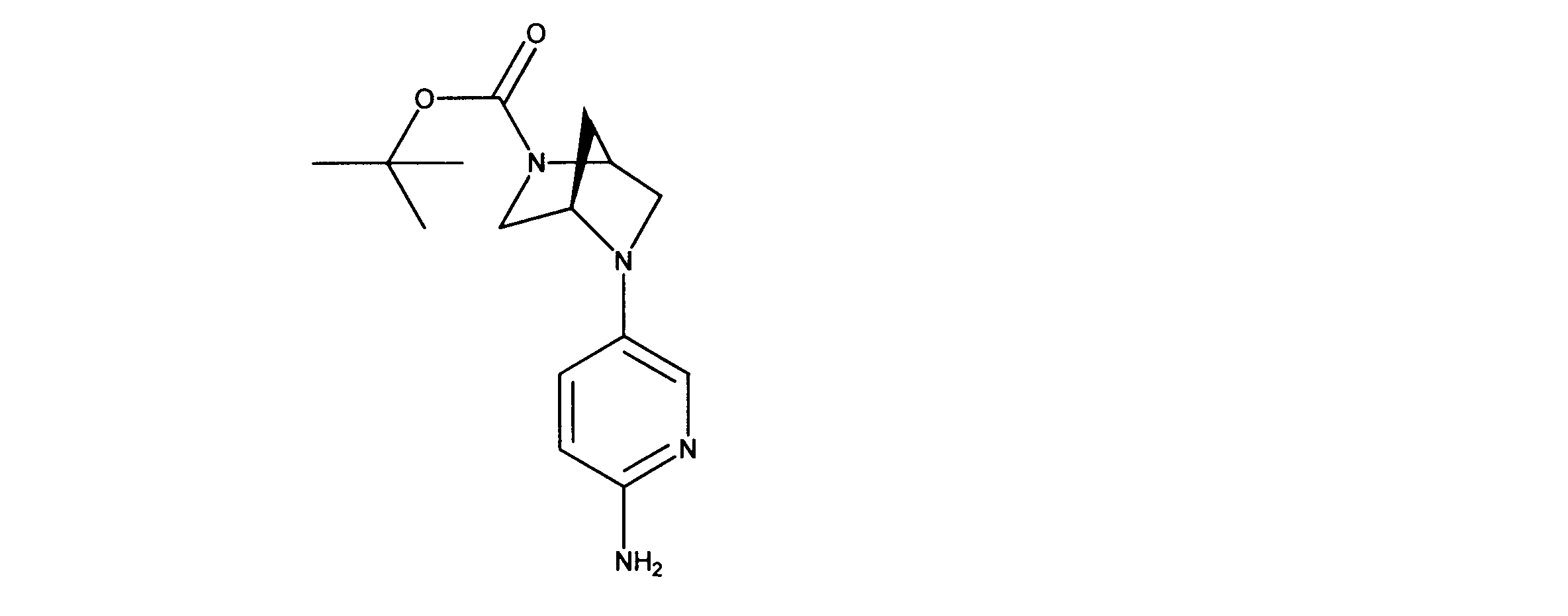
1HNMR (600 MHz, DMSO-d6) δ ppm 1.31 (d, J=31.91 Hz, 11 H) 1.83 (m, 2 H) 2.71 - 2.82 (m, 1 H) 3.44 (m,1 H) 4.30 (d, 2H) 5.08 (s, 2 H) 6.35 (d, J=8.78 Hz, 1 H) 6.77 - 6.91 (m, 1 H) 7.33 (s, 1 H). LCMS (ESI) 291 (M + H).
N,N-ジメチル-1-(6-ニトロ-3-ピリジル)ピペリジン-4-アミンの合成、化合物36
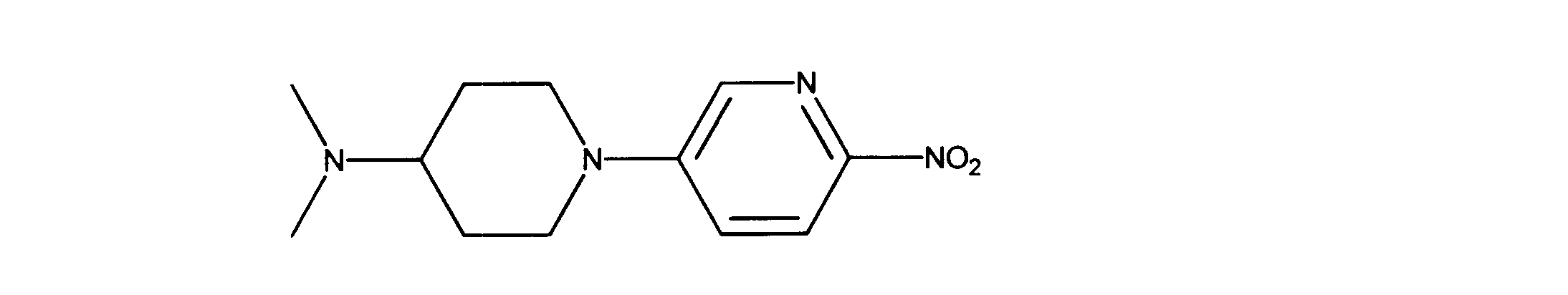
1HNMR (600 MHz, DMSO-d6) δ ppm 1.30 - 1.45 (m, 2 H) 1.79 (m, 2 H) 2.14 (s, 6 H) 2.33 (m, 1 H) 2.92 - 3.04 (m, 2 H) 4.03 (d, J=13.76 Hz, 2 H) 7.42 (dd, J=9.22, 3.07 Hz, 1 H) 8.04 - 8.11 (m, 1 H) 8.21 (d, J=2.93 Hz, 1 H).
5-[4-(ジメチルアミノ)-1-ピペリジル]ピリジン-2-アミンの合成、化合物37
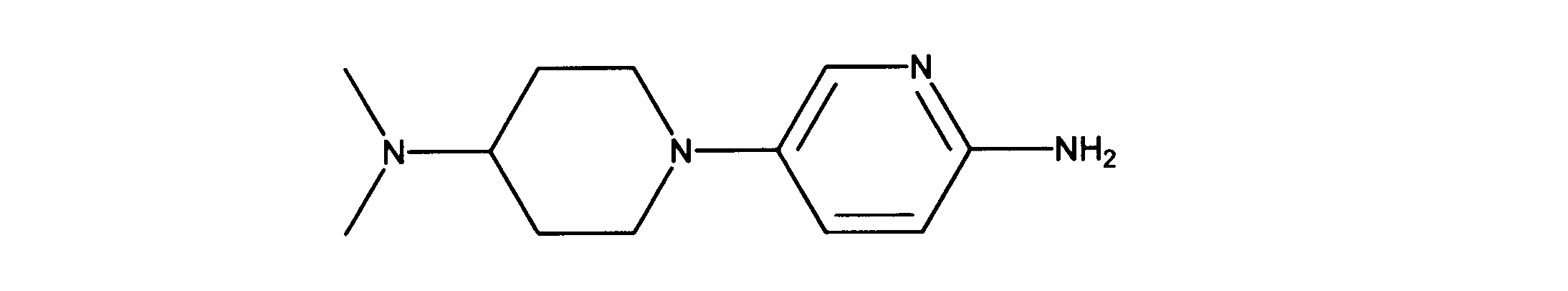
1HNMR (600 MHz, DMSO-d6) δ ppm 1.35 - 1.50 (m, 2 H) 1.69 - 1.81 (m, 2 H) 2.00 - 2.10 (m, 1 H) 2.11 - 2.22 (s, 6 H) 3.17 - 3.36 (m, 4 H) 5.19 - 5.38 (s, 2 H) 6.34 (d, J=8.78 Hz, 1 H) 7.10 (dd, J=8.78, 2.93 Hz, 1 H) 7.55 (d, J=2.63 Hz, 1 H). LCMS (ESI) 221 (M + H).
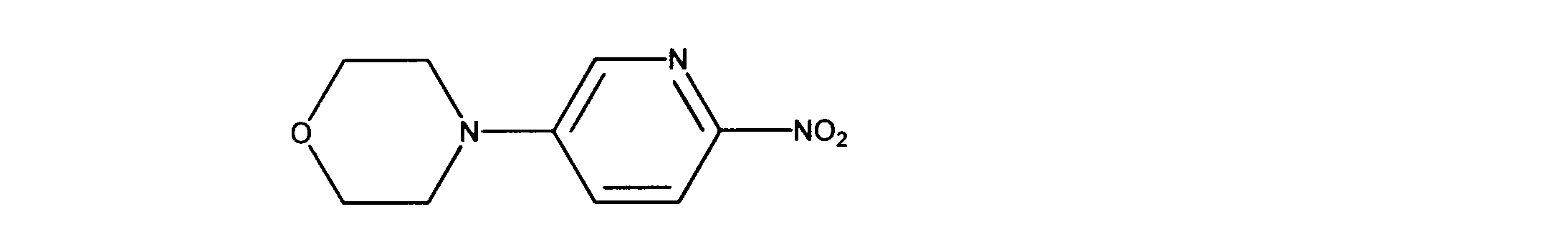
4-(6-ニトロ-3-ピリジル)モルホリンの合成、化合物38
5-モルホリノピリジン-2-アミンの合成、化合物39
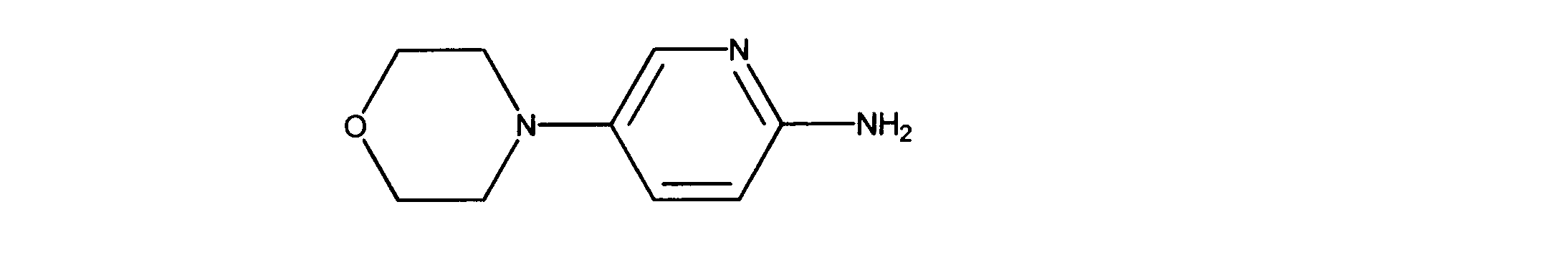
1HNMR (600 MHz, CHLOROFORM-d) δ ppm 2.91 - 3.00 (m, 4 H) 3.76 - 3.84 (m, 4 H) 4.19 (br. s., 2 H) 6.45 (d, J=8.78 Hz, 1 H) 7.12 (dd, J=8.78, 2.93 Hz, 1 H) 7.72 (d, J=2.93 Hz, 1 H).
5-(4-イソブチルピペラジン-1-イル)ピリジン-2-アミンの合成、化合物40
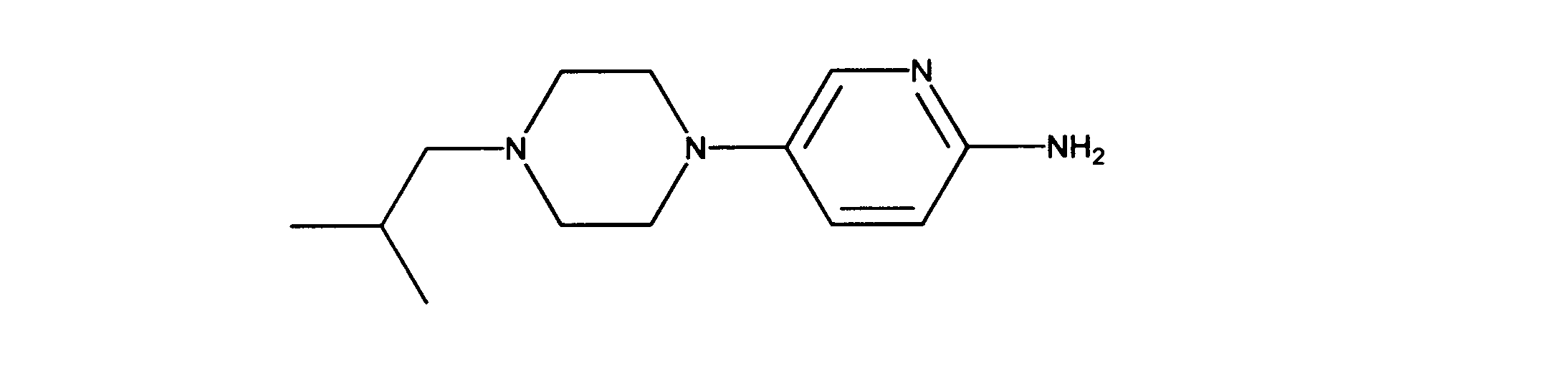
1HNMR (600 MHz, CHLOROFORM-d) δ ppm 0.88 (d, J=6.73 Hz, 6 H) 1.71 - 1.84 (m, 1 H) 2.10 (d, J=7.32 Hz, 2 H) 2.46 - 2.58 (m, 4 H) 2.97 - 3.07 (m, 4 H) 4.12 (s, 2 H) 6.45 (d, J=8.78 Hz, 1 H) 7.14 (dd, J=8.78, 2.93 Hz, 1 H) 7.75 (d, J=2.93 Hz, 1 H). LCMS (ESI) 235 (M + H).
5-(4-イソプロピルピペラジン-1-イル)ピリジン-2-アミンの合成、化合物41
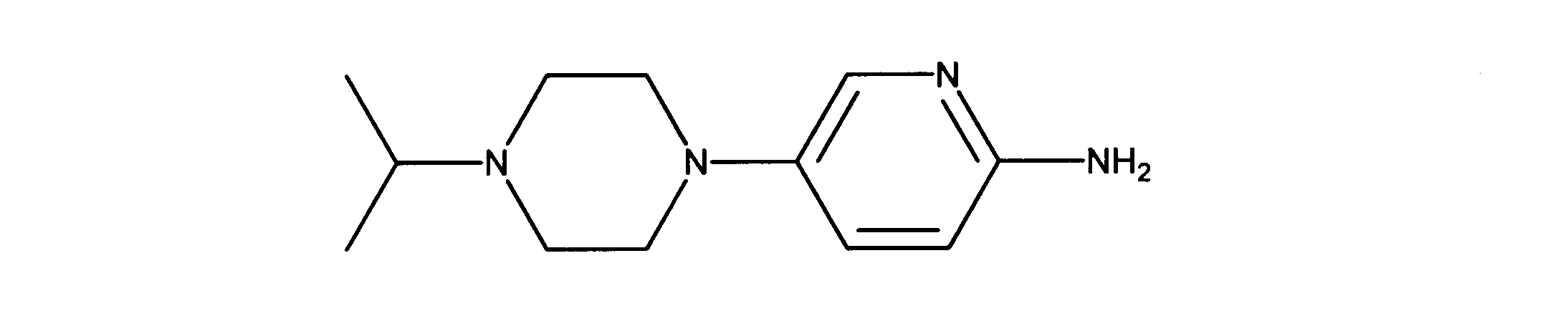
1HNMR (600 MHz, CHLOROFORM-d) δ ppm 1.06 (d, J=6.44 Hz, 6 H) 2.59 - 2.75 (m, 5 H) 2.97 - 3.10 (m, 4 H) 4.13 (s, 2 H) 6.45 (d, J=8.78 Hz, 1 H) 7.15 (dd, J=9.08, 2.93 Hz, 1 H) 7.76 (d, J=2.93 Hz, 1 H). LCMS (ESI) 221 (M + H).
5-[(2R,6S)-2,6-ジメチルモルホリン-4-イル]ピリジン-2-アミンの合成、化合物42
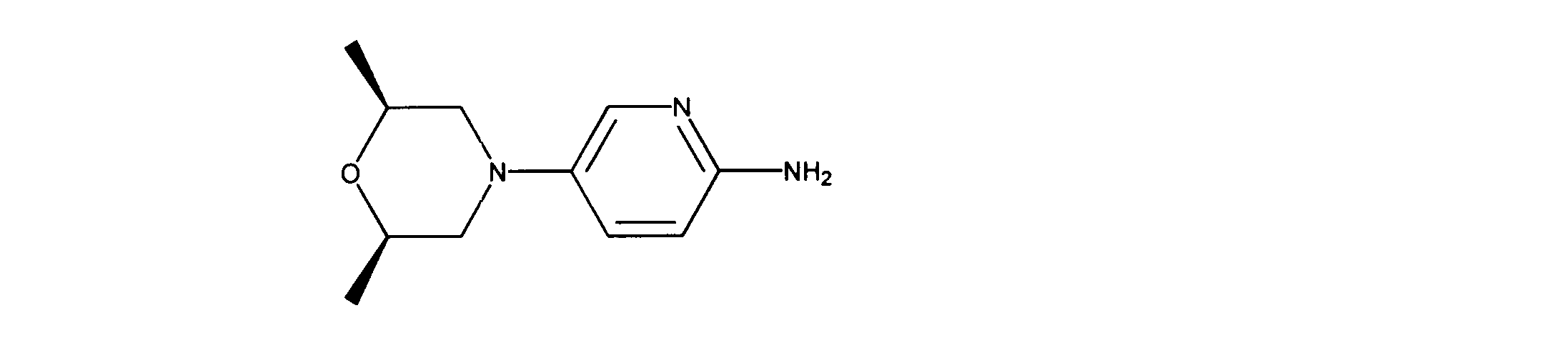
1HNMR (600 MHz, CHLOROFORM-d) δ ppm 1.20 (d, J=6.44 Hz, 6 H) 2.27 - 2.39 (m, 2 H) 3.11 - 3.21 (m, 2 H) 3.70 - 3.84 (m, 2 H) 4.15 (s, 2 H) 6.45 (d, J=8.78 Hz, 1 H) 7.12 (dd, J=8.78, 2.93 Hz, 1 H) 7.72 (d, J=2.63 Hz, 1 H). LCMS (ESI) 208 (M + H).
5-[(3R,5S)-3,5-ジメチルピペラジン-1-イル]ピリジン-2-アミンの合成、化合物43
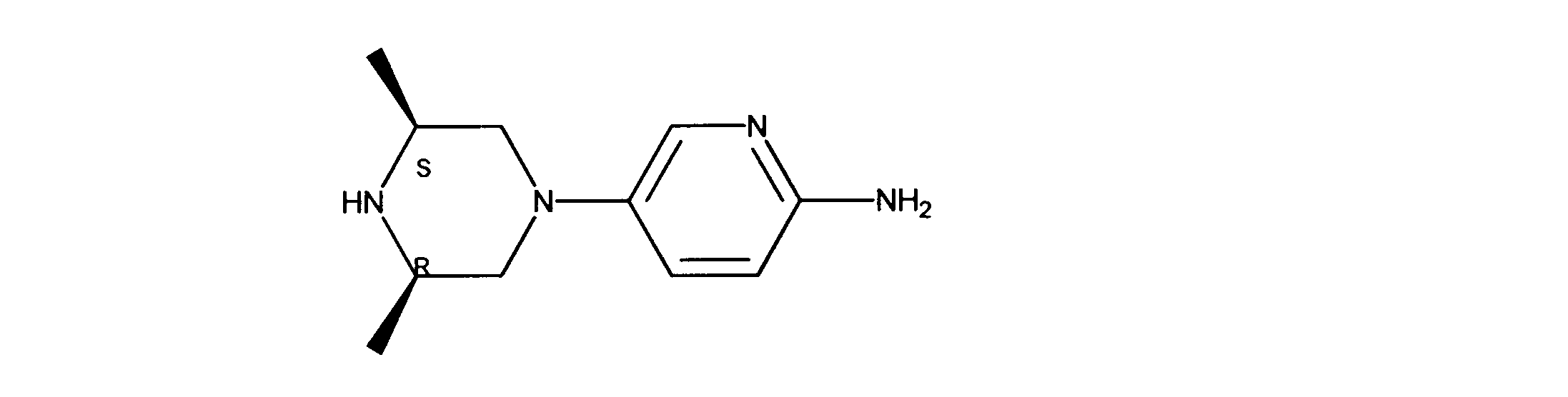
1HNMR (600 MHz, CHLOROFORM-d) δ ppm 1.09 (d, J=6.44 Hz, 6 H) 2.20 (t, J=10.83 Hz, 2 H) 2.95 - 3.08 (m, 2 H) 3.23 (dd, J=11.71, 2.05 Hz, 2 H) 4.13 (s, 2 H) 6.45 (d, J=8.78 Hz, 1 H) 7.14 (dd, J=8.78, 2.93 Hz, 1 H) 7.73 (d, J=2.63 Hz, 1 H). LCMS (ESI) 207 (M + H).
化合物44の合成
1HNMR (600 MHz, DMSO-d6) δ ppm 0.77 - 0.85 (d, J=6.5 Hz, 3 H) 0.87 (d, J=6.73 Hz, 3 H) 1.31 - 1.39 (m, 9 H) 1.82 - 1.93 (m, 1 H) 2.94 (d, J=5.56 Hz, 1 H) 3.08 - 3.22 (m, 2 H) 3.98 (d, J=8.20 Hz, 1 H) 6.96 (d, J=8.78 Hz, 1 H) 8.21 (s, 1 H). LCMS (ESI) 393 (M + H).
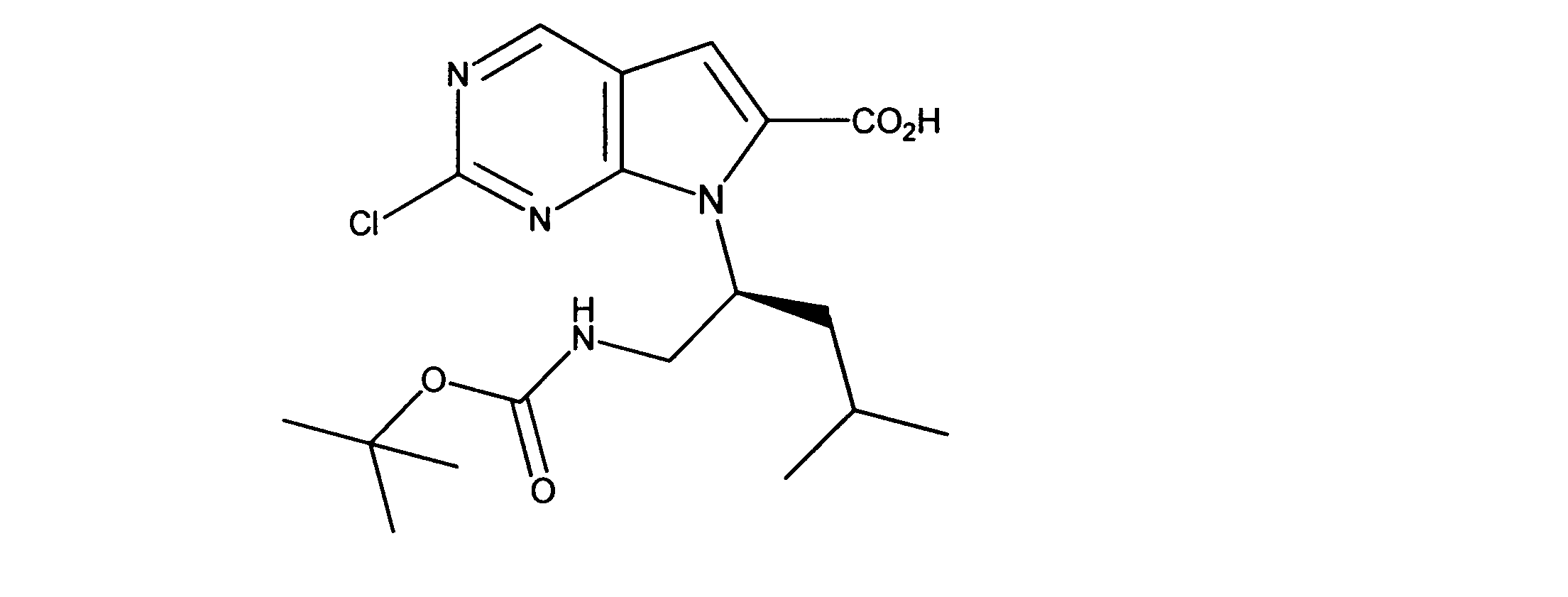
1HNMR (600 MHz, DMSO-d6) δ ppm 1.11 (d, J=6.44 Hz, 3 H) 1.18 (t, J=7.03 Hz, 6 H) 1.21 - 1.26 (m, 12 H) 2.88 (br. s., 1 H) 3.43 - 3.78 (m, 6 H) 3.97 - 4.08 (m, 1 H) 5.61 (s, 1 H) 6.65 (s, 1 H) 6.71 - 6.78 (m, 1 H) 8.87 (s, 1 H). LCMS (ESI) 441 (M + H).
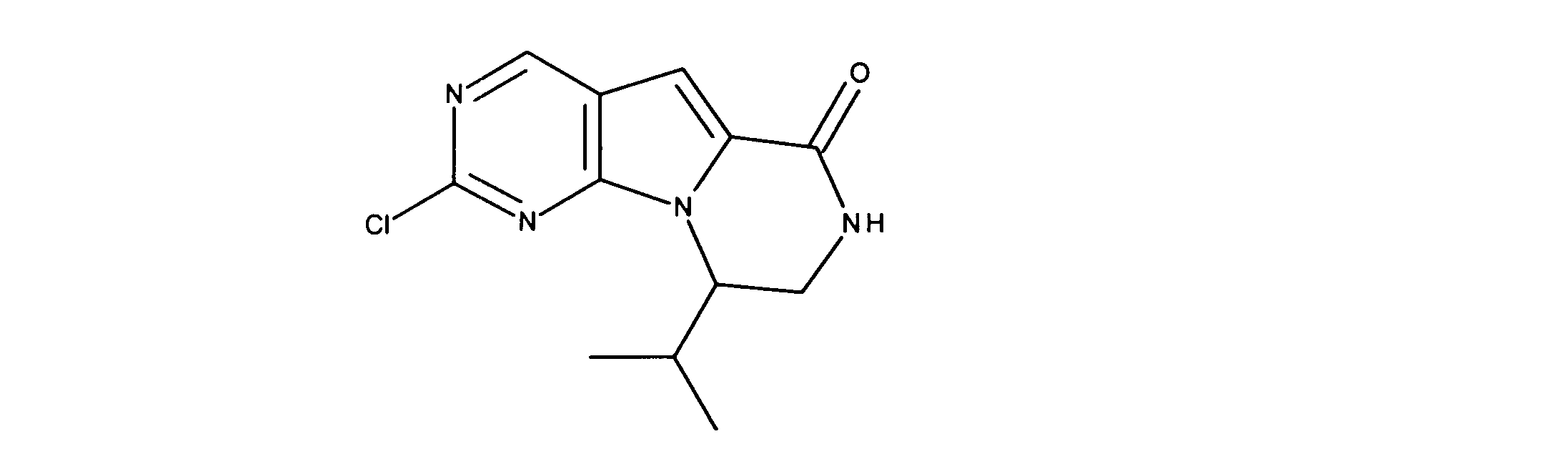
有機層を分離し、乾燥し、その後真空下で濃縮した。このように得られた粗反応生成物を、DMF中に溶解し、その後オキソンを添加し、内容物を3時間撹拌した。酢酸エチルの添加の後、CELITETMを通して反応物混合物をろ過し、真空下で濃縮した。粗製品を、ヘキサン/酢酸エチル(0~100%)を用いたシリカゲル上のカラムクロマトグラフィーにかけて、7-[1-[(第三級ブトキシカルボニルアミノ)メチル]-2メチル-プロピル]-2-クロロ-ピロロ[2,3-d]ピリミジン-6-カルボン酸を提供した。
1HNMR (600 MHz, DMSO-d6) δ ppm 0.85 (d, J=7.03 Hz, 3 H) 0.97 (d, J=6.73 Hz, 3 H) 1.52 (s, 9 H) 1.99 - 2.23 (m, 1 H) 3.98 (dd, J=14.05, 3.51 Hz, 1 H) 4.47 - 4.71 (m, 2 H) 7.47 (s, 1 H) 9.17 (s, 1 H). LCMS (ESI) 383 (M + H).
DCM(1.5mL)中の7-[1-[(第三級ブトキシカルボニルアミノ)メチル]-2メチル-プロピル]-2-クロロ-ピロロ[2,3-d]ピリミジン-6カルボン酸(0.050g、0.00013モル)に、DIC(32.7mg)及びDMAP(10mg)を添加した。内容物を、2時間撹拌した。その後、トリフルオロ酢酸(0.4mL)を添加し、さらに撹拌を30分間続けた。過剰な酸を中和する飽和NaHCO3の添加の後、酢酸エチルを添加し、有機層を分離して、硫酸マグネシウムを用いて乾燥し、その後真空下で濃縮した。ヘキサン/酢酸エチル(0- 100 %)を用いたシリカゲルカラムクロマトグラフィーにより、粗製品を精製し、製品を提供した。
1HNMR (600 MHz, DMSO-d6) δ ppm 0.72 (d, J=6.73 Hz, 3 H) 0.97 (d, J=6.73 Hz, 3 H) 2.09 - 2.22 (m, 1 H) 3.57 (dd, J=13.18, 4.98 Hz, 1 H) 3.72 (dd, J=13.61, 4.25 Hz, 1 H) 4.53 (dd, J=8.05, 3.95 Hz, 1 H) 7.20 (s, 1 H) 8.34 (d, J=4.98 Hz, 1 H) 9.08 (s, 1 H). LCMS (ESI) 265 (M + H).
化合物45の合成
化合物46の合成
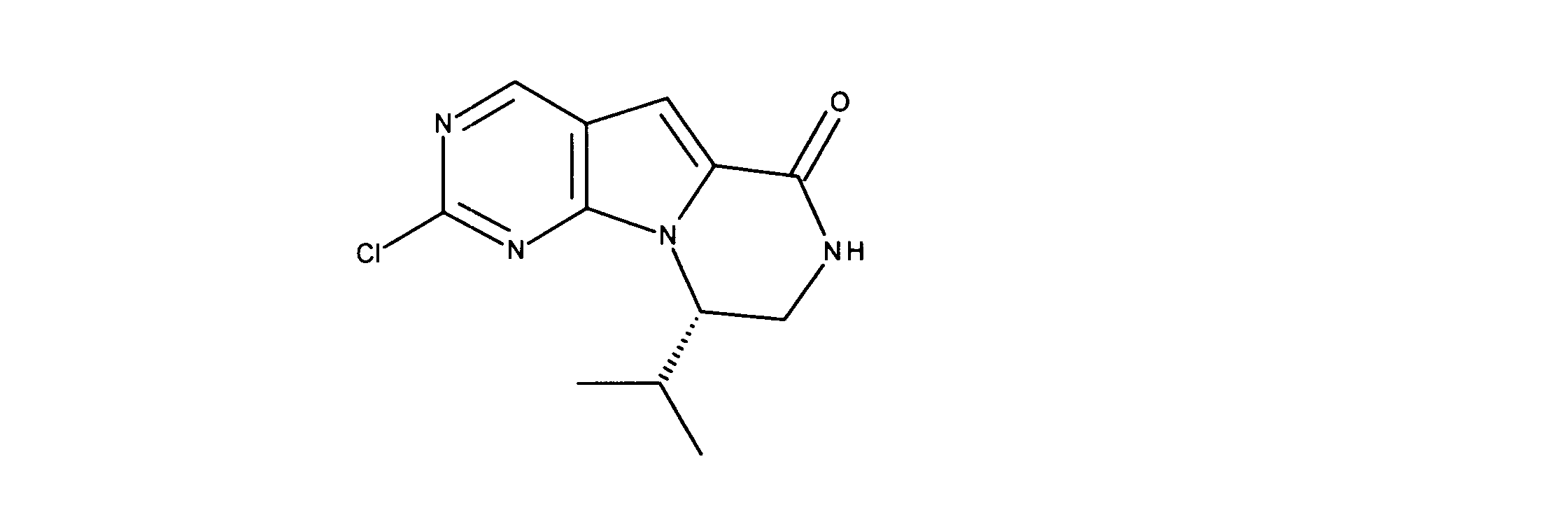
化合物47の合成
1HNMR (600 MHz, DMSO-d6) δ ppm 0.74 (d, J=6.73 Hz, 3 H) 0.91 (d, J=6.73 Hz, 3 H) 2.04 - 2.20 (m, 1 H) 3.04 (s, 3 H) 3.69 (dd, J=13.76, 1.17 Hz, 1 H) 3.96 (dd, J=13.76, 4.68 Hz, 1 H) 4.58 (dd, J=7.32, 3.51 Hz, 1 H) 7.16 (s, 1 H) 9.05 (s, 1 H). LCMS (ESI) 279 (M + H).
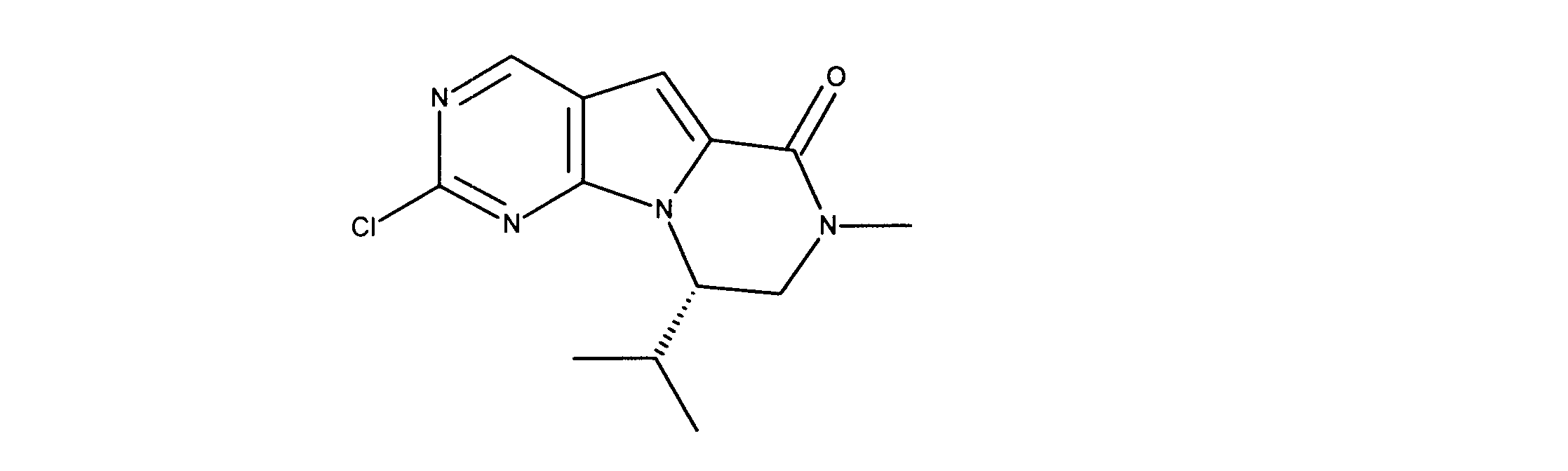
化合物48の合成
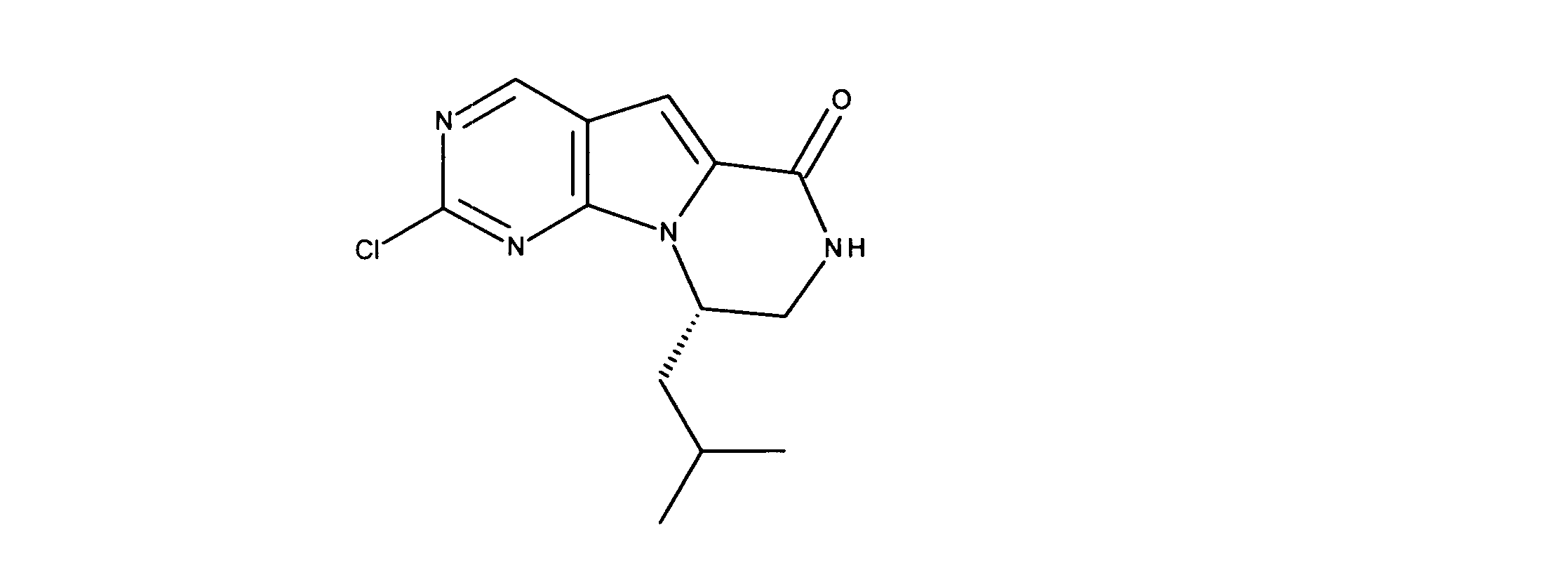
1HNMR (600 MHz, CHLOROFORM-d) δ ppm 0.91 (d, J=6.44 Hz, 3 H) 0.94 (d, J=6.44 Hz, 3 H) 1.32 - 1.51 (m, 11 H) 1.55 - 1.67 (m, 1 H) 3.28 (t, J=5.86 Hz, 2 H) 4.21 - 4.42 (m, 1 H) 4.84 (s, 1 H) 5.84 (d, J=7.32 Hz, 1 H) 8.07 (s, 1 H). LCMS (ESI) 407 (M + H).
LCMS (ESI) 455 (M + H).
1HNMR (600 MHz, DMSO-d6) δ ppm 0.88 (d, J=6.44 Hz, 3 H) 0.97 (d, J=6.44 Hz, 3 H) 1.47 (s, 9 H) 1.49 - 1.54 (m, 1 H) 1.56 (t, J=7.17 Hz, 2 H) 3.98 (dd, J=13.91, 3.07 Hz, 1 H) 3.76 (dd, J=13.31, 4.13 Hz, 1 H) 4.38 (d, J=14.05 Hz, 1 H) 4.90 (t, J=7.17 Hz, 1 H) 7.41 (s, 1 H) 9.11 (s, 1 H). LCMS (M + H) 397.
1HNMR (600 MHz, DMSO-d6) δ ppm 0.82 (d, J=6.73 Hz, 3 H) 0.97 (d, J=6.44 Hz, 3 H) 1.34 - 1.46 (m, 1 H) 1.48 - 1.65 (m, 2 H) 3.40 (dd, J=13.32, 5.42 Hz, 1 H) 3.76 (dd, J=13.47, 4.10 Hz, 1 H) 4.76 - 4.92 (m, 1 H) 7.17 (s, 1 H) 8.34 (d, J=5.27 Hz, 1 H) 9.04 (s, 1 H). LCMS (ESI) 279 (M + H) .
化合物49の合成
1HNMR (600 MHz, DMSO-d6) δ ppm 0.82 (d, J=6.44 Hz, 3 H) 0.97 (d, J=6.44 Hz, 3 H) 1.37 - 1.68 (m, 3 H) 3.04 (s, 3 H) 3.56 (d, J=13.47 Hz, 1 H) 4.00 (dd, J=13.32, 4.25 Hz, 1 H) 4.82 - 4.94 (m, 1 H) 7.16 (s, 1 H) 9.03 (s, 1 H). LCMS (ESI) 293 (M + H).
化合物50の合成
1HNMR (600 MHz, CHLOROFORM-d) δ ppm 0.88 - 0.95 (m, 6 H) 1.11 - 1.20 (m, 1 H) 1.34 (s, 9 H) 1.44 - 1.54 (m, 1 H) 1.64 - 1.72 (m, 1 H) 3.17 - 3.27 (m, 1 H) 3.33 - 3.43 (m, 1 H) 4.11 - 4.21 (m, 1 H) 4.81 (s, 1 H) 5.92 (d, J=8.20 Hz, 1 H) 8.05 (s, 1 H). LCMS (ESI) 407.
1HNMR (600 MHz, DMSO-d6) δ ppm 0.76 - 0.89 (m, 6 H) 1.03 (q, J=7.22 Hz, 3 H) 1.10 - 1.17 (m, 3 H) 1.25 - 1.42 (m, 11 H) 1.59 - 1.73 (m, 1 H) 3.35 - 3.47 (m, 4 H) 3.51 - 3.73 (m, 2 H) 3.99 - 4.11 (m, 1 H) 5.52 - 5.56 (m, 1 H) 6.76 - 7.03 (m, 2 H) 8.12 - 8.23 (m, 1 H). LCMS (ESI) 455 (M + H).
1HNMR (600 MHz, DMSO-d6) δ ppm 0.80 (t, J=7.47 Hz, 3 H) 0.86 (d, J=7.03 Hz, 3 H) 1.06 - 1.30 (m, 2 H) 1.48 (s, 9 H) 1.79 - 1.96 (m, 1 H) 3.95 (dd, J=14.05, 3.22 Hz, 1 H) 4.52 (d, J=14.35 Hz, 1 H) 4.61 - 4.73 (m, 1 H) 7.43 (s, 1 H) 9.13 (s, 1 H). LCMS (ESI) 397 (M + H).
1HNMR (600 MHz, DMSO-d6) δ ppm 0.74 (t, J=7.32 Hz, 3 H) 0.89 (d, J=6.73 Hz, 3 H) 1.00 - 1.12 (m, 2 H) 1.82 - 1.94 (m, 1 H) 3.55 (dd, J=13.91, 4.83 Hz, 1 H) 3.70 (dd, J=13.61, 4.25 Hz, 1 H) 4.57 (dd, J=7.91, 4.10 Hz, 1 H) 7.17 (s, 1 H) 8.31 (d, J=5.27 Hz, 1 H) 9.05 (s, 1 H). LCMS (ESI) 279 (M + H).
化合物51の合成
1HNMR (600 MHz, DMSO-d6) δ ppm 0.77 (t, J=7.47 Hz, 3 H) 0.84 (d, J=6.73 Hz, 3 H) 1.07 - 1.16 (m, 2 H) 1.82 - 1.95 (m, 1 H) 3.03 (s, 3 H) 3.68 (d, J=13.76 Hz, 1 H) 3.96 (dd, J=13.76, 4.39 Hz, 1 H) 4.59 - 4.70 (m, 1 H) 7.16 (s, 1 H) 9.04 (s, 1 H). LCMS (ESI) 293 (M + H).
化合物52の合成
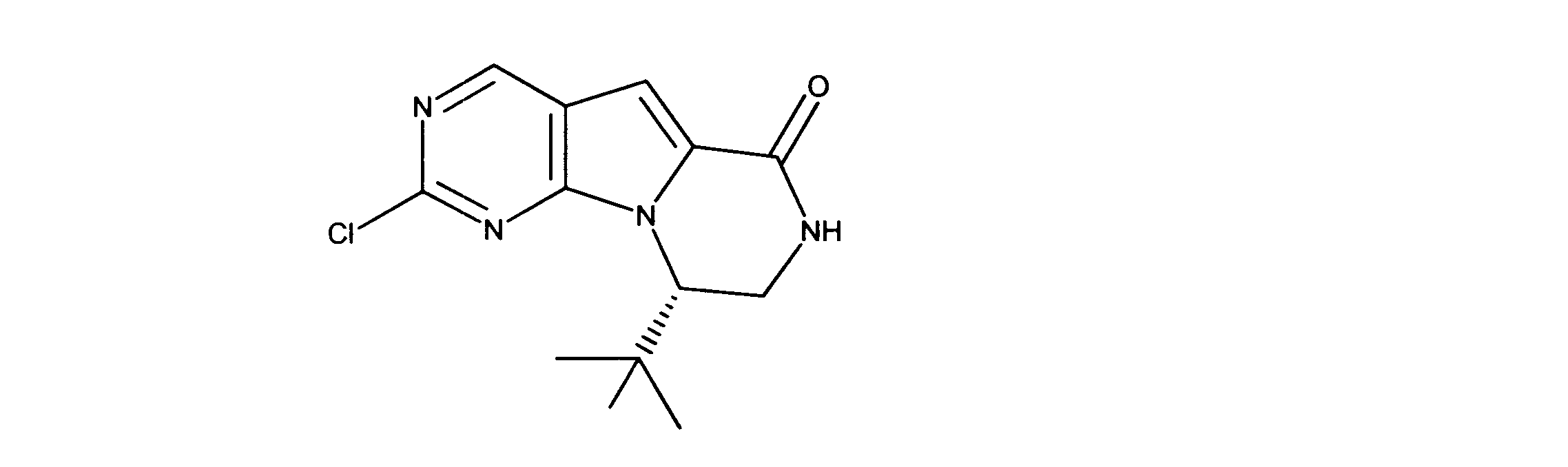
LCMS (ESI) 407 (M + H).
LCMS (ESI) 455 (M + H).
LCMS (ESI) 397 (M + H).
LCMS (ESI) 279 (M + H).
化合物53の合成

LCMS (ESI) 293 (M + H).
化合物54の合成
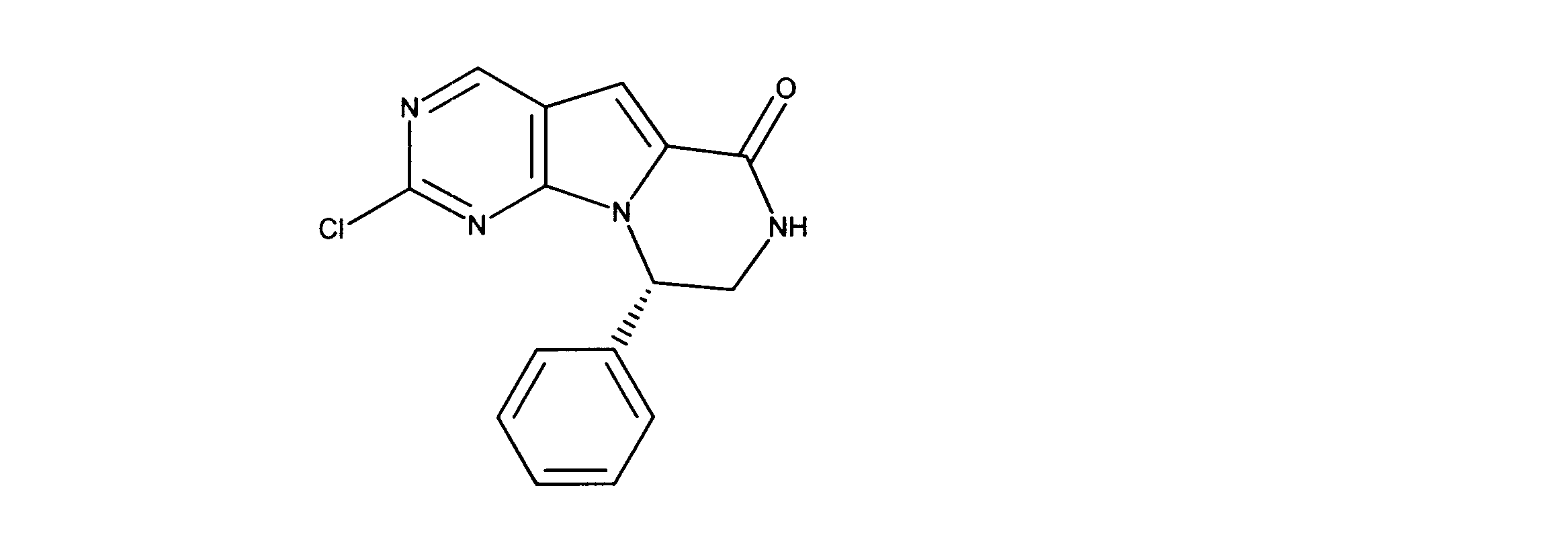
1HNMR (600 MHz, DMSO-d6) δ ppm 1.32 (s, 9 H) 3.29 - 3.50 (m, 2 H) 5.12 - 5.24 (m, 1 H) 7.10 (t, J=5.27 Hz, 1 H) 7.21 (t, J=6.88 Hz, 1 H) 7.26 - 7.34 (m, 4 H) 7.89 (d, J=7.32 Hz, 1 H) 8.24 (s, 1 H). LCMS (ESI) 427 (M + H).
1HNMR (600 MHz, DMSO-d6) δ ppm 1.14 (t, J=7.03 Hz, 6 H) 1.32 (s, 9 H) 3.39 (s, 2 H) 3.52 - 3.61 (m, 2 H) 3.64 - 3.73 (m, 2 H) 5.17 - 5.26 (m, 1 H) 5.57 (s, 1 H) 7.07 - 7.14 (m, 1 H) 7.20 - 7.25 (m, 1 H) 7.26 - 7.33 (m, 4 H) 7.90 (d, J=7.61 Hz, 1 H) 8.19 (s, 1 H). LCMS (ESI) 475 (M + H).
LCMS (ESI) 417 (M + H).
化合物44について記載されたと類似の合成シーケンスを用いて、化合物54を合成した。
1HNMR (600 MHz, DMSO-d6) δ ppm 3.58 - 3.69 (m, 1 H) 4.13 (dd, J=13.47, 4.39 Hz, 1 H) 6.07 (d, J=3.81 Hz, 1 H) 6.85 (d, J=7.32 Hz, 2 H) 7.19 - 7.31 (m, 3 H) 7.34 (s, 1 H) 8.27 (d, J=5.27 Hz, 1 H) 9.13 (s, 1 H). LCMS (ESI) 299 (M + H).
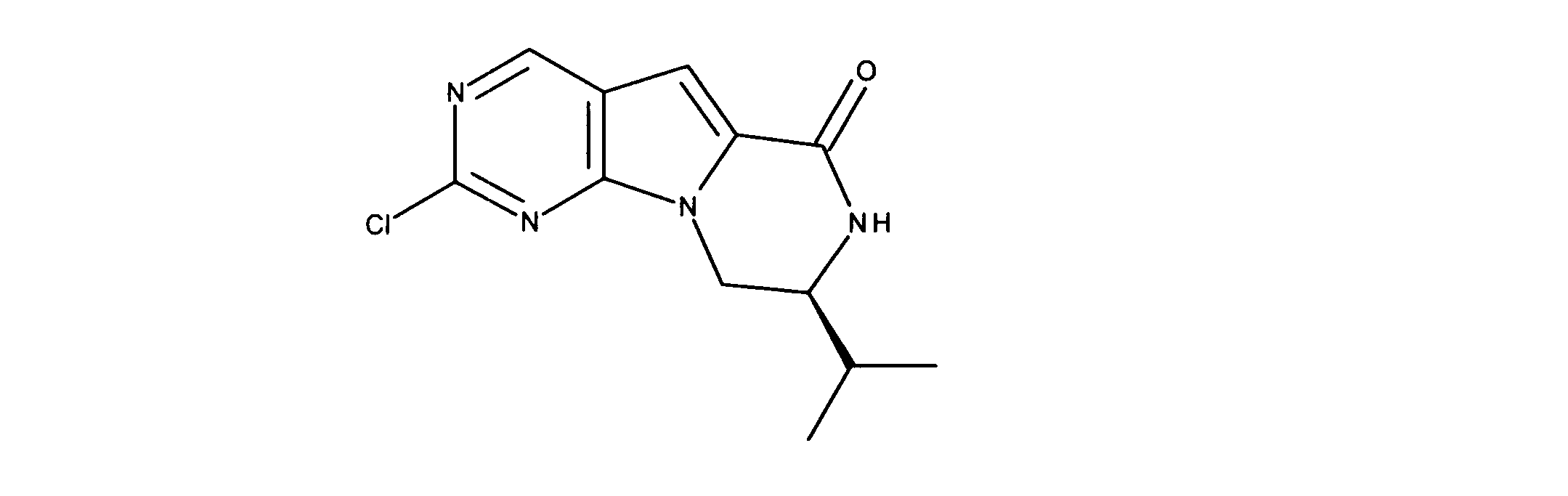
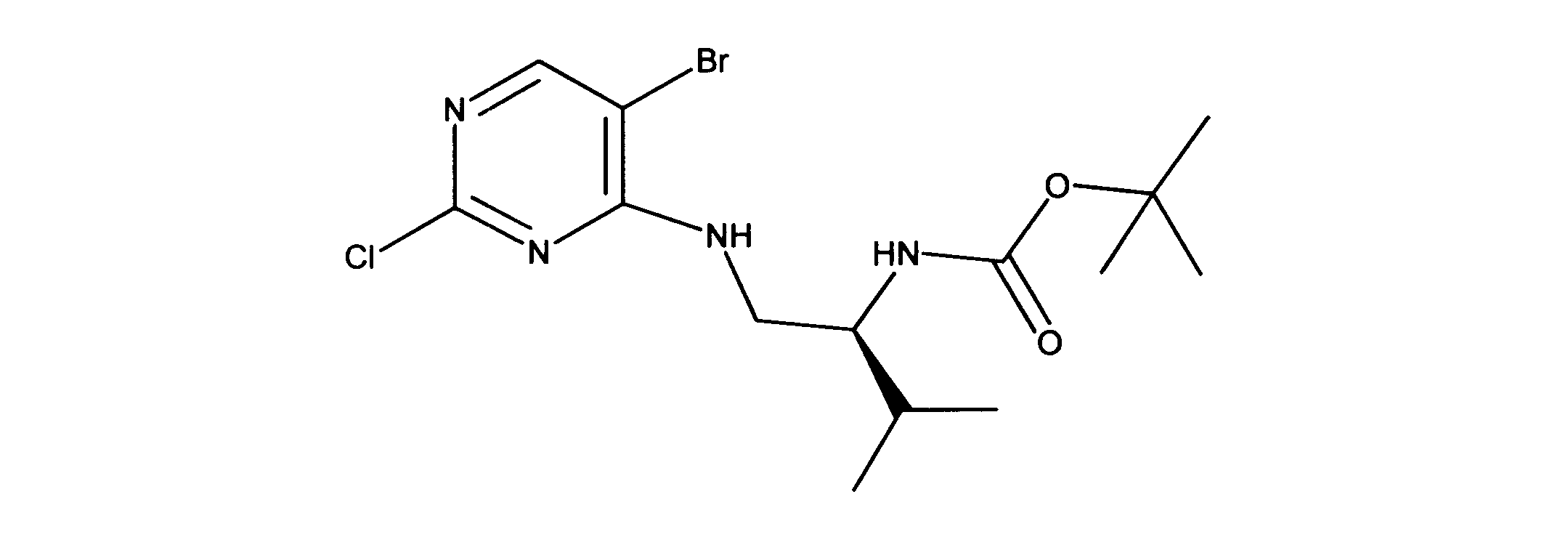
化合物55の合成
1HNMR (600 MHz, CHLOROFORM-d) δ ppm 0.95 - 1.02 (m, 6 H) 1.35 - 1.45 (m, 9 H) 1.75 - 1.90 (m, 1 H) 3.35 - 3.48 (m, 1 H) 3.52 - 3.61 (m, 1 H) 3.64 - 3.76 (m, 1 H) 4.56 (d, J=8.49 Hz, 1 H) 6.47 (s, 1 H) 8.07 (s, 1 H). LCMS (ESI) 393 (M + H).

1HNMR (600 MHz, CHLOROFORM-d) δ ppm 0.90 - 1.00 (m, 6 H) 1.18 - 1.25 (m, 6 H) 1.34 - 1.36 (m, 9 H) 1.69 - 1.90 (m, 1 H) 3.34 - 3.82 (m, 6 H) 4.53 - 4.77 (m, 1 H) 5.45 - 5.55 (m, 1 H) 6.37 (dd, J=15.37, 6.59 Hz, 1 H) 6.56 (s, 1 H) 8.05 (s, 1 H). LCMS (ESI) 441 (M + H).
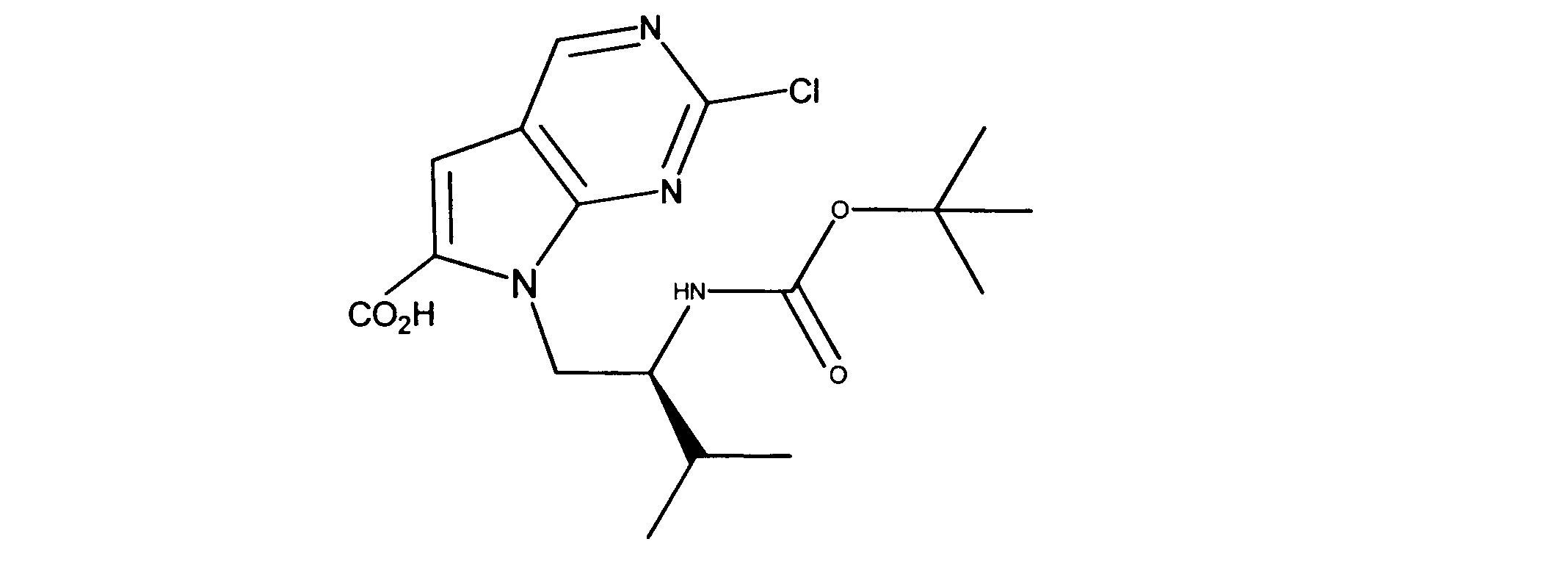
化合物44について記載されたものと類似の合成シーケンスを用いて、化合物55を合成した。
LCMS (ESI) 265 (M + H).
化合物56の合成
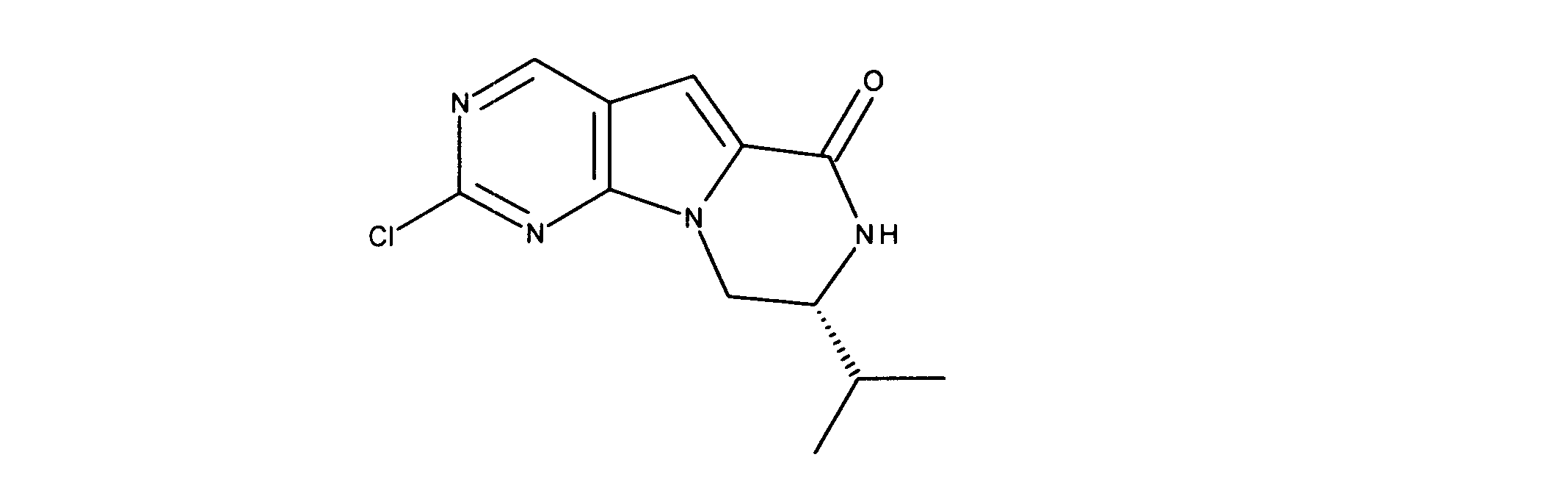
1HNMR (600 MHz, DMSO-d6) δ ppm 0.88 (d, J=6.44 Hz, 6 H) 1.73 - 1.86 (m, 1 H) 3.67 - 3.76 (m, 2 H) 4.11 - 4.21 (m, 1 H) 7.13 - 7.19 (m, 1 H) 8.56 (s, 1 H) 9.05 (s, 1 H). LCMS (ESI) 265 (M + H).
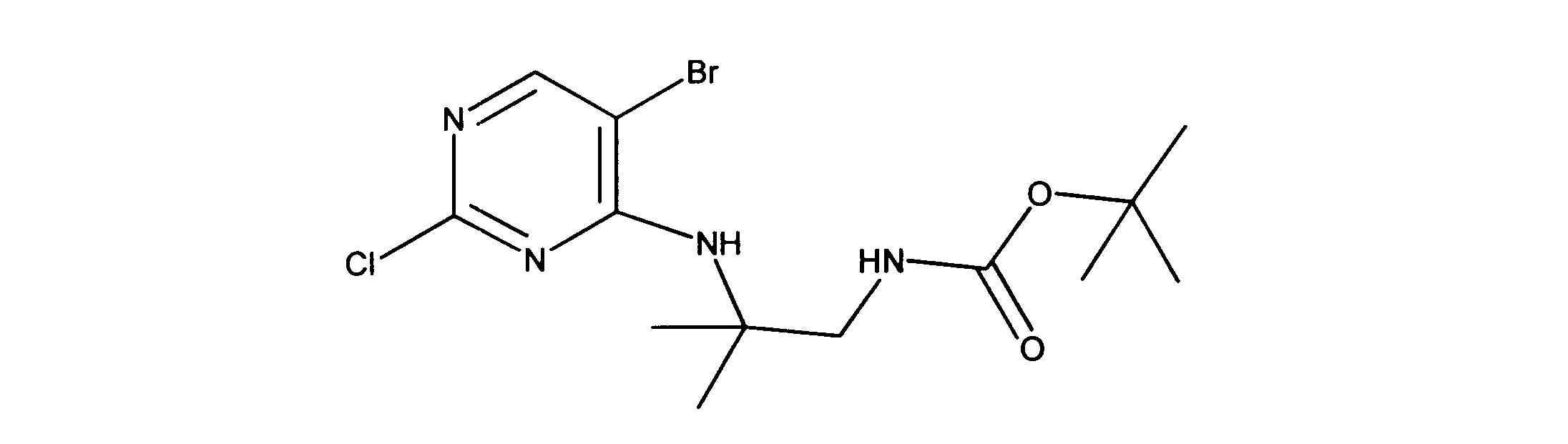
化合物57の合成
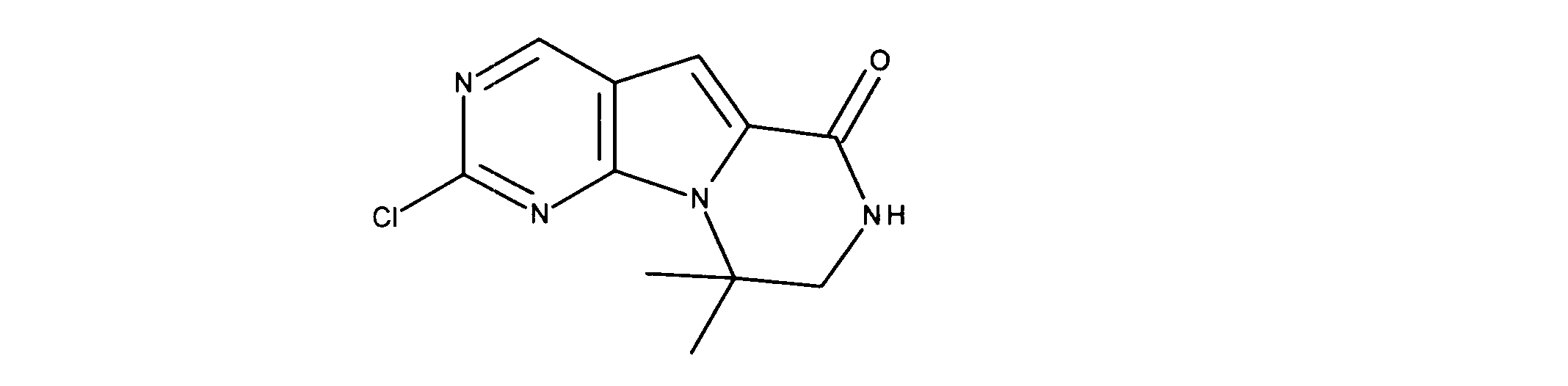
LCMS (ESI) 379 (M + H).
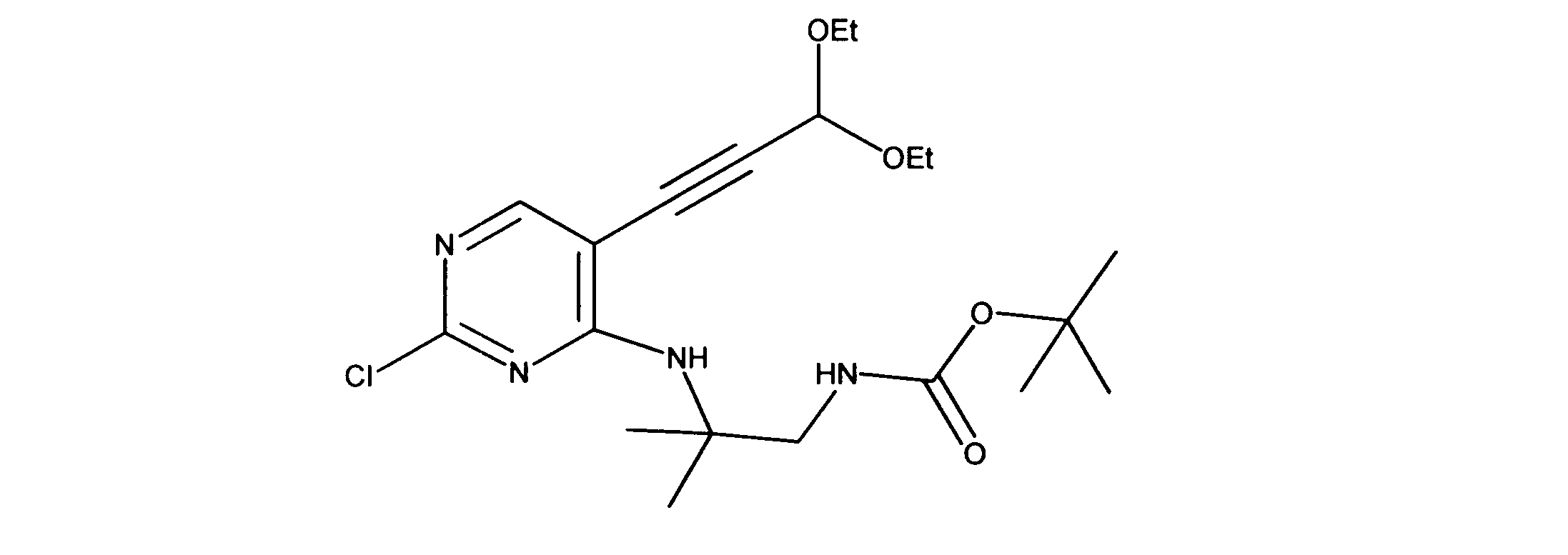
1HNMR (600 MHz, DMSO-d6) δ ppm 1.11 - 1.22 (m, 6 H) 1.31 - 1.45 (m, 15 H) 3.10 - 3.24 (m, 2 H) 3.51 - 3.76 (m, 4 H) 5.60 (s, 1 H) 6.94 (s, 1 H) 7.33 (t, J=6.44 Hz, 1 H) 8.18 (s, 1 H). LCMS (ESI) 427 (M + H).
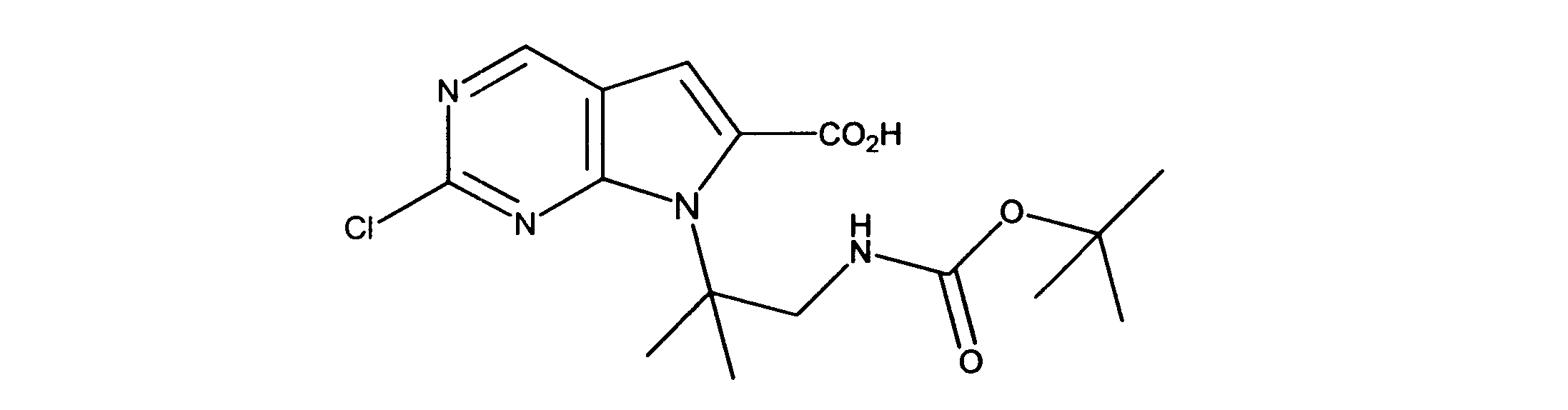
1HNMR (600 MHz, DMSO-d6) δ ppm 1.43 (s, 9H) 1.73 (s, 6 H) 4.06 (s, 2 H) 7.46 (s, 1 H) 9.23 (s, 1H). LCMS (ESI) 369 (M + H).
化合物44について記載されたものと類似の合成シーケンスを用いて、化合物57を合成した。
1HNMR (600 MHz, DMSO-d6) δ ppm 1.73 (s, 6 H) 3.50 (d, J=2.93 Hz, 2 H) 7.25 (s, 1 H) 8.46 - 8.55 (m, 1 H) 9.07 (s, 1 H). LCMS (ESI) 251 (M + H).
化合物58の合成
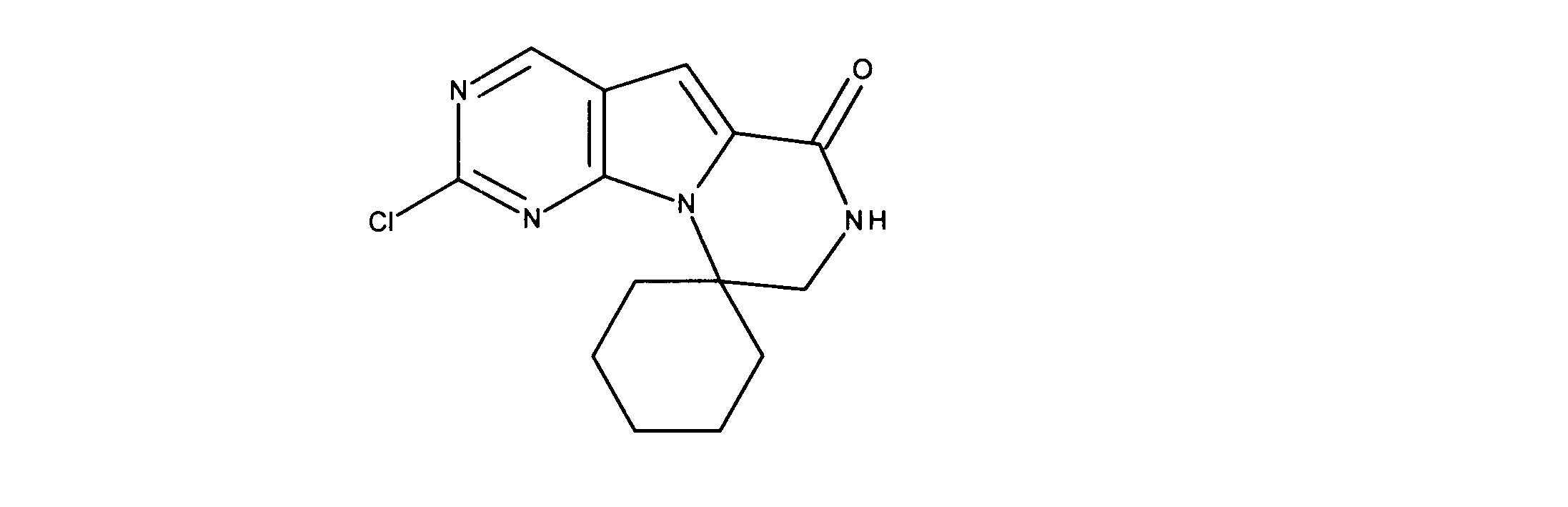
1HNMR (600 MHz, DMSO-d6) δ ppm 1.18 - 1.54 (m, 17 H) 2.23 (d, J=14.35 Hz, 2 H) 3.36 (d, J=6.44 Hz, 2 H) 5.82 (s, 1 H) 6.93 (s, 1 H) 8.22 (s, 1 H). LCMS (ESI) 419 (M + H).
1HNMR (600 MHz, DMSO-d6) δ ppm 1.08 - 1.16 (m, 6 H) 1.17 - 1.54 (m, 17 H) 2.13 (br. s., 2 H) 3.36 (d, J=6.73 Hz, 2 H) 3.50 - 3.69 (m, 4 H) 5.72 (s, 1 H) 6.94 (s, 1 H) 5.72 (br. s., 1H) 8.17 (s, 1 H). LCMS (ESI) 467 (M + H).

1HNMR (600 MHz, DMSO-d6) δ ppm 1.37 - 1.54 (m, 13 H) 1.75 (br. s., 4 H) 2.74 (br. s., 2 H) 3.78 - 3.84 (m, 2 H) 7.44 - 7.51 (m, 1 H) 8.23 (s, 1 H) 9.11 (s, 1 H). LCMS (ESI) 409 (M + H).
化合物44について記載されたものと類似の合成シーケンスを用いて、化合物58を合成した。
1HNMR (600 MHz, DMSO-d6) δ ppm 1.28 (br. s., 2 H) 1.42 (br. s., 2 H) 1.70 (br. s., 4 H) 1.85 - 1.95 (m, 2 H) 2.69 (m, 2 H) 7.16 - 7.25 (m, 1 H) 8.41 (br. s., 1 H) 9.04 (s, 1 H). LCMS 291 (M + H).
化合物59の合成
1HNMR (600 MHz, DMSO-d6) δ ppm 1.34 (s, 9 H) 1.50 - 1.58 (m, 2 H) 1.63 - 1.78 (m, 4 H) 1.96 - 2.06 (m, 2 H) 3.25 (d, J=6.15 Hz, 2 H) 6.71 (s, 1 H) 7.18 (t, J=6.29 Hz, 1 H) 8.20 (s, 1 H). LCMS (ESI) 405 (M + H).
LCMS (ESI) 453 (M + H).
化合物44について記載されたものと類似の合成シーケンスを用いて、化合物59を合成した。
1HNMR (600 MHz, DMSO-d6) δ ppm 1.72 (br. s., 2 H) 1.86 - 1.93 (m, 2 H) 1.99 (d, J=3.81 Hz, 2 H) 2.40 (br. s., 2 H) 3.48 (d, J=2.34 Hz, 2 H) 7.22 (s, 1 H) 8.53 (br. s., 1 H) 9.05 (s, 1 H). LCMS (ESI) 277 (M + H).
化合物60の合成
1HNMR (600 MHz, CHLOROFORM-d) δ ppm 1.21 - 1.31 (m, 12 H) 1.38 - 1.46 (m, 11 H) 1.70 (m, 1H) 3.24 (m, 2 H) 3.65 - 3.82 (m, 4 H) 4.86 (br s., 1H), 5.65 (s, 1 H) 5.85 (br s., 1H) 6.94 (s, 1 H) 8.21 (s, 1 H). LCMS (ESI) 455 (M + H).
化合物44のために記載されたものと類似の合成シーケンスを用いて、化合物60を合成した。分析データは、L-異性体について記載されたものと一致していた。
化合物61の合成
飽和NaHCO3の添加の後、酢酸エチルを添加した。有機層の分離の後、硫酸マグネシウムによる乾燥及び真空下での濃縮により、製品を提供した。分析データは、化合物49と同様だった。
化合物62の合成
1HNMR (600 MHz, DMSO-d6) δ ppm 1.27 (s, 9 H) 1.42 - 1.54 (m, 2 H) 1.56 - 1.65 (m, 2 H) 1.80 - 1.88 (m, 1 H) 1.96 - 2.01 (m, 1 H) 3.88 - 3.96 (m, 1 H) 4.03 - 4.09 (m, 1 H) 6.91 (d, J=8.20 Hz, 1 H) 7.41 (d, J=7.32 Hz, 1 H) 8.18 (s, 1 H). LCMS (ESI) 391 (M + H).
1HNMR (600 MHz, DMSO-d6) δ ppm 1.13 (t, 6 H) 1.28 (s, 9 H) 1.42 - 1.52 (m, 2 H) 1.58 - 1.65 (m, 2 H) 1.81 - 1.90 (m, 1 H) 1.99 - 2.08 (m, 1 H) 3.49 - 3.60 (m, 2 H) 3.63 - 3.71 (m, 2 H) 3.84 - 3.93 (m, 1 H) 3.96 - 4.04 (m, 1 H) 5.53 (s, 1 H) 6.96 (d, J=7.90 Hz, 1 H) 7.34 (d, J=7.03 Hz, 1 H) 8.14 (s, 1 H). LCMS (ESI) 439 (M + H).
1HNMR (600 MHz, DMSO-d6) δ ppm 1.41 - 1.52 (m, 9 H) 1.55 - 1.68 (m, 1 H) 1.88 - 2.00 (m, 2 H) 2.05 - 2.15 (m, 1 H) 2.26 - 2.35 (m, 1 H) 2.71 - 2.89 (m, 1 H) 4.01 - 4.16 (m, 1 H) 4.28 - 4.45 (m, 1 H) 7.41 (s, 1 H) 9.11 (s, 1 H). LCMS (ESI) 381 (M + H).
化合物44に対して記載されたものと類似の合成シーケンスを用いて、化合物62を合成した。
1HNMR (600 MHz, DMSO-d6) δ ppm 1.48 - 1.60 (m, 1 H) 1.88 - 1.98 (m, 3 H) 1.99 - 2.08 (m, 1 H) 2.66 - 2.75 (m, 1 H) 3.63 - 3.74 (m, 1 H) 3.99 - 4.12 (m, 1 H) 7.21 (s, 1 H) 8.89 (s, 1 H) 9.04 (s, 1 H). LCMS (ESI) 263 (M + H).
化合物63の合成
1HNMR (d6-DMSO) δ ppm 11.13 (brs, 1H), 9.07 (s, 1H), 8.42 (s, 1H), 8.03 (br m 1H), 7.99 (s, 1H), 7.67 (brm, 1H), 7.18 (s, 1H), 4.33 (m, 2H), 3.79 (m, 2H), 3.64 (m, 2H), 3.50 (m, 2H), 3.16 (m, 4H), 2.79 (s, 3H). LCMS (ESI) 379 (M + H).
化合物64の合成
1HNMR (d6-DMSO) δ ppm 9.48 (s, 1H), 8.84 (s, 1H), 8.29 (s, 1H), 8.18 (s, 1H), 7.99 (s, 1H), 7.42 (m, 1H), 6.98 (s, 1H), 4.23 (m, 2H), 3.59 (m, 2H), 3.45 (m, 4H), 3.50 (m, 2H), 3.05 (m, 4H). LCMS (ESI) 465 (M + H).
化合物65の合成
内容物を、16時間撹拌した。
反応物混合物を濃縮し、塩酸塩を提供した。
1HNMR (d6-DMSO) δ ppm 9.01 (s, 1H), 7.94 (m, 1H), 7.86 (m, 1H), 7.23 (s, 1H), 4.30 (m, 2H), 3.64 (m, 2H), 3.36 (m, 4H), 3.25 (m, 4H). LCMS (ESI) 465 (M + H).
化合物66の合成
シリカゲルカラムクロマトグラフィーを用いて、ジクロロメタン/メタノール(0~5%)の溶出剤で、粗製品を精製し、所望の製品(44mg)を提供した。
1HNMR (d6-DMSO) δ ppm 9.49 (s, 1H), 8.85 (s, 1H), 8.32 (m, 1H), 8.02 (s, 1H), 7.44 (m, 1H), 7.00 (s, 1H), 4.33 (m, 2H), 3.80 (m, 2H), 3.48 (m, 4H), 3.07 (m, 4H), 3.05 (s, 3H), 1.42 (s, 9H). LCMS (ESI) 479 (M + H)
化合物67の合成
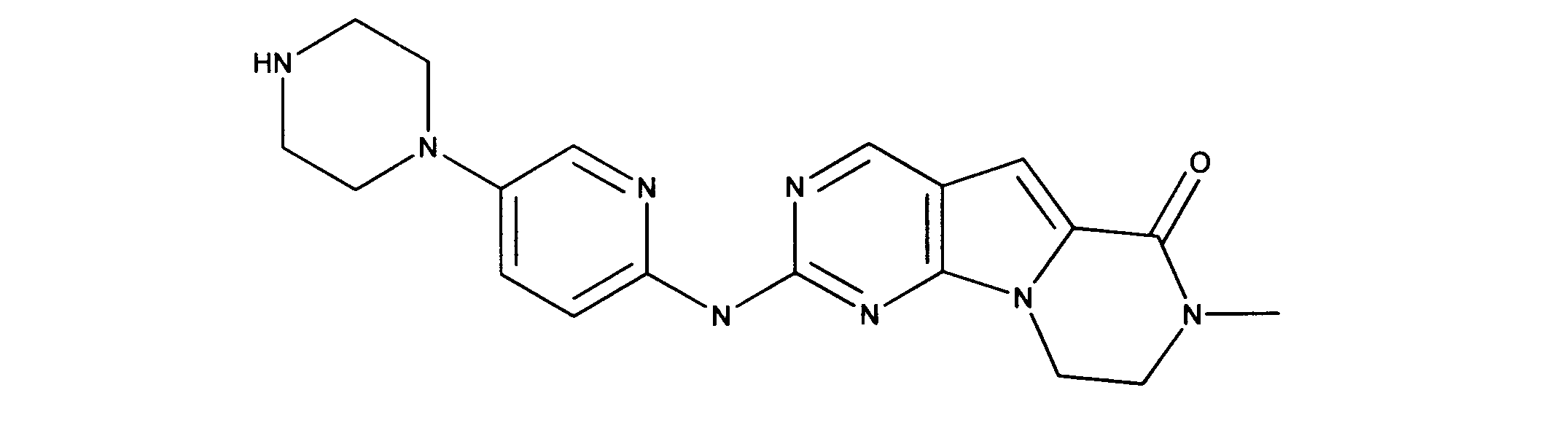
濃縮により、塩酸塩が提供された。
1HNMR (d6-DMSO) δ ppm 9.13 (m, 2H), 8.11 (m, 1H), 8.10 (s, 1H), 7.62 (m, 1H), 7.21 (s, 1H), 4.43 (m, 2H), 3.85 (m, 2H), 3.41 (m, 4H), 3.28 (m, 4H), 3.08 (s, 3H). LCMS (ESI) 379 (M + H).
化合物68の合成
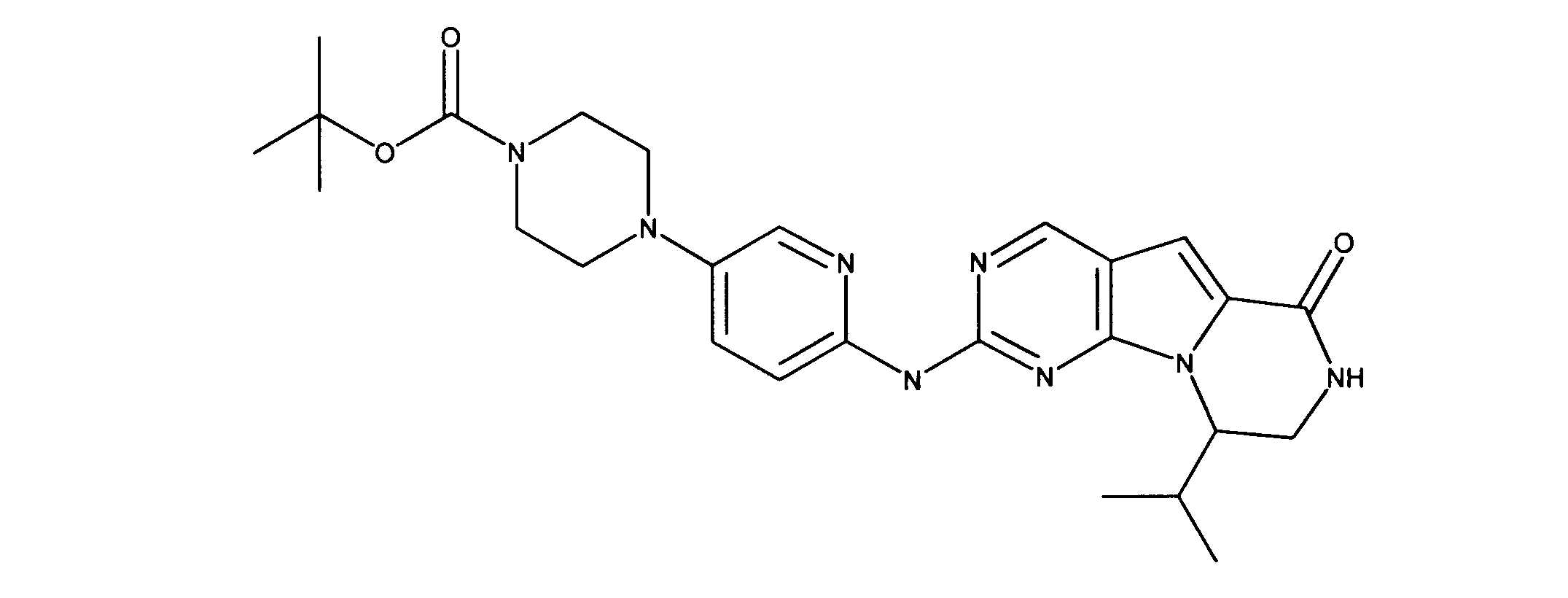
1HNMR (600 MHz, DMSO-d6) δ ppm 0.79 (d, J=7.03 Hz, 3 H) 1.01 (d, J=6.73 Hz, 3 H) 1.35 - 1.48 (m, 9 H) 2.16 (dd, J=14.64, 6.73 Hz, 1 H) 3.00 - 3.14 (m, 4 H) 3.40 - 3.51 (m, 4 H) 3.51 - 3.60 (m, 1 H) 3.63 - 3.74 (m, 1 H) 4.44 (dd, J=7.90, 3.81 Hz, 1 H) 6.99 (s, 1 H) 7.46 (dd, J=8.93, 2.78 Hz, 1 H) 7.94 - 8.09 (m, 2 H) 8.31 (dd, J=9.08, 1.46 Hz, 1 H) 8.85 (s, 1 H) 9.46 (s, 1 H). LCMS (ESI) 507 (M + H)
化合物69の合成
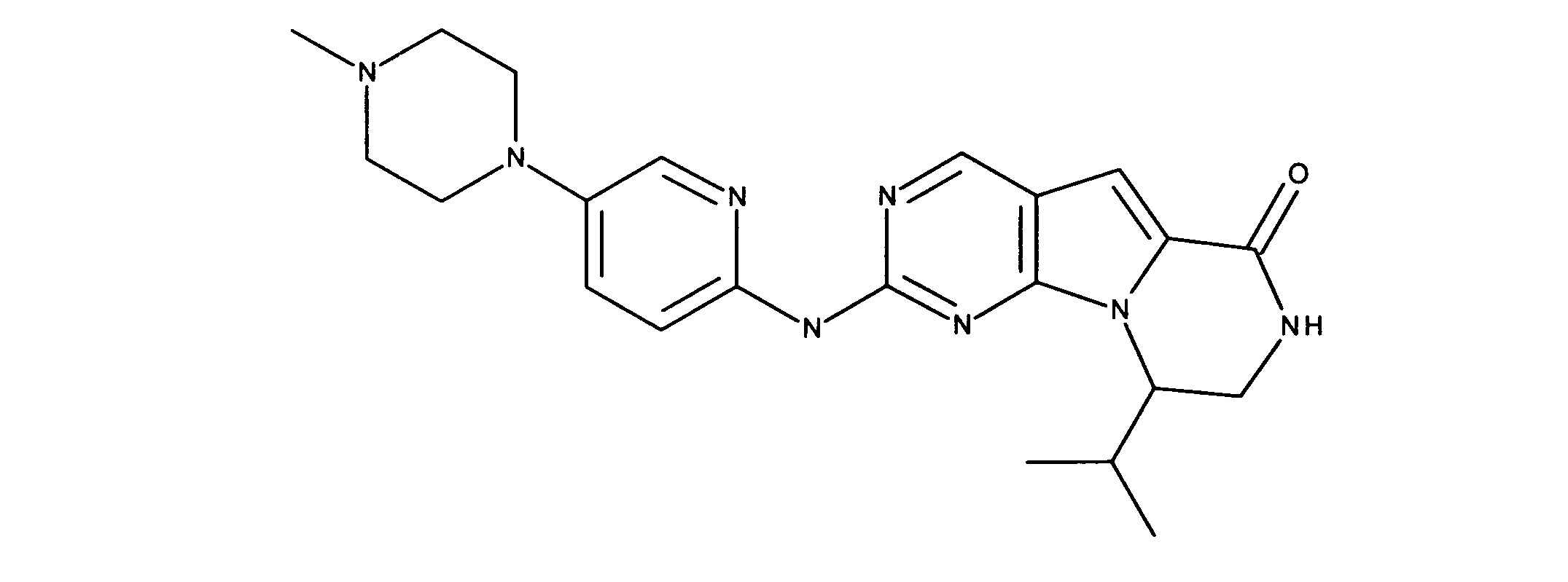
1HNMR (600 MHz, DMSO-d6) δ ppm 0.77 - 0.86 (m, 3 H) 0.96 (d, J=7.03 Hz, 3 H) 2.10 - 2.24 (m, 1 H) 3.07 (s, 3 H) 3.37 - 3.79 (m, 8 H) 4.00 (dd, J=13.61, 4.54 Hz, 2 H) 4.63 - 4.73 (m, 1 H) 7.20 (s, 1 H) 7.58 - 7.71 (m, 1 H) 7.99 (d, J=2.34 Hz, 1 H) 8.12 (d, J=9.37 Hz, 1 H) 9.11 (s, 1 H) 9.41 (br. s., 2 H) 11.76 (br. s., 1 H). LCMS (ESI) 421 (M + H).
化合物70の合成
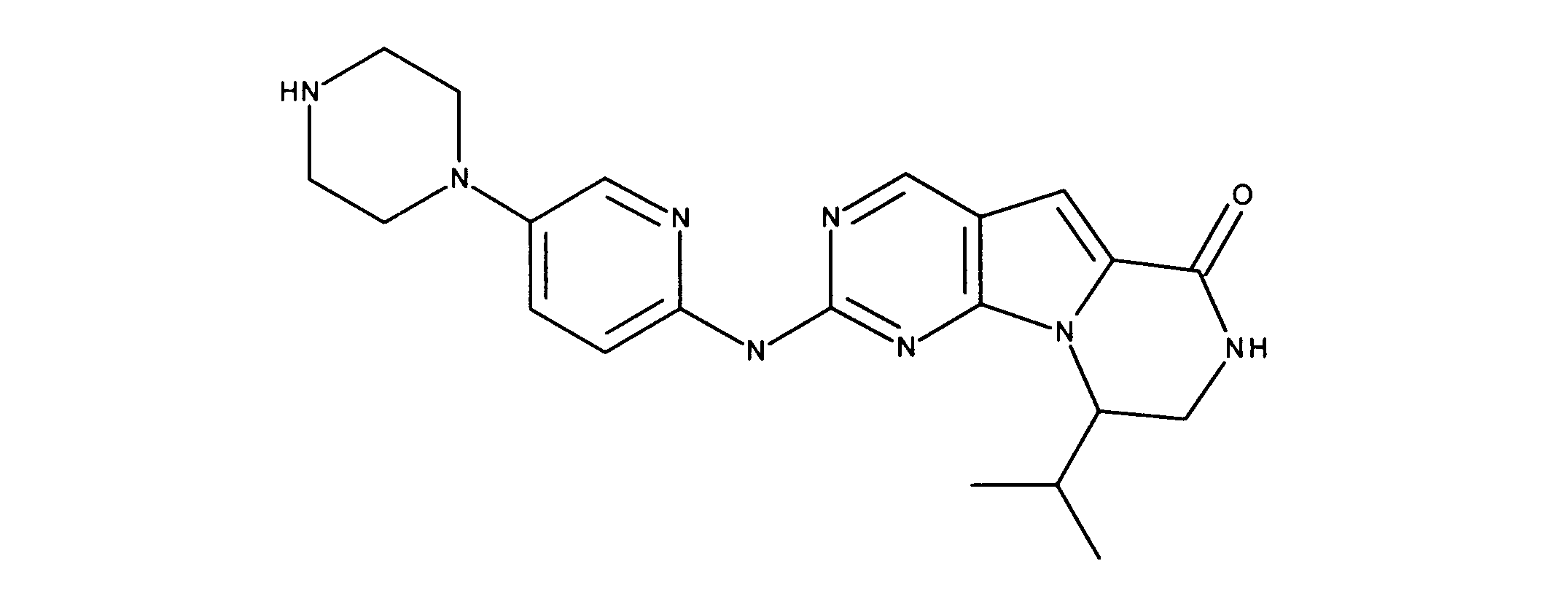
化合物71の合成
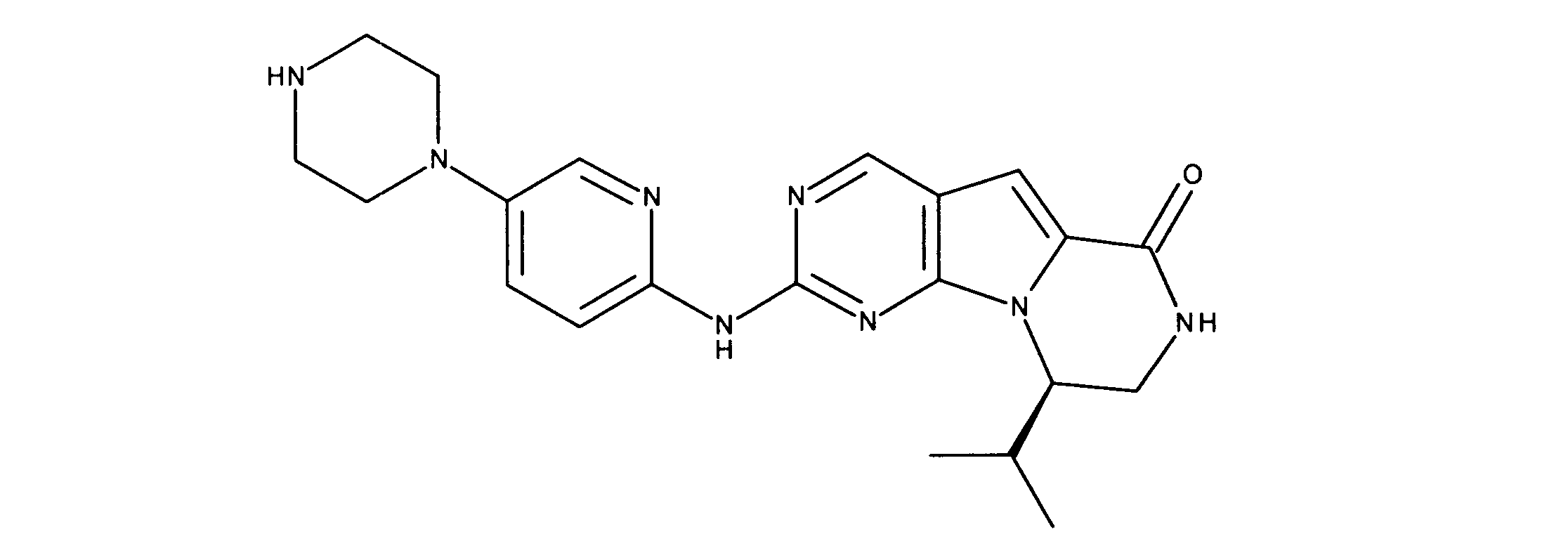
1HNMR (600 MHz, DMSO-d6) δ ppm 0.79 (d, J=6.73 Hz, 3 H) 1.01 (d, J=6.73 Hz, 3 H) 2.18 (dd, J=14.49, 7.17 Hz, 1 H) 3.18 - 3.84 (m, 10 H) 4.53 - 4.71 (m, 1 H) 7.24 (s, 1 H) 7.65 (d, J=9.37 Hz, 1 H) 8.01 (d, J=2.64 Hz, 1 H) 8.14 (d, J=1.46 Hz, 1 H) 8.35 (d, J=5.27 Hz, 1 H) 9.14 (s, 1 H) 9.46 (s, 2 H) 11.80 (s, 1 H) LCMS (ESI) 407 (M+H).
化合物72(化合物UUU)の合成
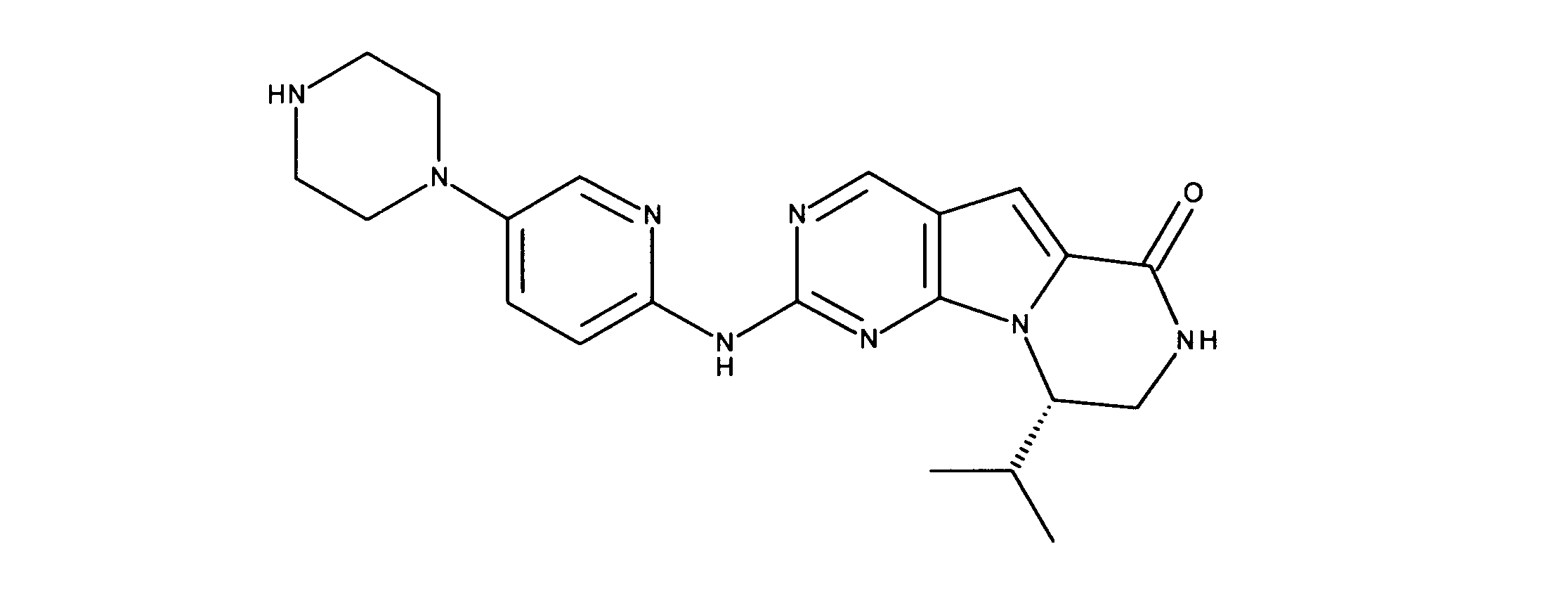
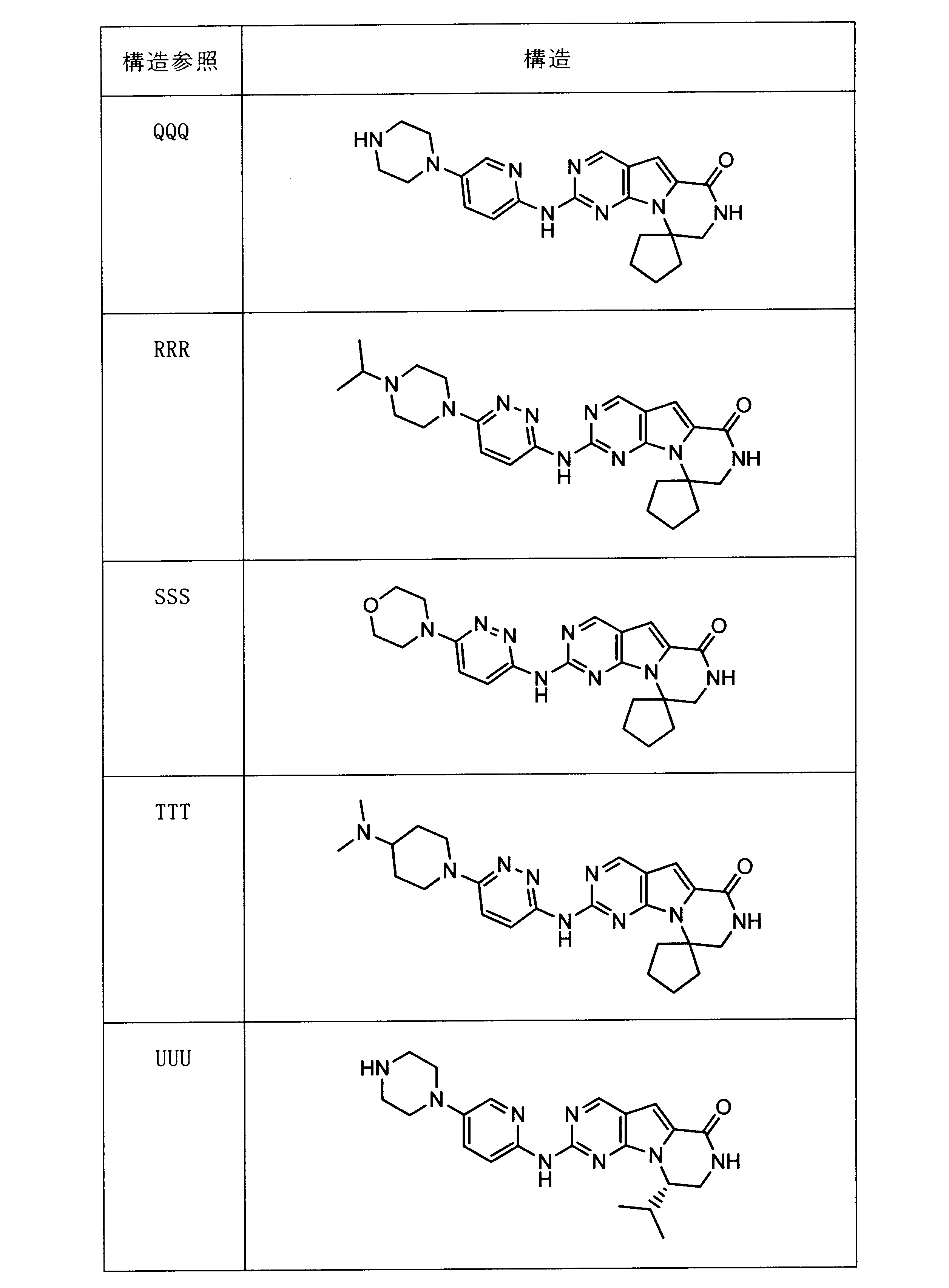
1HNMR (600 MHz, DMSO-d6) δ ppm 0.77 (d, J=7.03 Hz, 3 H) 0.99 (d, J=6.73 Hz, 3 H) 2.10 - 2.24 (m, 1 H) 3.18 - 3.81 (m, 10 H) 4.54 - 4.69 (m, 1 H) 7.22 (s, 1 H) 7.63 (d, J=9.08 Hz, 1 H) 7.99 (d, J=2.63 Hz, 1 H) 8.11 (s, 1 H) 8.33 (d, J=5.27 Hz, 1 H) 9.12 (s, 1 H) 9.43 (s, 2 H) 11.77 (s, 1 H). LCMS (ESI) 407 (M+H).
化合物73の合成
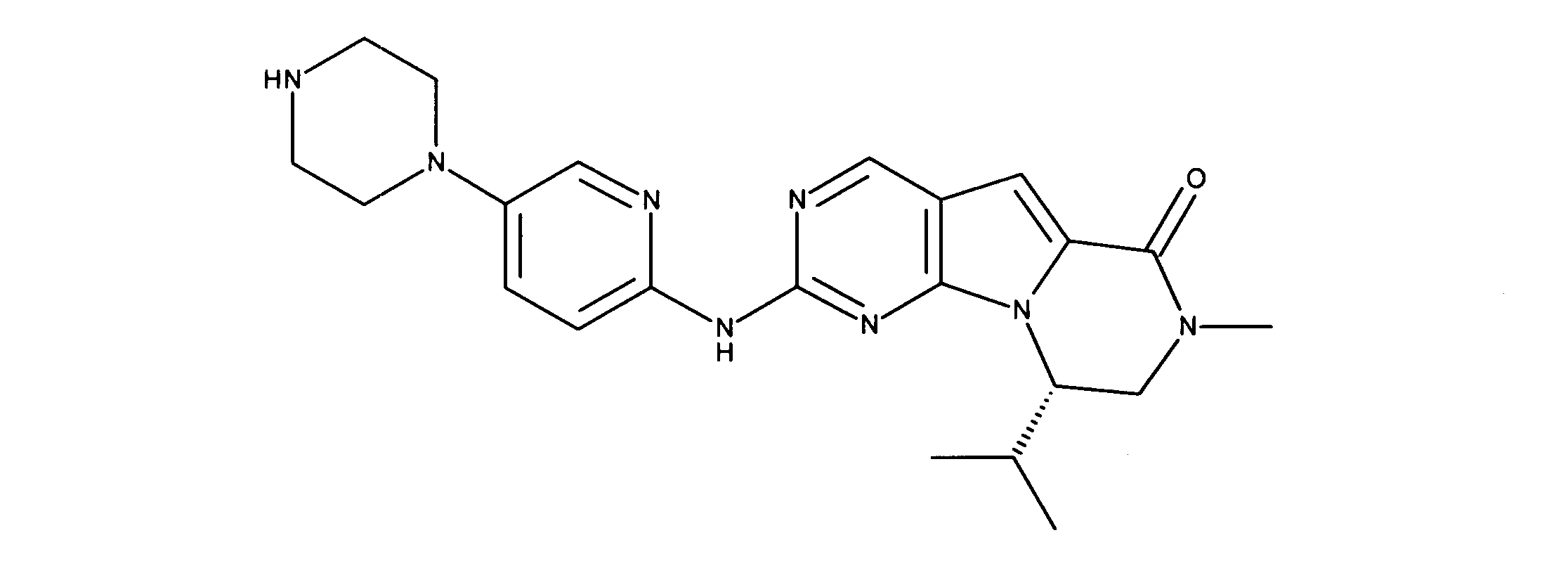
1HNMR (600 MHz, DMSO-d6) δ ppm 0.84 (d, J=6.73 Hz, 3 H) 0.98 (d, J=6.73 Hz, 3 H) 2.12 - 2.26 (m, 1 H) 3.09 (s, 3 H) 3.22 - 3.81 (m, 8 H) 4.01 (dd, J=13.61, 4.25 Hz, 2 H) 4.59 - 4.72 (m, 1 H) 7.19 (s, 1 H) 7.74 (s, 1 H) 7.96 - 8.10 (m, 2 H) 9.08 (s, 1 H) 9.22 (s, 2 H). LCMS (ESI) 421 (M+H).
化合物74の合成
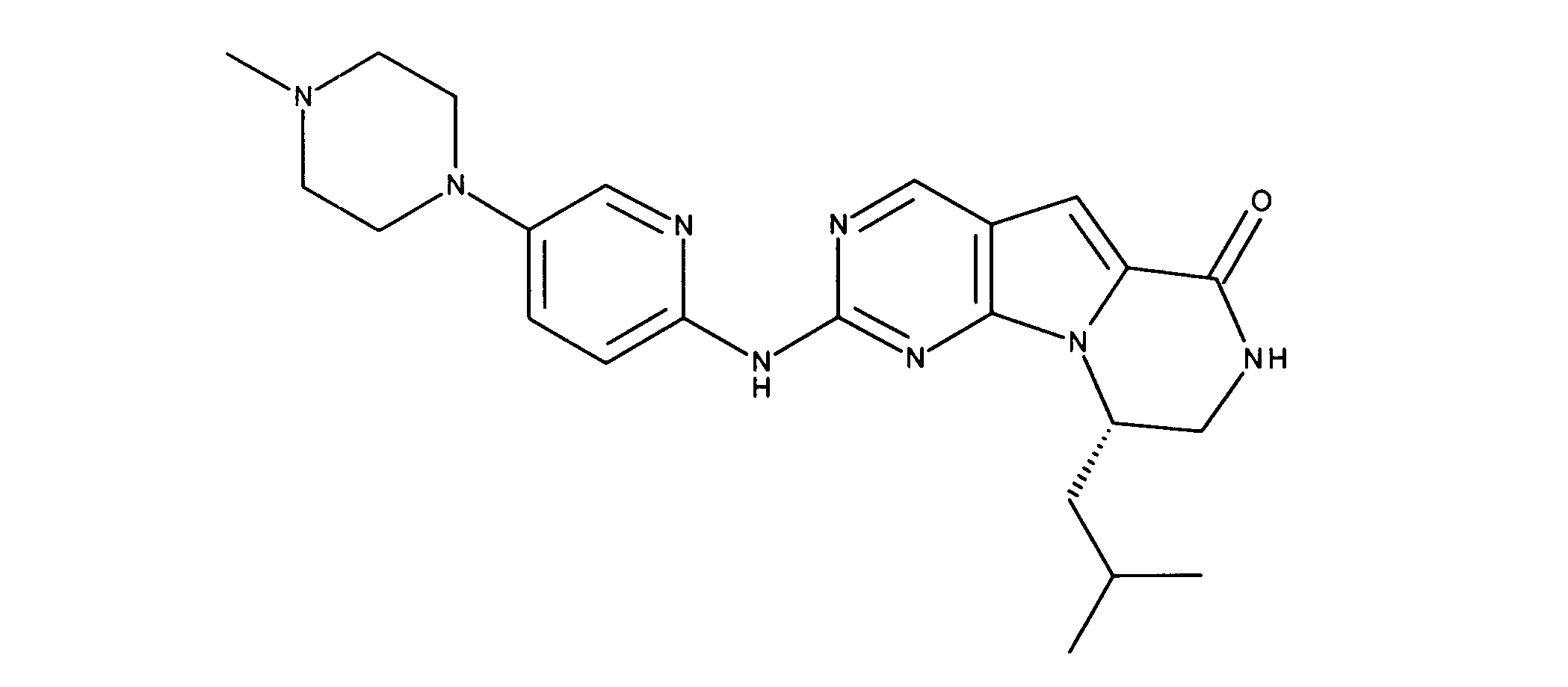
1HNMR (600 MHz, DMSO-d6) δ ppm 0.85 (d, J=4.98 Hz, 3 H) 0.95 (d, J=4.98 Hz, 3 H) 1.42 - 1.70 (m, 3 H) 2.77 (d, J=2.93 Hz, 3 H) 3.07 - 4.14 (m, 10 H) 4.95 (s, 1 H) 7.20 (s, 1 H) 7.66 (d, J=9.66 Hz, 1 H) 7.94 (s, 1 H) 8.08 - 8.16 (m, 1 H) 8.33 (d, J=4.68 Hz, 1 H) 9.09 (s, 1 H) 11.38 (s, 1 H) 11.71 (s, 1 H). LCMS (ESI) 435 (M+H).
化合物75の合成
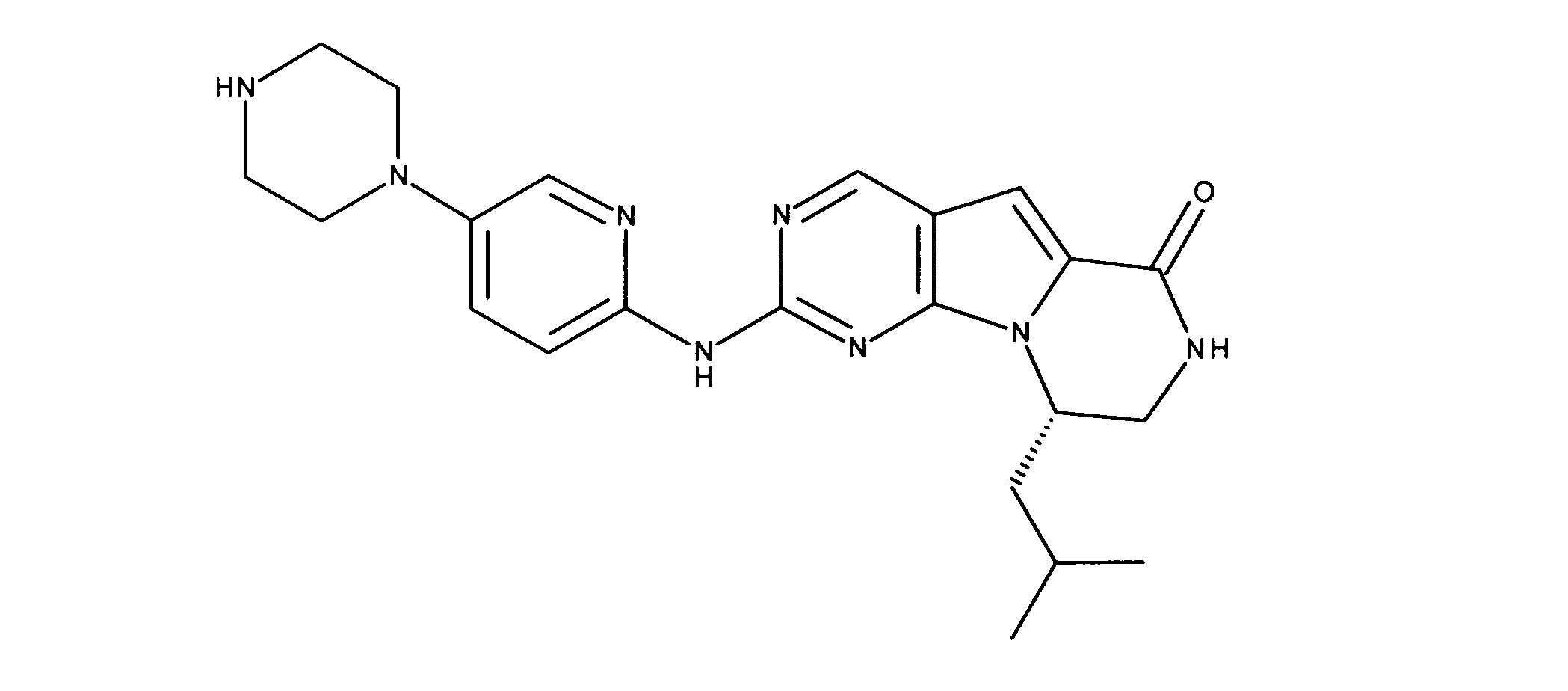
1HNMR (600 MHz, DMSO-d6) δ ppm 0.87 (d, J=6.15 Hz, 3 H) 0.94 (d, J=6.15 Hz, 3 H) 1.57 (d, J=84.61 Hz, 3 H) 3.05 (s, 3 H) 3.13 - 3.55 (m, 8 H) 3.69 (d, J=78.17 Hz, 2 H) 4.90 (s, 1 H) 7.15 (s, 1 H) 7.63 - 7.85 (m, 1 H) 7.93 (s, 1 H) 8.26 (s, 1 H) 9.03 (s, 1 H) 9.20 (s, 2 H). LCMS (ESI) 421 (M+H).
化合物76の合成
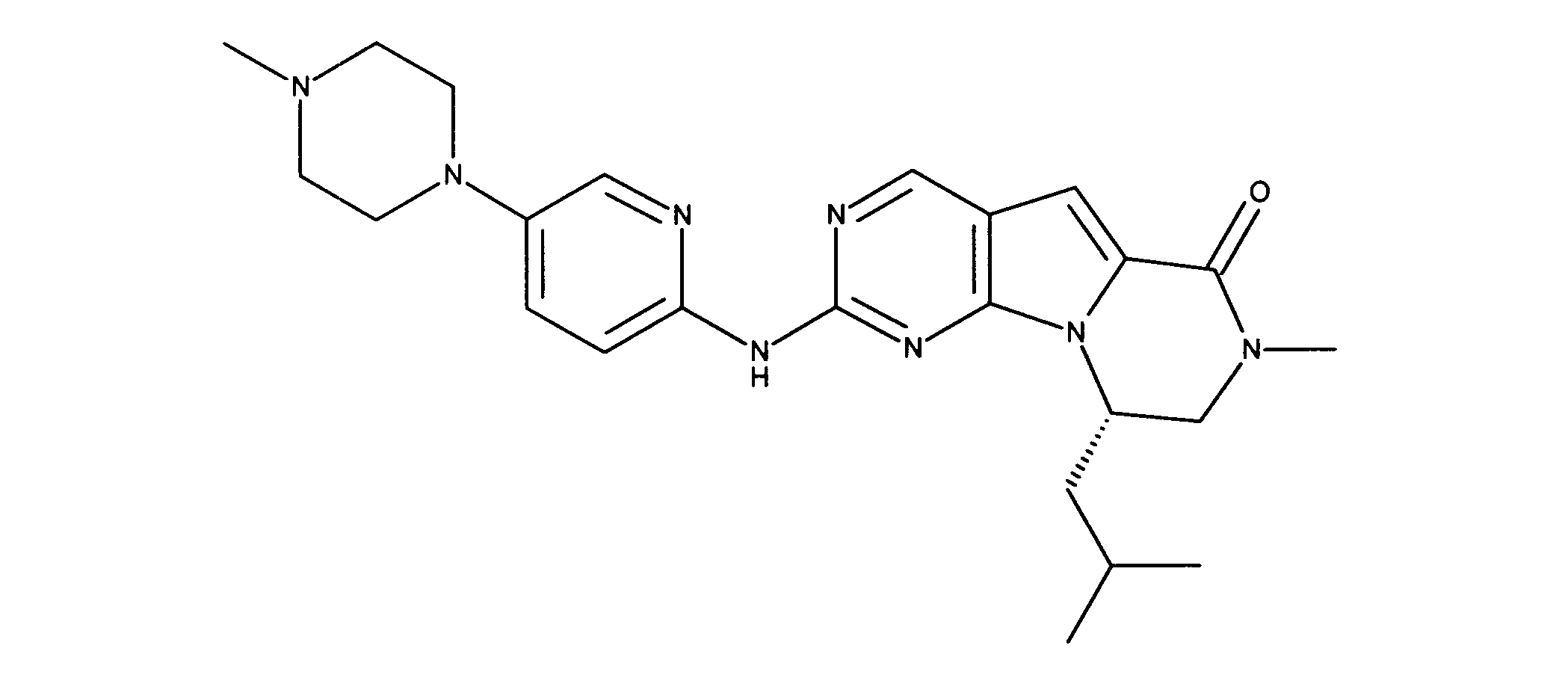
1HNMR (600 MHz, DMSO-d6) δ ppm 0.85 (d, J=6.44 Hz, 3 H) 0.95 (d, J=6.44 Hz, 3 H) 1.43 - 1.70 (m, 3 H) 2.78 (d, J=2.93 Hz, 3 H) 3.05 (s, 3 H) 3.24 - 3.84 (m, 8 H) 4.01 (d, J=9.66 Hz, 2 H) 4.89 - 5.01 (m, 1 H) 7.15 (s, 1 H) 7.77 (s, 1 H) 7.91 - 8.05 (m, 2 H) 9.03 (s, 1 H) 10.96 - 11.55 (m, 2 H). LCMS (ESI) 449 (M+H).
化合物77の合成
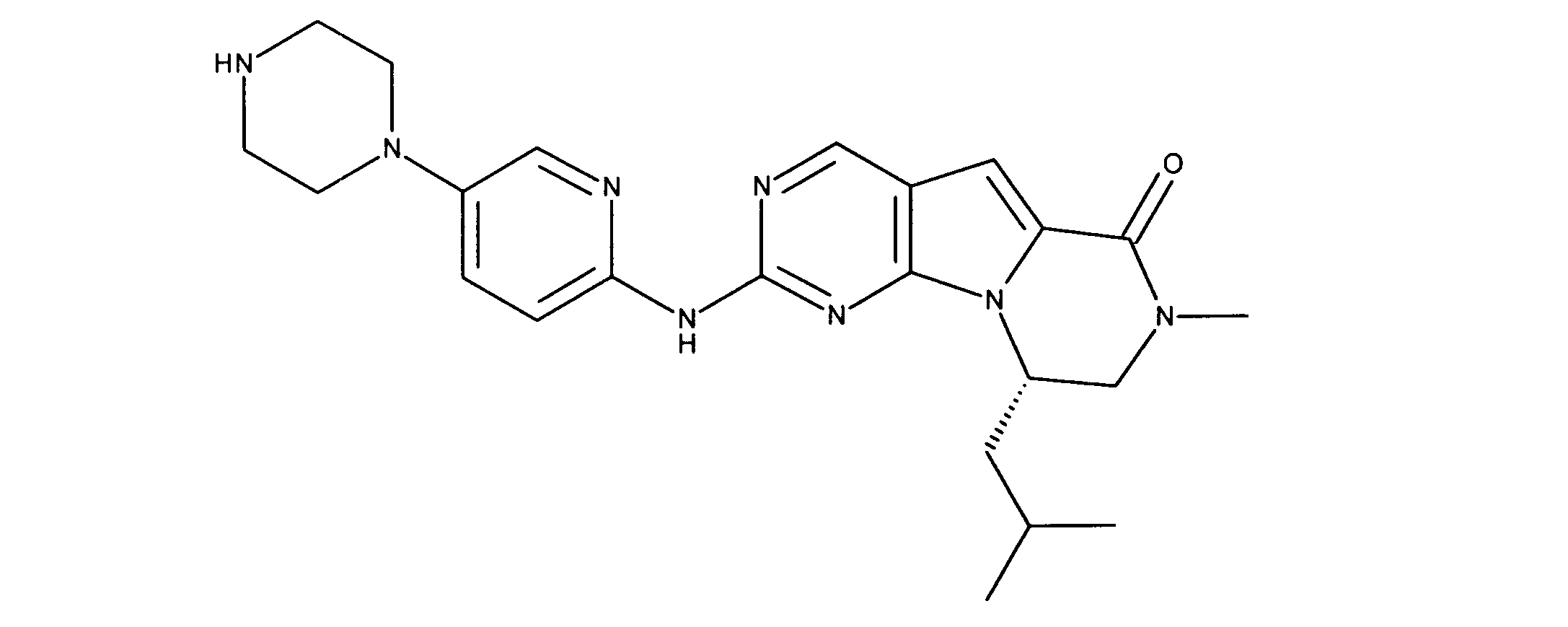
1HNMR (600 MHz, DMSO-d6) δ ppm 0.83 - 0.88 (d, J=6.15 Hz, 3 H) 0.95 (d, J=6.15 Hz, 3 H) 1.40 - 1.71 (m, 3 H) 3.28 - 3.83 (m, 8 H) 4.00 (d, J=3.22 Hz, 2 H) 4.91 - 5.08 (m, 1 H) 7.17 (s, 1 H) 7.68 (d, J=9.66 Hz, 1 H) 7.93 (s, 1 H) 8.07 (s, 1 H) 9.06 (s, 1 H) 9.40 (s, 2 H) 11.59 (s, 1 H). LCMS (ESI) 435 (M+H).
化合物78の合成
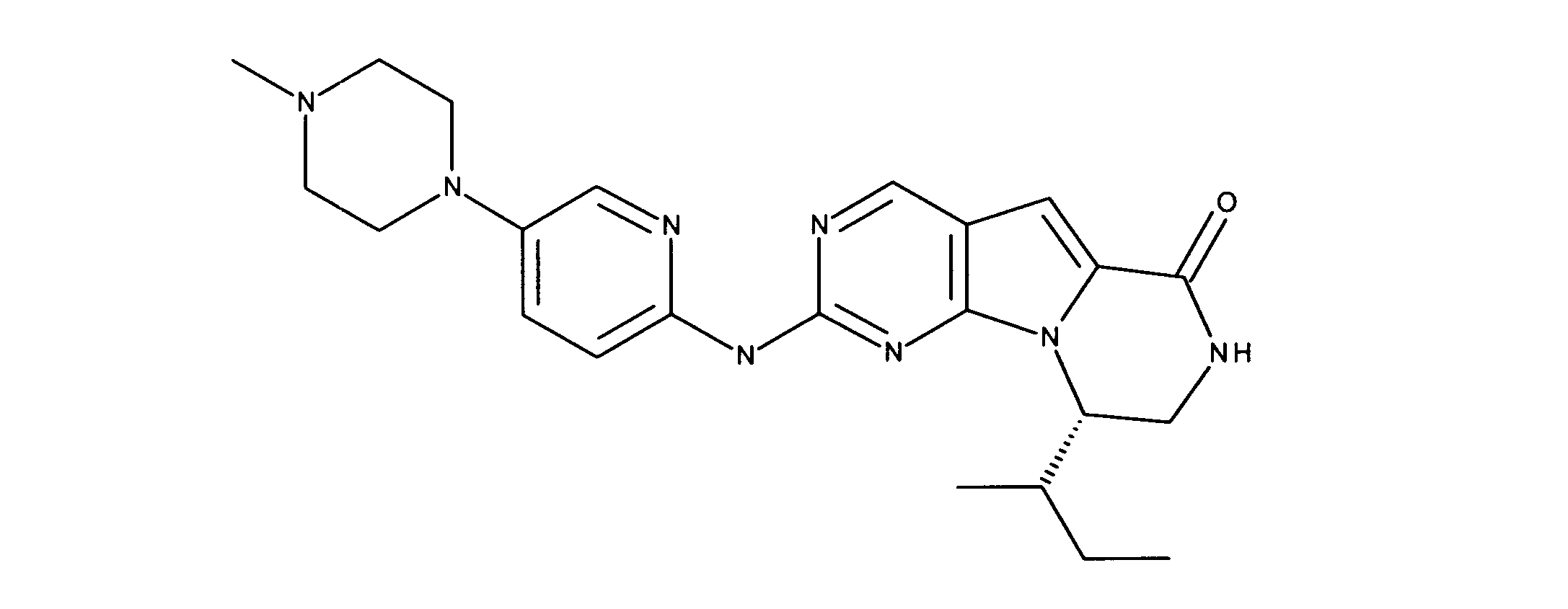
1HNMR (600 MHz, DMSO-d6) δ ppm 0.75 (t, J=7.47 Hz, 3 H) 0.91 (d, J=6.73 Hz, 3 H) 1.04 - 1.20 (m, 2 H) 1.80 - 1.98 (m, 1 H) 2.77 (d, J=3.81 Hz, 3 H) 2.94 - 3.90 (m, 10 H) 4.54 - 4.68 (m, 1 H) 7.06 - 7.23 (m, 2 H) 7.56 - 7.75 (m, 1 H) 7.90 - 8.12 (m, 2 H) 8.29 (s, 1 H) 9.07 (s, 1 H) 10.98 - 11.74 (m, 2 H). LCMS (ESI) 435 (M + H).
化合物79の合成
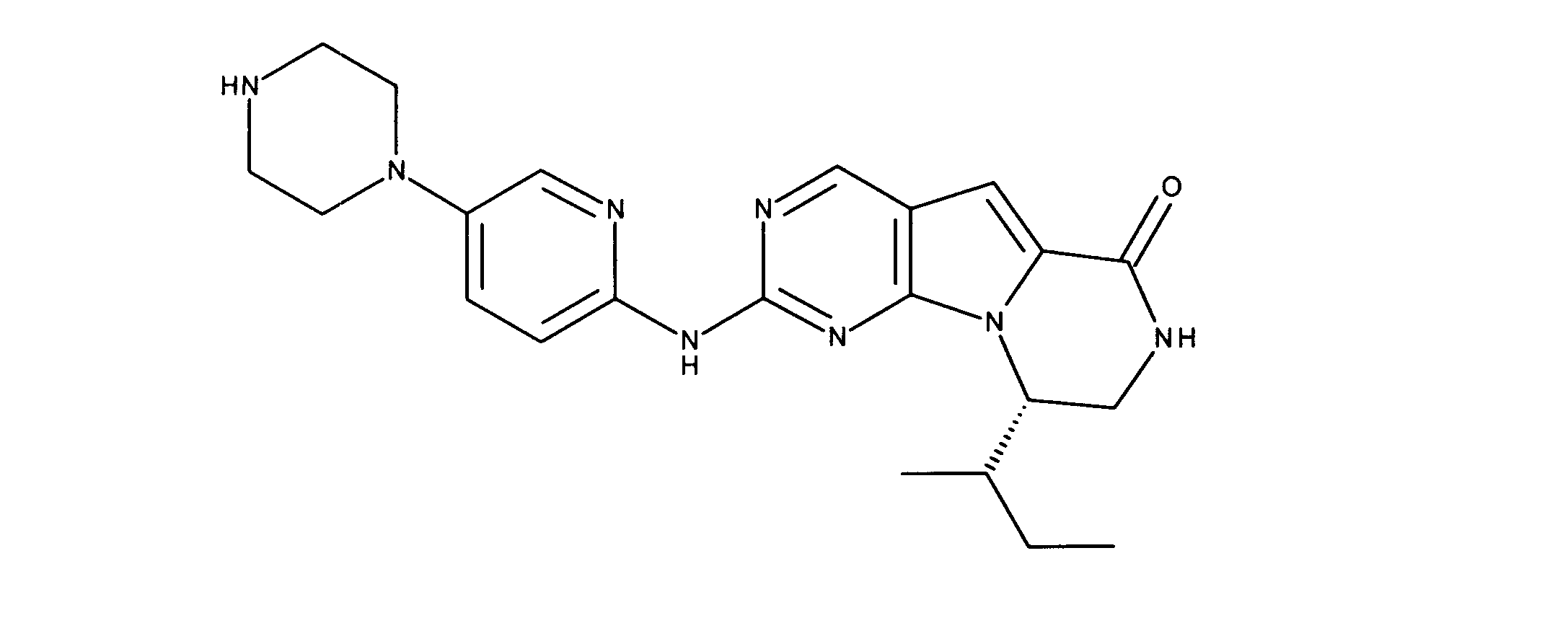
1HNMR (600 MHz, DMSO-d6) δ ppm 0.75 (t, J=7.32 Hz, 3 H) 0.90 (d, J=6.73 Hz, 3 H) 1.07 - 1.15 (m, 2 H) 1.85 - 1.94 (m, 1 H) 3.17 - 3.75 (m, 10 H) 4.58 - 4.67 (m, 1 H) 7.17 (s, 1 H) 7.71 (s, 1 H) 7.96 (s, 1 H) 7.98 - 8.05 (m, 1 H) 8.28 (d, J=4.10 Hz, 1 H) 9.06 (s, 1 H) 9.39 (s, 2 H). LCMS (ESI) 421 (M+H).
化合物80の合成
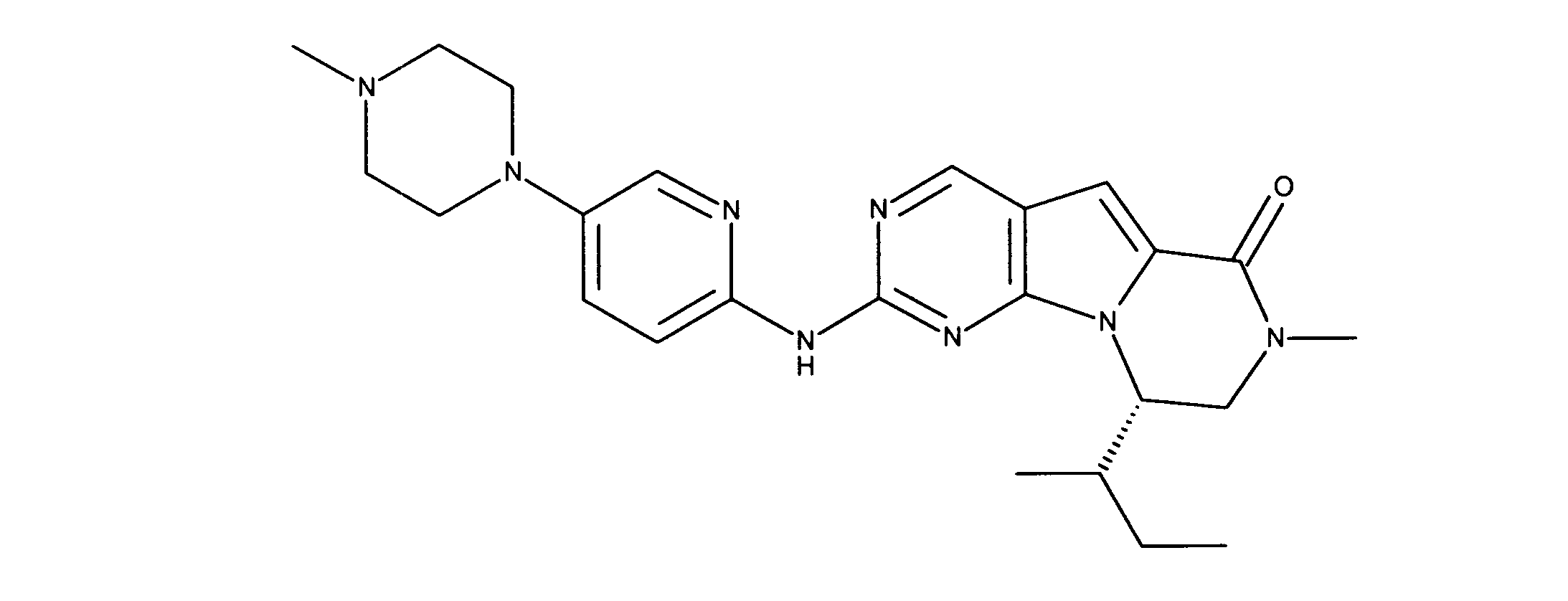
1HNMR (600 MHz, DMSO-d6) δ ppm 0.78 (t, J=7.32 Hz, 3 H) 0.86 (d, J=6.73 Hz, 3 H) 1.13 - 1.21 (m, 2 H) 1.84 - 1.96 (m, 1 H) 2.77 (d, J=4.39 Hz, 3 H) 3.04 (s, 3 H) 3.11 - 3.84 (m, 8 H) 3.98 (dd, J=13.61, 4.25 Hz, 2 H) 4.66 - 4.74 (m, 1 H) 7.17 (s, 1 H) 7.64 (s, 1 H) 7.96 (d, J=2.34 Hz, 1 H) 8.03 - 8.13 (m, 1 H) 9.08 (s, 1 H) 11.26 (s, 1 H) 11.66 (s, 1 H). LCMS (ESI) 449 (M+H).
化合物81の合成
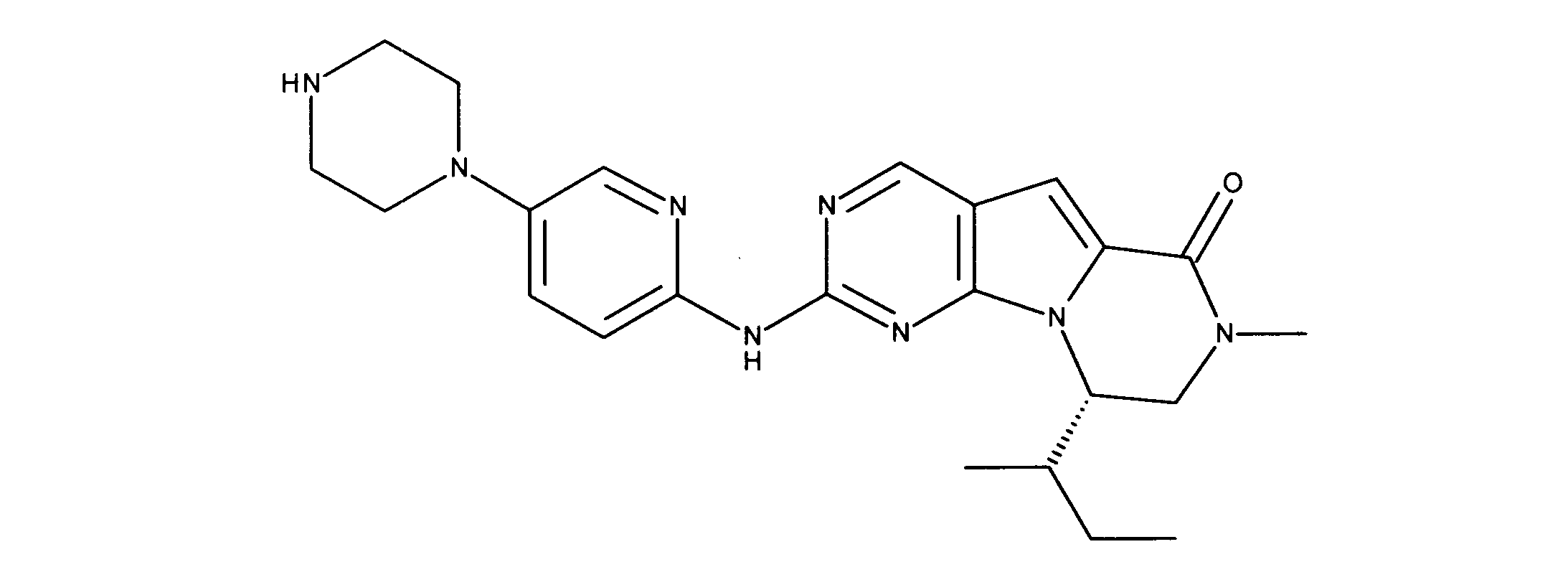
1HNMR (600 MHz, DMSO-d6) δ ppm 0.78 (t, J=7.32 Hz, 3 H) 0.85 (d, J=6.73 Hz, 3 H) 1.10 - 1.27 (m, 2 H) 1.82 - 1.99 (m, 1 H) 3.04 (s, 3 H) 3.28 - 3.77 (m, 8 H) 3.97 (dd, J=13.91, 4.54 Hz, 2 H) 4.62 - 4.75 (m, 1 H) 7.07 - 7.24 (m, 1 H) 7.62 - 7.75 (m, 1 H) 7.94 (d, J=2.34 Hz, 1 H) 7.97 - 8.08 (m, 1 H) 9.05 (s, 1 H) 9.29 (s, 2 H). LCMS (ESI) 435 (M+H).
化合物82の合成
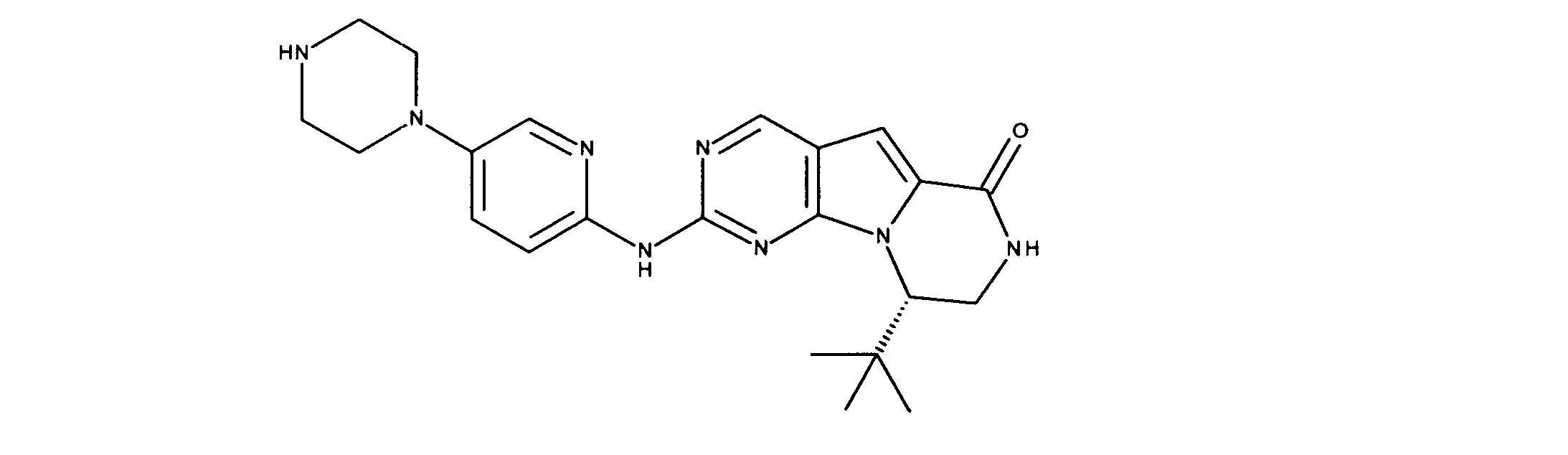
1HNMR (600 MHz, DMSO-d6) δ ppm 0.96 (s, 9 H) 3.15 - 3.87 (m, 10 H) 4.42 - 4.53 (m, 1 H) 6.99 (s, 1 H) 7.24 (s, 1 H) 8.06 (s, 1 H) 8.11 - 8.21 (m, 1 H) 8.79 - 8.98 (m, 2 H) 9.25 (s, 2 H) 9.88 (s, 1 H). LCMS (ESI) 421 (M+H).
化合物83の合成
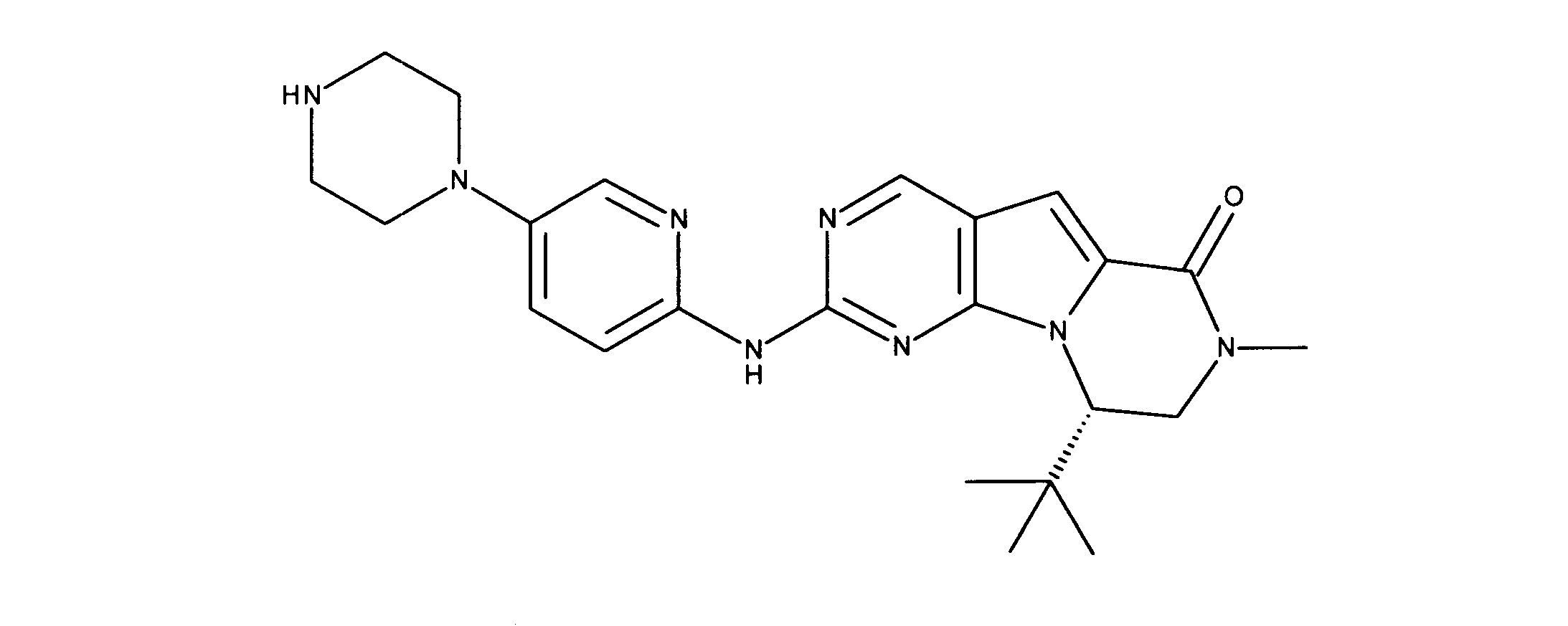
化合物84の合成
1HNMR (600 MHz, DMSO-d6) δ ppm 2.75 - 2.81 (m, 3 H) 3.12 - 3.16 (m, 2 H) 3.46 - 3.54 (m, 4 H) 3.60 - 3.69 (m, 2 H) 3.72 - 3.79 (m, 1 H) 4.07 - 4.18 (m, 2 H) 6.06 - 6.09 (m, 1 H) 6.90 (d, J=7.61 Hz, 2 H) 7.20 - 7.31 (m, 3 H) 7.33 (s, 1 H) 7.49 - 7.55 (m, 1 H) 7.62 - 7.70 (m, 1 H) 7.92 (d, J=2.93 Hz, 1 H) 8.22 (s, 1 H) 9.14 (s, 1 H). LCMS (ESI) 455 (M + H).
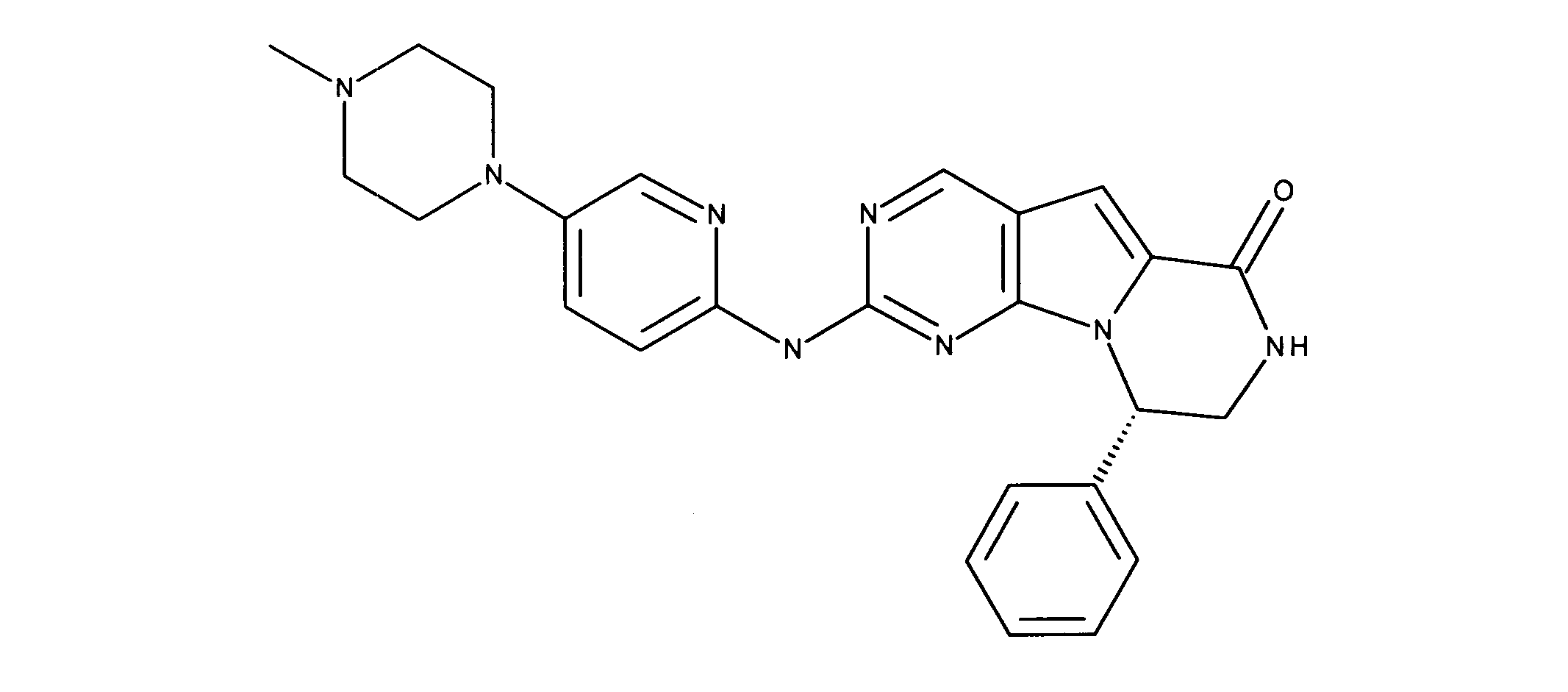
化合物85の合成
1HNMR (600 MHz, DMSO-d6) δ ppm 3.21 (s, 4 H) 3.35 - 3.67 (m, 5 H) 4.07 - 4.20 (m, 2 H) 6.13 (s, 1 H) 6.90 (d, J=7.32 Hz, 2 H) 7.22 - 7.31 (m, 3 H) 7.36 (s, 1 H) 7.48 (d, J=9.37 Hz, 1 H) 7.93 (d, J=2.34 Hz, 1 H) 8.04 - 8.11 (m, 1 H) 8.25 (d, J=4.98 Hz, 1 H) 9.17 (s, 1 H) 11.77 (br, s., 1H). LCMS (ESI) 441 (M + H).
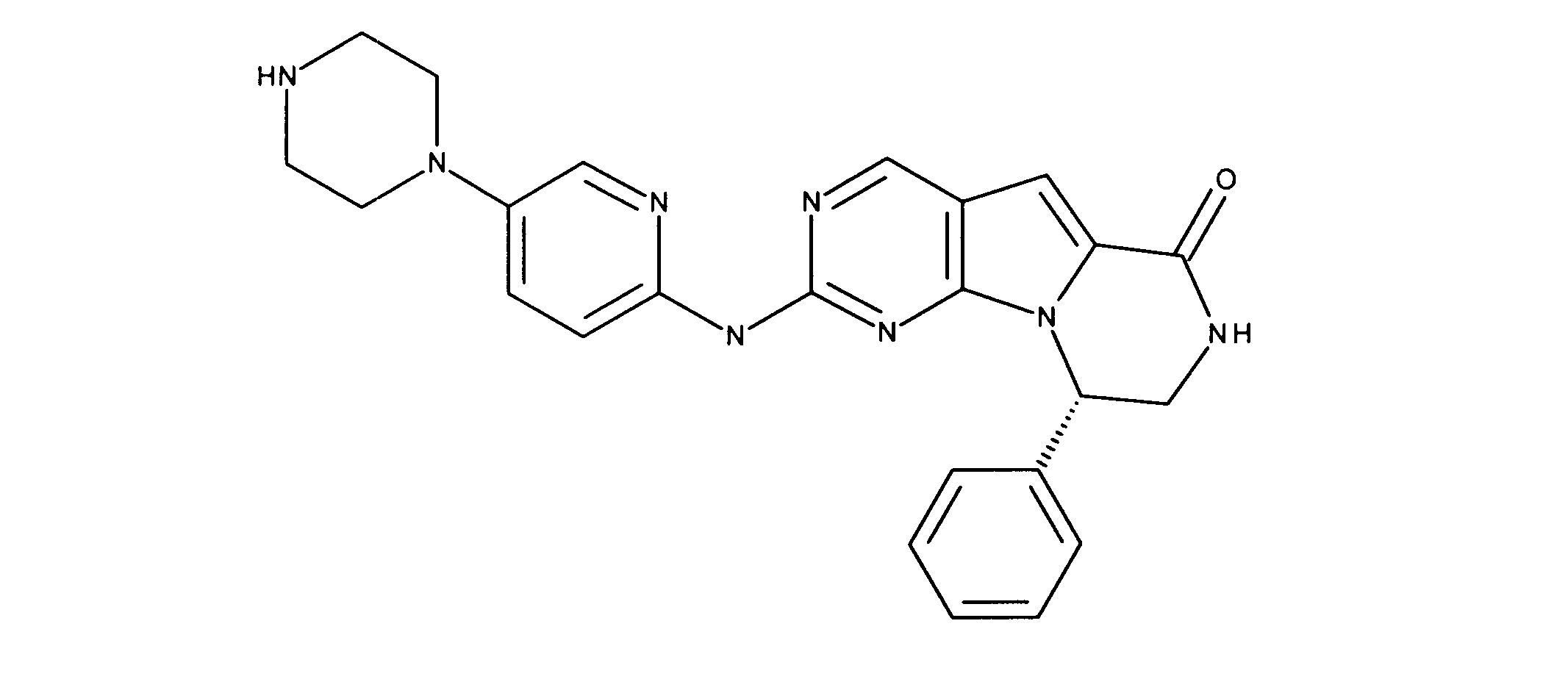
化合物86の合成
1HNMR (600 MHz, DMSO-d6) δ ppm 0.90 (d, J=6.15 Hz, 6 H) 1.72 - 1.89 (m, 1 H) 3.15 - 3.92 (m, 9 H) 4.10 - 4.46 (m, 2 H) 7.18 (s, 1 H) 7.59 (d, J=8.78 Hz, 1 H) 8.00 (s, 1 H) 8.13 (d, J=9.37 Hz, 1 H) 8.55 (s, 1 H) 9.09 (s, 1 H) 9.67 (s, 2 H) 11.91 (s, 1 H). LCMS (ESI) 407 (ESI).
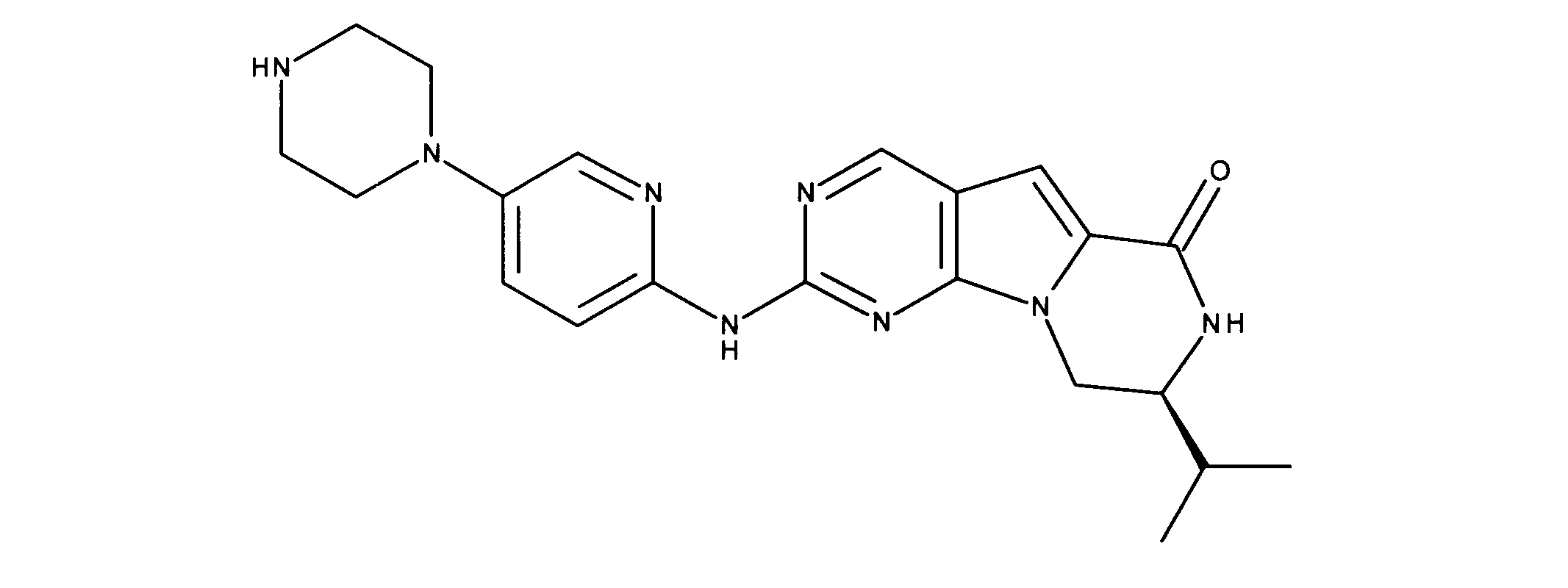
化合物87の合成
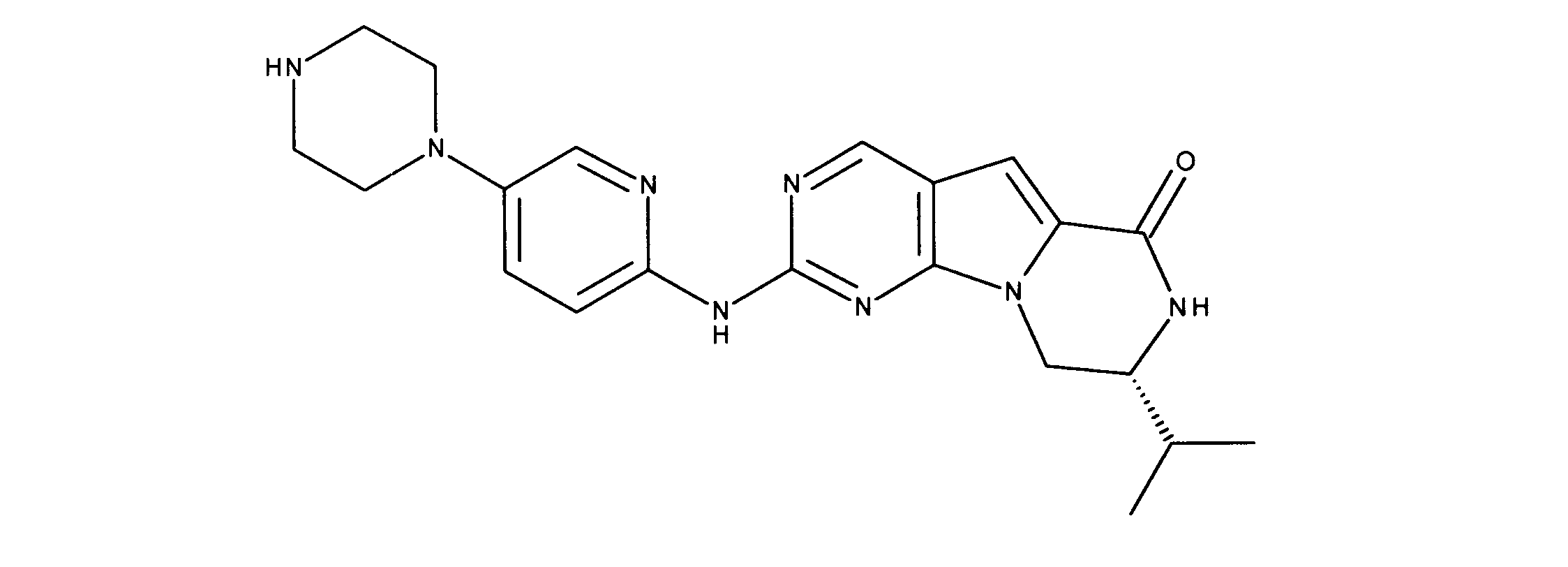
化合物88の合成
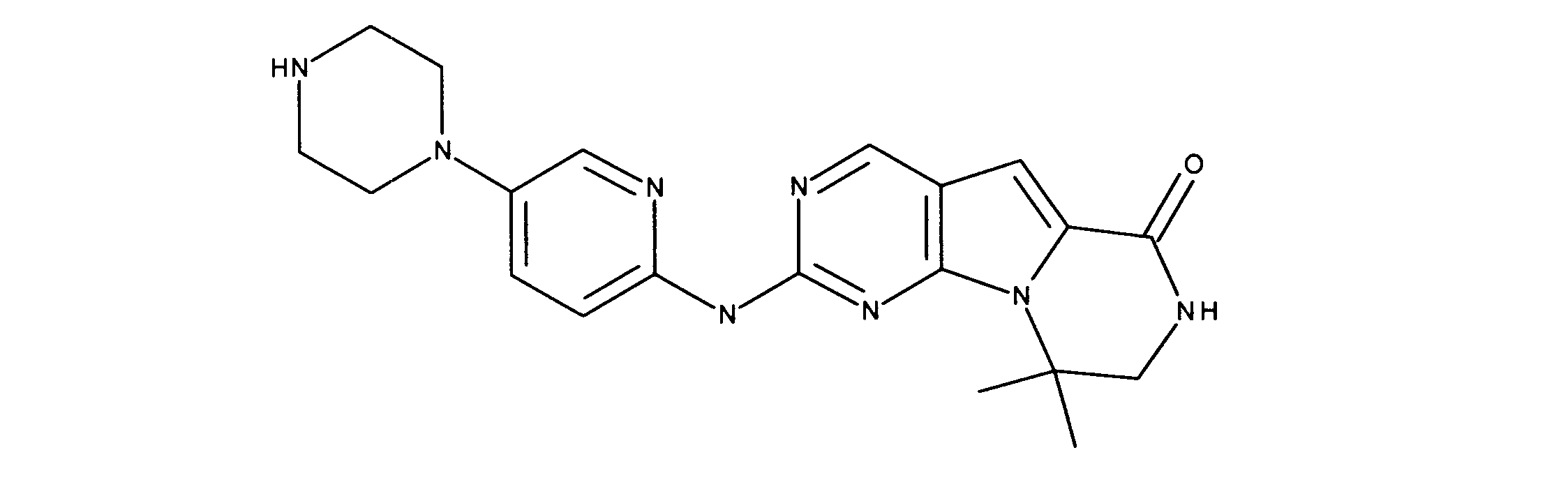
1HNMR (600 MHz, DMSO-d6) δ ppm 1.78 (s, 6 H) 3.40 - 3.53 (m, 6 H) 3.64 - 3.73 (m, 4 H) 7.27 (s, 1 H) 7.66 (d, J=9.37 Hz, 1 H) 7.98 (d, J=2.34 Hz, 1 H) 8.12 (br. s., 1 H) 8.47 (br. s., 1 H) 9.11 (s, 1 H) 9.45 (br. s., 2 H) 11.62 (br. s., 1 H). LCMS (ESI) 393 (M + H).
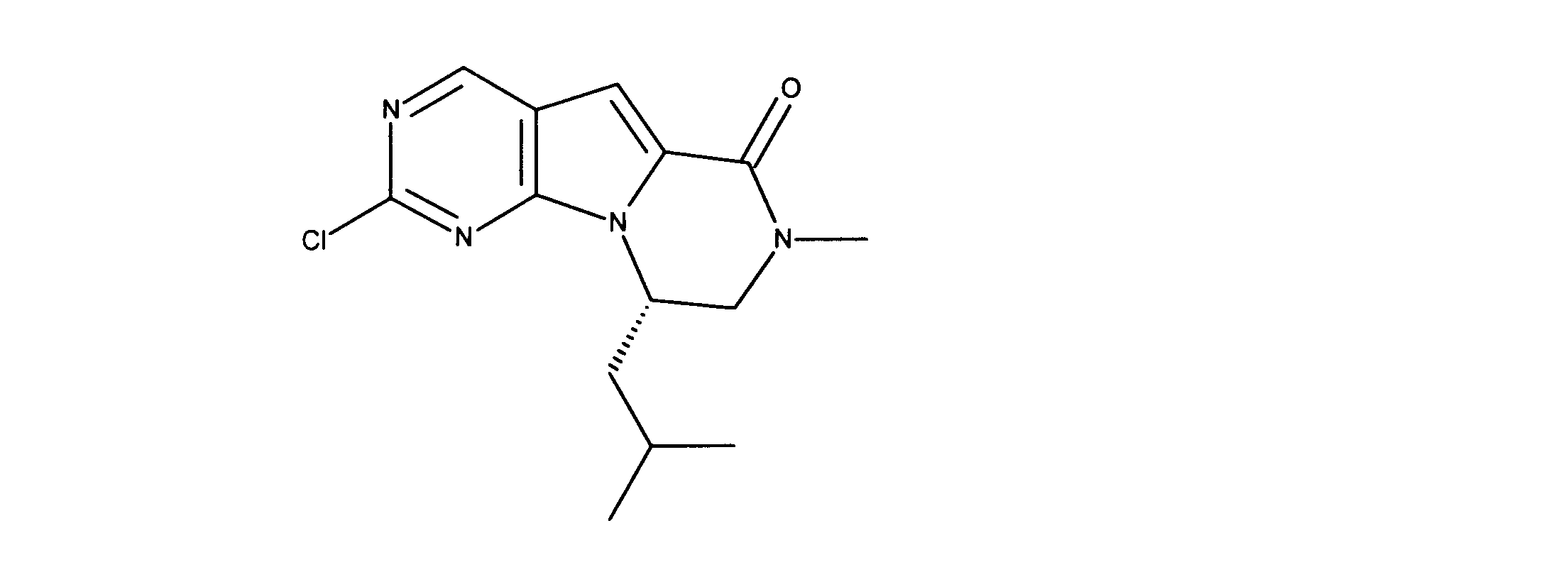
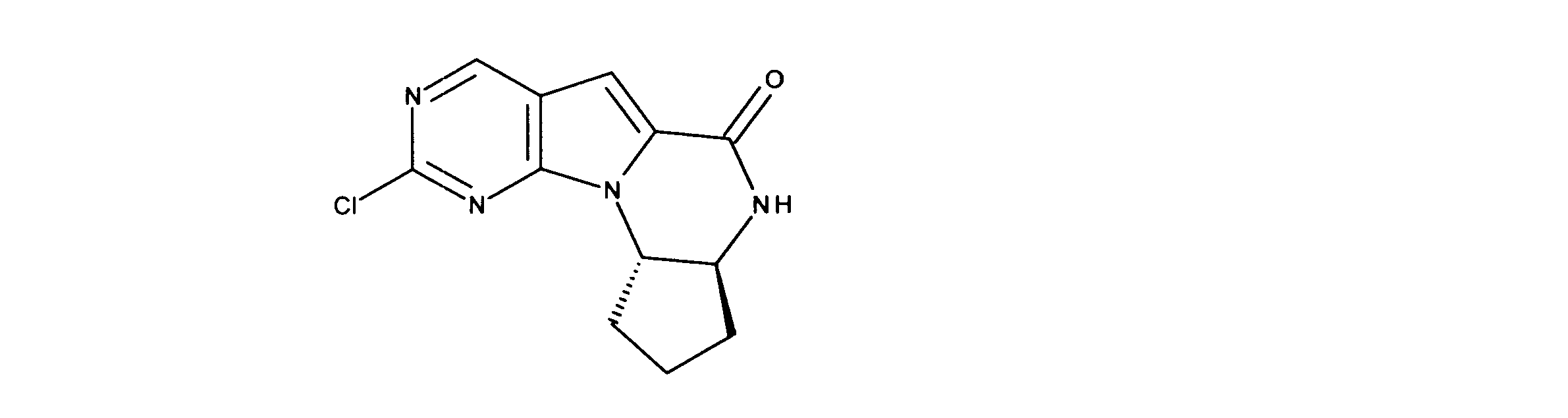
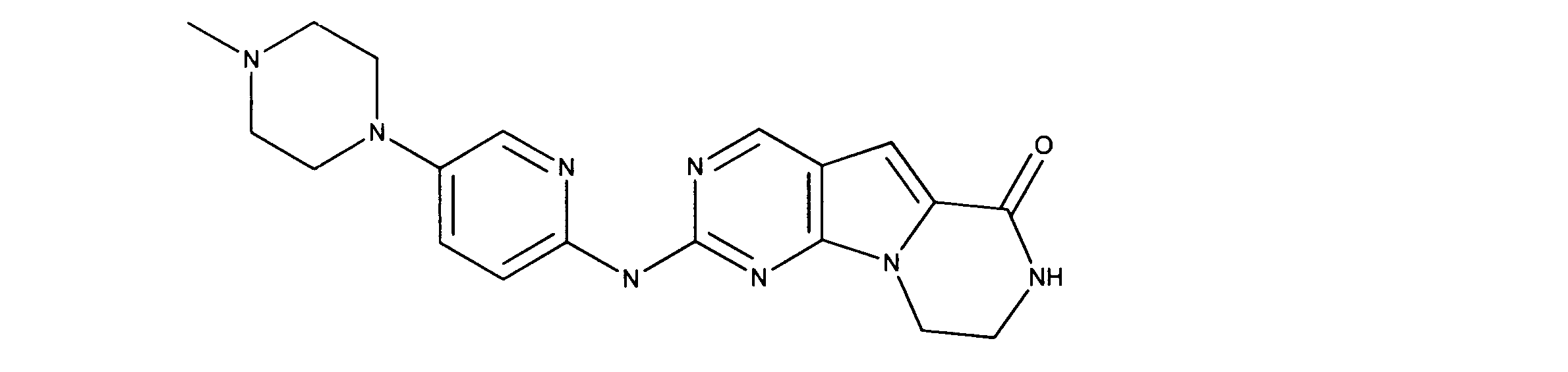
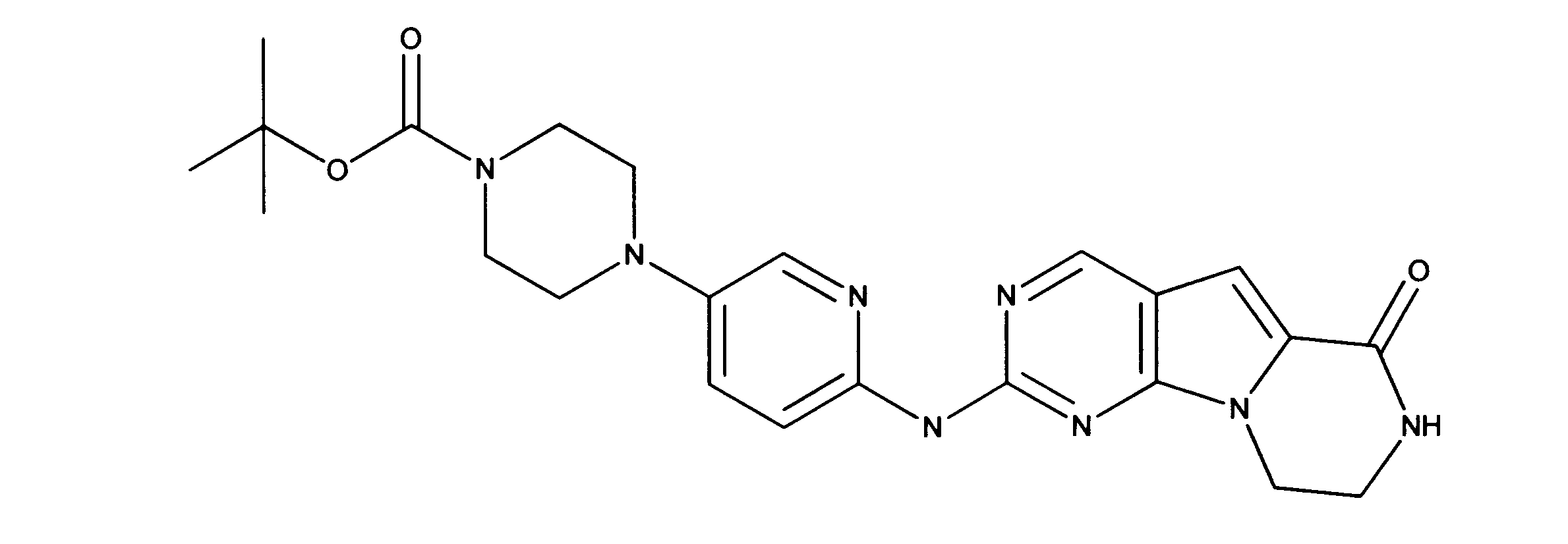
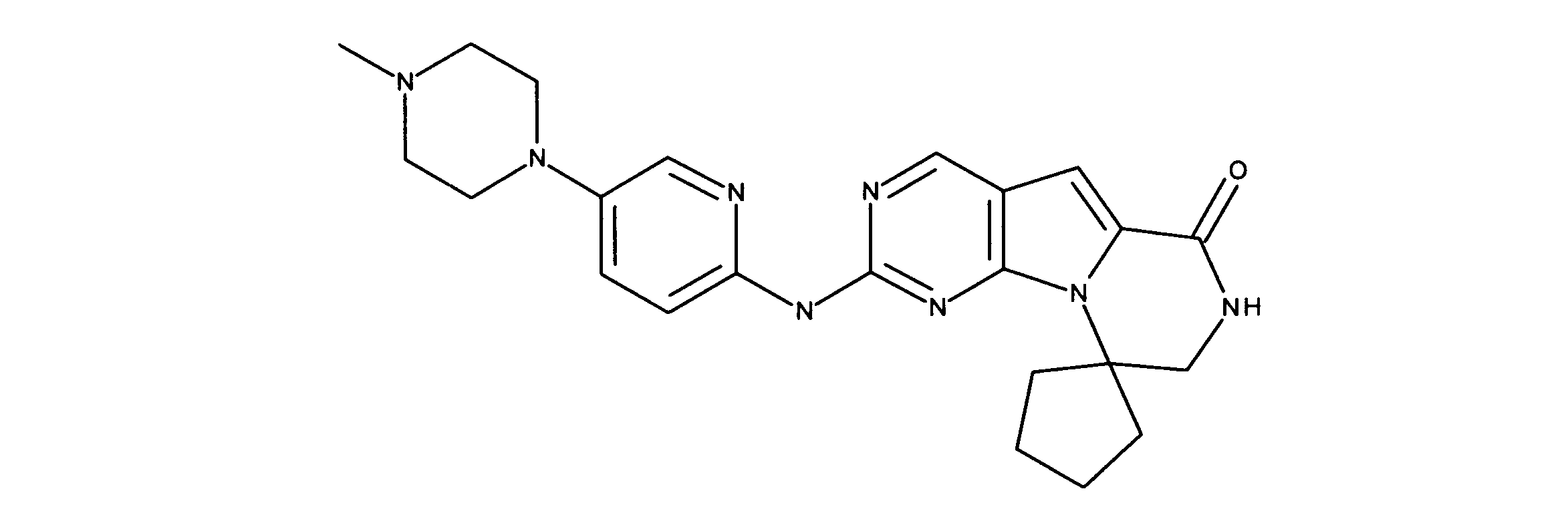
化合物89(化合物Tとも呼ばれる)の合成
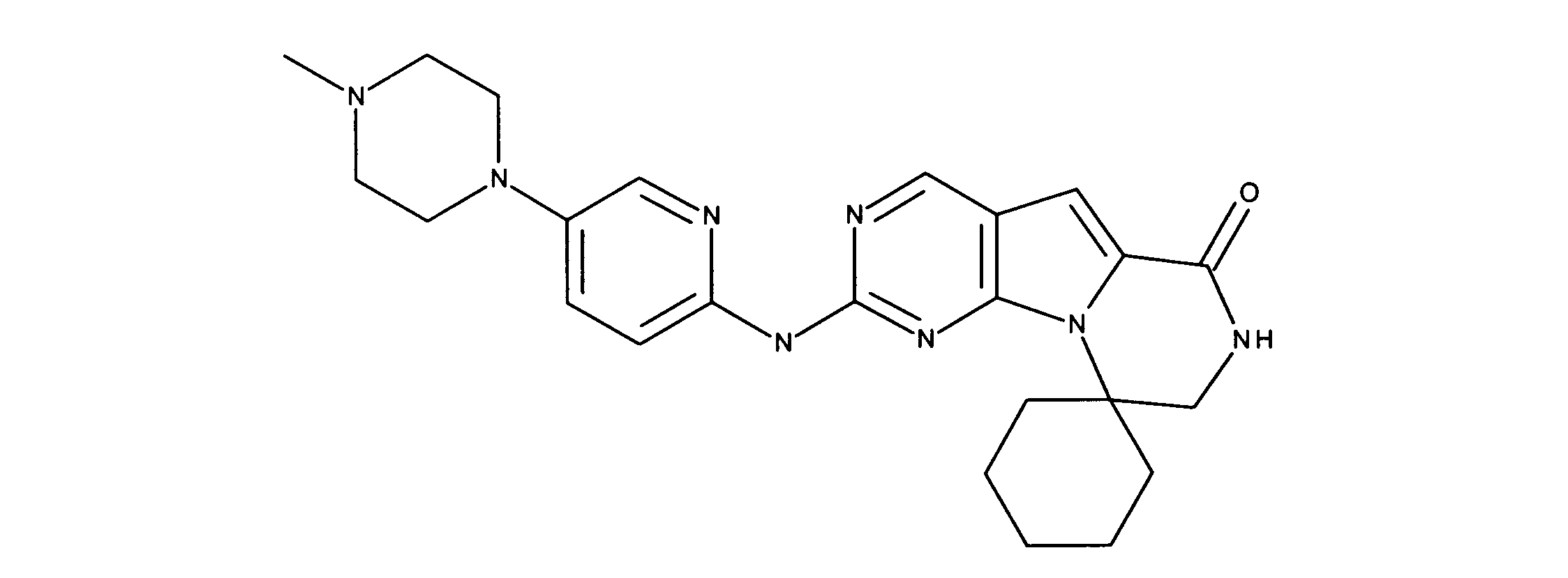
1HNMR (600 MHz, DMSO-d6) δ ppm 1.47 (br. s., 6 H) 1.72 (br. s., 2 H) 1.92 (br. s., 2 H) 2.77 (br. s., 3 H) 3.18 (br. s., 2 H) 3.46 (br. s., 2 H) 3.63 (br. s., 2 H) 3.66 (d, J=6.15 Hz, 2 H) 3.80 (br. s., 2 H) 7.25 (s, 1 H) 7.63 (br. s., 2 H) 7.94 (br. s., 1 H) 8.10 (br. s., 1 H) 8.39 (br. s., 1 H) 9.08 (br. s., 1 H) 11.59 (br. s., 1 H). LCMS (ESI) 447 (M + H).
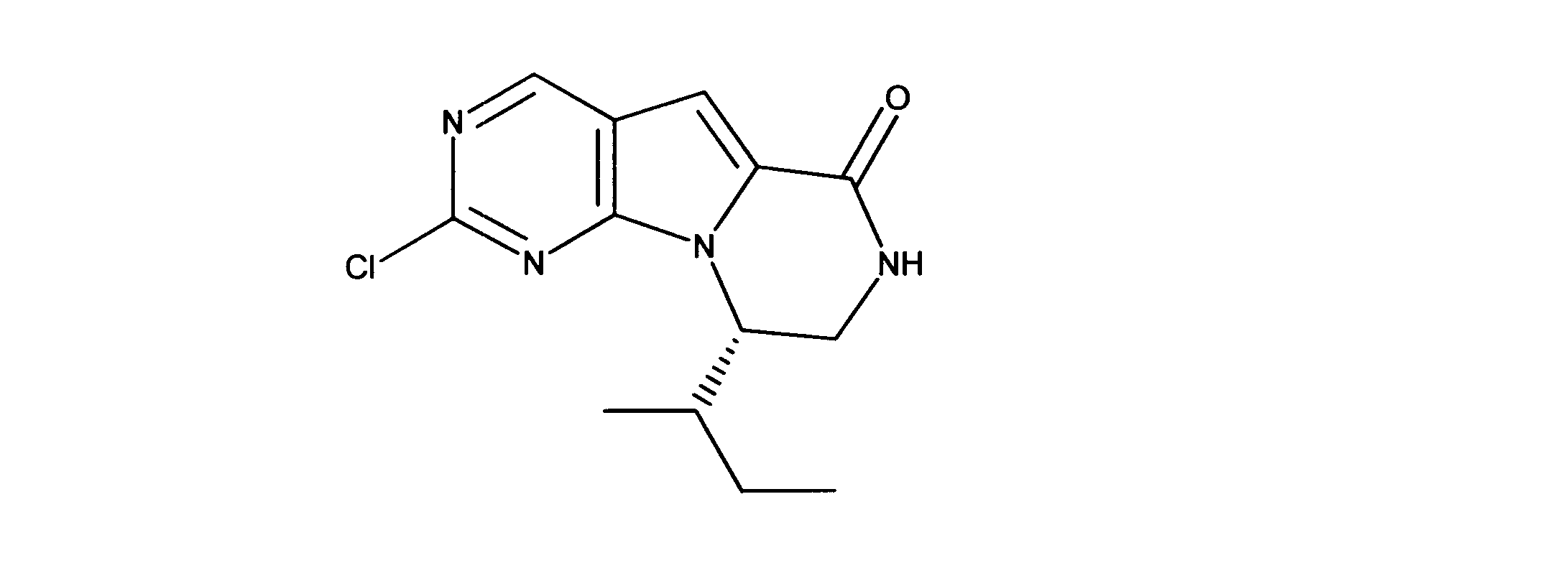
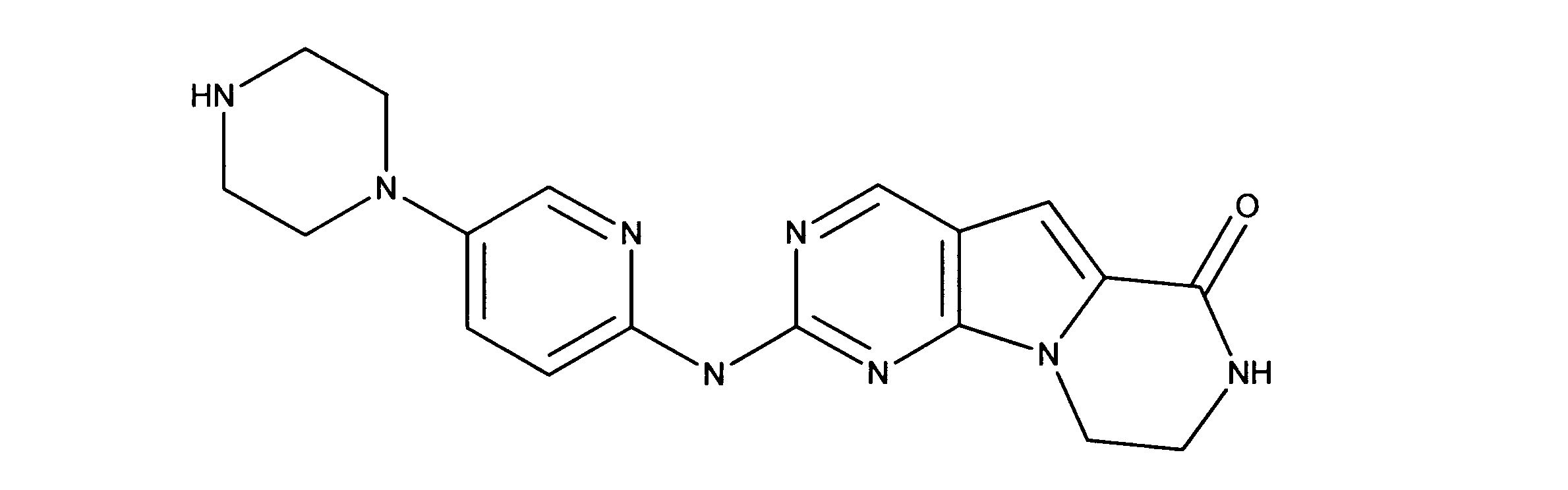
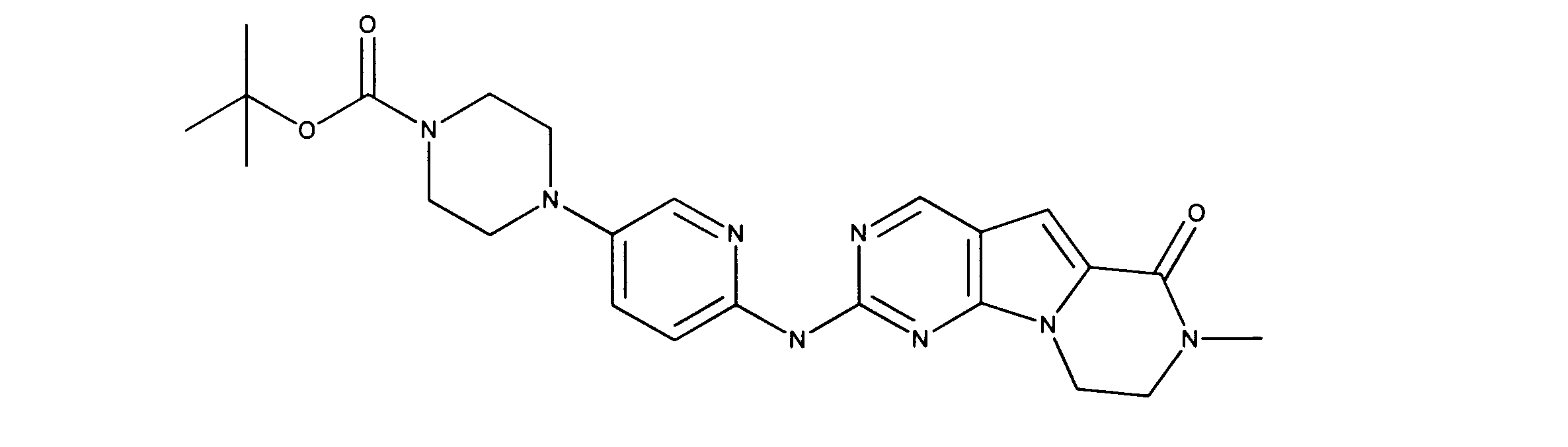
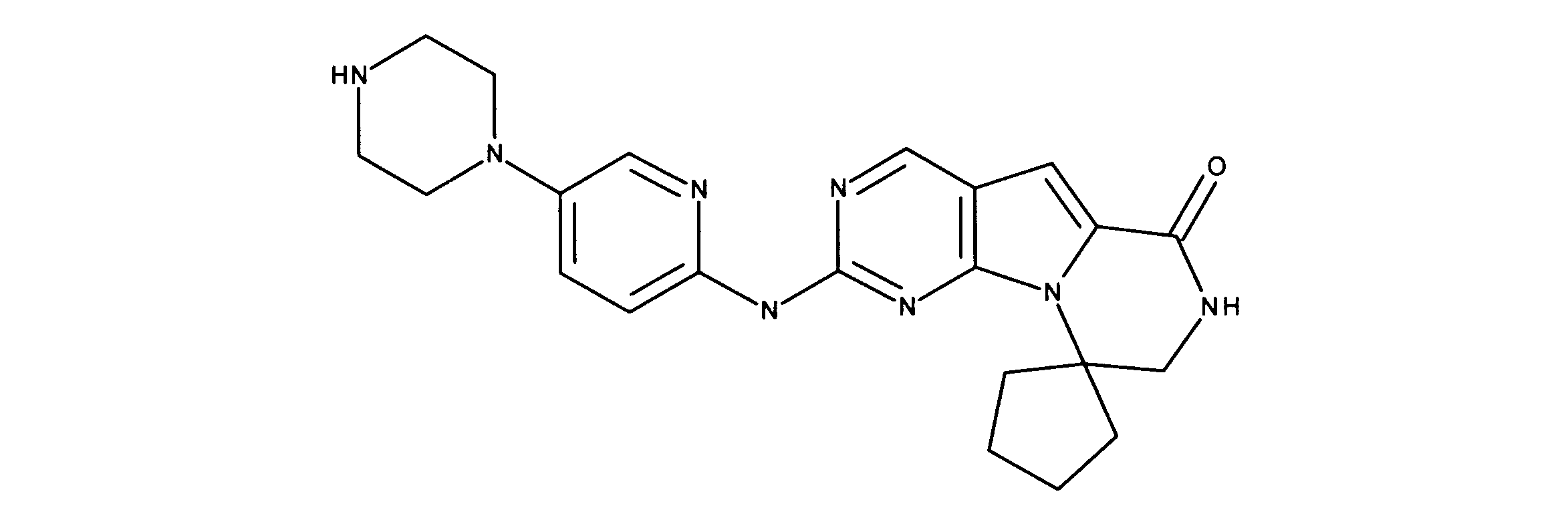
化合物90(化合物Qとも呼ばれる)の合成
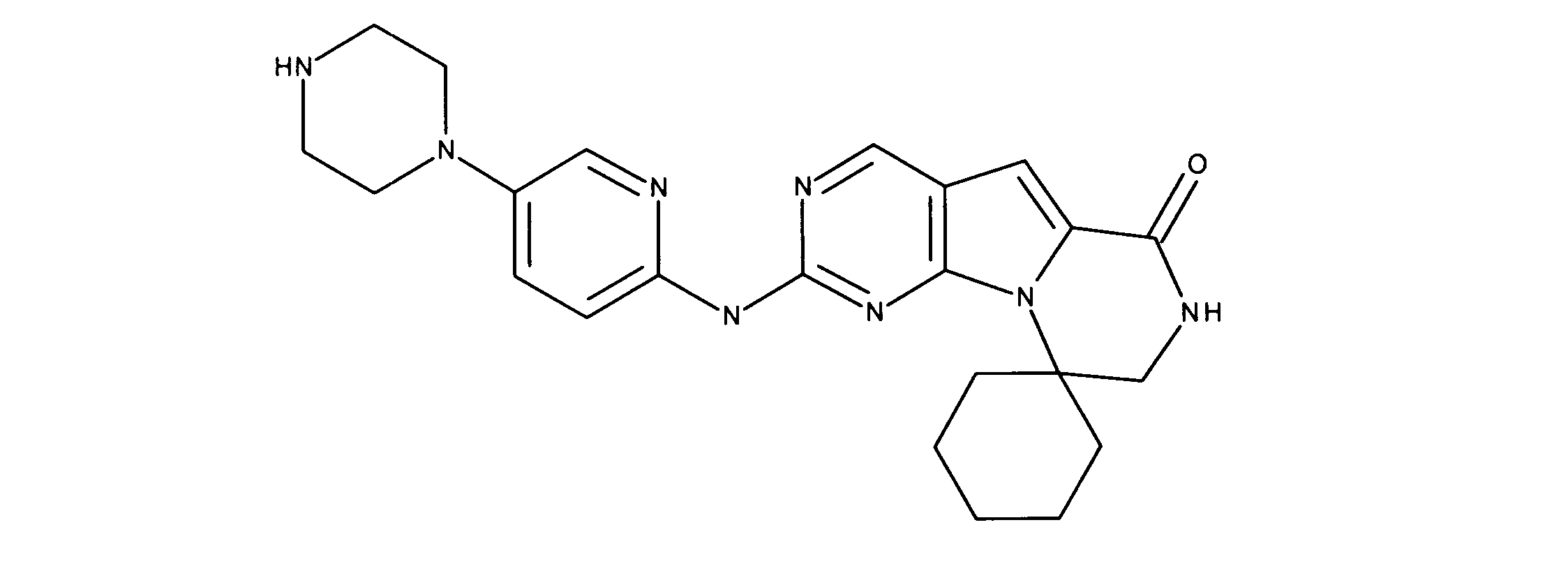
1HNMR (600 MHz, DMSO-d6) δ ppm 1.27 - 1.64 (m, 6 H) 1.71 (br. s., 2 H) 1.91 (br. s., 2 H) 2.80 (br. s., 1 H) 3.17 - 3.24 (m, 2 H) 3.41 (br. s., 4 H) 3.65 (br. s., 4 H) 7.26 (br. s., 1 H) 7.63 (br. s., 1 H) 7.94 (br. s., 1 H) 8.13 (br. s., 1 H) 8.40 (br. s., 1 H) 9.09 (br. s., 1 H) 9.62 (br. s., 1 H) 11.71 (br. s., 1 H). LCMS (ESI) 433 (M + H).
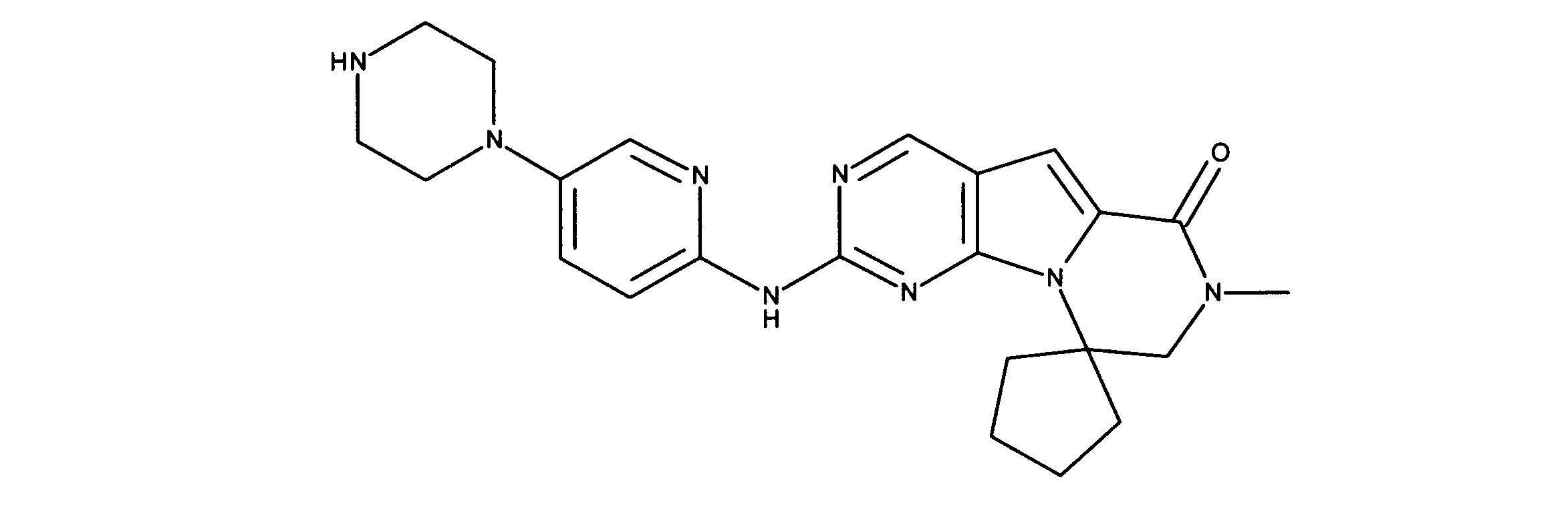
化合物91(化合物ZZとも呼ばれる)の合成
1HNMR (600 MHz, DMSO-d6) δ ppm 1.64 - 1.75 (m, 2 H) 1.83 - 1.92 (m, 2 H) 1.96 - 2.06 (m, 2 H) 2.49 - 2.58 (m, 2 H) 2.79 (d, J=3.81 Hz, 3 H) 3.06 - 3.18 (m, 4 H) 3.59 - 3.69 (m, 2 H) 3.73 - 3.83 (m, 2 H) 4.04 - 4.12 (m, 2 H) 7.17 (br. s., 1 H) 7.60 - 7.70 (m, 2 H) 7.70 - 7.92 (m, 2 H) 7.96 (br. s., 1 H) 8.41 (br. s., 1 H) 8.98 (br. s., 1 H) 10.77 (br. s., 1 H). LCMS (ESI) 433 (M + H).
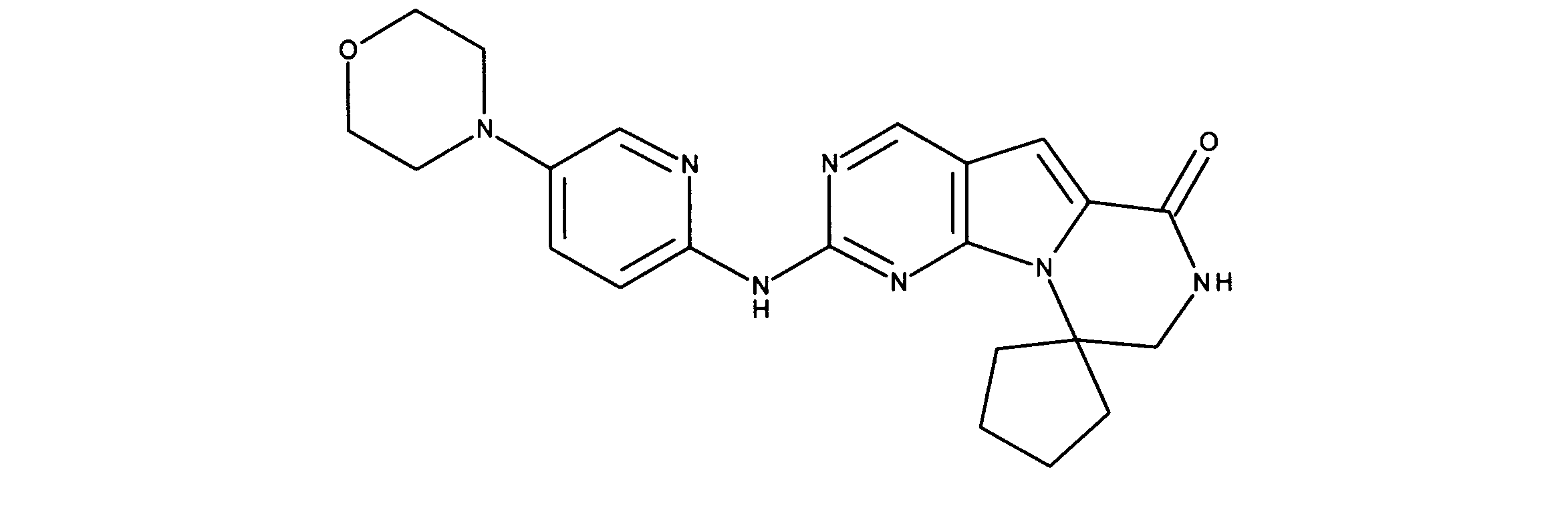
化合物92の合成
1HNMR (600 MHz, DMSO-d6) δ ppm 1.64 - 1.75 (m, 2 H) 1.84 - 1.92 (m, 2 H) 1.96 - 2.05 (m, 2 H) 2.48 - 2.56 (m, 2 H) 3.22 (br. s., 4 H) 3.42 - 3.48 (m, 4 H) 3.60 - 3.69 (m, 2 H) 4.05 - 4.13 (m, 1 H) 7.18 (s, 1 H) 7.65 (d, J=13.47 Hz, 1 H) 7.70 - 7.77 (m, 1 H) 7.94 (d, J=1.76 Hz, 1 H) 8.42 (br. s., 1 H) 9.00 (s, 1 H) 9.15 (br. s., 2 H). LCMS (ESI) 419 (M + H).
化合物93の合成
1HNMR (600 MHz, DMSO-d6) δ ppm 1.76 (br. s., 2 H) 1.89 (br. s., 2 H) 2.03 (br. s., 2 H) 2.47 - 2.58 (m, 2 H) 3.04 (s, 3 H) 3.22 (br. s., 4 H) 3.39 (br. s., 4 H) 3.66 (s, 2 H) 7.21 (s, 1 H) 7.67 (d, J=9.37 Hz, 1 H) 7.93 (br. s., 1 H) 7.98 - 8.09 (m, 1 H) 9.04 (s, 1 H) 9.34 (br. s., 2 H) 11.31 (br. s., 1 H). LCMS (ESI) 433 (M + H).
化合物94の合成
1HNMR (600 MHz, DMSO-d6) δ ppm 1.66 - 1.77 (m, 2 H) 1.84 - 1.94 (m, 2 H) 1.96 - 2.08 (m, 2 H) 2.48 - 2.57 (m, 2 H) 3.36 - 3.52 (m, 4 H) 3.60 - 3.80 (m, 6 H) 7.21 (s, 1 H) 7.53 - 7.74 (m, 2 H) 7.86 (s, 1 H) 8.02 (s, 1 H) 8.45 (s, 1 H) 9.03 (s, 1 H) 11.19 (br. s., 1 H). LCMS (ESI) 420 (M+H).
化合物95の合成
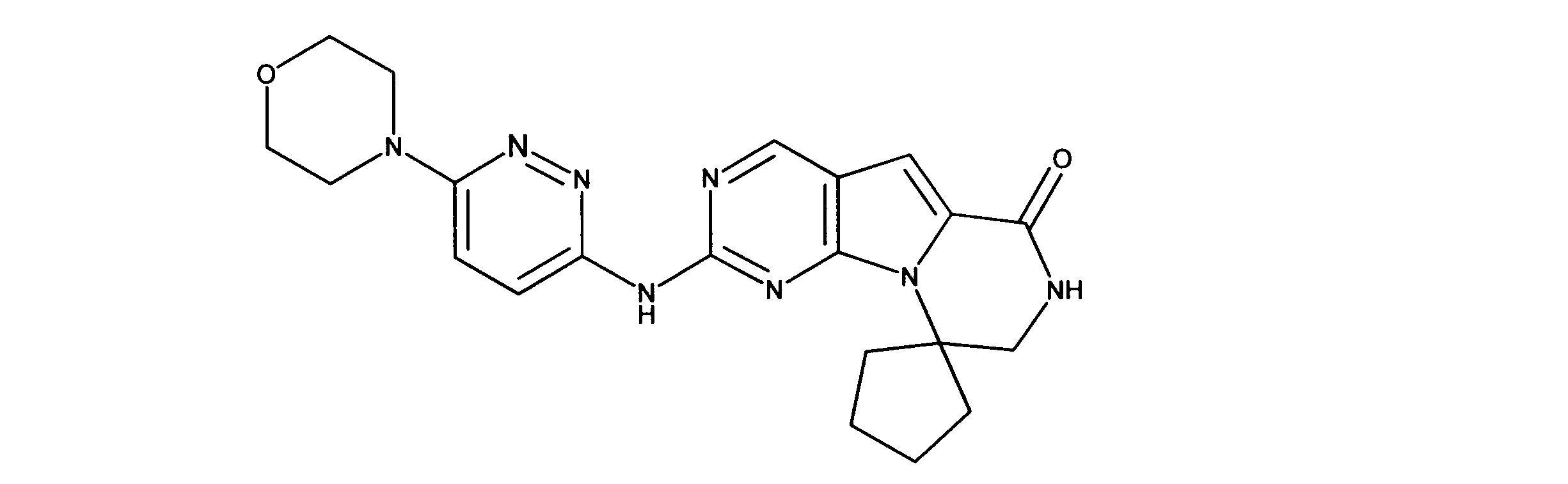
1HNMR (600 MHz, DMSO-d6) δ ppm 1.65 - 1.79 (m, 2 H) 1.85 - 1.95 (m, 2 H) 1.97 - 2.08 (m, 2 H) 2.47 - 2.54 (m, 2 H) 3.40 - 3.58 (m, 5 H) 3.65 (dd, J=21.67, 5.56 Hz, 1 H) 3.69 - 3.78 (m, 4 H) 7.24 (s, 1 H) 7.97 - 8.17 (m, 2 H) 8.48 (s, 1 H) 9.08 (s, 1 H) 11.81 (s, 1 H). LCMS (ESI) 421 (M+H).
化合物96の合成
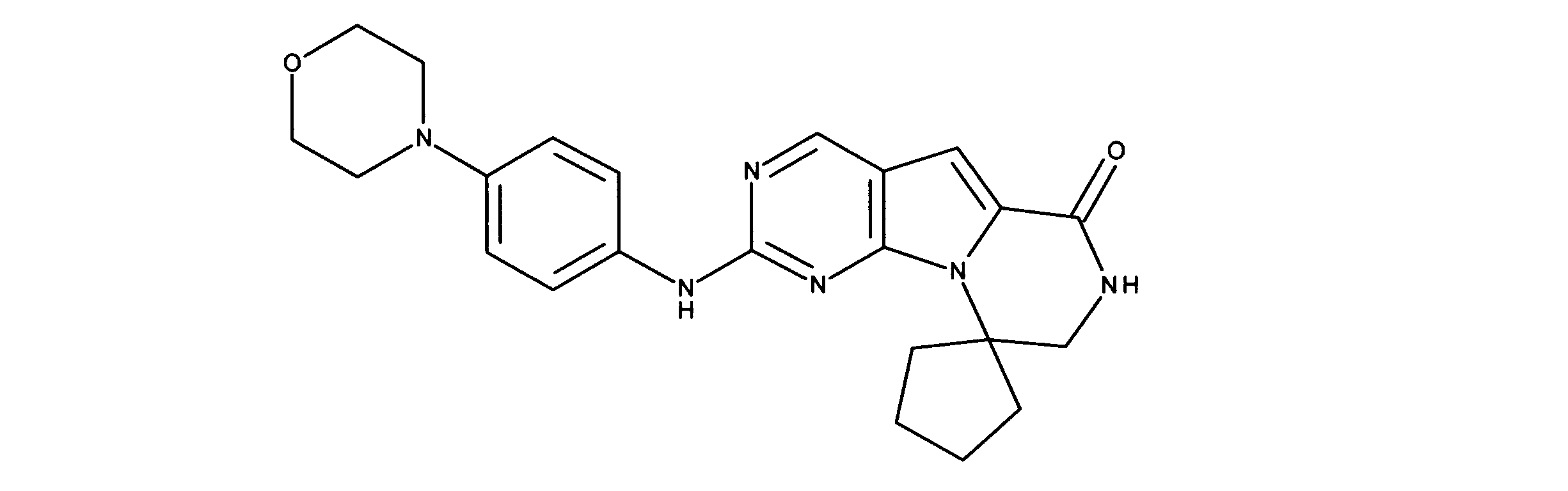
1HNMR (600 MHz, DMSO-d6) δ ppm 1.55 - 1.74 (m, 2 H) 1.80 - 1.98 (m, 4 H) 2.48 - 2.60 (m, 2 H) 3.40 - 3.50 (m, 4 H) 3.57 - 3.72 (m, 2 H) 3.90 - 4.20 (m, 4 H) 7.08 (s, 1 H) 7.37 - 7.57 (m, 2 H) 7.70 (m, 2 H) 8.32 (s, 1 H) 8.88 (s, 1 H) 9.98 (s, 1 H). LCMS (ESI) 419 (M+H).
化合物97(化合物IIIとも呼ばれる)の合成
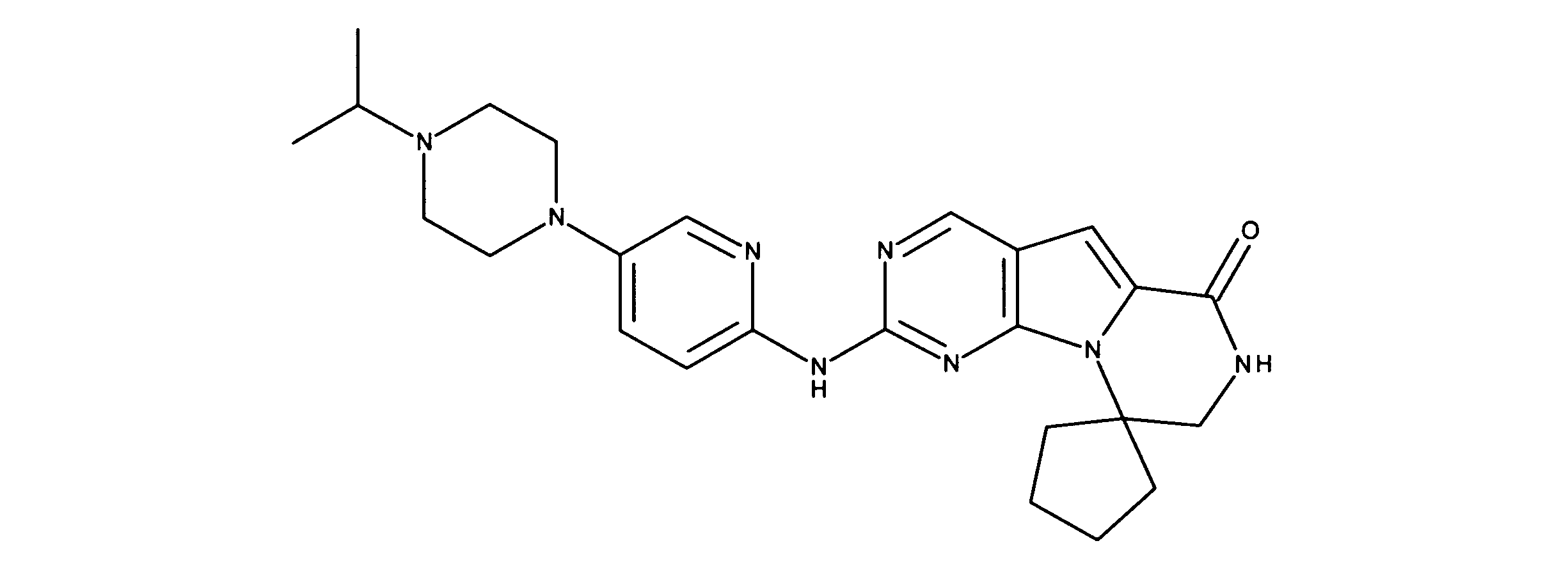
1HNMR (600 MHz, DMSO-d6) δ ppm 1.30 (d, J=5.27 Hz, 6 H) 1.65 - 1.78 (m, 2 H) 1.83 - 1.95 (m, 2 H) 1.97 - 2.10 (m, 2 H) 2.45 - 2.55 (m, 2H) 3.25 - 3.36 (m, 1 H) 3.39 - 3.48 (m, 4 H) 3.60 - 3.70 (m, 4 H) 3.75 - 4.15 (m, 2 H) 7.24 (s, 1 H) 7.54 - 7.75 (m, 2 H) 7.95 (s, 1 H) 8.10 (s, 1 H) 8.49 (s, 1 H) 9.07 (s, 1 H) 11.25 (s, 1 H) 11.48 (s, 1 H). LCMS (ESI) 461 (M+H).
化合物98の合成

1HNMR (600 MHz, DMSO-d6) δ ppm 0.99 (d, J=6.15 Hz, 6 H) 1.65 - 1.78 (m, 2 H) 1.90 (m, 2 H) 1.97 - 2.08 (m, 2 H) 2.08 - 2.17 (m, 1 H) 2.45 - 2.55 (m, 2H) 2.88 - 3.02 (m, 2 H) 3.33 - 3.48 (m, 4 H) 3.50 - 3.90 (m, 6 H) 7.24 (s, 1 H) 7.67 (s, 2 H) 7.94 (s, 1 H) 8.12 (s, 1 H) 8.49 (s, 1 H) 9.07 (s, 1 H) 10.77 (s, 1 H) 11.51 (s, 1 H). LCMS (ESI) 475 (M+H).
化合物99の合成
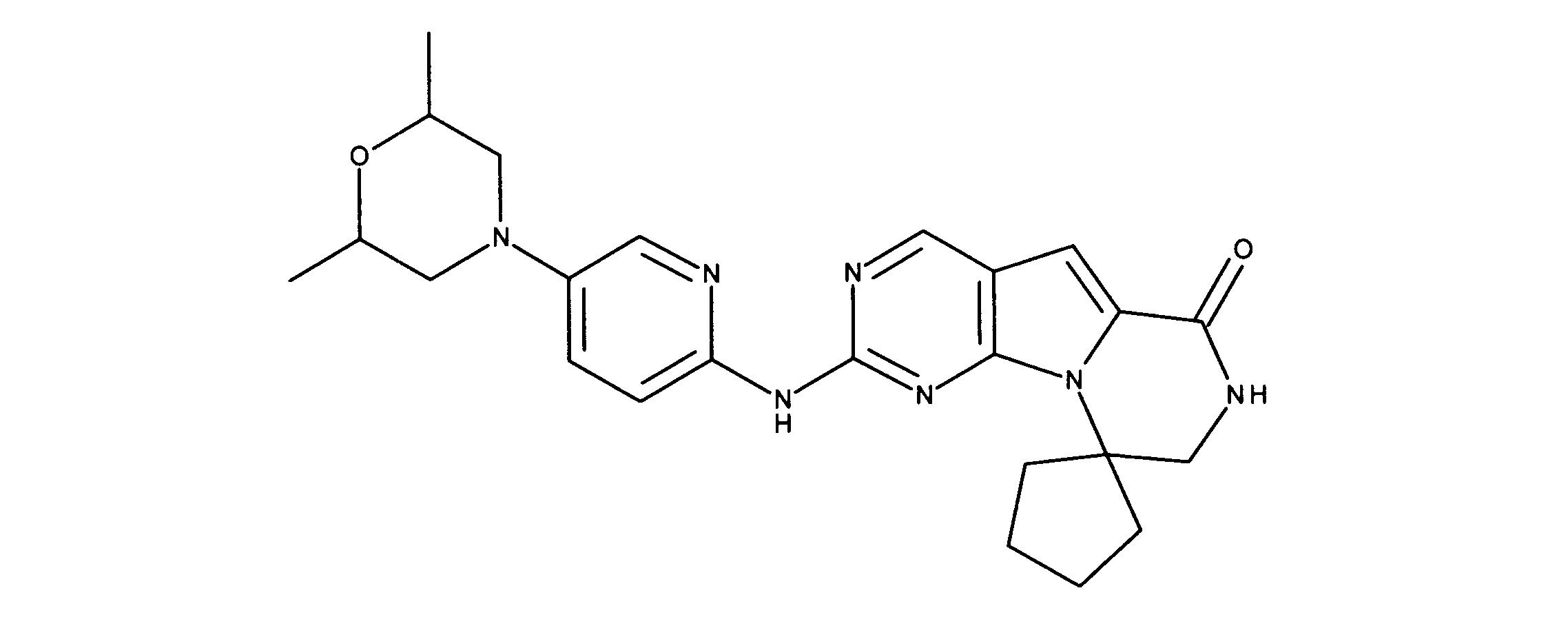
1HNMR (600 MHz, DMSO-d6) δ ppm 1.13 (d, J=5.86 Hz, 6 H) 1.66 - 1.77 (m, 2 H) 1.84 - 1.94 (m, 2 H) 1.97 - 2.09 (m, 2 H) 2.40 - 2.53 (m, 2 H) 3.37 - 3.49 (m, 2 H) 3.50 - 3.59 (m, 2 H) 3.59 - 3.73 (m, 4 H) 7.23 (s, 1 H) 7.64 (m, 3 H) 7.85 (s, 1 H) 8.11 (s, 1 H) 8.47 (s, 1 H) 9.05 (s, 1 H). 11.35 (br s., 1H). LCMS (ESI) 448 (M+H).
化合物100の合成
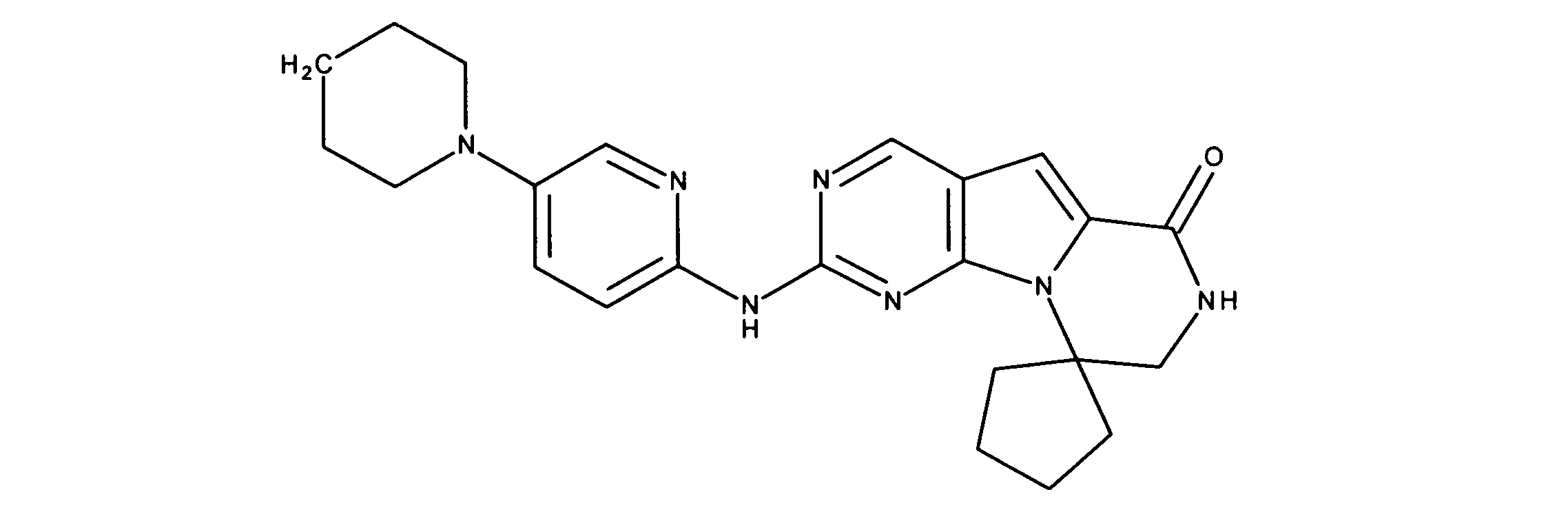
1HNMR (600 MHz, DMSO-d6) δ ppm 1.50 - 1.57 (m, 2 H) 1.62 - 1.68 (m, 3 H) 1.68 - 1.75 (m, 2 H) 1.84 - 1.92 (m, 2 H) 1.97 - 2.08 (m, 2 H) 2.48 - 2.53 (m, 2 H) 3.14 - 3.23 (m, 4 H) 3.43 - 3.47 (m, 2 H) 3.58 - 3.70 (m, 2 H) 7.22 (s, 1 H) 7.58 - 7.70 (m, 2 H) 7.85 - 8.00 (m, 1 H) 8.16 (d, 1 H) 8.46 (s, 1 H) 9.04 (s, 1 H) 11.37 (br s., 1H). LCMS (ESI) 418 (M + H).
化合物101(化合物WWとも呼ばれる)の合成

1HNMR (600 MHz, DMSO-d6) δ ppm 1.72 (s, 2 H) 1.90 (s, 4 H) 2.03 (s, 2 H) 2.21 (s, 2 H) 2.48 - 2.54 (m, 2 H) 2.73 (s, 2 H) 3.03 (s, 2 H) 3.25 - 3.35 (m, 1 H) 3.38 - 3.48 (m, 4 H) 3.65 - 3.99 (m, 5 H) 7.23 (s, 1 H) 7.63 (d, J=9.66 Hz, 1 H) 7.90 (s, 1 H) 8.13 (s, 1 H) 8.47 (s, 1 H) 9.06 (s, 1 H) 10.50 (br s., 1H). LCMS (ESI) 503 (M + H).
化合物102(化合物HHHとも呼ばれる)の合成
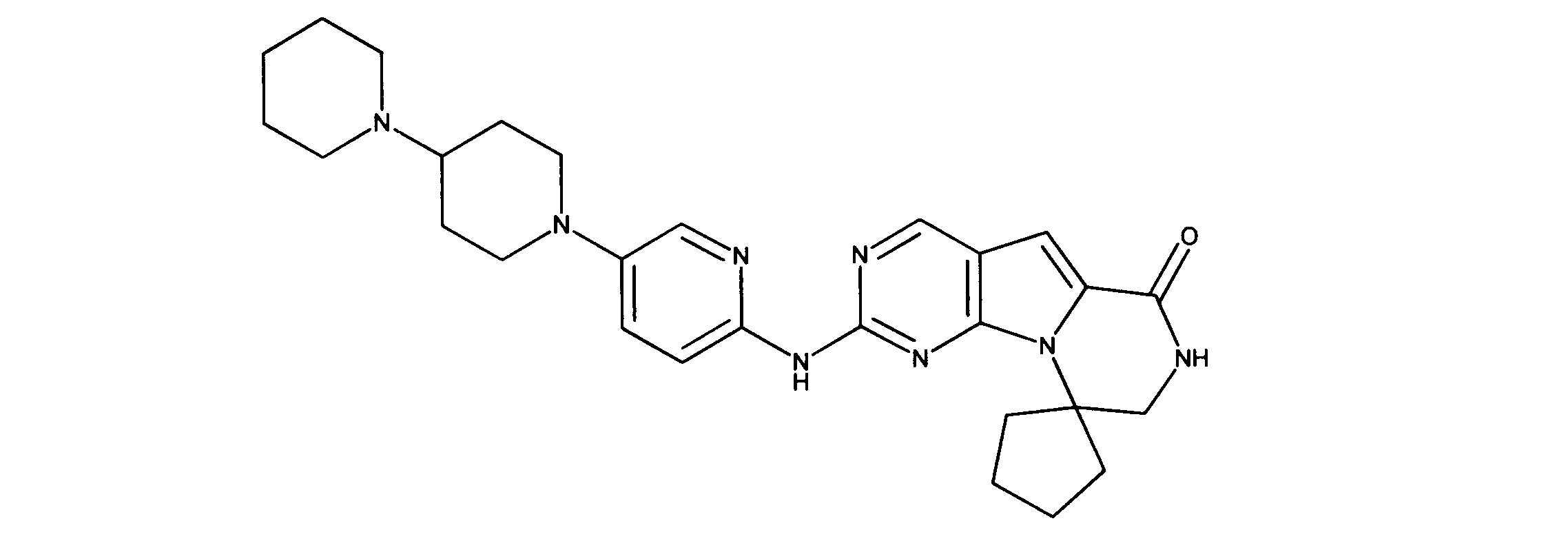
1HNMR (600 MHz, DMSO-d6) δ ppm 1.63 - 1.85 (m, 6 H) 1.87 - 1.92 (m, 2 H) 1.99 - 2.06 (m, 2 H) 2.15 - 2.23 (m, 2 H) 2.47 - 2.53 (m, 1 H) 2.69 - 2.79 (m, 2 H) 2.81 - 2.91 (m, 2 H) 2.98 - 3.08 (m, 2 H) 3.32 - 3.48 (m, 4 H) 3.57 - 3.72 (m, 4 H) 3.77 - 3.85 (m, 2 H) 7.22 (s, 1 H) 7.60 - 7.68 (m, 2 H) 7.90 (s, 1 H) 8.07 (s, 1 H) 8.46 (s, 1 H) 9.04 (s, 1 H). 11.41 (br s., 1H). LCMS (ESI) 501 (M + H).
化合物103の合成
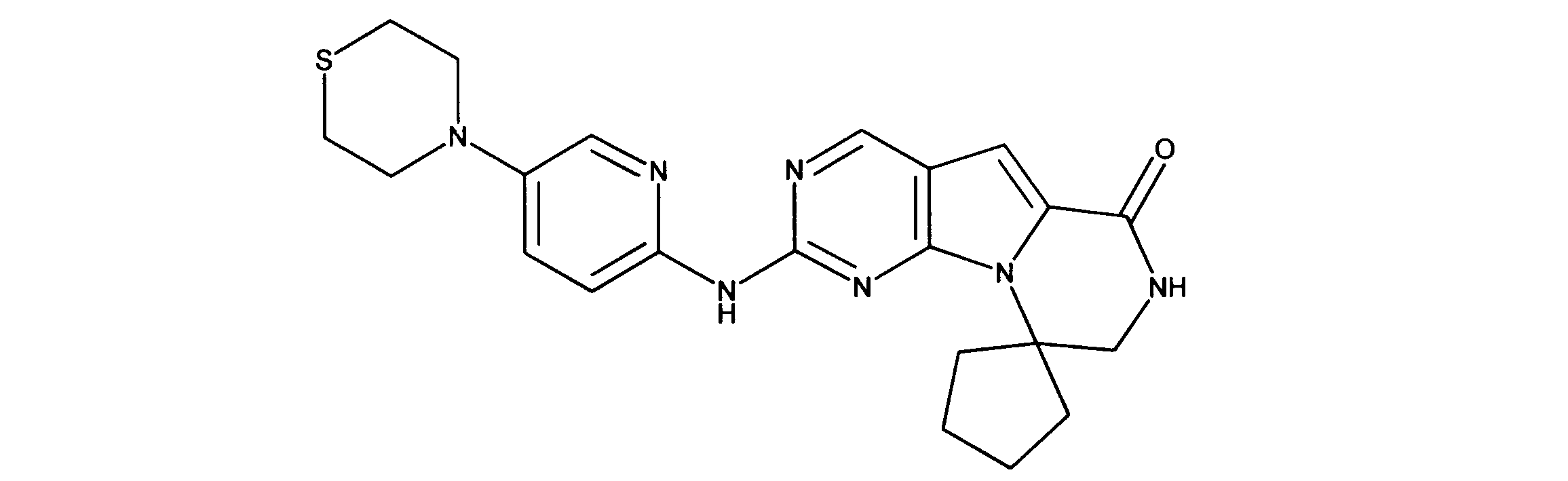
1HNMR (600 MHz, DMSO-d6) δ ppm 1.64 - 1.76 (m, 2 H) 1.87 - 1.93 (m, 2 H) 2.00 - 2.07 (m, 2 H) 2.48 - 2.53 (m, 2 H) 2.67 - 2.72 (m, 4 H) 3.44 - 3.47 (m, 2 H) 3.50 - 3.55 (m, 4 H) 7.24 (s, 1 H) 7.61 (d, J=9.37 Hz, 2 H) 7.86 (d, J=2.63 Hz, 1 H) 8.09 (d, J=12.88 Hz, 1 H) 8.48 (s, 1 H) 9.06 (s, 1 H) 11.41 (br s., 1H). LCMS (ESI) 436 (M + H).
化合物104の合成
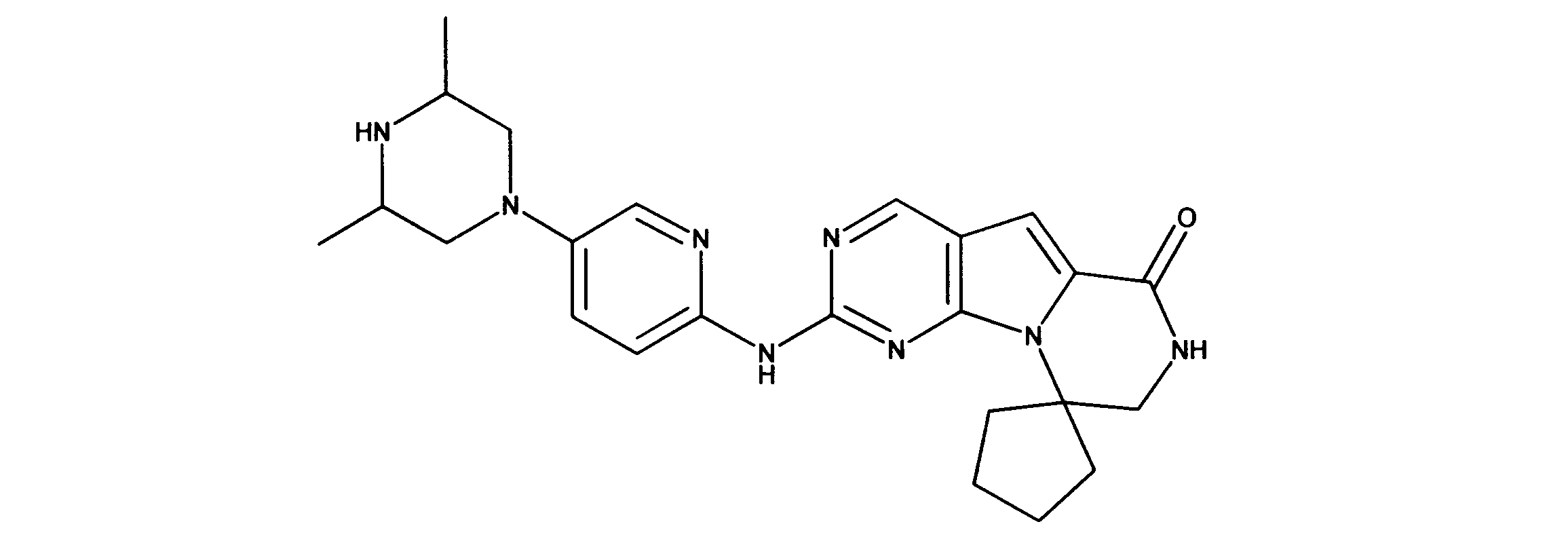
1HNMR (600 MHz, DMSO-d6) δ ppm 1.29 (d, J=6.73 Hz, 6 H) 1.66 - 1.79 (m, 2 H) 1.84 - 1.95 (m, 2 H) 1.98 - 2.09 (m, 2 H) 2.46 - 2.55 (m, 2 H) 3.29 - 3.39 (m, 2H) 3.58 - 3.70 (m, 4H) 3.77 - 3.86 (m, 4H) 7.24 (s, 1 H) 7.66 (d, J=9.37 Hz, 1 H) 7.96 (d, J=2.93 Hz, 1 H) 8.08 (s, 1 H) 8.48 (s, 1 H) 9.06 (s, 1 H) 9.28 (s, 1 H) 9.67 (s, 1 H) 11.36 (s, 1H). LCMS (ESI) 447 (M + H).
化合物105の合成
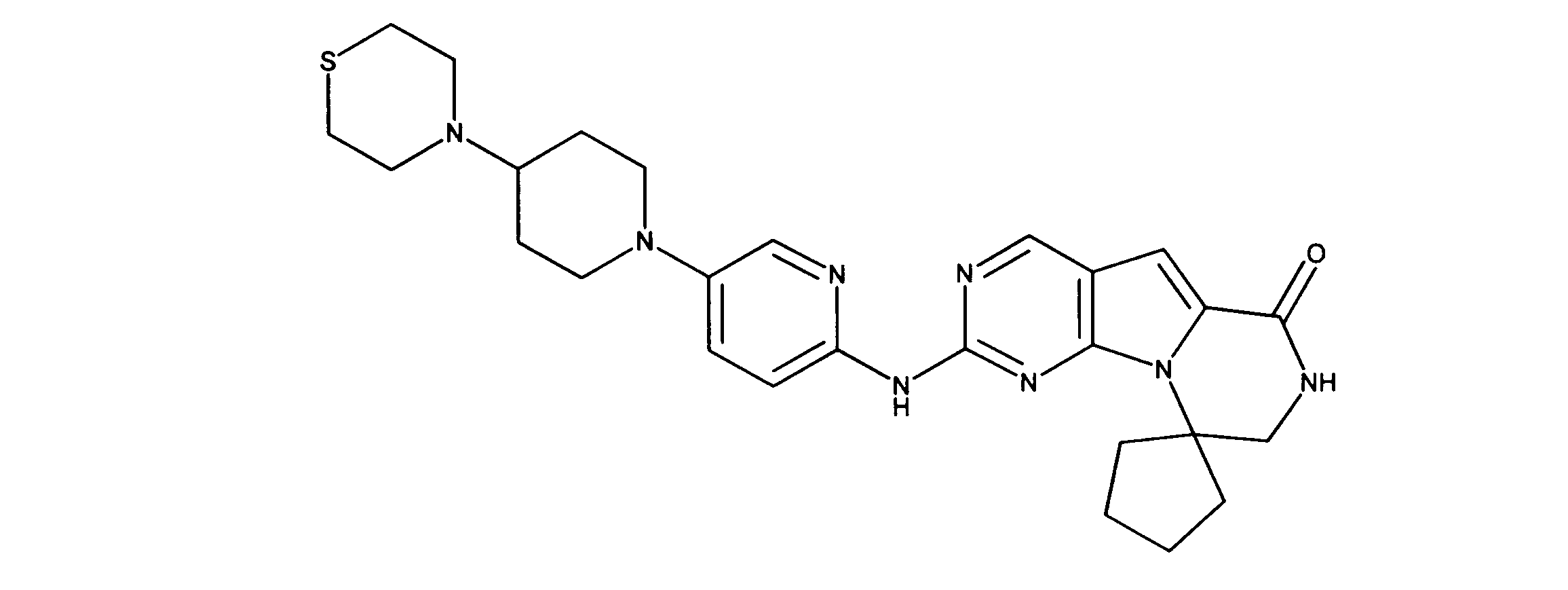
1HNMR (600 MHz, DMSO-d6) δ ppm 1.73 (s, 2 H) 1.76 - 1.85 (m, 2 H) 1.85 - 1.94 (m, 2 H) 1.98 - 2.07 (m, 2 H) 2.19 - 2.26 (m, 2 H) 2.48 - 2.52 (m, 1 H) 2.70 - 2.81 (m, 4 H) 3.13 - 3.20 (m, 1 H) 3.30 - 3.48 (m, 3 H) 3.58 - 3.71 (m, 4 H) 3.78 - 3.84 (m, 4 H) 7.24 (s, 1 H) 7.62 (d, J=9.37 Hz, 2 H) 7.89 (d, J=1.17 Hz, 1 H) 8.09 - 8.18 (m, 1 H) 8.48 (s, 1 H) 9.06 (s, 1 H) 11.46 (br s., 1H). LCMS (ESI) 519 (M + H).
化合物106の合成
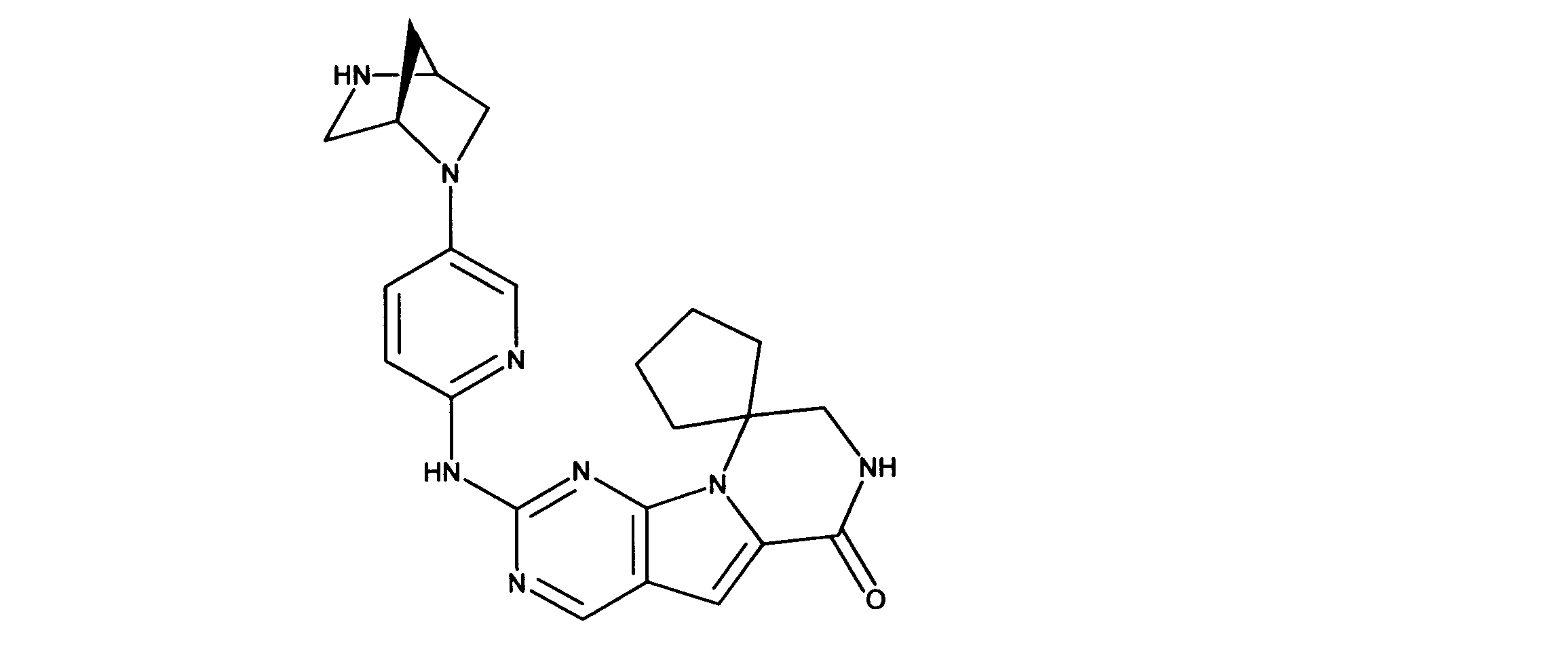
1HNMR (600 MHz, DMSO-d6) δ ppm 1.65 - 1.75 (m, 2 H) 1.85 - 1.93 (m, 2 H) 1.93 - 1.99 (m, 1 H) 2.00 - 2.06 (m, 2 H) 2.08 - 2.14 (m, 1 H) 2.47 - 2.55 (m, 2 H) 3.07 - 3.25 (m, 2 H) 3.25 - 3.69 (m, 5 H) 4.46 (s, 1 H) 4.67 (s, 1 H) 7.22 (s, 1 H) 7.58 - 7.69 (m, 2 H) 8.46 (s, 1 H) 9.02 (s, 1 H) 9.34 (s, 1 H) 9.65 (s, 1 H). LCMS (ESI) 431 (M + H).
化合物107(化合物YYとも呼ばれる)の合成
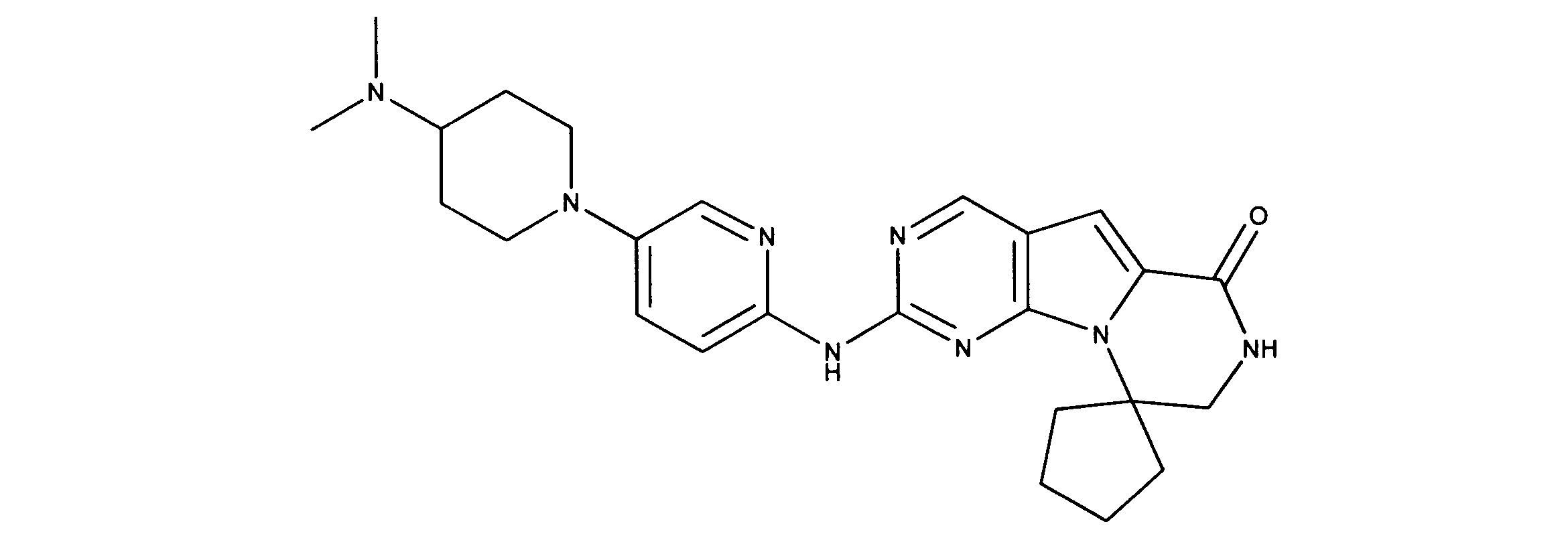
1HNMR (600 MHz, DMSO-d6) δ ppm 1.65 - 1.82 (m, 3 H) 1.89 (br. s., 2 H) 1.98 - 2.08 (m, 2 H) 2.13 (br. s., 2 H) 2.47 - 2.55 (m, 2 H) 2.68 (d, J=4.98 Hz, 6 H) 2.71 - 2.80 (m, 2 H) 3.29 - 3.71 (m, 10 H) 7.16 - 7.26 (m, 1 H) 7.67 (d, J=9.66 Hz, 2 H) 7.91 (d, J=2.05 Hz, 1 H) 8.14 (br. s., 1 H) 8.48 (br. s., 1 H) 9.05 (s, 1 H) 11.14 (br. s., 1 H) 11.43 (br. s., 1 H). LCMS (ESI) 461 (M + H).
化合物108の合成
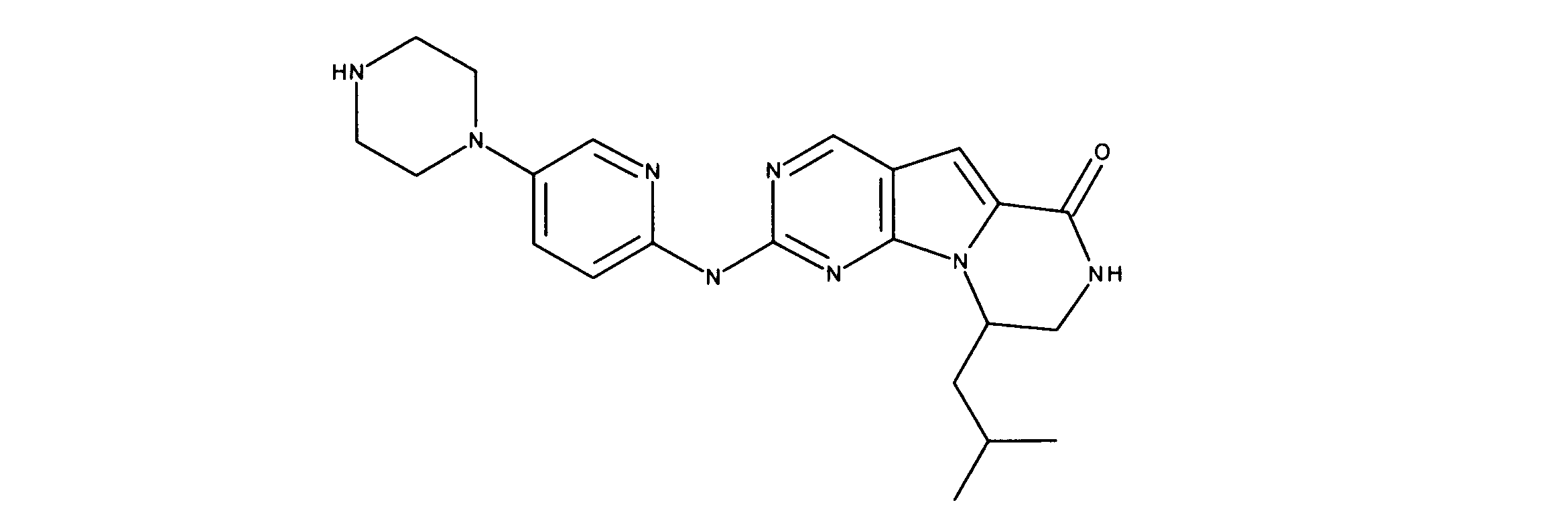
化合物109の合成
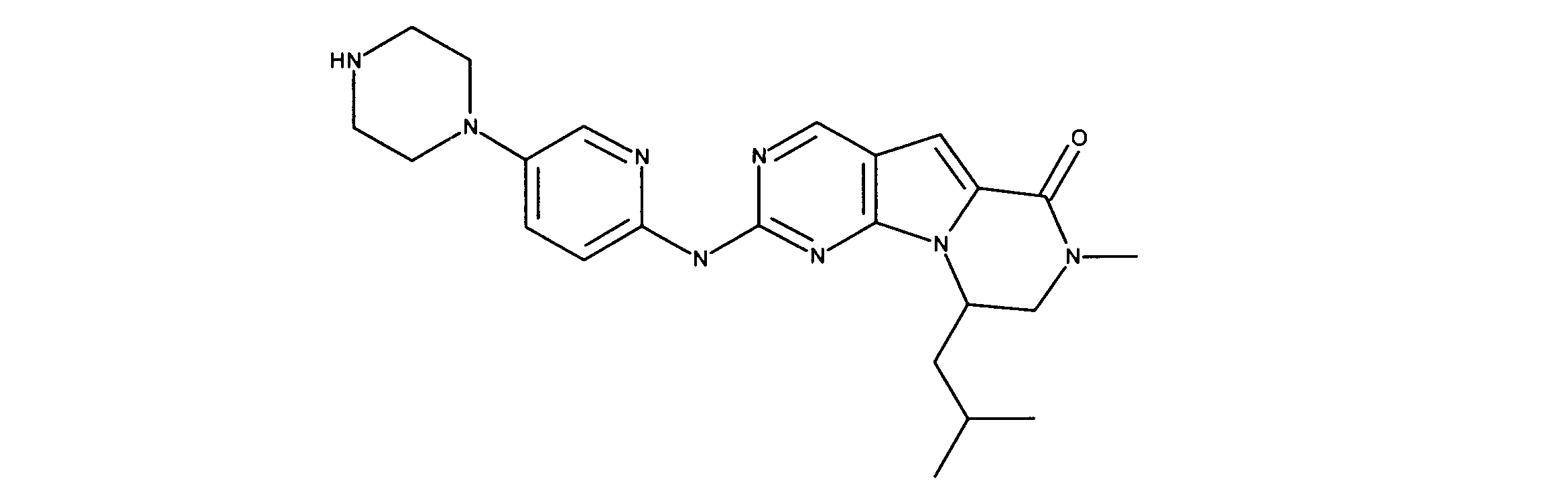
化合物110の合成
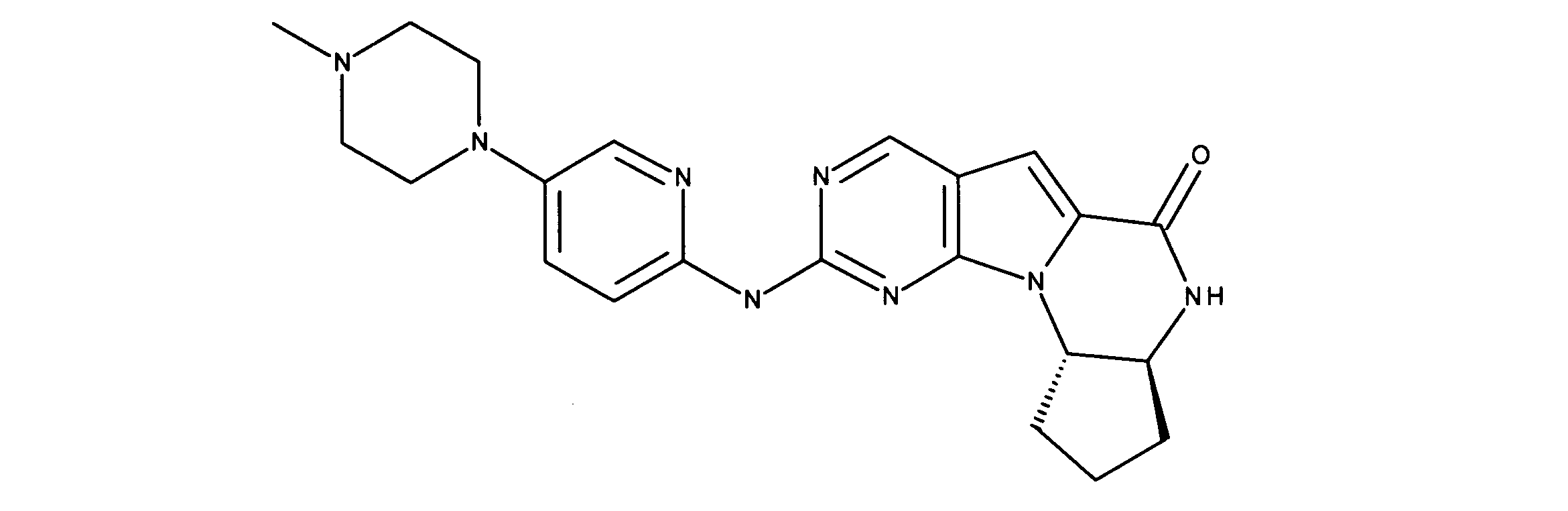
1HNMR (600 MHz, DMSO-d6) δ ppm 1.50 - 1.65 (m, 1 H) 1.92 - 2.02 (m, 3 H) 2.06 - 2.15 (m, 1 H) 2.78 (d, J=3.81 Hz, 4 H) 3.10 - 3.20 (m, 4 H) 3.47 - 3.51 (m, 2 H) 3.64 - 3.71 (m, 1 H) 3.76 - 3.83 (m, 2 H) 3.98 - 4.14 (m, 1 H) 7.20 (s, 2 H) 7.77 (s, 1 H) 7.97 (s, 2 H) 8.81 (s, 1 H) 9.03 (s, 1 H) 10.97 (br s., 1H). LCMS (ESI) 419 (M + H).
化合物111の合成
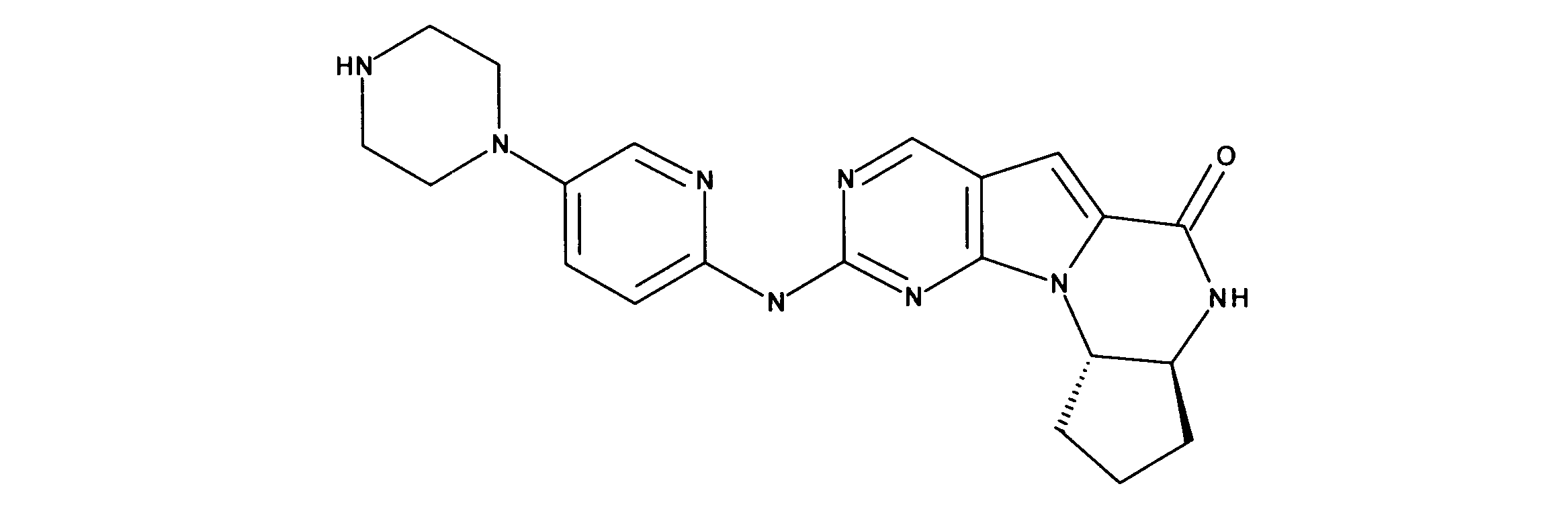
1HNMR (600 MHz, DMSO-d6) δ ppm 1.54 - 1.59 (m, 1 H) 1.92 - 2.01 (m, 3 H) 2.06 - 2.15 (m, 1 H) 2.76 - 2.84 (m, 1 H) 3.17 - 3.24 (m, 6 H) 3.64 - 3.71 (m, 2 H) 4.02 - 4.11 (m, 2 H) 7.22 (s, 2 H) 7.64 (s, 1 H) 7.97 (s, 2 H) 8.75 (s, 1 H) 8.97 (s, 1 H) 9.21 (s, 1 H). LCMS (ESI) 405 (M + H).
化合物112の合成

tert-ブチルN-[2-[(5-ブロモ2クロロピリミジン-4イル)アミノ]エチル]カルバメートの合成、化合物113
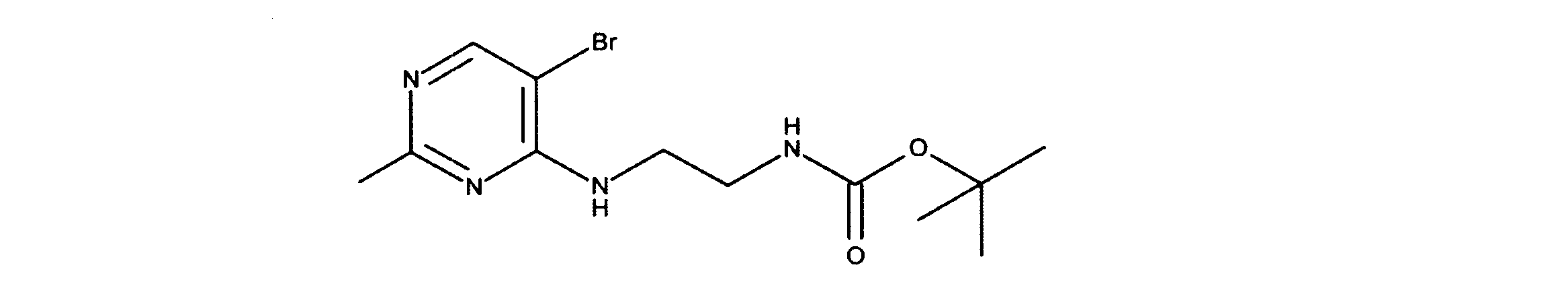
LCMS (ESI) 351 (M + H).
tert-ブチルN-[2-[[2-クロロ-5-(3、3-ジエトキシプロプ-1-イニル)ピリミジン-4イル]アミノ]エチル]カルバメートの合成、化合物114
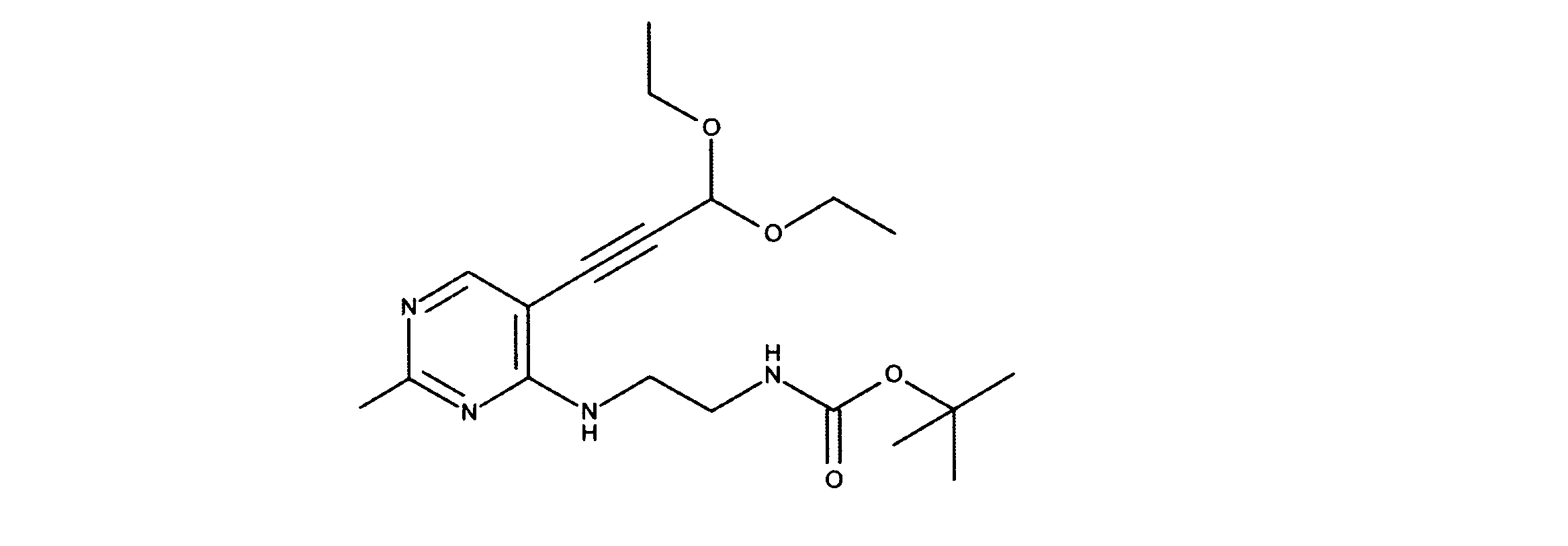
内容物を、70度で24時間加熱した。CELITE(登録商標)を通して濾過した後、ヘキサン/酢酸エチル(0~20%)を用いて、粗反応物をカラムに充填し、所望の製品(3.9g)を提供した。ヘキサン/酢酸エチル(0~30%)を用いて、得られた残留物のカラムクロマトグラフィーを行い、tert-ブチルN-[2-[[2-クロロ-5-(3、3-ジエトキシプロプ-1-イニル)ピリミジン-4-イル]アミノ]エチル]カルバメートを提供した。
LCMS (ESI) 399 (M + H).
tert-ブチルN-[2-[2-クロロ-6-(ジエトキシメチル)ピロロ[2,3-d]ピリミジン-7-イル]エチル]カルバメートの合成、化合物115
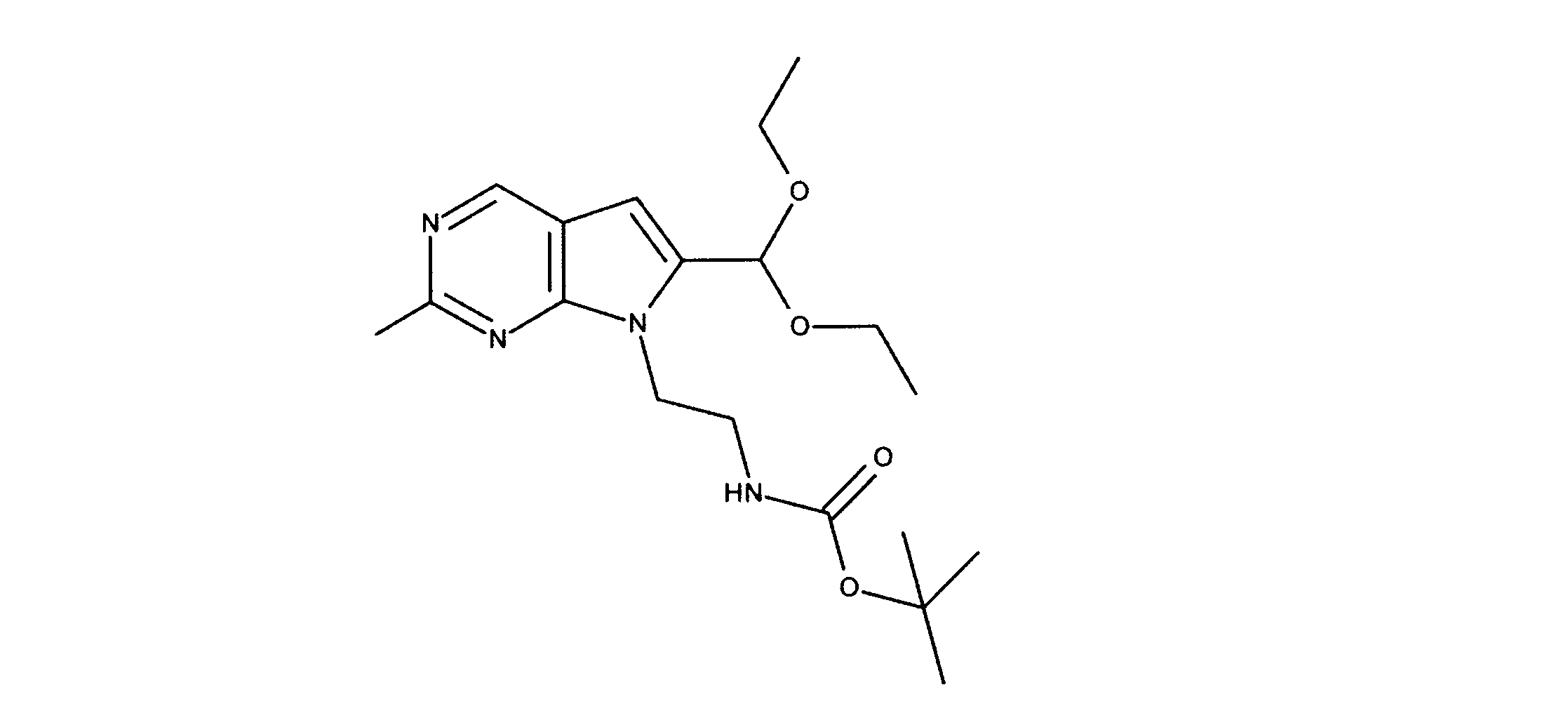
1HNMR (d6-DMSO) δ ppm 8.88 (s, 1H), 6.95 (brs, 1H), 6.69 (s, 1H), 5.79 (s, 1H), 4.29 (m, 2H), 3.59 (m, 4H), 3.34 (m, 1H), 3.18 (m, 1H), 1.19 (m, 9H), 1.17 (m, 6H). LCMS (ESI) 399 (M+H).
tert-ブチルN-[2-[2-クロロ6-(ジエトキシメチル)-5ヨードピロロ[2,3-d]ピリミジン-7-イル]エチル]カルバメートの合成、化合物116
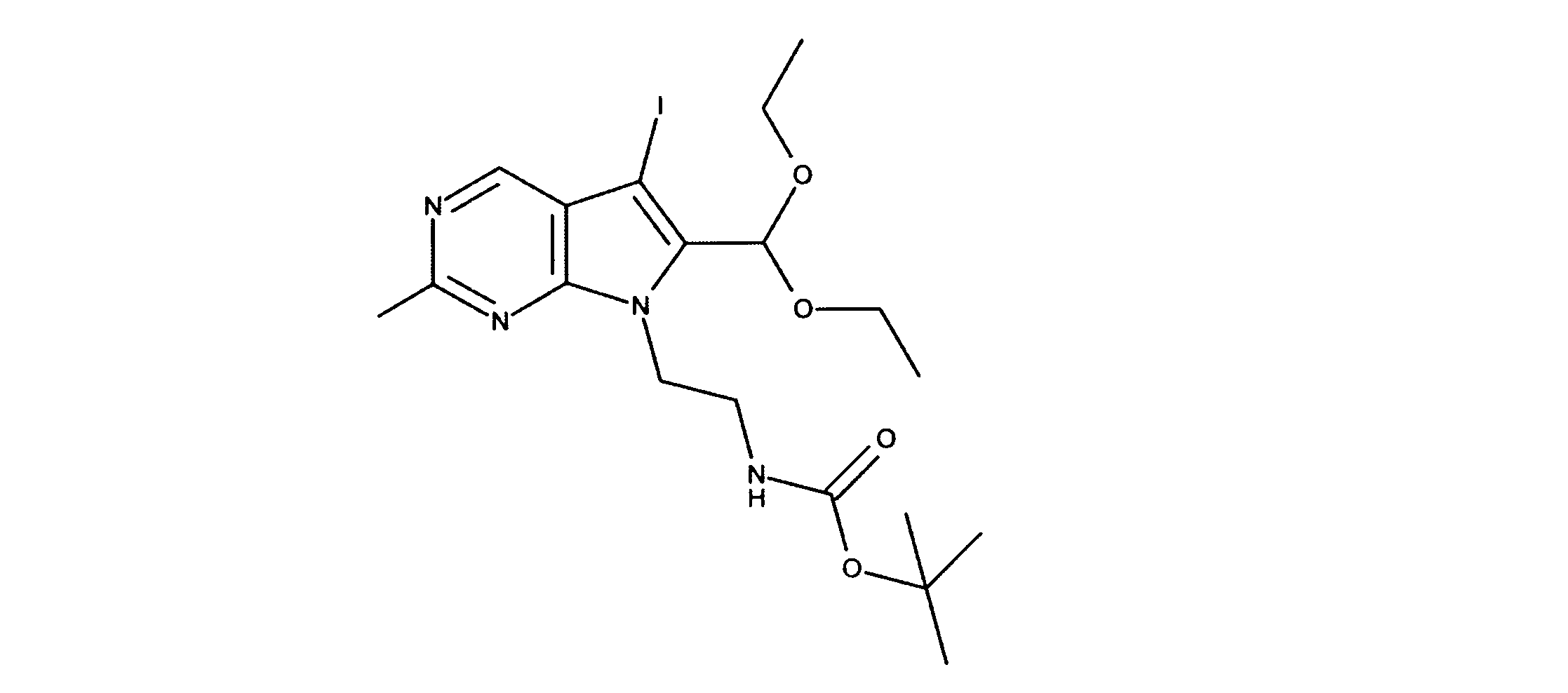
LCMS (ESI) 525 (M + H).
tert-ブチルN-[2-[2-クロロ-6-(ジエトキシメチル)-5-(o-トリル)ピロロ[2,3-d]ピリミジン-7-イル]エチル]カルバメートの合成、化合物117
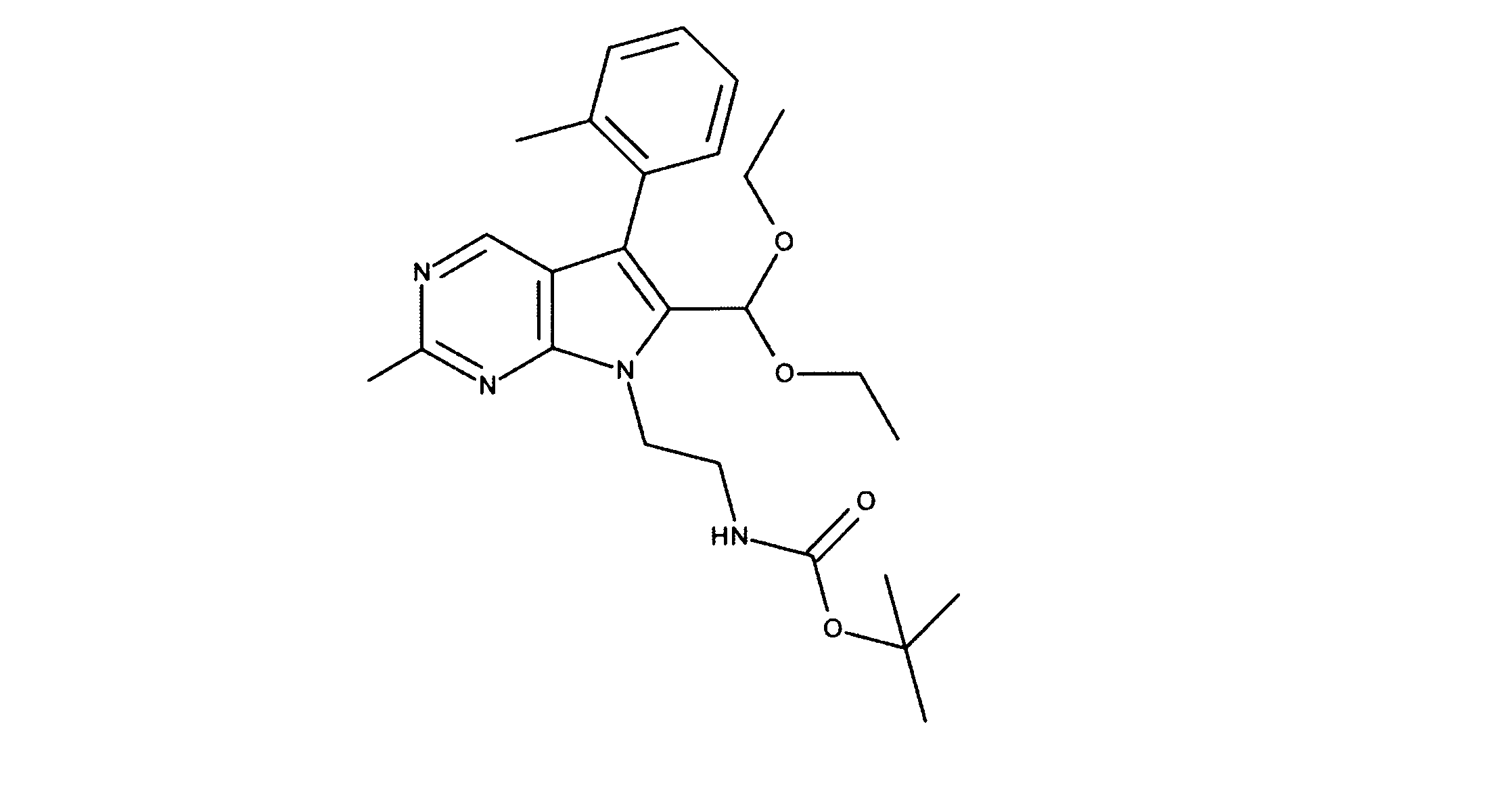
LCMS (ESI) 489 (M + H).
7-[2-(tert-ブトキシカルボニルアミノ)エチル]-2-クロロ-5-(o-トリル)ピロロ[2,3-d]ピリミジン-6カルボン酸の合成、化合物118
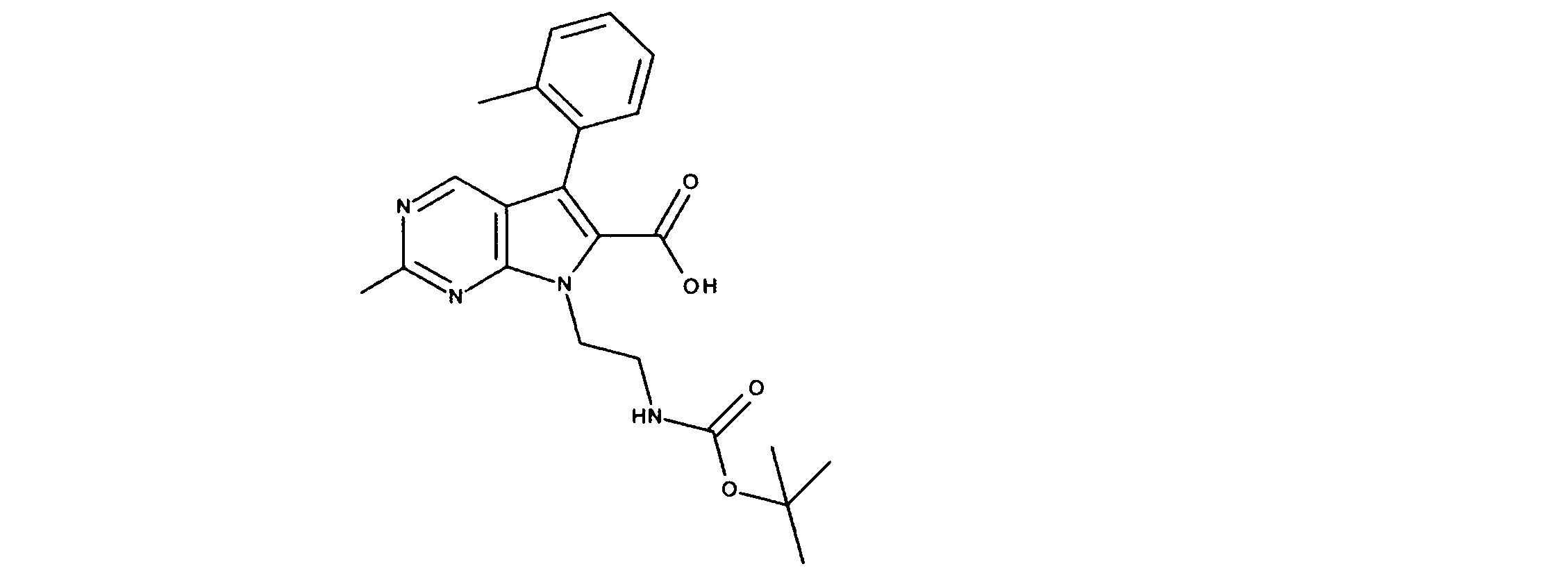
LCMS (ESI) 431 (M + H).
化合物119の合成
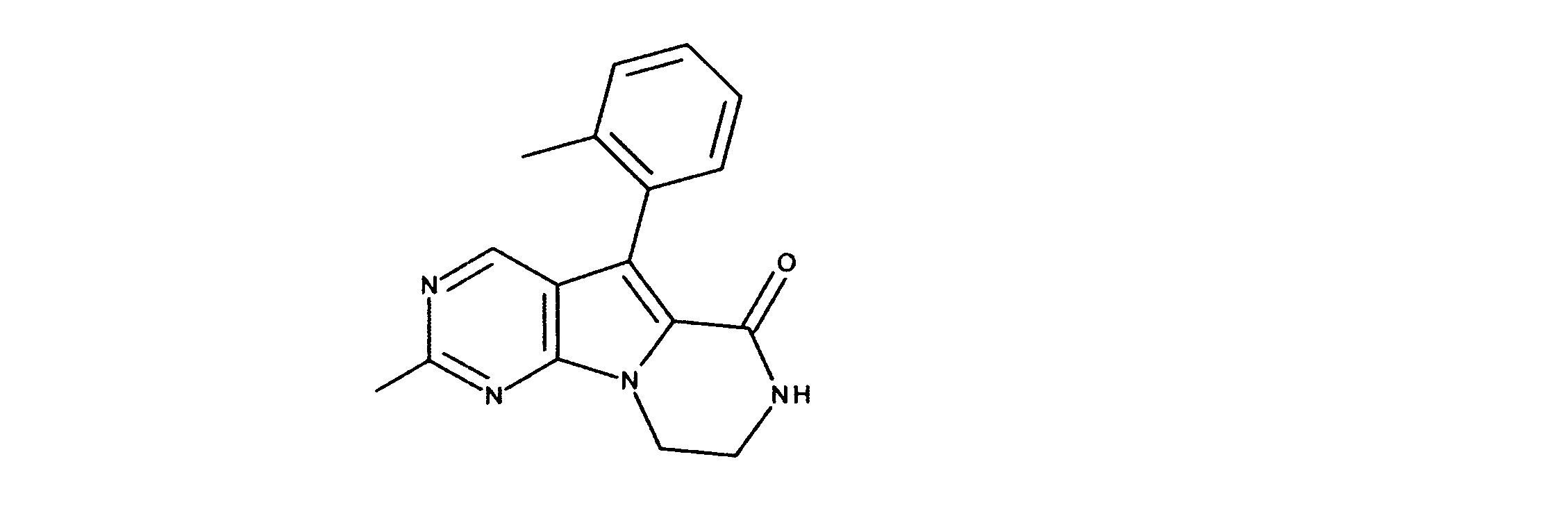
LCMS (ESI) 313 (M + H).
化合物120の合成
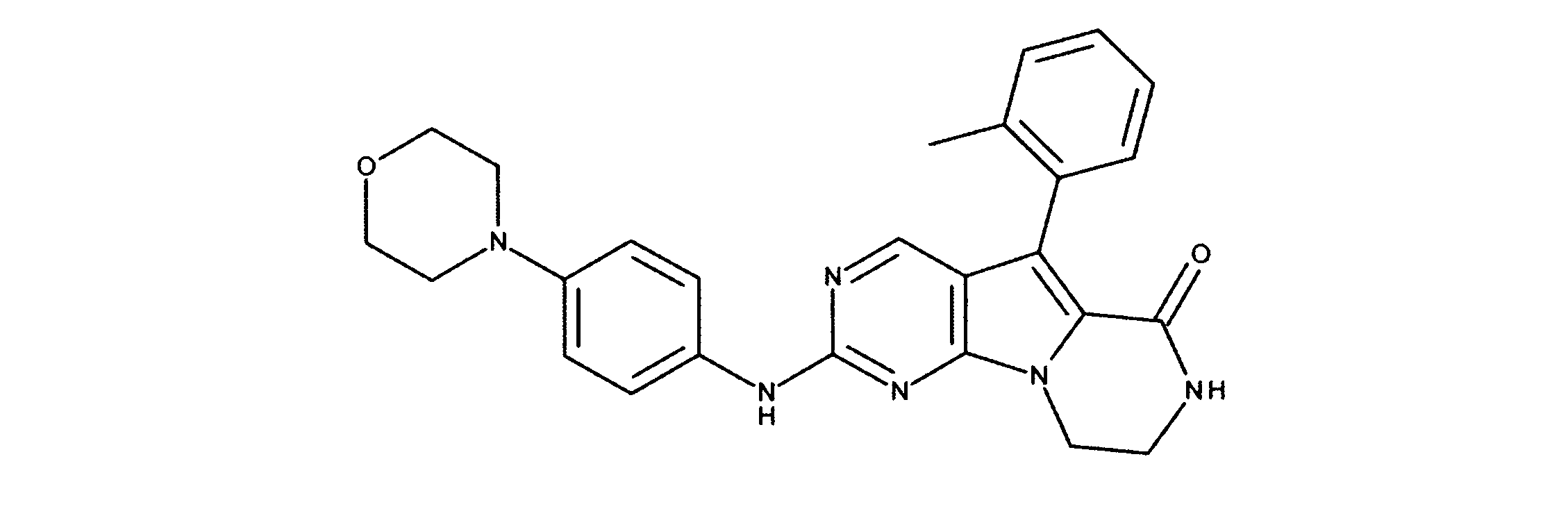
LCMS (ESI) 455 (M + H). 1HNMR (600 MHz, DMSO-d6) δ ppm 2.14 (s, 3 H) 3.23 - 3.50 (m, 2 H) 3.57 - 3.73 (m, 2 H), 3.81 - 3.92 (m, 8H), 7.11 - 7.31 (m, 4 H) 7.31 - 7.48 (m, 1 H) 7.58 - 7.73 (m, 1 H) 7.77 - 7.95 (m, 2 H) 8.05 - 8.21 (m, 1 H) 8.44 (s, 1 H) 9.85 - 10.01 (m, 1 H).
化合物121の合成
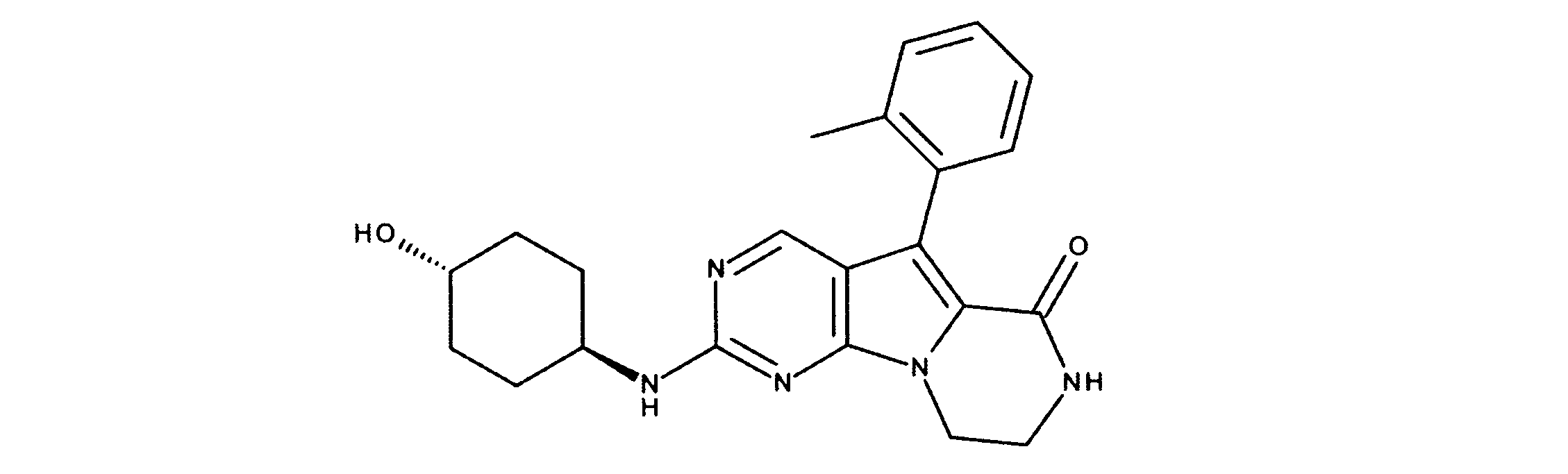
LCMS (ESI) 392 (M + H). 1HNMR (600 MHz, DMSO-d6) δ ppm 1.23 (d, J=8.78 Hz, 4 H) 1.84 (br. s., 4 H) 2.11 (s, 3 H) 3.34 - 3.43 (m, 1 H) 3.55 (br. s., 2 H) 3.72 (br. s., 1 H) 4.13 (br. s., 2 H) 4.50 (br. s., 1 H) 7.03 (br. s., 1 H) 7.12 - 7.28 (m, 4 H) 7.96 (br. s., 1 H) 8.18 (br. s., 1 H).
7-[2-(tert-ブトキシカルボニルアミノ)エチル]-2-クロロ-ピロロ[2,3-d]ピリミジン-6カルボン酸の合成、化合物122
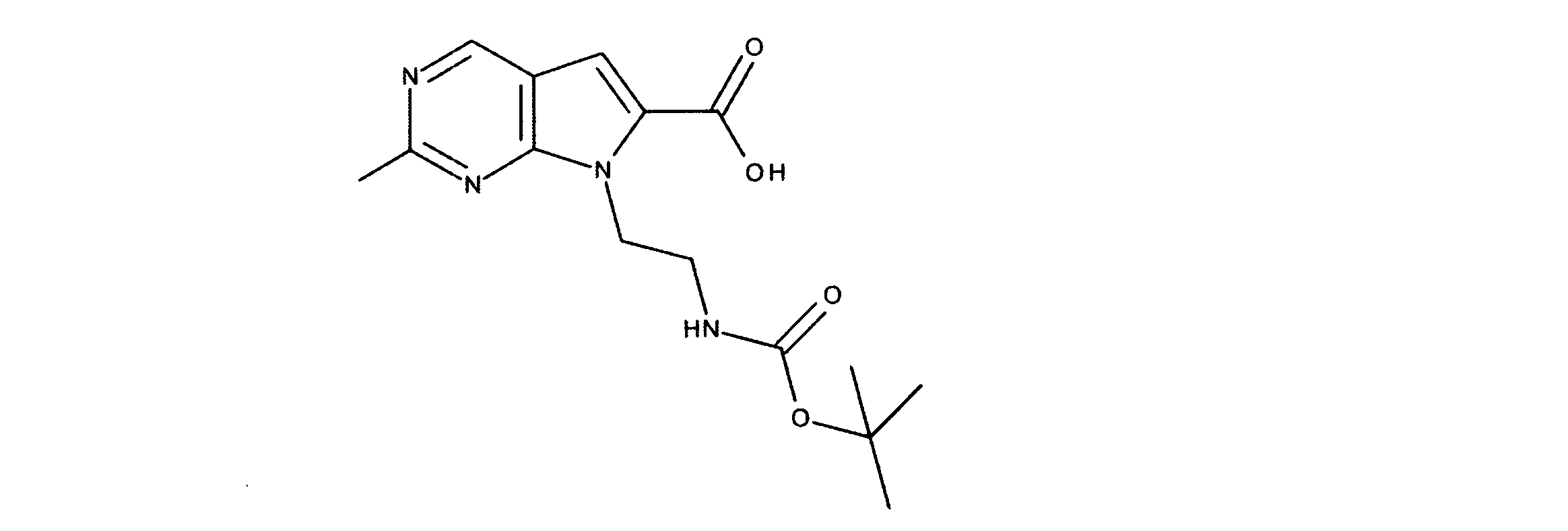
LCMS (ESI) 341 (M + H).
化合物123の合成
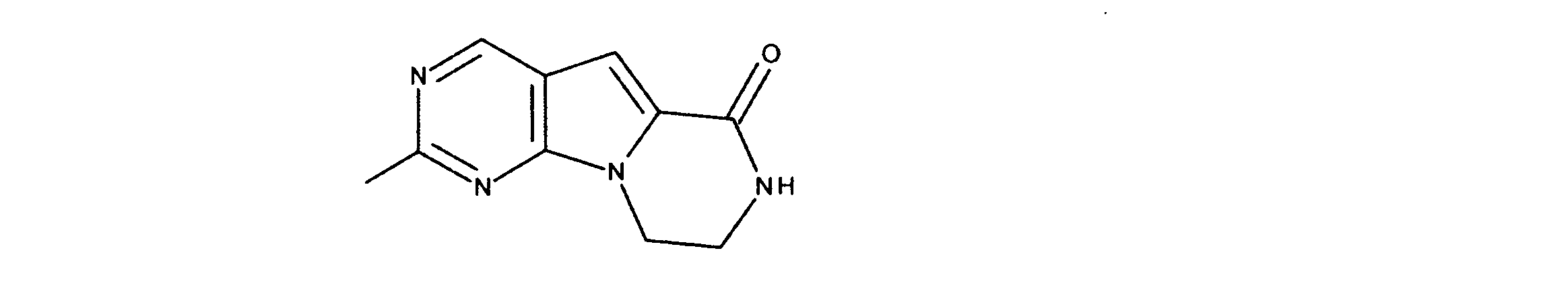
LCMS (ESI) 223 (M + H).
化合物124の合成
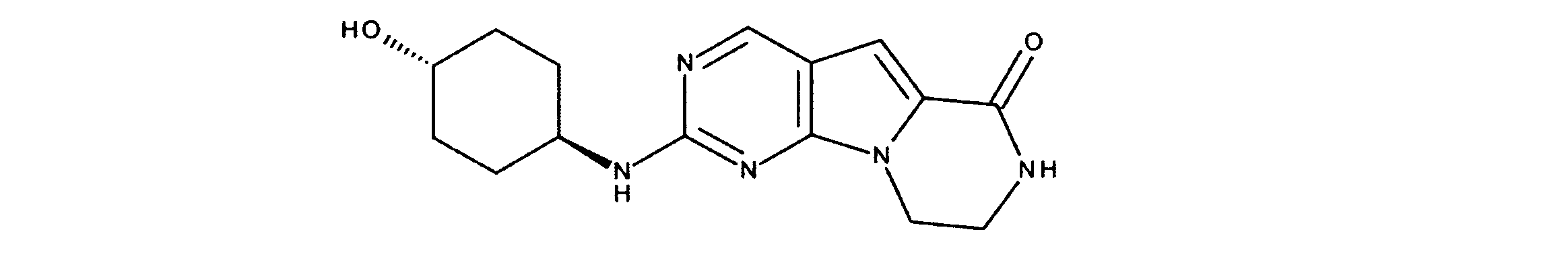
LCMS (ESI) 302 (M + H).
tert-ブチルN-[2-[(5-ブロモ-2-クロロ-ピリミジン-4-イル)アミノ]-2メチル-プロピル]カルバメートの合成、化合物125

LCMS (ESI) (M+H) 379.
tert-ブチルN-[2-[[2-クロロ-5-(3、3-ジエトキシプロプ-1-イニル)ピリミジン-4-イル]アミノ]-2メチル-プロピル]カルバメートの合成、化合物126
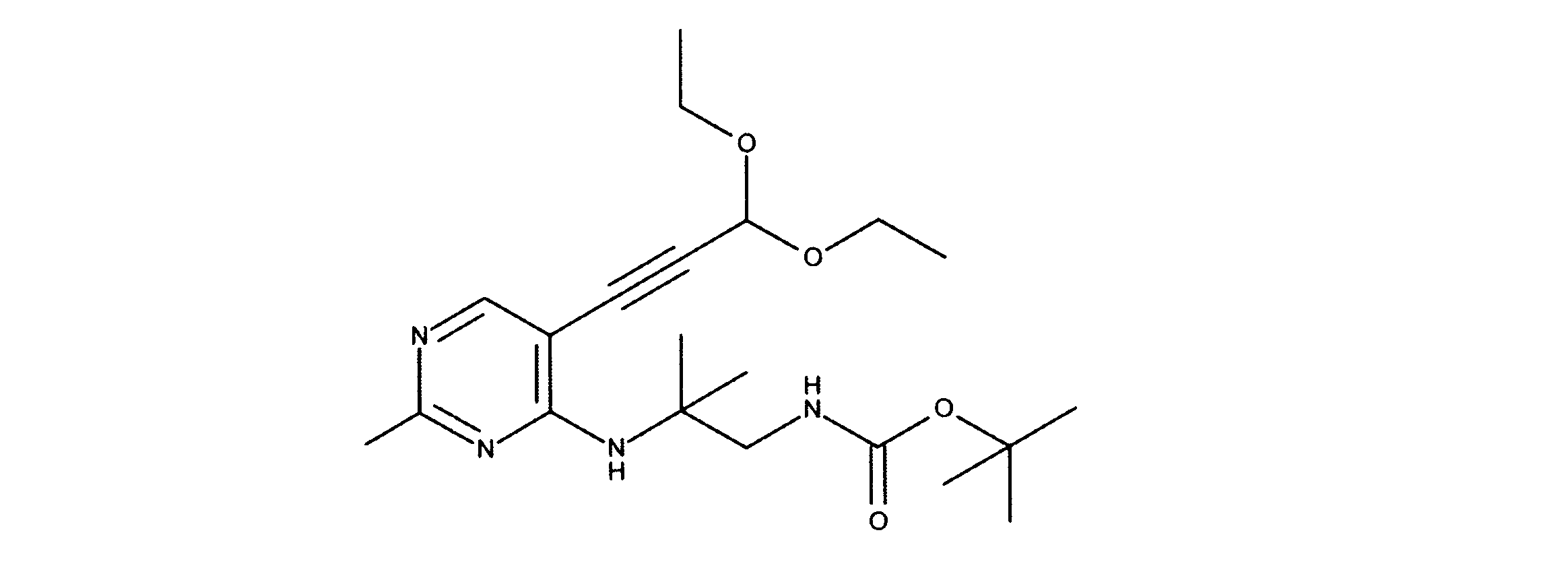
LCMS (ESI) (M+H) 427.
tert-ブチルN-[2-[2-クロロ-6-(ジエトキシメチル)ピロロ[2,3-d]ピリミジン-7-イル]-2メチル-プロピル]カルバメートの合成、化合物127
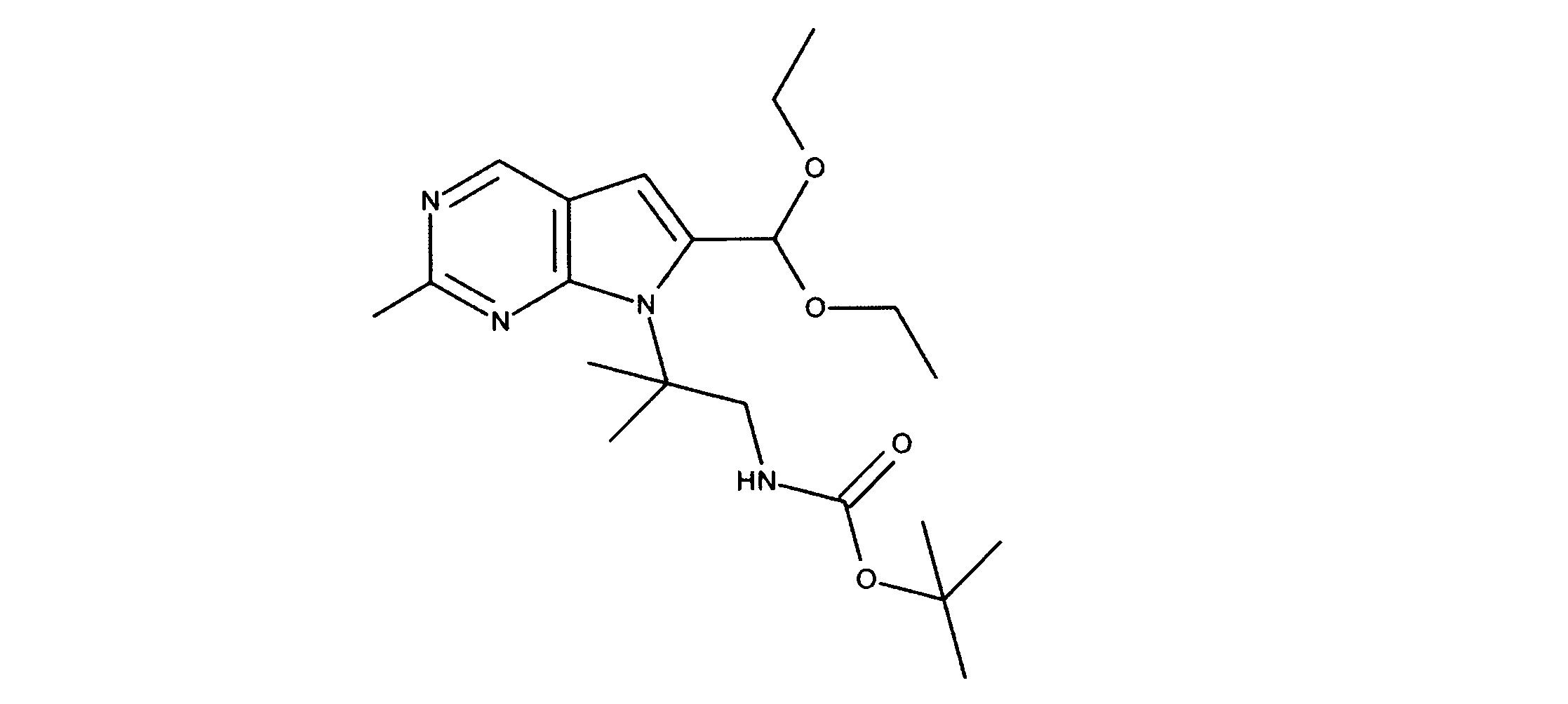
LCMS (ESI) (M+H) 427.
7-[2-(tert-ブトキシカルボニルアミノ)-1,1-ジメチルエチル]-2-クロロ-ピロロ[2,3-d]ピリミジン-6カルボン酸の合成、化合物128

LCMS (ESI) 369 (M + H).
化合物129の合成
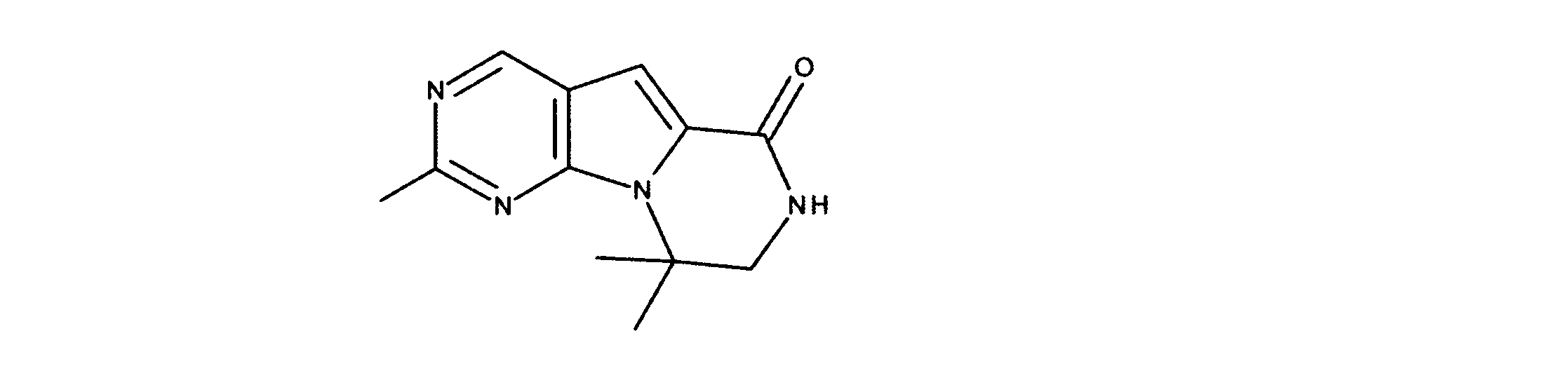
LCMS (ESI) 251 (M + H).
化合物130の合成
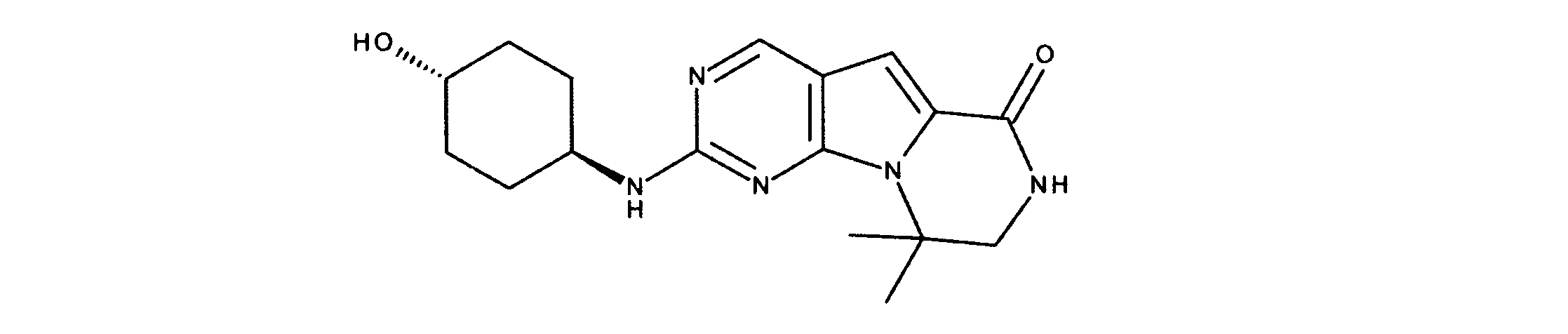
LCMS (ESI) 330 (M + H). 1HNMR (600 MHz, DMSO-d6) δ ppm 1.07 - 1.34 (m, 4 H) 1.47 - 2.05 (m, 10 H) 3.09 (m, 1H) 3.51 (d, J = 2.91 Hz, 2 H) 3.57 (m, 1H) 4.50 (br. s., 1 H) 6.89 (s, 1 H) 6.94 - 7.05 (m, 1 H) 8.04 (br. s., 1 H) 8.60 (s, 1 H) 9.00 (br. s., 1 H).
ベンジルN-[1-[[(5-ブロモ-2-クロロ-ピリミジン-4-イル)アミノ]メチル]プロピル]カルバメートの合成、化合物131
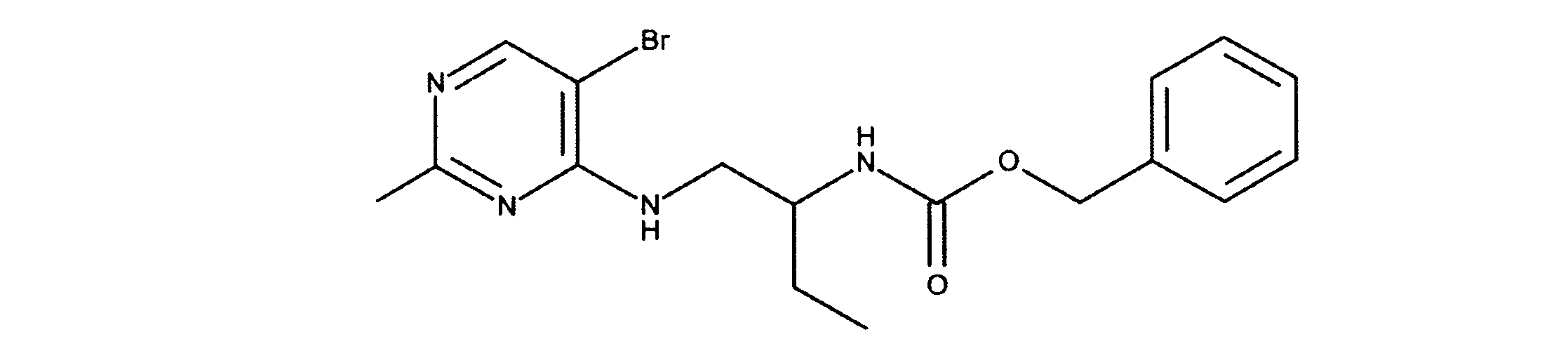
LCMS (ESI) (M+H) 413.
ベンジルN-[1-[[[2-クロロ-5-(3、3-ジエトキシプロプ-1-イニル)ピリミジン-4-イル]アミノ]メチル]プロピル]カルバメートの合成、化合物132

LCMS (ESI) (M+H) 461.
カルバミン酸ベンジルN-[1-[[2-クロロ-6-(ジエトキシメチル)ピロロ[2,3-d]ピリミジン-7-イル]メチル]プロピル]の合成、化合物133
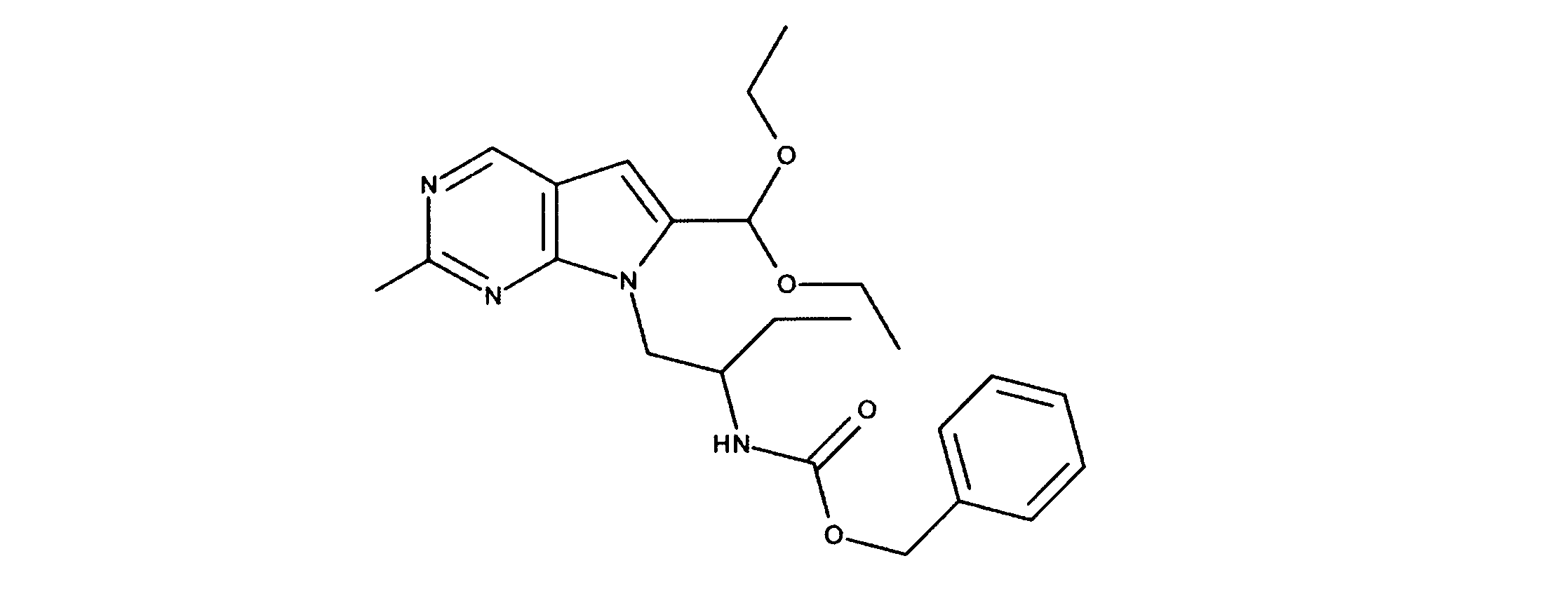
LCMS (ESI) (M+H) 461.
7-[2-(ベンジルオキシカルボニルアミノ)ブチル]-2-クロロ-ピロロ[2,3-d]ピリミジン-6カルボン酸の合成、化合物134
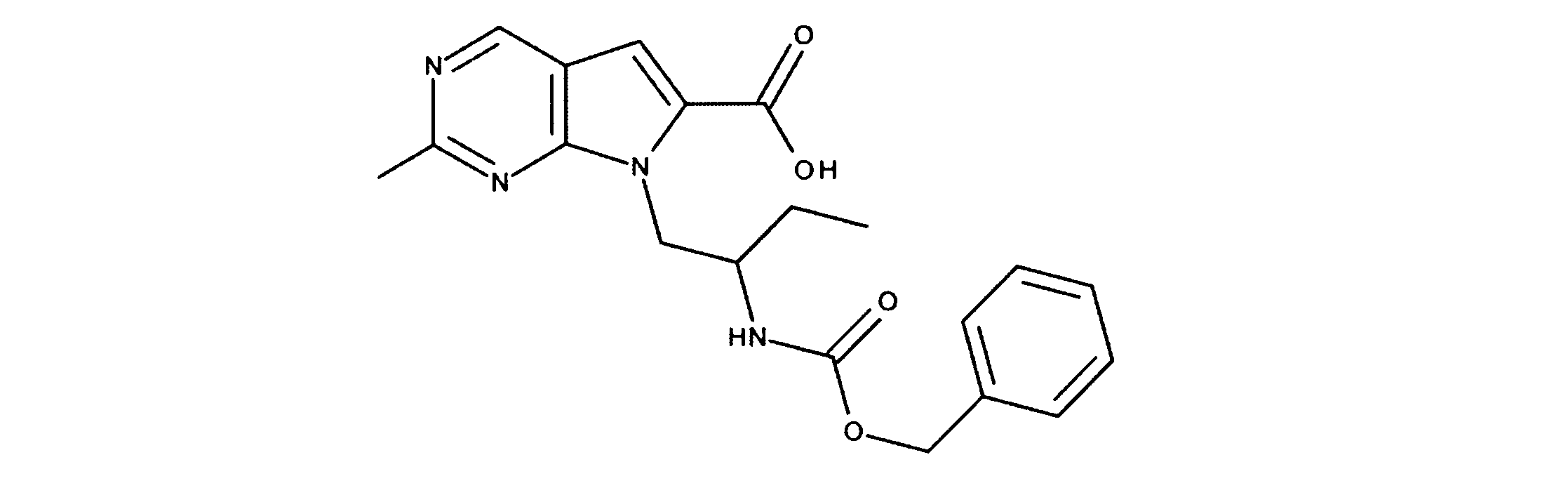
LCMS (ESI) 403 (M + H).
化合物135の合成
LCMS (ESI) 251 (M + H).
化合物136の合成
LCMS (ESI) 330 (M + H). 1HNMR (600 MHz, DMSO-d6) δ ppm 0.80 - 0.95 (m, 3 H) 1.35 - 1.92 (m, 10 H) 3.66 (br. m., 3 H) 4.17 (br. s., 2 H) 4.47 (br. s., 1 H) 6.85 (s, 1 H) 6.96 (br. s., 1 H) 8.15 (br. s., 1 H) 8.62 (br. s., 1 H).
tert-ブチルN-[1-[[(5-ブロモ-2-クロロ-ピリミジン-4-イル)アミノ]メチル]シクロペンチル]カルバメートの合成、化合物137

LCMS (ESI) 405 (M+H).
tert-ブチルN-[1-[[[2-クロロ-5-(3、3-ジエトキシプロプ-1-イニル)ピリミジン-4-イル]アミノ]メチル]シクロペンチル]カルバメートの合成、化合物138

LCMS (ESI) 453 (M+H).
tert-ブチルN-[1-[[2-クロロ-6-(ジエトキシメチル)ピロロ[2,3-d]ピリミジン-7-イル]メチル]シクロペンチル]カルバメートの合成、化合物139
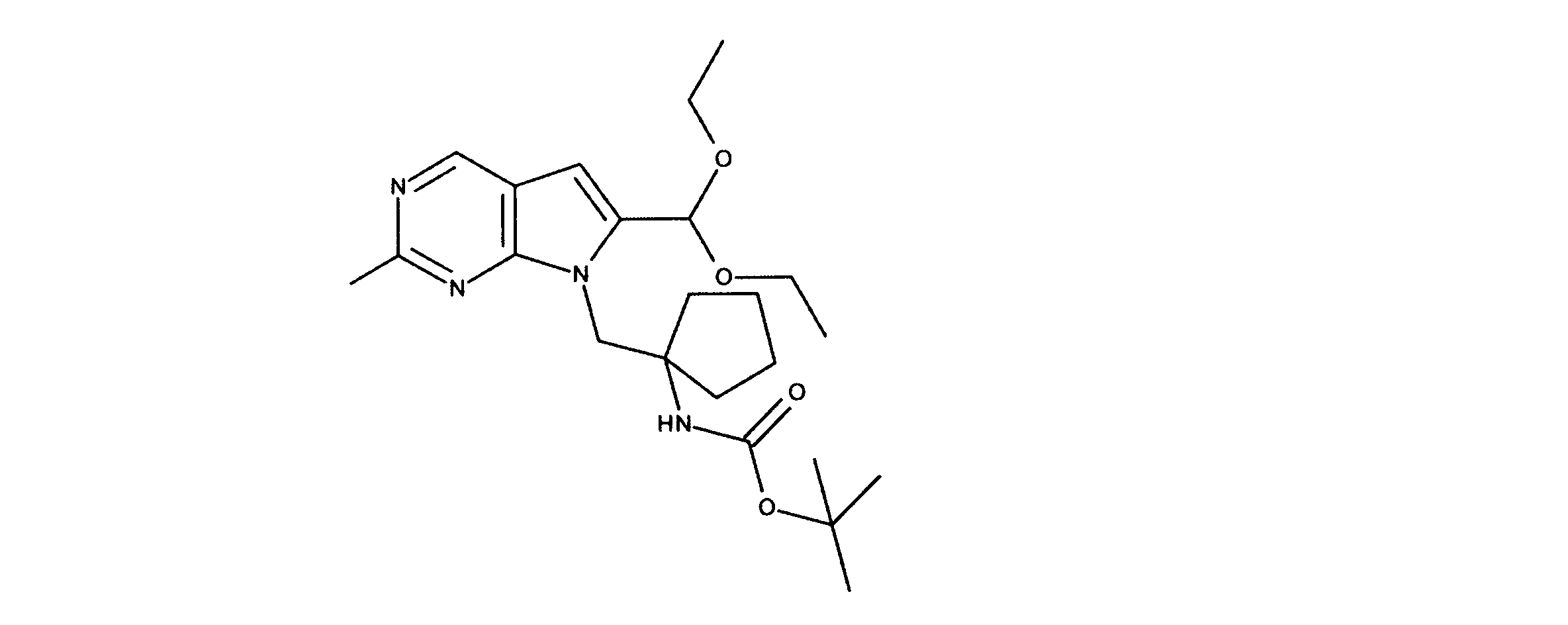
LCMS (ESI) 453 (M+H).
7-[[1-(tert-ブトキシカルボニルアミノ)シクロペンチル]メチル]-2-クロロ-ピロロ[2,3-d]ピリミジン-6カルボン酸の合成、化合物140
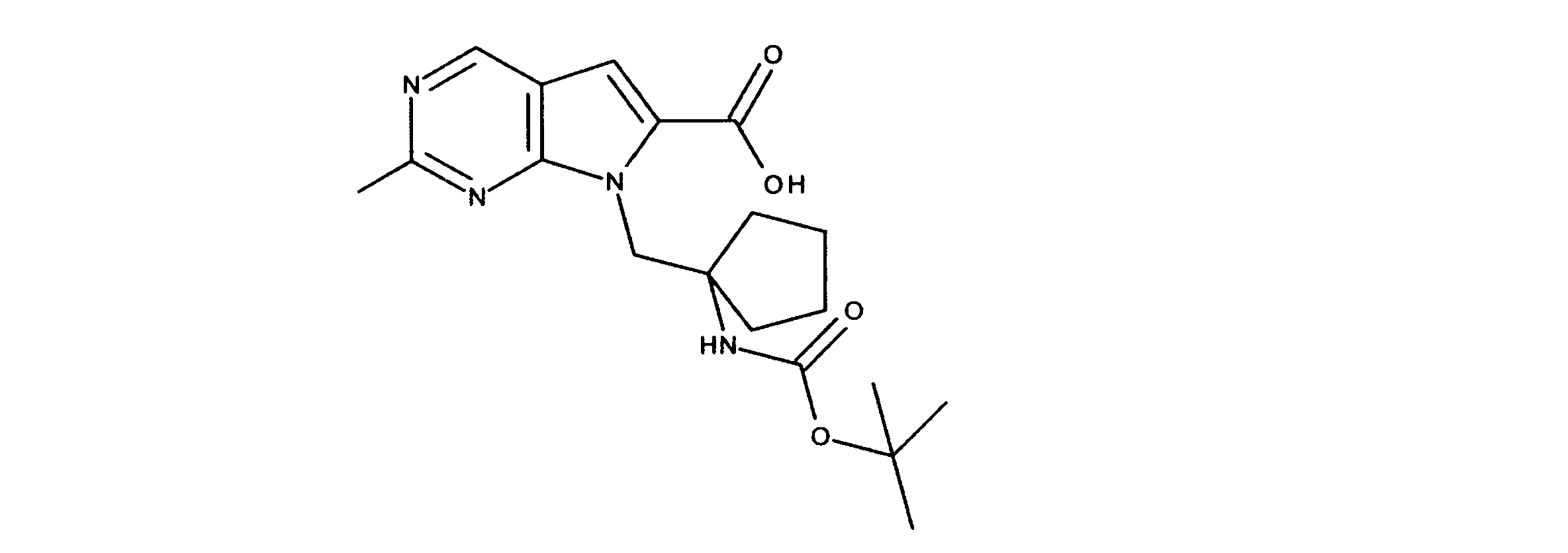
LCMS (ESI) 395 (M + H).
化合物141の合成
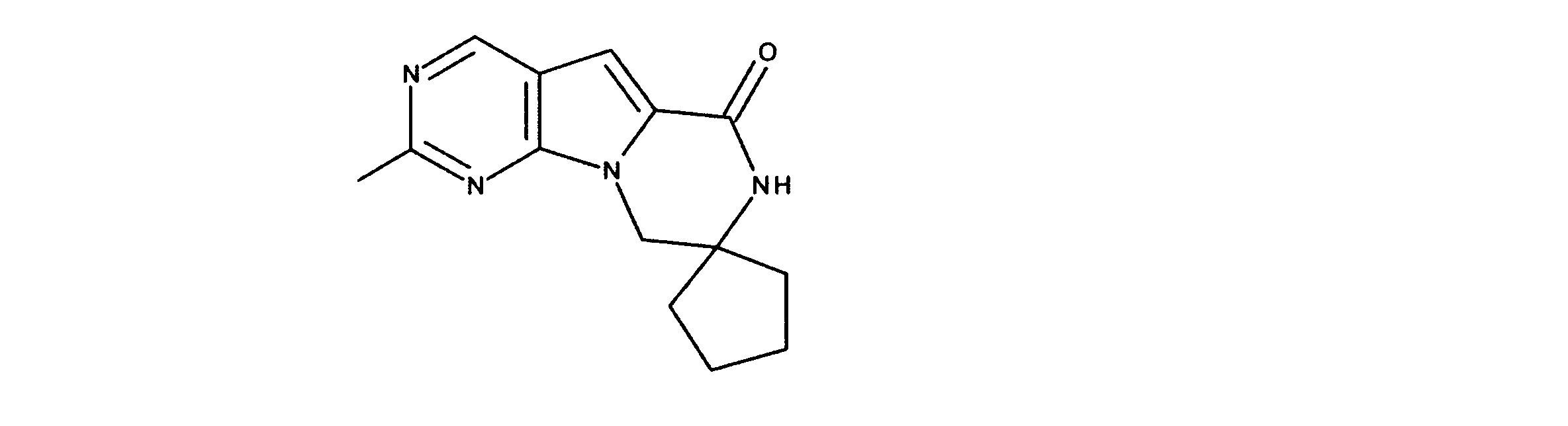
LCMS (ESI) 277 (M + H).
化合物142の合成
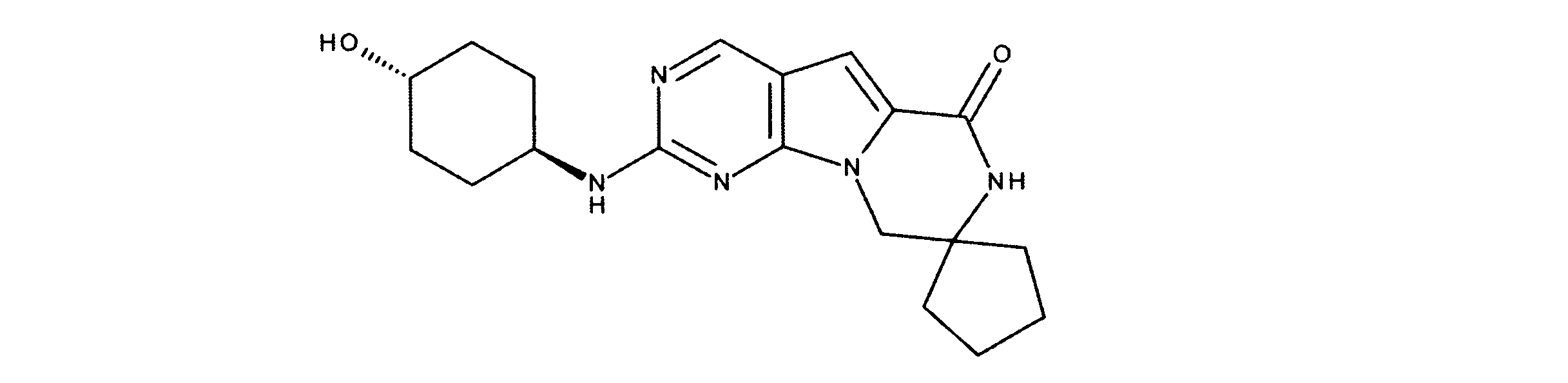
LCMS (ESI) 356 (M + H). 1HNMR (600 MHz, DMSO-d6) δ ppm 1.08 - 1.32 (m, 8 H) 1.60 - 2.09 (m, 8 H) 3.03 - 3.17 (m, 1 H) 3.35 (s, 2 H) 3.54 - 3.62 (m, 1 H) 4.51 (d, J=4.39 Hz, 1 H) 6.88 (s, 1 H) 6.96 (br. s., 1 H) 8.07 (br. s., 1 H) 8.58 (s, 1 H).
tert-ブチルN-[[1-[(5-ブロモ-2-クロロ-ピリミジン-4-イル)アミノ]シクロペンチル]メチル]カルバメートの合成、化合物143
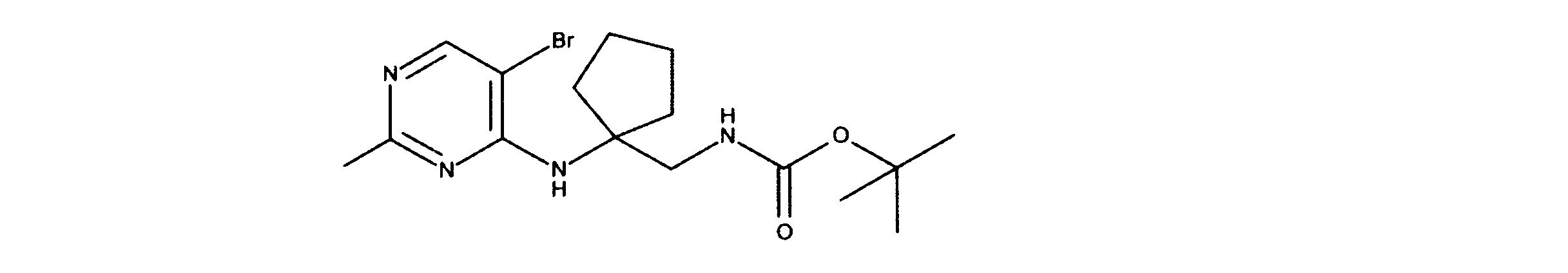
LCMS (ESI) 405 (M+H).
tert-ブチルN-[2-[[2-クロロ-5-(3、3-ジエトキシプロプ-1-イニル)ピリミジン-4-イル]アミノ]-2メチル-プロピル]カルバメートの合成、化合物144
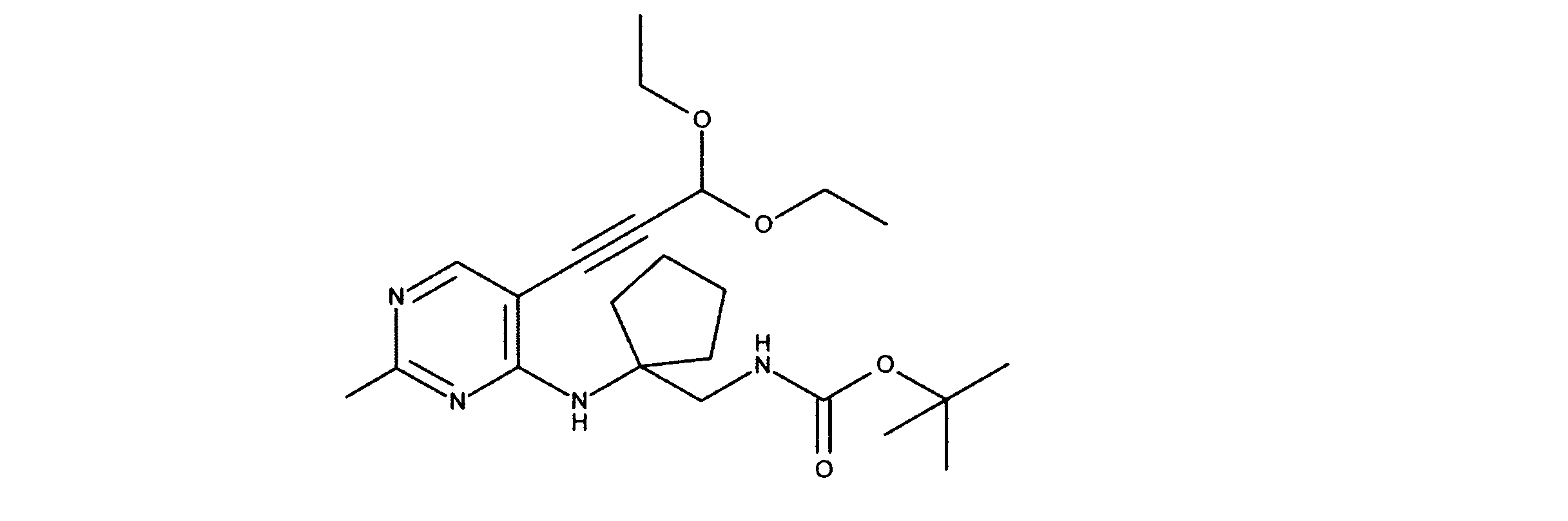
LCMS (ESI) 453 (M+H).
tert-ブチルN-[[1-[2-クロロ-6-(ジエトキシメチル)ピロロ[2,3-d]ピリミジン-7-イル]シクロペンチル]メチル]カルバメートの合成、化合物145
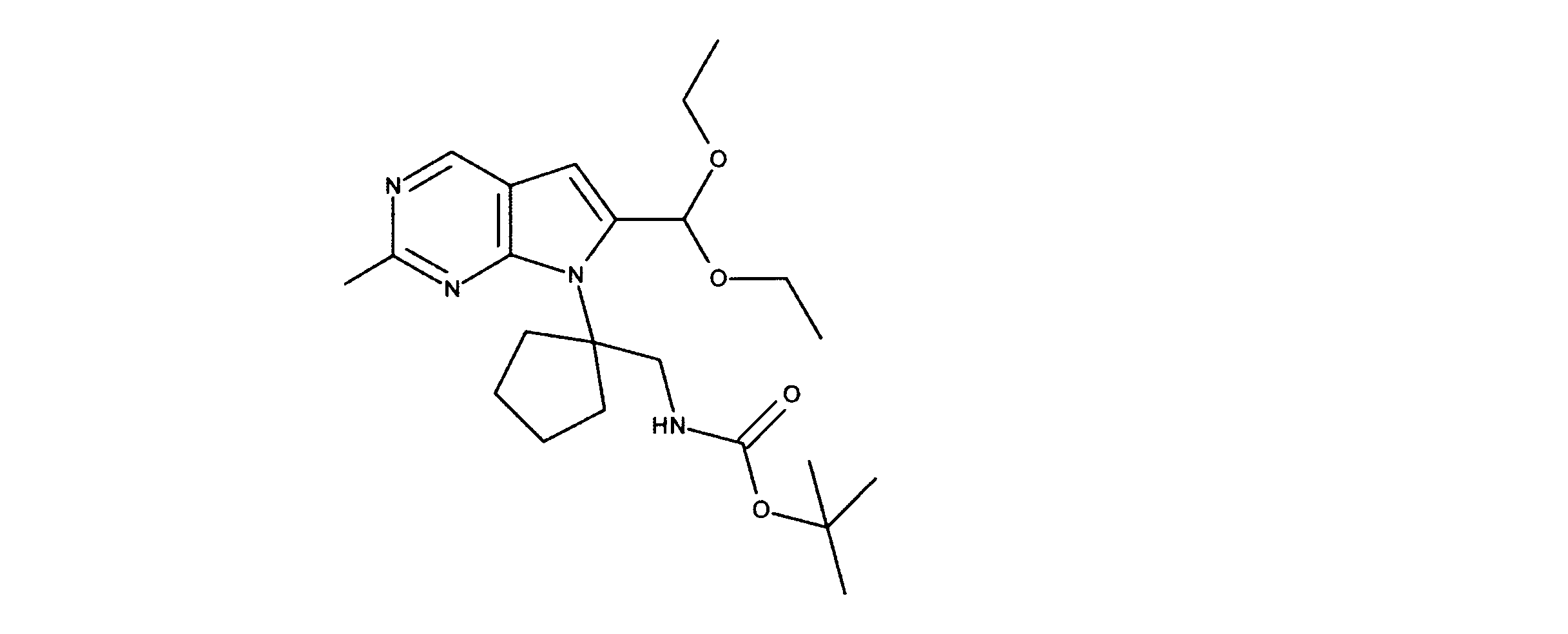
LCMS (ESI) 4534 (M+H).
7-[2-(tert-ブトキシカルボニルアミノ)-1,1-ジメチルエチル]-2-クロロ-ピロロ[2,3-d]ピリミジン-6カルボン酸の合成、化合物146
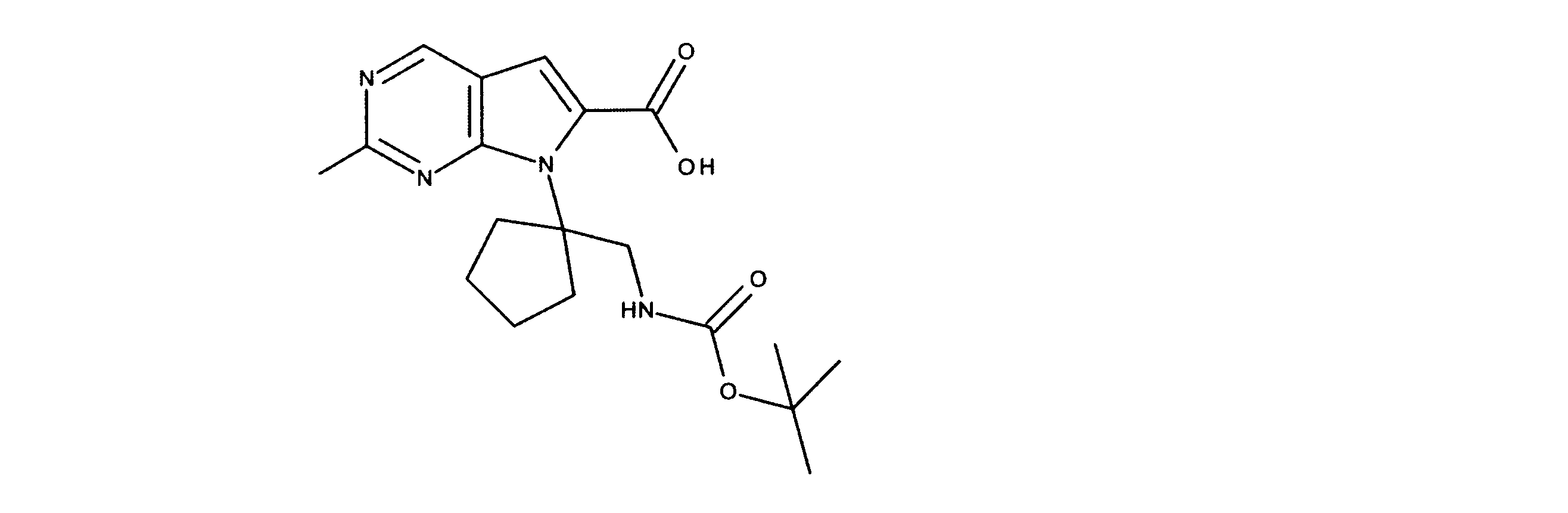
LCMS (ESI) 395 (M + H).
化合物147の合成

LCMS (ESI) 277 (M + H).
化合物148の合成

LCMS (ESI) 356 (M + H). 1HNMR (600 MHz, DMSO-d6) δ ppm 1.06 - 1.35 (m, 8 H) 1.45 - 1.95 (m, 8 H) 3.10 (m, 1 H) 3.58 (br. s., 2 H) 3.95 (br. s., 1 H) 4.49 (br. s., 1 H) 6.84 (s, 1 H) 6.85 - 6.93 (m, 1 H) 8.29 (s, 1 H) 8.61 (br. s., 1 H).
化合物149の合成

ステップ2:Boc保護された化合物59を、CO2(1気圧)の下、DMI中の5mol% NiCl2(Ph3)2、0.1eqトリフェニルホスフィン、3eq Mn、0.1eqヨウ化テトラエチルアンモニウムで、25℃で20時間処理して、ハロゲン化アリール誘導体をカルボン酸に変換する。
ステップ3:ステップ2からのカルボン酸を、標準的な条件を用いて、対応する酸塩化物に変換する。
ステップ4:ステップ3からの酸塩化物を、Nメチルピペラジンと反応させて、対応するアミドを生成する。
ステップ5:ステップ4からのアミドを、ジクロロメタン中のトリフルオロ酢酸を用いて脱保護して、ターゲット化合物を生成する。
化合物149を、シリカゲルカラムクロマトグラフィーにより、ジクロロメタン-メタノール勾配で溶出させて、精製して、化合物149を提供する。
化合物119~147のそれぞれ及び様々なR8、R1及びZの定義を含む対応する各化合物は、水素化ナトリウム及びハロゲン化アルキル又は他のハロゲン化物と反応してもよく、これにより、化合物120の合成に対して上記したように、アミンによる反応物への前に所望のR置換を挿入して、式I、II、III、IV又はVの所望の製品を生成させてもよい。
CDK4/6阻害インビトロアッセイ
ここに開示される化合物を選択して、Nanosyn(米国カリフォルニア州サンタクララ)によって、CDK4/cyclinD1、CDK2/CycA及びCDK2/cyclinEキナーゼアッセイの試験を行い、これらのCDKに対するそれらの抑制性効果を決定した。マイクロ流体キナーゼ検出技術(Caliper Assay Platform)を用いて、アッセイを実行した。singlicateの12点ドーズ反応フォーマットで、ATPに対してKmで化合物の試験を行った。全てのアッセイで用いたホスホアクセプタ基体ペプチド濃度は、1μMであり、全てのアッセイに対する参照化合物として、スタウロスポリンを用いた。
CDK2/CyclinA:酵素濃度:0.2nM;ATP濃度:50μM;インキュベーション時間:3時間
CDK2/CyclinE:酵素濃度:0.28nM;ATP濃度:100μM;インキュベーション時間:1時間
CDK4/CyclinD1:酵素濃度:1nM;ATP濃度:200μM;インキュベーション時間:10時間
CDK4/CycD1、CDK2/CycE、CDK2/CycAに関して表1の化合物に対する抑制性IC50値、ならびに、倍選択性が、表2に示される。

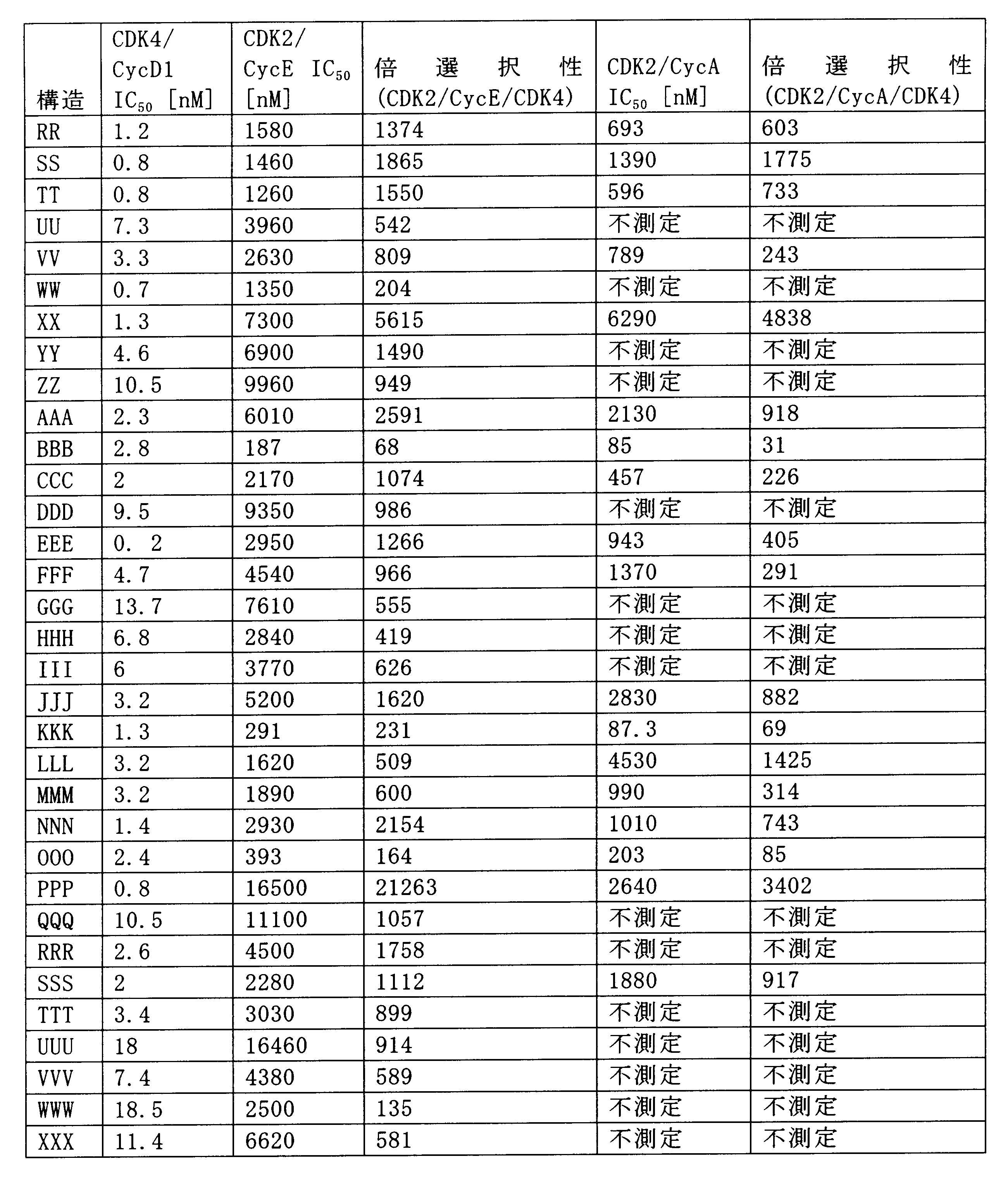
CDK4キナーゼ活性に加えて、いくつかの化合物は、CDK6キナーゼ活性に対しても試験された。CDK6/CycD3キナーゼアッセイの結果を、CDK4/cyclinD1、CDK2/CycA及びCDK2/cyclinEキナーゼアッセイとともに、PD0332991(参照)及び化合物T、Q、GG及びUに対して、表3に示した。CDK4/cyclinD1に対する10nM及びCDK12/cyclinEに対する10uMののIC50は、PD0332991に対してあらかじめ発表されたレポートと、良く合致する(Fry et al. Molecular Cancer Therapeutics (2004) 3(11)1427-1437、 Toogood et al. Journal of Medicinal Chemistry (2005) 48, 2388-2406)。化合物T、Q、GG及びUは、参照化合物(PD0332991)に関して、より有能(IC50が低い)であり、そして、参照化合物に対して、より高い倍選択性(CDK2/CycEのIC50を、CDK4/CycD1のIC50で除す)を示す。
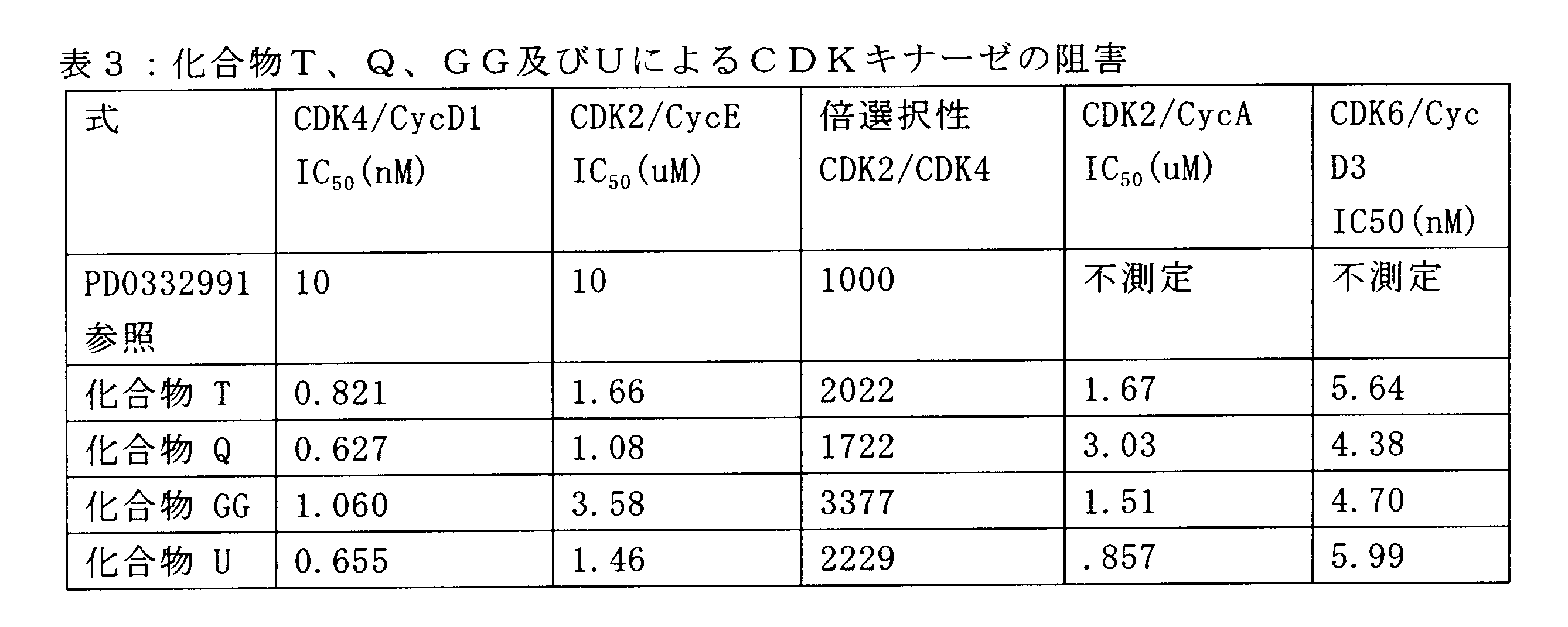
G1期停止(細胞G1及びS期)アッセイ
様々な処理の後の細胞サイクルの様々な段階における細胞画分の決定のために、HS68細胞(ヒト皮膚繊維芽細胞系統(Rbポジティブな))を、プロピジウムヨウ化物染色液で染色し、Dako Cyan Flow Cytometerで実行した。G0-G1DNA細胞サイクルの細胞の画分対S期DNA細胞サイクルの画分を、FlowJo7.2.2分析により測定した。
表1にリストされる化合物を、細胞サイクルのG1期でHS68細胞を停止する能力に関して、試験した。細胞G1期停止アッセイの結果から、HS68細胞のG1期停止のために必要な抑制性EC50値の範囲は、22nM~1500nMであった(表4の「細胞G1期停止EC50」というタイトルの欄を参照)。
細胞増殖の阻害
細胞増殖アッセイは、以下の癌細胞系統を用いて実行された:
MCF7(胸腺癌 ― Rbポジティブ)、ZR-75-1(胸腺管癌 ― Rbポジティブ)、H69(ヒト小細胞肺癌 ― Rb-ネガティブ)細胞、又はA2058(ヒト転移メラノーマ細胞 ― Rb-ネガティブ)。
これらの細胞を、Coster社(米国マサチューセッツ州テュークスバリー)3093号、96ウェル組織培養処理白壁/透明底板、に播種した。
細胞を、10uM~1nMの9点ドーズ応答希釈物系列として、表1の化合物で処理した。
細胞を化合物に曝露し、その後、製造者の推奨に従い、示される通り、4日後(H69)又は6日後(MCF7、ZR75-1、A2058)に、CellTiter-Glo(登録商標)発光細胞生存率測定を用いて細胞生存能力を決定した。
細胞を化合物に曝露し、その後、示される通り、4日後(H69)又は6日後(A2058)に、CellTiter-Glo(登録商標)発光細胞生存率測定を用いて、製造者(CTG; Promega、マディソン、ウィスコンシン、アメリカ合衆国)の推奨に従い、細胞生存能力を決定した。
プレートを、BioTek社(バーモント州Winooski)のSyngergy2マルチモードプレートリーダで読み込んだ。
可変モル濃度から相対発光量(RLU)をプロットし、Graphpad社の(カリフォルニア州LaJolla)プリズム5統計ソフトウェアを用いてデータを分析し、各化合物についてEC50を決定した。
2つのRbポジティブ乳癌細胞系統(MCF7及びZR75-1)に対する細胞阻害アッセイの結果が、表4に示される。
MCF7乳癌細胞増殖の阻害のために必要な抑制性EC50値の範囲は、28nM~257nMであった。
ZR75-1乳癌細胞増殖の阻害のために必要な抑制性EC50値の範囲は、24nM~581nMであった。
MCF7胸腺癌細胞の増殖に対して高活性な代表的な化合物の例が、図21~24に示される。
図21~24でテストされた化合物(化合物T、Q、GG、U、H、MM、OO、及びPD-332991)の全てが、MCF-7細胞の細胞増殖の大きな阻害を示した。
図21から分かるように、化合物Tは、MCF-7細胞に対して、PD0332991より有力な活性を示す。
ZR75-1(胸腺管癌(Rbポジティブ))細胞の増殖に対して高活性な代表的な化合物の例が、図25~28に示される。
図25-28でテストされた化合物(化合物T、Q、GG、U、H、MM、OO及びPD-332991)全てが、ZR75-1細胞の細胞増殖の大きな阻害を示した。
図25から分かるように、化合物Tは、ZR75-1細胞に対して、PD0332991より有力な活性を示す。
乳癌細胞系統に加えて、ここに開示される多数の化合物についても、小細胞肺癌細胞系統(H69)及びヒト転移性メラノーマ細胞系統(A2058)、2つのRb欠損(Rb-ネガティブ)細胞系統に対して評価した。
これらの細胞阻害アッセイの結果を、表4に示す。
H69小細胞肺癌細胞の阻害のために必要な抑制性EC50値の範囲は、2040nM~>3000nMであった。
A2058悪性黒色腫細胞増殖の阻害のために必要な抑制性EC50値の範囲は、1313nM~>3000nMであった。
2つのRbポジティブ乳癌細胞系統に認められる大きな阻害とは対照的に、試験された化合物は、小細胞肺癌又はメラノーマ細胞の増殖を抑制することに対してさほど有効ではなかったことが、見出された。
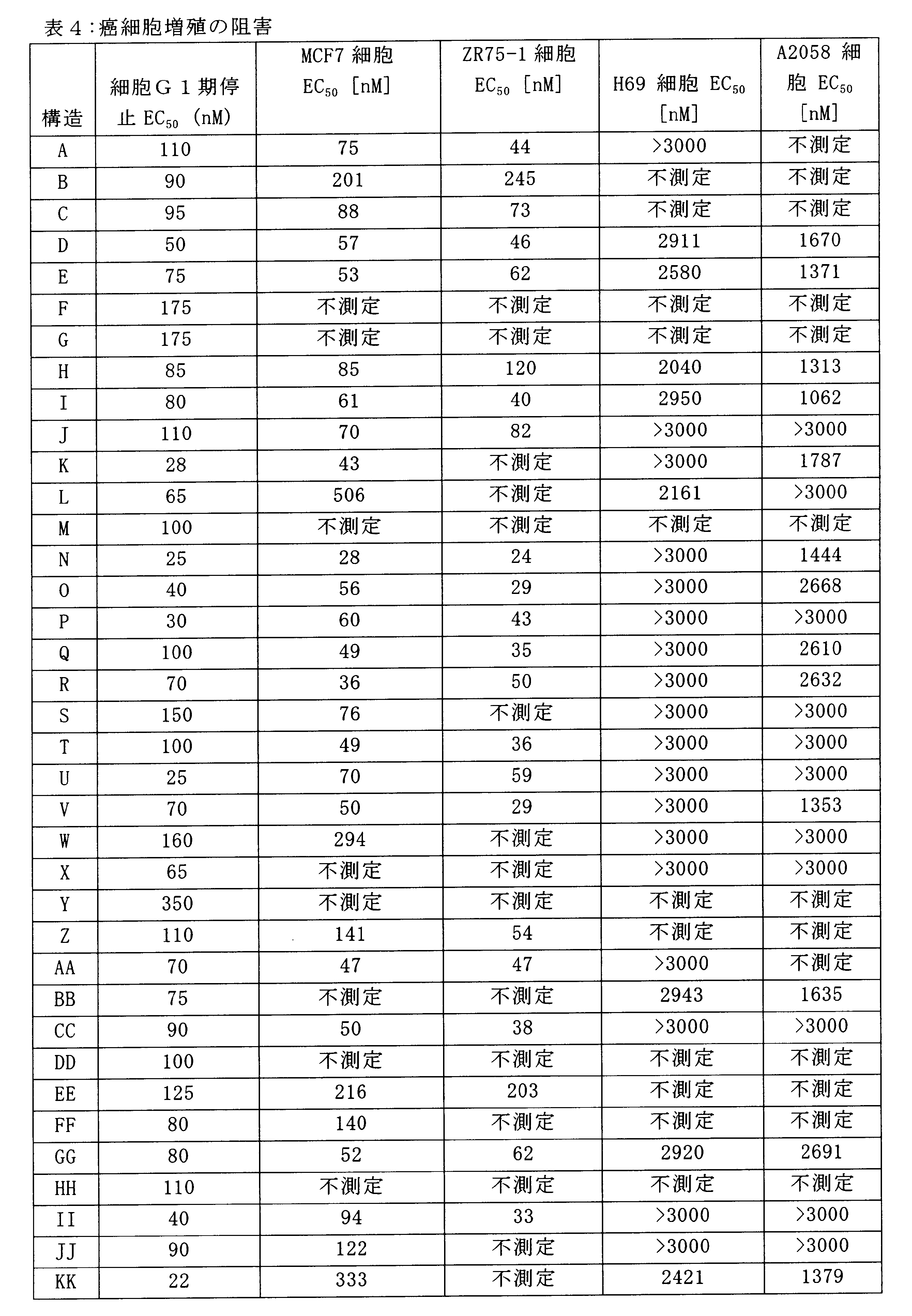
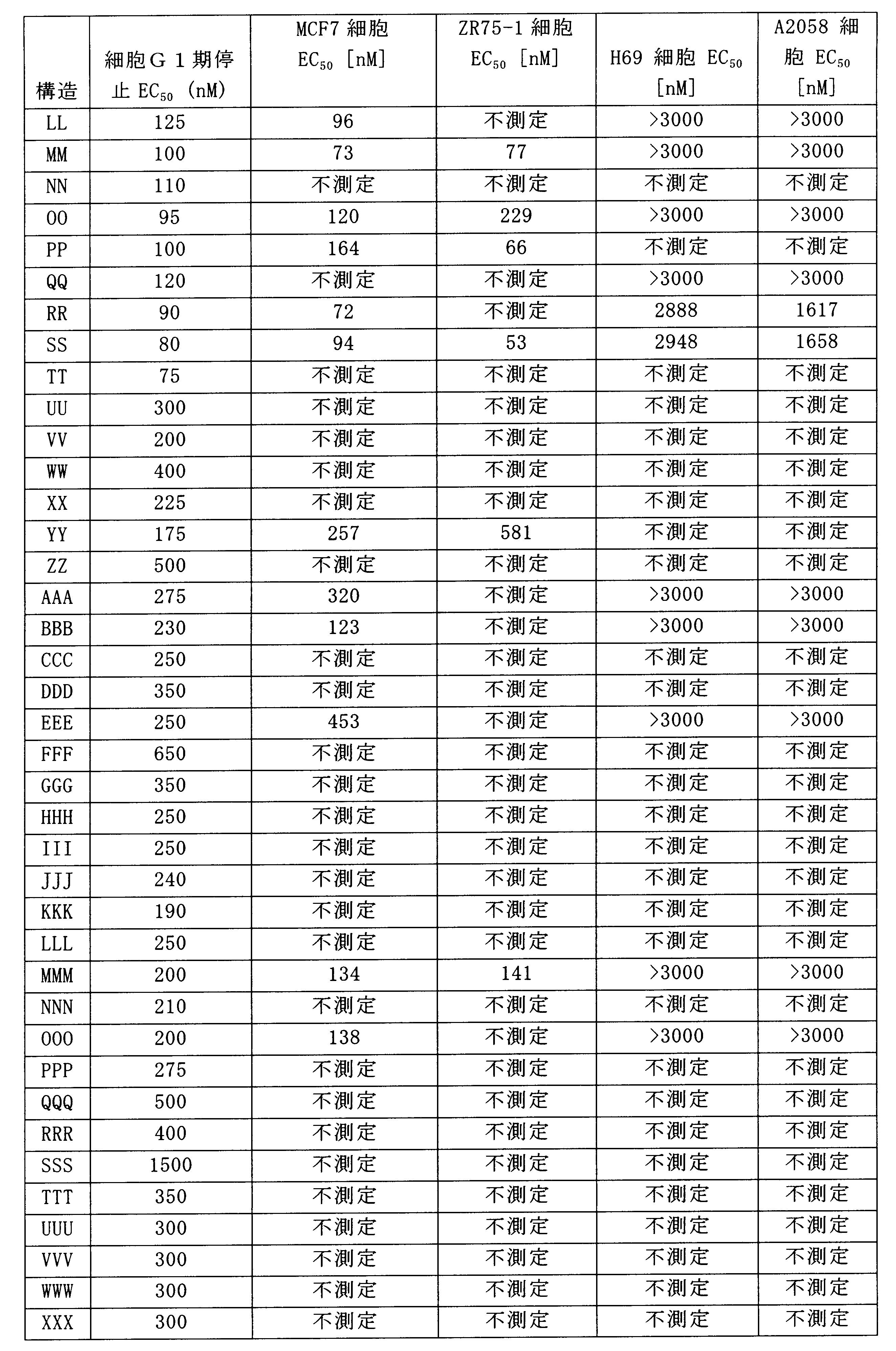
HSPC成長抑制の研究
HSPC上のPD0332991の効果を、前もって実証した。
図2は、マウスHSPC及び脊髄前駆細胞のEdU取込みを示し、Roberts et al. Multiple Roles of Cyclin-Dependent Kinase 4/6 Inhibitors in Cancer Therapy. JCNI 2012、104(6):476-487(図2A)で報告されるように、経口薬強制飼養による150mg/kgのPD0332991の単一ドーズの後、骨髄停止に関して過渡的CDK4/6阻害の時間効果を評価する。
図2から分かるように、PD0332991の単一経口薬ドーズにより、36時間を超えてHSPC(LKS+)及び脊髄前駆細胞(LKS-)の低下が維持された。
経口薬ドーズ後48時間経過前に、HSPC及び脊髄前駆細胞は、ベースライン細胞分割に戻っている。
抗新生物性化合物の薬物動態及び薬力学的特性
本発明の化合物は、良好な薬物動態及び薬力学的特性を実証する。
化合物T、Q、GG及びUは、経口強制飼養によって30mg/kgで、又は静脈内注射により10mg/kgで、マウスにドーズされた。
血液サンプルを、ドーズ後0、0.25、0.5、1.0、2.0、4.0及び8.0時間で取得し、化合物T、Q、GG又はUの血漿濃度を、HPLCによって決定した。
化合物T、GG及びUが、表5で示すように優れた経口薬物動態及び薬力学的特性を有することが、実証された。
これは、経口生体有用性(F(%))が52 %~80 %と非常に高いこと、及び、経口薬投与後のプラズマ半減期が3~5時間であることを含む。
化合物T、Q、GG及びUは、静脈内投与によって供給された場合に、優れた薬物動態及び薬力学的特性を有することが実証された。
4つの化合物全てに対する代表的なIV及び経口薬PKカーブが、図3に示される。
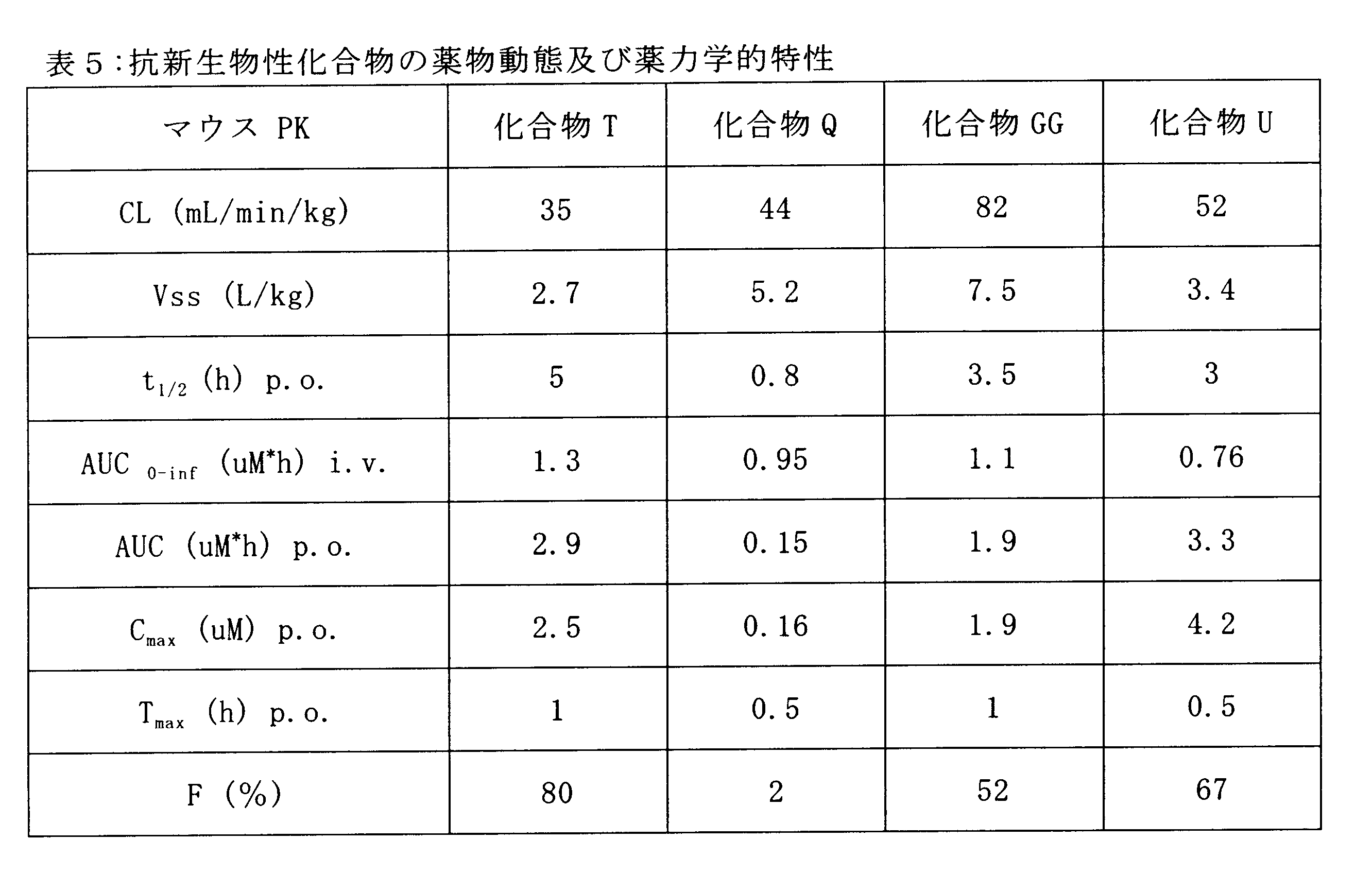
細胞ウォッシュアウト実験
HS68細胞は、10 %のウシ胎児血清、100U/mlのペニシリン/ストレプトマイシン及び記載される1x Glutamax(インビトロジェン)を含んだDMEM中で、1日目に60mmのディッシュ中40,000の細胞/ウェルで播種された(Brookes et al. EMBO J, 21(12)2936-2945 (2002) 及び Ruas et al. Mol Cell Biol, 27(12)4273-4282 (2007))。
シーディング24時間後、細胞は、化合物T、化合物Q、化合物GG、化合物U、PD0332991、又はDMSOビヒクル単独で、これら試験化合物の終濃度300nMで、処理された。
3日目に、1セットの処理細胞試料を、三組(0時間のサンプル)で採取した。
残留細胞をPBS-CMF中で2回洗浄し、試験化合物が無い培養基に戻された。
サンプルのセットは、24、40及び48時間に、三組で採取された。
代替的には、同じ実験を、米国菌培養収集所(ATCC、バージニア州マナサス)から得られる通常の腎近位尿細管上皮細胞(Rb-ポジティブ)を用いて、行った。
細胞は、37℃加湿インキュベーター内での腎臓上皮細胞成長キット(ATCC)を追加した腎臓上皮細胞基底培地(ATCC)により、37℃のインキュベーター内で5 %CO2加湿空気中培養された。
細胞を採取して即座に、サンプルを、プロピジウムヨウ化物染色液で染色し、サンプルはDako Cyan Flow Cytometerで実行された。
G0-G1DNA細胞サイクルの細胞の画分対S期DNA細胞サイクルの画分を、FlowJo7.2.2分析により測定した。
図4は、細胞ウォッシュアウト実験を示し、本発明の阻害剤化合物が、異なる細胞型において、短期かつ過渡的なG1期停止効果を有することを、実証した。
ヒト繊維芽細胞細胞(Rb-ポジティブ)(図4A及び4B)又はヒト腎近位尿細管上皮細胞(Rb-ポジティブ)(図4C及び4D)について、化合物T、Q、GG及びUを、PD0332991と比較し、化合物のウォッシュアウト後の細胞サイクルに対する効果を、24、36、40及び48時間で決定した。
これは、図4A及び図4Bで認められ、G0-G1画分又は細胞分裂のS期のそれぞれに対して、DMSOコントロールに対する等価な値が得られた。
対照的に、本発明の化合物で処理されたHS68細胞は、わずか24時間又は40時間で、通常のベースライン細胞分裂に戻り、これらの同じ時点におけるPD0332991とは異なっていた。
また、ヒト腎近位尿細管上皮細胞を用いた結果(図4C及び4D)では、PD0332991処理した細胞は、化合物T、Q、GG又はUで処理された細胞と比較して、細胞分裂のベースラインレベルに戻るためには著しく長い時間を要したことが示された。
EdU取込み及びフローサイトメトリー分析法を用いて評価される骨髄増殖
HSPC増殖実験のため、若い成熟した雌のFVB/Nマウスを、経口強制飼養により、化合物T、化合物Q、化合物GG又はPD0332991で示される単一ドーズで処理した。
その後、指示時間(化合物投与後の0、12、24、36又は48時間)にマウスを解剖し、前述のとおり(Johnson et al. J. Clin. Invest. (2010) 120(7), 2528-2536)、骨髄を採取した(時点当たりのマウス数n = 3)。
骨髄採取の4時間前に、マウスを、腹こう内投与(インビトロジェン)により、100μgのEdUで処理した。
骨髄単核細胞を採取し、前記の方法を用いて免疫表現型を特定し、その後、EdU陽性細胞パーセンテージを決定した(Johnson et al. J. Clin. Invest. (2010) 120(7), 2528-2536)。
手短に言えば、HSPCは、系統マーカー(Lin-)、Sca1(S+)及びc-Kit(K+)の発現によって特定された。
マウスの分析により、化合物T、化合物Q及び化合物GGが、骨髄幹細胞(HSPC)(図5)のドーズ依存的、過渡滴及び可逆的G1期停止を実証したことが決定された。
グループ当たり6匹のマウスに対し、150mg/kgの化合物T、化合物Q、化合物GG又はビヒクルのみ、を経口強制飼養によって投薬した。
4時間前に動物を解剖して骨髄を採取し、マウスを、腹こう内投与によってEdU100μgで処理した。
グループ当たり3匹のマウスを12時間で解剖し、グループ当たり残りの3匹の動物を24時間で解剖した。
化合物T及びGGは、12時間でEdU取込みが低下し、それは24時間に正常に戻り始めていたことを、実証した。
また、化合物Qでは、12時間で若干低下し、化合物Qの経口薬生体有用性が低いという事実にもかかわらず24時間でベースラインに戻り始めたことを実証した。
追実験は、化合物Tで完了され、化合物処理のドーズ応答及びより長い期間を検討した。
化合物Tは、経口強制飼養によって50、100又は150mg/kgでドーズされ、骨髄へのEdU取込みは、上記のように12及び24時間で決定された。
代替的には、化合物Tは150mg/kgで経口強制飼養によってドーズされ、骨髄へのEdU取込みは12、24、36及び48時間で決定された。
図5Bおよび5Cから分かるように、また、細胞ウォッシュアウト実験と同様に、多数のドーズの経口強制飼養後24時間でのEdU取込みにより測定されたように、骨髄細胞、及び特にHSPCは正常細胞分裂に戻っていた。
図5C中の化合物Tの150mg/kgの経口薬ドーズは、24及び36時間で細胞は依然分裂せず(EdU取込みが低かったと測定された)48時間でようやくに正常値に戻った図2に示されるPD0332991の同じドーズの結果と、直接比較できる。
化合物T及びPD0332991を比較したHSPC成長抑制研究
図6は、化合物の投与(時間)の後の、PD0332991(三角)又は化合物T(逆三角)のいずれかで処理されたマウスのEdUポジティブなHSPC細胞のパーセンテージの時間に対するグラフである。
両方の化合物は、経口強制飼養によって150mg/kgで投与された。
骨髄採取の1時間の前、EdUを腹腔内に注入し、サイクリング細胞を標識した。
骨髄は、化合物処理の後12、24、36及び48時間で採取し、EdU陽性HSPC細胞のパーセンテージを各時点で決定した。
図6で理解されるように、PD0332991の単一経口薬ドーズにより、36時間を超えて、HSPCの低下が維持された。
対照的に、化合物Tの単一経口薬ドーズでは、12時間でHSPC増殖が当初低下したが、HSPCの増殖は、化合物Tのドーズの後、24時間で再開した。
代謝安定性
化合物Tの代謝安定性を、PD0332991と比較し、ヒト、犬、ネズミ、猿及びマウス肝ミクロソームで決定した。
ヒト、マウス及び犬肝ミクロソームは、Xenotech社から購入し、スピローグ-ドーリーネズミ肝ミクロソームを、Absorption Systemsにより調製した。
0.5mg/mLの肝ミクロソーム、100mMのリン酸カリウム、pH7.4、5mMのマグネシウムクロリド、及び1uMのテスト化合物を含む反応物混合物を調製した。 テスト化合物を、1uM終濃度で、反応物混合物に加えた。
反応物混合物(共同因子なし)の均等量を、37℃で3分間水浴を振盪して培養した。 コントロール化合物、テストステロン、を、別々の反応物中で、テスト化合物と同時に行った。
共同因子(NADPH)の添加により、反応を開始し、その後、混合物を、37℃で振盪中の水浴中で培養した。
テスト化合物に対しては0、10、20、30及び60分、テストステロンに対しては0、10、30及び60分で、均等量(100μL)を回収した。
テスト化合物サンプルを、内部標準を含む氷冷アセトニトリル100μLと直ちに混合し、反応を終了させた。
テストステロンサンプルは、0.1 %の蟻酸及び内部標準を含む800μLの氷冷50/50アセトニトリル/dH2Oと直ちに混合し、反応を終了させた。
サンプルは、確認されたLC-MS/MS方法を用いてアッセイされた。
Orbitrap高分解能質量分析計を用いてテスト化合物サンプルを分析し、ペアレント試験化合物の消失を定量化するとともに、代謝生成物の出現を検出した。
内部標準に対するピーク面積応答割当量(PARR)を、時間0におけるPARRと比較し、テスト化合物のパーセンテージ又はその時点で残っているポジティブコントロールを決定した。
GraphPadソフトウェアを用いて半減期を計算し、単一相指数関数減衰方程式に当てはめた。
半減期を、t1/2 = 0.693kのベースで計算し、式中、kは、インキュベーション時間に対する残留する自然対数パーセンテージの勾配プロットに基づく排出速度定数である。
計算された半減期が実験の所要時間より長かったとき、半減期は>最も長いインキュベーション時間、と表現された。
計算された半減期は、括弧内にもリストされる。
計算された半減期が>2xの場合は、実験の所要時間、半減期は報告されなかった。
細胞増殖のタイムリーな再開は、組織修復のために必要であり、したがって、あまりに長い期間の停止は、HSPC等の正常細胞に好ましくない。
その停止持続時間に影響するCDK4/6阻害物質の特性は、その薬物動態(PK)及び酵素の半減期である。
一旦開始されれば、循環する化合物が抑制性レベルにとどまる限り、及び、化合物が酵素に係わる限り、G1期停止は生体内で維持される。
PD032991は、たとえば、全PK半減期が長く、酵素オフ速度が非常に低速である。
ヒトでは、PD0332991は、PK半減期は27時間と示される(Schwartz,GK et al.(2011)BJC,104:1862-1868を参照)。
ヒトでは、PD0332991の単一投与は、約1週間続いたHSPCの細胞サイクル進行を停止させる。
これは、化合物(5半減期×27時間の半減期)が消失するために6日かかること、ならびに、酵素のCDK4/6機能の阻害に付加的に1.5~2日がかかることを反映している。
この計算は、通常の骨髄機能が戻るためには合計7+日を要し、その間、新しい血液の生成が低下することを示唆する。
これらの観察は、診療室レベルでPD0332991に認められる深刻な顆粒球減少を説明することができる。
追実験は、化合物T及びPD0332991で完了され、ヒト、犬、ネズミ、猿及びマウス肝ミクロソームの代謝安定性(半減期)を比較した。
さらに、上記及び図4中で説明されるように、PD0332991はまた、酵素の半減期が長期であるように思われ、これは、ヒト細胞中に、はっきりした細胞サイクル進行の停止の発生によって明示され、化合物が細胞培養株培地から除去された後(すなわち、インビトロウォッシュアウト実験で)でさえ、40時間以上続いた。
図4にさらに示すように、ここに記載される化合物を培養基から除去すれば、増殖が迅速な再開し、これは高い酵素オフ速度と一致する。
図2、5C及び6に示すように、酵素のオフ速度のこれらの差は、薬力学的(PD)効果の著しい差につながる。
示されるように、PD0332991の単一経口薬ドーズは、ネズミ骨髄中の造血幹細胞及び前駆細胞(HSPC)の36+時間成長停止を生じさせ、これは、マウスにおけるPD0332991の6時間のPK半減期で説明したものよりも長い。
対照的に、化合物Tの効果は非常に短期であり、細胞サイクルへの迅速なリエントリーを与え、HSPC増殖の鋭いインヴィヴォコントロールを提供する。
CDK4/6阻害物質の有効性、HER2による胸腫瘍中の化合物T
MMTVプロモータによって誘導されるc-neu(ヒトHER2のマウスオーソログ)を発現する乳癌(Rbポジティブ)のHER2誘導モデル(Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 1988、54: 105-15)が、下記の例で用いられた。
腫瘍が容易にできる継代評価を許した標準サイズ(50-60のmm3)に着いたとき、マウスは治療研究の中に登録された。
腫瘍保持マウスは、それらの食事に加えられる化合物Tで、連続的に処理された(100mg/kg/d又は150mg/kg/d)。
MMTV-c-neu(コントロール、n = 9;化合物T 100mg/kg、n = 7;化合物T 150mg/kg(n = 6))マウスは、触感によって腫瘍発生を評価するために、毎週検討された。
腫瘍体積は、次の式で計算した。
体積=[(幅)2x長さ]/2。
腫瘍保持マウスは、所定の罹患率、腫瘍潰瘍又は直径1.5cm以上の腫瘍サイズとなった指示時間に安楽死させた。
図8及び9に示すように、化合物Tによる連続治療(100mg/kg/d又は150mg/kg/d)により、28日の治療過程の間、腫瘍体積に際立った減少が現れた。
いくつかの腫瘍では、完全な腫瘍後退を示し、治療の28日の過程の間、化合物tへの耐性が見られなかった。
このHER2誘導マウスモデルは増殖のためのCDK4/6活性に『中毒になって』おり、化合物TはCDK4/6依存的(Rbポジティブ腫瘍)に有効な薬剤であることが、これらのデータから示された。
HER2誘導胸腫瘍におけるCDK4/6阻害物質(化合物T、化合物GG及び化合物U)の有効性
化合物T、化合物GG及び化合物Uの臨床前キャラクタリゼーションによれば、それらは、CDK4及びCDK6を、IC50でそれぞれ0.7~1.0nM及び5~6nMに抑制することが示された。
機能性Rbタンパク質による腫瘍細胞においては、これらの化合物は、G1期停止中に得られたRbリン酸化を強力に抑制する。
CDK4/6阻害物質化合物T、化合物GG及び化合物Uのインヴィヴォ有効性が、管腔乳癌の遺伝子的加工マウスモデルに対して試験された。
腫瘍は、ノギス測定を使用して毎週順次評価された。
腫瘍が40~64mm3に到達すれば、治療的介入が開始された。
腫瘍体積は、式((幅2)×長さ)/2を用いて計算された。
3つの化合物全ては、医薬用食餌(100mg/kg/d)を介して経口で投与された。 医薬用食餌は28日間連続で投与され、その後停止された。
RECIST基準が、客観的奏効率を評価するために用いられた。
客観的奏効率(ORR)は、腫瘍体積のパーセンテージ変化に基づき、以下のカテゴリーを使用して分類された。
CR(完全な応答)= 100 %の応答;
PR(一部分の応答)=少なくとも30 %の低下;
SD(安定した疾病)=変化無し(PRでない及びPDでない);
PD(進行性の疾病)=20 %の上昇。
治療グループは、適切に認容され、毒性の臨床徴候が無く、体重損失が無く、毒性に関係した死が無かった。
線型回帰t検定を用いて、経時的な腫瘍体積成長の統計的有意差が決定された。
3つの一団全ては、未処置の一団と比較して統計的に有意だった。化合物T、p < 0.0001;化合物GG、p=0.0001;化合物U、p < 0.0001。
図10で示すように、各目的のカテゴリー中の動物の数が決定された。
化合物Tは、100%の客観的奏効率(n=7)を有することが見出され、化合物GGは、85%の客観的奏効率(n=7)を有することが見出され、化合物Uは、100%の客観的奏効率(n=8)を有することが見出された。
図11には、化合物T、化合物GG及び化合物Uで処理されるMMTV-Neuマウスからの腫瘍体積が示され、腫瘍体積は7日間毎日計測された。
図12には、最高の応答(14日又はそれ以後)のデータが、個々の腫瘍ごとに示される。
これらを纏めれば、化合物T(100mg/kg)(化合物GG)(100mg/kg)又は化合物U(100mg/kg)を用いた連続治療を行えば、28日間の治療過程の間、腫瘍体積に際立った減少が見られたということを、これらのデータが示している。
CDK4/6依存的細胞における化合物Tによる細胞サイクル進行の停止
完全なG1期停止を誘発する CDK4/6阻害剤の能力を試験するため、2つのCDK4/6依存的細胞系統(tHS68及びWM2664;Rb-ポジティブ)及び1つのCDK4/6-非依存的細胞系統(A2058; Rb-ネガティブ)からなる細胞ベースのスクリーニング方法を用いた。
表面被覆の24時間後、用量依存的方法で、各細胞系統を化合物Tで24時間処理した。
実験の末尾で、細胞を採取し、固定し、そして、488nmのライトで励起されると強く赤い蛍光を発する(最大発光637nm)プロピジウムヨウ化物(DNAインターカレータ)で染色した。
サンプルを、Dako Cyanflowcytometerで実行し、そして、>10,000イベントを各サンプルに対して収集した。
TreeStar社によって開発されたFlowJo2.2ソフトウェアを用いて、データを分析した。
図29Aでは、CDK4/6依存的細胞系統において、化合物Tは、強いG1細胞サイクル進行停止を誘発し、tHS68細胞のEC50が80nMであり、これに対応してS期での低下が、示した最も高い濃度でベースラインの28 %から6 %までの範囲にあったことを、この結果が示している。
化合物T(300nM)による治療に際して、S期個体群に同様の低下があり、CDK4/6依存的細胞系統の両方(tHS68(図29Bと29Eを比較)及びWM2664(図29Cと29Fを比較))に、G1停止細胞の増加が見られたが、CDK4/6-非依存的(A2058;図29Dと29Gを比較)細胞系統には見られなかった。
CDK4/6-非依存的細胞系統は、阻害剤の存在下での効果を示さない。
化合物TがRBのリン酸化を抑制する
CDK4/6-サイクリンD錯体は、DNA細胞サイクルのG1からS期まで進行のために必須である。
この錯体は、網膜芽細胞腫腫瘍サプレッサタンパク質(Rb)をリン酸化する。
CDK4/6阻害のRbリン酸化(pRb)への影響を実証するため、化合物Tは、3つの細胞系統、すなわち2つのCDK4/6依存的(tHS68、WM2664; Rb-ポジティブ)及び1つのCDK4/6非依存的(A2058; Rb-ネガティブ)、に曝露された。
シーディングの24時間後、細胞は、4、8、16及び24時間、300nM終濃度で化合物Tにより処理された。
サンプルは溶解され、タンパク質はウェスタンブロット解析によってアッセイされた。 Rbリン酸化は、CDK4/6-サイクリンD錯体、Ser780及びSer807/811によってターゲットにされる2つのサイトで、種特異抗体を用いて計測された。
結果によれば、化合物Tは、16時間のポスト曝露によってRb依存的細胞系統におけるRbリン酸化をブロックする一方で、Rb-非依存的細胞(図30)に対する効果を有していないということが、実証される。
この明細書は、本発明の実施形態に関して記載された。本発明は様々な実施形態に関して記載され、それは付随する実施例により例示される。しかしながら、本発明は、これらとは異なる形態で表されてもよく、ここに記載された実施形態に限定されるように解釈されてはならない。ここに与えられた教示により、当業者は所望された目的のために本発明を変更することができ、この変更は本発明の範囲内であると考えられる。
Claims (32)
- ヒトにおける網膜芽細胞腫タンパク質(Rb)ポジティブ癌を、造血幹細胞及び前駆細胞(HSPC)における有害作用を最小にしつつ、治療するための医薬組成物であって、
有効量の、下記構造を有する選択的サイクリン依存的キナーゼ(CDK)4/6阻害物質又はその薬理学的に許容される塩:
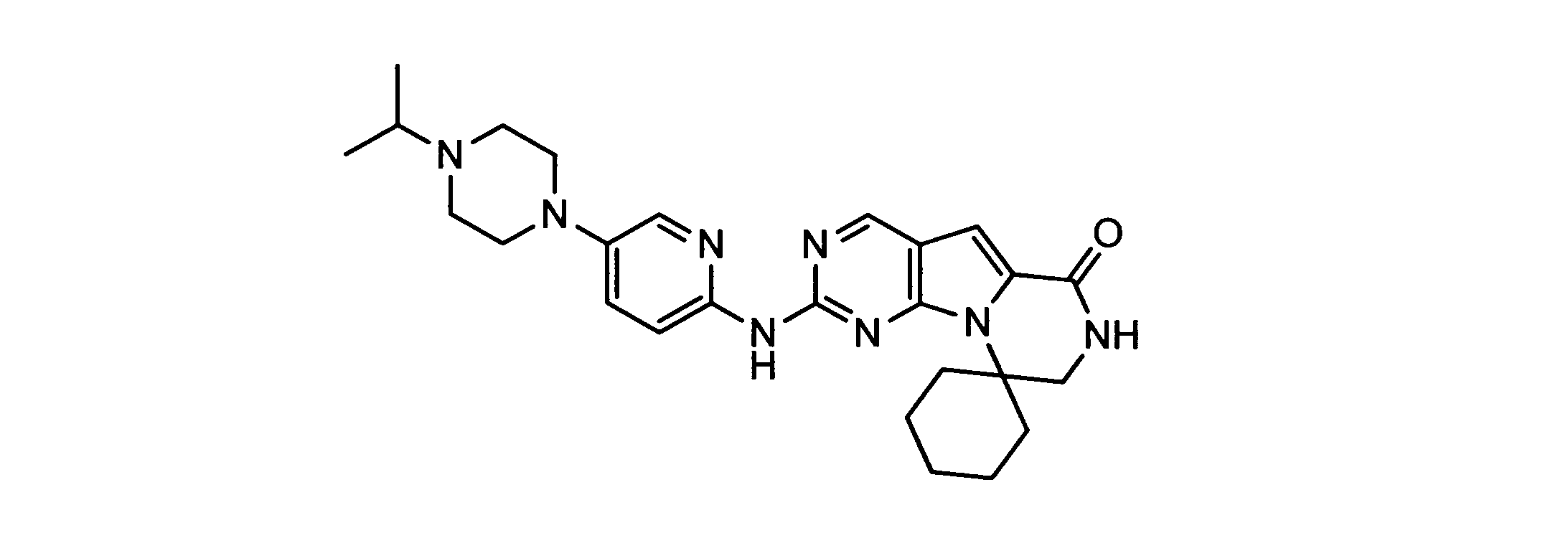
- 前記CDK4/6阻害物質が他の化学療法剤と組み合わせて投与される、請求項1に記載の医薬組成物。
- 前記化学療法剤がmTOR阻害剤である、請求項2に記載の医薬組成物。
- 前記mTOR阻害剤が、ラパマイシン、エベロリムス、テムシロリムス、リダフォロリムス、シロリムス及びデフォロリムスからなる群から選択される、請求項3に記載の医薬組成物。
- 前記化学療法剤がPI3K阻害剤である、請求項2に記載の医薬組成物。
- 前記PI3K阻害剤が、イデラリシブ、ピクチリシブ、ブパルリシブ、IPI-145、BAY80-6946、BEZ235、TGR1202(RP5264)、MLN1117(INK1117)、SAR245408(XL147)、SAR245409(XL765)、パロミド529、ZSTK474、PWT33597、RP6530、CUDC-907及びAEZS-136からなる群から選択される、請求項5に記載の医薬組成物。
- 前記化学療法剤がデュアルmTOR-PI3K阻害剤である、請求項2に記載の医薬組成物。
- 前記化学療法剤がMEK阻害剤である、請求項2に記載の医薬組成物。
- 前記MEK阻害剤が、トラメチニブ、セルメチニブ、MEK162及びGDC-0973(XL518)からなる群から選択される、請求項8に記載の医薬組成物。
- 前記化学療法剤がRAS阻害剤である、請求項2に記載の医薬組成物。
- 前記化学療法剤がALK阻害剤である、請求項2に記載の医薬組成物。
- 前記ALK阻害剤が、クリゾチニブ、AP26113及びLDK378からなる群から選択される、請求項11に記載の医薬組成物。
- 前記化学療法剤がHSP阻害剤である、請求項2に記載の医薬組成物。
- 前記HSP阻害剤が、ゲルダナマイシン及び17-N-アリルアミノ-17-デメトキシゲルダナマイシン(17AAG)からなる群から選択される、請求項13に記載の医薬組成物。
- 前記化学療法剤がPD-1阻害剤である、請求項2に記載の医薬組成物。
- 前記PD-1阻害剤が、ニボルマブ、CT-011及びMK-3475からなる群から選択される、請求項15に記載の医薬組成物。
- 前記化学療法剤がAKT阻害剤である、請求項2に記載の医薬組成物。
- 前記AKT阻害剤が、MK-2206、ペリホシン、GDC-0068及びAZD5363からなる群から選択される、請求項17に記載に医薬組成物。
- 前記化学療法剤がFLT-3阻害剤である、請求項2に記載の医薬組成物。
- 前記FLT-3阻害剤が、ドビチニブ、クイザルチニブ(AC220)、アムバチニブ(MP-470)及びタンデュチニブからなる群から選択される、請求項19に記載の医薬組成物。
- 前記化学療法剤が、ゲフィチニブ、エルロチニブ、セツキシマブ及びベバシズマブからなる群から選択される、請求項2に記載の医薬組成物。
- 前記化学療法剤が、イマチニブメシル酸塩、ダサチニブ、ニロチニブ、ボスチニブ、トラスツズマブ、ペルツズマブ、ラパチニブ、パニツムマブ、バンデタニブ、ベムラフェニブ、ボリノスタット、ロミデプシン、ベキサロテン、アリトレチノイン、トレチノイン、カーフィルゾミブ、プララトレキサート、ソラフェニブ、スニチニブ、パゾパニブ、レゴラフェニブ及びカボザンチニブからなる群から選択される、請求項2に記載の医薬組成物。
- 前記化学療法剤が、バタラニブ、ボロシキシマブ、エタラシズマブ、シレンギチド、アタシセプト、リツキシマブ、アレムツズマブ、アルデスロイキン、アトリズマブ、トシリズマブ、ルカツムマブ、マリゾミブ、タネスピマイシン、サキナビルメシル酸塩、リトナビル、ネルフィナビルメシル酸塩、インジナビル硫酸塩、ベリノスタット、パノビノスタット、マパツムマブ、レクサツムマブ、ドゥラネルミン、オブリメルセン、プリチデプシン、ダサチニブ、レナリドマイド、サリドマイド、シンバスタチン及びセレコキシブからなる群から選択される、請求項2に記載の医薬組成物。
- 前記Rbポジティブ癌がRbポジティブクラス変異体非小細胞性肺癌である、請求項1又は2に記載の医薬組成物。
- 前記Rbポジティブ癌がRbポジティブ子宮内膜癌である、請求項1又は2に記載の医薬組成物。
- 前記Rbポジティブ癌がRbポジティブ膵癌である、請求項1又は2に記載の医薬組成物。
- 前記Rbポジティブ癌がRbポジティブメラノーマである、請求項1又は2に記載の医薬組成物。
- 前記Rbポジティブ癌がRbポジティブ結腸直腸癌である、請求項1又は2に記載の医薬組成物。
- 前記Rbポジティブ癌がRbポジティブ脂肪肉腫である、請求項1又は2に記載の医薬組成物。
- 前記CDK4/6阻害物質が、連続する21日以上の間、1日に少なくとも1回、投与される、請求項1~29のいずれか一項に記載の医薬組成物。
- 前記CDK4/6阻害物質が、連続する24日以上の間、1日に少なくとも1回、投与される、請求項1~29のいずれか一項に記載の医薬組成物。
- 前記CDK4/6阻害物質が、連続する28日以上の間、1日に少なくとも1回、投与される、請求項1~29のいずれか一項に記載の医薬組成物。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023044746A JP2023078341A (ja) | 2013-03-15 | 2023-03-20 | Rbポジティブ異常細胞増殖に対するhspc温存治療 |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361798772P | 2013-03-15 | 2013-03-15 | |
| US61/798,772 | 2013-03-15 | ||
| US201361861374P | 2013-08-01 | 2013-08-01 | |
| US61/861,374 | 2013-08-01 | ||
| US201361911354P | 2013-12-03 | 2013-12-03 | |
| US61/911,354 | 2013-12-03 | ||
| US201461949786P | 2014-03-07 | 2014-03-07 | |
| US61/949,786 | 2014-03-07 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019077828A Division JP6738933B6 (ja) | 2013-03-15 | 2019-04-16 | Rbポジティブ異常細胞増殖に対するhspc温存治療 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2023044746A Division JP2023078341A (ja) | 2013-03-15 | 2023-03-20 | Rbポジティブ異常細胞増殖に対するhspc温存治療 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2020183422A JP2020183422A (ja) | 2020-11-12 |
| JP7250737B2 true JP7250737B2 (ja) | 2023-04-03 |
Family
ID=51527871
Family Applications (9)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016502870A Active JP6430483B2 (ja) | 2013-03-15 | 2014-03-14 | 化学療法の間の正常細胞の一時的な保護 |
| JP2016503092A Active JP6337083B2 (ja) | 2013-03-15 | 2014-03-14 | Rbポジティブ異常細胞増殖に対するhspc温存治療 |
| JP2018089382A Active JP6517401B2 (ja) | 2013-03-15 | 2018-05-07 | Rbポジティブ異常細胞増殖に対するhspc温存治療 |
| JP2018204234A Active JP6767456B2 (ja) | 2013-03-15 | 2018-10-30 | 化学療法の間の正常細胞の一時的な保護 |
| JP2019077828A Active JP6738933B6 (ja) | 2013-03-15 | 2019-04-16 | Rbポジティブ異常細胞増殖に対するhspc温存治療 |
| JP2020123189A Active JP7250737B2 (ja) | 2013-03-15 | 2020-07-17 | Rbポジティブ異常細胞増殖に対するhspc温存治療 |
| JP2020155691A Active JP7213854B2 (ja) | 2013-03-15 | 2020-09-16 | 化学療法の間の正常細胞の一時的な保護 |
| JP2023005413A Ceased JP2023052449A (ja) | 2013-03-15 | 2023-01-17 | 化学療法の間の正常細胞の一時的な保護 |
| JP2023044746A Withdrawn JP2023078341A (ja) | 2013-03-15 | 2023-03-20 | Rbポジティブ異常細胞増殖に対するhspc温存治療 |
Family Applications Before (5)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016502870A Active JP6430483B2 (ja) | 2013-03-15 | 2014-03-14 | 化学療法の間の正常細胞の一時的な保護 |
| JP2016503092A Active JP6337083B2 (ja) | 2013-03-15 | 2014-03-14 | Rbポジティブ異常細胞増殖に対するhspc温存治療 |
| JP2018089382A Active JP6517401B2 (ja) | 2013-03-15 | 2018-05-07 | Rbポジティブ異常細胞増殖に対するhspc温存治療 |
| JP2018204234A Active JP6767456B2 (ja) | 2013-03-15 | 2018-10-30 | 化学療法の間の正常細胞の一時的な保護 |
| JP2019077828A Active JP6738933B6 (ja) | 2013-03-15 | 2019-04-16 | Rbポジティブ異常細胞増殖に対するhspc温存治療 |
Family Applications After (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2020155691A Active JP7213854B2 (ja) | 2013-03-15 | 2020-09-16 | 化学療法の間の正常細胞の一時的な保護 |
| JP2023005413A Ceased JP2023052449A (ja) | 2013-03-15 | 2023-01-17 | 化学療法の間の正常細胞の一時的な保護 |
| JP2023044746A Withdrawn JP2023078341A (ja) | 2013-03-15 | 2023-03-20 | Rbポジティブ異常細胞増殖に対するhspc温存治療 |
Country Status (18)
| Country | Link |
|---|---|
| US (15) | US9487530B2 (ja) |
| EP (4) | EP3653209A1 (ja) |
| JP (9) | JP6430483B2 (ja) |
| CN (4) | CN108434149B (ja) |
| CA (2) | CA2906166C (ja) |
| CY (1) | CY1122434T1 (ja) |
| DK (1) | DK2968290T3 (ja) |
| ES (2) | ES2761406T3 (ja) |
| HR (1) | HRP20192168T1 (ja) |
| HU (1) | HUE046653T2 (ja) |
| LT (1) | LT2968290T (ja) |
| ME (1) | ME03557B (ja) |
| PL (1) | PL2968290T3 (ja) |
| PT (1) | PT2968290T (ja) |
| RS (1) | RS59790B1 (ja) |
| SI (1) | SI2968290T1 (ja) |
| SM (1) | SMT201900681T1 (ja) |
| WO (2) | WO2014144847A2 (ja) |
Families Citing this family (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009061345A2 (en) | 2007-11-07 | 2009-05-14 | Cornell Research Foundation, Inc. | Targeting cdk4 and cdk6 in cancer therapy |
| US8691830B2 (en) * | 2010-10-25 | 2014-04-08 | G1 Therapeutics, Inc. | CDK inhibitors |
| CA2868966C (en) | 2012-03-29 | 2021-01-26 | Francis Xavier Tavares | Lactam kinase inhibitors |
| CA2870019C (en) | 2012-04-26 | 2020-08-18 | Francis Xavier Tavares | Synthesis of lactams |
| ME03557B (me) | 2013-03-15 | 2020-07-20 | G1 Therapeutics Inc | Privremena zaštiтa normalnih ćelija током hemoterapije |
| CA2906157C (en) | 2013-03-15 | 2022-05-17 | G1 Therapeutics, Inc. | Highly active anti-neoplastic and anti-proliferative agents |
| WO2015161288A1 (en) | 2014-04-17 | 2015-10-22 | G1 Therapeutics, Inc. | Tricyclic lactams for use as anti-neoplastic and anti-proliferative agents |
| WO2016040848A1 (en) | 2014-09-12 | 2016-03-17 | G1 Therapeutics, Inc. | Treatment of rb-negative tumors using topoisomerase inhibitors in combination with cyclin dependent kinase 4/6 inhibitors |
| EP3191098A4 (en) | 2014-09-12 | 2018-04-25 | G1 Therapeutics, Inc. | Combinations and dosing regimes to treat rb-positive tumors |
| WO2016126889A1 (en) * | 2015-02-03 | 2016-08-11 | G1 Therapeutics, Inc. | Cdk4/6 inhibitor dosage formulations for the protection of hematopoietic stem and progenitor cells during chemotherapy |
| CN107787322B (zh) | 2015-06-17 | 2023-07-07 | 辉瑞大药厂 | 三环化合物以及它们作为磷酸二酯酶抑制剂的用途 |
| WO2017037575A1 (en) * | 2015-08-28 | 2017-03-09 | Novartis Ag | A pharmaceutical combination comprising the pi3k inhibitor alpelisib and the cdk4/6 inhibitor ribociclib, and the use thereof in the treatment/prevention of cancer |
| JP6635374B2 (ja) * | 2015-12-18 | 2020-01-22 | 京都府公立大学法人 | 癌におけるリン酸化rbタンパク質を指標としたイリノテカン感受性予測法 |
| WO2017161253A1 (en) * | 2016-03-18 | 2017-09-21 | Tufts Medical Center | Compositions and methods for treating and preventing metabolic disorders |
| MX388576B (es) | 2016-06-07 | 2025-03-20 | Jacobio Pharmaceuticals Co Ltd | Derivados heterociclicos novedosos utiles como inhibidores de shp2. |
| WO2018005863A1 (en) * | 2016-07-01 | 2018-01-04 | G1 Therapeutics, Inc. | Pyrimidine-based compounds for the treatment of cancer |
| WO2018005533A1 (en) | 2016-07-01 | 2018-01-04 | G1 Therapeutics, Inc. | Antiproliferative pyrimidine-based compounds |
| WO2018005865A1 (en) * | 2016-07-01 | 2018-01-04 | G1 Therapeutics, Inc. | Synthesis of n-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines |
| RU2019102647A (ru) * | 2016-07-01 | 2020-08-03 | Г1 Терапьютикс, Инк. | Антипролиферационные средства на основе пиримидина |
| BR112019003722A2 (pt) | 2016-08-23 | 2019-05-28 | Eisai R&D Man Co Ltd | terapias de combinação para o tratamento de carcinoma hepatocelular |
| US11865176B2 (en) | 2016-11-08 | 2024-01-09 | Dana-Farber Cancer Institute, Inc. | Compositions and methods of modulating anti-tumor immunity |
| KR20190092478A (ko) | 2016-12-05 | 2019-08-07 | 쥐원 쎄라퓨틱스, 인크. | 화학요법 레지멘 동안의 면역 반응의 보존 |
| MX2019008158A (es) | 2017-01-06 | 2019-12-09 | G1 Therapeutics Inc | Terapia de combinacion para el tratamiento del cancer. |
| US11395821B2 (en) | 2017-01-30 | 2022-07-26 | G1 Therapeutics, Inc. | Treatment of EGFR-driven cancer with fewer side effects |
| EP3585389A4 (en) | 2017-02-22 | 2020-12-23 | G1 Therapeutics, Inc. | EGFR-CAUSED CANCER TREATMENT WITH LESS SIDE EFFECTS |
| MX2019010981A (es) | 2017-03-16 | 2020-09-07 | Eisai R&D Man Co Ltd | Terapias de combinacion para el tratamiento de cancer de mama. |
| EA202190196A1 (ru) | 2017-03-23 | 2021-08-31 | Джакобио Фармасьютикалс Ко., Лтд. | Новые гетероциклические производные, применимые в качестве ингибиторов shp2 |
| WO2019006393A1 (en) | 2017-06-29 | 2019-01-03 | G1 Therapeutics, Inc. | Morphic forms of git38 and methods of manufacture thereof |
| CN111094291B (zh) | 2017-08-11 | 2022-06-21 | 晟科药业(江苏)有限公司 | 1h-吡唑并[4,3-h]喹唑啉类化合物作为蛋白激酶抑制剂 |
| EP3738084A4 (en) | 2018-01-08 | 2021-11-17 | G1 Therapeutics, Inc. | SUPERIOR G1T38 DOSING SCHEMES |
| JP2021514359A (ja) | 2018-02-15 | 2021-06-10 | ニューベイション・バイオ・インコーポレイテッドNuvation Bio Inc. | キナーゼ阻害剤としての複素環式化合物 |
| WO2019199883A1 (en) * | 2018-04-09 | 2019-10-17 | G1 Therapeutics, Inc. | Treatment of cancers having driving oncogenic mutations |
| MX2021000895A (es) | 2018-07-27 | 2021-08-24 | California Inst Of Techn | Inhibidores de cinasas dependientes de ciclinas (cdk) y usos de los mismos. |
| AU2019325685B2 (en) | 2018-08-24 | 2025-07-24 | Pharmacosmos Holding A/S | Improved synthesis of 1,4-diazaspiro(5.5)undecan-3-one |
| US20210393623A1 (en) | 2018-09-26 | 2021-12-23 | Jacobio Pharmaceuticals Co., Ltd. | Novel Heterocyclic Derivatives Useful as SHP2 Inhibitors |
| CN113271977A (zh) * | 2018-11-09 | 2021-08-17 | G1治疗公司 | 使用艾日布林和选择性cdk4/6抑制剂组合治疗癌症的治疗方案 |
| WO2020206035A1 (en) * | 2019-04-01 | 2020-10-08 | G1 Therapeutics, Inc. | Treatment of cdk4/6 inhibitor resistant neoplastic disorders |
| TW202114684A (zh) * | 2019-06-18 | 2021-04-16 | 美商G1治療公司 | 增強癌症病患之抗腫瘤免疫之方法 |
| TW202128174A (zh) * | 2019-10-09 | 2021-08-01 | 美商G1治療公司 | 失調之纖維母細胞生長因子受體訊息傳遞的癌症之標靶性治療 |
| KR20230012547A (ko) | 2020-05-19 | 2023-01-26 | 쥐원 쎄라퓨틱스, 인크. | 의학적 장애의 치료를 위한 시클린-의존성 키나제 억제 화합물 |
| EP4165046B1 (en) * | 2020-06-11 | 2025-09-17 | Lunella Biotech, Inc. | Selective cdk4/6 inhibitor cancer therapeutics |
| US10988479B1 (en) | 2020-06-15 | 2021-04-27 | G1 Therapeutics, Inc. | Morphic forms of trilaciclib and methods of manufacture thereof |
| EP4164653A4 (en) * | 2020-06-15 | 2024-06-19 | G1 Therapeutics, Inc. | MORPHIC FORMS OF TRILACICLIB AND METHODS OF PREPARING THE SAME |
| CN113788837B (zh) * | 2021-08-02 | 2022-08-26 | 深圳湾实验室坪山生物医药研发转化中心 | Trilaciclib的合成方法 |
| JP2023148228A (ja) * | 2022-03-30 | 2023-10-13 | 東芝ライテック株式会社 | 照明制御装置及び照明制御システム |
| WO2024073507A1 (en) | 2022-09-28 | 2024-04-04 | Theseus Pharmaceuticals, Inc. | Macrocyclic compounds and uses thereof |
| EP4626866A1 (en) * | 2022-11-28 | 2025-10-08 | Assia Chemical Industries Ltd. | Novel trilaciclib intermediates, method of preparation and use thereof |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6337083B2 (ja) | 2013-03-15 | 2018-06-06 | ジー1、セラピューティクス、インコーポレイテッドG1 Therapeutics, Inc. | Rbポジティブ異常細胞増殖に対するhspc温存治療 |
Family Cites Families (97)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5591855A (en) | 1994-10-14 | 1997-01-07 | Cephalon, Inc. | Fused pyrrolocarbazoles |
| US5628984A (en) | 1995-07-31 | 1997-05-13 | University Of North Carolina At Chapel Hill | Method of detecting lung disease |
| WO1998033798A2 (en) | 1997-02-05 | 1998-08-06 | Warner Lambert Company | Pyrido[2,3-d]pyrimidines and 4-amino-pyrimidines as inhibitors of cell proliferation |
| GB9718913D0 (en) | 1997-09-05 | 1997-11-12 | Glaxo Group Ltd | Substituted oxindole derivatives |
| US20040006074A1 (en) | 1998-04-28 | 2004-01-08 | The Government Of The United States Of America | Cyclin dependent kinase (CDK)4 inhibitors and their use for treating cancer |
| JP4555476B2 (ja) | 1998-06-16 | 2010-09-29 | ザ ガバメント オブ ザ ユナイテッド ステイツ オブ アメリカ, アズ レプレゼンテッド バイ ザ セクレタリー, デパートメント オブ ヘルス アンド ヒューマン サービシーズ | 縮合アゼピノン型サイクリン依存性キナーゼ阻害物質 |
| IL144294A0 (en) | 1999-01-29 | 2002-05-23 | Univ Illinois | P53 inhibitors and therapeutic use of the same |
| CA2380389A1 (en) | 1999-07-26 | 2001-02-01 | Banyu Pharmaceutical Co., Ltd. | Biarylurea derivatives |
| US6387900B1 (en) | 1999-08-12 | 2002-05-14 | Pharmacia & Upjohn S.P.A. | 3(5)-ureido-pyrazole derivatives process for their preparation and their use as antitumor agents |
| US6291504B1 (en) | 1999-10-20 | 2001-09-18 | Dupont Pharmaceuticals Company | Acylsemicarbazides and their uses |
| EP1242420A2 (en) | 1999-12-16 | 2002-09-25 | Eli Lilly And Company | Agents and methods for the treatment of proliferative diseases |
| US7053070B2 (en) | 2000-01-25 | 2006-05-30 | Warner-Lambert Company | Pyrido[2,3-d]pyrimidine-2,7-diamine kinase inhibitors |
| ATE423120T1 (de) | 2000-06-26 | 2009-03-15 | Pfizer Prod Inc | Pyrroloä2,3-düpyrimidin verbindungen als immunosuppressive wirkstoffe |
| AU2001292579A1 (en) | 2000-09-29 | 2002-04-15 | Eli Lilly And Company | Methods and compounds for treating proliferative diseases |
| WO2002044174A2 (en) | 2000-12-01 | 2002-06-06 | Bristol-Myers Squibb Pharma Company | 3-(2,4-dimethylthiazol-5-yl) indeno[1,2-c]pyrazol-4-one derivatives as cdk inhibitors |
| AU2002305942B2 (en) | 2001-02-28 | 2006-10-26 | Onconova Therapeutics, Inc. | Method for protecting cells and tissues from ionizing radiation toxicity with Alpha, Beta unsaturated aryl sulfones |
| KR100863624B1 (ko) | 2001-05-11 | 2008-10-15 | 보드 오브 리전츠, 더 유니버시티 오브 텍사스 시스템 | Cd26을 발현하는 세포와 연관된 질환의 치료제로사용되는 항-cd26 단클론항체 |
| MXPA04005939A (es) | 2002-01-22 | 2005-01-25 | Warner Lambert Co | 2-(piridin-2-ilamino)-pirido[2,3-d]pirimidin-7-onas. |
| WO2003093469A2 (en) | 2002-05-01 | 2003-11-13 | Chromos Molecular Systems, Inc. | Methods for delivering nucleic acid molecules into cells and assessment thereof |
| MXPA05007503A (es) | 2003-01-17 | 2005-09-21 | Warner Lambert Co | Heterociclicos 2-aminopiridina sustituidos como inhibidores de proliferacion celular. |
| BR122019010200B8 (pt) | 2003-05-22 | 2021-07-27 | Nerviano Medical Science S R L | compostos de pirazol-quinazolina, seus sais, produtos ou kits e composições farmacêuticas |
| AU2004255934B2 (en) | 2003-07-11 | 2010-02-25 | Warner-Lambert Company Llc | Isethionate salt of a selective CDK4 inhibitor |
| ES2297502T3 (es) | 2003-10-23 | 2008-05-01 | F. Hoffmann-La Roche Ag | Derivados de triaza-espiropiperidinas para uso como inhibidores de glyt-1 en el tratamiento de trastornos neurologicos y neuropsiquiatricos. |
| GB0327380D0 (en) | 2003-11-25 | 2003-12-31 | Cyclacel Ltd | Method |
| WO2005094830A1 (en) | 2004-03-30 | 2005-10-13 | Pfizer Products Inc. | Combinations of signal transduction inhibitors |
| WO2005100999A2 (en) | 2004-04-08 | 2005-10-27 | Cornell Research Foundation, Inc. | Functional immunohistochemical cell cycle analysis as a prognostic indicator for cancer |
| ITMI20040874A1 (it) | 2004-04-30 | 2004-07-30 | Ist Naz Stud Cura Dei Tumori | Derivati indolici ed azaindolici con azione antitumorale |
| WO2005117908A2 (en) | 2004-05-28 | 2005-12-15 | Gemin X Biotechnologies, Inc. | Triheterocyclic compounds compositions, and methods for treating cancer or viral diseases |
| EP1846408B1 (en) | 2005-01-14 | 2013-03-20 | Janssen Pharmaceutica NV | 5-membered annelated heterocyclic pyrimidines as kinase inhibitors |
| EP1845975A1 (en) | 2005-01-21 | 2007-10-24 | Astex Therapeutics Limited | Combinations of pyrazole kinase inhibitors and further antitumor agents |
| EP2354140A1 (en) | 2005-05-20 | 2011-08-10 | Vertex Pharmaceuticals Incorporated | Pyrrolopyridines useful as inhibitors of protein kinase |
| US20070049591A1 (en) | 2005-08-25 | 2007-03-01 | Kalypsys, Inc. | Inhibitors of MAPK/Erk Kinase |
| EP1779848A1 (en) | 2005-10-28 | 2007-05-02 | Nikem Research S.R.L. | V-ATPase inhibitors for the treatment of inflammatory and autoimmune diseases |
| DE602006010433D1 (de) | 2005-12-09 | 2009-12-24 | Hoffmann La Roche | Für die behandlung von obesitas geeignete tricyclische amidderivate |
| AU2006331765A1 (en) | 2005-12-22 | 2007-07-05 | Wyeth | Substituted isoquinoline-1,3(2H,4H)-diones, 1-thioxo-1,4-dihydro-2H-isoquinoline-3-ones and 1,4-dihydro-3(2H)-isoquinolones and use thereof as kinase inhibitor |
| US20090098137A1 (en) | 2006-04-05 | 2009-04-16 | Novartis Ag | Combinations of therapeutic agents for treating cancer |
| US20070270362A1 (en) | 2006-05-18 | 2007-11-22 | The University Of Washington | Methods and compositions for prevention or treatment of inflammatory-related diseases and disorders |
| EP2043651A2 (en) | 2006-07-05 | 2009-04-08 | Exelixis, Inc. | Methods of using igf1r and abl kinase modulators |
| EP2061469B8 (en) | 2006-09-11 | 2014-02-26 | Curis, Inc. | Quinazoline based egfr inhibitors |
| EP2109450A2 (en) | 2006-12-14 | 2009-10-21 | Panacea Pharmaceuticals, Inc. | Methods of neuroprotection by cyclin-dependent kinase inhibition |
| CN101568529A (zh) | 2006-12-22 | 2009-10-28 | 诺瓦提斯公司 | 作为cdk抑制剂、用于治疗癌症、炎症和病毒感染的杂芳基-杂芳基化合物 |
| CA2690748A1 (en) | 2007-06-25 | 2008-12-31 | Neurogen Corporation | Piperazinyl oxoalkyl tetrahydro-beta-carbolines and related analogues |
| WO2009061345A2 (en) | 2007-11-07 | 2009-05-14 | Cornell Research Foundation, Inc. | Targeting cdk4 and cdk6 in cancer therapy |
| EA201001030A1 (ru) | 2007-12-19 | 2011-02-28 | Амген Инк. | Конденсированные соединения пиридина, пиримидина и триазина в качестве ингибиторов клеточного цикла |
| EP2320903B1 (en) | 2008-07-29 | 2017-01-18 | Nerviano Medical Sciences S.r.l. | THERAPEUTIC COMBINATION COMPRISING A CDKs INHIBITOR AND AN ANTINEOPLASTIC AGENT |
| ES2522346T3 (es) | 2008-08-22 | 2014-11-14 | Novartis Ag | Compuestos de pirrolopirimidina como inhibidores de CDK |
| JP2012504646A (ja) * | 2008-10-01 | 2012-02-23 | ザ ユニバーシティ オブ ノース カロライナ アット チャペル ヒル | 選択的サイクリン依存性キナーゼ4/6阻害剤を用いた化学療法化合物に対する造血系の防護 |
| CN102231983A (zh) * | 2008-10-01 | 2011-11-02 | 北卡罗来纳大学查珀尔希尔分校 | 使用选择性细胞周期蛋白依赖性激酶4/6抑制剂对抗电离辐射的造血防护 |
| PA8852901A1 (es) | 2008-12-22 | 2010-07-27 | Lilly Co Eli | Inhibidores de proteina cinasa |
| JP2012526850A (ja) * | 2009-05-13 | 2012-11-01 | ザ ユニバーシティ オブ ノース カロライナ アット チャペル ヒル | サイクリン依存性キナーゼ阻害剤及びその用法 |
| US9040519B2 (en) | 2010-02-18 | 2015-05-26 | Medivation Technologies, Inc. | Fused tetracyclic pyrido [4,3-B] indole and pyrido [3,4-B] indole derivatives and methods of use |
| UY33227A (es) | 2010-02-19 | 2011-09-30 | Novartis Ag | Compuestos de pirrolopirimidina como inhibidores de la cdk4/6 |
| UY33226A (es) | 2010-02-19 | 2011-09-30 | Novartis Ag | Compuestos de pirrolopirimidina deuterada como inhibidores de la cdk4/6 |
| FR2957748B1 (fr) * | 2010-03-16 | 2012-09-07 | St Microelectronics Grenoble 2 | Composant electronique a montage en surface |
| CN102299073A (zh) | 2010-06-25 | 2011-12-28 | 无锡华润上华半导体有限公司 | Vdmos器件及其制作方法 |
| MX338327B (es) | 2010-10-25 | 2016-04-12 | G1 Therapeutics Inc | Inhibidores de cdk. |
| US8691830B2 (en) | 2010-10-25 | 2014-04-08 | G1 Therapeutics, Inc. | CDK inhibitors |
| WO2012061477A1 (en) | 2010-11-02 | 2012-05-10 | Promega Corporation | Coelenterazine derivatives and methods of using same |
| WO2012068381A2 (en) | 2010-11-17 | 2012-05-24 | The University Of North Carolina At Chapel Hill | Protection of renal tissues from schema through inhibition of the proliferative kisses cdk4 and cdk6 |
| DK2655375T3 (en) | 2010-12-23 | 2015-03-09 | Sanofi Sa | PYRIMIDINON DERIVATIVES, PREPARATION AND PHARMACEUTICAL USE THEREOF |
| EP2937349B1 (en) | 2011-03-23 | 2016-12-28 | Amgen Inc. | Fused tricyclic dual inhibitors of cdk 4/6 and flt3 |
| TW201733984A (zh) | 2011-04-13 | 2017-10-01 | 雅酶股份有限公司 | 經取代之苯化合物 |
| EP2726074B1 (en) | 2011-07-01 | 2018-04-04 | Novartis AG | Combination therapy comprising a cdk4/6 inhibitor and a pi3k inhibitor for use in the treatment of cancer |
| EP4119551A1 (en) | 2011-07-27 | 2023-01-18 | Astrazeneca AB | 2-(2,4,5-substituted-anilino)pyrimidine compounds |
| CA2868966C (en) | 2012-03-29 | 2021-01-26 | Francis Xavier Tavares | Lactam kinase inhibitors |
| CA2870019C (en) | 2012-04-26 | 2020-08-18 | Francis Xavier Tavares | Synthesis of lactams |
| AU2013296237B2 (en) | 2012-08-03 | 2019-05-16 | Foundation Medicine, Inc. | Human papilloma virus as predictor of cancer prognosis |
| US9241941B2 (en) | 2012-09-20 | 2016-01-26 | Memorial Sloan-Kettering Cancer Center | Methods for treatment of lymphomas with mutations in cell cycle genes |
| EP2925365A1 (en) | 2012-11-28 | 2015-10-07 | Novartis AG | Combination therapy |
| US20140274896A1 (en) | 2013-03-15 | 2014-09-18 | G1 Therapeutics, Inc. | Transient Protection of Hematopoietic Stem and Progenitor Cells Against Ionizing Radiation |
| CA2906157C (en) * | 2013-03-15 | 2022-05-17 | G1 Therapeutics, Inc. | Highly active anti-neoplastic and anti-proliferative agents |
| EP2983670A4 (en) | 2013-04-08 | 2017-03-08 | Pharmacyclics LLC | Ibrutinib combination therapy |
| CA2914284A1 (en) | 2013-05-30 | 2014-12-04 | Infinity Pharmaceuticals, Inc. | Treatment of cancers using pi3 kinase isoform modulators |
| US20160257688A1 (en) | 2013-10-24 | 2016-09-08 | Francis Xavier Tavares | Process for Synthesis of Lactams |
| WO2015084892A1 (en) | 2013-12-02 | 2015-06-11 | Cornell University | Methods for treating b cell proliferative disorders |
| US9796701B2 (en) | 2013-12-31 | 2017-10-24 | Xuanzhu Pharma Co., Ltd. | Kinase inhibitor and use thereof |
| PE20170255A1 (es) | 2014-01-24 | 2017-03-22 | Dana Farber Cancer Inst Inc | Moleculas de anticuerpo que se unen a pd-1 y usos de las mismas |
| WO2015161288A1 (en) | 2014-04-17 | 2015-10-22 | G1 Therapeutics, Inc. | Tricyclic lactams for use as anti-neoplastic and anti-proliferative agents |
| WO2016040848A1 (en) * | 2014-09-12 | 2016-03-17 | G1 Therapeutics, Inc. | Treatment of rb-negative tumors using topoisomerase inhibitors in combination with cyclin dependent kinase 4/6 inhibitors |
| EP3191098A4 (en) * | 2014-09-12 | 2018-04-25 | G1 Therapeutics, Inc. | Combinations and dosing regimes to treat rb-positive tumors |
| US9374037B2 (en) | 2014-10-30 | 2016-06-21 | M/A-Com Technology Solutions Holdings, Inc. | Voltage-controlled oscillator with mask-selectable performance |
| WO2016126889A1 (en) | 2015-02-03 | 2016-08-11 | G1 Therapeutics, Inc. | Cdk4/6 inhibitor dosage formulations for the protection of hematopoietic stem and progenitor cells during chemotherapy |
| WO2018005865A1 (en) * | 2016-07-01 | 2018-01-04 | G1 Therapeutics, Inc. | Synthesis of n-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines |
| WO2018005863A1 (en) | 2016-07-01 | 2018-01-04 | G1 Therapeutics, Inc. | Pyrimidine-based compounds for the treatment of cancer |
| KR20190092478A (ko) * | 2016-12-05 | 2019-08-07 | 쥐원 쎄라퓨틱스, 인크. | 화학요법 레지멘 동안의 면역 반응의 보존 |
| WO2018106870A1 (en) * | 2016-12-08 | 2018-06-14 | Icahn School Of Medicine At Mount Sinai | Compositions and methods for treating cdk4/6-mediated cancer |
| MX2019008158A (es) * | 2017-01-06 | 2019-12-09 | G1 Therapeutics Inc | Terapia de combinacion para el tratamiento del cancer. |
| US11395821B2 (en) * | 2017-01-30 | 2022-07-26 | G1 Therapeutics, Inc. | Treatment of EGFR-driven cancer with fewer side effects |
| KR20190117582A (ko) * | 2017-02-10 | 2019-10-16 | 쥐원 쎄라퓨틱스, 인크. | 벤조티오펜 에스트로겐 수용체 조정제 |
| EP3585389A4 (en) * | 2017-02-22 | 2020-12-23 | G1 Therapeutics, Inc. | EGFR-CAUSED CANCER TREATMENT WITH LESS SIDE EFFECTS |
| WO2019006393A1 (en) * | 2017-06-29 | 2019-01-03 | G1 Therapeutics, Inc. | Morphic forms of git38 and methods of manufacture thereof |
| WO2019136244A1 (en) | 2018-01-04 | 2019-07-11 | G1 Therapeutics, Inc. | Heterocyclic compounds for the treatment of abnormal cellular proliferation |
| EP3738084A4 (en) * | 2018-01-08 | 2021-11-17 | G1 Therapeutics, Inc. | SUPERIOR G1T38 DOSING SCHEMES |
| WO2019199883A1 (en) * | 2018-04-09 | 2019-10-17 | G1 Therapeutics, Inc. | Treatment of cancers having driving oncogenic mutations |
| US11091522B2 (en) * | 2018-07-23 | 2021-08-17 | Aileron Therapeutics, Inc. | Peptidomimetic macrocycles and uses thereof |
| TW202114684A (zh) * | 2019-06-18 | 2021-04-16 | 美商G1治療公司 | 增強癌症病患之抗腫瘤免疫之方法 |
| TW202128174A (zh) * | 2019-10-09 | 2021-08-01 | 美商G1治療公司 | 失調之纖維母細胞生長因子受體訊息傳遞的癌症之標靶性治療 |
-
2014
- 2014-03-14 ME MEP-2019-338A patent/ME03557B/me unknown
- 2014-03-14 HU HUE14762438A patent/HUE046653T2/hu unknown
- 2014-03-14 CN CN201810443050.5A patent/CN108434149B/zh active Active
- 2014-03-14 CN CN201810253672.1A patent/CN108283644B/zh active Active
- 2014-03-14 SM SM20190681T patent/SMT201900681T1/it unknown
- 2014-03-14 CN CN201480027269.7A patent/CN105407889B/zh active Active
- 2014-03-14 US US14/212,430 patent/US9487530B2/en active Active
- 2014-03-14 PL PL14762438T patent/PL2968290T3/pl unknown
- 2014-03-14 WO PCT/US2014/029429 patent/WO2014144847A2/en not_active Ceased
- 2014-03-14 EP EP19198057.2A patent/EP3653209A1/en active Pending
- 2014-03-14 CA CA2906166A patent/CA2906166C/en active Active
- 2014-03-14 ES ES14762438T patent/ES2761406T3/es active Active
- 2014-03-14 ES ES14765105T patent/ES3032492T3/es active Active
- 2014-03-14 LT LT14762438T patent/LT2968290T/lt unknown
- 2014-03-14 JP JP2016502870A patent/JP6430483B2/ja active Active
- 2014-03-14 US US14/212,911 patent/US9464092B2/en active Active
- 2014-03-14 US US14/214,048 patent/US9527857B2/en active Active
- 2014-03-14 EP EP25170501.8A patent/EP4596051A3/en active Pending
- 2014-03-14 SI SI201431380T patent/SI2968290T1/sl unknown
- 2014-03-14 EP EP14762438.1A patent/EP2968290B1/en active Active
- 2014-03-14 CA CA2906156A patent/CA2906156C/en active Active
- 2014-03-14 WO PCT/US2014/028685 patent/WO2014144326A1/en not_active Ceased
- 2014-03-14 CN CN201480027281.8A patent/CN105473140B/zh active Active
- 2014-03-14 PT PT147624381T patent/PT2968290T/pt unknown
- 2014-03-14 RS RS20191537A patent/RS59790B1/sr unknown
- 2014-03-14 HR HRP20192168TT patent/HRP20192168T1/hr unknown
- 2014-03-14 EP EP14765105.3A patent/EP2968291B1/en active Active
- 2014-03-14 JP JP2016503092A patent/JP6337083B2/ja active Active
- 2014-03-14 DK DK14762438T patent/DK2968290T3/da active
-
2016
- 2016-10-07 US US15/288,878 patent/US9931345B2/en active Active
- 2016-11-03 US US15/342,990 patent/US10085992B2/en active Active
- 2016-12-21 US US15/387,083 patent/US10076523B2/en active Active
-
2018
- 2018-04-02 US US15/943,278 patent/US10660896B2/en active Active
- 2018-05-07 JP JP2018089382A patent/JP6517401B2/ja active Active
- 2018-08-24 US US16/112,362 patent/US10434104B2/en active Active
- 2018-08-24 US US16/112,360 patent/US10966984B2/en active Active
- 2018-10-30 JP JP2018204234A patent/JP6767456B2/ja active Active
- 2018-11-01 US US16/178,419 patent/US10925878B2/en active Active
-
2019
- 2019-04-16 JP JP2019077828A patent/JP6738933B6/ja active Active
- 2019-12-11 CY CY20191101306T patent/CY1122434T1/el unknown
-
2020
- 2020-05-28 US US16/886,309 patent/US11040042B2/en active Active
- 2020-07-17 JP JP2020123189A patent/JP7250737B2/ja active Active
- 2020-09-16 JP JP2020155691A patent/JP7213854B2/ja active Active
-
2021
- 2021-02-22 US US17/181,638 patent/US11654148B2/en active Active
- 2021-04-05 US US17/222,873 patent/US11717523B2/en active Active
-
2023
- 2023-01-17 JP JP2023005413A patent/JP2023052449A/ja not_active Ceased
- 2023-03-06 US US18/117,983 patent/US20230364098A1/en active Pending
- 2023-03-20 JP JP2023044746A patent/JP2023078341A/ja not_active Withdrawn
- 2023-08-07 US US18/231,091 patent/US20230381189A1/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6337083B2 (ja) | 2013-03-15 | 2018-06-06 | ジー1、セラピューティクス、インコーポレイテッドG1 Therapeutics, Inc. | Rbポジティブ異常細胞増殖に対するhspc温存治療 |
| JP6738933B2 (ja) | 2013-03-15 | 2020-08-12 | ジー1、セラピューティクス、インコーポレイテッドG1 Therapeutics, Inc. | Rbポジティブ異常細胞増殖に対するhspc温存治療 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7250737B2 (ja) | Rbポジティブ異常細胞増殖に対するhspc温存治療 | |
| US20230241059A1 (en) | Anti-neoplastic combinations and dosing regimens using cdk4/6 inhibitor compounds to treat rb-positive tumors | |
| US12285431B2 (en) | Treatment of Rb-negative tumors using topoisomerase inhibitors in combination with cyclin dependent kinase 4/6 inhibitors | |
| HK1260112A1 (en) | Hspc-sparing treatments for rb-positive abnormal cellular proliferation | |
| HK1260112B (en) | Hspc-sparing treatments for rb-positive abnormal cellular proliferation | |
| HK1222792B (en) | Hspc-sparing treatments for rb-positive abnormal cellular proliferation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200727 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20200727 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20210713 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20211012 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220113 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20220506 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20220726 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20221107 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20230120 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20230216 |
|
| R155 | Notification before disposition of declining of application |
Free format text: JAPANESE INTERMEDIATE CODE: R155 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20230322 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7250737 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313111 Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| R371 | Transfer withdrawn |
Free format text: JAPANESE INTERMEDIATE CODE: R371 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313111 Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |