-
GEBIET DER ERFINDUNG
-
Die
vorliegende Erfindung stellt neue Verbindungen, neue Zusammensetzungen,
Verfahren zu ihrer Verwendung und Verfahren zu ihrer Erzeugung bereit,
derartige Verbindungen sind im Allgemeinen pharmakologisch als Mittel
in jenen Erkrankungszuständen
nützlich,
die durch die Änderung
von Mitogen-aktivierten Signalübertragungswegen
im Allgemeinen und im Besonderen der Hemmung oder dem Antagonismus
von Proteinkinasen, welche pathologischerweise aberrante zelluläre Proliferation
einbeziehen, gelindert werden, wobei derartige Erkrankungszustände Tumorwachstum
einschließen.
Die zuvor erwähnten
pharmakologischen Aktivitäten
sind nützlich
bei der Behandlung von Säugern.
Insbesondere betrifft die Erfindung Benzylidenoxindolderivate, welche
cRaf-1-Kinase-Hemmung zeigen, zur Behandlung von Erkrankungen, die
mit Zellproliferation in Zusammenhang stehen.
-
Genauer
gesagt können
die erfindungsgemäßen Verbindungen
bei der Behandlung von bestimmten Krebsformen verwendet werden,
können
verwendet werden, um additive oder synergistische Wirkungen mit bestimmten
existierenden Krebschemotherapien bereitzustellen und/oder verwendet
werden, um die Effektivität
bestimmter existierender Krebschemotherapien und Strahlung wieder
herzustellen. Gegenwärtig
gibt es einen Bedarf für
derartige therapeutische Mittel in den Bereichen von Erkrankungen,
die durch Zellproliferation gekennzeichnet sind.
-
HINTERGRUND DER ERFINDUNG
-
Krebs
ergibt sich aus der Deregulierung der normalen Vorgänge, die
Zellteilung, -differenzierung und apoptotischen Zelltod kontrollieren.
Proteinkinasen spielen eine entscheidende Rolle bei diesem regulatorischen
Vorgang. Eine teilweise, nicht beschränkende Liste derartiger Kinasen
schließen
ab1, ATK, bcr-ab1, Blk, Brk, Btk, c-kit, c-met, c-src, CDK1, CDK2,
CDK4, CDK6, cRaf1, CSF1R, CSK, EGFR, ErbB2, ErbB3, ErbB4, ERK, Fak,
fes, FGFR1, FGFR2, FGFR3, FGFR4, FGFR5, Fgr, FLK4, flt-1, Fps, Frk,
Fyn, Hck, IGF-1R, INS-R, Jak,
KDR, Lck, Lyn, MEK, p38, PDGFR, PIK, PKC, PYK2, ros, tie1, tie2, TRK, Yes
und Zap70 ein. In der Säugerbiologie
umfassen derartige Proteinkinasen Mitogen-aktivierte Proteinkinase-(MAPK)-Signalübertragungswege.
MAPK Signalübertragungswege
werden fehlerhaft durch eine Vielzahl von üblichen mit Erkrankung in Zusammenhang
stehenden Mechanismen, wie Mutation von ras-Genen und Deregulierung
von Wachstumsfaktorrezeptoren (Magnuson et al, Seminars in Cancer
Biology; 1994 (5), 247 – 252)
aktiviert. Daher ist die Hemmung von Proteinkinasen eine Aufgabe
der vorliegenden Erfindung.
-
Zusätzlich sind
Proteinkinasen als Ziele bei Störungen
des Zentralnervensystems (wie Alzheimer Krankheit), entzündlichen
Störungen
(wie Psoriasis), Knochenerkrankung (wie Osteoporose), Atherosklerose, Restenose,
Thrombose, metabolische Störungen
(wie Diabetes) und infektiösen
Erkrankungen (wie Virus- und Pilzinfektionen) in Verbindung gebracht
worden.
-
Einer
der üblichsten
untersuchten Übertragungswege,
welche Kinaseregulierung einbeziehen, ist zelluläres Signalübertragen von Rezeptoren an
der Zelloberfläche
in den Kern (Crews und Erikson, 1993). Ein Beispiel für diesen Übertragungsweg
schließt
eine Kaskade von Kinasen ein, bei welcher die Mitglieder der Tyrosinkinasen
des Wachstumsfaktorrezeptors (wie EGF-R, PDGF-R, VEGF-R, IGF1-R,
der Insulinrezeptor) Signale durch Phosphorylierung an andere Kinasen,
wie Src-Tyrosinkinase und die Raf-, Mek- und Erk-Serin/Threoninkinasefamilien
(Crews und Erikson, 1993; Ihle et al., 1994) abgeben. Jede dieser
Kinasen wird durch mehrere Familienmitglieder (Pelech und Sanghera,
1992) repräsentiert,
welche miteinander verwandte, aber funktionell unterschiedlichen
Rollen spielen. Der Verlust der Regulierung des Signalübertragungsweges für den Wachstumsfaktor
ist eine häufige
Erscheinung bei Krebs ebenso wie bei anderen Erkrankungszuständen.
-
Von
den durch Kinasen vermittelten Signalen ist auch gezeigt worden,
dass sie Wachstum, Tod und Differenzierung in der Zelle durch das
Regulieren der Vorgänge
des Zellzyklus (Massague und Roberts, 1995) kontrollieren. Fortschreiten
durch den eukaryotischen Zellzyklus wird durch eine Familie von
Kinasen, welche Cyclin-abhängige
Kinasen (CDKs) genannt wird, kontrolliert (Myerson et al., 1992).
Die Regulierung der CDK Aktivierung ist komplex, erfordert aber
die Assoziation des CDK's
mit einem Mitglied der Cyclinfamilie von regulatorischen Untereinheiten
(Draetta, 1993; Murray und Kirschner, 1989; Solomon et al., 1992).
Eine weitere Ebene der Regulierung findet durch sowohl Aktivierung
als auch Inaktivierung der Phosphorylierung der CDK-Untereinheit
statt (Draetta, 1993; Ducommun et al., 1991; Gautier et al., 1989;
Gould und Nurse, 1989; Krek und Nigg, 1991; Murray und Kirschner,
1989; Solomon et al., 1992; Solomon et al., 1990). Die koordinierte Aktivierung
und Inaktivierung von verschiedenen Cyclin/CDK Komplexen ist für das normale
Fortschreiten durch den Zellzyklus notwendig (eines, 1993; Sherr,
1993). Sowohl die entscheidenden G1-S- als auch G2-M-Übergänge werden durch die Aktivierung
von verschiedenen Cyclin/CDK Aktivitäten kontrolliert. In G1 wird
von sowohl Cyclin D/CDK4 als auch Cyclin E/CDK2 angenommen, dass
sie den Beginn der S-Phase vermitteln (Matsushime et al., 1994;
Ohtsubo und Roberts, 1993; Quelle et al., 1993; Resnitzky et al.,
1994). Fortschreiten durch die S-Phase erfordert die Aktivität von Cyclin
A/CDK2 (Girard et al., 1991; Pagano et al., 1992; Rosenblatt et
al., 1992; Walker und Maller, 1991; Zindy et al., 1992), wohingegen
die Aktivierung von Cyclin A/cdc2 (CDK1) und Cyclin B/cdc2 für den Beginn
der Metaphase erforderlich sind (Draetta, 1993; Girard et al., 1991;
Murray und Kirschner, 1989; Pagano et al., 1992; Rosenblatt et al.,
1992; Solomon et al., 1992; Walker und Maller, 1991; Zindy et al.,
1992). Es ist daher nicht überraschend,
dass der Verlust der Kontrolle der CDK Regulierung ein häufiges Ereignis
bei hyperproliferativen Erkrankungen und Krebs ist (Hunter und eines,
1994; Lees, 1995; eines, 1992).
-
Die
Kinase cRaf1 reguliert zelluläre
Proliferation auf zwei Weisen. Das Enzym reguliert Zellteilung durch
die Raf/MEK/ERK Proteinkinasenkaskade positiv. Diese Aktivität ist das
Ergebnis von cRaf1 katalysierter Phosphorylierung der Proteinkinase
MEK1. MEK1 phosphoryliert und aktiviert die Proteinkinase ERK. ERK phosphoryliert
und reguliert Transkriptionsfaktoren, welche für die Zellteilung erforderlich
sind (Avruch et al, TIBS; 1994 (19) 279 – 283). cRaf1 reguliert den
Zelltod negativ durch Modulierung der Aktivität von Bcl-2, einem entscheidenden
Regulator von Apoptose. Diese Regulierung bezieht die direkte Phosphorylierung
von Mitgliedern der Bcl-2 Familie ein (Gajewski und Thompson, Cell:
1996 (87) 619 – 628).
Diese beiden Aspekte von cRaf1-vermittelter Regulierung von zellulärer Proliferation
erfordern die Kinaseaktivität
von cRaf1.
-
cRaf1
wird durch Ereignisse, welche bei menschlichem Krebs üblich sind,
dereguliert. Zum Beispiel sind ras-Gene mit den folgenden Häufigkeiten
in den folgenden repräsentativen
primären
menschlichen Tumoren mutiert: Lunge (Adenkarzinom), 30 %; Kolon
(Adenkarzinom), 50 %; Bauchspeicheldrüsenkarzinom, 90 %; Seminom,
40 %; Schilddrüse,
50 % (McCormick, Ras oncogenes in Oncogenes and the molecular origins of
cancer: 1989, 125 – 146).
cRaf1 wird auch durch Deregulierung der Tyrosinkinasen einschließlich, cSrc, ErbB2,
EGFR und bcr/abl aktiviert. Diese Ereignisse werden mit Brust-,
Kolon- und Lungenkarzinomen und chronischer, myeloischer Leukämie in Zusammenhang
gebracht (Fearon, Genetic lesions in human cancer, in Molecular
oncology; 1996, 143 – 178).
Außerdem
lehrt die Raf-Antisense Literatur, dass die Verringerung von Raf-Proteinspiegeln
mit einer Verringerung der Tumorwachstumsgeschwindigeit bei in vivo
Tumormodellen der Maus korreliert. Inhibitoren der Kinaseaktivität von cRaf1
sollten daher eine wirksame Behandlung für eine breite Vielfalt üblicher
menschlicher Krebsarten bereitstellen.
-
Inhibitoren
von Kinasen, welche an der Vermittlung oder Erhaltung dieser Erkrankungszustände beteiligt
sind, repräsentieren
neue Therapien für
diese Störungen.
Beispiele für
derartige Kinasen schließen:
(1) Hemmung von Src (Brickell, 1992; Courtneidge, 1994), raf (Powis,
1994) und den Cyclin-abhängigen
Kinasen (CDKs) 1, 2 und 4 bei Krebs (Hunter und Pines, 1994; Lees,
1995; Pines, 1992), (2) Hemmung von CDK2 oder PDGF-R Kinase bei
Restenose (Buchdunger et al., 1995), (3) Hemmung von CDK5 und GSK3
Kinasen bei Alzheimer-Krankheit (Aplin et al., 1996; Hosoi et al.,
1995), (4) Hemmung von c-Src Kinase bei Osteoporose (Tanaka et al.,
1996), (5) Hemmung von GSK-3 Kinase bei Typ-2 Diabetes (Borthwick
et al., 1995); (6) Hemmung der p38 Kinase bei Entzündung (Radger
et al., 1996); (7) Hemmung von VEGF-R 1 – 3 und TIE-1 und -2 Kinasen
bei Angiogenese (Shawver et al., 1997); (8) Hemmung von UL97 Kinase
bei Virusinfektionen (He et al., 1997); (9) Hemmung von CSF-1R Kinase
bei Knochen- und hämatopoetischen
Erkrankungen (Myers et al., 1997) und (10) Hemmung von Lck Kinase
bei Autoimmunerkrankungen und Organabstoßungen nach Transplantationen
(Myers et al., 1997) ein, sind aber nicht beschränkt darauf.
-
Die
vorliegende Erfindung betrifft bestimmte Benzylidenoxindolderivate,
welche nicht nur antineoplastische Eigenschaften aufweisen, sondern
ebenfalls selektive und starke Inhibitoren der Serin/Threoninkinase cRaf1
sind, wobei dadurch selektive Verringerung oder Eliminierung von
bestimmten Erkrankungsgeweben erlaubt wird. Einige der erfindungsgemäßen Verbindungen
können
selektiv eine andere therapeutisch relevante Kinase hemmen.
-
Es
ist eine Aufgabe der vorliegenden Erfindung, starke, spezifische
oral, intravenös
oder subcutan wirksame kleine Inhibitoren der Signalübertragungsaktivität von Raf-Kinasen
zur Behandlung von menschlichen Malignitäten, zum Beispiel, einer oder
mehr von Brust-, Magen-, Eierstock-, Kolon-, Lungen-, Gehirn-, Larynxtumoren,
Tumoren des lymphatischen Systems, des Urogenitaltrakts (einschließlich Blase
und Prostata), Eierstock-, Magen-, Knochen- oder Bauchspeicheldrüsentumoren,
vorzugsweise jene, welche über
cRaf-1 signalisieren, unter Verwendung der erfindungsgemäßen Verbindungen,
Verfahren ihrer Verabreichung, Verfahren für ihre Formulierung und Verfahren
für ihre
Synthese bereitzustellen.
-
Die
erfindungsgemäßen Verbindungen
sind zusätzlich
bei der Behandlung von einer oder mehreren Erkrankungen, die Säuger heimsuchen,
nützlich,
welche durch zelluläre
Proliferation in Bereichen von proliferativen Störungen von Blutgefäßen, fibrotischen
Störungen,
proliferativen Störungen
von Mesangialzellen und metabolischen Erkrankungen gekennzeichnet
sind. Proliferative Störungen
der Blutgefäße schließen Arthritis und
Restenose ein. Fibrotische Störungen
schließen
Leberzirrhose und Atherosklerose ein. Proliferative Störungen der
Mesangialzellen schließen
Glomerulonephritis, diabetische Nephropathie, maligne Nephrosklerose,
thrombotische Mikroangiophatie-Syndrome, Organabstoßungen nach
Transplantationen und Glomerulopathien ein. Metabolische Störungen schließen Psoriasis,
Diabetes mellitus, Heilung einer chronischen Wunde, Entzündung und
neurodegenerative Krankheiten ein.
-
ZUSAMMENFASSUNG DER ERFINDUNG
-
Zusammenfassend
schließt
die Erfindung eine Familie von Verbindungen der allgemeinen Strukturformel
(I):
wobei:
R
1 H
ist oder gegebenenfalls mit R
2 verbunden
ist, um einen kondensierten Ring zu bilden, ausgewählt aus fünf- bis
zehngliedrigen Aryl-, Heteroaryl- oder Heterocyclylringen, wobei
die Heteroaryl- oder die Heterocyclylringe ein bis drei Heteroatome
aufweisen, wobei null bis drei der Heteroatome N sind und null bis
ein Heteroatom O oder S ist und wobei der kondensierte Ring gegebenenfalls
mit ein bis drei R
9 substituiert ist, wobei R
2 und R
9 wie nachstehend
definiert sind;
R
2 und R
3 unabhängig voneinander
H, HET, Aryl, ein C
1-12 aliphatischer Rest,
CN, NO
2, Halogen, R
10,
-OR
10, -SR
10, -S(O)R
10, -SO
2R
10, -NR
10R
11, -NR
11R
12, -NR
12COR
11, R
12CO
2R
11, -NR
12CONR
11R
12, -NR
12SO
2R
11, -NR
12C(NR
12)NHR
11, -COR
11, -CO
2R
11, -CONR
12R
11, -SO
2NR
12R
11,
-OCONR
12R
11, C(NR
12)NR
12R
11 sind,
wobei der C
1-12 aliphatische Rest gegebenenfalls
ein oder zwei Einfügungen
aus einer oder zwei Gruppen aufweist, ausgewählt aus C(O), O, S, S(O), SO
2 oder NR
12; wobei
HET, Aryl oder der C
1-12 aliphatische Rest
gegebenenfalls mit einem bis drei R
10 substituiert
sind; und wobei R
2 gegebenenfalls mit R
3 verbunden ist, um einen kondensierten Ring
zu bilden, ausgewählt
aus fünf-
bis zehngliedrigen Aryl-, Heteroaryl- oder Heterocyclylringen, wobei
die Heteroaryl- oder die Heterocyclylringe null bis drei Heteroatome
aufweisen, wobei null bis drei der Heteroatome N sind und null bis
ein Heteroatom O oder S ist und wobei der kondensierte Ring gegebenenfalls mit
einem bis drei R
9 substituiert ist, wobei
HET, R
9, R
10, R
11 und R
12 wie nachstehend
definiert sind;
R
4 H, Halogen, NO
2 oder CN ist;
R
5 H
oder ein C
1-12 aliphatischer Rest ist, gegebenenfalls
substituiert mit einem bis drei aus Halogen, Hydroxyl, Heteroaryl
oder Aryl;
R
6 und R
7 Halogen
sind;
R
9 jeweils unabhängig Halogen,
ein C
1-12 aliphatischer Rest, CN, NO
2, R
10, -OR
11, -SR
11, -S(O)R
10, -SO
2R
10, -NR
10R
11, -N
11R
12, -NR
12COR
11, -NR
12CO
2R
11, R
12CONR
11R
12, -NR
12SO
2R
11,
-NR
12C(NR
12)NHR
11, -CO
2R
11, -CONR
12R
11, -SO
2NR
12R
11, -OCONR
12R
11 oder C(NR
12)NR
12R
11 ist,
wobei R
10, R
11 und
R
12 wie nachstehend definiert sind;
R
10 jeweils unabhängig H, Halogen, ein C
1-12 aliphatischer Rest, Aryl oder HET ist,
wobei der aliphatische C
1-12-Rest gegebenenfalls
eine oder zwei eingefügte
Gruppen aufweist, ausgewählt
aus O, S, S(O), SO
2 oder NR
12,
wobei der C
1-12 aliphatische Rest, Aryl
oder HET gegebenenfalls substituiert sind mit einem bis drei aus Halogen,
einem weiteren HET, Aryl, CN, -SR
12, -OR
12, -N(R
12)
2, -S(O)R
12, -SO
2R
12, -SO
2N(R
12)–
2, -NR
12COR
12, -NR
12CO
2R
12, -S(O)R
12, -SO
2R
12, -SO
2N(R
12)
2, -NR
12COR
12, -NR
12CO
2R , -NR
12CON(R
12)
2, -NR
12(NR
12)NHR
12, -CO
2R
12, -CON(R
12)
2, NR
12SO
2R
12, -OCON(R
12)
2, wobei HET und
R
12 wie nachstehend definiert sind;
R
11 H oder R
10 ist;
R
12 H, ein C
1-12 aliphatischer
Rest oder HET ist, wobei der C
1-12 aliphatische
Rest gegebenenfalls mit einem bis drei aus Halogen oder OH substituiert
ist, wobei HET wie nachstehend definiert ist; und
HET ein fünf- bis
zehngliedriger gesättigter
oder ungesättigter
heterocyclischer Ring ist, ausgewählt aus Benzofuran, Benzoxazol,
Dioxin, Dioxan, Dioxolan, Dithian, Dithiazin, Dithiazol, Dithiolan,
Furan, Imidazol, Indol, Indazol, Morpholin, Oxazol, Oxadiazol, Oxathiazol,
Oxathiazolidin, Oxazin, Oxadiazin, Piperazin, Piperidin, Pyran,
Pyrazin, Pyrazol, Pyridin, Pyrimidin, Pyrrol, Pyrrolidin, Chinolin,
Chinazolin, Tetrahydrofuran, Tetrazin, Tetrazol, Thiophen, Thiadiazin,
Thiadiazol, Thiatriazol, Thiazin, Thiazol, Thiomorpholin, Thianaphthalin, Thiopyran,
Triazin und Triazol,
und die pharmazeutisch verträglichen
Salze oder Solvate davon.
-
Eine
bevorzugte Gruppe von erfindungsgemäßen Verbindungen ist jene der
allgemeinen Formel (I)
wobei,
R
1 H
ist oder gegebenenfalls mit R
2 verbunden
ist, um einen kondensierten Ring zu bilden, ausgewählt aus
der für
HET nachstehend definierten Gruppe, und wobei der kondensierte Ring
gegebenenfalls mit einem bis drei R
9 substituiert
ist, wobei R
2 und R
9 wie
nachstehend definiert sind;
R
2 und
R
3 unabhängig
voneinander H, HET, Aryl, ein C
1-6 aliphatischer
Rest, CN, NO
2, Halogen, R
10,
-OR
10, -SR
10, -S(O)R
10, -SO
2R
10, -NR
10R
11, -NR
11R
12, -NR
12COR
11, -NR
12CO
2R
11, -NR
12CONR
11R
12, -NR
12SO
2R
11, -NR
12C(NR
12)NHR
11, -COR
11, -CO
2R
11, -CONR
12R
11, -SO
2NR
12R
11,
-OCONR
12R
11, C(NR
12)NR
12R
11 sind,
wobei der C
1-6 aliphatische Rest gegebenenfalls
ein oder zwei Einfügungen
aus einer oder zwei Gruppen aufweist, ausgewählt aus C(O), O, S, S(O), SO
2 oder NR
12; wobei
HET, Aryl oder der C
1-6 aliphatische Rest
gegebenenfalls mit einem bis drei R
10 substituiert
sind; und wobei R
2 gegebenenfalls mit R
3 verbunden ist, um einen kondensierten Ring
zu bilden, ausgewählt
aus der nachstehend definierten Gruppe und wobei der kondensierte Ring
gegebenenfalls mit einem bis drei R
9 substituiert
ist, wobei HET, R
9, R
10,
R
11 und R
12 wie
nachstehend definiert sind;
R
4 H, Halogen,
NO
2 oder CN ist;
R
5 H
oder ein C
1-6 aliphatischer Rest ist, gegebenenfalls
substituiert mit einem bis drei aus Halogen, OH oder Aryl;
R
6 und R
7 Halogen
sind;
R
9 jeweils unabhängig Halogen,
ein aliphatischer C
1-6-Rest, CN, -NO
2, R
10, -OR
11, -SR
11, -S(O)R
10, -SO
2R
10, -NR
10R
11, -N
11R
12, -NR
12COR
11, -NR
12CO
2R
11, -NR
12CONR
11R
12, -NR
12SO
2R
11, -NR
12C(NR
12)NHR
11, -CO
2R
11, -CONR
12R
11, -SO
2NR
12R
11, -OCONR
12R
11 oder C(NR
12)NR
12R
11 ist,
wobei R
10, R
11 und
R
12 wie nachstehend definiert sind;
R
10 jeweils unabhängig H, Halogen, ein C
1-6 aliphatischer Rest, Aryl oder HET ist,
wobei der C
1-6 aliphatische Rest gegebenenfalls
ein oder zwei eingefügte
Gruppen aufweist, ausgewählt
aus O, S, S(O), SO
2 oder NR
12, wobei
der C
1-6 aliphatische Rest, Aryl oder HET
gegebenenfalls substituiert sind mit einem bis drei aus Halogen,
einem weiteren HET, Aryl, CN, -SR
12, -OR
12, N(R
12)
2, -S(O)R
12, -SO
2R
12, -SO
2N(R
12)
2,
-NR
12COR
12, -NR
12CO
2R
12,
-NR
12CON(R
12)
2, -NR
12(NR
12)NHR
12, -CO
2R
12, -CON(R
12)
2, -NR
12SO
2R
12,
-OCON(R
12)
2, wobei HET
und R
12 wie nachstehend definiert sind;
R
11 H oder R
10 ist;
R
12 H, ein C
1-6 aliphatischer
Rest oder HET ist, wobei der aliphatische C
1-6-Rest
gegebenenfalls mit einem bis drei aus Halogen oder OH substituiert
ist, wobei HET wie nachstehend definiert ist;
HET ein fünf- bis
zehngliedriger gesättigter
oder ungesättigter
heterocyclischer Ring ist, ausgewählt aus Benzofuran, Benzoxazol,
Dioxin, Dioxan, Dioxolan, Dithian, Dithiazin, Dithiazol, Dithiolan,
Furan, Imidazol, Indol, Indazol, Morpholin, Oxazol, Oxadiazol, Oxathiazol,
Oxathiazolidin, Oxazin, Oxadiazin, Piperazin, Piperidin, Pyran,
Pyrazin, Pyrazol, Pyridin, Pyrimidin, Pyrrol, Pyrrolidin, Chinolin,
Chinazolin, Tetrahydrofuran, Tetrazin, Tetrazol, Thiophen, Thiadiazin,
Thiadiazol, Thiatriazol, Thiazin, Thiazol, Thiomorpholin, Thianaphthalin, Thiopyran,
Triazin und Triazol,
und die pharmazeutisch verträglichen
Salze oder Solvate davon.
-
Eine
stark bevorzugte erfindungsgemäße Gruppe
von Verbindungen ist jene der allgemeinen Formel (I)
wobei,
R
1 H
ist oder gegebenenfalls mit R
2 verbunden
ist, um einen kondensierten Ring zu bilden, ausgewählt aus kondensiertem
Pyridin, kondensiertem Triazol, kondensiertem Thiazol oder kondensiertem
Amino-substituiertem Thiazol;
R
2 und
R
3 unabhängig
voneinander H, HET, Aryl, ein C
1-6 aliphatischer
Rest, -R
12NH
2, -R
12-Halogen,
CN NO
2, Halogen, R
10,
-OR
10, -SR
10, -S(O)R
10, -SO
2R
10, -NR
10R
11, -NR
11R
12, -R
12COR
11, -NR
12CO
2R
11, -NR
12CONR
11R
12, -NR
12SO
2R
11, -NR
12C(NR
12)NHR
11, -COR
11, -COR
11NR
12R
11,
-CO
2R
11, -CONR
12R
11, -SO
2NR
12R
11, -OCONR
12R
11, -C(NH)R
11, -C(NR
12)NR
12R
11 sind, wobei
der C
1-6 aliphatische Rest gegebenenfalls
eine Einfügung
aus einer C(O)-Gruppe aufweist, wobei HET, Aryl oder der C
1-6 aliphatische Rest gegebenenfalls mit
einem bis drei R
10 substituiert sind; und
wobei R
2 gegebenenfalls mit R
3 verbunden
ist, um einen kondensierten Ring zu bilden, ausgewählt aus
der für
HET nachstehend definierten Gruppe und wobei der kondensierte Ring gegebenenfalls
mit einem bis drei R
9 substituiert ist,
wobei HET, R
9, R
10,
R
11 und R
12 wie
nachstehend definiert sind;
R
4 H, Halogen,
NO
2 oder CN ist;
R
5 H
oder ein C
1-6 aliphatischer Rest ist, gegebenenfalls
substituiert mit ein bis drei aus Halogen, OH oder Aryl;
R
6 und R
7 Halogen
sind;
R
9 jeweils unabhängig Halogen,
ein C
1-6 aliphatischer Rest, CN, NO
2, R
10, -OR
11, -SR
11, -S(O)R
10, -SO
2R
10, -NR
10R
11, -N
11R
12, -NR
11COR
11, -NR
12CO
2R
11, -R
12CONR
11R
12, -NR
12SO
2R
11,
-NR
12C(NR
12)NHR
11, -CO
2R
11, -CONR
12R
11, -SO
2NR
12R
11, -OCONR
12R
11 oder C(NR
12)NR
12R
11 ist,
wobei R
10, R
11 und
R
12 wie nachstehend definiert sind;
R
10 jeweils unabhängig H, Halogen, ein C
1-6 aliphatischer Rest, Aryl oder HET ist,
wobei der C
1-6-aliphatische Rest gegebenenfalls
ein oder zwei eingefügte
Gruppen aufweist, ausgewählt
aus O, S, S(O), SO
2 oder NR
12, wobei
der C
1-6 aliphatische Rest, Aryl oder HET
gegebenenfalls substituiert sind mit einem bis drei aus Halogen,
einem weiteren HET, Aryl, CN, NO
2, -R
12, -SR
12, -OR
12, -N(R
12)
2, –R
12N(R
12)
2,
-S(O)R
12, -SO
2R
12, -SO
2N(R
12)
2, -NR
12COR
12, -NR
12CO
2R
12,
-NR
12CON(R
12)
2, -NR
12(NR
12)NHR
12, -CO
2R
12, -CON(R
12)
2, -NR
12SO
2R
12,
-OCON(R
12)
2 oder
Trifluor, wobei HET und R
12 wie nachstehend
definiert sind;
R
11 H oder R
10 ist;
R
12 H,
ein C
1-6 aliphatischer Rest, NO
2,
C
1-6-Alkoxy, Halogen, Aryl oder HET ist,
wobei der C
1-6 aliphatische Rest gegebenenfalls
mit einem bis drei aus Halogen oder OH substituiert ist, wobei HET
wie nachstehend definiert ist;
HET ein fünf- oder sechsgliedriger gesättigter
oder ungesättigter
Heterocyclyl-Ring ist, ausgewählt
aus Dioxin, Dioxan, Dioxolan, Dithian, Dithiazin, Dithiazol, Dithiolan,
Furan, Imidazol, Imidazopyridinyl, Morpholin, Oxazol, Oxadiazol,
Oxathiazol, Oxathiazolidin, Oxazin, Oxadiazin, Piperazin, Piperidin,
Pyran, Pyrazin, Pyrazol, Pyridin, Pyrimidin, Pyrrol, Pyrrolidin,
Tetrahydrofuran, Tetrazin, Thiophen, Thiadiazin, Thiadiazol, Thiatriazol,
Thiazin, Thiazol, Thiomorpholin, Thiopyran, Thioxotriazin, Triazin
und Triazol,
und die pharmazeutisch verträglichen Salze oder Solvate
davon.
-
Ebenfalls
stark bevorzugt wird eine Verbindung der Formel (I), bei der R1 und R2 zusätzlich einen
kondensierten Ring umfassen, welcher ein mit Methyl-substituiertes
kondensiertes Pyridin ist.
-
Eine
andere Gruppe von Verbindungen, welche in Bezug auf ihre Substituenten
an Position R
6 und R
7 bevorzugt
sind, sind die Verbindungen der Formel
wobei:
R
1 H
ist oder gegebenenfalls mit R
2 verbunden
ist, um einen kondensierten Ring zu bilden, ausgewählt aus fünf- bis
zehngliedrigen Aryl-, Heteroaryl- oder Heterocyclylringen, wobei
die Heteroaryl- oder die Heterocyclylringe ein bis drei Heteroatome
aufweisen, wobei null bis drei der Heteroatome N sind und null bis
ein Heteroatom O oder S ist und wobei der kondensierte Ring gegebenenfalls
mit ein bis drei R
9 substituiert ist, wobei R
2 und R
9 wie nachstehend
definiert sind;
R
2 und R
3 unabhängig voneinander
H, HET, Aryl, ein C
1-12 aliphatischer Rest,
CN, NO
2, Halogen, R
10,
-OR
10, -SR
10, -S(O)R
10, -SO
2R
10, -NR
18R
11, -NR
11R
12, -NR
12COR
11, -NR
12CO
2R
11, -NR
12CONR
11R
12, -NR
12SO
2R
11, -NR
12C(NR
12)NHR
11, -COR
11, -CO
2R
11, -CONR
12R
11, -SO
2NR
12R
11,
-OCONR
12R
11, C(NR
12)NR
12R
11 sind,
wobei der C
1-12 aliphatische Rest gegebenenfalls
ein oder zwei Einfügungen
aus einer oder zwei Gruppen aufweist, ausgewählt aus C(O), O, S, S(O), SO
2 oder NR
12; wobei
HET, Aryl oder der C
1-12 aliphatische Rest
gegebenenfalls mit einem bis drei R
10 substituiert
sind; und wobei R
2 gegebenenfalls mit R
3 verbunden ist, um einen kondensierten Ring
zu bilden, ausgewählt
aus fünf-
bis zehngliedrigen Aryl-, Heteroaryl- oder Heterocyclylringen, wobei
die Heteroaryl- oder die Heterocyclylringe null bis drei Heteroatome
aufweisen, wobei null bis drei der Heteroatome N sind und null bis
ein Heteroatom O oder S ist und wobei der kondensierte Ring gegebenenfalls mit
einem bis drei R
9 substituiert ist, wobei
HET, R
9, R
10, R
11 und R
12 wie nachstehend
definiert sind;
R
4 H, Halogen, NO
2 oder CN ist;
R
5 H
oder ein C
1-12 aliphatischer Rest ist, gegebenenfalls
substituiert mit einem bis drei aus Halogen, Hydroxyl oder Aryl;
R
6 und R
7 unabhängig voneinander
Brom oder Chlor sind;
R
9 jeweils unabhängig Halogen,
ein C
1-12 aliphatischer Rest, CN, -NO
2, R
10, -OR
11, -SR
11, -S(O)R
10, -SO
2R
18, -NR
10R
11, -N
11R
12, -NR
12COR
11, -NR
12CO
2R
11, -R
12CONR
11R
12, -NR
12SO
2R
11,
-NR
12C(NR
12)NHR
11, -CO
2R
11, -CONR
12R
11, -SO
2NR
12R
11, -OCONR
12R
11 oder C(NR
12)NR
12R
11 ist,
wobei R
10, R
11 und
R
12 wie nachstehend definiert sind;
R
10 jeweils unabhängig H, Halogen, ein C
1-12 aliphatischer Rest, Aryl oder HET ist,
wobei der aliphatische C
1-12-Rest gegebenenfalls
eine oder zwei eingefügte
Gruppen aufweist, ausgewählt
aus O, S, S(O), SO
2 oder NR
12,
wobei der C
1-12 aliphatische Rest, Aryl
oder HET gegebenenfalls substituiert sind mit einem bis drei aus Halogen,
einem weiteren HET, Aryl, CN, -SR
12, -S(O)R
12, -S(O)R
12, -SO
2N(R
12)
2,
-NR
12CO
2R, -NR
12CO
2R
12, -NR
12CON(R
12), -NR
12(NR
12)NHR
12, -CO
2R
12, -CON(R
12)
2, -NR
12SO
2R
12, -OCON(R
12)
2, wobei HET und
R
12 wie nachstehend definiert sind;
R
11 H oder R
10 ist;
R
12 H, ein C
1-12 aliphatischer
Rest oder HET ist, wobei der C
1-12 aliphatische
Rest gegebenenfalls mit einem bis drei aus Halogen oder OH substituiert
ist, wobei HET wie nachstehend definiert ist; und
HET ein fünf- bis
zehngliedriger gesättigter
oder ungesättigter
heterocyclischer Ring ist, ausgewählt aus Benzofuran, Benzoxazol,
Dioxin, Dioxan, Dioxolan, Dithian, Dithiazin, Dithiazol, Dithiolan,
Furan, Imidazol, Indol, Indazol, Morpholin, Oxazol, Oxadiazol, Oxathiazol,
Oxathiazolidin, Oxazin, Oxadiazin, Piperazin, Piperidin, Pyran,
Pyrazin, Pyrazol, Pyridin, Pyrimidin, Pyrrol, Pyrrolidin, Chinolin,
Chinazolin, Tetrahydrofuran, Tetrazin, Tetrazol, Thiophen, Thiadiazin,
Thiadiazol, Thiatriazol, Thiazin, Thiazol, Thiomorpholin, Thianaphthalin, Thiopyran,
Triazin und Triazol,
und die pharmazeutisch verträglichen
Salze oder Solvate davon.
-
Noch
eine andere Gruppe von Verbindungen, welche in Bezug auf ihre Substituenten
an Position R
6 und R
7 bevorzugt
sind, sind die Verbindungen der Formel
wobei:
R
1 H
ist oder gegebenenfalls mit R
2 verbunden
ist, um einen kondensierten Ring zu bilden, ausgewählt aus fünf- bis
sechsgliedrigen Heteroarylringen, wobei der Heteroarylring ein bis
zwei Heteroatome aufweist, wobei null bis zwei Heteroatome N sind
und null bis zwei Heteroatome O oder S sind, wobei der kondensierte
Ring gegebenenfalls mit einem bis drei R
9 substituiert
ist, wobei R
2 und R
9 wie
nachstehend definiert sind;
R
2 und
R
3 unabhängig
voneinander H, HET, Phenyl, ein C
1-6 aliphatischer
Rest, -NR
10R
11,
-COR
11, -CO
2R
11, -CONR
12R
11, -SO
2NR
12R
11 sind, wobei
HET, Phenyl oder der C
1-6 aliphatische Rest
gegebenenfalls mit R
10 substituiert sind;
und wobei R
2 gegebenenfalls mit
R
3 verbunden ist, um einen kondensierten fünfgliedrigen
Heterocyclylring zu bilden, wobei der Heterocyclylring null bis
ein Heteroatom aufweist, wobei das Heteroatom N ist, und O bis ein
Heteroatom aufweist, wobei das Heteroatom O oder S ist, und wobei
der kondensierte Ring gegebenenfalls mit R
9 substituiert
ist, wobei HET,
R9
, R10 , R
11 und
R
12 wie nachstehend definiert sind;
R
4 H ist;
R
5 H
ist;
R
6 und R
7 unabhängig voneinander
Brom oder Chlor sind;
R
9 H, ein C
1-6 aliphatischer Rest oder –COR
10 ist, wobei R
10 wie
nachstehend definiert ist;
R
10 H, ein
C
1-6 aliphatischer Rest oder Amino ist;
R
11 H, ein C
1-6 aliphatischer
Rest, ein Hydroxy-C
1-6 aliphatischer Rest,
Phenyl, ein Phenyl-C
1-6 aliphatischer Rest
oder HET ist;
R
12 H, ein C
1-6 aliphatischer
Rest, ein Hydroxy-C
1-6 aliphatischer Rest
oder ein (R
11)
2N–C
1-6 aliphatischer Rest ist; und
HET
ein heterocyclischer Ring ist, ausgewählt aus Oxazol, Pyridin, Tetrazol
und Thiazol; und die pharmazeutisch verträglichen Salze oder Solvate
davon.
-
Noch
eine andere Gruppe von Verbindungen, welche in Bezug auf ihre Substituenten
an Position R
6 und R
7 bevorzugt
sind, sind die Verbindungen der Formel:
wobei:
R
1 H
ist;
R
2 und R
3 unabhängig voneinander
H, HET, Phenyl, ein C
1-6 aliphatischer Rest,
Cyano, Halogen, -COR
11 oder -CONR
12R
11 sind, wobei
HET, Phenyl oder der C
1-6 aliphatische Rest
gegebenenfalls mit R
10 substituiert sind, wobei
HET, R
10, R
11 und
R
12 wie nachstehend definiert sind;
R
4 H ist;
R
5 H
ist;
R
6 und R
7 unabhängig voneinander
Brom oder Chlor sind;
R
10 H, ein C
1-6 aliphatischer Rest, Oxo oder Cyano ist;
R
11 H, ein C
1-6 aliphatischer
Rest, ein Trihalogen-C
1-6 aliphatischer
Rest, Phenyl oder Nitro substituiertes Phenyl ist;
R
12 H, ein C
1-6 aliphatischer
Rest, ein Hydroxy-C
1-6 aliphatischer Rest
ist; und
HET Thiophen oder Pyridin ist;
und die pharmazeutisch
verträglichen
Salze oder Solvate davon.
-
Bestimmte
Verbindungen von vorstehender Formel (I) können in stereoisomeren Formen
existieren (z. B. können
sie ein oder mehrere asymmetrische Kohlenstoffatome enthalten oder
können
cis-trans-Isomere zeigen). Die einzelnen Stereoisomere (Enantiomere
und Diastereoisomere) und Gemische von diesen sind in den Umfang
der vorliegenden Erfindung eingeschlossen. Gleichermaßen ist
es selbstverständlich,
dass Verbindungen der Formel (I) in tautomeren Formen existieren
können,
die sich von denen in der Formel gezeigten unterscheiden, und diese
sind ebenfalls in den Umfang der vorliegenden Erfindung eingeschlossen.
-
Aufgrund
der Gegenwart einer Doppelbindung sind in den Umfang der Erfindung
ebenfalls ihre einzelnen reinen geometrischen E- und Z-Isomere,
ebenso wie Gemische von E- und Z-Isomeren eingeschlossen.
-
-
Die
Erfindung wie beschrieben und beansprucht setzt keinerlei beschränkende Verhältnisse
auf das Vorherrschen von Z- zu E-Isomeren.
-
Daher
wird die Verbindung 3-(3,5-Dibrom-4-hydroxybenzyliden)-5-pyrid-3-yl-1,3-dihydroindol-2-on, Verbindungsnummer
138 in den nachstehenden Tabellen als das geometrische E-Isomer davon, das
geometrische Z-Isomer davon und ein Gemisch aus den E- und Z-Isomeren
davon offenbart und beansprucht, aber nicht durch jedwede(s) gegebene(s)
Verhältnis(se)
beschränkt.
-
Bestimmte
Verbindungen wie beschrieben, werden ein oder mehrere chirale Kohlenstoffatome
enthalten und werden daher rechtsdrehend oder linksdrehend sein.
Ebenfalls in die erfindungsgemäßen Verbindungen
eingeschlossen sind die einzelnen rechtsdrehenden oder linksdrehenden
reinen Präparate
und racemischen Gemische davon.
-
Salze
der erfindungsgemäßen Verbindungen
können
Säureadditionssalze,
welche von einem Stickstoff an einem Substituenten in der Verbindung
der Formel (I) abgeleitet sind, umfassen. Die therapeutische Aktivität bleibt
in der Einheit, die von der erfindungsgemäßen Verbindung, wie hierin
definiert, abgeleitet ist und die Identität von einer anderen Komponente
ist weniger wichtig, obwohl sie für therapeutische und prophylaktische
Zwecke vorzugsweise pharmazeutisch verträglich für den Patienten ist.
-
Stark
bevorzugte biohydrolysierbare Carbamate umfassen Verbindungen der
Formel (I), wobei die aromatische OH-Gruppe, welche von R6 und R7 flankiert
ist, mit einem Carbamoylkonjugat konjugiert ist, um ein biohydrolysierbare
Carbamat zu erhalten, wobei das Carbamoylkonjugat ausgewählt ist
aus Diethylaminocarbonyl, N-(2-Hydroxyethyl)aminocarbonyl, N,N,-Bis(2-hydroxyethyl)aminocarbonyl,
Hydroxyethyloxyethylaminocarbonyl, 4-Morpholinocarbonyl und 4-Methyl-1-piperazinylcarbonyl.
-
Stark
bevorzugte biohydrolysierbare Carbonate umfassen Verbindungen der
Formel (I), wobei die aromatische OH-Gruppe, welche von R6 und R7 flankiert
ist, mit einem Carbonatkonjugat konjugiert ist, um ein biohydrolysierbares
Carbonat zu erhalten, wobei das Carbonatkonjugat ausgewählt ist
aus Phenylmethyloxycarbonyl, Ethyloxycarbonyl, Isobutyloxycarbonyl
und Pyridinmethyloxycarbonyl.
-
Stark
bevorzugte biohydrolysierbare Ester umfassen Verbindungen der Formel
(I), wobei die aromatische OH-Gruppe, welche von R6 und
R7 flankiert ist, mit einem Esterkonjugat
konjugiert ist, um einen biohydrolysierbaren Ester zu erhalten,
wobei das Esterkonjugat ausgewählt
ist aus t-Butylcarbonyloxymethyl.
-
Die
Erfindung schließt
ferner eine Verbindung der Formel (I) oder eines seiner pharmazeutisch
verträglichen
Salze, Prodrugs, biohydrolysierbaren Ester, Amide, Carbonate, Amine,
Ureide oder Carbamate zur Verwendung bei der Herstellung eines Medikaments
zur Behandlung von Störungen,
die durch Proteinkinaseaktivität
vermittelt werden, ein.
-
Die
Erfindung schließt
ferner eine Verbindung der Formel (I) oder eines seiner pharmazeutisch
verträglichen
Salze, Prodrugs, biohydrolysierbaren Ester, Amide, Carbonate, Amine,
Ureide oder Carbamate zur Verwendung bei der Herstellung eines Medikaments
zur Behandlung von Störungen,
die durch Störungen
vermittelt werden, welche durch ein mutiertes ras-Gen verursacht
werden, ein.
-
Die
Erfindung schließt
ferner eine Verbindung der Formel (I) oder eines seiner pharmazeutisch
verträglichen
Salze, Prodrugs, biohydrolysierbaren Ester, Amide, Carbonate, Amin,
Ureide oder Carbamate zur Verwendung bei der Herstellung eines Medikaments
zur Behandlung von Störungen,
die durch einen hochregulierten Tyrosinekinase-Signalübertragungsweg
vermittelt werden, ein.
-
Die
Erfindung schließt
ferner eine Verbindung der Formel (I) oder eines seiner pharmazeutisch
verträglichen
Salze, Prodrugs, biohydrolysierbaren Ester, Amide, Carbonate, Amine,
Ureide oder Carbamate zur Verwendung bei der Herstellung eines Medikaments
zur Behandlung von Störungen,
die durch eine Mitogen-aktivierte Proteinkinase vermittelt werden,
ein.
-
Die
Erfindung schließt
ferner eine Verbindung der Formel (I) oder eines seiner pharmazeutisch
verträglichen
Salze, Prodrugs, biohydrolysierbaren Ester, Amide, Carbonate, Amine,
Ureide oder Carbamate zur Verwendung bei der Herstellung eines Medikaments
zur Behandlung von Störungen,
die durch cRaf-Kinase vermittelt werden, ein.
-
Eine
Gruppe von bevorzugten erfindungsgemäßen Verbindungsarten umfasst
die Gruppe:
-
Eine
andere Gruppe von bevorzugten erfindungsgemäßen Verbindungen umfasst die
Gruppe:
-
Noch
eine andere Gruppe von bevorzugten Verbindungen umfasst die Gruppe:
-
Eine
besonders bevorzugte Gruppe von Verbindungen umfasst die Gruppe:
-
Unabhängige
Substituenten
-
Die
Erfindung offenbart sieben verschiedene Punkte der Substitution
an der Strukturformel (I). Jeder dieser Punkte der Substitution
trägt einen
Substituenten, dessen Wahl und Synthese als Teil dieser Erfindung unabhängig von
allen anderen Punkten der Substitution an Formel (I) war. Daher
wird nun jeder Punkt der Substitution einzeln weiter beschrieben.
-
R1 ist Wasserstoff. Gegebenenfalls kann R1 mit einem Substituenten R2 verbunden
sein, um einen kondensierten Ring zu bilden. Derartige kondensierte
Ringe können
fünf- bis
zehngliedrige Aryl-, Heteroaryl- oder Heterocyclylringe oder –ringsysteme
sein mit 1 bis 3 Heteroatomen. Diese Heteroatome können Stickstoff, Sauerstoff
oder Schwefel sein. Derartige kondensierte Ringe können gegebenenfalls
mit einem bis drei Resten Halogen, Cyano, Nitro, substituiertem
Amid, substituiertem Sulfonamid, substituiertem Amin, substituiertem Ether
oder Hydroxyl substituiert sein. Substituenten für Amide, Sulfonamide, Amine
oder Ether schließen
Wasserstoff, Halogen, einen aliphatischen 1 bis 12 Kohlenstoffrest
(welcher einen eingefügten
Rest irgendwo entlang seiner Kettenlänge von einem Sauerstoff, einem
Schwefel, einem Sulfoxid, einem Sulfon, einem Sulfin oder einem
sekundärem
Amin tragen kann), Arylringe, heterocyclische Ringe ein. Substituenten
an diesen aliphatischen, Aryl- oder heterocyclischen Resten schließen 1 bis
3 Substitutionen mit einem Halogen, einem anderen heterocyclischen
Ring, einem anderen Arylring, Cyano, substituiertem Sulfo, substituiertem
Oxy, substituiertem Amin, substituiertem Sulfoxid, substituiertem
Sulfin, substituiertem Sulfon, substituiertem Sulfonamid, substituiertem
Amid, substituiertem Ureid, substituiertem Ester, substituiertem
Carbamat ein. Diese Substituenten wiederum können ein aliphatischer 1 bis
12 Kohlenstoffrest oder ein heterocyclischer Ring sein, wobei der
aliphatische 1 bis 12 Kohlenstoffrest selbst durch 1 bis 3 Vorkommnisse
eines Halogens oder Hydroxyls substituiert sein kann.
-
In
einer anderen Ausführungsform
kann R1 Wasserstoff sein oder gegebenenfalls
kann R1 mit einem Substituenten R2 verbunden sein, um einen kondensierten
Ring zu bilden. Derartige kondensierte Ringe können aus Benzofuran, Benzoxazol,
Dioxin, Dioxan, Dioxolan, Dithian, Dithiazin, Dithiazol, Dithiolan,
Furan, Imidazol, Indol, Indazol, Morpholin, Oxazol, Oxadiazol, Oxathiazol,
Oxathiazolidin, Oxazin, Oxadiazin, Piperazin, Piperidin, Pyran,
Pyrazin, Pyrazol, Pyridin, Pyrimidin, Pyrrol, Pyrrolidin, Chinolin,
Chinazolin, Tetrahydrofuran, Tetrazin, Tetrazol, Thiophen, Thiadiazin,
Thiadiazol, Thiatriazol, Thiazin, Thiazol, Thiomorpholin, Thianaphthalin,
Thiopyran, Triazin und Triazol sein. Jeder beliebige dieser Ringe
kann wiederum mit einem Rest der Substituenten, umfassend 1 bis
3 Substitutionen mit einem Halogen, einem anderen heterocyclischen
Ring, einem anderen Arylring, Cyano, substituiertem Sulfo, substituiertem
Oxy, substituiertem Amin, substituiertem Sulfoxid, substituiertem
Sulfin, substituiertem Sulfon, substituiertem Sulfonamid, substituiertem
Amid, substituiertem Ureid, substituiertem Ester, substituiertem
Carbamat substituiert sein. Diese Substituenten wiederum können ein
aliphatischer 1 bis 12 Kohlenstoffrest oder ein heterocyclischer
Ring sein, wobei der aliphatische 1 bis 12 Kohlenstoffrest selbst
durch 1 bis 3 Vorkommnisse eines Halogens oder Hydroxyls substituiert
sein kann.
-
Vorzugsweise
ist R1 Wasserstoff oder mit R2 kondensiert,
um kondensiertes Pyridin, kondensiertes Triazol, kondensiertes Thiazol
oder kondensiertes Amino-substitutiertes Thiazol zu bilden.
-
Am
meisten bevorzugt ist R1 Wasserstoff.
-
R2 ist Wasserstoff, ein Arylring, ein heterocyclischer
Ring, ein aliphatischer 1 bis 12 Kohlenstoffrest, Cyano, Nitro,
Halogen, substituierter Ether, substituierter Thioether, substituiertes
Sulfin, substituiertes Sulfon, substituiertes Amin, disubstituiertes
Amin, substituiertes Amid, substituiertes Carbamat, substituiertes
Sulfonamid, substituiertes Carbonyl oder substituierter Ester. Diese
Substituenten können
Wasserstoff, Halogen, ein aliphatischer 1 bis 12 Kohlenstoffrest
(welcher einen eingefügten
Rest irgendwo entlang seiner Kettenlänge von einem Sauerstoff, einem
Schwefel, einem Sulfoxid, einem Sulfon, einem Sulfin oder einem
sekundärem Amin
tragen kann), Arylringe, heterocyclische Ringe sein. Substituenten
an diesen aliphatischen, Aryl- oder heterocyclischen Resten schließen 1 bis
3 Substitutionen mit einem Halogen, einem anderen heterocyclischen Ring,
einem anderen Arylring, Cyano, substituiertem Sulfo, substituiertem
Oxy, substituiertem Amin, substituiertem Sulfoxid, substituiertem
Sulfin, substituiertem Sulfon, substituiertem Sulfonamid, substituiertem
Amid, substituiertem Ureid, substituiertem Ester, substituiertem
Carbamat ein. Diese Substituenten wiederum können ein aliphatischer 1 bis
12 Kohlenstoffrest oder ein heterocyclischer Ring sein, wobei der
aliphatische 1 bis 12 Kohlenstoffrest selbst durch 1 bis 3 Vorkommnisse
eines Halogens oder Hydroxyls substituiert sein kann.
-
R2 kann mit R3 verbunden
sein, um einen kondensierten Ring ausgewählt aus Benzofuran, Benzoxazol,
Dioxin, Dioxan, Dioxolan, Dithian, Dithiazin, Dithiazol, Dithiolan,
Furan, Imidazol, Indol, Indazol, Morpholin, Oxazol, Oxadiazol, Oxathiazol,
Oxathiazolidin, Oxazin, Oxadiazin, Piperazin, Piperidin, Pyran,
Pyrazin, Pyrazol, Pyridin, Pyrimidin, Pyrrol, Pyrrolidin, Chinolin,
Chinazolin, Tetrahydrofuran, Tetrazin, Tetrazol, Thiophen, Thiadiazin,
Thiadiazol, Thiatriazol, Thiazin, Thiazol, Thiomorpholin, Thianaphthalin,
Thiopyran, Triazin und Triazol zu bilden.
-
R2 kann stärker
bevorzugt Wasserstoff, ein heterocyclischer Ring, Phenyl, ein aliphatischer
1 bis 6 Kohlenstoffrest, ein substituiertes Amin, ein substituiertes
Carbonyl, ein substituierter Ester, ein substituiertes Amid, ein
substituiertes Sulfonamid sein. Der heterocyclische Ring, das Phenyl
oder der aliphatische Rest sind gegebenenfalls mit Amino oder einem
aliphatischen 1 bis 6 Kohlenstoffrest substituiert. Das Amin, Carbonyl, Esteramid
oder Sulfonamid sind gegebenenfalls mit einem aliphatischen 1 bis
6 Kohlenstoffrest, Amino, Hydroxy-aliphatischen 1 bis 6 Kohlenstoffresten,
Phenyl, Phenyl-aliphatischen 1 bis 6 Kohlenstoffresten, Amino-aliphatischen 1 bis
12 Kohlenstoffresten oder heterocyclischen Ringen, wie Oxazol, Pyridin,
Tetrazol oder Thiazol, substituiert.
-
R2 kann stärker
bevorzugt mit R3 verbunden sein, um einen
fünf-gliedrigen
kondensierten Ring mit einem Heteroatom von entweder Stickstoff,
Sauerstoff oder Schwefel zu bilden. Diese kondensierten Ringe können mit
einem aliphatischen 1 bis 6 Kohlenstoffrest oder einem 1 bis 6 Acylkohlenstoffrest
substituiert sein.
-
R2 kann ebenfalls stärker bevorzugt Wasserstoff,
Thiophen, Pyridin, Phenyl, ein aliphatischer 1 bis 6 Kohlenstoffrest,
Cyano, Halogen, substituiertes Acyl oder substituiertes Amid sein.
Diese Substituenten können
ein aliphatischer 1 bis 6 Kohlenstoffrest, ein Trihalogen-aliphatischer
1 bis 6 Kohlenstoffrest, Phenyl, Nitrosubstituiertes Phenyl oder
Hydroxy-aliphatische 1 bis 6 Kohlenstoffreste sein.
-
R2 ist Wasserstoff, ein Arylring, ein heterocyclischer
Ring, ein aliphatischer 1 bis 12 Kohlenstoffrest, Cyano, Nitro,
Halogen, substituierter Ether, substituierter Thioether, substituiertes
Sulfin, substituiertes Sulfon, substituiertes Amin, disubstituiertes
Amin, substituiertes Amid, substituiertes Carbamat, substituiertes
Sulfonamid, substituiertes Carbonyl oder substituierter Ester. Diese
Substituenten können
Wasserstoff, Halogen, ein aliphatischer 1 bis 12 Kohlenstoffrest
(welcher einen eingefügten
Rest irgendwo entlang seiner Kettenlänge von einem Sauerstoff, einem
Schwefel, einem Sulfoxid, einem Sulfon, einem Sulfin oder einem
sekundärem Amin
tragen kann), Arylringe, heterocyclische Ringe sein. Substituenten
an diesen aliphatischen, Aryl- oder heterocyclischen Resten schließen 1 bis
3 Substitutionen mit einem Halogen, einem weiteren heterocyclischen Ring,
einem weiteren Arylring, Cyano, substituiertem Sulfo, substituiertem
Oxy, substituiertem Amin, substituiertem Sulfoxid, substituiertem
Sulfin, substituiertem Sulfon, substituiertem Sulfonamid, substituiertem
Amid, substituiertem Ureid, substituiertem Ester, substituiertem
Carbamat ein. Diese Substituenten wiederum können ein aliphatischer 1 bis
12 Kohlenstoffrest oder ein heterocyclischer Ring sein, wobei der
aliphatische 1 bis 12 Kohlenstoffrest selbst mit 1 bis 3 Vorkommnissen
eines Halogens oder Hydroxyls substituiert sein kann.
-
R3 kann mit R2 verbunden
sein, um einen kondensierten Ring ausgewählt aus Benzofuran, Benzoxazol,
Dioxin, Dioxan, Dioxolan, Dithian, Dithiazin, Dithiazol, Dithiolan,
Furan, Imidazol, Indol, Indazol, Morpholin, Oxazol, Oxadiazol, Oxathiazol,
Oxathiazolidin, Oxazin, Oxadiazin, Piperazin, Piperidin, Pyran,
Pyrazin, Pyrazol, Pyridin, Pyrimidin, Pyrrol, Pyrrolidin, Chinolin,
Chinazolin, Tetrahydrofuran, Tetrazin, Tetrazol, Thiophen, Thiadiazin,
Thiadiazol, Thiatriazol, Thiazin, Thiazol, Thiomorpholin, Thianaphthalin,
Thiopyran, Triazin und Triazol zu bilden.
-
R3 kann stärker
bevorzugt Wasserstoff, ein heterocyclischer Ring, Phenyl, ein aliphatischer
1 bis 6 Kohlenstoffrest, ein substituiertes Amin, ein substituiertes
Carbonyl, ein substituierter Ester, ein substituiertes Amid, ein
substituiertes Sulfonamid sein. Der heterocyclische Ring, das Phenyl
oder der aliphatische Rest sind gegebenenfalls mit Amino oder einem
aliphatischen 1 bis 6 Kohlenstoffrest substituiert. Das Amin, Carbonyl, Esteramid
oder Sulfonamid sind gegebenenfalls mit einem aliphatischen 1 bis
6 Kohlenstoffrest, Amino, Hydroxy-aliphatischen 1 bis 6 Kohlenstoffresten,
Phenyl, Phenyl-aliphatischen 1 bis 6 Kohlenstoffresten, Amino-aliphatischen 1 bis
12 Kohlenstoffresten oder heterocyclischen Ringen, wie Oxazol, Pyridin,
Tetrazol oder Thiazol, substituiert.
-
R3 kann stärker
bevorzugt mit R2 verbunden sein, um einen
fünf-gliedrigen
kondensierten Ring mit einem Heteroatom von entweder Stickstoff,
Sauerstoff oder Schwefel zu bilden. Diese kondensierten Ringe können mit
einem aliphatischen 1 bis 6 Kohlenstoffrest oder einem 1 bis 6 Acylkohlenstoffrest
substituiert sein.
-
R3 kann ebenfalls stärker bevorzugt Wasserstoff,
Thiophen, Pyridin, Phenyl, ein aliphatischer 1 bis 6 Kohlenstoffrest,
Cyano, Halogen, substituiertes Acyl oder substituiertes Amid sein.
Diese Substituenten können
ein aliphatischer 1 bis 6 Kohlenstoffrest, ein Trihalogen-aliphatischer
1 bis 6 Kohlenstoffrest, Phenyl, Nitrosubstituiertes Phenyl oder
Hydroxy-aliphatische 1 bis 6 Kohlenstoffreste sein.
-
R4 ist Wasserstoff, Nitro, Cyano oder Halogen.
-
Vorzugsweise
ist R4 Wasserstoff.
-
R5 ist Wasserstoff oder ein aliphatischer
1 bis 12 Kohlenstoffrest, welcher gegebenenfalls an 1 bis 3 Positionen
mit einem Halogen, Hydroxyl, Heteroaryl oder einem Arylring substituiert
ist.
-
R5 ist in einer anderen Ausführungsform
Wasserstoff oder ein aliphatischer 1 bis 6 Kohlenstoffrest, welcher
gegebenenfalls an 1 bis 3 Positionen mit einem Halogen, Hydroxyl
oder einem Arylring substituiert ist.
-
Vorzugsweise
ist R5 Wasserstoff.
-
R6 ist ein Halogen.
-
R6 ist am meisten bevorzugt ein Brom.
-
In
einer anderen Ausführungsform
ist R6 am meisten bevorzugt ein Chlor.
-
R7 ist ein Halogen.
-
R7 ist am meisten bevorzugt ein Brom.
-
In
einer anderen Ausführungsform
ist R7 am meisten bevorzugt ein Chlor.
-
In
einem weiteren Aspekt stellt die vorliegende Erfindung ein Verfahren
zur Herstellung einer Verbindung der Formel (I) bereit, wobei das
Verfahren die Umsetzung einer Verbindung der Formel (II)
mit einer Verbindung der
Formel (III)
umfasst.
-
Die
Umsetzung wird zweckmäßigerweise
in Gegenwart einer katalytischen Säure in Gegenwart eines geeigneten
inerten Lösungsmittels
durchgeführt,
zum Beispiel einem aromatischen Kohlenwasserstoff oder einem halogenierten
Kohlenwasserstoff bei einer nicht extremen Temperatur, zum Beispiel
von 0 °C
bis 150 °C, vorzugsweise
80 °C bis
110 °C.
Gegebenenfalls wird diese Umsetzung in Gegenwart einer starken Säure, zum Beispiel
Salzsäure
oder Schwefelsäure,
in Essigsäure
als Lösungsmittel
durchgeführt.
-
Die
Herstellung der Verbindungen (II) und (III) ist Fachleuten wohlbekannt
und viele Verbindungen der Formel (II) sind im Handel erhältlich.
(P. G. Gassman; T. J. van Bergen, Oxindols. A New General Method
of Synthesis. Journal of the American Chemical Society, 96 (17),
1974, 5508 – 5512)
(Jutz, Adv. Org. Chem., 9, 225 – 342,
1975; Truce, Org. React., 9, 37 – 72, 1957).
-
Zusätzlich zum
Vorstehenden kann eine Verbindung der Formel (I) in eine andere
Verbindung der Formel (I) durch chemische Transformation des geeigneten
Substituenten oder der geeigneten Substituenten umgewandelt werden.
-
Die
vorliegende Erfindung stellt ebenfalls Verbindungen der Formel (I)
und pharmazeutisch verträgliche
Salze, Prodrugs, biohydrolysierbare Ester, Amide, Carbonate, Amine,
Ureide oder Carbamate davon (hierin nachstehende als die „wirksamen
Verbindungen" definiert)
zur Verwendung bei der medizinischen Therapie und insbesondere bei
der Behandlung von Störungen,
die durch Proteinkinaseaktivität
vermittelt werden, wie menschlichen Malignitäten, bereit. Die Verbindungen
sind besonders für
die Behandlung von Störungen,
die durch mutierte ras- und hochregulierte Tyrosinkinase-Signalübertragungswege
verursacht werden, wie Brust-, Kolon-, Lungen-, Bauchspeicheldrüsen-, Prostata-
und Magenkrebs, nützlich.
-
Die
vorliegende Erfindung stellt ebenfalls die Verwendung einer wie
vorstehend definierten wirksamen Verbindung bei der Herstellung
eines Medikaments zur Behandlung einer Erkrankung, die durch eine
Kinase, ausgewählt
aus ab1, ATK , bcr-ab1, Blk, Brk, Btk, c-kit, c-met, c-src, CDK1, CDK2, CDK4, CDK6,
CSF1R, CSK, EGFR, ErbB2, ErbB3, ErbB4, ERK, Fak, fes, FGFR1, FGFR2,
FGFR3, FGFR4, FGFR5, Fgr, FLK4, flt-1, Fps, Frk, Fyn, Hck, IGF-1R, INS-R, Jak, KDR,
Lck, Lyn, MEK, cRaf1, p38, PDGFR, PIK, PKC, PYK2, ros, tie1, tie2, TRK, Yes
und Zap70 vermittelt wird, bereit.
-
Ein
weiterer Aspekt der vorliegenden Erfindung stellt die Verwendung
einer wie vorstehend definierten wirksamen Verbindung bei der Herstellung
eines Medikaments zur Verwendung bei der Behandlung eines menschlichen
oder tierischen Körpers,
welcher an einer Erkrankung leidet, die durch eine Mitogen-aktivierte Proteinkinase
vermittelt wird, bereit.
-
Die
vorliegende Erfindung stellt insbesondere die Verwendung einer wirksamen
Verbindung wie vorstehend definiert bei der Herstellung eines Medikaments
zur Verwendung bei der Behandlung einer Erkrankung, die durch die
cRaf1-Kinase vermittelt wird, bereit.
-
Die
vorliegende Erfindung stellt ebenfalls die Verwendung einer wirksamen
Verbindung wie vorstehend definiert bei der Herstellung eines Medikaments
zur Verwendung bei der Hemmung von Tumorwachstum, bei der Verhinderung
von Organabstoßungen
nach Transplantationen, bei der Heilung chronischer Wunden oder
bei der Behandlung eines Erkrankungszustands, ausgewählt aus
Restenose, rheumatoider Arthritis, Angiogenese, Leberzirrhose, Atherosklerose, Glomerulonephritis,
diabetischer Nephropathie, maligner Nephrosklerose, thrombotischen
Mikroangiophatie-Syndromen, Glomerulopathie, Psoriasis, Diabetes
mellitus, Entzündung
und neurodegenerativer Krankheit bereit.
-
Ein
weiterer Aspekt der vorliegenden Erfindung stellt die Verwendung
einer wirksamen Verbindung der Formel (I) bei der Herstellung eines
Medikaments zur Behandlung von malignen Tumoren bereit.
-
Ein
weiterer Aspekt der vorliegenden Erfindung stellt die Verwendung
einer wirksamen Verbindung der Formel (I), in Coverabreichung mit
zuvor bekannten Tumor-hemmenden Therapien für eine effektivere Behandlung
derartiger Tumore, bereit.
-
Die
wirksamen Verbindungen der Formel (I) weisen eine antineoplastische
Aktivität
auf wie hierin nachstehend durch ihre Hemmung des Proteins Serin/Threoninkinase
c-Raf1 Enzym demonstriert wird. Es ist daher festgestellt worden,
dass die erfindungsgemäßen Verbindungen
in der Medizin von Nutzen sind und insbesondere bei der Behandlung
von bestimmten menschlichen Malignitäten, zum Beispiel Brust-, Eierstockkrebs,
nicht klein-zelligem Lungenkrebs, Bauchspeicheldrüsen-, Magen-
und Kolonkrebs. Demgemäß stellt
die vorliegende Erfindung ein Verfahren zur Behandlung von anfälligen Malignitäten bei
einem Tier, z. B. einem Menschen, bereit, was das Verabreichen einer
therapeutisch wirksamen Menge einer wirksamen Verbindung an das
Tier wie vorstehend definiert umfasst.
-
Verbindungen,
welche wir als Teil der vorliegenden Erfindung synthetisiert haben,
welche zurzeit bevorzugt werden, sind in den nachstehenden Tabellen
1A, 1B und 1C aufgelistet. Die Verbindungen werden durch die in
der ersten Spalte gezeigten Zahlen identifiziert; nachstehende Variablen
im Rest der Spalten sind unter Bezugnahme auf die allgemeine Struktur
(I). Die entsprechende IUPAC Nomenklatur wird in den nachstehenden
Tabellen 2A, 2B bzw. 2C offenbart. Da alle Substituenten an jedem
Punkt der Substitution zur voneinander unabhängigen Synthese in der Lage
sind, können
die Tabellen 1A, 1B und 1C ebenfalls als eine Matrix gelesen werden,
bei der jede beliebige Substituentenkombination im Umfang der erfindungsgemäßen Offenbarung
und den erfindungsgemäßen Ansprüchen ist.
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
Salze,
die den Begriff „pharmazeutisch
verträgliche
Salze" umspannen,
bezeichnen nicht toxische Salze der erfindungsgemäßen Verbindungen,
welche im Allgemeinen durch Umsetzen einer freien Base mit einer geeigneten
organischen oder anorganischen Säure
oder durch Umsetzen der Säure
mit einer geeigneten organischen oder anorganischen Base hergestellt
werden. Repräsentative
Salze schließen
die folgenden Salze ein: Acetat, Aluminium, Benzolsulfonat, Benzoat,
Bicarbonat, Eisulfat, Bitartrat, Borat, Bromid, Calcium, Calciumedetat,
Camsylat, Carbonat, Chlorid, Chlorprocain, Cholin, Clavulanat, Citrat,
Dibenzylethylendiamin, Diethanolamin, Dihydrochlorid, Edetat, Edisylat,
Estolat, Esylat, Ethylendiamin, Fumarat, Gluceptat, Gluconat, Glutamat,
Glycolylarsanilat, Hexylresorcinat, Hydrabamin, Hydrobromid, Hydrochlorid,
Hydroxynaphthoat, Iodid, Isethionat, Lactat, Lithium, Lactobionat,
Laurat, Malat, Maleat, Magnesium, Mandelat, Mesylat, Methylbromid, Methylnitrat,
Methylsulfat, Monokaliummaleat, Mucat, Napsylat, Nitrat, N-Methylglucamin,
Oxalat, Pamoat (Embonat), Palmitat, Pantothenat, Phosphat/Diphosphat,
Polygalacturonat, Kalium, Procain, Salicylat, Natrium, Stearat,
Subacetat, Succinat, Sulfat, Tannat, Tartrat, Teoclat, Tosylat,
Triethanolamin, Triethiodid, Trimethylammonium und Valerat.
-
Salze,
welche nicht pharmazeutisch verträglich sind, können bei
der Herstellung von Verbindungen der Formel (I) nützlich sein,
und diese bilden einen weiteren erfindungsgemäßen Aspekt.
-
Ebenfalls
in den erfindungsgemäßen Umfang
eingeschlossen sind die einzelnen Isomere der vorstehenden Verbindungen
der Formel (I), ebenso wie jede beliebige der völlig oder teilweise äquilibrierten
Gemische davon. Die vorliegende Erfindung deckt ebenfalls die einzelnen
Isomere der Verbindungen der vorstehenden Formeln als Gemische mit
Isomeren davon, bei welchen eine oder mehrere chirale Zentren invertiert sind,
ab.
-
Für die folgenden
definierten Begriffe sollen diese Definitionen angewendet werden,
außer
es wird eine andere Definition in den Ansprüchen oder anderswo in dieser
Beschreibung gegeben.
-
Wie
hierin verwendet, bezeichnet der Begriff "aliphatisch" die Begriffe Alkyl, Alkylen, Alkenyl,
Alkenylen, Alkinyl und Alkinylen.
-
Wie
hierin verwendet, bezeichnet der Begriff "Nieder-" einen Rest mit zwischen einem und sechs
Kohlenstoffen.
-
Wie
hierin verwendet, bezeichnet der Begriff "Alkyl" einen Kohlenwasserstoff mit gerader
oder verzweigter Kette mit einer spezifizierten Anzahl von Kohlenstoffatomen,
gegebenenfalls mit Substituenten, ausgewählt aus Niederalkyl, Niederalkoxy,
Niederalkylsulfanyl, Niederalkylsulfenyl, Niederalkylsulfonyl, Oxo,
Hydroxy, Mercapto, Amino, gegebenenfalls substituiert mit Alkyl,
Carboxy, Carbamoyl, gegebenenfalls substituiert mit Alkyl, Aminosulfonyl,
gegebenenfalls substituiert mit Alkyl, Nitro, Cyano, Halogen oder
Niederperfluoralkyl, wobei mehrere Grade der Substitution erlaubt
sind. Beispiele für "Alkyl", wie hierin verwendet,
schließen n-Butyl,
n-Pentyl, Isobutyl und Isopropyl und dergleichen ein, sind aber
nicht beschränkt
darauf. Der Begriff "Alkyl", wie hierin verwendet,
bezeichnet ebenfalls allgemein die nachstehend definierten Begriffe "Alkylen", "Alkenyl", "Alkenylen", "Alkinyl" und "Alkinylen".
-
Wie
hierin verwendet, bezeichnet der Begriff "Alkylen" einen divalenten Kohlenwasserstoffrest
mit gerader oder verzweigter Kette mit einem bis zehn Kohlenstoffatomen,
gegebenenfalls substituiert mit Substituenten, ausgewählt aus
Niederalkyl, Niederalkoxy, Niederalkylsulfanyl, Niederalkylsulfenyl,
Niederalkylsulfonyl, Oxo, Hydroxy, Mercapto, Amino, gegebenenfalls
substituiert mit Alkyl, Carboxy, Carbamoyl, gegebenenfalls substituiert
mit Alkyl, Aminosulfonyl, gegebenenfalls substituiert mit Alkyl,
Nitro, Cyano, Halogen oder Niederperfluoralkyl, wobei mehrere Grade
der Substitution erlaubt sind. Beispiele für "Alkylen" wie hierin verwendet, schließen Methylen,
Ethylen und dergleichen ein, sind aber nicht beschränkt darauf.
-
Wie
hierin verwendet, bezeichnet der Begriff "Alkenyl" einen Kohlenwasserstoffrest mit zwei
bis zehn Kohlenstoffen und mindestens einer Kohlenstoff-Kohlenstoffdoppelbindung,
gegebenenfalls substituiert mit Substituenten, ausgewählt aus
Niederalkyl, Niederalkoxy, Niederalkylsulfanyl, Niederalkylsulfenyl,
Niederalkylsulfonyl, Oxo, Hydroxy, Mercapto, Amino, gegebenenfalls
substituiert mit Alkyl, Carboxy, Carbamoyl, gegebenenfalls substituiert
mit Alkyl, Aminosulfonyl, gegebenenfalls substituiert mit Alkyl,
Nitro, Cyano, Halogen oder Niederperfluoralkyl, wobei mehrere Grade
der Substitution erlaubt sind.
-
Wie
hierin verwendet, bezeichnet der Begriff "Alkenylen" einen divalenten Kohlenwasserstoffrest
mit gerader oder verzweigter Kette mit zwei bis zehn Kohlenstoffatomen
und einer oder mehrerer Kohlenstoff-Kohlenstoffdoppelbindungen,
gegebenenfalls substituiert mit Substituenten, ausgewählt aus
Niederalkyl, Niederalkoxy, Niederalkylsulfanyl, Niederalkylsulfenyl,
Niederalkylsulfonyl, Oxo, Hydroxy, Mercapto, Amino, gegebenenfalls
substituiert mit Alkyl, Carboxy, Carbamoyl, gegebenenfalls substituiert
mit Alkyl, Aminosulfonyl, gegebenenfalls substituiert mit Alkyl,
Nitro, Cyano, Halogen oder Niederperfluoralkyl, wobei mehrere Grade
der Substitution erlaubt sind. Beispiele für "Alkenylen" wie hierin verwendet, schließen Ethen-1,2-diyl,
Propen-1,3-diyl, Methylen-diyl und dergleichen ein, sind aber nicht
beschränkt
darauf.
-
Wie
hierin verwendet, bezeichnet der Begriff "Alkinyl" einen Kohlenwasserstoffrest mit zwei
bis zehn Kohlenstoffen und mindestens einer Kohlenstoff-Kohlenstoffdreifachbindung,
gegebenenfalls substituiert mit Substituenten, ausgewählt aus
Niederalkyl, Niederalkoxy, Niederalkylsulfanyl, Niederalkylsulfenyl,
Niederalkylsulfonyl, Oxo, Hydroxy, Mercapto, Amino, gegebenenfalls
substituiert mit Alkyl, Carboxy, Carbamoyl, gegebenenfalls substituiert
mit Alkyl, Aminosulfonyl, gegebenenfalls substituiert mit Alkyl,
Nitro, Cyano, Halogen oder Niederperfluoralkyl, wobei mehrere Grade
der Substitution erlaubt sind.
-
Wie
hierin verwendet, bezeichnet der Begriff "Alkinylen" einen divalenten Kohlenwasserstoffrest
mit gerader oder verzweigter Kette mit zwei bis zehn Kohlenstoffatomen und
einer oder mehrerer Kohlenstoff-Kohlenstoffdreifachbindungen, gegebenenfalls
substituiert mit Substituenten, ausgewählt aus Niederalkyl, Niederalkoxy,
Niederalkylsulfanyl, Niederalkylsulfenyl, Niederalkylsulfonyl, Oxo,
Hydroxy, Mercapto, Amino, gegebenenfalls substituiert mit Alkyl,
Carboxy, Carbamoyl, gegebenenfalls substituiert mit Alkyl, Aminosulfonyl,
gegebenenfalls substituiert mit Alkyl, Nitro, Cyano, Halogen oder
Niederperfluoralkyl, wobei mehrere Grade der Substitution erlaubt
sind. Beispiele für "Alkinylen" wie hierin verwendet,
schließen
Ethin-1,2-diyl, Propin-1,3-diyl und dergleichen ein, sind aber nicht
beschränkt
darauf.
-
Wie
hierin verwendet, bezeichnet "Cycloalkyl" einen alicyclischen
Kohlenwasserstoffrest mit einem oder mehreren Graden von Ungesättigtheit,
mit drei bis zwölf
Kohlenstoffatomen, gegebenenfalls substituiert mit Substituenten,
ausgewählt
aus Niederalkyl, Niederalkoxy, Niederalkylsulfanyl, Niederalkylsulfenyl,
Niederalkylsulfonyl, Oxo, Hydroxy, Mercapto, Amino, gegebenenfalls
substituiert mit Alkyl, Carboxy, Carbamoyl, gegebenenfalls substituiert
mit Alkyl, Aminosulfonyl, gegebenenfalls substituiert mit Alkyl,
Nitro, Cyano, Halogen oder Niederperfluoralkyl, wobei mehrere Grade
der Substitution erlaubt sind. "Cycloalkyl" schließt als Beispiel
Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl, Cycloheptyl oder
Cyclooctyl und dergleichen ein. Der Begriff "Cycloalkyl", wie hierin verwendet, bezeichnet ebenfalls
allgemein die nachstehend definierten Begriffe "Cycloalkylen", "Cycloalkenyl" und "Cycloalkenylen".
-
Wie
hierin verwendet, bezeichnet der Begriff "Cycloalkylen" nicht aromatische alicyclische divalente Kohlenwasserstoffreste
mit drei bis zwölf
Kohlenstoffatomen, gegebenenfalls substituiert mit Substituenten, ausgewählt aus
Niederalkyl, Niederalkoxy, Niederalkylsulfanyl, Niederalkylsulfenyl,
Niederalkylsulfonyl, Oxo, Hydroxy, Mercapto, Amino, gegebenenfalls
substituiert mit Alkyl, Carboxy, Carbamoyl, gegebenenfalls substituiert
mit Alkyl, Aminosulfonyl, gegebenenfalls substituiert mit Alkyl,
Nitro, Cyano, Halogen oder Niederperfluoralkyl, wobei mehrere Grade
der Substitution erlaubt sind. Beispiele für "Cycloalkylen" wie hierin verwendet, schließen Cyclopropyl-1,1-diyl,
Cyclopropyl-1,2-diyl, Cyclobutyl-1,2-diyl, Cyclopentyl-1,3-diyl,
Cyclohexyl-1,4-diyl, Cycloheptyl-1,4-diyl oder Cyclooctyl-1,5-diyl
und dergleichen ein, sind aber nicht beschränkt darauf.
-
Wie
hierin verwendet, bezeichnet der Begriff "Cycloalkenyl" einen alicyclisches Kohlenwasserstoffrest mit
drei bis zwölf
Kohlenstoffatomen und mit mindestens einer Kohlenstoff-Kohlenstoffdoppelbindung
im Ringsystem, gegebenenfalls substituiert mit Substituenten, ausgewählt aus
Niederalkyl, Niederalkoxy, Niederalkylsulfanyl, Niederalkylsulfenyl,
Niederalkylsulfonyl, Oxo, Hydroxy, Mercapto, Amino, gegebenenfalls
substituiert mit Alkyl, Carboxy, Carbamoyl, gegebenenfalls substituiert
mit Alkyl, Aminosulfonyl, gegebenenfalls substituiert mit Alkyl,
Nitro, Cyano, Halogen oder Niederperfluoralkyl, wobei mehrere Grade
der Substitution erlaubt sind. Beispiele für "Cycloalkenylen" wie hierin verwendet, schließen 1-Cyclopenten-3-yl,
1-Cyclohexen-3-yl, 1-Cyclohepten-4-yl
und dergleichen ein, sind aber nicht beschränkt darauf.
-
Wie
hierin verwendet, bezeichnet der Begriff "Cycloalkenylen" einen substituierten alicyclischen
divalenten Kohlenwasserstoffrest mit drei bis zwölf Kohlenstoffatomen und mit
mindestens einer Kohlenstoff-Kohlenstoffdoppelbindung im Ringsystem,
gegebenenfalls substituiert mit Substituenten, ausgewählt aus
Niederalkyl, Niederalkoxy, Niederalkylsulfanyl, Niederalkylsulfenyl,
Niederalkylsulfonyl, Oxo, Hydroxy, Mercapto, Amino, gegebenenfalls
substituiert mit Alkyl, Carboxy, Carbamoyl, gegebenenfalls substituiert
mit Alkyl, Aminosulfonyl, gegebenenfalls substituiert mit Alkyl,
Nitro, Cyano, Halogen oder Niederperfluoralkyl, wobei mehrere Grade
der Substitution erlaubt sind. Beispiele für "Cycloalkenylen" wie hierin verwendet, schließen 4,5-Cyclopenten-1,3-diyl,
3,4-Cyclohexen-1,1-diyl
und dergleichen ein, sind aber nicht beschränkt darauf.
-
Wie
hierin verwendet, bezeichnet der Begriff "heterocyclisch" oder der Begriff "Heterocyclyl" einen drei bis zwölf-gliedrigen heterocyclischen
Ring mit einem oder mehreren Graden von Ungesättigtheit, welcher ein oder
mehrere Substitutionen mit Heteroatomen enthält, ausgewählt aus S, SO, SO2,
O oder N, gegebenenfalls substituiert mit Substituenten, ausgewählt aus
Niederalkyl, Niederalkoxy, Niederalkylsulfanyl, Niederalkylsulfenyl,
Niederalkylsulfonyl, Oxo, Hydroxy, Mercapto, Amino, gegebenenfalls
substituiert mit Alkyl, Carboxy, Carbamoyl, gegebenenfalls substituiert
mit Alkyl, Aminosulfonyl, gegebenenfalls substituiert mit Alkyl,
Nitro, Cyano, Halogen, Niederperfluoralkyl oder andere wie durch
diese Beschreibung und Ansprüche
hindurch identifizierten, wobei mehrere Grade der Substitution erlaubt
sind. Ein derartiger Ring kann gegebenenfalls an einen oder mehrere
weitere "heterocyclische" Ring(e) oder Cycloalkylring(e)
kondensiert sein. Beispiele für "heterocyclisch" schließen Tetrahydrofuran,
Pyran, 1,4-Dioxan, 1,3-Dioxan, Piperidin, Pyrrolidin, Morpholin,
Tetrahydrothiopyran, Tetrahydrothiophen und dergleichen ein, sind
aber nicht beschränkt
darauf. Eine umfassendere Auflistung derartiger Ringe wird in der
Zusammenfassung der Erfindung gefunden. Der Begriff "heterocyclisch" bezeichnet ebenfalls
allgemein den nachstehend definierten Begriff "Heterocyclylen".
-
Wie
hierin verwendet, bezeichnet der Begriff "Heterocyclylen" einen drei bis zwölf-gliedrigen divalenten heterocyclischen
Ringrest mit einem oder mehreren Graden von Ungesättigtheit,
welcher eine oder mehrere Heteroatome enthält, ausgewählt aus S, SO, SO2,
O oder N, gegebenenfalls substituiert mit Substituenten, ausgewählt aus
Niederalkyl, Niederalkoxy, Niederalkylsulfanyl, Niederalkylsulfenyl,
Niederalkylsulfonyl, Oxo, Hydroxy, Mercapto, Amino, gegebenenfalls
substituiert mit Alkyl, Carboxy, Carbamoyl, gegebenenfalls substituiert
mit Alkyl, Aminosulfonyl, gegebenenfalls substituiert mit Alkyl,
Nitro, Cyano, Halogen, Niederperfluoralkyl oder andere wie durch
diese Beschreibung und Ansprüche
hindurch identifizierten, wobei mehrere Grade der Substitution erlaubt
sind. Ein derartiger Ring kann gegebenenfalls an einen oder mehrere
andere Benzolringe oder an einen oder mehrere andere "heterocyclische" Ringe oder Cycloalkylringe
kondensiert sein. Beispiele für "Heterocyclylen" schließen Tetrahydrofuran-2,5-diyl,
Morpholin-2,3-diyl, Pyran-2,4-diyl, 1,4-Dioxan-2,3-diyl, 1,3-Dioxan-2,4-diyl,
Piperidin-2,4-diyl, Piperidin-1,4-diyl, Pyrrolidin-1,3-diyl, Morpholin-2,4-diyl und dergleichen
ein, sind aber nicht beschränkt
darauf. Eine umfassendere Auflistung derartiger Ringe wird in der
Zusammenfassung der Erfindung gefunden.
-
Wie
hierin verwendet, bezeichnet der Begriff "Aryl" einen
Benzolring oder ein gegebenenfalls substituiertes Benzolringsystem,
welches an einen oder mehrere gegebenenfalls substituierte Benzolringe
kondensiert ist, gegebenenfalls substituiert mit Substituenten,
ausgewählt
aus Niederalkyl, Niederalkoxy, Niederalkylsulfanyl, Niederalkylsulfenyl,
Niederalkylsulfonyl, Oxo, Hydroxy, Mercapto, Amino, gegebenenfalls
substituiert mit Alkyl, Carboxy, Tetrazolyl, Carbamoyl, gegebenenfalls
substituiert mit Alkyl, Aminosulfonyl, gegebenenfalls substituiert
mit Alkyl, Acyl, Aroyl, Heteroaroyl, Acyloxy, Aroyloxy, Heteroaroyloxy,
Alkoxycarbonyl, Nitro, Cyano, Halogen, Niederperfluoralkyl, Heteroaryl
oder Aryl, wobei mehrere Grade der Substitution erlaubt sind. Beispiele
für Aryl
schließen
Phenyl, 2-Naphthyl, 1-Naphthyl, Biphenyl und dergleichen ein, sind
aber nicht beschränkt
darauf.
-
Wie
hierin verwendet, bezeichnet der Begriff "Arylen" einen divalenten Benzolring oder ein
divalentes Benzolringsystem, welches an einen oder mehrere gegebenenfalls
substituierten Benzolringe kondensiert ist, gegebenenfalls substituiert
mit Substituenten, ausgewählt
aus Niederalkyl, Niederalkoxy, Niederalkylsulfanyl, Niederalkylsulfenyl,
Niederalkylsulfonyl, Oxo, Hydroxy, Mercapto, Amino, gegebenenfalls
substituiert mit Alkyl, Carboxy, Tetrazolyl, Carbamoyl, gegebenenfalls
substituiert mit Alkyl, Aminosulfonyl, gegebenenfalls substituiert
mit Alkyl, Acyl, Aroyl, Heteroaroyl, Acyloxy, Aroyloxy, Heteroaroyloxy,
Alkoxycarbonyl, Nitro, Cyano, Halogen, Niederperfluoralkyl, Heteroaryl
oder Aryl, wobei mehrere Grade der Substitution erlaubt sind. Beispiele für "Arylen" schließen Benzol-1,4-diyl,
Naphthalin-1,8-diyl, Anthrazin-1,4-diyl und dergleichen ein, sind
aber nicht beschränkt
darauf.
-
Wie
hierin verwendet, bezeichnet der Begriff "Heteroaryl" einen fünf- bis siebengliedrigen aromatischen
Ring oder einen polycyclischen heterocyclischen aromatischen Ring,
welcher ein oder mehrere Stickstoff-, Sauerstoff- oder Schwefelheteroatome
enthält,
wobei N-Oxide und Schwefelmonoxide und Schwefeldioxide zulässige heteroaromatische
Substitutionen sind, gegebenenfalls substituiert mit Substituenten,
ausgewählt
aus Niederalkyl, Niederalkoxy, Niederalkylsulfanyl, Niederalkylsulfenyl,
Niederalkylsulfonyl, Oxo, Hydroxy, Mercapto, Amino, gegebenenfalls
substituiert mit Alkyl, Carboxy, Tetrazolyl, Carbamoyl, gegebenenfalls substituiert
mit Alkyl, Aminosulfonyl, gegebenenfalls substituiert mit Alkyl,
Acyl, Aroyl, Heteroaroyl, Acyloxy, Aroyloxy, Heteroaroyloxy, Alkoxycarbonyl,
Nitro, Cyano, Halogen, Niederperfluoralkyl, Heteroaryl oder anderen
wie durch diese Beschreibung und Ansprüche hindurch identifizierten,
wobei mehrere Grade der Substitution erlaubt sind. Für polycyclische
aromatische Ringsysteme können
ein oder mehrere der Ringe ein oder mehrere Heteroatome enthalten.
Beispiele für
hierin verwendetes "Heteroaryl" sind Furan, Thiophen,
Pyrrol, Imidazol, Pyrazol, Triazol, Tetrazol, Thiazol, Oxazol, Isoxazol,
Oxadiazol, Thiadiazol, Isothiazol, Pyridin, Pyridazin, Pyrazin,
Pyrimidin, Chinolin, Isochinolin, Benzofuran, Benzothiophen, Indol
und Indazol, und dergleichen. Eine umfassendere Auflistung derartiger
Ringe wird in der Zusammenfassung der Erfindung gefunden. Der Begriff "Heteroaryl" bezeichnet ebenfalls
allgemein den nachstehend definierten Begriff "Heteroarylen".
-
Wie
hierin verwendet, bezeichnet der Begriff "Heteroarylen" einen fünf- bis siebengliedrigen divalenten aromatischen
Ring oder einen divalenten polycyclischen heterocyclischen aromatischen
Ring, welcher ein oder mehrere Stickstoff-, Sauerstoff- oder Schwefelheteroatome
enthält,
wobei N-Oxide und Schwefelmonoxide und Schwefeldioxide zulässige heteroaromatische
Substitutionen sind, gegebenenfalls substituiert mit Substituenten,
ausgewählt
aus Niederalkyl, Niederalkoxy, Niederalkylsulfanyl, Niederalkylsulfenyl,
Niederalkylsulfonyl, Oxo, Hydroxy, Mercapto, Amino, gegebenenfalls
substituiert mit Alkyl, Carboxy, Tetrazolyl, Carbamoyl, gegebenenfalls
substituiert mit Alkyl, Aminosulfonyl, gegebenenfalls substituiert
mit Alkyl, Acyl, Aroyl, Heteroaroyl, Acyloxy, Aroyloxy, Heteroaroyloxy,
Alkoxycarbonyl, Nitro, Cyano, Halogen, Niederperfluoralkyl, Heteroaryl oder
Aryl, wobei mehrere Grade der Substitution erlaubt sind. Für divalente
polycyclische aromatische Ringsysteme können ein oder mehrere der Ringe
ein oder mehrere Heteroatome enthalten. Beispiele für hierin
verwendetes "Heteroarylen" sind Furan-2,5-diyl,
Thiophen-2,4-diyl, 1,3,4-Oxadiazol-2,5-diyl, 1,3,4-Thiadiazol-2,5-diyl,
1,3-Thiazol-2,4-diyl, 1,3-Thiazol-2,5-diyl, Pyridin-2,4-diyl, Pyridin-2,3-diyl, Pyridin-2,5-diyl,
Pyrimidin-2,4-diyl, Chinolin-2,3-diyl und dergleichen.
-
Wie
hierin verwendet, bezeichnet der Begriff "Alkoxy" den Rest RaO-,
wobei Ra Alkyl, Alkenyl oder Alkinyl ist.
-
Wie
hierin verwendet, bezeichnet der Begriff "Alkylsulfanyl" den Rest RaS-,
wobei Ra Alkyl, Alkenyl oder Alkinyl ist.
-
Wie
hierin verwendet, bezeichnet der Begriff "Alkenylsulfanyl" den Rest RaS-,
wobei Ra Alkenyl oder Alkinyl ist.
-
Wie
hierin verwendet, bezeichnet der Begriff "Alkylsulfenyl" den Rest RaS(O)-,
wobei Ra Alkyl, Alkenyl oder Alkinyl ist.
-
Wie
hierin verwendet, bezeichnet der Begriff "Alkylsulfonyl" den Rest RaSO2-, wobei Ra Alkyl,
Alkenyl oder Alkinyl ist.
-
Wie
hierin verwendet, bezeichnet der Begriff "Acyl" den
Rest RaC(O)-, wobei Ra Alkyl,
Alkenyl, Alkinyl, Cycloalkyl, Cycloalkenyl oder Heterocyclyl ist.
-
Wie
hierin verwendet, bezeichnet der Begriff "Aroyl" den Rest RaC(O)-,
wobei Ra Aryl ist.
-
Wie
hierin verwendet, bezeichnet der Begriff "Heteroaroyl" den Rest RaC(O)-,
wobei Ra Heteroaryl ist.
-
Wie
hierin verwendet, bezeichnet der Begriff "Alkoxycarbonyl" den Rest RaOC(O)-,
wobei Ra Alkyl ist.
-
Wie
hierin verwendet, bezeichnet der Begriff "Carbamat" oder "Carbamoyl" den Rest RaRbNC(O)-, wobei Ra und
Rb Wasserstoff, Alkyl, Aryl, Heterocyclyl
oder Heteroaryl sind.
-
Wie
hierin verwendet, bezeichnet der Begriff "Alkylcarbonyloxy" den Rest RaC(O)O-,
wobei Ra Alkyl, Alkenyl, Alkinyl, Cycloalkyl,
Cycloalkenyl oder Heterocyclyl ist.
-
Wie
hierin verwendet, bezeichnet der Begriff "Aroyloxy" den Rest RaC(O)O-,
wobei Ra Aryl ist.
-
Wie
hierin verwendet, bezeichnet der Begriff "Heteroaroyloxy" den Rest RaC(O)O-,
wobei Ra Heteroaryl ist.
-
Wie
hierin verwendet, bedeutet der Begriff "gegebenenfalls", dass das/die anschließend beschriebene(n)
Ereignis(se) stattfinden können
oder nicht stattfinden können
und schließt
beide Situationen, bei denen das Ereignis stattgefunden oder nicht
stattgefunden hat, ein.
-
Wie
hierin verwendet, bezeichnet der Begriff "substituiert" eine Substitution mit dem genannten
Substituenten, wobei mehrere Grade der Substitution erlaubt sind.
-
Wie
hierin verwendet, können
die Begriffe "enthalten" oder "enthaltend" "in-line" Substitutionen an jeder beliebigen
Position auf den vorstehend definierten Alkyl-, Alkenyl-, Alkinyl-
oder Cycloalkylsubstituenten, mit einem oder mehreren von jedem
von O, S, SO, SO2, N oder N-Alkyl, einschließlich zum
Beispiel, -CH2-O-CH2-,
-CH2-SO2-CH2-, -CH2-NH-CH3 usw. bezeichnen.
-
Wie
hierin verwendet, ist der Begriff "Solvat" ein Komplex mit variabler Stöchiometrie,
welcher durch einen gelösten
Stoff (in dieser Erfindung eine Verbindung der Formel (I)) und ein
Lösungsmittel
gebildet wird. Derartige Lösungsmittel
für den
erfindungsgemäßen Zweck
dürfen
nicht mit der biologischen Aktivität des gelösten Stoffs Wechselwirken.
Lösungsmittel
können
als Beispiel Wasser, Ethanol oder Essigsäure sein.
-
Wie
hierin verwendet, sind die Begriffe "biohydrolysierbares Carbamat", "biohydrolysierbares
Carbonat" und "biohydrolysierbares
Ureid" ein Carbamat,
Carbonat bzw. Ureid eines Arzneistoffs (in dieser Erfindung eine
Verbindung der allgemeinen Formel (I)), welcher entweder a) nicht
mit der biologischen Aktivität
der Stammverbindung wechselwirkt, aber jenem Stoff vorteilhafte
Eigenschaften in vivo, wie Wirkungsdauer, Wirkungsbeginn und dergleichen
verleiht oder b) biologisch inaktiv ist, aber in vivo leicht durch
das Versuchsobjekt in den biologisch aktiven Grundbestandteil umgewandelt
wird. Der Vorteil ist jener, dass zum Beispiel das biohydrolysierbare
Carbamat oral vom Darm absorbiert wird und im Plasma in (I) transformiert
wird. Viele Beispiele für
derartiges sind auf dem Fachgebiet bekannt und schließen als
Beispiel Carbamate von Niederalkylaminen, substituierten Ethylendiaminen,
Aminosäuren,
Hydroxyalkylaminen, heterocyclischen und heteroaromatischen Aminen,
Polyetheraminen und dergleichen ein. Ein Beispiel für eine derartiges
biohydrolysierbares Carbamat, welches auf die allgemeine Formel
(I) angewendet wird, wird nachstehend in der allgemeinen Formel
(A) veranschaulicht.
-
-
Andere
Beispiele für
biohydrolysierbare Carbamate schließen jene Situationen ein, bei
denen die aromatische OH-Gruppe, welche von R6 und
R7 flankiert ist, mit einem Carbamoylkonjugat
konjugiert ist, um ein biohydrolysierbares Carbamat zu erhalten,
wobei das Carbamoylkonjugat ausgewählt ist aus Diethylaminocarbonyl,
N-(2-Hydroxyethyl)aminocarbonyl, N,N,-Bis(2-hydroxyethyl)aminocarbonyl, 4-Morpholinocarbonyl
und 4-Methyl-1-piperazinylcarbonyl.
-
Wie
hierin verwendet ist der Begriff "biohydrolysierbarer Ester" ein Ester eines
Arzneistoffs (in dieser Erfindung eine Verbindung der allgemeinen
Formel (I)), welcher entweder a) nicht mit der biologischen Aktivität der Stammverbindung
wechselwirkt, aber jenem Stoff vorteilhafte Eigenschaften in vivo,
wie Wirkungsdauer, Wirkungsbeginn und dergleichen verleiht oder
b) biologisch inaktiv ist, aber in vivo leicht durch das Versuchsobjekt
in den biologisch aktiven Grundbestandteil umgewandelt wird. Der
Vorteil ist jener, dass zum Beispiel der biohydrolysierbare Ester
oral vom Darm absorbiert wird und im Plasma in (I) transformiert
wird. Viele Beispiele für
derartiges sind auf dem Fachgebiet bekannt und schließen als
Beispiel Niederalkylester, Niederacyloxy-alkylester, Niederalkoxyacyloxyalkylester,
Alkoxyacyloxyester, Alkylacylaminoalkylester und Cholinester ein.
-
Wie
hierin verwendet ist der Begriff "biohydrolysierbares Amid" ein Amid eines Arzneistoffs
(in dieser Erfindung eine Verbindung der allgemeinen Formel (I),
welches entweder a) nicht mit der biologischen Aktivität der Stammverbindung
wechselwirkt, aber jenem Stoff vorteilhafte Eigenschaften in vivo,
wie Wirkungsdauer, Wirkungsbeginn und dergleichen verleiht oder
b) biologisch inaktiv ist, aber in vivo leicht durch das Versuchsobjekt
in den biologisch aktiven Grundbestandteil umgewandelt wird. Der
Vorteil ist jener, dass zum Beispiel das biohydrolysierbare Amid
oral vom Darm absorbiert wird und im Plasma in (I) transformiert
wird. Viele Beispiele für
derartiges sind auf dem Fachgebiet bekannt und schließen als
Beispiel Niederalkylamide, α-Aminosäureamide,
Alkoxyacylamide und Alkylaminoalkylcarbonylamide ein.
-
Wie
hierin verwendet schließt
der Begriff "Prodrug" biohydrolysierbare
Amide und biohydrolysierbare Ester und biohydrolysierbare Carbamate,
Carbonate und Ureide ein und nimmt ebenfalls a) Verbindungen, bei welchen
die biohydrolysierbare Funktionalität in einem derartigen Prodrug
in der Verbindung der Formel (I) aufgenommen ist: zum Beispiel das
Lactam, welches durch eine Carboxylgruppe in R1 und
ein Amin in R2 gebildet wird, und b) Verbindungen,
welche biologisch bei einer gegebenen funktionellen Gruppe oxidiert
oder reduziert werden können,
um Arzneistoffe der Formel (I) zu erhalten, auf. Beispiele für diese
funktionellen Gruppen sind 1,4-Dihydropyridin, N-Alkylcarbonyl-1,4-dihydropyridin,
1,4-Cyclohexadien,
tert-Butyl und dergleichen, sind aber nicht beschränkt darauf.
-
Wie
hierin verwendet ist der Begriff "Affinitätsreagens" ein Rest, der an die Verbindung der
Formel (I) angelagert ist, welcher seine biologische in vitro Aktivität nicht
beeinflusst, was erlaubt, die Verbindung an ein Ziel zu binden,
jedoch bindet ein derartiger Rest stark an eine dritte Verbindung,
was a) Charakterisierung des Ziels, bezüglich der Lokalisierung innerhalb
einer Zelle oder anderen Komponenten des Organismus, etwa durch
Visualisierung durch Fluoreszenz oder Radiographie, oder b) mühelose Abtrennung
des Ziels von einem unbekannten Gemisch von Zielen, egal ob proteinös oder nicht
proteinös
erlaubt. Ein Beispiel für
ein Affinitätsreagens
gemäß b) würde Biotin
sein, das entweder direkt an (I) angelagert ist oder über einen
Spacer von ein bis 50 Atome, ausgewählt aus C, H, O, N, S oder
P in jeder beliebigen Kombination, gebunden ist. Ein Beispiel für ein Affinitätsreagens
gemäß a) würde Fluoreszein
sein, das entweder direkt an (I) angelagert ist oder über einen
Spacer von ein bis 50 Atome, ausgewählt aus C, H, O, N, S oder
P in jeder beliebigen Kombination, gebunden ist.
-
Der
Begriff "pharmakologisch
wirksame Menge" soll
jene Menge eines Arzneistoffs oder Arzneimittels bedeuten, welche
die biologische oder medizinische Reaktion eines Gewebes, Systems,
Tiers oder Menschen, welche von einem Forscher oder Arzt begehrt
wird, auszulösen.
-
Warm
immer die Begriffe "Alkyl" oder "Aryl" oder jede ihrer
Präfixbasis
in einem Namen eines Substituenten erscheinen (z. B. Arylalkoxyaryloxy),
sollen sie so interpretiert werden, dass sie jene Beschränkungen, die
vorstehend für "aliphatisch" und "Aryl" gegeben wurden,
einschließen.
Alkyl- oder Cycloalkylsubstituenten sollen so verstanden werden,
dass sie funktionell zu jenen mit einem oder mehreren Graden an
Ungesättigtheit äquivalent
sind. Vorgesehene Anzahlen von Kohlenstoffatomen (z. B. C1-10) sollen unabhängig voneinander die Anzahl
von Kohlenstoffatomen in einer aliphatischen oder cycloaliphatischen
Einheit oder den aliphatischen Anteil eines größeren Substituenten bezeichnen.
-
Wie
hierin verwendet, soll der Begriff "Oxo" den
Substituenten =O bezeichnen.
-
Wie
hierin verwendet, soll der Begriff "Halogen" oder "Halo" Iod,
Brom, Chlor und Fluor einschließen.
-
Wie
hierin verwendet, soll der Begriff "Mercapto" den Substituenten -SH bezeichnen.
-
Wie
hierin verwendet, soll der Begriff "Carboxy" den Substituenten -COOH bezeichnen.
-
Wie
hierin verwendet, soll der Begriff "Cyano" den Substituenten -CN bezeichnen.
-
Wie
hierin verwendet, soll der Begriff "Aminosulfonyl" den Substituenten -SO2NH2 bezeichnen.
-
Wie
hierin verwendet, soll der Begriff "Carbamoyl" den Substituenten -C(O)NH2 bezeichnen.
-
Wie
hierin verwendet, soll der Begriff "Sulfanyl" den Substituenten -S- bezeichnen.
-
Wie
hierin verwendet, soll der Begriff "Sulfenyl" den Substituenten -S(O)- bezeichnen.
-
Wie
hierin verwendet, soll der Begriff "Sulfonyl" den Substituenten -S(O)2-
bezeichnen.
-
Herstellung
-
Die
Verbindungen der Formel (I) können
leicht gemäß den folgenden
Umsetzungsschemen (bei welchen alle Variablen wie vorstehend definiert
sind) und Beispielen oder Modifikationen davon unter Verwendung von
leicht erhältlichen
Ausgangsmaterialien, Reagenzien und üblichen Syntheseverfahren hergestellt
werden Bei diesen Umsetzungen ist es ebenfalls möglich, von Varianten, welche
an sich Fachleuten bekannt sind, Gebrauch zu machen, welche aber
nicht detaillierter erwähnt
sind.
-
Die
am meisten bevorzugten erfindungsgemäßen Verbindungen sind alle
die beliebigen, welche spezifisch in diesen Beispielen dargelegt
werden. Diese Verbindungen sollen allerdings nicht so ausgelegt
werden, dass sie die einzige Gattung bilden, die in der Erfindung
in Betracht gezogen wird, und jede beliebige Kombination der Verbindungen
oder ihrer Einheiten kann an sich eine Gattung bilden. Die folgenden
Beispiele veranschaulichen Details für die Herstellung der erfindungsgemäßen Verbindungen.
Fachleute werden leicht verstehen, dass bekannte Variationen der
Bedingungen und der Verfahren der folgenden präparativen Verfahren verwendet
werden können,
um diese Verbindungen herzustellen. Alle Temperaturen sind Grad
Celsius, außer
es ist anderweitig angegeben.
-
In
den Beispielen verwendete Abkürzungen
sind wie folgt:
- g
- = Gramm
- mg
- = Milligramm
- l
- = Liter
- ml
- = Milliliter
- μl
- = Mikroliter
- M
- = molar
- N
- = normal
- mM
- = millimolar
- i.v.
- = intravenös
- p.o.
- = per oral
- s.c.
- = subcutan
- Hz
- = Hertz
- mol
- = Mol
- mmol
- = Millimol
- mbar
- = Millibar
- psi
- = Pfund pro Quadratinch
- rt
- = Raumtemperatur
- min
- = Minuten
- Std.
- = Stunden
- Schmp.
- = Schmelzpunkt
- DC
- = Dünnschichtchromatographie
- Rf
- = relative DC Mobilität
- MS
- = Massenspektrometrie
- NMR
- = magnetische Kernresonanzspektroskopie
- APCI
- = chemische Ionisation
bei atmosphärischen
Druck
- ESI
- = Elektrosprühionisation
- m/z
- = Verhältnis Masse
zu Ladung
- HPLC
- = Hochddruckflüssigchromatographie
- tr
- = Retentionszeit
- Pd/C
- = Palladium auf aktiviertem
Kohlenstoff
- Ether
- = Diethylether
- MeOH
- = Methanol
- EtOAc
- = Ethylacetat
- TEA
- = Triethylamin
- DIEA
- = Diisopropylethylamin
- THF
- = Tetrahydrofuran
- DMF
- = N,N-Dimethylformamid
- DMSO
- = Dimethylsulfoxid
- DDQ
- = 2,3-Dichlor-5,6-dicyano-1,4-benzochinon
- LAH
- = Lithiumaluminiumhydrid
- TFA
- = Trifluoressigsäure
- HCl
- = Salzsäure
- LDA
- = Lithiumdiisopropylamid
- THP
- = Tetrahydropyranyl
- NMM
- = N-Methylmorpholin,
4-Methylmorpholin
- HMPA
- = Hexamethylphosphorsäuretriamid
- DMPU
- = 1,3-Dimethylpropylenharnstoff
- d
- = Tage
- ppm
- = Teile pro Million
- kD
- = Kilodalton
- LPS
- = Lipopolysaccharid
- PMA
- = Phorbolmyristatacetat
- SPA
- = Szintillationsproximitätsassay
- EDTA
- = Ethylendiamintetraessigsäure
- FBS
- = fötales Rinderserum
- PBS
- = Phosphat-gepufferte
Kochsalzlösung
-
Einige
der folgenden Beispiele repräsentieren
einzelne E-Isomere, einzelne Z-Isomere und Gemische von E/Z-Isomeren.
Bestimmung der E- und Z-Isomere kann durch analytische Verfahren,
wie Röntgenkristallographie, 1H NMR und 13C NMR,
getätigt
werden.
-
ALLGEMEINE UMSETZUNGSSCHEMEN
-
Erfindungsgemäße Verbindungen
können
durch Verfahren, die auf dem Fachgebiet bekannt sind, hergestellt
werden, wobei ein derartiges Verfahren in Umsetzungsschema I gezeigt
wird.
-
-
R1, R2, R3,
R4, R5, R6, R7 sind wie in
Formel (I) definiert.
-
Die
Umwandlung von II und III in I bezieht Verfahren, die als die Aldolkondensation
bekannt sind, gefolgt von Eliminierung ein, was in "Advanced Organic
Chemistry," Carey
und Sundberg, 3. Ausgabe, Plenum Press, 1990, hauptsächlich im
Kapitel 2 von Teil B enthalten, gut beschrieben ist. Die Umsetzung
kann unter Verwendung von Säure
(zum Beispiel konzentrierte HCl) in Kombination mit einem geeigneten
Lösungsmittel, wie
Essigsäure,
ausgeführt
werden. In einer anderen Ausführungsform
können
katalytische saure Bedingungen, wie die Verwendung einer katalytischen
Menge von para-Toluolsulfonsäure
in einem geeigneten Lösungsmittel,
wie Toluol, verwendet werden.
-
Benzaldehyde
der Formel (II) sind im Handel erhältlich oder können durch
veröffentlichte
Verfahren oder Variationen von veröffentlichten Verfahren hergestellt
werden. Umsetzungsschema 2 stellt zwei Wege, substituierte Benzaldehyde,
die nicht im Handel erhältlich
sind, leicht zu synthetisieren, anschaulich dar.
-
-
Darstellung
der substituierten Verbindungen der Formel (II) können von
Fachleuten durch eine Vielfalt an Verfahren erhalten werden. Zum
Beispiel kann die Umwandlung von (IV) in (II) durch Behandeln von
(IV) in einem geeigneten Lösungsmittel,
wie Essigsäure,
mit Hexamethylentetramin bei einer Temperatur von 90 °C bis 130 °C ausgeführt werden.
In einer anderen Ausführungsform
kann (V) in (II) durch Behandeln von (V) in einem geeigneten Lösungsmittel,
wie Dioxan, mit einer kleinen Menge Wasser mit DDQ bei einer Temperatur von
0 °C bis
140 °C umgewandelt
werden. Zusätzlich
zum Vorstehenden kann eine Verbindung der Formel (II) in eine andere
Verbindung der Formel (II) durch eine chemische Transformation des
geeigneten Substituenten oder der geeignete Substituenten umgewandelt
werden. Zum Beispiel wird in Bezug auf das aromatische Hydroxyl
in (II) die Umwandlung in ein Carbamat, Carbonat und einen Ether
durch Behandeln von (II) in einem geeigneten Lösungsmittel, wie THF mit einem
Alkylierungsmittel, wie Chlormethyl-R oder einem Acylierungsmittel,
wie Alkylchlorformiate und Alkylcarbamoylchloride, in einem geeigneten
Lösungsmittel,
wie Dichlormethan, ausgeführt.
Oxindole der Formel (III) sind im Handel erhältlich oder können durch
veröffentlichte
Verfahren oder Variationen von veröffentlichten Verfahren hergestellt werden.
Umsetzungsschema 3 stellt einige Wege, Verbindungen der Formel (III)
zu synthetisieren, anschaulich dar.
-
-
R1, R2, R3,
R4 sind wie in Formel (I) definiert.
-
Das
Anilin der Formel (VI) kann in das Isatin der Formel (VII) unter
Benutzung einer bekannten Transformation, welche die Sandmeyer Isonitrosoacetanilid
Isatinsynthese (T. Sandmeyer, Helv. Chim. Acta 2, 234 (1919)) genannt
wird, umgewandelt werden, wobei (VI) mit Chloralhydrat und Hydroxylamin
kondensiert werden kann, gefolgt von Cyclisierung mit konzentrierter
Schwefelsäure
und bei Verdünnung
mit Wasser quantitativer Hydrolyse in ein substituiertes Isatin
der Formel (VII). Die Umwandlung von (VII) in (III) kann unter Benutzung
einer bekannten Transformation, welche die Wolf-Kishner Reduktion
genannt wird, durch Behandeln mit Hydrazinhydrat in einem geeigneten
Lösungsmittel,
wie Ethanol, bei einer Temperatur von 20 °C bis 80 °C ausgeführt werden, um (VIII) zu bilden.
Die Umwandlung von (VIII) in (III) kann durch Behandlung mit Natriumethoxid
in einem geeigneten Lösungsmittel,
wie Ethanol, bei einer Temperatur von 0 °C bis 80 °C ausgeführt werden.
-
In
einer anderen Ausführungsform
kann ein substituiertes Anilin der Formel (VI) in eine Verbindung
der Formel (III) unter Benutzung bekannter Chemie umgewandelt werden.
(P. G. Gassman und T. J. van Bergen, Journal of the American Chemical
Society, 1974, 96(17), Seiten 5508 – 5512). Das Amin der Formel
(VI) kann in eine Verbindung der Formel (XI) durch Behandlung mit
t-Butylhypochlorit, gefolgt von Behandlung mit Ethylmethylthioacetat,
gefolgt von Behandlung mit Triethylamin in einem geeigneten Lösungsmittel,
wie wasserfreiem Dichlormethan, bei einer Temperatur von 78 °C bis 22 °C umgewandelt
werden. Die Umwandlung von (XI) in (III) kann durch Behandlung von
(XI) mit W-2 Raney Nickel in einem geeigneten Lösungsmittel, wie Ethanol, oder
Behandlung mit einer gesättigten
Lösung
aus Ammoniumchlorid, gefolgt von Behandlung mit aktiviertem Zink
in einem geeigneten Lösungsmittel,
wie THF, ausgeführt
werden.
-
Ein
Indol der Formel (IX) kann in (X) unter Benutzung eines Verfahrens,
das in der Literatur gut beschrieben ist (A. Marfat und M. Carta,
Tetrahedron letters, 28 (35), S. 4027 – 4030, 1987) durch Behandlung mit
Pyridiniumperbromid in einem geeigneten Lösungsmittel, wie t-Butylalkohol, bei
einer Temperatur von 25 °C
umgewandelt werden. Eine Verbindung der Formel (X) kann in (III)
durch Behandlung mit 10 % Pd/C in einem geeigneten Lösungsmittel,
wie wasserfreies Ethanol, bei 30 bis 50 psi Wasserstoff oder durch
Behandlung mit einer gesättigten
Lösung
aus Ammoniumchlorid, gefolgt von Behandlung mit aktiviertem Zink
in einem geeigneten Lösungsmittel,
wie THF, umgewandelt werden.
-
-
Zusätzlich zur
Aufnahme von Substitutionen in die anfänglichen Stadien der Synthese
kann eine Verbindung der Formel (III) in eine andere Verbindung
der Formel (III) durch eine chemische Transformation des geeigneten
Substituenten oder der geeigneten Substituenten umgewandelt werden.
Zum Beispiel demonstriert Umsetzungsschema 4 einige gut etablierte
Transformationen zur Funktionalisierung eines Oxindols der Formel
(III). Oxindol kann in ein Sulfonsäurederivat (IIIa) durch Behandlung
von (III) mit Chlorsulfonsäure
bei einer Temperatur von 0 °C
bis 60 °C
umgewandelt werden. Eine Verbindung der Formel (IIIa) kann in (IIIb), wobei
R substituiertes oder nicht substituiertes Amino ist, durch Behandlung
mit einem Set verschiedener Amine umgewandelt werden. Als Beispiel
kann (IIIa) mit Ammoniumhydroxid behandelt werden, um ein Sulfonamidderivat
der Formel (IIIb) bereitzustellen. Verbindung (III) kann in (IIIc)
durch Behandlung mit einem Säurechlorid,
in Gegenwart von Aluminiumchlorid in einem geeigneten Lösungsmittel,
wie Dichlormethan oder Kohlenstoffdisulfid, bei einer Temperatur
von 0 °C
bis 45 °C
umgewandelt werden. Wenn R in (IIIc) OH ist, welches gemäß Schema
3 synthetisiert wird, bezieht die Umwandlung von Carbonsäure (IIIc)
in Ester und Amide der Formel (IIId) in der Peptidchemie bekannte
Verfahren ein, zum Beispiel kann die Umsetzung unter Verwendung von
HOBt in Kombination mit einem Dehydratisierungsmittel, Dicyclohexylcarbodiimid
in einem geeigneten Lösungsmittel,
wie DMF, ausgeführt
werden. (III) kann in (IIIe) durch Behandeln von (III) mit Chloracetylchlorid
in Gegenwart von Aluminumchlorid in einem geeigneten Lösungsmittel,
wie Dichlormethan oder Kohlenstoffdisulfid, bei einer Temperatur
von 0 °C
bis 45 °C
umgewandelt werden. Weitere Funktionalisierungen zu verschiedenen
heterocyclischen Resten können
durch Behandlung von (IIIe) mit diversen substituierten Amidinen,
Thioamiden, Harnstoffen und substituierten Aminopyridinen erreicht
werden. Zum Beispiel kann (IIIe) in (IIIf) durch Behandeln von (IIIe)
mit Thioacetamid in einem geeigneten Lösungsmittel, wie Essigsäure, bei
einer Temperatur von 22 °C
bis 100 °C
umgewandelt werden. Verbindungen der Formel (IIIg), wobei R zum Beispiel
ein Alkyl oder cyclisches Amin ist, können durch Behandeln von (IIIe)
mit diversen Nukleophilen, wie Aminen, in einem geeigneten Lösungsmittel,
wie THF, bei einer Temperatur von 22 °C bis 80 °C erhalten werden.
-
-
Eine
chemische Transformation eines halogenierten Oxindols der Formel
III in eine andere Verbindung der Formel III wird in Umsetzungsschema
5 beschrieben. Zum Beispiel kann eine Verbindung der Formel (IIIh), wobei
X Brom oder Iod ist, mit einem Tributylzinnheterocyclus, zum Beispiel
3-Pyridyltributylzinn, in Gegenwart eines Palladiumkatalysators,
zum Beispiel Bistriphenylphosphindichlorpalladium, in einem geeigneten
Lösungsmittel,
wie Acetonitril, behandelt werden, um (IIIj) zu bilden. Alternativ
dazu kann (IIIh) in (IIIj) durch Behandlung mit einer heterocyclischen
oder aromatischen Boronsäure,
zum Beispiel Thiophen-3-boronsäure,
in Gegenwart einer Base, zum Beispiel Tetrakis-triphenylphosphinpalladium,
in einem geeigneten Lösungsmittel, wie
Toluol, bei einer Temperatur von 22 °C bis 125 °C umgewandelt werden.
-
-
R1, R2, R3,
R4 sind wie in Formel (I) definiert.
-
Eine
Verbindung der Formel (III) kann ebenfalls unter Verwendung eines
durch Umsetzungsschema 6 beschriebenen Verfahrens synthetisiert
werden. Ein substituiertes 2-Nitrotoluol
(XII) kann in eine Verbindung der Formel (XIII) durch Behandlung
mit Diethyloxalat und Natriumethoxid in einem geeigneten Lösungsmittel, wie
Ethanol, umgewandelt werden. Das Produkt dieser Umsetzung kann direkt
mit Wasser behandelt werden, um (XIII) zu bilden, was durch Behandlung
mit einer Wasserstoffperoxidlösung
und Natriumhydroxid in Wasser bei Temperaturen zwischen 0 °C und 100 °C in eine
Verbindung der Formel (XIV) umgewandelt werden könnte. Behandlung von (XIV)
mit Zink in Schwefelsäure
und einem geeigneten Lösungsmittel,
wie Ethanol, kann eine Verbindung der Formel (III) bereitstellen.
Alternativ dazu kann eine Verbindung der Formel XIV aus einer Verbindung
der Formel XV, wobei X ein Halogen ist, synthetisiert werden. Verbindung
XV kann mit einer Lösung, welche
Diethylmalonat und Natriumethoxid enthält, in einem geeigneten Lösungsmittel,
wie Ethanol, bei einer Temperatur von 0 °C bis 78 °C behandelt werden, um eine
Verbindung der Formel XVI bereitzustellen. Diese Verbindung der
Formel XVI kann hydrolysiert und decarboxyliert werden, um XIV unter
Verwendung von Standardbedingungen, wie Behandlung von XVI mit wässrigem
Natriumhydroxid, gefolgt von der Behandlung von XVI mit wässriger
Salzsäure,
bereitzustellen.
-
ARZNEIMITEL UND DOSEN
-
Die
erfindungsgemäßen Verbindungen
können
in derartigen oralen (einschließlich
buccalen und sublingualen) Dosierungsformen, wie Tabletten, Kapseln
(jeweils einschließlich
zeitlich festgelegt freisetzende und verlängert freisetzende Formulierungen),
Pillen, Pulvern, Granulatkörner,
Elixieren, Tinkturen, Suspensionen, Sirupen und Emulsionen verabreicht
werden. Gleichermaßen
können
sie ebenfalls in nasaler, ophthalmischer, otischer, rektaler, topischer,
intravenöser
(sowohl Bolus als auch Infusion), intraperitonealer, intraartikulärer, subcutaner
oder intramuskulärer,
Inhalierungs- oder Insufflationsform verabreicht werden, wobei alle
verwendeten Formen Fachleuten der Pharmazie wohlbekannt sind.
-
Die
Dosierungsregimen, welche die erfindungsgemäßen Verbindungen benutzen,
wird in Übereinstimmung
mit einer Vielfalt von Faktoren, einschließlich der Art, der Spezies,
dem Alter, Gewicht, Geschlecht und dem medizinischen Zustand des
Patienten; dem Schweregrad des zu behandelnden Zustands; dem Verabreichungsweg;
der Nieren- und Leberfunktion des Patienten; und der bestimmten
angewendeten Verbindung oder dem Salz davon, ausgewählt. Ein
praktischer Arzt oder Tierarzt kann leicht die wirksamen Menge des
Arzneistoffes, welcher zur Vermeidung, Entgegenwirkung oder zum
Arretieren des Fortschritts des Zustands erforderlich ist, bestimmen
und verschreiben.
-
Eine
therapeutisch wirksame Menge einer erfindungsgemäßen Verbindung oder eines Salzes
wird von etlichen Faktoren, einschließlich zum Beispiel dem Alter
und dem Gewicht des Tieres oder des Patienten, dem genauen Zustand,
der Behandlung erfordert und seinem Schweregrad, der Beschaffenheit
der Formulierung und dem Verabreichungsweg abhängen und wird letztendlich
im Ermessen des behandelnden Arztes oder Tierarztes liegen.
-
Orale
erfindungsgemäße Dosierungen
werden, wenn sie für
die angezeigten Wirkungen verwendet werden, von zwischen etwa 0,1
bis 300 mg/kg Körpergewicht
pro Tag, und insbesondere 1 bis 100 mg/kg Körpergewicht pro Tag reichen.
Orale Dosierungseinheiten werden im Allgemeinen im Bereich von 1
bis etwa 250 mg und stärker
bevorzugt von etwa 25 bis 250 mg verabreicht. Die tägliche Dosis
für einen
Säuger
mit 70 kg wird im Allgemeinen im Bereich von etwa 10 mg bis 5 Gramm
einer Verbindung der Formel I sein. Eine wirksame Menge eines erfindungsgemäßen Salzes
kann als ein Anteil der wirksamen Menge der Verbindung selbst bestimmt
werden.
-
Topische
Anwendungen können
auf ähnliche
Weise ein oder mehrmals am Tag sein, abhängig von den gewöhnlichen
medizinischen Betrachtungsweisen. Vorteilhafterweise können erfindungsgemäße Verbindungen
in einer einzigen täglichen
Dosis verabreicht werden oder die gesamte tägliche Dosierung kann in verteilten
Dosen von ein, zwei, drei oder vier Mal täglich verabreicht werden. Darüber hinaus
können
bevorzugte erfindungsgemäße Verbindungen
in intranasaler Form über
topische Verwendung von geeigneten intranasalen Vehikeln oder über transdermale
Wege, unter Verwendung jener Formen von transdermalen Hautpflastern, die
Fachleuten wohlbekannt sind, verabreicht werden. Um in Form eines
transdermalen Abgabesystems verabreicht zu werden, wird die Verabreichungsdosierung
selbstverständlich
eher kontinuierlich sein als durch das Dosierungsregimen unterbrochen
sein.
-
In
den erfindungsgemäßen Verfahren
können
die hierin detailliert beschriebenen Verbindungen den Wirkstoff
bilden und werden typischerweise als Beimischung mit geeigneten
pharmazeutischen Verdünnungsmitteln,
Exzipienten oder Trägern
(hierin gesammelt als „Trägermaterialien" bezeichnet), die
geeigneterweise unter Bezugnahme auf die beabsichtigte Verabreichungsform,
d. h. orale Tabletten, Kapseln, Elixiere, Sirupe und dergleichen,
ausgewählt
werden und die mit herkömmlichen
pharmazeutischen Durchführungen
konsistent sind, verabreicht.
-
Beispielsweise
kann für
eine orale Verabreichung in Form einer Tablette oder Kapsel die
wirksame Arzneistoffkomponente mit einem oralen, nicht toxischen
pharmazeutisch verträglichen
inerten Träger,
wie Ethanol, Glycerol, Wasser und dergleichen, kombiniert werden.
Pulver werden durch Zerkleinern der Verbindung auf eine geeignete
feine Größe und Mischen
mit einem ähnlich
zerkleinerten pharmazeutisch verträglichen Träger, wie ein essbares Kohlenhydrat,
wie zum Beispiel Stärke
oder Mannitol, hergestellt. Aromastoffe, Konservierungs-, Dispersion-
und Farbmittel können
ebenfalls vorliegen.
-
Kapseln
werden durch Herstellung eines Pulvergemisches wie vorstehend beschrieben
und durch Befüllen
geformter Gelatinehüllen
erzeugt. Schmiermittel und Gleitmittel, wie kolloidales Silica,
Talk, Magnesiumstearat, Calciumstearat oder festes Polyethylenglycol
können
dem Pulvergemisch vor dem Befüllungsvorgang zugegeben
werden. Ein Sprengmittel oder Solubilisierungsmittel, wie Agar-Agar,
Calciumcarbonat oder Natriumcarbonat können ebenfalls zugegeben werden,
um die Verfügbarkeit
des Medikaments bei Aufnahme der Kapsel zu verbessern.
-
Überdies
können,
falls gewünscht
oder notwendig, geeignete Bindemittel, Gleitmittel, Sprengmittel und
Farbmittel ebenfalls in das Gemisch einbezogen werden. Geeignete
Bindemittel schließen
Stärke,
Gelatine, natürliche
Zucker, wie Glucose oder beta-Lactose, Maissüßungsmittel, natürliche und
synthetische Gummis, wie Gummi arabicum, Traganth oder Natriumalginat,
Carboxymethylcellulose, Polyethylenglycol, Wachse und dergleichen
ein. In diesen Dosierungsformen verwendete Gleitmittel schließen Natriumoleat,
Natriumstearat, Magnesiumstearat, Natriumbenzoat, Natriumacetat,
Natriumchlorid und dergleichen ein. Sprengmittel schließen ohne
Beschränkung
Stärke,
Methylcellulose, Agar, Bentonit, Xanthangummi und dergleichen ein. Tabletten
werden zum Beispiel durch Herstellung eines Pulvergemisches, Granulierung
oder Slugging, Zugabe eines Gleitmittels und Sprengmittels und Verpressen
zu Tabletten erzeugt. Ein Pulvergemisch wird durch Mischen der geeigneterweise
zerkleinerten Verbindung mit einem Verdünnungsmittel oder einer Base
wie vorstehend beschrieben, und gegebenenfalls mit einem Bindemittel,
wie Carboxymethylcellulose, einem Aliginat, Gelatine oder Polyvinylpyrrolidon,
einem Lösungsverzögerer, wie
Paraffin, einem Resorptionsbeschleuniger, wie ein quaternäres Salz
und/oder einem Absorptionsmittel, wie Bentonit, Kaolin oder Dicalciumphosphat,
hergestellt. Das Pulvergemisch kann durch Benetzen mit einem Bindemittel,
wie Sirup, Stärkepaste,
Acadia-Mukilago oder Lösungen
von Cellulosematerialien oder polymeren Materialen, und durch Zwingen
durch ein Sieb granuliert werden. Als eine Alternative zum Granulieren
kann das Pulvergemisch durch die Tablettiermaschine laufen gelassen
werden und das Ergebnis sind nicht perfekt geformte Briketts, die
in Granulatkörner
gebrochen sind. Die Granulatkörner
können
mittels der Zugabe von Stearinsäure,
einem Stearatsalz, Talk oder Mineralöl geschmiert werden, um das
Kleben an die Tabletten formenden Matrizen zu vermeiden. Das geschmierte
Gemisch wird dann zu Tabletten verpresst. Die erfindungsgemäßen Verbindungen
können
ebenfalls mit einem frei fließendem
inerten Träger
kombiniert werden und direkt zu Tabletten verpresst werden, ohne durch
die Granulierungs- oder Sluggingschritte zu gehen. Ein klarer oder
undurchsichtiger Schutzüberzug,
der aus einem Versiegelungslack von Shellak, einem Überzug aus
Zucker oder polymerem Material und einem Polierüberzug aus Wachs besteht, kann
bereitgestellt werden. Farbstoffe können diesen Überzügen zugegeben werden,
um verschiedene Einheitsdosierungen zu unterscheiden.
-
Orale
Flüssigkeiten,
wie Lösung,
Sirupe und Elixiere können
in Dosierungseinheitsformen hergestellt werden, sodass eine gegebene
Menge eine vorher bestimmte Menge der Verbindung enthält. Sirupe
können durch
Lösen der
Verbindung in einer geeigneten aromatisierten wässrigen Lösung hergestellt werden, während Elixiere
durch die Verwendung eines nicht toxischen Alkohol-Vehikels hergestellt
werden. Suspensionen können
durch Dispergieren der Verbindung in einem nicht toxischen Vehikel
formuliert werden. Lösungsvermittler
und Emulgatoren, wie ethoxylierte Isostearylalkohole und Polyoxyethylensorbitether,
Konservierungsmittel, Aromazusatzstoffe, wie Pfefferminzöl oder Saccharin
und dergleichen können
ebenfalls zugegeben werden.
-
Wo
geeignet, können
Dosierungseinheitsformulierungen zur oralen Verabreichung mikroverkapselt werden.
Die Formulierung kann ebenfalls hergestellt werden, um die Freisetzung
auszudehnen oder zu verlängern,
wie zum Beispiel durch Überziehen
oder Einbetten teilchenförmigen
Materials in Polymere, Wachs oder dergleichen.
-
Die
erfindungsgemäßen Verbindungen
können
ebenfalls in Form von Liposomenabgabesystemen, wie kleinen unilamellaren
Vesikeln, großen
unilamellaren Vesikeln und multilamellaren Vesikeln, verabreicht werden.
Liposomen können
aus einer Vielfalt von Phospholipiden, wie Cholesterol, Stearylamin
oder Phosphatidylcholinen, gebildet werden.
-
Erfindungsgemäße Verbindungen
können
ebenfalls durch die Verwendung von monoklonalen Antikörpern als
einzelne Träger,
an welche die Moleküle
der Verbindung gekoppelt sind, abgegeben werden. Die erfindungsgemäßen Verbindungen
können
ebenfalls mit geeigneten löslichen
Polymeren als abzielbare Arzneistoffträger gekoppelt werden. Derartige
Polymere können
Polyvinylpyrrolidon, Pyrancopolymer, Polyhydroxypropylmethacrylamid-phenol,
Polyhydroxyethylaspartamidphenol oder Polyethylenoxidpolylysin,
substituiert mit Palmitoylresten, einschließen. Darüber hinaus können die
erfindungsgemäßen Verbindungen
an eine Klasse von biodegenerierbaren Polymeren gekoppelt sein,
die beim Erreichen von kontrollierter Freisetzung eines Arzneistoffs
nützlich
sind, zum Beispiel Polymilchsäure,
Polepsiloncaprolacton, Polyhydroxybuttersäure, Polyorthoester, Polyacetale,
Polydihydropyrane, Polycyanoacrylate und vernetzte oder amphipatische
Blockcopolymere von Hydrogelen.
-
Die
vorliegende Erfindung schließt
Arzneimittel, die 0,1 bis 99,5 %, ganz besonders 0,5 bis 90 % einer Verbindung
der Formel (I) enthalten, in Kombination mit einem pharmazeutisch
verträglichen
Träger
ein.
-
Parenterale
Verabreichung kann durch Benutzen flüssiger Dosierungseinheitsformen,
wie sterile Lösungen
und Suspensionen, die zur subcutanen, intramuskulären oder
intravenösen
Injektion gedacht sind, bewirkt werden. Diese werden durch Suspendieren
oder Lösen
in einer abgemessenen Menge der Verbindung in einem nicht toxischen
flüssigen
Vehikel, das zur Injektion geeignet ist, wie wässriges öliges Medium, und Sterilisieren
der Suspension oder Lösung
hergestellt.
-
In
einer anderen Ausführungsform
wird eine abgemessene Menge der Verbindung in ein Fläschchen gegeben
und das Fläschchen
und sein Inhalt werden sterilisiert und versiegelt. Ein begleitendes
Fläschchen oder
Vehikel kann zum Mischen vor der Verabreichung bereitgestellt werden.
Nicht toxische Salze und Lösungen
können
zugegeben werden, um die Injektion isotonisch zu erhalten. Stabilisatoren,
Konservierungsmittel und Emulgatoren können ebenfalls zugegeben werden.
-
Rektale
Verabreichung kann durch Benutzen von Zäpfchen bewirkt werden, bei
welchen die Verbindung mit niedrig schmelzenden wasserlöslichen
oder wasserunlöslichen
Feststoffen, wie Polyethylenglycol, Kakaobutter, höhere Ester,
wie zum Beispiel eine aromatisierte wässrige Lösung, vermengt werden, während Elixiere
durch Myristylpalmitat oder Gemische davon hergestellt werden.
-
Topische
erfindungsgemäße Formulierungen
können
beispielsweise als Salben, Cremes oder Lotionen, Augensalben und
Augen- oder Ohrentropfen, imprägnierte
Verbände
und Aerosole präsentiert
werden und können
geeignete herkömmliche
Zusatzstoffe, wie Konservierungsmittel, Lösungsmittel, um die Arzneistoffpenetration
zu unterstützen,
und erweichende Mittel bei Salben und Cremes enthalten. Die Formulierungen
können
ebenfalls kompatible herkömmliche
Träger,
wie Grundlagen für
Cremes oder Salben und Ethanol oder Oleylalkohol für Lotionen
enthalten. Derartige Träger
können
in von etwa 1 % bis zu etwa 98 % der Formulierung vorliegen. Gewöhnlicher
werden sie bis zu 80 % der Formulierung bilden.
-
Zur
Verabreichung durch Inhalation werden die erfindungsgemäßen Verbindungen
zweckmäßigerweise
in Form einer Aerosolsprühdarstellung
aus Packungen unter Druck oder einem Zerstäuber abgegeben, durch die Verwendung
eines geeigneten Treibgases, z. B. Dichlordifluormethan, Trichlorfluormethan,
Dichlortetrafluorethan, TetRaf1uorethan, Heptafluorpropan, Kohlendioxid
oder einem anderen geeigneten Gas. Im Fall eines Aerosols unter
Druck kann die Dosierungseinheit durch Bereitstellen eines Ventils
zur Abgabe einer abgemessenen Menge bestimmt werden. Kapseln und
Kartuschen von Z. B. Gelatine zur Verwendung in einem Inhalator
oder Insufflator können
formuliert werden, dass sie ein Pulvergemisch einer erfindungsgemäßen Verbindung
und einer geeigneten Pulverbasis, wie Lactose oder Stärke, enthalten.
-
Die
bevorzugten Arzneimittel sind jene in einer zur oralen Verabreichung
geeigneten Form, wie Tabletten und Flüssigkeiten und dergleichen
und topische Formulierungen.
-
SYNTHESEBEISPIELE
-
Wir
legen nun eine ausgewählte
Zahl von Synthesebeispielen dar, welche die zum Erhalten der erfindungsgemäßen Verbindungen
verwendeten Techniken veranschaulichen. Man nimmt an, dass ein Fachmann in
Anbetracht der vorstehend dargelegten Syntheseschemen in der Lage
sein wird, diesen Verfahren zu folgen oder sie entsprechend ohne übermäßige Experimentieren
zu modifizieren, um jede der vorstehend offenbarten Substitutionen
zu erhalten. Die folgenden Beispiele sind veranschaulichende erfindungsgemäße Ausführungsformen,
welche den Umfang der Erfindung auf keine Weise einschränken. Reagenzien
sind im Handel erhältlich
oder werden gemäß den Literaturverfahren
hergestellt. Beispielnummern bezeichnen jene Verbindungen, welche
in den vorstehenden Tabellen aufgelistet sind. 1H-NMR
Spektren wurden auf VARIAN Unity Plus NMR Spektrophotometern bei
300 oder 400 MHz erhalten. Massenspektren wurden auf Micromass Platform
II Massenspektrometern von Micromass Ltd. Altrincham, UK, entweder
unter Verwendung von Atmosphärischer Chemischer
Ionisation (APCI) oder Elektrosprühionisation (ESI) erhalten.
Analytische Dünnschichtchromatographie
(DC) wurde verwendet, um die Reinheit einiger Zwischenprodukte,
welche nicht isoliert werden konnten oder welche zur vollständigen Charakterisierung
zu instabil waren, zu verifizieren und um dem Fortschritt der Umsetzungen
zu folgen. Wenn nicht anderweitig angegeben, wurde dies unter Verwendung
von Silicagel (Merck Silica Gel 60 F254) getätigt. Wenn nicht anderweitig
angegeben, verwendete die Säulenchromatographie
zur Reinigung einiger Verbindungen Merck Silicagel 60 (Mesh 230 – 400) und
das angegebene Lösungsmittelsystem
unter Druck.
-
BEISPIEL
1; 3-(3,5-Dibrom-4-hydroxy-benzylidin)-2-oxo-2,3-dihydro-1H-indol-5-carbonitril
-
Beispiel 1a; 5-Cyano-3-methylthiooxindol
-
Eine
Lösung
aus 4-Cyanoanilin (5,0 g, 42 mmol) in trockenem Dichlormethan (100
ml) unter N2 wurde auf etwa –78 °C gekühlt. Zu
dieser gerührten
Lösung
wurde eine Lösung
aus tert-Butylhypochlorit
(4,6 g, 42 mmol) in trockenem Dichlormethan (10 ml) über 5 min
hinweg gegeben und die sich ergebende Lösung 10 Minuten lang gerührt. Eine
Lösung
aus Ethylmethylthioacetat (5,45 ml, 5,69 g, 42 mmol) in trockenem
Dichlormethan (10 ml) wurde dann tropfenweise zugegeben und das
Gemisch 1 Stunde lang gerührt.
Triethylamin (5,9 ml, 4,28, 42 mmol) wurde tropfenweise zugegeben,
und man ließ die
Lösung über 1 Stunde
hinweg sich auf Raumtemperatur erwärmen. Die Umsetzungslösung wurde
mit Wasser (3 × 20
ml) und Salzlösung
(1 × 20 ml)
gewaschen, über
wasserfreiem MgSO4 getrocknet und das Lösungsmittel
unter Vakuum eingedampft, um ein oranges Öl zurückzulassen. Dieses Öl wurde
in Diethylether gelöst
(100 ml) und 2 N wässrige
Salzsäure (5
ml) wurde zugegeben und das Gemisch heftig bei Raumtemperatur 18
Stunden lang gerührt.
Die sich ergebende braune Lösung
wurde durch Filtration gesammelt und mit Diethylether gewaschen
und unter Vakuum getrocknet, um 5-Cyano-3-methylthiooxindol als einen
weißen
Feststoff zu geben, (6,4 g, 76 %). 1H NMR (CDCl3) δ 9,04
(br s, 1H) 7,64 (s, 1H), 7,57 (d, 1H, J = 8,4 Hz), 6,99 (d, 1H,
J = 8,4 Hz), 4,28 (s, 1H), 2,04 (s, 3H). MS (-ve ES) 203 (100),
(M-H).
-
Beispiel 1b; 5-Cyanooxindol
-
Eine
Lösung
aus 5-Cyano-3-methylthiooxindol (6,0 g, 29 mmol) in THF (100 ml)
wurde bei Raumtemperatur gerührt
und eine gesättigte
wässrige
Lösung
aus NH4Cl (100 ml), gefolgt von aktiviertem
Zink (25 g, 0,38 mol) zugegeben. Das sich ergebende Gemisch wurde
18 Stunden lang gerührt.
Das Gemisch wurde durch ein Pod aus Diatomeenerde filtriert und
das Pod mit THF (20 ml) gewaschen. Die organische Phase wurde abgetrennt, über wasserfreiem
MgSO4 getrocknet, und das Lösungsmittel
eingedampft, um einen braunen Feststoff zurückzulassen.
-
Verreiben
dieses Feststoffes mit Diethylether gab 5-Cyanooxindol, einen weißen Feststoff,
(4,1 g, 88 %). 1H NMR (DMSO-d6) δ 7,63 (d,
1H, J = 8,4 Hz), 7,62 (s, 1H), 6,93 (d, 1H, J = 8,4 Hz), 3,55 (s,
2H). MS (-ve ES) 157 (100), (M-H).
-
Beispiel 1; 3-(3,5-Dibrom-4-hydrogy-benzylidin)-2-oxo-2,3-dihydro-1H-indol-5-carbonitril
-
Die
Titelverbindung wurde auf eine zu Beispiel 2 identische Weise synthetisiert,
mit Ausnahme dass 5-Cyano-oxindol anstelle von 5-(2-Methyl-thiazol-4-yl)-1,3-dihydro-indol-2-on-hydrochlorid verwendet
wurde. 1H NMR (DMSO-d6) δ 11,17 (s,
1H), 8,75 (s, 2H), 8,11 (s, 1H), 7,91 (s, 1H), 7,64 (d, 1H, J =
8,4 Hz), 6,96 (d, 1H), J = 8,4 Hz). MS (AP-ve) 419 (20) (M-H).
-
Beispiel
2; 3-(3,5-Dibrom-4-hydroxubenzyliden)-5-(2-methyl-thiazol-4-yl)-1,3-dihydroindol-2-on
-
Beispiel 2a; 5-(2-Chlor-acetyl)-1,3-dihydro-indol-2-on
-
Die
folgenden Reagenzien wurden in der aufgelisteten Reihenfolge unter
Stickstoff bei Raumtemperatur vereinigt: Aluminiumchlorid (17 g,
0,130 mol), Kohlenstoffdisulfid (40 ml), Chloracetylchlorid (3,01
g, 0,027 mol) und Oxindol (2,73 g, 0,021 mol). Das Umsetzungsgemisch
wurde auf Rückfluss
erwärmt
und das Rühren bei
dieser Temperatur 3 Stunden lang fortgeführt. Das Reaktionsgemisch wurde
auf Raumtemperatur gekühlt und
die Flüssigkeit
wurde vorsichtig abdekantiert. Eiswasser wurde tropfenweise zu dem
restlichen Rückstand unter
Stickstoff (langsam und vorsichtig) gegeben. Die Zugabe von Wasser
wurde beendet, als im Gesamten 50 ml zugegeben worden waren, und
das Reaktionsgemisch wurde 1 Stunde lang bei Raumtemperatur gerührt. Der
braune Feststoff wurde durch Filtration gesammelt, 3 Mal mit Wasser
gewaschen und im Vakuum bei 72 °C
getrocknet, um einen hellbraunen Feststoff (3,4 g, 79 % Ausbeute)
zu geben. 1H NMR (DMSO-d6) δ,3,62 (s,
2H); 5,15 (s, 2H); 6,96 (d, 1H); 7,84 (s, 1H); 7,91 (d, 2H); 10,84
(bs, 1H). APCI-MS m/z 208 (M-H).
-
Beispiel 2b; 5-(2-Methyl-thiazol-4-yl)-1,3-dihydro-indol-2-on-hydrochlorid
-
Thioacetamid
(90 mg, 1,2 mmol) wurde zu einer Aufschlämmung aus 5-(2-Chlor-acetyl)-1,3-dihydro-indol-2-on
(250 mg, 1,2 mmol) in Essigsäure
(3 ml) gegeben. Die Umsetzungstemperatur wurde auf 80 °C erhöht und bei
dieser Temperatur 16 Std. lang gerührt. Das Umsetzungsgemisch
wurde auf Raumtemperatur abgekühlt
und der sich ergebende Niederschlag wurde durch Filtration gesammelt.
Der Feststoff wurde mit EtOAc (2 × 20 ml) und Ether (2 × 20 ml)
gewaschen und im Vakuum getrocknet, um einen cremefarbenen Feststoff
(300 mg, 94 % Ausbeute) zu ergeben. 1H NMR
(DMSO-d6) δ 2,74 (s, 3H); 3,58 (s, 2H);
6,87 (d, 1H); 7,79 (m, 3H); 10,57 (s, 1H). APCI-MS (-ve) m/z 229
(M-H), APCI-MS (+ve) m/z 231 (M+H). Analytisch berechnet für C12H10N2OS
HCl: C, 54,02; H, 4,16; N, 10,50; S, 12,02.
Gemessen: C, 53,73;
H, 4,16; N, 10,17; S, 11,63.
-
Beispiel 2; 3-(3,5-Dibrom-4-hydroxybenzyliden)-5-(2-methyl-thiazol-4-yl)-1,3-dihydroindol-2-on
-
5-(2-Methyl-thiazol4-yl)-1,3-dihydro-indol-2-on-hydrochlorid
(0,030 g, 0,13 mmol) und 3,5-Dibrom-4-hydroxy-benzaldehyd
(0,037 g, 0,13 mmol) wurden vereinigt und in Essigsäure (1,0
ml) aufgeschlämmt. Konzentrierte
Salzsäure
(0,25 ml) wurde dem Umsetzungsgemisch zugegeben, was das Lösen der
Feststoffe verursachte. Ein gelber Niederschlag wurde durch Filtration
gesammelt, nachdem das Reaktionsgemisch 4 Std. lang rührte. Der
Feststoff wurde mit EtOAc (2 × 20
ml) und Ether (2 × 20
ml) gewaschen und im Vakuum getrocknet, um einen glänzend gelben
Feststoff (0,045 mg, 64 % Ausbeute) zu ergeben. 1H
NMR (DMSO-d6) δ 2,80 (s, 3H); 6,89 (d, 1H);
7,77 (s, 1H); 7,82 (d, 1H); 7,86 (s, 1H); 8,27 (s, 1H); 8,87 (s,
2H); 10,82 (s, 1H). Electrosprüh
MS m/z (-ve) 491. Analytisch berechnet für C19H12N2O2Br2S·HCl:
C, 43,17; H, 2,48; N, 5,30. Gemessen: C, 42,82; H, 2,66; N, 5,14.
-
Beispiel
7; 3-(3,5-Dichlor-4-hydroxybenzyliden)-5-(3-methyl-butanoyl)-1,3-dihydro-indol-2-on
-
Beispiel 7a; 5-(3-Methyl-butanoyl)-1,3-dihydro-indol-2-on
-
Aluminiumtrichlorid
(10,7 g, 7,5 mmol) wurde in einen Rundkolben unter Stickstoff bei
Raumtemperatur gestellt. Dimethylformamid (1,7 ml) wurde tropfenweise
zugegeben, was eine exotherme Umsetzung erzeugte. Das Reaktionsgemisch
stand 15 Minuten lang vor der Zugabe von Oxindol (1 g, 7,5 mmol),
gefolgt von 3-Methyl-butanoylchlorid (0,96 g, 8 mmol). Das Reaktionsgemisch
wurde 60 Minuten lang auf 70 °C
erhitzt. Das Reaktionsgemisch wurde auf zerstoßenes Eis (100 g) mit zugegebener
konzentrierter Salzsäure
(10 ml) gegossen. Die wässrige
Schicht wurde mit EtOAc (100 ml) extrahiert. Die organische Schicht
wurde mit einer gesättigten
(wässrigen)
NaCl-Lösung
gewaschen, über
MgSO4 getrocknet. Die flüchtigen Bestandteile wurden im
Vakuum entfernt, um die gewünschte
Verbindung (1,38 g, 85 %) zu ergeben. 1H
NMR (DMSO-d6): δ 10,71 (s, 1H); 7,82 (d, J =
8, 1H); 7,77 (s, 1H); 6,86 (d, J = 8, 1H); 3,51 (s, 2H); 2,76 (d,
J = 7, 2H); 2,13 – 2,05
(m, 1H); 0,88 (d, J = 7, 6H) ESI-MS: m/z 216 (m-H)-.
-
Beispiel 7; 3-(3,5-Dichlor-4-hydroxybenzvliden)-5-(3-methyl-butanoyl)-1,3-dihydro-indol-2-on
-
Die
Titelverbindung wurde auf eine zu Beispiel 2 identische Weise synthetisiert,
mit Ausnahme dass 5-(3-Methyl-butanoyl)-1,3-dihydro-indol-2-on anstelle
von 5-(2-Methyl-thiazol-4-yl)-1,3-dihydro-indol-2-on verwendet wurde und
3,5-Dichlor-4-hydroxy-benzaldehyd anstelle von 3,5-Brom-4-hydroxy-benzaldehyd
verwendet wurde. 1H NMR (DMSO-d6): δ 11,05 (s,
1H); 10,99 (s, 1H); 8,63 (s, 1H); 8,28 (s, 1H); 7,92 (s, 1H); 7,85
(d, J = 8,2, 1H); 7,77 (s, 111); 6,89 (d, J = 8,2, 1H); 2,84 (d,
J = 6,8, 2H); 2,21 – 2,08
(m, 1H); 0,92 (d, J = 6,8, 6H). ESI-MS: m/z 388 (m-H)-.
-
Beispiel
18; 5-Cyclopropancarbonyl-3-(3,5-dibrom-4-hydroxybenzyliden)-1,3-dihydroindol-2-on
-
Beispiel 18a; 5-Cyclopropancarbonyl-1,3-dihydro-indol-2-on
-
Diese
Verbindung wurde auf eine zu Beispiel 7a identische Weise hergestellt,
mit Ausnahme dass Cyclopropancarbonylchlorid anstelle von 3-Methyl-butanoylchlorid
verwendet wurde. 1H NMR (DMSO-d6): δ 10,73 (s,
1H); 7,93 (d, J = 8,2, 1H); 7,85 (s, 1H); 6,88 (d, J = 8,2, 1H);
3,53 (s, 2H); 2,79 (t, J = 6,2, 1H); 0,94 (d, J = 6,2, 4H) ESI-MS:
m/z 200 (m-H)-.
-
Beispiel 18; 5-Cyclopropancarbonyl-3-(3,5-dibrom-4-hydroxybenzyliden)-1,3-dihydroindol-2-on
-
Diese
Verbindung wurde auf eine zu Beispiel 150 identische Weise hergestellt,
mit Ausnahme dass 5-Cyclopropancarbonyl-1,3-dihydro-indol-2-on anstelle
von 6-Cyano-1,3-dihydro-indol-2-on
verwendet wurde. 1H NMR (DMSO-d6): δ 11,11 (s,
1H); 10,80 (bs, 1H); 8,86 (s, 2H); 8,46 (s, 1H); 8,02 – 7,98 (m,
2H); 6,98 (d, J = 8,1, 1H); 3,0 – 2,9 (m, 1H); 1,05 (d, J =
6, 4H) ESI-MS: m/z 462 (m-H)-.
-
Beispiel
19; 5-Aminomethyl-3-(3,5-dibrom-4-hydroxybenzyliden)-1,3-dihydro-indol-2-on
-
Beispiel 25a; 5-Aminomethyloxindol
-
Eine
Aufschlämmung
aus 5-Cyanooxindol (1,0 g, 6,3 mmol) und 10 Palladium auf Kohlenstoff
(0,05 g) in Eisessig (50 ml) bei Raumtemperatur wurde unter 40 psi
Druck 24 Stunden lang hydriert. Der Katalysator wurde durch Filtration
durch ein Pod aus Diatomeenerde entfernt und das Lösungsmittel
aus dem Filtrat eingedampft, um ein oranges Öl von 5-Aminomethyloxindol
als ein Acetatsalz (1,16 g) zurückzulassen. 1H NMR (DMSO-d6) δ 7,21 (s,
1H), 7,14 (d, 1H, J = 7,6 Hz), 6,75 (d, 1H, J = 7,6 Hz), 3,74 (s,
2H), 3,44 (s, 2H). MS (+ve ES) 146 (100), (M-NH2).
-
Beispiel 19; 5-Aminomethyl-3-(3.5-dibrom-4-hydroxybenzyliden)-1,3-dihydro-indol-2-on
-
Die
Titelverbindung wurde auf eine zu Beispiel 2 identische Weise synthetisiert,
mit Ausnahme dass 5-Aminomethyl-oxindolacetat anstelle von 5-(2-Methyl-thiazol-4-yl)-1,3-dihydro-indol-2-on-hydrochlorid verwendet
wurde. 1H NMR (DMSO-d6) δ 10,80 (s, 1H), 8,76 (s, 2H),
8,14 (br s, 3H), 7,71 (s, 1H), 7,65 (s, 1H), 7,31 (d, 1H, J = 7,6
Hz), 6,88 (d, 1H, J = 7,6 Hz), 3,96 (br q, 2H, J = 6 Hz). MS (AP+ve)
408 (100) (M-NH2).
-
Beispiel
92; 1-(3,5-Dibrom-4-hydroxybenzyliden)-1,3-dihydro-pyrrolo[3,2-f]chinolin-2-on
-
Beispiel 92a; 2-Hydroxyimino-N-(6-chinolinyl)acetamid-hydrochlorid
-
Zu
einer gerührten
Lösung
aus 10,0 g (60,0 mmol) Chloralhydrat in 250 ml Wasser wurde 70,0
g (220 mmol) Natriumsulfatdecahydrat, gefolgt von einer Lösung aus
11,8 g (170 mmol) Hydroxylamin-hydrochlorid in 100 ml Wasser gegeben.
Eine Lösung
aus 7,8 g (54 mmol) 6-Aminochinolin
in 200 ml 1,0 N HCl wurde dann unter Rühren zugegeben. Die sich ergebende
Suspension wurde erwärmt,
und 400 ml 95 % EtOH wurde zugegeben, um die Suspension zu lösen. Die
Lösung
wurde 0,75 Std. lang unter Rückfluss
erhitzt und dann auf Umgebungstemperatur gekühlt. Die Lösung wurde durch Zugabe von
festem Natriumbicarbonat neutralisiert, und der sich ergebende Feststoff
wurde durch Vakuumfiltration gesammelt und luftgetrocknet, um 8,1
g (60 %) 2-Hydroxyimino-N-(6-chinolinyl)acetamid als einen Feststoff
zu ergeben: 1H NMR (DMSO-d6):
7,76 (s, 1H); 7,80 (dd, 1H, J = 3,7, 8,4 Hz); 8,14 (s, 2H); 8,68
(s, 1H); 8,77 (d, 1H, J = 8,4 Hz); 9,02 (d, 1H, J = 3,7 Hz); 10,73 (s,
1H); 12,34 (s, 1H). Massenspektrum (chemische Ionisation negativer
Ionen): m/z = 214 (60 %).
-
Beispiel 92b; 3-H-Pyrrolo[3,2-f]chinolin-1,2-dion
-
2-Hydroxyimino-N-(6-chinolinyl)acetamid
(7,00 g, 32,5 mmol) wurde mit 70 ml konzentrierter Schwefelsäure unter
Rühren
vereinigt und auf eine Umsetzungstemperatur von 120 °C 20 min lang,
gefolgt von einer Stunde bei 95 °C
erhitzt. Man ließ das
Reaktionsgemisch sich auf Raumtemperatur abkühlen und dann wurde es tropfenweise
zu einem Gemisch aus 155 g (1,25 mol) Natriumcarbonatmonohydrat
und 200 g Eis gegeben. Nachdem die Zugabe vollständig war, wurde allmählich Wasser
(600 ml) zu dem Gemisch unter Rühren
gegeben, bis der gesamte anorganische weiße Feststoff gelöst war.
Das wässrige
Gemisch wurde mit 1 M Salzsäure
auf einen pH-Wert von 7 neutralisiert, und das Produkt wurde durch
Filtration gesammelt. Der gesammelte Feststoff wurde zu 200 ml Wasser
gegeben und durch tropfenweise Zugabe von 1 M Salzsäure in freie
eingeschlossene Salze gelöst.
Das Produkt wurde dann durch Zugabe von gesättigtem wässrigem Natriumbicarbonat auf
pH-Wert 7 gefällt.
Das Produkt wurde durch Filtration gesammelt und unter Vakuum bei
55 °C getrocknet,
um 3,89 g (60 %) 3-H-Pyrrolo[3,2-f]chinolin-1,2-dion
als einen rotbraunen Feststoff zu geben. Massenspektrum (chemische
Ionisation negativer Ionen): m/z = 197 (30 %). NMR (DMSO-d6): δ 7,43
(d, 1H, J = 8,8 Hz), 7,66 (dd, 1H, J = 4,1, 8,4 Hz); 8,29 (d, 1H,
J = 8,8 Hz); 8,72 (d, 1H, J = 8,4 Hz); 8,82 (d, 1H, J = 3,3 Hz); 11,2
(s, 1H).
-
Beispiel 92c; 1-Hydrazono-1,3-dihydropyrrolo[3,2-f]chinolin-2-on
-
3-H-Pyrrolo[3,2-f]chinolin-1,2-dion
(3,89 g, 19,6 mmol) wurde mit 12,0 ml wasserfreiem Hydrazin und 10,25
ml Wasser vereinigt und auf 100 °C
unter einem Kühler
und Stickstoffatmosphäre
unter einstündigem Rühren erhitzt.
Das Reaktionsgemisch schäumte
gelegentlich in den Kühler
und Hitze wurde nach Bedarf entfernt, um das Schäumen abklingen zu lassen. Das
Reaktionsgemisch wurde abgekühlt
und in 200 ml Wasser gegossen. Das Produkt wurde durch Filtration
gesammelt und unter Vakuum bei 55 °C getrocknet, um 2,86 g (69
%) 1-Hydrazono-1,3-dihydropyrrolo[3,2-f]chinolin-2-on
als einen braunen Feststoff zu geben. Massenspektrum (Elektrosprühung positiver
Ionen): m/z = 213. NMR (DMSO-d6): δ 7,37 (d,
J = 8,8 Hz, 1H), 7,47 (dd, J = 8,4, 4,2 Hz, 1H), 7,81 (d, J = 8,8
Hz, 1H), 8,71 (dd, J = 4,2, 1,6 Hz, 1H), 8,80 (d, J = 8,4 Hz, 1H),
9,90 (br d, J = 14,7 Hz, 1H), 10,89 (br d, J = 14,7 Hz, 1H), 10,95
(br s, 1H).
-
Beispiel 92d; 6-Aminochinolin-5-carbonsäurehydrazid
-
1-Hydrazono-1,3-dihydro-pyrrolo[3,2-f]chinolin-2-on
(1,07 g, 5,05 mmol) wurde mit 6,0 ml wasserfreiem Hydrazin und 5,0
ml Wasser in einem 100 ml Kolben (überdimensional, um Schäumen zu
erlauben) vereinigt und auf Rückfluss
(145 °C Ölbad) unter
einem Kühler
und Stickstoffatmosphäre
unter Rühren
erhitzt. Nach 4,5 Std. zeigte eine analytische HPLC, dass das gesamte
Hydrazon verbraucht worden war. Das Reaktionsgemisch wurde gekühlt, mit
75 ml Wasser verdünnt
und filtriert, um 0,45 g (luftgetrocknet) 6-Aminochinolin-5-carbonsäurehydrazid
als einen olivbraunen Feststoff zu geben. Massenspektrum (Elektrosprühung negativer
Ionen): m/z = 215 (100 %). NMR (DMSO-d6): δ, 3,64 (s,
2H), 4,22 (s, 2H), 5,66 (s, 2H), 7,25 (d, 1H, J = 9,1 Hz), 7,34
(m, 1H), 7,65 (d, 1H, J = 9 Hz), 8,34 (d, 1H, J = 8,5 Hz), 8,50
(s, 1H), 9,27 (s, 1H).
-
Beispiel 92e; 1,3-Dihydro-pyrrolo[3,2-f]chinolin-2-on
-
6-Aminochinolin-5-carbonsäurehydrazid
wurde in 10 ml 2 M Salzsäure
gelöst
und kurz auf einer heißen
Platte erhitzt. Das Reaktionsgemisch wurde durch die allmähliche Zugabe
von festem Natriumbicarbonat neutralisiert und filtriert. Das gesammelte
Produkt wurde unter Vakuum bei 55 °C getrocknet, um 416 mg (45 %)
1,3-Dihydro-pyrrolo[3,2-f]chinolin-2-on als einen braunen Feststoff
zu geben. Massenspektrum (chemische Ionisation negativer Ionen):
m/z = 183 (60 %). 1H NMR (DMSO-d6): δ,
3,80 (s, 2H), 7,35 (d, J = 8,8 Hz, 1H), 7,44 (dd, J = 8,4, 4,2 Hz,
1H), 7,88 (d, J = 8,8 Hz, 1H), 8,08 (d, J = 8,4 Hz, 1H), 8,70 (dd,
J = 4,2, 1,6 Hz, 1H), 10,57 (br s, 1H).
-
Beispiel 92; 1-(3'5-Dibrom-4-hydroxybenzyliden)-1,3-dihydro-pyrrolo[3,2-f]chinolin-2-on
-
1,3-Dihydro-pyrrolo[3,2-f]chinolin-2-on
(552 mg, 3,00 mmol) wurde mit 855 mg (3,05 mmol) 3,5-Dibrom-4-hydroxybenzaldehyd
(TCI Chemicals) in 6 ml Eisessig mit 1 ml konzentrierter Salzsäure vereinigt.
Das Reaktionsgemisch wurde bei 115 °C 8 Std. lang gerührt, gekühlt und
filtriert, wobei mit Ethylacetat gewaschen wurde, um 1,32 g 1-(3,5-Dibrom-4-hydroxybenzyliden)-1,3-dihydro-pyrrolo[3,2-f]chinolin-2-on-hydrochlorid
als einen braunen Feststoff, der aus einem 9/1 Gemisch von Z/E Isomeren
bestand, zu geben. Massenspektrum (chemische Ionisation negativer
Ionen): m/z = 443 (40 %), 445 (100 %), 447 (40 %). 1H
NMR (DMSO-d6): δ 7,69 (d, J = 8,8 Hz, 1H); 7,93
(dd, J = 8,7, 4,9 Hz, 1H); 8,24 (d, J = 8,8 Hz, 1H); 8,27 (s, 1H);
8,80 (s, 1H); 9,04 (d, J = 4,7 Hz, 1H); 9,51 (d, J = 8,6 Hz, 1H);
11,3 (s, 1H).
-
Beispiel
104; 6-Brom-3-(3,5-dichlor-4-hydroxybenzyliden)-1,3-dihydro-indol-2-on
-
Beispiel 104a; 4-Brom-2-nitrophenylbrenztraubensäure
-
Diethyloxalat
(29,2 g, 0,2 mol) und 4-Brom-2-nitrotoluol (21,6 g, 0,1 mol, Lancaster)
wurden in eine gekühlte
Natriumethoxidlösung,
die aus Natrium (4,6 g, 0,2 mol) und absolutem Ethanol (90 ml) hergestellt
wurde, gegossen. Das Gemisch wurde über Nacht bei Raumtemperatur
gerührt
und dann 10 Minuten lang am Ende der Umsetzung unter Rückfluss
erhitzt. Wasser (30 ml) wurde zugegeben und das Reaktionsgemisch
2,5 Std. lang unter Rückfluss
erhitzt. Das Umsetzungsgemisch wurde gekühlt und konzentriert, um Ethanol
im Überschuss
zu entfernen. Der Niederschlag wurde durch Filtration gesammelt,
mit Ethanol gewaschen und getrocknet. Das rohe Natriumsalz wurde
in Wasser gelöst
und mit konz. HCl angesäuert.
Der Feststoff fällte
aus und wurde durch Filtration gesammelt. Das Rohprodukt wurde aus
Hexan und Ethylacetat umkristallisiert, um 12,5 g (43 %) als einen
federnen Kitt-farbenen Feststoff zu geben; 1H
NMR (CDCl3): δ 8,40 (d, 1H, J = 1,9 Hz), 7,84 (dd,
1H, J = 8,0, 2,0 Hz), 7,30 (d, 1H, J = 2 Hz) 4,65 (s, 2H).
-
Beispiel 104b; 4-Brom-2-nitrophenylessigsäure
-
Eine
30 % Wasserstoffperoxidlösung
(4,95 ml, 0,04 mol) wurde tropfenweise zu einer Lösung aus 4-Brom-2-nitrophenylbrenztraubensäure (0,04
mol) und Natriumhydroxid (5,3 g 0,1 mol) in Wasser (175 ml) gegeben,
wobei bei 0 °C
gerührt
wurde. Die Umsetzungslösung
wurde 1 Std. lang bei 5 °C
gerührt
und dann mit verdünnter
HCl angesäuert.
Der gelbe Niederschlag wurde filtriert und das Rohprodukt aus Hexan
und Ethylacetat umkristallisiert, um 8,4 g (75 %) als einen hellbeigen
Feststoff zu erhalten; 1H-NMR (DMSO-d6): δ 12,67
(br s, 1H), 8,28 (d, J = 2,0 Hz), 7,97 (dd, 1H, J = 9,0 Hz, 2,0
Hz), 7,55 (d, 1H, J = 8,0 Hz) 4,00 (s, 3H). MS (-ve) m/z: 259 (M-H).
-
Beispiel 104c; 6-Bromoxindol
-
Zinkstaub
(8,5 g, 0,13 mol) wurde langsam zu einer Lösung aus 4-Brom-2-nitrophenylessigsäure (8,4 g,
0,03 mol) in 50 % Schwefelsäure
(200 ml) und absolutem Ethanol (300 ml) bei 90 °C über 0,75 Std. hinweg gegeben.
Das Gemisch wurde auf dieser Temperatur 2 Std. lang unter Rühren erhitzt.
Der Überschuss
an Ethanol wurde durch Eindampfen im Vakuum entfernt, und das Gemisch
wurde filtriert. Das Filtrat wurde mit Diethylether extrahiert.
Die vereinigten organischen Portionen wurde mit gesättigtem
Natriumbicarbonat, gesättigtem
Natriumchlorid gewaschen, mit Whatman 1 PS Phasentrennungspapier
filtriert und im Vakuum eingedampft, um 3,8 g (56 %) eines blass
pfürsichfarbenen
Feststoffs zu geben. 1H-NMR (DMSO-d6): δ 10,57 (brs,
1H), 7,14 (m, 2H), 6,98 (s, 1H), 3,47 (s, 2H).
-
Beispiel 104; 6-Brom-3-(3,5-dichlor-4-hydroxybenzyliden)-1,3-dihydro-indol-2-on
-
Die
Titelverbindung wird auf eine zu Beispiel 2 identische Weise synthetisiert,
mit Ausnahme dass 6-Brom-oxindol anstelle von 5-(2-Methyl-thiazol-4-yl)-1,3-dihydro-indol-2-on-hydrochlorid verwendet
wurde und 3,5-Dichlor-4-hydroxy-benzaldehyd anstelle von 3,5-Dibrom-4-hydroxy-benzaldehyd
verwendet wurde. 1H NMR (DMSO-d6) δ 7,05 (s,
1H); 7,12 (d, 1H); 7,43 (d, 1H); 7,56 (s, 1H); 7,76 (s, 2H); 10,78
(bs, 1H); 10,91 (bs, 1H). Elektrosprüh MS (ve) 384.
-
Beispiel
109; 3-(3,5-Dibrom-4-hydroxybenzyliden)-5-pyrid-3-yl-1,3-dihydro-indol-2-on
-
Beispiel 109a; 5-Pyrid-3-yl-1,3-dihydro-indol-2-on
-
Ein
Gemisch aus 0,736 g (2 mmol) 3-Tributylzinnpyridin, 0,259 g (1 mmol)
5-Iod-oxindol, 0,497 g (3 mmol) Tetraethylammoniumchlorid und 0,035
g (0,05 mmol) Bis(triphenylphospin)palladium(II)chlorid in 4 ml Acetonitril
wurde unter Rückfluss
24 Std. lang erhitzt. Nach dem Abkühlen auf Umgebungstemperatur
wurde das Gemisch mit 20 ml CHCl3 verdünnt und
50 ml 10 % Kaliumfluoridlösung
(wässr.)
wurde zugegeben. Das Gemisch wurde durch ein 1-Inch Celitepod filtriert
und in Schichten abgetrennt. Die organische Schicht wurde im Vakuum
konzentriert, und der Rückstand
wurde auf Silicagel (EtOAc/MeOH 5 %) chromatographiert, um 5-Pyrid-3-yl-1,3-dihydro-indol-2-on
als weißen
Feststoff (0,033 g, 16 %) zu ergeben: 1H
NMR (DMSO-d6): δ 10,51 (s, 1H); 8,83 (d, J =
2,2, 1H); 8,51 (dd, J1 = 1,3, J2 =
4,6, 1H); 8,02 – 7,97
(m, 1H); 7,59 (s, 1H); 7,54 (d, J = 8,1, 1H); 7,44 (dd, J1 = 4,7, J2 = 7,9,
1H); 6,93 (d, J = 8,1, 1H); 3,55 (s, 2H). APCI-MS: m/z 211 (m+H)+.
-
Beispiel 109; 3-(3,5-Dibrom-4-hydroxybenzyliden)-5-pyrid-3-yl-1,3-dihydro-indol-2-on
-
Die
Titelverbindung wurde auf eine zu Beispiel 2 identische Weise hergestellt,
mit Ausnahme dass 5-Pyrid-3-yl-1,3-dihydro-indol-2-on anstelle von
5-(2-Methyl-thiazol-4-yl)-1,3-dihydroindol-2-on verwendet wurde.
1H NMR (DMSO-d6): δ 10,93 (s, 1H); 9,13 (s, 1H);
8,81 (s, 2H); 8,8 – 8,7
(m, 1H); 8,6 – 8,5
(m, 1H); 8,23 (s, 1H); 7,94 (s, 1H); 7,9 – 7,8 (m, 1H); 7,72 (d, J =
8, 1H); 7,02 (d, J = 8, 1H). APCI-MS: m/z 471 (M-H)-.
-
Beispiel
115; N[bis(2-Hydroxyethyl)]-carbaminsäure-2,6-dibrom-4-[5-(2-methyl-thiazol-4-yl)-2-oxo-1,2-dihydro-indol-3-ylidenmethyl]-phenylester
-
3-(3,5-Dibrom-4-hydroxybenzyliden)-5-(2-methyl-thiazol-4-yl)-1,3-dihydro-indol-2-on
(0,50 g, 0,95 mmol) wurde in 10 ml wasserfreiem THF unter Stickstoff
mit 4A Molekularsieben 4 Std. lang aufgeschlämmt. Diisopropylethylamin (0,33
ml, 1,9 mmol) wurde zugegeben, um eine gelborange Lösung zu
geben. In einem getrennten Kolben wurden 0,50 ml Phosgenlösung (1,9
M in Toluol, 0,95 mmol) und 5 ml wasserfreies THF in einem Eisbad
unter einem druckausgleichenden Tropftrichter unter Stickstoffatmosphäre gekühlt. Die
gelborange Lösung
des Phenoxidanions wurde in den Tropftrichter über eine Spritze überführt, und
die Lösung
wurde tropfenweise über
30 min hinweg zu der Phosgenlösung
gegeben. Man ließ das
Reaktionsgemisch sich über
1 Std. hinweg auf Raumtemperatur wärmen. Das Reaktionsgemisch
wurde wieder in einem Eisbad gekühlt,
und eine Lösung
aus 135 mg (1,28 mmol) Diethanolamin und 0,165 ml Diisopropylethylamin
(0,95 mmol) in 2 ml wasserfreiem THF wurde in einer Portion zugegeben.
Man ließ das
Reaktionsgemisch sich über
Nacht auf Raumtemperatur wärmen.
Das orange Reaktionsgemisch wurde mit 100 ml Ethylacetat verdünnt, mit
50 ml 0,2 M wässrigem
Natriumbicarbonat gewaschen und über
Natriumsulfat getrocknet. Eindampfen des Lösungsmittels gab 0,6 g des
Rohprodukts als einen orangen Feststoff. Das Produkt wurde durch
Chromatographie auf Silicagel unter Verwendung von 1 : 1 Hexan/Ethylacetat,
gefolgt von Ethylacetat gereinigt, um 209 mg 3-(3,5-Dibrom-4-hydroxybenzyliden)-5-(2-methyl-thiazol-4-yl)-1,3-dihydro-indol-2-on
als einen orangen Feststoff zu geben. Massenspektrum (chemisch Ionisation
positiver Ionen): m/z = 644 (M+23) für 79Br
mit Isotopensignalen.
NMR (DMSO-d6):
(Gemisch von E/Z Isomeren ist etwa im Verhältnis von ~ 2: 1) δ 2,70 und
2,76 (2s, 3H); 3,47 (m, 2H); 3,62 (m, 4H); 3,8 (m, 2H); 4,88 (m,
1H); 4,96 (m, 1H); 6,92 und 6,96 (2d, 1H, J = 8 Hz); 7,62 (d, 1,3H, J
= 8 Hz); 7,76 – 7,96
(m, 2H); 8,15 – 8,33
(m, 2H); 8,87 (s, 0,7H); 10,81 und 10,88 (2s, 1H).
-
Beispiel
118; 3-(3,5-Dibrom-4-ethoxycarbonat-benzyliden)-2-oxo-2,3-dihydro-5-chlor-1H-indol
-
Ein
heterogenes Gemisch aus 3-(3,5-Dibrom-4-hydroxybenzyliden)-2-oxo-2,3-dihydro-5-chlor-1H-indol (0,41 g,
1,0 mmol) in trockenem Dichlormethan (15 ml) unter Stickstoff wurde
mit Diisopropylethylamin (0,70 ml, 4,0 mmol) bei Raumtemperatur
behandelt. Zu der sich ergebenden homogenen Lösung wurde Ethylchlorformiat
(0,19 ml, 2,0 mmol) auf eine tropfenweise Art gegeben und das Gemisch
drei Stunden lang gerührt.
Das Reaktionsgemisch wurde zuerst mit einer gesättigten Natriumbicarbonatlösung, dann
mit einer gesättigten
Ammoniumchloridlösung
gewaschen. Die organische Schicht wurde über Magnesiumsulfat getrocknet,
filtriert und konzentriert. Der Rückstand wurde in Dichlormethan
gelöst,
in einem Eisbad gekühlt,
und das Produkt wurde mit Hexan gefällt. Die Feststoffe wurden
auf einem Filter gesammelt, mit Hexan gewaschen und luftgetrocknet,
um die Titelverbindung (0,34 g, 68 %) bereitzustellen. 1H
NMR (DMSO-d6): δ 10,90 (s, 1H), 8,12 (s, 2H),
7,70 (s, 1H), 7,35 – 7,40 (m,
2H), 6,96 (d, 1H), 4,43 (q, 2H), 1,40 (t, 3H). Anal. berechnet für C18H12NO4Br2Cl: C, 43,11; H 2,41; N, 2,79. Gemessen:
C, 43,01; H, 2,47; N, 2,73. MS (API+): 502(5) (M+1).
-
Beispiel
121; 3-(3,5-Dibrom-4-pivalouloxymethoxy-benzyliden)-2-oxo-2,3-dihydro-5-chlor-1H-indol
-
Eine
Lösung
aus 3-(3,5-Dibrom-4-hydroxybenzyliden)-2-oxo-2,3-dihydro-5-chlor-1H-indol
(0,43 g, 1,0 mmol) in trockenen Acetonitril (20 ml) unter Stickstoff
wurde mit Kalium-tert-butoxid
(0,12 g, 1,1 mmol) bei Raumtemperatur behandelt. Das sich ergebende
orange heterogene Gemisch wurde mit 18-Crown-6 (0,053 g, 0,20 mmol)
behandelt und 15 Minuten lang vor der Zugabe von Chlormethylpivalat
(0,40 ml, 2,8 mmol) gerührt.
Das Reaktionsgemisch wurde auf siebzig Grad Celsius erhitzt, drei
Stunden lang gerührt,
dann während es
heiß was,
filtriert. Man ließ das
Filtrat auf Raumtemperatur kühlen
und rührte über Nacht.
Die sich ergebenden Feststoffe wurden auf einem Filter gesammelt,
mit Acetonitril gewaschen und unter Vakuum getrocknet, um die Titelverbindung
(0,24 g, 44 %) als ein Gemisch von E/Z Isomeren zu geben. 1H NMR (DMSO-d6):
(Gemisch von E/Z Isomeren) δ 10,93
(s, 1H), 10,87 (s, 1H), 8,85 (s, 2H), 8,09 (s, 2H), 7,94 (s, 1H),
7,84 (d, 1H), 7,67 (s, 1H), 7,32 – 7,39 (m, 3H), 6,96 (d, 1H),
6,91 (d, 1H), 5,91 (s, 2H), 5,88 (s, 2H), 1,23 (s, 9H), 1,22 (s, 9H).
MS (ES-): 542 (60) (M-1). Anal. berechnet für C21H18NO4ClBr2: C, 46,40; H, 3,34; N, 2,58. Gemessen:
C, 46,33; H, 3,30; N, 2,55.
-
Beispiel
123; 2,6-Dibrom-4-[(5-iod-2-oxo-1,2-dihydro-3H-indol-3-yliden)methyl]phenyl-N-[2-(2-hydroxyethoxy)ethyl]carbamat
-
Zu
einer Lösung
aus 3-(3,5-Dibrom-4-hydroxybenzyliden)-5-iod-1,3-dihydro-indol-2-on
(210 mg, 0,40 mmol) in 20 ml THF unter Stickstoff wurde 1 M Kalium-t-butoxid
in THF (0,40 ml, 0,40 mmol) tropfenweise über eine Spritze gegeben. Die
Lösung
wurde auf 0 °C
gekühlt
und 1,97 M Phosgen in Toluol (0,21 ml, 0,41 mmol) wurde tropfenweise über eine
Spritze zugegeben, und das Umsetzungsgemisch wurde 10 Minuten lang
gerührt.
2-(2-Aminoethoxy)ethanol
(40 μl,
0,40 mmol) wurde dann über
eine Spritze zugegeben, gefolgt von N-Methylmorpholin (~ 45 mg,
~ 45 mmol). Das Umsetzungsgemisch wurde 10 Minuten lang bei 0 °C gerührt und
dann ließ man
es auf Raumtemperatur wärmen.
Die Lösung
wurde mit einem gleichen Volumen Ether verdünnt, durch eine feine Fritte
zur Klärung
filtriert und weiter mit 10 ml Ether verdünnt. Das Produkt wurde dann durch
die Zugabe von ~ 80 ml Hexan gefüllt
und wurde filtriert und mit mehr Hexan gewaschen, um 0,21 g (79 %)
der Titelverbindung als einen gelben Feststoff zu geben. 1H NMR (400 MHz, DMSO-d6)
d: 10,77 (s, 1H), 8,24 (t, J = 5,6 Hz, 1H), 8,02 (s, 2H), 7,67 (s,
1H), 7,57 (s, 1H), 7,55 (dd, J = 8,2, 1,6 Hz, 1H), 6,71 (d, J =
8,2 Hz, 1H), 4,58 (br t, 1H), 3,52 – 3,42 (m, 6H), 3,27 – 3,20 (m,
2H). ESI-MS m/z 673, 675, 677 (M+23). Anal. berechnet für C20H17Br2IN2O5: C, 36,84; H,
2,63; N, 4,30. Gemessen: C, 36,75; H, 2,60; N, 4,22.
-
Beispiel 127; 3-(3,5-Dibrom-4-hydroxybenzyliden)-2-oxo-2,3-dihydro-1H-indol-5-carbonsäure
-
3-(3,5-Dibrom-4-hydroxybenzyliden)-2-oxo-2,3-dihydro-1H-indol-5-carbonsäure wurde
aus 5-Carbonsäureoxindol
und 3,5-Dibrom-4-hydroxybenzaldehyd, dem zu Beispiel 2 identischen
Verfahren folgend, hergestellt, mit Ausnahme dass 3-(3,5-Dibrom-4-hydroxybenzyliden)-2-oxo-2,3-dihydro-1H-indol-5-carbonsäure anstelle
von 5-(2-Methyl-thiazol-4-yl)-1,3-dihydro-indol-2-on-hydrochlorid verwendet
wurde. Ausbeute 79 %. 1H-NMR (DMSO-d6): δ 12,6
(bs, 1H), 10,96 (s, 1H), 10,61 (bs, 1H), 8,17 (s, 1H), 7,93 (s,
2H), 7,84 (d, J = 8,2 Hz, 1H), 7,54 (s, 1H), 6,93 (d, J = 8,2 Hz,
1H). Massenspektrum (APCI negativer Ionen): m/z = 436 (M-1, 5 %), 438
(M-1, 10 %), 440 (M-1, 8 %).
-
Beispiel
130; N[3-(3,5-Dibram-4-hydroxy-benzylidin)]-2-oxo-2,3-dihydro-1H-indol-5-yl)-acetamid
-
Beispiel 130a; 5-Nitro-1,3-dihydro-indol-2-on
-
Eine
Lösung
aus 5 g (37,6 mmol) Oxindol in 75 ml konz. H2SO4 wurde auf –5 °C in einem Eis/EtOH-Bad abgekühlt. Eine
Lösung
aus 3,83 g (45,1 mmol) NaNO3 in 25 ml konz.
H2SO4 wurde über 30 min
hinweg tropfenweise zugegeben. Das Gemisch wurde bei –5 °C 1 Std.
lang gerührt.
Das Eisbad wurde dann entfernt, und man ließ das Gemisch langsam auf Umgebungstemperatur
wärmen.
Das Gemisch wurde auf 500 g zerstoßenes Eis gegossen. Der Feststoff
wurde durch Vakuumfiltration gesammelt und luftgetrocknet. Der Feststoff wurde
in warmen Methanol gerührt
und durch Vakuumfiltration gesammelt, um 5-Nitro-1,3-dihydroindol-2-on (1,7
g, 25 %) zu erhalten: 1H NMR (DMSO-d6): δ 3,6
(s, 2H), 6,95 (d, J = 8,5 Hz, 1H), 8,1 (s, 1H), 8,12 (d, J = 8,7
Hz, 1H), 11,01 (s, 1H). APCI-MS: m/z 177 (m-H)-.
-
Beispiel 130b; 5-Amino-1,3-dihydro-indol-2-on
-
Ein
Gemisch aus 1,5 g (8,4 mmol) 5-Nitro-1,3-dihydro-indol-2-on, 150
mg Pd/C 10 % und 50 ml MeOH in 100 ml EtOAc wurde auf ein Parr® Hydriergerät gestellt
und mit 45 psi Wasserstoffgas beladen. Das Gemisch wurde 2 Std.
lang geschüttelt,
Das Gemisch wurde filtriert und das Lösungsmittel wurde im Vakuum
entfernt, um 5-Amino-1,3-dihydro-indol-2-on (1,22 g, 98 %) zu erhalten:
1H NMR (DMSO-d6): δ 3,27 (s, 2H), 4,6 (s, 2H), 6,34
(dd, J1 = 2 Hz, J2 =
8,1 Hz, 1H), 6,45 (m, 2H), 9,88 (s, 1H). APCI-MS: m/z 147 (m-H)-.
-
Beispiel 130c; N-(2-Oxo-2,3-dihydro-1H-indol-5-yl)-acetamid
-
Ein
Gemisch aus 200 mg (1,35 mmol) 5-Amino-1,3-dihydro-indol-2-on in
6 ml Essigsäureanhydrid
wurde 30 Minuten lang unter Rückfluss
erhitzt. Das Umsetzungsgemisch wurde auf 50 g zerstoßenes Eis
gegossen. Das Gemisch wurde gut gerührt, und der Feststoff wurde
durch Vakuumfiltration gesammelt. Der Feststoff wurde mit 200 ml
H2O gewaschen und luftgetrocknet, um N-(2-Oxo-2,3-dihydro-1H-indol-5-yl)-acetamid
(119 mg, 46 %) zu erhalten:
1H NMR
(DMSO-d6): δ,1,97 (s, 3H), 3,41 (s, 2H),
6,68 (d, J = 8,4 Hz, 1H),), 7,26 (dd, J1 =
2 Hz, J2 = 8,4 Hz, 1H), 7,46 (d, J = 2 Hz,
1H), 9,73 (s, 1H). 10,23 (s, 1H), ESI-MS: m/z 189 (m-H).
-
Beispiel 130; N[3-(3,5-Dibrom-4-hydroxy-benzylidin)]-2-oxo-2,3-dihydro-1H-indol-5-yl)-acetamid.
-
Ein
Gemisch aus 0,050 g (0,26 mmol) N-(2-Oxo-2,3-dihydro-1H-indol-5-yl)-acetamid
und 0,081 g (0,29 mmol) 3,5-Dibrom-4-hydroxybenzaldehyd wurde in
2 ml HOAc gerührt.
100 ?1 konzentrierter HCl wurde zugegeben, und das Gemisch wurde
3 Std. lang 80 °C
erhitzt. Nach dem Abkühlen
auf Umgebungstemperatur wurde der Feststoff durch Vakuumfiltration
gesammelt und mit EtOAc und Et
2O gewaschen
und in einem Vakuumofen getrocknet, um N-[3-(3,5-Dibrom-4-hydroxy-benzylidin)]-2-oxo-2,3-dihydro-1H-indol-5-yl)-acetamid
als einen gelben Feststoff (0,76 g, 65 %) zu erhalten: 1H NMR (DMSO-d
6): δ 1,99
(s, 3H), 6,72 (d, J = 8,3 Hz, 1H), 7,17 (dd, J
1 =
1,8 Hz, J
2 = 8,3 Hz, 1H), 7,54 (s, 1H),
7,86 (d, J = 1,5 Hz, 1H), 8,79 (s, 2H), 9,74 (s, 1H), 10,54 (s,
1H). APCI-MS: m/z 475 (m+Na)
+ Beispiel
143; 3-(3,5-Dibrom-4-hydroxybenzyliden)-2-oxo-2,3-dihydro-1H-indol-5-carbonsäure-(2-hydroxyethyl)amid
-
Beispiel 143a; 5-Carbonsäure-1,3-dihydro-indol-2-on
-
5-Carbonsäure-1,3-dihydro-indol-2-on-methylester
(5,6 g, 29,3 mmol) wurde in heißem
Acetonitril (500 ml) gelöst
und Aluminiumiodid (25 g, 61,3 mmol) langsam zugegeben. Das Reaktionsgemisch
wurde 0,5 Std. lang unter Rückfluss
erhitzt und dann auf Eiswasser gegossen und zwei Mal mit Ethylacetat
(200 ml) extrahiert. In beiden Phasen unlösliches Material wurde abfiltriert
und mit wässriger
Natriumthiosulfatlösung,
gefolgt von Wasser gewaschen und getrocknet. Ausbeute von 5-Carbonsäure-1,3-dihydro-indol-2-on:
1,9 g. Die Ethylacetatlösung
wurde mit wässriger
Natriumthiosulfatlösung
gewaschen und zur Trockene konzentriert.
-
Ausbeute
von 5-Carbonsäure-1,3-dihydro-indol-2-on:
0,2 g. Die wässrige
Phase wurde mit Natriumthiosulfatlösung behandelt und beim Stehen
setzte sich mehr Produkt als Niederschlag ab. Dieser wurde abfiltriert,
mit Wasser gewaschen und getrocknet. Ausbeute von 5-Carbonsäure-1,3-dihydro-indol-2-on:
2,8 g; 1H-NMR (DMSO-d6): δ 12,60 (bs,
1H), 10,73 (bs, 1H), 7,84 (d, J = 8,1 Hz, 1H), 7,77 (s, 1H), 6,90
(d, J = 8,1 Hz, 1H), 3,56 (s, 2H). Massenspektrum (Elektronsprühung negativer
Ionen): m/z = 176 (M-1, 6 %).
-
Beispiel 143; 3-(3,5-Dibrom-4-hydroxybenzyliden)-2-oxo-2,3-dihydro-1H-indol-5-carbonsäure(2-hydroxyethyl)amid
-
3-(3‚5-Dibrom-4-hydroxybenzyliden)-2-oxo-2,3-dihydro-1H-indol-5-carbonsäure (0,34
9, 0,77 mmol) (Beispiel 156) und Ethanolamin (0,08 g, 1,3 mmol)
wurden in DMF (2 ml) gelöst
und unter Rühren
auf 5 °C gekühlt. Diethylcyanophosphonat
(0,172 g, 1 mmol), gefolgt von Triethylamin (0,25 g, 2,5 mmol) wurden
zugegeben und das Rühren
0,5 Std. lang bei 50 °C
fortgeführt,
und dann ließ man
das Reaktionsgemisch sich auf Raumtemperatur wärmen. Nach 1,5 Std. wurde die
Umsetzung mit Wasser (10 ml) gequencht und vier Mal mit einem 4/1
Gemisch aus Chloroform/Isopropanol (25 ml) extrahiert. Die organischen
Phasen wurden vereinigt, über
Magnesiumsulfat getrocknet und zur Trockene konzentriert. Umkristallisierung
aus Ethanol gab 22 mg 3-(3,5-Dibrom-4-hydroxybenzyliden)-2-oxo-2,3-dihydro-1H-indol-5-carbonsäure-(2-hydroxyethyl)amid; 1H-NMR (DMSO-d6): δ 10,9 (s,
1H), 10,7 (bs, 1H), 8,81 (s, 2H), 8,25 (t, J = 5,6 Hz, 1H), 8,19
(s, 1H), 7,77 (s, 1H), 7,72 (d, J = 8,0 Hz, 1H), 6,86 (d, J = 8,0
Hz, 1H), 4,73 (bs, 1H), 3,52 (m, 2H), 3,37 (m, 2H). Massenspektrum
(Elektronsprühung
negativer Ionen): m/z = 479 (M-1, 22 %), 481 (M-1, 35 %), 483 (M-1,
30 %).
-
Beispiel
150; 6-Cyano-3-(3,5-dibrom-4-hydroxybenzyliden)-1,3-dihydro-indol-2-on
-
Beispiel 150a; 6-Cyano-1,3-dihydro-indol-2-on
-
6-Brom-1,3-dihydro-indol-2-on
(0,621 g, 2,93 mmol), Tributylzinncyanid (1,11 g, 3,5 mmol), Tetraammoniumchloridhydrat
(0,97 g, 5,9 mmol), Dichlor-bis(triphenylphosphin)palladium(II)
(0,21 g, 0,3 mmol) und Tetrakis(triphenylphosphin)palladium(0) (0,342
g, 0,3 mmol) wurden mit Dichlorethan (100 ml) behandelt und das Reaktionsgemisch
16 Std. lang unter Stickstoff unter Rühren unter Rückfluss
erhitzt. Eine weitere Zugabe von Tetrakis(triphenylphosphin)palladium(0)
(0,23 g, 0,2 mmol) wurde getätigt
und der Rückfluss
weitere 6 Std. lang fortgeführt.
Eine dritte Zugabe von Tetrakis(triphenylphosphin)palladium(0) (0,345
9, 0,3 mmol) wurde getätigt
und der Rückfluss
weitere 16 Std. lang fortgeführt.
Nach dem Kühlen
wurde die Umsetzungslösung
zwei Mal mit einer wässrigen
Lösung
aus Kaliumfluorid (50 ml) gewaschen und die organische Phase über Magnesiumsulfat
getrocknet, filtriert und zur Trockene eingedampft. Das Produkt
wurde einer Chromatographie auf Silicagel unter Verwendung von Dichlormethan
unterzogen, um 140 mg 6-Cyano-1,3-dihydro-indol-2-on, das mit Triphenylphosphinoxid
kontaminiert war, zu geben. Eine zweite chromatographische Reinigung
auf Silicagel unter Verwendung von Dichlormethan gab 61 mg reines
6-Cyano-1,3-dihydro-indol-2-on; 1H-NMR (DMSO-d6): δ 10,64 (bs,
1H), 7,37 (s, 2H), 7,10 (s, 1H), 3,56 (s, 2H). Massenspektrum (Elektronsprühung negativer Ionen):
m/z = 157 (M-1, 100 %).
-
Beispiel 150; 6-Cyano-3-(3,5-dibrom-4-hydroxybenzyliden)-1,3-dihydro-indol-2-on
-
6-Cyano-1,3-dihydro-indol-2-on
(46,3 mg, 0,29 mmol) und 3,5-Dibrom-4-hydroxybenzaldehyd (80 mg, 0,39
mmol) und p-Toluolsulfonsäuremonohydrat
(1 mg, 0,005 mmol) wurden mit Toluol (30 ml) behandelt, und das
Reaktionsgemisch unter Rückfluss
mit Rühren
mit einer Dean-Stark-Wasserfalle, welche 1,5 Std. lang befestigt
war, erhitzt. Während
dieser Zeit setzte sich ein oranger Feststoff ab, welcher nach dem
Abkühlen
abfiltriert, mit Toluol gewaschen und im Vakuum 3 Tage lang bei
125 °C getrocknet
wurde, um 80 mg 6-Cyano-3-(3,5-dibrom-4-hydroxybenzyliden)-1,3-dihydro-indol-2-on
zu geben; 1H-NMR (DMSO-d6): δ 11,05 (bs, 1H),
8,86 (s, 2H), 7,97 (s, 1H), 7,85 (d, J = 7,9 Hz, 1H), 7,50 (d, J
= 7,9 Hz, 1H), 7,20 (s, 1H). Massenspektrum (Elektronsprühung negativer
Ionen): m/z = 417 (M-1, 48 %), 419 (M-1, 100 %), 421 (M-1, 48 %).
-
NUTZEN
-
Kinase-Signalübertragung
führt unter
anderen Reaktionen zu Zellproliferation, -differenzierung und -metabolismus.
Abnormale Zellproliferation kann zu einem weiten Feld von Störungen und
Erkrankungen führen,
einschließlich
der Entwicklung von Neoplasie, wie Karzinom, Sarkom, Leukämie, Glioblastom,
Hämangiom;
Psoriasis, Arteriosklerose, Arthritis und diabetischer Retinopathie
oder anderen Störungen,
die mit unkontrollierter Angiogenese und oder Vaskulogenese in Zusammenhang
stehen.
-
Die
Effizienz von erfindungsgemäßen Verbindungen
als Inhibitoren der Raf-Kinaseaktivität kann unter Verwendung von
auf dem Fachgebiet bekannten pharmakologischen Verfahren oder wie
detailliert nachstehend beschrieben, beruhend auf der Ähnlichkeit
etablierter Methoden, evaluiert werden.
-
Die
Stärke
der cRaf1-abhängigen
Kinaseaktivität
wurde unter Verwendung von einem von zwei Assayformaten gemessen.
Das erste maß die
durch cRaf1 katalysierte Phosphorylierung von MEK1, dem natürlichen
Substrat für
cRaf1. Dieser Assay wird als der cRaf1-Assay bezeichnet. Das zweite
maß die
Fähigkeit
von cRaf1, MEK1 zu phosphorylieren und zu aktivieren. Dieser Assay
wird als der Raf/MEK-Kaskadenassay bezeichnet. Das Raf/MEK-Kaskadenassayformat
wurde als der primäre
Screen angewendet, da ein größeres Signal
mit weniger Enzym erreicht wurde. Das cRaf1-Assayformat wurde verwendet,
um zu bestätigen,
dass cRaf1 das Enzym war, das durch die erfindungsgemäßen Verbindungen
beeinflusst wurde.
-
A. cRaf1-Assay
-
Menschliches
Raf1, das mit Polyhistidin am Carboxyterminus markiert war, wurde
in einem Baculovirus-Expressionssystem exprimiert und durch Ni-Chelat-Affinitätschromatographie
gereinigt. Menschliches MEK1 wurde in E. coli als ein Fusionsprotein
mit Glutathion-S-Transferase
exprimiert und durch Glutathion-Sepharose-Affinitätschromatographie
gereinigt. Typische Assays wurde in einem Endvolumen von 40 – 100 ml mit
und ohne Inhibitoren durchgeführt.
Reaktionsgemische enthielten cRaf1 (20 nM), MEK1 (100 – 500 nM), [γ-32P]ATP (10 – 20 mM), Mg2+ (10
mM), MOPS (50 mM, pH 7,5). Reaktionsgemische wurden bei Raumtemperatur
für Zeiträume, die
von zwischen 20 – 120
Minuten reichen, inkubiert. Inhibitoren wurden vor dem Assay in
100 % DMSO verdünnt.
Umsetzungen wurde mit einem gleichen Volumen an 0,5 % Phosphorsäure beendet.
MEK1-Phosphorylation wurde durch Szintillationszählung detektiert, gefolgt von
Sammlung des Proteins auf Phosphocellulosefiltern.
-
B. Raf/MEK-Kaskadesassay
-
Menschliches
cRaf1 und MEK1 wurden wie vorstehend beschrieben gereinigt. Ein
durch MEK1 phosphoryliertes Peptidsubstrat wurde als der letzte
Phosphorylgruppenakzeptor verwendet. Die Sequenz des Peptids HTGFLTEYVATRWKK–OH wurde
von der Stelle in ERK2, welche durch MEK1 phosphoryliert wird, abgeleitet.
Assaybedingungen waren die gleichen wie jene vorstehend beschriebenen,
mit Ausnahme der folgenden Modifikationen. Die Reaktionsgemische
enthielten cRaf1 (1 – 5
nM), MEK1 (60 nM) und Peptid (250 mM).
-
C. CDK1 und CDK2
-
Cyclin-abhhängige Proteinkinaseassays
benutzten die Peptide Biotin-Aminohexyl-AAKAKKTPKKAKK und Biotin-Aminohexyl-ARRPMSPKKKA-NH2 als Phosphorylgruppenakzeptoren. Sowohl
CDK1 als auch CDK2 wurden unter Benutzung eines Bacluovirus-Expressionssystems
exprimiert und wurden teilweise gereinigt, um 20 – 80 % Gesamtprotein
zu umfassen, mit keinen detektierbaren kompetitierenden vorliegenden
Umsetzungen. Typischerweise wurde die Assays durch Inkubieren eines
der beiden Enzyme (0,2 – 10
nM), mit und ohne Inhibitor, eines der beiden Peptidsubstrate (1 – 10 μM), [γ-32P]ATP (1 – 20 μM), und 10 – 20 mM Mg2+ für Zeiträume, die
im Allgemeinen im Bereich von 10 – 120 Minuten liegen, durchgeführt. Umsetzungen
wurden mit 0,2 – 2
Volumina von entweder 20 % Essigsäure oder 50 – 100 mM
EDTA, gepuffert auf pH 7 (Substratverbrauch < 20 %) terminiert. Der in den Enzymassays
angewendete Puffer war entweder 30 mM HEPES 7,4, welcher 0,15 M
NaCl und 5 % DMSO enthält,
der Puffer 50 mM MOPS 7,0 welcher 0,15 M NaCl und 5 % DMSO enthält oder
der Puffer 100 mM HEPES pH 7,5, welcher 0,1 mg/ml BSA und 5 % DMSO
enthält.
Inhibitoren wurden vor dem Assay in 100 % DMSO verdünnt. Detektion
der Peptidphosphorylierung wurde durch Szintillationszählen, gefolgt
von entweder Sammlung von Peptid auf Phosphocellulosefiltern (für mit Essigsäure gestoppte
Umsetzungen), Sammlung von Peptid in Vertiefungen von Platten mit
96 Vertiefungen, die mit Streptavidin (Pierce) beschichtet sind
(Umsetzungen wurden mit EDTA gestoppt) oder Zugabe von mit Avidin
beschichteten mit Szintillationssubstanz imprägnierten Kügelchen (Scintillation Proximity
Assays von Amersham, Umsetzungen wurden mit EDTA gestoppt) erreicht.
Von der Zählrate,
die durch eines dieser Verfahren detektiert wurden, minus dem geeigneten
Hintergrund (Assays mit zusätzlich
40 mM EDTA oder Fehlen des Peptidsubstrats) wurde angenommen, dass
sie proportional zu Umsetzungsinitiationsgeschwindigkeiten ist,
und IC50-Werte wurde durch die Anpassung
durch das Verfahren der kleinsten Quadrate an die Gleichung CPM
= Vmax·(1 – ([I]/(K
+ [I]))) + nsb bestimmt oder pIC50-Werte
wurden durch eine Anpassung an die Gleichung CPM = nsb + (Vmax - nsb)/(1 + (x/10x – pIC50))
bestimmt, wobei nsb die Hintergrundzählrate ist.
-
D. UL97
-
UL97
wurde als ein GST-Fusionsprotein aus einem Baculovirusvektor, der
in sf9-Zellen wie von He (He et al., 1997) beschrieben exprimiert
wurde, erzeugt. UL97 wurde als eine Proteinkinase unter Verwendung
von 32P-Transfer von ATP auf Histon H2B
unter Detektion von radiomarkiertem Histon, das an Phosphocellulose gebunden
ist, analysiert. Assaygemische für
das Testen der Inhibitoren der UL97-Aktivität enthielten 2 mM [γ32P]-ATP,
15 mM Histon H2B, 50 mM Natrium-CHES, pH 9,5, 1 M NaCl, 2 mM Dithiothreitol
und 10 mM MgCl2. Inhibitoren wurden in DMSO
verdünnt,
um eine DMSO-Endkonzentration in dem Reaktionsgemisch von 1 % DMSO
zu geben. Nach Inkubation bei 20 °C
wurden die Inkubationen durch Zugabe von 10 Volumina 75 mM Phosphorsäure, 30
mM ATP, 1 mM EDTA beendet, dann auf Phosphocellulosefilter getupft
und vier Mal mit 75 mM Phosphorsäure
gewaschen. Die Radioaktivität
wurde durch Flüssigszintillationszählen bestimmt.
-
E. SRC/lck Enzym Assay
-
Die
in den Src- und Lck-Assays verwendeten Proteinsubstrate waren Biotin-Aminohexyl-EEIYGEF-NH2 (Src) und Biotin-Aminohexyl-EAIYGVLFAKKK-NH2 (Lck). Die src- und lck-Proteine wurde zu Homogenität aus einem
Baculovirus-Expressionssystem gereinigt und vor der Zugabe zum Assaygemisch voraktiviert.
Die maximale Aktivierung wurde durch 40 min lange Inkubation von
konzentriertem Enzym (10 – 30 μM) auf Eis
in Gegenwart von 1 μM
ATP und 10 mM MgCl2 in 100 mM HEPES, pH
7,5, erreicht. Das aktivierte Enzym wurde auf 2 nM in ein 50 ml
Umsetzungsgemisch verdünnt,
welches 100 mM HEPES, pH 7,5, 5 μM
ATP, 10 mM MgCl2, 2 μM Peptid, 0,05 mg/ml BSA und
einen Inhibitor bei variierenden Konzentrationen und mit oder ohne
8 mCi/ml [γ-33 P]ATP, abhängig vom Analyseverfahren für das Ausmaß der Umsetzung,
enthielt. Die Kontrollen waren Umsetzungen in Gegenwart (negative
Kontrollen) oder Abwesenheit (positive Kontrollen) von 50 mM EDTA.
Man ließ Umsetzungen
30 min lang bei Raumtemperatur ablaufen und quenchte durch Zugabe
von EDTA auf 50 mM in 220 μl.
Das Ausmaß von
Umsetzungen wurde in einem der zwei Assays analysiert: ein auf Elisa-beruhender
und ein auf radioaktivem Isotop beruhender. Die gequenchten Proben
(200 μl) wurden
in eine mit Neutravidin beschichtete Platte (Perice) übertragen
und bei Raumtemperatur 40 min lang inkubiert, um biotinyliertem
Peptid zu erlauben, an Neutravidin zu binden. Das ungebundene Peptid
und der Rest der Lösung
wurden unter Verwendung eines Plattenwaschgeräts weggewaschen. Im Elisa Format
wurde eine 200 μl
HRP-PY20 anti-Phosphotyrosin-Antikörperkonjugat-Lösung zugegeben.
Nach etwa 30 min langer Inkubation wurde die Platte gewaschen, um
ungebundenes Antikörper-HRP-Konjugat
zu entfernen. Ein Elisa-Substrat, K-Blue (Neogen), wurde zugegeben und die
Elisa-Umsetzung mit Red-Stop (Neogen) nach 15 min gequencht. Die
Platte wurde bei A625 in einem Plattenlesegerät gelesen.
In dem auf Isotopen beruhenden Format waren die Umsetzungen in Gegenwart
von [γ-33P]ATP durchgeführt worden. 200 ml Scintiverce
DB wurden jeder Vertiefung der Platte mit gebundenem Biotin-Peptid
zugegeben. Die Platte wurde versiegelt und in einem Micro-b-Zählgerät (Wallac)
gezählt.
IC50-Werte
wurde durch Anpassen der Rohdaten an A625 (cpm)
= Vmax·(1 – ([I]/(IC50
+ [I]))) + b erhalten, wobei b der Hintergrund ist.
-
F. VEGFR-2
-
Das
im VEGFR-2-Assay verwendete Peptidsubstrat war Biotin-Aminohexyl-EEEEYFELVAKKKK-NH2. Die Kinasedomäne des Enzyms wurde zu Homogenität aus einem
Baculovirus-Expressionssystem gereinigt. Das aktivierte Enzym wurde
auf 0,4 nM in eine 60 μl
Umsetzung verdünnt,
welche 100 mM HEPES, pH 7,5, 5 μM
ATP, 10 mM MgCl2, 5 μM Peptid, 0,1 mM DTT, 0,05 mg/ml
BSA und einen Inhibitor bei variierenden Konzentrationen enthält. Die
Kontrollumsetzungen waren in Gegenwart (negative Kontrollen) oder
Abwesenheit (positive Kontrollen) von 50 mM EDTA. Reaktionsgemische
wurden 30 min lang bei Raumtemperatur inkubiert und dann durch die
Zugabe von EDTA auf 60 mM in 210 μl
gequencht. Die gequenchten Proben (190 μl) wurden in eine mit Neutravidin
beschichtete Platte (Pierce) überführt und
bei Raumtemperatur 40 min lang inkubiert, um biotinyliertem Peptid
zu erlauben, an Neutravidin zu binden. Das ungebundene Peptid und
der Rest der Lösung
wurden unter Verwendung eines Plattenwaschgeräts weggewaschen, dann wurden
200 μl HRP-PY20
anti-Phosphotyrosin-Antikörperkonjugat
zu jeder Vertiefung gegeben. Nach 40 Minuten langer Inkubation wurde
die Platte gewaschen, um jedweden ungebundenen Antikörper zu
entfernen.
-
Ein
HRP-Substrat, K-Blue (Neogen) wurde zugegeben, und die Umsetzung
wurde mit Red-Stop (Neogen) nach 20 min gequencht. Die Absorption
der Vertiefungen wurde bei A650 in einem
Plattenlesegerät
gelesen.
-
IC50-Werte wurden durch Anpassen der Rohdaten
an A650 = VM·(1 – [I]/IC50 + [I])))+b erhalten,
wobei b der Hintergrund ist.
-
Repräsentative
Daten werden in Tabelle 3 zusammengefasst. Table 3 veranschaulicht
die Hemmungsaktivität
von erfindungsgemäßen Verbindungen
gegen eine repräsentative
Kinase (raf).
-
-
-
-
Zell-basierende Effizienz (MTT-Assay)
-
Die
Wirksamkeit der erfindungsgemäßen Verbindungen
wurde auf ihre Fähigkeit,
die Zellproliferation und Zellüberlebensfähigkeit
zu hemmen, getestet. Die metabolische Umwandlung von 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromid
(MTT, Sigma #M2128) in eine reduzierte Form ist ein üblich verwendetes
Maß für zelluläre Überlebensfähigkeit.
Es folgt das Verfahren:
Zellen werden in einem 75 cm2 Gewebekulturkolben gehalten, bis sie zur
Verwendung bereit waren. Die Zellen wurden gezüchtet und für den Assay in Dulbecco's modifiziertem Eagle's Medium, welches
10 % fötales
Rinderserum enthält,
ausplattiert. Zum Beispiel können
die folgenden Zelllinien verwendet werden: a) menschliche Vorhautfibroblasten
(HFF), b) HT29 (menschliche Kolonkarzinomzelllinie), c) MDA-MB-468
(menschliche Brustkarzinomzelllinie), d) RKO (menschliche Kolonadenokarzinomzelllinie),
e) SW620 (menschliche Kolonadenokarzinomzelllinie), f) A549 (menschliche
Lungenkarzinomzelllinie) und g) MIA PACA (menschliche Bauchspeicheldrüsenkarzinomzelllinie).
Die Zellen werden bei 37 °C
in 10 % CO2, 90 % befeuchtete Luft gehalten. Die
Zellen werden in Gewebekulturplatten mit 96 Vertiefungen bei den
nachstehend aufgelisteten Dichten gehalten. 100 μl Zellsuspension wird jeder
Vertiefung auf der Platte mit 96 Vertiefungen zugegeben, mit Ausnahme
der obersten Reihe der Platte, welche keine Zellen enthält, und
als eine Referenz für
das Spektrophotometer dient.
-
-
Die
Zellen werden über
Nacht in Dulbecco's
modifiziertem Eagle's
Medium, welches 10 % fötales
Rinderserum enthält,
in 10 % CO2, 90 % befeuchtet, bei 37 °C vor dem
Dosieren inkubiert. Die Zellen werden in 10 aufeinander folgenden
3-fach Verdünnungen,
ausgehend von 30 μM,
abhängig
von der Löslichkeit
der Verbindung, dosiert. Verbindungen mit Löslichkeiten von weniger als
30 μM werden
bei der höchsten
löslichen Konzentration
dosiert. Stammlösungen
von Verbindungen werden in 100 % Dimethylsulfoxid (DMSO) gemacht.
Stammlösungen
werden in Dulbecco's
modifiziertem Eagle's
Medium verdünnt,
welches 100 μg/ml Gentamicin
und 0,3 bis 0,6 % DMSO bei der zweithöchsten Konzentration, die auf
die Zellen zu platzieren sind, enthält. Falls Verbindungen in DMSO
gelöst
worden sind, wird die Endkonzentration von DMSO auf den Zellen unter
0,3 % gehalten. 3-fache Verdünnungsreihen
werden mit jeder Verbindung durchgeführt, um 10 Konzentrationen
der Verbindung für
das Dosieren herzustellen. 100 μl
verdünnte
Verbindung wird zu den 100 μl
des Mediums, das gegenwärtig
auf der Platte ist, gegeben. Für
jede Konzentration der Verbindung werden 2 – 4 Replikatvertiefungen hergestellt.
-
Die
Zellen werden wieder in den Inkubator gegeben, und man lässt sie
in Gegenwart der Verbindung 72 Stunden lang vor der Zugabe von MTT
proliferierten. MTT wird in mit Phosphat gepufferter Kochsalzlösung (Irvine
Scientific #9240) bei einer Konzentration von 2 mg/ml hergestellt.
50 μl MTT-Lösung pro
Vertiefung wird zu dem 200 μl
Medium gegeben, um eine Endkonzentration von 0,4 mg/ml zu erhalten,
und die Platten werden wieder 4 Stunden lang in den Inkubator gegeben.
Nach 4 Stunden langer Inkubation wird (ins Gemisch aus Medium, der
Verbindung und MTT von den Platten abgesaugt, und 100 μl 100 % DMSO
wird jeder Vertiefung zusätzlich
zu 25 μl
Sorenson's Puffer
(0,1 M Glycin, 0,1 M NaCl, pH 10,5) zugegeben. Quantifizierung der
metabolischen Reduktion von MTT in jeder Platte wird durch Lesen
der optischen Dichte bei einer Wellenlänge von 570 nm auf einem Molecular
Devices UVmax Mikroplattenlesegerät durchgeführt. Wachstumshemmungskurven
und 50 % Hemmkonzentrationen werden unter Verwendung von Microsoft
Excel bestimmt.
-
Repräsentative
Daten werden in Tabelle 4 zusammengefasst. Tabelle 4 veranschaulicht
die Hemmungsaktivität
von erfindungsgemäßen Verbindungen
gegen eine repräsentative
Kinase (raf) und die Cytotoxizität
von erfindungsgemäßen Verbindungen
gegen einen breiten Bereich von menschlichen Tumorzelllinien.
-
-
IN VIVO ASSAYS
-
Studien zur Tumor-Hemmung: Tiere
-
Mäuse werden
von Taconic Farms erworben und werden in Microisolator Käfigen bei
72 ± 2 °F mit einem
12 Stunden hell/dunkel Zyklus gehalten. Die Tiere werden zu 4 Mäusen pro
Käfig (28 × 17 × 12 cm)
beherbergt, und ihnen wird Nahrung und Wasser nach Belieben gegeben.
Die Tiere werden durch die Verwendung eines Ohrstempels oder einer
Schwanztätowierung
nummeriert. Die gesamte Tierhandhabung wird in einem Laminarströmungsabzug
getätigt.
-
Zellwachstum
-
SW620,
erhältlich
von der American Type Culture Collection, werden in Medium gezüchtet, das
aus RPMI 1640 mit fötalem
Rinderserum (10 %), Natriumpyruvat (1,0 mM) und Glutamin (2,0 mM)
besteht. Zellen werden bei 37 °C
in 5 % CO2 inkubiert. Zellen werden mit
Trypsin (0,05 %) geerntet, zentrifugiert und in PBS : Matrigel (1
: 1) bei 1 × 107 Zellen/ml resuspendiert.
-
Tumorimplantation
-
Eine
der verwendeten Tumorzelllinien ist die Darmlinie SW620. Tumore
werden durch subcutane Injektion einer Zellsuspension in die rechte
Lende jeder Maus injiziert. Das Inokulum besteht aus 2 × 106 Zellen/Maus/0,2 ml in PBS : Matrigel (1
: 1).
-
Tumormessung
-
Feste
Tumoren werden durch eine Tastermessung durch die Haut gemessen.
Die Tastermessungen werden typischerweise zwei Mal pro Woche getätigt. Das
Tumorgewicht wird unter Verwendung der Gleichung (Länge × Breite2/2) = mg Tumorgewicht, berechnet.
-
Messung des Körpergewichts
-
Mäuse werden
zwei Mal pro Woche zum Zeitpunkt der Tumormessung gewogen.
-
Herstellung der Verbindung
-
Verbindungen
werden in einem Vehikel, das aus DMSO, Cremophor und PBS besteht,
hergestellt.
-
Experimentelle Therapie
-
Die
Arzneistofftherapie beginnt, wenn die durchschnittliche Tumorgröße ungefähr 40 – 50 mg
ist, was gewöhnlich
an Tag 7 nach dem Implantieren ist. Das Dosisschema besteht aus
einer Dosis/Tag für
5 aufeinander folgende Tage. Arzneistoffe werden bei 3 oder 4 Dosisspiegeln,
beruhend auf der zuvor bestimmten maximalen tolerierten Dosis verabreicht.
Eine Kontrollgruppe aus Vehikel wird ebenfalls eingeschlossen. Die
Arzneistoffe können
entweder auf i.v., i.p., s.c. oder oralen (p.o.), transdermalen
Wegen oder anderen alternativen Wegen verabreicht werden. Die Arzneistoffe
können über eine
Infusion in die Schwanzvene verabreicht werden. Das verabreichte
Injektionsvolumen für
jede Maus ist gewöhnlich
0,01 – 0,02
ml/g Körpergewicht.
Im Fall von i.v. Injektionen und Infusion in die Schwanzvene werden
die Tiere in einem Broome-Behälter
während
der Handhabung behalten. Tiere fasten über Nacht vor der p.o. Dosierung.
Die Dauer von jedem Experiment ist typischerweise 28 Tage ab dem
Tumorimplantieren.
-
Repräsentative
Ergebnisse werden in Tabelle 5 aufgelistet.
-
-
Während die
Erfindung unter Bezugnahme auf bestimmte bevorzugte Ausführungsformen
davon beschrieben und veranschaulicht worden ist, ist für Fachleute
ersichtlich, dass verschiedene Änderungen,
Modifikationen und Substitutionen getätigt werden können, ohne
vom Geist und Umfang der Erfindung abzugehen. Zum Beispiel können wirksame
Dosierungen, die sich von den hierin vorstehend dargelegten Dosierungen
unterscheiden, als eine Folge von Variationen auf die Reaktivität des Säugers, der
für Krebszustände oder
für andere
Indikationen für
die vorstehend angegebenen erfindungsgemäßen Verbindungen behandelt
wird, angewendet werden. Gleichermaßen können die spezifischen beobachteten
pharmakologischen Reaktionen, gemäß und abhängig von der bestimmten ausgewählten wirksamen
Verbindung oder falls es pharmazeutische Träger gibt, ebenso von der Art
der Formulierung und dem angewendeten Verabreichungsmodus variieren, und
derartige erwartete Variationen oder Unterschiede in den Ergebnissen
werden gemäß den erfindungsgemäßen Aufgaben
und Praktiken betrachtet. Man beabsichtigt daher, dass die Erfindung
lediglich durch den Umfang der Ansprüche, die folgen, beschränkt werden
sollen, und dass derartige Ansprüche
so breit, wie es angemessen ist, interpretiert werden sollen.
















 umfasst.
umfasst.








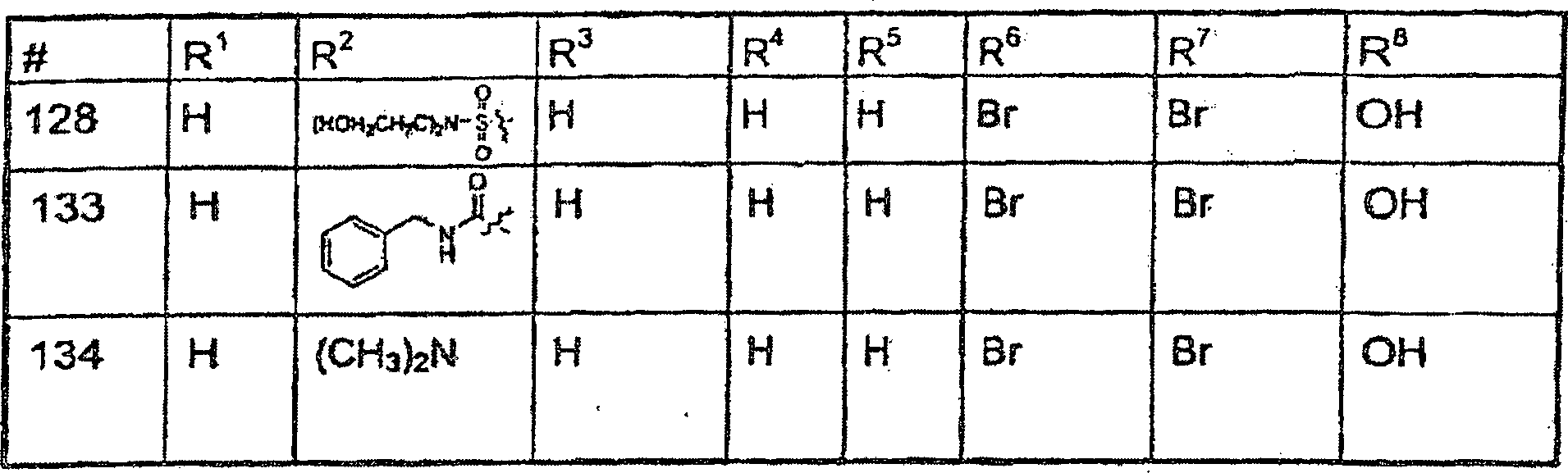




























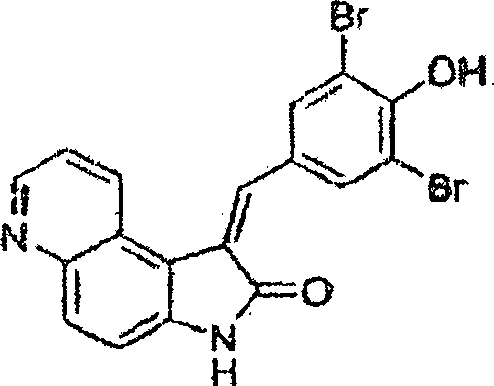


















 und
und

 und
und


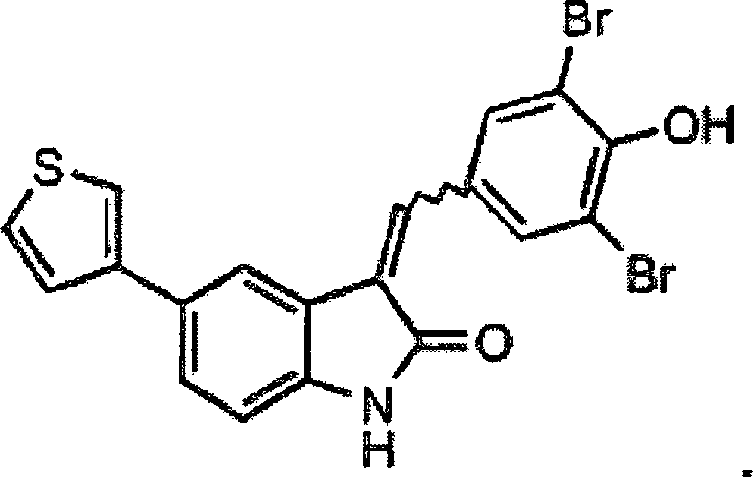




 wobei R1, R2, R3 und R4 wie in Anspruch 1 definiert sind, umfasst.
wobei R1, R2, R3 und R4 wie in Anspruch 1 definiert sind, umfasst.