BRPI0818533B1 - composto, composição farmacêutica, uso de um composto, e, processo para a preparação de um composto - Google Patents
composto, composição farmacêutica, uso de um composto, e, processo para a preparação de um composto Download PDFInfo
- Publication number
- BRPI0818533B1 BRPI0818533B1 BRPI0818533-6A BRPI0818533A BRPI0818533B1 BR PI0818533 B1 BRPI0818533 B1 BR PI0818533B1 BR PI0818533 A BRPI0818533 A BR PI0818533A BR PI0818533 B1 BRPI0818533 B1 BR PI0818533B1
- Authority
- BR
- Brazil
- Prior art keywords
- chlorophenyl
- mmol
- pyrimidin
- amino
- piperidine
- Prior art date
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
- C07D473/34—Nitrogen atom attached in position 6, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Oncology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
COMPOSTO, COMPOSIÇÃO FARMACÊUTICA, USO DE UM COMPOSTO E, PROCESSO PARA A PREPARAÇÃO DE UM COMPOSTO. A invenção diz respeito a um novo grupo de compostos da Fórmula (I) ou sais dos mesmos: em que Y, Z1, Z2, R1, R4, R5 e n são como descritos no relatório descritivo, que pode ser útil no tratamento ou prevenção de uma doença ou condição médica mediada através da proteína quinase B (PKB) tais como o câncer. A invenção também diz respeito às composições farmacêuticas que compreendem os ditos compostos, métodos de tratamento de doenças mediadas pelo PKB usando os ditos compostos e métodos para preparar os compostos da Fórmula (I)
Description
[0001] A presente invenção diz respeito a um novo grupo de heterociclos bicíclicos que pode ser útil no tratamento ou prevenção de uma doença ou condição médica mediadas através da proteína quinase B (PKB, ‘também conhecida coma AKT). Tais compostos portanto podem ser úteis no tratamento ou prevenção de vários cânceres diferentes. A invenção também diz respeito às composições farmacêuticas que compreendem os ditos compostos, aos processos para a fabricação dos ditos compostos e aos métodos de tratamento de doenças mediadas pela PKB usando os ditos compostos.
[0002] PKB é um componente do caminho da sinalização de fosfatidil 3-quinase (PI3K) que desempenha uma parte importante na proliferação e sobrevivência celulares, incluindo respostas celulares aos fatores de crescimento. Na ligação de um fator de crescimento, por exemplo o fator de crescimento epidérmico (EGF), à sua tirosina quinase receptora de superfície celular, por exemplo receptor de EGF (EGFR), o receptor dimeriza e sofre autofosforilação. Este evento de autofosforilação permite que a subunidade reguladora de 85 kDa de PI3K (p85) interaja com o receptor diretamente ou via uma proteína adaptadora, por exemplo proteína 2 ligada ao receptor do fator de crescimento (GRB2), e deste modo ative a subunidade catalítica de 110 kDa de PI3K (p110). Na ativação, p110 catalisa a fosforilação de fosfatidilinositol-4,5-bisfosfato (PIP2) para produzir fosfatidilinositol- 3,4,5-trifosfato (PIP3), uma segunda molécula mensageira que recruta tanto a quinase 1 dependente de fosfatidilinositol (PDK1) quanto PKB para a membrana plasmática onde PDK1 fosforila e ativa PKB.
[0003] Existem três isoformas conhecidas de PKB (PKBa/AKT1, PKBβ/AKT2 e PKBy/AKT3), derivadas de três genes distintos. A ativação de PKBα está associada com eventos de sinalização celular que medeiam a proliferação e sobrevivência celulares, ao passo que a ativação de PKBβ está associada com a invasão, motilidade e processos metabólico mediados por insulina. PKB ativado protege as células da apoptose pela inativação de fatores pró-apoptóticos, por exemplo os fatores de transcrição BAD, procaspase-9 e fatores de transcrição forkhead (FKHR), e ativando fatores de transcrição que super regulam genes antiapoptóticos, por exemplo proteína de ligação de elemento de resposta de AMP cíclica (CREB). PKB também pode contribuir para a sobrevivência da célula pela inativação de p53 via fosforilação de MDM2. Similarmente, PKB ativado induz a proliferação celular pela ativação de proteínas envolvida no crescimento e metabolismo celulares, por exemplo por um caminho que leva à ativação do alvo mamífero de rapamicina (mTOR) e via glicogênio sintase quinase-3 (GSK3).
[0004] A estimulação mediada por PKB da proliferação e proteção celulares da apoptose portanto favorecem a tumorigênese e perturbações genéticas de componentes dentro do caminho PI3K são habitualmente encontradas no câncer. Por exemplo, mutação ou amplificação dos genes que codificam as isoformas p110 de PI3K são verificados nos cânceres de mama, câncer do intestino, câncer ovariano, cânceres da cabeça e pescoço e cervical escamoso, cânceres gástricos e pulmonares, oligodendrogliomas angioplásticos, astrocitomas anaplásticos, glioblastoma multiforme e meduloblastomas. Similarmente, a amplificação de mutação e/ou super expressão dos genes que codificam as isoformas PKB são encontradas em tumores pancreáticos, mamários e ovarianos. Além disso, o gene que codifica para PTEN (uma fosfatase que tem um papel reverso para PI3K, que catalisa a conversão de PIP3 para PIP2) é inativado em muitos tipos de tumor, incluindo ovariano, colorretal, mamário, glioma, melanoma, pulmonar, leucemias e linfomas; isto resulta na ativação de PKB/AKT.
[0005] Em vista da importância do caminho da sinalização de PI3K na proliferação celular e sobrevivência de tumor, qualquer composto que rompa este caminho, incluindo inibidores de PKB, podem ser úteis no tratamento de câncer. Revisões detalhadas do caminho de sinalização de PI3K e seu envolvimento na tumorigênese são fornecidos por Hennessy et al., Nature Reviews / Drug Discovery (Dezembro de 2005) Vol. 4, 988-1004. e Cully et al., Nature reviews / Cancer (Março de 2006) Vol. 6, 184-192.
[0006] O canal de potássio dependente de potássio codificado pelo gene relacionado com o éter-a-go-go humano (hERG) é acreditado desempenhar um papel chave na repolarização do potencial de ação cardíaca ventricular. Mudanças na sua atividade, causadas pelas mutações herdadas da sequência de gene ou modificação farmacológica, pode levar à prolongação da duração potencial da ação. Isto pode levar à prolongação do intervalo QT registrado no ser humano em um eletrocardiograma e a uma arritmia cardíaca potencialmente fatal conhecida como Torsades de Pointes (Vandenberg et al. (2001). Trends Pharmacol. Sci. 22, 240-246). As diretrizes regulatórias recentes (CPMP/ICH/539/00) recomendaram que um ensaio in vitro que investiga os efeitos de compostos de teste no canal hERG poderia ser um elemento de uma estratégia pré-clínica objetivando prognosticar a probabilidade de que novas entidades químicas prolongarão o intervalo QT registrado no ser humano em um eletrocardiograma. Como tal, a eliminação da atividade bloqueadora de hERG permanece uma consideração importante no desenvolvimento de qualquer novo medicamento.
[0007] Vários compostos foram descritos que alvejam o caminho PI3K. Por exemplo a WO 2006/046023 e WO 2006/046024 (Astex Therapeutics Limited) descrevem compostos de purina, purinona e desazapurinona que inibem ou modulam a atividade da proteína quinase B (PKB) e proteína quinase A (PKA). Entretanto, ainda existe a necessidade quanto a agentes ainda mais melhorados tendo potência superior contra PKB e/ou propriedades químicas vantajosas (por exemplo, solubilidade aquosa mais alta, permeabilidade mais alta, e/ou ligação de proteína plasmática mais baixa) e/ou perfil de toxicidade favorável (por exemplo uma tendência para bloquear hERG diminuída) e/ou perfis metabólicos favoráveis em comparação com outros inibidores de PKB conhecidos.
[0008] Os requerentes surpreendentemente verificaram que certos derivados de heterociclo bicíclico são particularmente eficazes na inibição da atividade de PKB e portanto podem ser úteis no tratamento de estados de doença em que a atividade de PKB é implicada, por exemplo câncer.
[0009] De acordo com um primeiro aspecto da invenção, é portanto fornecido um composto da Fórmula (I), ou um sal farmaceuticamente aceitável do mesmo: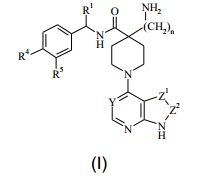 R1 representa alquila C1-4, alquenila C2-4, alquinila C2-4, alcóxi C1-4, alcóxi C1-4 alquila C1-4, fluoroalquila C1-4, aminoalquila C1-4, hidroxialquila C1-4, ciano, cianoalquila C1-4, cicloalquila C3-6, -(CH2)pNHCOCH3, -(CH2)pNHSO2CH3, -(CH2)pNHCONH2, -(CH2)p NHCONR2R3, -(CH2)pNR2R3, -(CH2)pSO2NH2, -(CH2)pSO2NR2R3, -(CH2)pCONH2, -(CH2)pCONR2R3 ou -(CH2)p-R7; onde p é 0, 1, 2 ou 3; R2 representa hidrogênio ou alquila C1-3; R3 representa alquila C1-3; e R7 representa fenila; R7 representa um anel heteroarila de 5 ou 6 membros monocíclico que compreende 1, 2 ou 3 heteroátomos selecionados de O, N ou S; ou R7 representa um anel heterocíclico monocíclico de 4, 5 ou 6 membros que compreende 1, 2 ou 3 heteroátomos selecionados de O, N ou S; em que R7 é opcionalmente substituído por 1 ou 2 substituintes selecionados de alquila C1-4, trifluorometila, alcóxi C1-4, flúor, cloro, bromo, e ciano; R4 representa hidrogênio, flúor, cloro, bromo, ciano ou trifluorometila; e R5 representa hidrogênio, flúor, cloro ou bromo.
R1 representa alquila C1-4, alquenila C2-4, alquinila C2-4, alcóxi C1-4, alcóxi C1-4 alquila C1-4, fluoroalquila C1-4, aminoalquila C1-4, hidroxialquila C1-4, ciano, cianoalquila C1-4, cicloalquila C3-6, -(CH2)pNHCOCH3, -(CH2)pNHSO2CH3, -(CH2)pNHCONH2, -(CH2)p NHCONR2R3, -(CH2)pNR2R3, -(CH2)pSO2NH2, -(CH2)pSO2NR2R3, -(CH2)pCONH2, -(CH2)pCONR2R3 ou -(CH2)p-R7; onde p é 0, 1, 2 ou 3; R2 representa hidrogênio ou alquila C1-3; R3 representa alquila C1-3; e R7 representa fenila; R7 representa um anel heteroarila de 5 ou 6 membros monocíclico que compreende 1, 2 ou 3 heteroátomos selecionados de O, N ou S; ou R7 representa um anel heterocíclico monocíclico de 4, 5 ou 6 membros que compreende 1, 2 ou 3 heteroátomos selecionados de O, N ou S; em que R7 é opcionalmente substituído por 1 ou 2 substituintes selecionados de alquila C1-4, trifluorometila, alcóxi C1-4, flúor, cloro, bromo, e ciano; R4 representa hidrogênio, flúor, cloro, bromo, ciano ou trifluorometila; e R5 representa hidrogênio, flúor, cloro ou bromo.
[00010] Em uma forma de realização da invenção, é fornecido um composto da Fórmula (I), ou um sal farmaceuticamente aceitável do mesmo: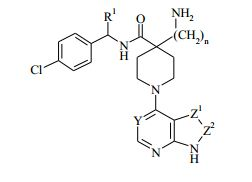 (I) em que: Y representa CH ou N; Z -Z representam um grupo selecionado de CH=CH, N=CH e CH=N; n é 0, 1 ou 2; e R1 representa alquila C1-4, aminoalquila C1-4, hidróxi-alquila C1-4, -(CH2)pNHCOCH3, -(CH2)qNR2R ou cicloalquila C3-6; onde R2 representa hidrogênio ou alquila C1-3; R3 representa alquila C1-3; e p e q independentemente representam 2 ou 3.
(I) em que: Y representa CH ou N; Z -Z representam um grupo selecionado de CH=CH, N=CH e CH=N; n é 0, 1 ou 2; e R1 representa alquila C1-4, aminoalquila C1-4, hidróxi-alquila C1-4, -(CH2)pNHCOCH3, -(CH2)qNR2R ou cicloalquila C3-6; onde R2 representa hidrogênio ou alquila C1-3; R3 representa alquila C1-3; e p e q independentemente representam 2 ou 3.
[00011] Em uma outra forma de realização da invenção, é portanto fornecido um composto da Fórmula (I), ou um sal farmaceuticamente aceitável do mesmo: pode ser de cadeia reta ou ramificada. Entretanto, as referências aos grupos alquila individuais tais como “propila” são específicos apenas para a versão da cadeia reta e as referências aos grupos alquila de cadeia ramificada individuais tais como t-butila são específicos apenas para a versão da cadeia ramificada. Por exemplo, “alquila C1-4” inclui metila, etila, propila, isopropila e t-butila.
pode ser de cadeia reta ou ramificada. Entretanto, as referências aos grupos alquila individuais tais como “propila” são específicos apenas para a versão da cadeia reta e as referências aos grupos alquila de cadeia ramificada individuais tais como t-butila são específicos apenas para a versão da cadeia ramificada. Por exemplo, “alquila C1-4” inclui metila, etila, propila, isopropila e t-butila.
[00012] O termo “alquila C1-4” é intencionado a significar uma cadeia de carbono saturada de 1 a 4 átomos de carbono no comprimento que
[00013] O termo “alquenila C2-4” é intencionado a significar uma cadeia de carbono não saturada de 2 a 4 átomos de carbono no comprimento, que pode ser de cadeia reta ou ramificada, contendo pelo menos uma ligação dupla de carbono para carbono. Entretanto, referências aos grupos alquila individuais tais como “propenila” são específicos apenas para a versão da cadeia reta e as referências aos grupos alquila de cadeia ramificada individuais tais como terc-butenila são específicos apenas para a versão da cadeia ramificada. Por exemplo, “alquenila C2-4” inclui, mas não é limitado a, etenila, propenila, isopropenila, butenila e terc-butenila.
[00014] O termo “alquinila C2-4” é intencionado a significar uma cadeia de carbono não saturada de 2 a 4 átomos de carbono no comprimento, que pode ser de cadeia reta ou ramificada, contendo pelo menos uma ligação tripla de carbono para carbono. Entretanto, as referências aos grupos alquinila individuais tais como “propinila” são específicos apenas para a versão da cadeia reta e as referências aos grupos alquila de cadeia ramificada individuais tais como terc-butinila são específicos apenas para a versão da cadeia ramificada. Por exemplo, “alquinila C2-4” inclui, mas não é limitado a, etinila, propinila, isopropinila, butinila e terc-butinila.
[00015] O termo “alcóxi C1-4” é intencionado a significar uma cadeia de carbono saturada de 1 a 4 átomos de carbono no comprimento, que pode ser de cadeia reta ou ramificada, ligada ao oxigênio. Por exemplo, “alcóxi C1-6” inclui, mas não é limitado a, metóxi, etóxi, propóxi e butóxi.
[00016] O termo “alcóxi C1-4 alquila C1-4” é intencionado a significar uma cadeia de carbono saturada de 1 a 4 átomos de carbono no comprimento, que pode ser de cadeia reta ou ramificada, ligada por intermédio de oxigênio a uma outra cadeia de carbono saturado de 1 a 4 átomos de carbono no comprimento, que pode ser de cadeia reta ou ramificada. Por exemplo, “alcóxi C1-4 alquila C1-4” inclui, mas não é limitado a, metoxietila, metoxipropila, etoxipropila, propoxietila e butoxipropila.
[00017] O termo “fluoroalquila C1-4” é intencionado a significar uma cadeia de carbono saturada de 1 a 4 átomos de carbono no comprimento que pode ser de cadeia reta ou ramificada em que pelo menos um dos átomos de hidrogênio foi substituído por flúor. Por exemplo, “fluoroalquila C1-4” inclui, mas não é limitado a, fluorometila, fluoroetila, fluoropropila, fluoroisopropila, fluorobutila, fluoroisobutila, fluoro-terc-butila, trifluorometila, pentafluoroetila, heptafluoropropila e nonafluorobutila.
[00018] O termo “aminoalquila C1-4” é intencionado a significar uma cadeia de carbono saturada de 1 a 4 átomos de carbono no comprimento, que pode ser de cadeia reta ou ramificada, que compreendem um grupo amino primário. Por exemplo “aminoalquila C1-4” inclui aminometila, aminoetila, 2-aminopropila, 3-aminopropila, 1- aminoisopropila e 4-aminobutila.
[00019] O termo “hidroxialquila C1-4” é intencionado a significar uma cadeia de carbono saturada de 1 a 4 átomos de carbono no comprimento, que pode ser de cadeia reta ou ramificada, que compreendem um grupo hidroxila. Por exemplo “hidroxialquila C1-4” inclui hidroximetila, hidroxietila, 2-hidroxipropila, 3-hidroxipropila, 1- hidroxiisopropila e 4-hidroxibutila.
[00020] O termo “cicloalquila C3-6” é intencionado a significar um anel de carbono monocíclico saturado de 3 a 6 membros. Por exemplo “cicloalquila C3-6” inclui ciclopropila, ciclobutila, ciclopentila e ciclo-hexila.
[00021] O termo “anel heteroarila” é intencionado a significar um anel monocíclico de 5 ou 6 membros, totalmente insaturado, aromático que compreende 1, 2 ou 3 heteroátomos independentemente selecionados de nitrogênio, oxigênio ou enxofre, ligados por intermédio de um átomos de carbono no anel ou um átomo de nitrogênio do anel onde um uma ligação de nitrogênio é possível, por exemplo nenhuma ligação é possível ao nitrogênio de um anel de piridina, mas uma ligação é possível através do 1-nitrogênio de um anel de pirazol. Os exemplos de anéis heteroarila de 5 ou 6 membros incluem, mas não são limitados a, pirrol, furano imidazol, triazol, tetrazol, pirazina, pirimidina, piridazina, piridina, pirazol, isoxazol, oxazol, 1,2,4 oxadiazol, isotiazol, tiazol, 1,2,4-triazol e tiofeno.
[00022] O termo “anel heterocíclico” é intencionado a significar um anel monocíclico de 4, 5 ou 6 membros completamente saturado ou parcialmente saturado que compreende 1, 2 ou 3 heteroátomos selecionados de nitrogênio, oxigênio ou enxofre ligado por intermédio de um átomos de carbono no anel ou um átomo de nitrogênio do anel. Os exemplos de anéis heterocíclicos de 4, 5 ou 6 membros incluem azetidina, tetraidrofurano tetraidropirano, pirrolina, pirrolidina, tiazolidina, morfolina, piperidina, piperazina, diidropiridina, diidro- pirimidina e azepano.
[00023] Em uma outra forma de realização da invenção, cada uma das seguintes invenções de Y, Z1-Z2, R1, R4, R5, R6, n e p em parágrafos de (1) a (26) a seguir podem ser usados individualmente ou em combinação com uma das outras seguintes definições para limitar a definição mais ampla das Fórmulas (I), (IA) ou (IB) como apropriado. (1) Y representa N; (2) Z1-Z2 representam CH=CH; (3) Z1-Z2 representam C(Cl)=CH; (4) Z1-Z2 representam C(Br)=CH; (5) R1 representa alquila C1-4; (6) R1 representa aminoalquila C1-4; (7) R1 representa hidroxialquila C1-4; (8) R1 representa cicloalquila C3-6; (9) R1 representa alcóxi C1-4 alquila C1-4, fluoroalquila C1-4, aminoalquila C1-4, hidroxialquila C1-4, cianoalquila C1-4, cicloalquila C3- 6,-(CH2)pNHCOCH3,-(CH2)pNHSO2CH3, -(CH2)pNHCONH2, - (CH2)pNHCONR2R3, -(CH2)pNR2R3, -(CH2)pSO2NH2, -(CH2)pCONH2, - (CH2)pCONR2R3 ou -(CH2)p-R7; (10) R1 representa -(CH2)p-R7 em que R7 é selecionado de fenila, piperidinila, piperazinila, pirrolidinila, morfolinila, imidazolila, isoxazolila, pirazolila e tiazolila e R7 é opcionalmente substituído por um grupo metila único; (11) R1 representa hidroxietila; (12) n é 0; (13) n é 1; (14) n é 1 ou 2; (15) n é 0 ou 1; (16) p é 1, 2 ou 3; (17) R4 representa cloro, bromo ou ciano; (18) R4 representa cloro, bromo; (19) R4 representa cloro; (20) R4 representa bromo; (21) R5 representa hidrogênio; (22) R5 representa cloro; (23) R6 representa hidrogênio; (24) R6 representa metila; (25) R6 representa difluorometila; (26) R6 representa trifluorometila.
[00024] De acordo com uma outra forma de realização da invenção, é fornecido um composto da Fórmula (I), ou um sal farmaceuticamente aceitável do mesmo, que é: 4-amino-N-(1-(4-clorofenil)etil)-1-(7H-pirrolo[2,3-d]pirimidin- 4-il)- piperidina-4-carboxamida; (S)-4-amino-N-(1-(4-clorofenil)etil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)- piperidina-4-carboxamida; 4-amino-N-(1-(4-clorofenil)propil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)- piperidina-4-carboxamida; (S)-4-amino-N-(1-(4-clorofenil)propil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida; (R)-4-amino-N-(1-(4-clorofenil)propil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-((4-clorofenil)(ciclolopropil)metil)-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(2-amino-1-(4-clorofenil)etil)-1-(7H-pirrolo[2,3- d]pirimidin- 4-il)piperidina-4-carboxamida; (S)-4-(aminometil)-N-(1-(4-clorofenil)etil)-1-(7H-pirrolo[2,3- d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(1-(4-clorofenil)-4-hidroxibutil)-1-(7H-pirrolo[2,3- d]- pirimidin-4-il)piperidina-4-carboxamida; (S)-4-amino-N-(1-(4-clorofenil)-4-hidroxibutil)-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; (R)-4-amino-N-(1-(4-clorofenil)-4-hidroxibutil)-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(1-(4-clorofenil)-2-hidroxietil)-1-(7H-pirrolo[2,3- d]pirimidin -4-il)piperidina-4-carboxamida; (S)-4-amino-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; N-(3-acetamido-1-(4-clorofenil)propil)-4-amino-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(1-(4-clorofenil)-3-(dimetilamino)propil)-1-(7H- pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida; (S)-4-amino-N-(1-(4-clorofenil)-3-(dimetilamino)propil)-1- (7H-pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida; (R)-4-amino-N-(1-(4-clorofenil)-3-(dimetilamino)propil)-1- (7H-pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida; 4-(aminometil)-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(7H- pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(7H-pirrolo[2,3- d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(3-amino-1-(4-clorofenil)propil)-1-(7H-pirrolo[2,3- d]- pirimidin-4-il)piperidina-4-carboxamida; (R)-4-amino-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; (R)-4-amino-N-(1-(4-clorofenil)etil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)- piperidina-4-carboxamida; (R)-4-(aminometil)-N-(1-(4-clorofenil)etil)-1-(7H-pirrolo[2,3- d]- pirimidin-4-il)piperidina-4-carboxamida; (S)-4-amino-N-(1-(4-cianofenil)-3-hidroxipropil)-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; (S)-4-amino-1-(5-bromo-7H-pirrolo[2,3-d]pirimidin-4-il)-N-(1- (4-cloro- fenil)-3-hidroxipropil)piperidina-4-carboxamida; (S)-4-amino-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(1H- pirazolo[3,4-d]- pirimidin-4-il)piperidina-4-carboxamida; (S)-4-amino-N-(3-hidróxi-1-fenilpropil)-1-(7H-pirrolo[2,3- d]pirimidin- 4-il)piperidina-4-carboxamida; (S)-4-amino-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(9H-purin- 6-il)- piperidina-4-carboxamida; (S)-4-(aminometil)-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(7H- pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida; (S)-4-amino-N-(1-(4-bromofenil)-3-hidroxipropil)-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(1-(4-clorofenil)-4-(dimetilamino)butil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida; (S)-4-amino-N-(1-(4-clorofenil)-3-(dietilamino)propil)-1-(7H- pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida; (S)-4-amino-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(5- ciclolopropil-7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(1-(4-clorofenil)-3-(metilamino)propil)-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-[(4-clorofenil)(fenil)metil]-1-(7H-pirrolo[2,3- d]pirimidin-4- il)piperidina-4-carboxamida; 4-amino-N-[2-amino-1-(4-clorofenil)-2-oxoetil]-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-1-(3-bromo-1H-pirazolo[3,4-d]pirimidin-4-il)-N- [(1S)-1-(4- clorofenil)etil]piperidina-4-carboxamida; 4-amino-N-[(1S)-1-(4-clorofenil)etil]-1-(5-cloro-7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-1-(3-bromo-1H-pirazolo[3,4-d]pirimidin-4-il)-N- [(1S)-1-(4- clorofenil)-3-hidroxipropil]piperidina-4-carboxamida; 4-amino-N-[(1S)-1-(4-clorofenil)-3-hidroxipropil]-1-(5-cloro- 7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-1-(5-bromo-7H-pirrolo[2,3-d]pirimidin-4-il)-N-[(1S)- 1-(4- clorofenil)etil]piperidina-4-carboxamida; 4-amino-N-[(1S)-1-(4-clorofenil)-3-hidroxipropil]-1-(5-metil- 7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-[(1S)-1-(4-clorofenil)etil]-1-(5-metil-7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-[(1S)-1-(4-clorofenil)-3-hidróxi-3-metilbutil]-1- (7H-pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-[(1S)-1-(4-cianofenil)etil]-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-[(1S)-1-(3-clorofenil)-3-hidroxipropil]-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-1-(7H-pirrolo[2,3-d]pirimidin-4-il)-N-{(1S)-1-[4- (trifluoro- metil)fenil]etil}piperidina-4-carboxamida; 4-amino-N-[(1R)-1-(4-bromofenil)etil]-1-(7H-pirrolo[2,3- d]pirimidin-4- il)piperidina-4-carboxamida; 4-amino-N-[1-(4-clorofenil)-2-feniletil]-1-(7H-pirrolo[2,3- d]pirimidin-4- il)piperidina-4-carboxamida; 4-amino-N-[1-(4-fluorofenil)etil]-1-(7H-pirrolo[2,3-d]pirimidin- 4-il)- piperidina-4-carboxamida; 4-amino-N-[(4-clorofenil)(ciano)metil]-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(1-feniletil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxamida; 4-amino-N-[1-(4-clorofenil)-4-pirrolidin-1-ilbutil]-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-[1-(4-clorofenil)-4-morfolin-4-ilbutil]-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-[1-(4-clorofenil)-4-piperidin-1-ilbutil]-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-[(1S)-1-(4-clorofenil)-4-piperidin-1-ilbutil]-1-(7H- pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-[(1R)-1-(4-clorofenil)-4-piperidin-1-ilbutil]-1-(7H- pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-[(1S)-1-(4-clorofenil)-3-(4-metilpiperazin-1- il)propil]-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-[(1S)-1-(4-clorofenil)-3-morfolin-4-ilpropil]-1-(7H- pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-[(1S)-1-(4-clorofenil)-3-piperidin-1-ilpropil]-1-(7H- pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-[(1S)-1-(4-clorofenil)-3-piperazin-1-ilpropil]-1- (7H-pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-[(1S)-1-(4-clorofenil)-3-(1H-imidazol-1-il)propil]- 1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-[(1S)-1-(4-clorofenil)-3-pirrolidin-1-ilpropil]-1-(7H- pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(1-(4-clorofenil)-2-sulfamoiletil)-1-(7H-pirrolo[2,3- d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(1-(4-clorofenil)-2-sulfamoiletil)-1-(7H-pirrolo[2,3- d]- pirimidin-4-il)piperidina-4-carboxamida; N-(2-acetamido-1-(4-clorofenil)etil)-4-amino-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(1-(4-clorofenil)-2-(1H-imidazol-2-il)etil)-1-(7H- pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida; 4-Amino-N-[1-(4-clorofenil)-2-(1H-pirazol-1-il)etil]-1-(7H- pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-[1-(4-clorofenil)-2-(3-metilisoxazol-5-il)etil]-1-(7H- pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(1-(4-clorofenil)-2-(tiazol-2-il)etil)-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(1-(4-clorofenil)-3-(dimetilamino)-3-oxopropil)-1- (7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(1-(4-clorofenil)-3-metóxipropil)-1-(7H-pirrolo[2,3- d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(1-(4-clorofenil)-3-sulfamoilpropil)-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(3-amino-1-(4-clorofenil)-3-oxopropil)-1-(7H- pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(1-(4-clorofenil)-3-ureidopropil)-1-(7H-pirrolo[2,3- d]- pirimidin-4-il)piperidina-4-carboxamida; 4-amino-N-(1-(4-clorofenil)-2-cianoetil)-1-(7H-pirrolo[2,3- d]pirimidin- 4-il)piperidina-4-carboxamida; ou 4-amino-N-(1-(4-clorofenil)-3-(metilsulfonamido)propil)-1- (7H-pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida.
[00025] Deve ser entendido que, à medida que os compostos da Fórmula (I) definidos acima existem em formas opticamente ativas ou racêmicas em virtude do átomo de carbono assimétrico, a invenção inclui em sua definição qualquer uma de tais formas opticamente ativas ou racêmicas que possua a propriedade da atividade inibidora de PKB. A síntese das formas opticamente ativas podem ser realizadas pelas técnicas padrão da química orgânica bem conhecida na técnica, por exemplo pela síntese de materiais opticamente ativos e materiais de partida ou pela resolução de uma forma racêmica. Os compostos racêmicos e intermediários racêmicos destes são desenhados aqui como estruturas planas considerando os compostos estereo-específicos e intermediários estereoespecíficos destes são desenhados com a estereoquímica apropriada indicada.
[00026] A invenção também diz respeito a qualquer uma e todas as formas dos compostos da Fórmula (I) que são inibidores da atividade de PKB.
[00027] Em uma forma de realização da invenção, o composto da Fórmula (I) tem a configuração mostrada na Fórmula (IA):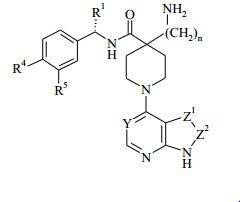 em que Y, Z1, Z2, R1, R4, R5 e n são como definidos mais acima.
em que Y, Z1, Z2, R1, R4, R5 e n são como definidos mais acima.
[00028] Em uma outra forma de realização da invenção, o composto da Fórmula (I) tem a configuração mostrada na Fórmula (IB) : (IB) em que Y, Z1, Z2, R1, R4, R5 e n são como definidos mais acima.
: (IB) em que Y, Z1, Z2, R1, R4, R5 e n são como definidos mais acima.
[00029] A referência aqui a um composto da Fórmula (I) deve ser entendida referir-se igualmente a um composto das Fórmulas (I), (IA) ou (IB).
[00030] Em uma forma de realização da invenção, é fornecido um composto da Fórmula (IA), ou um sal farmaceuticamente aceitável do Mesmo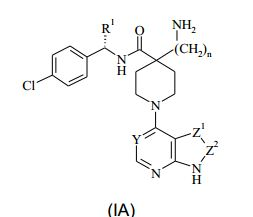 : (IA) em que: Y representa CH ou N; Z1-Z2 representam um grupo selecionado de CH=CH, N=CH e CH=N; n é 0, 1 ou 2; e R1 representa alquila C1-4, aminoalquila C1-4, hidróxi-alquila C1-4, -(CH2)pNHCOCH3, -(CH2)qNR2R3 ou cicloalquila C3-6; onde R2 representa hidrogênio ou alquila C1-3; R3 representa alquila C1-3; e p e q independentemente representam 2 ou 3.
: (IA) em que: Y representa CH ou N; Z1-Z2 representam um grupo selecionado de CH=CH, N=CH e CH=N; n é 0, 1 ou 2; e R1 representa alquila C1-4, aminoalquila C1-4, hidróxi-alquila C1-4, -(CH2)pNHCOCH3, -(CH2)qNR2R3 ou cicloalquila C3-6; onde R2 representa hidrogênio ou alquila C1-3; R3 representa alquila C1-3; e p e q independentemente representam 2 ou 3.
[00031] Em uma outra forma de realização da invenção, é portanto fornecido um composto da Fórmula (IA) ou um sal farmaceuticamente aceitável do mesmo: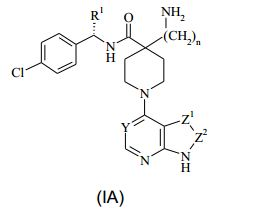 (IA) em que: Y representa CH ou N; Z1-Z2 representam um grupo selecionado de CH=CH, N=CH e CH=N; R1 representa alquila C1-4, aminoalquila C1-4 ou ciclo-alquila C3-6; e n é 0, 1 ou 2.
(IA) em que: Y representa CH ou N; Z1-Z2 representam um grupo selecionado de CH=CH, N=CH e CH=N; R1 representa alquila C1-4, aminoalquila C1-4 ou ciclo-alquila C3-6; e n é 0, 1 ou 2.
[00032] Em uma forma de realização da invenção, é fornecido um composto das Fórmulas (I), (IA) ou (IB) como definido mais acima, ou um sal farmaceuticamente aceitável do mesmo, em que o composto das Fórmulas (I), (IA) ou (IB) é outro que não (S)-4-amino-N-(1-(4- clorofenil)-3-hidroxipropil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)-piperidina- 4-carboxamida.
[00033] Um sal farmaceuticamente aceitável adequado de um composto da Fórmula (I) é, por exemplo, um sal de adição de ácido de um composto da invenção que é suficientemente básico, por exemplo, um sal de adição de ácido com, por exemplo, um ácido inorgânico ou orgânico, por exemplo ácido clorídrico, bromídrico, sulfúrico, fosfórico, trifluoroacético, cítrico ou maleico.
[00034] Será entendido que certos compostos da presente invenção podem existir nas formas solvatadas, por exemplo hidratadas, assim como não solvatadas. Deve ser entendido que a presente invenção abrange todas as tais formas solvatadas que são inibidores da atividade de PKB.
[00035] Os compostos da Fórmula (I) podem ser administrados na forma de um pró medicamento que é decomposto no corpo humano ou animal para dar um composto da Fórmula (I). Os exemplos de pró medicamentos incluem ésteres hidrolisáveis in vivo de um composto da Fórmula (I). várias formas de pró medicamentos são conhecidos na técnica. Para exemplos de tais derivados de pró medicamento, ver: a) Design of Prodrugs, editado por H. Bundgaard, (Elsevier, 1985) e Methods in Enzymology, Vol. 42, p. 309-396, editado por K. Widder, et al. (Academic Press, 1985); b) A Textbook of Drug Design and Development, editado por Krogsgaard-Larsen e H. Bundgaard, Capítulo 5 “Design and Application of Prodrugs”, por H. Bundgaard p. 113-191 (1991); c) H. Bundgaard, Advanced Drug Delivery Reviews, 8, 1-38 (1992); d) H. Bundgaard, et al., Journal of Pharmaceutical Sciences, 77, 285 (1988); e
[00036] N. Kakeya, et al., Chem Pharm Bull, 32, 692 (1984).
[00037] De acordo com um outro aspecto da invenção é fornecida uma composição farmacêutica, que compreende um composto da Fórmula (I), ou um sal farmaceuticamente aceitável do mesmo, como definido mais acima em associação com um diluente ou carreador farmaceuticamente aceitáveis.
[00038] As composições da invenção podem estar em uma forma adequada para o uso oral (por exemplo como tabletes, comprimidos, cápsulas douras ou moles, suspensões aquosas ou oleosas, emulsões, pós ou grânulos dispersáveis, xaropes ou elixires), para o uso tópico (por exemplo como cremes, unguentos, géis, ou soluções ou suspensões aquosas ou oleosas), para administração pela inalação (por exemplo como um pó finamente dividido ou um aerossol líquido), para administração pela insuflação (por exemplo como um pó finamente dividido) ou para a administração parenteral (por exemplo como uma solução aquosa ou oleosa estéril para dosagem intravenosa, subcutânea, intramuscular ou intramuscular ou como um supositório para a dosagem retal).
[00039] As composições da invenção podem ser obtidas pelos procedimentos convencionais usando excipientes farmacêuticos convencionais, bem conhecida na técnica. Assim, as composições intencionadas para o uso oral podem conter, por exemplo, um ou mais agentes de coloração, adoçante, flavorizante e/ou conservante.
[00040] O composto da Fórmula (I) será normalmente administrado a um animal de sangue quente em uma dose unitária dentro da faixa de 5 a 5000 mg/m2 de área corporal do animal, isto é aproximadamente de 0,1 a 100 mg/kg, e isso normalmente fornece uma dose terapeuticamente eficaz. Uma forma de dose unitária tal como um tablete ou cápsula usualmente conterá, por exemplo 1 a 250 mg do gradiente ativo. Preferivelmente uma dose diária na faixa de 1 a 50 mg/kg é utilizada, por exemplo de 4 a 7 mg/kg duas vezes ao dia. Entretanto, a dose diária necessariamente será variada dependendo do hospedeiro tratado, a via particular de administração, e a severidade da doença a ser tratada. Consequentemente o médico que está tratando qualquer paciente particular pode determinar a dosagem ótima.
[00041] Por exemplo, uma composição farmacêutica da presente invenção adequada para a administração oral pode compreender de 1 a 200 mg/ ml de um composto da Fórmula (I), ou um sal farmaceuticamente aceitável do mesmo, (tal como (S)-4-amino-N-(1- (4-clorofenil)-3-hidroxipropil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxamida) em 0,5 %de hidroxipropilmetilcelulose (HPMC).
[00042] No contexto do presente relatório descritivo, o termo “terapia” também inclui “profilaxia” a menos que existam indicações específicas ao contrário. Os termos “terapêutico” e “terapeuticamente” devem ser interpretados consequentemente.
[00043] Como aqui usado, o termo “tratamento” é intencionado a ter seus significados normal de todo dia de lidar com um doença de modo a aliviar total ou parcialmente um, alguns ou todos dos seus sintomas, ou corrigir ou compensar quanto à patologia subjacente.
[00044] Como aqui usado, o termo “profilaxia” é intencionado a ter seu significado normal de todo dia e inclui profilaxia primária para prevenir o desenvolvimento da doença e profilaxia secundária por meio da qual a doença já desenvolveu e o paciente é temporária ou permanentemente protegido contra exacerbação ou piora da doença ou o desenvolvimento de novos sintomas associados com a doença.
[00045] Como um resultado da sua atividade inibidora da PKB, os compostos da Fórmula (I) da presente invenção são esperados serem úteis no tratamento de doenças ou condições médicas mediadas sozinhas ou em parte pela atividade de PKB, por exemplo câncer. Os tipos de cânceres que podem ser suscetíveis ao tratamento usando os compostos da Fórmula (I) da presente invenção incluem, mas não são limitados a, câncer ovariano, câncer cervical, câncer colorretal, câncer de mama, câncer pancreático, glioma, glioblastoma, melanoma, câncer prostático, leucemia, linfoma, linfoma de Não Hodgkins, câncer gástrico, câncer pulmonar, câncer hepatocelular, câncer gástrico, tumor estrômico gastrointestinal (GIST), glioma, câncer tiroidal, câncer do duto biliar, câncer endometrial, câncer renal, linfoma de célula grande anaplástica, leucemia mielóide aguda (A ML), mieloma múltiplo, melanoma e mesotelioma. Câncer de mama, e mais especificamente câncer de mama luminal, podem ser particularmente suscetíveis para o tratamento usando os compostos da presente invenção.
[00046] É considerado que para os métodos de tratamento de câncer aqui mencionados, o composto da Fórmula (I) será administrado a um mamífero, mais particularmente um ser humano. Similarmente, para o uso de um composto da Fórmula (I) para o tratamento do câncer aqui mencionado, é considerado que o composto da Fórmula (I) será administrado a um mamífero, mais particularmente a um ser humano.
[00047] De acordo com um outro aspecto da invenção, é portanto fornecido um composto da Fórmula (I) como definido mais acima, ou um sal farmaceuticamente aceitável do mesmo, para o uso como um medicamento.
[00048] De acordo com um outro aspecto da invenção, é fornecido um composto da Fórmula (I) como definido mais acima, ou um sal farmaceuticamente aceitável do mesmo para o uso no tratamento de uma doença mediada através de PKB. Em uma forma de realização da invenção, a dita doença mediada através de PKB é câncer. Em uma outra forma de realização da invenção, o dito câncer é selecionado de câncer ovariano, câncer cervical, câncer colorretal, câncer de mama, câncer pancreático, glioma, glioblastoma, melanoma, câncer prostático, leucemia, linfoma, linfoma de Não Hodgkins, câncer gástrico, câncer pulmonar, câncer hepatocelular, câncer gástrico, tumor estrômico gastrointestinal (GIST), glioma, câncer tiroidal, câncer do duto biliar, câncer endometrial, câncer renal, linfoma de célula grande anaplástica, leucemia mielóide aguda (AML), mieloma múltiplo, melanoma e mesotelioma. Em uma forma de realização da invenção, o dito câncer é selecionado de câncer de mama, linfoma de Não Hodgkins, o dito câncer pancreático, câncer hepato-celular, câncer gástrico, câncer prostático e câncer pulmonar. Em uma forma de realização particular, o dito câncer é câncer de mama, mais particularmente câncer de mama luminal.
[00049] De acordo com um outro aspecto da invenção, é fornecido o uso de um composto da Fórmula (I) como definido mais acima, ou um sal farmaceuticamente aceitável do mesmo para a preparação de um medicamento para o tratamento de uma doença mediada através de PKB. Em uma forma de realização da invenção, a dita doença mediada através de PKB é câncer. Em uma outra forma de realização da invenção, o dito câncer é selecionado de câncer ovariano, câncer cervical, câncer colorretal, câncer de mama, câncer pancreático, glioma, glioblastoma, melanoma, câncer prostático, leucemia, linfoma, linfoma de Não Hodgkins, câncer gástrico, câncer pulmonar, câncer hepatocelular, câncer gástrico, tumor estrômico gastrointestinal (GIST), glioma, câncer tiroidal, câncer do duto biliar, câncer endometrial, câncer renal, linfoma de célula grande anaplástica, leucemia mielóide aguda (A ML), mieloma múltiplo, melanoma e mesotelioma. Em uma forma de realização da invenção, o dito câncer é selecionado de câncer de mama, linfoma de Não Hodgkins, câncer pancreático, câncer hepatocelular, câncer gástrico, câncer prostático e câncer pulmonar. Em uma forma de realização particular, o dito câncer é câncer de mama, mais particularmente câncer de mama luminal.
[00050] De acordo com um outro aspecto da invenção, é fornecido o uso de um composto da Fórmula (I) como definido mais acima, ou um sal farmaceuticamente aceitável do mesmo, para a preparação de um medicamento para o tratamento de câncer. Em uma forma de realização da invenção, o dito câncer é selecionado de câncer ovariano, câncer cervical, câncer colorretal, câncer de mama, câncer pancreático, glioma, glioblastoma, melanoma, câncer prostático, leucemia, linfoma, linfoma de Não Hodgkins, câncer gástrico, câncer pulmonar, câncer hepato-celular, câncer gástrico, tumor estrômico gastrointestinal (GIST), glioma, câncer tiroidal, câncer do duto biliar, câncer endometrial, câncer renal, linfoma de célula grande anaplástica, leucemia mielóide aguda (A ML), mieloma múltiplo, melanoma e mesotelioma. Em uma forma de realização da invenção, o dito câncer é selecionado de câncer de mama, linfoma de Não Hodgkins, câncer pancreático, câncer hepatocelular, câncer gástrico, câncer prostático e câncer pulmonar. Em uma forma de realização particular, o dito câncer é câncer de mama, mais particularmente câncer de mama luminal.
[00051] De acordo com um outro aspecto da invenção, é fornecido um método de usar um composto da Fórmula (I) como definido mais acima, ou um sal farmaceuticamente aceitável do mesmo, para o tratamento de câncer. Em uma forma de realização da invenção, o dito câncer é selecionado de câncer ovariano, câncer cervical, câncer colorretal, câncer de mama, câncer pancreático, glioma, glioblastoma, melanoma, câncer prostático, leucemia, linfoma, linfoma de Não Hodgkins, câncer gástrico, câncer pulmonar, câncer hepatocelular, câncer gástrico, tumor estrômico gastrointestinal (GIST), glioma, câncer tiroidal, câncer do duto biliar, câncer endometrial, câncer renal, linfoma de célula grande anaplástica, leucemia mielóide aguda (A ML), mieloma múltiplo, melanoma e mesotelioma. Em uma forma de realização da invenção, o dito câncer é selecionado de câncer de mama, linfoma de Não Hodgkins, câncer pancreático, câncer hepatocelular, câncer gástrico, câncer prostático e câncer pulmonar. Em uma forma de realização particular, o dito câncer é câncer de mama, mais particularmente câncer de mama luminal.
[00052] De acordo com um outro aspecto da invenção, é fornecido um método de tratar um ser humano que sofre de uma doença na qual a inibição de PKB é benéfico, que compreendem as etapas de administrar a uma pessoa em necessidade deste de uma quantidade terapeuticamente eficaz de um composto da Fórmula (I) como definido mais acima, ou um sal farmaceuticamente aceitável do mesmo. Em uma forma de realização da invenção, a doença na qual a inibição de PKB é benéfico é câncer. Em uma outra forma de realização da invenção, o dito câncer é selecionado de câncer ovariano, câncer cervical, câncer colorretal, câncer de mama, câncer pancreático, glioma, glioblastoma, melanoma, câncer prostático, leucemia, linfoma, linfoma de Não Hodgkins, câncer gástrico, câncer pulmonar, câncer hepatocelular, câncer gástrico, tumor estrômico gastrointestinal (GIST), glioma, câncer tiroidal, câncer do duto biliar, câncer endometrial, câncer renal, linfoma de célula grande anaplástica, leucemia mielóide aguda (A ML), mieloma múltiplo, melanoma e mesotelioma. Em uma forma de realização da invenção, o dito câncer é selecionado de câncer de mama, linfoma de Não Hodgkins, câncer pancreático, câncer hepatocelular, câncer gástrico, câncer prostático e câncer pulmonar. Em uma forma de realização particular, o dito câncer é câncer de mama, mais particularmente câncer de mama luminal.
[00053] O tratamento de câncer definido mais acima pode ser aplicado como uma terapia única ou pode envolver, além do composto da invenção, cirurgia ou radioterapia ou quimioterapia convencionais. Tal quimioterapia pode incluir uma ou mais das seguintes categorias de agentes anti-tumor:- (i) outros medicamentos e combinações de antiproliferativo/ antineoplástico destes, como usados na medicina oncológica, tais como agentes de alquilação (por exemplo cis-platina, oxaliplatina, carboplatina, ciclolofosfamida, mostarda nitrogenada, melfalano, clorambucila, bussulfano, temozolamida e nitrosouréias); antimetabólitos (por exemplo gencitabina e antifoliatos tais como fluoropirimidinas como 5-fluorouracila e tegafur, raltitrexed, metotrexato, citosina arabinosida, e hidroxiuréia); antibióticos antitumorais (por exemplo antraciclolinas como adriamicina, bleomicina, doxorrubicina, daunomicina, epirrubicina, idarrubicina, mitomicina-C, dactinomicina e mitramicina); agentes antimitóticos (por exemplo alcalóides vinca como vincristina, vimblastina, vindesina e vinorrelbina e taxóides como taxol e taxotere e inibidores da poloquinase); e inibidores da topoisomerase (por exemplo epipodofilotoxinas como etoposida e teniposida, ansacrina, topotecano e camptotecina); (ii) agentes citostáticos tais como antiestrogênios (por exemplo tamoxifeno, fulvestrant, toremifeno, raloxifeno, droloxifeno e iodoxifeno), antiandrogênios (por exemplo bicalutamida, flutamida, nilutamida e acetato de ciproterona), antagonistas de LHRH ou agonistas de LHRH (por exemplo goserelina, leuprorelina e buserelina), progestogênios (por exemplo acetato de megestrol), inibidores de aromatase (por exemplo como anastrozol, letrozol, vorazol e exemestano) e inibidores de 5α-redutase tal como finasterida; (iii) agentes anti-invasão (por exemplo inibidores da família c-Src quinase como 4-(6-cloro-2,3-metilenodioxianilino)-7-[2-(4-metil- piperazin-1-il)etóxi]-5-tetraidropiran-4-iloxiquinazolina (AZD0530; Pedido de Patente Internacional WO 01/94341) e N-(2-cloro-6-metil- fenil)-2-{6-[4-(2-hidroxietil)piperazin-1-il]-2-metilpirimidin-4-ilamino}- tiazol-5-carboxamida (dasatinib, BMS-354825; J. Med. Chem., 2004, 47, 6658-6661), e inibidores de metaloproteinase como inibidores de marimastato, da função receptora do ativador de da uroquinase de plasminogênio ou anticorpos para Heparanase); (iv) inibidores da função do fator de crescimento: por exemplo tais inibidores incluem anticorpos do fator de crescimento e anticorpos do receptor do fator de crescimento (por exemplo o anticorpo anti-erbB2 trastuzumab [Herceptin®], o anticorpo anti-EGFR panitumumab, o anticorpo anti-erbB1 cetuximab [Erbitux, C225] e qualquer fator de crescimento ou anticorpos do receptor do fator de crescimento divulgado por Stern et al. Critical reviews in oncology/ haematology, 2005, Vol. 54, pp 11-29); tais inibidores também incluem inibidores da tirosina quinase, por exemplo inibidores da família do fator de crescimento epidérmico (por exemplo inibidores da tirosina quinase da família EGFR tais como N-(3-cloro-4-fluorofenil)-7-metóxi- 6-(3-morfolinopropóxi)quinazolin-4-amina (gefitinib, ZD1839), N-(3- etinil -fenil)-6,7-bis(2-metoxietóxi)quinazolin-4-amina (erlotinib, OSI-774) e 6-acrilamido-N-(3-cloro-4-fluorofenil)-7-(3-morfolino- propóxi)-quinazolin-4-amina (CI 1033), inibidores da tirosina quinase de erbB2 tais como lapatinib, inibidores da família do fator de crescimento de hepatócito, inibidores da família do fator de crescimento derivado de plaqueta tais como imatinib, inibidores da serina/treonina quinases (por exemplo inibidores da sinalização de Ras/Raf tais como inibidores da farnesil transferase, por exemplo sorafenib (BAY 43-9006)), inibidores da sinalização celular através de MEK e/ou AKT quinases, inibidores da família do fator de crescimento de hepatócito, inibidores de c-kit, inibidores de ab1 quinase, inibidores do receptor da IGF (fator de crescimento equivalente a insulina) quinase; inibidores da aurora quinase (por exemplo AZD1152, PH739358, VX-680, MLN8054, R763, MP235, MP529, VX-528 E AX39459) e inibidores da quinase dependentes de ciclolina tais como inibidores de CDK2 e/ou CDK4; (v) agentes antiangiogênicos tais como aqueles que inibem os efeitos do fator de crescimento endotelial vascular, [por exemplo o anticorpo anti-fator de crescimento celular endotelial vascular bevacizumab (Avastin®) e inibidores do receptor da tirosina quinase de VEGF tais como 4-(4-bromo-2-fluoroanilino)-6-metóxi-7-(1-metil- piperidin-4-ilmetóxi)quinazolina (ZD6474; Exemplo 2 dentro da WO 01/32651), 4-(4-fluoro-2-metilindol-5-ilóxi)-6-metóxi-7-(3-pirrolidin-1- ilpropóxi)quinazolina (AZD2171; Exemplo 240 dentro da WO 00/47212), vatalanib (PTK787; WO 98/35985) e SU11248 (sunitinib; WO 01/60814), compostos tais como aqueles divulgados nos Pedidos de Patente Internacional WO 97/22596, WO 97/30035, WO 97/32856 e WO 98/13354 e compostos que trabalham por outros mecanismos (por exemplo linomida, inibidores da função de integrina αvβ3 e angiostatina)]; (vi) agentes de dano vascular tais como Combretastatina A4 e compostos divulgados nos Pedidos de Patente Internacional WO 99/02166, WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 e WO 02/08213; (vii) terapias de anti-sentido, por exemplo aquelas que são direcionadas ao alvo listado acima, tais como ISIS 2503, um anti- sentido anti-ras; (viii) métodos de terapia de gene, incluindo por exemplo métodos para repor agentes aberrantes tais como p53 aberrante ou BRCA1 ou BRCA2 aberrantes, métodos GDEPT (terapia de pró- medicamento de enzima direcionado a gene) tais como aqueles usando citosina desaminase, timidina quinase ou uma enzima de nitrorredutase bacteriana e métodos para aumentar a tolerância do paciente à quimioterapia ou radioterapia tal como terapia de gene de resistência a medicamento múltiplo; e (ix) métodos de imunoterapia, incluindo por exemplo métodos ex vivo e in vivo para aumentar a imunogenicidade de células de tumor do paciente, tal como transfecção com citocinas tais como interleucina 2, interleucina 4 ou fator estimulador de colônia de granulócito-macrófago, métodos para diminuir a anergia de célula T, métodos usando células imunes transfectadas tais como células dendríticas transfectadas com citocina, métodos usando linhagens de célula de tumor transfectadas com citocina e métodos usando anticorpos anti-idiotípicos.
[00054] Um composto da invenção, ou um sal deste, pode ser preparado por qualquer processo conhecido ser aplicável para a preparação de tais compostos ou compostos estruturalmente relacionados. Os grupos funcionais podem ser protegidos e desprotegidos usando métodos convencionais. Para os exemplos de grupos de proteção tais como os grupos de proteção de amino e ácido carboxílico (assim como meios de formação e eventual desproteção), ver T. W. Greene e P. G. M. Wuts, “Protective Groups in Organic Synthesis”, Segunda Edição, John Wiley & Sons, Nova Iorque, 1991.
[00055] Certos processos para a síntese dos compostos da Fórmula (I) são fornecidos como uma outra característica da invenção. Assim, de acordo com um outro aspecto da invenção é fornecido um processo para a preparação de um composto da Fórmula (I) ou um sal farmaceuticamente aceitável do mesmo, que compreende um processo (a), (b), (c) ou (d) (em que as variáveis são como definidos mais acima para o composto da Fórmula (I) a menos que de outro modo definido): (a) reação de um ácido da Fórmula (II) com uma amina da Fórmula (III) : em que P1 representa um grupo de proteção adequado, por exemplo terc-butoxicarbonila; (b) reação de uma carboxamida da Fórmula (IV) com um heterociclo bicíclico da Fórmula (V)
: em que P1 representa um grupo de proteção adequado, por exemplo terc-butoxicarbonila; (b) reação de uma carboxamida da Fórmula (IV) com um heterociclo bicíclico da Fórmula (V)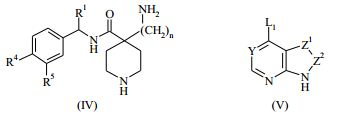 : em que L1 representa um grupo de partida adequado, por exemplo cloro; (c) quando n é 1, a hidrogenação de um composto da Fórmula (VI): ou
: em que L1 representa um grupo de partida adequado, por exemplo cloro; (c) quando n é 1, a hidrogenação de um composto da Fórmula (VI): ou (VI) (d) quando R1 representa aminometila, a hidrogenação de um composto da Fórmula (VII)
(VI) (d) quando R1 representa aminometila, a hidrogenação de um composto da Fórmula (VII)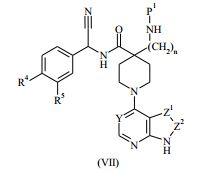 : em que P1 representa um grupo de proteção adequado, por exemplo terc-butoxicarbonila; e depois disso, se necessário: (i) converter um composto da Fórmula (I) em um outro composto da Fórmula (I); (ii) remover quaisquer grupos de proteção; e/ou (iii) formar um sal farmaceuticamente aceitável do mesmo.
: em que P1 representa um grupo de proteção adequado, por exemplo terc-butoxicarbonila; e depois disso, se necessário: (i) converter um composto da Fórmula (I) em um outro composto da Fórmula (I); (ii) remover quaisquer grupos de proteção; e/ou (iii) formar um sal farmaceuticamente aceitável do mesmo.
[00056] Os exemplos de conversões de um composto da Fórmula (I) em um outro composto da Fórmula (I), são bem conhecidos por aqueles habilitados na técnica, e incluem interconversões do grupo funcional tais como hidrólise, hidrogenação, hidrogenólise, oxidação ou redução e/ou outra funcionalização pelas reações padrão tais como ligação catalisada por amida ou metal, ou reações de deslocamento nucleofílico.
[00057] Condições de reação específica para processos (a), (b), (c) e (d) acima são como segue:
[00058] Processo (a) - ácidos da Fórmula (II) e aminas da Fórmula (III) podem ser reagidos juntos na presença de um reagente de ligação adequado, por exemplo O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio hexa-fluorofosfato (HATU), e uma base adequada, por exemplo N,N’-diiso-propiletilamina (DIPEA), em um solvente adequado, por exemplo dimetilacetamida (DMA), e em uma temperatura adequada, por exemplo de 50 a 70° C, mais adequadamente de cerca de 60° C;
[00059] Processo (b) - carboxamidas da Fórmula (IV) e heterociclos da Fórmula (V) podem ser reagidos juntos na presença de uma base adequada, por exemplo N,N’-diisopropiletilamina (DIPEA), em um solvente adequado, por exemplo butan-1-ol, e em uma temperatura adequada, por exemplo 50 a 70° C, mais adequadamente de cerca de 60° C;
[00060] Processos (c) e (d) - compostos das Fórmulas (VI) ou (VII) dissolvidos em um solvente adequado, por exemplo etanol, pode ser hidrogenado sob uma atmosfera de hidrogênio na presença de um catalisador adequado, por exemplo níquel de Raney®, e uma base adequada, por exemplo hidróxido de amônio.
[00061] Os compostos da Fórmula (II) podem ser preparados de acordo com o Esquema 1 : Esquema 1
: Esquema 1
[00062] em que P1 é um grupo de proteção adequado, por exemplo terc-butoxicarbonila, L1 é um grupo de partida adequado, por exemplo cloro, e todas as outras variáveis são como definidas mais acima.
[00063] Os compostos da Fórmula (IV) podem ser preparados de acordo com o Esquema 2 : Esquema 2
: Esquema 2
[00064] em que P1 e P2 são grupos de proteção adequados, por exemplo terc-butoxicarbonila, e todas as outras variáveis são como definidas mais acima.
[00065] Os compostos da Fórmula (VI) podem ser preparados de acordo com o Esquema 3 : Esquema 3
: Esquema 3
[00066] em que P1 é um grupo de proteção adequado, por exemplo terc-butoxicarbonila, e todas as outras variáveis são como definidas mais acima.
[00067] O compostos da Fórmula (VII) podem ser preparados de acordo com o Esquema 4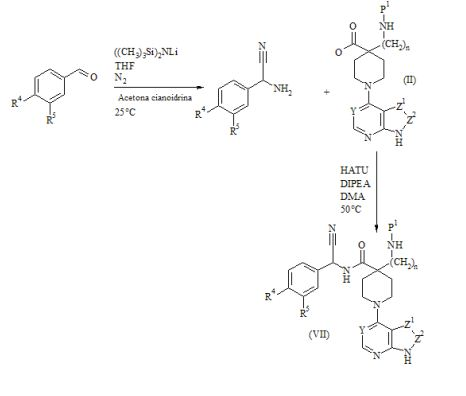 : Esquema 4
: Esquema 4
[00068] em que P1 representa um grupo de proteção adequado, por exemplo terc-butoxicarbonila, e todas as outras variáveis são como definidas mais acima.
[00069] Os compostos das Fórmulas (III), (V), (VIII) e (IX) são comercialmente disponíveis, conhecidos na literatura, preparados pelos processos padrão conhecidos na técnica, ou podem ser preparados de acordo com os aqui descritos.
[00070] Os seguintes exemplos são para propósito de ilustração e não são intencionados a limitar o escopo deste pedido. Cada composto exemplificado representa um aspecto particular e independente da invenção. Todos os materiais de partida são compostos comercialmente disponíveis, ou são conhecidos na literatura, ou são preparados pelos processos padrão conhecidos na técnica.
[00071] No geral, com respeito aos seguintes exemplos: (i) as temperaturas são dadas em graus Celsius (°C); as operações foram realizadas na temperatura da sala ou ambiente, isto é, em uma temperatura na faixa de 18 a 25° C; (ii) as soluções orgânicas foram secadas em sulfato de magnésio anidro ou sulfato de sódio anidro; evaporação do solvente foi realizado usando um evaporador rotativo sob pressão reduzida (600 a 4000 Pascais; de 4,5 a 30 mmHg) com um temperatura de banho de até 60° C; (iii) cromatografia significa cromatografia por vaporização instantânea em sílica gel; cromatografia de camada fina (TLC) foi realizada em placas de sílica gel; (iv) no general, o curso das reações foi seguido pela TLC e / ou LC-MS analítica, e os tempos de reação quando dados são apenas para ilustração. (v) os produtos finais têm espectros de ressonância magnética nuclear de próton (RMN) e/ou dados espectrais de massa satisfatórios; (vi) os rendimentos são dados apenas para ilustração e não são necessariamente aqueles que podem ser obtidos pelo desenvolvimento do processo diligente; as preparações foram repetidas se mais material foi requerido; (vii) quando dados, os dados de RMN estão na forma de valores delta para prótons de diagnóstico principais, dados em partes por milhão (ppm) relativo ao tetra metilsilano (TMS) como um padrão interno, determinado a 500 MHz usando sulfóxido de dimetila perdeutério (DMSO-d6) como solvente a menos que de outro modo indicado; as seguintes abreviações foram usadas: s, singleto; d, dubleto; t, tripleto; q, quarteto; m, multipleto; bs, singleto amplo; (viii) os símbolos químicos têm seus significados usuais; unidades SI e símbolos são usados; (ix) Espectro de massa (MS) e os dados de LC-MS foram gerados em um sistema de LC-MS onde o componente HPLC no geral compreende um equipamento Agilent 1100, Waters Alliance HT (2790 & 2795) ou uma bomba HP1100 e Série de Diodo com autoamostrador CTC e foi conduzido em uma coluna Phenomenex Gemini C18 5 μm, 50 x 2 mm (ou similar) eluindo com eluente ácido (por exemplo, usando um gradiente entre 0 e 95 % de água / acetonitrila com 5 % de um ácido fórmico a 1 % em mistura 50:50 água:acetonitrila (v/v)), ou eluente básico (por exemplo, usando um gradiente entre 0 e 95 % de água / acetonitrila com 5 % de uma mistura a 0,1 % de Amônia 880 em acetonitrila); e o componente MS compreendeu no geral de um espectrômetro de massa Waters ZQ que varre em uma faixa de massa apropriada. Cromatogramas para Eletropulverização de Intensidade de Pico Base (ESI) positiva e negativa, e Cromatograma de Absorção de UV Total de 220 a 300 nm, são gerados e valores para m/z são dados; no geral, apenas os íons que indicam a massa precursora são relatados e a menos que de outro modo estabelecido o valor quotado é o (M + H)+ para o modo de íon positivo e (M - H)- para o modo de íon negativo; (x) a menos que de outro modo estabelecido os compostos contendo um átomo de carbono e/ou enxofre assimetricamente substituído não foi resolvido; (xi) quaisquer reações em microonda foram realizados em um microonda Biotage Optimizer EXP, ou um CEM Explorer; (xii) a cromatografia líquida de alto desempenho preparativa (HPLC) foi realizada em um instrumento Gilson usando as seguintes condições:
[00072] Coluna: sílica de fase reversa C18, por exemplo, Waters ‘Xbridge’, 5 μm sílica, 19 x 100 mm, ou 30 x 100 mm, usando misturas decrescentemente polares como eluente (diminuindo a razão do solvente A para o Solvente B) Solvente A: Água com 1 % de hidróxido de amônio Solvente B: Acetonitrila Vazão: 28 ml / min ou 61 ml / min Gradiente: Feito de encomenda para se adequar a cada composto - no geral 7 a 10 min de duração Comprimento de onda: 254 nm Abreviações Boc Terc-butoxicarbonila CAS® Chemical Abstracts Service DCM diclorometano DIPEA N,N’-diisopropiletilamina DEA dietilamina DMA dimetilacetamida DMF dimetilformamida HATU hexafluorofosfato de O-(7-Azabenzotriazol-1- il)-N,N,N’,N’ -tetrametilurônio LCMS espectroscopia de massa de cromatografia líquida LDA diisopropilamida de lítio MPLC cromatografia líquida de pressão média NMP N-metilpirrolidinona OBD densidade de leito ótima PTFE politetrafluoroetileno SCX troca catiônica forte SFC cromatografia de fluxo superficial TBME éter metílico de t-butila TEA trietilamina TFA ácido trifluoroacético THF tetraidrofurano Exemplo 1: 4-amino-N-(1 -(4-clorofenil)etil)-1-(7H-pirrolo[2,3-d] pirimidin-4-il)piperidina-4-carboxamida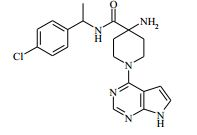
[00073] Ácido 4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxílico (Intermediário 1) (362 mg), 1-(4- clorofenil)etanamina (172 mg), N-(3-dimetilaminopropil)-3-etilcarbo- diimida (231 mg) e 1-hidroxibenzotriazol (163 mg) foram agitados juntos em DMF (2 ml) sob nitrogênio por 16 horas. A mistura de reação foi dividida entre EtOAc (20 ml) e salmoura (4 x 20 ml). As formas orgânicas combinadas, secadas em MgSO4 e evaporadas a vácuo. O sólido branco resultante foi dissolvido em 1,4-dioxano (5 ml) e uma solução 4 M de HCl em 1,4-dioxano (5 ml) foi adicionada. A mistura resultante foi agitada por 16 horas, depois diluída com éter dietílico (50 ml). O produto bruto foi isolado pela filtração como o sal de HCl que foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 7 M e as frações puras foram evaporadas até a secura. Este material foi purificado pela LCMS preparativa usando misturas decrescentemente polares de água (contendo 1 % de NH3) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura para produzir 4-amino-N-(1-(4-clorofenil)etil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida como um sólido branco (168 mg, 42 %).
[00074] 1H RMN (d6-dmso, 400 MHz) 1,33 - 1,49 (m, 5H), 1,84 - 2,04 (m, 2H), 2,12 - 2,22 (br s, 2H), 3,54 (t, 2H), 4,39 (t, 2H), 4,81 - 4,92 (m, 1H), 6,55 - 6,59 (m, 1H), 7,13 - 7,18 (m, 1H), 7,31 - 7,39 (m, 4H), 8,12 (s, 1H), 8,30 (d, 1H), 11,62 (s, 1H). MS m/e MH+ 399. Exemplo 2: (S)-4-amino-N-(1 -(4-clorofenil)etil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida
[00075] Hexafluorofosfato de O-(7-Azabenzotriazol-1-il)-N,N,N’,N’ - tetrametilurônio (0,418 g) foi adicionado em uma porção ao ácido 4- (terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina - 4-carboxílico (Intermediário 1) (0,361 g), (S)-1-(4-clorofenil)etanamina (0,140 ml) e DIPEA (0,524 ml) em DMA (10 ml) a 25° C sob nitrogênio. A solução resultante foi agitada a 60° C por 4 horas. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 7 M e as frações puras foram evaporadas até a secura. Este material bruto foi depois tratado com uma solução a 20 % de TFA em DCM (10 ml) e agitados na temperatura ambiente. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 7 M e as frações puras foram evaporadas até a secura. Este material foi purificado pela LCMS preparativa usando misturas decrescentemente polares de água (contendo 1 % de NH3) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura para produzir (S)-4-amino-N-(1-(4-clorofenil)etil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)- piperidina-4-carboxamida como um sólido branco (0,281 g, 70,4 %).
[00076] 1H RMN (400,13 MHz, DMSO-d6) δ 1,37 (3H, d), 1,42 - 1,45 (2H, m), 1,88 - 2,01 (2H, m), 2,27 (2H, s), 3,49 - 3,59 (2H, m), 4,34 - 4,44 (2H, m), 4,83 - 4,90 (1H, m), 6,57 - 6,58 (1H, m), 7,14 - 7,16 (1H, m), 7,32 - 7,38 (4H, m), 8,12 (1H, s), 8,30 (1H, d), 11,62 (1H, s). MS m/e MH+ 399. Exemplo 3: 4-amino-N-(1-(4-clorofenil)propil)-1-(7H-pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida
[00077] Hexafluorofosfato de O-(7-Azabenzotriazol-1-il)-N,N,N’,N’ - tetrametilurônio (0,209 g) foi adicionado em uma porção ao ácido 4- (terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina - 4-carboxílico (Intermediário 1) (0,181 g), 1-(4-clorofenil)propan-1- amina (0,085 g) e DIPEA (0,262 ml) em DMA (10 ml) a 25° C sob nitrogênio. A solução resultante foi agitada a 60° C por 4 horas. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 7 N e as frações puras foram evaporadas até a secura. Este material bruto foi depois tratado com uma solução a 20 % de TFA em DCM (10 ml) e agitados na temperatura ambiente por 2 horas. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 7 N e as frações puras foram evaporadas até a secura. Este material foi purificado pela LCMS preparativa usando misturas decrescentemente polares de água (contendo 1 % de amônia) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura para produzir 4-amino-N-(1-(4- clorofenil)-propil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxamida como um sólido branco (0,138 g, 66,8 %).
[00078] 1H RMN (400,13 MHz, DMSO-d6) δ 0,87 (3H, t), 1,42 - 1,55 (2H, m), 1,72 - 1,79 (2H, m), 1,91 - 2,05 (2H, m), 2,21 (2H, s), 3,54 - 3,62 (2H, m), 4,38 - 4,45 (2H, m), 4,65 - 4,70 (1H, m), 6,61 (1H, dd), 7,18 (1H, dd), 7,32 - 7,37 (4H, m), 8,31 (1H, d), 8,12 (1H, s). MS m/e MH+ 413. Exemplos 3A e 3B: (S)-4-amino-N-(1-(4-clorofenil)propil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida e (R)-4-amino-N- (1 -(4-clorofenil) propil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxamida
[00079] 4-amino-N-(1-(4-clorofenil)propil)-1-(7H-pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida racêmica (Exemplo 3) foi purificada pela HPLC quiral preparativa. As frações contendo o composto desejado foram evaporadas até a secura para produzir Isômero 1 (primeiro para eluir, 41 mg) como um sólido branco, e o Isômero 2 (segundo para eluir, 41 mg) como um sólido branco. Os dados analíticos foram idênticos à amostra original. A análise de HPLC analítica quiral (usando uma coluna Chiralpak AS 20 μm (250 mm x 4,6 mm), com uma mistura eluente de iso-hexano/(EtOH/MeOH 50/50)/TEA 90/10/0,1, 1 ml/min a 25° C, injetando 10 μl de uma solução a 1 mg/ ml em EtOH) mostrou cada enanciômero como sendo distinto um do outro e enanciomericamente puro (e.e. = 100 %). Exemplo 4: 4-amino-N-((4-clorofenil)(ciclolopropil)metil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida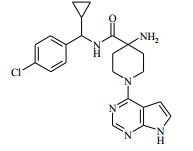
[00080] Hexafluorofosfato de O-(7-Azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (0,228 g) foi adicionado ao ácido 4-(terc-butóxi- carbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxílico (Intermediário 1) (0,181 g), (4-clorofenil)(ciclolopropil)-metanamina (Intermediário 3) (0,091 g) e DIPEA (0,261 ml) em DMA (5 ml) a 25° C. A solução resultante foi agitada a 50° C por 1 hora. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 7 N e as frações puras foram evaporadas até a secura. Este material bruto foi colocado em suspensão em diclorometano (25 ml), e TFA (5 ml) foi adicionado. A mistura de reação foi agitada por 1 hora, depois o produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 7 N e as frações puras foram evaporadas até a secura para produzir o material bruto como um sólido branco. Este triturado sob metanol frio para dar o produto puro, 4-amino-N-((4- clorofenil)(ciclolopropil)metil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)- piperidina-4-carboxamida como um sólido branco (0,167 g, 79 %).
[00081] 1H RMN (400,13 MHz, DMSO-d6) δ 0,27 - 0,37 (2H, m), 0,48 - 0,52 (2H, m), 1,18 - 1,24 (1H, m), 1,40 - 1,48 (2H, m), 1,88 - 2,02 (2H, m), 2,20 (2H, s), 3,50 - 3,59 (2H, m), 4,15 (1H, t), 4,36 - 4,42 (2H, m), 6,57 - 6,58 (1H, m), 7,14 - 7,16 (1H, m), 7,35 - 7,40 (4H, m), 8,12 (1H, s), 8,47 (1H, d), 11,62 (1H, s). MS m/e MH+ 425. Exemplo 5: 4-amino-N-(2-amino-1-(4-clorofenil)etil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida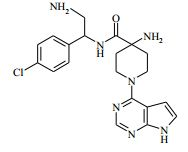
[00082] Ácido trifluoroacético (3 ml) foi adicionado ao 4-(2-amino-1- (4-clorofenil)etilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)-piperidin-4- ilcarbamato de terc-butila (Intermediário 6) (0,514 g) em DCM (100 ml) a 25° C. A solução resultante foi agitada a 25° C por 3 horas. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 7 M e as frações puras foram evaporadas até a secura. O produto bruto foi purificado pela HPLC preparativa (coluna Waters XBridge Prep C18 OBD, sílica 5μ, diâmetro 19 mm, comprimento 100 mm), usando misturas decrescentemente polares de água (contendo 1 % de NH3) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura. Este material foi depois purificado ainda pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 0 a 10 % de amônia metanólica (7 N) em DCM. As frações puras foram evaporadas até a secura para produzir 4-amino-N-(2-amino-1-(4-clorofenil)etil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida como uma goma incolor (0,047 g, 11,4 %).
[00083] 1H RMN (400,13 MHz, DMSO-d6) δ 1,41 - 1,51 (2H, m), 1,86 - 1,94 (2H, m), 1,97 - 2,05 (2H, m), 2,77 - 2,86 (2H, m), 3,18 (2H, s), 3,51 - 3,59 (2H, m), 4,35 - 4,44 (2H, m), 4,70 (1H, t), 6,58 (1H, d), 7,15 (1H, d), 7,30 - 7,32 (2H, m), 7,35 - 7,37 (2H, m), 8,12 (1H, s), 8,41 (1H, s), 11,62 (1H, s). MS m/e MH+ 414. Exemplo 6: (S)-4-(aminometil)-N-(1-(4-clorofenil)etil)-1-(7H-pirrolo-[2,3- d]pirimidin-4-il)piperidina-4-carboxamida
[00084] Ácido trifluoroacético (2 ml, 25,96 mmol) foi adicionado ao (4-(1-(4-clorofenil)etilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)- piperidin-4-il)metilcarbamato de (S)-terc-butila (Intermediário 13) (0,257 g, 0,5 mmol) em DCM (20 ml) a 25° C. A solução resultante foi agitada a 25° C por 1 hora. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 7 M e as frações puras foram evaporadas até a secura. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 0 a 10 % de amônia/MeOH 7 N em DCM. As frações puras foram evaporadas até a secura para produzir (S)-4-(aminometil)-N-(1- (4-clorofenil)etil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxamida (0,095 g, 46,0 %) como um sólido branco.
[00085] 1H RMN (400,13 MHz, DMSO-d6) δ 1,39 (3H, d), 1,43 - 1,50 (4H, m), 2,05 - 2,14 (2H, m), 2,69 (2H, s), 3,37 - 3,47 (2H, m), 4,20 - 4,25 (2H, m), 4,96 - 5,04 (1H, t), 6,55 (1H, d), 7,15 (1H, d), 7,37 (4H, s), 8,11 (1H, s), 8,49 (1H, d), 11,61 (1H, s). MS m/e MH+ 413. Exemplo 7: 4-amino-N-(1-(4-clorofenil)-4-hidroxibutil)-1-(7H-pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida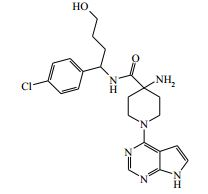
[00086] Ácido trifluoroacético (2 ml, 25,96 mmol) foi adicionado ao 4-(1-(4-clorofenil)-4-hidroxibutilcarbamoil)-1-(7H-pirrolo[2,3-d]-pirimidin- 4-il)piperidin-4-ilcarbamato de terc-butila (Intermediário 17) (140 mg, 0,26 mmol) a 20° C e a solução resultante agitada por 1 hora. A solução foi depois diluída com metanol, aplicada a uma coluna SCX 10 g e eluída com metanol seguida por NH3/MeOH 2 N. As frações contendo o produto foram combinadas, concentradas pela evaporação e purificadas pela HPLC preparativa (coluna Waters XBridge Prep C18 OBD, 5 μm sílica, diâmetro 19 mm, comprimento 100 mm), usando misturas decrescentemente polares de água (contendo 1 % de NH3) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura para produzir 4-amino-N-(1-(4- clorofenil)-4-hidroxibutil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxamida (58,0 mg, 50,8 %) como um sólido incolor.
[00087] 1H RMN (399,902 MHz, DMSO) δ 1,28 - 1,51 (4H, m), 1,69 - 1,80 (2H, m), 1,90 - 2,03 (2H, m), 3,37 - 3,41 (2H, m), 3,50 - 3,58 (2H, m), 4,37 - 4,43 (3H, m), 4,71 - 4,76 (1H, m), 6,59 (1H, m), 7,16 (1H, m), 7,36 (4H, m), 8,13 (1H, s), 8,33 (1H, d), 11,64 (1H, s). MS m/e MH+443. Exemplos 7A e 7B: (S)-4-amino-N-(1-(4-clorofenil)-4-hidroxibutil)-1- (7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida e (R)-4-amino- N-(1-(4-clorofenil)-4-hidroxibutil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxamida
[00088] DIPEA (2,85 ml, 16,0 mmol) foi adicionado ao 4-amino-N- (1-(4-clorofenil)-4-hidroxibutil)piperidina-4-carboxamida (Intermediário 72) (1,04 g, 3,19 mmol) e 4-cloro-7H-pirrolo[2,3-d]pirimidina (0,490 g, 3,19 mmol) em etanol (15,96 ml) a 25° C. A solução resultante foi agitada a 65° C durante a noite. O produto bruto foi analisado pela LCMS e evaporado até a secura. O sólido bruto foi depois purificado pela cromatografia de troca iônica, usando uma coluna SCX-2. O produto foi eluído da coluna usando 20 % de amônia 7 N em metanol / DCM. A mistura bruta foi depois re-purificada pela cromatografia por vaporização instantânea em sílica (eluente de 0 a 10 % de amônia 7 N / MeOH em DCM) e as frações puras evaporadas até a secura para produzir 4-amino-N-(1-(4-clorofenil)-4-hidroxibutil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)-piperidina-4-carboxamida (63,9 %) (racemato) como um sólido branco fino. O racemato foi quiralmente resolvido pela cromatografia de fluxo superficial (SFC) para produzir os enanciômeros puros em rendimentos de 272 mg (19 %) e 245 mg (17 %) respectivamente. Os espetros de RMN para ambos dos enanciômeros foram idênticos.
[00089] 1H RMN (400,13 MHz, DMSO) δ 1,38-1,42 (2H, m), 1,46 - 1,49 (2H, d), 1,74 (2H, s), 1,92 - 2,03 (2H, m), 2,19 (2H, s), 3,55 - 3,58 (2H, d), 4,38 (1H, s), 4,41 (2H, s), 4,75 - 4,76 (1H, d), 6,59 (1H, s), 7,17 (1H, s), 7,36 (4H, s), 8,15 (1H, s), 8,32 - 8,34 (1H, d), 11,65 (1H, s, troca); MS m/e MH+ 443; HPLC tR = 1,66 min. Exemplo 8: 4-amino-N-(1-(4-clorofenil)-2-hidroxietil)-1-(7H-pirrolo-[2,3- d]pirimidin-4-il)piperidina-4-carboxamida
[00090] 4-(1-(4-clorofenil)-2-hidroxietilcarbamoil)-1-(7H-pirrolo-[2,3- d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (Intermediário 18) (137 mg, 0,27 mmol) foi tratado com ácido trifluoroacético (2 ml). A solução foi agitada por 1 hora na temperatura ambiente. A mistura foi concentrada sob pressão reduzida. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O resíduo foi carregado na coluna em metanol e lavado com metanol. O produto desejado foi eluído da coluna usando amônia 2 M em metanol e as frações puras foram evaporadas até a secura para produzir 4-amino-N- (1-(4-clorofenil)-2-hidroxietil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxamida (111 mg, quant.) como um sólido cristalino incolor.
[00091] 1H RMN (399,9 MHz, DMSO-d6) δ 1,40 - 1,49 (2H, m), 1,85 - 2,09 (2H, m), 3,48 - 3,69 (4H, m), 4,35 - 4,48 (2H, m), 4,72 - 4,81 (1H, m), 4,90 - 4,96 (1H, m), 6,58 (1H, br, s), 7,12 - 7,18 (1H, m), 7,30 - 7,40 (4H, m), 8,13 (1H, s), 8,45 - 8,53 (1H, m), 11,64 (1H, s) m/z (ESI+) (M + H)+ = 415; HPLC tR = 1,57 min. Exemplo 9: (S)-4-amino-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida (E9)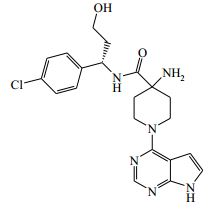
[00092] HCl (4 M em dioxano) (3,00 ml, 12,00 mmol) foi adicionado ao 4-(1-(4-clorofenil)-3-hidroxipropilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidin-4-ilcarbamato de (S)-terc-butila (Intermediário 22) (1,27 g, 2,40 mmol) em diclorometano (20 ml). A suspensão resultante foi agitada a 20° C por 16 horas. A mistura de reação foi filtrada através um copo de filtragem de PTFE e o sólido bruto foi purificada pela HPLC preparativa (coluna Waters XTerra C18, sílica 5μm, diâmetro 19 mm, comprimento 100 mm), usando misturas decrescentemente polares de água (contendo 1 % de TFA) e MeCN como eluentes. As frações contendo o composto desejado foram purificadas pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 7 M e as frações puras foram evaporadas até a secura para produzir (S)-4-amino-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)-piperidina-4-carboxamida (0,200 g, 19,4 %) como um sólido branco. 1H RMN (399,9 MHz, DMSO-d6) δ 1,45 (2H, d), 1,86 (1H, d), 1,90 - 1,93 (1H, m), 2,19 (2H, s), 3,38 (2H, q), 3,51 - 3,58 (2H, m), 4,35 - 4,38 (2H, m), 4,53 (1H, t), 4,88 (1H, d), 6,58 (1H, t), 7,16 (1H, t), 7,32 - 7,38 (4H, m), 8,12 (1H, s), 8,43 (1H, d), 11,63 (1H, s), m/z (ESI+) (M + H)+ = 429; HPLC tR = 1,46 min. Exemplo 9 via alternativa 1: (S)-4-amino-N-(1-(4-clorofenil)-3- hidroxipropil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida

[00093] N-Etildiisopropilamina (1,676 ml, 9,62 mmol) foi adicionada ao (S)-4-amino-N-(1-(4-clorofenil)-3-hidroxipropil)-piperidina-4-carboxamida (Intermediário 49) (1g, 3,21 mmol) e 4-cloro-7H-pirrolo[2,3-d]pirimidina (0,493 g, 3,21 mmol) em butan-1-ol (15 ml). A solução resultante foi agitada a 60° C por 18 horas. A mistura de reação foi diluída com EtOAc (50 ml), e lavada sequencialmente com água (25 ml) e salmoura saturada (25 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 0 a 6 % de MeOH com amônia em DCM. As frações puras foram evaporadas até a secura para produzir (S)-4-amino-N-(1-(4- clorofenil)-3-hidroxipropil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)-piperidina-4- carboxamida (842 mg) como uma espuma branca. (S)-4-amino-N-(1-(4- clorofenil)-3-hidroxipropil)-1-(7H-pirrolo[2,3-d]-pirimidin-4-il)piperidina-4- carboxamida foi agitada em acetato de etila (7 ml) por 18 horas. O sólido foi coletado pela filtração, lavado com uma quantidade pequena de acetato de etila e secado durante a noite a vácuo a 55° C por 18 horas para produzir (S)-4-amino-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida (0,585 g, 42,5 %) como um sólido branco. m/z (ES+) (M + H)+ = 429; HPLC tR = 1,60 min.
[00094] 1H RMN (400,13 MHz, DMSO-d6) δ 1,39 - 1,47 (2H, m), 1,80 - 2,02 (4H, m), 2,17 (2H, s), 3,35 - 3,40 (2H, m), 3,50 - 3,59 (2H, m), 4,34 - 4,41 (2H, m), 4,53 (1H, t), 4,88 (1H, d), 6,57 (1H, m), 7,14 - 7,16 (1H, m), 7,31 - 7,37 (4H, m), 8,12 (1H, s), 8,42 (1H, d), 11,62 (1H, s) Exemplo 9 via alternativa 2: (S)-4-amino-N-(1-(4-clorofenil)-3- hidroxipropil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida

[00095] (S)-3-Amino-3-(4-clorofenil)propan-1-ol (Intermediário 47) (2,055 g, 11,07 mmol) foi adicionado em uma porção ao ácido 4-(terc- butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxílico (Intermediário 1) (4g, 11,07 mmol) e DIPEA (5,80 ml, 33,20 mmol) em DMA (40 ml). HATU (4,63 g, 12,18 mmol) foi adicionado e a solução resultante foi agitada a 20° C por 24 horas. A mistura de reação foi evaporada até a secura depois diluída com EtOAc (300 ml), e lavada sequencialmente com água (50 ml) e salmoura saturada (50 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 2 a 6 % de MeOH com amônia em DCM. As frações puras foram evaporadas até a secura e trituradas com dioxano (40 ml) para produzir 4-(1-(4-clorofenil)-3-hidroxipropilcarbamoil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidin-4-ilcarbamato de (S)-terc-butila (Intermediário 22) (4,82 g, 82 %) como um sólido branco. 4-(1-(4-clorofenil)-3- hidroxipropilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4- ilcarbamato de (S)-terc-butila (Intermediário 22) (4,82 g, 82 %) foi colocado em suspensão em dioxano (40,0 ml) e cloreto de hidrogênio 4 M em dioxano (7,69 ml, 221,36 mmol) adicionado. A reação foi agitada na temperatura ambiente por 2 horas. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 3,5 M e as frações puras foram evaporadas até a secura. O produto bruto foi purificado pela HPLC preparativa (coluna Waters XBridge Prep C18 OBD, sílica 5μm, diâmetro 19 mm, comprimento 100 mm), usando misturas decrescentemente polares de água (contendo 1 % de NH3) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura para produzir (S)-4-amino-N-(1-(4- clorofenil)-3-hidroxipropil)-1-(7H-pirrolo-[2,3-d]pirimidin-4-il)piperidina- 4-carboxamida (1,200 g, 25,3 %) como um sólido branco. m/z (ES+) (M + H)+ = 429; HPLC tR = 1,67 min.
[00096] 1H RMN se igual com o anterior. Exemplo 10: N-(3-acetamido-1 -(4-clorofenil)propil)-4-amino-1-(7H- pirrolo[2,3-d]pirimidin-4-ilpiperidina-4-carboxamida
[00097] Cloreto de hidrogênio (4 M) em 1,4-dioxano (0,430 ml, 1,72 mmol) foi adicionado ao 4-(3-acetamido-1-(4-clorofenil)propil- carbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (Intermediário 28) (98 mg, 0,17 mmol) em DCM (4 ml) a 20° C. A suspensão resultante foi agitada a 20° C por 70 horas. A mistura de reação foi evaporada até a secura. O produto bruto foi purificado pela HPLC preparativa (coluna Waters XTerra C18, sílica 5μ, diâmetro 19 mm, comprimento 100 mm), usando misturas decrescentemente polares de água (contendo 1 % de NH3) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura para produzir N-(3-acetamido-1-(4-clorofenil)propil)-4-amino-1-(7H- pirrolo-[2,3-d]pirimidin-4-il)piperidina-4-carboxamida (15,00 mg, 18,5 %) como uma película seca branca. 1H RMN (399,9 MHz, DMSO-d6) δ 1,42 -1,47 (2H, m), 1,79 (3H, s), 1,85 (1H, t), 1,89 - 1,93 (1H, m), 2,10 (2H, s), 3,00 (2H, t), 3,55 (2H, d), 4,36 - 4,40 (2H, m), 4,80 (1H, d), 6,58 - 6,60 (1H, m), 7,16 (1H, t), 7,34 - 7,39 (4H, m), 7,80 (1H, t), 8,13 (1H, s), 8,39 (1H, s), 11,64 (1H, s) Nenhum NH2 visível. m/z (ESI+) (M + H)+ = 470; HPLC tR = 1,56 min. Exemplo 11: 4-amino-N-(1-(4-clorofenil)-3-(dimetilamino)propil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[00098] Cloreto de hidrogênio (4 M em 1,4-dioxano, 1,01 ml, 4,05 mmol) foi adicionado ao 4-(1-(4-clorofenil)-3-(dimetilamino)propil- carbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (Intermediário 34) (0,045 g, 0,08 mmol) em uma mistura de DCM (5 ml) e metanol (2 ml) a 22° C. A solução resultante foi agitada a 22° C por 16 horas. A mistura foi evaporada e o resíduo foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 2 M e as frações puras foram evaporadas até a secura para produzir 4-amino- N-(1-(4-cloro-fenil)-3-(dimetilamino)propil)-1-(7H-pirrolo[2,3-d]pirimidin- 4-il)-piperidina-4-carboxamida (0,034 g, 92 %) como uma goma incolor.
[00099] 1H RMN (399,902 MHz, CDCl3) δ 1,57 (2H, m), 1,66 (2H, br.s), 1,81 (1H, m), 2,02 (1H, m), 2,18 (6H, s), 2,18 - 2,36 (4H, m), 3,67 (3H, m), 4,50 (2H, m), 5,00 (1H, dt), 6,52 (1H, d), 7,05 (1H, d), 7,18 (2H, d), 7,29 (2H, d), 8,33 (1H, s), 9,07 (1H, d), 9,61 (1H, s). MS m/e MH+456 Exemplos 11A e 11B: (S)-4-amino-N-(1-(4-clorofenil)-3-(dimetil- amino)propil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxamida e (R)-4-amino-N-(1-(4-clorofenil)-3-(dimetilamino)propil)- 1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[000100] 4-amino-N-(1-(4-clorofenil)-3-(dimetilamino)propil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida racêmica (Exemplo 11) (231 mg, 0,51 mmol) foi quiralmente separada em uma coluna Chiralpak AD-H SFC (250 mm x 20 mm), usando cromatografia de fluído supercrítico, solvente de eluição 7:3 CO2/(EtOH + 0,1 % de DEA). As soluções apropriadas para o primeiro Isômero eluído foram evaporadas e o resíduo triturado com éter dietílico para dar 4-amino-N- (1-(4-clorofenil)-3-(dimetilamino)propil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxamida (59 mg, 25 %) como um sólido branco.
[000101] 1H RMN (399,902 MHz, DMSO) δ 1,43 (2H, ddd), 1,83 (2H, dt), 1,86 - 2,01 (2H, m), 2,11 (6H, s), 2,14 (2H, t), 3,56 (2H, ddd), 4,39 (2H, ddd), 4,83 (1H, dt), 6,58 (1H, dd), 7,16 (1H, dd), 7,33 (2H, d), 7,37 (2H, d), 8,13 (1H, s), 8,61 (1H, d), 11,63 (1H, s). MS m/e MH+456,5.
[000102] As soluções apropriadas para o segundo Isômero eluído foram evaporadas e o resíduo triturado com éter dietílico para dar 4- amino-N-(1-(4-clorofenil)-3-(dimetilamino)propil)-1-(7H-pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida (43 mg, 19 %) como um sólido branco.
[000103] 1H RMN (399,902 MHz, DMSO) δ 1,43 (2H, ddd), 1,83 (2H, dt), 1,86 - 2,01 (2H, m), 2,11 (6H, s), 2,14 (2H, t), 3,56 (2H, ddd), 4,39 (2H, ddd), 4,83 (1H, q), 6,58 (1H, dd), 7,16 (1H, dd), 7,33 (2H, d), 7,37 (2H, d), 8,13 (1H, s), 8,61 (1H, d), 11,63 (1H, s). MS m/e MH+456,4, 458,4 Exemplo 12: 4-(aminometil)-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[000104] Hexafluorofosfato de O-(7-Azabenzotriazol-1-il)-N,N,N’,N’ - tetrametilurônio (106 mg, 0,28 mmol) foi adicionado ao ácido 4-((terc- butoxicarbonilamino)metil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)-piperidina- 4-carboxílico (Intermediário 12) (100 mg, 0,27 mmol) e N,N- diisopropiletilamina (0,055 ml, 0,32 mmol) em NMP (5 ml) a 22° C. A solução resultante foi agitada a 50° C por 10 minutos depois esfriada até a temperatura ambiente. 3-Amino-3-(4-clorofenil)propan-1-ol (Intermediário 29) (49,5 mg, 0,27 mmol) foi adicionado como uma solução em NMP (2 ml) e a mistura foi agitada a 22° C por 16 horas. HCl 4 M em dioxano (1 ml) foi adicionado e a mistura foi agitada a 22° C por mais 24 horas. A mistura foi concentrada e o resíduo foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando 30 % (NH3 2 M em MeOH) em DCM e as frações puras foram evaporadas até a secura para produzir o produto bruto. O produto bruto foi purificado pela HPLC preparativa (coluna Waters XBridge Prep C18 OBD, sílica 5μ, diâmetro 19 mm, comprimento 100 mm), usando misturas decrescentemente polares de água (contendo 1 % de NH3) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura para produzir 4-(aminometil)-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida (0,095 g, 81 %) como um sólido branco.
[000105] 1H RMN (399,902 MHz, DMSO) δ 1,47 (2H, m), 1,80 - 1,96 (2H, m), 2,09 (2H, m), 2,69 (1H, s), 3,27 - 3,45 (4H, m), 4,26 (2H, ddd), 5,02 (1H, dd), 6,56 (1H, d), 7,15 (1H, d), 7,33 - 7,39 (4H, m), 8,12 (1H, s), 8,44 (1H, d), 11,63 (1H, s). MS m/e MH+556 Exemplo 13: 4-amino-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[000106] TFA (0,7 ml) foi adicionado a uma suspensão de 4-(1-(4- clorofenil)-3-hidroxipropilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidin-4-ilcarbamato de terc-butila (Intermediário 36) (175 mg, 0,33 mmol) em diclorometano (7 ml) sob argônio. A solução resultante foi agitada a 20° C por 16 horas. Os solventes foram removidos a vácuo e a mistura de reação foi purificada pela HPLC preparativa usando uma coluna de fase reversa Waters X-Bridge (C-18, sílica de 5 mícrons, diâmetro 19 mm, comprimento 100 mm) com misturas decrescentemente polares de água (contendo 0,2 % de carbonato de amônio) e acetonitrila como eluente. As frações puras foram evaporadas até a secura para produzir 4-amino-N-(1-(4-cloro-fenil)-3- hidroxipropil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida (99 mg, 69,8 %) como um pó branco.
[000107] 1H RMN (500 MHz, DMSO-d6) δ 1,38 -1,46 (2H, m), 1,81 - 1,91 (4H, m), 2,25 (2H, br s), 3,48 (2H, m), 3,35 - 3,54 (2H, m), 4,35 - 4,41 (2H, m), 4,57 (1H, t), 4,87 (1H, m), 6,58 (1H, d), 7,16 (1H, d), 7,31 - 7,37 (4H, m), 8,11 (1H, s), 8,45 (1H, d), 11,65 (1H, s). m/z (ESI+) (M + H)+ = 429; HPLC tR = 1,58 min. Exemplo 14: 4-amino-N-(3-amino-1-(4-clorofenil)propil)-1-(7H-pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[000108] Ácido trifluoroacético (0,05 ml) foi adicionado a uma suspensão agitada de 4-(3-amino-1-(4-clorofenil)propilcarbamoil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (Intermediário 38) (7,0 mg) em DCM (1 ml) sob argônio a 25° C. A suspensão resultante foi agitada a 25° C por 2 dias. A mistura de reação foi evaporada até a secura para produzir o sal de di-TFA de 4-amino-N- (3-amino-1-(4-clorofenil)propil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)- piperidina-4-carboxamida como um óleo parcialmente sólido (13,0 mg).
[000109] 1H RMN (500 MHz, DMSO-d6) δ 1,86 (1H, d), 2,01 (2H, m), 2,08 (1H, m) 2,41 (2H, m), 2,69 (2H, m), 2,85 (1H, m), 3,61 (2H, t), 4,63 (2H, t), 5,02 (1H, q), 6,82 (1H, s), 7,36 (1H, t), 7,41 (2H, d), 7,49 (2H, d), 7,93 (3H, s br), 8,32 (1H, s), 8,59 (3H, s br), 8,96 (1H, d). MS m/e MH+ 428. Exemplo 15: (R)-4-amino-N-(1 -(4-clorofenil)-3-hidroxipropil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[000110] HCl (4 M em dioxano) (0,378 ml, 1,51 mmol) foi adicionado ao 4-(1-(4-clorofenil)-3-hidroxipropilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidin-4-ilcarbamato de (R)-terc-butila (Intermediário 42) (0,160 g, 0,30 mmol) em DCM (3 ml). A suspensão resultante foi agitada a 20° C por 3 horas. A mistura de reação foi evaporada. O produto bruto foi purificado pela HPLC preparativa (coluna Waters XTerra C18, sílica 5μm, diâmetro 19 mm, comprimento 100 mm), usando misturas decrescentemente polares de água (contendo 1 % de amônia) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura para produzir (R)-4-amino-N-(1-(4-clorofenil)-3- hidroxipropil)-1-(7H-pirrolo[2,3-d]-pirimidin-4-il)piperidina-4-carboxamida (0,051 g, 39,3 %) como um sólido branco.
[000111] 1H RMN (399,9 MHz, DMSO-d6) δ 1,46 (2H, d), 1,86 (1H, d), 1,90 - 1,93 (1H, m), 2,10 (2H, m), 3,37 (1H, t), 3,55 (2H, d), 4,40 (2H, d), 4,53 (2H, m), 4,88 (1H, d), 6,58 (1H, t), 7,16 (1H, t), 7,32 - 7,38 (4H, m), 8,14 (1H, d), 8,43 (1H, d), 11,63 (1H, s) nenhum NH2 visível. MS m/e MH+ 429; HPLC tR = 1,46 min. Exemplo 16: (R)-4-amino-N-(1 -(4-clorofenil)etil)-1-(7H-pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida
[000112] Hexafluorofosfato de O-(7-Azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (0,418 g, 1,10 mmol) foi adicionado em uma porção ao ácido 4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)- piperidina-4-carboxílico (Intermediário 1) (0,361 g, 1 mmol), (R)-1-(4- clorofenil)etanamina (0,156 g, 1,00 mmol) e DIPEA (0,524 ml, 3,00 mmol) em DMA (10 ml) a 25° C sob nitrogênio. A solução resultante foi agitada a 60° C por 4 horas. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 7 N e as frações puras foram evaporadas até a secura. O material bruto foi tratado com uma solução a 10 % de TFA em DCM (5 ml) e agitado na temperatura ambiente. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 7 N e as frações puras foram evaporadas até a secura. O produto bruto foi purificado pela LCMS preparativa usando misturas decrescentemente polares de água (contendo 1 % de amônia) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura para produzir (R)-4-amino-N-(1-(4-clorofenil)etil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxamida (0,211 g, 52,9 %) como um sólido branco.
[000113] 1H RMN (400 MHz, DMSO) δ 1,37 (3H, d), 1,39 - 1,48 (2H, m), 1,86 - 2,02 (2H, m), 2,19 (2H, s), 3,49 - 3,58 (2H, m), 4,34 - 4,43 (2H, m), 4,83 - 4,91 (1H, m), 6,56 - 6,59 (1H, m), 7,14 - 7,16 (1H, m), 7,32 - 7,38 (4H, m), 8,12 (1H, s), 8,30 (1H, d), 11,62 (1H, s). MS m/e MH+ 399. Exemplo 17: (R)-4-(aminometil)-N-(1-(4-clorofenil)etil)-1-(7H-pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[000114] Níquel de Raney® (293 mg, 1,71 mmol) foi adicionado a uma solução de (R)-N-(1-(4-clorofenil)etil)-4-ciano-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida (Intermediário 45) (466 mg, 1,14 mmol), em etanol (30 ml). Hidróxido de amônio (10 ml) foi adicionado. Esta mistura foi primeiro purgada com nitrogênio, depois colocada sob um balão de hidrogênio e agitada por 36 horas. A mistura de reação foi filtrada através de celite e o solvente evaporado até a secura. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 0 a 10 % de amônia/MeOH 7 N em DCM. As frações puras foram evaporadas até a secura para produzir (R)-4-(aminometil)-N-(1-(4-clorofenil)etil)-1- (7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida (276 mg, 58,6 %) como um sólido branco.
[000115] 1H RMN (400 MHz, DMSO) δ 1,38 (3H, d), 1,41 - 1,51 (2H, m), 1,61 (2H, s), 2,09 (2H, d), 2,68 (2H, s), 3,37 - 3,50 (2H, m), 4,18 - 4,28 (2H, m), 4,95 - 5,04 (1H, m), 6,56 (1H, d), 7,15 (1H, d), 7,26 - 7,50 (4H, m), 8,11 (1H, s), 8,52 (1H, d), 11,66 (1H, s). MS m/e MH+ 413. Exemplo 18: (S)-4-amino-N-(1 -(4-cianofenil)-3-hidroxipropil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[000116] (S)-4-(1-amino-3-hidroxipropil)benzonitrila (Intermediário 46) (195 mg, 1,11 mmol) foi adicionada em uma porção ao ácido 4- (terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxílico (Intermediário 1) (400 mg, 1,11 mmol) e DIPEA (0,580 ml, 3,32 mmol) em DMA (5 ml). Hexafluorofosfato de O-(7-azabenzo- triazol-1-il)-N,N,N’,N’-tetrametilurônio (463 mg, 1,22 mmol) foi adicionado e a solução resultante foi agitada a 20° C por 24 horas. A mistura de reação foi evaporada até a secura depois diluída com EtOAc (300 ml), e lavada sequencialmente com água (50 ml) e salmoura saturada (50 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 2 a 6 % de MeOH com amônia em DCM. As frações puras foram evaporadas até a secura para produzir 4-(1-(4- cianofenil)-3-hidroxipropilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidin-4-ilcarbamato de (S)-terc-butila (267 mg, 46,4 %) como um sólido branco. 4-(1-(4-cianofenil)-3-hidroxipropilcarbamoil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de (S)-terc-butila (267 mg, 0,51 mmol) foi colocado em suspensão em dioxano (5,00 ml) e cloreto de hidrogênio 4 M em dioxano (0,769 ml, 22,14 mmol) adicionado. A reação foi agitada em temperatura ambiente por 2 horas. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 3,5 N e as frações puras foram evaporadas até a secura. O produto foi depois purificado pela HPLC preparativa (coluna Waters XBridge Prep C18 OBD, sílica 5μ, 21 mm de diâmetro, comprimento 100 mm), usando misturas decrescentemente polares de água (contendo 1 % de amônia) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura para produzir (S)-4-amino-N-(1-(4- cianofenil)-3-hidroxipropil)-1-(7H-pirrolo[2,3-d] pirimidin-4-il)piperidina- 4-carboxamida (70,0 mg, 15,1 %) como um sólido branco.
[000117] 1H RMN (400,13 MHz, DMSO-d6) δ 1,36 - 1,47 (2H, m), 1,80 - 2,01 (4H, m), 2,17 (2H, s), 3,39 (2H, q), 3,52 - 3,59 (2H, m), 4,34 - 4,40 (2H, m), 4,58 (1H, t), 4,94 (1H, s), 6,57 (1H, d), 7,15 (1H, d), 7,50 (2H, d), 7,76 - 7,79 (2H, d), 8,12 (1H, s), 8,52 (1H, s), 11,61 (1H, s). MS m/e MH+ 420. Exemplo 19: (S)-4-amino-1-(5-bromo-7H-pirrolo[2,3-d]pirimidin-4-il)-N- (1-(4-clorofenil)-3-hidroxipropil)piperidina-4-carboxamida
[000118] DIPEA (0,670 ml, 3,85 mmol) foi adicionada ao (S)-4- amino-N-(1-(4-clorofenil)-3-hidroxipropil)piperidina-4-carboxamida (Intermediário 49) (400 mg, 1,28 mmol) e 5-bromo-4-cloro-7H- pirrolo[2,3-d]pirimidina (Intermediário 50) (298 mg, 1,28 mmol) em butan-1-ol (6 ml). A solução resultante foi agitada a 60° C por 18 horas. A mistura de reação foi diluída com EtOAc (50 ml), e lavada sequencialmente com água (25 ml) e salmoura saturada (25 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 0 a 6 % de MeOH com amônia em DCM. O produto foi depois purificado pela HPLC preparativa (coluna Waters XBridge Prep C18 OBD, sílica 5μ, diâmetro 19 mm, comprimento 100 mm), usando misturas decrescentemente polares de água (contendo 1 % de amônia) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura e trituradas com éter dietílico para produzir (S)-4-amino-1-(5-bromo-7H-pirrolo[2,3-d]pirimidin-4-il)-N- (1-(4-clorofenil) -3-hidroxipropil)piperidina-4-carboxamida (70,0 mg, 10,8 %) como um sólido branco.
[000119] 1H RMN (400,13 MHz, DMSO-d6) δ 1,41 - 1,50 (2H, m), 1,81 - 1,92 (2H, m), 2,04 - 2,20 (4H, m), 3,37 - 3,44 (4H, m), 3,91 (2H, t), 4,54 (1H, t), 4,90 (1H, m), 7,33 - 7,38 (4H, m), 7,50 (1H, s), 8,25 (1H, s), 8,47 (1H, d), 12,18 (1H, s). MS m/e MH+ 510. Exemplo 20: (S)-4-amino-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(1H- pirazolo[3,4-d]pirimidin-4-il)piperidina-4-carboxamida
[000120] DIPEA (0,419 ml, 2,41 mmol) foi adicionada ao (S)-4- amino-N-(1-(4-clorofenil)-3-hidroxipropil)piperidina-4-carboxamida (Intermediário 49) (250 mg, 0,80 mmol) e 4-cloro-1H-pirazolo[3,4-d]- pirimidina (124 mg, 0,80 mmol) em butan-1-ol (5 ml). A solução resultante foi agitada a 60° C por 6 horas. A mistura de reação foi diluída com EtOAc (50 ml), e lavada sequencialmente com água (25 ml) e salmoura saturada (25 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 0 a 6 % de MeOH com amônia em DCM. As frações puras foram evaporadas até a secura e trituradas com éter dietílico para produzir (S)-4-amino-N-(1-(4-clorofenil)-3- hidroxipropil)-1-(1H-pirazolo[3,4-d]-pirimidin-4-il)piperidina-4- carboxamida (112 mg, 32,5 %) como um sólido branco.
[000121] 1H RMN (399,9 MHz, DMSO-d6) δ 1,49 (2H, t), 1,79 - 2,01 (4H, m), 3,39 (2H, m), 3,62 (2H, s), 4,40 (2H, s), 4,57 (1H, t), 4,88 (1H, m), 7,32 - 7,38 (4H, m), 8,22 (1H, d), 8,29 (1H, s), 8,45 (1H, d), 13,52 (1H, s). MS m/e MH+ 430. Exemplo 21: (S)-4-amino-N-(3-hidróxi-1-fenilpropil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida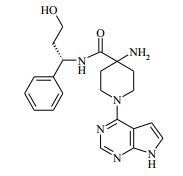
[000122] Cloridreto de (S)-3-amino-3-fenilpropan-1-ol (260 mg, 1,38 mmol) foi adicionado em uma porção ao ácido 4-(terc-butóxi- carbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxílico (Intermediário 21) (500 mg, 1,38 mmol) e DIPEA (0,967 ml, 5,53 mmol) em DMA (5 ml). Hexafluorofosfato de O-(7-azabenzo-triazol-1-il)- N,N,N’,N’-tetrametilurônio (579 mg, 1,52 mmol) foi adicionado e a solução resultante foi agitada a 20° C por 24 horas. A mistura de reação foi evaporada até a secura depois diluída com EtOAc (300 ml), e lavada sequencialmente com água (50 ml) e salmoura saturada (50 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 2 a 6 % de MeOH com amônia em DCM. As frações foram evaporadas para produzir 4-(3-hidróxi-1-fenilpropilcarbamoil)-1-(7H- pirrolo[2,3-d]-pirimidin-4-il)piperidin-4-ilcarbamato de (S)-terc-butila (334 mg, 48,8 %) como um sólido branco. O produto (334 mg, 0,67 mmol) foi colocado em suspensão em dioxano (5,00 ml) e cloreto de hidrogênio 4 M em dioxano (0,961 ml, 27,67 mmol) adicionado. A reação foi agitada na temperatura ambiente por 2 horas. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 3,5 N e as frações puras foram evaporadas até a secura. A goma resultante foi triturada com EtOAc para dar um sólido que foi coletado pela filtração e secado sob vácuo para dar (S)-4- amino-N-(3-hidróxi-1-fenilpropil)-1-(7H-pirrolo-[2,3-d]pirimidin-4- il)piperidina-4-carboxamida (93 mg, 17,0 %) como um sólido branco amarelado.
[000123] 1H RMN (400,13 MHz, DMSO-d6) δ 1,44 (2H, t), 1,82 - 2,03 (4H, m), 3,37 (2H, t), 3,50 - 3,58 (2H, m), 4,40 (2H, t), 4,49 (1H, t), 4,90 (1H, d), 6,58 (1H, s), 7,15 (1H, t), 7,21 - 7,23 (1H, m), 7,30 - 7,31 (4H, m), 8,12 (1H, s), 8,41 (1H, d), 11,62 (1H, s). MS m/e MH+ 395. Exemplo 22: (S)-4-amino-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(9H- purin-6-il)piperidina-4-carboxamida
[000124] DIPEA (0,335 ml, 1,92 mmol) foi adicionada ao (S)-4- amino-N-(1-(4-clorofenil)-3-hidroxipropil)piperidina-4-carboxamida (Intermediário 49) (200 mg, 0,64 mmol) e 6-cloro-9H-purina (99 mg, 0,64 mmol) em butan-1-ol (4 ml). A solução resultante foi agitada a 60° C por 18 horas. A mistura de reação foi diluída com EtOAc (50 ml), e lavada sequencialmente com água (25 ml) e salmoura saturada (25 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 0 a 6 % de MeOH com amônia em DCM. As frações puras foram evaporadas até a secura para produzir (S)-4-amino-N-(1-(4- clorofenil)-3-hidroxipropil)-1-(9H-purin-6-il)piperidina-4-carboxamida (141 mg, 51,1 %) como um sólido branco.
[000125] 1H RMN (400,13 MHz, DMSO-d6) δ 1,42 (2H, t), 1,78 - 2,00 (4H, m), 3,34 - 3,41 (2H, m), 3,61 (2H, s), 4,53 (1H, t), 4,88 (1H, d), 5,02 (2H, s), 7,30 - 7,39 (4H, m), 8,08 (1H, s), 8,17 - 8,22 (1H, s), 8,42 (1H, d), 13,02 (1H, s). MS m/e MH+ 430. Exemplo 23: (S)-4-(aminometil)-N-(1-(4-clorofenil)-3-hidroxipropil)-1- (7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida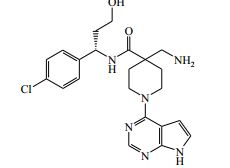
[000126] (S)-3-Amino-3-(4-clorofenil)propan-1-ol (Intermediário 47) (247 mg, 1,33 mmol) foi adicionado em uma porção ao ácido 4-((terc- butoxicarbonilamino)metil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)-piperidina- 4-carboxílico (Intermediário 12) (500 mg, 1,33 mmol) e DIPEA (0,698 ml, 4,00 mmol) em DMA (6 ml). Hexafluorofosfato de O-(7- azabenzotriazol-1-il)-N,N,N’,N’-tetrametilurônio (557 mg, 1,47 mmol) foi adicionado e a solução resultante foi agitada a 20° C por 2 horas depois 20° C por 18 horas. A mistura de reação foi diluída com EtOAc (50 ml), e lavada sequencialmente com água (20 ml) e salmoura saturada (20 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 2 a 6 % de MeOH com amônia em DCM. As frações puras foram evaporadas até a secura para produzir (4-(1-(4-clorofenil)-3- hidroxipropilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4- il)metilcarbamato de (S)-terc-butila (428 mg, 59,2 %) como uma goma incolor. O produto (428 mg) foi dissolvido em dioxano (6,00 ml) e cloreto de hidrogênio 4 M em dioxano (0,925 ml, 26,64 mmol) adicionado. A reação foi agitada na temperatura ambiente por 2 horas. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 3,5 N e as frações puras foram evaporadas até a secura. O produto bruto foi purificado pela HPLC preparativa (coluna Waters XBridge Prep C18 OBD, sílica 5 μm, diâmetro 19 mm, comprimento 100 mm), usando misturas decrescentemente polares de água (contendo 1 % de amônia) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura para produzir (S)-4-(aminometil)-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)-piperidina-4-carboxamida (192 mg, 32,5 %) como uma película seca branca.
[000127] 1H RMN (400,13 MHz, DMSO-d6) δ 1,45 - 1,47 (2H, m), 1,79 - 1,84 (1H, m), 1,89 - 1,99 (1H, m), 2,09 (2H, s), 2,67 (2H, s), 3,36 - 3,42 (4H, m), 4,25 (2H, d), 4,57 (1H, s), 5,01 (1H, d), 6,55 (1H, s), 7,14 (1H, s), 7,33 - 7,36 (4H, m), 8,11 (1H, s), 8,44 (1H, d), 11,61 (1H, s). MS m/e MH+ 443. Exemplo 24: (S)-4-amino-N-(1 -(4-bromofenil)-3-hidroxipropil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[000128] (S)-3-Amino-3-(4-bromofenil)propan-1-ol (Intermediário 51) (191 mg, 0,83 mmol) foi adicionado em uma porção ao ácido 4-(terc- butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)-piperidina-4- carboxílico (Intermediário 1) (300 mg, 0,83 mmol) e DIPEA (0,435 ml, 2,49 mmol) em DMA (5 ml). Hexafluorofosfato de O-(7- azabenzotriazol-1-il)-N,N,N’,N’-tetrametilurônio (347 mg, 0,91 mmol) foi adicionado e a solução resultante foi agitada a 20° C por 24 horas. A mistura de reação foi evaporada até a secura depois diluída com EtOAc (300 ml), e lavada sequencialmente com água (50 ml) e salmoura saturada (50 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 2 a 6 % de MeOH com amônia em DCM. As frações foram evaporadas para produzir 4-(1-(4-bromofenil)-3- hidroxipropilcarbamoil)-1-(7H-pirrolo-[2,3-d]pirimidin-4-il)piperidin-4- ilcarbamato de (S)-terc-butila (212 mg, 44,5 %) como um sólido branco. O produto (212 mg, 0,36 mmol) foi colocado em suspensão em dioxano (5,00 ml) e cloreto de hidrogênio 4 M em dioxano (0,577 ml, 16,6 mmol) adicionado. A reação foi agitada na temperatura ambiente por 2 horas. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 3,5 N e as frações puras foram evaporadas até a secura. O produto foi depois purificado pela HPLC preparativa (coluna Waters XBridge Prep C18 OBD, sílica 5μm, diâmetro 19 mm, comprimento 100 mm), usando misturas decrescentemente polares de água (contendo 1 % de amônia) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura para produzir (S)-4-amino-N-(1-(4- bromofenil)-3-hidroxipropil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina- 4-carboxamida (51,0 mg, 13,0 %) como um sólido branco.
[000129] 1H RMN (400,13 MHz, DMSO-d6) δ 1,38 - 1,46 (2H, m), 1,79 - 2,01 (4H, m), 2,15 (2H, s), 3,37 (2H, q), 3,51 - 3,58 (2H, m), 4,37 (2H, t), 4,52 (1H, t), 4,86 (1H, d), 6,57 (1H, d), 7,15 (1H, d), 7,27 (2H, d), 7,48 - 7,51 (2H, m), 8,12 (1H, s), 8,42 (1H, d), 11,62 (1H, s). MS m/e MH+ 473. Exemplo 25: 4-amino-N-(1-(4-clorofenil)-4-(dimetilamino)butil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida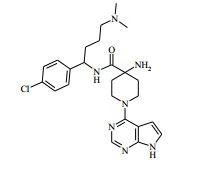
[000130] 1-(4-Clorofenil)-N4,N4-dimetilbutano-1,4-diamina (Intermediário 57) (330 mg, 1,46 mmol) foi adicionada em uma porção ao ácido 4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxílico (Intermediário 1) (526 mg, 1,46 mmol) e DIPEA (0,763 ml, 4,37 mmol) em DMA (5 ml). Hexafluorofosfato de O- (7-azabenzotriazol-1-il)-N,N,N’,N’-tetrametilurônio (609 mg, 1,60 mmol) foi adicionado e a solução resultante foi agitada a 50° C por 2 horas. A mistura de reação foi diluída com EtOAc (25 ml), e lavada sequencialmente com água (20 ml) e salmoura saturada (20 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 5 a 10 % de MeOH com amônia em isoexano. As frações puras foram evaporadas até a secura para produzir 4-(1-(4-clorofenil)- 4-(dimetilamino)butil-carbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidin-4-ilcarbamato de terc-butila (423 mg, 51,0 %) como uma goma incolor. 4-(1-(4-cloro-fenil)-4-(dimetilamino)butilcarbamoil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (423 mg, 0,74 mmol) foi dissolvido em DCM (5,00 ml) e TFA (1 ml) adicionado. A reação foi agitada na temperatura ambiente por 2 horas. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 3,5 N e as frações puras foram evaporadas até a secura para produzir o produto bruto que foi triturado com éter dietílico para produzir 4-amino-N-(1-(4-clorofenil)-4- (dimetilamino)butil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)-piperidina-4- carboxamida (245 mg, 35,8 %) como um sólido branco.
[000131] 1H RMN (400,13 MHz, DMSO-d6) δ 1,26 - 1,33 (2H, m), 1,38 - 1,47 (2H, m), 1,65 - 1,75 (2H, m), 1,87 - 2,01 (2H, m), 2,08 (6H, s), 2,18 (2H, t), 3,50 - 3,58 (2H, m), 4,35 - 4,41 (2H, m), 4,73 (1H, m), 6,57 (1H, d), 7,14 - 7,16 (1H, m), 7,32 - 7,37 (4H, m), 8,12 (1H, s), 8,31 (1H, d), 11,62 (1H, s). MS m/e MH+ 470. Exemplo 26: (S)-4-amino-N-(1 -(4-clorofenil)-3-(dietilamino)propil)-1 - (7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida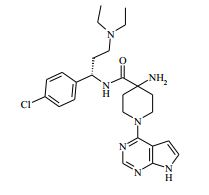
[000132] (S)-1-(4-Clorofenil)-N3,N3-dietilpropano-1,3-diamina (Intermediário 60) (119 mg, 0,49 mmol) foi adicionada em uma porção ao ácido 4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxílico (Intermediário 1) (179 mg, 0,49 mmol) e DIPEA (0,259 ml, 1,48 mmol) em DMA (5 ml). Hexafluorofosfato de O-(7- azabenzotriazol-1-il)-N,N,N’,N’-tetrametilurônio (207 mg, 0,54 mmol) foi adicionado e a solução resultante foi agitada a 50° C por 2 horas. A mistura de reação foi diluída com EtOAc (25 ml), e lavada sequencialmente com água (20 ml) e salmoura saturada (20 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 5 a 10 % de MeOH com amônia em DCM. As frações puras foram evaporadas até a secura para produzir 4-(1-(4-clorofenil)-3-(dietilamino)propilcarbamoil)- 1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de (S)-terc-butila (189 mg, 65,5 %) como uma goma incolor. 4-(1-(4-clorofenil)-3- (dietilamino)propilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)-piperidin-4- ilcarbamato de (S)-terc-butila (189 mg, 0,33 mmol) foi dissolvido em DCM (5,00 ml) e TFA (1 ml) adicionado. A reação foi agitada na temperatura ambiente por 2 horas. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 7 N e as frações puras foram evaporadas até a secura para produzir o produto bruto que foi triturado com éter dietílico para produzir (S)-4-amino-N-(1-(4-clorofenil)-3- (dietilamino)propil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxamida (70,0 mg, 29,3 %) como uma espuma branca.
[000133] 1H RMN (400,13 MHz, DMSO-d6) δ 0,94 (6H, d), 1,42 - 1,49 (2H, m), 1,86 - 2,01 (4H, m), 2,43 (2H, m), 3,51 - 3,59 (2H, m), 4,37 - 4,43 (2H, m), 4,84 (1H, t), 6,58 (1H, d), 7,15 - 7,16 (1H, d), 7,32 - 7,38 (4H, m), 8,12 (1H, s), 8,59 (1H, s), 11,63 (1H, s). MS m/e MH+ 484. Exemplo 27: (S)-4-amino-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(5- ciclolopropil-7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida
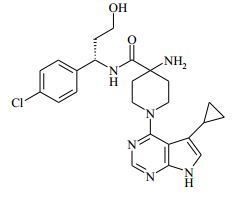
[000134] Cloreto de hidrogênio 4 M em dioxano (0,923 ml, 3,69 mmol) foi adicionado ao 4-(1-(4-clorofenil)-3-hidroxipropilcarbamoil)-1-(5- ciclolopropil-7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-il-carbamato de (S)- terc-butila (Intermediário 65) (420 mg, 0,74 mmol) em dioxano (25 ml). A solução resultante foi agitada na temperatura ambiente por 2 horas. A mistura de reação foi dissolvida em metanol e purificada pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 3,5 N e as frações puras foram evaporadas até a secura. O produto foi depois purificado pela LCMS preparativa (coluna Waters XBridge Prep C18 OBD, sílica 5μ, diâmetro 19 mm, comprimento 100 mm), usando misturas decrescentemente polares de água (contendo 1 % de amônia) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura para produzir (S)-4-amino-N-(1-(4-clorofenil)-3- hidroxipropil)-1-(5-ciclolopropil-7H-pirrolo[2,3-d]pirimidin-4-il)-piperidina-4- carboxamida (25,0 mg, 7,2 %) como um sólido branco.
[000135] 1H RMN (400,13 MHz, DMSO-d6) δ 0,66 - 0,70 (2H, m), 0,86 - 0,91 (2H, m), 1,41 - 1,50 (2H, m), 1,81 - 2,13 (5H, m), 3,35 - 3,43 (4H, m), 3,99 - 4,07 (2H, m), 4,54 (1H, t), 4,90 (1H, d), 6,90 (1H, s), 7,32 - 7,38 (4H, m), 8,18 (1H, s), 8,47 (1H, d), 11,46 (1H, s). MS m/e MH+ 470. Exemplo 28: 4-amino-N-(1 -(4-clorofenil)-3-(metilamino)propil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[000136] Cloreto de hidrogênio (4 M em dioxano, 0,461 ml, 1,84 mmol) foi adicionado ao 4-(1-(4-clorofenil)-3-(metilamino)propil- carbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (Intermediário 68) (10 mg, 0,02 mmol) em uma mistura de DCM (3 ml) e metanol (1 ml) a 22° C. A solução resultante foi agitada a 22° C por 2 horas. A mistura foi evaporada e o resíduo foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando 30 % (NH3 2 M em MeOH) em DCM e as frações puras foram evaporadas até a secura para produzir 4-amino-N-(1-(4-clorofenil)-3-(metilamino)propil)-1-(7H- pirrolo[2,3-d]-pirimidin-4-il)piperidina-4-carboxamida (7 mg, 86 %) como uma goma incolor.
[000137] 1H RMN (399,902 MHz, DMSO) δ 1,44 (2H, m), 1,83 (2H, dt), 1,87 - 2,01 (2H, m), 2,15 (2H, m), 2,25 (2H, br.s), 2,42 (2H, m), 3,56 (2H, m), 4,38 (2H, m), 4,84 (1H, br.s), 6,58 (1H, d), 7,16 (1H, d), 7,32 - 7,38 (4H, m), 8,13 (1H, s), 8,57 (1H, s), 11,63 (1H, s). MS m/e MH+442,4.
[000138] Os compostos da invenção listados na Tabela A abaixo foram fabricados a partir de materiais de partida apropriados usando um processo análogo a aquele descrito no Exemplo 2. Tabela A
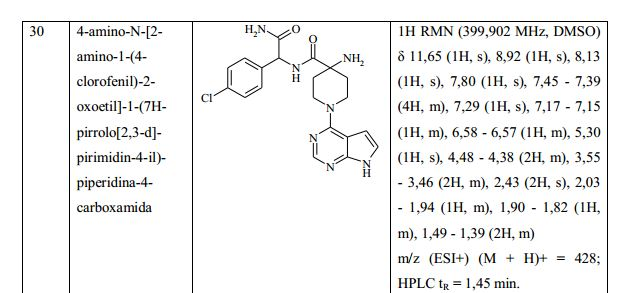
[000139] Os compostos da invenção listados na Tabela B abaixo foram fabricados a partir de materiais de partida apropriados usando um processo análogo a aquele descrito no Exemplo 9 via alternativa 1. Tabela B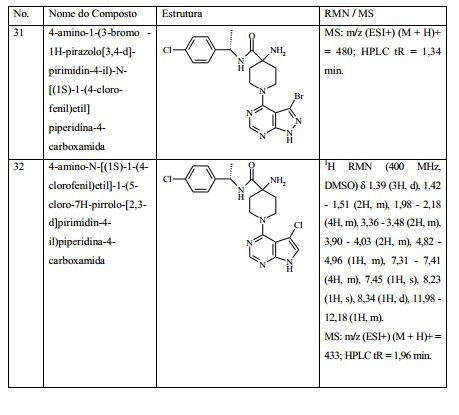
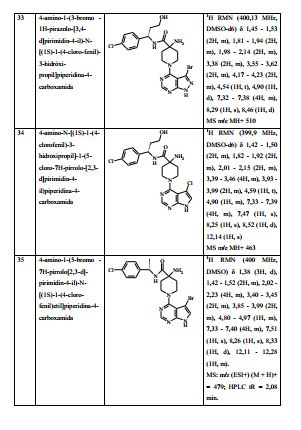

[000140] Os compostos da invenção listados na Tabela C abaixo foram fabricados a partir de materiais de partida apropriados usando um processo análogo a aquele descrito no Exemplo 9 via alternativa 2. Tabela C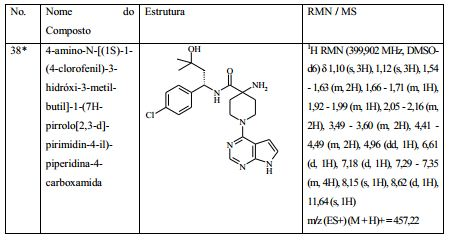
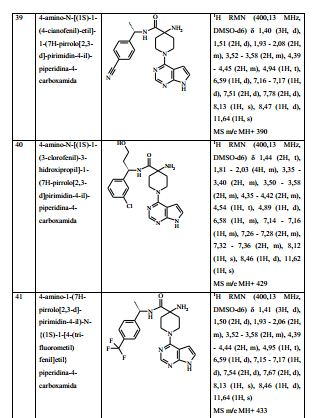 * Fabricada pela mesma metodologia de ligação de HATU como aquela descrita no Exemplo 9 via alternativa 2 e desprotegida com TFA usando a mesma metodologia como descrita para o exemplo 8.
* Fabricada pela mesma metodologia de ligação de HATU como aquela descrita no Exemplo 9 via alternativa 2 e desprotegida com TFA usando a mesma metodologia como descrita para o exemplo 8.
[000141] Os compostos da invenção listados na Tabela D abaixo foram fabricados a partir de materiais de partida apropriados usando um processo análogo a aquele descrito no Exemplo 16. Tabela D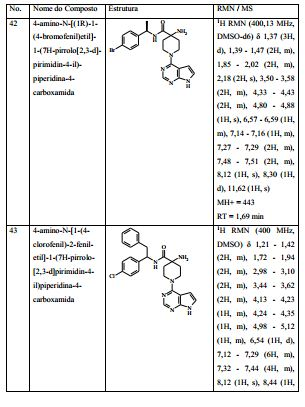
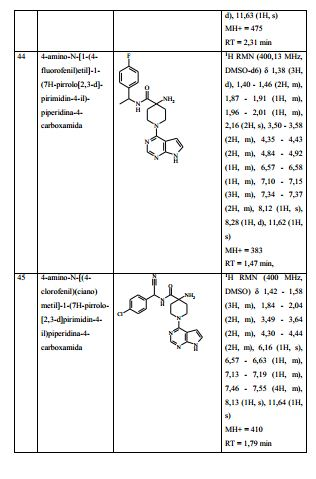

[000142] Os compostos da invenção listados na Tabela E abaixo foram fabricados a partir de materiais de partida apropriados usando um processo análogo a aquele descrito no Exemplo 25. Tabela E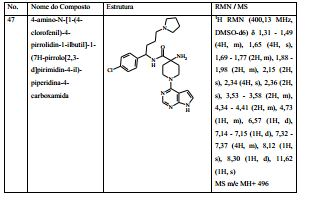
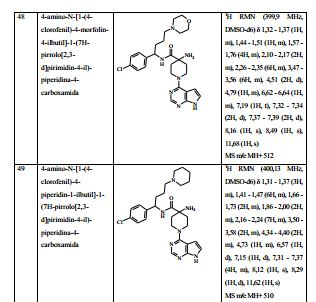 Exemplo 49A e 49B: 4-amino-N-[(1S)-1-(4-clorofenil)-4-piperidin-1- ilbutil]-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida e 4- amino-N-[(1R)-1-(4-clorofenil)-4-piperidin-1-ilbutil]-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida
Exemplo 49A e 49B: 4-amino-N-[(1S)-1-(4-clorofenil)-4-piperidin-1- ilbutil]-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida e 4- amino-N-[(1R)-1-(4-clorofenil)-4-piperidin-1-ilbutil]-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida
[000143] Os estereoisômeros individuais do racemato do Exemplo 49 foram separados usando a mesma metodologia como aquela descrita para os Exemplos 11A e 11B.
[000144] 1H RMN (500,13 MHz, DMSO-d6) δ 1,31 - 1,41 (4H, m), 1,44 - 1,48 (6H, m), 1,68 - 1,71 (2H, m), 1,90 - 1,98 (4H, m), 2,21 (6H, m), 3,54 - 3,56 (2H, m), 4,38 (2H, t), 4,73 (1H, d), 6,58 (1H, q), 7,15 (1H, q), 7,32 - 7,37 (4H, m), 8,12 (1H, s), 8,29 (1H, s), 11,63 (1H, s) MS m/e MH+ 510
[000145] 1H RMN (500,13 MHz, DMSO-d6) δ 1,31 - 1,41 (4H, m), 1,44 - 1,48 (7H, m), 1,68 - 1,71 (2H, m), 1,90 - 1,98 (2H, m), 2,21 (6H, m), 3,54 - 3,56 (2H, m), 4,38 (2H, t), 4,73 (1H, d), 6,58 (1H, q), 7,15 (1H, q), 7,32 - 7,37 (4H, m), 8,12 (1H, s), 8,29 (1H, s), 11,63 (1H, s) MS m/e MH+ 510
[000146] Os compostos da invenção listados na Tabela F abaixo foram fabricados a partir de materiais de partida apropriados usando um processo análogo a aquele descrito no Exemplo 26. Tabela F

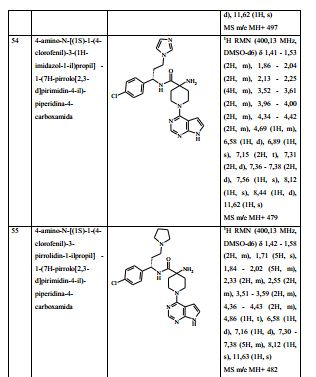 * Fabricada pela mesma metodologia como aquela descrita para o Exemplo 26 partindo de 1-(2,4-dimetoxibenzil)piperazina. A desproteção de TFA na etapa final foi aquecida a 80° C para remover o Boc e grupos dimetoxibenzila. Exemplo 56: 4-amino-N-(1 -(4-clorofenil)-2-sulfamoiletil)-1-(7H-pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida
* Fabricada pela mesma metodologia como aquela descrita para o Exemplo 26 partindo de 1-(2,4-dimetoxibenzil)piperazina. A desproteção de TFA na etapa final foi aquecida a 80° C para remover o Boc e grupos dimetoxibenzila. Exemplo 56: 4-amino-N-(1 -(4-clorofenil)-2-sulfamoiletil)-1-(7H-pirrolo- [2,3-d]pirimidin-4-il)piperidina-4-carboxamida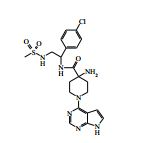
[000147] 4-(1-(4-clorofenil)-2-(metilsulfonamido)etilcarbamoil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (Intermediário 76) (100 mg, 0,17 mmol) foi tratado com ácido trifluoroacético (2 ml). A solução foi agitada por 1 hora na temperatura ambiente. A mistura foi concentrada sob pressão reduzida. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O resíduo foi carregado na coluna em metanol e lavado com metanol. O produto desejado foi eluído da coluna usando amônia 2 M em metanol e as frações puras foram evaporadas até a secura para produzir 4-amino-N-(1-(4-clorofenil)-2-(metilsulfonamido)etil)-1- (7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida (79 mg, 95 %) como um sólido incolor.
[000148] 1H RMN (399,9 MHz, DMSO-d6) δ 1,38 - 1,53 (2H, m), 1,85 - 2,07 (2H, m), 2,20 (2H, br, s), 2,85 (3H, s), 3,57 (2H, m), 4,34 - 4,46 (2H, m), 4,87 - 4,94 (1H, m), 6,57 - 6,60 (1H, m), 7,12 - 7,19 (2H, m), 7,35 - 7,43 (4H, m), 8,13 (1H, s), 8,46 (1H, br, s), 11,64 (1H, s) MS m/e MH+ = 492 Exemplo 57: 4-amino-N-(1-(4-clorofenil)-2-sulfamoiletil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida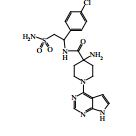
[000149] 4-(1-(4-clorofenil)-2-sulfamoiletilcarbamoil)-1-(7H-pirrolo- [2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (Intermediário 81) (153 mg, 0,26 mmol) foi tratado com ácido trifluoroacético (4 ml). A solução foi agitada por 30 minutos na temperatura ambiente. A mistura foi concentrada sob pressão reduzida e o resíduo foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia em metanol (2 M) e as frações puras foram evaporadas até a secura para produzir 4-amino- N-(1-(4-clorofenil)-2-sulfamoiletil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxamida (125 mg, 99 %) como um sólido incolor.
[000150] 1H RMN (399,9 MHz, DMSO-d6) δ 1,35 - 1,53 (2H, m), 1,86 - 2,04 (2H, m), 3,35 - 3,40 (1H, m), 3,52 - 3,62 (2H, m), 3,68 (1H, dd), 4,33 - 4,41 (2H, m), 5,24 - 5,29 (1H, m), 6,56 - 6,60 (1H, m), 6,88 (2H, s), 7,13 - 7,17 (1H, m), 7,39 (4H, s), 8,13 (1H, s), 8,68 (1H, br, s), 11,63 (1H, s)MS m/e MH+ = 478 Exemplo 58: N-(2-acetamido-1-(4-clorofenil)etil)-4-amino-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[000151] 4-(2-acetamido-1-(4-clorofenil)etilcarbamoil)-1-(7H-pirrolo [2,3- d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (Intermediário 84) (97 mg, 0,17 mmol) foi tratado com ácido trifluoroacético (2 ml). A solução foi agitada por 1 horas na temperatura ambiente. A mistura foi concentrada sob pressão reduzida. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O resíduo foi carregado na coluna em metanol e lavado com metanol. O produto desejado foi eluído da coluna usando amônia 2 M em metanol e as frações puras foram evaporadas até a secura para produzir N-(2-acetamido-1-(4- clorofenil)etil)-4-amino-1-(7H-pirrolo-[2,3-d]pirimidin-4-il)piperidina-4- carboxamida (80 mg, quant.) como um uma película creme.
[000152] 1H RMN (399,9 MHz, DMSO-d6) δ 1,43 (2H, t), 1,79 (3H, s), 1,83 - 2,04 (2H, m), 2,20 (2H, br, s), 3,32 - 3,38 (2H, m), 3,58 (2H, q), 4,32 - 4,42 (2H, m), 4,82 - 4,88 (1H, m), 6,56 - 6,60 (1H, m), 7,14 - 7,18 (1H, m), 7,33 (2H, d), 7,38 (2H, d), 7,94 (1H, t), 8,13 (1H, s), 8,42 - 8,50 (1H, m), 11,63 (1H, s) MS m/e MH+ = 456 Exemplo 59: 4-amino-N-(1-(4-clorofenil)-2-(1H-imidazol-2-il)etil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[000153] Ácido trifluoroacético (5 ml, 64,90 mmol) foi adicionado ao 4- (1-(4-clorofenil)-2-(1-tritil-1H-imidazol-2-il)etilcarbamoil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (Intermediário 89) (310 mg, 0,38 mmol) na temperatura ambiente e a solução resultante agitada por 30 minutos. Água (5 a 6 gotas) foi depois adicionada e a agitação continuada por mais 30 minutos. A solução resultante foi diluída com metanol e aplicada a uma coluna SCX 10 g que foi depois eluída com MeOH seguida pelo NH3 2 N (em MeOH). As frações contendo o produto foram combinadas e concentradas pela evaporação depois trituradas com MeCN / DMF (1:1) para dar um ppt. amarelo claro. O precipitado foi coletado pela filtração, lavado com MeCN e secado sob vácuo para produzir 4-amino-N-(1-(4-clorofenil)-2-(1H-imidazol-2-il)etil)-1- (7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida (130 mg, 72,8 %) como um sólido creme.
[000154] 1H RMN (399,902 MHz, DMSO) δ 1,36 - 1,40 (2H, m), 1,85 - 1,93 (2H, m), 3,08 (2H, d), 3,55 (2H, m), 4,29 - 4,35 (2H, m), 5,18 (1H, q), 6,57 (1H, d), 6,85 (2H, s), 7,16 (1H, d), 7,25 (2H, d), 7,31 (2H, d), 8,12 (1H, s), 8,89 (1H, d), 11,63 (1H, br s) m/z (ESI+) (M + H)+ = 465; HPLC tR = 1,62 min. Exemplo 60: 4-amino-N-[1-(4-clorofenil)-2-(1H-pirazol-1-il)etil]-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[000155] TFA (0,7 ml) foi adicionado a uma suspensão de 4-(1-(4- clorofenil)-2-(1H-pirazol-1-il)etilcarbamoil)-1-(7H-pirrolo[2,3-d]-pirimidin- 4-il)piperidin-4-ilcarbamato de terc-butila (180 mg, 0,32 mmol) em diclorometano (Intermediário 93) (7 ml) sob argônio. A solução resultante foi agitada na temperatura ambiente a noite. Os solventes foram removidos a vácuo e a mistura de reação foi purificada pela HPLC preparativa usando uma coluna de fase reversa Waters X- Bridge (C-18, sílica de 5 mícrons, diâmetro 19 mm, comprimento 100 mm) e misturas decrescentemente polares de água (contendo 0,2 % de carbonato de amônio) e acetonitrila como eluente. As frações foram evaporadas até a secura para produzir 4-amino-N-[1-(4-clorofenil)-2- (1H-pirazol-1-il)etil]-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxamida como um pó branco.
[000156] 1H RMN (500 MHz, DMSO-d6) δ 1,33 (2H, m), 1,83 (2H, m), 2,14 (2H, s), 3,53 (2H, m), 4,28 (2H, m), 4,43 (2H, m), 5,22 (1H, d), 6,15 (1H, t), 6,56 (1H, q), 7,15 (1H, t), 7,30 (2H, d), 7,36 (2H d), 7,44 (1H, d), 7,55 (1H, d), 8,11 (1H, s), 8,81 (1H, br d), 11,64 (1H, s); m/z (ESI+) (M + H)+ = 465. Exemplo 61: 4-amino-N-[1-(4-clorofenil)-2-(3-metilisoxazol-5-il)etil]-1- (7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida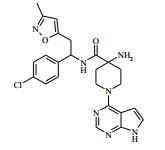
[000157] 4-Amino-N-[1-(4-clorofenil)-2-(3-metilisoxazol-5-il)etil]-1- (7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida foi fabricada a partir de materiais de partida apropriados usando um processo análogo a aquele descrito por exemplo 60.
[000158] 1H RMN (500 MHz, DMSO-d6) δ 1,35 (2H, d), 1,87 (2H, m), 2,14 (2H, s), 2,15 (3H, s), 3,17 - 3,33 (2H, m), 3,53 (2H, m), 4,31 (2H, m), 5,15 (1H, d), 6,06 (1H, s), 6,56 (1H, d), 7,16 (1H, t), 7,39 (4H, m), 8,12 (1H, s), 8,60 (1H, br d), 11,65 (1H, s). m/z (ESI+) (M + H)+ = 480 Exemplo 62: 4-amino-N-(1 -(4-clorofenil)-2-(tiazol-2-il)etil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida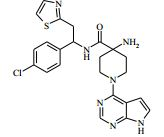
[000159] TFA (2,0 ml) foi adicionado a uma suspensão de 4-(1-(4- clorofenil)-2-(tiazol-2-il)etilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidin-4-ilcarbamato de terc-butila (Intermediário 96) (918 mg, 0,95 mmol) em diclorometano (20 ml) sob argônio. A solução resultante foi agitada na temperatura ambiente por uma noite. Os solventes foram removidos a vácuo e a mistura de reação foi purificada pela HPLC preparativa usando uma coluna de fase reversa Waters X-Bridge (C-18, sílica de 5 mícrons, diâmetro 19 mm, comprimento 100 mm) e misturas decrescentemente polares de água (contendo 0,2 % de carbonato de amônio) e acetonitrila como eluente. As frações foram evaporadas até a secura para produzir 4-amino-N-(1- (4-clorofenil)-2-(tiazol-2-il)etil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxamida (259 mg, 56,8 %) como um pó branco.
[000160] 1H RMN (500 MHz, DMSO-d6) δ 1,35 (2H, d), 1,85 (2H, m), 2,07 (2H, br s), 3,41 - 3,56 (4H, m), 4,30 (2H, m), 5,20 (1H, d), 6,56 (1H, d), 7,15 (1H, t), 7,36 (4H, m), 7,54 (1H, d), 7,70 (1H, d), 8,11 (1H, s), 8,75 (1H, br d), 11,70 (1H, br s); m/z (ESI+) (M + H)+ = 482 Exemplo 63: 4-amino-N-(1-(4-clorofenil)-3-(dimetilamino)-3-oxopropil) - 1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[000161] TFA (1,0 ml) foi adicionado a uma suspensão de 4-(1-(4- clorofenil)-3-(dimetilamino)-3-oxopropilcarbamoil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (Intermediário 98) (770 mg, 0,24 mmol) em diclorometano (10 ml) sob argônio. A solução resultante foi agitada na temperatura ambiente por uma noite. Os solventes foram removidos a vácuo e a mistura de reação foi purificada pela HPLC preparativa usando uma coluna de fase reversa Waters X-Bridge (C-18, sílica de 5 mícrons, diâmetro 19 mm, comprimento 100 mm) e misturas decrescentemente polares de água (contendo 0,2 % de carbonato de amônio) e acetonitrila como eluente. As frações foram evaporadas até a secura para produzir 4-amino-N-(1- (4-clorofenil)-3-(dimetilamino)-3-oxopropil)-1-(7H-pirrolo[2,3-d]pirimidin- 4-il)-piperidina-4-carboxamida (74,0 mg, 64,8 %) como um pó branco.
[000162] 1H RMN (500 MHz, DMSO-d6) δ 1,43 (2H, m), 1,94 (2H, m), 2,75 (3H, s), 2,8 (2H, m), 2,89 (3H, s), 3,52 (2H, m), 4,40 (2H, m), 5,15 (1H, d), 6,58 (1H, d), 7,16 (1H, t), 7,35 (4H, s), 8,12 (1H, s), 8,76 (1H, br d), 11,66 (1H, br s); m/z (ESI+) (M + H)+ = 470 Exemplo 64: 4-amino-N-(1-(4-clorofenil)-3-metóxi-propil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[000163] Cloreto de hidrogênio (4 M em 1,4-dioxano, 1,151 ml, 4,60 mmol) foi adicionado ao 4-(1-(4-clorofenil)-3-metóxi-propilcarbamoil)-1- (7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (Intermediário 101) (50 mg, 0,09 mmol) em uma mistura de DCM (5 ml) e metanol (2 ml) a 22° C. A solução resultante foi agitada a 22° C por 2 dias. A mistura foi evaporada e o resíduo foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 2 M; as frações puras foram evaporadas até a secura e o resíduo foi triturado com éter dietílico para produzir 4-amino-N-(1-(4-clorofenil)-3-metóxi-propil)-1- (7H-pirrolo [2,3-d]pirimidin-4-il)piperidina-4-carboxamida (39,0 mg, 96 %) como um sólido branco.
[000164] 1H RMN (399,902 MHz, DMSO) δ 1,44 (2H, m), 1,88 - 2,02 (5H, m), 2,46 (2H, s), 3,21 (3H, s), 3,28 (2H, t), 3,55 (2H, m), 4,39 (2H, m), 4,87 (1H, dt), 6,59 (1H, dd), 7,16 (1H, dd), 7,33 (2H, d), 7,37 (2H, d), 8,13 (1H, s), 8,45 (1H, d), 11,63 (1H, s); m/z (ESI+) (M + H)+ = 443,4; HPLC tR = 1,87 min. Exemplo 65: 4-amino-N-(1 -(4-clorofenil)-3-sulfamoilpropil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[000165] Cloreto de hidrogênio mmol) foi adicionado ao 4-(1-(4-clorofenil)-3-sulfamoilpropil-carbamoil)- 1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (Intermediário 105) (16 mg, 0,03 mmol) em uma mistura de DCM (5 ml) e metanol (5 ml) a 22° C. A solução resultante foi agitada a 22° C por 20 horas. A mistura foi evaporada e o resíduo foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando 30 % (NH3 2 M em MeOH) em DCM e as frações puras foram evaporadas até a secura para produzir 4-amino-N-(1-(4-clorofenil)-3-sulfamoilpropil)-1-(7H-pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamida (12,00 mg, 90 %) como uma goma incolor.
[000166] 1H RMN (399,902 MHz, DMSO) δ 1,48 (2H, m), 1,90 - 2,06 (2H, m), 2,09 - 2,24 (2H, m), 2,87 (1H, ddd), 3,02 (1H, ddd), 3,56 (2H, m), 3,56 (2H, d), 4,41 (2H, m), 4,91 (1H, br.s), 6,59 (1H, dd), 6,80 (2H, s), 7,16 (1H, dd), 7,38 - 7,43 (4H, m), 8,13 (1H, s), 8,46 (1H, s), 11,64 (1H, s); m/z (ESI+) (M + H)+ = 492,4, 494,3; HPLC tR = 1,60 min. Exemplo 66: 4-amino-N-(3-amino-1-(4-clorofenil)-3-oxopropil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida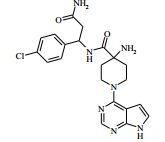
[000167] HCl em 1,4-dioxano (4 M) (0,228 ml, 0,91 mmol) foi adicionado ao 4-(3-amino-1-(4-clorofenil)-3-oxopropilcarbamoil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (Intermediário 108) (99 mg, 0,18 mmol) em DCM (4 ml) a 20° C. A solução resultante foi agitada a 20° C por 3 horas. A mistura de reação foi filtrada através de um copo de PTFE e o sólido coletado foi purificado pela HPLC preparativa (coluna Waters XTerra C18, sílica 5μ, diâmetro 30 mm, comprimento 100 mm), usando misturas decrescentemente polares de água (contendo 1 % de NH3) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura para produzir 4-amino-N-(3-amino-1-(4- clorofenil)-3-oxo-propil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxamida (9,00 mg, 11,1 %) como um sólido creme.
[000168] 1H RMN (399,9 MHz, DMSO-d6) δ 1,40 - 1,43 (2H, m), 1,93 - 1,96 (2H, m), 2,18 (2H, s), 3,54 - 3,56 (2H, m), 4,36 - 4,40 (2H, m), 5,12 (1H, d), 6,58 (1H, d), 6,81 (1H, s), 7,15 - 7,16 (1H, m), 7,32 - 7,37 (5H, m), 8,13 (1H, s), 8,76 (1H, d), 11,63 (1H, s), (nenhum NH2 visível). m/z (ESI+) (M + H)+ = 442; HPLC tR = 1,47 min. Exemplo 67: 4-amino-N-(1-(4-clorofenil)-3-ureidopropil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida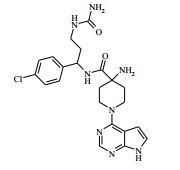
[000169] TFA (3 ml) foi adicionado ao 4-(1-(4-clorofenil)-3-ureído- propilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-il- carbamato de terc-butila (Intermediário 112) (379 mg, 0,66 mmol). A reação foi deixada agitar a 20° C por 4 horas e depois submetida a vácuo até a secura. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 0,35 M e as frações puras foram evaporadas até a secura para produzir material bruto. O produto bruto foi purificado pela HPLC preparativa (coluna Waters XBridge Prep C18 OBD, sílica 5μ, diâmetro 19 mm, comprimento 100 mm), usando misturas decrescentemente polares de água (contendo 1 % de NH3) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura para produzir 4-amino-N-(1-(4- clorofenil)-3-ureidopropil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxamida (14,00 mg, 5,60 %) como um sólido branco.
[000170] 1H RMN (399,9 MHz, DMSO-d6) δ 1,41 - 1,49 (2H, m), 1,78 - 1,88 (2H, m), 2,01 (1H, d), 2,91 (1H, t), 2,97 (2H, t), 3,55 (2H, d), 4,37 (2H, d), 4,78 (1H, d), 5,40 (2H, s), 5,95 (1H, t), 6,58 - 6,59 (1H, m), 7,15 - 7,16 (1H, m), 7,33 - 7,39 (4H, m), 8,13 (1H, s), 8,36 (1H, d), 11,63 (1H, s); m/z (ESI+) (M + H)+ = 471; HPLC tR = 1,50 min. Exemplo 68: 4-amino-N-(1-(4-clorofenil)-2-cianoetil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida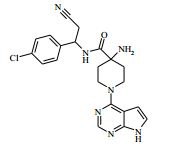
[000171] HCl (4 M) em 1,4-dioxano (2,011 ml, 8,04 mmol) foi adicionado ao 4-(1-(4-clorofenil)-2-cianoetilcarbamoil)-1-(7H-pirrolo- [2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (Intermediário 114) (0,281 g, 0,54 mmol) em DCM (8 ml) a 20° C. A solução resultante foi agitada a 20° C por 18 horas. A reação foi submetida a vácuo até a secura e foi purificada pela HPLC preparativa (coluna Waters XTerra C18, sílica 5μ, diâmetro 19 mm, comprimento 100 mm), usando misturas decrescentemente polares de água (contendo 1 % de NH3) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura para produzir 4-amino-N-(1- (4-clorofenil)-2-cianoetil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxamida (0,096 g, 42,2 %) como um sólido branco.
[000172] 1H RMN (399,9 MHz, DMSO-d6) δ 1,42 - 1,46 (2H, m), 1,52 (2H, d), 1,95 (1H, d), 1,98 - 2,01 (1H, m), 3,08 (1H, t), 3,56 - 3,59 (2H, m), 4,38 - 4,42 (2H, m), 5,18 (1H, s), 6,59 - 6,60 (1H, m), 7,16 (1H, t), 7,44 (4H, m), 8,13 (1H, s), 11,65 (1H, s), (nenhum NH2 visível), m/z (ESI+) (M + H)+ = 424; HPLC tR = 1,82 min. Exemplo 69: 4-amino-N-(1 -(4-clorofenil)-3-(metilsulfonamido)propil)-1- (7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[000173] HCl (4 M) em 1,4-dioxano (1,615 ml, 6,46 mmol) foi adicionado ao 4-(1-(4-clorofenil)-3-(metilsulfonamido)propilcarbamoil)- 1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (Intermediário 117) (261 mg, 0,43 mmol), A suspensão resultante foi agitada a 20° C por 1 hora. Nenhuma reação. TFA (1 ml) foi depois adicionado à reação e agitado por 16 horas. A reação foi submetida a vácuo até a secura e foi purificada pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 0,35 M e as frações puras foram evaporadas até a secura para produzir material bruto. O produto bruto foi purificado pela HPLC preparativa (coluna Waters XBridge Prep C18 OBD, sílica 5μ, diâmetro 19 mm, comprimento 100 mm), usando misturas decrescentemente polares de água (contendo [Tampão de AP-HPLC]) e MeCN como eluentes. As frações contendo o composto desejado foram evaporadas até a secura para produzir 4-amino-N-(1-(4- clorofenil)-3-(metilsulfonamido)propil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxamida (142 mg, 65,2 %) como um sólido branco.
[000174] 1H RMN (399,9 MHz, DMSO-d6) δ 1,40 - 1,48 (2H, m), 1,86 - 1,90 (2H, m), 1,93 - 1,97 (2H, m), 2,17 (2H, s), 2,88 (3H, s), 2,93 - 2,97 (2H, m), 3,53 - 3,60 (2H, m), 4,37 (2H, t), 4,87 (1H, d), 6,57 - 6,59 (1H, m), 7,00 (1H, t), 7,15 - 7,16 (1H, m), 7,35 - 7,40 (4H, m), 8,13 (1H, s), 8,38 (1H, d), 11,63 (1H, s), m/z (ESI+) (M + H)+ = 506; HPLC tR = 1,62 min. Intermediário 1: ácido 4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxílico
[000175] A uma mistura de ácido 4-[(2-metilpropan-2-il)oxicarbonil- amino]piperidina-4-carboxílico (115,6 g) em CH3CN-H2O (6 L) foi adicionado NaHCO3 (181 g), seguido pelo 4-cloro 7H-pirrol[2,3-d]- pirimidina (72,7 g). A mistura foi aquecida ao refluxo durante a noite sob nitrogênio por 24 horas e depois extraída com EtOAc (1L x 4). A camada aquosa foi concentrada e MeOH (1,5 L) foi adicionado. A mistura foi agita por 30 min a 45° C e filtrada. O filtrado foi concentrado mais uma vez e dissolvido em H2O (300 ml). HCl 6 N foi adicionado até que o pH atingisse 4,5 (cerca de 80 ml). A mistura foi filtrada e a torta foi secada sob vácuo para produzir o produto bruto, que foi purificado ainda pela cromatografia em sílica gel (eluindo com MeOH: DCM = 1:3) para produzir ácido 4-[(2-metilpropan-2-il)oxicarbonilamino]-1- (3,5,7-triazabiciclolo[4,3,0]nona-2,4,8,10-tetraen-2-il)piperidina-4- carboxílico como sólido cinza claro (105 g, 63 %).
[000176] 1H RMN (400,13 MHz, DMSO-d6) δ 1,40 (9H, s), 1,88 - 1,95 (2H, m), 2,02 - 2,06 (2H, m), 3,44 - 3,51 (2H, m), 4,30 (2H, d), 6,60 - 6,61 (1H, m), 7,16 - 7,18 (1H, m), 7,29 (1H, s), 8,14 (1H, s), 11,68 (1H, s). MS m/e MH+ 362. Intermediário 2: (4-clorofenil)(ciclolopropil)metanona O-metil oxima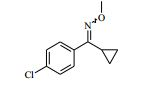
[000177] Cloridreto de metoxilamina (2,004 g) foi adicionado à (4- clorofenil)(ciclolopropil)metanona (2,71 g) em piridina (60 ml) a 25° C. A solução resultante foi agitada a 25° C por 24 horas. A piridina foi removida a vácuo, e o sólido residual extraído com éter. A filtração e a evaporação do solvente deu a (4-clorofenil)(ciclolopropil)metanona O- metil oxima bruta (2,75 g, 87 %) como um óleo amarelo. Este material foi usado diretamente na etapa seguinte sem outra purificação. MS m/e MH+ 210. Intermediário 3: (4-clorofenil)(ciclolopropil)metanamina
[000178] Complexo de borano tetraidrofurano (1 N em THF, 65,6 ml) foi adicionado à (4-clorofenil)(ciclolopropil)metanona O-metil oxima (Intermediário 2) (2,75 g) em THF (100 ml) a 25° C sob nitrogênio. A solução resultante foi agitada ao refluxo por 3 horas, depois esfriada a 0° C. Água foi cuidadosamente adicionada, seguida pelo NaOH aquoso (20 %, 100 ml). A mistura resultante foi agitada a refluxo durante a noite, depois deixada esfriar até a temperatura ambiente. O produto foi extraído em hexano, depois secado em sulfato de sódio, filtrado e evaporado para produzir (4-clorofenil)(ciclolopropil)metanamina como um óleo claro (2,205 g, 93 %).
[000179] 1H RMN (400,13 MHz, CDCl3) δ 0,24 - 0,29 (1H, m), 0,30 - 0,13 (1H, m), 0,45 - 0,50 (1H, m), 0,58 - 0,64 (1H, m), 1,01 - 1,09 (1H, m), 1,77 (2H, s), 3,20 (1H, d), 7,28 - 7,32 (2H, m), 7,33 - 7,36 (2H, m). Intermediário 4: 2-amino-2-(4-clorofenil)acetonitrile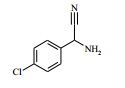
[000180] Bis(trimetilsilil)amida de lítio (42,7 ml) foi adicionada ao 4- clorobenzaldeído (5 g) em THF (100 ml) a -40° C sob nitrogênio. A solução resultante foi aquecida até a temperatura ambiente e agitada por 4 horas. α-Hidroxiisobutironitrila (acetona cianoidrina, 6,50 ml) foi depois adicionada, e a mistura de reação foi agitada a 25° C por mais 12 horas, depois extinta com NaHCO3 saturado (50 ml), extraído com EtOAc (3 x 100 ml), a camada orgânica foi secadas em MgSO4, filtrada e evaporada. O material bruto foi depois purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição 0 to 100 % de EtOAc em isoexano. As frações puras foram evaporadas até a secura para produzir 2-amino-2-(4-clorofenil)acetonitrila como um óleo incolor, que solidificou no repouso para dar um sólido branco (3,40 g, 57,4 %).
[000181] 1H RMN (400,13 MHz, CDCl3) δ 1,85 (2H, s), 4,82 (1H, s), 7,30 - 7,34 (2H, m), 7,39 - 7,43 (2H, m). Intermediário 5: 4-((4-clorofenil)(ciano)metilcarbamoil)-1-(7H-pirrolo- [2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila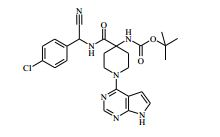
[000182] Hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (HATU, 0,456 g) foi adicionado ao ácido 4-(tercbutoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxílico (Intermediário 1) (0,361 g), 2-amino-2-(4-clorofenil)-acetonitrila (Intermediário 4) (0,167 g) e N-etildiisopropilamina (0,523 ml, 3,00 mmol) em DMA (5 ml) a 25º C. A solução resultante foi agitada a 50° C por 1 hora. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 7 M e as frações puras foram evaporadas até a secura para produzir 4-((4-clorofenil)(ciano)metilcarbamoil)-1-(7Hpirrolo[2,3-d]pirimidin-4-il) piperidin-4-ilcarbamato de terc-butila como um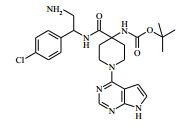
[000183] Níquel de Raney® (0,257 g), foi adicionado ao 4-((4-cloro- fenil)(ciano)metilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)-piperidin- 4-ilcarbamato de terc-butila (Intermediário 5) (0,510 g) em etanol (30 ml). Hidróxido de amônio (0,039 ml) foi adicionado. Esta mistura foi colocada sob um balão de hidrogênio e agitada por 48 horas. A mistura de reação foi filtrada através de celite e o solvente evaporado até a secura. A mistura de reação foi evaporada até a secura e redissolvida em DCM (200 ml), e lavada com água (125 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto, 4-(2-amino-1-(4-clorofenil)etilcarbamoil)-1-(7H-pirrolo- [2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila como uma goma incolor (0,514 g, 100 %). Este material foi usado bruto na etapa seguinte sem outra purificação. MS m/e MH+ 514. Intermediário 7: 4-etil 4-cianopiperidina-1,4-dicarboxilato de 1-terc- butila
[000184] Uma solução de LDA (107 ml, 214,01 mmol) foi adicionada a uma solução agitada de 4-cianopiperidina-1-carboxilato de terc-butila (30 g, 143 mmol) em THF (250 ml) a -78° C, sob nitrogênio. A solução resultante foi agitada a -78° C por 30 minutos. Cloroformiato de etila (16,37 ml, 171,2 mmol) foi adicionado. A solução resultante foi agitada e deixada aquecer até a temperatura ambiente. A mistura de reação foi extinta com NaHCO3 saturado (250 ml), extraída com DCM, e a camada orgânica foi lavada com salmoura saturada (100 ml) depois secadas em MgSO4, filtrada e evaporada para produzir o material bruto como um óleo laranja. Este material foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição 10 % de EtOAc em isoexano. As frações puras foram evaporadas até a secura para produzir 4-etil 4-cianopiperidina-1,4- dicarboxilato de 1-terc-butila (20,8 g, 51,6 %) como um óleo amarelo.
[000185] 1H RMN (400,13 MHz, CDCl3) δ 1,33 (3H, t), 1,46 (9H, s), 1,96 - 2,00 (2H, m), 2,04 - 2,08 (2H, m), 3,12 (2H, s), 4,09 - 4,14 (2H, m), 4,29 (2H, q). Intermediário 8: 4-etil 4-(aminometil)piperidina-1,4-dicarboxilato de 1- terc-butila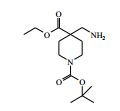
[000186] Óxido de paládio (IV) (0,724 g, 3,19 mmol) e 4-etil 4- cianopiperidina-1,4-dicarboxilato de 1-terc-butila (Intermediário 7) (9g, 31,9 mmol) em ácido acético (100 ml) foram agitados sob uma atmosfera de hidrogênio a 5 bar e 25° C por 1 dia. O produto bruto foi filtrado através de celite e o filtrado purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 7 M e as frações puras foram evaporadas até a secura para produzir 4-etila 4-(aminometil)piperidina- 1,4-dicarboxilato de 1-terc-butila (7,59 g, 83 %) como um óleo incolor.
[000187] 1H RMN (400,13 MHz, CDCl3) δ 1,27 - 1,28 (3H, m), 1,30 - 1,37 (2H, m), 1,41 (2H, s), 1,45 (9H, s), 2,10 (2H, d), 2,78 (2H, s), 2,91 - 2,97 (2H, m), 3,89 (2H, s), 4,21 (2H, q). Intermediário 9: 4-(aminometil)piperidina-4-carboxilato de etila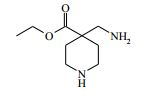
[000188] Cloreto de hidrogênio 4 M em dioxano (33,2 ml, 132,7 mmol) foi adicionado ao 4-etila 4-(aminometil)piperidina-1,4- dicarboxilato de 1-terc-butila (Intermediário 8) (7,6 g, 26,5 mmol) em dioxano (35 ml). A solução resultante foi agitada a 20° C por 3 horas. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 7 M e as frações puras foram evaporadas até a secura para produzir 4-(aminometil)piperidina-4-carboxilato de etila (3,34 g, 67,6 %) como um líquido amarelo.
[000189] 1H RMN (400,13 MHz, CDCl3) δ 1,23 - 1,30 (3H, m), 1,26 - 1,37 (2H, m), 2,12 (2H, d), 2,65 - 2,72 (2H, m), 2,77 (2H, s), 2,94 - 2,99 (2H, m), 4,21 (2H, q). Intermediário 10: 4-(aminometil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)- piperidina-4-carboxilato de etila
[000190] N-Etildiisopropilamina (3,70 ml, 21,26 mmol) foi adicionada ao 4-(aminometil)piperidina-4-carboxilato de etila (Intermediário 9) (3,3g, 17,7 mmol) e 4-cloro-7H-pirrolo[2,3-d]-pirimidina (2,72 g, 17,72 mmol) em DMA (35 ml). A solução resultante foi agitada a 60° C por 18 horas. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 7 M e as frações puras foram evaporadas até a secura para produzir 4-(aminometil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxilato de etila (5,08 g, 95 %) como um sólido bege.
[000191] 1H RMN (400,13 MHz, DMSO-d6) δ 1,22 (3H, t), 1,44 - 1,51 (2H, m), 2,04 - 2,07 (2H, m), 2,67 (2H, d), 3,23 - 3,30 (2H, m), 4,15 (2H, q), 4,39 - 4,44 (2H, m), 6,59 (1H, t), 7,16 - 7,17 (1H, m), 8,12 (1H, s), 11,67 (1H, s) MS m/e MH+ 304. Intermediário 11: 4-((terc-butoxicarbonilamino)metil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxilato de etila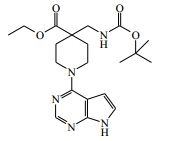
[000192] Dicarbonato de di-terc-butila (470 mg, 2,15 mmol) foi adicionado ao 4-(aminometil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)- piperidina-4-carboxilato de etila (Intermediário 10) (653 mg, 2,15 mmol) e trietilamina (0,300 ml, 2,15 mmol) em DCM (10 ml). A suspensão resultante foi agitada na temperatura ambiente por 2 horas. A mistura de reação foi diluída com DCM (50 ml), e lavada sequencialmente com água (50 ml) e salmoura saturada (50 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição 20 a 100 % de EtOAc em isoexano. As frações puras foram evaporadas até a secura para produzir 4-((terc- butoxicarbonilamino)metil)-1-(7H-pirrolo[2,3-d]-pirimidin-4-il)piperidina- 4-carboxilato de etila (468 mg, 53,9 %) como um óleo incolor que solidificou no repouso.
[000193] 1H RMN (400,13 MHz, DMSO-d6) δ 1,22 (3H, t), 1,36 - 1,38 (9H, m), 1,42 - 1,49 (2H, m), 2,05 (2H, d), 3,13 (2H, d), 3,20 (2H, t), 4,09 - 4,14 (2H, m), 4,45 (2H, d), 6,58 (1H, d), 6,94 (1H, t), 7,16 (1H, d), 8,13 (1H, d), 11,65 (1H, s). MS m/e MH+404. Intermediário 12: ácido 4-((terc-butoxicarbonilamino)metil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxílico
[000194] Hidróxido de lítio monoidratado (0,556 g, 13,26 mmol) foi adicionado ao 4-((terc-butoxicarbonilamino)metil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxilato de etila (Intermediário 11) (1,07g, 2,65 mmol) em água (6,25 ml), THF (25 ml) e etanol (25,00 ml). A solução resultante foi agitada a 20° C por 1 dia. A mistura de reação foi diluída com EtOAc (20 ml) e lavada com água (20 ml).O aquoso foi ajustado ao pH 5 com solução 1 M de ácido cítrico depois extraído com EtOAc (3 x 50 ml). Os extratos orgânicos foram lavados com salmoura saturada (25 ml) depois secados em MgSO4, filtrados e evaporados para produzir o produto desejado ácido 4-((terc- butoxicarbonilamino)metil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina- 4-carboxílico (0,628 g, 63,1 %) como uma espuma branca.
[000195] 1H RMN (400,13 MHz, DMSO-d6) δ 1,36 (9H, s), 1,44 - 1,51 (2H, m), 1,99 - 2,04 (2H, m), 3,14 (2H, d), 3,25 (2H, s), 4,43 - 4,46 (2H, m), 6,64 (1H, s), 6,84 (1H, t), 7,21 (1H, s), 8,16 (1H, s), 11,82 (1H, s) MS m/e MH+ 376. Intermediário 13: (4-(1-(4-clorofenil)etilcarbamoil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidin-4-il)metilcarbamato de (S)-terc-butila
[000196] HATU (0,251 g, 0,66 mmol) foi adicionado em uma porção ao ácido 4-((terc-butoxicarbonilamino)metil)-1-(7H-pirrolo[2,3-d]-pirimidin-4- il)piperidina-4-carboxílico (Intermediário 12) (0,225 g, 0,6 mmol), (S)-1-(4- clorofenil)etanamina (0,093 g, 0,60 mmol) e DIPEA (0,314 ml, 1,80 mmol) em DMA (10 ml) a 25° C sob nitrogênio. A solução resultante foi agitada a 60° C por 4 horas. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando metanol. HATU residual foi removido pela passagem da solução de metanol através de uma coluna de carbonato sustentada em sílica. O produto bruto assim obtido foi evaporado até a secura para produzir a (4-(1-(4-clorofenil)etilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidin-4-il)metilcarbamato de (S)-terc-butila (0,257 g, 83 %) como uma goma incolor. Este material foi usado diretamente na etapa seguinte sem outra purificação.
[000197] 1H RMN (400,13 MHz, DMSO-d6) δ 1,37 (9H, s), 1,38 (3H, d), 1,48 - 1,55 (2H, m), 2,17 (2H, d), 3,12 - 3,36 (4H, m), 4,28 - 4,34 (2H, m), 4,95 - 5,03 (1H, m), 6,65 (2H, s), 7,23 - 7,24 (1H, m), 7,35 (4H, s), 8,10 (1H, d), 8,18 (1H, s), 11,94 (1H, s). MS m/e MH+513. Intermediário 14: 4-(4-clorofenil)-4-(metoxiimino)butanoato de etila
[000198] Ácido 4-(4-Clorofenil)-4-oxobutanóico (2,0 g, 9,41 mmol), cloridreto de metoxilamina (0,982 g, 11,76 mmol) e bicarbonato de sódio (0,472 ml, 11,29 mmol) em etanol (30 ml) foram agitados e aquecidos a 80° C por 4 horas. A mistura resultante foi deixada esfriar até a temperatura ambiente e filtrada. O filtrado foi concentrado pela evaporação depois purificado pela cromatografia por vaporização instantânea em sílica, eluindo com 15 % de TBME em isoexano. As frações puras foram evaporadas até a secura para produzir 4-(4- clorofenil)-4-(metóxi-imino)butanoato de etila (1,790 g, 70,6 %) como um óleo incolor.
[000199] 1H RMN (399,902 MHz, CDCl3) δ 1,23 (3H, t), 2,53 (2H, t), 3,01 (2H, t), 3,98 (3H, s), 4,11 (2H, q), 7,33 (2H, d), 7,58 (2H, d). MS m/e MH+270. Intermediário 15: ácido 4-(4-clorofenil)-4-(metoxiimino)butanóico
[000200] Hidróxido de lítio (0,632 g, 26,40 mmol) foi adicionado em uma porção a 20° C a uma solução de 4-(4-clorofenil)-4-(metoxiimino)- butanoato de etila (Intermediário 14) (1,78 g, 6,60 mmol) em THF (30 ml) e água (20 ml). A solução resultante foi agitada na temperatura ambiente por 6 horas depois acidificada com HCl diluído e extraída com TBME (2 X). Os extratos combinados foram lavados com salmoura, secados em MgSO4 e evaporados para dar ácido 4-(4-clorofenil)-4- (metoxiimino)butanóico (1,520 g, 95 %) como um sólido incolor.
[000201] 1H RMN (399,902 MHz, CDCl3) δ 2,61 (2H, t), 3,01 (2H, t), 3,99 (3H, s), 7,34 (2H, d), 7,58 (2H, d). Intermediário 16: 4-amino-4-(4-clorofenil)butan-1-ol
[000202] Ácido 4-(4-clorofenil)-4-(metoxiimino)butanóico (Intermediário 15) (6,28 g, 26,00 mmol) em tetraidrofurano (8 ml) foi esfriado, sob uma atmosfera de N2, em um banho de metanol gelado depois tratado às gotas com complexo de borano-tetraidrofurano (1,0 M em THF) (26 ml, 26,00 mmol) em um período de 20 minutos. A solução resultante foi deixada aquecer até a temperatura ambiente, agitada por 1 hora depois aquecida ao refluxo por mais 6 horas. Depois de esfriar em água gelada a mistura foi tratada com água (6 ml) às gotas com agitação em 10 minutos. A mistura foi mais uma vez deixada aquecer até a temperatura ambiente e agitada por 2 horas antes da evaporação do volume do solvente. O resíduo foi depois esfriado em água gelada e 50 % de NaOH (aq.) (6 ml) adicionado às gotas com agitação. A mistura resultante foi agitada e aquecida a 90° C por 4 horas depois de esfriada até a temperatura ambiente e extraída com Et2O (3 X). Os extratos combinados foram lavados com água seguidos pela salmoura, secados em MgSO4 e evaporados para dar 4-amino-4-(4-clorofenil)butan-1-ol (1,100 g, 21,2 %) como um óleo viscoso incolor que foi usado sem outra purificação.
[000203] 1H RMN (399,902 MHz, CDCl3) δ 1,58 - 1,84 (4H, m), 2,39 (3H, br. s), 3,57 - 3,67 (2H, m), 3,88 - 3,91 (1H, m), 7,23 (2H, d), 7,30 (2H, d). MS m/e MH+200. Intermediário 17: 4-(1-(4-clorofenil)-4-hidroxibutilcarbamoil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila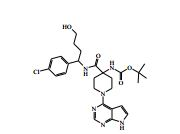
[000204] Hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (166 mg, 0,44 mmol) foi adicionado às porções ao ácido 4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)-piperidina- 4-carboxílico (Intermediário 1) (150 mg, 0,42 mmol), 4-amino-4-(4- clorofenil)butan-1-ol (Intermediário 16) (83 mg, 0,42 mmol) e N- etildiisopropilamina (0,087 ml, 0,50 mmol) em DMF (2,0 ml) a 20° C. A solução resultante foi agitada a 20° C por 3 horas depois extinta em água (10 ml) para dar um ppt. amarelo claro. O precipitado foi coletado pela filtração, lavado com água e secado sob vácuo para produzir 4-(1-(4- clorofenil)-4-hidroxibutilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidin-4-ilcarbamato de terc-butila (152 mg, 67,4 %) como um sólido creme, que foi usado sem outra purificação.
[000205] 1H RMN (399,902 MHz, DMSO) δ 1,40 (11H, s), 1,69 - 1,74 (2H, m), 1,93 - 2,09 (4H, m), 3,38 - 3,43 (2H, m), 3,51 - 3,61 (2H, m), 4,19 - 4,27 (2H, m), 4,38 (1H, br s), 4,76 (1H, q), 6,65 (1H, m), 6,95 (1H, s), 7,20 (1H, m), 7,33 (4H, s), 7,89 (1H, d), 11,78 (1H, s). MS m/e MH+ 543. Intermediário 18: 4-(1 -(4-clorofenil)-2-hidroxietilcarbamoil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila
[000206] Hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (148 mg, 0,39 mmol) foi adicionado em uma porção a uma solução saturada de ácido 4-(terc-butoxicarbonilamino)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxílico (Intermediário 1) (134 mg, 0,37 mmol) e N-etildiisopropilamina (0,077 ml, 0,44 mmol) em NMP (3 ml). A mistura foi tratada com 2-amino-2-(4-clorofenil)etanol (70 mg, 0,41 mmol) (CAS® no. 179811-64-4, ver US 2006/0004045 para preparação). A solução escura foi agitada por 16 horas na temperatura ambiente. A mistura foi dividida entre acetato de etila e solução aquosa de bicarbonato de sódio. A camada orgânica foi lavada duas vezes com água e depois salmoura. A solução orgânica foi secadas em sulfato de magnésio, filtrada e evaporada. O resíduo foi purificado pela cromatografia por vaporização instantânea em sílica em sílica usando eluição de gradiente (1 % metanol / DCM a 15 % de metanol / DCM). O produto contendo as frações foi combinado para dar 4-(1-(4-clorofenil)-2- hidroxietil-carbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4- ilcarbamato de terc-butila (152 mg, 80 %) como sólido incolor.
[000207] 1H RMN (399,9 MHz, DMSO-d6) δ 1,42 (9H, s), 1,93 - 2,12 (4H, m), 3,45 - 3,63 (4H, m), 4,22 - 4,33 (2H, m), 4,75 - 4,88 (2H, m), 6,59 - 6,61 (1H, m), 7,14 - 7,24 (2H, m), 7,33 (4H, s), 7,76 (1H, d), 8,14 (1H, s), 11,65 (1H, br, s) m/z (ESI+) (M + H)+ = 515; HPLC tR = 1,99 min. Intermediário 19: 3-(terc-butoxicarbonilamino)-3-(4-clorofenil)- propanoato de (S)-metila
[000208] O iodometano (1,038 ml, 16,68 mmol) foi adicionado em uma porção ao ácido (S)-3-(terc-butoxicarbonilamino)-3-(4-clorofenil)- propiônico (1 g, 3,34 mmol) e carbonato de potássio (0,922 g, 6,67 mmol) em DMF (15 ml). A suspensão resultante foi agitada a 80° C por 24 horas. A mistura de reação foi concentrada e diluída com EtOAc (50 ml) com água (50 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir 3-(terc-butoxicarbonilamino)-3-(4-clorofenil)- propanoato de (S)-metila (1,340 g, 128 %) como um sólido laranja. 1H RMN (399,9 MHz, DMSO-d6) δ 1,36 (9H, s), 2,71 - 2,74 (1H, m), 2,74 - 2,80 (1H, m), 3,57 (3H, s), 4,91 (1H, d), 7,33 (2H, d), 7,39 (2H, d), 7,49 (1H, d). m/z (ESI-) (M - H)- = 312; HPLC tR = 2,57 min. Intermediário 20: 3-amino-3-(4-clorofenil)propanoato de (S)-metila (cloridreto)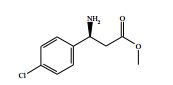
[000209] Ácido clorídrico (4,0 M em 1,4 Dioxano) (4,18 ml, 16,73 mmol) foi adicionado em uma porção ao 3-(terc-butoxicarbonilamino)- 3-(4-clorofenil)propanoato de (S)-metila (Intermediário 19) (1,05 g, 3,35 mmol) em DCM (20 ml) a 20° C. A solução resultante foi agitada a 20° C por 5 horas. A mistura de reação foi evaporada para dar) 3-amino-3- (4-clorofenil)propanoato de (S)-metila (cloridreto (0,850 g, 102 %) como um sólido branco. 1H RMN (399,9 MHz, DMSO-d6) δ 2,98 - 3,04 (1H, m), 3,16 - 3,21 (1H, m), 3,58 (3H, s), 4,62 - 4,66 (1H, m), 7,50 - 7,52 (2H, m), 7,57 - 7,59 (2H, m), 8,66 (3H, s). m/z (ESI+) (M + H)+ = 214; HPLC tR = 1,71 min. Intermediário 21: 3-(4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamido)-3-(4-clorofenil)propanoato de (S)-metila
[000210] Hexafluoro-fosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (1,157 g, 3,04 mmol) foi adicionado em uma porção ao ácido 4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxílico (Intermediário 1) (1 g, 2,77 mmol) e N,N- diisopropiletilamina (1,006 ml, 6,09 mmol) em NMP (10 ml) a 20° C. A solução resultante foi agitada a 20° C por 5 minutos. 3-amino-3-(4- clorofenil)propanoato de (S)-metila (cloridreto) (Intermediário 20) (0,692 g, 2,77 mmol) foi depois adicionado à solução e agitado na temperatura ambiente por 3 horas. A mistura de reação foi diluída com EtOAc (100 ml), e lavada sequencialmente com água (2 x 50 ml) e salmoura (50 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 0 a 5 % de MeOH em DCM. As frações puras foram evaporadas até a secura para produzir 3-(4-(terc-butoxicarbonilamino)- 1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamido)-3-(4- clorofenil) propanoato de (S)-metila de (1,27 g, 82 %) como um sólido branco. 1H RMN (399,9 MHz, DMSO-d6) δ 1,40 (9H, s), 1,99 (2H, s), 2,04 - 2,07 (2H, m), 2,79 - 2,84 (2H, m), 3,56 (3H, s), 3,60 - 3,66 (1H, m), 3,67 - 3,70 (1H, m), 4,20 (2H, t), 5,20 - 5,26 (1H, m), 6,73 (1H, d), 7,13 (1H, s), 7,27 (1H, t), 7,35 (4H, q), 8,14 (1H, d), 8,21 (1H, s), 11,98 (1H, s). m/z (ESI+) (M + H)+ = 557; HPLC tR = 2,12 min. Intermediário 22: 4-(1 -(4-clorofenil)-3-hidroxipropilcarbamoil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de (S)-terc-butila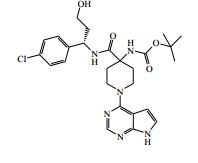
[000211] LiAlH4 (2,280 ml, 2,28 mmol) foi adicionado às gotas ao 3- (4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)- piperidina-4-carboxamido)-3-(4-clorofenil)propanoato de (S)-metila de (Intermediário 21) (1,27g, 2,28 mmol) em THF (70 ml) esfriado a 0° C sob nitrogênio. A solução resultante foi agitada a 20° C por 1 hora. A mistura de reação foi extinta com hidróxido de sódio (2 M) (2 ml) e água (1 ml). A solução foi filtrada e foi diluída com EtOAc (200 ml), e lavada sequencialmente com água (100 ml), água (100 ml), e salmoura saturada (100 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir 4-(1-(4-clorofenil)-3- hidroxipropilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4- ilcarbamato de (S)-terc-butila (1,04 g, 86 %) como um sólido branco. M/z (ESI+) (M + H)+ = 529; HPLC tR = 2,00 min. Intermediário 23: Metanossulfonato de 2-(terc-butoxicarbonilamino)-2- (4-clorofenil)etila
[000212] Cloreto de metanossulfonila (1,451 ml, 18,74 mmol) foi adicionada ao 1-(4-clorofenil)-2-hidroxietilcarbamato de terc-butila (4,63 g, 17,04 mmol) e N,N-diisopropiletilamina (6,23 ml, 35,78 mmol) em DCM (40 ml) adicionada a 0° C em um período de 5 minutos sob nitrogênio. A solução resultante foi agitada a 20° C por 2 horas. A mistura de reação foi diluída com DCM (100 ml) e lavada sequencialmente com água (100 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 0 a 10 % de EtOAc em DCM. As frações puras foram evaporadas até a secura para produzir o metanossulfonato de 2-(terc-butoxicarbonilamino)-2-(4-clorofenil)etila (3,12 g, 52,3 %) como um sólido branco. 1H RMN (399,9 MHz, DMSO- d6) δ 1,39 (9H, s), 3,17 (3H, s), 4,22 - 4,28 (2H, m), 4,90 (1H, d), 7,40 - 7,46 (4H, m), 7,68 (1H, d). m/z (ESI+) (M - H)- = 348; HPLC tR = 2,32 min.
[000213] O 1-(4-clorofenil)-2-hidroxietilcarbamato de terc-butila usado na reação acima foi preparado como segue. Ácido 2-amino-2- (4-clorofenil)acético (12 g, 64,65 mmol) foi agitado em THF (200 ml) e boroidreto de sódio (5,82 g, 153,87 mmol) foi adicionado em porções à mistura agitada sob nitrogênio. Uma solução de iodo (16,41 g, 64,65 mmol) em THF (20 ml) foi adicionado às gotas mantendo a temperatura abaixo de 15° C usando um banho de gelo. A mistura resultante foi aquecida até a temperatura ambiente e agitada a refluxo durante a noite. A reação foi extinta pela adição do metanol (40 ml). Uma porção desta solução foi removida (50 ml) e dividida entre acetato de etila e água. A camada orgânica foi concentrada sob pressão reduzida. O resíduo foi purificado pela MPLC em sílica usando eluição de gradiente (de 0 a 10 % de metanol / DCM). O produto desejado, 2-amino-2-(4-clorofenil)etanol (1,318 g, 11,88 %), foi assim isolado como um sólido incolor.
[000214] 1H RMN (399,9 MHz, CDCl3) δ 2,00 (3H, br, s), 3,48 - 3,58 (1H, m), 3,68 - 3,76 (1H, m), 4,02 - 4,08 (1H, m), 7,23 - 7,39 (4H, m). m/z (ESI-) (M - H)- = 284,286; HPLC tR = 2,20 min.
[000215] O resto da solução foi tratado com trietilamina (18,02 ml, 129,31 mmol) e dicarbonato de di-terc-butila (14,11 g, 64,65 mmol). A mistura foi agitada por 2 horas na temperatura ambiente antes sendo dividida entre acetato de etila e água. A camada orgânica foi secada com sulfato de magnésio, filtrada, e concentrada sob pressão reduzida. O resíduo foi purificado pela MPLC em sílica usando eluição de gradiente (10 % de acetato de etila / DCM a 50 % de acetato de etila / DCM). O produto desejado, 1-(4-clorofenil)-2-hidroxietilcarbamato de terc-butila (7,91 g, 45,0 %), foi assim isolado como um sólido incolor. As frações impuras foram repurificadas pela MPLC para dar uma segunda safra (2,41 g, 14 %).
[000216] 1H RMN (399,9 MHz, DMSO-d6) δ 1,37 (9H, s), 3,41 - 3,52 (2H, m), 4,42 - 4,58 (1H, m), 4,79 (1H, t), 7,23 (1H, d), 7,31 (2H, d), 7,37 (2H, d). Intermediário 24: 1-(4-clorofenil)-2-cianoetilcarbamato de terc-butila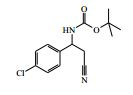
[000217] Cianeto de sódio (105 mg, 2,14 mmol) foi adicionado ao metanossulfonato de 2-(terc-butoxicarbonilamino)-2-(4-clorofenil)etila (Intermediário 23) (300 mg, 0,86 mmol) em DMF (5 ml) a 20° C. A suspensão resultante foi agitada a 80° C por 3 horas. A mistura de reação foi evaporada até a secura e redissolvida em água (10 ml) e lavada sequencialmente com DCM 3 X (10 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 0 a 25 % de EtOAc em isoexano. As frações puras foram evaporadas até a secura para produzir 1-(4-clorofenil)-2-cianoetilcarbamato de terc-butila (209 mg, 87 %) como um sólido branco. 1H RMN (399,9 MHz, DMSO- d6) δ 1,38 - 1,42 (9H, s), 2,82 - 2,89 (2H, m), 4,89 (1H, d), 7,38 - 7,45 (4H, m), 7,76 (1H, d). m/z (ESI+) (M - H)- = 279; HPLC tR = 2,42 min. Intermediário 25: 3-amino-1-(4-clorofenil)propilcarbamato de terc-butila
[000218] Alumino hidreto de lítio (0,712 ml, 0,71 mmol) foi adicionado às gotas ao 1-(4-clorofenil)-2-cianoetilcarbamato de terc-butila de (Intermediário 24) (200 mg, 0,71 mmol) em THF (4 ml) a 20° C sob nitrogênio. A solução resultante foi agitada a 20° C por 2 horas. A mistura de reação foi extinta com NaOH (1 M) (1 ml) e a solução foi filtrada. A solução foi diluída com EtOAc (20 ml), e lavada com água 2 x (10 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir 3-amino-1-(4-clorofenil)propilcarbamato de terc-butila (203 mg, 100 %) como uma goma. m/z (ESI+) (M + H)+ = 285; HPLC tR = 2,33 min. Intermediário 26: 3-acetamido-1-(4-clorofenil)propilcarbamato de terc- butila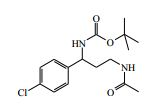
[000219] Anidrido acético (0,084 ml, 0,89 mmol) foi adicionado às gotas ao 3-amino-1-(4-clorofenil)propilcarbamato de terc-butila (Intermediário 25) (203 mg, 0,71 mmol) e N,N-diisopropiletilamina (0,248 ml, 1,43 mmol) em DCM (5 ml) a 20° C. A solução resultante foi agitada a 20° C por 16 horas. A mistura de reação foi extinta com NaHCO3 (2 M) (10 ml) e água (10 ml) e extraída com DCM (20 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 0 a 2,5 % de MeOH em DCM. As frações puras foram evaporadas até a secura para produzir 3-acetamido-1-(4- clorofenil)propilcarbamato de terc-butila (142 mg, 61,0 %) como um sólido branco. 1H RMN (399,9 MHz, DMSO-d6) δ 1,36 (9H, s), 1,67 - 1,82 (2H, m), 1,79 (3H, s), 2,97 (2H, q), 4,50 (1H, d), 7,32 (2H, d), 7,38 (2H, d), 7,43 - 7,45 (1H, m), 7,79 (1H, s). m/z (ESI+) (M + H)+ = 327; HPLC tR = 2,03 min. Intermediário 27: N-(3-amino-3-(4-clorofenil)propil)acetamida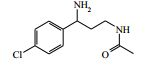
[000220] 3-acetamido-1-(4-clorofenil)propilcarbamato de terc-butila (Intermediário 26) (142 mg, 0,43 mmol) foi adicionado ao TFA (2 ml) a 20° C. A solução resultante foi agitada a 20° C por 40 minutos. A mistura de reação foi evaporada e purificada pela cromatografia de troca iônica usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 7 M e as frações puras foram evaporadas até a secura para produzir N-(3-amino-3-(4-clorofenil)propil)acetamida (39,0 mg, 39,6 %) como uma goma incolor. m/z (ESI+) (M + H)+ = 227; HPLC tR = 1,32 min. Intermediário 28: 4-(3-acetamido-1-(4-clorofenil)propilcarbamoil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila
[000221] Hexafluoro-fosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’ - tetrametilurônio (98 mg, 0,26 mmol) foi adicionado em uma porção ao ácido 4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)- piperidina-4-carboxílico (Intermediário 1) (62,2 mg, 0,17 mmol) e N,N- diisopropiletilamina (0,085 ml, 0,52 mmol) em NMP (2 ml) a 20° C sob nitrogênio. A solução resultante foi agitada a 20° C por 5 minutos. N- (3-amino-3-(4-clorofenil)propil)acetamida (Intermediário 27) (39 mg, 0,17 mmol) em NMP (2 ml) foi depois adicionada à reação e agitada por 1 hora. A mistura de reação foi concentrada e diluída com EtOAc (20 ml) e lavada com água (20 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir 4-(3-acetamido-1-(4- clorofenil)-propilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4- il-carbamato de terc-butila (1,04g, 86 %) como um sólido branco. m/z (ESI+) (M + H)+ = 570; HPLC tR = 1,95 min. Intermediário 29: 3-amino-3-(4-clorofenil)propan-1-ol
[000222] Complexo de borano-tetraidrofurano de (94,0 ml, 93,92 mmol) foi adicionado às gotas à uma suspensão agitada de ácido 3- amino-3-(4-clorofenil)propiônico (2,50 g, 12,52 mmol) em THF (75 ml) a 0° C em um período de 20 minutos sob nitrogênio. A suspensão resultante foi agitada a 0° C por 30 minutos depois a 22° C por 5 horas. A mistura de reação foi adicionada às porções ao metanol (500 ml). A mistura foi concentrada, redissolvida em metanol (250 ml) e reconcentrada (este processo foi repetido três vezes). O resíduo foi dissolvido em DCM (200 ml) e lavado com NaOH 1 N (150 ml). A camada aquosa foi extraída com DCM (5 x 100 ml) e os extratos combinados com a camada orgânica. Os combinados orgânicos foram lavados com salmoura saturada (2 x 150 ml), secados em MgSO4 e concentrados para produzir um sólido semi branco. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição 5 a 7 % (MeOH 10:1 / NH3 conc. (aq)) em DCM. As frações puras foram evaporadas até a secura para produzir 3-amino-3- (4-clorofenil)propan-1-ol (1,320 g, 56,8 %) como um sólido branco.
[000223] 1H RMN (399,902 MHz, CDCl3) δ 1,87 (2H, m), 2,34 (2H, br.s), 3,79 (2H, m), 4,13 (1H, t), 7,24 (2H, d), 7,32 (2H, d). MS m/e MH+ 169 Intermediário 30: 1-(4-clorofenil)-3-hidroxipropilcarbamato de terc- butila
[000224] Dicarbonato de di-terc-butila (0,705 g, 3,23 mmol) foi adicionado ao 3-amino-3-(4-clorofenil)propan-1-ol (Intermediário 29) (0,500 g, 2,69 mmol) em DCM (30 ml) a 22° C. A solução resultante foi agitada a 22° C por 2 horas. A mistura foi concentrada e o resíduo foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição 0 a 4 % (MeOH 10:1 / NH3 conc. (aq)) em DCM. As frações puras foram evaporadas até a secura para produzir 1-(4- clorofenil)-3-hidroxipropil-carbamato de terc-butila (0,759 g, 99 %) como um sólido branco.
[000225] 1H RMN (399,902 MHz, CDCl3) δ 1,43 (9H, s), 1,81 (1H, m), 2,04 (1H, m), 2,74 (1H, br.s), 3,69 (2H, m), 4,88 (1H, br.s), 5,04 (1H, d), 7,23 (2H, d), 7,32 (2H, d). MS m/e MH- 284, 286 Intermediário 31: Metanossulfonato de 3-(terc-butoxicarbonilamino)-3- (4-clorofenil)propila
[000226] Cloreto de metanossulfonila (0,097 ml, 1,25 mmol) foi adicionado às gotas ao 1-(4-clorofenil)-3-hidroxipropilcarbamato de terc-butila (Intermediário 30) (0,326 g, 1,14 mmol) e trietilamina (0,191 ml, 1,37 mmol) em DCM (15 ml) a 22° C. A solução resultante foi agitada a 22° C por 2 horas. A mistura foi concentrada e o resíduo foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição 20 a 40 % de EtOAc em isoexano. As frações puras foram evaporadas até a secura para produzir metanossulfonato de 3-(terc-butoxicarbonilamino)-3-(4-clorofenil)propila (0,366 g, 88 %) como um sólido branco.
[000227] 1H RMN (399,902 MHz, CDCl3) δ 1,42 (9H, s), 2,19 (2H, m), 3,01 (3H, s), 4,24 (2H, m), 4,82 (2H, m), 7,22 (2H, d), 7,33 (2H, d). MS m/e MH- 362, 364 Intermediário 32: 1-(4-clorofenil)-3-(dimetilamino)propilcarbamato de terc-butila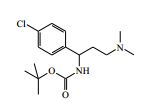
[000228] Metanossulfonato de 3-(terc-butoxicarbonilamino)-3-(4- clorofenil)propila (Intermediário 31) (0,075 g, 0,21 mmol) e iodeto de tetrabutilamônio (0,015 g, 0,04 mmol) foram dissolvidos em uma solução de dimetilamina em THF (2M, 5,153 ml, 10,31 mmol) e selados em um tubo de microonda. A reação foi aquecida a 150° C por 30 minutos no reator de microonda e esfriada até a temperatura ambiente. A mistura de reação foi concentrada, diluída com DCM (25 ml) e lavada com água (25 ml). A camada orgânica foi filtrada através de um papel de filtro de separação de fase e evaporada. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição 4 to 8 % (MeOH 10:1/ NH3 conc. (aq)) em DCM. As frações puras foram evaporadas até a secura para produzir 1-(4-clorofenil)-3-(dimetil-amino)propilcarbamato de terc-butila (0,054 mg, 84 %) como um óleo incolor.
[000229] 1H RMN (399,902 MHz, CDCl3) δ 1,40 (9H, s), 1,80 (1H, br.s), 1,94 (1H, m), 2,23 (6H, s), 2,26 (2H, m), 4,71 (1H, br.s), 6,16 (1H, br.s), 7,21 (2H, d), 7,29 (2H, d). MS m/e MH+313 Intermediário 33: 1-(4-clorofenil)-N3,N3-dimetilpropano-1,3-diamina
[000230] Cloreto de hidrogênio (4 M em dioxano, 1,132 ml, 32,61 mmol) foi adicionado ao 1-(4-clorofenil)-3-(dimetilamino)propil- carbamato de terc-butila (Intermediário 32) (0,051 g, 0,16 mmol) em uma mistura de DCM (5 ml) e metanol (2 ml) a 22° C. A solução resultante foi agitada a 22° C por 4 horas. A mistura foi concentrada e o resíduo foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 2 M e as frações puras foram evaporadas até a secura para produzir 1-(4-clorofenil)-N3,N3-dimetilpropano-1,3-diamina (0,032 g, 92 %) como um óleo incolor.
[000231] 1H RMN (399,902 MHz, CDCl3) δ 1,72 - 1,85 (2H, m), 2,19 - 2,32 (2H, m), 2,21 (6H, s), 3,99 (1H, t), 7,25 - 7,31 (4H, m). MS m/e MH+213 Intermediário 34: 4-(1 -(4-clorofenil)-3-(dimetilamino)propilcarbamoil)-1- (7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila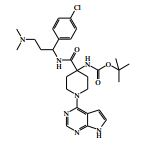
[000232] Hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (0,044 g, 0,12 mmol) foi adicionado ao ácido 4-(terc- butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxílico (Intermediário 1) (0,040 g, 0,11 mmol) e N,N-diisopropil- etilamina (0,023 ml, 0,13 mmol) em NMP (5 ml) a 22° C. A suspensão resultante foi agitada a 50° C por 10 minutos. A mistura foi esfriada até a temperatura ambiente e 1-(4-clorofenil)-N3,N3-dimetilpropano-1,3- diamina (Intermediário 33) (0,023 g, 0,11 mmol) foi adicionado como uma solução em NMP (2 ml). A mistura foi agitada a 22° C por 3 dias. A mistura foi diluída com metanol e purificada pela cromatografia de troca iônica usando uma coluna SCX; o produto desejado foi eluído da coluna usando 30 % (2 M NH3 em MeOH) em DCM e as frações contendo o produto foram evaporadas até a secura. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 2 a 6 % (MeOH 10:1/ NH3 conc. (aq)) em DCM. As frações puras foram evaporadas até a secura para produzir 4-(1-(4-clorofenil)-3-(dimetil- amino)propilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4- ilcarbamato de terc-butila (0,047 g, 76 %) como um sólido branco.
[000233] 1H RMN (399,902 MHz, DMSO) δ 1,43 (9H, s), 1,82 (2H, m), 1,90 - 2,04 (5H, m), 2,14 (6H, s), 2,16 (2H, m), 3,54 (2H, m), 4,25 (2H, m), 4,86 (1H, dt), 6,61 (1H, dd), 7,09 (1H, br.s), 7,17 (1H, dd), 7,33 (4H, s (efeito de cobertura)), 8,14 (1H, s), 8,45 (1H, d), 11,65 (1H, s). MS m/e MH+556 Intermediário 35: 3-(4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamido)-3-(4-clorofenil)propanoato de Metila
[000234] Hexafluoro-fosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’ - tetrametilurônio (84 mg, 0,22 mmol) foi adicionado em uma porção ao ácido 4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)- piperidina-4-carboxílico (Intermediário 1) (72,3 mg, 0,20 mmol), cloridreto do éster metílico do ácido 3-amino-3-(4-cloro-fenil)- propiônico (50 mg, 0,20 mmol) e N,N-diisopropiletilamina (0,104 ml, 0,60 mmol) em N-metil-2-pirrolidinona (1,5 ml) a 20° C sob argônio. A solução resultante foi agitada por 5 horas. A mistura de reação foi purificada pela HPLC preparativa usando uma coluna de fase reversa Waters X-Bridge (C-18, sílica de 5 mícrons, diâmetro 19 mm, comprimento 100 mm, vazão de 40 ml / minutos) com misturas decrescentemente polares de água (contendo 0,2 % de carbonato de amônio) e acetonitrila como eluente. As frações contendo o composto desejado foram evaporadas até a secura para produzir 3-(4-(terc- butoxicarbonilamino)-1-(7H-pirrolo-[2,3-d]pirimidin-4-il)piperidina-4- carboxamido)-3-(4-clorofenil)-propanoato de metila (71,0 mg, 63,8 %) como um sólido cristalino branco.
[000235] 1H RMN (500 MHz, DMSO-d6) δ 1,36 (9H, s), 1,94 - 2,00 (4H, m), 2,79 - 2,86 (2H, dq), 3,54 (3H, s), 3,52 - 3,61 (2H, m), 4,19 (2H, t), 5,22 (1H, q), 6,56 (1H, d), 6,9 (1H br s), 7,13 (1H, d), 7,33 (4H, q), 7,98 (1H, d), 8,12 (1H, s), 11,53 (1H, s). m/z (ESI+) (M + H)+ = 557; HPLC tR = 3,18 min. Intermediário 36: 4-(1 -(4-clorofenil)-3-hidroxipropilcarbamoil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila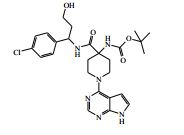
[000236] Boroidreto de sódio (1,019 g, 26,93 mmol) foi adicionado em uma porção à uma suspensão agitada de 3-(4-(terc-butoxicarbonil- amino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamido)-3- (4-clorofenil)propanoato de metila (Intermediário 35) (1,0 g, 1,80 mmol) dissolvido em EtOH (400 ml) sob argônio. A suspensão resultante foi agitada a 20° C por 70 horas. Umas poucas gotas de água foram adicionadas para extinguir o excesso de hidreto e a mistura agitada por 1 hora. A mistura foi evaporada até a secura e o resíduo foi dividido entre CH2Cl2 e água saturada com NaCl. A fase orgânica foi secada e concentrada para fornecer o produto bruto como cristais brancos. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica gel eluindo com 0 a 10 % de metanol em diclorometano. As frações puras foram evaporadas até a secura para produzir 4-(1-(4-clorofenil)-3-hidroxipropilcarbamoil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (286 mg, 30,1 %) como um sólido branco.
[000237] 1H RMN (500 MHz, DMSO-d6) δ 1,41 (9H, s), 1,79 - 1,99 (6H, m), 3,31 - 3,37 (2H, m), 3,51 (2H, m), 4,21 - 4,28 (2H, m), 4,55 (1H, s), 4,90 (1H, q), 6,59 (1H, d), 7,06 (1H, s), 7,16 (1H, d), 7,31 (4H, s), 8,01 (1H, d), 8,12 (1H, s), 11,67 (1H, s). m/z (ESI+) (M - H)- = 529; HPLC tR = 2,93 min. Intermediário 37: 4-(1-(4-clorofenil)-3-(1,3-dioxoisoindolin-2-il)propil- carbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila
[000238] Trifenilfosfina (158 mg) e ftalimida (22,11 mg) foram adicionados em uma porção a uma solução saturada de 4-(1-(4-cloro- fenil)-3-hidroxipropilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)- piperidin-4-ilcarbamato de terc-butila (Intermediário 36) (53 mg) dissolvido em THF (10 ml) e esfriado a 0° C sob argônio. A solução resultante foi agitada a 0° C por 30 minutos. Azodicarboxilato de dietila (0,093 ml) foi adicionado às gotas a 0° C sob argônio. A suspensão resultante foi agitada a 0° C por 60 minutos e durante a noite na temperatura ambiente. A extinta com água. O THF foi removido a vácuo e a fase aquosa extraída com DCM. A fase orgânica foi secada e concentrada para fornecer o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica gel eluindo com 0 a 10 % de metanol em DCM. O solvente foi evaporado até a secura para produzir 4-(1-(4-clorofenil)-3-(1,3-dioxoisoindolin-2-il) propilcarbamoil) -1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4- ilcarbamato de terc-butila como um sólido branco (14,00 mg, 21,23 %).
[000239] 1H RMN (500 MHz, DMSO-d6) δ 1,37 (9H, s), 2,01 (4H, m), 2,09 (2H, m), 3,58 (4H, m), 4,23 (2H, m), 4,84 (1H, q), 6,60 (1H, d), 7,00 (1H, m), 7,16 (1H, t), 7,27 (2H, d), 7,33 (2H, d), 7,82 (4H, m), 8,12 (1H, d), 8,13 (1H, s), 11,67 (1H, s). MS m/e MH+ 658. Intermediário 38: 4-(3-amino-1-(4-clorofenil)propilcarbamoil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila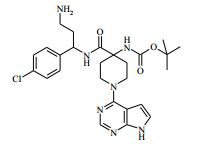
[000240] Hidrazina monoidratada (10,32 μl) foi adicionada a uma suspensão agitada de 4-(1-(4-clorofenil)-3-(1,3-dioxoisoindolin-2- il)propil-carbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4- ilcarbamato de terc-butila (Intermediário 37) (14 mg) em etanol (1,0 ml) sob argônio. A suspensão resultante foi agitada na temperatura ambiente por 3 dias. A mistura foi filtrada e foi purificada pela HPLC preparativa usando uma coluna de fase reversa Waters X-Bridge (C18, sílica de 5 mícrons, diâmetro 19 mm, comprimento 100 mm) e misturas decrescentemente polares de água (contendo 0,2 % de carbonato de amônio) e acetonitrila como eluente. As frações puras foram evaporadas até a secura para produzir 4-(3-amino-1-(4-cloro- fenil)propilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-il- carbamato de terc-butila como um pó branco (5,00 mg, 44,5 %).
[000241] 1H RMN (500 MHz, CDCl3 + CD3OD ) δ 1,43 (9H, s), 1,93 (2H, s), 2,19 (4H, m), 2,74 (2H, s) 3,57 (4H, m), 4,39 (2H, t), 5,01 (1H, t), 6,53 (1H, d), 7,07 (1H, d), 7,28 (4H, q), 8,19 (1H, s). MS m/e MH+ 528. Intermediário 39:3-(terc-butoxicarbonilamino)-3-(4- clorofenil)propanoato de (R)-metila
[000242] Iodometano (0,987 ml, 15,85 mmol) foi adicionado em uma porção ao ácido boc-(R)-3-amino-3-(4-cloro-fenil)-propiônico (950 mg, 3,17 mmol) e carbonato de potássio (876 mg, 6,34 mmol) em DMF (15 ml). A suspensão resultante foi agitada a 80° C por 24 horas. A mistura de reação foi concentrada e diluída com EtOAc (25 ml) com água (25 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir 3-(terc-butoxicarbonilamino)-3-(4-clorofenil)propanoato de (R)-metila (990 mg, 100 %) como um sólido laranja.
[000243] 1H RMN (399,9 MHz, DMSO-d6) δ 1,36 (9H, s), 2,67 - 2,70 (1H, m), 2,71 - 2,80 (1H, m), 3,57 (3H, s), 4,91 (1H, d), 7,32 - 7,34 (2H, m), 7,38 - 7,40 (2H, m), 7,49 (1H, d).MS m/e MH+ 312; HPLC tR = 2,57 min. Intermediário 40: 3-amino-3-(4-clorofenil)propanoato de (R)-metila (cloridreto)
[000244] Ácido clorídrico (4,0 M em dioxano) (7,89 ml, 31,55 mmol) foi adicionado em uma porção ao 3-(terc-butoxicarbonilamino)-3-(4- clorofenil)-propanoato de (R)-metila (Intermediário 39) (990 mg, 3,16 mmol) em DCM (20 ml) a 20° C. A solução resultante foi agitada a 20° C por 3 horas. A mistura de reação foi evaporada para produzir 3- amino-3-(4-clorofenil)-propanoato de (R)-metila (cloridreto) (777 mg, 98 %) como um solido amarelo.
[000245] 1H RMN (399,9 MHz, DMSO-d6) δ 2,99 - 3,05 (1H, m), 3,18 - 3,23 (1H, m), 3,58 (3H, d), 4,62 - 4,65 (1H, m), 7,49 - 7,53 (2H, m), 7,57 - 7,61 (2H, m), 8,72 (3H, s). MS m/e MH+ 214; HPLC tR = 1,71 min. Intermediário 41: 3-(4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamido)-3-(4-clorofenil)propanoato de (R)-metila
[000246] Hexafluoro-fosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (0,579 g, 1,52 mmol) foi adicionado em uma porção ao ácido 4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxílico (Intermediário 1) (0,5 g, 1,38 mmol) e DIPEA (0,503 ml, 3,04 mmol) em NMP (5 ml) a 20° C. A solução resultante foi agitada a 20° C por 5 minutos. 3-amino-3-(4-clorofenil)propanoato de (R)-metila (cloridreto) (Intermediário 40) (0,346 g, 1,38 mmol) foi depois adicionado à solução e agitado na temperatura ambiente por 3 horas. A mistura de reação foi diluída com EtOAc (100 ml) e lavada sequencialmente com água (50 ml), água (50 ml) e água (50 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição 0 a 5 % de MeOH em DCM. As frações puras foram evaporadas até a secura para 3-(4-(terc-butoxicarbonilamino)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamido)-3-(4- clorofenil)propanoato de produzir (R)-metila (0,800 g, 100 %) como um sólido branco.
[000247] 1H RMN (399,9 MHz, DMSO-d6) δ 1,40 (9H, s), 1,94 - 1,98 (2H, m), 2,00 - 2,01 (2H, m), 2,83 (1H, d), 2,85 - 2,87 (2H, m), 3,55 (3H, s), 3,60 (2H, s), 4,21 (2H, s), 5,23 (1H, d), 6,60 - 6,61 (1H, m), 7,16 - 7,18 (1H, m), 7,35 (4H, q), 8,09 - 8,14 (2H, m), 11,65 (1H, s). MS m/e MH+ 557; HPLC tR = 2,29 min. Intermediário 42: 4-(1 -(4-clorofenil)-3-hidroxipropilcarbamoil)-1-(7H- pirrolo-[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de (R)-terc-butila
[000248] LiAlH4 (1,398 ml, 1,40 mmol) foi adicionado às gotas ao 3- (4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxamido)-3-(4-clorofenil)propanoato de (R)-metila (Intermediário 41) (779 mg, 1,40 mmol) em THF (40 ml) esfriado a 0° C sob nitrogênio. A solução resultante foi agitada a 20° C por 3 horas. A mistura de reação foi extinta com água (1 ml) e neutralizada com HCl 2 M. A mistura de reação foi evaporada até a secura e redissolvida em DCM (100 ml) e lavada com água (100 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 0 a 10 % de MeOH em DCM. As frações puras foram combinadas e evaporadas para dar 4-(1-(4-clorofenil)-3-hidroxipropilcarbamoil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidin-4-il-carbamato de (R)-terc-butila (160 mg, 21,63 %) como um sólido.
[000249] 1H RMN (399,9 MHz, DMSO-d6) δ 1,37 - 1,41 (9H, s), 1,86 - 1,92 (2H, m), 1,94 (2H, d), 2,00 (2H, d), 3,38 (2H, s), 3,53 - 3,55 (2H, m), 4,25 (2H, t), 4,52 (1H, s), 4,91 (1H, d), 6,60 - 6,62 (1H, m), 7,01 (1H, s), 7,16 - 7,18 (1H, m), 7,33 (4H, m), 7,98 - 8,00 (1H, m), 8,14 (1H, s), 11,67 (1H, s). MS m/e MH+ 529; HPLC tR = 2,01 min. Intermediário 43: Ácido 1-(terc-butoxicarbonil)-4-cianopiperidina-4- carboxílico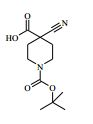
[000250] Uma solução aquosa de hidróxido de lítio (2 M, 39,0 ml, 77,92 mmol) foi adicionada a uma solução agitada de 4-etil 4- cianopiperidina-1,4-dicarboxilato de 1-terc-butila (Intermediário 7) (5,5 g, 19,48 mmol) em THF (78 ml) a 25º C. A mistura resultante foi agitada a 25° C por 3 horas e monitorada pela TLC. A mistura de reação foi diluída com éter dietílico (150 ml) e lavada com água (100 ml). As camadas aquosas foram combinadas e depois acidificadas com ácido cítrico (1 N, 200 ml). O produto foi extraído em DCM. A camada orgânica foi secada em sulfato de magnésio, filtrada e evaporada para produzir o ácido 1-(terc-butoxicarbonil)-4- cianopiperidina-4-carboxílico (4,50 g, 91 %). Este material foi usado na etapa seguintes sem outra purificação.
[000251] 1H RMN (400 MHz, CDCl3) δ 1,40 (9H, s), 1,87 - 1,97 (2H, m), 2,04 (2H, d), 3,08 (2H, t), 4,05 (2H, s), 8,23 (1H, s). Intermediário 44: (R)-N-(1-(4-clorofenil)etil)-4-cianopiperidina-4- Carboxamida
[000252] Hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (1,255 g, 3,30 mmol) foi adicionado em uma porção ao ácido 1-(terc-butoxicarbonil)-4-cianopiperidina-4-carboxílico (Intermediário 43) (0,763 g, 3 mmol), (R)-1-(4-clorofenil)etanamina (0,462 ml, 3,00 mmol) e DIPEA (1,572 ml, 9,00 mmol) em DMA (20 ml) a 25° C sob nitrogênio. A solução resultante foi agitada a 60° C por 4 horas. A mistura de reação foi evaporada até a secura e redissolvida em DCM (150 ml) e lavada sequencialmente com ácido cítrico 1 M aquoso (50 ml), água (50 ml) e hidrogeno carbonato de sódio saturado (100 ml). A camada orgânica foi secadas em sulfato de magnésio, filtrada e evaporada para produzir o produto bruto. O produto bruto assim obtido foi concentrado, depois purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 0 a 100 % de EtOAc em isoexano. As frações puras foram evaporadas para produzir 4-(1-(4-clorofenil)etilcarbamoil)-4-cianopiperidina-1-carboxilato de (R)-terc-butila (1,100 g, 94 %) como uma goma incolor que solidificou na secagem sob alto vácuo. Este material foi depois redissolvido em DCM (20,00 ml) e ácido trifluoroacético (2,311 ml, 30,00 mmol) foi adicionado. A solução foi agitada na temperatura ambiente por 3 horas, depois que o produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 7 N e as frações puras foram evaporadas até a secura para produzir (R)-N-(1-(4- clorofenil)etil)-4-cianopiperidina-4-carboxamida (0,720 g, 82 %) como uma goma incolor. Este material foi usado diretamente na etapa seguinte sem outra purificação. MS m/e MH+ 292. Intermediário 45: (R)-N-(1-(4-clorofenil)etil)-4-ciano-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidina-4-carboxamida
[000253] DIPEA (1,101 ml, 6,17 mmol) foi adicionada ao (R)-N-(1-(4- clorofenil)etil)-4-cianopiperidina-4-carboxamida (Intermediário 44) (720 mg, 2,47 mmol) e 6-cloro-7-desazapurina (379 mg, 2,47 mmol) em DMA (50 ml) a 25° C. A solução resultante foi agitada a 90° C por 3 horas. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 7 N e as frações puras foram evaporadas até a secura para produzir (R)-N-(1-(4-clorofenil)etil)-4- ciano-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida (46,2 %) como um sólido laranja. Esta foi usada na etapa seguinte sem outra purificação.
[000254] 1H RMN (400 MHz, DMSO) δ 1,38 (3H, d), 1,94 - 2,09 (3H, m), 2,19 (3H, t), 4,72 (2H, d), 4,92 (1H, dd), 6,61 - 6,72 (1H, m), 7,23 (1H, dd), 7,30 - 7,41 (4H, m), 8,18 (1H, s), 8,77 (1H, d), 11,77 (1H, s). MS m/e MH+ 409. Intermediário 46: (S)-4-(1-amino-3-hidroxipropil)benzonitrila
[000255] Boroidreto de sódio (1,039 ml, 29,48 mmol) foi adicionado às porções ao 3-amino-3-(4-cianofenil)propanoato de (S)-metila (2,23 g, 10,92 mmol) em metanol (20 ml) a 0° C em um período de 5 minutos. A solução resultante foi agitada a 20° C por 24 horas. A mistura de reação foi extinta com NaHCO3 saturado (50 ml) extraída com EtOAc (3 x 100 ml), a camada orgânica foi lavada com salmoura saturada (75 ml), secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 0 a 8 % de MeOH com amônia em DCM. As frações puras foram evaporadas até a secura para produzir (S)-4-(1-amino-3- hidroxipropil)benzonitrila (0,368 g, 19,13 %) como um óleo incolor.
[000256] 1H RMN (400,13 MHz, CDCl3) δ 1,84 - 1,93 (2H, m), 3,77 - 3,80 (2H, m), 4,22 - 4,25 (1H, m), 7,43 (2H, m), 7,63 - 7,66 (2H, m). MS m/e MH+ 177. Intermediário 47: (S)-3-amino-3-(4-clorofenil)propan-1-ol
[000257] Complexo de borano-tetraidrofurano (376 ml, 375,69 mmol) foi adicionado às gotas à uma suspensão agitada de ácido (S)-3- amino-3-(4-clorofenil)propiônico (10 g, 50,09 mmol) em THF (500 ml) a 0° C em um período de 45 minutos sob nitrogênio. A suspensão resultante foi agitada a 0° C por 30 minutos depois a 22° C por 5 horas. A mistura de reação foi adicionada às porções ao metanol (500 ml). A solução foi agitada na temperatura ambiente por três dias. A mistura foi concentrada, redissolvida em metanol (250 ml) e reconcentrada (este processo foi repetido três vezes). O resíduo foi dissolvido em DCM (75 ml) e lavado com NaOH 1 N (50 ml). A camada aquosa foi extraída com DCM (3 x 100 ml) e os extratos combinados com a camada orgânica. Os combinados orgânicos foram lavados com salmoura saturada (50 ml), secados em MgSO4 e concentrados para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 2 a 6 % de MeOH/amônia em DCM. As frações puras foram evaporadas até a secura para produzir (S)-3-amino-3-(4- clorofenil)propan-1-ol (5,76 g, 61,9 %) como um sólido branco.
[000258] 1H RMN (400,13 MHz, CDCl3) δ 1,84 - 1,93 (2H, m), 3,76 - 3,81 (2H, m), 4,13 (1H, t), 7,23 - 7,25 (2H, m), 7,30 - 7,34 (2H, m). MS m/e MH+ 186. Intermediário 48: 4-(terc-butoxicarbonilamino)-4-(1 -(4-clorofenil)-3- hidróxi-propilcarbamoil)piperidina-1-carboxilato de (S)-terc-butila
[000259] (S)-3-amino-3-(4-clorofenil)propan-1-ol (Intermediário 47) (1,09 g, 5,87 mmol) foi adicionado em uma porção ao ácido 1-(terc- butoxicarbonil)-4-(terc-butoxicarbonilamino)piperidina-4-carboxílico (Intermediário 70) (2,022 g, 5,87 mmol) e DIPEA (3,08 ml, 17,61 mmol) em DMA (20 ml). Hexafluorofosfato de O-(7-azabenzotriazol-1-il)- N,N,N’,N’-tetrametilurônio (2,456 g, 6,46 mmol) foi adicionado e a solução resultante foi agitada a 20° C por 24 horas. A mistura de reação foi evaporada até a secura depois diluída com EtOAc (300 ml) e lavada sequencialmente com água (50 ml) e salmoura saturada (50 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada depois triturada com éter dietílico para produzir 4-(terc- butoxicarbonilamino)-4-(1-(4-clorofenil)-3-hidroxipropilcarbamoil)- piperidina-1-carboxilato de (S)-terc-butila bruto (4,66 g, 155 %) como um sólido branco.
[000260] 1H RMN (400,13 MHz, DMSO-d6) δ 1,39 (18H, s), 1,71 - 1,92 (6H, m), 3,06 (2H, s), 3,36 (2H, t), 3,54 - 3,65 (2H, m), 4,52 (1H, t), 4,89 (1H, q), 6,89 (1H, s), 7,29 - 7,34 (4H, m). MS m/e M+Na+ 534. Intermediário 49:(S)-4-amino-N-(1 -(4-clorofenil)-3- hidroxipropil)piperidina-4-carboxamida
[000261] Cloreto de hidrogênio 4 M em dioxano (11,72 ml, 46,87 mmol) foi adicionado ao 4-(terc-butoxicarbonilamino)-4-(1-(4- clorofenil)-3-hidróxi-propilcarbamoil)piperidina-1-carboxilato de (S)- terc-butila (Intermediário 48) (3g, 5,86 mmol) em dioxano (30 ml). A solução resultante foi agitada a 20° C por 2 horas. A mistura de reação foi dissolvida em metanol e purificada pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 3,5 N e as frações puras foram evaporadas até a secura para produzir (S)-4-amino-N-(1-(4-clorofenil)- 3-hidroxipropil)piperidina-4-carboxamida (1,970 g, 108 %) como uma goma incolor.
[000262] 1H RMN (400,13 MHz, DMSO-d6) δ 1,20 - 1,28 (2H, m), 1,75 - 1,91 (4H, m), 2,67 - 2,83 (2H, m), 3,30 (2H, m), 4,87 (1H, s), 7,30 - 7,37 (4H, m). MS m/e MH+ 312. Intermediário 50: 5-bromo-4-cloro-7H-pirrolo[2,3-d]pirimidina
[000263] N-Bromossuccinimida (6,84 g, 38,42 mmol) foi adicionada às porções à 4-cloro-7H-pirrolo[2,3-d]pirimidina (5 g, 32,56 mmol) em DCM, seca (125 ml) a 20° C sob nitrogênio. A suspensão resultante foi agitada a 20° C por 1 hora. A mistura de reação foi evaporada e o sólido marrom resultante foi triturado com água para dar um sólido púrpura que foi coletado pela filtração. O sólido bruto foi triturado com MeOH quente para dar um sólido que foi coletado pela filtração. A trituração quente foi repetida para dar 5-bromo-4-cloro-7H-pirrolo[2,3- d]pirimidina (5,23 g, 69,1 %) como um sólido creme.
[000264] 1H RMN (400,13 MHz, DMSO-d6) δ 7,94 (1H, s), 8,63 (1H, s), 12,95 (1H, s) MS m/e MH+ 234. Intermediário 51: (S)-3-amino-3-(4-bromofenil)propan-1-ol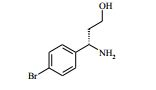
[000265] Complexo de borano-tetraidrofurano (71,7 ml, 71,70 mmol) foi adicionado às gotas à uma suspensão agitada de ácido (S)-3- amino-3-(4-bromofenil)propiônico (3,5 g, 14,34 mmol) em THF (80 ml) a 0° C em um período de 30 minutos sob nitrogênio. A suspensão resultante foi agitada a 0° C por 30 minutos depois a 22° C por 5 horas. A mistura de reação foi adicionada às porções ao metanol (250 ml). A solução foi agitada na temperatura ambiente por 24 horas. A mistura foi concentrada, redissolvida em metanol (250 ml) e reconcentrada (este processo foi repetido três vezes). O resíduo foi dissolvido em DCM (75 ml) e lavado com NaOH 1 N (50 ml). A camada aquosa foi extraída com DCM (3 x 100 ml) e os extratos combinados com a camada orgânica. Os combinados orgânicos foram lavados com salmoura saturada (50 ml), secados em MgSO4 e concentrados para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 2 a 6 % de MeOH/amônia em DCM. As frações puras foram evaporadas até a secura para produzir (S)-3-amino-3-(4- bromofenil)propan-1-ol (2,160 g, 65,5 %) como uma goma incolor.
[000266] 1H RMN (400,13 MHz, CDCl3) δ 1,82 - 1,90 (2H, m), 3,76 - 3,82 (2H, m), 4,12 (1H, t), 7,17 - 7,20 (2H, m), 7,45 - 7,49 (2H, m). MS m/e MH+ 230. Intermediário 52: 4-(4-clorofenil)-4-(metoxiimino)butanoato de etila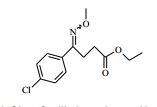
[000267] Ácido 4-(4-Clorofenil)-4-oxobutanóico (10 g, 47,03 mmol), cloridreto de metoxilamina (4,91 g, 58,79 mmol) e bicarbonato de sódio (5,98 g, 56,44 mmol) em etanol (150 ml) foram agitados por 4 horas a 80° C. A mistura foi depois esfriada até a temperatura ambiente antes de ser filtrada. O filtrado foi concentrado pela evaporação e purificado pela cromatografia por vaporização instantânea em sílica (eluente de 20 a 50 % de EtOAc / iso-hexano). As frações puras foram evaporadas até a secura para produzir 4-(4-clorofenil)-4-(metóxi-imino)butanoato de etila (12,33 g, 97 %) como um líquido transparente laranja.
[000268] 1H RMN (400,13 MHz, DMSO) 1,13 - 1,17 (3H, t), 2,26 - 2,50 (2H, t), 2,50 - 2,52 (1H, qu), 2,94 - 2,98 (2H, t), 3,94 (3H, s), 4,00 - 4,05 (2H, q), 7,47 - 7,49 (2H, d), 7,66 - 7,69 (2H, d). Intermediário 53: ácido 4-(4-clorofenil)-4-(metoxiimino)butanóico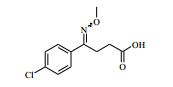
[000269] Hidróxido de lítio monoidratado (9,59 g, 228,57 mmol) foi adicionado ao 4-(4-clorofenil)-4-(metoxiimino)butanoato de etila (Intermediário 52) (12,33 g, 45,71 mmol) em água (25,4 ml), THF (102 ml) e etanol (102 ml). A solução resultante foi agitada a 20° C por 1 dia. A mistura de reação foi diluída com EtOAc (200 ml) e lavada com água (200 ml). O aquoso foi ajustado ao pH 5 com solução 1 M de ácido cítrico depois extraído com EtOAc (3 x 150 ml). Os extratos orgânicos foram lavados com salmoura saturada (25 ml) depois secados em MgSO4, filtrados e evaporados para produzir o produto desejado de ácido 4-(4-clorofenil)-4-(metoxiimino)butanóico (7,63 g, 69,1 %) como um sólido amarelo claro.
[000270] 1H RMN (400,13 MHz) 2,39 - 2,43 (2H, t), 2,91 - 2,95 (2H, t), 3,94 (3H, s), 7,47 - 7,49 (2H, d), 7,67 - 7,69 (2H, d), 12,21 (1H, amplo). MS m/e MH+ 242. Intermediário 54: 4-amino-4-(4-clorofenil)butan-1-ol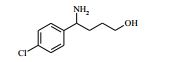
[000271] Ácido 4-(4-clorofenil)-4-(metoxiimino)butanóico (Intermediário 53) (6,43 g, 26,61 mmol) em tetraidrofurano (30 ml) foi esfriado, sob uma atmosfera de nitrogênio em um banho de metanol gelado. O Complexo de borano-tetraidrofurano (1,0 M em THF) (93 ml, 93,12 mmol) foi adicionado às gotas em um período de 20 minutos. A solução resultante foi deixada aquecer até a temperatura ambiente, agitada por 1 hora depois de aquecer ao refluxo por mais 6 horas. Depois de esfriar em água gelada a mistura foi tratada com água (20 ml) às gotas com agitação em 10 minutos. A mistura foi mais uma vez deixada aquecer até a temperatura ambiente e agitada por 2 horas antes da evaporação do volume do solvente. O resíduo foi depois esfriado em água gelada e 50 % NaOH (aq.) (20 ml) adicionado às gotas com agitação. A mistura resultante foi agitada e aquecida a 90° C por 4 horas depois esfriada até a temperatura ambiente e extraída três vezes com Et2O. Os extratos combinados foram lavados com água seguidos pela salmoura, secados em MgSO4 e evaporados até a secura. O produto bruto foi depois purificado pela cromatografia por vaporização instantânea em sílica (eluente de 0 a 10 % de amônia 7 N em MeOH / DCM) e as frações puras evaporadas para produzir 4- amino-4-(4-clorofenil)butan-1-ol (3,88 g, 73,0 %) como uma goma transparente incolor.
[000272] 1H RMN (400,13 MHz) 1,25 - 1,47 (2H, dm), 1,51 - 1,61 (2H, m), 3,34 - 3,38 (2H, t), 3,76 - 3,79 (1H, t), 7,33 - 7,38 (4H, m). Intermediário 55: 1-(4-clorofenil)-4-hidroxibutilcarbamato de terc-butila
[000273] Dicarbonato de di-terc-butila (1,588 ml, 6,91 mmol) foi adicionado em uma porção ao 4-amino-4-(4-clorofenil)butan-1-ol (Intermediário 54) (1,38 g, 6,91 mmol) em DCM (20 ml) a 25° C sob nitrogênio. A solução resultante foi agitada na temperatura ambiente por 24 horas. A mistura resultante foi evaporada até a secura e o óleo bruto foi triturado com isoexano para dar um sólido que foi coletado pela a filtração e secado sob vácuo para dar 1-(4-clorofenil)-4- hidroxibutilcarbamato de terc-butila (1,800 g, 87 %) como um sólido branco.
[000274] 1H RMN (400,13 MHz, CDCl3) δ 1,40 (9H, s), 1,48 - 1,66 (2H, m), 1,78 - 1,83 (2H, m), 3,63 - 3,68 (2H, m), 4,61 (1H, s), 4,84 (1H, s), 7,21 (2H, m), 7,29 - 7,31 (2H, m). Intermediário 56: metanossulfonato de 4-(terc-butoxicarbonilamino)-4- (4-clorofenil)butila
[000275] Cloreto de metanossulfonila (0,511 ml, 6,60 mmol) foi adicionado às gotas ao 1-(4-clorofenil)-4-hidroxibutilcarbamato de terc- butila (Intermediário 55) (1,80 g, 6,00 mmol) e trietilamina (1,004 ml, 7,20 mmol) em DCM (30,0 ml) a 25° C em um período de 15 minutos sob nitrogênio. A solução resultante foi agitada na temperatura ambiente por 30 minutos. A mistura resultante foi evaporada até a secura e o resíduo foi colocado em suspensão em DCM, filtrado e o filtrado purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição 20 a 100 % de EtOAc em isoexano. As frações puras foram evaporadas até a secura para produzir metanossulfonato 4-(terc-butoxicarbonilamino)-4-(4-clorofenil)butila (2,270 g, 100 %) como um sólido branco.
[000276] 1H RMN (400,13 MHz, CDCl3) δ 1,41 (9H, s), 1,67 - 1,87 (4H, m), 2,99 (3H, s), 4,23 (2H, t), 4,62 (1H, s), 4,75 (1H, s), 7,20 (2H, d), 7,30 - 7,33 (2H, d). MS m/e M+Na+ 400. Intermediário 57: 1-(4-clorofenil)-N4,N4-dimetilbutano-1,4-diamina
[000277] Dimetilamina (84 mg, 1,85 mmol) e metanossulfonato de 4- (terc-butoxicarbonilamino)-4-(4-clorofenil)butila (Intermediário 56) (700 mg, 1,85 mmol) foram dissolvidos em THF (10 ml) e selados em um tubo de microonda. A reação foi aquecida a 120° C por 40 minutos no reator de microonda e esfriada até a temperatura ambiente. A mistura de reação foi concentrada e depois dissolvida em DCM (10,00 ml) e TFA (2 ml). A reação foi agitada a 20° C por 2 horas. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 3,5 N e as frações puras foram evaporadas até a secura para produzir 1-(4-clorofenil)-N4,N4-dimetilbutano-1,4-diamina (366 mg, 87 %) como uma goma amarela.
[000278] 1H RMN (400,13 MHz, CDCl3) δ 0,70 - 0,79 (1H, m), 0,82 - 0,91 (1H, m), 0,93 - 1,09 (2H, m), 1,21 (6H, s), 1,64 (2H, t), 3,23 (1H, t), 6,58 - 6,66 (4H, m). MS m/e MH+ 227. Intermediário 58: 1-(4-clorofenil)-3-hidroxipropilcarbamato de (S)-terc- butila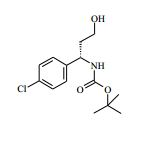
[000279] Dicarbonato de di-terc-butila (8,04 ml, 35,01 mmol) foi adicionado em uma porção ao (S)-3-amino-3-(4-clorofenil)propan-1-ol (Intermediário 47) (6,5 g, 35,01 mmol) em DCM (200 ml) a 25° C sob nitrogênio. A solução resultante foi agitada na temperatura ambiente por 24 horas. A mistura resultante foi evaporada até a secura e o óleo bruto foi triturado com isoexano para dar um sólido que foi coletado pela filtração e secado sob vácuo para dar 1-(4-clorofenil)-3- hidroxipropilcarbamato de (S)-terc-butila (9,20 g, 92 %) como um sólido branco.
[000280] 1H RMN (399,9 MHz, CDCl3) δ 1,43 (9H, s), 1,78 - 1,84 (1H, m), 2,05 (1H, d), 2,74 (1H, s), 3,67 - 3,71 (2H, m), 4,88 (1H, s), 5,04 (1H, d), 7,21 - 7,24 (2H, m), 7,30 - 7,33 (2H, m). MS m/e M+Na+ 308. Intermediário 59:Metanossulfonato de (S)-3-(terc- butoxicarbonilamino)-3-(4-clorofenil)propila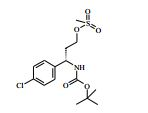
[000281] Cloreto de metanossulfonila (2,74 ml, 35,41 mmol) foi adicionado às gotas ao 1-(4-clorofenil)-3-hidroxipropilcarbamato de (S)-terc-butila (Intermediário 58) (9,2 g, 32,19 mmol) e trietilamina (5,38 ml, 38,63 mmol) em DCM (161 ml) a 25° C em um período de 15 minutos sob nitrogênio. A solução resultante foi agitada na temperatura ambiente por 30 minutos. A mistura resultante foi evaporada até a secura e o resíduo foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição 40 a 100 % de EtOAc em isoexano. As frações puras foram evaporadas até a secura para produzir metanossulfonato de (S)-3-(terc- butoxicarbonilamino)-3-(4-clorofenil)propila (10,03 g, 86 %) como um sólido branco.
[000282] 1H RMN (399,9 MHz, CDCl3) δ 1,36 - 1,48 (9H, m), 2,20 (2H, s), 3,01 (3H, s), 4,19 - 4,29 (2H, m), 4,81 (2H, s), 7,21 - 7,23 (2H, d), 7,32 - 7,35 (2H, d). MS m/e M+Na+ 386. Intermediário 60: (S)-1-(4-clorofenil)-N3,N3-dietilpropano-1,3-diamina
[000283] Dietilamina (0,426 ml, 4,12 mmol) e metanossulfonato de (S)-3-(terc-butoxicarbonilamino)-3-(4-clorofenil)propila (Intermediário 59) (500 mg, 1,37 mmol) foram dissolvidos em THF (10 ml) e selados em um tubo de microonda. A reação foi aquecida a 120° C por 50 minutos no reator de microonda e esfriada até a temperatura ambiente. A mistura de reação foi concentrada e diluída com DCM (5 ml). TFA (1 ml) foi adicionado e a reação foi agitada a 20° C por 1 hora. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando amônia/MeOH 3,5 N e as frações puras foram evaporadas até a secura para produzir (S)-1-(4-clorofenil)-N3,N3-dietilpropano-1,3- diamina (119 mg, 36,0 %) como um óleo amarelo.
[000284] 1H RMN (400,13 MHz, CDCl3) δ 1,00 (6H, t), 1,75 - 1,82 (2H, m), 2,40 - 2,54 (6H, m), 3,97 (1H, t), 7,27 - 7,31 (4H, m). MS m/e MH+ 241. Intermediário 61: 5-bromo-4-cloro-7-tosil-7H-pirrolo[2,3-d]pirimidina
[000285] 5-Bromo-4-cloro-7H-pirrolo[2,3-d]pirimidina (Intermediário 50) (2 g, 8,60 mmol) e cloreto de p-toluenossulfonila (1,640 g, 8,60 mmol) foi absorvido em acetona (12 ml) e agitado sob nitrogênio e esfriado entre -5° C e 5° C. À solução foi adicionada solução 2,0 M de NaOH (5,50 ml, 10,99 mmol) enquanto de mantém a temperatura interna a menos do que 5° C. A reação foi deixada aquecer até a temperatura ambiente e agitada por 1 hora. O sólido branco foi separado por filtração e bem lavado com acetona para produzir 5- bromo-4-cloro-7-tosil-7H-pirrolo[2,3-d]pirimidina (3,28 g, 99 %) como um sólido creme claro.
[000286] 1H RMN (400,13 MHz, DMSO-d6) δ 2,38 (3H, s), 7,48 - 7,50 (2H, m), 8,06 - 8,08 (2H, m), 8,41 (1H, s), 8,84 (1H, s). MS m/e MH+ 387. Intermediário 62: 1 -(5-bromo-7-tosil-7H-pirrolo[2,3-d]pirimidin-4-il)-4- (terc-butoxicarbonilamino)piperidina-4-carboxilato de metila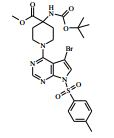
[000287] Trietilamina (3,55 ml, 25,45 mmol) foi adicionada à 5- bromo-4-cloro-7-tosil-7H-pirrolo[2,3-d]pirimidina (Intermediário 61) (3,28 g, 8,48 mmol) e 4-(terc-butoxicarbonilamino)piperidina-4- carboxilato metila de (WO 2008075109) (3,07 g, 11,88 mmol) em DMA (50 ml) a 20° C. A suspensão resultante foi agitada a 70° C por 2 horas. A mistura de reação foi concentrada e diluída com EtOAc (200 ml) e lavada sequencialmente com água (75 ml) e salmoura saturada (75 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição 30 a 70 % de EtOAc em isoexano. As frações puras foram evaporadas até a secura para produzir 1-(5-bromo-7-tosil-7H- pirrolo[2,3-d]pirimidin-4-il)-4-(terc-butoxicarbonilamino)-piperidina-4- carboxilato de metila (3,60 g, 69,7 %) como um sólido branco.
[000288] 1H RMN (400,13 MHz, DMSO-d6) δ 1,38 (9H, s), 2,03 (2H, s), 2,38 (3H, s), 3,33 - 3,37 (2H, m), 3,59 (3H, s), 3,84 (2H, d), 7,41 (1H, s), 7,46 (2H, d), 8,00 (1H, s), 8,03 (2H, d), 8,40 (1H, s). MS m/e MH+ 610. Intermediário 63: 4-(terc-butoxicarbonilamino)-1-(5-ciclolopropil-7-tosil- 7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxilato de metila
[000289] 1-(5-bromo-7-tosil-7H-pirrolo[2,3-d]pirimidin-4-il)-4-(terc- butoxicarbonilamino)piperidina-4-carboxilato de metila (Intermediário 62) (1,1 g, 1,81 mmol), fosfato de potássio tribásico (1,343 g, 6,33 mmol), tricicloloexil fosfina (0,101 g, 0,36 mmol) e ácido ciclopropilborônico (0,438 g, 4,34 mmol) foram absorvidos em tolueno (10 ml) e água (0,400 ml). A mistura foi purgada com nitrogênio por 30 minutos depois acetato de paládio (II) (0,041 g, 0,18 mmol) foi adicionado. A mistura foi aquecida a 75° C por 6 horas e depois deixada esfriar e agitar na temperatura ambiente durante a noite. A mistura de reação foi diluída com EtOAc (75 ml) e lavada sequencialmente com água (50 ml) e salmoura saturada (50 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 10 a 30 % de EtOAc em isoexano. As frações puras foram evaporadas até a secura para produzir 4-(terc-butoxicarbonilamino)-1- (5-ciclolopropil-7-tosil-7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxilato de metila (0,447 g, 43,4 %) como um sólido creme.
[000290] 1H RMN (400,13 MHz, CDCl3) δ 0,72 - 0,76 (2H, m), 0,99 - 1,04 (2H, m), 1,43 (9H, s), 1,90 - 1,94 (1H, m), 2,04 - 2,10 (2H, m), 2,17 - 2,25 (2H, m), 2,38 (3H, s), 3,34 - 3,41 (2H, m), 3,74 (3H, s), 3,98 - 4,03 (2H, m), 4,76 (1H, s), 7,12 (1H, s), 7,28 (2H, m), 8,03 - 8,05 (2H, m), 8,46 (1H, s). MS m/e MH+ 570. Intermediário 64: ácido 4-(terc-butoxicarbonilamino)-1-(5-ciclolopropil- 7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxílico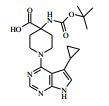
[000291] Solução 2 M de hidróxido de sódio (5,00 ml) foi adicionada ao 4-(terc-butoxicarbonilamino)-1-(5-ciclolopropil-7-tosil-7H-pirrolo[2,3- d]-pirimidin-4-il)piperidina-4-carboxilato de metila (Intermediário 63) (780 mg, 1,37 mmol) em THF (10 ml) a 20° C. A solução resultante foi agitada a 40° C por 24 horas. A mistura de reação foi diluída com EtOAc (50 ml) e a camada aquosa foi coletada e acidificada até o pH 4 com solução 2 M HCl. A camada aquosa foi extraída com DCM (2 x 50 ml) e a camada orgânicas combinada e lavada com salmoura saturada (50 ml). A camada orgânica foi secada em MgSO4, filtrada e evaporada para produzir o produto ácido 4-(terc-butoxicarbonilamino)- 1-(5-ciclolopropil-7H-pirrolo[2,3-d]pirimidin-4-il)-piperidina-4-carboxílico bruto (375 mg, 68,2 %) como um sólido creme.
[000292] 1H RMN (400,13 MHz, DMSO-d6) δ 0,66 - 0,73 (2H, m), 0,88 - 1,00 (2H, m), 1,40 (9H, s), 1,94 - 2,09 (5H, m), 3,41 - 3,53 (2H, m), 3,92 - 4,01 (2H, m), 6,92 (1H, d), 7,18 - 7,22 (1H, m), 8,21 (1H, s), 11,51 (1H, s), 12,29 (1H, s). MS m/e MH+ 402. Intermediário 65: 4-(1-(4-clorofenil)-3-hidroxipropilcarbamoil)-1-(5- ciclolo-propil-7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato (S)- terc-butila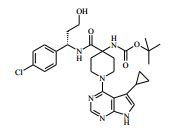
[000293] (S)-3-amino-3-(4-clorofenil)propan-1-ol (173 mg, 0,93 mmol) foi adicionado em uma porção ao ácido 4-(terc-butoxicarbonilamino)-1- (5-ciclolopropil-7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxílico (Intermediário 64) (375 mg, 0,93 mmol) e DIPEA (0,489 ml, 2,80 mmol) em DMA (5 ml). Hexafluorofosfato de O-(7-Azabenzotriazol-1-il)- N,N,N’,N’-tetrametilurônio (391 mg, 1,03 mmol) foi adicionado e a solução resultante foi agitada a 20° C por 24 horas. A mistura de reação foi evaporada até a secura depois diluída com EtOAc (75 ml) e lavada sequencialmente com água (50 ml) e salmoura saturada (50 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 2 a 6 % de MeOH com amônia em DCM. As frações foram evaporadas para produzir 4-(1-(4-clorofenil)-3-hidroxipropil carbamoil)- 1-(5-ciclolopropil-7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de (S)-terc-butila (420 mg, 79 %) como uma goma amarela.
[000294] 1H RMN (400,13 MHz, DMSO-d6) δ 0,66 - 0,70 (2H, m), 0,86 - 0,91 (2H, m), 1,40 (9H, s), 1,81 - 2,06 (7H, m), 3,33 - 3,40 (4H, m), 3,90 - 3,94 (2H, m), 4,53 (1H, t), 4,91 - 4,93 (1H, m), 6,92 (1H, s), 7,30 - 7,38 (4H, m), 7,96 (1H, d), 8,20 (1H, s), 11,49 (1H, s). MS m/e MH+ 569. Intermediário 66: 1-(4-clorofenil)-3-(metilamino)propilcarbamato de terc-butila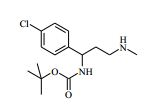
[000295] O gás de metilamina foi borbulhado em uma solução de metanossulfonato de 3-(terc-butoxicarbonilamino)-3-(4- clorofenil)propila (Intermediário 31) (70 mg, 0,19 mmol) em THF (5 ml) a 22º C em um período de 5 minutos. A mistura foi selada em um tubo de microonda. A reação foi aquecida a 125° C por 30 minutos no reator de microonda e esfriada até a temperatura ambiente. A mistura foi evaporada e o resíduo foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 4 a 8 % (MeOH 10:1 / NH3 conc. (aq)) em DCM. As frações puras foram evaporadas até a secura para produzir 1-(4-clorofenil)-3- (metilamino)propilcarbamato de terc-butila (52 mg, 90 %) como um óleo incolor.
[000296] 1H RMN (399,902 MHz, CDCl3) δ 1,41 (9H, s), 1,80 (1H, br.m), 1,93 (1H, br.m), 2,40 (3H, s), 2,59 (2H, m), 4,73 (1H, br.s), 5,94 (1H, br.s), 7,21 (2H, d), 7,29 (2H, d). MS m/e MH+299,5. Intermediário 67: 1-(4-clorofenil)-N3-metilpropano-1,3-diamina
[000297] Cloreto de hidrogênio (4 M em 1,4-dioxano, 0,50 ml, 2,00 mmol) foi adicionado ao 1-(4-clorofenil)-3-(metilamino)propilcarbamato de terc-butila (Intermediário 66) (50 mg, 0,17 mmol) em uma mistura de DCM (5 ml) e metanol (2 ml) a 22° C. A solução resultante foi agitada a 22° C por 4 horas. A mistura foi concentrada e o resíduo foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 2 M e as frações puras foram evaporadas até a secura para produzir 1-(4-clorofenil)-N3- metilpropano-1,3-diamina (30 mg, 90 %) como um óleo incolor.
[000298] 1H RMN (399,902 MHz, CDCl3) δ 1,81 (2H, dt), 2,40 (3H, s), 2,58 (2H, m), 4,01 (1H, t), 7,25 - 7,31 (4H, m). MS m/e MH+ 199,3. Intermediário 68: 4-(1-(4-clorofenil)-3-(metilamino)propilcarbamoil)-1- (7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila
[000299] Hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (44 mg, 0,12 mmol) foi adicionado ao ácido 4-(terc- butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxílico (Intermediário 1) (40 mg, 0,11 mmol) e N,N- diisopropiletilamina (0,023 ml, 0,13 mmol) em NMP (5 ml) a 22° C. A suspensão resultante foi agitada a 50° C por 10 minutos. A mistura foi esfriada até a temperatura ambiente e 1-(4-clorofenil)-N3- metilpropano-1,3-diamina (Intermediário 67) (22 mg, 0,11 mmol) foi adicionado como uma solução em NMP (2 ml) e a mistura agitada a 22° C por 3 dias. A mistura de reação foi diluída com EtOAc (50 ml) e lavada com água (50 ml). A camada aquosa foi passada através de uma coluna SCX. A coluna foi eluída com água e metanol para remover as impurezas seguidas por 30 % (2 M de NH3 em metanol) em DCM para eluir o produto. As soluções apropriadas foram evaporadas até a secura para produzir 4-(1-(4-clorofenil)-3- (metilamino)propilcarbamoil)-1-(7H-pirrolo[2,3-d]-pirimidin-4-il)piperidin- 4-ilcarbamato de terc-butila (10,00 mg, 17 %) como um sólido branco. MS m/e MH+ 542,5. Intermediário 69: 4-metil 4-(terc-butoxicarbonilamino)piperidina-1,4- dicarboxilato de 1-terc-butila
[000300] 4-(terc-butoxicarbonilamino)piperidina-4-carboxilato metila (WO 2008075109) (10 g, 38,71 mmol) foi dissolvido em DCM (194 ml). A este foi adicionado dicarbonato de di-terc-butila (10,67 ml, 46,46 mmol) às porções. Depois da adição a reação foi agitada a 25° C por 1 hora. O produto bruto foi lavado com bicarbonato de sódio saturado (3 x 50 ml) e as camadas orgânicas secadas em MgSO4 antes de serem evaporadas até a secura para produzir 4-metil 4-(terc- butoxicarbonilamino)piperidina-1,4-dicarboxilato de 1-terc-butila (13,6 g, 98 %) como um sólido branco.
[000301] 1H RMN (400,13 MHz, DMSO) δ 1,39 (9H, s), 1,40 (9H, s), 1,70 - 1,77 (2H, td), 1,88 - 1,92 (2H, d), 3,05 (2H, amplo), 3,61 (3H, s), 3,62 - 3,65 (2H, m), 7,33 (amplo, troca). Intermediário 70:ácido 1-(terc-butoxicarbonil)-4-(terc- butoxicarbonilamino)-piperidina-4-carboxílico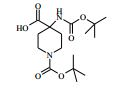
[000302] Hidróxido de lítio monoidratado (8,13 g, 193,62 mmol) foi adicionado ao 4-metil 4-(terc-butoxicarbonilamino)piperidina-1,4- dicarboxilato de 1-terc-butila (Intermediário 69) (13,88 g, 38,72 mmol) em água (21,51 ml), THF (86 ml) e metanol (86 ml). A solução resultante foi agitada a 20° C por 1 dia. A mistura de reação foi diluída com EtOAc (75 ml) e lavada com água (75 ml). O aquoso foi ajustado ao pH 5 com solução 1 M de ácido cítrico, depois extraída com EtOAc (3 x 200 ml). Os extratos orgânicos foram lavados com salmoura saturada (50 ml), depois secados em MgSO4, filtrados e evaporados para produzir o produto desejado de ácido 1-(terc-butoxicarbonil)-4- (terc-butoxicarbonilamino)piperidina-4-carboxílico (10,36 g, 78 %) como um sólido branco fino.
[000303] 1H RMN (400,13 MHz, DMSO) δ 1,39 (9H, s), 1,40 (9H, s), 1,68 - 1,76 (2H, td), 1,89 - 1,92 (2H, d), 3,03 (2H, amplo), 3,62 - 3,65 (2H, d), 7,16 (troca), 12,36 (troca). Intermediário 71: 4-(terc-butoxicarbonilamino)-4-(1-(4-clorofenil)-4- hidróxi-butilcarbamoil)piperidina-1-carboxilato de terc-butila
[000304] Hexafluorofosfato de O-(7-Azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (5,24 g, 13,77 mmol) foi adicionado em uma porção de ácido 1-(terc-butoxicarbonil)-4-(terc-butoxicarbonilamino)piperidina-4- carboxílico (Intermediário 70) (4,31 g, 12,5 mmol), 4-amino-4-(4- clorofenil)butan-1-ol (Intermediário 16) (2,5 g, 12,5 mmol) e DIPEA (6,56 ml, 37,6 mmol) em DMA (62,6 ml) sob nitrogênio. A solução resultante foi agitada a 60° C por 3 horas e evaporada até a secura. A mistura de reação bruta foi dissolvida em 100 ml de EtOAc e depois basificada com NaHCO3 saturado (150 ml) e a fração orgânica secada em MgSO4, filtrada e evaporada até a secura. A fração orgânica foi purificada pela cromatografia por vaporização instantânea em sílica (eluente de 0 a 10 % de amônia 7 N em MeOH / DCM). As frações puras foram depois evaporadas para produzir 4-(terc- butoxicarbonilamino)-4-(1-(4-clorofenil)-4-hidroxibutilcarbamoil) piperidina-1-carboxilato de terc-butila (5,06 g, 77 %) como um sólido branco amarelado.
[000305] 1H RMN (400,13 MHz, DMSO) δ 1,39 (18H, s), 1,42 - 1,50 (2H, sex), 1,67 - 1,73 (2H, q), 1,74 - 1,82 (2H, m), 1,87 (2H, amplo), 3,07 (2H, amplo), 3,34 - 3,41 (2H, m), 3,53 - 3,61 (2H, t), 4,71 - 4,76 (1H, q), 6,79 (3 x troca, amplo), 7,29 - 7,34 (4H, m), 7,80 - 7,82 (troca, d). MS m/e MH+ = 527; HPLC tR = 2,48 min. Intermediário 72: 4-amino-N-(1-(4-clorofenil)-4-hidroxibutil)piperidina-4- carboxamida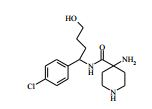
[000306] 4-(terc-butoxicarbonilamino)-4-(1-(4-clorofenil)-4-hidroxibutil carbamoil)piperidina-1-carboxilato de terc-butila (Intermediário 71) (1,98 g, 3,76 mmol) foi agitado em THF (18,82 ml) e cloreto de hidrogênio 4 M (14,11 ml, 56,46 mmol) em dioxano foi adicionado com agitação. A solução resultante foi agitada na temperatura ambiente durante a noite. A mistura bruta foi purificada pela cromatografia de SCX (eluente 20 % amônia 7 N / MeOH em DCM). As frações puras foram evaporadas até a secura para produzir 4-amino-N-(1-(4- clorofenil)-4-hidroxibutil)piperidina-4-carboxamida (1,04 g, 85 %), como uma goma transparente incolor.
[000307] 1H RMN (400,13 MHz) δ 1,16 - 1,27 (2H, dd), 1,29 - 1,51 (2H, sepd), 1,70 - 1,76 (2H, q), 1,75 - 1,89 (2H, m), 2,65 - 2,72 (2H, m), 2,74 - 2,83 (2H, dq), 3,37 - 3,39 (2H, m), 4,39 (troca, amplo) 4,70 - 4,76 (1H, q), 7,32 - 7,35 (2H, d), 7,36 - 7,39 (2H, d), 8,25 - 8,27 (troca, d) MS m/e MH+ = 326; HPLC tR = 1,70 min. Intermediário 73: 2-amino-1-(4-clorofenil)etilcarbamato de terc-butila
[000308] Uma solução de metanossulfonato 2-(terc- butoxicarbonilamino)-2-(4-clorofenil)etila (Intermediário 23) (535 mg, 1,53 mmol) em DMF (8 ml) foi tratado com azida de sódio (199 mg, 3,06 mmol) e a mistura foi aquecida a 80° C por 1 hora. A mistura foi esfriada e deixada agitar na temperatura ambiente durante a noite. A solução foi dividido entre acetato de etila e água. A camada orgânica foi lavada duas vezes com água depois secada com sulfato de magnésio, filtrada e concentrada até que o volume final foi de aproximadamente 5 ml. O Etanol (20 ml) e em paládio a 10 % em carbono (75 mg, 0,07 mmol) foram adicionados. A suspensão resultante foi agitada sob uma atmosfera de hidrogênio na pressão e temperatura ambiente por 1 hora. A mistura foi filtrada e o filtrado foi concentrado sob pressão reduzida para dar 2-amino-1-(4- clorofenil)etilcarbamato de terc-butila como uma goma (410 mg, 99 %). MS m/e MH+ = 271 Intermediário 74: 1-(4-clorofenil)-2-(metilsulfonamido)etilcarbamato de terc-butila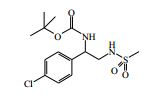
[000309] Uma solução de 2-amino-1-(4-clorofenil)etilcarbamato de terc- butila (Intermediário 73) (220 mg, 0,81 mmol) e N-etildiisopropilamina (0,281 ml, 1,63 mmol) em THF (5 ml) foi tratada com cloreto de metanossulfonila (0,075 ml, 0,98 mmol). A solução resultante foi agitada na temperatura ambiente por 2 horas. A mistura foi dividida entre DCM e solução de bicarbonato de sódio. A camada orgânica foi concentrada e o resíduo foi purificado pela cromatografia em coluna de vaporização instantânea em sílica usando eluição de gradiente (10 % de acetato de etila / DCM a 30 % de acetato de etila / DCM). O produto desejado, 1-(4- clorofenil)-2-(metilsulfonamido)etilcarbamato de terc-butila, foi assim isolado como um sólido incolor (154 mg, 54,3 %).
[000310] 1H RMN (399,9 MHz, CDCl3) δ 1,43 (9H, s), 2,92 (3H, s), 3,38 - 3,52 (2H, m), 4,68 - 4,84 (2H, m), 5,20 - 5,28 (1H, m), 7,23 (2H, d), 7,35 (2H, d) MS m/e (M - H)- = 347 Intermediário 75: N-(2-Amino-2-(4-clorofenil)etil)metanossulfonamida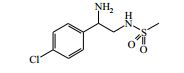
[000311] 1-(4-clorofenil)-2-(metilsulfonamido)etilcarbamato de terc- butila (Intermediário 74) (151 mg, 0,43 mmol) foi tratado com ácido trifluoroacético (2 ml). A solução foi agitada por 1 hora na temperatura ambiente. A mistura foi concentrada sob pressão reduzida. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O resíduo foi carregado na coluna em metanol e lavado com metanol. O produto desejado foi eluído da coluna usando amônia 2 M em metanol e as frações puras foram evaporadas até a secura para produzir N-(2-amino-2-(4-clorofenil)etil)-metanossulfonamida (93 mg, 86 %) como um sólido cristalino incolor.
[000312] 1H RMN (399,9 MHz, CDCl3) δ 2,89 (3H, s), 3,17 (1H, dd), 3,33 (1H, dd), 4,12 (1H, dd), 4,74 (1H, br, s), 7,29 (2H, d), 7,34 (2H, d) MS m/e (M - H)- = 247 Intermediário 76: 4-(1-(4-clorofenil)-2-(metilsulfonamido)etilcarbamoil)- 1 -(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila
[000313] Hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (88 mg, 0,23 mmol) foi adicionado em uma porção a uma solução saturada de ácido 4-(terc-butoxicarbonilamino)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxílico (Intermediário 1) (79 mg, 0,22 mmol) e N-etildiisopropilamina (0,046 ml, 0,26 mmol) em NMP (3 ml). A mistura foi tratada com N-(2-amino-2-(4- clorofenil)etil)metanossulfonamida (Intermediário 75) (60 mg, 0,24 mmol). A solução foi agitada por 65 horas na temperatura ambiente. A mistura foi dividida entre acetato de etila e água. A camada orgânica foi lavada duas vezes com água. A solução orgânica foi concentrada sob pressão reduzida. O resíduo foi purificado pela cromatografia em coluna de vaporização instantânea em sílica usando eluição de gradiente (1 % de metanol / DCM a 15 % de metanol / DCM). O produto contendo as frações foi combinado para dar 4-(1-(4-clorofenil)- 2-(metilsulfonamido)etil-carbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidin-4-ilcarbamato de terc-butila (103 mg, 79 %) como sólido incolor.
[000314] 1H RMN (399,9 MHz, DMSO-d6) δ 1,41 (9H, s), 2,00 (4H, br, s), 2,81 (3H, s), 3,52 - 3,69 (2H, m), 4,18 - 4,27 (2H, m), 4,96 (1H, q), 6,60 (1H, s), 6,97 (1H, br, s), 7,10 - 7,22 (2H, m), 7,37 (4H, s), 8,01 (1H, d), 8,14 (1H, s), 11,65 (1H, s) MS m/e MH+ = 592 Intermediário 77: 2-(terc-butoxicarbonilamino)-2-(4-clorofenil)etila etano-tioato
[000315] Uma solução de metanossulfonato de 2-(terc- butoxicarbonil-amino)-2-(4-clorofenil)etila (Intermediário 23) (600 mg, 1,72 mmol) em DMF (10 ml) foi tratada com tioacetato de potássio (392 mg, 3,43 mmol) e a mistura foi aquecida a 50° C por 1 hora. A mistura foi esfriada e dividido entre acetato de etila e água. A camada orgânica foi lavada duas vezes com água depois secada com sulfato de magnésio, filtrada e concentrada até a secura. O resíduo foi purificado pela cromatografia em coluna de vaporização instantânea em sílica usando eluição de gradiente (acetato de etila a 10 % / isoexano a 20 % de acetato de etila / isoexano). O produto desejado, etanotioato de 2-(terc-butoxicarbonilamino)-2-(4-clorofenil)etila (509 mg, 90 %), foi assim isolado como um sólido cristalino cremoso.
[000316] 1H RMN (399,9 MHz, CDCl3) δ 1,40 (9H, s), 2,35 (3H, s), 3,15 - 3,28 (2H, m), 4,78 (1H, br, s), 5,07 (1H, br, s), 7,24 (2H, d), 7,31 (2H, d) MS m/e (M - H-CH3CO)- = 286 Intermediário 78: 1-(4-clorofenil)-2-(clorossulfonil)etilcarbamato de terc- butila
[000317] N-Clorossuccinimida (819 mg, 6,14 mmol) foi adicionada a uma solução de ácido clorídrico 2 M (0,8 ml) em acetonitrila (10 ml). O frasco de reação foi esfriado com um banho de gelo a 10° C e etanotioato de 2-(terc-butoxicarbonilamino)-2-(4-clorofenil)etila (Intermediário 77) (506 mg, 1,53 mmol) foi adicionado às porções. A mistura aquecida durante a adição e foi agitada por 10 minutos na temperatura ambiente. A mistura foi dividida entre acetato de etila e água. A camada orgânica foi lavada com salmoura, secada com sulfato de magnésio, filtrada e concentrada até a secura. 1-(4- clorofenil)-2-(clorossulfonil)etilcarbamato de terc-butila (602 mg, quant.) foi assim obtido como um sólido incolor.
[000318] 1H RMN (399,9 MHz, CDCl3) δ 1,44 (9H, s), 2,77 (1H, s), 4,06 (1H, dd), 4,36 (1H, br, s), 5,15 - 5,23 (1H, m), 5,29 - 5,37 (1H, m), 7,29 (2H, d), 7,38 (2H, d) Intermediário 79: 1-(4-clorofenil)-2-sulfamoiletilcarbamato de terc-butila
[000319] Amônia (1,5 ml, 31,50 mmol) foi adicionada a uma suspensão de 1-(4-clorofenil)-2-(clorossulfonil)etilcarbamato de terc- butila (Intermediário 78) (0,542 g, 1,53 mmol) em acetonitrila (10 ml). A mistura foi agitada por 16 horas na temperatura ambiente. A mistura foi dividida entre acetato de etila e água e a camada orgânica foi lavada com salmoura. A solução orgânica foi secada com sulfato de magnésio, filtrada e concentrada sob pressão reduzida. O resíduo foi purificado pela cromatografia em coluna de vaporização instantânea em sílica usando eluição de gradiente (acetato de etila a 10 % / DCM a acetato de etila a 30 % / DCM). O produto desejado, 1-(4-clorofenil)-2- sulfamoiletil-carbamato de terc-butila (0,351 g, 68,5 %), foi assim isolado como um sólido incolor. 1H RMN (399,9 MHz, DMSO-d6) δ 1,36 (9H, s), 3,21 - 3,28 (1H, m), 3,47 - 3,56 (1H, m), 5,02 (1H, br, s), 6,88 (2H, s), 7,35 (2H, d), 7,41 (2H, d), 7,49 - 7,60 (1H, m) MS m/e (M - H-)- = 333 Intermediário 80: 2-amino-2-(4-clorofenil)etanossulfonamida
[000320] 1-(4-clorofenil)-2-sulfamoiletilcarbamato de terc-butila (Intermediário 79) (325 mg, 0,97 mmol) foi tratado com ácido trifluoroacético (8 ml). A solução resultante foi agitada por 15 minutos na temperatura ambiente. A mistura foi concentrada sob pressão reduzida e o resíduo foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. A coluna foi lavada com metanol e o produto desejado foi eluído usando amônia em metanol (2 M) e as frações puras foram evaporadas até a secura para produzir 2-amino-2-(4- clorofenil)etanossulfonamida (221 mg, 97 %) como um sólido quase incolor.
[000321] 1H RMN (399,9 MHz, DMSO-d6) δ 3,13 - 3,25 (2H, m), 4,39 (1H, dd), 7,35 - 7,48 (4H, m) MS m/e (M - H-)- = 233 Intermediário 81: 4-(1-(4-clorofenil)-2-sulfamoiletilcarbamoil)-1-(7H- pirrolo-[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila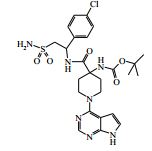
[000322] Hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (128 mg, 0,34 mmol) foi adicionado em uma porção a uma solução saturada de ácido 4-(terc-butoxicarbonilamino)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxílico (Intermediário 1) (115 mg, 0,32 mmol) e N-etildiisopropilamina (0,066 ml, 0,38 mmol) em NMP (2,5 ml). A mistura foi tratada com 2-amino-2-(4- clorofenil)etanossulfonamida (Intermediário 80) (75 mg, 0,32 mmol). A solução foi agitada por 1 hora na temperatura ambiente. A mistura foi tratada com água (5 ml) e o precipitado incolor resultante foi isolado pela filtração, lavado com água e depois acetonitrila para dar 4-(1-(4- clorofenil)-2-sulfamoiletilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidin-4-ilcarbamato de terc-butila (157 mg, 85 %) como sólido incolor.
[000323] 1H RMN (399,9 MHz, DMSO-d6) δ 1,40 (9H, s), 1,90 - 2,10 (4H, m), 3,35 - 3,39 (1H, m), 3,51 - 3,59 (1H, m), 3,62 - 3,71 (2H, m), 4,12 - 4,26 (2H, m), 5,30 - 5,39 (1H, m), 6,59 (1H, s), 6,82 (2H, s), 7,09 - 7,18 (2H, m), 7,30 - 7,43 (4H, m), 8,13 (1H, s), 8,19 (1H, d), 11,65 (1H, s) MS m/e MH+ = 578 Intermediário 82: 2-acetamido-1-(4-clorofenil)etilcarbamato de terc- butila
[000324] Uma solução de 2-amino-1-(4-clorofenil)etilcarbamato de terc-butila (Intermediário 73) (0,208 g, 0,77 mmol) e N- etildiisopropilamina (0,266 ml, 1,54 mmol) em THF (5 ml) foi tratada com anidrido acético (0,102 ml, 1,08 mmol). A solução resultante foi agitada na temperatura ambiente por 2 horas. A mistura foi dividida entre DCM e solução de bicarbonato de sódio. A camada orgânica foi concentrada e o resíduo foi purificado pela cromatografia em coluna de vaporização instantânea em sílica usando eluição de gradiente (acetato de etila a 10 % / DCM acetato de etila a 40 % / DCM). O produto desejado, 2-acetamido-1-(4-clorofenil)etilcarbamato de terc- butila (0,151 g, 62,7 %), foi assim isolado como um sólido incolor.
[000325] 1H RMN (399,9 MHz, CDCl3) δ 1,41 (9H, s), 1,98 (3H, s), 3,46 - 3,67 (2H, m), 4,74 (1H, br, s), 4,97 - 5,56 (1H, m), 5,89 (1H, br, s), 7,22 (2H, d), 7,32 (2H, d) MS m/e MH+ = 313 Intermediário 83: N-(2-amino-2-(4-clorofenil)etil)acetamida
[000326] 2-acetamido-1-(4-clorofenil)etilcarbamato de terc-butila (Intermediário 82) (148 mg, 0,47 mmol) foi tratado com ácido trifluoroacético (2 ml). A solução foi agitada por 1 horas na temperatura ambiente. A mistura foi concentrada sob pressão reduzida. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O resíduo foi carregado na coluna em metanol e lavado com metanol. O produto desejado foi eluído da coluna usando amônia 2 M em metanol e as frações puras foram evaporadas até a secura para produzir N-(2-amino-2-(4-clorofenil)etil)-acetamida (98 mg, 97 %) como um sólido cristalino amarelo claro.
[000327] 1H RMN (399,9 MHz, CDCl3) δ 1,61 (2H, br, s), 1,97 (3H, s), 3,28 - 3,37 (1H, m), 3,44 - 3,52 (1H, m), 4,05 - 4,11 (1H, m), 5,78 (1H, br, s), 7,28 - 7,36 (4H, m) MS m/e MH+ = 213 Intermediário 84: 4-(2-acetamido-1-(4-clorofenil)etilcarbamoil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila
[000328] Hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (89 mg, 0,23 mmol) foi adicionado em uma porção a uma solução saturada de ácido 4-(terc-butoxicarbonilamino)-1-(7H- pirrolo[2,3-d]-pirimidin-4-il)piperidina-4-carboxílico (Intermediário 1) (80 mg, 0,22 mmol) e N-etildiisopropilamina (0,046 ml, 0,27 mmol) em NMP (3 ml). A mistura foi tratada com N-(2-amino-2-(4- clorofenil)etil)acetamida (Intermediário 83) (52 mg, 0,24 mmol). A solução foi agitada por 65 horas na temperatura ambiente. A mistura foi dividida entre acetato de etila e água. A camada orgânica foi lavada duas vezes com água. A solução orgânica foi concentrada sob pressão reduzida. O resíduo foi purificado pela cromatografia em coluna de vaporização instantânea em sílica usando eluição de gradiente (1 % de metanol / DCM a 15 % de metanol / DCM). O produto contendo as frações foi combinado para dar 4-(2-acetamido-1- (4-clorofenil)etilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4- ilcarbamato de terc-butila (100 mg, 81 %) como sólido incolor.
[000329] 1H RMN (399,9 MHz, DMSO-d6) δ 1,42 (9H, s), 1,78 (3H, s), 1,98 (4H, br, s), 3,36 - 3,43 (1H, m), 3,50 - 3,68 (2H, m), 4,18 - 4,28 (2H, m), 4,85 - 4,93 (1H, m), 6,58 - 6,61 (1H, m), 7,14 - 7,23 (2H, m), 7,30 - 7,37 (4H, m), 7,84 (1H, br, s), 8,12 - 8,17 (2H, m), 11,65 (1H, br, s) MS m/e MH+ = 556 Intermediário 85: 2-metil-1-tritil-1H-imidazol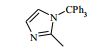
[000330] Trietilamina (11,54 ml, 82,82 mmol) foi adicionado às gotas ao 2-metil-1H-imidazol (4,0 g, 48,72 mmol) e clorotrifenilmetano (14,94 g, 53,59 mmol) em acetonitrila (120 ml) na temperatura ambiente em um período de 20 minutos sob nitrogênio. A suspensão resultante foi agitada na temperatura ambiente por 18 horas. Água (120 ml) e i- hexano (12 ml) foram adicionados e a pasta fluida agitada por 30 minutos antes da filtração. O sólido filtrado foi lavado com água (3 x 15 ml) e secado sob vácuo para produzir 2-metil-1-tritil-1H-imidazol (15,41 g, 97 %) como um sólido creme que foi usado sem outra purificação.
[000331] 1H RMN (399,902 MHz, CDCl3) δ 1,65 (3H, s), 6,70 (1H, d), 6,90 (1H, d), 7,11 - 7,16 (6H, m), 7,29 - 7,35 (9H, m); m/z (ESI+) (M + H)+ = 325; HPLC tR = 2,88 min. Intermediário 86: 1-(4-clorofenil)-2-(1-tritil-1H-imidazol-2-il)etanol
[000332] Butil-lítio (1,6 M em hexanos) (4,24 ml, 6,78 mmol) foi adicionado às gotas ao 2-metil-1-tritil-1H-imidazol (Intermediário 85) (2,0 g, 6,16 mmol) em THF (30 ml) esfriado a -78° C em um período de 20 minutos sob nitrogênio. A solução vermelho escura resultante foi agitada a -78° C por 30 minutos depois uma solução de 4- Clorobenzaldeído (0,867 g, 6,16 mmol) em THF (10 ml) adicionado às gotas. A reação foi deixada aquecer lentamente a 0° C depois extinta com NH4Cl saturado (50 ml) e extraída com TBME (2 X). Os extratos combinados foram lavados com salmoura, secados em MgSO4, concentrado pela evaporação depois purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição 20 a 30 % de EtOAc em isoexano. As frações puras foram evaporadas até a secura para produzir 1-(4-clorofenil)-2-(1-tritil-1H-imidazol-2-il)etanol (1,810 g, 63,1 %) como um sólido incolor.
[000333] 1H RMN (399,902 MHz, DMSO) δ 2,05 (2H, d), 4,59 - 4,63 (1H, m), 5,99 (1H, d), 6,66 (1H, d), 6,96 - 7,02 (9H, m), 7,26 (2H, d), 7,37 - 7,40 (9H, m);
[000334] m/z (ESI+) (M + H)+ = 465; HPLC tR = 3,54 min. Intermediário 87: 2-(1-(4-clorofenil)-2-(1-tritil-1H-imidazol-2-il)etil)iso- indolino-1,3-diona
[000335] Azodicarboxilat de di-terc-butila (0,808 g, 3,51 mmol) foi a 1-(4-clorofenil)-2-(1-tritil-1H-imidazol-2- il)etanol (Intermediário 86) (1,36 g, 2,92 mmol), ftalimida (0,473 g, 3,22 mmol) e trifenilfosfina (0,921 g, 3,51 mmol) em THF (25 ml) na temperatura resultante foi vaporização instantânea em sílica, gradiente de eluição 20 a 40 % de EtOAc em isoexano. As frações puras foram evaporadas até a secura para produzir 2-(1-(4-clorofenil)-2-(1-tritil-1H-imidazol-2- il)etil)isoindolino-1,3-diona (1,78 g, 102 %) como uma espuma incolor. m/z (ESI+) (M + H)+ = 594; HPLC tR = 2,80 min. Intermediário 88: 1 -(4-clorofenil)-2-(1 -tritil-1H-imidazol-2-il)etanamina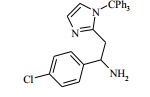
[000336] 2-(1-(4-clorofenil)-2-(1-tritil-1H-imidazol-2-il)etil)isoindolino- 1,3-diona (Intermediário 87) (1,78 g, 3,00 mmol) e hidrato de hidrazina (0,204 ml, 4,19 mmol) em metanol (30 ml) foram refluxados por 3 horas. A solução clara resultante foi deixada esfriar até a temperatura ambiente durante a noite depois filtrada. O filtrado foi aplicado a um cartucho 20 g de SCX e eluído com metanol seguido por NH3(MeOH) 2N. As frações contendo o produto foram combinadas e concentradas pela evaporação depois purificadas pela cromatografia por vaporização instantânea em sílica, gradiente de eluição 2 a 5 % de MeOH em DCM. As frações puras foram evaporadas até a secura para produzir 1-(4-clorofenil)-2-(1-tritil-1H-imidazol-2-il)etanamina (0,942 g, 67,8 %) como um sólido incolor.
[000337] 1H RMN (399,902 MHz, CDCl3) δ 2,16 (2H, d), 4,01 (1H, t), 6,74 (1H, d), 6,90 (2H, d), 6,99 (1H, d), 7,08 - 7,13 (7H, m), 7,29 - 7,32 (10H, m); m/z (ESI+) (M + H)+ = 464; HPLC tR = 3,13 min. Intermediário 89: 4-(1-(4-clorofenil)-2-(1-tritil-1H-imidazol-2-il)etil- carbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila
[000338] Hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (166 mg, 0,44 mmol) foi adicionado às porções ao ácido 4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxílico (Intermediário 1) (150 mg, 0,42 mmol), 1-(4- clorofenil)-2-(1-tritil-1H-imidazol-2-il)etanamina (Intermediário 88) (193 mg, 0,42 mmol) e N-etildiisopropilamina (0,087 ml, 0,50 mmol) em DMF (2,0 ml) na temperatura ambiente. A solução resultante foi agitada na temperatura ambiente por 3 horas depois extinta em água (10 ml) para dar um ppt. amarelo claro. O precipitado foi coletado pela filtração, lavado com água e secado sob vácuo para produzir 4-(1-(4- clorofenil)-2-(1-tritil-1H-imidazol-2-il)etilcarbamoil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (320 mg, 95 %) como um sólido creme, que foi usado sem outra purificação.
[000339] 1H RMN (399,902 MHz, CDCl3) δ 1,40 (9H, s), 2,05 - 2,29 (6H, m), 3,56 - 3,61 (2H, m), 4,35 - 4,42 (2H, m), 5,33 (1H, s), 6,50 (1H, d), 6,78 (1H, m), 6,93 (2H, d), 6,97 - 7,01 (7H, m), 7,13 (2H, d), 7,26 - 7,35 (10H, m), 8,27 (1H, s), 9,25 (1H, br s), 10,22 (1H, br s). m/z (ESI+) (M + H)+ = 807; HPLC tR = 2,28 min. Intermediário 90: 1-(4-Clorofenil)-2-(1H-pirazol-1-il)etanona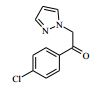
[000340] Uma solução de pirazol (3,40 g, 49,98 mmol) e 2-bromo-4’- cloroacetofenona (11,44 g, 49 mmol) dissolvida em DME (70 ml) foi agitada na temperatura ambiente sob argônio por 5 dias. Éter dietílico foi adicionado e o precipitado foi filtrado, lavado com éter dietílico e secado a vácuo durante a noite para dar o produto bruto, 8,842 g de cristais brancos puros. Amônia aquoso concentrada (30 %; 36 ml) foi adicionada a uma suspensão de 8,76 g do produto bruto em 7 ml de água. A reação foi agitada por 40 min depois que os cristais amarelos foram filtrado, lavado com um pouco de água e secado a vácuo para produzir 1-(4-clorofenil)-2-(1H-pirazol-1-il)etanona (5,04 g, 22,84 mmol, 46,6 %) como um sólido amarelo claro. A reextração da lavagem deu mais produto. Este foi purificado pela cromatografia por vaporização instantânea em sílica gel eluindo com 0 a 10 % de acetato de etila em diclorometano. O solvente foi evaporado até a secura para produzir 1- (4-clorofenil)-2-(1H-pirazol-1-il)etanona (2,78 g, 12,60 mmol, 25,7 %) como um sólido cristalino incolor. RMN & MS = EN00228-12-01 m/z (ESI+) (M + H)+ = 221.
[000341] 1H RMN (500 MHz, DMSO-d6) δ 5,83 (2H, s), 6,61 (1H, dd), 7,48 (1H, d), 7,66 (2H, d), 7,73 (1H, d), 8,04 (2H, d) Intermediário 91: (Z) e (E)-1-(4-clorofenil)-2-(1H-pirazol-1-il)etanona O- metil oxima
[000342] Cloridreto de metoxilamina (0,668 g, 8,00 mmol), foi adicionado a uma solução agitada de 1-(4-clorofenil)-2-(1H-pirazol-1- il)etanona (Intermediário 90) (1,103 g, 5 mmol), na temperatura ambiente sob argônio. A solução resultante foi agitada na temperatura ambiente a noite. A piridina foi evaporada a vácuo e o sólido residual foi triturado com uma solução aquosa saturada de hidrogeno carbonato de sódio (50-100 ml). O sólido foi filtrado, lavado com água e secado para produzir (Z) e (E)-1-(4-clorofenil)-2-(1H-pirazol-1- il)etanona O-metil oxima (1,080 g, 87 %) como um sólido amarelo escuro. RMN & MS = EN00228-18-01 m/z (ESI+) (M + H)+ = 250.
[000343] 1H RMN (500 MHz, DMSO-d6) δ 3,99 (3H, s), 5,44 (2H, s), 6,19 (1H, dd), 7,36 (1H, d), 7,42 (2H, d), 7,65 (2H, d), 7,71 (1H, d) Intermediário 92: 1-(4-clorofenil)-2-(1H-pirazol-1-il)etanamina
[000344] O complexo de borano tetraidrofurano (15,00 ml, 15,00 mmol) foi adicionado a uma solução agitada de (Z) e (E)-1-(4- clorofenil)-2-(1H-pirazol-1-il)etanona O-metil oxima (Intermediário 91) (0,749 g, 3 mmol) dissolvida em THF (30 ml) na temperatura ambiente sob argônio. A solução resultante foi agitada sob refluxo por 3 horas. A mistura foi esfriada em um banho de água/gelo e água (25 ml) foi cuidadosamente agitada seguida por 20 % de NaOH (25 ml). A mistura bifásica resultante foi refluxada durante a noite com agitação magnética vigorosa e deixada esfriar. Éter dietílico foi adicionado e as camadas foram separadas. A camada orgânica foi ainda extraída com éter dietílico secada em MgSO4, filtrada e evaporada para dar o produto bruto (741 mg, 55 %). Isto foi usado diretamente na reação seguinte. RMN = EN00228-27-01
[000345] 1H RMN (500 MHz, CDCl3) δ 4,23 (1H, dd), 4,42 (1H, dd), 4,61 (1H, dd), 6,54 (1H, dd), 7,14 (2H, d), 7,34 (2H, d), 7,60 (1H, d), 7,88 (1H, d) Intermediário 93: 4-(1 -(4-clorofenil)-2-(1 H-pirazol-1-il)etilcarbamoil)-1- (7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butil
[000346] HATU (0,209 g, 0,55 mmol) foi adicionado em uma porção ao ácido 4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)- piperidina-4-carboxílico (Intermediário 1) (0,181 g, 0,50 mmol), 1-(4- clorofenil)-2-(1H-pirazol-1-il)etanamina (Intermediário 92) (0,202 g, 0,5 mmol) e N,N-diisopropiletilamina (0,174 ml, 1,00 mmol) em N-metil-2- pirrolidinona (3,0 ml) na temperatura ambiente sob argônio. A solução resultante foi agitada durante a noite. A mistura de reação foi purificada pela HPLC preparativa usando uma coluna de fase reversa Waters X-Bridge (C-18, sílica de 5 mícrons, diâmetro 19 mm, comprimento de 100 mm, vazão de 40 ml / minutos) e misturas decrescentemente polares de água (contendo 0,2 % de carbonato de amônio) e acetonitrila como eluente. As frações contendo o composto desejado foram evaporadas até a secura para produzir 4-(1-(4- clorofenil)-2-(1H-pirazol-1-il)etilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin- 4-il)piperidin-4-ilcarbamato de terc-butila (0,216 g, 76 %) como um sólido cristalino branco. m/z (ESI+) (M + H)+ = 565. RMN = EN00228-28-01
[000347] 1H RMN (500 MHz, DMSO-d6) δ 1,40 (9H, s), 1,72 (1H, m), 1,87 (2H, m), 2,00 (1H, m), 3,53 (1H, m), 3,52 (1H, m), 3,94 (1H, m), 4,13 (1H, m), 4,38 (1H, dd), 4,74 (1H, dd), 5,30 (1H, dt), 6,14 (1H, dd), 6,57 (1H, d), 7,16 (1H, d), 7,25 (1H, s), 7,29 (4H, s), 7,43 (1H, d), 7,53 (1H, s), 8,12 (1H, s), 8,35 (1H, d), 11,67 (1H, s) Intermediário 94: (Z)-1-(4-Clorofenil)-2-(tiazol-2-il)etenamina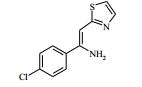
[000348] Uma solução 2,5 M de N-butilítio (8,00 ml, 20,00 mmol) em hexanos, foi adicionada às gotas ao 2-metiltiazol (1,983 g, 20 mmol) dissolvida em THF (35,00 ml) em um período de 5 minutos sob argônio a -70° C. A pasta fluida amarela clara resultante foi agitada a -70° C por 2,5 horas. 4-clorobenzonitrila (2,75 g, 20,00 mmol) em THF (35,00 ml) foi adicionada às gotas à suspensão, que foi agitada a -70° C por 90 minutos depois da fim da adição. A mistura de reação foi deixada aquecer até a temperatura ambiente sob agitação e escurecimento. O bruto foi concentrado até a secura e diluído com acetato de etila depois lavado com água, secado em sulfato de magnésio e concentrado. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica gel eluindo com 10 a 100 % de acetato de etila em diclorometano. O solvente foi evaporado até a secura para produzir (Z)-1-(4-clorofenil)-2-(tiazol-2-il)etenamina (1,466 g, 31,0 %) como um sólido amarelo claro. RMN & MS = EN00228-45-01 m/z (ESI+) (M + H)+ = 237
[000349] 1H RMN (500 MHz, CDCl3) δ 5,72 (1H, s), 6,54 (2H, m), 6,99 (1H, d), 7,38 (d, 2H), 7,55 (2H, d), 7,68 (1H, d) Intermediário 95: 1-(4-Clorofenil)-2-(tiazol-2-il)etanamina
[000350] Uma solução 2 N de ácido clorídrico em MeOH na temperatura ambiente, foi adicionadas às gotas à (Z)-1-(4-clorofenil)-2- (tiazol-2-il)etenamina (Intermediário 94) (1,45 g, 6,13 mmol) e traços de verde de bromocresol dissolvido em MeOH (45 ml) até que a solução virou de azul escuro para laranja. Cianoboroidreto de sódio (0,481 g, 7,66 mmol) foi adicionado em uma porção à solução agitada e a cor laranja foi mantida pela adição às gotas da solução 2 N de ácido clorídrico em metanol na temperatura ambiente. Depois de 1 hora de agitação, o solvente foi removido sob vácuo. o bruto foi absorvido em água, o pH foi ajustado em torno de 2 pela adição de umas poucas gotas da solução 2 N de ácido clorídrico em metanol. A fase aquosa foi lavada com éter dietílico duas vezes depois o pH foi ajustado a 14 pela adição de uma solução aquosa 6 N de hidróxido de sódio e a fase aquosa básica foi extraída com éter dietílico duas vezes. As fases orgânicas foram lavadas com salmoura secadas em sulfato de magnésio e concentradas para produzir 1-(4-clorofenil)-2- (tiazol-2-il)etanamina (1,310 g, 90 %) como um óleo incolor. RMN = EN00228-47-01
[000351] 1H RMN (500 MHz, DMSO-d6) δ 2,10 (2H, m), 3,22 (1H, dd), 3,26 (1H, dd), 4,22 (1H, t), 7,32 (2H, d), 7,37 (2H, d), 7,51 (1H, d), 7,66 (1H, d) Intermediário 96: Terc-butila 4-(1-(4-clorofenil)-2-(tiazol-2- il)etilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato
[000352] HATU (0,418 g, 1,10 mmol) foi adicionado em uma porção ao ácido 4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)- piperidina-4-carboxílico (Intermediário 1) (0,361 g, 1,0 mmol), 1-(4- clorofenil)-2-(tiazol-2-il)etanamina (Intermediário 95) (0,318 g, 1,00 mmol) e N,N-diisopropiletilamina (0,348 ml, 2,00 mmol) em N-metil-2- pirrolidinona (5,0 ml) na temperatura ambiente sob argônio. A solução resultante foi agitada durante a noite. Et2O foi adicionado ao precipitado o produto bruto, que foi filtrado, lavado abundantemente com Et2O e secado a vácuo para dar terc-butila 4-(1-(4-clorofenil)-2- (tiazol-2-il)etilcarbamoil)-1-(7H-pirrolo[2,3-d]-pirimidin-4-il)piperidin-4- ilcarbamato (0,550 g, 94 %). RMN & MS = EN00228-48-01
[000353] 1H RMN (500 MHz, DMSO-d6) δ 1,23 (9H, s), 1,74 (1H, m), 1,89 (2H, m), 2,00 (1H, m), 3,10 (2H, dd), 3,52 (1H, m), 3,58 (1H, m), 4,00 (1H, m), 4,19 (1H, m), 5,29 (1H, dt), 6,57 (1H, d), 7,17 (1H, d), 7,30 (2H, d), 7,37 (2H, d), 7,54 (1H), d), 7,69 (1H, d), 8,13 (1H, s), 8,33 (1H, d), 8,47 (1H, d), 11,68 (1H, s) m/z (ESI+) (M + H)+ = 582 Intermediário 97: 3-(4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]- pirimidin-4-il)piperidina-4-carboxamido)-3-(4-clorofenil)propanoato de lítio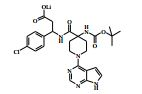
[000354] Hidróxido de lítio (1,436 ml, 2,87 mmol) 2 N em água foi adicionado a uma suspensão agitada de 3-(4-(terc-butoxicarbonilamino)- 1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamido)-3-(4- clorofenil)-propanoato de metila (Intermediário 35) (400 mg, 0,72 mmol) em THF (40 ml) na temperatura ambiente. A suspensão resultante rapidamente foi em solução e foi depois agitada na temperatura ambiente durante a noite. Na manhã seguinte um precipitado branco formou-se. Este foi filtrado e secado a vácuo para dar 3-(4-(terc- butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxamido)-3-(4-clorofenil)propanoato de lítio (396 mg, 100 %). RMN & MS = EN00228-39-01
[000355] 1H RMN (500 MHz, DMSO-d6) δ 1,41 (9H, s), 1,89 (2H, m), 2,06 (2H, m), 2,27 (2H, m), 3,30 (2H, m parcialmente escondido pela H2O), 4,33 (2H, m), 4,83 (1H, m), 6,57 (1H, s), 7,14 (1H, s), 7,22 (2H, d), 7,32 (2H, d), 7,58 (1H, m), 8,10 (1H, s), 10,02 (1H, s); m/z (ESI-) (M-Li)- = 541 Intermediário 98: 4-(1 -(4-clorofenil)-3-(dimetilamino)-3-oxopropil carbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila
[000356] HATU (152 mg, 0,40 mmol) foi adicionado em uma porção ao 3-(4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il) piperidina-4-carboxamido)-3-(4-clorofenil)propanoato de lítio (Intermediário 97) (200 mg, 0,36 mmol), cloridreto de dimetilamina (48,0 mg, 0,59 mmol) e N,N-diisopropiletilamina (0,190 ml, 1,09 mmol) em N- metil-2-pirrolidinona (3,0 ml) na temperatura ambiente sob argônio. A solução resultante foi agitada durante a noite. Éter dietílico adicionado para precipitar o produto bruto que formou como uma goma. Este foi enxaguado com Et2O e secado para dar a goma bruta (770 mg) que foi usado em reações subsequentes sem outra purificação. Intermediário 99: 1-(4-clorofenil)-3-metoxipropilcarbamato de terc- butila
[000357] Hidreto de sódio (35,0 mg, 0,87 mmol) foi adicionado ao 1- (4-clorofenil)-3-hidroxipropilcarbamato de terc-butila (Intermediário 30) (200 mg, 0,70 mmol) em THF (10 ml) a 0° C sob nitrogênio. A mistura foi agitada a 0° C por 15 minutos. Iodeto de metila (0,044 ml, 0,70 mmol) foi adicionado às gotas e a suspensão resultante foi agitada a 22° C por 4 horas. A reação foi extinta com solução KHSO4 (1 M, 0,5 ml) e água (15 ml). A mistura foi extraída com éter dietílico (3 x 20 ml); Os extratos combinados foram lavados com salmoura saturada (20 ml), secados em MgSO4, filtrados e evaporados para dar o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição 20 a 60 % de EtOAc em isoexano. As frações puras foram evaporadas até a secura para produzir 1-(4-clorofenil)-3-metoxipropilcarbamato de terc-butila (80 mg, 38,1 %) como um sólido branco.
[000358] 1H RMN (399,902 MHz, CDCl3) δ 1,40 (9H, s), 1,91 (1H, s), 2,01 (1H, s), 3,30 (3H, s), 3,32 (2H, m), 4,79 (1H, br.s), 5,45 (1H, br.s), 7,20 (2H, d), 7,29 (2H, d);
[000359] m/z (ESI+) (M + H)+ = 300, 302 (M + H+), 244,3, 246,3 (M + H+ -C4H8); HPLC tR = 2,58 min. Intermediário 100: 1-(4-clorofenil)-3-metoxipropan-1-amina
[000360] Cloreto de hidrogênio (4 M em 1,4-dioxano, 0,667 ml, 2,67 mmol) foi adicionado ao 1-(4-clorofenil)-3-metoxipropilcarbamato de terc-butila (Intermediário 99) (80 mg, 0,27 mmol) em uma mistura de DCM (5 ml) e metanol (2 ml) a 22° C. A solução resultante foi agitada a 22° C por 5 horas. A mistura foi concentrada e o resíduo foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 2 M e as frações puras foram evaporadas até a secura para produzir 1-(4- clorofenil)-3-metoxipropan-1-amina (47,0 mg, 88 %) como um óleo incolor.
[000361] 1H RMN (399,902 MHz, CDCl3) δ 1,80 - 1,96 (2H, m), 3,31 (3H, s), 3,32 (1H, m), 3,43 (1H, m), 4,09 (1H, t), 7,27 - 7,31 (4H, m);
[000362] m/z (ESI+) (M + H)+ = 200,2, 202,3 (M + H+); HPLC tR = 1,70 min. Intermediário 101: 4-(1 -(4-clorofenil)-3-metoxipropilcarbamoil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila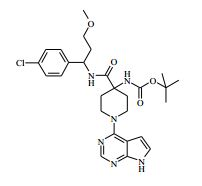
[000363] Hexafluorofosfato de O-(7-Azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (55,2 mg, 0,15 mmol) foi adicionado ao ácido 4-(terc- butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxílico (Intermediário 1) (50 mg, 0,14 mmol) e N- etildiisopropilamina (0,029 ml, 0,17 mmol) em NMP (5 ml). A solução resultante foi agitada a 50° C por 10 minutos depois esfriada até a temperatura ambiente. 1-(4-clorofenil)-3-metoxipropan-1-amina (Intermediário 100) (27,6 mg, 0,14 mmol) foi adicionado como uma solução em NMP (2 ml). A mistura resultante foi agitada a 22° C por 16 horas. A mistura de reação foi diluída com EtOAc (75 ml) e lavada sequencialmente com água (6 x 75 ml) e salmoura saturada (50 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição 1 a 4 % (MeOH 10:1 / NH3 conc. (aq)) em DCM. As frações puras foram evaporadas até a secura para produzir terc-butila 4-(1-(4- clorofenil)-3-metoxipropilcarbamoil)-1-(7H-pirrolo[2,3-d]-pirimidin-4- il)piperidin-4-ilcarbamato (52,0 mg, 69,2 %) como um sólido branco.
[000364] 1H RMN (399,902 MHz, DMSO) δ 1,42 (9H, s), 1,90 (2H, t), 1,92 - 2,05 (4H, m), 3,22 (3H, s), 3,29 (2H, t), 3,55 (2H, m), 4,24 (2H, m), 4,89 (1H, dt), 6,60 (1H, dd), 7,05 (1H, br.), 7,16 (1H, dd), 7,33 (4H, m), 7,99 (1H, d), 8,13 (1H, s), 11,65 (1H, s);
[000365] m/z (ESI+) (M + H)+ = 543,4, 545,3; HPLC tR = 2,31 min. Intermediário 102: S-3-(terc-butoxicarbonilamino)-3-(4-clorofenil)propila etanotioato
[000366] Tioacetato de potássio (43,9 mg, 0,38 mmol) foi adicionado ao metanossulfonato 3-(terc-butoxicarbonilamino)-3-(4- clorofenil)propila (Intermediário 31) (70 mg, 0,19 mmol) em DMF (5 ml) a 22° C. A solução resultante foi agitada a 50° C por 2 horas. A mistura de reação foi diluída com EtOAc (50 ml) e lavada sequencialmente com 20 % de salmoura saturada (4 x 50 ml) e salmoura saturada (25 ml). A camada orgânica foi secada em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, solvente de eluição 15 % de EtOAc em isoexano. As frações puras foram evaporadas até a secura para produzir o etanotioato de S-3-(terc- butoxicarbonilamino)-3-(4-cloro-fenil)propila (61,0 mg, 92 %) como um sólido branco.
[000367] 1H RMN (399,902 MHz, CDCl3) δ 1,41 (9H, s), 1,98 (2H, dt), 2,32 (3H, s), 2,83 (2H, m), 4,67 (1H, br.s), 4,88 (1H, br.s), 7,20 (2H, d), 7,30 (2H, d);
[000368] m/z (ESI+) (M + H+ -C4H8) = 288,3, 290,3; HPLC tR = 2,80 min. Intermediário 103: 1-(4-clorofenil)-3-sulfamoilpropilcarbamato de terc- butila
[000369] N-Clorossuccinimida (90 mg, 0,67 mmol) foi adicionada a uma mistura de acetonitrila (3 ml) e ácido clorídrico 2 M (0,1 ml) esfriado a 10° C. Etanotioato de S-3-(terc-butoxicarbonilamino)-3-(4- clorofenil)propila (Intermediário 102) (58 mg, 0,17 mmol) foi adicionado às gotas como uma solução em acetonitrila (2 ml) e a solução resultante foi agitada a 22° C por 1 hora. A solução de amônia aquoso concentrada (3 ml, 63,00 mmol) foi adicionada às gotas e a mistura agitada por 3 dias. A mistura foi evaporada e o resíduo foi dividido entre DCM (30 ml) e água. A camada aquosa foi extraída com DCM (30 ml) e os extratos combinados orgânicos separação de fase e cromatografia por vaporização instantânea em eluição de 30 a 80 % de EtOAc em isoexano. As frações contendo o produto foram evaporadas até a secura para produzir 1-(4-clorofenil)- 3-sulfamoilpropilcarbamato de terc-butila (40,0 mg, 68,0 %) como um sólido branco.
[000370] 1H RMN (399,902 MHz, CDCl3) δ 1,42 (9H, s), 2,75 - 2,91 (2H, m), 3,11 - 3,17 (2H, m), 4,74 (1H, br.s), 4,93 (1H, t), 7,23 (2H, d), 7,34 (2H, d);
[000371] m/z (ESI+) (M + H+ -C4H8) = 293,2, 295,2; HPLC tR = 2,11 min. Intermediário 104: 3-amino-3-(4-clorofenil)propano-1-sulfonamida
[000372] Cloreto de hidrogênio (4 M em dioxano, 1,0 ml, 4,00 mmol) foi adicionado ao 1-(4-clorofenil)-3-sulfamoilpropilcarbamato de terc- butila (Intermediário 103) (38 mg, 0,11 mmol) em uma mistura de DCM (5 ml) e metanol (2 ml) a 22° C. A solução resultante foi agitada a 22° C por 24 horas. A mistura foi evaporada até a secura e o resíduo foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando 30 % (NH3 2 M em MeOH) em DCM e as frações puras foram evaporadas até a secura para produzir 3-amino-3-(4-clorofenil)propano-1-sulfonamida (20,00 mg, 73,8 %) como uma goma incolor.
[000373] m/z (ESI+) (M + H)+ = 249,2, 251,2; HPLC tR = 1,32 min. Intermediário 105 4-(1-(4-clorofenil)-3-sulfamoilpropilcarbamoil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila :
:
[000374] Hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (30,9 mg, 0,08 mmol) foi adicionado ao ácido 4-(terc- butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxílico (Intermediário 1) (28 mg, 0,08 mmol) e N,N- diisopropiletilamina (0,016 ml, 0,09 mmol) em NMP (5 ml) a 22° C. A suspensão resultante foi agitada a 50° C por 10 minutos. A mistura foi esfriada até a temperatura ambiente e 3-amino-3-(4- clorofenil)propano-1-sulfonamida (Intermediário 104) (19,27 mg, 0,08 mmol) foi adicionado como uma solução em NMP (2 ml). A mistura foi agitada a 22° C por 3 dias. A mistura foi diluída com metanol e carregada em uma coluna SCX. O produto bruto foi purificado pela cromatografia de troca iônica; o produto desejado foi eluído da coluna usando NH3/MeOH 2 M e as frações puras foram evaporadas até a secura. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 2 a 6 % (MeOH 10:1 / NH3 conc. (aq)) em DCM. As frações puras foram evaporadas até a secura para produzir 4-(1-(4-clorofenil)-3- sulfamoilpropil-carbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4- ilcarbamato de terc-butila (16,00 mg, 34,9 %) como um sólido branco.
[000375] m/z (ESI+) (M + H)+ = 592,4, 594,3; HPLC tR = 1,99 min. Intermediário 106: 3-amino-1-(4-clorofenil)-3-oxopropilcarbamato de terc-butila
[000376] Hexafluoro-fosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (466 mg, 1,23 mmol) foi adicionado em uma porção ao ácido terc-butil-3-amino-3-(4-clorofenil)propiônico (245 mg, 0,82 mmol), cloreto de amônio (131 mg, 2,45 mmol) e N,N- diisopropiletilamina (0,811 ml, 4,90 mmol) em DMF (4 ml) a 20° C sob nitrogênio. A suspensão resultante foi agitada a 20° C por 18 horas. A mistura de reação foi evaporada até a secura e redissolvida em EtOAc (25 ml) e lavada sequencialmente com água (25 ml) e salmoura saturada (25 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir 3-amino-1-(4-clorofenil)-3-oxopropilcarbamato de terc-butila (200 mg, 82 %) como um sólido branco. m/z (ESI-) (M - H)- = 297; HPLC tR = 1,91 min. Intermediário 107: 3-amino-3-(4-clorofenil)propanamida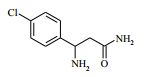
[000377] HCl (4 M) em 1,4 dioxano (1,674 ml, 6,69 mmol) foi adicionado ao 3-amino-1-(4-clorofenil)-3-oxopropilcarbamato de terc- butila (Intermediário 106) (200 mg, 0,67 mmol) em DCM (20 ml) a 20° C. A solução resultante foi agitada a 20° C por 4 horas. A mistura de reação foi evaporada e foi purificada pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 7 M e as frações puras foram evaporadas até a secura para produzir 3-amino-3-(4-clorofenil)propanamida (46,0 mg, 34,6 %) como um sólido branco.
[000378] 1H RMN (399,9 MHz, DMSO-d6) δ 0,99 (2H, t), 2,33 - 2,37 (2H, m), 3,38 (2H, s), 4,24 (1H, t), 7,35 - 7,42 (4H, m),
[000379] m/z (ESI+) (M + H)+ = 199; HPLC tR = 1,14 min. Intermediário 108: 4-(3-amino-1-(4-clorofenil)-3-oxopropilcarbamoil)-1- (7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila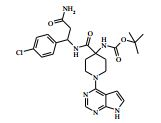
[000380] Hexafluoro-fosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (129 mg, 0,34 mmol) foi adicionado em uma porção ao ácido 4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxílico (Intermediário 1) (82 mg, 0,23 mmol) e N,N- diisopropiletilamina (0,112 ml, 0,68 mmol) em DMF (4 ml) a 20° C sob nitrogênio. A solução resultante foi agitada a 20° C por 5 minutos. 3- amino-3-(4-clorofenil)-propanamida (Intermediário 107) (45 mg, 0,23 mmol) foi depois adicionada à reação e agitada por 3 horas. A mistura de reação foi concentrada e diluída com EtOAc (20 ml) e lavada com água (20 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir 4-(3-amino-1-(4-clorofenil)-3- oxopropilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)-piperidin-4- ilcarbamato de terc-butila (99 mg, 100 %).
[000381] m/z (ESI+) (M + H)+ = 542; HPLC tR = 1,88 min. Intermediário 109: N-[1 -(4-clorofenil)-3-(fenoxicarbonilamino)-propil]- carbamato de terc-butila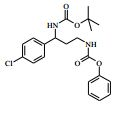
[000382] Cloroformiato de fenila (0,132 ml, 1,05 mmol) foi adicionado às gotas ao 3-amino-1-(4-clorofenil)propilcarbamato de terc-butila (Intermediário 25) (300 mg, 1,05 mmol) e bicarbonato de sódio (133 mg, 1,58 mmol) em dioxano (10 ml) a 20° C sob nitrogênio. A solução resultante foi agitada a 20° C por 2 horas. A mistura de reação foi concentrada e diluída com EtOAc (20 ml) e lavada com água (20 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir N-[1-(4-clorofenil)-3-(fenóxi-carbonilamino)-propil]carbamato de terc-butila (427 mg, 100 %) como uma goma.
[000383] m/z (ESI+) (M + H)+ = 405; HPLC tR = 2,85 min. Intermediário 110: 1-(4-clorofenil)-3-ureidopropilcarbamato de terc- butyl
[000384] Cloreto de amônio (113 mg, 2,11 mmol) foi adicionado em uma porção ao N-[1-(4-clorofenil)-3- (fenoxicarbonilamino)propil]carbamato de terc-butila (Intermediário 109) (427 mg, 1,05 mmol) e trietilamina (0,882 ml, 6,33 mmol) em DMF (3 ml). A solução resultante foi agitada a 0,352 molar por 24 horas. A mistura de reação foi concentrada e diluída com EtOAc (20 ml) e lavada com água (20 ml). A camada orgânica foi secada em MgSO4, filtrada e evaporada para produzir 1-(4-clorofenil)-3-ureidopropilcarbamato de terc-butila (346 mg, 100 %), m/z (ESI+) (M + H)+ = 328; HPLC tR = 1,91 min. Intermediário 111: 1-(3-amino-3-(4-clorofenil)propil)uréia
[000385] 1-(4-clorofenil)-3-ureidopropilcarbamato de terc-butila (Intermediário 110) (346 mg, 1,06 mmol) foi dissolvido em TFA (3 ml) e agitado a 20° C por 2 horas. A reação foi submetida a vácuo até a secura e foi purificada pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 0,35 M e as frações puras foram evaporadas até a secura para produzir 1-(3-amino-3-(4-clorofenil)propil)uréia (151 mg, 62,8 %) como uma goma amarela.
[000386] 1H RMN (399,9 MHz, DMSO-d6) δ 1,61 (2H, q), 2,98 (2H, m), 3,82 (1H, t), 4,05 (2H, s), 5,38 (2H, s), 5,94 (1H, s), 7,37 (4H, m),
[000387] m/z (ESI+) (M + H)+ = 228; HPLC tR = 1,25 min. Intermediário 112: 4-(1-(4-clorofenil)-3-ureidopropilcarbamoil)-1-(7H- pirrolo[2,3-d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila
[000388] Hexafluoro-fosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (378 mg, 0,99 mmol) foi adicionado em uma porção ao ácido 4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxílico (Intermediário 1) (240 mg, 0,66 mmol) e N,N- diisopropiletilamina (0,329 ml, 1,99 mmol) em NMP (2 ml) a 20° C sob nitrogênio. A solução resultante foi agitada a 20° C por 5 minutos. 1-(3- amino-3-(4-clorofenil)-propil)uréia (Intermediário 111) (151 mg, 0,66 mmol) em NMP (2 ml) foram depois adicionados à reação e agitados por 18 horas. A mistura de reação foi diluída com EtOAc (20 ml) e lavada com água (20 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir 4-(1-(4-clorofenil)-3- ureidopropilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4- ilcarbamato de terc-butila (379 mg, 100 %) como um sólido branco. m/z (ESI+) (M + H)+ = 571; HPLC tR = 1,88 min. Intermediário 113: 3-amino-3-(4-clorofenil)propanonitrila
[000389] HCl (4M) em 1,4-dioxano (0,668 ml, 2,67 mmol) foi adicionado em uma porção ao 1-(4-clorofenil)-2-cianoetilcarbamato de terc-butila (Intermediário 24) (150 mg, 0,53 mmol) em DCM (4 ml) a 20° C. A solução resultante foi agitada a 20° C por 18 horas. A mistura de reação foi evaporada para produzir 3-amino-3-(4-clorofenil)propanonitrila (sal de HCl) (115 mg, 99 %) como um sólido branco. Intermediário 114: (4-clorofenil)-2-cianoetilcarbamoil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidin-4-ilcarbamato
[000390] Hexafluoro-fosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (306 mg, 0,81 mmol) foi adicionado em uma porção ao ácido 4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxílico (194 mg, 0,54 mmol) e N,N- diisopropiletilamina (0,533 ml, 3,22 mmol) em NMP (3 ml) a 20° C sob nitrogênio. A solução resultante foi agitada a 20° C por 5 minutos. 3- amino-3-(4-clorofenil)-propanonitrila (Intermediário 113) (97 mg, 0,54 mmol) em NMP (3 ml) foi depois adicionada à reação e agitada por 1 hora. A mistura de reação foi concentrada e diluída com EtOAc (50 ml) e lavada com água (50 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir (4-clorofenil)-2- cianoetilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4- ilcarbamato (0,281 g, 100 %). m/z (ESI+) (M + H)+ = 524; HPLC tR = 2,19 min. Intermediário 115: 1-(4-clorofenil)-3-(metilsulfonamido)propilcarbamato de terc-butila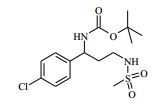
[000391] Cloreto de metanossulfonila (0,082 ml, 1,05 mmol) foi adicionado às gotas ao 3-amino-1-(4-clorofenil)propilcarbamato de terc-butila (Intermediário 25) (300 mg, 1,05 mmol) e N,N- diisopropiletilamina (0,367 ml, 2,11 mmol) em DCM (4 ml) a 20° C. A solução resultante foi agitada a 20° C por 18 horas. A mistura de reação foi concentrada e diluída com Et2O (25 ml) e lavada com água (25 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir o produto bruto. O produto bruto foi purificado pela cromatografia por vaporização instantânea em sílica, gradiente de eluição de 0 a 20 % de EtOAc em DCM. As frações puras foram evaporadas até a secura para produzir 1-(4-clorofenil)-3- (metilsulfonamido) propilcarbamato de terc-butila (275 mg, 71,9 %) como um sólido branco.
[000392] 1H RMN (399,9 MHz, DMSO-d6) δ 1,37 (9H, s), 1,76 (1H, m), 1,82 - 1,88 (1H, m), 2,87 (3H, s), 2,89 - 2,91 (2H, m), 4,58 (1H, d), 7,00 (1H, t), 7,32 (2H, d), 7,39 (2H, d), 7,48 (1H, d), m/z (ESI+) (M + H)+ = 361; HPLC tR = 2,25 min. Intermediário 116: N-(3-amino-3-(4-clorofenil)propil)metanossulfonamida
[000393] TFA (4 ml) foi adicionado ao 1-(4-clorofenil)-3-(metil- sulfonamido)propilcarbamato de terc-butila (Intermediário 115) (275 mg, 0,76 mmol) e agitado a 20° C por 2 horas. A reação foi submetida a vácuo até a secura. O produto bruto foi purificado pela cromatografia de troca iônica, usando uma coluna SCX. O produto desejado foi eluído da coluna usando NH3/MeOH 7 M e as frações puras foram evaporadas até a secura para produzir N-(3-amino-3-(4- clorofenil)propil)metanossulfonamida (113 mg, 56,7 %) como uma goma incolor.
[000394] 1H RMN (399,9 MHz, DMSO-d6) δ 1,69 - 1,72 (2H, m), 2,87 (3H, s), 2,94 - 2,98 (2H, m), 3,18 - 3,19 (1H, m), 3,87 (1H, t), 7,35 - 7,40 (4H, m), m/z (ESI+) (M + H)+ = 262; HPLC tR = 1,43 min. Intermediário 117:4-(1-(4-clorofenil)-3- (metilsulfonamido)propilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidin-4-ilcarbamato de terc-butila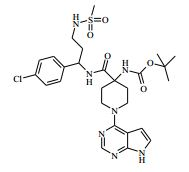
[000395] Hexafluoro-fosfato de O-(7-azabenzotriazol-1-il)-N,N,N’,N’- tetrametilurônio (245 mg, 0,65 mmol) foi adicionado em uma porção ao ácido 4-(terc-butoxicarbonilamino)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxílico (Intermediário 1) (155 mg, 0,43 mmol) e N,N- diisopropiletilamina (0,213 ml, 1,29 mmol) em NMP (2 ml) a 20° C sob nitrogênio. A solução resultante foi agitada a 20° C por 5 minutos. N- (3-amino-3-(4-clorofenil)-propil)metanossulfonamida (Intermediário 116) (113 mg, 0,43 mmol) em NMP (2 ml) foi depois adicionada à reação e agitada por 18 horas. A mistura de reação foi concentrada e diluída com EtOAc (20 ml) e lavada com água (20 ml). A camada orgânica foi secadas em MgSO4, filtrada e evaporada para produzir 4- (1-(4-clorofenil)-3-(metilsulfonamido) propilcarbamoil)-1-(7H-pirrolo[2,3- d]pirimidin-4-il)piperidin-4-ilcarbamato de terc-butila (261 mg, 100 %).
[000396] m/z (ESI+) (M + H)+ = 606; HPLC tR = 2,09 min.
[000397] Os efeitos biológicos dos compostos da Fórmula (I) podem ser testados como apresentados abaixo. Ensaio de Fosforilação de GSK-3 de adenocarcinoma de mama humano MDA-MB-468 In Vitro
[000398] Este ensaio determina a capacidade dos compostos de teste para inibir a fosforilação do resíduo de Serina-9 em Glicogênio Sintase Quinase-3beta (GSK-3β) como um marcador substituto da atividade de PKB (Akt) celular, como avaliado usando a tecnologia Acumen Explorer Fluorescent Plate-Reader. Uma linhagem de célula de adenocarcinoma mamário humano MDA-MB-468 (LGC Promochem, Teddington, Middlesex, UK, Catálogo No HTB-132) foi rotineiramente mantido em meio de cultivo de Eagle modificado por Dulbecco (DMEM; Invitrogen Limited, Paisley, UK Catálogo No 11966025) contendo 10 % de soro de bezerro fetal inativado por calor (FCS; Sigma, Poole, Dorset, UK, Catálogo No F0392) e 1 % de L-glutamina (Gibco, Catálogo No 25030-024) a 37° C com 5 % de CO2 até uma confluência de 70 a 90 %.
[000399] Para o ensaio de fosforilação, as células foram descoladas do frasco de cultura usando Tripsina-EDTA (Invitrogen Limited, Catálogo No 25300-062) e semeado nos reservatórios de uma placa de 96 reservatórios de Poliestireno Corning Costar preta de fundo transparente (Fisher Scientific UK, Loughborough, Leicestershire, UK; Catálogo No 3904 e DPS-130-020K) a uma densidade de 5000 células por reservatório em 100 μl de meio de cultivo completo. As células foram incubadas durante a noite a 37° C com 5 % de CO2 para permitir que elas adiram.
[000400] No dia 2, as células foram tratadas com compostos de teste e incubadas por 2 horas a 37° C com 5 % de CO2. Os compostos de teste foram preparados como soluções de estoque de 10 mM em DMSO e dosados diretamente até a concentração requerida dentro dos reservatórios de teste usando a tecnologia de dosagem ECHO de não contato (dispensa acústica de gotículas de 2,5 nl múltiplas diretamente nos reservatórios de ensaio) (Labcyte Inc. Sunnyvale, Califórnia, USA). Cada placa conteve reservatórios de controle sem composto de teste.
[000401] 20 μl de tampão de fixação (Solução Salina Tamponada com Fosfato (PBS) contendo 10 % de formaldeído; Sigma; Catálogo No F1635) foram depois adicionados a cada reservatórios para dar uma concentração de reservatório final de 1,6 %. As placas foram depois incubadas por 30 minutos na temperatura ambiente antes do fixativo ser removido. Cada reservatório foi lavado uma vez com 250 μl de PBS e depois 50 μl de PBS adicionados a cada reservatório. PBS foi depois aspirado e as células permeabilizadas e bloqueadas pela incubação de cada reservatório com 50 μl de tampão de permeabilização/bloqueio (PBS contendo 0,5 % de Tween 20 (Sigma; Catálogo No P5927) e 5 % de Leite em Pó Marvel (Andrews Pharmacy Ltd, Macclesfield, Cheshire, UK; Catálogo No APC100199)) por 1 hora na temperatura ambiente antes do tingimento.
[000402] A seguir da remoção de tampão Perm/Block, 50 μl de anticorpo anti-fosfo-GSK-3β primário (Cell Signalling Technology (New England Biolabs (Uk) Ltd.), Hitchin, Hertfordshire, UK; Catálogo No 9336 diluído 1:400 em tampão de bloqueio (PBS contendo 5 % de Marvel e 0,05 % de Tween/Polissorbato 20) foram adicionados a cada reservatório e incubados durante a noite a 4° C.
[000403] Cada reservatório foi lavado três vezes em 250 μl de tampão de lavagem (PBS contendo 0,05 % de polissorbato 20), e células incubadas por 1 hora na temperatura ambiente com 50 μl de anticorpo secundário fluorescentemente rotulado anti-coelho Alexa Fluor 488 (Molecular Probes, Invitrogen Limited, Catálogo No A11008) diluídos 1:750 e tampão de bloqueio. As placas foram lavadas três vezes em 250 μl de tampão de lavagem e armazenada contendo 50 μl de PBS a 4° C até requerido.
[000404] As placas foram analisadas usando uma leitora de placa Acumen Explorer para quantificar o nível de sinal fluorescente que representa quantidade de GSK-3β fosforilado. Os compostos ativos causaram uma diminuição na fosforilação fosfo-GSK-3β em relação ao controle máximo (não dosado) para cada ensaio, que é medido pelo número de objetos fosforilados por reservatório, e permitiu que a potência dos inibidores de PKB (Akt) fosse determinada.
[000405] Cálculo de IC50 - IC50 é a concentração de composto requerido para dar 50 % de efeito na faixa de atividade afetada pelo composto, entre os dados de controle de resposta máxima (nenhum composto) e mínima (nível em excesso de composto). Os valores de IC50 foram determinados ajustando-se o fundo corrigido, os dados de ensaio de resposta de dose para um modelo de equação de ajuste de curva logística de 4 parâmetros com a resposta mínima ajustada a zero. Isto foi feito usando um algoritmo desenvolvido em casa dentro do Origin graphing software package (OriginLab Corporation, Northampton, MA, USA).
[000406] Os exemplos da invenção foram testados no ensaio acima e seus valores IC50 médios calculados. Estes valores são mostrados na Tabela G. Ensaio de AKT1 quinase in vitro
[000407] Este ensaio detecta a atividade de inibidores de AKT1 (PKBα) quinase usando Caliper LabChip LC3000. O ensaio de mudança de mobilidade na incubação fora do chip Caliper usa um chip microfluídico para medir a conversão de um peptídeo de rótulo fluorescente a um produto fosforilado por uma respectiva quinase. A reação enzimática completa é realizada em placas de microtítulo e depois extinta. As soluções paradas resultantes são ‘tragadas’ em série através de um capilar sobre o chip, onde o substrato de peptídeo e o produto fosforilado são separados pela eletroforese. Estes são depois detectados por intermédio da fluorescência induzida por laser. O substrato e o produto são separados em dois picos pela aplicação de um campo elétrico alto e diretamente detectado usando fluorescência. A assinatura do sinal fluorescente revela o grau da reação.
[000408] Para cada dosagem Echo o solvente foi 100 % de DMSO. Uma placa mestra foi preparada com 40 μl do estoque de 10 mM de nosso Depósito de Líquido Primário no quadrante 1 de uma placa Labcyte de 384 reservatórios. Uma diluição 1 em 100 foi feita a partir do quadrante 1 para o quadrante 2 pela remoção de 0,4 μl e adicionando-o a 39,6 μl de DMSO. As diluições 1 em 100 subsequentes foram feitas no quadrante 3 a partir do quadrante 2 e quadrante 4 a partir do quadrante 3.
[000409] Gotículas de 2,5 nl múltiplas foram dispensadas de cada quadrante da placa mestra usando a tecnologia de dosagem ECHO (Labcyte Inc. Sunnyvale, Califórnia, USA) para gerar a faixa de dose que foi requerida no teste. A faixa de dose mais habitualmente usada foi como segue: 100 uM, 30 uM, 10 uM, 3 uM, 1 uM, 0,3 uM, 0,1 uM, 0,03 uM, 0,01 uM, 0,003 uM, 0,001 uM, 0,0001 uM. Cada reservatório foi retroenchido com Sulfóxido de Dimetila (DMSO) a um volume total de 120 nl, tal que quando a mistura de enzima e substrato foi adicionada a concentração de DMSO final foi de 1 %. DMSO foi adicionado aos reservatórios de controle max como 120 nl, os reservatórios de controle mínimo foram tratados com 120 nl de composto a uma concentração que inibiu a atividade de enzima em 100 %.
[000410] A seguir da adição de composto ou controle para a placa de ensaio, mistura de peptídeo de 6 μl contendo 3 μM de substrato (5- FAM-GRPRTSSFAEG-CONH2; CRB) e 40 μM de ATP em tampão base de Quinase (100 mM de Hepes pH 7,5, 0,015 % Brij-35) e 6 μl de mistura de enzima contendo 8 nM de enzima ativa de AKT1/PKBα (Upstate Biotechnology, Cat No 14 - 276), 8 mM de DTT e 20 mM de MgCl2 em tampão base de quinase foram adicionados. Todos os tampões foram fabricados com água a 18 MQ. As placas foram seladas e incubadas na temperatura ambiente por 50 minutos. A reação foi interrompida pela adição de 10 μl de tampão de parada (100 mM de Hepes pH 7,5, 0,015 % de solução de Brij-35, 0,1 % de reagente de revestimento #3, 40 mM de EDTA, 5 % de DMSO) a cada reservatório (N.B. as placas podem ser congeladas depois de interrompidas e lidas posteriormente). As placas foram depois analisadas usando o Caliper LabChip LC3000 Drug Discovery System (Caliper Life Sciences, 1 Wellfield, Preston Brook, Runcorn, WA7 3AZ) usando as seguintes condições de separação; -1,8 PSI, -500 de voltagem a montante, -1700 de voltagem a jusante, tempo de trago de amostra de 0,2 s, tempo de trago após a amostra de 30 s e uma demora final de 120 s. A integração do substrato e picos de produto foi realizada usando o software Caliper LabChip e curvas de IC50 foram calculadas usando o software Origin® (OriginLab Corporation, Northampton, MA, USA). Os exemplos da invenção foram testados no ensaio de enzima AKT1 in vitro e os valores IC50 médios obtidos são apresentados na Tabela G. Tabela G

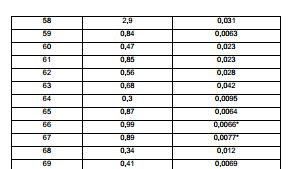 * = testado apenas uma vez Análise de hERG Cultura de célula
* = testado apenas uma vez Análise de hERG Cultura de célula
[000411] As células K1 do ovário do hamster chinês (CHO) que expressam hERG descritas por (Persson, Carlsson, Duker, & Jacobson, 2005) foram cultivadas até a semi-confluência a 37° C em um ambiente umidificado (5 % de CO2) em meio F-12 de Ham contendo L-glutamina, 10 % de soro de bezerro fetal (FCS) e 0,6 mg/ml de higromicina (todos da Sigma-Aldrich). Antes do uso, a monocamada foi lavada usando uma alíquota de 3 ml pré-aquecida (37° C) de Versene 1:5.000 (Invitrogen). Depois da aspiração desta solução o frasco foi incubado a 37° C em um incubador com mais 2 ml de Versene 1:5.000 por um período de 6 minutos. As células foram depois descoladas do fundo do frasco batendo-se suavemente e 10 ml de Solução Salina Tamponada com Fosfato de Dulbecco contendo cálcio (0,9 mM) e magnésio (0,5 mM) (PBS; Invitrogen) foram depois adicionados ao frasco e aspirados em um tubo de centrífuga de 15 ml antes da centrifugação (50 g, por 4 mins). O sobrenadante resultante foi descartado e a pelota suavemente re-colocada em suspensão em 3 ml de PBS. Uma alíquota de 0,5 ml da suspensão de célula foi removida e o número de células viáveis (com base na exclusão do azul de tripano) foi determinado em uma leitora automatizada (Cedex; Innovatis) de modo que o volume de recolocação em suspensão das células pôde ser ajustado com PBS para dar a concentração de célula final desejada. É a concentração de célula neste ponto do ensaio que é quotada quando da referência a este parâmetro. As células CHO- Kv1.5, que foram usadas para ajustar a compensação da voltagem no IonWorks® HT, foram mantidas e preparadas para o uso do mesmo modo. Eletrofisiologia
[000412] Os princípios e operação deste dispositivo foram descritos por (Schroeder, Neagle, Trezise, & Worley, 2003). Em resumo, a tecnologia está fundamentada em uma placa de 384 reservatórios (PatchPlate®) em que um registro é tentado em cada reservatório usando-se sucção para posicionar e manter uma célula em um furo pequeno separando duas câmaras fluídicas isoladas. Uma vez que a selagem ocorra, a solução no lado de baixo da PatchPlate® é trocada para uma contendo anfotericina B. Isto permeabiliza o embutimento de membrana celular que cobre o furo em cada reservatório e, em funcionamento, permite que um registro de clampe de voltagem perfurado, de célula inteira seja feito.
[000413] Um teste β IonWorks® HT da Essen Instrument foi usado. Não há capacidade para aquecer soluções neste dispositivo por este motivo o mesmo foi operado na temperatura ambiente (~21° C), como segue. O reservatório na posição “Tampão” foi carregado com 4 ml de PBS e este na posição “Células” com a suspensão de célula CHO- hERG descrita acima. Uma placa de 96 reservatórios (fundo em V, Greiner Bio-one) contendo os compostos a serem testados (em 3 vezes acima da sua concentração de teste final) foi colocada na posição “Placa 1” e um PatchPlate® foi clampeado na estação PatchPlate®. Cada placa de composto foi depositada em 12 colunas para permitir que dez curvas de concentração-efeito de 8 pontos fossem construídas; as duas colunas remanescentes na placa foram tomadas com veículo (concentração final 0,33 % de DMSO), para definir a linha de base do ensaio, e uma concentração de bloqueio supra-máxima de cisaprida (concentração final de 10 μM) para definir o nível de inibição de 100 %. A ponta fluídica (F-Head) do IonWorks® HT depois adicionou 3,5 μl de PBS a cada reservatório do PatchPlate® e seu lado de baixo foi perfusado com solução “interna” que teve a seguinte composição (em mM): K-Gliconato 100, KCl 40, MgCl2 3,2, EGTA 3 e HEPES 5 (todos da Sigma-Aldrich; pH 7,25 a 7,30 usando 10 M de KOH). Depois da imprimação e desfazimento das bolhas, a ponta eletrônica (E-head) depois movida em torno do PatchPlate® para realizar um teste de furo (isto é aplicar um pulso de voltagem para determinar se o furo em cada reservatório foi aberto). A F-head depois dispensou 3,5 μl da suspensão de célula descrita acima em cada reservatório do PatchPlate® e às células foram dados 200 segundos para atingir e selar o furo em cada reservatório. A seguir disto, a Ehead movida em torno do PatchPlate® para determinar a resistência do selo obtido em cada reservatório. Em seguida, a solução no lado de baixo da PatchPlate® foi trocada para “acessar” a solução que teve a seguinte composição (em mM): KCl 140, EGTA 1, MgCl2 1 e HEPES 20 (pH 7,25 a 7,30 usando 10 M de KOH) mais 100 μg/ ml de anfotericina B (Sigma-Aldrich). Depois de deixar 9 minutos para que a perfuração do emplastro ocorresse, a E-head movida em torno da PatchPlate® de 48 reservatórios em uma vez para obter medições de corrente hERG do pré-composto. A F-head depois adicionou 3,5 μl de solução de cada reservatório da placa de composto a 4 reservatórios na PatchPlate® (a concentração de DMSO final foi de 0,33 % em cada reservatório). Isto foi obtido movendo-se do reservatório mais diluído para o mais concentrado da placa de composto para minimizar o impacto de qualquer arraste de composto. Depois de aproximadamente 3,5 min de incubação, a E-head depois moveu em torno de todos os 384 reservatórios da PatchPlate® para obter medições de corrente hERG pós-composto. Deste modo, curvas de concentração-efeito não acumulativas puderam ser produzidas onde, com a condição de que os critérios de aceitação fossem obtidos em uma percentagem suficiente de reservatórios (ver abaixo), o efeito de cada concentração de composto de teste foi fundamentado no registro entre 1 e 4 células.
[000414] A corrente hERG no pré- e pós-composto foi evocada por um pulso de voltagem único consistindo de uma contenção de período de 20 s a -70 mV, uma etapa de 160 ms a -60 mV (para obter uma estimativa de vazamento), uma etapa de 100 ms de volta para -70 mV, uma etapa de 1 s a + 40 mV, uma etapa de 2 s a -30 mV e finalmente uma etapa de 500 ms a -70 mV. Entre os pulsos de voltagem pré- e pós-composto não houve nenhum clampeamento do potencial de membrana. As correntes foram subtraídas do vazamento com base na estimativa de corrente evocada durante a etapa de +10 mV no início do protocolo de pulso de voltagem. Quaisquer compensações de voltagem em IonWorks® HT foram ajustadas em um de dois modos. Quando da determinação da potência do composto, uma rampa de voltagem despolarizante foi aplicada às células CHO-Kv1.5 e a voltagem mencionada na qual houve um ponto de inflexão no traço de corrente (isto é o ponto no qual a ativação de canal foi observada com um protocolo de rampa). A voltagem na qual isto ocorreu foi previamente determinada usando o mesmo comando de voltagem na eletrofisiologia convencional e verificado ser -15 mV (dados não mostrados); assim um potencial de compensação pôde ser introduzido no software IonWorks® HT usando este valor como um ponto de referência. Quando da determinação das propriedades eletrofisiológicas básicas de hERG, qualquer compensação foi ajustada determinando-se o potencial reverso de corrente de cauda de hERG no IonWorks® HT, comparando o com aquele encontrado na eletrofisiologia convencional (-82 mV) e depois fazendo o ajuste de compensação necessário no software IonWorks® HT. O sinal de corrente foi amostrado a 2,5 kHz.
[000415] A magnitude de corrente hERG pré- e pós-varredura foi medida automaticamente dos traços subtraídos do vazamento pelo software IonWorks® HT tomando-se uma média de 40 ms da corrente durante o período de contenção inicial a -70 mV (corrente de referência) e subtraindo esta do pico da resposta de corrente de cauda. Os critérios de aceitação para as correntes evocadas em cada reservatório foram: resistência de selo pré-varredura >60 MQ, amplitude de corrente de cauda de hERG na pré-varredura >150 pA; resistência de selo pós-varredura >60 MQ. O grau de inibição da corrente hERG foi avaliado dividindo-se a corrente de hERG pós- varredura pela respectiva corrente hERG na pré-varredura para cada reservatório. Referências
[000416] Persson, F., Carlsson, L., Duker, G., & Jacobson, I. (2005). Blocking characteristics of hERG, hNav1.5, and hKvLQT1/hminK after administration of the novel anti-arrhythmic compound AZD7009. J Cardiovasc. Electrophysiol., 16, 329-341.
[000417] Schroeder, K., Neagle, B., Trezise, D. J., & Worley, J. (2003). Ionworks HT: a new high-throughput electrophysiology measurement platform. J Biomol Screen., 8, 50-64. Resultados da Análise de hERG
[000418] Exemplo 9, (S)-4-amino-N-(1-(4-clorofenil)-3-hidroxipropil)- 1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida, foi testada até 100 μM de acordo com o procedimento descrito acima e um valor IC50 de hERG médio de 177 μM foi derivado pela extrapolação da curva. Experimentos In Vivo Análise farmacodinâmica de proteínas de substrato PKB GSK3β e PRAS40 em resposta à (S)-4-amino-N-(1-(4-clorofenil)-3-hidróxi- propil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida
[000419] 2,5 x 106 células U87- MG (ATCC número HTB-14®) + 50 % de matrigel foram injetados s.c. (subcutaneamente) no flanco de camundongos nus. Quando os tumores atingiram um volume de cerca de 0,5 cm3 uma dose aguda de 150 ou 300 mg/kg de (S)-4-amino-N- (1-(4-clorofenil)-3-hidroxipropil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)- piperidina-4-carboxamida (E9) foi dada p.o. (gavagem oral). Os animais foram eutanizados no ponto de tempo especificado e os tumores dissecados e subitamente congelados em nitrogênio líquido.
[000420] Lisados de tumor ex-vivo foram preparados em tampão de Lise a 10 % de triton-X-100 Tris contendo inibidores de protease e fosfatase usando a metodologia ‘FastPrep 24’ (MP Biomedicals matrix #6910-500). As concentrações de proteína foram estimadas contra uma curva padrão de BSA usando o kit Pierce BCA (#23225). pGSK3β foi medido pelo western blotting e pPRAS40 pelo ELISA intercalado de fase sólida (Biosource KHO0421).
[000421] Para western blotting, quantidades equivalentes de proteína foram resolvidas por gradiente de 4 a 12 % de géis de Bis-Tris poliacrilamida pré-moldados (Invitrogen NP0323), transferidos para membranas de nitrocelulose Hybond C Extra (Invitrogen LC2001) e incubados com anti-soro primário (pGSK3β ser9, Cell Signaling Technology #9336; GSK3β total, BD Transduction Labs #610202) e subsequentemente com IgG anti-coelho conjugado com rábano peroxidase (Cell Signaling Technology #7074) ou IgG anti- camundongo (Cell Signalling Technology #7076). As proteínas imunorreativas foram detectadas pela quimioluminescência realçada (#34076 Pierce Supersignal Dura) e as faixas quantificadas com um ChemiGenius II (Syngene). Os valores plotados mostra a inibição percentual de pGSK3β comparada com os controles de veículo, a seguir da normalização para pGSK3β total.
[000422] Para o ELISA (Ensaio Imunossorvente Ligado à Enzima), quantidades equivalentes de proteína foram adicionados a uma placa de 96 reservatórios pré-revestidos com um anticorpo monoclonal específico de pPRAS40 de ‘captura’. Um anticorpo de ‘detecção’ específico para pPRAS40(Thr 246) é depois adicionado seguido por um anticorpo IgG anti-coelho rotulado com HRP. O ensaio é depois quantificado usando TMB (tetrametilbenzidina) estabilizada a 450 nm em uma leitora de placa. A intensidade de cor é proporcional à concentração de pPRAS40 (Thr 246). Os resultados são mostrados na Figura 1. Estudo anti-tumor MCF-7 usando (S)-4-amino-N-(1-(4-clorofenil)-3- hidroxipropil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida (E9)
[000423] Camundongos Imuno-Deficientes Combinados Graves (SCID) foram obtidos da Charles River Laboratories. Os camundongos foram alojados e mantidos em condições isentas de patógeno específicas. Para o implante in vivo, as células foram colhidas de frascos de cultura de tecido T225 por um tratamento de 2 a 5 minutos com 3 x tripsina (Invitrogen) em solução de EDTA seguido por suspensão em meio básico e três lavagens em solução salina tamponada com fosfato (Invitrogen). Apenas as suspensões de célula única de mais do que 90 % de viabilidade, como determinado pela exclusão de azul de tripano, foram usados para injeção. As células de tumor mamário MCF-7 (ATCC número HTB-22®) (5 x 106 células + 50 % de Matrigel®) foram injetadas subcutaneamente no flanco esquerdo dos animais em um volume de 0,1 ml. Antes da implantação das células MCF-7, os camundongos SCID foram anestesiados e implantados com uma pelota de estrogênio de 0,5 mg/21 dias de duração. Quando o tamanho de tumor médio atingiu ~ 0,3 cm3, os camundongos foram randomizados em grupos de controle e tratamento. O grupo de tratamento recebeu 300 mg/kg de E9 solubilizado em um veículo consistindo de 10 % (v/v) de DMSO, 25 % (p/v) de Kleptose® em água, pela gavagem oral. O grupo de controle recebeu apenas o veículo, uma vez ao dia pela gavagem oral. Os volumes de tumor (medidos pelo calibrador) foram registrados em intervalos para a duração do estudo. Os camundongos foram sacrificados pela eutanásia com CO2. O volume do tumor foi calculado (tomando o comprimento como sendo o diâmetro maior através do tumor e a largura como sendo o diâmetro perpendicular correspondente usando a fórmula: (comprimento x largura) x ^(comprimento x largura) x (π/6). A inibição do crescimento a partir do início do tratamento foi avaliado pela comparação das diferenças no volume de tumor entre grupos de controle e tratados. Porque a variação nos dados de volume de tumor médio aumenta proporcionalmente com o volume (e é portanto desproporcionado entre grupos), os dados foram transformados em log para remover qualquer dependência de tamanho antes da avaliação estatística. A significância estatística foi avaliada usando um teste t mono-caudado, de duas amostras. Os resultados são mostrados na Figura 2. Prognóstico de dose humana
[000424] O prognóstico de dose humana para estudos clínicos requer uma estimativa de parâmetros de farmacocinética humana (PK) importantes para a definição do T1/2 de eliminação e a forma e magnitude do perfil concentração plasmática vs tempo em uma dose particular. Estes parâmetros estimados incluem Depuração, Volume de Distribuição em Estado Constante (Vss), constante de taxa de absorção (Ka), biodisponibilidade (F) e frequência de dosagem. Os prognósticos de dose humana também requerem alguma evidência farmacológica como a exposição se relaciona com a eficácia (McGinnity, Collington, Austin & Riley, Current Drug Metabolism 2007 8 463-479).
[000425] Para a (S)-4-amino-N-(1-(4-clorofenil)-3-hidroxipropil)-1- (7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4-carboxamida (E9), a depuração humana foi estimada a partir dos dados da depuração intrínseca (Clint) determinados em hepatócitos humanos corrigidos para a depuração in vivo pela incorporação do modelo bem agitado (Riley, McGinnity & Austin, Drug Metabolism & Disposition 2005 33(9) 1304-1311). Diferenças menores foram observadas na ligação da proteína plasmática do composto através do rato, cão e ser humano; consequentemente, os valores de Vss de rato e cão observados foram corrigidos pelo fator Fu, ser humano/Fu (rato ou cão) (onde Fu = fração não ligada no plasma). A Vss humana foi estimada ser de 3,3 L/kg usando este método. A fração absorvida (Fabs) em seres humanos foi tomada como a média através do rato e cão. Este parâmetro Fabs foi depois ajustado para a biodisponibilidade (F) pela correção quanto a extração hepática. A taxa de absorção (Ka) foi ajustada para garantir que a Tmax fosse 1 h para E9, um valor relativamente conservativo dado que pré-clinicamente observado, e deve igualmente resultar em um valor Cmax conservativo para o uso no cálculo de margens com respeito aos estudos de segurança. A frequência de dosagem clínica desejada foi assumida ser duas vezes ao dia (BID).
[000426] Tendo definido a forma e a magnitude da curva concentração plasmática humana vs tempo, a etapa final foi ajustar a dose de modo que as concentrações plasmáticas fossem atingidas que são prováveis de alcançar eficácia. Em estudos PD de camundongo, a inibição de pGSK (um ponto final substituto para a atividade anti-tumor) foi observada quando concentrações plasmáticas livres excederam 1 vez IC50 para cada composto como medido no ensaio de célula da atividade inibidora de AKT, corrigida quanto a ligação devida ao Soro de Bezerro Fetal presente no ensaio. Consequentemente, o modelo PK humano assumiu que a exposição livre ao E9 deve exceder a PKB IC50 livre para garantir a eficácia clínica.
[000427] Colocando todos estes dados juntos, a (S)-4-amino-N-(1-(4- clorofenil)-3-hidroxipropil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxamida (E9) é prognosticada requerer uma dose humana de cerca de 7 mg/kg BID para alcançar a eficácia, com uma meia vida estimada ser em torno de 6 horas. Lista das Figuras
[000428] Figura 1: Níveis de fosforilação de GSK3β e PRAS40 em amostras tiradas de tumores U87- MG cultivados em camundongos nus a seguir de uma dose aguda de 150 ou 300 mg/kg de (S)-4-amino- N-(1-(4-clorofenil)-3-hidroxipropil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)- piperidina-4-carboxamida (E9), n = 5 por ponto de tempo.
[000429] Figura 2: Resultados do estudo antitumor de MCF-7 em camundongos SCID usando uma dose de uma vez ao dia (QD) de 300 mg/kg de (S)-4-amino-N-(1-(4-clorofenil)-3-hidroxipropil)-1-(7H-pirrolo[2,3- d]-pirimidin-4-il)piperidina-4-carboxamida (E9).
Claims (19)
1. Composto, caracterizado pelo fato de ser (S)-4-amino-N- (1-(4-clorofenil)-3-hidroxipropil)-1-(7H-pirrolo[2,3-d]pirimidin-4- il)piperidina-4-carboxamida: ou um sal farmaceuticamente aceitável do mesmo.
ou um sal farmaceuticamente aceitável do mesmo.
2. Composição farmacêutica, caracterizada pelo fato de que compreende um composto como definido na reivindicação 1 ou um sal farmaceuticamente aceitável do mesmo, junto com um diluente ou carreador farmaceuticamente aceitável.
3. Composto de acordo com a reivindicação 1, ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que é para o uso como um medicamento.
4. Composto de acordo com a reivindicação 1 ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que é para o uso no tratamento de câncer.
5. Composto de acordo com a reivindicação 1 ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que o câncer é selecionado dentre câncer de mama, linfoma não- Hodgkin, câncer pancreático, câncer hepatocelular, câncer gástrico, câncer de próstata e câncer de pulmão.
6. Composto de acordo com a reivindicação 1 ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que é para o uso no tratamento de câncer de mama.
7. Uso de um composto como definido na reivindicação 1 ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que é para a preparação de um medicamento para o tratamento de câncer.
8. Uso de um composto como definido na reivindicação 1 ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que é para a preparação de um medicamento para o tratamento de câncer, em que o câncer é selecionado dentre câncer de mama, linfoma não-Hodgkin, câncer pancreático, câncer hepatocelular, câncer gástrico, câncer de próstata e câncer de pulmão
9. Uso de um composto como definido na reivindicação 1 ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que é para a preparação de um medicamento para o tratamento de câncer de mama.
10. Composto de acordo com a reivindicação 1, caracterizado pelo fato de que é (S)-4-amino-N-(1-(4-clorofenil)-3- hidroxipropil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidina-4- carboxamida.
11. Composto de acordo com a reivindicação 10 caracterizada pelo fato de que é para o uso no tratamento de câncer.
12. Composto de acordo com a reivindicação 10 caracterizada pelo fato de que é para o uso no tratamento de câncer, em que o câncer é selecionado dentre câncer de mama, linfoma não-Hodgkin, câncer pancreático, câncer hepatocelular, câncer gástrico, câncer de próstata e câncer de pulmão
13. Composto de acordo com a reivindicação 10 caracterizada pelo fato de que é para o uso no tratamento de câncer de mama.
14. Processo para a preparação de um composto de Formula(I) como definido na reivindicação 1 ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que compreende a reação de um ácido da Fórmula (II) com (S)-3- amino-3-(4-clorofenil)propan-1-ol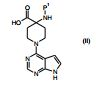 : em que P1 representa um grupo de proteção adequado; e depois disso, se necessário: (i) remover os grupos de proteção; e/ou (ii) formar um sal farmaceuticamente aceitável do mesmo.
: em que P1 representa um grupo de proteção adequado; e depois disso, se necessário: (i) remover os grupos de proteção; e/ou (ii) formar um sal farmaceuticamente aceitável do mesmo.
15. Processo de acordo com a reivindicação 14, caracterizado pelo fato de que P1 representa um grupo de proteção terc- butoxicarbonila.
16. Composto, caracterizado pelo fato de ser 4-(1-(4- clorofenil)-3-hidroxipropilcarbamoil)-1-(7H-pirrolo[2,3-d]pirimidin-4-il) piperidin-4-ilcarbamato de (S)-terc-butila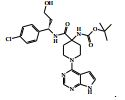 :
:
17. Processo para a preparação de um composto como definido na reivindicação 1 ou um sal farmaceuticamente aceitável do mesmo, caracterizado pelo fato de que compreende a reação de (S)-4- amino-N-(1-(4-clorofenil)-3-hidroxipropil)piperidina-4-carboxamida com heterociclo bicíclico de Fórmula (V)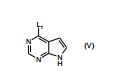 : em que L1 representa um grupo de proteção adequado; e depois disso, se necessário, formar um sal farmaceuticamente aceitável do mesmo.
: em que L1 representa um grupo de proteção adequado; e depois disso, se necessário, formar um sal farmaceuticamente aceitável do mesmo.
18. Processo de acordo com a reivindicação 17, caracte rizado pelo fato de que LI é cloro.
19. Composto, caracterizado pelo fato de ser (S)-4-amino- N-(1-(4-clorofenil)-3-hidroxipropil)piperidina-4-carboxamida
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US97919207P | 2007-10-11 | 2007-10-11 | |
| US60/979,192 | 2007-10-11 | ||
| US4786208P | 2008-04-25 | 2008-04-25 | |
| US61/047,862 | 2008-04-25 | ||
| PCT/GB2008/050925 WO2009047563A1 (en) | 2007-10-11 | 2008-10-09 | Pyrrolo [2, 3 -d] pyrimidin derivatives as protein kinase b inhibitors |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| BRPI0818533A2 BRPI0818533A2 (pt) | 2017-06-06 |
| BRPI0818533B1 true BRPI0818533B1 (pt) | 2020-12-01 |
| BRPI0818533B8 BRPI0818533B8 (pt) | 2021-05-25 |
Family
ID=40251789
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0818533A BRPI0818533B8 (pt) | 2007-10-11 | 2008-10-09 | composto, composição farmacêutica, uso de um composto, e, processo para a preparação de um composto |
Country Status (38)
| Country | Link |
|---|---|
| US (8) | US8101623B2 (pt) |
| EP (1) | EP2201012B1 (pt) |
| JP (2) | JP4705695B2 (pt) |
| KR (1) | KR101494734B1 (pt) |
| CN (1) | CN101861321B (pt) |
| AR (1) | AR068846A1 (pt) |
| AU (1) | AU2008309383B2 (pt) |
| BR (1) | BRPI0818533B8 (pt) |
| CA (1) | CA2701057C (pt) |
| CL (1) | CL2008003023A1 (pt) |
| CO (1) | CO6270328A2 (pt) |
| CR (1) | CR11359A (pt) |
| CU (1) | CU23886B1 (pt) |
| CY (1) | CY1116929T1 (pt) |
| DK (1) | DK2201012T3 (pt) |
| DO (1) | DOP2010000103A (pt) |
| EA (1) | EA018512B1 (pt) |
| ES (1) | ES2522365T3 (pt) |
| GT (1) | GT201000082A (pt) |
| HK (1) | HK1143154A1 (pt) |
| HN (1) | HN2010000653A (pt) |
| HR (1) | HRP20140807T1 (pt) |
| IL (1) | IL204721A (pt) |
| ME (1) | ME01999B (pt) |
| MX (1) | MX2010003927A (pt) |
| MY (1) | MY150059A (pt) |
| NI (1) | NI201000050A (pt) |
| NZ (1) | NZ585261A (pt) |
| PE (2) | PE20130152A1 (pt) |
| PL (1) | PL2201012T3 (pt) |
| PT (1) | PT2201012E (pt) |
| RS (1) | RS53552B1 (pt) |
| SA (1) | SA08290625B1 (pt) |
| SI (1) | SI2201012T1 (pt) |
| TW (1) | TWI453021B (pt) |
| UY (1) | UY31384A1 (pt) |
| WO (1) | WO2009047563A1 (pt) |
| ZA (1) | ZA201002318B (pt) |
Families Citing this family (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MY179032A (en) | 2004-10-25 | 2020-10-26 | Cancer Research Tech Ltd | Ortho-condensed pyridine and pyrimidine derivatives (e.g.purines) as protein kinase inhibitors |
| WO2007125321A2 (en) | 2006-04-25 | 2007-11-08 | Astex Therapeutics Limited | Purine and deazapurine derivatives as pharmaceutical compounds |
| EP2057187B1 (en) | 2006-08-10 | 2016-12-28 | Oncotherapy Science, Inc. | Genes and polypeptides relating to breast cancers |
| RS53552B1 (en) | 2007-10-11 | 2015-02-27 | Astrazeneca Ab | DERIVATI PIROLO [2,3-D] PIRIMIDINA KAO INHIBITORI PROTEIN KINAZE B |
| WO2011002772A1 (en) * | 2009-06-29 | 2011-01-06 | Oncotherapy Science, Inc. | Imidazopyridine derivatives and pbk inhibitors containing the same |
| GB201020161D0 (en) | 2010-11-26 | 2011-01-12 | Almac Discovery Ltd | Pharmaceutical compounds |
| WO2012080735A1 (en) | 2010-12-16 | 2012-06-21 | Convergence Pharmaceuticals Limited | Ask1 inhibiting pyrrolopyrimidine derivatives |
| SG10201600077RA (en) | 2011-01-11 | 2016-02-26 | Glaxosmithkline Llc | Combination |
| AU2012235902B2 (en) * | 2011-04-01 | 2015-08-27 | Astrazeneca Ab | Therapeutic treatment |
| JP6309454B2 (ja) | 2011-11-30 | 2018-04-11 | アストラゼネカ アクチボラグ | 癌の併用処置 |
| AU2013204533B2 (en) * | 2012-04-17 | 2017-02-02 | Astrazeneca Ab | Crystalline forms |
| RU2644769C2 (ru) | 2013-01-23 | 2018-02-14 | Астразенека Аб | Химические соединения |
| EP2815749A1 (en) | 2013-06-20 | 2014-12-24 | IP Gesellschaft für Management mbH | Solid form of 4-amino-2-(2,6-dioxopiperidine-3-yl)isoindoline-1,3-dione having specified X-ray diffraction pattern |
| CA2938626A1 (en) | 2013-07-26 | 2015-01-29 | John Rothman | Compositions to improve the therapeutic benefit of bisantrene |
| JP6295578B2 (ja) * | 2013-09-30 | 2018-03-20 | 凸版印刷株式会社 | 反応容器、核酸解析装置、および核酸解析方法 |
| RU2016116915A (ru) * | 2013-10-01 | 2017-11-13 | Новартис Аг | Комбинация |
| EP3392244A1 (en) | 2014-02-13 | 2018-10-24 | Incyte Corporation | Cyclopropylamines as lsd1 inhibitors |
| AU2015217073B2 (en) | 2014-02-13 | 2019-08-22 | Incyte Holdings Corporation | Cyclopropylamines as LSD1 inhibitors |
| WO2015123437A1 (en) | 2014-02-13 | 2015-08-20 | Incyte Corporation | Cyclopropylamines as lsd1 inhibitors |
| EP3105226B1 (en) | 2014-02-13 | 2019-09-04 | Incyte Corporation | Cyclopropylamines as lsd1 inhibitors |
| ES2691223T3 (es) * | 2014-05-28 | 2018-11-26 | Astrazeneca Ab | Proceso para la preparación de AZD5363 e intermedio novedoso utilizado en el mismo |
| US9695167B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted triazolo[1,5-a]pyridines and triazolo[1,5-a]pyrazines as LSD1 inhibitors |
| TWI687419B (zh) | 2014-07-10 | 2020-03-11 | 美商英塞特公司 | 作為lsd1抑制劑之咪唑并吡啶及咪唑并吡嗪 |
| US9695180B2 (en) | 2014-07-10 | 2017-07-04 | Incyte Corporation | Substituted imidazo[1,2-a]pyrazines as LSD1 inhibitors |
| US9758523B2 (en) | 2014-07-10 | 2017-09-12 | Incyte Corporation | Triazolopyridines and triazolopyrazines as LSD1 inhibitors |
| EA201792205A1 (ru) | 2015-04-03 | 2018-02-28 | Инсайт Корпорейшн | Гетероциклические соединения как ингибиторы lsd1 |
| WO2017027678A1 (en) * | 2015-08-12 | 2017-02-16 | Incyte Corporation | Salts of an lsd1 inhibitor |
| TW201726140A (zh) | 2015-09-17 | 2017-08-01 | 瑞典商阿斯特捷利康公司 | 治療癌症之新型生物標記及方法 |
| BR112018071585B1 (pt) | 2016-04-22 | 2024-01-02 | Incyte Corporation | Formulações de um inibidor de lsd1, seus usos e método de preparação das mesmas |
| WO2020047198A1 (en) | 2018-08-31 | 2020-03-05 | Incyte Corporation | Salts of an lsd1 inhibitor and processes for preparing the same |
| CN114728003A (zh) | 2019-11-04 | 2022-07-08 | 阿斯利康(瑞典)有限公司 | 治疗b细胞恶性肿瘤的阿卡替尼和卡帕塞替尼的治疗组合 |
| EP4271678A1 (en) * | 2021-01-04 | 2023-11-08 | Teva Pharmaceuticals International GmbH | Solid state forms of capivasertib and process for preparation thereof |
| WO2023187037A1 (en) | 2022-03-31 | 2023-10-05 | Astrazeneca Ab | Epidermal growth factor receptor (egfr) tyrosine kinase inhibitors in combination with an akt inhibitor for the treatment of cancer |
| CN114601836B (zh) * | 2022-04-12 | 2022-12-06 | 四川大学华西医院 | 一种akt抑制剂在制备治疗乳腺癌的药物中的应用 |
| WO2024083716A1 (en) | 2022-10-17 | 2024-04-25 | Astrazeneca Ab | Combinations of a serd for the treatment of cancer |
| WO2024100236A1 (en) | 2022-11-11 | 2024-05-16 | Astrazeneca Ab | Combination therapies for the treatment of cancer |
| WO2024104561A1 (en) | 2022-11-15 | 2024-05-23 | Astrazeneca Ab | Therapeutic combinations of capivasertib and venetoclax |
Family Cites Families (70)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE637271A (pt) | 1963-04-04 | 1900-01-01 | ||
| GB9312853D0 (en) | 1993-06-22 | 1993-08-04 | Euro Celtique Sa | Chemical compounds |
| GB9624482D0 (en) | 1995-12-18 | 1997-01-15 | Zeneca Phaema S A | Chemical compounds |
| KR19990082463A (ko) | 1996-02-13 | 1999-11-25 | 돈 리사 로얄 | 혈관 내피 성장 인자 억제제로서의 퀴나졸린유도체 |
| CN1116286C (zh) | 1996-03-05 | 2003-07-30 | 曾尼卡有限公司 | 4-苯胺基喹唑啉衍生物 |
| CA2249601A1 (en) | 1996-04-03 | 1997-10-23 | Thorsten E. Fisher | Inhibitors of farnesyl-protein transferase |
| GB9718972D0 (en) | 1996-09-25 | 1997-11-12 | Zeneca Ltd | Chemical compounds |
| WO1998035985A1 (en) | 1997-02-12 | 1998-08-20 | The Regents Of The University Of Michigan | Protein markers for lung cancer and use thereof |
| US6432947B1 (en) | 1997-02-19 | 2002-08-13 | Berlex Laboratories, Inc. | N-heterocyclic derivatives as NOS inhibitors |
| GB9714249D0 (en) | 1997-07-08 | 1997-09-10 | Angiogene Pharm Ltd | Vascular damaging agents |
| DE69830409T2 (de) | 1997-08-05 | 2006-01-26 | Pfizer Products Inc., Groton | 4-aminopyrrole[3,2-d]pyrimidine als neuropeptid rezeptor antagonisten |
| US6162804A (en) | 1997-09-26 | 2000-12-19 | Merck & Co., Inc. | Tyrosine kinase inhibitors |
| BR9910864A (pt) | 1998-06-04 | 2002-02-05 | Abbott Lab | Compostos anti-inflamatórios para inibição de aderência celular |
| PA8474101A1 (es) | 1998-06-19 | 2000-09-29 | Pfizer Prod Inc | Compuestos de pirrolo [2,3-d] pirimidina |
| US6262066B1 (en) | 1998-07-27 | 2001-07-17 | Schering Corporation | High affinity ligands for nociceptin receptor ORL-1 |
| GB9900334D0 (en) | 1999-01-07 | 1999-02-24 | Angiogene Pharm Ltd | Tricylic vascular damaging agents |
| GB9900752D0 (en) | 1999-01-15 | 1999-03-03 | Angiogene Pharm Ltd | Benzimidazole vascular damaging agents |
| KR100838617B1 (ko) | 1999-02-10 | 2008-06-16 | 아스트라제네카 아베 | 혈관형성 억제제로서의 퀴나졸린 유도체 |
| EP1181296A1 (en) | 1999-06-03 | 2002-02-27 | Abbott Laboratories | Cell adhesion-inhibiting antiinflammatory compounds |
| PT1244647E (pt) | 1999-11-05 | 2006-10-31 | Astrazeneca Ab | Derivados de quinazolina como inibidores de vegf |
| US7160890B2 (en) | 1999-12-02 | 2007-01-09 | Osi Pharmaceuticals, Inc. | Compounds specific to adenosine A3 receptor and uses thereof |
| ME00415B (me) | 2000-02-15 | 2011-10-10 | Pharmacia & Upjohn Co Llc | Pirol supstituisani 2-indol protein kinazni inhibitori |
| IL152682A0 (en) | 2000-05-31 | 2003-06-24 | Astrazeneca Ab | Indole derivatives with vascular damaging activity |
| UA73993C2 (uk) | 2000-06-06 | 2005-10-17 | Астразенека Аб | Хіназолінові похідні для лікування пухлин та фармацевтична композиція |
| PT1294724E (pt) | 2000-06-26 | 2006-07-31 | Pfizer Prod Inc | Compostos pirrolo (2,3-d ) pirimidina como agentes imunosupressivos |
| PL359181A1 (en) | 2000-07-07 | 2004-08-23 | Angiogene Pharmaceuticals Limited | Colchinol derivatives as angiogenesis inhibitors |
| CN1255391C (zh) | 2000-07-07 | 2006-05-10 | 安吉奥金尼药品有限公司 | 作为血管破坏剂的colchinol衍生物 |
| BR0113585A (pt) | 2000-08-31 | 2003-07-29 | Hoffmann La Roche | Derivados quinazolina como antagonistas adrenérgicos alfa-1 |
| US6680324B2 (en) | 2000-12-01 | 2004-01-20 | Osi Pharmaceuticals, Inc. | Compounds specific to adenosine A1 receptors and uses thereof |
| PL363245A1 (en) | 2000-12-01 | 2004-11-15 | Osi Pharmaceuticals, Inc. | Compounds specific to adenosine a1 |
| US6673802B2 (en) | 2000-12-01 | 2004-01-06 | Osi Pharmaceuticals, Inc. | Compounds specific to adenosine A3 receptor and uses thereof |
| CA2445568A1 (en) * | 2001-04-27 | 2002-11-07 | Vertex Pharmaceuticals Incorporated | Triazole-derived kinase inhibitors and uses thereof |
| DK1474425T3 (da) | 2002-01-07 | 2006-09-25 | Eisai Co Ltd | Deazapuriner og anvendelser deraf |
| TW200403058A (en) | 2002-04-19 | 2004-03-01 | Bristol Myers Squibb Co | Heterocyclo inhibitors of potassium channel function |
| WO2004002531A1 (ja) | 2002-06-26 | 2004-01-08 | Ono Pharmaceutical Co., Ltd. | 血管の収縮または拡張による疾患治療剤 |
| US20040138238A1 (en) | 2002-08-08 | 2004-07-15 | Dhanoa Dale S. | Substituted aminopyrimidine compounds as neurokinin antagonists |
| US20030139427A1 (en) | 2002-08-23 | 2003-07-24 | Osi Pharmaceuticals Inc. | Bicyclic pyrimidinyl derivatives and methods of use thereof |
| US20050288503A1 (en) | 2002-09-06 | 2005-12-29 | Adams Jerry L | Novel compounds |
| AU2003290673B2 (en) | 2002-11-08 | 2011-01-06 | Massachusetts Institute Of Technology | Small technetium-99m and rhenium labeled agents and methods for imaging tissues, organs and tumors |
| NZ540495A (en) | 2002-12-04 | 2007-09-28 | Eisai R&D Man Co Ltd | 1,3-Dihydro-imidazole fused-ring compound |
| EP1444982A1 (de) | 2003-02-06 | 2004-08-11 | Merckle Gmbh | Verwendung von Purinderivaten als selektive Kinase-Inhibitoren |
| EP1601357A4 (en) | 2003-03-10 | 2007-10-03 | Schering Corp | HETEROCYCLIC KINASE INHIBITORS: METHOD OF USE AND SYNTHESIS |
| AU2004232392A1 (en) | 2003-04-21 | 2004-11-04 | Ustav Organicke Chemie A Biochemie Akademie Ved Ceske Republiky | (Purin-6-yl) amino acid and production method thereof |
| FR2856685B1 (fr) | 2003-06-25 | 2005-09-23 | Merck Sante Sas | Derives de thiazolylpiperidine, leurs procedes de preparation et les compositions pharmaceutiques qui les contiennent |
| WO2005020921A2 (en) | 2003-08-29 | 2005-03-10 | Exelixis, Inc. | C-kit modulators and methods of use |
| WO2005044181A2 (en) | 2003-09-09 | 2005-05-19 | Temple University-Of The Commonwealth System Of Higher Education | Protection of tissues and cells from cytotoxic effects of ionizing radiation by abl inhibitors |
| TW200526626A (en) | 2003-09-13 | 2005-08-16 | Astrazeneca Ab | Chemical compounds |
| NZ547327A (en) | 2003-11-21 | 2009-08-28 | Array Biopharma Inc | AKT protein kinase inhibitors |
| EP1698375B1 (en) | 2003-12-25 | 2014-04-02 | Ono Pharmaceutical Co., Ltd. | Azetidine ring compounds and drugs comprising the same |
| AU2005228899A1 (en) * | 2004-03-30 | 2005-10-13 | Novartis Vaccines And Diagnostics, Inc. | Substituted thiophene derivatives as anti-cancer agents |
| CA2563699C (en) * | 2004-04-23 | 2014-03-25 | Exelixis, Inc. | Kinase modulators and method of use |
| MY179032A (en) * | 2004-10-25 | 2020-10-26 | Cancer Research Tech Ltd | Ortho-condensed pyridine and pyrimidine derivatives (e.g.purines) as protein kinase inhibitors |
| UY29177A1 (es) * | 2004-10-25 | 2006-05-31 | Astex Therapeutics Ltd | Derivados sustituidos de purina, purinona y deazapurina, composiciones que los contienen métodos para su preparación y sus usos |
| JP5274842B2 (ja) * | 2004-12-28 | 2013-08-28 | エグゼリクシス, インコーポレイテッド | 免疫疾患、炎症疾患および増殖疾患の処置のためのセリン−スレオニンキナーゼモジュレーター(p70S6K、Akt−1およびAkt−2)としての[1H−ピペラゾ[3,4−d]ピリミジン−4−イル]−ピペラジンまたは[1H−ピペラゾ[3,4−d]ピリミジン−4−イル]−ピペラジン化合物 |
| FR2880540B1 (fr) | 2005-01-13 | 2008-07-11 | Aventis Pharma Sa | Utilisation de derives de la purine comme inhibiteurs de la proteine hsp90 |
| FR2880626B1 (fr) | 2005-01-13 | 2008-04-18 | Aventis Pharma Sa | Derives de la purine, compositions les contenant et utilisation |
| WO2006091450A1 (en) | 2005-02-18 | 2006-08-31 | Lexicon Genetics Incorporated | 4-piperidin-1-yl-7h-pyrrolo[2,3-d]pyrimidine compounds |
| US20060281768A1 (en) | 2005-06-10 | 2006-12-14 | Gaul Michael D | Thienopyrimidine and thienopyridine kinase modulators |
| JP5071374B2 (ja) | 2005-07-14 | 2012-11-14 | アステラス製薬株式会社 | ヘテロ環ヤヌスキナーゼ3阻害剤 |
| WO2007025090A2 (en) | 2005-08-25 | 2007-03-01 | Kalypsys, Inc. | Heterobicyclic and - tricyclic inhibitors of mapk/erk kinase |
| US20070208053A1 (en) | 2006-01-19 | 2007-09-06 | Arnold Lee D | Fused heterobicyclic kinase inhibitors |
| WO2007125315A2 (en) | 2006-04-25 | 2007-11-08 | Astex Therapeutics Limited | Pharmaceutical compounds |
| WO2007125321A2 (en) | 2006-04-25 | 2007-11-08 | Astex Therapeutics Limited | Purine and deazapurine derivatives as pharmaceutical compounds |
| WO2007125320A1 (en) | 2006-04-25 | 2007-11-08 | Astex Therapeutics Limited | Pharmaceutical compounds |
| US20090082370A1 (en) | 2006-04-25 | 2009-03-26 | Neil Thomas Thompson | Pharmaceutical Combinations of PK Inhibitors and Other Active Agents |
| EP2029592A1 (en) * | 2006-04-25 | 2009-03-04 | Astex Therapeutics Limited | Pharmaceutical compounds |
| AR064416A1 (es) | 2006-12-21 | 2009-04-01 | Cancer Rec Tech Ltd | Derivados de purina, piridina y pirimidina condensadas con heterociclos, moduladores de pka y/o pkb, composiciones farmaceuticas que los contienen, y usos para el tratamiento de enfermedades hiperproliferativas. |
| AR064415A1 (es) | 2006-12-21 | 2009-04-01 | Cancer Rec Tech Ltd | Derivados de pirrolo-piperidinas y purinas,composiciones farmaceuticas que los contienen y usos en trastornos y/o enfermedades mediadas por pka y pkb. |
| JP5406039B2 (ja) | 2006-12-21 | 2014-02-05 | バーテックス ファーマシューティカルズ インコーポレイテッド | タンパク質キナーゼ阻害剤として有用な5−シアノ−4−(ピロロ[2,3b]ピリジン−3−イル)−ピリミジン誘導体 |
| RS53552B1 (en) | 2007-10-11 | 2015-02-27 | Astrazeneca Ab | DERIVATI PIROLO [2,3-D] PIRIMIDINA KAO INHIBITORI PROTEIN KINAZE B |
-
2008
- 2008-10-09 RS RS20140457A patent/RS53552B1/en unknown
- 2008-10-09 WO PCT/GB2008/050925 patent/WO2009047563A1/en active Application Filing
- 2008-10-09 JP JP2010528485A patent/JP4705695B2/ja active Active
- 2008-10-09 PL PL08806741T patent/PL2201012T3/pl unknown
- 2008-10-09 TW TW097139021A patent/TWI453021B/zh active
- 2008-10-09 ES ES08806741.8T patent/ES2522365T3/es active Active
- 2008-10-09 MX MX2010003927A patent/MX2010003927A/es active IP Right Grant
- 2008-10-09 AU AU2008309383A patent/AU2008309383B2/en active Active
- 2008-10-09 PT PT88067418T patent/PT2201012E/pt unknown
- 2008-10-09 EP EP08806741.8A patent/EP2201012B1/en active Active
- 2008-10-09 ME MEP-2014-102A patent/ME01999B/me unknown
- 2008-10-09 DK DK08806741.8T patent/DK2201012T3/da active
- 2008-10-09 BR BRPI0818533A patent/BRPI0818533B8/pt active IP Right Grant
- 2008-10-09 CN CN200880116651XA patent/CN101861321B/zh active Active
- 2008-10-09 SI SI200831281T patent/SI2201012T1/sl unknown
- 2008-10-09 CA CA2701057A patent/CA2701057C/en active Active
- 2008-10-09 NZ NZ585261A patent/NZ585261A/en unknown
- 2008-10-09 KR KR1020107010107A patent/KR101494734B1/ko active Protection Beyond IP Right Term
- 2008-10-09 EA EA201000552A patent/EA018512B1/ru not_active IP Right Cessation
- 2008-10-09 MY MYPI2010001617A patent/MY150059A/en unknown
- 2008-10-09 UY UY31384A patent/UY31384A1/es not_active Application Discontinuation
- 2008-10-10 PE PE2012002041A patent/PE20130152A1/es not_active Application Discontinuation
- 2008-10-10 CL CL2008003023A patent/CL2008003023A1/es unknown
- 2008-10-10 AR ARP080104450A patent/AR068846A1/es active IP Right Grant
- 2008-10-10 US US12/249,477 patent/US8101623B2/en active Active
- 2008-10-10 PE PE2008001748A patent/PE20090964A1/es active IP Right Grant
- 2008-10-11 SA SA8290625A patent/SA08290625B1/ar unknown
-
2010
- 2010-03-25 IL IL204721A patent/IL204721A/en active IP Right Grant
- 2010-03-31 ZA ZA2010/02318A patent/ZA201002318B/en unknown
- 2010-04-09 NI NI201000050A patent/NI201000050A/es unknown
- 2010-04-09 HN HN2010000653A patent/HN2010000653A/es unknown
- 2010-04-09 GT GT201000082A patent/GT201000082A/es unknown
- 2010-04-09 CR CR11359A patent/CR11359A/es unknown
- 2010-04-09 CU CU20100062A patent/CU23886B1/es active IP Right Grant
- 2010-04-09 DO DO2010000103A patent/DOP2010000103A/es unknown
- 2010-05-06 CO CO10054272A patent/CO6270328A2/es active IP Right Grant
- 2010-10-12 HK HK10109649.4A patent/HK1143154A1/xx unknown
-
2011
- 2011-03-09 JP JP2011051117A patent/JP5330430B2/ja active Active
- 2011-12-13 US US13/324,191 patent/US20120190679A1/en not_active Abandoned
-
2014
- 2014-08-26 HR HRP20140807AT patent/HRP20140807T1/hr unknown
- 2014-08-28 CY CY20141100689T patent/CY1116929T1/el unknown
-
2015
- 2015-02-19 US US14/626,303 patent/US9492453B2/en active Active
-
2016
- 2016-11-15 US US15/351,481 patent/US10059714B2/en active Active
-
2018
- 2018-07-06 US US16/028,979 patent/US10654855B2/en active Active
-
2020
- 2020-04-07 US US16/841,820 patent/US11236095B2/en active Active
-
2021
- 2021-12-16 US US17/644,654 patent/US11760760B2/en active Active
-
2023
- 2023-08-04 US US18/365,286 patent/US20240109902A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11760760B2 (en) | Protein kinase B inhibitors | |
| CA2957046C (en) | Optionally fused heterocyclyl-substituted derivatives of pyrimidine useful for the treatment of inflammatory, metabolic, oncologic and autoimmune diseases | |
| KR20190129936A (ko) | Prmt5-매개성 질환의 치료 또는 예방에 유용한 화합물 | |
| CN101952286A (zh) | 用于治疗和雄激素受体有关的病症的双环衍生物 | |
| CN110621675A (zh) | 用于治疗增殖性疾病的三环化合物 | |
| US20240270753A1 (en) | Heterocyclic compound for inhibiting shp2 activity, preparation method therefor and use thereof | |
| AU2016214131B2 (en) | Autotaxin inhibitors | |
| AU2019296085A1 (en) | Heterocyclic compound as TRK inhibitor | |
| US20100292222A1 (en) | Chemical compounds 751 | |
| BR112018015501B1 (pt) | Derivados de pirazolo[1,5-a]pirazin-4-ila como inibidores de jak, seu uso e composição farmacêutica que os compreende |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] | ||
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B07E | Notification of approval relating to section 229 industrial property law [chapter 7.5 patent gazette] | ||
| B06U | Preliminary requirement: requests with searches performed by other patent offices: procedure suspended [chapter 6.21 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 10 (DEZ) ANOS CONTADOS A PARTIR DE 01/12/2020, OBSERVADAS AS CONDICOES LEGAIS. |
|
| B16C | Correction of notification of the grant [chapter 16.3 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 09/10/2008 OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF |