ES2931460T3 - Compuestos de pirazol sustituido como inhibidores de serina proteasa - Google Patents
Compuestos de pirazol sustituido como inhibidores de serina proteasa Download PDFInfo
- Publication number
- ES2931460T3 ES2931460T3 ES16756553T ES16756553T ES2931460T3 ES 2931460 T3 ES2931460 T3 ES 2931460T3 ES 16756553 T ES16756553 T ES 16756553T ES 16756553 T ES16756553 T ES 16756553T ES 2931460 T3 ES2931460 T3 ES 2931460T3
- Authority
- ES
- Spain
- Prior art keywords
- methyl
- pyrazol
- carbonyl
- chlorothiophen
- amino
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/4155—1,2-Diazoles non condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0048—Eye, e.g. artificial tears
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Abstract
Se proporcionan, entre otros, compuestos aromáticos multisustituidos útiles para la inhibición de trombina y/o calicreína, cuyos compuestos incluyen pirazolilo sustituido. También se proporcionan composiciones farmacéuticas. Además, se proporcionan métodos para tratar y prevenir ciertas enfermedades o trastornos, cuyas enfermedades o trastornos son susceptibles de tratamiento o prevención mediante la inhibición de la trombina y/o la calicreína. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Compuestos de pirazol sustituido como inhibidores de serina proteasa
Antecedentes de la invención
La presente descripción se refiere a compuestos, p. ej., determinados compuestos de pirazol sustituido que presentan actividad biológica, p. ej., acción inhibidora, frente a las serina proteasas, incluidas la trombina y la calicreína plasmática.
Las serina proteasas son una gran familia de enzimas con diversas funciones biológicas, siendo su característica común la presencia y función fundamental del resto de serina del sitio activo. Su función central es la escisión catalítica de sustratos de enlaces peptídicos a través de una tríada Ser, His, Asp dentro del sitio activo (Kraut, J. Annual Review of Biochemistry 1977, 46, 331-358). La presente descripción se refiere a compuestos, p. ej., compuestos de pirazolilo sustituido con heterocicloalquilo, que presentan actividad biológica, p. ej., acción inhibidora, frente a serina proteasas, incluida la trombina y diversas calicreínas.
En los sistemas de los mamíferos, las lesiones de los vasos sanguíneos provocan hemorragias que son tratadas por la cascada de la coagulación de la sangre. La cascada incluye las vías extrínsecas e intrínsecas que implica la activación de al menos 13 factores interconectados y una variedad de cofactores y otras proteínas reguladoras. Tras la lesión vascular, el factor VII plasmático interactúa con el factor tisular (TF, Tissue Factor) expuesto y el complejo de TF-fVIIa resultante inicia una serie compleja de acontecimientos. El factor Xa se produce directamente "en dirección 3'" del complejo TF-fVIIa y se amplifica de forma múltiple a través de la vía intrínseca. El FXa después sirve como catalizador para la formación de trombina (fIIa) que, a su vez, es el precursor directo de la fibrinólisis. El resultado es un coágulo fibrinolítico que detiene el sangrado. La fibrinólisis del coágulo polimérico en monómeros de fibrina conduce a la disolución y al retorno del sistema al estado previo al coágulo. La cascada es un equilibrio complejo de factores y cofactores, y está muy bien regulada.
En las patologías, la regulación positiva o negativa no deseada de cualquier factor conduce a afecciones tales como hemorragia o trombosis. Históricamente, se han utilizado anticoagulantes en pacientes con riesgo de padecer complicaciones trombóticas, tales como angina, accidente cerebrovascular e infarto de miocardio. La warfarina ha sido dominante como anticoagulante terapéutico de primera línea. Desarrollado en la década de los 40 del siglo pasado, es un antagonista de la vitamina K e inhibe los factores II, VII, IX y X, entre otros. Se administra por vía oral, pero su facilidad de uso se ve atenuada por otros efectos: tiene una semivida muy larga (> 2 días) y tiene interacciones farmacológicas graves. Es importante destacar que, dado que la vitamina K es un cofactor ubicuo dentro de la cascada de la coagulación, el antagonismo produce la inhibición simultánea de muchos factores de la coagulación y, por lo tanto, puede provocar complicaciones hemorrágicas significativas.
Se ha prestado mucha atención a la heparina, el polisacárido natural que activa la AT III, el inhibidor endógeno de muchos de los factores en la cascada de la coagulación. La necesidad de la administración parenteral de los productos terapéuticos derivados de la heparina y los inconvenientes requisitos de una estrecha supervisión de la warfarina disponible por vía oral han impulsado el descubrimiento y desarrollo de fármacos disponibles por vía oral con amplios márgenes terapéuticos en cuanto a seguridad y eficacia.
De hecho, la posición de la trombina en la cascada de la coagulación la ha convertido en una diana popular para el descubrimiento de fármacos. La trombina es una proteína central en el proceso de coagulación, que se activa y amplifica con la lesión vascular. La generación de trombina provoca una cascada de varios factores en la cascada de la coagulación, depositando finalmente fibrina, el marco para un coágulo. El coágulo provoca el cese del episodio hemorrágico que acompaña a la lesión vascular. La trombina y las proteínas asociadas finalmente provocan la disolución del coágulo a través de la “fibrinólisis” , devolviendo el sistema al estado anterior a la lesión. En un estado “ normal” de lesión, se desea esta generación de trombina y este depósito de coágulos. En una patología, no se desea la deposición de coágulos. Los episodios trombóticos generales son el resultado clínico de la deposición y acumulación de coágulos en las arterias, venas o dentro del corazón. La rotura final de la estructura del coágulo acumulado en el sistema vascular hace que el coágulo se desplace al cerebro y/o los pulmones, lo que provoca un accidente cerebrovascular, infarto de miocardio (ataque al corazón), embolia pulmonar, parálisis y la consiguiente muerte. Se ha demostrado en la bibliografía que los compuestos que inhiben la trombina son útiles como anticoagulantes in vitro e in vivo, y han demostrado cubrir en la clínica de pacientes una necesidad médica fundamental. Se puede encontrar un análisis exhaustivo de la trombina y sus funciones en el proceso de coagulación en una variedad de referencias, incluidas las siguientes que se incorporan en la presente memoria por referencia en su totalidad y para todos los fines: Wieland, H. A., y col., 2003, Curr Opin Investig Drugs, 4 : 264-71; Gross, P. L. y Weitz, J. I., 2008, Arterioscler Thromb Vasc Biol, 28: 380-6; Hirsh, J., y col., 2005, Blood, 105: 453-63; Prezelj, A., y col., 2007, Curr Pharm Des, 13: 287-312. Sin pretender limitarse más a una teoría en particular, se cree que el uso de inhibidores directos de la trombina (IDT) tiene muy buenos precedentes, tal como los anticoagulantes a base de hirudina, y por lo tanto, existe un gran interés en el descubrimiento y desarrollo de nuevos IDT, particularmente aquellos con selectividad para inhibir la trombina frente a otras serina proteasas relacionadas. Las calicreínas son un subgrupo de serina proteasas, divididas en calicreína plasmática y calicreína tisular. La calicreína
plasmática (KLKB1) libera cininas (bradicinina y calidina) de los cininógenos, péptidos responsables de la regulación de la presión arterial y la activación de la inflamación. En la vía de activación por contacto de la cascada de la coagulación, la calicreína plasmática ayuda en la conversión del factor XII en factor XIIa (Keel, M.; Trentz, O. Injury 2005, 36, 691-709). El factor XIIa convierte el factor XI en factor XIa, que a su vez activa el factor IX, que, con su cofactor VIIIa, forma el complejo de tenasa, que finalmente activa el factor X en factor Xa. En la parte de la fibrinólisis de la cascada de la coagulación, la calicreína plasmática sirve para convertir el plasminógeno en plasmina. Por lo tanto, se ha propuesto que los inhibidores de la calicreína plasmática pueden ser útiles en el tratamiento de enfermedades y afecciones trombóticas y fibrinolíticas (patente de EE. UU. n.° 7.625.944; Bird y col. Thrombosis and Hemostasis 2012, 107, Dhaval Kolte, M. D. y col., Cardiology in Review, 2015).
Las calicreínas tisulares (KLK, por ejemplo, KLK1) se subdividen en varios tipos y se han investigado ampliamente en la biología del cáncer y la inflamación. Se ha encontrado que varios KLK de calicreína están regulados positiva o negativamente en varios tipos de cáncer, tales como el adenocarcinoma de pulmón no microcítico, testicular y cervicouterino (Caliendo y col. J. Med. Chem., 2012, 55, 6669). Además, la sobreexpresión de varias KLK en la piel ha conducido al reconocimiento de que determinados inhibidores de la calicreína pueden ser útiles para determinadas afecciones dermatológicas, tales como la dermatitis atópica, la psoriasis y enfermedades raras de la piel tales como el síndrome de Netherton (Freitas y col. Bioorganic & Medicinal Chemistry Letters 2012, 22, 6072 6075). Se puede encontrar un análisis exhaustivo de las calicreínas tisulares, la calicreína plasmática, sus funciones y posibles papeles en varias enfermedades en una variedad de referencias, incluidas las siguientes que se incorporan en la presente memoria por referencia en su totalidad y para todos los fines: Renné, T.; Gruber, A. Thromb Haemost 2012, 107, 1012-3; Sotiropoulou, G.; Pampalakis, G. Trends in Pharmacological Sciences 2012, 33, 623-634; Pampalakis, G.; Sotiropoulou, G., Capítulo 9: “ Pharmacological Targeting of Human Tissue Kallikrein-Related Peptidases” . En Proteinases as Drug Targets, Dunn, B., Ed. The Royal Society of Chemistry: 2012; pág.
199-228; Caliendo, G.; Santagada, V.; Perissutti, E.; Severino, B.; Fiorino, F.; Frecentese, F.; Juliano, L. J Med Chem 2012, 55, 6669-86.
Breve sumario de la invención
Las realizaciones de la invención engloban compuestos con la siguiente estructura:

o una sal, un éster, solvato o profármaco farmacéuticamente aceptable de los mismos, en donde:
L1, L2 y L4 son, independientemente entre sí, un enlace, alquileno sustituido o no sustituido, heteroalquileno sustituido o no sustituido, -C(O)-, -S-, -SO-, -SO2-, -O-, -NHSO2-, -NHC(O)- o -NR5-;
R1, R2 y R4 son, independientemente entre sí, hidrógeno, halógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, o heteroarilo sustituido o no sustituido;
R5 es hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, o heteroarilo sustituido o no sustituido;
V es hidrógeno o alquilo sustituido o no sustituido;
W está ausente, o es hidrógeno, halógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, heteroarilo sustituido o no sustituido, - C(O)R6, -C(O)OR 6, -
C(O)NR6R7, -SR6 SOR6, -SO2 R0, -SO2NR6R', -OR6 -NHSO2 R6 o -NR6R7, donde R6 y R7 son, independientemente entre sí, hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o
no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido, en donde R6 y R7 se pueden combinar, si ambos están presentes, para formar un alquileno sustituido o no sustituido, o heteroalquileno sustituido o no sustituido;
X es un enlace, alquileno sustituido o no sustituido, -O- o -NR8-;
Y es un enlace, alquileno sustituido o no sustituido, -O- o -N-, siempre que cuando Y sea -O-, W esté ausente; y
Z es un enlace, -C(O)-, alquileno sustituido o no sustituido, -O- o -NR9-;
en donde R8 y R9 son, independientemente entre sí, hidrógeno, halógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, heteroarilo sustituido o no sustituido, -C(O)R6, -C(O)OR6, -
C(O)NR6R7, -SR6, -SOR6 -SO2R6 -SO2NR6R7, -OR6, -NHSO2 R6 o -n r 6r 7, en donde R6 y R7 son como se han definido anteriormente; y
siempre que al menos uno de X sea -O- o -NR8-, Y sea -O- o -N-, o Z sea -O- o -NR9
En algunas realizaciones, el compuesto puede ser una sal, un éster, un solvato o un profármaco farmacéuticamente aceptable de un compuesto de Fórmula (IV). En algunas realizaciones, el compuesto no es un éster ni un solvato ni un profármaco.
En algunas realizaciones, X puede ser un enlace, o un alquileno sustituido o no sustituido. En algunas realizaciones, Z puede ser un enlace, o un alquileno sustituido o no sustituido. En algunas realizaciones, X puede ser un enlace, o alquileno sustituido o no sustituido, y Z puede ser un enlace, o alquileno sustituido o no sustituido.
Algunas realizaciones incluyen compuestos donde X es un enlace, o alquileno sustituido o no sustituido, y Z es un enlace, o alquileno sustituido o no sustituido, Y puede ser -N- y W puede ser hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, arilo sustituido o no sustituido,-C(O)R6, -C(O)OR6, - C(O)NR6R7, -SO2 R6 o -SO2NR6R7, en donde R6 y R7 pueden ser, independientemente entre sí, alquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido, o R6 y R7 se pueden combinar, si ambos están presentes, para formar un alquileno sustituido o no sustituido, o heteroalquileno sustituido o no sustituido.
Algunas realizaciones incluyen compuestos en donde X puede ser metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido, o pentileno sustituido o no sustituido, y Z puede ser un enlace. En algunas realizaciones, X puede ser un enlace y Z puede ser metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido, o pentileno sustituido o no sustituido. En algunas realizaciones, X puede ser metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido, o pentileno sustituido o no sustituido, y Z puede ser metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido, o pentileno sustituido o no sustituido. En algunas realizaciones, tanto X como Z pueden ser alquileno ramificado, y X y Z pueden estar unidos covalentemente. En algunas realizaciones, Z puede ser metileno sustituido, etileno sustituido, propileno sustituido, butileno sustituido o pentileno sustituido, que tiene uno o más grupos sustituyentes que pueden ser -OH, -NH2 , -SH, -CN, -CF3 , -NO2 , oxo, halógeno, -COOH, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido.
Algunas realizaciones incluyen compuestos en donde X puede ser metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido, o pentileno sustituido o no sustituido, y Z puede ser -C(O)-. En algunas realizaciones, W puede ser hidrógeno. En algunas realizaciones, W puede ser alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, -C(O)R6 , -C(O)OR6 , - C(O)NR6 R7 , -SR6 , -SOR6 , -SO2 R6 o -SO2NR6. En algunas realizaciones, W puede ser alquilo sustituido, heteroalquilo sustituido, alquenilo sustituido, heteroalquenilo sustituido, cicloalquilo sustituido o heterocicloalquilo sustituido, que tiene uno o más sustituyentes que pueden ser -OH, -NH2 , -SH, -CN, -CF3 , -NO2 , oxo, halógeno, -COOH, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido. En algunas realizaciones, W puede ser -COR6, -C(O)OR6, -C(O)NR R , -SO2 R o -SO2NR R , donde R y R se pueden seleccionar del grupo que consiste en alquilo sustituido
o no sustituido, heteroalquilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido, o R6 y R7 se pueden combinar para formar un alquileno sustituido o no sustituido.
Algunas realizaciones incluyen compuestos en donde W puede estar ausente, X puede ser -NR8-, Y puede ser un enlace, o alquileno sustituido o no sustituido, y Z puede ser -NR9-. En algunas realizaciones en donde W puede estar ausente, X puede ser -NR8-, Y puede ser un enlace, o alquileno sustituido o no sustituido, y Z puede ser -NR9-, Y puede ser metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido, o pentileno sustituido o no sustituido. En algunas realizaciones, R8 puede ser alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, -COR6, -C(O)OR6, - C(O)NR6R7, -SR6, -SOR6, -SO2 R6 o -SO2 NR6R7, y en donde R9 puede ser alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, -COR6, -C(O)OR6, - C(O)NR6R7, -SR6, -SOR6, -SO2 R6 o -SO2NR6R7.
Algunas realizaciones incluyen compuestos en donde Y puede ser -O- y W puede estar ausente. En algunas realizaciones en donde Y puede ser -O- y W puede estar ausente, X puede ser metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido, o pentileno sustituido o no sustituido, y Z puede ser un enlace. En algunas realizaciones, X puede ser un enlace y Z puede ser metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido, o pentileno sustituido o no sustituido. En algunas realizaciones, X puede ser metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido, o pentileno sustituido o no sustituido, y en donde Z puede ser metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido, o pentileno sustituido o no sustituido.
En algunas realizaciones, V puede ser hidrógeno, o metilo sustituido o no sustituido.
En algunas realizaciones, L1 puede ser -S-, -O-, -NR5-, alquileno sustituido o no sustituido, o heteroalquileno sustituido o no sustituido; R1 puede ser alquilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, heteroarilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido; y R5 puede ser hidrógeno, alquilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido. En algunas realizaciones, L1 puede ser -NR5-, o heteroalquilo sustituido o no sustituido, y IR1 puede ser alquilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido. En algunas realizaciones, L1 puede ser -NR5-, y R1 puede ser alquilo sustituido que tiene uno o más grupos sustituyentes que pueden ser heteroarilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido. En algunas realizaciones, R1 puede ser alquilo sustituido con tiofenilo sustituido con cloro. En algunas realizaciones, L1 puede ser heteroalquilo sustituido o no sustituido, y R1 puede ser heteroarilo sustituido o no sustituido.
En algunas realizaciones, L2 puede ser enlace, alquileno sustituido o no sustituido, -C(O)- o -SO2-, y R2 puede ser hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, heteroarilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido. En algunas realizaciones, L2 puede ser enlace, y R2 es hidrógeno. En algunas realizaciones, L2 puede ser -C(O)-, y R2 puede ser alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, heteroarilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido.
En algunas realizaciones, L4 puede ser un enlace, y R4 puede ser hidrógeno, halógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido.
En algunas realizaciones, los compuestos pueden estar incluidos entre los expuestos en la Tabla A, Tabla B, Tabla C o Tabla D.
Las realizaciones de la invención también se refieren a composiciones farmacéuticas que comprenden uno o más compuestos como se ha expuesto anteriormente, o uno o más compuestos incluidos entre los expuestos en la Tabla A, Tabla B, Tabla C o Tabla D, y un excipiente farmacéuticamente aceptable.
Las realizaciones de la invención también incluyen métodos para tratar y/o prevenir una o más enfermedades o trastornos en un sujeto, que comprenden administrar un compuesto como se ha expuesto anteriormente, o una composición farmacéutica que incluye dicho compuesto, a un sujeto que lo necesita en una cantidad eficaz para tratar o prevenir dicha(s) enfermedad(es) o trastorno(s).
En algunas realizaciones de los métodos descritos en la presente memoria, la enfermedad o el trastorno que se va a tratar puede incluir una o más enfermedades o trastornos trombóticos y/o puede implicar un trombo de coágulo sanguíneo o la posible formación de un trombo de coágulo sanguíneo. En algunas realizaciones, la enfermedad o el
trastorno trombótico puede ser síndrome coronario agudo, tromboembolia y/o trombosis. En algunas realizaciones, la tromboembolia puede ser tromboembolia venosa, tromboembolia arterial y/o tromboembolia cardiogénica. En algunas realizaciones, la tromboembolia venosa puede incluir trombosis venosa profunda y/o embolia pulmonar. En algunas realizaciones, la trombosis venosa profunda y/o la embolia pulmonar pueden ocurrir después de una intervención médica. En algunas realizaciones, la enfermedad o el trastorno trombótico puede implicar coagulación disfuncional o coagulación intravascular diseminada. En algunas realizaciones, el sujeto con coagulación disfuncional puede someterse a una Intervención Coronaria Percutánea (ICP). En algunas realizaciones, la enfermedad o el trastorno trombótico puede implicar un trombo de coágulo sanguíneo o la posible formación de un trombo de coágulo sanguíneo, y además puede implicar accidente cerebrovascular y/o uno o más ataques isquémicos transitorios (AIT). En algunas realizaciones, la enfermedad o el trastorno trombótico que implica un trombo de coágulo sanguíneo o la posible formación de un trombo de coágulo sanguíneo puede implicar además un accidente cerebrovascular, en donde el sujeto puede tener fibrilación auricular no valvular. En algunas realizaciones, la enfermedad o el trastorno trombótico puede implicar un trombo de coágulo sanguíneo o la posible formación de un trombo de coágulo sanguíneo y además puede implicar hipertensión pulmonar. En algunas realizaciones, la hipertensión pulmonar puede estar provocada por uno o más trastornos de las cavidades izquierdas del corazón y/o enfermedad tromboembólica crónica. En algunas realizaciones, la hipertensión pulmonar puede estar asociada con una o más enfermedades pulmonares, incluida la fibrosis pulmonar (idiopática o de otro tipo) y/o hipoxia.
En algunas realizaciones, la tromboembolia venosa puede estar asociada con la formación de un trombo dentro de una vena asociado con uno o más factores de riesgo adquiridos o heredados y/o la embolia de venas periféricas provocada por un trombo desprendido. En algunas realizaciones, uno o más factores de riesgo pueden incluir una tromboembolia venosa previa. En algunas realizaciones, la tromboembolia cardiogénica puede deberse a la formación de un trombo en el corazón asociado con arritmia cardíaca, defecto de válvula cardíaca, válvulas cardíacas protésicas o cardiopatía y/o embolia de arterias periféricas provocada por un trombo desprendido. En algunas realizaciones, el trombo desprendido puede estar en el cerebro (accidente cerebrovascular isquémico). En algunas realizaciones, el trombo desprendido puede provocar un ataque isquémico transitorio (AIT). En algunas realizaciones, la tromboembolia cardiogénica puede deberse a una fibrilación auricular no valvular. En algunas realizaciones, la trombosis puede ser una trombosis arterial. En algunas realizaciones, la trombosis arterial puede deberse a uno o más procesos ateroescleróticos subyacentes en las arterias. En algunas realizaciones, uno o más procesos ateroescleróticos subyacentes en las arterias pueden obstruir u ocluir una arteria, provocar isquemia miocárdica (angina de pecho, síndrome coronario agudo), provocar infarto de miocardio, obstruir u ocluir una arteria periférica (enfermedad arterial periférica isquémica), y/u obstruir u ocluir la arteria después de una operación en un vaso sanguíneo (reoclusión o reestenosis después de angioplastia coronaria transluminal, reoclusión o reestenosis después de angioplastia transluminal percutánea de arterias periféricas).
En algunas realizaciones, la enfermedad o el trastorno puede incluir fibrosis, enfermedad de Alzheimer, esclerosis múltiple, dolor, cáncer, inflamación y/o diabetes mellitus de tipo I. En algunas realizaciones, la enfermedad o el trastorno puede implicar episodios cardíacos recurrentes después de un infarto de miocardio.
En algunas realizaciones, el tratamiento o la prevención pueden incluir una terapia adyuvante. En algunas realizaciones, el sujeto puede tener un infarto de miocardio y la terapia adyuvante puede ser junto con la terapia trombolítica. En algunas realizaciones, el sujeto puede tener angina de pecho inestable, trombosis y/o trombocitopenia inducida por heparina, y la terapia adyuvante puede estar junto con tratamiento antiplaquetario. En algunas realizaciones, el sujeto puede tener fibrilación auricular no valvular, y la terapia adyuvante puede darse junto con una o más terapias diferentes.
En algunas realizaciones de los métodos descritos en la presente memoria, la enfermedad o el trastorno puede ser un trastorno relacionado con la calicreína. En algunas realizaciones, el trastorno relacionado con la calicreína puede ser una enfermedad trombótica, una enfermedad fibrinolítica, un trastorno fibrótico, un tipo de cáncer, una afección inflamatoria o una afección dermatológica.
En algunas realizaciones, el trastorno relacionado con la calicreína puede ser una enfermedad oftálmica. En algunas realizaciones, el compuesto o la composición farmacéutica se puede administrar en forma de una composición oftálmica aplicada por vía tópica en el ojo. En algunas realizaciones, la composición oftálmica puede estar en forma de colirio. En algunas realizaciones, el compuesto o la composición farmacéutica se puede administrar en forma de una composición oftálmica mediante inyección intravítrea. En algunas realizaciones, la enfermedad oftálmica puede ser edema macular diabético, angioedema hereditario, degeneración macular relacionada con la edad o retinopatía diabética.
En algunas realizaciones en donde la enfermedad o el trastorno puede ser un tipo de cáncer, dicho tipo de cáncer puede ser adenocarcinoma de pulmón no microcítico, de cuello uterino o testicular. En algunas realizaciones, el cáncer puede ser cáncer de pulmón microcítico limitado. En algunas realizaciones, el cáncer puede ser un glioma. En algunas realizaciones, el cáncer puede ser cáncer de mama maligno. En algunas realizaciones, el cáncer puede ser una micrometástasis. En algunas realizaciones, la micrometástasis puede ser de la sangre o del hígado. En algunas realizaciones, el cáncer puede ser una metástasis de pulmón. En algunas realizaciones, el cáncer puede ser cáncer de próstata.
En algunas realizaciones en donde la enfermedad o el trastorno puede ser una afección inflamatoria, dicha afección inflamatoria puede ser sepsis, enfermedad intestinal inflamatoria, síndrome de respuesta inflamatoria sistémica, artritis inflamatoria o artritis reumatoide.
En algunas realizaciones en donde la enfermedad o el trastorno puede ser una afección dermatológica, dicha afección dermatológica puede ser dermatitis atópica, psoriasis o síndrome de Netherton.
En algunas realizaciones, el compuesto puede actuar inhibiendo la trombina y/o la calicreína. En algunas realizaciones, el compuesto puede actuar inhibiendo la calicreína tisular y/o la calicreína plasmática. En algunas realizaciones, el compuesto puede tener actividad inhibidora contra la trombina y/o la calicreína plasmática dentro de un intervalo de 1-10 nM, 10-100 nM, 0,1-1 pM, 1-10 pM, 10-100 pM, 100-200 pM, 200-500 pM o 500-1000 pM, o mayor.
En algunas realizaciones, la cantidad de compuesto administrado puede ser una dosis terapéuticamente eficaz suficiente para alcanzar una concentración plasmática del compuesto o su(s) metabolito(s) activo(s) dentro de un intervalo de 1-10 nM, 10-100 nM, 0,1-1 pM, 1-10 pM, 10-100 pM, 100-200 pM, 200-500 pM o 500-1000 pM, o mayor.
Las realizaciones de la invención también se refieren a compuestos o composiciones farmacéuticas como se describe en la presente memoria, para su uso en métodos para tratar y/o prevenir una o más enfermedades o trastornos en un sujeto, como se describe en la presente memoria.
Breve descripción de los dibujos
No procede.
Descripción detallada de la invención
I. Definiciones
Las abreviaturas utilizadas en la presente memoria tienen su significado convencional dentro de las técnicas químicas y biológicas. Las estructuras y fórmulas químicas expuestas en la presente memoria se construyen según las reglas convencionales de valencias químicas conocidas en las técnicas químicas.
Cuando los grupos sustituyentes se especifican mediante sus fórmulas químicas convencionales, escritas de izquierda a derecha, también engloban los sustituyentes químicamente idénticos que resultarían de escribir la estructura de derecha a izquierda, por ejemplo, -CH2O- es equivalente a -OCH2-.
Como se utiliza en la presente memoria, el término “unido” significa un enlace covalente estable, siendo evidentes para los expertos en la técnica determinados puntos preferidos de unión.
Los términos “ halógeno” o “ halo” incluyen flúor, cloro, bromo y yodo. Además, se pretende que términos tales como “ haloalquilo” incluyan monohaloalquilo y polihaloalquilo. Por ejemplo, el término “ haloalquilo (C1-C4)” incluye, pero no se limita a, fluorometilo, difluorometilo, trifluorometilo, 2,2,2-trifluoroetilo, 4-clorobutilo, 3-bromopropilo y similares.
El término “alquilo” , solo o como parte de otro sustituyente, significa, salvo que se indique lo contrario, una cadena lineal (es decir, no ramificada) o ramificada, o una combinación de las mismas, que puede estar completamente saturada, monoinsaturada o poliinsaturada y que puede incluir radicales divalentes y multivalentes, que tienen el número de átomos de carbono designado (es decir, C1-C10 significa de uno a diez carbonos). Los ejemplos de radicales de hidrocarburos saturados incluyen, aunque no de forma limitativa, grupos tales como metilo, etilo, npropilo, /so-propilo, n-butilo, f-butilo, /so-butilo, sec-butilo, (ciclohexil)metilo, homólogos e isómeros de, por ejemplo, n-pentilo, n-hexilo, n-heptilo, n-octilo, y similares. Un grupo alquilo insaturado es aquel que tiene uno o más dobles o triples enlaces. Los ejemplos de grupos alquilo insaturados incluyen, aunque no de forma limitativa, vinilo, 2-propenilo, crotilo, 2-isopentenilo, 2-(butadienilo), 2,4-pentadienilo, 3-(1,4-pentadienilo), etinilo, 1- y 3-propinilo, 3-butinilo y los homólogos e isómeros superiores. Por consiguiente, el término “ alquilo” puede referirse a grupos de hidrocarburo C1-C16 de cadena lineal saturados, C1-C16 ramificados saturados, C3-C8 cíclicos saturados, C3-C8 cíclicos insaturados y C1-C16 de cadena lineal o ramificados alifáticos saturados o insaturados sustituidos con grupos de hidrocarburo C3-C8 cíclicos alifáticos saturados o insaturados que tienen el número especificado de átomos de carbono, y similares. Los ejemplos de grupos alquilo cíclicos incluyen, aunque no de forma limitativa, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, ciclooctilo, ciclopropilmetilo y similares.
El término “ alquileno” , por sí mismo o como parte de otro sustituyente, significa, salvo que se indique lo contrario, un radical divalente derivado de un alquilo saturado o insaturado, como se ha definido anteriormente y como se ilustra, aunque no de forma limitativa, mediante -CH2CH2CH2CH2-, y similares. Normalmente, un grupo alquilo (o alquileno) tendrá de 1 a 24 átomos de carbono, prefiriéndose aquellos grupos que tienen 10 átomos de carbono o menos en
los compuestos descritos en la presente memoria. Un “ alquilo inferior” o “alquileno inferior” es un grupo alquilo o alquileno de cadena más corta, que generalmente tiene ocho átomos de carbono o menos.
El término “ heteroalquilo” , solo o junto con otro término, significa, salvo que se indique lo contrario, una cadena lineal o ramificada estable, o combinaciones de las mismas, que consiste en al menos un átomo de carbono y al menos un heteroátomo seleccionado del grupo que consiste en O, N, P, Si y S, y en donde los átomos de nitrógeno y azufre pueden oxidarse opcionalmente y el heteroátomo de nitrógeno puede cuaternizarse opcionalmente. Los heteroátomos O, N, P, S y Si se pueden colocar en cualquier posición interior del grupo heteroalquilo o en la posición en donde el grupo alquilo está unido al resto de la molécula. El grupo heteroalquilo puede estar completamente saturado, monoinsaturado o poliinsaturado, y puede incluir radicales divalentes y multivalentes, con el número de átomos designado. Por consiguiente, el término “ heteroalquilo” puede referirse a cadenas lineales o ramificadas saturadas o insaturadas que contienen de 2 a 16 átomos a lo largo de la cadena, grupos cíclicos saturados o insaturados que contienen de 3 a 8 átomos en el ciclo, y similares. Los ejemplos incluyen, aunque no de forma limitativa: -CH2-CH2-O-CH3 , -CH2-CH2-NH-CH3 , -CH2-CH2-N(CH3)-CH3 , -CH2-S-CH2-CH 3 , -CH2-CH2 , -S(O)-CH3 , -CH2-CH2-S(O)2-CH3 , -CH=CH-O-CH3 , -Si(CH3)3 , -CH2-C H=N-OCH3 , -CH=CH-N(CH3)-CH3 , -O-CH3 , -O-CH2-CH3 , -CN, y similares. Pueden ser consecutivos hasta dos heteroátomos, tal como, por ejemplo, -CH2-NH-OCH3.
De manera similar, el término “ heteroalquileno” , por sí mismo o como parte de otro sustituyente, significa, salvo que se indique lo contrario, un radical divalente derivado de heteroalquilo, como se ha definido anteriormente y como se ilustra, aunque no de forma limitativa, mediante -CH2-CH2-S-CH2-CH2- y -CH2-S-CH2-CH2-NH-CH2-, y similares. Para los grupos heteroalquileno, los heteroátomos también pueden ocupar uno o ambos extremos de la cadena (p. ej., alquilenoxi, alquilendioxi, alquilenamino, alquilendiamino y similares). Aún más, para los grupos enlazadores alquileno y heteroalquileno, la dirección en la que se escribe la fórmula del grupo enlazador no implica ninguna orientación del grupo enlazador. Por ejemplo, la fórmula -C(O)2R'- representa tanto -C(O)2R'- como -R'C(O)2-. Como se ha descrito anteriormente, los grupos heteroalquilo, como se utilizan en la presente memoria, incluyen aquellos grupos que están unidos al resto de la molécula a través de un heteroátomo, tal como -C(O)R', -C(O)NR', -NR'R", -OR, -SR' y/o -SO2R'. Cuando se menciona “ heteroalquilo” , seguido de enumeraciones de grupos heteroalquilo específicos, tales como -NR'R" o similares, se entenderá que los términos heteroalquilo y -NR'R" no son redundantes ni excluyentes entre sí. En cambio, los grupos heteroalquilo específicos se enumeran para añadir claridad. Por lo tanto, el término “ heteroalquilo” no debe interpretarse en la presente memoria como excluyente de grupos heteroalquilo específicos, tales como -NR'R" o similares.
Los términos “cicloalquilo” y “ heterocicloalquilo” , solos o junto con otros términos, significan, salvo que se indique lo contrario, versiones cíclicas de “ alquilo” y “ heteroalquilo” , respectivamente. Los grupos “cicloalquilo” y “ heterocicloalquilo” incluyen, por ejemplo, anillos monocíclicos que tienen de 3 a 8 miembros en el anillo, así como anillos bicíclicos que tienen de 4 a 16 miembros en el anillo, anillos tricíclicos que tienen de 5 a 24 miembros en el anillo, etc. Además, para heterocicloalquilo, un heteroátomo puede ocupar la posición en la que el heterociclo está unido al resto de la molécula. Los ejemplos de cicloalquilo incluyen, aunque no de forma limitativa, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, 1-ciclohexenilo, 3-ciclohexenilo, cicloheptilo y similares. Los ejemplos de heterocicloalquilo incluyen, aunque no de forma limitativa, 1 -(1,2,5,6-tetrahidropiridilo), 1 -piperidinilo, 2-piperidinilo, 3-piperidinilo, 4-morfolinilo, 3-morfolinilo, tetrahidrofuran-2-ilo, tetrahidrofuran-3-ilo, tetrahidrotien-2-ilo, tetrahidrotien-3-ilo, 1 -piperazinilo, 2-piperazinilo y similares. Un “cicloalquileno” y un “ heterocicloalquileno” , solos o como parte de otro sustituyente, significan un radical divalente derivado de un cicloalquilo y un heterocicloalquilo, respectivamente.
El término “alquenilo” incluye grupos de hidrocarburo C2-C16 de cadena lineal insaturados, C2-C11 ramificados insaturados, C5-C8 cíclicos insaturados, C2-C16 de cadena lineal o ramificados alifáticos insaturados sustituidos con grupos de hidrocarburo C3-C8 cíclicos alifáticos saturados o insaturados que tienen el número especificado de átomos de carbono, y similares. Los dobles enlaces pueden ocurrir en cualquier punto estable a lo largo de la cadena y los dobles enlaces carbono-carbono pueden tener la configuración cis o trans. Por ejemplo, esta definición incluirá, pero no se limitará a, etenilo, propenilo, butenilo, pentenilo, hexenilo, heptenilo, octenilo, nonenilo, decenilo, undecenilo, 1,5-octadienilo, 1,4,7-nonatrienilo, ciclopentenilo, ciclohexenilo, cicloheptenilo, ciclooctenilo, etilciclohexenilo, butenilciclopentilo, 1-pentenil-3-ciclohexenilo y similares. De manera similar, “ heteroalquenilo” se refiere a heteroalquilo que tiene uno o más dobles enlaces, en donde heteroalquilo es como se ha definido anteriormente.
El término “alquinilo” se refiere en el sentido habitual a alquilo, como se ha definido anteriormente, que tiene además uno o más triples enlaces. El término “cicloalquenilo” se refiere a cicloalquilo, como se ha definido anteriormente, que tiene además uno o más dobles enlaces. El término “ heterocicloalquenilo” se refiere a heterocicloalquilo, como se ha definido anteriormente, que tiene además uno o más dobles enlaces.
El término “acilo” significa, salvo que se indique lo contrario, -C(O)R donde R es un alquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido.
El término “arilo” significa, salvo que se indique lo contrario, un sustituyente de hidrocarburo aromático, poliinsaturado, que puede ser un solo anillo o varios anillos (preferiblemente de 1 a 3 anillos) que están
condensados entre sí (es decir, un arilo de anillo condensado) o enlazados covalentemente, en donde cada anillo contiene entre 4 y 20 átomos, y preferiblemente entre 5 y 10 átomos. Un arilo de anillo condensado se refiere a varios anillos condensados entre sí, en donde al menos uno de los anillos condensados es un anillo de arilo. El término “ heteroarilo” se refiere a grupos (o anillos) de arilo, como se ha definido anteriormente, que contienen de uno a cuatro heteroátomos seleccionados de N, O y S, en donde los átomos de nitrógeno y azufre están opcionalmente oxidados, y el átomo o átomos de nitrógeno) están opcionalmente cuaternizados. Por lo tanto, el término “ heteroarilo” incluye grupos heteroarilo de anillos condensados (es decir, varios anillos condensados entre sí en donde al menos uno de los anillos condensados es un anillo heteroaromático). Un heteroarileno de anillos condensados en 5,6 se refiere a dos anillos condensados, en donde un anillo tiene 5 miembros y el otro anillo tiene 6 miembros, y en donde al menos un anillo es un anillo de heteroarilo. Del mismo modo, un heteroarileno de anillos condensados en 6,6 se refiere a dos anillos condensados, en donde un anillo tiene 6 miembros y el otro anillo tiene 6 miembros, y en donde al menos un anillo es un anillo de heteroarilo. Y un heteroarileno de anillos condensados en 6,5 se refiere a dos anillos condensados, en donde un anillo tiene 6 miembros y el otro anillo tiene 5 miembros, y en donde al menos un anillo es un anillo de heteroarilo. Un grupo heteroarilo se puede unir al resto de la molécula a través de un carbono o un heteroátomo. Los ejemplos no limitantes de grupos arilo y heteroarilo incluyen fenilo, 1-naftilo, 2-naftilo, 4-bifenilo, 1 -pirrolilo, 2-pirrolilo, 3-pirrolilo, 3-pirazolilo, 2-imidazolilo, 4-imidazolilo, pirazinilo, 2-oxazolilo, 4-oxazolilo, 2-fenil-4-oxazolilo, 5-oxazolilo, 3-isoxazolilo, 4-isoxazolilo, 5-isoxazolilo, 2-tiazolilo, 4-tiazolilo, 5-tiazolilo, 2-furilo, 3-furilo, 2-tienilo, 3-tienilo, 2-piridilo, 3-piridilo, 4-piridilo, 2-pirimidilo, 4-pirimidilo, 5-benzotiazolilo, purinilo, 2-bencimidazolilo, 5-indolilo, 1 -isoquinolilo, 5-isoquinolilo, 2-quinoxalinilo, 5-quinoxalinilo, 3-quinolilo y 6-quinolilo, y similares. Los sustituyentes para cada uno de los sistemas de anillos de arilo y heteroarilo indicados anteriormente se seleccionan del grupo de sustituyentes aceptables que se describen a continuación. Un “ arileno” y un “ heteroarileno” , solos o como parte de otro sustituyente, significan un radical divalente derivado de un arilo y heteroarilo, respectivamente. Por consiguiente, el término “arilo” puede representar grupos aromáticos monocíclicos, policíclicos, biarílicos y heterocíclicos no sustituidos, mono-, di- o tri-sustituidos, unidos covalentemente en cualquier posición del anillo capaz de formar un enlace covalente estable, siendo evidentes determinados puntos de unión preferidos para los expertos en la materia (p. ej., 3-indolilo, 4-imidazolilo). Los sustituyentes arilo se seleccionan, independientemente entre sí, del grupo que consiste en halo, nitro, ciano, trihalometilo, alquilo C1-16, arilalquilo C1-16, alquiloxi Cü-16-alquilo C0-16, arilalquiloxi Cü-16-alquilo C0-16, alquiltio Cü-16-alquilo C0-16, arilalquiltio Cü-16-alquilo C0-16, alquilamino Cü-16-alquilo C0-16, arilalquilamino Cü-16-alquilo C0-16, di(arilalquil C1-16)amino-alquilo C0-16, alquilcarbonil C1-16-alquilo C0-16, arilalquilcarbonil C1-16-alquilo C0-16, alquilcarboxi C1-16-alquilo C0-16, arilalquilcarboxi C1-16-alquilo C0-16, alquilcarbonilamino C1-16-alquilo C0-16, arilalquilcarbonilamino C1-16-alquilo C0-16, -alquil C0-16-COOR4 , alquil C0-16-CONR5 R6 , en donde R4 , R5 y R6 se seleccionan, independientemente entre sí, de hidrógeno, alquilo C1-C11, arilalquilo C0-C11, o R5 y R6 se toman junto con el nitrógeno al que están unidos formando un sistema cíclico que contiene de 3 a 8 átomos de carbono con o sin un sustituyente alquilo C1-16, arilalquilo C0-C16 o alquilarilo C0-C16. Arilo incluye, pero no se limita a, pirazolilo y triazolilo.
Para abreviar, el término “arilo” cuando se utiliza junto con otros términos (p. ej., ariloxi, ariltioxi, arilalquilo) incluye tanto anillos arilo tales como heteroarilo como se ha definido anteriormente. Por lo tanto, los términos “arilalquilo” , “aralquilo” y similares incluyen aquellos radicales en donde un grupo arilo está unido a un grupo alquilo (p. ej., bencilo, fenetilo, piridilmetilo y similares), incluidos aquellos grupos alquilo en donde un átomo de carbono (p. ej., un grupo metileno) ha sido reemplazado, por ejemplo, por un átomo de oxígeno (p. ej., fenoximetilo, 2-piridiloximetilo, 3-(1-naftiloxi)propilo y similares), o un átomo de azufre. Por consiguiente, los términos “ arilalquilo” y similares (p. ej., (4-hidroxifenil)etilo, (2-aminonaftil)hexilo, piridilciclopentilo) representan un grupo arilo como se ha definido anteriormente unido a través de un grupo alquilo como se ha definido anteriormente que tiene el número indicado de átomos de carbono.
Cada uno de los términos anteriores (p. ej., “ alquilo” , “ heteroalquilo” , “arilo” y “ heteroarilo” ) incluye formas sustituidas y no sustituidas del radical indicado. Los sustituyentes preferidos para cada tipo de radical se proporcionan en la presente memoria.
Los sustituyentes para los radicales alquilo y heteroalquilo (incluidos los grupos a menudo denominados alquileno, alquenilo, heteroalquileno, heteroalquenilo, alquinilo, cicloalquilo, heterocicloalquilo, cicloalquenilo y heterocicloalquenilo) pueden ser uno o más de una variedad de grupos seleccionados, aunque no de forma limitativa, de -OR', =O, =NR', =N-OR', -NR'R", -SR', -halógeno, -SiR'R"R"', -OC(O)R', -C(O)R', -CO2 R', -CONR'R", -OC(O)NR'R", -NR"C(O)R', -NR'-C(O)NR"R"', -NR"C(O)2 R', -NR-C(NR'R")=NR"', - S(O)R', -S(O)2 R', -S(O)2NR'R", -NRSO2 R', -CN y -NO2 en un número que varía de cero a (2m'+1), donde m' es el número total de átomos de carbono en dicho radical. Cada uno de R', R" y R"' se refiere preferiblemente, independientemente entre sí, a hidrógeno, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido (p. ej., arilo sustituido con 1-3 halógenos), grupos alquilo, alcoxi o tioalcoxi sustituidos o no sustituidos, o grupos arilalquilo. Cuando un compuesto descrito en la presente memoria incluye más de un grupo R, por ejemplo, cada uno de los grupos R se selecciona, independientemente entre sí, como lo son cada grupo R', R" y R"' cuando está presente más de uno de estos grupos. Cuando R' y R" están unidos al mismo átomo de nitrógeno, pueden combinarse con el átomo de nitrógeno para formar un anillo de 4, 5, 6 o 7 miembros. Por ejemplo, -NR'R" incluye, pero no se limita a, 1 -pirrolidinilo y 4-morfolinilo. A partir del análisis anterior de los sustituyentes, un experto en la materia entenderá que el término “ alquilo” incluye grupos que incluyen átomos de
carbono unidos a grupos distintos de los grupos de hidrógeno, tales como haloalquilo (p. ej., -CF3 y -CH2CF3) y acilo (p. ej., -C(O)CH3 , -C(O)CF3 , -C(O)CH2OCH3 y similares).
Similar a los sustituyentes descritos para el radical alquilo, los sustituyentes para los grupos arilo y heteroarilo varían y se seleccionan, por ejemplo, de: -Or ', -NR'R", -SR', -halógeno, -SiR'R"R'", -OC(O)R', -C(O)R', -CO2 R', -CO NR'R", -OC(O)NR'R", -NR"C(O)R', -NR'-C(O)NR"R'", -NR"C(O)2R', -NR-C(NR'R") =NR"', -S(O)R', -S(O)2R', -S(O)2NR'R", -NRSO2 R', -CN, -NO2 , -R', -N3 , -CH(Ph)2 , fluoro-alcoxi (C1-C4) y fluoro-alquilo (C1-C4), en un número que varía de cero al número total de valencias abiertas en el sistema de anillos aromáticos; y donde R', R" y R"' se seleccionan preferiblemente, independientemente entre sí, de hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido y heteroarilo sustituido o no sustituido. Cuando un compuesto descrito en la presente memoria incluye más de un grupo R, por ejemplo, cada uno de los grupos R se selecciona, independientemente entre sí, como lo son cada grupo R', R" y R"' cuando está presente más de uno de estos grupos.
Se pueden unir opcionalmente dos o más sustituyentes para formar grupos arilo, heteroarilo, cicloalquilo o heterocicloalquilo. Dichos sustituyentes, denominados formadores de anillos, normalmente, aunque no necesariamente, se encuentran unidos a una estructura base cíclica. En una realización, los sustituyentes formadores del anillo están unidos a miembros adyacentes de la estructura base. Por ejemplo, dos sustituyentes formadores de anillos unidos a miembros adyacentes de una estructura base cíclica crean una estructura de anillos condensados. En otra realización, los sustituyentes formadores de anillos están unidos a un solo miembro de la estructura base. Por ejemplo, dos sustituyentes formadores de anillos unidos a un solo miembro de una estructura base cíclica crean una estructura espirocíclica. En otra realización más, los sustituyentes formadores de anillos están unidos a miembros no adyacentes de la estructura base.
Dos de los sustituyentes en átomos adyacentes del anillo arilo o heteroarilo pueden formar opcionalmente un anillo de fórmula -T-C(O)-(CRR')q-U-, en donde T y U son, independientemente entre sí, -NR-, -O-, -CRR'-, o un enlace simple, y q es un número entero de 0 a 3. Como alternativa, dos de los sustituyentes en los átomos adyacentes del anillo arilo o heteroarilo se pueden reemplazar opcionalmente por un sustituyente de fórmula -A-(CH2)r-B-, en donde A y B son, independientemente entre sí, -CRR'-, -O-, -NR-, -S-, -S(O) -, -S(O)2-, -S(O)2NR'-, o un enlace simple, y r es un número entero de 1 a 4. Uno de los enlaces simples del nuevo anillo así formado se puede reemplazar opcionalmente por un doble enlace. Como alternativa, dos de los sustituyentes en los átomos adyacentes del anillo arilo o heteroarilo se pueden reemplazar opcionalmente por un sustituyente de fórmula -(CRR')s-X'- (C"R"')d-, en donde s y d son, independientemente entre sí, números enteros de 0 a 3, y X' es -O-, -NR'-, -S-, -S(O)-, -S(O)2- o -S(O)2NR'-. Los sustituyentes R, R', R" y R'' se seleccionan preferiblemente, independientemente entre sí, de hidrógeno, alquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido y heteroarilo sustituido o no sustituido.
Como se utilizan en la presente memoria, los términos “ heteroátomo” o “ heteroátomo de anillo” incluyen oxígeno (O), nitrógeno (N), azufre (S), fósforo (P) y silicio (Si).
El término “alquiloxi” (p. ej., metoxi, etoxi, propiloxi, aliloxi, ciclohexiloxi) representa un grupo alquilo como se ha definido anteriormente que tiene el número indicado de átomos de carbono unidos a través de un puente de oxígeno (-O-).
El término “ alquiltio” (p. ej., metiltio, etiltio, propiltio, ciclohexiltio y similares) representa un grupo alquilo como se ha definido anteriormente que tiene el número indicado de átomos de carbono unidos a través de un puente de azufre (-S-).
El término “ alquilamino” representa uno o dos grupos alquilo como se ha definido anteriormente que tienen el número indicado de átomos de carbono unidos a través de un puente de amina. Los dos grupos alquilo pueden tomarse junto con el nitrógeno al que están unidos formando un sistema cíclico que contiene de 3 a 8 átomos de carbono con o sin un sustituyente alquilo C1-C16, arilalquilo C0-C16 o alquil Cü-C16-arilo.
El término “ alquilaminoalquilo” representa un grupo alquilamino unido a través de un grupo alquilo como se ha definido anteriormente que tiene el número indicado de átomos de carbono.
El término “ alquiloxi(alquil)amino” (p. ej., metoxi(metil)amina, etoxi(propil)amina) representa un grupo alquiloxi como se ha definido anteriormente unido a través de un grupo amino, teniendo el propio grupo amino un sustituyente alquilo.
El término “ alquilcarbonilo” (p. ej., ciclooctilcarbonilo, pentilcarbonilo, 3-hexilcarbonilo) representa un grupo alquilo como se ha definido anteriormente que tiene el número indicado de átomos de carbono unidos a través de un grupo carbonilo.
El término “ alquilcarboxi” (p. ej., heptilcarboxi, ciclopropilcarboxi, 3-pentenilcarboxi) representa un grupo alquilcarbonilo como se ha definido anteriormente, en donde el carbonilo está unido a su vez a través de un oxígeno.
El término “ alquilcarboxialquilo” representa un grupo alquilcarboxi unido a través de un grupo alquilo como se ha definido anteriormente que tiene el número indicado de átomos de carbono.
El término “ alquilcarbonilamino” (p. ej., hexilcarbonilamino, ciclopentilcarbonilaminometilo, metilcarbonilaminofenilo) representa un grupo alquilcarbonilo como se ha definido anteriormente, en donde el carbonilo está unido a su vez a través del átomo de nitrógeno de un grupo amino.
El propio grupo nitrógeno puede estar sustituido con un grupo alquilo o arilo.
El término “oxo” , como se utiliza en la presente memoria, significa un oxígeno que tiene un doble enlace con un átomo de carbono.
El término “alquilsulfonilo” , como se utiliza en la presente memoria, significa una fracción que tiene la fórmula -S(O2)-R', donde R' es un grupo alquilo como se ha definido anteriormente. R' puede tener un número específico de carbonos (p. ej., “alquilsulfonilo C1-C4” ).
El término “carboniloxi” representa un grupo carbonilo unido a través de un puente de oxígeno.
En las definiciones anteriores, los términos “alquilo” y “ alquenilo” se pueden usar indistintamente en la medida en que se forme una entidad química estable, como será evidente para los expertos en la materia.
El término “enlazador” se refiere a grupos de unión interpuestos entre sustituyentes, por ejemplo, R1, R2, R3 o R4 descrito en la presente memoria, por ejemplo, Fórmula (Ia) y denominado genéricamente Rn , y el grupo que está sustituido, p. ej., el grupo “anillo A” de, p. ej., la Fórmula (Ia). En algunas realizaciones, el enlazador incluye fracciones enlazadoras de amido (-CONH-Rn o -NHCO-Rn), tioamido (-CSNH-Rn o -NHCS-Rn), carboxilo (-CO2-Rn o -OCORn), carbonilo (-CO-Rn), urea (-NHCONH-Rn), tiourea (-NHCSNH-Rn), sulfonamido (-NHSO2-Rn o -SO2NH-Rn), éter (-O-Rn), sulfonilo (-SO2-Rn), sulfoxilo (-SO-Rn), carbamoílo (-NHCO2-Rn o -OCONH-Rn) o amino (-NHRn).
Un “grupo sustituyente” , como se utiliza en la presente memoria, significa un grupo seleccionado de las siguientes fracciones:
(A) -OH, -NH2 , -SH, -CN, -CF3 , -NO2 , oxo, halógeno, -COOH, alquilo no sustituido, heteroalquilo no sustituido, cicloalquilo no sustituido, heterocicloalquilo no sustituido, arilo no sustituido, heteroarilo no sustituido y
(B) alquilo, heteroalquilo, cicloalquilo, heterocicloalquilo, arilo y heteroarilo, sustituido con al menos un sustituyente seleccionado de:
(i) oxo, -OH, -NH2 , -SH, -CN, -CF3 , -NO2 , halógeno, -COOH, alquilo no sustituido, heteroalquilo no sustituido, cicloalquilo no sustituido, heterocicloalquilo no sustituido, arilo no sustituido, heteroarilo no sustituido y
(ii) alquilo, heteroalquilo, cicloalquilo, heterocicloalquilo, arilo y heteroarilo, sustituido con al menos un sustituyente seleccionado de:
(a) oxo, -OH, -NH2 , -SH, -CN, -CF3 , -NO2 , halógeno, -COOH, alquilo no sustituido, heteroalquilo no sustituido, cicloalquilo no sustituido, heterocicloalquilo no sustituido, arilo no sustituido, heteroarilo no sustituido y
(b) alquilo, heteroalquilo, cicloalquilo, heterocicloalquilo, arilo o heteroarilo, sustituido con al menos un sustituyente seleccionado de: oxo,-OH, -NH2 , -SH, -CN, -CF3, -NO2 , halógeno, -COOH, alquilo no sustituido, heteroalquilo no sustituido, cicloalquilo no sustituido, heterocicloalquilo no sustituido, arilo no sustituido y heteroarilo no sustituido. Un “sustituyente de tamaño limitado” o “grupo sustituyente de tamaño limitado” , como se utiliza en la presente memoria, significa un grupo seleccionado de todos los sustituyentes descritos anteriormente para un “grupo sustituyente” , en donde cada alquilo sustituido o no sustituido es un grupo alquilo C1-C20 sustituido o no sustituido, cada heteroalquilo sustituido o no sustituido es un heteroalquilo de 2-20 miembros sustituido o no sustituido, cada cicloalquilo sustituido o no sustituido es un cicloalquilo C4-C8 , y cada heterocicloalquilo sustituido o no sustituido es un heterocicloalquilo de 4-8 miembros sustituido o no sustituido.
Un “sustituyente inferior” o “grupo sustituyente inferior” , tal como se utiliza en la presente memoria, significa un grupo seleccionado de todos los sustituyentes descritos anteriormente para un “grupo sustituyente” , en donde cada alquilo sustituido o no sustituido es un grupo alquilo C1-C8 sustituido o no sustituido, cada heteroalquilo sustituido o no sustituido es un heteroalquilo de 2-8 miembros sustituido o no sustituido, cada cicloalquilo sustituido o no sustituido es un cicloalquilo C5-C7 , y cada heterocicloalquilo sustituido o no sustituido es un heterocicloalquilo de 5-7 miembros sustituido o no sustituido.
El término “aproximadamente” utilizado en el contexto de un valor numérico indica un intervalo de /-10 % del valor numérico, salvo que se indique expresamente lo contrario.
II. Compuestos
En un aspecto, se proporciona un compuesto con estructura de Fórmula (Ia):

o una sal, un éster, un solvato o un profármaco farmacéuticamente aceptable del mismo. El anillo A es pirazolilo sustituido o no sustituido. L1, L2 y L4 son, independientemente entre sí, un enlace, alquileno sustituido o no sustituido, heteroalquileno sustituido o no sustituido, -S-, -SO-, -SO2-, -O-, -NHSO2- o -NR5-. L3 es un enlace, alquileno sustituido o no sustituido, o heteroalquileno sustituido o no sustituido. R1, R2 y R4 son, independientemente entre sí, hidrógeno, halógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, heteroarilo sustituido o no sustituido, o arilo de anillo condensado sustituido o no sustituido. R3 es heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, o heterocicloalquenilo sustituido o no sustituido. R5 es hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, alquileno sustituido o no sustituido, heteroalquileno sustituido o no sustituido, cicloalquilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, o heteroarilo sustituido o no sustituido.
En algunas realizaciones, el compuesto es una sal, un éster, un solvato o un profármaco farmacéuticamente aceptable de un compuesto de Fórmula (Ia). En algunas realizaciones, el compuesto no es un éster ni un solvato ni un profármaco.
En algunas realizaciones, L4 y R4 están ausentes, proporcionando un compuesto con la siguiente estructura de Fórmula (Ib).

En algunas realizaciones, se proporciona un compuesto según la Fórmula (Ib) con la siguiente estructura de Fórmula (II).

En algunas realizaciones, el compuesto tiene la estructura de Fórmula (II), en donde L3 es un enlace, o alquileno sustituido o no sustituido, y R3 es heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido o heterocicloalquilo sustituido o no sustituido. En algunas realizaciones, R3 es heteroalquilo sustituido o no sustituido. En algunas realizaciones, R3 es cicloalquilo sustituido o no sustituido. En algunas realizaciones, R3 es ciclohexilo sustituido o no sustituido, ciclopentilo sustituido o no sustituido, cicloheptilo
sustituido o no sustituido. En algunas realizaciones, R3 es cicloalquenilo sustituido o no sustituido. En algunas realizaciones, R3 es ciclohexenilo sustituido o no sustituido. En algunas realizaciones, R3 es heterocicloalquilo sustituido o no sustituido. En algunas realizaciones, R3 es piperidinilo sustituido o no sustituido. En algunas realizaciones, R3 es pirrolidinilo sustituido o no sustituido. En algunas realizaciones, R3 es pirrolidinilo sustituido o no sustituido. En algunas realizaciones, R3 es azetidinilo sustituido o no sustituido. En algunas realizaciones, R3 es oxetanilo sustituido o no sustituido. En algunas realizaciones, R3 es oxolanilo sustituido o no sustituido. En algunas realizaciones, R3 es oxanilo sustituido o no sustituido.
Además de cualquier realización anterior en donde el compuesto tiene la estructura de Fórmula (II), en algunas realizaciones, L1 es -S-, -O-, -NR5-, alquileno sustituido o no sustituido, o heteroalquileno sustituido o no sustituido, donde R5 es como se describe en la fórmula la, y R1 es hidrógeno, alquilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, heteroarilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido. En algunas realizaciones, R1 es fenilo sustituido o no sustituido. En algunas realizaciones, R1 es fenilo no sustituido. En algunas realizaciones, R1 es un piridilo sustituido o no sustituido. En algunas realizaciones, R1 es un piridazinilo sustituido o no sustituido. En algunas realizaciones, R1 es un pirimidinilo sustituido o no sustituido. En algunas realizaciones, R1 es un tienilo sustituido o no sustituido. En algunas realizaciones, R1 es un furilo sustituido o no sustituido. En algunas realizaciones, R1 es un piridilo no sustituido. En algunas realizaciones, R1 es un piridazinilo no sustituido. En algunas realizaciones, R1 es un pirimidinilo no sustituido. En algunas realizaciones, R1 es un tienilo no sustituido. En algunas realizaciones, R1 es un tienilo sustituido con cloro. En algunas realizaciones, R1 es un furilo no sustituido. En algunas realizaciones, R1 es un morfolinilo sustituido o no sustituido. En algunas realizaciones, R1 es un oxanilo sustituido o no sustituido. En algunas realizaciones, R1 es un oxetanilo sustituido o no sustituido. En algunas realizaciones, R1 es un morfolinilo no sustituido. En algunas realizaciones, R1 es un oxanilo no sustituido. En algunas realizaciones, R1 es un oxetanilo no sustituido. En algunas realizaciones, R1 es benzodioxinilo sustituido o no sustituido. En algunas realizaciones, R1 es naftilo sustituido o no sustituido. En algunas realizaciones, R1 es benzodioxinilo no sustituido. En algunas realizaciones, R1 es naftilo no sustituido. En algunas realizaciones, L2 y R2 están ausentes. En algunas realizaciones, L2 es un enlace. En algunas realizaciones, L2 es un enlace y R2 es hidrógeno.
Además de cualquier realización anterior en donde el compuesto tiene la estructura de Fórmula (II), R2 es hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, o heteroarilo sustituido o no sustituido. En algunas realizaciones, R2 es fenilo sustituido o no sustituido. En algunas realizaciones, R2 es fenilo no sustituido. En algunas realizaciones, R2 es un piridilo sustituido o no sustituido. En algunas realizaciones, R2 es un piridazinilo sustituido o no sustituido. En algunas realizaciones, R2 es un pirimidinilo sustituido o no sustituido. En algunas realizaciones, R2 es un tienilo sustituido o no sustituido. En algunas realizaciones, R2 es un furilo sustituido o no sustituido. En algunas realizaciones, R2 es un piridilo no sustituido. En algunas realizaciones, R2 es un piridazinilo no sustituido. En algunas realizaciones, R2 es un pirimidinilo no sustituido. En algunas realizaciones, R2 es un tienilo no sustituido. En algunas realizaciones, R2 es un tienilo sustituido con cloro. En algunas realizaciones, R2 es un furilo no sustituido. En algunas realizaciones, R2 es un morfolinilo sustituido o no sustituido. En algunas realizaciones, R2 es un oxanilo sustituido o no sustituido. En algunas realizaciones, R2 es un oxetanilo sustituido o no sustituido. En algunas realizaciones, R2 es un morfolinilo no sustituido. En algunas realizaciones, R2 es un oxanilo no sustituido. En algunas realizaciones, R2 es un oxetanilo no sustituido. En algunas realizaciones, R2 es benzodioxinilo sustituido o no sustituido. En algunas realizaciones, R2 es naftilo sustituido o no sustituido. En algunas realizaciones, R2 es benzodioxinilo no sustituido. En algunas realizaciones, R2 es naftilo no sustituido.
En algunas realizaciones, se proporciona un compuesto según la fórmula (Ia) con la siguiente estructura de fórmula (III).

En algunas realizaciones, se proporciona un compuesto según la fórmula (III) en donde L4 es un enlace; y R4 es hidrógeno, halógeno, alquilo sustituido o no sustituido, o heteroalquilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido. En algunas realizaciones, R4 es halógeno. En algunas realizaciones, R4 es alquilo no sustituido. Además de cualquier realización en donde el compuesto tiene la estructura
de Fórmula (III), en algunas realizaciones L3 es un enlace, o alquileno sustituido o no sustituido, y R3 es heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido. En algunas realizaciones, R3 es heteroalquilo sustituido o no sustituido. En algunas realizaciones, R3 es cicloalquilo sustituido o no sustituido. En algunas realizaciones, R3 es ciclohexilo sustituido o no sustituido, ciclopentilo sustituido o no sustituido, cicloheptilo sustituido o no sustituido. En algunas realizaciones, R3 es cicloalquenilo sustituido o no sustituido. En algunas realizaciones, R3 es ciclohexenilo sustituido o no sustituido. En algunas realizaciones, R3 es heterocicloalquilo sustituido o no sustituido. En algunas realizaciones, R3 es piperidinilo sustituido o no sustituido. En algunas realizaciones, R3 es pirrolidinilo sustituido o no sustituido. En algunas realizaciones, R3 es pirrolidinilo sustituido o no sustituido. En algunas realizaciones, R3 es azetidinilo sustituido o no sustituido. En algunas realizaciones, R3 es oxetanilo sustituido o no sustituido. En algunas realizaciones, R3 es oxolanilo sustituido o no sustituido. En algunas realizaciones, R3 es oxanilo sustituido o no sustituido.
Además de cualquier realización anterior en donde el compuesto tiene la estructura de Fórmula (III), en algunas realizaciones, L1 es -S-, -O-, -NR5-, alquileno sustituido o no sustituido, o heteroalquileno sustituido o no sustituido, donde R5 es como se describe en la fórmula la, y R1 es hidrógeno, alquilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, heteroarilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido. En algunas realizaciones, R1 es fenilo sustituido o no sustituido. En algunas realizaciones, R1 es fenilo no sustituido. En algunas realizaciones, R1 es un piridilo sustituido o no sustituido. En algunas realizaciones, R1 es un piridazinilo sustituido o no sustituido. En algunas realizaciones, R1 es un pirimidinilo sustituido o no sustituido. En algunas realizaciones, R1 es un tienilo sustituido o no sustituido. En algunas realizaciones, R1 es un furilo sustituido o no sustituido. En algunas realizaciones, R1 es un piridilo no sustituido. En algunas realizaciones, R1 es un piridazinilo no sustituido. En algunas realizaciones, R1 es un pirimidinilo no sustituido. En algunas realizaciones, R1 es un tienilo no sustituido. En algunas realizaciones, R1 es un tienilo sustituido con cloro. En algunas realizaciones, R1 es un furilo no sustituido. En algunas realizaciones, R1 es un morfolinilo sustituido o no sustituido. En algunas realizaciones, R1 es un oxanilo sustituido o no sustituido. En algunas realizaciones, R1 es un oxetanilo sustituido o no sustituido. En algunas realizaciones, R1 es un morfolinilo no sustituido. En algunas realizaciones, R1 es un oxanilo no sustituido. En algunas realizaciones, R1 es un oxetanilo no sustituido. En algunas realizaciones, R1 es benzodioxinilo sustituido o no sustituido. En algunas realizaciones, R1 es naftilo sustituido o no sustituido. En algunas realizaciones, R1 es benzodioxinilo no sustituido. En algunas realizaciones, R1 es naftilo no sustituido. En algunas realizaciones, L2 y R2 están ausentes. En algunas realizaciones, L2 es un enlace. En algunas realizaciones, L2 es un enlace y R2 es hidrógeno.
Además de cualquier realización anterior en donde el compuesto tiene la estructura de Fórmula (III), R2 es hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, o heteroarilo sustituido o no sustituido. En algunas realizaciones, R2 es fenilo sustituido o no sustituido. En algunas realizaciones, R2 es fenilo no sustituido. En algunas realizaciones, R2 es un piridilo sustituido o no sustituido. En algunas realizaciones, R2 es un piridazinilo sustituido o no sustituido. En algunas realizaciones, R2 es un pirimidinilo sustituido o no sustituido. En algunas realizaciones, R2 es un tienilo sustituido o no sustituido. En algunas realizaciones, R2 es un furilo sustituido o no sustituido. En algunas realizaciones, R2 es un piridilo no sustituido. En algunas realizaciones, R2 es un piridazinilo no sustituido. En algunas realizaciones, R2 es un pirimidinilo no sustituido. En algunas realizaciones, R2 es un tienilo no sustituido. En algunas realizaciones, R2 es un tienilo sustituido con cloro. En algunas realizaciones, R2 es un furilo no sustituido. En algunas realizaciones, R2 es un morfolinilo sustituido o no sustituido. En algunas realizaciones, R2 es un oxanilo sustituido o no sustituido. En algunas realizaciones, R2 es un oxetanilo sustituido o no sustituido. En algunas realizaciones, R2 es un morfolinilo no sustituido. En algunas realizaciones, R2 es un oxanilo no sustituido. En algunas realizaciones, R2 es un oxetanilo no sustituido. En algunas realizaciones, R2 es benzodioxinilo sustituido o no sustituido. En algunas realizaciones, R2 es naftilo sustituido o no sustituido. En algunas realizaciones, R2 es benzodioxinilo no sustituido. En algunas realizaciones, R2 es naftilo no sustituido.
En algunas realizaciones, se proporciona un compuesto según la fórmula (III) en donde L3 es un enlace, R3 es heterocicloalquilo sustituido o no sustituido, o heterocicloalquenilo sustituido o no sustituido, con la siguiente estructura de Fórmula (IV).

en donde: L1, L2 y L4 son, independientemente entre sí, un enlace, alquileno sustituido o no sustituido, heteroalquileno sustituido o no sustituido, -C(O)-, -S-, -SO-, -SO2-, -O-, -NHSO2-, -NHC(O)- o -NR5-; R1, R2 y R4 son, independientemente entre sí, hidrógeno, halógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, o heteroarilo sustituido o no sustituido; R5 es hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, o heteroarilo sustituido o no sustituido; V es hidrógeno o alquilo sustituido o no sustituido; W está ausente, o es hidrógeno, halógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, heteroarilo sustituido o no sustituido, -C(O)R6 , -C(O)OR6 , - C(O)NR6 R7 , -SR6 , -SOR6 , -SO2 R6 , -SO2NR6 R7 , -OR6 , -NHSO2 R6 o -NR6 R7 , donde R6 y R7 son, independientemente entre sí, hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido, en donde R6 y R7 se pueden combinar, si ambos están presentes, para formar un alquileno sustituido o no sustituido, o heteroalquileno sustituido o no sustituido; X es un enlace, alquileno sustituido o no sustituido, -O- o -NR8-; Y es un enlace, alquileno sustituido o no sustituido, -O- o -N-, siempre que cuando Y sea -O-, W esté ausente; y Z es un enlace, -C(O)-, alquileno sustituido o no sustituido, -O- o -NR9-; en donde R8 y R9 son, independientemente entre sí, hidrógeno, halógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, heteroarilo sustituido o no sustituido,-C(O)R6 , -C(O)OR6 , - C(O)NR6 R7 , -SR6 , -SOR6 , -SO2 R6 , -SO2NR6 R7 , -OR6 , -NHSO2 R6 o -NR6 R7 , en donde R6 y R7 son como se han definido anteriormente; y siempre que al menos uno de X sea -O- o -NR8-, Y es -O- o -N-, o Z es -O- o -NR9-. En algunas realizaciones, el compuesto es una sal, un éster, un solvato o un profármaco farmacéuticamente aceptable de un compuesto de Fórmula (IV). En algunas realizaciones, el compuesto no es un éster ni un solvato ni un profármaco.
En algunas realizaciones, X es un enlace, o alquileno sustituido o no sustituido. En algunas realizaciones, Z es un enlace, o alquileno sustituido o no sustituido. En algunas realizaciones, X es un enlace, o alquileno sustituido o no sustituido, y Z es un enlace, o alquileno sustituido o no sustituido.
En algunas realizaciones, en donde X es un enlace, o alquileno sustituido o no sustituido y Z es un enlace, o alquileno sustituido o no sustituido, Y es -N- y W es hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, arilo sustituido o no sustituido, -C(O)R6 , -C(O)OR6 , -C(O)NR6 R7 , -SO2 R6 o SO2NR6 R7 , en donde R6 y R7son, independientemente entre sí, alquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido, o R6 y R7 se pueden combinar, si ambos están presentes, para formar un alquileno sustituido o no sustituido, o heteroalquileno sustituido o no sustituido.
En algunas realizaciones, X se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido, y Z es un enlace. En algunas realizaciones, X es un enlace y Z se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido. En algunas realizaciones, X se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido, y en donde Z se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido. En algunas realizaciones, X y Z son ambos alquileno ramificado, y X y Z están unidos covalentemente. En algunas realizaciones, Z es metileno sustituido, etileno sustituido, propileno sustituido, butileno sustituido o pentileno sustituido, que tiene uno o más grupos sustituyentes seleccionados del grupo que consiste en -OH, -NH2 , -SH, -CN, -CF3 , -NO2 , oxo, halógeno, -COOH, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido.
En algunas realizaciones, X se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido, y en donde Z es -C(O)-. En algunas realizaciones, W es hidrógeno. En algunas realizaciones, W se selecciona del grupo que consiste en alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido,
heterocicloalquilo sustituido o no sustituido, -C(O)R6, - C(O)OR6, -C(O)NR6R7, -SR6, -SOR6, -SO2 R6 y -SO2NR6. En algunas realizaciones, W es alquilo sustituido, heteroalquilo sustituido, alquenilo sustituido, heteroalquenilo sustituido, cicloalquilo sustituido o heterocicloalquilo sustituido, que tiene uno o más grupos sustituyentes seleccionados del grupo que consiste en -OH, -NH2 , -SH, -CN, -CF3 , -NO2 , oxo, halógeno, -COOH, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido. En algunas realizaciones, W es -COR6, -C(O)OR6, -C(O)NR6R7, -SO2 R6 o -SO2 NR6R7, y en donde R6 y R7 se seleccionan del grupo que consiste en alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, arilo sustituido o no sustituido y heteroarilo sustituido o no sustituido, o R6 y R7 se combinan para formar un alquileno sustituido o no sustituido.
En algunas realizaciones, W está ausente, X es -NR8-, Y es un enlace, o alquileno sustituido o no sustituido, y Z es -NR9-. En algunas realizaciones en donde W está ausente, X es -NR8-, Y es un enlace, o alquileno sustituido o no sustituido, y Z es -NR9-, Y se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido. En algunas realizaciones, R8 se selecciona del grupo que consiste en alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, -COR6, -C(O)OR6, - C(O)NR6R7, -SR6, -SOR6, -SO2 R6 y -SO2NR6R7, y en donde R9 se selecciona del grupo que consiste en alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, -COR6, -C(O)OR6, -C(O)NR6R7, -SR6, -SOR6, -SO2 R6 y -SO2NR6R7.
En algunas realizaciones, Y es -O- y W está ausente. En algunas realizaciones en donde Y es -O- y W está ausente, X se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido, y Z es un enlace. En algunas realizaciones, X es un enlace y Z se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido. En algunas realizaciones, X se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido, y en donde Z se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido.
En algunas realizaciones, V es hidrógeno o metilo sustituido o no sustituido.
Además de cualquier realización anterior en donde el compuesto tiene la estructura de Fórmula (IV), en algunas realizaciones, L1 es -S-, -O-, -NR5-, alquileno sustituido o no sustituido, o heteroalquileno sustituido o no sustituido; R1 es alquilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, heteroarilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido; y R5 es hidrógeno, alquilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido. En algunas realizaciones, L1 es -NR5-o heteroalquilo sustituido o no sustituido, y R1 es alquilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido. En algunas realizaciones, L1 es -NR5-, y R1 es alquilo sustituido que tiene uno o más grupos sustituyentes seleccionados del grupo que consiste en heteroarilo sustituido o no sustituido y heterocicloalquilo sustituido o no sustituido. En algunas realizaciones, R1 es alquilo sustituido con tiofenilo sustituido con cloro. En algunas realizaciones, L1 es heteroalquilo sustituido o no sustituido, y R1 es heteroarilo sustituido o no sustituido. En algunas realizaciones, L1 y R1 puede ser cualquier grupo específico como se ha expuesto anteriormente para cualquiera de las Fórmulas (Ia), (Ib), (II) o (III).
Además de cualquier realización anterior en donde el compuesto tiene la estructura de Fórmula (IV), en algunas realizaciones, L2 es enlace, alquileno sustituido o no sustituido, -C(O)-, o -SO2-, y R2 es hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, heteroarilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido. En algunas realizaciones, L2 es enlace, y R2 es hidrógeno. En algunas realizaciones, L2 es -C(O)-, y R2 es alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, heteroarilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido. En algunas realizaciones, L2 y R2 pueden ser cualquier grupo específico como se ha expuesto anteriormente para cualquiera de las Fórmulas (Ia), (Ib), (II) o (III).
Además de cualquier realización anterior en donde el compuesto tiene la estructura de fórmula (IV), en algunas realizaciones, L4 es un enlace, y R4 es hidrógeno, halógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido. En algunas realizaciones, L4 y R4 pueden ser cualquier grupo específico como se ha expuesto anteriormente para cualquiera de las Fórmulas (la), (Ib), (II) o (III).
En algunas realizaciones, se proporciona un compuesto según la Fórmula (IV) y sus realizaciones enumeradas, en donde L2 es un enlace, y R2 es hidrógeno, proporcionando un compuesto con la siguiente estructura de Fórmula (V).

En algunas realizaciones, el compuesto es una sal, un éster, un solvato o un profármaco farmacéuticamente aceptable de un compuesto de Fórmula (V). En algunas realizaciones, el compuesto no es un éster ni un solvato ni un profármaco.
En la presente memoria, se proporcionan ejemplos de compuestos, p. ej., compuestos aromáticos multisustituidos, según la presente descripción. En las siguientes Tablas A y B, se describen el número de entrada, el nombre químico (es decir, el nombre de la Unión Internacional de Química Pura y Aplicada [IUPAC]), el peso molecular calculado (PM) y la actividad biológica (es decir, la actividad de inhibición en los ensayos de trombina y KLKB1). En la siguiente Tabla C, se describen los nombres químicos.
Para la siguiente Tabla A, se ensayó la inhibición de la actividad proteasa de la trombina en los compuestos descritos como se describe en la presente memoria. En la Tabla A, el nivel de inhibición en el ensayo de trombina se indica de la siguiente manera: una CI50 á 0,1 pM; b: 0,1 pM < CI50 < 1 pM; c: 1 pM < CI50 < 10 pM; d: 10 pM < CI50 < 100 pM; e: CI50 ^ 100 pM. Por consiguiente, en algunas realizaciones, se proporciona un compuesto como se expone expresamente en la siguiente Tabla A.
Tabla A
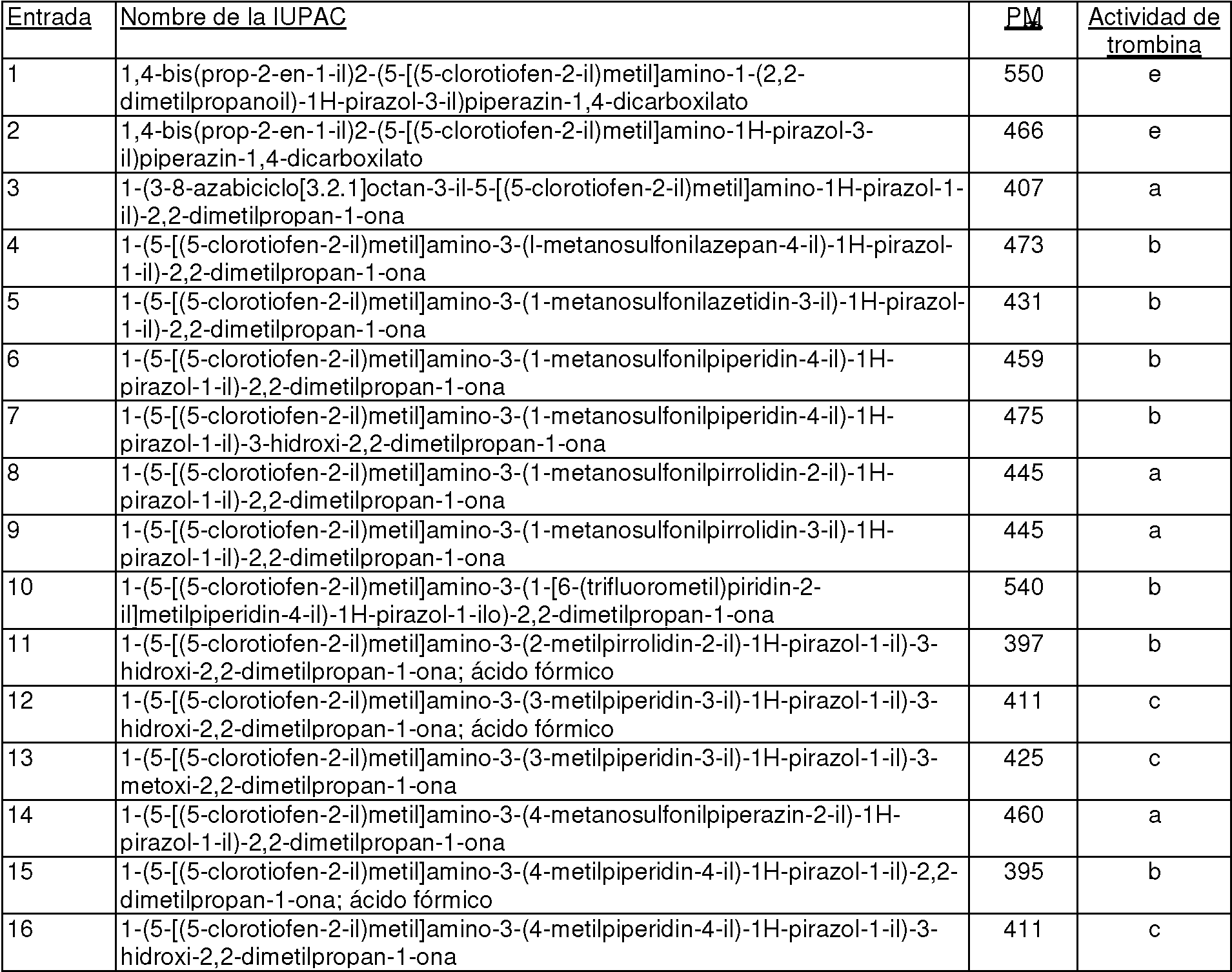









Para la siguiente Tabla B, se ensayó la inhibición de la actividad proteasa de KLKB1 en los compuestos descritos como se describe en la presente memoria. En la siguiente Tabla B, se indica el nivel de inhibición del ensayo de KLKB1 de la siguiente manera: a: CI50 á 1 pM; b: 1 pM < CI50 < 10 pM; c: CI50 s 10 pM. Por consiguiente, en algunas realizaciones, se proporciona un compuesto como se expone expresamente en la siguiente Tabla B.
Tabla B

1 -(5-[(5-clorotiofen-2-il)metil]amino-3-(piperidin-4-il)-1 H-pirazol-1 -il)-2,2- 381 c dimetilpropan-1-ona
1 -(5-[(5-clorotiofen-2-il)metil]amino-3-(pirrolidin-3-il)-1 H-pirazol-1 -il)-2,2- 367 c dimetilpropan-1-ona
1 -(5-[(5-clorotiofen-2-il)metil]amino-3-[1 -(1,3-oxazol-4-ilmetil)piperidin-4-il]- 462 c pirazol-1 -il)-2,2-dimetilpropan-1 -ona
1 -(5-[(5-clorotiofen-2-il)metil]amino-3-[1 -(1H-1,2,3,4-tetrazol-5-ilmetil)piper 463 c il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona
1 -(5-[(5-clorotiofen-2-il)metil]amino-3-[1 -(1 H-1,2,3-triazol-5-ilmetil)piperidi 462 c 1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona
1 -(5-[(5-clorotiofen-2-il)metil]amino-3-[1 -(1 H-imidazol-4-ilmetil)piperidin-4-il 461 c pirazol-1 -il)-2,2-dimetilpropan-1 -ona
1 -(5-[(5-clorotiofen-2-il)metil]amino-3-[1 -(2-ciclopropoxietil)piperidin-4-il]-1 465 c pirazol-1 -il)-2,2-dimetilpropan-1 -ona
1 -(5-[(5-clorotiofen-2-il)metil]amino-3-[1 -(2-ciclopropoxietil)piperidin-4-il]-1 481 c pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona
1 -(5-[(5-clorotiofen-2-il)metil]amino-3-[1 -(2-metoxietil)piperidin-4-il]-1 H-pir 439 c il)-2,2-dimetilpropan-1-ona
439 c il)-2,2-dimetilpropan-1-ona
1 -(5-[(5-clorotiofen-2-il)metil]amino-3-[1 -(ciclopropilmetil)piperidin-4-il]-1 H-pirazol- 435 c 1 -il)-2,2-dimetilpropan-1 -ona
1-(5-[(5-clorotiofen-2-il)metil]amino-3-[l-(morfolin-4-carbonil)azetidin-3-il]-1 H-  466 c pirazol-1 -il)-2,2-dimetilpropan-1 -ona
466 c pirazol-1 -il)-2,2-dimetilpropan-1 -ona
1 -(5-[(5-clorotiofen-2-il)metil]amino-3-[1 -(oxetan-3-ilmetil)piperidin-4-il]-1 H-pirazol- 451 c 1 -il)-2,2-dimetilpropan-1 -ona
1 -(5-[(5-clorotiofen-2-il)metil]amino-3-[1 -(piridin-2-ilmetil)piperidin-4-il]-1 H-pirazol- 472 c 1 -il)-2,2-dimetilpropan-1 -ona
1 -(5-[(5-clorotiofen-2-il)metil]amino-3-[1 -(piridin-3-ilmetil)piperidin-4-il]-1 H-pirazol- 472 c 1 -il)-2,2-dimetilpropan-1 -ona
1 -(5-[(5-clorotiofen-2-il)metil]amino-3-1 -[(4-metoxifenil)metil]piperidin-4-il-1 H- 501 c pirazol-1 -il)-2,2-dimetilpropan-1 -ona
1 -(5-[(5-clorotiofen-2-il)metil]amino-3-1 -[2-(morfolin-4-il)etil]piperidin-4-il-1 H-  494 c pirazol-1-il)-2,2-dimetilpropan-1-ona; ácido fórmico
494 c pirazol-1-il)-2,2-dimetilpropan-1-ona; ácido fórmico
1 -(5-[(5-clorotiofen-2-il)metil]amino-3-1 -[2-(piridin-2-il)etil]piperidin-4-il-1 H-pirazol- 486 c 1 -il)-2,2-dimetilpropan-1 -ona
1-(5-[(5-clorotiofen-2-il)metil]amino-4-fluoro-3-(piperidin-4-il)-1 H-pirazol-1 - 399 b dimetilpropan-1-ona
1-(5-[(5-clorotiofen-2-il)metil]amino-4-fluoro-3-(piperidin-4-il)-1 H-pirazol-1 - 415 c hidroxi-2,2-dimetilpropan-1 -ona
1 -[3-(1 -bencilpiperidin-4-il)-5-[(5-clorotiofen-2-il)metil]amino-1 H-pirazol-1 -i 471 c dimetilpropan-1-ona
1 -[4-(5-[(5-clorotiofen-2-il)metil]amino-1 -(tiofen-3-carbonil)-1 H-pirazol-3-  449 c il)piperidin-1 -iletan-1-ona clorhidrato de 1 -benzoil-N-[(5-clorotiofen-2-il)metil]-3-(piperidin-4-il)-1 H-pirazol-5- 401 b amina
449 c il)piperidin-1 -iletan-1-ona clorhidrato de 1 -benzoil-N-[(5-clorotiofen-2-il)metil]-3-(piperidin-4-il)-1 H-pirazol-5- 401 b amina
3-(5-[(5-clorotiofen-2-il)metil]amino-1-(tiofen-3-carbonil)-1 H-pirazol-3-il)-3- 435 c metilpiperidin-2-ona
3-(5-[(5-clorotiofen-2-il)metil]amino-1-(tiofen-3-carbonil)-1 H-pirazol-3-il)-3- 421 c metilpirrolidin-2-ona
5-[(5-clorotiofen-2-il)metil]amino-1-(2,2-dimetilpropanoil)-3-[l-(morfolin-4- 519 b carbonil)piperidin-4-il]-1 H-pirazol-4-carbonitrilo clorhidrato de N-[(5-clorotiofen-2-il)metil]-1 -(2,3-dihidro-1,4-benzodioxin-5- 459 c carbonil)-3-(piperidin-4-il)-1 H-pirazol-5-amina
N-[(5-clorotiofen-2-il)metil]-1-(2-metoxibenzoil)-3-(piperidin-3-il)-1 H-pirazol-5 431 c amina
431 c amina
clorhidrato de N-[(5-clorotiofen-2-il)metil]-1 -(2-metoxibenzoil)-3-(piperidin-4-il)-1 H- 431 c pirazol-5-amina
N-[(5-clorotiofen-2-il)metil]-1-(4-metiloxano-4-carbonil)-3-(piperidin-3-il)-1 H-  423 c pirazol-5-amina
423 c pirazol-5-amina
clorhidrato de N-[(5-clorotiofen-2-il)metil]-1 -(furan-3-carbonil)-3-(piperidin-4-il)-1 H- 391 c pirazol-5-amina
N-[(5-clorotiofen-2-il)metil]-3-(1 -metanosulfonilazepan-4-il)-1 -(1,3-tiazol-4- 500 b carbonil)-1 H-pirazol-5-amina
N-[(5-clorotiofen-2-il)metil]-3-(1 -metanosulfonilazetidin-3-il)-1 -(1,3-tiazol-4- 458 b carbonil)-1 H-pirazol-5-amina 

En algunas realizaciones, se proporciona un compuesto como se expone expresamente en la siguiente Tabla C.
Tabla C _________________________________________________________________________________________________ ácido 1 -(2,2-dimetilpropanoil)-3-[1 -(2,2-dimetilpropanoil)-4-fluoro-5-{[(4-fluorofenil)metil]amino}-1 H-pirazol-3-illpiperidin-2-carboxílico__________________________ '_________________________________________________ 1 -(2,2-dimetilpropanoil)-4-(5-{[(4-fluorofenil)metil](metil)amino}-4-metoxi-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-3-(trifluorometil)piperidin-2-ona________________________________________________________________________ 1-(2-clorobenzoil)-5-[(5-clorotiofen-2-il)metoxi]-3-[l-metanosulfonil-4-(trifluorometil)pirrolidin-3-il]-1 H-pirazol 1-(2-clorobenzoil)-5-{[(4-fluorofenil)metil](metil)amino}-3-{1-[(3-hidroxipirrolidin-1-il)sulfonil]-6-oxo-4-(trifluorometil)piperidin-3-il}-1 H-pirazol-4-carbonitrilo ________________________________________________


na
lin-4-folin-

til]-

nil)-
folin-

fonil-




azol-

zol-

razolda

}-1 H-
onil)-

-4-



Para la siguiente Tabla D, se ensayó la inhibición de la actividad proteasa de la quimotripsina y del Factor XIa de los compuestos descritos como se describe en la presente memoria. En la Tabla D, el nivel de inhibición en el ensayo se indica de la siguiente manera: una CI50 ^ 0,1 pM; b: 0,1 pM < CI50 < 1 pM; c: 1 pM <CI50 << 10 pM; d: 10 pM < CI50 < 100 pM; e: CI50 s 100 pM. Por consiguiente, en algunas realizaciones, se proporciona un compuesto como se expone expresamente en la siguiente Tabla D.
Tabla D

Los compuestos descritos en la presente memoria también incluyen mezclas racémicas, estereoisómeros y mezclas de los compuestos, incluidos compuestos marcados isotópicamente y radiomarcados. Véase, p. ej., Goding, 1986, “ MONOCLONAL ANTIBODIES PRINCIPLES AND PRACTICE” ; Academic Press, pág. 104. Dichos isómeros pueden aislarse mediante técnicas de resolución convencionales, que incluyen, p. ej., cristalización fraccionada, cromatografía quiral y similares. Véase, p. ej., Eliel, E. L. y Wilen S. H., 1993, “STEREOCHEMISTRY IN ORGANIC COMPOUNDS” ; John Wiley & Sons, Nueva York.
En algunas realizaciones, los compuestos descritos en la presente memoria tienen centros asimétricos y pueden presentarse en forma de racematos, mezclas racémicas y como enantiómeros o diastereoisómeros individuales, estando todas las formas isoméricas, así como sus mezclas contempladas para su uso en los compuestos y los métodos descritos en la presente memoria. Los compuestos contemplados para su uso en los compuestos y métodos descritos en la presente memoria no incluyen aquellos conocidos en la técnica por ser demasiado inestables para su síntesis y/o aislamiento.
Los compuestos descritos en la presente memoria también pueden contener proporciones no naturales de isótopos atómicos en uno o más de los átomos que constituyen dichos compuestos. Por ejemplo, los compuestos se pueden radiomarcar con isótopos radiactivos tales como, por ejemplo, tritio (3H), yodo-125 ( 25I) o carbono-14 (14C). Todas las variaciones isotópicas de los compuestos descritos en la presente memoria, ya sean radiactivas o no, se incluyen dentro del alcance contemplado.
En algunas realizaciones, los metabolitos de los compuestos descritos en la presente memoria son útiles para los métodos descritos en la presente memoria.
En algunas realizaciones, los compuestos contemplados en la presente memoria se proporcionan en forma de profármaco. El término “ profármaco” se refiere a un compuesto que se puede convertir en un compuesto (p. ej., un compuesto biológicamente activo) descrito en la presente memoria in vivo. Los profármacos pueden ser útiles por una variedad de razones conocidas en la técnica, que incluyen, por ejemplo, la facilidad de administración debido, p. ej., a la biodisponibilidad potenciada en la administración oral y similares. El profármaco también puede tener una solubilidad mejorada en composiciones farmacéuticas sobre los compuestos biológicamente activos. Un ejemplo, sin limitación, de un profármaco es un compuesto que se administra como un éster (es decir, el “profármaco” ) para facilitar la transmisión a través de una membrana celular donde la solubilidad en agua es perjudicial para la
movilidad pero que después se hidroliza metabólicamente al ácido carboxílico, la entidad activa, una vez dentro de la célula donde la solubilidad en agua es beneficiosa. Los procedimientos convencionales para la selección y preparación de derivados de profármacos adecuados se describen, por ejemplo, en “ DESIGN OF PRODRUGS” , (ed. H. Bundgaard, Elsevier, 1985), que se incorpora en la presente memoria por referencia para el fin limitado de describir procedimientos y preparación de derivados de profármacos adecuados.
Por consiguiente, en algunas realizaciones, los compuestos contemplados en la presente memoria se proporcionan en forma de éster de profármaco. La expresión “éster de profármaco” se refiere a derivados de los compuestos descritos en la presente memoria formados por la adición de cualquiera de una variedad de grupos formadores de éster, p. ej., grupos conocidos en la técnica, que se hidrolizan en condiciones fisiológicas. Los ejemplos de grupos éster de profármacos incluyen pivaloiloximetilo, acetoximetilo, ftalidilo, indanilo y metoximetilo, así como otros grupos conocidos en la técnica, incluido un grupo (5-R-2-oxo-1,3-dioxolen-4-il)metilo. Pueden encontrarse otros ejemplos de grupos éster de profármacos, por ejemplo, en T. Higuchi y V. Stella, en “ PRO-DRUGS AS NOVEL De L iVERY SYSTEMS” , Vol. 14, A.C.S. Symposium Series, American Chemical Society (1975); y “ BIOREVERSIBLE CARRIERS IN DRUG DESIGN: THEORY AND APPLICATION” , editado por E. B. Roche, Pergamon Press: Nueva York, 14-21 (1987) (que proporciona ejemplos de ésteres útiles como profármacos para compuestos que contienen grupos carboxilo). Cada una de las referencias mencionadas anteriormente se incorpora en la presente memoria por referencia con el fin limitado de describir grupos formadores de ésteres que pueden formar ésteres de profármacos.
En algunas realizaciones, los profármacos se pueden convertir lentamente en los compuestos descritos en la presente memoria útiles para los métodos descritos en la presente memoria cuando se colocan en un depósito de parche transdérmico con una enzima o reactivo químico adecuado.
Determinados compuestos descritos en la presente memoria pueden existir en formas no solvatadas, así como en formas solvatadas, incluidas las formas hidratadas. En general, las formas solvatadas son equivalentes a las formas no solvatadas y están incluidas dentro del alcance de los compuestos contemplados. Determinados compuestos de la presente invención pueden existir en múltiples formas cristalinas o amorfas. En general, todas las formas físicas son equivalentes para los compuestos y métodos contemplados en la presente memoria y se pretende que estén dentro del alcance descrito en la presente memoria.
III. Actividades biológicas
En algunas realizaciones, los compuestos descritos en la presente memoria presentan actividad inhibidora contra la trombina con actividades > 1 pM, p. ej., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 16, 18, 20, 22, 24, 26, 28, 30, 32, 34, 36, 38, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100 pM, o incluso mayores. En algunas realizaciones, los compuestos presentan actividad inhibidora contra la trombina con actividades entre 0,1 pM y 1 pM, p. ej., aproximadamente 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9 o 1,0 pM. En algunas realizaciones, los compuestos descritos en la presente memoria presentan actividad inhibidora contra la trombina con actividades < 0,1 pM, p. ej., aproximadamente 1, 2, 5, 10, 15, 20, 30, 40, 50, 60, 70, 80, 90 o 100 nM. También se contemplan intervalos de valores que utilizan una combinación de cualquiera de los valores indicados en la presente memoria como límites superior y/o inferior, por ejemplo, aunque no de forma limitativa, 1-10 nM, 10-100 nM, 0,1-1 pM, 1-10 pM, 10 100 pM, 100-200 pM, 200-500 pM o incluso 500-1000 pM. En algunas realizaciones, la actividad inhibidora está en el intervalo de aproximadamente 1-10 nM, 10-100 nM, 0,1-1 pM, 1-10 pM, 10-100 pM, 100-200 pM, 200-500 pM, o incluso 500-1000 pM. Se entiende que para fines de cuantificación, los términos “ actividad” , “ actividad inhibidora” , “actividad biológica” , “ actividad de trombina y similares en el contexto de un compuesto inhibidor descrito en la presente memoria pueden cuantificarse en una variedad de formas conocidas en la técnica. Salvo que se indique lo contrario, como se utilizan en la presente memoria, dichos términos se refieren a CI50 en el sentido habitual (es decir, concentración para lograr la mitad de la inhibición máxima).
La actividad inhibidora contra la trombina inhibe a su vez el proceso de coagulación de la sangre. Por consiguiente, los compuestos descritos en la presente memoria están indicados en el tratamiento o abordaje de trastornos trombóticos. En algunas realizaciones, una dosis o una dosis terapéuticamente eficaz de un compuesto descrito en la presente memoria será la que sea suficiente para alcanzar una concentración plasmática del compuesto o su(s) metabolito(s) activo(s) dentro de un intervalo establecido en la presente memoria, p. ej., aproximadamente 1-10 nM, 10-100 nM, 0,1-1 pM, 1-10 pM, 10-100 pM, 100-200 pM, 200-500 pM, o incluso 500-1000 pM, preferiblemente aproximadamente 1-10 nM, 10-100 nM o 0,1-1 pM. Sin pretender limitarse a ninguna teoría, se cree que dichos compuestos están indicados en el tratamiento o abordaje de trastornos trombóticos.
En algunas realizaciones, los compuestos descritos en la presente memoria presentan actividad inhibidora frente a KLKB1 con actividades entre 1 pM y 10 pM, por ejemplo, aproximadamente 1, 2, 3, 4, 5, 6, 7, 8, 9 o 10 pM. En algunas realizaciones, los compuestos descritos en la presente memoria presentan actividad inhibidora contra KLKB1 con actividades > 10 pM, p. ej., aproximadamente 10, 20, 50, 100, 150, 200, 300, 400, 500, 600, 700, 800, 900, 1000 pM o incluso mayores. En algunas realizaciones, los compuestos descritos en la presente memoria muestran actividad inhibidora frente a KLKB1 con actividades < 1 pM, por ejemplo, aproximadamente 900, 800, 700, 600, 500, 400, 300, 200, 100, 50 nM o incluso menores. También se contemplan intervalos de valores que utilizan una combinación de cualquiera de los valores indicados en la presente memoria como límites superior y/o inferior,
por ejemplo, aunque no de forma limitativa, 1-10 nM, 10-100 nM, 0,1-1 pM, 1-10 pM, 10-100 pM, 100-200 pM, 200 500 pM o incluso 500-1000 pM. En algunas realizaciones, la actividad inhibidora está en el intervalo de aproximadamente 1-10 nM, 10-100 nM, 0,1-1 pM, 1-10 pM, 10-100 pM, 100-200 pM, 200-500 pM, o incluso 500 1000 pM. Se entiende que para fines de cuantificación, el término “ actividad” , y las expresiones “ actividad inhibidora” , “ actividad biológica” , “ actividad de KLKB1” y similares en el contexto de un compuesto inhibidor descrito en la presente memoria pueden cuantificarse en una variedad de formas conocidas en la técnica. Salvo que se indique lo contrario, como se utilizan en la presente memoria, dichos términos se refieren a CI50 en el sentido habitual (es decir, concentración para lograr la mitad de la inhibición máxima).
La actividad inhibidora contra KLKB1 tiene un efecto sobre la cascada de coagulación y la respuesta inflamatoria. Por tanto, se ha propuesto que los inhibidores de KLKB1 pueden ser útiles en el tratamiento de enfermedades y estados patológicos trombóticos y fibrinolíticos.
Por consiguiente, los compuestos descritos en la presente memoria están indicados en el tratamiento o abordaje de una variedad de enfermedades o trastornos. En algunas realizaciones, una dosis o una dosis terapéuticamente eficaz de un compuesto descrito en la presente memoria será la que sea suficiente para alcanzar una concentración plasmática del compuesto o su(s) metabolito(s) activo(s) dentro de un intervalo establecido en la presente memoria, p. ej., aproximadamente 1 - 1 0 nM, 1 0 - 1 0 0 nM, 0 ,1 - 1 pM, 1 - 1 0 pM, 1 0 - 10 0 pM, 1 0 0 - 2 0 0 pM, 200-500 pM, o incluso 500-1000 pM, preferiblemente aproximadamente 1-10 nM, 10-100 nM o 0,1-1 pM. Sin querer limitarse a ninguna teoría, se cree que dichos compuestos están indicados en el tratamiento o abordaje de enfermedades asociadas con la trombina o la calicreína.
En algunas realizaciones, los compuestos inhiben selectivamente la trombina y/o la calicreína plasmática sobre serina proteasas relacionadas tales como tripsina, quimotripsina, factor XlIa, factor Xla, factor Xa y factor VlIa. En algunas realizaciones, los compuestos inhiben la quimotripsina con una CI50 superior a 1 pM. En algunas realizaciones, los compuestos inhiben la quimotripsina con una CI50 superior a 10 pM. En algunas realizaciones, los compuestos inhiben la quimotripsina con una IC50 superior a 100 pM. En algunas realizaciones, los compuestos inhiben el Factor XIa con una CI50 superior a 1 pM. En algunas realizaciones, los compuestos inhiben el Factor XIa con una CI50 superior a 10 pM. En algunas realizaciones, los compuestos inhiben el Factor XIa con una CI50 superior a 100 pM.
En algunas realizaciones, los compuestos permanecen en el plasma sanguíneo después de la infusión intravenosa. En algunas realizaciones, más del 50 % de la concentración inicial del compuesto permanece en el plasma sanguíneo de los ratones 1 hora después de la inyección intravenosa. En algunas realizaciones, más del 50 % de la concentración inicial del compuesto permanece en el plasma sanguíneo de ratones 3 horas o más después de la inyección intravenosa.
IV. Métodos de tratamiento y prevención de enfermedades
Enfermedades y afecciones relacionadas con la trombina (p. ej., trombosis). Las enfermedades trombóticas son las indicaciones primarias para la inhibición de la trombina, debido a la ubicación de la trombina en la cascada de la coagulación y, a su vez, la importancia de la cascada de la coagulación en la progresión de los procesos de coagulación de la sangre. Sin embargo, sin querer limitarse a ninguna teoría, se cree que la cascada de la coagulación en general, y la trombina en particular, es importante en una variedad de otras patologías.
Se ha descubierto que los compuestos descritos en la presente memoria, p. ej., compuestos aromáticos multisustituidos, muestran una acción inhibidora contra la trombina (factor II de coagulación sanguínea activado; CE 3.4.21.5). Esto, a su vez, inhibe el proceso de coagulación de la sangre.
Esta acción inhibidora es útil en el tratamiento de una variedad de trastornos trombóticos, tales como, pero sin limitación, enfermedades vasculares agudas tales como síndromes coronarios agudos; tromboembolias venosas, arteriales y cardiogénicas; la prevención de otros estados tales como coagulación intravascular diseminada u otras afecciones que implican la presencia o la posible formación de un trombo de coágulo sanguíneo. Otras indicaciones para los métodos descritos en la presente memoria incluyen las siguientes.
Durante mucho tiempo se ha reconocido que la progresión del cáncer se acompaña de trombosis venosa, pero no se ha entendido cómo se relaciona cada enfermedad. A partir de varios ensayos clínicos que estudian el tratamiento de TEV, los metanálisis han demostrado que las heparinas de bajo peso molecular (HBPM) mejoran la supervivencia general en subgrupos de pacientes con cáncer. Véase, p. ej., Zacharski, L. R. y Lee, A. Y., 2008, Expert Opin Investig Drugs, 17: 1029-1037; Falanga, A. y Piccioli, A., 2005, Current Opinión in Pulmonary Medicine, 11: 403-407; Smorenburg, S. M., y col., 1999, Thromb Haemost, 82: 1600-1604; Hettiarachchi, R. J., y col., 1999, Thromb Haemóst , 82: 947-952. Este hallazgo se confirmó en ensayos clínicos posteriores que midieron específicamente la supervivencia de los pacientes con cáncer. Véase, p. ej., Lee, A. Y. y col., 2005, J Clin Oncol, 23: 2123-2129; Klerk, C. P. y col., J Clin Oncol 2005, 23: 2130-2135; Kakkar, A. K., y col., 2004, J Clin OncoI, 22: 1944-1948; Altinbas, M., y col., 2004, J Thromb Haemost, 2: 1266-1271.
Más recientemente, los investigadores se han centrado en el efecto anticancerígeno específico de los IDT. Por ejemplo, se demostró que la heparina prolongó significativamente la supervivencia de pacientes con cáncer de pulmón microcítico limitado. Véase, p. ej., Akl, E. A., y col., 2008, J Exp Clin Cáncer Res , 27: 4. Otros investigadores encontraron que el uso sistémico de dabigatrán redujo la masa tumoral y prolongó el tiempo de supervivencia en modelos de glioma de rata, lo que llevó a la conclusión de que el dabigatrán debe considerarse como una nueva terapia para el glioma, un tipo de cáncer notoriamente difícil de tratar. Véase, p. ej., Hua, Y., y col., 2005, Acta Neurochir, Supl. 2005, 95: 403-406; Hua, Y., y col., 2005, J Thromb Haemost, 3: 1917-1923. Muy recientemente, se demostró que el etexilato de dabigatrán, un IDT recientemente aprobado por la FDA (véase, p. ej., Hughes, B., 2010, Nat Rev Drug Discovery, 9: 903-906) para indicaciones de DVT, inhibió tanto la invasión como la metástasis de tumores de mama malignos. Véase, p. ej., DeFeo, K. y col., 2010, Thrombosis Research, 125 (Suplemento 2): S188-S188; Defeo, K., y col., 2010, Cancer Biol Ther, 10: 1001-1008. Por lo tanto, el tratamiento con etexilato de dabigatrán condujo a una reducción del 50 % en el volumen del tumor a las 4 semanas sin pérdida de peso en los ratones tratados. El etexilato de dabigatrán también redujo las células tumorales en la sangre y las micrometástasis hepáticas en un 50-60 %. Estos investigadores concluyeron que el etexilato de dabigatrán puede ser beneficioso no solo para prevenir episodios trombóticos en pacientes con cáncer, sino también como terapia adyuvante para tratar tumores malignos.
Además, la hirudina y la HBPM nadroparina redujeron drásticamente el número de metástasis pulmonares cuando se administraron antes de la inoculación de células cancerosas. Véase, p. ej., Hu, L., y col., 2004, Blood, 104: 2746 51.
El inhibidor de trombina de novo d-Arg-Oic-Pro-d-Ala-Phe(p-Me) bloquea la invasión estimulada por trombina de la estirpe celular de cáncer de próstata PC-3 de una manera dependiente de la concentración. Véase, p. ej., Nieman, M. T., y col., 2008, J Thromb Haemost, 6 : 837-845. Se observó una velocidad reducida de crecimiento tumoral en ratones que recibieron dosis del pentapéptido a través del agua potable. Los ratones también mostraron un aumento reducido en el tamaño del tumor y un peso total del tumor reducido en comparación con los ratones no tratados. El examen microscópico de los tumores tratados mostró un número reducido de vasos sanguíneos grandes, concluyendo así que el pentapéptido interfería con la angiogénesis tumoral. Nieman, M. T., y col., Thromb Haemost, 104: 1044-8.
En vista de estos y otros estudios relacionados, se sugiere que los anticoagulantes afectan a la metástasis tumoral; es decir, procesos de angiogénesis, adhesión de células cancerosas, migración e invasión. Véase p. ej., Van Noorden, C. J., y col., 2 0 1 0 , Thromb Res, 125 Supl 2 : S77-79.
Enfermedad de Alzheimer. Experimentos muy recientes confirmaron niveles más altos de trombina en las células endoteliales del cerebro de pacientes con enfermedad de Alzheimer. Si bien los niveles de trombina “ normales” están relacionados con las funciones reguladoras del SNC, la acumulación de trombina en el cerebro es tóxica. También se ha encontrado que el inhibidor de la trombina neuronal Proteasa Nexina 1 (PN-1) se reduce significativamente en el cerebro con enfermedad de Alzheimer, a pesar de que los niveles de ARNm de PN-1 no varían. Estas observaciones han llevado a algunos investigadores a sugerir que la reducción de la trombina residente en el SNC resultará útil en el tratamiento de la enfermedad de Alzheimer (EA). Véase, p. ej., Vaughan, P. J., y col., 1994, Brain Res, 6 6 8 : 160-170; Yin, X., y col., 2010, Am J Pathol, 176: 1600-1606; Akiyama, H., y col., 1992, Neurosci Lett, 146: 152-154.
Esclerosis múltiple. Los investigadores encontraron que el tratamiento con hirudina en un modelo animal de esclerosis múltiple (EM) mostró una mejora espectacular en la gravedad de la enfermedad. Véase, p. ej., Han, M. H., y col., 2008, Nature, 451: 1076-1081. Se obtuvieron resultados similares tras el tratamiento con heparina (un IDT) y sulfato de dermatán, otro inhibidor de la coagulación. Véase, por ejemplo, Chelmicka-Szorc, E. y Arnason, BG, 1972, arco neurol, 27: 153-158; Inaba, Y., y col., 1999, Cell Inmunol, 198: 96-102. Otra evidencia muestra que la antitrombina III natural tiene efectos antiinflamatorios en enfermedades tales como la endotoxemia y otras afecciones relacionadas con la sepsis. Véase, por ejemplo, Wiedermann, C. J. y Romisch, J., 2002, Acta Med Austriaca, 29: 89-92. Los inhibidores de la trombina de origen natural presumiblemente se sintetizan in situ y tienen funciones protectoras en la inflamación del SNC. Por lo tanto, la inhibición terapéutica de la trombina se ha propuesto como un posible tratamiento para la EM. Véase p. ej., Luo, W., y col., 2009, en: THROMBIN, Maragoudakis, M. E.; Tsopanoglou, N. E., Eds. Springer New York: 2009; págs. 133-159.
Dolor. En un modelo de dolor en ratas con lesión parcial del nervio ciático, la hirudina intratecal impidió el desarrollo de dolor neuropático y redujo las respuestas al dolor durante 7 días. Los investigadores encontraron que después de la lesión, el dolor neuropático estaba mediado por la generación de trombina que, a su vez, activaba el receptor PAR-1 en la médula espinal. La hirudina inhibió la generación de trombina y finalmente condujo al alivio del dolor. Véase, p. ej., Garcia, P. S., y col., 2010, Thromb Haemost, 103: 1145-1151; Narita, M., y col., 2005, J Neurosci, 25: 10000-10009. Los investigadores plantean la hipótesis de que la trombina y los PAR no solo participan como parte de la cascada de la coagulación, sino también en la inflamación, la nocicepción y el neurodesarrollo. El desarrollo de un IDT para cruzarse con una farmacología no explotada dará lugar a tratamientos del dolor distintos de los opioides y los AINE, cuyas deficiencias están bien documentadas. Véase, p. ej., Garcia 2010, Id. Se ha informado que los inhibidores de la trombina conocidos son útiles para prevenir el accidente cerebrovascular en individuos con
fibrilación auricular. El inhibidor selectivo de la trombina ximelagatrán se estudió en dos ensayos clínicos de fase III (SPORTIF III y SPORTIF V), que compararon el ximelagatrán con la warfarina para la prevención de episodios cardioembólicos en pacientes con fibrilación auricular no valvular. Los investigadores del ensayo clínico SPORTIF III encontraron que el ximelagatrán, administrado a una dosis fija sin control de la coagulación, protege a los pacientes de alto riesgo con fibrilación auricular contra la tromboembolia, al menos con la misma eficacia que la warfarina bien controlada y se asocia con un menor sangrado. Cuando se combinaron los resultados de SPORTIF III y V, el ximelagatrán se asoció con una reducción del riesgo relativo del 16 % en el criterio de valoración compuesto de todos los accidentes cerebrovasculares (isquémicos o hemorrágicos), episodios embólicos sistémicos, hemorragia mayor y muerte. (Olsson, S. B. Lancet 2003, 362 (9397), 1691-1698; Hirsh, J. y col. Blood 2005, 105 (2), 453-463; Clemens, A. y col. Solicitud de patente WIPO WO/2008/009638). Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la trombina puede ser útil para prevenir el accidente cerebrovascular en individuos con fibrilación auricular.
Se ha informado que los inhibidores de la trombina conocidos son útiles en el tratamiento y la prevención del síndrome coronario agudo (Clemens, A. y col. Solicitud de patente WIPO WO/2008/009638). El SCA es un grupo de síntomas provocado por la isquemia miocárdica. El fármaco se podría utilizar como profilaxis del infarto de miocardio, o un determinado tiempo después del episodio (p. ej., después de un infarto de miocardio, después de una IM; es decir, terapia crónica, prevención secundaria). Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la trombina puede ser útil para tratar y prevenir el síndrome coronario agudo.
Se ha informado que los inhibidores de la trombina conocidos son útiles en la prevención de episodios cardíacos recurrentes después de un infarto de miocardio. El inhibidor selectivo de la trombina ximelagatrán se estudió en un ensayo clínico de fase II titulado ESTEEM, midiendo la eficacia y seguridad del inhibidor directo de la trombina oral ximelagatrán en pacientes con daño miocárdico reciente. El resultado del ensayo ESTEEM respalda la idea de que el tratamiento a largo plazo con un inhibidor directo de la trombina oral reduce los episodios trombóticos arteriales. El ximelagatrán oral junto con el ácido acetilsalicílico fue más eficaz que el ácido acetilsalicílico solo para reducir la frecuencia de los episodios cardiovasculares importantes durante 6 meses de tratamiento en pacientes con infarto de miocardio reciente. (Hirsh, J. y col. Blood 2005, 105 (2), 453-463.). Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la trombina puede ser útil para prevenir episodios cardíacos recurrentes después de un infarto de miocardio.
Se ha informado que los inhibidores de la trombina conocidos son útiles en la profilaxis postoperatoria de la trombosis venosa profunda. Se descubrió que el inhibidor selectivo de la trombina ximelagatrán es eficaz para la prevención de la tromboembolia venosa después de una intervención médica tal como el reemplazo total de cadera o rodilla (Francis, C.W. y col. Ann Intern Med 2002; 137: 648-55; Heit, J. A. y col. Arch Intern Med 2001; 161: 2215 21; Eriksson B. I. y col. Thromb Haemost 2003; 89: 288-96). Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la trombina puede ser útil en la profilaxis postoperatoria de la trombosis venosa profunda.
Se ha informado que los inhibidores de la trombina conocidos como el dabigatrán son útiles en el tratamiento a largo plazo de la embolia pulmonar. (Robertson L., Kesteven P., McCaslin J. E. Cochrane Database Syst Rev. 4 de diciembre de 2015; 12). Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la trombina puede ser útil en el tratamiento de la embolia pulmonar.
Se ha informado que los inhibidores de la trombina conocidos son útiles para la prevención de la coagulación en pacientes que se someten a una intervención coronaria percutánea. La intervención coronaria percutánea (ICP) requiere una terapia anticoagulante agresiva e, históricamente, se lograba con heparina no fraccionada. Sin embargo, en muchos pacientes, está contraindicada la heparina, especialmente en pacientes con trombocitopenia inducida por heparina (TIH). En dichos casos, la interrupción endovascular y el estado de hipercoagulabilidad que caracterizan a la TIH significan que los pacientes corren el riesgo de sufrir trombosis durante la ICP. (Lewis, B.E. y col. Catheterization and cardiovascular interventions 2002, 57 (2), 177-184; Kokolis, S. y col. Progress in cardiovascular diseases 2004, 46 (6 ), 506-523.) El dabigatrán, que ya había sido reivindicado como un inhibidor de la trombina y un anticoagulante útil en el ámbito clínico, también se publicó como medicación secundaria en el cateterismo cardíaco intervencionista percutáneo. (Reilly y col. Solicitud de patente WIPO WO/2010/020602). Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la trombina puede ser útil para prevenir la coagulación en pacientes que se someten a una intervención coronaria percutánea.
Se ha informado que los inhibidores de la trombina conocidos son útiles para el tratamiento de la hipertensión arterial pulmonar. El dabigatrán, un inhibidor selectivo de la trombina, se ha publicado como un fármaco útil para el tratamiento de la hipertensión arterial pulmonar (HAP). Además, el dabigatrán había encontrado uso como tratamiento de: (i); la hipertensión pulmonar provocada por trastornos de las cavidades izquierdas del corazón, (ii); la hipertensión pulmonar asociada a enfermedades pulmonares tales como fibrosis pulmonar, particularmente fibrosis pulmonar idiopática y/o hipoxia, (iii); la hipertensión pulmonar provocada por enfermedades tromboembólicas crónicas. (Feuring, M. Solicitud de patente WIPO WO/2010/020600). Sin desear quedar más ligados a teoría alguna,
es razonable creer que, en general, la inhibición de la trombina puede ser útil para el tratamiento de la hipertensión arterial pulmonar.
Se ha informado que los inhibidores de la trombina conocidos son útiles para el tratamiento de la hipertensión arterial pulmonar provocada por trastornos de las cavidades izquierdas del corazón (Feuring, M. Solicitud de patente WIPO W o /2010/020600). Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la trombina puede ser útil para el tratamiento de la hipertensión arterial pulmonar provocada por trastornos de las cavidades izquierdas del corazón.
Se ha informado que los inhibidores de la trombina conocidos son útiles para el tratamiento de la hipertensión arterial pulmonar asociada con enfermedades pulmonares tales como la fibrosis pulmonar, particularmente la fibrosis pulmonar idiopática y/o hipoxia (Feuring, M. Solicitud de patente WIPO WO/2010/020600). Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la trombina puede ser útil para el tratamiento de la hipertensión arterial pulmonar asociada con enfermedades pulmonares.
Se ha informado que los inhibidores de la trombina conocidos son útiles para el tratamiento de la hipertensión pulmonar provocada por enfermedades tromboembólicas crónicas (Feuring, M. Solicitud de patente WIPO WO/2010/020600). Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la trombina puede ser útil para el tratamiento de la hipertensión arterial pulmonar provocada por enfermedades tromboembólicas crónicas.
La fibrilación auricular no valvular es una alteración cardíaca sostenida que se suele asociar con cardiopatías. Se ha informado que los inhibidores de la trombina conocidos, como el ximelagatrán, son útiles para la prevención del accidente cerebrovascular en pacientes con fibrilación auricular no valvular (Diener H.-C. Cerebrovasc Dis 2006; 21: 279-293). Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la trombina puede ser útil para la prevención del accidente cerebrovascular en pacientes con fibrilación auricular no valvular.
Un ataque isquémico transitorio (AIT) es un episodio agudo de disfunción neurológica temporal que suele durar menos de una hora; se debe a una isquemia cerebral focal, de la médula espinal o de la retina; y no se asocia con el infarto tisular agudo. En las personas que tienen un AIT, la incidencia de un accidente cerebrovascular subsiguiente es tan alta como del 11 % durante los 7 siguientes días y del 24-29 % durante los 5 siguientes años. En vista del alto riesgo de accidente cerebrovascular a corto plazo después de un AIT, muchos médicos creen que la terapia antitrombótica debe iniciarse tan pronto como se haya descartado la hemorragia intracraneal. Los medicamentos para la prevención de accidentes cerebrovasculares que normalmente se recomiendan para el AIT cardioembólico son los siguientes: Para pacientes con fibrilación auricular después de AIT, anticoagulación a largo plazo con warfarina (aspirina a 325 mg/día para aquellos que no pueden tomar anticoagulantes orales); En el infarto agudo de miocardio (IM) con trombo en el ventrículo izquierdo, anticoagulación oral con warfarina; aspirina concurrente hasta 162 mg/día para Enfermedad Arterial Coronaria Isquémica [EACI]); En una miocardiopatía dilatada, anticoagulación oral con warfarina o tratamiento antiplaquetario; En la enfermedad reumática de la válvula mitral, anticoagulación oral con warfarina. Para los pacientes con AIT y accidente cerebrovascular isquémico de origen cardíaco debido a fibrilación auricular, los antagonistas de la vitamina K (AVK) son muy eficaces para prevenir el accidente cerebrovascular isquémico recurrente, pero tienen limitaciones importantes y, por lo tanto, están infrautilizados. El tratamiento antiplaquetario es mucho menos eficaz que los AVK. El inhibidor directo de la trombina, el etexilato de dabigatrán, ha demostrado eficacia sobre la warfarina en un ensayo reciente. Otros nuevos anticoagulantes, incluidos los inhibidores orales del factor Xa, rivaroxabán, apixabán y edoxabán, el inhibidor parenteral del factor Xa, idrabiotaparinux, y el nuevo AVK, tecarfarina, se estaban evaluando en 2010. (Hankey, G. J.; Eikelboom, J. W. “Antithrombotic Drugs for Patients with Ischaemic Stroke and Transient Ischaemic Attack to Prevent Recurrent Major Vascular Events” . The Lancet Neurology 2010, 9 (3), 273-284.)
Se ha informado que los inhibidores de la trombina conocidos son útiles para el tratamiento de la tromboembolia venosa debida a la formación de un trombo dentro de una vena (trombosis venosa) asociada con factores de riesgo adquiridos (descanso en cama prolongado, cirugía, lesión, malignidad, embarazo y posparto) o heredados (deficiencia de inhibidores naturales de la coagulación) (Marsic, L. P. y col. Solicitud de Patente WIPO WO/2003/048155). Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la trombina puede ser útil para el tratamiento de la tromboembolia venosa debida a la formación de un trombo dentro de una vena asociada con factores de riesgo adquiridos o heredados y/o la embolia de venas periféricas provocada por un trombo desprendido. Un ejemplo de factor de riesgo adquirido sería una tromboembolia venosa previa y/o embolia de venas periféricas provocada por un trombo desprendido. Un ejemplo de factor de riesgo adquirido sería una tromboembolia venosa previa.
Se ha informado que los inhibidores de la trombina conocidos son útiles para el tratamiento de la tromboembolia cardiogénica debida a la formación de un trombo en el corazón asociada con arritmia cardíaca, defecto de válvula cardíaca, válvulas cardíacas protésicas o cardiopatía, embolia de arterias periféricas provocada por un trombo desprendido, más comúnmente en el cerebro (accidente cerebrovascular isquémico). Véase Marsic, L. P. y col.
Solicitud de Patente WIPO WO/2003/048155. Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la trombina puede ser útil para el tratamiento de la tromboembolia cardiogénica.
Se ha informado que los inhibidores de la trombina conocidos son útiles para el tratamiento de la trombosis arterial debida a procesos ateroescleróticos subyacentes en las arterias que obstruyen u ocluyen una arteria y provocan isquemia miocárdica (angina de pecho, síndrome coronario agudo) o infarto de miocardio, que obstruyen u ocluyen una arteria periférica arterial (enfermedad arterial periférica isquémica) y que obstruyen u ocluyen la arteria después de la intervención en el vaso sanguíneo (reoclusión o reestenosis después de angioplastia coronaria transluminal, reoclusión o reestenosis después de angioplastia transluminal percutánea de arterias periféricas). Véase Marsic, L. P. y col. Solicitud de Patente WIPO WO/2003/048155. Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la trombina puede ser útil para el tratamiento de la trombosis arterial.
Se ha informado que los inhibidores de la trombina conocidos son útiles para el tratamiento de la coagulación intravascular diseminada en varios estados (p. ej., en complicaciones en el embarazo, en enfermedades malignas metastásicas, después de lesiones extensas, en sepsis bacteriana) cuando la activación trombogénica provoca coagulación disfuncional con formación generalizada de trombos dentro del sistema vascular. Véase Marsic, L. P. y col. Solicitud de Patente WIPO WO/2003/048155. Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la trombina puede ser útil para el tratamiento de la coagulación intravascular diseminada.
Se ha informado que los inhibidores de la trombina conocidos son útiles como terapia adyuvante junto con la terapia trombolítica en infarto de miocardio reciente, junto con la aspirina en pacientes con angina de pecho inestable que van a someterse a angioplastia transluminal percutánea y en el tratamiento de pacientes con trombosis y con trombocitopenia inducida por heparina (Marsic, L.P. y col. Solicitud de Patente WIPO WO/2003/048155). Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la trombina puede ser útil como terapia adyuvante con otras terapias antitrombóticas.
Se ha informado que los inhibidores de la trombina conocidos son útiles para el tratamiento de la inflamación (Kirk, I. Solicitud de patente WIPO WO/2000/041716), la diabetes mellitus de tipo I (Korsgren, O.; Nillson, B. Solicitud de patente WIPO WO/2003/061682), el cáncer (Kakkar, A. K. y col. J Clin Oncol 2004, 2 2 , (1 0 ), 1944-8; Hua, Y. y col. Acta Neurochir Suppl 2005, 95, 403-6; Nieman, M. T. y col. J Thromb Haemost, 6 (2008), 837-845; Van Ryn, J.; Clemens, A. Solicitud de patente WIPO WO/2010/020601), la fibrosis (Duplantier, J. G. y col. Gut, 2004, 53: 1682 1687; Seijo, S. y col. J Hepatol, 2007, 46: 286-294; Assy, N. y col. Dig Dis Sci, 2007, 52: 1187-1193; Bogatkevich, G. S. y col. Arthritis Rheum, 2009, 60: 3455-3464) y el dolor (Garcia, P.S. y col. Thromb Haemost, 103: 1145-1151; Narita, M. y col. J Neurosci, 2005, 25: 10000-10009). Los metanálisis de ensayos clínicos que estudiaron el uso de anticoagulantes en pacientes oncológicos mostraron que las heparinas de bajo peso molecular (HBPM), inhibidores selectivos de la trombina, mejoran la supervivencia general en subgrupos de pacientes con cáncer. Este hallazgo se comprobó en ensayos clínicos posteriores, en particular, los ensayos clínicos FAMOUS, que midieron específicamente la supervivencia de los pacientes con cáncer.
Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la trombina puede ser útil para el tratamiento de enfermedades o trastornos trombóticos y/o enfermedades o trastornos que implican un trombo de coágulo sanguíneo o la posible formación de un trombo de coágulo sanguíneo y/o implica además accidente cerebrovascular y/o uno o más ataques isquémicos transitorios (AIT) y/o hipertensión pulmonar. Dichas afecciones incluyen, por ejemplo, síndrome coronario agudo, tromboembolia, trombosis, inflamación, diabetes mellitus, cáncer, fibrosis, enfermedad de Alzheimer, esclerosis múltiple, dolor, episodios cardíacos recurrentes después de un infarto de miocardio o similares.
Enfermedades y afecciones relacionadas con la calicreína. Las enfermedades o los trastornos relacionados con la calicreína son condiciones biológicas asociadas o moderadas por la calicreína. Incluyen, aunque no de forma limitativa, aquellas condiciones asociadas con vías biológicas que son moderadas por la calicreína plasmática. Un ejemplo de dicha vía es el sistema calicreína-cinina (Moreau, M. E. 2005, Journal of Pharmacological Sciences, 99, 6 ). Las enfermedades o los trastornos relacionados con la calicreína incluyen, aunque no de forma limitativa, fibrosis, inflamación, trombosis, angioedema hereditario, trastornos de la piel, cáncer y enfermedades oftálmicas. Las enfermedades oftálmicas incluyen, aunque no de forma limitativa, edema macular diabético, retinopatía diabética y degeneración macular relacionada con la edad.
Edema macular diabético. En modelos de roedores, se ha demostrado que la activación de KLKB1 en el ojo aumenta la permeabilidad vascular de la retina; mientras que la inhibición del sistema calicreína-cinina reduce la fuga retiniana inducida por la diabetes y la hipertensión. Estos hallazgos sugieren que la activación intraocular de la vía KLKB1 puede contribuir a una permeabilidad vascular retiniana excesiva que puede provocar edema macular diabético. Por lo tanto, las pruebas sugieren que los inhibidores de KLKB1 pueden brindar una nueva oportunidad terapéutica para reducir la permeabilidad vascular de la retina (Feener, E. P. 2010, Curr Diab Rep 10, 270).
Angioedema hereditario. Ecallantide (Kalbitor) es una proteína recombinante de 60 aminoácidos que actúa como un potente inhibidor reversible de KLKB1 (Schneider L., y col. 2007, J Allergy Clin Immunol, 120, 416) y ha sido
aprobado por la FDA para el tratamiento de ataques agudos de AngioEdema Hereditario (AEH). Por lo tanto, la inhibición de la calicreína plasmática puede ser un tratamiento útil para el AEH y existe un gran interés en el desarrollo de inhibidores de la calicreína plasmática como terapia para el AEH.
Las personas hiperglucémicas y diabéticas tienen un riesgo elevado de hemorragia durante la terapia trombolítica. En modelos de roedores de Hemorragia IntraCerebral (HIC), se ha demostrado que la inhibición o eliminación de KLKB1 reduce este efecto. Si bien el mecanismo no se comprende por completo, estas pruebas sugieren que los inhibidores de la calicreína plasmática pueden ser útiles en el tratamiento de la hemorragia cerebral (Feener, E. P.
Curr Diab Rep 2010, 10, 270).
Se ha demostrado que los inhibidores de la calicreína plasmática y el factor XIIa son neuroprotectores en modelos animales de accidente cerebrovascular isquémico agudo y lesión cerebral traumática, reduciendo la formación de edemas, la inflamación y la trombosis (Albert-WeiBenberger C., Siren A. L., Kleinschnitz C. Prog Neurobiol. 2013, 101-102, 65-82). Por lo tanto, la evidencia sugiere que los inhibidores de la calicreína plasmática pueden ser útiles en el tratamiento del accidente cerebrovascular isquémico agudo y la lesión cerebral traumática.
La calicreína plasmática también puede escindir el péptido 1 similar al glucagón (GLP-1) y el neuropéptido Y (NPY), ambos sustratos de la dipeptidil peptidasa-4 (DPP-4), una diana farmacológica validada para la diabetes. En el caso del GLP-1, la escisión por KLKB1 reduce tanto su potencia como la estabilidad del plasma. En el caso del NPY, la escisión por KLKB1 reduce su afinidad hacia los receptores Y2 e Y5. Por lo tanto, la evidencia sugiere que los inhibidores de la calicreína plasmática pueden ser útiles en la modulación de la homeostasis energética y en el tratamiento de la diabetes. (Feener, E. P. Curr Diab Rep 2010, 10, Feener, E. P. y col., Biol. Chem. 2013, 394, 319).
El sistema calicreína-cinina participa en la regulación del factor de crecimiento endotelial vascular (VEGF, Vascular Endothelial Growth Factor), la NO sintasa endotelial y el factor de crecimiento de fibroblastos 2, la totalidad de los cuales participa en la angiogénesis (Bader M. 2009, Arteriosclerosis, Thrombosis, and Vascular Biology, 29: 617). La calicreína tisular (KLK1) se ha relacionado con el crecimiento de los vasos sanguíneos (Miura S., 2003, Hypertension, 41, 1118). Se han propuesto terapias que moderan la angiogénesis para el tratamiento tanto del Edema Macular Diabético (EMD) como de la Degeneración Macular Relacionada con la Edad (DMRE) (Syed, B. A.; Evans, J. B.; Bielory, L., 2012, Nature Reviews Drug Discovery, 11, 827). Sin desear quedar más ligados a teoría alguna, es razonable concluir que los inhibidores de KLK1 pueden ser útiles en el tratamiento de la retinopatía diabética, el EMD y la DMRE.
Los estudios han demostrado que la inflamación desempeña un papel importante en el origen y desarrollo de la DMRE, y el tratamiento suele incluir antiinflamatorios tales como los corticoesteroides (Telander, D., 2011, Seminars in Ophthalmology, 26(3), 192). La conexión entre el sistema calicreína-cinina y la inflamación también está bien establecida (Duchene, 2 0 1 1 , “ Kallikrein-kinin system in inflammatory diseases” . Kinins. De Gruyter. 261). Sin desear quedar más ligados a teoría alguna, es razonable concluir que la naturaleza antiinflamatoria de los inhibidores de la calicreína (p. ej., KLK1 y KLKB1) puede ser útil en el tratamiento de la DMRE.
PF-04886847 es un inhibidor de la calicreína plasmática y ha demostrado ser eficaz para reducir niveles plasmáticos de la 6 -ceto-PGF1a en ratas tratadas con lipopolisacárido (LPS) (Kolte, D. y col. Cardiovascular & Hematological Agents in Medicinal Chemistry, 2012, 10, 154-166). Sin desear quedar más ligados a teoría alguna, es razonable creer que los inhibidores de la calicreína plasmática pueden ser útiles en el tratamiento del choque hipotensivo durante la sepsis.
Daiichi Seiyaku Co Ltd recibió la aprobación en Japón para comercializar el cetraxato para la gastritis y las úlceras pépticas. El cetraxato se informa como un inhibidor de la calicreína plasmática (solicitud de patente WIPO WO/2006/108643). Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la calicreína plasmática puede ser útil en el tratamiento de la gastritis y las úlceras pépticas.
Fibrosis. Las calicreínas son un subgrupo de las serina proteasas, divididas en calicreínas plasmáticas (KLKB1) y calicreínas tisulares. La KLKB1 libera cininas (bradicinina y calidina) de los cininógenos, péptidos responsables de la regulación de la presión arterial y la activación de la inflamación. En la vía de activación por contacto de la cascada de coagulación, la KLKB1 ayuda en la conversión del factor XII al factor XIIa (Keel, M.; Trentz, O. Injury 2005, 36, 691-709). El factor XIIa convierte FXI en FXIa que, a su vez, activa FIX, que, con su cofactor FVIIIa, forma el complejo de tenasa, que finalmente activa FX a FXa. En la parte de la fibrinólisis de la cascada de coagulación, KLKB1 sirve para convertir el plasminógeno en plasmina. Por tanto, se ha propuesto que los inhibidores de KLKB1 pueden ser útiles en el tratamiento de enfermedades y afecciones trombóticas y fibrinolíticas (patente de EE. UU. n.° 7.625.944; Bird y col. Thrombosis and Hemostasis 2012, 107, 1141).
Varios estudios han demostrado la utilidad de la terapia anticoagulante en los trastornos fibróticos. Por ejemplo, en un modelo de rata de lesión hepática crónica inducida por CCU, el IDT SSR182289 disminuyó significativamente la fibrogénesis hepática tras 7 semanas de administración. Se hicieron observaciones similares en otros estudios que utilizaron las HBPM nadroparina, tinzaparina, enoxaparina y dalteparina sódica. Véase, p. ej., Duplantier, J. G., y col., 2004, Gut, 53: 1682-1687; Abdel-Salam, O. M., y col., 2005, Pharmacol Res, 51: 59-67; Assy, N., y col., 2007,
Dig Dis Sci, 52:1187-1193; Abe, W., y col., 2007, J Hepatol, 46:286-294. Por tanto, puede ser útil un inhibidor de trombina como anticoagulante en el tratamiento de enfermedades fibrinolíticas.
En otro ejemplo, el IDT melagatrán redujo en gran medida el daño por isquemia-reperfusión en un modelo de trasplante de riñón en el cerdo blanco grande. Esto condujo a una mejora drástica de la supervivencia del injerto renal a los 3 meses. Véase, p. ej., Favreau, F., y col., 2010, Am J Transplant, 10: 30-39.
Estudios recientes han demostrado que, en un modelo de ratón de fibrosis pulmonar inducida por bleomicina, el tratamiento con etexilato de dabigatrán redujo los episodios profibróticos importantes en los fibroblastos pulmonares, incluida la producción de colágeno y factor de crecimiento del tejido conjuntivo. Véase, p. ej., Silver, R. M., y col., 2010, Am. J. Respir. Crit. Care Med., 181: A6780; Bogatkevich, G. S., y col., 2009, Arthritis Rheum, 60: 3455-3464.
Las anteriores pruebas experimentales apuntan a una estrecha relación entre la trombina y la fibrosis, y sugieren nuevas oportunidades terapéuticas para la fibrosis mediante el uso de inhibidores de la trombina. Véase, p. ej., Calvaruso, V., y col., 2008, Gut, 57: 1722-1727; Chambers, R. C., 2008, Br J Pharmacol, 153 Supl 1: S367-378; Chambers, R. C. y Laurent, G. J., 2002, Biochem Soc Trans, 30: 194-200; Howell, D. C., y col., 2001, Am J Pathol, 159: 1383-1395.
Inflamación. La calicreína se ha relacionado durante mucho tiempo con la inflamación (Clements, J. A. “THE MOLECULAR BIOLOGY OF THE KALLIKREINS AND THEIR ROLES IN INFLAMMATION” , Academic Press: San Diego, CA, 1997; Vol. 5). Existen pruebas experimentales de que la KLKB1 está asociada con la sepsis y la artritis inflamatoria (Colman, R. W., 1998, Clinical Reviews in Allergy and Immunology, 16: 365). Por lo tanto, un inhibidor de KLKB1 puede ser útil en el tratamiento de afecciones inflamatorias asociadas con el sistema calicreína-cinina, tales como el síndrome de respuesta inflamatoria sistémica, sepsis, artritis reumatoide y enfermedad intestinal inflamatoria.
Sin desear quedar más ligados a teoría alguna, es razonable creer que, en general, la inhibición de la calicreína puede ser útil para el tratamiento de enfermedades o trastornos relacionados con la calicreína y/o enfermedades o trastornos. Dichos trastornos incluyen, por ejemplo, enfermedades trombóticas, enfermedades fibrinolíticas, trastornos fibróticos, cáncer, afecciones inflamatorias, afecciones dermatológicas o similares.
Por consiguiente, en otro aspecto, se proporciona un método para tratar una enfermedad o un trastorno en un sujeto que lo necesite. El método incluye administrar un compuesto de cualquiera de las Fórmulas (la), (Ib), (II), (III), (IV) o (V) como se describe en la presente memoria, un compuesto como se expone en la Tabla A, Tabla B, la Tabla C, o la Tabla D, una sal, un éster, un solvato o un profármaco farmacéuticamente aceptable del mismo, o una composición farmacéutica del mismo, a un sujeto que lo necesite en una cantidad eficaz para tratar la enfermedad o el trastorno. Las expresiones “cantidad terapéuticamente eficaz” , “cantidad eficaz para tratar” , “cantidad eficaz para prevenir” y similares se refieren a la cantidad de fármaco o agente farmacéutico (p. ej., compuesto o composición farmacéutica descritos en la presente memoria) que provocará la respuesta biológica o médica de un tejido, sistema, animal o ser humano que un investigador, veterinario, médico u otro clínico esté buscando obtener.
Los compuestos útiles para los métodos descritos en la presente memoria incluyen los compuestos establecidos para las Fórmulas (Ia), (Ib), (II), (III), (IV) o (V) y los compuestos establecidos en la Tabla A, Tabla B, Tabla C o Tabla D anteriores.
En algunas realizaciones de los métodos descritos en la presente memoria, la enfermedad o el trastorno que se va a tratar puede incluir una o más enfermedades o trastornos trombóticos y/o puede implicar un trombo de coágulo sanguíneo o la posible formación de un trombo de coágulo sanguíneo. En algunas realizaciones, la enfermedad o el trastorno trombótico puede ser síndrome coronario agudo, tromboembolia y/o trombosis. En algunas realizaciones, la tromboembolia puede ser tromboembolia venosa, tromboembolia arterial y/o tromboembolia cardiogénica. En algunas realizaciones, la tromboembolia venosa puede incluir trombosis venosa profunda y/o embolia pulmonar. En algunas realizaciones, la trombosis venosa profunda y/o la embolia pulmonar pueden ocurrir después de una intervención médica. En algunas realizaciones, la enfermedad o el trastorno trombótico puede implicar coagulación disfuncional o coagulación intravascular diseminada. En algunas realizaciones, el sujeto con coagulación disfuncional puede someterse a una Intervención Coronaria Percutánea (ICP). En algunas realizaciones, la enfermedad o el trastorno trombótico puede implicar un trombo de coágulo sanguíneo o la posible formación de un trombo de coágulo sanguíneo y además puede implicar accidente cerebrovascular y/o uno o más ataques isquémicos transitorios (AIT). En algunas realizaciones, la enfermedad o el trastorno trombótico que implica un trombo de coágulo sanguíneo o la posible formación de un trombo de coágulo sanguíneo puede implicar además un accidente cerebrovascular, en donde el sujeto puede tener fibrilación auricular no valvular. En algunas realizaciones, la enfermedad o el trastorno trombótico puede implicar un trombo de coágulo sanguíneo o la posible formación de un trombo de coágulo sanguíneo y además puede implicar hipertensión pulmonar. En algunas realizaciones, la hipertensión pulmonar puede estar provocada por uno o más trastornos de las cavidades izquierdas del corazón y/o enfermedad tromboembólica crónica. En algunas realizaciones, la hipertensión pulmonar puede estar asociada con una o más enfermedades pulmonares, incluida la fibrosis pulmonar (idiopática o de otro tipo) y/o hipoxia.
En algunas realizaciones, la tromboembolia venosa puede estar asociada con la formación de un trombo dentro de una vena asociado con uno o más factores de riesgo adquiridos o heredados y/o la embolia de venas periféricas provocada por un trombo desprendido. En algunas realizaciones, uno o más factores de riesgo pueden incluir una tromboembolia venosa previa. En algunas realizaciones, la tromboembolia cardiogénica puede deberse a la formación de un trombo en el corazón asociado con arritmia cardíaca, defecto de válvula cardíaca, válvulas cardíacas protésicas o cardiopatía y/o embolia de arterias periféricas provocada por un trombo desprendido. En algunas realizaciones, el trombo desprendido puede estar en el cerebro (accidente cerebrovascular isquémico). En algunas realizaciones, el trombo desprendido puede provocar un ataque isquémico transitorio (AIT). En algunas realizaciones, la tromboembolia cardiogénica puede deberse a una fibrilación auricular no valvular. En algunas realizaciones, la trombosis puede ser una trombosis arterial. En algunas realizaciones, la trombosis arterial puede deberse a uno o más procesos ateroescleróticos subyacentes en las arterias. En algunas realizaciones, uno o más procesos ateroescleróticos subyacentes en las arterias pueden obstruir u ocluir una arteria, provocar isquemia miocárdica (angina de pecho, síndrome coronario agudo), provocar infarto de miocardio, obstruir u ocluir una arteria periférica (enfermedad arterial periférica isquémica), y/u obstruir u ocluir la arteria después de una operación en un vaso sanguíneo (reoclusión o reestenosis después de angioplastia coronaria transluminal, reoclusión o reestenosis después de angioplastia transluminal percutánea de arterias periféricas).
En algunas realizaciones, la enfermedad o el trastorno puede incluir fibrosis, enfermedad de Alzheimer, esclerosis múltiple, dolor, cáncer, inflamación y/o diabetes mellitus de tipo I. En algunas realizaciones, la enfermedad o el trastorno puede implicar episodios cardíacos recurrentes después de un infarto de miocardio.
En algunas realizaciones, el tratamiento o la prevención pueden incluir una terapia adyuvante. En algunas realizaciones, el sujeto puede tener un infarto de miocardio y la terapia adyuvante puede ser junto con la terapia trombolítica. En algunas realizaciones, el sujeto puede tener angina de pecho inestable, trombosis y/o trombocitopenia inducida por heparina, y la terapia adyuvante puede estar junto con tratamiento antiplaquetario. En algunas realizaciones, el sujeto puede tener fibrilación auricular no valvular, y la terapia adyuvante puede darse junto con una o más terapias diferentes.
En algunas realizaciones de los métodos descritos en la presente memoria, la enfermedad o el trastorno puede ser un trastorno relacionado con la calicreína. En algunas realizaciones, el trastorno relacionado con la calicreína puede ser una enfermedad trombótica, una enfermedad fibrinolítica, un trastorno fibrótico, un tipo de cáncer, una afección inflamatoria o una afección dermatológica.
En algunas realizaciones, el trastorno relacionado con la calicreína puede ser una enfermedad oftálmica. En algunas realizaciones, el compuesto o la composición farmacéutica se puede administrar en forma de una composición oftálmica aplicada por vía tópica en el ojo. En algunas realizaciones, la composición oftálmica puede estar en forma de colirio. En algunas realizaciones, el compuesto o la composición farmacéutica se puede administrar en forma de una composición oftálmica mediante inyección intravítrea. En algunas realizaciones, la enfermedad oftálmica puede ser edema macular diabético, angioedema hereditario, degeneración macular relacionada con la edad o retinopatía diabética.
En algunas realizaciones en donde la enfermedad o el trastorno puede ser un tipo de cáncer, dicho tipo de cáncer puede ser adenocarcinoma de pulmón no microcítico, de cuello uterino o testicular. En algunas realizaciones, el cáncer puede ser cáncer de pulmón microcítico limitado. En algunas realizaciones, el cáncer puede ser un glioma. En algunas realizaciones, el cáncer puede ser cáncer de mama maligno. En algunas realizaciones, el cáncer puede ser una micrometástasis. En algunas realizaciones, la micrometástasis puede ser de la sangre o del hígado. En algunas realizaciones, el cáncer puede ser una metástasis de pulmón. En algunas realizaciones, el cáncer puede ser cáncer de próstata.
En algunas realizaciones en donde la enfermedad o el trastorno puede ser una afección inflamatoria, dicha afección inflamatoria puede ser sepsis, enfermedad intestinal inflamatoria, síndrome de respuesta inflamatoria sistémica, artritis inflamatoria o artritis reumatoide.
En algunas realizaciones en donde la enfermedad o el trastorno puede ser una afección dermatológica, dicha afección dermatológica puede ser dermatitis atópica, psoriasis o síndrome de Netherton.
En otro aspecto, se proporciona un método para prevenir una enfermedad o un trastorno en un sujeto. El método incluye administrar un compuesto de cualquiera de las Fórmulas (Ia), (Ib), (II), (III), (IV) o (V) como se describe en la presente memoria, compuesto como se expone en cualquiera de las Tablas A, Tabla B, Tabla C o Tabla D de la presente memoria, una sal, un éster, un solvato o un profármaco farmacéuticamente aceptable del mismo, o una composición farmacéutica del mismo, a un sujeto que lo necesite en una cantidad eficaz para prevenir la enfermedad o el trastorno.
V. Ensayos
Los compuestos descritos en la presente memoria se pueden ensayar mediante una variedad de métodos conocidos en la técnica y descritos en la presente memoria, para la inhibición de la actividad biológica, p. ej., la actividad proteasa, de una variedad de proteínas, p. ej., trombina y KLKB1.
La actividad de la trombina que se informa en la presente memoria (p. ej., Tabla A) se obtuvo de la siguiente manera. Se obtuvo trombina humana de Haematologic Technologies Inc. El sustrato cromogénico S-2238 se obtuvo de DiaPharma. Se analizó la trombina en tampón que contenía Tris 0,05 M (pH 7,4), NaCl 0,015 M y PEG-8000 al 0,01 %. La concentración final de enzima utilizada fue trombina 3 nM. La concentración final de sustrato utilizada fue 125 pM S-2238 para la trombina. Todos los ensayos se realizaron en placas de microtitulación de 96 pocillos a temperatura ambiente (TA). La enzima y el inhibidor se preincubaron durante 10 minutos, después se añadió el sustrato y se leyó a 405 nm en un espectrofotómetro SpectraMax Plus (Molecular Devices). Los valores de CI 50 inhibidora se determinaron mediante la adición del compuesto de ensayo como diluciones en serie triples de diez puntos en solución tampón, como se conoce en la técnica. La placa se leyó 10 minutos después de la adición del sustrato. La CI50 se calculó representando el porcentaje (%) de inhibición frente a la concentración del compuesto y ajustando los datos a una curva sigmoidea restringida de cuatro parámetros, como se conoce en la técnica.
La actividad de la calicreína KLKB1 que se informa en la presente memoria (p. ej., Tabla B) se obtuvo de la siguiente manera. La proteína KLKB1 humana se obtuvo de Enzyme Research Labs. El sustrato cromogénico S-2302 se obtuvo de DiaPharma. Se analizó KLKB1 en tampón que contenía T ris 0,05 M (pH 7,4), NaCl 0,01 M y / PEG-8000 al 0,2 % p/v. La concentración final de enzima utilizada fue KLKB1 3 nM. La concentración final de sustrato utilizada fue S-2302 250 pM para KLKB1. Todos los ensayos se realizaron en placas de microtitulación de 96 pocillos a temperatura ambiente (TA). La enzima y el inhibidor se preincubaron durante 10 minutos, después se añadió el sustrato y se leyó a 405 nm en un espectrofotómetro SpectraMax Plus (Molecular Devices). Los valores de CI50 inhibidora se determinaron mediante la adición del compuesto de ensayo como diluciones en serie triples de diez puntos en solución tampón, como se conoce en la técnica. La placa se leyó 10 minutos después de la adición del sustrato. La CI50 se calculó representando el porcentaje (%) de inhibición frente a la concentración del compuesto y ajustando los datos a una curva sigmoidea restringida de cuatro parámetros, como se conoce en la técnica.
La actividad de la quimotripsina indicada en la presente memoria (p. ej., Tabla D) se obtuvo de la siguiente manera. La a-quimotripsina de páncreas humano se obtuvo de Sigma. El sustrato cromogénico S-7388 se obtuvo de Sigma. La concentración final de sustrato utilizada fue de 250 pM para la quimotripsina. Todos los ensayos se realizaron en placas de microtitulación de 96 pocillos a temperatura ambiente (TA). La enzima y el inhibidor se preincubaron durante 10 minutos, después se añadió el sustrato y se leyó a 405 nm en un espectrofotómetro SpectraMax Plus (Molecular Devices). Los valores de CI 50 inhibidora se determinaron mediante la adición del compuesto de ensayo como diluciones en serie triples de diez puntos en solución tampón, como se conoce en la técnica. La placa se leyó 5 minutos después de la adición del sustrato. La CI50 se calculó representando el porcentaje (%) de inhibición frente a la concentración del compuesto y ajustando los datos a una curva sigmoidea restringida de cuatro parámetros, como se conoce en la técnica.
La actividad del Factor Xla que se informa en la presente memoria (p. ej., Tabla D) se obtuvo de la siguiente manera. El factor humano Xla se obtuvo de Enzyme Research. El sustrato cromogénico S-2366 se obtuvo de DiaPharma. La concentración final de sustrato utilizada fue de 10 mM para el Factor Xla. Todos los ensayos se realizaron en placas de microtitulación de 96 pocillos a temperatura ambiente (TA). La enzima y el inhibidor se preincubaron durante 10 minutos, después se añadió el sustrato y se leyó a 405 nm en un espectrofotómetro SpectraMax Plus (Molecular Devices). Los valores de CI 50 inhibidora se determinaron mediante la adición del compuesto de ensayo como diluciones en serie triples de diez puntos en solución tampón, como se conoce en la técnica. La placa se leyó 10 minutos después de la adición del sustrato. La CI50 se calculó representando el porcentaje (%) de inhibición frente a la concentración del compuesto y ajustando los datos a una curva sigmoidea restringida de cuatro parámetros, como se conoce en la técnica.
VI. Composiciones farmacéuticas
En otro aspecto, se proporciona una composición farmacéutica que comprende un compuesto descrito en la presente memoria y un excipiente farmacéuticamente aceptable. El compuesto es un compuesto de cualquiera de las Fórmulas (Ia), (Ib), (II), (III), (IV) o (V) como se describe en la presente memoria, un compuesto como se expone en la Tabla A, Tabla B, Tabla C o Tabla D de la presente memoria, o una sal, un éster, un solvato o un profármaco farmacéuticamente aceptable. En algunas realizaciones, el compuesto se expone en la Tabla A, Tabla B, Tabla C o Tabla D de la presente memoria.
La expresión “sales farmacéuticamente aceptables” pretende incluir sales de los compuestos activos que se preparan con ácidos o bases relativamente no tóxicos, dependiendo de los sustituyentes particulares encontrados en los compuestos descritos en la presente memoria. Cuando los compuestos descritos en la presente memoria contienen funcionalidades relativamente ácidas, se pueden obtener sales de adición de base poniendo en contacto la forma neutra de dichos compuestos con una cantidad suficiente de la base deseada, ya sea pura o en un disolvente inerte adecuado. Los ejemplos de sales de adición de base farmacéuticamente aceptables incluyen sal de
sodio, potasio, calcio, amonio, amino orgánico o magnesio, o una sal similar. Cuando los compuestos descritos en la presente memoria contienen funcionalidades relativamente básicas, se pueden obtener sales de adición de ácido poniendo en contacto la forma neutra de dichos compuestos con una cantidad suficiente del ácido deseado, ya sea puro o en un disolvente inerte adecuado. Los ejemplos de sales de adición de ácido farmacéuticamente aceptables incluyen las derivadas de ácidos inorgánicos tales como los ácidos clorhídrico, bromhídrico, nítrico, carbónico, monohidrogenocarbónico, fosfórico, monohidrogenofosfórico, dihidrogenofosfórico, sulfúrico, monohidrogenosulfúrico, yodhídrico o fosforoso y similares, así como las sales derivadas de ácidos orgánicos relativamente no tóxicos tales como acético, propiónico, isobutírico, maleico, malónico, benzoico, succínico, subérico, fumárico, láctico, mandélico, ftálico, bencenosulfónico, p-tolilsulfónico, cítrico, tartárico, oxálico, metanosulfónico y similares. También se incluyen sales de aminoácidos tales como arginato y similares, y sales de ácidos orgánicos como ácidos glucurónicos o galacturónicos y similares (véase, por ejemplo, Berge y col., “ Pharmaceutical Salts” , Journal of Pharmaceutical Science, 1977, 6 6 , 1-19). Determinados compuestos específicos descritos en la presente memoria contienen funcionalidades tanto básicas como ácidas que permiten que los compuestos se conviertan en sales de adición de bases o de ácidos.
Los compuestos descritos en la presente memoria pueden existir en forma de sales, tales como con ácidos farmacéuticamente aceptables. Por consiguiente, los compuestos contemplados en la presente memoria incluyen dichas sales. Los ejemplos de dichas sales incluyen clorhidratos, bromhidratos, sulfatos, metanosulfonatos, nitratos, maleatos, acetatos, citratos, fumaratos, tartratos (p. ej., (+)-tartratos, (-)-tartratos o mezclas de los mismos, incluidas las mezclas racémicas), succinatos, benzoatos, y sales con aminoácidos tales como ácido glutámico. Estas sales se pueden preparar mediante métodos conocidos por los expertos en la materia.
Las formas neutras de los compuestos se regeneran preferiblemente poniendo en contacto la sal con una base o un ácido y aislando el compuesto original de la manera convencional. La forma original del compuesto difiere de las diversas formas de sal en determinadas propiedades físicas, tal como la solubilidad en disolventes polares.
Las sales farmacéuticamente aceptables de los compuestos anteriores, donde está presente un grupo básico o ácido en la estructura, también se incluyen dentro del alcance de los compuestos contemplados en la presente memoria. Cuando está presente un sustituyente ácido, tal como NHSO3H, -COOH and -P(O)(OH)2 , se puede formar la sal de amonio, sodio, potasio, calcio y similares, para su uso como forma farmacéutica. Se pueden utilizar grupos básicos, tales como radicales amino o heteroarilo básico, o piridilo y sales ácidas, tales como clorhidrato, bromhidrato, acetato, maleato, palmoato, metanosulfonato, p-toluenosulfonato, y similares, como forma farmacéutica.
Además, en las realizaciones en donde está presente R-COOH, se pueden emplear ésteres farmacéuticamente aceptables, p. ej., metilo, etilo, terc-butilo, pivaloiloximetilo y similares, y los ésteres conocidos en la técnica para modificar las características de solubilidad o hidrólisis para su uso como formulaciones de liberación sostenida o profármaco.
A. Formulaciones
Los compuestos descritos en la presente memoria se pueden preparar y administrar en una amplia variedad de formas farmacéuticas oftálmicas, orales, parenterales y tópicas. Los compuestos descritos en la presente memoria se pueden administrar mediante un colirio. Además, los compuestos descritos en la presente memoria se pueden administrar mediante inyección (p. ej., por vía intravenosa, intramuscular, intravítrea, intracutánea, subcutánea, intraduodenal o intraperitoneal). Como tales, los compuestos descritos en la presente memoria también se pueden administrar mediante inyección intravítrea. Además, los compuestos descritos en la presente memoria se pueden administrar por inhalación, por ejemplo, por vía intranasal. Además, los compuestos descritos en la presente memoria se pueden administrar por vía transdérmica. También se prevé que se puedan utilizar múltiples vías de administración (p. ej., intramuscular, oral, ocular) para administrar los compuestos descritos en la presente memoria.
En algunas realizaciones, los compuestos descritos en la presente memoria se pueden preparar en composiciones farmacéuticas líquidas para la administración ocular. La composición para uso ocular puede contener uno o más agentes seleccionados del grupo de agentes tamponadores, agentes solubilizantes, agentes colorantes, agentes potenciadores de la viscosidad y agentes conservantes para producir preparaciones convenientes y farmacéuticamente atractivas.
En algunas realizaciones, la composición para uso ocular puede contener conservantes para la protección contra la contaminación microbiológica, incluidos, aunque no de forma limitativa, cloruro de benzalconio y/o EDTA. Otros posibles conservantes incluyen, aunque no de forma limitativa, alcohol bencílico, metilparabenos, propilparabenos y clorobutanol. Preferiblemente, se empleará un conservante, o una combinación de conservantes, para impartir protección microbiológica además de la protección contra la oxidación de los componentes.
En algunas realizaciones, los compuestos descritos en la presente memoria se pueden administrar por vía oral en forma de comprimidos, suspensiones acuosas u oleosas, gominolas, pastillas, polvos, gránulos, emulsiones, cápsulas, jarabes o elixires. La composición para uso oral puede contener uno o más agentes seleccionados del
grupo de agentes edulcorantes, agentes saborizantes, agentes colorantes y agentes conservantes para producir preparaciones farmacéuticamente atractivas y agradables al paladar. Por consiguiente, también se proporcionan composiciones farmacéuticas que comprenden un vehículo o excipiente farmacéuticamente aceptable y uno o más compuestos descritos en la presente memoria.
En algunas realizaciones, los comprimidos contienen el principio activo mezclado con excipientes no tóxicos farmacéuticamente aceptables que son adecuados para la fabricación de comprimidos. Estos excipientes pueden ser, por ejemplo, (1) diluyentes inertes, tales como carbonato de calcio, lactosa, fosfato de calcio, carboximetilcelulosa o fosfato de sodio; (2) agentes de granulación y disgregación, tales como almidón de maíz o ácido algínico; (3) agentes aglutinantes, tales como almidón, gelatina o goma arábiga; y (4) agentes lubricantes, tales como estearato de magnesio, ácido esteárico o talco. Estos comprimidos pueden estar sin recubrir o se pueden recubrir mediante técnicas conocidas para retardar la disgregación y la absorción en el tubo gastrointestinal y, por lo tanto, proporcionar una acción sostenida durante un período más prolongado. Por ejemplo, puede emplearse un material de tiempo de retardo tal como monoestearato de glicerilo o diestearato de glicerilo.
Para preparar composiciones farmacéuticas a partir de los compuestos descritos en la presente memoria, los vehículos farmacéuticamente aceptables pueden ser sólidos o líquidos. Las preparaciones en forma sólida incluyen polvos, comprimidos, píldoras, cápsulas, obleas, supositorios y gránulos dispersables. Un vehículo sólido puede ser una o más sustancias que también pueden actuar como diluyentes, agentes saborizantes, aglutinantes, conservantes, agentes de disgregación de comprimidos o material de encapsulación.
Un compuesto descrito en la presente memoria, en forma de un compuesto libre o un profármaco, metabolito, análogo, derivado, solvato o sal farmacéuticamente aceptable, puede administrarse para aplicación in vivo, por vía parenteral mediante inyección o mediante perfusión gradual a lo largo del tiempo. La administración puede ser por vía intravenosa, intraperitoneal, intramuscular, subcutánea, intracavitaria o transdérmica. Para estudios in vitro, los compuestos pueden añadirse o disolverse en un tampón biológicamente aceptable apropiado y añadirse a una célula o a un tejido.
En polvo, el vehículo es un sólido finamente dividido en una mezcla con el componente activo finamente dividido. En comprimidos, el componente activo se mezcla con el vehículo que tiene las propiedades aglutinantes necesarias en proporciones adecuadas y se compacta en la forma y el tamaño deseados.
Los polvos y los comprimidos contienen preferiblemente del 5 % al 70 % del compuesto activo. Los vehículos adecuados son carbonato de magnesio, estearato de magnesio, talco, azúcar, lactosa, pectina, dextrina, almidón, gelatina, tragacanto, metilcelulosa, carboximetilcelulosa de sodio, cera de bajo punto de fusión, manteca de cacao y similares. El término “ preparación” pretende incluir la formulación del compuesto activo con material de encapsulación como vehículo que proporciona una cápsula en donde el componente activo con o sin otros vehículos está rodeado por un vehículo, que por lo tanto está asociado con el mismo. Del mismo modo, se incluyen obleas y gominolas. Los comprimidos, los polvos, las cápsulas, las píldoras, las obleas y las gominolas se pueden utilizar como formas farmacéuticas sólidas adecuadas para la administración oral.
Para preparar supositorios, primero se funde una cera de bajo punto de fusión, tal como una mezcla de glicéridos de ácidos grasos o manteca de cacao, y el componente activo se dispersa homogéneamente en la misma, como por agitación. A continuación, se vierte la mezcla homogénea fundida en moldes de tamaño conveniente, se deja enfriar y, por lo tanto, se solidifica.
Las preparaciones en forma líquida incluyen soluciones, suspensiones y emulsiones, por ejemplo, agua o soluciones de agua/propilenglicol. Para inyección parenteral, las preparaciones líquidas se pueden formular en solución, en solución acuosa de polietilenglicol.
Cuando se necesita o se desea la aplicación parenteral, las mezclas particularmente adecuadas para los compuestos descritos en la presente memoria son soluciones estériles inyectables, preferiblemente soluciones oleosas o acuosas, así como suspensiones, emulsiones o implantes, incluidos los supositorios. En particular, los vehículos para la administración parenteral incluyen soluciones acuosas de dextrosa, solución salina, agua pura, etanol, glicerol, propilenglicol, aceite de cacahuete, aceite de sésamo, polímeros de bloques de polioxietileno y similares. Las ampollas son dosis unitarias convenientes. Los compuestos descritos en la presente memoria también pueden incorporarse a liposomas o administrarse mediante bombas o parches transdérmicos. Las mezclas farmacéuticas adecuadas para su uso en las composiciones farmacéuticas y los métodos descritos en la presente memoria incluyen los descritos, por ejemplo, en “ PHARMACEUTICAL SCIENCES” (17.a Ed., Mack Pub. Co., Easton, PA) y el documento WO 96/05309, cuyas enseñanzas se incorporan en la presente memoria por referencia.
En algunas realizaciones, las preparaciones para la administración parenteral incluyen soluciones, suspensiones y emulsiones acuosas o no acuosas estériles. Algunos ejemplos de disolventes no acuosos son propilenglicol, polietilenglicol, aceites vegetales tales como el aceite de oliva y ésteres orgánicos inyectables tales como el oleato de etilo. Los vehículos acuosos incluyen agua, soluciones alcohólicas/acuosas, emulsiones o suspensiones, incluidos medios salinos y tamponados. Los vehículos parenterales incluyen solución de cloruro de sodio, dextrosa
de Ringer, dextrosa y cloruro de sodio, los vehículos intravenosos de Ringer lactato incluyen reponedores de fluidos y nutrientes, reponedores de electrolitos (tales como los basados en dextrosa de Ringer) y similares. También pueden estar presentes conservantes y otros aditivos como, por ejemplo, agentes antimicrobianos, antioxidantes, agentes quelantes, factores de crecimiento y gases inertes y similares.
Las soluciones acuosas adecuadas para uso oral se pueden preparar disolviendo el componente activo en agua y añadiendo colorantes, sabores, estabilizantes y agentes espesantes adecuados según se desee. Se pueden preparar suspensiones acuosas adecuadas para uso oral dispersando el componente activo finamente dividido en agua con material viscoso, tal como gomas naturales o sintéticas, resinas, metilcelulosa, carboximetilcelulosa sódica y otros agentes de suspensión bien conocidos.
También se incluyen preparaciones en forma sólida que están destinadas a convertirse, poco antes de su uso, en preparaciones en forma líquida para administración oral. Dichas formas líquidas incluyen soluciones, suspensiones y emulsiones. Estas preparaciones pueden contener, además del componente activo, colorantes, sabores, estabilizantes, tampones, edulcorantes artificiales y naturales, dispersantes, espesantes, agentes solubilizantes y similares.
La preparación farmacéutica está preferiblemente en forma farmacéutica unitaria. En dicha forma, la preparación se subdivide en dosis unitarias que contienen cantidades apropiadas del componente activo. La forma farmacéutica unitaria puede ser una preparación envasada, conteniendo el envase cantidades diferenciadas de preparación, tales como comprimidos, cápsulas y polvos empaquetados en viales o ampollas. Además, la forma farmacéutica unitaria puede ser una cápsula, un comprimido, una oblea o una gominola, o puede ser el número apropiado de cualquiera de estos en forma envasada.
La cantidad de componente activo en una preparación de dosis unitaria se puede variar o ajustar de 0,1 mg a 10000 mg, más normalmente de 1,0 mg a 1000 mg, más normalmente de 10 mg a 500 mg, según la aplicación particular y la potencia del componente activo. La composición puede, si se desea, contener también otros agentes terapéuticos compatibles.
Algunos compuestos pueden tener una solubilidad limitada en agua y, por lo tanto, pueden requerir un tensioactivo u otro codisolvente apropiado en la composición. Dichos codisolventes incluyen: polisorbato 20, 60 y 80; Pluronic F-6 8 , F-84 y P-103; ciclodextrina; y aceite de ricino polioxil 35. Dichos codisolventes normalmente se emplean a un nivel entre aproximadamente el 0 , 01 % y aproximadamente el 2 % en peso.
Puede ser deseable una viscosidad mayor que la de las soluciones acuosas simples para disminuir la variabilidad en la dispensación de las formulaciones, para disminuir la separación física de los componentes de una suspensión o emulsión de la formulación y/o para mejorar la formulación. Dichos agentes formadores de la viscosidad incluyen, por ejemplo, alcohol polivinílico, polivinilpirrolidona, metilcelulosa, hidroxipropilmetilcelulosa, hidroxietilcelulosa, carboximetilcelulosa, hidroxipropilcelulosa, sulfato de condroitina y sus sales, ácido hialurónico y sus sales, y combinaciones de los anteriores. Dichos agentes normalmente se emplean a un nivel de entre aproximadamente el 0 , 01 % y aproximadamente el 2 % en peso.
Las composiciones descritas en la presente memoria pueden incluir adicionalmente componentes para proporcionar liberación sostenida y/o comodidad. Dichos componentes incluyen polímeros mucomiméticos aniónicos de alto peso molecular, polisacáridos gelificantes y sustratos portadores de fármacos finamente divididos. Estos componentes se analizan con mayor detalle en la patente de EE. UU. n.° 4.911.920; 5.403.841; 5.212.162; y 4.861.760. El contenido completo de estas patentes se incorpora en la presente memoria por referencia en su totalidad para todos los propósitos.
Por el momento, se proporcionan métodos para mejorar la cicatrización de heridas y para mediar en la reparación de tejidos (que incluyen, aunque no de forma limitativa, el tratamiento de enfermedades vasculares periféricas y coronarias). Según estos métodos, un sujeto que tiene una herida o necesita reparación de tejido, se trata en el sitio de la herida o tejido dañado, o se trata sistémicamente, con un compuesto descrito en la presente memoria en forma de un compuesto libre o un profármaco, metabolito, análogo, derivado, solvato o sal farmacéuticamente aceptable.
Generalmente, los términos “tratar” , “ tratamiento” y similares se utilizan en la presente memoria para referirse a afectar a un sujeto, tejido o célula a fin de obtener un efecto farmacológico y/o fisiológico deseado. El efecto puede ser profiláctico en términos de prevención total o parcial de una enfermedad o trastorno, o signo o síntoma de los mismos, y/o puede ser terapéutico en términos de una cura parcial o completa de un trastorno y/o efecto adverso atribuible al mismo, p. ej., embolia pulmonar después de una intervención médica. “Tratar” , como se utiliza en la presente memoria, cubre cualquier tratamiento o prevención de una enfermedad o trastorno en un vertebrado, un mamífero, particularmente un ser humano, e incluye: (a) prevenir que la enfermedad o el trastorno ocurra en un sujeto que puede estar predispuesto a la enfermedad o el trastorno, pero que aún no ha sido diagnosticado de tenerlo; (b) inhibir la enfermedad o el trastorno, es decir, detener su desarrollo; o (c) aliviar o mejorar la enfermedad o el trastorno, es decir, provocar la regresión de la enfermedad o del trastorno.
Se proporcionan diferentes composiciones farmacéuticas útiles para mejorar determinadas enfermedades y trastornos. Las composiciones farmacéuticas según una realización se preparan formulando un compuesto descrito en la presente memoria en forma de un compuesto libre o un profármaco, metabolito, análogo, derivado, solvato o sal farmacéuticamente aceptable, ya sea solo o junto con otros agentes farmacéuticos, adecuados para la administración a un sujeto mediante vehículos, excipientes y aditivos o sustancias auxiliares. Los vehículos o las sustancias auxiliares de uso frecuente incluyen carbonato de magnesio, dióxido de titanio, lactosa, manitol y otros azúcares, talco, proteína de leche, gelatina, almidón, vitaminas, celulosa y sus derivados, aceites animales y vegetales, polietilenglicoles y disolventes, tales como agua estéril, alcoholes, glicerol y alcoholes polihídricos. Los vehículos intravenosos incluyen reponedores de líquidos y nutrientes.
Los conservantes incluyen agentes antimicrobianos, antioxidantes, agentes quelantes y gases inertes. Otros vehículos farmacéuticamente aceptables incluyen soluciones acuosas, excipientes no tóxicos, incluyendo sales, conservantes, tampones y similares, como se describe, por ejemplo, en “ REMINGTON'S PHARMACEUTICAL SCIENCES” , 15.a ed. Easton: Mack Publishing Co., 1405-1412, 1461-1487 (1975) y “THE NATIONAL FORMULARY XIV” , 14.a ed. Washington: American Pharmaceutical Association (1975), cuyo contenido se incorpora en la presente memoria por referencia. El pH y la concentración exacta de los diferentes componentes de la composición farmacéutica se ajustan según los conocimientos habituales en la técnica. Véase, p. ej., Goodman y Gilman (eds.), 1990, “THE PHARMACOLOGICAL BASIS FOR THERAPEUTICS” (7.a ed.).
Las composiciones farmacéuticas se preparan y administran preferiblemente en dosis unitarias. Las dosis unitarias sólidas son comprimidos, cápsulas y supositorios. Para el tratamiento de un sujeto, dependiendo de la actividad del compuesto, la forma de administración, la naturaleza y gravedad de la enfermedad o del trastorno, la edad y el peso corporal del sujeto, se pueden utilizar diferentes dosis diarias.
En determinadas circunstancias, sin embargo, pueden ser apropiadas dosis diarias más altas o más bajas. La administración de la dosis diaria se puede llevar a cabo tanto mediante una única administración en forma de una dosis unitaria individual o también en varias unidades de dosis más pequeñas, así como mediante varias administraciones de dosis subdivididas a intervalos específicos.
Las composiciones farmacéuticas contempladas en la presente memoria pueden administrarse local o sistémicamente a una dosis terapéuticamente eficaz. Las cantidades eficaces para este uso dependerán, por supuesto, de la gravedad de la enfermedad o del trastorno y del peso y estado general del sujeto. Normalmente, las dosis utilizadas in vitro pueden proporcionar una guía útil en las cantidades útiles para la administración in situ de la composición farmacéutica, y se pueden usar modelos animales para determinar las dosis eficaces para el tratamiento de trastornos particulares.
Se describen diferentes consideraciones, p. ej., en Langer, 1990, Science, 249: 1527; Goodman and Gilman's (eds.), 1990, Id., cada uno de los cuales se incorpora en la presente memoria por referencia y para todos los fines. Las dosis para administración parenteral de agentes farmacéuticos activos pueden convertirse en dosis correspondientes para administración oral multiplicando las dosis parenterales por factores de conversión apropiados. En cuanto a las aplicaciones generales, la dosis parenteral en mg/ml multiplicada por 1,8 = la dosis oral correspondiente en miligramos (“ mg” ). En cuanto a las aplicaciones oncológicas, la dosis parenteral en mg/ml multiplicada por 1,6 = la dosis oral correspondiente en mg. Un adulto promedio pesa aproximadamente 70 kg. Véase, p. ej., Miller-Keane, 1992, ENCYCLOPEDIA & DICTIONARY OF MEDICINE, NURSING & ALLIED HEALTH, 5th Ed., (W. B. Saunders Co.), págs. 1708 y 1651.
El método mediante el que se puede administrar el compuesto descrito en la presente memoria para uso oral sería, por ejemplo, en una cápsula de gelatina dura, en donde el principio activo se mezcla con un diluyente sólido inerte, o en una cápsula de gelatina blanda, en donde el principio activo se mezcla con un mezcla codisolvente, tal como PEG 400 que contiene Tween-20. Un compuesto descrito en la presente memoria también se puede administrar en forma de una solución o suspensión inyectable estéril acuosa u oleaginosa. El compuesto generalmente se puede administrar por vía intravenosa o como una dosis oral de 0,1 pg a 20 mg/kg administrada, por ejemplo, cada 3 a 24 horas.
Las formulaciones para uso oral pueden estar en forma de cápsulas de gelatina dura, en donde el principio activo se mezcla con un diluyente sólido inerte, por ejemplo, carbonato de calcio, fosfato de calcio o caolín. También pueden presentarse en forma de cápsulas de gelatina blanda, en donde el principio activo se mezcla con agua o un medio oleoso, tal como aceite de cacahuete, parafina líquida o aceite de oliva.
Las suspensiones acuosas normalmente contienen los materiales activos mezclados con excipientes adecuados para la fabricación de suspensiones acuosas. Dichos excipientes pueden ser (1) un agente de suspensión, tal como carboximetilcelulosa sódica, metilcelulosa, hidroxipropilmetilcelulosa, alginato sódico, polivinilpirrolidona, goma de tragacanto y goma arábiga; (2 ) agentes dispersantes o humectantes que pueden ser (a) fosfátidos naturales tales como lecitina; (b) un producto de condensación de un óxido de alquileno con un ácido graso, por ejemplo, estearato de polioxietileno; (c) un producto de condensación de óxido de etileno con un alcohol alifático de cadena larga, por ejemplo, heptadecaetilenoxietanol; (d) un producto de condensación de óxido de etileno con un éster parcial
derivado de un ácido graso y hexitol tal como monooleato de sorbitol polioxietilenado, o (e) un producto de condensación de óxido de etileno con un éster parcial derivado de ácidos grasos y anhídridos de hexitol, por ejemplo, monooleato de sorbitán polioxietilenado.
Las composiciones farmacéuticas pueden presentarse en forma de suspensión acuosa u oleosa inyectable estéril. Esta suspensión se puede formular según métodos conocidos mediante aquellos agentes dispersantes o humectantes y agentes de suspensión adecuados que se han mencionado anteriormente. La preparación inyectable estéril también puede ser una solución o suspensión inyectable estéril en un diluyente o disolvente no tóxico aceptable por vía parenteral, por ejemplo, en forma de una solución en 1,3-butanodiol. Entre los vehículos y disolventes aceptables que se pueden emplear están el agua, la solución de Ringer y la solución isotónica de cloruro de sodio. Además, los aceites fijos estériles se emplean convencionalmente como disolvente o medio de suspensión. Para este fin, se puede emplear cualquier aceite fijo suave, incluidos los monoglicéridos o diglicéridos sintéticos. Además, los ácidos grasos tales como el ácido oleico encuentran uso en la preparación de inyectables.
Un compuesto descrito en la presente memoria también se puede administrar en forma de composiciones oftálmicas aplicadas por vía tópica al ojo, preferiblemente en forma de colirio. Un compuesto descrito en la presente memoria también se puede administrar en forma de inyección intravítrea.
Un compuesto descrito en la presente memoria también se puede administrar en forma de supositorios para la administración rectal del fármaco. Estas composiciones se pueden preparar mezclando el fármaco con un excipiente no irritante adecuado que sea sólido a temperatura normal, pero líquido a temperatura rectal y, por lo tanto, que se funda en el recto para liberar el fármaco. Dichos materiales incluyen manteca de cacao y polietilenglicoles.
Los compuestos descritos en la presente memoria, como se utilizan en los métodos descritos en la presente memoria, también se pueden administrar en forma de sistemas de administración de liposomas, tales como vesículas unilamelares pequeñas, vesículas unilamelares grandes y vesículas multilamelares. Los liposomas se pueden formar a partir de una variedad de fosfolípidos, tales como colesterol, estearilamina o fosfatidilcolinas.
Para uso tópico, se emplean cremas, pomadas, jaleas, soluciones o suspensiones, etc., que contienen los compuestos descritos en la presente memoria.
Además, algunos de los compuestos descritos en la presente memoria pueden formar solvatos con agua o disolventes orgánicos comunes. Dichos solvatos están incluidos dentro del alcance de los métodos contemplados en la presente memoria.
B. Dosis eficaces
Las composiciones farmacéuticas proporcionadas en la presente memoria incluyen composiciones en donde el principio activo está contenido en una cantidad terapéuticamente eficaz, es decir, en una cantidad eficaz para lograr el fin previsto. La cantidad real eficaz para una aplicación en particular dependerá, entre otros factores, de la afección que se esté tratando.
La posología y frecuencia (dosis únicas o múltiples) del compuesto administrado pueden variar dependiendo de una variedad de factores, incluida la vía de administración; tamaño, edad, sexo, estado de salud, peso corporal, índice de masa corporal y dieta del receptor; la naturaleza y extensión de los síntomas de la enfermedad que se esté tratando (p. ej., la enfermedad que responde a la inhibición de la trombina y/o KLKB1); la presencia de otras enfermedades u otros problemas relacionados con la salud; el tipo de tratamiento concurrente; y complicaciones de cualquier enfermedad o pauta de tratamiento. Se pueden utilizar otras pautas o agentes terapéuticos junto con los métodos y compuestos descritos en la presente memoria.
Para cualquier compuesto descrito en la presente memoria, la cantidad terapéuticamente eficaz se puede determinar inicialmente a partir de una variedad de técnicas conocidas en la técnica, p. ej., la caracterización bioquímica de la inhibición de la enzima (trombina o KLKB1), ensayos de cultivo celular y similares. Las concentraciones diana serán aquellas concentraciones de compuesto(s) activo(s) que son capaces de disminuir la actividad enzimática medida, por ejemplo, mediante los métodos descritos.
Las cantidades terapéuticamente eficaces para uso en seres humanos pueden determinarse a partir de modelos animales. Por ejemplo, se puede formular una dosis para seres humanos para lograr una concentración que haya resultado ser eficaz en animales. La posología en seres humanos se puede ajustar controlando la inhibición enzimática y ajustando las dosis de forma creciente o decreciente, como se ha descrito anteriormente.
Las dosis pueden variar dependiendo de las necesidades del paciente y del compuesto que se emplee. La dosis administrada a un paciente, en el contexto de los métodos descritos en la presente memoria, debe ser suficiente para afectar a una respuesta terapéutica beneficiosa en el paciente a lo largo del tiempo. El tamaño de la dosis también estará determinado por la existencia, la naturaleza y el alcance de cualquier efecto secundario adverso. Generalmente, el tratamiento se inicia con dosis más pequeñas, que son inferiores a la dosis óptima del compuesto.
Después se va aumentando la dosis en pequeños incrementos hasta que se alcanza el efecto óptimo según las circunstancias. En algunas realizaciones de un método descrito en la presente memoria, el intervalo de dosis es del 0,001 % al 10 % p/v. En algunas realizaciones, el intervalo de dosis es del 0,1 % al 5 % p/v.
Las cantidades posológicas y los intervalos se pueden ajustar individualmente para proporcionar niveles eficaces del compuesto administrado para la indicación clínica particular que se esté tratando. Esto proporcionará una pauta terapéutica acorde a la gravedad de la patología del individuo.
Mediante las enseñanzas proporcionadas en la presente memoria, se puede planificar una pauta de tratamiento profiláctico o terapéutico eficaz que no provoque una toxicidad sustancial y, sin embargo, sea completamente eficaz para tratar los síntomas clínicos experimentados por el paciente en particular. Esta planificación debe implicar la elección cuidadosa del compuesto activo considerando factores como la potencia del compuesto, la biodisponibilidad relativa, el peso corporal del paciente, la presencia y la gravedad de los efectos secundarios adversos, el modo de administración preferido y el perfil de toxicidad del agente seleccionado.
Por consiguiente, en algunas realizaciones, los niveles posológicos de los compuestos descritos en la presente memoria como se utilizan en los presentes métodos son del orden de, p. ej., de aproximadamente 0,1 mg a aproximadamente 1 mg, de aproximadamente 1 mg a aproximadamente 10 mg, de aproximadamente 0,5 mg a aproximadamente 20 mg por kilogramo de peso corporal, un adulto promedio que pesa 70 kilogramos, con un intervalo posológico preferido de aproximadamente 0,1 mg a aproximadamente 20 mg por kilogramo de peso corporal al día (de aproximadamente 7,0 mg a aproximadamente 1,4 g por paciente al día). La cantidad del compuesto descrito en la presente memoria que se puede combinar con los materiales portadores para producir una dosis única variará dependiendo del hospedador tratado y del modo de administración en particular. Por ejemplo, una formulación destinada a la administración oral a seres humanos puede contener de aproximadamente 5 pg a 1 g de un compuesto descrito en la presente memoria con una cantidad apropiada y conveniente de material portador que puede variar entre aproximadamente el 5 y el 95 por ciento de la composición total. En general, las formas farmacéuticas unitarias contendrán de aproximadamente 0,1 mg a 500 mg de un compuesto descrito en la presente memoria.
Sin embargo, se entenderá que el nivel de dosis específico para cualquier paciente en particular dependerá de una variedad de factores, incluida la actividad del compuesto específico empleado, la edad, el peso corporal, el estado de salud general, el sexo, la dieta, el momento de la administración, la vía de administración, la tasa de excreción, la combinación de fármacos y la gravedad de la enfermedad particular objeto de terapia.
C. Toxicidad
La relación entre la toxicidad y efecto terapéutico de un compuesto en particular es su índice terapéutico, y se puede expresar como la relación entre DL50 (la cantidad de compuesto letal en el 50 % de la población) y la DE50 (la cantidad de compuesto eficaz en el 50 % de la población). Se prefieren los compuestos que presentan altos índices terapéuticos. Los datos del índice terapéutico obtenidos de ensayos in vitro, ensayos de cultivos celulares y/o estudios en animales se pueden utilizar para formular una selección de dosis para uso en seres humanos. La posología de dichos compuestos se encuentra preferiblemente dentro de un intervalo de concentraciones plasmáticas que incluyen la DE50 con poca o ninguna toxicidad. La posología puede variar dentro de este intervalo dependiendo de la forma farmacéutica empleada y de la vía de administración utilizada. Véase, p. ej., Fingl y col., en: THE PHARMACOLOGICAL BASIS OF THErAp EUTICS, capítulo 1, pág. l, 1975. Cada médico puede escoger la formulación exacta, la vía de administración y la posología en vista del estado del paciente y del método en particular en donde se utilice el compuesto. Para las formulaciones in vitro, cada médico puede escoger la formulación y la posología exactas en vista del estado del paciente y del método en particular en donde se utilice el compuesto.
VIII. Ejemplos
Los siguientes ejemplos pretenden ilustrar determinadas realizaciones de la invención y no limitar el alcance de la misma. Las abreviaturas utilizadas en la presente memoria tienen su significado convencional en la técnica, salvo que se indique lo contrario. Las abreviaturas específicas incluyen las siguientes: Á = Angstrom; Ac2O = anhídrido acético; AcOH = ácido acético; ac = acuoso; Bt = benzotriazol; COB = N-terc-butoxicarbonilo; a = ancho; t-BuOH = terc-butanol; 0C = grado Celsius; d = doblete; DABCO = 1,4-diazabiciclo[2.2.2]octano; DCE = 1,2-dicloroetano; DCM = diclorometano; dd = doblete de dobletes; DIEA = diisopropiletilamina; DMAP = 4-dimetilaminopiridina; DMF = N,N-dimetilformamida; DMSO = dimetilsulfóxido; 5 = desplazamiento químico (dado en ppm, salvo que se indique lo contrario); EDCI = 1-etil-3-(3-dimetilaminopropil)carbodiimida; eq = equivalente/s; Et2O = éter dietílico; Et3N = trietilamina; EtOAc = acetato de etilo; EtOH = etanol; g = gramo/s; h = hora/s; HOBt = hidroxibenzotriazol; HPLC = cromatografía líquida de alta resolución (High Performance Liquid Chromatography); Hz = Hercios; CI50 = concentración inhibidora al 50 % de inhibición; J = constante de acoplamiento (dada en Hz, salvo que se indique lo contrario); CL = Cromatografía Líquida; LHMDS = hexametildisilazida de litio; m = multiplete; M = molar; [M+H]+ = máximo del espectro de masas principal más H+; EM = Espectro de Masas; tm = tamices moleculares; Pf = punto de fusión; Me2NH = dimetilamina; MeOH = metanol; mg = miligramo/s; ml = mililitro/s; mM = milimolar; mmol =
milimol/es; min = minuto/s; = microlitro/s; = micromolar; ng = nanogramo/s; nM = nanomolar; RMN = Resonancia Magnética Nuclear; ppm = partes por millón; c = cuarteto; Fr = Factor de retención; TA = Temperatura Ambiente; s = singlete; t = triplete; TFA = ácido trifluoroacético; THF = tetrahidrofurano; TLC = cromatografía en capa fina (Thin Layer Chromatography).
Lista de procedimientos generales
Procedimiento general 1

Procedimiento general 2

Procedimiento general 3

Procedimiento general 4

Procedimiento general 5a

Procedimiento general 5b

Procedimiento general 5c

Procedimiento general 5d

Procedimiento general 6 a

Procedimiento general 6 b

Procedimiento general 6 c

Procedimiento general 6 d

Procedimiento general 7

Procedimiento general 8 a

Procedimiento general 8 b

Procedimiento general 9

Procedimiento general 10

Procedimiento general 11

Procedimiento general 12

Procedimiento general 13

Procedimiento general 14

P ro ce d im ie n to g e n e ra l 15

Procedimiento general 16

Procedimiento general 17

Procedimiento general 18

Procedimiento general 19

Procedimiento general 20

Procedimiento general 21

Procedimiento general 22

P ro ce d im ie n to g e n e ra l 23

Procedimiento general 24
Mo(CO)6, Pd(OAc)2,

, , , ,
tubo sellado, 5 h
Procedimiento general 25
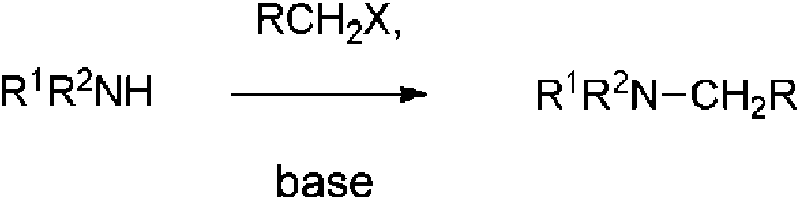
Procedimiento general 26

Ejemplos sintéticos
Ejemplo 1-Preparación del producto intermedio 1
La síntesis del Producto intermedio 1 siguió el siguiente Procedimiento general 1.

A una solución de diisopropilamina (6,55 g, 64,7 mmol, 2,1 eq) en THF frío (-78 0C, 80 ml), se añadió n-butillitio (4,04 g, 63,1 mmol, 2,05 eq) y, a continuación, se agitó durante 1 hora a 0 °C. A continuación, se enfrió la mezcla de reacción hasta -78 °C y, después, se añadió 1-(ferc-butil)-3-metilpiperidin-1,3-dicarboxilato (7,5 g, 30,8 mmol, 1 eq). Se agitó la mezcla durante 1 hora y se añadió yoduro de metilo (13,12 g, 97,4 mmol, 3,0 eq). Se calentó la mezcla de reacción hasta la temperatura ambiente durante la noche. Se controló mediante TLC y CL-EM. Una vez completada, se inactivó la mezcla de reacción con cloruro de amonio y se extrajo con acetato de etilo (2 x 150 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera, a continuación, se desecaron sobre sulfato de sodio y se concentraron a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida utilizando gel de sílice (tamaño de malla 60-120), eluyendo con acetato de etilo al 5-10 % en n-hexano, dando el producto (Producto intermedio 1, 4,8 g, rendimiento: 60,5 %) m/z 202 [M-56]+ RMN de 1H (400 MHz, CDC^) 5 3,87 (d, J = 13,3 Hz, 1H), 3,70 (s, 3H), 3,46 (s, 1H), 3,27 (d, J = 5,9 Hz, 1H), 3,15 (d, J = 13,3 Hz, 1H), 2,04 (dd, J = 12,7, 6,2 Hz, 1H), 1,58 (s, 1H), 1,49 (m, 11H), 1,18 (s, 3H) ppm.
Ejemplo 2-Preparación del producto intermedio 2
La síntesis del Producto intermedio 2 siguió el procedimiento del siguiente Procedimiento general 2.

A una solución fría (-78 0C) de acetonitrilo (1,67 g, 40,8 mmol, 1,5 eq) en tetrahidrofurano (70 ml), se añadió n-BuLi (23 % en hexano, 2,61 g, 40,8 mmol, 1,5 eq) en atmósfera de N2 inerte durante un período de 20 minutos. Una vez completada la adición, la reacción se agitó durante otros 60 minutos. A continuación, se añadió a la mezcla fría ( 78 0C) el Producto intermedio 1 (7,0 g, 27,2 mmol, 1,0 eq) poco a poco y se agitó la mezcla de reacción durante 3 horas. Se inactivó la mezcla de reacción con una solución saturada de cloruro de amonio y se extrajo el producto con acetato de etilo. La fase orgánica se desecó con sulfato de sodio y se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna utilizando gel de sílice (tamaño de malla 60-120), eluyendo con acetato de etilo al 10-40 % en n-hexano, dando el producto 3-(2-cianoacetil)-3-metilpiperidin-1-carboxilato terc- butílico (Producto intermedio 2; 4,775 g, rendimiento: 65,9 %) m/z 211,0 [M-56]+ RMN de 1H (400 MHz, DMSO) 5 4,26 (c, J = 20,1 Hz, 2H), 3,74 (d, J = 12,6 Hz, 1H), 3,38-3,32 (m, 1H), 3,21-3,06 (m, 2H), 1,94-1,77 (m, 1H), 1,56 1,31 (m, 12H), 1,06 (s, 3H) ppm.
Ejemplo 3-Preparación del compuesto 1
La síntesis del Compuesto 1 siguió el procedimiento del siguiente Procedimiento general 3.
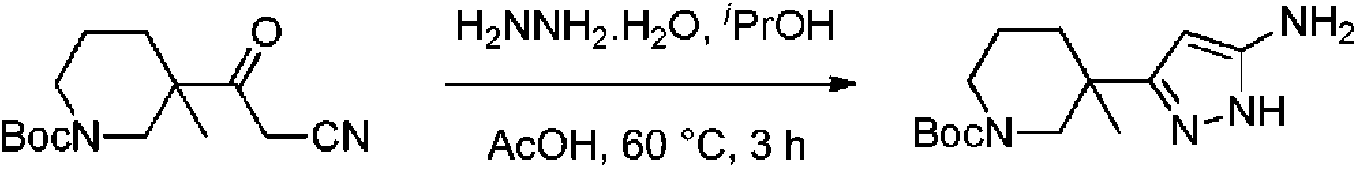
Producto intermedio 2 Compuesto 1
A una solución de 3-(2-cianoacetil)-3-metilpiperidin-1-carboxilato terc-butílico (Producto intermedio 2, 9,5 g, 35 mmol, 1,0 eq) en ácido acético (0,2 ml) e isopropanol (80 ml), se añadió monohidrato de hidracina (2,67 g, 53 mmol, 1,5 eq) gota a gota. Se agitó la mezcla de reacción a 60 0C durante 3 horas. La mezcla de reacción se controló mediante CL-EM y, una vez completada, se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna utilizando gel de sílice (malla 60-120), eluyendo con metanol al 5-10 % en diclorometano, produciendo 3-(5-amino-1H-pirazol-3-il)-3-metilpiperidin-1-carboxilato terc-butílico (Compuesto 1; 9,005 g, rendimiento: 90,0 %) m/z 225,05 [M-56]+ RMN de 1H (400 MHz, DMSO) 5 11,26 (s, 1H), 4,62 (s, 2H), 3,38 (d, J = 22,7 Hz, 2H), 3,32 (d, J = 12,2 Hz, 1H), 3,16 (d, J = 12,5 Hz, 1H), 1,58-1,43 (m, 2H), 1,38 (d, J = 9,7 Hz, 11H), 1,09 (d, J = 8,6 Hz, 3H) ppm.
Ejemplo 4-Preparación del compuesto 2
La síntesis del Compuesto 2 siguió el procedimiento del siguiente Procedimiento general 4.

A una solución fría (0 0C) de 3-(5-amino-1 H-pirazol-3-il)-3-metilpiperidin-1-carboxilato terc-butílico (Compuesto 1, 9,0 g, 32,1 mmol, 1,0 eq) y 5-clorotiofen-2-carbaldehído (5,64 g, 38,5 mmol, 1,2 eq) en metanol (100 ml), se añadió ácido acético (1,92 g, 32,1 mmol, 1,0 eq). Después de agitar durante 3 horas, se añadió cianoborohidruro de sodio (4,03 g, 64,2 mmol, 2,0 eq). Se calentó la mezcla de reacción hasta la temperatura ambiente durante la noche y se controló mediante TLC. Se vertió la mezcla de reacción en agua (200 ml) y se extrajo con acetato de etilo ( 2 x 150 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera, se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida utilizando gel de sílice (tamaño de malla 60-120), eluyendo con acetato de etilo al 10-40 % en n-hexano, produciendo 3-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)-3-metilpiperidin-1-carboxilato terc-butílico (Compuesto 2, 7,0 g,
rendimiento: 53,0 %) m/z 411,40 [M+1]+ RMN de 1H (400 MHz, DMSO) 56,92 (d, J = 3,7 Hz, 1H), 6,84 (s, 1H), 5,67 (s, 1H), 5,35 (s, 1H), 4,28 (d, J = 5,5 Hz, 2H), 3,44-3,34 (m, 2H), 3,29-3,26 (m, 2H), 1,90 (s, 1H), 1,54-1,43(m, 3H), 1,39 (s, 9H), 1,12 (s, 3H) ppm.
Ejemplo 5-Preparación del compuesto 3
La síntesis del Compuesto 3 siguió el procedimiento del siguiente Procedimiento general 5a.

A una solución de ácido piválico (0,373 g, 3,65 mmol, 1,5 eq) en THF frío (0 0C) (15 ml) en atmósfera de nitrógeno, se añadieron clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 0,701 g, 3,65 mmol, 1,5 eq) y trietilamina (0,74 g, 7,31 mmol, 3,0 eq). Se agitó la mezcla de reacción durante 30 minutos y se añadió hidroxibenzotriazol (HOBt, 0,065 g, 0,48 mmol, 0,2 eq), seguido de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)-3-metilpiperidin-1 -carboxilato ferc-butílico (Compuesto 2, 1,0 g, 3,43 mmol, 1,0 eq). Se agitó la mezcla durante la noche a temperatura ambiente. Se controló la reacción mediante CL-EM y, una vez finalizada, se vertió en agua (20 ml), se extrajo con acetato de etilo (2 x 20 ml), se lavó con agua y salmuera, se desecó sobre sulfato de sodio, se filtró y se evaporó a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida utilizando gel de sílice (tamaño de malla 60-120), eluyendo con acetato de etilo al 0-20 % en n-hexano, produciendo 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 -pivaloil-1 H-pirazol-3-il)-3-metilpiperidin-1 -carboxilato terc-butílico (Compuesto 3, 0,500 g, rendimiento: 41,4 %) m/z 494,88 [M+1]+ RMN de 1H (400 MHz, DMSO) 57,63 (d, J = 5,6 Hz, 1H), 6,97 (s, 2H), 5,47 (s, 1H), 4,41 (s, 2H), 3,44 (d, J = 45,8 Hz, 2H), 3,24 (s, 2H), 1,94 (s, 1H), 1,50 (d, J = 47,4 Hz, 3H), 1,40 (s, 9H), 1,34 (s, 9H), 1,12 (s, 3H) ppm.
Ejemplo 6-Preparación del compuesto 4
La síntesis del Compuesto 4 siguió el siguiente procedimiento de Procedimiento general 5b.

A una solución fría (0 0C) de ácido 3-hidroxi-2,2-dimetilpropanoico (0,576 g, 4,85 mmol, 2,0 eq) en THF (25 ml), se añadió anhídrido 1-propanofosfónico (T3P, 1,551 g, 4,85 mmol, 2,0 eq), seguido de diisopropiletilamina (DIPEA, 0,986 g, 9,75 mmol, 4,0 eq) bajo nitrógeno. Se agitó la mezcla de reacción durante 1 hora y se añadió 3-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)-3-metilpiperidin-1-carboxilato ferc-butílico (Compuesto 2 , 1,0 g, 3,43 mmol, 1,0 eq). Se agitó la reacción durante 48 horas y se controló mediante TLC. Una vez completada la reacción, se vertió la mezcla en agua (50 ml), se extrajo con acetato de etilo (2 x 20 ml), se lavó con agua y, a continuación, con salmuera, después se desecó sobre sulfato de sodio, se filtró y se evaporó a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida utilizando gel de sílice (tamaño de malla 60-120) eluyendo con acetato de etilo al 10-20 % en n-hexano, produciendo 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-(3-hidroxi-2,2-dimetilpropanoil)-1H-pirazol-3-il)-3-metilpiperidin-1-carboxilato ferc-butílico (Compuesto 4 , 0,410 g, rendimiento: 32,9 %) m/z 511,8 [M+1]+ RMN de 1H (400 MHz, DMSO) 57,63 (s, 1H), 6,92 (d, J = 37,7 Hz, 2H), 5,46 (s, 1H), 4,83 (t, J = 5,3 Hz, 1H), 4,41 (s, 2H), 3,87 (s, 2H), 3,42 (s, 2H), 3,19 (d, J = 5,2 Hz, 2H), 1,91 (s, 1H), 1,55 (s, 2H), 1,36 (s, 3H), 1,30 (s, 6H), 1,12 (s, 3H) ppm.
Ejemplo 7-Preparación del compuesto 5
La s ín te s is de l
Compuesto 5
s ig u ió el p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 6a
.

A una solución fría (0 0C) de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-(3-hidroxi-2,2-dimetilpropanoil)-1H-pirazol-3-il)-3-metilpiperidin-1-carboxilato ferc-butílico (Compuesto 4 , 0,4 g, 0,78 mmol, 1,0 eq) en acetato de etilo (600 ml), se añadió gota a gota yodotrimetilsilano (0,5 ml). La mezcla de reacción se dejó a temperatura ambiente y se agitó durante 30 horas. Se controló la reacción mediante CL-EM y TLC. Una vez completada, se lavó la mezcla de reacción con solución de bicarbonato de sodio (20 %, 150 ml), agua (150 ml), seguida de salmuera (150 ml). La capa orgánica se desecó sobre sulfato de sodio y se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando amoníaco-agua como fase móvil, dando 1-(5-(((5-clorotiofen-2-il)metil)amino)-3-(3-metilpiperidin-3-il)-1H-pirazol-1-il)-3-hidroxi-2,2-dimetilpropan-1-ona (Compuesto 5 , 0,030 g, rendimiento: 9,3 %) m/z 410,90 [M+1]+ RMN de 1H (400 MHz, DMSO) 5 7,71 (s, 1H), 7,00 (s, 2H), 5,57 (s, 1H), 4,89-4,75 (m, 1H), 4,44 (s, 2H), 3,85 (s, 2H), 2,89 (s, 4H), 2,02-1,99 (m, 1H), 1,62 (s, 3H), 1,32 (s, 6H), 1,19 (s, 3H) ppm.
Ejemplo 8-Preparación del compuesto 6
La síntesis del Compuesto 6 siguió el procedimiento del siguiente Procedimiento general 5a.

A una solución fría (0 0C) de ácido 3-metoxi-2,2-dimetilpropanoico (0,483 g, 3,65 mmol, 1,5 eq) en THF (15 ml), se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 0,701 g, 3,65 mmol, 1,5 eq) y, a continuación, trietilamina (TEA, 0,74 g, 7,31 mmol, 3,0 eq) bajo nitrógeno. Después de agitar durante 30 minutos, se añadió a la mezcla hidroxibenzotriazol (HOBt, 0,065 g, 0,48 mmol, 0,2 eq), seguido de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)-3-metilpiperidin-1-carboxilato ferc-butílico (Compuesto 2, 1,0 g, 3,43 mmol, 1,0 eq). La mezcla de reacción se controló mediante CL-EM y se agitó durante 72 horas a temperatura ambiente. Una vez completada, se vertió la mezcla de reacción en agua (20 ml) y se extrajo con acetato de etilo (2 x 20 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera, a continuación, se desecaron sobre sulfato de sodio, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida utilizando gel de sílice (tamaño de malla 60-120), eluyendo con acetato de etilo al 0-20 % en n-hexano, produciendo 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 -(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-3-metilpiperidin-1 -carboxilato fercbutílico (Compuesto 6 , 0,625 g, rendimiento: 48,9 %) m/z 524,93 [M+1]+ r Mn de 1H (400 MHz, DMSO) 57,60 (t, J = 5,6 Hz, 1H), 6,97 (s, 2H), 5,47 (s, 1H), 4,41 (s, 2H), 3,84 (s, 2H), 3,52 (d, J = 13,0 Hz, 1H), 3,24 (s, 3H), 3,13 (s, 3H), 1,95 (s, 1H), 1,57 (s, 3H), 1,35 (d, J = 4,6 Hz, 6H), 1,33 (s, 9H), 1,13 (s, 3H) ppm.
Ejemplo 9-Preparación del compuesto 7
La s ín te s is de l
Compuesto 7
s ig u ió el s ig u ie n te p ro ce d im ie n to de l
Procedimiento general 6b
.

A una solución fría (0 0C) de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-(3-metoxi-2,2-dimetilpropanoil)-1H-pirazol-3-il)-3-metilpiperidin-1-carboxilato ferc-butílico (Compuesto 6 , 0,4 g (0,76 mmol, 1,0 eq) en diclorometano (300 ml), se añadió una solución de ácido trifluoroacético (TFA, 2,5 ml) en diclorometano (6 ml). Se controló la reacción mediante CL-EM y TLC y, una vez completada (12 horas), se lavó la mezcla de reacción con bicarbonato de sodio al 20 % (200 ml), agua (200 ml) y salmuera (200 ml). La capa orgánica se desecó sobre sulfato de sodio y se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando amoníaco-agua como fase móvil, produciendo 1 -(5-((5-clorotiofen-2-il)metil)amino)-3-(3-metilpiperidin-3-il)-1 H-pirazol-1 -il)-3-metoxi-2,2-dimetilpropan-1-ona (Compuesto 7, 0,22 g, rendimiento: 83,4 %) m/z 425,36 [M+1]+ RMN de 1H (400 MHz, DMSO) 58,27 (s, 1H), 7,63 (d, J = 6,2 Hz, 1H), 6,98 (c, J = 3,8 Hz, 2H), 5,52 (s, 1H), 4,42 (d, J = 6,3 Hz, 2H), 3,82 (dd, J = 20,7, 8,4 Hz, 4H), 3,14 (s, 4H), 2,86-2,61 (m, 3H), 1,98 (s, 2H), 1,41 (t, J = 44,2 Hz, 6H), 1,16 (s, 3H) ppm.
Ejemplo 10-Preparación del compuesto 8
La síntesis del Compuesto 8 siguió el procedimiento del siguiente Procedimiento general 5a.

A una solución enfriada (0 0C) de ácido 2-metoxibenzoico (0,278 g, 1,82 mmol, 1,5 eq) en THF (12 ml), se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 0,351 g, 1,82 mmol, 1,5 eq) y, a continuación, trietilamina (TEA, 0,369 g, 3,65 mmol, 3,0 eq) bajo nitrógeno. Después de agitar durante 30 minutos, se añadieron hidroxibenzotriazol (HOBt, 0,032 g, 0,24 mmol, 0,2 eq) y luego 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)-3-metilpiperidin-1-carboxilato ferc-butílico (Compuesto 2, 0,5 g, 1,21 mmol, 1,0 eq). Se controló la reacción mediante CL-EM y, después de agitar durante la noche, se vertió la mezcla de reacción en agua (20 ml) y se extrajo con acetato de etilo (2 x 20 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se desecaron sobre sulfato de sodio, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida utilizando gel de sílice (tamaño de malla 60-120), eluyendo con 0-20 % de acetato de etilo en n-hexano, produciendo 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 -(2-metoxibenzoil)-1 H-pirazol-3-il)-3-metilpiperidin-1 -carboxilato ferc-butílico (Compuesto 8 , 0,25 g, rendimiento: 46 %) m/z 545,49 [M+1] RMN de 1H (400 MHz, Dm SO) 57,60 (t, J = 6,2 Hz, 1H), 7,47 (dt, J = 35,7, 14,2 Hz, 1H), 7,38 (dd, J = 7,5, 1,6 Hz, 1H), 7,13 (d, J = 8,4 Hz, 1H), 7,05-6,98 (m, 3H), 6,50-6,50 (m, 1H), 5,55 (s, 2H), 4,43 (d, J = 38,0 Hz, 2H), 3,73 (s, 3H), 3,48-3,11 (m, 4H), 1,73 (s, 1H), 1,60 1,06 (m, 13H), 1,03 (s, 3H) ppm.
Ejemplo 11-Preparación del compuesto 9
La s ín te s is de l
Compuesto 9
s ig u ió el s ig u ie n te p ro ce d im ie n to de l
Procedimiento general 6b
:

A una solución enfriada (0 0C) de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-(2-metoxibenzoil)-1H-pirazol-3-il)-3-metilpiperidin-1-carboxilato ferc-butílico (Compuesto 8 , 0,4 g, 0,73 mmol, 1,0 eq) en diclorometano (250 ml), se añadió gota a gota una solución de TFA (2,5 ml) en DCM (10 ml). Se controló la reacción mediante CL-EM y TLC y, después de agitar a 0 0C durante 12 horas, se lavó la mezcla con bicarbonato de sodio al 20 % (200 ml), agua (200 ml) y, después, con salmuera (200 ml). La capa orgánica se desecó sobre sulfato de sodio, se filtró y se evaporó a presión reducida. El residuo se purificó mediante HPLC preparativa eluyendo con amoníaco-agua como fase móvil, obteniéndose el producto (5-(((5-clorotiofen-2-il)metil)amino)-3-(3-metilpiperidin-3-il)-1 H-pirazol-1-il)(2-metoxifenil)metanona (Compuesto 9 , 0,08 g, rendimiento: 24 %) m/z 444,81 [M+1]+ RMN de 1H (400 MHz, DMSO) 5 7,71 (t, J = 6,4 Hz, 1H), 7,50 (dd, J = 11,4, 4,4 Hz, 1H), 7,44 (dd, J = 7,5, 1,6 Hz, 1H), 7,14 (d, J = 8,2 Hz, 1H), 7,02 (dd, J = 12,5, 4,5 Hz, 3H), 5,64 (s, 1H), 4,49 (d, J = 6,3 Hz, 2H), 3,07 (d, J = 13,1 Hz, 1H), 2,81 (s, 1H), 2,69 (d, J = 12,1 Hz, 2H), 1,85 (s, 1H), 1,49 (d, J = 11,6 Hz, 3H), 1,09 (s, 3H) ppm.
Ejemplo 12-Preparación del compuesto 10
La síntesis del Compuesto 10 siguió el procedimiento del siguiente Procedimiento general 5a.

A una solución enfriada (0 0C) de ácido tiofen-3-carboxílico (0,374 g, 2,9 mmol, 1,5 eq) en THF (20 ml), se añadieron clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 0,561 g, 2,9 mmol, 1,5 eq) y trietilamina (0,591 g, 5,8 mmol, 3,0 eq) bajo nitrógeno. Se agitó la mezcla de reacción durante 30 minutos y se añadió hidroxibenzotriazol (HOBt, 0,052 g, 0,39 mmol, 0,2 eq), seguido de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)-3-metilpiperidin-1 -carboxilato ferc-butílico (Compuesto 2 , 0,8 g, 1,9 mmol, 1,0 eq). Se controló la reacción mediante CL-EM y, después de agitar durante la noche a temperatura ambiente, se vertió la mezcla de reacción en agua (20 ml) y se extrajo con acetato de etilo (2 x 20 ml). Las fases orgánicas combinadas se lavaron con agua y, a continuación, con salmuera, se desecaron sobre sulfato de sodio, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida utilizando gel de sílice (tamaño de malla 60-120) eluyendo con 0-20 % de acetato de etilo en n-hexano, produciendo el producto 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-(tiofen-3-carbonil)-1H-pirazol-3-il)-3-metilpiperidin-1-carboxilato ferc-butílico (Compuesto 10, 0,4 g, rendimiento: 49 %) m/z 521,77 [M+1]+ RMN de 1H (400 MHz, DMSO) 58,95 (d, J = 58,3 Hz, 1H), 7,78 (dd, J = 12,2, 5,9 Hz, 2H), 7,66 (d, J = 3,0 Hz, 1H), 6,99 (dd, J = 11,6, 3,7 Hz, 2H), 5,61 (s, 1H), 4,49 (s, 2H), 3,70-3,53 (m, 1H), 3,29-3,15 (m, 3H), 1,98 (s, 1H), 1,59 (d, J = 14,8 Hz, 3H), 1,33 (s, 9H), 1,16 (s, 3H) ppm.
Ejemplo 13-Preparación del compuesto 11
La s ín te s is de l
Compuesto 11
s ig u ió e l s ig u ie n te p ro ce d im ie n to de l
Procedimiento general 6b
:

A una solución enfriada (0 0C) de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-(tiofen-3-carbonil)-1H-pirazol-3-il)-3-metilpiperidin-1-carboxilato ferc-butílico (Compuesto 10, 0,4 g, 0,76 mmol, 1,0 eq) en diclorometano (250 ml), se añadió gota a gota una solución de TFA (2,5 ml) en DCM (10 ml). Se controló la reacción mediante CL-EM y TLC, y después de agitar durante 12 horas a 0 0C, se lavó la solución con bicarbonato de sodio (20 %, 200 ml), agua (200 ml) y salmuera (200 ml). La capa orgánica se desecó sobre sulfato de sodio, se filtró y se evaporó a presión reducida. El residuo se concentró y se purificó mediante HPLC preparativa, eluyendo con amoníaco-agua como fase móvil, dando el producto deseado (5-(((5-clorotiofen-2-il)metil)amino)-3-(3-metilpiperidin-3-il)-1 H-pirazol-1-il)(tiofen-3-il)metanona (Compuesto 11, 0,075 g, rendimiento: 23 %) m/z 420,80 [M+1]+ RMN de 1H (400 MHz, DMSO) 5 8,90 (d, J = 1,8 Hz, 1H), 8,31 (s, 1H), 7,85-7,77 (m, 2H), 7,66 (dd, J = 5,1,3,0 Hz, 1H), 7,01 (t, J = 7,5 Hz, 1H), 6,98 (d, J = 3,7 Hz, 1H), 5,68 (s, 1H), 4,50 (d, J = 6,2 Hz, 2H), 3,34 (d, J = 12,3 Hz, 1H), 2,81 (d, J = 12,0 Hz, 3H), 2,00 (s, 1H), 1,60 (d, J = 8,9 Hz, 2H), 1,48 (s, 1H), 1,19 (d, J = 12,7 Hz, 3H) ppm.
Ejemplo 14-Preparación del compuesto 12
La síntesis del Compuesto 12 siguió el procedimiento del siguiente Procedimiento general 5a.

A una solución enfriada (0 0C) de ácido 4-metiltetrahidro-2H-piran-4-carboxílico (0,527 g, 3,6 mmol, 1,5 eq) en THF (20 ml), se añadieron clorhidrato de 1 -(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 0,701 g, 3,6 mmol, 1,5 eq) y trietilamina (0,74 g, 7,3 mmol, 3,0 eq) bajo nitrógeno. Después de agitar durante 30 minutos a 0 0C, se añadió a la mezcla de reacción hidroxibenzotriazol (HOBt, 0,065 g, 4,8 mmol, 0,2 eq), seguido de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)-3-metilpiperidin-1-carboxilato ferc-butílico (Compuesto 2 , 1,0 g, 2,4 mmol, 1,0 eq). Se agitó la mezcla de reacción durante 48 horas a temperatura ambiente y se controló mediante CL-EM. Se vertió la solución en agua (20 ml) y se extrajo con acetato de etilo (2 x 20 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera y a continuación, se desecaron sobre sulfato de sodio, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida utilizando gel de sílice (tamaño de malla 60-120), eluyendo con acetato de etilo al 0-20 % en n-hexano, produciendo 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-(4-metiltetrahidro-2H-pirano-4-carbonil)-1 H-pirazol-3-il)-3-metilpiperidin-1-carboxilato ferc-butílico (Compuesto 12, 0,610 g, rendimiento: 47 %) m/z 536,89 [M+1]+ RMN de 1H (400 MHz, DMSO) 5 7,67 (s, 1H), 6,97 (s, 2H), 5,48 (s, 1H), 4,42 (s, 2H), 3,67 (s, 2H), 3,47 (s, 3H), 3,22 (s, 1H), 2,34 (s, 2H), 1,91 (s, 1H), 1,71 (s, 2H), 1,51 (s, 5H), 1,33 (s, 9H), 1,11 (s, 3H) ppm.
Ejemplo 15-Preparación del compuesto 13
La s ín te s is de l
Compuesto 13
s ig u ió e l s ig u ie n te p ro ce d im ie n to de l
Procedimiento general 6b
.

A una solución enfriada (0 0C) de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-(4-metiltetrahidro-2H-pirano-4-carbonil)-1H-pirazol-3-il)-3-metilpiperidin-1 -carboxilato ferc-butílico (Compuesto 12, 0,4 g, 0,74 mmol, 1,0 eq) en diclorometano (300 ml), se añadió gota a gota una solución de TFA (2,5 ml) en diclorometano (6 ml). Se controló la reacción mediante CL-EM y TLC y, después de agitar a 0 0C durante 12 horas, se lavó la mezcla de reacción con bicarbonato de sodio (20 %, 200 ml), agua (200 ml) y salmuera (200 ml). La capa orgánica se desecó sobre sulfato de sodio, se filtró y se evaporó a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando amoníaco-agua como fase móvil, dando (5-(((5-clorotiofen-2-il)metil)amino)-3-(3-metilpiperidin-3-il)-1 H-pirazol-1-il)(4-metiltetrahidro-2H-piran-4-il)metanona (Compuesto 13, 0,09 g, rendimiento: 28 %) m/z 436,81 [M+1]+ RMN de 1H (400 MHz, DMSO) 58,29 (s, 1H), 7,67 (s, 1H), 6,99 (d, J = 5,4 Hz, 2H), 5,53 (s, 1H), 4,44 (d, J = 6,2 Hz, 2H), 3,69 (s, 2H), 3,48 (s, 2H), 3,10 (s, 1H), 2,72 (s, 2H), 2,34 (s, 2H), 1,94 (s, 1H), 1,70 (s, 2H), 1,52 (s, 5H), 1,14 (s, 3H) ppm.
Ejemplo 16-Preparación del producto intermedio 3
La síntesis del Producto intermedio 3 siguió el procedimiento del siguiente Procedimiento general 2 :

Producto intermedio 3
A una solución enfriada (-78 0C) de acetonitrilo (3,86 ml, 69,8 mmol, 1,7 eq) en tetrahidrofurano (150 ml), se añadió n-BuLi (2,5 M en hexano, 27,9 ml, 69,8 mmol, 1,7 eq) gota a gota durante un período de 30 minutos, seguido de agitación a -78 0C durante 30 minutos más. A esto se añadió entonces 4-metil-piperidin-1,4-dicarboxilato ^(fercbutílico) (10,0 g, 41,1 mmol, 1,0 eq) poco a poco, y se agitó la mezcla a -78 0C durante 3 horas más. Se inactivó la mezcla de reacción con solución acuosa saturada de cloruro de amonio y después se extrajo con acetato de etilo. La fase orgánica se desecó (sulfato de sodio), se filtró y se evaporó, obteniéndose un aceite, que se utilizó sin mayor purificación (Producto intermedio 3, 9,28 g, rendimiento: 89 %) m/z 252,15.
Ejemplo 17-Preparación del compuesto 14
La síntesis del Compuesto 14 siguió el procedimiento del Procedimiento general 3 :

A una solución de 4-(2-cianoacetil)piperidin-1-carboxilato) ferc-butílico (Producto intermedio 3 , 7,0 g, 27,6 mmol, 1,0 eq) en isopropanol (210 ml), se añadió gota a gota monohidrato de hidrazina (1,65 ml, 33,2 mmol, 1,2 eq), seguida de la adición de ácido acético (1,65 ml, 27,6 mmol, 1,0 eq). Se agitó la mezcla de reacción a 85 0C durante 4-5 horas. Una vez completada, se evaporó la mezcla de reacción a presión reducida. El residuo se purificó mediante cromatografía en columna utilizando gel de sílice (malla 60-120), eluyendo con metanol al 10-15 % en diclorometano, produciendo 4-(5-amino-1H-pirazol-3-il)piperidin-1-carboxilato ferc-butílico (Compuesto 14, 6,2 g, rendimiento: 84 %) m/z 266,17. RMN de 1H (DMSO-da, 400 MHz) 55,182 (1H, s), 3,935-3,965 (2H, d), 3,172 (2H, s), 2,791 (1H, m), 1,747-1,802 (2H, d), 1,424(9H, s), 1,361 -1,402 (2H, d), 1,341 -1,351 (1H, d) ppm.
Ejemplo 18-Preparación del compuesto 15
La s ín te s is de l
Compuesto 15
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 4
:

A una solución de 4-(5-amino-1H-pirazol-3-il)piperidin-1-carboxilato ferc-butílico (Compuesto 14, 3,0 g, 11,3 mmol, 1,0 eq) en metanol (30 ml), se añadió ácido acético (0,67 ml, 11,3 mmol, 1,0 eq), seguido de 5-clorotiofen-2-carbaldehído (1,81 g, 12,4 mmol, 1,1 eq) poco a poco. Se agitó la reacción durante 2 horas a temperatura ambiente. Después, se añadió a la mezcla de reacción cianoborohidruro de sodio (1,42 g, 22,6 mmol, 1,5 eq) poco a poco durante un período de 45 minutos. Se agitó la mezcla de reacción durante 3 horas más. Una vez completada la reacción, se concentró la mezcla de reacción a presión reducida, y el residuo se vertió en agua helada agitada y se extrajo con acetato de etilo. La capa orgánica se desecó sobre sulfato de sodio, se filtró y se evaporó a presión reducida. El residuo se purificó mediante cromatografía en columna utilizando gel de sílice neutro, eluyendo con metanol al 5-10 % en diclorometano, proporcionando 4-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)piperidin-1-carboxilato ferc-butílico (Compuesto 15, 3,0 g, rendimiento: 68 %) m/z[M+H]+ 396,14 RMN de 1H (DMSO-d6, 400 MHz) 5 11,34 (1H, s), 6,87-6,94 (1H, c), 6,834-6,843 (1H, c), 5,68 (1H, s), 4,275-4,288 (1H, s), 3,945-3,975 (2H, d),2,786-2,796 (2H, d), 2,623-2,681 (1H, d), 1,725-1,995 (2H, d), 1,402-1,559 (12H, m) ppm.
Ejemplo 19-Preparación del compuesto 16
La síntesis del Compuesto 16 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución enfriada (0 0C) de ácido piválico (0,19 g, 1,9 mmol, 1,5 eq) en THF (10 ml), se añadió diisopropiletilamina (DIPEA, 0,24 ml, 1,4 mmol, 1,5 eq) y, a continuación, clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 0,365 g, 1,9 mmol, 1,5 eq). Después de agitar durante 20 minutos más, se añadió a la mezcla de reacción 4-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)piperidin-1-carboxilato ferc-butílico (Compuesto 15, 0,5 g, 1,3 mmol, 1,0 eq), seguido de hidroxibenzotriazol (HOBt, 0,035 g, 0,25 mmol, 0,2 eq). Se agitó la reacción durante 16 horas a temperatura ambiente. Una vez completada, se diluyó la mezcla de reacción con agua helada agitada y se extrajo el producto con acetato de etilo. La capa orgánica se desecó sobre sulfato de sodio, se filtró y se evaporó a presión reducida. El residuo se purificó mediante cromatografía en columna utilizando gel de sílice (malla 60-120, previamente lavado con trietilamina) eluyendo con 20-40 % de acetato de etilo en hexano, produciendo 4-(5-(((5-clorotiofen-2-il)metil)amino)-1 -pivaloil-1 H-pirazol-3-il)piperidin-1 -carboxilato fercbutílico (Compuesto 16, 0,5 g, rendimiento: 83 %) m/z[M+H]+ 480,20 RMN de 1H (DMSO-d6, 400 MHz) 57,691-7,66 (1H, t), 6,957-6,984 (2H, t), 5,415 (1H, s), 4,4-4,416 (2H, d), 3,916-3,948 (2H, d), 2,867 (2H, s), 2,624-2,681 (2H, m),1,818-1,852(2H, d), 1,402-1,484(16H, s) ppm.
Ejemplo 20-Preparación del compuesto 17
La síntesis del Compuesto 17 siguió el siguiente procedimiento del Procedimiento general 6a:

A una solución enfriada (0 0C) de 4-(5-(((5-clorotiofen-2-il)metil)amino)-1-pivaloil-1H-pirazol-3-il)piperidin-1-carboxilato terc-butílico (Compuesto 16, 0,5 g, 1,0 mmol, 1,0 eq) en acetato de etilo, se añadió gota a gota yodotrimetilsilano (0,148 g, 1,0 mmol, 1,0 eq) diluido en acetato de etilo. Se agitó la reacción durante 15 minutos con la temperatura por debajo de 10 0C. Una vez completada la reacción, se diluyó la mezcla con solución de bicarbonato de sodio con agitación y se extrajo con acetato de etilo. La capa orgánica se desecó sobre sulfato de sodio, se filtró y se concentró a presión reducida. El residuo se purificó mediante el método de HPLC preparativa, produciendo 1 -(5-(((5-clorotiofen-2-il)metil)amino)-3-(piperidin-4-il)-1 H-pirazol-1 -il)etan-1 -ona (Compuesto 17, 0,042 g, rendimiento: 10 %) m/z[M+H]+ 380,14 RMN de 1H (DMSO-d6, 400 MHz) 5 8,473 (1H, s), 8,189 (1H, s), 7,733-7,764 (1H, t), 6,945-6,971 (2H, s), 5,39 (1H, d), 4,421-4,436 (2H, d),3,289-3,344 (2H, d), 2,985-3,035 (2H, d),2,896-2,736(1 H, m),2,029-2,057(2H, d), 1,715-1,782(2H,d), 1,366-1,413(9H, s)1,247-1,127 (1H, m) ppm.
Ejemplo 21-Preparación del producto intermedio 4
La síntesis del Producto intermedio 4 siguió el procedimiento del siguiente Procedimiento general 7 :

A una solución enfriada (0 0C) de piperidin-4-carboxilato de etilo (10,0 g, 64,1 mmol, 1,0 eq) y trietilamina (6,4 g, 64,1 mmol, 1,0 eq) en diclorometano (300 ml), se añadió cloroformiato de alilo (7,7 g, 64,1 mmol, 1,0 eq) gota a gota. Se agitó la reacción a temperatura ambiente durante 16 horas. Una vez completada, la mezcla se diluyó con agua helada con agitación y se extrajo con diclorometano (3 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna utilizando gel de sílice (malla 60-120), proporcionando 4-etil-piperidin-1,4-dicarboxilato de 1 -alilo (10,0 g, rendimiento: 89 %) m/z 241,13 RMN de 1H (400 MHz, CDCls) 55,96-5,81 (m, 1H), 5,32-5,10 (m, 2H), 4,53 (dt, J = 5,5, 1,4 Hz, 2H), 4,16-4,05 (m, 2H), 4,06-3,93 (m, 2H), 2,88 (s, 2H), 2,42 (tt, J = 10,9, 3,9 Hz, 1H), 1,82 (dd, J = 24,3, 8,9 Hz, 2H), 1,60 (dtd, J = 13,6, 11,2, 4,3 Hz, 2H), 1,21 (t, J = 7,1 Hz, 3H) ppm.
Ejemplo 22-Preparación del producto intermedio 5
La síntesis del Producto intermedio 5 siguió el procedimiento del siguiente Procedimiento general 2 :

A una solución enfriada (-78 0C) de acetonitrilo (3,9 ml, 70,5 mmol, 1,7 eq) en tetrahidrofurano (150 ml), se añadió n-BuLi (2,5 M en hexano, 28,2 ml, 70,5 mmol, 1,7 eq) gota a gota durante un período de 30 minutos, seguido de otro período de agitación durante 30 minutos. A esta mezcla, se añadió 4-etil-piperidin-1,4-dicarboxilato de 1 -alilo (Producto intermedio 4 , 10,0 g, 41,5 mmol, 1,0 eq) poco a poco y la mezcla de reacción se mantuvo a -78 0C durante 3 horas. Se inactivó la reacción con la adición de cloruro de amonio acuoso saturado y se extrajo el producto con acetato de etilo. Tras la desecación (sulfato de sodio), el producto se agitó en una mezcla de acetato de etilo/hexano y, a continuación, se desecó al vacío, produciendo 4-(2-cianoacetil)piperidin-1-carboxilato de alilo (Producto intermedio 5, 9,8 g, rendimiento: 99 %) m/z 236,12 RMN de 1H (400 MHz, CDC^) 55,95 (ddd, J = 22,7, 10,8, 5,6 Hz, 1H), 5,27 (ddd, J = 13,8, 11,7, 1,4 Hz, 2H), 4,60 (d, J = 5,5 Hz, 2H), 4,22 (s, 2H), 3,56 (s, 2H), 2,92 (s, 2H), 2,77 (tt, J = 11,3, 3,7 Hz, 1H), 1,92 (d, J = 11,4 Hz, 2H), 1,68-1,54 (m, 2H) ppm.
Ejemplo 23-Preparación del compuesto 18
La síntesis del Compuesto 18 siguió el procedimiento del siguiente Procedimiento general 3 :

A una solución de 4-(2-cianoacetil)piperidin-1-carboxilato de alilo (Producto intermedio 5, 9,5 g, 40,1 mmol, 1,0 eq) en isopropanol (285 ml), se añadió gota a gota monohidrato de hidrazina (2,0 ml, 40,1 mmol, 1,0 eq), seguido de ácido acético (2,88 ml, 48,1 mmol, 1,2 eq). Se agitó la reacción a temperatura ambiente durante 3-4 horas. Una vez completada, se concentró la mezcla de reacción a presión reducida y el residuo se purificó mediante cromatografía en columna utilizando gel de sílice (malla 60-120), eluyendo con metanol al 10-15 % en diclorometano, produciendo 4-(5-amino-1 H-pirazol-3-il)piperidin-1-carboxilato de alilo (Compuesto 18, 7,0 g, rendimiento: 70 %) m/z 250,14. RMN de 1H (400 MHz, DMSO) 5 11,84-10,78 (m, 1H), 5,94 (ddd, J = 22,4, 10,5, 5,2 Hz, 1H), 5,28 (d, J = 17,3 Hz, 1H), 5,19 (d, J = 6,0 Hz, 2H), 4,53 (d, J = 5,1 Hz, 2H), 4,01 (d, J = 13,1 Hz, 2H), 3,17 (s, 2H), 2,89 (s, 2H), 2,62 (dd, J = 34,1,22,6 Hz, 1H), 1,82 (d, J = 12,7 Hz, 2H), 1,52-1,21 (m, 2H) ppm.
Ejemplo 24-Preparación del compuesto 19
La síntesis del Compuesto 19 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución enfriada (0 °C) de 4-(5-amino-1H-pirazol-3-il)piperidin-1-carboxilato de alilo (Compuesto 18, 7,0 g, 28 mmol, 1,0 eq) en MeOH (140 ml), se añadió ácido acético (2,52 ml, 42 mmol, 1,5 eq), seguido de 5-clorotiofen-2-carbaldehído (4,9 g, 33,6 mmol, 1,2 eq) poco a poco. Se agitó la reacción durante 4-5 horas a temperatura ambiente. A esto, se añadió después cianoborohidruro de sodio (3,5 g, 56 mmol, 2,0 eq) poco a poco durante un período de 45 minutos, y se agitó la mezcla de reacción durante 4-5 horas más. Una vez completada la reacción, se concentró la mezcla de reacción a presión reducida y se vertió la masa residual en agua helada con agitación. A continuación, se extrajo el producto con acetato de etilo y la capa orgánica se desecó sobre sulfato de sodio, se filtró y se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna utilizando gel de sílice neutro, eluyendo con metanol al 2-4 % en diclorometano, dando 4-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)piperidin-1 -carboxilato de alilo puro (Compuesto 19, 4,5 g, rendimiento: 45 %) m/z[M+H]+ 380,11 RMN de 1H (400 MHz, DMSO) 5 11,41 (s, 1H), 6,88 (dd, J = 30,0, 3,7 Hz, 2H), 5,94 (ddd, J = 22,4, 10,5, 5,2 Hz, 1H), 5,68 (d, J = 6,2 Hz, 1H), 5,28 (dd, J = 16,4, 2,5 Hz, 2H), 5,19 (dd, J = 10,5, 1,5 Hz, 1H), 4,60-4,46 (m, 2H), 4,28 (d, J = 6,1 Hz, 2H), 4,12 (d, J = 4,8 Hz, 1H), 4,02 (d, J = 13,2 Hz, 2H), 3,17 (d, J = 4,0 Hz, 2H), 2,90 (s, 2H), 2,69 (t, J = 11,6 Hz, 1H) ppm.
Ejemplo 25-Preparación del compuesto 20
La síntesis del Compuesto 20 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución enfriada (0 0C) de ácido piválico (0,644 g, 6,3 mmol, 1,2 eq) en THF (10 ml), se añadió diisopropiletilamina (DIEA, 1,38 ml, 7,9 mmol, 1,5 eq) y, a continuación, 1-(3-clorhidrato de dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 1,52 g, 7,9 mmol, 1,5 eq). Después de agitar durante 20 minutos, se añadió 4-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)piperidin-1-carboxilato de alilo (Compuesto 19, 2,0 g, 5,3 mmol, 1,0 eq), seguido de hidroxibenzotriazol (HOBt, 0,142 g, 1,05 mmol, 0,2 eq). Se agitó la reacción durante 16 horas a
temperatura ambiente. Una vez completada, se diluyó la mezcla de reacción con agua helada agitada y después se extrajo con acetato de etilo. La capa orgánica se desecó sobre sulfato de sodio, se filtró y se evaporó a presión reducida. El residuo se purificó mediante cromatografía en columna utilizando gel de sílice (malla 60-120) previamente lavado con trietilamina, y el producto se eluyó con acetato de etilo al 2-5 % en hexano, dando 4-(5-(((5-clorotiofen-2-il)metil)amino)-1-pivaloil-1H-pirazol-3-il)piperidin-1-carboxilato de alilo (Compuesto 20, 1,5 g, rendimiento: 61 %) m/z[M+H]+ 464,16 RMN de 1H (400 MHz, DMSO) 5 7,69 (t, J = 6,2 Hz, 1H), 6,97 (dd, J = 8,1, 3,7 Hz, 2H), 5,94 (ddd, J = 22,4, 10,4, 5,2 Hz, 1H), 5,42 (s, 1H), 5,28 (dd, J = 17,2, 1,5 Hz, 1H), 5,19 (d, J = 10,5 Hz, 1H), 4,53 (d, J = 5,1 Hz, 2H), 4,41 (d, J = 6,1 Hz, 2H), 3,99 (d, J = 13,2 Hz, 2H), 2,96 (s, 2H), 2,69 (t, J = 11,0 Hz, 1H), 1,86 (d, J = 11,1 Hz, 2H), 1,56-1,44 (m, 2H), 1,40 (s, 9H) ppm.
Ejemplo 26-Preparación del compuesto 21
La síntesis del Compuesto 21 siguió el siguiente procedimiento del Procedimiento general 8a :

Se desgasificó una solución de 4-(5-(((5-clorotiofen-2-il)metil)amino)-1-pivaloil-1H-pirazol-3-il)piperidin-1-carboxilato de alilo (Compuesto 20, 0,1 g, 0,22 mmol, 1,0 eq) y cloruro de cinc (0,003 g, 21,6 mmol, 0,1 eq) en diclorometano (10 ml) con una corriente de nitrógeno durante 15 minutos. A continuación, se añadió a la mezcla fr/s(dibencilidenacetona)dipaladio (0) (Pd2(dba)3 , 0,0098 g, 0,011 mmol, 0,05 eq), y la solución se desgasificó de nuevo con una corriente de nitrógeno durante 15 minutos. A continuación, se añadió gota a gota a temperatura ambiente poli(metilhidrosiloxano) (PMHS, 0,818 g, 0,43 mmol, 2,0 eq) y se agitó la mezcla de reacción durante 3 4 horas a temperatura ambiente. Una vez completada, se filtró la mezcla de reacción a través de Celite®, y después se evaporó el disolvente bajo presión reducida. Al residuo, se añadió hexano y, a continuación, acetonitrilo, y después se agitó la mezcla durante 10 minutos. Se filtraron las partículas negras, y se separaron las capas de acetonitrilo y hexano. Se lavó la capa de acetonitrilo con hexano 3 veces y se evaporó el disolvente. El residuo se purificó mediante HPLC preparativa, produciendo 1-(5-(((5-clorotiofen-2-il)metil)amino)-3-(piperidin-4-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 21, 0,0375 g, rendimiento: 32 %) m/z[M+H]+ 380,14 RMN de 1H (400 MHz, DMSO) 5 8,36 (s, 1H), 7,72 (t, J = 6,2 Hz, 1H), 6,97 (s, 2H), 5,38 (s, 1H), 4,42 (d, J = 6,1 Hz, 2H), 3,21 (d, J = 12,6 Hz, 2H), 2,88 (dd, J = 12,2, 9,6 Hz, 2H), 2,75 (dd, J = 12,9, 9,4 Hz, 1H), 1,98 (d, J = 11,2 Hz, 2H), 1,76-1,61 (m, 2H), 1,41 (s, 9H) ppm.
Ejemplo 27-Preparación del producto intermedio 6
La síntesis del Producto intermedio 6 siguió el procedimiento del siguiente Procedimiento general 9 :

A una solución enfriada (-78 0C) de diisopropilamina (8,74 g, 86,4 mmol, 1,6 eq) en THF (200 ml), se añadió n-BuLi (34,5 ml, 86,4 mmol, 1,6 eq), y se agitó la mezcla durante 1 hora a 0 0C. La mezcla se enfrió hasta -78 0C y se añadió 2-oxopiperidin-1-carboxilato ferc-butílico (10,0 g, 54,0 mmol, 1,0 eq). Se agitó la mezcla durante 1 hora y se añadió cloroformiato de metilo (6,12 g, 64,8 mmol, 1,2 eq). La mezcla de reacción se dejó calentar hasta la temperatura ambiente durante la noche y se controló mediante TLC y CL-EM. Una vez completada, se inactivó la mezcla de reacción con cloruro de amonio y se evaporó a presión reducida. El residuo se extrajo con acetato de etilo (2 x 150 ml) y las fases orgánicas combinadas se lavaron con agua y salmuera, se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida utilizando gel de sílice (malla 60-120) eluyendo con acetato de etilo al 40 % en n-hexano, produciendo 1-(ferc-butil)-3-metil-2-oxopiperidin-1,3-dicarboxilato (Producto intermedio 6 , 9,2 g, rendimiento: 70 %) m/z 202,13 [M-56]+; RMN de 1H (400 MHz, DMSO) 53,70 (s, 1H), 3,62-3,50 (m, 3H), 2,10-1,89 (m, 2H), 1,88-1,72 (m, 2H), 1,42 (s, 9H) ppm.
E je m p lo 28 -P re p a ra c ió n de l p ro d u c to in te rm ed io 7
La síntesis del Producto intermedio 7 siguió el procedimiento del siguiente Procedimiento general 1:

A una solución enfriada (0 0C) de 1-(ferc-butil)-3-metil-2-oxopiperidin-1,3-dicarboxilato (Producto intermedio 6 , 10,0 g, 38,9 mmol, 1,0 eq) en DMF (100 ml), se añadió carbonato de potasio (27,0 g, 194 mmol, 5,0 eq) y se agitó la mezcla de reacción durante 1 hora. A la mezcla, se añadió yoduro de metilo (6,62 g, 46,7 mmol, 1,2 eq). Se dejó que la mezcla de reacción alcanzara la temperatura ambiente y se agitó durante la noche. Una vez completada, se vertió la mezcla de reacción en agua helada y se filtró, produciendo el producto 1-(ferc-butil)-3-metil-3-metil-2-oxopiperidin-1,3-dicarboxilato (Producto intermedio 7 , 7,91 g, rendimiento: 75 %) m/z 272,2 [M+1]+; RMN de 1H (400 MHz, DMSO) 5 3,72-3,61 (m, 4H), 3,56-3,45 (m, 1H), 2,17 (d, J = 8,0 Hz, 1H), 1,78 (dd, J = 17,4, 12,4 Hz, 3H), 1,45 (s, 9H), 1,36 (s, 3H) ppm.
Ejemplo 29-Preparación del producto intermedio 8
La síntesis del Producto intermedio 8 siguió el siguiente procedimiento del Procedimiento general 6b :

A una solución enfriada (0 °C) de 1-(ferc-butil)-3-metil-3-metil-2-oxopiperidin-1,3-dicarboxilato (Producto intermedio 7, 5,0 g, 18,4 mmol, 1,0 eq) en diclorometano (50 ml), se añadió ácido trifluoroacético (2,5 ml) y se agitó la mezcla de reacción durante 1 hora a 0 °C. La mezcla de reacción se dejó calentar hasta la temperatura ambiente durante la noche. Una vez completada la reacción, se trató la mezcla de reacción con solución saturada de NaHCO3 y se extrajo con diclorometano (2 x 50 ml). Las capas orgánicas combinadas se lavaron con salmuera, se desecaron sobre sulfato de sodio anhidro y se evaporaron a presión reducida, produciendo 3-metil-2-oxopiperidin-3-carboxilato de metilo (Producto intermedio 8 , 2,8 g rendimiento: 89 %); m/z 173,16 [M+2]+; RMN de 1H (400 MHz, DMSO) 57,94 (s, 1H), 3,63 (s, 3H), 3,32-3,14 (m, 2H), 2,58-2,37 (m, 2H), 2,04-1,99 (m, 2H), 1,26 (s, 3H) ppm.
Ejemplo 30-Preparación del producto intermedio 9
La síntesis del Producto intermedio 9 siguió el procedimiento del siguiente Procedimiento general 2 :

A una solución enfriada (-78 °C) de acetonitrilo (1,0 g, 24,3 mmol, 1,6 eq) en tetrahidrofurano (30 ml), se añadió n-BuLi (2,5 M en hexano, 9,73 ml, 24,3 mmol, 1,6 eq) gota a gota durante un período de 20 minutos. Se agitó la reacción durante otros 60 minutos a -78 °C. A la mezcla, se añadió 3-metil-2-oxopiperidin-3-carboxilato de metilo (Producto intermedio 8 , 2,6 g, 15,2 mmol, 1,0 eq) poco a poco y se agitó la solución a -78 0C durante 3 horas. Se inactivó la mezcla con una solución saturada de cloruro de amonio y se extrajo con acetato de etilo. La capa orgánica se desecó sobre sulfato de sodio anhidro, se filtró y se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna utilizando gel de sílice (tamaño de malla 60-120), eluyendo con acetato de etilo al 70 % en n-hexano, dando 3-(3-metil-2-oxopiperidin-3-il)-3-oxopropanonitrilo (Producto intermedio 9 , 2,41 g, rendimiento: 88 %) m/z 181,11 [M+1]+; RMN de 1H (400 MHz, DMSO) 5 7,89 (s, 1H), 4,39-4,05 (m, 2H), 3,27-3,06 (m, 2H), 2,14 (ddd, J=19,7, 10,0, 5,2 Hz, 1H), 1,80-1,60 (m, 2H), 1,60-1,43 (m, 1H), 1,32 (s, 3H) ppm.
E je m p lo 31 -P re p a ra c ió n de l c o m p u e s to 22
La síntesis del Compuesto 22 siguió el procedimiento del siguiente Procedimiento general 3 :

A una solución de 3-(3-metil-2-oxopiperidin-3-il)-3-oxopropanonitrilo (Producto intermedio 9 , 2,0 g, 11,0 mmol, 1,0 eq) en isopropanol (10 ml) y ácido acético (2,0 ml), se añadió gota a gota monohidrato de hidrazina (0,825 g, 16,5 mmol, 1,5 eq), y la reacción se agitó a 60 0C durante 3 horas. La mezcla de reacción se controló mediante TLC y CL-EM y, una vez completada, se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna utilizando gel de sílice (malla 60-120), eluyendo con metanol al 0-5 % en diclorometano, dando 3-(5-amino-1 H-pirazol-3-il)-3-metilpiperidin-2-ona (Compuesto 22, 1,16 g, rendimiento: 54 %); m/z 195,19 [M+1]+.
Ejemplo 32-Preparación del compuesto 23
La síntesis del Compuesto 23 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución enfriada (0 0C) de 3-(5-amino-1 H-pirazol-3-il)-3-metilpiperidin-2-ona (Compuesto 22, 1,0 g, 5,15 mmol, 1,0 eq) en metanol (20 ml), se añadió gota a gota ácido acético (0,3 g, 5,15 mmol, 1,0 eq). A la mezcla, se añadió gota a gota 5-clorotiofen-2-carbaldehído (0,91 g, 6,18 mmol, 1,2 eq) y la mezcla se calentó hasta la temperatura ambiente durante 45 minutos. Se añadió cianoborohidruro de sodio (0,644 g, 10,3 mmol, 2,0 eq) poco a poco durante un período de 15 minutos y se agitó la mezcla de reacción a temperatura ambiente durante 12 horas. Una vez completada la reacción, la mezcla se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna utilizando gel de sílice (malla 60-100), eluyendo con metanol al 0-5 % en diclorometano, dando 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona (Compuesto 23, 0,686 g, rendimiento: 41 %); m/z 325,25; RMN de 1H (400 MHz, DMSO) 5 11,26 (s, 1H), 7,50 (s, 1H), 6,88 (dd, J = 31,3, 3,6 Hz, 2H), 5,66 (s, 1H), 5,30 (s, 1H), 4,29 (d, J = 6,2 Hz, 2H), 3,14 (s, 2H), 2,10 (s, 1H), 1,65 (s, 3H), 1,37 (s, 3H) ppm.
Ejemplo 33-Preparación del compuesto 24
La síntesis del Compuesto 24 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución enfriada (0 0C) de ácido piválico (0,188 g, 1,84 mmol, 1,5 eq) en THF (15 ml) bajo nitrógeno, se añadió clorhidrato de 1 -(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 0,352 g, 1,84 mmol, 1,5 eq), seguida de trietilamina (TEA, 0,373 g, 3,69 mmol, 3,0 eq). Se agitó la mezcla de reacción durante 30 minutos y, a continuación, se añadió a la mezcla hidroxibenzotriazol (HOBt, 0,033 g, 0,24 mmol, 0,2 eq), seguido de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona (Compuesto 23, 0,4 g, 1,23 mmol, 1,0 eq). Se controló la
reacción mediante CL-EM y, una vez completada, se vertió la mezcla de reacción en agua (50 ml) y se extrajo con acetato de etilo (3 x 25 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera, se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua como fase móvil, dando el producto 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-pivaloil-1 H-pirazol-3-il)-3-metilpiperidin-2-ona (Compuesto 24, 0,21 g, rendimiento: 42 %) m/z 408,95 [M+1]+; RMN de 1H (400 MHz, DMSO) 57,62 (t, J = 6,2 Hz, 1H), 7,53 (s, 1H), 6,96 (dd, J = 12,1,3,7 Hz, 2H), 5,42 (s, 1H), 4,41 (d, J = 6,1 Hz, 2H), 3,25-3,06 (m, 2H), 2,29-2,17 (m, 1H), 1,73 (ddd, J = 21,5, 11,1, 7,2 Hz, 3H), 1,41 (s, 9H), 1,37 (s, 3H) ppm.
Ejemplo 34-Preparación del compuesto 25
La síntesis del Compuesto 25 siguió el procedimiento del siguiente Procedimiento general 5c:

A una solución enfriada (0 0C) de ácido 3-hidroxi-2,2-dimetilpropanoico (0,130 g, 1,1 mmol, 1,2 eq) en THF (10 ml) bajo nitrógeno, se añadieron tetrafluoroborato de W,W,W',W'-tetrametil-0-(benzotriazol-1-il)uronio (TBTU, 0,320 g, 1,0 mmol, 1,1 eq) y diisopropiletilamina (DIEA, 0,280 g, 2,9 mmol, 3,0 eq). Se agitó la mezcla de reacción durante 30 minutos y se añadió 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona (Compuesto 23, 0,3 g, 0,9 mmol, 1,0 eq). Se controló la reacción mediante CL-EM y, una vez completada, se vertió la mezcla en agua (40 ml) y se extrajo con acetato de etilo (3 x 25 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera, se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua como fase móvil, dando el producto deseado 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona (Compuesto 25, 0,0129 g, rendimiento: 3 %) m/z 425,30 [M+1]+; RMN de 1H (400 MHz, CD3CN) 5 7,48 (s, 1H), 6,88 (c, J = 3,8 Hz, 2H), 6,10 (s, 1H), 5,44 (s, 1H), 4,45 (d, J = 6,2 Hz, 2H), 3,89 (d, J = 6,2 Hz, 2H), 3,43 (t, J = 6,2 Hz, 1H), 3,27 (d, J = 6,6 Hz, 2H), 2,34-2,23 (m, 1H), 1,91 -1,70 (m, 3H), 1,49-1,34 (m, 3H), 1,30 (s, 6H) ppm.
Ejemplo 35-Preparación del compuesto 26
La síntesis del Compuesto 26 siguió el procedimiento del siguiente Procedimiento general 5c:

A una solución enfriada (0 0C) de ácido 3-metoxi-2,2-dimetilpropanoico (0,134 g, 1,0 mmol, 1,1 eq) en THF (12 ml) bajo nitrógeno, se añadieron tetrafluoroborato de W,W,W',W'-tetrametil-0-(benzotriazol-1-il)uronio (TBTU, 0,326 g, 1,0 mmol, 1,1 eq) y diisopropiletilamina (DIPEA, 0,280 g, 2,8 mmol, 3,0 eq). Se agitó la mezcla de reacción durante 30 minutos y se añadió 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona (Compuesto 23, 0,3 g, 0,92 mmol, 1,0 eq). Se controló la reacción mediante CL-EM y, una vez completada, se vertió la mezcla en agua (30 ml) y se extrajo con acetato de etilo (3 x 25 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera, se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua como fase móvil, dando 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-(3-metoxi-2,2-dimetilpropanoil)-1H-pirazol-3-il)-3-metilpiperidin-2-ona (Compuesto 26, 0,061 g, rendimiento: 15 %); m/z 439,21 [M+1]+; RMN de 1H (400 MHz, DMSO) 5 7,59 (s, 1H), 7,54 (s, 1H), 6,96 (t, J = 6,9 Hz, 2H), 5,42 (s, 1H), 4,41 (d, J = 6,0 Hz, 2H), 3,83 (d, J = 5,1 Hz, 2H), 3,13 (s, 3H), 2,22 (s, 1H), 1,70 (s, 3H), 1,40-1,29 (m, 6H) ppm.
E je m p lo 36 -P re p a ra c ió n de l c o m p u e s to 27
La síntesis del Compuesto 27 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución enfriada (0 0C) de ácido 2-metoxibenzoico (0,351 g, 2,3 mmol, 1,5 eq) en THF (15 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 0,441 g, 2,3 mmol, 1,5 eq) y, a continuación, trietilamina (TEA, 0,467 g, 4,6 mmol, 3,0 eq). Se agitó la mezcla de reacción durante 30 minutos y se añadió hidroxibenzotriazol (HOBt, 0,041 g, 0,3 mmol, 0,2 eq), seguido de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona (Compuesto 23, 0,5 g, 1,53 mmol, 1,0 eq). Se controló la reacción mediante CL-EM y, una vez completada, se vertió la mezcla de reacción en agua (5 ml) y se extrajo con acetato de etilo (3 x 25 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera, se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando amoníaco-agua como fase móvil, dando el producto deseado 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-(2-metoxibenzoil)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona (Compuesto 27, 0,23 g, rendimiento: 32 %); m/z 458,91 [M+1]+; RMN de 1H (400 MHz, DMSO) 5 7,59 (t, J = 6,0 Hz, 1H), 7,55-7,34 (m, 3H), 7,12 (d, J = 8,3 Hz, 1H), 7,01 (dd, J = 13,4, 5,8 Hz, 2H), 5,48 (s, 1H), 4,48 (d, J = 6,0 Hz, 2H), 3,73 (s, 3H), 3,07 (s, 2H), 2,15-1,94 (m, 2H), 1,73 1,45 (m, 3H), 1,25 (s, 3H) ppm.
Ejemplo 37-Preparación del compuesto 28
La síntesis del Compuesto 28 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución enfriada (0 0C) de ácido 3-tiofenocarboxílico (0,177 g, 1,38 mmol, 1,5 eq) en THF (15 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI. HCl, 0,264 g, 1,38 mmol, 1,5 eq) y, a continuación, trietilamina (TEA, 0,28 g, 2,77 mmol, 3,0 eq). Se agitó la mezcla de reacción durante 30 minutos y se añadió hidroxibenzotriazol (HOBt, 0,024 g, 0,18 mmol, 0,2 eq), seguido de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona (Compuesto 23, 0,3 g, 0,92 mmol, 1,0 eq). Se controló la reacción mediante CL-EM y, una vez completada, se vertió la mezcla de reacción en agua (30 ml) y se extrajo con acetato de etilo (3x25 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera, se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando amoníaco-agua como fase móvil, produciendo el producto deseado 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-(tiofen-3-carbonil)-1H-pirazol-3-il)-3-metilpiperidin-2-ona (Compuesto 28, 0,12 g, rendimiento: 30 %) m/z 436,06 [M+1]+; RMN de 1H (400 MHz, DMSO) 58,90 (d, J = 2,4 Hz, 1H), 7,90-7,49 (m, 4H), 6,98 (s, 2H), 5,55 (s, 1H), 4,49 (d, J = 6,2 Hz, 2H), 3,18 (s, 2H), 2,23 (d, J = 10,4 Hz, 1H), 1,75 (d, J = 10,8 Hz, 3H), 1,42 (s, 3H) ppm.
Ejemplo 38-Preparación del compuesto 29
La s ín te s is de l
Compuesto 29
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5a
:
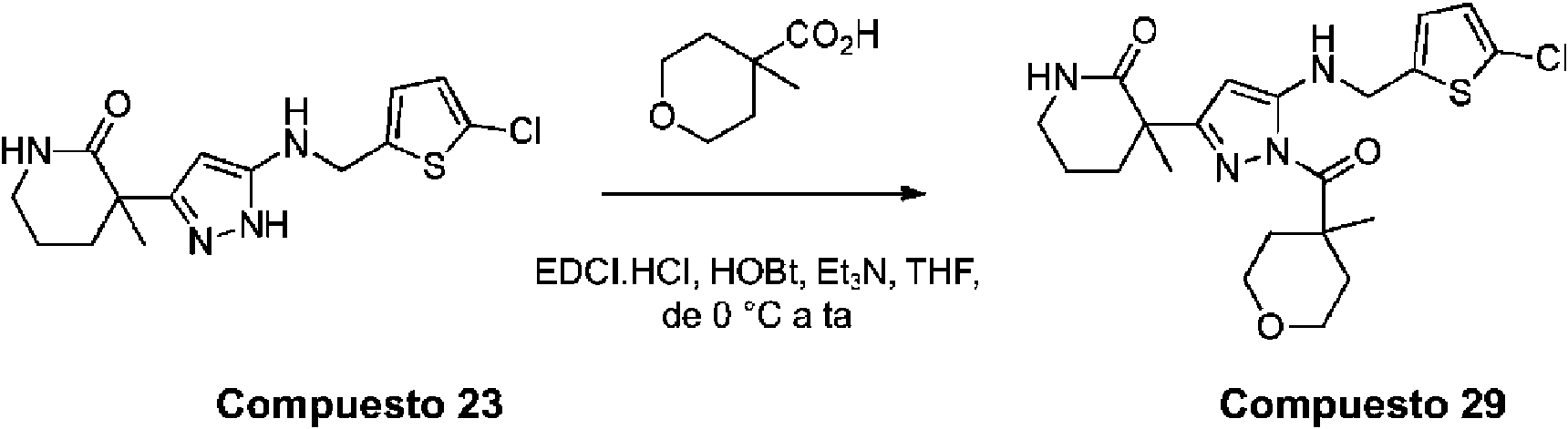
A una solución enfriada (0 °C) de ácido 4-metiltetrahidro-2H-piran-4-carboxílico (0,2 g, 1,4 mmol, 1,5 eq) en THF (15 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 0,265 g, 1,4 mmol, 1,5 eq) y, después, trietilamina (TEA, 0,283 g, 2,8 mmol, 3,0 eq). Se agitó la mezcla de reacción durante 30 minutos y se añadieron hidroxibenzotriazol (HOBt, 0,025 g, 0,19 mmol, 0,2 eq) y 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona (Compuesto 23, 0,3 g, 0,92 mmol, 1,0 eq). Se agitó la reacción durante 2 días y se controló mediante CL-EM. Una vez completada la reacción, se vertió la mezcla en agua (40 ml) y se extrajo con acetato de etilo (3 x 25 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera, se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua como fase móvil, dando el producto deseado 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 -(1 -metilciclohexano-1 -carbonil)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona (Compuesto 29, 0,052 g, rendimiento 13 %) m/z 450,96 [M+2]+; RMN de 1H (400 MHz, DMSO) 57,65 (t, J = 6,3 Hz, 1H), 7,53 (s, 1H), 6,96 (dd, J = 11,0, 3,7 Hz, 2H), 5,43 (s, 1H), 4,42 (d, J = 6,1 Hz, 2H), 3,81-3,60 (m, 2H), 3,45 (dd, J = 11,8, 8,8 Hz, 2H), 3,14 (s, 2H), 2,34 (s, 2H), 2,18 (d, J = 9,3 Hz, 1H), 1,70 (d, J = 8,5 Hz, 5H), 1,51 (s, 3H), 1,36 (s, 3H) ppm.
Ejemplo 39-Preparación del producto intermedio 10
La síntesis del Producto intermedio 10 siguió el procedimiento del siguiente Procedimiento general 9 :

A una solución enfriada (-78 °C) de diisopropilamina (8,74 g, 86,4 mmol, 1,6 eq) en THF anhidro (200 ml), se añadió n-BuLi (34,5 ml, 86,4 mmol, 1,6 eq), seguido de agitación a 0 °C durante 1 hora. La mezcla se volvió a enfriar hasta -78 °C y se añadió ferc-butil-2-oxopirrolidin-1-carboxilato (10,0 g, 54 mmol, 1,0 eq) poco a poco, se agitó durante 1 hora y se añadió cloroformiato de metilo (6,12 g, 64,8 mmol, 1,2 eq). Se dejó que la mezcla de reacción alcanzara la temperatura ambiente y se agitó durante la noche. Se controló mediante TLC y CL-EM. Una vez completada, se inactivó la mezcla de reacción con cloruro de amonio y se evaporó hasta obtener un residuo, que se extrajo con acetato de etilo (2 x 150 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera, se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida utilizando gel de sílice (malla 60-120) eluyendo con acetato de etilo al 40 % en n-hexano, dando 3-metil 2-oxopirrolidin-1,3-dicarboxilato 1-(íerc-butílico) (Producto intermedio 10, 9,46 g, rendimiento: 45 %) m/z 188,17 [M-56]+; RMN de 1H (400 MHz, CDCls) 53,90 (ddd, J = 10,8, 8,5, 5,4 Hz, 1H), 3,81 (s, 3H), 3,73 (ddd, J = 10,8, 8,1,6,6 Hz, 1H), 3,62-3,52 (m, 1H), 2,50-2,34 (m, 1H), 2,25 (dddd, J = 13,3, 9,1,8,1,5,4 Hz, 1H), 1,55 (s, 9H) ppm.
Ejemplo 40-Preparación del producto intermedio 11
La síntesis del Producto intermedio 11 siguió el siguiente procedimiento del Procedimiento general 1:

A una solución enfriada (0 °C) de 1-(íerc-butil)-3-metil-2-oxopirrolidin-1,3-dicarboxilato (Producto intermedio 10, 10,0 g, 41,1 mmol, 1,0 eq) en DMF (100 ml), se añadió carbonato de potasio (28,4 g, 205,5 mmol, 5,0 eq). Después
de agitar durante 1 hora a 0 °C, se añadió yoduro de metilo (7,02 g, 49,3 mmol, 1,2 eq). Se dejó que la mezcla de reacción alcanzara la temperatura ambiente y se agitó durante la noche. Se controló mediante TLC y CL-EM. Una vez completada, se vertió la mezcla de reacción en agua enfriada con hielo y se filtró, produciendo 1-(íerc-butil)-3-metil-3-metil-2-oxopirrolidin-1,3-dicarboxilato (Producto intermedio 11, 7,3 g, rendimiento: 69 %) m/z 202,13 [M-56]+ RMN de 1H (400 MHz, CDCls) ó RMN de 1H (400 MHz, CDCls) ó 3,78 (d, J = 6,0 Hz, 5H), 2,54 (ddd, J = 13,0, 7,4, 4,3 Hz, 1H), 1,91 (dt, J = 13,1,8,2 Hz, 1H), 1,54 (d, J = 11,7 Hz, 9H), 1,49 (d, J = 11,6 Hz, 3H) ppm.
Ejemplo 41-Preparación del producto intermedio 12
La síntesis del Producto intermedio 12 siguió el siguiente procedimiento del Procedimiento general 6b:

A una solución enfriada (0 °C) de 1-(ferc-butil)-3-metil-3-metil-2-oxopiperidin-1,3-dicarboxilato (Producto intermedio 11, 5,0 g, 19,4 mmol, 1,0 eq) en diclorometano (50 ml), se añadió ácido trifluoroacético (2,5 ml). Después de agitar durante 1 hora, se dejó que la mezcla alcanzara la temperatura ambiente durante la noche. Se controló mediante TLC y CL-EM. Una vez completada, se trató la mezcla de reacción con solución saturada de NaHCO3 y se extrajo con diclorometano (2 x 50 ml). Las capas orgánicas combinadas se lavaron con salmuera, se desecaron sobre sulfato de sodio anhidro, se filtraron y se concentraron, dando 3-metil-2-oxopirrolidin-3-carboxilato de metilo (Producto intermedio 12, 2,26 g, rendimiento: 74 %) m/z 158,04 [M+1]+ RMN de 1H (400 MHz, DMSO) ó RMN de 1H (400 MHz, DMSO) ó 7,94 (s, 1H), 3,63 (s, 3H), 3,32-3,14 (m, 2H), 2,58-2,37 (m, 1H), 2,04-1,86 (m, 1H), 1,26 (s, 3H) ppm.
Ejemplo 42-Preparación del producto intermedio 13
La síntesis del Producto intermedio 13 siguió el procedimiento del siguiente Procedimiento general 2 :

A una solución enfriada (-78 °C) de acetonitrilo (1,2 g, 26,5 mmol, 1,6 eq) en tetrahidrofurano anhidro (30 ml), se añadió n-BuLi (2,5 M en hexano, 10,6 ml, 26,5 mmol, 1,6 eq) gota a gota durante un período de 20 minutos. Después de agitar durante 1 hora, se añadió metil-3-metil-2-oxopirrolidin-3-carboxilato (Producto intermedio 12, 2,6 g, 16,5 mmol, 1,0 eq) poco a poco y se agitó la mezcla de reacción a -78 °C durante 3 horas. Se inactivó la reacción con solución saturada de cloruro de amonio y se extrajo el producto con acetato de etilo. La capa orgánica se desecó sobre sulfato de sodio anhidro, se filtró y se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna utilizando gel de sílice (tamaño de malla 60-120), eluyendo con acetato de etilo al 70 % en n-hexano, dando el producto deseado 3-(3-metil-2-oxopirrolidin-3-il)-3-oxopropanonitrilo (Producto intermedio 13, 2,17 g, rendimiento: 79 %) m/z 167,1 [M+1]+; RMN de 1H (400 MHz, DMSO) ó 8,09 (s, 1H), 4,27 (c, J = 20,3 Hz, 2H), 3,35 (s, 3H), 3,20 (dd, J = 13,0, 5,6 Hz, 2H), 1,81 (ddd, J = 13,1,7,7, 5,6 Hz, 1H), 1,35-1,20 (m, 1H) ppm.
Ejemplo 43-Preparación del compuesto 30
La síntesis del Compuesto 30 siguió el procedimiento del siguiente Procedimiento general 3 :

A una solución de 3-(3-metil-2-oxopirrolidin-3-il)-3-oxopropanonitrilo (Producto intermedio 13, 0,5 g, 3,0 mmol, 1,0 eq) en isopropanol (10 ml) y ácido acético (2,0 ml), se añadió gota a gota monohidrato de hidrazina (0,25 g,
4,5 mmol, 1,5 eq). Se agitó la reacción a 60 °C durante 3 horas y se controló mediante TLC y CL-EM. Una vez completada, se concentró la mezcla de reacción a presión reducida, obteniéndose un residuo que se purificó mediante cromatografía en columna utilizando gel de sílice (malla 60-120). El producto se eluyó con metanol al 0 5 % en diclorometano, dando 3-(5-amino-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona (Compuesto 30, 0,435 g, rendimiento: 80 %) m/z 181,19 [M+1]+.
Ejemplo 44-Preparación del compuesto 31
La síntesis del Compuesto 31 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución enfriada (0 °C) de 3-(5-amino-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona (Compuesto 30, 1,0 g, 5,5 mmol, 1,0 eq) en metanol (20 ml), se añadió gota a gota ácido acético (0,3 g, 5,5 mmol, 1,0 eq), seguido de 5-clorotiofen-2-carbaldehído (0,92 g, 6,6 mmol, 1,2 eq). Se agitó la reacción durante 30-45 minutos a temperatura ambiente. A continuación, se añadió poco a poco cianoborohidruro de sodio (0,65 g, 11,0 mmol, 2,0 eq) durante un período de 15 minutos. Se agitó la reacción a temperatura ambiente durante 12 horas. Una vez completada la reacción, se concentró la mezcla de reacción a presión reducida. El residuo se purificó mediante cromatografía en columna utilizando gel de sílice (malla 60-100, eluyendo con metanol al 0-5 % en diclorometano, dando 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona (Compuesto 31, 0,21 g, rendimiento: 12 %) m/z 311,3 [M+1]+; RMN de 1H (400 MHz, DMSO) 5 11,43 (s, 1H), 7,70 (s, 1H), 6,89 (dd, J = 29,1, 3,5 Hz, 2H), 5,77 (s, 1H), 5,35 (s, 1H), 4,29 (s, 2H), 3,20 (t, J = 6,5 Hz, 2H), 2,33 (s, 1H), 2,01 (d, J = 11,9 Hz, 1H), 1,32 (s, 3H) ppm.
Ejemplo 45-Preparación del compuesto 32
La síntesis del Compuesto 32 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución enfriada (0 °C) de ácido piválico (0,147 g, 1,45 mmol, 1,5 eq) en THF (15 ml), se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 0,276 g, 1,45 mmol, 1,5 eq) seguido de trietilamina (TEA, 0,4 ml, 2,89 mmol, 3,0 eq). Después de agitar durante 30 minutos, se añadieron hidroxibenzotriazol (HOBt, 0,026 g, 0,2 mmol, 0,2 eq) y 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona (Compuesto 31, 0,3 g, 0,96 mmol, 1,0 eq). Se controló la reacción mediante CL-EM y, una vez completada, se vertió la mezcla de reacción en agua (50 ml) y se extrajo con acetato de etilo (3 x 25 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera, se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua como fase móvil, dando 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 -pivaloil-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona (Compuesto 32, 0,05 g, rendimiento: 13 %) m/z 395,7 [M+1]+; RMN de 1H (400 MHz, DMSO) 57,74 (s, 1H), 7,66 (t, J = 6,2 Hz, 1H), 6,97 (s, 2H), 5,43 (s, 1H), 4,43 (d, J = 6,2 Hz, 2H), 3,30-3,18 (m, 2H), 2,54 (s, 1H), 2,00 (dt, J = 12,6, 7,7 Hz, 1H), 1,41 (s, 9H), 1,34 (s, 3H) ppm.
Ejemplo 46-Preparación del compuesto 33
La s ín te s is de l
Compuesto 33
s ig u ió el p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5a
:

A una solución enfriada (0 0C) de ácido 2-metoxibenzoico (0,219 g, 1,45 mmol, 1,5 eq) en THF (15 ml), se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 0,276 g, 1,45 mmol, 1,5 eq) seguido de trietilamina (TEA, 0,4 ml, 2,89 mmol, 3,0 eq). Después de agitar durante 30 minutos, se añadieron hidroxibenzotriazol (HOBt, 0,026 g, 0,19 mmol, 0,2 eq) y 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)-3-metilpirrolidin-2 -ona (Compuesto 31, 0,3 g, 0,96 mmol, 1 , 0 eq). Se controló la reacción mediante CL-EM y, una vez completada, se vertió la mezcla en agua (40 ml) y se extrajo con acetato de etilo (3 x 25 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera, se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua como fase móvil, dando 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 -(2-metoxibenzoil)-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona (Compuesto 33, 0,05 g, rendimiento: 12 %) m/z 447,18; RMN de 1H (400 MHz, DMSO) 5 7,70-7,58 (m, 2H), 7,55 7 , 4 4 (m, 1 H), 7,38 (dd, J = 7,5, 1,7 Hz, 1H), 7,13 (d, J = 8,2 Hz, 1 H), 7,09-6,95 (m, 3H), 5,51 (s, 1H), 4,49 (d, J = 6,2 Hz, 2H), 3,74 (s, 3H), 3,18-3,00 (m, 2H), 2,35 (ddd, J = 12,0, 7,1, 4,7 Hz, 1H), 1,84 (dt, J = 12,5, 7,5 Hz, 1H), 1,27-1,18 (m, 3H) ppm.
Ejemplo 47-Preparación del compuesto 34
La síntesis del Compuesto 34 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución fría (0 0C) de ácido tiofen-3-carboxílico (0,123 g, 0,96 mmol, 1,5 eq) en THF (12 ml), se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 0,187 g, 0,96 mmol, 1,5 eq) y, a continuación, trietilamina (TEA, 0,26 ml, 1,93 mmol, 3,0 eq) bajo nitrógeno. Después de agitar durante 30 minutos, se añadieron hidroxibenzotriazol (HOBt, 0,0173 g, 0,138 mmol, 0,2 eq) y, a continuación, 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona (Compuesto 31, 0,2 g, 0,643 mmol, 1,0 eq). Se agitó la reacción durante 2 días y se controló mediante CL-EM. Una vez completada, se vertió la mezcla de reacción en agua (40 ml) y se extrajo con acetato de etilo (3 x 25 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera, se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua como fase móvil, dando 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-(tiofen-3-carbonil)-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona (Compuesto 34, 0,05 g, rendimiento: 19 %) m/z 421,01 [M+1]+; RMN de 1H (400 MHz, DMSO) 58,90 (dd, J = 3,0, 1,1 Hz, 1H), 7,86-7,73 (m, 3H), 7,66 (dd, J = 5,1, 3,0 Hz, 1H), 6,98 (c, J = 3,8 Hz, 2H), 5,57 (s, 1H), 4,50 (d, J = 6,2 Hz, 2H), 3,32-3,17 (m, 2H), 2,54-2,60 (m, 1H), 2,12-1,96 (m, 2H), 1,38 (s, 3H) ppm.
Ejemplo 48-Preparación del compuesto 35
La s ín te s is de l
Compuesto 35
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5a
:

A una solución fría (0 °C) de ácido 4-metiltetrahidro-2H-piran-4-carboxílico (0,184 g, 1,45 mmol, 1,5 eq) en THF (15 ml), se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 0,276 g, 1,45 mmol, 1,5 eq), seguido de trietilamina (TEA, 0,3 g, 2,9 mmol, 3,0 eq) bajo nitrógeno. Después de agitar durante 30 minutos, se añadió hidroxibenzotriazol (HOBt, 0,026 g, 0,19 mmol, 0,2 eq), seguido de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)-3-metilpirrolidin-2-ona (Compuesto 31, 0,3 g, 0,96 mmol, 1,0 eq). Se agitó la reacción durante 2 días y se controló mediante CL-EM y, una vez completada, se vertió la mezcla de reacción en agua (40 ml) y se extrajo con acetato de etilo (3 x 25 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera, se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua como fase móvil, dando 3-(5-((5-clorotiofen-2-il)metil)amino)-1-(4-metiltetrahidro-2H-pirano-4-carbonil)-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona (Compuesto 35, 0,05 g, rendimiento: 12 %) m/z 437,39 [M+1]+; RMN de 1H (400 MHz, DMSO) 57,71 (dd, J = 15,0, 8,8 Hz, 2H), 6,96 (d, J = 4,3 Hz, 2H), 5,44 (s, 1H), 4,44 (d, J = 6,2 Hz, 2H), 3,69 (d, J = 11,6 Hz, 2H), 3,48 (t, J = 8,6 Hz, 2H), 3,24 (dd, J = 9,2, 5,0 Hz, 2H), 2,35 (d, J = 6,8 Hz, 3H), 1,99 (dt, J = 12,5, 7,5 Hz, 1H), 1,69 (s, 2H), 1,52 (s, 3H), 1,33 (s, 3H) ppm.
Ejemplo 49-Preparación del producto intermedio 14
La síntesis del Producto intermedio 14 siguió el procedimiento del siguiente Procedimiento general 10:

A una solución agitada de diclorhidrato de ácido (+/-)-piperazin-2-carboxílico (25 g, 123,2 mmol) en agua (200 ml) a temperatura ambiente, se añadió hidróxido de sodio acuoso (2,5 M, 200 ml) seguido de solución de sulfato de cobre pentahidratado (CuSO4.5H2O, 15,88 g, 61,5 mmol en 100 ml de H2O) gota a gota. La mezcla se enfrió hasta 0 °C y se añadió bicarbonato de sodio sólido (12,5 g, 148,8 mmol). Después de agitar a 0 °C durante 15 minutos, se añadió una solución de CBZ-Cl en 1,4-dioxano. Se agitó la mezcla de reacción a temperatura ambiente durante 16 horas. (NOTA: El pH se mantuvo > 7 durante toda la reacción). Se evaporaron las sustancias volátiles a presión reducida, produciendo ácido 4-(benciloxicarbonil)piperazin-2-carboxílico (Producto intermedio 14, 45 g, en bruto) en forma de un sólido de complejo de cobre de color azul. Se consideró para la siguiente etapa sin ninguna purificación; TLC: MeOH al 5 % en diclorometano, Fr: 0,4.
Ejemplo 50-Preparación del producto intermedio 15
La síntesis del Producto intermedio 15 siguió el procedimiento del siguiente Procedimiento general 11:

A una solución agitada de complejo de cobre-ácido 4-(benciloxicarbonil)piperazin-2-carboxílico (Producto intermedio 15, 40 g, 122,6 mmol) en agua (200 ml), se añadió ácido etilendiaminotetraacético (EDTA, 20 g, 61,4 mmol en 200 ml de agua), y la mezcla se calentó hasta 100 °C durante 16 horas. Se concentró la mezcla de reacción a presión reducida, produciendo el ácido 4-(benciloxicarbonil)piperazin-2-carboxílico en bruto (Producto intermedio 15, 30 g) en forma de un sólido de color azul que se llevó a la siguiente etapa sin mayor purificación; Sistema de TLC: MeOH al 5 % en diclorometano, Fr: 0,2.
Ejemplo 51-Preparación del producto intermedio 16
La síntesis del Producto intermedio 16 siguió el procedimiento del siguiente Procedimiento general 12:

,
Producto intermedio 15 Producto intermedio 16
A una solución enfriada (0 °C) de ácido 4-(benciloxicarbonil)piperazin-2-carboxílico (Producto intermedio 15, 48 g, 181,8 mmol) en agua (10 volúmenes, 480 ml), se añadió NaHCO3 sólido (45,8 g, 545,4 mmol) seguido de dicarbonato di-terc-butílico ((Boc)2O, 109 ml, 490,9 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 17 horas. Después de enfriar hasta la temperatura ambiente, se extrajo en EtOAc (4 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 50 %/n-hexano, produciendo ácido 4-(benciloxicarbonil)-1-(terc-butoxicarbonil)piperazin-2-carboxílico (Producto intermedio 16, 25 g, rendimiento: 55 %) en forma de un sólido blanquecino; TLC: metanol al 5 %-diclorometano, Fr: 0,3.
Ejemplo 52-Preparación del producto intermedio 17
La síntesis del Producto intermedio 17 siguió el procedimiento del siguiente Procedimiento general 13:

A una solución enfriada (0 °C) de ácido 4-(benciloxicarbonil)-1-(terc-butoxicarbonil)piperazin-2-carboxílico (Producto intermedio 16, 20 g, 54,9 mmol) en DMF seco (200 ml), se añadió carbonato de cesio (Cs2CO3 , 35,8 g, 109,9 mmol) seguido de yoduro de metilo (10,26 ml, 164,83 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 4 horas. Después de enfriar hasta 0 °C, se inactivó la mezcla con agua helada (90 ml) y se extrajo con EtOAc (2 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 20 %/n-hexano, produciendo 2-metil-piperazin-1,2,4-tricarboxilato 4-bencil-1-terc- butílico (Producto intermedio 17, 25 g, rendimiento: 55 %) en forma de un sólido blanquecino. TLC: acetato de etilo al 30 % en hexano. Fr: 0,5.
Ejemplo 53-Preparación del producto intermedio 18
La s ín te s is de l
Producto intermedio 18
s ig u ió el p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 14
:
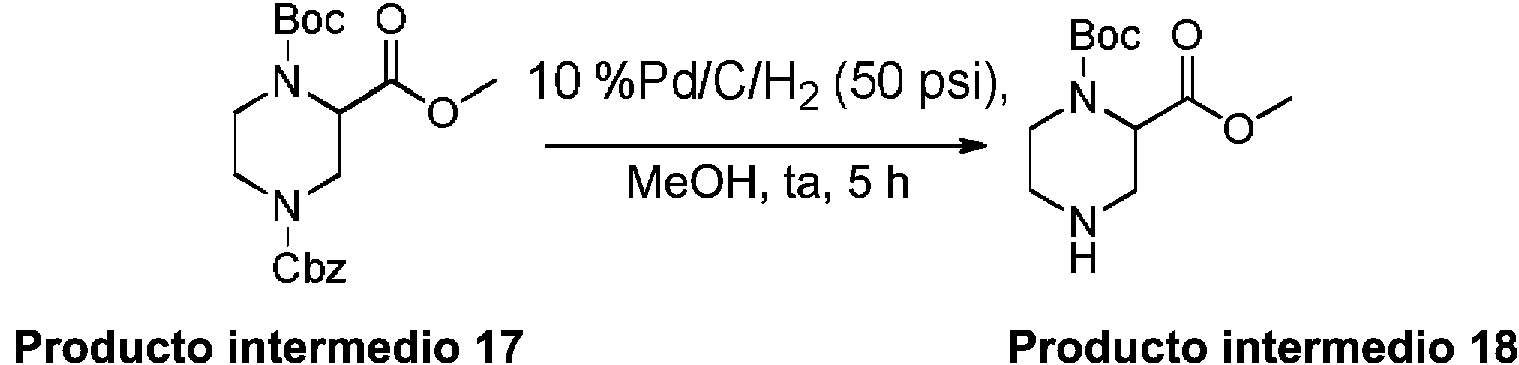
A una solución agitada de piperazin-1,2,4-tricarboxilato de 4-bencilo 1-íerc-butil-2-metilo (Producto intermedio 17, 10 g, 26,5 mmol) en MeOH (100 ml) en un recipiente de acero, se añadió Pd al 10 %/C (30 % p/p, 3,0 g) y se agitó la mezcla a una presión de hidrógeno de 344,738 kPa (50 psi) a temperatura ambiente durante 5 horas. Se filtró la mezcla de reacción a través de un lecho corto de Celite y se concentraron las sustancias volátiles a presión reducida, produciendo 2-metil piperazin-1,2-dicarboxilato 1-íerc-butílico (Producto intermedio 18, 5,6 g, rendimiento: 88 %) en forma de un líquido amarillo claro; TLC: metanol al 5 %-diclorometano. Fr: 0,3.
Ejemplo 54-Preparación del producto intermedio 19
La síntesis del Producto intermedio 19 siguió el procedimiento del siguiente Procedimiento general 15:

A una solución enfriada (0 °C) de 2-metil piperazin-1,2-dicarboxilato 1-íerc-butílico (Producto intermedio 18, 3,5 g, 14,3 mmol) en diclorometano seco (70 ml), se añadió trietilamina (TEA, 5 ml, 35,9 mmol) seguida de cloruro de morfolin-4-carbonilo (3,35 ml, 28,7 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 3 horas. A la mezcla, se añadió agua helada (20 ml), y después se extrajo en EtOAc (2 x 50 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo en bruto se purificó mediante cromatografía ultrarrápida utilizando EtOAc al 30 %/n-hexano, produciendo 2-metil-4-(morfolin-4-carbonil)piperazin-1,2-dicarboxilato de 1-íerc-butilo (Producto intermedio 19, 3,6 g, rendimiento: 72 %) en forma de un sólido blanquecino; TLC: acetato de etilo al 50 % en hexano. Fr: 0,5.
Ejemplo 55-Preparación del producto intermedio 20
La síntesis del Producto intermedio 20 siguió el siguiente procedimiento del Procedimiento general 6c:

A una solución enfriada (0 °C) de 1-íerc-butil-2-metil-4-(morfolin-4-carbonil)piperazin-1,2-dicarboxilato (Producto intermedio 19, 1,0 g, 2,8 mmol) en 1,4-dioxano (10 ml), se añadió HCl (4 M en dioxano, 10 ml) y, a continuación, se agitó la mezcla a temperatura ambiente durante 5 horas. Se concentró la mezcla de reacción y se trituró con éter dietílico, produciendo la sal clorhidrato de 4-(morfolin-4-carbonil)piperazin-2-carboxilato de metilo (Producto intermedio 20, 800 mg, rendimiento: 86 %) en forma de un sólido blanquecino; TLC: metanol al 5 %-diclorometano. Fr: 0,3.
E je m p lo 56 -P re p a ra c ió n de l p ro du c to in te rm e d io 21
La síntesis del Producto intermedio 21 siguió el procedimiento del siguiente Procedimiento general 7 :

A una solución agitada enfriada (0 0C) de 4-(morfolin-4-carbonil)piperazin-2-carboxilato de metilo (Producto intermedio 20, 8,2 g, 28,0 mmol) en H2O (300 ml) a 0 °C, se añadió NaHCO3sólido (7,05 g, 83,2 mmol), seguido de cloroformiato de alilo ( 8 ml, 30,8 mmol) en THF (50 ml). Después de agitar a temperatura ambiente durante 16 horas, se añadió agua helada (100 ml) a la mezcla de reacción y se extrajo en EtOAc (2 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 40-50 %/nhexano, produciendo 4-(morfolin-4-carbonil)piperazin-1,2-dicarboxilato de 1 -alilo 2-metilo (Producto intermedio 21, 7,2 g, rendimiento: 84 %) en forma de un líquido de color amarillo pálido; Sistema de TLC: metanol al 5 %-diclorometano, Fr: 0 ,6.
Ejemplo 57-Preparación del producto intermedio 22
La síntesis del Producto intermedio 22 siguió el procedimiento del siguiente Procedimiento general 2 :

A una solución enfriada (-78 °C) de acetonitrilo (4,04 ml, 77 mmol) en THF seco (120 ml), se añadió n-BuLi (2,5 M en hexano, 26 ml, 65 mmol). Después de agitar a -78 °C durante 30 minutos, se añadió a la mezcla una solución de 1-alil-2-metil-4-(morfolin-4-carbonil)piperazin-1,2-dicarboxilato (Producto intermedio 21, 7,5 g, 22 mmol) en THF seco (30 ml). Se agitó la mezcla a -78 0C durante 30 minutos más. Se inactivó la mezcla de reacción con solución saturada de NH4Cl (80 ml) y se extrajo en EtOAc (2 x 120 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 50 %/n-hexano, produciendo alil-2-(2-cianoacetil)-4-(morfolin-4-carbonil)piperazin-1-carboxilato (Producto intermedio 22, 6,5 g, rendimiento -85 %) en forma de un líquido amarillo claro; Sistema de TLC: metanol al 5 %-diclorometano, Fr: 0,5.
Ejemplo 58-Preparación del compuesto 36
La síntesis del Compuesto 36 siguió el procedimiento del siguiente Procedimiento general 3 :

A una solución de 2-(2-cianoacetil)-4-(morfolin-4-carbonil)piperazin-1-carboxilato de alilo (Producto intermedio 22, 6,5 g, 18,6 mmol) en etanol (65 ml), se añadió N2H4 (1,1 ml, 22,3 mmol) y se calentó la mezcla de reacción a 80 0C
durante 5 horas. Se enfrió la mezcla de reacción hasta la temperatura ambiente, se evaporaron las sustancias volátiles y el residuo resultante se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con metanol al 3 % en diclorometano, produciendo alil-2-(5-amino-1H-pirazol-3-il)-4-(morfolin-4-carbonil)piperazin-1-carboxilato (Compuesto 36, 4,5 g, rendimiento: 67 %) en forma de un semisólido de color amarillo pálido; Sistema de TLC: metanol al 5 %-diclorometano, Fr: 0,3; m/z 365,28 [M+1]+ RMN de 1H (400 MHz, DMSO) 5 11,2 (s, 1H), 5,9 (m, 1H), 5,0-5,3 (m, 4H), 4,8 (s a, 2H), 4,6 (s a, 2H), 3,85 (m, 2H), 3,4-3,6 (m, 4H), 3,0-3,2 (m, 6H), 2,8 (m, 1H) ppm.
Ejemplo 59-Preparación del compuesto 37
La síntesis del Compuesto 37 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución enfriada (0 0C) de 2-(5-amino-1H-pirazol-3-il)-4-(morfolin-4-carbonil)piperazin-1-carboxilato de alilo (Compuesto 36, 3,5 g, 9,6 mmol) en MeOH seco (70 ml), se añadió 5-clorotiofen-2-carbaldehído (2,1 ml, 19,2 mmol), AcOH (0,2 ml) y tamices moleculares en polvo de 4 Á. Se agitó la mezcla de reacción a temperatura ambiente durante 1 hora. (La formación de imina se observó como una mancha menos polar mediante TLC). Se añadió a la mezcla de reacción cianoborohidruro de sodio (NaCNBH3 , 0,73 g, 11,5 mmol) poco a poco y se agitó la mezcla a temperatura ambiente durante 1 hora. A continuación, se añadió a la mezcla agua helada (50 ml), después se filtró a través de un lecho corto de Celite y el filtrado se extrajo en EtOAc (4 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con MeOH al 3 %-DCM, produciendo el producto deseado (1,95 g, rendimiento: 41 %) en forma de un líquido amarillo claro que se utilizó en la siguiente etapa. Se purificó una muestra (120 mg) adicionalmente mediante HPLC preparativa, produciendo 2-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)-4-(morfolin-4-carbonil)piperazin-1-carboxilato de alilo (Compuesto 37; 55 mg) en forma de un líquido gomoso. Rm N de 1H (400 MHz, DMSO-d6) 5 = 11,55-11,41 (s a, 1 H), 6,96-6,77 (m, 2H), 6,15-5,71 (m, 2H), 5,41-4,97 (m, 4H), 4,56 (s a, 2H), 4,27 (s a, 2H), 3,83 (t a, J = 12,5 Hz, 2H), 3,67-3,32 (m, 5H), 3,25-2,85 (m, 6H), 2,81 -2,64 (m, 1H); Sistema de TLC: metanol al 5 %-diclorometano, Fr: 0,5.
Ejemplo 60-Preparación del compuesto 38
La síntesis del Compuesto 38 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 0C) de 2-(5-((5-clorotiofen-2-il)metilamino)-1 H-pirazol-3-il)-4-(morfolin-4-carbonil)piperazin-1-carboxilato de alilo (Compuesto 37, 1,0 g, 2,0 mmol) en diclorometano seco (20 ml), se añadió trietilamina (TEA, 0,4 ml, 3,0 mmol), seguida de cloruro de trimetilacetilo (0,23 ml, 1,8 mmol). Se agitó la mezcla a temperatura ambiente durante 4 horas y después se inactivó con agua (25 ml). Esto se extrajo en diclorometano (3 x 40 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 20 %/n-hexano, produciendo 2-(5-((5-clorotiofen-2-il)metilamino)-1 -pivaloil-1 H-pirazol-3-il)-4-(morfolin-4-carbonil)piperazin-1-carboxilato de alilo (Compuesto 38, 600 mg, rendimiento: 69 %) en forma de un sólido amarillo pálido. RMN de 1H (400 MHz, DMSO-da) 5 = 7,71 (t, J = 6,1 Hz, 1H), 6,97-6,91 (m, 2H), 5,87 (s a, 1H), 5,37 (s, 1H), 5,14 (s a, 1H), 5,05 (d a, J = 2,0 Hz, 1H), 4,53 (s a, 2H), 4,40 (d, J = 6,4 Hz, 2H), 3,94-3,79 (m, 2H), 3,60-3,36 (m, 6H), 3,22-3,15 (m, 1H), 3,19 (dd a, J = 4,4, 13,2 Hz, 2H), 2,93-2,78 (m, 3H), 1,35 (s, 9H) ppm; Sistema de TLC: acetato de etilo al 30 % en hexano, Fr: 0,5.
E je m p lo 61 -P re p a ra c ió n de l c o m p u e s to 39
La síntesis del Compuesto 39 siguió el siguiente procedimiento del Procedimiento general 8b :
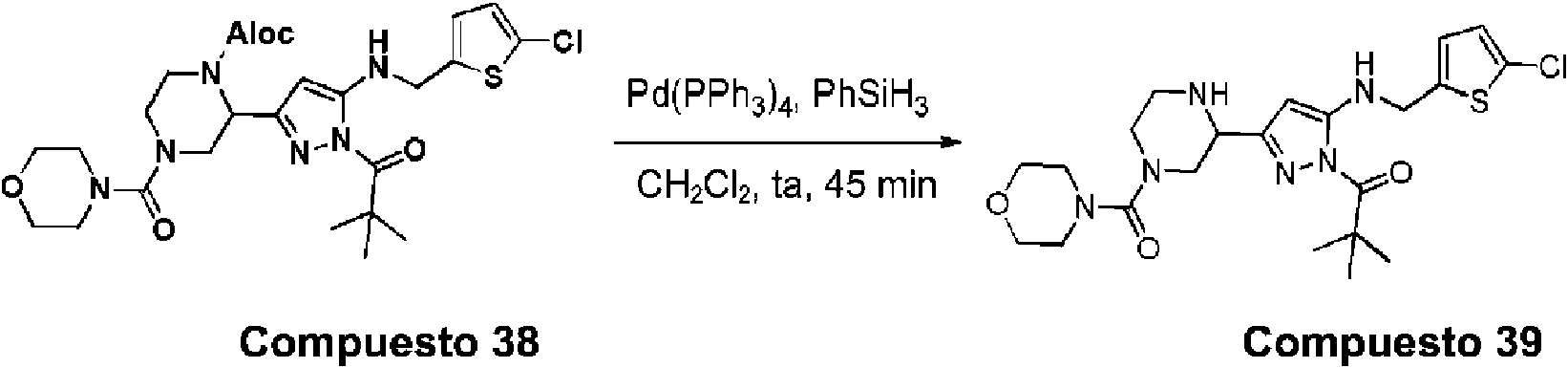
Se desgasificó una solución agitada de 2-(5-((5-clorotiofen-2-il)metilamino)-1-pivaloil-1H-pirazol-3-il)-4-(morfolin-4-carbonil)piperazin-1-carboxilato de alilo (Compuesto 38, 500 mg, 0,86 mmol) en diclorometano (10 ml) con una corriente de argón durante 15 minutos, a continuación, se añadió a la solución tetraquis(trifenilfosfin)paladio (0) (Pd(PPh3)4 , 100 mg, 0,087 mmol) seguido de fenilsilano (PhS¡H3 , 0,64 ml, 5,2 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 45 minutos, a continuación, se filtró a través de un lecho corto de Celite y se evaporó. El residuo en bruto se purificó por CombiFlash® cromatografía eluyendo con MeOH al 4 %-diclorometano, produciendo 1 -(5-((5-clorotiofen-2-il)metilamino)-3-(4-(morfolin-4-carbonil)piperazin-2-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona (Compuesto 39, 130 mg, rendimiento: 31 %) en forma de un sólido blanquecino. RMN de 1H (400 MHz, DMSO-d6) 5 = 7,72 (t, J = 6,4 Hz, 1H), 6,98-6,94 (m, 2H), 5,47 (s, 1H), 4,42 (d, J = 6,4 Hz, 2H), 3,67 (d a, J = 12,7 Hz, 1H), 3,61-3,53 (m, 5H), 3,43 (d a, J = 11,7 Hz, 1H), 3,11 (dd, J= 3,7, 5,1 Hz, 4H), 2,92-2,63 (m, 5H), 1,41 (s, 9H) ppm; Sistema de TLC: metanol al 5 %-diclorometano, Fr: 0,5.
Ejemplo 62-Preparación del compuesto 40
La síntesis del Compuesto 40 siguió el siguiente procedimiento del Procedimiento general 6c:

A una solución enfriada (0 0C) de 1-(5-((5-clorotiofen-2-il)metilamino)-3-(4-(morfolin-4-carbonil)piperazin-2-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 39, 100 mg, 0,2 mmol) en 1,4-dioxano (10 ml), se añadió HCl (10 ml en dioxano) y, a continuación, la reacción se agitó a temperatura ambiente durante 5 horas. Se concentró la mezcla de reacción a presión reducida y se trituró con éter dietílico, dando 100 mg de una muestra semipura. Este se purificó mediante HPLC preparativa, produciendo clorhidrato de (3-(5-((5-clorotiofen-2-il)metilamino)-1 H-pirazol-3-il)piperazin-1-il)(morfolino)metanona (Compuesto 40, 28 mg, rendimiento: 34 %) en forma de un sólido blanquecino. RMN de 1H (400 MHz, DMSO-da) 5 = 12,02 (s a, 1H), 11,61 (s a, 1H), 9,06 (s, 2H), 6,92 (d a, J = 13,7 Hz, 2H), 6,39 (s a, 1H), 5,81 (s a, 1H), 5,53 (s a, 1H), 4,32 (d a, J = 5,9 Hz, 2H), 4,17 (s a, 1H), 3,95-3,40 (m, 6H), 3,27-2,57 (m, 8H) ppm; Sistema de TLC: metanol al 10 %-diclorometano, Fr: 0,5.
Ejemplo 63-Preparación del compuesto 41
La síntesis del Compuesto 41 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 0C) de 2-(5-((5-clorotiofen-2-il)metilamino)-1 H-pirazol-3-il)-4-(morfolin-4-carbonil)piperazin-1-carboxilato de alilo (Compuesto 40, 2,0 g, 4 mmol) en diclorometano seco (20 ml), se añadió trietilamina (TEA, 1,1 ml, 8,1 mmol) seguida de cloruro de 1,3-tiazol-4-carbonilo (896 mg, 1,8 mmol). Después de agitar a temperatura
ambiente durante 4 horas, la mezcla de reacción se combinó con agua (25 ml) y se extrajo con diclorometano (3 x 40 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 20 %/n-hexano, produciendo 2-(5-((5-clorotiofen-2-il)metilamino)-1-(tiazol-4-carbonil)-1H-pirazol-3-il)-4-(morfolin-4-carbonil)piperazin-1-carboxilato de alilo (Compuesto 41, 600 mg, rendimiento: 69 %) en forma de un líquido amarillo claro. Se purificaron adicionalmente 300 mg de esta muestra mediante HPLC preparativa, dando el compuesto puro en forma de un sólido de color amarillo claro. RMN de 1H (400 MHz, DMSO-d6) 5 = 9,22 (d, J = 2,0 Hz, 1H), 8,98 (d, J = 2,0 Hz, 1H), 7,87 (t a, J = 6,1 Hz, 1H), 6,96 (s, 2H), 5,88 (s a, 1H), 5,48 (s, 1H), 5,12 (d a, J = 2,4 Hz, 3H), 4,59-4,48 (m, 4H), 4,02 (d a, J = 13,7 Hz, 1H), 3,84 (d a, J = 12,7 Hz, 1H), 3,56 (d a, J = 13,2 Hz, 1H), 3,39 (d a, J = 2,9 Hz, 2H), 3,33 (s a, 2H), 3,23-3,18 (m, 2H), 3,06-2,99 (m, 2H), 2,90-2,81 (m, 3H) ppm; Sistema de TLC: metanol al 10 %-diclorometano, Fr: 0,5.
Ejemplo 64-Preparación del producto intermedio 23
La síntesis del Producto intermedio 23 siguió el procedimiento del siguiente Procedimiento general 16:

A una solución fría (-78 0C) de 4-oxopiperidin-1-carboxilato de bencilo (20 g, 8,6 mmol) en éter dietílico (Et2O, 250 ml), se añadió diazoacetato de etilo (11,6 ml, 11,1 mmol), seguido de dietileterato de trifluoruro de boro (BF3OEt2 , 10,7 ml, 8,6 mmol). La mezcla de reacción se dejó en agitación a -78 0C durante 1 h, a continuación, se agitó a temperatura ambiente durante 1 hora y después se inactivó con solución saturada de K2CO3. La solución se agitó hasta que cesó el desprendimiento de nitrógeno, después se extrajo con EtOAc (300 ml). La capa orgánica combinada se desecó sobre sulfato de sodio, se filtró y se concentró a presión reducida, obteniéndose 4-etil-5-oxoazepan-1,4-dicarboxilato de 1-bencilo (Producto intermedio 23, 26 g) en forma de un líquido amarillo. El compuesto en bruto se llevó a la siguiente etapa sin mayor purificación. EM (IEN): m/z 320,1 [M+H]+; Sistema de TLC: acetato de etilo al 20 % en hexano. Fr: 0,3.
Ejemplo 65-Preparación del producto intermedio 24
La síntesis del Producto intermedio 24 siguió el procedimiento del siguiente Procedimiento general 17:

A una solución fría (0 0C) de 4-etil-5-oxoazepan-1,4-dicarboxilato de 1-bencilo (Producto intermedio 23, 26 g, 81,5 mmol) en EtOH (250 ml), se añadió borohidruro de sodio (NaBH4 , 3 g, 81,5 mmol) poco a poco. Después de 10 minutos, la reacción se elevó a temperatura ambiente y se agitó durante 30 minutos más. La reacción se concentró y se diluyó con EtOAc (350 ml) y se lavó con agua (2 x 150 ml), se desecó sobre Na2SO4 , se filtró y se concentró a presión reducida. El residuo se purificó con gel de sílice (malla 100-200) eluyendo con EtOAc al 20 % en hexano, dando 4-etil-5-hidroxiazepan-1,4-dicarboxilato de 1-bencilo (Producto intermedio 24, 12 g, 46 %) en forma de un aceite incoloro. EM (IEN): m/z 321,96 ([M+H]+); Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,3.
Ejemplo 66-Preparación del producto intermedio 25
La síntesis del Producto intermedio 25 siguió el procedimiento del siguiente Procedimiento general 18:
Procedimiento general 18

A una solución agitada de 4-etil-5-hidroxiazepan-1,4-dicarboxilato de 1-bencilo (Producto intermedio 24, 12 g, 37,3 mmol) en THF (270 ml), se añadieron cloruro de metanosulfonilo (MsCl, 7,1 ml, 93,4 mmol) y trietilamina (TEA, 15,5 ml, 112,1 mmol) en dos porciones y se agitó la mezcla durante 12 horas. A continuación, la mezcla se concentró a presión reducida, se diluyó con EtOAc (300 ml) y se lavó con solución acuosa saturada de NaHCÜ3 (2 x 100 ml), se desecó (Na2SO4), se filtró y se concentró a presión reducida. El residuo se disolvió en THF (270 ml), se añadió 1,8-diazabicicloundec-7-eno (DBU, 8,4 ml, 56,3 mmol) y la mezcla resultante se calentó a 80 0C durante 1 hora. A continuación, la reacción se concentró, se diluyó con EtOAc (300 ml), se lavó con solución acuosa saturada de NaHCO3 (3 x 80 ml) y agua (200 ml), se desecó (Na2SO4), se filtró y se concentró a presión reducida. El residuo se purificó mediante cromatografía en gel de sílice (malla 100-200) eluyendo con EtOAc al 20 % en hexano, dando 4-etil-2,3-dihidro-1 H-azepin-1,4(6H,7H)-dicarboxilato de 1-bencilo (Producto intermedio 25, 10,5 g, 92 %) en forma de un aceite incoloro; e M (IEN): m/z 304 ([M+H]+); Sistema de TLC: acetato de etilo al 20 % en hexano. Fr: 0,9.
Ejemplo 67-Preparación del producto intermedio 26
La síntesis del Producto intermedio 26 siguió el procedimiento del siguiente Procedimiento general 19:

Una suspensión de 4-etil-2,3-dihidro-1 H-azepin-1,4(6 H,7H)-dicarboxilato de 1-bencilo (Producto intermedio 25, 10,5 g, 34,6 mmol) y Pd/C al 20 % (2,1 g) en etanol y acetato de etilo (1:1, 200 ml) se agitó en una atmósfera de hidrógeno de 551,581 kPa (80 psi) durante 12 horas. A continuación, la mezcla se filtró sobre un lecho corto de Celite y el lecho de Celite se lavó con metanol (2 x 50 ml). Las capas orgánicas combinadas se concentraron, dando azepan-4-carboxilato de etilo en bruto (Producto intermedio 26, 5,6 g, 94 %) en forma de un aceite amarillo; Sistema de TlC: MeOH al 10 % en diclorometano. Fr: 0,1.
Ejemplo 68-Preparación del producto intermedio 27
La síntesis del Producto intermedio 27 siguió el procedimiento del siguiente Procedimiento general 7 :

A una solución enfriada (0 0C) de azepan-4-carboxilato de etilo (Producto intermedio 26, 5,6 g, 32,9 mmol) en diclorometano (100 ml), se añadió trietilamina (TEA, 13,7 ml, 98,8 mmol) seguida de cloroformiato de alilo (7 ml, 65,8 mmol), y, a continuación, se agitó la mezcla a temperatura ambiente durante 12 horas. Se inactivó la mezcla de reacción con agua (100 ml) y se extrajo en diclorometano (2 x 200 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 20 %/n-hexano, produciendo 4
etil-azepan-1,4-dicarboxilato de 1-alilo (Producto intermedio 27, 6,5 g, 77 %) en forma de un líquido incoloro; EM (IEN): m/z 256,59 [M+H]+; Sistema de TLC: acetato de etilo al 20 % en hexano. Fr: 0,7.
Ejemplo 69-Preparación del producto intermedio 28
La síntesis del Producto intermedio 28 siguió el procedimiento del siguiente Procedimiento general 2 :
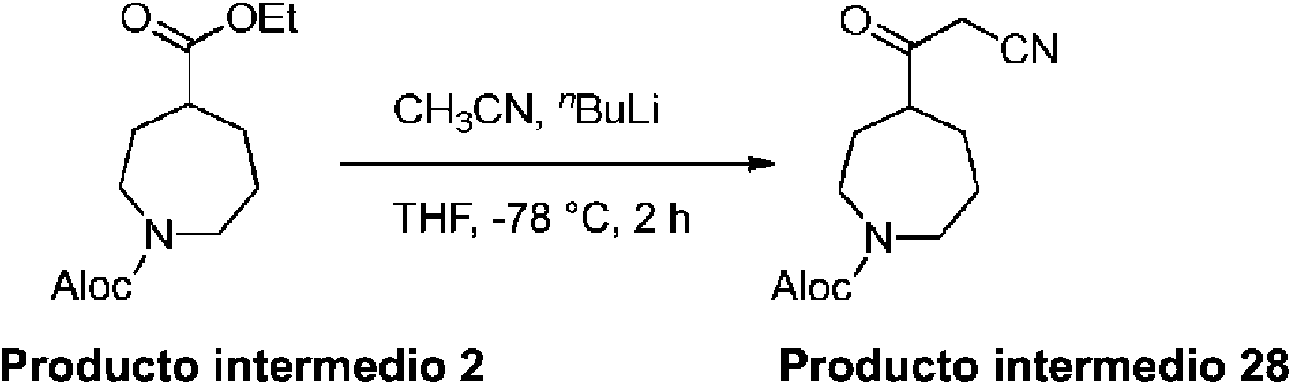
A una solución enfriada (-78 °C) de acetonitrilo (1,2 ml, 23,5 mmol) en THF seco (30 ml), se añadió n-BuLi en THF (2,5 M, 9,4 ml, 23,5 mmol). Después de agitar durante 30 minutos, una solución de 4-etil azepan-1,4-dicarboxilato de 1-alilo (Producto intermedio 27, 5 g, 19,6 mmol) en THF seco (10 ml). Se agitó la mezcla de reacción a -78 °C durante 1 hora, a continuación, se inactivó con solución saturada de NH4O (10 ml) y se extrajo en EtOAc (2 x 120 ml). La capa orgánica combinada se desecó sobre sulfato de sodio, se filtró y se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 50 %/n-hexano, produciendo 4-(2-cianoacetil)azepan-1-carboxilato de alilo (Producto intermedio 28, 3,1 g, 63 %) en forma de un líquido amarillo claro. EM (IEN): m/z 251,55 [M+H]+; Sistema de TLC: acetato de etilo al 50 % en hexano, Fr: 0,5.
Ejemplo 70-Preparación del compuesto 42
La síntesis del Compuesto 42 siguió el procedimiento del siguiente Procedimiento general 3 :

A una solución agitada de 4-(2-cianoacetil)azepan-1-carboxilato de alilo (Producto intermedio 28, 3,1 g, 12,4 mmol) en etanol (30 ml), se añadió hidrato de hidrazina (N2H4.H2O, 0,74 ml, 14,8 mmol) y, a continuación, se calentó la mezcla de reacción hasta 80 °C durante 5 horas. La mezcla de reacción se enfrió hasta la temperatura ambiente y se evaporaron las sustancias volátiles. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con MeOH al 5 % en diclorometano, produciendo 4-(5-amino-1 H-pirazol-3-il)azepan-1-carboxilato de alilo (Compuesto 42, 2,8 g, 87 %) en forma de un líquido naranja. EM (IEN): m/z 265,13 [M+H]+; Sistema de TLC: metanol al 10 %-diclorometano. Fr: 0,3.
Ejemplo 71-Preparación del compuesto 43
La síntesis del Compuesto 43 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución fría (0 °C) de 4-(5-amino-1 H-pirazol-3-il)azepan-1-carboxilato de alilo (Compuesto 42, 2,8 g, 10,6 mmol) en MeOH seco (28 ml), se añadieron 5-clorotiofen-2-carbaldehído (1,2 ml, 12,6 mmol), AcOH (0,6 ml) y, a continuación, tamices moleculares en polvo de 4 Á (1 , 6 g). Se agitó la mezcla de reacción a temperatura ambiente durante 1 hora. (La formación de imina se observó como una mancha menos polar en TLC). A esto, se añadió
cianoborohidruro de sodio (NaCNBH3 , 0,8 g, 12,6 mmol) poco a poco a 0 0C y se siguió agitando a temperatura ambiente durante 2 horas. Se inactivó la mezcla de reacción con agua helada (50 ml), se filtró a través de un lecho corto de Celite y el filtrado se extrajo en EtOAc (4 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 60 %/n-hexano, produciendo el producto deseado (2,5 g, 60 %) en forma de un líquido amarillo claro. Se purificó adicionalmente una porción (200 mg) de muestra semipura mediante HPLC preparativa, produciendo 4-(5-((5-clorotiofen-2-il)metilamino)-1 H-pirazol-3-il)azepan-1-carboxilato de alilo (Compuesto 43, 110 mg) en forma de una goma. EM (lEN): m/z 395,05 [M+H]+. RMN de 1H (300 MHz, DMSO-d6): 511,23 (s, 1H), 6,89-6,91 (d, J = 6,0 Hz, 1H), 6,83 (d, J = 3,0 Hz, 1H), 5,89-5,91 (m, 1H), 5,62 (s a, 1H), 5,16-5,29 (m, 3H), 4,54 (d, J = 6,0 Hz, 2H), 4,27 (d, J = 6,1 Hz, 2H), 3,23-3,61 (m, 4H), 2,63 (t, J = 9,0 Hz, 1H), 1,83-1,97 (m, 4H), 1,70-1,97 (m, 2H) ppm; Sistema de t Lc : acetato de etilo. Fr: 0,5.
Ejemplo 72-Preparación del compuesto 44
La síntesis del Compuesto 44 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 °C) de 4-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)azepan-1-carboxilato de alilo (Compuesto 43, 2,5 g, 6,3 mmol) en diclorometano seco (100 ml), se añadió trietilamina (TEA, 1,3 ml, 9,5 mmol), seguida de cloruro de trimetilacetilo (0,7 ml, 5,7 mmol). Después de agitar a temperatura ambiente durante 4 horas, se diluyó la mezcla de reacción con agua (25 ml) y se extrajo en diclorometano (3 x 40 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 10 %/n-hexano, produciendo el producto deseado 4-(5-((5-clorotiofen-2-il)metilamino)-1-pivaloil-1 H-pirazol-3-il)azepan-1-carboxilato de alilo (Compuesto 44, 1,8 g, 60 %) en forma de un líquido incoloro. Em (IEN): m/z 479,26 [M+H]+. RMN de 1H (400 MHz, DMSO-da): 57,63 (t, J = 6,0 Hz, 1H), 6,95 (s, 2H), 5,88-5,96 (m, 1H), 5,37 (d, J = 3,2 Hz, 1H), 5,14-5,28 (m, 2H), 4,53 (d, J = 5,2 Hz, 2H), 4,40 (d, J = 6,0 Hz, 2H), 3,28-3,61 (m, 4H), 2,64-2,61 (m, 1H), 1,84-1,88 (m, 4H), 1,54-1,72 (m, 2H), 1,39 (s, 9H).
Sistema de TLC: acetato de etilo al 20 % en hexano. Fr: 0,5.
Ejemplo 73-Preparación del compuesto 45
La síntesis del Compuesto 45 siguió el siguiente procedimiento del Procedimiento general 8b :

Se desgasificó una solución agitada de 4-(5-((5-clorotiofen-2-il)metilamino)-1-pivaloil-1H-pirazol-3-il)azepan-1-carboxilato de alilo (Compuesto 44, 400 mg, 0,8 mmol) en diclorometano (30 ml) con corriente de argón durante 15 minutos, a continuación, se añadió fenilsilano (PhSiH3 , 0,6 ml, 4,8 mmol) seguido de tetraq uis(trife nilfosfin)palad io (0) (Pd(PPh3)4 , 93 mg, 0,08 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 45 minutos, a continuación, la mezcla se filtró a través de un lecho corto de Celite y después se evaporó. El residuo en bruto se purificó mediante cromatografía ultrarrápida utilizando MeOH al 30 %-DCM, produciendo la 1-(3-(azepan-4-il)-5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona semipura (Compuesto 45, 200 mg; 94 % mediante CL-EM). EM (IEN) m/z 395,19 [M+H]+, RMN de 1H (300 MHz, DMSO-da) 57,62 (t, J = 6,0 Hz, 1H), 6,96 (s, 2H), 5,34 (s, 1H), 4,40 (d, J = 5,7 Hz, 2H), 3,18-2,68 (m, 6H), 2,05-1,62 (m, 6H), 1,38 (s, 9H) ppm; Sistema de TLC: MeOH al 20 %-diclorometano, Fr: 0,2.
E je m p lo 74 -P re p a ra c ió n de l c o m p u e s to 46
La síntesis del Compuesto 46 siguió el procedimiento del siguiente Procedimiento general 15:

A una solución fría (0 0C) de 1-(3-(azepan-4-il)-5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 45, 317 mg, 0,8 mmol) en diclorometano seco (50 ml), se añadió trietilamina (TEA, 0,28 ml, 2 mmol), seguida de cloruro de morfolina carbamilo (0,14 ml, 1,2 mmol). Se agitó la mezcla a temperatura ambiente durante 3 horas. Se diluyó la mezcla de reacción con agua (20 ml) y se extrajo con diclorometano (2 x 20 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó por CombiFlash, eluyendo con EtOAc al 30 %/n-hexano, produciendo 1-(5-((5-clorotiofen-2-il)metilamino)-3-(1-(morfolin-4-carbonil)azepan-4-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 46, 140 mg, 34 %) en forma de un semisólido blanquecino; EM (IEN): m/z 508,28 [M+H]+. RMN de 1H (400 MHz, DMSO-afe): 5 7,63 (t, J = 6,4 Hz, 1H), 6,95 (s, 2H), 5,36 (s, 1H), 4,40 (d, J = 6,0 Hz, 2H), 3,57-3,55 (m, 4H), 3,51-3,41 (m, 2H), 3,31-3,21 (m, 2H), 3,06-2,98 (m, 4H), 2,64-2,62 (m, 1H), 2,00-1,83 (m, 5H), 1,81-1,51 (m, 1H), 1,39 (s, 9H) ppm; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,5.
Ejemplo 75-Preparación del compuesto 47
La síntesis del Compuesto 47 siguió el procedimiento del siguiente Procedimiento general 6d :

A una solución agitada de 1-(5-((5-clorotiofen-2-il)metilamino)-3-(1-(morfolin-4-carbonil)azepan-4-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 46, 200 mg, 0,39 mmol) en metanol (5 ml), se añadió bicarbonato de sodio (NaHCO3 , 100 mg, 1,1 mmol). Se agitó la mezcla de reacción a 50 0C durante 3 horas. La mezcla de reacción se enfrió hasta la temperatura ambiente y se evaporaron las sustancias volátiles, y el residuo resultante se diluyó con EtOAc (20 ml) y se lavó con agua (2 x 10 ml). La capa orgánica se desecó sobre sulfato de sodio, se filtró y se concentró a presión reducida. El sólido resultante se trituró con éter dietílico y pentano, y se desecó a alto vacío durante 1 hora, produciendo (4-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)azepan-1-il)(morfolino)metanona (Compuesto 47, 150 mg, 90 %) en forma de un sólido blanquecino. CL-EM: m/z 424,24 [M+H]+; r Mn de 1H (400 MHz, DMSO-de) 5 11,21 (s, 1H), 6,90-6,82 (dd, J = 10,0, 3,2 Hz, 2H), 5,58 (s a, 1H), 5,25 (s, 1H), 4,27 (d, J = 5,6 Hz, 2H), 3,57-3,53 (m, 4H), 3,50-3,42 (m, 2H), 3,27-3,17 (m, 2H), 3,08-2,98 (m, 4H), 2,66-2,62 (m, 1H), 1,69-1,92 (m, 5H), 1,46-1,49 (m, 1H) ppm; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,2.
Ejemplo 76-Preparación del compuesto 48
La síntesis del Compuesto 48 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 °C) de (4-(5-((5-clorotiofen-2-il)metilamino)-1 H-pirazol-3-il)azepan-1-il)(morfolino)metanona (Compuesto 47, 187 mg, 0,44 mmol) en diclorometano seco (10 ml), se añadió trietilamina (TEA, 1,84 ml, 1,32 mmol), seguida de cloruro de tiazol-4-carbonilo (78 mg, 0,53 mmol). Se agitó la mezcla a temperatura ambiente durante 3 horas. Se diluyó la mezcla de reacción con agua (15 ml) y se extrajo con diclorometano (3 x 40 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó por CombiFlash, eluyendo con EtOAc al 60 %/n-hexano, produciendo (4-(5-((5-clorotiofen-2-il)metilamino)-1 -(tiazol-4-carbonil)-1 H-pirazol-3-il)azepan-1 -il)(morfolino)metanona (Compuesto 48, 60 mg, 25 %) en forma de un sólido amarillo. EM (IEN): m/z 535,21 [M+H]+; RMN de 1H (400 MHz, DMSO-ak) 59,19 (d, J = 1,2 Hz, 1H), 9,03 (d, J = 1,2 Hz, 1H), 7,77 (t, J = 6,4 Hz, 1H), 6,98 (dd, J = 9,2, 3,2 Hz, 2H), 5,52 (s, 1H), 4,50 (d, J = 6,4 Hz, 2H), 3,56-3,42 (m, 6H), 3,31-3,21 (m, 2H), 3,06-2,96 (m, 4H), 2,68-2,64 (m, 1H), 2,04-1,85 (m, 4H), 1,72-1,67 (m, 1H), 1,60-1,59 (m, 1H) ppm; Sistema de TLC: acetato de etilo al 80 % en hexano. Fr: 0,7.
Ejemplo 77-Preparación del producto intermedio 29
La síntesis del Producto intermedio 29 siguió el procedimiento del siguiente Procedimiento general 20:

A una solución agitada de 4-(3-amino-cianoacetil)piperidin-1-carboxilato de alilo (Producto intermedio 5, 2,5 g, 10,6 mmol) en etanol (30 ml), se añadieron tricloroacetonitrilo (2,1 ml, 21,2 mmol) y trietilamina (TEA, 0,2 ml). Se agitó la mezcla de reacción a temperatura ambiente durante 2 horas. Se evaporaron las sustancias volátiles y el residuo resultante se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 50 %/n-hexano, produciendo el producto deseado (Z)-4-(3-amino-4,4,4-tricloro-2-cianobut-2-enoil)piperidin-1-carboxilato de alilo (Producto intermedio 29, 3,2 g, 80 %) en forma de un líquido marrón. EM (IEN): m/z 380,13 [M+H]+; Sistema de TLC: acetato de etilo al 30 % en hexano, Fr: 0,5.
Ejemplo 78-Preparación del compuesto 49
La síntesis del Compuesto 49 siguió el procedimiento del siguiente Procedimiento general 21:

A una solución agitada de 4-(3-amino-4,4,4-tricloro-2-cianobut-2-enoil)piperidina-1-carboxilato de (Z)-alilo (Producto intermedio 29, 3,2 g, 8,4 mmol) en tolueno (30 ml), se añadió N2H4 (1 M en THF) (16,8 ml, 16,8 mmol) y se calentó la mezcla de reacción a 100 °C durante 15 horas. Se enfrió la mezcla de reacción hasta la temperatura ambiente, se evaporaron las sustancias volátiles y el residuo resultante se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con MeOH al 5 % en diclorometano, produciendo el producto deseado 4-(5-amino-4-ciano-1 H-pirazol-3-il)piperidin-1-carboxilato de alilo (Compuesto 49, 1,4 g, 61 %) en forma de un sólido blanquecino. EM (IEN): m/z 275,13 [M+H]+. Sistema de TLC: metanol al 10 %-diclorometano. Fr: 0,3.
Ejemplo 79-Preparación del compuesto 50
La síntesis del Compuesto 50 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución fría (0 0C) de 4-(5-amino-4-ciano-1 H-pirazol-3-il)piperidin-1-carboxilato de alilo (Compuesto 49, 3,2 g, 11,6 mmol) en MeOH seco (30 ml), se añadieron 5-clorotiofen-2-carbaldehído (1,8 ml, 14 mmol), AcOH (2 ml) y tamices moleculares de 4 Á en polvo (1,6 g). Se agitó la mezcla de reacción a temperatura ambiente durante 1 hora. La formación de imina se observó como una mancha menos polar en TLC. A continuación, se añadió a la mezcla cianoborohidruro de sodio (NaCNBH3 , 1,08 g, 17,5 mmol) poco a poco a 0 0C, y la reacción se agitó a temperatura ambiente durante 2 horas. Se inactivó la mezcla de reacción con agua helada (50 ml), se filtró a través de un lecho corto de Celite y el lecho de Celite se enjuagó varias veces con EtOAc. Se separaron las dos capas, y la capa acuosa se extrajo en EtOAc (4 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 60 %/n-hexano, produciendo el producto deseado (2,5 g, 60 %) en forma de un líquido amarillo claro. Se purificó una porción de la muestra (200 mg) adicionalmente mediante HPLC preparativa, produciendo 4-(5-((5-clorotiofen-2-il)metilamino)-4-ciano-1H-pirazol-3-il)piperidin-1-carboxilato de alilo (Compuesto 50, 110 mg) en forma de un líquido gomoso. EM (IEN): m/z 405,05 [M+H]+; RMN de 1H (300 MHz, CDCls) 56,81-6,74 (m, 2H), 5,98-5,89 (m, 1H), 5,32-5,21 (m, 2H), 4,60 (d, J = 8,0 Hz, 2H), 4,52 (s, 2H), 4,48-4,42 (s a, 1H), 4,40-4,24 (m, 2H), 3,00-2,84 (m, 3H), 2,04-1,96 (m, 2H), 1,76-1,67 (m, 2H) ppm; Sistema de TLC: acetato de etilo al 80 % en hexano, Fr: 0,4.
Ejemplo 80-Preparación del compuesto 51
La síntesis del Compuesto 51 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 °C) del Compuesto 50 (2,1 g, 5,2 mmol) en diclorometano seco (100 ml), se añadió trietilamina (2,2 ml, 15,5 mmol), seguida de cloruro de pivaloílo (0,66 ml, 6,2 mmol) y se agitó la mezcla a temperatura ambiente durante 4 horas. Se diluyó la mezcla de reacción con agua (25 ml) y se extrajo con diclorometano (3 x 40 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 10 %/n-hexano, produciendo el producto deseado (1,8 g, 72 %) en forma de un líquido incoloro. Se purificó una porción de muestra de 200 mg adicionalmente mediante HPLC preparativa, produciendo 4-(5-((5-clorotiofen-2-il)metilamino)-4-ciano-1 -pivaloil-1 H-pirazol-3-il)piperidin-1 -carboxilato de alilo (Compuesto 51, 104 mg, 99 %) en forma de un líquido gomoso. EM (IEN): m/z 479,26 [M+H]+. RMN de 1H (400 MHz, CDCls) 5 8,44 (t, J = 5,6 Hz, 1H), 6,90 (d, J = 4,0 Hz, 1H), 6,80 (d, J = 4,0 Hz, 1H), 6,00-5,91 (m, 1H), 5,33-5,20 (m, 2H), 4,85 (d, J = 4,0 Hz, 2H), 4,61 (d, J = 4,0 Hz, 2H), 4,30-4,14 (m, 2H), 3,04-2,90 (m, 2H), 2,89-2,81 (m, 1H), 2,01 (d, J = 10,8 Hz, 2H), 1,73 (d, J = 10,8 Hz, 2H), 1,42 (s, 9H); Sistema de TLC: acetato de etilo al 20 % en hexano. Fr: 0,5.
Ejemplo 81-Preparación del compuesto 52
La síntesis del Compuesto 52 siguió el siguiente procedimiento del Procedimiento general 8b :

Se desgasificó una solución agitada de Compuesto 51 (250 mg, 0,51 mmol) en diclorometano (10 ml) con una corriente de argón durante 15 minutos, después se añadió fenilsilano (PhSiH3 , 0,19 ml, 1,53 mmol), seguido de tetraquis(trifenilfosfin)paladio (0) (Pd(PPh3)4 , 58 mg, 0,05 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 45 minutos, a continuación, se filtró a través de un lecho corto de Celite y se evaporó el filtrado. El residuo se purificó mediante cromatografía ultrarrápida utilizando MeOH al 30 % en diclorometano, produciendo el producto deseado 1 -(3-(piperdina-4-il)-5-((5-clorotiofen-2-il)metilamino)-4-ciano-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona semipura (Compuesto 52, 280 mg/74 % mediante CL-EM), que se utilizó directamente para la siguiente reacción sin purificación adicional; Sistema de TLC: MeOH al 20 %-diclorometano, Fr: 0,2.
Ejemplo 82-Preparación del compuesto 53
La síntesis del Compuesto 53 siguió el procedimiento del siguiente Procedimiento general 15:

A una solución enfriada (0 °C) de 1-(3-(piperdina-4-il)-5-((5-clorotiofen-2-il)metilamino)-4-ciano-1 H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 52, 280 mg, 0,69 mmol) en diclorometano seco (50 ml), se añadió trietilamina (TEA, 0,28 ml, 2,1 mmol), seguida de cloruro de carbamilo de morfolina (0,12 mg, 0,83 mmol). Se agitó la mezcla a temperatura ambiente durante 3 horas. Se diluyó la mezcla de reacción con agua (20 ml) y se extrajo con diclorometano (2 x 20 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó por CombiFlash, eluyendo con EtOAc al 30 %/n-hexano seguido de HPLC preparativa, produciendo el producto deseado 1-(5-((5-clorotiofen-2-il)metilamino)-3-(1-(morfolin-4-carbonil)piperdina-4-il)-4-ciano-1 H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 53, 140 mg, 34 %) en forma de un semisólido blanquecino. EM (IEN): m/z 508,28 [M+H]+. RMN de 1H (400 MHz, CDCls) 58,45 (t, J = 6,0 Hz, 1H), 6,90 (d, J = 4,0 Hz, 1H), 6,80 (d, J = 4,0 Hz, 1H), 4,85 (d, J = 6,0 Hz, 2H), 3,80 (d, J = 13,2 Hz, 2H), 3,71-3,68 (m, 4H), 3,28-3,25 (m, 4H), 2,97-2,90 (m, 2H), 2,87-2,81 (m, 1H), 2,04-2,00 (m, 2H), 1,80-1,71 (m, 2H), 1,42 (s, 9H) ppm; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,5.
Ejemplo 83-Preparación del producto intermedio 31
La síntesis del Producto intermedio 31 siguió el procedimiento del siguiente Procedimiento general 2 :

A una solución enfriada (-40 °C) de propionitrilo (5,8 ml, 74,7 mmol) en THF seco (120 ml), se añadió hexametilsilazida de litio (1HMDS, 1 M en THF, 99 ml, 99,6 mmol). Después de agitar a-40 °C durante 15 minutos, se añadió una solución de 1 -alil-4-etil-piperidin-1,4-dicarboxilato (Producto intermedio 5, 12 g, 49,8 mmol) en THF
seco (30 ml) y se agitó la mezcla de reacción a la misma temperatura durante 30 minutos. Se inactivó la mezcla de reacción con solución saturada de NH4Cl (80 ml) y se extrajo con EtOAc (2 x 120 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 50 %/n-hexano, produciendo 4-(2-cianopropanoil)piperidin-1-carboxilato de alilo (Producto intermedio 31, 9 g, rendimiento: 72 %) en forma de un líquido amarillo claro. m/z 250,87 [M+H]+; Sistema de TLC: acetato de etilo al 30 % en hexano, Fr: 0,5.
Ejemplo 84-Preparación del compuesto 54
La síntesis del Compuesto 54 siguió el procedimiento del siguiente Procedimiento general 3 :

A una solución agitada de 4-(2-cianopropanoil)piperidin-1-carboxilato de alilo (Producto intermedio 31, 9 g, 36 mmol) en etanol (90 ml), se añadió N2H4 (1,6 ml, 32,4 mmol) y se calentó la mezcla de reacción a 70 0C durante 5 horas. Se enfrió la mezcla de reacción hasta la temperatura ambiente, se evaporaron las sustancias volátiles y el residuo resultante se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con MeOH al 3 % en diclorometano, produciendo 4-(5-amino-4-metil-1H-pirazol-3-il)piperidin-1-carboxilato de alilo (Compuesto 54, 8 g, rendimiento: 84 %) líquido incoloro. m/z 265,13 [M+H]+; Sistema de TLC: metanol al 5 %-diclorometano. Fr: 0,5.
Ejemplo 85-Preparación del compuesto 55
La síntesis del Compuesto 55 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución enfriada (0 0C) de 4-(5-amino-4-metil-1H-pirazol-3-il)piperidin-1-carboxilato de alilo (Compuesto 54, 5 g, 18,9 mmol) en MeOH seco (50 ml), se añadieron 5-clorotiofen-2-carbaldehído (4,2 ml, 37,8 mmol), AcOH (0,2 ml) y, a continuación, tamices moleculares de 4 Á en polvo. Se agitó la mezcla de reacción a temperatura ambiente durante 1 hora. La formación de imina se observó como una mancha menos polar mediante TLC. A continuación, se añadió a la mezcla de reacción cianoborohidruro de sodio (NaCNBH3 , 1,4 g, 22,7 mmol) poco a poco y se agitó la mezcla a temperatura ambiente durante 2 horas. A la mezcla, se añadió agua helada (50 ml), después se filtró a través de un lecho corto de Celite y el filtrado se extrajo con EtOAc (4 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 50 %/n-hexano, produciendo el producto deseado (3,5 g, rendimiento: 46 %) en forma de un líquido amarillo claro. Una porción (300 mg) de muestra semipura se purificó adicionalmente mediante HPLC preparativa, produciendo 4-(5-((5-clorotiofen-2-il)metilamino)-4-metil-1H-pirazol-3-il)piperidin-1-carboxilato de alilo (Compuesto 55, 11 0 mg) en forma de un líquido gomoso. m/z 395,16 [M+H]+. RMN de 1H (400 MHz, CDCta) 5 11,08 (s a, 1 H), 6,89 (d, J = 3,9 Hz, 1H), 6,82 (d, J = 3,4 Hz, 1H), 5,94 (tdd, J = 5,2, 10,5, 17,2 Hz, 1H), 5,38-5,26 (m, 2H), 5,19 (dd, J = 1,5, 10,3 Hz, 1H), 4,56-4,50 (m, 2H), 4,33 (d, J = 6,4 Hz, 2H), 4,08 (d a, J = 13,2 Hz, 2H), 2,86 (s a, 2H), 2,73 (tt, J = 3,5, 12 , 2 Hz, 1 h ), 1,79-1,71 (m, 3H), 1,70-1,63 (m, 2 H), 1,59-1,48 (m, 2 H) ppm; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,5.
E je m p lo 86 -P re p a ra c ió n de l c o m p u e s to 56
La síntesis del Compuesto 56 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 0C) de 4-(5-((5-clorotiofen-2-il)metilamino)-4-metil-1H-pirazol-3-il)piperidin-1-carboxilato de alilo (Compuesto 55, 1,9 g, 4,8 mmol) en diclorometano seco (20 ml), se añadió cloruro de trimetilacetilo (0,57 ml, 4,3 mmol), seguido de trietilamina (TEA, 1 ml, 7,2 mmol) y se agitó la mezcla a temperatura ambiente durante 4 horas. La mezcla de reacción se combinó con agua (25 ml) y se extrajo con diclorometano (3 x 40 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 20 %/n-hexano, produciendo el producto deseado (1,3 g, rendimiento: 69 %) en forma de un líquido amarillo claro. Se purificó una porción (300 mg) de muestra semipura adicionalmente mediante HPLC preparativa, produciendo 4-(5-((5-clorotiofen-2-il)metilamino)-4-metil-1-pivaloil-1H-pirazol-3-il)piperidin-1-carboxilato de alilo (Compuesto 56, 70 mg) en forma de un líquido amarillo claro. m/z 479,23 [M+H]+ RMN de 1H (400 MHz, CDC^) 5 = 7,26 (s, 1H), 6,74 (d, 3,9 Hz, 1H), 6,69 (d, 3,4 Hz, 1H), 6,02-5,91 (m, 1H), 5,32 (dd, 1,5, 17,1 Hz, 1H), 5,22 (dd, 1,2, 10,5 Hz, 1H), 4,61 (d a, 5,4 Hz, 2H), 4,51 (d a, 5,4 Hz, 2H), 4,20 (d a, 10,8 Hz, 2H), 2,98 (t a, 10,8 Hz, 2H), 2,68 (tt, 3,7, 10,8 Hz, 1H), 2,01 -1,97 (m, 3H), 1,86 (d a, 11,2 Hz, 2 H), 1,78-1,69 (m, 2H), 1,43 (s, 9h) ppm; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,5.
Ejemplo 87-Preparación del compuesto 57
La síntesis del Compuesto 57 siguió el procedimiento de Procedimiento general 8b :

Se desgasificó una solución agitada de 4-(5-((5-clorotiofen-2-il)metilamino)-4-metil-1-pivaloil-1H-pirazol-3-il)piperidin-1-carboxilato de alilo (Compuesto 56, 1 g, 2,1 mmol) en diclorometano (10 ml) con una corriente de argón durante 15 minutos, después se añadió a la solución tetraquis(trifenilfosfin)paladio (0) (Pd(PPh3)4 , 240 mg, 0,21 mmol), seguido de fenilsilano (PhS¡H3 , 1,6 ml, 12,5 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 45 minutos, a continuación, se filtró a través de un lecho corto de Celite y se evaporó el filtrado. El residuo en bruto se purificó mediante cromatografía ultrarrápida eluyendo con MeOH al 4 %-diclorometano, produciendo el producto deseado semipuro (600 mg (91 % mediante CL-EM), rendimiento: 72 %). Se purificó una porción (130 mg) de muestra semipura adicionalmente mediante HPLC preparativa, produciendo 1-(5-((5-clorotiofen-2-il)metilamino)-4-metil-3-(piperidin-4-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 57, 40 mg) en forma de un sólido blanquecino. m/z 395,19 [M+H]+, RMN de 1H (400 MHz, DMSO-da) 5 = 8,37 (s a, 1H), 7,26 (t a, J = 7,1 Hz, 1H), 6,96 (d, J=3,9 Hz, 1H), 6,82 (d, J=3,9 Hz, 1H), 4,55 (d, J=6,8 Hz, 2H), 3,16 (d a, J=12,2 Hz, 2H), 2,86-2,72 (m, 3H), 1,92 (s, 3H), 1,83 (d a, J=12,7 Hz, 2H), 1,67 (c, J=10,9 Hz, 2H), 1,38 (s, 9H) ppm; Sistema de TLC: MeOH al 10 %-diclorometano, Fr: 0,5.
Ejemplo 88-Preparación del compuesto 58
La síntesis del Compuesto 58 siguió el procedimiento del siguiente Procedimiento general 6d :

A una solución de 1-(5-((5-clorotiofen-2-il)metilamino)-3-(1-(morfolin-4-carbonil)piperidin-4-il)-4-ciano-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 53, 350 mg, 0,67 mmol) en metanol (15 ml), se añadió bicarbonato de sodio (NaHCO3 , 113 mg, 1,3 mmol). Se agitó la mezcla de reacción a 50 0C durante 3 horas. La mezcla de reacción se enfrió hasta la temperatura ambiente, se evaporaron las sustancias volátiles, y el residuo resultante se diluyó con EtOAc (20 ml) y se lavó con agua (2 x 10 ml). La capa orgánica se desecó sobre sulfato de sodio, se filtró y se concentró a presión reducida. El sólido obtenido se trituró con éter dietílico y pentano, y se desecó a alto vacío durante 1 h, produciendo 5-((5-clorotiofen-2-il)metilamino)-3-(1-(morfolin-4-carbonil)piperidin-4-il)-1H-pirazol-4-carbonitrilo (Compuesto 58, 250 mg, 90 %) en forma de un sólido blanquecino. m/z 424,24 [M+H]+; RMN de 1H (400 MHz, DMSO-da, mezcla de rotámeros) 5 12,30 (s, 0,7H), 12,12 (s, 0,3H), 7,45 (s a, 0,3H), 7,00-6,80 (m, 2H), 6,60 (t, J = 5,2 Hz, 0,7H), 4,48-4,32 (m, 2H), 3,74-3,62 (m, 2H), 3,56 (s a, 4H), 3,11 (s a, 4H), 2,93-2,66 (3H), 1,86 1,78 (m, 2H), 1,70-1,52 (m, 2H) ppm; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,2.
Ejemplo 89-Preparación del producto intermedio 32
La síntesis del Producto intermedio 32 siguió el siguiente procedimiento del Procedimiento general 6c:

A una solución enfriada (0 0C) de d/-1-ferc-butil-3-metilpirrolidin-1,3-dicarboxilato (12 g, 52,4 mmol) en 1,4-dioxano (30 ml), se añadió HCl 4 M en dioxano (24 ml). La solución se dejó calentar a temperatura ambiente durante 4 horas. Se concentró la mezcla de reacción a presión reducida, obteniéndose clorhidrato de c//-metil-pirrolidin-3-carboxilato (8,6 g, rendimiento: 100 %). Se utilizó “tal cual” para la siguiente etapa sin ninguna purificación.
Ejemplo 90-Preparación del producto intermedio 33
La síntesis del Producto intermedio 33 siguió el procedimiento del siguiente Procedimiento general 7 :

A una solución enfriada (0 °C) de clorhidrato de d/-metilpirrolidin-3-carboxilato (Producto intermedio 32, 14,4 g, 87,3 mmol) en THF (150 ml), se añadieron piridina (28,1 ml, 349,3 mmol) y 4-(dimetilamino)piridina (DMAP, 1,07 g, 8,7 mmol), seguida de cloroformiato de alilo (18,5 ml, 174,7 mmol). Después de calentar hasta la temperatura ambiente durante 16 horas, se añadió agua (100 ml) a la mezcla de reacción y después se extrajo en diclorometano (2 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), se eluyó con EtOAc al 20-25 %/n-hexanos, obteniéndose d/-1-alil-3-metilpirrolidin-1,3-dicarboxilato (Producto intermedio 33, 15 g, rendimiento: 80 %) en forma de un líquido incoloro. m/z 214,07 [M+H]+; Sistema de TLC: acetato de etilo al 50 % en hexano, Fr: 0,5.
Ejemplo 91-Preparación del producto intermedio 34
La síntesis del Producto intermedio 34 siguió el procedimiento del siguiente Procedimiento general 2 :

A una solución enfriada (-78 0C) de acetonitrilo (5,5 ml, 105,6 mmol) en THF seco (25 ml), se añadió n-butillitio (2,5 M en n-hexano, 42,2 ml, 105,6 mmol). Después de agitar durante 45 minutos, se añadió una solución de dl-1- alil-3-metilpirrolidin-1,3-dicarboxilato (Producto intermedio 33, 15 g, 70,4 mmol) en THF seco (30 ml) y se agitó a -78 °C durante 30 minutos. Se inactivó la mezcla de reacción con solución saturada de NH4Cl (50 ml) y se extrajo en EtOAc (2 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con 30-35 % de EtOAc/n-hexano, produciendo c//-alil-3-(2-cianoacetil)pirrolidin-1-carboxilato (Producto intermedio 34, 11 g, rendimiento: 70 %) en forma de un líquido amarillo claro; Sistema de TLC: acetato de etilo al 50 % en hexano, Fr: 0,2.
Ejemplo 92-Preparación del compuesto 59
La síntesis del Compuesto 59 siguió el procedimiento del siguiente Procedimiento general 3 :

A una solución agitada de d/-alil-3-(2-cianoacetil)pirrolidin-1 -carboxilato (Producto intermedio 34, 15 g, 67,6 mmol) en etanol (150 ml), se añadió monohidrato de hidrazina (2,9 ml, 60,8 mmol) y, a continuación, se calentó la mezcla de reacción hasta 70 0C durante 5 horas. Se enfrió la mezcla hasta la temperatura ambiente, se evaporaron las sustancias volátiles y el residuo resultante se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con metanol al 3 % en diclorometano, produciendo d/-alil-3-(5-amino-1H-pirazol-3-il)pirrolidin-1-carboxilato (Compuesto 59, 12 g, rendimiento: 75 %) en forma de un líquido incoloro. m/z 237,54 [M+H]+; Sistema de TLC: metanol al 5 % en diclorometano, Fr: 0,3.
Ejemplo 93-Preparación del compuesto 60
La síntesis del Compuesto 60 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución enfriada (0 0C) de d/-alil-3-(5-amino-1H-pirazol-3-il)pirrolidin-1-carboxilato (Compuesto 59, 3 g, 12,7 mmol) en MeOH seco (30 ml), se añadieron 5-clorotiofen-2-carbaldehído (2,7 ml, 25,4 mmol), AcOH (0,1 ml) y tamices moleculares de 4 Á en polvo. Se agitó la mezcla de reacción a temperatura ambiente durante 1 hora (se observó la formación de imina como una mancha menos polar en la TLC). Se añadió cianoborohidruro de sodio (NaCNBH3 , 1,18 g, 19,1 mmol) poco a poco. Después de que la mezcla de reacción se agitara a temperatura ambiente durante 2 horas, se añadió agua helada (30 ml), la mezcla se filtró a través de un lecho corto de Celite y el filtrado se extrajo en EtOAc (3 x 50 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante una columna de fase inversa Reveleris
C-18 utilizando acetonitrilo al 35 %-40 % en ácido fórmico acuoso (0,1 %), obteniéndose d/-alil-3-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)pirrolidin-1-carboxilato (Compuesto 60, 1,6 g, rendimiento: 34 %) en forma de un sólido blanquecino. m/z 367,10 [M+H]+; RMN de 1H (400 MHz, DMSO-da) 5 11,43 (s a, 1H), 6,91 (d, J = 3,9 Hz, 1H), 6,84 (s a, 1H), 5,98-5,88 (m, 1H), 5,67 (s a, 1H, intercambiable), 5,37-5,24 (m, 2H), 5,18 (d a, J = 10,3 Hz, 1H), 4,52 (td, J = 1,5, 3,3 Hz, 2H), 4,28 (d, J = 6,4 Hz, 2H), 3,67 (s a, 1H), 3,49-3,35 (m, 2H), 3,30-3,21 (m, 2H), 2,16 (s a, 1H), 1,99 1,85 (m, 1H) ppm; Sistema de TLC: metanol al 10 % en diclorometano, Fr: 0,4.
Ejemplo 94-Preparación del compuesto 61
La síntesis del Compuesto 61 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 0C) de d/-alil-3-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)pirrolidin-1-carboxilato (Compuesto 60, 900 mg, 2,45 mmol) en diclorometano seco (10 ml), se añadió cloruro de trimetilacetilo (0,27 ml, 2,2 mmol) seguido de trietilamina (TEA, 0,51 ml, 3,7 mmol). Después de agitar a temperatura ambiente durante 3 horas, se diluyó la mezcla de reacción con agua (25 ml) y se extrajo en diclorometano (3 x 40 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 10 %/n-hexano, produciendo d/-alil-3-(5-((5-clorotiofen-2-il)metilamino)-1 -pivaloil-1 H-pirazol-3-il)pirrolidin-1 -carboxilato (Compuesto 61, 450 mg, rendimiento: 40 %) en forma de un líquido gomoso incoloro. m/z 451,47 [M+H]+. RMN de 1H (400 MHz, DMSO-da) 5 7,69 (t, J = 6,4 Hz, 1H), 6,98-6,94 (m, 2H), 5,97-5,86 (m, 1H), 5,44 (s, 1H), 5,27 (dd, J = 1,5, 17,1 Hz, 1H), 5,17 (dd, J = 1,7, 10,5 Hz, 1H), 4,51 (d a, J = 5,4 Hz, 2H), 4,41 (d, J = 6,4 Hz, 2H), 3,65-3,55 (m, 1H), 3,53-3,37 (m, 3H), 3,36-3,33 (m, 1H), 2,24-2,12 (m, 1H), 2,03-1,94 (m, 1H), 1,39 (s, 9H) ppm; Sistema de TLC: acetato de etilo al 50 % en hexano, Fr: 0,7.
Ejemplo 95-Preparación del compuesto 62
La síntesis del Compuesto 62 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución helada de d/-alil-3-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)pirrolidin-1-carboxilato (Compuesto 60, 300 mg, 0,8 mmol) en diclorometano seco (10 ml), se añadió cloruro de 1,3-tiazol-4-carbonilo (158 mg, 1,1 mmol), seguido de trietilamina (TEA, 0,28 ml, 2,0 mmol). Después de agitar a 0 0C durante 2 horas, se añadió agua helada (15 ml) y se extrajo en diclorometano (3 x 20 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice ultrarrápida (malla 100-200), eluyendo con MeOH al 2-4 % en diclorometano, obteniéndose un producto semipuro (270 mg, rendimiento: 69 %). Posteriormente, se purificó mediante HPLC preparativa, dando d/-alil-3-(5-((5-clorotiofen-2-il)metilamino)-1-(tiazol-4-carbonil)-1H-pirazol-3-il)pirrolidin-1 -carboxilato (Compuesto 62, 62 mg) en forma de un sólido amarillo pálido. m/z 478,17 [M+H]+. RMN de 1H (400 MHz, CDCl3) 59,12 (d, J = 8,8 Hz, 1H), 8,88 (s, 1H), 7,70 (t, J = 5,6 Hz, 1H), 6,82-6,76 (m, 2H), 6,00-5,90 (m, 1H), 5,35-5,26 (m, 2H), 5,21 (dd, J = 1,2, 10,5 Hz, 1H), 4,62 (d, J = 5,4 Hz, 2H), 4,47 (d, J = 5,9 Hz, 2H), 3,86 3,76 (m, 1H), 3,68-3,50 (m, 3H), 3,36 (td, J = 7,0, 13,5 Hz, 1H), 2,28 (s a, 1H), 2,10 (dt, J = 4,2, 8,2 Hz, 1H) ppm; Sistema de TLC: metanol al 5 %-diclorometano. Fr: 0,6.
E je m p lo 96 -P re p a ra c ió n de l c o m p u e s to 63
La síntesis del Compuesto 63 siguió el siguiente procedimiento del Procedimiento general 8b :

Se desgasificó solución agitada de alil-3-(5-((5-clorotiofen-2-il)metilamino)-1-pivaloil-1H-pirazol-3-il)pirrolidin-1-carboxilato (Compuesto 61, 1,5 g, 3,3 mmol) en diclorometano seco (30 ml) con una corriente de argón durante 15 minutos, después se añadió tetraquis(trifenilfosfin)paladio (0) (Pd(PPh3)4 , 385 mg, 0,33 mmol) seguido de fenilsilano (PhS¡H3 , 1,2 ml, 10 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 45 minutos. Se filtró la mezcla de reacción a través de un lecho corto de Celite y se evaporó el filtrado. El residuo en bruto se purificó mediante cromatografía en sílice CombiFlash utilizando MeOH al 4 % en diclorometano, lo que proporcionó 1-(5-((5-clorotiofen-2-il)metilamino)-3-(pirrolidin-3-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 63, 1,1 g, rendimiento: 91 %) en forma de un sólido blanquecino. m/z 367,26 [M+H]+, RMN de 1H (400 MHz, DMSO-d6) 5 = 9,11 (s a, 2H, intercambiable), 7,78 (t a, J = 6,1 Hz, 1H, intercambiable), 7,02-6,95 (m, 2H), 5,50 (s, 1H), 4,42 (d a, J = 5,9 Hz, 2H), 3,57-3,34 (m, 2H), 3,32-3,16 (m, 3H), 2,34-2,20 (m, 1H), 2,05-1,90 (m, 1H), 1,41 (s, 9H) ppm; Sistema de TLC: metanol al 10 % en diclorometano, Fr: 0,1.
Ejemplo 97-Preparación del compuesto 64
La síntesis del Compuesto 64 siguió el procedimiento del siguiente Procedimiento general 15:

A una solución enfriada (a 0 0C) de c//-1-(5-((5-clorotiofen-2-il)metilamino)-3-(pirrolidin-3-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 63, 450 mg, 1,2 mmol) en diclorometano seco (10 ml), se añadió cloruro de morfolin-4-carbonilo (366 mg, 2,45 mmol) seguido de trietilamina (0,31 ml, 3,0 mmol). Después de agitar la mezcla a temperatura ambiente durante 2 horas, se añadió agua helada (10 ml) a la mezcla. La mezcla se extrajo en diclorometano (2 x 15 ml), y las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 50 %-hexanos, produciendo el producto deseado (370 mg, rendimiento: 62 %) en forma de un semisólido de color amarillo claro. Se purificó adicionalmente una porción (160 mg) de material semipuro (85 % mediante CL-EM) mediante HPLC preparativa, obteniéndose d/-1-(5-((5-clorotiofen-2-il)metilamino)-3-(1 -(morfolin-4-carbonil)pirrolidin-3-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona (Compuesto 64, 60 mg) en forma de un sólido blanquecino. m/z 480,16 [M+H]+. RMN de 1H (400 MHz, DMSO-de) 5 7,70 (t, J = 6,1 Hz, 1H, intercambiable), 6,96 (s, 2H), 5,42 (s, 1H), 4,41 (d, J = 6,4 Hz, 2H), 3,58-3,47 (m, 6H), 3,36 (d, J = 3,9 Hz, 2H), 3,24 3,19 (m, 1H), 3,15-3,10 (m, 4H), 2,11 (dd, J = 6,1, 12,5 Hz, 1H), 1,97-1,89 (m, 1H), 1,39 (s, 9H) ppm; Sistema de TLC: acetato de etilo al 70 % en n-hexano-Fr: 0,6.
Ejemplo 98-Preparación del compuesto 65
La síntesis del Compuesto 65 siguió el procedimiento del siguiente Procedimiento general 6d :

A una solución de d/-1-(5-((5-clorotiofen-2-il)metilamino)-3-(1-(morfolin-4-carbonil)pirrolidin-3-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 64, 500 mg, 1,0 mmol) en metanol (10 ml), se añadió bicarbonato de sodio (438 mg, 5,2 mmol) a temperatura ambiente. A continuación, se agitó la mezcla a 50 0C durante 4 horas. La mezcla de reacción se enfrió de nuevo a temperatura ambiente, se añadió agua (20 ml) y después se extrajo en diclorometano (3 x 20 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó parcialmente mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con metanol al 5-7 % en diclorometano, obteniéndose el producto deseado (300 mg, rendimiento: 72 %) en forma de un semisólido de color amarillo claro. Se purificó adicionalmente una porción (150 mg, 72 % mediante CL-EM) de material semipuro mediante HPLC preparativa, produciendo d1-(3-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)pirrolidin-1-il)(morfolino)metanona (Compuesto 65, 62 mg) en forma de un sólido blanquecino. m/z 396,21 [M+H]+. RMN de 1H (400 MHz, DMSO-de) 5 11,42 (s a, 1H, intercambiable), 6,91 (d, J = 3,4 Hz, 1H), 6,83 (s a, 1H), 5,67 (s a, 1H, intercambiable), 5,35 (s a, 1H), 4,28 (d, J = 5,9 Hz, 2H), 3,56 (td, J =3,8, 5,6 Hz, 5H), 3,37 (td, J = 3,7, 7,7 Hz, 2H), 3,21-3,07 (m, 6H), 2,12 (s a, 1H), 1,84 (dd, J = 8,6, 12,0 Hz, 1H) ppm; Sistema de TLC: metanol al 10 % en diclorometano-Fr: 0,3.
Ejemplo 99-Preparación del compuesto 66
La síntesis del Compuesto 66 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 0C) de d/-(3-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)pirrolidin-1-il)(morfolino)metanona (Compuesto 65, 150 mg, 0,37 mmol) en diclorometano seco (10 ml), se añadió cloruro de 1,3-tiazol-4-carbonilo (73 mg, 0,5 mmol), seguido de trietilamina (TEA, 0,13 ml, 0,9 mmol) y la mezcla se agitó a 0 0C durante 2 horas. Se añadió agua helada (10 ml) a la mezcla de reacción y después se extrajo en diclorometano (2 x 15 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo en bruto se purificó inicialmente mediante cromatografía ultrarrápida utilizando MeOH al 4-5 % en diclorometano, dando el producto deseado (146 mg, rendimiento: 76 %) en forma de un semisólido de color amarillo claro. Se purificó adicionalmente mediante HPLC preparativa, produciendo dí-(3-(5-((5-clorotiofen-2-il)metilamino)-1-(tiazol-4-carbonil)-1H-pirazol-3-il)pirrolidin-1-il)(morfolino)metanona (Compuesto 66, 55 mg) en forma de un sólido amarillo pálido. m/z: 507,22 [M+H]+. RMN de 1H (400 MHz, CDC^) 59,13 (d, J = 2,4 Hz, 1H), 8,88 (d, J = 2,0 Hz, 1H), 7,70 (t, J = 5,9 Hz, 1H, intercambiable), 6,82-6,77 (m, 2H), 5,28 (s, 1H), 4,47 (d, J = 5,4 Hz, 2H), 3,73 3,67 (m, 5H), 3,62-3,53 (m, 3H), 3,36-3,28 (m, 5H), 2,27-2,22 (m, 1H), 2,10-2,03 (m, 1H) ppm; Sistema de TLC: metanol al 10 %-diclorometano. Fr: 0,6.
Ejemplo 100-Preparación del compuesto 67
La síntesis del Compuesto 67 siguió el procedimiento del siguiente Procedimiento general 22:

A una solución fría (0 0C) de d/-1-(5-((5-clorotiofen-2-il)metilamino)-3-(pirrolidin-3-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 63, 650 mg, 1,77 mmol) en diclorometano seco (20 ml), se añadió cloruro de metanosulfonilo (304,4 mg, 2,65 mmol) seguido de trietilamina (TEA, 0,62 ml, 4,42 mmol). Se agitó la mezcla a temperatura ambiente durante 2 horas. Se inactivó la mezcla de reacción con solución de NaHCO3 (10 ml) y se extrajo en diclorometano (3 x 20 ml). El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 20-25 % en n-hexano, obteniéndose el producto deseado (550 mg, rendimiento: 69 %). Se purificó adicionalmente una porción de la muestra (170 mg) mediante HPLC preparativa, produciendoc//-1-(5-((5-clorotiofen-2-il)metilamino)-3-(1-(metilsulfonil)pirrolidin-3-il)-1 H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 67, 60 mg) en forma de un líquido amarillo pálido. m/z 445,20 [M+H]+. RMN de 1H (400 MHz, CDCls) 57,50 (t, J =5,4 Hz, 1H, intercambiable), 6,89-6,62 (m, 2H), 5,16 (s, 1H), 4,36 (d, J = 5,9 Hz, 2H), 3,73 (dd, J = 7,3, 9,8 Hz, 1H), 3,61-3,42 (m, 3H), 3,40-3,28 (m, 1H), 2,80 (s, 3H), 2,39-2,10 (m, 2H), 1,45 (s, 9H) ppm; Sistema de TLC: acetato de etilo al 30 % en n-hexano-Fr: 0,6.
Ejemplo 101-Preparación del compuesto 68
La síntesis del Compuesto 68 siguió el procedimiento del siguiente Procedimiento general 6d :

A una solución enfriada (0 0C) de d/-1-(5-((5-clorotiofen-2-il)metilamino)-3-(1-(metilsulfonil)pirrolidin-3-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 67, 380 mg, 0,86 mmol) en metanol seco (10 ml), se añadió bicarbonato de sodio (431,2 mg, 5,13 mmol) y se agitó la mezcla a 70 0C durante 4 horas. La mezcla de reacción se enfrió hasta la temperatura ambiente, después se añadió agua (20 ml) y se extrajo en diclorometano (3 x 20 ml). El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con metanol al 5-7 % en diclorometano, obteniéndose el compuesto deseado (300 mg, rendimiento: 72 %) en forma de un semisólido de color amarillo claro. La purificación mediante HPLC preparativa de una porción de este material (130 mg; 89 % de pureza CL-EM) produjo c//-W-((5-clorotiofen-2-il)metil)-3-(1-(metilsulfonil)pirrolidin-3-il)-1H-pirazol-5-amina (Compuesto 68, 30 mg) en forma de un sólido blanco. m/z 360,8 [M+H]+. Rm N de 1H (400 MHz, DMSO-d6) 5 11,44 (s a, 1H, intercambiable), 6,94-6,82 (m, 2H), 5,69 (s a, 1H, intercambiable), 5,41 (s a, 1H), 4,29 (d, J = 6,4 Hz, 2H), 3,59 (d a, J = 4,4 Hz, 1H), 3,36 (d a, J = 6,8 Hz, 2H), 3,28 (s a, 1H), 3,15 (s a, 1H), 2,88 (s a, 3H), 2,20 (s a, 1H), 2,01 -1,89 (m, 1H) ppm; Sistema de TLC: metanol al 5 % en diclorometano-Fr: 0,3.
Ejemplo 102-Preparación del compuesto 69
La s ín te s is de l
Compuesto 69
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5d
:

A una solución enfriada (0 0C) de d/-W-((5-clorotiofen-2-il)metil)-3-(1-(metilsulfonil)pirrolidin-3-il)-1H-pirazol-5-amina (Compuesto 68, 170 mg, 0,47 mmol) en diclorometano seco (10 ml), se añadió cloruro de 1,3-tiazol-4-carbonilo (83,4 mg, 0,56 mmol) seguido de trietilamina (TEA, 0,13 ml, 0,94 mmol). Después de agitar la mezcla a temperatura ambiente durante 2 horas, se inactivó la mezcla de reacción con solución de NaHCO3 (10 ml) y se extrajo en diclorometano (3 x 20 ml). Los extractos orgánicos combinados se desecaron sobre sulfato sódico, se filtraron y se concentraron a presión reducida, produciendo el producto en bruto (200 mg, rendimiento: 89 %) en forma de un semisólido de color amarillo claro. La purificación mediante HPLC preparativa de una porción del material (200 mg, 87 % de pureza mediante CL-EM) produjo dí-('5-((5-clorotiofen-2-il)metilamino)-3-(1-(metilsulfonil)pirrolidin-3-il)-1H-pirazol-1-il)(tiazol-4-il)metanona (Compuesto 69, 65 mg) en forma de un sólido amarillo pálido. m/z 472,10 [M+H]+. RMN de 1H (400 MHz, DMSO-da) 59,20 (d, J = 1,5 Hz, 1H), 9,04 (d, J = 1,5 Hz, 1H), 7,85 (t, J = 6,4 Hz, 1H, intercambiable), 7,03-7,00 (d, 1H), 6,97 (d, J = 3,9 Hz, 1H), 5,64 (s, 1H), 4,51 (d, J = 5,9 Hz, 2H), 3,66-3,59 (m, 1H), 3,42-3,33 (m, 4H), 2,87 (s, 3H), 2,29-2,20 (m, 1H), 2,12-2,02 (m, 1H) ppm; Sistema de TlC: metanol al 5 % en diclorometano-Fr: 0,6.
Ejemplo 103-Preparación del compuesto 70
La síntesis del Compuesto 70 siguió el procedimiento del siguiente Procedimiento general 6d :

A una solución de 1-(3-(azepan-4-il)-5-((5-clorotiofen-2-il)metilamino)-7H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 45, 100 mg, 0,25 mmol) en metanol (5 ml), se añadió NaHCO3 (63 mg, 0,76 mmol). Se agitó la mezcla de reacción a 50 °C durante 3 horas. La mezcla de reacción se enfrió hasta la temperatura ambiente, se evaporaron las sustancias volátiles a presión reducida y el residuo resultante se diluyó con iPrOH-CHCl3 al 30 % (20 ml) y se lavó con agua (2 x 10 ml). La capa orgánica se desecó sobre sulfato de sodio, se filtró y se concentró a presión reducida. El residuo resultante (70 mg) se purificó mediante HPLC preparativa, produciendo 3-(azepan-4-il)-n-((5-clorotiofen-2-il)metil)-1 H-pirazol-5-amina (Compuesto 70, 15 mg, rendimiento: 19 %) en forma de un sólido marrón. EM: m/z 311,15 [M+H]+. RMN de 1H (400 MHz, DMSO-cfe): 5 11,24 (s, 1H), 6,90 (d, J = 3,6 Hz, 1H), 6,83 (d, J = 4,0 Hz, 1H), 5,60-5,57 (m, 1H), 5,24 (s, 1H), 4,27 (d, J = 6,0 Hz, 2H), 3,97-3,18 (m, 2H), 2,86-2,64 (m, 4H), 1,90-1,80 (m, 2H), 1,71-1,47 (m, 4H) ppm; Sistema de TLC: acetato de etilo al 10 % en hexano. Fr: 0,1.
Ejemplo 104-Preparación del compuesto 71
La s ín te s is de l
Compuesto 71
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 22
:

A una solución enfriada (0 °C) de 1-(3-(azepan-4-il)-5-((5-clorotiofen-2-il)metilamino)-1 H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 45, 280 mg, 0,71 mmol) en diclorometano seco (10 ml), se añadió trietilamina (TEA, 0,25 ml, 1,77 mmol), seguida de cloruro de metanosulfonilo (MsCl, 0,082 ml, 1,06 mmol). Después de agitar la reacción a temperatura ambiente durante 2 horas, la mezcla se diluyó con agua (10 ml) y se extrajo en diclorometano (2 x 20 ml). La capa orgánica combinada se desecó sobre sulfato de sodio, se filtró y se concentró a presión reducida. El residuo se purificó por CombiFlash, eluyendo con EtOAc al 10 %/n-hexano, produciendo el producto deseado 1 -(5-((5-clorotiofen-2-il)metilamino)-3-(1 -(metilsulfonil)azepan-4-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona (Compuesto 71, 100 mg, rendimiento: 40 %) en forma de un sólido blanquecino. EM (IEN): m/z 473,08 [M+H]+. RMN de 1H (300 MHz, DMSO-de) 5 7,65 (t, J = 6,3 Hz, 1H), 6,96 (s, 2H), 5,39 (s, 1H), 4,41 (d, J = 6,0 Hz, 2H), 3,49-3,34 (m, 2H), 3,32-3,17 (m, 2H), 2,97 (s, 3H), 2,72-2,50 (m, 1H), 2,07-1,60 (m, 6H), 1,40 (s, 9H) ppm; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,7.
Ejemplo 105-Preparación del compuesto 72
La síntesis del Compuesto 72 siguió el procedimiento del siguiente Procedimiento general 6d:

A una solución agitada de 1-(5-((5-clorotiofen-2-il)metilamino)-3-(1-(metilsulfonil)azepan-4-il)-1 H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 71, 80 mg, 0,17 mmol) en metanol (5 ml), se añadió NaHCO3 (37 mg, 0,51 mmol). Se agitó la mezcla de reacción a 50 °C durante 3 horas. La mezcla de reacción se enfrió hasta la temperatura ambiente, se evaporaron las sustancias volátiles, y el residuo resultante se diluyó con EtOAc (20 ml) y se lavó con agua (2 x 10 ml). La capa orgánica se desecó sobre sulfato de sodio, se filtró y se concentró a presión reducida. El sólido resultante se trituró con éter dietílico y pentano, y se desecó a presión reducida durante 1 hora, produciendo W-((5-clorotiofen-2-il)metil)-3-(1-(metilsulfonil)azepan-4-il)-1H-pirazol-5-amina (Compuesto 72, 55 mg, rendimiento: 84 %) en forma de un sólido blanquecino. EM (IEN): m/z 389,15 [M+H]+. Rm N de 1H (400 MHz, DMSO-ak) 5 11,24 (s, 1H), 6,90-6,83 (m, 2H), 5,60 (s a, 1H), 5,28 (s, 1H), 4,28 (s, 2H), 3,42-3,31 (m, 2H), 3,26-3,14 (m, 2H), 2,87 (s, 3H), 2,80-2,70 (m, 1H), 2,04-1,60 (m, 3H), 1,72-1,62 (m, 3H) ppm; Sistema de TLC: acetato de etilo al 70 % en hexano. Fr: 0,2.
Ejemplo 106-Preparación del compuesto 73
La s ín te s is de l
Compuesto 73
s ig u ió el p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5d
:
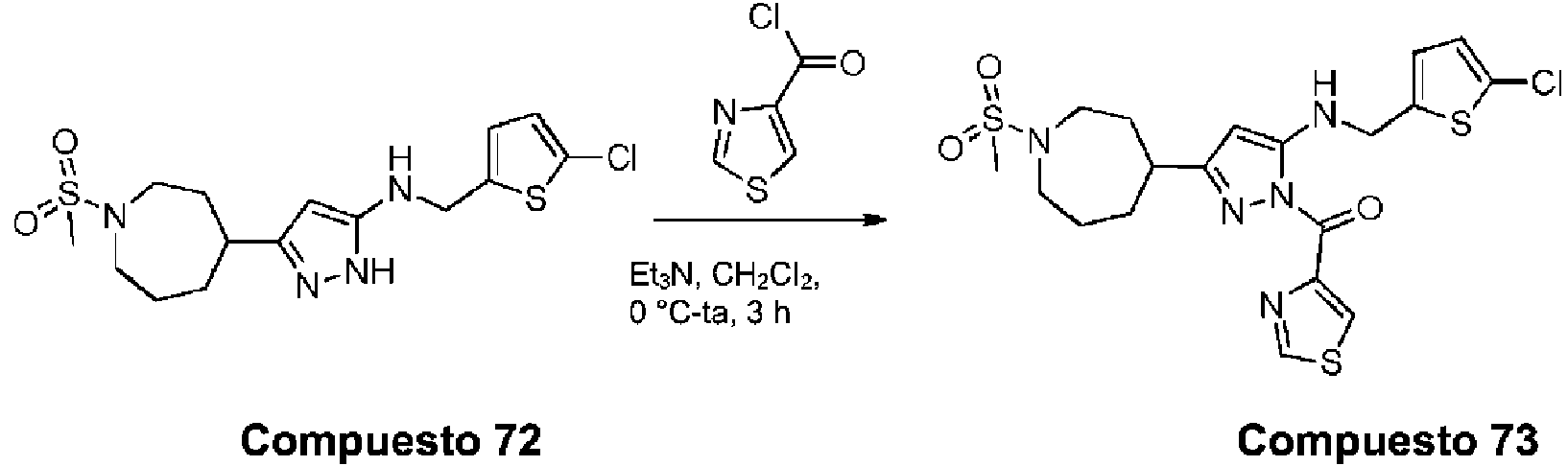
A una solución enfriada (0 0C) de W-((5-clorotiofen-2-il)metil)-3-(1-(metilsulfonil)azepan-4-il)-1H-pirazol-5-amina (Compuesto 72, 110 mg, 0,28 mmol) en diclorometano seco (10 ml), se añadió trietilamina (TEA, 0,12 ml, 0,85 mmol) seguida de cloruro de tiazol-4-carbonilo (54 mg, 0,36 mmol). Después de agitar la mezcla a temperatura ambiente durante 3 horas, se diluyó la mezcla de reacción con agua (10 ml) y se extrajo en diclorometano (3 x 10 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó por CombiFlash, eluyendo con EtOAc al 50 %/n-hexano, produciendo (5-((5-clorotiofen-2-il)metilamino)-3-(1-(metilsulfonil)azepan-4-il)-1H-pirazol-1-il)(tiazol-4-il)metanona (Compuesto 73, 60 mg, rendimiento: 41 %) en forma de un sólido blanquecino. EM (lEN): m/z 500,13 [M+H]+. RMN de 1H (400 MHz, DMSO- de) 5 9,18 (s, 1H), 9,03 (s, 1H), 7,78 (s a, 1H), 6,98 (d, J = 12,8 Hz, 2H), 5,55 (s, 1H), 4,50 (d, J = 5,2 Hz, 2H), 3,47 3,39 (m, 2H), 3,24-3,22 (m, 2H), 2,88 (s, 3H), 2,75 (s a, 1H), 2,07-1,67 (m, 6H) ppm; Sistema de TLC: acetato de etilo al 80 % en hexano. Fr: 0,7.
Ejemplo 107-Preparación del compuesto 74
La síntesis del Compuesto 74 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 °C) de 4-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)azepan-1-carboxilato de alilo (Compuesto 43, 250 mg, 0,634 mmol) en diclorometano seco (10 ml), se añadió trietilamina (TEA, 0,26 ml, 1,9 mmol), seguida de cloruro de tiazol-4-carbonilo (112 mg, 0,76 mmol). Después de agitar la mezcla a temperatura ambiente durante 3 horas, se diluyó la mezcla de reacción con agua (10 ml) y se extrajo en diclorometano (3 x 10 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó por CombiFlash, eluyendo con EtOAc al 30 %/n-hexano, después trituración con éter dietílico y pentano, produciendo 4-(5-((5-clorotiofen-2-il)metilamino)-1-(tiazol-4-carbonil)-1H-pirazol-3-il)azepan-1-carboxilato de alilo (Compuesto 74, 180 mg, rendimiento: 61 %) en forma de un sólido amarillo. m/z 506,13 [M+H]+. RMN de 1H (400 MHz, DMSO-de): 59,18 (d, J = 1,6 Hz, 1H), 9,02-9,01 (m, 1H), 7,77 (t, J = 6,0 Hz, 1H), 7,00 (d, J = 4,0 Hz, 1H), 6,97 (d, J = 3,6 Hz, 1H), 5,93-5,92 (m, 1H), 5,53 (d, J = 2,8 Hz, 1H), 5,28-5,23 (m, 1H), 5,18-5,15 (m, 1H), 4,53-4,48 (m, 4H), 3,64-3,58 (m, 1H), 3,48-3,31 (m, 3H), 2,68-2,64 (m, 1H), 2,04-1,86 (m, 3H), 1,76-1,54 (m, 3H) ppm; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,8.
Ejemplo 108-Preparación del producto intermedio 35
La síntesis del Producto intermedio 35 siguió el procedimiento del siguiente Procedimiento general 23 :

A una solución enfriada (-78 0C) de -3-oxo-8-azabiciclo[3.2.1]octan-8-carboxilato ferc-butílico (30 g, 133,3 mmol) en THF seco (300 ml), se añadió LiHMDS (hexametildisilazida de litio, 1 M en THF, 146 ml, 146,7 mmol). Después de agitar durante 30 minutos, se añadió una solución de N-fenil-bis(trifluorometanosulfonimida) (PhNTf2 , 52 g, 146,7 mmol) en THF seco (30 ml) y se agitó la mezcla a temperatura ambiente durante 5 horas. Se inactivó la mezcla de reacción con una solución saturada de cloruro de amonio (80 ml) y se extrajo con EtOAc (2 x 300 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 3 5 %/n-hexano, produciendo ferc-butil-3-(trifluorometilsulfoniloxi)-8-azabiciclo[3.2.1]oct-3-eno-8-carboxilato (Producto intermedio 35, 40 g, rendimiento: 84 %) en forma de un líquido amarillo pálido; Sistema de TLC: acetato de etilo al 20 % en hexano. Fr: 0,5.
Ejemplo 109-Preparación del producto intermedio 36
La síntesis del Producto intermedio 36 siguió el procedimiento del siguiente Procedimiento general 24:

A una solución agitada de ferc-butil-3-(trifluorometilsulfoniloxi)-8-azabiciclo[3.2.1]oct-3-eno-8-carboxilato (Producto intermedio 35, 21 g, 58,8 mmol) en 1,4-dioxano:agua (5:1, 125 ml) en un tubo sellado, se añadieron hexacarbonilo de molibdeno (Mo(cO)6, 7,7 g, 29,4 mmol), 4-dimetilaminopiridina (DMAP, 14,3 g, 117,6 mmol) y N,N- diisopropiletilamina (DIEA, 25 ml, 141,2 mmol). Después de desgasificar con una corriente de argón durante 15 minutos, se añadió acetato de paladio (Pd(OAc)2, 1,3 g, 5,9 mmol), seguido de 1,1'-¿>/s(difenilfosfin)ferroceno (dppf, 3,3 g, 5,9 mmol). Se agitó la mezcla de reacción a 120 0C durante 5 horas. Se filtró la mezcla de reacción a través de un lecho corto de Celite y se vertió en bicarbonato de sodio acuoso (60 ml) y se extrajo con EtOAc (200 ml). La capa acuosa se acidificó con HCl (2 N, a pH = 2) y se extrajo con EtOAc (2 x 200 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida, produciendo ácido 8-(ferc-butoxicarbonil)-8-azabiciclo[3.2.1]oct-3-en-3-carboxílico (Producto intermedio 36, 14 g, rendimiento: 94 %) en forma de un líquido marrón claro; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,2.
Ejemplo 110-Preparación del producto intermedio 37
La síntesis del Producto intermedio 37 siguió el procedimiento del siguiente Procedimiento general 13:
A una solución enfriada (0 0C) de 8-(ferc-butoxicarbonil)-8-azabiciclo[3.2.1]oct-3-eno-3-ácido carboxílico (Producto intermedio 36, 14 g, 55,3 mmol) en acetonitrilo (105 ml), se añadió carbonato de cesio (36 g, 110,7 mmol). Después de agitar durante 15 minutos, se añadió yodometano (Mel, 14 ml, 221,3 mmol) y después se agitó a temperatura ambiente durante 16 horas. Después de enfriar hasta 0 0C, se inactivó con agua helada (50 ml) y se extrajo con EtOAc (2 x 150 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 5-10 %/n-hexano, dando 8-ferc-butil-3-metil-8-azabiciclo[3.2.1]oct-3-eno-3,8-dicarboxilato (Producto intermedio 37, 11,8 g, rendimiento: 80 %) en forma de un sólido blanquecino; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,6.
Ejemplo 111-Preparación del producto intermedio 38
La síntesis del Producto intermedio 38 siguió el procedimiento del siguiente Procedimiento general 19:

A una solución de 8-ferc-butil-3-metil-8-azabiciclo[3.2.1]oct-3-eno-3,8-dicarboxilato (Producto intermedio 37, 11,8 g, 44,2 mmol) en MeOH (120 ml) en un recipiente de acero, se añadió Pd al 10 %/C (2,7 g) y se agitó a una presión de hidrógeno de 551,581 kPa (80 psi) a temperatura ambiente durante 16 horas. Se filtró la mezcla de reacción a través de un lecho corto de Celite y se concentraron las sustancias volátiles a presión reducida, dando 8-ferc-butil-3-metil-8-azabiciclo[3.2.1]octan-3,8-dicarboxilato (Producto intermedio 38, 11 g, rendimiento: 92 %) en forma de un sólido blanco; Sistema de TLC: acetato de etilo al 10 % en hexano. Fr: 0,5.
Ejemplo 112-Preparación del producto intermedio 39
La síntesis del Producto intermedio 39 siguió el siguiente procedimiento del Procedimiento general 6c:

A una solución enfriada (0 °C) de 8-ferc-butil-3-metil-8-azabiciclo[3.2.1]octan-3,8-dicarboxilato (Producto intermedio 38, 11 g, 40,9 mmol) en 1,4-dioxano (33 ml), se añadió HCl (4 M en 1,4-dioxano, 66 ml) a 0 °C y, a continuación, se calentó hasta la temperatura ambiente durante 3 horas. Se concentró la mezcla de reacción y se trituró con éter dietílico, produciendo clorhidrato de 8-azabiciclo[3.2.1]octan-3-carboxilato de metilo (producto intermedio 39, 8 g, rendimiento: 95 %) en forma de un sólido blanco; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,1.
Ejemplo 113-Preparación del producto intermedio 40
La síntesis del Producto intermedio 40 siguió el procedimiento del siguiente Procedimiento general 7 :

A una solución enfriada (0 °C) de clorhidrato de 8-azabiciclo[3.2.1]octan-3-carboxilato (Producto intermedio 39, 10 g, 48,8 mmol) en agua (130 ml), se añadió bicarbonato de sodio (12,3 g, 146,3 mmol), seguido de una solución de cloroformiato de alilo (5,7 ml, 53,7 mmol) en THF (60 ml). Después de agitar a temperatura ambiente durante 16 horas, se añadió agua helada (100 ml) y después se extrajo en EtOAc (2 x 150 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 15 %/n-hexano, dando 8-alil-3-metil-8-azabiciclo[3.2.1]octan-3,8-dicarboxilato (Producto intermedio 40, 8 g, rendimiento: 64 %) en forma de un líquido incoloro; Sistema de TLC: metanol al 5 %-diclorometano. Fr: 0,5.
Ejemplo 114-Preparación del producto intermedio 41
La síntesis del Producto intermedio 41 siguió el procedimiento del siguiente Procedimiento general 2 :

A una solución enfriada (-78 °C) de acetonitrilo (4,1 ml, 79,1 mmol) en THF seco (50 ml), se añadió n-BuLi (2,5 M en hexano, 28 ml, 69,2 mmol). Después de agitar durante 1 hora, una solución de 8-alil-3-metil-8-azabiciclo[3.2.1]octan-3,8-dicarboxilato (Producto intermedio 40, 5 g, 19,8 mmol) en THF seco (10 ml) y después se agitó a -78 °C durante 2 horas. Se inactivó la mezcla de reacción con solución saturada de NH4O (80 ml) y se extrajo en EtOAc (2 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 25 %/n-hexano, dando 3-(2-cianoacetil)-8-azabiciclo[3.2.1]octan-8-carboxilato de alilo (Producto intermedio 41, 2,7 g, rendimiento: 52 %) en forma de un líquido amarillo claro; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,3.
Ejemplo 115-Preparación del compuesto 74
La síntesis del Compuesto 74 siguió el procedimiento del siguiente Procedimiento general 3 :

A una solución agitada de alil-3-(2-cianoacetil)-8-azabiciclo[3.2.1]octan-8-carboxilato (Producto intermedio 41, 6 g, 22,9 mmol) en etanol (60 ml), se añadió hidrazina (1 ml, 20,6 mmol) y se calentó la mezcla de reacción a 75 °C durante 3 horas. Se enfrió la mezcla de reacción hasta la temperatura ambiente, se evaporaron las sustancias volátiles y el residuo resultante se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con MeOH al 2-3 %-diclorometano, dando alil-3-(5-amino-1 H-pirazol-3-il)-8-azabiciclo[3.2.1]octan-8-carboxilato (Compuesto 74, 4 g, rendimiento: 63 %) en forma de un semisólido de color amarillo pálido; Sistema de TLC: metanol al 5 %-diclorometano, Fr: 0,2.
Ejemplo 116-Preparación del compuesto 75
La síntesis del Compuesto 75 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución enfriada (0 °C) de alil-3-(5-amino-1 H-pirazol-3-il)-8-azabiciclo[3.2.1]octan-8-carboxilato (Compuesto 74, 3,2 g, 11,6 mmol) en MeOH seco (35 ml), se añadió 5-clorotiofen-2-carbaldehído (2,6 ml, 23,18 mmol), seguido de AcOH (0,2 ml) y, a continuación, tamices moleculares de 4 Á en polvo. Se agitó la mezcla de reacción a temperatura ambiente durante 1 hora (se observó la formación de imina como una mancha menos polar mediante TLC). A esto se añadió cianoborohidruro de sodio (NaCNBH3 , 575 mg, 9,3 mmol) poco a poco y se agitó la mezcla a temperatura ambiente durante 2 horas. A la mezcla, se añadió agua helada (50 ml), después se filtró a través de un lecho corto de Celite y el filtrado se extrajo con EtOAc (4 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con MeOH al 2-5 % en diclorometano, produciendo el producto deseado (2,1 g, rendimiento: 44 %) en forma de un sólido amarillo claro. Se purificó adicionalmente una porción (170 mg de muestra semipura) mediante HPLC preparativa, produciendo 3-(5-((5-clorotiofen-2-il)metilamino)-1 H-pirazol-3-il)-8-azabiciclo[3.2.1]octan-8-carboxilato de alilo (Compuesto 75, 50 mg) en forma de un sólido blanquecino. m/z 407,27 [M+H]+. RMN de 1H (300 MHz, DMSO-dfí) 5 = 11,28 (s, 1H, intercambiable), 6,90 (d, J = 4,0 Hz, 1H), 6,82 (d, J = 3,7 Hz, 1H), 5,93 (tdd, J = 5,3, 10,4, 17,2 Hz, 1H), 5,61 (s a, 1H, intercambiable), 5,33-5,14 (m, 3H), 4,54 (d a, J = 4,4 Hz, 2H), 4,31-4,15 (m, 4H), 3,11 (s a, 1H), 1,92 (s a, 2H), 1,82-1,57 (m, 6H) ppm; Sistema de TLC: metanol al 5 %-diclorometano, Fr: 0,4.
Ejemplo 117-Preparación del compuesto 76
La síntesis del Compuesto 76 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 0C) de 3-(5-((5-clorotiofen-2-il)metilamino)-1 H-pirazol-3-il)-8-azabiciclo[3.2.1]octan-8-carboxilato de alilo (Compuesto 75, 1 g, 2,5 mmol) en diclorometano seco (15 ml), se añadió trietilamina (TEA, 0,52 ml, 3,7 mmol), seguida de cloruro de trimetilacetilo (0,27 ml, 2,2 mmol). Después de agitar a temperatura ambiente durante 2 horas, se añadió la mezcla de reacción a agua (25 ml) y se extrajo con diclorometano (2 x 40 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con MeOH al 1-2 %-diclorometano, dando el producto deseado (900 mg, rendimiento: 75 %) en forma de un líquido amarillo claro. Se purificó adicionalmente una porción (250 mg) de material semipuro mediante HPLC preparativa, dando 3-(5-((5-clorotiofen-2-il)metilamino)-1-pivaloil-1 H-pirazol-3-il)-8-azabiciclo[3.2.1]octan-8-carboxilato alilo (Compuesto 76, 60 mg) en forma de un sólido blanco. m/z 491,21 [M+H]+. RMN de 1H (400 MHz, DMSO-de) 5 = 7,65 (t, J = 6,4 Hz, 1H, intercambiable), 6,97-6,93 (m, 2H), 5,97-5,86 (m, 1H), 5,33 (s, 1H), 5,26 (dd, J = 1,5, 17,1 Hz, 1H), 5,16 (dd, J = 1,5, 10,3 Hz, 1H), 4,61-4,47 (m, 2H), 4,40 (d, J = 5,9 Hz, 2H), 4,21 (s a, 2H), 3,11 3,01 (m, 1H), 1,93 (s a, 2 H), 1,83-1,65 (m, 6H), 1,39 (s, 9H) ppm; Sistema de TLC: metanol al 5 %-diclorometano, Fr: 0,7.
Ejemplo 118-Preparación del compuesto 77
La síntesis del Compuesto 77 siguió el siguiente procedimiento del Procedimiento general 8b :

Se desgasificó una solución agitada de 3-(5-((5-clorotiofen-2-il)metilamino)-1-pivaloil-1H-pirazol-3-il)-8-azabiciclo[3.2.1]octan-8-carboxilato de alilo (Compuesto 76, 500 mg, 1 mmol) en diclorometano (10 ml) con una corriente de argón durante 15 minutos, a continuación, se añadió a la solución tetraquis(trifenilfosfin)paladio (0) (Pd(PPh3)4 , 70 mg, 0,06 mmol) seguido de fenilsilano (PhSiH3 , 0,5 ml, 4,1 mmol). Después de agitar durante 45 minutos, se filtró la mezcla de reacción a través de un lecho corto de Celite y se evaporó el filtrado. El residuo en bruto se purificó mediante cromatografía en columna de gel de sílice eluyendo con MeOH al 5-10 %-diclorometano, dando 1-(3-(8-azabiciclo[3.2.1]octan-3-il)-5-((5-clorotiofen-2-il)metilamino)-1 H-pirazol-1-il)-2,2-dimetilpropan-1-ona (60 mg) en forma de un sólido blanquecino. m/z 407,24 [M+H]+. RMN de 1H (400 MHz, DMSO-de) 5 = 8,89 (s a, 2H, intercambiable), 7,72 (t, J = 6,1 Hz, 1H), 6,98-6,93 (m, 2H), 5,36 (s, 1H), 4,41 (d, J = 6,4 Hz, 2H), 4,00 (s a, 2H), 3,00 (tt, J = 5,9, 11,5 Hz, 1 H), 2,03-1,86 (m, 8H), 1,41 (s, 9H) ppm; Sistema de TLC: metanol al 10 %-diclorometano, Fr: 0,2.
Ejemplo 119-Preparación del compuesto 78
La síntesis del Compuesto 78 siguió el siguiente procedimiento del Procedimiento general 8b :

Se desgasificó una solución de alil-3-(5-((5-clorotiofen-2-il)metilamino)-1 H-pirazol-3-il)-8-azabiciclo[3.2.1]octan-8-carboxilato (Compuesto 75, 250 mg, 0,6 mmol) en diclorometano seco (10 ml) con una corriente de argón durante 15 minutos, a continuación, se añadió a la solución tetraquis(trifenilfosfin)paladio (0) (Pd(PPh3)4 , 42 mg, 0,03 mmol) seguido de fenilsilano (PhSiH3 , 0,3 ml, 2,5 mmol). Después de agitar a temperatura ambiente durante 45 minutos, se filtró la mezcla de reacción a través de un lecho corto de Celite y se evaporó el filtrado. El residuo en bruto se purificó mediante cromatografía en columna de gel de sílice utilizando MeOH al 10 %-diclorometano, dando el producto deseado (150 mg, rendimiento: 75 %) en forma de un sólido marrón claro. Se purificó adicionalmente mediante HPLC preparativa, dando 3-(8-azabiciclo[3.2.1]octan-3-il)-n-((5-clorotiofen-2-il)metil)-1 H-pirazol-5-amina (25 mg) en forma de un sólido amarillo claro como la sal de ácido fórmico; m/z 323,23 [M+H]+. Rm N de 1H (400 MHz, DMSO-d6) 5 = 12,89-8,82 (m, 3H, intercambiable), 8,40 (s a, 1H), 6,91 (d, J = 3,9 Hz, 1H), 6,83 (d, J = 3,4 Hz, 1H), 5,71 (s a, 1H), 5,25 (s, 1H), 4,28 (s a, 2H), 3,79 (s a, 2H), 2,97 (ddd, J = 5,9, 10,9, 17,0 Hz, 1H), 1,95-1,74 (m, 8H) ppm; Sistema de TLC: metanol al 10 %-diclorometano, Fr: 0,1.
Ejemplo 120-Preparación del compuesto 79
La síntesis del Compuesto 79 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 0C) de alil-3-(5-((5-clorotiofen-2-il)metilamino)-1 H-pirazol-3-il)-8-azabiciclo[3.2.1]octan-8-carboxilato (Compuesto 75, 300 mg, 0,73 mmol) en diclorometano seco (15 ml), se añadió trietilamina (TEA, 0,26 ml, 1,84 mmol), y a continuación, cloruro de 1,3-tiazol-4-carbonilo (218 mg, 1,47 mmol). Después de agitar a temperatura ambiente durante 2 horas, se añadió agua helada (10 ml) y se extrajo con diclorometano (2 x 20 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida utilizando MeOH al 1-2 % en diclorometano, dando el producto deseado (300 mg, rendimiento: 78 %) en forma de un líquido marrón claro. Se purificó adicionalmente una porción mediante HPLC preparativa, dando alil-3-(5-((5-clorotiofen-2-il)metilamino)-1-(tiazol-4-carbonil)-1 H-pirazol-3-il)-8-azabiciclo[3,2,1]octan-8-carboxilato (Compuesto 79, 75 mg) en forma de un sólido amarillo pálido. m/z 518,18 [M+H]+. RMN de 1H (400 MHz, DMSO-de) 5 = 9,19 (d, J = 2,0 Hz, 1H), 9,02 (d, J = 2,0 Hz, 1H), 7,79 (t, J = 6,4 Hz, 1H), 7,00-6,95 (m, 2H), 5,93 (tdd, J = 5,3, 10,4, 17,2 Hz, 1H), 5,50 (s, 1H), 5,27 (dd, J = 1,5, 17,1 Hz, 1H), 5,16 (dd, J = 1,5, 10,3 Hz, 1H), 4,57-4,47 (m, 4H), 4,23 (s a, 2H), 3,16-3,08 (m, 1H), 1,96-1,66 (m, 8H) ppm; Sistema de TLC: metanol al 5 %-diclorometano. Fr: 0,7.
Ejemplo 121-Preparación del compuesto 80
La síntesis del Compuesto 80 siguió el procedimiento del siguiente Procedimiento general 15:

A una solución enfriada (0 0C) de 1-(3-(8-azabiciclo[3.2.1]octan-3-il)-5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 77, 350 mg, 0,86 mmol) en diclorometano seco (10 ml), se añadió trietilamina (TEA, 0,3 ml, 2,15 mmol) y, a continuación, cloruro de morfolin-4-carbonilo (0,2 ml, 1,72 mmol). Después de agitar a temperatura ambiente durante 2 horas, se añadió agua helada (5 ml) y se lavó con diclorometano (2 x 20 ml). Las capas orgánicas combinadas se desecaron con sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida utilizando MeOH al 1-2 % en diclorometano, dando el producto semipuro deseado (320 mg, rendimiento: 71 %) en forma de un líquido marrón claro. Se purificó
adicionalmente una porción (120 mg) mediante HPLC preparativa, produciendo 1-(5-((5-clorotiofen-2-il)metilamino)-3-(8-(morfolin-4-carbonil)-8-azabiciclo[3.2.1]octan-3-il)-1 H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 80, 60 mg) en forma de un sólido blanquecino. m/z 520,25 [M+H]+. r Mn de 1H (400 MHz, DMSO-d6) 5 = 7,65 (t, J = 6,1 Hz, 1H, intercambiable), 6,97-6,93 (m, 2H), 5,35 (s, 1H), 4,40 (d, J = 6,4 Hz, 2H), 4,03 (s a, 2H), 3,58-3,52 (m, 4H), 3,25-3,18 (m, 4H), 2,93 (tt, J = 5,7, 11,7 Hz, 1H), 1,85-1,66 (m, 8H), 1,39 (s, 9H) ppm; Sistema de TLC: metanol al 5 %-diclorometano. Fr: 0,6.
Ejemplo 122-Preparación del compuesto 81
La síntesis del Compuesto 81 siguió el procedimiento del siguiente Procedimiento general 6d :

A una solución de 1-(5-((5-clorotiofen-2-il)metilamino)-3-(8-(morfolin-4-carbonil)-8-azabiciclo[3.2.1]octan-3-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 80, 140 mg, 0,26 mmol) en metanol (5 ml) a temperatura ambiente, se añadió bicarbonato de sodio (113 mg, 1,34 mmol). Después de agitar a 50 0C durante 4 horas, se concentró la mezcla de reacción y se lavó con diclorometano (2 x 15 ml). Las capas orgánicas combinadas se desecaron con sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante HPLC preparativa, dando (3-(5-((5-clorotiofen-2-il)metilamino)-1 H-pirazol-3-il)-8-azabiciclo[3.2.1]octan-8-il)(morfolino)metanona (Compuesto 81, 30 mg) en forma de un sólido blanquecino. m/z 436,22 [M+H]+. Rm N de 1H (400 MHz, DMSO-de) 5 = 11,25 (s, 1H), 6,90 (d, J = 3,4 Hz, 1H), 6,82 (d a, J = 2,9 Hz, 1H), 5,59 (s a, 1H, intercambiable), 5,26 (s a, 1H), 4,27 (d, J = 6,4 Hz, 2H), 4,01 (s a, 2H), 3,59-3,52 (m, 4H), 3,27-3,19 (m, 4H), 2,99 (s a, 1H), 1,84-1,64 (m, 8H) ppm; Sistema de TLC: metanol al 5 %-diclorometano. Fr: 0,2.
Ejemplo 123-Preparación del compuesto 82
La síntesis del Compuesto 82 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 0C) de 3-(5-((5-clorotiofen-2-il)metilamino)-1 H-pirazol-3-il)-8-azabiciclo[3.2.1]octan-8-il)(morfolino)metanona (Compuesto 81, 175 mg, 0,4 mmol) en diclorometano seco (10 ml), se añadió trietilamina (TEA, 0,14 ml, 1 mmol), seguida de cloruro de 1,3-tiazol-4-carbonilo (119 mg, 0,8 mmol). Después de agitar a temperatura ambiente durante 2 horas, se añadió agua helada (5 ml) y después se lavó con diclorometano (2 x 15 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna ultrarrápida, eluyendo con MeOH al 1 2 % en diclorometano, dando el producto deseado (165 mg, rendimiento: 75 %) en forma de un líquido marrón claro. Se purificó adicionalmente mediante HPLC preparativa, dando (3-(5-((5-clorotiofen-2-il)metilamino)-1-(tiazol-4-carbonil)-1 H-pirazol-3-il)-8-azabiciclo[3.2.1]octan-8-il)(morfolino)metanona (Compuesto 82, 55 mg) en forma de un sólido blanquecino. m/z 547,23 [M+H]+. RMN de 1H (400 MHz, DMSO-de) 5 = 9,19 (d, J = 2,0 Hz, 1H), 9,04 (d, J = 2,0 Hz, 1H), 7,78 (t a, J = 6,4 Hz, 1H, intercambiable), 6,98 (c, J = 3,9 Hz, 2H), 5,52 (s, 1H), 4,50 (d a, J = 5,9 Hz, 2H), 4,04 (s a, 2H), 3,58-3,53 (m, 4H), 3,25-3,21 (m, 4H), 2,98 (dd a, J = 5,9, 11,7 Hz, 1H), 1,86-1,69 (m, 8H) ppm; Sistema de TLC: metanol al 5 %-diclorometano. Fr: 0,5.
Ejemplo 124-Preparación del compuesto 83
La síntesis del Compuesto 83 siguió el procedimiento del siguiente Procedimiento general 22:

A una solución enfriada (0 0C) de 1-(3-(8-azabiciclo[3.2.1]octan-3-il)-5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 77, 350 mg, 0,86 mmol) en diclorometano seco (10 ml), se añadió trietilamina (TEA, 0,3 ml, 2,15 mmol), seguida de cloruro de metanosulfonilo (0,1 ml, 1,3 mmol). Después de agitar a temperatura ambiente durante 2 horas, se añadió agua helada (5 ml) y se lavó con diclorometano (2 x 20 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna ultrarrápida utilizando MeOH al 1 %-diclorometano, dando 1-(5-((5-clorotiofen-2-il)metilamino)-3-(8-(metilsulfonil)-8-azabiciclo[3.2.1]octan-3-il)-1 H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 83, 310 mg, rendimiento: 74 %) en forma de un sólido blanquecino. m/z 485,15 [M+H]+. RMN de 1H (400 MHz, DMSO-de) 5 = 7,67 (t a, J = 6,1 Hz, 1H, intercambiable), 6,96 (s, 2H), 5,36 (s, 1H), 4,41 (d, J = 5,9 Hz, 2H), 4,18 (s a, 2H), 3,00-2,90 (m, 4H), 2,05-1,96 (m, 2H), 1,89-1,69 (m, 6H), 1,40 (s, 9H) ppm; Sistema de TLC: metanol al 5 %-diclorometano. Fr: 0,6.
Ejemplo 125-Preparación del compuesto 84
La síntesis del Compuesto 84 siguió el procedimiento del siguiente Procedimiento general 6d :

A una solución agitada de 1-(5-((5-clorotiofen-2-il)metilamino)-3-(8-(metilsulfonil)-8-azabiciclo[3.2.1]octan-3-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 83, 145 mg, 0,3 mmol) en metanol (5 ml) a temperatura ambiente, se añadió bicarbonato de sodio (126 mg, 1,5 mmol). Después de agitar a 50 0C durante 4 horas, se concentró la mezcla de reacción y se extrajo en diclorometano (2 x 15 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante HPLC preparativa, dando n-((5-clorotiofen-2-il)metil)-3-(8-(metilsulfonil)-8-azabiciclo[3.2.1]octan-3-il)-1 H-pirazol-5-amina (Compuesto 84, 25 mg) en forma de un sólido blanquecino. m/z 401,20 [M+H]+. RMN de 1H (400 mHz, DMSO-de) 5 = 11,28 (s, 1H), 6,90 (d, J = 3,9 Hz, 1H), 6,83 (d a, J = 3,4 Hz, 1H), 5,62 (s a, 1H, intercambiable), 5,27 (s a, 1H), 4,27 (d, J = 6,4 Hz, 2H), 4,16 (s a, 2H), 2,94 (s, 4H), 2,05-1,97 (m, 2H), 1,86-1,74 (m, 4H), 1,73-1,64 (m, 2H) ppm; Sistema de TLC: metanol al 5 %-diclorometano. Fr: 0,2.
Ejemplo 126-Preparación del compuesto 85
La síntesis del Compuesto 85 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 0C) de W-((5-clorotiofen-2-il)metil)-3-(8-(metilsulfonil)-8-azabiciclo[3.2.1]octan-3-il)-1 H- pirazol-5-amina (Compuesto 84, 170 mg, 0,42 mmol) en diclorometano seco (10 ml), se añadió trietilamina (TEA, 0,15 ml, 1,06 mmol) y, a continuación, cloruro de 1,3-tiazol-4-carbonilo (125 mg, 0,85 mmol). Después de agitar a temperatura ambiente durante 2 horas, se añadió agua helada (5 ml) y después se lavó con diclorometano (2 x 15 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna ultrarrápida, eluyendo con MeOH al 1 2 % en diclorometano, dando el producto deseado (170 mg, rendimiento: 78 %) en forma de un líquido marrón claro. Se purificó adicionalmente mediante HPLC preparativa, dando 50 mg de (5-((5-clorotiofen-2-il)metilamino)-3-(8-(metilsulfonil)-8-azabiciclo[3.2.1]octan-3-il)-1H-pirazol-1-il)(tiazol-4-il)metanona pura (Compuesto 85, 50 mg) en forma de un sólido amarillo pálido. m/z 512,14 [M+H]+. RMN de 1H (400 MHz, DMSO-de) 5 = 9,19 (s, 1H), 9,04 (s, 1H), 7,80 (s a, 1H), 7,19 (s a, 1H), 7,05-6,93 (m, 2H), 5,54 (s, 1H), 4,50 (d a, J = 5,4 Hz, 2H), 4,19 (s a, 2H), 3,07 2,90 (m, 4H), 2,01 (s a, 2H), 1,92-1,73 (m, 6H) ppm; Sistema de TLC: metanol al 5 %-diclorometano. Fr: 0,5.
Ejemplo 127-Preparación del producto intermedio 42
La síntesis del Producto intermedio 42 siguió el procedimiento del siguiente Procedimiento general 7 :

A una solución enfriada (0 0C) de clorhidrato de azetidin-3-carboxilato de metilo (10 g, 66 mmol) en THF/H2O (3:6, 100 ml), se añadió bicarbonato de sodio (16,6 g, 198 mmol) poco a poco, seguido de cloroformiato de alilo (7,73 ml, 72,6 mmol). Después de agitar a temperatura ambiente durante 5 horas, la mezcla se extrajo en EtOAc (3 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con MeOH al 2 %/diclorometano, dando 3-metil-azetidin-1,3-dicarboxilato de 1 -alilo (Producto intermedio 42, 11 g, rendimiento: 83 %) en forma de un jarabe incoloro. m/z 200,03 [M+H]+; Sistema de TLC: metanol al 5 %-diclorometano; Fr: 0,7.
Ejemplo 128-Preparación del producto intermedio 43
La síntesis del Producto intermedio 43 siguió el procedimiento del siguiente Procedimiento general 2 :

A una solución enfriada (-78 0C) de acetonitrilo (3,1 ml, 60,3 mmol) en THF seco (60 ml), se añadió n-BuLi (2,5 M en THF, 24,1 ml, 60,3 mmol), después se agitó durante 1 hora y, a continuación, se añadió una solución de 1 -alil-3-metil azetidin-1,3-dicarboxilato (Producto intermedio 42, 10 g, 50,3 mmol) en THF seco (20 ml). Después de agitar a -78 °C durante 1 hora, Se inactivó la mezcla con solución saturada de NH4Cl (20 ml) y se extrajo en EtOAc (2 x 120 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 50 %/n-hexano, produciendo el 3-(2-cianoacetil)azetidin-1-carboxilato de alilo deseado (Producto intermedio 43, 5,1 g, 49 %) en forma de un líquido amarillo claro; Sistema de TLC: acetato de etilo al 60 % en hexano. Fr: 0,3.
Ejemplo 129-Preparación del compuesto 86
La s ín te s is de l
Compuesto 86
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 3
:

NC e uo,
Producto intermedio 43 Compuesto 86
A una solución de 3-(2-cianoacetil)azetidin-1-carboxilato de alilo (Producto intermedio 43, 5 g, 24 mmol) en etanol (30 ml), se añadió hidrato de hidrazina (N2H4.H2O, 1,2 ml, 24 mmol) y, a continuación, se calentó la mezcla de reacción a 80 0C durante 5 horas. La mezcla de reacción se enfrió hasta la temperatura ambiente y se evaporaron las sustancias volátiles. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 1 0 0 -2 0 0 ), eluyendo con MeOH al 4 %-diclorometano, produciendo 3-(5-amino-1H-pirazol-3-il)azetidin-1-carboxilato de alilo (Compuesto 86, 3,6 g, 68 %) en forma de un líquido naranja; m/z 223,11 [M+H]+; Sistema de TLC: metanol al 5 %-diclorometano. Fr: 0,3.
Ejemplo 130-Preparación del compuesto 87
La síntesis del Compuesto 87 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución enfriada (0 0C) de 3-(5-amino-1H-pirazol-3-il)azetidin-1-carboxilato de alilo (Compuesto 86, 3,5 g, 15,8 mmol) en EtOH seco (40 ml), se añadieron 5-clorotiofen-2-carbaldehído (1,9 ml, 18,9 mmol), ácido acético (0,5 ml) y, a continuación, tamices moleculares de 4 Á en polvo (1 g). Se agitó la mezcla de reacción a temperatura ambiente durante 1 hora. (La formación de imina se observó como una mancha menos polar en TLC). A continuación, se añadió a la mezcla cianoborohidruro de sodio (1,19 g, 18,9 mmol) poco a poco y se siguió agitando a temperatura ambiente durante 2 horas. Se inactivó la mezcla de reacción con agua helada (50 ml), se filtró a través de un lecho corto de Celite y el filtrado se extrajo en EtOAc (3 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con MeOH al 5 %/diclorometano, produciendo 3-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)azetidin-1-carboxilato de alilo (Compuesto 87, 3,2 g, 58 %) en forma de un líquido gomoso de color amarillo claro; m/z 353,17 [M+H]+. RMN de 1H (400 MHz, DMSO-d6) 5 11,54 (s a, 1H), 6,97-6,79 (m, 2H), 5,98-5,84 (m, 1H), 5,73 (s a, 1H), 5,47 (s a, 1H), 5,28 (dd, J = 1,5, 17,1 Hz, 1H), 5,19 (dd, J = 1 ,2 , 10,5 Hz, 1 H), 4,50 (d, J = 5,4 Hz, 2 H), 4,36-4,10 (m, 4H), 3,97-3,64 (m, 3h) ppm; Sistema de TLC: metanol al 5 % en diclorometano, Fr: 0,4.
Ejemplo 131-Preparación del compuesto 88
La síntesis del Compuesto 88 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 °C) de 3-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)azetidin-1-carboxilato de alilo (Compuesto 87, 3 g, 8,5 mmol) en diclorometano seco (45 ml), se añadió trietilamina (TEA, 1,84 ml, 12,8 mmol),
seguida de cloruro de trimetilacetilo (1,0 ml, 8,5 mmol). Después de agitar a temperatura ambiente durante 2 horas, se diluyó la mezcla de reacción con agua (50 ml) y se extrajo en diclorometano (3 x 50 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 15 %/hexanos, produciendo 3-(5-((5-clorotiofen-2-il)metilamino)-1-pivaloil-1H-pirazol-3-il)azetidin-1-carboxilato de alilo (Compuesto 88, 1,9 g, 51 %) en forma de un líquido incoloro. EM (IEN): m/z 437,44 [M+H]+. RMN de 1H (400 MHz, DMsO-d6) 5 7,75 (t, J = 6,1 Hz, 1H), 6,96 (c, J = 3,4 Hz, 2H), 5,90 (tdd, J = 5,3, 10,4, 17,1 Hz, 1H), 5,52 (s, 1H), 5,31 -5,23 (m, 1H), 5,18 (dd, J = 1,5, 10,8 Hz, 1H), 4,53-4,47 (m, 2H), 4,43 (d, J = 5,9 Hz, 2H), 4,24 (s a, 2H), 3,96 (s a, 2H), 3,70 (tt, J = 5,9, 8,8 Hz, 1H), 1,41 (s, 9H) ppm; Sistema de TLC: acetato de etilo al 30 % en hexano. Fr: 0,6.
Ejemplo 132-Preparación del compuesto 89
La síntesis del Compuesto 89 siguió el siguiente procedimiento de los Procedimientos Generales 8b y 15:

Una solución enfriada (0 0C) de 3-(5-((5-clorotiofen-2-il)metilamino)-1-pivaloil-1H-pirazol-3-il)azetidin-1-carboxilato de alilo (Compuesto 88, 600 mg, 1,4 mmol) en diclorometano (30 ml) se desgasificó con una corriente de argón durante 15 minutos, a continuación, se añadió fenilsilano (PhSiH3 , 0,5 ml, 4,1 mmol), seguido de tetraquis(trifenilfosfin)paladio (0) (Pd(PPh3)4, 79,4 mg, 0,068 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 3 horas. Una vez consumido por completo el material de partida (controlado mediante TLC), se volvió a enfriar la mezcla hasta 0 0C y se añadió trietilamina (TEA, 0,39 ml, 2,75 mmol), seguida de cloruro de morfolin-4-carbamilo (0,16 ml, 1,4 mmol) y se agitó la mezcla a temperatura ambiente durante 3 horas. Se diluyó la mezcla de reacción con agua (20 ml) y se extrajo con diclorometano (2 x 20 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna CombiFlash, eluyendo con MeOH al 2 %/diclorometano, dando 1-(5-((5-clorotiofen-2-il)metilamino)-3-(1 -(morfolin-4-carbonil)azetidin-3-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona (Compuesto 89, 200 mg, 31 % en dos etapas) en forma de un sólido blanquecino. EM (IEN) m/z 466,49 [M+H]+. RMN de 1H (400 MHz, DMSO-da) 5 7,75 (t, J = 6,1 Hz, 1H), 6,96 (s, 2H), 5,47 (s, 1H), 4,43 (d, J = 5,9 Hz, 2H), 4,20 (t, J = 8,3 Hz, 2H), 3,94 (dd, J = 6,1, 7,6 Hz, 2H), 3,64 (ddd, J = 6,4, 8,7, 14,8 Hz, 1H), 3,57-3,45 (m, 4H), 3,25-3,14 (m, 4H), 1,40 (s, 9H) ppm; Sistema de TLC: metanol al 5 % en diclorometano, Fr: 0,6.
Ejemplo 133-Preparación del compuesto 90
La síntesis del Compuesto 90 siguió el procedimiento del siguiente Procedimiento general 6d :

A una solución de 1-(5-((5-clorotiofen-2-il)metilamino)-3-(1-(morfolin-4-carbonil)azetidin-3-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 89, 200 mg, 0,43 mmol) en metanol (5 ml), se añadió bicarbonato de sodio (NaHCO3 , 108 mg, 1,3 mmol). Tras agitar la mezcla de reacción a 50 0C durante 4 horas, la mezcla de reacción se enfrió hasta la temperatura ambiente, se evaporaron las sustancias volátiles, y el residuo resultante se diluyó con EtOAc (20 ml) y se lavó con agua (2 x 10 ml). Las capas orgánicas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El sólido resultante se trituró con éter dietílico y n-pentano, y se desecó a alto vacío durante 1 hora, produciendo (3-(5-((5-clorotiofen-2-il)metilamin)-1H-pirazol-3-il)azetidin-1-il)(morfolino)metanona (Compuesto 90, 150 mg, 92 %) en forma de un sólido gomoso de color amarillo pálido; Sistema de TLC: metanol al 5 % en diclorometano, Fr: 0,3.
Ejemplo 134-Preparación del compuesto 91
La síntesis del Compuesto 91 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 0C) de (3-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)azetidin-1-il)(morfolino)metanona (Compuesto 90, 120 mg, 0,31 mmol) en diclorometano seco (5 ml), se añadió trietilamina (TEA, 0,09 ml, 0,63 mmol), seguida de cloruro de tiazol-4-carbonilo (51 mg, 0,35 mmol). Después de agitar a temperatura ambiente durante 3 horas, se diluyó la mezcla de reacción con agua (10 ml) y se extrajo en diclorometano (3 x 20 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna CombiFlash, eluyendo con acetona/diclorometano al 20 %, dando (3-(5-((5-clorotiofen-2-il)metilamino)-1-(tiazol-4-carbonil)-1H-pirazol-3-il)azetidin-1-il)(morfolino)metanona (Compuesto 91, 50 mg, 32 %) en forma de un sólido amarillo pálido. m/z 493,16 [MH]+. RMN de 1H (400 MHz, DMSO-de) 59,20 (d, J = 1,5 Hz, 1H), 9,04 (s, 1H), 7,87 (t, J = 6,1 Hz, 1H), 6,99 (dd, J = 3,4, 13,2 Hz, 2H), 5,65 (s, 1H), 4,53 (d, J = 6,4 Hz, 2H), 4,22 (t, J = 8,6 Hz, 2H), 3,98 (t, J = 6,8 Hz, 2H), 3,75-3,64 (m, 1H), 3,59-3,49 (m, 4H), 3,26-3,17 (m, 4H) ppm; Sistema de TLC: acetona al 30 % en diclorometano, Fr: 0,6.
Ejemplo 135-Preparación del compuesto 92
La síntesis del Compuesto 92 siguió el siguiente procedimiento del Procedimiento general 8b :

Se desgasificó una solución enfriada (0 0C) de 3-(5-((5-clorotiofen-2-il)metilamino)-1-pivaloil-1H-pirazol-3-il)azetidin-1-carboxilato de alilo (Compuesto 88, 100 mg, 0,23 mmol) en diclorometano (10 ml) con una corriente de argón durante 15 minutos, después se añadió fenilsilano (PhSiH3 , 0,075 ml, 0,69 mmol) seguido de tetraquis(trifenilfosfin)paladio (0) (Pd(PPh3)4 , 13 mg, 0,01 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 3 horas, a continuación, se filtró a través de un lecho corto de Celite y se evaporó el filtrado. El residuo se purificó mediante cromatografía ultrarrápida, eluyendo con MeOH al 4 %-diclorometano, dando 1-(3-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)azetidin-1-il)-2,2-dimetilpropan-1-ona (Compuesto 92, 30 mg, 37 %) en forma de un líquido gomoso de color amarillo pálido. EM (IEN) m/z 353,27 [M+H]+; RMN de 1H (400 MHz, DMSO-d6) 5 11,54 (s, 1H), 7,01-6,70 (m, 2H), 5,71 (s a, 1H), 5,48 (s, 1H), 4,65 (s, 1H), 4,40-3,97 (m, 4H), 3,71 (s, 2H), 1,30 0,83 (s, 9H) ppm; Sistema de TLC: MeOH al 5 % en diclorometano, Fr: 0,4.
Ejemplo 136-Preparación del compuesto 93
La s ín te s is de l
Compuesto 93
s ig u ió los p ro ce d im ie n to s de los
Procedimientos generales 8b y 5d
:

Se desgasificó una solución enfriada (0 0C) de alilo 3-(5-((5-clorotiofen-2-il)metilamino)-1-pivaloil-1H-pirazol-3-il)azetidin-1-carboxilato (Compuesto 88, 200 mg, 0,46 mmol) en diclorometano (15 ml) con una corriente de argón durante 15 minutos, después se añadió fenilsilano (PhSiH3 , 0,16 ml, 1,38 mmol) seguido de tetraquis(trifenilfosfin)paladio (0) (Pd(PPh3)4 , 26 mg, 0,02 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 3 horas. Una vez consumido por completo el material de partida (controlado mediante TLC), la mezcla se enfrió hasta 0 0C y se añadió trietilamina (TEA, 0,16 ml, 1,1 mmol), seguida de cloruro de pivaloílo (0,06 ml, 0,46 mmol). Después de agitar a temperatura ambiente durante 2 horas, se diluyó la mezcla de reacción con agua (10 ml) y se extrajo en diclorometano (3 x 15 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna CombiFlash, eluyendo con EtOAc al 20 %/hexano, produciendo 1-(3-(5-((5-clorotiofen-2-il)metilamino)-1-pivaloil-1H-pirazol-3-il)azetidin-1-il)-2,2-dimetilpropan-1-ona (Compuesto 93, 40 mg, 20 %; en dos etapas) en forma de un material gomoso de color amarillo pálido. m/z: 437,21 [M+H]+. RMN de 1H (400 MHz, DMSO-da) 5 7,72 (t, J = 6,4 Hz, 1H), 6,94-6,90 (m, 2H), 5,44 (s, 1H), 4,63 (s, 1H), 4,43-4,28 (m, 3H), 4,10 (s, 1H), 3,79 (s, 1H), 3,63-3,58 (m, 1H), 1,37 (s, 9H), 1,07 (s, 9H) ppm; Sistema de TLC: acetato de etilo al 30 % en hexano, Fr: 0,4.
Ejemplo 137-Preparación del compuesto 94
La síntesis del Compuesto 94 siguió los procedimientos de los Procedimientos Generales 8b y 22:

Se desgasificó una solución enfriada (0 0C) de 3-(5-((5-clorotiofen-2-il)metilamino)-1-pivaloil-1H-pirazol-3-il)azetidin-1-carboxilato de alilo (Compuesto 88, 550 mg, 1,3 mmol) en diclorometano (25 ml) con una corriente de argón durante 15 minutos, después se añadió fenilsilano (PhSiH3 , 0,46 ml, 3,8 mmol) seguido de tetraquis(trifenilfosfin)paladio (0) (Pd(PPh3)4 , 72 mg, 0,06 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 3 horas. Una vez consumido por completo el material de partida (controlado mediante TLC), la mezcla se enfrió hasta 0 0C y se añadió trietilamina (TEA, 0,35 ml, 2,52 mmol), seguida de cloruro de metanosulfonilo (MsCl, 0,1 ml, 1,3 mmol). Se agitó la mezcla a temperatura ambiente durante 2 horas. Se diluyó la mezcla de reacción con agua (20 ml) y se extrajo con diclorometano (3 x 20 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna CombiFlash, eluyendo con diclorometano al 70 %/hexano, produciendo 1-(5-((5-clorotiofen-2-il)metilamino)-3-(1 -(metilsulfonil)azetidin-3-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona (Compuesto 94, 160 mg, 29 % en dos etapas) en forma de un sólido blanquecino; m/z 431,17 [M+H]+; RMN de 1H (400 MHz, DMSO-da) 57,78 (t, J = 6,1 Hz, 1H), 7,01-6,93 (m, 2H), 5,51 (s, 1H), 4,44 (d, J = 6,4 Hz, 2H), 4,18-4,08 (m, 2H), 3,96 (t, J = 7,6 Hz, 2H), 3,78-3,65 (m, 1H), 3,02 (s, 3h ), 1,42 (s, 9H) ppm; Sistema de TLC: diclorometano, Fr: 0,4.
Ejemplo 138-Preparación del compuesto 95
La s ín te s is de l
Compuesto 95
s ig u ió el p ro ce d im ie n to 6 de
Procedimiento general 6d
:

A una solución de 1-(5-((5-clorotiofen-2-il)metilamino)-3-(1-(metilsulfonil)azetidin-3-il)-1 H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 94, 140 mg, 0,33 mmol) en metanol (5 ml), se añadió bicarbonato de sodio (NaHCO3 , 82 mg, 0,98 mmol) y se agitó la mezcla de reacción a temperatura ambiente durante 4 horas. Se evaporaron las sustancias volátiles y el residuo resultante se diluyó con EtOAc (20 ml) y se lavó con agua (2 x 10 ml). La capa orgánica se desecó sobre sulfato de sodio, se filtró y se concentró a presión reducida. El residuo se trituró con éter dietílico y n-pentano, y se desecó a alto vacío, produciendo W-((5-clorotiofen-2-il)metil)-3-(1-(metilsulfonil)azetidin-3-il)-1 H-pirazol-5-amina (Compuesto 95, 90 mg, 80 %) en forma de un sólido amarillo pálido; m/z 347,19 [M+H]+; RMN de 1H (400 MHz, DMSO-da) 5 11,54 (s a, 1H), 6,96-6,78 (m, 2H), 5,85-5,44 (m, 2H), 4,30 (d, J = 6,4 Hz, 2H), 4,10 (t, J = 7,8 Hz, 2H), 3,87 (t, J = 7,3 Hz, 2H), 3,73 (s a, 1H), 3,03 (s, 3H) ppm; Sistema de TLC: metanol al 5 % en diclorometano, Fr: 0,4.
Ejemplo 139-Preparación del compuesto 96
La síntesis del Compuesto 96 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 0C) de W-((5-clorotiofen-2-il)metil)-3-(1-(metilsulfonil)azetidin-3-il)-1H-pirazol-5-amina (Compuesto 95, 70 mg, 0,202 mmol) en diclorometano seco (4 ml), se añadió trietilamina (TEA, 0,07 ml, 0,51 mmol), seguida de cloruro de tiazol-4-carbonilo (32,7 mg, 0,22 mmol). A continuación, se agitó la mezcla a temperatura ambiente durante 3 horas. Se diluyó la mezcla de reacción con agua (10 ml) y se extrajo en diclorometano (3 x 15 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo resultante se purificó mediante cromatografía en columna CombiFlash, eluyendo con acetona al 5 %/diclorometano, produciendo (5-((5-clorotiofen-2-il)metilamino)-3-(1-(metilsulfonil)azetidin-3-il)-1H-pirazol-1-il)(tiazol-4-il)metanona (Compuesto 96, 50 mg, 54 %) en forma de un sólido amarillo pálido; m/z 458,14 [M+H]+; RMN de 1H (400 MHz, DMSO-d6) 59,20 (d, J = 2,0 Hz, 1H), 9,08 (d, J = 2,0 Hz, 1H), 7,91 (t, J = 6,1 Hz, 1H), 7,07-6,85 (m, 2H), 5,66 (s, 1H), 4,53 (d, J = 5,9 Hz, 2H), 4,21-4,11 (m, 2H), 4,02 (t, J = 7,3 Hz, 2H), 3,83-3,72 (m, 1H), 3,03 (s, 3H) ppm; Sistema de TLC: acetona al 10 % en diclorometano, Fr: 0,5.
Ejemplo 140-Preparación del compuesto 97
La s ín te s is de l
Compuesto 97
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5d
:

A una solución enfriada (0 °C) de 3-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)azetidin-1-carboxilato de alilo (Compuesto 87, 100 mg, 0,28 mmol) en diclorometano seco (5 ml), se añadió trietilamina (TEA, 0,1 ml, 0,71 mmol), seguida de cloruro de tiazol-4-carbonilo (41,8 mg, 0,28 mmol). Se agitó la mezcla a temperatura ambiente durante 3 horas. Se diluyó la mezcla de reacción con agua (10 ml) y se extrajo en diclorometano (3 x 15 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna CombiFlash, eluyendo con MeOH al 2 %/diclorometano, produciendo 3-(5-((5-clorotiofen-2-il)metilamino)-1 -(tiazol-4-carbonil)-1 H-pirazol-3-il)azetidin-1 -carboxilato de alilo (Compuesto 97, 60 mg, 45 %) en forma de un sólido amarillo pálido; m/z 464,15 [M+H]+. RMN de 1H (400 MHz, DMSO-afe) 59,20 (d, J = 1,5 Hz, 1H), 9,03 (d, J = 2,0 Hz, 1H), 7,88 (t, J = 6,4 Hz, 1H), 7,02 (d, J = 3,4 Hz, 1H), 6,97 (d, J = 3,9 Hz, 1H), 5,98-5,84 (m, 1H), 5,70 (s, 1H), 5,27 (dd, J = 1,7, 17,4 Hz, 1H), 5,18 (dd, J = 1,5, 10,3 Hz, 1H), 4,51 (dd, J = 5,9, 10,3 Hz, 4H), 4,25 (s a, 2H), 4,00 (s a, 2H), 3,76 (tt, J = 6,2, 8,7 Hz, 1H) ppm; Sistema de TLC: MeOH al 5 % en diclorometano, Fr: 0,6.
Ejemplo 141-Preparación del producto intermedio 44
La síntesis del Producto intermedio 44 siguió el procedimiento del siguiente Procedimiento general 13:

A una solución enfriada (0 0C) de ácido 1-(ferc-butoxicarbonil)pirrolidin-2-carboxílico (20 g, 92,9 mmol) en dimetilformamida seca (DMF, 100 ml), se añadió carbonato de cesio (Cs2CO3 , 103 g, 316 mmol) seguido de yodometano (Mel, 12 ml, 186 mmol). Después de agitar a temperatura ambiente durante 16 horas, se añadió agua (100 ml) y se extrajo en EtOAc (2 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 10-15 %/n-hexanos, obteniéndose 1-ferc-butil-2-metil pirrolidin-1,2-dicarboxilato (Producto intermedio 44, 19 g, rendimiento: 90 %) en forma de un líquido amarillo pálido; Sistema de TLC: acetato de etilo al 20 % en hexano, Fr: 0,5.
Ejemplo 142-Preparación del producto intermedio 45
La síntesis del Producto intermedio 45 siguió el siguiente procedimiento del Procedimiento general 6c:

A una solución enfriada (0 0C) de 1-ferc-butil-2-metil pirrolidin-1,2-dicarboxilato (Producto intermedio 44, 19 g, 83 mmol) en 1,4-dioxano (120 ml), se añadió HCl (4 M en 1,4-dioxano, 95 ml) y, a continuación, la reacción se agitó a temperatura ambiente durante 5 horas. Se concentró la mezcla de reacción a presión reducida y se trituró con éter dietílico, produciendo clorhidrato de pirrolidin-2-carboxilato de metilo (producto intermedio 45, 12 g, rendimiento: 88 %) en forma de un sólido blanco.
Ejemplo 143-Preparación del producto intermedio 46
La síntesis del Producto intermedio 46 siguió el procedimiento del siguiente Procedimiento general 7 :

A una solución enfriada (0 °C) de clorhidrato de metilpirrolidin-2-carboxilato (Producto intermedio 45, 12 g, 72,7 mmol) en THF (150 ml), se añadió piridina (23,5 ml, 290,9 mmol), y después 4-(dimetilamino)piridina (DMAP, 889 mg, 7,3 mmol), seguida de cloroformiato de alilo (15,5 ml, 145,4 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 16 horas, a continuación, se añadió agua ( 10 0 ml) y se extrajo en diclorometano ( 2 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 10-15 %/n-hexanos, obteniéndose 1 -alil-2-metil-pirrolidin-1,2-dicarboxilato (Producto intermedio 46, 13 g, rendimiento: 80 %) en forma de un líquido incoloro. m/z 236,00 [M+Na]+; Sistema de TLC: acetato de etilo al 20 % en hexano, Fr: 0,5.
Ejemplo 144-Preparación del producto intermedio 47
La síntesis del Producto intermedio 47 siguió el procedimiento del siguiente Procedimiento general 2 :

A una solución fría (-78 °C) de acetonitrilo (1,8 ml, 35,2 mmol) en THF seco (50 ml), se añadió n-butillitio (2,5 M en nhexano, 14 ml, 35,2 mmol). Después de agitar a -78 °C durante 45 minutos, se añadió una solución de 1 -alil-2-metilpirrolidin-1,2-dicarboxilato (Producto intermedio 46, 5 g, 23,5 mmol) en THF seco (10 ml) y la reacción se agitó a -78 °C durante 1 hora. Se inactivó la mezcla de reacción con solución saturada de NH4O (20 ml) y se extrajo en EtOAc (2 x 50 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 20-25 %/n-hexano, dando alil-2-(2-cianoacetil)pirrolidin-1 -carboxilato (Producto intermedio 47, 4,3 g, rendimiento: 82 %) en forma de un líquido de color marrón; Sistema de TLC: acetato de etilo al 50 % en hexano, Fr: 0,2.
Ejemplo 145-Preparación del compuesto 98
La síntesis del Compuesto 98 siguió el procedimiento del siguiente Procedimiento general 3 :

A una solución agitada de alil-2-(2-cianoacetil)pirrolidin-1 -carboxilato (Producto intermedio 47, 4,3 g, 19,4 mmol) en etanol (40 ml), se añadió monohidrato de hidrazina (0,85 ml, 17,4 mmol). A continuación, se calentó la mezcla de reacción a 70 °C durante 5 horas. A continuación, la mezcla de reacción se enfrió hasta la temperatura ambiente y se evaporaron las sustancias volátiles. El residuo resultante se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con MeOH al 3 %-diclorometano, dando alil-2-(5-amino-1H-pirazol-3-il)pirrolidin-1-carboxilato (Compuesto 98, 4 g, rendimiento: 87 %) en forma de un líquido incoloro. m/z 237,14 [M+H]+; Sistema de TLC: metanol al 5 % en diclorometano, Fr: 0,3.
Ejemplo 146-Preparación del compuesto 99
La síntesis del Compuesto 99 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución enfriada (0 0C) de alil-2-(5-amino-1H-pirazol-3-il)pirrolidin-1-carboxilato (Compuesto 98, 4 g, 16,9 mmol) en MeOH seco (70 ml), se añadieron 5-clorotiofen-2-carbaldehído (3,9 ml, 33,9 mmol) y ácido acético (0,1 ml), seguidos de tamices moleculares de 4 Á en polvo. Se agitó la mezcla de reacción a temperatura ambiente durante 1 hora (se observó la formación de imina como una mancha menos polar en la TLC). Después de enfriar la mezcla de reacción nuevamente a 0 0C, se añadió cianoborohidruro de sodio (NaCNBH3 , 1,52 g, 8,5 mmol) poco a poco. A continuación, se agitó la mezcla a temperatura ambiente durante 2 horas. Se inactivó la mezcla de reacción con agua helada (30 ml), se filtró a través de un lecho corto de Celite y se extrajo el filtrado en EtOAc (3 x 50 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando una columna de fase inversa Reveleris C-18, eluyendo con acetonitrilo al 35-40 % en ácido fórmico acuoso (0,1 %), obteniéndose el compuesto necesario (3 g, rendimiento: 45 %). Se purificó adicionalmente una de sus porciones (100 mg) mediante HPLC preparativa, dando alil-2-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)pirrolidin-1-carboxilato (Compuesto 99, 50 mg) en forma de un líquido gomoso de color amarillo pálido. m/z 367,07 [M+H]+. RMN de 1H (300 MHz, DMSO-de) 5 11,39 (s a, 1H, intercambiable), 7,02-6,92 (m, 1H), 6,83 (d a, J = 3,7 Hz, 1 H, intercambiable), 6,09-5,47 (m, 2H), 5,38-4,97 (m, 3H), 4,81 (s a, 1H), 4,47 (d a, J = 15,0 Hz, 2H), 4,28 (d, J = 6,2 Hz, 2H), 3,48 (s a, 2H), 2,08 (d a, J = 6,2 Hz, 1H), 1,95 1,58 (m, 3H) ppm; Sistema de TLC: metanol al 5 % en diclorometano, Fr: 0,4.
Ejemplo 147-Preparación del compuesto 100
La síntesis del Compuesto 100 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 0C) de alil-2-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)pirrolidin-1-carboxilato (Compuesto 99, 3,5 g, 9,6 mmol) en diclorometano seco (50 ml), se añadió cloruro de trimetilacetilo (1,1 ml, 8,6 mmol), seguido de trietilamina (TEA, 1,9 ml, 14,3 mmol). Después de agitar a temperatura ambiente durante 2 horas, se añadió agua (25 ml) y la mezcla se extrajo con diclorometano (3 x 40 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 10 %/n-hexano, produciendo el compuesto deseado (3 g, rendimiento: 69 %) en forma de un líquido gomoso de color amarillo pálido. Se purificó adicionalmente una porción (120 mg) mediante HPLC preparativa, produciendo alil-2-(5-((5-clorotiofen-2-il)metilamino)-1-pivaloil-1H-pirazol-3-il)pirrolidin-1-carboxilato (Compuesto 100, 50 mg) en forma de un líquido gomoso de color amarillo pálido; m/z 451,17 [M+H]+. RMN de 1H (400 MHz, CDC^) 5 7,49-7,31 (m, 1H), 6,83-6,66 (m, 2H), 6,07-5,74 (m, 1H), 5,38-5,26 (m, 1H), 5,24-5,06 (m, 2H), 4,98-4,77 (m, 1H), 4,64-4,46 (m, 2H), 4,34 (d, J = 5,9 Hz, 2H), 3,67-3,41 (m, 2 H), 2,27-1,80 (m, 4 H), 1,44 (s, 9H) ppm; Sistema de TLC: acetato de etilo al 50 % en hexano, Fr: 0,7.
Ejemplo 148-Preparación del compuesto 101
La s ín te s is de l
Compuesto 101
s ig u ió e l s ig u ie n te p ro ce d im ie n to de l
Procedimiento general 8b
:

Se desgasificó una solución agitada de alil-2-(5-((5-clorotiofen-2-il)metilamino)-1-pivaloil-1H-pirazol-3-il)pirrolidin-1-carboxilato (Compuesto 100, 1,5 g, 3,3 mmol) en diclorometano (30 ml) con una corriente de argón durante 15 minutos, después se añadió a la mezcla tetraquis(trifenilfosfin)paladio (0) (Pd(PPh3)4 , 193 mg, 0,16 mmol) seguido de fenilsilano (PhSiH3 , 1,2 ml, 10 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 45 minutos, a continuación, se filtró a través de un lecho corto de Celite y se evaporó el filtrado. El residuo en bruto se purificó mediante cromatografía en sílice CombiFlash eluyendo con MeOH al 4 %-diclorometano, produciendo 1-(5-((5-clorotiofen-2-il)metilamino)-3-(pirrolidin-3-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 101, 1 g, rendimiento: 81 %) en forma de un líquido gomoso amarillo pálido. m/z 367,26 [M+H]+, Rm N de 1H (300 MHz, DMSO-da) 57,72 (t a, J = 6,1 Hz, 1H, intercambiable), 6,96 (s, 2H), 5,44 (s, 1H), 4,42 (d a, J = 6,2 Hz, 2H), 4,05 (t a, J = 6,6 Hz, 1H), 3,11-2,82 (m, 2H), 2,13-1,95 (m, 1H), 1,87-1,65 (m, 3H), 1,41 (s, 9H) ppm; Sistema de TLC: metanol al 10 % en diclorometano, Fr: 0,3
Ejemplo 149-Preparación del compuesto 102
La síntesis del Compuesto 102 siguió el procedimiento del siguiente Procedimiento general 5d :
A una solución enfriada (0 0C) de 1-(5-((5-clorotiofen-2-il)metilamino)-3-(pirrolidin-2-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 101, 200 mg, 1,2 mmol) en diclorometano seco (10 ml), se añadió cloruro de ácido 4-morfolineacético (94 mg, 0,66 mmol), seguido de trietilamina (TEA, 0,22 ml, 1,6 mmol). Después de agitar a temperatura ambiente durante 2 horas, se añadió agua helada (10 ml), y a continuación, se lavó con diclorometano (2 x 15 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 50 %-hexanos, dando el producto deseado (120 mg, rendimiento: 81 %) en forma de un semisólido de color amarillo claro. Se purificó una porción (160 mg) de este material (85 % mediante CL-EM) mediante HPLC preparativa, dando 1-(5-((5-clorotiofen-2-il)metilamino)-3-(1-(2-morfolinoacetil) pirrolidin-2-il)-1 H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 102, 60 mg) en forma de un semisólido gomoso blanquecino; m/z 494,22 [M+H]+. RMN de 1H (400 MHz, DMSO-de) 57,49 (s a, 1H), 6,90 (s, 2H), 5,34 (s a, 1H), 5,10 (s a, 2H), 4,42 (s a, 2H), 3,49 (s a, 8H), 2,98 (s, 2H), 2,48 (s a, 3H), 1,94 (s a, 2H), 1,39 (s, 9H) ppm; Sistema de TLC: acetato de etilo al 70 % en n-hexano-Fr: 0,6.
Ejemplo 150-Preparación del compuesto 103
La s ín te s is de l
Compuesto 103
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 6d
:

A una solución a temperatura ambiente de 1-(5-((5-clorotiofen-2-il)metilamino)-3-(1-(2-morfolinoacetil)pirrolidin-2-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 102, 230 mg, 0,54 mmol) en metanol (10 ml), se añadió bicarbonato de sodio (NaHCO3 , 230 mg, 2,7 mmol). Se agitó la mezcla a 50 0C durante 4 horas, después se enfrió de nuevo hasta la temperatura ambiente, se añadió agua (20 ml) y se lavó con diclorometano (3 x 20 ml). Las fases orgánicas combinadas se desecaron con sulfato de sodio, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con metanol al 5-7 % en diclorometano, produciendo el producto deseado (130 mg, rendimiento: 67 %). Se purificó adicionalmente una porción (130 mg, 72 % mediante CL-EM) de este material mediante HPLC preparativa, produciendo 1-(2-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)pirrolidin-1-il)-2-morfolinoetanona (Compuesto 103, 25 mg) en forma de un sólido amarillo pálido; m/z 410,15 [M+H]+. RMN de 1H (400 MHz, DMSO-de) 5 11,84-10,62 (m, 1H, intercambiable), 7,46-5,87 (m, 2H), 5,27 (s a, 2H), 5,04 (s a, 1H), 4,29 (d a, J = 6,6 Hz, 2H), 3,53 (s a, 6H), 3,07 (d a, J = 6,6 Hz, 1H), 2,97-2,71 (m, 1H), 2,48 (d a, J = 1,5 Hz, 4H), 2,07 (s a, 1H), 1,85 (s a, 3H) ppm; Sistema de TLC: metanol al 10 % en diclorometano-Fr: 0,3.
Ejemplo 151-Preparación del compuesto 104
La síntesis del Compuesto 104 siguió el procedimiento del siguiente Procedimiento general 22:

A una solución fría (0 0C) de 1-(5-((5-clorotiofen-2-il)metilamino)-3-(pirrolidin-2-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 101, 700 mg, 1,9 mmol) en diclorometano seco (20 ml), se añadió cloruro de metanosulfonilo (MsCl, 0,22 ml, 2,8 mmol), seguido de trietilamina (TEA, 0,6 ml, 4,4 mmol). Después de agitar la mezcla a temperatura ambiente durante 1 hora, se añadió agua helada (20 ml) y se extrajo en diclorometano (3 x 20 ml). Las fases orgánicas combinadas se desecaron con sulfato de sodio, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 20-25 % en n-hexano, dando el producto deseado (550 mg, rendimiento: 64 %). Una porción (170 mg) se purificó adicionalmente mediante cromatografía HPLC preparativa, produciendo 1-(5-((5-clorotiofen-2-il)metilamino)-3-(1-(metilsulfonil)pirrolidin-2-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 104, 50 mg) en forma de un líquido gomoso de color amarillo pálido; m/z 445,17 [M+h ]+; RMN de 1H (400 MHz, CDC^) 5 7,52-7,35 (m, 1H, intercambiable), 6,91-6,61 (m, 2H), 5,37 (s, 1H), 4,83 (dd, J = 2,7, 7,1 Hz, 1H), 4,37 (d, J = 5,9 Hz, 2H), 3,63-3,36 (m, 2H), 2,75 (s, 3H), 2,33-1,89 (m, 4 H), 1,45 (s, 9 H) ppm; Sistema de TLC: metanol al 5 % en diclorometano-Fr: 0,6.
Ejemplo 152-Preparación del compuesto 105
La s ín te s is de l
Compuesto 105
s ig u ió e l p ro ce d im ie n to
del Procedimiento general 6d
:

A una solución enfriada (0 0C) de 1-(5-((5-clorotiofen-2-il)metilamino)-3-(1-(metilsulfonil)pirrolidin-2-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 104, 200 mg, 0,45 mmol) en metanol seco (10 ml), se añadió bicarbonato de sodio (189 mg, 2,25 mmol). Tras calentar la mezcla a 50 0C durante 4 horas, la mezcla de reacción se enfrió hasta la temperatura ambiente, se añadió agua (15 ml) y se extrajo en diclorometano (3 x 20 ml). Las fases orgánicas combinadas se desecaron con sulfato de sodio, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con metanol al 5-7 % en diclorometano, dando el compuesto semipuro requerido (145 mg, rendimiento: 89 %) en forma de un sólido blanquecino. Se purificó adicionalmente una porción (130 mg de 89 % de pureza mediante CL-EM) mediante HPLC preparativa, produciendo W-((5-clorotiofen-2-il)metil)-3-(1 -(metilsulfonil)pirrolidin-2-il)-1 H-pirazol-5-amina (Compuesto 105, 50 mg) en forma de un sólido blanco; m/z 361,16 [M+H]+; RMN de 1H (400 MHz, DMSO-de) 5 11,52-11,30 (m, 1H, intercambiable), 6,99-6,80 (m, 2H), 5,68 (s a, 1H, intercambiable), 5,41 (s a, 1H), 4,74 (s a, 1H), 4,29 (d, J = 6,4 Hz, 2H), 3,46-3,24 (m, 2H), 2,84 (s a, 3H), 2,21-2,06 (m, 1H), 1,89 (d a, J = 5,4 Hz, 3H) ppm; Sistema de TLC: metanol al 5 % en diclorometano-Fr: 0,3.
Ejemplo 153-Preparación del compuesto 106
La síntesis del Compuesto 106 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 0C) de W-((5-clorotiofen-2-il)metil)-3-(1-(metilsulfonil)pirrolidin-2-il)-1H-pirazol-5-amina (Compuesto 105, 200 mg, 0,56 mmol) en diclorometano seco (10 ml), se añadió cloruro de 1,3-tiazol-4-carbonilo (148 mg, 1,11 mmol) seguido de trietilamina (TEA, 0,18 ml, 1,4 mmol). Después de agitar a temperatura ambiente durante 2 horas, se inactivó la mezcla de reacción con solución de NaHCO3 (10 ml) y se extrajo en diclorometano (3 x 20 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio y se concentraron a presión reducida, produciendo el compuesto deseado (150 mg, rendimiento: 57 %) en forma de un semisólido de color amarillo claro. Se purificó adicionalmente una porción (100 mg con un 87 % de pureza según la CL-EM) mediante cromatografía de HPLC preparativa, produciendo (5-((5-clorotiofen-2-il)metilamino)-3-(1-(metilsulfonil)pirrolidin-2-il)-1 H-pirazol-1-il)(tiazol-4-il)metanona (Compuesto 106, 40 mg) en forma de un sólido blanquecino. m/z 472,10 [M+H]+. RMN de 1H (400 MHz, CDCls) 59,13 (d, J = 2,0 Hz, 1H), 8,88 (d, J = 2,0 Hz, 1H), 7,65 (t a, J = 5,6 Hz, 1H, intercambiable), 6,89-6,69 (m, 2H), 5,44 (s, 1H), 4,88 (dd, J = 3,2, 8,1 Hz, 1H), 4,49 (d, J = 5,9 Hz, 2H), 3,66-3,39 (m, 2H), 2,78 (s, 3H), 2,37-2,16 (m, 2H), 2,14-1,95 (m, 2 H) ppm; Sistema de TLC: metanol al 5 % en diclorometano-Fr: 0,5.
Ejemplo 154-Preparación del compuesto 107
La síntesis del Compuesto 107 siguió el procedimiento del siguiente Procedimiento general 15:

A una solución enfriada (0 0C) de 1-(5-((5-clorotiofen-2-il)metilamino)-3-(pirrolidin-2-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 101, 320 mg, 0,87 mmol) en diclorometano seco (10 ml), se añadió cloruro de morfolin-4-carbonilo (0,2 ml, 1,7 mmol), seguido de trietilamina (TEA, 0,29 ml, 2,1 mmol). Después de agitar a temperatura ambiente durante 1 hora, se añadió agua helada (10 ml) y después se extrajo en diclorometano (2 x 15 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con EtOAc al 50 %-hexanos, dando el producto deseado (170 mg, rendimiento: 40 %) en forma de un semisólido de color amarillo claro. Se purificó una porción (160 mg, 85 % mediante CL-EM) mediante HPLC preparativa, obteniéndose 1 -(5-((5-clorotiofen-2-il)metilamino)-3-(1 -(morfolin-4-carbonil)pirrolidin-2-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona (Compuesto 107, 45 mg) en forma de un líquido gomoso de color amarillo pálido; m/z 480,25 [M+H]+. RMN de ]H (400 MHz, CDC^) 57,40 (t a, J = 5,6 Hz, 1H, intercambiable), 6,79-6,71 (m, 2H), 5,17 (s, 1H), 5,04 (t, J = 6,8 Hz, 1H), 4,34 (d, J = 5,4 Hz, 2H), 3,71-3,58 (m, 4H), 3,57-3,41 (m, 2H), 3,36 (ddd, J = 3,4, 6,4, 13,2 Hz, 2H), 3,20 (ddd, J = 2,9, 6,4, 13,2 Hz, 2H), 2,32-2,19 (m, 1H), 2,06-1,79 (m, 3H), 1,45 (s, 9H) ppm; Sistema de TLC: metanol al 5 %-diclorometano. Fr: 0,5.
Ejemplo 155-Preparación del compuesto 108
La síntesis del Compuesto 108 siguió el procedimiento del siguiente Procedimiento general 6d :

A una solución a temperatura ambiente de 1-(5-((5-clorotiofen-2-il)metilamino)-3-(1-(morfolin-4-carbonil)pirrolidin-2-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 107, 200 mg, 0,42 mmol) en metanol (10 ml), se añadió bicarbonato de sodio (176 mg, 2,1 mmol), seguido de calentamiento de la mezcla a 50 0C durante 4 horas. La mezcla de reacción se enfrió hasta la temperatura ambiente, se añadió agua (20 ml) y se extrajo en diclorometano (3 x 20 ml). El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con metanol al 5-7 % en diclorometano, dando el producto deseado (100 mg, rendimiento: 65 %) en forma de un semisólido de color amarillo claro. Se purificó adicionalmente una porción (150 mg, 70 % mediante CL-EM) de material mediante cromatografía HPLc preparativa, dando (2-(5-((5-clorotiofen-2-il)metilamino)-1 H-pirazol-3-il)pirrolidin-1-il)(morfolino)metanona (Compuesto 108, 30 mg) en forma de un sólido blanquecino. m/z 396,21 [M+H]+. RMN de 1H (400 MHz, CDCls) 10,04 (s a, 1H, intercambiable), 6,83-6,56 (m, 2H), 5,47 (s, 1H), 5,05 (dd, J = 6,8, 9,3 Hz, 1H), 4,42 (d a, J = 4,9 Hz, 2H), 3,95 (s a, 1H, intercambiable), 3,76-3,51 (m, 4H), 3,50-3,31 (m, 4H), 3,20 (ddd, J = 2,9, 6,4, 13,2 Hz, 2H), 2,51-2,15 (m, 1H), 2,10-1,95 (m, 2H), 1,93-1,73 (m, 1H) ppm; Sistema de TLC: metanol al 5 % en diclorometano-Fr: 0,3.
Ejemplo 156-Preparación del compuesto 109
La s ín te s is de l
Compuesto 109
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5d
:

A una solución enfriada (0 0C) de (2-(5-((5-clorotiofen-2-il)metilamino)-1H-pirazol-3-il)pirrolidin-1-il)(morfolino)metanona (Compuesto 108, 260 mg, 0,66 mmol) en diclorometano seco (15 ml), se añadió cloruro de 1,3-tiazol-4-carbonilo (194 mg, 1,3 mmol), seguido de trietilamina (TEA, 0,22 ml, 0,94 mmol) y se agitó la mezcla a 0 0C durante 2 horas más. A continuación, se añadió agua helada (15 ml) y se lavó la mezcla con diclorometano (2 x 15 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía ultrarrápida, eluyendo con THF al 5-10 %-EtOAc, produciendo ((2-(5-((5-clorotiofen-2-il)metilamino)-1 -(tiazol-4-carbonil)-1 H-pirazol-3-il)pirrolidin-1 -il)(morfolino)metanona (Compuesto 109, 75 mg) en forma de un sólido amarillo pálido. m/z: 507,22 [M+H]+. RMN de 1H (400 MHz, CDCls) 5 = 9,18 (d, J = 2,0 Hz, 1H), 8,87 (d, J = 2,0 Hz, 1H), 7,64 (t, J = 6 Hz, 1H, intercambiable), 6,95-6,65 (m, 2H), 5,27 (s, 1H), 5,07 (t, J = 7,2 Hz, 1H), 4,46 (d, J = 5,9 Hz, 2H), 3,73-3,46 (m, 6H), 3,38 (ddd, J = 2,9, 6,4, 13,2 Hz, 2H), 3,20 (ddd, J = 2,9, 6,4, 13,2 Hz, 2H), 2,43-2,25 (m, 1H), 2,10-1,79 (m, 3H) ppm; Sistema de TLC: THF al 10 %-EtOAc. Fr: 0,5.
Ejemplo 157-Preparación del producto intermedio 48
La síntesis del Producto intermedio 48 siguió el procedimiento del siguiente Procedimiento general 25:

Se disolvió piperidin-4-carboxilato de etilo (1,0 g, 6,4 mmol, 1,0 eq) en acetonitrilo anhidro (25,0 ml), seguido de la adición de carbonato de cesio (4,15 g, 12,7 mmol, 2,0 eq) y se agitó la mezcla de reacción a temperatura ambiente durante 30 minutos. A continuación, la reacción se enfrió hasta 0 °C y se añadió clorhidrato de 4-(2-cloroetil)morfolina (1,42 g, 7,6 mmol, 1,2 eq). La mezcla se llevó lentamente a temperatura ambiente y se agitó a 80 0C durante 12 horas. Una vez completada la reacción, controlada mediante TLC y CL-EM, se diluyó la mezcla de reacción con agua fría (10 ml) y se extrajo con acetato de etilo (3 x 20 ml). La capa orgánica se lavó con agua y se concentró a presión reducida, obteniéndose un residuo amarillo que se purificó mediante cromatografía en columna (sílice; malla 100-200) eluyendo con acetato de etilo al 45 % en hexano, obteniéndose 1-(2-morfolinoetil)piperidin-4-carboxilato de etilo (intermedio 48, 1,07 g, rendimiento: 62 %).
Ejemplo 158-Preparación del producto intermedio 49
La síntesis del Producto intermedio 49 siguió el procedimiento del siguiente Procedimiento general 2 :

En una atmósfera inerte con flujo de gas de N2 , se añadió acetonitrilo (1,14 g, 27,8 mmol, 1,5 eq) en tetrahidrofurano (30,0 ml) y se enfrió hasta -78 0C. A esto, se añadió n-BuLi (2,5 M en n-hexano, 1,77 ml, 27,8 mmol, 1,5 eq) gota a gota durante un período de 1 hora y la reacción se agitó durante otros 60 minutos. Se añadió etil-1 -(2-morfolinoetil)piperidin-4-carboxilato (Producto intermedio 48, 5,0 g, 18,5 mmol, 1,0 eq) poco a poco a la mezcla de reacción y se agitó a -78 °C durante 3 horas. Se inactivó la reacción con una solución saturada de cloruro de amonio y se extrajo el producto con acetato de etilo (3 x 20 ml). La capa orgánica se desecó sobre sulfato de sodio anhidro y se concentró a presión reducida, obteniéndose un residuo que se lavó con acetato de etilo y se utilizó “tal cual” para la siguiente etapa ((3-(1-(2-morfolinoetil)piperidin-4-il)-3-oxopropanonitrilo, Producto intermedio 49, 4,5 g, rendimiento: 92 %) m/z 266,03;
Ejemplo 159-Preparación del compuesto 110
La síntesis del Compuesto 110 siguió el procedimiento del siguiente Procedimiento general 3 :

A una solución de 3-(1-(2-morfolinoetil)piperidin-4-il)-3-oxopropanonitrilo (Producto intermedio 49, 4,5 g, 17,1 mmol, 1.0 eq) en isopropanol (50 ml) y ácido acético (1,02 g), se añadió gota a gota monohidrato de hidrazina (0,9 g, 18,2 mmol, 1,1 eq) y la reacción se agitó a 80 °C durante 5 horas. La mezcla de reacción se controló mediante TLC y CL-EM y, una vez finalizada, se concentró la mezcla de reacción a presión reducida, obteniéndose un residuo que se purificó mediante cromatografía en columna utilizando gel de sílice (malla 100-200). El producto se eluyó con metanol al 10 % en diclorometano, dando 3-(1-(2-morfolinoetil)piperidin-4-il)-1H-pirazol-5-amina (Compuesto 110, 4.0 g, rendimiento: 83 %) m/z 280,13 [M+1]+.
Ejemplo 160-Preparación del compuesto 111
La síntesis del Compuesto 111 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución enfriada (0 °C) de 3-(1-(2-morfolinoetil)piperidin-4-il)-1H-pirazol-5-amina (Compuesto 110, 2,0 g, 7,2 mmol, 1,0 eq) en MeOH (40,0 ml) y ácido acético glacial (0,431 g), se añadió gota a gota 5-clorotiofen-2-carbaldehído (1,265 g, 8,6 mmol, 1,2 eq). La mezcla de reacción se llevó lentamente a temperatura ambiente y después se agitó durante un día. Una vez completada, controlada mediante TLC y CL-EM, la mezcla se enfrió de nuevo hasta 0 0C y se añadió cianoborohidruro de sodio (0,68 g, 10,7 mmol, 1,5 eq). Se agitó la mezcla a temperatura ambiente durante 4 horas. Se concentró la mezcla de reacción a presión reducida, obteniéndose un residuo amarillo que se diluyó con agua fría (20 ml), se extrajo con acetato de etilo (3 x 20 ml) y se purificó mediante cromatografía en columna (malla 60-120) eluyendo con MeOH al 6 % en diclorometano como fase móvil, dando n-((5-clorotiofen-2-il)metil)-3-(1-(2-morfolinoetil)piperidin-4-il)-1H-pirazol-5-amina (Compuesto 111, 0,60 g, rendimiento: 20 %); m/z 412,00 [M+2]+ RMN de 1H (400 MHz, DMSO) 5 11,33 (s, 1H), 6,91 (d, J = 2,9 Hz, 1H), 6,84 (s, 1H), 5,67 (s, 1H), 5,28 (s, 1H), 4,28 (d, J = 5,4 Hz, 2H), 3,56 (s, 5H), 2,98 (s, 2H), 2,42 (d, J = 24,9 Hz, 7H), 2,10 (s, 2H), 1,81 (s, 2H), 1,58 (s, 2H) ppm.
Ejemplo 161-Preparación del compuesto 112
La s ín te s is de l
Compuesto 112
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5a
:

A una solución de ácido piválico (49,0 mg, 0,49 mmol, 1,0 eq) en THF (10,0 ml), se añadió DIEA (130 mg, 1,0 mmol, 2,0 eq), HOBt (66,0 mg, 0,49 mmol, 1,0 eq) y EDCI-HCl (143 mg, 0,75 mmol, 1,5 eq), y se agitó la mezcla de reacción a temperatura ambiente durante 30 minutos. Después, se añadió W-((5-clorotiofen-2-il)metil)-3-(1-(2-morfolinoetil)piperidin-4-il)-1 H-pirazol-5-amina (Compuesto 111, 200 mg, 0,49 mmol, 1,0 eq) a la mezcla de reacción y se dejó agitar a temperatura ambiente durante otras 12 horas. Una vez completada, se concentró la mezcla de reacción, se vertió en agua helada y se extrajo con acetato de etilo (3 x 50 ml). Las capas orgánicas combinadas se concentraron a presión reducida, dando un residuo que se purificó mediante HPLC preparativa utilizando MeCN al 100 % y ácido fórmico al 0,1 % en agua, dando 1-(5-(((5-clorotiofen-2-il)metil) amino)-3-(1-(2-morfolinoetil) piperidin-4-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 112, 60,0 mg, rendimiento: 25 %) m/z 495,53 [M+1]+; RMN de 1H (400 MHz, DMSO) 58,20 (s, 1H), 7,67 (t, J = 6,2 Hz, 1H), 7,07-6,86 (m, 2H), 5,39 (s, 1H), 4,41 (d, J = 6,0 Hz, 2H), 3,62-3,51 (m, 4H), 2,93 (d, J = 11,7 Hz, 2H), 2,50-2,31 (m, 9H), 2,10 (dd, J = 13,3, 7,1 Hz, 2H), 1,83 (d, J = 12,3 Hz, 2H), 1,66-1,53 (m, 2H), 1,40 (s, 9H), 1,35 (s, 1H) ppm.
Ejemplo 162-Preparación del compuesto 113
La síntesis del Compuesto 113 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución de ácido hidroxipiválico (60,0 mg, 0,49 mmol, 1,0 eq) en THF (10 ml), se añadieron DIPEA (130,0 mg, 0,98 mmol, 2,0 eq), HOBt (66,0 mg, 0,49 mmol, 1,0 eq) y, a continuación, EDCI-HCl (143,0 mg, 0,75 mmol, 1,5 eq). Se agitó la mezcla a temperatura ambiente durante 30 minutos. Se añadió N-((5-clorotiofen-2-il)metil)-3-(1-(2-morfolinoetil)piperidin-4-il)-1 H-pirazol-5-amina (Compuesto 111, 200,0 mg, 0,49 mmol, 1,0 eq) y la mezcla se dejó en agitación a temperatura ambiente durante otras 12 horas. Una vez completada, se concentró la mezcla de reacción y se vertió sobre agua helada y se extrajo con acetato de etilo (3 x 50 ml). Las capas orgánicas combinadas se concentraron a presión reducida, dando un residuo que se purificó mediante HPLC preparativa utilizando MeCN al 100 % y ácido fórmico al 0,1 % en agua, dando 1-(5-(((5-clorotiofen-2-il)metilo)amino)-3-(1-(2-morfolinoetil)piperidin-4-il)-1H-pirazol-1-il)-3-hidroxi-2,2-dimetilpropan-1-ona (Compuesto 113, 55,0 mg, rendimiento: 22 %) m/z 510,78 [M+1]+; RMN de 1H (400 MHz, DMSO) 58,21 (s, 1H), 7,67 (t, J = 6,2 Hz, 1H), 6,97 (c, J = 3,8 Hz, 2H), 5,38 (s, 1H), 4,40 (d, J = 6,2 Hz, 2H), 3,86 (s, 2H), 3,59-3,53 (m, 4H), 2,95 (d, J = 11,2 Hz, 2H), 2,43 (d, J = 6,7 Hz, 3H), 2,38 (s, 4H), 2,12 (t, J = 11,1 Hz, 2H), 1,83 (d, J = 11,8 Hz, 2H), 1,67-1,51 (m, 2H), 1,30 (s, 6H), 1,03 (s, 1H) ppm.
Ejemplo 163-Preparación del producto intermedio 50
La síntesis del Producto intermedio 50 siguió el procedimiento del siguiente Procedimiento general 5d :

A una solución enfriada (0 0C) de piperidin-4-carboxilato de etilo (0,2 g, 1,3 mmol, 1,0 eq) en diclorometano (2,0 ml), se añadió trietilamina (TEA, 0,34 ml, 2,6 mmol, 2,0 eq), seguida de cloruro de cloroacetilo (0,13 ml, 1,5 mmol, 1,2 eq) gota a gota. A continuación se agitó a temperatura ambiente durante 6 horas. Se controló la reacción mediante TLC y CL-EM y, una vez completada, se diluyó la mezcla de reacción con agua fría (10 ml) y se extrajo el producto utilizando diclorometano (2 x 10 ml). Las capas orgánicas se desecaron sobre sulfato de sodio y se evaporaron al vacío, obteniéndose un residuo que se purificó mediante cromatografía en columna, eluyendo con metanol al 3 % en diclorometano, dando 1-(2-cloroacetil)piperidin-4-carboxilato de etilo (Producto intermedio 50, 210 mg, rendimiento: 70 %) m/z 234,21 [M+H]+ RMN de 1H (400 MHz, CDCls) 5 4,39-4,32 (m, 1H), 4,17 (c, J = 7,1 Hz, 2H), 4,09 (d, J = 1,4 Hz, 2H), 3,85 (d, J = 13,6 Hz, 1H), 3,22 (tt, J = 16,0, 7,9 Hz, 1H), 2,99-2,86 (m, 1H), 2,58 (tt, J = 10,5, 4,0 Hz, 1H), 2,05-1,94 (m, 1H), 1,87-1,76 (m, 1H), 1,69 (dt, J = 11,0, 5,3 Hz, 2H), 1,28 (t, J = 7,1 Hz, 3H) ppm.
Ejemplo 164-Preparación del producto intermedio 51
La síntesis del Producto intermedio 51 siguió el procedimiento del siguiente Procedimiento general 25:

A una solución de morfolina (0,08 ml, 1,0 mmol, 1,2 eq) en acetonitrilo (2,0 ml), se añadió carbonato de potasio (0,355 g, 2,4 mmol, 3,0 eq) y se agitó a temperatura ambiente durante 10-15 minutos. A la mezcla, se añadió una solución de etil-1-(2-cloroacetil)piperidin-4-carboxilato (Producto intermedio 50, 0,2 g, 0,8 mmol, 1,0 eq) en acetonitrilo (2,0 ml) y después se agitó a temperatura ambiente durante 14 horas. Una vez completada la reacción, controlada por TLC y CL-EM, se diluyó la mezcla de reacción con acetato de etilo (20 ml) y se lavó con agua (50 ml). Las capas orgánicas se desecaron sobre sulfato de sodio y se evaporaron al vacío, obteniéndose un producto en bruto. Este se purificó mediante cromatografía en columna a través de sílice (malla 60-120), eluyendo con metanol al 4 % en diclorometano, dando 1-(2-morfolinoacetil)piperidin-4-carboxilato de etilo (Producto intermedio 51, 0,141 g, rendimiento: 69 %) m/z 285,42 [M+H]+ RMN de 1H (400 MHz, DMSO) 54,19 (d, J = 13,1 Hz, 1H), 4,11-4,02 (m, 2H), 3,97 (d, J = 13,7 Hz, 1H), 3,60-3,50 (m, 4H), 3,21 (d, J = 13,3 Hz, 1H), 3,07 (dd, J = 21,2, 12,3 Hz, 2H), 2,76-2,65 (m, 1H), 2,59 (tt, J = 11,0, 3,9 Hz, 1H), 2,41-2,33 (m, 4H), 1,83 (t, J = 12,5 Hz, 2H), 1,54 (ddd, J = 24,5, 11,5, 4,0 Hz, 1H), 1,35 (dt, J = 11,3, 7,5 Hz, 1H), 1,22-1,15 (m, 3H) ppm.
Ejemplo 165-Preparación del producto intermedio 52
La síntesis del Producto intermedio 52 siguió el procedimiento del siguiente Procedimiento general 2 :

En una atmósfera inerte con flujo de gas de N2 , se añadió acetonitrilo (0,12 ml, 2,9 mmol, 1,7 eq) a THF (7,5 ml) y se enfrió hasta -78 0C. Se añadió n-BuLi (2,5 M en n-hexano, 1,2 ml, 2,9 mmol, 1,7 eq) gota a gota durante un período de 20 minutos y se agitó la reacción durante otros 60 minutos. Se añadió etil-1 -(2-morfolinoacetil)piperidin-4-carboxilato (Producto intermedio 51, 0,5 g, 1,7 mmol, 1,0 eq) en una porción a la mezcla de reacción y se mantuvo la temperatura a -78 0C durante 1 hora, y después se agitó a temperatura ambiente durante 14 horas. Se inactivó la reacción con una solución saturada de cloruro de amonio y se extrajo el producto con acetato de etilo (3 x 20 ml). La capa orgánica se desecó sobre sulfato de sodio anhidro y se concentró a presión reducida, obteniéndose un producto en bruto que se lavó con acetato de etilo y se utilizó para la siguiente etapa (3-(1-(2-morfolinoacetil)piperidin-4-il)-3-oxopropanonitrilo; Producto intermedio 52, 0,294 g, rendimiento: 60 %) m/z 280,44 [M+H]+ RMN de 1H (400 MHz, DMSO) 54,39-4,25 (m, 1H), 4,07-3,97 (m, 2H), 3,60-3,52 (m, 4H), 3,20-3,12 (m, 1H), 3,02 (d, J = 13,4 Hz, 2H), 2,91 -2,83 (m, 1H), 2,37 (s, 4H), 1,99 (s, 2H), 1,51 (d, J = 15,6 Hz, 1H), 1,45-1,36 (m, 1H), 1,17 (t, J = 7,1 Hz, 2H) ppm.
Ejemplo 166-Preparación del compuesto 114
La s ín te s is de l
Compuesto 114
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 3
:

A una solución de 3-(1-(2-morfolinoacetil)piperidin-4-il)-3-oxopropanonitrilo (Producto intermedio 52, 0,29 g, 1 mmol, 1.0 eq) en isopropanol (9,0 ml) y ácido acético (0,6 ml), se añadió gota a gota monohidrato de hidracina (0,5 ml, 1.0 mmol, 1,0 eq), y la reacción se agitó a 80 0C durante 12 horas. La mezcla de reacción se controló mediante TLC y CL-EM y, una vez completada, la mezcla se concentró a presión reducida, obteniéndose un residuo en bruto. Este se purificó mediante cromatografía en columna utilizando gel de sílice (malla 100-200), eluyendo con metanol al 8 % en diclorometano, dando 1-(4-(5-amino-1 H-pirazol-3-il)piperidin-1-il)-2-morfolinoetan-1-ona (Compuesto 114, 0,164 g, rendimiento: 54 %) m/z 294,03 [M+1]+ RMN de 1H (400 MHz, DMSO) 5 5,17 (s, 1H), 4,34 (d, J = 13,1 Hz, 1H), 4,10-4,01 (m, 1H), 3,56 (s, 4H), 3,30-3,22 (m, 1H), 3,05 (t, J = 14,3 Hz, 2H), 2,73-2,54 (m, 2H), 2,51 (s, 2H), 2,36 (d, J = 19,7 Hz, 3H), 1,89 (d, J = 15,2 Hz, 2H), 1,84 (t, J = 13,0 Hz, 2H), 1,50 (dd, J = 22,8, 10,4 Hz, 1H), 1,37 1,19 (m, 1H) ppm.
Ejemplo 167-Preparación del compuesto 115
La síntesis del Compuesto 115 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución enfriada (0 0C) de 1-(4-(5-amino-1H-pirazol-3-il)piperidin-1-il)-2-morfolinoetan-1-ona (Compuesto 114, 1,5 g, 5,0 mmol, 1,0 eq) en metanol (30 ml), se añadió gota a gota ácido acético glacial (0,33 ml) y, a continuación, 5-clorotiofen-2-carbaldehído (0,75 g, 5,0 mmol, 1,0 eq). Se agitó la mezcla de reacción a temperatura ambiente durante 16 horas. Una vez completada, controlada mediante TLC y CL-EM, la mezcla se enfrió de nuevo hasta 0 0C, y después se añadió cianoborohidruro de sodio (0,63 g, 10,0 mmol, 2,0 eq) y se agitó a temperatura ambiente durante 14 horas. Se concentró la mezcla de reacción lentamente a presión reducida, obteniéndose un residuo amarillo, a continuación, se diluyó con acetato de etilo (100 ml) y después se lavó con agua (25 ml). Tras desecar con sulfato de sodio y filtrar, se concentró la fase orgánica a presión reducida, dando un residuo que se purificó mediante cromatografía en columna (malla 60-120), eluyendo con metanol al 5 % en diclorometano, dando 1-(4-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)piperidin-1 -il)-2-morfolinoetan-1 -ona (Compuesto 115, 0,75 g, rendimiento: 60 %); m/z 424,67 [M+1]+ RMN de 1H (400 MHz, DMSO) 5 11,41 (s, 1H), 6,88 (dd, J = 29,5, 3,7 Hz, 2H), 5,69 (t, J = 6,1 Hz, 1H), 5,28 (s, 1H), 4,32 (dd, J = 27,0, 9,8 Hz, 2H), 4,09 (dd, J = 21,6, 9,2 Hz, 1H), 3,65-3,52 (m, 4H), 3,28-3,10 (m, 1H), 3,04 (d, J = 13,2 Hz, 2H), 2,76 (d, J = 11,6 Hz, 1H), 2,62 (dd, J = 25,3, 12,3 Hz, 2H), 2,36 (d, J = 22,6 Hz, 4H), 1,87 (dd, J = 23,9, 12,8 Hz, 2H), 1,51 (dd, J = 21,4, 12,1 Hz, 1H), 1,40-1,24 (m, 1H) ppm.
Ejemplo 168-Preparación del compuesto 116
La síntesis del Compuesto 116 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución de ácido piválico (0,10 g, 1,0 mmol, 1,1 eq) en THF (2,5 ml), se añadieron DIEA (0,21 ml, 1,2 mmol, 1,5 eq), HOBt (0,021 g, 0,16 mmol, 0,2 eq) y, a continuación, EDCI.HCl (0,23 g, 1,2 mmol, 1,5 eq), y se agitó la
mezcla a temperatura ambiente durante 30 minutos. A continuación, se añadió 1-(4-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)piperidin-1-il)-2-morfolinoetan-1-ona (Compuesto 115, 0,35 g, 0,8 mmol, 1,0 eq) a la mezcla y se dejó agitar a temperatura ambiente durante 14 horas. Una vez completada, se concentró la mezcla de reacción y se vertió en agua helada (5,0 ml) y se extrajo con acetato de etilo (3 x 10 ml). Las capas orgánicas combinadas se concentraron a presión reducida, dando un residuo que se purificó mediante cromatografía en columna (malla 100-200), eluyendo con acetato de etilo al 50 % en hexano, dando 1-(5-(((5-clorotiofen-2-il)metil)amino)-3-(1-(2-morfolinoacetil)piperidin-4-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 116, 46 mg, rendimiento: 11 %) m/z 509,03 [M+H]+; RMN de 1H (400 MHz, DMSO) 57,69 (t, J = 6,2 Hz, 1H), 6,96 (t, J = 3,6 Hz, 2H), 5,39 (s, 1H), 4,41 (d, J = 6,4 Hz, 2H), 4,29 (d, J = 14,3 Hz, 1H), 4,03 (d, J = 12,2 Hz, 1H), 3,56 (s, 4H), 3,25 (d, J = 13,2 Hz, 1H), 3,10 (d, J = 11,6 Hz, 1H), 3,04 (d, J = 13,3 Hz, 1H), 2,70 (d, J = 22,7 Hz, 3H), 2,39 (s, 3H), 1,88 (s, 2H), 1,61 (s, 2H), 1,40 (s, 9H) ppm.
Ejemplo 169-Preparación del compuesto 117
La síntesis del Compuesto 117 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución de ácido hidroxipiválico (0,1 g, 0,9 mmol, 1,0 eq) en THF (6,0 ml), se añadieron DIEA (0,18 ml, 1.0 mmol, 1,5 eq), HOBt (0,018 g, 0,14 mmol, 0,2 eq) y, a continuación, EDCl.HCl (0,13 g, 1,0 mmol, 1,5 eq), y se agitó la mezcla a temperatura ambiente durante 30 minutos. A continuación, se añadió a la mezcla 1-(4-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)piperidin-1-il)-2-morfolinoetan-1-ona (Compuesto 115, 0,30 g, 0,7 mmol, 1.0 eq) y se agitó la mezcla de reacción a temperatura ambiente durante 16 horas. Una vez completada, se concentró la mezcla de reacción y se vertió en agua helada (5,0 ml) y se extrajo con acetato de etilo (3 x 10 ml). Las capas orgánicas combinadas se concentraron a presión reducida, dando un residuo que se purificó mediante HPLC preparativa utilizando MeCN al 100 % y agua como fase móvil, dando 1-(5-(((5-clorotiofen-2-il)metil) amino)-3-(1-(2-morfolinoacetil)piperidin-4-il)-1H-pirazol-1-il)-3-hidroxi-2,2-dimetilpropan-1-ona (Compuesto 117, 38,0 mg, rendimiento: 10 %) m/z 524,83 [M+H]+; RMN de 1H (400 MHz, DMSO) 57,69 (t, J = 6,0 Hz, 1H), 6,96 (c, J = 3,7 Hz, 2H), 5,39 (s, 1H), 4,83 (t, J = 5,4 Hz, 1H), 4,41 (d, J = 6,1 Hz, 2H), 4,28 (d, J = 12,3 Hz, 1H), 4,03 (d, J = 13,0 Hz, 1H), 3,86 (d, J = 5,5 Hz, 2H), 3,56 (s, 4H), 3,24 (d, J = 13,3 Hz, 1H), 3,18-3,01 (m, 2H), 2,73 (dd, J = 23,5, 12,4 Hz, 2H), 2,36 (d, J = 22,9 Hz, 4H), 1,87 (s, 2H), 1,60 (d, J = 12,5 Hz, 1H), 1,37 (d, J = 11,6 Hz, 1H), 1,31 (s, 6H) ppm.
Ejemplo 169a-Preparación del compuesto 118
La síntesis del Compuesto 118 siguió el procedimiento del siguiente Procedimiento general 15:

A una solución enfriada (0 0C) de W-((5-clorotiofen-2-il)metil)-3-(piperidin-4-il)-1 H-pirazol-5-amina (0,5 g, 1,7 mmol, 1,0 eq) en diclorometano (10 ml), se añadió trietilamina (TEA, 0,26 g, 2,6 mmol, 1,5 eq) y se agitó durante 10 15 minutos. A continuación, se añadió cloruro de morfolin-4-carbonilo (0,25 g, 1,7 mmol, 1,0 eq) y se agitó a temperatura ambiente durante 2 horas. Se controló la reacción mediante TLC y CL-EM. Una vez completada, se evaporó el disolvente, dando un residuo que se purificó mediante cromatografía en columna utilizando gel de sílice (malla 60-120) eluyendo con metanol al 5 % en diclorometano, dando (4-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)piperidin-1-il)(morfolino)metanona (Compuesto 118, 0,24 g, rendimiento: 34 %); m/z 410,37 [M+1]+ RMN de 1H (400 MHz, DMSO) 5 11,34 (s, 1H), 6,91 (d, J = 3,7 Hz, 1H), 6,84 (d, J = 3,3 Hz, 1H), 5,66 (s, 1H), 5,29 (s, 1H),
4,28 (d, J = 6,1 Hz, 2H), 3,62 (d, J = 13,1 Hz, 2H), 3,57 (dd, J = 13,4, 9,1 Hz, 4H), 3,18-3,01 (m, 4H), 2,80 (t, J = 11,7 Hz, 2H), 2,66 (s, 1H), 1,81 (d, J = 11,0 Hz, 2H), 1,53-1,32 (m, 2H) ppm.
Ejemplo 170-Preparación del compuesto 119
La síntesis del Compuesto 119 siguió el siguiente procedimiento del Procedimiento general 5c:

A una solución enfriada (0 °C) de ácido 4-hidroxi-1-metilciclohexano-1-carboxílico (0,15 g, 0,95 mmol, 1,3 eq) en THF (5,0 ml), se añadieron TBTU (0,35 g, 1,1 mmol, 1,5 eq) y DIEA (0,38 ml, 2,2 mmol, 3,0 eq) bajo nitrógeno. Se agitó la mezcla de reacción durante 30 minutos y se añadió (4-(5-(((5-clorotiofen-2-il)metil)amino)-1-(4-hidroxi-1-metilciclohexano-1-carbonil)-1H-pirazol-3-il)piperidin-1-il)(morfolino)metanona (Compuesto 118, 0,298 g, 0,73 mmol, 1,0 eq) y, a continuación, se agitó la mezcla a temperatura ambiente durante 14 horas. Se controló la reacción mediante CL-EM y, una vez finalizada, la mezcla se concentró, dando un residuo que se purificó mediante HPLC preparativa utilizando ácido fórmico al 0,1 % en agua-acetonitrilo como fase móvil, dando (4-(5-(((5-clorotiofen-2-il)metil)amino)-1 -(4-hidroxi-1 -metil ciclohexano-1 -carbonil)-1 H-pirazol-3-il)piperidin-1 -il)(morfolino)metanona (Compuesto 119, 0,053 g, rendimiento: 10 %); m/z 550,68 [M+1]+ RMN de 1H (400 MHz, DMSO) 57,68 (s, 1H), 6,95 (s, 2H), 5,39 (s, 1H), 4,40 (t, 1H), 4,40 (d, 2H), 3,52 (d, J = 29,7 Hz, 6H), 3,11 (m, 4H), 2,84 (m, 2H), 2,58 (m, 1H), 2,53 (m, 3H), 1,98 (d, J = 36,1 Hz, 2H), 1,84 (m, 3H), 1,54 (d, J = 30,8 Hz, 2H), 1,42 (m,6H) ppm.
Ejemplo 171-Preparación del compuesto 120
La síntesis del Compuesto 120 siguió el procedimiento del siguiente Procedimiento general 25:

A una solución enfriada (0 0C) de W-((5-clorotiofen-2-il)metil)-3-(piperidin-4-il)-1 H-pirazol-5-amina (0,1 g, 2,4 mmol, 1 eq) en acetonitrilo (10 ml), se añadió 2-cloro-1-morfolinoetan-1-ona (1,23 ml, 2,4 mmol, 1,0 eq) y, a continuación, carbonato de potasio (0,102 g, 7,2 mmol, 3,0 eq). Se agitó la mezcla de reacción a temperatura ambiente durante 12 horas. El progreso de la reacción se controló mediante TLC. Se diluyó la mezcla de reacción con agua (10 ml), se extrajo con diclorometano (2 x 10 ml) y las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro y se concentraron, dando el producto en bruto. Este residuo se purificó mediante cromatografía CombiFlash, eluyendo con metanol al 6 % en diclorometano, dando 2-(4-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)piperidin-1 -il)-1 -morfolinoetan-1 -ona (Compuesto 120, 0,60 g, rendimiento: 57 %) m/z 424,42[M+1]+, r Mn de 1H (400 MHz, DMSO) 5 11,29 (s, 1H), 6,91 (d, J = 3,7 Hz, 1H), 6,84 (d, J = 3,6 Hz, 1H), 5,62 (s, 1H), 5,28 (s, 1H), 4,29 (d, J = 6,2 Hz, 2H), 3,50-3,56 (m, 4H), 3,40-3,43 (m,4H), 3,14 (s, 2H), 2,84 (d, J = 11,3 Hz, 2H), 2,44 (s, 1H), 2,07 (t, J = 10,7 Hz, 2H), 1,81 (d, J = 11,3 Hz, 2H), 1,53 (dd, J = 21,3, 11,9 Hz, 2H) ppm;
Ejemplo 172-Preparación del compuesto 121
La s ín te s is de l
Compuesto 121
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5d
:

A una solución enfriada (0 0C) de 2-(4-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il) piperidin-1 -il)-1 -morfolinoetan-1-ona (Compuesto 120, 0,15 g, 3,5 mmol, 1,0 eq) en diclorometano (10 ml), se añadió trietilamina (TEA, 0,11 g, 11 mmol, 3 eq), seguida de cloruro de pivaloílo (0,042 g, 3,5 mmol, 1 eq). Se agitó la mezcla de reacción a temperatura ambiente durante 14 horas. Una vez completada, la mezcla se concentró a presión reducida, dando un residuo en bruto. Este se purificó mediante cromatografía en columna, eluyendo con acetato de etilo al 60 % en hexano, dando 1-(5-(((5-clorotiofen-2-il)metil)amino)-3-(1-(2-morfolino-2-oxoetil)piperidin-4-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 121, 0,15 g, rendimiento: 83 %) m/z 508,48[M+1]+, RMN de 1H (400 MHz, DMSO) 57,68 (d, J = 6,3 Hz, 1H), 6,96 (d, J = 3,8 Hz, 2H), 5,39 (s, 1H), 4,41 (d, J = 5,9 Hz, 2H), 3,50-3,55 (m, 5H), 3,41 (d, J = 11,7 Hz, 2H), 3,13 (s, 2H), 2,83 (d, J = 10,6 Hz, 2H), 2,38 (d, J = 34,4 Hz, 1H), 2,09 (t, J = 10,9 Hz, 2H), 1,84 (d, J = 11,3 Hz, 2H), 1,56 (d, J = 9,2 Hz, 2H), 1,40 (s, 9H), 1,11 (s, 1H) ppm.
Ejemplo 173-Preparación del compuesto 122
La síntesis del Compuesto 122 siguió el procedimiento del siguiente Procedimiento general 5c:

A una solución de ácido hidroxipiválico (92 mg, 0,78 mmol, 1,0 eq) en acetonitrilo (10 ml), se añadió DIEA (301 mg, 2,3 mmol, 3,0 eq) y, a continuación, TBTU (375 mg, 0,78 mmol, 1,0 eq) y, después, se agitó la mezcla a temperatura ambiente durante 30 minutos. A esto, se añadió 2-(4-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)piperidin-1-il)-1-morfolinoetan-1-ona (Compuesto 120, 0,33 g, 0,78 mmol, 1,0 eq) y la reacción se agitó a temperatura ambiente durante 12 horas. Una vez completada, la mezcla se concentró a presión reducida, dando un producto en bruto, que se purificó mediante HPLC preparativa utilizando MeCN al 100 % y ácido fórmico al 0,1 % en agua, dando 1-(5-(((5-clorotiofen-2-il)metil)amino)-3-(1 -(2-morfolino-2-oxoetil)piperidin-4-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona (Compuesto 122, 50 mg, rendimiento: 12 %) m/z 524,63 [M+1]+; RMN de 1H (400 MHz, DMSO) 5 7,74 (d, J = 6,1 Hz, 1H), 6,97 (s, 2H), 5,39 (s, 1H), 4,85 (t, J = 5,4 Hz, 1H), 4,42 (d, J = 6,2 Hz, 2H), 4,30-4,06 (m, 1H), 3,87 (d, J = 5,4 Hz, 2H), 3,66-3,54 (m, 4H), 3,50 (s, 2H), 3,38 (d, J = 17,7 Hz, 2H), 3,03 (s, 2H), 2,68 (s, 2H), 2,38-1,74 (m, 6H), 1,32 (s, 6H) ppm.
Ejemplo 174-Preparación del compuesto 123
La síntesis del Compuesto 123 siguió el procedimiento del siguiente Procedimiento general 22 :

A una solución enfriada (0 0C) de W-((5-clorotiofen-2-il)metil)-3-(piperidin-4-il)-1 H-pirazol-5-amina (0,1 g, 0,24 mmol, 1,0 eq) en diclorometano (4 ml), se añadió trietilamina (TEA, 0,1 g, 0,98 mmol, 4,0 eq). Después de agitar durante 15 minutos, se añadió cloruro de metanosulfonilo (MsCl, 0,025 g, 0,22 mmol, 0,9 eq). Se agitó la mezcla de reacción durante 4 horas a temperatura ambiente y se controló mediante TLC y CL-EM. Una vez completada la reacción, se vertió la mezcla de reacción en agua (10 ml) y se extrajo con acetato de etilo (2 x 10 ml). Las fases orgánicas
combinadas se lavaron con agua y salmuera, y se desecaron sobre sulfato de sodio, dando un residuo que se purificó mediante HPLC preparativa utilizando agua-acetonitrilo como fase móvil, dando N-((5-clorotiofen-2-il)metil)-3-(1-(metilsulfonil)piperidin-4-il)-1H-pirazol-5-amina (Compuesto 123, 0,05 g, rendimiento: 40 %) m/z 375,31 [M+1]+ 1347 RMN de 1H (400 MHz, DMSO) 5 11,39 (s, 1H), 6,97-6,81 (m, 2H), 5,73 (d, J = 32,5 Hz, 1H), 5,32 (s, 1H), 4,29 (d, J = 6,0 Hz, 2H), 3,57 (d, J = 12,6 Hz, 2H), 2,87 (s, 3H), 2,78 (t, J = 11,0 Hz, 2H), 2,61 (s, 1H), 1,93 (d, J = 13,0 Hz, 2H), 1,66-1,52 (m, 2H) ppm.
Ejemplo 175-Preparación del compuesto 124
La síntesis del Compuesto 124 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución enfriada (0 0C) de ácido piválico (0,098 g, 0,96 mmol, 1,5 eq) en THF (8 ml), se añadió EDCI.HCl (0,18 g, 0,96 mmol, 1,5 eq) y, a continuación, trietilamina (TEA, 0,194 g, 1,92 mmol, 3,0 eq). Se agitó la mezcla de reacción durante 30 minutos, a continuación, se añadieron HOBt (0,017 g, 0,13 mmol, 0,2 eq) y W-((5-clorotiofen-2-il)metil)-3-(1-(metilsulfonil)piperidin-4-il)-1H-pirazol-5-amina (Compuesto 123, 0,24 g, 0,64 mmol, 1 eq). Se agitó la mezcla de reacción a temperatura ambiente durante la noche. Una vez completada (controlada mediante CL-EM), se vertió la mezcla de reacción en agua (15 ml) y se extrajo con acetato de etilo (2 x 15 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, y se desecaron sobre sulfato de sodio, dando un compuesto en bruto. Este se purificó mediante HPLC preparativa utilizando agua-acetonitrilo como fase móvil, dando 1-(5-(((5-clorotiofen-2-il)metil)amino)-3-(1-(metilsulfonil)piperidin-4-ilo)-1 H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 124, 0,035 g, rendimiento: 11 %) m/z 459,43 [M+]+ RMN de 1H (400 MHz, DMSO) 5 7,70 (t, J = 6,2 Hz, 1H), 7,04-6,92 (m, 2H), 5,46 (d, J = 13,5 Hz, 1H), 4,42 (d, J = 6,1 Hz, 2H), 3,57 (d, J = 11,8 Hz, 2H), 2,99-2,73 (m, 5H), 2,60 (dd, J = 24,9, 13,6 Hz, 1H), 1,98 (d, J = 10,9 Hz, 2H), 1,62 (td, J = 15,1,3,7 Hz, 2H), 1,51-1,35 (m, 9H) ppm.
Ejemplo 176-Preparación del compuesto 125
La síntesis del Compuesto 125 siguió el siguiente procedimiento del Procedimiento general 5b :

A una solución de ácido hidroxipiválico (0,119 g, 1,1 mmol, 1,5 eq) en THF (8 ml), se añadieron anhídrido propilfosfónico (T3 P en solución al 50 % en acetato de etilo, 0,318 g, 1,0 mmol, 1,5 eq), DIEA (0,20 g, 2 mmol, 3 eq) y, a continuación, W-((5-clorotiofen-2-il)metil)-3-(1-(metilsulfonil)piperidin-4-il)-1H-pirazol-5-amina (Compuesto 123, 0,25 g, 0,66 mmol, 1 eq). Se agitó la mezcla de reacción a temperatura ambiente durante la noche. Una vez completada la reacción (controlada mediante CL-EM), se vertió la mezcla en agua (20 ml) y se extrajo con acetato de etilo (2 x 30 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, y se desecaron sobre sulfato de sodio, dando un residuo que se purificó mediante HPLC preparativa mediante agua-acetonitrilo como fase móvil, dando 1 -(5-(((5-clorotiofen-2-il)metil)amino)-3-(1 -(metilsulfonil)piperidin-4-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona (Compuesto 125, 0,051 g, rendimiento: 16 %) m/z 475,43 [M+]+ Rm N de 1H (400 MHz, DMSO) 5 7,70 (d, J = 6,0 Hz, 1H), 6,97 (t, J = 4,0 Hz, 2H), 5,43 (s, 1H), 4,83 (s, 1H), 4,41 (d, J = 6,4 Hz, 2H), 3,87 (d, J = 5,1 Hz, 2H), 3,56 (d, J = 11,2 Hz, 2H), 3,12-2,74 (m, 5H), 2,64 (d, J = 27,0 Hz, 2H), 1,97 (d, J = 12,0 Hz, 2H), 1,62 (d, J = 9,3 Hz, 2H), 1,27 (d, J = 28,3 Hz, 6H) ppm.
Ejemplo 177-Preparación del compuesto 126
La síntesis del Compuesto 126 siguió el procedimiento del siguiente Procedimiento general 22:

A una solución enfriada (0 0C) de W-((5-clorotiofen-2-il)metil)-3-(piperidin-4-il)-1 H-pirazol-5-amina (0,1 g, 0,24 mmol, 1,0 eq) en diclorometano (4 ml), se añadió trietilamina (TEA, 0,1 g, 1,0 mmol, 4,0 eq). Después de agitar durante 15 minutos, se añadió cloruro de bencenosulfonilo (0,039 g, 0,22 mmol, 0,9 eq). Se agitó la mezcla de reacción a temperatura ambiente durante la noche. Se controló la reacción mediante TLC y CL-EM. Una vez completada la reacción, se vertió la mezcla de reacción en agua (10 ml) y se extrajo con acetato de etilo (2 x 10 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, y se desecaron sobre sulfato de sodio, dando un compuesto en bruto. Este se purificó mediante HPLC preparativa utilizando agua-acetonitrilo como fase móvil, dando N-((5-clorotiofen-2-il)metil)-3-(1-(fenilsulfonil)piperidin-4-il)-1 H-pirazol-5-amina (Compuesto 126, 0,045 g, rendimiento: 31 %) m/z 437,2 [M+1]+ RMN de 1H (400 MHz, DMSO) 5 11,33 (s, 1H), 8,30-7,26 (m, 5H), 7,08-6,63 (m, 2H), 5,68 (s, 1H), 5,24 (s, 1H), 4,26 (d, J = 6,2 Hz, 2H), 3,65 (d, J = 11,1 Hz, 2H), 2,32 (t, J = 11,2 Hz, 2H), 1,89 (d, J = 11,4 Hz, 2H), 1,72-1,38 (m, 2H) ppm.
Ejemplo 178-Preparación del compuesto 127
La síntesis del Compuesto 127 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución enfriada (0 0C) de ácido piválico (0,080 g, 0,78 mmol, 1,5 eq) en THF (8 ml), se añadieron EDCI.HCl (0,15 g, 0,78 mmol, 1,5 eq) y trietilamina (TEA, 0,16 g, 1,6 mmol, 3,0 eq). Se agitó la mezcla de reacción durante 30 minutos y, a continuación, se añadieron HOBt (0,014 g, 0,1 mmol, 0,2 eq) y W-((5-clorotiofen-2-il)metil)-3-(1-(fenilsulfonil)piperidin-4-il)-1 H-pirazol-5-amina (Compuesto 126, 0,23 g, 0,5 mmol, 1 eq). Se agitó la mezcla de reacción a temperatura ambiente durante la noche. Una vez completada (controlada mediante CL-EM), se vertió la mezcla de reacción en agua (15 ml), se extrajo con acetato de etilo (2 x 20 ml) y, a continuación, se lavaron las fases orgánicas combinadas con agua, salmuera, se desecaron sobre sulfato de sodio, se filtraron y se evaporaron a presión reducida, dando un residuo. Este se purificó mediante HPLC preparativa utilizando agua-acetonitrilo como fase móvil, dando 1-(5-(((5-clorotiofen-2-il)metil)amino)-3-(1-(fenilsulfonil)piperidin-4-il) -1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 127, 0,056 g, rendimiento: 20 %) m/z 521,48 [M+]+ RMN de 1H (400 MHz, d Ms O) 5 7,83-7,51 (m, 6H), 6,95 (s, 2H), 5,34 (s, 1H), 4,38 (d, J = 6,2 Hz, 2H), 3,55 (d, J = 12,0 Hz, 2H), 1,91 (d, J = 10,6 Hz, 2H), 1,58 (dt, J = 34,4, 17,4 Hz, 2H), 1,34 (s, 9H) ppm.
Ejemplo 179-Preparación del compuesto 128
La síntesis del Compuesto 128 siguió el siguiente procedimiento del Procedimiento general 5b :

A una solución de ácido hidroxipiválico (0,102 g, 0,85 mmol, 1,5 eq) en THF (6 ml), se añadieron anhídrido propilfosfónico (T3 P en solución al 50 % en acetato de etilo, 0,273 g, 0,85 mmol, 1,5 eq), DIEA (0,173 g, 1,7 mmol, 3 eq) seguidos de W-((5-chlorotiofen-2-il)metil)-3-(1-(fenilsulfonil)piperidin-4-il)-1H-pirazol-5-amina (Compuesto 126, 0,27 g, 0,57 mmol, 1 eq). Se agitó la mezcla de reacción a temperatura ambiente durante la noche. Una vez completada (controlada mediante CL-EM), se vertió la mezcla en agua (20 ml) y se extrajo con acetato de etilo (2 x 25 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera, y se desecaron sobre sulfato de sodio y, a continuación, se filtraron y se evaporaron a presión reducida, dando un residuo. Este se purificó mediante HpLC preparativa utilizando agua-acetonitrilo como fase móvil, dando 1-(5-(((5-clorotiofen-2-il)metil)amino)-3-(1-(fenilsulfonil)piperidin-4-il) -1H-pirazol-1-il)-3-hidroxi-2,2-dimetilpropan-1-ona (Compuesto 128, 0,054 g, rendimiento: 16 %) m/z 537,54 [M+]+ RMN de 1H (400 MHz, DMSO) 57,70 (ddd, J = 29,6, 11,6, 7,1 Hz, 5H), 6,95 (s, 2H), 5,33 (s, 1H), 4,79 (t, J = 5,3 Hz, 1H), 4,38 (d, J = 6,2 Hz, 2H), 3,81 (d, J = 5,2 Hz, 2H), 3,59 (d, J = 11,7 Hz, 2H), 1,91 (d, J = 10,8 Hz, 2H), 1,59 (dd, J = 21,2, 10,6 Hz, 2H), 1,25 (s, 6H) ppm.
Ejemplo 179a-Preparación del compuesto 129
La síntesis del Compuesto 129 siguió el procedimiento del siguiente Procedimiento general 15:

A una solución enfriada (0 0C) de W-((5-clorotiofen-2-il)metil)-3-(piperidin-4-il)-1 H-pirazol-5-amina (0,8 g, 2,7 mmol, 1,0 eq) en diclorometano (10 ml), se añadió trietilamina (TEA, 1,36 g, 13,5 mmol, 5,0 eq). Después de agitar durante 10-15 minutos, se añadió a la mezcla cloruro de W,W-dimetilcarbamoílo (0,22 g, 2,7 mmol, 1,0 eq) y se agitó la mezcla a temperatura ambiente durante 2 horas. Se controló la reacción mediante TLC y CL-EM. Una vez completada, se evaporó el disolvente, dando un residuo que se purificó mediante cromatografía en columna utilizando gel de sílice (malla 100-200), eluyendo con metanol al 5 % en diclorometano como fase móvil, dando 4-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)-W,W-dimetilpiperidin-1-carboxamida, (Compuesto 129, 0,3 g, rendimiento: 30 %); m/z 368,31[M+1]+; RMN de 1H (400 MHz, DMSO) 5 11,37 (s, 1H), 6,92 (d, J = 3,7 Hz, 1H), 6,84 (d, J = 3,6 Hz, 1H), 5,70 (s, 1H), 5,29 (s, 1H), 4,28 (s, 2H), 3,56 (d, J = 13,1 Hz, 2H), 3,35 (s, 2H), 3,15-3,02 (m, 1H), 2,80-2,67 (m, 8H), 2,62 (d, J = 11,8 Hz, 1H), 1,81 (d, J = 11,6 Hz, 2H), 1,48 (dt, J = 12,1, 8,9 Hz, 2H), 1,18 (t, J = 7,3 Hz, 1H) ppm.
Ejemplo 180-Preparación del compuesto 130
La s ín te s is de l
Compuesto 130
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5a
:

A una solución de ácido piválico (50 mg, 0,49 mmol, 1,0 eq) en THF (10 ml), se añadieron DIEA (100 mg, 0,6 mmol, 1,5 eq), HOBt (11 mg, 0,08 mmol, 0,2 eq) y finalmente Ed C i.HCI (118 mg, 0,6 mmol, 1,5 eq). La solución se agitó a temperatura ambiente durante 30 minutos. A continuación, se añadió 4-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)-N,N-dimetilpiperidin-1-carboxamida (Compuesto 129, 150 mg, 0,41 mmol, 1,0 eq) y se agitó la solución a temperatura ambiente durante otras 12 horas. Una vez completada, se concentró la mezcla de reacción y se vertió en agua helada y se extrajo con acetato de etilo (3 x 50 ml). Las capas orgánicas combinadas se concentraron a presión reducida, dando un residuo. Este se purificó mediante HPLC preparativa utilizando acetonitrilo al 100 % y ácido fórmico al 0,1 % en agua, dando 4-(5-(((5-clorotiofen-2-il)metil)amino)-1-pivaloil-1H-pirazol-3-il)-N,N-dimetilpiperidin-1-carboxamida (Compuesto 130, 52 mg, rendimiento: 28 %) m/z 454,38 [M+1]+; RMN de 1H (400 MHz, DMSO) 57,68 (t, J = 6,3 Hz, 1H), 6,96 (t, J = 3,0 Hz, 2H), 5,41 (s, 1H), 4,41 (d, J = 6,1 Hz, 2H), 3,54 (d, J = 13,2 Hz, 2H), 2,79 (t, J = 11,4 Hz, 2H), 2,73 (s, 6H), 2,62 (d, J = 11,3 Hz, 1H), 1,85 (d, J = 11,0 Hz, 2H), 1,52 (dd, J = 21,3, 11,6 Hz, 2H), 1,41 (s, 9H) ppm.
Ejemplo 181-Preparación del compuesto 131
La síntesis del Compuesto 131 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución de ácido hidroxipiválico (154 mg, 1,3 mmol, 1,0 eq) en THF (10 ml), se añadieron DIEA (280 mg, I , 6 mmol, 1,5 eq), HOBt (29 mg, 0,22 mmol, 0,2 eq) y finalmente e Dc I.HCI (315 mg, 1,6 mmol, 1,5 eq). Después de agitar a temperatura ambiente durante 30 minutos, se añadió 4-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)-N,N-dimetilpiperidin-1-carboxamida (Compuesto 129, 400 mg, 1,1 mmol, 1,0 eq) y se agitó la mezcla de reacción a temperatura ambiente durante otras 12 horas. Una vez completada, se concentró la mezcla de reacción a presión reducida, se vertió en agua helada y se extrajo con acetato de etilo (3 x 50 ml). Las capas orgánicas combinadas se concentraron a presión reducida, dando un residuo que se purificó mediante HPLC preparativa utilizando acetonitrilo al 100 % y ácido fórmico al 0,1 % en agua, dando 4-(5-(((5-clorotiofen-2-il)metilo)amino)-1-(3-hidroxi-2,2-dimetilpropanoil)-1H-pirazol-3-il)-N,N-dimetilpiperidin-1-carboxamida (Compuesto 131, 56 mg, rendimiento: 11 %) m/z 468,63 [M+1]+; RMN de 1H (400 MHz, DMSO) 5 7,68 (t, J = 6,2 Hz, 1H), 6,97 (c, J = 3,8 Hz, 2H), 5,40 (s, 1H), 4,83 (t, J = 5,3 Hz, 1H), 4,41 (d, J = 6,1 Hz, 2H), 3,87 (d, J = 5,2 Hz, 2H), 3,55 (d, J = 13,0 Hz, 2H), 2,80 (t, J = I I , 4 Hz, 2H), 2,73 (s, 6H), 2,63 (t, J = 11,3 Hz, 1H), 1,85 (d, J = 11,3 Hz, 2H), 1,51 (dt, J = 15,0, 7,7 Hz, 2H), 1,31 (s, 6H) ppm.
Ejemplo 182-Preparación del compuesto 132
La s ín te s is de l
Compuesto 132
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 15
:

A una solución enfriada (0 0C) de W-((5-clorotiofen-2-il)metil)-3-(piperidin-4-il)-1 H-pirazol-5-amina (100 mg, 2,7 mmol, 1,0 eq) en diclorometano (10 ml), se añadió trietilamina (TEA, 200 mg, 1,6 mmol, 6,0 eq). Después de 10 15 minutos, se añadió cloruro W,W-dimetilcarbámico (87 mg, 0,82 mmol, 3,0 eq) y se agitó la mezcla a temperatura ambiente durante 16 horas. Se controló la reacción mediante TLC y CL-EM. Una vez completada, se evaporó el disolvente, dando un residuo que se purificó mediante cromatografía en columna utilizando gel de sílice (malla 60 120), eluyendo con metanol al 5 % en diclorometano, dando 4-(5-(((5-clorotiofen-2-il)metil)amino)-1-(dimetilcarbamoil)-1H-pirazol-3-il)-W,W-dimetilpiperidin-1-carboxamida (Compuesto 132, 0,65 g, rendimiento: 41 %); m/z 439,57 [M+1]+; RMN de 1H (400 MHz, DMSO) 57,00-6,91 (m, 2H), 6,75 (t, J = 6,0 Hz, 1H), 5,40 (s, 1H), 4,36 (d, J = 6,0 Hz, 2H), 3,54 (d, J = 13,2 Hz, 2H), 2,98 (d, J = 66,8 Hz, 6H), 2,78 (d, J = 12,1 Hz, 2H), 2,72 (s, 6H), 2,64-2,53 (m, 1H), 1,80 (d, J = 10,9 Hz, 2H), 1,59-1,40 (m, 2H) ppm.
Ejemplo 182a-Preparación del producto intermedio 53
La síntesis del Producto intermedio 53 siguió el procedimiento del siguiente Procedimiento general 26:

A una solución enfriada (0 °C) de piperidin-4-carboxilato de etilo (0,93 g, 5,9 mmol, 3,0 eq) en 1,2-dicloroetano (50 ml), se añadieron ácido acético (0,12 g, 2 mmol, 1,0 eq) y tetrahidro-4H-piran-4-ona (0,2 g, 2 mmol, 1 eq). Después de agitar durante 1 hora a temperatura ambiente, se añadió triacetoxiborohidruro de sodio (NaBH(OAc)3 , 0,63 g, 3 mmol, 1,5 eq) y se agitó la mezcla a temperatura ambiente durante otras 12 horas. El progreso de la reacción se controló mediante TLC. Se añadió la mezcla de reacción a agua (20 ml), se extrajo con diclorometano (3 x 30 ml), las capas orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El producto en bruto se purificó mediante cromatografía en columna (malla 100-200) eluyendo con metanol al 3 % en diclorometano, dando 1-(tetrahidro-2H-piran-4-il)piperidin-4-carboxilato de etilo (Producto intermedio 53, 0,13 g, rendimiento: 42 %);RMN de 1H (400 MHz, CDC^) 5 4,15 (c, J = 7,1 Hz, 2H), 4,05 (dd, J = 11,3, 4,3 Hz, 2H), 3,45-3,32 (m, 2H), 3,08-2,96 (m, 2H), 2,68 (s, 1H), 2,41 (dd, J = 12,2, 7,1 Hz, 3H), 2,06 (d, J = 11,2 Hz, 2H), 1,95-1,80 (m, 4H), 1,67 (ddd, J = 24,4, 12,2, 4,4 Hz, 2H), 1,26 (t, J = 7,1 Hz, 3H) ppm.
Ejemplo 183-Preparación del producto intermedio 54
La síntesis del Producto intermedio 54 siguió el procedimiento del siguiente Procedimiento general 2 :

A una solución enfriada (-78 °C) de acetonitrilo (0,97 g, 24 mmol, 1,5 eq) en THF (50 ml), se añadió n-BuLi (2,5 M en n-hexano, 9,5 ml, 2,4 mmol, 1,5 eq) gota a gota durante un período de 20 minutos. Después de agitar durante 60 minutos a -78 0C, se añadió 1-(tetrahidro-2)H-piran-4-il)piperidin-4-carboxilato de etilo (Producto intermedio 53,
3,8 g, 15,7 mmol, 1,0 eq) en una porción. A continuación, la mezcla se calentó hasta la temperatura ambiente durante 3 horas. Se inactivó la reacción con una solución saturada de cloruro de amonio y se extrajo el producto con acetato de etilo (3 x 20 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se concentraron a presión reducida, obteniéndose un producto en bruto. Este se lavó con acetato de etilo, produciendo 3-oxo-3-(1-(tetrahidro-2H-piran-4-il)piperidin-4-il)propanonitrilo (Producto intermedio 54, 1,5 g, rendimiento: 40 %) y se utilizó “ tal cual” para la siguiente etapa; m/z 237,04 [M+H]+; RMN de 1H (400 MHz, DMSO) 5 4,16 (s, 2H), 3,88 (d, J = 11,0 Hz, 2H), 3,25 (t, J = 11,0 Hz, 2H), 2,91 (d, J = 11,4 Hz, 2H), 2,45 (s, 1H), 2,14 (d, J = 10,7 Hz, 2H), 1,81 (d, J = 11,9 Hz, 3H), 1,65 (d, J = 12,0 Hz, 2H), 1,49-1,33 (m, 4H) ppm.
Ejemplo 184-Preparación del compuesto 132
La síntesis del Compuesto 132 siguió el procedimiento del siguiente Procedimiento general 3 :

A una solución de 3-oxo-3-(1-(tetrahidro-2H-piran-4-il)piperidin-4-il)propanonitrilo (Producto intermedio 54, 1,3 g, 5,7 mmol, 1,0 eq) en isopropanol (15 ml) y ácido acético (0,34 g, 5,7 mmol, 1,0 eq), se añadió gota a gota monohidrato de hidrazina (0,31 g, 6,2 mmol, 1,1 eq). Se agitó la mezcla de reacción a 80 °C durante 12 horas. La mezcla de reacción se controló mediante TLC y CL-EM y, una vez completada, se concentró la mezcla de reacción, obteniéndose un producto en bruto. Este se purificó mediante cromatografía en columna utilizando gel de sílice (malla 60-120), eluyendo con metanol al 10 % en diclorometano, dando 3-(1-(tetrahidro-2H-piran-4-il)piperidin-4-il)-1 H-pirazol-5-amina (Compuesto 132, 1,3 g, rendimiento: 95 %) m/z 251,14 [M+1]+ RMN de 1H (400 MHz, DMSO) 5 8,95 (s, 1H), 5,18 (s, 1H), 3,88 (dd, J = 10,8, 3,6 Hz, 2H), 3,26 (t, J = 11,1 Hz, 2H), 3,17 (s, 2H), 2,90 (d, J = 11,4 Hz, 2H), 2,40 (dd, J = 21,3, 11,5 Hz, 2H), 2,15 (t, J = 10,6 Hz, 2H), 1,80 (d, J = 12,4 Hz, 2H), 1,71-1,60 (m, 2H), 1,57 1,33 (m, 4H) ppm.
Ejemplo 185-Preparación del compuesto 133
La síntesis del Compuesto 133 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución enfriada (0 °C) de 3-(1-(tetrahidro-2H-piran-4-il)piperidin-4-il)-1 H-pirazol-5-amina (Compuesto 132, 1,7 g, 6,8 mmol, 1,0 eq) en metanol anhidro (30 ml), se añadió ácido acético glacial (0,41 g, 6,8 mmol, 1,0 eq) y, a continuación, 5-clorotiofen-2-carbaldehído (1,19 g, 8,2 mmol, 1,2 eq) gota a gota. La mezcla de reacción se llevó lentamente a temperatura ambiente y después se agitó durante 1 hora. Una vez completada, controlada mediante TLC y CL-EM, la mezcla se enfrió de nuevo hasta 0 °C y se añadió cianoborohidruro de sodio (2,16 g, 10 mmol, 1,5 eq). Se agitó la mezcla a temperatura ambiente durante 4 horas. Se controló la reacción mediante TLC. Se concentró la mezcla de reacción lentamente a presión reducida, obteniéndose un residuo amarillo que se diluyó con acetato de etilo (100 ml) y se lavó con agua (3 x 25 ml). La capa orgánica se desecó con sulfato de sodio, se filtró y se concentró a presión reducida, dando un residuo que se purificó mediante cromatografía en columna (gel de sílice, malla 60-120) eluyendo con metanol al 5 % en diclorometano, dando /V-((5-clorotiofen-2-il)metil)-3-(1 -(tetrahidro-2H-piran-4-il)piperidin-4-il)-1 H-pirazol-5-amina (Compuesto 133, 0,5 g, rendimiento: 19 %); m/z 381,36[M+1]+; RMN de 1H (400 MHz, DMSO) 511,33 (s, 1H), 6,99-6,90 (m, 1H), 6,84 (d, J = 3,5 Hz, 1H), 5,66 (s, 1H), 5,28 (s, 1 h ), 4,28 (d, J = 6,2 Hz, 2H), 3,90 (d, J = 7,4 Hz, 2H), 3,33-3,22 (m, 4H), 2,98 (s, 2H), 2,31-2,08 (m, 2H), 2,02-1,64 (m, 4H), 1,49 (s, 4H) ppm.
Ejemplo 186-Preparación del compuesto 134
La síntesis del Compuesto 134 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución de ácido piválico (44,7 mg, 0,44 mmol, 1,2 eq) en THF (3 ml), se añadieron DIEA (0,09 ml, 0,55 mmol, 1,5 eq), HOBt (9,8 mg, 0,07 mmol, 0,2 eq) y, a continuación, e Dc I.h C i (106 mg, 0,55 mmol, 1,5 eq). Después de agitar durante 30 minutos, se añadió (4-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)piperidin-1-il)(morfolino)metanona (Compuesto 118, 150 mg, 0,37 mmol, 1,0 eq) y se dejó la mezcla de reacción en agitación a temperatura ambiente durante otras 16 horas. Una vez completada, la mezcla se concentró y se vertió en agua helada y se extrajo con diclorometano (3 x 50 ml). Las capas orgánicas combinadas se desecaron con sulfato de sodio, se desecaron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo al 100 % y ácido fórmico al 0,1 % en agua, dando 1-(5-(((5-clorotiofen-2-il)metil)amino)-3-(1-(morfolin-4 -carbonil)piperidin-4-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 134, 53,3 mg, rendimiento: 29 %) m/z 494,48 [M+1]+; RMN de 1H (400 MHz, DMSO) 5 7,69 (d, J = 6,1 Hz, 1H), 6,97 (s, 2H), 5,41 (s, 1H), 4,41 (d, J = 6,1 Hz, 2H), 3,61 (d, J = 13,2 Hz, 2H), 3,60-3,53 (m, 4H), 3,12 (d, J = 4,6 Hz, 4H), 2,86 (t, J = 11,7 Hz, 2H), 2,66 (d, J = 11,7 Hz, 1H), 1,85 (d, J = 10,9 Hz, 2H), 1,57-1,44 (m, 2H), 1,40 (s, 9H) ppm.
Ejemplo 187-Preparación del compuesto 135
La síntesis del Compuesto 135 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución de ácido hidroxipiválico (138 mg, 1,2 mmol, 1,2 eq) en THF (8 ml), se añadieron DIEA (0,25 ml, 1,46 mmol, 1,5 eq), HOBt (26 mg, 0,2 mmol, 0,2 eq) y, a continuación, EDCI.h Ci (282 mg, 1,5 mmol, 1,5 eq). Después de agitar durante otros 30 minutos, se añadió (4-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)piperidin-1-il)(morfolino)metanona (Compuesto 118, 400 mg, 0,98 mmol, 1,0 eq) y la mezcla se dejó en agitación a temperatura ambiente durante 16 horas. Una vez completada, se concentró la mezcla de reacción, se vertió en agua helada y se extrajo con acetato de etilo (3 x 50 ml). Las capas orgánicas combinadas se concentraron a presión reducida, dando un residuo que se purificó mediante HPLC preparativa utilizando acetonitrilo al 100 % y agua al 100 %, dando 1 -(5-(((5-clorotiofen-2-il)metil)amino)-3-(1 -(morfolin-4-carbonil)piperidin-4-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona (Compuesto 135, 58 mg, rendimiento: 12 %) m/z 510,53 [M+1]+; r Mn de 1H (400 MHz, DMSO) 57,68 (t, J = 6,2 Hz, 1H), 6,96 (d, J = 3,8 Hz, 2H), 5,40 (s, 1H), 4,83 (t, J = 5,4 Hz, 1H), 4,41 (d, J = 6,1 Hz, 2H), 3,87 (d, J = 5,3 Hz, 2H), 3,61 (d, J = 13,5 Hz, 2H), 3,59-3,53 (m, 4H), 3,15-3,08 (m, 4H), 2,86 (t, J = 11,6 Hz, 2H), 2,66 (d, J = 11,5 Hz, 1H), 1,85 (d, J = 10,6 Hz, 2H), 1,50 (dd, J = 21,0, 11,8 Hz, 2H), 1,31 (s, 6H) ppm.
Ejemplo 188-Preparación del compuesto 136
La s ín te s is de l
Compuesto 136
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 15
:

A una solución enfriada (0 0C) de W-((5-clorotiofen-2-il)metil)-3-(piperidin-4-il)-1 H-pirazol-5-amina (0,1 g, 0,24 mmol, 1,0 eq) en diclorometano (2 ml), se añadió trietilamina (TEA, 0,2 ml, 1,46 mmol, 6,0 eq). Después de agitar durante 15 minutos, se añadió cloruro de morfolin-4-carbonilo (0,1 g, 0,73 mmol, 3,0 eq) y la reacción se agitó a temperatura ambiente durante 16 horas. Se controló la reacción mediante TLC y CL-EM. Una vez completada, se evaporó el disolvente, dando un residuo que se purificó mediante HPLC preparativa utilizando acetonitrilo al 100 % y agua al 100 %, dando (4-(5-(((5-clorotiofen-2-il)metil)amino)-1 -(morfolin-4-carbonil)-1 H-pirazol-3-il)piperidin-1 -il)(morfolino)metanona (Compuesto 136, 15 mg, rendimiento: 2 %) m/z 523,63 [M+1]+; RMN de 1H (400 MHz, DMSO) 5 6,99-6,93 (m, 2H), 6,85 (t, J = 6,2 Hz, 1H), 5,42 (s, 1H), 4,37 (d, J = 6,1 Hz, 2H), 3,74-3,54 (m, 14H), 3,15-3,07 (m, 4H), 2,82 (t, J = 11,5 Hz, 2H), 2,60 (t, J = 11,5 Hz, 1H), 1,80 (d, J = 10,9 Hz, 2H), 1,48 (dd, J = 21,0, 11,9 Hz, 2H) ppm.
Ejemplo 189-Preparación del compuesto 137
La síntesis del Compuesto 137 siguió el procedimiento del siguiente Procedimiento general 15:

A una solución enfriada (0 0C) de W-((5-clorotiofen-2-il)metil)-3-(piperidin-4-il)-1 H-pirazol-5-amina (0,7 g, 2,4 mmol, 1,0 eq) en diclorometano (10 ml), se añadió trietilamina (TEA, 1,2 g, 12 mmol, 5 eq), seguida de cloruro de dimetilsulfamoílo (0,34 g, 2,3 mmol, 1,0 eq). Se agitó la mezcla a temperatura ambiente durante 3 horas. El progreso de la reacción se controló mediante TLC. Se añadió la mezcla de reacción a agua fría (10 ml), se extrajo con diclorometano (3 x 25 ml), y las capas orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se concentraron, dando un producto en bruto. Este se purificó mediante cromatografía en columna sobre gel de sílice (tamaño de malla 60-120) con acetato de etilo al 45 % en n-hexano, dando 4-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)-W,W-dimetilpiperidin-1-sulfonamida (Compuesto 137, 0,62 g, rendimiento: 65 %) m/z 404,30[M+H]+ RMN de 1H (DMSO-d6, 400 MHz, 12092) 5: 11,36 (s, 1H), 7,03-6,76 (m, 2H), 5,65 (s, 1H), 5,31 (s, 1H), 4,29 (d, J = 6,3 Hz, 2H), 3,58 (d, J = 12,5 Hz), 2H), 2,90 (td, J = 12,3, 2,3 Hz, 2H), 2,76 (d, J = 5,7 Hz, 6H), 2,64 (s, 1H), 1,89 (d, J = 11,2 Hz, 2H), 1,54 (cd, J = 12,6, 4,0 Hz, 2H) ppm.
Ejemplo 189-Preparación del compuesto 138
La síntesis del Compuesto 138 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución enfriada (0 0C) de ácido piválico (0,085 g, 0,85 mmol, 1,5 eq) en acetonitrilo (12 ml), se añadieron EDCI.HCl (0,16 g, 0,85 mmol, 1,5 eq), HOBt (0,022 g 0,17 mmol, 0,3 eq) y DIEA (0,23 ml, 1,7 mmol, 3,0 eq). Después de agitar durante 30 minutos, se añadió 4-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)-W,A/-dimetilpiperidin-1-sulfonamida (Compuesto 137, 0,27 g, 0,56 mmol, 1,0 eq), y se agitó la mezcla a temperatura ambiente durante 16 horas. Se controló la reacción mediante CL-EM y, una vez completada la reacción, se añadió la mezcla de reacción a agua (10 ml), se extrajo con acetato de etilo (3 x 20 ml), se desecó con sulfato de sodio, se filtró y se concentró, dando el producto en bruto. El residuo se purificó mediante cromatografía CombiFlash, eluyendo con acetato de etilo al 30 % en n-hexano, dando 4-(5-(((5-clorotiofen-2-il)metil)amino)-1-pivaloil-1H-pirazol-3-il)-W,W-dimetilpiperidin-1-sulfonamida (Compuesto 138, 0,09 g, rendimiento: 28 %) m/z 488,53 [M+H]+ RMN de 1H (DMSO-d6, 400 MHz, 12445) 5: 7,69 (t, J = 6,2 Hz, 1H), 6,97 (c, J = 3,7 Hz, 2H), 5,42 (s, 1H), 4,42 (d, J = 6,1 Hz, 2H), 3,57 (d, J = 12,3 Hz, 2H), 2,95 (t, J = 10,9 Hz, 2H), 2,75 (s, 6H), 2,69-2,59 (m, 1H), 2,01 -1,89 (m, 2H), 1,58 (td, J = 15,2, 3,7 Hz, 2H), 1,41 (s, 9H) ppm.
Ejemplo 190-Preparación del compuesto 139
La síntesis del Compuesto 139 siguió el procedimiento del siguiente Procedimiento general 5c:

A una solución enfriada (0 0C) de ácido 3-hidroxi-2,2-dimetilpropanoico (Compuesto 137, 0,15 g, 1,3 mmol, 1,5 eq) en acetonitrilo (12 ml), se añadió tetrafluoroborato de W,W,W',W'-tetrametil-O-(benzotriazol-1-il)uronio (TBTU, 0,42 g, I , 3 mmol, 1,5 eq) y, a continuación, (diisopropil)etilamina (DIEA, 0,24 ml, 2,6 mmol, 3,0 eq). Después de agitar durante 30 minutos, se añadió a la mezcla 4-(5-(((5-clorotiofen-2-il)metil)amino)-1-(3-hidroxi-2,2-dimetilpropanoil)-1H-pirazol-3-il)-W,W-dimetilpiperidin-1-sulfonamida (Compuesto 137, 0,35 g, 0,9 mmol, 1,0 eq), y se agitó la mezcla a temperatura ambiente durante 12 horas. El progreso de la reacción se controló mediante TLC. Una vez completada, se vertió la mezcla en agua (10 ml) y se extrajo con acetato de etilo (3 x 20 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se evaporaron, obteniéndose un residuo. El compuesto en bruto se purificó mediante HPLC preparativa eluyendo con agua y acetonitrilo como fase móvil en una columna Sunfire C18 de 5 micrómetros. Las fracciones purificadas se liofilizaron, dando 4-(5-(((5-clorotiofen-2-il)metil)amino)-1-(3-hidroxi-2,2-dimetilpropanoil)-1H-pirazol-3-il)-W,A/-dimetilpiperidin-1-sulfonamida (Compuesto 139, 0,05 g, rendimiento: 11 %) m/z 504,48 [M+H]+ RMN de 1H (DMSO-d6, 400 MHz, 13050) 5: 7,69 (t, J = 6,1 Hz, 1H), 6,97 (d, J = 2,3 Hz, 2H), 5,41 (s, 1H), 4,84 (t, J = 5,3 Hz, 1H), 4,41 (d, J = 6,0 Hz, 2H), 3,87 (d, J = 5,3 Hz, 2H), 3,57 (d, J = 12,1 Hz, 2H), 2,95 (t, J = 10,9 Hz, 2H), 2,75 (s, 6H), 2,62 (dd, J = 22,7, 11,4 Hz, 1H), 1,92 (d, J = 11,5 Hz, 2H), 1,57 (dd, J = 21,0, I I , 1 Hz, 2H), 1,31 (s, 6H) ppm.
Ejemplo 191-Preparación del compuesto 140
La síntesis del Compuesto 140 siguió el procedimiento del siguiente Procedimiento general 25:

A una solución de W-((5-clorotiofen-2-il)metil)-3-(piperidin-4-il)-1 H-pirazol-5-amina (1,0 g, 2,5 mmol, 1,0 eq) en acetonitrilo (15 ml), se añadió carbonato de potasio (2,0 g, 14,7 mmol, 6,0 eq). Después de agitar la mezcla de reacción a temperatura ambiente durante 30 minutos, se añadió trifluorometanosulfonato de 2-ciclopropoxietilo (0,576 g, 2,5 mmol, 1 eq) y se dejó la mezcla de reacción en agitación a temperatura ambiente durante 16 horas. Una vez completada (controlada mediante CL-EM), se concentró la mezcla de reacción, dando un producto en bruto que se purificó mediante HPLC preparativa utilizando agua-acetonitrilo como fase móvil, dando N-((5-clorotiofen-2-il)metil)-3-(1 -(2-ciclopropoxietil)piperidin-4-il)-1 H-pirazol-5-amina (Compuesto 140, 0,61 g, rendimiento: 65 %) m/z 381,56[M+1]+ RMN de 1H (400 MHz, DMSO) 5 11,65 (s, 1H), 6,97-6,81 (m, 2H), 5,80 (s, 1H), 5,35 (d, J = 39,2 Hz,
1H), 4,29 (d, J = 6,0 Hz, 2H), 3,74 (s, 2H), 3,41-3,31 (m, 5H), 3,23 (s, 2H), 2,98 (s, 1H), 2,08-2,01 (m, 2H), 1,78 (s, 2H), 0,60-0,42 (m, 4H) ppm.
Ejemplo 192-Preparación del compuesto 141
La síntesis del Compuesto 141 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución enfriada (0 0C) de ácido piválico (0,2 g, 0,64 mmol, 1,2 eq) en THF ( 6 ml), se añadieron EDCI.HCl (0,15 g, 0,78 mmol, 1,5 eq) y trietilamina (TEA, 0,16 g, 1 , 6 mmol, 3,0 eq). Después de agitar durante 30 minutos, se añadieron HOBt (0,014 g, 0,1 mmol, 0,2 eq) y N-((5-clorotiofen-2-il)metil)-3-(1-(2-ciclopropoxietil)piperidin-4-il)-1H-pirazol-5-amina (Compuesto 140, 0,2 g, 0,52 mmol, 1 eq). Se agitó la mezcla de reacción a temperatura ambiente durante 16 horas. Una vez completada (controlada mediante CL-EM), se vertió la mezcla de reacción en agua (15 ml) y se extrajo con acetato de etilo (2 x 15 ml). Las fases orgánicas combinadas se lavaron con agua y salmuera, se desecaron sobre sulfato sódico, se filtraron y se evaporaron. El residuo se purificó mediante HPLC preparativa utilizando agua-acetonitrilo como fase móvil, dando 1-(5-((5-clorotiofen-2-il)metil)amino)-3-(1-(2-ciclopropoxietil)piperidin-4-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 141, 0,06 g, rendimiento: 25 %) m/z 465,02 [M+H]+ RMN de 1H (400 MHz, DMSO) 5 7,64 (t, J = 6,1 Hz, 1H), 6,96 (c, J = 3,8 Hz, 2H), 5,37 (s, 1H), 4,40 (d, J = 6,2 Hz, 2H), 3,52 (t, J = 6,0 Hz, 2H), 3,26 (ddd, J = 9,1, 5,9, 3,2 Hz, 1H), 2,85 (d, J = 11,6 Hz, 2H), 2,45 2,31 (m, 3H), 2,13-1,95 (m, 3H), 1,79 (d, J = 10,6 Hz, 2H), 1,56 (dd, J = 21,1, 11,8 Hz, 2H), 1,39 (s, 9H), 0,46-0,33 (m, 4H) ppm.
Ejemplo 193-Preparación del compuesto 142
La síntesis del Compuesto 142 siguió el siguiente procedimiento del Procedimiento general 5b :

A una solución de ácido hidroxipiválico (0,093 g, 0,78 mmol, 1,5 eq) en THF ( 6 ml), se añadieron anhídrido propilfosfónico (T3 P en solución al 50 % en acetato de etilo, 0,25 g, 0,8 mmol, 1,5 eq), DIEA (0,2 g, 1, 6 mmol, 3 eq), seguido de n-((5-clorotiofen-2-il)metil)-3-(1-(2-ciclopropoxietil)piperidin-4-il)-1 H-pirazol-5-amina (Compuesto 140, 0,2 g, 0,5 mmol, 1,0 eq). Se agitó la mezcla de reacción a temperatura ambiente durante 16 horas. Una vez completada (controlada mediante CL-EM), se vertió la mezcla de reacción en agua (10 ml) y se extrajo con acetato de etilo (2 x 25 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se desecaron sobre sulfato de sodio, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando agua-acetonitrilo como fase móvil, dando 1-(5-((5-clorotiofen-2-il)metil)amino)-3-(1-(2-ciclopropoxietil)piperidin-4-il)-1H-pirazol-1-il)-3-hidroxi-2,2-dimetilpropan-1-ona (Compuesto 142, 0,032 g, rendimiento: 12 %) m/z 481,1 [M+H]+ RMN de 1H (400 MHz, DMSO) 5 7,65 (d, J = 6,2 Hz, 1H), 6,96 (t, J = 3,4 Hz, 2H), 5,38 (s, 1H), 4,82 (t, J = 5,2 Hz, 1H), 4,40 (d, J = 6,2 Hz, 2H), 3,86 (d, J = 5,3 Hz, 2H), 3,52 (t, J = 5,9 Hz, 2H), 3,26 (d, J = 3,2 Hz, 1H), 2,87 (d, J = 11,1 Hz, 2H), 2,40 (dd, J = 28,5, 14,2 Hz, 3H), 2,04 (dd, J = 22,2, 11,0 Hz, 3H), 1,78 (s, 2H), 1,56 (d, J = 9,6 Hz, 2H), 1,30 (s, 6 H), 0,51-0,28 (m, 4H) ppm.
Ejemplo 194-Preparación del compuesto 143
La síntesis del Compuesto 143 siguió el procedimiento del siguiente Procedimiento general 25:

A una solución de W-((5-clorotiofen-2-il)metil)-3-(piperidin-4-il)-1 H-pirazol-5-amina (1,0 g, 3,4 mmol, 1,0 eq) en acetonitrilo (15 ml), se añadió carbonato de potasio (K2CO3 , 3,26 g, 23,7 mmol, 7 eq), seguido de trifluorometanosulfonato de 2,2-difluoro-2-(piridin-2-il)etilo (1,1 g, 3,7 mmol, 1,1 eq). Se agitó la mezcla a 80 0C durante 12 horas. El progreso de la reacción se controló mediante CL-EM. Se diluyó la mezcla de reacción con agua (50 ml), se extrajo con diclorometano (3 x 80 ml) y las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se concentraron, dando el producto en bruto. Este se purificó mediante cromatografía en columna, eluyendo con metanol al 2 % en diclorometano, dando W-((5-clorotiofen-2-il)metil)-3-(1-(2,2-difluoro-2-(piridin-2-il)etil)piperidin-4-il)-1 H-pirazol-5-amina (Compuesto 143, 0,4 g, 27 %); RMN de 1H (400 MHz, DMSO) 5 11,26 (s, 1H), 8,68 (d, J = 3,9 Hz, 1H), 7,98 (t, J = 7,7 Hz, 1H), 7,70 (d, J = 7,7 Hz, 1H), 7,59-7,49 (m, 1H), 6,90 (d, J = 3,7 Hz, 1H), 6,82 (d, J = 3,6 Hz, 1H), 5,62 (s, 1H), 5,23 (s, 1H), 4,26 (d, J = 6,1 Hz, 2H), 3,21 (t, J = 14,8 Hz, 2H), 2,81 (d, J = 11,7 Hz, 2H), 2,32 (dd, J = 29,7, 18,6 Hz, 3H), 1,78-1,62 (m, 2H), 1,39 (dd, J = 21,2, 12,6 Hz, 2H) ppm.
Ejemplo 195-Preparación del compuesto 144
La síntesis del Compuesto 144 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución enfriada (0 °C) de ácido piválico (77 mg, 0,76 mmol, 1,1 eq) en THF (10 ml), se añadieron DIPEA (221 mg, 1,7 mmol, 2,5 eq), HoBt (46 mg, 0,34 mmol, 0,5 eq) y, a continuación, EDCI.HCl (197 mg, 1,0 mmol, 1,5 eq). Después de agitar a 0 0C durante 30 minutos, se añadió a la mezcla W-((5-clorotiofen-2-il)metil)-3-(1-(2,2-difluoro-2-(piridin-2-il)etil)piperidin-4-il)-1 H-pirazol-5-amina (Compuesto 143, 300 mg, 0,7 mmol, 1,0 eq), y la mezcla se dejó en agitación a temperatura ambiente durante 12 horas. Una vez completada, se concentró la mezcla de reacción y se vertió en agua helada y se extrajo con acetato de etilo (3 x 50 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo/agua, dando 1-(5-(((5-clorotiofen-2-il)metil)amino)-3-(1-(2,2-difluoro-2-(piridina -2-il)etil)piperidin-4-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 144, 74,5 mg, rendimiento: 17 %) m/z 522,10 [M+1]+; RMN de 1H (400 MHz, DMSO) 5 8,68 (d, J = 4,8 Hz, 1H), 7,98 (t, J = 7,8 Hz, 1H), 7,70 (d, J = 7,8 Hz, 1H), 7,64 (t, J = 6,4 Hz, 1H), 7,56-7,49 (m, 1H), 6,95 (d, J = 3,8 Hz, 2H), 5,36 (s, 1H), 4,39 (d, J = 6,2 Hz, 2H), 3,21 (t, J = 14,8 Hz, 2H), 2,81 (d, J = 11,5 Hz, 2H), 2,33 (t, J = 11,5 Hz, 4H), 1,72 (d, J = 12,2 Hz, 2H), 1,51-1,40 (m, 2H), 1,38 (s, 9H) ppm.
Ejemplo 196-Preparación del compuesto 145
La síntesis del Compuesto 145 siguió el procedimiento del siguiente Procedimiento general 5a:

A una solución enfriada (0 0C) de ácido hidroxipiválico (89 mg, 0,76 mmol, 1,1 eq) en THF (10 ml), se añadieron DIPEA (221 mg, 1,7 mmol, 2,5 eq), HOBt (46 mg, 0,34 mmol, 0,5 eq) y, a continuación, EDCI.HCl (197 mg, 1,0 mmol, 1,5 eq). Después de agitar a 0 0C durante 30 minutos, se añadió a la mezcla W-((5-clorotiofen-2-il)metil)-3-(1-(2,2-difluoro-2-(piridin-2-il)etil)piperidin-4-il)-1H-pirazol-5-amina (Compuesto 143, 300 mg, 0,68 mmol, 1,0 eq) y se agitó la mezcla a temperatura ambiente durante otras 12 horas. Una vez completada, se concentró la mezcla de reacción, se vertió en agua helada y se extrajo con acetato de etilo (3 x 50 ml). Las capas orgánicas combinadas se concentraron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo al 100 % y agua al 100 %, dando 1-(5-(((5-clorotiofen-2-il)metil)amino)-3-(1-(2,2-difluoro-2-(piridin-2-il)etil)piperidin-4-il)-1H-pirazol-1-il)-3-hidroxi-2,2-dimetilpropan-1-ona (Compuesto 145, 76 mg, rendimiento: 21 %) m/z 540,63 [M+1]+; RMN de 1H (400 MHz, DMSO) 5 8,68 (d, J = 4,4 Hz, 1H), 7,97 (td, J = 7,8, 1,7 Hz, 1H), 7,71 (s, 1H), 7,63 (d, J = 6,3 Hz, 1H), 7,54 (dd, J = 7,4, 4,8 Hz, 1H), 7,03-6,89 (m, 2H), 5,35 (s, 1H), 4,81 (t, J = 5,4 Hz, 1H), 4,39 (d, J = 6,2 Hz, 2H), 3,84 (d, J = 5,4 Hz, 2H), 3,21 (t, J = 14,8 Hz, 2H), 2,82 (d, J = 11,5 Hz, 2H), 2,34 (dd, J = 23,2, 11,6 Hz, 4H), 1,72 (d, J = 11,4 Hz, 2H), 1,50-1,35 (m, 2H), 1,29 (s, 6H) ppm.
Ejemplo 197-Preparación del compuesto 146
La síntesis del Compuesto 146 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de ácido ciclohexanocarboxílico (181 mg, 1,51 mmol) en diclorometano (10 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 361 mg, 1,89 mmol), seguido de hidroxibenzotriazol (HOBt, 511 mg, 3,78 mmol). Se agitó la mezcla de reacción durante 10 minutos y, a continuación, se añadió a la mezcla diisopropiletilamina (DIEA, 0,64 ml, 3,78 mmol), seguida de 4-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)piperidin-1-carboxilato ferc-butílico (Compuesto 15, 500 mg, 1,26 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 30 %/éter de petróleo, produciendo el producto 4-(5-(((5-clorotiofen-2-il)metil)amino)-1 -(ciclohexanocarbonil)-1 H-pirazol-3-il)piperidin-1 -carboxilato fercbutílico (Compuesto 146, 487 mg, rendimiento: 60 %) m/z 508,16 [M+1]+; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,7.
Ejemplo 198-Preparación del compuesto 147
La síntesis del Compuesto 147 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de 4-(5-(((5-clorotiofen-2-il)metil)amino)-1-(ciclohexanocarbonil)-1H-pirazol-3-il)piperidin-1 -carboxilato ferc-butílico (200 mg, 0,39 mmol) en éter dietílico (2 ml), se añadió una solución de cloruro de hidrógeno (1 M) en éter dietílico (2 ml). Se agitó la mezcla de reacción durante 2 h y, a continuación, se evaporó a presión reducida, se trituró con éter dietílico y acetona, produciendo el producto clorhidrato de (5-(((5-clorotiofen-2-il)metil)amino)-3-(piperidin-4-il)-1H-pirazol-1-il)(ciclohexil)metanona (Compuesto 147, 100 mg, rendimiento: 64 %) en forma de un sólido blanquecino. m/z 407,27 [(M-HCl)+1]+; RMN de *H (400 MHz, DMSO-d6) 58,70 (s a, 1H), 8,35 (s
a, 1H), 7,65-7,63 (m, 1H), 6,95 (s, 2H), 5,39 (s, 1H), 4,43-4,41 (m, 2H), 3,40-3,26 (m, 3H), 2,98-2,77 (m, 3H), 2,01 1,67 (m, 8H), 1,41-1,24 (m, 4H); Sistema de TLC: metanol al 10 % en diclorometano. Fr: 0,15.
Ejemplo 199-Preparación del compuesto 148
La síntesis del Compuesto 148 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de ácido ciclopentanocarboxílico (172 mg, 1,51 mmol) en diclorometano (10 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 361 mg, 1,89 mmol), seguido de hidroxibenzotriazol (HOBt, 511 mg, 3,78 mmol). Se agitó la mezcla de reacción durante 10 minutos y, a continuación, se añadió a la mezcla diisopropiletilamina (DIEA, 0,64 ml, 3,78 mmol), seguida de 4-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)piperidin-1-carboxilato ferc-butílico (Compuesto 15, 500 mg, 1,26 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 30 %/éter de petróleo, produciendo el producto 4-(5-(((5-clorotiofen-2-il)metil)amino)-1 -(ciclopentanocarbonil)-1 H-pirazol-3-il)piperidin-1 -carboxilato fercbutílico (Compuesto 148, 323 mg, rendimiento: 52 %) m/z 494,16 [M+1]+; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,7.
Ejemplo 200-Preparación del compuesto 149
La síntesis del Compuesto 149 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de 4-(5-(((5-clorotiofen-2-il)metil)amino)-1-(ciclopentanocarbonil)-1H-pirazol-3-il)piperidin-1 -carboxilato ferc-butílico (200 mg, 0,40 mmol) en éter dietílico (2 ml), se añadió una solución de cloruro de hidrógeno (1 M) en éter dietílico (2 ml). Se agitó la mezcla de reacción durante 2 h y, a continuación, se evaporó a presión reducida, se trituró con éter dietílico y acetona, produciendo el producto (5-(((5-clorotiofen-2-il)metil)amino)-3-(piperidin-4-il)-1H-pirazol-1-il)(ciclopentil)metanona (Compuesto 149, 60 mg, rendimiento: 38 %) en forma de un sólido blanquecino. m/z 393,25 [(M-HCl)+1]+; RMN de 1H (400 MHz, DMSO-da) 58,70 (s a, 1H), 8,35 (s a, 1H), 7,65 7,63 (m, 1H), 6,95 (s, 2H), 5,39 (s, 1H), 4,43-4,41 (m, 2H), 3,40-3,26 (m, 3H), 2,98-2,77 (m, 3H), 2,01-1,67 (m, 8H), 1,41-1,24 (m, 4H); Sistema de TLC: metanol al 10 % en diclorometano. Fr: 0,15.
Ejemplo 201-Preparación del compuesto 150
La s ín te s is de l
Compuesto 150
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5
:

A una solución enfriada (0 0C) de ácido benzoico (184 mg, 1,51 mmol) en diclorometano (10 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 361 mg, 1,89 mmol), seguido de hidroxibenzotriazol (HOBt, 511 mg, 3,78 mmol). Se agitó la mezcla de reacción durante 10 minutos y, a continuación, se añadió a la mezcla diisopropiletilamina (DIEA, 0,64 ml, 3,78 mmol), seguida de 4-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)piperidin-1-carboxilato ferc-butílico (Compuesto 15, 500 mg, 1,26 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 30 %/éter de petróleo, produciendo el producto 4-(1 -benzoil-5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)piperidin-1 -carboxilato ferc-butílico (Compuesto 150, 487 mg, rendimiento: 60 %) m/z 502,16 [M+1]+; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,6.
Ejemplo 202-Preparación del compuesto 151
La síntesis del Compuesto 151 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de 4-(1-benzoil-5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)piperidin-1-carboxilato ferc-butílico (200 mg, 0,40 mmol) en acetato de etilo (3 ml), se añadió una solución de cloruro de hidrógeno (1 M) en acetato de etilo (3 ml). Se agitó la mezcla de reacción durante 6 h y, a continuación, se evaporó a presión reducida y se trituró con éter dietílico y acetona. El residuo se recristalizó en alcohol isopropílico, dando el producto (5-(((5-clorotiofen-2-il)metil)amino)-3-(piperidin-4-il)-1 H-pirazol-1 -il)(fenil) clorhidrato de metanona (Compuesto 151, 40 mg, rendimiento: 25 %) en forma de un sólido blanquecino. m/z 401,26 [(M-HCl)+1]+; RMN de ^H (400 MHz, DMSO-da) 5 8,79 (s a, 1H), 8,48 (s a, 1H), 7,97-7,94 (m, 2H), 7,81-7,79 (m, 1H), 7,64-6,97 (s, 3H), 5,51 (s, 1H), 4,51-4,49 (m, 2H), 3,31-3,23 (m, 2H), 2,99-2,77 (m, 3H), 2,02-1,96 (m, 2H), 1,78-1,66 (m, 2H); Sistema de TLC: metanol al 10 % en diclorometano. Fr: 0,15.
Ejemplo 203-Preparación del compuesto 152
La s ín te s is de l
Compuesto 152
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5
:

A una solución enfriada (0 0C) de ácido 2,3-dihidrobenzo[b][1,4]dioxin-5-carboxílico (273 mg, 1,51 mmol) en diclorometano (10 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 361 mg, 1,89 mmol), seguido de hidroxibenzotriazol (HOBt, 511 mg, 3,78 mmol). Se agitó la mezcla de reacción durante 10 minutos y, a continuación, se añadió a la mezcla diisopropiletilamina (DIEA, 0,64 ml, 3,78 mmol), seguida de 4-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)piperidin-1-carboxilato ferc-butílico (Compuesto 15, 500 mg, 1,26 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 30 %/éter de petróleo, produciendo el producto 4-(5-(((5-clorotiofen-2-il)metil)amino)-1-(2,3-dihidrobenzo[b][1,4]dioxin-5-carbonil)-1 H-pirazol-3-il)piperidin-1 -carboxilato ferc-butílico (Compuesto 152, 318 mg, rendimiento: 45 %) m/z 560,16 [M+1]+; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,6.
Ejemplo 204-Preparación del compuesto 153
La síntesis del Compuesto 153 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de 4-(5-(((5-clorotiofen-2-il)metil)amino)-1-(2,3-dihidrobenzo[b][1,4]dioxin-5-carbonil)-1 H-pirazol-3-il)piperidin-1 -carboxilato ferc-butílico (250 mg, 0,45 mmol) en acetato de etilo (3 ml), se añadió una solución de cloruro de hidrógeno (1 M) en acetato de etilo (3 ml). Se agitó la mezcla de reacción durante 7 h y, a continuación, se evaporó a presión reducida y se trituró con éter dietílico y acetona. El residuo se recristalizó en alcohol isopropílico, dando el producto clorhidrato de (5-(((5-clorotiofen-2-il)metil)amino)-3-(piperidin-4-il)-1H-pirazol-1-il)(2,3-dihidrobenzo[b][1,4]dioxin-5-il)metanona (Compuesto 153, 150 mg, rendimiento: 67 %) en forma de un sólido blanquecino. m/z 401,26 [(M-HCl)+1]+; RMN de 1H (400 MHz, DMSO-d6) 58,79 (s a, 1H), 8,48 (s a, 1H), 7,97 7,94 (m, 2H), 7,81-7,79 (m, 1H), 7,04-6,97 (m, 5H), 5,51 (s, 1H), 4,51-4,49 (m, 2H), 3,31-3,23 (m, 2H), 2,99-2,77 (m, 3H), 2,02-1,96 (m, 2H), 1,78-1,66 (m, 2 H); Sistema de TLC: metanol al 5 % en diclorometano. Fr: 0,1.
Ejemplo 205-Preparación del compuesto 154
La s ín te s is de l
Compuesto 154
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5
:
A una solución enfriada (0 °C) de ácido furan-3-carboxílico (169 mg, 1,51 mmol) en diclorometano (10 ml) bajo nitrógeno se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 361 mg, 1,89 mmol), seguida de hidroxibenzotriazol (HOBt, 511 mg, 3,78 mmol). Se agitó la mezcla de reacción durante 10 minutos y, a continuación, se añadió a la mezcla diisopropiletilamina (DIEA, 0,64 ml, 3,78 mmol), seguida de 4-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)piperidin-1-carboxilato ferc-butílico (Compuesto 15, 500 mg, 1,26 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 30 %/éter de petróleo, produciendo el producto 4-(5-(((5-clorotiofen-2-il)metil)amino)-1 -(furan-3-carbonil)-1 H-pirazol-3-il)piperidin-1 -carboxilato ferc-butílico (Compuesto 154, 235 mg, rendimiento: 38 %) m/z 492,16 [M+1]+; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,6.
Ejemplo 206-Preparación del compuesto 155
La síntesis del Compuesto 155 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de 4-(5-(((5-clorotiofen-2-il)metil)amino)-1-(furan-3-carbonil)-1H-pirazol-3-il)piperidin-1-carboxilato ferc-butílico (100 mg, 0,20 mmol) en acetato de etilo (3 ml), se añadió una solución de cloruro de hidrógeno (1 M) en acetato de etilo (4 ml). Se agitó la mezcla de reacción durante 6 h y, a continuación, se evaporó a presión reducida y se trituró con éter dietílico y acetona. El residuo se recristalizó en alcohol isopropílico, dando el producto (5-(((5-clorotiofen-2-il)metil)amino)-3-(piperidin-4-il)-1 H-pirazol-1 -il)(furan-Clorhidrato de 3-il)metanona (Compuesto 155, 30 mg, rendimiento: 34 %) en forma de un sólido blanquecino. m/z 391,28 [(M-HCl)+1]+; RMN de ]H (400 MHz, DMSO-da) 5 8,82 (s a, 1H), 8,75 (s, 1H), 8,52 (s a, 1H), 7,86-7,84 (m, 1H), 7,82-7,77 (m, 1H), 7,04 6,95 (m, 3H), 5,50 (s, 1H), 4,50-4,48 (m, 2H), 3,33-3,27 (m, 2H), 2,99-2,85 (m, 3H), 2,11-2,07 (m, 2H), 1,81-1,77 (m, 2H); Sistema de TLC: metanol al 5 % en diclorometano. Fr: 0,1.
Ejemplo 207-Preparación del compuesto 156
La s ín te s is de l
Compuesto 156
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5
:

A una solución enfriada (0 0C) de ácido 2,4-dimetoxibenzoico (274 mg, 1,51 mmol) en diclorometano (10 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 361 mg, 1,89 mmol), seguido de hidroxibenzotriazol (HOBt, 511 mg, 3,78 mmol). Se agitó la mezcla de reacción durante 10 minutos y, a continuación, se añadió a la mezcla diisopropiletilamina (DIEA, 0,64 ml, 3,78 mmol), seguida de 4-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)piperidin-1-carboxilato ferc-butílico (Compuesto 15, 500 mg, 1,26 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 30 %/éter de petróleo, produciendo el producto 4-(5-(((5-clorotiofen-2-il)metil)amino)-1 -(2,4-dimetoxibenzoil)-1 H-pirazol-3-il)piperidin-1 -carboxilato fercbutílico (Compuesto 156, 324 mg, rendimiento: 45 %) m/z 562,16 [M+1]+; Sistema de t LC: acetato de etilo al 50 % en hexano. Fr: 0,6.
Ejemplo 208-Preparación del compuesto 157
La síntesis del Compuesto 157 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (-78 0C) de 4-(5-(((5-clorotiofen-2-il)metil)amino)-1-(2,4-dimetoxibenzoil)-1H-pirazol-3-il)piperidin-1 -carboxilato ferc-butílico (250 mg, 0,45 mmol) en éter dietílico (3 ml), se añadió una solución de cloruro de hidrógeno (1 M) en éter dietílico (3 ml). Se agitó la mezcla de reacción durante 1 h y, a continuación, se evaporó a presión reducida y se trituró con éter dietílico y acetona. El residuo se recristalizó en alcohol isopropílico, dando el producto clorhidrato de (5-(((5-clorotiofen-2-il)metil)amino)-3-(piperidin-4-il)-1 H-pirazol-1 -il)(2,4-dimetoxifenil)metanona (Compuesto 157, 10 mg, rendimiento: 4 %) en forma de un sólido blanquecino. m/z 461,26 [(M-HCl)+1]+; RMN de 1H (400 MHz, CDCls) 57,45-7,43 (m, 2H), 6,81-6,76 (m, 2H), 6,53-6,50 (m, 2H), 5,23 (s, 1H), 4,42-4,41 (m, 2H), 3,85 (s, 3H), 3,80 (s, 3H), 3,37-3,33 (m, 2H), 2,99-2,75 (m, 3H), 2,07-2,05 (m, 2H), 1,95-1,92 (m, 2H); Sistema de TLC: metanol al 5 % en diclorometano. Fr: 0,1.
Ejemplo 209-Preparación del compuesto 158
La síntesis del Compuesto 158 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 °C) de ácido 2,6-dimetilciclohexano-1-carboxílico (235 mg, 1,51 mmol) en diclorometano (10 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 361 mg, 1,89 mmol), seguido de hidroxibenzotriazol (HOBt, 511 mg, 3,78 mmol). Se agitó la mezcla de reacción durante 10 minutos y, a continuación, se añadió a la mezcla diisopropiletilamina (DIEA, 0,64 ml, 3,78 mmol), seguida de 4-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)piperidin-1-carboxilato terc-butílico (Compuesto 15, 500 mg, 1,26 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 30 %/éter de petróleo, produciendo el producto 4-(5-(((5-clorotiofen-2-il)metil)amino)-1-(2,6-dimetilciclohexano-1-carbonil)-1 H-pirazol-3-il)piperidin-1 -carboxilato terc-butílico (Compuesto 158, 371 mg, rendimiento: 55 %) m/z 536,16 [M+1 ]+; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,8.
Ejemplo 210-Preparación del compuesto 159
La síntesis del Compuesto 159 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (-60 °C) de 4-(5-(((5-clorotiofen-2-il)metil)amino)-1-(2,6-dimetilciclohexano-1-carbonil)-1H-pirazol-3-il)piperidin-1 -carboxilato terc-butílico (15 mg, 0,03 mmol) en éter dietílico (3 ml), se añadió una solución de cloruro de hidrógeno (1 M) en éter dietílico (0,5 ml). Se agitó la mezcla de reacción durante 1 h y, a continuación, se evaporó a presión reducida, se trituró con éter dietílico y acetona, produciendo el producto clorhidrato de (5-(((5-clorotiofen-2-il)metil)amino)-3-(piperidin-4-il)-1H-pirazol-1-il)(2,6-dimetilciclohexil)metanona (Compuesto 159, 10 mg, rendimiento: 71 %) en forma de un sólido blanquecino. m/z 435,34 [(M-HCl)+1]+; RMN de 1H (400 MHz, DMSO-d6) 5 8,66 (s a, 1H), 8,38 (s a, 1H), 7,72-7,69 (m, 1H), 6,96-6,94 (m, 2H), 5,40 (s, 1H), 4,44-4,43 (m, 2H), 3,40-3,26 (m, 3H), 3,00-2,77 (m, 3H), 2,32-2,28 (m, 1H), 2,01-1,87 (m, 3H), 1,75-1,65 (m, 3H), 1,61-1,42 (m, 4H), 0,82 (s, 3H), 0,81 (s, 3H); Sistema de TLC: metanol al 10 % en diclorometano. Fr: 0,15.
Ejemplo 211-Preparación del compuesto 160
La síntesis del Compuesto 160 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 °C) de 4-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)piperidin-1-carboxilato terc- butílico (3 g, 7,5 mmol) en éter dietílico (20 ml), se añadió una solución de cloruro de hidrógeno (1 M) en éter dietílico (20 ml). Se agitó la mezcla de reacción durante 2 h y, a continuación, se evaporó a presión reducida, se trituró con éter dietílico y acetona, produciendo el producto clorhidrato de N-((5-clorotiofen-2-il)metil)-3-(piperidin-4-il)-1H-pirazol-5-amina (Compuesto 160, 2,8 g, rendimiento: 91 %) en forma de un sólido blanquecino. m/z 297,21 ((M-HCl)+1 ]+; Sistema de TlC: metanol al 10 % en cloroformo. Fr: 0,1.
Ejemplo 212-Preparación del compuesto 161
La síntesis del Compuesto 161 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de clorhidrato de W-((5-clorotiofen-2-il)metil)-3-(piperidin-4-il)-1H-pirazol-5-amina (2,8 g, 3,01 mmol) en dioxano (70 ml), se añadió una solución de carbonato de sodio (0,63 g, 6,02 mmol) en agua (42 ml), seguida de succinimida Fmoc (0,81 g, 2,4 mmol). Se agitó la mezcla de reacción durante 2 h y se dejó que alcanzara la temperatura ambiente, a continuación, se vertió en agua (100 ml) y se extrajo con acetato de etilo (2 x 50 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo, produciendo el producto (9H-fluoren-9-il)metil-4-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)piperidin-1 -carboxilato (Compuesto 161, 1,2 g, rendimiento: 76 %) en forma de un sólido esponjoso blanquecino. m/z 505,3 [M+1]+; Sistema de TLC: metanol al 10 % en diclorometano. Fr: 0,5.
Ejemplo 213-Preparación del compuesto 162
La síntesis del Compuesto 162 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución de (9H-fluoren-9-il)metil-4-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)piperidin-1-carboxilato (Compuesto 161, 300 mg, 0,48 mmol) y ácido 3-metoxi-2,2-dimetilpropanoico (76 mg, 0,58 mmol) en cloroformo (5 ml) bajo nitrógeno a temperatura ambiente, se añadió tetrafluoroborato de O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio (TBTU, 171 mg, 0,53 mmol), seguido de trietilamina (TEA, 0,1 ml, 0,72 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 4 h, se diluyó con agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 20 %/éter de petróleo, produciendo (9H-fluoren-9-il)metil-4-(5-(((5-clorotiofen-2-il)metil)amino)-1-(3-metoxi-2,2-dimetilpropanoil)-1H-pirazol-3-il)piperidin-1-carboxilato (Compuesto 162, 300 mg, rendimiento: 82 %). m/z 605,19 [M+1]+; Sistema de TLC: metanol al 10 % en diclorometano. Fr: 0,9.
Ejemplo 214-Preparación del compuesto 163
La s ín te s is de l
Compuesto 163
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5
:

A una solución de (9H-fluoren-9-il)metil-4-(5-(((5-clorotiofen-2-il)metil)amino)-1-(3-metoxi-2,2-dimetilpropanoil)-1H-pirazol-3-il)piperidin-1 -carboxilato (200 mg, 0,31 mmol) en tetrahidrofurano (2 ml), se añadió piperidina (0,8 ml). Se agitó la mezcla de reacción durante 30 min y, a continuación, se evaporó a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con metanol al 10 %/diclorometano, produciendo el producto 1 -(5-(((5-clorotiofen-2-il)metil)amino)-3-(piperidin-4-il)-1 H-pirazol-1-il)-3-metoxi-2,2-dimetilpropan-1-ona (Compuesto 163, 40 mg, rendimiento: 31 %) en forma de un sólido blanquecino. m/z 411,22 [(M+1]+; RMN de 1H (400 MHz, CDCls) 59,80 (s a, 1H), 8,38 (s a, 1H), 7,49 (s, 1H), 6,75 (s, 2H), 5,14 (s, 1H), 4,35 4,34 (m, 2H), 3,50-3,47 (m, 2H), 3,27 (s, 3H), 3,10-3,07 (m, 2H), 2,86 (s a, 1H), 2,21-2,00 (m, 4H), 1,41 (s, 6H); Sistema de TLC: metanol al 10 % en diclorometano. Fr: 0,15.
Ejemplo 215-Preparación del compuesto 164
La síntesis del Compuesto 164 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución de (9H-fluoren-9-il)metil 4-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)piperidin-1-carboxilato (Compuesto 161, 300 mg, 0,48 mmol) y ácido 3-hidroxi-2,2-dimetilpropanoico (59 mg, 0,58 mmol) en cloroformo (5 ml) bajo nitrógeno a temperatura ambiente, se añadió tetrafluoroborato de O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio (TBTU, 171 mg, 0,53 mmol), seguido de trietilamina (TEA, 0,1 ml, 0,72 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 4 h, se diluyó con agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 20 %/éter de petróleo, produciendo (9H-fluoren-9-il)metil-4-(5-(((5-clorotiofen-2-il)metil)amino)-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)piperidin-1 -carboxilato (Compuesto 164, 250 mg, rendimiento: 71 %). m/z 620,19 [M+1]+; Sistema de TLC: metanol al 10 % en diclorometano. Fr: 0,85.
Ejemplo 216-Preparación del compuesto 165
La síntesis del Compuesto 165 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución de (9H-fluoren-9-il)metil-4-(5-(((5-clorotiofen-2-il)metil)amino)-1-(3-hidroxi-2,2-dimetilpropanoil)-1H-pirazol-3-il)piperidin-1 -carboxilato (250 mg, 0,31 mmol) en tetrahidrofurano (10 ml), se añadió piperidina (1 ml). Se agitó la mezcla de reacción durante 30 min y, a continuación, se evaporó a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con metanol al 10 %/diclorometano, produciendo el producto 1 -(5-(((5-clorotiofen-2-il)metil)amino)-3-(piperidin-4-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2
dimetilpropan-1-ona (Compuesto 165, 60 mg, rendimiento: 48 %) en forma de un sólido blanquecino. m/z 397,31 [(M+1]+; RMN de 1H (400 MHz, CDC^) 59,82 (s a, 1H), 7,49-7,47 (m, 1H), 6,77-6,76 (m, 2H), 5,21 (s, 1H), 4,38-4,36 (m, 2H), 4,00 (s a, 1H), 3,78 (s, 2H), 3,51-3,48 (m, 2H), 3,07-3,02 (m, 2H), 2,87-2,82 (m, 1H), 2,23-2,12 (m, 4H), 1,43 (s, 6H); Sistema de TLC: metanol al 10 % en diclorometano. Fr: 0,15.
Ejemplo 217 -Caracterización de Compuesto 166
La síntesis del Compuesto 166 siguió el procedimiento del siguiente Procedimiento general 5.

m/z 531,30 [M+H]+. RMN de 1H (400 MHz, CDCls) 5 7,4 (m, 3H), 7,0 (m, 2H), 6,8 (m, 2H), 5,2 (s, 1H), 4,4 (m, 2H), 4,1 (m, 2H), 3,8 (s, 3H), 2,8 (m, 2H), 2,6 (m, 2H), 1,8 (m, 2H), 1,4 (s, 9H).
Ejemplo 218 -Caracterización de Compuesto 167
La síntesis del Compuesto 167 siguió el siguiente procedimiento del General Procedimiento 6c.

m/z 431,25 [M+H]+. RMN de 1H (400 MHz, DMSO-da) 58,2-8,8 (m a, 3H), 7,7 (m, 1H), 7,5 (m, 1H), 7,4 (m, 1H), 7,1 (m, 1H), 7,0 (m, 3H), 5,4 (s, 1H), 4,5 (m, 2H), 3,7 (s, 3H), 3,2 (m, 2H), 2,9 (m, 2H), 2,7 (m, 2H), 1,9 (m, 2H), 1,6 (m, 2H).
Ejemplo 219 -Caracterización de Compuesto 168
La síntesis del Compuesto 168 siguió el procedimiento del siguiente Procedimiento general 7.

m/z 473,27 [M+H]+. RMN de 1H (400 MHz, CDCls) 5 7,4 (m, 3H), 7,0 (m, 2H), 6,8 (m, 2H), 5,2 (s, 1H), 4,5 (m, 1H), 4,4 (m, 2H), 3,8 (s, 3H), 3,8 (m, 1H), 3,1 (m, 1H), 2,7 (m, 2H), 2,1 (s, 3H), 1,8 (m, 2H), 1,5 (m, 1H).
Ejemplo 220 -Caracterización de Compuesto 169
La s ín te s is de l
Compuesto 169
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 7
.

m/z 515,24 [M+H]+. RMN de 1H (400 MHz, CDCI3) 5 7,5 (m, 3H), 7,0 (m, 2H), 6,8 (m, 2H), 5,2 (s, 1H), 4,4 (m, 1H), 4,3 (m, 2H), 3,8 (s, 3H), 2,9 (m, 2H), 2,7 (m, 1H), 1,9 (m, 2H), 1,6 (m, 1H), 1,5 (m, 1H), 1,3 (s, 9H).
Ejemplo 221-Caracterización de Compuesto 170
La síntesis del Compuesto 170 siguió el procedimiento del siguiente Procedimiento general 5.

m/z 507,24 [M+H]+. RMN de 1H (400 MHz, CDC^) 5 8,9 (s, 1H), 7,9 (m, 1H), 7,6 (m, 1H), 7,3 (m, 1H), 6,8 (m, 2H), 5,3 (s, 1H), 4,4 (m, 2H), 4,2 (s a, 2H), 2,9 (m, 2H), 2,7 (m, 1H), 1,9 (m, 2H), 1,6 (m, 2H), 1,4 (s, 9H).
Ejemplo 222-Caracterización de Compuesto 171
La síntesis del Compuesto 171 siguió el siguiente procedimiento del Procedimiento general 6c.

m/z 407,14 [M+H]+. RMN de 1H (400 MHz, DMSO-d6) 5 8,9 (s, 1H), 8,6 (s a, 1H), 8,3 (s a, 1H), 7,9 (m, 1H), 7,8 (m, 1H), 7,6 (m, 1H), 7,0 (m, 2H), 5,5 (s, 1H), 4,5 (m, 2H), 3,2 (m, 2H), 3,0 (m, 2H), 2,8 (m, 1H), 2,1 (m, 2H), 1,8 (m, 2H). Ejemplo 223-Caracterización de Compuesto 172
La s ín te s is de l
Compuesto 172
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 7
.

m/z 449,16 [M+H]+. RMN de 1H (400 MHz, CDCI3) 5 8,9 (s, 1H), 7,9 (m, 1H), 7,6 (m, 1H), 7,3 (m, 1H), 6,8 (m, 2H), 5,2 (s, 1H), 4,6 (m, 1H), 4,4 (m, 2H), 3,9 (m, 1H), 3,2 (m, 1H), 2,7-2,9 (m, 2H), 2,1 (s, 3H), 2,0 (m, 2H), 1,7 (m, 3H). Ejemplo 224 -Caracterización de Compuesto 173
La síntesis del Compuesto 173 siguió el procedimiento del siguiente Procedimiento general 7.

m/z 491,18 [M+H]+. RMN de 1H (400 MHz, CDC^) 5 8,9 (s, 1H), 7,9 (m, 1H), 7,6 (m, 1H), 7,3 (m, 1H), 6,8 (m, 2H), 5,2 (s, 1H), 4,4-4,5 (m, 4H), 3,0 (m, 2H), 2,8 (m, 1H), 2,0 (m, 2H), 1,7 (m, 2H), 1,3 (s, 9H).
Ejemplo 225-Caracterización de Compuesto 174
La síntesis del Compuesto 174 siguió el procedimiento del siguiente Procedimiento general 5.

m/z 481,33 [M+H]+. RMN de 1H (400 MHz, DMSO-ak) 57,7 (m, 1H), 7,0 (m, 2H), 5,4 (s, 1H), 4,4 (m, 2H), 3,9 (m, 2H), 2,8-2,9 (s a, 2H), 2,7 (m, 1H), 1,8 (m, 2H), 1,4 (m, 18H).
Ejemplo 226 -Caracterización de Compuesto 175
La síntesis del Compuesto 175 siguió el siguiente procedimiento de los Procedimientos Generales 6c y 7.

m/z 423,26 [M+H]+. RMN de 1H (400 MHz, CDCI3) 57,5 (m, 1H), 6,8 (m, 2H), 5,1 (s, 1H), 4,5 (m, 1H), 4,4 (m, 2H), 3,8 (m, 1H), 3,2 (m, 1H), 2,7-2,9 (m, 2H), 2,1 (s, 3H), 2,0 (m, 2H), 1,6 (m, 2H), 1,4 (s, 9H).
Ejemplo 227 -Caracterización de Compuesto 176
La síntesis del Compuesto 176 siguió el siguiente procedimiento de los Procedimientos Generales 6c y 7.

m/z 465,34 [M+H]+. RMN de 1H (400 MHz, CDCls) 5 7,5 (m, 1H), 6,8 (m, 2H), 5,1 (s, 1H), 4,3-4,4 (m, 4H), 3,0 (m, 2H), 2,8 (m, 1H), 2,0 (m, 2H), 1,6 (m, 2H), 1,5 (s, 9H), 1,3 (s, 9H).
Ejemplo 228-Preparación del producto intermedio 55
La síntesis del Producto intermedio 55 siguió el procedimiento del siguiente Procedimiento general 7 :

A una solución enfriada (0 °C) de ácido tetrahidrofurano-2-carboxílico (5,8 g, 43 mmol) en etanol (40 ml), se añadió cloruro de tionilo (9,6 ml, 129 mmol). Después de agitar a esta temperatura durante 2 horas, la mezcla se concentró a presión reducida. La mezcla se diluyó con agua (20 ml) y se extrajo en éter dietílico (3 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida, dando tetrahidrofurano-2-carboxilato de etilo (Producto intermedio 55, 6 g, rendimiento: 96 %) en forma de un residuo aceitoso que se utilizó sin mayor purificación en la siguiente etapa. m/z 145,03 [M+H]+; Sistema de TLC: metanol al 10 %-diclorometano; Fr: 0,6.
Ejemplo 229-Preparación del producto intermedio 56
La síntesis del Producto intermedio 56 siguió el procedimiento del siguiente Procedimiento general 2 :

A una solución enfriada (0 °C) de tetrahidrofuran-2-carboxilato de etilo (Producto intermedio 55, 6 g, 41,66 mmol en THF (100 ml), se añadió acetonitrilo seco (3,4 ml, 83,33 mmol). Después de 10 min, se añadió LHDMS (1 M en THF, 13,9 g, 83,3 mmol). Después de agitar a 0 °C durante 2 horas, se inactivó la mezcla con solución saturada de ácido cítrico hasta pH = 5 y se extrajo en acetato de etilo (3 x 200 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida, dando 3-oxo-3-(tetrahidrofuran-2-il)propanonitrilo (Producto intermedio 56, 7,5 g, rendimiento: 99 %) en forma de un residuo aceitoso que se utilizó sin mayor purificación en la siguiente etapa. m/z 140,02 [M+H]+; Sistema de TLC: metanol al 10 %-diclorometano; Fr: 0,4.
Ejemplo 230-Preparación del compuesto 177
La s ín te s is de l
Compuesto 177
s ig u ió el p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 3
:

A una solución de 3-oxo-3-(tetrahidrofuran-2-il)propanonitrilo (Producto intermedio 56, 7,5 g, 53,95 mmol) en etanol (90 ml), se añadió hidrato de hidrazina (N2H4.H2O, 4,04 ml, 24 mmol) y, a continuación, se calentó la mezcla de reacción a 90 °C durante 5 horas. La mezcla de reacción se enfrió hasta la temperatura ambiente y se evaporaron las sustancias volátiles. El residuo se purificó mediante cromatografía en columna de alúmina neutra (malla 100 300), eluyendo con MeOH al 2 %-diclorometano, produciendo 3-(tetrahidrofuran-2-il)-1H-pirazol-5-amina (Compuesto 177, 2 g, 24 %) en forma de un líquido naranja; m/z 154,13 [M+H]+; Sistema de TLC: metanol al 5 %-diclorometano. Fr: 0,5.
Ejemplo 231-Preparación del compuesto 178
La síntesis del Compuesto 178 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución enfriada (0 °C) de 3-(tetrahidrofuran-2-il)-1H-pirazol-5-amina (Compuesto 177, 2 g, 13,1 mmol) en DMF seco (20 ml), se añadieron 5-clorotiofen-2-carbaldehído (1,97 ml, 19,6 mmol) y ácido acético (2 ml). Se agitó la mezcla de reacción a temperatura ambiente durante 2 horas. (La formación de imina se observó como una mancha menos polar en TLC). La mezcla de reacción se enfrió (0 °C) y se añadió poco a poco cianoborohidruro de sodio (1,64 g, 26,1 mmol) y una cantidad catalítica de ácido acético. Después de 2 horas, se inactivó la mezcla de reacción con agua helada (50 ml) y se extrajo en acetato de etilo (3 x 100 ml). Las capas orgánicas combinadas se lavaron con NaHCO3 (2 x 50 ml), agua (2 x 50 ml) y salmuera (2 x 50 ml), y se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con éter dietílico al 10 %/diclorometano, produciendo N-((5-clorotiofen-2-il)metil)-3-(tetrahidrofuran-2-il)-1H-pirazol-5-amina (Compuesto 178, 650 mg, 6,2 %) en forma de un líquido gomoso de color amarillo claro; m/z 284,13 [M+H]+. RMN de 1H (400 MHz, DMSO-d6) 5 11,50 (s a, 1H), 6,91-6,84 (m, 2H), 5,74 (s a, 1H), 5,37 (s, 1H), 4,70 (s, 2H), 4,29 (s a, 2H), 3,84-3,79 (m, 1H), 3,71 -3,66 (m, 1H), 2,13-2,11 (m, 1H), 1,91 -1,79 (m, 3H) ppm; Sistema de TLC: acetato de etilo al 50 % en hexano, Fr: 0,5.
Ejemplo 232-Preparación del compuesto 179
La síntesis del Compuesto 179 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 °C) de N-((5-clorotiofen-2-il)metil)-3-(tetrahidrofuran-2-il)-1H-pirazol-5-amina (Compuesto 178, 200 mg, 0,70 mmol) en diclorometano seco (8 ml), se añadió trietilamina (TEA, 0,10 ml, 0,7 mmol), seguida de cloruro de trimetilacetilo (0,06 ml, 0,56 mmol). Después de agitar a temperatura ambiente durante 2 horas, se diluyó
la mezcla de reacción con agua (50 ml) y se extrajo en diclorometano (2 x 25 ml). Las capas orgánicas combinadas se lavaron con NaHCÜ3 saturado (2 x l0 ml) y agua (2 x 10 ml), y se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante CCF preparativa sobre gel de sílice (malla 100 200), eluyendo con acetato de etilo al 15 %/hexanos, produciendo 1-(5-(((5-clorotiofen-2-il)metil)amino)-3-(tetrahidrofuran-2-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 179, 35 mg, 14 %) en forma de un líquido incoloro. EM (IEN): m/z 368,2 [M+H]+. RMN de 1H (400 MHz, DMSO-de) 5 7,71 (s, 1H), 6,96 (s, 2H), 5,41 (s, 1H), 4,66 (s, 1H), 4,43 (s, 3H), 3,82-3,80 (m, 1H), 3,73-3,71 (m, 1H), 2,12-2,11 (m, 1H), 1,94-1,89 (m, 3H) ppm; Sistema de TLC: acetato de etilo al 30 % en hexano. Fr: 0,6.
Ejemplo 233-Preparación del compuesto 180
La síntesis del Compuesto 180 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de ácido tiofen-3-carboxílico (95 mg, 0,84 mmol) en diclorometano (10 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 201 mg, 1,05 mmol), seguido de hidroxibenzotriazol (HOBt, 283 mg, 2,1 mmol). Se agitó la mezcla de reacción durante 10 minutos y, a continuación, se añadió a la mezcla diisopropiletilamina (DIEA, 0,48 ml, 2,8 mmol), seguida de N-((5-clorotiofen-2-il)metil)-3-(tetrahidrofurano-2-il)-1H-pirazol-5-amina (Compuesto 178, 200 mg, 0,7 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua como fase móvil, dando el producto (5-(((5-clorotiofen-2-il)metil)amino)-3-(tetrahidrofuran-2-il)-1H-pirazol-1-il)(tiofen-3-il)metanona (Compuesto 180, 38 mg, rendimiento: 15 %) m/z 394,16 [M+1]+; RMN de 1H (400 MHz, DMSO) 5 8,87 (s, 1H), 7,84-7,83 (m, 1H), 7,81-7,77 (m, 1H), 7,66-7,64 (m, 1H), 7,00-6,96 (m, 2H), 5,54 (s, 1H), 4,73-4,70 (m, 1H), 4,51 (s, 2H), 3,88-3,83 (m, 1H), 3,77-3,72 (m, 1H), 2,21-2,14 (m, 1H), 1,99-1,87 (m, 3H) ppm; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,6.
Ejemplo 234-Preparación del compuesto 181
La síntesis del Compuesto 181 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de ácido 2-metoxibenzoico (127 mg, 0,84 mmol) en diclorometano (10 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 201 mg, 1,05 mmol), seguido de hidroxibenzotriazol (HOBt, 283 mg, 2,1 mmol). Se agitó la mezcla de reacción durante 10 minutos y, a continuación, se añadió a la mezcla diisopropiletilamina (DIEA, 0,48 ml, 2,8 mmol), seguida de N-((5-clorotiofen-2il)metil)-3-(tetrahidrofurano-2-il)-1H-pirazol-5-amina (Compuesto 178, 200 mg, 0,7 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua como fase móvil, dando el producto (5-(((5-clorotiofen-2-il)metil)amino)-3-(tetrahidrofuran-2-il)-1 H-pirazol-1-il)(2-metoxifenil)metanona (Compuesto 181, 32 mg, rendimiento: 11 %) m/z 394,16 [M+1]+; RMN de 1H (400 MHz, DMSO) 5 7,71-7,68 (m, 1H), 7,49-7,45 (m, 1H), 7,36-7,34 (m, 1H), 7,14-7,12 (m, 2H), 7,03-6,98 (m, 3H), 5,49 (s, 1H), 4,50-4,46 (m, 3H), 3,83-3,77 (m, 1H), 3,73 (s, 3H), 3,69-3,64 (m, 1H), 2,06-2,02 (m, 1H), 1,88-1,68 (m, 3H); Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,6.
Ejemplo 235-Preparación del producto intermedio 57
La síntesis del Producto intermedio 57 siguió el procedimiento del siguiente Procedimiento general 7 :

A una solución enfriada (0 0C) de ácido tetrahidro-2H-piran-4-carboxílico (10 g, 76 mmol) en etanol (100 ml), se añadió cloruro de tionilo (17,1 ml, 230 mmol). Después de agitar a esta temperatura durante 3 horas, la mezcla se concentró a presión reducida. La mezcla se diluyó con solución saturada de NaHCO3 (20 ml) y se extrajo en éter dietílico (3 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida, dando tetrahidro-2H-piran-4-carboxilato de etilo (Producto intermedio 57, 11 g, rendimiento: 91 %) en forma de un residuo aceitoso que se utilizó sin mayor purificación en la siguiente etapa. m/z 145,03 [M+H]+; Sistema de TLC: acetato de etilo al 30 % en hexano; Fr: 0,7.
Ejemplo 236-Preparación del producto intermedio 58
La síntesis del Producto intermedio 58 siguió el procedimiento del siguiente Procedimiento general 2 :

A una solución enfriada (0 0C) de tetrahidro-2H-piran-4-carboxilato de etilo (Producto intermedio 57, 3 g, 18,98 mmol) en THF (50 ml), se añadió acetonitrilo seco (1,5 ml, 37,97 mmol). Después de 10 min, se añadió LHDMS (1 M en THF, 63,4 g, 37,9 mmol). Después de agitar a 0 0C durante 2 horas, se inactivó la mezcla con solución saturada de ácido cítrico hasta p5 y se extrajo en acetato de etilo (3 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida, dando 3-oxo-3-(tetrahidro-2H-piran-4-il)propanonitrilo (Producto intermedio 58, 3,3 g, rendimiento: 99 %) en forma de un residuo aceitoso que se utilizó sin mayor purificación en la siguiente etapa. m/z 153,12 [M+H]+; Sistema de TLC: acetato de etilo al 50 % en hexano; Fr: 0,2.
Ejemplo 237-Preparación del compuesto 182
La síntesis del Compuesto 182 siguió el procedimiento del siguiente Procedimiento general 3 :

A una solución de 3-oxo-3-(tetrahidro-2H-piran-4-il)propanonitrilo (Producto intermedio 58, 3,3 g, 21,5 mmol) en etanol (90 ml), se añadió hidrato de hidrazina (N2 H4.H2O, 1,56 ml, 32,3 mmol) y, a continuación, se calentó la mezcla
de reacción hasta 90 0C durante 5 horas. La mezcla de reacción se enfrió hasta la temperatura ambiente y se evaporaron las sustancias volátiles. El residuo se purificó mediante cromatografía en columna de alúmina neutra (malla 100-300), eluyendo con MeOH al 2 %-diclorometano, produciendo 3-(tetrahidro-2H-piran-4-il)-1H-pirazol-5-amina (Compuesto 182, 1,7 g, 47 %) en forma de un líquido naranja; m/z 168,15 [M+H]+; Sistema de TLC: metanol al 10 %-diclorometano. Fr: 0,3.
Ejemplo 238-Preparación del compuesto 183
La síntesis del Compuesto 183 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución enfriada (0 0C) de 3-(tetrahidro-2H-piran-4-il)-1H-pirazol-5-amina (Compuesto 182, 2,1 g, 12,5 mmol) en DMF seca (20 ml), se añadió 5-clorotiofen-2-carbaldehído (1,96 ml, 18,8 mmol) y ácido acético (2 ml). Se agitó la mezcla de reacción a temperatura ambiente durante 2 horas. (La formación de imina se observó como una mancha menos polar en TLC). La mezcla de reacción se enfrió (0 0C) y se añadió poco a poco cianoborohidruro de sodio (2,58 g, 25,1 mmol) y una cantidad catalítica de ácido acético. Después de 2 horas, se inactivó la mezcla de reacción con agua helada (25 ml) y se extrajo en acetato de etilo (3 x 50 ml). Las capas orgánicas combinadas se lavaron con NaHCO3 (2 x 50 ml), agua (2 x 50 ml) y salmuera (2 x 50 ml), y se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con éter dietílico al 10 %/diclorometano, produciendo N-((5-clorotiofen-2-il)metil)-3-(tetrahidro-2H-pirano-4-il)-1 H-pirazol-5-amina (Compuesto 183, 600 mg, 16 %) en forma de un líquido gomoso de color amarillo claro; m/z 298,19 [M+H]+. RMN de 1H (400 MHz, DMSO-da) 5 11,50 (s a, 1H), 6,90 (s, 1H), 6,83 (s, 1H), 5,69 (s a, 1H), 5,29 (s, 1H), 4,28 (s, 2H), 3,88-3,84 (m, 2H), 3,39-3,34 (m, 3H), 2,76-2,68 (m, 1H), 1,76-1,72 (m, 2H), 1,60-1,50 (m, 2H) ppm; Sistema de TLC: acetato de etilo al 50 % en hexano, Fr: 0,5.
Ejemplo 239-Preparación del compuesto 184
La síntesis del Compuesto 184 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de N-((5-clorotiofen-2-il)metil)-3-(tetrahidro-2H-piran-4-il)-1H-pirazol-5-amina (Compuesto 183, 100 mg, 0,33 mmol) en diclorometano seco (8 ml), se añadió trietilamina (TEA, 0,06 ml, 0,7 mmol), seguida de cloruro de trimetilacetilo (0,03 ml, 0,26 mmol). Después de agitar a temperatura ambiente durante 3 horas, se diluyó la mezcla de reacción con agua (50 ml) y se extrajo en diclorometano (2 x 25 ml). Las capas orgánicas combinadas se lavaron con NaHCO3 saturado (2 x 10 ml) y agua (2 x 10 ml), y se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante CCF preparativa sobre gel de sílice (malla 100-200), eluyendo con acetato de etilo al 15 %/hexanos, produciendo 1-(5-(((5-clorotiofen-2-il)metil)amino)-3-(tetrahidrofuran-2-il)-1H-pirazol-1-il)-2,2-dimetilpropan-1-ona (Compuesto 184, 15 mg, 11 %) en forma de un líquido incoloro. EM (IEN): m/z 382,2 [M+H]+. r Mn de 1H (400 MHz, DMSO-ak) 5 7,69-7,66 (m, 1H), 6,96 (s, 2H), 5,40 (s, 1H), 4,41 (s, 1H), 4,43 (s, 3H), 3,88-3,85 (m, 2H), 3,43-3,34 (m, 2H), 2,74-2,67 (m, 1H), 1,79 1,76 (m, 2H), 1,64-1,54 (m, 2H), 1,40 (s, 9H) ppm; Sistema de t Lc : acetato de etilo al 50 % en hexano. Fr: 0,7.
E je m p lo 240 -P re p a ra c ió n de l c o m p u e s to 185
La s ín te s is de l
Compuesto 185
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5
:

A una solución enfriada (0 0C) de ácido tiofen-3-carboxílico (103 mg, 0,80 mmol) en diclorometano (10 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 192 mg, 1,00 mmol), seguido de hidroxibenzotriazol (HOBt, 271 mg, 2,01 mmol). Se agitó la mezcla de reacción durante 10 minutos y, a continuación, se añadió a la mezcla diisopropiletilamina (DIEA, 0,46 ml, 2,68 mmol), seguida de N-((5-clorotiofen-2-il)metil)-3-(tetrahidro-2H-piran-4-il)-1H-pirazol-5-amina (Compuesto 183, 200 mg, 0,67 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua como fase móvil, dando el producto (5-(((5-clorotiofen-2-il)metil)amino)-3-(tetrahidrofuran-2-il)-1H-pirazol-1-il)(tiofen-3-il)metanona (Compuesto 185, 30 mg, rendimiento: 11 %) m/z 408,23 [M+1]+; RMN de 1H (400 MHz, DMSO) 5 8,90 (s, 1H), 7,81-7,78 (m, 2H), 7,65-7,63 (m, 1H), 7,01-6,96 (m, 2H), 5,54 (s, 1H), 4,49-4,47 (m, 2H), 3,91-3,88 (m, 2H), 3,44-3,41 (m, 2H), 2,80-2,67 (m, 1H), 1,83-1,79 (m, 2H), 1,68-1,58 (m, 2H) ppm; Sistema de TLC: acetato de etilo al 50 % en hexano. Fr: 0,5.
Ejemplo 241-Preparación del compuesto 186
La síntesis del Compuesto 186 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de ácido 2-metoxibenzoico (91 mg, 0,60 mmol) en diclorometano (10 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 144 mg, 0,75 mmol), seguido de hidroxibenzotriazol (HOBt, 202 mg, 1,5 mmol). Se agitó la mezcla de reacción durante 10 minutos y, a continuación, se añadió a la mezcla diisopropiletilamina (DIEA, 0,34 ml, 1,5 mmol), seguida de N-((5-clorotiofen-2-il)metil)-3-(tetrahidro-2H-piran-4-il)-1H-pirazol-5-amina (Compuesto 183, 150 mg, 0,50 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua como fase móvil, dando el producto (5-(((5-clorotiofen-2-il)metil)amino)-3-(tetrahidrofuran-2-il)-1 H-pirazol-1-il)(2-metoxifenil)metanona (Compuesto 186, 25 mg, rendimiento: 12 %) m/z 432,27 [M+1]+; RMN de 1H (400 MHz, DMSO) 5 7,67-7,64 (m, 1H), 7,49-7,44 (m, 1H), 7,36-7,33 (m, 1H), 7,13-7,11 (m, 2H), 7,03-6,98 (m, 3H), 5,50 (s, 1H), 4,48-4,47 (m, 2H), 3,82-3,74 (m, 2H), 3,73 (s, 3H), 3,34-3,28 (m, 1H), 2,55-2,49 (m, 1H), 1,61-1,44 (m, 4H); Sistema de TLC: acetato de etilo al 30 % en hexano. Fr: 0,5.
Ejemplo 242-Preparación del producto intermedio 59
La s ín te s is de l
Producto intermedio 59
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 2
:

A una solución enfriada (-20 °C) de 2-etil pirrolidin-1,2-dicarboxilato 1-(terc-butílico) (15 g, 61,7 mmol) en THF (150 ml), se añadió acetonitrilo seco (2,5 ml, 61,7 mmol). Después de 10 min, se añadió LHDMS (1 M en THF, 15 g, 92,5 mmol). Después de agitar a 0 °C durante 2 horas, se inactivó la mezcla con solución saturada de ácido cítrico hasta p = 5 y se extrajo en acetato de etilo (3 x 100 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida, dando 2-(2-cianoacetil)pirrolidin-1-carboxilato de terc-butilo (Producto intermedio 59, 15 g, rendimiento: 99 %) en forma de un residuo aceitoso que se utilizó sin mayor purificación en la siguiente etapa. m/z 238,12 [M+H]+; Sistema de TLC: acetato de etilo al 50 % en éter de petróleo; Fr: 0,3.
Ejemplo 243-Preparación del compuesto 187
La síntesis del Compuesto 187 siguió el procedimiento del siguiente Procedimiento general 3 :

A una solución de 2-(2-cianoacetil)pirrolidin-1-carboxilato terc-butílico (Producto intermedio 59, 15 g, 61,7 mmol) en etanol (300 ml), se añadió hidrato de hidrazina (N2H4.H2O, 4,68 ml, 32,3 mmol) y, a continuación, se calentó la mezcla de reacción a 80 °C durante 16 horas. La mezcla de reacción se enfrió hasta la temperatura ambiente y se evaporaron las sustancias volátiles. El residuo se purificó mediante cromatografía en columna de alúmina neutra (malla 100-300), eluyendo con MeOH al 3 %-diclorometano, produciendo 2-(5-amino-1H-pirazol-3-il)pirrolidin-1-carboxilato terc-butílico (Compuesto 187, 5 g, 32 %) en forma de un sólido marrón claro; m/z 252,15 [M+H]+; Sistema de TLC: metanol al 5 %-diclorometano. Fr: 0,2.
Ejemplo 244-Preparación del compuesto 188
La síntesis del Compuesto 188 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución enfriada (0 °C) de 2-(5-amino-1H-pirazol-3-il)pirrolidin-1-carboxilato terc-butílico (Compuesto 187, 5 g, 19,8 mmol) en DMF seco (10 ml), se añadieron 5-clorotiofen-2-carbaldehído (2,75 ml, 25,8 mmol) y ácido acético (1 ml). Se agitó la mezcla de reacción a temperatura ambiente durante 2 horas. (La formación de imina se observó como una mancha menos polar en TLC). La mezcla de reacción se enfrió (0 °C) y se añadió poco a poco cianoborohidruro de sodio (2,58 g, 25,1 mmol) y una cantidad catalítica de ácido acético. Después de 2 horas, se inactivó la mezcla de reacción con agua helada (25 ml) y se extrajo en acetato de etilo (3 x 50 ml). Las capas orgánicas combinadas se lavaron con salmuera (2 x 50 ml) y se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con diclorometano al 1 %/MeOH, produciendo 2-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)pirrolidin-1 -carboxilato terc-butílico (Compuesto 188, 2,2 g, 27 %) en forma de un sólido marrón rojizo; m/z 383,29 [M+H]+. RMN de 1H (400 MHz, DMSO-d6) 5 11,34 (s a, 1H), 6,89 (s, 1H), 6,82 (s, 1H), 5,63 (s a, 1H), 5,24 (s, 1H),
4,29-4,27 (m, 2H), 3,38-3,24 (m, 2H), 2,1 (s a, 1H), 1,84 (s a, 3H), 1,38-1,22 (m a, 9H) ppm; Sistema de TLC: metanol al 5 %-diclorometano, Fr: 0,5.
Ejemplo 245-Preparación del compuesto 189
La síntesis del Compuesto 189 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de ácido 2-metoxibenzoico (1 g, 6,59 mmol) en diclorometano (20 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 2,1 g, 10,99 mmol), seguido de hidroxibenzotriazol (HOBt, 742 mg, 5,49 mmol). Se agitó la mezcla de reacción durante 10 minutos, se enfrió (0 °C) y, a continuación, se añadió a la mezcla diisopropiletilamina (DIEA, 2,8 ml, 16,49 mmol), seguida de 2-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)pirrolidin-1-carboxilato ferc-butílico (Compuesto 188, 2,1 g, 5,49 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 30 %/éter de petróleo, produciendo 2-(5-(((5-clorotiofen-2-il)metil)amino)-1 -(2-metoxibenzoil)-1 H-pirazol-3-il)pirrolidin-1 -carboxilato fercbutílico (Compuesto 189, 1,5 g, rendimiento: 53 %) en forma de un sólido blanquecino. m/z 417,37 [M+1]+; RMN de 1H (400 MHz, CDCls) 5 7,47-7,40 (m, 3H), 7,02-6,96 (m, 2H), 6,82-6,77 (m, 2H), 5,34 (s, 1H), 4,45-4,43 (m, 2H), 4,03-3,99 (m, 1H), 3,81 (s, 3H), 3,06-3,02 (m, 1H), 2,93-2,87 (m, 1H), 2,08-2,03 (m, 1H), 1,87-1,72 (m, 3H); Sistema de TLC: acetato de etilo al 70 % en éter de petróleo. Fr: 0,8.
Ejemplo 246-Preparación del compuesto 190
La síntesis del Compuesto 190 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (-10 0C) de 2-(5-(((5-clorotiofen-2-il)metil)amino)-1-(2-metoxibenzoil)-1H-pirazol-3-il)pirrolidin-1-carboxilato ferc-butílico (500 mg, 0,96 mmol) en acetato de etilo (10 ml), se añadió una solución de cloruro de hidrógeno (1 M) en acetato de etilo (2 ml). Se agitó la mezcla de reacción durante 1 hora y, a continuación, se evaporó a presión reducida, dando el producto clorhidrato de (5-(((5-clorotiofen-2-il)metil)amino)-3-(pirrolidin-2-il)-1H-pirazol-1-il)(2-metoxifenil)metanona (Compuesto 190, 350 mg, rendimiento: 74 %) en forma de un sólido blanquecino. m/z 417,37 [(M-HCl)+1]+; RMN de 1H (400 MHz, CDCls) 5 7,47-7,40 (m, 3H), 7,02-6,96 (m, 2H), 6,82 6,77 (m, 2H), 5,34 (s, 1H), 4,45-4,43 (m, 2H), 4,03-3,99 (m, 1H), 3,81 (s, 3H), 3,06-3,02 (m, 1H), 2,93-2,87 (m, 1H), 2,08-2,03 (m, 1H), 1,87-1,72 (m, 3H); Sistema de TLC: acetato de etilo al 70 % en éter de petróleo. Fr: 0,8.
E je m p lo 247 -P re p a ra c ió n de l c o m p u e s to 191
La s ín te s is de l
Compuesto 191
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5
:

A una solución enfriada (0 °C) de clorhidrato de (5-(((5-clorotiofen-2-il)metil)amino)-3-(pirrolidin-2-il)-1 H-pirazol-1 -il)(2-metoxifenil)metanona (170 mg, 0,39 mmol) en diclorometano (10 ml), se añadió trietilamina (0,1 ml, 0,74 mmol) seguido de cloruro de pivaloílo (36 mg, 0,29 mmol). Se agitó la mezcla de reacción durante 4 h a temperatura ambiente y, a continuación, se vertió en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se lavaron con salmuera (2 x 25 ml), se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100 200), eluyendo con acetato de etilo al 50 %/éter de petróleo, produciendo el producto 1-(2-(5-(((5-clorotiofen-2-il)metil) amino)-1-(2-metoxibenzoil)-1H-pirazol-3-il)pirrolidin-1-il)-2,2-dimetilpropan-1-ona (Compuesto 191, 80 mg, rendimiento: 43 %) en forma de un sólido blanquecino. m/z 501,29 [M+1]+; RMN de 1H (400 MHz, CDC^) 57,41 -7,32 (m, 3H), 6,99-6,93 (m, 2H), 6,80-6,76 (m, 2H), 5,18 (s, 1H), 5,17-5,13 (m, 1H), 4,42-4,40 (m, 2H), 3,80 (s, 3H), 3,62 (s, 2H), 1,96-1,87 (m, 4H), 1,56 (s, 9H); Sistema de TLC: metanol al 5 % en cloroformo. Fr: 0,6.
Ejemplo 248-Preparación del compuesto 192
La síntesis del Compuesto 192 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 °C) de clorhidrato de (5-(((5-clorotiofen-2-il)metil)amino)-3-(pirrolidin-2-il)-1 H-pirazol-1 -il)(2-metoxifenil)metanona (150 mg, 0,33 mmol) en diclorometano (15 ml), se añadió trietilamina (0,09 ml, 0,66 mmol) seguida de cloruro de isobutirilo (35 mg, 0,33 mmol). Se agitó la mezcla de reacción durante 4 h a temperatura ambiente y, a continuación, se vertió en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se lavaron con salmuera (2 x 25 ml), se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100 200), eluyendo con acetato de etilo al 60 %/éter de petróleo, produciendo el producto 1-(2-(5-(((5-clorotiofen-2-il)metil) amino)-1-(2-metoxibenzoil)-1H-pirazol-3-il)pirrolidin-1-il)-2-metilpropan-1-ona (Compuesto 192, 90 mg, rendimiento: 56 %) en forma de un sólido blanquecino. m/z 487,35 [M+1]+; RMN de 1H (400 MHz, CDCl3) 5 rotámeros 7,49-7,32 (m, 3H), 7,04-6,91 (m, 2H), 6,79-6,75 (m, 2H), 5,16 (s, 1H), 4,80-4,78 (m, 1H), 4,42-4,39 (m, 2H), 3,80 (s, 3H), 3,62-3,48 (m, 2H), 2,63-2,53 (m, 1H), 2,02-1,86 (m, 4H), 1,09-0,86 (m, 6H); Sistema de TLC: metanol al 5 % en cloroformo. Fr: 0,5.
E je m p lo 249 -P re p a ra c ió n de l c o m p u e s to 193
La s ín te s is de l
Compuesto 193
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5
:

A una solución enfriada (0 °C) de clorhidrato de (5-(((5-clorotiofen-2-il)metil)amino)-3-(pirrolidin-2-il)-1 H-pirazol-1 -il)(2-metoxifenil)metanona (130 mg, 0,28 mmol) en diclorometano (15 ml), se añadió trietilamina (0,08 ml, 0,57 mmol) seguida de cloruro de acetilo (22 mg, 0,28 mmol). Se agitó la mezcla de reacción durante 4 h a temperatura ambiente y, a continuación, se vertió en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se lavaron con salmuera (2 x 25 ml), se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100 200), eluyendo con metanol/diclorometano al 1 %, produciendo el producto 1-(2-(5-(((5-clorotiofen-2-il)metil)amino)-1-(2-metoxibenzoil)-1H-pirazol-3-il)pirrolidin-1-il)etan-1-ona (Compuesto 193, 15 mg, rendimiento: 12 %) en forma de un líquido gomoso incoloro. m/z 459,27 [M+1]+; RMN de 1H (400 MHz, CDC^) 5 rotámeros 7,52-7,28 (m, 3H), 7,04 6,94 (m, 2H), 6,82-6,77 (m, 2H), 5,31-5,06 (m, 2H), 4,79-4,76 (m, 1H), 4,48-4,38 (m, 2H), 3,81 (s, 3H), 3,62-3,48 (m, 2H), 2,29-2,01 (m, 1H), 1,99-1,86 (m, 6H); Sistema de TLC: metanol al 5 % en cloroformo. Fr: 0,2.
Ejemplo 250-Preparación del compuesto 194
La síntesis del Compuesto 194 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de ácido 2,3-dihidrobenzo[b][1,4]dioxin-5-carboxílico (282 mg, 1,57 mmol) en diclorometano (15 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 191 mg, 3,91 mmol), seguido de hidroxibenzotriazol (HOBt, 135 mg, 1,3 mmol). Se agitó la mezcla de reacción durante 10 minutos y, a continuación, se añadió a la mezcla diisopropiletilamina (DIEA, 0,69 ml, 3,91 mmol), seguido de 2-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)pirrolidin-1-carboxilato ferc-butílico (Compuesto 188, 500 mg, 1,30 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 25 %/éter de petróleo, produciendo el producto 2-(5-(((5-clorotiofen-2-il)metil)amino)-1-(2,3-dihidrobenzo[b][1,4]dioxin-5-carbonil)-1 H-pirazol-3-il)pirrolidin-1 -carboxilato ferc-butílico (Compuesto 194, 200 mg, rendimiento: 32 %) m/z 545,16 [M+1]+; Sistema de TLC: acetato de etilo al 70 % en éter de petróleo. Fr: 0,7.
Ejemplo 251-Preparación del compuesto 195
La s ín te s is de l
Compuesto 195
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5
:

A una solución enfriada (0 0C) de 2-(5-(((5-clorotiofen-2-il)metil)amino)-1-(2,3-dihidrobenzo[b][1,4]dioxin-5-carbonil)-1 H-pirazol-3-il)pirrolidin-1 -carboxilato ferc-butílico (200 mg, 0,96 mmol) en éter dietílico (2 ml), se añadió una solución de cloruro de hidrógeno (1 M) en éter dietílico (2 ml). Se agitó la mezcla de reacción durante 2 h y, a continuación, se evaporó a presión reducida, se trituró con éter dietílico y acetona, produciendo el producto clorhidrato de (5-(((5-clorotiofen-2-il)metil)amino)-3-(pirrolidin-2-il)-1 H-pirazol-1 -il)(2,3-dihidrobenzo[b][1,4]dioxin-5-il)metanona (Compuesto 195, 50 mg, rendimiento: 15 %) en forma de un sólido blanquecino. m/z 445,19 [(M-HCl)+1 ]+; RMN de 1H (400 MHz, DMSO-da) 59,85 (s a, 1H), 8,95 (s a, 1H), 7,86 (s, 1H), 7,03-6,88 (m, 5H), 5,78-5,74 (m, 1H), 4,51-4,49 (m, 1H), 4,41 (s, 1H), 4,27-4,21 (m, 4H), 3,17 (s, 1H), 2,24-2,18 (m, 1H), 1,98-1,88 (m, 3H); Sistema de TLC: metanol al 10 % en diclorometano. Fr: 0,25.
Ejemplo 252-Preparación del compuesto 196
La síntesis del Compuesto 196 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 °C) de ácido furan-3-carboxílico (175 mg, 1,57 mmol) en diclorometano (15 ml) bajo nitrógeno se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 191 mg, 3,91 mmol), seguida de hidroxibenzotriazol (HOBt, 135 mg, 1,3 mmol). Se agitó la mezcla de reacción durante 10 minutos y, a continuación, se añadió a la mezcla diisopropiletilamina (DIEA, 0,69 ml, 3,91 mmol), seguido de 2-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)pirrolidin-1 -carboxilato ferc-butílico Compuesto 188, 500 mg, 1,30 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente a temperatura ambiente, se vertió la mezcla de reacción en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 15 %/éter de petróleo, produciendo el producto 2-(5-(((5-clorotiofen-2-il)metil)amino)-1-(furan-3-carbonil)-1H-pirazol-3-il)pirrolidin-1-carboxilato ferc-butílico (Compuesto 196, 280 mg, rendimiento: 45 %) m/z 477,16 [M+1]+; Sistema de TLC: acetato de etilo al 70 % en éter de petróleo. Fr: 0,75.
Ejemplo 253-Preparación del compuesto 197
La s ín te s is de l
Compuesto 197
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5
:

A una solución enfriada (-30 0C) de 2-(5-(((5-clorotiofen-2-il)metil)amino)-1-(furan-3-carbonil)-1H-pirazol-3-il)pirrolidin-1-carboxilato ferc-butílico (100 mg, 0,20 mmol) en dioxano (3 ml), se añadió una solución de cloruro de hidrógeno (1 M) en éter dietílico (2 ml). Se agitó la mezcla de reacción durante 2 h y, a continuación, se evaporó a presión reducida. Se lavó la mezcla con n-pentano, éter dietílico y n-pentano, produciendo el producto clorhidrato de (5-(((5-clorotiofen-2-il)metil)amino)-3-(pirrolidin-2-il)-1H-pirazol-1-il)(furan-3-il)metanona (Compuesto 197, 30 mg, rendimiento: 38 %) en forma de un sólido blanquecino. m/z 377,21 [(M-HCl)+1]+; RMN de 1H (400 MHz, DMSO-d6) 5 9,64 (s a, 1H), 8,95 (s a, 1H), 9,01 (s, 1H), 8,99 (s, 1H), 8,00-7,98 (m, 1H), 7,88 (s, 1H), 7,06 (s, 1H), 6,99-6,97 (m, 2H), 5,69 (s, 1H), 4,69-4,51 (m, 3H), 3,32-3,29 (m, 2H), 2,39-2,33 (m, 1H), 1,96-1,92 (m, 3H); Sistema de TLC: metanol al 10 % en cloroformo. Fr: 0,1.
Ejemplo 254-Preparación del compuesto 198
La síntesis del Compuesto 198 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 °C) de 2-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)pirrolidin-1-carboxilato ferc- butílico (200 mg, 0,52 mmol) en dioxano (5 ml), se añadió una solución de cloruro de hidrógeno (1 M) en dioxano (5 ml). Se agitó la mezcla de reacción durante 8 h a temperatura ambiente y, a continuación, se evaporó a presión reducida. La mezcla se diluyó con solución saturada de NaHCO3 (20 ml) y se extrajo en acetato de etilo (3 x 50 ml). Las capas orgánicas combinadas se lavaron con salmuera (50 ml), se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con metanol/diclorometano al 5 %, produciendo el producto N-((5-clorotiofen-2-il)metil)-3-(pirrolidin-2-il)-1 H-pirazol-5-amina (Compuesto 198, 130 mg, rendimiento: 91 %) en forma de un sólido blanquecino. m/z 283,21 (M+1]+; Sistema de TLC: metanol al 10 % en cloroformo. Fr: 0,2.
Ejemplo 255-Preparación del compuesto 199
La síntesis del Compuesto 199 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de clorhidrato de N-((5-clorotiofen-2-il)metil)-3-(pirrolidin-2-il)-1 H-pirazol-5-amina (3,5 g, 11,0 mmol) en diclorometano (20 ml), se añadió trietilamina (0,72 ml, 16,5 mmol) seguida de cloruro de Fmoc (3,13 g, 12,1 mmol). Se agitó la mezcla de reacción durante 2 horas y, a continuación, se vertió en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de
sílice (malla 100-200), eluyendo con metanol/diclorometano al 5 %, produciendo el producto (9H-fluoren-9-il)metil-2-(5-(((5-clorot¡ofen-2-¡l)met¡í)amino)-1H-p¡razol-3-¡l)p¡rrol¡d¡n-1-carbox¡lato (Compuesto 199, 3,5 g, rendimiento: 63 %) en forma de un sólido esponjoso blanquecino. m/z 505,3 [M+1]+; Sistema de TLC: metanol al 10 % en cloroformo. Fr: 0,4.
Ejemplo 256-Preparación del compuesto 200
La síntesis del Compuesto 200 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución de 2-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)pirrolidin-1-carboxilato ferc-butílico (Compuesto 199, 500 mg, 0,99 mmol) y ácido 3-metoxi-2,2-dimetilpropanoico (180 mg, 1,18 mmol) en diclorometano (5 ml) bajo nitrógeno a temperatura ambiente, se añadió hexafluorofosfato de 3-óxido de 1-[¿>/s(dimetilamino)metileno]-1H-1,2,3-triazolo[4,5-b]piridinio(HATU, 564 mg, 1,48 mmol), seguido de diisopropiletilamina (DIEA, 0,32 ml, 1,48 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 16 h, se diluyó con agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua como fase móvil, dando el producto (9H-fluoren-9-il)metil-2-(5-(((5-clorotiofen-2-il)metil)amino)-1 -(2,4-dimetoxibenzoil)-1 H-pirazol-3-il)pirrolidin-1 -carboxilato (Compuesto 200, 140 mg, rendimiento: 21 %). m/z 669,19 [M+1]+; Sistema de t Lc : acetona al 10 % en diclorometano. Fr: 0,85.
Ejemplo 257-Preparación del compuesto 201
La síntesis del Compuesto 201 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución de (9H-fluoren-9-il)metil-2-(5-(((5-clorotiofen-2-il)metil)amino)-1-(2,4-dimetoxibenzoil)-1H-pirazol-3-il)pirrolidin-1 -carboxilato (140 mg, 0,20 mmol) en dimetilformamida (2 ml), se añadió diisopropiletilamina (DIEA, 142 mg, 1,70 mmol). Se agitó la mezcla de reacción durante 2 h y, a continuación, se evaporó a presión reducida. La mezcla se diluyó con agua y se extrajo con acetato de etilo. Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con metanol/diclorometano al 5 %, produciendo el producto (5-(((5-clorotiofen-2-il)metil)amino)-3-(pirrolidina -2-¡l)-1 H-pirazol-1 -il)(2,4-dimetoxifenil)metanona
(Compuesto 201, 45 mg, rendimiento: 48 %) en forma de un líquido gomoso. m/z 447,24 [(M+1]+; RMN de 1H (400 MHz, DMSO-d6) 5 9,64 (s a, 1H), 8,95 (s a, 1H), 9,11 (s, 1H), 7,84-7,81 (m, 1H), 7,39-7,37 (m, 1H), 7,02-6,98 (m, 2H), 6,67 (s, 1H), 6,60-6,57 (m, 1H), 5,72 (s, 1H), 4,50-4,37 (m, 3H), 3,83 (s, 3H), 3,74 (m, 3H), 3,19-3,14 (m, 2H), 2,22-2,17 (m, 1H), 1,96-1,88 (m, 3H); Sistema de TLC: metanol al 10 % en cloroformo. Fr: 0,3.
Ejemplo 258-Preparación del compuesto 202
La síntesis del Compuesto 202 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución de 2-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)pirrolidin-1-carboxilato ferc-butílico (Compuesto 199, 500 mg, 0,99 mmol) y ácido piválico (121 mg, 1,18 mmol) en diclorometano (5 ml) bajo nitrógeno a temperatura ambiente, se añadió hexafluorofosfato de 3-óxido de 1-[bis(dimetilamino)metileno]-1H-1,2,3-triazolo[4,5-b]piridinio (HATU, 564 mg, 1,48 mmol), seguido de diisopropiletilamina (DIEA, 0,32 ml, 1,48 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 16 h, se diluyó con agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua como fase móvil, dando el producto (9H-fluoren-9-il)metil-2-(5-(((5-clorotiofen-2-il)metil)amino)-1-pivaloil-1H-pirazol-3-il)pirrolidin-1 -carboxilato (Compuesto 202, 200 mg, rendimiento: 35 %). m/z 590,19 [M+1]+; Sistema de TLC: acetona al 10 % en diclorometano. Fr: 0,8.
Ejemplo 259-Preparación del compuesto 203
La síntesis del Compuesto 203 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución de (9H-fluoren-9-il)metil-2-(5-(((5-clorotiofen-2-il)metil)amino)-1 -pivaloil-1 H-pirazol-3-il)pirrolidin-1 -carboxilato (100 mg, 0,17 mmol) en metanol (2 ml), se añadió bicarbonato de sodio (142 mg, 1,70 mmol). Se agitó la mezcla de reacción durante 16 horas y, a continuación, se evaporó a presión reducida. La mezcla se diluyó con agua y se extrajo con acetato de etilo. Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con metanol/diclorometano al 8 %, produciendo el producto 1-(5-(((5-clorotiofen-2-il)metil)amino)-3-(pirrolidin-2-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona (Compuesto 203, 35 mg, rendimiento: 56 %) en forma de un líquido gomoso. m/z 367,23 [(M+1]+; RMN de 1H (400 MHz, DMSO-d6) 5 7,72-7,62 (m, 1H), 6,99 (s, 1H), 5,4 (s, 1H), 4,45-4,38 (m, 2H), 3,93-3,82 (m, 1H), 2,85-2,97 (m, 2H), 1,98-1,88 (m, 1H), 1,78-1,62 (m, 3H), 1,42 (s, 9H); Sistema de TLC: metanol al 10 % en cloroformo. Fr: 0,35.
Ejemplo 260-Preparación del compuesto 204
La s ín te s is de l
Compuesto 204
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5
:

A una solución de (9H-fluoren-9-il)metil-2-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)pirrolidin-1-carboxilato (Compuesto 199, 500 mg, 0,99 mmol) y ácido 3-hidroxi-2,2-dimetilpropanoico (146 mg, 1,2 mmol) en cloroformo (25 ml) bajo nitrógeno a temperatura ambiente, se añadió tetrafluoroborato de 0-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio (TBTU, 365 mg, 1,1 mmol), seguido de diisopropiletilamina (DIEA, 0,3 ml, 7,4 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 4 h, se diluyó con agua (50 ml) y se extrajo con cloroformo (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua como fase móvil, dando el producto (9H-fluoren-9-il)metil-2-(5-(((5-clorotiofen-2-il)metil)amino)-1 -(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)pirrolidin-1 -carboxilato (Compuesto 204, 250 mg, rendimiento: 42 %). m/z 605,19 [M+1]+; Sistema de TLC: metanol al 10 % en diclorometano. Fr: 0,9.
Ejemplo 261-Preparación del compuesto 205
La síntesis del Compuesto 205 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución de (9H-fluoren-9-il)metil-2-(5-(((5-clorotiofen-2-il)metil)amino)-1-(3-hidroxi-2,2-dimetilpropanoil)-1H-pirazol-3-il)pirrolidin-1 -carboxilato (100 mg, 0,16 mmol) en metanol (2 ml), se añadió bicarbonato de sodio (139 mg, 1,65 mmol). Se agitó la mezcla de reacción durante 16 horas y, a continuación, se evaporó a presión reducida. La mezcla se diluyó con agua y se extrajo con acetato de etilo. Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con metanol/diclorometano al 5 %, produciendo el producto 1 -(5-(((5-clorotiofen-2-il)metil)amino)-3-(pirrolidin-2-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona (Compuesto 205, 15 mg, rendimiento: 24 %) en forma de un semisólido. m/z 383,23 [(M+1]+; RMN de 1H (400 MHz, DMSO-da) 5 7,72-7,62 (m, 1H), 6,99 (s, 1H), 5,4 (s, 1H), 4,84-4,83 (m, 1H), 4,45-4,38 (m, 2H), 3,93-3,82 (m, 1H), 2,85-2,97 (m, 2H), 2,78-2,81 (m, 1H), 1,98-1,88 (m, 1H), 1,78-1,62 (m, 3H), 1,42 (s, 6H); Sistema de TLC: metanol al 5 % en cloroformo. Fr: 0,3.
Ejemplo 262-Preparación del compuesto 206
La s ín te s is de l
Compuesto 206
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5
:

A una solución de (9H-fluoren-9-il)metil-2-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)pirrolidin-1-carboxilato (Compuesto 199, 500 mg, 0,99 mmol) y ácido 4-metiltetrahidro-2H-piran-4-carboxílico (143 mg, 0,99 mmol) en diclorometano (5 ml) bajo nitrógeno a temperatura ambiente, se añadió hexafluorofosfato de 3-óxido de 1-[¿>/s(dimetilamino)metilen]-1H-1,2,3-triazolo[4,5-b]piridinio (HATU, 564 mg, 1,48 mmol), seguido de diisopropiletilamina (DIEA, 0,32 ml, 1,48 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 16 h, se diluyó con agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua como fase móvil, dando el producto (9H-fluoren-9-il)metil-2-(5-(((5-clorotiofen-2-il)metil)amino)-1-(4-metiltetrahidro-2H-piran-4-carbonil)-1 H-pirazol-3-il)pirrolidin-1 -carboxilato (Compuesto 206, 110 mg, rendimiento: 18 %). m/z 632,19 [M+1]+; Sistema de TLC: acetona al 10 % en diclorometano. Fr: 0,9.
Ejemplo 263-Preparación del compuesto 207
La síntesis del Compuesto 207 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución de (9H-fluoren-9-il)metil-2-(5-(((5-clorotiofen-2-il)metil)amino)-1-(4-metiltetrahidro-2H-piran-4-carbonil)-1 H-pirazol-3-il)pirrolidin-1 -carboxilato (100 mg, 0,16 mmol) en metanol (2 ml), se añadió bicarbonato de sodio (139 mg, 1,65 mmol). Se agitó la mezcla de reacción durante 16 horas y, a continuación, se evaporó a presión reducida. La mezcla se diluyó con agua y se extrajo con acetato de etilo. Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con metanol/diclorometano al 5 %, produciendo el producto (5-(((5-clorotiofen-2-il)metil)amino)-3-(pirrolidina-2-il)-1 H-pirazol-1 -il)(4-metiltetrahidro-2H-piran-4-il)metanona (Compuesto 207, 15 mg, rendimiento: 24 %) en forma de un semisólido. m/z 410,31 [(M+1]+; RMN de 1H (400 MHz, DMSO-da) 5 8,81 (s a, 1H), 7,85 (s a, 1H), 6,96 (s, 2H), 5,60 (s, 1H), 4,54-4,43 (m, 2H), 3,78 (s a, 2H), 3,45 (s a, 2H), 3,22 (s a, 2H), 2,43-2,21 (s a, 3H), 1,98-1,89 (m, 3H), 1,75 (s a, 2H), 1,54-1,50 (m, 3H); Sistema de TLC: metanol al 10 % en cloroformo. Fr: 0,35.
Ejemplo 264-Preparación del compuesto 208
La s ín te s is de l
Compuesto 208
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5
:

A una solución enfriada (0 °C) de ácido 3-metoxi-2,2-dimetilpropanoico (200 mg, 1,57 mmol) en diclorometano (15 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 191 mg, 3,91 mmol), seguido de hidroxibenzotriazol (HOBt, 135 mg, 1,3 mmol). Se agitó la mezcla de reacción durante 10 minutos y, a continuación, se añadió a la mezcla diisopropiletilamina (DIEA, 0,69 ml, 3,91 mmol), seguida de (9H-fluoren-9-il)metil-2-(5-(((5-clorotiofen-2-il)metil)amino)-1 H-pirazol-3-il)pirrolidin-1 -carboxilato (Compuesto 199, 500 mg, 1,30 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (50 ml) y se extrajo con diclorometano (2 x 25 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 20 %/éter de petróleo, produciendo el producto (9H-fluoren-9-il)metil-2-(5-(((5-clorotiofeno -2-il)metil)amino)-1-(3-metoxi-2,2-dimetilpropanoil)-1H-pirazol-3-il)pirrolidin-1-carboxilato (Compuesto 208, 200 mg, rendimiento: 31 %) m/z 497,26 [M+1]+; Sistema de TLC: acetato de etilo al 70 % en éter de petróleo. Fr: 0,7.
Ejemplo 265-Preparación del compuesto 209
La síntesis del Compuesto 209 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución de (9H-fluoren-9-il)metil-2-(5-(((5-clorotiofen-2-il)metil)amino)-1-(3-metoxi-2,2-dimetilpropanoil)-1H-pirazol-3-il)pirrolidin-1 -carboxilato (180 mg, 0,29 mmol) en metanol (2 ml), se añadió bicarbonato de sodio (200 mg, 2,91 mmol). Se agitó la mezcla de reacción durante 16 horas y, a continuación, se evaporó a presión reducida. La mezcla se diluyó con agua y se extrajo con acetato de etilo. Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con metanol/diclorometano al 8 %, produciendo el producto 1 -(5-(((5-clorotiofen-2-il)metil)amino)-3-(pirrolidin-2-il)-1 H-pirazol-1 -il)-3-metoxi-2,2-dimetilpropan-1 -ona (Compuesto 209, 15 mg, rendimiento: 24 %) en forma de un semisólido. m/z 397,25 [(M+1]+; RMN de 1H (400 MHz, CDCls) 57,51-7,50 (m, 1H), 6,80-6,79 (m, 1H), 6,76-6,75 (m, 1H), 5,42 (s, 1H), 4,64-4,61 (m, 1H), 4,36-4,35 (m, 2H), 3,79 (s, 2H), 3,41-3,40 (m, 1H), 3,27 (s, 3H), 2,37-2,32 (m, 1H), 2,17-1,97 (m, 4H), 1,42 (s, 6H); Sistema de TLC: metanol al 10 % en cloroformo. Fr: 0,2.
Ejemplo 266-Preparación del producto intermedio 60
La síntesis del Producto intermedio 60 siguió el procedimiento del siguiente Procedimiento general 7 :

A una solución enfriada (0 °C) de ácido azetidin-3-carboxílico (50 g, 494 mmol) en etanol (500 ml), se añadió gota a gota cloruro de tionilo (110 ml, 1483 mmol). Después de agitar a esta temperatura durante 2 horas, la mezcla se concentró a presión reducida. La mezcla se destiló junto con benceno (3 x 50 ml) y se concentró a presión reducida, dando clorhidrato de azetidin-3-carboxilato de etilo (Producto intermedio 60, 70 g, rendimiento: 96 %) en forma de un líquido gomoso que se utilizó sin mayor purificación en la siguiente etapa. m/z 130,03 [(M-HCl)+1 ]+; Sistema de TLC: metanol al 10 %-cloroformo; Fr: 0,6.
Ejemplo 267-Preparación del producto intermedio 61
La síntesis del Producto intermedio 61 siguió el procedimiento del siguiente Procedimiento general 7 :
Boc20 , TEA,

Producto intermedio 60 Producto intermedio 61
A una solución enfriada (0 °C) de clorhidrato de azetidin-3-carboxilato de etilo (70 g, 424 mmol) en diclorometano (700 ml), se añadió gota a gota trietilamina (119 ml, 848 mmol) durante 30 min, seguida de dicarbonato di-tercbutílico (111 g, 509 mmol). Después de agitar a esta temperatura durante 3 horas, la mezcla se concentró a presión reducida. Se diluyó la mezcla con agua (20 ml) y se extrajo con diclorometano (3 x 100 ml). Las capas orgánicas combinadas se lavaron con salmuera (2 x 10 ml), se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida, dando 3-etil-azetidin-1,3-dicarboxilato 1-(terc-butílico) (Producto intermedio 61,40 g, rendimiento: 42 %) en forma de un residuo aceitoso que se utilizó sin mayor purificación en la siguiente etapa. m/z 230,23 [M+H]+; Sistema de TLC: metanol al 10 %-diclorometano; Fr: 0,7.
Ejemplo 268-Preparación del producto intermedio 62
La síntesis del Producto intermedio 62 siguió el procedimiento del siguiente Procedimiento general 2 :

A una solución enfriada (0 °C) de 3-etil-azetidin-1,3-dicarboxilato 1-(terc-butílico) (Producto intermedio 61, 1 g, 4,36 mmol en THF (50 ml), se añadió acetonitrilo seco (0,32 ml, 8,7 mmol). Después de 10 min, se añadió LHDMS (1 M en THF, 8,7 ml, 8,7 mmol). Después de agitar a 0 durante 2 horas, se inactivó la mezcla con solución saturada de ácido cítrico hasta p5 y se extrajo en acetato de etilo (3 x 50 ml). Las capas orgánicas combinadas se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida, dando 3-(2-cianoacetil)azetidin-1-carboxilato de terc-butilo (Producto intermedio 62, 1,2 g, rendimiento: 99 %) en forma de un residuo aceitoso que se utilizó sin mayor purificación en la siguiente etapa. m/z 225,02 [M+H]+; Sistema de TLC: metanol al 10 %-diclorometano; Fr: 0,5.
Ejemplo 269-Preparación del compuesto 210
La síntesis del Compuesto 210 siguió el procedimiento del siguiente Procedimiento general 3 :

A una solución de 3-(2-cianoacetil)azetidin-1-carboxilato terc-butílico (Producto intermedio 62, 1,2 g, 5,35 mmol) en etanol (12 ml), se añadió hidrato de hidrazina (N2H4.H2O, 1,34 ml, 8,0 mmol) y, a continuación, se calentó la mezcla de reacción hasta 90 °C durante 5 horas. La mezcla de reacción se enfrió hasta la temperatura ambiente y se evaporaron las sustancias volátiles. El residuo se purificó mediante cromatografía en columna de alúmina neutra
(malla 100-300), eluyendo con MeOH al 2 % en diclorometano, produciendo 3-(5-amino-1H-pirazol-3-il)azetidin-1-carboxilato ferc-butílico (Compuesto 210, 600 mg, 47 %) en forma de un sólido blanquecino; m/z 239,13 [M+H]+; Sistema de TLC: metanol al 10 %-diclorometano. Fr: 0,4.
Ejemplo 270-Preparación del compuesto 211
La síntesis del Compuesto 211 siguió el procedimiento del siguiente Procedimiento general 4 :

A una solución enfriada (0 0C) de 3-(5-amino-1H-pirazol-3-il)azetidin-1-carboxilato ferc-butílico (Compuesto 210, 11 g, 46,2 mmol) en DMF seco (40 ml), se añadió 5-clorotiofen-2-carbaldehído (7,12 ml, 69,3 mmol) y ácido acético (12 ml). Se agitó la mezcla de reacción a temperatura ambiente durante 2 horas. (La formación de imina se observó como una mancha menos polar en TLC). Se enfrió la mezcla de reacción (0 0C), y se añadió poco a poco cianoborohidruro de sodio (6,18 g, 92,4 mmol) y una cantidad catalítica de ácido acético. Después de 2 horas, se inactivó la mezcla de reacción con agua helada (50 ml) y se extrajo en acetato de etilo (3 x 100 ml). Las capas orgánicas combinadas se lavaron con NaHCO3 (2 x 50 ml), agua (2 x 50 ml) y salmuera (2 x 50 ml), y se desecaron sobre sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con metanol/diclorometano al 2 %, produciendo 3-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)azetidin-1-carboxilato ferc-butílico (Compuesto 211, 7 g, 41 %); m/z 368,13 [M+H]+; Sistema de TLC: metanol al 10 % en diclorometano, Fr: 0,5.
Ejemplo 271-Preparación del compuesto 212
La síntesis del Compuesto 212 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 °C) de ácido furan-3-carboxílico (182 mg, 1,63 mmol) en diclorometano (10 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 388 mg, 2,0 mmol), seguido de hidroxibenzotriazol (HOBt, 135 mg, 4 mmol). Se agitó la mezcla de reacción durante 10 minutos, se enfrió (0 0C) y después se añadió a la mezcla diisopropiletilamina (DIEA, 0,9 ml, 5,4 mmol), seguida de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)azetidin-1-carboxilato ferc-butílico (Compuesto 211, 500 mg, 1,35 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (25 ml) y se extrajo con diclorometano (2 x 10 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 10 %/hexano, produciendo 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 -(furan-3-carbonil)-1 H-pirazol-3-il)azetidin-1 -carboxilato fercbutílico (Compuesto 212, 230 mg, rendimiento: 35 %) en forma de un sólido blanquecino. m/z 463,37 [M+1]+; Sistema de TLC: acetato de etilo al 30 % en hexano. Fr: 0,5.
Ejemplo 272-Preparación del compuesto 213
La s ín te s is de l
Compuesto 213
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5
:

A una solución enfriada (-10 0C) de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-(furan-3-carbonil)-1H-pirazol-3-il)azetidin-1-carboxilato ferc-butílico (230 mg, 0,49 mmol) en acetato de etilo (5 ml), se añadió una solución de cloruro de hidrógeno (1 M) en acetato de etilo (8 ml). Se agitó la mezcla de reacción durante 2 h y, a continuación, se evaporó a presión reducida, se trituró con éter dietílico y acetona, y el residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua-TFA como fase móvil, dando el producto trifluoroacetato de (3-(azetidin-3-2,2,2-il)-5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-1-il)(furan-3-il)metanona (Compuesto 213, 10 mg, rendimiento: 8 %) en forma de un sólido blanquecino. m/z 363,14 [(M-TfA)+1]+; RMN de 1H (400 MHz, DMSO-d6) 58,94 (s, 1H), 8,74 (s a, 1H), 7,96-7,92 (m, 1H), 7,89-7,88 (m, 1H), 7,07 (s, 1H), 6,98-6,96 (m, 2H), 5,58 (s, 1H), 4,50-4,49 (m, 2H), 4,26-4,22 (m, 2H), 4,13-3,95 (m, 3H); Sistema de TLC: acetato de etilo al 10 % en éter de petróleo. Fr: 0,1.
Ejemplo 273-Preparación del compuesto 214
La síntesis del Compuesto 214 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de ácido tiofen-3-carboxílico (208 mg, 1,63 mmol) en diclorometano (10 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 388 mg, 2,0 mmol), seguido de hidroxibenzotriazol (HOBt, 135 mg, 4 mmol). Se agitó la mezcla de reacción durante 10 minutos, se enfrió (0 0C) y después se añadió a la mezcla diisopropiletilamina (DIEA, 0,9 ml, 5,4 mmol), seguida de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)azetidin-1-carboxilato ferc-butílico (Compuesto 211, 500 mg, 1,35 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (25 ml) y se extrajo con diclorometano (2 x 10 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 10 %/hexano, produciendo 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 -(tiofen-3-carbonil)-1 H-pirazol-3-il)azetidin-1 -carboxilato fercbutílico (Compuesto 214, 230 mg, rendimiento: 36 %) en forma de un sólido blanquecino. m/z 479,37 [M+1]+; Sistema de TLC: acetato de etilo al 30 % en hexano. Fr: 0,5.
Ejemplo 274-Preparación del compuesto 215
La s ín te s is de l
Compuesto 215
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5
:

A una solución enfriada (-10 0C) de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-(tiofen-3-carbonil)-1H-pirazol-3-il)azetidin-1-carboxilato ferc-butílico (230 mg, 0,48 mmol) en acetato de etilo (5 ml), se añadió una solución de cloruro de hidrógeno (1 M) en acetato de etilo (8 ml). Se agitó la mezcla de reacción durante 2 h y, a continuación, se evaporó a presión reducida, se trituró con éter dietílico y acetona y el residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua-TFA como fase móvil, dando el producto 2,2,2-trifluoroacetato de (3-(azetidin-3-il)-5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-1-il)(tiofen-3-il)metanona (Compuesto 215, 10 mg, rendimiento: 7 %) en forma de un sólido blanquecino. m/z 379,13 [(M-TFA)+1]+; RMN de 1H (400 MHz, DMSO-d6) 59,01 (s, 1H), 9,00 (s a, 1H), 7,98-7,94 (m, 1H), 7,81-7,79 (m, 1H), 7,68-7,67 (m, 1H), 7,09-6,97 (m, 2H), 5,63 (s, 1H), 4,51-4,49 (m, 2H), 4,26-4,21 (m, 2H), 4,12-3,93 (m, 3H); Sistema de TLC: acetato de etilo al 10 % en éter de petróleo. Fr: 0,1.
Ejemplo 275-Preparación del compuesto 216
La síntesis del Compuesto 216 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de ácido 2-metoxibenzoico (247 mg, 1,63 mmol) en diclorometano (10 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 388 mg, 2,0 mmol), seguido de hidroxibenzotriazol (HOBt, 135 mg, 4 mmol). Se agitó la mezcla de reacción durante 10 minutos, se enfrió (0 0C) y después se añadió a la mezcla diisopropiletilamina (DIEA, 0,9 ml, 5,4 mmol), seguida de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)azetidin-1-carboxilato ferc-butílico (Compuesto 211, 500 mg, 1,35 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (25 ml) y se extrajo con diclorometano (2 x 10 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 10 %/hexano, produciendo 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 -(2-metoxibenzoil)-1 H-pirazol-3-il)azetidin-1 -carboxilato fercbutílico (Compuesto 216, 150 mg, rendimiento: 25 %) en forma de un sólido blanquecino. m/z 503,37 [M+1]+; Sistema de TLC: acetato de etilo al 30 % en hexano. Fr: 0,5.
Ejemplo 276-Preparación del compuesto 217
La s ín te s is de l
Compuesto 217
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5
:

A una solución enfriada (-10 0C) de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-(2-metoxibenzoil)-1H-pirazol-3-il)azetidin-1-carboxilato ferc-butílico (150 mg, 0,29 mmol) en acetato de etilo (5 ml), se añadió una solución de cloruro de hidrógeno (1 M) en acetato de etilo (8 ml). Se agitó la mezcla de reacción durante 2 h y, a continuación, se evaporó a presión reducida, se trituró con éter dietílico y acetona, y el residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua como fase móvil, dando el producto clorhidrato de (3-(azetidin-3-il)-5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-1-il)(2-metoxifenil)metanona (Compuesto 217, 8 mg, rendimiento: 5 %) en forma de un sólido blanquecino. m/z 403,24 [(M-HCl)+1]+; RMN de ^H (400 MHz, CDCls) 5 7,51-7,38 (m, 3H), 7,06-6,94 (m, 2H), 6,86 (s, 1H), 6,78 (s, 1H), 5,51 (s, 1H), 4,46-4,43 (m, 2H), 4,25-4,16 (m, 4H), 3,82 (s, 3H), 3,81-3,76 (m, 1H); Sistema de TLC: acetato de etilo al 50 % en éter de petróleo. Fr: 0,1.
Ejemplo 277-Preparación del compuesto 218
La síntesis del Compuesto 218 siguió el procedimiento del siguiente Procedimiento general 5 :

A una solución enfriada (0 0C) de ácido 2-metoxibenzoico (293 mg, 1,63 mmol) en diclorometano (10 ml) bajo nitrógeno, se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI.HCl, 388 mg, 2,0 mmol), seguido de hidroxibenzotriazol (HOBt, 135 mg, 4 mmol). Se agitó la mezcla de reacción durante 10 minutos, se enfrió (0 0C) y después se añadió a la mezcla diisopropiletilamina (DIEA, 0,9 ml, 5,4 mmol), seguida de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-3-il)azetidin-1-carboxilato ferc-butílico (Compuesto 211, 500 mg, 1,35 mmol). Se controló la reacción mediante CL-EM y, después de 16 horas a temperatura ambiente, se vertió la mezcla de reacción en agua (25 ml) y se extrajo con diclorometano (2 x 10 ml). Las fases orgánicas combinadas se desecaron sobre sulfato de sodio anhidro, se filtraron y se evaporaron a presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (malla 100-200), eluyendo con acetato de etilo al 10 %/hexano, produciendo 3-(5-(((5-clorotiofen-2-il)metil)amino)-1 -(2-metoxibenzoil)-1 H-pirazol-3-il)azetidin-1 -carboxilato fercbutílico (Compuesto 218, 100 mg, rendimiento: 18 %) en forma de un sólido blanquecino. m/z 531,37 [M+1]+; Sistema de TLC: acetato de etilo al 30 % en hexano. Fr: 0,5.
Ejemplo 278-Preparación del compuesto 219
La s ín te s is de l
Compuesto 219
s ig u ió e l p ro ce d im ie n to de l s ig u ie n te
Procedimiento general 5
:

A una solución enfriada (-10 0C) de 3-(5-(((5-clorotiofen-2-il)metil)amino)-1-(2-metoxibenzoil)-1H-pirazol-3-il)azetidin-1-carboxilato ferc-butílico (100 mg, 0,19 mmol) en acetato de etilo (5 ml), se añadió una solución de cloruro de hidrógeno (1 M) en acetato de etilo (8 ml). Se agitó la mezcla de reacción durante 2 h y, a continuación, se evaporó a presión reducida, se trituró con éter dietílico y acetona, y el residuo se purificó mediante HPLC preparativa utilizando acetonitrilo-agua-TFA como fase móvil, dando el producto 2,2,2-trifluoroacetato de (3-(azetidin-3-il)-5-(((5-clorotiofen-2-il)metil)amino)-1H-pirazol-1-il)(2-metoxifenil)metanona (Compuesto 219, 2 mg, rendimiento: 2 %) en forma de un líquido aceitoso. m/z 431,2 [(M-TFA)+1]+; Rm N de 1H (400 MHz, DMSO-d6) 5 7,84-7,81 (m, 1H), 7,02 6,87 (m, 5H), 5,63 (s, 1H), 4,50-4,49 (m, 4H), 4,14-4,09 (m, 2H), 3,94-3,85 (m, 2H); Sistema de TLC: acetato de etilo al 50 % en éter de petróleo. Fr: 0,1.
Claims (1)
- REIVINDICACIONESUn compuesto con la siguiente estructura:
 o una sal, un éster o un solvato farmacéuticamente aceptable del mismo, en donde:L1 es alquilenamino;L2 y L4 son, independientemente entre sí, un enlace, alquileno sustituido o no sustituido, heteroalquileno sustituido o no sustituido, -C(O)-, -S-, -SO-, -SO2-, -O-, -NHSO2-, -NHC(O)- o -NR5; R1 es alquilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, heteroarilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido;R2 es hidrógeno, halógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, o heteroarilo sustituido o no sustituido;R4 es, independientemente entre sí, hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, o heteroarilo sustituido o no sustituido;R5 es hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, o heteroarilo sustituido o no sustituido;V es hidrógeno o alquilo sustituido o no sustituido;W está ausente, o es hidrógeno, halógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, heteroarilo sustituido o no sustituido, -C(O)R6, - C(O)NR6R7, -SR6, -SOR6, -SO2 R6, -SO2NR6R7, -OR6, -NHSO2 R6 o -NR6R7, en donde R6 y R7 son, independientemente entre sí, hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido, en donde R6 y R7 se pueden combinar, si ambos están presentes, para formar un alquileno sustituido o no sustituido, o heteroalquileno sustituido o no sustituido;X es un enlace, alquileno sustituido o no sustituido o -NR8-;Y es un enlace, alquileno sustituido o no sustituido o -N-; yZ es un enlace, -C(O)-, alquileno sustituido o no sustituido o -NR9-;en donde R8 y R9 son, independientemente entre sí, hidrógeno, halógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, heteroarilo sustituido o no sustituido, -C(O)R6, -C(O)NR6R7, -SR6, -SOR6, -SO2 R6, -SO2NR6R7, -OR6, -NHSO2 R6 o - NR6R7, en donde R6 y R7 son como se han definido anteriormente; ysiempre que al menos uno de X sea -NR8-, Y es -N- o Z es -NR9-.2. El compuesto de la reivindicación 1, en donde X es un enlace, o alquileno sustituido o no sustituido; opcionalmente, en donde Z es un enlace, o alquileno sustituido o no sustituido.3. El compuesto de la reivindicación 2, en donde Y es -N- y W es hidrógeno, alquilo sustituido o no sustituido, heteroajquilo sustituido o no sustituido, arilo sustituido o no sustituido,-C(O)R6, -C(O)NR6R7, -SO2R6 o SO2 NR6R7, en donde R6 y R7 son independientemente entre sí, alquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido, o R6 y R7 se pueden combinar, si ambos están presentes, para formar un alquileno sustituido o no sustituido, o heteroalquileno sustituido o no sustituido.4. El compuesto de la reivindicación 3, en donde:X se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido, y Z es un enlace o -C(O)-; oX es un enlace y Z se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido; opcionalmente, en donde, opcionalmente, en donde Z se selecciona del grupo que consiste en metileno sustituido, etileno sustituido, propileno sustituido, butileno sustituido y pentileno sustituido, que tiene uno o más grupos sustituyentes seleccionados del grupo que consiste en -OH, -NH2 , -SH, -CN, -CF3 , -NO2 , oxo, halógeno, -COOH, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido; oX se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido, y Z se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido; opcionalmente, en donde X y Z son ambos alquileno ramificado, y X y Z están unidos covalentemente; opcionalmente, en donde Z se selecciona del grupo que consiste en metileno sustituido, etileno sustituido, propileno sustituido, butileno sustituido y pentileno sustituido, que tiene uno o más grupos sustituyentes seleccionados del grupo que consiste en -OH, -NH2 , -SH, -CN, -CF3 , -NO2 , oxo, halógeno, -COOH, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido.5. El compuesto de la reivindicación 3 o reivindicación 4, en donde W es hidrógeno, o en donde W se selecciona del grupo que consiste en alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido, -C(O)R6, -C(O)NR6R7, -SR6, -SOR6, -SO2 R6 y -SO2NR6 ; en donde, opcionalmente, W es alquilo sustituido, heteroalquilo sustituido, alquenilo sustituido, heteroalquenilo sustituido, cicloalquilo sustituido o heterocicloalquilo sustituido, que tiene uno o más grupos sustituyentes seleccionados del grupo que consiste en -OH, -NH2 , -SH, -CN, -CF3 , -NO2, oxo, halógeno, -COOH, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido o heteroarilo sustituido o no sustituido; además, opcionalmente, en donde W es -COR6, -C(O)NR6R7, -SO2 R6 o -SO2 NR6R7, y en donde R6 y R7 se seleccionan del grupo que consiste en alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, arilo sustituido o no sustituido y heteroarilo sustituido o no sustituido, o R6 y R7 se combinan para formar un alquileno sustituido o no sustituido.6. El compuesto de la reivindicación 1, en donde W está ausente, X es -NR8-, Y es un enlace, o alquileno sustituido o no sustituido, y Z es -NR9-; opcionalmente, en donde Y se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido; además, opcionalmente, en donde R8 se selecciona del grupo que consiste en alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, -COR6, -C(O)NR6R7, -Sr6, -SOR6, -SO2 R6 y -SO2NR6R7, y en donde R9 se selecciona del grupo que consiste en alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, -COR6 -C(O)NR6R7 SR6, -SOR6 -SO2 R6 y -SO2NR6R7.7. El compuesto de cualquiera de las reivindicaciones 1-6, en donde V es hidrógeno, o metilo sustituido o no sustituido.El compuesto según cualquiera de las reivindicaciones 1-7, en donde L1 es -NR5-, o heteroalquileno sustituido o no sustituido, y R1 es alquilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido; además, opcionalmente, en donde R1 es heteroarilo sustituido o no sustituido.El compuesto según cualquiera de las reivindicaciones 1 -8, en donde L2 es enlace, alquileno sustituido o no sustituido, -C(O)- o -SO2-, y R2 es hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, heteroarilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido; opcionalmente, en donde L2 es enlace, y R2 es hidrógeno, o en donde L2 es -C(O)-, y R2 es alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, heteroarilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido.El compuesto según cualquiera de las reivindicaciones 1-9, en donde L4 y R4 se seleccionan de manera que a) L4 es un enlace, y R4 es hidrógeno, halógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido;b) L4 y R4 están ausentes; oc) R4 es alquilo no sustituido.El compuesto según cualquiera de las reivindicaciones 1 a 10, seleccionándose el compuesto de:1-(3-{8-azabiciclo[3.2.1]octan-3-il}-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(1 -metanosulfonilazepan-4-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(1 -metanosulfonilazetidin-3-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(1 -metanosulfonilpiperidin-4-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(1 -metanosulfonilpiperidin-4-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(1 -metanosulfonilpirrolidin-2-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amonio}-3-(1 -metanosulfonilpirrolidin-3-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-(1-{[6-(trifluorometil)piridin-2-il]metil}piperidin-4-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-(2-metilpirrolidin-2-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona; ácido fórmico;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-(3-metilpiperidin-3-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona; ácido fórmico;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-(3-metilpiperidin-3-il)-1 H-pirazol-1-il)-3-metoxi-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-(4-metanosulfonilpiperazin-2-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(4-metilpiperidin-4-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; ácido fórmico;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-(4-metilpiperidin-4-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-(4-metilpiperidin-4-il)-1 H-pirazol-1-il)-3-metoxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(piperazin-2-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; 1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(piperidin-3-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; ácido fórmico;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(piperidin-3-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona; ácido fórmico;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(piperidin-3-il)-1 H-pirazol-1 -il)-3-metoxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(piperidin-4-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; 1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(piperidin-4-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(piperidin-4-il)-1 H-pirazol-1 -il)-3-metoxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(pirrolidin-2-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; 1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(pirrolidin-2-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(pirrolidin-2-il)-1 H-pirazol-1 -il)-3-metoxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(pirrolidin-3-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; 1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(1,2,4-oxadiazol-3-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2.2- dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(1,3-oxazol-4-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(1,3-oxazol-5-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(1H-1,2,3,4-tetrazol-5-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(1 H-1,2,3-triazol-5-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2.2- dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(1 H-imidazol-4-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(2,2,2-trifluoroetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1-(2,2,2-trifluoroetil)piperidin-4-il]-1-il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(2,2-dimetilpropanoil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(2-ciclopropoxietil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(2-ciclopropoxietil)piperidin-4-il]-1 H-pirazol-1 -il)-3-hidroxi-2.2- dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(2-metoxietil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(2-metoxietil)piperidin-4-il]-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(ciclopropilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1-(ciclopropilmetil)piperidin-4-il]-1-il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(morfolin-4-carbonil)azepan-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(morfolin-4-carbonil)azetidin-3-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino-3-[1 -(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(morfolin-4-carbonil)pirrolidin-2-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(morfolin-4-carbonil)pirrolidin-3-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(morfolin-4-sulfonil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(morfolin-4-sulfonil)piperidin-4-il]-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(oxetan-3-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(oxetan-3-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-3-hidroxi-2.2- dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(piridin-2-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1-(piridin-2-ilmetil)piperidin-4-il]-1-il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(piridin-3-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[4-(morfolin-4-carbonil)piperazin-2-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[8-(morfolin-4-carbonil)-8-azabiciclo[3.2.1]octan-3-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[(1 -metilazetidin-3-il)metil]piperidin-4-il}-1 H-pirazol-1 -il)-2.2- dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[(2-metoxifenil)metil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[(3-metoxifenil)metil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[(4-metoxifenil)metil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[(6-metilpiridin-2-il)metil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; ácido fórmico;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2,2,2-trifluoro-1 -(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2,2,2-trifluoro-1 -(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2,2-difluoro-2-(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2,2-difluoro-2-(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)-2-oxoetil]piperidin-4-il}-1 H-pirazol-1 -il)-2.2- dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)-2-oxoetil]piperidin-4-il}-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)acetil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)acetil]piperidin-4-il}-1 H-pirazol-1 -il)-2-(morfolin-4-il)etan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)acetil]piperidin-4-il}-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)acetil]pirrolidin-2-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)acetil]pirrolidin-3-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)etil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; ácido fórmico;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)etil]piperidin-4-il}-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona; ácido fórmico;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-1 -il)-3-hidroxi-2.2- dimetilpropan-1 -ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-{8-metanosulfonil-8-azabiciclo[3.2.1]octan-3-il}-1 H-pirazol-1-il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-4-metil-3-(piperidin-4-il)-1 H-pirazol-1-il)-2,2-dimetilpropan-1-ona; ácido fórmico;1 -[2-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2-metoxibenzoil)-1 H-pirazol-3-il)pirrolidin-1 -il]-2,2-dimetilpropan-1-ona;1 -[2-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2-metoxibenzoil)-1 H-pirazol-3-il)pirrolidin-1 -il]-2-metilpropan-1-ona;1 -[2-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2-metoxibenzoil)-1 H-pirazol-3-il)pirrolidin-1 -il]etan-1 -ona; 1 -[2-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il)pirrolidin-1 -il]-2-(morfolin-4-il)etan-1 -ona; 1 -[3-(1 -acetilpiperidin-4-il)-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-1 -il]-2,2-dimetilpropan-1 -ona;1 -[3-(1 -bencilpiperidin-4-il)-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-1-il]-2,2-dimetilpropan-1 -ona;1 -[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2,2-dimetilpropanoil)-1 H-pirazol-3-il)azetidin-1 -il]-2,2-dimetilpropan-1-ona;1 -[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il)azetidin-1 -il]-2,2-dimetilpropan-1 -ona;1 -[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il)pirrolidin-1 -il]-2-(morfolin-4-il)etan-1 -ona; 1 -[4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2-metoxibenzoil)-1 H-pirazol-3-il)piperidin-1 -il]-2,2-dimetilpropan-1-ona;1 -[4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2-metoxibenzoil)-1 H-pirazo1 -3-il)piperidin-1 -il]etan-1 -ona;1 -[4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(tiofen-3-carbonil)-1 H-pirazol-3-il)piperidin-1 -il]-2,2-dimetilpropan-1-ona;1 -[4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(tiofen-3-carbonil)-1 H-pirazol-3-il)piperidin-1 -il]etan-1 -ona; 1 -[4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il)piperidin-1 -il]-2-(morfolin-4-il)etan-1 -ona; clorhidrato de 1 -benzoil-N-[(5-clorotiofen-2-il)metil]-3-(piperidin-4-il)-1 H-pirazol-5-amina;1 -{3-[1 -(bencenosulfonil)piperidin-4-il]-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-1 -il}-2,2-dimetilpropan-1-ona;1 -{3-[1 -(bencenosulfonil)piperidin-4-il]-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-1 -il}-3-hidroxi-2,2-dimetilpropan-1 -ona;ácido 2-[4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2,2-dimetilpropanoil)-1 H-pirazol-3-il)piperidin-1 -il]acético;ácido 2-[4-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)piperidin-1-il]acético;2- [4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il)piperidin-1 -il]-1 -(morfolin-4-il)etan-1 -ona; ácido 2-[4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il)piperidin -1 -il]acético;3- (1-bencilpiperidin-4-il)-N-[(5-clorotiofen-2-il)metil]-1 H-pirazol-5-amina;3-(1-bencilpirrolidin-3-il)-N-[(5-clorotiofen-2-il)metil]-1-(furan-3-carbonil)-1 H-pirazol-5-amina;3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(2,2-dimetilpropanoil)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona; 3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(2,2-dimetilpropanoil)-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona; 3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(2-metoxibenzoil)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona; 3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(2-metoxibenzoil)-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona; 3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona;3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona;3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(4-metiloxano-4-carbonil)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona;3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(4-metiloxano-4-carbonil)-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona;3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(tiofen-3-carbonil)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona; 3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(tiofen-3-carbonil)-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona; 3-(azepan-4-il)-N-[(5-clorotiofen-2-il)metil]-1 H-pirazol-5-amina;3-(azetidin-3-il)-N-[(5-clorotiofen-2-il)metil]-1-(2,3-dihidro-1,4-benzodioxin-5-carbonil)-1 H-pirazol-5-amina; ácido trifluoroacético;clorhidrato de 3-(azetidin-3-il)-N-[(5-clorotiofen-2-il)metil]-1 -(2-metoxibenzoil)-1 H-pirazol-5-amina; 3-(azetidin-3-il)-N-[(5-clorotiofen-2-il)metil]-1-(furan-3-carbonil)-1 H-pirazol-5-amina; ácido trifluoroacético;3-(azetidin-3-il)-N-[(5-clorotiofen-2-il)metil]-1-(tiofen-3-carbonil)-1 H-pirazol-5-amina; ácido trifluoroacético;3-[1-(bencenosulfonil)piperidin-4-il]-N-[(5-clorotiofen-2-il)metil]-1 H-pirazol-5-amina;3- {8-azabiciclo[3.2.1]octan-3-il}-N-[(5-clorotiofen-2-il)metil]-1 H-pirazol-5-amina;4- (5-{[(5-clorotiofen-2-il)metil]amino}-1-(2,2-dimetilpropanoil)-1 H-pirazol-3-il)-N,N-dimetilpiperidin-1-carboxamida;4-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(2,2-dimetilpropanoil)-1 H-pirazol-3-il)-N,N-dimetilpiperidin-1-sulfonamida;4-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-N,N-dimetilpiperidin-1 -carboxamida;4-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-N,N-dimetilpiperidin-1 -sulfonamida;4-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(dimetilcarbamoil)-1 H-pirazol-3-il)-N,N-dimetilpiperidin-1-carboxamida;4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il)-N,N-dimetilpiperidin-1-carboxamida;4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il)-N,N-dimetilpiperidin-1-sulfonamida;4-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1-(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-1-carbonil)-4- metilciclohexan-1 -ol;5- {[(5-clorotiofen-2-il)metil]amino}-1-(2,2-dimetilpropanoil)-3-[1-(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil]amino}-3-[1-(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-4-carbonitrilo; N-[(5-clorotiofen-2-il)metil]-1-(1,4-dimetilpiperidin-4-carbonil)-3-[1-(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-5-amina; ácido fórmico;clorhidrato de N-[(5-clorotiofen-2-il)metil]-1 -(2,3-dihidro-1,4-benzodioxin-5-carbonil)-3-(piperidin-4-il)-1 H-pirazol-5-amina;clorhidrato de N-[(5-clorotiofen-2-il)metil]-1 -(2,3-dihidro-1,4-benzodioxin-5-carbonil)-3-(pirrolidin-2-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(2,4-dimetoxibenzoil)-3-(piperidin-4-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(2,4-dimetoxibenzoil)-3-(pirrolidin-2-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(2,6-dimetilciclohexanocarbonil)-3-(piperidin-4-il)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-1-(2-metoxibenzoil)-3-(2-metilpirrolidin-2-il)-1 H-pirazol-5-amina; ácido fórmico;N-[(5-clorotiofen-2-il)metil]-1-(2-metoxibenzoil)-3-(3-metilpiperidin-3-il)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-1-(2-metoxibenzoil)-3-(4-metilpiperidin-4-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(2-metoxibenzoil)-3-(piperidin-3-il)-1 H-pirazol-5-amina; clorhidrato de N-[(5-clorotiofen-2-il)metil]-1 -(2-metoxibenzoil)-3-(piperidin-4-il)-1 H-pirazol-5-amina; clorhidrato de N-[(5-clorotiofen-2-il)metil]-1 -(2-metoxibenzoil)-3-(pirrolidin-2-il)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-1-(2-metoxibenzoil)-3-[1-(2,2,2-trifluoroetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(4-metiloxano-4-carbonil)-3-(2-metilpirrolidin-2-il)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-1-(4-metiloxano-4-carbonil)-3-(3-metilpiperidin-3-il)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-1-(4-metiloxano-4-carbonil)-3-(4-metilpiperidin-4-il)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-1-(4-metiloxano-4-carbonil)-3-(piperidin-3-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(4-metiloxano-4-carbonil)-3-(pirrolidin-2-il)-1 H-pirazol-5-amina; clorhidrato de N-[(5-clorotiofen-2-il)metil]-1 -(furan-3-carbonil)-3-(piperidin-4-il)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-1-(furan-3-carbonil)-3-(pirrolidin-2-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(morfolin-4-carbonil)-3-[1-(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-5-amina;clorhidrato de N-[(5-clorotiofen-2-il)metil]-1 -ciclohexanocarbonil-3-(piperidin-4-il)-1 H-pirazol-5-amina;clorhidrato de N-[(5-clorotiofen-2-il)metil]-1-ciclopentanocarbonil-3-(piperidin-4-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1-metanosulfonilazepan-4-il)-1H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1-metanosulfonilazetidin-3-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1-metanosulfonilpiperidin-4-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1-metanosulfonilpirrolidin-2-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1-metanosulfonilpirrolidin-3-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1-{[6-(trifluorometil)piridin-2-il]metil}piperidin-4-il)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-3-(2-metilpirrolidin-2-il)-1-(tiofen-3-carbonil)-1 H-pirazol-5-amina; ácido fórmico;N-[(5-clorotiofen-2-il)metil]-3-(3-metilpiperidin-3-il)-1-(tiofen-3-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(4-metanosulfonilpiperazin-2-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(4-metilpiperidin-4-il)-1-(tiofen-3-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(piperidin-3-il)-1-(tiofen-3-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(piperidin-3-il)-1 H-pirazol-5-amina;clorhidrato de N-[(5-clorotiofen-2-il)metil]-3-(piperidin-4-il)-1 -(tiofen-3-carbonil)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-3-(piperidin-4-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(pirrolidin-2-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(pirrolidin-3-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(1,2,4-oxadiazol-3-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(1,2,4-oxadiazol-5-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(1,3-oxazol-2-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(1,3-oxazol-5-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(1 H-1,2,3,4-tetrazol-5-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-3-[1-(1 H-1,2,3-triazol-5-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(1 H-imidazol-4-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(2,2,2-trifluoroetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(2-ciclopropoxietil)piperidin-4-il]-1 H-pirazol-5-amina; ácido fórmico; N-[(5-clorotiofen-2-il)metil]-3-[1-(2-metoxietil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(ciclopropilmetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-carbonil)azepan-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-carbonil)pirrolidin-2-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-carbonil)pirrolidin-3-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-carbonil)pirrolidin-3-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-sulfonil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(oxan-4-il)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(oxetan-3-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina; ácido fórmico; N-[(5-clorotiofen-2-il)metil]-3-[1-(piridin-2-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(piridin-3-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina;clorhidrato de N-[(5-clorotiofen-2-il)metil]-3-[4-(morfolin-4-carbonil)piperazin-2-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[8-(morfolin-4-carbonil)-8-azabiciclo[3.2.1]octan-3-il]-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[8-(morfolin-4-carbonil)-8-azabiciclo[3.2.1]octan-3-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-{1-[(1-metilazetidin-3-il)metil]piperidin-4-il}-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-{1-[(2-metoxifenil)metil]piperidin-4-il}-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-3-{1-[(3-metoxifenil)metil]piperidin-4-il}-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-{1-[(4-metoxifenil)metil]piperidin-4-il}-1-(tiofen-3-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-{1-[(4-metoxifenil)metil]piperidin-4-il}-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-{1 -[(6-metilpiridin-2-il)metil]piperidin-4-il}-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-{1 -[2,2,2-trifluoro-1 -(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-3-{1-[2,2-difluoro-2-(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-3-{1-[2-(morfolin-4-il)etil]piperidin-4-il}-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-{1-[2-(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-{8-metanosulfonil-8-azabiciclo[3.2.1]octan-3-il}-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-18-metanosulfonil-8-azabiciclo[3.2.1]octan-3-il}-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-4-metil-3-[1-(1,3-tiazol-4-carbonil)piperidin-4-il]-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-4-metil-3-[1-(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-N-{3-[1-(morfolin-4-sulfonil)piperidin-4-il]-1 H-pirazol-5-il}morfolin-4-sulfonamida;N-[(5-clorotiofen-2-il)metil]-3-(1 -metanosulfonilazepan-4-il)-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1 -metanosulfonilazetidin-3-il)-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1 -metanosulfonilpirrolidin-2-il)-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1 -metanosulfonilpirrolidin-3-il)-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-1 -(morfolin-4-carbonil)azepan-4-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-carbonil)azetidin-3-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-carbonil)pirrolidin-2-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;1-(2,2-dimetilpropanoil)-4-(5-{[(4-fluorofenil)metil](metil)amino}-4-metoxi-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-3-(trifluorometil)piperidin-2-ona;1-(2-clorobenzoil)-5-[(5-clorotiofen-2-il)metoxi]-3-[1-metanosulfonil-4-(trifluorometil)pirrolidin-3-il]-1 H-pirazol;1-(2-clorobenzoil)-5-{[(4-fluorofenil)metil](metil)amino}-3-{1-[(3-hidroxipirrolidin-1-il)sulfonil]-6-oxo-4-(trifluorometil)piperidin-3-il}-1 H-pirazol-4-carbonitrilo;1-(2-clorobenzoil)-5-{[(4-fluorofenil)metil]sulfanil}-3-{4-[2-(morfolin-4-il)-2-oxoetil]-3-(trifluorometil)piperazin-2-il}-1 H-pirazol-4-carbonitrilo;1-(2-clorobenzoil)-N-[(4-fluorofenil)metil]-N-metil-3-[1-(morfolin-4-carbonil)-3-(trifluorometil)piperazin-2-il]-1 H-pirazol-5-amina;1-(2-fluorobenzoil)-N-[(4-fluorofenil)metil]-3-[3-(trifluorometil)azetidin-2-il]-1 H-pirazol-5-amina; 1 -(2-{5-[(5-clorotiofen-2-il)metoxi]-1 -(furan-2-carbonil)-4-metoxi-1 H-pirazol-3-il}-3-metilpiperazin-1 -il)-2,2-dimetilpropan-1 -ona;1 -(3-15-[(4-fluorofenil)metoxi]-4-metil-1 -(4-metilfuran-3-carbonil)-1 H-pirazol-3-il}-2-(trifluorometil)azetidin-1 -il)-2-(morfolin-4-il)etan-1 -ona;1 -(3-{5-[(5-clorotiofen-2-il)metoxi]-1 -(2-metoxibenzoil)-4-metil-1 H-pirazol-3-il}-2-(trifluorometil)piperazin-1 -il)-2-(morfolin-4-il)etan-1 -ona;1 -(3-{5-[(5-clorotiofen-2-il)metoxi]-4-metoxi-1 H-pirazol-3-il}piperazin-1 -il)-2,2-dimetilpropan-1 -ona; 1-(5-{[(4-fluorofenil)metil]amino}-3-[4-(morfolin-4-carbonil)piperazin-2-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(4-fluorofenil)metil]sulfanil}-3-(1 -metanosulfonil-4-metilpiperidin-3-il)-1 H-pirazol-1 -il)-3-metoxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(4-fluorofenil)metil]sulfanil}-3-[5-hidroxi-2-metil-1 -(pirrolidin-1 -sulfonil)pirrolidin-3-il]-4-metil-1 H-pirazol-1-il)-3-metoxi-2,2-dimetilpropan-1 -ona;1-(5-{[(4-fluorofenil)metil]sulfanil}-4-metoxi-3-{4-metil-1-[2-(morfolin-4-il)-2-oxoetil]piperidin-3-il}-1 H-pirazol-1 -il)-3-metoxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil](metil)amino}-3-[5-hidroxi-1 -(pirrolidin-1 -carbonil)pirrolidin-3-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona;1-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-3-{5-hidroxi-2-metil-1-[2-(morfolin-4-il)-2-oxoetil]pirrolidin-3-il}-4-metoxi-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona;1-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-metoxi-3-(morfolin-3-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[(3-hidroxipirrolidin-1-il)sulfonil]-4-metilpirrolidin-3-il}-4-metil-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]sulfanil}-3-[1 -(3-hidroxipirrolidin-1 -carbonil)-2-metilpiperidin-3-il]-1 H-pirazol-1 -il)-3-metoxi-2,2-dimetilpropan-1 -ona;1 -(5-{[4-(aminometil)fenil]metoxi}-3-{3-metil-1 -[2-(morfolin-4-il)-2-oxoetil]piperidin-4-il}-1 H-pirazol-1 -il)-3-metoxi-2,2-dimetilpropan-1-ona;ácido 1 -(dimetilcarbamoil)-3-(5-{[(4-fluorofenil)metil]amino}-1 -(tiofen-3-carbonil)-1 H-pirazol-3-il)-4-metilpiperidin-2-carboxílico;ácido 1 -(dimetilsulfamoil)-3-(5-{[(4-fluorofenil)metil](metil)amino}-1 -(furan-3-carbonil)-1 H-pirazol-3-il)-4-metilpirrolidin-2-carboxílico;1-({3-[5-({[4-(aminometil)fenil]metil}amino)-1-(2-fluorobenzoil)-4-metoxi-1 H-pirazol-3-il]-2-(trifluorometil)piperazin-1-il}sulfonil)pirrolidin-3-ol;1 -[2-(5-{[(4-fluorofenil)metil](metil)amino}-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-3-(trifluorometil)piperazin-1 -il]-2-(morfolin-4-il)etan-1 -ona;1 -[2-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(4-metilfuran-3-carbonil)-1 H-pirazol-3-il)-3-(trifluorometil)azetidin-1 -il]-2,2-dimetilpropan-1 -ona;1 -[2-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(tiofen-3-carbonil)-1 H-pirazol-3-il)-3-metilpiperazin-1 -il]-2.2- dimetilpropan-1 -ona;1 -[3-(1 -benzoil-5-{[(4-fluorofenil)metil]amino}-4-metoxi-1 H-pirazol-3-il)-2-metilpiperidin-1 -carbonil]pirrolidin-3-ol;1 -[3-(5-{[(4-fluorofenil)metil](metil)amino}-1 -(furan-2-carbonil)-1 H-pirazol-3-il)-4-metilpirrolidin-1 -carbonil]pirrolidin-3-ol;1-[3-(5-{[(4-fluorofenil)metil](metil)amino}-1-(furan-2-carbonil)-4-metil-1 H-pirazol-3-il)-4-metilpiperidin-1 -il]-2-(morfolin-4-il)etan-1 -ona;1 -[3-(5-{[(4-fluorofenil)metil]amino}-1 -(4-metilfuran-3-carbonil)-1 H-pirazol-3-il)-4-(trifluorometil)piperidin-1-carbonil]pirrolidin-3-ol;1-[3-(5-{[(4-fluorofenil)metil]sulfanil}-1-(furan-3-carbonil)-4-metoxi-1 H-pirazol-3-il)-2-metilpiperazin-1 -carbonil]pirrolidin-3-ol;1-[3-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-1-(4-metilfuran-3-carbonil)-1 H-pirazol-3-il)-2-(trifluorometil)piperidin-1 -il]-2,2-dimetilpropan-1 -ona;1 -[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(5-metilfuran-3-carbonil)-1 H-pirazol-3-il)piperazin-1 -il]-2.2- dimetilpropan-1 -ona;1-[3-(5-{[(5-clorotiofen-2-il)metil]amino}-4-metil-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il)pirrolidin-1 -il]-2,2-dimetilpropan-1 -ona;1-[3-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-metoxi-1-(2-metoxibenzoil)-1 H-pirazol-3-il)-2-(trifluorometil)pirrolidin-1 -il]-2-(morfolin-4-il)etan-1 -ona;1 -[3-(5-{[4-(aminometil)fenil]metoxi}-4-metoxi-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-4-(trifluorometil)piperidin-1-carbonil]pirrolidin-3-ol;1 -[4-(1 -benzoil-5-{[(4-fluorofenil)metil]amino}-4-metil-1 H-pirazol-3-il)-3-metilpiperidin-1 -carbonil]pirrolidin-3-ol;1 -[5-({[4-(aminometil)fenil]metil}(metil)amino)-3-(1 -metanosulfonilazetidin-2-il)-1 H-pirazol-1 -il]-2,2-dimetilpropan-1-ona;1-[5-({[4-(aminometil)fenil]metil}(metil)amino)-4-metoxi-3-{4-metil-1-[2-(morfolin-4-il)acetil]pirrolidin-3-il}-1 H-pirazol-1 -il]-3-hidroxi-2,2-dimetilpropan-1-ona;1 -[5-({[4-(aminometil)fenil]metil}amino)-3-[1 -(pirrolidin-1 -sulfonil)piperidin-4-il]-1 H-pirazol-1 -il]-2,2-dimetilpropan-1-ona;1-[5-({[4-(aminometil)fenil]metil}amino)-4-fluoro-3-[5-hidroxi-1-(3-hidroxipirrolidin-1-carbonil)-2-metilpirrolidin-3-il]-1 H-pirazol-1 -il]-3-hidroxi-2,2-dimetilpropan-1 -ona;1-benzoil-5-{[(4-fluorofenil)metil]amino}-3-(4-metil-2-oxopirrolidin-3-il)-1 H-pirazol-4-carbonitrilo; 1-benzoil-5-{[(5-clorotiofen-2-il)metil](metil)amino}-3-[1-(2,2-dimetilpropanoil)-2-metilpirrolidin-3-il]-1 H-pirazol-4-carbonitrilo;1 -{2-[1 -(2-fluorobenzoil)-5-{[(4-fluorofenil)metil]amino}-4-metoxi-1 H-pirazol-3-il]-3-(trifluorometil)piperazin-1-carbonil}pirrolidin-3-ol;1-{2-[5-({[4-(aminometil)fenil]metil}amino)-4-metil-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il]azetidin-1 -il}-2-(morfolin-4-il)etan-1 -ona;1-{2-[5-({[4-(aminometil)fenil]metil}sulfanil)-1-(furan-3-carbonil)-4-metoxi-1 H-pirazol-3-il]-3-metilazetidin-1 -carbonil}pirrolidin-3-ol;1 -{3-[1 -(2,2-dimetilpropanoil)-5-[(4-fluorofenil)metoxi]-4-metil-1 H-pirazol-3-il]azetidin-1 -il}-2,2-dimetilpropan-1-ona;1-{3-[1-(2-clorobenzoil)-5-[(5-clorotiofen-2-il)metoxi]-4-metoxi-1 H-pirazol-3-il]-5-hidroxi-2-(trifluorometil)pirrolidin-1 -il}-2-(morfolin-4-il)etan-1 -ona;1-{3-[5-({[4-(aminometil)fenil]metil}sulfanil)-1-(2-metoxibenzoil)-4-metil-1 H-pirazol-3-il]-4-(trifluorometil)piperidin-1 -il}-2,2-dimetilpropan-1 -ona;1-{4-[5-({[4-(aminometil)fenil]metil}(metil)amino)-4-metil-1-(2-metilfuran-3-carbonil)-1 H-pirazol-3-il]-3-(trifluorometil)piperidin-1 -il}-2,2-dimetilpropan-1 -ona;1 -{5-[(4-fluorofenil)metoxi]-3-(1-metanosulfonil-2-metilazetidin-3-il)-4-metoxi-1 H-pirazol-1 -il}-3-hidroxi-2,2-dimetilpropan-1-ona;1-{5-[(5-clorotiofen-2-il)metoxi]-3-[1-(2,2-dimetilpropanoil)-2-metilpirrolidin-3-il]-4-metoxi-1 H-pirazol-1 -il}-3-metoxi-2,2-dimetilpropan-1 -ona;1 -{[2-(5-{[(4-fluorofenil)metil]sulfanil}-1 -(2-metoxibenzoil)-4-metil-1 H-pirazol-3-il)-3-(trifluorometil)piperazin-l-il]sulfonil}pirrolidin-3-ol;1 -{[3-(5-{[4-(aminometil)fenil]metoxi}-1 -(tiofen-2-carbonil)-1 H-pirazol-3-il)pirrolidin-1 -il]sulfonil}pirrolidin-3-ol;1 -{[4-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-metoxi-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-3-(trifluorometil)piperidin-1-il]sulfonil}pirrolidin-3-ol;2-(3-{5-[(4-fluorofenil)metoxi]-1-(tiofen-3-carbonil)-1 H-pirazol-3-il}-2-metilazetidin-1 -il)-1 -(morfolin-4-il)etan-1-ona;2-(3-{5-[(5-clorotiofen-2-il)metoxi]-1-(furan-3-carbonil)-1 H-pirazol-3-il}-5-hidroxi-2-metilpirrolidin-1-il)-1 -(morfolin-4-il)etan-1 -ona;2-(5-{[(4-carbamimidoilfenil)metil]sulfanil}-4-metoxi-1-(tiofen-2-carbonil)-1 H-pirazol-3-il)-N,N-dimetil-4-oxoazetidin-1 -carboxamida;2-(5-{[(4-fluorofenil)metil](metil)amino}-4-metil-1-(tiofen-2-carbonil)-1 H-pirazol-3-il)-N,N-dimetilpiperazin-1 -carboxamida;2-(5-{[(4-fluorofenil)metil]sulfanil}-1-(furan-2-carbonil)-4-metil-1 H-pirazol-3-il)-N,N,3-trimetilazetidin-1- carboxamida;2- (5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-ciano-1-(tiofen-3-carbonil)-1 H-pirazol-3-il)-N,N,3-trimetilazetidin-1 -carboxamida;2-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-1-(furan-3-carbonil)-4-metil-1 H-pirazol-3-il)-N,N,3-trimetilazetidin-1 -sulfonamida;2-[3-(1 -benzoil-5-{[(5-clorotiofen-2-il)metil](metil)amino}-1 H-pirazol-3-il)-4-metilpiperidin-1 -il]-1 -(morfolin-4-il)etan-1 -ona;2-[3-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-1 -(tiofen-2-carbonil)-1 H-pirazol-3-il)pirrolidin-1 -il]-1 -(morfolin-4-il)etan-1 -ona;2-[4-ciano-1-(2,2-dimetilpropanoil)-5-[(4-fluorofenil)metoxi]-1 H-pirazol-3-il]-N,N-dimetilpiperazin-1 -sulfonamida;2-[5-({[4-(aminometil)fenil]metil}amino)-1-(2-fluorobenzoil)-1 H-pirazol-3-il]-N,N-dimetil-4-oxo-3-(trifluorometil)azetidin-1-sulfonamida;ácido 2-cloro-3-(5-{[(4-fluorofenil)metil](metil)amino}-3-{1-[(3-hidroxipirrolidin-1-il)sulfonil]-5-oxopirrolidin-3-il}-4-metoxi-1 H-pirazol-1 -carbonil)benzoico;ácido 2-cloro-3-(5-{[(4-fluorofenil)metil]sulfanil}-3-(2-oxoazetidin-3-il)-1 H-pirazol-1 -carbonil)benzoico;ácido 2-cloro-3-(5-{[(4-fluorofenil)metil]sulfanil}-4-metil-3-[1-(morfolin-4-carbonil)-6-oxopiperidin-3-il]-1 H-pirazol-1 -carbonil)benzoico;ácido 2-cloro-3-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-ciano-3-[1-(morfolin-4-carbonil)-2-oxopirrolidin-3-il]-1 H-pirazol-1 -carbonil)benzoico;ácido 2-cloro-3-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-metoxi-3-[4-(pirrolidin-1-carbonil)piperazin-2-il]-1 H-pirazol-1 -carbonil)benzoico;ácido 2-cloro-3-{3-[1-(2,2-dimetilpropanoil)-2-oxoazetidin-3-il]-5-{[(4-fluorofenil)metil](metil)amino}-1 H-pirazol-1-carbonil}benzoico;ácido 2-cloro-3-{3-[1-(2,2-dimetilpropanoil)azetidin-3-il]-4-fluoro-5-{[(4-fluorofenil)metil](metil)amino}-1 H-pirazol-1-carbonil}benzoico;ácido 2-cloro-3-{5-[(5-clorotiofen-2-il)metoxi]-3-(morfolin-3-il)-1 H-pirazol-1-carbonil}benzoico;2- {4-[5-({[4-(aminometil)fenil]metil}(metil)amino)-1-benzoil-4-metoxi-1 H-pirazol-3-il]-3-metilpiperidin-1 -il}-1 -(morfolin-4-il)etan-1 -ona;ácido 3-(1 -benzoil-5-{[(4-carbamimidoilfenil)metil](metil)amino}-4-metoxi-1 H-pirazol-3-il)-4-metil-1 -(morfolin-4-carbonilo)piperidin-2-carboxílico;3- (1-benzoil-5-{[(4-carbamimidoilfenil)metil]amino}-4-fluoro-1 H-pirazol-3-il)-N,N,2-trimetil-4-oxopirrolidin-1 -carboxamida;ácido 3-(1 -benzoil-5-{[(5-clorotiofen-2-il)metil]amino}-4-metil-1 H-pirazol-3-il)-1 -(dimetilcarbamoil)-4-metilpirrolidin-2-carboxílico;ácido 3-(4-ciano-5-{[(4-fluorofenil)metil]amino}-3-{1-[2-(morfolin-4-il)acetil]-5-oxopirrolin-il}-1 H-pirazol-1 -carbonil)benzoico;3-(4-ciano-5-{[(4-fluorofenil)metil]sulfanil}-1-(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-N,N,2-trimetilpirrolidin-1-carboxamida;3-(4-ciano-5-{[(4-fluorofenil)metil]sulfanil}-1-(furan-2-carbonil)-1 H-pirazol-3-il)-N,N,2-trimetilpiperidin-1-sulfonamida;3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-1-(2-fluorobenzoil)-4-metoxi-1 H-pirazol-3-il)-N,N-dimetil-2-(trifluorometil)piperidin-1-carboxamida;3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-5-hidroxi-N,N,2-trimetilpirrolidin-1-carboxamida;ácido 3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-3-[1-(3-hidroxipirrolidin-1-carbonil)-4-oxopirrolidin-3-il]-4-metoxi-1 H-pirazol-1 -carbonil)benzoico;ácido 3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-3-[1-(dimetilsulfamoil)piperidin-4-il]-4-fluoro-1 H-pirazol-1-carbonil)benzoico;ácido 3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-4-ciano-1-(5-metilfuran-3-carbonil)-1H-pirazol-3-il)-1 -(pirrolidin-1 -carbonil)piperidin-2-carboxílico;ácido 3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-4-ciano-3-[1-(pirrolidin-1-carbonil)pirrolidin-3-il]-1 H-pirazol-1 -carbonil)benzoico;ácido 3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-4-metoxi-1-(2-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1 -[2-(morfolin-4-il)acetil]-4-(trifluorometil)piperidin-2-carboxílico;ácido 3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-4-metil-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1 -(pirrolidin-1 -sulfonil)piperidin-2-carboxílico;3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-4-metil-1-(tiofen-3-carbonil)-1;ácido 3-(5-{[(4-carbamimidoilfenil)metil]amino}-1 -(3-carboxibenzoil)-1 H-pirazol-3-il)-1 -[2-(morfolin-4-il)-2-oxoetil]pirrolidin-2-carboxílico;ácido 3-(5-{[(4-carbamimidoilfenil)metil]amino}-3-[1-(dimetilcarbamoil)piperazin-2-il]-4-fluoro-1 H-pirazol-1 -carbonil)benzoico;ácido 3-(5-{[(4-carbamimidoilfenil)metil]amino}-3-[1-(dimetilsulfamoil)-5-hidroxipirrolidin-3-il]-4-metil-1 H-pirazol-1-carbonil)benzoico;3-(5-{[(4-carbamimidoilfenil)metil]amino}-4-fluoro-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-N,N,2-trimetilpiperazin-1-carboxamida;3- (5-{[(4-carbamimidoilfenil)metil]amino}-4-metil-1-(tiofen-3-carbonil)-1 H-pirazol-3-il)-N,N,2-trimetil-4- oxoazetidin-1 -carboxamida;3-(5-{[(4-carbamimidoilfenil)metil]sulfanil}-4-metoxi-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-N,N-dimetil-2-(trifluorometil)azetidin-1-carboxamida;ácido 3-(5-{[(4-carbamimidoilfenil)metil]sulfanil}-4-metoxi-1 H-pirazol-3-il)-1 -[(3-hidroxipirrolidin-1 -il)sulfonil]piperidin-2-carboxílico;ácido 3-(5-{[(4-carbamimidoilfenil)metil]sulfanil}-4-metil-3-{1-[2-(morfolin-4-il)acetil]-2-oxoazetidin-3-il}-1 H-pirazol-1 -carbonil)-2-clorobenzoico;3-(5-{[(4-fluorofenil)metil](metil)amino}-1-(2-metoxibenzoil)-4-metil-1 H-pirazol-3-il)-1-[(3-hidroxipirrolidin-1 -il)sulfonil]-4-(trifluorometil)piperidin-2-ona;3-(5-{[(4-fluorofenil)metil](metil)amino}-1-(3-metoxi-2,2-dimetilpropanoil)-4-metil-1 H-pirazol-3-il)-N,N,2-trimetil-4-oxopirrolidin-1-carboxamida;3- (5-{[(4-fluorofenil)metil](metil)amino}-4-metoxi-1-(2-metoxibenzoil)-1 H-pirazol-3-il)-1-[2-(morfolin-4- il)-2-oxoetil]-4-(trifluorometil)azetidin-2-ona;3-(5-{[(4-fluorofenil)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-4-metoxi-1 H-pirazol-3-il)-1-metanosulfonil-4-metilpirrolidin-2-ona;3-(5-{[(4-fluorofenil)metil]amino}-4-metoxi-1 -(2-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1 -(pirrolidin-1 -sulfonil)-4-(trifluorometil)pirrolidin-2-ona;ácido 3-(5-{[(4-fluorofenil)metil]amino}-4-metoxi-3-{1-[2-(morfolin-4-il)-2-oxoetil]pirrolidin-3-il}-1 H-pirazol-1 -carbonil)benzoico;3-(5-{[(4-fluorofenil)metil]amino}-4-metil-1-(4-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1-(3-hidroxipirrolidin-1-carbonil)-4-(trifluorometil)azetidin-2-ona;ácido 3-(5-{[(4-fluorofenil)metil]amino}-4-metil-1 -(tiofen-3-carbonil)-1 H-pirazol-3-il)-4-metil-1 -(morfolin-4-carbonil)pirrolidin-2-carboxílico;3-(5-{[(4-fluorofenil)metil]sulfanil}-1 -(tiofen-2-carbonil)-1 H-pirazol-3-il)-1 -(3-hidroxipirrolidin-1 -carbonil)piperidin-2-ona;3-(5-{[(4-fluorofenil)metil]sulfanil}-4-metil-1 H-pirazol-3-il)-1-[2-(morfolin-4-il)-2-oxoetil]pirrolidin-2-ona;ácido 3-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-1-(2,2-dimetilpropanoil)-4-metil-1 H-pirazol-3-il)pirrolidin-2-carboxílico;3-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-1-(tiofen-3-carbonil)-1 H-pirazol-3-il)-4-metil-1 -(pirrolidin-1 -sulfonil)azetidin-2-ona;ácido 3-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-3-[1-(3-hidroxipirrolidin-1-carbonil)-5-oxopirrolidin-3-il]-4-metil-1 H-pirazol-1 -carbonil)benzoico;ácido 3-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-ciano-3-(3-oxopiperidin-4-il)-1 H-pirazol-1 -carbonil)benzoico;3- (5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-metoxi-1-(tiofen-3-carbonil)-1 H-pirazol-3-il)-N,N,2-trimetilpiperidin-1-sulfonamida;ácido 3-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-metil-1-(2-metilfuran-3-carbonil)-1 H-pirazol-3-il)-4- (trifluorometil)piperidin-2-carboxílico;ácido 3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2,2-dimetilpropanoil)-4-metoxi-1 H-pirazol-3-il)-1 -[(3-hidroxipirrolidm-1 -il)sulfonil]pirrolidin-2-carboxílico;ácido 3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1 -metanosulfonil-4-(trifluorometil)piperidin-2-carboxílico;ácido 3-(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[(3-hidroxipirrolidin-1-il)sulfonil]azetidin-3-il}-4-metoxi-1 H-pirazol-1 -carbonil)benzoico;ácido 3-(5-{[(5-clorotiofen-2-il)metil]amino}-4-ciano-1 -(2,2-dimetilpropanoil)-1 H-pirazol-3-il)-1 -(2,2)-dimetilpropanoil)pirrolidin-2-carboxílico;3- (5-{[(5-clorotiofen-2-il)metil]amino}-4-ciano-1-(4-metiliuran-3-carbonil)-1 H-pirazol-3-il)-N,N-dimetil-4- (trifluorometil)piperidin-1-sulfonamida;ácido 3-(5-{[(5-clorotiofen-2-il)metil]amino}-4-ciano-3-[1 -(dimetilcarbamoy 1 )-4-oxoazetidin-2-il]-1 H-pirazol-1 -carbonil)benzoico;3-(5-{[(5-clorotiofen-2-il)metil]amino}-4-metoxi-1-(4-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1-(pirrolidin-1-carbonil)-4-(trifluorometil)azetidin-2-ona;ácido 3-(5-{[(5-clorotiofen-2-il)metil]amino}-4-metil-3-[1 -(pirrolidin-1 -sulfonil)pirrolidin-3-il]-1 H-pirazol-1 -carbonil)benzoico;3-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-1 -(2-metoxibenzoil)-1 H-pirazol-3-il)-1 -(pirrolidin-1 -sulfonil)-4-(trifluorometil)pirrolidin-2-ona;3-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-1-(3-metoxi-2,2-dimetilpropanoil)-4-metil-1 H-pirazol-3-il)-1-[(3-hidroxipirrolidin-1-il)sulfonil]-4-metilazetidin-2-ona;3-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-1-(furan-3-carbonil)-4-metoxi-1 H-pirazol-3-il)-5-hidroxi-N,N,2-trimetilpirrolidin-1-carboxamida;ácido 3-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-ciano-1 -(furan-2-carbonil)-1 H-pirazol-3-il)-4-metil-1 -(morfolin-4-carbonil)pirrolidin-2-carboxílico;ácido 3-(5-{[4-(aminometil)fenil]metoxi}-1 -(2-clorobenzoil)-1 H-pirazol-3-il)-1 -(3-hidroxipirrolidin-1 -carbonil)-4-(trifluorometil)piperidin-2-carboxílico;3-(5-{[4-(aminometil)fenil]metoxi}-1-(2-clorobenzoil)-4-metil-1 H-pirazol-3-il)-N,N-dimetil-2-(trifluorometil)piperazin-1-sulfonamida;3-(5-{[4-(aminometil)fenil]metoxi}-1 -(2-metoxibenzoil)-1 H-pirazol-3-il)-1 -(3-hidroxipirrolidin-1 -carbonil)-4-(trifluorometil)pirrolidin-2-ona;ácido 3-(5-{[4-(aminometil)fenil]metoxi}-1 -(3-carboxi-2-clorobenzoil)-4-ciano-1 H-pirazol-3-il)-1 -[2-(morfolin-4-il)acetil]pirrolidin-2-carboxílico;3-(5-{[4-(aminometil)fenil]metoxi}-1-(3-metoxi-2,2-dimetilpropanoil)-4-metil-1 H-pirazol-3-il)-4-metilpirrolidin-2-ona;ácido 3-(5-{[4-(aminometil)fenil]metoxi}-1 -(furan-2-carbonil)-4-metoxi-1 H-pirazol-3-il)-1 -(dimetilcarbamoil)-4-metilpiperidin-2-carboxílico;3-(5-{[4-(aminometil)fenil]metoxi}-4-metoxi-1-(2-metoxibenzoil)-1 H-pirazol-3-il)-N,N-dimetil-4-(trifluorometil)pirrolidin-1-carboxamida;3-(5-{[4-(aminometil)fenil]metoxi}-4-metoxi-1-(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-4-metil-1 -[2-(morfolin-4-il)acetil]pirrolidin-2-ona;3-(5-{[4-(aminometil)fenil]metoxi}-4-metoxi-1 H-pirazol-3-il)-1 -(pirrolidin-1 -sulfonil)piperidin-2-ona; ácido 3-(5-{[4-(aminometil)fenil]metoxi}-4-metoxi-3-[4-(morfolin-4-carbonil)piperazin-2-il]-1 H-pirazol-1 -carbonil)-2-clorobenzoico;3- (5-{[4-(aminometil)fenil]metoxi}-4-metil-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-1 -metanosulfonil-4- (trifluorometil)azetidin-2-ona;ácido 3-(5-{[4-(aminometil)fenil]metoxi}-4-metil-3-{1-[2-(morfolin-4-il)-2-oxoetil]piperazin-2-il}-1 H-pirazol-1-carbonil)-2-clorobenzoico;3-[1-(2,2-dimetilpropanoil)-5-[(4-fluorofenil)metoxi]-4-metoxi-1 H-pirazol-3-il]azetidin-2-ona; ácido 3-[1 -(2,2-dimetilpropanoil)-5-{[(4-fluorofenil)metil]amino}-4-metoxi-1 H-pirazol-3-il]-1 -[2-(morfolin-4-il)acetil]pirrolidin-2-carboxílico;3-[1-(2,2-dimetilpropanoil)-5-{[(4-fluorofenil)metil]amino}-4-metil-1 H-pirazol-3-il]-1-(morfolin-4-carbonil)pirrolidin-2-ona;3-[1-(2-clorobenzoil)-5-{[(4-fluorofenil)metil](metil)amino}-4-metoxi-1 H-pirazol-3-il]-N,N-dimetil-2-(trifluorometil)azetidin-1-sulfonamida;3-[1 -(2-clorobenzoil)-5-{[(4-fluorofenil)metil]sulfanil}-1 H-pirazol-3-il]-1 -[(3-hidroxipirrolidin-1 -il)sulfonil]-4-(trifluorometil)pirrolidin-2-ona;3-[1-(2-clorobenzoil)-5-{[(4-fluorofenil)metil]sulfanil}-4-metoxi-1 H-pirazol-3-il]-N,N-dimetil-2-(trifluorometil)pirrolidin-1 -sulfonam ida;3-[1-(2-clorobenzoil)-5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-ciano-1 H-pirazol-3-il]-N,N-dimetil-2-(trifluorometil)piperidin-1-carboxamida;3-[1 -(2-clorobenzoil)-5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-metil-1 H-pirazol-3-il]-1 -(pirrolidin-1 -carbonil)-4-(trifluorometil)pirrolidin-2-ona;ácido 3-[1-(3-carboxi-2-clorobenzoil)-4-fluoro-5-{[(4-fluorofenil)metil]sulfanil}-1 H-pirazol-3-il]pirrolidin-2-carboxílico;ácido 3-[1 -(3-carboxi-2-clorobenzoil)-5-[(5-clorotiofen-2-il)metoxi]-4-metil-1 H-pirazol-3-il]-1 -(2,2-dimetilpropanoil)pirrolidin-2-carboxílico;ácido 3-[1 -(3-carboxi-2-clorobenzoil)-5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-metil-1 H-pirazol-3-il]-1 -(2,2-dimetilpropanoil)piperidin-2-carboxílico;3-[4-ciano-1-(2-fluorobenzoil)-5-[(4-fluorofenil)metoxi]-1 H-pirazol-3-il]-N,N-dimetil-2-(trifluorometil)pirrolidin-1 -sulfonam ida;3-[4-ciano-1-(2-fluorobenzoil)-5-{[(4-fluorofenil)metil]amino}-1 H-pirazol-3-il]-N,N-dimetil-2-(trifluorometil)piperazin-1-sulfonamida;ácido 3-[5-({[4-(aminometil)fenil]metil}(metil)amino)-1 -(2-fluorobenzoil)-4-metoxi-1 H-pirazol-3-il]-1 -(3-hidroxipirrolidin-1-carbonil)-4-(trifluorometil)piperidin-2-carboxílico;3-[5-({[4-(aminometil)fenil]metil}(metil)amino)-1-(4-metilfuran-3-carbonil)-1 H-pirazol-3-il]-N,N-dimetil-2-(trifluorometil)azetidin-1-carboxamida;ácido 3-[5-({[4-(aminometil)fenil]metil}(metil)amino)-1 -(5-metilfuran-3-carbonil)-1 H-pirazol-3-il]-1 -[(3-hidroxipirrolidin-1 -il)sulfonil]piperidin-2-carboxílico;ácido 3-[5-({[4-(aminometil)fenil]metil}(metil)amino)-3-[1-(2,2-dimetilpropanoil)-5-hidroxipirrolidin-3-il]-4-metil-1 H-pirazol-1 -carbonil]benzoico;ácido 3-[5-({[4-(aminometil)fenil]metil}(metil)amino)-3-{1-[(3-hidroxipirrolidin-1-il)sulfonil]pirrolidin-3-il}-4-metboxi-1 H-pirazol-1 -carbonil]benzoico;3-[5-({[4-(aminometil)fenil]metil}(metil)amino)-4-ciano-1-(2-metilfuran-3-carbonil)-1 H-pirazol-3-il]-N,N-dimetil-4-(trifluorometil)pirrolidin-1-carboxamida;ácido 3-[5-({[4-(amometil)fenil]metil}(metil)amino)-4-ciano-3-[1-(dimetilsulfamoil)-4-oxopirrolidin-3-il]-1 H-pirazol-1 -carbonil]benzoico;3-[5-({[4-(aminometil)fenil]metil}(metil)amino)-4-metoxi-1-(4-metilfuran-3-carbonil)-1 H-pirazol-3-il]-1-metanosulfonil-4-(trifluorometil)piperidin-2-ona;3-[5-({[4-(aminometil)fenil]metil}(metil)amino)-4-metil-1-(tiofen-3-carbonil)-1 H-pirazol-3-il]-4-metil-1-(morfolin-4-carbonil)azetidin-2-ona;ácido 3-[5-({[4-(aminometil)fenil]metil}amino)-1 -(2-metilfuran-3-carbonil)-1 H-pirazol-3-il]-1 -(dimetilsulfamoil)-4-(trifluorometil)piperidin-2-carboxílico;ácido 3-[5-({[4-(aminometil)fenil]metil}amino)-1 -(3-carboxibenzoil)-4-metoxi-1 H-pirazol-3-il]-1 -[2-(morfolin-4-il)-2-oxoetil]piperidin-2-carboxílico;3-[5-({[4-(aminometil)fenil]metil}amino)-1-(3-hidroxi-2,2-dimetilpropanoil)-4-metil-1 H-pirazol-3-il]-N,N,4-trimetil-2-oxopiperidin-1-sulfonamida;3-[5-({[4-(aminometil)fenil]metil}amino)-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il]-N,N-dimetil-2-oxopiperidin-1-carboxamida;ácido 3-[5-({[4-(aminometil)fenil]metil}amino)-1 -benzoil-1 H-pirazol-3-il]-1 -(3-hidroxipirrolidin-1 -carbonil)-4-metilpirrolidin-2-carboxílico;ácido 3-[5-({[4-(aminometil)fenil]metil}amino)-3-[1-(dimetilsulfamoil)-3-oxopiperidin-4-il]-1 H-pirazol-1- carbonil]benzoico;3-[5-({[4-(aminometil)fenil]metil}amino)-4-metil-1 -(tiofen-3-carbonil)-1 H-pirazol-3-il]-4-metilpiperidin-2- ona;3- [5-({[4-(aminometil)fenil]metil}sulfanil)-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il]-N,N-dimetil-2-oxo-4- (trifluorometil)azetidin-1-sulfonamida;ácido 3-[5-({[4-(aminometil)fenil]metil}sulfanil)-1 -(3-carboxi-2-clorobenzoil)-1 H-pirazol-3-il]-1 -metanosulfonilpirrolidin-2-carboxílico;3-[5-({[4-(aminometil)fenil]metil}sulfanil)-1-(furan-2-carbonil)-4-metoxi-1 H-pirazol-3-il]-4-metil-1-[2-(morfolin-4-il)-2-oxoetil]piperidin-2-ona;3-[5-({[4-(aminometil)fenil]metil}sulfanil)-1-(furan-3-carbonil)-1 H-pirazol-3-il]-N,N,2-trimetilpiperazin-1- carboxamida;3-[5-({[4-(aminometil)fenil]metil}sulfanil)-4-ciano-1-(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il]-N,N,4-trimetilpiperidin-1-carboxamida;3-[5-({[4-(aminometil)fenil]metil}sulfanil)-4-ciano-1-(furan-3-carbonil)-1 H-pirazol-3-il]-N,N,4-trimetil-2- oxopiperidin-1 -sulfonamida;ácido 3-[5-({[4-(aminometil)fenil]metil}sulfanil)-4-ciano-3-[5-oxo-1 -(pirrolidin-1 -sulfonil)pirrolidin-3-il]-1 H-pirazol-1 -carbonil]-2-clorobenzoico;ácido 3-[5-({[4-(aminometil)fenil]metil}sulfanil)-4-metoxi-1 -(tiofen-2-carbonil)-1 H-pirazol-3-il]-1 -[2-(morfolin-4-il)-2-oxoetil]pirrolidin-2-carboxílico;ácido 3-[5-({[4-(aminometil)fenil]metil}sulfanil)-4-metoxi-3-{1-[2-(morfolin-4-il)acetil]piperidin-3-il}-1 H-pirazol-1 -carbonil]-2-clorobenzoico;3- {1-benzoil-5-[(4-fluorofenil)metoxi]-4-metil-1 H-pirazol-3-il}-N,N,2-trimetilpirrolidin-1-carboxamida; ácido 3-{3-[1 -(dimetilcarbamoil)piperidin-4-il]-5-{[(4-fluorofenil)metil]amonio}-1 H-pirazol-1 -carbonil}benzoico;ácido 3-{3-[1 -(dimetilsulfamoil)-5-oxopirrolidin-3-il]-5-[(4-fluorofenil)metoxi]-1 H-pirazol-1 -carbonil}benzoico;ácido 3-{3-[1 -(dimetilsulfamoil)-6-oxopiperidin-3-il]-5-[(4-fluorofenil)metoxi]-4-metil-1 H-pirazol-1 -carbonil}benzoico;ácido 3-{5-[(4-carbamimidoilfenil)metoxi]-1 -(2-metoxibenzoil)-1 H-pirazol-3-il}-1 -[2-(morfolin-4-il)acetil]-4-(trifluorometil)piperidin-2-carboxílico;ácido 3-{5-[(4-carbamimidoilfenil)metoxi]-1-(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il}-4-metil-1- (morfolin-4-carbonil)piperidin-2-carboxílico;ácido 3-{5-[(4-carbamimidoilfenil)metoxi]-3-[1 -(dimetilsulfamoil)pirrolidin-3-il]-1 H-pirazol-1 -carbonil}-2- clorobenzoico;ácido 3-{5-[(4-carbamimidoilfenil)metoxi]-4-ciano-1 -(tiofen-2-carbonil)-1 H-pirazol-3-il}-1 -[2-(morfolin-4-il)-2-oxoetil]piperidin-2-carboxílico;ácido 3-{5-[(4-carbamimidoilfenil)metoxi]-4-ciano-3-[2-oxo-1 -(pirrolidin-1 -carbonil)piperidin-3-il]-1 H-pirazol-1-carbonil}-2-clorobenzoico;ácido 3-{5-[(4-carbamimidoilfenil)metoxi]-4-metoxi-1 H-pirazol-3-il}-1 -(pirrolidin-1 -carbonil)piperidin-2- carboxílico;ácido 3-{5-[(4-carbamimidoilfenil)metoxi]-4-metoxi-3-[1 -(pirrolidin-1 -sulfonil)piperidin-4-il]-1 H-pirazol-1-carbonil}-2-clorobenzoico;3- {5-[(4-fluorofenil)metoxi]-1-(2-metilfuran-3-carbonil)-1 H-pirazol-3-il}-1-[2-(morfolin-4-il)-2-oxoetil]-4- (trifluorometil)azetidin-2-ona;3-{5-[(5-clorotiofen-2-il)metoxi]-1-(2-metoxibenzoil)-1 H-pirazol-3-il}-1-(morfolin-4-carbonil)-4-(trifluorometil)piperidin-2-ona;3-{5-[(5-clorotiofen-2-il)metoxi]-1-(furan-2-carbonil)-4-metil-1H-pirazol-3-il}-1-metanosulfonil-2-metilpiperazina;3-{5-[(5-clorotiofen-2-il)metoxi]-1 -(furan-3-carbonil)-4-metil-1 H-pirazol-3-il}-2-metil-1 -(pirrolidin-1 -carbonil)piperidina;ácido 3-{5-[(5-clorotiofen-2-il)metoxi]-1 -(tiofen-2-carbonil)-1 H-pirazol-3-il}-1 -(pirrolidin-1 -sulfonil)pirrolidin-2-carboxílico;ácido 3-{5-[(5-clorotiofen-2-il)metoxi]-4-ciano-1 -(2-metoxibenzoil)-1 H-pirazol-3-il}-4-(trifluorometil)piperidin-2-carboxílico;ácido 3-{5-[(5-clorotiofen-2-il)metoxi]-4-ciano-1 H-pirazol-3-il}-1 -(pirrolidin-1 -carbonil)pirrolidin-2-carboxílico;ácido 3-{5-[(5-clorotiofen-2-il)metoxi]-4-metoxi-1 -(2-metoxibenzoil)-1 H-pirazol-3-il}-1 -metanosulfonil-4-(trifluorometil)piperidin-2-carboxílico;3- {5-[(5-clorotiofen-2-il)metoxi]-4-metil-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il}-N,N-dimetil-4-(trifluorometil)piperidin-1-sulfonamida;4- (4-ciano-5-{[(4-fluorofenil)metil](metil)amino}-1-(tiofen-2-carbonil)-1 H-pirazol-3-il)-2-hidroxi-N,N-dimetilpirrolidin-1-sulfonamida;4-(5-{[(4-carbamimidoilfenil)metil]sulfanil}-4-ciano-1-(2-metoxibenzoil)-1 H-pirazol-3-il)-N,N-dimetil-2-oxo-3-(trifluorometil)pirrolidin-1-carboxamida;4-(5-{[(4-fluorofenil)metil](metil)amino}-1-(furan-3-carbonil)-4-metoxi-1 H-pirazol-3-il)-3-metil-1-[2-(morfolin-4-il)-2-oxoetil]azetidin-2-ona;4-(5-{[(4-fluorofenil)metil](metil)amino}-4-metoxi-1-(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona;4-(5-{[(4-nuorofenil)metil](metil)amino}-4-metoxi-1-(tiofen-2-carbonil)-1 H-pirazol-3-il)-N,N-dimetil-2-oxopirrolidin-1 -sulfonamida;4-(5-{[(4-fluorofenil)metil](metil)amino}-4-metil-1 H-pirazol-3-il)-1-[2-(morfolin-4-il)acetil]azetidin-2-ona;4-(5-{[(4-fluorofenil)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-1-[(3-hidroxipirrolidin-1 -il)sulfonil]-5-metilpiperidin-3-ona;4-(5-{[(4-fluorofenil)metil]sulfanil}-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-1 -[2-(morfolin-4-il)-2-oxoetil]-3-(trifluorometil)pirrolidin-2-ona;4-(5-{[(4-fluorofenil)metil]sulfanil}-4-metoxi-1 H-pirazol-3-il)-1-(3-hidroxipirrolidin-1-carbonil)piperidin-2-ona;4-(5-{[(4-fluorofenil)metil]sulfanil}-4-metil-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-3-(trifluorometil)azetidin-2-ona;4-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-4-metil-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona;4-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1-(3-hidroxipirrolidin-1 -carbonil)piperidin-2-ona;4-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-metoxi-1-(4-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1-[2-(morfolin-4-il)acetil]-5-(trifluorometil)pirrolidin-3-ona;4-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-metoxi-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1-[2-(morfolin-4-il)-2-oxoetil]pirrolidin-3-ona;4-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-3-metil-1-(pirrolidin-1 -sulfonil)azetidin-2-ona;4-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-4-metoxi-1 H-pirazol-3-il)-3-metil-1-(morfolin-4-carbonil)pirrolidin-2-ona;4-(5-{[(5-clorotiofen-2-il)metil]amino}-4-metil-1-(2-metilfuran-3-carbonil)-1 H-pirazol-3-il)-N,N-dimetil-2- oxo-3-(trifluorometil)pirrolidin-1-carboxamida;4-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-1-(2-metoxibenzoil)-4-metil-1 H-pirazol-3-il)-1-[2-(morfolin-4-il)acetil]-3-(trifluorometil)piperidin-2-ona;4-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-1 -(furan-2-carbonil)-1 H-pirazol-3-il)-5-metil-1 -(pirrolidin-1 -carbonil)pirrolidin-3-ona;4-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-1-(furan-2-carbonil)-4-metoxi-1 H-pirazol-3-il)-5-metilpirrolidin-3- ona;4- (5-{[(5-clorotiofen-2-il)metil]sulfanil}-1 H-pirazol-3-il)-1 -(pirrolidin-1 -sulfonil)piperidin-3-ona; 4-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-3-[1 -(pirrolidin-1 -carbonil)-3-(trifluorometil)azetidin-2-il]-1 H-pirazol-1 -carbonil)-1,3-tiazol;4-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-fluoro-1-(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-1-metanosulfonil-3-metilpiperidin-2-ona;4-(5-{[4-(aminometil)fenil]metoxi}-1-(furan-2-carbonil)-4-metil-1 H-pirazol-3-il)-N,N,3-trimetil-5-oxopiperidin-1 -carboxamida;4-(5-{[4-(aminometil)fenil]metoxi}-1 H-pirazol-3-il)-1 -(pirrolidin-1 -carbonil)piperidin-3-ona;4-(5-{[4-(aminometil)fenil]metoxi}-4-ciano-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-N,N-dimetil-2-oxo-3-(trifluorometil)piperidin-1-carboxamida;4-(5-{[4-(aminometil)fenil]metoxi}-4-metil-1-(tiofen-2-carbonil)-1 H-pirazol-3-il)-1-[2-(morfolin-4-il)acetil]pirrolidin-2-ona;4-({[1-(2,2-dimetilpropanoil)-3-[1-(2,2-dimetilpropanoil)-2-oxoazetidin-3-il]-4-metoxi-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({[1 -(2,2-dimetilpropanoil)-3-[4-(pirrolidin-1 -carbonil)piperazin-2-il]-1 H-pirazol-5-il](metil)amino}metil)benceno-1-carboximidamida;4-({[1 -(2,2-dimetilpropanoil)-4-metil-3-[2-oxo-1 -(pirrolidin-1 -carbonil)piperidin-3-il]-1 H-pirazol-5-il](metil)amino}metil)benceno-1-carboximidamida;4-({[1-(2,2-dimetilpropanoil)-4-metil-3-{1-[2-(morfolin-4-il)acetil]-3-oxopiperidin-4-il}-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({[1-(2-clorobenzoil)-3-[1-(dimetilsulfamoil)-4-oxo-3-(trifluorometil)azetidin-2-il]-4-metoxi-1 H-pirazol-5-il]oxi}metil)benceno-1-carboximidamida;4-({[1 -(2-clorobenzoil)-3-[1 -(pirrolidin-1 -sulfonil)-4-(trifluorometil)piperidin-3-il]-1 H-pirazol-5-il]sulfanil}metil)benceno-1-carboximidamida;4-({[1 -(2-clorobenzoil)-3-{1-[(3-hidroxipirrolidin-1 -il)sulfonil]-3-(trifluorometil)azetidin-2-il}-4-metil-1 H-pirazol-5-il]oxi}metil)benceno-1-carboximidamida;4-({[1-(2-clorobenzoil)-4-ciano-3-{4-[(3-hidroxipirrolidin-1-il)sulfonil]-3-(trifluorometil)piperazin-2-il}-1 H-pirazol-5-il]oxi}metil)benceno-1 -carboximidamida;4-({[1 -(2-clorobenzoil)-4-metil-3-[1 -(pirrolidin-1 -sulfonil)-3-(trifluorometil)piperazin-2-il]-1 H-pirazol-5-il]sulfanil}metil)benceno-1-carboximidamida;4-({[1-(2-fluorobenzoil)-3-{1-[(3-hidroxipirrolidin-1-il)sulfonil]-6-oxo-4-(trifluorometil)piperidin-3-il}-4-metil-1 H-pirazol-5-il]amino}metil)benceno-1 -carboximidamida;4-({[1-(2-fluorobenzoil)-3-{1-[2-(morfolin-4-il)-2-oxoetil]-2-(trifluorometil)piperidin-3-il}-1 H-pirazol-5-il](metil)amino}metil)benceno-1-carboximidamida;4-({[1-(2-metoxibenzoil)-3-[6-oxo-4-(trifluorometil)piperidin-3-il]-1 H-pirazol-5-il]sulfanil}metil)benceno-1-carboximidamida;4-({[1 -(2-metilfuran-3-carbonil)-3-[1 -(pirrolidin-1 -sulfonil)-2-(trifluorometil)pirrolidin-3-il]-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({[1-(3-hidroxi-2,2-dimetilpropanoil)-3-(1-metanosulfonil-4-metil-5-oxopirrolidin-3-il)-4-metil-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({[1-(3-hidroxi-2,2-dimetilpropanoil)-3-[1-(3-hidroxipirrolidin-1-carbonil)-2-metilpirrolidin-3-il]-4-metil-1 H-pirazol-5-il](metil)amino}metil)benceno-1-carboximidamida;4-({[1-(3-hidroxi-2,2-dimetilpropanoil)-4-metoxi-3-[3-metil-4-oxo-1-(pirrolidin-1-carbonil)azetidin-2-il]-1 H-pirazol-5-il](metil)amino}metil)benceno-1 -carboximidamida;4-({[1 -(5-metilfuran-3-carbonil)-3-[2-oxo-1 -(pirrolidin-1 -sulfonil)piperidin-4-il]-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({[1 -(furan-2-carbonil)-3-[1 -(3-hidroxipirrolidin-1 -carbonil)-2-metilazetidin-3-il]-4-metil-1 H-pirazol-5-il]sulfanil}metil)benceno-1-carboximidamida;4-({[1 -(furan-2-carbonil)-3-[3-metil-1 -(pirrolidin-1 -carbonil)piperazin-2-il]-1 H-pirazol-5-il]oxi}metil)benceno-1-carboximidamida;4-({[1-(furan-2-carbonil)-4-metoxi-3-[2-metil-1-(morfolin-4-carbonil)pirrolidin-3-il]-1 H-pirazol-5-il]oxi}metil)benceno-1-carboximidamida;4-({[1 -(furan-3-carbonil)-3-[1 -(3-hidroxipirrolidin-1 -carbonil)-3-metil-5-oxopiperidin-4-il]-4-metil-1 H-pirazol-5-il]oxi}metil)benceno-1-carboximidamida;4-({[1-(furan-3-carbonil)-3-[4-metil-1-(morfolin-4-carbonil)-5-oxopirrolidin-3-il]-1 H-pirazol-5-il]sulfanil}metil)benceno-1-carboximidamida;4-({[1 -(furan-3-carbonil)-3-[5-hidroxi-1 -(3-hidroxipirrolidin-1 -carbonil)-2-metilpirrolidin-3-il]-1 H-pirazol-5-il]oxi}metil)benceno-1-carboximidamida;4-({[3-(1-metanosulfonil-3-metilpiperidin-4-il)-1-(3-metoxi-2,2-dimetilpropanoil)-4-metil-1 H-pirazol-5-il]oxi}metil)benceno-1-carboximidamida;4-({[4-ciano-1 -(2,2-dimetilpropanoil)-3-{1 -[(3-hidroxipirrolidin-1 -il)sulfonil]-5-oxopirrolidin-3-il}-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({[4-ciano-1-(2-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)acetil]-2-oxo-3-(trifluorometil)piperidin-4-il}-1 H-pirazol-5-il](metil)amino}metil)benceno-1-carboximidamida;4-({[4-ciano-1-(3-hidroxi-2,2-dimetilpropanoil)-3-{3-metil-1-[2-(morfolin-4-il)-2-oxoetil]-4-oxoazetidin-2-il}-1 H-pirazol-5-il]amino}metil)benceno-1 -carboximidamida;4-({[4-ciano-1-(3-metoxi-2,2-dimetilpropanoil)-3-(2-metilazetidin-3-il)-1 H-pirazol-5-il]oxi}metil)benceno-1-carboximidamida;4-({[4-ciano-1 -(furan-2-carbonil)-3-[2-metil-1 -(pirrolidin-1 -sulfonil)azetidin-3-il]-1 H-pirazol-5-il]sulfanil}metil)benceno-1-carboximidamida;4- ({[4-ciano-1 -(furan-3-carbonil)-3-[1 -(3-hidroxipirrolidin-1 -carbonil)-2-metilpirrolidin-3-il]-1 H-pirazol-5- il]oxi}metil)benceno-1-carboximidamida;4-({[4-ciano-1-(furan-3-carbonil)-3-{4-metil-1-[2-(morfolin-4-il)acetil]pirrolidin-3-il}-1 H-pirazol-5-il]sulfanil}metil)benceno-1-carboximidamida;4-({[4-ciano-3-(3-metilpiperazin-2-il)-1-(tiofen-3-carbonil)-1 H-pirazol-5-il]amino}-carboximidamida; 4-({[4-metoxi-1-(2-metilfuran-3-carbonil)-3-[2-(trifluorometil)oxetan-3-il]-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({[4-metoxi-1-(4-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)acetil]-3-(trifluorometil)piperazin-2-il}-1 H-pirazol-5-il]amino}metil)benceno-1 -carboximidamida;4-({[4-metoxi-1-(5-metilfuran-3-carbonil)-3-(piperidin-3-il)-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({[4-metoxi-1-(5-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)-2-oxoetil]-6-oxopiperidin-3-il}-1 H-pirazol-5-il](metil)amino}metil)benceno-1-carboximidamida;4-({[4-metil-1-(4-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)acetil]-3-(trifluorometil)piperidin-4-il}-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({metil[1-(2-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)-2-oxoetil]-3-oxo-5-(trifluorometil)piperidin-4-il}-1 H-pirazol-5-il]amino}metil)benceno-1 -carboximidamida;4-({metil[4-metil-1-(2-metilfuran-3-carbonil)-3-[1-(morfolin-4-carbonil)-2-(trifluorometil)piperidin-3-il]-1 H-pirazol-5-il]amino}metil)benceno-1 -carboximidamida;4-({metil[4-metil-1 -(4-metilfliran-3-carbonil)-3-[2-oxo-1 -(pirrolidin-1 -carbonil)-3-(trifluorometil)piperidin-4-il]-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-[({1-benzoil-3-[2-metil-4-oxo-1 -(pirrolidin-1 -sulfonil)pirrolidin-3-il]-1 H-pirazol-5-il}amino)metil]benceno-1-carboximidamida;4-[({3-[1-(2,2-dimetilpropanoil)-2-oxo-4-(trifluorometil)piperidin-3-il]-4-metil-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-il}sulfanil)metil]benceno-1 -carboximidamida;4-[({3-[1 -(2,2-dimetilpropanoil)-3-(trifluorometil)azetidin-2-il]-4-fluoro-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-il}sulfanil)metil]benceno-1-carboximidamida;4-[({3-[1-(2,2-dimetilpropanoil)-3-oxo-5-(trifluorometil)piperidin-4-il]-1-(2-metoxibenzoil)-4-metil-1 H-pirazol-5-il}oxi)metil]benceno-1-carboximidamida;4- [({3-[1-(2,2-dimetilpropanoil)-4-metil-6-oxopiperidin-3-il]-1-(furan-3-carbonil)-4-metoxi-1 H-pirazol-5- il}oxi)metil]benceno-1-carboximidamida;4-[({3-[1 -(3-hidroxipirrolidin-1 -carbonil)-4-metilpirrolidin-3-il]-4-metoxi-1 -(tiofen-3-carbonil)-1 H-pirazol-5-il}amino)metil]benceno-1-carboximidamida;4-[({3-[1-(dimetilsulfamoil)-3-metil-2-oxopiperidin-4-il]-1-(furan-2-carbonil)-4-metoxi-1 H-pirazol-5-il}sulfanil)metil]benceno-1-carboximidamida;4-[({3-[1-(dimetilsulfamoil)-4-oxopirrolidin-3-il]-4-metil-1-(tiofen-2-carbonil)-1 H-pirazol-5-il}sulfanil)metil]benceno-1-carboximidamida;4-[({3-[1 -(dimetilsulfamoil)piperidin-4-il]-4-metil-1 -(tiofen-2-carbonil)-1 H-pirazol-5-il}oxi)metil]benceno-1-carboximidamida;4-[({3-[1 -(morfolin-4-carbonil)-4-oxo-3-(trifluorometil)azetidin-2-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-il}oxi)metil]benceno-1-carboximidamida;4-[({4-ciano-3-[1-(2,2-dimetilpropanoil)-2-oxo-3-(trifluorometil)piperidin-4-il]-1-(4-metilfuran-3-carbonil)-1 H-pirazol-5-il}amino)metil]benceno-1-carboximidamida;4-[({4-ciano-3-[1-(2,2-dimetilpropanoil)-4-(trifluorometil)piperidin-3-il]-1-(2-metilfuran-3-carbonil)-1 H-pirazol-5-il}amino)metil]benceno-1-carboximidamida;4-[({4-ciano-3-[1-(2,2-dimetilpropanoil)-4-oxoazetidin-2-il]-1-(5-metilfuran-3-carbonil)-1 H-pirazol-5-il}amino)metil]benceno-1-carboximidamida;4-[({4-ciano-3-[1-(2,2-dimetilpropanoil)pirrolidin-3-il]-1 H-pirazol-5-il}sulfanil)metil]benceno-1-carboximidamida;4-[({4-ciano-3-[1 -metanosulfonil-2-oxo-4-(trifluorometil)piperidin-3-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-il}sulfanil)metil]benceno-1-carboximidamida;4-[({4-ciano-3-[2-metil-1-(morfolin-4-carbonil)pirrolidin-3-il]-1-(tiofen-3-carbonil)-1 H-pirazol-5-il}(metil)amino)metil]benceno-1-carboximidamida;4-[({4-ciano-3-[2-oxo-1 -(pirrolidin-1 -carbonil)-3-(trifluorometil)piperidin-4-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-il}oxi)metil]benceno-1 -carboximidamida;4-[5-({[4-(aminometil)fenil]metil}(metil)amino)-1-(2,2-dimetilpropanoil)-4-metoxi-1 H-pirazol-3-il]-1-metanosulfonilazetidin-2-ona;4-[5-({[4-(aminometil)fenil]metil}(metil)amino)-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il]-3-metil-1-[2-(morfolin-4-il)-2-oxoetil]piperidin-2-ona;4-[5-({[4-(aminometil)fenil]metil}(metil)amino)-1-(tiofen-3-carbonil)-1 H-pirazol-3-il]-N,N,3-trimetil-2-oxopiperidin-1 -sulfonamida;4-[5-({[4-(aminometil)fenil]metil}(metil)amino)-1-benzoil-4-metil-1 H-pirazol-3-il]-3-metil-1-(morfolin-4-carbonil)piperidin-2-ona;4-[5-({[4-(aminometil)fenil]metil}(metil)amino)-4-metil-1-(4-metilfuran-3-carbonil)-1 H-pirazol-3-il]-N,N-dimetil-2-oxo-3-(trifluorometil)piperidin-1-carboxamida;4-[5-({[4-(aminometil)fenil]metil}(metil)amino)-4-metil-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il]pirrolidin-2-ol;4-[5-({[4-(aminometil)fenil]metil}amino)-1-(2,2-dimetilpropanoil)-4-metil-1 H-pirazol-3-il]-1 -(pirrolidin-1 -sulfonil)pirrolidin-2-ona;4-[5-({[4-(aminometil)fenil]metil}amino)-4-metoxi-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il]-1-(2,2-dimetilpropanoil)pirrolidin-3-ona;4-[5-({[4-(aminometil)fenil]metil}amino)-4-metoxi-1-(tiofen-3-carbonil)-1 H-pirazol-3-il]-1-[(3-hidroxipirrolidin-1 -il)sulfonil]-5-metilpirrolidin-2-ol;4-[5-({[4-(aminometil)fenil]metil}sulfanil)-1-(2-clorobenzoil)-4-metil-1 H-pirazol-3-il]-1-(2,2-dimetilpropanoilo)-3-(trifluorometil)pirrolidin-2-ona;4-[5-({[4-(aminometil)fenil]metil}sulfanil)-1 -(furan-2-carbonil)-1 H-pirazol-3-il]-1 -[(3-hidroxipirrolidin-1 -il)sulfonil]-5-metilpirrolidin-2-ol;4-[5-({[4-(aminometil)fenil]metil}sulfanil)-1 H-pirazol-3-il]-1-(2,2-dimetilpropanoil)pirrolidin-3-ona; 4- [5-({[4-(aminometil)fenil]metil}sulfanil)-4-metoxi-1-(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il]-5- metil-1 -(pirrolidin-1 -sulfonil)pirrolidin-3-ona;4-[5-({[4-(aminometil)fenil]metil}sulfanil)-4-metil-1-(tiofen-2-carbonil)-1 H-pirazol-3-il]-1-(morfolin-4-carbonil)piperidin-3-ona;4-[5-({[4-(aminometil)fenil]metil}sulfanil)-4-metil-1 H-pirazol-3-il]-1-(2,2-dimetilpropanoil)azetidin-2-ona;4-{1-benzoil-5-[(4-fluorofenil)metoxi]-1 H-pirazol-3-il}-3-metilpiperidin-2-ona;4-{5-[(4-fluorofenil)metoxi]-1 -(3-hidroxi-2,2-dimetilpropanoil)-4-metil-1 H-pirazol-3-il}-1 -metanosulfonil-5-metilpiperidin-3-ona;4-{5-[(4-fluorofenil)metoxi]-1 -(4-metilfuran-3-carbonil)-1 H-pirazol-3-il}-1 -(3-hidroxipirrolidin-1 -carbonil)-3-(trifluorometil)azetidin-2-ona;4-{5-[(4-fluorofenil)metoxi]-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il}-1 -metanosulfonilpirrolidin-3-ona;4-{5-[(4-fluorofenil)metoxi]-4-metoxi-1-(4-metilfuran-3-carbonil)-1 H-pirazol-3-il}-1-[2-(morfolin-4-il)-2-oxoetil]-3-(trifluorometil)pirrolidin-2-ona;4-{5-[(4-fluorofenil)metoxi]-4-metoxi-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il}piperidina;4-{5-[(5-clorotiofen-2-il)metoxi]-1 -(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il}-1 -[(3-hidroxipirrolidin-1 -il)sulfonil]-3-metilpiperidin-2-ona;4-{5-[(5-clorotiofen-2-il)metoxi]-4-metoxi-1-(tiofen-2-carbonil)-1 H-pirazol-3-il}piperidin-3-ona;4-{[(1 -benzoil-3-{1 -[(3-hidroxipirrolidin-1 -il)sulfonil]-4-metilpiperidin-3-il}-1 H-pirazol-5-il)(metil)amino]metil}benceno-1-carboximidamida;4-{[(1 -benzoil-4-ciano-3-{1 -[(3-hidroxipirrolidin-1 -il)sulfonil]-2-metil-4-oxoazetidin-3-il}-1 H-pirazol-5-il)(metil)amino]metil}benceno-1-carboximidamida;4-{[(3-{1 -[(3-hidroxipirrolidin-1 -il)sulfonil]-2-metilpiperidin-3-il}-1 -(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-5-il)sulfanil]metil}benceno-1-carboximidamida;4-{[(3-{1-[(3-hidroxipirrolidin-1-il)sulfonil]-3-(trifluorometil)piperidin-4-il}-1-(4-metilfuran-3-carbonil)-1 H-pirazol-5-il)(metil)amino]metil}benceno-1 -carboximidamida;4-{[(3-{1 -[(3-hidroxipirrolidin-1 -il)sulfonil]-4-oxoazetidin-2-il}-4-metil-1 H-pirazol-5-il)sulfanil]metil}benceno-1-carboximidamida;4-{[(3-{1 -[(3-hidroxipirrolidin-1 -il)sulfonil]azetidin-3-il}-1 -(tiofen-2-carbonil)-1 H-pirazol-5-il)sulfanil]metil}benceno-1-carboximidamida;4-{[(3-{1-[2-(morfolin-4-il)-2-oxoetil]-6-oxopiperidin-3-il}-1 H-pirazol-5-il)oxi]metil}benceno-1-carboximidamida;4-{[(3-{3-metil-1-[2-(morfolin-4-il)-2-oxoetil]azetidin-2-il}-1-(tiofen-3-carbonil)-1 H-pirazol-5-il)amino]metil}benceno-1-carboximidamida;4- {[(4-metil-3-{1-[2-(morfolin-4-il)acetil]-2-oxopiperidin-3-il}-1 H-pirazol-5-il)oxi]metil}benceno-1-carboximidamida;5- (1 -benzoil-5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-metil-1 H-pirazol-3-il)-4-metil-1 -(pirrolidin-1 -carbonil)piperidin-2-ona;5-(4-ciano-5-{[(4-fluorofenil)metil]sulfanil}-1-(tiofen-2-carbonil)-1 H-pirazol-3-il)-N,N-dimetil-2-oxopiperidin-1 -sulfonamida;5-(5-{[(4-fluorofenil)metil]sulfanil}-4-metoxi-1 -(tiofen-2-carbonil)-1 H-pirazol-3-il)-1 -(pirrolidin-1 -sulfonil)piperidin-2-ona;5-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-ciano-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-N,N,4-trimetil-2-oxopiperidin-1-carboxamida;5-(5-{[4-(aminometil)fenil]metoxi}-1-(furan-2-carbonil)-1 H-pirazol-3-il)-1-metanosulfonil-4-metilpiperidin-2-ona;5-({[4-(aminometil)fenil]metil}(metil)amino)-1-(2-fluorobenzoil)-3-[1-(pirrolidin-1-sulfonil)-3-(trifluorometil)piperazin-2-il]-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}(metil)amino)-1-(5-metilfuran-3-carbonil)-3-[2-oxo-1-(pirroldina-1-sulfonil)piperidin-3-il]-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}(metil)amino)-1-benzoil-3-{4-metil-1-[2-(morfolin-4-il)acetil]-2-oxopirrolidin-3-il}-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}(metil)amino)-3-[1-(2,2-dimetilpropanoil)-4-metil-2-oxopirrolidin-3-il]-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}amino)-1-(2,2-dimetilpropanoil)-3-{1-[2-(morfolin-4-il)acetil]piperidin-3-il}-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}amino)-1-(4-metilfuran-3-carbonil)-3-[1-(morfolin-4-carbonil)-3-(trifluorometil)piperidin-4-il]-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}amino)-1-(5-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)acetil]-2-oxopiperidin-3-il}-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}amino)-1-benzoil-3-(1-metanosulfonil-3-metilpiperidin-4-il)-1 H-pirazol-4- carbonitrilo;5- ({[4-(aminometil)fenil]metil}amino)-3-[1-(2,2-dimetilpropanoil)-3-oxo-5-(trifluorometil)piperidin-4-il]-1 -(2-metilfuran-3-carbonil)-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}amino)-3-{4-metil-1 -[2-(morfolin-4-il)-2-oxoetil]-2-oxopiperidin-3-il}-1 -(tiofen-3-carbonil)-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}sulfanil)-1 -(2-clorobenzoil)-3-[1 -(pirrolidin-1 -carbonil)-3-(trifluorometil)piperidin-4-il]-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}sulfanil)-1-(2-metoxibenzoil)-3-[2-(trifluorometil)oxetan-3-il]-1 H-pirazol-4- carbonitrilo;5- ({[4-(aminometil)fenil]metil}sulfanil)-3-{1-[(3-hidroxipirrolidin-1-il)sulfonil]-4-oxopirrolidin-3-il}-1-(tiofen-2-carbonil)-1 H-pirazol-4-carbonitrilo;5-[(4-fluorofenil)metoxi]-1-(3-hidroxi-2,2-dimetilpropanoil)-3-[4-(3-hidroxipirrolidin-1-carbonil)-3-metilpiperazin-2-il]-1 H-pirazol-4-carbonitrilo;5-[(4-fluorofenil)metoxi]-1-(4-metilfuran-3-carbonil)-3-[1-(morfolin-4-carbonil)-3-(trifluorometil)azetidin-2-il]-1 H-pirazol-4-carbonitrilo;5-[(4-fluorofenil)metoxi]-1-(5-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)acetil]-4-oxoazetidin-2-il}-1 H-pirazol-4-carbonitrilo;5-[(4-fluorofenil)metoxi]-3-{4-metil-1-[2-(morfolin-4-il)acetil]piperidin-3-il}-1-(tiofen-3-carbonil)-1 H-pirazol-4-carbonitrilo;5-[(4-fluorofenil)metoxi]-4-metoxi-1-(2-metilfuran-3-carbonil)-3-[2-(trifluorometil)oxan-3-il]-1H-pirazol;5-[(5-clorotiofen-2-il)metoxi]-1-(3-metoxi-2,2-dimetilpropanoil)-3-[4-metil-6-oxo-1-(pirrolidin-1-carbonil)piperidin-3-il]-1 H-pirazol-4-carbonitrilo;5-[(5-clorotiofen-2-il)metoxi]-1 -(furan-2-carbonil)-3-[4-metil-1 -(pirrolidin-1 -carbonil)piperidin-3-il]-1 H-pirazol-4-carbonitrilo;5-[(5-clorotiofen-2-il)metoxi]-3-[1 -(3-hidroxipirrolidin-1 -carbonil)-5-oxopirrolidin-3-il]-1 -(tiofen-2-carbonil)-1 H-pirazol-4-carbonitrilo;5-[(5-clorotiofen-2-il)metoxi]-3-[2-oxo-1 -(pirrolidin-1 -carbonil)-4-(trifluorometil)azetidin-3-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-4-carbonitrilo;5-[(5-clorotiofen-2-il)metoxi]-4-metil-3-(pirrolidin-3-il)-1 H-pirazol;5-[5-({[4-(aminometil)fenil]metil}(metil)amino)-4-fluoro-1-(2-metilfuran-3-carbonil)-1 H-pirazol-3-il]-4-(trifluorometil)piperidin-2-ona;5-{1 -benzoil-5-[(4-fluorofenil)metoxi]-4-metoxi-1 H-pirazol-3-il}-1 -(3-hidroxipirrolidin-1 -carbonil)-4-metilpiperidin-2-ona;5-{[(4-fluorofenil)metil](metil)amino}-1-(furan-3-carbonil)-3-(1-metanosulfonil-4-metil-5-oxopirrolidin-3-il)-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil](metil)amino}-3-(piperidin-4-il)-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil](metil)amino}-3-[1-(3-hidroxipirrolidin-1-carbonil)-4-metil-6-oxopiperidin-3-il]-1 -(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil]amino}-1-(2-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)acetil]-2-(trifluorometil)pirrolidina-3-il}-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil]amino}-1-(5-metilfuran-3-carbonil)-3-[5-oxo-1-(pirrolidin-1-carbonil)pirrolidin-3-il]-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil]amino}-3-(2-metil-4-oxopirrolidin-3-il)-1-(tiofen-3-carbonil)-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil]sulfanil}-1-(2-metoxibenzoil)-3-{1-[2-(morfolin-4-il)acetil]-6-oxo-4-(trifluorometil)piperidin-3-il}-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil]sulfanil}-1-(furan-3-carbonil)-3-(1-metanosulfonil-2-metilazetidin-3-il)-1 H-pirazol;5-{[(4-fluorofenil)metil]sulfanil}-3-{1-[2-(morfolin-4-il)-2-oxoetil]-4-oxopirrolidin-3-il}-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil]sulfanil}-3-{1-[2-(morfolin-4-il)acetil]-4-oxo-2-(trifluorometil)pirrolidin-3-il}-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil]sulfanil}-4-metoxi-1-(2-metoxibenzoil)-3-[3-(trifluorometil)oxetan-2-il]-1H-pirazol;5-{[(5-clorotiofen-2-il)metil](metil)amino}-1-(2,2-dimetilpropanoil)-3-[1-(morfolin-4-carbonil)-6-0xopiperidin-3-il]-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil](metil)amino}-1-(2-fluorobenzoil)-3-{5-hidroxi-1-[2-(morfolin-4-il)acetil]-2-(trifluorometil)pirrolidin-3-il}-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil](metil)amino}-1-(4-metilfuran-3-carbonil)-3-[4-oxo-3-(trifluorometil)azetidin-2-il]-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil]amino}-1-(2-fluorobenzoil)-3-[5-hidroxi-1-(morfolin-4-carbonil)-2-(trifluorometil)pirrolidin-3-il]-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil]amino}-1-(2-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)-2-oxoetilo]-4-(trifluorometil)pirrolidin-3-il}-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-3-[2-metil-1-(pirrolidin-1-carbonil)piperidin-3-il]-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil]sulfanil}-1-(furan-3-carbonil)-3-[3-metil-4-oxo-1-(pirrolidin-1-carbonil)azetidin-2-il]-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil]sulfanil}-3-[1-(3-hidroxipirrolidin-1-carbonil)-2-oxo-4-(trifluorometil)azetidin-3-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil]sulfanil}-3-[1-(3-hidroxipirrolidin-1-carbonil)-4-oxopirrolidin-3-il]-1-(tiofen-2- carbonil)-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-metil-3-[1 -(pirrolidin-1 -carbonil)pirrolidin-3-il]-1 -(tiofen-2-carbonil)-1 H-pirazol;5-{[4-(aminometil)fenil]metoxi}-1 -(2-clorobenzoil)-3-[1 -(3-hidroxipirrolidin-1 -carbonil)-3-(trifluorometil)piperazin-2-il]-1 H-pirazol-4-carbonitrilo;5-{[4-(aminometil)fenil]metoxi}-1-(furan-2-carbonil)-3-[2-metil-1-(morfolin-4-carbonil)-4-oxoazetidin-3- il]-1 H-pirazol-4-carbonitrilo;5-{[4-(aminometil)fenil]metoxi}-1-(furan-3-carbonil)-3-(1-metanosulfonil-4-metil-2-oxopirrolidin-3-il)-1 H-pirazol-4-carbonitrilo;5-{[4-(aminometil)fenil]metoxi}-3-(5-hidroxipirrolidin-3-il)-1 H-pirazol-4-carbonitrilo;N-[(4-fluorofenil)metil]-3-[1-metanosulfonil-2-(trifluorometil)piperidin-3-il]-4-metil-1-(2-metiliuran-3-carbonil)-1 H-pirazol-5-amina;N-[(4-fluorofenil)metil]-4-metil-1-(5-metilfuran-3-carbonil)-3-(morfolin-2-il)-1 H-pirazol-5-amina; N-[(4-fluorofenil)metil]-N,4-dimetil-3-[1-(morfolin-4-carbonil)-3-(trifluorometil)azetidin-2-il]-1-(1,3-tiazol)-4-carbonil)-1 H-pirazol-5-amina;N-[(4-fluorofenil)metil]-N-metil-3-(pirrolidin-3-il)-1-(tiofen-2-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(2-fluorobenzoil)-3-[1-metanosulfonil-4-(trifluorometil)pirrolidin-3-il]-4-metoxi-N-metil-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1 -(2-fluorobenzoil)-4-metoxi-3-[1 -(pirrolidin-1 -sulfonil)-4-(trifluorometil)piperidin-3-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(2-fluorobenzoil)-4-metil-3-[1-(morfolin-4-carbonil)-4-(trifluorometil)piperidin-3-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1 -(2-fluorobenzoil)-N,4-dimetil-3-[1 -(pirrolidin-1 -sulfonil)-2-(trifluorometil)piperidin-3-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-4-metoxi-3-[4-metil-1 -(pirrolidin-1 -carbonil)piperidin-3-il]-1 -(tiofen-3-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-4-metil-3-[2-metil-1 -(pirrolidin-1 -sulfonil)azetidin-3-il]-1 -(tiofen-3-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-N-metil-1-(2-metilfuran-3-carbonil)-3-[3-(trifluorometil)oxetan-2-il]-1 H-pirazol-5-amina;N-{[4-(aminometil)fenil]metil}-1-(2-fluorobenzoil)-3-[1-metanosulfonil-2-(trifluorometil)pirrolidin-3-il]-N-metil-1 H-pirazol-5-amina;N-{[4-(aminometil)fenil]metil}-1 -(2-fluorobenzoil)-4-metil-3-[1 -(pirrolidin-1 -carbonil)-3-(trifluorometil)piperidin-4-il]-1 H-pirazol-5-amina;N-{[4-(aminometil)fenil]metil}-1-(2-fluorobenzoil)-N,4-dimetil-3-[3-(trifluorometil)piperazin-2-il]-1 H-pirazol-5-amina;N-{[4-(aminometil)fenil]metil}-1-benzoil-4-metoxi-3-(2-metilazetidin-3-il)-1 H-pirazol-5-amina; N-{[4-(aminometil)fenil]metil}-4-metoxi-1-(2-metilfuran-3-carbonil)-3-[1-(morfolin-4-carbonil)-2-(trifluorometil)azetidin-3-il]-1 H-pirazol-5-amina;N-{[4-(aminometil)fenil]metil}-4-metil-1-(4-metilfuran-3-carbonil)-3-[4-(pirrolidin-1-sulfonil)-3-(trifluorometil)piperazin-2-il]-1H-pirazol-5-amina;[4-({[1-(2-clorobenzoil)-3-[4-(trifluorometil)piperidin-3-il]-1 H-pirazol-5-il]sulfanil}metil)fenil]metanamina;[4-({[1-(2-metoxibenzoil)-3-[1-(morfolin-4-carbonil)-2-(trifluorometil)azetidin-3-il]-1 H-pirazol-5-il]sulfanil}metilo)fenil]metanamina;[4-({[1-(furan-2-carbonil)-4-metil-3-(3-metilpiperazin-2-il)-1 H-pirazol-5-il]sulfanil}metil)fenil]metanamina;[4-({[1 -(furan-3-carbonil)-4-mctil-3-[2-metil-1 -(pirrolidin-1 -carbonil)azetidin-3-il]-1 H-pirazol-5-il]oxi}metil)fenil]metanamina;{4-[({4-metoxi-3-[1 -(morfolin-4-carbonil)-3-(trifluorometil)piperidin-4-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-il}sulfanil)metil]fenil}metanamina;N-[(5-clorotiofen-2-il)metil]-3-(piperazin-2-il)-1 H-pirazol-5-amina;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-{4-[2-(morfolin-4-il)acetil]piperazin-2-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazo1 -3-il)piperazin-1 -il]-2-(morfolin-4-il)etan-1 -ona; 1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[4-(morfolin-4-sulfonil)piperazin-2-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[4-(2,2,2-trifluoroetil)piperazin-2-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;N-[(5-clorotiofen-2-il)metil]-3-(pirrolidin-2-il)-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;1 -[2-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)pirrolidin-1 -il]-2-(morfolin-4-il)etan-1 -ona;N-[(5-clorotiofen-2-il)metil]-3-(pirrolidin-3-il)-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;1 -[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)pirrolidin-1 -il]-2-(morfolin-4-il)etan-1 -ona;N-[(5-clorotiofen-2-il)metil]-3-(piperazin-2-il)-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;1 -[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)piperazin-1 -il]-2-(morfolin-4-il)etan-1 -ona;N-[(5-clorotiofen-2-il)metil]-3-(4-metanosulfonilpiperazin-2-il)-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[4-(morfolin-4-carbonil)piperazin-2-il]-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;1 -[3-(azepan-4-il)-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-1 -il]-2,2-dimetilpropan-1 -ona; 3-(azepan-4-il)-N-[(5-clorotiofen-2-il)metil]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;1 -[3-(azetidin-3-il)-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-1 -il]-2,2-dimetilpropan-1 -ona; N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-carbonil)azetidin-3-il]-1 H-pirazol-5-amina;3-(azetidin-3-il)-N-[(5-clorotiofen-2-il)metil]-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;3-[1-(2-clorobenzoil)-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il]-N,N-dimetilpirrolidin-1-sulfonamida;1 -(2-clorobenzoil)-N-[(5-clorotiofen-2-il)metil]-3-[1 -(pirrolidin-1 -sulfonil)pirrolidin-3-il]-1 H-pirazol-5-amina;1 -(2-clorobenzoil)-N-[(5-clorotiofen-2-il)metil]-3-[1 -(piperazin-1 -sulfonil)pirrolidin-3-il]-1 H-pirazol-5-amina;1 -(2-clorobenzoil)-N-[(5-clorotiofen-2-il)metil]-3-[1 -(piperazin-1 -carbonil)pirrolidin-3-il]-1 H-pirazol-5-amina;3-[1-(2-clorobenzoil)-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il]-N,N-dimetilazetidin-1-sulfonamida;1 -(2-clorobenzoil)-N-[(5-clorotiofen-2-il)metil]-3-[1 -(pirrolidin-1 -sulfonil)azetidin-3-il]-1 H-pirazol-5-amina;1 -(2-clorobenzoil)-N-[(5-clorotiofen-2-il)metil]-3-[1 -(piperazin-1 -sulfonil)azetidin-3-il]-1 H-pirazol-5-amina;1 -(2-clorobenzoil)-N-[(5-clorotiofen-2-il)metil]-3-[1 -(piperazin-1 -carbonil)azetidin-3-il]-1 H-pirazol-5-amina;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[4-(2-ciclopropoxietil)piperazin-2-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;ácido 4-[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2,2-dimetilpropanoil)-1 H-pirazol-3-il)piperazin-1 -carbonil]morfolin-3-carboxílico;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[4-(3-metilmorfolin-4-carbonil)piperazin-2-il]-1 H-pirazol-1 -il)-2.2- dimetilpropan-1 -ona;1-(5-1[(5-clorotiofen-2-il)metil]amino}-3-[4-(3,5-dimetilmorfolin-4-carbonil)piperazin-2-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-{4-[2-(morfolin-4-il)-2-oxoetil]piperazin-2-il}-1 H-pirazol-1 -il)-2.2- dimetilpropan-1 -ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-{4-[2-(morfolin-4-il)etil]piperazin-2-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;ácido 4-{2-[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2,2-dimetilpropanoil)-1 H-pirazol-3-il)piperazin-1 -il]etil}morfolin-3-carboxílico;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-{4-[4-(hidroximetil)-1,3-oxazol-2-il]piperazin-2-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-(4-{[4-(hidroximetil)-1,3-oxazol-2-il]metil}piperazin-2-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; yácido 2-[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2,2-dimetilpropanoil)-1 H-pirazol-3-il)piperazin-1 -iljacético.12. Una composición farmacéutica que comprende un compuesto según cualquiera de las reivindicaciones 1 a 11, y un excipiente farmacéuticamente aceptable.13. El compuesto según cualquiera de las reivindicaciones 1 a 11, para usar en el tratamiento de una enfermedad o un trastorno relacionado con la trombina, o una enfermedad o un trastorno relacionado con la calicreína; opcionalmente, en donde el tratamiento tiene un efecto profiláctico.14. El compuesto para el uso según la reivindicación 13, en donde el compuesto es para usar en el tratamiento de una enfermedad o un trastorno trombótico, y/o una enfermedad o un trastorno que implica un trombo de coágulo sanguíneo o la posible formación de un trombo de coágulo sanguíneo, o en donde la enfermedad o el trastorno implica episodios cardíacos recurrentes después de un infarto de miocardio.15. El compuesto para el uso según la reivindicación 14, en donde dicha enfermedad o dicho trastorno trombótico comprende síndrome coronario agudo, tromboembolia y/o trombosis; opcionalmente, en donde la tromboembolia comprende tromboembolia venosa, tromboembolia arterial y/o tromboembolia cardiogénica.16. El compuesto para el uso según la reivindicación 15, en donde:la tromboembolia comprende tromboembolia venosa; opcionalmente, en donde la tromboembolia venosa comprende trombosis venosa profunda y/o embolia pulmonar, o en donde la tromboembolia venosa está asociada con la formación de un trombo dentro de una vena asociado con uno o más factores de riesgo adquiridos o heredados, y/o embolia de venas periféricas provocada por un trombo desprendido; además, opcionalmente, en donde la trombosis venosa profunda y/o la embolia pulmonar se producen después de una intervención médica, o en donde uno o más factores de riesgo comprenden una tromboembolia venosa previa; o la tromboembolia comprende tromboembolia cardiogénica; opcionalmente, en donde la tromboembolia cardiogénica se debe a fibrilación auricular no valvular o formación de un trombo en el corazón asociado con arritmia cardíaca, defecto de válvula cardíaca, válvulas cardíacas protésicas o cardiopatía, y/o embolia de arterias periféricas provocada por un trombo desprendido; además, opcionalmente, en donde el trombo desprendido está en el cerebro (accidente cerebrovascular isquémico); además, opcionalmente, en donde el trombo desprendido provoca un ataque isquémico transitorio (AIT); ola trombosis comprende trombosis arterial; opcionalmente, en donde la trombosis arterial se debe a uno o más procesos ateroescleróticos subyacentes en las arterias; además, opcionalmente, en donde el uno o más procesos ateroescleróticos subyacentes en las arterias obstruyen u ocluyen una arteria, provocar isquemia miocárdica (angina de pecho, síndrome coronario agudo), provocar infarto de miocardio, obstruir u ocluir una arteria periférica (enfermedad arterial periférica isquémica), y/u obstruir u ocluir la arteria después de una operación en un vaso sanguíneo (reoclusión o reestenosis después de angioplastia coronaria transluminal, reoclusión o reestenosis después de angioplastia transluminal percutánea de arterias periféricas); ola enfermedad o el trastorno trombótico implica coagulación disfuncional o coagulación intravascular diseminada; opcionalmente, en donde el sujeto se somete a una intervención coronaria percutánea (ICP).17. El compuesto para el uso según la reivindicación 14, en donde dicha enfermedad o dicho trastorno trombótico implica un trombo de coágulo sanguíneo o la posible formación de un trombo de coágulo sanguíneo, y además implica un accidente cerebrovascular y/o uno o más ataques isquémicos transitorios (AIT) y/o hipertensión pulmonar; además, opcionalmente, en donde dicha enfermedad o dicho trastorno que implica un trombo de coágulo sanguíneo o la posible formación de un trombo de coágulo sanguíneo implica además un accidente cerebrovascular, y en donde el sujeto tiene fibrilación auricular no valvular; además, opcionalmente, en donde dicha enfermedad o dicho trastorno que implica un trombo de coágulo sanguíneo o la posible formación de un trombo de coágulo sanguíneo implica además hipertensión pulmonar provocada por uno o más trastornos de las cavidades izquierdas del corazón y/o enfermedad tromboembólica crónica o asociada con una o más enfermedades pulmonares, incluida la fibrosis pulmonar (idiopática o no) y/o la hipoxia.18. El compuesto para el uso según la reivindicación 13, en donde dicha enfermedad o dicho trastorno comprende fibrosis, enfermedad de Alzheimer, esclerosis múltiple, dolor, cáncer, inflamación y/o diabetes mellitus de tipo I.19. El compuesto para el uso según la reivindicación 14, en donde el tratamiento o la prevención comprende una terapia adyuvante; opcionalmente, en donde el sujeto tiene un infarto de miocardio y la terapia adyuvante se realiza junto con terapia trombolítica, o en donde el sujeto tiene angina de pecho inestable, trombosis y/o trombocitopenia inducida por heparina, y la terapia adyuvante se realiza junto con tratamiento antiplaquetario, o en donde el sujeto tiene fibrilación auricular no valvular, y la terapia adyuvante se realiza junto con una o más terapias diferentes.20. El compuesto para el uso según la reivindicación 13, en donde dicha enfermedad o dicho trastorno es un trastorno relacionado con la calicreína; opcionalmente, en donde dicho trastorno relacionado con la calicreína es una enfermedad trombótica, una enfermedad fibrinolítica, un trastorno fibrótico, un tipo de cáncer, una afección inflamatoria, una afección dermatológica o una enfermedad oftálmica.21. El compuesto para el uso según la reivindicación 20, en donde:la enfermedad o el trastorno comprende una enfermedad oftálmica; opcionalmente, en donde dicha enfermedad oftálmica comprende edema macular diabético, degeneración macular relacionada con la edad o retinopatía diabética; ola enfermedad o el trastorno comprende un tipo de cáncer; opcionalmente, en donde dicho tipo de cáncer se selecciona del grupo que consiste en adenocarcinoma de pulmón no microcítico, testicular o cervicouterino, cáncer de pulmón microcítico limitado, glioma, cáncer de mama maligno, micrometástasis (p. ej., micrometástasis de sangre o hígado), metástasis de pulmón y cáncer de próstata; ola enfermedad o el trastorno comprende un estado inflamatorio; opcionalmente, en donde dicha afección inflamatoria es sepsis, enfermedad intestinal inflamatoria, artritis inflamatoria, síndrome de respuesta inflamatoria sistémica, angioedema hereditario o artritis reumatoide; o la enfermedad o el trastorno comprende una afección dermatológica; opcionalmente, en donde dicha afección dermatológica es dermatitis atópica, psoriasis o síndrome de Netherton.22. El compuesto para el uso según la reivindicación 21, la enfermedad o el trastorno comprende una enfermedad oftálmica, y en donde dicho compuesto o dicha composición farmacéutica se administra en forma de una composición oftálmica aplicada por vía tópica en el ojo, o en donde dicho compuesto o dicha composición farmacéutica se administra en forma de una composición oftálmica mediante inyección intravítrea; además, opcionalmente, en donde la composición oftálmica está en forma de colirio.23. El compuesto para el uso según la reivindicación 13, en donde dicho compuesto actúa mediante la inhibición de la trombina y/o la calicreína; opcionalmente, en donde dicho compuesto actúa mediante la inhibición de la calicreína tisular y/o la calicreína plasmática.24. El compuesto para el uso según la reivindicación 13, en donde la cantidad de compuesto administrado es una dosis terapéuticamente eficaz suficiente para alcanzar una concentración inicial del compuesto o su(s) metabolito(s) activo(s) en plasma dentro de un intervalo de 1-10 nM, 10-100 nM, 0,1-1 mM, 1-10 mM, 10 100 mM, 100-200 mM, 200-500 mM o 500-1000 mM, o mayor, o en donde el compuesto tiene actividad inhibidora contra la trombina y/o la calicreína plasmática dentro de un intervalo de 1-10 nM, 10-100 nM, 0,1 1 mM, 1-10 mM, 10-100 mM, 100-200 mM, 200-500 mM o 500-1000 mM, o mayor; opcionalmente, en donde más del 50 % de la concentración inicial del compuesto permanece en el plasma durante 1 hora, 3 horas o más, después de la inyección intravenosa.
o una sal, un éster o un solvato farmacéuticamente aceptable del mismo, en donde:L1 es alquilenamino;L2 y L4 son, independientemente entre sí, un enlace, alquileno sustituido o no sustituido, heteroalquileno sustituido o no sustituido, -C(O)-, -S-, -SO-, -SO2-, -O-, -NHSO2-, -NHC(O)- o -NR5; R1 es alquilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, heteroarilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido;R2 es hidrógeno, halógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, o heteroarilo sustituido o no sustituido;R4 es, independientemente entre sí, hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, o heteroarilo sustituido o no sustituido;R5 es hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, o heteroarilo sustituido o no sustituido;V es hidrógeno o alquilo sustituido o no sustituido;W está ausente, o es hidrógeno, halógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, heteroarilo sustituido o no sustituido, -C(O)R6, - C(O)NR6R7, -SR6, -SOR6, -SO2 R6, -SO2NR6R7, -OR6, -NHSO2 R6 o -NR6R7, en donde R6 y R7 son, independientemente entre sí, hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido, en donde R6 y R7 se pueden combinar, si ambos están presentes, para formar un alquileno sustituido o no sustituido, o heteroalquileno sustituido o no sustituido;X es un enlace, alquileno sustituido o no sustituido o -NR8-;Y es un enlace, alquileno sustituido o no sustituido o -N-; yZ es un enlace, -C(O)-, alquileno sustituido o no sustituido o -NR9-;en donde R8 y R9 son, independientemente entre sí, hidrógeno, halógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquenilo sustituido o no sustituido, heterocicloalquenilo sustituido o no sustituido, arilo sustituido o no sustituido, heteroarilo sustituido o no sustituido, -C(O)R6, -C(O)NR6R7, -SR6, -SOR6, -SO2 R6, -SO2NR6R7, -OR6, -NHSO2 R6 o - NR6R7, en donde R6 y R7 son como se han definido anteriormente; ysiempre que al menos uno de X sea -NR8-, Y es -N- o Z es -NR9-.2. El compuesto de la reivindicación 1, en donde X es un enlace, o alquileno sustituido o no sustituido; opcionalmente, en donde Z es un enlace, o alquileno sustituido o no sustituido.3. El compuesto de la reivindicación 2, en donde Y es -N- y W es hidrógeno, alquilo sustituido o no sustituido, heteroajquilo sustituido o no sustituido, arilo sustituido o no sustituido,-C(O)R6, -C(O)NR6R7, -SO2R6 o SO2 NR6R7, en donde R6 y R7 son independientemente entre sí, alquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido, o R6 y R7 se pueden combinar, si ambos están presentes, para formar un alquileno sustituido o no sustituido, o heteroalquileno sustituido o no sustituido.4. El compuesto de la reivindicación 3, en donde:X se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido, y Z es un enlace o -C(O)-; oX es un enlace y Z se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido; opcionalmente, en donde, opcionalmente, en donde Z se selecciona del grupo que consiste en metileno sustituido, etileno sustituido, propileno sustituido, butileno sustituido y pentileno sustituido, que tiene uno o más grupos sustituyentes seleccionados del grupo que consiste en -OH, -NH2 , -SH, -CN, -CF3 , -NO2 , oxo, halógeno, -COOH, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido; oX se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido, y Z se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido; opcionalmente, en donde X y Z son ambos alquileno ramificado, y X y Z están unidos covalentemente; opcionalmente, en donde Z se selecciona del grupo que consiste en metileno sustituido, etileno sustituido, propileno sustituido, butileno sustituido y pentileno sustituido, que tiene uno o más grupos sustituyentes seleccionados del grupo que consiste en -OH, -NH2 , -SH, -CN, -CF3 , -NO2 , oxo, halógeno, -COOH, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido.5. El compuesto de la reivindicación 3 o reivindicación 4, en donde W es hidrógeno, o en donde W se selecciona del grupo que consiste en alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido, -C(O)R6, -C(O)NR6R7, -SR6, -SOR6, -SO2 R6 y -SO2NR6 ; en donde, opcionalmente, W es alquilo sustituido, heteroalquilo sustituido, alquenilo sustituido, heteroalquenilo sustituido, cicloalquilo sustituido o heterocicloalquilo sustituido, que tiene uno o más grupos sustituyentes seleccionados del grupo que consiste en -OH, -NH2 , -SH, -CN, -CF3 , -NO2, oxo, halógeno, -COOH, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, heterocicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido o heteroarilo sustituido o no sustituido; además, opcionalmente, en donde W es -COR6, -C(O)NR6R7, -SO2 R6 o -SO2 NR6R7, y en donde R6 y R7 se seleccionan del grupo que consiste en alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, arilo sustituido o no sustituido y heteroarilo sustituido o no sustituido, o R6 y R7 se combinan para formar un alquileno sustituido o no sustituido.6. El compuesto de la reivindicación 1, en donde W está ausente, X es -NR8-, Y es un enlace, o alquileno sustituido o no sustituido, y Z es -NR9-; opcionalmente, en donde Y se selecciona del grupo que consiste en metileno sustituido o no sustituido, etileno sustituido o no sustituido, propileno sustituido o no sustituido, butileno sustituido o no sustituido y pentileno sustituido o no sustituido; además, opcionalmente, en donde R8 se selecciona del grupo que consiste en alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, -COR6, -C(O)NR6R7, -Sr6, -SOR6, -SO2 R6 y -SO2NR6R7, y en donde R9 se selecciona del grupo que consiste en alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, heteroalquenilo sustituido o no sustituido, -COR6 -C(O)NR6R7 SR6, -SOR6 -SO2 R6 y -SO2NR6R7.7. El compuesto de cualquiera de las reivindicaciones 1-6, en donde V es hidrógeno, o metilo sustituido o no sustituido.El compuesto según cualquiera de las reivindicaciones 1-7, en donde L1 es -NR5-, o heteroalquileno sustituido o no sustituido, y R1 es alquilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido; además, opcionalmente, en donde R1 es heteroarilo sustituido o no sustituido.El compuesto según cualquiera de las reivindicaciones 1 -8, en donde L2 es enlace, alquileno sustituido o no sustituido, -C(O)- o -SO2-, y R2 es hidrógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, heteroarilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido; opcionalmente, en donde L2 es enlace, y R2 es hidrógeno, o en donde L2 es -C(O)-, y R2 es alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido, arilo de anillo condensado sustituido o no sustituido, heteroarilo sustituido o no sustituido, o heterocicloalquilo sustituido o no sustituido.El compuesto según cualquiera de las reivindicaciones 1-9, en donde L4 y R4 se seleccionan de manera que a) L4 es un enlace, y R4 es hidrógeno, halógeno, alquilo sustituido o no sustituido, heteroalquilo sustituido o no sustituido, arilo sustituido o no sustituido, o heteroarilo sustituido o no sustituido;b) L4 y R4 están ausentes; oc) R4 es alquilo no sustituido.El compuesto según cualquiera de las reivindicaciones 1 a 10, seleccionándose el compuesto de:1-(3-{8-azabiciclo[3.2.1]octan-3-il}-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(1 -metanosulfonilazepan-4-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(1 -metanosulfonilazetidin-3-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(1 -metanosulfonilpiperidin-4-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(1 -metanosulfonilpiperidin-4-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(1 -metanosulfonilpirrolidin-2-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amonio}-3-(1 -metanosulfonilpirrolidin-3-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-(1-{[6-(trifluorometil)piridin-2-il]metil}piperidin-4-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-(2-metilpirrolidin-2-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona; ácido fórmico;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-(3-metilpiperidin-3-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona; ácido fórmico;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-(3-metilpiperidin-3-il)-1 H-pirazol-1-il)-3-metoxi-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-(4-metanosulfonilpiperazin-2-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(4-metilpiperidin-4-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; ácido fórmico;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-(4-metilpiperidin-4-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-(4-metilpiperidin-4-il)-1 H-pirazol-1-il)-3-metoxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(piperazin-2-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; 1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(piperidin-3-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; ácido fórmico;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(piperidin-3-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona; ácido fórmico;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(piperidin-3-il)-1 H-pirazol-1 -il)-3-metoxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(piperidin-4-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; 1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(piperidin-4-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(piperidin-4-il)-1 H-pirazol-1 -il)-3-metoxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(pirrolidin-2-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; 1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(pirrolidin-2-il)-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(pirrolidin-2-il)-1 H-pirazol-1 -il)-3-metoxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-(pirrolidin-3-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; 1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(1,2,4-oxadiazol-3-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2.2- dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(1,3-oxazol-4-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(1,3-oxazol-5-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(1H-1,2,3,4-tetrazol-5-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(1 H-1,2,3-triazol-5-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2.2- dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(1 H-imidazol-4-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(2,2,2-trifluoroetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1-(2,2,2-trifluoroetil)piperidin-4-il]-1-il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(2,2-dimetilpropanoil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(2-ciclopropoxietil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(2-ciclopropoxietil)piperidin-4-il]-1 H-pirazol-1 -il)-3-hidroxi-2.2- dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(2-metoxietil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(2-metoxietil)piperidin-4-il]-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(ciclopropilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1-(ciclopropilmetil)piperidin-4-il]-1-il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(morfolin-4-carbonil)azepan-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(morfolin-4-carbonil)azetidin-3-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino-3-[1 -(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(morfolin-4-carbonil)pirrolidin-2-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(morfolin-4-carbonil)pirrolidin-3-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(morfolin-4-sulfonil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(morfolin-4-sulfonil)piperidin-4-il]-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(oxetan-3-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(oxetan-3-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-3-hidroxi-2.2- dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(piridin-2-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1-(piridin-2-ilmetil)piperidin-4-il]-1-il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1 -(piridin-3-ilmetil)piperidin-4-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[4-(morfolin-4-carbonil)piperazin-2-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[8-(morfolin-4-carbonil)-8-azabiciclo[3.2.1]octan-3-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[(1 -metilazetidin-3-il)metil]piperidin-4-il}-1 H-pirazol-1 -il)-2.2- dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[(2-metoxifenil)metil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[(3-metoxifenil)metil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[(4-metoxifenil)metil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[(6-metilpiridin-2-il)metil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; ácido fórmico;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2,2,2-trifluoro-1 -(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2,2,2-trifluoro-1 -(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2,2-difluoro-2-(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2,2-difluoro-2-(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)-2-oxoetil]piperidin-4-il}-1 H-pirazol-1 -il)-2.2- dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)-2-oxoetil]piperidin-4-il}-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)acetil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)acetil]piperidin-4-il}-1 H-pirazol-1 -il)-2-(morfolin-4-il)etan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)acetil]piperidin-4-il}-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)acetil]pirrolidin-2-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)acetil]pirrolidin-3-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)etil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; ácido fórmico;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(morfolin-4-il)etil]piperidin-4-il}-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona; ácido fórmico;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[2-(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-1 -il)-3-hidroxi-2.2- dimetilpropan-1 -ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-{8-metanosulfonil-8-azabiciclo[3.2.1]octan-3-il}-1 H-pirazol-1-il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-4-metil-3-(piperidin-4-il)-1 H-pirazol-1-il)-2,2-dimetilpropan-1-ona; ácido fórmico;1 -[2-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2-metoxibenzoil)-1 H-pirazol-3-il)pirrolidin-1 -il]-2,2-dimetilpropan-1-ona;1 -[2-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2-metoxibenzoil)-1 H-pirazol-3-il)pirrolidin-1 -il]-2-metilpropan-1-ona;1 -[2-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2-metoxibenzoil)-1 H-pirazol-3-il)pirrolidin-1 -il]etan-1 -ona; 1 -[2-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il)pirrolidin-1 -il]-2-(morfolin-4-il)etan-1 -ona; 1 -[3-(1 -acetilpiperidin-4-il)-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-1 -il]-2,2-dimetilpropan-1 -ona;1 -[3-(1 -bencilpiperidin-4-il)-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-1-il]-2,2-dimetilpropan-1 -ona;1 -[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2,2-dimetilpropanoil)-1 H-pirazol-3-il)azetidin-1 -il]-2,2-dimetilpropan-1-ona;1 -[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il)azetidin-1 -il]-2,2-dimetilpropan-1 -ona;1 -[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il)pirrolidin-1 -il]-2-(morfolin-4-il)etan-1 -ona; 1 -[4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2-metoxibenzoil)-1 H-pirazol-3-il)piperidin-1 -il]-2,2-dimetilpropan-1-ona;1 -[4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2-metoxibenzoil)-1 H-pirazo1 -3-il)piperidin-1 -il]etan-1 -ona;1 -[4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(tiofen-3-carbonil)-1 H-pirazol-3-il)piperidin-1 -il]-2,2-dimetilpropan-1-ona;1 -[4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(tiofen-3-carbonil)-1 H-pirazol-3-il)piperidin-1 -il]etan-1 -ona; 1 -[4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il)piperidin-1 -il]-2-(morfolin-4-il)etan-1 -ona; clorhidrato de 1 -benzoil-N-[(5-clorotiofen-2-il)metil]-3-(piperidin-4-il)-1 H-pirazol-5-amina;1 -{3-[1 -(bencenosulfonil)piperidin-4-il]-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-1 -il}-2,2-dimetilpropan-1-ona;1 -{3-[1 -(bencenosulfonil)piperidin-4-il]-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-1 -il}-3-hidroxi-2,2-dimetilpropan-1 -ona;ácido 2-[4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2,2-dimetilpropanoil)-1 H-pirazol-3-il)piperidin-1 -il]acético;ácido 2-[4-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)piperidin-1-il]acético;2- [4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il)piperidin-1 -il]-1 -(morfolin-4-il)etan-1 -ona; ácido 2-[4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il)piperidin -1 -il]acético;3- (1-bencilpiperidin-4-il)-N-[(5-clorotiofen-2-il)metil]-1 H-pirazol-5-amina;3-(1-bencilpirrolidin-3-il)-N-[(5-clorotiofen-2-il)metil]-1-(furan-3-carbonil)-1 H-pirazol-5-amina;3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(2,2-dimetilpropanoil)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona; 3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(2,2-dimetilpropanoil)-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona; 3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(2-metoxibenzoil)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona; 3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(2-metoxibenzoil)-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona; 3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona;3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona;3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(4-metiloxano-4-carbonil)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona;3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(4-metiloxano-4-carbonil)-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona;3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(tiofen-3-carbonil)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona; 3-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(tiofen-3-carbonil)-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona; 3-(azepan-4-il)-N-[(5-clorotiofen-2-il)metil]-1 H-pirazol-5-amina;3-(azetidin-3-il)-N-[(5-clorotiofen-2-il)metil]-1-(2,3-dihidro-1,4-benzodioxin-5-carbonil)-1 H-pirazol-5-amina; ácido trifluoroacético;clorhidrato de 3-(azetidin-3-il)-N-[(5-clorotiofen-2-il)metil]-1 -(2-metoxibenzoil)-1 H-pirazol-5-amina; 3-(azetidin-3-il)-N-[(5-clorotiofen-2-il)metil]-1-(furan-3-carbonil)-1 H-pirazol-5-amina; ácido trifluoroacético;3-(azetidin-3-il)-N-[(5-clorotiofen-2-il)metil]-1-(tiofen-3-carbonil)-1 H-pirazol-5-amina; ácido trifluoroacético;3-[1-(bencenosulfonil)piperidin-4-il]-N-[(5-clorotiofen-2-il)metil]-1 H-pirazol-5-amina;3- {8-azabiciclo[3.2.1]octan-3-il}-N-[(5-clorotiofen-2-il)metil]-1 H-pirazol-5-amina;4- (5-{[(5-clorotiofen-2-il)metil]amino}-1-(2,2-dimetilpropanoil)-1 H-pirazol-3-il)-N,N-dimetilpiperidin-1-carboxamida;4-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(2,2-dimetilpropanoil)-1 H-pirazol-3-il)-N,N-dimetilpiperidin-1-sulfonamida;4-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-N,N-dimetilpiperidin-1 -carboxamida;4-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-N,N-dimetilpiperidin-1 -sulfonamida;4-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(dimetilcarbamoil)-1 H-pirazol-3-il)-N,N-dimetilpiperidin-1-carboxamida;4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il)-N,N-dimetilpiperidin-1-carboxamida;4-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il)-N,N-dimetilpiperidin-1-sulfonamida;4-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[1-(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-1-carbonil)-4- metilciclohexan-1 -ol;5- {[(5-clorotiofen-2-il)metil]amino}-1-(2,2-dimetilpropanoil)-3-[1-(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil]amino}-3-[1-(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-4-carbonitrilo; N-[(5-clorotiofen-2-il)metil]-1-(1,4-dimetilpiperidin-4-carbonil)-3-[1-(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-5-amina; ácido fórmico;clorhidrato de N-[(5-clorotiofen-2-il)metil]-1 -(2,3-dihidro-1,4-benzodioxin-5-carbonil)-3-(piperidin-4-il)-1 H-pirazol-5-amina;clorhidrato de N-[(5-clorotiofen-2-il)metil]-1 -(2,3-dihidro-1,4-benzodioxin-5-carbonil)-3-(pirrolidin-2-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(2,4-dimetoxibenzoil)-3-(piperidin-4-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(2,4-dimetoxibenzoil)-3-(pirrolidin-2-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(2,6-dimetilciclohexanocarbonil)-3-(piperidin-4-il)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-1-(2-metoxibenzoil)-3-(2-metilpirrolidin-2-il)-1 H-pirazol-5-amina; ácido fórmico;N-[(5-clorotiofen-2-il)metil]-1-(2-metoxibenzoil)-3-(3-metilpiperidin-3-il)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-1-(2-metoxibenzoil)-3-(4-metilpiperidin-4-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(2-metoxibenzoil)-3-(piperidin-3-il)-1 H-pirazol-5-amina; clorhidrato de N-[(5-clorotiofen-2-il)metil]-1 -(2-metoxibenzoil)-3-(piperidin-4-il)-1 H-pirazol-5-amina; clorhidrato de N-[(5-clorotiofen-2-il)metil]-1 -(2-metoxibenzoil)-3-(pirrolidin-2-il)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-1-(2-metoxibenzoil)-3-[1-(2,2,2-trifluoroetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(4-metiloxano-4-carbonil)-3-(2-metilpirrolidin-2-il)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-1-(4-metiloxano-4-carbonil)-3-(3-metilpiperidin-3-il)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-1-(4-metiloxano-4-carbonil)-3-(4-metilpiperidin-4-il)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-1-(4-metiloxano-4-carbonil)-3-(piperidin-3-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(4-metiloxano-4-carbonil)-3-(pirrolidin-2-il)-1 H-pirazol-5-amina; clorhidrato de N-[(5-clorotiofen-2-il)metil]-1 -(furan-3-carbonil)-3-(piperidin-4-il)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-1-(furan-3-carbonil)-3-(pirrolidin-2-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(morfolin-4-carbonil)-3-[1-(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-5-amina;clorhidrato de N-[(5-clorotiofen-2-il)metil]-1 -ciclohexanocarbonil-3-(piperidin-4-il)-1 H-pirazol-5-amina;clorhidrato de N-[(5-clorotiofen-2-il)metil]-1-ciclopentanocarbonil-3-(piperidin-4-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1-metanosulfonilazepan-4-il)-1H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1-metanosulfonilazetidin-3-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1-metanosulfonilpiperidin-4-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1-metanosulfonilpirrolidin-2-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1-metanosulfonilpirrolidin-3-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1-{[6-(trifluorometil)piridin-2-il]metil}piperidin-4-il)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-3-(2-metilpirrolidin-2-il)-1-(tiofen-3-carbonil)-1 H-pirazol-5-amina; ácido fórmico;N-[(5-clorotiofen-2-il)metil]-3-(3-metilpiperidin-3-il)-1-(tiofen-3-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(4-metanosulfonilpiperazin-2-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(4-metilpiperidin-4-il)-1-(tiofen-3-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(piperidin-3-il)-1-(tiofen-3-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(piperidin-3-il)-1 H-pirazol-5-amina;clorhidrato de N-[(5-clorotiofen-2-il)metil]-3-(piperidin-4-il)-1 -(tiofen-3-carbonil)-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-3-(piperidin-4-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(pirrolidin-2-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(pirrolidin-3-il)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(1,2,4-oxadiazol-3-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(1,2,4-oxadiazol-5-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(1,3-oxazol-2-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(1,3-oxazol-5-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(1 H-1,2,3,4-tetrazol-5-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-3-[1-(1 H-1,2,3-triazol-5-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(1 H-imidazol-4-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(2,2,2-trifluoroetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(2-ciclopropoxietil)piperidin-4-il]-1 H-pirazol-5-amina; ácido fórmico; N-[(5-clorotiofen-2-il)metil]-3-[1-(2-metoxietil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(ciclopropilmetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-carbonil)azepan-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-carbonil)pirrolidin-2-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-carbonil)pirrolidin-3-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-carbonil)pirrolidin-3-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-sulfonil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(oxan-4-il)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(oxetan-3-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina; ácido fórmico; N-[(5-clorotiofen-2-il)metil]-3-[1-(piridin-2-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(piridin-3-ilmetil)piperidin-4-il]-1 H-pirazol-5-amina;clorhidrato de N-[(5-clorotiofen-2-il)metil]-3-[4-(morfolin-4-carbonil)piperazin-2-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[8-(morfolin-4-carbonil)-8-azabiciclo[3.2.1]octan-3-il]-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[8-(morfolin-4-carbonil)-8-azabiciclo[3.2.1]octan-3-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-{1-[(1-metilazetidin-3-il)metil]piperidin-4-il}-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-{1-[(2-metoxifenil)metil]piperidin-4-il}-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-3-{1-[(3-metoxifenil)metil]piperidin-4-il}-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-{1-[(4-metoxifenil)metil]piperidin-4-il}-1-(tiofen-3-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-{1-[(4-metoxifenil)metil]piperidin-4-il}-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-{1 -[(6-metilpiridin-2-il)metil]piperidin-4-il}-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-{1 -[2,2,2-trifluoro-1 -(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-3-{1-[2,2-difluoro-2-(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-3-{1-[2-(morfolin-4-il)etil]piperidin-4-il}-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-{1-[2-(piridin-2-il)etil]piperidin-4-il}-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-{8-metanosulfonil-8-azabiciclo[3.2.1]octan-3-il}-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-18-metanosulfonil-8-azabiciclo[3.2.1]octan-3-il}-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-4-metil-3-[1-(1,3-tiazol-4-carbonil)piperidin-4-il]-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-4-metil-3-[1-(morfolin-4-carbonil)piperidin-4-il]-1 H-pirazol-5-amina; N-[(5-clorotiofen-2-il)metil]-N-{3-[1-(morfolin-4-sulfonil)piperidin-4-il]-1 H-pirazol-5-il}morfolin-4-sulfonamida;N-[(5-clorotiofen-2-il)metil]-3-(1 -metanosulfonilazepan-4-il)-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1 -metanosulfonilazetidin-3-il)-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1 -metanosulfonilpirrolidin-2-il)-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-(1 -metanosulfonilpirrolidin-3-il)-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-1 -(morfolin-4-carbonil)azepan-4-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-carbonil)azetidin-3-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-carbonil)pirrolidin-2-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;1-(2,2-dimetilpropanoil)-4-(5-{[(4-fluorofenil)metil](metil)amino}-4-metoxi-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-3-(trifluorometil)piperidin-2-ona;1-(2-clorobenzoil)-5-[(5-clorotiofen-2-il)metoxi]-3-[1-metanosulfonil-4-(trifluorometil)pirrolidin-3-il]-1 H-pirazol;1-(2-clorobenzoil)-5-{[(4-fluorofenil)metil](metil)amino}-3-{1-[(3-hidroxipirrolidin-1-il)sulfonil]-6-oxo-4-(trifluorometil)piperidin-3-il}-1 H-pirazol-4-carbonitrilo;1-(2-clorobenzoil)-5-{[(4-fluorofenil)metil]sulfanil}-3-{4-[2-(morfolin-4-il)-2-oxoetil]-3-(trifluorometil)piperazin-2-il}-1 H-pirazol-4-carbonitrilo;1-(2-clorobenzoil)-N-[(4-fluorofenil)metil]-N-metil-3-[1-(morfolin-4-carbonil)-3-(trifluorometil)piperazin-2-il]-1 H-pirazol-5-amina;1-(2-fluorobenzoil)-N-[(4-fluorofenil)metil]-3-[3-(trifluorometil)azetidin-2-il]-1 H-pirazol-5-amina; 1 -(2-{5-[(5-clorotiofen-2-il)metoxi]-1 -(furan-2-carbonil)-4-metoxi-1 H-pirazol-3-il}-3-metilpiperazin-1 -il)-2,2-dimetilpropan-1 -ona;1 -(3-15-[(4-fluorofenil)metoxi]-4-metil-1 -(4-metilfuran-3-carbonil)-1 H-pirazol-3-il}-2-(trifluorometil)azetidin-1 -il)-2-(morfolin-4-il)etan-1 -ona;1 -(3-{5-[(5-clorotiofen-2-il)metoxi]-1 -(2-metoxibenzoil)-4-metil-1 H-pirazol-3-il}-2-(trifluorometil)piperazin-1 -il)-2-(morfolin-4-il)etan-1 -ona;1 -(3-{5-[(5-clorotiofen-2-il)metoxi]-4-metoxi-1 H-pirazol-3-il}piperazin-1 -il)-2,2-dimetilpropan-1 -ona; 1-(5-{[(4-fluorofenil)metil]amino}-3-[4-(morfolin-4-carbonil)piperazin-2-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -(5-{[(4-fluorofenil)metil]sulfanil}-3-(1 -metanosulfonil-4-metilpiperidin-3-il)-1 H-pirazol-1 -il)-3-metoxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(4-fluorofenil)metil]sulfanil}-3-[5-hidroxi-2-metil-1 -(pirrolidin-1 -sulfonil)pirrolidin-3-il]-4-metil-1 H-pirazol-1-il)-3-metoxi-2,2-dimetilpropan-1 -ona;1-(5-{[(4-fluorofenil)metil]sulfanil}-4-metoxi-3-{4-metil-1-[2-(morfolin-4-il)-2-oxoetil]piperidin-3-il}-1 H-pirazol-1 -il)-3-metoxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil](metil)amino}-3-[5-hidroxi-1 -(pirrolidin-1 -carbonil)pirrolidin-3-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona;1-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-3-{5-hidroxi-2-metil-1-[2-(morfolin-4-il)-2-oxoetil]pirrolidin-3-il}-4-metoxi-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona;1-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-metoxi-3-(morfolin-3-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[(3-hidroxipirrolidin-1-il)sulfonil]-4-metilpirrolidin-3-il}-4-metil-1 H-pirazol-1 -il)-3-hidroxi-2,2-dimetilpropan-1 -ona;1 -(5-{[(5-clorotiofen-2-il)metil]sulfanil}-3-[1 -(3-hidroxipirrolidin-1 -carbonil)-2-metilpiperidin-3-il]-1 H-pirazol-1 -il)-3-metoxi-2,2-dimetilpropan-1 -ona;1 -(5-{[4-(aminometil)fenil]metoxi}-3-{3-metil-1 -[2-(morfolin-4-il)-2-oxoetil]piperidin-4-il}-1 H-pirazol-1 -il)-3-metoxi-2,2-dimetilpropan-1-ona;ácido 1 -(dimetilcarbamoil)-3-(5-{[(4-fluorofenil)metil]amino}-1 -(tiofen-3-carbonil)-1 H-pirazol-3-il)-4-metilpiperidin-2-carboxílico;ácido 1 -(dimetilsulfamoil)-3-(5-{[(4-fluorofenil)metil](metil)amino}-1 -(furan-3-carbonil)-1 H-pirazol-3-il)-4-metilpirrolidin-2-carboxílico;1-({3-[5-({[4-(aminometil)fenil]metil}amino)-1-(2-fluorobenzoil)-4-metoxi-1 H-pirazol-3-il]-2-(trifluorometil)piperazin-1-il}sulfonil)pirrolidin-3-ol;1 -[2-(5-{[(4-fluorofenil)metil](metil)amino}-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-3-(trifluorometil)piperazin-1 -il]-2-(morfolin-4-il)etan-1 -ona;1 -[2-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(4-metilfuran-3-carbonil)-1 H-pirazol-3-il)-3-(trifluorometil)azetidin-1 -il]-2,2-dimetilpropan-1 -ona;1 -[2-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(tiofen-3-carbonil)-1 H-pirazol-3-il)-3-metilpiperazin-1 -il]-2.2- dimetilpropan-1 -ona;1 -[3-(1 -benzoil-5-{[(4-fluorofenil)metil]amino}-4-metoxi-1 H-pirazol-3-il)-2-metilpiperidin-1 -carbonil]pirrolidin-3-ol;1 -[3-(5-{[(4-fluorofenil)metil](metil)amino}-1 -(furan-2-carbonil)-1 H-pirazol-3-il)-4-metilpirrolidin-1 -carbonil]pirrolidin-3-ol;1-[3-(5-{[(4-fluorofenil)metil](metil)amino}-1-(furan-2-carbonil)-4-metil-1 H-pirazol-3-il)-4-metilpiperidin-1 -il]-2-(morfolin-4-il)etan-1 -ona;1 -[3-(5-{[(4-fluorofenil)metil]amino}-1 -(4-metilfuran-3-carbonil)-1 H-pirazol-3-il)-4-(trifluorometil)piperidin-1-carbonil]pirrolidin-3-ol;1-[3-(5-{[(4-fluorofenil)metil]sulfanil}-1-(furan-3-carbonil)-4-metoxi-1 H-pirazol-3-il)-2-metilpiperazin-1 -carbonil]pirrolidin-3-ol;1-[3-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-1-(4-metilfuran-3-carbonil)-1 H-pirazol-3-il)-2-(trifluorometil)piperidin-1 -il]-2,2-dimetilpropan-1 -ona;1 -[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(5-metilfuran-3-carbonil)-1 H-pirazol-3-il)piperazin-1 -il]-2.2- dimetilpropan-1 -ona;1-[3-(5-{[(5-clorotiofen-2-il)metil]amino}-4-metil-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il)pirrolidin-1 -il]-2,2-dimetilpropan-1 -ona;1-[3-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-metoxi-1-(2-metoxibenzoil)-1 H-pirazol-3-il)-2-(trifluorometil)pirrolidin-1 -il]-2-(morfolin-4-il)etan-1 -ona;1 -[3-(5-{[4-(aminometil)fenil]metoxi}-4-metoxi-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-4-(trifluorometil)piperidin-1-carbonil]pirrolidin-3-ol;1 -[4-(1 -benzoil-5-{[(4-fluorofenil)metil]amino}-4-metil-1 H-pirazol-3-il)-3-metilpiperidin-1 -carbonil]pirrolidin-3-ol;1 -[5-({[4-(aminometil)fenil]metil}(metil)amino)-3-(1 -metanosulfonilazetidin-2-il)-1 H-pirazol-1 -il]-2,2-dimetilpropan-1-ona;1-[5-({[4-(aminometil)fenil]metil}(metil)amino)-4-metoxi-3-{4-metil-1-[2-(morfolin-4-il)acetil]pirrolidin-3-il}-1 H-pirazol-1 -il]-3-hidroxi-2,2-dimetilpropan-1-ona;1 -[5-({[4-(aminometil)fenil]metil}amino)-3-[1 -(pirrolidin-1 -sulfonil)piperidin-4-il]-1 H-pirazol-1 -il]-2,2-dimetilpropan-1-ona;1-[5-({[4-(aminometil)fenil]metil}amino)-4-fluoro-3-[5-hidroxi-1-(3-hidroxipirrolidin-1-carbonil)-2-metilpirrolidin-3-il]-1 H-pirazol-1 -il]-3-hidroxi-2,2-dimetilpropan-1 -ona;1-benzoil-5-{[(4-fluorofenil)metil]amino}-3-(4-metil-2-oxopirrolidin-3-il)-1 H-pirazol-4-carbonitrilo; 1-benzoil-5-{[(5-clorotiofen-2-il)metil](metil)amino}-3-[1-(2,2-dimetilpropanoil)-2-metilpirrolidin-3-il]-1 H-pirazol-4-carbonitrilo;1 -{2-[1 -(2-fluorobenzoil)-5-{[(4-fluorofenil)metil]amino}-4-metoxi-1 H-pirazol-3-il]-3-(trifluorometil)piperazin-1-carbonil}pirrolidin-3-ol;1-{2-[5-({[4-(aminometil)fenil]metil}amino)-4-metil-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il]azetidin-1 -il}-2-(morfolin-4-il)etan-1 -ona;1-{2-[5-({[4-(aminometil)fenil]metil}sulfanil)-1-(furan-3-carbonil)-4-metoxi-1 H-pirazol-3-il]-3-metilazetidin-1 -carbonil}pirrolidin-3-ol;1 -{3-[1 -(2,2-dimetilpropanoil)-5-[(4-fluorofenil)metoxi]-4-metil-1 H-pirazol-3-il]azetidin-1 -il}-2,2-dimetilpropan-1-ona;1-{3-[1-(2-clorobenzoil)-5-[(5-clorotiofen-2-il)metoxi]-4-metoxi-1 H-pirazol-3-il]-5-hidroxi-2-(trifluorometil)pirrolidin-1 -il}-2-(morfolin-4-il)etan-1 -ona;1-{3-[5-({[4-(aminometil)fenil]metil}sulfanil)-1-(2-metoxibenzoil)-4-metil-1 H-pirazol-3-il]-4-(trifluorometil)piperidin-1 -il}-2,2-dimetilpropan-1 -ona;1-{4-[5-({[4-(aminometil)fenil]metil}(metil)amino)-4-metil-1-(2-metilfuran-3-carbonil)-1 H-pirazol-3-il]-3-(trifluorometil)piperidin-1 -il}-2,2-dimetilpropan-1 -ona;1 -{5-[(4-fluorofenil)metoxi]-3-(1-metanosulfonil-2-metilazetidin-3-il)-4-metoxi-1 H-pirazol-1 -il}-3-hidroxi-2,2-dimetilpropan-1-ona;1-{5-[(5-clorotiofen-2-il)metoxi]-3-[1-(2,2-dimetilpropanoil)-2-metilpirrolidin-3-il]-4-metoxi-1 H-pirazol-1 -il}-3-metoxi-2,2-dimetilpropan-1 -ona;1 -{[2-(5-{[(4-fluorofenil)metil]sulfanil}-1 -(2-metoxibenzoil)-4-metil-1 H-pirazol-3-il)-3-(trifluorometil)piperazin-l-il]sulfonil}pirrolidin-3-ol;1 -{[3-(5-{[4-(aminometil)fenil]metoxi}-1 -(tiofen-2-carbonil)-1 H-pirazol-3-il)pirrolidin-1 -il]sulfonil}pirrolidin-3-ol;1 -{[4-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-metoxi-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-3-(trifluorometil)piperidin-1-il]sulfonil}pirrolidin-3-ol;2-(3-{5-[(4-fluorofenil)metoxi]-1-(tiofen-3-carbonil)-1 H-pirazol-3-il}-2-metilazetidin-1 -il)-1 -(morfolin-4-il)etan-1-ona;2-(3-{5-[(5-clorotiofen-2-il)metoxi]-1-(furan-3-carbonil)-1 H-pirazol-3-il}-5-hidroxi-2-metilpirrolidin-1-il)-1 -(morfolin-4-il)etan-1 -ona;2-(5-{[(4-carbamimidoilfenil)metil]sulfanil}-4-metoxi-1-(tiofen-2-carbonil)-1 H-pirazol-3-il)-N,N-dimetil-4-oxoazetidin-1 -carboxamida;2-(5-{[(4-fluorofenil)metil](metil)amino}-4-metil-1-(tiofen-2-carbonil)-1 H-pirazol-3-il)-N,N-dimetilpiperazin-1 -carboxamida;2-(5-{[(4-fluorofenil)metil]sulfanil}-1-(furan-2-carbonil)-4-metil-1 H-pirazol-3-il)-N,N,3-trimetilazetidin-1- carboxamida;2- (5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-ciano-1-(tiofen-3-carbonil)-1 H-pirazol-3-il)-N,N,3-trimetilazetidin-1 -carboxamida;2-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-1-(furan-3-carbonil)-4-metil-1 H-pirazol-3-il)-N,N,3-trimetilazetidin-1 -sulfonamida;2-[3-(1 -benzoil-5-{[(5-clorotiofen-2-il)metil](metil)amino}-1 H-pirazol-3-il)-4-metilpiperidin-1 -il]-1 -(morfolin-4-il)etan-1 -ona;2-[3-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-1 -(tiofen-2-carbonil)-1 H-pirazol-3-il)pirrolidin-1 -il]-1 -(morfolin-4-il)etan-1 -ona;2-[4-ciano-1-(2,2-dimetilpropanoil)-5-[(4-fluorofenil)metoxi]-1 H-pirazol-3-il]-N,N-dimetilpiperazin-1 -sulfonamida;2-[5-({[4-(aminometil)fenil]metil}amino)-1-(2-fluorobenzoil)-1 H-pirazol-3-il]-N,N-dimetil-4-oxo-3-(trifluorometil)azetidin-1-sulfonamida;ácido 2-cloro-3-(5-{[(4-fluorofenil)metil](metil)amino}-3-{1-[(3-hidroxipirrolidin-1-il)sulfonil]-5-oxopirrolidin-3-il}-4-metoxi-1 H-pirazol-1 -carbonil)benzoico;ácido 2-cloro-3-(5-{[(4-fluorofenil)metil]sulfanil}-3-(2-oxoazetidin-3-il)-1 H-pirazol-1 -carbonil)benzoico;ácido 2-cloro-3-(5-{[(4-fluorofenil)metil]sulfanil}-4-metil-3-[1-(morfolin-4-carbonil)-6-oxopiperidin-3-il]-1 H-pirazol-1 -carbonil)benzoico;ácido 2-cloro-3-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-ciano-3-[1-(morfolin-4-carbonil)-2-oxopirrolidin-3-il]-1 H-pirazol-1 -carbonil)benzoico;ácido 2-cloro-3-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-metoxi-3-[4-(pirrolidin-1-carbonil)piperazin-2-il]-1 H-pirazol-1 -carbonil)benzoico;ácido 2-cloro-3-{3-[1-(2,2-dimetilpropanoil)-2-oxoazetidin-3-il]-5-{[(4-fluorofenil)metil](metil)amino}-1 H-pirazol-1-carbonil}benzoico;ácido 2-cloro-3-{3-[1-(2,2-dimetilpropanoil)azetidin-3-il]-4-fluoro-5-{[(4-fluorofenil)metil](metil)amino}-1 H-pirazol-1-carbonil}benzoico;ácido 2-cloro-3-{5-[(5-clorotiofen-2-il)metoxi]-3-(morfolin-3-il)-1 H-pirazol-1-carbonil}benzoico;2- {4-[5-({[4-(aminometil)fenil]metil}(metil)amino)-1-benzoil-4-metoxi-1 H-pirazol-3-il]-3-metilpiperidin-1 -il}-1 -(morfolin-4-il)etan-1 -ona;ácido 3-(1 -benzoil-5-{[(4-carbamimidoilfenil)metil](metil)amino}-4-metoxi-1 H-pirazol-3-il)-4-metil-1 -(morfolin-4-carbonilo)piperidin-2-carboxílico;3- (1-benzoil-5-{[(4-carbamimidoilfenil)metil]amino}-4-fluoro-1 H-pirazol-3-il)-N,N,2-trimetil-4-oxopirrolidin-1 -carboxamida;ácido 3-(1 -benzoil-5-{[(5-clorotiofen-2-il)metil]amino}-4-metil-1 H-pirazol-3-il)-1 -(dimetilcarbamoil)-4-metilpirrolidin-2-carboxílico;ácido 3-(4-ciano-5-{[(4-fluorofenil)metil]amino}-3-{1-[2-(morfolin-4-il)acetil]-5-oxopirrolin-il}-1 H-pirazol-1 -carbonil)benzoico;3-(4-ciano-5-{[(4-fluorofenil)metil]sulfanil}-1-(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-N,N,2-trimetilpirrolidin-1-carboxamida;3-(4-ciano-5-{[(4-fluorofenil)metil]sulfanil}-1-(furan-2-carbonil)-1 H-pirazol-3-il)-N,N,2-trimetilpiperidin-1-sulfonamida;3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-1-(2-fluorobenzoil)-4-metoxi-1 H-pirazol-3-il)-N,N-dimetil-2-(trifluorometil)piperidin-1-carboxamida;3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-5-hidroxi-N,N,2-trimetilpirrolidin-1-carboxamida;ácido 3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-3-[1-(3-hidroxipirrolidin-1-carbonil)-4-oxopirrolidin-3-il]-4-metoxi-1 H-pirazol-1 -carbonil)benzoico;ácido 3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-3-[1-(dimetilsulfamoil)piperidin-4-il]-4-fluoro-1 H-pirazol-1-carbonil)benzoico;ácido 3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-4-ciano-1-(5-metilfuran-3-carbonil)-1H-pirazol-3-il)-1 -(pirrolidin-1 -carbonil)piperidin-2-carboxílico;ácido 3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-4-ciano-3-[1-(pirrolidin-1-carbonil)pirrolidin-3-il]-1 H-pirazol-1 -carbonil)benzoico;ácido 3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-4-metoxi-1-(2-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1 -[2-(morfolin-4-il)acetil]-4-(trifluorometil)piperidin-2-carboxílico;ácido 3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-4-metil-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1 -(pirrolidin-1 -sulfonil)piperidin-2-carboxílico;3-(5-{[(4-carbamimidoilfenil)metil](metil)amino}-4-metil-1-(tiofen-3-carbonil)-1;ácido 3-(5-{[(4-carbamimidoilfenil)metil]amino}-1 -(3-carboxibenzoil)-1 H-pirazol-3-il)-1 -[2-(morfolin-4-il)-2-oxoetil]pirrolidin-2-carboxílico;ácido 3-(5-{[(4-carbamimidoilfenil)metil]amino}-3-[1-(dimetilcarbamoil)piperazin-2-il]-4-fluoro-1 H-pirazol-1 -carbonil)benzoico;ácido 3-(5-{[(4-carbamimidoilfenil)metil]amino}-3-[1-(dimetilsulfamoil)-5-hidroxipirrolidin-3-il]-4-metil-1 H-pirazol-1-carbonil)benzoico;3-(5-{[(4-carbamimidoilfenil)metil]amino}-4-fluoro-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-N,N,2-trimetilpiperazin-1-carboxamida;3- (5-{[(4-carbamimidoilfenil)metil]amino}-4-metil-1-(tiofen-3-carbonil)-1 H-pirazol-3-il)-N,N,2-trimetil-4- oxoazetidin-1 -carboxamida;3-(5-{[(4-carbamimidoilfenil)metil]sulfanil}-4-metoxi-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-N,N-dimetil-2-(trifluorometil)azetidin-1-carboxamida;ácido 3-(5-{[(4-carbamimidoilfenil)metil]sulfanil}-4-metoxi-1 H-pirazol-3-il)-1 -[(3-hidroxipirrolidin-1 -il)sulfonil]piperidin-2-carboxílico;ácido 3-(5-{[(4-carbamimidoilfenil)metil]sulfanil}-4-metil-3-{1-[2-(morfolin-4-il)acetil]-2-oxoazetidin-3-il}-1 H-pirazol-1 -carbonil)-2-clorobenzoico;3-(5-{[(4-fluorofenil)metil](metil)amino}-1-(2-metoxibenzoil)-4-metil-1 H-pirazol-3-il)-1-[(3-hidroxipirrolidin-1 -il)sulfonil]-4-(trifluorometil)piperidin-2-ona;3-(5-{[(4-fluorofenil)metil](metil)amino}-1-(3-metoxi-2,2-dimetilpropanoil)-4-metil-1 H-pirazol-3-il)-N,N,2-trimetil-4-oxopirrolidin-1-carboxamida;3- (5-{[(4-fluorofenil)metil](metil)amino}-4-metoxi-1-(2-metoxibenzoil)-1 H-pirazol-3-il)-1-[2-(morfolin-4- il)-2-oxoetil]-4-(trifluorometil)azetidin-2-ona;3-(5-{[(4-fluorofenil)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-4-metoxi-1 H-pirazol-3-il)-1-metanosulfonil-4-metilpirrolidin-2-ona;3-(5-{[(4-fluorofenil)metil]amino}-4-metoxi-1 -(2-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1 -(pirrolidin-1 -sulfonil)-4-(trifluorometil)pirrolidin-2-ona;ácido 3-(5-{[(4-fluorofenil)metil]amino}-4-metoxi-3-{1-[2-(morfolin-4-il)-2-oxoetil]pirrolidin-3-il}-1 H-pirazol-1 -carbonil)benzoico;3-(5-{[(4-fluorofenil)metil]amino}-4-metil-1-(4-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1-(3-hidroxipirrolidin-1-carbonil)-4-(trifluorometil)azetidin-2-ona;ácido 3-(5-{[(4-fluorofenil)metil]amino}-4-metil-1 -(tiofen-3-carbonil)-1 H-pirazol-3-il)-4-metil-1 -(morfolin-4-carbonil)pirrolidin-2-carboxílico;3-(5-{[(4-fluorofenil)metil]sulfanil}-1 -(tiofen-2-carbonil)-1 H-pirazol-3-il)-1 -(3-hidroxipirrolidin-1 -carbonil)piperidin-2-ona;3-(5-{[(4-fluorofenil)metil]sulfanil}-4-metil-1 H-pirazol-3-il)-1-[2-(morfolin-4-il)-2-oxoetil]pirrolidin-2-ona;ácido 3-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-1-(2,2-dimetilpropanoil)-4-metil-1 H-pirazol-3-il)pirrolidin-2-carboxílico;3-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-1-(tiofen-3-carbonil)-1 H-pirazol-3-il)-4-metil-1 -(pirrolidin-1 -sulfonil)azetidin-2-ona;ácido 3-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-3-[1-(3-hidroxipirrolidin-1-carbonil)-5-oxopirrolidin-3-il]-4-metil-1 H-pirazol-1 -carbonil)benzoico;ácido 3-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-ciano-3-(3-oxopiperidin-4-il)-1 H-pirazol-1 -carbonil)benzoico;3- (5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-metoxi-1-(tiofen-3-carbonil)-1 H-pirazol-3-il)-N,N,2-trimetilpiperidin-1-sulfonamida;ácido 3-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-metil-1-(2-metilfuran-3-carbonil)-1 H-pirazol-3-il)-4- (trifluorometil)piperidin-2-carboxílico;ácido 3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2,2-dimetilpropanoil)-4-metoxi-1 H-pirazol-3-il)-1 -[(3-hidroxipirrolidm-1 -il)sulfonil]pirrolidin-2-carboxílico;ácido 3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1 -metanosulfonil-4-(trifluorometil)piperidin-2-carboxílico;ácido 3-(5-{[(5-clorotiofen-2-il)metil]amino}-3-{1-[(3-hidroxipirrolidin-1-il)sulfonil]azetidin-3-il}-4-metoxi-1 H-pirazol-1 -carbonil)benzoico;ácido 3-(5-{[(5-clorotiofen-2-il)metil]amino}-4-ciano-1 -(2,2-dimetilpropanoil)-1 H-pirazol-3-il)-1 -(2,2)-dimetilpropanoil)pirrolidin-2-carboxílico;3- (5-{[(5-clorotiofen-2-il)metil]amino}-4-ciano-1-(4-metiliuran-3-carbonil)-1 H-pirazol-3-il)-N,N-dimetil-4- (trifluorometil)piperidin-1-sulfonamida;ácido 3-(5-{[(5-clorotiofen-2-il)metil]amino}-4-ciano-3-[1 -(dimetilcarbamoy 1 )-4-oxoazetidin-2-il]-1 H-pirazol-1 -carbonil)benzoico;3-(5-{[(5-clorotiofen-2-il)metil]amino}-4-metoxi-1-(4-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1-(pirrolidin-1-carbonil)-4-(trifluorometil)azetidin-2-ona;ácido 3-(5-{[(5-clorotiofen-2-il)metil]amino}-4-metil-3-[1 -(pirrolidin-1 -sulfonil)pirrolidin-3-il]-1 H-pirazol-1 -carbonil)benzoico;3-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-1 -(2-metoxibenzoil)-1 H-pirazol-3-il)-1 -(pirrolidin-1 -sulfonil)-4-(trifluorometil)pirrolidin-2-ona;3-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-1-(3-metoxi-2,2-dimetilpropanoil)-4-metil-1 H-pirazol-3-il)-1-[(3-hidroxipirrolidin-1-il)sulfonil]-4-metilazetidin-2-ona;3-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-1-(furan-3-carbonil)-4-metoxi-1 H-pirazol-3-il)-5-hidroxi-N,N,2-trimetilpirrolidin-1-carboxamida;ácido 3-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-ciano-1 -(furan-2-carbonil)-1 H-pirazol-3-il)-4-metil-1 -(morfolin-4-carbonil)pirrolidin-2-carboxílico;ácido 3-(5-{[4-(aminometil)fenil]metoxi}-1 -(2-clorobenzoil)-1 H-pirazol-3-il)-1 -(3-hidroxipirrolidin-1 -carbonil)-4-(trifluorometil)piperidin-2-carboxílico;3-(5-{[4-(aminometil)fenil]metoxi}-1-(2-clorobenzoil)-4-metil-1 H-pirazol-3-il)-N,N-dimetil-2-(trifluorometil)piperazin-1-sulfonamida;3-(5-{[4-(aminometil)fenil]metoxi}-1 -(2-metoxibenzoil)-1 H-pirazol-3-il)-1 -(3-hidroxipirrolidin-1 -carbonil)-4-(trifluorometil)pirrolidin-2-ona;ácido 3-(5-{[4-(aminometil)fenil]metoxi}-1 -(3-carboxi-2-clorobenzoil)-4-ciano-1 H-pirazol-3-il)-1 -[2-(morfolin-4-il)acetil]pirrolidin-2-carboxílico;3-(5-{[4-(aminometil)fenil]metoxi}-1-(3-metoxi-2,2-dimetilpropanoil)-4-metil-1 H-pirazol-3-il)-4-metilpirrolidin-2-ona;ácido 3-(5-{[4-(aminometil)fenil]metoxi}-1 -(furan-2-carbonil)-4-metoxi-1 H-pirazol-3-il)-1 -(dimetilcarbamoil)-4-metilpiperidin-2-carboxílico;3-(5-{[4-(aminometil)fenil]metoxi}-4-metoxi-1-(2-metoxibenzoil)-1 H-pirazol-3-il)-N,N-dimetil-4-(trifluorometil)pirrolidin-1-carboxamida;3-(5-{[4-(aminometil)fenil]metoxi}-4-metoxi-1-(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-4-metil-1 -[2-(morfolin-4-il)acetil]pirrolidin-2-ona;3-(5-{[4-(aminometil)fenil]metoxi}-4-metoxi-1 H-pirazol-3-il)-1 -(pirrolidin-1 -sulfonil)piperidin-2-ona; ácido 3-(5-{[4-(aminometil)fenil]metoxi}-4-metoxi-3-[4-(morfolin-4-carbonil)piperazin-2-il]-1 H-pirazol-1 -carbonil)-2-clorobenzoico;3- (5-{[4-(aminometil)fenil]metoxi}-4-metil-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-1 -metanosulfonil-4- (trifluorometil)azetidin-2-ona;ácido 3-(5-{[4-(aminometil)fenil]metoxi}-4-metil-3-{1-[2-(morfolin-4-il)-2-oxoetil]piperazin-2-il}-1 H-pirazol-1-carbonil)-2-clorobenzoico;3-[1-(2,2-dimetilpropanoil)-5-[(4-fluorofenil)metoxi]-4-metoxi-1 H-pirazol-3-il]azetidin-2-ona; ácido 3-[1 -(2,2-dimetilpropanoil)-5-{[(4-fluorofenil)metil]amino}-4-metoxi-1 H-pirazol-3-il]-1 -[2-(morfolin-4-il)acetil]pirrolidin-2-carboxílico;3-[1-(2,2-dimetilpropanoil)-5-{[(4-fluorofenil)metil]amino}-4-metil-1 H-pirazol-3-il]-1-(morfolin-4-carbonil)pirrolidin-2-ona;3-[1-(2-clorobenzoil)-5-{[(4-fluorofenil)metil](metil)amino}-4-metoxi-1 H-pirazol-3-il]-N,N-dimetil-2-(trifluorometil)azetidin-1-sulfonamida;3-[1 -(2-clorobenzoil)-5-{[(4-fluorofenil)metil]sulfanil}-1 H-pirazol-3-il]-1 -[(3-hidroxipirrolidin-1 -il)sulfonil]-4-(trifluorometil)pirrolidin-2-ona;3-[1-(2-clorobenzoil)-5-{[(4-fluorofenil)metil]sulfanil}-4-metoxi-1 H-pirazol-3-il]-N,N-dimetil-2-(trifluorometil)pirrolidin-1 -sulfonam ida;3-[1-(2-clorobenzoil)-5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-ciano-1 H-pirazol-3-il]-N,N-dimetil-2-(trifluorometil)piperidin-1-carboxamida;3-[1 -(2-clorobenzoil)-5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-metil-1 H-pirazol-3-il]-1 -(pirrolidin-1 -carbonil)-4-(trifluorometil)pirrolidin-2-ona;ácido 3-[1-(3-carboxi-2-clorobenzoil)-4-fluoro-5-{[(4-fluorofenil)metil]sulfanil}-1 H-pirazol-3-il]pirrolidin-2-carboxílico;ácido 3-[1 -(3-carboxi-2-clorobenzoil)-5-[(5-clorotiofen-2-il)metoxi]-4-metil-1 H-pirazol-3-il]-1 -(2,2-dimetilpropanoil)pirrolidin-2-carboxílico;ácido 3-[1 -(3-carboxi-2-clorobenzoil)-5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-metil-1 H-pirazol-3-il]-1 -(2,2-dimetilpropanoil)piperidin-2-carboxílico;3-[4-ciano-1-(2-fluorobenzoil)-5-[(4-fluorofenil)metoxi]-1 H-pirazol-3-il]-N,N-dimetil-2-(trifluorometil)pirrolidin-1 -sulfonam ida;3-[4-ciano-1-(2-fluorobenzoil)-5-{[(4-fluorofenil)metil]amino}-1 H-pirazol-3-il]-N,N-dimetil-2-(trifluorometil)piperazin-1-sulfonamida;ácido 3-[5-({[4-(aminometil)fenil]metil}(metil)amino)-1 -(2-fluorobenzoil)-4-metoxi-1 H-pirazol-3-il]-1 -(3-hidroxipirrolidin-1-carbonil)-4-(trifluorometil)piperidin-2-carboxílico;3-[5-({[4-(aminometil)fenil]metil}(metil)amino)-1-(4-metilfuran-3-carbonil)-1 H-pirazol-3-il]-N,N-dimetil-2-(trifluorometil)azetidin-1-carboxamida;ácido 3-[5-({[4-(aminometil)fenil]metil}(metil)amino)-1 -(5-metilfuran-3-carbonil)-1 H-pirazol-3-il]-1 -[(3-hidroxipirrolidin-1 -il)sulfonil]piperidin-2-carboxílico;ácido 3-[5-({[4-(aminometil)fenil]metil}(metil)amino)-3-[1-(2,2-dimetilpropanoil)-5-hidroxipirrolidin-3-il]-4-metil-1 H-pirazol-1 -carbonil]benzoico;ácido 3-[5-({[4-(aminometil)fenil]metil}(metil)amino)-3-{1-[(3-hidroxipirrolidin-1-il)sulfonil]pirrolidin-3-il}-4-metboxi-1 H-pirazol-1 -carbonil]benzoico;3-[5-({[4-(aminometil)fenil]metil}(metil)amino)-4-ciano-1-(2-metilfuran-3-carbonil)-1 H-pirazol-3-il]-N,N-dimetil-4-(trifluorometil)pirrolidin-1-carboxamida;ácido 3-[5-({[4-(amometil)fenil]metil}(metil)amino)-4-ciano-3-[1-(dimetilsulfamoil)-4-oxopirrolidin-3-il]-1 H-pirazol-1 -carbonil]benzoico;3-[5-({[4-(aminometil)fenil]metil}(metil)amino)-4-metoxi-1-(4-metilfuran-3-carbonil)-1 H-pirazol-3-il]-1-metanosulfonil-4-(trifluorometil)piperidin-2-ona;3-[5-({[4-(aminometil)fenil]metil}(metil)amino)-4-metil-1-(tiofen-3-carbonil)-1 H-pirazol-3-il]-4-metil-1-(morfolin-4-carbonil)azetidin-2-ona;ácido 3-[5-({[4-(aminometil)fenil]metil}amino)-1 -(2-metilfuran-3-carbonil)-1 H-pirazol-3-il]-1 -(dimetilsulfamoil)-4-(trifluorometil)piperidin-2-carboxílico;ácido 3-[5-({[4-(aminometil)fenil]metil}amino)-1 -(3-carboxibenzoil)-4-metoxi-1 H-pirazol-3-il]-1 -[2-(morfolin-4-il)-2-oxoetil]piperidin-2-carboxílico;3-[5-({[4-(aminometil)fenil]metil}amino)-1-(3-hidroxi-2,2-dimetilpropanoil)-4-metil-1 H-pirazol-3-il]-N,N,4-trimetil-2-oxopiperidin-1-sulfonamida;3-[5-({[4-(aminometil)fenil]metil}amino)-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il]-N,N-dimetil-2-oxopiperidin-1-carboxamida;ácido 3-[5-({[4-(aminometil)fenil]metil}amino)-1 -benzoil-1 H-pirazol-3-il]-1 -(3-hidroxipirrolidin-1 -carbonil)-4-metilpirrolidin-2-carboxílico;ácido 3-[5-({[4-(aminometil)fenil]metil}amino)-3-[1-(dimetilsulfamoil)-3-oxopiperidin-4-il]-1 H-pirazol-1- carbonil]benzoico;3-[5-({[4-(aminometil)fenil]metil}amino)-4-metil-1 -(tiofen-3-carbonil)-1 H-pirazol-3-il]-4-metilpiperidin-2- ona;3- [5-({[4-(aminometil)fenil]metil}sulfanil)-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il]-N,N-dimetil-2-oxo-4- (trifluorometil)azetidin-1-sulfonamida;ácido 3-[5-({[4-(aminometil)fenil]metil}sulfanil)-1 -(3-carboxi-2-clorobenzoil)-1 H-pirazol-3-il]-1 -metanosulfonilpirrolidin-2-carboxílico;3-[5-({[4-(aminometil)fenil]metil}sulfanil)-1-(furan-2-carbonil)-4-metoxi-1 H-pirazol-3-il]-4-metil-1-[2-(morfolin-4-il)-2-oxoetil]piperidin-2-ona;3-[5-({[4-(aminometil)fenil]metil}sulfanil)-1-(furan-3-carbonil)-1 H-pirazol-3-il]-N,N,2-trimetilpiperazin-1- carboxamida;3-[5-({[4-(aminometil)fenil]metil}sulfanil)-4-ciano-1-(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il]-N,N,4-trimetilpiperidin-1-carboxamida;3-[5-({[4-(aminometil)fenil]metil}sulfanil)-4-ciano-1-(furan-3-carbonil)-1 H-pirazol-3-il]-N,N,4-trimetil-2- oxopiperidin-1 -sulfonamida;ácido 3-[5-({[4-(aminometil)fenil]metil}sulfanil)-4-ciano-3-[5-oxo-1 -(pirrolidin-1 -sulfonil)pirrolidin-3-il]-1 H-pirazol-1 -carbonil]-2-clorobenzoico;ácido 3-[5-({[4-(aminometil)fenil]metil}sulfanil)-4-metoxi-1 -(tiofen-2-carbonil)-1 H-pirazol-3-il]-1 -[2-(morfolin-4-il)-2-oxoetil]pirrolidin-2-carboxílico;ácido 3-[5-({[4-(aminometil)fenil]metil}sulfanil)-4-metoxi-3-{1-[2-(morfolin-4-il)acetil]piperidin-3-il}-1 H-pirazol-1 -carbonil]-2-clorobenzoico;3- {1-benzoil-5-[(4-fluorofenil)metoxi]-4-metil-1 H-pirazol-3-il}-N,N,2-trimetilpirrolidin-1-carboxamida; ácido 3-{3-[1 -(dimetilcarbamoil)piperidin-4-il]-5-{[(4-fluorofenil)metil]amonio}-1 H-pirazol-1 -carbonil}benzoico;ácido 3-{3-[1 -(dimetilsulfamoil)-5-oxopirrolidin-3-il]-5-[(4-fluorofenil)metoxi]-1 H-pirazol-1 -carbonil}benzoico;ácido 3-{3-[1 -(dimetilsulfamoil)-6-oxopiperidin-3-il]-5-[(4-fluorofenil)metoxi]-4-metil-1 H-pirazol-1 -carbonil}benzoico;ácido 3-{5-[(4-carbamimidoilfenil)metoxi]-1 -(2-metoxibenzoil)-1 H-pirazol-3-il}-1 -[2-(morfolin-4-il)acetil]-4-(trifluorometil)piperidin-2-carboxílico;ácido 3-{5-[(4-carbamimidoilfenil)metoxi]-1-(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il}-4-metil-1- (morfolin-4-carbonil)piperidin-2-carboxílico;ácido 3-{5-[(4-carbamimidoilfenil)metoxi]-3-[1 -(dimetilsulfamoil)pirrolidin-3-il]-1 H-pirazol-1 -carbonil}-2- clorobenzoico;ácido 3-{5-[(4-carbamimidoilfenil)metoxi]-4-ciano-1 -(tiofen-2-carbonil)-1 H-pirazol-3-il}-1 -[2-(morfolin-4-il)-2-oxoetil]piperidin-2-carboxílico;ácido 3-{5-[(4-carbamimidoilfenil)metoxi]-4-ciano-3-[2-oxo-1 -(pirrolidin-1 -carbonil)piperidin-3-il]-1 H-pirazol-1-carbonil}-2-clorobenzoico;ácido 3-{5-[(4-carbamimidoilfenil)metoxi]-4-metoxi-1 H-pirazol-3-il}-1 -(pirrolidin-1 -carbonil)piperidin-2- carboxílico;ácido 3-{5-[(4-carbamimidoilfenil)metoxi]-4-metoxi-3-[1 -(pirrolidin-1 -sulfonil)piperidin-4-il]-1 H-pirazol-1-carbonil}-2-clorobenzoico;3- {5-[(4-fluorofenil)metoxi]-1-(2-metilfuran-3-carbonil)-1 H-pirazol-3-il}-1-[2-(morfolin-4-il)-2-oxoetil]-4- (trifluorometil)azetidin-2-ona;3-{5-[(5-clorotiofen-2-il)metoxi]-1-(2-metoxibenzoil)-1 H-pirazol-3-il}-1-(morfolin-4-carbonil)-4-(trifluorometil)piperidin-2-ona;3-{5-[(5-clorotiofen-2-il)metoxi]-1-(furan-2-carbonil)-4-metil-1H-pirazol-3-il}-1-metanosulfonil-2-metilpiperazina;3-{5-[(5-clorotiofen-2-il)metoxi]-1 -(furan-3-carbonil)-4-metil-1 H-pirazol-3-il}-2-metil-1 -(pirrolidin-1 -carbonil)piperidina;ácido 3-{5-[(5-clorotiofen-2-il)metoxi]-1 -(tiofen-2-carbonil)-1 H-pirazol-3-il}-1 -(pirrolidin-1 -sulfonil)pirrolidin-2-carboxílico;ácido 3-{5-[(5-clorotiofen-2-il)metoxi]-4-ciano-1 -(2-metoxibenzoil)-1 H-pirazol-3-il}-4-(trifluorometil)piperidin-2-carboxílico;ácido 3-{5-[(5-clorotiofen-2-il)metoxi]-4-ciano-1 H-pirazol-3-il}-1 -(pirrolidin-1 -carbonil)pirrolidin-2-carboxílico;ácido 3-{5-[(5-clorotiofen-2-il)metoxi]-4-metoxi-1 -(2-metoxibenzoil)-1 H-pirazol-3-il}-1 -metanosulfonil-4-(trifluorometil)piperidin-2-carboxílico;3- {5-[(5-clorotiofen-2-il)metoxi]-4-metil-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il}-N,N-dimetil-4-(trifluorometil)piperidin-1-sulfonamida;4- (4-ciano-5-{[(4-fluorofenil)metil](metil)amino}-1-(tiofen-2-carbonil)-1 H-pirazol-3-il)-2-hidroxi-N,N-dimetilpirrolidin-1-sulfonamida;4-(5-{[(4-carbamimidoilfenil)metil]sulfanil}-4-ciano-1-(2-metoxibenzoil)-1 H-pirazol-3-il)-N,N-dimetil-2-oxo-3-(trifluorometil)pirrolidin-1-carboxamida;4-(5-{[(4-fluorofenil)metil](metil)amino}-1-(furan-3-carbonil)-4-metoxi-1 H-pirazol-3-il)-3-metil-1-[2-(morfolin-4-il)-2-oxoetil]azetidin-2-ona;4-(5-{[(4-fluorofenil)metil](metil)amino}-4-metoxi-1-(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-3-metilpiperidin-2-ona;4-(5-{[(4-nuorofenil)metil](metil)amino}-4-metoxi-1-(tiofen-2-carbonil)-1 H-pirazol-3-il)-N,N-dimetil-2-oxopirrolidin-1 -sulfonamida;4-(5-{[(4-fluorofenil)metil](metil)amino}-4-metil-1 H-pirazol-3-il)-1-[2-(morfolin-4-il)acetil]azetidin-2-ona;4-(5-{[(4-fluorofenil)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-1-[(3-hidroxipirrolidin-1 -il)sulfonil]-5-metilpiperidin-3-ona;4-(5-{[(4-fluorofenil)metil]sulfanil}-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-1 -[2-(morfolin-4-il)-2-oxoetil]-3-(trifluorometil)pirrolidin-2-ona;4-(5-{[(4-fluorofenil)metil]sulfanil}-4-metoxi-1 H-pirazol-3-il)-1-(3-hidroxipirrolidin-1-carbonil)piperidin-2-ona;4-(5-{[(4-fluorofenil)metil]sulfanil}-4-metil-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-3-(trifluorometil)azetidin-2-ona;4-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-4-metil-1 H-pirazol-3-il)-3-metilpirrolidin-2-ona;4-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1-(3-hidroxipirrolidin-1 -carbonil)piperidin-2-ona;4-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-metoxi-1-(4-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1-[2-(morfolin-4-il)acetil]-5-(trifluorometil)pirrolidin-3-ona;4-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-metoxi-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il)-1-[2-(morfolin-4-il)-2-oxoetil]pirrolidin-3-ona;4-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-3-metil-1-(pirrolidin-1 -sulfonil)azetidin-2-ona;4-(5-{[(5-clorotiofen-2-il)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-4-metoxi-1 H-pirazol-3-il)-3-metil-1-(morfolin-4-carbonil)pirrolidin-2-ona;4-(5-{[(5-clorotiofen-2-il)metil]amino}-4-metil-1-(2-metilfuran-3-carbonil)-1 H-pirazol-3-il)-N,N-dimetil-2- oxo-3-(trifluorometil)pirrolidin-1-carboxamida;4-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-1-(2-metoxibenzoil)-4-metil-1 H-pirazol-3-il)-1-[2-(morfolin-4-il)acetil]-3-(trifluorometil)piperidin-2-ona;4-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-1 -(furan-2-carbonil)-1 H-pirazol-3-il)-5-metil-1 -(pirrolidin-1 -carbonil)pirrolidin-3-ona;4-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-1-(furan-2-carbonil)-4-metoxi-1 H-pirazol-3-il)-5-metilpirrolidin-3- ona;4- (5-{[(5-clorotiofen-2-il)metil]sulfanil}-1 H-pirazol-3-il)-1 -(pirrolidin-1 -sulfonil)piperidin-3-ona; 4-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-3-[1 -(pirrolidin-1 -carbonil)-3-(trifluorometil)azetidin-2-il]-1 H-pirazol-1 -carbonil)-1,3-tiazol;4-(5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-fluoro-1-(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-1-metanosulfonil-3-metilpiperidin-2-ona;4-(5-{[4-(aminometil)fenil]metoxi}-1-(furan-2-carbonil)-4-metil-1 H-pirazol-3-il)-N,N,3-trimetil-5-oxopiperidin-1 -carboxamida;4-(5-{[4-(aminometil)fenil]metoxi}-1 H-pirazol-3-il)-1 -(pirrolidin-1 -carbonil)piperidin-3-ona;4-(5-{[4-(aminometil)fenil]metoxi}-4-ciano-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)-N,N-dimetil-2-oxo-3-(trifluorometil)piperidin-1-carboxamida;4-(5-{[4-(aminometil)fenil]metoxi}-4-metil-1-(tiofen-2-carbonil)-1 H-pirazol-3-il)-1-[2-(morfolin-4-il)acetil]pirrolidin-2-ona;4-({[1-(2,2-dimetilpropanoil)-3-[1-(2,2-dimetilpropanoil)-2-oxoazetidin-3-il]-4-metoxi-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({[1 -(2,2-dimetilpropanoil)-3-[4-(pirrolidin-1 -carbonil)piperazin-2-il]-1 H-pirazol-5-il](metil)amino}metil)benceno-1-carboximidamida;4-({[1 -(2,2-dimetilpropanoil)-4-metil-3-[2-oxo-1 -(pirrolidin-1 -carbonil)piperidin-3-il]-1 H-pirazol-5-il](metil)amino}metil)benceno-1-carboximidamida;4-({[1-(2,2-dimetilpropanoil)-4-metil-3-{1-[2-(morfolin-4-il)acetil]-3-oxopiperidin-4-il}-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({[1-(2-clorobenzoil)-3-[1-(dimetilsulfamoil)-4-oxo-3-(trifluorometil)azetidin-2-il]-4-metoxi-1 H-pirazol-5-il]oxi}metil)benceno-1-carboximidamida;4-({[1 -(2-clorobenzoil)-3-[1 -(pirrolidin-1 -sulfonil)-4-(trifluorometil)piperidin-3-il]-1 H-pirazol-5-il]sulfanil}metil)benceno-1-carboximidamida;4-({[1 -(2-clorobenzoil)-3-{1-[(3-hidroxipirrolidin-1 -il)sulfonil]-3-(trifluorometil)azetidin-2-il}-4-metil-1 H-pirazol-5-il]oxi}metil)benceno-1-carboximidamida;4-({[1-(2-clorobenzoil)-4-ciano-3-{4-[(3-hidroxipirrolidin-1-il)sulfonil]-3-(trifluorometil)piperazin-2-il}-1 H-pirazol-5-il]oxi}metil)benceno-1 -carboximidamida;4-({[1 -(2-clorobenzoil)-4-metil-3-[1 -(pirrolidin-1 -sulfonil)-3-(trifluorometil)piperazin-2-il]-1 H-pirazol-5-il]sulfanil}metil)benceno-1-carboximidamida;4-({[1-(2-fluorobenzoil)-3-{1-[(3-hidroxipirrolidin-1-il)sulfonil]-6-oxo-4-(trifluorometil)piperidin-3-il}-4-metil-1 H-pirazol-5-il]amino}metil)benceno-1 -carboximidamida;4-({[1-(2-fluorobenzoil)-3-{1-[2-(morfolin-4-il)-2-oxoetil]-2-(trifluorometil)piperidin-3-il}-1 H-pirazol-5-il](metil)amino}metil)benceno-1-carboximidamida;4-({[1-(2-metoxibenzoil)-3-[6-oxo-4-(trifluorometil)piperidin-3-il]-1 H-pirazol-5-il]sulfanil}metil)benceno-1-carboximidamida;4-({[1 -(2-metilfuran-3-carbonil)-3-[1 -(pirrolidin-1 -sulfonil)-2-(trifluorometil)pirrolidin-3-il]-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({[1-(3-hidroxi-2,2-dimetilpropanoil)-3-(1-metanosulfonil-4-metil-5-oxopirrolidin-3-il)-4-metil-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({[1-(3-hidroxi-2,2-dimetilpropanoil)-3-[1-(3-hidroxipirrolidin-1-carbonil)-2-metilpirrolidin-3-il]-4-metil-1 H-pirazol-5-il](metil)amino}metil)benceno-1-carboximidamida;4-({[1-(3-hidroxi-2,2-dimetilpropanoil)-4-metoxi-3-[3-metil-4-oxo-1-(pirrolidin-1-carbonil)azetidin-2-il]-1 H-pirazol-5-il](metil)amino}metil)benceno-1 -carboximidamida;4-({[1 -(5-metilfuran-3-carbonil)-3-[2-oxo-1 -(pirrolidin-1 -sulfonil)piperidin-4-il]-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({[1 -(furan-2-carbonil)-3-[1 -(3-hidroxipirrolidin-1 -carbonil)-2-metilazetidin-3-il]-4-metil-1 H-pirazol-5-il]sulfanil}metil)benceno-1-carboximidamida;4-({[1 -(furan-2-carbonil)-3-[3-metil-1 -(pirrolidin-1 -carbonil)piperazin-2-il]-1 H-pirazol-5-il]oxi}metil)benceno-1-carboximidamida;4-({[1-(furan-2-carbonil)-4-metoxi-3-[2-metil-1-(morfolin-4-carbonil)pirrolidin-3-il]-1 H-pirazol-5-il]oxi}metil)benceno-1-carboximidamida;4-({[1 -(furan-3-carbonil)-3-[1 -(3-hidroxipirrolidin-1 -carbonil)-3-metil-5-oxopiperidin-4-il]-4-metil-1 H-pirazol-5-il]oxi}metil)benceno-1-carboximidamida;4-({[1-(furan-3-carbonil)-3-[4-metil-1-(morfolin-4-carbonil)-5-oxopirrolidin-3-il]-1 H-pirazol-5-il]sulfanil}metil)benceno-1-carboximidamida;4-({[1 -(furan-3-carbonil)-3-[5-hidroxi-1 -(3-hidroxipirrolidin-1 -carbonil)-2-metilpirrolidin-3-il]-1 H-pirazol-5-il]oxi}metil)benceno-1-carboximidamida;4-({[3-(1-metanosulfonil-3-metilpiperidin-4-il)-1-(3-metoxi-2,2-dimetilpropanoil)-4-metil-1 H-pirazol-5-il]oxi}metil)benceno-1-carboximidamida;4-({[4-ciano-1 -(2,2-dimetilpropanoil)-3-{1 -[(3-hidroxipirrolidin-1 -il)sulfonil]-5-oxopirrolidin-3-il}-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({[4-ciano-1-(2-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)acetil]-2-oxo-3-(trifluorometil)piperidin-4-il}-1 H-pirazol-5-il](metil)amino}metil)benceno-1-carboximidamida;4-({[4-ciano-1-(3-hidroxi-2,2-dimetilpropanoil)-3-{3-metil-1-[2-(morfolin-4-il)-2-oxoetil]-4-oxoazetidin-2-il}-1 H-pirazol-5-il]amino}metil)benceno-1 -carboximidamida;4-({[4-ciano-1-(3-metoxi-2,2-dimetilpropanoil)-3-(2-metilazetidin-3-il)-1 H-pirazol-5-il]oxi}metil)benceno-1-carboximidamida;4-({[4-ciano-1 -(furan-2-carbonil)-3-[2-metil-1 -(pirrolidin-1 -sulfonil)azetidin-3-il]-1 H-pirazol-5-il]sulfanil}metil)benceno-1-carboximidamida;4- ({[4-ciano-1 -(furan-3-carbonil)-3-[1 -(3-hidroxipirrolidin-1 -carbonil)-2-metilpirrolidin-3-il]-1 H-pirazol-5- il]oxi}metil)benceno-1-carboximidamida;4-({[4-ciano-1-(furan-3-carbonil)-3-{4-metil-1-[2-(morfolin-4-il)acetil]pirrolidin-3-il}-1 H-pirazol-5-il]sulfanil}metil)benceno-1-carboximidamida;4-({[4-ciano-3-(3-metilpiperazin-2-il)-1-(tiofen-3-carbonil)-1 H-pirazol-5-il]amino}-carboximidamida; 4-({[4-metoxi-1-(2-metilfuran-3-carbonil)-3-[2-(trifluorometil)oxetan-3-il]-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({[4-metoxi-1-(4-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)acetil]-3-(trifluorometil)piperazin-2-il}-1 H-pirazol-5-il]amino}metil)benceno-1 -carboximidamida;4-({[4-metoxi-1-(5-metilfuran-3-carbonil)-3-(piperidin-3-il)-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({[4-metoxi-1-(5-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)-2-oxoetil]-6-oxopiperidin-3-il}-1 H-pirazol-5-il](metil)amino}metil)benceno-1-carboximidamida;4-({[4-metil-1-(4-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)acetil]-3-(trifluorometil)piperidin-4-il}-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-({metil[1-(2-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)-2-oxoetil]-3-oxo-5-(trifluorometil)piperidin-4-il}-1 H-pirazol-5-il]amino}metil)benceno-1 -carboximidamida;4-({metil[4-metil-1-(2-metilfuran-3-carbonil)-3-[1-(morfolin-4-carbonil)-2-(trifluorometil)piperidin-3-il]-1 H-pirazol-5-il]amino}metil)benceno-1 -carboximidamida;4-({metil[4-metil-1 -(4-metilfliran-3-carbonil)-3-[2-oxo-1 -(pirrolidin-1 -carbonil)-3-(trifluorometil)piperidin-4-il]-1 H-pirazol-5-il]amino}metil)benceno-1-carboximidamida;4-[({1-benzoil-3-[2-metil-4-oxo-1 -(pirrolidin-1 -sulfonil)pirrolidin-3-il]-1 H-pirazol-5-il}amino)metil]benceno-1-carboximidamida;4-[({3-[1-(2,2-dimetilpropanoil)-2-oxo-4-(trifluorometil)piperidin-3-il]-4-metil-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-il}sulfanil)metil]benceno-1 -carboximidamida;4-[({3-[1 -(2,2-dimetilpropanoil)-3-(trifluorometil)azetidin-2-il]-4-fluoro-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-il}sulfanil)metil]benceno-1-carboximidamida;4-[({3-[1-(2,2-dimetilpropanoil)-3-oxo-5-(trifluorometil)piperidin-4-il]-1-(2-metoxibenzoil)-4-metil-1 H-pirazol-5-il}oxi)metil]benceno-1-carboximidamida;4- [({3-[1-(2,2-dimetilpropanoil)-4-metil-6-oxopiperidin-3-il]-1-(furan-3-carbonil)-4-metoxi-1 H-pirazol-5- il}oxi)metil]benceno-1-carboximidamida;4-[({3-[1 -(3-hidroxipirrolidin-1 -carbonil)-4-metilpirrolidin-3-il]-4-metoxi-1 -(tiofen-3-carbonil)-1 H-pirazol-5-il}amino)metil]benceno-1-carboximidamida;4-[({3-[1-(dimetilsulfamoil)-3-metil-2-oxopiperidin-4-il]-1-(furan-2-carbonil)-4-metoxi-1 H-pirazol-5-il}sulfanil)metil]benceno-1-carboximidamida;4-[({3-[1-(dimetilsulfamoil)-4-oxopirrolidin-3-il]-4-metil-1-(tiofen-2-carbonil)-1 H-pirazol-5-il}sulfanil)metil]benceno-1-carboximidamida;4-[({3-[1 -(dimetilsulfamoil)piperidin-4-il]-4-metil-1 -(tiofen-2-carbonil)-1 H-pirazol-5-il}oxi)metil]benceno-1-carboximidamida;4-[({3-[1 -(morfolin-4-carbonil)-4-oxo-3-(trifluorometil)azetidin-2-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-il}oxi)metil]benceno-1-carboximidamida;4-[({4-ciano-3-[1-(2,2-dimetilpropanoil)-2-oxo-3-(trifluorometil)piperidin-4-il]-1-(4-metilfuran-3-carbonil)-1 H-pirazol-5-il}amino)metil]benceno-1-carboximidamida;4-[({4-ciano-3-[1-(2,2-dimetilpropanoil)-4-(trifluorometil)piperidin-3-il]-1-(2-metilfuran-3-carbonil)-1 H-pirazol-5-il}amino)metil]benceno-1-carboximidamida;4-[({4-ciano-3-[1-(2,2-dimetilpropanoil)-4-oxoazetidin-2-il]-1-(5-metilfuran-3-carbonil)-1 H-pirazol-5-il}amino)metil]benceno-1-carboximidamida;4-[({4-ciano-3-[1-(2,2-dimetilpropanoil)pirrolidin-3-il]-1 H-pirazol-5-il}sulfanil)metil]benceno-1-carboximidamida;4-[({4-ciano-3-[1 -metanosulfonil-2-oxo-4-(trifluorometil)piperidin-3-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-il}sulfanil)metil]benceno-1-carboximidamida;4-[({4-ciano-3-[2-metil-1-(morfolin-4-carbonil)pirrolidin-3-il]-1-(tiofen-3-carbonil)-1 H-pirazol-5-il}(metil)amino)metil]benceno-1-carboximidamida;4-[({4-ciano-3-[2-oxo-1 -(pirrolidin-1 -carbonil)-3-(trifluorometil)piperidin-4-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-il}oxi)metil]benceno-1 -carboximidamida;4-[5-({[4-(aminometil)fenil]metil}(metil)amino)-1-(2,2-dimetilpropanoil)-4-metoxi-1 H-pirazol-3-il]-1-metanosulfonilazetidin-2-ona;4-[5-({[4-(aminometil)fenil]metil}(metil)amino)-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il]-3-metil-1-[2-(morfolin-4-il)-2-oxoetil]piperidin-2-ona;4-[5-({[4-(aminometil)fenil]metil}(metil)amino)-1-(tiofen-3-carbonil)-1 H-pirazol-3-il]-N,N,3-trimetil-2-oxopiperidin-1 -sulfonamida;4-[5-({[4-(aminometil)fenil]metil}(metil)amino)-1-benzoil-4-metil-1 H-pirazol-3-il]-3-metil-1-(morfolin-4-carbonil)piperidin-2-ona;4-[5-({[4-(aminometil)fenil]metil}(metil)amino)-4-metil-1-(4-metilfuran-3-carbonil)-1 H-pirazol-3-il]-N,N-dimetil-2-oxo-3-(trifluorometil)piperidin-1-carboxamida;4-[5-({[4-(aminometil)fenil]metil}(metil)amino)-4-metil-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il]pirrolidin-2-ol;4-[5-({[4-(aminometil)fenil]metil}amino)-1-(2,2-dimetilpropanoil)-4-metil-1 H-pirazol-3-il]-1 -(pirrolidin-1 -sulfonil)pirrolidin-2-ona;4-[5-({[4-(aminometil)fenil]metil}amino)-4-metoxi-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il]-1-(2,2-dimetilpropanoil)pirrolidin-3-ona;4-[5-({[4-(aminometil)fenil]metil}amino)-4-metoxi-1-(tiofen-3-carbonil)-1 H-pirazol-3-il]-1-[(3-hidroxipirrolidin-1 -il)sulfonil]-5-metilpirrolidin-2-ol;4-[5-({[4-(aminometil)fenil]metil}sulfanil)-1-(2-clorobenzoil)-4-metil-1 H-pirazol-3-il]-1-(2,2-dimetilpropanoilo)-3-(trifluorometil)pirrolidin-2-ona;4-[5-({[4-(aminometil)fenil]metil}sulfanil)-1 -(furan-2-carbonil)-1 H-pirazol-3-il]-1 -[(3-hidroxipirrolidin-1 -il)sulfonil]-5-metilpirrolidin-2-ol;4-[5-({[4-(aminometil)fenil]metil}sulfanil)-1 H-pirazol-3-il]-1-(2,2-dimetilpropanoil)pirrolidin-3-ona; 4- [5-({[4-(aminometil)fenil]metil}sulfanil)-4-metoxi-1-(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il]-5- metil-1 -(pirrolidin-1 -sulfonil)pirrolidin-3-ona;4-[5-({[4-(aminometil)fenil]metil}sulfanil)-4-metil-1-(tiofen-2-carbonil)-1 H-pirazol-3-il]-1-(morfolin-4-carbonil)piperidin-3-ona;4-[5-({[4-(aminometil)fenil]metil}sulfanil)-4-metil-1 H-pirazol-3-il]-1-(2,2-dimetilpropanoil)azetidin-2-ona;4-{1-benzoil-5-[(4-fluorofenil)metoxi]-1 H-pirazol-3-il}-3-metilpiperidin-2-ona;4-{5-[(4-fluorofenil)metoxi]-1 -(3-hidroxi-2,2-dimetilpropanoil)-4-metil-1 H-pirazol-3-il}-1 -metanosulfonil-5-metilpiperidin-3-ona;4-{5-[(4-fluorofenil)metoxi]-1 -(4-metilfuran-3-carbonil)-1 H-pirazol-3-il}-1 -(3-hidroxipirrolidin-1 -carbonil)-3-(trifluorometil)azetidin-2-ona;4-{5-[(4-fluorofenil)metoxi]-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il}-1 -metanosulfonilpirrolidin-3-ona;4-{5-[(4-fluorofenil)metoxi]-4-metoxi-1-(4-metilfuran-3-carbonil)-1 H-pirazol-3-il}-1-[2-(morfolin-4-il)-2-oxoetil]-3-(trifluorometil)pirrolidin-2-ona;4-{5-[(4-fluorofenil)metoxi]-4-metoxi-1-(5-metilfuran-3-carbonil)-1 H-pirazol-3-il}piperidina;4-{5-[(5-clorotiofen-2-il)metoxi]-1 -(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il}-1 -[(3-hidroxipirrolidin-1 -il)sulfonil]-3-metilpiperidin-2-ona;4-{5-[(5-clorotiofen-2-il)metoxi]-4-metoxi-1-(tiofen-2-carbonil)-1 H-pirazol-3-il}piperidin-3-ona;4-{[(1 -benzoil-3-{1 -[(3-hidroxipirrolidin-1 -il)sulfonil]-4-metilpiperidin-3-il}-1 H-pirazol-5-il)(metil)amino]metil}benceno-1-carboximidamida;4-{[(1 -benzoil-4-ciano-3-{1 -[(3-hidroxipirrolidin-1 -il)sulfonil]-2-metil-4-oxoazetidin-3-il}-1 H-pirazol-5-il)(metil)amino]metil}benceno-1-carboximidamida;4-{[(3-{1 -[(3-hidroxipirrolidin-1 -il)sulfonil]-2-metilpiperidin-3-il}-1 -(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-5-il)sulfanil]metil}benceno-1-carboximidamida;4-{[(3-{1-[(3-hidroxipirrolidin-1-il)sulfonil]-3-(trifluorometil)piperidin-4-il}-1-(4-metilfuran-3-carbonil)-1 H-pirazol-5-il)(metil)amino]metil}benceno-1 -carboximidamida;4-{[(3-{1 -[(3-hidroxipirrolidin-1 -il)sulfonil]-4-oxoazetidin-2-il}-4-metil-1 H-pirazol-5-il)sulfanil]metil}benceno-1-carboximidamida;4-{[(3-{1 -[(3-hidroxipirrolidin-1 -il)sulfonil]azetidin-3-il}-1 -(tiofen-2-carbonil)-1 H-pirazol-5-il)sulfanil]metil}benceno-1-carboximidamida;4-{[(3-{1-[2-(morfolin-4-il)-2-oxoetil]-6-oxopiperidin-3-il}-1 H-pirazol-5-il)oxi]metil}benceno-1-carboximidamida;4-{[(3-{3-metil-1-[2-(morfolin-4-il)-2-oxoetil]azetidin-2-il}-1-(tiofen-3-carbonil)-1 H-pirazol-5-il)amino]metil}benceno-1-carboximidamida;4- {[(4-metil-3-{1-[2-(morfolin-4-il)acetil]-2-oxopiperidin-3-il}-1 H-pirazol-5-il)oxi]metil}benceno-1-carboximidamida;5- (1 -benzoil-5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-metil-1 H-pirazol-3-il)-4-metil-1 -(pirrolidin-1 -carbonil)piperidin-2-ona;5-(4-ciano-5-{[(4-fluorofenil)metil]sulfanil}-1-(tiofen-2-carbonil)-1 H-pirazol-3-il)-N,N-dimetil-2-oxopiperidin-1 -sulfonamida;5-(5-{[(4-fluorofenil)metil]sulfanil}-4-metoxi-1 -(tiofen-2-carbonil)-1 H-pirazol-3-il)-1 -(pirrolidin-1 -sulfonil)piperidin-2-ona;5-(5-{[(5-clorotiofen-2-il)metil](metil)amino}-4-ciano-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-3-il)-N,N,4-trimetil-2-oxopiperidin-1-carboxamida;5-(5-{[4-(aminometil)fenil]metoxi}-1-(furan-2-carbonil)-1 H-pirazol-3-il)-1-metanosulfonil-4-metilpiperidin-2-ona;5-({[4-(aminometil)fenil]metil}(metil)amino)-1-(2-fluorobenzoil)-3-[1-(pirrolidin-1-sulfonil)-3-(trifluorometil)piperazin-2-il]-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}(metil)amino)-1-(5-metilfuran-3-carbonil)-3-[2-oxo-1-(pirroldina-1-sulfonil)piperidin-3-il]-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}(metil)amino)-1-benzoil-3-{4-metil-1-[2-(morfolin-4-il)acetil]-2-oxopirrolidin-3-il}-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}(metil)amino)-3-[1-(2,2-dimetilpropanoil)-4-metil-2-oxopirrolidin-3-il]-1-(3-hidroxi-2,2-dimetilpropanoil)-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}amino)-1-(2,2-dimetilpropanoil)-3-{1-[2-(morfolin-4-il)acetil]piperidin-3-il}-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}amino)-1-(4-metilfuran-3-carbonil)-3-[1-(morfolin-4-carbonil)-3-(trifluorometil)piperidin-4-il]-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}amino)-1-(5-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)acetil]-2-oxopiperidin-3-il}-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}amino)-1-benzoil-3-(1-metanosulfonil-3-metilpiperidin-4-il)-1 H-pirazol-4- carbonitrilo;5- ({[4-(aminometil)fenil]metil}amino)-3-[1-(2,2-dimetilpropanoil)-3-oxo-5-(trifluorometil)piperidin-4-il]-1 -(2-metilfuran-3-carbonil)-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}amino)-3-{4-metil-1 -[2-(morfolin-4-il)-2-oxoetil]-2-oxopiperidin-3-il}-1 -(tiofen-3-carbonil)-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}sulfanil)-1 -(2-clorobenzoil)-3-[1 -(pirrolidin-1 -carbonil)-3-(trifluorometil)piperidin-4-il]-1 H-pirazol-4-carbonitrilo;5-({[4-(aminometil)fenil]metil}sulfanil)-1-(2-metoxibenzoil)-3-[2-(trifluorometil)oxetan-3-il]-1 H-pirazol-4- carbonitrilo;5- ({[4-(aminometil)fenil]metil}sulfanil)-3-{1-[(3-hidroxipirrolidin-1-il)sulfonil]-4-oxopirrolidin-3-il}-1-(tiofen-2-carbonil)-1 H-pirazol-4-carbonitrilo;5-[(4-fluorofenil)metoxi]-1-(3-hidroxi-2,2-dimetilpropanoil)-3-[4-(3-hidroxipirrolidin-1-carbonil)-3-metilpiperazin-2-il]-1 H-pirazol-4-carbonitrilo;5-[(4-fluorofenil)metoxi]-1-(4-metilfuran-3-carbonil)-3-[1-(morfolin-4-carbonil)-3-(trifluorometil)azetidin-2-il]-1 H-pirazol-4-carbonitrilo;5-[(4-fluorofenil)metoxi]-1-(5-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)acetil]-4-oxoazetidin-2-il}-1 H-pirazol-4-carbonitrilo;5-[(4-fluorofenil)metoxi]-3-{4-metil-1-[2-(morfolin-4-il)acetil]piperidin-3-il}-1-(tiofen-3-carbonil)-1 H-pirazol-4-carbonitrilo;5-[(4-fluorofenil)metoxi]-4-metoxi-1-(2-metilfuran-3-carbonil)-3-[2-(trifluorometil)oxan-3-il]-1H-pirazol;5-[(5-clorotiofen-2-il)metoxi]-1-(3-metoxi-2,2-dimetilpropanoil)-3-[4-metil-6-oxo-1-(pirrolidin-1-carbonil)piperidin-3-il]-1 H-pirazol-4-carbonitrilo;5-[(5-clorotiofen-2-il)metoxi]-1 -(furan-2-carbonil)-3-[4-metil-1 -(pirrolidin-1 -carbonil)piperidin-3-il]-1 H-pirazol-4-carbonitrilo;5-[(5-clorotiofen-2-il)metoxi]-3-[1 -(3-hidroxipirrolidin-1 -carbonil)-5-oxopirrolidin-3-il]-1 -(tiofen-2-carbonil)-1 H-pirazol-4-carbonitrilo;5-[(5-clorotiofen-2-il)metoxi]-3-[2-oxo-1 -(pirrolidin-1 -carbonil)-4-(trifluorometil)azetidin-3-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-4-carbonitrilo;5-[(5-clorotiofen-2-il)metoxi]-4-metil-3-(pirrolidin-3-il)-1 H-pirazol;5-[5-({[4-(aminometil)fenil]metil}(metil)amino)-4-fluoro-1-(2-metilfuran-3-carbonil)-1 H-pirazol-3-il]-4-(trifluorometil)piperidin-2-ona;5-{1 -benzoil-5-[(4-fluorofenil)metoxi]-4-metoxi-1 H-pirazol-3-il}-1 -(3-hidroxipirrolidin-1 -carbonil)-4-metilpiperidin-2-ona;5-{[(4-fluorofenil)metil](metil)amino}-1-(furan-3-carbonil)-3-(1-metanosulfonil-4-metil-5-oxopirrolidin-3-il)-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil](metil)amino}-3-(piperidin-4-il)-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil](metil)amino}-3-[1-(3-hidroxipirrolidin-1-carbonil)-4-metil-6-oxopiperidin-3-il]-1 -(3-metoxi-2,2-dimetilpropanoil)-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil]amino}-1-(2-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)acetil]-2-(trifluorometil)pirrolidina-3-il}-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil]amino}-1-(5-metilfuran-3-carbonil)-3-[5-oxo-1-(pirrolidin-1-carbonil)pirrolidin-3-il]-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil]amino}-3-(2-metil-4-oxopirrolidin-3-il)-1-(tiofen-3-carbonil)-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil]sulfanil}-1-(2-metoxibenzoil)-3-{1-[2-(morfolin-4-il)acetil]-6-oxo-4-(trifluorometil)piperidin-3-il}-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil]sulfanil}-1-(furan-3-carbonil)-3-(1-metanosulfonil-2-metilazetidin-3-il)-1 H-pirazol;5-{[(4-fluorofenil)metil]sulfanil}-3-{1-[2-(morfolin-4-il)-2-oxoetil]-4-oxopirrolidin-3-il}-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil]sulfanil}-3-{1-[2-(morfolin-4-il)acetil]-4-oxo-2-(trifluorometil)pirrolidin-3-il}-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-4-carbonitrilo;5-{[(4-fluorofenil)metil]sulfanil}-4-metoxi-1-(2-metoxibenzoil)-3-[3-(trifluorometil)oxetan-2-il]-1H-pirazol;5-{[(5-clorotiofen-2-il)metil](metil)amino}-1-(2,2-dimetilpropanoil)-3-[1-(morfolin-4-carbonil)-6-0xopiperidin-3-il]-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil](metil)amino}-1-(2-fluorobenzoil)-3-{5-hidroxi-1-[2-(morfolin-4-il)acetil]-2-(trifluorometil)pirrolidin-3-il}-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil](metil)amino}-1-(4-metilfuran-3-carbonil)-3-[4-oxo-3-(trifluorometil)azetidin-2-il]-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil]amino}-1-(2-fluorobenzoil)-3-[5-hidroxi-1-(morfolin-4-carbonil)-2-(trifluorometil)pirrolidin-3-il]-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil]amino}-1-(2-metilfuran-3-carbonil)-3-{1-[2-(morfolin-4-il)-2-oxoetilo]-4-(trifluorometil)pirrolidin-3-il}-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil]amino}-1-(3-hidroxi-2,2-dimetilpropanoil)-3-[2-metil-1-(pirrolidin-1-carbonil)piperidin-3-il]-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil]sulfanil}-1-(furan-3-carbonil)-3-[3-metil-4-oxo-1-(pirrolidin-1-carbonil)azetidin-2-il]-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil]sulfanil}-3-[1-(3-hidroxipirrolidin-1-carbonil)-2-oxo-4-(trifluorometil)azetidin-3-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil]sulfanil}-3-[1-(3-hidroxipirrolidin-1-carbonil)-4-oxopirrolidin-3-il]-1-(tiofen-2- carbonil)-1 H-pirazol-4-carbonitrilo;5-{[(5-clorotiofen-2-il)metil]sulfanil}-4-metil-3-[1 -(pirrolidin-1 -carbonil)pirrolidin-3-il]-1 -(tiofen-2-carbonil)-1 H-pirazol;5-{[4-(aminometil)fenil]metoxi}-1 -(2-clorobenzoil)-3-[1 -(3-hidroxipirrolidin-1 -carbonil)-3-(trifluorometil)piperazin-2-il]-1 H-pirazol-4-carbonitrilo;5-{[4-(aminometil)fenil]metoxi}-1-(furan-2-carbonil)-3-[2-metil-1-(morfolin-4-carbonil)-4-oxoazetidin-3- il]-1 H-pirazol-4-carbonitrilo;5-{[4-(aminometil)fenil]metoxi}-1-(furan-3-carbonil)-3-(1-metanosulfonil-4-metil-2-oxopirrolidin-3-il)-1 H-pirazol-4-carbonitrilo;5-{[4-(aminometil)fenil]metoxi}-3-(5-hidroxipirrolidin-3-il)-1 H-pirazol-4-carbonitrilo;N-[(4-fluorofenil)metil]-3-[1-metanosulfonil-2-(trifluorometil)piperidin-3-il]-4-metil-1-(2-metiliuran-3-carbonil)-1 H-pirazol-5-amina;N-[(4-fluorofenil)metil]-4-metil-1-(5-metilfuran-3-carbonil)-3-(morfolin-2-il)-1 H-pirazol-5-amina; N-[(4-fluorofenil)metil]-N,4-dimetil-3-[1-(morfolin-4-carbonil)-3-(trifluorometil)azetidin-2-il]-1-(1,3-tiazol)-4-carbonil)-1 H-pirazol-5-amina;N-[(4-fluorofenil)metil]-N-metil-3-(pirrolidin-3-il)-1-(tiofen-2-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(2-fluorobenzoil)-3-[1-metanosulfonil-4-(trifluorometil)pirrolidin-3-il]-4-metoxi-N-metil-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1 -(2-fluorobenzoil)-4-metoxi-3-[1 -(pirrolidin-1 -sulfonil)-4-(trifluorometil)piperidin-3-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1-(2-fluorobenzoil)-4-metil-3-[1-(morfolin-4-carbonil)-4-(trifluorometil)piperidin-3-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-1 -(2-fluorobenzoil)-N,4-dimetil-3-[1 -(pirrolidin-1 -sulfonil)-2-(trifluorometil)piperidin-3-il]-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-4-metoxi-3-[4-metil-1 -(pirrolidin-1 -carbonil)piperidin-3-il]-1 -(tiofen-3-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-4-metil-3-[2-metil-1 -(pirrolidin-1 -sulfonil)azetidin-3-il]-1 -(tiofen-3-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-N-metil-1-(2-metilfuran-3-carbonil)-3-[3-(trifluorometil)oxetan-2-il]-1 H-pirazol-5-amina;N-{[4-(aminometil)fenil]metil}-1-(2-fluorobenzoil)-3-[1-metanosulfonil-2-(trifluorometil)pirrolidin-3-il]-N-metil-1 H-pirazol-5-amina;N-{[4-(aminometil)fenil]metil}-1 -(2-fluorobenzoil)-4-metil-3-[1 -(pirrolidin-1 -carbonil)-3-(trifluorometil)piperidin-4-il]-1 H-pirazol-5-amina;N-{[4-(aminometil)fenil]metil}-1-(2-fluorobenzoil)-N,4-dimetil-3-[3-(trifluorometil)piperazin-2-il]-1 H-pirazol-5-amina;N-{[4-(aminometil)fenil]metil}-1-benzoil-4-metoxi-3-(2-metilazetidin-3-il)-1 H-pirazol-5-amina; N-{[4-(aminometil)fenil]metil}-4-metoxi-1-(2-metilfuran-3-carbonil)-3-[1-(morfolin-4-carbonil)-2-(trifluorometil)azetidin-3-il]-1 H-pirazol-5-amina;N-{[4-(aminometil)fenil]metil}-4-metil-1-(4-metilfuran-3-carbonil)-3-[4-(pirrolidin-1-sulfonil)-3-(trifluorometil)piperazin-2-il]-1H-pirazol-5-amina;[4-({[1-(2-clorobenzoil)-3-[4-(trifluorometil)piperidin-3-il]-1 H-pirazol-5-il]sulfanil}metil)fenil]metanamina;[4-({[1-(2-metoxibenzoil)-3-[1-(morfolin-4-carbonil)-2-(trifluorometil)azetidin-3-il]-1 H-pirazol-5-il]sulfanil}metilo)fenil]metanamina;[4-({[1-(furan-2-carbonil)-4-metil-3-(3-metilpiperazin-2-il)-1 H-pirazol-5-il]sulfanil}metil)fenil]metanamina;[4-({[1 -(furan-3-carbonil)-4-mctil-3-[2-metil-1 -(pirrolidin-1 -carbonil)azetidin-3-il]-1 H-pirazol-5-il]oxi}metil)fenil]metanamina;{4-[({4-metoxi-3-[1 -(morfolin-4-carbonil)-3-(trifluorometil)piperidin-4-il]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-il}sulfanil)metil]fenil}metanamina;N-[(5-clorotiofen-2-il)metil]-3-(piperazin-2-il)-1 H-pirazol-5-amina;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-{4-[2-(morfolin-4-il)acetil]piperazin-2-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1 -[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazo1 -3-il)piperazin-1 -il]-2-(morfolin-4-il)etan-1 -ona; 1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[4-(morfolin-4-sulfonil)piperazin-2-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[4-(2,2,2-trifluoroetil)piperazin-2-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;N-[(5-clorotiofen-2-il)metil]-3-(pirrolidin-2-il)-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;1 -[2-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)pirrolidin-1 -il]-2-(morfolin-4-il)etan-1 -ona;N-[(5-clorotiofen-2-il)metil]-3-(pirrolidin-3-il)-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;1 -[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)pirrolidin-1 -il]-2-(morfolin-4-il)etan-1 -ona;N-[(5-clorotiofen-2-il)metil]-3-(piperazin-2-il)-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;1 -[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-3-il)piperazin-1 -il]-2-(morfolin-4-il)etan-1 -ona;N-[(5-clorotiofen-2-il)metil]-3-(4-metanosulfonilpiperazin-2-il)-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;N-[(5-clorotiofen-2-il)metil]-3-[4-(morfolin-4-carbonil)piperazin-2-il]-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;1 -[3-(azepan-4-il)-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-1 -il]-2,2-dimetilpropan-1 -ona; 3-(azepan-4-il)-N-[(5-clorotiofen-2-il)metil]-1 -(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;1 -[3-(azetidin-3-il)-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-1 -il]-2,2-dimetilpropan-1 -ona; N-[(5-clorotiofen-2-il)metil]-3-[1-(morfolin-4-carbonil)azetidin-3-il]-1 H-pirazol-5-amina;3-(azetidin-3-il)-N-[(5-clorotiofen-2-il)metil]-1-(1,3-tiazol-4-carbonil)-1 H-pirazol-5-amina;3-[1-(2-clorobenzoil)-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il]-N,N-dimetilpirrolidin-1-sulfonamida;1 -(2-clorobenzoil)-N-[(5-clorotiofen-2-il)metil]-3-[1 -(pirrolidin-1 -sulfonil)pirrolidin-3-il]-1 H-pirazol-5-amina;1 -(2-clorobenzoil)-N-[(5-clorotiofen-2-il)metil]-3-[1 -(piperazin-1 -sulfonil)pirrolidin-3-il]-1 H-pirazol-5-amina;1 -(2-clorobenzoil)-N-[(5-clorotiofen-2-il)metil]-3-[1 -(piperazin-1 -carbonil)pirrolidin-3-il]-1 H-pirazol-5-amina;3-[1-(2-clorobenzoil)-5-{[(5-clorotiofen-2-il)metil]amino}-1 H-pirazol-3-il]-N,N-dimetilazetidin-1-sulfonamida;1 -(2-clorobenzoil)-N-[(5-clorotiofen-2-il)metil]-3-[1 -(pirrolidin-1 -sulfonil)azetidin-3-il]-1 H-pirazol-5-amina;1 -(2-clorobenzoil)-N-[(5-clorotiofen-2-il)metil]-3-[1 -(piperazin-1 -sulfonil)azetidin-3-il]-1 H-pirazol-5-amina;1 -(2-clorobenzoil)-N-[(5-clorotiofen-2-il)metil]-3-[1 -(piperazin-1 -carbonil)azetidin-3-il]-1 H-pirazol-5-amina;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[4-(2-ciclopropoxietil)piperazin-2-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;ácido 4-[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2,2-dimetilpropanoil)-1 H-pirazol-3-il)piperazin-1 -carbonil]morfolin-3-carboxílico;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-[4-(3-metilmorfolin-4-carbonil)piperazin-2-il]-1 H-pirazol-1 -il)-2.2- dimetilpropan-1 -ona;1-(5-1[(5-clorotiofen-2-il)metil]amino}-3-[4-(3,5-dimetilmorfolin-4-carbonil)piperazin-2-il]-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-{4-[2-(morfolin-4-il)-2-oxoetil]piperazin-2-il}-1 H-pirazol-1 -il)-2.2- dimetilpropan-1 -ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-{4-[2-(morfolin-4-il)etil]piperazin-2-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1-ona;ácido 4-{2-[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2,2-dimetilpropanoil)-1 H-pirazol-3-il)piperazin-1 -il]etil}morfolin-3-carboxílico;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-{4-[4-(hidroximetil)-1,3-oxazol-2-il]piperazin-2-il}-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona;1-(5-{[(5-clorotiofen-2-il)metil]amino}-3-(4-{[4-(hidroximetil)-1,3-oxazol-2-il]metil}piperazin-2-il)-1 H-pirazol-1 -il)-2,2-dimetilpropan-1 -ona; yácido 2-[3-(5-{[(5-clorotiofen-2-il)metil]amino}-1 -(2,2-dimetilpropanoil)-1 H-pirazol-3-il)piperazin-1 -iljacético.12. Una composición farmacéutica que comprende un compuesto según cualquiera de las reivindicaciones 1 a 11, y un excipiente farmacéuticamente aceptable.13. El compuesto según cualquiera de las reivindicaciones 1 a 11, para usar en el tratamiento de una enfermedad o un trastorno relacionado con la trombina, o una enfermedad o un trastorno relacionado con la calicreína; opcionalmente, en donde el tratamiento tiene un efecto profiláctico.14. El compuesto para el uso según la reivindicación 13, en donde el compuesto es para usar en el tratamiento de una enfermedad o un trastorno trombótico, y/o una enfermedad o un trastorno que implica un trombo de coágulo sanguíneo o la posible formación de un trombo de coágulo sanguíneo, o en donde la enfermedad o el trastorno implica episodios cardíacos recurrentes después de un infarto de miocardio.15. El compuesto para el uso según la reivindicación 14, en donde dicha enfermedad o dicho trastorno trombótico comprende síndrome coronario agudo, tromboembolia y/o trombosis; opcionalmente, en donde la tromboembolia comprende tromboembolia venosa, tromboembolia arterial y/o tromboembolia cardiogénica.16. El compuesto para el uso según la reivindicación 15, en donde:la tromboembolia comprende tromboembolia venosa; opcionalmente, en donde la tromboembolia venosa comprende trombosis venosa profunda y/o embolia pulmonar, o en donde la tromboembolia venosa está asociada con la formación de un trombo dentro de una vena asociado con uno o más factores de riesgo adquiridos o heredados, y/o embolia de venas periféricas provocada por un trombo desprendido; además, opcionalmente, en donde la trombosis venosa profunda y/o la embolia pulmonar se producen después de una intervención médica, o en donde uno o más factores de riesgo comprenden una tromboembolia venosa previa; o la tromboembolia comprende tromboembolia cardiogénica; opcionalmente, en donde la tromboembolia cardiogénica se debe a fibrilación auricular no valvular o formación de un trombo en el corazón asociado con arritmia cardíaca, defecto de válvula cardíaca, válvulas cardíacas protésicas o cardiopatía, y/o embolia de arterias periféricas provocada por un trombo desprendido; además, opcionalmente, en donde el trombo desprendido está en el cerebro (accidente cerebrovascular isquémico); además, opcionalmente, en donde el trombo desprendido provoca un ataque isquémico transitorio (AIT); ola trombosis comprende trombosis arterial; opcionalmente, en donde la trombosis arterial se debe a uno o más procesos ateroescleróticos subyacentes en las arterias; además, opcionalmente, en donde el uno o más procesos ateroescleróticos subyacentes en las arterias obstruyen u ocluyen una arteria, provocar isquemia miocárdica (angina de pecho, síndrome coronario agudo), provocar infarto de miocardio, obstruir u ocluir una arteria periférica (enfermedad arterial periférica isquémica), y/u obstruir u ocluir la arteria después de una operación en un vaso sanguíneo (reoclusión o reestenosis después de angioplastia coronaria transluminal, reoclusión o reestenosis después de angioplastia transluminal percutánea de arterias periféricas); ola enfermedad o el trastorno trombótico implica coagulación disfuncional o coagulación intravascular diseminada; opcionalmente, en donde el sujeto se somete a una intervención coronaria percutánea (ICP).17. El compuesto para el uso según la reivindicación 14, en donde dicha enfermedad o dicho trastorno trombótico implica un trombo de coágulo sanguíneo o la posible formación de un trombo de coágulo sanguíneo, y además implica un accidente cerebrovascular y/o uno o más ataques isquémicos transitorios (AIT) y/o hipertensión pulmonar; además, opcionalmente, en donde dicha enfermedad o dicho trastorno que implica un trombo de coágulo sanguíneo o la posible formación de un trombo de coágulo sanguíneo implica además un accidente cerebrovascular, y en donde el sujeto tiene fibrilación auricular no valvular; además, opcionalmente, en donde dicha enfermedad o dicho trastorno que implica un trombo de coágulo sanguíneo o la posible formación de un trombo de coágulo sanguíneo implica además hipertensión pulmonar provocada por uno o más trastornos de las cavidades izquierdas del corazón y/o enfermedad tromboembólica crónica o asociada con una o más enfermedades pulmonares, incluida la fibrosis pulmonar (idiopática o no) y/o la hipoxia.18. El compuesto para el uso según la reivindicación 13, en donde dicha enfermedad o dicho trastorno comprende fibrosis, enfermedad de Alzheimer, esclerosis múltiple, dolor, cáncer, inflamación y/o diabetes mellitus de tipo I.19. El compuesto para el uso según la reivindicación 14, en donde el tratamiento o la prevención comprende una terapia adyuvante; opcionalmente, en donde el sujeto tiene un infarto de miocardio y la terapia adyuvante se realiza junto con terapia trombolítica, o en donde el sujeto tiene angina de pecho inestable, trombosis y/o trombocitopenia inducida por heparina, y la terapia adyuvante se realiza junto con tratamiento antiplaquetario, o en donde el sujeto tiene fibrilación auricular no valvular, y la terapia adyuvante se realiza junto con una o más terapias diferentes.20. El compuesto para el uso según la reivindicación 13, en donde dicha enfermedad o dicho trastorno es un trastorno relacionado con la calicreína; opcionalmente, en donde dicho trastorno relacionado con la calicreína es una enfermedad trombótica, una enfermedad fibrinolítica, un trastorno fibrótico, un tipo de cáncer, una afección inflamatoria, una afección dermatológica o una enfermedad oftálmica.21. El compuesto para el uso según la reivindicación 20, en donde:la enfermedad o el trastorno comprende una enfermedad oftálmica; opcionalmente, en donde dicha enfermedad oftálmica comprende edema macular diabético, degeneración macular relacionada con la edad o retinopatía diabética; ola enfermedad o el trastorno comprende un tipo de cáncer; opcionalmente, en donde dicho tipo de cáncer se selecciona del grupo que consiste en adenocarcinoma de pulmón no microcítico, testicular o cervicouterino, cáncer de pulmón microcítico limitado, glioma, cáncer de mama maligno, micrometástasis (p. ej., micrometástasis de sangre o hígado), metástasis de pulmón y cáncer de próstata; ola enfermedad o el trastorno comprende un estado inflamatorio; opcionalmente, en donde dicha afección inflamatoria es sepsis, enfermedad intestinal inflamatoria, artritis inflamatoria, síndrome de respuesta inflamatoria sistémica, angioedema hereditario o artritis reumatoide; o la enfermedad o el trastorno comprende una afección dermatológica; opcionalmente, en donde dicha afección dermatológica es dermatitis atópica, psoriasis o síndrome de Netherton.22. El compuesto para el uso según la reivindicación 21, la enfermedad o el trastorno comprende una enfermedad oftálmica, y en donde dicho compuesto o dicha composición farmacéutica se administra en forma de una composición oftálmica aplicada por vía tópica en el ojo, o en donde dicho compuesto o dicha composición farmacéutica se administra en forma de una composición oftálmica mediante inyección intravítrea; además, opcionalmente, en donde la composición oftálmica está en forma de colirio.23. El compuesto para el uso según la reivindicación 13, en donde dicho compuesto actúa mediante la inhibición de la trombina y/o la calicreína; opcionalmente, en donde dicho compuesto actúa mediante la inhibición de la calicreína tisular y/o la calicreína plasmática.24. El compuesto para el uso según la reivindicación 13, en donde la cantidad de compuesto administrado es una dosis terapéuticamente eficaz suficiente para alcanzar una concentración inicial del compuesto o su(s) metabolito(s) activo(s) en plasma dentro de un intervalo de 1-10 nM, 10-100 nM, 0,1-1 mM, 1-10 mM, 10 100 mM, 100-200 mM, 200-500 mM o 500-1000 mM, o mayor, o en donde el compuesto tiene actividad inhibidora contra la trombina y/o la calicreína plasmática dentro de un intervalo de 1-10 nM, 10-100 nM, 0,1 1 mM, 1-10 mM, 10-100 mM, 100-200 mM, 200-500 mM o 500-1000 mM, o mayor; opcionalmente, en donde más del 50 % de la concentración inicial del compuesto permanece en el plasma durante 1 hora, 3 horas o más, después de la inyección intravenosa.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562126424P | 2015-02-27 | 2015-02-27 | |
| PCT/US2016/020116 WO2016138532A1 (en) | 2015-02-27 | 2016-02-29 | Substituted pyrazole compounds as serine protease inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2931460T3 true ES2931460T3 (es) | 2022-12-29 |
Family
ID=56789853
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16756553T Active ES2931460T3 (es) | 2015-02-27 | 2016-02-29 | Compuestos de pirazol sustituido como inhibidores de serina proteasa |
Country Status (22)
| Country | Link |
|---|---|
| US (2) | US10532995B2 (es) |
| EP (1) | EP3261639B1 (es) |
| JP (3) | JP2018506563A (es) |
| KR (1) | KR20170122767A (es) |
| CN (1) | CN107405333A (es) |
| AU (2) | AU2016224974B2 (es) |
| BR (1) | BR112017018092A2 (es) |
| CA (1) | CA2977993A1 (es) |
| DK (1) | DK3261639T3 (es) |
| ES (1) | ES2931460T3 (es) |
| HK (1) | HK1246164A1 (es) |
| HR (1) | HRP20221287T1 (es) |
| HU (1) | HUE060104T2 (es) |
| IL (1) | IL253876A0 (es) |
| LT (1) | LT3261639T (es) |
| MX (1) | MX2017011000A (es) |
| PL (1) | PL3261639T3 (es) |
| PT (1) | PT3261639T (es) |
| RU (1) | RU2017131562A (es) |
| SG (2) | SG11201706411YA (es) |
| SI (1) | SI3261639T1 (es) |
| WO (1) | WO2016138532A1 (es) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI636047B (zh) | 2013-08-14 | 2018-09-21 | 英商卡爾維斯塔製藥有限公司 | 雜環衍生物 |
| GB201421085D0 (en) | 2014-11-27 | 2015-01-14 | Kalvista Pharmaceuticals Ltd | New enzyme inhibitors |
| GB201421083D0 (en) | 2014-11-27 | 2015-01-14 | Kalvista Pharmaceuticals Ltd | Enzyme inhibitors |
| EA202191548A1 (ru) | 2015-10-01 | 2021-09-02 | Байокрист Фармасьютикалз, Инк. | Ингибиторы плазменного калликреина человека |
| KR102424709B1 (ko) | 2016-05-31 | 2022-07-22 | 칼비스타 파마슈티컬즈 리미티드 | 혈장 칼리크레인 저해제로서 피라졸 유도체 |
| GB201609603D0 (en) | 2016-06-01 | 2016-07-13 | Kalvista Pharmaceuticals Ltd | Polymorphs of N-[(6-cyano-2-fluoro-3-methoxyphenyl)Methyl]-3-(methoxymethyl)-1-({4-[(2-ox opyridin-1-YL)Methyl]phenyl}methyl)pyrazole-4-carboxamide |
| GB201609607D0 (en) | 2016-06-01 | 2016-07-13 | Kalvista Pharmaceuticals Ltd | Polymorphs of N-(3-Fluoro-4-methoxypyridin-2-yl)methyl)-3-(methoxymethyl)-1-({4-((2-oxopy ridin-1-yl)methyl)phenyl}methyl)pyrazole-4-carboxamide and salts |
| CA3070717A1 (en) | 2017-08-04 | 2019-02-07 | Dyax Corp. | Inhibitors of plasma kallikrein and uses thereof |
| GB201719881D0 (en) | 2017-11-29 | 2018-01-10 | Kalvista Pharmaceuticals Ltd | Solid forms of plasma kallikrein inhibitor and salts thereof |
| PL3716952T3 (pl) | 2017-11-29 | 2022-05-02 | Kalvista Pharmaceuticals Limited | Postacie dawkowania zawierające inhibitor kalikreiny osoczowej |
| HU231206B1 (hu) | 2017-12-15 | 2021-10-28 | Richter Gedeon Nyrt. | Triazolobenzazepinek |
| US11459296B2 (en) | 2018-05-16 | 2022-10-04 | Infex Therapeutics Limited | Antibacterial compounds |
| US11034669B2 (en) | 2018-11-30 | 2021-06-15 | Nuvation Bio Inc. | Pyrrole and pyrazole compounds and methods of use thereof |
| WO2021028645A1 (en) | 2019-08-09 | 2021-02-18 | Kalvista Pharmaceuticals Limited | Plasma kallikrein inhibitors |
| WO2023137469A2 (en) * | 2022-01-13 | 2023-07-20 | Children's Hospital Medical Center | Thrombin and fibrinogen as targets for atopic dermatitis diagnosis and treatment |
Family Cites Families (123)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE627394A (es) | 1962-01-23 | |||
| BE793955A (fr) | 1972-01-15 | 1973-07-12 | Merck Patent Gmbh | Arylpiperazines et leur procede de preparation |
| DE2409753A1 (de) | 1974-03-01 | 1975-09-11 | Basf Ag | Substituierte pyrazole |
| US4256108A (en) | 1977-04-07 | 1981-03-17 | Alza Corporation | Microporous-semipermeable laminated osmotic system |
| US4160452A (en) | 1977-04-07 | 1979-07-10 | Alza Corporation | Osmotic system having laminated wall comprising semipermeable lamina and microporous lamina |
| US4265874A (en) | 1980-04-25 | 1981-05-05 | Alza Corporation | Method of delivering drug with aid of effervescent activity generated in environment of use |
| US4911920A (en) | 1986-07-30 | 1990-03-27 | Alcon Laboratories, Inc. | Sustained release, comfort formulation for glaucoma therapy |
| FR2588189B1 (fr) | 1985-10-03 | 1988-12-02 | Merck Sharp & Dohme | Composition pharmaceutique de type a transition de phase liquide-gel |
| IL82542A0 (en) | 1986-05-23 | 1987-11-30 | Smithkline Beckman Corp | Triazole derivatives,their preparation and pharmaceutical compositions containing them |
| JPH089541B2 (ja) | 1988-03-07 | 1996-01-31 | 三井東圧化学株式会社 | ピラゾール類を主成分とする脳浮腫抑制剤 |
| JP2784925B2 (ja) | 1988-09-07 | 1998-08-13 | 日本農薬株式会社 | 3又は5−フェニルピラゾール類又はその塩及び除草剤 |
| DE69212850T2 (de) | 1991-01-15 | 1997-03-06 | Alcon Lab Inc | Verwendung von Karrageenan in topischen ophthalmologischen Zusammensetzungen |
| US5212162A (en) | 1991-03-27 | 1993-05-18 | Alcon Laboratories, Inc. | Use of combinations gelling polysaccharides and finely divided drug carrier substrates in topical ophthalmic compositions |
| JP3810097B2 (ja) | 1993-01-15 | 2006-08-16 | 明治製菓株式会社 | ピロリジン−2−イルカルボニル複素環式化合物誘導体 |
| CN1061036C (zh) | 1993-11-30 | 2001-01-24 | G·D·瑟尔公司 | 用于治疗炎症的取代的吡唑基苯磺酰胺类化合物 |
| US5466823A (en) | 1993-11-30 | 1995-11-14 | G.D. Searle & Co. | Substituted pyrazolyl benzenesulfonamides |
| US6309853B1 (en) | 1994-08-17 | 2001-10-30 | The Rockfeller University | Modulators of body weight, corresponding nucleic acids and proteins, and diagnostic and therapeutic uses thereof |
| ATE262902T1 (de) | 1994-11-10 | 2004-04-15 | Millennium Pharm Inc | Verwendung von pyrazole verbindungen zur behandlung von glomerulonephritis, krebs, atherosklerose oder restenose |
| US5733955A (en) | 1995-02-10 | 1998-03-31 | The Goodyear Tire & Rubber Company | Asphalt cement modification |
| JP3663522B2 (ja) | 1995-08-12 | 2005-06-22 | 日本農薬株式会社 | 植物生育調節用組成物及びその使用方法 |
| US5756529A (en) | 1995-09-29 | 1998-05-26 | G.D. Searle & Co. | Substituted pyrazolyl benzenesulfonamides for use in veterinary therapies |
| US5739083A (en) | 1995-10-13 | 1998-04-14 | Otsuka Kagaku Kabushiki Kaisha | Pyrazole derivatives and insecticidal compositions containing the derivative as active component |
| US5792761A (en) | 1996-08-12 | 1998-08-11 | Merck & Co., Inc. | Thrombin inhibitors |
| DE19632773A1 (de) | 1996-08-14 | 1998-02-19 | Basf Ag | Neue Thrombininhibitoren |
| GB9622370D0 (en) | 1996-10-28 | 1997-01-08 | Merck Sharp & Dohme | Therapeutic agents |
| US5869500A (en) | 1996-12-13 | 1999-02-09 | Hoffmann-La Roche Inc. | Pyridone compounds useful in treating Alzheimer's disease |
| HUP0000735A3 (en) | 1996-12-23 | 2002-03-28 | Bristol Myers Squibb Pharma Co | Nitrogen containing heteroaromatics as factor xa inhibitors |
| KR20010005578A (ko) | 1997-03-21 | 2001-01-15 | 한스 루돌프 하우스, 헨리테 브룬너, 베아트리체 귄터 | 제초제로서의 피라졸 유도체 |
| US6310049B1 (en) | 1998-08-11 | 2001-10-30 | Nihon Bayer Agrochem K.K. | Nematicidal pyrazoles |
| KR20000047461A (ko) | 1998-12-29 | 2000-07-25 | 성재갑 | 트롬빈 억제제 |
| SE9900070D0 (sv) | 1999-01-13 | 1999-01-13 | Astra Ab | New use |
| AR023510A1 (es) | 1999-04-21 | 2002-09-04 | Astrazeneca Ab | Un equipo de partes, formulacion farmaceutica y uso de un inhibidor de trombina. |
| WO2000071536A1 (en) | 1999-05-20 | 2000-11-30 | E.I. Du Pont De Nemours And Company | Heteroaryloxypyrimidine insecticides and acaricides |
| BR0013143A (pt) | 1999-08-12 | 2002-06-11 | Pharmacia Italia Spa | Derivados de 3 (5) amino pirazol, processo para sua preparação e uso dos mesmos como agentes antitumorais |
| IL148698A0 (en) | 1999-09-17 | 2002-09-12 | Cor Therapeutics Inc | INHIBITORS OF FACTOR Xa |
| US6632815B2 (en) | 1999-09-17 | 2003-10-14 | Millennium Pharmaceuticals, Inc. | Inhibitors of factor Xa |
| US6673820B2 (en) | 1999-12-02 | 2004-01-06 | Novartis Animal Health Us, Inc. | Aminoheterocyclylamides as pesticides and antiparasitic agents |
| WO2002000651A2 (en) | 2000-06-27 | 2002-01-03 | Bristol-Myers Squibb Pharma Company | Factor xa inhibitors |
| JP4316893B2 (ja) | 2001-05-16 | 2009-08-19 | バーテックス ファーマシューティカルズ インコーポレイテッド | Srcおよび他のプロテインキナーゼのインヒビター |
| US6589997B2 (en) | 2001-06-29 | 2003-07-08 | North Shore-Long Island Jewish Health System | Small-molecule modulators of hepatocyte growth factor/scatter factor activities |
| SI21097A (sl) | 2001-12-04 | 2003-06-30 | Univerza V Ljubljani | Inhibitorji trombina |
| EP1509505A2 (en) | 2002-01-23 | 2005-03-02 | Arena Pharmaceuticals, Inc. | SMALL MOLECULE MODULATORS OF THE 5−HT2A SEROTONIN RECEPTOR USEFUL FOR THE PROPHYLAXIS AND TREATMENT OF DISORDERS RELATED THERETO |
| SE0200198D0 (sv) | 2002-01-23 | 2002-01-23 | Astrazeneca Ab | New use |
| JP2003313103A (ja) | 2002-02-20 | 2003-11-06 | Sankyo Agro Kk | 4−アシルアミノピラゾール誘導体を有効成分として含有する農薬 |
| GB0214139D0 (en) | 2002-06-19 | 2002-07-31 | Glaxo Group Ltd | Chemical compounds |
| DE10229070A1 (de) * | 2002-06-28 | 2004-01-15 | Merck Patent Gmbh | Phenylderivate 5 |
| KR20050039846A (ko) | 2002-08-09 | 2005-04-29 | 아스트라제네카 에이비이 | 메타보트로픽 글루타메이트 수용체-5의 조절제로서의옥사디아졸 |
| AU2003274025A1 (en) | 2002-10-17 | 2004-05-04 | Syngenta Participations Ag | Pyridine derivatives useful as herbicides |
| US7192976B2 (en) | 2002-12-21 | 2007-03-20 | Angion Biomedica Corporation | Small molecule modulators of hepatocyte growth factor (scatter factor) activity |
| DE60329529D1 (de) | 2002-12-23 | 2009-11-12 | Sanofi Aventis Deutschland | Pyrazolderivate als faktor-xa-inhibitoren |
| US20060229335A1 (en) | 2002-12-24 | 2006-10-12 | Bradley Teegarden | Diarylamine and arylheteroarylamine pyrazole derivatives as modulators of 5ht2a |
| US20040147561A1 (en) | 2002-12-27 | 2004-07-29 | Wenge Zhong | Pyrid-2-one derivatives and methods of use |
| JP2004231528A (ja) | 2003-01-28 | 2004-08-19 | Sankyo Agro Kk | アミド誘導体 |
| GB0308318D0 (en) | 2003-04-10 | 2003-05-14 | Merck Sharp & Dohme | Therapeutic agents |
| PL1618092T3 (pl) | 2003-05-01 | 2011-02-28 | Bristol Myers Squibb Co | Związki amidopirazolowe z podstawnikami arylowymi jako inhibitory kinazy |
| TW200509910A (en) | 2003-05-02 | 2005-03-16 | Elan Pharm Inc | Substituted pyrazole derivatives and related compounds as bradykinin B1 receptor antagonists |
| US7223780B2 (en) | 2003-05-19 | 2007-05-29 | Sanofi-Aventis Deutschland Gmbh | Triazole-derivatives as blood clotting enzyme factor Xa inhibitors |
| EP1479679A1 (en) | 2003-05-19 | 2004-11-24 | Aventis Pharma Deutschland GmbH | Triazole-derivatives as factor Xa inhibitors |
| US20050065144A1 (en) | 2003-09-08 | 2005-03-24 | Syrrx, Inc. | Dipeptidyl peptidase inhibitors |
| JP4895811B2 (ja) | 2003-09-11 | 2012-03-14 | ケミア,インコーポレイテッド | サイトカイン阻害剤 |
| US20060281803A1 (en) | 2003-09-23 | 2006-12-14 | Lindsley Craig W | Pyrazole modulators of metabotropic glutamate receptors |
| TW200526588A (en) | 2003-11-17 | 2005-08-16 | Smithkline Beecham Corp | Chemical compounds |
| WO2005066162A1 (en) | 2003-12-23 | 2005-07-21 | Human Biomolecular Research Institute | Synthetic compounds and derivatives as modulators of smoking or nicotine ingestion and lung cancer |
| AU2005225471B2 (en) | 2004-03-26 | 2011-05-12 | Methylgene Inc. | Inhibitors of histone deacetylase |
| US7429604B2 (en) | 2004-06-15 | 2008-09-30 | Bristol Myers Squibb Company | Six-membered heterocycles useful as serine protease inhibitors |
| US7235530B2 (en) | 2004-09-27 | 2007-06-26 | Dyax Corporation | Kallikrein inhibitors and anti-thrombolytic agents and uses thereof |
| US8084451B2 (en) | 2005-01-10 | 2011-12-27 | University Of Connecticut | Heteropyrrole analogs acting on cannabinoid receptors |
| WO2006109846A1 (ja) | 2005-04-06 | 2006-10-19 | Takeda Pharmaceutical Company Limited | トリアゾール誘導体およびその用途 |
| GB0507577D0 (en) | 2005-04-14 | 2005-05-18 | Novartis Ag | Organic compounds |
| CN100427481C (zh) | 2005-05-26 | 2008-10-22 | 沈阳化工研究院 | 一种芳基醚类化合物及其制备与应用 |
| WO2007067836A2 (en) | 2005-12-05 | 2007-06-14 | Boehringer Ingelheim International Gmbh | Substituted pyrazole compounds useful as soluble epoxide hydrolase inhibitors |
| US20090118336A1 (en) | 2006-05-03 | 2009-05-07 | Laurent David | Pyrazole derivatives and their use as pi3k inhibitors |
| AU2007257959A1 (en) | 2006-06-09 | 2007-12-21 | Kemia, Inc. | Therapy using cytokine inhibitors |
| AU2007263655A1 (en) * | 2006-06-30 | 2008-01-03 | Astrazeneca Ab | Pyrimidine derivatives useful in the treatment of cancer |
| DE102006032824A1 (de) | 2006-07-14 | 2008-01-17 | Bayer Healthcare Ag | Substituierte Indazole |
| JP2009543842A (ja) | 2006-07-17 | 2009-12-10 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | 心臓血管分野における直接トロンビン阻害薬のための新規適応 |
| JP5322935B2 (ja) | 2006-07-31 | 2013-10-23 | アクティベサイト ファーマシューティカルズ インコーポレイティッド | 血漿カリクレインの阻害薬 |
| WO2008023235A1 (en) | 2006-08-25 | 2008-02-28 | Pfizer Products Inc. | Pyrazole derivatives as anti-platelet and anti-thrombotic agents |
| WO2008062739A1 (fr) | 2006-11-20 | 2008-05-29 | Japan Tobacco Inc. | Pyrazoles et leur utilisation en tant que médicaments |
| WO2008063888A2 (en) | 2006-11-22 | 2008-05-29 | Plexxikon, Inc. | Compounds modulating c-fms and/or c-kit activity and uses therefor |
| WO2008061796A2 (en) | 2006-11-24 | 2008-05-29 | Ac Immune Sa | Compounds for the treatment of diseases associated with amyloid or amyloid-like proteins |
| EP2125778A1 (en) | 2006-12-22 | 2009-12-02 | Millennium Pharmaceuticals, Inc. | Certain pyrazoline derivatives with kinase inhibitory activity |
| UY30892A1 (es) | 2007-02-07 | 2008-09-02 | Smithkline Beckman Corp | Inhibidores de la actividad akt |
| WO2008105383A1 (ja) | 2007-02-26 | 2008-09-04 | Toray Industries, Inc. | ピラゾール誘導体およびその医薬用途 |
| US20090011833A1 (en) * | 2007-07-03 | 2009-01-08 | Seelig Jerald C | Descending Qualification Community Game |
| GB0714129D0 (en) | 2007-07-19 | 2007-08-29 | Smithkline Beecham Corp | compounds |
| AU2008284746A1 (en) | 2007-08-06 | 2009-02-12 | Dainippon Sumitomo Pharma Co., Ltd. | Aminopyrazole amide derivative |
| US8334301B2 (en) | 2007-09-28 | 2012-12-18 | Takeda Pharmaceutical Company Limited | 5-Membered heterocyclic compound |
| WO2009086303A2 (en) | 2007-12-21 | 2009-07-09 | University Of Rochester | Method for altering the lifespan of eukaryotic organisms |
| EP2259679A4 (en) | 2008-01-31 | 2011-09-14 | Joslin Diabetes Ct | METHOD FOR THE TREATMENT OF CALLIQUE CLEANING DISORDERS |
| CN101990433B (zh) | 2008-02-07 | 2014-11-05 | 马萨诸塞眼科耳科诊所 | 提高Atoh1表达的化合物 |
| WO2009140621A2 (en) | 2008-05-15 | 2009-11-19 | Duke University | Compositions and methods relating to heat shock transcription factor activating compounds and targets thereof |
| CL2009001214A1 (es) | 2008-05-19 | 2010-12-31 | Schering Corp | Compuestos derivados de heterociclo, moduladores de serina proteasas composicion fa<rmacaeutica que los comprende; y su uso en el tratamiento de trastornos tromboembolicos |
| JP5775452B2 (ja) | 2008-07-10 | 2015-09-09 | アンジオン バイオメディカ コーポレイション | 肝細胞増殖因子(分散因子)活性の低分子モジュレーターの組成物および方法 |
| TW201004941A (en) | 2008-07-16 | 2010-02-01 | Wyeth Corp | Alpha7 nicotinic acetylcholine receptor inhibitors |
| US8273900B2 (en) | 2008-08-07 | 2012-09-25 | Novartis Ag | Organic compounds |
| WO2010017902A1 (de) | 2008-08-14 | 2010-02-18 | Bayer Cropscience Aktiengesellschaft | Insektizide 4-phenyl-1h-pyrazole |
| WO2010020600A1 (en) | 2008-08-19 | 2010-02-25 | Boehringer Ingelheim International Gmbh | Use of dabigatranetexilate for treating patients with pulmonary hypertension |
| CA2734794A1 (en) | 2008-08-19 | 2010-02-25 | Boehringer Ingelheim International Gmbh | Dabigatran for percutaneous interventional cardiac catheterisation |
| WO2010020601A1 (en) | 2008-08-19 | 2010-02-25 | Boehringer Ingelheim International Gmbh | Dabigatran in tumour therapy |
| AR076753A1 (es) | 2009-05-07 | 2011-07-06 | Gruenenthal Chemie | Derivados de carboxamida y urea aromaticas sustituidas como ligandos del receptor de vanilloides. |
| SG10201502484SA (en) * | 2010-03-30 | 2015-05-28 | Verseon Corp | Multisubstituted aromatic compounds as inhibitors of thrombin |
| CN101851207B (zh) | 2010-06-11 | 2012-01-25 | 扬州康伊尔医药科技有限公司 | 抗病毒化合物中间体1-酰基-吡唑-3-羧酸及其制备方法 |
| ES2571334T3 (es) * | 2010-06-24 | 2016-05-24 | Gilead Sciences Inc | Pirazolo[1,5-a]pirimidinas y -triazinas como agentes antivirales |
| US20130137865A1 (en) | 2010-08-11 | 2013-05-30 | Taisho Pharmaceutical Co., Ltd. | Heteroaryl-pyrazole derivative |
| CN105360131A (zh) | 2010-11-03 | 2016-03-02 | 陶氏益农公司 | 杀虫组合物和与其相关的方法 |
| WO2012065019A2 (en) | 2010-11-12 | 2012-05-18 | Exelixis, Inc. | Pyridopyrimidinone inhibitors of p13k alpha |
| US9169260B2 (en) | 2011-03-22 | 2015-10-27 | Merck Sharp & Dohme Corp. | Amidopyrazole inhibitors of interleukin receptor-associated kinases |
| US8691861B2 (en) | 2011-04-13 | 2014-04-08 | Activesite Pharmaceuticals, Inc. | Prodrugs of inhibitors of plasma kallikrein |
| WO2012154880A1 (en) | 2011-05-09 | 2012-11-15 | Proteostasis Therapeutics, Inc. | Proteostasis regulators for treating cystic fibrosis and other protein misfolding diseases |
| RU2631487C2 (ru) | 2011-05-17 | 2017-09-22 | Плексксикон Инк. | Модуляция киназ и показания к её применению |
| WO2013026914A1 (en) * | 2011-08-25 | 2013-02-28 | F. Hoffmann-La Roche Ag | Serine/threonine pak1 inhibitors |
| WO2013039985A2 (en) | 2011-09-12 | 2013-03-21 | The Johns Hopkins University | Serine protease inhibitors |
| WO2013049591A2 (en) | 2011-09-29 | 2013-04-04 | Verseon Corporation | Dual inhibitor compounds and methods of use thereof |
| CN106117194A (zh) | 2011-12-27 | 2016-11-16 | 铁木医药有限公司 | 可用作sgc刺激剂的2‑苄基、3‑(嘧啶‑2‑基)取代的吡唑类 |
| WO2013111108A1 (en) | 2012-01-27 | 2013-08-01 | Novartis Ag | 5-membered heteroarylcarboxamide derivatives as plasma kallikrein inhibitors |
| CA2876382A1 (en) | 2012-06-14 | 2013-12-19 | Daiichi Sankyo Company, Limited | Piperidinylpyrazolopyridine derivative |
| BR112015016395A2 (pt) * | 2013-01-18 | 2017-07-11 | Hoffmann La Roche | pirazóis 3-substituídos e uso como inibidores de dlk |
| MX2015012867A (es) | 2013-03-15 | 2016-06-10 | Verseon Corp | Compuestos aromaticos multisustituidos como inhibidores de serina proteasa. |
| AU2014238478B2 (en) * | 2013-03-15 | 2018-07-19 | Verseon Corporation | Halogenopyrazoles as inhibitors of thrombin |
| WO2015087995A1 (ja) * | 2013-12-13 | 2015-06-18 | 第一三共株式会社 | シクロアルキル又はヘテロシクリルピラゾロピリジン誘導体 |
| US9670183B2 (en) * | 2014-02-06 | 2017-06-06 | Heptares Therapeutics Limited | Bicyclic AZA compounds as muscarinic M1 receptor and/or M4 receptor agonists |
| EP3368529B1 (en) | 2015-10-27 | 2022-04-06 | Boehringer Ingelheim International GmbH | Heteroarylcarboxamide derivatives as plasma kallikrein inhibitors |
-
2016
- 2016-02-29 CN CN201680011748.9A patent/CN107405333A/zh active Pending
- 2016-02-29 SG SG11201706411YA patent/SG11201706411YA/en unknown
- 2016-02-29 PL PL16756553.0T patent/PL3261639T3/pl unknown
- 2016-02-29 KR KR1020177025488A patent/KR20170122767A/ko not_active Application Discontinuation
- 2016-02-29 PT PT167565530T patent/PT3261639T/pt unknown
- 2016-02-29 RU RU2017131562A patent/RU2017131562A/ru not_active Application Discontinuation
- 2016-02-29 SI SI201631632T patent/SI3261639T1/sl unknown
- 2016-02-29 DK DK16756553.0T patent/DK3261639T3/da active
- 2016-02-29 EP EP16756553.0A patent/EP3261639B1/en active Active
- 2016-02-29 SG SG10201907699YA patent/SG10201907699YA/en unknown
- 2016-02-29 AU AU2016224974A patent/AU2016224974B2/en active Active
- 2016-02-29 HR HRP20221287TT patent/HRP20221287T1/hr unknown
- 2016-02-29 JP JP2017545289A patent/JP2018506563A/ja active Pending
- 2016-02-29 MX MX2017011000A patent/MX2017011000A/es unknown
- 2016-02-29 BR BR112017018092A patent/BR112017018092A2/pt not_active Application Discontinuation
- 2016-02-29 WO PCT/US2016/020116 patent/WO2016138532A1/en active Application Filing
- 2016-02-29 ES ES16756553T patent/ES2931460T3/es active Active
- 2016-02-29 HU HUE16756553A patent/HUE060104T2/hu unknown
- 2016-02-29 US US15/553,540 patent/US10532995B2/en active Active
- 2016-02-29 LT LTEPPCT/US2016/020116T patent/LT3261639T/lt unknown
- 2016-02-29 CA CA2977993A patent/CA2977993A1/en active Pending
-
2017
- 2017-08-07 IL IL253876A patent/IL253876A0/en active IP Right Grant
-
2018
- 2018-05-02 HK HK18105646.7A patent/HK1246164A1/zh unknown
-
2019
- 2019-11-26 US US16/696,817 patent/US20200109132A1/en not_active Abandoned
-
2020
- 2020-01-02 AU AU2020200016A patent/AU2020200016A1/en not_active Abandoned
- 2020-10-23 JP JP2020177836A patent/JP2021020939A/ja active Pending
-
2023
- 2023-01-05 JP JP2023000535A patent/JP2023040150A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| HRP20221287T1 (hr) | 2023-02-03 |
| HUE060104T2 (hu) | 2023-01-28 |
| AU2016224974A1 (en) | 2017-08-31 |
| SI3261639T1 (sl) | 2023-01-31 |
| SG11201706411YA (en) | 2017-09-28 |
| JP2023040150A (ja) | 2023-03-22 |
| DK3261639T3 (da) | 2022-10-31 |
| HK1246164A1 (zh) | 2018-09-07 |
| US20200109132A1 (en) | 2020-04-09 |
| MX2017011000A (es) | 2017-10-20 |
| AU2016224974B2 (en) | 2019-09-26 |
| RU2017131562A (ru) | 2019-03-27 |
| WO2016138532A1 (en) | 2016-09-01 |
| EP3261639B1 (en) | 2022-08-24 |
| PT3261639T (pt) | 2022-11-25 |
| AU2020200016A1 (en) | 2020-02-06 |
| US20180237421A1 (en) | 2018-08-23 |
| JP2018506563A (ja) | 2018-03-08 |
| BR112017018092A2 (pt) | 2018-04-10 |
| PL3261639T3 (pl) | 2022-12-19 |
| US10532995B2 (en) | 2020-01-14 |
| CA2977993A1 (en) | 2016-09-01 |
| LT3261639T (lt) | 2022-11-25 |
| EP3261639A4 (en) | 2018-07-04 |
| RU2017131562A3 (es) | 2019-07-24 |
| SG10201907699YA (en) | 2019-09-27 |
| IL253876A0 (en) | 2017-10-31 |
| KR20170122767A (ko) | 2017-11-06 |
| CN107405333A (zh) | 2017-11-28 |
| JP2021020939A (ja) | 2021-02-18 |
| EP3261639A1 (en) | 2018-01-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2931460T3 (es) | Compuestos de pirazol sustituido como inhibidores de serina proteasa | |
| AU2017258834B2 (en) | Multisubstituted aromatic compounds as serine protease inhibitors | |
| AU2020202167A1 (en) | Pyrazolyl-substituted pyridone compounds as serine protease inhibitors | |
| ES2312472T3 (es) | Compuesto alifatico de cinco miembros de anillo que contiene nitrogeno. | |
| CA3033239A1 (en) | Spiro bicyclic inhibitors of menin-mll interaction | |
| US20160251341A1 (en) | Substituted triazole compounds as serine protease inhibitors | |
| NO324934B1 (no) | Pyrrolo[2,3-d]pyramidinforbindelser som immunosuppresive midler | |
| CA2939326A1 (en) | Condensed 5-oxazolidinone derivative | |
| ES2791749T3 (es) | Halogenopirazoles como inhibidores de la trombina | |
| WO2013049591A2 (en) | Dual inhibitor compounds and methods of use thereof | |
| AU2018377993A1 (en) | Pyrazolopyrimidines having activity against the respiratory syncytial virus (rsv) | |
| MX2007015644A (es) | Compuesto heterociclico. |