WO2022260168A1 - ヒドロキシチエノイミダゾール誘導体、ビニルスルフィド誘導体、n-ブチリデンスルフィド誘導体、及び飽和直鎖炭化水素置換チエノイミダゾール誘導体の製造方法 - Google Patents
ヒドロキシチエノイミダゾール誘導体、ビニルスルフィド誘導体、n-ブチリデンスルフィド誘導体、及び飽和直鎖炭化水素置換チエノイミダゾール誘導体の製造方法 Download PDFInfo
- Publication number
- WO2022260168A1 WO2022260168A1 PCT/JP2022/023481 JP2022023481W WO2022260168A1 WO 2022260168 A1 WO2022260168 A1 WO 2022260168A1 JP 2022023481 W JP2022023481 W JP 2022023481W WO 2022260168 A1 WO2022260168 A1 WO 2022260168A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- derivative
- represented
- hydroxythienoimidazole
- mol
- Prior art date
Links
- TWLVBDIMPJJOPK-UHFFFAOYSA-N 1,3-dihydrothieno[2,3-d]imidazol-2-one Chemical class S1C=CC2=C1N=C(O)N2 TWLVBDIMPJJOPK-UHFFFAOYSA-N 0.000 title claims abstract description 56
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 39
- 150000008127 vinyl sulfides Chemical class 0.000 title claims abstract description 22
- -1 n-butylidene sulfide derivative Chemical class 0.000 title claims abstract description 14
- JCZAVVUIFWZMQI-UHFFFAOYSA-N 1h-thieno[2,3-d]imidazole Chemical class N1C=NC2=C1C=CS2 JCZAVVUIFWZMQI-UHFFFAOYSA-N 0.000 title claims abstract description 13
- 229920006395 saturated elastomer Polymers 0.000 title claims abstract description 13
- 239000007818 Grignard reagent Substances 0.000 claims abstract description 58
- 150000004795 grignard reagents Chemical class 0.000 claims abstract description 58
- 150000001879 copper Chemical class 0.000 claims abstract description 46
- FAYOCELKCDKZCA-UHFFFAOYSA-N 5-hydroxy-2,4-dimethylthiophen-3-one Chemical class CC1SC(O)=C(C)C1=O FAYOCELKCDKZCA-UHFFFAOYSA-N 0.000 claims abstract description 36
- 125000000217 alkyl group Chemical group 0.000 claims description 25
- 125000003118 aryl group Chemical group 0.000 claims description 24
- 238000000034 method Methods 0.000 claims description 21
- 239000003054 catalyst Substances 0.000 claims description 20
- 125000001424 substituent group Chemical group 0.000 claims description 17
- 239000003153 chemical reaction reagent Substances 0.000 claims description 15
- 125000005843 halogen group Chemical group 0.000 claims description 14
- 238000002156 mixing Methods 0.000 claims description 14
- 239000010949 copper Substances 0.000 claims description 12
- 229910052739 hydrogen Inorganic materials 0.000 claims description 11
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 claims description 9
- 239000001257 hydrogen Substances 0.000 claims description 9
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 8
- 229910052802 copper Inorganic materials 0.000 claims description 7
- 125000003107 substituted aryl group Chemical group 0.000 claims description 7
- 125000000547 substituted alkyl group Chemical group 0.000 claims description 6
- 150000001875 compounds Chemical class 0.000 description 37
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 34
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 22
- 239000000203 mixture Substances 0.000 description 19
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 18
- 239000011777 magnesium Substances 0.000 description 18
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 17
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 16
- 229910052749 magnesium Inorganic materials 0.000 description 16
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 15
- 125000004432 carbon atom Chemical group C* 0.000 description 14
- 239000007810 chemical reaction solvent Substances 0.000 description 14
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 13
- 238000006243 chemical reaction Methods 0.000 description 12
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 12
- 239000000243 solution Substances 0.000 description 12
- 229960002685 biotin Drugs 0.000 description 11
- 235000020958 biotin Nutrition 0.000 description 11
- 239000011616 biotin Substances 0.000 description 11
- 239000000543 intermediate Substances 0.000 description 11
- 238000005481 NMR spectroscopy Methods 0.000 description 10
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 10
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 9
- 229910021591 Copper(I) chloride Inorganic materials 0.000 description 8
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical compound [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 description 8
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 7
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 7
- 239000012046 mixed solvent Substances 0.000 description 7
- 239000012044 organic layer Substances 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 238000010306 acid treatment Methods 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 229910052801 chlorine Inorganic materials 0.000 description 6
- 229910052736 halogen Inorganic materials 0.000 description 6
- 150000002367 halogens Chemical class 0.000 description 6
- 238000010438 heat treatment Methods 0.000 description 6
- 238000006460 hydrolysis reaction Methods 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 5
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 5
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 5
- 238000004809 thin layer chromatography Methods 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 4
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- VMQMZMRVKUZKQL-UHFFFAOYSA-N Cu+ Chemical compound [Cu+] VMQMZMRVKUZKQL-UHFFFAOYSA-N 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 4
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 239000007868 Raney catalyst Substances 0.000 description 4
- 229910000564 Raney nickel Inorganic materials 0.000 description 4
- 239000003377 acid catalyst Substances 0.000 description 4
- 239000012190 activator Substances 0.000 description 4
- 125000003545 alkoxy group Chemical group 0.000 description 4
- 230000003197 catalytic effect Effects 0.000 description 4
- 125000001309 chloro group Chemical group Cl* 0.000 description 4
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 4
- 229910052731 fluorine Inorganic materials 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 4
- 125000002950 monocyclic group Chemical group 0.000 description 4
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 4
- UIYCHXAGWOYNNA-UHFFFAOYSA-N vinyl sulfide Chemical class C=CSC=C UIYCHXAGWOYNNA-UHFFFAOYSA-N 0.000 description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 3
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- 238000007259 addition reaction Methods 0.000 description 3
- 239000012267 brine Substances 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 239000011593 sulfur Substances 0.000 description 3
- 125000004434 sulfur atom Chemical group 0.000 description 3
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- 235000011054 acetic acid Nutrition 0.000 description 2
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 150000001450 anions Chemical class 0.000 description 2
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 150000001615 biotins Chemical class 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- 229910017052 cobalt Inorganic materials 0.000 description 2
- 239000010941 cobalt Substances 0.000 description 2
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 2
- 150000004699 copper complex Chemical class 0.000 description 2
- ORTQZVOHEJQUHG-UHFFFAOYSA-L copper(II) chloride Chemical compound Cl[Cu]Cl ORTQZVOHEJQUHG-UHFFFAOYSA-L 0.000 description 2
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 2
- BERDEBHAJNAUOM-UHFFFAOYSA-N copper(i) oxide Chemical compound [Cu]O[Cu] BERDEBHAJNAUOM-UHFFFAOYSA-N 0.000 description 2
- OPQARKPSCNTWTJ-UHFFFAOYSA-L copper(ii) acetate Chemical compound [Cu+2].CC([O-])=O.CC([O-])=O OPQARKPSCNTWTJ-UHFFFAOYSA-L 0.000 description 2
- QTMDXZNDVAMKGV-UHFFFAOYSA-L copper(ii) bromide Chemical compound [Cu+2].[Br-].[Br-] QTMDXZNDVAMKGV-UHFFFAOYSA-L 0.000 description 2
- 230000018044 dehydration Effects 0.000 description 2
- 238000006297 dehydration reaction Methods 0.000 description 2
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- SKTCDJAMAYNROS-UHFFFAOYSA-N methoxycyclopentane Chemical compound COC1CCCC1 SKTCDJAMAYNROS-UHFFFAOYSA-N 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- FDPIMTJIUBPUKL-UHFFFAOYSA-N pentan-3-one Chemical compound CCC(=O)CC FDPIMTJIUBPUKL-UHFFFAOYSA-N 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 235000019260 propionic acid Nutrition 0.000 description 2
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 239000008096 xylene Substances 0.000 description 2
- SEQRDAAUNCRFIT-UHFFFAOYSA-N 1,1-dichlorobutane Chemical compound CCCC(Cl)Cl SEQRDAAUNCRFIT-UHFFFAOYSA-N 0.000 description 1
- SPEUIVXLLWOEMJ-UHFFFAOYSA-N 1,1-dimethoxyethane Chemical compound COC(C)OC SPEUIVXLLWOEMJ-UHFFFAOYSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- 238000004009 13C{1H}-NMR spectroscopy Methods 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- CPPQOSMKJDPRID-UHFFFAOYSA-M 2-carboxy-6-methylphenolate;copper(1+) Chemical compound [Cu+].CC1=CC=CC(C([O-])=O)=C1O CPPQOSMKJDPRID-UHFFFAOYSA-M 0.000 description 1
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 1
- 229910021590 Copper(II) bromide Inorganic materials 0.000 description 1
- 229910021592 Copper(II) chloride Inorganic materials 0.000 description 1
- JPVYNHNXODAKFH-UHFFFAOYSA-N Cu2+ Chemical compound [Cu+2] JPVYNHNXODAKFH-UHFFFAOYSA-N 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- DSVGQVZAZSZEEX-UHFFFAOYSA-N [C].[Pt] Chemical compound [C].[Pt] DSVGQVZAZSZEEX-UHFFFAOYSA-N 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- QWLDGFJOWIBTBQ-UHFFFAOYSA-M copper(1+);2,2-dimethylpropanoate Chemical compound [Cu+].CC(C)(C)C([O-])=O QWLDGFJOWIBTBQ-UHFFFAOYSA-M 0.000 description 1
- RFKZUAOAYVHBOY-UHFFFAOYSA-M copper(1+);acetate Chemical compound [Cu+].CC([O-])=O RFKZUAOAYVHBOY-UHFFFAOYSA-M 0.000 description 1
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 1
- 229910000366 copper(II) sulfate Inorganic materials 0.000 description 1
- NKNDPYCGAZPOFS-UHFFFAOYSA-M copper(i) bromide Chemical compound Br[Cu] NKNDPYCGAZPOFS-UHFFFAOYSA-M 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- SFJMFSWCBVEHBA-UHFFFAOYSA-M copper(i)-thiophene-2-carboxylate Chemical compound [Cu+].[O-]C(=O)C1=CC=CS1 SFJMFSWCBVEHBA-UHFFFAOYSA-M 0.000 description 1
- KAVVFFBJUZQIFN-UHFFFAOYSA-N copper;1,3,5-trimethylbenzene Chemical group [Cu].CC1=CC(C)=CC(C)=C1 KAVVFFBJUZQIFN-UHFFFAOYSA-N 0.000 description 1
- ITFUHOHJQIDNQW-UHFFFAOYSA-L copper;2,2-dimethylpropanoate Chemical compound [Cu+2].CC(C)(C)C([O-])=O.CC(C)(C)C([O-])=O ITFUHOHJQIDNQW-UHFFFAOYSA-L 0.000 description 1
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 238000006264 debenzylation reaction Methods 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 229910052741 iridium Inorganic materials 0.000 description 1
- GKOZUEZYRPOHIO-UHFFFAOYSA-N iridium atom Chemical compound [Ir] GKOZUEZYRPOHIO-UHFFFAOYSA-N 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- MUMZUERVLWJKNR-UHFFFAOYSA-N oxoplatinum Chemical compound [Pt]=O MUMZUERVLWJKNR-UHFFFAOYSA-N 0.000 description 1
- 229910003445 palladium oxide Inorganic materials 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- JQPTYAILLJKUCY-UHFFFAOYSA-N palladium(ii) oxide Chemical compound [O-2].[Pd+2] JQPTYAILLJKUCY-UHFFFAOYSA-N 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229910003446 platinum oxide Inorganic materials 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- FVSKHRXBFJPNKK-UHFFFAOYSA-N propionitrile Chemical compound CCC#N FVSKHRXBFJPNKK-UHFFFAOYSA-N 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 229910052707 ruthenium Inorganic materials 0.000 description 1
- YBCAZPLXEGKKFM-UHFFFAOYSA-K ruthenium(iii) chloride Chemical compound [Cl-].[Cl-].[Cl-].[Ru+3] YBCAZPLXEGKKFM-UHFFFAOYSA-K 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- DANYXEHCMQHDNX-UHFFFAOYSA-K trichloroiridium Chemical compound Cl[Ir](Cl)Cl DANYXEHCMQHDNX-UHFFFAOYSA-K 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 150000003722 vitamin derivatives Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
Definitions
- the present invention relates to methods for producing hydroxythienoimidazole derivatives, vinyl sulfide derivatives, n-butylidene sulfide derivatives, and saturated straight-chain hydrocarbon-substituted thienoimidazole derivatives.
- biotin shown below is considered a type of vitamin useful as animal feed and pharmaceuticals.
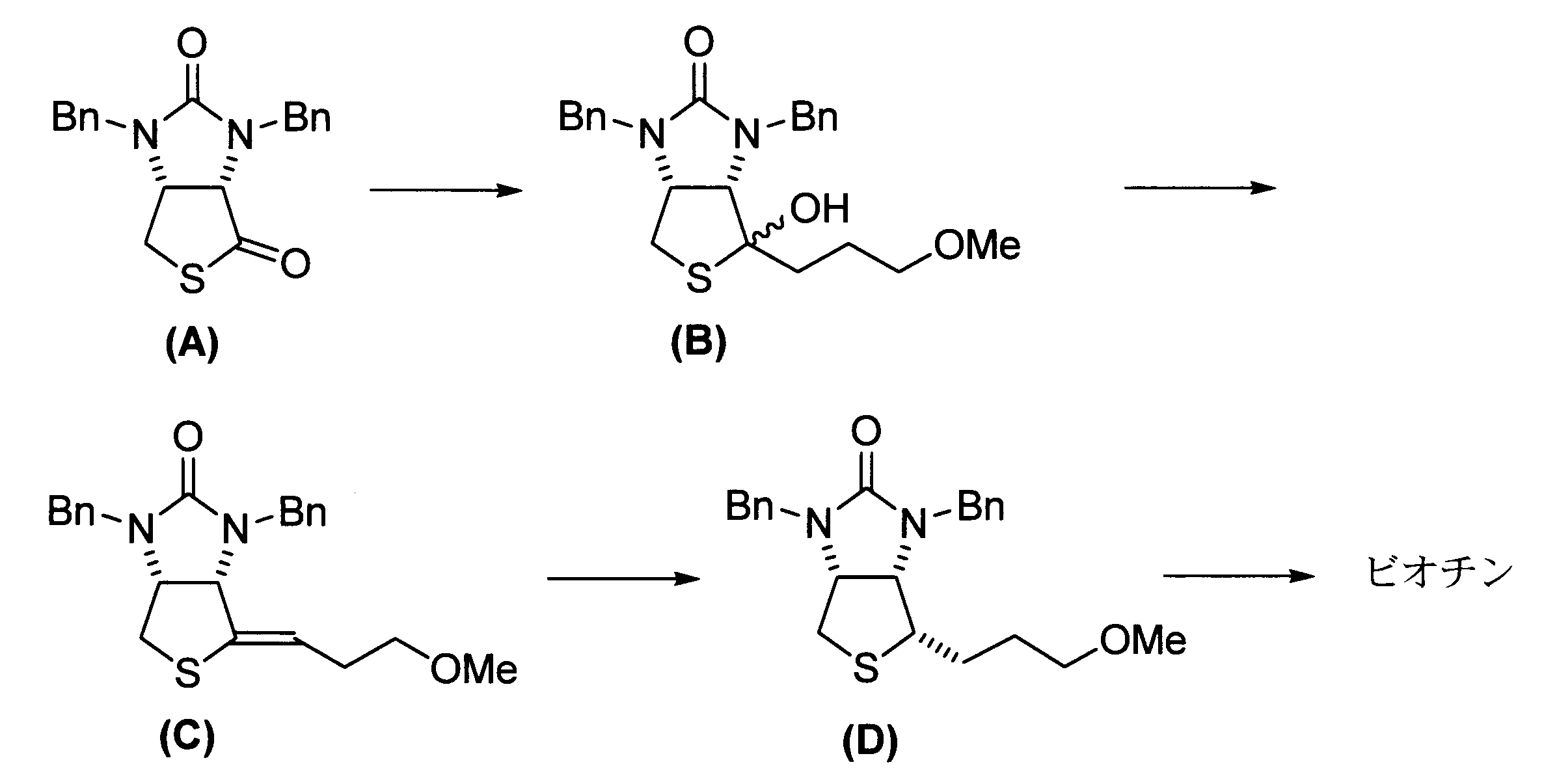
- Non-Patent Document 1 describes a biotin synthesis method using the Grignard reagent ClMg(CH 2 ) 3 OMe. Specifically, as shown below, the thiolactone compound (A) is subjected to an addition reaction with the Grignard reagent, and then the product is hydrolyzed to obtain an intermediate (B), which is then dehydrated to obtain a compound (C). ) is obtained, the compound (C) is hydrogenated to obtain the compound (D), and finally the compound (D) is subjected to coupling with a malonate ester and deprotection to obtain biotin is stated to obtain "Bn” represents a benzyl group, and "Me” represents a methyl group.
- An object of the present invention is to provide a method for producing a hydroxythienoimidazole derivative, a vinyl sulfide derivative, an n-butylidene sulfide derivative, and a saturated linear hydrocarbon-substituted thienoimidazole derivative in high yield.
- a method for producing a hydroxythienoimidazole derivative represented by formula (II) below is provided.
- a thiolactone derivative represented by the following formula (I) a Grignard reagent represented by the following formula (1), and a copper salt are mixed to obtain a hydroxythienoimidazole derivative represented by the following formula (II).
- a thiolactone derivative represented by the following formula (I) a Grignard reagent represented by the following formula (1), and a copper salt are mixed to obtain a hydroxythienoimidazole derivative represented by the following formula (II).
- a hydroxythienoimidazole derivative represented by the following formula (II) including the step of obtaining
- R 1 and R 2 are each independently an alkyl group, a substituted alkyl group, an aryl group, or a substituted aryl group.
- R 3 is an alkyl group, an alkyl group having a substituent, an aryl group, or an aryl group having a substituent.
- X 1 is a halogen atom.
- R 1 and R 2 have the same definitions as in formula (I).
- R 3 has the same definition as in formula (1).
- a method for producing a hydroxythienoimidazole derivative represented by formula (IV) below comprises mixing a thiolactone derivative represented by the above formula (I), a Grignard reagent represented by the following formula (2), and a copper salt to obtain a hydroxythienoimidazole derivative represented by the following formula (IV). including the step of obtaining
- each X2 is independently a halogen atom.
- R 1 and R 2 have the same definitions as in formula (I).
- a method for producing a vinyl sulfide derivative represented by the following formula (III) includes the steps of obtaining a hydroxythienoimidazole derivative represented by the above formula (II) by the method according to the above embodiment, dehydrating the obtained hydroxythienoimidazole derivative, and obtaining a product represented by the following formula (III). and obtaining a vinyl sulfide derivative.
- R 1 and R 2 have the same definitions as in formula (I).
- R 3 has the same definition as in formula (1).
- This production method includes the steps of obtaining the vinyl sulfide derivative represented by the above formula (III) by the method according to the above embodiment, and bringing the obtained vinyl sulfide derivative into contact with hydrogen in the presence of a catalyst to obtain the following formula ( obtaining a saturated linear hydrocarbon-substituted thienoimidazole derivative represented by VI).
- R 1 and R 2 have the same definitions as in formula (I).
- R 3 has the same definition as in formula (1).
- This production method includes the steps of obtaining a hydroxythienoimidazole derivative represented by the above formula (IV) by the method according to the above embodiment, dehydrating the obtained hydroxythienoimidazole derivative, and obtaining a product represented by the following formula (V). obtaining the n-butylidene sulfide derivative.
- R 1 and R 2 have the same definitions as in formula (I).
- a method for producing hydroxythienoimidazole derivatives, vinyl sulfide derivatives, n-butylidene sulfide derivatives, and saturated linear hydrocarbon-substituted thienoimidazole derivatives with high yield is provided.
- a production method comprises mixing a thiolactone derivative represented by the above formula (I) and a Grignard reagent represented by the above formula (1) or (2) in the presence of a copper salt, and obtaining a hydroxythienoimidazole derivative represented by formula (II) or (IV) above.
- Hydroxythienoimidazole derivatives can be used, for example, as intermediates for the synthesis of biotin and biotin derivatives described above.
- a hydroxythienoimidazole derivative can be obtained in high yield. That is, copper (Cu) has a high affinity with sulfur (S). Therefore, Cu in the copper salt is easily coordinated to the S atom of the thiolactone derivative, and as a result activates the S atom site of the thiolactone derivative. It is believed that this makes it easier for the Grignard reagent to react with the carbon atom having a carbonyl group adjacent to the S atom of the thiolactone derivative.
- a thiolactone derivative is represented by the following formula (I).
- Thiolactone derivatives can be used as intermediates for biotin synthesis as described above.
- R 1 and R 2 are each independently an alkyl group, a substituted alkyl group, an aryl group, or a substituted aryl group.
- R 1 and R 2 may be the same functional group, or different types of functional groups.
- R 1 and R 2 are each independently preferably an alkyl group having a substituent, more preferably an alkyl group having a phenyl group, and even more preferably a benzyl group.
- the alkyl group represented by R 1 or R 2 may be linear or branched.
- the number of carbon atoms in the alkyl group represented by R 1 or R 2 is, for example, 1 or more and 20 or less, preferably 1 or more and 10 or less, more preferably 1 or more and 8 or less, still more preferably 1 or more and 6 or less, still more preferably 1 or more and 4 or less, more preferably 1 or more and 3 or less, still more preferably 1 or 2, and still more preferably 1.
- the alkyl group represented by R 1 or R 2 may have a substituent.
- Substituents that the alkyl group represented by R 1 or R 2 may have include, for example, those having 3 to 20 carbon atoms (preferably 6 to 20 carbon atoms, more preferably 6 to 14 carbon atoms, still more preferably 6 or more 10 or less), an alkoxy group having 1 or more and 6 or less carbon atoms (preferably 1 or more and 4 or less, more preferably 1 or more and 3 or less, still more preferably 1 or 2), and a halogen atom.
- Aryl groups can be monocyclic, bicyclic or tricyclic aromatic hydrocarbon ring groups.
- the aryl group is preferably a monocyclic 3- to 8-membered ring, more preferably a phenyl group.
- An alkoxy group may be linear or branched.
- Halogen atoms can be selected from fluorine, chlorine, bromine and iodine atoms.
- a substituent which the alkyl group represented by R 1 or R 2 may have an aryl group having 3 or more and 8 or less carbon atoms is preferable, and a phenyl group is more preferable.
- the alkyl group represented by R 1 or R 2 has a substituent, the number of substituents is, for example, 1 or more and 5 or less, preferably 1 or more and 3 or less, more preferably 1 or 2, still more preferably 1. be.
- the aryl group represented by R 1 or R 2 can be a monocyclic, bicyclic or tricyclic aromatic hydrocarbon ring group.
- the aryl group represented by R 1 or R 2 is preferably a 3- to 8-membered monocyclic ring.
- the number of carbon atoms in the aryl group represented by R 1 or R 2 is, for example, 3 or more and 30 or less, preferably 3 or more and 20 or less, more preferably 6 or more and 20 or less, still more preferably 6 or more and 14 or less, still more preferably It is 6 or more and 10 or less.
- the aryl group represented by R 1 or R 2 is preferably a phenyl group.
- the aryl group represented by R 1 or R 2 may have a substituent.
- Substituents that the aryl group represented by R 1 or R 2 may have include, for example, 1 to 6 carbon atoms (preferably 1 to 4, more preferably 1 to 3, still more preferably 1 or 2) alkyl groups, alkoxy groups having 1 to 6 carbon atoms (preferably 1 to 4 carbon atoms, more preferably 1 to 3 carbon atoms, still more preferably 1 or 2 carbon atoms), carboxyl groups, halogen atoms, and the like.
- Alkyl groups and alkoxy groups may be linear or branched.
- Halogen atoms can be selected from fluorine, chlorine, bromine and iodine atoms.
- the number of substituents is, for example, 1 or more and 5 or less, preferably 1 or more and 3 or less, more preferably 1 or 2, and still more preferably 1. be.
- the first Grignard reagent is represented by the following formula (1).
- R 3 is an alkyl group, an alkyl group having a substituent, an aryl group, or an aryl group having a substituent.
- the above descriptions of the alkyl group, substituted alkyl group, aryl group, and substituted aryl group represented by R 1 or R 2 refer to the alkyl group and substituted aryl group represented by R 3 It also applies to alkyl groups, aryl groups, and aryl groups having substituents. Examples of the alkyl group, substituted alkyl group, aryl group, and substituted aryl group represented by R 3 include the same groups as those exemplified for R 1 and R 2 .
- R 3 is preferably an alkyl group having 1 to 6 carbon atoms, more preferably a methyl group or an ethyl group.
- X 1 is a halogen atom.
- a halogen atom can be selected from a fluorine atom, a chlorine atom, a bromine atom and an iodine atom, preferably a chlorine atom or a bromine atom.
- the first Grignard reagent is obtained, for example, by bringing an organic halogen derivative represented by the following formula (1a) into contact with magnesium.
- R 3 and X 1 have the same definitions as in formula (1).
- Magnesium may be powdered or strip-shaped.
- the contact temperature between the organohalogen derivative represented by the above formula (1a) and magnesium is, for example, 40°C or higher and 150°C or lower, preferably 60°C or higher and 100°C or lower.
- the contact time between the organohalogen derivative represented by the formula (1a) and magnesium is, for example, 10 minutes or more and 10 hours or less, preferably 1 hour or more and 5 hours or less.
- the amount of the organic halogen derivative represented by formula (1a) per 1 mol of magnesium is, for example, 0.1 mol or more and 2 mol or less, preferably 0.5 mol or more and 1.5 mol or less.
- the contact between the organohalogen derivative represented by the above formula (1a) and magnesium is preferably carried out in the presence of a magnesium activator.
- a magnesium activator for example, at least one selected from the group consisting of 1,2-dibromoethane, bromine, iodine, and trimethylsilyl chloride can be used.
- the amount of the magnesium activator to 1 mol of magnesium is, for example, 0.01 mol or more and 1.5 mol or less, preferably 0.2 mol or more and 0.8 mol or less.
- the contact between the organohalogen derivative represented by the above formula (1a) and magnesium is preferably carried out in the presence of the first reaction solvent.
- the first reaction solvent include acetonitrile, propionitrile, tetrahydrofuran (THF), 2-methyl-tetrahydrofuran, 1,4-dioxane, tert-butyl methyl ether, diisopropyl ether, dimethyloxyethane, diglyme, acetone, and methyl ethyl ketone.
- the amount of the first reaction solvent used in the production of the first Grignard reagent is, for example, 0.001 mL or more and 10 mL or less, preferably 0.01 mL or more and 1 mL or less, relative to 1 mg of magnesium.
- the first Grignard reagent When using the first reaction solvent, it is preferable to prepare the first Grignard reagent by the following method. First, the organic halogen derivative represented by the formula (1a) and half of the first reaction solvent are mixed to prepare an organic halogen derivative solution. Next, magnesium, magnesium activator, and half of the first reaction solvent are mixed to obtain a first mixture. An organohalogen derivative solution is added dropwise to this first mixture to obtain a second mixture. After the second mixture is heated to the above contact temperature, it is stirred for the above contact time to obtain the first Grignard reagent.
- Copper salt The valence of the copper atom contained in the copper salt is preferably monovalent or divalent, more preferably monovalent.
- a copper salt in which the valence of the copper atom is monovalent has excellent catalytic activity.
- Copper salts include, for example, copper (I) chloride (CuCl), copper (II) chloride (CuCl 2 ), copper (I) bromide (CuBr), copper (II) bromide (CuBr 2 ), copper cyanide (I) (CuCN), copper (I) 3-methylsalicylate, copper mesitylene (I) (MesCu), copper (I) isopropoxy (iPrOCu), copper (I) iodide (CuI), copper (II) iodide ) (CuI 2 ), copper(I) acetate (CuOAc), copper(II) acetate (Cu(OAc) 2 ), copper(II) sulfate (CuSO 4 ), copper(I) oxide (Cu 2
- Copper (I) thiophene-2-carboxylate is preferably used as the sulfur-containing copper salt.
- CuCl, CuI or CuBr is particularly preferred among the copper salts in which the valence of the copper atom is monovalent.
- CuCl, CuI and CuBr are particularly catalytic.
- a method for producing a hydroxythienoimidazole derivative represented by formula (II) comprises mixing a thiolactone derivative represented by formula (I), a first Grignard reagent, and a copper salt.
- the amount of copper salt used is preferably 0.05 mol or more and 1 mol or less per 1 mol of the first Grignard reagent.
- the amount of the copper salt used is more preferably 0.5 mol or more and 0.8 mol or less, still more preferably 0.6 mol or more and 0.72 mol or less, relative to 1 mol of the first Grignard reagent. be.
- the amount of the copper salt used is generally 0.1 mol or more and 10 mol or less, preferably 0.5 mol or more and 5 mol or less, more preferably 0.5 mol or more and 2 mol or less, relative to 1 mol of the thiolactone derivative.
- the amount of the first Grignard reagent to be used is generally 0.5 to 10 mol, preferably 1.0 to 5 mol, more preferably 1.0 to 2.0 mol, per 1 mol of the thiolactone derivative. It is below.
- the thiolactone derivative, the first Grignard reagent, and the copper salt may be mixed in the presence of the second reaction solvent.
- the second reaction solvent include tetrahydrofuran (THF), 2-methyl-tetrahydrofuran, 1,4-dioxane, tert-butyl methyl ether, cyclopentyl methyl ether, dimethoxyethane, diglyme, methylene chloride, toluene, xylene, hexane, At least one selected from heptane and the like can be used.
- the second reaction solvent may be used singly or in combination of two or more as a mixed solvent.
- the second reaction solvent is preferably THF, toluene or a mixed solvent thereof.
- the amount of the second reaction solvent used is, for example, 1 mL or more and 100 mL or less, preferably 2 mL or more and 50 mL or less, per 1 g of the thiolactone derivative.
- the temperature at which the thiolactone derivative, the first Grignard reagent, and the copper salt are mixed is, for example, within the range of -40°C or higher and 100°C or lower.
- the temperature during mixing is preferably -20°C or higher and 40°C or lower, more preferably -10°C or higher and 20°C or lower. Within this temperature range, the yield of the hydroxythienoimidazole derivative tends to be higher.
- the time for mixing the thiolactone derivative, the first Grignard reagent and the copper salt is usually 0.5 to 72 hours, preferably 1 to 48 hours.
- the mixing of the thiolactone derivative, the first Grignard reagent and the copper salt is preferably carried out by the following method.
- a first Grignard reagent and a copper salt are mixed to obtain an organic copper reagent.
- a first Grignard reagent solution obtained by dissolving the first Grignard reagent in the first or second reaction solvent may be used.
- a copper salt solution obtained by dissolving the copper salt in the second reaction solvent may be used.
- the temperature for mixing the first Grignard reagent and the copper salt may be within the range of the temperature for mixing the thiolactone derivative, the first Grignard reagent, and the copper salt.
- the mixing time of the first Grignard reagent and the copper salt is, for example, 1 minute or more and 1 hour or less.
- the first Grignard reagent and the copper salt are thought to form a copper complex represented by the following formula (3).
- R 3 and X 1 have the same definitions as in formula (1).
- Y represents an anion of the copper salt.
- Each of m and n is an integer of 1 or more and 3 or less.
- this organocopper reagent is brought into contact with the thiolactone derivative represented by formula (I).
- a thiolactone derivative solution obtained by dissolving this thiolactone derivative in a second reaction solvent may be used.
- the contact temperature of the organocopper reagent and the thiolactone derivative may be within the range of the mixing temperature of the thiolactone derivative, the first Grignard reagent, and the copper salt.
- the contact time of the organocopper reagent and the thiolactone derivative may be within the range of the mixing time of the thiolactone derivative, the first Grignard reagent, and the copper salt.
- hydroxythienoimidazole derivative represented by formula (II)>
- a hydroxythienoimidazole derivative is represented by the following formula (II). Hydroxythienoimidazole derivatives can be used as intermediates for biotin synthesis as described above.
- R 1 and R 2 have the same definitions as in formula (I).
- R 3 has the same definition as in formula (1).
- a hydroxythienoimidazole derivative is derived into biotin by a known method.
- a vinyl sulfide derivative represented by the following formula (III) is obtained by dehydrating the hydroxythienoimidazole derivative represented by the formula (II).
- R 1 and R 2 have the same definitions as in formula (I).
- R 3 has the same definition as in formula (1).
- Examples of dehydration methods for hydroxythienoimidazole derivatives include acid treatment and heat treatment.
- the acid treatment includes, for example, contacting the hydroxythienoimidazole derivative represented by formula (II) with an acid catalyst.
- Acid catalysts include, for example, sulfuric acid, hydrochloric acid, or mixtures thereof.
- the temperature of the heat treatment is, for example, -20 to 120°C, preferably 0 to 70°C. Acid treatment and heat treatment may be combined.
- the vinyl sulfide derivative represented by formula (III) is hydrogenated, for example, in the presence of a Pd catalyst to obtain a compound represented by formula (VI) below.
- a Pd catalyst for example, a Pd catalyst
- the obtained compound is reacted with CH 2 (COOEt) 2 to obtain a compound represented by the following formula (XI).
- Biotin is obtained by debenzylation of the resulting compound followed by treatment with, for example, hydrogen bromide.
- "Et" represents an ethyl group.
- R 1 and R 2 have the same definitions as in formula (I).
- R 3 has the same definition as in formula (1).
- This saturated straight-chain hydrocarbon-substituted thienoimidazole derivative is obtained by, for example, contacting the vinyl sulfide derivative represented by the formula (III) obtained by the method according to the above embodiment with hydrogen (H 2 ) in the presence of a catalyst. obtained by
- catalysts include platinum catalysts such as platinum carbon and platinum oxide; palladium catalysts such as palladium black, palladium carbon, palladium acetate, palladium chloride, and palladium oxide; nickel catalysts such as Raney nickel, cobalt catalysts such as Raney cobalt, and ruthenium chloride catalysts. Ruthenium catalysts, iridium catalysts such as iridium chloride, and iron catalysts such as iron powder may be used. At least one of Raney nickel and palladium carbon is preferred.
- the amount of the catalyst is, for example, 0.001 to 1000 mol%, preferably 0.1 to 800 mol%, relative to the substrate vinyl sulfide derivative.
- the hydrogen pressure is, for example, 1 to 150 atmospheres, preferably 1 to 50 atmospheres.
- the contact temperature is, for example, 10 to 200°C, preferably 25 to 150°C.
- the contact time is, for example, 0.5 to 100 hours, preferably 1 to 72 hours.
- the contact between the vinyl sulfide derivative represented by formula (III) and hydrogen in the presence of a catalyst may be carried out in the presence of a solvent.
- Solvents include methanol, ethanol, isopropanol, butanol, 2-butanol, ethylene glycol, 1,2-dimethoxyethane, methyl cellosolve, ethyl acetate, methyl acetate, THF, cyclopentyl methyl ether, 1,4-dioxane, acetic acid, water. , or a mixed solvent thereof can be used.
- methanol or a mixed solvent of methanol and water is used.
- the amount of the solvent used is, for example, 1-200 mL, preferably 3-100 mL, per 1 g of the vinyl sulfide derivative that is the substrate.
- the hydroxythienoimidazole derivative represented by formula (IV) is produced by the same method as the method for producing the hydroxythienoimidazole derivative represented by formula (II) above, except that the second Grignard reagent is used instead of the first Grignard reagent. It can be manufactured by the method of
- a method for producing a hydroxythienoimidazole derivative represented by formula (IV) includes mixing a thiolactone derivative represented by formula (I), a Grignard reagent represented by formula (2), and a copper salt. Including process.
- R 1 and R 2 have the same definitions as in formula (I).
- X2 has the same definition as in formula ( 2 ).
- the second Grignard reagent is represented by the following formula (2).
- each X2 is independently a halogen atom.
- a halogen atom can be selected from a fluorine atom, a chlorine atom, a bromine atom and an iodine atom, preferably a chlorine atom or a bromine atom.
- Two X2's may be the same kind of halogen atom or different kinds of halogen atoms.
- an organic halogen derivative represented by the following formula (2a) is used. It can be manufactured by the same method as the method.
- a second Grignard reagent and a copper salt are mixed in the same manner as in the method for producing the hydroxythienoimidazole derivative represented by formula (II). It is preferred to use an organocopper reagent prepared by
- the second Grignard reagent and the copper salt are thought to form a copper complex represented by the following formula (4).
- X2 has the same meaning as in formula ( 2 ).
- Z represents an anion of the copper salt.
- p and q are each an integer of 1 or more and 3 or less.
- the amount of copper salt used is preferably 0.1 mol or more and 2 mol or less per 1 mol of the second Grignard reagent.
- the amount of the copper salt used is more preferably 0.5 mol or more and 1.5 mol or less, still more preferably 0.6 mol or more and 1.2 mol or less, relative to 1 mol of the second Grignard reagent.
- the amount of the copper salt used is generally 0.1 mol or more and 10 mol or less, preferably 0.5 mol or more and 5 mol or less, more preferably 0.5 mol or more and 2 mol or less, relative to 1 mol of the thiolactone derivative.
- the amount of the second Grignard reagent used is generally 0.5 mol or more and 10 mol or less, preferably 1.0 mol or more and 5 mol or less, more preferably 1.0 mol or more and 2 mol or less, per 1 mol of the thiolactone derivative. be.
- a hydroxythienoimidazole derivative is represented by the following formula (IV). This hydroxythienoimidazole derivative can be used as an intermediate for the synthesis of biotin derivatives.
- R 1 and R 2 have the same definitions as in formula (I).
- n-butylidene sulfide derivative represented by the following formula (V) is obtained.
- This n-butylidene sulfide derivative can be used as an intermediate for biotin synthesis.
- R 1 and R 2 have the same definitions as in formula (I).
- Examples of dehydration methods for hydroxythienoimidazole derivatives include acid treatment and heat treatment.
- Acid treatment includes, for example, contacting the hydroxythienoimidazole derivative represented by formula (IV) with an acid catalyst.
- Acid catalysts include, for example, sulfuric acid, hydrochloric acid, or mixtures thereof.
- the temperature of the heat treatment is, for example, -20 to 120°C, preferably 0 to 70°C. Acid treatment and heat treatment may be combined.
- first Grignard reagent was prepared by the following method. Mg (24.3 mg, 1.00 mmol, 2.0 eq) was activated by adding THF (1.00 mL), 1,2-dibromoethane (0.05 mL), followed by 1-chloro-3-methoxy A solution of propane (54.3 mg, 0.500 mmol, 1.00 eq) in THF (1.00 mL) was slowly added dropwise. After the addition was completed, the mixture was stirred at 80°C for 3 hours.
- the mixture was stirred at a temperature of 0° C. for 2 hours to obtain a reactant.
- the reaction was developed by thin layer chromatography (TLC) to confirm completion of the reaction.
- TLC thin layer chromatography
- a developing solvent a mixed solvent in which ethyl acetate and n-hexane were mixed at a volume ratio of 1:1 was used.
- the Rf value of the compound represented by formula (II') was 0.11.
- the plate used in TLC is coated with silica gel, and this silica gel acts as an acid, and hydrolysis of the intermediate (see formula (Ia)) yields the compound represented by formula (II'). .
- Example 2 The amount of 1,2-dibromoethane was 0.025 mL, the amount of CuCl was 18.6 mg (0.188 mmol, 0.75 equivalents), and the amount of THF solution of the first Grignard reagent was 1 .10 mL (0.275 mmol, 1.1 equivalents) was used to convert the compound represented by formula (I′) to formula (III′) in the same manner as described in Example 1. A compound was obtained. The yield of the compound represented by formula (III') was 100%.
- a second Grignard reagent was prepared by the following method. Mg (48.6 mg, 2.00 mmol, 4.0 eq) was activated by addition of THF (1.00 mL), 1,2-dibromoethane (0.05 mL, 0.58 mmol), followed by 1,4 - A solution of dichlorobutane (63.5 mg, 0.500 mmol, 1.00 eq) in THF (1.00 mL) was slowly added dropwise. After the addition was completed, the mixture was stirred at 80°C for 3 hours.
- reaction product was stirred at a temperature of 0° C. for 1 hour to obtain a reaction product.
- the reaction was developed by thin layer chromatography (TLC) to confirm completion of the reaction.
- TLC thin layer chromatography
- a developing solvent a mixed solvent in which ethyl acetate and n-hexane were mixed at a volume ratio of 1:1 was used.
- the Rf value of the compound represented by formula (IV') was 0.15.
- the plate used in TLC is coated with silica gel, and this silica gel acts as an acid, and the hydrolysis of the intermediate (see formula (Ib)) yields the compound represented by formula (IV'). .
- Raney nickel (92.5 wt% in water, 100 mg, 1.58 mmol, 6.7 eq) was quickly weighed into a glass autoclave test tube and methanol (5.00 mL) was added. After adding a methanol solution (5.00 mL) of the compound represented by formula (III') (92.6 mg, 0.235 mmol, 1.0 equivalent), the mixture was stirred at 40°C under a hydrogen pressure of 20 atm for 20 hours. The reaction was developed by thin layer chromatography (TLC) to confirm completion of the reaction. As a developing solvent, a mixed solvent in which ethyl acetate and n-hexane were mixed at a volume ratio of 1:1 was used. The Rf value of the compound represented by formula (VI') was 0.51.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description


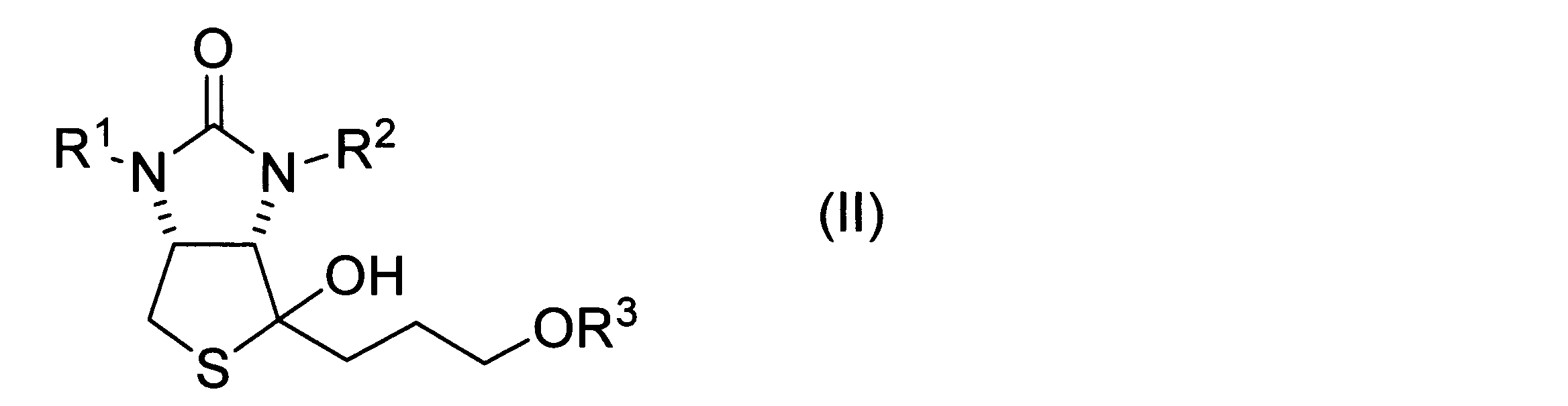


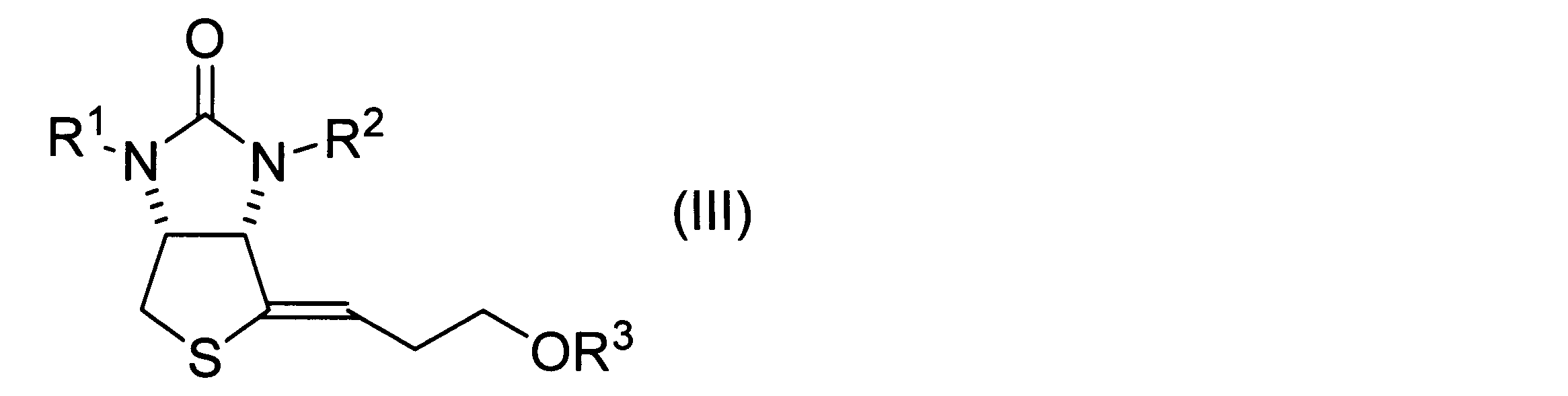


チオラクトン誘導体は、下記式(I)に表される。チオラクトン誘導体は、上述したビオチン合成のための中間体として用い得る。

第1グリニャール試薬は、下記式(1)に表される。
銅塩に含まれる銅原子の価数は、1価又は2価であることが好ましく、1価であることがより好ましい。銅原子の価数が1価である銅塩は、触媒作用が優れている。銅塩としては、例えば、塩化銅(I)(CuCl)、塩化銅(II)(CuCl2)、臭化銅(I)(CuBr)、臭化銅(II)(CuBr2)、シアン化銅(I)(CuCN)、3-メチルサリチル酸銅(I)、メシチレン銅(I)(MesCu)、イソプロポキシ銅(I)(iPrOCu)、ヨウ化銅(I)(CuI)、ヨウ化銅(II)(CuI2)、酢酸銅(I)(CuOAc)、酢酸銅(II)(Cu(OAc)2)、硫酸銅(II)(CuSO4)、酸化銅(I)(Cu2O)、酸化銅(II)(CuO)、ピバル酸銅(I)(CuOPiv)、ピバル酸銅(II)(Cu(OPiv)2)、硫黄含有銅塩等から選ばれる少なくとも1種を用いることができる。硫黄含有銅塩としては、銅(I)チオフェン-2-カルボン酸塩を用いることが好ましい。銅原子の価数が1価である銅塩のうち、CuCl、CuI又はCuBrが特に好ましい。CuCl、CuI及びCuBrは、触媒作用が特に優れている。
式(II)に表されるヒドロキシチエノイミダゾール誘導体を製造する方法は、式(I)に表されるチオラクトン誘導体と、第1グリニャール試薬と、銅塩とを混合する工程を含む。


ヒドロキシチエノイミダゾール誘導体は、下記式(II)に表される。ヒドロキシチエノイミダゾール誘導体は、上述したビオチン合成のための中間体として用い得る。
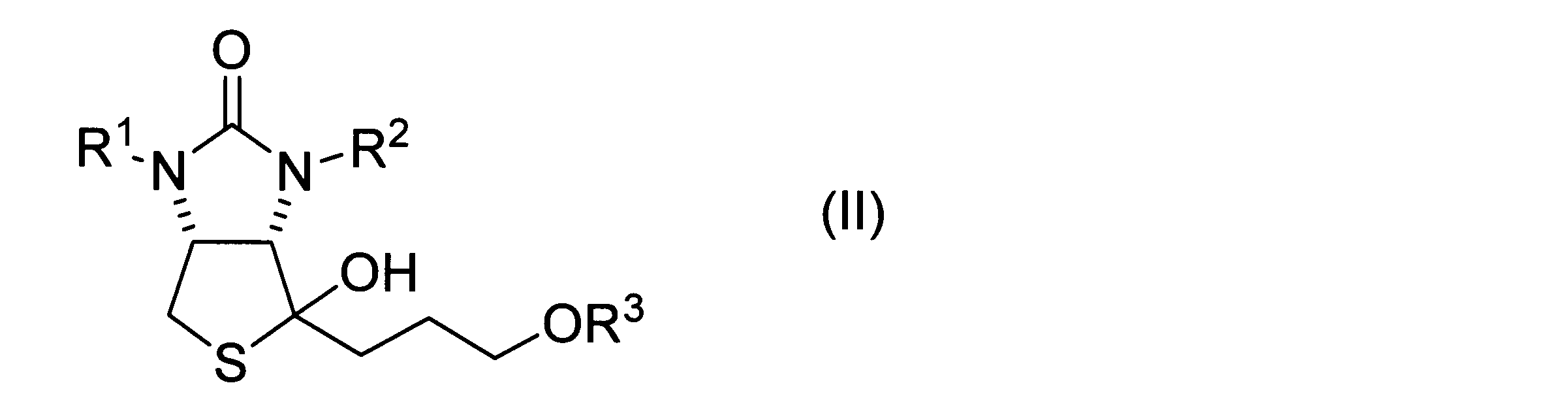
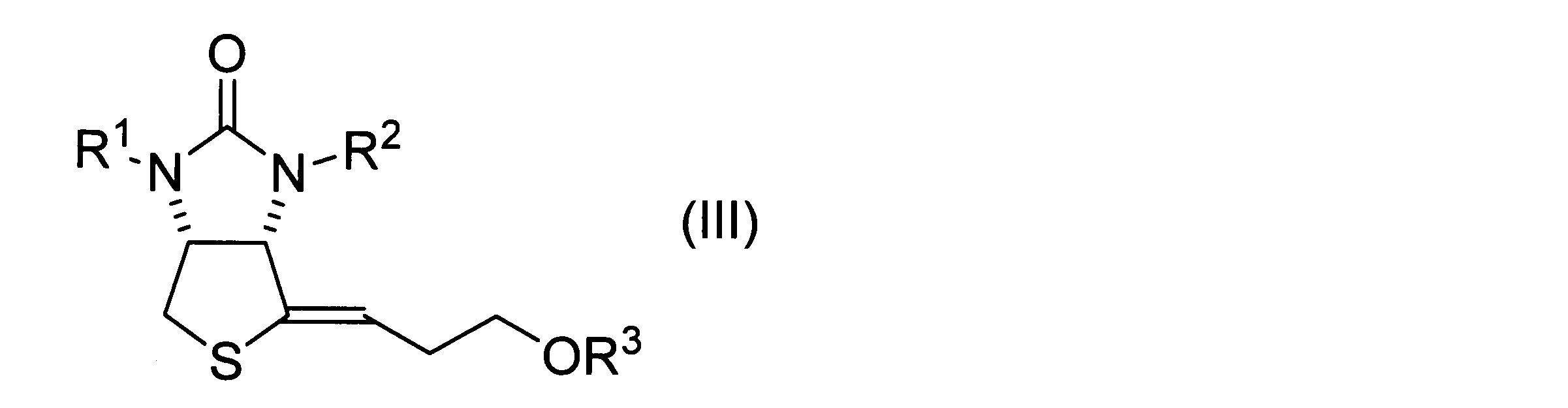

飽和直鎖炭化水素置換チエノイミダゾール誘導体は、下記式(VI)に表される。飽和直鎖炭化水素置換チエノイミダゾール誘導体は、上述したビオチン合成のための中間体として用い得る。

式(IV)に表されるヒドロキシチエノイミダゾール誘導体は、第1グリニャール試薬の代わりに第2グリニャール試薬を用いること以外は、上述した式(II)に表されるヒドロキシチエノイミダゾール誘導体の製造方法と同一の方法で製造できる。

第2グリニャール試薬は、下記式(2)に表される。
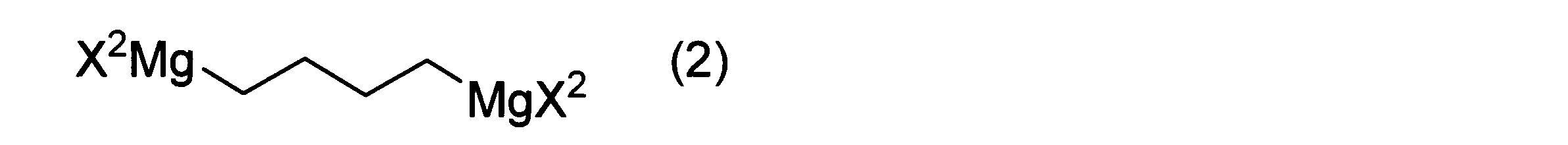

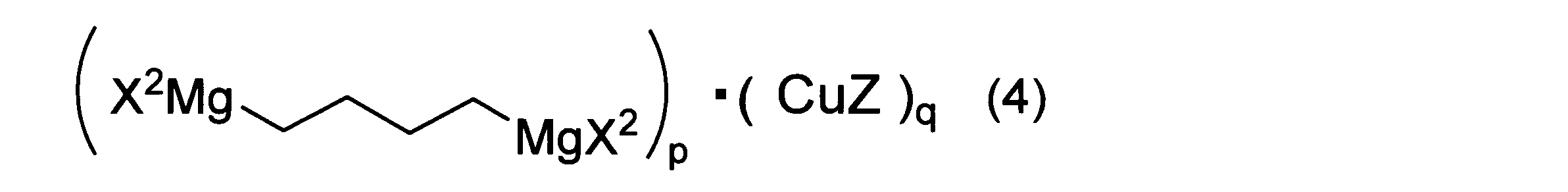
ヒドロキシチエノイミダゾール誘導体は、下記式(IV)に表される。このヒドロキシチエノイミダゾール誘導体は、ビオチン誘導体の合成のための中間体として用い得る。


下記反応式に示すように、式(I’)に表される化合物から、式(III’)に表される化合物を得た。なお、「Bn」はベンジル基を表し、「Me」はメチル基を表す。
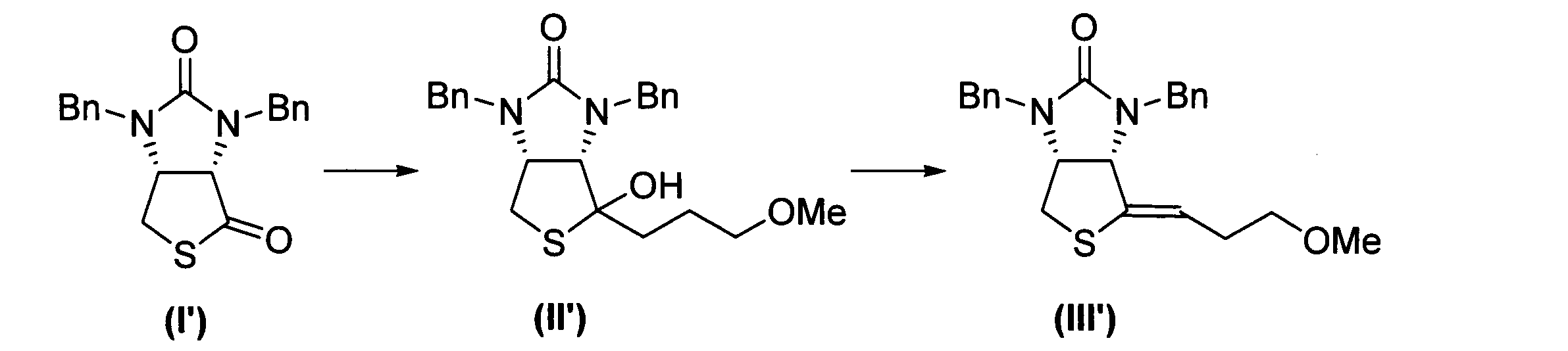
先ず、以下の方法で第1グリニャール試薬を準備した。Mg(24.3mg、1.00mmol、2.0当量)にTHF(1.00mL)、1,2-ジブロモエタン(0.05mL)を加えて活性化させた後、1-クロロ-3-メトキシプロパン(54.3mg、0.500mmol、1.00当量)のTHF(1.00mL)溶液をゆっくり滴下した。すべて加え終わった後、80℃で3時間撹拌した。
CuCl(24.8mg、0.250mmol、1.0当量)の乾燥THF(1.50mL)懸濁液に、上記の方法で得られた第1グリニャール試薬(0.25M)のTHF溶液(1.50mL、0.375mmol、1.5当量)を0℃の温度下で5分間にわたって滴下した後、0℃の温度で10分間攪拌して有機銅試薬を得た。この有機銅試薬に、式(I’)に表される化合物(84.6mg、0.250mmol、1.0当量)のTHF溶液(2.00mL)を0℃の温度下で5分間にわたって滴下した後、0℃の温度で2時間にわたって攪拌して反応物を得た。反応物を薄層クロマトグラフィー(TLC)で展開し、反応終了を確認した。展開溶媒としては、酢酸エチルとn-ヘキサンと1:1の体積比で混合した混合溶媒を用いた。式(II’)に表される化合物のRf値は0.11であった。なお、TLCで使用されるプレートにはシリカゲルが塗布されており、このシリカゲルが酸として働き、中間体(式(Ia)参照)の加水分解により、式(II’)に表される化合物が生じる。反応物に10%H2SO4溶液(2mL)とトルエン(5mL)とを0℃の温度下で加えた後、室温で30分にわたって攪拌し、水層と有機層とに分離させて有機層を得た。
1H NMR (400MHz,CDCl3,30℃) δ 7.35-7.22(m,10H),5.16-5.10(m,1H),4.86-4.78(m,1H),4.43(s,1H),4.17-3.96(m,3H),3.67(dd,J=15.7,9.3Hz,1H),3.61-3.32(m,5H),3.04-2.77(m,2H),2.38-2.31(m,1H),2.01-1.67(m,3H)。
次に、この有機層に1滴(触媒量)の濃硫酸を加えた後、60℃で1時間にわたって攪拌して混合物を得た。この混合物を上記と同様の方法でTLCで展開した。式(III’)に表される化合物のRf値は0.50であった。混合物を5mLの1M塩酸で3回洗浄し、更に5mLの食塩水で洗浄した後、Na2SO4を用いて乾燥させて残留物を得た。この残留物についてNMRを用いて分析して、上記式(III’)に表される化合物を含むことを確認した。収率は97%であった。式(III’)に表される化合物のNMR結果は下記のとおりであった。
1H NMR (400MHz,CDCl3,30℃) δ 7.36-7.27(m,10H),5.51(t,J=7.0Hz,1H),4.93(d,J=15.6Hz,1H),4.79(d,J=15.3Hz,1H),4.30(d,J=7.7Hz,1H),4.23(d,J=15.3Hz,1H),4.10-4.04(m,2H),3.36(dt,J=12.9,2.1Hz,2H),3.32(s,3H),3.00-2.92(m,2H),2.41-2.23(m,2H)。
1,2-ジブロモエタンの量を0.025mLとしたこと、CuClの量を18.6mg(0.188mmol、0.75当量)としたこと、及び、第1グリニャール試薬のTHF溶液の量を1.10mL(0.275mmol、1.1当量)としたこと以外は、実施例1に記載したのと同様の方法で、式(I’)に表される化合物から式(III’)に表される化合物を得た。式(III’)に表される化合物の収率は100%であった。
下記反応式に示すように、式(I’)に表される化合物から、式(V’)に表される化合物を得た。なお、「Bn」はベンジル基を表す。
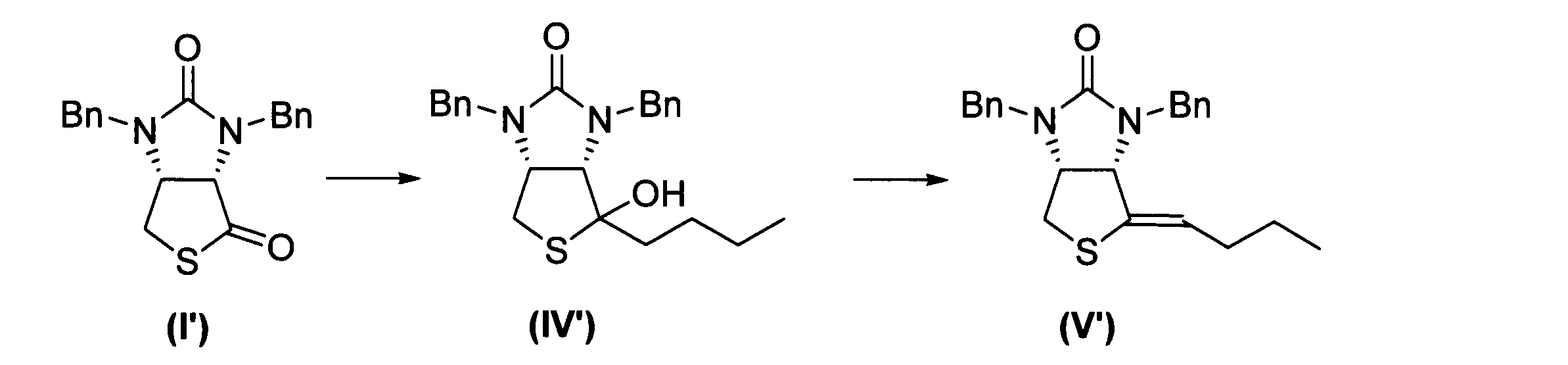
先ず、以下の方法で第2グリニャール試薬を準備した。Mg(48.6mg、2.00mmol、4.0当量)にTHF(1.00mL)、1,2-ジブロモエタン(0.05mL、0.58mmol)を加えて活性化させた後、1,4-ジクロロブタン(63.5mg、0.500mmol、1.00当量)のTHF(1.00mL)溶液をゆっくり滴下した。すべて加え終わった後、80℃で3時間撹拌した。
CuCl(27.2mg、0.275mmol、1.1当量)の乾燥THF(2.00mL)懸濁液に、上記の方法で得られた第2グリニャール試薬(0.25M)のTHF溶液(1.10mL、0.275mmol、1.1当量)を0℃の温度下で5分間にわたって滴下した後、0℃の温度で10分間攪拌して有機銅試薬を得た。この有機銅試薬に、式(I’)に表される化合物(84.6mg、0.250mmol、1.0当量)のTHF溶液(2.00mL)を0℃の温度下で5分間にわたって滴下した後、0℃の温度で1時間にわたって攪拌して反応物を得た。反応物を薄層クロマトグラフィー(TLC)で展開し、反応終了を確認した。展開溶媒としては、酢酸エチルとn-ヘキサンとを1:1の体積比で混合した混合溶媒を用いた。式(IV’)に表される化合物のRf値は0.15であった。なお、TLCで使用されるプレートにはシリカゲルが塗布されており、このシリカゲルが酸として働き、中間体(式(Ib)参照)の加水分解により、式(IV’)に表される化合物が生じる。反応物に10%H2SO4溶液(2mL)とトルエン(5mL)とを0℃の温度下で加えた後、室温で30分にわたって攪拌し、水層と有機層とに分離させて有機層を得た。
次に、この有機層に1滴(触媒量)の濃硫酸を加えた後、60℃で1時間にわたって攪拌して混合物を得た。この混合物を上記と同様の方法でTLCで展開した。式(V’)に表される化合物のRf値は0.6であった。混合物を5mLの1M塩酸で3回洗浄し、更に5mLの食塩水で洗浄した後、Na2SO4を用いて乾燥させて残留物を得た。この残留物についてNMRを用いて分析して、上記式(V’)に表される化合物を含むことを確認した。収率は36%であった。式(V’)に表される化合物のNMR結果は下記のとおりであった。
1H NMR (400MHz,CDCl3,30℃) δ 7.36-7.24(m,10H),5.46(t,J=7.2Hz,1H),4.97(d,J=15.8Hz,1H),4.81(d,J=15.2Hz,1H),4.31-4.20(m,2H),4.15-4.01(m,2H),3.00-2.92(m,2H),2.04(hept,J=8.0Hz,2H),1.39(sext,J=7.3Hz,1H),1.26(t,J=7.1Hz,1H),0.91(t,J=7.3Hz,3H)。
下記反応式に示すように、下記式(III’)に表される化合物から、式(VI’)に表される化合物を得た。なお、「Bn」はベンジル基を表し、「Me」はメチル基を表す。
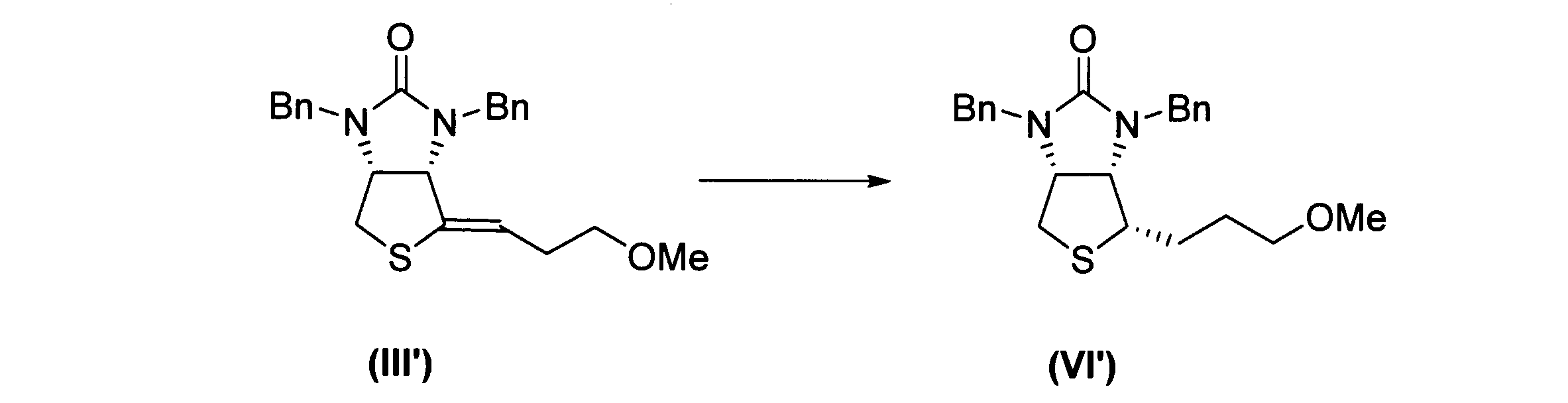
1H NMR (400 MHz,CDCl3,30°C) δ 7.34-7.24(m,10H),5.10(d,J=15.2Hz,1H),4.75(d,J=15.1Hz,1H),4.14(d,J=15.2Hz,1H),3.99-3.95(m,2H),3.87(dd,J=9.5,5.6Hz,1H),3.43-3.35(m,2H),3.34(s,3H),3.335-3.25(m,1H),3.17-3.11(m,1H),2.77-2.66(m,1H),1.90-1.74(m,2H),1.61-1.45(m,2H);13C{1H} NMR (100MHz,CDCl3,30℃) δ 161.1,137.1,137.0,128.8,128.80,128.4,127.8,72.2,62.7,61.3,58.7,54.2,47.9,46.8,34.9,29.2,25.6; HRMS (FAB+) m/z calcd. for C23H29N2O2S 397.1950 ([M+H]+) found 397.1946。
Claims (9)
- 下記式(I):

に表されるチオラクトン誘導体、
下記式(1):

に表されるグリニャール試薬、及び
銅塩
を混合して、下記式(II):
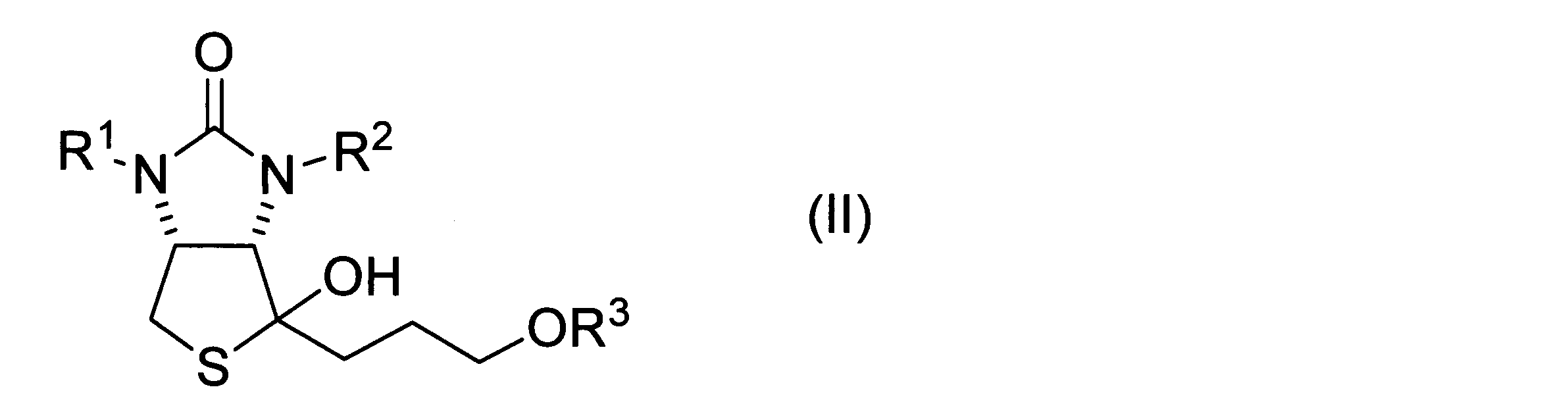
に表されるヒドロキシチエノイミダゾール誘導体を得る工程を含む、ヒドロキシチエノイミダゾール誘導体の製造方法。 - 前記工程において、前記式(1)に表されるグリニャール試薬及び前記銅塩を混合して有機銅試薬を形成させた後、前記有機銅試薬と前記式(I)に表されるチオラクトン誘導体とを接触させて、前記式(II)に表されるヒドロキシチエノイミダゾール誘導体を得る、請求項1に記載のヒドロキシチエノイミダゾール誘導体の製造方法。
- 1モルの前記式(1)に表されるグリニャール試薬に対する前記銅塩の量は、0.05モル以上1モル以下である、請求項1に記載のヒドロキシチエノイミダゾール誘導体の製造方法。
- 請求項1乃至3の何れか一項に記載の方法で前記ヒドロキシチエノイミダゾール誘導体を得る工程と、
前記ヒドロキシチエノイミダゾール誘導体を脱水して、下記式(III):

に表されるビニルスルフィド誘導体を得る工程と
を含む、ビニルスルフィド誘導体の製造方法。 - 請求項4に記載の方法で前記ビニルスルフィド誘導体を得る工程と、
触媒存在下で前記ビニルスルフィド誘導体と水素とを接触させて、下記式(VI):

に表される飽和直鎖炭化水素置換チエノイミダゾール誘導体を得る工程と
を含む、飽和直鎖炭化水素置換チエノイミダゾール誘導体の製造方法。 - 下記式(I):

に表されるチオラクトン誘導体、
下記式(2):

に表されるグリニャール試薬、及び
銅塩
を混合して、下記式(IV):

- 前記工程において、前記式(2)に表されるグリニャール試薬及び前記銅塩を混合して有機銅試薬を形成させた後、前記有機銅試薬と前記式(I)に表されるチオラクトン誘導体とを接触させて、前記式(IV)に表されるヒドロキシチエノイミダゾール誘導体を得る、請求項6に記載のヒドロキシチエノイミダゾール誘導体の製造方法。
- 1モルの前記式(2)に表されるグリニャール試薬に対する前記銅塩の量は、0.1モル以上2モル以下である、請求項6に記載のヒドロキシチエノイミダゾール誘導体の製造方法。
- 請求項6乃至8の何れか一項に記載の方法で前記ヒドロキシチエノイミダゾール誘導体を得る工程と、
前記ヒドロキシチエノイミダゾール誘導体を脱水して、下記式(V):
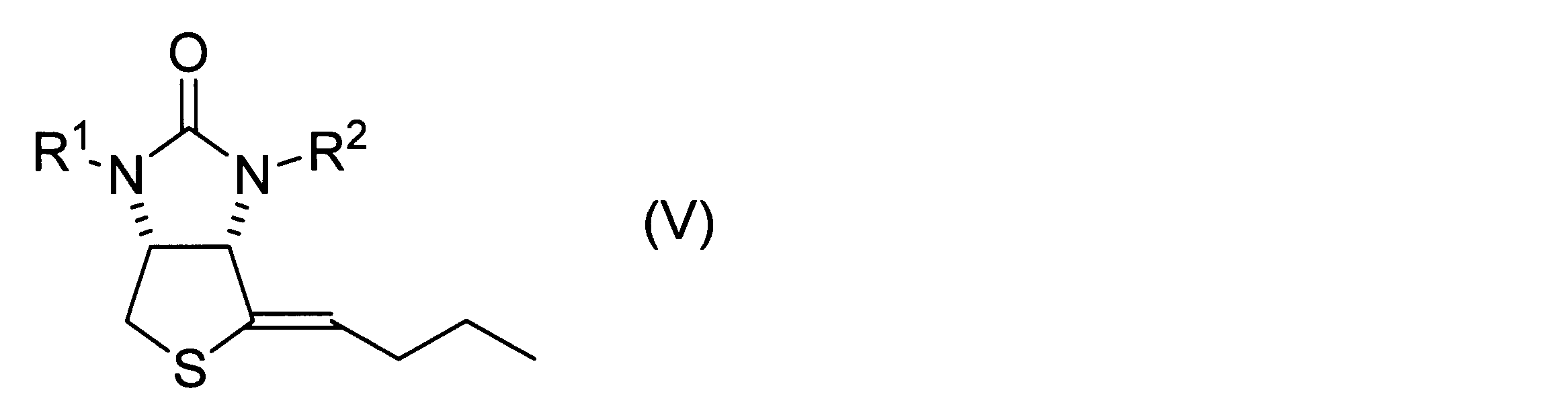
を含む、n-ブチリデンスルフィド誘導体の製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022557190A JP7229434B1 (ja) | 2021-06-11 | 2022-06-10 | ヒドロキシチエノイミダゾール誘導体、ビニルスルフィド誘導体、n-ブチリデンスルフィド誘導体、及び飽和直鎖炭化水素置換チエノイミダゾール誘導体の製造方法 |
| CN202280008852.8A CN116685567A (zh) | 2021-06-11 | 2022-06-10 | 羟基噻吩并咪唑衍生物、乙烯基硫醚衍生物、正丁亚基硫醚衍生物、及饱和直链烃取代噻吩并咪唑衍生物的制造方法 |
| EP22820346.9A EP4353728A1 (en) | 2021-06-11 | 2022-06-10 | Hydroxy thienoimidazole derivative, vinyl sulfide derivative, n-butylidene sulfide derivative, and production method for saturated straight-chain hydrocarbon-substituted thienoimidazole derivative |
| US18/287,236 US20240217986A1 (en) | 2021-06-11 | 2022-06-10 | Method of producing hydroxythienoimidazole derivative, vinyl sulfide derivative, n-butylidene sulfide derivative, and thienoimidazole derivative substituted with straight-chain saturated hydrocarbon |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021098332 | 2021-06-11 | ||
| JP2021-098332 | 2021-06-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022260168A1 true WO2022260168A1 (ja) | 2022-12-15 |
Family
ID=84424576
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/023481 WO2022260168A1 (ja) | 2021-06-11 | 2022-06-10 | ヒドロキシチエノイミダゾール誘導体、ビニルスルフィド誘導体、n-ブチリデンスルフィド誘導体、及び飽和直鎖炭化水素置換チエノイミダゾール誘導体の製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20240217986A1 (ja) |
| EP (1) | EP4353728A1 (ja) |
| JP (1) | JP7229434B1 (ja) |
| CN (1) | CN116685567A (ja) |
| TW (1) | TW202317584A (ja) |
| WO (1) | WO2022260168A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024080317A1 (ja) * | 2022-10-13 | 2024-04-18 | 株式会社トクヤマ | ヒドロキシビオチン誘導体及びビニルビオチン誘導体の製造方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5327279B1 (ja) * | 1969-11-29 | 1978-08-07 | ||
| WO2008124922A1 (en) * | 2007-04-12 | 2008-10-23 | Endorecherche, Inc. | 17alpha-substituted steroids as systemic antiandrogens and selective androgen receptor modulators |
| US20130065935A1 (en) * | 2011-07-15 | 2013-03-14 | Michael P. Kavanaugh | Novel Inhibitors of the Amino Acid Transporters ASCT1 and ASCT2 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5327279B2 (ja) | 2011-06-13 | 2013-10-30 | 株式会社デンソー | 超音波センサ装置 |
| CN105418634B (zh) * | 2015-12-10 | 2018-07-10 | 蚌埠丰原医药科技发展有限公司 | 生物素中间体杂质的制备方法 |
| CN107973806A (zh) * | 2016-10-21 | 2018-05-01 | 大丰海嘉诺药业有限公司 | 一种制备d-生物素溴盐中间体的方法 |
| CN110577547B (zh) * | 2019-08-05 | 2021-05-11 | 浙江工业大学 | 一种生物素中间体的合成方法 |
-
2022
- 2022-06-10 WO PCT/JP2022/023481 patent/WO2022260168A1/ja active Application Filing
- 2022-06-10 TW TW111121650A patent/TW202317584A/zh unknown
- 2022-06-10 EP EP22820346.9A patent/EP4353728A1/en active Pending
- 2022-06-10 US US18/287,236 patent/US20240217986A1/en active Pending
- 2022-06-10 CN CN202280008852.8A patent/CN116685567A/zh active Pending
- 2022-06-10 JP JP2022557190A patent/JP7229434B1/ja active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5327279B1 (ja) * | 1969-11-29 | 1978-08-07 | ||
| WO2008124922A1 (en) * | 2007-04-12 | 2008-10-23 | Endorecherche, Inc. | 17alpha-substituted steroids as systemic antiandrogens and selective androgen receptor modulators |
| US20130065935A1 (en) * | 2011-07-15 | 2013-03-14 | Michael P. Kavanaugh | Novel Inhibitors of the Amino Acid Transporters ASCT1 and ASCT2 |
Non-Patent Citations (5)
| Title |
|---|
| DATABASE CAPLUS 1 January 1900 (1900-01-01), LEE H. L, RAGGIOLINI E G, USKOKOVIC M R: "Synthesis of D-biotin from cysteine", XP002375145, Database accession no. 1988-492607 * |
| ISAKA ICHIRO, KAZUO KUBO, MUTSUO TAKASHIMA, MASUO MURAKAMI: "Studies on the synthesis of biotin. 3. Grignard-reaction of 3,4-(1',3'-dibenzyl-2'-oxoimidazolido)-2-oxothiophane", PHARMACY MAGAZINE, vol. 88, no. 8, 31 August 1968 (1968-08-31), pages 1068 - 1073, XP093014488, DOI: 10.1248/yakushi1947.88.8_1068 * |
| KATO DAIKI, TOMOYA MURASE, JALINDAR TALODE, HARUKI NAGAE, HAYATO TSURUGI, MASAHIKO SEKI, KAZUSHI MASHIMA: "Diarylcuprates for Selective Syntheses of Multifunctionalized Ketones from Thioesters under Mild Conditions", CHEMISTRY - A EUROPEAN JOURNAL, vol. 28, no. 26, 15 March 2022 (2022-03-15), pages e202200474, XP093014490, DOI: 10.1002/chem.202200474 * |
| M. GERECKEJ. -P. ZIMMERMANNW. ASCHWANDE: "116. Versuche zur Biotinsynthese. Herstellung von (3aS, 6aR)-1,3-Dibenzyl-tetrahydro-4H-thieno[3,4-d]imidazol-2,4(1H)-dion", HELV. CHIM. ACTA., vol. 53, 1970, pages 991 - 999 |
| WARM ALEKSANDER, NAUGHTON ANDREW B., SAIKALI ELIE A.: "Process Development Implications of Biotin Production Scale-Up", ORGANIC PROCESS RESEARCH & DEVELOPMENT, AMERICAN CHEMICAL SOCIETY, US, vol. 7, no. 3, 1 May 2003 (2003-05-01), US , pages 272 - 284, XP093014489, ISSN: 1083-6160, DOI: 10.1021/op020089o * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024080317A1 (ja) * | 2022-10-13 | 2024-04-18 | 株式会社トクヤマ | ヒドロキシビオチン誘導体及びビニルビオチン誘導体の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP7229434B1 (ja) | 2023-02-27 |
| US20240217986A1 (en) | 2024-07-04 |
| CN116685567A (zh) | 2023-09-01 |
| JPWO2022260168A1 (ja) | 2022-12-15 |
| EP4353728A1 (en) | 2024-04-17 |
| TW202317584A (zh) | 2023-05-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN107417505B (zh) | α-卤代四甲基环己酮及其与(2,3,4,4-四甲基环戊基)甲基羧酸酯的制备方法 | |
| JP7229434B1 (ja) | ヒドロキシチエノイミダゾール誘導体、ビニルスルフィド誘導体、n-ブチリデンスルフィド誘導体、及び飽和直鎖炭化水素置換チエノイミダゾール誘導体の製造方法 | |
| CN102432485B (zh) | 一种α,β-二氨基酸衍生物及其合成方法和应用 | |
| TWI540119B (zh) | 化合物、及其製造方法、以及磷酸奧司他韋之製造方法 | |
| US6476250B1 (en) | Optically active fluorinated binaphthol derivative | |
| CN113173908A (zh) | 一种噻吩类化合物的制备方法 | |
| CN111499600A (zh) | 一种多取代2,3-二氢呋喃类化合物的合成方法 | |
| JP4667593B2 (ja) | 2−アルキル−2−アダマンチル(メタ)アクリレート類の製造法 | |
| CN112010884A (zh) | 一种苯基(1-苯基乙基)硅烷的合成方法 | |
| JP4789108B2 (ja) | 光学活性なα−トリフルオロメチルケトン化合物の製造方法 | |
| CN111205184A (zh) | 一种合成(9z,12e)-十四碳-9,12-二烯-1-醇乙酸酯的方法 | |
| JP2917552B2 (ja) | α−メチレンシクロペンタノン誘導体の製造法 | |
| KR102467497B1 (ko) | 유기금속 화합물을 제조하기 위한 리간드의 합성 방법 | |
| JP7470080B2 (ja) | (6z,9z)-6,9-ドデカジエン-1-イン及びその製造方法 | |
| JPH0959290A (ja) | フェロセニルジフェニルホスフィン誘導体、該配位子金属錯体によるヒドロシリル化 | |
| JP4034040B2 (ja) | 含フッ素ジエン化合物 | |
| US8211820B2 (en) | Catalyst composition, and process for production of cross-coupling compound using the same | |
| CN106496005B (zh) | 一种4-(4-氯苯基)环己酮的合成方法 | |
| JP2000044509A (ja) | 含フッ素カルボン酸誘導体及びその製造方法 | |
| JP2008069104A (ja) | ヘリセン誘導体、トリイン誘導体、ヘリセン誘導体の製造方法 | |
| JP4839678B2 (ja) | ジハロゲン化ビアリール誘導体の製造方法 | |
| JP2003055285A (ja) | 4−tert−ブトキシ−4’−ハロゲノビフェニルおよびその製法、並びに4−ハロゲノ−4’−ヒドロキシビフェニルの製法 | |
| JP2004292340A (ja) | α−ペンタフルオロエチルアクリル酸誘導体およびその製造方法 | |
| JPH08311020A (ja) | β−カロテンの製造方法 | |
| JP2018108942A (ja) | アセト酢酸エステルアルカリ塩の製造方法および脂肪族ジケトンの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2022557190 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22820346 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202280008852.8 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18287236 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202317079338 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022820346 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2022820346 Country of ref document: EP Effective date: 20240111 |