ES2900263T3 - Modulador de regulador de conductancia transmembrana de fibrosis quística, composiciones farmacéuticas, métodos de tratamiento y proceso de fabricación del modulador - Google Patents
Modulador de regulador de conductancia transmembrana de fibrosis quística, composiciones farmacéuticas, métodos de tratamiento y proceso de fabricación del modulador Download PDFInfo
- Publication number
- ES2900263T3 ES2900263T3 ES17792230T ES17792230T ES2900263T3 ES 2900263 T3 ES2900263 T3 ES 2900263T3 ES 17792230 T ES17792230 T ES 17792230T ES 17792230 T ES17792230 T ES 17792230T ES 2900263 T3 ES2900263 T3 ES 2900263T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- formula
- salt
- independently selected
- pharmaceutically acceptable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims description 150
- 239000008194 pharmaceutical composition Substances 0.000 title claims description 77
- 108010079245 Cystic Fibrosis Transmembrane Conductance Regulator Proteins 0.000 title description 7
- 102000012605 Cystic Fibrosis Transmembrane Conductance Regulator Human genes 0.000 title description 6
- 238000004519 manufacturing process Methods 0.000 title description 3
- 150000001875 compounds Chemical class 0.000 claims abstract description 392
- 150000003839 salts Chemical class 0.000 claims abstract description 335
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 145
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 89
- 150000002367 halogens Chemical class 0.000 claims abstract description 81
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 39
- 125000001424 substituent group Chemical group 0.000 claims abstract description 39
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 28
- 125000006376 (C3-C10) cycloalkyl group Chemical group 0.000 claims abstract description 25
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 20
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 19
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 12
- 238000006243 chemical reaction Methods 0.000 claims description 149
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 124
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 119
- 201000003883 Cystic fibrosis Diseases 0.000 claims description 95
- NLFBCYMMUAKCPC-KQQUZDAGSA-N ethyl (e)-3-[3-amino-2-cyano-1-[(e)-3-ethoxy-3-oxoprop-1-enyl]sulfanyl-3-oxoprop-1-enyl]sulfanylprop-2-enoate Chemical compound CCOC(=O)\C=C\SC(=C(C#N)C(N)=O)S\C=C\C(=O)OCC NLFBCYMMUAKCPC-KQQUZDAGSA-N 0.000 claims description 44
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 24
- 229910052731 fluorine Inorganic materials 0.000 claims description 24
- 229910052801 chlorine Inorganic materials 0.000 claims description 22
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 21
- 229910052799 carbon Inorganic materials 0.000 claims description 14
- 239000003937 drug carrier Substances 0.000 claims description 14
- 239000003153 chemical reaction reagent Substances 0.000 claims description 10
- 229910052739 hydrogen Inorganic materials 0.000 claims description 9
- 238000005859 coupling reaction Methods 0.000 claims description 7
- 230000008878 coupling Effects 0.000 claims description 6
- 238000010168 coupling process Methods 0.000 claims description 6
- 150000003840 hydrochlorides Chemical class 0.000 claims description 6
- 230000003301 hydrolyzing effect Effects 0.000 claims description 5
- 150000001602 bicycloalkyls Chemical group 0.000 claims description 4
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 claims description 4
- 230000007062 hydrolysis Effects 0.000 claims description 3
- 238000006460 hydrolysis reaction Methods 0.000 claims description 3
- 229910052717 sulfur Inorganic materials 0.000 claims description 3
- 125000004434 sulfur atom Chemical group 0.000 claims description 3
- HTSGKJQDMSTCGS-UHFFFAOYSA-N 1,4-bis(4-chlorophenyl)-2-(4-methylphenyl)sulfonylbutane-1,4-dione Chemical compound C1=CC(C)=CC=C1S(=O)(=O)C(C(=O)C=1C=CC(Cl)=CC=1)CC(=O)C1=CC=C(Cl)C=C1 HTSGKJQDMSTCGS-UHFFFAOYSA-N 0.000 claims 3
- 125000005843 halogen group Chemical group 0.000 abstract description 12
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 abstract description 2
- 235000002639 sodium chloride Nutrition 0.000 description 267
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 233
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 141
- 230000035772 mutation Effects 0.000 description 135
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 134
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 126
- 239000000243 solution Substances 0.000 description 121
- 125000004353 pyrazol-1-yl group Chemical group [H]C1=NN(*)C([H])=C1[H] 0.000 description 120
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 119
- 239000000203 mixture Substances 0.000 description 119
- 239000007787 solid Substances 0.000 description 112
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 97
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 86
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 84
- 229910001868 water Inorganic materials 0.000 description 84
- 235000019439 ethyl acetate Nutrition 0.000 description 82
- 230000014759 maintenance of location Effects 0.000 description 78
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 76
- -1 tricyclic Chemical group 0.000 description 72
- 239000002253 acid Substances 0.000 description 68
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 66
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 60
- 238000003786 synthesis reaction Methods 0.000 description 58
- 239000011541 reaction mixture Substances 0.000 description 57
- 230000015572 biosynthetic process Effects 0.000 description 56
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 55
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 54
- 238000005160 1H NMR spectroscopy Methods 0.000 description 52
- 239000012267 brine Substances 0.000 description 51
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 51
- 239000000047 product Substances 0.000 description 48
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 47
- 239000012071 phase Substances 0.000 description 47
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 46
- 238000003756 stirring Methods 0.000 description 46
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 44
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 44
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 44
- 239000010410 layer Substances 0.000 description 43
- 235000005152 nicotinamide Nutrition 0.000 description 42
- 239000011570 nicotinamide Substances 0.000 description 42
- 230000002829 reductive effect Effects 0.000 description 41
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 41
- 101150029409 CFTR gene Proteins 0.000 description 39
- 206010064571 Gene mutation Diseases 0.000 description 39
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 39
- 229910052938 sodium sulfate Inorganic materials 0.000 description 39
- 235000011152 sodium sulphate Nutrition 0.000 description 39
- 238000007792 addition Methods 0.000 description 38
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 36
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 36
- 239000000725 suspension Substances 0.000 description 36
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 36
- 241000124008 Mammalia Species 0.000 description 33
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 33
- 239000000741 silica gel Substances 0.000 description 33
- 229910002027 silica gel Inorganic materials 0.000 description 33
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 30
- 239000002585 base Substances 0.000 description 30
- 239000012044 organic layer Substances 0.000 description 30
- DRSHXJFUUPIBHX-UHFFFAOYSA-N COc1ccc(cc1)N1N=CC2C=NC(Nc3cc(OC)c(OC)c(OCCCN4CCN(C)CC4)c3)=NC12 Chemical compound COc1ccc(cc1)N1N=CC2C=NC(Nc3cc(OC)c(OC)c(OCCCN4CCN(C)CC4)c3)=NC12 DRSHXJFUUPIBHX-UHFFFAOYSA-N 0.000 description 29
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 29
- 239000000284 extract Substances 0.000 description 29
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 29
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 29
- 239000011734 sodium Substances 0.000 description 29
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 28
- 229910052805 deuterium Inorganic materials 0.000 description 28
- 239000003921 oil Substances 0.000 description 28
- 235000019198 oils Nutrition 0.000 description 28
- 230000032258 transport Effects 0.000 description 28
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 27
- 102200128219 rs75527207 Human genes 0.000 description 25
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 24
- 230000000694 effects Effects 0.000 description 23
- 239000011664 nicotinic acid Substances 0.000 description 23
- 235000001968 nicotinic acid Nutrition 0.000 description 23
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 22
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 21
- 108700028369 Alleles Proteins 0.000 description 19
- 229910000027 potassium carbonate Inorganic materials 0.000 description 19
- 102200128591 rs78655421 Human genes 0.000 description 19
- 238000010438 heat treatment Methods 0.000 description 18
- 238000010992 reflux Methods 0.000 description 18
- 102200128204 rs121909005 Human genes 0.000 description 18
- 229910052708 sodium Inorganic materials 0.000 description 18
- 238000001816 cooling Methods 0.000 description 17
- 235000019441 ethanol Nutrition 0.000 description 17
- 239000000463 material Substances 0.000 description 17
- 108090000623 proteins and genes Proteins 0.000 description 17
- FKLJPTJMIBLJAV-UHFFFAOYSA-N Compound IV Chemical compound O1N=C(C)C=C1CCCCCCCOC1=CC=C(C=2OCCN=2)C=C1 FKLJPTJMIBLJAV-UHFFFAOYSA-N 0.000 description 16
- 101000907783 Homo sapiens Cystic fibrosis transmembrane conductance regulator Proteins 0.000 description 16
- 150000003857 carboxamides Chemical class 0.000 description 16
- 102000056427 human CFTR Human genes 0.000 description 16
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 15
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 15
- 238000004587 chromatography analysis Methods 0.000 description 15
- 238000000746 purification Methods 0.000 description 15
- 102200128203 rs121908755 Human genes 0.000 description 15
- 102200132025 rs150212784 Human genes 0.000 description 15
- 102200132105 rs193922525 Human genes 0.000 description 15
- 102200132035 rs200321110 Human genes 0.000 description 15
- 102200132017 rs267606723 Human genes 0.000 description 15
- 102200132015 rs74503330 Human genes 0.000 description 15
- 102200030785 rs749758687 Human genes 0.000 description 15
- 229940125904 compound 1 Drugs 0.000 description 14
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 14
- 239000000706 filtrate Substances 0.000 description 14
- 150000002500 ions Chemical class 0.000 description 14
- 102200132013 rs121909041 Human genes 0.000 description 14
- 102220020559 rs397508453 Human genes 0.000 description 14
- 239000000523 sample Substances 0.000 description 14
- 239000000377 silicon dioxide Substances 0.000 description 14
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 13
- 238000005481 NMR spectroscopy Methods 0.000 description 13
- 201000010099 disease Diseases 0.000 description 13
- 102200128220 rs121909013 Human genes 0.000 description 13
- 108091006146 Channels Proteins 0.000 description 12
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 12
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 12
- 239000013058 crude material Substances 0.000 description 12
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 12
- AJMDMNYTTSGFEL-LURJTMIESA-N (4s)-2,2,4-trimethylpyrrolidine Chemical compound C[C@@H]1CNC(C)(C)C1 AJMDMNYTTSGFEL-LURJTMIESA-N 0.000 description 11
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 11
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 11
- 239000003814 drug Substances 0.000 description 11
- 238000001914 filtration Methods 0.000 description 11
- 230000006870 function Effects 0.000 description 11
- 230000001965 increasing effect Effects 0.000 description 11
- 102200132008 rs75541969 Human genes 0.000 description 11
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 10
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 10
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 10
- 239000008186 active pharmaceutical agent Substances 0.000 description 10
- 210000004027 cell Anatomy 0.000 description 10
- 238000012217 deletion Methods 0.000 description 10
- 230000037430 deletion Effects 0.000 description 10
- 239000000499 gel Substances 0.000 description 10
- 239000011521 glass Substances 0.000 description 10
- 238000010348 incorporation Methods 0.000 description 10
- 239000002245 particle Substances 0.000 description 10
- 102220020608 rs186045772 Human genes 0.000 description 10
- 102200132034 rs202179988 Human genes 0.000 description 10
- 102200132016 rs34911792 Human genes 0.000 description 10
- 102220020548 rs397508442 Human genes 0.000 description 10
- 102220020628 rs397508537 Human genes 0.000 description 10
- 102200086162 rs61754278 Human genes 0.000 description 10
- 238000010898 silica gel chromatography Methods 0.000 description 10
- 238000010922 spray-dried dispersion Methods 0.000 description 10
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 9
- 239000007864 aqueous solution Substances 0.000 description 9
- 238000004440 column chromatography Methods 0.000 description 9
- 125000000753 cycloalkyl group Chemical group 0.000 description 9
- 229940079593 drug Drugs 0.000 description 9
- 230000009977 dual effect Effects 0.000 description 9
- 238000002451 electron ionisation mass spectrometry Methods 0.000 description 9
- 239000004615 ingredient Substances 0.000 description 9
- 210000004379 membrane Anatomy 0.000 description 9
- 239000012528 membrane Substances 0.000 description 9
- 229910052700 potassium Inorganic materials 0.000 description 9
- 239000011591 potassium Substances 0.000 description 9
- 102220020411 rs397508256 Human genes 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- 239000000126 substance Substances 0.000 description 9
- MSFQEZBRFPAFEX-UHFFFAOYSA-N 4-methoxybenzenesulfonamide Chemical compound COC1=CC=C(S(N)(=O)=O)C=C1 MSFQEZBRFPAFEX-UHFFFAOYSA-N 0.000 description 8
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 8
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 8
- 238000004128 high performance liquid chromatography Methods 0.000 description 8
- 229960004592 isopropanol Drugs 0.000 description 8
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 8
- 238000001565 modulated differential scanning calorimetry Methods 0.000 description 8
- 239000002773 nucleotide Substances 0.000 description 8
- 125000003729 nucleotide group Chemical group 0.000 description 8
- 238000000634 powder X-ray diffraction Methods 0.000 description 8
- 102200132106 rs11971167 Human genes 0.000 description 8
- 102200128182 rs74551128 Human genes 0.000 description 8
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 7
- 125000004429 atom Chemical group 0.000 description 7
- 239000012043 crude product Substances 0.000 description 7
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 7
- 239000007789 gas Substances 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- 230000000269 nucleophilic effect Effects 0.000 description 7
- 235000018102 proteins Nutrition 0.000 description 7
- 102000004169 proteins and genes Human genes 0.000 description 7
- 102200128186 rs121908752 Human genes 0.000 description 7
- 102200128167 rs121908753 Human genes 0.000 description 7
- 102200128612 rs368505753 Human genes 0.000 description 7
- 229920006395 saturated elastomer Polymers 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M sodium chloride Inorganic materials [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 6
- 208000035467 Pancreatic insufficiency Diseases 0.000 description 6
- 150000001450 anions Chemical class 0.000 description 6
- 125000000131 cyclopropyloxy group Chemical group C1(CC1)O* 0.000 description 6
- 238000003818 flash chromatography Methods 0.000 description 6
- 231100000221 frame shift mutation induction Toxicity 0.000 description 6
- 230000037433 frameshift Effects 0.000 description 6
- 238000000338 in vitro Methods 0.000 description 6
- 238000003780 insertion Methods 0.000 description 6
- 230000037431 insertion Effects 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
- 238000010926 purge Methods 0.000 description 6
- 230000002441 reversible effect Effects 0.000 description 6
- 102200128244 rs397508288 Human genes 0.000 description 6
- 238000001228 spectrum Methods 0.000 description 6
- 208000024891 symptom Diseases 0.000 description 6
- KHBQMWCZKVMBLN-UHFFFAOYSA-N Benzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC=C1 KHBQMWCZKVMBLN-UHFFFAOYSA-N 0.000 description 5
- 101100485158 Salmonella typhimurium (strain LT2 / SGSC1412 / ATCC 700720) wzzE gene Proteins 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 238000003556 assay Methods 0.000 description 5
- 239000012298 atmosphere Substances 0.000 description 5
- 230000008859 change Effects 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 239000012065 filter cake Substances 0.000 description 5
- 230000000155 isotopic effect Effects 0.000 description 5
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 5
- 235000019341 magnesium sulphate Nutrition 0.000 description 5
- 230000001404 mediated effect Effects 0.000 description 5
- 101150089110 metN gene Proteins 0.000 description 5
- 150000004702 methyl esters Chemical class 0.000 description 5
- 229960003966 nicotinamide Drugs 0.000 description 5
- 239000012299 nitrogen atmosphere Substances 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 230000009467 reduction Effects 0.000 description 5
- 102200128582 rs113993958 Human genes 0.000 description 5
- 102200128619 rs115545701 Human genes 0.000 description 5
- 102200092599 rs33971270 Human genes 0.000 description 5
- 102200132028 rs78194216 Human genes 0.000 description 5
- 229910000104 sodium hydride Inorganic materials 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- 238000003828 vacuum filtration Methods 0.000 description 5
- 238000005406 washing Methods 0.000 description 5
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 4
- AJPKQSSFYHPYMH-UHFFFAOYSA-N 2,6-dichloropyridine-3-carboxylic acid Chemical compound OC(=O)C1=CC=C(Cl)N=C1Cl AJPKQSSFYHPYMH-UHFFFAOYSA-N 0.000 description 4
- XRPDDDRNQJNHLQ-UHFFFAOYSA-N 2-ethyl-1h-pyrrole Chemical compound CCC1=CC=CN1 XRPDDDRNQJNHLQ-UHFFFAOYSA-N 0.000 description 4
- NIXOWILDQLNWCW-UHFFFAOYSA-N Acrylic acid Chemical compound OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 4
- 108091006515 Anion channels Proteins 0.000 description 4
- 102000037829 Anion channels Human genes 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- 206010016654 Fibrosis Diseases 0.000 description 4
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 4
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 4
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 239000012230 colorless oil Substances 0.000 description 4
- 230000002950 deficient Effects 0.000 description 4
- 238000006073 displacement reaction Methods 0.000 description 4
- 210000000981 epithelium Anatomy 0.000 description 4
- 230000004761 fibrosis Effects 0.000 description 4
- 238000005187 foaming Methods 0.000 description 4
- 238000005227 gel permeation chromatography Methods 0.000 description 4
- 125000005842 heteroatom Chemical group 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 4
- 239000005457 ice water Substances 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- 239000012280 lithium aluminium hydride Substances 0.000 description 4
- 229910052751 metal Inorganic materials 0.000 description 4
- 239000002184 metal Substances 0.000 description 4
- 239000008185 minitablet Substances 0.000 description 4
- 210000003739 neck Anatomy 0.000 description 4
- 239000012074 organic phase Substances 0.000 description 4
- 230000000241 respiratory effect Effects 0.000 description 4
- 102200128243 rs1800098 Human genes 0.000 description 4
- 102200128273 rs1800100 Human genes 0.000 description 4
- 102200146268 rs397516881 Human genes 0.000 description 4
- 102200084783 rs749452002 Human genes 0.000 description 4
- 102200128617 rs75961395 Human genes 0.000 description 4
- 102200128169 rs77932196 Human genes 0.000 description 4
- 102200132108 rs80034486 Human genes 0.000 description 4
- 230000028327 secretion Effects 0.000 description 4
- 159000000000 sodium salts Chemical class 0.000 description 4
- 239000011343 solid material Substances 0.000 description 4
- 229910021653 sulphate ion Inorganic materials 0.000 description 4
- IMNIMPAHZVJRPE-UHFFFAOYSA-N triethylenediamine Chemical compound C1CN2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 description 4
- 239000003039 volatile agent Substances 0.000 description 4
- JMLPZBGYYFXYBO-UHFFFAOYSA-N (5,5-dimethylpyrrolidin-3-yl)methanol Chemical compound CC1(C)CC(CO)CN1 JMLPZBGYYFXYBO-UHFFFAOYSA-N 0.000 description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 3
- GEGLCEMEPSOJRT-UHFFFAOYSA-N 1-methyl-1-prop-2-ynoxycyclopropane Chemical compound CC1(CC1)OCC#C GEGLCEMEPSOJRT-UHFFFAOYSA-N 0.000 description 3
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 3
- ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 2,3-dimethylbutane Chemical group CC(C)C(C)C ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 0.000 description 3
- VGUWZCUCNQXGBU-UHFFFAOYSA-N 3-[(4-methylpiperazin-1-yl)methyl]-5-nitro-1h-indole Chemical compound C1CN(C)CCN1CC1=CNC2=CC=C([N+]([O-])=O)C=C12 VGUWZCUCNQXGBU-UHFFFAOYSA-N 0.000 description 3
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 3
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 3
- 206010011224 Cough Diseases 0.000 description 3
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 239000007868 Raney catalyst Substances 0.000 description 3
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 3
- 229910000564 Raney nickel Inorganic materials 0.000 description 3
- 244000269722 Thea sinensis Species 0.000 description 3
- 240000007313 Tilia cordata Species 0.000 description 3
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 3
- 150000001413 amino acids Chemical group 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 150000001768 cations Chemical class 0.000 description 3
- ZCDOYSPFYFSLEW-UHFFFAOYSA-N chromate(2-) Chemical compound [O-][Cr]([O-])(=O)=O ZCDOYSPFYFSLEW-UHFFFAOYSA-N 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 230000007547 defect Effects 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 238000007876 drug discovery Methods 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 239000003623 enhancer Substances 0.000 description 3
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical class CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 3
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Substances CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 238000010943 off-gassing Methods 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 230000036961 partial effect Effects 0.000 description 3
- 239000000376 reactant Substances 0.000 description 3
- 102200128264 rs139468767 Human genes 0.000 description 3
- 102200128163 rs147422190 Human genes 0.000 description 3
- 102200128210 rs74571530 Human genes 0.000 description 3
- 102200122254 rs764549054 Human genes 0.000 description 3
- 102200128229 rs80055610 Human genes 0.000 description 3
- 239000002002 slurry Substances 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 description 3
- 229940124530 sulfonamide Drugs 0.000 description 3
- 150000003456 sulfonamides Chemical class 0.000 description 3
- 210000004243 sweat Anatomy 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- NJBGXPJITDKQPS-UHFFFAOYSA-N tert-butyl 5-oxo-1h-pyrazole-2-carboxylate Chemical compound CC(C)(C)OC(=O)N1C=CC(O)=N1 NJBGXPJITDKQPS-UHFFFAOYSA-N 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- GUBYTKCYAJWUCF-YFKPBYRVSA-N (2S)-2,4-dimethyl-4-nitropentanoic acid Chemical compound C[C@@H](CC(C)(C)[N+]([O-])=O)C(O)=O GUBYTKCYAJWUCF-YFKPBYRVSA-N 0.000 description 2
- JJXUKXQTFKDOSH-YFKPBYRVSA-N (3S)-3,5,5-trimethylpyrrolidin-2-one Chemical compound C[C@@H]1C(NC(C1)(C)C)=O JJXUKXQTFKDOSH-YFKPBYRVSA-N 0.000 description 2
- ICKWICRCANNIBI-UHFFFAOYSA-N 2,4-di-tert-butylphenol Chemical compound CC(C)(C)C1=CC=C(O)C(C(C)(C)C)=C1 ICKWICRCANNIBI-UHFFFAOYSA-N 0.000 description 2
- IQUSQNGRCXYILE-UHFFFAOYSA-N 2-[1-(trifluoromethyl)cyclopropyl]ethanol Chemical compound FC(C1(CC1)CCO)(F)F IQUSQNGRCXYILE-UHFFFAOYSA-N 0.000 description 2
- YCMLQMDWSXFTIF-UHFFFAOYSA-N 2-methylbenzenesulfonimidic acid Chemical compound CC1=CC=CC=C1S(N)(=O)=O YCMLQMDWSXFTIF-UHFFFAOYSA-N 0.000 description 2
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 2
- FGLBSLMDCBOPQK-UHFFFAOYSA-N 2-nitropropane Chemical compound CC(C)[N+]([O-])=O FGLBSLMDCBOPQK-UHFFFAOYSA-N 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- CRINBBOGNYCAOV-UHFFFAOYSA-N 3-fluorobenzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC(F)=C1 CRINBBOGNYCAOV-UHFFFAOYSA-N 0.000 description 2
- ILNJBIQQAIIMEY-UHFFFAOYSA-N 4-oxo-1h-quinoline-3-carboxylic acid Chemical compound C1=CC=CC2=C(O)C(C(=O)O)=CN=C21 ILNJBIQQAIIMEY-UHFFFAOYSA-N 0.000 description 2
- FLDSMVTWEZKONL-AWEZNQCLSA-N 5,5-dimethyl-N-[(3S)-5-methyl-4-oxo-2,3-dihydro-1,5-benzoxazepin-3-yl]-1,4,7,8-tetrahydrooxepino[4,5-c]pyrazole-3-carboxamide Chemical compound CC1(CC2=C(NN=C2C(=O)N[C@@H]2C(N(C3=C(OC2)C=CC=C3)C)=O)CCO1)C FLDSMVTWEZKONL-AWEZNQCLSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 2
- 102000011045 Chloride Channels Human genes 0.000 description 2
- 108010062745 Chloride Channels Proteins 0.000 description 2
- 241000207199 Citrus Species 0.000 description 2
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- 102220478371 Interphotoreceptor matrix proteoglycan 1_G463V_mutation Human genes 0.000 description 2
- 239000004367 Lipase Substances 0.000 description 2
- 102000004882 Lipase Human genes 0.000 description 2
- 108090001060 Lipase Proteins 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 229910002651 NO3 Inorganic materials 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- UQSIEHVSHVIAPB-UHFFFAOYSA-N [1-(trifluoromethyl)cyclobutyl]methanol Chemical compound OCC1(C(F)(F)F)CCC1 UQSIEHVSHVIAPB-UHFFFAOYSA-N 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000010933 acylation Effects 0.000 description 2
- 238000005917 acylation reaction Methods 0.000 description 2
- 229940024606 amino acid Drugs 0.000 description 2
- 235000001014 amino acid Nutrition 0.000 description 2
- 229920006125 amorphous polymer Polymers 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 238000002306 biochemical method Methods 0.000 description 2
- 239000006172 buffering agent Substances 0.000 description 2
- 102220384479 c.1358T>C Human genes 0.000 description 2
- 102220430005 c.1570T>C Human genes 0.000 description 2
- 102220384478 c.44T>C Human genes 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- NEHMKBQYUWJMIP-UHFFFAOYSA-N chloromethane Chemical compound ClC NEHMKBQYUWJMIP-UHFFFAOYSA-N 0.000 description 2
- 235000020971 citrus fruits Nutrition 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- AIMMVWOEOZMVMS-UHFFFAOYSA-N cyclopropanecarboxamide Chemical compound NC(=O)C1CC1 AIMMVWOEOZMVMS-UHFFFAOYSA-N 0.000 description 2
- 230000034994 death Effects 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 2
- 229940043264 dodecyl sulfate Drugs 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 239000003792 electrolyte Substances 0.000 description 2
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 210000002919 epithelial cell Anatomy 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- PJFXDPZWCVXNQZ-UHFFFAOYSA-N ethyl 3-oxo-1,2-dihydropyrazole-4-carboxylate Chemical compound CCOC(=O)C1=CNNC1=O PJFXDPZWCVXNQZ-UHFFFAOYSA-N 0.000 description 2
- 125000004494 ethyl ester group Chemical group 0.000 description 2
- MMXKVMNBHPAILY-UHFFFAOYSA-N ethyl laurate Chemical compound CCCCCCCCCCCC(=O)OCC MMXKVMNBHPAILY-UHFFFAOYSA-N 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 230000004907 flux Effects 0.000 description 2
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 2
- 229910052737 gold Inorganic materials 0.000 description 2
- 239000010931 gold Substances 0.000 description 2
- 230000005283 ground state Effects 0.000 description 2
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 2
- 125000004464 hydroxyphenyl group Chemical group 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 208000015181 infectious disease Diseases 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 230000005445 isotope effect Effects 0.000 description 2
- 235000019421 lipase Nutrition 0.000 description 2
- 238000011068 loading method Methods 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- 229910000000 metal hydroxide Inorganic materials 0.000 description 2
- 150000004692 metal hydroxides Chemical class 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- CXHHBNMLPJOKQD-UHFFFAOYSA-N methyl hydrogen carbonate Chemical compound COC(O)=O CXHHBNMLPJOKQD-UHFFFAOYSA-N 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 210000003097 mucus Anatomy 0.000 description 2
- APVPOHHVBBYQAV-UHFFFAOYSA-N n-(4-aminophenyl)sulfonyloctadecanamide Chemical compound CCCCCCCCCCCCCCCCCC(=O)NS(=O)(=O)C1=CC=C(N)C=C1 APVPOHHVBBYQAV-UHFFFAOYSA-N 0.000 description 2
- GYHSDCAQMDKVKI-SFHVURJKSA-N n-cyclopropyl-4-hydroxy-n-[(2r)-2-hydroxy-2-phenylpropyl]benzenesulfonamide Chemical compound C([C@@](O)(C)C=1C=CC=CC=1)N(S(=O)(=O)C=1C=CC(O)=CC=1)C1CC1 GYHSDCAQMDKVKI-SFHVURJKSA-N 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N nickel Substances [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical class CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 2
- 229940049964 oleate Drugs 0.000 description 2
- ZQPPMHVWECSIRJ-KTKRTIGZSA-M oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC([O-])=O ZQPPMHVWECSIRJ-KTKRTIGZSA-M 0.000 description 2
- 239000004006 olive oil Substances 0.000 description 2
- 235000008390 olive oil Nutrition 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 230000004783 oxidative metabolism Effects 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000008363 phosphate buffer Substances 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 102200155144 rs121908409 Human genes 0.000 description 2
- 102200128232 rs121908758 Human genes 0.000 description 2
- 102200128252 rs121909008 Human genes 0.000 description 2
- 102200132109 rs121909015 Human genes 0.000 description 2
- 102200128198 rs121909016 Human genes 0.000 description 2
- 102200132029 rs121909019 Human genes 0.000 description 2
- 102200128251 rs121909034 Human genes 0.000 description 2
- 102200132021 rs121909036 Human genes 0.000 description 2
- 102200128217 rs121909044 Human genes 0.000 description 2
- 102220020811 rs138338446 Human genes 0.000 description 2
- 102220020488 rs140455771 Human genes 0.000 description 2
- 102200128256 rs141033578 Human genes 0.000 description 2
- 102220289395 rs141482808 Human genes 0.000 description 2
- 102200075212 rs143010236 Human genes 0.000 description 2
- 102200075393 rs1432273 Human genes 0.000 description 2
- 102200128608 rs1800073 Human genes 0.000 description 2
- 102200128620 rs1800076 Human genes 0.000 description 2
- 102200128255 rs1800110 Human genes 0.000 description 2
- 102200128253 rs1800111 Human genes 0.000 description 2
- 102200132026 rs1800112 Human genes 0.000 description 2
- 102200132006 rs1800120 Human genes 0.000 description 2
- 102220088961 rs189437004 Human genes 0.000 description 2
- 102200128192 rs191456345 Human genes 0.000 description 2
- 102220008545 rs193922500 Human genes 0.000 description 2
- 102200128238 rs201124247 Human genes 0.000 description 2
- 102220020508 rs201386642 Human genes 0.000 description 2
- 102220020540 rs201759207 Human genes 0.000 description 2
- 102200128240 rs201978662 Human genes 0.000 description 2
- 102200042493 rs28909982 Human genes 0.000 description 2
- 102200132009 rs36210737 Human genes 0.000 description 2
- 102200132012 rs36210737 Human genes 0.000 description 2
- 102200018965 rs371518124 Human genes 0.000 description 2
- 102200128170 rs397508146 Human genes 0.000 description 2
- 102200128183 rs397508195 Human genes 0.000 description 2
- 102220020390 rs397508224 Human genes 0.000 description 2
- 102200128209 rs397508225 Human genes 0.000 description 2
- 102200128233 rs397508280 Human genes 0.000 description 2
- 102220020443 rs397508300 Human genes 0.000 description 2
- 102200128241 rs397508306 Human genes 0.000 description 2
- 102220098132 rs397508357 Human genes 0.000 description 2
- 102220020566 rs397508462 Human genes 0.000 description 2
- 102220020591 rs397508498 Human genes 0.000 description 2
- 102220020599 rs397508510 Human genes 0.000 description 2
- 102220020602 rs397508513 Human genes 0.000 description 2
- 102200132011 rs397508531 Human genes 0.000 description 2
- 102220020625 rs397508533 Human genes 0.000 description 2
- 102200132007 rs397508567 Human genes 0.000 description 2
- 102220020672 rs397508602 Human genes 0.000 description 2
- 102200132113 rs397508616 Human genes 0.000 description 2
- 102200128592 rs397508718 Human genes 0.000 description 2
- 102220020767 rs397508721 Human genes 0.000 description 2
- 102200128185 rs397508783 Human genes 0.000 description 2
- 102200144543 rs397515439 Human genes 0.000 description 2
- 102200125616 rs397515481 Human genes 0.000 description 2
- 102200093459 rs397517963 Human genes 0.000 description 2
- 102200131525 rs57045855 Human genes 0.000 description 2
- 102220123600 rs747754623 Human genes 0.000 description 2
- 102200132111 rs77902683 Human genes 0.000 description 2
- 102200128588 rs78655421 Human genes 0.000 description 2
- 102200132033 rs78769542 Human genes 0.000 description 2
- 102200132030 rs79635528 Human genes 0.000 description 2
- 102200128599 rs80282562 Human genes 0.000 description 2
- 102220220450 rs80358570 Human genes 0.000 description 2
- 239000012047 saturated solution Substances 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 239000007962 solid dispersion Substances 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 238000005507 spraying Methods 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 230000029305 taxis Effects 0.000 description 2
- FQFILJKFZCVHNH-UHFFFAOYSA-N tert-butyl n-[3-[(5-bromo-2-chloropyrimidin-4-yl)amino]propyl]carbamate Chemical compound CC(C)(C)OC(=O)NCCCNC1=NC(Cl)=NC=C1Br FQFILJKFZCVHNH-UHFFFAOYSA-N 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 125000004205 trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 2
- ONDSBJMLAHVLMI-UHFFFAOYSA-N trimethylsilyldiazomethane Chemical compound C[Si](C)(C)[CH-][N+]#N ONDSBJMLAHVLMI-UHFFFAOYSA-N 0.000 description 2
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- SNICXCGAKADSCV-JTQLQIEISA-N (-)-Nicotine Chemical compound CN1CCC[C@H]1C1=CC=CN=C1 SNICXCGAKADSCV-JTQLQIEISA-N 0.000 description 1
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 1
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 1
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 1
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- UKRZNDDXXIOZJH-UHFFFAOYSA-N (2,4-ditert-butyl-5-nitrophenyl) hydrogen carbonate Chemical compound CC(C)(C)C1=CC(C(C)(C)C)=C([N+]([O-])=O)C=C1OC(O)=O UKRZNDDXXIOZJH-UHFFFAOYSA-N 0.000 description 1
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 1
- AJMDMNYTTSGFEL-ZCFIWIBFSA-N (4R)-2,2,4-trimethylpyrrolidine Chemical compound CC1(NC[C@@H](C1)C)C AJMDMNYTTSGFEL-ZCFIWIBFSA-N 0.000 description 1
- AJMDMNYTTSGFEL-FYFSCIFKSA-N (4S)-2,2-dimethyl-4-(trideuteriomethyl)pyrrolidine Chemical compound [2H]C([2H])([2H])[C@@H]1CNC(C)(C)C1 AJMDMNYTTSGFEL-FYFSCIFKSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- STDJRYDBFFDKCR-UHFFFAOYSA-N 1,2,2-trimethylpyrrolidine Chemical compound CN1CCCC1(C)C STDJRYDBFFDKCR-UHFFFAOYSA-N 0.000 description 1
- SCZNXLWKYFICFV-UHFFFAOYSA-N 1,2,3,4,5,7,8,9-octahydropyrido[1,2-b]diazepine Chemical compound C1CCCNN2CCCC=C21 SCZNXLWKYFICFV-UHFFFAOYSA-N 0.000 description 1
- FTTATHOUSOIFOQ-UHFFFAOYSA-N 1,2,3,4,6,7,8,8a-octahydropyrrolo[1,2-a]pyrazine Chemical compound C1NCCN2CCCC21 FTTATHOUSOIFOQ-UHFFFAOYSA-N 0.000 description 1
- LZDKZFUFMNSQCJ-UHFFFAOYSA-N 1,2-diethoxyethane Chemical compound CCOCCOCC LZDKZFUFMNSQCJ-UHFFFAOYSA-N 0.000 description 1
- IMNIMPAHZVJRPE-UHFFFAOYSA-P 1,4-diazoniabicyclo[2.2.2]octane Chemical compound C1C[NH+]2CC[NH+]1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-P 0.000 description 1
- ZFPGARUNNKGOBB-UHFFFAOYSA-N 1-Ethyl-2-pyrrolidinone Chemical compound CCN1CCCC1=O ZFPGARUNNKGOBB-UHFFFAOYSA-N 0.000 description 1
- NCTCZGRRDXIGIY-UHFFFAOYSA-N 1-methylcyclopropan-1-ol Chemical compound CC1(O)CC1 NCTCZGRRDXIGIY-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- KWTSXDURSIMDCE-UHFFFAOYSA-N 1-phenylpropan-2-amine Chemical compound CC(N)CC1=CC=CC=C1 KWTSXDURSIMDCE-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- WTMJHBZSSSDBFQ-UHFFFAOYSA-N 2,3,4-trimethyl-1h-pyrrole Chemical compound CC1=CNC(C)=C1C WTMJHBZSSSDBFQ-UHFFFAOYSA-N 0.000 description 1
- CHHHXKFHOYLYRE-UHFFFAOYSA-M 2,4-Hexadienoic acid, potassium salt (1:1), (2E,4E)- Chemical compound [K+].CC=CC=CC([O-])=O CHHHXKFHOYLYRE-UHFFFAOYSA-M 0.000 description 1
- GUBYTKCYAJWUCF-UHFFFAOYSA-N 2,4-dimethyl-4-nitropentanoic acid Chemical compound OC(=O)C(C)CC(C)(C)[N+]([O-])=O GUBYTKCYAJWUCF-UHFFFAOYSA-N 0.000 description 1
- IZJAXMGHAFLFMH-UHFFFAOYSA-N 2-(anilinomethylidene)propanedioic acid Chemical compound OC(=O)C(C(O)=O)=CNC1=CC=CC=C1 IZJAXMGHAFLFMH-UHFFFAOYSA-N 0.000 description 1
- SLUFXNGFJSNQMD-UHFFFAOYSA-N 2-(hydroxymethyl)-4-methyl-4-nitropentanoic acid Chemical compound OCC(C(=O)O)CC(C)([N+](=O)[O-])C SLUFXNGFJSNQMD-UHFFFAOYSA-N 0.000 description 1
- SLCITTDQGXZACP-VIFPVBQESA-N 2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide Chemical compound CC1(N(C[C@H](C1)C)C1=NC=CC=C1C(=O)N)C SLCITTDQGXZACP-VIFPVBQESA-N 0.000 description 1
- JWVHOPCZDUVEAV-UHFFFAOYSA-N 2-[1-(trifluoromethyl)cyclopropyl]acetic acid Chemical compound OC(=O)CC1(C(F)(F)F)CC1 JWVHOPCZDUVEAV-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 1
- KQCZCINJGIRLCD-UHFFFAOYSA-N 2-bromo-n-[4-chloro-3-(1-methylpyrrolidin-3-yl)oxyphenyl]-4,5-dimethoxybenzenesulfonamide Chemical compound C1=C(OC)C(OC)=CC(Br)=C1S(=O)(=O)NC1=CC=C(Cl)C(OC2CN(C)CC2)=C1 KQCZCINJGIRLCD-UHFFFAOYSA-N 0.000 description 1
- LDLCZOVUSADOIV-UHFFFAOYSA-N 2-bromoethanol Chemical compound OCCBr LDLCZOVUSADOIV-UHFFFAOYSA-N 0.000 description 1
- WFLBWYLZCQOPCA-UHFFFAOYSA-N 2-fluorobenzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC=C1F WFLBWYLZCQOPCA-UHFFFAOYSA-N 0.000 description 1
- 102100027324 2-hydroxyacyl-CoA lyase 1 Human genes 0.000 description 1
- GQUFSEOISDLWMN-UHFFFAOYSA-N 2-methoxy-4-methylbenzenesulfonamide Chemical compound COC1=CC(C)=CC=C1S(N)(=O)=O GQUFSEOISDLWMN-UHFFFAOYSA-N 0.000 description 1
- YRZWAACJIAJKCD-UHFFFAOYSA-N 2-methyl-4-nitropentanoic acid Chemical compound OC(=O)C(C)CC(C)[N+]([O-])=O YRZWAACJIAJKCD-UHFFFAOYSA-N 0.000 description 1
- ZOQUFUUTAALPJD-UHFFFAOYSA-N 2-nitropentanoic acid Chemical compound CCCC(C(O)=O)[N+]([O-])=O ZOQUFUUTAALPJD-UHFFFAOYSA-N 0.000 description 1
- ZSLUVFAKFWKJRC-IGMARMGPSA-N 232Th Chemical compound [232Th] ZSLUVFAKFWKJRC-IGMARMGPSA-N 0.000 description 1
- JVLFMTZUPSBCNJ-UHFFFAOYSA-N 3,5-difluoropyridin-2-amine Chemical compound NC1=NC=C(F)C=C1F JVLFMTZUPSBCNJ-UHFFFAOYSA-N 0.000 description 1
- BJCFKHYEUXXLJQ-UHFFFAOYSA-N 3-(hydroxymethyl)-5,5-dimethylpyrrolidin-2-one Chemical compound OCC1C(NC(C1)(C)C)=O BJCFKHYEUXXLJQ-UHFFFAOYSA-N 0.000 description 1
- LOODSWOUDUWCDB-UHFFFAOYSA-N 3-[3-(2-carboxyethyl)-2,4,5,6-tetramethylphenyl]propanoic acid Chemical compound CC1=C(C)C(CCC(O)=O)=C(C)C(CCC(O)=O)=C1C LOODSWOUDUWCDB-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- ZQDPJFUHLCOCRG-UHFFFAOYSA-N 3-hexene Chemical group CCC=CCC ZQDPJFUHLCOCRG-UHFFFAOYSA-N 0.000 description 1
- VBKIEQKVSHDVGH-UHFFFAOYSA-N 3-methoxybenzenesulfonamide Chemical compound COC1=CC=CC(S(N)(=O)=O)=C1 VBKIEQKVSHDVGH-UHFFFAOYSA-N 0.000 description 1
- RTZZCYNQPHTPPL-UHFFFAOYSA-N 3-nitrophenol Chemical compound OC1=CC=CC([N+]([O-])=O)=C1 RTZZCYNQPHTPPL-UHFFFAOYSA-N 0.000 description 1
- UZVAVNYJJAUYRH-UHFFFAOYSA-N 3-oxo-1,2-dihydropyrazole-4-carboxylic acid Chemical compound OC(=O)C1=CNNC1=O UZVAVNYJJAUYRH-UHFFFAOYSA-N 0.000 description 1
- IMBBXSASDSZJSX-UHFFFAOYSA-N 4-Carboxypyrazole Chemical compound OC(=O)C=1C=NNC=1 IMBBXSASDSZJSX-UHFFFAOYSA-N 0.000 description 1
- UZECCNDOASGYNH-UHFFFAOYSA-N 4-cyanobenzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=C(C#N)C=C1 UZECCNDOASGYNH-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- UZFMOKQJFYMBGY-UHFFFAOYSA-N 4-hydroxy-TEMPO Chemical compound CC1(C)CC(O)CC(C)(C)N1[O] UZFMOKQJFYMBGY-UHFFFAOYSA-N 0.000 description 1
- DIRCLGLKRZLKHG-UHFFFAOYSA-N 4-hydroxybenzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=C(O)C=C1 DIRCLGLKRZLKHG-UHFFFAOYSA-N 0.000 description 1
- QXAUPUZBANXHAK-UHFFFAOYSA-N 5-oxo-1h-pyrazole-2-carboxylic acid Chemical compound OC(=O)N1C=CC(=O)N1 QXAUPUZBANXHAK-UHFFFAOYSA-N 0.000 description 1
- USQCUKQZXOWUDF-YWZLYKJASA-N 6-chloro-n-[(3s)-1-[(2s)-1-(4-methyl-5-oxo-1,4-diazepan-1-yl)-1-oxopropan-2-yl]-2-oxopyrrolidin-3-yl]naphthalene-2-sulfonamide Chemical compound O=C([C@@H](N1C([C@@H](NS(=O)(=O)C=2C=C3C=CC(Cl)=CC3=CC=2)CC1)=O)C)N1CCN(C)C(=O)CC1 USQCUKQZXOWUDF-YWZLYKJASA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 240000002234 Allium sativum Species 0.000 description 1
- FBEHFRAORPEGFH-UHFFFAOYSA-N Allyxycarb Chemical compound CNC(=O)OC1=CC(C)=C(N(CC=C)CC=C)C(C)=C1 FBEHFRAORPEGFH-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 238000012935 Averaging Methods 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- AFUFJPXCBRPOPS-UHFFFAOYSA-N CC1(C(C1(C)C)CON2C=CC=N2)C Chemical compound CC1(C(C1(C)C)CON2C=CC=N2)C AFUFJPXCBRPOPS-UHFFFAOYSA-N 0.000 description 1
- BQXUPNKLZNSUMC-YUQWMIPFSA-N CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 Chemical compound CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 BQXUPNKLZNSUMC-YUQWMIPFSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- 244000223760 Cinnamomum zeylanicum Species 0.000 description 1
- 235000008733 Citrus aurantifolia Nutrition 0.000 description 1
- 229940126657 Compound 17 Drugs 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 102000034534 Cotransporters Human genes 0.000 description 1
- 108020003264 Cotransporters Proteins 0.000 description 1
- 244000110556 Cyclopia subternata Species 0.000 description 1
- LVZWSLJZHVFIQJ-UHFFFAOYSA-N Cyclopropane Chemical compound C1CC1 LVZWSLJZHVFIQJ-UHFFFAOYSA-N 0.000 description 1
- 241000065675 Cyclops Species 0.000 description 1
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 1
- GUUVPOWQJOLRAS-UHFFFAOYSA-N Diphenyl disulfide Chemical compound C=1C=CC=CC=1SSC1=CC=CC=C1 GUUVPOWQJOLRAS-UHFFFAOYSA-N 0.000 description 1
- 101100346656 Drosophila melanogaster strat gene Proteins 0.000 description 1
- 208000000059 Dyspnea Diseases 0.000 description 1
- 206010013975 Dyspnoeas Diseases 0.000 description 1
- 241001331845 Equus asinus x caballus Species 0.000 description 1
- 240000006890 Erythroxylum coca Species 0.000 description 1
- 101100322888 Escherichia coli (strain K12) metL gene Proteins 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- KIWBPDUYBMNFTB-UHFFFAOYSA-N Ethyl hydrogen sulfate Chemical compound CCOS(O)(=O)=O KIWBPDUYBMNFTB-UHFFFAOYSA-N 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 241000463109 Haloprofundus marisrubri Species 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101001009252 Homo sapiens 2-hydroxyacyl-CoA lyase 1 Proteins 0.000 description 1
- 101100166894 Homo sapiens CFTR gene Proteins 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- 108090000862 Ion Channels Proteins 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 229910010199 LiAl Inorganic materials 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 1
- 241000145847 Moria Species 0.000 description 1
- 108010021466 Mutant Proteins Proteins 0.000 description 1
- 102000008300 Mutant Proteins Human genes 0.000 description 1
- 108010052185 Myotonin-Protein Kinase Proteins 0.000 description 1
- 102100022437 Myotonin-protein kinase Human genes 0.000 description 1
- NUGPIZCTELGDOS-QHCPKHFHSA-N N-[(1S)-3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-pyridin-3-ylpropyl]cyclopentanecarboxamide Chemical compound C(C)(C)C1=NN=C(N1C1CCN(CC1)CC[C@@H](C=1C=NC=CC=1)NC(=O)C1CCCC1)C NUGPIZCTELGDOS-QHCPKHFHSA-N 0.000 description 1
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 1
- LIMFPAAAIVQRRD-BCGVJQADSA-N N-[2-[(3S,4R)-3-fluoro-4-methoxypiperidin-1-yl]pyrimidin-4-yl]-8-[(2R,3S)-2-methyl-3-(methylsulfonylmethyl)azetidin-1-yl]-5-propan-2-ylisoquinolin-3-amine Chemical compound F[C@H]1CN(CC[C@H]1OC)C1=NC=CC(=N1)NC=1N=CC2=C(C=CC(=C2C=1)C(C)C)N1[C@@H]([C@H](C1)CS(=O)(=O)C)C LIMFPAAAIVQRRD-BCGVJQADSA-N 0.000 description 1
- LFZAGIJXANFPFN-UHFFFAOYSA-N N-[3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-thiophen-2-ylpropyl]acetamide Chemical compound C(C)(C)C1=NN=C(N1C1CCN(CC1)CCC(C=1SC=CC=1)NC(C)=O)C LFZAGIJXANFPFN-UHFFFAOYSA-N 0.000 description 1
- JVFWBZSKHJQVEH-UHFFFAOYSA-N N1C(=C(C=C1)C(=O)OC(C)C)C(=O)OC(C)C Chemical compound N1C(=C(C=C1)C(=O)OC(C)C)C(=O)OC(C)C JVFWBZSKHJQVEH-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- IOVCWXUNBOPUCH-UHFFFAOYSA-M Nitrite anion Chemical compound [O-]N=O IOVCWXUNBOPUCH-UHFFFAOYSA-M 0.000 description 1
- YZZVIWCCFCTJEX-UHFFFAOYSA-N O.O.O.O.O.O.O.O.O.O.[Na] Chemical compound O.O.O.O.O.O.O.O.O.O.[Na] YZZVIWCCFCTJEX-UHFFFAOYSA-N 0.000 description 1
- LOTSWFGMBUDZBP-UHFFFAOYSA-N OCN1C(CCC1(C)C)=O Chemical compound OCN1C(CCC1(C)C)=O LOTSWFGMBUDZBP-UHFFFAOYSA-N 0.000 description 1
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium on carbon Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 1
- 206010033645 Pancreatitis Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- SCKXCAADGDQQCS-UHFFFAOYSA-N Performic acid Chemical compound OOC=O SCKXCAADGDQQCS-UHFFFAOYSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 102000007327 Protamines Human genes 0.000 description 1
- 108010007568 Protamines Proteins 0.000 description 1
- 241000287531 Psittacidae Species 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 1
- 241000235402 Rhizomucor Species 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- 244000000231 Sesamum indicum Species 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- RAHZWNYVWXNFOC-UHFFFAOYSA-N Sulphur dioxide Chemical compound O=S=O RAHZWNYVWXNFOC-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- 229910052776 Thorium Inorganic materials 0.000 description 1
- 235000011941 Tilia x europaea Nutrition 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 102100040106 Vesicle transport protein USE1 Human genes 0.000 description 1
- 108010046377 Whey Proteins Proteins 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- NJVHJTQSGGRHGP-UHFFFAOYSA-K [Li].[Al+3].[Cl-].[Cl-].[Cl-] Chemical compound [Li].[Al+3].[Cl-].[Cl-].[Cl-] NJVHJTQSGGRHGP-UHFFFAOYSA-K 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- ZUAAPNNKRHMPKG-UHFFFAOYSA-N acetic acid;butanedioic acid;methanol;propane-1,2-diol Chemical compound OC.CC(O)=O.CC(O)CO.OC(=O)CCC(O)=O ZUAAPNNKRHMPKG-UHFFFAOYSA-N 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 230000002862 amidating effect Effects 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 239000000728 ammonium alginate Substances 0.000 description 1
- 235000010407 ammonium alginate Nutrition 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000002924 anti-infective effect Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229960005475 antiinfective agent Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 1
- 125000005228 aryl sulfonate group Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 1
- 125000005604 azodicarboxylate group Chemical group 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- KXDAEFPNCMNJSK-UHFFFAOYSA-N benzene carboxamide Natural products NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- KGNDCEVUMONOKF-UGPLYTSKSA-N benzyl n-[(2r)-1-[(2s,4r)-2-[[(2s)-6-amino-1-(1,3-benzoxazol-2-yl)-1,1-dihydroxyhexan-2-yl]carbamoyl]-4-[(4-methylphenyl)methoxy]pyrrolidin-1-yl]-1-oxo-4-phenylbutan-2-yl]carbamate Chemical compound C1=CC(C)=CC=C1CO[C@H]1CN(C(=O)[C@@H](CCC=2C=CC=CC=2)NC(=O)OCC=2C=CC=CC=2)[C@H](C(=O)N[C@@H](CCCCN)C(O)(O)C=2OC3=CC=CC=C3N=2)C1 KGNDCEVUMONOKF-UGPLYTSKSA-N 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 230000002146 bilateral effect Effects 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 238000010504 bond cleavage reaction Methods 0.000 description 1
- 150000001649 bromium compounds Chemical group 0.000 description 1
- 229940124630 bronchodilator Drugs 0.000 description 1
- 239000000168 bronchodilator agent Substances 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 210000001217 buttock Anatomy 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 102220355060 c.1546C>G Human genes 0.000 description 1
- 102220347284 c.64G>A Human genes 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 150000004651 carbonic acid esters Chemical class 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 239000012159 carrier gas Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 238000004296 chiral HPLC Methods 0.000 description 1
- 239000012320 chlorinating reagent Substances 0.000 description 1
- 150000001805 chlorine compounds Chemical group 0.000 description 1
- 235000017803 cinnamon Nutrition 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 235000008957 cocaer Nutrition 0.000 description 1
- ZPUCINDJVBIVPJ-LJISPDSOSA-N cocaine Chemical compound O([C@H]1C[C@@H]2CC[C@@H](N2C)[C@H]1C(=O)OC)C(=O)C1=CC=CC=C1 ZPUCINDJVBIVPJ-LJISPDSOSA-N 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 229940125758 compound 15 Drugs 0.000 description 1
- 229940125833 compound 23 Drugs 0.000 description 1
- 229940125961 compound 24 Drugs 0.000 description 1
- 238000013329 compounding Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 239000010779 crude oil Substances 0.000 description 1
- 239000002178 crystalline material Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- YMGUBTXCNDTFJI-UHFFFAOYSA-N cyclopropanecarboxylic acid Chemical compound OC(=O)C1CC1 YMGUBTXCNDTFJI-UHFFFAOYSA-N 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 150000001975 deuterium Chemical group 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- AQEFLFZSWDEAIP-UHFFFAOYSA-N di-tert-butyl ether Chemical compound CC(C)(C)OC(C)(C)C AQEFLFZSWDEAIP-UHFFFAOYSA-N 0.000 description 1
- 239000012973 diazabicyclooctane Substances 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 235000020930 dietary requirements Nutrition 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 230000001079 digestive effect Effects 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- 235000019797 dipotassium phosphate Nutrition 0.000 description 1
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical class [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000004815 dispersion polymer Substances 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- 238000007337 electrophilic addition reaction Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000010931 ester hydrolysis Methods 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- GCFHZZWXZLABBL-UHFFFAOYSA-N ethanol;hexane Chemical compound CCO.CCCCCC GCFHZZWXZLABBL-UHFFFAOYSA-N 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- DQYBDCGIPTYXML-UHFFFAOYSA-N ethoxyethane;hydrate Chemical compound O.CCOCC DQYBDCGIPTYXML-UHFFFAOYSA-N 0.000 description 1
- KKYBVLDQTWQETN-UHFFFAOYSA-N ethyl 2,6-dichloropyridine-3-carboxylate Chemical compound CCOC(=O)C1=CC=C(Cl)N=C1Cl KKYBVLDQTWQETN-UHFFFAOYSA-N 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 239000003172 expectorant agent Substances 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 230000035558 fertility Effects 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 1
- 235000004611 garlic Nutrition 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- TZBJGXHYKVUXJN-UHFFFAOYSA-N genistein Natural products C1=CC(O)=CC=C1C1=COC2=CC(O)=CC(O)=C2C1=O TZBJGXHYKVUXJN-UHFFFAOYSA-N 0.000 description 1
- 238000003205 genotyping method Methods 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 229960002449 glycine Drugs 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- KWHDXJHBFYQOTK-UHFFFAOYSA-N heptane;toluene Chemical compound CCCCCCC.CC1=CC=CC=C1 KWHDXJHBFYQOTK-UHFFFAOYSA-N 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- 239000002044 hexane fraction Substances 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 230000001744 histochemical effect Effects 0.000 description 1
- 239000012456 homogeneous solution Substances 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- ZTUJDPKOHPKRMO-UHFFFAOYSA-N hydron;2,2,2-trifluoroethanamine;chloride Chemical compound Cl.NCC(F)(F)F ZTUJDPKOHPKRMO-UHFFFAOYSA-N 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 1
- 208000021267 infertility disease Diseases 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 230000037427 ion transport Effects 0.000 description 1
- KQNPFQTWMSNSAP-UHFFFAOYSA-M isobutyrate Chemical compound CC(C)C([O-])=O KQNPFQTWMSNSAP-UHFFFAOYSA-M 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000004816 latex Substances 0.000 description 1
- 229920000126 latex Polymers 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 239000004571 lime Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 230000003908 liver function Effects 0.000 description 1
- 235000001055 magnesium Nutrition 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- OBGLKIIPIZLFRH-UHFFFAOYSA-K magnesium sodium hydrogen carbonate Chemical compound [Na+].[Mg+2].OC([O-])=O.OC([O-])=O.OC([O-])=O OBGLKIIPIZLFRH-UHFFFAOYSA-K 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- CKJNUZNMWOVDFN-UHFFFAOYSA-N methanone Chemical compound O=[CH-] CKJNUZNMWOVDFN-UHFFFAOYSA-N 0.000 description 1
- AUTCCPQKLPMHDN-ONEGZZNKSA-N methyl (e)-3-methoxyprop-2-enoate Chemical compound CO\C=C\C(=O)OC AUTCCPQKLPMHDN-ONEGZZNKSA-N 0.000 description 1
- RFUCOAQWQVDBEU-UHFFFAOYSA-N methyl 2-(hydroxymethyl)prop-2-enoate Chemical class COC(=O)C(=C)CO RFUCOAQWQVDBEU-UHFFFAOYSA-N 0.000 description 1
- VUQUOGPMUUJORT-UHFFFAOYSA-N methyl 4-methylbenzenesulfonate Chemical compound COS(=O)(=O)C1=CC=C(C)C=C1 VUQUOGPMUUJORT-UHFFFAOYSA-N 0.000 description 1
- XMJHPCRAQCTCFT-UHFFFAOYSA-N methyl chloroformate Chemical compound COC(Cl)=O XMJHPCRAQCTCFT-UHFFFAOYSA-N 0.000 description 1
- QNPUPNARQKQABL-UHFFFAOYSA-N methyl pyrazole-1-carboxylate Chemical compound COC(=O)N1C=CC=N1 QNPUPNARQKQABL-UHFFFAOYSA-N 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 238000005065 mining Methods 0.000 description 1
- 229940111688 monobasic potassium phosphate Drugs 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- 229940066491 mucolytics Drugs 0.000 description 1
- PURKAOJPTOLRMP-ASMGOKTBSA-N n-[2-tert-butyl-4-[1,1,1,3,3,3-hexadeuterio-2-(trideuteriomethyl)propan-2-yl]-5-hydroxyphenyl]-4-oxo-1h-quinoline-3-carboxamide Chemical compound C1=C(O)C(C(C([2H])([2H])[2H])(C([2H])([2H])[2H])C([2H])([2H])[2H])=CC(C(C)(C)C)=C1NC(=O)C1=CNC2=CC=CC=C2C1=O PURKAOJPTOLRMP-ASMGOKTBSA-N 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- GFAGHCPSFAXPDV-UHFFFAOYSA-N n-oxo-1,2-dihydroquinoline-3-carboxamide Chemical compound C1=CC=C2NCC(C(=O)N=O)=CC2=C1 GFAGHCPSFAXPDV-UHFFFAOYSA-N 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-M nicotinate Chemical compound [O-]C(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-M 0.000 description 1
- 229960002715 nicotine Drugs 0.000 description 1
- SNICXCGAKADSCV-UHFFFAOYSA-N nicotine Natural products CN1CCCC1C1=CC=CN=C1 SNICXCGAKADSCV-UHFFFAOYSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 230000008518 non respiratory effect Effects 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- 238000001208 nuclear magnetic resonance pulse sequence Methods 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 238000007344 nucleophilic reaction Methods 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 230000004203 pancreatic function Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- DYUMLJSJISTVPV-UHFFFAOYSA-N phenyl propanoate Chemical compound CCC(=O)OC1=CC=CC=C1 DYUMLJSJISTVPV-UHFFFAOYSA-N 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- GNSKLFRGEWLPPA-UHFFFAOYSA-M potassium dihydrogen phosphate Chemical compound [K+].OP(O)([O-])=O GNSKLFRGEWLPPA-UHFFFAOYSA-M 0.000 description 1
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical compound [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 1
- 239000004302 potassium sorbate Substances 0.000 description 1
- 235000010241 potassium sorbate Nutrition 0.000 description 1
- 229940069338 potassium sorbate Drugs 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 230000036515 potency Effects 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229950008679 protamine sulfate Drugs 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 239000008213 purified water Substances 0.000 description 1
- WHYQCQRHPQBZHM-UHFFFAOYSA-N pyrazole-1-carboxylic acid Chemical compound OC(=O)N1C=CC=N1 WHYQCQRHPQBZHM-UHFFFAOYSA-N 0.000 description 1
- BGCSUJHSGHKXKF-UHFFFAOYSA-N pyrrole-1,3-dicarboxylic acid Chemical compound OC(=O)C=1C=CN(C(O)=O)C=1 BGCSUJHSGHKXKF-UHFFFAOYSA-N 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 238000005956 quaternization reaction Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- PMZDQRJGMBOQBF-UHFFFAOYSA-N quinolin-4-ol Chemical compound C1=CC=C2C(O)=CC=NC2=C1 PMZDQRJGMBOQBF-UHFFFAOYSA-N 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 210000001533 respiratory mucosa Anatomy 0.000 description 1
- 208000023504 respiratory system disease Diseases 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 238000002390 rotary evaporation Methods 0.000 description 1
- 102200160628 rs1042393 Human genes 0.000 description 1
- 102200044453 rs1114167515 Human genes 0.000 description 1
- 102200111565 rs121908418 Human genes 0.000 description 1
- 102220002718 rs121908745 Human genes 0.000 description 1
- 102200128615 rs121908750 Human genes 0.000 description 1
- 102200128270 rs121908759 Human genes 0.000 description 1
- 102220020418 rs121909006 Human genes 0.000 description 1
- 102200128180 rs121909009 Human genes 0.000 description 1
- 102220002744 rs121909021 Human genes 0.000 description 1
- 102200128230 rs121909047 Human genes 0.000 description 1
- 102200101626 rs121964978 Human genes 0.000 description 1
- 102220080376 rs138211345 Human genes 0.000 description 1
- 102200132039 rs139304906 Human genes 0.000 description 1
- 102220008547 rs139573311 Human genes 0.000 description 1
- 102220020598 rs140883683 Human genes 0.000 description 1
- 102200132023 rs142394380 Human genes 0.000 description 1
- 102220020551 rs142773283 Human genes 0.000 description 1
- 102220008594 rs144476686 Human genes 0.000 description 1
- 102200128280 rs145449046 Human genes 0.000 description 1
- 102220020635 rs146521846 Human genes 0.000 description 1
- 102200065833 rs148044781 Human genes 0.000 description 1
- 102220020832 rs148519623 Human genes 0.000 description 1
- 102220020585 rs149279509 Human genes 0.000 description 1
- 102220020371 rs151020603 Human genes 0.000 description 1
- 102200128257 rs151048781 Human genes 0.000 description 1
- 102200128227 rs1800097 Human genes 0.000 description 1
- 102200128278 rs1800103 Human genes 0.000 description 1
- 102200127595 rs281864970 Human genes 0.000 description 1
- 102220008566 rs386134230 Human genes 0.000 description 1
- 102200036929 rs387907087 Human genes 0.000 description 1
- 102220020354 rs397508144 Human genes 0.000 description 1
- 102220020359 rs397508154 Human genes 0.000 description 1
- 102220020391 rs397508224 Human genes 0.000 description 1
- 102200128222 rs397508267 Human genes 0.000 description 1
- 102200128611 rs397508272 Human genes 0.000 description 1
- 102220020421 rs397508272 Human genes 0.000 description 1
- 102200128223 rs397508276 Human genes 0.000 description 1
- 102200128226 rs397508277 Human genes 0.000 description 1
- 102220020440 rs397508297 Human genes 0.000 description 1
- 102220020453 rs397508308 Human genes 0.000 description 1
- 102220020456 rs397508310 Human genes 0.000 description 1
- 102200128276 rs397508378 Human genes 0.000 description 1
- 102220020543 rs397508435 Human genes 0.000 description 1
- 102220020544 rs397508436 Human genes 0.000 description 1
- 102220020567 rs397508463 Human genes 0.000 description 1
- 102200128586 rs397508464 Human genes 0.000 description 1
- 102220020568 rs397508467 Human genes 0.000 description 1
- 102200128254 rs397508479 Human genes 0.000 description 1
- 102200128260 rs397508480 Human genes 0.000 description 1
- 102220020586 rs397508488 Human genes 0.000 description 1
- 102220020614 rs397508522 Human genes 0.000 description 1
- 102220020626 rs397508535 Human genes 0.000 description 1
- 102220020649 rs397508573 Human genes 0.000 description 1
- 102220020670 rs397508598 Human genes 0.000 description 1
- 102220020671 rs397508600 Human genes 0.000 description 1
- 102220020679 rs397508609 Human genes 0.000 description 1
- 102200132110 rs397508621 Human genes 0.000 description 1
- 102200128607 rs397508635 Human genes 0.000 description 1
- 102220020713 rs397508653 Human genes 0.000 description 1
- 102220457918 rs397508678 Human genes 0.000 description 1
- 102220020773 rs397508729 Human genes 0.000 description 1
- 102220020788 rs397508744 Human genes 0.000 description 1
- 102220020824 rs397508784 Human genes 0.000 description 1
- 102220020833 rs397508796 Human genes 0.000 description 1
- 102220000257 rs62514891 Human genes 0.000 description 1
- 102200132019 rs75389940 Human genes 0.000 description 1
- 102200128215 rs75549581 Human genes 0.000 description 1
- 102200128196 rs75763344 Human genes 0.000 description 1
- 102220020447 rs77284892 Human genes 0.000 description 1
- 102220405937 rs773569201 Human genes 0.000 description 1
- 102200128193 rs77409459 Human genes 0.000 description 1
- 102200128207 rs77646904 Human genes 0.000 description 1
- 102200128590 rs77834169 Human genes 0.000 description 1
- 102220248941 rs778632674 Human genes 0.000 description 1
- 102200128171 rs77932196 Human genes 0.000 description 1
- 102200128589 rs78655421 Human genes 0.000 description 1
- 102220065656 rs793888531 Human genes 0.000 description 1
- 102220085748 rs864309723 Human genes 0.000 description 1
- 210000003296 saliva Anatomy 0.000 description 1
- 229910052706 scandium Inorganic materials 0.000 description 1
- HZXJVDYQRYYYOR-UHFFFAOYSA-K scandium(iii) trifluoromethanesulfonate Chemical compound [Sc+3].[O-]S(=O)(=O)C(F)(F)F.[O-]S(=O)(=O)C(F)(F)F.[O-]S(=O)(=O)C(F)(F)F HZXJVDYQRYYYOR-UHFFFAOYSA-K 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 208000013220 shortness of breath Diseases 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 208000017520 skin disease Diseases 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 235000011006 sodium potassium tartrate Nutrition 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 125000003003 spiro group Chemical group 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 229950008188 sulfamidochrysoidine Drugs 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- IWJDYMDQNRKMAN-UHFFFAOYSA-N sulfonylmethanol Chemical compound OC=S(=O)=O IWJDYMDQNRKMAN-UHFFFAOYSA-N 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 239000007916 tablet composition Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- GTWXYRAFRYVGNS-UHFFFAOYSA-N tert-butyl 2,6-dichloropyridine-3-carboxylate Chemical compound CC(C)(C)OC(=O)C1=CC=C(Cl)N=C1Cl GTWXYRAFRYVGNS-UHFFFAOYSA-N 0.000 description 1
- BKRYJISKTILWAE-UHFFFAOYSA-N tert-butyl 2-chloropyridine-3-carboxylate Chemical compound CC(C)(C)OC(=O)C1=CC=CN=C1Cl BKRYJISKTILWAE-UHFFFAOYSA-N 0.000 description 1
- QOGNBDVPMQPHBY-UHFFFAOYSA-N tert-butyl 3-ethenoxypyrazole-1-carboxylate Chemical compound C(=C)OC1=NN(C=C1)C(=O)OC(C)(C)C QOGNBDVPMQPHBY-UHFFFAOYSA-N 0.000 description 1
- OFRLMTIBUAXBGV-UHFFFAOYSA-N tert-butyl 6-chloropyridine-3-carboxylate Chemical compound CC(C)(C)OC(=O)C1=CC=C(Cl)N=C1 OFRLMTIBUAXBGV-UHFFFAOYSA-N 0.000 description 1
- BSJFXAFLSWDUPK-UHFFFAOYSA-N tert-butyl pyrazole-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1C=CC=N1 BSJFXAFLSWDUPK-UHFFFAOYSA-N 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000167 toxic agent Toxicity 0.000 description 1
- 239000003440 toxic substance Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 102000035160 transmembrane proteins Human genes 0.000 description 1
- 108091005703 transmembrane proteins Proteins 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- VLPFTAMPNXLGLX-UHFFFAOYSA-N trioctanoin Chemical compound CCCCCCCC(=O)OCC(OC(=O)CCCCCCC)COC(=O)CCCCCCC VLPFTAMPNXLGLX-UHFFFAOYSA-N 0.000 description 1
- FIQMHBFVRAXMOP-UHFFFAOYSA-N triphenylphosphane oxide Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)(=O)C1=CC=CC=C1 FIQMHBFVRAXMOP-UHFFFAOYSA-N 0.000 description 1
- 238000005292 vacuum distillation Methods 0.000 description 1
- 238000009834 vaporization Methods 0.000 description 1
- 230000008016 vaporization Effects 0.000 description 1
- 210000001177 vas deferen Anatomy 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 230000004584 weight gain Effects 0.000 description 1
- 235000019786 weight gain Nutrition 0.000 description 1
- 235000021119 whey protein Nutrition 0.000 description 1
- 210000002268 wool Anatomy 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5365—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/12—Mucolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/002—Heterocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/05—Isotopically modified compounds, e.g. labelled
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Pulmonology (AREA)
- Gastroenterology & Hepatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Medicinal Preparation (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
Un compuesto de Fórmula I: **(Ver fórmula)** una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores, en donde: - uno de Y1 e Y2 es N y el otro es CH; - X se elige entre los grupos O, NH y N(C1-C4 alquilo); - R1 se elige entre grupos -(CR2)k-O-(CR2)m(CR)n(Anillo A)n+1, en donde cada Anillo A se elige independientemente entre grupos C3-C10 cicloalquilo opcionalmente sustituidos con uno o más sustituyentes elegidos cada uno independientemente entre grupos C1-C2 alquilo, grupos C1-C2 alquilo halogenados y halógenos, y en los que cada R se elige independientemente entre H, OH y grupos C1-C2 alquilo opcionalmente sustituidos con uno o más halógenos; - cada R2 se elige independientemente entre grupos C1-C2 alquilo, OH, grupos C1-C2 alcoxi, halógenos y ciano; - cada R3 se elige independientemente entre grupos C1-C2 alquilo opcionalmente sustituidos con uno o más grupos OH; - cada R4 se elige independientemente entre halógenos; - k es 0 o 1; - r es 0 o 1; - m es 0, 1, 2 o 3; - n es 0 o 1; - p es 0, 1, 2, 3, 4 o 5; y - q es 0, 1, 2, 3, 4, 5, 6, 7 u 8.
Description
DES
Modulador de regulador de conductancia transmembran de tratamiento y proceso de fabricación del modulador
[0001] En el presente documento se decribe un modula quistica (CFTR), composiciones farmaceuticas que conti fibrosis quistica y un proceso para fabricar el modulador.
[0002] La fibrosis quística (FQ) es una enfermedad gen adultos en todo el mundo. A pesar de los avances en el t
[0003] En pacientes con FQ, las mutaciones en CFTR conducen a una secreción aniónica apical reducida, lo qu La disminución resultante en el transporte de aniones c las infecciones microbianas que lo acompañan que final la enfermedad respiratoria, los pacientes con FQ suelen que, si no se tratan, provocan la muerte. Además, la m fertilidad se reduce entre las mujeres con fibrosis quístic
[0004] El análisis de secuencia del gen CFTR ha revel (Cutting, GR et al. (1990) Nature 346: 366-369; Dean, M Science 245: 1073-1080; Kerem, B-S y otros (1990) Proc han identificado más de 2000 mutaciones en el gen información sobre solo 322 de estas mutaciones identifi como causantes de enfermedad. La mutación causante d en la posición 508 de la secuencia de aminoácidos de mutación ocurre en aproximadamente el 70% de los ca grave.
[0005] La supresión de residuo 508 en CFTR impide que resultado la incapacidad de la proteína mutante para salir plasmática. Como resultado, el número de canales CFT mucho menor que el observado en células que expre mutaciones. Además del tráfico deficiente, la mutación d número reducido de canales en la membrana y la compu aniones y líquidos a través del epitelio. (Quinton, P. M. debido a la mutación F508del siguen siendo funcionale salvaje. (Dalemans y col. (1991), Nature Lond. 354: 526 50). Además de F508del, otras mutaciones que causan síntesis y/o compuerta de canal defectuosos podrían reg aniones y modificar la progresión y/o la gravedad de la e
[0006] El CFTR es un canal de aniones mediado por AM incluidas las células epiteliales de absorción y secreción así como la actividad de otros canales de iones y proteí CFTR es fundamental para el mantenimiento del tran respiratorio y digestivo. El CFTR está compuesto por apr que está formada por una repetición en tándem de dom hélices transmembrana y un dominio de unión a nucleót gran dominio polar regulador (R) con múltiples sitios d celular.
[0007] El transporte de cloruro tiene lugar por la activid apical y la bomba de Na+-K+-ATPasa y los canales de transporte activo secundario de cloruro desde el lado lu luego puede salir pasivamente de la célula a través de lo disposición del cotransportador de Na+/2ClVK+, la bom basolateral en la superficie basolateral y CFTR en el lad en el lado luminal. Dado que el agua probablemente nun depende de pequeños gradientes osmóticos transepiteli
[0008] El documento US2016095858A1 se refiere a un por CFTR, y también se refiere a composiciones farmac tales enfermedades.
CIÓN
fibrosis quística, composiciones farmacéuticas, métodos
e regulador de conductancia transmembrana de fibrosis n el modulador, su uso en metodos de tratamiento de la
recesiva que afecta aproximadamente a 70.000 niños y miento de la FQ, no existe cura.
presadas de forma endógena en el epitelio respiratorio voca un desequilibrio en el transporte de iones y líquidos. buye a una mayor acumulación de moco en el pulmón y e causan la muerte en los pacientes con FQ. Además de r problemas gastrointestinales e insuficiencia pancreática ía de los hombres con fibrosis quística son infértiles y la
una variedad de mutaciones que causan enfermedades al. (1990) Cell 61: 863: 870; y Kerem, B-S y otros (1989) tl. Acad. Sci. EE. UU. 87: 8447-8451). Hasta la fecha, se la FQ; actualmente, la base de datos CFTR2 contiene s, con evidencia suficiente para definir 281 mutaciones fermedad más prevalente es una deleción de fenilalanina R, y se denomina comúnmente mutación F508del. Esta de fibrosis quística y está asociada con una enfermedad
roteína naciente se pliegue correctamente. Esto da como retículo endoplásmico (RE) y transitar hacia la membrana ra el transporte de aniones presentes en la membrana es CFTR de tipo salvaje, es decir, CFTR que no tienen mo resultado una puerta de canal defectuosa. Juntos, el defectuosa conducen a una reducción del transporte de 90), FASEB J. 4: 2709-2727). Los canales defectuosos nque menos funcionales que los canales CFTR de tipo ; Pasyk y Foskett (1995), J. Cell. Biochem. 270: 12347 rmedades en CFTR que dan como resultado un tráfico, se hacia arriba o hacia abajo para alterar la secreción de medad.
TP que se expresa en una variedad de tipos de células, nde regula el flujo de aniones a través de la membrana, . En las células epiteliales, el funcionamiento normal de rte de electrolitos en todo el cuerpo, incluido el tejido adamente 1480 aminoácidos que codifican una proteína s transmembrana, cada uno de los cuales contiene seis . Los dos dominios transmembrana están unidos por un forilación que regulan la actividad del canal y el tráfico
oordinada de ENaC y CFTR presentes en la membrana expresados en la superficie basolateral de la célula. El al conduce a la acumulación de cloruro intracelular, que nales de Cl-, lo que resulta en un transporte vectorial. La e Na+-K+-ATPasa y los canales de K+ de la membrana inal coordinan la secreción de cloruro a través de CFTR e transporta de forma activa, su flujo a través del epitelio generados por el flujo masivo de sodio y cloruro.
puesto para el tratamiento de enfermedades mediadas as, métodos de tratamiento y kits para el tratamiento de
[0009] Por consiguiente, existe la necesidad de nuevos [0010] En el presente documento se describen compu sales farmacéuticamente aceptables de los mismos. Por como:
mientos de enfermedades mediadas por CFTR.
s nuevos, que incluyen Compuestos de Fórmulas I-IV y plo, los compuestos de Fórmula I se pueden representar

o una sal farmacéuticamente aceptable del mismo,
en la que:
- uno de Y1 e Y2 es N y el otro es CH;
- X se elige entre los grupos O, NH y N(C1-C4 alquilo);
- R 1 se elige entre grupos -(CR 2)k-O-(CR 2)m(CR)n(AniMo
en donde cada Anillo A se elige independi sustituidos con uno o más sustituyentes elegid grupos C1-C2 alquilo halogenados y halógenos,
en los que cada R se elige independientemente con uno o más halógenos;
- cada R 2 se elige independientemente entre grupos C1-- cada R 3 se elige independientemente entre grupos C OH;
- cada R 4 es independientemente un halógeno;
- k es 0 o 1 ;
- r es 0 o 1 ;
- m es 0, 1,2 o 3;
- n es 0 o 1 ;
- p es 0, 1,2, 3, 4 o 5; y
- q es 0, 1,2, 3, 4, 5, 6 , 7, u 8.
[0011] También se describen en el presente documento de los nuevos compuestos descritos en este document mismos, cuyas composiciones pueden incluir además menos un vehículo. También se describen métodos para que comprenden administrar al menos uno de los nuevo sal farmacéuticamente aceptable del mismo, opciona comprende al menos un componente adicional, para un
[0012] También se describen métodos de tratamiento comprende administrar al menos uno de los nuevos com aceptable del mismo, (R)-1-(2,2-difluorobenzo[d][1 metilpropan-2-il)-1H-indol-5-il)ciclopropanocarboxamida dihidro-4-oxoquinolin-3-carboxamida (Compuesto III), farmacéutica que comprende al menos un componente
Breve descripción de los dibujos
[0013]
+1,
mente entre grupos C3-C10 cicloalquilo opcionalmente ada uno independientemente entre grupos C1-C2 alquilo,
re H, OH y grupos C1-C2 alquilo opcionalmente sustituidos
lquilo, OH, grupos C1-C2 alcoxi, halógenos y ciano;
alquilo opcionalmente sustituidos con uno o más grupos
posiciones farmacéuticas que comprenden al menos uno o al menos una sal farmacéuticamente aceptable de los enos un ingrediente farmacéutico activo adicional y/o al ar la fibrosis quística de la enfermedad mediada por CFTR mpuestos descritos en este documento y/o al menos una te como parte de una composición farmacéutica que to que lo necesita.
fibrosis quística de enfermedad mediada por CFTR que tos descrito aquí y/o al menos una sal farmacéuticamente oxol-5-il)-N-(1-(2,3-dihidroxipropil)-6-fluoro-2-(1-hidroxi-2-mpuesto II) y N-[2,4-bis(1,1-dimetiletil)-5-hidroxifenil]-1,4-onalmente como parte de al menos una composición ional, a un paciente que lo necesite.
FIG. 1 muestra las estructuras de ejemplos documento.
FIG. 2 es un difractograma de rayos X en polvo del compuesto 1 al 50% en HPMCAS-HG.
FIG. 3 es un espectro que muestra un espectro una dispersión secada por pulverización (SDD) FIG. 4 es una lista representativa de mutaciones FIG. 5 es un XRPD de una muestra de la sal d ejemplo de la sal de sodio del Compuesto 1.
Definiciones
[0014] Tal como se utiliza aquí, el término "alquilo" se r ramificado que contiene átomos de carbono (como, por e 18, 19 o 20 átomos de carbono). Los grupos alquilo pued [0015] El término "alcoxi" como se usa aquí, se refiere a oxígeno. Los grupos alcoxi pueden estar sustituidos o no [0016] Como se usa en este documento, "cicloalquilo" s bicíclicos, tricíclicos o policíclicos que tienen de 3 a 12 ca "cicloalquilo" abarcan anillos monocíclicos, bicíclicos, tricíc mono espiro y diespiro. Los ejemplos no limitantes de ciclohexilo, adamantilo, norbornilo y diespiro[2.0.2.1]hept sustituidos.
[0017] "Sustituido", ya sea precedido por el término "opci "sustituido" se sustituye por un sustituyente. A menos qu puede tener un sustituyente adecuado en cada posició cualquier estructura dada puede estar sustituida con m sustituyente puede ser iguales o diferentes en cada posic [0018] Como se usa en este documento, "derivado(s) d uno o más átomos de hidrógeno sustituidos por un átomo [0019] Como se usa en el presente documento, "CFTR" fibrosis quística.
[0020] Como se usa en este documento, "mutaciones" CFTR. Una "mutación del gen CFTR' se refiere a una mut se refiere a una mutación en la proteína CFTR. Un defec de un gen en general, da como resultado una mutación marco.
[0021] El término "F508del" se refiere a una proteína CF la posición 508.
[0022] Como se usa en el presente documento, un pa particular tiene la misma mutación en cada alelo.
[0023] Como se usa en este documento, un paciente que esta mutación en un alelo, y una mutación diferente en el [0024] Como se usa en este documento, el término "mod de un compuesto biológico tal como una proteína. Por eje la actividad de CFTR. El aumento de actividad resultan compuestos que corrigen, potencian, estabilizan y/o ampl [0025] Como se usa en este documento, el término "c procesamiento y el tráfico de CFTR para aumentar la ca Fórmulas (I), (II), (III), (IV) y (V) y el compuesto II y sus s limitantes de nuevos compuestos descritos en este
RPD") de una dispersión secada por pulverización (SDD)
calorimetría diferencial de barrido modulado (MDSC) de 50% del Compuesto 1 en HPMCAS-HG.
néticas CFTR.
odio del Compuesto 1 preparado como se informa en el
re a un hidrocarburo alifático saturado, ramificado o no plo, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, estar sustituidos o no sustituidos.
alquilo o cicloalquilo unido covalentemente a un átomo de stituidos.
efiere a grupos de hidrocarburos no aromáticos cíclicos, nos (tales como, por ejemplo 3-10 carbonos). Los grupos os, con puente, condensados y espiro, incluidos los anillos pos cicloalquilo son ciclopropilo, ciclobutilo, ciclopentilo, o. Los grupos cicloalquilo pueden estar sustituidos o no
lmente" o no, indica que al menos un hidrógeno del grupo indique lo contrario, un grupo "opcionalmente sustituido" ustituible del grupo, y cuando más de una posición en de un sustituyente elegido de un grupo especificado, el .
rado(s)" significa la misma estructura química, pero con deuterio.
gnifica regulador de conductancia de transmembrana de
de referirse a mutaciones en el gen CFTR o la proteína ión en el gen CFTR y una "mutación de la proteína CFTR" genético o una mutación, o un cambio en los nucleótidos la proteína CFTR traducida de ese gen, o un cambio de
mutante que carece de la fenilalanina de aminoácido en
te que es "homocigoto" para una mutación del gen en
"heterocigoto" para una mutación del gen particular tiene ro alelo.
dor" se refiere a un compuesto que aumenta la actividad lo, un modulador de CFTR es un compuesto que aumenta de un modulador de CFTR incluye, pero no se limita a an CFTR.
ector CFTR" se refiere a un compuesto que facilita el ad de CFTR en la superficie celular. Los Compuestos de s farmacéuticamente aceptables descritas en el presente
documento son correctores de CFTR.
[0026] Como se usa en este documento, el término "pot la actividad de los canales de la proteína CFTR situada e mejorado. El compuesto III descrito en el presente docum
[0027] Tal como se utiliza aquí, el término "ingredient biológicamente activo.
[0028] Tal como se utiliza aquí, el término "sal farmacé compuesto de esta descripción en la que la sal no e compuestos de esta divulgación incluyen las derivadas sales farmacéuticamente aceptables son bien conocidas farmacéuticamente aceptables en detalle en J. Pharmace
[0029] Como se usa en este documento, el término "amor alcance en la posición de sus moléculas. Los sólidos am las moléculas están dispuestas de forma aleatoria de m empaquetamiento molecular, ni un orden de largo alcance. exhiben propiedades similares en todas las direcciones y amorfo es un material sólido que no tiene picos cristalinos de rayos X (XRPD) (es decir, no es cristalino según lo picos anchos (p. ej., halos) en su patrón XRPD. Los picos 2004/0006237 para una comparación de XRPD de un m
[0030] Tal como se utiliza aquí, el término "sustancialme ningún orden de largo alcance en la posición de sus mol tienen menos del 15% de cristalinidad (por ejemplo, cristalinidad). También se observa que el término "sust refiere a materiales que no tienen (0%) cristalinidad.
[0031] Como se usa en este documento, el término "dispe una sustancia, la fase dispersa, en unidades discretas, vehículo). El tamaño de la fase dispersa puede variar dimensión nanométrica, hasta varios micrones de tam líquidas o gaseosas. En el caso de una dispersión sóli aplicaciones farmacéuticas, una dispersión sólida puede amorfo (fase continua); o alternativamente, un fármac continua). En algunas realizaciones, una dispersión sóli fármaco constituye la fase continua. O, una dispersión s polímero que constituye la fase continua.
[0032] Los términos "paciente" y "sujeto" se utilizan in humanos.
[0033] Los términos "dosis eficaz" y "cantidad eficaz" se cantidad de un compuesto que produce el efecto desead un síntoma de FQ, o disminución de la gravedad de la F eficaz dependerá del propósito del tratamiento y la pod conocidas (véase, por ejemplo, Lloyd (1999) The Art, Sci
[0034] Como se usa en este documento, los términos "t mejora de FQ o sus síntomas o disminuir la gravedad de usa en este documento, incluye, pero no se limita a lo sigu reducción de la mucosidad en los pulmones, mejora de l del pecho y/o reducción de la tos o falta de aire. Las mej síntomas pueden evaluarse fácilmente de acuerdo con m
[0035] Tal como se utiliza aquí, el término "en combinació o ingredientes farmacéuticos activos adicionales, signific ingredientes farmacéuticos activos al paciente antes sim
[0036] Los términos "sobre" y "aproximadamente", cuan en peso de ingredientes de una composición o una forma cantidad o por ciento en peso o un intervalo de dosis, cant en la técnica para proporcionar un efecto farmacológic iador de CFTR" se refiere a un compuesto que aumenta superficie celular, lo que resulta en el transporte de iones to es un potenciador de CFTR.
armacéutico activo" ("API") se refiere a un compuesto
amente aceptable" se refiere a una forma de sal de un tóxica. Las sales farmacéuticamente aceptables de los ácidos y bases inorgánicos y orgánicos adecuados. Las la técnica. Por ejemplo, S. M. Berge, et al. describen sales cal Sciences, 1977, 66, 1-19.
se refiere a un material sólido que no tiene orden de largo os son generalmente líquidos superenfriados en los que que no hay una disposición bien definida, por ejemplo, os sólidos amorfos son generalmente isotrópicos, es decir, tienen puntos de fusión definidos. Por ejemplo, un material racterísticos nítidos en su patrón de difracción de potencia erminado por XRPD). En cambio, aparecen uno o varios chos son característicos de un sólido amorfo. Véase, US ial amorfo y un material cristalino.
amorfo" se refiere a un material sólido que tiene poco o las. Por ejemplo, los materiales sustancialmente amorfos enos del 10% de cristalinidad o menos del 5% de ialmente amorfo" incluye el descriptor "amorfo", que se
n" se refiere a un sistema disperso en el que se distribuye lo largo de una segunda sustancia (la fase continua o nsiderablemente (por ejemplo, partículas coloidales de ). En general, las fases dispersas pueden ser sólidas, , las fases dispersa y continua son ambas sólidas. En cluir un fármaco cristalino (fase dispersa) en un polímero amorfo (fase dispersa) en un polímero amorfo (fase incluye el polímero que constituye la fase dispersa y el a incluye el fármaco que constituye la fase dispersa y el
tintamente y se refieren a un animal, incluyendo seres
an indistintamente en este documento y se refieren a la ara el que se administra (por ejemplo, la mejora de FQ o o un síntoma de la FQ). La cantidad exacta de una dosis determinar un experto en la técnica utilizando técnicas e and Technology of Pharmaceutical Compounding).
amiento", "tratar", y similares generalmente significan la o de sus síntomas en un sujeto. "Tratamiento", como se te: aumento del crecimiento del sujeto, aumento de peso, nción pancreática y/o hepática, reducción de infecciones s o la disminución de la gravedad de cualquiera de estos dos y técnicas estándar conocidos en la técnica.
con", cuando se refiere a dos o más compuestos, agentes la administración de dos o más compuestos, agentes, o neos o posteriores entre sí.
se usa en relación con las dosis, cantidades, o por ciento dosificación, incluyen el valor de una dosis especificada, d o porcentaje en peso que es reconocido por un experto equivalente al obtenido a partir de la dosis, cantidad o
porcentaje en peso especificados.
[0037] Cada uno de los Compuestos de Fórmulas (I), farmacéuticamente aceptables de los mismos descritos forma independiente se puede administrar una vez realizaciones, al menos un compuesto elegido de los farmacéuticamente aceptables y sus derivados deutera al menos un compuesto elegido de los Compuestos de F aceptables y sus derivados deuterados se administran compuesto elegido del Compuesto II y sus sales farma algunas realizaciones, al menos un compuesto elegido del mismo se administran dos veces al día. En algunas r III y sus sales farmacéuticamente aceptables se admini compuesto elegido del Compuesto III y las sales farmac al día. En algunas realizaciones, al menos un compuest aceptables se administran una vez al día. En algunas re IV y las sales farmacéuticamente aceptables del mismo emplea un derivado deuterado del Compuesto II, III y/ cualquiera de estas realizaciones.
[0038] En algunas realizaciones, 10 mg a 1500 mg de farmacéuticamente aceptable del mismo, o un derivado
[0039] Un experto en la técnica reconocería que, cuand farmacéuticamente aceptable del mismo", la cantidad de es la cantidad equivalente a la concentración de la base de los compuestos o sus sales farmacéuticamente acept base libre. Por ejemplo, "10 mg de al menos un comp farmacéuticamente aceptables" incluye 10 mg de un farmacéuticamente aceptable de compuestos de Fórmul
[0040] Como se indicó anteriormente, descritos en esta II), (III), (IV) y (V), y Compuestos II, III, IV, y las sales sus derivados deuterados en el presente documento de día, dos veces al día o tres veces al día. En algunas puestos de Fórmulas (I), (II), (III), (IV) y (V) y sus sales se administran una vez al día. En algunas realizaciones, mulas (I), (II), (III), (IV) y (V) y sus sales farmacéuticamente os veces al día. En algunas realizaciones, al menos un uticamente aceptables se administran una vez al día. En l Compuesto II y las sales farmacéuticamente aceptables lizaciones, al menos un compuesto elegido del Compuesto an una vez al día. En algunas realizaciones, al menos un ticamente aceptables del mismo se administran dos veces elegido del Compuesto IV y sus sales farmacéuticamente izaciones, al menos un compuesto elegido del Compuesto administran dos veces al día. En algunas realizaciones, se IV o una sal farmacéuticamente aceptable del mismo en
n compuesto descrito en el presente documento, una sal terado de tal compuesto o sal se administran diariamente.
se da a conocer una cantidad de "un compuesto o una sal forma de sal farmacéuticamente aceptable del compuesto re del compuesto. Se observa que las cantidades descritas les de los mismos en la presente se basan en su forma de sto elegido entre compuestos de Fórmula (I) y sus sales puesto de Fórmula (I) y una concentración de una sal I) equivalente a 10 mg de compuestos de Fórmula (I).
moria son compuestos de fórmula (I):

y sales farmacéuticamente aceptables de los mismos,
en donde:
- uno de Y1 e Y2 es N y el otro es CH;
- X se elige entre los grupos O, NH y N(C-i-C4 alquilo);
- R1 se elige entre grupos -(CR 2)k-O-(CR 2)m(CR)n(AniMo entre grupos C3-C10 cicloalquilo opcionalmente susti independientemente entre grupos C1-C2 alquilo, grupos elige independientemente entre H, OH y grupos C1-C2 al - cada R2 se elige independientemente entre grupos C1-- cada R3 se elige independientemente entre grupos C1 OH;
- cada R4 se elige independientemente entre halógenos; - k es 0 o 1;
)n+1, en donde cada Anillo A se elige independientemente dos con uno o más sustituyentes, cada uno elegido C2 alquilo halogenados y halógenos, y en donde cada R se ilo opcionalmente sustituidos con uno o más halógenos;
alquilo, OH, grupos C1-C2 alcoxi, halógenos y ciano;
2 alquilo opcionalmente sustituidos con uno o más grupos
- r es 0 o 1 ;
- m es 0, 1,2 o 3;
- n es 0 o 1 ;
- p es 0, 1,2, 3, 4 o 5; y
- q es 0, 1,2, 3, 4, 5, 6 , 7, u 8.
[0041] También se describen en el presente document mpuestos de fórmula (II):

y sales farmacéuticamente aceptables de los mismos,
en donde:
- X se elige de grupos O, NH y N(C-i-C4 alquilo);
- R 1 se elige entre grupos -(CR 2 )k-O-(CR 2)m(CR)n(AniMo
en donde cada Anillo A se elige independie sustituidos con uno o más sustituyentes, cada grupos C1-C2 alquilo halogenados y halógenos, en donde cada R se elige independientemente con uno o más halógenos;
- cada R 2 se elige independientemente entre grupos C1-- cada R 3 se elige independientemente entre grupos C1-OH;
- cada R 4 se elige independientemente entre halógenos; - k es 0 o 1 ;
- r es 0 o 1 ;
- m es 0, 1,2 o 3;
- n es 0 o 1 ;
- p es 0, 1,2, 3, 4 o 5; y
- q es 0, 1,2, 3, 4, 5, 6 , 7, u 8.
[0042] Incluyen dentro del alcance de las Fórmulas (I) y ( n+1,
mente entre grupos C3-C10 cicloalquilo opcionalmente elegido independientemente entre grupos C1-C2 alquilo,
e H, OH y grupos C1-C2 alquilo opcionalmente sustituidos
lquilo, OH, grupos C1-C2 alcoxi, halógenos y ciano;
alquilo opcionalmente sustituidos con uno o más grupos
son compuestos que comprenden un

grupo (donde R' es H o C1-C4 alquilo), es decir, en do ejemplos no limitantes de tales compuestos incluyen co X se elige entre los grupos NH y N(C1-C4 alquilo). Los estos que tienen la siguiente estructura:

y sales farmacéuticamente aceptables de los mis enantioenriquecidos (por ejemplo, >90% ee, >95% ee o [0043] También se describen en el presente document , ya sea como una mezcla isomérica o isómeros % ee).
mpuestos de fórmula (III):

y sales farmacéuticamente aceptables de los mismos,
en donde:
- R1 se selecciona de entre grupos -(CR 2)K-O-(CR 2)m(CR
en los que cada Anillo A se elige independi sustituidos con uno o más sustituyentes, cada grupos C1-C2 alquilo halogenados y halógenos, en donde cada R se elige independientemente con uno o más halógenos;
- cada R2 se elige independientemente entre grupos C1-- cada R3 se elige independientemente entre grupos C1-OH;
- cada R4 se elige independientemente entre halógenos; - k es 0 o 1;
- r es 0 o 1;
- m es 0, 1,2 o 3;
- n es 0 o 1;
- p es 0, 1,2, 3, 4 o 5; y
- q es 0, 1,2, 3, 4, 5, 6, 7, u 8.
[0044] En algunas realizaciones, en los compuestos de del mismo, si R2 es ciano, entonces R2 es meta o para c
[0045] En algunas realizaciones, en los compuestos de de los mismos:
- cada anillo A se elige independientemente de grupos niMo A )n+1,
mente entre grupos C3-C10 cicloalquilo opcionalmente elegido independientemente entre grupos C1-C2 alquilo,
e H, OH y grupos C1-C2 alquilo opcionalmente sustituidos
lquilo, OH, grupos C1-C2 alcoxi, halógenos y ciano;
alquilo opcionalmente sustituidos con uno o más grupos
rmula (I), (II), (III), y sales farmacéuticamente aceptables especto al átomo de azufre.
rmula (I), (II), (III), y sales farmacéuticamente aceptables
C10 cicloalquilo opcionalmente sustituidos con uno o más
sustituyentes cada uno elegido independientemente entr halógenos, y
- cada R se elige independientemente entre H y OH;
- cada R2 se elige independientemente entre grupos C1-- R4 es F;
- k es 0;
- p es 0, 1 o 2;
- q es 0, 1,2, 3 o 4;
- r es 0; y
- m y n no son 0 al mismo tiempo.
[0046] En algunas realizaciones, en los compuestos de de los mismos:
- R1 se elige entre grupos -O-(CR2)m-AniMo A ,
en donde el Anillo A se elige entre grupos C3-C10 cicloal cada uno elegidos independientemente entre grupos C1-- m es 1 o 2.
[0047] En algunas realizaciones, en los compuestos de F de los mismos, cada R3 es un grupo metilo y q es 3 o 4.
[0048] También se describen en el presente documento rupos C1-C2 alquilo, grupos C1-C2 alquilo halogenados y
lquilo, OH, grupos C1-C2 alcoxi y halógenos;
rmula (I), (II), (III), y sales farmacéuticamente aceptables
lo opcionalmente sustituidos con uno o más sustituyentes alquilo, grupos C1-C2 alquilo halogenados, y halógenos, y
ula (I), (II), (III), y las sales farmacéuticamente aceptables
puestos de Fórmula (IV):

y sales farmacéuticamente aceptables de los mismos, en
- Anillo A se elige de grupos C3-C10 cicloalquilo opcional uno independientemente entre grupos C1-C2 alquilo, gru - cada R2 se elige independientemente entre grupos C1-- m es 1 o 2; y
- p es 0, 1, ó 2. En algunas realizaciones, p es 0 o 1. En
[0049] También se describen en el presente document nde:
nte sustituidos con uno o más sustituyentes elegidos cada C1-C2 alquilo halogenados y halógenos; y
lquilo, OH, F, Cl y grupos C1-C2 alcoxi;
unas realizaciones, p es 0.
ompuestos de Fórmula V:

y sales farmacéuticamente aceptables de los mismos, en donde:
- Anillo A se elige entre grupos C3-C10 cicloalquilo opcio elegido independientemente entre grupos C1-C2 alquilo, - cada R2 se elige independientemente entre grupos C1-- m es 1 o 2; y
- p es 0, 1, o 2.
mente sustituidos con uno o más sustituyentes, cada uno pos C1-C2 alquilo halogenados y halógenos; y
lquilo, OH, F, Cl y grupos C1-C2 alcoxi;
[0050] En algunas realizaciones, en los compuestos d aceptables de los mismos, cada uno de R2 se elige i realizaciones, p es 0 o 1. En algunas realizaciones, p es
[0051] En algunas realizaciones, en los compuestos d aceptables de los mismos, el Anillo A es un grupo cicl grupo C2 alquilo halogenado. En algunas realizaciones, CF3.
[0052] En algunas realizaciones, en los compuestos d aceptables de los mismos, m es 1, el Anillo A es un gru si está presente, es un grupo metilo, un grupo hidroxi o A es un grupo ciclopropilo sustituido con un grupo CF3,
[0053] En algunas realizaciones, en los compuestos de aceptables, m es 2, el Anillo A es un grupo C3 cicloal presente, es un grupo metilo, un grupo hidroxi, o un grup grupo ciclopropilo sustituido con un grupo CF3 , y p es 0.
[0054] En algunas realizaciones, m es 2, el Anillo A es
[0055] en algunas realizaciones, en los compuestos d aceptables de los mismos, el Anillo A se elige entre gru sustituyentes, cada uno elegido independientemente en halógenos. En algunas realizaciones, el Anillo A es halógeno.
[0056] En algunas realizaciones, en los compuestos d aceptables de los mismos, el Anillo A se elige entre gru sustituidos con uno o más sustituyentes, cada uno elegi C2 alquilo halogenados y halógenos. En algunas realizac
[0057] También se describe en la presente invención cualquiera de las fórmulas representadas en la FIG. 1 y
[0058] También se describen en el presente documento 64, y las sales farmacéuticamente aceptables de los mis
[0059] También se describen en el presente docume farmacéuticamente aceptables de los mismos.
[0060] También se describen en el presente docume aceptables de los mismos.
[0061] También se describen en la presente memoria d 5, 8, 10-16, 18-65, y sales farmacéuticamente aceptable
[0062] También se describe en la presente invención un órmula (I), (II), (III), (IV), (V), y sales farmacéuticamente ependientemente entre CH3 , OH, F y OCH3. En algunas
órmula (I), (II), (III), (IV), (V), y sales farmacéuticamente ropilo sustituido con un grupo C1 alquilo halogenado o un Anillo A es un grupo ciclopropilo sustituido con un grupo
órmula (I), (II), (III), (IV), (V), y sales farmacéuticamente ciclopropilo sustituido con un grupo CF3, p es 0 o 1, y R2 , grupo metoxi. En algunas realizaciones, m es 2, el Anillo es 0.
rmula (I), (II), (III), (IV), (V), y sus sales farmacéuticamente lo sustituido con un grupo CF3, p es 0 o 1, y R2 , si está etoxi. En algunas realizaciones, m es 2, el Anillo A es un
grupo ciclopropilo sustituido con un grupo CF3, y p es 0.
órmula (I), (II), (III), (IV), (V), y sales farmacéuticamente C5 bicicloalquilo sustituidos opcionalmente con uno o más grupos C1-C2 alquilo, grupos C1-C2 alquilo halogenados y grupo C5 bicicloalquilo opcionalmente sustituido con un
órmula (I), (II), (III), (IV), (V), y sales farmacéuticamente s C7 bicicloalquilo y grupos C7 tricicloalquilo opcionalmente independientemente entre grupos C1-C2 alquilo, grupos C1-es, el Anillo A es un grupo C7 tricicloalquilo no sustituido.
compuestos que tienen una fórmula elegida a partir de s sales farmacéuticamente aceptables.
mpuestos 1-5, 8, 10-16, 18-30, 32, 33, 35-37, 39-60, 63, y s.
compuestos 9, 31, 34, 38, 61, 62, y 65, y las sales
compuestos 6, 7, y 17, y las sales farmacéuticamente
ados deuterados de uno cualquiera de los compuestos 1 e los mismos.
mpuesto que tiene la siguiente fórmula:

y sales farmacéuticamente aceptables del mismo.
[0063] También se describe en la presente invención un mpuesto que tiene la siguiente fórmula:

y sales farmacéuticamente aceptables del mism [0064] También se describe en el presente docu nto un compuesto que tiene la siguiente fórmula:
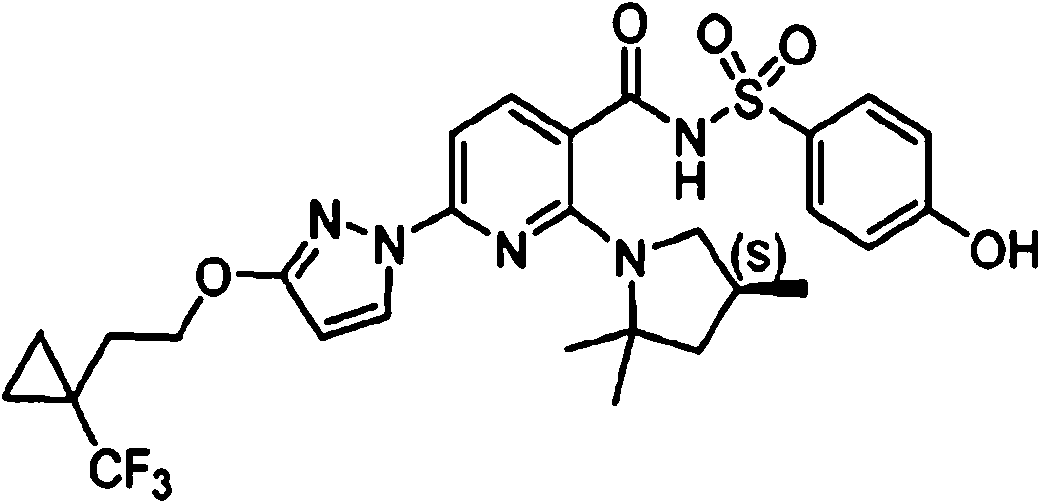
y sales farmacéuticamente aceptables del mism [0065] También se describe en la presente inven n un compuesto que tiene la siguiente fórmula:

y sales farmacéuticamente aceptables del mism [0066] También se describe en la presente inven n un compuesto que tiene la siguiente fórmula:

y sales farmacéuticamente aceptables del mism [0067] También se describe en la presente inven n un compuesto que tiene la siguiente fórmula:

y sales farmacéuticamente aceptables del mismo.
[0068] También se describe en el presente documento un mpuesto que tiene la siguiente fórmula:

y sales farmacéuticamente aceptables del mismo.
[0069] En algunas realizaciones, al menos un nuevo com del mismo y/o al menos un derivado deuterado de tal com menos un ingrediente farmacéutico activo adicional. En al activo adicional se elige entre:
(a) Compuesto II:
esto (y/o al menos una sal farmacéuticamente aceptable esto o sal) se puede administrar en combinación con al as realizaciones, al menos un ingrediente farmacéutico

y sales farmacéuticamente aceptables del mismo. Un nombre químico para el Compuesto II dihidroxipropil)-6-fluoro-2-(1-hidroxi-2-metilpropan (b) Compuesto III:
es (R/M-(2,2-difluorobenzo[d][1,3]dioxol-5-¡l)-A/-(1-(2,3-il)-1H-indol-5-il)ciclopropanocarboxamida;

y sus sales farmacéuticamente aceptables. Un no ferc-butilfenil)-4-oxo-1H-quinolin-3-carboxamida; re químico para el Compuesto III es A/-(5-hidroxi-2,4-di-(c) Compuesto IV:

y sus sales farmacéuticamente aceptables.
Un nombre químico para el Compuesto il)cidopropanocarboxamido)-3-metilpiridin-2-il)be
[0070] Las sales farmacéuticamente aceptables son, por Sciences, 1977, 66, 1-19. Por ejemplo, la Tabla 1 de ese aceptables:
Ta es ácido 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-ico.
mplo, las descritas en S. M. Berge, et al. J. Pharmaceutical ículo proporciona las siguientes sales farmacéuticamente
1:
[0071] Los ejemplos no limitantes de sales farmacéutica sales formadas con ácidos inorgánicos, tales como ácido o ácido perclórico; sales formadas con ácidos orgánicos, t tartárico, ácido cítrico, ácido succínico o ácido malónico; y tales como intercambio iónico. Los ejemplos no limitativo alginato, ascorbato, aspartato, bencenosulfonato, benzo citrato, ciclopentanopropionato, digluconato, dodecilsul hemisulfato, heptanoato, hexanoato, hidroyoduro, 2-hidro malato, maleato, malonato, metanosulfonato, 2-naftale pamoato, pectinato, persulfato, 3-fenilpropionato, fosfato, tartrato, tiocianato, p-toluenosulfonato y sales de valera nte aceptables derivadas de ácidos apropiados incluyen: hídrico, ácido bromhídrico, ácido fosfórico, ácido sulfúrico s como ácido acético, ácido oxálico, ácido maleico, ácido les formadas usando otros métodos usados en la técnica, e sales farmacéuticamente aceptables incluyen adipato, , bisulfato, borato, butirato, canforato, canforsulfonato, o, etanosulfonato, formiato, fumarato, glucocerofato., tanosulfonato, lactobionato, lactato, laurato, lauril sulfato, ulfonato, nicotinato, nitrato, oleato, oxalato, palmitato, icrato, pivalato, propionato, estearato, succinato, sulfato, Las sales farmacéuticamente aceptables derivadas de
bases apropiadas incluyen sales de metales alcalinos, divulgación también prevé la cuaternización de cualquie descritos en este documento. Los ejemplos no limitantes incluyen sodio, litio, potasio, calcio y magnesio. Otros eje incluyen amonio, amonio cuaternario y cationes amina f carboxilato, sulfato, fosfato, nitrato, alquilsulfonato inferio sales farmacéuticamente aceptables incluyen sales de b
[0072] En algunas realizaciones, al menos un compuest presente documento, sales farmacéuticamente aceptabl se administran en combinación con al menos un farmacéuticamente aceptables de los mismos, y derivado menos un compuesto elegido entre los nuevos compuesto aceptables y los derivados deuterados de los anteriores elegido entre el Compuesto III y sus sales farmacéutic compuesto elegido entre los nuevos compuestos des aceptables y los derivados deuterados de los anteriores elegido entre el Compuesto IV y sus sales farmacéutic compuesto elegido de los nuevos compuestos descritos y los derivados deuterados de los anteriores se admi farmacéuticamente aceptable o derivado deuterado del m sales farmacéuticamente aceptables del mismo y deriva realizaciones, al menos un compuesto elegido de los nu farmacéuticamente aceptables y derivados deuterados de con al menos un compuesto elegido del Compuesto III, deuterados de cualquiera de los anteriores y al m farmacéuticamente aceptables del mismo y derivados de
[0073] Cualquiera de los compuestos según la invenció Fórmula (I), (II), (III), (IV), (V), y sus sales farmacéuticam tales compuestos y sales pueden estar comprendidos farmacéuticas separadas en combinación con otros ingr Compuesto II, III o IV, o su sal farmacéuticamente acepta sal). Dichas composiciones farmacéuticas se pueden ad al día. En algunas realizaciones, la divulgación presenta compuesto elegido entre cualquiera de los compuestos d aceptables, y al menos un vehículo farmacéuticamente a
[0074] En algunas realizaciones, la divulgación presenta compuesto elegido entre los nuevos compuestos descr aceptables de los mismos, al menos un compuesto elegi aceptables de los mismos, y al menos un vehículo farma
[0075] En algunas realizaciones, la divulgación presenta compuesto elegido entre los nuevos compuestos descrito aceptables de los mismos, al menos un compuesto ele aceptables de los mismos, y al menos un vehículo farma
[0076] En algunas realizaciones, la divulgación presenta compuesto elegido entre los nuevos compuestos descr aceptables de los mismos, al menos un compuesto elegi aceptables de los mismos, al menos un compuesto e aceptables, y al menos un vehículo farmacéuticamente a
[0077] En algunas realizaciones, la divulgación presenta compuesto elegido entre los nuevos compuestos descrito aceptables de los mismos, al menos un compuesto ele aceptables del mismo, al menos un compuesto elegido d y al menos un vehículo farmacéuticamente aceptable.
[0078] En algunas realizaciones, las composiciones farm al menos un ingrediente adicional farmacéutico activ farmacéutico activo adicional es un modulador de CF farmacéutico activo adicional es un corrector de CFT farmacéutico activo adicional es un potenciador de CF comprende (i) un compuesto de fórmulas (I), (II), (III), (IV) etales alcalinotérreos, amonio y N+(C-m alquilo)4. Esta rupo que contenga nitrógeno básico de los compuestos ecuados de sales de metales alcalinos y alcalinotérreos los no limitantes de sales farmacéuticamente aceptables ados usando contraiones tales como haluro, hidróxido, arilsulfonato. Otros ejemplos adecuados, no limitantes de ato y glucosamina.
elegido entre los compuestos novedosos descritos en el de los mismos, y derivados deuterados de los anteriores mpuesto seleccionado entre el Compuesto II, sales euterados de los anteriores. En algunas realizaciones, al escritos en este documento, sus sales farmacéuticamente administran en combinación con al menos un compuesto nte aceptables. En algunas realizaciones, al menos un tos en este documento, sus sales farmacéuticamente administran en combinación con al menos un compuesto ente aceptables. En algunas realizaciones, al menos un este documento, las sales farmacéuticamente aceptables tran en combinación con los Compuestos II o una sal o y al menos un compuesto elegido entre Compuesto III, deuterados de cualquiera de los anteriores. En algunas s compuestos descritos en el presente documento, sales alquiera de los anteriores se administran en combinación es farmacéuticamente aceptables del mismo y derivados os un compuesto elegido del Compuesto IV, sales rados de cualquiera de los anteriores.
escritos aquí, tales como, por ejemplo, compuestos de te aceptables de los mismos, y derivados deuterados de una sola composición farmacéutica o composiciones ientes farmacéuticos activos adicionales (por ejemplo, el del mismo, o un derivado deuterado de tal compuesto o strar una vez al día o varias veces al día, como dos veces a composición farmacéutica que comprende al menos un critos en este documento y sus sales farmacéuticamente table.
a composición farmacéutica que comprende al menos un en el presente documento y sales farmacéuticamente a partir del Compuesto II y las sales farmacéuticamente ticamente transportista aceptable.
a composición farmacéutica que comprende al menos un n el presente documento, y las sales farmacéuticamente o a partir del Compuesto III y sales farmacéuticamente ticamente aceptable.
a composición farmacéutica que comprende al menos un en el presente documento y sales farmacéuticamente a partir del Compuesto II y las sales farmacéuticamente ido del Compuesto III y sus sales farmacéuticamente table.
a composición farmacéutica que comprende al menos un n el presente documento, y las sales farmacéuticamente o a partir del Compuesto III y sales farmacéuticamente ompuesto IV y sus sales farmacéuticamente aceptables,
éuticas descritas en el presente documento comprenden n algunas realizaciones, el al menos un ingrediente En algunas realizaciones, el al menos un ingrediente En algunas realizaciones, el al menos un ingrediente En algunas realizaciones, la composición farmacéutica (V), o una sal farmacéuticamente aceptable del mismo, o
un derivado deuterado de dicho compuesto. o sal; y (ii) al uno de los cuales es un corrector de CFTR y uno de los c
[0079] En algunas realizaciones, al menos un ingr mucolíticos agentes, broncodilatadores, antibióticos, age
[0080] Una composición farmacéutica puede compre aceptable. En algunas realizaciones, el al menos un veh farmacéuticamente aceptables y adyuvantes farmacéutic vehículo farmacéuticamente aceptable se elige entre car farmacéuticamente aceptables.
[0081] También se apreciará que una composición farm farmacéutica que comprende combinaciones descrit combinación; es decir, las composiciones pueden admini un ingrediente farmacéutico activo adicional o procedimie
[0082] Las composiciones farmacéuticas que compren quística.
[0083] Como se ha descrito anteriormente, las composi opcionalmente comprender además al menos un vehíc farmacéuticamente aceptable se puede elegir entr farmacéuticamente aceptable, como se usa en este d diluyentes, otros vehículos líquidos, auxiliares de dispersi isotónicos, agentes espesantes, agentes emulsionantes, adapte a la forma de dosificación particular deseada. Re 2005, ed. D. B. Troy, Lippincott Williams & Wilkins, Phil eds. J. Swarbrick y J. C. Boylan, 1988-1999, Marcel De formulación de composiciones farmacéuticas y técnicas c cualquier vehículo convencional sea incompatible con l producir cualquier efecto biológico indeseable o al interac otro componente (s) de la composición farmacéutica, se c alcance de esta divulgación. Los ejemplos no limitante incluyen, entre otros, intercambiadores de iones, alúmin albúmina de suero humano), sustancias tampón (como f de glicéridos parciales de ácidos grasos vegetales satur hidrogenofosfato de disodio, hidrogenofosfato de potasio, magnesio, polivinilpirrolidona, poliacrilatos, ceras, políme azúcares (como lactosa, glucosa y sacarosa), almidones derivados (como carboximetilcelulosa de sodio, etilcelulos talco, excipientes (como manteca de cacao y ceras para semilla de algodón, aceite de oliva, aceite de sésamo, ac propilenglicol y polietilenglicol), ésteres (como oleato de hidróxido de magnesio e hidróxido de aluminio), ácido alg de Ringer, alcohol etílico, soluciones tampón de fosfato, sodio y estearato de magnesio), agentes colorantes, edulcorantes, agentes aromatizantes, agentes perfumant
[0084] También se apreciará que una composición farm farmacéutica que comprende cualquiera de las combinaci de combinación; es decir, las composiciones pueden a menos un ingrediente farmacéutico activo o procedimient
[0085] En algunas realizaciones, los métodos de la divulg del mismo de al menos un compuesto seleccionado de documento y las sales farmacéuticamente aceptables de Compuesto II, Compuesto III, Compuesto IV y sales farm
[0086] Cualesquiera composiciones farmacéuticas adecu compuestos descritos en el presente documento,
farmacéuticamente aceptables de los mismos. En los Ej ejemplares para el Compuesto 1 y sus sales farmacéuti ejemplares para el Compuesto II y sus sales farmacéutic W o 2011/119984 y WO 2014/015841. Algunas composic sales farmacéuticamente aceptables se pueden encontr enos dos ingredientes farmacéuticos activos adicionales, les es un potenciador de CFTR.
nte adicional farmacéutico activo se selecciona de s anti-infecciosos, y agentes anti-inflamatorios.
r además al menos un vehículo farmacéuticamente lo farmacéuticamente aceptable se elige entre vehículos nte aceptables. En algunas realizaciones, el al menos un , desintegrantes, tensioactivos, aglutinantes, lubricantes
éutica de esta descripción, que incluye una composición anteriormente, se pueden emplear en terapias de arse simultáneamente con, antes o después de al menos s médicos.
estas combinaciones son útiles para tratar la fibrosis
nes farmacéuticas descritas en este documento pueden farmacéuticamente aceptable. El al menos un vehículo adyuvantes y vehículos. El al menos un vehículo umento, incluye todos y cada uno de los disolventes, auxiliares de suspensión, agentes tensioactivos, agentes nservantes, aglutinantes sólidos y lubricantes, según se gton: La ciencia y la práctica de la farmacia, 21a edición, lphia, and Encyclopedia of Pharmaceutical Technology, r, Nueva York describen varios vehículos usados en la ocidas para su preparación. Excepto en la medida en que compuestos de esta divulgación, como por ejemplo al r de otra manera de una manera perjudicial con cualquier templa que su uso esté dentro de los límites establecidos. de vehículos farmacéuticamente aceptables adecuados stearato de aluminio, lecitina, proteínas del suero (como tos, glicina, ácido sórbico y sorbato de potasio), mezclas s, agua, sales y electrolitos (como sulfato de protamina, ruro de sodio y sales de zinc), sílice coloidal, trisilicato de de bloque de polietileno-polioxipropileno, grasa de lana, mo almidón de maíz y almidón de patata), celulosa y sus acetato de celulosa), tragacanto en polvo, malta, gelatina, positorios), aceites (como aceite de cacahuete, aceite de de oliva, aceite de maíz y aceite de soja), glicoles (como o y laurato de etilo), agar, agentes tamponadores (como o, agua sin pirógenos, solución salina isotónica, solución bricantes compatibles no tóxicos (como lauril sulfato de ntes de liberación, agentes de recubrimiento, agentes conservantes y antioxidantes.
éutica de esta descripción, que incluye una composición s descritas anteriormente, se pueden emplear en terapias inistrarse simultáneamente con, antes o después de al médicos.
ón emplean la administración a un paciente en necesidad e cualquiera de los compuestos descritos en el presente s mismos, y al menos un compuesto seleccionado entre uticamente aceptables de cualquiera de los anteriores.
s conocidas en la técnica se pueden usar para los nuevos mpuesto II, Compuesto III, IV Compuesto, y sales plos se describen algunas composiciones farmacéuticas ente aceptables. Algunas composiciones farmacéuticas ente aceptables se pueden encontrar en los documentos es farmacéuticas ejemplares para el Compuesto III y sus en los documentos WO 2007/134279, WO 2010/019239,
WO 2011/019413, WO 2012/027731 y WO 2013/13066 Compuesto IV y sus sales farmacéuticamente aceptable WO 2011/127241, WO 2013/112804 y WO 2014/071122 [0087] En algunas realizaciones, una composición farm los nuevos compuestos descritos aquí, y las sales farm composición farmacéutica que comprende el Compuesto comprenden el Compuesto II y el Compuesto III se descr En la siguiente Tabla se muestra una realización ejempla Tabla 2. Comprimido ejemplar que comprende 1 unas composiciones farmacéuticas ejemplares para el ueden encontrar en los documentos W o 2010/037066,
ca que comprende al menos un compuesto elegido entre icamente aceptables del mismo se administra con una l Compuesto III. Las composiciones farmacéuticas que en la Publicación PCT N° WO 2015/160787.
g del Compuesto II y 150 mg del Compuesto III.

[0088] En algunas realizaciones, una composición farm los compuestos novedosos descritos en este docum composición farmacéutica que comprende el Compuest Compuesto III se describen en la publicación PCT N° WO en la siguiente Tabla:
Tabla 3: Ingredientes para un co ca que comprende al menos un compuesto elegido entre y sus sales farmacéuticas se administran con una Las composiciones farmacéuticas que comprenden el /019239. Una forma de realización ejemplar se muestra
ido ejemplar del Compuesto III.

[0089] Las composiciones farmacéuticas adicionales qu PCT N° WO 2013/130669. Se formularon minicomprimid minicomprimido con un peso de aproximadamente 6,9 m III por 26 minicomprimidos y aproximadamente 75 mg de de ingredientes enumeradas en la Tabla 4, a continuació
Tabla 4 : Ingredientes para minicompr prenden el Compuesto III se describen en la publicación mplares (~ 2 mm de diámetro, ~ 2 mm de grosor, cada ra que tuvieran aproximadamente 50 mg de Compuesto uesto III por 39 minicomprimidos usando las cantidades
os para potencias de 50 mg y 75 mg

[0090] En algunas realizaciones, las composiciones far comprimidos son adecuados para la administración oral
[0091] Estas combinaciones son útiles para tratar la fibr
[0092] En algunas realizaciones, se describe aquí un m tratamiento de la fibrosis quística en un paciente que composición farmacéutica de la presente divulgación p tiene fibrosis quística y se elige entre paciente con genotipos F508de//F508de/, pacientes con genotip residual (RF).
[0093] En algunas realizaciones, el paciente es heter cualquier mutación causante de FQ, y se espera que s compuestos descritos en este documento, tales como los genotipos del Compuesto IV basados en datos in vit
[0094] Los pacientes con una F508de//genotipo de heterocigotos F508de/CFTR con un segundo alelo de en una proteína CFTR con función de mínimo y que no combinación de Compuesto II y Compuesto III. Est principales:
• plausibilidad biológica para que responda la mutación • evidencia de gravedad clínica sobre una base poblaci 15 de febrero de 2016)
- cloruro de sudor promedio >86 mmol/L, y
- prevalencia de insuficiencia pancreática (IP)
• Pruebas in vitro
- mutaciones que resultan en el transporte de función mínima
- las mutaciones que dan como resultado el tra la adición del Compuesto II y/o el Compuesto I
[0095] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación posee una mutación genética CFTR G551D. En algun genética G551D. En algunas realizaciones, el paciente realizaciones, el paciente es heterocigoto para la mutac y cualquier otra mutación que cause FQ en el otro alelo. mutación genética G551D en un alelo y la otra mutaci F508del, G542X, N1303K, W1282X, R117H, R553X R1162X, G85E, 3120 1G->A, AI507, 1898 1G->A, 3 >T. En algunas realizaciones, el paciente es heterocigot CFTR es F508del. En algunas realizaciones, el pacient mutación genética CFTR es R117H.
éuticas son un comprimido. En algunas realizaciones, los
quística.
o para tratar, reducir la gravedad de, o sintomáticamente prende administrar una cantidad eficaz de al menos una l paciente, tal como un humano, en donde dicho paciente n genotipos F508de//función mínima (MF), pacientes 508de//gating y pacientes con genotipos F508de//función
to para F508del, y la otra mutación genética CFTR es /o es sensible a cualquier combinación de (i) los nuevos mpuesto 1 y (ii) el Compuesto II y/o el Compuesto III y/o o clínicos.
ción mínima se definen como pacientes que son que contiene una mutación que se predice para resultar spera que responden al Compuesto II, Compuesto III, o la utaciones de CFTR se definieron utilizando 3 fuentes
decir, clase de mutación)
(según el registro de pacientes de CFTR2; consultado el
ro basal <10% de CFTR de tipo salvaje se consideraron
rte de cloruro <10% de CFTR de tipo salvaje después de consideraron que no respondían.
método para tratar, reducir la gravedad de, o tratar e comprende administrar una cantidad eficaz de una el paciente, tal como un humano, en donde el paciente alizaciones, el paciente es homocigoto para la mutación eterocigoto para la mutación genética G551D. En algunas enética G551D, teniendo la mutación G551D en un alelo algunas realizaciones, el paciente es heterocigoto para la enética que causa FQ en el otro alelo es cualquiera de 17-1G->A, 621 1G->T, 2789+5G->A, 3849+10kbC->T, elC, R347P, R560T, R334W, A455E, 2184delA o 711+1G-ra la mutación genética G551D y la otra mutación genética heterocigoto para la mutación genética G551D y la otra
[0096] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética CFTR F508del. En algun genética F508del. En algunas realizaciones, el paciente el paciente tiene la mutación genética F508del en un alelo. En algunas realizaciones, el paciente es heteroci cualquier mutación que cause FQ, que incluye, entre otr 1G->A, 621 1G->T, 2789+5G->A, 3849+10kbC->T, R R347P, R560T, R334W, A455E, 2184delA o 711 1G-> F508del y la otra mutación genética de CFTR es G551 F508del y la otra mutación genética de CFTR es R117H
[0097] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética de CFTR seleccionada de S549R, S1251N, E193K, F1052V, G1069R, R117C, D1 F1074L, D110E, D1270N, D1152H, 1717-1G->A, 621 1 405+1G->A, 406-1G->A, 4005+1G->A, 1812-1G->A, 15 >A, 4374+1G->T, 3850-1G->A, 2789+5G->A, 3849 1811 1.6kbA->G, 711+3A->G, 1898+3A->G, 1717-8 1898+5G->T, 3850-3T->G, IVS14b+5G->A, 1898+1G-> 3199del6, 3905InsT, 4209TGTT->A, A1006E, A120T, A2 D924N, D979V, E116K, E547, E22K, F1099L, F191V, G149R, G194R, G194V, G27R, G314E, G458V, G463V, H1375P, H139R, H199R, H609R, H939R, I1005R, I1234 L10280R, L8013, L101F, I618T, L8013K, L15P, L165S, M1V, M265R, M952I, M952T, P574H, P5L, P750L, P R117L, R117P, R1283M, R1283S, R170H, R258G, R3 R792P, R913F, S5933G->C), S549R (T->G), S589N, V1240G, V1293G, V201M, V232D, V456A, V456F, V Y1014C, Y1032C, Y109N, Y161D, Y161S, Y563D, Y5 tiene al menos una mutación combinada elegida entre: S549R, S1251N, E193K, F1052V, G1069R, R117C, D1 D579, D1270N, D1152H, 1717-1G->A, 621 1G->T, 31 >A, 406-1G->A, 4005+1G->A, 1812-1G->A, 1525-1G 4374+1G->T, 3850-1G->A, 2789+5G->A, 3849+10kbC-711+3A->G, 1898+3A->G, 1717-8G->A, 1342-2A->C, 40 IVS14b+5G->A, 1898+1G->T, 4005+2T->C y 621+3A->
[0098] En algunas realizaciones, el paciente tiene al m 3141del9, 3195del6, 3199del6, 3905insT, 4209TGTT-> D443Y, D513G, D836Y, D924N, D979V, E116K, E403 F311del, F311L, F508C, F575Y, G1061R, G1249R, G12 >A), G91R, G970D, H1054D, H1085P, H1085R, H1375 I506T, I601F, I618T, I807M, I980K, L102R, L1324P, L1 L571S, L967S, M1101R, M152T, M152V, M57V495, P5 Q452P, Q98R, R1066C, R1066H, R117G, R117L, R R553Q, R751L, R792G, R933G, S1118F, S1159F, S1 S912L, T1036N, T1053I, T1246I, T604I, V1153E, V1240 W1098R, W1282R, Y10G321R, Y95710, Y161S, Y563D
[0099] En algunas realizaciones, el paciente tiene al m
D443Y;G576A; R668C,
F508C;S1251N,
G576A;R668C,
G970R;M470V,
R74W;D1270N,
étodo para tratar, reducir la gravedad de, o tratar comprende administrar una cantidad eficaz de una paciente, tal como un mamífero, en donde el paciente lizaciones, el paciente es homocigoto para la mutación terocigoto para la mutación genética F508del en donde cualquier mutación genética que causa FQ en el otro para F508del, y la otra mutación genética de CFTR es 51D, G542X, N1303K, W1282X, R117H, R553X, 1717-, G85E, 3120+1G->A, AI507, 1898+1G->A, 3659delC, algunas realizaciones, el paciente es heterocigoto para algunas realizaciones, el paciente es heterocigoto para
étodo para tratar, reducir la gravedad de, o tratar comprende administrar una cantidad eficaz de una paciente, tal como un mamífero, en donde el paciente R, G551S, G970R, G1244E, S1255P, G1349D, S549N, R347H, R352W67L, E455K, S1235R, S945L, R1070W, 3120+1G->A, 1898+1G->A, 711 1G->T, 2622+1G->A, ->A, 712-1G->T, 1248+1G->A, 1341 1G->A, 3121-1G-0 kbC->T, 3272- 26A->G, 711+5G->A, 3120G->A, 1342-2A->C, 405+3A->C, 1716G/A, 1811 1G->C, 5+2T->C, 621+3A->G, 1949del84, 3141del9, 3195del6, 349V, A613T, C524R, D192G, D443Y, D513G, D836Y, el, F311L, F508C, F575Y, G1061R, G1249R, G126D, C, G622D, A28R70D, G91-, H1054D, H1085P, H1085R, 69N, I1366N, I175V, I502T, I506S, I506T, I601F, I618T, , L346P, L453S, L571S, L967S, M1101R, M152V, M1T, 1100P, Q2E91, Q1291H, R1066C, R1066H, R117G, 34L, R334Q, R347L, R352W, R516G, R553Q, R751L, F, S912L, T1036N, T1053I, T1246I, T604I, V1153E, W1098C, W1098R, W1282R, W361R, W57G, W57R, 569C y Y913C. En algunas realizaciones, el paciente , G551S, G970R, G1244E, S1255P, G1349D, S549N, R347H, R352Q, E56K, P67L, L206W, R10G70L, S125, ->A, 1898+1G->A, 711 1G->T, 2622+1G->A, 405+1G-712-1G->T, 1248+1G->A, 1341 1G->A, 3121-1G->A, 72-26A->G, 711+5G->A, 3120G->A, 1811 1,6 kbA->G, >C, 1716G/A, 1811 1G->C, 1898+5G->T, 3850-3T->G,
na mutación de combinación elegida entre: 1949del84, 006E, A120T, A234D, A349V, A613T, C524R, D192G, 74K, E588V, E60K, E822K, F1016S, F1099L, F191V, V, G149R, G463V, G480C, G622D, G628R, G628R (G-9R, H199R, H609R, H939R, I1005R, I1234V, I506502, L138ins, L1480P, L15P, L165S, L320V, L346P, L453S, 0L, P99L, Q1100P, Q1291H, Q1291R, Q237E, Q237H, R1283M, R1283S, R170H, R258G, R334L, R31L34, S13F, S549R (A->C), S549R (T->G), S589N, S737F, 293G, V201M, V232D, V456A, V456F, V562I, W1098C, 3N, Y569C y Y913C.
na mutación de combinación elegida entre:
R74W;V201M y
R74W;V201M;D1270N.
[0100] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética CFTR seleccionada entre S549R, S1251N, E193K, F1052V y G1069R. En alguna tratar CFTR que comprende administrar un compuesto aceptable del mismo a un paciente que posee una m G970R, G1244E, S1255P, G1349D, S549N, S549R documento un método para tratar, disminuir la graveda que comprende administrar una cantidad eficaz de un como un mamífero, en donde el paciente posee una m G1069R. En algunas realizaciones, el método produc transporte de cloruro inicial del paciente.
[0101] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética CFTR seleccionada entre D579G, S1235R, S945L, R1070W, F1074L, D110E, D1 un aumento en el transporte de cloruro por encima del tr
[0102] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética CFTR seleccionada e 711 1G->T, 2622+1G->A, 405+1G->A, 406-1G->A, 40 >A, 1341 1G->A, 3121-1G->A, 4374+1G->T, 3850-1G >A, 3120G->A, 1811 1.6kbA->G, 711+3A->G, 1898 1811 1G->C, 1898+5G->T, 3850-3T->G, IVS14b+5G-realizaciones, se describe en este documento un métod la fibrosis quística en un paciente que comprende admi de esta divulgación al paciente, como un mamífero, seleccionada entre 1717-1G->A, 1811 1,6kbA->G, 2 realizaciones, se describe en este documento un métod la fibrosis quística en un paciente que comprende admi de esta divulgación al paciente, como un mamífero, seleccionada entre 2789+5G->A y 3272-26A->G.
[0103] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética de CFTR seleccionada de S549R, S1251N, E193K, F1052V, G1069R, R117C, D1 F1074L, D110E, D1270N, D1152H, 1717-1G->A, 621 405+1G->A, 406-1G->A, 4005+1G->A, 1812-1G->A, 15 >A, 4374+1G->T, 3850-1G->A, 2789+5G->A, 3849+10 k >G, 711+3A->G, 1898+3A->G, 1717-8G->A, 1342-2A-> 3T->G, IVS14b+5G->A, 1898+1G->T, 4005+2T->C y 6 F508del, R117H y G551D.
[0104] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética de CFTR seleccionada de S549R, S1251N, E193K, F1052V y G1069R, y una mut G551. En algunas realizaciones, se describe en este do sintomáticamente la fibrosis quística en un paciente composición farmacéutica de esta divulgación al paci mutación genética de CFTR seleccionada de G178R,
S1251N, y una mutación de CFTR humana seleccionad describe en este documento un método para tratar, dis en un paciente que comprende administrar una cantida étodo para tratar, reducir la gravedad de, o tratar comprende administrar una cantidad eficaz de una paciente, tal como un mamífero, en donde el paciente R, G551S, G970R, G1244E, S1255P, G1349D, S549N, izaciones, esta divulgación proporciona un método para rmula (I), (II), (III), (IV), (V), o una sal farmacéuticamente n de CFTR humana seleccionada de G178R, G551S, 51N. En algunas realizaciones, se describe en este tar sintomáticamente la fibrosis quística en un paciente posición farmacéutica de esta divulgación al paciente, n genética CFTR seleccionada entre E193K, F1052V y umento en el transporte de cloruro en relación con el
étodo para tratar, reducir la gravedad de, o tratar comprende administrar una cantidad eficaz de una paciente, tal como un mamífero, en donde el paciente , D110H, R347H, R352Q, E56K, P67L, L206W, A455E, y D1152H. En algunas realizaciones, el método produce rte de cloruro de referencia del paciente.
étodo para tratar, reducir la gravedad de, o tratar comprende administrar una cantidad eficaz de una paciente, tal como un mamífero, en donde el paciente 1717-1G->A, 621 1G->T, 3120+1G->A, 1898+1G->A, ->A, 1812-1G->A, 1525-1G->A, 712-1G->T, 1248+1G-789+5G->A, 3849+10kbC->T, 3272-26A->G, 711+5G-G, 1717-8G->A, 1342-2A->C, 405+3A->C, 1716G/A, 898+1G->T, 4005+2T->C y 621+3A->G. En algunas a tratar, disminuir la gravedad o tratar sintomáticamente r una cantidad eficaz de una composición farmacéutica nde el paciente posee una mutación genética CFTR G->A, 3272-26A->G y 3849+10kbC->T. En algunas a tratar, disminuir la gravedad o tratar sintomáticamente r una cantidad eficaz de una composición farmacéutica nde el paciente posee una mutación genética CFTR
étodo para tratar, reducir la gravedad de, o tratar comprende administrar una cantidad eficaz de una paciente, tal como un mamífero, en donde el paciente R, G551S, G970R, G1244E, S1255P, G1349D, S549N, R347H, R352W67L, E455K, S1235R, S945L, R1070W, , 3120+1G->A, 1898+1G->A, 711 1G->T, 2622+1G->A, ->A, 712-1G->T, 1248+1G->A, 1341 1G->A, 3121-1G-, 3272-26A->G, 711+5G->A, 3120G->A, 1811 1.6kbA-5+3A->C, 1716G/A, 1811 1G->C, 1898+5G->T, 3850-->G, y una mutación CFTR humana seleccionada de
étodo para tratar, reducir la gravedad de, o tratar comprende administrar una cantidad eficaz de una paciente, tal como un mamífero, en donde el paciente R, G551S, G970R, G1244E, S1255P, G1349D, S549N, de CFTR humana seleccionada de F508del, R117DH y nto un método para tratar, disminuir la gravedad o tratar comprende administrar una cantidad eficaz de una como un mamífero, en donde el paciente posee una , G970R, G1244E, S1255P, G1349D, S549N, S549R y F508del, R117H y G551D. En algunas realizaciones, se la gravedad o tratar sintomáticamente la fibrosis quística z de una composición farmacéutica de esta divulgación
al paciente, como un mamífero, en donde el paciente E193K, F1052V y G1069R, y una mutación de CFTR algunas realizaciones, el método produce un aumento cloruro inicial del paciente.
[0105] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética de CFTR seleccionada de D579G, S1235R, S945L, R1070W, F1074L, D110E, D1 G551D. En algunas realizaciones, el método produce u transporte de cloruro de referencia del paciente.
[0106] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética CFTR seleccionada e 711 1G->T, 2622+1G->A, 405+1G->A, 406-1G->A, 40 >A, 1341 1G->A, 3121-1G->A, 4374+1G->T, 3850-1G >A, 3120G->A, 1811 1.6kbA->G, 711+3A->G, 1898 1811 1G->C, 1898+5G->T, 3850-3T->G, IVS14b+5G-> de CFTR humana seleccionada entre F508del, R117 documento un método para tratar, disminuir la graveda que comprende administrar una cantidad eficaz de un como un mamífero, en donde el paciente posee una 1811 1.6kbA->G, 2789+5G->A, 3272-26A->G y 3849+1 F508del, R117H y G551D. En algunas realizaciones, se la gravedad o tratar sintomáticamente la fibrosis quísti eficaz de una composición farmacéutica de esta divulg posee una mutación genética de CFTR seleccionada humana seleccionada de F508del, R117H.
[0107] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética de CFTR seleccionada de S549R, S1251N, E193K, F1052V, G1069R, R117C, D1 F1074L, D110E, D1270N, D1152H, 1717-1G->A, 621 405+1G->A, 406-1G->A, 4005+1G->A, 1812-1G->A, 15 >A, 4374+1G->T, 3850-1G->A, 2789+5G->A, 3849+10 k >G, 711+3A->G, 1898+3A->G, 1717-8G->A, 1342-2A-> 3T->G, IVS14b+5G->A, 1898+1G->T, 4005+2T->C y 6 F508del, R117H y G551D.
[0108] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética CFTR seleccionada entre S549R, S1251N, E193K, F1052V y G1069R. En algun para tratar, disminuir la gravedad o tratar sintomátic administrar una cantidad eficaz de una composición far en donde el paciente posee una mutación genética CFT G1349D, S549N, S549R y S1251N. En algunas realizac disminuir la gravedad o tratar sintomáticamente la fibr cantidad eficaz de una composición farmacéutica de e paciente posee una mutación genética CFTR selecciona el método produce un aumento en el transporte de cloru
[0109] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética CFTR seleccionada entre D579G, S1235R, S945L, R1070W, F1074L, D110E, D1 un aumento en el transporte de cloruro que está por en
[0110] En algunas realizaciones, se describe aquí e una mutación genética de CFTR seleccionada entre na seleccionada entre F508del, R117H y G551D. En transporte de cloruro en relación con el transporte de
étodo para tratar, reducir la gravedad de, o tratar comprende administrar una cantidad eficaz de una paciente, tal como un mamífero, en donde el paciente , D110H, R347H, R352Q, E56K, P67L, L206W, A455E, y D1152H, y una mutación de CFTR humana R117H y ento en el transporte de cloruro que está por encima del
étodo para tratar, reducir la gravedad de, o tratar comprende administrar una cantidad eficaz de una paciente, tal como un mamífero, en donde el paciente 1717-1G->A, 621 1G->T, 3120+1G->A, 1898+1G->A, ->A, 1812-1G->A, 1525-1G->A, 712-1G->T, 1248+1G-789+5G->A, 3849+10kbC->T, 3272-26A->G, 711+5G-G, 1717-8G->A, 1342-2A->C, 405+3A->C, 1716G/A, 98+1G->T, 4005+2T->C y 621+3A->G, y una mutación 551D. En algunas realizaciones, se describe en este tar sintomáticamente la fibrosis quística en un paciente posición farmacéutica de esta divulgación al paciente, ción genética de CFTR seleccionada de 1717-1G->A, ->T, y una mutación de CFTR humana seleccionada de ribe en este documento un método para tratar, disminuir un paciente que comprende administrar una cantidad al paciente, como un mamífero, en donde el paciente 89+5G->A y 3272-26A->G, y una mutación de CFTR
étodo para tratar, reducir la gravedad de, o tratar comprende administrar una cantidad eficaz de una paciente, tal como un mamífero, en donde el paciente R, G551S, G970R, G1244E, S1255P, G1349D, S549N, R347H, R352W67L, E455K, S1235R, S945L, R1070W, , 3120+1G->A, 1898+1G->A, 711 1G->T, 2622+1G->A, ->A, 712-1G->T, 1248+1G->A, 1341 1G->A, 3121-1G-, 3272-26A->G, 711+5G->A, 3120G->A, 1811 1.6kbA-5+3A->C, 1716G/A, 1811 1G->C, 1898+5G->T, 3850-->G, y una mutación CFTR humana seleccionada de
étodo para tratar, reducir la gravedad de, o tratar comprende administrar una cantidad eficaz de una paciente, tal como un mamífero, en donde el paciente R, G551S, G970R, G1244E, S1255P, G1349D, S549N, lizaciones, se describe en este documento un método e la fibrosis quística en un paciente que comprende tica de esta divulgación al paciente, como un mamífero, ccionada de G178R, G551S, G970R, G1244E, S1255P, , se describe en este documento un método para tratar, uística en un paciente que comprende administrar una vulgación al paciente, como un mamífero, en donde el tre E193K, F1052V y G1069R. En algunas realizaciones, relación con el transporte de cloruro inicial del paciente.
étodo para tratar, reducir la gravedad de, o tratar comprende administrar una cantidad eficaz de una paciente, tal como un mamífero, en donde el paciente , D110H, R347H, R352Q, E56K, P67L, L206W, A455E, y D1152H. En algunas realizaciones, el método produce el transporte de cloruro de referencia del paciente.
étodo para tratar, reducir la gravedad de, o tratar
sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética CFTR seleccionada e 711 1G->T, 2622+1G->A, 405+1G->A, 406-1G->A, 40 >A, 1341 1G->A, 3121-1G->A, 4374+1G->T, 3850-1G >A, 3120G->A, 1811 1.6kbA->G, 711+3A->G, 1898 1811 1G->C, 1898+5G->T, 3850-3T->G, IVS14b+5G-realizaciones, se describe en este documento un métod la fibrosis quística en un paciente que comprende adm de esta divulgación al paciente, como un mamífero, seleccionada entre 1717-1G->A, 1811 1,6 kbA->G, realizaciones, se describe en este documento un métod la fibrosis quística en un paciente que comprende adm de esta divulgación al paciente, como un mamífero, seleccionada entre 2789+5G->A y 3272-26A->G.
[0111] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética de CFTR seleccionada de S549R, S1251N, E193K, F1052V, G1069R, R117C, D1 F1074L, D110E, D1270N, D1152H, 1717-1G->A, 621 405+1G->A, 406-1G->A, 4005+1G->A, 1812-1G->A, 15 >A, 4374+1G->T, 3850-1G->A, 2789+5G->A, 3849+10 >G, 711+3A->G, 1898+3A->G, 1717-8G->A, 1342-2A-3T->G, IVS14b+5G->A, 1898+1G->T, 4005+2T->C y 6 F508del, R117H y G551D, y una o más mutaciones de
[0112] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética de CFTR seleccionada de S549R, S1251N, E193K, F1052V y G1069R, y una o m R117DH y G551DH. En algunas realizaciones, se des gravedad o tratar sintomáticamente la fibrosis quística e de una composición farmacéutica de esta divulgación al mutación genética de CFTR seleccionada de G178R, S1251N, y una o más mutaciones CFTR humanas realizaciones, se describe en este documento un métod la fibrosis quística en un paciente que comprende adm de esta divulgación al paciente, como un mamífero, e seleccionada entre E193K, F1052V y G1069R, y una o R117H y G551D. En algunas realizaciones, el método p el transporte de cloruro inicial del paciente.
[0113] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética CFTR seleccionada entre D579G, S1235R, S945L, R1070W, F1074L, D110E, D F508del, R117H y G551D. En algunas realizaciones, el está por encima del transporte de cloruro de referencia
[0114] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética CFTR seleccionada e 711 1G->T, 2622+1G->A, 405+1G->A, 406-1G->A, 40 >A, 1341 1G->A, 3121-1G->A, 4374+1G->T, 3850-1G >A, 3120G->A, 1811 1.6kbA->G, 711+3A->G, 1898 1811 1G->C, 1898+5G->T, 3850-3T->G, IVS14b+5G-mutaciones CFTR humanas seleccionadas de F508del, un método para tratar, reducir la gravedad de, o tr comprende administrar una cantidad eficaz de una c paciente, tal como un mamífero, en donde el paciente p >A, 1811 1.6kbA->G, 2789+5G->A, 3272-26A->G y comprende administrar una cantidad eficaz de una paciente, tal como un mamífero, en donde el paciente 1717-1G->A, 621 1G->T, 3120+1G->A, 1898+1G->A, ->A, 1812-1G->A, 1525-1G->A, 712-1G->T, 1248+1G-789+5G->A, 3849+10kbC->T, 3272-26A->G, 711+5G-G, 1717-8G->A, 1342-2A->C, 405+3A->C, 1716G/A, 898+1G->T, 4005+2T->C y 621+3A->G. En algunas a tratar, disminuir la gravedad o tratar sintomáticamente r una cantidad eficaz de una composición farmacéutica nde el paciente posee una mutación genética CFTR 5G->A, 3272-26A->G y 3849+10kbC->T. En algunas a tratar, disminuir la gravedad o tratar sintomáticamente r una cantidad eficaz de una composición farmacéutica nde el paciente posee una mutación genética CFTR
étodo para tratar, reducir la gravedad de, o tratar comprende administrar una cantidad eficaz de una paciente, tal como un mamífero, en donde el paciente R, G551S, G970R, G1244E, S1255P, G1349D, S549N, R347H, R352W67L, E455K, S1235R, S945L, R1070W, , 3120+1G->A, 1898+1G->A, 711 1G->T, 2622+1G->A, ->A, 712-1G->T, 1248+1G->A, 1341 1G->A, 3121-1G-, 3272-26A->G, 711+5G->A, 3120G->A, 1811 1.6kbA-5+3A->C, 1716G/A, 1811 1G->C, 1898+5G->T, 3850-->G, y una mutación CFTR humana seleccionada de humana seleccionadas entre F508del, R117H y G551D.
étodo para tratar, reducir la gravedad de, o tratar comprende administrar una cantidad eficaz de una paciente, tal como un mamífero, en donde el paciente R, G551S, G970R, G1244E, S1255P, G1349D, S549N, taciones de CFTR humanas seleccionadas de F508del, en este documento un método para tratar, disminuir la aciente que comprende administrar una cantidad eficaz nte, como un mamífero, en donde el paciente posee una , G970R, G1244E, S1255P, G1349D, S549N, S549R y cionadas de F508del, R117H y G551D. En algunas a tratar, disminuir la gravedad o tratar sintomáticamente r una cantidad eficaz de una composición farmacéutica de el paciente posee una mutación genética de CFTR utaciones CFTR humanas seleccionadas entre F508del, un aumento en el transporte de cloruro en relación con
étodo para tratar, reducir la gravedad de, o tratar comprende administrar una cantidad eficaz de una paciente, tal como un mamífero, en donde el paciente , D110H, R347H, R352Q, E56K, P67L, L206W, A455E, y D1152H, y una o más mutaciones CFTR humanas do produce un aumento en el transporte de cloruro que ciente.
étodo para tratar, reducir la gravedad de, o tratar comprende administrar una cantidad eficaz de una paciente, tal como un mamífero, en donde el paciente 1717-1G->A, 621 1G->T, 3120+1G->A, 1898+1G->A, ->A, 1812-1G->A, 1525-1G->A, 712-1G->T, 1248+1G-789+5G->A, 3849+10kbC->T, 3272-26A->G, 711+5G-G, 1717-8G->A, 1342-2A->C, 405+3A->C, 1716G/A, 898+1G->T, 4005+2T->C y 621+3A->G, y una o más H y G551D. En algunas realizaciones, se describe aquí intomáticamente fibrosis quística en un paciente que ición farmacéutica de la presente divulgación para el una mutación genética CFTr seleccionada de 1717-1G-10kbC->T, y una o más mutaciones CFTR humanas
seleccionado entre F508del, R117H y G551D. En algun para tratar, disminuir la gravedad o tratar sintomátic administrar una cantidad eficaz de una composición far en donde el paciente posee una mutación genética CF más mutaciones CFTR humanas seleccionadas entre F
[0115] En algunas realizaciones, el paciente es heteroc otra mutación causante de FQ en el otro alelo. En algun la otra mutación genética de CFTR es cualquier mutació de CFTR y una mutación de CFTR en el segundo alelo función residual de CFTR o un defecto en la actividad d
[0116] En algunas realizaciones, la mutación causante d el paciente es heterocigoto y tiene una mutación que ca enumeradas en la tabla de la FIG. 4 y otra mutación q mutaciones CFTR enumeradas en la Tabla 5A.
Ta
Mutaciones CFT Q39X
W57X
E60X
R75X
E92X
Q98X
Y122X
L218X
Q220X
C276X
Q290X
G330X
W401X
Q414X
S434X
S466X
S489X
Q493X
W496X
Q525X
G542X
Q552X
R553X
E585X
G673X
R709X
K710X
L732X
R764X
R785X
R792X
E822X
W846X
R851X
Q890X
S912X
W1089X
Y1092X
E1104X
R1158X
R1162X
S1196X
W1204X
S1255X
W1282X
realizaciones, se describe en este documento un método nte la fibrosis quística en un paciente que comprende éutica de esta divulgación al paciente, como un mamífero, seleccionada entre 2789+5G->A y 3272-26A->G, y una o del, R117H y G551D.
to que tiene una mutación causante de FQ en un alelo y realizaciones, el paciente es heterocigoto para F508del, y ue cause FQ, que incluye, entre otros, F508del en un alelo e CFTR que está asociado con función mínima de CFTR, ompuerta del canal CFTR.
Q se selecciona de la Tabla 5A. En algunas realizaciones, FQ en un alelo de CFTR seleccionado de las mutaciones causa FQ en el otro alelo de CFTR se selecciona de las
5A.
Mutacione Q1313X__
621 1G ^T 711 1G ^T 711+5G ^A 712-1G^T 405+1G ^A 405+3A^C 406-1G ^A 621 1G ^T 1248+1G^A 1341 1G ^A 1717-1G^A 1811 1.6kb 1811 1G ^C 1812-1G^A 1898+1G^A 2622+1G^A 3120+1G^A 3120G ^A 3850-1G^A 4005+1G^A 4374+1G^T
663delT
2183AA^G CFTRdel2,3 3659delC
394delTT
2184insA
3905insT
2184delA
1078delT
1154insTC 2183delAA^ 2143delT
1677delTA 3876delA
2307insA
4382delA
4016insT
2347delG
3007delG
574delA
2711delT
3791delC
CFTRdele22 457TAT^G 2043delG
2869insG
3600+2insT 3737delA
4040delA
541delC
A46D
T338I
R347P
L927P
G85E
S341P
L467P
ontinuación)
TR
__
(Con
I507de
V520F
A559T
R560T
R560S
A561E
Y569D
L1065P
R1066C
R1066M
L1077P
H1085R
M1101K
N1303K
2789+5G^A
3849+10kbC^
T
3272-26A+G
711+3A+G
E56K
P67L
R74W
D110E
D110H
R117C
L206W
R347H
R352Q
A455E
D579G
E831X
S945L
S977F
F1052V
R1070W
F1074L
D1152H
D1270N
R117H
G178R
S549N
S549R
G551D
G551S
G1244E
S1251N
S1255P
G1349D
tinuación)
Tabla 5B: Criterios Mutación
taciones de CFTR
•ausente o poco 405+1G—-A 48+1G 1811 1G—C 3040G -C

mutaciones de cambio ,23
de marco 124del23bp 852del22 •%PI >50% y/o SwCl- 991del5
>86 mmol/L
• proteína confusa y/o
truncada
24del7 4209TGTT-55del9 AA
A 2105-17del13i
AGAAA
21del11
(Con
Criterios Mutación
Mutaciones de Clase II, A46Db V5 III, IV que no responden G85E A5 al Compuesto III solo o R347P R5 en combinación con L467Pb R5 Compuesto II o I507del A5 Compuesto IV
•%PI>50% y/o SwCl
>86 mmol/L Y
• No sensibles in vitro al
Compuesto III solo o en
combinación con
Compuesto II o
Compuesto IV___________________________ Nota:%PI: porcentaje de pacientes heterocig que son pancreáticos insuficientes; SwCl-: clor CFTR en el registro de pacientes CFTR2
a También conocido como 2183delAA^G.
b Datos no publicados.____________________
[0117] La Tabla 5B anterior incluye ciertas mutaciones f por un ensayo de genotipado aclarado por la FDA, pero
[0118] En algunas realizaciones, se describe aquí sintomáticamente la fibrosis quística en un paciente co una mutación de MF que no se espera que resp con genotipo F508del/F508del (F/F) (homocigoto
(heterocigotos para F508del y una mutación gating que que responde al Compuesto III). En algunas realizacion mutación MF que no se espera que responda al Co Compuesto III. En algunas realizaciones, un paciente mutaciones de MF de la Tabla 5B.
[0119] En algunas realizaciones, el paciente es heteroc cualquier mutación que cause FQ, incluidas mutaciones mutaciones de cambio de marco (<3 nucleótidos) de in no pequeñas (>3 nucleótidos) de inserción o deleción (i Compuesto III solo o en combinación con el Compuesto
[0120] En algunas realizaciones, el paciente es heteroc mutación de truncamiento. En algunas realizaciones e truncamiento listada en la Tabla 5B.
[0121] En algunas realizaciones, el paciente es heteroc mutación de empalme. En algunas realizaciones especí enumerada en la Tabla 5B.
[0122] En algunas realizaciones, el paciente es heteroc pequeña mutación de cambio de marco (<3 nucleótido específicas, la pequeña mutación de cambio de marc pequeña mutación de cambio de marco (<3 nucleótidos)
[0123] En algunas realizaciones, el paciente es heteroc cualquier mutación causante de FQ que se espera qu cualquier combinación de (i) un nuevo compuesto el compuestos de Fórmula (I), (II), (III), (IV) o (V), y sus sale y (ii) Compuesto II y/o Compuesto III y/o Compuesto IV.
[0124] En algunas realizaciones, el paciente es heteroc cualquier mutación causante de FQ que se espera que ación)
Y569Db N1303K
b L1065P
R1066C
L1077Pb
M1101K
_____________________________________________
s F508del-CFTR en el registro de pacientes CFTR2
en sudor medio de pacientes heterocigotos F508del-
_____________________________________________
ionales mínimas de CFTR ejemplares, que son detectables incluye una lista exhaustiva
método para tratar, reducir la gravedad de, o tratar enotipos F508del/MF (F/MF) (heterocigoto para F508del y a a moduladores de CFTR, como el Compuesto III);
F508del); y/o con genotipos F508del/gating (F/G) sabe que responde al modulador de CFTR (por ejemplo, un paciente con genotipos F508del/MF (F/MF) tienen una esto II, Compuesto III y tanto al Compuesto II como al los genotipos F508del/MF (F/MF) tiene cualquiera de los
to para F508del, y la otra mutación genética de CFTR es truncamiento, mutaciones de corte y empalme, pequeñas ción o deleción (ins/del); mutaciones de cambio de marco del); y mutaciones de Clase II, III, IV que no responden al o el Compuesto IV.
to para F508del, y la otra mutación genética CFTR es una cíficas, la mutación de truncamiento es una mutación de
to para F508del, y la otra mutación genética CFTR es una s, la mutación de empalme es una mutación de empalme
to para F508del, y la otra mutación genética CFTR es una e inserción o deleción (ins/del). En algunas realizaciones ins/del) de inserción o deleción (<3 nucleótidos) es una inserción o deleción (ins/del) enumerada en la Tabla 5B.
to para F508del, y la otra mutación genética de CFTR es ea y/o responda a, en base a datos in vitro y/o clínicos, o entre los descritos en este documento (por ejemplo, rmacéuticamente aceptables y sus derivados deuterados),
to para F508del, y la otra mutación genética de CFTR es a y/o es sensible, en base a datos in vitro/clínicos y, a la
triple combinación de un nuevo compuesto elegido entr de Fórmula (I), (II), (III), (IV) o (V), y sus sales farma Compuesto II y Compuesto III.
[0125] En algunas realizaciones, el paciente es heteroci mutación de cambio de marco no pequeña (>3 nucleótid específicas, la mutación de cambio de marco de inserc mutación de cambio de marco de lectura (ins/del) no enumerada en la Tabla 5B.
[0126] En algunas realizaciones, el paciente es hetero mutaciones de Clase II, III, IV que no responden al Co Compuesto IV. En algunas realizaciones específicas, Compuesto III solo o en combinación con el Compuesto no responden al Compuesto III solo o en combinación c
[0127] En algunas realizaciones, el paciente es hetero cualquier mutación enumerada en la Tabla 5B.
[0128] En algunas realizaciones, el paciente es hetero cualquier mutación enumerada en la Tabla 5A, 5B, y la
[0129] En algunas realizaciones, el paciente es hetero cualquier mutación enumerada en la Tabla 5A. En algun la otra mutación genética de CFTR es cualquier mutaci paciente es heterocigoto para F508del, y la otra mutació FIG. 4.
[0130] En algunas realizaciones, el paciente es homocig
[0131] En algunas realizaciones, el paciente es heteroci CFTR seleccionado de las mutaciones enumeradas en l alelo de CFTR se selecciona de las mutaciones CFTR e
[0132] Los pacientes con un genotipo de la mutaci heterocigotos F508delCFTR con un segundo alelo de gating y clínicamente demostró ser sensible al Compu S549N, S549R, G551D, G551S, G1244E, S1251N, S12
[0133] Los pacientes con un genotipo de F508del heterocigotos F508delCFTR con un segundo alelo de C de la cantidad o la función de proteínas en la superfi mutaciones del gen CFTR que se sabe que dan como r realizaciones, una mutación de función residual de C 26A4^G, 711+3A^G, E56K, P67L, R74W, D110E, D11 S945L, S977F, F1052V, R1070W, F1074L, D1152H, D12 de la función residual de CFTR se selecciona de R117 R1070Q, R31C, D614G, G1069R, R1162L, E56K, A106 de la función residual de CFTR se selecciona de R117 R1070Q, R31C, D614G, G1069R, R1162L, E56K o A10
[0134] En algunas realizaciones, se describe aquí
sintomáticamente la fibrosis quística en un paciente composición farmacéutica de la presente divulgación p posee una mutación genética CFTR seleccionada de las
[0135] En algunas realizaciones, la composición según l sintomáticamente la fibrosis quística en pacientes que e epitelios respiratorios y no respiratorios. La presencia d detectarse fácilmente usando métodos conocidos en la t o histoquímicas estándar. Dichos métodos i electrofisiológicas in vivo o ex vivo, medición de concent histoquímicas ex vivo para controlar la densidad de la s de CFTR se puede detectar fácilmente en pacientes q mutaciones diferentes, incluidos pacientes heterocigot mutaciones como la mutación G551D o la mutación R11 s descritos en este documento (por ejemplo, compuestos ticamente aceptables y sus derivados deuterados), y el
o para F508del, y la otra mutación genética CFTR es un de inserción o deleción (ins/del). En algunas realizaciones o deleción (ins/del) no pequeña (>3 nucleótidos) es una ueña (>3 nucleótidos) de inserción o deleción (ins/del)
to para F508del, y la otra mutación genética CFTR son esto III solo o en combinación con el Compuesto II o el mutaciones de Clase II, III, IV que no responden al el Compuesto IV son mutaciones de Clase II, III, IV que l Compuesto II o Compuesto IV listado en la Tabla 5B.
to para F508del, y la otra mutación genética CFTR es
to para F508del, y la otra mutación genética CFTR es 4.
to para F508del, y la otra mutación genética CFTR es realizaciones, el paciente es heterocigoto para F508del y enumerada en la Tabla 5B. En algunas realizaciones, el enética de CFTR es cualquier mutación enumerada en la
para F508del.
o que tiene una mutación causante de FQ en un alelo de bla de la FIG. 4 y otra mutación que causa FQ en el otro eradas en la Tabla 5B.
F508del/gating se definen como pacientes que son R que contiene una mutación asociada con un defecto III. Los ejemplos de tales mutaciones incluyen: G178R, y G1349D.
ción residual se definen como pacientes que son que contiene una mutación que resulta en la reducción celular que puede producir actividad CFTR parcial. Las ltado un fenotipo de función residual incluyen, en algunas seleccionada de 2789+5G^A, 3849+l0kbC^T, 3272-R117C, L206W, R347H, R352Q, A455E, D579G, E831X, , E193K y K1060T. En algunas realizaciones, la mutación S1235R, I1027T, R668C, G576A, M470V, L997F, R75Q, E193K o K1060T. En algunas realizaciones, la mutación S1235R, I1027T, R668C, G576A, M470V, L997F, R75Q, .
método para tratar, reducir la gravedad de, o tratar e comprende administrar una cantidad eficaz de una el paciente, tal como un mamífero, en donde el paciente taciones enumeradas en la FIG. 4.
vención es útil para tratar, reducir la gravedad de, o tratar en actividad CFTR residual en la membrana apical de los ctividad residual de CFTR en la superficie epitelial puede ica, por ejemplo, técnicas electrofisiológicas, bioquímicas tifican la actividad de CFTR usando técnicas ones de Cl en sudor o salivares, o técnicas bioquímicas o rficie celular. Usando tales métodos, la actividad residual son heterocigotos u homocigotos para una variedad de para la mutación más común, F508del, así como otras En algunas realizaciones, las composiciones descritas en
el presente documento son útiles para tratar, disminuir l pacientes que presentan poca o ninguna actividad resid descritas en el presente documento son útiles para tratar quística en pacientes que presentan poca o ninguna acti respiratorio.
[0136] En algunas realizaciones, las composiciones de disminuir la gravedad de la fibrosis quística en paciente farmacológicos. Dichos métodos aumentan la cantidad d actividad CFTR hasta ahora ausente en un paciente o a un paciente.
[0137] En algunas realizaciones, las composiciones de disminuir la gravedad de la fibrosis quística en paciente residual.
[0138] En algunas realizaciones, las composiciones desc la gravedad de, o tratar sintomáticamente la fibrosis quí ejemplo, un fenotipo clínico leve a moderado que norm residual en la membrana apical del epitelio. Dichos pancreática.
[0139] En algunas realizaciones, las composiciones desc la gravedad de, o tratar sintomáticamente pacientes diagn y ausencia bilateral congénita de los vasos deferentes, actividad residual de CFTR.
[0140] En algunas realizaciones, la presente divulgación del canal de aniones in vitro o in vivo, comprendiendo la invención. En algunas realizaciones, el canal de aniones realizaciones, el canal de aniones es un canal de cloruro.
[0141] La cantidad exacta de una composición farmacéu especie, edad, y estado general del sujeto, la graved administración, y similares. Los compuestos de esta dosificación para facilitar la administración y uniformid dosificación" como se usa en este documento se refiere el paciente a ser tratado. Se entenderá, sin embargo, compuestos y composiciones de esta descripción dentro específico para cualquier paciente u organismo particul trastorno que se está tratando y la gravedad del trast composición específica empleada; la edad, el peso cor momento de administración, la vía de administración y la la duración del tratamiento; fármacos usados en com empleado, y factores similares bien conocidos en la té documento, significa un animal, como un mamífero, e incl
[0142] En algunas realizaciones, la divulgación también métodos de tratamiento utilizando compuestos mar mencionados, que tienen las mismas estructuras como se han sido reemplazados por un átomo o átomos que tiene masa atómica o número de masa del átomo que normal Los ejemplos de isótopos que están disponibles comercial de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, flúor 35S, 18F y 36Cl, respectivamente.
[0143] Los compuestos y sales marcados con isótopos Pueden ser adecuados para medicamentos y/o varios tip de sustrato. Por ejemplo, los compuestos marcados con t varios tipos de ensayos, tales como ensayos de dist relativamente simple y su excelente detectabilidad. Por ej útiles con ventajas terapéuticas potenciales sobre los co y sales marcados con deuterio (2H) pueden tener una m están marcados con isótopos debido al efecto isotópico ci metabólica se traduce directamente en un aumento de la s Los compuestos y sales marcados con isótopos se puede ravedad o tratar sintomáticamente la fibrosis quística en de CFTR. En algunas realizaciones, las composiciones isminuir la gravedad o tratar sintomáticamente la fibrosis ad residual de CFTR en la membrana apical del epitelio
itas en el presente documento son útiles para tratar o ue exhiben actividad CFTR residual utilizando métodos FTR presente en la superficie celular, induciendo así una entando el nivel existente de actividad CFTR residual en
itas en el presente documento son útiles para tratar o con ciertos genotipos que presentan actividad de CFTR
s en el presente documento son útiles para tratar, reducir ca en pacientes dentro de ciertos fenotipos clínicos, por ente se correlaciona con la cantidad de actividad CFTR notipos incluyen pacientes que presentan suficiencia
s en el presente documento son útiles para tratar, reducir ticados con suficiencia pancreática, pancreatitis idiopática nfermedad pulmonar leve en donde el paciente presenta
refiere a un método para aumentar o inducir la actividad esta en contacto el canal con una composición según la un canal de cloruro o un canal de bicarbonato. En algunas
requerida variará de sujeto a sujeto, dependiendo de la de la enfermedad, el agente particular, su modo de ulgación se pueden formular en forma de unidad de de la dosificación. La expresión "forma de unidad de na unidad de agente físicamente discreta apropiada para e el médico tratante decidirá el uso diario total de los l alcance del buen juicio médico. El nivel de dosis eficaz dependerá de una variedad de factores que incluyen el o; la actividad del compuesto específico empleado; la ral, la salud general, el sexo y la dieta del paciente; el ocidad de excreción del compuesto específico empleado; ción o coincidentemente con el compuesto específico ca médica. El término "paciente", como se usa en este o más como un ser humano.
refiere a compuestos y composiciones para su uso en os con isótopos de los compuestos anteriormente scribe aquí, excepto que uno o más átomos de los mismos na masa atómica o un número de masa que difiere de la nte se produce de forma natural (marcado con isótopos). nte y son adecuados para la divulgación incluyen isótopos loro, por ejemplo 2H, 3H, 13C, 14C, 15N, 18O, 17O, 31P, 32P,
pueden utilizar en un número de maneras beneficiosas. de ensayos, tales como ensayos de distribución de tejido (3H) y/o carbono 14 (14C) son particularmente útiles para ución de tejido de sustrato, debido a su preparación plo, los marcados con deuterio (2H) son terapéuticamente uestos no marcados con 2H. En general, los compuestos or estabilidad metabólica en comparación con los que no ico que se describe a continuación. Una mayor estabilidad ivida in vivo o en dosis más bajas, lo que podría desearse. reparar normalmente llevando a cabo los procedimientos
descritos en los esquemas de síntesis y la descripción rel del presente texto, reemplazando un reactivo no marc reactivo marcado con isótopos fácilmente disponible.
[0144] En algunas realizaciones, los compuestos y sales En algunas realizaciones específicas, los compuestos y (2H), en los que uno o más átomos de hidrógeno en el químicas, el deuterio se representa como "2H" o "D".
[0145] Los compuestos marcados con deuterio (2H) y sal a modo del efecto isotópico cinético primario. El efecto is reacción química que resulta del intercambio de núcleo energías del estado fundamental necesarias para la for isotópico. El intercambio de un isótopo más pesado gen del estado fundamental para un enlace químico y, por lo t la velocidad. Si la rotura de la unión se produce en o en l la coordenada de una reacción de múltiples productos, sustancialmente. Para una explicación: si el deuterio intercambiable, las diferencias de velocidad de kM/ko = 2-y R. D. Tung, Deuterium In Drug Discovery and Develop "Using deuterium in drug discovery: leaving the label in t
[0146] La concentración del (de los) isótopo(s) (por eje con isótopos y la sal de la descripción puede ser definid de enriquecimiento isotópico" como se usa en este docu abundancia natural de un isótopo especificado. En algu divulgación se denota deuterio, dicho compuesto tiene deuterio designado de al menos 3500 (52,5% de incorpo menos 4000 (60% de incorporación de deuterio), al men (75% de incorporación de deuterio), al menos 5500 (82 incorporación de deuterio), al menos 6333,3 (95% d incorporación de deuterio), al menos 6600 (99% de incorporación de deuterio).
[0147] Cuando descubre y desarrolla agentes terapéutic farmacocinéticos conservando las propiedades in vitr compuestos con perfiles farmacocinéticos deficientes so
[0148] Una persona de experiencia ordinaria en la técn metabólicamente lábiles en un compuesto o metabolito de DMPK superiores mientras se mantiene la actividad de hidrógeno. La propiedad o propiedades superiores media, el aclaramiento, el metabolismo y/o incluso l medicamento. La deuteración también puede cambia compuesto deuterado.
[0149] En algunas realizaciones, la divulgación incluye d este documento y de sus sales farmacéuticamente acept se describen en la FIG. 1.
[0150] En algunas realizaciones, el Compuesto III' como Patente de Estados Unidos N° 8.865.902, y CTP-656.
[0151] En algunas realizaciones, el Compuesto III' es: nada, en la parte de ejemplo y en la parte de preparación con isótopos por un reactivo no marcado con isótopos.
rcados con isótopos son los marcados con deuterio (2H). s marcados con isótopos están marcados con deuterio an sido reemplazados por deuterio. En las estructuras
ueden manipular el metabolismo oxidativo del compuesto ico cinético primario es un cambio en la velocidad de una tópicos, que a su vez es causada por el cambio en las ión de enlaces covalentes después de este intercambio ente da como resultado una disminución de la energía , provoca una reducción en la rotura del enlace que limita roximidades de una región de punto de silla a lo largo de elaciones de distribución de productos pueden alterarse unido a un átomo de carbono en una posición no n típicas. Para más información, consulte S. L. Harbeson , Ann. Rep. Med. Chem. 2011,46, 403-417; y T. G. Gant rug" J. Med. Chem. 2014, 57, 3595-3611.
, deuterio) incorporado(s) en los compuestos marcados el factor de enriquecimiento isotópico. El término "factor to significa la relación entre la abundancia isotópica y la realizaciones, si un sustituyente en un compuesto de la actor de enriquecimiento isotópico para cada átomo de n de deuterio en cada átomo de deuterio designado), al 00 (67,5% de incorporación de deuterio), al menos 5000 de incorporación de deuterio), al menos 6000 (90% de corporación de deuterio), al menos 6466,7 (97% de rporación de deuterio) o al menos 6633,3 (99,5% de
el experto en la técnica intenta optimizar los parámetros seables. Puede ser razonable suponer que muchos sceptibles al metabolismo oxidativo.
entenderá que la deuteración de una o más posiciones o puede conducir a la mejora de una o más propiedades gica en comparación con los correspondientes análogos MPK pueden tener un impacto en la exposición, la vida equisitos alimentarios para una absorción óptima del metabolismo en otras posiciones no deuteradas del
dos deuterados de los nuevos compuestos descritos en s. Los ejemplos no limitantes de compuestos deuterados
tiliza aquí incluye el compuesto deuterado descrito en la

[0152] Las realizaciones ejemplares de la divulgación luyen:Los nuevos compuestos descritos en el presente
documento (por ejemplo, Compuestos de Fórmulas (I) -derivados deuterados de cualquiera de los anteriores, in específicamente en el presente documento) se puede pre Por ejemplo, se pueden preparar de acuerdo con los pro mediante las síntesis ejemplar descrita a continuación en nuevos Compuestos de Fórmulas (I) -(V) y sus sales far similar a los Compuestos de Fórmulas (I) -(V) y sus sales reactivos en los que uno o más átomos de hidrógeno deuterium in drug discovery: leaving the label in the drug"
[0153] En algunas realizaciones, los Compuestos de aceptables de los mismos, y derivados deuterados de cu los Esquemas 1-2, en donde las variables de la misma son (I), (II), (III), (IV) o (V) anteriores, y en donde cada Ra se e Xa se elige independientemente entre F o Cl. Pueden emp cada paso representado en los esquemas. En algunas re D-1 y F-1 en los Esquemas 2-4 es independientemente Cl. O y P en el Esquema 6 es independientemente F.
[0154] En algunas realizaciones, como se muestra en el compuesto de fórmula (F) o una sal del mismo con un co un compuesto de Fórmula (IIIa), una sal farmacéutica cualquiera de los anteriores.
Esquem , sales farmacéuticamente aceptables de los mismos y idos los compuestos de la Figura 1 y los representados ar mediante métodos adecuados conocidos en la técnica.
imientos descritos en el documento WO2016/057572 y Ejemplos. Por ejemplo, los derivados deuterados de los éuticamente aceptables se pueden preparar de manera rmacéuticamente aceptables empleando intermedios y/o reemplazan con deuterio. Por ejemplo, consulte Using Med. Chem. 2014, 57, 3595-3611.
mulas (III), (IV) y (V) y las sales farmacéuticamente uiera de los anteriores se preparan como se muestra en da uno e independientemente son como los de la Fórmula independientemente entre grupos C1-C4 alquilo; y cada rse condiciones adecuadas conocidas en la técnica para aciones, cada Xa para las Fórmulas B, C, D, F, B-1, C-1, n algunas realizaciones, cada Xa para las Fórmulas D, L,
quema 1, los métodos comprenden hacer reaccionar un uesto de Fórmula (G) o una sal del mismo para generar te aceptable del mismo o un derivado deuterado de

[0155] Cualesquiera condiciones adecuadas, tales como técnica pueden ser utilizadas. En algunas realizaciones, presencia de una base, tal como un carbonato metálico (p
[0156] En algunas realizaciones, compuestos de Fórmula derivados deuterados de cualquiera de los anteriores, en (F), (G) y (IIIa), se preparó mediante los procedimientos e
[0157] En algunas realizaciones, se emplea una sal de u emplea una sal de HCl de un compuesto de Fórmula (G).
[0158] Un compuesto de fórmula (F) o una sal del mism puede preparar por cualquier método adecuado conocido de la síntesis ejemplares descritas a continuación en los
[0159] En algunas realizaciones, como se muestra en farmacéuticamente aceptable del mismo, o un derivado d un método que comprende hacer reaccionar un compuest Fórmula (E) o una sal del mismo. En algunas realizaciones deuterados de cualquiera de los anteriores se preparan compuesto de Fórmula (A) o una sal del mismo con un co un compuesto de Fórmula (C) o una sal del mismo; e h generar un compuesto de Fórmula (D) o una sal del mism la técnica para las etapas (a), (b) y (c) del Esquema 2 acoplamiento entre ácido carboxílico y sulfonamida o aque de hidrólisis del éster para el paso (b) y los de una reacció
[0160] En algunas realizaciones, la etapa (a) del Esquema En algunas realizaciones específicas, la etapa (a) se re realizaciones, en el paso (a), la reacción de un compuesto Fórmula (E) o una sal del mismo comprende hacer reacci un reactivo de acoplamiento, tal como carbonildiimidazol ( una sal del mismo en presencia de una base, tal como una de Fórmula (D) o una sal del mismo se hace reaccionar c (E) o una sal del mismo, y luego posteriormente con un c de una reacción nucleofílica de amina, conocida en la reacción representada en el Esquema 1 se realiza en ejemplo, Na2CO3 o K2CO3).
a), sales farmacéuticamente aceptables de los mismos o que Y2 es N e Y1 es CH en cada una de las Fórmulas l esquema 1.
ompuesto de Fórmula (G). En algunas realizaciones, se
un compuesto de Fórmula (G) o una sal del mismo se la técnica, por ejemplo, aquellos en WO2016/57572 y los plos.
Esquema 2, un compuesto de Fórmula (F), una sal terado de cualquiera de lo anterior se prepara mediante e Fórmula (D) o una sal del mismo con un compuesto de s compuestos de Fórmula (D), sus sales o los derivados diante un método que comprende hacer reaccionar un uesto de Fórmula (B) o una sal del mismo para generar olizar el -C(O)ORa del compuesto de Fórmula (C) para e puede usar cualquier condición adecuada conocida en ontinuación, tales como aquellas para una reacción de s para una acilación de sulfonamida para la etapa (a), los ucleofílica de amina para el paso (c).
a continuación se lleva a cabo en presencia de una base. a en presencia de una base no nucleófila. En algunas Fórmula (D) o una sal del mismo con un compuesto de r un compuesto de Fórmula (D) o una sal del mismo con I), y posteriormente con un compuesto de Fórmula (E) o se no nucleófila. En algunas realizaciones, un compuesto CDI antes de la reacción con un compuesto de Fórmula uesto de Fórmula (E) o una sal del mismo en presencia
de una base, tal como DBU (1,8-Diazabiciclo(5.4.0)undec-
[0161] En algunas realizaciones, la etapa (b) del Esquema En algunas realizaciones, la etapa (b) se realiza en pres algunas realizaciones, la etapa (b) se realiza en presencia
[0162] En algunas realizaciones, la etapa (c) del Esquema En algunas realizaciones, la etapa (c) se realiza en pre K2C 03).
no).
a continuación se lleva a cabo en presencia de una base. ia de una base acuosa, tal como hidróxido acuoso. En un hidróxido metálico acuoso, como NaOH acuoso.
a continuación se lleva a cabo en presencia de una base. cia de un carbonato metálico (por ejemplo, Na2CO3 o

[0163] En algunas realizaciones, se describe aquí un mét de preparación de un compuesto de la siguiente fórmula:

o una sal farmacéuticamente aceptable del mismo, o un d comprende hacer reaccionar un compuesto de fórmula (F 1) o una sal del mismo, en donde Xa es F o Cl, como se ado deuterado de cualquiera de los anteriores. El método o una sal del mismo con un compuesto de Fórmula (G-stra en el Esquema 3:

[0164] Se pueden usar cualesquiera condiciones adecu conocidas en la técnica. En algunas realizaciones, la reac de una base, tal como un carbonato metálico (por ejemplo s, tales como las de una reacción nucleofílica de amina, n representada en el Esquema 3 se realiza en presencia a2CO3 o K2CO3).
[0165] En algunas realizaciones, una sal del compuest emplea una sal de HCl de un compuesto de Fórmula (G
[0166] Un compuesto de fórmula (F-1) o una sal del mis puede preparar por cualquier método adecuado conocid de las síntesis ejemplares descritas a continuación en l
[0167] En algunas realizaciones, como se muestra en mismo, o un derivado deuterado de cualquiera de los ant reaccionar un compuesto de Fórmula (D-1 ) o una sal mismo. En algunas realizaciones, los compuestos de preparan mediante un método que comprende hacer re con un compuesto de Fórmula (B-1) o una sal del mis mismo; e hidrolizar el -C(O)ORa del compuesto de Fór Fórmula (D-1) o una sal del mismo. Se puede usar c etapas (a-1), (b-1) y (c-1) del Esquema 4 a continuación carboxílico y sulfonamida o las de una acilación de sulf paso (b-1 ) y los de una reacción nucleofílica de amina p
[0168] En algunas realizaciones, la etapa (a-1) del Esq base. En algunas realizaciones, la etapa (a-1) del Es nucleófila. En algunas realizaciones, en el paso (a-1), l mismo con un compuesto de Fórmula (E-1) o una sa Fórmula (D-1) o una sal del mismo con un reactivo de a con un compuesto de Fórmula (E-1) o una sal del mism algunas realizaciones, (i) un compuesto de Fórmula (D-la reacción con un compuesto de Fórmula (E-1 ) o una sal de la etapa (i) se hace reaccionar con un compuesto de tal como DBU (1,8-diazabiciclo(5.4.0)undec-7-eno).
[0169] En algunas realizaciones, la etapa (b-1) del Esq base. En algunas realizaciones, la etapa (b-1) se realiza En algunas realizaciones, la etapa (b-1) se realiza en pre
[0170] En algunas realizaciones, el paso (c-1) del Es algunas realizaciones, el paso (c-1) se realiza en presen e Fórmula se emplea (G-1). En algunas realizaciones, se .
y un compuesto de Fórmula (G-1) o una sal del mismo se n la técnica, por ejemplo, aquellos en WO2016/57572 y los jemplos.
Esquema 4, un compuesto de fórmula (F-1) o una sal del ores se prepara mediante un método que comprende hacer mismo con un compuesto de Fórmula (E-1 ) o una sal del rmula (D-1) o sus sales, o sus derivados deuterados se ionar un compuesto de Fórmula (A-1) o una sal del mismo para generar un compuesto de fórmula (C-1) o una sal del (C-1) o una sal del mismo para generar un compuesto de uier condición adecuada conocida en la técnica para las les como las de una reacción de acoplamiento entre ácido mida para la etapa (a-1 ), los de hidrólisis del éster para el el paso (c-1 ).
ma 4 a continuación se lleva a cabo en presencia de una ma 4 siguiente se realiza en presencia de una base no eacción de un compuesto de Fórmula (D-1) o una sal del el mismo comprende hacer reaccionar un compuesto de lamiento, como carbonildiimidazol (CDI), y posteriormente n presencia de una base, como una base no nucleófila. En o una sal del mismo se hace reaccionar con CDI antes de l mismo, y luego posteriormente (ii) el producto de reacción rmula (E-1) o una sal del mismo en presencia de una base,
ma 4 a continuación se lleva a cabo en presencia de una presencia de una base acuosa, tal como hidróxido acuoso. ncia de un hidróxido metálico acuoso, como NaOH acuoso.
ma 4 siguiente se realiza en presencia de una base. En de un carbonato metálico (por ejemplo, Na2CO3 o K2CO3 ).

[0171] En el Esquema 4, Ra se selecciona de entre grup F o Cl.
C1-C4 alquilo; y cada Xa se elige independientemente entre
[0172] En algunas realizaciones, los métodos de prepar es NH o N(C-i-C4 alquilo) o una sal farmacéuticamente a de los anteriores, comprenden hacer reaccionar un comp R * es H o C1-C4 alquilo, como se representa en los Esqu n de un compuesto de las Fórmulas (I) y (II), en donde X table del mismo, o un derivado deuterado de cualquiera to de Fórmula (L) o una sal del mismo con NR * 3 donde as 5 y 6 :

[0173] Cualquier condición adecuada conocida en la técni por ejemplo, para las adiciones electrofílicas de aminas.
realiza en presencia de un agente oxidante o clorante, tal [0174] En algunas realizaciones, un compuesto de Fórm comprende la oxidación de la unidad de azufre
puede ser utilizada para la reacción de sulfoxaminación, algunas realizaciones, la reacción de sulfoxaminación se mo N-clorosuccinimida (NCS).
(L) o una sal del mismo se prepara por un método que

del grupo de un compuesto de Fórmula (M) o sal del mis como se muestra en el Esquema 7 a continuación:
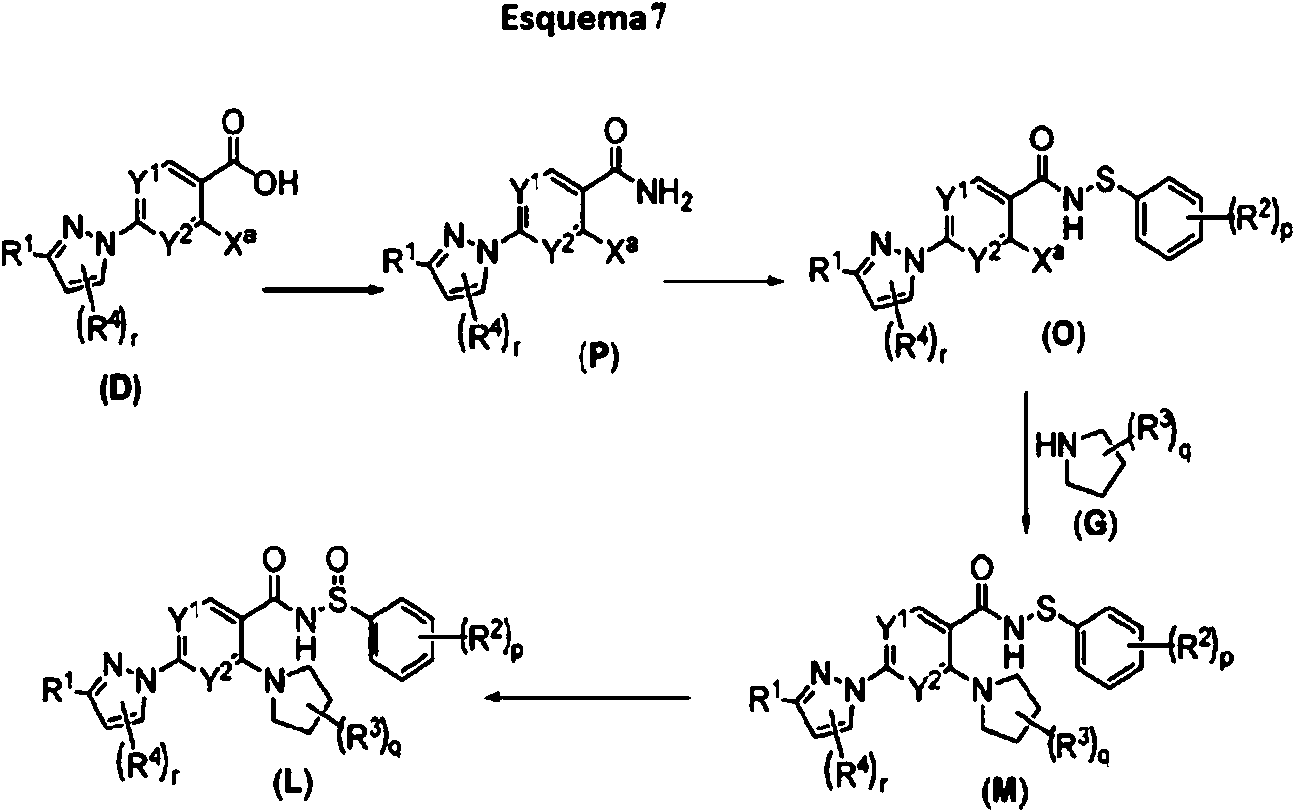
[0175] Se puede usar cualquier condición adecuada con a en la técnica para la reacción de oxidación. En algunas
realizaciones, la oxidación se realiza en presen metacloroperoxibenzoico (m-CPBA).
[0176] En algunas realizaciones, un compuesto de Fórm que comprende hacer reaccionar un compuesto de Fór mismo. Puede usarse cualquier condición adecuada con [0177] En algunas realizaciones, un compuesto de Fórm comprende hacer reaccionar un compuesto de Fórmula ( (Q):
de un ácido peroxicarboxílico, tal como ácido
(M) o una sal del mismo se prepara mediante un método la (O) con un compuesto de Fórmula (G) o una sal del a en la técnica.
(O) o una sal se prepara de la misma por un método que o una sal del mismo con un disulfuro de fenilo de Fórmula

En algunas realizaciones, se prepara un compuesto de F de un compuesto de Fórmula (D) o una sal del mismo. técnica.
[0178] Las realizaciones adicionales incluyen:
1. Un compuesto a de la fórmula I:
ula (P) o una sal del mismo amidando el grupo -C(O)OH de usarse cualquier condición adecuada conocida en la

una sal farmacéuticamente aceptable del mismo, donde:
- uno de Y1 e Y2 es N y el otro es CH;
- X se elige entre los grupos O, NH y N(C1-C4 al - R1 se elige entre grupos -(CR 2)k-O-(CR2)m(CR
en donde cada Anillo A se elige indepe sustituidos con uno o más sustituyentes alquilo, grupos C1-C2 alquilo halogenad en los que cada R se elige independie sustituidos con uno o más halógenos;
- cada R2 se elige independientemente entre g ciano;
- cada R3 se elige independientemente entre gru grupos OH;
- cada R4 se elige independientemente entre hal - k es 0 o 1 ;
- r es 0 o 1 ;
- m es 0, 1,2 o 3;
- n es 0 o 1 ;
- p es 0, 1,2, 3, 4 o 5; y
un derivado deuterado de cualquiera de los anteriores, en
o);
niMo A)n+1,
entemente entre grupos C3-C10 cicloalquilo opcionalmente ada uno elegido independientemente entre grupos C1-C2 y halógenos, y
mente entre H, OH y grupos C1-C2 alquilo opcionalmente
os C1-C2 alquilo, OH, grupos C1-C2 alcoxi, halógenos y s C1-C2 alquilo opcionalmente sustituidos con uno o más nos;
- q es 0, 1,2, 3, 4, 5, 6 , 7 u 8.

una sal farmacéuticamente aceptable del mismo, o un donde:
- X se elige entre los grupos O, NH y N(C-i-C4 alquilo); - R1 se elige entre grupos -(CR 2)k-O-(CR2)m(CR)n(Ani
en donde cada Anillo A se elige independient sustituidos con uno o más sustituyentes elegi alquilo, grupos C1-C2 alquilo halogenados y h
en los que cada R se elige independienteme sustituidos con uno o más halógenos;
- cada R2 se elige independientemente entre grupos ciano;
- cada R3 se elige independientemente entre grupos grupos OH;
- cada R4 se elige independientemente entre halógeno - k es 0 o 1 ;
- r es 0 o 1 ;
- m es 0, 1,2 o 3;
- n es 0 o 1 ;
- p es 0, 1,2, 3, 4 o 5; y
- q es 0, 1,2, 3, 4, 5, 6 , 7 u 8.
3. Un compuesto de la realización 1, en donde el com rivado deuterado de cualquiera de los anteriores, en
A)n+1,
ente entre grupos C3-C10 cicloalquilo opcionalmente s cada uno independientemente entre grupos C1-C2 genos, y
entre H, OH y grupos C1-C2 alquilo opcionalmente
1-C2 alquilo, OH, grupos C1-C2 alcoxi, halógenos y C2 alquilo opcionalmente sustituidos con uno o más
esto es de Fórmula III:

una sal farmacéuticamente aceptable del mismo, o un donde:
rivado deuterado de cualquiera de los anteriores, en
- R1 se elige entre grupos -(CR 2)kO -(CR 2)m(CR)n(
en donde cada Anillo A se elige independi sustituidos con uno o más sustituyentes, c alquilo, grupos C1-C2 alquilo halogenados y
donde cada R se elige independientemen sustituido con uno o más halógenos;
- cada R2 se elige independientemente entre grup ciano;
- cada R3 se elige independientemente entre grupos grupos OH;
- cada R4 se elige independientemente entre halóge - k es 0 o 1 ;
- r es 0 o 1 ;
- m es 0, 1,2 o 3;
- n es 0 o 1 ;
- p es 0, 1,2, 3, 4 o 5; y
- q es 0, 1,2, 3, 4, 5, 6 , 7 u 8.
4. Un compuesto según cualquiera de las realizacio o un derivado deuterado de cualquiera de los an es meta o para con respecto al átomo de azufre.
5. Un compuesto según cualquiera de las realizacio o un derivado deuterado de cualquiera de los anteri
- cada An illo A se elige independientemente entre uno o más sustituyentes elegidos cada uno indepe alquilo halogenados y halógenos, y
- cada R se elige independientemente entre H y OH; - cada R2 se elige independientemente entre grupos - R4 es F;
- k es 0 ;
- p es 0 , 1 o 2 ;
- q es 0, 1,2, 3 o 4;
- r es 0 ; y
donde m y n no son 0 al mismo tiempo.
6. Un compuesto de acuerdo con la realización 5, derivado deuterado de cualquiera de los anteriores,
- R1 se elige entre los grupos -O-(CR2 )m-Anillo A,
en donde el Anillo A es elegido entre grupos C3-C sustituyentes, cada uno elegido independienteme halogenados y halógenos, y
- m es 1 o 2.
7. Un compuesto según la realización 6 , una sal f deuterado de cualquiera de los anteriores, en donde
8. Un compuesto según la realización 7 que tiene la A)n+i,
mente de grupos C3-C10 cicloalquilo opcionalmente no elegido independientemente entre grupos C1-C2 genos, y
ntre H, OH y grupos C1-C2 alquilo opcionalmente
1-C2 alquilo, OH, grupos C1-C2 alcoxi, halógenos y C2 alquilo opcionalmente sustituidos con uno o más
-3, una sal farmacéuticamente aceptable del mismo res, en donde si R2 es ciano, entonces dicho R2
-3, una sal farmacéuticamente aceptable del mismo en donde:
os C3-C10 cicloalquilo opcionalmente sustituidos con ntemente entre grupos C1-C2 alquilo, grupos C1-C2
C2 alquilo, OH, grupos C1-C2 alcoxi y halógenos;
sal farmacéuticamente aceptable del mismo, o un onde:
cloalquilo opcionalmente sustituidos con uno o más entre grupos C1-C2 alquilo, grupos C1-C2 alquilo
céuticamente aceptable del mismo, o un derivado a R3 es un grupo metilo y q es 3 o 4.
ula IV:
una sal farmacéuticamente aceptable del mismo, o un donde:
- El Anillo A se elige entre grupos C3-C10 cicloalquilo cada uno elegido independientemente entre grupos halógenos; y
- cada R2 se elige independientemente entre grupos - m es 1 o 2 ; y
- p es 0 , 1 o 2.
9. Un compuesto según la realización 8 , en donde p e 10. Un compuesto según la realización 8 , en donde p 11. Un compuesto según la realización 8 que tiene Fó rivado deuterado de cualquiera de los anteriores, en
ionalmente sustituidos con uno o más sustituyentes, 1-C2 alquilo, grupos C1-C2 alquilo halogenados y
C2 alquilo, OH, F, Cl y grupos C1-C2 alcoxi;
0 o 1.
0.
ula V:

donde:
- El Anillo A se elige entre grupos C3-C10 cicloalquilo cada uno elegido independientemente entre grupos halógenos; y
- cada R2 se elige independientemente entre grupos - m es 1 o 2 ; y
- p es 0 , 1 o 2.
12. Un compuesto de acuerdo con una cualquiera aceptable del mismo o un derivado deuterado de cu independientemente de CH3 , OH, F y OCH3.
13. Un compuesto según la realización 12, una sal fa deuterado de cualquiera de los anteriores, en donde 14. Un compuesto según la realización 13, una sal fa deuterado de cualquiera de los anteriores, en donde 15. Un compuesto de acuerdo con la realización 11, derivado deuterado de cualquiera de los anteriores, e con un grupo C1 alquilo halogenado o un grupo C2 alq 16. Un compuesto según la realización 15, una sal f deuterado de cualquiera de los anteriores, en donde grupo CF3.
17. Un compuesto de acuerdo con la realización 11, derivado deuterado de cualquiera de los anteriores, ionalmente sustituidos con uno o más sustituyentes, 1-C2 alquilo, grupos C1-C2 alquilo halogenados y
C2 alquilo, OH, F, Cl y grupos C1-C2 alcoxi;
las realizaciones 1-11, una sal farmacéuticamente uiera de los anteriores, en donde cada R2 se elige
acéuticamente aceptable del mismo, o un derivado s 0 o 1.
acéuticamente aceptable del mismo, o un derivado s 0.
a sal farmacéuticamente aceptable del mismo, o un donde el An illo A es un grupo ciclopropilo sustituido lo halogenado.
acéuticamente aceptable del mismo o un derivado An illo A es un grupo ciclopropilo sustituido con un
a sal farmacéuticamente aceptable del mismo, o un onde m es 1, el An illo A es un grupo ciclopropilo
sustituido con un grupo CF3, p es 0 o 1, y R2, si está p metoxi.
18. Un compuesto de acuerdo con la realización 11, derivado deuterado de cualquiera de los anteriores, sustituido con un grupo CF3, p es 0 o 1, y R2, si está p metoxi.
19. Un compuesto según la realización 17 o 18, una s deuterado de cualquiera de los anteriores, en donde con un grupo CF3 y p es 0,
20. Un compuesto de acuerdo con la realización 11, derivado deuterado de cualquiera de los anterior bicicloalquilo opcionalmente sustituidos con uno o m entre grupos C1-C2 alquilo, grupos C1-C2 alquilo halo
21. Un compuesto según la realización 20, una sal deuterado de cualquiera de los anteriores, en donde sustituido con un halógeno.
22. Un compuesto de acuerdo con la realización 11, derivado deuterado de cualquiera de los anterior bicicloalquilo y grupos C7 tricicloalquilo opcionalment uno independientemente de grupos C1-C2 alquilo, gr
23. Un compuesto según la realización 22, una sal f deuterado de cualquiera de los anteriores, en donde
24. Un compuesto que tiene una fórmula elegida entr una sal farmacéuticamente aceptable del mismo, o u
25. Un compuesto de acuerdo con la realización 1 q ente, es un grupo metilo, un grupo hidroxi o un grupo
a sal farmacéuticamente aceptable del mismo, o un nde m es 2, el An illo A es un grupo C3 cicloalquilo ente, es un grupo metilo, un grupo hidroxi o un grupo
rmacéuticamente aceptable del mismo o un derivado es 2, el An illo A es un grupo ciclopropilo sustituido
a sal farmacéuticamente aceptable del mismo, o un en donde el Anillo A se elige entre grupos C5 sustituyentes, cada uno elegido independientemente nados y halógenos.
macéuticamente aceptable del mismo o un derivado An illo A es un grupo C5 bicicloalquilo opcionalmente
a sal farmacéuticamente aceptable del mismo, o un en donde el Anillo A se elige entre grupos C7 ustituidos con uno o más sustituyentes elegidos cada s C1-C2 alquilo halogenados y halógenos.
acéuticamente aceptable del mismo, o un derivado An illo A es un grupo C7 tricicloalquilo no sustituido.
ualquiera de las fórmulas representadas en la FIG. 1, erivado deuterado de cualquiera de los anteriores.
tiene la siguiente fórmula:

una sal farmacéuticamente aceptable del mismo, o u 26. Un compuesto de acuerdo con la realización 1 q erivado deuterado de cualquiera de los anteriores. iene la siguiente fórmula:

una sal farmacéuticamente aceptable del mismo, o u erivado deuterado de cualquiera de los anteriores.
27. Un compuesto según la realización 1 que tien siguiente fórmula:

una sal farmacéuticamente aceptable del mismo, o 28. Un compuesto de acuerdo con la realización 1 derivado deuterado de cualquiera de los anteriores. e tiene la siguiente fórmula:

una sal farmacéuticamente aceptable del mismo, o 29. Un compuesto según la realización 1 que tien derivado deuterado de cualquiera de los anteriores. siguiente fórmula:

una sal farmacéuticamente aceptable del mismo, o 32. Un compuesto de acuerdo con la realización 1 derivado deuterado de cualquiera de los anteriores. tiene la siguiente fórmula:

o una sal farmacéuticamente aceptable del mismo.
33. Un compuesto que tiene la siguiente fórmula:

o una sal farmacéuticamente aceptable del mismo.
34. Un compuesto según la realización 1 que tiene la uiente fórmula:

o una sal farmacéuticamente aceptable del mismo.
35. Una composición farmacéutica que comprende cualquiera de las realizaciones 1-34, una sal far deuterado de cualquiera de los anteriores, y opcional (a) Compuesto II:
enos un compuesto elegido entre los compuestos de éuticamente aceptable del mismo, o un derivado nte uno o más de:

una sal farmacéuticamente aceptable del anteriores;
(b) Compuesto III:
mo, o un derivado deuterado de cualquiera de los

una sal farmacéuticamente aceptable del
anteriores; y
(c) un portador farmacéuticamente aceptabl mo, o un derivado deuterado de cualquiera de los
36. Una composición farmacéutica según la realizaci fibrosis quística.
37. Un método para preparar un compuesto de Fórmu 35 para su uso en un método de tratamiento de la
(Mía):

una sal farmacéuticamente aceptable del mismo, o u que comprende hacer reaccionar un compuesto de F Fórmula (G) o una sal del mismo para generar dicho co aceptable del mismo, o un derivado deuterado de cual derivado deuterado de cualquiera de los anteriores, ula (F) o una sal del mismo con un compuesto de uesto de Fórmula (IMa) o una sal farmacéuticamente iera de los anteriores:

- uno de Y1 e Y2 es N y el otro es CH;
- cada R1 se elige independientemente entre grupos -( en donde cada Anillo A se elige independient sustituidos con uno o más sustituyentes, cad alquilo, grupos C1-C2 alquilo halogenados y h donde cada R se elige independientemente sustituidos con uno o más halógenos;
- cada R2 se elige independientemente entre grupos ciano;
- cada R3 se elige independientemente entre grupos grupos OH;
- cada R4 se elige independientemente entre halógeno - Xa se elige entre F o Cl;
- cada k es independientemente 0 o 1 ;
- cada r es independientemente 0 o 1 ;
- cada m es independientemente 0, 1, 2 o 3;
- cada n es independientemente 0 o 1 ;
- cada p es independientemente 0, 1, 2, 3, 4 o 5; y
- cada q es independientemente 0, 1, 2, 3, 4, 5, 6 , 7 u R 2)k-O-(CR2 )m(CR)n(AniMo A)n+i,
ente entre grupos C3-C10 cicloalquilo opcionalmente no elegido independientemente entre grupos C1-C2 genos, y
ntre H, OH y grupos C1-C2 alquilo opcionalmente
1-C2 alquilo, OH, grupos C1-C2 alcoxi, halógenos y
C2 alquilo opcionalmente sustituidos con uno o más
38. El método de la realización 37, en donde independientemente CH.
39. El método de la realización 37 o 38, en donde dic del mismo con un compuesto de Fórmula (G) o una s 40. El método de una cualquiera de las realizaciones Fórmula (G).
41. El método de la realización 40, en donde dicha s un compuesto de Fórmula (G).
42. Un método para preparar un compuesto de Fórm da Y2 es independientemente N; y cada Y1 es
reacción de un compuesto de Fórmula (F) o una sal el mismo se realiza en presencia de una base.
-39, en donde se emplea una sal del compuesto de
el compuesto de Fórmula (G) es una sal de HCl de
(F) o una sal del mismo:

o un derivado deuterado de cualquiera de los anterior Fórmula (D) o una sal del mismo con un compuest compuesto de Fórmula (F) o una sal del mismo:
, que comprende hacer reaccionar un compuesto de e Fórmula (E) o un sal del mismo para generar un

- uno de Y1 e Y2 es independientemente N y el otro e - cada R1 se elige independientemente entre grupos -en donde cada Anillo A se elige independien sustituidos con uno o más sustituyentes, cad alquilo, grupos C1-C2 alquilo halogenados y donde cada R se elige independientemente sustituidos con uno o más halógenos;
- cada R2 se elige independientemente entre grupos ciano;
- cada R4 se elige independientemente entre halógen - Xa se elige entre F o Cl;
- cada k es independientemente 0 o 1 ;
- cada r es independientemente 0 o 1 ;
- cada m es independientemente 0, 1, 2 o 3;
- cada n es independientemente 0 o 1 ; y
dependientemente CH;
R 2)k-O-(CR2 )m(CR)n(AniMo A)n+1,
ente entre grupos C3-C10 cicloalquilo opcionalmente uno elegido independientemente entre grupos C1-C2 ógenos, y
ntre H, OH y grupos C1-C2 alquilo opcionalmente
1-C2 alquilo, OH, grupos C1-C2 alcoxi, halógenos y
- cada p es independientemente 0, 1,2, 3, 4 o 5.
43. El método de la realización 42, en donde independientemente CH.
44. El método de la realización 42 o 43, en donde di del mismo con un compuesto de Fórmula (E) o una s 45. El método de la realización 42 o 43, en donde di del mismo con un compuesto de Fórmula (E) o una s de Fórmula (D-1 ) con un reactivo de acoplamiento y presencia de una base.
46. El método de la realización 37, que es un métod da Y2 es independientemente N; y cada Y1 es
reacción de un compuesto de Fórmula (D) o una sal del mismo se realiza en presencia de una base.
reacción de un compuesto de Fórmula (D) o una sal el mismo comprende hacer reaccionar un compuesto steriormente con un compuesto de Fórmula (E-1 ) en
ara preparar un compuesto de la siguiente fórmula:

o una sal farmacéuticamente aceptable del mismo, o que comprende hacer reaccionar un compuesto de entre F o Cl, con un compuesto de Fórmula (G-1) o sal farmacéuticamente aceptable del mismo, o un de derivado deuterado de cualquiera de los anteriores, mula (F-1) o un sal del mismo, en donde Xa se elige sal del mismo para generar dicho compuesto o una ado deuterado de cualquiera de los anteriores:

47. El método de la realización 46, en donde dicha r mismo con un compuesto de Fórmula (G-1) o una sa 48. El método de la realización 46 o 47, en donde se 49. El método de la realización 48, en donde dicha s un compuesto de Fórmula (G-1).
50. El método de la realización 42, que es un métod sal del mismo:
ción de un compuesto de Fórmula (F-1) o una sal del el mismo se realiza en presencia de una base.
plea una sal del compuesto de Fórmula (G-1).
el compuesto de Fórmula (G-1) es una sal de HCl de
para preparar un compuesto de Fórmula (F-1) o una

o un derivado deuterado de cualquiera de los anterio Fórmula (D-1) y un compuesto de Fórmula (E-1) par mismo:
, que comprende hacer reaccionar un compuesto de enerar un compuesto de Fórmula (F-1) o una sal del

51. El método de la realización 50, en donde dicha re mismo con un compuesto de Fórmula (E-1) o una sal
52. El método de la realización 50, en donde dicha re mismo con un compuesto de Fórmula (E-1) o una sa de Fórmula (D-1 ) con un reactivo de acoplamiento y presencia de una base.
53. Un método para preparar un compuesto de Fórm ción de un compuesto de Fórmula (D-1) o una sal del l mismo se realiza en presencia de una base.
ción de un compuesto de Fórmula (D-1) o una sal del l mismo comprende hacer reaccionar un compuesto steriormente con un compuesto de Fórmula (E-1 ) en
(D) o una sal del mismo:

o un derivado deuterado de cualquiera de los anterio (i) hacer reaccionar un compuesto de Fórmul (B) o una sal del mismo para generar un co , que comprende:
A) o una sal del mismo con un compuesto de Fórmula uesto de Fórmula (C) o una sal del mismo:

y
(ii) hidrolizar el grupo -C(O)ORa de un com Fórmula (D) o una sal del mismo, en donde - uno de Y1 e Y2 es independiente - cada R1 se elige independientem en donde cada Anillo cicloalquilo opcionalment esto de Fórmula (C) para generar un compuesto de cada una de dichas fórmulas:
te N y el otro es independientemente CH;
e entre grupos -(CR2)k-O-(CR2 )m(CR)n(AniMo A)n+1, se elige independientemente entre grupos C3-C10 ustituidos con uno o más sustituyentes, cada uno
elegido independienteme halogenados y halógenos, donde cada R se elige in opcionalmente sustituidos - cada R4 se elige independientem - cada Ra se elige independientem - cada Xa se elige independienteme - cada k es independientemente 0 - cada r es independientemente 0 - cada m es independientemente 0, - cada n es independientemente 0 54. El método de la realización 53, en donde independientemente CH.
55. El método de la realización 53 o 54, en donde la una base.
56. El método de una cualquiera de las realizacione Fórmula (A) o una sal del mismo con un compuesto de una base.
57. El método de una cualquiera de las realizaciones 58. El método de la realización 53, que es un métod sal del mismo:
entre grupos C1-C2 alquilo, grupos C1-C2 alquilo
ndientemente entre H, OH y grupos C1-C2 alquilo uno o más halógenos;
entre halógenos;
entre C1-C4 alquilo;
entre F o Cl;
o 3;
a Y2 es independientemente N; y cada Y1 es
lisis del grupo -C(O)ORa se realiza en presencia de
-55, en donde dicha reacción de un compuesto de órmula (B) o sal del mismo se realiza en presencia
56, en donde Ra es etilo o í-butilo.
ra preparar un compuesto de Fórmula (D-1) o una

o un derivado deuterado de cualquiera de los anterio (i) hacer reaccionar un compuesto de Fórmul (B-1) o una sal del mismo para generar un c que comprende:
-1) o una sal del mismo y un compuesto de Fórmula uesto de Fórmula (C-1) o una sal del mismo:

y
(ii) hidrolizar el grupo -C(O)OR3 de un comp de Fórmula (C-1) o una sal del mismo para generar
un compuesto de Fórmula (D-1) o una sal d en donde cada Ra se elige independientemente entre entre F o Cl.
59. El método de la realización 58, en donde la hidról base.
60. El método de 58 o 59, en donde dicha reacción y un compuesto de Fórmula (B-1) o una sal del mism 61. El método de una cualquiera de las realizaciones 62. Un método para preparar un compuesto de Fórmu o un derivado deuterado de cualquiera de los anterio Fórmula (I) o una sal del mismo con NR*3 :
ismo,
-C4 alquilo; y cada - Xa se elige independientemente
del grupo -C(O)ORa se realiza en presencia de una
n compuesto de Fórmula (A-1) o una sal del mismo e realiza en presencia de una base.
-60, en donde Ra es etilo o í-butilo.
I) o una sal farmacéuticamente aceptable del mismo, que comprende hacer reaccionar un compuesto de

en donde en cada una de dichas fórmulas:
- X es NH o N(C1-C4 alquilo);
- uno de Y1 e Y2 es independientemente N y el otro e - cada R1 se elige independientemente entre grupos en donde cada Anillo A se elige independie sustituidos con uno o más sustituyentes, ca alquilo, grupos C1-C2 alquilo halogenados y donde cada R se elige independientement sustituidos con uno o más halógenos;
- cada R2 se elige independientemente entre grupo ciano;
- cada R3 se elige independientemente entre grupos grupos OH;
- cada R4 se elige independientemente entre halógen - R* es H o C1-C4 alquilo.
- Xa se elige entre F o Cl;
- cada k es independientemente 0 o 1 ;
- cada r es independientemente 0 o 1 ;
- cada m es independientemente 0, 1, 2 o 3;
- cada n es independientemente 0 o 1 ;
- cada p es independientemente 0, 1, 2, 3, 4 o 5; y - cada q es independientemente 0, 1,2, 3, 4, 5, 6 , 7 dependientemente CH;
2)k-O-(CR2 )m(CR)n(AniMo A)n+1,
ente entre grupos C3-C10 cicloalquilo opcionalmente no elegido independientemente entre grupos C1-C2 genos, y
ntre H, OH y grupos C1-C2 alquilo opcionalmente
1-C2 alquilo, OH, grupos C1-C2 alcoxi, halógenos y
C2 alquilo opcionalmente sustituidos con uno o más
63. Al menos un compuesto elegido entre los farmacéuticamente aceptable de los mismos, opcionalmente uno o más de:
(a) Compuesto II:
mpuestos de cualquiera de las realizaciones 1-34, una sal un derivado deuterado de cualquiera de los anteriores, y

una sal farmacéuticamente aceptable anteriores; y
(b) Compuesto III:
l mismo, o un derivado deuterado de cualquiera de los

una sal farmacéuticamente aceptable anteriores;
para el tratamiento de la fibrosis quística.
Métodos de preparación de compuestos
Procedimientos experimentales generales
[0179] Los reactivos y materiales de partida se obtuviero y se usaron sin purificación. Los espectros de RMN de Bruker Biospin DRX de 400 MHz que funcionaba a un respectivamente, o en un espectrómetro de RMN de 300 y carbono utilizando una sonda de observación de band resolución digital de 0,1834 y 0,9083 Hz/Pt, respecti adquirieron con control de temperatura a 30°C utilizan parámetros de procesamiento de rutina. La pureza fina reversa usando una columna Acquity UPLC BEH C18 ( 186002350), y un gradiente dual desde el 1-99% de la CF3CO2H). Fase móvil B = CH3CN (0,035% CF3CO2H). |jL y temperatura de la columna = 60°C. La pureza final trazas de UV (220 nm, 254 nm). Los espectros de mas obtenidas utilizando un espectrómetro de masas de c electropulverización (ESI) capaz de lograr una precisió unidades en resolución) en todo el rango de detección. metilo se determinó mediante análisis de cromatografía 5975C, utilizando un Restek Rt-pDEXcst (30m X 0,25m de H2 ), a una temperatura de inyección de 220°C y una t
Ejemplo 1: Preparación de una dispersión secada p
[0180] Se preparó una dispersión secada por pulverizaci B290 de Buchi. Se disolvió HPMCAS-HG (6,0 gramos) e añadió el Compuesto 1 (6,0 gramos) y se agitó durante resultante se secó por pulverización en las siguientes c l mismo, o un derivado deuterado de cualquiera de los
e fuentes comerciales a menos que se indique lo contrario tón y carbono se adquirieron en un espectrómetro FTNMR recuencia de resonancia de 1H y 13C de 400 y 100 MHz z. Se adquirieron espectros unidimensionales de protones ncha (BBFo ) con una rotación de muestra de 2 0 Hz a una mente. Todos los espectros de protones y carbono se secuencias de pulsos estándar previamente publicadas y e los compuestos se determinó mediante UPLC de fase X 2,1 mm, partícula de 1,7 mm) fabricada por Waters (pn: e móvil B sobre 3,0 minutos. Fase móvil A = H2O (0,05% locidad de flujo = 1,2 mL/min, volumen de inyección = 1,5 calculó promediando el área bajo la curva (AUC) de dos de baja resolución se informaron como especies [M+H]+ rupolo único equipado con una fuente de ionización por e masa de 0,1 Da y una resolución mínima de 1000 (sin a pureza óptica del (2S)-2,4-dimetil-4-nitro-pentanoato de gases (CG) quiral en un instrumento Agilent 7890A/MSD X 0,25um_df), con un caudal de 2,0 mL/min (gas portador peratura del horno de 120°C, 15 minutos.
ulverización (SDD) del Compuesto 1
del Compuesto 1 usando el mini secador por pulverización 00 ml de MeOH (metanol)/DCM (diclorometano) (1/1) y se minutos formando una solución transparente. La solución iciones dando como resultado una dispersión secada por
pulverización de 50% de Compuesto 1/50% de HPMCAS(Rendimiento: 80%, Carga sólida: 6 %).

Difracción de rayos X en polvo
[0181] Las mediciones de difracción de rayos X en po PANalytical a temperatura ambiente con radiación de cobr por una rendija de divergencia variable para asegurar una haz difractado; se utilizó un detector de estado sólido lin medida en un modo de exploración. La muestra de polvo s fondo cero y se realizó un centrifugado para lograr mejore grados 2 theta con un tamaño de paso de 0,017 grados y [0182] FIG. 2 muestra el espectro XRPD de una SDD del Compuesto 1 es amorfo en la SDD.
Calorimetría diferencial de barrido modulada (MDSC) [0183] Se usó MDSC para determinar la temperatura de utilizando el calorímetro diferencial de barrido TA Discover calibró con indio. Se pesaron muestras de aproximadame tapas con un orificio. La muestra de MDSC se escaneó 2°C/min con /- 1°C de modulación en 1 minuto. Los datos Software (TA Instruments, New Castle, DE).
[0184] FIG. 3 muestra un espectro MDSC de un SDD del SDD tiene una temperatura de inicio de aproximad aproximadamente 82,7°C y una temperatura de compens Ejemplo 2: Síntesis del compuesto II: (R)-1-(2,2-Difl fluoro-2-(1-hidroxi-2-metilpropan-2-il)-1H-indol-5-il)cicl [0185]
se realizaron usando el difractómetro X-pert Pro de ,54060 A). La óptica del haz incidente estaba compuesta gitud iluminada constante en la muestra y en el lado del rápido con una longitud activa de 2 , 1 2 grados 2 theta mpaquetó en el área dentada de un soporte de silicio de stadísticas. Se midió una exploración simétrica de 4 a 40 tiempo de paso de exploración de 15,5 s.
% del Compuesto 1 en HPMCAS-HG, y muestra que el
nsición vitrea del material amorfo. La MDSC se realizó SC (TA Instruments, New Castle, DE). El instrumento se 1-3 mg en recipientes herméticos que se rizaron usando de -20°C a 210°C a una velocidad de calentamiento de eron recolectados y analizados por TA Instruments Trios
% del Compuesto 1 en HPMCAS-HG, y muestra que el ente 75,6°C, una temperatura de punto medio de n de aproximadamente 89,7°C.
obenzo[d][1,3]dioxol-5-il)-N-(1-(2,3-dihidroxipropil)-6-ropanocarboxamida

Paso 1: (R)-bencil 2-(1 -((2,2-dimetil-1,3-dioxolan-4-il ((S)-2,2-dimetN-1,3-dioxolan-4-N)metN 2-(1-(((R)-2,2 il)-2 -metilpropanoato
[0186] Se añadió carbonato de cesio (8,23 g, 25,3 mm metilpropanoato (3,0 g, 8,4 mmol) y (S)-(2,2-dimetil-1 mmol) en DMF (N, N-dimetilformamida) (17 ml). La rea nitrógeno. Después, la mezcla se repartió entre acetat etilo. Las capas de acetato de etilo combinadas se lava concentraron. El producto crudo, un aceite pardo viscos llevó directamente al siguiente paso sin purificación adi fluoro-5-nitro-1H-indol-2-il)-2-metilpropanoato, IEN-EM 2,20 minutos. ((S)-2,2-Dimetil-1,3-dioxolan-4-il)metil 2-indol-2-il)-2-metilpropanoato, IEN-EM m/z calc. 494,5, e
Paso 2: (R)-2-(1-((2,2-dimetil-1,3-dioxolan-4-il)metil)-
[0187] La mezcla de reacción cruda obtenida en la etap en un baño de agua helada. Se añadió gota a gota LiAl la adición, la mezcla se agitó durante 5 minutos más. L solución de NaOH al 15% (1 ml) y luego agua (3 ml). La y acetato de etilo. El filtrado se concentró y se purificó etilo-hexanos) para obtener (R)-2-(1-((2,2-dimetiM,3-di 1 -ol como un aceite marrón (2,68 g, 87% en 2 pasos). I retención 1,68 minutos. 1H RMN (400 MHz, DMSO-d6) 1 H), 4,94 (t, J = 5,4 Hz, 1H), 4,64 - 4,60 (m, 1H), 4,52 3,53 (m, 2H), 1,42 (s, 3 H), 1,38 - 1,36 (m, 6 H) y 1,19 (s
Paso 3: (R)-2-(5-amino-1-((2,2-dimetil-1,3-dioxolan-4
[0188] (R)-2-(1-((2,2-dimetil-1,3-dioxolan-4-il)metil)-6-flu mmol) se disolvió en etanol (70 ml) y la reacción se lavó en peso). La reacción se lavó de nuevo con nitrógeno LCMS solo se observó una conversión parcial en el pro )-6-fluoro-5-nitro-1H-indol-2-ilo)-2-metilpropanoato y tN-1,3-dioxolan-4-N)metN)-6-fluoro-5-mtro-1H-mdol-2-
na mezcla de bencil 2-(6-fluoro-5-nitro-1H-indol-2-il)-2-xolan-4-il)metilo 4-metilbencenosulfonato (7,23 g, 25,3 e agitó a 80°C durante 46 horas bajo una atmósfera de tilo y agua. La capa acuosa se extrajo con acetato de on salmuera, se secaron sobre MgSO4, se filtraron y se contiene los dos productos mostrados anteriormente, se l. (R)-Bencil 2-(1-((2,2-dimetil-1,3)-dioxolan-4-N)metN)-6-lc. 470,2, encontrado 471,5 (M+1)+ Tiempo de retención )-2,2-dimetiM,3-dioxolan-4-N)metN)-6-fluoro-5-nitro-1H-rado 495,7 (M+1)+. Tiempo de retención 2,01 minutos.
ro-5-nitro-1H-indol-2-il)-2-metilpropan-1-ol
se disolvió en THF (tetrahidrofurano) (42 ml) y se enfrió ,8 ml de solución 1 M, 16,8 mmol). Una vez completada ción se detuvo mediante la adición de agua (1 ml), una la se filtró sobre Celite y los sólidos se lavaron con THF ante cromatografía en columna (30-60% de acetato de -4-N)metN)-6-fluoro-5-nitro-1H-indol-2-N)-2-metNpropan-m/z calc. 366,4, encontrado 367,3 (M+1)+. Tiempo de (d, J = 7,6 Hz, 1 H), 7,65 (d, J = 13,4 Hz, 1 H), 6,57 (s, (m, 2H), 4,16 - 4,14 (m, 1H), 3,76 - 3,74 (m, 1H), 3,63 -ppm. (DMSO es dimetilsulfóxido).
til)-6-fluoro-1H-indol-2-ilo)-2-metilpropan-1-ol
-nitro-1H-indol-2-il)-2-metilpropan-1-ol (2,5 g, 6,82 dantemente con N2. Luego se añadió Pd-C (250 mg, 5% o se agitó en H2 (atm). Después de 2,5 horas, mediante . La reacción se filtró a través de Celite y se concentró.
El residuo se volvió a someter a las condiciones anteri completa en producto. La mezcla de reacción se filtró producto (1,82 g, 79%). IEN-EM m/z calc. 336,2, encontr (400 MHz, DMSO-d6 J 6 7,17 (d, J = 12,6 Hz, 1H), 6,76 ( 2H), 4,37 - 4,31 (m, 3H), 4,06 (dd, J = 6,1, 8,3 Hz, 1H), 6 H) y 1,21 (s, 3H) ppm.
Paso 4: (R)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-i hidroxi-2-metilpropan-2-il)-1H-indol-5-il)ciclopropano
[0189] Se añadió DMF (3 gotas) a una mezcla c il)ciclopropanocarboxílico (1,87 g, 7,7 mmol) y cloruro de solución transparente. La solución se concentró al vací nuevamente. La etapa de tolueno se repitió una vez m continuación, el cloruro de ácido se disolvió en diclorom ((2,2-dimetil-1,3-dioxolan- 4-il)metil)-6-fluoro-1H-indol-2-i 16,1 mmol) en diclorometano (45 ml). La reacción se agit con una solución de 1N HCl, una solución saturada de para producir el producto (3 g, 100%). IEN-EM m/z calc. minutos. 1H RMN (400 MHz, DMSO-d6J 6 8,31 (s, 1H), 1H), 4,51 - 4,48 (m, 1H), 4,39 - 4,34 (m, 2H), 4,08 (dd, 2H), 1,48 - 1,45 (m, 2H), 1,39 (s, 3H), 1,34 - 1,33 (m, 6 H
Paso 5: (R)-1-(2,2-difluorobenzo[d][1,3]dio metilpropan-2-il)-1H-indol-5-il)ciclopropanocarboxam
[0190] (R)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-il)-N-(1-(( metilpropan-2-il)-1H-indol-5-il)ciclopropanocarboxamida agua (5,2 ml) seguido de p-TsOH.H2O (ácido p-toluenosu a 80°C durante 45 minutos. La solución se concentró y lu de NaHCO3. La capa de acetato de etilo se secó sob cromatografía en columna (50-100% de acetato de etilo por SFC). IEN-EM m/z calc. 520,5, encontrado 521,7 (M DMSO-d6 j 6 8,31 (s, 1H), 7,53 (s, 1H), 7,42 - 7,38 (m, 2 4,90 (t, J = 5,5 Hz, 1H), 4,75 (t, J = 5,8 Hz, 1H), 4,40 (d (s, 1H), 3,65 - 3,54 (m, 2H), 3,48 - 3,33 (m, 2H), 1,48 -ppm.
Ejemplo 3: Síntesis del Compuesto MI: N-(2 carboxamida
Parte A: Síntesis de ácido 4-oxo-1,4-dihidroquinolin-
[0191]
. Después de 2 horas, la LCMS indicó una conversión vés de Celite. El filtrado se concentró para producir el 37,5 (M+1)+. Tiempo de retención 0,86 minutos. 1H RMN = 9,0 Hz, 1H), 6,03 (s, 1H), 4,79 - 4,76 (m, 1H), 4,46 (s, - 3,67 (m, 1H), 3,55 - 3,52 (m, 2H), 1,41 (s, 3H), 1,32 (s,
(1-((2,2-dimetil-1,3-dioxolan-4)-il)metil)-6-fluoro-2-(1-oxamida
gitación de ácido 1-(2,2-difluorobenzo[d][1,3]dioxol-5-o (1,30 ml, 17,9 mmol). Después de 1 hora se formó una ego se añadió tolueno (3 mL) y la mezcla se concentró el residuo se colocó a alto vacío durante 10 minutos. A (10 ml) y se añadió a una mezcla de (R)-2-(5-amino-1-etilpropan-1-ol (1,8 g, 5,4 mmol) y trietilamina (2,24 ml, mperatura ambiente durante 1 hora. La reacción se lavó CO3 y salmuera, se secó sobre MgSO4 y se concentró ,6, encontrado 561,7 (M+1)+. Tiempo de retención 2,05 (s, 1H), 7,42 - 7,40 (m, 2H), 7,34 - 7,30 (m, 3H), 6,24 (s, ,0, 8,3 Hz, 1H), 3,69 (t, J = 7,6 Hz, 1H), 3,58 - 3,51 (m, 8 (s, 3H) y 1,14 -1,12 (m, 2H) ppm.
-N)-N-(1-(2,3-dihidroxipropN)-6-fluoro-2-(1-hidroxi-2-
imetil-1,3-dioxolan-4-il)metil)-6-fluoro-2-(1-hidroxi-2-g, 5,4 mmol) se disolvió en metanol (52 ml). Se añadió co hidratado) (204 mg, 1,1 mmol). La reacción se calentó se repartió entre acetato de etilo y una solución saturada gSO4 y se concentró. El residuo se purificó mediante xanos) para producir el producto. (1,3 g, 47%, ee> 98% Tiempo de retención 1,69 minutos. 1H RMN (400 MHz, ,33 - 7,30 (m, 2H), 6,22 (s, 1H), 5,01 (d, J = 5,2 Hz, 1H), 2,6, 15,1 Hz, 1H), 4,10 (dd, J = 8,7, 15,1 Hz, 1H), 3,90 (m, 2H), 1,35 (s, 3H), 1,32 (s, 3H) y 1,14 - 1,11 (m, 2H)
-terc-butil-5-hidroxifenil)-4-oxo-1,4-dihidroquinolin-3-
rboxílico

Paso 1: Éster d ietílico del ácido 2-fenilaminometilen [0192] Una mezcla de anilina (25,6 g, 0,275 mol) y 2-(et a 140-150°C durante 2 h. La mezcla se enfrió a temperat éster dietílico del ácido 2 -fenilaminometilen-malónico co adicional. 1H RMN (D M S O d j 6 11,00 (d, 1H), 8,54 (d, 4,33 (m, 4H), 1,18-1,40 (m, 6 H).
Paso 2: Éster etílico del ácido 4-hidroxiquinolin-3-ca [0193] Se cargó un matraz de tres bocas de 1 L equipa fenilaminometilen-malónico (26,3 g, 0,100 mol), ácido po calentó a 70°C y se agitó durante 4 h. La mezcla se enf nico
etilen)malonato de dietilo (62,4 g, 0,288 mol) se calentó mbiente y se secó a presión reducida para proporcionar sólido, que se usó en la siguiente etapa sin purificación 3,6 Hz, 1H), 7,36-7,39 (m, 2H), 7,13-7,17 (m, 3H), 4,17
ílico
on un agitador mecánico con éster dietílico del ácido 2-órico (270 g) y cloruro de fosforilo (750 g). La mezcla se temperatura ambiente y se filtró. El residuo se trató con
una solución acuosa de Na2CO3 , se filtró, se lavó con agu 3-carboxílico como un sólido marrón pálido (15,2 g, 70 purificación.
Paso 3: Ácido 4-oxo-1,4-dihidroquinolina-3-carboxílic
[0194] Se suspendió el éster etílico del ácido 4-hidroxi hidróxido de sodio (2 N, 150 ml) y se agita durante 2 h a acidificó a pH 4 con 2N HCl. El precipitado resultante s vacío para dar ácido 4-oxo-1,4-dihidroquinolin-3-carboxíli 1H RMN (DMSO-daj 8 15,34 (s, 1H), 13,42 (s, 1H), 8,89 ( Hz, 1H), 7,60 (m, 1H).
Parte B: Síntesis de N-(2,4-di-terc-butil-5-hidroxifenil)
[0195]
secó. Se obtuvo éster etílico del ácido 4-hidroxiquinolin-El producto crudo se usó en el siguiente paso sin más
olin-3-carboxílico (15 g, 69 mmol) en una solución de jo. Después de enfriar, la mezcla se filtró y el filtrado se ogió mediante filtración, se lavó con agua y se secó al forma de un sólido de color blanco pálido (10,5 g, 92%). ), 8,28 (d, J = 8,0 Hz, 1H), 7,88 (m, 1H), 7,81 (d, J = 8,4
xo-1,4-dihidroquinolin-3-carboxamida

Paso 1: Ácido carbónico éster metílico del éster 2,4-
[0196] Se añadió gota a gota cloroformiato de metilo (58 g, 500 mmol), Et3N (139 ml, 1000 mmol) y DMAP (3,05 g, agua helada a 0°C. La mezcla se dejó calentar a temperat se filtró a través de gel de sílice (aprox. 1 l) utilizando 10 filtrados combinados se concentraron para producir carbónico en forma de un aceite amarillo (132 g, cuant.) 7,29 (dd, J = 8,5, 2,4 Hz, 1H), 7,06 (d, J = 8,4 Hz, 1H), 3,
Paso 2: Ácido carbónico éster metílico de éster 2,4-d del éster 2,4-di-íerc-butil-6-nitro-fenilo
[0197] A una mezcla con agitación de ácido carbónico d mmol) en ácido sulfúrico conc. (2 mL), enfriado en un ba sulfúrico (2 mL) y ácido nítrico (2 mL). La adición se real excediera de 50°C. Se dejó agitar la reacción durante 2 h mezcla de reacción se añadió a agua helada y se extr concentró y se purificó mediante cromatografía en colum mezcla de ácido carbónico éster metílico de éster 2,4-di éster 2,4-di-terc-butil-6-nitrofenílico como un sólido ama siguiente.
Paso 3: 2,4-Di-íerc-butil-5-nitrofenol y 2,4-Di-íerc-buti
[0198] La mezcla de ácido carbónico éster metílico del metílico del éster 2,4-di-terc-butil-6-nitrofenílico (4,2 g, 14, (2,0 g, 36 mmol). La mezcla se agitó a temperatura amb aciduló (pH 2-3) mediante la adición de HCl conc. y se (MgSO4), se concentró y se purificó mediante cromatogr rc-butil-fenílico
50 mmol) a una solución de 2,4-di-terc-butil-fenol (103,2 mmol) en diclorometano (400 ml) enfriado en un baño de ambiente mientras se agitaba durante la noche, después e acetato de etilo - hexanos (~ 4 L) como eluyente. Los metílico del éster 2,4-di- tere -butil-fenílico del ácido RMN (400 MHz, DMSO-cfe) 8 7,35 (d, J = 2,4 Hz, 1H), , 3H), 1,30 (s, 9H), 1,29 (s, 9H).
c-butil-5-nitro-femlo y ácido carbónico éster metílico
er de éster metílico de 2,4-di-terc-butil-fenilo (4,76 g, 180 e agua helada, se añadió una mezcla enfriada de ácido lentamente de modo que la temperatura de reacción no ntras se calentaba a temperatura ambiente. Después, la on éter dietílico. La capa de éter se secó (MgSO4 ), se cetato de etilo al 0 - 10 % - hexanos) para producir una -butil-5-nitro-fenilo. y ácido carbónico éster metílico del pálido (4,28 g), que se utilizó directamente en el paso
itrofenol
r 2,4-di-terc-butil-5-nitrofenílico y ácido carbónico éster mol) se disolvió en MeOH (65 ml) antes de añadirse KOH durante 2 h. A continuación, la mezcla de reacción se rtió entre agua y éter dietílico. La capa de éter se secó en columna (acetato de etilo - hexanos al 0 - 5%) para
proporcionar 2,4-di-ferc-butil-5-nitrofenol (1,31 g, 29% s 5-nitrofenol; 1H RMN (400 MHz, DMSOC) 8 10,14 (s, 2,4-Di-ferc-butil-6-nitrofenol: 1H RMN (400 MHz, CDCI3 ) 1H), 1,47 (s, 9H), 1,34 (s, 9H).
Paso 4: 5-Amino-2,4-di-ferc-butN-fenol
[0199] A una solución a reflujo de 2,4-di-ferc-butil-5-nitr etanol (75 ml) se añadió Pd-5% en peso. sobre carbón durante 2 h, se enfrió a temperatura ambiente y se filtró combinados se concentraron para producir 5-amino-2,4-(400 MHz, DMSO-cfe) 8 8,64 (s, 1H, OH), 6,84 (s, 1H), 6, de HPLC 2,72 min, CH3CN al 10-99%, ejecución de 5 m
Paso 5: N-(5-hidroxi-2,4-di-íerc-butN-fenN)-4-oxo-1H-
[0 2 0 0 ]
pasos) y 2,4-di-ferc-butil-6-nitrofenol. 2,4-Di-ferc-butil-), 7,34 (s, 1H), 6,83 (s, 1H), 1,36 (s, 9H), 1,30 (s, 9H).
48 (s, 1H), 7,98 (d, J = 2,5 Hz, 1H), 7,66 (d, J = 2,4 Hz,
l (1,86 g, 7,40 mmol) y formiato de amonio (1,86 g) en do (900 mg). La mezcla de reacción se agitó a reflujo s de Celite. La Celite se lavó con metanol y los filtrados -butil-fenol como un sólido gris (1,66 g, cuant.). 1H RMN 1H), 4,39 (s, 2H, NH2), 1,27 (m, 18H); HPLC tiempo ret. -EM 222,4 m/z [M+H]+.
Mn-3-carboxamida
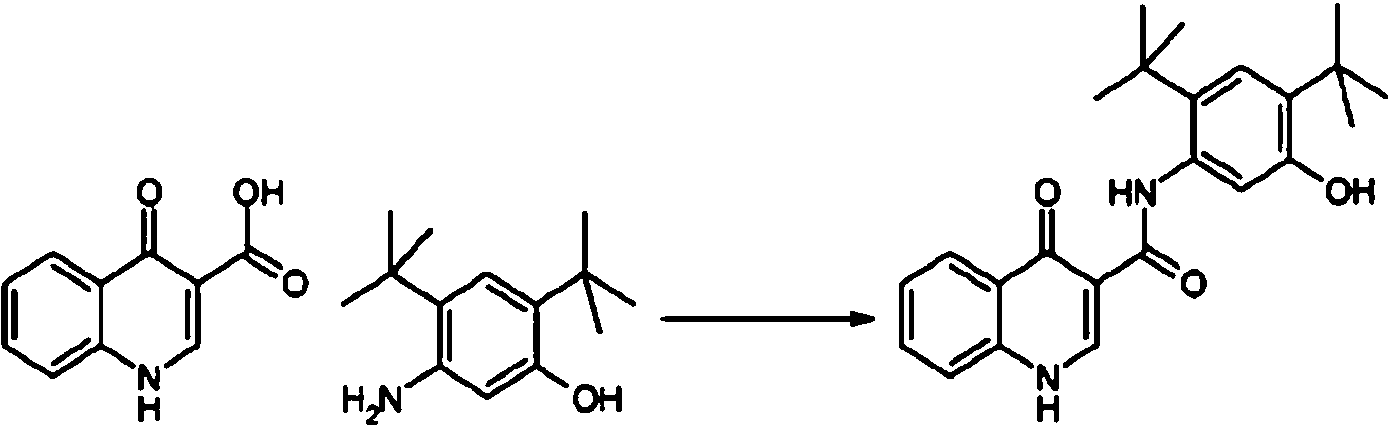
[0201] A una suspensión de ácido 4-oxo-1,4-dihidroq mmol) en DMF ( 28 0 ml) se añadió Et3N (63,0 ml, 451 m y se dejó agitar durante 10 min antes de añadir 5-a porciones. Se dejó agitar la mezcla durante la noche a t transcurso de la reacción. Después de que se consu disolvente se eliminó al vacío. Se añadió EtOH (alcohol e La mezcla se agitó en un rotavapor (temperatura del b mezcla se filtró y el sólido capturado se lavó con hexan de EtOH. Se añadió Et2O (éter dietílico) al sólido obte mezcla se agitó en un rotavapor (temperatura del baño la mezcla y se capturó el sólido. Este procedimiento se r la quinta precipitación se colocó bajo vacío durante la oxo-1H-quinolina-3-carboxamida (38 g, 52%). Tiempo d min; 1H RMN (400 MHz, DMSO-cfe) 8 12,88 (s, 1H), 11,8 1H), 7,83-7,79 (m, 1H), 7,76 (d, J = 7,7 Hz, 1H), 7,54-7 9H); IEN-EM m/z calculado 392,21; encontrado 393,3 [
Ejemplo 4: Síntesis de los compuestos 1-65
Ejemplo sintético 1: Síntesis de N-(bencenosulfom [(4S)-2,2,4-trimetilpirrolidin-1-il]piridin-3-carboxamid
Parte A: Síntesis de clorhidrato de (4S)-2,2,4-trimetil
[0 2 0 2 ]
-3-carboxílico (35,5 g, 188 mmol) y HBTU (85,7 g, 226 temperatura ambiente. La mezcla se volvió homogénea ,4-di-ferc-butil-fenol (50,0 g, 226 mmol) en pequeñas atura ambiente. La mezcla se volvió heterogénea en el do el ácido (análisis CL-EM, MH+ 190, 1,71 min), el al material sólido naranja para producir una suspensión. °C) durante 15 min sin colocar el sistema al vacío. La ra proporcionar un sólido blanco que era el cristalizado nteriormente hasta que se formó una suspensión. La durante 15 min sin colocar el sistema al vacío. Se filtró un total de cinco veces. El sólido obtenido después de para proporcionar N-(5-hidroxi-2,4-di-ferc-butil-fenil)-4-de HpLC 3,45 min, CH3CN al 10-99%, ejecución de 5 1H), 9,20 (s, 1H), 8,87 (s, 1H), 8,33 (dd, J = 8,2, 1,0 Hz, , 1H), 7,17 (s, 1H), 7,10 (s, 1H), 1,38 (s, 9H), 1,37 (s,
-[2-[1-(trifluorometN)ciclopropN]etoxi]pirazoM-M]-2-mpuesto 1)
idina
Paso 1: Síntesis de metilo-2,4-dimetil-4-nitropentano [0203]

[0204] Se añadió tetrahidrofurano (THF, 4,5 L) a un react A continuación, se cargaron en el reactor 2-nitropropa (DBU) (1,282 kg, 8,42 mol) y se aumentó la temperatur estuvo próximo a 50°C, se añadió lentamente metacrila temperatura de reacción se mantuvo en o cerca de 50° vacío, luego se transfirió de nuevo al reactor y se diluyó 2 M (7,5 L) y esta mezcla se agitó durante 5 minuto transparentes: una fase acuosa amarilla inferior y una f agitó nuevamente la capa orgánica con 2 M HCl (3 L). De y se agitaron con MTBE (3 L) durante 5 minutos. Se elimi en el reactor y se agitaron con agua (3 L) durante 5 concentraron al vacío para proporcionar un aceite verde t 2,4-dimetil-4-nitro-pentanoato como un aceite verde c cloroformo- d) 8 3,68 (s, 3H), 2,56 - 2,35 (m, 2H), 2,11 -3H).
Paso 2: Síntesis de (2S)-2,4-dimetil-4-nitro-pentanoat
[0205]
vidrio de 20 L y se agitó bajo N2 a temperatura ambiente. ,5 kg, 16,83 mol) y 1,8-diazabiciclo[5.4.0]undec-7-eno la camisa a 50°C. Una vez que el contenido del reactor e metilo (1,854 kg, 18,52 mol) durante 100 minutos. La rante 21 horas. La mezcla de reacción se concentró al éter ferc-butílico de metilo (MTBE) (14 l). Se añadió HCl luego se dejó sedimentar. Fueron visibles dos capas rgánica verde superior. Se eliminó la capa acuosa y se s de la separación, los lavados con HCl se recombinaron capa acuosa y todas las capas orgánicas se combinaron os. Después de la separación, las capas orgánicas se . Este se secó con MgSO4 y se filtró para producir metil-(3,16 kg, rendimiento del 99%). 1H RMN (400 MHz, (m, 1H), 1,57 (s, 3H), 1,55 (s, 3H), 1,19 (d, J = 6 , 8 Hz,
metilo

[0206] Se cargó un reactor con agua purificada (2090 L; moles; 13 g/L para carga de agua). El pH del contenid carbonato de potasio al 20% (p/v). El reactor se cargó con moles) y lipasa Palatase 20000L (13 L, 15,8 kg; 0,06 vol)
[0207] La mezcla de reacción se ajustó a 32 ±2°C y se a de pH con la adición automática de solución de carbonat se convirtió en >98% de ee del enantiómero S, deter Después, el reactor se cargó con MTBE (35 L; 5 vol) y l Los extractos orgánicos combinados se lavaron con Na2 2,5 vol) y NaCl acuoso al 10% (314 L, 1,5 vol). La cap dimetil-4-nitropentanoato de metilo como un aceite amari
Paso 3: Síntesis de (3S)-3,5,5-trimetilpirrolidin-2-ona
[0208]
ol) y luego fosfato de potasio monobásico (27 kg, 198,4 reactor se ajustó a pH 6,5 (± 0 ,2 ) con una solución de il-2,4-dimetil-4-nitropentanoato racémico (209 kg; 1104,6
urante 15-21 horas, y pH 6,5 se mantuvo usando un stat potasio al 20%. Cuando el material de partida racémico do por GC quiral, se apagó el calentamiento externo. pa acuosa se extrajo con MTBE (3 veces, 400-1000 L). acuoso (4 veces, 522 L, 18% p/p 2,5 vol), agua (523 L; ánica se concentró al vacío para proporcionar (2S)-2,4-óvil (>98% ee, 94,4 kg; 45% de rendimiento).

[0209] Se purgó un reactor de 20 L con N2. El recipient g) húmedo, enjuagado con agua DI, (2S)-2,4-dimetil-4-ni vol). La reacción se agitó a 900 rpm y el reactor se lavó la mezcla de reacción se calentó a 60°C durante 5 hora níquel Raney y la torta sólida se enjuagó con etanol (3,5 cargó secuencialmente con Raney® Ni (grado 2800, 250 ntanoato de metilo (1741 g, 9,2 mol) y etanol (13,9 L, 8 dantemente con H2 y se mantuvo a ~ 2,5 bar. Después, mezcla de reacción se enfrió y se filtró para eliminar el vol). La solución etanólica del producto se combinó con
un segundo lote de igual tamaño y se concentró al volúmenes). Se añadió heptano (2,5 L) y la suspensión veces; la suspensión resultante se enfrió a 0-5°C, se fil secó al vacío durante 20 minutos, luego se transfirió a durante la noche para producir (3S)-3,5,5-trimetilpirrolidi 87%). 1H RMN (400 MHz, cloroformo d) 8 6,39 (s, 1H), 1H), 1,56 (dd, J = 12,5, 9,9 Hz, 1H), 1,31 (s, 3H), 1,25 (
Paso 4: Síntesis de clorhidrato de (4S)-2,2,4-trimetil
[0 2 1 0 ]
para reducir a un volumen mínimo de etanol (~ 1,5 ncentró de nuevo a ~ 1,5 volúmenes. Esto se repitió 3 n succión y se lavó con heptano (2,5 L). El producto se jas de secado y se secó en un horno de vacío a 40°C na como un sólido cristalino blanco (2,042 kg, 16,1 mol, dq, J = 9,9, 8 ,6 , 7,1 Hz, 1H), 2,17 (dd, J = 12,4, 8 , 6 Hz, 1,20 (d, J = 7,1 Hz, 3H).
dina

[0211] Se cargó un reactor de 120 L revestido de vidrio THF seco (60 L) y calentado a 30°C. La suspensión re 54 mol) en THF (25 L) durante 2 horas mientras se
completada la adición, se aumentó la temperatura de re de reacción se enfrió a 22°C, luego se inactivó cuidado moles), seguido de una mezcla de THF (3,4 L) y agua hidróxido de sodio acuoso al 50% (750 g, 2 equiv. de aluminio), seguido de 7,5 L de agua. Una vez complet ambiente y el sólido se eliminó por filtración y se lavó trataron con 5,0 L (58 moles) de HCl acuoso al 37% (1, 30°C. La solución resultante se concentró mediante des ( 8 L) y la solución se concentró hasta casi sequedad
producto se suspendió calentando a aproximadamente El producto se recogió por filtración y se enjuagó con torr/sangrado de N2 ) para producir (4S)-2,2,4-trimetilpirr de rendimiento). 1H RMN (400 MHz, DMSO-d6) 8 9,34 11,4, 8 , 6 Hz, 1H), 2,50 - 2,39 (m, 1H), 1,97 (dd, J = 12, 1,31 (s, 3H), 1,05 (d, J = 6 , 6 Hz, 3H).
Parte B: Síntesis de N-(bencenosulfonil)-6-[3-[2-[ trim etilpirrolid in-1-il]pirid in-3-carboxam ida
[0 2 1 2 ]
gránulos de hidruro de litio y aluminio (2,5 kg, 6 6 mol) y te se cargó con (S)-3,5,5-trimetilpirrolidin-2-ona (7,0 kg, ía la temperatura de reacción de 30 a 40°C. Una vez a 60 - 63°C y se mantuvo durante la noche. La mezcla te con la adición de acetato de etilo (EtOAc) (1,0 L, 10 kg, 2,0 eq) y luego una mezcla de agua (1,75 kg) con con 1,4 equiv. de hidróxido de sodio con respecto al adición, la mezcla de reacción se enfrió a temperatura F (3 x 25 L). El filtrado y los lavados se combinaron y iv.) mientras se mantenía la temperatura por debajo de n a vacío hasta una suspensión. Se añadió isopropanol te destilación al vacío. Se añadió isopropanol (4 L) y el Se añadió MTBE ( 6 L) y la suspensión se enfrió a 2-5°C. de MTBE y se secó en un horno de vacío (55°C/300 a ■ HCl como un sólido cristalino blanco. (6 , 21 kg, 75% cho, 2H), 3,33 (dd, J = 11,4, 8,4 Hz, 1H), 2,75 (dd, J = Hz, 1H), 1,42 (s, 3H), 1,38 (dd, J = 12,8, 10,1 Hz, 1H),
luorometil)ciclopropil]etoxilpirazol-1-il]-2-[(4S)-2,2,4- 
Síntesis de materiales de partida:
Síntesis de 2,6-dicloropiridin-3-carboxilato de tere-b [0213]

[0214] Una solución de ácido 2,6-dicloropiridin-3-car sucesivamente con dicarbonato de di-ferc-butilo (17 g, 7 se agitó durante la noche a temperatura ambiente. En vigorosamente durante aproximadamente 10 minutos.
capas orgánicas combinadas se lavaron con agua (300 y se concentraron a presión reducida para dar 12,94 g ferc-butilo como aceite incoloro. IEN-EM m/z calc. 247 minutos. 1H RMN (300 MHz, CDCla) ppm 1,60 (s, 9H), 7,
Síntesis de 3-oxo-2,3-dihidro-1H-pirazol-1-carboxilat
[0215]
lico (10 g, 52,08 mmol) en THF (210 ml) se trató mmol) y 4-(dimetilamino) piridina (3,2 g, 26,19 mmol) y punto, se añadió 1N HCl (400 ml) y la mezcla se agitó oducto se extrajo con acetato de etilo (2X300 mL) y las y salmuera (150 mL), se secaron sobre sulfato de sodio dimiento del 96%) de 2,6-dicloropiridin-3-carboxilato de encontrado 248,1 (M+1)+; Tiempo de retención: 2,27 d, J = 7,9 Hz, 1H), 8,05 (d, J = 8,2 Hz, 1H).
tere-butilo

[0216] Se puso en marcha un reactor de 50 L y se pus camisa a 20°C, con agitación a 150 rpm, condensador de
reflujo (10°C) y purga de nitrógeno. Se añadieron MeO 22,76 mol) y se tapó el reactor. La reacción se calentó mantener la temperatura de la camisa a 40°C. Se añadió mol) mediante un embudo de adición durante 30 min. La se enfrió a 20°C y se añadió en porciones trietilamina (2 reacción <30°C. Se añadió en porciones una solución 5,228 L, 22,76 mol) en MeOH (2,860 L) manteniendo l durante 16 h. La solución de reacción se concentró pa aceite de color ámbar claro, claro. El aceite resultante (7,150 L) y heptano (7,150 L). Las adiciones hicieron acuosa se drenó en un recipiente limpio y la interfaz y la La capa acuosa se transfirió de nuevo al reactor y el sóli la capa acuosa. Se añadió un embudo de goteo al react y se añadió gota a gota. La camisa se ajustó a 0°C para la adición (pH = 5), la mezcla de reacción se agitó dura (7,150 L) y se lavó una segunda vez con agua (3,575 rotatorio de 20 L y se añadió heptano (7,150 L). La me por destilación 1-2 volúmenes de disolvente. Se filtró la los sólidos con heptano (3,575 L). El sólido se secó adici 2-carboxilato de ferc-butilo (2921 g, 71%) como un sólid 1H), 7,98 (d, J = 2,9 Hz, 1H), 5,90 (d, J = 2,9 Hz, 1H), 1,
Síntesis de 2-[1-(trifluorometil)ciclopropil]etanol
[0217]
60 L) y (E)-3-metoxiprop-2-enoato de metilo (2,643 kg, a temperatura interna de 40°C y el sistema se fijó para orciones hidrato de hidrazina (1300 g de 55% p/p, 22,31 ión se calentó a 60°C durante 1 h. La mezcla de reacción kg, 3,420 L, 24,54 mol), manteniendo la temperatura de hídrido de Boc (dicarbonato de di-ferc-butilo) (4,967 kg, peratura <45°C. La mezcla de reacción se agitó a 20°C ente para eliminar el MeOH, dando como resultado un nsfirió al reactor de 50 L, se agitó y se añadieron agua recipitara una pequeña cantidad del producto. La capa de heptano se filtraron para separar el sólido (producto). cogido se colocó de nuevo en el reactor y se mezcló con e cargó con ácido acético (1,474 kg, 1,396 L, 24,54 mol) rber la exotermia de enfriamiento. Una vez completada h. El sólido se recogió por filtración y se lavó con agua sólido cristalino se transfirió a un bulbo de evaporador e suspendió a 45°C durante 30 minutos y se separaron nsión en el matraz de evaporación rotatoria y se lavaron ente al vacío (50°C, 15 mbar) para dar 5-oxo-1H-pirazoltalino grueso. 1H RMN (400 MHz, DMSO-cfe) 8 10,95 (s, 9H).

[0218] A una solución de hidruro de litio y aluminio (29 ácido 2-[1-(trifluorometil)ciclopropil]acético (1,002 g, 5,9 período de 30 minutos manteniendo la temperatura de re gradualmente a temperatura ambiente y se agitó durant secuencialmente con agua (294 mg, 295 pL, 16,36 mm |jL, 49,07 mmol) para producir un sólido granular en la precipitado se lavó con éter. El filtrado se secó adicion proporcionar el producto con THF residual y éter. L purificación.
Paso 1: 3-[2-[1-(trifluorometil)ciclopropil]etoxi]pirazo
[0219]
7,732 mmol) en THF (10,00 mL) en un baño de hielo, mol) en THF (3,0 mL) se añadió gota a gota durante un por debajo de 20°C. Se dejó que la mezcla se calentara . La mezcla se enfrió con un baño de hielo y se inactivó aOH (297 pL de 6 M, 1,784 mmol) y luego agua (884,0 cla. El sólido se separó por filtración usando celite y el nte con MgSO4 y se filtró y se concentró a vacío para zcla se llevó directamente al siguiente paso sin más
arboxilato de íerc-Butilo
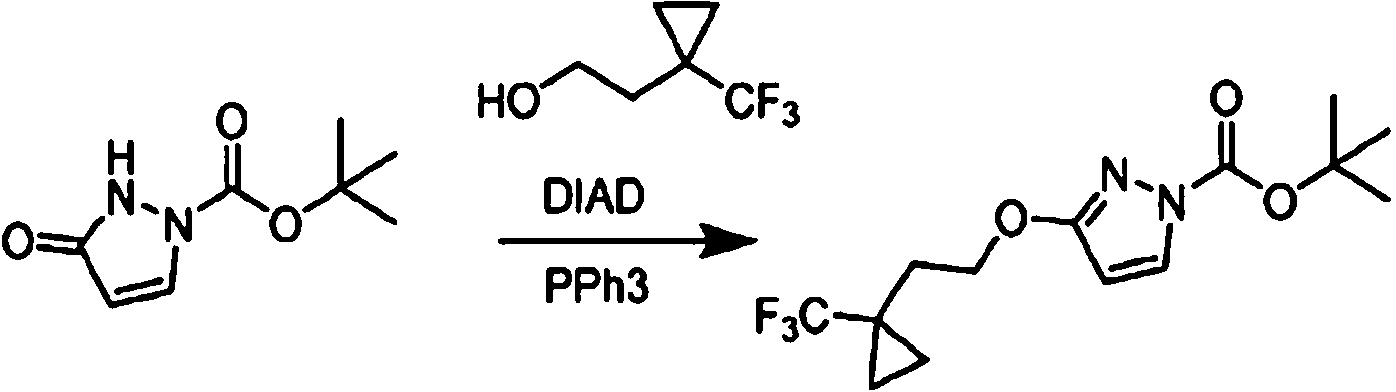
[0220] 5-oxo-1H-pirazol-2-carboxilato de ferc-butilo (1, mg, 5,943 mmol) y trifenilfosfina (1,637 g, 6,243 mmol) baño de hielo. Se añadió gota a gota azodicarboxilato d reacción y la reacción se dejó calentar a temperatura a resultante se repartió entre acetato de etilo (30 mL) e hi lavó con salmuera (30 mL), se secó sobre sulfato de , 5,660 mmol), 2-[1-(trifluorometil)ciclopropil]etanol (916 mbinaron en THF (10,48 mL) y la reacción se enfrió en opropilo (1,288 g, 1,254 ml, 6,368 mmol) a la mezcla de te durante 16 horas. La mezcla se evaporó y el material o de sodio 1 N (30 ml). La capa orgánica se separó, se y se concentró. El material crudo se purificó mediante
cromatografía en gel de sílice eluyendo con un gradie (trifluorometil)ciclopropil]etoxi]pirazol-1-carboxilato de te 321,1 (M+1)+; Tiempo de retención: 0,72 minutos.
Paso 2: 3-[2-[1-(Trifluorometil)ciclopropil]etoxi]-1H-p [0 2 2 1 ]
e acetato de etilo en hexanos (0-30%) para dar 3-[2-[1-tilo (1,03 g, 57%). IEN-EM m/z calc. 320,13, encontrado ol

[0222] terc-Butil-3-[2-[1-(trifluorometil)ciclopropil]etoxi]p diclorometano (10,30 mL) con ácido trifluoroacético (2 ambiente durante 2 horas. La reacción se evaporó y el una solución saturada de bicarbonato de sodio. La ca sulfato de sodio y se evaporó para dar 3-[2-[1-(trifl EM m/z calc. 220,08, encontrado 221,0 (M+1)+; Tiempo 11,86 (s, 1H), 7,50 (t, J = 2,1 Hz, 1H), 5,63 (t, J = 2,3 H 0,88 (m, 2H), 0,88 -0,81 (m, 2H).
Paso 3: 2-doro-6-[3-[2-[1-(trifluorometM)ddopropM]e
[0223]
l-1-carboxilato (1,03 g, 3,216 mmol) se disolvió en ml, 32,16 mmol) y la reacción se agitó a temperatura ite resultante se repartió entre acetato de etilo (10 mL) y gánica se separó, se lavó con salmuera, se secó sobre metil)ciclopropil]etoxi]-1H-pirazol (612 mg, 8 6 %). IEN-retención: 0,5 minutos. 1H RMN (400 MHz, DMSO-cfe) 8 ), 4,14 (t, J = 7,1 Hz, 2H), 2,01 (t, J = 7,1 Hz, 2H), 0,96 -
pirazoM-M]piridma-3-carboxMato de tere-butilo

[0224] 2,6-dicloropiridin-3-carboxilato de terc-butilo (68 pirazol (610 mg, 2,770 mmol) y carbonato de potasio r anhidro (13,75 ml). Se añadió 1,4-diazabiciclo[2.2.2]oc mmol) y la mezcla se agitó a temperatura ambiente bajo con agua (20 mL) y se agitó durante 15 minutos. El sólid en diclorometano y se secó sobre sulfato de magnesio. (trifluorometil)ciclopropil]etoxi]pirazol-1-il]piridina-3-carb encontrado 432,1 (M+1)+; Tiempo de retención: 0,88 mi Paso 4: Ácido 2-doro-6-[3-[2-[1-(trifluorometil)cidop [0225]
, 2,770 mmol), 3-[2-[1-(trifluorometil)ciclopropil]etoxi]-1H-molido (459 mg, 3,324 mmol) se combinaron en DMSO (DABc O (1,4-diazabiciclo[2.2.2]octano), 62 mg, 0,5540 ógeno durante 16 horas. La mezcla de reacción se diluyó ultante se recogió y se lavó con agua. El sólido se disolvió ezcla se filtró y se concentró para dar 2-cloro-6-[3-[2-[1-o de terc-butilo (1,01 g, 84%). IEN-EM m/z calc. 431,12, .
l]etoxi]pirazol-1-il]piridin-3-carboxílico

[0226] 2-cloro-6-[3-[2-[1-(trifluorometil)ciclopropil]etoxi] mmol) y ácido trifluoroacético (1,8 mL, 23,39 mmol) se durante 3 h. La reacción se concentró. Se añadieron h ol-1-il]piridin-3-carboxilato de terc-butilo (1,01 g, 2,339 binaron en diclorometano (10 mL) y se calentó a 40°C os y la mezcla se concentró de nuevo para dar ácido 2-cloro-6-[3-[2-[1-(trifluorometil)ciclopropil]etoxi]pirazol-1-il] encontrado 376,1 (M+1)+; Tiempo de retención: 0,69 mi Paso 5: N-(bencenosulfoml)-2-doro-6-[3-carboxamida
[0227]
-3-carboxílico (873 mg, 99%) IEN-EM m/z calc. 375,06,
(trifluorometM)ddopropM]etoxi]pirazoM-M]piridm-3-

[0228] Una solución de ácido 2-cloro-6-[3-[2-[1-(trifluor 0,3992 mmol) y carbonil diimidazol (77 mg, 0, 4790 mm bencenosulfonamida (81 mg, 0,50190 mmol) y DBU (72 acidificó con ácido cítrico acuoso 1 M y se extrajo con sulfato de sodio y se evaporaron. El residuo se purific gradiente de metanol en diclorometano (0 (trifluorometil)ciclopropil]etoxi]pirazol 1-il]piridin-3-carbox 515,1 (M+1)+; Tiempo de retención: 0,74 minutos.
Paso 6 : N-(bencenosulfoml)-6-[3-[2-[1 trim etilpirrolid in-1-il]pirid in-3carboxam ida
[0229]
ciclopropil]etoxi]pirazol-1-il]piridin-3-carboxílico (0,15 g, THF (2,0 mL) se agitó durante una hora y se añadieron ,4790 mmol). La reacción se agitó durante 16 horas, se o de etilo. Los extractos combinados se secaron sobre diante cromatografía en gel de sílice eluyendo con un para dar N-(bencenosulfonil)-2-cloro-6-[3-[2-[1-(160 mg, 78%). IEN-EM m/z calc. 514,07, encontrado
orometN)ddopropN]etoxi]pirazoM-N]-2-[(4S)-2,2,4-

[0230] Una mezcla de sal de
(trifluorometil)ciclopropil]etoxi]pirazol-1-il]piridina-3-carbo (139 mg, 0,9321 mmol) y carbonato de potasio (258 mg, horas. La mezcla de reacción se acidificó con ácido cítri combinados se secaron sobre sulfato de sodio y se ev HPLC de fase inversa utilizando un gradiente de ace (bencenosulfonil)-6-[3-[2-[1-(trifluorometil)ciclopropil]eto carboxamida (87 mg, 47%). IEN-EM m/z calc. 591,21, e 1H RMN (400 MHz, DMSO-d6) 8 12,48 (s, 1H), 8,19 (d, 7,77 -7,70 (m, 1H), 7,70 -7,62 (m, 2H), 6,92 (d, J = 8,2 (t, J = 10,5 Hz, 1H), 2,28 (dd, J = 10,2, 7,0 Hz, 1H), 2,1 9,4 Hz, 6 H), 1,36 (t, J = 12,1 Hz, 1H), 1,01 -0,92 (m, 2H),
Síntesis de sal sódica de N-(bencenosulfoml)-6-[ 2,2,4trimetilpirrolidin-1-il]piridin-3-carboxamida (sal rato de N-(bencenosulfonil)-2-cloro-6-[3-[2-[1-a (160 mg, 0,3107 mmol), (4S)-2,2,4-trimetilpirrolidina mmol) en DMSO (1,5 mL) se agita a 130°C durante 17 oso 1 M y se extrajo con acetato de etilo. Los extractos on para producir un producto crudo que se purificó por lo al 10-99% en HCl acuoso 5 mM para producir N-zol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-il]piridin-3-rado 592,3 (M+1)+; Tiempo de retención: 2,21 minutos.
8 Hz, 1H), 8,04 - 7,96 (m, 2H), 7,81 (d, J = 8,2 Hz, 1H), ), 6,10 (d, J = 2,8 Hz, 1H), 4,31 (t, J = 7,0 Hz, 2H), 2,42 1 (m, 3H), 1,82 (dd, J = 11,9, 5,5 Hz, 1H), 1,52 (d, J = -0,85 (m, 2H), 0,65 (d, J = 6,3 Hz, 3H). pKa: 4,9560,06.
-(trifluorometN)ddopropN]etoxi]pirazoM-N]-2-[(4S)-a del Compuesto 1)
[0231] N-(bencenosulfonil)-6-[3-[2-[1-(trifluorometil)ciclopr il]piridin-3-carboxamida (1000 mg, 1,679 mmol) se di transparente a través de un filtro de jeringa (0,2 mm), se l con 1M NaOH (1,679 ml, 1,679 mmol). La solución se e mL), para dar un sólido, que se secó durante la noche nitrógeno para dar 951 mg de un sólido de color crema. secado a 45°C con una purga de nitrógeno durante (bencenosulfonil)-6-[3-[2-[1-(trifluorometil)ciclopropil]etoxi carboxamida se obtuvo como un sólido amorfo blanqueci 7,81 (dd, J = 6,7, 3,1 Hz, 2H), 7,61 (d, J = 7,9 Hz, 1H), (d, J = 2,6 Hz, 1H), 4,29 (t, J = 7,0 Hz, 2H), 2,93 -2,78 (m, 1,52 (d, J = 13,6 Hz, 6 H), 1,33 (t, J = 12,0 Hz, 1H), 1,00 Hz, 3H). EST-MS m/z calc. 591,2127, encontrado 592,0 la FIG. 5).
Síntesis alternativa de ácido 2-doro-6-[3-[2-[1-(trifluo
Paso 1: 3-hidroxi-1H-pirazol-4-carboxilato de etilo
[0232]
l]etoxi]pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirroMdin-1-ió en etanol (19,87 ml) con calentamiento, se filtró con etanol caliente (10 mL) y la solución caliente se trató oró a 30-35°C, se co-evaporó 3 veces con etanol (~ 20 cío en una estufa de secado a 45°C con una purga de sólido se secó adicionalmente al vacío en una estufa de n de semana. 930 mg (89%) de la sal sódica de N-azol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-il]piridin-3-1H RMN (400 MHz, DMSO-cfe) 8 8,15 (d, J = 2,7 Hz, 1H), (dd, J = 4,9, 2,0 Hz, 3H), 6,74 (d, J = 7,9 Hz, 1H), 6,01 ), 2,07 (t, J = 7,1 Hz, 3H), 1,78 (dd, J = 11,8, 5,6 Hz, 1H), 92 (m, 2H), 0,89 (q, J = 5,3, 4,6 Hz, 2H), 0,71 (d, J = 6,3 1)+; Tiempo de retención: 3,28 minutos. XRPD (consulte
etil)ddopropil]etoxi]p irazoM -il]p iridm -3-carboxnico

[0233] Se añadió una mezcla de EtOH (20,00 L, 10 vol) y 1,0 equiv.) bajo purga de nitrógeno a a un reactor de 50 ajustada a 40°C. La mezcla se agitó y luego se añadió p/p, 9,249 mol, 1,00 equiv) en porciones mediante un e reacción se calentó a 75°C durante 22 h para producir u se usó directamente en el siguiente paso.
Paso 2: 1-(terc-butil) 4-etil 3-hidroxi-1H-pirazol-1,4-di
[0234]
etoximetilen)propanodioato de dietilo (2000 g, 9,249 mol, uipado con un condensador de reflujo (10°C) y la camisa to de hidrazina (538,9 g de 55% p/p, 523,7 mL de 55% udo de adición. Una vez que se completó la adición, la olución de 3-hidroxi-1H-pirazol-4-carboxilato de etilo que
oxilato

[0235] La solución de 3-hidroxi-1H-pirazol-4-carboxilato (TEA) (46,80 g, 64,46 ml, 462,5 mmol, 0,05 eq.). Se añadi equiv.) en EtOH (2.000 L, 1 equiv.) al reactor durante 35 reacción; luego se añadió agua (10,00 L, 5,0 vol) duran completar la cristalización del producto. Se dejó que los cr El sólido se lavó con una mezcla de EtOH (4.000 L, 2,0 vacío para proporcionar 1 -(terc-butil)-4-etil-3-hidroxi-1 cristalino incoloro de aguja fina. 1H RMN (400 MHz, DMS 1,56 (s, 9H), 1,25 (t, J = 7,1 Hz, 3H).
Paso 3: 1-(terc-butil) 4-etil 3-(2-(1-(trifluorometil)ciclo [0236]
tilo se enfrió de 75°C a 40°C, luego se añadió trietilamina na solución de anhídrido de Boc (2,119 kg, 9,711 mol1,05 n. La mezcla se agitó durante 4 horas para completar la 15 minutos. La mezcla resultante se enfrió a 20°C para les envejecieran durante 1 hora y luego se filtró la mezcla.
y agua (2.000 L, 1,0 vol). Después, el sólido se secó al azol-1,4-dicarboxilato (1530 g, 65%) como un sólido fe) 8 11,61 (s, 1H), 8,40 (s, 1H), 4,20 (q, J = 7,1 Hz, 2H),
il)etoxi)-1H-pirazol-1,4-dicarboxilato

[0237] Se puso en marcha un reactor de 5 L con la ca reflujo a temperatura ambiente y se purgó con nitróge (trifluorometil)cidopropil]etanol (100,0 g, 648,8 mmol, dicarboxilato (166,3 g, 648,8 mmol) y la mezcla se agitó 746,1 mmol, 1,15 equiv.), luego se ajustó el reactor azoldicarboxilato de diisopropilo (150,9 g, 746,1 mmol, 1 mientras se mantenía la temperatura de reacción entre controlada) y se agitó durante un total de de 2,5 horas. añadió heptano (400 mL, 4 vol), la solución se enfrió a 2 de óxido de trifenilfosfina-DIAD (TPPO-DIAD). Una vez con heptano (400 mL, 4,0 vol) y se secó. El filtrado se u sin purificación adicional.
Paso 4: 3-(2-(1-(trifluorometil)cidopropil)etoxi)-1H-pi
[0238]
e puso a 40°C, se agitó a 450 rpm, el condensador de recipiente se cargó con tolueno (1,0 L, 10,0 vol), 2-[1-equiv.) y 1-(terc-butil) 4-etil 3-hidroxi-1H-pirazol-1,4-ezcla de reacción se cargó con trifenilfosfina (195,7 g, antener una temperatura interna de 40°C. Se añadió uiv.) en un embudo de adición y se añadió a la reacción 50°C (la adición fue exotérmica, la adición exotérmica ez que la reacción se consideró completa por HPLC, se rante 60 minutos y cristalizó la mayor parte del complejo peratura ambiente, la mezcla se filtró y el sólido se lavó el siguiente paso como una solución en tolueno-heptano
4-carboxilato de etilo
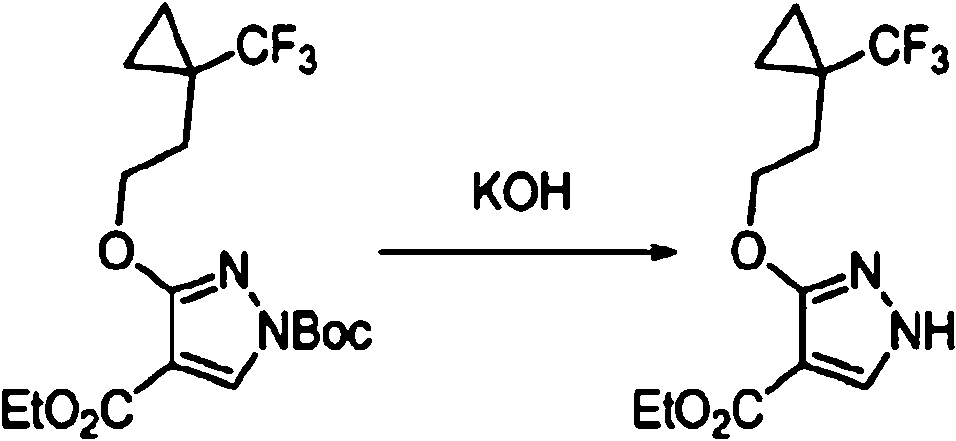
[0239] Se puso en marcha un reactor de 500 mL con la reflujo a temperatura ambiente y purga de nitrógeno. El de aproximadamente 160 mmol, 65,0 g de 1 -(terc-butil dicarboxilato en 3 vol de tolueno (preparado concentra hasta 4 volúmenes en un rotavapor). La reacción se fij KOH (33,1 g, 1,5 eq. de solución acuosa de KOH al 4 exotérmica suave, mientras que se generó CO2 al elim controlada por HPLC, con el producto cristalizando parc vol) a la mezcla de reacción y la reacción se enfrió a te se filtró y el sólido se lavó con heptano (80,00 ml, 1, obtuvieron 52,3 g de 3-(2-(1-(trifluorometil)ciclopropil)et en crudo que se utilizó sin purificación adicional.
Paso 5: Ácido 3-(2-(1-(trifluorometil)ciclopropil)etoxi)
[0240]
a ajustada a 40°C, agitando a 450 rpm, condensador de nte se cargó con una solución de tolueno que constaba l 3-(2-(1-(trifluorometil)ciclopropil)etoxi)-1H-pirazol-1,4 -na porción del filtrado del 25% de la reacción anterior mantener una temperatura interna a 40°C y se añadió n una porción, lo que dio como resultado una adición l grupo protector. La reacción prosiguió durante 1,5 h, nte durante la reacción. Se añadió heptano (160 ml, 2,5 tura ambiente durante 30 minutos. La mezcla resultante l), se secó y luego se secó al vacío (55°C, vacío). Se H-pirazol-4-carboxilato de etilo como un sólido incoloro
irazol-4-carboxílico
[0241] Se puso en marcha un reactor de 500 mL con la reflujo a temperatura ambiente y purga de nitrógeno. solución de 3-(2-(1-(trifluorometil)ciclopropil)etoxi)-1H-pi la reacción se agitó para suspender los sólidos. El reac mezcla se le añadió KOH (96 g de KOH acuoso al temperatura interna <50°C. Una vez que se completó la 50°C y la reacción prosiguió durante 23 horas, monitori 10°C y luego se concentró parcialmente en un evaporad resultante se diluyó con agua (250 ml, 5,0 vol) y 2-M temperatura ambiente, luego se detuvo y se dejó que la TPPO-DIAD restante en la capa orgánica y el producto Me-THF (100 mL, 2,0 vol), las capas se separaron y la marcha el agitador y se ajustó a 450 rpm, y la camisa de la adición de HCl acuoso 6 M (427 ml, 15 equiv.) en po El producto comenzó a cristalizar cerca de pH neutro y s añadió el ácido lentamente, y después se añadió más había terminado. A la suspensión resultante se le añad disolviera en la capa orgánica. Se detuvo la agitación, s se agitó y se volvió a extraer con 2-Me-THF (100 mL, 2, agitaron a temperatura ambiente, se lavaron con salmu través de celite y el sólido se lavó con 2-Me-THF (50 limpio, se agitó, se calentó a 50°C y se añadió heptano adición de heptano (300 mL, 6,0 vol) y luego se sembr 1H-pirazol-4-carboxílico), y el producto cristalizó durante se hubo destilado la mayor parte del 2-Me-THF. Se apa y enfriar la mezcla a temperatura ambiente. La mezcla mL, 2,0 vol) y el sólido se recogió y se secó al vacío (5 (2-(1-(trifluorometil)ciclopropil)etoxi)-1H-pirazol-4-carbox DMSO-cfe) 8 12,45 (s, 2H), 8,01 (s, 1H), 4,26 (t, J = 7,0
Paso 6 : 3-(2-(1-(trifluorometM)ddopropM)etoxi)-1H-pi
[0242]
a ajustada a 40°C, agitando a 450 rpm, condensador de ipiente se cargó con metanol (150,0 ml, 3,0 vol), una 4-carboxilato de etilo (50,0 g, 171,1 mmol, 1,0 equiv.), y fijó para mantener la temperatura interna a 40°C. A la 1,71 mol, 10,0 equiv.) en porciones manteniendo la n, la reacción se ajustó para mantener la temperatura a por HPLC. Una vez completada, la reacción se enfrió a torio para eliminar la mayor parte del MeOH. La solución (150 ml, 3,0 vol) y se transfirió al reactor, se agitó a s se separaran. Se probaron las capas, con el complejo capa acuosa. La capa acuosa se lavó de nuevo con 2-acuosa se devolvió al recipiente del reactor. Se puso en tor se ajustó a 0°C. El pH se ajustó a pH ácido mediante s, manteniendo la temperatura interna entre 10 y 30°C. mpañó con un fuerte desprendimiento de gases, y así se para alcanzar pH 1 una vez que la liberación de gases e-THF (400 mL, 8,0 vol) y se dejó que el producto se raron las capas y la capa acuosa se devolvió al reactor, . Las capas orgánicas se combinaron en el reactor y se 00 mL, 2 vols), se secaron sobre Na2SO4, se filtraron a 0 vol). El filtrado se transfirió a un matraz de rotavapor mL, 4,0 vol), y luego se concentró parcialmente con la 50 mg de ácido 3-(2-(1-(trifluorometil)ciclopropil)etoxi)-minación del disolvente. La destilación se detuvo cuando calentador del baño, se eliminó el vacío y se dejó agitar ó (velocidad lenta) y el sólido se lavó con heptano (100 vaporador rotatorio). Se obtuvieron 22,47 g de ácido 3-en forma de un sólido blanquecino. 1H RMN (400 MHz, ), 2,05 (t, J = 7,0 Hz, 2H), 0,92 (m, 4H).

[0243] Una mezcla de tolueno (490,0 mL), ácido 3-(2-(1-g, 264,9 mmol) y DMSO (70,00 mL) se colocó en un (aproximadamente 20,16 g, 19,80 ml, 132,4 mmol) al r completar la reacción y luego se enfrió a 20°C. La mezc (280,0 mL), luego con agua (2 x 140,0 mL) y finalmen Na2SO4 y luego se añadió carbón activado (5 g, Darco través de celite y el sólido se lavó con tolueno (140,0 (50°C, vacío) para producir 3-[2-[1-(trifluorometil)ciclopro RMN (400 MHz, DMSO-cfe) 8 11,87 (s, 1H), 7,50 (d, J = (t, J = 7,1 Hz, 2H), 1,00 - 0,77 (m, 4H).
Paso 7: 2-doro-6-[3-[2-[1-(trifluorometM)ddopropM]et
[0244]
rometil)ciclopropilo)etoxi)-1H-pirazol-4-carboxílico (70,0 r y se calentó a 100°C con agitación. Se añadió DBU r durante 15 min. La mezcla se agitó durante 20 h para lavó con agua (350,0 mL), luego con HCl acuoso 0,5 N n salmuera (210,0 ml). La capa orgánica se secó con esh) a la suspensión agitada. La mezcla seca se filtró a luego se secó. El filtrado se concentró en un rotavapor oxi]-1H-pirazol (30,89 g, 53%) como un aceite ámbar. 1H z, 1H), 5,63 (d, J = 2,4 Hz, 1H), 4,23 - 4,06 (m, 2H), 2,01
irazoM-M]piridm-3-carboxMato de etilo

[0245] Una mezcla de DMF (180,0 mL), 2,6-dicloropiri mmol), 3-[2-[1-(tnfluorometN)ddopropN]etoxi]-1H-pirazol 24,48 g, 177,1 mmol) se añadió a un reactor agitado a 20,43 mmol) al reactor, y la mezcla se agitó a 20°C dur mezcla se agitó durante 24 horas para completar l lentamente agua (360 ml). Después, la mezcla se drenó se lavó con agua (2 X 150 mL) y luego el sólido se (trifluorometil)ciclopropil]etoxi]pirazol-1-il]piridin-3-carbox RMN (400 MHz, DMSO-da) 8 8,44 (d, J = 2,9 Hz, 1H), 2,9 Hz, 1H), 4,34 (m, 4H), 2,09 (t, J = 7,1 Hz, 2H), 1,34 (
Paso 8 : Ácido 2-doro-6-[3-[2-[1-(trifluorometN)cidopr
[0246]
-carboxilato de etilo (aproximadamente 29,97 g, 136,2 g, 136,2 mmol) y K2CO3, (325 mesh, aproximadamente . Luego se agregó DABCO (aproximadamente 2,292 g, 1 hora, y luego se aumentó la temperatura a 30°C, y la cción. La mezcla se enfrió a 20°C; luego se añadió reactor y el sólido se aisló por filtración. Luego, el sólido al vacío a 55°C para producir etil 2-cloro-6-[3-[2-[1-(51,37 g, 93%) como un sólido fino de color beige. 1H d, J = 8,5 Hz, 1H), 7,75 (d, J = 8,5 Hz, 1H), 6,21 (d, J = 7,1 Hz, 3H), 1,00 - 0,84 (m, 4H).
]etoxi]pirazoM-N]piridm-3-carboxnico

[0247] Una solución de 2-cloro-6-[3-[2-[1-(trifluorometil) g, 123,8 mmol) en THF (300,0 mL) se preparó en un re acuoso (aproximadamente 59,44 g de 10% p/p, 148,6 reacción; luego se añadió lentamente HCl acuoso 1N (7 10°C y luego el sólido se aisló por filtración. El sólido s mediante vacío. A continuación, el sólido se secó adici cloro-6-[3-[2-[1-(trifluorometil)ciclopropil]etoxi]pirazol-1-il] DMSO-cfe) 8 13,63 (s, 1H), 8,48 - 8,35 (m, 2H), 7,73 (d, 2H), 2,09 (t, J = 7,1 Hz, 2H), 1,01 - 0,82 (m, 4H).
Ejemplo sintético 2: Síntesis del compuesto 2, (R)-1H-pirazol-1-il)-2-(2,2,4-trimetilpirrolidin-1 -il)nicotina
[0248]
ropil]etoxi]pirazol-1-il]piridin-3-carboxilato de etilo (50,0 a 20°C. Se añadió EtOH (150,0 mL), seguido de NaOH ). La mezcla se agitó durante 1 hora para completar la ml). La suspensión resultante se agitó durante 30 min a con agua (150 ml, luego 2 x 100 mL) y luego se secó ente al vacío con calentamiento para producir ácido 2-n-3-carboxílico (42,29 g, 91%). 1H Rm N (400 MHz, ,4 Hz, 1H), 6,20 (d, J = 2,9 Hz, 1H), 4,35 (t, J = 7,1 Hz,
Msulfoml)-6-(3-(2-(1-(trifluorometN)ddopropN)etoxi)-
[0249] (R)-N-(fenilsulfonil)-6-(3-(2-( 1 -(trifluorometil)ciclop il)nicotinamida se sintetizó de una manera análoga al (trifluorometil)cidopropil]etoxi]pirazol-1-il]piridin-3-carbox mmol), (4R)-2,2,4-trimetilpirrolidina (sal de clorhidrato) (1 dietoxietano (1,5 mL) proporcionando N-(bencenosu [(4R)-2,2,4-trimetilpirrolidin-1 -il]piridin-3-carboxamida (1, (M+1)+; Tiempo de retención: 2,3 minutos. 1H RMN (400 - 7,96 (m, 2H), 7,81 (d, J = 8,2 Hz, 1H), 7,76 - 7,69 (m, 6,11 (d, J = 2,8 Hz, 1H), 4,31 (t, J = 7,0 Hz, 2H), 2,41 (t, 3H), 1,82 (dd, J = 11,9, 5,5 Hz, 1H), 1,52 (d, J = 9,4 Hz, 3,9, 1,6 Hz, 2H), 0,64 (d, J = 6,3 Hz, 3H).
Ejemplo sintético 3: Síntesis del compuest (trifluorometM)cidopropM)etoxi)-1H-pirazoM-M)-2-(2,2
Paso A: 2-Cloro-N-((4-hidroxi-3-metoxifeml)sulfo il)nicotinamida
[0250]
etoxi)-1H-pirazol-1-il)-2-(2,2,4-trimetilpirroMdin-1-puesto 1 usando N-(bencenosulfonil)-2-cloro-6-[3-[2-[1-a (1,5 g, 2,91 mmol), carbonato de potasio (2,0 g, 14,56 6,7 mmol) en NMP (W-metil-2-pirrolidona) (7,5 mL) y 1,2--6-[3-[ 2-[1-(tnfluorometil)ciclopropil]etoxi]pirazol-1-il]-2-, 79%). IEN-EM m/z calc. 591,2127, encontrado 592,0 DMSO-da) 8 12,51 (s, 1H), 8,19 (d, J = 2,8 Hz, 1H), 8,03 7,66 (dd, J = 8,3, 6,7 Hz, 2H), 6,91 (d, J = 8,2 Hz, 1H), 10,5 Hz, 1H), 2,27 (t, J = 8,7 Hz, 1H), 2.07 (t, J = 7,1 Hz, 1,36 (t, J = 12,1 Hz, 1H), 0,99 -0,92 (m, 2H), 0,88 (tt, J =
, (S)-N-((4-hidroxi-3-metoxifeml)sulfoml)-6-(3-(2-(1-etMpirroMdm-1-M)mcotmamida
(3-(2-(1-(trifluorometN)ddopropN)etoxi)-1H-pirazoM-

[0251] Una solución de ácido 2-cloro-6-[3-[2-[1-(trifluoro 2,24 mmol) y carbonil diimidazol (434 mg, 2 , 68 mmol) en 3-metoxibencenosulfonamida (0,500 g, 2,46 mmol) y D durante 21 horas, se diluyó con acetato de etilo (5 mL) extrajo con acetato de etilo. Los extractos combinados se evaporaron. El residuo se purificó mediante cromatog etilo en hexanos (50-100%) para
(trifluorometil)ciclopropil)etoxi)-1H-pirazol-1-il)nicotinami 515,1 (M+1)+; Tiempo de retención: 0,74 minutos.
Paso B: (S)-N-((4-Hidroxi-3-metoxifenil)sulfonil)-6-(2,2,4-trimetilpirrelidin-1-il)nicotinamida
[0252]
ciclopropil]etoxi]pirazol-1-il]piridin-3-carboxílico (0,843 g, (2,5 mL) se agitó durante 2,5 horas y se agitó 4-hidroxi-0,5 mL, 3,35 mmol) se agregaron. La reacción se agitó acidificó con ácido clorhídrico acuoso 1 N (10 mL) y se aron con salmuera, se secaron sobre sulfato de sodio y en gel de sílice eluyendo con un gradiente de acetato de 2-cloro-N-((4-hidroxi-3-metoxifenil)sulfonil)-6-(3-(2-(1-06 mg, 72%). IEN-EM m/z calc. 560,07, encontrado
-(1-(trifluorometil)ciclopropil)etoxi)-1H-pirazol-1-il)-2-

[0253] Una mezcla de 2-cloro-N-((4-hidroxi-3-m pirazol-1-il)nicotinamida (906 mg, 1,62 mmol), sal de cl y se agitó carbonato de potasio (1,29 g, 9,33 mmol) e reacción se diluyó con 15 mL de agua y 5 mL de acet ácido clorhídrico acuoso 6 N y las capas se separaron. extractos combinados se lavaron con salmuera, se sec enil)sulfonil)-6-(3-(2-(1-(trifluorometil)ciclopropil)etoxi)-1H-ato de (4S)-2,2,4-trimetilpirrolidina (545 mg, 3,64 mmol) SO (5,5 mL) a 120°C durante 24 horas. La mezcla de etilo. Después, la mezcla de reacción se acidificó con pa acuosa se extrajo con 10 mL de acetato de etilo. Los sobre sulfato de sodio y se evaporaron para producir un
producto crudo que se purificó mediante cromatografía hexanos para producir (S)-N-((4-hidroxi-3- metoxifenil)s il)-2-(2,2,4-trimetilpirroMdin-1-il)nicotinamida (470 mg, Tiempo de retención: 10,07 minutos.
Ejemplo sintético 4: Síntesis de (trinuorometil)cicloprepil]etoxi]pirazol-1 -il]-2-[(4S)-2
Paso A: 2-Cloro-N-(o-tolilsulfonil)-6-[3-[2-[1-(trifluor
[0254]
l de sílice utilizando un gradiente de acetato de etilo en l)-6-(3-(2-(1-(trifluorometil)ciclopropil)etoxi)-1H-pirazol-1-. IEN-EM m/z calc. 637,2, encontrado 638,2 (M+1)+;
compuesto 4, N-(o-toMlsulfoml)-6[3-[2-[1-imetilpirrolidin-1-il]piridin-3-carboxamida
l)ciclopropil]etoxilpirazol-1-il]piridin-3-carboxamida

[0255] A ácido 2-cloro-6-[3-[2-[1 -(trifluorometil)cicloprop en THF (1,739 mL) se añadió 1,1'-carbonildiimidazol (ap durante una hora. Se añadió 2-metilbencenosulfonamid diazabiciclo(5,4.0)undec-7-eno (DBU) (aproximadamen durante 3 horas. La reacción se diluyó con acetato de capas orgánicas se secaron y concentraron y (trifluorometil)ciclopropil]etoxi]pirazol-1-il]piridin-3-carbo paso sin caracterización.
Paso B: N-(o-TolMsulfoml)-6-[3-[2-[ trimetilpirrolid ina-1-il]p iridin-3-carboxam ida
[0256]
i]pirazol-1-il]piridin-3-carboxílico (196 mg, 0,5217 mmol) adamente 106,6 mg, 0,6573 mmol) y la reacción se agitó oximadamente 89,32 mg, 0,5217 mmol), seguido de 1,8-2,2 mg, 257,6 ml, 1,722 mmol) y la reacción se agitó ácido cítrico acuoso 1 M y las capas se separaron. Las sólido resultante 2-cloro-N-(o-tolilsulfonil)-6-[3-[2-[1-a (aproximadamente 252 mg) se utilizó para el siguiente
uorometN)ciclopropN]etoxi]pirazoM-N]-2-[(4S)-2,2,4-

[0257] A 2-cloro-N-(o-tolilsulfonil)-6-[3-[2-[1-(t (aproximadamente 252 mg) y carbonato de potasio (39 trimetilpirrolidina (sal de hidrocloruro) (212 mg, 1,42 mm se enfrió, se diluyó con acetato de etilo y ácido cítrico ac concentraron y el residuo resultante se purificó con diclorometano para dar N-(o-tolilsulfonil)-6-[ trimetilpirrolidin-1-il]piridin-3-carboxamida (60,6 mg, 19 2,8 Hz, 1H), 8,04 (dd, J = 7,9, 1,4 Hz, 1H), 7,81 (d, J = 6,93 (d, J = 8,3 Hz, 1H), 6,10 (d, J = 2,7 Hz, 1H), 4,31 ( (ddt, J = 11,8, 9,0, 4,5 Hz, 1H), 2,08 (t, J = 7,0 Hz, 2H), Hz, 1H), 1,00 - 0,93 (m, 2H), 0,92 - 0,84 (m, 2H), 0,69 (d (M+1)+; Tiempo de retención: 1,92 minutos
Ejemplo sintético 5: Síntesis del
(trifluorom etil)ciclopropil]etoxi]pirazol-1 -il]-2-[(4S)-2 rometil)ciclopropil]etoxi]pirazol-1-il]piridin-3-carboxamida 2,84 mmol) en 0,4 mL de DMSO se añadió (4S)-2,2,4-reacción se agitó a 130°C durante 16 horas. La reacción 1 M y se separaron las capas. Los orgánicos se secaron, de sílice (24 g) eluyendo con metanol al 0-14% en -(trifluorometil)ciclopropil]etoxi]pirazol-1-il]-2-[(4S)-2,2,4-RMN (400 MHz, DMSO-d6) 8 12,63 (s, 1H), 8,19 (d, J = , 1H), 7,58 (td, J = 7,5, 1,5 Hz, 1H), 7,50 - 7,40 (m, 2H), 7,1 Hz, 2H), 2,64 (s, 3H), 2,39 (d, J = 8,8 Hz, 2H), 2,16 (dd, J = 11,9, 5,6 Hz, 1H), 1,52 (s, 6H), 1,35 (t, J = 12,1 6,2 Hz, 3H). IEN-EM m/z calc. 605,23, encontrado 606,4
puesto 5, N-(3-fluorofeml)sulfoml-6-[3-[2-[1-rimetilpirrolidin-1-il]piridin-3-carboxamida
Paso A: 2-C loro-N-(3-fluorofenil)sulfonil-6-[3 carboxamida
[0258]
-(trifluorometil)ciclopropil]etoxi]pirazol-1-il]piridin-3-

[0259] A ácido 2-cloro-6-[3-[2-[1-(trifluorometil)ciclopropi en THF (1,7 mL) se añadió 1,1'-carbonildiimidazol (108, añadió 3-fluorobencenosulfonamida (93,25 mg, 0,5323 (267,5 mg, 262,8 ml, 1,757 mmol) y la reacción se agitó ácido cítrico acuoso 1 M y se separaron las capas. Los cloro-N-(3-fluorofenil)sulfonil-6-[3-[2-[1-(trifluorometil)cicl (aproximadamente 259 mg) se utilizó en el siguiente pas
Paso B: N-(3-Fluorofenil)sulfonil-6-[3-[2-[1-(tri trim etilpirrolid in-1-il]pirid in-3-carboxam ida
[0260]
i]pirazol-1-il]piridin-3-carboxílico (0,200 g, 0,532 mmol) 0,6707 mmol) y la reacción se agitó durante 1 hora. Se , seguido de 1,8-diazabiciclo(5,4,0)undec-7-eno (DBU) te 2 horas. La reacción se diluyó con acetato de etilo y icos se secaron y concentraron y el sólido resultante 2-il]etoxi]pirazol-1-il]piridin-3-carboxamida
caracterización.
metil)ciclopropil]etoxi]pirazol-1-il]-2-[(4S)-2,2,4 -

[0261] A 2-cloro-N-(3-fluorofenil)sulfonil-6-[3-[2-[1-( (aproximadamente 259 mg, 0,486 mmol) y carbonato de (4S)-2,2,4-trimetilpirrolidina (sal de hidrocloruro) (211,0 horas. La reacción se enfrió, se diluyó con acetato de et orgánicos se secaron, se concentraron y el residuo resul al 0-14% en diclorometano para dar N-(3-fluorofenil) [(4S)-2,2,4-trimetilpirrolidin-1-il]piridin-3-carboxamida (50 2,7 Hz, 1H), 7,96 - 7,89 (m, 1H), 7,87 - 7,77 (m, 2H), 7,6 7,02 (d, J = 8,3 Hz, 1H), 5,95 (d, J = 2,8 Hz, 1H), 4,37 (t, 2,48 (m, 1H), 2,28 -2,16 (m, 1H), 2,10 (t, J = 7,0 Hz, 2H), (t, J = 12,1 Hz, 1H), 1,02 -0,96 (m, 2H), 0,86 -0,77 (m, 5 de retención: 0,81 minutos
Ejemplo sintético 6: Síntesis del compuesto 6,2-[(4S il]-N-(4-hidroxifenil)sulfonil-6-[3-[2-[1-(trifluorometil)c
Paso A: 2-cloro-N-(4-hidroxifenil)sulfonil-6-[3 carboxamida
rometil)ciclopropil]etoxi]pirazol-1-il]piridin-3-carboxamida io (389,6 mg, 2,819 mmol) en 0,4 ml de DMSO se añadió 1,41 mmol) y la reacción se agitó a 130°C durante 16 ácido cítrico acuoso 1 M y se separaron las capas. Los se purificó en gel de sílice (24 g) eluyendo con metanol il-6-[3-[2-[1-(trifluorometil)ciclopropil]etoxi]pirazol-1-il]-2-, 15%) 1H RMN (400 MHz), Metanol-d4) 88,23 (d, J = J = 8,1, 5,3 Hz, 1H), 7,46 (tdd, J = 8,5, 2,5, 1,0 Hz, 1H), ,0 Hz, 2H), 3,34 (s, 1H), 2,68 (t, J = 10,3 Hz, 1H), 2,56 -(dd, J = 11,9, 5,7 Hz, 1H), 1,59 (d, J = 9,7 Hz, 6H), 1,48 N-EM m/z calc.609,2, encontrado 610,3 (M+1)+; Tiempo
Dideuterio-2,2-dimetil-4-(trideutetiometil)pirrolidin-1-ropil]etoxilpirazol-1-il]piridin-3-carboxam ida
-(trifluorometil)ciclopropil]etoxi]pirazol-1-il]piridin-3-
[0262]

[0263] Ácido 2-Cloro-6-[3-[2-[1 -(trifluorometil)ciclopropil] CDI (aproximadamente 51,38 mg, 0,3169 mmol) se co ambiente durante 2 horas. Se añadió 4-hidroxibencen seguido de DBU (aproximadamente 54,41 mg, 53,45 j L, temperatura ambiente. La mezcla de reacción se diluyó porciones de 10 ml de acetato de etilo. Los extractos sobre sulfato de sodio y se concentraron para dar (trifluorometil)ciclopropil]etoxi]pirazol-1-il]piridin-3-carbox purificación adicional. IEN-EM m/z calc. 530,1, encontra
Paso B: 2-[(4S)-3,3-Dideuterio-2,2-dimetil-4-(trideute (trifluorometM)cidopropM]etoxMpirazoM-M]piridm-3-c
[0264]
pirazol-1 -il]piridin-3-carboxflico (0,100 g, 0,266 mmol) y aron en THF (600,0 mL) y se agitaron a temperatura fonamida (aproximadamente 50,69 mg, 0,2927 mmol) 74 mmol) y la reacción se agitó durante 16 horas más a 10 ml de ácido cítrico acuoso 1 M y se extrajo con tres icos combinados se lavaron con salmuera, se secaron ólido blanco 2-cloro-N-(4-hidroxifenil)sulfonil-6-[3-[2-[1-a (128 mg, 91%) que se usó en el siguiente paso sin 1,0 (M+1)+; Tiempo de retención: 0,69 minutos.
etil)pirrolidin-1-il]-N-(4-hidroxifenil)sulfonil-6-[3-[2-[1-xamida

[0265] 2-Cloro-N-(4-hidroxifenil)sulfonil-6-[3-[2-[1-(trifluor g, 1,9 mmol), sal de clorhidrato (S)-2,2-dimetil-4-(metilpotasio (1,55 g, 11,2 mmol) se combinaron en DMSO (6 se enfrió a temperatura ambiente y se diluyó con agua acetato de etilo (50 mL) a la mezcla. La mezcla se acidific se separaron. Los orgánicos se combinaron, se lavar concentraron. El material crudo obtenido se purificó
utilizando un gradiente de acetato de etilo al 0-30% en h las fracciones que cumplían con las especificaciones de una mezcla de acetato de etilo/MTBE 9:1. Los orgánicos secó durante la noche a alto vacío para proporcionar 2-il]-N-(4-hidroxifenil)sulfonil-6-[3-[2-[1-(trifluorometil)ciclopr |En -Em m/z calc. 612,2, encontrado 613,7 (M+1)+; Tiem
Ejemplo sintético 7: Síntesis del compuesto 7, (trideuteriometil)pirrolidin-1-il]-6-[3-[2-hidroxi-2-[1-(triflu
[0266]
til)ciclopropil]etoxi]pirazol-1-il]piridin-3-carboxamida (1,0 rrolidina-3,3-d2 (0,892 g, 5,66 mmol) y el carbonato de y se calentaron a 130°C durante 16 horas. La reacción mL). Después de agitar durante 15 minutos, se añadió n ácido cítrico acuoso 1 M (pH ~ 3-4) (30 mL) y las capas on salmuera, se secaron sobre sulfato de sodio y se nte cromatografía en columna (24 g de gel de sílice) o. Las fracciones individuales se analizaron por HPLC y za requeridas se combinaron, evaporaron y trituraron en vaporaron hasta el 10% y el sólido obtenido se filtró y se -3,3-dideuterio-2,2-dimetil-4-(trideuteriometil)pirrolidin-1-etoxi]pirazol-1 -il]piridin-3-carboxamida (0,38 g, 32%) retención: 1,40 minutos.
(bencenosulfoml)-2-[(4S)-3,3-dideuterio-2,2-dimetN-4-etil)ciclopropil]etoxi]pirazol-1-il]piridin-3-carboxamida
[0267] Se cargó un recipiente de reacció (trifluorometil)cidopropil]etoxi]pirazol-1-il]piridin-3-carbo 4-(trideuteriometil)pirroMdina (sal de clorhidrato) (320 m en una atmósfera de nitrógeno. Se añadió carbonato d calentó a 130°C. La mezcla de reacción se agitó duran agua (2.000 mL) y se ajustó el pH a <3 con HCl acuoso pH se ajustó adicionalmente con cloruro de hidrógeno con acetato de etilo (4 mL) dos veces y las capas orgán se secaron sobre sulfato de sodio. Después, la capa org de sílice utilizando un gradiente de acetato de etilo al 0-6 de heptanos y MTBE para producir N-(bencenosulfonN) 1 -il]-6-[3-[2-hidroxi-2-[1 -(trifluorometil)ciclopropil]etoxi]pir calc. 612,2, encontrado 613,1 (M+1)+; Tiempo de reten 1H), 8,20 (d, J = 2,8 Hz, 1H), 8,05 - 7,94 (m, 2H), 7,81 ( 2H), 6,90 (d, J = 8,2 Hz, 1H), 6,12 (d, J = 2,8 Hz, 1H), (m, 1H), 3,89 (d, J = 4,9 Hz, 1H), 2,39 (d, J = 10,5 Hz, (d, J = 9,7 Hz, 6H), 1,04 - 0,83 (m, 4H).
Síntesis de clorhidrato de (4S)-3,3-Dideuterio-2,2-di
[0268]
con N-(bencenosulfonil)-2-doro-6-[3-[2-hidroxi-2-[1-a (0,500 g, 0,942 mmol), (4S)-3,3-dideuterio-2,2-dimetilmmol), NMP (3000 mL) y 1,2-dietoxietano (500,0 mL) sio (650,8 mg, 4,709 mmol) y la mezcla de reacción se oche. La mezcla de reacción se enfrió y se diluyó con mL de 6 M, 7,800 mmol), que se añadió gota a gota. El mL de 6 M, 0,8760 mmol). La capa acuosa se extrajo ombinadas se lavaron dos veces con agua, salmuera y se concentró hasta un residuo que se purificó sobre gel hexanos. Luego, este material se trituró en una mezcla )-3,3-dideuterio-2,2-dimetN-4-(trideuteriometN)pirroNdin--il]piridin-3-carboxamida (266 mg, 46%) Ie N-EM m/z ,67 minutos. 1H RMN (400 MHz, DMSO-cfe) 8 12,51 (s, 8,3 Hz, 1H), 7,72 (d, J = 7,3 Hz, 1H), 7,65 (t, J = 7,6 Hz, d, J = 5,5, 2,7 Hz, 1H), 4,42 - 4,28 (m, 1H), 4,23 -4,09 ,37 - 2,22 (m, 1H), 2,06 (dd, J = 10,6, 7,0 Hz, 1H), 1,52
4(trideuteriometil)pirrolidina

Paso A: MetN-cfe 4-metN-2-(metN-d3)-4-mtropentanoat [0269]
d2

[0270] Un matraz de fondo redondo de tres bocas de línea de nitrógeno y un termopar J-Kem con manta cal metacrilato de metilo (50,0 g, 460 mmol), y se diazabiciclo[5.4.0]undec-7-eno (DBU, 1,47 g, 9,62 m exotermia de 20 a ~40°C y se dejó agitar sin calentar ni e (HPLC), por lo que la solución se calentó a 80°C duran se lava con 1 M HCl (15 mL), se seca sobre sulfato de el disolvente y cualquier material de partida residual par g, 99%). Se utilizó para el siguiente paso sin purificació
Paso B: metN-cfe SM-metM-2-(metN-d3)-4-mtropentan
[0271]
L equipado con una barra de agitación magnética, una ra se cargó con 2-nitropropano (34,3 g, 385 mmol), ds-a temperatura ambiente cuando se añadió 1,8-n una porción. La solución de reacción produjo una durante 16 h. La reacción se completó solo parcialmente . La mezcla de reacción se diluye con MTBE (170 mL), io, se filtra y se concentra (29” Hg a 60°C) para eliminar ucir el producto. como aceite de color amarillo claro (75 onal por destilación.
3,3-d2

[0272] Un matraz de fondo redondo de tres bocas de 5 nitrógeno y un termopar J-Kem con manto calefactor se c d2 (75 g, 380 mmol) y 2000 ml de tampón fosfato de sodi miehei (sigma L4277, palatase de Novozymes) (0,5 vol) 250 mm, 5 mm, 1,0 mL/min, heptano al 98%/IPA al 2%) de reacción se extrajo dos veces con MTBE (1 L cada v las extracciones. Los orgánicos combinados se lavaron (5 vol), salmuera (5 vol), se secaron sobre sulfato de sod deseado metil-d¿ ('SM-metil-2.-(metil-d3)-4-nitropentano rendimiento).
Paso C: (S)-5,5-Dimetil-3-(metN-d3)pirroMdm-2-ona-4,4-
[0273]
uipado con un agitador mecánico superior, una linea de con metil-cb 4-metilo-2-(metil-d3)-4-nitropentanoato-3,3-7,5 a 0,8 M. A esto se le añadió lipasa de Rhizomucor agitó a 30°C durante 25 h. La HPLC quiral (ADH 4,6 x tra una relación de enantiómeros de 99,8/0,2. La mezcla El orgánico incluyó cualquier emulsión formada durante eces con una solución acuosa de bicarbonato de sodio se concentraron al vacío para proporcionar el producto 3,3-d2 como aceite amarillo pálido (32,5 g, 43% de

[0274] Un recipiente de alta presión (botella de agitación se cargó secuencialmente con agua desionizada enjuag ('Sj-4-metil-2-(metil-d3)-4-nitropentanoato-3,3-d2 (32,5 g, evacuó/rellenó con N2 (3 veces). Sin agitación, se evacu Parr se agitó mientras se calentaba el contenido a 60°C recipiente se evacuó/rellenó con N2 (3 veces) y el conte Celite; manta de N2 ). El matraz/lecho filtrante se lavó c filtración humedecida con disolvente se transfirió a otro r ningún momento se debe secar completamente el cataliza El filtrado y los lavados se combinaron y concentraron (40 2-ona-4,4-d2 como un sólido blanco (20 g, 92%).
Paso D: Clorhidrato de (4S)-3,3-dideuterio-2,2-dimetil
[0275]
r, 500 mL) se purgó y se mantuvo bajo N2. El recipiente (3 veces) con Ni Raney®2800 húmedo (6,1 g), metil-d3 mmol) y etanol (290 ml). El recipiente se selló y se recipiente y se volvió a llenar con H2 (30 psi). La botella presión de H2 se mantuvo a 30 psi durante 8 horas. El se eliminó mediante filtración al vacío (almohadilla de tanol (3 x 50 ml). Después del lavado final, la torta de ptor y se cubrió con agua para su eliminación. Nota: En (mantener húmedo durante todo el proceso de filtración).
0 torr) para producir (S)-5,5-dimetil-3-(metil-d3)pirrolidin-
trideuteriometil)pirrolidina

[0276] Un matraz de fondo redondo de tres bocas de 1 nitrógeno y un termopar J-Kem se cargó con gránulos de 4 vol) calentado de 20 a 36°C (calor de mezclado). Se a ona-4,4-d2 (20 g, 150 mmol) en THF (120 ml, 6 vol) a la temperatura de reacción se elevara a ~60°C. La tempera se mantuvo allí durante 16 h. La mezcla de reacción se MTBE. La mezcla se inactivó lentamente con la adición sodio (1 vol) durante 2 h. Nota: Se observó una desgasific y la mezcla gris oscura se vuelve blanca. Una vez complet ambiente. El sólido se eliminó mediante filtración (lec uipado con un agitador mecánico superior, una línea de ruro de litio y aluminio (7,6 g, 202 mmol) en THF (80 ml, ó una solución de (S)-5,5-dimetil-3-(metil-d3)pirrolidin-2-pensión durante 30 minutos mientras se dejaba que la de reacción se aumentó hasta casi el reflujo (~ 68°C) y ó por debajo de 40°C y se diluyó con 200 ml (10 vol) de a gota de una solución acuosa saturada de sulfato de n vigorosa (H2), la mezcla se espesa, luego se adelgaza la adición, la mezcla de reacción se enfrió a temperatura e Celite) y se lavó con acetato de etilo (4 vol). Con
enfriamiento externo y una manta de N2, el filtrado y los l de 4 M HCl anhidro en dioxano (38 ml, 152 mmol) mient completada la adición (20 minutos), la suspensión result llenar con heptanos (4 vol) dos veces durante la conce el sólido se recogió por filtración bajo una capa de N2. a alto vacío a 45°C para producir clorhidrato de (4S)-3 75%). El producto es bastante higroscópico por lo que s
Ejemplo sintético 8: Síntesis del compuesto 8, tolilsulfonil)-2-[(4S)-2,2,4-trimetilpirrolidin-1-il]piridin-
[0277]
os se combinaron y se trataron con la adición gota a gota e mantenía la temperatura por debajo de 20°C. Una vez se concentró al vacío a 45°C. La suspensión se volvió a ión. La suspensión se enfrió por debajo de 30°C cuando ido se secó con succión de N2 y se secó adicionalmente deuterio-2,2-dimetil-4-(trideuteriometil)pirrolidina (17,5 g, nipuló bajo nitrógeno.
Diespiro[2.0.24.13]beptan-7-Nmetoxi)pirazoM-M]-N-(orboxamida

[0278] 2-Cloro-6-[3-(diespiro[2.0.2413]heptan-7-ilmetoxi) 0,341 mmol) y (4S)-2,2,4-trimetilpirrolidina (sal de clorhi DMSO (2 mL). Se añadió carbonato de potasio finament y se calentó durante la noche a 130°C. Después de enf con acetato de etilo (50 mL) y se lavó con ácido cítrico ac se secó sobre sulfato de sodio, se filtró y se concentró a en columna de gel de sílice eluyendo con un gradiente de sílice de 12 gramos para producir 6-[3-(diespiro[2.
2,2,4-trimetilpirrolidin-1-il]piridin-3-carboxamida (0,030 g Tiempo de retención: 2,46 minutos.
Ejemplo sintético 9: Síntesis del compuesto 9, N-(b M]-6-[3-[2-[1-(trifluorometN)ciclopropN]etoxi]pirazoM
[0279]
ol-1-il]-N-(o-tolilsulfonil)piridin-3-carboxamida (0,170 g, ) (0,116 g, 1,02 mmol) se combinaron y se disolvieron en lido (95 mg, 0,68 mmol). La mezcla de reacción se selló temperatura ambiente, la mezcla de reacción se diluyó (1 M, 2X 50 mL) y salmuera (1X 50 ml). La capa orgánica ón reducida. El producto se aisló mediante cromatografía etanol al 0-20% en diclorometano en una columna de gel 3]heptan-7-ilmetoxi)pirazol-1-il]-N-(o-tolilsulfonil)-2-[(4S)-). iEn -EM m/z calc. 575,26, encontrado 576,36 (M+1)+;
nosulfoml)-2-[4-(hidroximetN)-2,2-dimetN-pirroNdm-1-ridm-3-carboxamida

Paso A: Éster metílico del ácido 2-hidroximetil-4-metil [0280]
nitropentanoico

[0281] 1,8-diazabiciclo[5.4.0]undec-7-eno (3,6 ml, 24 m mezcla se calentó a 65°C y se apagó el calor y se añad mmol). A continuación, se volvió a encender el calor a 80 la reacción se agitó a temperatura ambiente durante la reacción se diluyó con acetato de etilo (250 mL) y se la acuoso (125 mL) y salmuera (125 ml). La mezcla de prod g de gel de sílice en hexanos al 0-60%: éter eluyendo a metil-4-nitro-pentanoico (29,68 g, 60%) como un aceite 206,1 (M+1)+. Tiempo de retención: 1,67 minutos. 1H RM 1H) 2,10 -2,23 (m, 1H) 2,36 -2,50 (m, 1H)2,60 (d, J = 5,
Paso B: 3-Hidroximetil-5,5-dimetil-pirrolidin-2-ona
[0282]
se añadió a 2-nitropropano (26,5 ml, 292 mmol). Esta ota a gota 2-(hidroximetil)acrilato de metilo (25 ml, 243 Después de calentar durante 1 hora, se apagó el calor y he antes de calentar a 80°C durante otras 2 horas. La on cloruro de hidrógeno 1 M (2 x 125 mL), bicarbonato os de reacción se cromatografió en una columna de 330 -60% para dar éster metílico del ácido 2-hidroximetil-4-color verde claro. IEN-EM m/z calc. 205,21, encontrado 50 MHz, CDCh) ppm 1,50 - 1,59 (m, 6H) 1,85 - 1,98 (m, Hz, 1H) 3,66 - 3,77 (s, 3H)

[0283] Éster metílico del ácido hidroximetil-4-metil-4-nitr (60 mL) seguido de níquel Raney (1,7 g, ~15% en peso). noche. Se añadió más níquel Raney (1,0 g, ~50% en pes 3,5 h. En este punto, se añadió más éster metílico del mmol) y la reacción se calentó durante 72 h rellenando H y se lavó con metanol. La reacción cruda se cromatografi metanol al 10%, dando como resultado 3-hidroximetil-5,5-1H RMN (250 MHz, CDCla) 8 ppm 1,31 (d, J = 9,01 Hz, 6H Hz, 1H) 2,73 - 2,91 (m, 1H) 3,31 (d, J = 4,72 Hz, 1H) 3,64
Paso C: (5,5-Dimetil- pirrolidin-3-il)-metanol
ntanoico (4,45 g, 21,7 mmol) se añadió a etanol absoluto reacción se calentó a 60°C bajo 2 bar de H2 durante la y la reacción se calentó a 60°C bajo 5 bar de H2 durante o 2-hidroximetil-4-metil-4-nitro-pentanoico (3,95 g, 19,3 ra mantener 5 bar. La reacción se filtró a través de celite bre gel de sílice y se eluyó con diclorometano al 0-10%: etil-pirrolidin-2-ona (3,69 g, 63%) como un sólido blanco.
72 (dd, J = 12,52, 10,33 Hz, 1H) 2,04 (dd, J = 12,58, 8,84 ,95 (m, 2H) 5,93 (ancho s., 1H)
[0284]

[0285] Se suspendió hidruro de litio y aluminio (3,90 g, gota a gota hidroximetil-5,5-dimetil-pirrolidin-2-ona (3,69 calentó a 65°C durante 40 h. La reacción se diluyó con de hielo antes de agregar gota a gota sal de Rochelle a metil-tetrahidrofurano (2 X 200 mL) y se secó sobre sulf (3,47 g, 104%). 1H RMN (250 MHz, CDCla 8 ppm 1,06 1,68 - 1,89 (sa, 1H)2,31 -2,52 (m, 1H)2,83 (dd, J = 11,
Paso D: 4-(terc-Butil-dimetilo-silaniloximetil)-2,2-dim
[0286]
mmol) en tetrahidrofurano (60 ml). Después se añadió ,77 mmol) en tetrahidrofurano (30 mL) y la reacción se l-tetrahidrofurano (125 mL) y luego se enfrió en un baño saturada (200 ml). La capa orgánica se extrajo con 2-sodio para dar (5,5-dimetil-pirrolidin-3-il)-metanol crudo (m, 6H) 1,29 (dd, J = 12,58, 7,20 Hz, 2H) 1,43 (s, 1H) 49 Hz, 1H) 3,05 -3,26 (m, 1H) 3,48 -3,71 (m, 1H)
irrolidina

[0287] A (5,5-dimetil-pirrolidin-3-il)-metanol (3,08 g, 23,8 acetonitrilo (24 mL) se añadió 1,8-Diazabiciclo[5.4.0]und h. La reacción se diluyó con cloroformo (250 mL) y se sobre sulfato de sodio. El crudo se cromatografió en ge 15-35% de metanol para dar 4-(terc-butil-dimetil-silanil amarillo después de dos columnas IEN-EM m/z calc. minutos. 1H RMN (250 MH z, CDCla) 8 ppm -0,05 -0,11 1H) 1,74 (dd, J = 12,63, 8,79 Hz, 1H) 1,92 (ancho s., 1 (dd, J = 11,48, 7,97 Hz, 1H) 3,45 - 3,61 (m, 2H)
Paso E: N-(bencenosulfo (trifluorom etil)ciclopropil]etoxi]pirazol-1 -il]piridin-3-c
[0288]
l), cloruro de tercbutildimetilsililo (4,31 g, 28,6 mmol) en no (5,3 ml, 35,7 mmol) La reacción se agitó durante 3,5 on agua (125 mL) y salmuera (125 mL) luego se secó ílice y se eluyó con diclorometano/metanol, eluyendo a til)-2,2-dimetil-pirrolidina (3,88 g, 67%) como un aceite , encontrado 244,2 (M+1)+ Tiempo de retención: 2,52 ) 0,89 (s, 9H) 1,19 (d, J = 18,02 Hz, 6H) 1,25 -1,32 (m, 2 - 2,50 (m, 1H) 2,81 (dd, J = 11,54, 6,37 Hz, 1H) 3,11
4-(hidroximetN)-2,2-dimetN-pirroNdm-1-N]-6-[3-[2-[1-xamida

[0289] N-(bencenosulfonil)-2-doro-6-[3-[2-[1-(trifluorome mg, 0,04855 mmol), terc-butilo-[(5,5-dimetilpirroMdin-3-il mmol) y K2CO3 (aproximadamente 33,56 mg, 0,2428 mm durante 16 h. La reacción se repartió entre una solució extractos orgánicos. Los orgánicos se lavaron con salm material crudo se purificó mediante cromatografía en ge para dar N-(bencenosulfonil)-2-[4-[[íerc-(trifluorometil)ciclopropil]etoxi]pirazol-1-il]piridin-3-carbox 722,4 (M+1)+; Tiempo de retención: 0,97 minutos.
Paso F: N-(bencenosulfoml (trifluorom etil)ciclopropil]etoxi]pirazol-1 -il]piridin-3-c
[0290]
dopropil]etoxi]pirazol-1-ilo]piridina-3-carboxamida (25 toxi]-dimetil-silano (aproximadamente 35,45 mg, 0,1456 e combinaron en DMSO (0,5 mL) y se calentaron a 130°C ácido cítrico 1 M y acetato de etilo y se separaron los , se secaron sobre sulfato de sodio y se evaporaron. El sílice eluyendo con metanol al 0 - 10 % en diclorometano o(dimetil)silil]oximetil]-2,2-dimetil-pirrolidin-1 -il]-6-[3-[2-[1 -a (15 mg, 43%) IEN-EM m/z calc. 721,2941, encontrado
[4-(hidroximetN)-2,2-dimetN-pirroNdm-1-N]-6-[3-[2-[1-xamida

[0291] N-(bencenosulfonil)-2-[4-[[íerc-butilo(dimetil)silil]o (trifluorometil)ciclopropil]etoxi]pirazol-1-il]piridin-3-carbox un baño de hielo. Se añadió fluoruro de tetra-n-butilamo dejó calentar a temperatura ambiente. La mezcla de reac de etilo y una solución de ácido cítrico 1M. Los orgánicos y se evaporaron. El material crudo se purificó mediante c en diclorometano para dar N-(bencenos (trifluorometil)ciclopropil]etoxi]pirazol-1-il]piridina-3-carbo encontrado 608,4 (M+1)+; Tiempo de retención: 1,9 minu
Ejemplo sintético 10: Síntesis del Compuesto metoxi]pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-il]
(1 -T rifluorometil-ciclobutil)-metanol
[0292]
til]-2,2-dimetil-pirrolidin-1-il]-6-[3-[2-[1-a (15 mg, 43%) se disolvió en THF (1 mL) y se enfrió en en THF (300 pL de 1 M, 0,3000 mmol) y la reacción se se agitó durante 1 hora y luego se repartió entre acetato lavaron con salmuera, se secaron sobre sulfato de sodio atografía en gel de sílice eluyendo con metanol al 0-10% nil)-2-[4-(hidroximetil)-2,2-dimetil-pirrolidin-1-il]-6-[3-[2-[1-ida (8,5 mg, 29%) IEN-EM m/z calc. 607,20764,
N-(bencenosulfonil)-6-[3-[[1-(trifluorometil)ciclobutil] in-3-carboxamida

[0293] Se disolvió ácido 1-trifluorometil-cidobutanocarbo 0°C. Se añadió gota a gota hidruro de litio y aluminio (38 temperatura ambiente durante la noche. La solución de r sodio decahidratado, lo que dio como resultado un d porciones hasta que no se observó más burbujeo a temp sobre un lecho de Celite, lavando con éter dietílico. El filtr mezcla que contenía el producto deseado y algo de re proporcionó 1-trifluorometil-ciclobutil-metanol (3,46 g, 7 (ppm): 3,82 (s, 2H), 2,39-2,14 (m, 2H), 2,10-1,85 (m, 4H).
Éster terc-butílico del ácido 3-(1-trifluorometil-ciclobu
[0294]
o (5,0 g, 30 mmol) en éter dietílico (60 mL) y se enfrió a ml, 1 M en éter dietílico) y la solución se dejó calentar a ción se enfrió a 0°C con agitación y se añadió sulfato de ndimiento gradual de gas. Se continuó la adición en tura ambiente. Después, la solución de reacción se filtró se concentró a presión reducida para dar 5,44 g de una o de éter dietílico (36% por integración de RMN). Esto como un aceite incoloro. 1H RMN (250 MHz, cD ch) 8
etoxi)-pirazol-1-carboxílico

[0295] 1-trifluorometil-ciclobutil-metanol (1,50 g, 9,73 m carboxílico (1,63 g, 8,85 mmol) se disolvieron en tetrahidr sonicación y se lavó abundantemente con gas nitrógeno. gota a gota azodicarboxilato de diisopropilo (1,92 ml, 9,73 a 50°C durante 16 horas. Después de enfriar a temperat mL) y se lavó con una solución de hidróxido de sodio 1 se secaron sobre sulfato de sodio, se filtraron y se concen mediante cromatografía flash usando un método de grad éster terc-butílico del ácido 3-(1-trifluorometil-ciclobutil blanquecino. IEN-EM m/z calc. 320,31, encontrado 321,1
Sal de h idrocloruro de 3-(1-trifluorometil-ciclobutilme
[0296]
y éster ferc-butílico del ácido 3-oxo-2,3-dihidro-pirazol-1-ano anhidro (32 ml). La solución se desgasificó mediante ñadió trifenilfosfina (2,55 g, 9,73 mmol) y luego se añadió ol). Una vez completada la adición, la reacción se calentó mbiente, la reacción se diluyó con acetato de etilo (100 x 100 mL) y luego con salmuera (125 ml). Los orgánicos n a presión reducida. El aceite amarillo crudo se purificó e de acetato de etilo al 0-10% en hexanos para producir xi)pirazol-1-carboxílico (2,48 g, 87%) como un sólido 1)+. Tiempo de retención: 3,74 minutos
-1H-pirazol

[0297] Éster terc-butílico del ácido 3-(1-Trifluorometil-ci disolvió en cloruro de hidrógeno 4 M en dioxano (77 ml). L seguido de la eliminación de los volátiles a presión reduc ciclobutilmetoxi)-1H-pirazol (1,95 g, 98%) como un polvo Tiempo de retención: 2,67 minutos.
2-cloro-6-(3-((1-(trifluorometil)ciclobutil)metoxi)-1H-pi
[0298]
utilmetoxi)-pirazol-1-carboxílico (2,48 g, 7,74 mmol) se lución se agitó durante la noche a temperatura ambiente, para producir la sal de hidrocloruro de 3-(1-trifluorometilco. IEN-EM m/z calc. 220,20, encontrado 221,2 (M+1)+.
l-1-il)nicotinato de terc-butilo

[0299] Sal de hidrocloruro de 3-(1-trifluorometil-cidobutil ácido 2,6-didoronicotínico (1,89 g, 7,62 mmol) se disolv potasio (4,21 g, 30,5 mmol) seguido de 1,4-diazabici temperatura ambiente durante la noche, luego se añadi etilo:hexanos 4:1 (100 mL). La fase orgánica se lavó concentró a presión reducida. El aceite crudo se purific de gradiente de acetato de etilo al 0-10% en hexano trifluorometil-ciclobutilmetoxi)-pirazol-1-il]-nicotínico (1,9 encontrado 432,2 (M+1)+. Tiempo de retención: 4,61 mi
Ácido 2-doro-6-[3-(1-tnfluorometN-ddobutNmetoxi)-
[0300]
i)-1H-pirazol (1,95 g, 7,61 mmol) y éster terc-butílico del en dimetilformamida (15 mL) y se añadió carbonato de 2]octano (0,43 g, 3,8 mmol). La reacción se agitó a a (150 mL) y la capa acuosa se extrajo con acetato de almuera (70 mL), se secó sobre sulfato de sodio y se iante cromatografía en gel de sílice usando un método a producir éster terc-butílico de ácido 2-cloro-6-[3-(1-6%) como un sólido blanco. IEN-EM m/z calc. 431,85,
oM-N]-mcotm ico

[0301] Se disolvió éster terc-butílico del ácido 2-cloro-6-g, 4,40 mmol) en diclorometano (20 mL) y se añadió áci temperatura ambiente durante la noche, después de lo c ácido 2-cloro-6-[3-(1-trifluorometil-ciclobutilmetoxi)-piraz EM m/z calc. 375,74, encontrado 376,2 (M+1)+. Tiempo Síntesis de N-(bencenosulfoml)-2-doro-6-[ carboxamida
[0302]
trifluorometil-ciclobutilmetoxi)-pirazol-1-il]-nicotínico (1,9 luoroacético (5,0 ml). La solución de reacción se agitó a volátiles se eliminaron a presión reducida para producir ]-nicotínico (1,61 g, 97%) como un sólido blanco. IEN-tención: 3,57 minutos.
trifluorometM)ddobutM]metoxi]pirazoM-M]piridm-3-

[0303] A una solución agitada de ácido 2-cloro-6-[3-[[ (0,150 g, 0,399 mmol) en tetrahidrofurano anhidro (3,0 solución se agitó a temperatura ambiente durante 2 h. Lu en una porción, seguida de DBU (183 mg, 1,20 mmol) y l 2 h más. A la mezcla de reacción se le añadió lentam salmuera (5 ml). Después de agitar durante 10 min, el m Los extractos orgánicos combinados se lavaron con sal filtraron y se concentraron a presión reducida. Después [3-[[1-(trifluorometil)ciclobutil]metoxi]pirazol-1-il]piridin-3-Contenía algo de impureza ácida de partida y se usó en orometil)ciclobutil]metoxi]pirazol-1-il]piridin-3-carboxílico e añadió CDI (78 mg, 0,4810 mmol) en una porción. La e añadió bencenosulfonamida sólida (76 mg, 0,48 mmol) ción de color té se agitó a temperatura ambiente durante cido cítrico (2,5 ml de 1,0 M, 2500 mmol), seguido de l homogéneo se extrajo con acetato de etilo (3 x 25 ml). (10 mL), se secaron sobre sulfato de sodio anhidro, se car al vacío durante 1 h, N-(bencenosulfonil)-2-cloro-6-xamida (181 mg, 88%) se obtuvo como un sólido blanco. so posterior sin purificación adicional. IEN-EM m/z calc.
514,0689, encontrado 515,1 (M+1)+; Tiempo de retenció Síntesis de N-(bencenosulfoml)-6-[3-[[1 trimetilpirrolid ina-1-il]p iridin-3-carboxam ida
[0304]
98 minutos
orometN)cidobutN]metoxi]pirazoM-N]-2-[(4S)-2,2,4-

[0305] Una mezcla de N-(bencenosulfonil)-2-clo carboxamida (0,160 g, 0,311 mmol), (4S)-2,2,4-trimetMpi de potasio (215 mg, 1,554 mmol) se agitó en en dimetil 130°C durante 18 h. La reacción se dejó enfriar a tempe acetato de etilo (3 x 25 ml). Los extractos orgánicos com M (310 |jL de 1,0 M, 0,3107 mmol) y salmuera, se secar para dar un material crudo de color amarillo. Se purific sílice de oro de 40 g y eluyendo con metanol al 0-5% e 25 min (metanol al 2,6%). Las fracciones deseadas se adicionalmente durante la noche a alto vacío, N-(bence 2-[(4S)-2,2,4-trimetilpirrolidin-1-il]piridin-3-carboxamida 591,2127, encontrado 592,3 (M+1)+; Tiempo de retenci 1H), 8,22 (d, J = 2,8 Hz, 1H), 8,00 (dd, J = 8,2, 2,1 Hz, 2 (dt, J = 8,2, 2,0 Hz, 2H), 6,95 (d, J = 8,2 Hz, 1H), 6,18 ( - 2,22 (m, 3H), 2,11 (td, J = 12,1, 5,7 Hz, 4H), 1,95 (qd, 3H), 1,51 (s, 3H), 1,37 (t, J = 12,2 Hz, 1H), 0,65 (d, J =
Ejemplo sintético 11: Síntesis del Com (trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-
Paso A: 2-Cloro-N-(4-ciano-2-metil-fenil)sulfonil-6-[ carboxamida
[0306]
3-[[1-(trifluorometil)ciclobutil]-metoxi]pirazol-1-il]piridin-3-na (sal de clorhidrato) (139 mg, 0,932 mmol) y carbonato ido anhidro (2,7 mL) bajo una atmósfera de nitrógeno a ambiente y se diluyó con agua (15 mL) y se extrajo con os se lavaron sucesivamente con ácido cítrico acuoso 1 bre sulfato de sodio anhidro, se filtraron y se evaporaron sistema CombiFlashRf usando una columna de gel de uro de metileno (durante 45 min). El producto salió a los binaron y concentraron a presión reducida. Tras secar lfonil)-6-[3-[[1-(trifluorometil)ciclobutil]metoxi]pirazol-1-il]-de HCl, 40 mg, 20%) se obtuvo. IEN-Em m/z calc. ,25 minutos. 1H RMN (400 MHz, DMSO-da) 8 12,49 (s, ,82 (d, J = 8,2 Hz, 1H), 7,72 (tt, J = 8,2, 2,0 Hz, 1H), 7,65 2,8 Hz, 1H), 4,48 (s, 2H), 2,42 (t, J = 10,5 Hz, 1H), 2,36 ,7, 4,3 Hz, 1H), 1,83 (dd, J = 12,0, 5,6 Hz, 1H), 1,54 (s, , 3H).
to 11, N-(4-Ciano-2-metil-fenil)sulfonil-6-[3-[[1--trimetilpirrolidin-1-il]piridin-3-carboxamida
(trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-3-

[0307] Una solución de ácido 2-cloro-6-[3-[[1-(trifluoro (186,4 mg, 0,5 mmol) y 1,1'-carbonildiimidazol (97,29 m se agitó 4-ciano-2-metil-bencenosulfonamida (127,5 mg, j L, 0,60 mmol) se añadieron. Después de 16 horas, la con acetato de etilo. Los extractos combinados se sec purificó mediante cromatografía en gel de sílice con met metil-fenil)sulfonil-6-[3-[[1-(trifluorometil)ciclopropilo]met m/z calc. 539,06, encontrado 540,1 (M+1)+; Tiempo de r
Paso B: N-(4-Ciano-2-metil-fenil)sulfonil-6-[3-[[1-trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0308]
ciclopropil]metoxi] ácido pirazol-1-il]piridin-3-carboxílico 0 mmol) en THF (2,5 mL) se agitó durante 30 minutos, y mmol) y 1,8-diazabiciclo(5,4.0)undec-7-eno (DBU) (89,7 ción se diluyó con ácido cítrico acuoso 1 M y se extrajo sobre sulfato de sodio y se evaporaron. El residuo se al 0-5% en diclorometano para dar 2-cloro-N-(4-ciano-2-razol-1-il]piridin-3-carboxamida (270 mg, 100%) IEN-EM ión: 0,73 minutos.
orometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4- 
[0309] Una mezcla de 2-doro-N-(4-dano-2-metN-f il]piridin-3-carboxamida (270 mg, 0,50 mmol), (4S)-2,2,4 y se agitó carbonato de potasio (310,4 mg, 2,246 mmol) se acidificaron con ácido cítrico acuoso 1 M y se extrajer con salmuera, se secaron sobre sulfato de sodio y se gel de sílice con metanol al 0-5% en diclorometano p purificar usando un método de HPCL-EM de fase invers partícula de 5 pm) vendida por Phenomenex (pn: 00C-la fase móvil B durante 15 minutos. Fase móvil A = H2O ml/min y temperatura de la columna = 25°C (trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4 IEN-EM m/z calc. 616,21, encontrado 617,3 (M+1)+; Tie cfe) 8 12,96 (s, 1H), 8,23 - 8,18 (m, 2H), 8,03 (d, J = 1,6 1H), 6,95 (d, J = 8,3 Hz, 1H), 6,16 (d, J = 2,7 Hz, 1H), 4, (s, 1H), 2,17 (dd, J = 11,3, 5,7 Hz, 1H), 1,83 (dd, J = 1 (dt, J = 5,5, 1,6 Hz, 4H), 0,70 (d, J = 6,0 Hz, 3H).
Ejemplo sintético 12: Síntesis del Com (trifluorometN)cidopropN]metoxi]pirazoM-N]-2-[(4S)-2
Paso A: 2-Cloro-N-(2-metoxi-4-metil-fenil)sulfonil-6 3-carboxamida
[0310]
ulfonN-6-[3-[[1-(trifluorometN)ddopropN]metoxi]pirazoM-tilpirrolidina (sal de clorhidrato) (168,1 mg, 1,123 mmol) SO (1,87 mL) a 130°C durante 15 horas. Las reacciones n acetato de etilo. Los extractos combinados se lavaron raron. El residuo se purificó mediante cromatografía en ar un producto impuro. El producto impuro se volvió a ndo una columna Luna C18 (2) (75 X 30 mm, tamaño de U0-AX), y un ciclo de gradiente dual desde el 1-99% de 5 mM). Fase móvil B = CH3CN. Velocidad de flujo = 50 proporcionar N-(4-dano-2-metilfenN)sulfonN-6-[3-[[1-tilpirrolidin-1-il]piridin-3-carboxamida (150 mg, 48%) de retención: 2,06 minutos. 1H RMN (400 MHz, DMSO-1H), 7,97 (dd, J = 8,0, 1,8 Hz, 1H)), 7,88 (d, J = 8,3 Hz, 4,32 (m, 2H), 2,67 (s, 3H), 2,27 (d, J = 3,5 Hz, 1H), 2,25 ,3 Hz, 1H), 1,52 (d, J = 4,4 Hz, 6H), 1,36 (s, 1H), 1,09
to 12, N-(2-metoxi-4-metN-feml)sulfoml-6-[3-[[1-trimetMpirroNdm-1-M]piridm-3-carboxamida
-(trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-

[0311] Una solución de ácido 2-doro-6-[3-[[1-(tnfluoro mg, 0,5 mmol) y 1,1'-carbonildiimidazol (97,29 mg, 0,60 2-metoxi-4-metil-bencenosulfonamida (130,8 mg, 0,65 0,60 mmol) se añadieron. Después de 16 horas, la rea acetato de etilo. Los extractos combinados se secaron mediante cromatografía en gel de sílice con metanol al fenil)sulfonil-6-[3-[[1-(trifluorometil)ciclopropilo]metoxi]pir calc. 544,1, encontrado 545,1 (M+1)+; Tiempo de retenci
Paso B: N-(2-metoxi-4-metil-fenil)sulfonil-6-[3-[[1-trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0312]
idopropM]metoxi]pirazol-1-M]pmdin-3-carboxihco (186,4 ) en THF (2,5 mL) se agitó durante 30 minutos y se agitó l) y 1,8-diazabiciclo(5,4.0)undec-7-eno (DBU) (89,7 pL, se diluyó con ácido cítrico acuoso 1 M y se extrajo con sulfato de sodio y se evaporaron. El residuo se purificó en diclorometano para dar 2-cloro-N-(2-metoxi-4-metil-1-il]piridin-3-carboxamida (210 mg, 77%) IEN-EM m/z ,73 minutos como sólido incoloro.
orometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4- 
[0313] Una mezcla de 2-doro-N-(2-metoxi-4-metil-f il]piridin-3-carboxamida (210 mg, 0,3854 mmol), (4S)-2 mmol) y se agitó carbonato de potasio (310,4 mg, 2,24 reacción se acidificó con ácido cítrico acuoso 1 M y s lavaron con salmuera, se secaron sobre sulfato de cromatografía en gel de sílice con metanol al 0-5% en di se volvió a purificar usando un método de HPCL-EM de f tamaño de partícula de 5 pm) vendida por Phenomenex 1-99% de la fase móvil B durante 15 minutos. Fase móv flujo = 50 ml/min y temperatura de la columna = 25° (trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4 IEN-EM m/z calc. 621,2, encontrado 622,3 (M+1)+; Tiem 8 12,39 (s, 1H), 8,20 (d, J = 2,8 Hz, 1H), 7,78 (t, J = 8,5 2H), 6,15 (d, J = 2,7 Hz, 1H), 4,43 - 4,30 (m, 2H), 3,89 6.1 Hz, 1H), 1,85 (dd, J = 11,9, 5,5 Hz, 1H), 1,53 (d, J = 6.2 Hz, 3H).
Ejemplo sintético 13: Síntesis del C (trifluorometM)ddopropM]metoxi]pirazoM-M]-2-[(4S)-2
Paso A: 2-Cloro-N-(2,4-dimetoxifeml)sulfoml-6-[3 carboxamida
[0314]
ulfonil-6-[3-[[1-(tnfluorometil)ddopropil]metoxi]pirazol-1-rimetilpirroMdina (sal de clorhidrato) (168,1 mg, 1,123 ol) en DMSO (1,87 mL) a 130°C durante 15 horas. La ajo con acetato de etilo. Los extractos combinados se y se evaporaron. El residuo se purificó mediante etano para dar un producto impuro. El producto impuro nversa usando una columna Luna C18 (2) (75 X 30 mm, 00C-4252-U0-AX), y un ciclo de gradiente dual desde el H2O (HCl 5 mM). Fase móvil B = CH3CN. Velocidad de a proporcionar N-(2-metoxi-4-metilfenil)sulfonil-6-[3-[[1-tilpirrolidin-1-il]piridin-3-carboxamida (95 mg, 39,25%) retención: 2,19 minutos. 1H RMN (400 MHz, DMSO-cfe) H), 7,10 (d, J = 1,4 Hz, 1H), 6,94 (dd, J = 10,1, 8,1 Hz, ), 2,49 - 2,38 (m, 2H), 2,37 (s, 3H), 2,21 (dd, J = 11,2, Hz, 6H), 1,37 (s, 1H), 1,12 - 1,04 (m, 4H), 0,78 (d, J =
esto 13: N-(2,4-dimetoxifeml)sulfoml-6-[3-[[1-rimetMpirroMdm-1-M]piridm-3-carboxamida
rifluorometM)ddopropM]metoxi]pirazoM-M]piridm-3-

[0315] Una solución de ácido 2-cloro-6-[3-[[1-(trifluorom mg, 0,5 mmol) y 1,1'-carbonildiimidazol (97,29 mg, 0,60 dimetoxibencenosulfonamida (141,2 mg, 0,65 mmol) y ml, 0,60 mmol). Después de 16 horas, la reacción se dil etilo. Los extractos combinados se secaron sobre sulfat cromatografía en gel de sílice con metanol al 0-5% en [3-[[1-(trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-3 encontrado 561,1 (M+1)+; Tiempo de retención: 0,71 mi
Paso B: N-(2,4-Dimetoxifenil)sulfonil-6-[3-[[1-( trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0316]
iclopropil]metoxi]pirazol-1-il]piridina-3-carboxílico (186,4 l) en THF (2,5 mL) se agitó durante 30 minutos, y 2,4-adieron 1,8-diazabiciclo(5,4.0)undec-7-eno (DBU) (89,7 on ácido cítrico acuoso 1 M y se extrajo con acetato de sodio y se evaporaron. El residuo se purificó mediante metano para dar 2-cloro-N-(2,4-dimetoxifenil)sulfonil-6-oxamida (210 mg, 75%) IEN-EM m/z calc. 560,1, como un sólido incoloro.
orometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4- 
[0317] Una mezcla de 2-doro-N-(2,4-dimetoxifenil)sulfo 3-carboxamida (210 mg, 0,3744 mmol), (4S)-2,2,4-trim carbonato de potasio (310,4 mg, 2,246 mmol) en DMSO se acidificaron con ácido cítrico acuoso 1 M y se extraje con salmuera, se secaron sobre sulfato de sodio y se gel de sílice con metanol al 0-5% en diclorometano p purificar usando un método de HPCL-EM de fase invers partícula de 5 pm) vendida por Phenomenex (pn: 00C-la fase móvil B durante 15 minutos. Fase móvil A = H2 ml/min y temperatura de la columna = 25° (trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2, IEN-EM m/z calc. 637,2, encontrado 638,3 (M+1)+; Tiem 8 12,34 (s, 1H), 8,20 (d, J = 2,8 Hz, 1H), 7,82 (d, J = 8 6,74 (d, J = 2,3 Hz, 1H), 6,70 (dd, J = 8,8, 2,3 Hz, 1H), 6 (s, 3H), 2,54 (s, 1H), 2,42 (dd, J = 10,5, 7,0 Hz, 1H), 2, 1,55 (s, 3H), 1,52 (s, 3H), 1,38 (s, 1H), 1,09 (dt, J = 5,9,
Ejemplo sintético 14: Síntesis del (trifluorometM)cidopropM]metoxi]pirazoM-M]-2-[(4S)-
[0318]
3-[[1-(trifluorometil)cidopropilo]metoxi]pirazol-1-il]piridinroMdina (sal de clorhidrato) (168,1 mg, 1,123 mmol) y mL) se agitó a 130°C durante 15 horas. Las reacciones n acetato de etilo. Los extractos combinados se lavaron raron. El residuo se purificó mediante cromatografía en ar un producto impuro. El producto impuro se volvió a ndo una columna Luna C18 (2) (75 X 30 mm, tamaño de U0-AX), y un ciclo de gradiente dual desde el 1-99% de 5 mM). Fase móvil B = CH3CN. Velocidad de flujo = 50 ra proporcionar N-(2,4-dimetoxifenil)sulfonN-6-[3-[[1-tilpirrolidin-1-il]piridin-3-carboxamida (110 mg, 46%) retención: 2,14 minutos. 1H RMN (400 MHz, DMSO-cfe) , 1H), 7,77 (d, J = 8,3 Hz, 1H), 6,92 (d, J = 8,2 Hz, 1H), , J = 2,7 Hz, 1H), 4,43 - 4,31 (m, 2H), 3,90 (s, 3H), 3,85 , J = 11,6, 5,9 Hz, 1H), 1,85 (dd, J = 11,9, 5,5 Hz, 1H), z, 4H), 0,80 (d, J = 6,3 Hz, 3H).
ompuesto 14: N-(bencenosulfonil)-6-[3-[[1-trimetMpirroMdm-1-M]piridm-3-carboxamida

Paso A: 3-[[1-(trifluorom etil)ciclopropil]metoxi]piraz [0319]
arboxilato de tere-butilo

[0320] Un matraz de fondo redondo de 3 bocas de calefactora, una sonda/controlador de temperatura J-Ke por agua y una entrada/salida de nitrógeno. El recipiente 2-carboxilato de terc-butilo (70 g, 0,3800 mol) y tetrah transparente de color amarillo pálido. Se inició la agitació el recipiente se cargó con [1-(trifluorometil)ciclopropil]m seguido de trifenilfosfina (109,6 g, 0,4180 mol) añadida color amarillo pálido resultante se trató luego con azodica rojizo) (82,3 ml, 0,4180 mol) añadido gota a gota puro dur a 40°C y una solución transparente de color ámbar claro. del recipiente de 50°C y la condición se mantuvo duran completo del material de partida. La mezcla de reacció aceite de color ámbar oscuro claro resultante se suspe durante 1 hora, tiempo durante el cual precipitó un sólido se filtró a través de un embudo Buchner de frita de vidrio (150 mL) y luego se extrajo durante 30 minutos. El filtra proporcionar un aceite de color ámbar claro. El material de sílice (carga sólida en una columna RediSep de 1,5 k EtOAc al 20% en hexano recogiendo fracciones de 450 hexano. Las fracciones deseadas se combinaron y con color amarillo pálido transparente como producto desea de terc-butilo (81 g, 0,264 mol, 70%). 1H RMN (400 MH 1H), 4,31 (s, 2H), 1,55 (s, 9H), 1,07 (dp, J = 4,9, 1,3 Hz, Tiempo de retención: 1,76 minutos
Paso B: 3-[[1-(Trifluorometil)ciclopropil]metoxi]-1H-p
[0321]
mL equipado con un agitador mecánico, una manta embudo de adición, un condensador de reflujo enfriado rgó en una atmósfera de nitrógeno con 5-oxo-1H-pirazolrano (840 ml, 12 ml/g) que proporcionó una solución registró la temperatura del recipiente a 19°C. Después, l (58,56 g, 0,4180 mol) añadido puro en una porción un sólido en una porción. La solución transparente de lato de diisopropilo (líquido transparente de color naranja 1 hora, lo que dio como resultado una exotermia gradual ués, la mezcla de reacción se calentó a una temperatura horas cuando el análisis por CL/EM indicó el consumo color ámbar claro se concentró a presión reducida y el n tolueno (560 mL) y se agitó a temperatura ambiente o de trifenilfosfina PM = 278,28). La suspensión espesa orta de filtración se lavó por desplazamiento con tolueno color ámbar claro se concentró a presión reducida para urificó mediante cromatografía flash en columna de gel Celite) eluyendo con un gradiente de hexano al 100% a l producto eluye aproximadamente el 5% de EtOAc en ron a presión reducida para proporcionar un aceite de -[[1-(trifluorometil)ciclopropil]metoxi]pirazol-1-carboxilato SO-d6) 8 8,10 (d, J = 2,9 Hz, 1H), 6,14 (d, J = 3,0 Hz, EN-EM m/z calc. 306,11914, encontrado 259,0 (M-48)+;
l

[0322] Se equipó un matraz de fondo redondo de 3 calefactora, una sonda de temperatura J-Kem, un conde una entrada/salida de nitrógeno. El recipiente (trifluorometil)ciclopropil]metoxi]pirazol-1-carboxilato de t y metil alcohol (320 mL, 4 mL/g) que proporcionó una agitación y se registró la temperatura del recipiente a 19° (195,9 ml, 0,7836 mol) que posteriormente se añadió exotermia gradual a 30°C. La solución de color amarillo del recipiente de 45°C y la condición se mantuvo durant se había completado. Se dejó enfriar la mezcla de reac reducida. El residuo restante se disolvió en éter metílico decantación y se repartió con una solución de hidróxid orgánica y se extrajo la fase acuosa residual con éter m se lavó con una solución saturada de cloruro de sodio (5 a través de un embudo Buchner de frita de vidrio. El f presión reducida para proporcionar un aceite de colo proporcionar un sólido blanco (49,5 g, 0,24 (trifluorometil)ciclopropilo]metoxi]-1H-pirazol. 1H RMN (4 s de 5000 mL con un agitador mecánico, una manta or de reflujo enfriado por agua, un embudo de adición y cargó en atmósfera de nitrógeno con 3-[[1-utilo (80 g, 0,2612 mol), diclorometano (320 ml, 4 mL/g) ción transparente de color amarillo pálido. Se inició la embudo de adición se cargó con 4 M HCl en 1,4-dioxano a gota durante 1 hora, lo que dio como resultado una o transparente resultante se calentó a una temperatura ora cuando el análisis por CL/EM indicó que la reacción temperatura ambiente y luego se concentró a presión rc-butilo (640 mL) y luego se transfirió a un embudo de sodio 2 M (391,8 ml, 0,7836 mol). Se eliminó la capa de terc-butilo (2 X 200 ml). La fase orgánica combinada ), se secó sobre sulfato de sodio (300 g) y luego se filtró de color amarillo pálido transparente se concentró a rillo claro transparente que solidificó al reposar para ol, 92%) como el producto deseado 3-[[1-Hz, DMSO-d6) 811,90 (s, 1H), 7,51 (d, J = 2,4 Hz, 1H),
5,67 (d, J = 2,4 Hz, 1H), 4,19 (s, 2H), 1,09 - 0,97 (m, Tiempo de retención: 1,07 minutos.
Paso C: 2-doro-6-[3-[[1-(trifluorometM)ddopropM]me [0323]
IEN-EM m/z calc. 206,0667, encontrado 207,0 (M+1)+;
irazoM-M]piridm-3-carboxMato de terc-butilo

[0324] Un matraz de fondo redondo de 3 bocas de 500 enfriamiento usado como contención secundaria, una enfriado por agua, un embudo de adición y una entrada de nitrógeno con 3-[[1-(trifluorometil)ciclopropil]metoxi]-1 12 mL/g) que proporcionó una solución transparente d temperatura del recipiente a 17°C. Después, el recipien (54,16 g, 0,2183 mol) añadido como un sólido en un resultante se trató luego con carbonato de potasio (39 seguido de 1,4-diazabiciclo[2.2.2]octano (3,67 g, 0,0327 de color amarillo pálido resultante se dejó agitar a temp enfrió a 10°C con un baño de enfriamiento de agua/hielo añadida gota a gota durante 45 minutos, lo que dio com La suspensión resultante se continuó agitando a 15°C Buchner de frita de vidrio. La torta de filtración se lavó p en el Buchner durante 2 horas. Después, el material se 0,175 mol, 80%) de un sólido granular blanco como terc 1 -il]piridin-3-carboxilato. IEN-EM m/z calc. 361,0441, en
Paso D: Ácido 2-doro-6-[3-[[1-(trifluorom etil)cidopro
[0325]
estaba equipado con un agitador mecánico, un baño de a de temperatura J-Kem, un condensador de reflujo a de nitrógeno. El recipiente se cargó en una atmósfera azol (45 g, 0,2183 mol) y N,N-dimetilformamida (540 ml, r amarillo pálido. Se inició la agitación y se registró la cargó con 2,6-dicloropiridin-3-carboxilato de terc-butilo ión. La solución de color amarillo pálido transparente , 0,2838 mol) añadido como un sólido en una porción ) añadido como un sólido en una porción. La suspensión a ambiente durante 24 horas. La mezcla de reacción se ado. El embudo de adición se cargó con agua (540 mL) ultado una suspensión espesa y una exotermia a 15°C. te 30 minutos y luego se filtró a través de un embudo plazamiento con agua (2 X 500 mL) y luego se introdujo secar al aire durante la noche para proporcionar (73 g, 2-cloro-6-[3-[[1-(trifluorometil)ciclopropil]metoxi]pirazoldo 361,9 (M+1)+; Tiempo de retención: 2,27 minutos.
etoxi]p irazol-1-il]p irid in-3-carboxílico
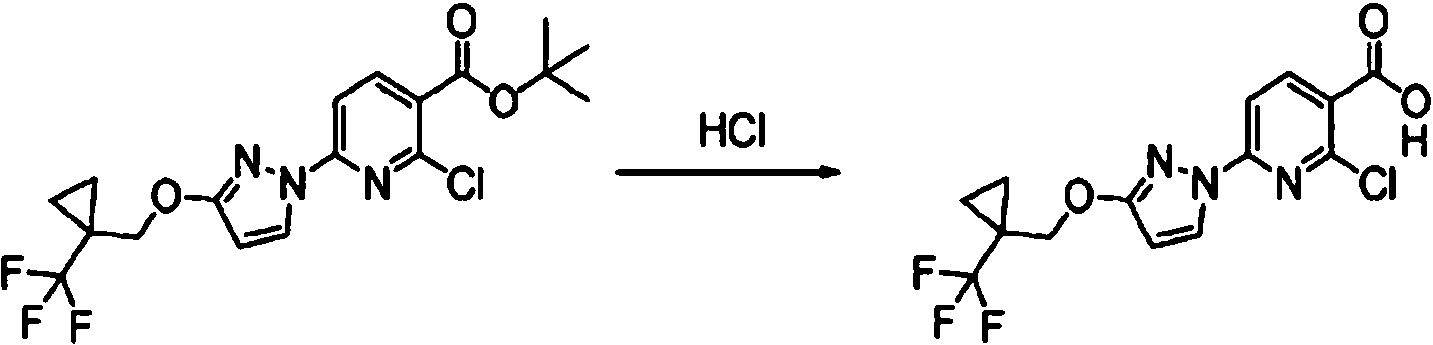
[0326] Un matraz de fondo redondo de 3 bocas de 1000 una sonda/controlador de temperatura J-Kem, un embu una entrada/salida de nitrógeno. El recipiente se c (trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-3-carb que proporcionó una suspensión blanquecina. Se inició l El embudo de adición se cargó con HCl acuoso 6 M ( minutos, lo que dio como resultado una exotermia a (temperatura del recipiente ~82°C). Tras calentar la s transparente (temperatura del recipiente ~75°C en est comenzó a precipitar un sólido. La suspensión se conti donde se añadió gota a gota agua (210 mL) durante 15 m la suspensión y se dejó enfriar lentamente a temperatur en un embudo Buchner de frita de vidrio y la torta de filtr (100 mL) seguido de agua (2 x 100 mL) y luego se intr adicionalmente en un horno de vacío a 45°C du (trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-3-carb (400 MHz, DMSO-d6) 813,64 (s, 1H), 8,44 (d, J = 2,9 H (d, J = 2,9 Hz, 1H), 4,41 (s, 2H), 1,16 - 1,07 (m, 4H). IE de retención: 3,23 minutos
uipado con un agitador mecánico, una manta calefactora, adición, un condensador de reflujo enfriado por agua y en una atmósfera de nitrógeno con 2-cloro-6-[3-[[1-de terc-butilo (70 g, 0,1675 mol) y 2-propanol (350 mL) tación y se registró la temperatura del recipiente a 19°C. ml, 0,8375 mol) que se añadió gota a gota durante 10 . La suspensión resultante se calentó luego a reflujo sión, se volvió una solución de color amarillo pálido to). Después de agitar a reflujo durante ~30 minutos, gitando a reflujo durante 30 minutos más, momento en . A continuación, se retiró el calor y se continuó agitando iente. El material se recogió mediante filtración al vacío se lavó por desplazamiento con 1:1 de agua/2-propanol en el Buchner durante 30 minutos. El material se secó 24 horas para proporcionar ácido 2-cloro-6-[3-[[1-. (56 g, 0,155 mol, 92%) como un sólido blanco. 1H r Mn , 8,41 (d, J = 8,4 Hz, 1H), 7,74 (d, J = 8,4 Hz, 1H), 6,24 m/z calc. 361,0441, encontrado 361,9 (M+1)+; Tiempo
Paso E: N-(bencenosulfoml)-2-doro-6-[3-carboxamida
[0327]
rifluorometM)ddopropM]metoxi]pirazoM-M]piridm-3-

[0328] Se disolvió acido 2-cloro-6-[3-[[1-(tnfluorome 0,4144 mmol) en THF (2.000 ml). Se añadió CDI (aprox se agitó a temperatura ambiente durante 1,5 horas. S 0,5387 mmol) seguida de DBU (aproximadamente 126 reacción a temperatura ambiente durante otras 1,5 hora se diluyó con diclorometano y se inyectó directamente un gradiente de metanol al 0-10% en diclorometano; pro deseado se combinaron y concentraro (trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-3-carb petróleo. IEN-EM m/z calc. 500,05328, encontrado 501, minutos).
Paso F: N-(bencenosulfoml)-6-[3-[[1-( trim etilpirrolid in-1-ilo]piridin-3-carboxam ida
[0329]
lopropil]metoxiprazol-1-ilpridin-3-carboxílico (150 mg, amente 80,64 mg, 0,4973 mmol). La mezcla de reacción dió bencenosulfonamida (aproximadamente 84,68 mg, g, 124,0 pL, 0,8288 mmol). Se dejó agitar la mezcla de mezcla de reacción se concentró a la mitad del volumen, a columna de gel de sílice de 12 gramos y se sometió a eluido al 10%. Las fracciones que contenían el producto Se obtuvo N-(bencenosulfonil)-2-cloro-6-[3-[[1-ida (168 mg, 81%) como un incoloro transparente 1)+; Tiempo de retención: 1,92 minutos (ejecución de 3
orometN)ddopropN]metoxi]pirazoM-N]-2-[(4S)-2,2,4-

[0330] N-(bencenosulfoml)-2-cloro-6-[3-[[1-(tnfluorome 0,3354 mmol) y (4S)-2,2,4-trimetilpirrolidina (sal de combinaron y se disolvieron en DMSo (0,5 ml). Se aña 278,1 mg, 2,012 mmol) y se dejó agitar la mezcla de r diluyó con EtOAc (50 mL) y se lavó con acido cítrico acu se secó sobre sulfato de sodio, se filtró y se concentr cromatografía en columna de gel de sílice: columna de el producto eluyó al 2,5%. Las fracciones puras se c azeotrópicamente con MeOH, para proporcionar N-(be 1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-il]piridin-3-carboxam 1H), 8,20 (d, J = 2,8 Hz, 1H), 8,05 - 7,95 (m, 2H), 7,82 Hz, 2H), 6,92 (d, J = 8,3 Hz, 1H), 6,15 (d, J = 2,7 Hz, 1 Hz, 1H), 2,09 (dt, J = 12,3, 6,4 Hz, 1H), 1,82 (dd, J = 12 1H), 1,15 - 1,04 (m, 4H), 0,64 (d, J = 6,2 Hz, 3H). IEN-retención: 2,16 minutos (ejecución de 3 minutos).
Ejemplo sintético 15: Síntesis del
tetrametilciclopropil)metoxi]pirazol-1-il]-2-[(4S)-2,2,
[0331]
lopropil]metoxiprazol-1-ilpridin-3-carboxamida (168 mg, idrato) (aproximadamente 150,6 mg, 1,006 mmol) se rbonato de potasio finamente molido (aproximadamente n a 130°C durante la noche. La mezcla de reacción se 1 M (2X 50 mL) y salmuera (1X 50 mL). La capa orgánica resión reducida. El producto crudo se purificó mediante sílice de 24 gramos, gradiente de MeOH/DCM al 0-5%; naron y concentraron a presión reducida, y se destiló osulfonil)-6-[3-[[1-(trifluorometil)ciclopropil]metoxi]pirazol-4,9 mg, 39%). 1H RMN (400 MHz, DMSO-cfe) 812,51 (s, = 8,2 Hz, 1H), 7,78 - 7,70 (m, 1H), 7,66 (dd, J = 8,3, 6,7 3 -4,30 (m, 2H), 2,40 (t, J = 10,5 Hz, 1H), 2,26 (t, J = 8,6 6 Hz, 1H), 1,53 (s, 3H), 1,51 (s, 3H), 1,36 (t, J = 12,1 Hz, /z calc. 577,1971, encontrado 578,3 (M+1)+; Tiempo de
mpuesto 15: N-(o-Tolilsulfonil)-6-[3-[(2,2,3,3-etilpirrolidin-1-il]piridin-3-carboxamida

Paso A: 3-[(2,2,3,3-tetrametilciclopropil)metoxi]pirazo [0332]
arboxilato de terc-butilo

[0333] A una solución desgasificada de Ph3P (aproxima nitrógeno a DIAD 0°C se añadió (diisopropilazodicarboxil a gota. La mezcla se agitó a 0°C durante 30 min proporci solución de (2,2,3,3-tetrame tilciclopropil)metanol (apr hidroxipirazol-1-carboxilato de terc-butilo (30 g, 162,9 mm La mezcla se dejó calentar a temperatura ambiente y se un total de 6 horas y luego se dejó enfriar a temperatura se agitó a temperatura ambiente durante 3 horas. La sus con 100 mL de heptano. El filtrado se concentró al vacío se cromatografió en una columna de gel de sílice de 75 gradiente de EtOAc/hexanos al 0-20%. Las fracciones r proporcionando un sólido blanquecino. Se obtuvo 3-[(2,2 butilo (30,1 g, 63%). 1H RMN (400 MHz, cloroformo-d) 8 7,7 Hz, 2H), 1,61 (s, 9H), 1,12 (s, 6H), 1,04 (s, 6H), 0,70 295,0 (M+1)+; Tiempo de retención: 2,19 minutos
Paso B: 3-[(2,2,3,3-tetrametilciclopropil)metoxi]-1H-pi
[0334]
nte 51,28 g, 195,5 mmol) en tolueno (360,0 mL) en gas aproximadamente 39,53 g, 37,86 mL, 195,5 mmol) gota do una suspensión blanca. A la mezcla se le añadió una damente 29,84 g de 70% p/p, 162,9 mmol) y 3-tolueno (600,0 mL) gota a gota a ~5°C durante 2 horas. durante 18 horas. La mezcla se calentó a 75°C durante nte. La suspensión se diluyó con heptano (900,0 mL) y ión se filtró sobre celite y el precipitado se lavó 3 veces rcionando un aceite amarillo espeso. El producto crudo mos cargándose con diclorometano y eluyendo con un as que contenían el producto se concentraron al vacío tetrametilciclopropil)metoxi]pirazol-1-carboxilato de terc-(d, J = 3,0 Hz, 1H), 5,88 (d, J = 2,9 Hz, 1H), 4,30 (d, J = = 7,8 Hz, 1H). IEN-EM m/z calc. 294,19434, encontrado
l

[0335] A una solución de 3-[(2,2,3,3-tetrametilciclopro mmol) en THF (317,5 mL) y alcohol etílico (635,0 mL) 431,4 ml de 2 M, 862,8 mmol) y se agitó a temperatura eliminó a presión reducida. El residuo acuoso se diluyó (762,0 ml). La fase orgánica se lavó dos veces con salm con éter t-butílico de metilo (250 ml). Las fases orgánica [(2,2,3,3-tetrametilciclopropil)metoxi]-1H-pirazol (75 g, 89 11,78 (s, 1H), 7,48 (t, J = 2,1 Hz, 1H), 5,65 (s, 1H), 4,05 Hz, 1H). IEN-EM m/z calc. 194,1419, encontrado 195,0 (
Paso C: 2-Cloro-6-[3-[(2,2,3,3-tetrametilciclopropil)m etoxi]pirazol-1-carboxilato de terc-butilo (127 g, 431,4 adió lentamente hidróxido de sodio (aproximadamente nte durante la noche. La mayor parte del disolvente se agua (400 mL) y se extrajo con éter t-butílico de metilo (2 X 300 mL) y las fases acuosas se extrajeron una vez binadas se secaron, filtraron y evaporaron para dar 3-omo un aceite viscoso. 1H Rm N (400 MHz, DMSO-cfe) 8 = 7,7 Hz, 2H), 1,08 (s, 6H), 1,00 (s, 6H), 0,67 (t, J = 7,7 ; Tiempo de retención: 1,43 minutos.
]pirazol-1-il]piridin-3-carboxilato de etilo
[0336]

[0337] Al 2,6-dicloropiridina-3-carboxilato de etilo (16,8 pirazol (aproximadamente 14,83 g, 76,35 mmol) (aproximadamente 13,72 g, 99,26 mmol) seguido de DA se agitó a temperatura ambiente durante 16 horas. La s (201,6 mL) y la suspensión espesa resultante se agitó superior. El precipitado se recogió usando una frita med al aire durante 30 minutos y luego se secó al vacío usa tetrametilciclopropil)metoxi]pirazol-1-il]piridin-3-carboxila EM m/z calc. 377,1506, encontrado 378,37 (M+1)+; Tiem 8 8,43 (dd, J = 2,9, 0,9 Hz, 1H), 8,39 (dd, J = 8,5, 0,9 Hz 1H), 4,34 (td, J = 7,5, 6,6 Hz, 2H), 4,28 (d, J = 7,8 Hz, 0,75 (t, J = 7,8 Hz, 1H).
Paso D: Ácido 2-doro-6-[3-[(2,2,3,3-tetrametNcidopr
[0338]
,35 mmol) y 3-[(2,2,3,3-tetrametilciclopropil)metoxi]-1H-MF (201,6 mL) se añadió carbonato de potasio (aproximadamente 1,284 g, 11,45 mmol). La suspensión sión fina de color crema se diluyó lentamente con agua peratura ambiente durante 30 minutos con un agitador y se lavó 3 veces con 25 mL de agua. El sólido se secó n azeótropo de EtOAc. Se obtuvo 2-cloro-6-[3-[(2,2,3,3-etilo (28,8 g, 100%) como un sólido blanquecino. IEN-retención: 2,47 minutos. 1H RMN (400 MHz, DMSO-cfe) 7,76 (dd, J = 8,5, 0,9 Hz, 1H), 6,24 (dd, J = 2,9, 0,9 Hz, 1,34 (td, J = 7,1, 0,9 Hz, 3H), 1,11 (s, 6H), 1,05 (s, 6H),
etoxi]pirazoM-M]piridm-3-carboxnico

[0339] 2-Cloro-6-[3-[(2,2,3,3-tetrametilciclopropil)metoxi] en THF (730,0 mL) y EtOH (292,0 mL) se se trató con solución se agitó a temperatura ambiente durante 5 hora y la solución se acidificó mediante la adición de ácido enfriando con hielo. La suspensión espesa formada (pH lavó con abundante agua y se secó en un armario de se días para dar ácido 2-cloro-6-[3-[(2,2,3,3-tetrametilciclo como un sólido blanquecino. Ie N-EM m/z calc. 349,1 minutos. 1H RMN (400 MHz, DMSO-d6) 813,64 (s, 1H), Hz, 1H), 4,28 (d, J = 7,8 Hz, 2H), 1,08 (d, J = 24,9 Hz, 1
Paso E: 2-Cloro-N-(o-toMlsulfoml)-6-[3-[(2,2,3,3-tetra
[0340]
ol-1-il]piridin-3-carboxilato de etilo (146 g, 386,4 mmol) (aproximadamente 772,8 ml de 1 M, 772,8 mmol) y la mayor parte del disolvente se eliminó a presión reducida co (aproximadamente 148,5 g, 89,19 ml, 772,8 mmol) se agitó en el baño de hielo durante 1 hora, se filtró, se al vacío a 45°C con una purga de nitrógeno durante dos il)metoxi]pirazol-1-il]piridin-3-carboxílico (128,2 g, 90%) encontrado 350,0 (M+1)+; Tiempo de retención: 2,11 - 8,22 (m, 2H), 7,73 (d, J = 8,4 Hz, 1H), 6,22 (d, J = 2,9 0,75 (t, J = 7,8 Hz, 1H).
cidopropM)metoxi]pirazoM-M]piridm-3-carboxamida

[0341] Ácido 2-cloro-6-[3-[(2,2,3,3-tetrametilciclopropil) se disolvió/suspendió en THF (2 mL) y se añadió c suspensión a temperatura ambiente durante 1,5 horas. mg, 0,429 mmol) seguido de DBU (59,2 ml, 0,396 mmol) 1,5 horas. Se evaporaron los volátiles. El residuo resta cítrico acuoso 1 M (132 ml). La capa orgánica se inyectó i]pirazol-1-il]piridin-3-carboxílico (150 mg 0,429 mmol) y ildiimidazol (64,2 mg, 0,396 mmol). Se dejó agitar la ntinuación, se añadió 2-metilbencenosulfonamida (73,4 ontinuación, la solución resultante se agitó durante otras e recogió en diclorometano (2 mL) y se lavó con ácido na columna de gel de sílice para cromatografía: columna
de gel de sílice de 12 gramos, gradiente de Me tetrametilcidopropil)metoxi]pirazol-1-il]piridin-3-carboxam 502,14417, encontrado 503,0 (M+1)+; Tiempo de retenci
Paso F: N-(o-ToMlsulfoml)-6-[3-[(2,2,3,3-tetrametMdd 1-il]piridin-3-carboxamida
[0342]
CM al 0-10%. 2-cloro-N-(o-tolilsulfonil)-6-[3-[(2,2,3,3-(115 mg, 53%) fue adquirido. IEN-EM m/z calc. ,25 minutos.
M)metoxi]pirazoM-M]-2-[(4S)-2,2,4-trimetMpirroMdm-
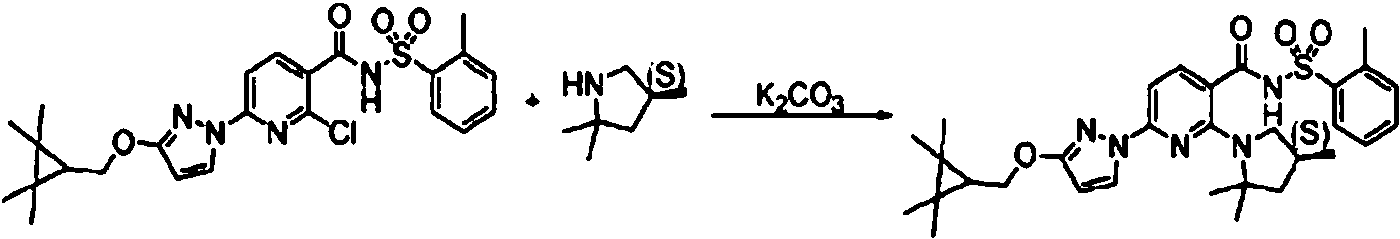
[0343] 2-Cloro-N-metilsulfonil-6-[3-[(2,2,3,3-tetrametilcicl 0,229 mmol) y (4S)-2,2,4-trimetilpirrolidina (sal de clorhi en DMSO (1 ml). Se añadió carbonato de potasio finam selló y se calentó durante la noche a 130°C. Después diluyó con EtOAc (50 mL) y se lavó con ácido cítrico acuo se secó sobre sulfato de sodio, se filtró y se concentró a en columna de gel de sílice eluyendo con un gradiente d gramos. N-(o-tolilsulfonil)-6-[3-[(2,2,3,3-tetrametil il]piridin-3-carboxamida (57,2 mg, 42%) se obtuvo. IEN-retención: 2,52 minutos. 1H RMN (400 MHz, DMSO-da) Hz, 1H), 7,79 (d, J = 8,2 Hz, 1H), 7,59 (td, J = 7,5, 1,5 H = 2,7 Hz, 1H), 4,24 (d, J = 7,8 Hz, 2H), 2,63 (s, 3H), 2,3 11,9, 5,5 Hz, 1H), 1,52 (d, J = 1,6 Hz, 6H), 1,35 (t, J = 1 (m, 4H).
Ejemplo sintético 16: Síntesis del Co tetrametilciclopropil)metoxi]pirazol-1-il]-2-[(4S)-2,2,4
Paso A: 2-Cloro-N-(3-fluorofeml)sulfoml-6-[3-[(2 carboxamida
[0344]
pil)metoxi]pirazol-1-ilo]piridin-3-carboxamida (115 mg, ) (106 mg, 0,935 mmol) se combinaron y se disolvieron molido (258 mg, 1,87 mmol). La mezcla de reacción se friar a temperatura ambiente, la mezcla de reacción se M, 2X 50 mL) y salmuera (1X 50 mL). La capa orgánica n reducida. El producto se aisló mediante cromatografía OH/DCM al 0-5% en una columna de gel de sílice de 12 ropil)metoxi]pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-/z calc. 579,2879, encontrado 580,3 (M+1)+; Tiempo de 2 (s, 1H), 8,18 (d, J = 2,8 Hz, 1H), 8,04 (dd, J = 8,0, 1,4 ), 7,50 - 7,40 (m, 2H), 6,93 (d, J = 8,2 Hz, 1H), 6,13 (d, J J = 8,8 Hz, 2H), 2,16 (d, J = 10,3 Hz, 1H), 1,82 (dd, J = z, 1H), 1,10 (s, 6H), 1,04 (d, J = 1,1 Hz, 6H), 0,77 - 0,67
esto 16: N-(3-fluorofeml)sulfoml-6-[3-[(2,2,3,3-etilpirrolidin-1-il]piridin-3-carboxamida
3-tetrametMddopropMo)metoxi]pirazoM-M]piridm-3-

[0345] Ácido 2-cloro-6-[3-[(2,2,3,3-tetrametilciclopropil)m se disolvió/suspendió en THF (2 mL) y se añadió car suspensión a temperatura ambiente durante 1,5 horas. L mmol) seguido de DBU (59,2 pL, 0,396 mmol). Despué evaporaron los volátiles. El residuo restante se recogió M (1X2 ml). La capa orgánica se inyectó en una colum columna de gel de sílice de 12 gramos, gradiente de [(2,2,3,3-tetrametilciclopropil)metoxi]pirazol-1-il]piridin-3-506,11908, encontrado 507,0 (M+1)+; Tiempo de retenci
Paso B: N-(3-fluorofenil)sulfonil-6-[3-[(2,2, trim etilpirrolid in-1-il]pirid in-3-carboxam ida
[0346]
i]pirazol-1-il]piridina-3-carboxílico (150 mg, 0,429 mmol) l diimidazol (64,2 mg, 0,396 mmol). Se dejó agitar la se añadió 3-fluorobencenosulfonamida (75,1 mg, 0,429 solución resultante se agitó durante otras 1,5 horas. Se lorometano (2 mL) y se lavó con ácido cítrico acuoso 1 e gel de sílice para purificarla mediante cromatografía: H/DCM al 0-10%. 2-cloro-N-(3-fluorofenil)sulfonil-6-[3-xamida (150 mg, 70%) fue obtenido. IEN-EM m/z calc. ,24 minutos
etrametilcidopropil)metoxi]pirazol-1-il]-2-[(4S)-2,2,4- 
[0347] 2-Cloro-N-(3-fluorofenil)sulfonil-6-[3-[(2,2,3,3-tetra (158 mg, 0,312 mmol) y (4S)-2,2,4-trimetilpirroMdina (sa disolvieron en DMSO (1 mL). Se añadió carbonato de p reacción se selló y se calentó durante la noche a 130°C reacción se diluyó con EtOAc (50 mL) y se lavó con ácid capa orgánica se secó sobre sulfato de sodio, se filtró y s cromatografía en columna eluyendo con un gradiente de gramos. N-(3-fluorofenil)sulfonil-6-[3-[(2,2,3,3-tetrametilc il]piridin-3-carboxamida (28,4 mg, 16%) se obtuvo. IEN-E retención: 2,46 minutos. 1H RMN (400 MHz, DMSO-da) 7,79 - 7,71 (m, 2H), 7,63 (tdd, J = 8,6, 2,6, 1,1 Hz, 1H), 6 7,7 Hz, 2H), 2,44 (t, J = 10,4 Hz, 1H), 2,36 - 2,26 (m, 1H 1H), 1,54 (s, 3H), 1,52 (s, 3H), 1,39 (t, J = 12,1 Hz, 1H), 6,6 Hz, 3H).
Ejemplo sintético 17: Síntesis del Compuesto 17: (trideuteriometM)pirroMdm-1-M]-6-[3-[2-[1-(trifluorome
[0348]
lcidopropil)metoxi]pirazol-1-il]piridin-3-carboxamida
clorhidrato) (105,9 mg, 0,935 mmol) se combinaron y o finamente molido (258 mg, 1,87 mmol). La mezcla de spués de enfriar a temperatura ambiente, la mezcla de rico acuoso (1 M, 2X 50 mL) y salmuera (1X 50 mL). La centró a presión reducida. El producto se aisló mediante H/DCM al 0-5% en una columna de gel de sílice de 12 ropil)metoxi]pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1 -/z calc. 583,2629, encontrado 584,6 (M+1)+; Tiempo de 61 (s, 1H), 8,19 (d, J = 2,8 Hz, 1H), 7,87 - 7,81 (m, 2H), (d, J = 8,2 Hz, 1H), 6,13 (d, J = 2,8 Hz, 1H), 4,24 (d, J = 13 (td, J = 11,8, 6,0 Hz, 1H), 1,84 (dd, J = 11,8, 5,5 Hz, (s, 6H), 1,04 (s, 6H), 0,74 (d, J = 7,7 Hz, 1H), 0,70 (t, J =
bencenosulfoml)-2-[(4S)-3,3-dideuterio-2,2-dimetN-4-dopropM]etoxi]pirazoM-M]piridm-3-carboxamida

[0349] Se disolvió N-(bencenosulfonil)-2-cloro carboxamida (2 g, 3,884 mmol) en NMP (10,00 mL) y 1,2 (aproximadamente 2,684 g, 19,42 mmol) y (4S)-3,3 clorhidrato) (aproximadamente 1,502 g, 9,710 mmol), y la la noche. La mezcla de reacción se enfrió y se vertió (aproximadamente 3,499 g, 3,313 ml, 58,26 mmol). Des fluye bastante uniformemente, los sólidos se separaron diclorometano y se separó la capa acuosa resultante e agua y salmuera, se secó sobre sulfato de sodio y se co hasta unos pocos mililitros. Se añadió gota a gota agua m una suspensión fina y se dejó enfriar durante 30 minutos. cantidad de etanol para dar N-(bencenosulfonil)-2-[(4S) [3-[2-[1-(trifluorometil)ciclopropil]etoxi]pirazol-1-il]piridin-3 encontrado 597,0 (M+1)+; Tiempo de retención: 2,29 min
Ejemplo sintético 18: Síntesis del
(trifluorometM)ddopropoxi]pirazoM-M]-2-[(4S)-2,2,4-tri
[0350]
-[2-[1-(trifluorometil)ciclopropil]etoxi]pirazol-1-il]piridin-3-oxietano (2.000 mL). Se añadieron carbonato de potasio euterio-2,2-dimetil-4-(trideuteriometil)pirrolidina (sal de pensión resultante se calentó a 130°C y se agitó durante hielo agitado rápidamente (60,00 mL) y ácido acético de agitar durante 20 minutos para formar un sólido que filtración y se lavaron con agua. La torta se disolvió en ada. La capa de diclorometano se lavó dos veces con tró. Se añadió etanol (20 mL) y la solución se concentró ntamente. La suspensión que se formó se calentó hasta sólidos cristalinos se filtraron y lavaron con una pequeña dideuterio-2,2-dimetil-4-(trideuteriometil)pirrolidin-1-il]-6-oxamida (600 mg, 26%). IEN-EM m/z calc. 596,24,
puesto 18: N-(Bencenosulfoml)-6-[3-[(ds)-2-pirroMdm-1-M]piridma-3-carboxamida

Paso A: 3-(2-bromoetoxi)-1H-pirazoM-carboxilato de [0351]
c-butilo

[0352] A la disolución de 2-bromoetanol (1,69 g, 13,53
11,28 mmol) y trifenilfosfina (3,55 g, 13,53 mmol) en tetr azodicarboxilato de diisopropilo (2,74 g, 13,53 mmol). Un a 0°C durante 1 hora, luego se calentó a temperatura am ml). La solución orgánica se lavó con solución acuosa s luego se secó sobre sulfato de magnesio, se filtró y se c por cromatografía en gel de sílice usando hexanos acet para proporcionar 3-(2-bromoetoxi)-1H-pirazol-1-carboxil RMN (250 MHz, CDCla) 8 (ppm): 7,85 (d, J = 3,0 Hz, 1H), 6,0 Hz, 2H), 1,64 (s, 9H). lEN-EM m/z calc. 292,0 encontr
Paso B: 3-(VmMoxi)-1H-pirazoM-carboxilato de terc-b
[0353]
l), terc-butil-2,3-dihidro-3-oxopirazol-1-carboxilato (2,08 g, idrofurano anhidro (45 mL) a 0°C se añadió gota a gota ez completada la adición, la solución de reacción se agitó nte y se agitó durante 2 horas más. Se añadió éter (400 rada de carbonato de sodio (80 mL), salmuera (50 mL), entró a presión reducida. El residuo obtenido se purificó de etilo método de gradiente (0 a 15% acetato de etilo) de terc-butilo (2,56 g, 78%) como un sólido blanco. 1H 92 (d, J = 3,0 Hz, 1H), 4,63 (t, J = 6,0 Hz, 2H), 3,68 (t, J = o 292,9 (M+1)+. Tiempo de retención: 4,91 minutos.
lo

[0354] A la solución de 3-(2-bromoetoxi)-1H-pirazol-1-ca anhidro (90 mL) se añadió terc-butóxido de potasio (1,4 horas, luego dicarbonato de diterc-butilo (5,67 g, 26,0 m (400 ml). Las capas orgánicas se lavaron con agua (5 magnesio, se filtraron y se concentraron a presión reduci xilato de terc-butilo (2,52 g, 8,66 mmol) en tetrahidrofurano , 13,0 mmol). La solución resultante se agitó durante 2 l) y se agitó durante 1 hora más. Se añadió éter dietílico L), salmuera (2 x 50 mL), se secaron sobre sulfato de El residuo obtenido se purificó por cromatografía en gel
de sílice usando método de gradiente de hexanos-acet butilo 3-(viniloxi)-1H-pirazol-1-carboxilato de metilo (1,1 (ppm): 7,89 (d, J = 3,0 Hz, 1H), 7,24 (dd, J = 6, 13,5 Hz, 4,50 (dd, J = 1,8, 6,0 Hz, 1H), 1,62 (s, 9H). IEN-EM m/ 4,74 minutos.
Paso C: 3-(2-(TnfluorometN)ciclopropoxi)-1H-pirazo
[0355]
etilo (0 a 10% acetato de etilo) para proporcionar terc-%) como aceite incoloro. 1H RMN (250 MHz, CDCh) 6 5,95 (d, J = 3,0 Hz, 1H), 4,88 (dd, J = 1,8, 13,5 Hz, 1H), 210,1 encontrado 211,0 (M+1)+. Tiempo de retención:
oxNato de terc-butilo

[0356] 3-(Viniloxi)-1H-pirazol-1-carboxilato de terc-butil se añadió agua (20 mL) y se burbujeó con argón durant mmol) seguido de 2,2,2-trifluoroetilamina clorhidrato (3 0,523 mmol). La solución se burbujeó con argón duran tetrametil-1,3-bencenodipropiónico)] (397 mg, 0,523 mm mientras se añadía una solución acuosa de nitrito de bomba de jeringa dentro de las 10 horas. Una vez co horas más. Se añadió éter dietílico (300 mL) y se sep salmuera (30 mL), se secó sobre sulfato de magnesio, s se purificó mediante cromatografía en gel de sílice utiliz 100% de diclorometano). El residuo obtenido se sometió de etilo, 0 a 10% acetato de etilo en gradiente) para pirazol-1-carboxilato de metilo y 3-(1,2-cis-2-(trifluorome trans-2-(trifluorometil)ciclopropoxi)-1H-pirazol-1-carboxil (250 MHz, CDCla) 6 (ppm): 7,84 (d, J = 2,8 Hz, 1H), 5,9 1,56-1,25 (m, 2H). IEN-e M m/z calc. 292,1 encontrado 2 (1,2-cis-2-(trifluorometil)ciclopropoxi)-1H-pirazol-1-carbo (250 MHz, CDCla) 6 (ppm): 7,90 (d, J = 2,8 Hz, 1H), 5,9 1,30 (m, 2H). IEN-e M m/z calc. 292,1 encontrado 293,1
Paso D: 3-(1,2-cis-2-(tnfluorometN)ciclopropoxi)-1H-
[0357]
g, 5,23 mmol) en un matraz en forma de pera (100 mL) nutos, luego se añadió acetato de sodio (85,8 mg, 1,05 26,17 mmol) y ácido sulfúrico concentrado (51,3 mg, os 5 minutos antes de añadir ácido bis[rodio(a,a,a',a'-solución de reacción se mantuvo bajo argón con balón (2,17 g, 31,4 mmol) en agua (12,8 mL) mediante una da la adición, la solución resultante se agitó durante 6 capa orgánica. Luego, la capa orgánica se lavó con y se concentró a presión reducida. El residuo obtenido el método de gradiente de hexanos-diclorometano (0 a matografía en gel de sílice de nuevo (hexanos y acetato erc-butilo 3-(1,2-trans-2-(trifluorometil)ciclopropoxi)-1H-opropoxi)-1H-pirazol-1-carboxilato de terc-butilo. 3-(1,2-terc-butilo: (366 mg, 24%); un sólido blanco. 1H RMN = 2,8 Hz, 1H), 4,49 (m, 1H), 1,75 (m, 1H), 1,62 (s, 9H), M+1)+ Tiempo de retención: 5,22 minutos. terc-butilo 3-de metilo: (314 mg, 21%); un sólido blanco. 1H RMN = 2,8 Hz, 1H), 4,49 (m, 1H), 1,94 (m, 1H), 1,62 (s, 9H), +. Tiempo de retención: 5,48 minutos.
l

[0358] El ácido trifluoroacético (2,76 g, 24,3 mmol) se a 1H-pirazol-1-carboxilato de terc-butilo (708 mg, 2,43 m se agitó a temperatura ambiente durante 16 horas. Se Todos los disolventes se eliminaron a presión reducida. con solución acuosa saturada de bicarbonato de sodi magnesio, se filtró y se concentró a presión reducida pa 1H-pirazol (461 mg, 99%) como aceite amarillo-marrón. sin más purificación. IEN-EM m/z calc. 192,1 encontrad
Paso E: 6-(3-(1,2-cis-2-(tnfluorometN)ciclopropoxi)-1
[0359]
a la solución de 3-(1,2-cis-2-(trifluorometil)ciclopropoxi)-diclorometano anhidro (24 ml). La solución resultante ió 1,2-dicloroetano (10 mL) a la solución de reacción. duo obtenido se disolvió en éter etílico (150 mL), se lavó mL). La solución orgánica se secó sobre sulfato de porcionar ácido 3-(1,2-cis-2-(trifluorometil)ciclopropoxi)-oducto crudo se usó directamente en el siguiente paso 0 (M+1)+. Tiempo de retención: 3,26 minutos.
zoM-M)-2-cloropiridma-3-carboxNato de terc-butilo

[0360] Para la solución de ácido 3-(1,2-cis-2-(tnfluo dimetilformamida (8 mL) se añadió 2,6-d¡dorop¡^d¡na-3-potasio (669 mg, 4,85 mmol) y 1,4-diazabiddo[2.2.2]oct ambiente durante 48 horas. La solución de reacción s salmuera (20 ml). La capa orgánica se secó sobre sulfat residuo obtenido se purificó por cromatografía en gel de (0 a 100% de diclorometano) para proporcionar cloropiridin-3-carboxilato de ferc-butilo (731 mg, 68%) co (d, J = 2,8 Hz, 1H), 8,22 (d, J = 8,5 Hz, 1H), 7,74 (d, J = 1H), 1,62 (s, 9H), 1,45-1,26 (m, 2H). IEN-EM m/z calc. minutos.
Paso F: Ácido 6-(3-(1,2-ds-2-(trifluorometN)ddoprop
[0361]
t¡l)ddopropox¡)-1H-p¡razol (461 mg, 2,43 mmol) en x¡lato de terc-butilo (659 mg, 2,67 mmol), carbonato de (55 mg, 0,49 mmol). La reacción se agitó a temperatura yó con éter (200 mL), se lavó con agua (4 x 20 mL) y magnesio, se filtró y se concentró a presión reducida. El usando método de gradiente de hexanos-diclorometano -(1,2-ds-2-(t^fluoromet¡l)ddopropox¡)-1H-p¡razol-1-¡l)-2-n sólido blanco. 1H RMN (250 MHz, CDCh) 8 (ppm): 8,39 Hz, 1H), 6,01 (d, J = 2,8 Hz, 1H), 4,33 (m, 1H), 1,93 (m, 1 encontrado 404,1 (M+1)+. Tiempo de retención: 7,29
]-1H-pirazoM-M)-2-doropiridm-3-carboxflico

[0362] Se añadió ácido trifluoroacético (2,03 g, 17,8 mm 1H-p¡razol-1-¡l)-2-dorop¡r¡d¡na-3-carbox¡lato de ferc-butil solución resultante se agitó a temperatura ambiente dura de reacción. Todos los disolventes se eliminaron a presi 10% en hexanos (25 mL) y se sonicó durante 30 minuto hexances (10 mL) y se secó a alto vacío para proporcion 1-¡l)-2-dorop¡r¡d¡na-3-carboxíl¡co (517 mg, 84%) como u 1H), 8,47 (d, J = 3,0 Hz, 1H), 8,42 (d, J = 8,8 Hz, 1H), 7, 2,40 (m, 1H), 1,47 (m, 1H), 1,32 (m, 1H). IEN-EM m/z ca minutos.
Paso G: N-(bencenosulfom l)-2-doro-6-[3-[(ds)-2-(trifl
[0363]
la solución de 6-(3-(1,2-ds-2-(trifluoromet¡l)ddopropox¡)-8 mg, 1,78 mmol) en diclorometano anhidro (18 ml). La 6 horas. Se añadió 1,2-dicloroetano (10 mL) a la solución ducida. El sólido crudo obtenido se añadió éter etílico al filtró, se lavó con éter etílico al 10% en hexanos (10 mL), do 6-(3-(1,2-ds-2-(t^fluoromet¡l)ddopropox¡)-1H-p¡razolo blanco. 1H RMN (500 MHz, Dm SO) 8 (ppm): 13,6 (bs, , J = 8,8 Hz, 1H), 6,27 (d, J = 3,0 Hz, 1H), 4,46 (m, 1H), 7,0 encontrado 347,9 (M+1)+. Tiempo de retención: 5,20
metM)ddopropoxi]pirazoM-M]piridm-3-carboxamida

[0364] Ácido 6-(3-(1,2-c¡s-2-(tnfluoromet¡l)c¡clopropox¡)-mmol) se d¡solv¡ó en THF (1 ml). Se añad¡ó 1,1'-carbon¡ld reacc¡ón a temperatura amb¡ente durante 1 hora. Se añ DBU (64,5 ml, 0,431 mmol). Se dejó ag¡tar la mezcla de volát¡les se el¡m¡naron por evaporac¡ón. Se recog¡ó en cítrico 1 M (2X 50 mL) y salmuera (1X 50 mL). La capa or a pres¡ón reduc¡da. Se obtuvo N-(bencenosulfon¡l)-2-clor 3-carboxam¡da (201 mg). IEN-e M m/z calc. 486,03763, (ejecuc¡ón de 1 m¡nuto).
Paso H; N-(bencenosulfonil)-6-[3-[(ci trim etilpirro lid in-1 -il]p irid ina -3-carboxamida
[0365]
razol-1-¡l)-2-clorop¡r¡d¡na-3-carboxíl¡co (125 mg, 0,360 azol (75,6 mg, 0,431 mmol). Se dejó ag¡tar la mezcla de bencenosulfonam¡da (67,8 mg, 0,431 mmol) segu¡do de c¡ón f¡nal durante la noche a temperatura amb¡ente. Los c (50 mL) y se lavó con una soluc¡ón acuosa de ác¡do a se secó sobre sulfato de sod¡o, se f¡ltró y se concentró -[(c¡s)-2-(tr¡fluoromet¡l)c¡clopropox¡]p¡razol-1-¡l] p¡r¡d¡metrado 486,9 (M+1)+; T¡empo de retenc¡ón: 0,67 m¡nutos
(trifluorometil)ciclopropoxi]pirazoM-il]-2-[(4S)-2,2,4-

[0366] N-(bencenosulfon¡l)-2-cloro-6-[3-[(c¡s)-2-(tr¡fluoro 0,3595 mmol) se d¡solv¡ó en DMSO (1 ml). Se añad¡ó ( mmol) segu¡do de carbonato de potas¡o (298 mg, 2,16 la noche. Después de enfr¡ar a temperatura amb¡ente, l con ác¡do cítrico acuoso (1 M, 2X 50 mL) y salmuera (1X f¡ltró y se concentró a pres¡ón reduc¡da. El producto se una columna de gel de síl¡ce de 12 gramos eluyendo con 6-[3-[(c¡s)-2-(tr¡fluoromet¡l)c¡clopropox¡]p¡razol-1-¡l]-2-[(4S) mg, 56%) se obtuvo. IEN-EM m/z calc. 563,1814, encont
Ejemplo sintético 19: Síntesis del
(trifluorometil)ciclopropoxi]pirazol-1-il]-2-[(4S)-2,2,4-t
[0367]
¡clopropox¡]p¡razol-1-¡l]p¡r¡d¡n-3-carboxam¡da (175 mg, ,2,4-tr¡met¡lp¡rrol¡d¡na (sal de clorh¡drato) (161 mg, 1,08 ). Se dejó ag¡tar la mezcla de reacc¡ón a 130°C durante zcla de reacc¡ón se d¡luyó con EtOAc (50 mL) y se lavó L). La capa orgán¡ca se secó sobre sulfato de sod¡o, se med¡ante cromatografía en columna de gel de síl¡ce en ad¡ente de EtOAc/hexano al 0-10%. N-(bencenosulfon¡l)-4-tr¡met¡lp¡rrol¡d¡n-1-¡l]p¡r¡d¡na-3-carboxam¡da (114,3 564,5 (M+1)+; T¡empo de retenc¡ón: 2,08 m¡nutos
uesto 19: N-(bencenosulfonil)-6-[3-[(trans)-2-tilpirrolidin-1-il]piridina-3-carboxamida

Paso A: 3-(1,2-trans-2-(TnfluorometN)cidopropoxi)-1 [0368]
irazol

[0369] El ácido trifluoroacético (3,15 g, 27,64
(trifluorometil)ciclopropoxi)-1H-pirazol-1-carboxilato de fe ml). La solución resultante se agitó a temperatura ambien solución de reacción. Todos los disolventes se eliminaro etílico (200 mL), se lavó con solución acuosa saturada de sobre sulfato de magnesio, se filtró y se conce (trifluorometil)ciclopropoxi)-1H-pirazol crudo (525 mg, 99 crudo se usó directamente en el siguiente paso sin más p Tiempo de retención: 2,97 minutos.
Paso B: 6-(3-(1,2-trans-2-(trifluorometil)ciclopropoxi) butilo
[0370]
ol) se añadió a la solución de 3-(1,2-trans-2-butilo (807 mg, 2,76 mmol) en diclorometano anhidro (28 durante 16 horas. Se añadió 1,2-dicloroetano (15 mL) a la presión reducida. El residuo obtenido se disolvió en éter carbonato de sodio (30 mL). La solución orgánica se secó ó a presión reducida para producir 3-(1,2-trans-2-como un aceite de color marrón amarillento. El producto icación. IEN-EM m/z calc. 192,1 encontrado 193,0 (M+1)+.
H pirazol-1-il)-2-chloropyiidine-3-carboxilato de terc- 
[0371] A la solución de 3-(1,2-trans-2-(tnfluoromet¡l)cido (9,2 mL) se añadió 2,6-d¡dorop¡^d¡na-3-carbox¡lato de fe 5,53 mmol) y 1,4-diazabiddo[2.2.2]odano (62 mg, 0,55 48 horas. La solución de reacción se diluyó con éter (2 capa orgánica se secó sobre sulfato de magnesio, se fil purificó por cromatografía en gel de sílice usando mét diclorometano) para proporcionar ferc-butilo 6 cloropiridin-3-carboxilato (314 mg, 21%) como un aceit Tiempo de retención: 6,92 minutos. 1H RMN (250 MHz, 1H), 7,73 (d, J = 8,5 Hz, 1H), 6,03 (d, J = 3,0 Hz, 1H), 4, 1H).
Paso C: Ácido 6-(3-(1,2-trans-2-(trifluorometN)ddopr
[0372]
x¡)-1H-p¡razol (525 mg, 2,76 mmol) en dimetilformamida tilo (751 mg, 3,04 mmol), carbonato de potasio (763 mg, l). La reacción se agitó a temperatura ambiente durante L), se lavó con agua (4 x 20 mL) y salmuera (20 ml). La se concentró a presión reducida. El residuo obtenido se de gradiente de hexanos-diclorometano (0 a 100% de ,2-trans-2-(t^fluoromet¡l)ddopropox¡)-1H-p¡razol-1-¡l)-2-oloro. IEN-EM m/z calc. 403,1 encontrado 404,1 (M+1)+. la) 8 (ppm): 8,38 (d, J = 3,0 Hz, 1H), 8,20 (d, J = 8,5 Hz, , 1H), 1,77 (m, 1H), 1,62 (s, 9H), 1,44 (m, 1H), 1,31 (m,
xi)-1H-pirazoM-N)-2-doropiridma-3-carboxflico
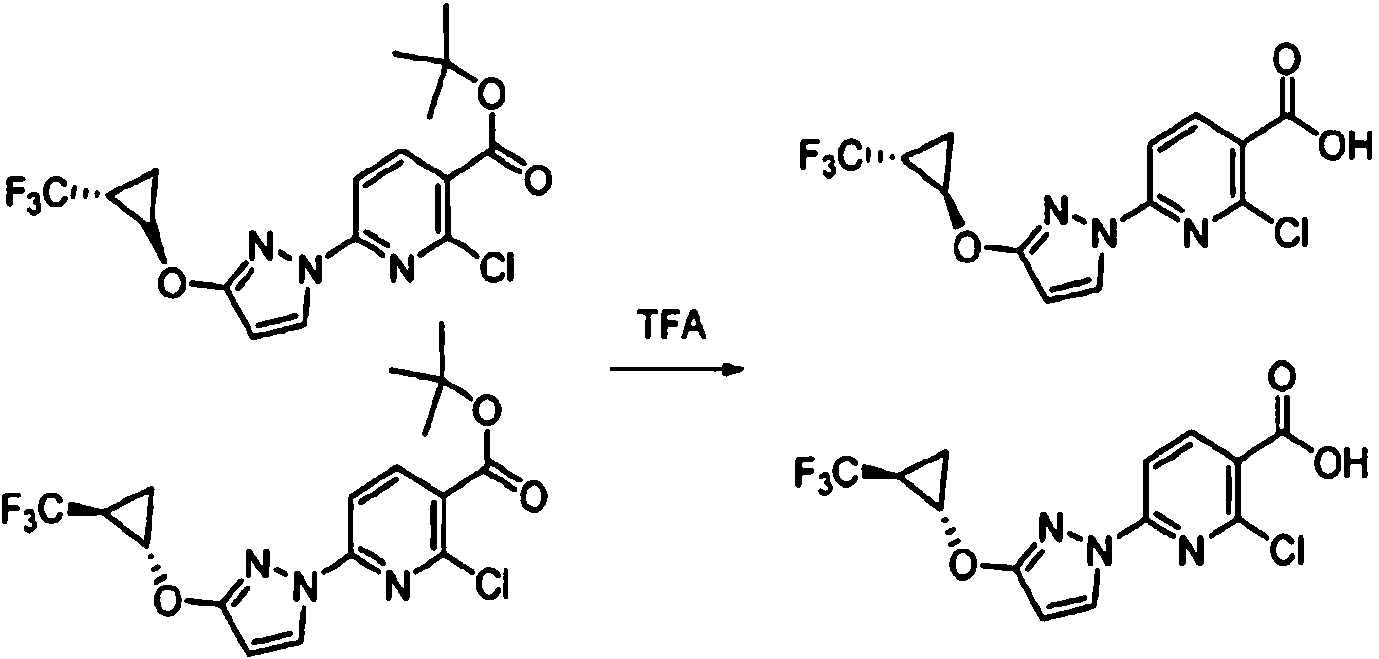
[0373] El ácido trifluoroacético (se añadió 2,39 (t^fluoromet¡l)ddopropox¡)-1H-p¡razol-1-¡l)-2-dorop¡^d¡n diclorometano anhidro (21 ml). La solución resultante s 1,2-dicloroetano (15 mL) a la mezcla de reacción. Todo crudo obtenido se añadió éter etílico al 10% en hexanos éter etílico al 10% en hexanos (10 mL), hexanos (10 trans-2-(tr¡fluoromet¡l)c¡dopropox¡)-1 H p¡razol-1-¡l)-2-dor IEN-EM m/z calc. 347,0 encontrado 347,9 (M+1)+. Tiem (ppm): 8,46 (d, J = 2,8 Hz, 1H), 8,41 (d, J = 8,3 Hz, 1H), 2,15 (m, 1H), 1,40 (m, 1H), 1,34 (m, 1H).
Paso D: N-(bencenosulfoml)-2-doro-6-[3-[ carboxamida
[0374]
21,0 mmol) a la solución de 6-(3-(1,2-trans-2-arbox¡lato de ferc-butilo (847 mg, 2,10 mmol) en tó a temperatura ambiente durante 20 horas. Se añadió disolventes se eliminaron a presión reducida. El sólido mL) y se sonicó durante 30 minutos, se filtró, se lavó con se secó a alto vacío para proporcionar ácido 6-(3-(1,2-d¡na-3-carboxíl¡co (600 mg, 82%) como un sólido blanco. e retención: 4,91 minutos. 1H r Mn (500 MHz, DMSO) 8 (d, J = 8,3 Hz, 1H), 6,30 (d, J = 2,8 Hz, 1H), 4,46 (m, 1H),
s)-2-(trifluorometM)ddopropoxi]pirazoM-M]piridm-3- 
[0375] Ácido 6-(3-(1,2-trans-2-(tnfluoromet¡l)c¡clopropox¡ mmol) se d¡solv¡ó en THF (1 ml). Se añad¡ó 1,1'-carbon¡l reacc¡ón a temperatura amb¡ente durante 1 hora. Se añ DBU (64,5 ml, 0,431 mmol). Se dejó ag¡tar la mezcla de volát¡les se el¡m¡naron por evaporac¡ón. Se recog¡ó en cítrico 1 M (2X 50 mL) y salmuera (1X 50 mL). La capa o a pres¡ón reduc¡da. Se obtuvo N-(bencenosulfon¡l)-2-clo 3-carboxam¡da (199 mg). IEN-EM m/z calc. 486,0, en (ejecuc¡ón de 1 m¡nuto)
Paso E: N-(Bencenosulfoml)-6-[3-[(tran trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0376]
p¡razol-1-¡l)-2-clorop¡r¡d¡na-3-carboxíl¡co (125 mg, 0,360 azol (75,6 mg, 0,431 mmol). Se dejó ag¡tar la mezcla de bencenosulfonam¡da (67,8 mg, 0,431 mmol) segu¡do de c¡ón f¡nal durante la noche a temperatura amb¡ente. Los c (50 mL) y se lavó con una soluc¡ón acuosa de ác¡do ca se secó sobre sulfato de sod¡o, se f¡ltró y se concentró 3-[(trans)-2-(tr¡fluoromet¡l)c¡clopropox¡]p¡razol-1-¡l]p¡r¡d¡nado 486,9 (M+1)+; T¡empo de retenc¡ón: 0,65 m¡nutos
(trifluorometN)cidopropoxi]pirazoM-N]-2-[(4S)-2,2,4-

[0377] N-(Bencenosulfon¡l)-2-cloro-6-[3-[(trans)-2-(tr¡fluor mg, 0,3595 mmol) se d¡solv¡ó en DMSO (1 ml). Se añ 1,08 mmol) segu¡do de carbonato de potas¡o (298 mg, durante la noche. Después de enfr¡ar a temperatura am se lavó con ác¡do cítr¡co acuoso (1 M, 2X 50 mL) y sal sod¡o, se f¡ltró y se concentró a pres¡ón reduc¡da. El pro síl¡ce en una columna de gel de síl¡ce de 12 gramos (bencenosulfon¡l)-6-[3-[(trans)-2-(tr¡fluoromet¡l)c¡clopropo carboxam¡da (115,7 mg, 57%) se obtuvo. IEN-EM m/z c 2,01 m¡nutos
Ejemplo sintético 20: Síntesis del
(trifluorometN)ddopropN]metoxi]pirazoM-N]-2-[(4S)-2
Paso A: 2-C loro-N-(2-hidroxifenil)sulfonil-6-[3-[ carboxamida
[0378]
¡l)c¡clopropox¡]p¡razol-1-¡l]p¡r¡d¡n-3-carboxam¡da (175 4S)-2,2,4-tr¡met¡lp¡rrol¡d¡na (sal de clorh¡drato) (161 mg, mmol). Se dejó ag¡tar la mezcla de reacc¡ón a 130°C e, la mezcla de reacc¡ón se d¡luyó con EtOAc (50 mL) y (1X 50 mL). La capa orgán¡ca se secó sobre sulfato de se a¡sló med¡ante cromatografía en columna de gel de ndo con un grad¡ente de EtOAc/hexano al 0-10%. N-azol-1-¡l]-2-[(4S)-2,2,4-tr¡met¡lp¡rrol¡d¡n-1-¡l]p¡r¡d¡na-3-63,1814, encontrado 564,5 (M+1)+; T¡empo de retenc¡ón:
puesto 20: N-(2-hidroxifenil)sulfonil-6-[3-[[1-trimetMpirroNdm-1-M]piridm-3-carboxamida
rifluorometil)ciclopropilo]metoxi]pirazol-1-il]piridin-3- 
[0379] Una solución de ácido 2-doro-6-[3-[[1-(trifluoro mg, 0,5 mmol) y carbonildiimidazol (aproximadamente 9 minutos. Se agitó una solución de 2-hidroxibenceno hexametildisilazida de sodio (aproximadamente 600 pL d dos soluciones se combinaron y se agitaron durante 15 acidificó con 10 ml de ácido cítrico acuoso 1 M y se extra se secaron sobre sulfato de sodio y se concentraron cromatografía en gel de sílice eluyendo con un gradiente hidroxifenil)sulfonil-6-[3-[[1-(trifluorometil)ciclopropil]meto m/z calc. 516,0, encontrado 517,2 (M+1)+; Tiempo de ret
Paso B: N-(2-Hidroxifenil)sulfonil-6-[3-[[1-(t trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0380]
cidopropil]metoxi]pirazol-1-il]piridina-3-carboxílico (181 mg, 0,60 mmol) en DMF (2,5 mL) se agitó durante 30 onamida (aproximadamente 113 mg, 0,65 mmol) y , 0,60 mmol) en DMF (2,5 mL) durante 30 minutos. Las as a temperatura ambiente. La mezcla de reacción se on 10 ml de acetato de etilo. Los extractos combinados sión reducida. El material crudo se purificó mediante etanol al 0-5% en diclorometano para dar 2-cloro-N-(2-azol-1-il]piridin-3-carboxamida (82 mg, 32%) IEN-EM n: 0,67 minutos.
orometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4-

[0381] 2-Cloro-N-(2-hidroxifenil)sulfonil-6-[3-[[1-(trifluoro (82 mg, 0,16 mmol), (4S)-2,2,4-trimetilpirrolidina (sal de cl de potasio (aproximadamente 132 mg, 0,95 mmol) se com 15 h. La reacción se filtró y purificó usando un método d (2) (75 X 30 mm, tamaño de partícula de 5 pm) vendida gradiente dual desde el 30-99% de la fase móvil B duran B = acetonitrilo; caudal = 50 ml/min y temperatura de la (trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4-t EM m/z calc. 593,2, encontrado 594,3 (M+1)+; Tiempo de
Ejemplo sintético 21: Síntesis del
(trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2
Paso A: 2-C loro-N-(3-hidroxifenil)sulfonil-6-[3-carboxamida
[0382]
ciclopropil]metoxi]pirazol-1-ilo]piridina-3-carboxamida
rato) (aproximadamente 71 mg, 0,48 mmol) y carbonato ron en DMSO (793 mL) y se calentaron a 130°C durante CL-EM de fase inversa usando una columna Luna C18 Phenomenex (pn: 00C-4252-U0-AX), y un desarrollo de 5,0 minutos [fase móvil A = H2O (HCl 5 mM); fase móvil mna = 25°C] para dar N-(2-hidroxifenil)sulfonil-6-[3-[[1-tilpirrolidin-1-il]piridin-3-carboxamida (44 mg, 46%) lEN-nción: 2,07 minutos.
puesto 21: N-(3-hidroxifenil)sulfonil-6-[3-[[1--trimetilpirrolidin-1-il]piridin-3-carboxamida
trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-3-
[0383] Ácido 2-doro-6-[3-[[1-(trifluorometil)cidopropil]me carbonildiimidazol (aproximadamente 97 mg, 0,60 mmo hidroxibencenosulfonamida (aproximadamente 113 mg, 0,60 mmol) en DMF (2,5 mL) durante 30 minutos. Las temperatura ambiente. La mezcla de reacción se acidifi ml de acetato de etilo. Los extractos combinados se s reducida. El material crudo se purificó mediante cromato al 0-8% en diclorometano para dar 2-cloro-N-(3-hidroxif il]piridin-3-carboxamida (250 mg, 97%) IEN-e M m/z cal 0,67 minutos.
Paso B: N-(3-Hidroxifenil)sulfonil-6-[3-[[1-( trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0384]
irazol-1-il]piridin-3-carboxílico (181 mg, 0,50 mmol) y DMF (2,5 mL) se agitó durante 30 minutos. Se agitó 3-mmol) y NaH (aproximadamente 24,0 mg al 60% p/p, soluciones se combinaron y se agitaron durante 4 h a n 10 ml de ácido cítrico acuoso 1 M y se extrajo con 10 n sobre sulfato de sodio y se concentraron a presión en gel de sílice eluyendo con un gradiente de metanol ulfonil-6-[3-[[1-(trifluorometil)ciclopropil]metoxi]pirazol-1-,0482, encontrado 517,2 (M+1)+; Tiempo de retención:
rometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4-

[0385] 2-Cloro-N-(3-hidroxifenil)sulfonil-6-[3-[[1-(trifluo (290 mg, 0,56 mmol), (4S)-2,2,4-trimetilpirrolidina (sal carbonato de potasio (aproximadamente 465 mg, 3,37 La reacción se filtró y purificó usando un método de HP X 30 mm, tamaño de partícula de 5 pm) vendida por Phe dual desde el 30-99% de la fase móvil B durante 15, acetonitrilo; caudal = 50 ml/min y temperatura de la (trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4 EM m/z calc. 593,2, encontrado 594,3 (M+1)+; Tiempo d
Ejemplo sintético 22: Síntesis del
(trifluorometM)ddopropM]metoxi]pirazoM-M]-2-[(4S)-2
Paso A: 2-Cloro-N-(4-hidroxifeml)sulfoml-6-[3-[ carboxamida
[0386]
til)ciclopropil]metoxi]pirazol-1-ilo]piridina-3-carboxamida lorhidrato) (aproximadamente 252 mg, 1,68 mmol) y en DMSO (2,80 mL) se calentó a 130°C durante 15 h. de fase inversa usando una columna Luna C18 (2) (75 nex (pn: 00C-4252-U0-AX), y un desarrollo de gradiente utos [fase móvil A = H2O (Hcl 5 mM); fase móvil B = na = 25°C] para dar N-(3-hidroxifenil)sulfonil-6-[3-[[1-tilpirrolidin-1-il]piridin-3-carboxamida (37 mg, 11%) iEn -nción: 1,98 minutos.
puesto 22: N-(4-hidroxifeml)sulfoml-6-[3-[[1-rimetMpirroMdm-1-M]piridm-3-carboxamida
ifluorometM)ddopropM]metoxi]pirazoM-M]piridm-3-

[0387] Ácido 2-cloro-6-[3-[[1-(trifluorometil)ciclopropil]m carbonildiimidazol (aproximadamente 97 mg, 0,60 mmo hidroxibencenosulfonamida (aproximadamente 113 mg, 0,60 mmol) en DMF (2,5 mL) durante 30 minutos. Las temperatura ambiente. La mezcla de reacción se acidifi ml de acetato de etilo. Los extractos combinados se s reducida. El material crudo se purificó mediante cromato al 0-8% en diclorometano para dar 2-cloro-N-(4-hidroxif il]piridin-3-carboxamida (210 mg, 81%) IEN-e M m/z calc pirazol-1-il]piridin-3-carboxílico (181 mg, 0,50 mmol) y DMF (2,5 mL) se agitó durante 30 minutos. Se agitó 4-mmol) y NaH (aproximadamente 24,0 mg al 60% p/p, soluciones se combinaron y se agitaron durante 4 h a n 10 ml de ácido cítrico acuoso 1 M y se extrajo con 10 n sobre sulfato de sodio y se concentraron a presión en gel de sílice eluyendo con un gradiente de metanol ulfonil-6-[3-[[1-(trifluorometil)ciclopropil]metoxi]pirazol-1-,0, encontrado 517,2 (M+1)+; Tiempo de retención: 0,64
minutos.
Paso B: N-(4-Hidroxifenil)sulfonil-6-[3-[[1-( trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0388]
orometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4-

[0389] 2-Cloro-N-(4-hidroxifenil)sulfonil-6-[3-[[1-(trifluo (220 mg, 0,42 mmol), (4S)-2,2,4-trimetilpirrolidina (sal carbonato de potasio (aproximadamente 353 mg, 2,56 La reacción se filtró y purificó usando un método de HP X 30 mm, tamaño de partícula de 5 pm) vendida por Phe dual desde el 30-99% de la fase móvil B durante 15, acetonitrilo; caudal = 50 ml/min y temperatura de la (trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4 EM m/z calc. 593,2, encontrado 594,3 (M+1)+; Tiempo d Ejemplo sintético 23: Síntesis del (trifluorometM)ddopropM]metoxi]pirazoM-M]-2-[(4S)-2 Paso A: 2-Cloro-N-(o-toMlsulfoml)-6-[3-[[1-(trifluorom [0390]
til)cidopropil]metoxi]pirazol-1-ilo]piridina-3-carboxamida clorhidrato) (aproximadamente 191 mg, 1,28 mmol) y ) en DMSO (2,13 mL) se calentó a 130°C durante 15 h.
de fase inversa usando una columna Luna C18 (2) (75 nex (pn: 00C-4252-U0-AX), y un desarrollo de gradiente utos [fase móvil A = H2O (Hcl 5 mM); fase móvil B = na = 25°C] para dar N-(4-hidroxifenil)sulfonil-6-[3-[[1-etilpirrolidin-1-il]piridin-3-carboxamida (48 mg, 19%) iEn -ención: 1,98 minutos.
Compuesto 23: N-(o-Tolilsulfonil)-6-[3-[[1-trimetMpirroMdm-1-M]piridm-3-carboxamida
ddopropM]metoxi]pirazoM-M]piridm-3-carboxamida

[0391] Ácido 2-cloro-6-[3-[[1-(trifluorometil)ciclopropil]me CDI (aproximadamente 107,6 mg, 0,6635 mmol) se comb durante 2 horas. Se añadió 2-metilbencenosulfonamida (aproximadamente 101,0 mg, 99,21 pL, 0,6635 mmol) ambiente. Se añadió una solución de ácido cítrico 1 M (1 se recogió mediante filtración al vacío (lavado con agua el siguiente paso sin purificación adicional. 2-cloro-N-(oil]piridin-3-carboxamida (280 mg, 98%) IEN-EM m/z calc minutos. 1H RMN (400 MHz, DMSO) 8 d 13,56 - 12,55 ( (d, J = 7,9 Hz, 1H), 7,71 (d, J = 8,3 Hz, 1H), 7,63 (t, J = 4,39 (s, 2H), 2,64 (s, 3H), 1,12 - 1,06 (m, 4H).
Paso B: N-(o-Tolilsulfonil)-6-[3-[[1-(trifluorometil)cicl ilo]piridina-3-carboxamida
[0392]
pirazol-1-il]piridin-3-carboxílico (200 mg, 0,5529 mmol) y n en THF (960 mL) y se agitaron a temperatura ambiente ximadamente 123,1 mg, 0,7188 mmol) seguido de DBU reacción se agitó durante 16 horas más a temperatura y la reacción se agitó durante 20 min. El sólido resultante e secó al vacío para dar un polvo blanco, que se usó en ulfonil)-6-[3-[[1-(trifluorometil)ciclopropil]metoxi]pirazol-1 -,1, encontrado 515,1 (M+1)+; Tiempo de retención: 0,73 ), 8,42 (d, J = 2,8 Hz, 1H), 8,12 (d, J = 8,3 Hz, 1H), 8,04 z, 1H), 7,47 (t, J = 7,6 Hz, 2H), 6,23 (d, J = 2,9 Hz, 1H),
pil]metoxi]pirazol-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1- 
[0393] 2-doro-N-(o-tolilsulfonil)-6-[3-[[1-(trifluorometil)cid 0,2227 mmol), (4S)-2,2,4-trimetilpirrolidina (sal de hidroc combinaron en DMSO (0,5 mL) en un tubo con tapón de enfriar a temperatura ambiente, la mezcla de reacción transfirió a un embudo de decantación. Se añadió una capa orgánica. La capa acuosa se extrajo dos veces combinados se lavaron con salmuera, se secaron sobre s se purificó mediante cromatografía flash en gel de s diclorometano para dar N-(o-tolilsulfonil)-6-[3 trimetilpirrolidin-1 -il]piridin-3-carboxamida ( 101 mg, 77%) de retención: 2,22 minutos. 1H RMN (400 MHz, DMSO) 8,21 (d, J = 2,8 Hz, 1H), 7,90 (d, J = 7,9 Hz, 1H), 7,89 - 7 4,41 -4,31 (m, 2H), 3,36 -3,29 (m, 3H), 2,40 (t, J = 10,4 1,88 - 1,81 (m, 1H), 1,53 (d, J = 9,8 Hz, 6 H), 1,39 (t, J Hz, 3H).
Ejemplo sintético 24: Síntesis del (trifluorometM)ddopropM]metoxi]pirazoM-M]-2-[(4S)-2
Paso A: 2-Cloro-N-(p-toMlsulfoml)-6-[3-[[1-(trifluorom
[0394]
pil]metoxi]pirazol-1-il]piridin-3-carboxamida (114,7 mg, o) ( l 00 mg, 0,6682) y K2CO3 (184,6 mg, 1,336 mmol) se a y se calentaron a 130°C durante 16 horas. Después de uyó con 2 0 ml de acetato de etilo y 10 ml de agua y se ión acuosa de 15 ml de ácido cítrico 1 M y se separó la on 15 ml de acetato de etilo y los extractos orgánicos o de sodio y se concentraron. El material crudo resultante eluyendo con un gradiente de metanol al 0 - 10 % en trifluorometil)ciclopropil]metoxi]pirazol-1 -il]-2-[(4S)-2,2,4--EM m/z calc. 591,21, encontrado 592,3 (M+1)+; Tiempo ,74 (s, 1H), 8,38 (t, J = 1,7 Hz, 1H), 8,33 - 8,22 (m, 2H), m, 1H), 6,93 (d, J = 8,3 Hz, 1H), 6,15 (d, J = 2,7 Hz, 1H), H), 2,27 (t, J = 8 , 6 Hz, 1H), 2,11 (tt, J = 12,1, 6,3 Hz, 1H), Hz, 1H), 1,09 (dt, J = 6,7, 2,2 Hz, 4H), 0,68 (d, J = 6,2
Compuesto 24: N-(p-tolilsulfonil)-6-[3-[[1-trimetMpirroMdm-1-M]piridm-3-carboxamida
ddopropM]metoxi]pirazoM-M]piridm-3-carboxamida

[0395] Ácido 2-cloro-6-[3-[[1-(trifluorometil)ciclopropil]me CDI (aproximadamente 107,6 mg, 0,6635 mmol) se c ambiente durante 2 horas. Se añadió 4-metilbencenosulf de DBU (aproximadamente 101,0 mg, 99,21 pL, 0,66 temperatura ambiente. La mezcla de reacción se diluyó ml de acetato de etilo. Los extractos orgánicos combin salmuera, se secaron sobre sulfato de sodio y se concen sílice, eluyendo con metanol al 0-10%/dicl (trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-3-carb encontrado 515,1 (M+1)+; Tiempo de retención: 0,74 mi
Paso B: N-(p-tolilsulfonil)-6-[3-[[1-(trifluorometil)cicl 1-il]piridin-3-carboxamida
[0396]
pirazol-1-il]piridin-3-carboxílico (200 mg, 0,5529 mmol) y aron en THF (1,200 mL) y se agitaron a temperatura ida (aproximadamente 123,1 mg, 0,7188 mmol) seguido mol) y la reacción se agitó durante 16 horas más a ácido cítrico acuoso 1 M y agua, luego se extrajo 3 x 20 se lavaron con 10 ml de ácido cítrico 1 M, seguido de n, luego se purificaron mediante cromatografía en gel de etano para dar 2-cloro-N-(p-tolilsulfonil)-6-[3-[[1-ida (262 mg, 92%) IEN-EM m/z calc. 514,0689, .
pil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-

[0397] 2-Cloro-N-(p-tolilsulfonil)-6-[3-[[1-(trifluorometil)ci ropil]metoxi]pirazol-1-il]piridina-3-carboxamida (114,7 mg,
0,2227), (4S)-2,2,4-trimetilpirroMdina (sal de clorhidrat combinaron en DMSO (0,5 mL) en un tubo con tapón de enfriar a temperatura ambiente, la mezcla de reacción transfirió a un embudo de decantación. Se añadió una capa orgánica. La capa acuosa se extrajo dos veces combinados se lavaron con salmuera, se secaron sobre s se purificó mediante cromatografía flash en gel de s diclorometano para dar N-(p-tolilsulfonil)-6-[3 trimetilpirrolidin-1 -il]piridin-3-carboxamida (65 mg, 49%). de retención: 2,25 minutos. 1H RMN (400 MHz, DMSO) 8,21 (d, J = 2,8 Hz, 1H), 7,90 (d, J = 7,9 Hz, 1H), 7,89 - 7 4,41 -4,31 (m, 2H), 3,36 -3,29 (m, 3H), 2,40 (t, J = 10,4 1,88 - 1,81 (m, 1H), 1,53 (d, J = 9,8 Hz, 6 H), 1,39 (t, J Hz, 3H).
Ejemplo sintético 25: Síntesis del
(trifluorometM)ddopropM]metoxi]pirazoM-M]-2-[(4S)-2
Paso A: 2-Cloro-N-(3-danofeml)sulfoml-6-[3-[ carboxamida
[0398]
0 mg, 0,6682) y K2CO3 (184,6 mg, 1,336 mmol) se y se calentaron a 130°C durante 16 horas. Después de yó con 2 0 ml de acetato de etilo y 10 ml de agua y se ón acuosa de 15 ml de ácido cítrico 1 M y se separó la on 15 ml de acetato de etilo y los extractos orgánicos de sodio y se concentraron. El material crudo resultante eluyendo con un gradiente de metanol al 0 - 10 % en rifluorometil)ciclopropil]metoxi]pirazol-1 -il]-2-[(4S)-2,2,4-EM m/z calc. 591,21, encontrado 592,3 (M+1)+; Tiempo 74 (s, 1H), 8,38 (t, J = 1,7 Hz, 1H), 8,33 - 8,22 (m, 2H), , 1H), 6,93 (d, J = 8,3 Hz, 1H), 6,15 (d, J = 2,7 Hz, 1H), H), 2,27 (t, J = 8 , 6 Hz, 1H), 2,11 (tt, J = 12,1, 6,3 Hz, 1H), Hz, 1H), 1,09 (dt, J = 6,7, 2,2 Hz, 4H), 0,68 (d, J = 6,2
puesto 25: N-(3-Cianofeml)sulfoml-6-[3-[[1-trimetMpirroMdm-1-M]piridm-3-carboxamida
ifluorometM)ddopropM]metoxi]pirazoM-M]piridm-3-
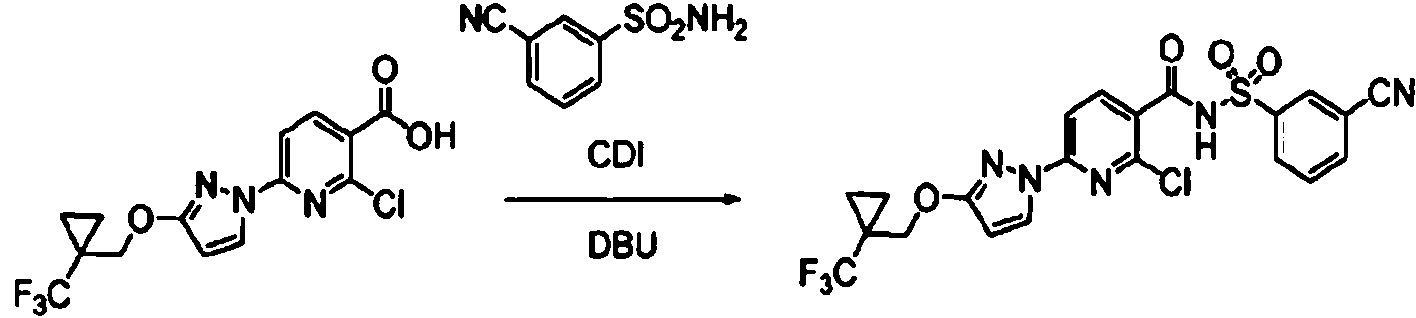
[0399] Ácido 2-cloro-6-[3-[[1-(trifluorometil)ciclopropil]me CDI (aproximadamente 107,6 mg, 0,6635 mmol) se c ambiente durante 2 horas. Se añadió 3-cianobencenosulf de DBU (aproximadamente 101,0 mg, 99,21 pL, 0,66 temperatura ambiente. La mezcla de reacción se diluyó acetato de etilo. Los extractos orgánicos combinados se luego se secaron sobre sulfato de sodio y se concentrar 2-cloro-N-(3-cianofenil)sulfonil-6-[3-[[1-(trifluorometil)cicl 78%) IEN-EM m/z calc. 525,0485, encontrado 526,0 (M+
Paso B: N-(3-danopbeml)sulfoml-6-[3-[[1-(t trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0400]
irazol-1-il]piridin-3-carboxílico (200 mg, 0,5529 mmol) y aron en THF (1,200 mL) y se agitaron a temperatura ida (aproximadamente 131,0 mg, 0,7188 mmol) seguido mol) y la reacción se agitó durante 16 horas más a cido cítrico acuoso 1 M y agua y se extrajo 3 x 20 ml de on con 10 ml de ácido cítrico 1 M, seguido de salmuera, e usaron en el siguiente paso sin purificación adicional. il]metoxi]pirazol-1-il]piridin-3-carboxamida (228 mg, iempo de retención: 0,7 minutos.
rometM)ddopropM]metoxi]pirazoM-M]-2-[(4S)-2,2,4-
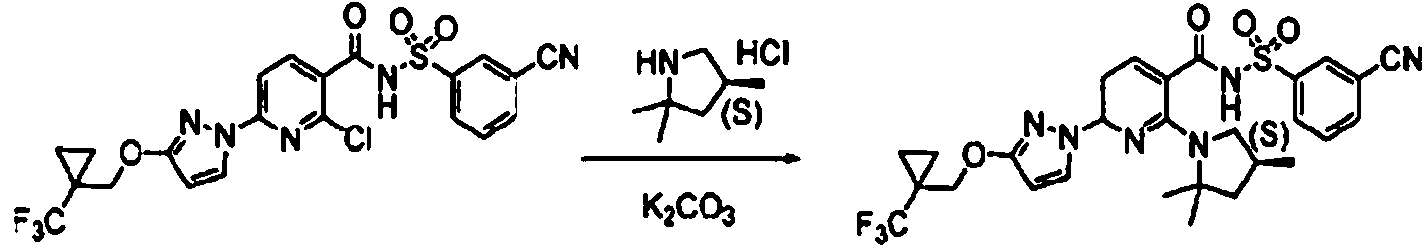
[0401] 2-Cloro-N-(3-cianofenil)sulfonil-6 carboxamida (117,1 mg, 0,2227), (4S)-2,2,4-trimetilpirrol mg, 1,336 mmol) se combinaron en DMSO (0,5 mL) en 16 horas. Después de enfriar a temperatura ambiente, la 10 ml de agua y se transfirió a un embudo de decantació M y se separó la capa orgánica. La capa acuosa se extra orgánicos combinados se lavaron con salmuera, se se crudo resultante se purificó mediante cromatografía flas 10% en diclorometano para dar N-(3-cianofenil)sulfon 2,2,4-trimetilpirrolidin-1-il]piridin-3-carboxamida, (73 mg, 1-(trifluorometil)ciclopropil]metoxi]pirazol-1-ilo]piridina-3-(sal de hidrocloruro) (100 mg, 0,6682) y K2CO3 (184,6 bo con tapón de rosca y se calentaron a 130°C durante cla de reacción se diluyó con 20 ml de acetato de etilo y añadió una solución acuosa de 15 ml de ácido cítrico 1 veces más con 15 ml de acetato de etilo y los extractos sobre sulfato de sodio y se concentraron. El material el de sílice, eluyendo con un gradiente de metanol al 0 --[[1-(trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-) Ie N-EM m/z calc. 602,19, encontrado 603,3 (M+1 )+;
Tiempo de retención: 2,04 minutos. 1H RMN (400 MHz, = 8,1, 1,9, 1,1 Hz, 1H), 8,24 (dt, J = 7,8, 1,3 Hz, 1H), 8,2 1H), 6,15 (d, J = 2,7 Hz, 1H), 4,42 -4,31 (m, 2H), 2,40 (t 6,3 Hz, 1H), 1,89 - 1,78 (m, 1H), 1,53 (d, J = 9,8 Hz, 6 H) (d, J = 6,2 Hz, 3H).
Ejemplo sintético 26: Síntesis del
(trifluorometN)ddopropN]metoxi]pirazoM-N]-2-[(4S)-2 Paso A: 2-C loro-N-(2-danofeml)sulfom l-6-[3 carboxamida
[0402]
O) 512,74 (s, 1H), 8,38 (t, J = 1,7 Hz, 1H), 8,30 (ddd, J J = 2,8 Hz, 1H), 7,92 - 7,84 (m, 2H), 6,93 (d, J = 8,3 Hz, 10,4 Hz, 1H), 2,27 (t, J = 8 , 6 Hz, 1H), 2,11 (tt, J = 12,1, (t, J = 12,1 Hz, 1H), 1,09 (dt, J = 6,7, 2,2 Hz, 4H), 0,68
mpuesto 26: N-(2-danofeml)sulfoml-6-[3-[[1-rimetMpirroNdm-1-M]piridm-3-carboxamida
rifluorometN)ddopropN]metoxi]pirazoM-N]piridm-3-

[0403] Ácido 2-doro-6-[3-[[1-(trifluorometil)cidopropil]me CDI (aproximadamente 107,6 mg, 0,6635 mmol) se c ambiente durante 2 horas. Se añadió 2-cianobencenosulf de DBU (aproximadamente 101,0 mg, 99,21 pL, 0,66 temperatura ambiente. Se añadió una solución de ácido El precipitado sólido resultante se recogió mediante filt que se secó al vacío y se usó en el siguiente paso sin (trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-3-carb encontrado 526,1 (M+1)+; Tiempo de retención: 0,69 mi 2,9 Hz, 1H), 8,46 - 8,39 (m, 1H), 8,35 (d, J = 8,3 Hz, 1H) 1H), 6,27 (d, J = 2,9 Hz, 1H), 4,42 (s, 2H), 1,11 (dt, J =
Paso B: N-(2-Cianofeml)sulfoml-6-[3-[[1-( trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0404]
irazol-1-il]piridin-3-carboxílico (200 mg, 0,5529 mmol) y aron en THF (1,200 mL) y se agitaron a temperatura ida (aproximadamente 131,0 mg, 0,7188 mmol) seguido mol) y la reacción se agitó durante 16 horas más a o 1 M (1 mL) y la reacción se agitó durante 20 minutos.
al vacío (lavado con agua) para dar un sólido blanco, ación adicional, 2-cloro-N-(2-cianofenil)sulfonil-6-[3-[[1-ida (279 mg, 96%) IEN-EM m/z calc. 525,0485, 1H RMN (400 MHz, DMSO) 512,23 (s, 1H), 8,49 (d, J = - 8,13 (m, 1H), 7,96 - 7,90 (m, 2H), 7,82 (d, J = 8,3 Hz, 2 Hz, 4H).
rometN)ddopropN]metoxi]pirazoM-N]-2-[(4S)-2,2,4-

[0405] 2-C loro-N-(2-cianofenil)sulfonil-6-[3-[[ carboxamida (117,1 mg, 0,2227), (4S)-2,2,4-trimetilpirr mg, 1,336 mmol) se combinaron en DMSO (0,5 mL) en 16 horas. Después de enfriar a temperatura ambiente, l 10 ml de agua y se transfirió a un embudo de decantaci ml y se separó la capa orgánica. La capa acuosa se extr orgánicos combinados se lavaron con salmuera, se se crudo resultante se purificó mediante cromatografía flas 0-10% en diclorometano para dar N-(2-cianofenil)sulfo 2,2,4-trimetilpirrolidin-1-ilo]piridin-3-carboxamida, (41 m Tiempo de retención: 2,12 minutos. 1H RMN (400 MHz, 8,17 - 8,11 (m, 1H), 8,06 (d, J = 8,3 Hz, 1H), 7,94 - 7,8 4,44-4,32 (m, 2H), 3,07 - 2,91 (m, 2H), 2,32 (d, J = 19, 3H), 1,57 (t, J = 10,4 Hz, 1H), 1,13 -1,06 (m, 4H), 1,02 (
Ejemplo sintético 27: Síntesis del luorometil)cidopropil]metoxi]pirazol-1-ilo]piridina-3-(sal de hidrocloruro) (100 mg, 0,6682) y K2CO3 (184,6 bo con tapón de rosca y se calentaron a 130°C durante cla de reacción se diluyó con 20 ml de acetato de etilo y añadió una solución acuosa de ácido cítrico 1 M de 15 s veces más con 15 ml de acetato de etilo y los extractos sobre sulfato de sodio y se concentraron. El material e gel de sílice, eluyendo con un gradiente de metanol al -[[1-(trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-) IEN-EM m/z calc. 602,19, encontrado 603,2 (M+1)+; ) 511,77 (s, 1H), 8,47 (s, 1H), 8,29 (d, J = 2,8 Hz, 1H), 2H), 6,97 (d, J = 8,3 Hz, 1H), 6,20 (d, J = 2,8 Hz, 1H), 1H), 1,98 (q, J = 5,9, 5,5 Hz, 1H), 1,67 (s, 3H), 1,63 (s, 6,3 Hz, 3H).
mpuesto 27: N-(4-cianofenil)sulfonil-6-[3-[[1-(trifluorometM)ddopropM]metoxi]pirazoM-M]-2-[(4S)-2 Paso A: 2-Cloro-N-(4-danofeml)sulfoml-6-[3-carboxamida
[0406]
trimetMpirroMdm-1-M]piridm-3-carboxamida
rifluorometM)ddopropM]metoxi]pirazoM-M]piridm-3-

[0407] Ácido 2-doro-6-[3-[[1-(trifluorometil)cidopropil]me CDI (81 mg, 0,4995 mmol) se combinaron en THF (900, Se añadió 4-cianobencenosulfonamida (98 mg, 0,5379 agitó a temperatura ambiente durante 2 horas. Se añadió una hora más a temperatura ambiente. La mezcla de re M y agua y se extrajo 3 x 20 ml de acetato de etilo. Los e cítrico 1 M, seguido de salmuera, luego se secaron sobr purificó adicionalmente mediante cromatografía en gel diclorometano, para dar un sóli (trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-3-carb encontrado 526,0 (M+1)+; Tiempo de retención: 0,71 mi
Paso B: N-(4-Cianofeml)sulfoml-6-[3-[[1-(t trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0408]
irazol-1-il]piridin-3-carboxílico (150 mg, 0,4147 mmol) y ) y se agitaron a temperatura ambiente durante 2 horas. ) seguido de DBU (75 ml, 0,5015 mmol) y la reacción se DBU (80 ml, 0,5350 mmol) y la reacción se agitó durante n se diluyó con 20 ml de una solución de ácido cítrico 1 tos orgánicos combinados se lavaron con 10 ml de ácido ato de sodio y se concentraron. El material resultante se lice eluyendo con un gradiente de metanol al 0 - 1 0 % en blanco; 2-cloroN-(4-cianofenil)sulfonil-6-[3-[[1-ida (192 mg, 8 8 %) IEN-EM m/z calc. 525,0485,
rometM)ddopropM]metoxi]pirazoM-M]-2-[(4S)-2,2,4-

[0409] 2-Cloro-N-(4-cianofenil)sulfonil-6-[3-[[1-(triflu (117,1 mg, 0,2227), (4S)-2,2,4-trimetilpirrolidina (sal de hi se combinaron en DMSO (0,5 mL) en un tubo con tapón de enfriar a temperatura ambiente, la mezcla de reacció transfirió a un embudo de decantación. Se añadió una capa orgánica. La capa acuosa se extrajo dos veces combinados se lavaron con salmuera, se secaron sobre se purificó mediante cromatografía flash sobre gel de diclorometano para dar N-(4-cianofenil)sulfonil-6-[3 trimetilpirrolidin-1 -il]piridin-3-carboxamida, (62 mg, 46%) de retención: 2,04 minutos. 1H RMN (400 MHz, DMSO) J = 8,3 Hz, 1H), 6,93 (d, J = 8,3 Hz, 1H), 6,15 (d, J = 2, (dd, J = 10,2, 6,9 Hz, 1H), 2,11 (tt, J = 11,9, 6,4 Hz, 1H), (t, J = 12,1 Hz, 1H), 1,13 -1,05 (m, 4H), 0,66 (d, J = 6,2
Ejemplo sintético 28: Síntesis del (trifluorom etil)cidopropil]metoxi]pirazol-1-il]-2-[(4S)-
Paso A: 2-C loro-N-(m-tolilsulfonil)-6-[3-[[1-(trifluoro
[0410]
til)ciclopropil]metoxi]pirazol-1-ilo]piridina-3-carboxamida loruro) (100 mg, 0,6682) y K2CO3 (184,6 mg, 1,336 mmol) sca y se calentaron a 130°C durante 16 horas. Después iluyó con 20 ml de acetato de etilo y 10 ml de agua y se ión acuosa de ácido cítrico 1 M de 15 ml y se separó la on 15 ml de acetato de etilo y los extractos orgánicos o de sodio y se concentraron. El material crudo resultante , eluyendo con un gradiente de metanol al 0 - 10 % en trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4-EM m/z calc. 602,19, encontrado 603,3 (M+1)+; Tiempo 77 (s, 1H), 8,20 (d, J = 2,8 Hz, 1H), 8,16 (s, 4H), 7,87 (d, 1H), 4,50 -4,17 (m, 2H), 2,33 (t, J = 10,3 Hz, 1H), 2,20 (dd, J = 11,8, 5,4 Hz, 1H), 1,52 (d, J = 5,6 Hz, 6 H), 1,37 H).
Compuesto 28: N-(m-tolilsulfonil)-6-[3-[[1--trimetilpirrolidin-1-il]piridin-3-carboxam ida
cidopropil]metoxilpirazol-1-il]piridin-3-carboxamida

[0411] Ácido 2-doro-6-[3-[[1-(trifluorometil)cidopropil]me CDI (aproximadamente l07,6 mg, 0,6635 mmol) se c ambiente durante 2 horas. Se añadió 3-metilbencenosulf de DBU (aproximadamente 101,0 mg, 99,21 ml, 0,66 temperatura ambiente. La mezcla de reacción se diluy extrajo 3 x 20 ml de acetato de etilo. Los extractos orgá seguido de salmuera, se secaron sobre sulfato de s cromatografía en gel de sílice eluyendo con metanol al tolilestdfonil)-6-[3-[[1-(trifluorometil)ciclopropil]metoxi]pir calc. 514,0689, encontrado 515,1 (m 1 )+; Tiempo de ret
Paso B: N-(m-TolMsulfoml)-6-[3-[[1-(trifluorometN)cid 1-il]piridin-3-carboxamida
[0412]
irazol-1-il]piridin-3-carboxílico (200 mg, 0,5529 mmol) y aron en THF (964,9 mL) y se agitaron a temperatura ida (aproximadamente 123,1 mg, 0,7188 mmol) seguido mol) y la reacción se agitó durante 16 horas más a una solución acuosa de ácido cítrico 1 M y agua y se combinados se lavaron con 10 ml de ácido cítrico 1 M, se concentraron y finalmente se purificaron mediante /diclorometano para dar un sólido blanco, 2-cloro-N-(m--il]piridin-3-carboxamida (178 mg, 63%) IEN-EM m/z n: 0,74 minutos
N]metoxi]pirazoM-N]-2-[(4S)-2,2,4-trimetMpirroNdm-

[0413] 2-Cloro-N-(m-tolilsulfonil)-6-[3-[[1-(trifluoromet carboxamida (114,7 mg, 0,2227 mmol), (4S)-2,2,4-trim (184,6 mg, 1,336 mmol) se combinaron en DMSO (0,5 durante 16 horas. Después de enfriar a temperatura am de etilo y 10 ml de agua y se transfirió a un embudo de 1 M de 15 ml y se separó la capa orgánica. La capa acu los extractos orgánicos combinados se lavaron con salm material crudo resultante se purificó mediante cromato metanol al 0-10% en diclorometano para dar N-(m-tolil [(4S)-2,2,4-trimetilpirrolidin-1-il]piridin-3-carboxamida, (4 Tiempo de retención: 2,24 minutos. 1H RMN (400 MHz, 6,0, 2,5 Hz, 3H), 7,57 - 7,50 (m, 2H), 6,91 (d, J = 8,2 Hz, 2,42 (s, 3H), 2,29 (t, J = 8 , 8 Hz, 1H), 2,11 (dt, J = 13,2, Hz, 6 H), 1,38 (t, J = 12,1 Hz, 1H), 1,09 (dd, J = 4,5, 3,2
Ejemplo sintético 29: Síntesis del Compu metilcidopropoxi)metil]pirazol-1-il]-2-[(4S)-2,2,4-trim
Paso A: (1-Metil-1-(prop-2-in-1-iloxi)ciclopropano
[0414]
propN]metoxi]pirazoM-N]piridma-3-rolidina (sal de hidrocloruro) (100 mg, 0,6682) y K2CO3 n un tubo con tapón de rosca y se calentaron a 130°C , la mezcla de reacción se diluyó con 20 ml de acetato tación. Se añadió una solución acuosa de ácido cítrico e extrajo dos veces más con 15 ml de acetato de etilo y se secaron sobre sulfato de sodio y se concentraron. El flash en gel de sílice, eluyendo con un gradiente de il)-6-[3-[[1-(trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-34%) iEn -EM m/z calc. 591,2, encontrado 592,2 (M+1)+; O) 8 12,41 (s, 1H), 8,20 (d, J = 2,8 Hz, 1H), 7,79 (tt, J = 6,15 (d, J = 2,7 Hz, 1H), 4,48 -4,24 (m, 2H), 2,46 (s, 1H), z, 1H), 1,83 (dd, J = 11,8, 5,5 Hz, 1H), 1,53 (d, J = 12,0 ), 0,66 (d, J = 6,2 Hz, 3H).
29: Síntesis de N-(bencenosulfonil)-6-[3-[(1-rolidin-1 -il]piridin-3-carboxamida

[0415] 1-Metilciclopropan-1-ol (1,0 g, 13,9 mmol) se dis NaH (50% en aceite, 0,67 g, 13,9 mmol). La mezcla s bromuro (80% en tolueno, 3,1 g, 20,9 mmol). La mezcla n Et2O (50 mL) y se enfrió a 0°C. Se añadió en porciones ó durante 10 min a 0°C antes de añadirse gota a gota itó durante 1 hora a 0°C. Al no proseguirse la reacción,
se añadió DMF (20 ml). La mezcla se agitó durante una La mezcla se extrajo con Et2O (2X50 ml). Las capas org se secaron sobre Na2SO4 y se concentraron (a 40°C, 50 crudo que se utilizó como tal en el siguiente paso. 1H 3H); 2,37 (s, 1H); 4,10 (s, 2H).
Paso B: 3-((1-metilcidopropoxi)metil)-1H-pirazol
[0416]
adicional a 0° y se inactivó con NH4CI acuoso saturado. combinadas se lavaron con agua dos veces y salmuera, r) para producir (1-metil-1-(prop-2-in-1-iloxi)ciclopropano cDch, 300 MHz): d 0,39 (m, 2H); 0,85 (m, 2H); 1,40 (s,

[0417] (1-Metil-1-(prop-2-in-1-iloxi)ciclopropano en cru trimetilsilil diazometano (2,0 M en hexano, 10 ml, 20 m La mezcla se enfrió a 40°C y se inactivó con MeOH ( heptanos/EtOAc 2:1) dio 3-((1-metilciclopropoxi)metil)-1 1H RMN (CDCla, 300 MHz): d 0,44 (m, 2H); 0,85 (m, 2 RMN (75 MHz, CDCla): d 13,4, 20,3, 58,4, 6 1 ,9 , 103,9,
Paso C: N-(bencenosulfonil)-6-[3-[(1-metilcido il]piridin-3-carboxamida
[0418]
varios lotes (máx. 27,8 mmol, 3,0 g) se mezcló con se agitó en un tubo sellado a 115°C durante 18 horas. ) y se concentró. La cromatografía en columna (sílice; zol como un aceite incoloro (1,2 g, 28% en dos etapas).
44 (s, 3H); 4,60 (s, 2H); 6,23 (s, 1H); 7,51 (s, 1H). 13C-(no se muestra un carbono cuaternario).
xi)metil]pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-

[0419] N-(Bencenosulfonil)-6-cloro-2-[(4S)-2,2,4-trimetil [(1-metilciclopropoxi)metil]-1H-pirazol (62 mg, 0,4074 combinaron en DMSO (1,660 ml). Se añadió NaH (41
minutos antes de sellarla y calentarla a 160°C durante 1 una solución de ácido cítrico 1 M. Los orgánicos se sep sodio. Después, los orgánicos se evaporaron a presi preparativa (1-99 C ^C N en agua con HCl 5 mM), dura diluyeron con agua y se extrajeron con acetato de etil metilciclopropoxi)metil]pirazol-1-il]-2-[(4S)-2,2,4-trimetilp calc. 523,22534, encontrado 524,2 (M+1 )+; Tiempo de r
Ejemplo sintético 30: Síntesis del
tetrametilcidopropil)metoxi]pirazol-1-il]-2-[(4S)-2,2,4
Paso A: N-(bencenosulfonil)-2-doro-6-[3-carboxamida
[0420]
din-1-il]piridin-3-carboxamida (83 mg, 0,2035 mmol), 3-l) y triflato de escandio (10 mg, 0,02032 mmol) se 60% p/p, 1,025 mmol) y la reacción se agitó durante 15 a reacción se enfrió y se repartió entre acetato de etilo y , se lavaron con salmuera y se secaron sobre sulfato de ducida y el material crudo se purificó mediante HPLC 0 minutos. Las fracciones que contenían el producto se a dar, tras concentración, N-(bencenosulfonil)-6-[3-[(1-in-1il]piridin-3-carboxamida ( 10 mg, 9%) IEN-e M m/z ión: 2,04 minutos.
puesto 30: N-(bencenosulfonil)-6-[3-[(2,2,3,3-etilpirrolidin-1-il]piridin-3-carboxamida
3,3-tetrametilcidopropil)metoxi]pirazol-1-il]piridin-3-
[0421] Ácido 2-doro-6-[3-[(2,2,3,3-tetrametilcidopropil)m y CDI (111 mg, 0,6846 mmol) se combinaron en THF (1, Se añadió bencenosulfonamida (117 mg, 0,7443 mmol) durante 6 h más a temperatura ambiente. La mezcla d agua y se extrajo 3 X 20 ml de acetato de etilo. Los e secaron sobre sulfato de sodio y se concentraron, luego un gradiente de metanol al 0-10% en diclorometano [(2,2,3,3-tetrametilciclopropil)metoxi]pirazol-1-il]piridin-3-encontrado 489,2 (M+1)+; Tiempo de retención: 0,81 mi
Paso B: N-(bencenosulfoml)-6-[3-[(2,2, trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0422]
i]pirazol-1-il]piridin-3-carboxílico (200 mg, 0,5717 mmol) ) y se agitaron a temperatura ambiente durante 2 horas. ido de DBU (102 ml, 0,6821 mmol) y la reacción se agitó cción se diluyó con una solución de ácido cítrico 1 M y tos orgánicos combinados se lavaron con salmuera, se rificaron mediante cromatografía en gel de sílice usando dar un polvo blanco. N-(bencenosulfonil)-2-cloro-6-[3-xamida (250 mg, 89%) IEN-EM m/z calc. 488,1285, .
etrametNddopropN)metoxi]pirazoM-N]-2-[(4S)-2,2,4-

[0423] N-(bencenosulfonil)-2-cloro-6-[3-[(2,2,3,3-tetrame mg, 0,2352 mmol), (4S)-2,2,4-trimetilpirrolidina (sal de carbonato de potasio (aproximadamente 195,6 mg, 1,41 a 130°C durante 16 h. La reacción se enfrió a temperat cítrico 1 M y 30 ml de acetato de etilo. Las capas acuo veces más con 30 ml de acetato de etilo, las capas org sobre sulfato de sodio y se concentraron. El sólido re eluyendo con metanol al 0-10% en diclorometano, y lueg usando acetato de etilo al 0-100% en diclor tetrametilciclopropil)metoxi]pirazol-1-il]-2-[(4S)-2,2,4-trim m/z calc. 565,2723, encontrado 566,3 (M+1)+; Tiempo d (s, 1H), 8,18 (d, J = 2,8 Hz, 1H), 8,02 - 7,95 (m, 2H), 7,7 6,92 (d, J = 8,3 Hz, 1H), 6,13 (d, J = 2,7 Hz, 1H), 4,24 (d, 7,1 Hz, 1H), 2,17 -2,03 (m, 1H), 1,82 (dd, J = 11,8, 5,5 H (s, 6H), 1,04 (s, 6H), 0,73 (t, J = 7,7 Hz, 1H), 0,65 (d, J =
Ejemplo sintético 31: Síntesis del Com (trifluorometil)ciclopropil]etoxi]pirazol-1-il]-2-[(4S)-2,
Paso A: 2-Cloro-N-(4-hidroxifeml)sulfoml-6-[3-[2 carboxamida
[0424]
lopropil)metoxi]pirazol-1-il]piridina-3-carboxamida (115 hidrato) (aproximadamente 105,9 mg, 0,7077 mmol) y ol) se combinaron en DMSO (575,0 mL) y se calentaron ambiente, se diluyó con 15 ml de agua, 15 ml de ácido orgánica se separaron y la capa acuosa se extrajo dos s se combinaron, se lavaron con salmuera, se secaron nte se purificó mediante cromatografía en gel de sílice purificó adicionalmente mediante cromatografía en sílice tano, para dar N-(bencenosulfonil)-6-[3-[(2,2,3,3-rrolidin-1-il]piridin-3-carboxamida (43 mg, 32%) IEN-EM nción: 2,43 minutos. 1H RMN (400 MHz, DMSO) 812,47 J = 8,3 Hz, 1H), 7,76 - 7,69 (m, 1H), 7,68 - 7,62 (m, 2H), 7,7 Hz, 2H), 2,42 (t, J = 10,5 Hz, 1H), 2,28 (dd, J = 10,2, ), 1,52 (d, J = 9,4 Hz, 6H), 1,36 (t, J = 12,1 Hz, 1H), 1,10 Hz, 3H).
to 31: N N-(4-hidroxifenil)sulfonil-6-[3-[2-[1-rimetilpirrolidin-1-il]piridin-3-carboxamida
trifluorometM)ddopropMo]etoxMpirazoM-M]piridm-3-

[0425] Ácido 2-Cloro-6-[3-[2-[1-(trifluorometil)ciclopropil y CDI (aproximadamente 51,38 mg, 0,3169 mmol) se ambiente durante 2 horas. Se añadió 4-hidroxibence seguido de DBU (aproximadamente 53,45 pL, 0,3574 m ambiente. La mezcla de reacción se diluyó con 10 mL d de etilo. Los orgánicos combinados se lavaron con sal para dar un sólido blanco, que se usó en el siguiente pa 6-[3-[2-[1-(trifluorometil)ciclopropil]etoxi]pirazol-1-il]piridin encontrado 531,0 (M+1)+; Tiempo de retención: 0,69 mi i]pirazol-1-il]piridina-3-carboxílico (100 mg, 0,2661 mmol) inaron en THF (600,0 mL) y se agitaron a temperatura fonamida (aproximadamente 50,69 mg, 0,2927 mmol) la reacción se agitó durante 16 horas más a temperatura do cítrico 1 M y se extrajo 3 veces con 10 mL de acetato a, se secaron sobre sulfato de sodio y se concentraron n purificación adicional. 2-cloro-N-(4-hidroxifenil)sulfonilrboxamida (128 mg, 91%) ESI -m S m/z calc. 530,06384, .
Paso B: N-(4-Hidroxifenil)sulfonil-6-[3-[2-[1 trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0426]
luorometil)ciclopropil]etoxi]pirazol-1-il]-2-[(4S)-2,2,4-

[0427] 2-Cloro-N-(4-hidroxifenN)sulfonN-6-[3-[2-[1-(tnfluo (134 mg, 0,2524 mmol), (4S)-2,2,4-trimetilpirrolidina (sal (210 mg, 1,519 mmol) se combinaron en dimetilsulfóxido se enfrió a temperatura ambiente y se añadió 1 ml de ag el contenido del vial, se eliminó la porción líquida con u acetato de etilo. Los orgánicos se lavaron con 15 ml de la capa acuosa se extrajo dos veces más con 15 ml de a salmuera, se secaron sobre sulfato de sodio y se co cromatografía en gel de sílice eluyendo con metanol al [3-[2-[1-(trifluorometil)ciclopropil]etoxi]pirazol-1-il]-2-[(4S) 28%) IEN-EM m/z calc. 607,20764, encontrado 608,2 (M DMSO) 812,25 (s, 1H), 10,58 (s, 1H), 8,19 (d, J = 2,8 Hz, (m, 2H), 6,89 (d, J = 8,2 Hz, 1H), 6,10 (d, J = 2,7 Hz, 1H) (m, 1H), 2,26 (t, J = 8,8 Hz, 1H), 2,07 (t, J = 7,0 Hz, 2H), (t, J = 12,1 Hz, 1H), 1,00 - 0,93 (m, 2H), 0,91 - 0,86 (m,
Ejemplo sintético 32: Síntesis del C (triMuorometM)ciclopropMo)]etoxi]pirazoM-il]-2-[(4S)-
[0428]
tN)cidopropil1]etoxi]pirazoM-N]piridin-3-carboxamida
rhidrato) (113 mg, 0,7550 mmol) y carbonato de potasio ,0 j L) y se calentaron a 130°C durante 16 h. La reacción espués de 15 minutos de agitación, se dejó sedimentar eta y se disolvieron los sólidos restantes con 20 ml de cítrico 1M. Se separaron las capas acuosa y orgánica y o de etilo. Los orgánicos se combinaron, se lavaron con raron. El sólido crudo resultante se purificó mediante en diclorometano para dar N-(4-hidroxifenil)sulfonil-6--trimetilpirrolidin-1-il]piridin-3-carboxamida (43 mg, Tiempo de retención: 2,07 minutos. 1H RMN (400 MHz, 7,87 -7,79 (m, 2H), 7,75 (d, J = 8,2 Hz, 1H), 6,97 -6,91 (t, J = 7,1 Hz, 2H), 2,44 (t, J = 10,4 Hz, 1H), 2,16 -2,09 dd, J = 11,9, 5,5 Hz, 1H), 1,54 (s, 3H), 1,51 (s, 3H), 1,38 ,69 (d, J = 6,2 Hz, 3H).
uesto 32: W-(bencenosulfoml)-6-[5-fluoro-3-[2-[1--trimetMpirrolidm-1-M]piridm-3-carboxamida

Paso A: W-(bencenosulfoml)-2,6-dicloropmdm-3-carb [0429]
ida
[0430] A Se equipó un matraz de fondo redondo de 3 enfriamiento, una sonda/controlador de temperatura J-Ke de adición y una entrada/salida de nitrógeno. El recipient de hidruro de sodio en aceite mineral (26,04 g, 0,6510 dimetilformamida (200 mL). Se inició la agitación y se embudo de adición se cargó con una solución de benceno (868 ml, ~8,5 ml/g, 0,75 M), requiriendo un poco de cale solución resultante de color amarillo pálido claro de bence 1 hora al matraz de fondo redondo, lo que dio como resul gas. Una vez completada la adición, se registró la temp recipiente con una manta calefactora y la mezcla grisáce 60°C durante 1 hora, momento en donde pareció haber mezcla mientras se dejaba enfriar la mezcla a temperatur 3 bocas de 1000 ml se equipó con un agitador mecánico, u J-Kem, un condensador de reflujo enfriado por agua y un atmósfera de nitrógeno con ácido 2,6-dicloropiridin-3-carb 5 mL/g) que proporcionó una solución transparente de temperatura del recipiente a 17°C. Después, el recipie añadido como un sólido en porciones durante 10 minutos, y desprendimiento de gas, no se observó exotermia. transparente resultante a temperatura ambiente dura bencenosulfonamida previamente formada en N,N-dimeti solución ámbar transparente de 2,6-dicloropiridin-3-il) (1H recipiente se equipó con una manta calefactora y la mez hora cuando el análisis por CL/EM indicó el consumo temperatura ambiente y luego se vertió en una solución d adicionalmente con agua (500 mL) y luego se transfirió a (1000 mL). Se eliminó la capa orgánica y se eliminó el ac orgánicas combinadas se lavaron con una solución satu sulfato de sodio (300 g) y luego se filtraron a través de un de color amarillo pálido se concentró a presión reducida residual transparente se diluyó con éter ferc-butílico de reducida, tiempo durante el cual comenzó a precipitar un suspensión resultante se dejó reposar a temperatura a embudo Buchner de frita de vidrio. La torta de filtración s x 150 mL) y luego se introdujo en el embudo Buchner du horno de vacío a 45°C durante 2 horas para proporciona como el producto deseado, N-(bencenosulfonil)-2,6-dicl encontrado 330,9 (M+1)+; Tiempo de retención: 1,22 minu
Paso B: W-(bencenosulfoml)-6-cloro-2-[(4S)-2,2,4-tr¡m
[0431]
as de 5000 mL con un agitador mecánico, un baño de un condensador de reflujo enfriado por agua, un embudo e cargó en una atmósfera de nitrógeno con 60% en peso l). Después, el recipiente se cargó lentamente con N,N-istró la temperatura del recipiente a 19°C. Después, el lfonamida (102,3 g, 0,6510 mol) en N,N-dimetilformamida miento suave para obtener una solución homogénea. La ulfonamida se añadió posteriormente gota a gota durante o una ligera formación de espuma y desprendimiento de tura del recipiente a 28°C. A continuación, se equipó el e calentó a 60°C. Se continuó la agitación de la mezcla a ado el desprendimiento de gas. Se continuó agitando la mbiente. Mientras tanto, un matraz de fondo redondo de manta calefactora, una sonda/controlador de temperatura ntrada/salida de nitrógeno. El recipiente se cargó en una lico (100 g, 0,5208 mol) y N,N-dimetilformamida (500 mL, lor amarillo claro. Se inició la agitación y se registró la se cargó con carbonildiimidazol (84,45 g, 0,5208 mol) que dio como resultado una ligera formación de espuma continuó agitando la solución de color ámbar claro 1 hora. El matraz que contenía la sal sódica de rmamida se trató gota a gota durante 45 minutos con la idazol-1-il)metanona. Después de la adición completa, el se calentó a 60°C y la condición se mantuvo durante 1 ompleto del intermedio. La reacción se dejó enfriar a M HCl helada (500 mL). La mezcla resultante se diluyó embudo de decantación y se repartió con acetato de etilo a se extrajo con acetato de etilo (2 X 500 mL). Las capas a de cloruro de sodio (3 X 500 mL), se secaron sobre budo Buchner de frita de vidrio. La solución transparente sta un volumen de aproximadamente 200 mL. El aceite tilo (1000 mL) y luego se concentró de nuevo a presión do. El volumen se redujo a aproximadamente 200 mL. La nte durante 30 minutos y luego se filtró a través de un vó por desplazamiento con éter ferc-butílico de metilo (2 te 30 minutos. El material se secó adicionalmente en un n sólido blanco (101 g, 0,305 mol, rendimiento del 58%) piridina-3-carboxamida. IEN-EM m/z calc. 329,96326, .
p¡rrol¡dm-1-¡l]pmdm-3-carboxam¡da

[0432] Un matraz RB de 3 bocas de 5000 mL estaba equ sonda/controlador de temperatura J-Kem, un condensa nitrógeno. El recipiente se cargó bajo una atmósfera carboxamida (100 g, 0,3020 mol), clorhidrato de (4S)-2,2 (500 ml, 5 mL/g) que proporcionó una solución transparen la temperatura del recipiente a 19°C. A continuación, se c 1,208 mol, 325 mesh) añadido como un sólido en por desprendimiento de gas menor y formación de espuma. L ambiente durante 10 minutos y luego se calentó a una te durante 24 horas. El análisis por CL/EM indicó la finaliza enfriar a temperatura ambiente. Se equipó un matraz RB de enfriamiento y una sonda de temperatura J-Kem. El r inició la agitación a una velocidad vigorosa. El baño de en do con un agitador mecánico, una manta calefactora, una r de reflujo enfriado por agua y una entrada/salida de nitrógeno con N-(bencenosulfonil)-2,6-dicloro-piridin-3-trimetilpirrolidina (54,24 g, 0,3624 mol) y dimetilsulfóxido de color amarillo pálido. Se inició la agitación y se registró ó el recipiente con polvo de carbonato de potasio (167 g, es durante 10 minutos, lo que dio como resultado un uspensión blanquecina resultante se agitó a temperatura ratura del recipiente de 115°C y se mantuvo la condición n de la reacción y la suspensión de color ámbar se dejó 3 bocas de 5000 mL con un agitador mecánico, un baño ipiente se cargó con 2 M HCl (1057 ml, 2,114 mol) y se miento se cargó con hielo triturado/agua y la temperatura
del recipiente se redujo a 0°C. La mezcla de reacción lentamente en porciones durante 30 minutos, lo que dio c a 8°C. Nota: Formación de espuma suave tras la adició suspensión resultante a ~5°C durante 1 hora y luego se r frita de vidrio. La torta del filtro se lavó por desplazamient en el embudo Buchner para proporcionar un sólido blanc con un agitador mecánico, una manta calefactora, una so reflujo enfriado por agua y una entrada/salida de nitrógen el producto aislado (150 g) y 2-propanol (1050 ml, 7 mL/ Se inició la agitación y se registró la temperatura del reci reflujo (~ 82°C) y la condición se mantuvo durante 10 min pálido transparente. Se continuó la agitación de la sol temperatura ambiente, tiempo durante el cual comenzó a f y se equipó el recipiente con un baño de enfriamiento qu del recipiente a 0°C y se continuó agitando la suspensión filtración al vacío en un embudo Buchner de frita de vidrio enfriado con hielo (2 x 50 mL) y luego se introdujo adicionalmente en un horno de vacío a 45°C durante 15 rendimiento del 81%) como el producto, N-(bence carboxamida como solvato de 2-propanol con 11% en pe 408,1 (M+1)+; Tiempo de retención: 1,9 minutos.
Paso C: 5-Fluoro-3-[2-[1-(trifluorometil)ciclopropil]eto
[0433]
suspensión de color ámbar se añadió posteriormente resultado la precipitación de un sólido y una exotermia na vez completada la adición, se continuó agitando la ió mediante filtración al vacío en un embudo Buchner de n agua (4 X 500 mL) y luego se extrajo durante 2 horas 50 g). Se equipó un matraz RB de 3 bocas de 5000 mL /controlador de temperatura J-Kem, un condensador de l recipiente se cargó en una atmósfera de nitrógeno con ue proporcionó una suspensión de color amarillo pálido.
te a 19°C. La temperatura del recipiente se aumentó a , lo que dio como resultado una solución de color ámbar n y se dejó que la solución se enfriara lentamente a arse un sólido. Se continuó la agitación de la suspensión cargó con hielo triturado/agua. Se bajó la temperatura sa a 0°C durante 1 hora. El material se recogió mediante torta del filtro se lavó por desplazamiento con 2-propanol el Buchner durante 30 minutos. El material se secó s para proporcionar un sólido blanco (100 g, 0,245 mol, lfonil)-6-cloro-2-[(4S)-2,2,4-trimetilpirrolidin-1-il]piridin-3-e 2-propanol. IEN-EM m/z calc. 407,10703, encontrado
H-pirazol

[0434] Una solución de 3-[2-[1-(trifluorometil)ciclopropil]et 1,4-diazoniabiciclo[2.2.2]octano; ditetrafluoroborato (1,3 17 horas. La reacción se diluyó con agua y se extrajo co agua, se secaron sobre sulfato de sodio y se evaporaron de gel de sílice eluyendo con un gradiente de acetato de e como un aceite marrón que se purificó adicionalmente u columna Luna C18 (2) (75 X 30 mm, (tamaño de partícul una corrida de gradiente dual desde 1-99% de fase móvi Fase móvil B = CH3CN Velocidad de flujo = 50 ml/min y (trifluorometil)ciclopropil]etoxi]-1H-pirazol (90 mg) como encontrado 239,1 (M+1)+; Tiempo de retención: 0,56 mi 1H), 4,30-4,16 (m, 2H), 2.04 (t, J = 7,1 Hz, 2H), 0,97 -0,
Paso D: W-(bencenosulfoml)-6-[5-fluoro-3-[2-[1-( trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0435]
1H-pirazol (0,68 g, 3,088 mmol) y 1-(clorometil)-4-fluoro-7 mmol) en acetonitrilo (15 mL) se agitó a 50°C durante etato de etilo. Los extractos combinados se lavaron con residuo se purificó mediante cromatografía en columna l 0-30% en hexanos para dar el producto todavía impuro o un método de HPCL-EM de fase inversa usando una pm) vendida por Phenomenex (pn: 00C-4252-U0-AX), y durante 15,0 minutos (Fase móvil A = H2O (HCl 5 mM). peratura de la columna = 25°C) dando 5-fluoro-3-[2-[1-aceite de color canela. IEN-EM m/z calc. 238,07292, . 1H RMN (400 MHz, DMSO-cfe) 57,70 (d, J = 4,5 Hz, m, 2H), 0,88 -0,81 (m, 2H).
orometN)ciclopropN]etoxi]pirazoM-N]-2-[(4S)-2,2,4-

[0436] Una mezcla de 5-fluoro-3-[2-[1-(trifluorometil) (bencenosulfonil)-6-cloro-2-[(4,S)-2,2,4-trimetilpirrolidin-1-escandio (10 mg, 0,020 mmol) e hidruro de sodio (38 mg durante 15 horas. La reacción se filtró y purificó usando u Luna C18 (2) (75 X 30 mm, tamaño de partícula de 5 pm) propil]etoxi]-1H-pirazol (87 mg, 0,3653 mmol), N-idin-3-carboxamida (79 mg, 0,19 mmol), triflato de % p/p, 0,95 mmol) en DMSO (0,92 mL) se agitó a 160°C todo de HPCL-EM de fase inversa usando una columna dida por Phenomenex (pn: 00C-4252-U0-AX), y un ciclo
de gradiente dual desde 50-99% de la fase móvil B dura = CH3CN. Velocidad de flujo = 50 ml/min y temperatura 3-[2-[1-(trifluorometil)ciclopropil]etoxi]pirazol-1-il]-2-[(4S) 10%). IEN-EM m/z calc. 609,2033, encontrado 610,3 ( DMSO-cfe) 8 12,53 (s, 1H), 8,26 (d, J = 4,5 Hz, 1H), 8,0 Hz, 1H), 7,73 (d, J = 7,4 Hz, 1H), 7,66 (dd, J = 8,2, 6,7 Hz, 1H), 2,29 -2,21 (m, 1H), 2,10 (q, J = 7,0 Hz, 3H), 1 (s, 1H), 0,96 (dd, J = 3,7, 2,4 Hz, 2H), 0,89 (dt, J = 3,7,
Ejemplo sintético 33: Síntesis del Co tetrametNddopropN)metoxi]pirazoM-M]-2-[(4S)-2,2,4-
Paso A: 2-Cloro-N-(4-hidroxifeml)sulfoml-6-[3-[( carboxamida
[0437]
minutos (fase móvil A = H2O (HCl 5 mM). Fase móvil B columna = 25°C) dando W-(toencenosulfoml)-6-[5-fluoro--trimetilpirrolidin-1-il]piridin-3-carboxamida (11 mg, Tiempo de retención: 2,3 minutos. 1H RMN (400 MHz, = 1,3 Hz, 1H), 7,98 (d, J = 1,5 Hz, 1H), 7,82 (d, J = 8,3 ), 6,90 (d, J = 8,3 Hz, 1H), 4,41 (s, 2H), 2,38 (d, J = 10,5 d, J = 12,0, 5,5 Hz, 1H), 1,52 (s, 3H), 1,50 (s, 3H), 1,36 , 2H), 0,64 (d, J = 6,3 Hz, 3H).
sto 33: N-(4-hidroxifeml)sulfoml-6-[3-[(2,2,3,3-tMpirroNdm-1-N]piridm-3-carboxamida
3-tetrametMddopropMo)metoxi]pirazoM-M]piridm-3-

[0438] Ácido 2-Cloro-6-[3-[(2,2,3,3-tetrametilciclopropil) y CDI (83 mg, 0,5119 mmol) se combinaron en THF (75 Se añadió 4-hidroxibencenosulfonamida (86 mg, 0,4966 agitó durante 16 h más a temperatura ambiente. La me extrajo 3 x 10 ml de acetato de etilo. Los extractos salmuera, se secaron sobre sulfato de sodio y se conce paso sin purificación adicional. 2-cloro-N-(4-hidroxif il]piridin-3-carboxamida (235 mg, 94%) IEN-Em m/z cal 0,75 minutos.
Paso 2: N-(4-Hidroxifenil)sulfonil-6-[3-[(2,2 trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0439]
i]pirazol-1-il]piridin-3-carboxílico (150 mg, 0,4288 mmol) y se agitaron a temperatura ambiente durante 2 horas. l) seguido de DBU (90 ml, 0,6018 mmol) y la reacción se e reacción se diluyó con 10 ml de ácido cítrico 1 M y se cos combinados se lavaron con agua, se lavaron con n para dar un sólido blanco, que se usó en el siguiente lfonil-6-[3-[(2,2,3,3-tetrametilciclopropil)metoxi]pirazol-1-,1234, encontrado 505,2 (M+1)+; Tiempo de retención:
etrametilcidopropil)metoxi]pirazol-1-il]-2-[(4S)-2,2,4-
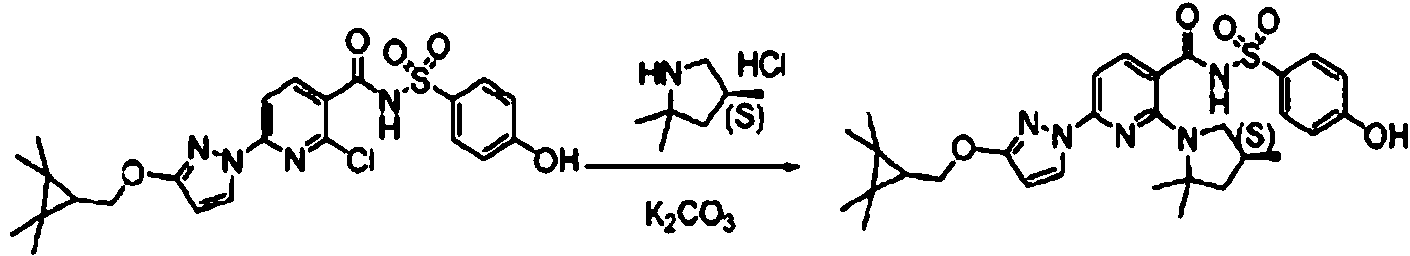
[0440] 2-Cloro-N-(4-hidroxifenil)sulfonil-6-[3-[(2,2,3,3-t (235 mg, 0,4654 mmol), (4S)-2,2,4-trimetilpirrolidina (sa carbonato de potasio (aproximadamente 387,4 mg, 2,80 a 130°C durante 16 h. La reacción se enfrió a temperatur de agitación, se dejó sedimentar el contenido del vial, s sólidos restantes con 20 ml de acetato de etilo, luego se acuosa y orgánica y la capa acuosa se extrajo dos v combinaron, se lavaron con salmuera, se secaron sobr purificó mediante cromatografía en gel de sílice eluye hidroxifenil)sulfonil-6-[3-[(2,2,3,3-tetrametilciclopropil)m carboxamida (30 mg, 11%) IEN-EM m/z calc. 581,2672, 1H RMN (400 MHz, DMSO) 812,24 (s, 1H), 10,58 (s, 1H Hz, 1H), 6,97 - 6,92 (m, 2H), 6,90 (d, J = 8,3 Hz, 1H), 6,1 Hz, 1H), 2,26 (t, J = 9,0 Hz, 1H), 2,10 (dt, J = 13,1, 6,8 3H), 1,37 (t, J = 12,1 Hz, 1H), 1,10 (s, 6H), 1,04 (s, 6H),
Ejemplo sintético 34: Síntesis del
tilciclopropil)metoxi]pirazol-1-il]piridin-3-carboxamida
lorhidrato) (aproximadamente 209,2 mg, 1,398 mmol) y ol) se combinaron en DMSO (775,7 mL) y se calentaron biente y se añadió 1 ml de agua. Después de 15 minutos inó la porción líquida con una pipeta y se disolvieron los n con 15 ml de ácido cítrico 1M. Se separaron las capas más con 15 ml de acetato de etilo. Los orgánicos se ato de sodio y se concentraron. El sólido resultante se on metanol al 0-10% en diclorometano para dar N-(4-irazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-il]piridin-3-trado 582,3 (m 1)+; Tiempo de retención: 2,26 minutos.
8 (d, J = 2,7 Hz, 1H), 7,86 -7,78 (m, 2H), 7,74 (d, J = 8,2 J = 2,6 Hz, 1H), 4,23 (d, J = 7,7 Hz, 2H), 2,43 (t, J = 10,4 ), 1,82 (dd, J = 11,9, 5,4 Hz, 1H), 1,54 (s, 3H), 1,51 (s, (t, J = 7,7 Hz, 1H), 0,69 (d, J = 6,2 Hz, 3H).
uesto 34: N-(2-hidroxifenil)sulfonil-6-[3-[2-[1-(trifluorom etil)ciclopropil]etoxi]pirazol-1 -il]-2-[(4S)-2 Paso A: 2-Cloro-N-(2-hidroxifeml)sulfoml-6-[3-carboxamida
[0441]
rimetilpirrolidin-1-il]piridin-3-carboxamida
(trifluorometN)cidopropN]etoxi]pirazoM-N]piridm-3-

[0442] Ácido 2-doro-6-[3-[2-[1-(trifluorometil)cidopropil] y CDI (51 mg, 0,3145 mmol) se combinaron en THF (60 Se añadió 2-hidroxibencenosulfonamida (51 mg, 0,2945 agitó durante 16 horas más a temperatura ambiente. La se extrajo 3 x 10 ml de acetato de etilo. Los orgánicos c sulfato de sodio y se concentraron para dar un sólido bla 2-cloro-N-(2-hidroxifenil)sulfonil-6-[3-[2-[1-(trifluorometil) 93%) ESI -MS m/z calc. 530,06384, encontrado 531,1 (
Paso B: N-(2-hidroxifeml)sulfoml-6-[3-[2-[1 trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0443]
irazol-1-il]piridina-3-carboxílico (100 mg, 0,2661 mmol) ) y se agitaron a temperatura ambiente durante 2 horas. l) seguido de DBU (55 ml, 0,3678 mmol) y la reacción se la de reacción se diluyó con 10 ml de ácido cítrico 1 M y ados se lavaron con agua, salmuera, se secaron sobre ue se usó en el siguiente paso sin purificación adicional. ropil]etoxi]pirazol-1-il]piridin-3-carboxamida (132 mg, ; Tiempo de retención: 0,7 minutos.
orometN)ddopropN]etoxi]pirazoM-N]-2-[(4S)-2,2,4-
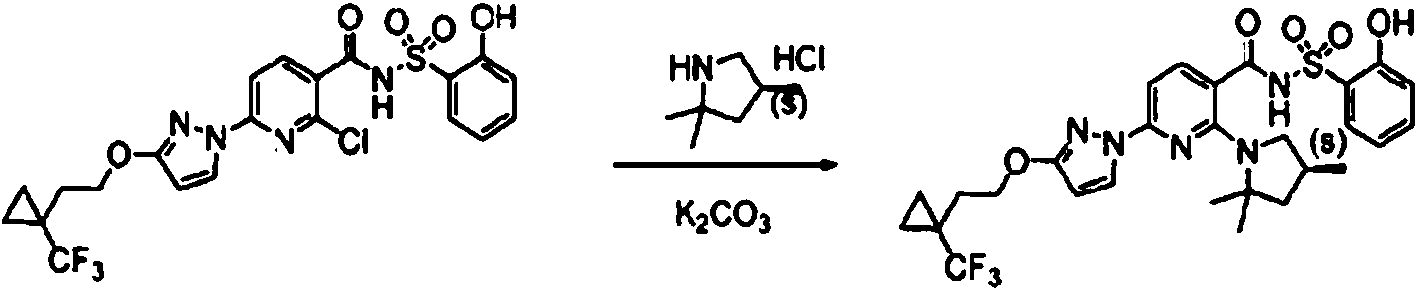
[0444] 2-Cloro-N-(2-hidroxifenil)sulfonil-6-[3-[2-[1-(trifluo mg, 0,2486 mmol), (4S)-2,2,4-trimetilpirrolidina (sal de hi mg, 2,496 mmol) se combinaron en DMSO (660,0 pL) y temperatura ambiente y se añadió 1 ml de agua. Despué del vial, se eliminó la porción líquida con una pipeta y se luego se lavaron con 15 ml de ácido cítrico 1M. Se separ dos veces más con 15 ml de acetato de etilo. Los org sobre sulfato de sodio y se concentraron. El sólido res gel de sílice eluyendo con metanol al 0-10% en (trifluorometil)ciclopropiletoxilpirazol-1 -il-2-[(4S)-2,2,4-tri m/z calc. 607,20764, encontrado 608,3 (M+1)+; Tiempo (s, 1H), 10,89 (s, 1H), 8,20 (d, J = 2,8 Hz, 1H), 7,79 (dd, 6,88 (d, J = 8,2 Hz, 1H), 6,11 (d, J = 2,7 Hz, 1H), 4,31 (t, 1H), 2,19 (d, J = 11,0 Hz, 1H), 2,08 (t, J = 7,0 Hz, 2H), 1 J = 12,1 Hz, 1H), 1,00 - 0,92 (m, 2H), 0,90 (d, J = 10,8
Ejemplo sintético 35: Síntesis del
(trifluorom etil)ciclopropil]etoxi]pirazol-1 -il]-2-[(4S)-2
Paso A: 2-Cloro-N-(3-hidroxifenil)sulfonil-6-[3 carboxamida
[0445]
til)ciclopropil]etoxi]pirazol-1-il]piridin-3-carboxamida (132 ruro) (190 mg, 1,270 mmol) y carbonato de potasio (345 alentaron a 130°C durante 16 h. La reacción se enfrió a 15 minutos de agitación, se dejó sedimentar el contenido ieron los sólidos restantes con 20 ml de acetato de etilo, las capas acuosa y orgánica y la capa acuosa se extrajo se combinaron, se lavaron con salmuera, se secaron se purificó adicionalmente mediante cromatografía en ometano para dar N-(2-hidroxifenil)sulfonil-6-[3-[2-[1-irrolidin-1 -il]piridin-3-carboxamida (51 mg, 31%) Ie N-Em nción: 2,14 minutos 1H RMN (400 MHz, DMSO) 812,41 ,1, 2,1 Hz, 2H), 7,56 - 7,43 (m, 1H), 7,07 - 6,95 (m, 2H), ,1 Hz, 2H), 2,64 (d, J = 8,1 Hz, 1H), 2,58 (d, J = 10,6 Hz, d, J = 11,9, 5,6 Hz, 1H), 1,54 (d, J = 8,1 Hz, 6H), 1,39 (t, ), 0,82 (d, J = 6,3 Hz, 3H).
uesto 35: N-(3-hidroxifenil)sulfonil-6-[3-[2-[1-rimetilpirrolidin-1-il]piridin-3-carboxamida
-(trifluorometil)ciclopropil]etoxi]pirazol-1-il]piridin-3- 
[0446] Ácido 2-Cloro-6-[3-[2-[1-(trifluorometil)ciclopropil] y CDI (51 mg, 0,3145 mmol) se combinaron en THF (6o0 Se añadió 3-hidroxibencenosulfonamida (51 mg, 0,2945 agitó durante 16 horas más a temperatura ambiente. La se extrajo con 3 x 10 ml de acetato de etilo. Los orgáni sobre sulfato de sodio y se concentraron para dar un s adicional. 2-cloroN-(3-hidroxifenil)sulfonil-6-[3-[2-[1-(t (135 mg, 96%) IEN-EM m/z calc. 530,06384, encontrado Paso B: N-(3-Hidroxifenil)sulfonil-6-[3-[2-[ trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0447]
irazol-1-il]piridina-3-carboxílico (100 mg, 0,2661 mmol) ) y se agitaron a temperatura ambiente durante 2 horas. ) seguido de DBU (55 ml, 0,3678 mmol) y la reacción se la de reacción se diluyó con 10 ml de ácido cítrico 1 M y ombinados se lavaron con agua, salmuera, se secaron lanco, que se usó en el siguiente paso sin purificación ometil)ciclopropil]etoxi]pirazol-1-il]piridin-3-carboxamida 2 (M+1)+; Tiempo de retención: 0,69 minutos.
uorometil)ciclopropil]etoxi]pirazol-1-il]-2-[(4S)-2,2,4-

[0448] 2-Cloro-N-(3-hidroxifenil)sulfonil-6-[3-[2-[1-(trifluo mg, 0,2543 mmol), (4S)-2,2,4-trimetilpirrolidina (sal de cl mg, 2,547 mmol) se combinaron en DMSO (508,6 j L) y temperatura ambiente y se añadió 1 ml de agua. Despué del vial y la porción líquida se retiró con una pipeta y s acetato de etilo y luego se lavaron con 15 ml de ácido cít acuosa se extrajo dos veces más con 15 ml de acetato d se secaron sobre sulfato de sodio y se concentraron. El sílice eluyendo con metanol al 0-10% en dic (trifluorometil)ciclopropil]etoxi]pirazol-1-il]-2-[(4S)-2,2,4-tr EM m/z calc. 607,20764, encontrado 608,3 (m 1) 1; Ti 8 12,44 (s, 1H), 10,19 (s, 1H), 8,20 (d, J = 2,8 Hz, 1H), 7, 2H), 7,06 (d, J = 7,9 Hz, 1H), 6,91 (d, J = 8,2 Hz, 1H), 10,0 Hz, 1H), 2,33 (s, 2H), 2,08 (m, J = 8,1, 7,0 Hz, 2H), (t, J = 12,1 Hz, 1H), 0,96 (td, J = 5,0, 3,3 Hz, 2H), 0,90 (
Ejemplo sintético 36: Síntesis del Compu tetrametMcidopropM)metoxi]pirazoM-M]-2-[(4S)-2,2,4-
Paso A: Dideuterio-(2,2,3,3-tetrametilciclopropil)met
[0449]
il)ciclopropil]etoxi]pirazol-1-il]piridin-3-carboxamida (135 ato) (193 mg, 1,290 mmol) y carbonato de potasio (352 lentaron a 130°C durante 16 h. La reacción se enfrió a 5 minutos de agitación, se dejó sedimentar el contenido echó. Los sólidos restantes se disolvieron en 20 ml de M. Se separaron las capas acuosa y orgánica y la capa Los orgánicos se combinaron, se lavaron con salmuera, resultante se purificó mediante cromatografía en gel de etano para dar N-(3-hidroxifenil)sulfonil-6-[3-[2-[1-pirrolidin-1-il]piridin-3-carboxamida (40 mg, 26%) IEN-de retención: 2,05 minutos. 1H RMN (400 MHz, DMSO) J = 8,2 Hz, 1H), 7,44 (t, J = 8,0 Hz, 1H), 7,40 -7,37 (m, d, J = 2,7 Hz, 1H), 4,31 (t, J = 7,1 Hz, 2H), 2,47 (d, J = dd, J = 11,8, 5,5 Hz, 1H), 1,54 (s, 3H), 1,52 (s, 3H), 1,38 11,1 Hz, 2H), 0,70 (d, J = 6,2 Hz, 3H).
36: N-(bencenosulfonil)-6-[3-[dideuterio-(2,2,3,3-MpirroMdm-1-M]piridm-3-carboxamida

[0450] Se disolvió ácido 2,2,3,3-Tetrametilciclopropano un matraz de fondo redondo de 100 ml purgado con n tetradeuterioalumanuido sólido (sal de litio) (420 mg, 10, xílico (1,077 g, 7,574 mmol) en éter dietílico anhidro en no. La mezcla de reacción se enfrió a 0°C. Se añadió mol) en 3 porciones. Se dejó que la mezcla de reacción
alcanzara gradualmente la temperatura ambiente y se a de reacción se enfrió de nuevo a 0°C. Se añadió gota a g fase acuosa se extrajo con éter dietílico (2 x 30 ml). Las f seguido de salmuera, luego se secaron sobre sulfato de s tetrametilciclopropil)metanol (920 mg, 93%). 1H RMN (40 (s, 1H).
Paso B: 3-[Dideuterio-(2,2,3,3-tetrametNcidopropN)m
[0451]
durante un total de 16 horas. A continuación, la mezcla HCl (acuoso, 0,2 N, 5 mL), seguido de 20 ml de agua. La orgánicas combinadas se lavaron con NaHCO3 acuoso, , se filtraron y se evaporaron para dar dideuterio-(2,2,3,3-z, DMSO) 8 4,11 (s, 1H), 1,04 (s, 6 H), 0,93 (s, 6 H), 0,37
i]pirazoM-carboxMato de terc-butilo

[0452] Se añadió gota a gota DIAD (1,4 ml, 7,111 mmol) ml de tolueno anhidro, a 0°C. Después de 30 minutos a butilo (1,155 g, 6,271 mmol) y dideuterio-(2,2,3,3-tetra tolueno se añadió lentamente con una jeringa. La reacci luego se calentó a 55°C durante 18 h. La mezcla se evap (30 mL) e hidróxido de sodio 1 N (30 ml). Los orgánicos sobre sulfato de sodio y se evaporaron. El material crudo con acetato de etilo al 0-30% en hexanos para dar un acei ferc-butilo 3-[dideuterio-(2,2,3,3-tetramelilciclopropilo)me 296,2069, encontrado 297,2 (M+1)+; Tiempo de retención
Paso C: 3-[Dideuterio-(2,2,3,3-tetrametilciclopropil)m
[0453]
a solución de trifenilfosfina (1,815 g, 6,920 mmol) en 40 , una solución de 3-hidroxipirazol-1-carboxilato de tercciclopropil)metanol (980 mg, 7,525 mmol) en 30 ml de e calentó a temperatura ambiente durante 45 minutos y y el material resultante se repartió entre acetato de etilo epararon, se lavaron con salmuera (30 mL), se secaron urificó mediante cromatografía en gel de sílice eluyendo ue finalmente solidificó en un sólido ligeramente amarillo: pirazol-1-carboxilato (820 mg, 44%) IEN-EM m/z calc.
9 minutos.
i]-1H-pirazol

[0454] A 3-[dideuterio-(2,2,3,3-tetrametilciclopropilo)meto en 1,2-dimetoxietano (10 mL) se añadió carbonato de s reacción se calentó hasta 90°C durante 16 horas en un temperatura ambiente y se diluyó con agua (50 mL) y ac acuosa se extrajo 2 X 25 ml de acetato de etilo. Los ext secaron sobre sulfato de sodio, luego se concentr tetrametilciclopropil)metoxi]-1H-pirazol (492 mg, 93%) IE de retención: 0,57 minutos, 1H RMN (400 MHz, DMSO) 1H), 1,08 (s, 6 H), 1,00 (s, 6 H), 0,66 (s, 1H).
Paso D: 2-cloro-6-[3-[dideuterio-(2,2,3,3-tetrametilcicl
[0455]
irazol-1-carboxilato de ferc-butilo (800 mg, 2,699 mmol) (460 mg, 4,340 mmol) en agua (3 mL) y la mezcla de con tapón de rosca. La mezcla de reacción se enfrió a de etilo (50 mL). Los orgánicos se separaron y la capa s orgánicos combinados se lavaron con salmuera y se para dar un aceite incoloro. 3-[dideuterio-(2,2,3,3-m/z calc. 196,15446, encontrado 197,1 (M+1)+; Tiempo ,78 (s, 1H), 7,48 (t, J = 2,1 Hz, 1H), 5,65 (t, J = 2,3 Hz,
opil)metoxi]pirazol-1-il]piridin-3-carboxilato de etilo

[0456] El matraz de fondo redondo se cargó bajo nitróge pirazol (485 mg, 2,471 mmol), 2,6-dicloropiridin-3-carbox mmol) (recién molido en un mortero) y DMF anhidro (4,12 se agitó a temperatura ambiente bajo nitrógeno durante 1 (50 mL) y agua (50 mL) y se separaron las dos fases. La on 3-[dideuterio-(2,2,3,3-tetrametilciclopropil)metoxi]- 1H-de etilo (545 mg, 2,477 mmol), K2CO3 (513 mg, 3,712 L). Se añadió DABCO (50 mg, 0,4457 mmol) y la mezcla ras. La mezcla de reacción se diluyó con acetato de etilo e acuosa se extrajo adicionalmente con acetato de etilo
(2 x 30 mL) y los extractos combinados se lavaron con cual se eliminó el disolvente a presión reducida. El materi un gradiente de acetato de etilo al 0-20% en hexanos eliminaron a presión reducida para proporcio tetrametilciclopropil)metoxi]pirazol-1-il]piridin-3-carboxila encontrado 380,1 (M+1)+; Tiempo de retención: 0,9 min 8,39 (d, J = 8,5 Hz, 1H), 7,76 (d, J = 8,4 Hz, 1H), 6,24 (d 3H), 1,11 (s, 6H), 1,04 (s, 6H), 0,74 (s, 1H).
Paso E: Ácido 2-doro-6-[3-[dideuterio-(2,2,3,3-tetra
[0457]
era y se secaron sobre sulfato de sodio, después de lo sometió a cromatografía flash sobre gel de sílice usando fracciones puras se combinaron y los disolventes se un sólido blanco; 2-cloro-6-[3-[dideuterio-(2,2,3,3-etilo (505 mg, 54%) IEN-EM m/z calc. 379,16318, 1H RMN (400 MHz, DMSO) 88,42 (d, J = 2,9 Hz, 1H), 2,9 Hz, 1H), 4,34 (q, J = 7,1 Hz, 2H), 1,33 (t, J = 7,1 Hz,
ddopropN)metoxi]pirazoM-N]piridm-3-carboxnico

[0458] Se añadió una solución de sodio hidróxido (275 cloro-6-[3-[dideuterio-(2,2,3,3-tetrametilciclopropil)metox isopropanol (2,500 mL) y se agitó a 90°C durante 45 mi se diluyó con 50 ml de acetato de etilo, 20 ml de ácido porción acuosa se extrajo 2 x 25 ml de acetato de etilo.
se secaron sobre sulfato de sodio y se concentraron. brevemente y se recogió por filtración y luego se secó pa tetrametilciclopropil)metoxi]pirazol-1-il]piridin-3-carboxílic 352,1 (M+1)+; Tiempo de retención: 0,77 minutos. 1H R 8,4 Hz, 1H), 7,74 (d, J = 8,4 Hz, 1H), 6,22 (d, J = 2,8 Hz,
Paso F: N-(Bencenosulfoml)-2-doro-6-[3-[dideuteriocarboxamida
[0459]
6,875 mmol) en agua (2,500 mL) a una solución de 2-zol-1-il]piridin-3-carboxilato (500 mg, 1,316 mmol) en . La reacción se enfrió a temperatura ambiente y luego 1 M y 10 ml de agua. Los orgánicos se separaron y la xtractos orgánicos combinados se lavaron con salmuera, lido resultante se trituró en 40 ml de agua, se sonicó r un sólido blanco, ácido 2-cloro-6-[3-[dideuterio-(2,2,3,3-04 mg, 87%) IEN-EM m/z calc. 351,13187, encontrado 00 MHz, DMSO) 88,41 (d, J = 2,9 Hz, 1H), 8,38 (d, J = 1,11 (s, 6H), 1,04 (s, 6H), 0,74 (s, 1H).
3,3-tetrametMddopropM)metoxi]pirazoM-M]piridm-3-

[0460] Ácido 2-cloro-6-[3-[dideuteno-(2,2,3,3-tetrametil 0,2842 mmol) y CDI (55 mg, 0,3392 mmol) se combinar durante 2 horas. Se añadió bencenosulfonamida (49 m reacción se agitó durante 16 horas más a temperatura a cítrico 1 M y se extrajo 3 x 10 ml de acetato de etilo. salmuera, se secaron sobre sulfato de sodio y se concent que se usó en el siguiente paso sin purificación a tetrametilciclopropil)metoxi]pirazol-1-il]piridin-3-carboxa Tiempo de retención: 0,81 minutos.
Paso G: N-(bencenosulfonil)-6-[3-[dideuterio-(2,2 trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0461]
ropil)metoxi]pirazol-1-il]piridin-3-carboxílico (100 mg, n THF (600,0 j L) y se agitaron a temperatura ambiente 117 mmol) seguido de DBU (57 ml, 0,3812 mmol) y la nte. La mezcla de reacción se diluyó con 10 ml de ácido extractos orgánicos combinados se lavaron con agua, para dar aproximadamente 135 mg de un sólido blanco, al. N-(bencenosulfonil)-2-cloro-6-[3-[dideuterio-(2,2,3,3-IEN-EM m/z calc. 490,14105, encontrado 491,2 (M+1)+;
tetrametilcidopropil)metoxi]pirazol-1-il]-2-[(4S)-2,2,4- 
[0462] N-(bencenosulfonil)-2-doro-6-[3-[dideuterio-(2,2, carboxamida (135 mg, 0,2749 mmol), (4S)-2,2,4-trim carbonato de potasio (237 mg, 1,715 mmol) se combina h. La reacción se enfrió a temperatura ambiente y se a dejó sedimentar el contenido del vial, la porción líquida s con 20 ml de acetato de etilo y luego se lavaron con orgánica y la capa acuosa se extrajo dos veces más co lavaron con salmuera, se secaron sobre sulfato de adicionalmente mediante cromatografía en gel de sílice (bencenosulfonil)-6-[3-[dideuterio-(2,2,3,3-tetrametilciclo il]piridin-3-carboxamida (112 mg, 72%) IEN-EM m/z calc 2,42 minutos. 1H RMN (400 MHz, DMSO) 8 12,51 (s, 1 (d, J = 8,3 Hz, 1H), 7,76 - 7,70 (m, 1H), 7,70 - 7,62 (m, J = 10,4 Hz, 1H), 2,26 (t, J = 8,7 Hz, 1H), 2,09 (dq, J = 9,5 Hz, 6 H), 1,36 (t, J = 12,2 Hz, 1H), 1,10 (s, 6 H), 1,04
Ejemplo sintético 37: Síntesis del (trifluorometM)cidopropM]metoximetM]pirazoM-M]-2-[(
Paso A: 3-(terc-butoximetil)-1H-pirazol
[0463]
trametilcidopropil)metoxi]pirazol-1-il]piridin-3-olidina (sal de clorhidrato) (128 mg, 0,8553 mmol) y n DMSO (458,2 j L) y se calentaron a 130°C durante l 6 1 ml de agua. Después de 15 minutos de agitación, se inó con una pipeta y los sólidos restantes se disolvieron de ácido cítrico 1M. Se separaron las capas acuosa y l de acetato de etilo. Los orgánicos se combinaron, se y se concentraron. El sólido resultante se purificó ndo con metanol al 0-10% en diclorometano para dar N-)metoxi]pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-,28485, encontrado 568,3 (M+1)+; Tiempo de retención: 8 (d, J = 2,8 Hz, 1H), 7,99 (dt, J = 7,1, 1,4 Hz, 2H), 7,80 ,92 (d, J = 8,3 Hz, 1H), 6,13 (d, J = 2,7 Hz, 1H), 2,40 (t, ,0 Hz, 1H), 1,82 (dd, J = 11,8, 5,4 Hz, 1H), 1,52 (d, J = ), 0,72 (s, 1H), 0,64 (d, J = 6,2 Hz, 3H).
ompuesto 37: N-(bencenosulfonil)-6-[3-[[1-,2,4-trimetMpirroMdm-1-M]piridma-3-carboxamida

[0464] aAcohol terc-Butilpropargil (2,5 g, 22,2 mmol) s 11,1 ml, 22,2 mmol) y se agitó en un tubo sellado a 115 con metanol (5 mL) y se concentró. La cromatografía e (tercbutoximetil)-1H-pirazol como un aceite incoloro (1,5 2H); 6,22 (s, 1H); 7,48 (s, 1H). 13C-NMR (75 MHz, CDC cuaternario).
Paso B: 6-[3-(terc-butoximetil)pirazol-1-il]-2-cloropiri
[0465]
cló con diazometano de trimetilsililo (2,0 M en hexano, rante 18 horas. La mezcla se enfrió a 40°C y se inactivó mna (sílice; heptanos/EtOAc 2:1 a 1:1) proporcionó 3-%). 1H RMN (CDCh, 300 MHz): 8 1,26 (s, 9H); 4,53 (s, 7,3, 57,2, 73,9,103,5, 134,0 (no se muestra un carbono
-carboxilato de terc-butilo

[0466] Se cargó un matraz de fondo redondo de 100 8,047 mmol), 2,6-dicloropiridin-3-carboxilato de terc-buti molido en un mortero) y DMF anhidra (12,41 ml). Se a temperatura ambiente bajo nitrógeno durante 16 horas. y agua y salmuera (50 mL) y se separaron las dos fas etilo (2 x 30 ml). Los extractos combinados se lavaron c se eliminó a presión reducida. El material se sometió a c acetato de etilo (0 a 10%) en hexanos. Las fracciones p reducida para proporcionar 6-[3-(terc-butoximetil)pirazol jo nitrógeno con 3-(terc-butoximetil)-1H-pirazol (1,241 g, 0 g, 8,061 mmol), K2CO3 (1,448 g, 10,48 mmol) (recién DABCO (163 mg, 1,453 mmol) y la mezcla se agitó a ezcla de reacción se diluyó con acetato de etilo (50 mL) fase acuosa se extrajo adicionalmente con acetato de muera, se secaron sobre sulfato de sodio y el disolvente tografía flash sobre gel de sílice usando un gradiente de se combinaron y los disolventes se eliminaron a presión -cloro-piridin-3-carboxilato de terc-butilo (1,956 g, 6 6 %)
como un aceite incoloro, que solidificó a un sólido blanc encontrado 366,2 (M+1)+; Tiempo de retención: 0,82 min Paso C: Ácido 2-cloro-6-[3-(hidroximetil)pirazol-1-il]p [0467]
ante la noche a alto vacío. IEN-EM m/z calc. 365,1506,
-3-carboxílico

[0468] 6-[3-(terc-butoximetil)pirazol-1-il]-2-cloro-piridin-3-HCl en dioxano (8,0 ml de 4 M, 32,00 mmol) y se calent enfrió a temperatura ambiente y se concentró hast (hidroximetil)pirazol-l-il]piridin-3-carboxílico (370 mg, 99 Tiempo de retención: 0,33 minutos
Paso D: Ácido 2-doro-6-[3-[[1-(trifluorometN)ddopro
[0469]
xilato de ferc-butilo (538 mg, 1,471 mmol) se disolvió en °C durante 2 horas. Después, la mezcla de reacción se quedad, dando un polvo blanco. Ácido 2-cloro-6-[3-EN-EM m/z calc. 253,02542, encontrado 254,1 (M+1)+;
etoximetN]pirazoM-N]piridm-3-carboxnico

[0470] [1-(Trifluorometil)ciclopropil]metil 4-metilbencen (hidroximetil)pirazol-1-il]piridin-3-carboxílico (370 mg, 1, añadió terc-butoxipotasio (660 mg, 5,882 mmol) y la me de 30 minutos, la mezcla de reacción se vertió en acido Los extractos orgánicos combinados se lavaron con sal El material resultante se purificó mediante cromatografía en diclorometano. Las fracciones que contenían el prod Ácido 2-cloro-6-[3-[[1-(trifluorometil)ciclopropil]metoximet calc. 375,05975, encontrado 376,1 (M+1)+; Tiempo de re
Paso E: N-(Bencenosulfonil)-2-doro-6-[3-[[1-(t carboxamida
[0471]
nato (1,3 g, 4,417 mmol) y acido 2-cloro-6-[3-mol), se combinaron en DMSO anhidro (9,250 ml). Se de reacción se agitó a temperatura ambiente. Después o 1 M (15 mL) y se extrajo 3 x 15 ml de acetato de etilo.
, se secaron sobre sulfato de sodio y se concentraron. e gel de sílice usando un gradiente de metanol al 0-10% e recogieron y concentraron para dar un sólido blanco. zol-1-il]piridin-3-carboxílico (292 mg, 53%) IEN-EM m/z ón: 0,62 minutos.
rom etil)cidopropil]metoxim etil]p irazol-1-il]pirid in-3-

[0472] Se combinaron ácido 2-cloro-6-[3-[[1-(trifluoro mg, 0,1597 mmol) y CDI (31 mg, 0,1912 mmol) en THF ( Se añadió bencenosulfonamida (28 mg, 0,1781 mmol) durante 16 horas más a temperatura ambiente. La mez extrajo con 3 x 10 ml de acetato de etilo. Los orgánicos c sulfato de sodio y se concentraron para dar un sólido bla N-(bencenosulfonil)-2-cloro-6-[3-[[1-(trifluorometil)ciclopr iclopropil]metoximetil]pirazol-1-il]piridin-3-carboxílico (60 j L) y se agitó a temperatura ambiente durante 2 horas. o de DBU (35 ml, 0,2340 mmol) y la reacción se agitó reacción se diluyó con 10 ml de ácido cítrico 1 M y se nados se lavaron con agua, salmuera, se secaron sobre ue se usó en el siguiente paso sin purificación adicional. etoximetil]pirazol-1-il]piridin-3-carboxamida
(aproximadamente 78 mg) IEN-EM m/z calc. 514,0689, Paso G: N-(bencenosulfoml)-6-[3-[[1-(trifluo trim etilpirrolid in-1-ilo]piridin-3-carboxam ida
[0473]
trado 515,1 (M+1)+; Tiempo de retención: 0,69 minutos. N)cidopropN]metoximetN]pirazoM-N]-2-[(4S)-2,2,4-

[0474] N-(Bencenosulfonil)-2-doro-6-[3-[[1-(trifluoromet mg, 0,1515 mmol), (4S)-2,2,4-trimetilpirrolidina (sal de cl mg, 1,519 mmol) se combinaron en DMSO (390,0 mL) y temperatura ambiente y se añadió 1 ml de agua. Despué del vial, se eliminó la porción líquida con una pipeta y se luego se lavaron con 15 ml de ácido cítrico 1M. Se separ dos veces más con 15 ml de acetato de etilo. Los org sobre sulfato de sodio y se concentraron. El sólido res gel de sílice eluyendo con metanol al 0-10% (trifluorometil)ciclopropil]metoximetil]pirazol-1-il]-2-[(4S)-IEN-EM m/z calc. 591,2127, encontrado 592,3 (M+1)+; Ti 8 12,56 (s, 1H), 8,33 (d, J = 2,6 Hz, 1H), 8,04 - 7,97 (m, (m, 2H), 7,07 (d, J = 8,2 Hz, 1H), 6,56 (d, J = 2,6 Hz, 1H J = 96 Hz, 1H), 2,18 -2,04 (m, 1H), 1,83 (dd, J = 11,7, 5 1,03 - 0,94 (m, 2H), 0,91 - 0,80 (m, 2H), 0,65 (d, J = 6,2
Ejemplo sintético 38: Síntesis del Co (trifluorom etil)ciclopropil]etoxi]pirazol-1 -il]-2-[(4S)-2
Pasos A-B: 1-[1-(Trifluorometil)ciclopropil]prop-2-en-
[0475]
propil]metoximetil]pirazol-1-il]piridin-3-carboxamida (78 to) (115 mg, 0,7684 mmol) y carbonato de potasio (210 lentaron a 130°C durante 16 h. La reacción se enfrió a 5 minutos de agitación, se dejó sedimentar el contenido ieron los sólidos restantes con 20 ml de acetato de etilo, las capas acuosa y orgánica y la capa acuosa se extrajo se combinaron, se lavaron con salmuera, se secaron se purificó adicionalmente mediante cromatografía en iclorometano para dar N-(bencenosulfonil)-6-[3-[[1-trimetilpirrolidin-1-il]piridin-3-carboxamida (46 mg, 51%) de retención: 2,11 minutos. 1H RMN (400 MHz, DMSO) 7,89 - 7,79 (m, 1H), 7,73 (t, J = 7,1 Hz, 1H), 7,70 - 7,60 (s, 2H), 3,59 (s, 2H), 2,42 (t, J = 10,4 Hz, 1H), 2,29 (d, 1H), 1,54 (d, J = 9,4 Hz, 6H), 1,37 (t, J = 12,1 Hz, 1H), ).
to 38: N-(bencenosulfoml)-6-[3-[2-hidroxi-2-[1-rimetilpirrolidin-1-il]piridin-3-carboxamida

[0476] Se agitó [1-(trifluorometil)ciclopropil]metanol (1 g en un baño de hielo. Se añadió cloro-hidroxi-dioxo-crom baño de hielo y la reacción se agitó durante 24 horas a t ml de éter dietílico y se filtró a través de un lecho de síli éter dietílico. El filtrado resultante se secó sobre sulfato siguiente paso sin concentración debido a la volatilida lentamente bromo(vinilo)magnesio (14,5 ml de 1 M, 14 calentara lentamente hasta casi la temperatura ambient 0°C, se inactivó con 1 M HCl y se diluyó con agua. Las éter dietílico. Los extractos orgánicos combinados se la concentraron parcialmente. El material crudo resultante (trifluorometil)ciclopropil]prop-2-en-1-ol (aproximadame restante) 1H RMN crudo (400 MHz, DMSO) 85,95 - 5,84 Hz, 1H), 5,18 (ddd, J = 10,4, 2,0, 1,3 Hz, 1H), 4,17 (tt, J
Paso C: terc-Butil-d im etil-[1-[1-(trifluorom etil)ciclopr
[0477]
8 mmol) en diclorometano seco (30 mL) y se enfrió a 0°C idina (2,77 g, 12,85 mmol) en una porción, y se retiró el atura ambiente. La mezcla de reacción se vertió en 100 n una capa de celite en la parte superior, eluyendo con odio, luego se filtró a través de algodón y se usó en el aldehído. El filtrado crudo se enfrió a 0°C y se añadió mol) (en THF). Se dejó que la mezcla de reacción se nte 2 horas. Después, la mezcla de reacción se enfrió a se separaron y la acuosa se extrajo 4 veces más con con salmuera, se secaron sobre sulfato de sodio y se só en el siguiente paso sin purificación adicional. 1-[1-25 g en crudo, con una cantidad sustancial de t Hf H), 5,36 (d, J = 5,2 Hz, 1H), 5,32 (ddd, J = 17,1,2,1, 1,4 1,4 Hz, 1H), 0,95 - 0,87 (m, 4H).
liloxi]silano
[0478] 1-[1-(trifluorometil)cidopropil]prop-2-en-1-ol (270 disolvieron en DMF (2 mL) y se enfriaron a 0°C en un b terc-butildimetilsililo) (370 mg, 2,455 mmol) en una sola p se dejó agitar la mezcla de reacción durante 16 horas a acuoso saturado (1 mL) y la mezcla de reacción se agitó d con 30 ml de éter dietílico y 20 ml de agua. Los extractos 15 ml de éter dietílico. Los extractos orgánicos combina sobre sulfato de sodio y se concentraron al vacío. El m eluyendo con hexanos, y se concentró hasta un aceite q pero se usó en el siguiente paso sin purificación adicional, (aproximadamente 317 mg, crudo).
Pasos D-F: 3-[2-[terc-butMo(dimetM)silM]oxi-2-[1-(trifl butilo
[0479]
, 1,625 mmol) e imidazol (220 mg, 3,232 mmol) se de hielo. A continuación, se añadió TBS-Cl (cloruro de ión y, después de 15 minutos, se retiró el baño de hielo y peratura ambiente. Luego se añadió cloruro de amonio nte 20 minutos. Después, la mezcla de reacción se diluyó ánicos se separaron y la capa acuosa se extrajo 23 con s se lavaron con agua, luego con salmuera, se secaron rial crudo se pasó luego a través de un tapón de sílice, contenía impurezas de sililo superpuestas sustanciales, rc-butil-dimetil-[1-[1-(trifluorometil)ciclopropil]aliloxi]silano
rometM)ddopropM]etoxi]pirazoM-carboxMato de terc-

[0480] Se combinó terc-butil-dimetil-[1-[1-(trifluorometil)ci dioxano (6 mL) y agua (2 mL) con 2,6-dimetilpiridina (185 Luego se añadió tetraoxoosmio (420 pL, 0,3139 mmol) y la ambiente. Después, la reacción se diluyó con 30 mL de orgánicos y se extrajo la capa acuosa 2 X 25 mL de diclo sobre sulfato de sodio, se concentraron hasta aproxi aislamiento.
[0481] La mezcla en crudo de la etapa anterior se diluyó c de sodio (90 mg, 2,379 mmol) y la mezcla de reacción s inactivó con ácido acético y se concentró. El material re sodio acuoso y se separaron las capas. La porción acu combinados se lavaron con salmuera, se secaron sobre s se usó en el siguiente paso sin purificación. El producto 3-hidroxipirazol-1-carboxilato de terc-butilo (145 mg, 0,78 enfrió a 0°C y se añadió lentamente DIAD (230 pL, 1,18 temperatura ambiente durante una hora y luego se calen concentró parcialmente, se disolvió en 100 mL de acetato sobre sulfato de sodio y se concentró. El material result con acetato de etilo al 0-50% en hexanos para dar butilo(dimetil)silil]oxi-2-[1 -(trifluorometil)ciclopropil]etoxi]pir calc. 450,21616, encontrado 451,3 (M+1)+; Tiempo de ret
Paso G: 3-(2-((terc-butMdimetMsiMl)oxi)-2-(1-(trifluoro
[0482]
propil]aliloxi]silano (220 mg, 0,7846 mmol) (en crudo) en 1,597 mmol) y peryodato de sodio (675 mg, 3,156 mmol). ezcla de reacción se agitó durante 20 horas a temperatura orometano y 30 mL de agua. Se separaron los extractos etano. Los extractos orgánicos combinados se secaron amente 8 mL y se usaron en el siguiente paso, sin
metanol (10 mL), y se enfrió a 0°C. Se añadió borohidruro gitó durante 1 hora. Después, la mezcla de reacción se tante se repartió entre acetato de etilo y bicarbonato de se extrajo 2 veces con acetato de etilo y los orgánicos to de sodio y se concentraron. El alcohol crudo resultante do se combinó con trifenilfosfina (300 mg, 1,144 mmol) y mmol) disuelto en THF (15 ml). La mezcla de reacción se mol). Se retiró el baño de hielo y la reacción se agitó a a 55°C durante 16 horas más. La mezcla de reacción se etilo, se lavó con 25 mL de IN NaOH, salmuera, se secó se purificó por cromatografía en gel de sílice eluyendo con una impureza activa UV superpuesta, 3-[2-[terel-1 -carboxilato de tere-butilo (63 mg, 18%) IEN-EM m/z ión: 0,95 minutos
M)ddopropM)etoxi)-1H-pirazol

[0483] Se disolvió 3-[2-[fere-butilo(dimetil)silil]oxi-2-[1-(trif (63 mg, 0,1398 mmol) (con una impureza principal trifluorracético) (aproximadamente 194,3 mg, 131,3 pL, rometil)ciclopropil]etoxi]pirazol-1 -carboxilato de tere-butilo sente) en diclorometano (1,312 mL) con TFA (ácido 04 mmol) y la reacción se agitó a temperatura ambiente
durante 30 minutos. Se añadieron hexanos (1 mL) y la acetato de etilo (10 mL) y una solución saturada de bica salmuera, se secaron sobre sulfato de sodio y se evapor 2-( 1 -(trifluorometil)ciclopropil)etoxi)-1H-pirazol (37 mg, Tiempo de retención: 0,81 minutos.
Paso H: 6-[3-[2-[terc-butMo(dimetM)siNl]oxi-2-[1-(tr carboxilato de tere-butilo
[0484]
ción se evaporó y el aceite resultante se repartió entre to de sodio. Los orgánicos se separaron, se lavaron con ara dar un aceite incoloro, 3-(2-((terc-butildimetilsMil)oxi)-IEN-EM m/z calc. 350,16373, encontrado 351,2 (M+1)+;
metN)ddopropN]etoxi]pirazoM-M]-2-doro-piridma-3-

[0485] Se cargó un matraz de fondo redondo bajo (trifluorometil)ciclopropil]etoxi]silano (37 mg, 0,1056
0,1048 mmol), K2CO3 (25 mg, 0,1809 mmol) (recién moli (2 mg, 0,01783 mmol) y la mezcla se agitó a temperat reacción se diluyó con acetato de etilo (15 mL) y agua (1 adicionalmente con acetato de etilo (2 x 15 ml). Los extr sulfato de sodio y el disolvente se eliminó a presión red de sílice usando un gradiente de acetato de etilo (0 a disolventes se eliminaron a presión reducida para propo [1-(trifluorometil)ciclopropil]etoxi]pirazol-1-il]-2-cloro-pirid 561,20374, encontrado 562,4 (M+1)+; Tiempo de reten 2,9 Hz, 1H), 8,34 (d, J = 8,4 Hz, 1H), 7,71 (d, J = 8,4 Hz, 0,13 - 0,03 (m, 6 H), 4,32 - 4,25 (m, 1H), 3,89 (s, 1H), 1,
Paso I: Ácido 6-[3-[2-[terc-butilo(d im etil)silil]o piridin-3-carboxílico
[0486]
geno con terc-butil-dimetil-[2-(1H-pirazol-3-iloxi)-1-[1-2,6-dicloropiridin-3-carboxilato de terc-butilo (26 mg, un mortero) y DMF anhidro (250 pL). Se añadió DABCO biente bajo nitrógeno durante 16 horas. La mezcla de y se separaron las dos fases. La fase acuosa se extrajo ombinados se lavaron con salmuera y se secaron sobre . El material se sometió a cromatografía flash sobre gel en hexanos. Las fracciones puras se combinaron y los r un aceite incoloro, 6-[3-[2-[terc-butilo(dimetil)silil]oxi-2-arboxilato de terc-butilo (20 mg, 34%). IEN-EM m/z calc. ,84 minutos, 1H RMN (400 MHz, DMSO) 8 8,44 (d, J = 6,17 (d, J = 2,9 Hz, 1H), 4,44 (dd, J = 10,7, 2,9 Hz, 1H), 9H), 0,98 (d, J = 35,8 Hz, 4H), 0,86 (s, 9H)
-(trifluorom etil)cidopropil]etoxi]p irazol-1-il]-2-doro-

[0487] Se combinaron 6-[3-[2-[terc-butilo(dimetil)silil]ox 3-carboxilato de terc-butilo (20 mg, 0,03558 mmol) y calentó a 45°C durante 3 h. Se evaporó la reacción. Se un ácido 6-[3-[2-[terc-butilo(dimetil)silil]oxi-2-[1-(trifluo sólido blanco (18 mg, 100%) IEN-EM m/z calc. 505,1 minutos.
Paso J: N-(Bencenosulfonil)-6-[3-[2-[terc-butilo(di il]-2-cloro-piridin-3-carboxamida
[0488]
-(trifluorometil)ciclopropil]etoxi]pirazol-1-il]-2-cloropiridin-0 mL, 0,6490 mmol) en diclorometano (0,75 mL) y se ron hexanos y la mezcla se evaporó de nuevo para dar il)ciclopropil]etoxi]pirazol-1-il]-2-cloropiridin-3-carboxílico encontrado 506,3 (M+1)+; Tiempo de retención: 0,59
ilil]oxi-2-[1-(trifluorometil)ciclopropil]etoxi]pirazol-1-
[0489] Se combinaron ácido 6-[3-[2-[íerc-butilo(dimetil)s piridin-3-carboxílico (19 mg, 0,03755 mmol) y CDI (8 mg, ambiente durante 2 horas. Se añadió bencenosulfonam mmol) y la reacción se agitó durante 2 h más a temperat 10 ml de ácido cítrico 1 M y se extrajo con 3 x 10 ml d agua, salmuera, se secaron sobre sulfato de sodio y s siguiente paso sin purificación adicional. (trifluorometil)ciclopropil]etoxi]pirazol-1-il]-2-cloro-piridin-644,1503, encontrado 645,3 (M+1)+; Tiempo de retenció
Paso O: N-(bencenosulfoml)-6-[3-[2-[terc-butMo(dim il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-il]piridin-3-carboxa
[0490]
i-2-[1 -(trifluorometil)ciclopropil]etoxi]pirazol-1 -il]-2-cloro-934 mmol) en THF (200 j L) y se agitaron a temperatura 7 mg, 0,04453 mmol) seguido de DBU (10 j L, 0,06687 ambiente. Después, la mezcla de reacción se diluyó con tato de etilo. Los orgánicos combinados se lavaron con centraron para dar un sólido blanco, que se usó en el -(bencenosulfonN)-6-[3-[2-[terc-butN(dimetN)silN]oxi-2-[1-boxamida (aproximadamente 22 mg) IEN-EM m/z calc.
minutos.
sMM]oxi-2-[1-(trifluorometM)cidopropM]etoxi]pirazoM-

[0491] N-(bencenosulfonil)-6-[3-[2-[terc-butilo(dimetil)silil piridin-3-carboxamida (22 mg, 0,03410 mmol), (4S)-2, 40,83 mg, 0,2728 mmol) y el carbonato de potasio (ap DMSO (180 j L) y se calentaron a 130°C durante 24 hor con 15 ml de ácido cítrico 1 M y 20 mL de acetato de etilo se extrajo dos veces más con 15 mL de acetato de etilo secaron sobre sulfato de sodio y se concentraron. El s sílice eluyendo con metanol al 0-10% en diclorometano N-(bencenosulfonil)-6-[3-[2-[terc-butil(dimetil)sililo]oxi-2-[ trimetilpirrolidin-1 -il]piridin-3-carboxamida (8 mg, 32%) I de retención: 0,88 minutos
Paso P: N-(Bencenosulfonil)-6-[3-[2-hidroxi-2-[ trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0492]
-2-[1 -(trifluorometil)ciclopropil]etoxi]pirazol-1 -il]-2-clororimetilpirrolidina (sal de clorhidrato) (aproximadamente adamente 75,40 mg, 0,5456 mmol) se combinaron en a reacción se enfrió a temperatura ambiente y se diluyó separaron las capas acuosa y orgánica y la capa acuosa orgánicos se combinaron, se lavaron con salmuera, se resultante se purificó mediante cromatografía en gel de dar el material protegido con TBS como un sólido blanco, fluorometil)ciclopropil]etoxi]pirazol-1-il]-2-[(4S)-2,2,4-M m/z calc. 721,2941, encontrado 722,4 (M+1)+; Tiempo
luorometil)ciclopropil]etoxi]pirazol-1-il]-2-[(4S)-2,2,4-

[0493] Luego se disolvió
(trifluorometil)ciclopropil]etoxi]pirazol-1-il]-2-[(4S)-2,2,4-tr mmol). en THF (0,4 mL), enfriado a 0°C, y se añadió aproximadamente 0,17 ml, 0,1705 mmol). Después de 5 ambiente. Después de 20 minutos a temperatura ambie 1 M y se extrajo con 3 x 10 ml de acetato de etilo. Los secaron sobre sulfato de sodio y se concentraron. El m cromatografía en gel de sílice eluyendo con metanol al hidroxi-2-[1-(trifluorometil)ciclopropil]etoxi]pirazol-1-il]-2-[ 14%) IEN-e M m/z calc. 607,20764, encontrado 608,3 (M DMSO) 812,51 (s, 1H), 8,19 (d, J = 2,8 Hz, 1H), 7,97 (d, 1H), 7,64 (d, J = 7,9 Hz, 2H), 6,89 (d, J = 8,2 Hz, 1H), 6 4,32 (m, 1H), 2,46 - 2,39 (m, 1H), 4,20 -4,12 (m, 1H), 3, Hz, 1H), 1,52 (d, J = 9,7 Hz, 6H), 1,43 - 1,36 (m, 1H), 1,
Ejemplo sintético 39: Síntesis del Compuesto (bencenosulfonN)-6-[3-[2-[ferc-butNo(dimetN)silN]oxi-2-[1-ilpirrolidin-1-il]piridin-3-carboxamida (8 mg, 0,0341 una jeringa fluoruro de tetrabutilamonio (1 M en THF, utos, se dejó que la reacción se calentara a temperatura la mezcla de reacción se vertió en 10 ml de ácido cítrico tos orgánicos combinados se lavaron con salmuera, se l crudo resultante se purificó luego dos veces mediante en diclorometano para dar N-(bencenosulfonil)-6-[3-[2--2,2,4-trimetilpirrolidin-1-il]piridin-3-carboxamida (3 mg, Tiempo de retención: 1,99 minutos. 1H RMN (400 MHz, 7,5 Hz, 2H), 7,79 (d, J = 8,2 Hz, 1H), 7,70 (d, J = 9,6 Hz, d, J = 2,8 Hz, 1H), 5,57 (dd, J = 5,5, 2,6 Hz, 1H), 4,40 -, 1H), 2,28 (s, 1H), 2,09 (s, 1H), 1,82 (dd, J = 12,1, 5,5 ,89 (m, 4H), 0,65 (d, J = 6,2 Hz, 3H).
N-(bencenosulfonil)-6-[3-[(1R,2S,4S)-norbornano-2-
il]oxipirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-il]pir Paso A: 3-(((1R,2S,4S)-biciclo[2.2.1]heptan-2-il)oxi)-[0494]
3-carboxamida
razol-1-carboxilato de terc-butilo

[0495] 3-hidroxipirazol-1-carboxilato de terc-butilo (1,63 fosfina de trifenilo (2,57 g, 9,798 mmol) se combinaron e A la mezcla se le añadió DIAD (2 ml, 10,16 mmol) gota y se agitó durante 16 h. La mezcla se evaporó y el m hidróxido de sodio 1 N (30 ml). Los orgánicos se separar de sodio y se evaporaron. El material crudo se purificó de etilo al 0-30% en hexanos para dar terc-butilo 3-(((1 (2,08 g, 84%) IEN-EM m/z calc. 278,16306, encontrado (400 MHz, DMSO) 88,05 (d, J = 3,0 Hz, 1H), 6,07 (d, J 2,32 - 2,22 (m, 1H), 1,75 (td, J = 6,7, 2,4 Hz, 1H), 1,54 2H), 1,18 -1,07 (m, 3H).
Paso B: 3-[(1R,2S,4S)-norbomano-2-il]oxi-1H-pirazol
[0496]
,860 mmol), (+)-endo-2-norborneol (1 g, 8,915 mmol) y F (21,98 mL) y la reacción se enfrió en un baño de hielo. a y la reacción se dejó calentar a temperatura ambiente l resultante se repartió entre acetato de etilo (30 mL) e lavaron con salmuera (30 mL), se secaron sobre sulfato nte cromatografía en gel de sílice eluyendo con acetato S)-biciclo[2.2.1]heptan-2-il)oxi)-1H-pirazol-1-carboxilato ,3 (M+1)+; Tiempo de retención: 0,72 minutos. 1H RMN 0 Hz, 1H), 4,47 (d, J = 6,8 Hz, 1H), 2,43 - 2,36 (m, 1H), H), 1,53 - 1,49 (m, 2H), 1,42 (ddt, J = 14,8, 7,8, 4,4 Hz,

[0497] 3-[(1R,2S,4S)-norbornano-2-ilo]oxipirazol-1-carb CH2Cl2 (20,80 mL) con ácido trifluoroacético (5,8 ml, durante 1 h. La reacción se evaporó a presión reducida y una solución saturada de bicarbonato de sodio (30 salmuera, se secaron sobre sulfato de sodio y se concen 2-il]oxi-1H-pirazol (1,29 g, 97%) IEN-EM m/z calc. 178, minutos.
Paso C: 2-Cloro-6-[3-[(1R,2S,4S)-norbornano-2-il]ox [0498]
o de terc-butilo (2,08 g, 7,473 mmol) se disolvió en mmol) y la reacción se agitó a temperatura ambiente ceite resultante se repartió entre acetato de etilo (50 mL) Los extractos orgánicos se separaron, se lavaron con n al vacío para dar un aceite, 3-[(1R,2S,4S)-norbornano-1, encontrado 179,2 (M+1)+; Tiempo de retención: 0,45
ol-1-il]piridin-3-carboxilato de terc-butilo

[0499] Se cargó un matraz de fondo redondo de 100 m butilo (1,796 g, 7,239 mmol), 3-[(1R,2S,4S)-norbomano 9,479 mmol) (recién molido en un mortero) y DMF anhi mezcla se agitó a temperatura ambiente bajo nitrógeno de etilo (50 mL), agua y salmuera (50 mL) y se separaron acetato de etilo (2 x 50 mL). Los extractos combinados presión reducida. El material se sometió a cromatografí etilo (0 a 20%) en hexanos. Las fracciones puras se co para proporcionar 2-cloro-6-[3-[(1R,2S,4S)-norbornano-2 64%) IEN-EM m/z calc. 389,1506, encontrado 390,3 (M DMSO) 8 8,40 (d, J = 2,9 Hz, 1H), 8,32 (d, J = 8,4 Hz, 1 J = 6 , 6 Hz, 1H), 1,88 - 1,78 (m, 1H), 2,45 (d, J = 4,6 Hz, 1,22 - 1,08 (m, 3H).
Paso D: Ácido 2-cloro-6-[3-[(1R,2S,4S)-norbomano-
[0500]
ajo nitrógeno con 2,6-didoropiridin-3-carboxNato de tercl]oxi-1H-pirazol (1,29 g, 7,238 mmol) y K2CO3 (1,310 g, (12 mL). Se añadió DABCO (146 mg, 1,302 mmol) y la nte 8 horas. La mezcla de reacción se diluyó con acetato dos fases. La fase acuosa se extrajo adicionalmente con ecaron sobre sulfato de sodio y el disolvente se eliminó a sh sobre gel de sílice usando un gradiente de acetato de naron y los disolventes se eliminaron a presión reducida xipirazol-1-il]piridina-3-carboxilato de terc-butilo (1,814 g, ; Tiempo de retención: 0,92 minutos 1H RMN (400 MHz, 7,72 (d, J = 8,4 Hz, 1H), 6,18 (d, J = 2,9 Hz, 1H), 4,53 (d, 2,29 (t, J = 4,3 Hz, 1H), 1,56 (s, 9H), 1,55 - 1,39 (m, 4H),
oxipirazol-1-il]piridin-3-carboxílico
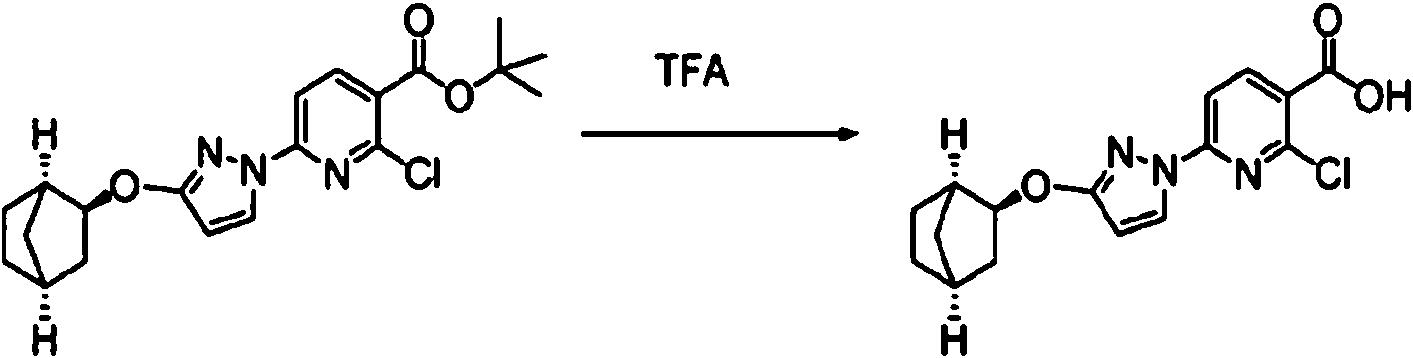
[0501] 2-cloro-6-[3-[(1R,2S,4S)-norbornano-2-il]oxipirazo y TFA (5 mL, 64,90 mmol) fueron combinados en diclo evaporó la reacción. Se agregaron hexanos y la mezcla usó en el siguiente paso sin purificación adicional. Ácido 3-carboxílico (1,47 g, 79%) IEN-EM m/z calc. 333,088, e
Paso E: N-(Bencenosulfoml)-2-cloro-6-[3-[(1R,2S,4S)
[0502]
il]piridin-3-carboxilato de tere-butilo (1,814 g, 4,653 mmol) etano (18,14 mL) y calentados a 40°C durante 2 h. Se evaporó nuevamente para dar un sólido blanco, que se ro-6-[3-[(1R,2S,4S)-norbornano-2-il]oxipirazol-1-il]piridinntrado 334,2 (M-1)+; Tiempo de retención: 0,71 minutos.
rbomano-2-M]oxipirazoM-M]piridm-3-carboxamida

[0503] Ácido 2-cloro-6-[3-[(1R,2S,4S)-norbornano-2-il] 0,323 mmol) se combinaron en THF y se agitaron du añadieron bencenosulfonamida (52 mg, 0,331 mmol) y D a temperatura ambiente. Después, la mezcla de reacció mL de acetato de etilo. Los extractos orgánicos combin sobre sulfato de sodio y se concentraron para dar apr irazol-1-il]piridin-3-carboxílico (100 mg)) y CDI (52 mg, e 2 horas a temperatura ambiente. A continuación, se (0,048 ml, 0,323 mmol) y la reacción se agitó 2 horas más e vertió en 20 mL de ácido cítrico 1 M y se extrajo 3 x 20 s se lavaron con agua, luego con salmuera, se secaron adamente 122 mg de N-(bencenosulfonil)-2-cloro-6-[3-
[(1R,2S,4S)-norbomano-2-il]oxipirazol-1-il]piridin-3-carb adicional. iEn -EM m/z calc. 490,12, encontrado 491,3 ( Paso F: N-(Bencenosulfonil)-6-[3-[(1R,2S,4S)-norbo il]piridin-3-carboxamida
[0504]
da que se usó en el siguiente paso sin purificación ; Tiempo de retención: 0,75 minutos.
-2-il]oxipirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidina-1-

[0505] N-(bencenosulfonil)-2-cloro-6-[3-[(1R,2S,4S)-nor 0,2537 mmol), (4S)-2,2,4-trimetilpirrolidina (sal de clorhi mg, 1,522 mmol) se combinaron en 0,423 ml de DMSO 16 horas. Después, la mezcla de reacción se enfrió a t como resultado la formación de un precipitado. Despué se desechó, y los sólidos restantes se disolvieron en 1 con 15 mL de ácido cítrico 1 M y la capa acuosa se ex extractos orgánicos combinados se lavaron con salmue material crudo se purificó mediante cromatografía en co 0-10% en diclorometano. Las fracciones puras se co [(1R,2S,4S)-norbornano-2-il]oxipirazol-1-il]-2-[(4S)-2,2,4 EM m/z calc. 549,24, encontrado 550,4 (M+1)+; Tiempo (s, 1H), 8,17 (d, J = 2,8 Hz, 1H), 8,02 - 7,97 (m, 2H), 7,8 6,90 (d, J = 8,2 Hz, 1H), 6,08 (d, J = 2,7 Hz, 1H), 4,49 ( Hz, 2H), 2,09 (dq, J = 11,6, 5,9, 5,4 Hz, 1H), 1,86 - 1,7 1,46 (dd, J = 13,8, 6,6 Hz, 3H), 1,36 (t, J = 12,1 Hz, 1H),
Ejemplo sintético 40: Síntesis del Compuest il]metoxi]pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-
Paso A: 3-[[(1S, 4R)-norbomano-2-M]metoxi]pirazoM
[0506]
no-2-il]oxipirazol-1-il]piridin-3-carboxamida (120 mg, (113,9 mg, 0,7611 mmol) y carbonato de potasio (210,3 vial con tapón de rosca y se calentaron a 130°C durante ratura ambiente y se añadieron 3 mL de agua, dando 0 minutos, la porción líquida se retiró con una jeringa y de acetato de etilo. Los extractos orgánicos se lavaron un tiempo adicional con 15 mL de acetato de etilo. Los e secaron sobre sulfato de sodio y se concentraron. El a sobre gel de sílice usando un gradiente de metanol al ron y concentraron para dar N-(bencenosulfonil)-6-[3-tilpirrolidin-1-il]piridin-3-carboxamida (59 mg, 40%). IEN-nción: 2,37 minutos 1H RMN (400 m Hz , DMSO) 812,50 J = 8,2 Hz, 1H), 7,77 - 7,70 (m, 1H), 7,70 - 7,63 (m, 2H), 6,7 Hz, 1H), 2,46 - 2,37 (m, 2H), 2,27 (q, J = 10,0, 7,6 1H), 1,56 (d, J = 6,9 Hz, 1H), 1,53 (s, 3H), 1,51 (s, 3H), - 1,06 (m, 4H), 0,64 (d, J = 6,3 Hz, 3H).
: N-(bencenosulfonil)-6-[3-[[(1R,4R)-norbornano-2-din-3-carboxamida
oxMato de terc-butilo

[0507] 3-hidroxipirazol-1-carboxilato de terc-butilo (1,327 mmol) (mezcla de endo y exo), y se combinaron trifenilf se enfrió en un baño de hielo. A la mezcla se le añadió calentar a temperatura ambiente y se agitó durante 72 entre acetato de etilo (50 mL) e hidróxido de sodio 1 N ( se secaron sobre sulfato de sodio y se evaporaron. El sílice eluyendo con acetato de etilo al 0-30% en he carboxilato de terc-butilo (1,698 g, 81%) IEN-EM m/z cal 0,77 minutos. (2 diastereómeros - mezcla de norbornan J = 2,9 Hz, 1H), 6,10 (dd, J = 2,9, 1,0 Hz, 1H), 4,23 -3,8 1,54 (d, J = 1,4 Hz, 9H), 1,51 - 1,03 (m, 7 H), 0,75 (dd, J
Paso B: 3-[[(1S,4R)-norbornano-2-il]metoxi]-1H-piraz
[0508]
204 mmol), [(1S,4R)-norbornano-2-il]metanol (1 g, 7,924 (2,09 g, 7,968 mmol) en THF (17,87 mL) y la reacción 1,627 mL, 8,263 mmol) gota a gota y la reacción se dejó mezcla se evaporó y el material resultante se repartió ). Los orgánicos se separaron, se lavaron con salmuera, rial crudo se purificó mediante cromatografía en gel de para dar 3-[[(1S,4R)-norbornano-2-il]metoxi]pirazol-1-,17868, encontrado 293,3 (M+1)+; Tiempo de retención:
y exo sustituido) 1H RMN (400 MHz, DMSO) 88,06 (d, 2H), 2,29 -2,15 (m, 2H), 1,69 (dq, J = 12,1, 4,2 Hz, 1H), , 2,4 Hz, 1H).

[0509] 3-[[(1S,4R)-Norbornano-2-ilo]metoxi]pirazol-1-ca CH2CI2 (16,98 mL) con ácido trifluoroacético (aproximad a temperatura ambiente durante 2 h. La reacción se ev (50 mL) y una solución saturada de bicarbonato de sod con salmuera, se secaron sobre sulfato de sodio y se co 2-il]metoxi]-1H-pirazol (1,11 g, 99%) IEN-EM m/z calc.
0,52 minutos.
Paso C: 2-Cloro-6-[3-[[(1S,4R)-norbomano-2-M]meto
[0510]
ato de terc-butilo (1,698 g, 5,808 mmol) se disolvió en te 6,622 g, 4,474 ml, 58,08 mmol) y la reacción se agitó y el aceite resultante se repartió entre acetato de etilo mL). Los extractos orgánicos se separaron, se lavaron aron al vacío para dar un aceite, 3-[[(1s,4R)-norbornano-12627, encontrado 193,2 (M+1)+; Tiempo de retención:
zoM-M]pindm-3-carboxMato de terc-butilo

[0511] Un matraz de fondo redondo se cargó bajo nitró g, 5,774 mmol) (mezcla de dos diastereómeros), 2,6-di K2CO3 (1,05 g, 7,597 mmol) (recién molido en un morter mmol) y la mezcla se agitó a temperatura ambiente bajo con acetato de etilo (50 mL) y agua (50 mL) y se separ con acetato de etilo (2 x 30 mL). Los extractos combinad y el disolvente se eliminó a presión reducida. El materia un gradiente de acetato de etilo (0 a 20%) en hexano eliminaron a presión reducida para proporcionar 2-clo carboxilato de terc-butilo (1,88 g, 81%) iEn-EM m/z calc 0,94 minutos
Paso D: Ácido 2-doro-6-[3-[[(1S,4R)-norbomano-2-M
[0512]
con 3-[[(1S,4R)-norbornano-2-il]metoxi]-1H-pirazol (1,11 iridin-3-carboxilato de terc-butilo (1,433 g, 5,776 mmol), MF anhidra (10 mL). Se añadió DABCO (117 mg, 1,043 geno durante 16 horas. La mezcla de reacción se diluyó las dos fases. La fase acuosa se extrajo adicionalmente lavaron con salmuera, se secaron sobre sulfato de sodio ometió a cromatografía flash sobre gel de sílice usando s fracciones puras se combinaron y los disolventes se [3-[[(1S,4R)-norbornano-2-il]metoxi]pirazol-1-il]piridina-3-,16626, encontrado 404,3 (M+1)+; Tiempo de retención:
oxi]pirazoM-N]piridm-3-carboxflico

[0513] 2-Cloro-6-[3-[[(1S,4R)-norbornano-2-il]metoxi]pir mmol) y TFA (5 ml, 64,90 mmol) se combinaron en dicl evaporó la reacción. Se añadieron hexanos y la mezcla norbornano-2-il]metoxi]pirazol-1-il]piridin-3-carboxílico s encontrado 348,2 (M+1)+; Tiempo de retención: 0,75 mi
Paso E: N-(bencenosulfonil)-2-cloro-6-[3-[[(1R,4R)-n
[0514]
-il]piridin-3-carboxilato de terc-butilo (1,88 g, 4,655 tano (18,80 mL) y se calentaron a 40°C durante 2 h. Se aporó de nuevo para dar un ácido 2-cloro-6-[3-[[(1S,4R)-blanco. (1,58 g, 98%) IEN-EM m/z calc. 347,10367, .
rnano-2-il]metoxi]pirazol-1-il]piridin-3-carboxamida

[0515] Ácido 2-doro-6-[3-[[(1S,4R)-norbomano-2-il]met CDI (60,59 mg, 0,3737 mmol) se agitaron en THF (0,5 bencenosulfonamida (50 mg, 0,3181 mmol), seguido de a temperatura ambiente. Después, la mezcla de reacció de ácido cítrico. La capa acuosa se extrajo con 25
combinados se lavaron con agua y luego con salmuera, N-(bencenosulfonil)-2-cloro-6-[3-[[(1R,4R)-norbornano-2 (aproximadamente 135 mg), que se usó en el siguient encontrado 487,2 (M+1)+; Tiempo de retención: 0,84 mi
Paso F: N-(bencenosulfonil)-6-[3-[[(1R,4R)-norboma il]piridin-3-carboxamida
[0516]
azol-1-il]piridin-3-carboxílico (100 mg, 0,2875 mmol) y emperatura ambiente durante 2 horas. Luego se añadió (0,05588 ml, 0,3737) y la reacción se agitó 4 horas más diluyó con 25 ml de acetato de etilo y se vertieron 25 ml ionales de acetato de etilo, y los extractos orgánicos caron sobre sulfato de sodio y se concentraron para dar toxi]pirazol-1-il]piridin-3-carboxamida
o sin purificación adicional. IEN-EM m/z calc. 486,11,
il]metoxi]pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-

[0517] N-(bencenosulfonil)-2-cloro-6-[3-[[(1R,4R)-norbor 0,2772 mmol), (4S)-2,2,4-trimetilpirrolidina (sal de clorhi mg, 1,663 mmol)) se combinaron en DMSO en un vial c Después, la mezcla de reacción se enfrió a temperatura la formación de un precipitado. Después de 30 minutos, sólidos restantes se disolvieron en 15 mL de acetato de M y la capa acuosa se extrajo un tiempo adicional con 1 se lavaron con salmuera, se secaron sobre sulfato de s cromatografía en columna sobre gel de sílice usando fracciones puras se combinaron y concentraron il]metoxi]pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-il]pir EM m/z calc. 563,26, encontrado 564,4 (M+1)+; Tiemp 12,50 (s, 1H), 8,18 (dd, J = 2,8, 1,0 Hz, 1H), 7,99 (dd, J (m, 1H), 7,66 (dd, J = 8,3, 6,7 Hz, 2H), 6,92 (dd, J = 8,3, (t, J = 10,5 Hz, 1H), 2,35 -2,16 (m, 4H), 2,09 (tt, J = 12 1,52 (d, J = 9,7 Hz, 7 H), 1,50 - 1,45 (m, 1H), 1,42 - 1,27 1H), 0,64 (d, J = 6,2 Hz, 3H).
Ejemplo s intético 41: Síntesis del Compuesto 41: N-2-[(4S)-2, 2,4-trimetilpirrolidin-1-il]piridin-3-carboxam
[0518]
-2-il]metoxi]pirazol-1-il]piridin-3-carboxamida, (135 mg, (124,5 mg, 0,8316 mmol) y carbonato de potasio (229,8 ón de rosca y se calentaron a 130°C durante 16 horas. nte y se añadieron 3 ml de agua, dando como resultado ción líquida se retiró con una jeringa y se desechó, y los Los orgánicos se lavaron con 15 mL de ácido cítrico 1 de acetato de etilo. Los extractos orgánicos combinados se concentraron. El material crudo se purificó mediante radiente de metanol al 0-10% en diclorometano. Las dar N-(bencenosulfonil)-6-[3-[[(1R,4R)-norbornano-2--carboxamida, (mezcla de endo y exo norbornano) IEN-retención: 2,45 minutos. 1H Rm N (400 MHz, Dm So ) 8 , 1,7 Hz, 2H), 7,80 (dd, J = 8,3, 1,1 Hz, 1H), 7,79 - 7,70 z, 1H), 6,12 (t, J = 2,9 Hz, 1H), 4,23 - 3,90 (m, 2H), 2,40 2 Hz, 1H), 1,82 (dd, J = 11,9, 5,5 Hz, 1H), 1,73 (s, 1H), H), 1,21 -1,08 (m, 2H), 0,75 (ddd, J = 12,5, 5,0, 2,2 Hz,
cenosulfoml)-6-[3-(2,2-dicidopropMetoxi)pirazoM-M]- 
Paso A: 3-(2,2-DiCidopropiletoxi)pirazol-1-carboxilat [0519]
tere-butilo

[0520] Una solución de 2,2-diCiclopropiletanol (500 mg, mg, 3,963 mmol) y trifenilfosfano (1,1 g, 4,194 mmol) en lentamente DIAD (800,0 pL, 4,063 mmol) en atmósfer temperatura ambiente y se agitó durante 16 h. La me bicarbonato de sodio acuoso saturado, salmuera, se se mediante cromatografía en gel de sílice con 100% de h (2,2-diciclopropiletoxi)pirazol-1-carboxilato de ferc-butil 292,17868, encontrado 293,3 (M+1)+; Tiempo de retenci J = 3,0 Hz, 1H), 5,67 (s, 1H), 4,13 (d, J = 5,3 Hz, 2H), 1 Hz, 1H), 0,32 - 0,12 (m, 4H) 0,10 - 0,08 (m, 4H).
Paso B: 3-(2,2-dicidopropiletoxi)-1H-pirazol:
[0521]
mmol), 3-hidroxipirazol-1-carboxilato de terc-butilo (730 seco (20,0 mL) se enfrió en un baño de hielo y se añadió N2. Se dejó que la reacción se calentara lentamente a de reacción se diluyó con acetato de etilo, se lavó con re sulfato de sodio y se concentró. El residuo se purificó s a 50% de acetato de etilo en hexanos para producir 3-mg, 68%) como un aceite incoloro. IEN-EM m/z calc. ,98 minutos. 1H RMN (400 MHz, cloroformo-d) 87,62 (d, , 9H), 0,58 (qt, J = 8,2, 5,0 Hz, 2H), 0,36 (tt, J = 8,9, 5,6

[0522] Una solución de 3-(2,2-diciclopropiletoxi)pirazol trifluoroacético (1,0 ml, 12,98 mmol) en diclorometano ( presión reducida y el residuo se basificó con bicarbonat Los extractos combinados se secaron sobre sulfato de pirazol como aceite incoloro que se usó tal cual sin purif 192,12627, encontrado 193,3 (M+1)+; Tiempo de retenc
Paso C: 2-Cloro-6-[3-(2,2-dicidopropiletoxi)pirazol-1
[0523]
rboxilato de terc-butilo (750 mg, 2,565 mmol) y acido se agitó durante 2,5 horas. Los volátiles se eliminaron a sodio acuoso saturado y se extrajo con acetato de etilo.
y se evaporaron para dar 3-(2,2-diciclopropiletoxi)-1H-n adicional para la siguiente reacción. IEN-Em m/z calc. ,32 minutos.
idina-3-carboxilato de tere-butilo

[0524] Una mezcla de 3-(2,2-dicidopropiletoxi)-1H-piraz tere-butilo (682,0 mg, 2,749 mmol), carbonato de potasi mg, 0,5349 mmol) en DMSO (20,0 mL) se agitó a temp agua y se extrajo con acetato de etilo. Los extractos co de sodio y se evaporaron. El residuo se purificó media hexanos a 20% de acetato de etilo en hexanos para pr carboxilato de tere-butilo (680 mg, 66%) como aceite inc Tiempo de retención: 2,49 minutos. 1H RMN (400 MHz, 1H), 7,70 (d, J = 8,5 Hz, 1H), 5,98 (d, J = 2,9 Hz, 1H), 4 - 0,56 (m, 1H), 0,54 - 0,36 (m, 4H), 0,32 - 0,13 (m, 4H).
Paso D: Ácido 2-doro-6-[3-(2,2-diddopropMetoxi)pir
[0525]
3,0 mg, 2,564 mmol), 2,6-dicloropiridina-3-carboxilato de 0,0 mg, 3,111 mmol), y 1,4-diazabiddo[2.2.2]octano (60 ra ambiente durante 15 horas. La reacción se diluyó con ados se lavaron con salmuera, se secaron sobre sulfato romatografía en columna de gel de sílice con 100% de r 2-cloro-6-[3-(2,2-diciclopropiletoxi)pirazol-1-il]piridina-3-. IEN-EM m/z calc. 403,16626, encontrado 404,4 (M+1)+; formo-d) 88,35 (d, J = 2,8 Hz, 1H), 8,18 (d, J = 8,4 Hz, , J = 5,6 Hz, 2H), 1,61 (s, 9H), 0,92 - 0,75 (m, 2H), 0,70
-N]piridm-3-carboxnico

[0526] Una solución de 2-cloro-6-[3-(2,2-diciclopropileto mmol) en ácido trifluoroacético (1,5 ml, 19,47 mmol) y d ambiente. El disolvente se evaporó y el residuo se rec ácido 2-cloro-6-[3-(2,2-diciclopropiletoxi)pirazol-1-il]piridi encontrado 348,3 (M+1)+; Tiempo de retención: 1,95 mi
Paso E: N-(Bencenosulfoml)-2-doro-6-[3-(2,2-diddo
[0527]
zol-1-il]piridin-3-carboxilato de itere-butilo (675 mg, 1,671 metano (4,5 mL) se agitó durante 4 horas a temperatura os veces en THF y se concentró al vacío para producir arboxílico (580 mg, 100%). IEN-EM m/z calc. 347,10367, .
etoxi)pirazoM-M]piridm-3-carboxamida

[0528] Una solución de ácido 2-cloro-6-[3-(2,2-dicicl mmol) y carbonil diimidazol (60,0 mg, 0,3700 mmol) en T bencenosulfonamida (50 mg, 0,3181 mmol) y DBU (60 horas más a temperatura ambiente. La mezcla de reacci y se extrajo con acetato de etilo. Los extractos orgáni sulfato de sodio y se evaporaron para producir N-il]piridin-3-carboxamida. que se usó tal cual para la sigui (M+1)+; Tiempo de retención: 0,79 minutos.
Paso F: N-(Bencenosulfonil)-6-[3-(2,2-dicidopropilet 3-carboxamida
[0529]
iletoxi)pirazol-1-il]piridin-3-carboxílico (100 mg, 0,2875 ,0 mL) se agitó durante 45 minutos. Luego, se añadieron ,4012 mmol) y la mezcla de reacción se agitó durante 2 inactivó con una solución saturada de cloruro de amonio ombinados se lavaron con salmuera, se secaron sobre enosulfonil)-2-cloro-6-[3-(2,2-diciclopropiletoxi)pirazol-1-reacción. IEN-EM m/z calc. 486,11285, encontrado 487,4
irazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-il]piridina-

[0530] Una mezcla de N-(bencenosulfonil)-2-doro-6-[3-mg, 0,2875 mmol), (4S)-2,2,4-trimetilpirrolidina (sal de (240,0 mg, 1,737 mmol) en DMSO (2 mL) se agitó a 13 de un disco de filtro Whatman (puradisc 25 TF) y el fil inversa usando un gradiente dual desde el 50-99% de l HCl) y fase móvil B = CH3CN) para producir N-(bencen trimetilpirrolidin-1 -il]piridin-3-carboxamida (75,9 mg, 45 encontrado 564,5 (M+1)+; Tiempo de retención: 2,3 min 1H), 8,21 (d, J = 2,8 Hz, 1H), 8,18 -8,08 (m, 2H), 7,66 -3,48 (dd, J = 10,4, 8,4 Hz, 1H), 3,08 (dd, J = 10,4, 7,6 Hz, 1H), 1,73 (dd, J = 12,4, 9,5 Hz, 1H), 1,36 (s, 3H), 1 2H), 0,61 (tt, J = 8,8, 5,6 Hz, 1H), 0,55 - 0,38 (m, 4H), 0
Ejemplo sintético 42: Síntesis del Compuesto 42: N-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-il]piridin-3-carbox
[0531]
icidopropiletoxi)pirazol-1-il]piridin-3-carboxamida (140,0 drato) (145,0 mg, 0,9689 mmol) y carbonato de potasio urante 15 horas. La mezcla de reacción se filtró a través se purificó mediante un método de HPCL-EM de fase móvil B durante 15 minutos (fase móvil A = H2O (5 mM nN)-6-[3-(2,2-diddopropNetoxi)pirazol-1-N]-2-[(4S)-2,2,4-o un sólido blanquecino. IEN-EM m/z calc. 563,25665, 1H RMN (400 MHz, cloroformo-d) 88,35 (d, J = 8,6 Hz, m, 5H), 5,96 (d, J = 2,8 Hz, 1H), 4,32 (d, J = 5,6 Hz, 2H), ), 2,61 (dt, J = 15,3, 7,8 Hz, 1H), 2,14 (dd, J = 12,4, 7,9 , 3H), 1,20 (d, J = 6,6 Hz, 3H), 0,81 (qt, J = 8,3, 5,0 Hz, , J = 4,8 Hz, 4H).
cenosulfoml)-6-[3-(3,3-dicidopropMpropoxi)pirazoM-

3,3-Diciclopropilpropan-1 -ol [0532]
[0533] A una solución de ácido 3,3-dicidopropilpropanoi hidruro de litio y aluminio (845,0 pL de 2 M, 1,690 mmol lentamente. Se dejó que la mezcla se calentara gradual matraz se enfrió de nuevo en un baño de hielo y se (lentamente), seguido de NaOH (70,0 pL de 6 M, 0,4200 sólido granular blanco en la mezcla. A esta mezcla se mezcla heterogénea blanca resultante se filtró a travé concentró para producir 3,3-diciclopropilpropan-1-ol (14 (M+1)+; Tiempo de retención: 0,5 minutos.
Paso A: 3-(3,3-DicidopropMpropoxi)pirazoM-carbox
[0534]
00 mg, 1,297 mmol) en THF seco (2,000 mL) se añadió un baño de hielo/agua bajo atmósfera de N2 gota a gota a temperatura ambiente y se agitó durante 16 horas. El tivó secuencialmente con agua (70,0 pL, 3,886 mmol) ol), luego agua (200 pL, 11,10 mmol) proporcionando un adió MgSO4 anhidro y se agitó durante 10 minutos. La celite y el precipitado se lavó con éter. El filtrado se , 77%). IEN-EM m/z calc. 140,12012, encontrado 141,2
de tere-butilo

[0535] Una solución de 3,3-diciclopropilpropan-1-ol (14 butilo (185,0 mg, 1,004 mmol) y trifenilfosfano (278 mg, hielo, y se añadió lentamente DIAD (200,0 pL, 1,016
calentara lentamente a temperatura ambiente y se agitó de etilo, se lavó con una solución acuosa saturada de bi y se evaporó al vacío. El residuo se purificó mediante cr de acetato de etilo en hexanos para producir 3-(3,3-dici 83%) como un aceite incoloro. IEN-EM m/z calc. 306, minutos.
Paso B: 3-(3,3-diciclopropilpropoxi)-1H-pirazol
[0536]
g, 0,9984 mmol), 3 -hidroxipirazol-1-carboxilato de terc-0 mmol) en THF seco (7,0 mL) se enfrió en un baño de bajo una atmósfera de N2. Se dejó que la reacción se te 16 horas. La mezcla de reacción se diluyó con acetato nato de sodio, salmuera, se secó sobre sulfato de sodio ografía en gel de sílice usando 100% de hexanos a 50% opilpropoxi)pirazol-1-carboxilato de terc-butilo (255 mg, , encontrado 307,4 (M+1)+; Tiempo de retención: 0,81

[0537] Una solución de 3-(3,3-diciclopropilpropoxi)piraz ácido trifluoroacético (325,0 pL, 4,218 mmol) en dicloro al vacío para proporcionar 3-(3,3-diciclopropilpropoxi)-1 se usó tal cual sin purificación adicional para la siguie (M+1)+; Tiempo de retención: 0,59 minutos.
Paso C: 2-Cloro-6-[3-(3,3-diciclopropilpropoxi)pirazo
[0538]
arboxilato de terc-butilo (255 mg, 0,8322 mmol) y se agitó no (1 mL) durante 2,5 horas. Los volátiles se eliminaron zol (sal de trifluoroacetato) como un aceite incoloro que acción. IEN-EM m/z calc. 206,1419, encontrado 207,2
]piridina-3-carboxilato de tere-butilo

diciclopropilpropoxi)-1H-pirazol (266,0 mg, 0,8305 m diazabiciclo[2.2.2]octano (20 mg, 0,1783 mmol) en DMS La reacción se diluyó con agua y se extrajo con acetato salmuera, se secaron sobre sulfato de sodio y se ev columna de gel de sílice usando 100% de hexanos a 20 cloro-6-[3-(3,3-diciclopropilpropoxi)pirazol-1-il]piridina-3carbonato de potasio (230 mg, 1,664 mmol) y 1,4-mL) se agitó a temperatura ambiente durante 15 horas. tilo. Los extractos orgánicos combinados se lavaron con ron. El residuo se purificó mediante cromatografía en acetato de etilo en hexanos para producir terc-butilo 2-xilato (245 mg, 71%) como aceite incoloro. IEN-EM m/z
calc. 417,18192, encontrado 418,4 (M+1)+; Tiempo de r Paso D: Ácido 2-doro-6-[3-(3,3-diddopropNpropoxi) [0540]
ción: 1,28 minutos.
zoM-N]piridm-3-carboxnico

[0541] Una solución de 2-cloro-6-[3-(3,3-diciclopropil mg, 0,5862 mmol) en ácido trifluoroacético (500,0 j L, 6, a temperatura ambiente. Se evaporó el disolvente y se para producir ácido 2-cloro-6-[3-(3,3-diciclopropilpropoxi blanco que se usó tal cual para la siguiente reacción. IE de retención: 0,8 minutos. 1H RMN (400 MHz, metanol-2,9 Hz, 1H), 4,45 (t, J = 6,7 Hz, 2H), 1,98 (q, J = 7,0 Hz, 1H), 0,26 - 0,19 (m, 2H), 0,15 - 0,06 (m, 2H).
Paso E: N-(bencenosulfoml)-2-doro-6-[3-(3,3-diddo
[0542]
oxi)pirazol-1-il]piridin-3-carboxilato de ferc-butilo (245,0 mmol) y diclorometano (1,5 mL) se agitó durante 4 horas gió dos veces el residuo en THF y se concentró al vacío zol-1-il]piridin-3-carboxílico (204 mg, 96%) como un sólido m/z calc. 361,11932, encontrado 362,3 (M+1)+; Tiempo 8,47 - 8,32 (m, 2H), 7,73 (d, J = 8,5 Hz, 1H), 6,03 (d, J = , 0,75 - 0,64 (m, 2H), 0,50 - 0,39 (m, 4H), 0,35 - 0,26 (m,
Npropoxi)pirazoM-M]piridm-3-carboxamida

[0543] Una solución de ácido 2-cloro-6-[3-(3,3-diciclop mmol) y carbonil diimidazol (30,0 mg, 0,1850 mmol) en T bencenosulfonamida (25,0 mg, 0,1590 mmol) y DBU (3 horas más a temperatura ambiente. La mezcla de reacci y se extrajo con acetato de etilo. Los extractos orgáni sulfato de sodio y se evaporaron para producir N-(b il]piridin-3-carboxamida. (84 mg, 121%) como aceite vis del 100%) como está para la siguiente reacción. IEN-retención: 0,83 minutos.
Paso F: N-(bencenosulfonil)-6-[3-(3,3-dicido il]piridina-3-carboxamida
[0544]
propoxi)pirazol-1-il]piridin-3-carboxílico (50,0 mg, 0,1382 2,0 mL) se agitó durante 45 minutos. Luego, se agregaron 0,2006 mmol). La mezcla de reacción se agitó durante 2 e inactivó con una solución saturada de cloruro de amonio combinados se lavaron con salmuera, se secaron sobre nosulfonil)-2-cloro-6-[3-(3,3-diciclopropilpropoxi)pirazol-1-marrón claro que se usó (considerando una conversión /z calc. 500,1285, encontrado 501,4 (M+1)+; Tiempo de
ilpropoxi)pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-

[0545] Una mezcla de N-(bencenosulfonil)-2-cloro(3,3-diciclopropilpropoxi)pirazol-1-il]piridin-3-carboxamida
(68,0 mg, 0,1357 mmol), (4S)-2,2,4-trimetilpirrolidina ( potasio (115,0 mg, 0,8321 mmol) en DMSO (1 mL) se a a través de un disco de filtro Whatman (puradisc 25 TF) fase inversa usando gradiente dual desde 50-99% de fa y fase móvil B = CH3CN para producir N-(bencenos trimetilpirrolidina-1-il]piridin-3-carboxamida (23,1 mg, 2 Tiempo de retención: 1,0 minutos. 1H RMN (400 MHz, 1H), 8,17 (d, J = 1,2 Hz, 1H), 8,15 (d, J = 1,6 Hz, 1H)), 1H), 4,43 (t, J = 6 , 8 Hz, 2H), 3,49 (dd, J = 10,3, 8,5 Hz, (dd, J = 12,4, 7,9 Hz, 1H), 1,97 (q, J = 6 , 8 Hz, 2H), 1,73 J = 6,7 Hz, 3H), 0,73 - 0,59 (m, 2H), 0,50 - 0,37 (m, 4H),
Ejemplo sintético 43: Síntesis del
(trifluorom etil)ciclopropil]etoxi]pirazol-1 -il]-2-[(4S)-2
Paso A: 2-C loro-N-(2-clorofenil)sulfonil-6-[3-carboxamida
[0546]
clorhidrato) (70,0 mg, 0,4677 mmol), y carbonato de 130°C durante 15 horas. La mezcla de reacción se filtró iltrado se purificó mediante un método de HPCL-EM de vil B durante 15 minutos (fase móvil A=H2O (5 mM HCl) )-6-[3-(3,3-diciclopropilpropoxi)pirazol-1-il]-2-[(4S)-2,2,4-IEN-e M m/z calc. 577,2723, encontrado 578,5 (M+1 )+; ormo-d) 8 8,36 (d, J = 8 , 6 Hz, 1H), 8,21 (d, J = 2,8 Hz, - 7,57 (m, 2H), 7,57 - 7,51 (m, 2H), 5,94 (d, J = 2,8 Hz, ,09 (dd, J = 10,4, 7,6 Hz, 1H), 2,70 -2,56 (m, 1H), 2,14 J = 12,4, 9,4 Hz, 1H), 1,36 (s, 3H), 1,28 (s, 3H), 1,21 (d, - 0,29 (m, 1H), 0,24 - 0,15 (m, 2H), 0,12 - 0,07 (m, 2H).
puesto 43: N-(2-clorofenil)sulfonil-6-[3-[2-[1-rimetilpirrolidin-1-il]piridin-3-carboxamida
(trifluorometil)ciclopropilo]etoxi]pirazol-1-il]piridin-3-

[0547] Paso 1: Se disolvió cloruro de 2-clorobencenosul de 7 M, 1,050 mmol) y se agitó a temperatura ambiente volvió a evaporar en diclorometano. Los sólidos se disol La mezcla se agitó a 70°C durante 30 minutos para libe
[0548] Paso 2: Ácido 2-cloro-6-[3-[2-[1-(trifluorometil)cicl mmol) y carbonil diimidazol (53 mg, 0,3269 mmol) se c En este punto, esta mezcla se añadió a la mezcla de sul a temperatura ambiente. La reacción se diluyó con ace seguido de salmuera. Los orgánicos se separaron, s producir 2-cloro-N-(2-clorofenil)sulfonil-6-[3-[2-[1-(trifl junto con material de partida y amida primaria. La mez EM m/z calc. 548,02997, encontrado 549,28 (M+1)+; Tie
Paso B: N-(2-Clorofeml)sulfoml-6-[3-[2-[1 trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0549]
(50 pL, 0,3667 mmol) en amoniaco en metanol (150 pL nte 30 minutos. La mezcla se evaporó a sequedad y se en THF (1 mL) y se añadió DBU (60 pL, 0,4012 mmol). alquier amoniaco restante de la reacción.
il]etoxi]pirazol-1 -il]piridina-3-carboxílico (100 mg, 0,2661 aron en THF (1,000 mL) y se agitaron durante 2 horas. ida (del paso-1) y la reacción se agitó durante la noche e etilo y se lavó con una solución de ácido cítrico 1 M, ron sobre sulfato de magnesio y se evaporaron para etil)ciclopropil]etoxi]pirazol-1-ilo]piridina-3-carboxamida utilizó de esta manera para la siguiente reacción. IEN-de retención: 0,76 minutos.
orometN)cidopropN]etoxi]pirazoM-N]-2-[(4S)-2,2,4-

[0550] Una mezcla de 2-cloro-N-(2-clorofenil)sulfo carboxamida (50,0 mg, 0,09102 mmol) (mezcla como l (50,0 mg, 0,3341 mmol) y se agitó carbonato potásico (8 horas. La mezcla de reacción se filtró a través de un di mediante un método de HPCL-EM de fase inversa us durante 15 minutos (fase móvil A = H2O (5 HCl mM) y f [3-[2-[1-(trifluorometil)ciclopropil]etoxi]pirazol-1-il]-2-[(4s) 31%). IEN-EM m/z calc. 625,17377, encontrado 626,5 ( cloroformo-d) 8 8,43 - 8,36 (m, 1H), 8,32 (d, J = 8,5 Hz, 3-[2-[1-(trifluorometil)ciclopropil]etoxi]pirazol-1-il]piridin-3-Paso A), (4S)-2,2,4-trimetilpirrolidina (sal de clorhidrato) g, 0,5788 mmol) en DMSO (2,0 mL) a 130°C durante 15 filtro Whatman (puradisc 25 TF) y el filtrado se purificó un gradiente dual desde el 50-99% de la fase móvil B óvil B = CH3CN) para producir N-(2-clorofenil)sulfonil-6--trimetilpirrolidin-1-il]piridin-3-carboxamida (18,7 mg, Tiempo de retención: 2,35 minutos. 1H RMN (400 MHz, 8,21 (d, J = 2,8 Hz, 1H), 7,55 (d, J = 8 , 6 Hz, 1H), 7,53 -7,44 (m, 3H), 5,94 (d, J = 2,7 Hz, 1H), 4,40 (t, J = 7,1 H 2,73 - 2,55 (m, 1H), 2,16 (dd, J = 12,4, 7,7 Hz, 1H), 2,0 3H), 1,41 (s, 3H), 1,20 (d, J = 6,6 Hz, 3H), 1,09 - 0,97 ( Ejemplo sintético 44: Síntesis del Compuest (trifluorometM)cidopropM)etoxi)-1H-pirazoM-M)-4-(2,2 Paso A: 6-(3-(2-(1-(trifluorometN)ddopropN)etoxi)-1 [0551]
, 3,51 (t, J = 9,5 Hz, 1H), 3,13 (dd, J = 10,6, 8,1 Hz, 1H), = 7,1 Hz, 2H), 1,77 (dd, J = 12,5, 9,6 Hz, 1H), 1,47 (s, , 0,81 - 0,64 (m, 2H).
4: Preparación de (S)-N-(fenMsulfoml)-6-(3-(2-(1-etMpirroMdm-1-M)mcotmamida
zoM-M)-4-doropiridm-3-carboxNato de metilo

[0552] A la solución de 3-(2-(1-(trifluorometil)ciclopropil) carboxilato de metilo (742 mg, 3,60 mmol) en N,N-dime g, 9,82 mmol) y 1,4-diazabiciclo[2.2.2]octano (110 mg, 16 horas. La solución de reacción se enfrió a temperatu añadió agua (50 mL) y se separaron las capas orgánica de cloruro de hidrógeno (15 mL), salmuera (3 x 15 m concentraron. El residuo se purificó mediante cromatog acetato de etilo para producir metilo 6-(3-(2-( 1 -(tr carboxilato (631 mg, 49%) como un sólido blanco. 1H Hz, 1H), 7,85 (s, 1H), 5,96 (d, J = 2,8 Hz, 1H), 4,42 (t, J 2H), 0,76 (m, 2h ). IEN-EM m/z calc. 389,1 encontrado
Paso B: Ácido 6-(3-(2-(1-(trifluorometil)cidopropil)et
[0553]
-1H-pirazol (720 mg, 3,27 mmol) y 4,6-dicloropiridina-3-amida (11 mL) se añadieron carbonato de potasio (9,36 mol). La solución resultante se calentó a 80°C durante biente y se diluyó con éter dietílico (400 mL). Luego se capas orgánicas se lavaron con una solución acuosa 1 secaron sobre sulfato de magnesio, se filtraron y se n columna de gel de sílice usando 0-20% de hexanos -ometil)ciclopropil)etoxi)-1H-pirazol-1-il)-4-cloropiridina-3-250 MHz, CDCh) 8 (ppm): 8,84 (s, 1H), 8,36 (d, J = 2,8 Hz, 2H), 3,96 (s, 3H), 2,11 (t, J = 7,0 Hz, 2H), 1,05 (m, (M1). Tiempo de retención: 7,08 minutos.
H-pirazol-1-il)-4-doropiridin-3-carboxílico

[0554] 6-(3-(2-(1-(trifluorometil)ciclopropil)etoxi)-1H-pira mmol) se disolvió en una mezcla de tetrahidrofurano ( solución acuosa de hidróxido de sodio 2 N (1,4 ml, 2,84 durante 3 horas. Todos los disolventes se eliminaron acuosa 1 de cloruro de hidrógeno hasta que el valor d mL). La capa orgánica se lavó con salmuera (2 x 20 mL para producir ácido 6-(3-(2-(1-(trifluorometil)cicloprop 97%) como un sólido blanco. 1H RMN (250 MHz, DMS 6,21 (d, J = 3,0 Hz, 1H), 4,36 (t, J = 7,0 Hz, 2H), 2,11 (t, 375,1 encontrado 376,0 (M+1)+ Tiempo de retención: 5,
Paso C: 4-Cloro-N-(fenilsulfonil)-6-(3-(2-(1-(trifluoro
[0555]
-il)-4-cloropiridin-3-carboxilato de metilo (553 mg, 1,42 L) y se añadió metanol (3,5 mL), luego se añadió una . La solución resultante se agitó a temperatura ambiente sión reducida. El residuo se acidificó con una solución lcanzó 2 y luego se extrajo con acetato de etilo (3 x 80 secó sobre sulfato de magnesio, se filtró y se concentró i)-1H-pirazol-1-il)-4-cloropiridin-3-carboxílico (534 mg, pm): 8,85 (s, 1H), 8,51 (d, J = 3,0 Hz, 1H), 7,76 (s, 1H), ,0 Hz, 2H), 0,95 (m, 2H), 0,90 (m, 2H). IEN-EM m/z calc. nutos.
cidopropil)etoxi)-1H-pirazol-1-il)nicotinamida

[0556] Ácido 6-(3-(2-(1-(tnfluorometil)ciclopropil)etoxi)-1 y 1,1-carbonildiimidazol (341 mg, 2,11 mmol) en tetrah luego se agitó bencenosulfonamida (220 mg, 1,40 mm La solución de reacción se agitó durante 16 horas más con una solución acuosa saturada de ácido tartárico (2 sulfato de magnesio, se filtró y se concentró. El residu acetato de etilo para producir 4-cloro-N-(fenilsulfo nicotinamida (411 mg, 57%) como un sólido blanco. 1H Hz, 1H), 7,94 (d, J = 6,8 Hz, 2H), 7,67 (s, 1H), 7,59 (m, = 7,0Hz, 2H), 0,94 (m, 2H), 0,89 (m, 2H). IEN-EM m/z c minutos.
Paso D: (S)-N-(fenMsulfoml)-6-(3-(2-( trimetilpirrolid in-1-il)nicotinam ida
[0557]
zoM-il)-4-cloropiridin-3-carboxílico (528 mg, 1,40 mmol) rano (9 mL) durante 2 horas a temperatura ambiente, ,8-diazabiciclo[5.4.0]undec-7-eno (641 mg, 4,21 mmol). iluyó con acetato de etilo (200 mL). La solución se lavó , agua (40 mL), salmuera (40 mL), luego se secó sobre urificó mediante cromatografía en gel de sílice usando (3-(2-(1-(trifluorometil)ciclopropil)etoxi)-1H-pirazol-1-ilo). (250 MHz, DMSO) 8 (ppm): 8,56 (s, 1H), 8,47 (d, J = 2,5 ,16 (d, J = 2,5Hz, 1H), 4,34 (t, J = 7,0Hz, 2H), 2,09 (t, J 14,1 encontrado 515,0 (M1). Tiempo de retención: 6,31
fluorometN)cidopropN)etoxi)-1H-pirazoM-M)-4-(2,2,4-

[0558] A la solución de 4-cloro-N-(fenilsulfonil)-6-(3-(2-( il)nicotinamida (54,6 mg, 0,11 mmol) en dimetilsulfóxido 0,64 mmol) y fluoruro de cesio (97 mg, 0,64 mmol). La mezcla se purificó por HPLC de fase inversa usando trifluoroacético). Las fracciones puras se combinaron trifluoroacético, que se volvió a disolver en 50% de agu nuevo para producir sal de cloruro de hidrógeno de (S) pirazol-1-il)-4-(2,2,4-trimetilpirrolidin-1-il)nicotinamida (2 1H), 8,40 (s, 1H), 8,08 (d, J = 1,3 Hz, 1H), 8,02 (d, J = 8, 1H), 4,34 (d, J = 7,0Hz, 2H), 2,50 (m, 3H), 2,09 (t, J = 7 2H), 0,84 (m, 2H), 0,64 (d, J = 6,0 Hz, 3H). IEN-EM m/z minutos.
Ejemplo sintético 45: Síntesis de los Compuestos 4 (trifluorometN)cidopropN)metoxi)-1H-pirazoM-N)-2-(( isómero 2
Paso A: 2,6-Difluoropiridin-3-carboxilato de tere-buti
[0559]
luorometil)ciclopropil)etoxi)-1H-pirazol- Se añadieron 1-mL) de clorhidrato de (S)-2,2,4-trimetilpirrolidina (96 mg, ión resultante se calentó a 120°C durante 48 horas. La 0% de agua-acetonitrilo (que contenía 0,1% de ácido filizaron para producir el producto como sal de ácido tonitrilo (0,1% de cloruro de hidrógeno) y se liofilizó de nilsulfonil)-6-(3-(2-(1-(trifluorometil)ciclopropil)etoxi)-1H-g, 42%). 1H RMN (250 MHz, DMSO) 8 (ppm): 12,65 (s, 2H), 7,70 (m, 3H), 7,24 (s, 1H), 6,05 (dd, J = 1,3, 2,5Hz, , 2H), 2,10 (m, 1H), 1,91 (m, 1H), 1,53 (s, 6H), 0,87 (m, 91,2 encontrado 592,6 (M1). Tiempo de retención: 2,88
y 47: N-(ammo(oxo)(feml)-A6-sulfanoMideno)-6-(3-((1-,4-trimetMpirroNdm-1-M)mcotmamida isómero 1 e
[0560] Se disolvió ácido 2,6-difluoropiridin-3-carboxílico Se añadió dicarbonato de di-ferc-butilo (1,5 g, 6,9 mmol) 3,78 mmol). La mezcla se convirtió en una suspensión, heterogénea se agitó a temperatura ambiente durante metilo (30 mL). La capa orgánica se lavó sucesivament bicarbonato de sodio al 5% p/v (10 mL), se secó sobr reducida. El residuo se purificó por cromatografía en gel para proporcionar 2,6-difluoropiridina-3-carboxilato de f color amarillo claro. 1H RMN (300 MHz, CDCh) ppm 1,6 1H). 19F RMN (282 MHz, CDCla) ppm -61,5 - -61,4 (m, I
Paso B: 2-Fluoro-6-[3-[[1-(trifluorometN)cidopropN]m
[0561]
, 6,3 mmol) en 2-metil tetrahidrofurano anhidro (12 mL). na porción seguido de 4-(dimetilamino)piridina (462 mg, na gran cantidad de desprendimiento de gas. La mezcla de semana y luego se diluyó con éter ferc-butílico de HCl acuoso 1 M (10 mL), solución acuosa saturada de to de sodio anhidro, se filtró y se concentró a presión ice, eluyendo con acetato de etilo al 0-30% en heptanos, tilo (360 mg, 26% de rendimiento) como un aceite de 9H), 6,88 (ddd, J = 8,4, 3,0, 0,5 Hz, 1H), 8,40 - 8,47 (m, ,3 (t, J = 8,6 Hz, 1 F).
i]pirazoM-N]piridma-3-carboxNato de tere-butilo

[0562] 2,6-Difluoropiridina-3-carboxilato de ferc-butilo pirazol (1,8 g, 8,7 mmol) y carbonato de potasio recién m (20 mL). La mezcla se agitó a 20°C bajo nitrógeno dura La capa orgánica se lavó con agua (3 x 30 mL), se se presión reducida. El residuo se purificó mediante crom 15% en heptanos, para producir 2-fluoro-6-[3-[[1-(trifluo ferc-butilo (1,9 g, rendimiento del 57%) como un sólido 1,13 - 1,18 (m, 2H), 1,60 (s, 9H), 4,40 (s, 2H), 6,00 (d, 1H), 8,37 (t, J = 8,4 Hz, 1H). 19F RMN (282 MHz, CDCla) 402,1.
Paso C: Ácido 2-fluoro-6-[3-[[1-(trifluorometil)cidopr
[0563]
g, 8,4 mmol), 3-[[1-(trifluorometil)ciclopropil]metoxi]-1H-(1,7 g, 12 mmol) se añadieron a dimetilsulfóxido anhidro 6 horas y luego se diluyó con acetato de etilo (100 mL). bre sulfato de sodio anhidro, se filtró y se concentró a fía en gel de sílice, eluyendo con acetato de etilo al 0-til)ciclopropil]metoxi]pirazol-1-ilo.]piridin-3-carboxilato de o. 1H Rm N (300 m Hz , CDCh) ppm 0,92 - 0,99 (m, 2H), 8 Hz, 1H), 7,61 (d, J = 8,4 Hz, 1H), 8,30 (d, J = 2,8 Hz, -69,7 (s, 3F), -62,2 (d, J = 9,2 Hz, 1F). CLEM: [M+H]+ =
etoxi]pirazol-1-il]p irid in-3-carboxílico

[0564] Se añadió ácido trifluoroacético
(trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridina-3-car mL). La mezcla se agitó a 40°C durante 4 horas, des mezcla se concentró a presión reducida y el residuo s proporcionar ácido 2-fluoro-6-[3-[[1-(trifluorometil)c rendimiento del 98%) como un sólido blanco. 1H Rm N 6,24 (d, J = 2,8 Hz, 1H), 7,66 (dd, J = 8,3, 1,0 Hz, 1H), 8, (282 MHz, DMSO-cfe) ppm -67,9 (s, 3F), -63,2 (d, J = 7,9
Paso D: 2-Fluoro-6-[3-[[1-(trifluorometil)ciclopropil]m
[0565]
mL) a una solución de 2-fluoro-6-[3-[[1-to de ferc-butilo (1,9 g, 4,7 mmol) en diclorometano (16 e lo cual la TLC mostró una conversión completa. La ró con heptanos, se filtró y se secó a alto vacío para opil]metoxi]pirazol-1-il]piridina-3-carboxílico (1,6 g, MHz, DMSO-cfe) ppm 1,06 - 1,11 (m, 4H), 4,39 (s, 2H), J = 8,3 Hz, 1H), 8,47 (dd, J = 9,6, 8,4 Hz, 1H). 19F RMN 1F). CLEM: [M+H]+ = 346,1.
]pirazol-1-il]piridin-3-carboxamida

[0566] A una suspensión de ácido 2-fluoro-6-[3-[[1-(triflu g, 4,6 mmol) en diclorometano (20 mL) se añadió, segui gota a gota de cloruro de oxalilo (0,52 ml, 6,0 mmol). La hasta que cesó el burbujeo. El disolvente se eliminó a tetrahidrofurano anhidro (10 mL) y se añadió a una mez (5 mL) enfriada con un baño de agua helada. La reacció diluyó con acetato de etilo (100 mL), se lavó con agua ( concentró a presión reducida para producir 2-fluor carboxamida (1,55 g, rendimiento del 98%) como un só 4H), 4,38 (s, 2H), 6,21 (d, J = 2,7 Hz, 1H), 7,61 (dd, J = 8 Hz, 1H), 8,4 (d, J = 2,7 Hz, 1H). 19F RMN (282 MHz, CD = 345,1.
Paso E: 2-fluoro-W-femlsulfaml-6-[3-[[1-(tr¡fluoromet¡l
[0567]
etil)cidopropil]metoxi]pirazol-1-il]piridin-3-carboxílico (1,6 una gota de W,A/-dimetilformamida mediante la adición ión se agitó a temperatura ambiente durante dos horas ón reducida. El sólido blanco resultante se disolvió en hidróxido de amonio al 28% (10 mL) y tetrahidrofurano gitó a temperatura ambiente durante 1 hora y luego se ), se secó sobre sulfato de sodio anhidro, se filtró y se [[1 -(trifluorometil)ciclopropil]metoxi]pirazol-1 -il]piridin-3-lanco. 1H RMN (300 MHz, CDCh) ppm 1,06 - 1,11 (m, 5 Hz, 1H), 7,76 (d, J = 8,2 Hz, 2H), 8,33 (dd, J = 9,4, 8,4 m -67,9 (s, 3F), -66,3 (d, J = 8,9 Hz, 1F). CLEM: [M+H]+
prop¡l]metox¡]p¡razol-1-¡l]pmdm-3-carboxam¡da

[0568] Bromo (0,14 ml, 2,7 mmol) se añadió lentamente en acetonitrilo anhidro (4 mL) a 0°C. La reacción se agitó solución. Esta solución se añadió a una solución de 2-flu 3-carboxamida (940 mg, 2,73 mmol) en acetonitrilo anhid se agitó a temperatura ambiente durante la noche, luego (10 mL). El sólido marrón residual se purificó mediante c 0-30% en heptanos, para dar 2-fluoro-W-fenilsulfan carboxamida (380 mg, 31% de rendimiento) como un s 2H), 1,14 - 1,19 (m, 2H), 4,41 (s, 2H), 6,03 (d, J = 3,0 H 8,4, 1,8 Hz, 1H), 7,84 (d, J = 15 Hz, 1H), 8,28 (d, J = 3,0
Paso F: rac-W-(bencenosulf¡ml)-2-fluoro-6-[3-carboxam¡da
[0569]
suspensión de disulfuro de difenilo (596 mg, 2,73 mmol) peratura ambiente durante 2 minutos para producir una [3-[[1-(trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridinmL) y piridina (4 mL) a 0°C. La mezcla oscura resultante ncentró a presión reducida y se co-evaporó con tolueno ografía en gel de sílice, eluyendo con acetato de etilo al -[[1-(trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-3-lanco. 1H RMN (300 Mh z , CDCla) ppm 0,93 - 1,00 (m, , 7,20 - 7,26 (m, 1H), 7,30 - 7,42 (m, 4H), 7,73 (dd, J = H), 8,63 (t, J = 9,0 Hz, 1H). CLEM: [M+H]+ = 453,0.
r¡fluoromet¡l)c¡cloprop¡l]metox¡]p¡razoM-¡l]p¡r¡dm-3-

[0570] Se añadió ácido mefa-cloroperoxibenzoico (469 6-[3-[[1-(trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin mL) a 0°C y la reacción se agitó a la misma tempera diclorometano (70 mL), se lavó sucesivamente con tiosu secó sobre sulfato de sodio anhidro, se filtró y se co cromatografía en gel de sílice, eluyendo con acetato de 2-fluoro-6-[3-[[1-(trifluorometil)ciclopropil]metoxi]pirazol-1 rendimiento) como un sólido blanco. 1H RMN (300 MHz (d, J = 12,1, 1H), 4,43 (d, J = 12,1, 1H), 6,03 (d, J = 3,0 (m, 2H), 8,24 (d, J = 3,0 Hz, 1H), 8,40 (d, J = 12 Hz, 1H 69,7 (s, 3F), -63,2 (t, J = 10,5 Hz, 1F). CLEM: [M+H]+ = 77%, 2,1 mmol) a una solución de 2-fluoro-W-fenilsulfanilrboxamida (860 mg, 1,90 mmol) en diclorometano (30 durante 1 hora. La mezcla de reacción se diluyó con de sodio al 10% p/v, bicarbonato de sodio al 5% p/v, se ó a presión reducida. El residuo se purificó mediante l 0-40% en heptanos, para producir W-(benzonesulfinil)-idin-3-carboxamida racémica (700 mg, 78% de h) ppm 0,92 - 1,00 (m, 2H), 1,13 - 1,20 (m, 2H), 4,38 H), 7,56 - 7,65 (m, 3H), 7,70 - 7,78 (m, 1H), 7,80 - 7,88 (t, J = 8,5 Hz, 1H). 19F RMN (282 MHz, CDCh) ppm -
Paso G: W-BencenosulfmN)-6-[3-[[1-( trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0571]
rometN)c¡cloprop¡l]metox¡]p¡razol-1-N]-2-[(4S)-2,2,4-

[0572] Se disolvió clorhidrato de (4S)-2,2,4-tri dimetilformamida anhidra (10 mL), la mezcla se enfrió c 60% en aceite mineral, 7,2 mmol). La reacción se agitó nuevo a 0°C. Una solución de N-(bencenosulfinN)-2-fluor carboxamida racémica (960 mg, 2,05 mmol) en N,N-temperatura ambiente durante 10 minutos, la mezcla de de conversión; había mucho hidruro de sodio sin consu se agitó a temperatura ambiente durante la noche. La con acetato de etilo (80 mL). La capa orgánica se lavó c de sodio anhidro, se filtró y se concentró a presión redu sílice, eluyendo con acetato de etilo al 0-50% en heptan partida se combinaron y se concentraron a presión redu y diclorometano (10 mL) durante 30 minutos y luego producir N-(bencenosulfinil)-6-[3-[[1-(trifluorometil)ciclopr il]piridin-3-carboxamida (500 mg, 8 8 % de pureza por CL en la siguiente etapa sin purificación adicional. CLEM: [
Paso H: Síntesis de W-(femlsulfomm¡do¡l)-6-[3-[[1-( trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0573]
irroMdina (400 mg, 2,67 mmol) en N,N-ua helada e hidruro de sodio (287 mg de dispersión al peratura ambiente durante 10 minutos y se enfrió de -[[1-(trifluorometN)cidopropN]metoxi]pirazol-1-N]piridin-3-lformamida (10 mL) se añadió. Después de agitar a ión se agitó a 50°C durante 3 horas (CLEM mostró 60% e añadió tetrahidrofurano anhidro (0,5 mL) y la reacción se inactivó con agua (10 mL) a 0°C y luego se extrajo a (3 x 20 mL), salmuera (10 mL), se secó sobre sulfato El residuo se purificó mediante cromatografía en gel de s fracciones que contenían el producto y el material de El residuo se agitó en una mezcla de heptanos (10 mL) ltró. El filtrado se concentró a presión reducida para etoxi]pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-8% de rendimiento) como un sólido amarillo que se usó = 562,2.
romet¡l)c¡cloprop¡l]metox¡]p¡razol-1-¡l]-2-[(4S)-2,2,4-

[0574] Se añadió amoníaco (7,8 ml de una solución 0,5 6-[3-[[1-(trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4 8 8 % de pureza, 0,78 mmol) en acetonitrilo anhidro (20
en una porción (la mezcla se volvió naranja) y la rea clorosuccinimida (12 mg, 0,090 mmol) y la reacción se inactivó con una solución acuosa de tiosulfato de sodio orgánica se lavó sucesivamente con una solución acuos sulfato de sodio anhidro, se filtró y se concentró a presió gel de sílice, eluyendo con acetato de etilo al 5-45% e trituró con acetonitrilo (3 mL) para proporcionar una mez (3-((1-(trifluorometil)ciclopropil)metoxi)-1H-pirazol-1-il)-2-(Compuesto 45) ( 18 0 mg, 97% de pureza por CLEM, 38 CDCla) ppm 0,89 - 1,06 (m, 5 H), 1,08 - 1,18 (m, 2H), 1 2,59 - 2,71 (m, 0,4 H), 2,82 - 2,96 (m, 0,6 H), 3,18 (t, J 2H), 5,90 (d, J = 2,6 Hz, 1H), 6,25 (ancho s., 2H), 6,90 -8.09 (m, 2,6 H), 8,16 (d, J = 8,2 Hz, 0,4 H), 8,20 -8,23 ( [M+H]+ = 577,2.
[0575] Los isómeros se separaron por cromatografía de fl Lux-1 (250 X 2 1 , 2 mm) de 5 pm y eluyendo con 2 0 % Me ioxano, 3,9 mmol) a una solución de N-(bencenosulfinil)-,4-trimetilpirrolidin-1-il]piridin-3-carboxamida (500 mg, °C. Se añadió N-clorosuccinimida (120 mg, 0,90 mmol) se agitó a 0°C durante una hora. Se añadió más N-a la misma temperatura durante 30 minutos y luego se p/v y se extrajo con acetato de etilo (50 mL). La capa icarbonato de sodio al 5% p/v, salmuera, se secó sobre ucida. El residuo se purificó mediante cromatografía en tanos, para producir un sólido blanco (300 mg) que se astereomérica de N-(amino(oxo)(fenN)-A6sulfaniliden)-6 -,2,4-trimetilpirrolidin-1-il)nicotinamida
rendimiento) como un sólido blanco. 1H RMN (300 MHz, 1,72 (m, 7 H), 1,81 - 1,95 (m, 1H), 2,07 - 2,40 (m, 1H), Hz, 0,4 H), 3,29 (t, J = 10,7 Hz, 0,6 H), 4,32 - 4,43 (m, (m, 1H), 7,48 -7,57 (m, 2H), 7,58 - 7,66 (m, 1H), 7,97 -). 19F RMN (282 MHz, CDCh) ppm -69,7 (s, 3F). CLEM:
quiral supercrítica utilizando una columna Phenomenex 0% de CO2 con una velocidad de flujo de 70 ml/minuto.
[0576] Diastereoisómero 1 (Compuesto 46): > 98% de I de retención: 1,82 minutos; 1H RMN (400 MHz, DMSO-7,75 (s, 2H), 7,68 - 7,49 (m, 3H), 6,85 (d, J = 8,1 Hz, 1 1H), 2,76 (dd, J = 10,6, 7,1 Hz, 1H), 2,21 (dq, J = 12,1, (t, J = 12,0 Hz, 1H), 1,09 (dd, J = 4,5,3,1 Hz, 4H), 0,92 ( [0577] Diastereoisómero 2 (Compuesto 47): > 98% de. I de retención: 1,81 minutos; 1H RMN (400 MHz, DMSO-d 7,69 - 7,55 (m, 3H), 6,87 (d, J = 8,1 Hz, 1H), 6,11 (d, J 2,21 - 2,03 (m, 1H)), 1,80 (dd, J = 11,8, 5,4 Hz, 1H), 1, 5,6, 2,1 Hz, 4H), 0,71 (d, J = 6,2 Hz, 3H).
Ejemplo sintético 46: Síntesis del Compuesto 48:
N-(Bencenosulfoml)-6-[3-(cidopropoxi)pirazoM-M]-2-[ Paso A: 3-C iclopropoxM H-pirazoM -carboxtato de t [0578]
m/z calc. 576,2131, encontrado 577,4 (M+1)+; Tiempo 8,19 (d, J = 2,7 Hz, 1H), 7,95 (dd, J = 8,1, 1,6 Hz, 3H), 2 (d, J = 2,7 Hz, 1H), 4,35 (s, 2H), 3,08 (t, J = 10,6 Hz, , 1H), 1,97 - 1,79 (m, 1H), 1,55 (d, J = 1,7 Hz, 6H), 1,44 6,2 Hz, 3H).
M m/z calc. 576,2131, encontrado 577,3 (M+1)+; Tiempo ,19 (d, J = 2,7 Hz, 1H), 7,99 - 7,88 (m, 3H), 7,79 (s, 2H), Hz, 1H), 4,44 - 4,28 (m, 2H), 2,63 (t, J = 10,8 Hz, 1H), J = 1,7 Hz, 6H), 1,33 (t, J = 12,2 Hz, 2H), 1,09 (dt, J =
2,2,4-trimetMpirroMdm-1-M]piridma-3-carboxamida
utilo
[0579] A una solución de ciclopropanol (30,8 mg, 0,53 (97,7 mg, 0,531 mmol) y trifenilfosfina (139,3 mg, 0,531 de di-terc-butilo (122,2 mg, 0,531 mmol). La solución s ambiente durante 30 minutos. Luego, la solución de r enfriarse a temperatura ambiente. La solución se diluy salmuera, se secó sobre sulfato de sodio, se filtró y se mediante cromatografía en gel de sílice (hexano y acet producir 3-ciclopropoxi-1H-pirazol-1-carboxilato de terc-224,116, encontrado 225,0 (M+1)+; Tiempo de retención = 2,8 Hz, 1H), 5,93 (d, J = 2,8 Hz, 1H), 4,20-4,15 (m, 1H
Paso B: 3-ciclopropoxM H-pirazol
[0580]
Boc
ol), 2,3-dihidro-3-oxopirazol-1-carboxilato de terc-butilo l) en tolueno anhidro (2 mL), se añadió azodicarboxilato ó con argón durante 1 minuto y se agitó a temperatura ón se calentó a 110°C durante 5 horas más antes de éter (50 mL), se lavó con solución acuosa de NaOH, ntró a presión reducida. El residuo obtenido se purificó etilo, gradiente de acetato de etilo del 0 al 10%) para (52 mg, 46%) como un sólido blanco. IEN-EM m/z calc.
minutos. 1H RMN (250 MHz, CDCh) 8 (ppm) 7,86 (d, J 1 (s, 9H)), 0,85-0,72 (m, 4H).

[0581] A una solución de 3-ciclopropoxi-1H-pirazol-1-car (6 mL) se añadió TFA (667 mg, 0,38 ml, 5,84 mmol). La 3 horas. Todos los disolventes se eliminaron a presión lavó con una solución acuosa saturada de bicarbonato concentró a presión reducida para producir 3-ciclopropo obtenido se utilizó directamente en el siguiente paso.
Paso 3: 2-Cloro-6-(3-ddopropoxMH-pirazoM-N)pirid
[0582]
to de terc-butilo (131 mg, 0,584 mmol) en diclorometano ión resultante se agitó a temperatura ambiente durante ida. El residuo obtenido se disolvió en éter (100 mL), se odio, se secó sobre sulfato de magnesio, se filtró y se pirazol como un aceite amarillo pálido. El producto crudo
-carboxNato de tere-butilo

[0583] 3-ciclopropoxi-1H-pirazol crudo (73 mg, 0,584 m 0,643 mmol), K2CO3 (162 mg, 1,17 mmol) y DABCO (13 solución de reacción se agitó a temperatura ambiente d (100 mL), se lavó con agua (3 x 25 mL) y salmuera (25 sulfato de magnesio, se filtraron y se concentraron a cromatografía en gel de sílice (hexano y diclorometano, cloro-6-(3-ciclopropoxi-1H-pirazol-1-il)piridina-3-carboxilat IEN-EM m/z calc. 335,104, encontrado 336,1 (M+1)+; Tie
Paso D: Ácido 2-cloro-6-(3-cidopropoxMH-pirazoM-
[0584]
), 2,6-didoropiridin-3-carboxNato de terc-butilo (159 mg, 0,117 mmol) se disolvieron en DMF anhidra (1,5 mL). La te 16 horas. La solución de reacción se diluyó con éter ). Las capas orgánicas se separaron, se secaron sobre ión reducida. El residuo obtenido se purificó mediante diente de diclorometano del 0 al 1 0 0 %) para producir 2 -e terc-butilo. (153 mg, 78%) como un aceite pegajoso.
de retención: 6,84 minutos.
dm-3-carboxflico
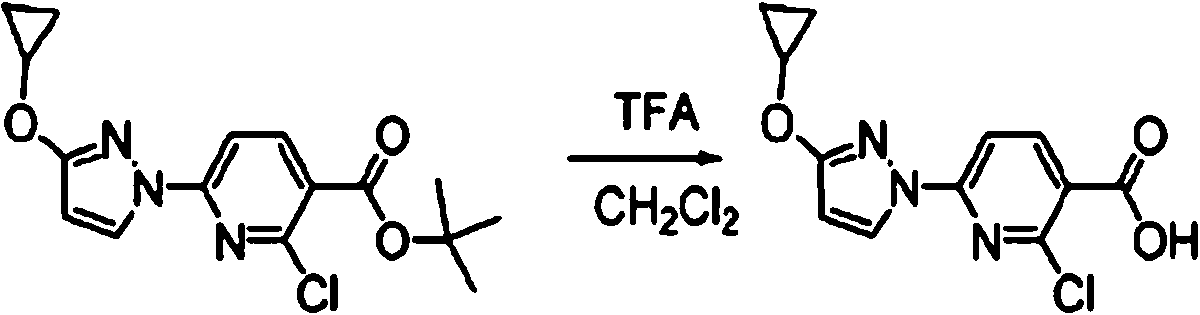
[0585] A una solución de 2-cloro-6-(3-ciclopropoxi-1 0,456 mmol) en diclorometano (2,2 mL) se añadió TFA ( a temperatura ambiente durante 48 horas. Luego se eliminaron a presión reducida. El sólido blanco obteni hexano/éter, 19/1), se sonicó, se filtró, se lavó con he ciclopropoxi-1H-pirazol-1-il)piridina-3-carboxílico (122 m encontrado 279,9 (M+1)+; Tiempo de retención: 4,43 minu (d, J = 3,0 Hz, 1H), 8,39 (d, J = 8,5 Hz, 1H), 7,72 (d, J = 8 0,71 (m, 4H).
Paso E: N-(bencenosulfoml)-2-doro-6-[3-(cidopropoxi [0586]
irazol-1-il)piridin-3-carboxilato de terc-butilo (153 mg, mg, 0,35 ml, 4,56 mmol). La solución resultante se agitó dió 1,2-dicloroetano (2 mL) y todos los disolventes se se suspendió en la mezcla de hexano y éter (10 ml, o (10 mL) y se secó para producir ácido 2-cloro-6-(3-7%) como un sólido blanco. IEN-EM m/z calc. 279,041, . 1H RMN (500 MHz, DMSO-cfe) 8 (ppm) 13,6 (s, 1H), 8,43 z, 1H), 6,28 (d, J = 3,0 Hz, 1H), 4,16-4,13 (m, 1H), 0,79
azoM-M]piridm-3-carboxamida

[0587] Ácido 2-cloro-6-[3-(ciclopropoxi)pirazol-1-il]piridi HATU (hexafluorofosfato de 3-óxido de 1-[bis(dimetilami mmol) y DIEA (diisopropiletilamina) (38 pL, 0,2182 mmol) 16 h. La mezcla de reacción se filtró y se purificó en HP 25-75% en agua que contenía HCl 5 mM para dar N-(ben carboxamida 1H RMN (400 MHz, DMSO-d6 ) 8 12,97 (s, 1 J = 9,4 Hz, 2H), 7,77 (s, 1H), 7,69 (d, J = 6,0 Hz, 3H), 6,3 carboxílico (30 mg, 0,1073 mmol) en DMF (600,0 pL), etileno]-1H-1,2,3-triazolo[4,5-b]piridinio) (85 mg, 0,2235 combinaron y se agitaron a temperatura ambiente durante de fase inversa utilizando un gradiente de acetonitrilo al osulfonil)-2-cloro-6-[3-(ciclopropoxi)pirazol-1-il]piridina-3-8,43 (d, J = 2,8 Hz, 1H), 8,12 (d, J = 8,3 Hz, 1H), 8,01 (d, ,26 (m, 1H), 4,16 (s, 1H), 0,76 (s, 4H). IEN-EM m/z calc.
418,05026, encontrado 419,0 (M+1)+; Tiempo de retenci Paso F: N-(Bencenosulfoml)-6-[3-(ddopropoxi carboxamida
[0588]
,63 minutos (ejecución de 3 min).
zoM-N]-2-[(4S)-2,2,4-trimetMpirroNdm-1-N]piridm-3-

[0589] Una mezcla de N-(bencenosulfonN)-2-doro-6-[ trimetilpirrolidina (sal de clorhidrato) (aproximadamente 0,2370 mmol), K2CO3 (aproximadamente 72,01 mg, 0,5 La reacción se filtró y se purificó en HPLC de fase inver en agua que contenía HCl 5 mM para dar N-trimetilpirrolidin-1 -il]piridin-3-carboxamida (5,6 mg, 1 1 % (M+1)+; Tiempo de retención: 1,98 minutos] (ejecución d
Ejemplo sintético 47: Síntesis del (trifluorometN)ddopropN]metoxi]pirazoM-Mo]-2-[(4S)-
Paso A: (1-(Trifluorometil)ciclopropil)metanol
[0590]
opropoxi)pirazol-1-N]piridin-3-carboxamida, (4S)-2,2,4-mg, 0,1604 mmol), CsF (aproximadamente 36,00 mg, mol) en DMSO (0,5 mL) se agitó a 140°C durante 16 h. rificada utilizando un gradiente de acetonitrilo al 25-75% enosulfonN)-6-[3-(cidopropoxi)pirazol-1-N]-2-[(4S)-2,2,4-pasos) IEN-e M m/z calc. 495,19403, encontrado 496,0 in)
ompuesto 49: W-(3-metoxifeml)sulfoml-6-[3-[[1--trimetMpirroNdm-1-N]piridm-3-carboxamida

[0591] Se disolvió ácido 1-(trifluorometil)ciclopropano-1-mL). La mezcla de reacción se enfrió a 0 °C. Se añadió 1,30 eq.). La mezcla de reacción se agitó durante la noch de reacción se enfrió a 0°C. Se añadió gota a gota H dietílico (2 x 25 mL). Las fases orgánicas combinadas s sodio, se filtraron y se evaporaron al vacío (Tbaño <30° mmol, 70% de rendimiento) como un aceite incoloro. 1 (m, 2H), 0,82-0,75 (m, 2H).
Paso B: 1-(3-hidroxipirazol-1-il)etanona
[0592]
xílico (858 mg, 5,57 mmol, 1,00 eq.) en éter dietílico (15 orciones hidruro de litio y aluminio (274 mg, 7,24 mmol, dejó que alcanzara la temperatura ambiente. La mezcla uoso, 1 N, 25 mL). La fase acuosa se extrajo con éter ron con salmuera (25 mL), se secaron sobre sulfato de a dar (1-(trifluorometil)ciclopropil)metanol (547 mg, 3,90 N (CDC13): d 3,73 (s, 2H), 1,58 (ancho, 1H), 1,07-1,01

[0593] Se cargó un matraz de fondo redondo de 100 1H-pirazol-5-ol (4,97 g, 59,11 mmol) y piridina (25 ml, 3 una solución de anhídrido acético (5,6 ml, 59,35 mmo minutos. Después, la mezcla se agitó a 95°C durante tres El residuo sólido se trituró en 40 ml de éter dietílico, hidroxipirazol-1-il)etanona (6,96 g, 93%). 1H RMN (400 (d, J = 3,0 Hz, 1H), 2,48 (s, 3H).
Paso C: 1-(3-((1-(trifluorometil)cidopropil)metoxi)-1H ipado con una barra de agitación y un condensador con mol). La mezcla se agitó a 95°C. Se añadió gota a gota piridina ( 10 ml, 123,6 mmol) durante un período de 3 s más. Los disolventes se eliminaron a presión reducida. ró, se lavó con éter dietílico y se secó para dar 1-(3-DMSO-cfe) 8 10,96 (s, 1H), 8,13 (d, J = 3,0 Hz, 1H), 6,01
ol-1-il)etan-1-ona
[0594]

[0595] 1-(3-H¡droxMH-pirazol-1-il)etan-1-ona (443 mg, (1-(trifluorometil)cidopropil)metanol (547 mg, 3,90 mm mezcla de reacción se enfrió a 0°C. Se añadió gota a mmol, 1,20 eq.) (manteniendo la temperatura <5°C). La el fin de semana. La evaporación de los volátiles al va crudo se purificó por gel de sílice cromatografía eluyen (t^fluoromet¡l)c¡cloprop¡l)metox¡)-1H-p¡razol-1-¡l)etan-1-u blanco. 1H RMN (CDCl3): 88,06 (d, 1H), 5,99 (d, 1H), 4, 19F RMN (CDCl3): 8 -69,77.
Paso D: 3-((1-(TrifluorometN)ddopropN)metoxi)-1H-
[0596]
mol, 1,00 eq.) se disolvió en THF (8 mL). Se añadieron eq.) y trifenilfosfina (1,10 g, 4,21 mmol, 1,20 eq.). La azodicarboxilato de diisopropilo (829 ml, 851 mg, 4,21 la de reacción se agitó a temperatura ambiente durante un aceite ligeramente amarillo (2,88 g). El material en 0-25% acetato de etilo en heptanos para dar 1-(3-((1-1 mg, 2,82 mmol, rendimiento del 80%) como un sólido 2H), 2,57 (s, 3H), 1,18-1,12 (m, 2H), 0,98-0,90 (m, 2H).
l

[0597] Se disolvió 1-(3-((1-(t^fluoromet¡l)c¡cloprop¡l)met en MeOH (30 mL). Se añadió NaOH (acuoso, 30%, 421 agitó a temperatura ambiente durante la noche. La evap El residuo se repartió entre acetato de etilo (25 mL) y a x 25 mL). Las fases orgánicas combinadas se lavaron co y se evaporaron a vacío para dar 3-((1-(t^fluoromet¡l)c¡ del 95%) como un aceite ligeramente amarillo. 1H RMN (s, 2H), 1,14-1,08 (m, 2H), 0,96-0,89 (m, 2H). 19F RMN
Paso E: 2-doro-6-[3-[[1-(trifluorometM)ddopropM]me
[0598]
H-p¡razol-1-¡l)etan-1-ona (695 mg, 2,80 mmol, 1,00 eq.) 60 mg, 4,20 mmol, 1,50 eq.). La mezcla de reacción se n de los volátiles a vacío dio un sólido blanco (940 mg).
5 mL). La fase acuosa se extrajo con acetato de etilo (2 uera (25 mL), se secó sobre sulfato de sodio, se filtraron p¡l)metox¡)-1H-p¡razol (548 mg, 2,66 mmol, rendimiento l3): 89,10 (ancho, 1H), 7,36 (d, 1H), 5,77 (d, 1H), 4,29 3): 8 -69,75.
irazoM-M]piridma-3-carboxMato de tere-butilo

[0599] 2,6-d¡clorop¡r¡d¡na-3-carbox¡lato de tere-butil (t^fluoromet¡l)c¡cloprop¡l]metox¡]-1H-p¡razol (375 mg, 1, mg, 2,183 mmol) (recién molidos) se combinar diazabiciclo[2.2.2]octano (aproximadamente 40,81 mg, bajo nitrógeno durante 16 horas. La mezcla de reacción separaron las dos fases. Los orgánicos se lavaron con El material crudo se purificó mediante cromatografía en para dar tere-butilo 2-cloro-6-[3-[[1-(tr¡fluoromet¡l)c¡clopro EM m/z calc. 417,1067, encontrado 418,1 (M+1)+; Tiem
Paso F: Ácido 2-doro-6-[3-[[1-(trifluorom etil)cidopro
[0600]
proximadamente 451,3 mg, 1,819 mmol), 3-[[1-mol), y carbonato de potasio (aproximadamente 301,7 n d Ms O anhidro (9,026 mL). Se añadió 1,4-8 mmol) y la mezcla se agitó a temperatura ambiente luyó con acetato de etilo (10 mL) y agua (2 x 5 mL) y se era, se secaron sobre sulfato de sodio y se evaporaron. sílice eluyendo con acetato de etilo al 0-30% en hexanos tox¡]p¡razol-1-¡l]p¡r¡d¡n-3-carbox¡lato (620 mg, 82%) IEN-retención: 0,85 minutos.
etoxi]p irazol-1-il]p irid in-3-carboxílico
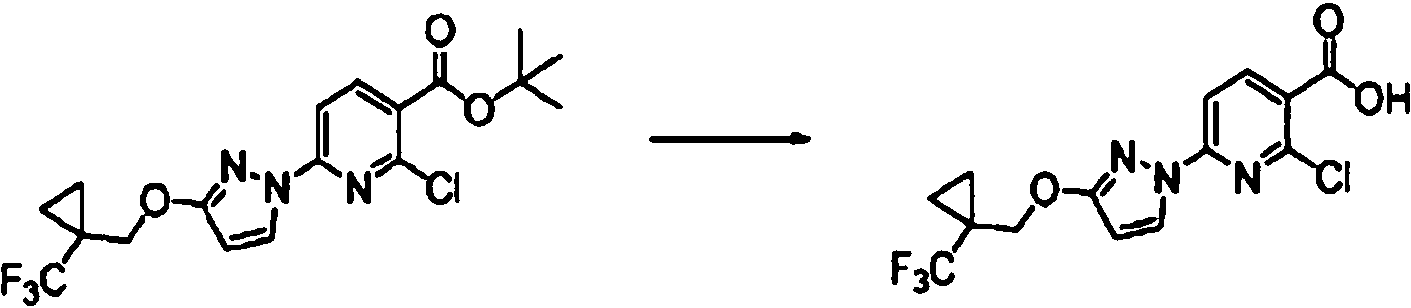
[0601] 2-Cloro-6-[3-[[1-(trifluorometil)cidopropil]metoxi] mmol) y TFA (aproximadamente 1,692 g, 1,143 mL, 1 40°C durante 16 h. La reacción se evaporó hasta un sól nuevo para dar ácido 2-cloro-6-[3-[[1-(trifluorometil)cicl IEN-EM m/z calc. 361,0441, encontrado 362,1 (M+1)+;
Paso G: 2-C loro-N-(3-metoxifenil)sulfonil-6-[3 carboxamida
[0602]
l-1-il]piridin-3-carboxilato de ferc-butilo (620 mg, 1,484 mol) se combinaron en DCM (5 mL) y se calentaron a lanco. Se añadieron hexanos y la mezcla se evaporó de il]metoxi]pirazol-1-il]piridin-3-carboxílico (500 mg, 93%) o de retención: 0,66 minutos.
(trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-3-

[0603] Se combinaron ácido 2-cloro-6-[3-[[1-(trifluorom 0,55 mmol) y carbonildiimidazol (110 mg, 0,66 mmol) horas. Se añadió 3-metoxibencenosulfonamida (104 mg, de reacción se agitó durante 2 ha temperatura ambient etilo y se lavó con 10 ml de ácido cítrico acuoso 1M. La extractos orgánicos combinados se lavaron con salmu presión reducida. El material crudo se purificó mediante metanol al 0-10% en diclorometano (trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-3-carb encontrado 531,1 (M+1)+; Tiempo de retención: 0,72 mi
Paso H: W-p-metoxifeml)sulfoml-6-[3-[[1-( trim etilpirrolid in-1-il]pirid in-3-carboxam ida
[0604]
lopropil]metoxi]pirazol-1-il]piridin-3-carboxílico (200 mg, F (2 mL) y se agitó a temperatura ambiente durante 2 mmol), seguido de DBU (0,25 ml, 1,66 mmol) y la mezcla mezcla de reacción se diluyó con 10 ml de acetato de acuosa se extrajo con acetato de etilo (2 x 10 mL) y los e secaron sobre sulfato de sodio y se concentraron a atografía en gel de sílice eluyendo con un gradiente de ra dar 2-cloro-N-(3-metoxifenil)sulfonil-6-[3-[[1-ida (217 mg, 74%) IEN-EM m/z calc. 530,06384,
rometil)ciclopropil]metoxi]pirazoM-il]-2-[(4S)-2,2,4-

[0605] 2-Cloro-N-(3-metoxifenil)sulfonil-6-[3-[[1-(trifluor (120 mg, 0,23 mmol), (4S)-2,2,4-trimetilpirrolidina (sal de mg, 1,25 mmol) se combinaron en DMSO. (600 mL) y entre acetato de etilo y agua. Los orgánicos se separaro se secaron sobre sulfato de sodio y se evaporaron. El sílice eluyendo con metanol al 0-10% en (trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4 EM m/z calc. 607,20764, encontrado 608,3 (M+1)+; tiem 8 12,43 (s, 1H), 8,20 (d, J = 2,8 Hz, 1H), 7,82 (d, J = 8,2 (m, 1H), 6,92 (d, J = 8,2 Hz, 1H), 6,15 (d, J = 2,8 Hz, 1 2,35 -2,28 (m, 1H), 2,20 -2,03 (m, 1H), 1,84 (dd, J = 11 1H), 1,12 -1,05 (m, 4H), 0,67 (d, J = 6,2 Hz, 3H).
Ejemplo sintético 48: Síntesis del (trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)l)ciclopropil]metoxi]pirazol-1-il]piridina-3-carboxamida
idrato) (107 mg, 0,71 mmol) y carbonato de potasio (173 lentaron a 130°C durante 16 h. La reacción se repartió lavaron con una solución de ácido cítrico 1 M, salmuera, rial crudo se purificó mediante cromatografía en gel de rometano para dar W-(3-metoxifen¡l)sulfonil-6-[3-[[1-tilpirrolidin-1-il]piridin-3-carboxamida (92 mg, 67%) lEN-retención: 2,17 minutos. 1H RMN (400 MHz, DMSO-cfe) 1H), 7,61 - 7,53 (m, 2H), 7,48 - 7,45 (m, 1H), 7,32 - 7,27 2 -4,31 (m, 2H), 3,84 (s, 3H), 2,45 (d, J = 10,5 Hz, 1H), 6 Hz, 1H), 1,53 (d, J = 10,9 Hz, 6H), 1,38 (t, J = 12,1 Hz,
Compuesto 50: W-(2-fluorofeml)sulfoml-6-[3-[[1--trimetilpirrolidin-1-il]piridin-3-carboxamida
Paso A: 2-Cloro-W-(2-fluorofeml)sulfoml-6-[3-[ carboxamida
[0606]
(trifluorometN)cidopropN]metoxi]pirazoM-N]piridm-3-

[0607] Ácido 2-doro-6-[3-[[1-(trifluorometil)cidopropil]me carbonildiimidazol (97 mg, 0,6 mmol) se combinaron en 30 minutos. Se añadió 2-fluorobencenosulfonamida (11 mezcla de reacción se agitó durante 3 h a temperatura a de etilo y se lavó con 10 ml de ácido cítrico acuoso concentraron a presión reducida. El material crudo se p un gradiente de metanol al 0-8% en dicloro (trifluorometil)ciclopropilo]metoxi]pirazol-1-il]piridin-3-carb encontrado 519,1 (M+1)+; Tiempo de retención: 0,70 min
Paso B: W-(2-fluorofeml)sulfoml-6-[3-[[1-(t trimetilpirrolid ina-1-il]p iridin-3-carboxam ida
[0608]
i]pirazol-1-il]piridin-3-carboxílico (181 mg, 0,5 mmol) y F (2,5 mL) y se agitaron a temperatura ambiente durante g, 0,65 mmol), seguido de DBU (0,09 ml, 0,6 mmol) y la nte. La mezcla de reacción se diluyó con 10 ml de acetato Los orgánicos se secaron sobre sulfato de sodio y se có mediante cromatografía en gel de sílice eluyendo con no para dar 2-cloro-N-(2-fluorofenil)sulfonil-6-[3-[[1-mida (180 mg, 69%) IEN-EM m/z calc. 518,0439, .
orometN)cidopropN]metoxi]pirazoM-N]-2-[(4S)-2,2,4-

[0609] 2-Cloro-N-(2-fluorofenil)sulfonil-6-[3-[[1-(trifluorom (78 mg, 0,15 mmol), (4S)-2,2,4-trimetilpirrolidina (sal de mg, 0,9 mmol) se combinaron en DMSo (600 mL) y se filtró y se purificó mediante CL/EM utilizando un gradient (2-fluorofenil)sulfonil-6-[3-[[1-(trifluorometil)ciclopropil]me carboxamida (28 mg, 31%) IEN-EM m/z calc. 595,1876,
Ejemplo sintético 49: Síntesis del
(trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-
Paso A: 2-C loro-W-p-fluorofem l)sulfom l-6-[3-[ carboxamida
[0610]
)ciclopropil]metoxi]pirazol-1-ilo]piridina-3-carboxamida
hidrato) (67 mg, 0,45 mmol) y carbonato de potasio (124 ntaron a 130°C durante 16 h. La mezcla de reacción se e acetonitrilo al 30-99% en HCl acuoso 5 mM para dar N-i]pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-il]piridin-3-ontrado 596,3 (M+1)+; tiempo de retención: 2,08 minutos.
mpuesto 51: N-(3-fluorofenil)sulfonil-6-[3-[[1-4-trimetilpirrolidin-1-il]piridin-3-carboxamida
(trifluorometil)ciclopropN]metoxi]pirazoM-N]piridm-3-

[0611] Ácido 2-cloro-6-[3-[[1-(trifluorometil)ciclopropil]m carbonildiimidazol (97 mg, 0,6 mmol) se combinaron en 30 minutos. Se añadió 3-fluorobencenosulfonamida (11 mezcla de reacción se agitó durante 3 h a temperatura a de etilo y se lavó con 10 ml de ácido cítrico acuoso i]pirazol-1-il]piridin-3-carboxílico (181 mg, 0,5 mmol) y (2,5 mL) y se agitaron a temperatura ambiente durante g, 0,65 mmol), seguido de DBU (0,09 ml, 0,6 mmol) y la nte. La mezcla de reacción se diluyó con 10 ml de acetato Los orgánicos se secaron sobre sulfato de sodio y se
concentraron a presión reducida. El material crudo se p un gradiente de metanol al 0-8% en dicloro (trifluorometil)cidopropil]metoxi]pirazol-1-il]piridin-3-carb encontrado 519,1 (m 1)+; Tiempo de retención: 0,72 mi
Paso B: N-(3-Fluorofeml)sulfoml-6-[3-[[1-(t trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0612]
ó mediante cromatografía en gel de sílice eluyendo con o para dar 2-cloro-N-(3-fluorofenil)sulfonil-6-[3-[[1-ida (190 mg, 73%) iEn -EM m/z calc. 518,0439, .
rometN)cidopropN]metoxi]pirazoM-N]-2-[(4S)-2,2,4-
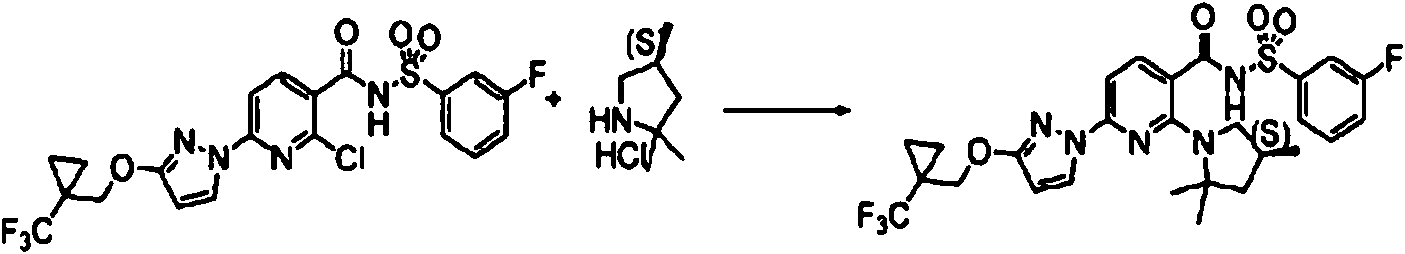
[0613] 2-Cloro-N-(3-fluorofenil)sulfonil-6-[3-[[1-(trifluorom mg, 0,15 mmol), (4,S)-2,2,4-trimetilpirrolidina (sal de hidr 0,9 mmol) fueron combinados en DMSO (600 mL) y cale y se purificó por CL-EM para dar /V-^3-fluorofenil)sulfo 2,2,4-trimetilpirrolidin-1-il]piridin-3-carboxamida (47 mg, tiempo de retención: 2,14 minutos 1H RMN (400 MHz, D - 7,69 (m, 2H), 7,68 - 7,59 (m, 1H), 6,93 (d, J = 8,2 Hz, 10,4 Hz, 1H), 2,30 (dd, J = 10,2, 7,0 Hz, 1H), 2,23 -2,08 6H), 1,39 (t, J = 12,2 Hz, 1H), 1,17 -1,02 (m, 4H), 0,69 (
Ejemplo sintético 50: Síntesis del
(trifluorometN)cidopropN]metoxi]pirazoM-N]-2-[(4S)-2
Paso A: 2-Cloro-W-(4-fluorofeml)sulfoml-6-13-carboxamida
[0614]
iclopropil]metoxi]pirazol-1-ilo]piridina-3-carboxamida (78 uro) (67 mg, 0,45 mmol) y carbonato de potasio (124 mg, os a 130°C durante 16 h. La mezcla de reacción se filtró 3-[[1-(trifluorometil)ciclopropM]metoxi]pirazol-1 -il]-2-[(4S)-) iEn-EM m/z calc. 595,1876, encontrado 596,3 (M+1)+; -d6) 88,21 (d, J = 2,8 Hz, 1H), 7,90 - 7,82 (m, 2H), 7,80 6,15 (d, J = 2,8 Hz, 1H), 4,43 -4,27 (m, 2H), 2,44 (t, J = 1H), 1,84 (dd, J = 11,9, 5,5 Hz, 1H), 1,53 (d, J = 9,7 Hz, 6,3 Hz, 3H).
mpuesto 52: N-(4-fluorofeml)sulfoml-6-[3-[[1-trimetMpirroNdm-1-M]piridm-3-carboxamida
trifluorometN)ciclopropN]metoxi]pirazoM-N]piridm-3-

[0615] Ácido 2-cloro-6-[3-[[1-(tnfluorometil)ciclopropil]m carbonildiimidazol (97 mg, 0,6 mmol) se combinaron en 30 minutos. Se añadió 4-fluorobencenosulfonamida (11 mezcla de reacción se agitó durante 3 h a temperatur acetato de etilo y se lavó con 10 mL de ácido cítrico acu concentraron a presión reducida. El material crudo se p un gradiente de metanol al 0-8% en dicloro (trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-3-carb encontrado 519,1 (m 1)+; tiempo de retención: 0,72 min
Paso B: N-(4-Fluorofenil)sulfonil-6-[3-[[1-( trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0616]
]pirazol-1-il]piridin-3-carboxílico (181 mg, 0,5 mmol) y (2,5 mL) y se agitaron a temperatura ambiente durante , 0,65 mmol), seguido de DBU (0,09 ml, 0,6 mmol) y la biente. La mezcla de reacción se diluyó con 10 mL de M. Los orgánicos se secaron sobre sulfato de sodio y se ó mediante cromatografía en gel de sílice eluyendo con o para dar 2-cloro-N-(4-fluorofenil)sulfonil-6-[3-[[1-ida (160 mg, 62%) Ie N-EM m/z calc. 518,0439,
orometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4- 
[0617] 2-Cloro-N-(4-fluorofenil)sulfonil-6-[3-[[1-(trifluoro mg, 0,15 mmol), (4S)-2,2,4-trimetilpirrolidina (sal de hidr 0,9 mmol) se combinaron en DMSO (600 mL) y se cale se purificó por CL/EM utilizando un gradiente de ac fluorofenil)sulfonil-6-[3-[[1 -(trifluorometil)ciclopropil]meto carboxamida (34 mg, 38%) IEN-EM m/z calc. 595,1876, 1H RMN (400 MHz, DMSO-d6) 88,20 (d, J = 2,8 Hz, 1H) 2H), 6,92 (d, J = 8,2 Hz, 1H), 6,15 (d, J = 2,7 Hz, 1H), 4, 7,0 Hz, 1H), 2,18 -2,05 (m, 1H), 1,83 (dd, J = 11,9, 5,5 - 1,00 (m, 4H), 0,67 (d, J = 6,3 Hz, 3H).
Ejemplo sintético 51: Síntesis del (trifluorometM)ddopropM]metoxi]pirazoM-M]-2-[(4S)-
Paso A: 2-doro-N-(2-metoxifeml)sulfoml-6-[3-carboxamida
[0618]
idopropil]metoxi]pirazol-1-ilo]piridina-3-carboxamida (78 ro) (67 mg, 0,45 mmol) y carbonato de potasio (124 mg, a 130°C durante 16 h. La mezcla de reacción se filtró y rilo al 30-99% en HCl acuoso 5 mM para dar N-(4-zol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-il]piridin-3-ntrado 596,3 (M+1)+; tiempo de retención: 2,16 minutos.
- 8,02 (m, 2H), 7,82 (d, J = 8,3 Hz, 1H), 7,59 - 7,45 (m, ,30 (m, 2H), 2,37 (t, J = 10,4 Hz, 1H), 2,22 (dd, J = 101, ), 1,52 (d, J = 8,8 Hz, 6H), 1,37 (t, J = 12,1 Hz, 1H), 1,15
ompuesto 53: W-(2-metoxifeml)sulfoml-6-[3-[[1-rimetMpirroMdm-1-M]piridm-3-carboxamida
ifluorometM)ddopropM]metoxi]pirazoM-M]piridm-3-

[0619] Se combinaron ácido 2-cloro-6-[3-[[1-(trifluorom 0,55 mmol) y carbonildiimidazol (110 mg, 0,66 mmol) e horas. Se añadió 2-metoxibencenosulfonamida (104 mg, de reacción se agitó durante 2 h a temperatura ambien etilo y se lavó con 10 ml de ácido cítrico acuoso 1M. La extractos orgánicos combinados se lavaron con salmu presión reducida. El material crudo se usó sin pu (trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-3-carb encontrado 531,1 (M+1)+; tiempo de retención: 0,70 min
Paso B: W-(2-metoxifeml)sulfoml-6-[3-[[1-( trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0620]
lopropil]metoxi]pirazol-1-il]piridin-3-carboxílico (200 mg, (2 mL) y se agitaron a temperatura ambiente durante 2 mmol), seguido de DBU (0,25 ml, 1,66 mmol) y la mezcla mezcla de reacción se diluyó con 10 ml de acetato de acuosa se extrajo con acetato de etilo (2 x 10 mL) y los e secaron sobre sulfato de sodio y se concentraron a ión adicional. 2-cloro-N-(2-metoxifenil)sulfonil-6-[3-[[1-ida (286 mg, 97%) Ie N-EM m/z calc. 530,06384,
rometN)ddopropN]metoxi]pirazoM-N]-2-[(4S)-2,2,4-

[0621] 2-Cloro-N-(2-metoxifenil)sulfonil-6-[3-[[1-(trifl (120 mg, 0,23 mmol), (4S)-2,2,4-trimetilpirrolidina (sal de mg, 1,25 mmol) se combinaron en DMSO (600 |jL) y se acetato de etilo y agua. Los orgánicos se separaron, se secaron sobre sulfato de sodio y se evaporaron. El mat eluyendo con metanol al 0-10% en d (trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4 EM m/z calc. 607,20764, encontrado 608,3 (M+1)+; tiem etil)ciclopropil]metoxi]pirazol-1-il]piridina-3-carboxamida idrato) (107 mg, 0,71 mmol) y carbonato de potasio (173 aron a 130°C durante 16 h. La reacción se repartió entre on con una solución de ácido cítrico 1 M, salmuera, se rudo se purificó mediante cromatografía en gel de sílice metano para dar N-(2-metoxifenil)sulfonil-6-[3-[[1-tilpirrolidin-1-il]piridin-3-cafboxafnida (82 mg, 60%) iEn -retención: 2,15 minutos.
Ejemplo sintético 52: Síntesis del (trifluorom etil)ciclopropil]metoxi]pirazoM-il]-2-[(4S)-Paso A: 2-Cloro-W-(4-metoxifem l)sulfom l-6-[3 carboxamida
[0622]
ompuesto 54: W-(4-metoxifenil)sulfoml-6-[3-[[1--trimetilpirrolidm-1-il]piridm-3-carboxamida
(trifluorometil)ciclopropil]metoxi]pirazoM-il]piridm-3-

[0623] Ácido 2-doro-6-[3-[[1-(trifluorometil)cidopropil]m carbonildiimidazol (162 mg, 1,0 mmol) se combinaron 30 minutos. Se añadió 4-metoxibencenosulfonamida (2 mezcla de reacción se agitó durante 2 h a temperatur acetato de etilo y se lavó con 10 mL de ácido cítrico acu concentraron a presión reducida. El material crudo se metanol al 0-8% en diclorometano (trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-3-carb encontrado 531,1 (M+1)+; tiempo de retención: 0,71 min
Paso B: W-(4-metoxifeml)sulfoml-6-[3-[[1-trim etilpirrolid in-1-il]pirid in-3-carboxam ida
[0624]
pirazol-1-il]piridin-3-carboxílico (300 mg, 0,83 mmol) y F (4 mL) y se agitaron a temperatura ambiente durante , 1,08 mmol), seguido de DBU (0,15 mL, 1,0 mmol) y la biente. La mezcla de reacción se diluyó con 10 mL de M. Los orgánicos se secaron sobre sulfato de sodio y se ó mediante cromatografía en gel de sílice eluyendo con ra dar 2-cloro-A/-(4-metoxifenil)sulfonil-6-[3-[[1-ida (430 mg, 97%) iEn-EM m/z calc. 530,06384,
orometil)ciclopropil]metoxi]pirazoM-il]-2-[(4S)-2,2,4-

[0625] 2-Cloro-N-(4-metoxifenil)sulfonil-6-[3-[[1-(trifluor (210 mg, 0,39 mmol), (4S)-2,2,4-trimetilpirrolidina (sal de mg, 2,39 mmol) se combinaron en DMSo (2 mL) y se c acetato de etilo y agua. Los orgánicos se separaron, se secaron sobre sulfato de sodio y se evaporaron. El mat eluyendo con metanol al 0-5% en diclorometano. El mat gradiente de acetonitrilo al 30-99% en HCl acu (trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4 EM m/z calc. 607,20764, encontrado 608,3 (M+1)+; tiem 8 8,19 (d, J = 2,8 Hz, 1H), 7,99 - 7,87 (m, 2H), 7,77 (d, 6,14 (d, J = 2,8 Hz, 1H), 4,44 -4,28 (m, 2H), 3,84 (s, 3H) 1H), 1,82 (dd, J = 11,9, 5,6 Hz, 1H), 1,52 (d, J = 10,7 Hz = 6,3 Hz, 3H).
Ejemplo sintético 53: Síntesis del (difluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-
Paso A: 3-((1-(Difluorometil)ciclopropil)metoxi)-1H-piraz
[0626]
il)ciclopropil]metoxi]pirazol-1-il]piridina-3-carboxamida
cloruro) (180 mg, 1,2 mmol) y carbonato de potasio (330 ron a 130°C durante 15 h. La reacción se repartió entre ron con una solución de ácido cítrico 1 M, salmuera, se rudo se purificó mediante cromatografía en gel de sílice se purificó adicionalmente mediante CL/EM utilizando un 5 mM para producir W-(4-metoxifenil)sulfonil-6-[3-[[1-etilpirrolidin-1-il]piridin-3-carboxamida (25 mg, 10%) lEN-retención: 2 , 1 6 minutos. 1H RMN (400 MHz, DMSO-cfe) ,2 Hz, 1H), 7,26 - 7,10 (m, 2H), 6,91 (d, J = 8,2 Hz, 1H), (t, J = 10,5 Hz, 1H), 2,29 -2,21 (m, 1H), 2,16 -2,00 (m, , 1,37 (t, J = 12,1 Hz, 1H), 1,16 -1,02 (m, 4H), 0,64 (d, J
Compuesto 55: N-(Bencenosulfonil)-6-[3-[1--trimetilpirrolidin-1-il]piridina-3-carboxamida
arboxilato de ferc-butilo
[0627] A la solución de (1-(difluorometil)cidopropil)m carboxilato de terc-butilo (1,19 g, 6,46 mmol) y trifenilfosfi añadió gota a gota azodicarboxilato de diisopropilo (1,44 dejó calentar a temperatura ambiente, luego se calentó temperatura ambiente y se añadió acetato de etilo (300 acuoso (20 mL, 1M), agua, salmuera y se secó sobre sulf se purificó por cromatografía en gel de sílice (hexano y di proporcionar 3-((1-(difluorometil)ciclopropil)metoxi)-1H-pir g, rendimiento del 80%). IEN-EM m/z calc. 288,1, encon RMN (250 MHz, CDCla) 8 (ppm): 7,84 (d, J = 3,0 Hz, 1H) 2H), 1,61 (s, 9H), 0,97 (m, 2H), 0,75 (m, 2H).
Paso B: 3-((1-(Difluorometil)ciclopropil)metoxi)-1H-pirazol
[0628]
ol (867 mg, 7,11 mmol), 2,3-dihidro-3-oxopirazol-1-1,86 g, 7,11 mmol) en tetrahidrofurano (22 mL) a 0°C se 1 mmol). Una vez completada la adición, la reacción se °C durante 1 hora. La solución de reacción se enfrió a ). Después, la solución se lavó con hidróxido de sodio e magnesio, se filtró y se concentró. El residuo obtenido metano, 0 a 100% de gradiente de diclorometano) para -1-carboxilato de ferc-butilo como un sólido blanco (1,50 o 289,2 M+1)+. Tiempo de retención: 3,08 minutos. 1H 7 (t, J = 57,8 Hz, 1H), 5,89 (d, J = 3,0 Hz, 1H), 4,32 (s,

[0629] Una solución de cloruro de hidrógeno en frí (difluorometil)ciclopropil)metoxi)-1H-pirazol-1-carboxilato redondo, y la solución de reacción se calentó a temperatu todos los disolventes a presión reducida, el residuo así ob La capa orgánica se separó y la capa acuosa se extrajo co se lavaron con salmuera (2 x 30 mL), se secaron sobre reducida. El residuo así obtenido se purificó por cromato acetato de etilo en gradiente) para dar 3-((1-(difluorometi mg, 90% de rendimiento). IEN-EM m/z calc. 188,1, enco RMN (250 MHz, DMSO) 8 (ppm): 11,87 (s, 1H), 7,51 (m, 0,80 (m, 4H).
Paso C: 2-Cloro-6-[3-[[1-(difluorometM)cidopropM]met
[0630]
30 mL, 4,0M en 1,4-dioxano) se añadió a 3-((1-erc-butilo (1,69 g, 5,88 mmol) en un matraz de fondo mbiente y se agitó durante 3 horas. Después de eliminar o se repartió entre agua (50 mL) y éter dietílico (80 mL). er dietílico (2 x 80 mL). Las capas orgánicas combinadas lfato de sodio, se filtraron y se concentraron a presión a en gel de sílice (hexanos y acetato de etilo, 0 a 40% lopropil)metoxi)-1H-pirazol como un sólido blanco (997 o 189,1 (M+1)+. Tiempo de retención: 1,94 minutos. 1H ), 5,98 (t, J = 57,0 Hz, 1H), 5,66 (m, 1H), 4,10 (s, 2H)),
irazoM-M]piridma-3-carboxMato de terc-butilo

[0631] 2,6-Dicloropiridina-3-carboxilato de terc-butilo (difluorometil)ciclopropil]metoxi]-1H-pirazol (500 mg, 2,6 mg, 3,188 mmol) (recién molidos) se combinaro diazabiciclo[2.2.2]octano (aproximadamente 59,61 mg, 0 bajo nitrógeno durante 16 horas. La mezcla de reacción s resultante se recogió y se lavó con agua. El sólido se dis capa acuosa. Los extractos orgánicos se secaron sobre (difluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-3-carbo 399,11612, encontrado 400,1 (M+1)+; tiempo de retención
Paso D: Ácido 2-cloro-6-[3 [[1-(difluorometM)ddoprop
[0632]
proximadamente 659,2 mg, 2,657 mmol), 3-[[1-mol) y carbonato de potasio (aproximadamente 440,6 n DMSO anhidro (13,18 mL). Se añadió 1,4-4 mmol) y la mezcla se agitó a temperatura ambiente yó con agua (20 mL) y se agitó durante 15 min. El sólido en diclorometano y se eliminó la pequeña cantidad de ato de sodio y se evaporaron para dar 2-cloro-6-[3-[[1-de terc-butilo (842 mg, 79%). IEN-EM m/z calc.
82 minutos.
etoxM]pirazoM-M]piridm-3-carboxíMco

[0633] 2-Cloro-6-[3-[[1-(difluorometil)ciclopropil]metoxi] mmol) y TFA (aproximadamente 2,401 g, 1,622 ml, 21 calenaron a 40°C durante 3 h. La reacción se evapor evaporar para dar ácido 2-doro-6-[3-[[1-(difluorometi 98%). IEN-EM m/z calc. 343,05353, encontrado 344,1 ( DMSO-d6 ) 8 13,59 (s, 1H), 8,43 (d, J = 2,9 Hz, 1H), 8,3 Hz, 1H), 5,98 (t, J = 56,4 Hz, 1H), 4,32 (s, 2H), 0,93 - 0,
Paso E: N-(bencenosulfoml)-2-doro-6-[3 carboxamida
[0634]
ol-1-il]piridin-3-carboxilato de ferc-butilo (842 mg, 2,106 mmol) se disolvieron en diclorometano (8,420 mL) y se l sólido resultante se trituró con hexanos y se volvió a opropil]metoxi]pirazol-1-il]piridin-3-carboxílico (710 mg, ; tiempo de retención: 0 , 62 minutos. 1H RMN (400 MHz, J = 8,4 Hz, 1H), 7,73 (d, J = 8,4 Hz, 1H), 6,22 (d, J = 2,9 , 4H).
(difluorometM)cidopropM]metoxi]pirazoM-Mpiridm-3-

[0635] Se combinaron ácido 2-doro-6-[3-[[1-(difluorome 0,5819 mmol) y carbonildiimidazol (aproximadamente 1 2 h. En este punto, se añadió bencenosulfonamida (a (aproximadamente 265,8 mg, 261,1 ml, 1,746 mmol) y l Se añadió una solución de ácido cítrico 1 M (5 mL) y la acetato de etilo. Los orgánicos se lavaron con salmuera, crudo se purificó mediante cromatografía en gel de sílic N-(bencenosulfonil)-2-cloro-6-[3-[[1-(difluorometil)ciclopr Ie N-EM m/z calc. 482,0627, encontrado 483,1 (M+1 )+; ti
Paso F: N-(bencenosulfonil)-6-[3-[[1-( trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0636]
dopropil]metoxi]pirazol-1-il]piridin-3-carboxílico (200 mg, mg, 0,6983 mmol) en THF (2,5 mL) y se agitaron durante imadamente 91,47 mg, 0,5819 mmol) seguido de DBU cción se agitó durante 2 h más a temperatura ambiente. ción se agitó durante 20 min. La solución se extrajo con ecaron sobre sulfato de sodio y se evaporaron. El material yendo con metanol al 0 -1 0 % en diclorometano para dar metoxi]pirazol-1-il]piridin-3-carboxamida (153,6 mg, 55%) o de retención: 0 , 68 minutos.
orometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4-

[0637] N-(Bencenosulfonil)-2-cloro-6-[3-[[1-(difluoromet 0,24 mmol), (4,S)-2,2,4-trimetilpirrolidina (sal de hidrocl 1,05 mmol) se combinaron en DMSO (600 mL) y se ca acetato de etilo y agua. Los orgánicos se separaron, se secaron sobre sulfato de sodio y se evaporaron. El ma sílice eluyendo con metanol al 0-10% en (difluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4 EM m/z calc. 559,2065, encontrado 560,3 (M+1)+; tiemp 12,47 (s, 1H), 8,19 (d, J = 2,8 Hz, 1H), 8,00 (t, J = 1,3 H -7,70 (m, 1H), 7,65 (tt, J = 6 ,8 , 1,6 Hz, 2H), 6,91 (d, J = -2,37 (m, 1H), 2,28 (dd, J = 10,3, 7,0 Hz, 1H), 2,09 (dq, lopropil]metoxi]pirazol-1-il]piridin-3-carboxamida (116 mg, o) (71 mg, 0,63 mmol) y carbonato de potasio (145 mg, ron a 130°C durante 16 h. La reacción se repartió entre ron con una solución de ácido cítrico 1 M, salmuera, se l en crudo se purificó mediante cromatografía en gel de lorometano para dar N-(bencenosulfonía)-6-[3-[[1-etilpirrolidin-1-il]piridin-3-carboxamida (87 mg, 65%) lEN-retención: 2,06 minutos. 1H RMN (400 MHz, DMSO-cfe) 8 ), 7,98 (d, J = 1,6 Hz, 1H), 7,80 (d, J = 8,2 Hz, 1H), 7,76 z, 1H), 6,13 (d, J = 2,8 Hz, 2H), 4,32 -4,23 (m, 2H), 2,47 11,9, 6,2 Hz, 1H), 1,82 (dd, J = 11,9, 5,6 Hz, 1H), 1,52 (d,
J = 9,5 Hz, 6H), 1,36 (t, J = 12,1 Hz, 1H), 0,87 (dt, J = 5, Ejemplo sintético 54: Síntesis del Compuesto 56: N [[1-(trifluorometN)cidopropN]metoxi]pirazoM-N]pirid Paso A: N-(2,4,4-Trimetilpentan-2-il)picolinamida
[0638]
Hz, 4H), 0,65 (d, J = 6,2 Hz, 3H).
ncenosulfoml)-2-(2,2,4,4-tetrametMpirroNdm-1-M)-6-[3-carboxamida

[0639] A 5°C, a ácido picolínico (20,5 g, 167 mmol) e seguido de 2,4,4-trimetilpentan-2-amina (27,0 mL, ~1,0 durante 1,0 h. La mezcla de reacción se vertió en agua h combinado se lavó con agua (2X300 mL) y salmuera ( producto crudo, que se purificó mediante filtración en ta 25% en hexanos, dando un aceite de color amarillo cla [M+1]: 235.
Paso B: Piridin-2-il(2,2,4,4-tetrametilpirrolidin-1-il)me
[0640]
F (200 mL) se añadió HATU (65 g, 171 mmol, ~1,0 eq), luego DIEA (65 mL, ~2,5 eq). La reacción se agitó a TA (350 mL) y se extrajo con EtOAc (2X600 mL). El extracto L), se secó sobre Na2SO4 y se concentró para dar un través de un lecho de gel de sílice, eluido con EtOAc al -(2,4,4-trimetilpentan-2-il)picolinamida (36 g, 92%); MS
na
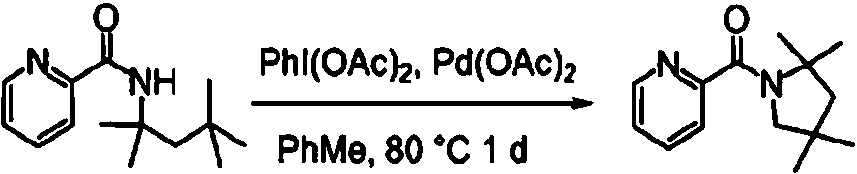
[0641] N-(2,4,4-trimetilpentan-2-il)picolinamida (36,0 g, mmol) se mezclaron en tolueno (600 mL) y se calentaro para eliminar la mayor parte del tolueno y el residuo se 50% en hexanos, dando como resultado un sólido de il)metanona (32 g, 90%); MS [M+1]: 233. 1H RMN (250 1H), 7,61 (d, J = 7,8 Hz, 1H), 7,35 - 7,25 (m, 1H), 3,36 (s
Paso C: 2,2,4,4-Tetrametilpirrolidina (sal de HCl)
[0642]
mmol), Pd(OAc)2 (1,72 g, 5%), PhI(OAc)2 (99,2 g, 308 0°C durante la noche (~18 h). La reacción se concentró en una columna de gel de sílice, se eluyó con EtOAc al r amarillo claro. piridin-2-il(2,2,4,4-tetrametilpirrolidin-1-CDCla) 8,56 (d, J = 4,8 Hz, 1H), 7,76 (td, J = 7,7, 1,7 Hz, ), 1,79 (s, 2H), 1,67 (s, 6H), 1,08 (s, 6H)

[0643] Se disolvió piridin-2-il(2,2,4,4-tetrametilpirroliding, 225 mmol) en agua (6 mL) y EtOH (18 mL) en un react 48 h. Se completó la reacción. La mezcla se disolvió con se lavó con agua (2X100 mL) y se secó sobre Mg2SO4. durante 5 min. Se formó un aceite en el fondo del matr aceite restante se lavó con éter (2X30 mL) y el lavado blanco, que se secó en un horno de vacío a 50°C tetrametilpirrolidina (sal de HCl) (4,0 g, 95%). EM [M+1]: 5,5 Hz, 2H), 1,69 (s, 2H), 1,40 (s, 6H), 1,16 (s, 6H).
Paso D: N-(b (trifluorometil)ciclopropil]metoxi]pirazol-1-il]p irid ina-
[0644]
etanona (6 g, 25,9 mmol) en una mezcla de NaOH (9,0 presión pequeño (~45 mL) y se calentó a 140°C durante L de agua y se extrajo con Et2O (3X200 mL). El extracto pués de filtrar, se burbujeó gas HCl a través del filtrado a capa superior de éter se decantó cuidadosamente, el cantó. El aceite final se evaporó para dar un semisólido ante 1 día, para dar un sólido blanquecino. 2,2,4,4-1H RMN (250 MHz, DMSO-d6) 9,39 (s, 2H), 3,01 (t, J =
nosulfonil)-2-(2,2,4,4-tetrametilpirrolidin-1-il)-6-13-[[1-rboxam ida

[0645] N-(bencenosulfonil)-2-doro-6-[3-[[1-(trifluorometil) 0,2 mmol), 2,2,4,4-tetrametilpirrolidina (sal de hidrocloru mmol) se combinaron en DMSO (500 mL) y se calentar mL) y se agitó durante 20 min. Se formó un sólido y se etilo y se lavó con una solución de ácido cítrico 1 M y lue sodio y se evaporaron. El material crudo se purificó med 0-10% en diclorometano para dar
(trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-3-carb encontrado 592,2 (M+1)+; tiempo de retención: 2,20 min J = 2,8 Hz, 1H), 8,04 - 7,97 (m, 2H), 7,82 - 7,72 (m, 2H), Hz, 1H), 4,36 (s, 2H), 2,38 (s, 2H), 1,72 (s, 2H), 1,58 (s,
Ejemplo sintético 55: Síntesis del Com (trifluorometM)cidopropM]metoxi]pirazoM-M]-2-[(4S)-2
Paso A: 2-Cloro-N-(4-metoxi-2-metil-fenil)sulfonil-6-3-carboxamida
[0646]
propil]metoxi]pirazol-1-il]piridin-3-carboxamida (100 mg, 98 mg, 0,6 mmol) y carbonato de potasio (138 mg, 1,0 130°C durante 16 h. La reacción se diluyó con agua (3 ntó el líquido acuoso. El sólido se disolvió en acetato de on salmuera. Los orgánicos se secaron sobre sulfato de cromatografía en gel de sílice eluyendo con metanol al cenosulfonN)-2-(2,2,4,4-tetrametNpirroNdin-1-N)-6-[3-[[1-ida (57 mg, 48%) IEN-EM m/z calc. 591,2127, 1H RMN (400 MHz, DMSO-d6) 812,52 (s, 1H), 8,20 (d, - 7,63 (m, 2H), 6,94 (d, J = 8,0 Hz, 1H), 6,14 (d, J = 2,8 1,14 -1,04 (m, 4H), 0,81 (s, 6H).
to 57: N-(4-metoxi-2-metM-feml)sulfoml-6-[3-[[1-trimetMpirroMdm-1-M]piridm-3-carboxamida
1-(trifluorometil)ciclopropil]metoxi]pirazol-1-il]piridin-

[0647] Ácido 2-Cloro-6-[3-[[1-(trifluorometil)ciclopropil]m carbonildiimidazol (82 mg, 0,5057 mmol) se combinaron se añadió 4-metoxi-2-metil-bencenosulfonamida (85 mg, reacción se agitó durante 2 h más a temperatura ambie una solución de ácido cítrico 1 M, seguido de salmuera. L y se evaporaron para dar 2-cloro-N-(4-metoxi-2-metil-f il]piridin-3-carboxamida (205 mg, 94%) IEN-EM m/z cal 0,73 minutos.
Paso B: N-(4-Metoxi-2-metil-fenil)sulfonil-6-[3-[[1-( trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0648]
pirazol-1-il]piridin-3-carboxílico (150 mg, 0,4023 mmol) y HF (1,299 mL) y se agitaron durante 2 h. En este punto, 224 mmol) seguido de DBU (200 mL, 1,337 mmol) y la La reacción se diluyó con acetato de etilo y se lavó con rgánicos se separaron, se secaron sobre sulfato de sodio ulfonil-6-[3-[[1-(trifluorometil)ciclopropil]metoxi]pirazol-1-4,0795, encontrado 545,0 (M+1)+; tiempo de retención:
orometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2,4-

[0649] 2-Cloro-N-(4-metoxi-2-metil-fenil)sulfonil-6-[3-[[1 carboxamida (100 mg, 0,18 mmol), (4S)-2,2,4-trimetilpirr de potasio (138 mg, 1,0 mmol) se combinaron en DMSo se diluyó con agua (3 mL) y se agitó durante 20 min. Se disolvió en acetato de etilo y se lavó con una solución secaron sobre sulfato de sodio y se evaporaron. El mate rometil)ciclopropil]metoxi]pirazol-1-il]piridin-3-a (sal de hidrocloruro) (90 mg, 0,6 mmol), y carbonato mL) y se calentaron a 130°C durante 16 h. La reacción ó un sólido y se decantó el líquido acuoso. El sólido se ido cítrico 1 M y luego con salmuera. Los orgánicos se rudo se purificó mediante cromatografía en gel de sílice
eluyendo con metanol al 0-10% en diclorom (trifluorometil)ciclopropil]metoxi]pirazol-1-il]-2-[(4S)-2,2, EM m/z calc. 621,22327, encontrado 622,3 (M+1)+; tiem 8 12,46 (s, 1H), 8,20 (d, J = 2,8 Hz, 1H), 7,98 (d, J = 8,7 J = 8,2 Hz, 1H), 6,14 (d, J = 2,7 Hz, 1H), 4,41 -4,32 (m, (m, 1H), 1,82 (dd, J = 11,9, 5,6 Hz, 1H), 1,52 (s, 6H), 1, 3H).
Ejemplo sintético 56: Síntesis de (trifluorometN)ddobutN]metoxi]pirazoM-N]-2-[(4S)-2,
Paso A: 2-Cloro-N-(o-toNlsulfoml)-6-[3-[[1-(trifluoro
[0650]
para dar N-(4-metoxi-2-metil-fenil)sulfonil-6-[3-[[1-tilpirrolidin-1-il]piridin-3-carboxamida (75 mg, 67%) IEN-retención: 2,23 minutos. 1H RMN (400 MHz, DMSO-cfe) H), 7,78 (d, J = 8,3 Hz, 1H), 7,01 -6,94 (m, 2H), 6,92 (d, ,82 (s, 3H), 2,59 (s, 3H), 2,41 -2,29 (m, 2H), 2,22 -2,10 = 12,1 Hz, 1H), 1,12 -1,06 (m, 4H), 0,70 (d, J = 6,2 Hz,
Compuesto 58: N-(o-ToMlsulfoml)-6-[3-[[1-imetMpirroNdm-1-M]piridm-3-carboxamida
ddobutN]metoxi]pirazoM-M]piridm-3-carboxamida

[0651] Ácido 2-cloro-6-[3-[[1-(trifluorometil)ciclobutil]met carbonil diimidazol (aproximadamente 81,69 mg, 0,50 durante 2 h. En este punto, se añadió 2-metilbence seguido de DBU (aproximadamente 202,6 mg, 199,0 temperatura ambiente. La reacción se diluyó con acet seguido de salmuera. Los orgánicos se separaron, se cloro-N-(o-tolilsulfonil)-6-[3-[[1-(trifluorometil)ciclobutil]m EM m/z calc. 528,0846, encontrado 529,0 (M+1)+; tiemp
Paso B: N-(o-tolilsulfonil)-6-[3-[[1-(trifluorometil)cid il]piridin-3-carboxamida
[0652]
irazol-1-il]piridin-3-carboxílico (150 mg, 0,3992 mmol) y mol) se combinaron en THF (1,339 mL) y se agitaron onamida (aproximadamente 71,77 mg, 0,4192 mmol), ,331 mmol) y la reacción se agitó durante 2 h más a etilo y se lavó con una solución de ácido cítrico 1 M, on sobre sulfato de sodio y se evaporaron para dar 2-pirazol-1-il]piridina-3-carboxamida (208 mg, 99%) IEN-retención: 0,77 minutos
l]metoxi]pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-

[0653] 2-Cloro-N-(o-tolilsulfonil)-6-[3-[[1-(trifluorometil)ci 0,19 mmol), (4S)-2,2,4-trimetilpirrolidina (sal de clorhidr mmol) se combinaron en DMSo (500 mL) y se calenta mL) y se agitó durante 20 min. Se formó un sólido y se etilo y se lavó con una solución de ácido cítrico 1 M y lu sodio y se evaporaron. El material crudo se purificó me 0-10% en diclorometano para dar N-(o-tolilsulfonil)-6-[ trimetilpirrolidin-1 -il]piridin-3-carboxamida (69 mg, 60 tiempo de retención: 2,33 minutos. 1H RMN (400 MHz, = 7,9 Hz, 1H), 7,82 (d, J = 8,2 Hz, 1H), 7,59 (td, J = 7,5, (d, J = 2,7 Hz, 1H), 4,48 (s, 2H), 2,63 (s, 3H), 2,39 (d, J 1,91 (m, 1H), 1,83 (dd, J = 11,9, 5,6 Hz, 1H), 1,53 (s, 6
Ejemplo sintético 57: Síntesis del
(trifluorometil)ciclobutil]metoxi]pirazol-1-il]-2-[(4S)-2
Paso A: 2-C loro-N-(3-fluorofenil)sulfonil-6-carboxamida
[0654]
til]metoxi]pirazol-1-il]piridina-3-carboxamida (100 mg, 90 mg, 0,6 mmol) y carbonato de potasio (138 mg, 1,0 130°C durante 16 h. La reacción se diluyó con agua (3 ntó el líquido acuoso. El sólido se disolvió en acetato de on salmuera. Los orgánicos se secaron sobre sulfato de cromatografía en gel de sílice eluyendo con metanol al (trifluorometil)ciclobutil]metoxi]pirazol-1-il].-2-[(4S)-2,2,4--EM m/z calc. 605,22833, encontrado 606,5 (M+1)+; -cfe) 812,63 (s, 1H), 8,22 (d, J = 2,8 Hz, 1H), 8,04 (d, J z, 1H), 7,49 -7,40 (m, 2H), 6,96 (d, J = 8,3 Hz, 1H), 6,18 Hz, 2H), 2,35 - 2,23 (m, 2H), 2,21 - 2,04 (m, 4H), 2,02 -5 (t, J = 12,1 Hz, 1H), 0,69 (d, J = 6,2 Hz, 3H).
mpuesto 59: N-(3-fluorofenil)sulfonil-6-[3-[[1-trimetilpirrolidin-1-il]piridin-3-carboxamida
-(trifluorometil)ciclobutil]metoxi]pirazol-1-il]piridin-3- 
[0655] Ácido 2-doro-6-[3-[[1-(trifluorometil)cidobutil]met carbonildiimidazol (aproximadamente 81,69 mg, 0,5038 2 h. En este punto, se añadió 3-fluorobencenosulfonam DBU (aproximadamente 202,6 mg, 199,0 ml, 1,331 m ambiente. La reacción se diluyó con acetato de etilo y salmuera. Los orgánicos se separaron, se secaron so fluorofenil)sulfonil-6-[3-[[1-(trifluorometil)ciclobutil]metoxi] EM mlz calc. 532,0595, encontrado 533,0 (M+1)+; tiemp
Paso B: N-(3-Fluoropbeml)sulfoml-6-[3-[[1-trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0656]
azol-1-il]piridin-3-carboxílico (150 mg, 0,3992 mmol) y se combinaron en THF (1,339 mL) y se agitaron durante aproximadamente 69,93 mg, 0,3992 mmol) seguido de la reacción se agitó durante 2 h más a temperatura vó con una solución de ácido cítrico 1 M, seguido de lfato de sodio y se evaporaron para dar 2-cloro-N-(3-ol-1-il]piridina-3-carboxamida (210 mg, 99%) IEN-etención: 0,77 minutos.
orometN)ciclobutN]metoxi]pirazoM-N]-2-[(4S)-2,2,4-

[0657] 2-Cloro-N-(3-fluorofenil)sulfonil-6-[3-[[1-(trifluorom mg, 0,19 mmol), (4S)-2,2,4-trimetilpirrolidina (sal de hidr 1,0 mmol) se combinaron en DMSO (500 mL) y se cale mL) y se agitó durante 20 min. Se formó un sólido y se etilo y se lavó con una solución de ácido cítrico 1 M y lu sodio y se evaporaron. El material crudo se purificó med 0-10% en diclorometano para dar N-(3-fluorofenil)sulfo 2,2,4-trimetilpirrolidin-1-il]piridin-3-carboxamida (73 mg, tiempo de retención: 2,27 minutos. 1H RMN (400 MHz, = 2,3 Hz, 1H), 7,84 (d, J = 2,0 Hz, 1H), 7,78 - 7,70 (m, 2 2,7 Hz, 1H), 4,48 (s, 2H), 2,43 (d, J = 10,4 Hz, 1H), 2,35 1H), 1,84 (dd, J = 11,8, 5,6 Hz, 1H), 1,55 (s, 3H), 1,52 (s,
Ejemplo sintético 58: Síntesis del Compue ilmetoxi)pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1 -il
Paso A: espiro[2.2]Pent-1-il-metanol
[0658]
iclobutil]metoxi]pirazol-1-il]piridina-3-carboxamida (100 ro) (90 mg, 0,6 mmol) y carbonato de potasio (138 mg, 130°C durante 16 h. La reacción se diluyó con agua (3 tó el líquido acuoso. El sólido se disolvió en acetato de n salmuera. Los orgánicos se secaron sobre sulfato de cromatografía en gel de sílice eluyendo con metanol al [3-[[1-(trifluorometil)ciclobutil]metoxi]pirazol-1-il]-2-[(4S)-Ie N-EM m/z calc. 609,2033, encontrado 610,2 (M+1)+; -cfe) 812,63 (s, 1H), 8,22 (d, J = 2,8 Hz, 1H), 7,86 (d, J ,66 - 7,59 (m, 1H), 6,96 (d, J = 8,2 Hz, 1H), 6,18 (d, J = (m, 3H), 2,19 -2.05 (m, 4H), 1,96 (td, J = 10,0, 5,3 Hz, 1,40 (t, J = 12,2 Hz, 1H), 0,69 (d, J = 6,2 Hz, 3H).
60: N-(bencenosulfonil)-6-[3-(espiro[2.2]pentan-2-in-3-carboxamida

[0659] A una suspensión de hidruro de litio y aluminio ácido espiro[2.2]pentano-1-carboxílico (1,75 g, 15,6 mm La reacción se calentó a 50°C durante 16 horas. La re sulfato de sodio sólido decahidratado. La mezcla se dil corto de celite y se concentró para dar espiro[2.2]pent-1 98,15 encontrado 98,8 (M+1)+. Tiempo de retención: 2, 4H) 0,91 -1,09 (m, 1H) 1,20 - 1,37 (m, 1H) 1,43 (m, 1H)
Paso B: Éster terc-butílico del ácido 3-(espiro[2.2]pe mg, 23,4 mmol) en tetrahidrofurano (30 mL) se añadió tetrahidrofurano (5 mL) gota a gota durante 5 minutos. n se diluyó con éter dietílico (20 mL) y se inactivó con on éter dietílico (100 mL), se filtró a través de un lecho tanol (793 mg, 52%) como un aceite. IEN-EM m/z calc. inutos. 1H RMN (250 MHz, CDCh) ppm 0,58 - 0,89 (m, (dd, J = 11,98, 6,37 Hz, 2H)
ilmetoxi)-pirazol-1-carboxílico
[0660]

[0661] A una solución de espiro[2.2]pent-1-il-metanol cr trifenilfosfina (2,58 g, 9,8 mmol), éster terc-butílico del mezcla de reacción se enfrió en un baño de hielo seguid mmol). Se retiró el baño de hielo y se agitó la reacción cruda se purificó mediante cromatografía en columna dar éster terc-butílico de ácido 3-(espiro[2.2]pent-1-il transparente. IEN-EM m/z calc. 264,33 encontrado 265,
Paso C: 3-(espiro[2.2]pent-1-ilmetoxi)-1H-pirazol
[0662]
966 mg, 9,8 mmol) en tetrahidrofurano (40 mL) se añadió o 3-hidroxi-pirazol-1-carboxílico (1,64 g, 8,9 mmol). La la adición de azodicarboxilato de diisopropilo (1,9 ml, 9,8 nte 2 horas. El disolvente se eliminó al vacío y la mezcla l de sílice usando hexanos-éter dietílico al 10-20% para xi)-pirazol-1-carboxílico (1,20 g, 44%) como un aceite 1)+. Tiempo de retención: 3,36 minutos

[0663] A ester terc-butílico del ácido 3-(espiro[2.2]pentdiclorometano (30 mL) y ácido trifluoroacético (3,4 ml, temperatura ambiente y se concentró hasta sequedad a 1,2-dicloroetano (15 mL) para dar 3-(espiro[2.2]pentamarillo. IEN-EM m/z calc. 164,09 encontrado 164,6 (M
Paso D: Éster metílico del ácido 2-cloro-6-[3-(espiro
[0664]
etoxi)-pirazol-1-carboxílico (1,2 g, 4,54 mmol) se añadió mol). La mezcla de reacción se agitó durante 2 horas a ío. El residuo se destiló azeotrópicamente dos veces con etoxi)-1H-pirazol crudo (1,87 g, 51%) como un aceite Tiempo de retención: 2,11 minutos
pent-1-ilmetoxi)pirazol-1-il]-nicotínico
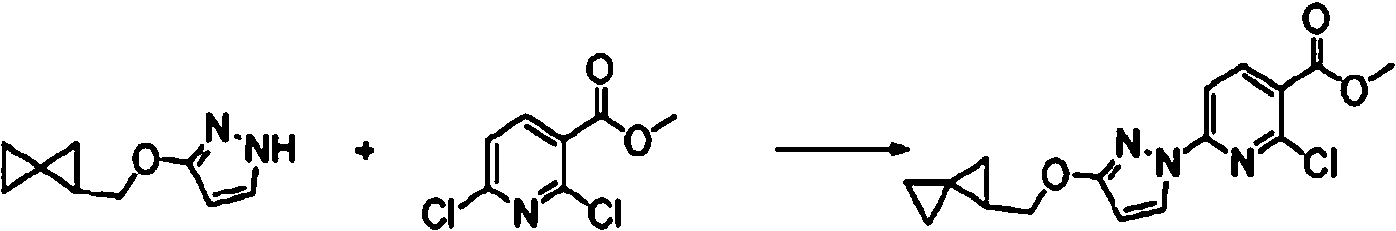
[0665] A 3-(espiro[2.2]pent-1-ilmetoxi)-1H-pirazol crudo de metilo (935 mg, 4,54 mmol), 1,4-diazabiciclo[2.2. carbonato de potasio (1,9 g, 13,6 mmol). La reacción se éter dietílico (75 mL) y se lavó con agua que contenía u mL). Esta capa orgánica se secó sobre sulfato de sod purificó mediante cromatografía en columna de gel de éster metílico de ácido 2-cloro-6-[3-(espiro[2.2]pent-1-i blanquecino. IEN-EM m/z calc. 333,09 encontrado 333,
Paso E: Ácido 2-cloro-6-[3-(espiro[2.2]pent-1-ilmeto
[0666]
7 g, asumido 4,54 mmol) se añadió 2,6-dicloronicotinato ano (102 mg, 0,91 mmol), dimetilformamida (8 mL) y durante 48 horas a temperatura ambiente, se diluyó con equeña cantidad de salmuera (3 x 50 mL) y salmuera (50 se concentró al vacío. La mezcla de reacción cruda se e usando hexanos al 0-15%: éter dietílico para producir oxi)pirazol-1-il]-nicotínico (1,02 g, 67%) como un sólido 1)+. Tiempo de retención: 3,85 minutos.
irazol-1-il]-nicotínico

[0667] Aéster metílico del ácido 2-cloro-6-[3-(espiro[2.2 añadió agua (6 mL), metanol (6 mL) y tetrahidrofurano reacción se agitó durante 1 hora y se añadió ácido clor filtración, se lavó con agua y hexanos para dar ácido (927 mg, 98%) como un sólido blanco. iEn -EM m/z calc minutos 1H RMN (250 MHz, CDC13) ppm: 0,76 - 0,88 ( 7,0, 3,3, Hz, 2H) 6.00 (d, J = 2,5 Hz, 1H), 7,76 (d, J = 8,
Paso F: N-(bencenosulfoml)-2-doro-6-[3-(espiro[2.2]
[0668]
1-ilmetoxi)pirazol-1-il]-nicotínico (990 mg, 2,97 mmol) se seguido de hidróxido de litio (285 mg, 11,88 mmol). La o 1 M (12 mL). El sólido blanco formado se separó por o-6-[3-(espiro[2.2]pent-1-ilmetoxi)-pirazol-1-il]-nicotínico 07 encontrado 320,0 (M+1)+. Tiempo de retención: 3,25 H), 1,11-1,13 (m, 1H), 1,60 - 1,75 (m, 1H), 4,22 (dd, J = 1H), 8,38 (d, J = 2,5 Hz, 1H), 8,43 (d, J = 8,5 Hz, 1H).
n-2-Mmetoxi)pirazoM-M]piridm-3-carboxamida

[0669] Ácido 2-cloro-6-[3-(espiro[2.2]pentan-2-ilmetoxi carbonildiimidazol (38 mg, 0,23 mmol) fueron combinad añadió bencenosulfonamida (25 mg, 0,16 mmol) seguid h más a temperatura ambiente. La reacción se diluyó c 1 M, seguido de salmuera. Los orgánicos se separaron N-(bencenosulfonil)-2-cloro-6-[3-(espiro[2.2]pentan-2-il EM m/z calc. 458,08154, encontrado 459,2 (M+1)+; tiem
Paso G: N-(bencenosulfonil)-6-[3-(espiro[2.2]pen il]piridin-3-carboxamida
[0670]
ol-1-il]piridin-3-carboxílico (50 mg, 0,16 mmol) y THF (1,5 mL) y agitados durante 2 h. En este punto, se BU (70 pL, 0,47 mmol) y la reacción se agitó durante 2 tato de etilo y se lavó con una solución de ácido cítrico ecaron sobre sulfato de sodio y se evaporaron para dar )pirazol-1-il]piridin-3-carboxamida (72 mg, 98%) IEN-retención: 0,75 minutos.
ilmetoxi)pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-

[0671] N-(bencenosulfonil)-2-cloro-6-[3-(espiro[2.2]pent mmol), (4S)-2,2,4-trimetilpirrolidina (sal de hidrocloruro) se combinaron en DMSO (1 mL) y se calentaron a 130 agitó durante 20 min. Se formó un sólido y se decantó lavó con una solución de ácido cítrico 1 M y luego con se evaporaron. El material crudo se purificó mediante c en diclorometano para dar N-(bencenosulfon trimetilpirrolidin-1 -il]piridin-3-carboxamida (38 mg, 45 tiempo de retención: 2,22 minutos.
Ejemplo sintético 59: Síntesis del Compuest (trifluorometil)ciclopropil]etoxi}-1H-pirazol-1-il)-2,7,1
[0672]
lmetoxi)pirazol-1-il]piridin-3-carboxamida (72 mg, 0,16 g, 0,42 mmol) y carbonato de potasio (97 mg, 0,7 mmol) rante 16 h. La reacción se diluyó con agua (3 mL) y se ido acuoso. El sólido se disolvió en acetato de etilo y se era. Los orgánicos se secaron sobre sulfato de sodio y tografía en gel de sílice eluyendo con metanol al 0-10% -(espiro[2.2]pentan-2-ilmetoxi)pirazol-1-il]-2-[(4S)-2,2,4--EM m/z calc. 535,22534, encontrado 536,1 (M+1)+;
: (5S)-7-(bencenosulfonil)-3,3,5-trimetil-12-(3-{2-[1-zatricido[7.4.0.02.6] trideca-1(9), 10,12-trien-8-ona

[0673] N-(bencenosulfonil)-6-[3-[2-[1-(trifluorometil)ciclop il]piridin-3-carboxamida (52 mg, 0,08789 mmol), Na [Ir{dF(CF3)ppy}2(dtbpy)]pF6 (5 mg, 0,004 mmol) se co reacción se colocó junto a una fuente de luz CFL de 23 columna de gel de sílice sin ningún tratamiento. La mez eluyendo con acetato de etilo al 0-100% en hexanos (trifluorometil)ciclopropil]etoxi}-1H-pirazol-1-il)-2,7,13-tria 19%) IEN-EM m/z calc. 589,1971, encontrado 590,3 (M DMSO-cfe) 88,26 (d, J = 2,8 Hz, 1H), 8,06 (d, J = 7,7 Hz, J = 7,6 Hz, 2H), 6,97 (d, J = 8,2 Hz, 1H), 6,07 (d, J = 2,7 (dt J = 12,2, 5,7 Hz, 1H), 2,17 (dd, J = 13,4, 7,3 Hz, 1H), 3H), 1,21 (d, J = 6,3 Hz, 3H), 0,97 (m, 2H), 0,88 (m, 2H)
Ejemplo sintético 60: Síntesis del (trifluorometil)ciclopropil]etoxi}-1H-pirazol-1 -il)-7-ox ona
Paso A: Ácido 6-[3-[2-[1-(trifluorometil)ciclopropil]e 3-carboxílico
[0674]
]etoxi]pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirroMdin-1-(14 mg, 0,1707 mmol), agua (16 pL, 0,89 mmol) y ron en DMA (dimetilacetamida) (0,9 pL) y la mezcla de rante 1,5 h. La reacción se inyectó directamente en una ruda se purificó mediante cromatografía en gel de sílice a dar (5S)-7-(bencenosulfonN)-3,3,5-trimetiM2-(3-{2-[1-ciclo[7.4.0.02.6]trideca-1(9), 10,12-trien-8-ona (10 mg, tiempo de retención: 2,51 minutos. 1H RMN (400 MHz, , 7,81 (d, J = 8,2 Hz, 1H), 7,73 (t, J = 7,4 Hz, 1H), 7,66 (t, H), 6,02 (d, J = 3,9 Hz, 1H), 4,36 (t, J = 7,0 Hz, 2H), 3,04 (m, 2H), 1,86 (d, J = 13,2 Hz, 1H), 1,71 (s, 3H), 1,61 (s,
ompuesto 62: (5S)-3,3,5-trimetiM2-(3-{2-[1-3-diazatriciclo[7.4.0.02.6]trideca-1(13),9,11-trien-8-
pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1-il]piridin-

[0675] A una mezcla de ácido 2-cloro-6-[3-[2-[1 -(triflu 2,661 mmol) y (4S)-2,2,4-trimetilpirrolidina (sal de clorhi 1,2-dietoxietano (1 mL) se añadió carbonato de potasio ( 68 h. CL/EM mostró una conversión del 40%. Se aña continuó durante 18 ha 135°C. Se enfrió la suspensión d una solución rápidamente agitada de HCl (2 ml de 6 marrón. La suspensión se extrajo con acetato de etilo.
de sodio y se concentró. El material crudo se cromatog etilo. El producto salió ~30% de acetato de etilo. Ácid 2,2,4-trimetilpirrolidin-1-il]piridin-3-carboxílico (667,8 m (M+1)+; tiempo de retención: 1,87 minutos. 1H RMN (400 Hz, 1H), 6,86 (dd, J = 32,9, 7,9 Hz, 1H), 6,29 - 6,01 (m, (s, 2H), 1,95 (s, 1H), 1,74 - 1,46 (m, 7 H), 1,12 -1,01 (m,
Paso B: (5S)-3,3,5-Trimetil-12-(3-{2-[1-( diazatriciclo[7.4.0.02.6]trideca-1(13),9,11-trien-8-ona
[0676]
etil)ciclopropil]etoxi]pirazol-1 -il]piridin-3-carboxílico (1 g, ) (620 mg, 4,143 mmol) en N-metilpirrolidinona (5 mL) y , 13,02 mmol). La suspensión se calentó a 125°C durante ás (4S)-2,2,4-trimetilpirrolidina (400 mg) y la reacción acción a temperatura ambiente y se añadió lentamente a ,00 mmol) en hielo (¡espumas!) dando una suspensión ánico se lavó con agua, salmuera, se secó sobre sulfato sobre sílice usando un gradiente de hexano/acetato de 3-[2-[1-(trifluorometil)ciclopropil]etoxi]pirazol-1-il]-2-[(4S)-5%). iEn-EM m/z calc. 452,20352, encontrado 453,0 , DMSO-d6) 812,68 (s, 1H), 8,24 (s, 1H), 7,92 (d, J = 8,2 4,31 (s, 2H), 3,54 (s, 1H), 2,89 (s, 1H), 2,33 (s, 1H), 2,08 , 0,92 (d, J = 29,3 Hz, 4H).
orometil)ciclopropil]etoxi}-1H-pirazol-1-il)-7-oxa-2,13- 
0677] Ácido 6-[3-[2-[1-(Trifluorometil)ciclopropi carboxílico (50 mg, 0,1105 mmol), agua (20 ml, 1,110 mm (4 mg, 0,003565 mmol) se disolvieron en DMA (0,9 mL) fuente de luz durante 1,5 h. La reacción se inyectó directa La mezcla cruda se purificó mediante cromatografía e hexanos para dar (5S)-3,3,5-trimetil-12-(3-{2-diazatriciclo[7.4.0.02.6]trideca-1(13),9,11-trien-8-ona (30 (M+1)+; tiempo de retención: 2,35 minutos.
Ejemplo sintético 61: Síntesis del C bicido[1.1.1]pentaml)metoxi]pirazoM-N]-2-[(4S)-2,2,4-
Paso A: 3-[(3-fluoro-1-biciclo[1.1.1]pentanil)metoxi]p
[0678]
toxi]pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirroMdin-1-il]piridin-3-, NaOAc (18 mg, 0,22 mmol) y [Ir{dF(CF3)ppy}2(dtbpy)]PF6 la mezcla de reacción se colocó junto a una CFL de 23 W nte en una columna de gel de sílice sin ningún tratamiento. el de sílice eluyendo con acetato de etilo al 0-100% en -(trifluorometil)ciclopropil]etoxi}-1H-pirazol-1 -il)-7-oxa-2,13-mg, 62%) IEN-EM m/z calc. 450,18787, encontrado 451,3
puesto 63: N-(bencenosulfoml)-6-[3-[(3-fluoro-1-etMpirroNdm-1-M]piridm-3-carboxamida
zol-1-carboxilato de terc-butilo

[0679] Una solución de (3-fluoro-1-biciclo[1.1.1]pentanil) terc-butilo (0,46 g, 2,5 mmol) y trifenilfosfina (0,67 g, 2, añadió lentamente N-isopropoxicarboniliminocarbamato lentamente a temperatura ambiente y se agitó durante tre de sodio acuoso saturado, se secó sobre sulfato de s cromatografía en gel de sílice con acetato de biciclo[1.1.1]pentanil)metoxi]pirazol-1-carboxilato de t encontrado 283,3 (M+1)+; tiempo de retención: 0,65 min
Paso B: 3-[(3-fluoro-1 -biciclo[1.1.1]pentanil)metoxi]-1
[0680]
tanol (0,27 g, 2,3 mmol), 3-hidroxipirazol-1-carboxilato de mol) en THF (12 mL) se enfrió en un baño de hielo y se isopropilo (0,50 ml, 2,6 mmol). La reacción se dejó calentar ías. Se diluyó con acetato de etilo, se lavó con bicarbonato o y se evaporó al vacío. El residuo se purificó mediante tilo al 0-40% en hexanos para dar 3-[(3-fluoro-1--butilo (0,43 g)., 66%) IEN-Em m/z calc. 282,13797, s.
pirazol

[0681] Una solución de 3-[(3-fluoro-1-biciclo[1.1.1]pent mmol) y ácido trifluoroacético (587 pL, 7,62 mmol) en di eliminaron al vacío y el residuo se basificó con bicarbona Los extractos combinados se secaron sobre sulfa biciclo[1.1.1]pentanil)metoxi]-1H-pirazol (0,28 g, 100%) I de retención: 0,39 minutos.
Paso C: 2-Cloro-6-[3-[(3-fluoro-1-biciclo[1.1.1]penta
[0682]
l)metoxi]pirazol-1-carboxilato de terc-butilo (0,43 g, 1,523 rometano (4 mL) se agitó durante 5 horas. Los volátiles se de sodio acuoso saturado y se extrajo con acetato de etilo.
de sodio y se evaporaron para dar 3-[(3-fluoro-1--EM m/z calc. 182,08554, encontrado 183,1 (M+1)+; tiempo
metoxi]pirazol-1-il]piridin-3-carboxilato de terc-butilo

[0683] Una mezcla de 3-[(3-fluoro-1-bicido[1.1.1]pentani carboxilato de terc-butilo (0,38 g, 1,5 mmol), carbonato (34 mg, 0,30 mmol) en DMSO (7,5 mL) se agitó a temper y se extrajo con acetato de etilo. Los extractos combinad de sodio y se evaporaron. El residuo se purificó media diclorometano para dar 2-cloro-6-[3-[(3-fluoro-1-biciclo[1 butilo (0,50 g, 85%) IEN-EM m/z calc. 393,12555, encont
Paso D: Ácido 2-doro-6-[3-[(3-fluoro-1-biddo[1.1.1]p
[0684]
toxi]-1H-pirazol (0,28 g, 1,5 mmol), 2,6-dicloropiridin-3-otasio (0,26 g, 1,9 mmol) y 1,4-diazabiciclo[2.2.2]octano a ambiente durante 16 h. La reacción se diluyó con agua e lavaron con salmuera y agua, se secaron sobre sulfato cromatografía en gel de sílice con metanol al 0-5% en ]pentanil)metoxi]pirazol-1-il]piridin-3-carboxilato de terc-394,2 (M+1)+; tiempo de retención: 0,86 minutos.
m l)metoxi]pirazoM-N]pmdm-3-carboxflico
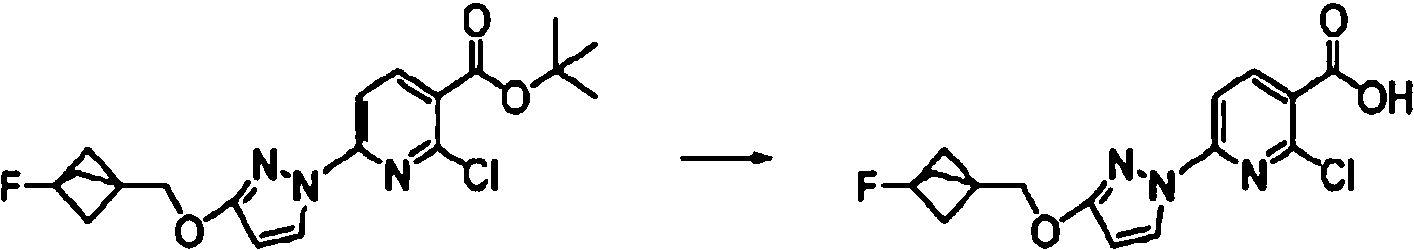
[0685] Una solución de 2-cloro-6-[3-[(3-fluoro-1-biciclo[1 butilo (0,50 g, 1,270 mmol) y se agitó ácido trifluoroacéti horas. Se evaporó el disolvente y se recogió el residuo en 6-[3-[(3-fluoro-1 -biciclo[1.1.1]pentanil)metoxi]pirazol-1-il]p 337,06296, encontrado 338,1 (M+1)+; tiempo de retenció J = 8,5 Hz, 1H), 8,39 (d, J = 2,9 Hz, 1H), 7,73 (d, J = 8,5 Hz, 6H).
Paso E: N-(bencenosulfom l)-2-doro-6-[3-[(3-fl carboxamida
[0686]
]pentanil)metoxi]pirazol-1-il]piridin-3-carboxilato de terc-978 pL, 12,7 mmol) en diclorometano (6 mL) durante 15 tonitrilo. El disolvente se evaporó para dar ácido 2-cloron-3-carboxílico (0,43 g, 100%) IEN-EM m/z calc. ,63 minutos 1H RMN (400 MHz, cloroformo-d) 88,43 (d, H), 6,00 (d, J = 2,8 Hz, 1H), 4,51 (s, 2H), 2,13 (d, J = 2,6
-1-biddo[1.1.1]pentam l)metoxi]pirazoM-M]pindm-3-

[0687] Ácido 2-cloro-6-[3-[(3-fluoro-1-biciclo[1.1.1]pentan y carbonildiimidazol (58 mg, 0,36 mmol) se combinaron añadió bencenosulfonamida (61 mg, 0,39 mmol) seguido h más a temperatura ambiente. La reacción se diluyó co 1 M, seguido de salmuera. Los orgánicos se separaron, N-(bencenosulfonil)-2-cloro-6-[3-[(3-fluoro-1-biciclo[1.1.1] 135%) IEN-EM m/z calc. 476,07214, encontrado 477,2 (
Paso F: N-(bencenosulfoml)-6-[3-[(3-fluoro trimetilpirrolid in-1-il]pirid in-3-carboxam ida
[0688]
etoxi]pirazol-1-il]piridin-3-carboxílico (100 mg, 0,3 mmol) HF (1,5 mL) y se agitaron durante 2 h. En este punto, se BU (54 ml, 0,36 mmol) y la reacción se agitó durante 16 etato de etilo y se lavó con una solución de ácido cítrico ecaron sobre sulfato de sodio y se evaporaron para dar tanil)metoxi]pirazol-1-il]piridin-3-carboxamida (190 mg, ; tiempo de retención: 0,69 minutos.
iddo[1.1.1]pentaml)metoxi]pirazoM-N]-2-[(4S)-2,2,4- 
[0689] N-(Bencenosulfonil)-2-cloro-6-[3-[(3-fluoro-1-bicicl (140 mg, 0,29 mmol), (4S)-2,2,4-trimetilpirrolidina (sal de mg, 1,76 mmol) se combinaron en DMSO (1,5 mL) y se filtró y se purificó por CL/EM utilizando un gradiente d (bencenosulfonil)-6-[3-[(3-fluoro-1 -biciclo[1.1.1]pentanil) carboxamida (89 mg, 54%) IEN-EM m/z calc. 553,2159, 1H RMN (400 MHz, DMSO-da) 88,19 (d, J = 2,8 Hz, 1H), 1H), 7,70 -7,61 (m, 2H), 6,91 (d, J = 8,2 Hz, 1H), 6,14 (d, (m, 1H), 2,15 -2,08 (m, 7 H), 1,82 (dd, J = 11,9, 5,5 Hz, (d, J = 6,2 Hz, 3H)
Ejemplo sintético 62: Síntesis del Compuesto ilmetoxi)pirazol-1-il]-2-[(4S)-2,2,4-trimetilpirrolidin-1 -il
Paso A: 3-(diespiro[2.0,2,1]heptan-7-il metoxi)-1H-pir
[0690]
.1]pentanilo)metoxi]pirazol-1-il]piridin-3-carboxamida
idrato) (131 mg, 0,88 mmol) y carbonato de potasio (243 ntaron a 130°C durante 16 h. La mezcla de reacción se etonitrilo al 30-99% en HCl acuoso 5 mM para dar N-i]pirazol-1 -il]-2-[(4S)-2,2,4-trimetilpirroMdin-1-il]piridin-3-ntrado 554,4 (M+1)+; tiempo de retención: 2,16 minutos.
- 7,95 (m, 2H), 7,81 (d, J = 8,3 Hz, 1H), 7,77 - 7,70 (m, 2,8 Hz, 1H), 4,47 (s, 2H), 2,45 -2,36 (m, 1H), 2,31 -2,22 1,52 (d, J = 9,2 Hz, 6H), 1,36 (t, J = 12,1 Hz, 1H), 0,64
W-(bencenosulfoml)-6-[3-(diespiro[2.0.2.1]heptan-7-in-3-carboxamida
1-carboxilato de íerc-butilo

[0691] Una solución de diespiro[2.0.2,1]heptan-7-il met 2002, 485-492), 3-hidroxi-pirazol-1-carboxilato de íerc-bu (28 mL) se enfrió en un baño de hielo y diisopropil azodi Se retiró el baño de enfriamiento y se agitó la reacción se lavó con bicarbonato de sodio acuoso saturado, se se purificó por cromatografía de gel de sílice eluyend (diespiro[2.0.2.1]heptan-7-il metoxi)-1H-pirazol-1-carboxi aceite incoloro. IEN-EM m/z calc. 290,16306, encontrado
Paso B: 3-(Diespiro[2.0.2.1]heptan-7-ilmetoxi)-1H-pir
[0692]
1,36 g, 11,0 mmol) (Meijere, et al., Eur. J. Org. Chem.
2,3 g, 12 mmol), y trifenilfosfina (3,2 g, 12 mmol) en THF xilato (DIAD) (2,4 mL, 12 mmol) se añadió lentamente. te 15 horas. La reacción se diluyó con acetato de etilo, bre sulfato de sodio y se evaporó al vacío. El residuo se n 0-20% acetato de etilo en hexanos para dar 3-de íerc-butilo (1,57 g, 49% de rendimiento) como un ,3 (M+1)+; tiempo de retención: 0,76 minutos.

[0693] Una solución de 3-(diespiro[2.0.2.1]heptan-7-il mmol) y ácido trifluoroacético (2,2 ml, 29 mmol) en diclor eliminaron al vacío y el residuo se basificó con bicarbona Los extractos combinados se secaron sobre sulfato de ilmetoxi)-1H-pirazol (0,94 g, 91% de rendimiento) co encontrado 191,1 (M+1)+; tiempo de retención: 0,52 min
Paso C: 2-Cloro-6-(3-(diespiro[2.0.2.1]heptan-7-ilmet
[0694]
i)-1H-pirazol-1-carboxilato de terc-butilo (1,57 g, 5,41 ano (20 mL) se agitó durante tres horas. Los volátiles se sodio acuoso saturado y se extrajo con acetato de etilo. y se evaporaron para dar 3-(diespiro[2.0.2.1]heptan-7-aceite amarillo pálido. IEN-Em m/z calc. 190,11061,
H-pirazol-1-il)nicotinato de etilo

[0695] Una mezcla de 3-(d¡espiro[2.0.2.1]heptan-7-¡l dicloropiridina-3-carboxNato de etilo (1,15 g, 5,23
d iazab iciclop^^octano (0,12 g, 1,1 mmol) en DMSO ( agua y se extrajo con acetato de etilo. Los extractos co sulfato de sodio y se evaporaron al vacío. El residuo se p con 0-20% acetato de etilo en hexanos para dar acetato d 1-il)nicotinato (1,39 g, rendimiento del 75%) como un sól (M+1)+; tiempo de retención: 0,87 minutos. 1H RMN (40 8,5 Hz, 1H), 7,72 (d, J = 8,5 Hz, 1H), 5,96 (d, J = 2,9 Hz (t, J = 7,0 Hz, 1H), 1,42 (t, J = 7,1 Hz, 3H), 1,02-0,89 (m,
Paso D: Ácido 2-Cloro-6-[3-(diespiro[2.0. 2,1] ácido h
[0696]

[0697] Una solución de 2-cloro-6-(3-(d¡esp¡ro[2.0.2.1]hep mmol) e hidróxido sódico (7,5 ml de solución 1 M, 7,5 mm Los volátiles se eliminaron al vacío y se añadió agua. La ácido clorhídrico (7,5 ml de solución 1 M, 7,5 mmol). La Los extractos combinados se lavaron con salmuera, se s 2-cloro-6-[3-(d¡esp¡ro[2.0.2.1]heptan-7-¡lmetox¡)p¡razol-1-sólido incoloro. IEN-EM m/z calc. 345,088, encontrado 3 MHz, DMSO-d6) 88,41 (d, J = 2,9 Hz, 1H), 8,38 (d, J = 4,27 (d, J = 7,0 Hz, 2H), 1,93 (t, J = 7,0 Hz, 1H), 0,97 - 0
Paso E: W-(bencenosulfoml)-2-cloro-6-[3-(d¡esp¡ro[2.
[0698]
¡)-1H-p¡razol (0,94 g, 4,9 mmol), acetato de 2,6-carbonato de potasio (0,83 g, 6,0 mmol) y 1,4-L) se agitó durante 24 horas. La reacción se diluyó con dos se lavaron con salmuera y agua, se secaron sobre por cromatografía en columna de gel de sílice eluyendo oro-6-(3-(d¡esp¡ro[2.0.2.1]heptan-7-¡lmetox¡)-1H-p¡razolcoloro. iEn-EM m/z calc. 373,11932, encontrado 374,2 z, cloroformo-d) 88,36 (d, J = 2,8 Hz, 1H), 8,27 (d, J = , 4,41 (q, J = 7,1 Hz, 2H), 4,30 (d, J = 7,0 Hz, 2H), 1,94 0,75-0,65 (m, 2H), 0,65-0,53 (m, 2H)
-7-ilmetoxi)pirazol-1-il]piridin-3-carboxílico
THF (6 mL) y etanol (3 mL) se agitó durante 90 minutos. ión se enfrió en un baño de hielo y se añadió lentamente ión se diluyó con agua y se extrajo con acetato de etilo. n sobre sulfato de sodio y se evaporaron para dar ácido ¡n-3-carboxíl¡co (1,16 g, rendimiento del 8 2 %) como un M+1)+; tiempo de retención: 0,73 minutos. 1H RMN (400 , 1H), 7,73 (d, J = 8,4 Hz, 1H), 6,19 (d, J = 2,8 Hz, 1H), , 4H), 0,76 - 0,66 (m, 2H), 0,65 - 0,56 (m, 2H)
heptan-7-¡lmetox¡)p¡razoM-M]p¡r¡dm-3-carboxam¡da

[0699] Una solución de ácido 2-cloro-6-[3-(d¡esp¡ro[2.0.
0,29 mmol) y se agitó carbonil diimidazol (0,06 g bencenosulfonamida (55 mg, 0,35 mmol) y I,8-d¡aza añadieron. Después de 15 horas, la reacción se diluyó c Los extractos combinados se lavaron con salmuera, se (bencenosulfon¡l)-2-cloro-6-[3-(d¡esp¡ro[2.0.2.1]heptan-7-¡ se usó en el siguiente paso de esta manera. IEN-EM retención: 0,81 minutos.
Paso F: W-(Bencenosulfoml)-6-[3-(d¡ tr¡met¡lp¡rrol¡d¡n-1-¡l]p¡r¡d¡n-3-carboxam¡da
eptan-7-¡lmetox¡)p¡razol-1-¡l]p¡r¡d¡n-3-carboxíl¡co (0,10 g, mmol) en t Hf (1,4 mL) durante 45 minutos, y lo(5,4.0)undec-7-eno (DBU) (130 pL, 0,87 mmol) se ido cítrico acuoso 1 M y se extrajo con acetato de etilo. ron sobre sulfato de sodio y se evaporaron para dar N-x¡)p¡razol-1-¡l]p¡r¡d¡n-3-carboxam¡da cruda (0,16 g) que calc. 484,0972, encontrado 485,2 (M+1)+; tiempo de
o[2.0.2.1]heptan-7-¡lmetox¡)p¡razol-1-¡l]-2-[(4S)-2,2,4-
[0700]

[0701] Una mezcla de W-(bencenosulfon¡l)-2-clorocarboxamida (0,14 g, 0,29 mmol), clorhidrato de (4S)-potas¡o (0,24 g, 1,7 mmol) en NMP (1,3 mL) se ag¡tó usando un método de HPCL-EM de fase ¡nversa usand 5 mM. A/-(bencenosulfon¡l)-6-[3-(d¡esp¡ro[2.0.2.1] ¡l]p¡r¡d¡n-3-carboxam¡da (65 mg, 40% de rend¡m¡ento) (M+1)+; t¡empo de retenc¡ón: 2,35 m¡nutos.
Ejemplo sintético 63: Síntesis del Compuesto 65: trimetilpirrolid in-1-il]pirid in-3-carboxam ida
Paso A: 3-((1-EtMcidopropN)metoxi)-1H-pirazoM-car [0702]
d¡esp¡ro[2.0.2.1]hieptan-7-¡lmetox¡)p¡razol-1-¡l]p¡r¡d¡na-3-tr¡met¡lp¡rrol¡d¡na (0,14 g, 0,94 mmol) y carbonato de °C durante 15 horas. La reacc¡ón se f¡ltró y se pur¡f¡có rad¡ente doble de aceton¡tr¡lo al 30-99% en HCl acuoso n-7-¡lmetox¡)p¡razol-1-¡l]-2-[(4S)-2,2,4-tr¡met¡lp¡rrol¡d¡n-1-tuvo. IEN-EM m/z calc. 561,24097, encontrado 562,3
encenosulfoml)-6-(3-hidroxipirazoM-N)-2-[(4S)-2,2,4-
ato de íerc-butilo

[0703] A la soluc¡ón de (1-et¡lc¡cloprop¡l)metanol (1,68 but¡lo (2,80 g, 15,21 mmol) y tr¡fen¡lfosf¡na (4,39 g, azod¡carbox¡lato de d¡¡soprop¡lo (3,38 g, 16,73 mmol) g luego se calentó a 50°C durante 21 horas. La soluc¡ón acetato de et¡lo (500 mL). La capa orgán¡ca se lavó con salmuera (2 x 50 mL) y se secó sobre sulfato de m cromatografía en gel de sílice usando método de grad¡e 3-((1-et¡lc¡cloprop¡l)metox¡)-1H-p¡razol-1-carbox¡lato de e M m/z calc. 266,2, encontrado 267,3 (M+1)+. T¡empo d 7,82 (d, J = 2,5Hz, 1H), 5,88 (d, J = 2,5Hz, 1H), 4,09 (s, 3H), 0,49 (m, 2H), 0,42 (m, 2H).
Paso B: 3-((1-EtMciclopropN)metoxi)-1H-pirazol
[0704]
73 mmol), 2,3-d¡h¡dro-3-oxop¡razol-l-carbox¡lato de terc-mmol) en tetrah¡drofurano (40 mL) a 0°C se añad¡ó gota. La reacc¡ón se calentó a temperatura amb¡ente y eacc¡ón se enfr¡ó a temperatura amb¡ente y se añad¡ó soluc¡ón acuosa de h¡dróx¡do de sod¡o 0,3 M (100 mL), o, se f¡ltró y se concentró. El res¡duo se pur¡f¡có por e 0 a 80% de hexanos/d¡clorometano para proporc¡onar ut¡lo (1,73 g, 43%) como un ace¡te amar¡llo. IEN-nc¡ón: 3,47 m¡nutos. 1H RMN (250MHz, CDCh) 8 (ppm): 1,60 (s, 9H), 1,48 (q, J = 7,5 Hz, 2H), 0,94 (t, J = 7,5 Hz,

[0705] Una soluc¡ón de cloruro de hidrógeno 4M en 1,4 p¡razol-1-carbox¡lato de terc-but¡lo (1,73 g, 6,49 mmol) durante 16 horas y se concentraron a sequedad para o un ace¡te que se usó d¡rectamente en el s¡gu¡ente paso. retenc¡ón: 0,60 m¡nutos.
Paso C: 2-Cloro-6-(3-((1-etilciclopropil)metoxi)-1H-pi
[0706]
no (65 mL) se añad¡ó a 3-((1-et¡lc¡cloprop¡l)metox¡)-1H-soluc¡ón de reacc¡ón se ag¡tó a temperatura amb¡ente r el 3-((1-et¡lc¡cloprop¡lo)metox¡)-1H-p¡razol crudo como M m/z calc. 166,1, encontrado 167,3 (M+1)+. T¡empo de
-1-il)nicotinato de metilo

[0707] Se añadieron 3-((1-etilcidopropil)metoxi)-1H-pira mmol) a dimetilformamida (20 mL). Se añadieron carbon (0,167 g, 1,5 mmol) a la mezcla de reacción que se dejó de reacción se diluyó con agua (60 mL) y se extrajo co secó sobre sulfato de sodio y se evaporó a presión redu sílice usando un método de gradiente de hexanos etilciclopropil)metoxi)-1H-pirazol-1-il)nicotinato de metilo encontrado 336,5 (M+1)+. Tiempo de retención: 4,29 min 1H), 8,41 (d, J = 8,4 Hz, 1H), 7,74 (d, J = 8,4 Hz, 1H), = 7,4Hz, 2H), 0,93 (t, J = 7,4Hz, 3H), 0,53 (m, 2H), 0,42
Paso D: Ácido 2-doro-6-(3-((1-etMddopropN)metoxi)-
[0708]
,49 mmol) y 2,6-dicloronicotinato de metilo (1,81 g, 8 potasio (2,8 g, 20 mmol) y 1,4-diazabicido[2.2.2]octano r a temperatura ambiente durante 16 horas. La mezcla r dietílico (3 x 60 mL). La capa orgánica combinada se l residuo se sometió a cromatografía flash sobre gel de rometano del 0 al 100% para dar 2-cloro-6-(3-((1-, 77%) como un sólido blanco. IEN-EM m/z calc. 335,10, 1H RMN (250 MHz, DMSO) 8 (ppm): 8,43 (d, J = 2,8 Hz, d, J = 2,8Hz, 1H), 4,08 (s, 2H), 3,88 (s, 3H), 1,44 (q, J ).
razol-1-N)mcotmico

[0709] 2-cloro-6-(3-((1-etilciclopropil)metoxi)-1H-pirazolmatraz de fondo redondo enfriado con hielo con tetrah hidróxido de sodio (0,4 g, 10 mmol) en agua (5 mL) y la 5 horas. La mezcla de reacción resultante se concentr acidificó con una solución acuosa 1 N de cloruro de hidr se separó por filtración para dar ácido 2-cloro-6-(3-((1-eti IEN-Em m/z calc. 321,09, encontrado 322,2 (M+1)+. Tie (ppm): 8,43 (d, J = 2,8 Hz, 1H), 8,42 (d, J = 8,4 Hz, 1H), 1,45 (q, J = 7,4Hz, 2H), 0,92 (t, J = 7,4Hz, 3H), 0,54 (m,
Paso E: N-(Bencenosulfonil)-2-doro-6-[3-[(1-etilcido
[0710]
cotinato de metilo (1,7 g, 50 mmol) se añadió en un rano (5 mL) y metanol (5 mL). Se añadió lentamente n de reacción se agitó a temperatura ambiente durante resión reducida para eliminar el tetrahidrofurano y se a pH = 1 en un baño de hielo. El sólido blanco formado ropil)metoxi)-1H-pirazol-1-il)-nicotínico (1,54 g, 95,0%). e retención: 3,59 minutos. 1H RMN (250 MHz, DMSO) 8 d, J = 8,4 Hz, 1H), 6,24 (d, J = 2,8Hz, 1H), 4,09 (s, 2H), ,44 (m, 2H).
)metoxi]pirazol-1-il]piridin-3-carboxamida

[0711] Se combinaron ácido 2-cloro-6-[3-[(1-etilciclop mmol) y carbonildiimidazol (100 mg, 0,6167 mmol) en T bencenosulfonamida (77 mg, 0,4899 mmol) seguido de a temperatura ambiente. La reacción se diluyó con acet seguido de salmuera. Los orgánicos se separaron, se (bencenosulfonil)-2-cloro-6-[3-[(1-etilciclopropil)metoxi]pi EM m/z calc. 460,0972, encontrado 461,1 (M+1)+; tiemp
Paso F: N-(Bencenosulfonil)-2-cloro-6-(3-hidroxipira etoxi]pirazol-1-il]piridin-3-carboxílico (157 mg, 0,4879 mL) y se agitaron durante 2 h. En este punto, se añadió (243 ml, 1,625 mmol) y la reacción se agitó 30 min más e etilo y se lavó con una solución de ácido cítrico 1 M, n sobre sulfato de sodio y se evaporaron para dar N-1-il]piridin-3-carboxamida cruda (224 mg, 100%) IEN-etención: 0,75 minutos.
il)piridin-3-carboxamida
[0712]

[0713] Se disolvió N-(bencenosulfonil)-2-doro-6-[3-[(1-et 100%) en diclorometano (2 mL) con TFA (1 ml, 12,98 m a sequedad para dar N-(bencenosulfonil)-2-cloro-6-(3-EM m/z calc. 378,01895, encontrado 379,1 (M+1)+; tiem Paso G: N-(bencenosulfonil)-6-(3-hidroxipirazol-il)-2 [0714]
opropil)metoxi]pirazol-1-il]piridin-3-carboxamida (224 mg, y la reacción se agitó durante 4 h. La reacción se evaporó xipirazol-1-il)piridin-3-carboxamida (136 mg, 74%) IEN-e retención: 0,54 minutos.
)-2,2,4-trimetilpirrolidin-1-il]piridin-3-carboxamida

[0715] N-(Bencenosulfonil)-2-cloro-6-(3-hidroxipirazol-1-trimetilpirrolidina (sal de clorhidrato) (110 mg, 0,76 mm en DMSO (0,5 mL) y se calentaron a 130°C durante 16 min. Se formó un sólido y se decantó el líquido acuos solución de ácido cítrico 1 M y luego con salmuera. Los El material crudo se purificó mediante cromatografía en para dar N-(bencenosulfonil)-6-(3-hidroxipirazol-1-il)-2-[ 4%) IEN-EM m/z calc. 455,16272, encontrado 456,3 (M Ejemplo 5: ENSAYOS Y DATOS
5A. Ensayos para detectar y medir el modulador F5 Métodos ópticos de potencial de membrana para en [0716] El ensayo utiliza tintes sensores fluorescentes utilizando un lector de placas fluorescentes (p. ej., FLIP de F508del funcional en células NIH 3T3. La fuerza imp cloruro junto con la activación del canal y al mismo tie de adición de líquido después de que las células se hay 5A-A1. Identificación de moduladores F508del-CFTR
[0717] Para identificar moduladores de F508del, fue de Este ensayo HTS utiliza tintes fluorescentes sensores d en el FLIPR III como una medida para el aumento de la fuerza impulsora de la respuesta es la creación de un g mismo tiempo que el tratamiento del compuesto medi células se hayan cargado previamente con un tinte se obtuvieron usando el ensayo descrito aquí se resumen e el Compuesto 1 tenía una CE50 de menos de 3 pM y un Soluciones
[0718] Solución de baño n.° 1: (en mM) NaCl 160, KC1 10.
[0719] Solución de baño sin cloruro: Sales de cloruro e gluconato.
ridin-3-carboxamida (100 mg, 0,26 mmol), (4S)-2,2,4-carbonato de potasio (180 mg, 1,3 mmol) se combinaron reacción se diluyó con agua (3 mL) y se agitó durante 20 sólido se disolvió en acetato de etilo y se lavó con una nicos se secaron sobre sulfato de sodio y se evaporaron. e sílice eluyendo con metanol al 0-10% en diclorometano -2,2,4-trimetilpirrolidin-1-il]piridin-3-carboxamida (4,6 mg, Tiempo de retención: 1,50 minutos.
l-CFTR Propiedades de los compuestos
r las propiedades de los moduladores F508del-CFTR
oltaje para medir cambios en el potencial de membrana I, Molecular Devices, Inc.) como una lectura del aumento a de la respuesta es la creación de un gradiente de iones que el tratamiento del compuesto mediante un solo paso argado previamente con un tinte sensor de voltaje.
llado un formato de ensayo HTS basado en fluorescencia. ltaje para medir los cambios en el potencial de membrana puerta (conductancia) de las células F508del NIH 3T3. La nte de iones cloruro junto con la activación del canal y al un solo paso de adición de líquido después de que las de voltaje. Los datos para los compuestos 1-65 que se Tabla 6 a continuación. Por ejemplo, usando este método, e eficacia de > 100% con respecto al Compuesto II.
CaCh 2, MgCh 1, HEPES 10, pH 7,4 con NaOH, glucosa
lución de baño n.° 1 (anterior) se sustituye con sales de
Cultivo de células
[0720] Fibroblastos de ratón NIH3T3 que expresan esta potencial de membrana. Las células se mantienen a 3 modificado de Dulbecco suplementado con glutamina pen/estrep y 25 mM HEPES en matraces de cultivo d sembraron a 12.000 células/pocillo en placas recubierta horas a 37°C para el ensayo de potenciador. Para los e compuestos durante 18 a 24 horas.
[0721] Ensayos electrofisiológicos para ensayar propied
Ensayo de cámara Ussing
[0722] Experimentos en cámara Ussing se realizaron e expresan F508del para caracterizar adicionalmente los aislaron epitelios de las vías respiratorias sin FQ y con anteriormente (Galietta, L.J.V., Lantero, S., Gazzolo,
(1998) In Vitro Cell. Dev. Biol. 34, 478-481) y se colo recubrieron previamente con medios acondicionados co las células se cultivaron en una interfaz aire-líquido dura de células columnares completamente diferenciadas qu los epitelios de las vías respiratorias. Se aislaron HBE pulmonar conocida. Se aislaron HBE con FQ de pacie para F508del con una enfermedad diferente que causab
[0723] HBE cultivadas en insertos de cultivo celular C (Physiologic Instruments, Inc., San Diego, CA), y la resist de un gradiente Cl- basolateral a apical (Isc) se midió Bioingeniería, Universidad de lowa, IA). Brevemente, los voltaje (Vretención = 0 mV) a 37°C. La solución basolateral MgCl2 , 1,2 CaCl2 , 10 Glucosa, 10 HEPES (pH ajustado NaGluconato, 1,2 MgCh, 1,2 CaCh, 10 glucosa, 10 HEP
5A-A2. Identificación de moduladores F508del-CFTR
[0724] El protocolo típico utilizaba un gradiente de co establecer este gradiente, se usaron anillos normales reemplazó por gluconato de sodio equimolar (titulado a p de Cl- a través del epitelio. Los moduladores se agregar durante el ensayo. Se añadió forskolina (10 mM) al lad mediado por CFTR.
Registros de pinza de parche
[0725] Se monitorizó la corriente cr total en células F5 perforado como se describió previamente (Rae, J., Coo 37, 15-26). Los registros de tensión de pinza se realiz Axopatch 200B (Axon Instruments lnc., Foster City, CA glucamina (NMDG)-Cl, 2 MgCh, 2 CaCh, 10 EGTA, 10 HCl). El medio extracelular contenía (en mM) 150 NMD HCl). La generación de pulsos, la adquisición de datos y interfaz Digidata 1320 A/D junto con Clampex 8 (Axon I forskolina 10 mM y genisteína 20 mM y se controló la rel
5A-A3. Identificación de moduladores F508del-CFTR
[0726] La capacidad de los moduladores F508del-CF (lF508del) en células NIH3T3 que expresan establemen de parche perforadas. Los moduladores identificados dependiente de la dosis en IAF508 con una potencia y e
Cultivo celular
[0727] Se usan fibroblastos de ratón NIH3T3 que e completas. Las células se mantienen a 37°C en 5% de ente F508del se utilizan para las mediciones ópticas del en 5% de CO2 y 90% de humedad en medio de Eagle M, suero bovino fetal al 10%, 1 X NEAA, p-ME, 1 X 5 cm2 Para todos los ensayos ópticos, las células se n matrigel de 384 pocillos y se cultivaron durante 18-24 s de corrección, las células se cultivan a 37°C con y sin
de modulación F508del de compuestos.
ulas epiteliales de las vías respiratorias polarizadas que ladores F508del identificados en los ensayos ópticos. Se partir de tejido bronquial, cultivados como se describió cco, 0., Romano, L., Rossi, G.A. y Zegarra-Moran, O. n en placas sobre filtros Costar® Snapwell™ que se 3T3. Después de cuatro días, se retiró el medio apical y 14 días antes de su uso. Esto resultó en una monocapa aban ciliadas, características que son características de Q de no fumadores que no tenían ninguna enfermedad homocigotos para F508del o compuestos heterocigotos a mutación en el otro alelo.
® Snapwell™ fueron montadas en una cámara Ussing a transepitelial y la corriente de cortocircuito en presencia ando un sistema de pinza de voltaje (Departamento de se examinaron en condiciones de registro de fijación de tenía (en mM) 145 NaCl, 0,83 K2HPO4 , 3,3 KH2PO4, 1,2 5 con NaOH) y la solución apical contenía (en mM) 145 pH ajustado a 7,35 con NaoH).
tración de Cl- de membrana basolateral a apical. Para membrana basolateral, mientras que el NaCl apical se con NaOH) para dar un gran gradiente de concentración lado basolateral 18-24 antes del ensayo o al lado apical al durante el ensayo para estimular el transporte de Cl-
l-NlH3T3 usando la configuración de registro de parche K., Gates, P., & Watsky, M. (1991) J. Neurosci. Methods a 22°C utilizando un amplificador de pinza de parche solución de la pipeta contenía (en mM) 150 N-metil-D-S y 240 pg/ml de anfotericina-B (pH ajustado a 7,35 con , 2 MgCh, 2 CaCh, 10 HEPES (pH ajustado a 7,35 con nálisis se realizaron utilizando una PC equipada con una ments lnc.). Para activar F508del, se añadieron al baño n corriente-voltaje cada 30 segundos.
para aumentar la corriente C F508del macroscópica 508del también se investigó usando técnicas de registro artir de los ensayos ópticos provocaron un aumento ia similares observadas en los ensayos ópticos.
an establemente F508del para registros de células y 90% de humedad en medio de Eagle modificado de
Dulbecco suplementado con glutamina 2 mM, suero bo HEPES en matraces de cultivo de 175 cm2 Para los regi en cubreobjetos de vidrio recubiertos de poli-L-lisina y s moduladores a 37°C.
Registros de canal único
[0728] La actividad de compuerta de F508del-CFTR exp se observó usando registros de parches de membrana Barbry, P., Champigny, G., Jallat, S., Dott, K., Dreyer, D., Nature 354, 526 - 528) usando un amplificador de pinza contenía (en mM): 150 NMDG, 150 ácido aspártico, 5 Tris). El baño contenía (en mM): 150 NMDG-Cl, 2 MgC HCl). Después de la escisión, tanto wt- como F508del s catalítica de la proteína quinasa dependiente de cAMP ( las proteínas fosfatasas, lo que evitó el resumen actual del canal se analizó a partir de parches de membrana aperturas simultáneas determinó el número de canales la amplitud de corriente de un solo canal, los datos re "fuera de línea" a 100 Hz y luego se usaron para co ajustaron con funciones multigaussianas utilizando el corriente microscópica total y la probabilidad de apertur del canal. El P0 se determinó utilizando el software Biomedia, i = amplitud de corriente de un solo canal y N =
Cultivo celular
[0729] Fibroblastos de ratón NIH3T3 que expresan esta de membrana extirpada. Las células se mantienen a 3 modificado de Dulbecco suplementado con glutamina pen/estrep y 25 mM HEPES en matraces de cultivo de 2500 a 5000 células en cubreobjetos de vidrio recubie presencia o ausencia de moduladores a 37°C.
5B. Determinación cromatográfica de ensayo de alb
[0730] La determinación cromatográfica de los valores UPCL-EM usando una columna ChiralPak® HSA (p/n: tampón de acetato de amonio 50 mM en agua ajustada de la columna se mantuvo a una temperatura constan columna HSA se realizó inyectando 3 mL de 0,5 mM d 30% B en 2,5 minutos, seguido de una retención a 30% 0% de B en 1,5 minutos, para un tiempo de ejecución tot el gradiente y se fijó en 1,8 mL/min. El tiempo de retenci de % HSA de acuerdo con un protocolo previamente p retención de la columna con los valores estándar de uni de diálisis. Los datos de HSA para ciertos compuestos s
[0731] Valko, K., Nunhuck, S., Bevan, C., Abraham, M. Compounds Binding to Human Serum Albumin. Relation Lipophilicity. J. of Pharm. Sci. 2003, 92, 2236-2248.
5C. Protocolo experimental para estudios de PK IV y
[0732] El compuesto ensayado se administró a ratas S única de 3,0 mg/kg como una solución en NMP al 10%, s compuesto probado también se administró a ratas mach como una solución en 5% de NMP, 30% de PEG400, 10 Los análisis de plasma y preparaciones de dosis se reali
[0733] Los perfiles de concentración-tiempo plasmático (nominales) de muestreo se analizaron por métodos f dentro del software Watson LIMS, Versión 7.4.2 (The calcularon utilizando la regla trapezoidal lineal.
etal al 10%, 1 X NEAA, p-ME, 1 X pen/estrep y 25 mM de células enteras, se sembraron de 2500 a 5000 células varon durante 18 a 24 horas en presencia o ausencia de
a en células NIH3T3 después del tratamiento modulador ados como se ha descrito previamente (Dalemans, W., tal, R.G., Pavirani, A., Lecocq, J.P., Lazdunski, M. (1991) arche Axopatch 200B (Axon Instruments Inc.). La pipeta , 2 MgCh y 10 HEp Es (pH ajustado a 7,35 con base EGTA, 10 TES y 14 Tris base (pH ajustado a 7,35 con varon añadiendo Mg-ATP 1 mM, 75 nM de la subunidad Promega Corp. Madison, WI) y NaF 10 mM para inhibir otencial de la pipeta se mantuvo a 80 mV. La actividad contenían < 2 canales activos. El número máximo de s durante el curso de un experimento. Para determinar dos de 120 segundos de actividad F508del se filtraron histogramas de amplitud de todos los puntos que se are Bio-Patch Analysis (Bio-Logic Comp. Francia). La ) se determinaron a partir de 120 segundos de actividad o a partir de la relación P0 = I/i (N), donde I = corriente o de canales activos en el parche.
nte F508del se utilizan para registros de pinza de parche n 5% de CO2 y 90% de humedad en medio de Eagle M, suero bovino fetal al 10%, 1 X NEAA, p-ME, 1 X m2. Para los registros de canal único, se sembraron de de poli-L-lisina y se cultivaron durante 18-24 horas en
sérica humana (HSA)
lbúmina sérica humana (HSA) se realizó en un sistema 9AST) de Sigma Aldrich. La fase móvil A consistió en = 7,4 y la fase móvil B fue 2-propanol. El compartimento 30°C. La determinación del tiempo de retención en la puesto (en DMSO) usando un gradiente lineal de 0% -rante 2 minutos y el paso de equilibrado final del 30% al 6 minutos. El caudal se mantuvo constante durante todo l compuesto en la columna HSA se convirtió en valores do (Valko, et. Al, 2003) que correlaciona los tiempos de proteínas plasmáticas (PPB) obtenidos de experimentos umen a continuación en la Tabla 8 a continuación.
ynolds, D. P. Fast Gradient HPLC Method to Determine with Octanol/Water and Immobilized Artificial Membrane
n ratas
e-Dawley macho como una dosis intravenosa nominal al 10%, EtOH al 15%, PEG400 al 35% y D5W al 30%. El ague-Dawley a una dosis oral nominal única de 3 mg/kg TPGS, 5% de PVP-K30 a 5 ml/kg de volumen de dosis. mediante CL/EM/EM.
compuesto ensayado en ratas Sprague-Dawley a horas ocinéticos no compartimentales usando la función PK Scientific Inc, Waltham, MA). Los valores de AUC se
5D. Protocolo experimental para el ensayo de PXR
[0734] La propensión a la inducción de CYP3A4 medi vitro. Esta línea celular, que ha obtenido la licencia de Pu de manera estable con genes que codifican PXR human a la región promotora CYP3A4 y potenciadores distales y [0735] El ensayo se realiza en formato de 384 pocillos y de 0,1 a 60 pM. El día 1, las células DPX-2 que previame se descongelan y se siembran en placas de cultivo de t cultivan en medio que contiene el artículo de prueba, el rifampicina inductora de CYP3A4 clínicamente validada. durante 48 horas y luego se evalúa la viabilidad celular Promega) con un lector de placas EnVision (PerkinEl proporcional a la actividad luciferasa, se mide mediante l Promega One-Glo utilizando el mismo lector de placas.
[0736] El procesamiento de datos dentro del paquete de de inducción en comparación con el control del vehículo, de respuesta de dosis de 11 puntos. Los pocillos con una y no se informan las placas en las que la respuesta del esperado, ya sea en potencia o inducción máxima.
5E. Datos CFTR de los Compuestos 1 - 65
[0737] Los compuestos de Fórmula (I) son útiles como m ilustra la CE50 de los compuestos de la Tabla 6 usando l anteriormente en el Ejemplo 5A-A1). En la Tabla 6 a cont significa <0,1 uM ;"++" significa entre 0,1 uM y 1 uM ;"+" Tabla 6. Ac por PXR se evalúa usando la línea celular DPX-2 in yp Inc., se derivó de células HepG2 y se ha transfectado así como un indicador de luciferasa modificado vinculado oximales relacionados.
da artículo de ensayo se administra en 11 dosis que van se han expandido internamente y se han criopreservado os. Al día siguiente, se cambia el medio y las células se ntrol del vehículo o el compuesto de control positivo, la s células se cultivan en presencia del artículo de prueba ndo un ensayo basado en fluorescencia (Cell TiterFluor, r). Posteriormente, la transactivación CYP3A4, que es ctura de luminiscencia usando el sistema de reactivos de
ware Genedata permite informar sobre el número máximo valor de CE50 para los inductores de CYP3A4 y una curva bilidad celular inferior al 70% no se utilizan para el análisis trol positivo de rifampicina se encuentra fuera del rango
uladores de la actividad CFTR. La Tabla 6 a continuación procedimientos descritos anteriormente (ensayo descrito ación, se aplican los siguientes significados. CE50: "+++" nifica más de 1 uM.
idad CFTR

(Conti ción)

5F. Metabolitos
[0738] Se ha determinado que el Compuesto 1 se me metabolismo oxidativo. Se prepararon y ensayaron el Co oliza tanto in vitro como in vivo, principalmente mediante esto 1 y los metabolitos mostrados en la siguiente tabla.

[0739] 5G. Acilsulfoxamidas. Se determinó que los co resto sulfonimidoilamida - en donde X en las fórmulas I como resultado una unión disminuida de albúmina de su con compuestos que comprenden un grupo acilsulfonam O). Los datos de HAS se midieron como se describió ant la albúmina de suero humano puede resultar en una may actividad biológica.
Tabla 8. Da estos que comprenden un resto acilsulfoxamida (es decir, I se elige entre aminas sustituidas o no sustituidas) dan humano y una fracción libre mejorada en comparación (es decir, en donde X en las fórmulas I o II se elige de rmente en el Ejemplo 5B. La disminución de la unión de antidad de fármaco libre (no unido) que puede afectar la
de la HSA

[0740] La Tabla 8 a continuación resume la actividad aclaramiento de IV de rata, AUC de PO de rata y anteriormente.
Tabla 9. Dato CFTR (CFTR dF508 CE50), inducción de PXR Max, s de PO de rata para ciertos compuestos descritos omparativos
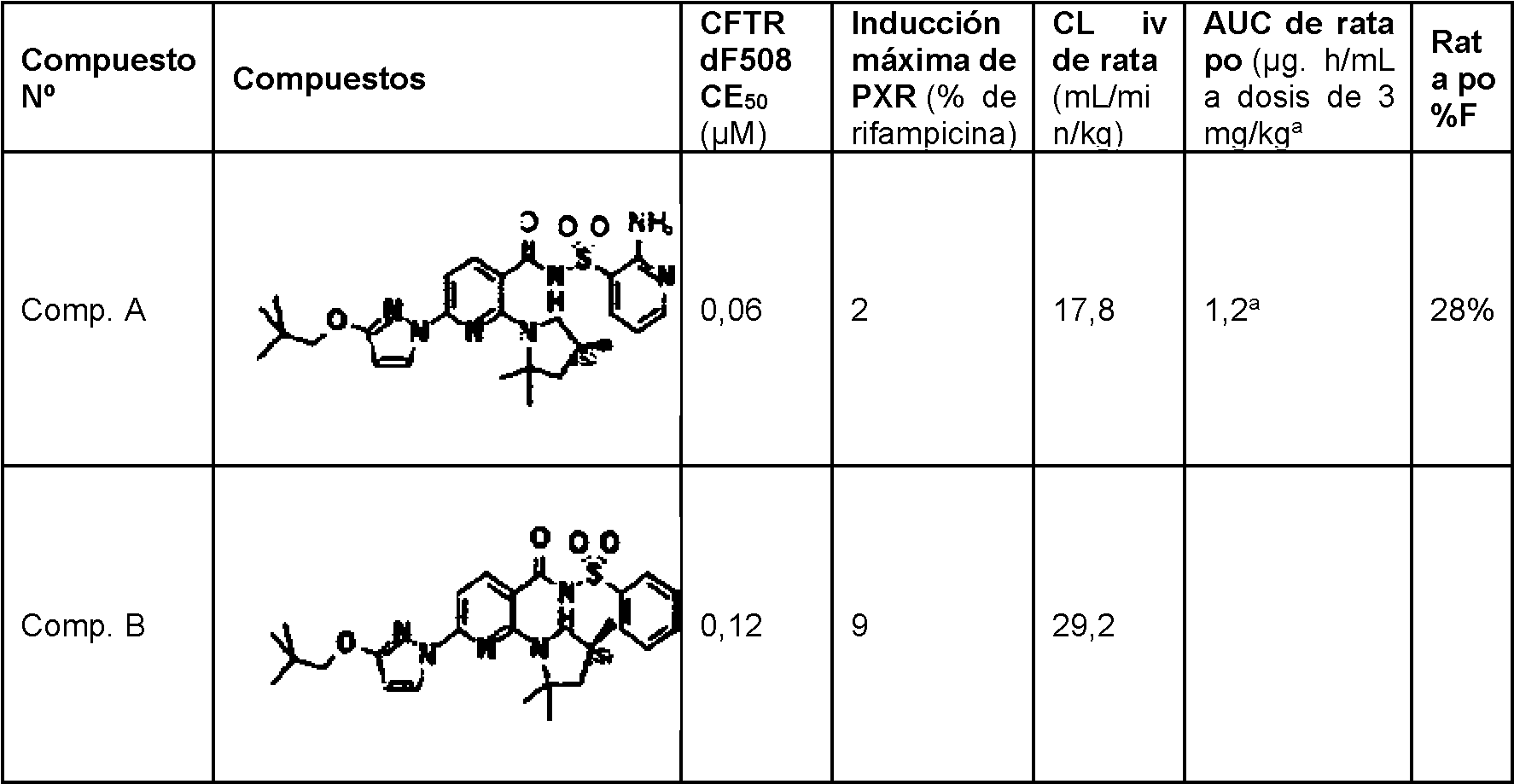
(Continua n)

(Continua n)

(Continua n)

(Continua n)

(Continua n)

(Conti ción)

Ejemplo 6: Experimentos de transporte de cloruro
[0741] En un experimento de Cámara de Ussing con transporte de cloruro. El efecto del Compuesto 1 sobre II. Además, el F508del-CFTR administrado a la superfici Compuesto II fue potenciado por el Compuesto III. La co III proporcionó un aumento superior (aproximadamente 3 regímenes duales en la mayoría de las condiciones proba Ejemplo 7: Experimentos con procesamiento y tráfico [0742] In vitro, el Compuesto 1 mejoró el procesamiento proteína F508del-CFTR funcional en la superficie celular. Compuesto 1 solo o en combinación con el Compue Compuesto III. En las células del epitelio bronquial hu Compuesto I, Compuesto II y Compuesto III (Compuest cloruro de CFTR más que cualquiera de las combin I/Compuesto III y Compuesto II/Compuesto III) o compon III) en la mayoría de las condiciones estudiadas.
[0743] El procesamiento y tráfico de F508del-CFTR fue 170 a 180 kDa. Dicho monitoreo estableció que el C procesamiento y tráfico de F508del-CFTR para aument celular.
[0744] La incubación de células F508del/F508del-HBE d combinación con el Compuesto II 3 pM dio como resu alcanzando 6,5 veces y 18,7 veces de niveles sin tratar, r Otras formas de realización
[0745] La discusión anterior da a conocer y describe forma Un experto en la técnica reconocerá fácilmente a partir de que se pueden realizar varios cambios, modificaciones y v como se define en las siguientes reivindicaciones.
ulas F508del/F508del-HBE, el Compuesto 1 mejoró el ansporte de cloruro fue aditivo al efecto del Compuesto celular por el Compuesto 1 solo o en combinación con el inación triple de Compuesto 1/Compuesto II/Compuesto es) en el transporte de cloruro en comparación con los 3 s.
F508del-CFTR in vitro
ráfico de F508del-CFTR, aumentando así la cantidad de proteína CFTR suministrada a la superficie celular por el II (Compuesto I/Compuesto II) fue potenciada por el no (HBE) estudiadas in vitro, la combinación triple de /Compuesto II/Compuesto III) aumentó el transporte de ones duales (Compuesto I/Compuesto II, Compuesto s individuales (Compuesto I, Compuesto II y Compuesto
itoreado directamente por la aparición de una banda de puesto I es un corrector de CFTR, ya que facilita el la cantidad de F508del-CFTR funcional en la superficie
nte 16 a 24 horas con el Compuesto I 1 mM solo o en do un aumento en los niveles de estado estacionario, ectivamente.
e realización meramente ejemplares de esta divulgación. ha discusión y de los dibujos y reivindicaciones adjuntos, aciones en los mismos sin apartarse de la descripción tal
Claims (50)
1. Un compuesto de Fórmula I:

una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores, en donde:
- uno de Y1 e Y2 es N y el otro es CH;
- X se elige entre los grupos O, NH y N(C1-C4 alquilo);
- R1 se elige entre grupos -(CR2)k-O-(CR2)m(CR)n(AniMo A)n+1,
en donde cada Anillo A se elige independientemente entre grupos C3-C10 cicloalquilo opcionalmente sustituidos con uno o más sustituyentes elegidos cada uno independientemente entre grupos C1-C2 alquilo, grupos C1-C2 alquilo halogenados y halógenos, y
en los que cada R se elige independientemente entre H, OH y grupos C1-C2 alquilo opcionalmente sustituidos con uno o más halógenos;
- cada R2 se elige independientemente entre grupos C1-C2 alquilo, OH, grupos C1-C2 alcoxi, halógenos y ciano;
- cada R3 se elige independientemente entre grupos C1-C2 alquilo opcionalmente sustituidos con uno o más grupos OH;
- cada R4 se elige independientemente entre halógenos;
- k es 0 o 1 ;
- r es 0 o 1 ;
- m es 0, 1, 2 o 3;
- n es 0 o 1 ;
- p es 0, 1,2, 3, 4 o 5; y
- q es 0, 1, 2, 3, 4, 5, 6 , 7 u 8.
2. Un compuesto de la reivindicación 1, en donde el compuesto es de Fórmula II:

una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores, en donde:
- X se elige entre los grupos O, NH y N(Ci -C4 alquilo);
- R1 se elige entre grupos -(CR2)k-O-(CR2)m(CR)n(AniMo A)n+i,
en donde cada Anillo A se elige independientemente entre grupos C3-C10 cicloalquilo opcionalmente sustituidos con uno o más sustituyentes elegidos cada uno independientemente entre grupos C1-C2 alquilo, grupos C1-C2 alquilo halogenados y halógenos, y
en los que cada R se elige independientemente entre H, OH y grupos C1-C2 alquilo opcionalmente sustituidos con uno o más halógenos;
- cada R2 se elige independientemente entre grupos C1-C2 alquilo, OH, grupos C1-C2 alcoxi, halógenos y ciano;
- cada R3 se elige independientemente entre grupos C1-C2 alquilo opcionalmente sustituidos con uno o más grupos OH;
- cada R4 se elige independientemente entre halógenos;
- k es 0 o 1;
- r es 0 o 1;
- m es 0, 1, 2 o 3;
- n es 0 o 1;
- p es 0, 1,2, 3, 4 o 5; y
- q es 0, 1, 2, 3, 4, 5, 6, 7 u 8.
3. Un compuesto de la reivindicación 1, en donde el compuesto es de Fórmula III:

una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores, en donde:
- R1 se elige entre grupos -(CR2)k-O-(CR2)m(CR)n(AniNo A)n+i,
en donde cada Anillo A se elige independientemente de grupos C3-C10 cicloalquilo opcionalmente sustituidos con uno o más sustituyentes, cada uno elegido independientemente entre grupos C1-C2 alquilo, grupos C1-C2 alquilo halogenados y halógenos, y
donde cada R se elige independientemente entre grupos H, OH y C1-C2 alquilo opcionalmente sustituidos con uno o más halógenos;
- cada R2 se elige independientemente entre grupos C1-C2 alquilo, OH, grupos C1-C2 alcoxi, halógenos y ciano;
- cada R3 se elige independientemente entre grupos C1-C2 alquilo opcionalmente sustituidos con uno o más grupos OH;
- cada R4 se elige independientemente entre halógenos;
- k es 0 o 1;
- r es 0 o 1;
- m es 0, 1, 2 o 3;
- n es 0 o 1;
- p es 0, 1, 2, 3, 4 o 5; y
- q es 0, 1, 2, 3, 4, 5, 6, 7 u 8.
4. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1-3, una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores, en donde si R2 es ciano, entonces dicho R2 es meta
o para con respecto al átomo de azufre.
5. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1-3, una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores, en donde:
- cada Anillo A se elige independientemente entre grupos C3-C10 cicloalquilo opcionalmente sustituidos con uno o más sustituyentes elegidos cada uno independientemente entre grupos C1-C2 alquilo, grupos C1-C2 alquilo halogenados y halógenos, y
- cada R se elige independientemente entre H y OH;
- cada R2 se elige independientemente entre grupos C1-C2 alquilo, OH, grupos C1-C2 alcoxi y halógenos; - R4 es F;
- k es 0;
- p es 0, 1 o 2;
- q es 0, 1, 2, 3 o 4;
- r es 0; y
en donde m y n no son 0 al mismo tiempo.
6. Un compuesto de acuerdo con la reivindicación 5, una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores, en donde:
- R1 se elige entre grupos -O-(CR2)m-AniNo A,
en donde el An illo A es elegido entre grupos C3-C10 cicloalquilo opcionalmente sustituidos con uno o más sustituyentes, cada uno elegido independientemente entre grupos C1-C2 alquilo, grupos C1-C2 alquilo halogenados y halógenos, y
- m es 1 o 2.
7. Un compuesto según la reivindicación 6, una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores, en donde cada R3 es un grupo metilo y q es 3 o 4.
8. Un compuesto según la reivindicación 7 que tiene la Fórmula IV:

una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores, en donde:
- El Anillo A se elige entre grupos C3-C10 cicloalquilo opcionalmente sustituidos con uno o más sustituyentes, cada uno elegido independientemente entre grupos C1-C2 alquilo, grupos C1-C2 alquilo halogenados y halógenos; y
- cada R2 se elige independientemente entre grupos C1-C2 alquilo, OH, F, Cl y grupos C1-C2 alcoxi;
- m es 1 o 2; y
- p es 0, 1 o 2.
9. Un compuesto según la reivindicación 8, en donde:
(a) p es 0 o 1; o
(b) p es 0.
10. Un compuesto de acuerdo con la reivindicación 8 que tiene la Fórmula V:

una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores, en donde:
- El An illo A se elige entre grupos C3-C10 cicloalquilo opcionalmente sustituido con uno o más sustituyentes, cada uno elegido independientemente entre grupos C1-C2 alquilo, grupos C1-C2 alquilo halogenados y halógenos; y
- cada R2 se elige independientemente entre grupos C1-C2 alquilo, OH, F, Cl y grupos C1-C2 alcoxi;
- m es 1 o 2 ; y
- p es 0 , 1 o 2.
11. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1-10, una sal farmacéuticamente aceptable del mismo o un derivado deuterado de cualquiera de los anteriores, en donde cada R2 se elige independientemente de CH3 , OH, F y OCH3.
12. Un compuesto de acuerdo con la reivindicación 11, una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores, en donde:
(a) p es 0 o 1 ; o
(b) p es 0.
13. Un compuesto según la reivindicación 10, una sal farmacéuticamente aceptable del mismo o un derivado deuterado de cualquiera de los anteriores, en donde:
(a) el An illo A es un grupo ciclopropilo sustituido con un grupo C1 alquilo halogenado o un grupo C2 alquilo halogenado; o
(b) el An illo A es un grupo ciclopropilo sustituido con un grupo CF3 ; o
(c) m es 1, el An illo A es un grupo ciclopropilo sustituido con un grupo CF3, p es 0 o 1, y R2, si está presente, es un grupo metilo, un grupo hidroxi o un grupo metoxi; o
(d) m es 2, el Anillo A es un grupo C3 cicloalquilo sustituido con un grupo CF3 , p es 0 o 1, y R2, si está presente, es un grupo metilo, un grupo hidroxi o un grupo metoxi; o
(e) m es 2, el An illo A es un grupo ciclopropilo sustituido con un grupo CF3 y p es 0; o
(f) el An illo A se elige entre grupos C5 bicicloalquilo opcionalmente sustituidos con uno o más sustituyentes elegidos cada uno independientemente entre grupos C1-C2 alquilo, grupos C1-C2 alquilo halogenados y halógenos; o
(g) el An illo A es un grupo C5 bicicloalquilo opcionalmente sustituido con un halógeno; o
(h) el Anillo A se elige entre grupos C7 bicicloalquilo y grupos C7 tricicloalquilo opcionalmente sustituidos con uno o más sustituyentes elegidos cada uno independientemente entre grupos C1-C2 alquilo, grupos C1-C2 alquilo halogenados y halógenos; o
(i) el An illo A es un grupo C7 tricicloalquilo no sustituido.
14. Un compuesto de acuerdo con la reivindicación 1 que tiene una fórmula elegida entre cualquiera de las fórmulas representadas a continuación:











una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores.
15. Un compuesto de acuerdo con la reivindicación 1 que tiene la siguiente fórmula:

una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores.
16. Un compuesto de acuerdo con la reivindicación 15 que tiene la siguiente fórmula:

en forma de una sal farmacéuticamente aceptable.
17. Un compuesto de acuerdo con la reivindicación 15 que tiene la siguiente fórmula:

18. Un compuesto de acuerdo con la reivindicación 1 que tiene:
(a) la siguiente fórmula:

una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores; o
(b) la siguiente fórmula:

una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores; o (c) la siguiente fórmula:

una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores; o (d) la siguiente fórmula:
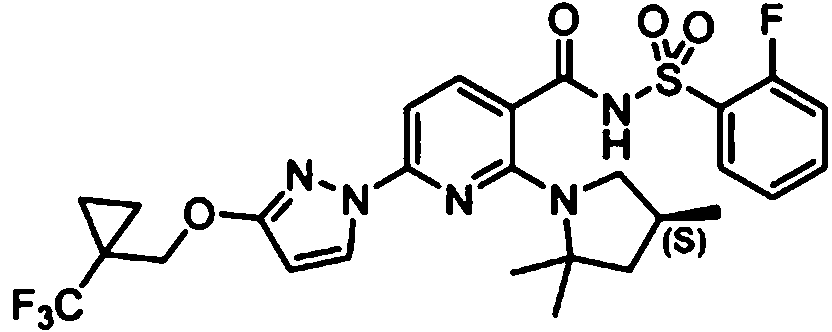
una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores; o (e) la siguiente fórmula:

o una sal farmacéuticamente aceptable del mismo.
(f) la siguiente fórmula:

o una sal farmacéuticamente aceptable del mismo; o
(g) la siguiente fórmula:

o una sal farmacéuticamente aceptable del mismo.
19. Una composición farmacéutica que comprende el compuesto, una sal farmacéuticamente aceptable del mismo o un derivado deuterado de cualquiera de los anteriores, como se define en la reivindicación 15.
20. Una composición farmacéutica que comprende la sal farmacéuticamente aceptable de la reivindicación 16.
21. Una composición farmacéutica que comprende el compuesto de la reivindicación 17.
22. Una composición farmacéutica que comprende al menos un compuesto elegido entre los compuestos de una cualquiera de las reivindicaciones 1-18, una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores, y opcionalmente uno o más de:
(a) Compuesto II:

una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores; (b) Compuesto III:
una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores; y (c) un portador farmacéuticamente aceptable.
23. La composición farmacéutica de la reivindicación 22 que comprende el compuesto de la reivindicación 15 o una sal farmacéuticamente aceptable del mismo, y opcionalmente uno o más de:
(a) Compuesto II:

o una sal farmacéuticamente aceptable del mismo;
(b) Compuesto III:

o una sal farmacéuticamente aceptable del mismo; y
(c) un portador farmacéuticamente aceptable.
24. La composición farmacéutica de la reivindicación 23, en donde el compuesto que tiene la fórmula:

está en forma de una sal farmacéuticamente aceptable.
25. La composición farmacéutica de acuerdo con la reivindicación 22 para su uso en un método de tratamiento de la fibrosis quística.
26. El compuesto, sal farmacéuticamente aceptable del mismo o derivado deuterado de cualquiera de los anteriores, según la reivindicación 15, para su uso en un método de tratamiento de la fibrosis quística.
27. Sal farmacéuticamente aceptable según la reivindicación 16 para su uso en un método de tratamiento de la fibrosis quística.
28. El compuesto de acuerdo con la reivindicación 17 para su uso en un método de tratamiento de la fibrosis quística.
29. La composición farmacéutica de acuerdo con la reivindicación 19 para su uso en un método de tratamiento de la fibrosis quística.
30. Composición farmacéutica según la reivindicación 20 para su uso en un método de tratamiento de la fibrosis quística.
31. La composición farmacéutica de acuerdo con la reivindicación 21 para su uso en un método de tratamiento de la fibrosis quística.
32. La composición farmacéutica según la reivindicación 22 para su uso en un método de tratamiento de la fibrosis quística.
33. Composición farmacéutica según la reivindicación 23 para su uso en un método de tratamiento de la fibrosis quística.
34. La composición farmacéutica según la reivindicación 24 para su uso en un método de tratamiento de la fibrosis quística.
35. Un método para preparar un compuesto de Fórmula (Mía):

una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores, que comprende hacer reaccionar un compuesto de Fórmula (F) o una sal del mismo con un compuesto de Fórmula (G) o una sal del mismo para generar dicho compuesto de Fórmula (Illa) o una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores:

en donde en cada una de dichas fórmulas:
- uno de Y1 e Y2 es N y el otro es CH;
- cada R1 se elige independientemente entre grupos -(CR 2)k-O-(CR2 )m(CR)n(AniMo A)n+i,
en donde cada Anillo A se elige independientemente entre grupos C3-C10 cicloalquilo opcionalmente sustituidos con uno o más sustituyentes, cada uno elegido independientemente entre grupos C1-C2 alquilo, grupos C1-C2 alquilo halogenados y halógenos, y
donde cada R se elige independientemente entre H, OH y grupos C1-C2 alquilo opcionalmente sustituidos con uno o más halógenos;
- cada R2 se elige independientemente entre grupos C1-C2 alquilo, OH, grupos C1-C2 alcoxi, halógenos y ciano;
- cada R3 se elige independientemente entre grupos C1-C2 alquilo opcionalmente sustituidos con uno o más grupos OH;
- cada R4 se elige independientemente entre halógenos;
- Xa se elige entre F o Cl;
- cada k es independientemente 0 o 1;
- cada r es independientemente 0 o 1;
- cada m es independientemente 0, 1, 2 o 3;
- cada n es independientemente 0 o 1;
- cada p es independientemente 0, 1,2, 3, 4 o 5; y
- cada q es independientemente 0, 1, 2, 3, 4, 5, 6, 7 u 8; opcionalmente, en donde cada Y2 es independientemente N; y cada Y1 es independientemente CH.
36. El método de la reivindicación 35, en donde dicha reacción de un compuesto de Fórmula (F) o una sal del mismo con un compuesto de Fórmula (G) o una sal del mismo se realiza en presencia de una base.
37. El método de las reivindicaciones 35 ó 36, en donde se emplea una sal del compuesto de Fórmula (G); opcionalmente, en donde dicha sal del compuesto de Fórmula (G) es una sal de HCl de un compuesto de Fórmula (G).
38. Un método para preparar un compuesto de Fórmula (F) o una sal del mismo:

o un derivado deuterado de cualquiera de los anteriores, que comprende hacer reaccionar un compuesto de Fórmula (D) o una sal del mismo con un compuesto de Fórmula (E) o un sal del mismo para generar un compuesto de Fórmula (F) o una sal del mismo:

en donde
- uno de Y1 e Y2 es independientemente N y el otro es independientemente CH;
- cada R1 se elige independientemente entre grupos -(CR 2)k-O-(CR2 )m(CR)n(AniMo A)n+1,
en donde cada Anillo A se elige independientemente entre grupos C3-C10 cicloalquilo opcionalmente sustituidos con uno o más sustituyentes, cada uno elegido independientemente entre grupos C1-C2 alquilo, grupos C1-C2 alquilo halogenados y halógenos, y
donde cada R se elige independientemente entre grupos H, OH y C1-C2 alquilo opcionalmente sustituidos con uno o más halógenos;
- cada R2 se elige independientemente entre grupos C1-C2 alquilo, OH, grupos C1-C2 alcoxi, halógenos y ciano;
- cada R4 se elige independientemente entre halógenos;
- Xa se elige entre F o Cl;
- cada k es independientemente 0 o 1;
- cada r es independientemente 0 o 1;
- cada m es independientemente 0, 1,2 o 3;
- cada n es independientemente 0 o 1; y
- cada p es independientemente 0, 1, 2, 3, 4 o 5; opcionalmente, en donde cada Y2 es independientemente N; y cada Y1 es independientemente CH.
39. El método de la reivindicación 38, en donde dicha reacción de un compuesto de Fórmula (D) o una sal del mismo con un compuesto de Fórmula (E) o una sal del mismo se realiza en presencia de una base; o comprende hacer reaccionar un compuesto de Fórmula (D-1) con un reactivo de acoplamiento y posteriormente con un compuesto de Fórmula (E-1) en presencia de una base.
40. El método de la reivindicación 35, que es un método para preparar un compuesto de la siguiente fórmula:

o una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores, que comprende hacer reaccionar un compuesto de Fórmula (F-1) o una sal del mismo, en donde Xa se elige entre F o Cl, con un compuesto de Fórmula (G-1) o una sal del mismo para generar dicho compuesto o una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores:

opcionalmente, en donde dicha reacción de un compuesto de Fórmula (F-1) o una sal del mismo con un compuesto de Fórmula (G-1) o una sal del mismo se realiza en presencia de una base.
41. El método de la reivindicación 40, en donde se emplea una sal del compuesto de Fórmula (G-1); opcionalmente, en donde dicha sal del compuesto de Fórmula (G-1) es una sal de HCl de un compuesto de Fórmula (G-1).
42. El método de la reivindicación 38, que es un método para preparar un compuesto de Fórmula (F-1) o una sal del mismo:

o un derivado deuterado de cualquiera de los anteriores, que comprende hacer reaccionar un compuesto de Fórmula (D-1) y un compuesto de Fórmula (E-1) para generar un compuesto de Fórmula (F-1) o una sal del mismo:

en donde cada Xa se elige independientemente entre F o Cl.
43. El método de la reivindicación 42, en donde dicha reacción de un compuesto de Fórmula (D-1) o una sal del mismo con un compuesto de Fórmula (E-1) o una sal del mismo se realiza en presencia de una base; o comprende hacer reaccionar un compuesto de Fórmula (D-1) con un reactivo de acoplamiento y posteriormente con un compuesto de Fórmula (E-1) en presencia de una base.
44. Un método para preparar un compuesto de Fórmula (D) o una sal del mismo:

o un derivado deuterado de cualquiera de los anteriores, que comprende:
(i) hacer reaccionar un compuesto de Fórmula (A) o una sal del mismo con un compuesto de Fórmula (B) o una sal del mismo para generar un compuesto de Fórmula (C) o una sal del mismo:

y
(ii) hidrolizar el grupo -C(O)ORa de un compuesto de Fórmula (C) para generar un compuesto de Fórmula (D) o una sal del mismo, en donde en cada una de dichas fórmulas:
- uno de Y1 e Y2 es independientemente N y el otro es independientemente CH;
- cada R1 se elige independientemente entre grupos -(CR 2)k-O-(CR2)m(CR)n(AniMo A)n+1,
en donde cada Anillo A se elige independientemente entre grupos C3-C10 cicloalquilo opcionalmente sustituidos con uno o más sustituyentes, cada uno elegido independientemente entre grupos C1-C2 alquilo, grupos C1-C2 alquilo halogenados y halógenos, y
donde cada R se elige independientemente entre H, OH y grupos C1-C2 alquilo opcionalmente sustituidos con uno o más halógenos;
- cada R4 se elige independientemente entre halógenos;
- cada Ra se elige independientemente entre C1-C4 alquilo;
- cada Xa se elige independientemente entre F o Cl;
- cada k es independientemente 0 o 1;
- cada r es independientemente 0 o 1;
- cada m es independientemente 0, 1,2 o 3, y
- cada n es independientemente 0 o 1;
opcionalmente, en donde cada Y2 es independientemente N; y cada Y1 es independientemente CH.
45. El método de la reivindicación 44, en donde la hidrólisis del grupo -C(O)ORa se realiza en presencia de una base; opcionalmente, en donde dicha reacción de un compuesto de Fórmula (A) o una sal del mismo con un compuesto de Fórmula (B) o una sal del mismo se realiza en presencia de una base.
46. El método de una cualquiera de las reivindicaciones 44 ó 45, en donde Ra es etilo o í-butilo.
47. El método de la reivindicación 44, que es un método para preparar un compuesto de Fórmula (D-1) o una sal del mismo:
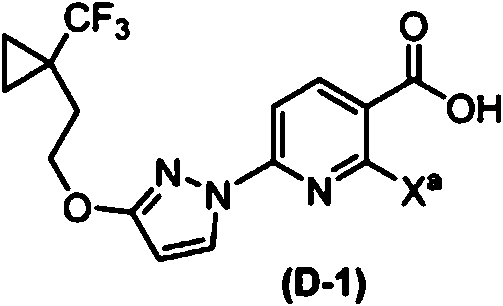
o un derivado deuterado de cualquiera de los anteriores, que comprende:
(i) hacer reaccionar un compuesto de Fórmula (A -1) o una sal del mismo y un compuesto de Fórmula (B-1) o una sal del mismo para generar un compuesto de Fórmula (C-1) o una sal del mismo:

(ii) hidrolizar el grupo -C(O)ORa de un compuesto de Fórmula (C-1) o una sal del mismo para generar un compuesto de Fórmula (D-1) o una sal del mismo,
en donde cada Ra se elige independientemente entre C1-C4 alquilo; y cada - Xa se elige independientemente entre F o Cl;
opcionalmente, en donde la hidrólisis del grupo -C(O)ORa se realiza en presencia de una base.
48. El método de la reivindicación 47, en donde dicha reacción de un compuesto de Fórmula (A-1) o una sal del mismo y un compuesto de Fórmula (B-1) o una sal del mismo se realiza en presencia de una base; opcionalmente, en donde Ra es etilo o í-butilo.
49. Un método para preparar un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores, que comprende hacer reaccionar un compuesto de Fórmula (L) o una sal del mismo con NR*3 :
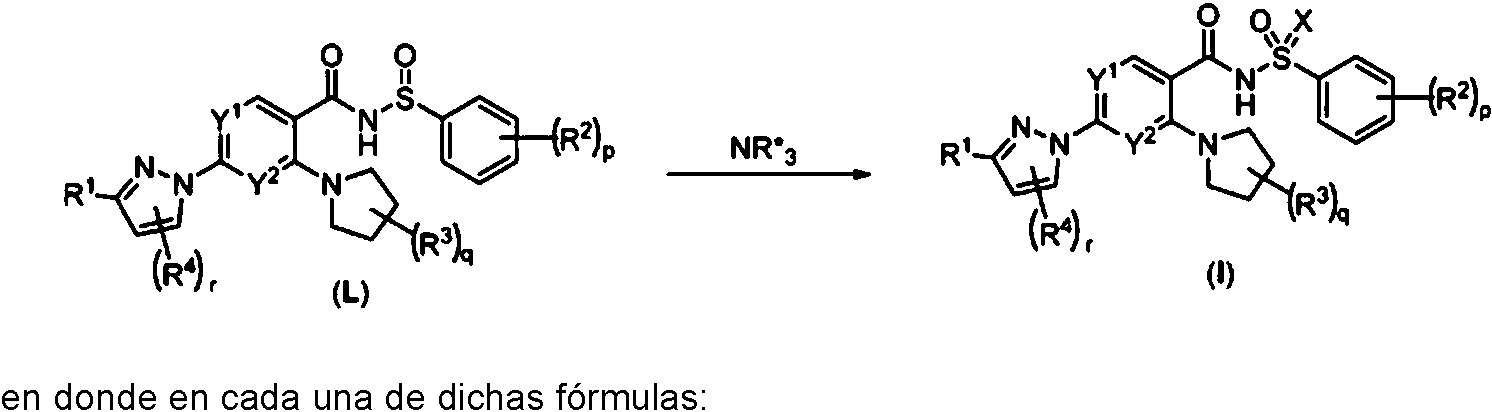
-X es NH o N(C1-C4 alquilo);
- uno de Y1 e Y2 es independientemente N y el otro es independientemente CH;
- cada R1 se elige independientemente entre grupos -(CR2)k-O-(CR2)m(CR)n(AniMo A)n+1,
en donde cada Anillo A se elige independientemente entre grupos C3-C10 cicloalquilo opcionalmente sustituidos con uno o más sustituyentes, cada uno elegido independientemente entre grupos C1-C2 alquilo, grupos C1-C2 alquilo halogenados y halógenos, y
donde cada R se elige independientemente entre H, OH y grupos C1-C2 alquilo opcionalmente sustituidos con uno o más halógenos;
- cada R2 se elige independientemente entre grupos C1-C2 alquilo, OH, grupos C1-C2 alcoxi, halógenos y ciano;
- cada R3 se elige independientemente entre grupos C1-C2 alquilo opcionalmente sustituidos con uno o más grupos OH;
- cada R4 se elige independientemente entre halógenos;
- R* es H o C1-C4 alquilo.
- Xa se elige entre F o Cl;
- cada k es independientemente 0 o 1;
- cada r es independientemente 0 o 1;
- cada m es independientemente 0, 1,2 o 3;
- cada n es independientemente 0 o 1;
- cada p es independientemente 0, 1,2, 3, 4 o 5; y
- cada q es independientemente 0, 1, 2, 3, 4, 5, 6, 7 u 8.
50. Al menos un compuesto elegido entre los compuestos de cualquiera de las reivindicaciones 1-18, una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores, y opcionalmente uno o más de:
(a) Compuesto II:

una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores; y (b) Compuesto III:

una sal farmacéuticamente aceptable del mismo, o un derivado deuterado de cualquiera de los anteriores; para su uso en el tratamiento de la fibrosis quística.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662402838P | 2016-09-30 | 2016-09-30 | |
| US201662410353P | 2016-10-19 | 2016-10-19 | |
| US201662415409P | 2016-10-31 | 2016-10-31 | |
| US201662419935P | 2016-11-09 | 2016-11-09 | |
| PCT/US2017/054611 WO2018064632A1 (en) | 2016-09-30 | 2017-09-29 | Modulator of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2900263T3 true ES2900263T3 (es) | 2022-03-16 |
Family
ID=61757799
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17792230T Active ES2900263T3 (es) | 2016-09-30 | 2017-09-29 | Modulador de regulador de conductancia transmembrana de fibrosis quística, composiciones farmacéuticas, métodos de tratamiento y proceso de fabricación del modulador |
Country Status (38)
| Country | Link |
|---|---|
| US (3) | US10570115B2 (es) |
| EP (1) | EP3519401B1 (es) |
| JP (1) | JP7061115B2 (es) |
| KR (1) | KR102531491B1 (es) |
| CN (1) | CN109803962B (es) |
| AU (1) | AU2017336156B2 (es) |
| CA (1) | CA3037986C (es) |
| CL (1) | CL2019000827A1 (es) |
| CO (1) | CO2019004065A2 (es) |
| CY (1) | CY1124862T1 (es) |
| DK (1) | DK3519401T3 (es) |
| DO (1) | DOP2019000081A (es) |
| EC (1) | ECSP19028690A (es) |
| ES (1) | ES2900263T3 (es) |
| GE (2) | GEP20217329B (es) |
| HR (1) | HRP20211683T1 (es) |
| HU (1) | HUE056716T2 (es) |
| IL (1) | IL265627B (es) |
| JO (1) | JOP20190042B1 (es) |
| LT (1) | LT3519401T (es) |
| MA (1) | MA46357B1 (es) |
| MD (1) | MD3519401T2 (es) |
| MX (1) | MX387060B (es) |
| NZ (1) | NZ752486A (es) |
| PE (1) | PE20191147A1 (es) |
| PL (1) | PL3519401T3 (es) |
| PT (1) | PT3519401T (es) |
| RS (1) | RS62670B1 (es) |
| SA (1) | SA519401446B1 (es) |
| SG (1) | SG10201913595YA (es) |
| SI (1) | SI3519401T1 (es) |
| SM (1) | SMT202100646T1 (es) |
| TN (1) | TN2019000079A1 (es) |
| TW (1) | TWI766886B (es) |
| UA (1) | UA124708C2 (es) |
| UY (1) | UY37428A (es) |
| WO (1) | WO2018064632A1 (es) |
| ZA (1) | ZA201902124B (es) |
Families Citing this family (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI735416B (zh) | 2014-10-06 | 2021-08-11 | 美商維泰克斯製藥公司 | 囊腫纖維化症跨膜傳導調節蛋白之調節劑 |
| ES2946970T3 (es) | 2016-03-31 | 2023-07-28 | Vertex Pharma | Regulador de conductancia transmembrana de moduladores de fibrosis quística |
| SI3519401T1 (sl) | 2016-09-30 | 2022-01-31 | Vertex Pharmaceuticals Incorporated | Modulator regulatorja transmembranske prevodnosti pri cistični fibrozi, farmacevtski sestavki, postopki zdravljenja in proces za izdelavo modulatorja |
| CN110267948B (zh) | 2016-12-09 | 2023-12-08 | 弗特克斯药品有限公司 | 囊性纤维化跨膜传导调控剂的调节剂、药物组合物、治疗方法和制备所述调节剂的方法 |
| AU2018279646B2 (en) * | 2017-06-08 | 2023-04-06 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| MA49631A (fr) | 2017-07-17 | 2020-05-27 | Vertex Pharma | Méthodes de traitement de la fibrose kystique |
| TWI799435B (zh) * | 2017-08-02 | 2023-04-21 | 美商維泰克斯製藥公司 | 製備化合物之製程 |
| US10654829B2 (en) | 2017-10-19 | 2020-05-19 | Vertex Pharmaceuticals Incorporated | Crystalline forms and compositions of CFTR modulators |
| KR102716244B1 (ko) | 2017-12-08 | 2024-10-14 | 버텍스 파마슈티칼스 인코포레이티드 | 낭포성 섬유증 막횡단 전도 조절자의 조정제의 제조 방법 |
| TWI810243B (zh) | 2018-02-05 | 2023-08-01 | 美商維泰克斯製藥公司 | 用於治療囊腫纖化症之醫藥組合物 |
| AU2019222758B2 (en) * | 2018-02-15 | 2022-07-07 | Vertex Pharmaceuticals Incorporated | Macrocycles as modulators of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions thereof, their use in the treatment of cystic fibrosis, and process for making them |
| US20210139514A1 (en) * | 2018-04-05 | 2021-05-13 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| EP3774825A1 (en) | 2018-04-13 | 2021-02-17 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| PT3880197T (pt) | 2018-11-14 | 2023-05-09 | Vertex Pharma | Métodos de tratamento para a fibrose quística |
| TWI848092B (zh) * | 2019-04-03 | 2024-07-11 | 美商維泰克斯製藥公司 | 囊腫纖維化跨膜傳導調節蛋白調節劑 |
| WO2020214921A1 (en) | 2019-04-17 | 2020-10-22 | Vertex Pharmaceuticals Incorporated | Solid forms of modulators of cftr |
| TWI867024B (zh) * | 2019-08-14 | 2024-12-21 | 美商維泰克斯製藥公司 | 囊腫纖維化跨膜傳導調節蛋白之調節劑 |
| TWI899097B (zh) | 2019-08-14 | 2025-10-01 | 美商維泰克斯製藥公司 | 製備cftr調節劑之方法 |
| CA3150162A1 (en) * | 2019-08-14 | 2021-02-18 | Vertex Pharmaceuticals Incorporated | Crystalline forms of cftr modulators |
| CN114585628B (zh) * | 2019-08-14 | 2024-03-26 | 弗特克斯药品有限公司 | 囊性纤维化跨膜传导调节因子的调节剂 |
| CR20230120A (es) | 2020-08-07 | 2023-09-01 | Vertex Pharma | Moduladores del regulador de la conductancia transmembrana de la fibrosis quística |
| IL300413A (en) | 2020-08-13 | 2023-04-01 | Vertex Pharma | Crystal forms of CFTR modulators |
| EP4225762A1 (en) | 2020-10-07 | 2023-08-16 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| MX2023004074A (es) * | 2020-10-07 | 2023-07-05 | Vertex Pharma | Moduladores del regulador de la conductancia transmembrana de la fibrosis quistica. |
| KR20230123522A (ko) * | 2020-11-18 | 2023-08-23 | 버텍스 파마슈티칼스 인코포레이티드 | 낭성 섬유증 막관통 전도도 조절제로서 사용하기 위한 1,3,4-옥사디아졸 고리를 함유하는 거대환 |
| US12324802B2 (en) | 2020-11-18 | 2025-06-10 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| AU2021397294A1 (en) | 2020-12-10 | 2023-07-06 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| JP2025505577A (ja) | 2022-02-03 | 2025-02-28 | バーテックス ファーマシューティカルズ インコーポレイテッド | (6a,12a)-17-アミノ-12-メチル-6,15-ビス(トリフルオロメチル)-13,19-ジオキサ-3,4,18-トリアザトリシクロ[12.3.1.12,5]ノナデカ-1(18),2,4,14,16-ペンタエン-6-オールの調製方法及び結晶形態 |
| IL314449A (en) * | 2022-02-03 | 2024-09-01 | Vertex Pharma | Methods of treatment for cystic fibrosis |
| WO2023154291A1 (en) | 2022-02-08 | 2023-08-17 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| EP4525858A1 (en) | 2022-05-16 | 2025-03-26 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| CA3253977A1 (en) | 2022-05-16 | 2023-11-23 | Vertex Pharmaceuticals Incorporated | SOLID FORMS OF MACROCYCLIC COMPOUNDS AS CFTR MODULATORS AND THEIR PREPARATION |
Family Cites Families (204)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0194599A3 (en) | 1985-03-14 | 1988-01-20 | Nissan Chemical Industries Ltd. | Benzamide derivatives, process for producing the same, and soil fungicides containing the same |
| GB9122590D0 (en) | 1991-10-24 | 1991-12-04 | Lilly Industries Ltd | Pharmaceutical compounds |
| DE4410453A1 (de) | 1994-03-25 | 1995-09-28 | Hoechst Ag | Substituierte heterocyclische Carbonsäureamidester, ihre Herstellung und ihre Verwendung als Arzneimittel |
| GB9514160D0 (en) | 1994-07-25 | 1995-09-13 | Zeneca Ltd | Aromatic compounds |
| AU4437296A (en) | 1995-01-19 | 1996-08-07 | Novartis Ag | Herbicidal composition |
| WO1997018712A1 (en) | 1995-11-23 | 1997-05-29 | Novartis Ag | Herbicidal composition |
| WO1997022586A1 (en) | 1995-12-15 | 1997-06-26 | Merck Frosst Canada Inc. | Tri-aryl ethane derivatives as pde iv inhibitors |
| JPH10114654A (ja) | 1996-10-09 | 1998-05-06 | Fujisawa Pharmaceut Co Ltd | 新規用途 |
| AU6209098A (en) | 1997-01-15 | 1998-08-07 | Novartis Ag | Herbicidal agent |
| GB9716657D0 (en) | 1997-08-07 | 1997-10-15 | Zeneca Ltd | Chemical compounds |
| DE19742951A1 (de) | 1997-09-29 | 1999-04-15 | Hoechst Schering Agrevo Gmbh | Acylsulfamoylbenzoesäureamide, diese enthaltende nutzpflanzenschützende Mittel und Verfahren zu ihrer Herstellung |
| DE19802697A1 (de) | 1998-01-24 | 1999-07-29 | Bayer Ag | Selektive Herbizide auf Basis von N-Aryl-triazolin(thi)onen und N-Arylsulfonylamino(thio)carbonyltriazolin(thi)onen |
| EP1069112A4 (en) | 1998-02-13 | 2001-04-18 | Kureha Chemical Ind Co Ltd | N- (PHENYLSULFONYL) PICOLINAMIDE DERIVATIVES, METHOD FOR THEIR PREPARATION AND HERBICIDES |
| KR20010052235A (ko) | 1998-04-06 | 2001-06-25 | 후지야마 아키라 | 인돌 유도체 |
| YU86801A (sh) | 1999-06-10 | 2004-07-15 | Warner-Lambert Company | Izoindol derivati korisni u inhibiciji agregacije amiloidnih proteina i snimanju amiloidnih naslaga |
| DE19936438A1 (de) | 1999-08-03 | 2001-02-08 | Aventis Cropscience Gmbh | Kombinationen von Herbiziden und Safenern |
| DE19940860A1 (de) | 1999-08-27 | 2001-03-01 | Bayer Ag | Selektive Herbizide auf Basis eines substituierten Phenylsulfonyl aminocarbonyltriazolinons und Safenern II |
| DE19958381A1 (de) | 1999-12-03 | 2001-06-07 | Bayer Ag | Herbizide auf Basis von N-Aryl-uracilen |
| AU2001286557A1 (en) | 2000-08-23 | 2002-03-04 | Merck Frosst Canada And Co. | Method of treating or preventing urinary incontinence using prostanoid ep1 receptor antagonists |
| US6720338B2 (en) | 2000-09-20 | 2004-04-13 | Abbott Laboratories | N-acylsulfonamide apoptosis promoters |
| AR031130A1 (es) | 2000-09-20 | 2003-09-10 | Abbott Lab | N-acilsulfonamidas promotoras de la apoptosis |
| US20020055631A1 (en) | 2000-09-20 | 2002-05-09 | Augeri David J. | N-acylsulfonamide apoptosis promoters |
| AU2002211901A1 (en) | 2000-10-10 | 2002-04-22 | Smithkline Beecham Corporation | Substituted indoles, pharmaceutical compositions containing such indoles and their use as PPAR-gamma binding agents |
| DE10119721A1 (de) | 2001-04-21 | 2002-10-31 | Bayer Cropscience Gmbh | Herbizide Mittel enthaltend Benzoylcyclohexandione und Safener |
| CN1529697B (zh) | 2001-05-31 | 2010-09-22 | 维科尔药物公司 | 用作血管紧张素ⅱ激动剂的三环化合物 |
| KR100575944B1 (ko) | 2001-06-28 | 2006-05-02 | 화이자 프로덕츠 인코포레이티드 | 미소체 트리글리세라이드 전달 단백질(mtp) 및/또는아포지방단백질 b(apo b)분비의 억제제로서의트리아미드-치환된 인돌, 벤조푸란 및 벤조티오펜 |
| DE10145019A1 (de) | 2001-09-13 | 2003-04-03 | Bayer Cropscience Gmbh | Kombinationen aus Herbiziden und Safenern |
| DE10146873A1 (de) | 2001-09-24 | 2003-04-17 | Bayer Cropscience Gmbh | Heterocyclische Amide und -Iminderivate, Verfahren zu ihrer Herstellung, sie enthaltende Mittel und ihre Verwendung als Schädlingsbekämpfungsmittel |
| AU2002354056A1 (en) | 2001-11-19 | 2003-06-10 | Ono Pharmaceutical Co., Ltd. | Remedies for urinary frequency |
| DE10157545A1 (de) | 2001-11-23 | 2003-06-12 | Bayer Cropscience Gmbh | Herbizide Mittel enthaltend Benzoylpyrazole und Safener |
| AU2003220558A1 (en) | 2002-03-27 | 2004-06-03 | Smithkline Beecham Corporation | Amide compounds and methods of using the same |
| GB0212785D0 (en) | 2002-05-31 | 2002-07-10 | Glaxo Group Ltd | Compounds |
| AU2003252537A1 (en) | 2002-06-08 | 2003-12-22 | Bayer Cropscience Gmbh | Combinations of herbicidal aromatic carboxylic acids and safeners |
| DE10237461A1 (de) | 2002-08-16 | 2004-02-26 | Bayer Cropscience Gmbh | Herbizide Mittel enthaltend Benzoylpyrazole und Safener |
| AU2003288901A1 (en) | 2002-09-06 | 2004-03-29 | Merck And Co., Inc. | Treatment of rheumatoid arthritis by inhibition of pde4 |
| EP1562914A1 (en) | 2002-10-22 | 2005-08-17 | Merck Frosst Canada & Co. | Nitric oxide releasing selective cyclooxygenase-2 inhibitors |
| GB0225548D0 (en) | 2002-11-01 | 2002-12-11 | Glaxo Group Ltd | Compounds |
| AU2003302106A1 (en) | 2002-11-21 | 2004-06-15 | Vicore Pharma Ab | New tricyclic angiotensin ii agonists |
| CA2510815A1 (en) | 2002-12-20 | 2004-07-08 | Pfizer Products Inc. | Microsomal triglyceride transfer protein inhibitors |
| MXPA05006744A (es) | 2002-12-20 | 2005-09-08 | Pfizer Prod Inc | Inhibidores de la proteina microsomal de transferencia de trigliceridos. |
| WO2004078114A2 (en) | 2003-02-28 | 2004-09-16 | Encysive Pharmaceuticals Inc. | Pyridine, pyrimidine, quinoline, quinazoline, and naphthalene urotensin-ii receptor antagonists. |
| AU2003219291A1 (en) | 2003-03-24 | 2004-10-18 | Vicore Pharma Ab | Bicyclic compounds useful as angiotensin ii agonists |
| JP4869072B2 (ja) | 2003-11-14 | 2012-02-01 | バーテックス ファーマシューティカルズ インコーポレイテッド | Atp結合カセットトランスポーターのモジュレーターとして有用なチアゾールおよびオキサゾール |
| GB0328024D0 (en) | 2003-12-03 | 2004-01-07 | Glaxo Group Ltd | Compounds |
| US20090131342A1 (en) | 2004-01-22 | 2009-05-21 | Nitromed, Inc. | Nitrosated and/or nitrosylated compounds, compositions and methods of use |
| MXPA06008606A (es) | 2004-01-30 | 2007-04-13 | Vertex Pharma | Moduladores de transportadores con casete de union a atp. |
| AP2006003685A0 (en) | 2004-02-04 | 2006-08-31 | Pfizer Prod Inc | Substituted quinoline compounds |
| US20050197376A1 (en) | 2004-03-02 | 2005-09-08 | Fujisawa Pharmaceutical Co. Ltd. | Concomitant drugs |
| AU2005219788B2 (en) | 2004-03-05 | 2010-06-03 | Nissan Chemical Corporation | Isoxazoline-substituted benzamide compound and noxious organism control agent |
| WO2005099705A2 (en) | 2004-03-24 | 2005-10-27 | Bayer Pharmaceuticals Corporation | Preparation of imidazole derivatives and methods of use |
| GB0410121D0 (en) | 2004-05-06 | 2004-06-09 | Glaxo Group Ltd | Compounds |
| US8354427B2 (en) | 2004-06-24 | 2013-01-15 | Vertex Pharmaceutical Incorporated | Modulators of ATP-binding cassette transporters |
| LT2502911T (lt) | 2004-06-24 | 2017-09-11 | Vertex Pharmaceuticals Incorporated | Atp surišančios kasetės transporterių moduliatoriai |
| JP4953297B2 (ja) | 2004-09-15 | 2012-06-13 | 塩野義製薬株式会社 | Hivインテグラーゼ阻害活性を有するカルバモイルピリドン誘導体 |
| EP1812429A4 (en) | 2004-09-29 | 2010-07-21 | Portola Pharm Inc | SUBSTITUTED 2H-1,3-BENZOXAZIN-4 (3H) -ONE |
| US20080287399A1 (en) | 2004-12-14 | 2008-11-20 | Astrazeneca Ab | Substituted Aminopyridines and Uses Thereof |
| EP1833795B1 (en) | 2004-12-23 | 2009-03-04 | Glaxo Group Limited | Pyridine compounds for the treatment of prostaglandin mediated diseases |
| GB0428173D0 (en) | 2004-12-23 | 2005-01-26 | Astrazeneca Ab | Compounds |
| WO2007019397A2 (en) | 2005-08-05 | 2007-02-15 | Genelabs Technologies, Inc. | 6-membered aryl and heteroaryl derivatives for the treatment of hepatitis c virus |
| ATE512145T1 (de) | 2005-08-11 | 2011-06-15 | Vertex Pharma | Modulatoren des cystic fibrosis transmembrane conductance regulators |
| WO2007053641A2 (en) | 2005-11-01 | 2007-05-10 | Mars, Incorporated | A-type procyanidins and inflammation |
| AU2006331565A1 (en) | 2005-12-27 | 2007-07-05 | Vertex Pharmaceuticals Incorporated | Compounds useful in CFTR assays and methods therewith |
| WO2007079139A2 (en) | 2005-12-28 | 2007-07-12 | Vertex Pharmaceuticals, Inc. | Solid forms of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| AU2006336504C9 (en) | 2005-12-28 | 2015-05-14 | Vertex Pharmaceuticals Incorporated | 1-(benzo [D] [1,3] dioxol-5-yl) -N- (phenyl) cyclopropane- carboxamide derivatives and related compounds as modulators of ATP-Binding Cassette transporters for the treatment of Cystic Fibrosis |
| US7671221B2 (en) | 2005-12-28 | 2010-03-02 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-Binding Cassette transporters |
| US7932283B2 (en) | 2006-04-05 | 2011-04-26 | Bayer Cropscience Ag | Fungicide N-cyclopropyl-sulfonylamide derivatives |
| NZ571803A (en) | 2006-04-07 | 2011-12-22 | Vertex Pharma | Amide indole derivatives as modulators of ATP-binding cassette transporters |
| EP2021797B1 (en) | 2006-05-12 | 2011-11-23 | Vertex Pharmaceuticals, Inc. | Compositions of n-ý2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl¨-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| EP2038272B8 (en) | 2006-06-30 | 2013-10-23 | Sunesis Pharmaceuticals, Inc. | Pyridinonyl pdk1 inhibitors |
| CA2668305C (en) | 2006-11-03 | 2017-01-03 | Vertex Pharmaceuticals Incorporated | Azaindole derivatives as cftr modulators |
| MX2009008439A (es) | 2007-02-12 | 2009-08-13 | Intermune Inc | Nuevos inhibidores de la replicacion del virus de hepatitis c. |
| WO2008141385A1 (en) | 2007-05-21 | 2008-11-27 | Biota Scientific Management Pty Ltd | Viral polymerase inhibitors |
| US8058299B2 (en) | 2007-05-22 | 2011-11-15 | Via Pharmaceuticals, Inc. | Diacylglycerol acyltransferase inhibitors |
| GB0716532D0 (en) | 2007-08-24 | 2007-10-03 | Angeletti P Ist Richerche Bio | Therapeutic compounds |
| WO2009032116A1 (en) | 2007-08-29 | 2009-03-12 | Schering Corporation | 2, 3-substituted indole derivatives for treating viral infections |
| WO2009038683A2 (en) | 2007-09-14 | 2009-03-26 | Vertex Pharmaceuticals Incorporated | Solid forms of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| MX2010005356A (es) | 2007-11-16 | 2010-05-27 | Schering Corp | Derivados de indol 3-heterociclico sustituidos y metodos de uso de los mismos. |
| GB0723794D0 (en) | 2007-12-05 | 2008-01-16 | Lectus Therapeutics Ltd | Potassium ion channel modulators and uses thereof |
| EP2639224B1 (en) | 2007-12-07 | 2016-08-24 | Vertex Pharmaceuticals Incorporated | Process for producing cycloalkylcarboxiamido-pyridine benzoic acids |
| SI2225230T1 (sl) | 2007-12-07 | 2017-03-31 | Vertex Pharmaceuticals Incorporated | Trdne oblike 3-(6-(1-2,2-difluorobenzo(d)(1,3)dioxol-5-il)ciklopropan- karboksamido)-3-metilpiridin-2-il) benzojske kisline |
| WO2009127822A2 (en) | 2008-04-16 | 2009-10-22 | Biolipox Ab | Bis-aryl compounds for use as medicaments |
| US20110112193A1 (en) | 2008-05-14 | 2011-05-12 | Peter Nilsson | Bis-aryl compounds for use as medicaments |
| EP2145537A1 (en) | 2008-07-09 | 2010-01-20 | Bayer CropScience AG | Plant growth regulator |
| UY31982A (es) | 2008-07-16 | 2010-02-26 | Boehringer Ingelheim Int | Derivados de 1,2-dihidropiridin-3-carboxamidas n-sustituidas |
| US20100074949A1 (en) | 2008-08-13 | 2010-03-25 | William Rowe | Pharmaceutical composition and administration thereof |
| HRP20180328T1 (hr) | 2008-08-13 | 2018-04-20 | Vertex Pharmaceuticals Inc. | FARMACEUTSKI PRIPRAVAK N-[2,4-bis(1,1-DIMETILETIL)-5-HIDROKSIFENIL]-1,4-DIHIDRO-4-OKSOKINOLIN-3-KARBOKSAMIDA I NJEGOVO DAVANJE |
| US20110160227A1 (en) | 2008-08-21 | 2011-06-30 | Antony Shaw | Prolyl Hydroxylase Inhibitors |
| TWI392673B (zh) | 2008-08-27 | 2013-04-11 | Calcimedica Inc | 調控細胞內鈣離子濃度之化合物 |
| EP2365972B1 (en) | 2008-11-06 | 2014-12-17 | Vertex Pharmaceuticals Incorporated | Modulators of atp-binding cassette transporters |
| UA104876C2 (uk) | 2008-11-06 | 2014-03-25 | Вертекс Фармасьютікалз Інкорпорейтед | Модулятори atф-зв'язувальних касетних транспортерів |
| US20100160322A1 (en) | 2008-12-04 | 2010-06-24 | Abbott Laboratories | Apoptosis-inducing agents for the treatment of cancer and immune and autoimmune diseases |
| UA108193C2 (uk) | 2008-12-04 | 2015-04-10 | Апоптозіндукуючий засіб для лікування раку і імунних і аутоімунних захворювань | |
| CA2747835A1 (en) | 2009-01-19 | 2010-07-22 | Abbott Laboratories | Apoptosis-inducing agents for the treatment of cancer and immune and autoimmune diseases |
| BRPI1008949B1 (pt) | 2009-03-11 | 2018-07-10 | Bayer Intellectual Property Gmbh | Cetoenóis haloalquilmetilenóxi-fenil-substituídos e seu uso, composição, seu uso e seu método de produção, métodos para combate de pestes animais e/ou crescimento de plantas indesejadas |
| SI2408750T1 (sl) | 2009-03-20 | 2015-11-30 | Vertex Pharmaceuticals Incorporated | Postopek za izdelavo modulatorjev cistično-fibroznega transmembranskega regulatorja prevodnosti |
| EP2412708A4 (en) | 2009-03-26 | 2014-07-23 | Shionogi & Co | SUBSTITUTED 3-HYDROXY-4-PYRIDONE DERIVATIVE |
| WO2010123822A1 (en) | 2009-04-20 | 2010-10-28 | Institute For Oneworld Health | Compounds, compositions and methods comprising pyridazine sulfonamide derivatives |
| WO2010138588A2 (en) | 2009-05-26 | 2010-12-02 | Abbott Laboratories | Apoptosis-inducing agents for the treatment of cancer and immune and autoimmune diseases |
| WO2011102514A1 (ja) | 2010-02-22 | 2011-08-25 | 武田薬品工業株式会社 | 芳香環化合物 |
| US8247436B2 (en) * | 2010-03-19 | 2012-08-21 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of CF |
| US8471029B2 (en) | 2010-03-19 | 2013-06-25 | Vertex Pharmaceutical Incorporated | Solid forms of N-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| US20110251253A1 (en) | 2010-03-25 | 2011-10-13 | Vertex Pharmaceuticals Incorporated | Solid forms of (r)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-n-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1h-indol-5-yl) cyclopropanecarboxamide |
| HRP20160682T1 (hr) | 2010-04-07 | 2016-07-29 | Vertex Pharmaceuticals Incorporated | Čvrsti oblici 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioksol-5-il)ciklopropankarboksiamido)-3-metilpiridin-2-il)benzojeve kiseline |
| HRP20211752T1 (hr) | 2010-04-07 | 2022-02-18 | Vertex Pharmaceuticals Incorporated | Farmaceutski pripravci 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioksol-5-il)ciklopropankarboksamido)-3-metilpiridin-2-il)benzojeve kiseline i njihova primjena |
| US8344137B2 (en) | 2010-04-14 | 2013-01-01 | Hoffman-La Roche Inc. | 3,3-dimethyl tetrahydroquinoline derivatives |
| ES2608474T3 (es) | 2010-04-22 | 2017-04-11 | Vertex Pharmaceuticals Incorporated | Proceso de producción de compuestos indol cycloalkylcarboxamido |
| CA2796642A1 (en) | 2010-04-22 | 2011-10-27 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions and administrations thereof |
| TWI520960B (zh) | 2010-05-26 | 2016-02-11 | 艾伯維有限公司 | 用於治療癌症及免疫及自體免疫疾病之細胞凋亡誘導劑 |
| US8563593B2 (en) | 2010-06-08 | 2013-10-22 | Vertex Pharmaceuticals Incorporated | Formulations of (R)-1-(2,2-difluorobenzo[D] [1,3] dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| CA2808501A1 (en) | 2010-08-23 | 2012-03-01 | Vertex Pharmaceuticals Incorporated | Pharmaceutical composition of (r)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-n-(1-(2,3-dihydroxy propyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1h-indol-5-yl) cyclopropanecarboxamide and administration therof |
| RU2013113627A (ru) | 2010-08-27 | 2014-10-10 | Вертекс Фармасьютикалз Инкорпорейтед | Фармацевтическая композиция и ее введения |
| US9394290B2 (en) | 2010-10-21 | 2016-07-19 | Universitaet Des Saarlandes Campus Saarbruecken | Selective CYP11B1 inhibitors for the treatment of cortisol dependent diseases |
| WO2012087938A1 (en) | 2010-12-20 | 2012-06-28 | Glaxosmithkline Llc | Quinazolinone derivatives as antiviral agents |
| EP2471363A1 (de) | 2010-12-30 | 2012-07-04 | Bayer CropScience AG | Verwendung von Aryl-, Heteroaryl- und Benzylsulfonamidocarbonsäuren, -carbonsäureestern, -carbonsäureamiden und -carbonitrilen oder deren Salze zur Steigerung der Stresstoleranz in Pflanzen |
| JPWO2012102297A1 (ja) | 2011-01-26 | 2014-06-30 | 杏林製薬株式会社 | ピラゾロピリジン誘導体、またはその薬理学的に許容される塩 |
| EP2675788A1 (de) | 2011-02-17 | 2013-12-25 | Bayer Intellectual Property GmbH | Substituierte 3-(biphenyl-3-yl)-8,8-difluor-4-hydroxy-1-azaspiro[4.5]dec-3-en-2-one zur therapie |
| US9204640B2 (en) | 2011-03-01 | 2015-12-08 | Bayer Intellectual Property Gmbh | 2-acyloxy-pyrrolin-4-ones |
| AR085585A1 (es) | 2011-04-15 | 2013-10-09 | Bayer Cropscience Ag | Vinil- y alquinilciclohexanoles sustituidos como principios activos contra estres abiotico de plantas |
| EP2714680B1 (en) | 2011-05-27 | 2015-11-25 | Amira Pharmaceuticals, Inc. | Heterocyclic autotaxin inhibitors and uses thereof |
| KR20140072037A (ko) | 2011-08-30 | 2014-06-12 | 씨에이치디아이 파운데이션, 인코포레이티드 | 키뉴레닌-3-모노옥시게나제 억제제, 약학적 조성물 및 이의 사용 방법 |
| WO2013038373A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine amide derivatives |
| UA115971C2 (uk) | 2011-09-16 | 2018-01-25 | Байєр Інтеллектуал Проперті Гмбх | Застосування ацилсульфонамідів для покращення врожайності рослин |
| BR112014006940A2 (pt) | 2011-09-23 | 2017-04-04 | Bayer Ip Gmbh | uso de derivados de ácido 1-fenilpirazol-3-carboxílico 4-substituído como agentes contra estresse abiótico em plantas |
| IN2014KN00885A (es) | 2011-11-08 | 2015-10-02 | Vertex Pharma | |
| US8426450B1 (en) | 2011-11-29 | 2013-04-23 | Helsinn Healthcare Sa | Substituted 4-phenyl pyridines having anti-emetic effect |
| WO2013112804A1 (en) | 2012-01-25 | 2013-08-01 | Vertex Pharmaceuticals Incorporated | Formulations of 3-(6-(1-(2.2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid |
| JP2015083542A (ja) | 2012-02-08 | 2015-04-30 | 大日本住友製薬株式会社 | 3位置換プロリン誘導体 |
| EP2819670A1 (en) | 2012-02-27 | 2015-01-07 | Vertex Pharmaceuticals Incorporated | Pharmaceutical composition and administration thereof |
| US8889730B2 (en) | 2012-04-10 | 2014-11-18 | Pfizer Inc. | Indole and indazole compounds that activate AMPK |
| AU2012377446A1 (en) | 2012-04-20 | 2014-10-23 | Vertex Pharmaceuticals Incorporated | Solid forms of N-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| SG11201408284VA (en) | 2012-05-22 | 2015-02-27 | Xenon Pharmaceuticals Inc | N-substituted benzamides and their use in the treatment of pain |
| AU2013270681A1 (en) | 2012-06-08 | 2014-12-18 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions for the treatment of CFTR -mediated disorders |
| WO2013185202A1 (en) | 2012-06-14 | 2013-12-19 | Beta Pharma Canada Inc | Apoptosis inducers |
| US10071957B2 (en) | 2012-07-06 | 2018-09-11 | Genentech, Inc. | N-substituted benzamides and methods of use thereof |
| WO2014014841A1 (en) | 2012-07-16 | 2014-01-23 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of (r)-1-(2,2-diflurorbenzo[d][1,3]dioxol-5-yl)-n-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1h-indol-5-yl) cyclopropanecarboxamide and administration thereof |
| JP2015178458A (ja) | 2012-07-25 | 2015-10-08 | 杏林製薬株式会社 | ベンゼン環縮合含窒素5員複素環式化合物、またはその薬理学的に許容される塩 |
| CA2879332C (en) | 2012-08-13 | 2022-08-30 | Abbvie Inc. | 1,2,3,4- tetrahydroisoquinoline-8-carboxamide compounds and their use as apoptosis-inducing agents |
| US9896443B2 (en) | 2012-08-21 | 2018-02-20 | Peter Maccallum Cancer Institute | Perforin inhibiting benzenesulfonamide compounds, preparation and uses thereof |
| EP2892534B8 (en) | 2012-09-06 | 2021-09-15 | Plexxikon Inc. | Compounds and methods for kinase modulation, and indications therefor |
| WO2014047427A2 (en) | 2012-09-21 | 2014-03-27 | Vanderbilt University | Substituted benzofuran, benzothiophene and indole mcl-1 inhibitors |
| LT3470063T (lt) | 2012-11-02 | 2025-02-10 | Vertex Pharmaceuticals Incorporated | Farmacinės kompozicijos, skirtos cftr nulemtų ligų gydymui |
| EP2917203B1 (en) | 2012-11-02 | 2019-04-03 | Dana-Farber Cancer Institute, Inc. | Method for identifying myc inhibitors |
| CA2890018A1 (en) | 2012-11-05 | 2014-05-08 | Nant Holdings Ip, Llc | Substituted indol-5-ol derivatives and their therapeutical applications |
| WO2014086751A1 (de) | 2012-12-05 | 2014-06-12 | Bayer Cropscience Ag | Verwendung substituierter 1-(arylethinyl)-, 1-(heteroarylethinyl)-, 1-(heterocyclylethinyl)- und 1-(cyloalkenylethinyl)-cyclohexanole als wirkstoffe gegen abiotischen pflanzenstress |
| BR112015013056A2 (pt) | 2012-12-05 | 2017-07-11 | Bayer Cropscience Ag | uso de 1-(ariletinil)-, 1-(heteroariletinil)-, 1-(heterocicliletinil)- substituídos e 1-(cicloalqueniletinil)-bicicloalcanóis como agentes ativos contra o estresse abiótico da planta |
| UA115576C2 (uk) | 2012-12-06 | 2017-11-27 | Байєр Фарма Акцієнгезелльшафт | Похідні бензимідазолу як антагоністи ер4 |
| GB201223265D0 (en) | 2012-12-21 | 2013-02-06 | Selvita Sa | Novel benzimidazole derivatives as kinase inhibitors |
| US20150353542A1 (en) | 2013-01-14 | 2015-12-10 | Amgen Inc. | Methods of using cell-cycle inhibitors to modulate one or more properties of a cell culture |
| WO2014152018A1 (en) | 2013-03-14 | 2014-09-25 | The Trustees Of Columbia University In The City Of New York | Octahydrocyclopentapyrroles, their preparation and use |
| EP3495357B1 (en) | 2013-03-14 | 2021-05-05 | The Trustees of Columbia University in the City of New York | 4-phenylpiperidines, their preparation and use |
| EA033168B1 (ru) | 2013-03-15 | 2019-09-30 | Сайклерион Терапьютикс, Инк. | СТИМУЛЯТОРЫ sGC |
| EA031647B1 (ru) | 2013-03-29 | 2019-02-28 | Такеда Фармасьютикал Компани Лимитед | 6-(5-гидрокси-1h-пиразол-1-ил)никотинамидные ингибиторы phd |
| EP2994463B1 (en) * | 2013-05-07 | 2017-10-25 | Galapagos NV | Novel compounds and pharmaceutical compositions thereof for the treatment of cystic fibrosis |
| WO2014181287A1 (en) | 2013-05-09 | 2014-11-13 | Piramal Enterprises Limited | Heterocyclyl compounds and uses thereof |
| CN105473578A (zh) | 2013-05-24 | 2016-04-06 | 加州生物医学研究所 | 用于治疗抗药性和持续性结核病的化合物 |
| WO2015010832A1 (en) | 2013-07-22 | 2015-01-29 | Syngenta Participations Ag | Microbiocidal heterocyclic derivatives |
| CA2922341C (en) | 2013-08-28 | 2022-06-07 | Vanderbilt University | Substituted indole mcl-1 inhibitors |
| US9663508B2 (en) | 2013-10-01 | 2017-05-30 | Amgen Inc. | Biaryl acyl-sulfonamide compounds as sodium channel inhibitors |
| WO2015069287A1 (en) | 2013-11-08 | 2015-05-14 | Allergan, Inc. | Compounds as tyrosine kinase modulators |
| JP6963896B2 (ja) | 2013-11-12 | 2021-11-10 | バーテックス ファーマシューティカルズ インコーポレイテッドVertex Pharmaceuticals Incorporated | Cftr媒介性疾患の処置のための医薬組成物を調製する方法 |
| ES2885181T3 (es) | 2014-04-15 | 2021-12-13 | Vertex Pharma | Composiciones farmacéuticas para el tratamiento de enfermedades mediadas por el regulador de la conductancia transmembrana de fibrosis quística |
| TWI735416B (zh) | 2014-10-06 | 2021-08-11 | 美商維泰克斯製藥公司 | 囊腫纖維化症跨膜傳導調節蛋白之調節劑 |
| KR20170063954A (ko) | 2014-10-07 | 2017-06-08 | 버텍스 파마슈티칼스 인코포레이티드 | 낭성 섬유증 막횡단 전도도 조절자의 조정제의 공-결정 |
| PT3221692T (pt) | 2014-11-18 | 2021-09-10 | Vertex Pharma | Processo de realização de testagem de alta produtividade por cromatografia líquida de alta eficiência |
| US10738011B2 (en) | 2014-12-23 | 2020-08-11 | Proteostasis Therapeutics, Inc. | Derivatives of 5-(hetero)arylpyrazol-3-carboxylic amide or 1-(hetero)aryltriazol-4-carboxylic amide useful for the treatment of inter alia cystic fibrosis |
| MA41253A (fr) | 2014-12-23 | 2017-10-31 | Proteostasis Therapeutics Inc | Composés, compositions et procédés pour augmenter l'activité du cftr |
| EP3259356B1 (en) * | 2015-02-20 | 2021-12-01 | Rosalind Franklin University of Medicine and Science | Antisense compounds targeting genes associated with cystic fibrosis |
| US20210401869A1 (en) * | 2015-02-20 | 2021-12-30 | Rosalind Franklin University Of Medicine And Science | Antisense Compounds Targeting Genes Associated with Cystic Fibrosis |
| HK1249893A1 (zh) | 2015-03-31 | 2018-11-16 | Vertex Pharmaceuticals (Europe) Limited | 氘代vx-661 |
| UA124619C2 (uk) | 2015-09-21 | 2021-10-20 | Вертекс Фармасьютікалз (Юроп) Лімітед | Введення дейтерованих підсилювачів cftr |
| EP3854782A1 (en) | 2016-03-30 | 2021-07-28 | Genentech, Inc. | Substituted benzamides and methods of use thereof |
| ES2946970T3 (es) | 2016-03-31 | 2023-07-28 | Vertex Pharma | Regulador de conductancia transmembrana de moduladores de fibrosis quística |
| BR112018070747B1 (pt) | 2016-04-07 | 2024-01-09 | Proteostasis Therapeutics, Inc | Átomos de silicone contendo análogos de ivacaftor, composições farmacêuticas e usos terapêuticos |
| AU2017256172A1 (en) | 2016-04-26 | 2018-09-06 | AbbVie S.à.r.l. | Modulators of cystic fibrosis transmembrane conductance regulator protein |
| US10138227B2 (en) | 2016-06-03 | 2018-11-27 | Abbvie S.Á.R.L. | Heteroaryl substituted pyridines and methods of use |
| MA45397A (fr) | 2016-06-21 | 2019-04-24 | Proteostasis Therapeutics Inc | Composés, compositions et procédés pour augmenter l'activité du cftr |
| SI3519401T1 (sl) | 2016-09-30 | 2022-01-31 | Vertex Pharmaceuticals Incorporated | Modulator regulatorja transmembranske prevodnosti pri cistični fibrozi, farmacevtski sestavki, postopki zdravljenja in proces za izdelavo modulatorja |
| US10399940B2 (en) | 2016-10-07 | 2019-09-03 | Abbvie S.Á.R.L. | Substituted pyrrolidines and methods of use |
| US9981910B2 (en) | 2016-10-07 | 2018-05-29 | Abbvie S.Á.R.L. | Substituted pyrrolidines and methods of use |
| CA3041675A1 (en) | 2016-10-26 | 2018-05-03 | Proteostasis Therapeutics, Inc. | N-phenyl-2-(3-phenyl-6-oxo-1,6-dihydropyridazin-1-yl)acetamide derivatives for treating cystic fibrosis |
| WO2018081381A1 (en) | 2016-10-26 | 2018-05-03 | Proteostasis Therapeutics, Inc | Pyridazine derivatives, compositions and methods for modulating cftr |
| WO2018081378A1 (en) | 2016-10-26 | 2018-05-03 | Proteostasis Therapeutics, Inc. | Compounds, compositions, and methods for modulating cftr |
| AU2017362350B2 (en) | 2016-11-18 | 2021-12-23 | Cystic Fibrosis Foundation | Pyrrolopyrimidines as CFTR potentiators |
| CN110267948B (zh) | 2016-12-09 | 2023-12-08 | 弗特克斯药品有限公司 | 囊性纤维化跨膜传导调控剂的调节剂、药物组合物、治疗方法和制备所述调节剂的方法 |
| US10131670B2 (en) | 2016-12-16 | 2018-11-20 | Cystic Fibrosis Foundation Therapeutics, Inc. | Bicyclic heteroaryl derivatives as CFTR potentiators |
| EP3558982A1 (en) | 2016-12-20 | 2019-10-30 | AbbVie S.À.R.L. | Deuterated cftr modulators and methods of use |
| EP3565815B1 (en) | 2017-01-07 | 2024-03-13 | Fochon Pharmaceuticals, Ltd. | Compounds as bcl-2-selective apoptosis-inducing agents |
| TW201831471A (zh) | 2017-02-24 | 2018-09-01 | 盧森堡商艾伯維公司 | 囊腫纖化症跨膜傳導調節蛋白的調節劑及其使用方法 |
| WO2018183367A1 (en) | 2017-03-28 | 2018-10-04 | Van Goor Fredrick F | Methods of treating cystic fibrosis in patients with residual function mutations |
| MA49061A (fr) | 2017-04-28 | 2021-04-21 | Proteostasis Therapeutics Inc | Dérivés 4-sulfonylaminocarbonylquinoline pour augmenter l'activité cftr |
| AU2018279646B2 (en) | 2017-06-08 | 2023-04-06 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| CA3068609A1 (en) | 2017-07-01 | 2019-01-10 | Vertex Pharmaceuticals Incorporated | Compositions and methods for treatment of cystic fibrosis |
| US20200171015A1 (en) | 2017-07-17 | 2020-06-04 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| MA49631A (fr) | 2017-07-17 | 2020-05-27 | Vertex Pharma | Méthodes de traitement de la fibrose kystique |
| US11427858B2 (en) | 2017-07-31 | 2022-08-30 | Technion Research & Development Foundation Limited | Methods of detecting modified and unmodified DNA |
| TWI799435B (zh) | 2017-08-02 | 2023-04-21 | 美商維泰克斯製藥公司 | 製備化合物之製程 |
| US10988454B2 (en) | 2017-09-14 | 2021-04-27 | Abbvie Overseas S.À.R.L. | Modulators of the cystic fibrosis transmembrane conductance regulator protein and methods of use |
| AU2018346602B2 (en) | 2017-10-06 | 2024-10-17 | Proteostasis Therapeutics, Inc. | Compounds, compositions and methods for increasing CFTR activity |
| US10654829B2 (en) | 2017-10-19 | 2020-05-19 | Vertex Pharmaceuticals Incorporated | Crystalline forms and compositions of CFTR modulators |
| WO2019113089A1 (en) | 2017-12-04 | 2019-06-13 | Vertex Pharmaceuticals Incorporated | Compositions for treating cystic fibrosis |
| KR102716244B1 (ko) | 2017-12-08 | 2024-10-14 | 버텍스 파마슈티칼스 인코포레이티드 | 낭포성 섬유증 막횡단 전도 조절자의 조정제의 제조 방법 |
| TWI810243B (zh) | 2018-02-05 | 2023-08-01 | 美商維泰克斯製藥公司 | 用於治療囊腫纖化症之醫藥組合物 |
| WO2019191620A1 (en) | 2018-03-30 | 2019-10-03 | Vertex Pharmaceuticals Incorporated | Crystalline forms of modulators of cftr |
| EP3774825A1 (en) | 2018-04-13 | 2021-02-17 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
-
2017
- 2017-09-29 SI SI201731014T patent/SI3519401T1/sl unknown
- 2017-09-29 MA MA46357A patent/MA46357B1/fr unknown
- 2017-09-29 PL PL17792230T patent/PL3519401T3/pl unknown
- 2017-09-29 MD MDE20190875T patent/MD3519401T2/ro unknown
- 2017-09-29 JP JP2019517075A patent/JP7061115B2/ja active Active
- 2017-09-29 TN TNP/2019/000079A patent/TN2019000079A1/en unknown
- 2017-09-29 DK DK17792230.9T patent/DK3519401T3/da active
- 2017-09-29 PT PT177922309T patent/PT3519401T/pt unknown
- 2017-09-29 US US15/721,390 patent/US10570115B2/en active Active
- 2017-09-29 RS RS20211432A patent/RS62670B1/sr unknown
- 2017-09-29 ES ES17792230T patent/ES2900263T3/es active Active
- 2017-09-29 GE GEAP201715057A patent/GEP20217329B/en unknown
- 2017-09-29 HU HUE17792230A patent/HUE056716T2/hu unknown
- 2017-09-29 SG SG10201913595YA patent/SG10201913595YA/en unknown
- 2017-09-29 JO JOP/2019/0042A patent/JOP20190042B1/ar active
- 2017-09-29 CN CN201780061062.5A patent/CN109803962B/zh active Active
- 2017-09-29 LT LTEPPCT/US2017/054611T patent/LT3519401T/lt unknown
- 2017-09-29 HR HRP20211683TT patent/HRP20211683T1/hr unknown
- 2017-09-29 PE PE2019000737A patent/PE20191147A1/es unknown
- 2017-09-29 AU AU2017336156A patent/AU2017336156B2/en active Active
- 2017-09-29 KR KR1020197012347A patent/KR102531491B1/ko active Active
- 2017-09-29 MX MX2019003620A patent/MX387060B/es unknown
- 2017-09-29 CA CA3037986A patent/CA3037986C/en active Active
- 2017-09-29 UA UAA201904551A patent/UA124708C2/uk unknown
- 2017-09-29 GE GEAP202115057A patent/GEAP202115057A/en unknown
- 2017-09-29 EP EP17792230.9A patent/EP3519401B1/en active Active
- 2017-09-29 WO PCT/US2017/054611 patent/WO2018064632A1/en not_active Ceased
- 2017-09-29 SM SM20210646T patent/SMT202100646T1/it unknown
- 2017-09-29 NZ NZ752486A patent/NZ752486A/en unknown
- 2017-09-30 TW TW106133964A patent/TWI766886B/zh active
- 2017-10-02 UY UY0001037428A patent/UY37428A/es active IP Right Grant
-
2019
- 2019-03-26 IL IL265627A patent/IL265627B/en unknown
- 2019-03-28 SA SA519401446A patent/SA519401446B1/ar unknown
- 2019-03-28 DO DO2019000081A patent/DOP2019000081A/es unknown
- 2019-03-28 CL CL2019000827A patent/CL2019000827A1/es unknown
- 2019-04-04 ZA ZA2019/02124A patent/ZA201902124B/en unknown
- 2019-04-24 EC ECSENADI201928690A patent/ECSP19028690A/es unknown
- 2019-04-24 CO CONC2019/0004065A patent/CO2019004065A2/es unknown
- 2019-12-04 US US16/702,891 patent/US11186566B2/en active Active
-
2021
- 2021-10-20 US US17/505,699 patent/US20220306606A1/en not_active Abandoned
- 2021-12-09 CY CY20211101083T patent/CY1124862T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2900263T3 (es) | Modulador de regulador de conductancia transmembrana de fibrosis quística, composiciones farmacéuticas, métodos de tratamiento y proceso de fabricación del modulador | |
| AU2021211993B2 (en) | Modulator of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator | |
| ES2968810T3 (es) | Agentes moduladores del regulador de la conductancia transmembrana de la fibrosis quística | |
| ES2737696T3 (es) | Pirazolopiridinas y pirazolopirimidinas | |
| BR112020015509A2 (pt) | macrociclos como moduladores de regulador da condutância transmembrana da fibrose cística, composições farmacêuticas dos mesmos, sua utilização no tratamento da fibrose cística, e processo para sua produção | |
| CA3123578A1 (en) | Methods of treatment for cystic fibrosis | |
| CN107709305A (zh) | 新型化合物 | |
| BR112019006213B1 (pt) | Modulador de regulador de condutância transmembranar de fibrose cística, seus usos e seu método de preparação, e composições farmacêuticas | |
| BR122024017810B1 (pt) | Derivado deuterado de um composto modulador do regulador de condutância transmembrana na fibrose cística, seu uso, e composições farmacêuticas | |
| BR122024017810A2 (pt) | Derivado deuterado de um composto modulador do regulador de condutância transmembrana na fibrose cística, seu uso, e composições farmacêuticas | |
| BR122021023793B1 (pt) | Forma cristalina a, m, e, x, y, p2 do composto 1, seus usos, solvato do composto 1, método de preparo da forma cristalina a do composto 1, dispersão sólida, seu uso, e composição farmacêutica | |
| BR112019011626B1 (pt) | Composto modulador do regulador de condutância transmembrana na fibrose cística, seu uso, e composições farmacêuticas | |
| HK40011778A (en) | Modulator of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator | |
| HK40011778B (en) | Modulator of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator | |
| OA19370A (en) | Modulator of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator. | |
| EA040454B1 (ru) | Модулятор муковисцидозного трансмембранного регулятора проводимости, фармацевтические композиции, способы лечения и способы получения модулятора | |
| NZ792410A (en) | Modulator of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |