EP0000718A2 - Neue Chinazolinderivate, Verfahren zu ihrer Herstellung, pharmazeutische Präparate und deren Herstellung - Google Patents
Neue Chinazolinderivate, Verfahren zu ihrer Herstellung, pharmazeutische Präparate und deren Herstellung Download PDFInfo
- Publication number
- EP0000718A2 EP0000718A2 EP78100471A EP78100471A EP0000718A2 EP 0000718 A2 EP0000718 A2 EP 0000718A2 EP 78100471 A EP78100471 A EP 78100471A EP 78100471 A EP78100471 A EP 78100471A EP 0000718 A2 EP0000718 A2 EP 0000718A2
- Authority
- EP
- European Patent Office
- Prior art keywords
- lower alkyl
- formula
- compounds
- hydrogen
- hydroxy
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 0 *C(C(N1)=O)N(C2*)C1=NC1=C2C=C*(*)C=C1 Chemical compound *C(C(N1)=O)N(C2*)C1=NC1=C2C=C*(*)C=C1 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
Definitions
- the present invention relates to new tricyclic compounds, namely imidazo-quinazolines of the formula wherein R 1 and R 2 are hydrogen, lower alkyl, hydroxy, lower alkoxy, hydroxy lower alkyl, lower alkoxy lower alkyl, halogen, phenyl, phenoxy, amino, lower alkylamino or di-lower alkylamino, and R 1 and R 2 on adjacent carbon atoms also together methylenedioxy; R is hydrogen, lower alkyl or phenyl; and R 4 is lower alkyl, hydroxy lower alkyl, lower alkoxy lower alkyl, aryl lower alkyl or aryl, their tautomers and salts of such compounds.
- alkyl radicals can be straight-chain or branched. Examples of alkyl radicals are: methyl, ethyl, propyl, isopropyl, butyl, isobutyl, pentyl and hexyl.
- Aryl means in particular phenyl or phenyl substituted by halogen, lower alkyl, hydroxy and / or lower alkoxy.
- R and R are hydrogen
- R 2 is halogen in the 6- or 7-position or lower alkyl in the 6-position, in particular 6-chloro, 7-bromo or 6-methyl
- R 4 lower alkyl, especially methyl are preferred.
- the invention further relates to a process for the preparation of the compounds mentioned and pharmaceutical preparations based on the compounds mentioned.
- the compounds of formula I can exist in various tautomeric forms.
- the invention is therefore not limited to compounds of the formula I shown above, but also includes the tautomers, for example those of the formula and
- the compounds of formula I and their tautomers e.g. Ia and Ib can also be present in the form of racemates or in optically active form, all of which are the subject of the invention.
- physiologically compatible salts are mineral acid salts, such as hydrochlorides, hydrobromides, sulfates and phosphates; Salts of organic sulfonic acids such as alkyl sulfates and aryl sulfonates; and carboxylic acid salts such as succinates, citrates, tartrates and maleates.
- reaction of a compound of formula II with cyanogen bromide is conveniently carried out with heating in a solvent such as a lower alcohol, e.g. Ethanol performed.
- reaction of a compound of formula III with ammonia is conveniently carried out with heating in a solvent such as a lower alcohol, e.g. Ethanol, and water.
- a compound of the formula I in which R 1 and / or R 2 is hydrogen can be halogenated in a manner known per se. For example, you can find a solution in the React positions 6, 7, 8 and 9 of unsubstituted compound in acetic acid with bromine to the 7-bromo compound.
- R 1 and R 2 are different from an optionally alkylated amino group
- R 11 and R 21 have the same meanings as R 1 and R 2 with the exception of optionally alkylated amino and R 3 and R 4 have the above meaning.
- the compounds of the formula I can furthermore be prepared according to the formula scheme II given below, in which Z represents oxygen or sulfur, M ammonium, potassium or sodium and the remaining symbols have the above meaning.
- the compounds of the formula I, their tautomers and physiologically tolerable salts of such compounds are to be used as medicaments. They inhibit e.g. platelet aggregation and can therefore be used to prevent thrombosis. They are also effective in the circulation. Because of their positive inotropic effects, they can be used without significant tachycardia for the treatment and prophylaxis of heart failure and heart failure.
- the compounds of formula I and their tautomers can be used as medicaments e.g. in the form of pharmaceutical preparations which they or their salts are mixed with a pharmaceutical, organic or inorganic inert carrier material suitable for enteral, percutaneous or parenteral administration, such as e.g. Contain water, gelatin, gum arabic, milk sugar, starch, magnesium stearate, talc, vegetable oils, polyalkylene glycols, petroleum jelly, etc.
- the pharmaceutical preparations can be in solid form, e.g. as tablets, dragees, suppositories, capsules; in semi-solid form, e.g. as ointments; or in liquid form, e.g. as solutions, suspensions or emulsions.
- auxiliary substances such as preservatives, stabilizers, wetting agents or emulsifiers, salts for changing the osmotic pressure or buffers. They can also contain other therapeutically valuable substances. Oral administration of the compounds according to the invention is preferred. For adults, an oral daily dose of 0.5 to 30 mg / kg and a parenteral daily dose of 0.05 to 10 mg / kg are possible.
- the aggregation-inhibiting effect was determined by the aggregometer method of BORN [Nature 194, 927 (1962)] and MICHAL and BORN [Nature 231, 220 proven.
- the maximum rate of aggregation was taken as the test parameter and the effective concentration (EC 50 ) was determined from dose-response curves.
- Human plasma was obtained from venous blood decomposed with citrate (10.6 mM) by centrifugation. 0.18 ml of plasma were mixed with 10 ⁇ l of aqueous suspension of the test compounds, incubated for 10 minutes at 37 ° C., whereupon the aggregation was initiated by adding 10 ⁇ l of collagen-fibril suspension.
- Rabbit plasma was obtained from arterial blood decomposed with citrate (9 mM) by centrifugation. 1 ml of plasma was mixed with 10 ⁇ l of test solution and incubated for 1 minute at 37 ° C., whereupon 8 ⁇ l of collagen-fibril suspension or 10 ⁇ l of adenosine diphosphate (ADP) in 10 ⁇ 4 M saline solution were added. Plasma incubated with dimethyl sulfoxide was used as a control value.
- ADP adenosine diphosphate
- the positive inotropic effect was measured after oral administration of the test substances to awake German shepherds.
- the animals are equipped with an implanted pressure telemetry system, the pressure sensor being fixed in the left ventricle.
- the left ventricular pressure is transmitted from the animal via the implanted radio transmitter and received, demodulated and amplified via a suitable antenna and receiver system.
- LVP left ventricular pressure
- dLVP / dtmax maximum rate of pressure rise
- the heart rate is recorded on a cardiotachograph.
- Inotropy is the percentage. Change ( ⁇ %) of dLVP / dt max and the duration of action in minutes (min) are given.
- Tachycardia shows the percentage changes in heart rate ( ⁇ %) after administration of the test substance and the duration of action in minutes (min). The results are shown in Table II below.
- N- (2-amino-3-methylbenzyl) -D-alanine ethyl ester was used to convert D-1,5-dihydro-3,9-dimethylimidazo [2,1-b] quinazolin-2 (3H ) -On hydrochloride obtained. Melting point 270-275 ° (dec.). The free base melts at 262-265 °.
- N- (2-amino-6-methylbenzyl) -L-alanine ethyl ester was converted into L-1,5-dihydro-3,6-dimethyl-imidazo [2, lb] quinazolin-2 (3H ) -On hydrochloride obtained. Colorless crystals with a melting point of 285-288 ° (dec.). The free base melts above 340 ° with decomposition.
- N- (2-amino-6-methylbenzyl) -D-alanine ethyl ester became D-1,5-dihydro-3,6-dimethyl-imidazo [2,1-b] quinazolin-2 Obtained (3H) -one hydrochloride. Light yellow crystals with a melting point of 287-290 ° (Dec.). The free base melts above 340 °.
- N- (2-amino-6-methylbenzyl) -L-serine ethyl ester became L-1,5-dihydro-3-hydroxymethyl-6-methylimidazo [2,1-b] quinazoline -2 (3H) -one hydrochloride obtained. Yellow crystals with melting point 320-325 ° (dec.).
- N- (2-amino-6-methylbenzyl) -Da-phenylglycine-ethyl ester became D-1,5-dihydro-3-phenyl-6-methyl-imidazo [2,1-b] quinazoline Obtained -2 (3H) -one hydrochloride. Light yellow crystals with a melting point of about 320 ° (dec.).
- N- (2-chloro-6-nitrobenzyl) -D-alanine ethyl ester was obtained analogously from ethyl D-alanine and a-bromo-2-chloro-6-nitrotoluene, .
- N- (2-amino-6-chlorobenzyl) -D-alanine ethyl ester was obtained analogously by hydrogenation of N- (2-chloro-6-nitrobenzyl) - D-alanine ethyl ester, .
- Gelatin capsules of the following composition are produced in the usual way:
- a solution for injection of the following composition is prepared in the usual way:
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Hospice & Palliative Care (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
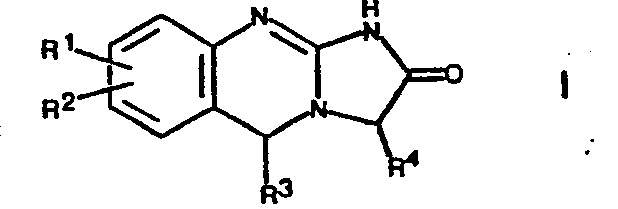
deren Tautomere und Salze solcher Verbindungen. Diese Verbindungen und Salze hemmen die Aggregation der Blutplättchen und haben positive inotrope Wirkund ohne wesentliche Tachycardie. Sie können in an sich bekannter Weise hergestellt werden, u.a. ausgehend von neuen N-(2-Aminobenzyl)- glycinderivaten.
Description
- Die vorliegende Erfindung betrifft neue tricyclische Verbindungen, nämlich Imidazo-chinazoline der Formel

- Der hier verwendete Ausdruck "nieder" bezieht sich vorzugsweise auf Reste mit 1 - 6, insbesondere 1 - 4 C-Atomen. Alkylreste können geradkettig oder verzweigt sein. Beispiele von Alkylresten sind: Methyl, Aethyl, Propyl, Isopropyl, Butyl, Isobutyl, Pentyl und Hexyl. Aryl bedeutet insbesondere Phenyl oder durch Halogen, nieder-Alkyl, Hydroxy und/oder nieder-Alkoxy substituiertes Phenyl.
- Unter den Verbindungen der Formel I sind diejenigen, die in D-Form vorliegen, bevorzugt. Bevorzugt sind ferner 3 die Verbindungen an Formel I, worin R und R Wasserstoff, R2 Halogen in 6- oder 7-Stellung oder nieder-Alkyl in 6-Stellung, insbesondere 6-Chlor, 7-Brom oder 6-Methyl, und R 4 nieder-Alkyl, insbesondere Methyl, darstellen, bevorzugt.
- Besonders bevorzugt sind
- D-6-Chlor-1,5-dihydro-3-methyl-imidazo[2,1-b]china- zolin-2(3H)-on,
- D-1,5-Dihydro-3,6-dimethyl-imidazo[2,1-b]chinazolin-2(3H)-on,
- D-7-Brom-l,5-dihydro-3-methyl-imidazo[2,l-b]china- zolin-2(3H)-on
- Beispiele von Verbindungen der Formel I sind
- L-6-Chlor-1,5-dihydro-3-hydroxymethyl-imidazo[2,1-b] chinazolin-2(3H)-on,
- L-6-Chlor-1,5-dihydro-3-phenyl-imidazo[2,1-b]china- zolin-2(3H)-on,
- L-6-Chlor-1,5-dihydro-3-isobutyl-imidazo[2,1-b] chinazolin-2(3H)-on,
- L-3-Benzyl-6-chlor-1,5-dihydro-imidazo[2,1-b] chinazolin-2(3H)-on
- Die Erfindung betrifft weiterhin ein Verfahren zur Herstellung der genannten Verbindungen sowie pharmazeutische Präparate auf der Basis der genannten Verbindungen.
- Die Verbindungen der Fprmel I können in verschiedenen tautomeren Formen vorliegen. Die Erfindung beschränkt sich daher nicht auf Verbindungen der oben dargestellten Formel I, sondern umfasst auch die Tautomeren, beispielsweise solche der Formel
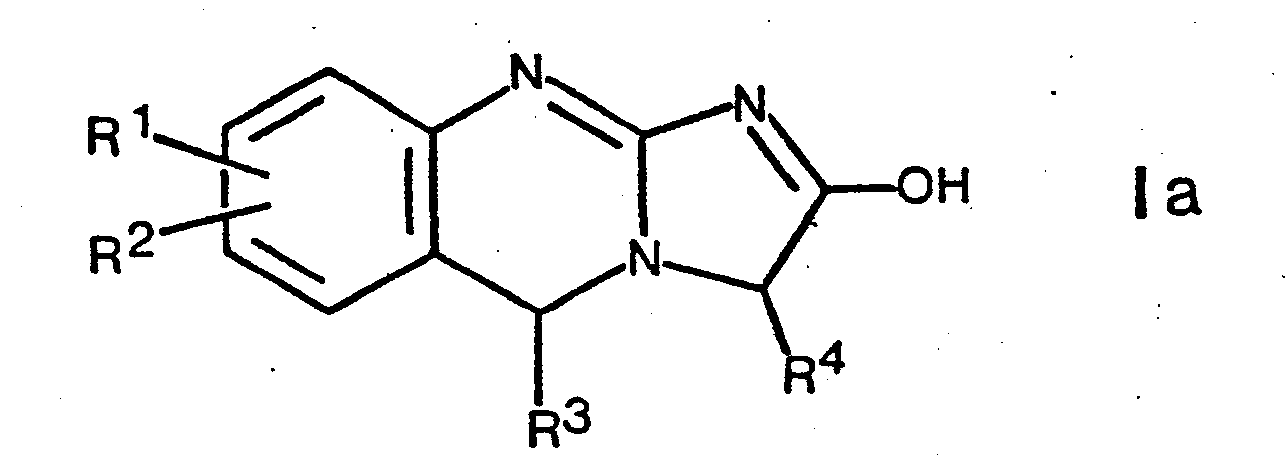

- Die Verbindungen der Formel I und deren Tautomeren, z.B. Ia und Ib können weiterhin in Form von Racematen oder in optisch aktiver Form vorliegen, wobei alle diese Formen Gegenstand der Erfindung sind.
- Beispiele physiologisch verträglicher Salze sind Mineralsäuresalze, wie Hydrochloride, Hydrobromide, Sulfate und Phosphate; Salze organischer Sulfonsäuren, wie Alkylsulfate und Arylsulfonate; und Carbonsäuresalze, wie Succinate, Citrate,Tartrate und Maleate.
- Die Verbindungen der Formel I und deren Tautomeren können erfindungsgemäss dadurch hergestellt werden, dass man
- a) eine Verbindung der Formel

- b) eine Verbindung der Formel

- Die Umsetzung einer Verbindung der Formel II mit Bromcyan wird zweckmässig unter Erwärmen in einem Lösungsmittel, wie einem niederen Alkohol, z.B. Aethanol durchgeführt. Die Umsetzung einer Verbindung der Formel III mit Ammoniak wird zweckmässig unter Erwärmen in einem Lösungsmittel, wie einem niederen Alkohol, z.B. Aethanol, und Wasser, durchgeführt.
- Eine Verbindung der Formel I, worin R 1 und/oder R 2 Wasserstoff ist, kann in an sich bekannter Weise halogeniert werden. So kann man z.B. eine Lösung einer in den Stellungen 6, 7, 8 und 9 unsubstituierten Verbindung in Essigsäure mit Brom zur 7-Bromverbindung umsetzen.
- Die Verbindungen der Formel I, worin R1 und R 2 von einer gegebenenfalls alkylierten Aminogruppe verschieden sind, können nach dem nachstehend wiedergegebenen Formelschema I hergestellt werden, worin Y Chlor oder Brom darstellt, R11 und R21 die gleichen Bedeutungen haben wie R 1 und R 2 mit Ausnahme von gegebenenfalls alkyliertem Amino und R3 und R4 obige Bedeutung haben.
- Die Verbindungen der Formel I können ferner nach dem nachstehend wiedergegebenen Formelschema II hergestellt werden, worin Z Sauerstoff oder Schwefel, M Ammonium, Kalium oder Natrium darstellen und die restlichen Symbole obige Bedeutung haben.


- Die Verbindungen der Formel II sind neu und als solche ebenfalls Gegenstand der vorliegenden Erfindung.
- Die Ausgangsverbindungen der Formeln II und III können nach dem nachstehend wiedergegebenen Formelschema III, worin X Halogen darstellt und die restlichen Symbole obige Be-deutung haben, bzw. in Analogie zu den in'den Beispielen angegebenen Methoden hergestellt werden.

- Die Verbindungen der Formel I, deren Tautomere und physiologisch verträgliche Salze solcher Verbindungen sollen als Heilmittel Verwendung finden. Sie hemmen z.B. die Aggregation der Blutplättchen und können daher zur Verhütung von Thrombosen verwendet werde-. Ausserdem sind sie kreislaufwirksam. So können sie auf Grund ihrer positiven inotropen Wirkung ohne wesentliche Tachycardie zur Behandlung und Prophylaxe von Herzversagen und Herzschwäche verwendet werden.
- Die Verbindungen der Formel I und deren Tautomere können als Heilmittel z.B. in Form pharmazeutischer Präparate Verwendung finden, welche sie oder ihre Salze in Mischung mit einem für die enterale, perkutane oder parenterale Applikation geeigneten pharmazeutischen, organischen oder anorganischen inerten Trägermaterial, wie z.B. Wasser, Gelatine, Gummi arabicum, Milchzucker, Stärke, Magnesiumstearat, Talk, pflanzliche Oele, Polyalkylenglykole, Vaseline, usw. enthalten. Die pharmazeutischen Präparate können in fester Form, z.B. als Tabletten, Dragees, Suppositorien, Kapseln; in halbfester Form, z.B. als Salben; oder in flüssiger Form, z.B. als Lösungen, Suspensionen oder Emulsionen, vorliegen. Gegebenenfalls sind sie sterilisiert und bzw. oder enthalten Hilfsstoffe, wie Konservierungs-, Stabilisierunga-, Netz- oder Emulgiermittel, Salze zur Veränderung des osmotischen Druckes oder Puffer. Sie können auch noch andere therapeutisch wertvolle Stoffe enthalten. Die orale Verabreichung der erfindungsgemässen Verbindungen ist bevorzugt. Für den Erwachsener kommen eine orale Tagesdosis von 0,5 bis 30 mg/kg und eine parenterale Tagesdosis von 0,05 bis 10 mg/kg in Frage.
- bie aggregationshemmende Wirkung wurde nach der Aggregometer-Methode von BORN [Nature 194, 927 (1962)] und MICHAL und BORN [Nature 231, 220nachgewiesen. Die maximale Aggregationsgeschwindigkeit wurde als Versuchsparameter genommen und die effektive Konzentration (EC50) aus Dosis-Wirkungskurven ermittelt.

- Menschliches Plasma wurde aus mit Citrat (10,6 mM) zersetztem venösem Blut durch Zentrifugieren erhalten. 0,18 ml Plasma wurden mit 10 µl wässriger Suspension der Testverbindungen versetzt, 10 Minuten bei 37°C inkubiert, worauf die Aggregation durch Zusatz von 10 µl Collagen-Fibril-Suspension eingeleitet wurde.
- Kaninchen-Plasma wurde aus mit Citrat (9 mM) zersetztem arteriellem Blut durch Zentrifugieren erhalten. 1 ml Plasma wurde mit 10 µl Test-Lösung versetzt und 1 Minute bei 37°C inkubiert, worauf 8 µl Collagen-Fibril-Suspension oder 10 µl Adenosindiphosphat (ADP) in 10-4 M Kochsalzlösung zugesetzt wurden. Als Kontrollwert diente mit Dimethylsulfoxid inkubiertes Plasma.
- Die Resultate sind in der nachstehenden Tabelle I wiedergegeben.
- Die positiv inotrope Wirkung wurde nach oraler Gabe der Prüfsubstanzen an wachen Schäferhunden gemessen. Die Tiere sind zu diesem Zweck mit einem implantierten Druck-Telemetrie-System ausgerüstet, wobei der Druckaufnehmer im linken Ventrikel fixiert ist. Der linksventrikuläre Druck wird über den implantierten Radiosender aus dem Tier gesendet und über ein geeignetes Antennen- und Empfängersystem empfangen, demoduliert und verstärkt. Durch Differenzierung des ansteigenden Schenkels des linksventrikulären Druckes (LVP) wird die maximale Druckanstiegsgeschwindigkeit (dLVP/dtmax) errechnet, was als Kontraktilitätsparameter gilt. Gleichzeitig wird die Herzfrequenz über einen Cardiotachographen aufgezeichnet. Unter Inotropie werden die prozentuale. Veränderung (Δ%) von dLVP/ dtmax und die Wirkungsdauer in Minuten (Min) angegeben.. Unter Tachycardie werden die prozentualen Veränderungen der Herzfrequenz (Δ%) nach Gabe der Prüfsubstanz und die Wirkungsdauer in Minuten (Min) angegeben. Die Resultate sind in der nachstehenden Tabelle II wiedergegeben.


- Die folgenden Beispiele erläutern die Erfindung. Die Temperaturen sind in °C angegeben.
- Zu einer Lösung von 11,8 g N-(2-Amino-3-methylbenzyl)-L-alanin-äthylester in 30 ml Aethanol wurde bei Raumtemperatur unter Rühren eine Lösung von.5,3 g Bromcyan in 10 ml Aethanol gegeben. Das Reaktionsgemisch wurde 1 Stunde zum Rückfluss erhitzt und dann unter vermindertem Druck zur Trockene eingedampft. Der Rückstand wurde mit 100 ml Wasser versetzt und durch Zusatz von 3 N Ammoniumhydroxyd unter Rühren alkalisch gestellt. Das Gemisch wurde dann weitere 30 Minuten gerührt und dreimal mit je 100 ml Methylenchlorid extrahiert. Die organischen Extrakte wurden zweimal mit je 150 ml Wasser gewaschen, über Natriumsulfat getrocknet und eingedampft. Der Rückstand wurde aus Aethanol umkristallisiert und lieferte L-1,5-Dihydro-3,9-dimethylimidazo[2,1-b]-chinazolin-2(3H)-on, Schmelzpunkt 259-261°, [α]D +15,5° (c = l% in Methanol).
- Durch Umkristallisation der so erhaltenen Base aus 1 N Salzsäure und Acetonitril (3:1) wurde das Hydrochlorid vom Schmelzpunkt 272-275° (Zers.) erhalten.
- Das Ausgangsmaterial wurde wie folgt hergestellt:
- Zu einer Lösung von 91,8 g L-Alanin-äthylester-hydrochlorid in 300 ml absolutem Aethanol wurde innerhalb von 30 Minuten eine Lösung von 120 ml Triäthylamin in 200 ml absolutem Aethanol getropft. Das Reaktionsgemisch wurde auf 60° erwärmt, wobei eine klare Lösung entstand. Zu dieser Lösung wurde innerhalb 1 Stunde eine Lösung von 55,5 g 3-(Chlormethyl)-2-nitrotoluol in 300 ml absolutem Aethanol getropft. Danach wurde die Temperatur auf 80° erhöht und das Reaktionsgemisch über Nacht bei dieser Temperatur gerührt. Danach wurde unter vermindertem Druck zur Trockene eingedampft und der Rückstand in 600 ml Wasser gelöst. Die Lösung wurde dreimal mit Methylenchlorid extrahiert und die Extrakte wurden nacheinander mit Wasser und mit gesättigter Natriumchloridlösung gewaschen, getrocknet und eingedampft. Das so erhaltene Rohprodukt wurde durch Chromatographie an Kieselgel mit Methylenchlorid/5% Methanol als Laufmittel gereinigt. Man erhielt N-(3-Methyl-2-nitrobenzyl)-L-alanin- äthylester als gelbes Oel, [α]D -36° (c = 1% in Methanol).
- Eine Lösung von 26,6 g N-(3-Methyl-2-nitrobenzyl)-L-alanin-äthylester in 100 ml absolutem Aethanol wurde in Gegenwart von 2 g 10%igem Pd/C hydriert. In 5 Stunden wurden 6,7 1 Wasserstoff aufgenommen. Nach beendeter Hydrierung wurde vom Katalysator abfiltriert und das Filtrat zur Trockene eingedampft. Man erhielt N-(2-Amino-3-methylbenzyl)-L-alanin-äthylester als gelbes Oel, [α]D -52,6° (c = 1% in Methanol).
- Analog wurden folgende Verbindungen hergestellt:
- aus 3-(Chlormethyl)-2-nitrotoluol und D-Alanin-äthylester-hydrochlorid der N-(3-methyl-2-nitrobenzyl)-D-alanin- äthylester, gelbes Oel, [α]D + 31,4° (c = 1% in Methanol);
- aus α3-Chlor-4-nitro-m-xylol und L-Alanin-äthylesterhydrochlorid der N-(5-Methyl-2-nitrobenzyl)-L-alanin-äthylester, rotes Oel, [α]D -12,6° (c = 1% in Methanol);
- aus α3-Chlor-4-nitro-m-xylol und D-Alanin-äthylesterhydrochlorid der N-(5-Methyl-2-nitrobenzyl)-D-alanin-äthylester, rotes Oel, [α]D + 11,4° (c = 1% in Methanol);
- aus α2-Chlor-3-nitro-o-xylol und L-Alanin-äthylesterhydrochlorid der N-(2-Methyl-6-nitrobenzyl)-L-alanin-äthylester, rotes Oel, [α]D +35,8° (c = 1% in Methanol);
- aus α2-Chlor-3-nitro-o-xylol und D-Alanin-äthylesterhydrochlorid der N-(2-Methyl-6-nitrobenzyl)-D-alanin-äthylester, rotes Oel, [α]D -34° (c = 1% in Methanol);
- aus α2-Chlor-3-nitro-o-xylol und L-Serin-äthylesterhydrochlorid der N-(2-Methyl-6-nitrobenzyl)-L-serin-äthylester, rotes Oel,

- aus a -Chlor-3-nitro-o-xylol und D-a-Phenylglycin- äthylester-hydrochlorid der N-(2-Methyl-6-nitrobenzyl)-D-a-phenylglycin-äthylester, rotes Oel,
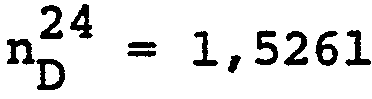
- aus 2-Nitrobenzylchlorid und L-Alanin-äthylester-hydrochlorid der 2-Nitrobenzyl-L-alanin-äthylester, dunkelrotes Oel, [a]D -5,4° (c = l% in Aethanol);
- aus 2-Nitrobenzylchlorid und D-Alaniri-äthylester-hydrochlorid der (2-Nitrobenzyl)-D-alanin-äthylester, rotes Oel, [a]D +5,4° (c = 1% in Aethanol);
- aus N-3-Methyl-2-nitrobenzyl-D-alanin-äthylester der N-(2-Amino-3-methylbenzyl)-D-alanin-äthylester, hellgelbes Oel, [α]D +51° (c = 1% in Methanol);
- aus N-(5-Methyl-2-nitrobenzyl)-L-alanin-äthylester der N-(2-Amino-5-methylbenzyl)-L-alanin-äthylester, [a]D -45° (c = 1% in Methanol);
- aus N-(5-Methyl-2-nitrobenzyl)-D-alanin-äthylester der N-(2-Amino-5-methylbenzyl)-D-alanin-äthylester, rotes Oel, [a]D +34,2° (c = 1% in Methanol);
- aus N-(2-Methyl-6-nitrobenzyl)-L-alanin-äthylester der N-(2-Amino-6-methylbenzyl)-L-alanin-äthylester, gelbes Oel, [α]D -34,7° (c = 1% in Methanol);
- aus N-(2-Methyl-6-nitrobenzyl)-D-alanin-äthylester der N-(2-Amino-6-methylbenzyl)-D-alanin-äthylester, rötliches Oel, [α]D +36,8° (c = 1% in Methanol);
- aus N-(2-Methyl-6-nitrobenzyl)-L-serin-äthylester der N-(2-Amino-6-methylbenzyl)-L-serin-äthylester, rotes Oel,

- aus N-(2-Methyl-6-nitrobenzyl)-D-α-phenylglycin-äthylester der N-(2-Amino-6-methylbenzyl)-D-a-phenylglycin- äthylester, gelbes Oel,

- aus (2-Nitrobenzyl)-L-alanin-äthylester der 2-Aminobenzyl-L-alanin-äthylester, rotes Oel, [a]D -55,1° (c = 1% in Aethanol);
- aus 2-Nitrobenzyl-D-alanin-äthylester der 2-Aminobenzyl-D-alanin-äthylester, dunkelrotes Oel, [a]D +57,20 (c = 1% in Aethanol).
- In Analogie zu Beispiel 1 wurde aus N-(2-Amino-3-methylbenzyl)-D-alanin-äthylester das D-1,5-Dihydro-3,9-dimethylimidazo[2,1-b]chinazolin-2(3H)-on-hydrochlorid erhalten. Schmelzpunkt 270-275° (Zers.). Die freie Base schmilzt bei 262-265°.
- In Analogie zu Beispiel 1 wurde aus N-(2-Amino-5- . methylbenzyl)-L-alanin-äthylester das L-1,5-Dihydro-2,7-dimethyl-imidazo[2,l-b]chinazolin-2(3H)-on-hydrochlorid erhalten. Hellgelbe Kristalle, Schmelzpunkt 173-176°. Die freie Base schmilzt unter Zersetzung über 300°.
- In Analogie zu Beispiel 1 wurde aus N-(2-Amino-5-methylbenzyl)-D-alanin-äthylester das D-1,5-Dihydro-3,7-dimethyl-imidazo[2,1-b]chinazolin-2(3H)-on-hydrochlorid erhalten. Hellgelbe Kristalle, Schmelzpunkt 173-1760 (Zers.). Die freie Base schmilzt unter Zersetzung bei 310-314°.
- In Analogie zu Beispiel 1 wurde aus N-(2-Amino-6-methylbenzyl)-L-alanin-äthylester das L-1,5-Dihydro-3,6-dimethyl-imidazo[2,l-b]chinazolin-2(3H)-on-hydrochlorid erhalten. Farblose Kristalle vom Schmelzpunkt 285-288° (Zers.). Die freie Base schmilzt oberhalb 340° unter Zersetzung.
- In Analogie zu Beispiel 1 wurde aus N-(2-Amino-6-methylbenzyl)-D-alanin-äthylester das D-1,5-Dihydro-3,6-dimethyl-imidazo[2,1-b]chinazolin-2(3H)-on-hydrochlorid erhalten. Hellgelbe Kristalle vom Schmelzpunkt 287-290° (Zers.). Die freie Base schmilzt oberhalb 340°.
- In Analogie zu Beispiel 1 wurde aus N-(2-Amino-6-methylbenzyl)-L-serin-äthylester das L-1,5-Dihydro-3-hydroxymethyl-6-methyl-imidazo[2,1-b]chinazolin-2(3H)-on- hydrochlorid erhalten. Gelbe Kristalle vom Schmelzpunkt 320-325° (Zers.).
- In Analogie zu Beispiel 1 wurde aus N-(2-Amino-6-methylbenzyl)-D-a-phenylglycin-äthylester das D-1,5-Dihydro-3-phenyl-6-methyl-imidazo[2,1-b]chinazolin-2(3H)-on-hydrochlorid erhalten. Hellgelbe Kristalle vom Schmelzpunkt etwa 320° (Zers.).
- In Analogie zu Beispiel 1 wurde aus 2-Amino-benzyl-L-alanin-äthylester das L-1,5-Dihydro-3-methyl-imidazo[2,1-b]-chinazolin-2(3H)-on-hydrochlorid erhalten. Braune Kristalle vom Schmelzpunkt 223-226°. Die freie Base schmilzt bei 300-305° unter Zersetzung.
- In Analogie zu Beispiel 1 wurde aus 2-Amino-benzyl-D-alanin-äthylester das D-1,5-Dihydro-3-methyl-imidazo[2,1-b]-chinazölin-2(3H)-on-hydrochlorid erhalten. Gelbe Kristalle vom Schmelzpunkt 225-227°. Die freie Base schmilzt bei etwa 300° unter Zersetzung.
- Zu einer Lösung von 11,9 g N-(2-Amino-6-chlorbenzyl)-L-alaninäthylester in 20 ml Aethanol wurde bei Zimmertemperatur unter Rühren eine Lösung von 5 g Bromcyan in 20 ml Aethanol zugetropft. Dann wurde das Reaktionsgemisch 1 Stunde am Rückfluss gekocht, zur Trockene eingedampft, der Rückstand mit 150 ml Wasser versetzt und unter Rühren mit 3N NH4OH alkalisch gestellt. Nach 30 Minuten Rühren wurde die Fällung abfiltriert und aus 1N HC1 und Acetonitril umkristallisiert. Man erhielt 9,1 g (68% d.Th.) L-6-Chlor-1,5-dihydro-3-methylimidazo[2,1-b]chinazolin-2(3H)-on-hydrochlorid als gelbe Kristalle vom Smp. 260-263°, [α]D +34,2° (DMSO).
- D-6-Chlor-1,5-dihydro-3-methylimidazo[2,1-b]china- zolin-2(3H)-on-hydrochlorid wurde analogerweise aus N-(2-Amino-6-chlorbenzyl)-D-alanin-äthylester erhalten, Smp. 263-266°, [a]D -23,9° (DMSO); Smp. der freien Base 275-280°.
- Das Ausgangsmaterial kann wie folgt hergestellt werden:
- 18,24 g L-Alaninäthylester-hydrochlorid in 60 ml Aethanol wurde eine Mischung von 25 ml Triäthylamin in 60 ml Aethanol zugetropft und das Gemisch auf 80° erhitzt. Der entstandenen Lösung wurde bei dieser Temperatur eine Lösung von 15 g a-Brom-2-chlor-6-nitrotoluol in 60 ml Aethanol zugetropft. Das Gemisch wurde über Nacht bei 80° gerührt und dann zur Trockene eingedampft, der Rückstand mit 150 ml ionenfreiem Wasser versetzt und zweimal mit 100 ml Methylenchlorid extrahiert. Die Methylenchloridextrakte wurden mit Wasser gewaschen, getrocknet und eingedampft. Das so erhaltene Produkt wurde durch Chromatographie an Kieselgel mit Methylenchlorid/5% Methanol gereinigt. Man erhielt 15,75 g (91% d.Th.) N-(2-Chlor-6-nitrobenzyl-L-alaninäthylester,

- N-(2-Chlor-6-nitrobenzyl)-D-alaninäthylester wurde analogerweise aus D-Alaninäthylester und a-Brom-2-chlor- 6-nitrotoluol erhalten,

- Eine Lösung von 14,3 g N-(2-Chlor-6-nitrobenzyl)-L-alaninäthylester in 50 ml absolutem Aethanol wurde in Gegenwart von 1 g Raney-Nickel hydriert. Nach beendeter Hydrierung wurde vom Katalysator abfiltriert und das Filtrat zur Trockene eingedampft. Man erhielt 12,6 g (99% d.Th.) N-(2-Amino-6-chlorbenzyl)-L-alaninäthylester,

- N-(2-Amino-6-chlorbenzyl)-D-alaninäthylester wurde analogerweise durch Hydrierung von N-(2-Chlor-6-nitrobenzyl)- D-alaninäthylester erhalten,

- In zu Beispiel 11 analoger Weise wurden folgende Verbindungen hergestellt:
- D-6,7-Dichlor-1,5-dihydro-3-methylimidazo[2,1-b] chinazolin-2(3H)-on-hydrochlorid, Smp. >280°,
- L-6,7-Dichlor-1,5-dihydro-3-methylimidazo[2,1-b]-chinazolin-2(3H)-on-hydrochlorid, Smp. > 290°,
- D-7-Brom-1,5-dihydro-3-methylimidazo[2,1-b]chinazolin-2(3H)-on-hydrochlorid, Smp. 268-270°,
- L-7-Brom-1,5-dihydro-3-methylimidazo[2,1-b]chinazolin-2(3H)-on-hydrochlorid, Smp. 280-284° (Zers.),
- L-6-Chlor-7-methoxy-1,5-dihydro-3-methylimidazo[2,1-b] chinazolin-2(3H)-on-hydrochlorid, Smp. >280°.
- In zu Beispiel 11 analoger Weise wurden folgende Verbindungen hergestellt:
- aus N-(2-Chlor-6-nitrobenzyl)-3-phenyl-D-alanin- äthylester, [α]D -21,2° (c = 1% in Aethanol) über N-(2- Amino-6-chlorbenzyl)-3-phenyl-D-alanin-äthylester, [a]D +40,7° (c = 1% in Aethanol), das D-3-Benzyl-6-chlor-1,5-dihydroimidazo[2,1-b]chinazolin-2(3H)-on-hydrochlorid, Smp. 260-265° (Zers.); Smp. der Base 270-275° (Zers.);
- aus N-(2-Chlor-6-nitrobenzyl)-D-leucin-äthylester über N-(2-Amino-6-chlorbenzyl)-D-leucin-äthylester, [α]D +8,5° (c = 1% in Aethanol) das D-6-Chlor-1,5-dihydro-3-isobutylimidazo [2,1-b]chinazolin-2(3H)-on-hydrochlorid, Smp. 290-293°; Smp. der Base 280-285°;
- aus N-(2-Chlor-6-nitrobenzyl)-D-serin-äthylester, [a]D 2,7° (c = 1% in Aethanol), über N-2-(Amino-6-chlorbenzyl)-D-serin-äthylester, Smp. 73-75°, [a]D +65,5° (c = 1% in Aethanol) das D-6-Chlor-3-hydroxymethyl-l,5-dihydro- imidazo[2,1-b]chinazolin-2(3H)-on-hydrochlorid, Smp. der Base > 300° (Zers.);
- aus D-N-(2-Chlor-6-nitrobenzyl)-2-phenylglycin- äthylester, [a]D -21° (c = 1% in Aethanol), über D-N-(2-Amino-6-chlorbenzyl)-2-phenylglycin-äthylester, [a]D -4,5° (c = l% in Aethanol) das D-6-Chlor-3-phenyl-1,5-dihydroimidazo[2,1-b]chinazolin-2(3H)-on-hydrochlorid, Smp. > 300° (Zers.); Smp. der Base 260-265° (Zers.).
- Ein Gemisch von 6 g Aethyl-D-2,5-dichlor-a-methyl-3-(4H)-chinazolin-acetat, 20 ml absolutem Aethanol und 25 ml 5%igem alkoholischem Ammoniak wurde über Nacht im Druckrohr auf 110° erhitzt. Das Druckrohr wurde im Eisbad abgekühlt und geöffnet. Der entstandene Kristallbrei wurde abgenutscht und mit kaltem Aethanol gewaschen. Die erhaltenen Kristalle wurden in 1N Salzsäure gelöst und filtriert. Das Filtrat wurde zur Trockene eingedampft und der Rückstand aus 1N Salzsäure und Acetonitril als D-6-Chlor-1,5-dihydro-3-methyl-imidazo[2,1-b]-chinazolin-2(3H)-on-hydrochlorid umkristallisiert (4,3 g, 74% d.Th.), Smp. 275-278°.
- Das Ausgangsmaterial kann wie folgt hergestellt werden:
- Einem Gemisch von 18,24 g D-Alaninäthylester-hydrochlorid in 60 ml Aethanol wurde ein Gemisch von 25 ml Triäthylamin in 60 ml Aethanol zugetropft und das Reaktionsgemisch auf 80° erhitzt. Der entstandenen Lösung wurde bei dieser Temperatur eine Lösung von 15 g 2-Chlor-6-nitro- benzylbromid in 60 ml Aethanol zugetropft. Das Gemisch wurde über Nacht bei 800 gerührt, dann zur Trockene eingedampft, der Rückstand mit 150 ml ionenfreiem Wasser versetzt und zweimal mit 100 ml Methylenchlorid extrahiert. Die Methylenchloridextrakte wurden mit Wasser gewaschen, über Natriumsulfat getrocknet und eingedampft. Das so erhaltene Produkt wurde durch Chromatographie an Kieselgel mit Methylenchlorid/5% Methanol gereinigt. Man erhielt 16 g (93% d.Th.) N-(2-Chlor-6-nitrobenzyl)-D-alaninäthylester,

- Eine Lösung von 14,3 g N-(2-Chlor-6-nitrobenzyl)-D-alaninäthylester in 50 ml Aethanol wurde in Gegenwart von 1 g Raney-Nickel hydriert. In 2 Stunden wurden 3,35 1 Wasserstoff aufgenommen. Dann wurde vom Katalysator abfiltriert und das Filtrat zur Trockene eingedampft. Man erhielt 12,6 g (99% d.Th.) N-(2-Amino-6-chlorbenzyl)-D-alaninäthylester,

- Zu einer Lösung von 71,75 g N-(2-Amino-6-chlorbenzyl)-D-alaninäthylester in 400 ml trockenem Tetrahydrofuran wurden unter Stickstoffbegasung und Rühren 52 g N,N'-Carbonyldiimidazol portionsweise zugegeben. Das Gemisch wurde 2 Stunden gerührt und 18 Stunden unter Rückfluss erhitzt und zur Trockene eingedampft und der Rückstand mit 1500 ml Methylenchlorid extrahiert, die organische Phase mit zweimal 400 ml 1N Salzsäure und dann mit 400 ml Wasser gewaschen, getrocknet und eingedampft. Das so erhaltene Oel wurde durch Chromatographie an Kieselgel mit Methylenchlorid/5% Methanol gereinigt. Ausbeute: 79 g (99% d.Th.) Aethyl-D-5-chlor-1,4-dihydro-a-methyl-2-oxo-3(2H)-chinazolinacetat,

- 50,9 g Aethyl-D-5-chlor-1,4-dihydro-α-methyl-2-oxo-3(2H)-chinazolinacetat wurden in 135 ml Phosphoroxychlorid gelöst und 3 Stunden unter Rühren auf 110° erhitzt. Nach dem Kühlen wurde das Reaktionsgemisch zur Trockene eingedampft, der Rückstand in 250 ml Chloroform gelöst, die Lösung mit 300 ml Eiswasser verdünnt und durch tropfenweise Zugabe von 40%igem Natriumhydroxyd bis auf pH 7-8 eingestellt. Die Chloroformphase wurde abgetrennt, getrocknet und eingedampft. Das Produkt wurde durch Chromatographie an Kieselgel mit Methylenchlorid/5% Methanol gereinigt. Ausbeute: 37,4 g (70% d.Th.) Aethyl-D-2,5-dichlor-α-methyl-3(4H)-chinazolin- acetat,

- Einer Lösung von 5 g D-l,5-Dihydro-3-methylimidazo-[2,l-b]chinazolin-2(3H)-on in 80 ml Eisessig werden tropfenweise 1,5 ml Brom zugesetzt. Das Gemisch wird 1 1/2 Stunden bei Raumtemperatur gerührt, mit 100 ml Wasser verdünnt, auf 30 ml eingeengt, nochmals mit 100 ml Wasser verdünnt, mit 3N Ammoniumhydroxyd alkalisch gestellt, gewaschen und filtriert. Das ausgefallene Produkt wird mit Wasser gewaschen und aus 100 ml 2N HCl umkristallisiert. Man erhält 3,9 g (56%) D-7-Brom-1,5-dihydro-3-methyl-imidazo[2,1-b]chinazolin-2(3H)-on-hydrochlorid, Smp. 268-270°.
- In zu Beispiel 15 analoger Weise werden 4,7 g L-1,5-Dihydro-3-methylimidazo[2,1-b]chinazolin-2(3H)-on zu 4,2 g (64%) L-7-Brom-1,5-dihydro-3-methyl-imidazo[2,1-b]china- zolin-2(3H)-on-hydrochlorid, Smp. 280-284° (Zers.) bromiert.
- In üblicher Weise werden Tabletten folgender Zusammensetzung hergestellt:

- In üblicher Weise werden Gelatinesteckkapseln folgender Zusammensetzung hergestellt:

- In üblicher Weise wird eine Injektionslösung folgender Zusammensetzung hergestellt:

Claims (14)





Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| LU77829 | 1977-07-25 | ||
| LU77829 | 1977-07-25 | ||
| CH5776/78 | 1978-05-26 | ||
| CH577678 | 1978-05-26 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| EP0000718A2 true EP0000718A2 (de) | 1979-02-21 |
| EP0000718A3 EP0000718A3 (en) | 1979-06-13 |
| EP0000718B1 EP0000718B1 (de) | 1982-03-24 |
Family
ID=25698353
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP78100471A Expired EP0000718B1 (de) | 1977-07-25 | 1978-07-21 | Neue Chinazolinderivate, Verfahren zu ihrer Herstellung, pharmazeutische Präparate und deren Herstellung |
Country Status (29)
| Country | Link |
|---|---|
| US (1) | US4256748A (de) |
| EP (1) | EP0000718B1 (de) |
| JP (1) | JPS5441894A (de) |
| AR (1) | AR218500A1 (de) |
| AT (1) | AT363479B (de) |
| AU (1) | AU519688B2 (de) |
| BR (1) | BR7804763A (de) |
| CA (1) | CA1094555A (de) |
| CS (1) | CS203014B2 (de) |
| DE (2) | DE2832138A1 (de) |
| DK (1) | DK144128C (de) |
| ES (2) | ES471981A1 (de) |
| FI (1) | FI63409C (de) |
| FR (1) | FR2398748A1 (de) |
| GB (1) | GB2001638B (de) |
| GR (1) | GR72968B (de) |
| HU (1) | HU177643B (de) |
| IE (1) | IE47280B1 (de) |
| IL (1) | IL55183A (de) |
| IT (1) | IT1097337B (de) |
| MC (1) | MC1199A1 (de) |
| MY (1) | MY8500249A (de) |
| NL (1) | NL7807507A (de) |
| NO (1) | NO150800C (de) |
| NZ (1) | NZ187921A (de) |
| PH (1) | PH14642A (de) |
| PT (1) | PT68342A (de) |
| SE (1) | SE7808111L (de) |
| YU (1) | YU177578A (de) |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0021338A1 (de) * | 1979-06-20 | 1981-01-07 | F. HOFFMANN-LA ROCHE & CO. Aktiengesellschaft | Neue Chinazolinderivate und pharmazeutische Präparate |
| EP0054180A3 (de) * | 1980-12-16 | 1982-12-01 | F. HOFFMANN-LA ROCHE & CO. Aktiengesellschaft | Verfahren zur Herstellung eines Imidazochinazolinonderivates |
| EP0116948A3 (en) * | 1983-02-16 | 1984-12-19 | Syntex (U.S.A.) Inc. | (2-oxo-1,2,3,5-tetrahydroimidazo-(2,1-b)quinazolinyl)-oxyalkyl-amides |
| EP0129258A1 (de) * | 1983-06-21 | 1984-12-27 | Daiichi Seiyaku Co., Ltd. | Imidazochinazolinverbindung |
| EP0133234A3 (en) * | 1983-07-14 | 1985-10-30 | Daiichi Seiyaku Co. Ltd. | Imidazoquinazolin-2-one compounds process for their production and pharmaceutical compositions containing said compounds |
| US4663320A (en) * | 1983-02-16 | 1987-05-05 | Syntex (U.S.A.) Inc. | (2-oxo-1,2,3,5-tetrahydroimidazo[2,1-b]quinoazolinyl)oxyalkylamides, compositions and the use thereof |
| US4670434A (en) * | 1985-11-14 | 1987-06-02 | Syntex (U.S.A.) Inc. | (2-oxo-3-methylene-1,2,3,5-tetrahydroimidazo[2,1-b]quinazolinyl)oxyalkylamides useful as cyclic AMP phosphodiesterase inhibitors |
| EP0153152A3 (de) * | 1984-02-15 | 1987-07-15 | Syntex (U.S.A.) Inc. | (2-Oxo-1,2,3,5-tetrahydroimidazo[2,1-b]chinazolinyl)oxyalkylamide, ihre Herstellung, sie enthaltende Zusammensetzungen und ihre Verwendung bei der Herstellung von Medikamenten |
| EP2088154A1 (de) | 2004-03-09 | 2009-08-12 | Ironwood Pharmaceuticals, Inc. | Verfahren und Zusammensetzungen zur Behandlung von Magen-Darm-Störungen |
| WO2011069038A2 (en) | 2009-12-03 | 2011-06-09 | Synergy Pharmaceuticals, Inc. | Agonists of guanylate cyclase useful for the treatment of hypercholesterolemia, atherosclerosis, coronary heart disease, gallstone, obesity and other cardiovascular diseases |
| WO2012118972A2 (en) | 2011-03-01 | 2012-09-07 | Synegy Pharmaceuticals Inc. | Process of preparing guanylate cyclase c agonists |
| WO2013138352A1 (en) | 2012-03-15 | 2013-09-19 | Synergy Pharmaceuticals Inc. | Formulations of guanylate cyclase c agonists and methods of use |
| WO2014131024A2 (en) | 2013-02-25 | 2014-08-28 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase and their uses |
| WO2014151200A2 (en) | 2013-03-15 | 2014-09-25 | Synergy Pharmaceuticals Inc. | Compositions useful for the treatment of gastrointestinal disorders |
| WO2014151206A1 (en) | 2013-03-15 | 2014-09-25 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase and their uses |
| EP2810951A2 (de) | 2008-06-04 | 2014-12-10 | Synergy Pharmaceuticals Inc. | Für die Behandlung von gastrointestinalen Erkrankungen, Entzündungen, Krebs und anderen Erkrankungen geeignete Agonisten von Guanylatcyclase |
| WO2014197720A2 (en) | 2013-06-05 | 2014-12-11 | Synergy Pharmaceuticals, Inc. | Ultra-pure agonists of guanylate cyclase c, method of making and using same |
| WO2015021358A2 (en) | 2013-08-09 | 2015-02-12 | Dominique Charmot | Compounds and methods for inhibiting phosphate transport |
| EP2998314A1 (de) | 2007-06-04 | 2016-03-23 | Synergy Pharmaceuticals Inc. | Für die behandlung von gastrointestinalen erkrankungen, entzündungen, krebs und anderen erkrankungen geeignete agonisten von guanylatcyclase |
| US20170204492A1 (en) * | 2014-08-08 | 2017-07-20 | Jfe Steel Corporation | Cold-rolled steel sheet having excellent spot weldability, and manufacturing method therefor |
| EP3241839A1 (de) | 2008-07-16 | 2017-11-08 | Synergy Pharmaceuticals Inc. | Zur behandlung von erkrankungen des magen-darm-trakts, entzündlichen erkrankungen, krebs und anderen erkrankungen geeignete agonisten von guanylatcyclase |
| WO2020237096A1 (en) | 2019-05-21 | 2020-11-26 | Ardelyx, Inc. | Combination for lowering serum phosphate in a patient |
| EP4201403A1 (de) | 2021-12-21 | 2023-06-28 | Som Innovation Biotech, S.L. | Verbindungen tirapazamin und quazinon zur verwendung bei der behandlung von gm2-gangliosidosen |
Families Citing this family (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL7807507A (nl) | 1977-07-25 | 1979-01-29 | Hoffmann La Roche | Tricyclische verbindingen. |
| US4146718A (en) * | 1978-04-10 | 1979-03-27 | Bristol-Myers Company | Alkyl 5,6-dichloro-3,4-dihydro-2(1h)-iminoquinazoline-3-acetate hydrohalides |
| EP0029559A3 (de) * | 1979-11-24 | 1981-09-30 | Sandoz Ag | 5,10 Dihydroimidazo (2,1-b) chinazoline, ihre Herstellung und sie enthaltende pharmazeutische Zusammensetzungen |
| US4444768A (en) * | 1980-03-04 | 1984-04-24 | Ciba-Geigy Corporation | Pyrimido[1,6-a]indoles, pharmaceutical preparations containing them, and methods of treating pain and inflammation with them |
| ZW16481A1 (en) * | 1980-08-15 | 1982-03-10 | Hoffmann La Roche | Novel imidazoquinazoline derivatives |
| JPS57178234U (de) * | 1981-05-06 | 1982-11-11 | ||
| US4455311A (en) * | 1981-08-28 | 1984-06-19 | Hoffmann-La Roche Inc. | Imidazoquinazoline derivatives which inhibit the aggregation of blood platelets, inhibit gastric secretion or have activity on the circulatory system |
| US4551459A (en) * | 1983-02-16 | 1985-11-05 | Syntex (U.S.A.) Inc. | Method of treating heart failure using (2-oxo-1,2,3,5-tetrahydroimidazo-[2,1-b]quinazolinyl)oxyalkylamides |
| GB8514207D0 (en) * | 1985-06-05 | 1985-07-10 | Pfizer Ltd | Cardiac stimulants |
| US4783467A (en) * | 1985-06-05 | 1988-11-08 | Pfizer Inc. | Tetrahydroimidazoquinazolinone inotropic agents |
| US4837239A (en) * | 1985-08-23 | 1989-06-06 | Syntex (U.S.A.) Inc. | Cardiotonic phosphodiesterase inhibitors complexed with water soluble vitamins |
| US4775674A (en) * | 1986-05-23 | 1988-10-04 | Bristol-Myers Company | Imidazoquinolinylether derivatives useful as phosphodiesterase and blood aggregation inhibitors |
| US4701459A (en) * | 1986-07-08 | 1987-10-20 | Bristol-Myers Company | 7-amino-1,3-dihydro-2H-imidazo[4,5-b]quinolin 2-ones and method for inhibiting phosphodiesterase and blood platelet aggregation |
| US5043327A (en) * | 1989-07-18 | 1991-08-27 | Janssen Pharmaceutica N.V. | Positive inotropic and lusitropic 3,5-dihydroimidazo[2,1-b]quinazolin-2(1H)-one derivatives, compositions and use |
| US4943573A (en) * | 1989-11-01 | 1990-07-24 | Bristol-Myers Squibb Company | Imidazo[4,5-b]quinolinyloxyalkanoic acid amides with enhanced water solubility |
| WO1993003034A1 (en) * | 1991-07-29 | 1993-02-18 | Warner-Lambert Company | Quinazoline derivatives as acetylcholinesterase inhibitors |
| US5208237A (en) * | 1992-04-03 | 1993-05-04 | Bristol-Meyers Squibb Company | 7-oxypropylsulfonamido-imidazo[4,5-b]quinolin-2-ones |
| US5348960A (en) * | 1992-04-03 | 1994-09-20 | Bristol-Myers Squibb Company | Imidazo[4,5-b]quinolinyl oxy alkyl tetrazolyl piperidine derivatives |
| US5196428A (en) * | 1992-04-03 | 1993-03-23 | Bristol-Myers Squibb Company | Imidazo[4,5-b]qinolinyl oxy alkyl ureas |
| US5158958A (en) * | 1992-04-03 | 1992-10-27 | Bristol-Myers Squibb Company | Imidazo[4,5-b]quinolinyl oxy alkyl sulfonyl piperidine derivatives |
| JPH06167328A (ja) * | 1992-05-26 | 1994-06-14 | Mayekawa Mfg Co Ltd | 氷厚測定装置 |
| US6585995B1 (en) * | 1999-09-21 | 2003-07-01 | Hanson Stephen R | Methods and compositions for treating platelet-related disorders |
| US6388073B1 (en) * | 2000-07-26 | 2002-05-14 | Shire Us Inc. | Method for the manufacture of anagrelide |
| HRP20050633A2 (en) * | 2003-01-23 | 2006-06-30 | Shire Holdings Ag | Formulation and methods for the treatment of thrombocythemia |
| US20060030574A1 (en) * | 2004-08-04 | 2006-02-09 | Shire Holdings Ag | Quinazoline derivatives useful for the treatment of peripheral arterial disease and as phosphodiesterase inhibitors |
| US7700608B2 (en) * | 2004-08-04 | 2010-04-20 | Shire Holdings Ag | Quinazoline derivatives and their use in the treatment of thrombocythemia |
| US8383637B2 (en) | 2004-08-06 | 2013-02-26 | Jansssen Pharmaceutica N.V. | 2-amino-quinazoline derivatives useful as inhibitors of β-secretase (BACE) |
| US8426429B2 (en) | 2004-08-06 | 2013-04-23 | Jansssen Pharmaceutica N.V. | 2-amino-quinazoline derivatives useful as inhibitors of β-secretase (BACE) |
| US8436006B2 (en) | 2004-08-06 | 2013-05-07 | Jansssen Pharmaceutica N.V. | 2-amino-quinazoline derivatives useful as inhibitors of β-secretase (BACE) |
| US8304420B2 (en) | 2006-11-28 | 2012-11-06 | Shire Llc | Substituted quinazolines for reducing platelet count |
| US7910597B2 (en) * | 2006-11-28 | 2011-03-22 | Shire Llc | Substituted quinazolines |
| GB0623750D0 (en) * | 2006-11-28 | 2007-01-10 | Shire Llc | Substituted quinazolines |
| GB0808948D0 (en) * | 2008-05-16 | 2008-06-25 | Shire Llc | Substituted quinazolines |
| GB0808944D0 (en) * | 2008-05-16 | 2008-06-25 | Shire Llc | Substituted quinazolines |
| GB0808947D0 (en) * | 2008-05-16 | 2008-06-25 | Shire Llc | Substituted quinazolines |
| GB0808950D0 (en) * | 2008-05-16 | 2008-06-25 | Shire Llc | Substituted quinazolines |
| GB0808967D0 (en) * | 2008-05-16 | 2008-06-25 | Shire Llc | Substitute quinazolines |
| GB0808951D0 (en) * | 2008-05-16 | 2008-06-25 | Shire Llc | Substituted quinazolines |
| GB0808968D0 (en) * | 2008-05-16 | 2008-06-25 | Shire Llc | Substitute quinazolines |
| GB0808952D0 (en) * | 2008-05-16 | 2008-06-25 | Shire Llc | Substituted quinazolines |
| GB0808953D0 (en) * | 2008-05-16 | 2008-06-25 | Shire Llc | substituted quinazolines |
| GB0810005D0 (en) * | 2008-06-02 | 2008-07-09 | Shire Llc | Substituted quinazolines |
| GB0822970D0 (en) | 2008-12-17 | 2009-01-21 | Shire Llc | Process for the preparation of anagrelide and analogues |
| GB201004491D0 (en) | 2010-03-18 | 2010-05-05 | Shire Llc | Quinazoline analogues |
| GB201004495D0 (en) | 2010-03-18 | 2010-05-05 | Shire Llc | Subtituted quinazolines |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1645917A1 (de) | 1962-10-26 | 1970-09-24 | American Home Prod | Verfahren zur Herstellung von 5-Aryl-3-acyloxy-1,2-dihydro-3H-1,4-benzodiazepin-2-onen |
| GB1037375A (en) * | 1964-08-07 | 1966-07-27 | Hoffmann La Roche | A process for the manufacture of benzodiazepine derivatives |
| US3932407A (en) * | 1973-11-19 | 1976-01-13 | Bristol-Myers Company | Optionally substituted 1,2,3,5-tetrahydroimidezo(2,1-b)-quinazolin-2-ones and 6(H)-1,2,3,4-tetrahydropyimido(2,1-b)quinazolin-2-ones |
| BE794964A (fr) * | 1972-02-04 | 1973-08-02 | Bristol Myers Co | Nouveaux agents hypotenseurs et procede pour les preparer |
| US3983119A (en) * | 1974-11-06 | 1976-09-28 | Bristol-Myers Company | Process for the preparation of optionally substituted 1,2,3,5-tetrahydroimidazo[2,1-b]quinazolin-2-ones |
| US3988340A (en) * | 1975-01-23 | 1976-10-26 | Bristol-Myers Company | 6-Alkoxymethyl-1,2,3,5-tetrahydroimidazo[2,1-b]quinazolin-2-ones and 7-alkoxymethyl-6-[H]-1,2,3,4-tetrahydropyrimido[2,1-b]quinazolin-2-ones |
| NL7807507A (nl) | 1977-07-25 | 1979-01-29 | Hoffmann La Roche | Tricyclische verbindingen. |
-
1978
- 1978-07-12 NL NL7807507A patent/NL7807507A/xx not_active Application Discontinuation
- 1978-07-13 MC MC781312A patent/MC1199A1/fr unknown
- 1978-07-14 FI FI782248A patent/FI63409C/fi not_active IP Right Cessation
- 1978-07-18 AU AU38127/78A patent/AU519688B2/en not_active Expired
- 1978-07-19 FR FR7821396A patent/FR2398748A1/fr active Granted
- 1978-07-19 HU HU78HO2088A patent/HU177643B/hu unknown
- 1978-07-20 CA CA307,767A patent/CA1094555A/en not_active Expired
- 1978-07-20 NZ NZ187921A patent/NZ187921A/xx unknown
- 1978-07-20 IL IL55183A patent/IL55183A/xx unknown
- 1978-07-21 DE DE19782832138 patent/DE2832138A1/de not_active Withdrawn
- 1978-07-21 EP EP78100471A patent/EP0000718B1/de not_active Expired
- 1978-07-21 DE DE7878100471T patent/DE2861688D1/de not_active Expired
- 1978-07-21 PH PH21412A patent/PH14642A/en unknown
- 1978-07-24 IT IT26019/78A patent/IT1097337B/it active
- 1978-07-24 DK DK328978A patent/DK144128C/da not_active IP Right Cessation
- 1978-07-24 ES ES471981A patent/ES471981A1/es not_active Expired
- 1978-07-24 GB GB787830868A patent/GB2001638B/en not_active Expired
- 1978-07-24 GR GR56847A patent/GR72968B/el unknown
- 1978-07-24 NO NO782541A patent/NO150800C/no unknown
- 1978-07-24 BR BR7804763A patent/BR7804763A/pt unknown
- 1978-07-24 IE IE1478/78A patent/IE47280B1/en unknown
- 1978-07-24 CS CS784914A patent/CS203014B2/cs unknown
- 1978-07-24 PT PT68342A patent/PT68342A/pt unknown
- 1978-07-24 AT AT0535178A patent/AT363479B/de active
- 1978-07-24 SE SE7808111A patent/SE7808111L/xx unknown
- 1978-07-24 JP JP8953478A patent/JPS5441894A/ja active Pending
- 1978-07-25 AR AR273064A patent/AR218500A1/es active
- 1978-07-25 YU YU01775/78A patent/YU177578A/xx unknown
-
1979
- 1979-01-18 ES ES476955A patent/ES476955A1/es not_active Expired
- 1979-06-20 US US06/050,395 patent/US4256748A/en not_active Expired - Lifetime
-
1985
- 1985-12-30 MY MY249/85A patent/MY8500249A/xx unknown
Cited By (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0021338A1 (de) * | 1979-06-20 | 1981-01-07 | F. HOFFMANN-LA ROCHE & CO. Aktiengesellschaft | Neue Chinazolinderivate und pharmazeutische Präparate |
| EP0054180A3 (de) * | 1980-12-16 | 1982-12-01 | F. HOFFMANN-LA ROCHE & CO. Aktiengesellschaft | Verfahren zur Herstellung eines Imidazochinazolinonderivates |
| EP0116948A3 (en) * | 1983-02-16 | 1984-12-19 | Syntex (U.S.A.) Inc. | (2-oxo-1,2,3,5-tetrahydroimidazo-(2,1-b)quinazolinyl)-oxyalkyl-amides |
| US4663320A (en) * | 1983-02-16 | 1987-05-05 | Syntex (U.S.A.) Inc. | (2-oxo-1,2,3,5-tetrahydroimidazo[2,1-b]quinoazolinyl)oxyalkylamides, compositions and the use thereof |
| EP0129258A1 (de) * | 1983-06-21 | 1984-12-27 | Daiichi Seiyaku Co., Ltd. | Imidazochinazolinverbindung |
| EP0133234A3 (en) * | 1983-07-14 | 1985-10-30 | Daiichi Seiyaku Co. Ltd. | Imidazoquinazolin-2-one compounds process for their production and pharmaceutical compositions containing said compounds |
| EP0153152A3 (de) * | 1984-02-15 | 1987-07-15 | Syntex (U.S.A.) Inc. | (2-Oxo-1,2,3,5-tetrahydroimidazo[2,1-b]chinazolinyl)oxyalkylamide, ihre Herstellung, sie enthaltende Zusammensetzungen und ihre Verwendung bei der Herstellung von Medikamenten |
| US4690925A (en) * | 1984-02-15 | 1987-09-01 | Syntex (U.S.A.) Inc. | (2-Oxo-1,2,3,5-tetrahydroimidazo-[2,1-B]quinazolinyl) oxyalkylamides properties having phosphodiosterase inhibiting |
| US4670434A (en) * | 1985-11-14 | 1987-06-02 | Syntex (U.S.A.) Inc. | (2-oxo-3-methylene-1,2,3,5-tetrahydroimidazo[2,1-b]quinazolinyl)oxyalkylamides useful as cyclic AMP phosphodiesterase inhibitors |
| EP2088154A1 (de) | 2004-03-09 | 2009-08-12 | Ironwood Pharmaceuticals, Inc. | Verfahren und Zusammensetzungen zur Behandlung von Magen-Darm-Störungen |
| EP2998314A1 (de) | 2007-06-04 | 2016-03-23 | Synergy Pharmaceuticals Inc. | Für die behandlung von gastrointestinalen erkrankungen, entzündungen, krebs und anderen erkrankungen geeignete agonisten von guanylatcyclase |
| EP2810951A2 (de) | 2008-06-04 | 2014-12-10 | Synergy Pharmaceuticals Inc. | Für die Behandlung von gastrointestinalen Erkrankungen, Entzündungen, Krebs und anderen Erkrankungen geeignete Agonisten von Guanylatcyclase |
| EP3241839A1 (de) | 2008-07-16 | 2017-11-08 | Synergy Pharmaceuticals Inc. | Zur behandlung von erkrankungen des magen-darm-trakts, entzündlichen erkrankungen, krebs und anderen erkrankungen geeignete agonisten von guanylatcyclase |
| EP2923706A1 (de) | 2009-12-03 | 2015-09-30 | Synergy Pharmaceuticals Inc. | Guanylatcyclaseagonisten zur behandlung von hypercholesterinämie |
| WO2011069038A2 (en) | 2009-12-03 | 2011-06-09 | Synergy Pharmaceuticals, Inc. | Agonists of guanylate cyclase useful for the treatment of hypercholesterolemia, atherosclerosis, coronary heart disease, gallstone, obesity and other cardiovascular diseases |
| WO2012118972A2 (en) | 2011-03-01 | 2012-09-07 | Synegy Pharmaceuticals Inc. | Process of preparing guanylate cyclase c agonists |
| EP3708179A1 (de) | 2012-03-15 | 2020-09-16 | Bausch Health Ireland Limited | Formulierungen von guanylatcyclase-c-agonisten und verfahren zur verwendung |
| WO2013138352A1 (en) | 2012-03-15 | 2013-09-19 | Synergy Pharmaceuticals Inc. | Formulations of guanylate cyclase c agonists and methods of use |
| EP4309673A2 (de) | 2012-03-15 | 2024-01-24 | Bausch Health Ireland Limited | Formulierungen von guanylatcyclase-c-agonisten und verfahren zur verwendung |
| WO2014131024A2 (en) | 2013-02-25 | 2014-08-28 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase and their uses |
| EP3718557A2 (de) | 2013-02-25 | 2020-10-07 | Bausch Health Ireland Limited | Guanylat-cyclase-rezeptoragonist sp-333 zur verwendung in der darmreinigung |
| WO2014151200A2 (en) | 2013-03-15 | 2014-09-25 | Synergy Pharmaceuticals Inc. | Compositions useful for the treatment of gastrointestinal disorders |
| WO2014151206A1 (en) | 2013-03-15 | 2014-09-25 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase and their uses |
| WO2014197720A2 (en) | 2013-06-05 | 2014-12-11 | Synergy Pharmaceuticals, Inc. | Ultra-pure agonists of guanylate cyclase c, method of making and using same |
| EP4424697A2 (de) | 2013-06-05 | 2024-09-04 | Bausch Health Ireland Limited | Ultrareine agonisten der guanylatcyclase c, verfahren zur herstellung und verwendung davon |
| EP3492106A1 (de) | 2013-08-09 | 2019-06-05 | Ardelyx, Inc. | Verbindungen und verfahren zur hemmung eines phosphattransports |
| EP3884935A1 (de) | 2013-08-09 | 2021-09-29 | Ardelyx, Inc. | Verbindungen und verfahren zur hemmung eines phosphattransports |
| WO2015021358A2 (en) | 2013-08-09 | 2015-02-12 | Dominique Charmot | Compounds and methods for inhibiting phosphate transport |
| US20170204492A1 (en) * | 2014-08-08 | 2017-07-20 | Jfe Steel Corporation | Cold-rolled steel sheet having excellent spot weldability, and manufacturing method therefor |
| WO2020237096A1 (en) | 2019-05-21 | 2020-11-26 | Ardelyx, Inc. | Combination for lowering serum phosphate in a patient |
| EP4706758A2 (de) | 2019-05-21 | 2026-03-11 | Ardelyx, Inc. | Verfahren zur hemmung des phosphattransports |
| EP4201403A1 (de) | 2021-12-21 | 2023-06-28 | Som Innovation Biotech, S.L. | Verbindungen tirapazamin und quazinon zur verwendung bei der behandlung von gm2-gangliosidosen |
| WO2023118113A1 (en) | 2021-12-21 | 2023-06-29 | Som Innovation Biotech, S.A. | Compounds tirapazamine and quazinone for use in the treatment of gm2 gangliosidoses |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0000718B1 (de) | Neue Chinazolinderivate, Verfahren zu ihrer Herstellung, pharmazeutische Präparate und deren Herstellung | |
| DE2305575C2 (de) | ||
| DE3614132A1 (de) | 1,3-dihydro-2h-imidazo(4,5-b)chinolin-2-one, verfahren zu deren herstellung und diese verbindungen enthaltende, pharmazeutische mittel | |
| EP0005205A1 (de) | Substituierte 5,6-Dimethylpyrrolo(2,3-d)pyrimidine, Verfahren zu ihrer Herstellung und diese enthaltende Arzneimittel | |
| DE60207759T2 (de) | Pyrimidotriazine als phosphataseinhibitoren | |
| EP0053767B1 (de) | Tricyclische Cytosinderivate zur Verwendung in Arzneimitteln und Verfahren zu ihrer Herstellung | |
| DE2505297B2 (de) | Verfahren zur Herstellung von in 2- und 6-Stellung des Phenylkerns substituierten 2-Phenylamino-2-imidazolin-Derivaten | |
| DD255344A5 (de) | Heteroaryl-oxy-beta-carbolinderivate, ihre herstellung und ihre verwendung als arzneimittel | |
| EP0073060A1 (de) | Neue Imidazochinazolinderivate, ihre Herstellung und diese Derivate enthaltende Arzneimittel | |
| EP0021338B1 (de) | Neue Chinazolinderivate und pharmazeutische Präparate | |
| EP0148431A1 (de) | Neue Benzimidazole, ihre Herstellung und diese Verbindungen enthaltende Arzneimittel | |
| EP0124893A2 (de) | Neue Pyrimidonderivate | |
| EP0046267A1 (de) | Imidazochinazolinderivate, ihre Herstellung und Arzneimittel enthaltend diese Derivate | |
| EP0103158A1 (de) | Neue Thieno-thiazol-Derivate, Verfahren zu ihrer Herstellung und diese Verbindungen enthaltende Arzneimittel | |
| Heymann et al. | Derivatives of p, p'-Diaminodiphenyl Sulfone1a | |
| Richter et al. | Studies in Purine Chemistry. New Routes to Certain 2, 1, 3-Triazoles, Pyrimidines and 2, 1, 3-Triazolo [4, 5-d] pyrimidines1, 2 | |
| EP0180833B1 (de) | 4-Oxo-pyrido[2,3]pyrimidin-Derivate, Verfahren zur deren Herstellung und diese ethaltende Arzneimittel | |
| EP0239667B1 (de) | 5- oder 6-substituierte-beta-carbolin-3-carbonsäureester | |
| DE2340217C2 (de) | 3-Benzoylthiophene, ihre Salze, Verfahren zu ihrer Herstellung und diese Verbindungen enthaltende Arzneimittel | |
| EP0175650A2 (de) | Benzazole, Verfahren zu ihrer Herstellung und pharmazeutische Präparate enthaltend solche Verbindungen und deren Verwendung | |
| US3468889A (en) | O-and/or s-nicotinoyl diacylthiamines and acylation process for preparing the same | |
| AT363481B (de) | Verfahren zur herstellung von neuen imidazo-chinazolinen deren tautomeren und salzen | |
| KR810001155B1 (ko) | 트리사이클 화합물의 제조방법 | |
| DE2215999A1 (de) | Nitroimidazolyl-triazolo-pyridazine und verfahren zu ihrer herstellung | |
| DE2137341A1 (de) | Neue thieno eckige klammer auf 3,2-d eckige klammer zu pyrimidine |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR LU NL SE |
|
| PUAL | Search report despatched |
Free format text: ORIGINAL CODE: 0009013 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR LU NL SE |
|
| 17P | Request for examination filed | ||
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR LU NL SE |
|
| REF | Corresponds to: |
Ref document number: 2861688 Country of ref document: DE Date of ref document: 19820429 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19820731 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: LU Payment date: 19830628 Year of fee payment: 6 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 19840704 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 19840709 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 19840711 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 19840731 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: SE Payment date: 19840930 Year of fee payment: 7 Ref country code: BE Payment date: 19840930 Year of fee payment: 7 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Effective date: 19850722 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CH Effective date: 19850731 Ref country code: BE Effective date: 19850731 |
|
| BERE | Be: lapsed |
Owner name: F. HOFFMANN-LA ROCHE & CO. A.G. Effective date: 19850731 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Effective date: 19860201 |
|
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee | ||
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19860328 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19860402 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| EUG | Se: european patent has lapsed |
Ref document number: 78100471.8 Effective date: 19860730 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |