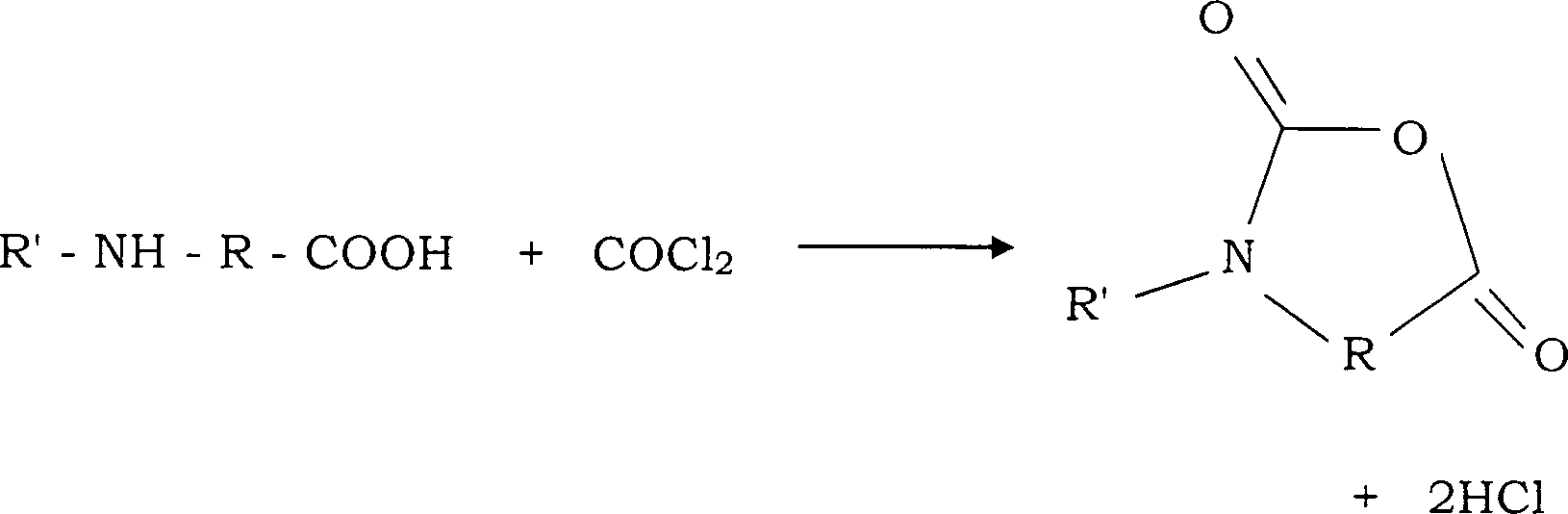-
Die
Erfindung betrifft ein verbessertes Verfahren zur Herstellung von
N-Carboxyanhydriden ausgehend von den entsprechenden Aminosäuren und
Phosgen, Diphosgen oder Triphosgen.
-
Die
N-Carboxyanhydride (abgekürzt
NCA), die aus α-, β- oder γ-Aminosäuren hergestellt
werden, sind wegen der Aktivierung ihrer Säurefunktion sehr zweckdienliche
Verbindungen. Es ist nämlich
möglich,
ihre Säurefunktion
mit beliebigen nukleophilen Einheiten umzusetzen. Durch Umsetzung
mit einer Aminofunktion kann daher die Amidfunktion leichter gebildet
werden. Daher polymerisieren sie leichter und werden zur Bildung
von Peptiden verwendet. Auch die Esterbindung bildet sich einfach
durch Umsetzung mit einem Alkohol. Außerdem sind sie interessant,
wenn eine Säurefunktion
reduziert werden soll.
-
Es
sind mehrere Verfahren zur Herstellung von N-Carboxyanhydriden bekannt.
Eines der geläufigsten und
direktesten Verfahren ist das Verfahren, in dem eine Aminosäure oder
ihr Hydrochlorid in einem Lösungsmittelmedium
mit Phosgen, Diphosgen oder Triphosgen umgesetzt wird.
-
Das
allgemeine Reaktionsschema mit Phosgen ist das Folgende:
worin
R das Gerüst
der α-, β- oder γ-Aminosäure bedeutet
und R' ein Wasserstoffatom
oder die Gruppe der sekundären
Aminogruppe der Aminosäure
bedeutet, wobei R' mit
R einen Ring bilden kann.
-
Man
stellt fest, dass sich neben dem N-Carboxyanhydrid auch Salzsäure in großen Mengen
bildet, d. h. 2 mol auf 1 mol NCA. Die Salzsäure ist sehr reaktiv. Ihre
Gegenwart in dem Medium führt
zu Nebenreaktionen und dem Auftreten von chlorierten Nebenprodukten.
Diese chlorierten Verunreinigungen, die in den gebildeten NCA verbleiben,
sind jedoch sowohl hinsichtlich der Qualität als auch im Hinblick auf
die Ausbeute unerwünscht.
Sie stören
nämlich
die Polymerisation der NCA enorm. Damit die Polymerisation in geeigneter Weise
abläuft,
ist es erforderlich, dass die Menge der in den NCA-Monomeren enthaltenen
chlorierten Verbindungen möglichst
gering ist. Der Gehalt an hydrolysierbarem Chlor sollte im Allgemeinen
unter 0,05 Gew.-% liegen.
-
Wenn
die Umsetzung in Gegenwart einer basischen Verbindung durchgeführt wird,
ist es gemäß den bekannten
Verfahren schwierig, reproduzierbar einen geringen Gehalt an hydrolysierbarem
Chlor zu erhalten. Wenn andererseits eine basische Verbindung zur
Neutralisation der Salzsäure
eingearbeitet wird, wird die Polymerisation der NCA, die in dieser
Stufe nicht gewünscht
wird, aktiviert und es besteht die Gefahr, dass sie in dem Medium
abläuft.
Nach dem in dem Patent GB 1 210 719 beschriebenen Verfahren handelt
es sich bei den zur Neutralisation der Salzsäure verwendeten Verbindungen
um anorganische Salze, beispielsweise Oxide oder Cyanide von Silber,
Blei oder Quecksilber.
-
Gemäß dem in
dem Patent FR 1 561 268 beschriebenen Verfahren wird die Umsetzung
in halogenierten Kohlenwasserstoffen als Lösungsmittel durchgeführt.
-
Nach
dem Patent FR 2 000 198 besteht eine Verbesserung des obigen Verfahrens
darin, ein Gemisch von Lösungsmitteln
zu verwenden, das aus aromatischen Kohlenwasserstoffen und organischen
Nitrilen besteht.
-
Im Übrigen besteht
eine der Schwierigkeiten der im Stand der Technik bekannten Verfahren
in der Wahl des Lösungsmittels.
Es hat sich nämlich
herausgestellt, dass in Lösungsmitteln
wie aliphatischen Estern, beispielsweise Ethylacetat, oder apolaren
aprotischen Lösungsmitteln,
wie Dichlormethan oder Toluol, die Bildung der NCA im Allgemeinen
sehr langsam und nicht vollständig
ist. In einem Lösungsmittel
aus der Gruppe der Ether, wie Tetrahydrofuran, oder Dioxan ist die
Umsetzung schneller, diese Lösungsmittel
sind jedoch gegenüber
Phosgen und Salzsäure
nicht vollständig
inert, wodurch weitere Verunreinigungen gebildet werden.
-
Es
gibt daher ein Bedürfnis,
das bestehende Verfahren, in dem die Aminosäure direkt mit Phosgen, Diphosgen
oder Triphosgen hergestellt wird, zu verbessern, um die NCA in höheren Ausbeuten
und mit einer besseren Reinheit zu erhalten, wobei sie insbesondere
einen Gehalt an hydrolysierbarem Chlor unter 0,05 % aufweisen sollten.
-
Die
Herabsetzung der Reaktionsdauer in den inertesten Lösungsmitteln
ist außerdem
wünschenswert.
-
Das
erfindungsgemäße Verfahren
wird diesen Bedürfnissen
gerecht. Gemäß diesem
Verfahren werden die N-Carboxyanhydride durch Umsetzung der entsprechenden α-, β- oder γ-Aminosäuren oder
einem ihrer Salze mit Phosgen, Diphosgen und/oder Triphosgen in
einem Lösungsmittelmedium
in Gegenwart einer ungesättigten
organischen Verbindung, die eine oder mehrere Doppelbindungen vom
ethylenischen Typ aufweist, deren Molekülrest gegenüber den in dem Medium vorliegenden
Verbindungen inert ist und bei der eines der Kohlenstoffatome mindestens
einer ethylenischen Doppelbindung vollständig mit Substituenten substituiert
ist, die von Halogenatomen verschieden sind, während der gesamten Reaktionsdauer
oder einem Teil der Reaktionsdauer hergestellt.
-
Durch
dieses neue Verfahren werden die Probleme, die im Stand der Technik
auftreten, gelöst.
Die Salzsäure,
die gebildet wird, bindet, in dem Maße wie sie gebildet wird, an
die ethylenische(n) Doppelbindungen) der ungesättigten Verbindung. Die zahlreichen
Nebenreaktionen, die durch die Salzsäure hervorgerufen werden, werden
daher unterdrückt
und als Folge treten keine störenden
Verunreinigungen auf. Außerdem
wird ferner das Reaktionsgleichgewicht in Richtung der gewünschten
NCA verschoben, wodurch auch die Kinetik der Reaktion schneller
ist.
-
Es
hat sich außerdem
herausgestellt, dass im Falle der Umformung von Aminosäuren, deren
Aminogruppe eine sekundäre
Aminogruppe ist, die Gegenwart der ungesättigten Verbindung den Zusatz
eines tertiären
Amins, wie Triethylamin oder N-Methylmorpholin, in das Medium überflüssig macht.
Ein solches Amin wurde vom Fachmann bis jetzt als unerlässlich angesehen,
um die Cyclisierung aus dem Carbamoylchlorid, das sich in dem Medium
zunächst
als Zwischenprodukt bildet, zu bewirken.
-
Durch
das erfindungsgemäße Verfahren
können
die N-Carboxyanhydride der meisten cyclischen oder nicht cyclischen,
natürlichen
oder synthetischen α-Aminosäuren und
ihrer Derivate, deren Aminofunktion primär oder sekundär sein kann,
und insbesondere aller Aminosäuren
hergestellt werden, die bereits dafür bekannt sind, dass sie mit
Phosgen, Diphosgen und/oder Triphosgen reagieren.
-
Das
Verfahren ist auch sehr nützlich,
um die N-Carboxyanhydride von β-
und γ-Aminosäuren und
deren Derivaten mit primärer
oder sekundärer
Aminofunktion herzustellen. Diese Verbindungen werden nämlich nach
den Verfahren des Standes der Technik als schwierig herzustellen
angesehen.
-
Die
Aminosäuren,
die als Ausgangsverbindungen verwendet werden, sind vorzugsweise α-, β- oder γ-Aminosäuren, deren α-, β- und gegebenenfalls γ-Kohlenstoffatom(e),
die sich zwischen der Säuregruppe und
der Aminogruppe, die reagieren, befinden, eine substituierte oder
unsubstituierte Alkylkette auf Kohlenwasserstoffbasis bilden, die
ganz oder teilweise in einer geradkettigen oder verzweigten, substituierten
oder unsubstituierten Alkylgruppe und/oder in einem substituierten
oder unsubstituierten Alkyl- oder Heteroalkylring enthalten sein
kann. Die Substituenten sind Gruppen oder Atome, die für Aminosäuren üblich sind,
wie beispielsweise die Gruppen Hydroxy, Carboxy, Mercapto, Alkylthio,
Alkyldithio, Alkyl, Cycloalkyl, Heterocycloalkyl, Aryl, Heteroaryl,
Alkyloxy, Aryloxy, Halogenatome, beispielswei se Fluor, Chlor, Brom
oder Iod, Aminogruppen, Guanidino oder Amidogruppen, die gegebenenfalls
mit Alkylgruppen substituiert sind.
-
In
den in Betracht gezogenen Aminosäuren
enthalten die Alkylgruppen genauer 1 bis 7 Kohlenstoffatome und
sie sind gegebenenfalls mit den genannten Substituenten substituiert.
Die Arylgruppen sind unsubstituiert oder mit Substituenten substituiert,
die unter den Halogenatomen, wie Fluor, Chlor, Brom oder Iod, und Alkyl,
Alkoxy, Aryloxy, Aryl, Mercapto, Alkylthio, Hydroxy, Carboxy, Amino,
Alkylamino, Dialkylamino, Nitro und Trifluormethyl ausgewählt sind.
Wenn solche Substituenten vorliegen, beträgt ihre Anzahl insbesondere
1 bis 3. Die Arylgruppen sind insbesondere substituierte oder unsubstituierte
Phenyl- oder Naphthylgruppen.
-
Die
Cycloalkylgruppen bestehen aus Ringen mit 3 bis 7 Kohlenstoffatomen,
die gegebenenfalls substituiert sind. Die Heterocyclen, die substituiert
oder unsubstituiert sein können,
sind Cycloalkyl- oder Arylgruppen, die im Ring mindestens ein Heteroatom
enthalten, das unter Stickstoff, Sauerstoff oder Schwefel ausgewählt ist.
-
Die
Substituenten der Cycloalkylgruppen oder Heterocycloalkylgruppen
sind unter den oben für
die Alkyl- und Arylgruppen angegebenen Substituenten ausgewählt. Die
Substituenten der Heteroarylgruppen sind insbesondere die für die Arylgruppen
angegebenen Substituenten.
-
Bei
den Heteroarylgruppen handelt es sich vorzugsweise um 2- oder 3-Furanyl,
2- oder 3-Thienyl, 2-, 3- oder 4-Pyridinyl, 4-Imidazolyl und 3-Indolyl,
wobei sie gegebenenfalls substituiert sein können.
-
Die
Aminosäuren
können
in ihren unterschiedlichen Formen vorliegen und insbesondere in
ihren verschiedenen enantiomeren Formen, als racemische Gemische
oder Gemische von Diastereoisomeren oder auch in Form der reinen
Stereoisomere vorliegen, wenn sie ein oder mehrere asymmetrische
Kohlenstoffatome besitzen.
-
Wenn
die Gruppe der Aminosäure
funktionelle Gruppen aufweist, die von der Aminogruppe und der Säuregruppe,
die den Anhydridring bilden, verschieden und befähigt sind, unter den Verfahrensbedingungen zu
reagieren, werden sie in bekannter Weise mit Schutzgruppen maskiert.
-
Als
Beispiele für
Aminosäuren
können
die geläufigsten
Aminosäuren,
wie beispielsweise Glycin, Alanin, Valin, Leucin, Isoleucin, Phenylalanin,
Serin, Threonin, Lysin, δ-Hydroxylysin,
Arginin, Ornithin, Asparaginsäure,
Asparagin, Glutaminsäure,
Glutamin, Cystein, Cystin, Methionin, Tyrosin, Thyroxin, Prolin,
Hydroxyprolin, Tryptophan, Histidin, und deren Derivate, genannt
werden.
-
Die
reaktive Aminogruppe kann eine primäre oder sekundäre Aminogruppe
sein. Das Stickstoffatom kann also eine aliphatische, cycloaliphatische
oder araliphatische Gruppe oder Arylgruppe tragen, die substituiert
oder unsubstituiert ist, wie dies für die Gruppe der Amine gewöhnlich der
Fall ist. Die Gruppe kann insbesondere mit den oben als Substituenten
genannten Gruppen substituiert sein.
-
Die
Gruppe an der Aminogruppe kann mit der restlichen Aminosäure auch
einen substituierten oder unsubstituierten Ring bilden, wie dies
beispielsweise bei Prolin der Fall ist.
-
Wenn
die Gruppe reaktive Gruppen enthält,
werden diese in herkömmlicher
Weise geschützt.
-
Von
den an der Aminogruppe vorhandenen Gruppen können insbesondere die Alkylgruppen,
Cycloalkylgruppen oder Aralkylgruppen angegeben werden, die unsubstituiert
vorliegen oder beispielsweise mit den in dem Patent
US 4 686 295 für die neuen, mit Phosgen gebildeten
NCA beschriebenen Gruppen substituiert sind, und insbesondere mit
einer oder mehreren Gruppen substituiert sind, die unter Alkoxycarbonyl,
Aryloxycarbonyl und Aralkyloxycarbonyl ausgewählt sind.
-
Anstelle
der Aminosäure
kann auch eines ihrer Salze als Ausgangsverbindung eingesetzt werden.
Unter Salzen von Aminosäuren
werden die Salze verstanden, die durch Umsetzung der Aminogruppe
mit organischen oder anorganischen Säuren erhalten werden, wie beispielsweise
Sulfate, Acetate, Toluolsulfonate, Methansulfonate und vorzugsweise
Hydrohalogenide, besonders Hydrochloride und Hydrobromide.
-
Die
Hydrochloride sind die bevorzugten Salze.
-
Das
Verfahren eignet sich gut zur Herstellung von N-Carboxyanhydriden
von Aminosäuren
wie N-(1-Ethoxycarbonyl-3-phenyl-propyl)alanin,
Leucin, Alanin, N-Trifluoracetyllysin, Glutaminsäure-γ-benzylester oder Glutaminsäure-γ-methylester.
-
Bei
der Durchführung
des Verfahrens können
zur Bildung des N-Carboxyanhydridrings Phosgen, Diphosgen und/oder
Triphosgen mit der Aminosäure
umgesetzt werden. Vorzugsweise wird Phosgen verwendet.
-
Im
Verhältnis
zur Aminosäure
ist kein großer Überschuss
an Phosgen erforderlich. Es werden vorzugsweise etwa 1 bis 2 mol
Phosgen auf 1 mol Aminosäure
oder dem Aminosäuresalz
zugegeben.
-
Diphosgen
oder Triphosgen werden in einer entsprechenden Menge eingearbeitet,
um die gleichen Phosgen/Aminosäure-Verhältnisse
einzustellen.
-
Die
Reaktion kann in einem aprotischen und polaren Lösungsmittel erfolgen. Es können Ether
und insbesondere Tetrahydrofuran und Dioxan verwendet werden, vorzugsweise
wird jedoch ein Lösungsmittel
aus der Gruppe der aliphatischen Ester gewählt.
-
Aprotische
und apolare Lösungsmittel
aus der Gruppe der aliphatischen und aromatischen, chlorierten oder
nicht chlorierten Kohlenwasserstoffe, beispielsweise Dichlormethan
oder Toluol, können
ebenfalls verwendet werden.
-
Die
Lösungsmittel
aus der Gruppe der Ester oder Kohlenwasserstoffe haben den Vorteil,
dass sie mit Phosgen oder Salzsäure
nicht reagieren. Ihre Verwendung ist daher vorteilhafter.
-
Alkylacetate
und insbesondere das Ethylacetat sind gut geeignet.
-
Die
Gegenwart einer ungesättigten
organischen Verbindung, die mindestens eine ethylenische Doppelbindung
aufweist und bei der eines der Kohlenstoffatome mindestens einer
der ethylenischen Doppelbin dungen vollständig mit Substituenten, die
von Halogenatomen verschieden sind, substituiert ist, in dem Reaktionsmedium
ist für
die Herstellung der NCA mit höherer
Reinheit und besseren Ausbeuten unerlässlich.
-
Es
können
alle Verbindungen eingesetzt werden, die mindestens eine ethylenische
Doppelbindung dieses Typs besitzen, an die die Salzsäure binden
kann. Die ungesättigte
Verbindung darf natürlich
keine anderen Gruppen und/oder Atome enthalten, wie insbesondere
die Nitrilgruppe und/oder Halogenatome, die mit den in dem Reaktionsmedium
vorhandenen Verbindungen reagieren können. Hieraus würden neue
Verunreinigungen entstehen und die Ausbeute gesenkt werden. Wenn
die Verbindung andere reaktive Gruppen aufweist, werden diese in
bekannter Weise geschützt.
-
Die
ungesättigten
Verbindungen mit einer Doppelbindung, bei der ein Kohlenstoffatom
vollständig
mit Kohlenwasserstoffgruppen substituiert ist, sind gut geeignet.
-
Vorzugsweise
wird eine Verbindung aus der Gruppe der Kohlenwasserstoffe eingesetzt.
Als Beispiele für
solche Verbindungen sind α-Pinen und Diisobuten
zu nennen. α-Pinen
ist die Verbindung, die bevorzugt wird.
-
Die
verwendete Menge der ungesättigten
Verbindung ist im Allgemeinen 1 bis 3 mol auf 1 mol Aminosäure oder
1,5 bis 4 mol auf 1 mol Aminosäuresalz,
wenn diese Verbindung als Ausgangsverbindung gewählt wird, und vorzugsweise
etwa 2 mol auf 1 mol Aminosäure
bzw. etwa 3 mol auf 1 mol Aminosäuresalz.
-
Die
ungesättigte
Verbindung kann in dem Reaktionsmedium bereits zu Beginn der Reaktion
vorliegen, sie kann jedoch auch im Laufe der Reaktion eingearbeitet
werden.
-
Die
Umsetzung erfolgt im Allgemeinen bei einer üblichen Temperatur im Bereich
von 0 bis 120 °C
oder bei 0 °C
oder 120 °C
und vorzugsweise im Bereich von etwa 40 °C bis etwa 90 °C.
-
Der
Druck, unter dem gearbeitet wird, ist im Allgemeinen Atmosphärendruck.
Man kann auch unter einem verminderten Druck insbesondere bis zu
etwa 500 mbar und besonders bei etwa 700 bis 800 mbar arbeiten.
-
Die
Umsetzung erfolgt vorzugsweise unter wasserfreien Bedingungen.
-
Einer
der Vorteile des erfindungsgemäßen Verfahrens
besteht darin, dass die Reaktionsdauer kürzer ist und im Vergleich mit
der Reaktionsdauer des Standes der Technik insbesondere in Lösungsmitteln
wie Estern sogar auf die Hälfte
vermindert werden kann. Da diese zuletzt genannten Lösungsmittel
außerdem
weniger teuer sind, führt
die Durchführung
des erfindungsgemäßen Verfahrens
zu einer wirklichen Kosteneinsparung.
-
Nach
beendigter Umsetzung werden die Produkte nach herkömmlichen
Verfahren abgetrennt. Das Phosgen und das Lösungsmittel werden im Allgemeinen
unter vermindertem Druck entfernt. Die ausgehend von den ungesättigten
Verbindungen erhaltenen chlorierten Derivate werden bei der Kristallisation
der NCA abgetrennt.
-
Die
in Bezug auf die NCA nach der Kristallisation erhaltenen Ausbeuten
sind deutlich besser und liegen oft über 90 %. Der Gehalt an hydrolysierbarem
Chlor liegt immer unter 0,05 %, wobei der Gehalt an chlorierten
Verunreinigungen meistens so gering ist, dass er nicht genau ermittelt
werden kann.
-
Die
gemäß dem oben
beschriebenen Verfahren hergestellten NCA können daher für zahlreiche
Anwendungen eingesetzt werden, für
die sehr reine Produkte erforderlich sind, insbesondere für die Herstellung von
pharmazeutischen Erzeugnissen.
-
Die
folgenden Beispiele erläutern
die Erfindung, ohne dass sie einschränkend zu verstehend sind.
-
Beispiel
1: Herstellung
des N-Carboxyanhydrid von Leucin (H-Leu-NCA)
-
In
einen temperaturgeregelten Reaktor mit einem Fassungsvermögen von
2,5 1, der zuvor mit Stickstoff unter Inertgas gesetzt wurde, werden
1 1 Ethylacetat und anschließend
100 g L-Leucin (0,76 mol, 1 Äquivalent)
gegeben. In diese mechanisch gerührte
Suspension werden 208, 0 g α-Pinen
(1, 52 mol, 2 Äquivalente) eingearbeitet,
worauf das Gemisch auf 5 °C
abgekühlt
wird. Dann werden während
einer Zeitspanne von 1 h 154,5 g Phosgen (1,56 mol, 2,05 Äquivalente)
in das Reaktionsmedium eingeleitet, wobei die Temperatur im Bereich
von 5 bis 10 °C
gehalten wird. Dann wird das Reaktionsmedium auf 60 bis 65 °C erwärmt. Nach
2 h in diesem Temperaturbereich wird das Reaktionsmedium unter vermindertem
Druck entgast, um das überschüssige Phosgen
zu entfernen und es schließlich
zu konzentrieren, indem das gesamte Ethylacetat entfernt wird.
-
Anschließend werden
in der Wärme
750 ml industrielles Heptan zu dem konzentrierten Medium gegeben.
Das H-Leu-NCA beginnt zu kristallisieren. Man kühlt das Reaktionsmedium auf
0 bis 5 °C
ab. Man filtriert unter Stickstoffatmosphäre. Nach dem Trocknen unter
Vakuum bei Raumtemperatur fallen 101,9 g (Ausbeute 85 %) L-H-Leu-NCA mit einer
Reinheit über
99,9% (durch HPLC bestimmt) an, dessen durch argentometrische Bestimmung
ermittelter Gehalt an hydrolysierbarem Chlor 0,018 Gew.-% beträgt.
-
Beispiel
2: Herstellung
des N-Carboxyanhydrid von Alanin (H-Ala-NCA)
-
125
g Alanin (H-Ala-OH) (1,4 mol) werden in einem Gemisch von 445 ml α-Pinen (382
g, 2,8 mol, 2 eq) und 937 ml Ethylacetat suspendiert. Die Suspension
wird auf Rückflusstemperatur
erwärmt, dann
leitet man 209 g (2,11 mol, 1,5 eq) gasförmiges Phosgen ein. Nach 12
h sind noch einige unlösliche
Teile übrig.
-
Man
destilliert, um von dem Reaktionsmedium 800 ml Ethylacetat/Phosgen-Gemisch
abzutrennen und filtriert das restliche Medium in der Wärme.
-
Zu
dem konzentrierten Medium werden in der Wärme 800 ml industrielles Heptan
gegeben, worauf über
Nacht auf –10 °C abgekühlt wird.
Das Produkt, das kristallisiert ist, wird filtriert und mit industriellem
Heptan gewaschen.
-
Nach
dem Trocknen erhält
man 111 g H-Ala-NCA mit einer Ausbeute von 68,8 %. Der Mengenanteil an
hydrolysierbarem Chlor ist zum Nachweis zu gering, da er unter der
Nachweisgrenze liegt, d. h. unter 0,01 %.
-
Beispiel
3: Herstellung
des N-Carboxyanhydrid von N-Trifluoracetyllysin (H-Lys(TFA)-NCA).
-
250
g H-TFA-Lys-O (1,03 mol) werden in einem Gemisch von 328 ml α-Pinen (281
g, 2,06 mol, 2 eq) und 1875 ml Ethylacetat suspendiert. Die Suspension
wird auf 65 °C
erwärmt,
worauf 154 g (1,55 mol, 1,5 eq) gasförmiges Phosgen eingeleitet
werden. Das Reaktionsmedium wird auf Rückflusstemperatur erwärmt und
3 h auf dieser Temperatur belassen.
-
Man
destilliert, um 1750 ml Ethylacetat/Phosgen-Gemisch abzutrennen.
Zu dem restlichen Medium gibt man in der Wärme 1750 ml industrielles Heptan
und kühlt über Nacht
auf –10 °C ab. Das
kristallisierte Produkt wird durch Filtration abgetrennt und mit
industriellem Heptan gewaschen.
-
Nach
dem Trocknen fallen 261 g H-Lys(TFA)-NCA an, wobei dies einer Ausbeute
von 94,48 % entspricht. Die Menge an hydrolysierbarem Chlor war
nicht zu ermitteln, da dieser Gehalt unter der Nachweisschwelle,
d. h. unter 0,01 % liegt.
-
Beispiel
4: Herstellung
des N-Carboxyanhydrid von Glutaminsäure-γ-benzylester (H-Glu(OBzl)-NCA).
-
250
g H-Flu(OBzl)-OH (1,05 mol) werden in einem Gemisch von 334 ml α-Pinen (287
g, 2,1 mol, 2 eq) und 1875 ml Ethylacetat suspendiert. Die Suspension
wird auf +5 °C
abgekühlt,
worauf 164 g (2,28 mol, 1,57 eq) gasförmiges Phosgen eingeleitet
werden. Man erwärmt
das Reaktionsmedium auf Rückflusstemperatur und
belässt
es 3 h bei dieser Temperatur.
-
Dann
wird destilliert, um 1500 ml Ethylacetat/Phosgen-Gemisch abzutrennen.
Man gibt zu dem restlichen Medium in der Wärme 1500 ml industrielles Heptan
und kühlt
2 h auf –10 °C ab. Das
kristallisierte Produkt wird durch Filtrieren abgetrennt und mit
industriellem Heptan gewaschen.
-
Nach
dem Trocknen erhält
man 253 g H-Glu(OBzl)-NCA mit einer Ausbeute von 91,3 %. Der Gehalt an
hydrolysierbarem Chlor ist nicht zu bestimmen, da er unter 0,01
% liegt (für
diese Methode Nachweisgrenze).
-
Vergleichsbeispiel:
-
Herstellung des N-Carboxyanhydrid
von Glutaminsäure-γ-benzylester
(N-Glu(OBzl)-NCA)
-
100
g H-Glu(OBzl)-OH (0,42 mol) werden in 885 ml Ethylacetat suspendiert.
Man kühlt
die Suspension auf +5 °C
ab und leitet dann 90 g (0,91 mol, 2,16 eq) gasförmiges Phosgen ein.
-
Das
Reaktionsmedium wird auf Rückflusstemperatur
erwärmt.
Trotz der Gegenwart eines Überschusses
von Phosgen, der über
der Menge des vorhergehenden Beispiels liegt, ist die Reaktion langsam
und das Reaktionsmedium muss anstelle von 3 h wie im vorhergehenden
Beispiel 6 h bei Rückflusstemperatur
belassen werden.
-
Man
destilliert dann, um 600 ml Ethylacetat/Phosgen-Gemisch abzutrennen.
Dann gibt man in der Wärme
600 ml industrielles Heptan zu und kühlt 2 h auf –10 °C ab. Das
kristallisierte Produkt wird durch Filtration abgetrennt und mit
industriellem Heptan gewaschen. Nach dem Trocknen fallen 88 g H-Glu(OBzl)-NCA mit
einer Ausbeute von 74,6 % an. Der Gehalt an hydrolysierbarem Chlor
ist 0,13 %.
-
BEISPIEL
5: Herstellung
des N-Carboxyanhydrid von Glutaminsäure-γ-methylester (H-Glu(OMe)-NCA).
-
250
g H-Glu(OMe)-OH (1,55 mol) werden in einem Gemisch von 493 ml α-Pinen (423
g, 3,1 mol, 2 eq) und 1875 ml Ethylacetat gelöst. Man erwärmt die Suspension auf 65 °C und leitet
dann 227 g (2,31 mol, 1,5 eq) gasförmiges Phosgen ein.
-
Man
erwärmt
das Reaktionsmedium auf Rückflusstemperatur
und belässt
es 6 h bei dieser Temperatur. Dann wird destilliert, um 1500 ml
Ethylacetat/Phosgen-Gemisch abzutrennen.
-
Man
gibt in der Wärme
zu dem restlichen Medium 1500 ml industrielles Heptan und kühlt das
Medium über
Nacht auf –10 °C ab. Das
kristallisierte Produkt wird durch Filtration abgetrennt und mit
industriellem Heptan gewaschen.
-
Nach
dem Trocknen erhält
man 269 g H-Glu(OMe)-NCA in einer Ausbeute von 92,6 %. Der Gehalt
an hydrolysierbarem Chlor liegt unter 0,01 % (Nachweisgrenze).
-
BEISPIEL
6: Herstellung
des N-Carboxyanhydrid von N-(1-Ethoxycarbonyl-3-phenylpropyl)alanin (EPAL-NCA).
-
In
einen temperaturgeregelten Reaktor mit einem Fassungsvermögen von
3 l, der zuvor mit Stickstoff unter Inertgas gesetzt wurde, werden
2,6 l wasserfreies Ethylacetat und anschließend 312 g EPAL (1,11 mol, 1 Äquivalent)
gegeben. Dann leitet man in diese mechanisch gerührte Suspension bei 40 °C während einer Zeitspanne
von 15 min 45 g HCL gasförmig
(1,22 mol, 1,1 Äquivalente
/ EPAL) ein.
-
Anschließend leitet
man in das Reaktionsmedium während
1 h 223 g gasförmiges
Phosgen (2,22 mol, 2,00 eq) ein. Das Reaktionsmedium wird auf 60
bis 65 °C
erwärmt;
nach 2 h bei dieser Temperatur gibt man 227 g α-Pinen (1,66 mol, 1,5 eq/EPAL)
zu. Nach einer zusätzlichen
Wartezeit von 30 min wird das Reaktionsmedium unter vermindertem
Druck entgast, um das überschüssige Phosgen
zu entfernen und das gesamte Ethylacetat abzutrennen.
-
Anschließend gibt
man 1385 ml Isopropylether zu dem konzentrierten Reaktionsmedium.
Das Medium wird auf 0 bis 5 °C
abgekühlt,
das EPAL-NCA kristallisiert. Es wird unter Stickstoffatmosphäre durch
Filtration abgetrennt.
-
Nach
dem Trocknen unter Vakuum bei Umgebungstemperatur fallen 312 g (Ausbeute
91,5 %) EPAL-NCA (weißer
Feststoff) mit einer Reinheit über
99,7 % (mit HPLC bestimmt) an, wobei der Gehalt an hydrolysierbarem
Chlor 0,04 % beträgt.