CN1073992C - 1,4-二氢-2h-3,1-苯并噁嗪-2-酮的有效合成方法 - Google Patents
1,4-二氢-2h-3,1-苯并噁嗪-2-酮的有效合成方法 Download PDFInfo
- Publication number
- CN1073992C CN1073992C CN98802445A CN98802445A CN1073992C CN 1073992 C CN1073992 C CN 1073992C CN 98802445 A CN98802445 A CN 98802445A CN 98802445 A CN98802445 A CN 98802445A CN 1073992 C CN1073992 C CN 1073992C
- Authority
- CN
- China
- Prior art keywords
- dihydro
- benzoxazine
- ketone
- alkyl
- preparation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D265/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom and one oxygen atom as the only ring hetero atoms
- C07D265/04—1,3-Oxazines; Hydrogenated 1,3-oxazines
- C07D265/12—1,3-Oxazines; Hydrogenated 1,3-oxazines condensed with carbocyclic rings or ring systems
- C07D265/14—1,3-Oxazines; Hydrogenated 1,3-oxazines condensed with carbocyclic rings or ring systems condensed with one six-membered ring
- C07D265/18—1,3-Oxazines; Hydrogenated 1,3-oxazines condensed with carbocyclic rings or ring systems condensed with one six-membered ring with hetero atoms directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/26—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atom of at least one of the carbamate groups bound to a carbon atom of a six-membered aromatic ring
- C07C271/28—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atom of at least one of the carbamate groups bound to a carbon atom of a six-membered aromatic ring to a carbon atom of a non-condensed six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
本发明公开了采用氨基醇中间体和烷基或芳基氯甲酸酯和碱的环化反应得到化合物(-)-6-氯-4-环丙基乙炔基-4-三氟甲基-1,4-二氢-2H-3,1-苯并嗪-2-酮,也称为DMP-266,一种逆转录酶抑制剂的有效制备方法。
Description
发明背景
逆转录酶抑制剂,(-)-6-氯-4-环丙基乙炔基-4-三氟甲基-1,4-二氢-2H-3,1-苯并噁嗪-2-酮,也称为DMP-266的合成的关键步骤是使用光气进行氨基醇的环化。
DMP-266和结构类似的逆转录酶的合成在US5519021和相应的1995年8月3公开的PCT国际专利申请WO95/20389中公开。此外,Thompson等的Tetrahedron Letters 1995,36,8937-8940以及1996年11月28日公开的PCT公开WO96/37457中描述了通过高对映选择性的乙炔化物加成和环化方法进行对映体苯并噁嗪的不对称合成。
本发明公开了使用氯甲酸酯和碱在溶剂中环化下式氨基醇: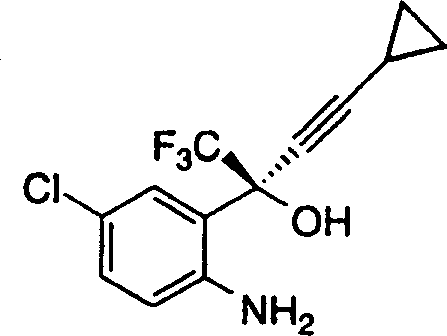 以得到下式的1,4-二氢-2H-3,1-苯并噁嗪-2-酮的有效方法。
以得到下式的1,4-二氢-2H-3,1-苯并噁嗪-2-酮的有效方法。
环化方法使用芳基或烷基氯甲酸酯,避免使用光气,一种需要特殊操作的有毒和十分有害的气体。
发明概述
本发明涉及使用氯甲酸酯和碱在溶剂中环化下式氨基醇:以得到下式的1,4-二氢-2H-3,1-苯并噁嗪-2-酮的有效方法。发明详述
本发明公开了制备下式1,4-二氢-2H-3,1-苯并噁嗪-2-酮的有效方法:其包括如下步骤:1)在约0℃-约25℃的温度和在惰性气氛下在下式的氨基醇在有机溶剂和碱中的搅拌混合物中加入芳基氯甲酸酯: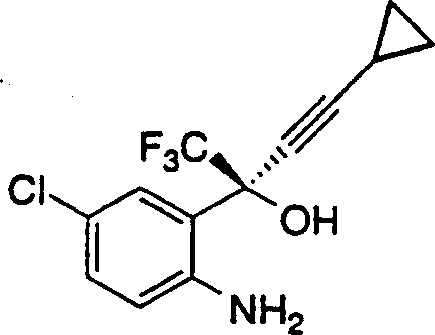 得到下式的氨基甲酸酯中间体:其中R表示氯甲酸酯的芳基侧链;2)在约20℃-约25℃下搅拌反应混合物约1-约6小时以完成氨基甲酸酯中间体的形成;3)用水或含水碱中止反应以得到在有机溶剂相中含有1,4-二氢-2H-3,1-苯并噁嗪-2-酮的两相溶液;4)在约20℃-约25℃下搅拌两相混合物约1-约6小时以完成环化反应,生成1,4-二氢-2H-3,1-苯并噁嗪-2-酮;和5)由有机相中分离1,4-二氢-2H-3,1-苯并噁嗪-2-酮。
得到下式的氨基甲酸酯中间体:其中R表示氯甲酸酯的芳基侧链;2)在约20℃-约25℃下搅拌反应混合物约1-约6小时以完成氨基甲酸酯中间体的形成;3)用水或含水碱中止反应以得到在有机溶剂相中含有1,4-二氢-2H-3,1-苯并噁嗪-2-酮的两相溶液;4)在约20℃-约25℃下搅拌两相混合物约1-约6小时以完成环化反应,生成1,4-二氢-2H-3,1-苯并噁嗪-2-酮;和5)由有机相中分离1,4-二氢-2H-3,1-苯并噁嗪-2-酮。
用于制备上述1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中芳基氯甲酸酯的芳基定义为苯基或萘基,它任选地被一个、两个或三个选自卤素(F、Cl、Br、I)、CF3、CO2C1-C6-烷基和NO2的取代基取代。用于本发明的芳基氯甲酸酯的实例定义为苯基氯甲酸酯,其中苯基任选地被一个、两个或三个选自卤素(F、Cl、Br、I)、CF3和NO2的取代基取代。用于本发明方法的优选芳基氯甲酸酯是4-硝基苯基氯甲酸酯。
用于制备上述1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中碱定义为氢氧化钾、氢氧化钠、氢氧化锂、碳酸钾、碳酸钠、碳酸锂、碳酸氢钾、碳酸氢钠、碳酸氢锂和它们的混合物的固体或溶液。用于制备上述1,2-二氢-2H-3,1-苯并噁嗪,其中优选的碱定义为氢氧化钾、氢氧化钠、碳酸钾、碳酸钠、碳酸氢钾、碳酸氢钠或所述碱的混合物的固体或溶液。
用于制备如上所述的1,4-二氢-2H-3,1-苯并噁嗪的-2-酮方法,其中有机溶剂选自甲基叔丁基醚、甲苯、四氢呋喃、乙腈、二甲基乙酰胺、N-甲基吡咯烷酮或所述溶剂的混合物。
本发明的优选实施方案是制备下式1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法: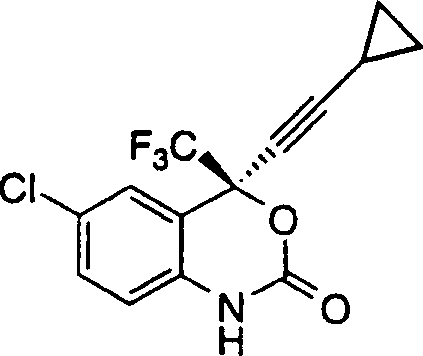 其包括如下步骤:1)在约25℃的温度和在氮气气氛下并保持pH约8.5-4之间下将4-硝基苯基氯甲酸酯分批加入下式的氨基醇在甲基叔丁基醚和碳酸氢钾水溶液中的搅拌混合物中:
其包括如下步骤:1)在约25℃的温度和在氮气气氛下并保持pH约8.5-4之间下将4-硝基苯基氯甲酸酯分批加入下式的氨基醇在甲基叔丁基醚和碳酸氢钾水溶液中的搅拌混合物中: 得到下式的氨基甲酸酯中间体;2)在约20℃-约25℃下搅拌反应混合物约2小时以完成氨基甲酸酯中间体的形成;3)用含水KOH调节pH至约11中止反应并加入水得到在有机溶剂相中含有1,4-二氢-2H-3,1-苯并噁嗪-2-酮的两相溶液;4)由有机相中分离1,4-二氢-2H-3,1-苯并噁嗪-2-酮。
得到下式的氨基甲酸酯中间体;2)在约20℃-约25℃下搅拌反应混合物约2小时以完成氨基甲酸酯中间体的形成;3)用含水KOH调节pH至约11中止反应并加入水得到在有机溶剂相中含有1,4-二氢-2H-3,1-苯并噁嗪-2-酮的两相溶液;4)由有机相中分离1,4-二氢-2H-3,1-苯并噁嗪-2-酮。
本发明制备下式1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法: 其包括如下步骤:1)在约0℃-约25℃的温度和在惰性气氛下将芳基氯甲酸酯加入下式的氨基醇在有机溶剂中的搅拌混合物中:
其包括如下步骤:1)在约0℃-约25℃的温度和在惰性气氛下将芳基氯甲酸酯加入下式的氨基醇在有机溶剂中的搅拌混合物中: 得到下式的氨基甲酸酯中间体:
得到下式的氨基甲酸酯中间体: 其中R表示氯甲酸酯的芳基侧链;2)在约20℃-约25℃下搅拌反应混合物约1-约6小时以完成氨基甲酸酯中间体的形成;3)用含水碱中止反应以得到在有机溶剂相中含有1,4-二氢-2H-3,1-苯并噁嗪-2-酮的两相溶液;4)在约20℃-约50℃搅拌两相混合物约1-约6小时以完成环化反应生成1,4-二氢-2H-3,1-苯并噁嗪-2-酮;和5)由有机相中分离1,4-二氢-2H-3,1-苯并噁嗪-2-酮。
其中R表示氯甲酸酯的芳基侧链;2)在约20℃-约25℃下搅拌反应混合物约1-约6小时以完成氨基甲酸酯中间体的形成;3)用含水碱中止反应以得到在有机溶剂相中含有1,4-二氢-2H-3,1-苯并噁嗪-2-酮的两相溶液;4)在约20℃-约50℃搅拌两相混合物约1-约6小时以完成环化反应生成1,4-二氢-2H-3,1-苯并噁嗪-2-酮;和5)由有机相中分离1,4-二氢-2H-3,1-苯并噁嗪-2-酮。
用于制备上述1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中芳基氯甲酸酯的芳基定义为苯基或萘基,它任选地被一个、两个或三个选自卤素(F、Cl、Br、I)、CF3、CO2C1-C6-烷基和NO2的取代基取代。
用于制备上述1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中碱定义为氢氧化钾、氢氧化钠、氢氧化锂、碳酸钾、碳酸钠、碳酸锂、碳酸氢钾、碳酸氢钠、碳酸氢锂和它们的混合物的固体或溶液。
用于制备如上所述的1,4-二氢-2H-3,1-苯并噁嗪的-2-酮方法,其中有机溶剂选自甲基叔丁基醚、甲苯或所述溶剂的混合物。
用于制备上述1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中芳基氯甲酸酯的芳基定义为苯基氯甲酸酯,其中苯基任选地被一个、两个或三个选自卤素(F、Cl、Br、I)、CF3和NO2的取代基取代。
用于制备上述1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中碱定义为氢氧化钾、氢氧化钠、碳酸钾、碳酸钠、碳酸氢钾、碳酸氢钠或所述碱的混合物的固体或液体。
用于制备如上所述的1,4-二氢-2H-3,1-苯并噁嗪的-2-酮方法,其中芳基氯甲酸酯的芳基定义为4-硝基苯基氯甲酸酯。
本发明的另一方面是制备下式1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法:其包括如下步骤:1)在约0℃-约25℃的温度和在惰性气氛下将烷基氯甲酸酯加入下式的氨基醇在第一种有机溶剂和第一种碱中的搅拌混合物中: 得到下式的氨基甲酸酯中间体:
得到下式的氨基甲酸酯中间体: 其中R表示氯甲酸酯的烷基侧链;2)在约20℃-约25℃下搅拌反应混合物约1-约30小时以完成氨基甲酸酯中间体的形成;3)分离含有烷基氨基甲酸酯的有机相;4)真空蒸馏出约90%-约95%的第一种有机溶剂并加入反萃溶剂以分离固体烷基氨基甲酸酯;5)在固体烷基氨基甲酸酯中加入第二种有机溶剂,形成烷基氨基甲酸酯溶液;6)在约20℃-约25℃的温度下使烷基氨基甲酸酯溶液与第二种碱反应约2小时至约30小时,产生1,4-二氢-2H-3,1-苯并噁嗪-2-酮;7)用酸中止反应混合物以得到在有机溶剂相中含有1,4-二氢-2H-3,1-苯并噁嗪-2-酮的两相溶液;8)由有机相中分离1,4-二氢-2H-3,1-苯并噁嗪-2-酮。
其中R表示氯甲酸酯的烷基侧链;2)在约20℃-约25℃下搅拌反应混合物约1-约30小时以完成氨基甲酸酯中间体的形成;3)分离含有烷基氨基甲酸酯的有机相;4)真空蒸馏出约90%-约95%的第一种有机溶剂并加入反萃溶剂以分离固体烷基氨基甲酸酯;5)在固体烷基氨基甲酸酯中加入第二种有机溶剂,形成烷基氨基甲酸酯溶液;6)在约20℃-约25℃的温度下使烷基氨基甲酸酯溶液与第二种碱反应约2小时至约30小时,产生1,4-二氢-2H-3,1-苯并噁嗪-2-酮;7)用酸中止反应混合物以得到在有机溶剂相中含有1,4-二氢-2H-3,1-苯并噁嗪-2-酮的两相溶液;8)由有机相中分离1,4-二氢-2H-3,1-苯并噁嗪-2-酮。
用于制备上述1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中烷基氯甲酸酯的烷基定义为C1-C10-烷基,它任选地被一个、两个或三个选自卤素(F、Cl、Br、I)、CF3、C3-C7-环烷基、CO2C1-C6-烷基和NO2的取代基取代。用于制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法的烷基氯甲酸酯的实例是甲基或乙基氯甲酸酯。
用于制备上述1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中第一种碱定义为氢氧化钾、氢氧化钠、氢氧化锂、碳酸钾、碳酸钠、碳酸锂、碳酸氢钾、碳酸氢钠、碳酸氢锂和它们的混合物的固体或溶液。用于制备上述1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中第二种碱定义为KOC1-C6-烷基、NaOC1-C6-烷基、LiOC1-C6-烷基、KC1-C6-烷基、NaC1-C6-烷基、LiC1-C6-烷基、KHMDS、NaHMDS、LiHMDS、LDA或所述碱的混合物的固体或溶液。
用于制备如上所述的1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中第一种有机溶剂选自甲基叔丁基醚、甲苯、四氢呋喃、乙腈或所述溶剂的混合物。用于制备如上所述的1,4-二氢-2H-3,1-苯并噁嗪的-2-酮方法,其中反萃溶剂是庚烷。用于制备如上所述的1,4-二氢-2H-3,1-苯并噁嗪的-2-酮方法,其中第二种有机溶剂选自甲基叔丁基醚、甲苯、四氢呋喃、C1-C6烷基醇或所述溶剂的混合物。
用于制备如上所述的1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中酸选自盐酸、硝酸、硫酸和乙酸。
用于制备如上所述的1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其包括附加步骤:由甲苯-庚烷或甲基叔丁基醚-庚烷中结晶在步骤4中产生的烷基氨基甲酸酯,以得到晶体烷基氨基甲酸酯。
本发明的另一方面是下式的烷基氨基甲酸酯: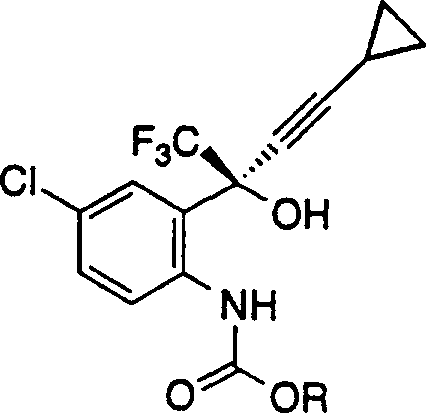 其中R表示C1-C10-烷基,它任选地被一个、两个或三个选自卤素(F、Cl、Br、I)、CF3、C3-C7-环烷基、CO2C1-C6-烷基和NO2的取代基取代。
其中R表示C1-C10-烷基,它任选地被一个、两个或三个选自卤素(F、Cl、Br、I)、CF3、C3-C7-环烷基、CO2C1-C6-烷基和NO2的取代基取代。
用于本文的术语“烷基”包括规定原子数目的直链、支链或环构型的烷基,它任选地被选自卤素(F、Cl、Br、I)、CF3、CO2C1-C6-烷基、C3-C7-环烷基和NO2的取代基取代。“烷基”的实例包括甲基、乙基、丙基、异丙基、丁基、仲和叔丁基、戊基、己基、庚基、环丙基、环丁基、环戊基、环己基、环庚基、降冰片基等。术语“芳基”定义为苯基或萘基环,它在任何可利用的碳原子上被一个、两个或三个选自卤素(F、Cl、Br、I)、CF3、CO2C1-C6-烷基和NO2的取代基取代。
术语惰性气氛理解为氩气或氮气,优选氮气气氛。
方案1说明了合成(-)-6-氯-4-环丙基乙炔基-4-三氟甲基-1,4-二氢-2H-3,1-苯并噁嗪-2-酮(DMP-266)的关键步骤。手性加成步骤在化合物1的三氟甲基酮上对映选择性地加成环丙基乙炔化物。生成的PMB保护的氨基醇2随后脱保护得到氨基醇3。该氨酸醇随后用氯甲酸酯和碱环化得到DMP-266。
方案1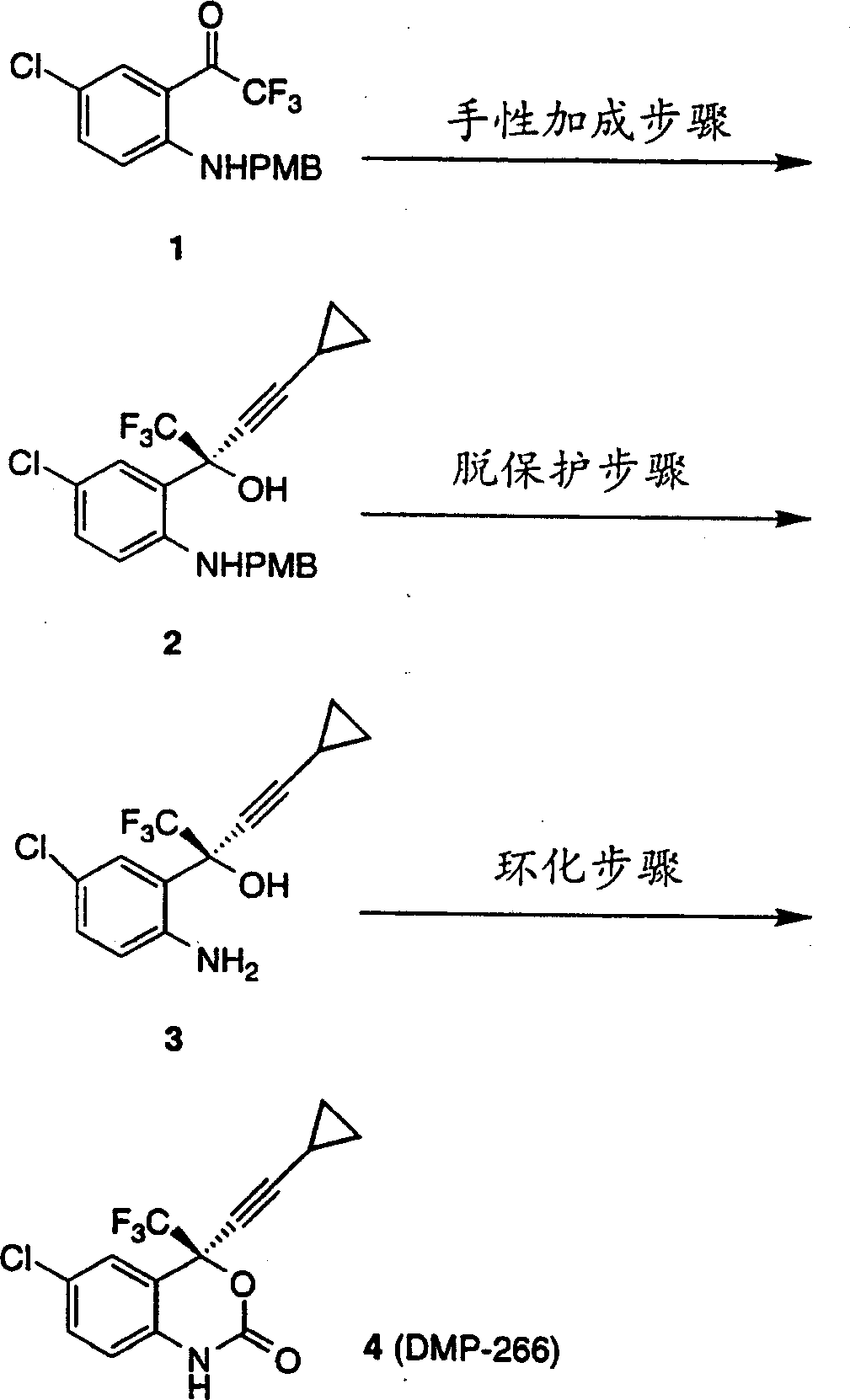
环化氨基醇3以生成1,4-二氢-2H-3,1-苯并噁嗪-2-酮的过程在方案2中说明。反应可作为一步方法进行或依据所使用的氯甲酸酯的可能的中间体氨基甲酸酯分离的两步方法。已表明在一步方法中芳基氯甲酸酯形成不太稳定的氨基甲酸酯,使得当它们用含水碱处理时它们环化成产物。此外,烷基氯甲酸酯得到烷基氨基甲酸酯,一种在进行环化步骤之前能够分离和纯化的关键中间体。根据烷基氨基甲酸酯的稳定性,已开发了可行的制备DMP-266的两步方法,其包括形成烷基氨基甲酸酯中间体5,随后环化氨基甲酸酯得到所需的产物4。
方案2
表1列出了当进行采用芳基氯甲酸酯的一步方法时可以使用的溶剂和碱。此外,可结合如下列出的碱,例如使用碳酸氢盐和氢氧化物或碳酸氢盐和碳酸盐。反应可在单相或两相反应混合物中进行。碱可以在开始加入氯甲酸酯时加入,或在芳基氯甲酸酯加完后加入。碱优选在开始加入氯甲酸酯时加入,以中和产生的盐酸。
| 表1:一步方法 | |
| 溶剂 | 碱 |
| ACN | 碳酸盐(s) |
| 碳酸氢盐(s) | |
| 氢氧化物(s) | |
| DMAC | 碳酸盐(s) |
| 碳酸氢盐(s) | |
| 氢氧化物(s) | |
| MTBE | 碳酸盐(aq) |
| 碳酸氢盐(aq) | |
| 氢氧化物(aq) | |
| NMP | 碳酸盐(s) |
| 碳酸氢盐(s) | |
| 氢氧化物(s) | |
| THF | 碳酸盐(s) |
| 碳酸氢盐(s) | |
| 氢氧化物(s) | |
| TOL | 碳酸盐(aq) |
| 碳酸氢盐(aq) | |
| 氢氧化物(aq) | |
表2列出了当进行采用烷基氯甲酸酯的两步方法时可以使用的溶剂和碱。两步骤方法的步骤1,烷基氨基甲酸酯的形成可在单相或两相反应混合物中进行。两步方法的步骤2,烷基氨基甲酸酯的环闭合可在标注的溶剂和碱中进行。
+使用乙腈作为第一种碱可能需要另一种分离方法,例如蒸馏乙腈并冷却以进行氨基甲酸酯的结晶,这不同于蒸馏大多数第一种有机溶剂,并随后加入反萃溶剂以结晶氨基甲酸酯。*两相反应混合物s是指固体,当在无水反应混合物中使用固体碱时,则随后用水中止反应。aq是指碱的含水溶液。用于溶剂的缩写:乙腈(ACN)、C1-C6-醇(醇)、二甲基乙酰胺(DMAC)、甲基叔丁基醚(MTBE)、N-甲基吡咯烷酮(NMP)、四氢呋喃(THF)和甲苯(TOL)。此外可以使用上述溶剂的混合物以使反应最佳化。用于碱的缩写:氢氧化锂、钠和钾;碳酸锂、钠和钾;碳酸氢锂、钠和钾;烷基锂、钠和钾(例如正丁基锂);六甲基二硅氨化锂、钠和钾(例如LiHMDS)和二异丙基氨化锂(LDA)。
| 表2:两步法 | |||
| 步骤1:氨基甲酸酯的形成 | 步骤2:环闭合 | ||
| 第一种溶剂 | 第一种碱 | 第二种溶剂 | 第二种碱 |
| ACN+ | 碳酸盐(s) | 醇 | 醇盐 |
| 碳酸氢盐(s) | THF | 醇盐 | |
| 氢氧化物(s) | 烷基碱金属 | ||
| THF | 碳酸盐(s) | 碱金属HMDS | |
| 碳酸氢盐(s) | LDA | ||
| 氢氧化物(s) | MTBE | 醇盐 | |
| MTBE* | 碳酸盐(aq) | 烷基碱金属 | |
| 碳酸氢盐(aq) | 碱金属HMDS | ||
| 氢氧化物(aq) | LDA | ||
| TOL* | 碳酸盐(aq) | TOL | 醇盐 |
| 碳酸氢盐(aq) | 烷基碱金属 | ||
| 氢氧化物(aq) | 碱金属HMDS | ||
| LDA | |||
如下实施例用于说明本发明,这些实施例用于举例说明本发明,而不构成对发明范围的限制。
实施例1
| FW | g | mL | mmol | 当量 | |
| 氨基醇12 | 289 | 7.0 | 24.2 | 1 | |
| 4-硝基苯基氯甲酸酯 | 201.6 | 5.9 | 29.3 | 1.2 | |
| 碳酸氢钾 | 100 | 7.26 | 72.6 | 3 | |
| 碳酸钾 | 138 | 10 | 72.5 | 3 | |
| 水 | 250 | ||||
| THF | 100 | ||||
| MTBE | 100 | ||||
| 乙醇 | 45 |
向装备有机械搅拌器、氮气管线和热电偶的三颈圆底烧瓶中加入固体氨基醇3、THF(100ml)和固体碳酸氢钾。将得到的混合物冷却至5℃,然后一次加入固体4-硝基苯基氯甲酸酯。反应是放热的。随着该硝基苯基氯甲酸酯的溶解/反应,温度上升了4℃(最终为9℃)。混合物在20-25℃搅拌2小时。分批取样,用ACN和5%碳酸氢钠稀释,得到黄色均匀溶液。在220nm的HPLC分析显示硝基苯酚(43%)、化合物4(49%)、硝基苯基碳酸酯(7%)和若干低含量(<0.3%)杂质。以这种方式分批取样和稀释对于可重复的数据是重要的。如果分批的取样用ACN稀释和用HPLC分析,主要的峰是硝基苯基氨基甲酸酯5。在加入含水碱后转化为化合物4。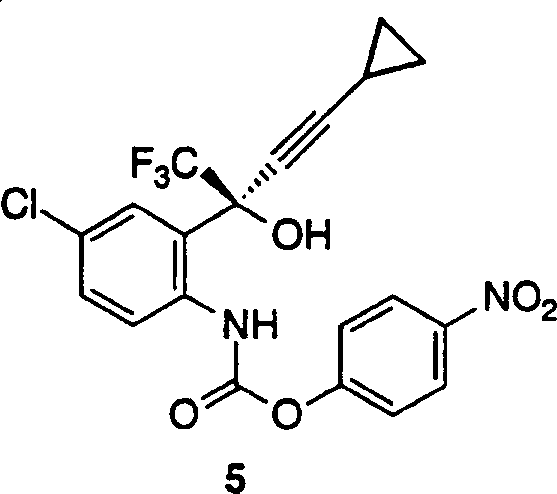
通过加入含水碳酸钾(在150ml水中的10g)中上反应,得到的两相混合物在25℃剧烈搅拌2小时。加入MTBE(100ml),分层,有机层用5%含水碳酸钾(2×50ml)和水(2×50ml)洗涤。溶剂转换为乙醇或异丙醇(IPA),由乙醇或IPA和水中结晶产物。参见实施例10和11。
实施例2
| FW | g | mL | mmol | 当量 | |
| 氨基醇12 | 289 | 7.0 | 24.2 | 1 | |
| 4-硝基苯基氯甲酸酯 | 201.6 | 5.9 | 29.3 | 1.2 | |
| 碳酸钾 | 138 | 20 | 145.2 | 6 | |
| 水 | 370 | ||||
| MTBE | 100 | ||||
| 乙醇 | 45 |
向装备有机械搅拌器、氮气管线和热电偶的三颈圆底烧瓶中加入固体氨基醇12、MTBE(100ml)和含水碳酸钾(在120ml水中的10g,3当量),在25℃一次加入固体4-硝基苯基氯甲酸酯。混合物在20-25℃搅拌2小时,此时将两相混合物加热至50℃3小时以有效转化硝基苯基碳酸酯为硝基苯酚。在冷却至25℃后分层,MTBE层用含水碳酸钾(在150ml水中的10g,分两次,各75ml)和水(2×50ml)洗涤。此时将混合物的溶剂转换为乙醇/IPA,实施例10和11中所述结晶。
实施例3
| FW | g | mL | mmol | 当量 | |
| 氨基醇12 | 289 | 100 | 346 | 1 | |
| 4-硝基苯基氯甲酸酯 | 201.6 | 80.2 | 398 | 1.15 | |
| 碳酸氢钾 | 100 | 45 | 450 | 1.3 | |
| 2N KOH | 56 | 346 | 692 | 2.0 | |
| 水 | 654 | ||||
| MTBE | 500 |
向装备有机械搅拌器、氮气管线和热电偶的三颈圆底烧瓶中加入固体氨基醇3、MTBE(500ml)和含水碳酸氢钾(在654ml水中的45g),在25℃一次加入固体4-硝基苯基氯甲酸酯。在加入过程中监测溶液pH,在反应过程中pH维持在8.5-4,最终为8.0。混合物在20-25℃搅拌2小时,在20分钟内加入含水KOH(2N),直至水层的pH达到11.0。
分层,MTBE层用pH7缓冲液(500ml)和盐水(500ml)洗涤。此时将混合物的溶剂转换为乙醇/IPA,并如实施例10和11中所述结晶。
实施例4
| FW | g | mL | mmol | 当量 | |
| 氨基醇3 | 289 | 100 | 346 | 1 | |
| 4-硝基苯基氯甲酸酯 | 201.6 | 73.2 | 363 | 1.05 | |
| 碳酸氢钾 | 100 | 45 | 450 | 1.3 | |
| 2N KOH | 56 | 346 | 692 | 2.0 | |
| 水 | 654 | ||||
| MTBE | 500 |
向装备有机械搅拌器、氮气管线和热电偶的三颈圆底烧瓶中加入固体氨基醇3、MTBE(500ml)和含水碳酸氢钾(在654ml水中的45g),在25℃分4次次加入固体4-硝基苯基氯甲酸酯。在加入过程中监测溶液pH,在反应过程中pH维持在8.5-4,最终为8.0.混合物在20-25℃搅拌2小时,在20分钟内加入含水KOH(2N),直至水层的pH达到11.0。
分层,在MTBE层中加入500mL盐水。加入0.1N乙酸直至pH为6-7。分层,有机层用盐水(500ml)洗涤。此时将混合物的溶剂转换为乙醇/IPA,并如实施例10和11中所述结晶。
实施例5步骤A:氨基甲酸酯5的制备
| MW | 密度 | mmol | 数量 | 当量 | |
| 12 | 289.7 | 35.38 | 10.25g | ||
| 碳酸氢钾 | 100.12 | 70.76 | 7.08g | 2 | |
| ClCO2Me | 94.50 | 1.223 | 70.76 | 5.46ml | 2 |
| MTBE | 100ml | ||||
| 水 | 100ml | ||||
| 盐水 | 100ml | ||||
| 庚烷 | 300ml |
在500ml 3颈烧瓶中装备顶部搅拌器、温度探针和氮气管线,加入氨基醇3(35.38mmol,10.25ml)和MTBE(100ml),形成浅色浆状物,加入水(100ml),然后加入碳酸氢钾(2当量,7.08g)。经注射管加入甲基氯甲酸酯(2当量,5.46ml),在20-25℃下剧烈搅拌两相混合物。用HPLC分析样品直至残留<0.5%氨基醇3(约8.5小时),分层,有机层用盐水(100ml)洗涤,用硫酸镁干燥。在过滤后,溶剂转换(50-60℃,真空)为总体积为102ml(10ml/g起始物料)的MTBE-庚烷混合物(用1H NMR测量约5%MTBE(体积))。在溶剂转换过程容易地结晶出甲基氨基甲酸酯5。浆状物在20-25℃陈化约30分钟后,过滤物质。固体用母液洗涤,随后用1滤饼体积的庚烷洗涤。以92%的收率分离出无水甲基氨基甲酸酯5(11.32g),1-2%损失在母液中。步骤B:环闭合化合物4的制备
| MW | 密度 | mmol | 数量 | 当量 | |
| 5 | 347.72 | 32.55 | 11.32g | ||
| 1M LiOtBu | 32.55 | 32.6ml | 1 | ||
| MTBE | 170ml | ||||
| 0.5N HCl | 150ml | ||||
| 盐水 | 150ml |
在500ml 3颈圆底烧瓶中装备顶部搅拌器、温度探针和氮气管线,将甲基氨基甲酸酯5(32.55mmol,11.32g)溶解在MTBE(170ml)中,加入LiOtBu(1当量,32.6ml)反应混合物立即变成浆状物,浆状物随着时间变稀,在30分钟内为透明黄色溶液。反应混合物在20-25℃陈化,用HPLC跟踪反应。在8小时后,残留<1%的甲基氯甲酸酯5。在约16小时后,残留少于0.3%的甲基氨基甲酸酯。用0.5N盐酸(150ml)中止反应,分层,有机层用盐水(150ml)洗涤,用硫酸镁干燥并过滤。HPLC分析化合物4的收率为96.0%,该物质在乙醇-水结晶的制备中溶剂转换为乙醇,参见实施例10和11。
实施例6 步骤A:氨基甲酸酯5的制备
步骤A:氨基甲酸酯5的制备
根据上述实施例5步骤A的方法,只是使用甲苯作为溶剂,由甲苯-庚烷混合物中分离氨基甲酸酯。步骤B:环闭合化合物4的制备
根据上述实施例5步骤B的方法,只是使用甲苯作为溶剂,通过由甲苯-庚烷混合物中结晶分离产物4。
实施例7 步骤A:氨基甲酸酯5的制备
步骤A:氨基甲酸酯5的制备
| MW | 密度 | mmol | 数量 | 当量 | |
| 3 | 289.7 | 35.38 | 10.25g | ||
| 碳酸氢钾 | 100.12 | 70.76 | 7.08g | 2 | |
| ClCO2Et | 108.52 | 1.135 | 70.76 | 6.76ml | 2 |
| MTBE | 100ml | ||||
| 水 | 100ml | ||||
| 盐水 | 100ml | ||||
| 庚烷 | 300ml |
在500ml 3颈烧瓶中装备顶部搅拌器、温度探针和氮气管线,加入氨基醇3(35.38mmol,10.25ml)和MTBE(100ml),形成浅色浆状物,加入水(100ml),然后加入碳酸氢钾(2当量,7.08g)。经注射管加入乙基氯甲酸酯(2当量,6.76ml),在20-25℃下剧烈搅拌两相混合物。用HPLC分析样品直至残留<2%氨基醇3(约30小时),分层,有机层用盐水(100ml)洗涤,用硫酸镁干燥。在过滤后,溶剂转换(50-60℃,真空)为总体积为102ml(10ml/g起始物料)的MTBE-庚烷混合物(用1HNMR测量约5%MTBE(体积))。在溶剂转换过程容易地结晶出甲基氨基甲酸酯5。浆状物在20-25℃陈化约30分钟后,过滤物质。固体用母液洗涤,随后用1滤饼体积的庚烷洗涤。以92%的收率分离出无水乙基氨基甲酸酯5(11.77g),1-2%损失在母液中。步骤B:环闭合化合物4的制备
| MW | 密度 | mmol | 数量 | 当量 | |
| 5 | 361.75 | 32.55 | 11.77g | ||
| 1M LiOtBu | 32.55 | 32.6ml | 1 | ||
| MTBE | 170ml | ||||
| 0.5N HC1 | 150ml | ||||
| 盐水 | 150ml |
在500ml 3颈圆底烧瓶中装备顶部搅拌器、温度探针和氮气管线,将乙基氨基甲酸酯5(32.55mmol,11.32g)溶解在MTBE(170ml)中,加入LiOtBu(1当量,32.6ml)反应混合物立即变成浆状物,浆状物随着时间变稀,在30分钟内为透明黄色溶液。反应混合物在20-25℃陈化,用HPLC跟踪反应。在26小时后,残留<1%的乙基氯甲酸酯5。用0.5盐酸(150ml)中止反应,分层,有机层用盐水(150ml)洗涤,用硫酸镁干燥并过滤。HPLC分析化合物4的收率为96.0%,该物质在乙醇-水结晶的制备中溶剂转换为乙醇,参见实施例10和11。
实施例8 步骤A:氨基甲酸酯5的制备
步骤A:氨基甲酸酯5的制备
根据上述实施例7步骤A的方法,只是使用甲苯作为溶剂,由甲苯-庚烷混合物中分离氨基甲酸酯。步骤B:环闭合化合物4的制备
根据上述实施例7步骤B的方法,只是使用甲苯作为溶剂,通过由甲苯-庚烷混合物中结晶分离产物4。
实施例9步骤A:氨基甲酸酯5的制备
根据上述实施例7步骤A的方法。步骤B:环闭合化合物4的制备
| MW | 密度 | mmol | 数量 | 当量 | |
| 15 | 361.75 | 32.55 | 11.77g | ||
| 2.5M nBuLi | 32.55 | 13.0ml | 1 | ||
| MTBE | 170ml | ||||
| pH7缓冲溶液 | 150ml | ||||
| 盐水 | 150ml |
在500ml 3颈圆底烧瓶中装备顶部搅拌器、温度探针和氮气管线,将乙基氨基甲酸酯5(32.55mmol,11.32g)溶解在MTBE(170ml)中,加入nBuLi(1当量,13.0ml),反应混合物在20-25℃陈化,用HPLC跟踪反应。在30小时后,残留<1%的乙基氯甲酸酯5。用pH7的缓冲溶液(150ml)中止反应,分层,有机层用盐水(150ml)洗涤,用硫酸镁干燥并过滤。HPLC分析化合物4的收率为96.0%,该物质在乙醇-水结晶的制备中溶剂转换为乙醇,参见实施例10和11。
实施例10控制的反溶剂添加结晶方法
将400g DMP-266粗物料溶解在2.400L乙醇中,过滤溶液以除了外来的物质。在30-60分钟内在溶液中加入2.088L去离子(DI)水。在溶液中加入20g DMP-266晶种,晶种层陈化1小时,优选使用Intermig搅拌器混合浆状物。如果需要(存在太长结晶或稠浆状物),湿磨浆状物15-约60秒。在4-6小时内加入1.512L DI水,如果需要(存在太长结晶或稠浆状物),在加入过程中湿磨浆状物15-约60秒。将浆状物陈化1-3小时,然后冷却至10℃3小时。将浆状物陈化2-16小时直至在上清液中产物的浓度保持恒定。过滤浆状物以分离结晶湿滤饼。湿滤饼用1-2床层体积的在水中的40%乙醇洗涤,然后用2L DI水洗涤两次,在50℃的真空下干燥湿滤饼。
实施例11半连续尾料结晶过程
将400g DMP-266粗物料溶解在2.400L乙醇中,通过将20g在0.3L的40%(v/v)乙醇水产生尾料浆状物。在6小时内将溶解的料和3.6LDI水同时加入尾料浆状物中以保持在结晶器中恒定的溶剂组成。在结晶过程中优选使用Intermig搅拌器,在添加过程中如果结晶长度变得过长或浆状物变得太稠,则湿磨浆状物。将浆状物陈化1-3小时,然后冷却至10℃3小时。将浆状物陈化2-16小时直至在上清液中产物的浓度保持恒定。过滤浆状物以分离结晶湿滤饼。湿滤饼用1-2床层体积的40%乙醇水洗涤,然后用2L DI水洗涤两次,在50℃的真空下干燥湿滤饼。
Claims (24)
1.一种制备下式1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法: 其包括如下步骤:1)在约0℃-约25℃的温度和在惰性气氛下将芳基氯甲酸酯加入下式的氨基醇在有机溶剂和碱中的搅拌混合物中:
其包括如下步骤:1)在约0℃-约25℃的温度和在惰性气氛下将芳基氯甲酸酯加入下式的氨基醇在有机溶剂和碱中的搅拌混合物中: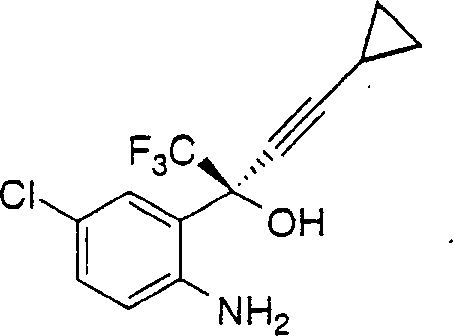 得到下式的氨基甲酸酯中间体:其中R表示氯甲酸酯的芳基侧链,其定义为苯基或萘基,它任选地被一个、两个或三个选自卤素、CF3、CO2C1-C6-烷基和NO2的取代基取代;2)在约20℃-约25℃下搅拌反应混合物约1-约6小时以完成氨基甲酸酯中间体的形成;3)用水或含水碱中止反应以得到在有机溶剂相中含有1,4-二氢-2H-3,1-苯并噁嗪-2-酮的两相溶液;4)在约20℃-约50℃搅拌两相混合物约1-约6小时以完成环化反应,生成1,4-二氢-2H-3,1-苯并噁嗪-2-酮;和5)由有机相中分离1,4-二氢-2H-3,1-苯并噁嗪-2-酮。
得到下式的氨基甲酸酯中间体:其中R表示氯甲酸酯的芳基侧链,其定义为苯基或萘基,它任选地被一个、两个或三个选自卤素、CF3、CO2C1-C6-烷基和NO2的取代基取代;2)在约20℃-约25℃下搅拌反应混合物约1-约6小时以完成氨基甲酸酯中间体的形成;3)用水或含水碱中止反应以得到在有机溶剂相中含有1,4-二氢-2H-3,1-苯并噁嗪-2-酮的两相溶液;4)在约20℃-约50℃搅拌两相混合物约1-约6小时以完成环化反应,生成1,4-二氢-2H-3,1-苯并噁嗪-2-酮;和5)由有机相中分离1,4-二氢-2H-3,1-苯并噁嗪-2-酮。
2.权利要求1的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中碱定义为氢氧化钾、氢氧化钠、氢氧化锂、碳酸钾、碳酸钠、碳酸锂、碳酸氢钾、碳酸氢钠、碳酸氢锂或它们的混合物的固体或溶液。
3.权利要求2的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中有机溶剂选自甲基叔丁基醚、甲苯、四氢呋喃、乙腈、二甲基乙酰胺、N-甲基吡咯烷酮或所述溶剂的混合物。
4.权利要求3的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中芳基氯甲酸酯的芳基定义为苯基氯甲酸酯,其中苯基任选地被一个、两个或三个选自卤素、CF3和NO2的取代基取代。
5.权利要求4的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中碱定义为氢氧化钾、氢氧化钠、碳酸钾、碳酸钠、碳酸氢钾、碳酸氢钠或所述碱的混合物的固体或溶液。
6.权利要求5的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中芳基氯甲酸酯的芳基定义为4-硝基苯基氯甲酸酯。
7.一种制备下式1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法: 其包括如下步骤:1)在约25℃的温度和在氮气气氛下并保持pH约8.5-4之间下将4-硝基苯基氯甲酸酯分批加入下式的氨基醇在甲基叔丁基醚和碳酸氢钾水溶液中的搅拌混合物中:
其包括如下步骤:1)在约25℃的温度和在氮气气氛下并保持pH约8.5-4之间下将4-硝基苯基氯甲酸酯分批加入下式的氨基醇在甲基叔丁基醚和碳酸氢钾水溶液中的搅拌混合物中: 得到下式的氨基甲酸酯中间体;
得到下式的氨基甲酸酯中间体; 2)在约20℃-约25℃下搅拌反应混合物约2小时以完成氨基甲酸酯中间体的形成;3)用含水KOH调节pH至约11中止反应并加入水得到在有机溶剂相中含有1,4-二氢-2H-3,1-苯并噁嗪-2-酮的两相溶液;4)由有机相中分离1,4-二氢-2H-3,1-苯并噁嗪-2-酮。
2)在约20℃-约25℃下搅拌反应混合物约2小时以完成氨基甲酸酯中间体的形成;3)用含水KOH调节pH至约11中止反应并加入水得到在有机溶剂相中含有1,4-二氢-2H-3,1-苯并噁嗪-2-酮的两相溶液;4)由有机相中分离1,4-二氢-2H-3,1-苯并噁嗪-2-酮。
8.根据权利要求1的方法,其中在步骤3)中,反应用含水碱中止。
9.权利要求8的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中碱定义为氢氧化钾、氢氧化钠、氢氧化锂、碳酸钾、碳酸钠、碳酸锂、碳酸氢钾、碳酸氢钠、碳酸氢锂或它们的混合物的固体或溶液。
10.权利要求9的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中有机溶剂选自甲基叔丁基醚、甲苯或所述溶剂的混合物。
11.权利要求10的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中芳基氯甲酸酯的芳基定义为苯基氯甲酸酯,其中苯基任选地被一个、两个或三个选自卤素、CF3和NO2的取代基取代。
12.权利要求11的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中碱定义为氢氧化钾、氢氧化钠、碳酸钾、碳酸钠、碳酸氢钾、碳酸氢钠或所述碱的混合物的固体或溶液。
13.权利要求12的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中芳基氯甲酸酯的芳基定义为4-硝基苯基氯甲酸酯。
14.一种制备下式1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法: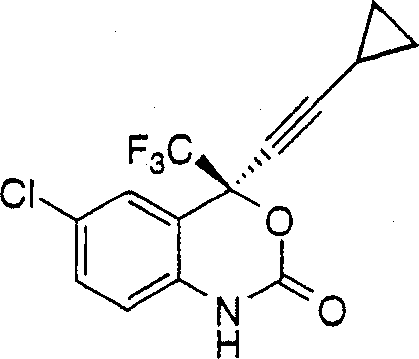 其包括如下步骤:1)在约0℃-约25℃的温度和在惰性气氛下将烷基氯甲酸酯加入下式的氨基醇在第一种有机溶剂和第一种碱中的搅拌混合物中:
其包括如下步骤:1)在约0℃-约25℃的温度和在惰性气氛下将烷基氯甲酸酯加入下式的氨基醇在第一种有机溶剂和第一种碱中的搅拌混合物中: 得到下式的氨基甲酸酯中间体:其中R表示氯甲酸酯的烷基侧链,其定义为C1-C10-烷基,它任选地被一个、两个或三个选自卤素、CF3、C3-C7-环烷基、CO2C1-C6-烷基和NO2的取代基取代;2)在约20℃-约25℃下搅拌反应混合物约1-约30小时以完成氨基甲酸酯中间体的形成;3)分离含有烷基氨基甲酸酯的有机相;4)真空蒸馏出约90%-约95%的第-种有机溶剂并加入反萃溶剂以分离固体烷基氨基甲酸酯;5)在固体烷基氨基甲酸酯中加入第二种有机溶剂以形成烷基氨基甲酸酯溶液;6)在约20℃-约25℃的温度下使烷基氨基甲酸酯溶液与第二种碱反应约2小时至约30小时,产生1,4-二氢-2H-3,1-苯并噁嗪-2-酮;7)用酸中止反应混合物以得到在有机溶剂相中含有1,4-二氢-2H-3,1-苯并噁嗪-2-酮的两相溶液;8)由有机相中分离1,4-二氢-2H-3,1-苯并噁嗪-2-酮。
得到下式的氨基甲酸酯中间体:其中R表示氯甲酸酯的烷基侧链,其定义为C1-C10-烷基,它任选地被一个、两个或三个选自卤素、CF3、C3-C7-环烷基、CO2C1-C6-烷基和NO2的取代基取代;2)在约20℃-约25℃下搅拌反应混合物约1-约30小时以完成氨基甲酸酯中间体的形成;3)分离含有烷基氨基甲酸酯的有机相;4)真空蒸馏出约90%-约95%的第-种有机溶剂并加入反萃溶剂以分离固体烷基氨基甲酸酯;5)在固体烷基氨基甲酸酯中加入第二种有机溶剂以形成烷基氨基甲酸酯溶液;6)在约20℃-约25℃的温度下使烷基氨基甲酸酯溶液与第二种碱反应约2小时至约30小时,产生1,4-二氢-2H-3,1-苯并噁嗪-2-酮;7)用酸中止反应混合物以得到在有机溶剂相中含有1,4-二氢-2H-3,1-苯并噁嗪-2-酮的两相溶液;8)由有机相中分离1,4-二氢-2H-3,1-苯并噁嗪-2-酮。
15.权利要求14的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中第一种碱定义为氢氧化钾、氢氧化钠、氢氧化锂、碳酸钾、碳酸钠、碳酸锂、碳酸氢钾、碳酸氢钠、碳酸氢锂或它们的混合物的固体或溶液。
16.权利要求15的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中第一种有机溶剂选自甲基叔丁基醚、甲苯、四氢呋喃、乙腈或所述溶剂的混合物。
17.权利要求16的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中反萃溶剂是庚烷。
18.权利要求17的制备1,4-二氢-2H-3,1-苯并噁嗪的-2-酮方法,其中第二种有机溶剂选自甲基叔丁基醚、甲苯、四氢呋喃、C1-C6链烷醇或所述溶剂的混合物。
19.权利要求18的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中第二种碱定义为KOC1-C6-烷基、NaOC1-C6-烷基、LiOC1-C6-烷基、KC1-C6-烷基、NaC1-C6-烷基、LiC1-C6-烷基、KHMDS、NaHMDS、LiHMDS、LDA或所述碱的混合物的固体或溶液。
20.权利要求19的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中酸选自盐酸、硝酸、硫酸和乙酸。
21.权利要求20的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其包括附加步骤:由甲苯-庚烷或甲基叔丁基醚-庚烷中结晶在步骤4中产生的烷基氨基甲酸酯,以得到烷基氨基甲酸酯结晶。
22.权利要求21的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中烷基氯甲酸酯定义为甲基或乙基氯甲酸酯。
23.权利要求22的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中有机溶剂或溶剂混合物选自甲基叔丁基醚、甲基叔丁基醚和四氢呋喃、和甲基叔丁基醚和乙醇。
24.权利要求23的制备1,4-二氢-2H-3,1-苯并噁嗪-2-酮的方法,其中碱定义为碳酸氢钾和氢氧化钾、碳酸氢钾和碳酸钾、或碳酸氢钾和LiOtBu的固体或溶液。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US3705997P | 1997-02-12 | 1997-02-12 | |
| US60/037,059 | 1997-02-12 | ||
| US60/037059 | 1997-02-12 | ||
| GB9706082.6 | 1997-03-24 | ||
| GBGB9706082.6A GB9706082D0 (en) | 1997-03-24 | 1997-03-24 | Efficient synthesis of A 1,4-dihydro-2H-3,1-benzoxazin-2-one |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1246852A CN1246852A (zh) | 2000-03-08 |
| CN1073992C true CN1073992C (zh) | 2001-10-31 |
Family
ID=26311250
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN98802445A Expired - Fee Related CN1073992C (zh) | 1997-02-12 | 1998-02-09 | 1,4-二氢-2h-3,1-苯并噁嗪-2-酮的有效合成方法 |
Country Status (13)
| Country | Link |
|---|---|
| EP (1) | EP0968197B1 (zh) |
| CN (1) | CN1073992C (zh) |
| AR (1) | AR011128A1 (zh) |
| AT (1) | ATE294165T1 (zh) |
| AU (1) | AU6152598A (zh) |
| BR (1) | BR9807213A (zh) |
| DE (1) | DE69829956T2 (zh) |
| EA (1) | EA001784B1 (zh) |
| ES (1) | ES2239798T3 (zh) |
| HR (1) | HRP980056A2 (zh) |
| SK (1) | SK108999A3 (zh) |
| TW (1) | TW480253B (zh) |
| WO (1) | WO1998034928A1 (zh) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AR011731A1 (es) * | 1997-05-16 | 2000-08-30 | Merck & Co Inc | Un proceso de reaccion de adicion enantioselectiva eficiente utilizando un reactivo de organozinc. |
| PE20060587A1 (es) * | 2004-09-02 | 2006-07-09 | Bristol Myers Squibb Co | Sintesis de (s)-6-cloro-4-ciclopropiletinil-4-trifluorometil-1,4-dihidro-2h-3,1-benzoxazin-2-ona |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996037457A1 (en) * | 1995-05-25 | 1996-11-28 | Merck & Co., Inc. | Asymmetric synthesis of (-) 6-chloro-4-cyclopropyl-ethynyl-4-trifluoromethyl-1,4-dihydro-2h-3,1-benzoxazin-2-one |
-
1998
- 1998-02-04 TW TW087101435A patent/TW480253B/zh not_active IP Right Cessation
- 1998-02-04 HR HR9706082.6A patent/HRP980056A2/hr not_active Application Discontinuation
- 1998-02-09 BR BR9807213-7A patent/BR9807213A/pt not_active IP Right Cessation
- 1998-02-09 EA EA199900736A patent/EA001784B1/ru not_active IP Right Cessation
- 1998-02-09 WO PCT/US1998/002495 patent/WO1998034928A1/en active IP Right Grant
- 1998-02-09 SK SK1089-99A patent/SK108999A3/sk unknown
- 1998-02-09 DE DE69829956T patent/DE69829956T2/de not_active Expired - Lifetime
- 1998-02-09 ES ES98906255T patent/ES2239798T3/es not_active Expired - Lifetime
- 1998-02-09 EP EP98906255A patent/EP0968197B1/en not_active Expired - Lifetime
- 1998-02-09 CN CN98802445A patent/CN1073992C/zh not_active Expired - Fee Related
- 1998-02-09 AU AU61525/98A patent/AU6152598A/en not_active Abandoned
- 1998-02-09 AT AT98906255T patent/ATE294165T1/de not_active IP Right Cessation
- 1998-02-10 AR ARP980100589A patent/AR011128A1/es not_active Application Discontinuation
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996037457A1 (en) * | 1995-05-25 | 1996-11-28 | Merck & Co., Inc. | Asymmetric synthesis of (-) 6-chloro-4-cyclopropyl-ethynyl-4-trifluoromethyl-1,4-dihydro-2h-3,1-benzoxazin-2-one |
Also Published As
| Publication number | Publication date |
|---|---|
| DE69829956D1 (de) | 2005-06-02 |
| AR011128A1 (es) | 2000-08-02 |
| TW480253B (en) | 2002-03-21 |
| HRP980056A2 (en) | 1998-12-31 |
| BR9807213A (pt) | 2000-05-23 |
| EP0968197A1 (en) | 2000-01-05 |
| SK108999A3 (en) | 2000-05-16 |
| DE69829956T2 (de) | 2006-03-02 |
| ATE294165T1 (de) | 2005-05-15 |
| CN1246852A (zh) | 2000-03-08 |
| WO1998034928A1 (en) | 1998-08-13 |
| EP0968197B1 (en) | 2005-04-27 |
| EA199900736A1 (ru) | 2000-02-28 |
| ES2239798T3 (es) | 2005-10-01 |
| AU6152598A (en) | 1998-08-26 |
| EA001784B1 (ru) | 2001-08-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI386212B (zh) | 硼酸酯及硼酸化合物之合成 | |
| Colson et al. | Asymmetric synthesis of. alpha.-alkyl-. alpha. amino acids from chromium-carbene-complex-derived. beta.-lactams | |
| Brinner et al. | A rapid and general method for the asymmetric synthesis of 2-substituted pyrrolidines using tert-butanesulfinamide | |
| JPH07501078A (ja) | プロリンボロネートエステルの製法 | |
| CN1665760A (zh) | 手性氨基腈的制备 | |
| CN101631781A (zh) | (s)-2-乙氧基-4-[n-[1-(2-哌啶苯基)-3-甲基-1-丁基]氨基羰基甲基]苯甲酸乙酯的改进的制备方法及其用于制备瑞格列奈的用途 | |
| CN1073992C (zh) | 1,4-二氢-2h-3,1-苯并噁嗪-2-酮的有效合成方法 | |
| CN1308319C (zh) | 合成hiv蛋白酶抑制剂的方法 | |
| CN1113877C (zh) | 用于合成新的il-1抑制剂的呋喃磺酰胺化合物的有效合成方法 | |
| KR20070047798A (ko) | 6,7-디하이드로-5H-이미다조[1,2-a]이미다졸-3-설폰산아미드의 합성 | |
| US5922864A (en) | Efficient synthesis of a 1,4-dihydro2H-3,1-benzoxazin-2-one | |
| CN1310884C (zh) | 对映体纯的n-甲基-n-[(1s)-1-苯基-2-((3s)-3-羟基吡咯烷-1-基)乙基]-2,2-二苯基乙酰胺的制备方法 | |
| CN1320114A (zh) | 新颖的6-(4-苯基丁氧基)己胺衍生物和获得沙美特罗的方法 | |
| CN1204337A (zh) | 棒酸盐的制备 | |
| CN1197836C (zh) | 一种不对称合成二级丙炔醇类化合物的方法 | |
| CN1442403A (zh) | 手性氨基醇配体及其在端炔对亚氨的不对称加成中的应用 | |
| CN1714075A (zh) | 1,3-取代的茚类的改进制备方法 | |
| CN1185235C (zh) | 双(氟代芳基)硼衍生物的制备方法 | |
| CN1028361C (zh) | 戊二酸衍生物的制备方法 | |
| CN106966940B (zh) | 一种西他列汀磷酸盐中间体n-芳甲基-2s-氰基甲基吖啶的制备方法 | |
| EP1663968A1 (en) | Process for the preparation of pure levetiracetam | |
| CN102432616A (zh) | 一种制备l-脯氨酰胺及其中间体的方法 | |
| CN1055460C (zh) | C-取代二亚乙基三胺的制备方法 | |
| CN1147810A (zh) | 原型血管紧张肽原酶抑制剂的合成 | |
| JPH0285235A (ja) | 光学活性アミノアルコール類の製法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: GR Ref document number: 1049731 Country of ref document: HK |