WO2016072491A1 - 分岐ポリカーボネートの製造方法 - Google Patents
分岐ポリカーボネートの製造方法 Download PDFInfo
- Publication number
- WO2016072491A1 WO2016072491A1 PCT/JP2015/081309 JP2015081309W WO2016072491A1 WO 2016072491 A1 WO2016072491 A1 WO 2016072491A1 JP 2015081309 W JP2015081309 W JP 2015081309W WO 2016072491 A1 WO2016072491 A1 WO 2016072491A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polycarbonate
- branched polycarbonate
- organic solvent
- group
- dihydric phenol
- Prior art date
Links
- 0 Oc1ccc(*c(cc2)ccc2O)cc1 Chemical compound Oc1ccc(*c(cc2)ccc2O)cc1 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
- C08G64/20—General preparatory processes
- C08G64/22—General preparatory processes using carbonyl halides
- C08G64/24—General preparatory processes using carbonyl halides and phenols
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
- C08G64/04—Aromatic polycarbonates
- C08G64/06—Aromatic polycarbonates not containing aliphatic unsaturation
Definitions
- the present invention relates to a method for producing a branched polycarbonate.
- it is related with the manufacturing method of the branched polycarbonate excellent in production efficiency by the interfacial polymerization method.
- Polycarbonate has excellent characteristics such as transparency, heat resistance, and mechanical properties, and is used in a wide range of applications such as OA / consumer housings, electrical / electronic components, and optical materials such as lenses.
- Many of the polycarbonates used in this wide range of applications are linear polymers obtained by reacting dihydric phenols with carbonate precursors such as phosgene, but these polymers exhibit Newtonian flow behavior under melt processing conditions.
- carbonate precursors such as phosgene
- this branched polycarbonate is produced using an interfacial polymerization method or a transesterification method.
- the transesterification method since the raw material components are melted at a high temperature and subjected to a transesterification reaction for polymerization, the resulting branched polycarbonate is easily colored due to the influence of the polymerization catalyst used. Therefore, it is not preferable to produce a branched polycarbonate using a transesterification method in applications where transparency is required.
- Patent Document 1 discloses a method for obtaining a branched polycarbonate by using a polycarbonate oligomer incorporating a branching agent and reacting the polycarbonate oligomer with dihydric phenols. And a method for producing a branched polycarbonate by using a polycarbonate oligomer not incorporating a branching agent and reacting the polycarbonate oligomer with a branching agent and dihydric phenols.
- the method of obtaining the branched polycarbonate by reacting this polycarbonate oligomer with dihydric phenols using the polycarbonate oligomer incorporating the former branching agent uses the polycarbonate oligomer not incorporating the latter branching agent.
- the unit derived from the branching agent in the branched polycarbonate becomes more uniform, and the physical properties are more uniform than the latter production method. It is.
- a method of obtaining a branched polycarbonate by reacting this polycarbonate oligomer with dihydric phenols using a polycarbonate oligomer incorporating a branching agent is used in the continuous production of a polycarbonate oligomer incorporating a branching agent.
- a reaction liquid containing a polycarbonate oligomer incorporating a branching agent is separated into an organic solvent phase containing the polycarbonate oligomer and an aqueous phase, an intermediate layer is generated and the separability deteriorates, resulting in a significant reduction in production efficiency. There was a problem.
- the present invention uses a polycarbonate oligomer incorporating a branching agent and reacts the polycarbonate oligomer with dihydric phenols to produce a branched polycarbonate with high production efficiency.
- An object of the present invention is to provide a method for producing the above.
- the present inventors have made the above-described relationship between the addition amount of the polymerization catalyst to be added and the chloroformate group amount of the polycarbonate oligomer at a specific ratio when producing the polycarbonate oligomer.
- the inventors have found that the object is achieved and have completed the present invention. That is, the present invention relates to the following [1] to [11].
- the amount of chloroformate group of the polycarbonate oligomer contained in the reaction solution obtained from the step (b) is y mol / hr
- (x / y) is 0.0035 or more.
- a method for producing polycarbonate. [2] The method for producing a branched polycarbonate according to the above [1], wherein the polycarbonate oligomer has a weight average molecular weight of 5000 or less. [3] The method for producing a branched polycarbonate according to the above [1] or [2], wherein the branching agent is a compound represented by the following general formula (I).
- R represents a hydrogen atom or an alkyl group having 1 to 5 carbon atoms
- R 1 to R 6 each represents a hydrogen atom, an alkyl group having 1 to 5 carbon atoms, or a halogen atom.
- R 11 and R 12 each independently represent a halogen atom, an alkyl group having 1 to 6 carbon atoms, or an alkoxy group having 1 to 6 carbon atoms.
- Z is a single bond, an alkylene group having 1 to 8 carbon atoms, an alkylidene group having 2 to 8 carbon atoms, a cycloalkylene group having 5 to 15 carbon atoms, a cycloalkylidene group having 5 to 15 carbon atoms, a fluorenediyl group, a carbon
- a reaction liquid containing a polycarbonate oligomer is separated into an organic solvent phase containing a polycarbonate oligomer and an aqueous phase, generation of an intermediate layer can be reduced. This improves the production efficiency of branched polycarbonate.
- the method for producing a branched polycarbonate of the present invention comprises a step (a) of obtaining a reaction solution by subjecting an alkaline aqueous solution of dihydric phenol, phosgene and a branching agent to a phosgenation reaction in the presence of an organic solvent, and the step (a).
- a method for producing a branched polycarbonate comprising a step (d) of obtaining a reaction solution containing
- the addition amount of the polymerization catalyst added to) is x mol / hr
- the chloroformate group amount of the polycarbonate oligomer contained in the reaction liquid extracted from the step (b) is y mol / hr
- x / y is set to 0.0035 or more.
- Step (a) is a step of obtaining a reaction solution by subjecting an alkaline aqueous solution of dihydric phenol, phosgene and a branching agent to a phosgenation reaction in the presence of an organic solvent.
- the raw materials and reaction conditions used in this step (a) will be described.
- dihydric phenol used for the production of polycarbonate is used.
- dihydric phenol it is preferable to use a dihydric phenol represented by the following general formula (1).
- R 11 and R 12 each independently represent a halogen atom, an alkyl group having 1 to 6 carbon atoms, or an alkoxy group having 1 to 6 carbon atoms.
- Z is a single bond, an alkylene group having 1 to 8 carbon atoms, an alkylidene group having 2 to 8 carbon atoms, a cycloalkylene group having 5 to 15 carbon atoms, a cycloalkylidene group having 5 to 15 carbon atoms, a fluorenediyl group, a carbon
- the dihydric phenol represented by the general formula (1) is not particularly limited, but 2,2-bis (4-hydroxyphenyl) propane [common name: bisphenol A] is preferable.
- dihydric phenols other than bisphenol A include bis (4-hydroxyphenyl) methane, 1,1-bis (4-hydroxyphenyl) ethane, 2,2-bis (4-hydroxyphenyl) butane, 2,2 -Bis (4-hydroxyphenyl) octane, bis (4-hydroxyphenyl) phenylmethane, bis (4-hydroxyphenyl) diphenylmethane, 2,2-bis (4-hydroxy-3-methylphenyl) propane, bis (4- Hydroxyphenyl) naphthylmethane, 1,1-bis (4-hydroxy-t-butylphenyl) propane, 2,2-bis (4-hydroxy-3-bromophenyl) propane, 2,2-bis (4-hydroxy-) 3,5-dimethylphenyl) propane, 2,2-bis (4-hydroxy-3-chlorophenyl) Bis
- the dihydric phenol is used as an aqueous alkali solution.
- the alkali used at this time include alkali hydroxides, particularly strong basic hydroxides such as sodium hydroxide and potassium hydroxide. it can.
- the alkali concentration of the alkaline aqueous solution usually 1 to 15% by mass is preferably used.
- the content of the dihydric phenol in the alkaline aqueous solution is usually selected in the range of 0.5 to 20% by mass.
- the phosgene used in step (a) is usually obtained by reacting chlorine and carbon monoxide with a ratio of 1.01 to 1.3 moles of carbon monoxide per mole of chlorine using activated carbon as a catalyst. Compound.
- phosgene gas when used as phosgene gas, phosgene gas containing about 1 to 30% by volume of unreacted carbon monoxide can be used. Also, phosgene in a liquefied state can be used.
- the branching agent used in the step (a) is not particularly limited, and a known branching agent can be used.
- known branching agents by using a compound represented by the following general formula (I), it is possible to obtain a branched polycarbonate that hardly causes drawdown during melting.
- R represents a hydrogen atom or an alkyl group having 1 to 5 carbon atoms
- R 1 to R 6 each represents a hydrogen atom, an alkyl group having 1 to 5 carbon atoms, or a halogen atom.
- examples of the alkyl group having 1 to 5 carbon atoms as R include a methyl group, an ethyl group, an n-propyl group, an n-butyl group, and an n-pentyl group.
- examples of the alkyl group having 1 to 5 carbon atoms in which R 1 to R 6 are 1 to 5 carbon atoms include a methyl group, an ethyl group, an n-propyl group, an n-butyl group, an n-pentyl group, and the like, and a halogen atom Is a chlorine atom, a bromine atom, a fluorine atom, or the like.
- branching agent of the general formula (I) examples include 1,1,1-tris (4-hydroxyphenyl) -methane; 1,1,1-tris (4-hydroxyphenyl) -ethane; , 1-tris (4-hydroxyphenyl) -propane; 1,1,1-tris (2-methyl-4-hydroxyphenyl) -methane; 1,1,1-tris (2-methyl-4-hydroxyphenyl) 1, ethane-tris (3-methyl-4-hydroxyphenyl) -methane; 1,1,1-tris (3-methyl-4-hydroxyphenyl) -ethane; 1,1,1-tris (3,5-dimethyl-4-hydroxyphenyl) -methane; 1,1,1-tris (3,5-dimethyl-4-hydroxyphenyl) -ethane; 1,1,1-tris (3-chloro-4 -Hydroxyphenyl)- 1,1,1-tris (3-chloro-4-hydroxyphenyl) ethane; 1,1,1
- branching agents represented by the general formula (I) 1,1,1-tris (4-hydroxyphenyl) -ethane [hereinafter sometimes referred to as THPE]. It is particularly preferable from the viewpoint of the branching property of the branched polycarbonate.
- organic solvent used in the step (a) examples include a solvent that dissolves the polycarbonate oligomer and the branched polycarbonate.
- organic solvent used in the step (a) examples include halogenated hydrocarbon solvents such as dichloromethane (methylene chloride), dichloroethane, trichloroethane, tetrachloroethane, pentachloroethane, hexachloroethane, dichloroethylene, chlorobenzene, and dichlorobenzene, with dichloromethane (methylene chloride) being particularly preferred.
- the alkaline aqueous solution of dihydric phenol and the branching agent and phosgene react vigorously and generate heat. Therefore, it is desirable to cool the reaction product to 0 to 50 ° C. in order to suppress side reactions. . Therefore, as the reactor used in the step (a), it is preferable to use a reactor equipped with a cooling facility for cooling the reaction product.
- the phosgenation reaction may be performed in such a state that the reaction solution becomes a turbulent state in the reactor. preferable.
- the mixed reactor is preferably a static mixer, that is, a static mixer.
- the static mixer is preferably a tubular reactor having therein an element having an action of dividing, converting, and inverting the fluid, and the element generally has a shape obtained by twisting a rectangular plate by 180 degrees. .
- the reaction mixture introduced into the reactor is divided into two parts each time it passes through one element. Further, the reaction mixture fluid or the reaction product fluid is rearranged from the tube center to the wall and from the tube wall to the center along the twisted surface in the element. In addition, the direction of rotation of the fluid is changed for each element, and the fluid is agitated by turbulent flow due to a sudden reversal of inertial force.
- the tubular static mixer described above bubbles in the liquid are miniaturized in the reactor, and the contact interface becomes larger, thereby dramatically increasing the reaction efficiency.
- step (a) an alkaline aqueous solution of dihydric phenol, phosgene, a branching agent and an organic solvent are introduced and mixed to cause a phosgenation reaction.
- the amount of the organic solvent used is the same as that of the organic solvent phase. It is desirable to select so that the volume ratio of the aqueous phase is 5/1 to 1/7, preferably 2/1 to 1/4.
- phosgene should be used in an excess amount so that the amount of phosgene used is usually 1.05 to 1.5 mol, preferably 1.1 to 1.3 mol, per mol of dihydric phenol. Is preferred.
- the molar ratio of dihydric phenol and branching agent used is usually a molar ratio of dihydric phenol: branching agent in the range of 99: 1 to 90:10, preferably 98: 2 to 92: 8. It is preferable to use at a molar ratio within the range.
- the branching agent differs depending on the branching agent used, the branching agent represented by the general formula (I) can be dissolved in an alkaline aqueous solution, so that it is desirable to introduce it by dissolving in an alkaline aqueous solution.
- the reaction of the dihydric phenol and branching agent end groups is chloroformated with phosgene and the reaction where phosgene is decomposed with alkali, generating heat and raising the temperature of the reaction product. It is preferable to cool the product so that the temperature is 0 to 80 ° C., preferably 5 to 70 ° C.
- the tubular static mixer described above is used as a reactor, an exothermic reaction starts from the junction of the alkaline aqueous solution of dihydric phenol and the branching agent and phosgene. Therefore, it is preferable to cool at this junction.
- reaction product flows through the reactor of the tubular static mixer to the outlet of the reactor, phosgene is consumed and no intense reaction heat is generated.
- the main purpose of the reaction in this step (a) is to chloroformate the end groups of the dihydric phenol and the branching agent with phosgene, and the oligomerization reaction hardly proceeds.
- Step (b) is a step of obtaining a reaction solution containing a polycarbonate oligomer by adding an alkaline aqueous solution of a dihydric phenol and a polymerization catalyst to the reaction solution obtained from the step (a).
- the raw materials and reaction conditions used in this step (b) will be described.
- the oligomerization reaction is performed in the step (b) to increase the molecular weight to produce a polycarbonate oligomer.
- an alkaline aqueous solution of a dihydric phenol and a polymerization catalyst are added to the reaction solution obtained from the step (a) to perform an oligomerization reaction.
- the alkaline aqueous solution of the dihydric phenol described in the step (a) is used.
- a known catalyst used during the interfacial polymerization of the polycarbonate resin can be used.
- a phase transfer catalyst such as a tertiary amine or a salt thereof, a quaternary ammonium salt, or a quaternary phosphonium salt can be preferably used.
- the tertiary amine include triethylamine, tributylamine, N, N-dimethylcyclohexylamine, pyridine, dimethylaniline and the like, and examples of the tertiary amine salt include hydrochlorides and bromates of these tertiary amines. Etc.
- Examples of the quaternary ammonium salt include trimethylbenzylammonium chloride, triethylbenzylammonium chloride, tributylbenzylammonium chloride, trioctylmethylammonium chloride, tetrabutylammonium chloride, and tetrabutylammonium bromide.
- Examples thereof include butylphosphonium chloride and tetrabutylphosphonium bromide.
- These catalysts may be used alone or in combination of two or more. Among the catalysts, tertiary amines are preferable, and triethylamine is particularly preferable. These catalysts can be introduced as they are in a liquid state or dissolved in an organic solvent or water. Moreover, a solid-state thing can be dissolved and introduce
- a stirring tank is generally used as the reactor used in the step (b).
- the stirring tank is not particularly limited as long as it is a tank-type stirring tank having a stirrer.
- the reaction liquid obtained from the step (a) is introduced into the reactor for proceeding with the oligomerization reaction.
- the reaction solution obtained from step (a) the remaining amount of unreacted dihydric phenol and the remaining alkali component is small, and in order to proceed with the oligomerization reaction, dihydric phenol and alkali component are added. It is necessary to react.
- the oligomerization reaction of the step (b) is a compound in which the dihydric phenol and the end group of the branching agent contained in the reaction solution obtained from the step (a) are chloroformated with phosgene in the reactor used. It proceeds by reacting with dihydric phenol in the presence of alkali.
- the dihydric phenol is introduced into the reactor with an alkaline aqueous solution of the dihydric phenol prepared in advance used in step (a), or in addition to this, The oligomerization reaction can be advanced by introducing the prepared aqueous alkaline solution into the reactor.
- the aqueous phase [the aqueous phase obtained in the step (e) described later] of the reaction solution containing the branched polycarbonate obtained after the polycondensation step is separated into an organic solvent phase and an aqueous phase. And it can introduce
- the aqueous phase obtained in the step (e) contains unreacted dihydric phenol and alkali, and the dihydric phenol and alkali can be effectively utilized by recycling the aqueous phase.
- the aqueous phase after the polycondensation step may include sodium carbonate generated by the decomposition reaction with sodium hydroxide without using the chloroformate group of the polycarbonate oligomer during the polycondensation step.
- the concentration of the dihydric phenol added to the step (b) is usually 0.05 to 0.15 mol / liter, preferably 0.06 to 0.12 mol / liter, more preferably 0.06 to 0.08 mol.
- the alkali concentration added in the step (b) is usually 0.03 to 0.25 mol / liter, preferably 0.05 to 0.22 mol / liter, more preferably 0. It is desirable to add as 10 to 0.22 mol / liter.
- the amount of the organic solvent used in the reaction solution in this step (b) is usually such that the volume ratio of the organic phase to the aqueous phase is preferably 5/1 to 1/7, more preferably 2/1 to 1/4. Is selected.
- Step (b) is a step of obtaining a reaction liquid containing a polycarbonate oligomer, but preferably the upper limit of the weight average molecular weight is 5000, and the lower limit is usually about 500.
- a terminal terminator so that the weight average molecular weight of the polycarbonate oligomer is 5000 or less. By adding the terminal stopper, it becomes easy to adjust the weight average molecular weight of the polycarbonate oligomer in the step (b) to 5000 or less.
- the end terminator is not particularly limited, and those used for the production of polycarbonate can be used.
- examples of the compound used for the terminal stopper include phenol, p-cresol, p-tert-butylphenol, p-tert-octylphenol, p-cumylphenol, 3-pentadecylphenol, bromophenol, Examples thereof include monohydric phenols such as tribromophenol and nonylphenol. Among these, at least one selected from p-tert-butylphenol, p-cumylphenol, and phenol is preferable from the viewpoints of economy and availability.
- These end terminators may be dissolved in an organic solvent such as methylene chloride, added to the reaction solution obtained from step (a), and introduced into step (b) or used in step (b). May be added directly to the reactor to be introduced.
- the temperature in the reactor is usually 5 to 50 ° C., preferably 5 to 40 ° C., and the reaction is carried out. Stirring is performed under relatively gentle conditions of laminar flow.
- the residence time of the reaction solution in the reactor varies depending on the molecular weight of the target polycarbonate oligomer, the properties of the reaction solution obtained from step (a), etc., but is generally 15 to 60 minutes.
- the amount of the polymerization catalyst added in the step (b) is x mol / hr, and the chloroformate group of the polycarbonate oligomer contained in the reaction solution obtained from the step (b).
- the amount is y mol / hr, (x / y) needs to be 0.0035 or more.
- (x / y) is less than 0.0035, when the reaction liquid obtained from the step (b) is separated into an organic solvent phase containing a polycarbonate oligomer and an aqueous phase in the step (c), the intermediate phase is This is unfavorable because a large amount of the reaction liquid is separated and the separation of the reaction liquid deteriorates, and the production efficiency of the branched polycarbonate is lowered.
- (X / y) is preferably 0.0038 or more, and more preferably 0.0042 or more.
- the upper limit of (x / y) is usually 0.023 or less from the viewpoint that the effect of reducing the amount of intermediate phase generated does not change.
- the step (b) is usually performed according to the amount of chloroformate group of the polycarbonate oligomer contained in the reaction liquid extracted from the step (b). What is necessary is just to adjust the addition amount of the polymerization catalyst added.
- the measurement of the amount of chloroformate groups of the polycarbonate oligomer contained in the reaction solution obtained from the step (b) is performed by sampling the reaction solution containing the polycarbonate oligomer obtained from the step (b), stationary separation, centrifugation, etc.
- the organic solvent phase containing the polycarbonate oligomer and the aqueous phase are separated to obtain the chloroformate group concentration of the polycarbonate oligomer contained in the obtained organic solvent phase, and the chloroformate group concentration and the step (b) are obtained.
- the molar amount per unit time can be determined from the amount (flow rate) of the organic solvent phase containing the polycarbonate oligomer separated from the reaction solution obtained.
- reaction liquid obtained from the step (b) is separated in the step (c), and the molar amount per unit time is determined from the extraction amount (flow rate) of the organic solvent phase containing the separated polycarbonate oligomer and the concentration of the chloroformate group. The amount can also be determined.
- Step (c) is a step of separating the reaction liquid containing the polycarbonate oligomer obtained in the step (b) into an organic solvent phase containing a polycarbonate oligomer and an aqueous phase.
- a stationary separation tank is preferably used as a device used in the step (c).
- the reaction liquid containing the polycarbonate oligomer obtained in the step (b) is introduced into a stationary separation tank and separated into an organic solvent phase containing the polycarbonate oligomer and an aqueous phase due to a difference in specific gravity.
- the organic solvent phase containing the lower layer polycarbonate oligomer is continuously or intermittently extracted from the lower side of the stationary separation tank.
- the upper water phase is withdrawn continuously or intermittently, and the level of each phase in the stationary separation tank is kept within a certain level range.
- the organic solvent phase containing the polycarbonate oligomer is continuously or intermittently extracted from the lower side of the stationary separation tank.
- the polycarbonate oligomer chloro contained in the organic solvent phase is extracted.
- the formate group concentration is obtained, and the value of (x / y) can be obtained from the extracted amount.
- Step (d) is a step of obtaining a reaction solution containing a branched polycarbonate by reacting the organic solvent phase containing the polycarbonate oligomer separated in step (c) with an alkaline aqueous solution of a dihydric phenol.
- a polycarbonate oligomer and a dihydric phenol are subjected to a polycondensation reaction in the presence of an alkaline aqueous solution and an organic solvent, and the molecular weight of the branched polycarbonate is adjusted to a target molecular weight range.
- the polycondensation reaction is carried out until the molecular weight of the obtained branched polycarbonate is usually in the range of about 10,000 to 50,000 in terms of viscosity average molecular weight.
- an organic solvent phase containing the polycarbonate oligomer separated in the step (c), an optionally used end terminator, an optionally used catalyst, an organic solvent, an alkaline aqueous solution, and a dihydric phenol An aqueous alkali solution is mixed and interfacial polycondensation is usually carried out at a temperature in the range of 0 to 50 ° C., preferably 20 to 40 ° C.
- Examples of the alkali, the organic solvent, the terminal terminator, and the catalyst in the aqueous alkali solution used in the step (d) include the same as those described in the step (a) or (b).
- the amount of the organic solvent used in the interfacial polycondensation is usually such that the volume ratio of the organic phase to the aqueous phase is preferably 7/1 to 1/1, more preferably 5/1 to It is selected to be 2/1.
- the reactor used in the step (d) can complete the reaction with only one reactor depending on the capability of the reactor, but if necessary, the reaction of the second subsequent reactor can be further performed. And a plurality of reactors such as a third reactor can be constructed and used.
- a stirring tank As these reactors, a stirring tank, a multi-stage tower type stirring tank, a non-stirring tank, a static mixer, a line mixer, an orifice mixer, piping, and the like can be used. These reactors may be arbitrarily combined and used as a plurality of reactors.
- a reaction liquid containing a branched polycarbonate is obtained by steps (a) to (d).
- the method for producing a branched polycarbonate according to the present invention further includes the following step (e), and at least a part of the aqueous phase containing unreacted dihydric phenol from the following step (e) is added to step (b). It is preferably used as an alkaline aqueous solution of dihydric phenol.
- step (e) will be described.
- Step (e) is a step of separating the reaction solution containing the branched polycarbonate obtained in the step (d) into an organic solvent phase containing the branched polycarbonate and an aqueous phase containing unreacted dihydric phenol.
- examples of the equipment used for separating the organic solvent phase containing the branched polycarbonate and the aqueous phase containing the unreacted dihydric phenol include a stationary separation tank and a centrifuge. .
- the organic solvent phase containing the branched polycarbonate separated in this step (e) is subjected to alkali washing, acid washing and pure water washing in this order to obtain an organic solvent phase containing the branched polycarbonate purified.
- the organic solvent phase containing the purified polycarbonate can be concentrated as necessary to obtain an organic solvent solution containing the purified polycarbonate, which can be kneaded or subjected to hot water granulation to obtain a branched polycarbonate powder. . Since the organic solvent remains in the obtained branched polycarbonate powder, a branched polycarbonate powder from which the organic solvent has been removed can be obtained by performing a drying treatment such as a heat treatment. The obtained branched polycarbonate powder can be pelletized using a pelletizer or the like to form various molded products.
- the aqueous phase separated in step (e) contains unreacted dihydric phenol and alkali, and from the viewpoint of effectively using raw materials, the whole or a part thereof is recycled in step (b). It is preferable.
- the weight average molecular weight (Mw) was determined by using THF (tetrahydrofuran) as a developing solvent, GPC [column: TOSOH TSK-GEL MULTIPIORE HXL-M (2) + Shodex KF801 (1)], temperature 40 ° C., flow rate 1.0 ml / Minute, detector: RI], and measured as a standard polystyrene equivalent molecular weight (weight average molecular weight: Mw).
- Example 1 ⁇ Production of polycarbonate oligomer> The polycarbonate oligomer was produced according to the flow shown in FIG. First, a 6.0% by mass sodium hydroxide aqueous solution was prepared, and further bisphenol A (abbreviated as BPA) was dissolved therein to obtain a 13.5% by mass (in terms of solids) BPA sodium hydroxide aqueous solution. Prepared. Next, p-tert-butylphenol (PTBP) was dissolved in methylene chloride to prepare a 24 mass% solution.
- BPA bisphenol A
- PTBP p-tert-butylphenol
- the BPA sodium hydroxide aqueous solution was flowed at a flow rate of 36 liters / hr, methylene chloride was flowed at a flow rate of 15.4 liters / hr, and 1,1 as a branching agent.
- 1-tris (4-hydroxyphenyl) -ethane (abbreviated as THPE) with 5.1% by weight sodium hydroxide was prepared, and 11% by weight (in terms of solids) of THPE sodium hydroxide aqueous solution was prepared. It was continuously supplied at a flow rate of 0.7 liter / hr.
- the molar ratio of dihydric phenol (BPA): branching agent (THPE) is 98.9: 1.1.
- BPA dihydric phenol
- THPE branching agent
- phosgene is continuously blown into the tubular reactor at a flow rate of 3.1 kg / hr to carry out a phosgenation reaction, a PTBP solution is supplied at a flow rate of 310 ml / hr, and a phosgenation reaction product is supplied. A containing reaction solution was obtained.
- the tubular reactor was cooled so that the temperature of the reaction product at the outlet of the tubular reactor was 30 ° C.
- phosgene was separately synthesized from carbon monoxide (CO) and chlorine (Cl 2 ).
- step (b) having a stirrer with an internal volume of 100 liters, 210 ml / hr of the above reaction solution and a 3% by mass aqueous solution of triethylamine (abbreviated as TEA) prepared in advance as a catalyst.
- TEA triethylamine
- step (d) is continuously supplied, and the reaction liquid obtained from the polycondensation reaction described later [step (d)] is separated into an aqueous phase and an organic solvent phase containing polycarbonate [ 15.7 liters / hr of the recycled aqueous phase obtained from the step (e)] was introduced into the oligomerization reactor to carry out the oligomerization reaction.
- BPA in the aqueous phase before introduction into the oligomerization reactor “the aqueous phase after joining the recycled aqueous phase from [Step (e)] and the additional aqueous phase (pure water) for concentration adjustment”]
- the concentration was 0.07 mol / liter
- the sodium hydroxide concentration was 0.13 mol / liter
- the sodium carbonate concentration was 0.08 mol / liter.
- the inside of this oligomerization reactor was rotated at 350 rpm, and the oligomerization reaction was performed in a laminar flow state.
- the reaction liquid extracted from the bottom of the oligomerization reactor is continuously passed through a transfer pipe [manufactured by SUS, pipe diameter 12.7 mm (1/2 inch)] into a horizontal stationary separation tank (inner diameter 350 mm, inner volume 100 liters).
- the aqueous phase and the organic solvent phase were separated [step (c)].
- the reaction liquid containing the polycarbonate oligomer was separated into an aqueous phase and an organic solvent phase in a horizontal stationary separation tank.
- the organic solvent phase was continuously withdrawn from the horizontal stationary separation tank at a flow rate of 20 l / hr, and the chloroformate group concentration in the withdrawn organic solvent phase was 0.72 mol / l.
- the weight average molecular weight of the polycarbonate oligomer in the organic solvent phase was 3,100.
- the aqueous phase and the organic solvent phase in the horizontal stationary separation tank were observed, and an intermediate phase was slightly generated between the aqueous phase and the organic solvent phase.
- the thickness of the phase is about 9.0 mm (corresponding to 3.2 liters), and the thickness of the intermediate phase does not increase even when continuous stationary separation is performed.
- the water content in the organic solvent phase after separation was 2000 mass ppm, and the solid content in the water phase after separation was less than 10 mass ppm.
- a polycondensation reaction was performed in the step (d) in the flow shown in FIG. 1 using an organic solvent phase (sometimes abbreviated as PCO) containing a polycarbonate oligomer separated from the horizontal stationary separation tank.
- the organic solvent phase (PCO) containing the polycarbonate oligomer was 20 liters / hr
- the sodium hydroxide solution of BPA used for the production of the polycarbonate oligomer
- a TEA aqueous solution having a concentration of 3% by mass as a catalyst was used.
- the polycondensation reaction was carried out by introducing into the polycondensation reactor of step (d) at a flow rate.
- the reactor used for the polycondensation reactor two reactors, a line mixer and a tower reactor, were used.
- the reaction mixture overflowed from the top of the column reactor was allowed to stand and separated into an aqueous phase and an organic solvent phase [step (e)].
- the total amount of the separated aqueous phase was introduced into the oligomerization reactor of step (b) and recycled.
- the obtained organic solvent phase was washed with a sodium hydroxide aqueous solution adjusted to a pH of 13.5, a hydrochloric acid aqueous solution adjusted to a pH of 1.5, and pure water, respectively.
- a methylene chloride solution of polycarbonate was obtained. Methylene chloride was removed by evaporation from the resulting methylene chloride solution of the branched polycarbonate with a kneader to obtain a branched polycarbonate powder. Furthermore, the remaining methylene chloride was dried by heating to remove it to 100 ppm or less to obtain a white branched polycarbonate powder. It was 23,000 when the viscosity average molecular weight (Mv) was measured about this powder.
- the weight average molecular weight of the polycarbonate oligomer in the organic solvent phase is 3,100, the water content in the separated organic solvent phase is 2000 ppm by mass, and the solid content in the separated aqueous phase is less than 10 ppm by mass. there were.
- the weight average molecular weight of the polycarbonate oligomer in the organic solvent phase is 3,100, the water content in the separated organic solvent phase is 2000 ppm by mass, and the solid content in the separated aqueous phase is less than 10 ppm by mass. there were.
- Example 1 the phosgenation reaction was performed without supplying a sodium hydroxide aqueous solution of THPE during the phosgenation reaction.
- a slight intermediate phase was generated between the aqueous phase and the organic solvent phase at that time, but the thickness of the intermediate phase was about 4 mm (equivalent to 1.4 liters), and the stationary separation was continuously performed.
- the separation of the reaction solution is good in the step of separating the reaction solution containing the polycarbonate oligomer, and the production efficiency of the branched polycarbonate can be increased.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Polyesters Or Polycarbonates (AREA)
Abstract
二価フェノールのアルカリ水溶液、ホスゲン及び分岐剤を有機溶媒の存在下でホスゲン化反応させて、反応液を得る工程(a)と、工程(a)から得られる該反応液に、二価フェノールのアルカリ水溶液及び重合触媒を添加し、ポリカーボネートオリゴマーを含む反応液を得る工程(b)と、工程(b)で得られたポリカーボネートオリゴマーを含む反応液を、ポリカーボネートオリゴマーを含む有機溶媒相と水相に分離する工程(c)と、工程(c)で分離されたポリカーボネートオリゴマーを含む有機溶媒相と二価フェノールのアルカリ水溶液を反応させて、分岐ポリカーボネートを含む反応液を得る工程(d)を有する分岐ポリカーボネートの製造方法であって、工程(b)に添加される重合触媒の添加量をxモル/hrとし、工程(b)から得られる反応液に含まれるポリカーボネートオリゴマーのクロロフォーメート基量をyモル/hrとするときに、(x/y)を0.0035以上とするものである。
Description
本発明は、分岐ポリカーボネートの製造方法に関する。詳しくは、界面重合法による、生産効率に優れた分岐ポリカーボネートの製造方法に関する。
ポリカーボネートは、透明性、耐熱性、機械特性など優れた特徴を有し、OA・家電の筐体や電気・電子分野の部材、レンズなどの光学材料など、幅広い用途に使用されている。この幅広い用途に使用されているポリカーボネートの多くは、二価フェノールとホスゲン等のカーボネート前駆物質を反応させて得られる線状重合体であるが、該重合体は溶融加工条件下ではニュートン流動挙動を示し、ブロー成形、押出し成形、発泡成形を行う際には自重によるドローダウンが生じ易く、特に大型成形の場合には問題が生じる。
このような問題点を改善するために、上記の成形用途には溶融加工条件下で非ニュートン流動性を示し溶融時にドローダウンを生じ難い分岐ポリカーボネートが好ましく採用されている。
このような問題点を改善するために、上記の成形用途には溶融加工条件下で非ニュートン流動性を示し溶融時にドローダウンを生じ難い分岐ポリカーボネートが好ましく採用されている。
この分岐ポリカーボネートを製造する方法として、界面重合法やエステル交換法を用いて製造することが知られている。エステル交換法を用いて製造する場合、原料成分を高温下で溶融させエステル交換反応させて重合させるため、使用される重合触媒による影響等により、得られる分岐ポリカーボネートが着色し易くなる。従って、透明性が必要とされる用途に、エステル交換法を用いて分岐ポリカーボネートを製造することは、好ましくない。
界面重合法を用いて、分岐ポリカーボネートを製造する方法として、特許文献1では、分岐剤を組み込んだポリカーボネートオリゴマーを使用して、このポリカーボネートオリゴマーと二価フェノール類とを反応させて分岐ポリカーボネートを得る方法と、分岐剤を組み込まないポリカーボネートオリゴマーを使用し、このポリカーボネートオリゴマーと分岐剤及び二価フェノール類とを反応させて分岐ポリカーボネートを得る方法の2つの製造方法が開示されている。
前者の分岐剤を組み込んだポリカーボネートオリゴマーを使用して、このポリカーボネートオリゴマーと二価フェノール類とを反応させて分岐ポリカーボネートを得る方法は、後者の分岐剤を組み込まないポリカーボネートオリゴマーを使用し、このポリカーボネートオリゴマーと分岐剤及び二価フェノール類とを反応させて分岐ポリカーボネートを得る方法に比較して、分岐ポリカーボネート中の分岐剤由来の単位がより均一となり、後者の製造方法より物性が均一となるので好ましい方法である。
しかしながら、分岐剤を組み込んだポリカーボネートオリゴマーを使用して、このポリカーボネートオリゴマーと二価フェノール類とを反応させて分岐ポリカーボネートを得る方法は、分岐剤を組み込んだポリカーボネートオリゴマーを連続的に製造するに当り、分岐剤を組み込んだポリカーボネートオリゴマーを含む反応液を、該ポリカーボネートオリゴマーを含む有機溶媒相と水相とに分離させる際に、中間層が発生して分離性が悪化し、生産効率が大きく低下するという問題点があった。
本発明は、界面重合法を用いて、分岐ポリカーボネートを製造するに当り、分岐剤を組み込んだポリカーボネートオリゴマーを使用して、このポリカーボネートオリゴマーと二価フェノール類とを反応させて、生産効率よく分岐ポリカーボネートを製造する方法を提供することを目的とするものである。
本発明者等は、鋭意検討した結果、該ポリカーボネートオリゴマーを製造する際に、添加する重合触媒の添加量と該ポリカーボネートオリゴマーのクロロフォーメート基量との関係を特定な比率とすることにより、上記目的が達成されることを見出し、本発明を完成させるに至った。
すなわち、本発明は、下記[1]~[11]に関する。
すなわち、本発明は、下記[1]~[11]に関する。
[1]二価フェノールのアルカリ水溶液、ホスゲン及び分岐剤を有機溶媒の存在下でホスゲン化反応させて、反応液を得る工程(a)と、前記工程(a)から得られる該反応液に、二価フェノールのアルカリ水溶液及び重合触媒を添加し、ポリカーボネートオリゴマーを含む反応液を得る工程(b)と、前記工程(b)で得られたポリカーボネートオリゴマーを含む反応液を、ポリカーボネートオリゴマーを含む有機溶媒相と水相に分離する工程(c)と、前記工程(c)で分離されたポリカーボネートオリゴマーを含む有機溶媒相と二価フェノールのアルカリ水溶液を反応させて、分岐ポリカーボネートを含む反応液を得る工程(d)を有する分岐ポリカーボネートの製造方法であって、前記工程(b)に添加される重合触媒の添加量をxモル/hrとし、前記工程(b)から得られる反応液に含まれるポリカーボネートオリゴマーのクロロフォーメート基量をyモル/hrとするときに、(x/y)が0.0035以上である、分岐ポリカーボネートの製造方法。
[2]前記ポリカーボネートオリゴマーの重量平均分子量が5000以下である、上記[1]に記載の分岐ポリカーボネートの製造方法。
[3]分岐剤が下記一般式(I)で表わされる化合物である、上記[1]又は[2]に記載の分岐ポリカーボネートの製造方法。
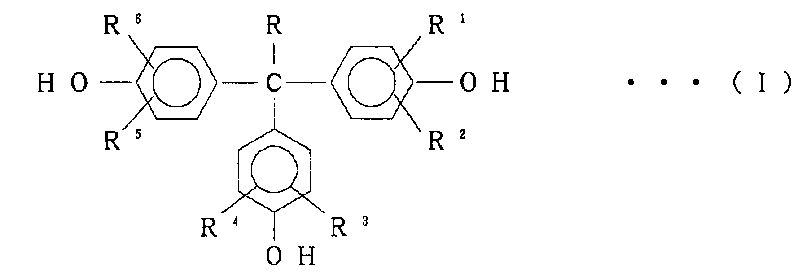
[式中、Rは水素原子又は炭素数1~5のアルキル基であり、R1~R6は、それぞれ水素原子又は炭素数1~5のアルキル基あるいはハロゲン原子を示す。]
[4]前記一般式(I)で表わされる化合物が、1,1,1-トリス(4-ヒドロキシフェニル)エタンである、上記[3]に記載の分岐ポリカーボネートの製造方法。
[5]前記二価フェノールが、下記一般式(1)で表わされる化合物である、上記[1]~[4]のいずれかに記載の分岐ポリカーボネートの製造方法。

[式中、R11及びR12は、それぞれ独立にハロゲン原子、炭素数1~6のアルキル基又は炭素数1~6のアルコキシ基を示す。Zは、単結合、炭素数1~8のアルキレン基、炭素数2~8のアルキリデン基、炭素数5~15のシクロアルキレン基、炭素数5~15のシクロアルキリデン基、フルオレンジイル基、炭素数7~15のアリールアルキレン基、炭素数7~15のアリールアルキリデン基、-S-、-SO-、-SO2-、-O-又は-CO-を示す。a及びbは、それぞれ独立に0~4の整数を示す。]
[6]前記一般式(1)で表わされる化合物が、2,2-ビス(4-ヒドロキシフェニル)プロパンである、上記[5]に記載の分岐ポリカーボネートの製造方法。
[7]前記工程(a)から得られる該反応液に、さらに末端停止剤を添加する、上記[1]~[6]のいずれかに記載の分岐ポリカーボネートの製造方法。
[8]前記工程(d)で得られた分岐ポリカーボネートを含む反応液を、分岐ポリカーボネートを含む有機溶媒相と未反応の二価フェノールを含む水相に分離する工程(e)を有する、上記[1]~[7]のいずれかに記載の分岐ポリカーボネートの製造方法。
[9]前記工程(b)に添加される二価フェノールのアルカリ水溶液として、前記工程(e)で分離された未反応の二価フェノールを含む水相の少なくとも一部を使用する、上記[8]に記載の分岐ポリカーボネートの製造方法。
[10]前記工程(b)から抜き出される反応液に含まれるポリカーボネートオリゴマーのクロロフォーメート基量を、前記工程(c)で分離されたポリカーボネートオリゴマーを含む有機溶媒相の単位時間当たりのモル量とする、上記[1]~[9]のいずれかに記載の分岐ポリカーボネートの製造方法。
[11]前記重合触媒がトリエチルアミンである、上記[1]~[10]のいずれかに記載の分岐ポリカーボネートの製造方法。
[2]前記ポリカーボネートオリゴマーの重量平均分子量が5000以下である、上記[1]に記載の分岐ポリカーボネートの製造方法。
[3]分岐剤が下記一般式(I)で表わされる化合物である、上記[1]又は[2]に記載の分岐ポリカーボネートの製造方法。
[式中、Rは水素原子又は炭素数1~5のアルキル基であり、R1~R6は、それぞれ水素原子又は炭素数1~5のアルキル基あるいはハロゲン原子を示す。]
[4]前記一般式(I)で表わされる化合物が、1,1,1-トリス(4-ヒドロキシフェニル)エタンである、上記[3]に記載の分岐ポリカーボネートの製造方法。
[5]前記二価フェノールが、下記一般式(1)で表わされる化合物である、上記[1]~[4]のいずれかに記載の分岐ポリカーボネートの製造方法。
[式中、R11及びR12は、それぞれ独立にハロゲン原子、炭素数1~6のアルキル基又は炭素数1~6のアルコキシ基を示す。Zは、単結合、炭素数1~8のアルキレン基、炭素数2~8のアルキリデン基、炭素数5~15のシクロアルキレン基、炭素数5~15のシクロアルキリデン基、フルオレンジイル基、炭素数7~15のアリールアルキレン基、炭素数7~15のアリールアルキリデン基、-S-、-SO-、-SO2-、-O-又は-CO-を示す。a及びbは、それぞれ独立に0~4の整数を示す。]
[6]前記一般式(1)で表わされる化合物が、2,2-ビス(4-ヒドロキシフェニル)プロパンである、上記[5]に記載の分岐ポリカーボネートの製造方法。
[7]前記工程(a)から得られる該反応液に、さらに末端停止剤を添加する、上記[1]~[6]のいずれかに記載の分岐ポリカーボネートの製造方法。
[8]前記工程(d)で得られた分岐ポリカーボネートを含む反応液を、分岐ポリカーボネートを含む有機溶媒相と未反応の二価フェノールを含む水相に分離する工程(e)を有する、上記[1]~[7]のいずれかに記載の分岐ポリカーボネートの製造方法。
[9]前記工程(b)に添加される二価フェノールのアルカリ水溶液として、前記工程(e)で分離された未反応の二価フェノールを含む水相の少なくとも一部を使用する、上記[8]に記載の分岐ポリカーボネートの製造方法。
[10]前記工程(b)から抜き出される反応液に含まれるポリカーボネートオリゴマーのクロロフォーメート基量を、前記工程(c)で分離されたポリカーボネートオリゴマーを含む有機溶媒相の単位時間当たりのモル量とする、上記[1]~[9]のいずれかに記載の分岐ポリカーボネートの製造方法。
[11]前記重合触媒がトリエチルアミンである、上記[1]~[10]のいずれかに記載の分岐ポリカーボネートの製造方法。
本発明の分岐ポリカーボネートの製造方法によれば、ポリカーボネートオリゴマーを含む反応液を、ポリカーボネートオリゴマーを含む有機溶媒相と水相に分離させる際に、中間層の発生を低減することができるので、分離性がよくなり、分岐ポリカーボネートの生産効率を上げることができる。
本発明の分岐ポリカーボネートの製造方法は、二価フェノールのアルカリ水溶液、ホスゲン及び分岐剤を有機溶媒の存在下でホスゲン化反応させて、反応液を得る工程(a)と、前記工程(a)から得られる該反応液に、二価フェノールのアルカリ水溶液及び重合触媒を添加し、ポリカーボネートオリゴマーを含む反応液を得る工程(b)と、前記工程(b)で得られたポリカーボネートオリゴマーを含む反応液を、ポリカーボネートオリゴマーを含む有機溶媒相と水相に分離する工程(c)と、前記工程(c)で分離されたポリカーボネートオリゴマーを含む有機溶媒相と二価フェノールのアルカリ水溶液を反応させて、分岐ポリカーボネートを含む反応液を得る工程(d)を有する分岐ポリカーボネートの製造方法であって、前記工程(b)に添加される重合触媒の添加量をxモル/hrとし、前記工程(b)から抜き出される反応液に含まれるポリカーボネートオリゴマーのクロロフォーメート基量をyモル/hrとするときに、(x/y)を0.0035以上とするものである。
以下、本発明の分岐ポリカーボネートの製造方法について詳細に説明する。なお、本明細書において、好ましいとされている規定は任意に採用することができ、好ましいもの同士の組み合わせはより好ましいと言える。
以下、本発明の分岐ポリカーボネートの製造方法について詳細に説明する。なお、本明細書において、好ましいとされている規定は任意に採用することができ、好ましいもの同士の組み合わせはより好ましいと言える。
[工程(a)]
工程(a)は、二価フェノールのアルカリ水溶液、ホスゲン及び分岐剤を有機溶媒の存在下でホスゲン化反応させて、反応液を得る工程である。この工程(a)に使用される原料、反応条件について説明する。
工程(a)は、二価フェノールのアルカリ水溶液、ホスゲン及び分岐剤を有機溶媒の存在下でホスゲン化反応させて、反応液を得る工程である。この工程(a)に使用される原料、反応条件について説明する。
<二価フェノールのアルカリ水溶液>
二価フェノールとしては、ポリカーボネートの製造に使用される二価フェノールが用いられる。前記二価フェノールとしては、下記一般式(1)で表される二価フェノールを用いることが好ましい。

上記一般式(1)中、R11及びR12は、それぞれ独立にハロゲン原子、炭素数1~6のアルキル基又は炭素数1~6のアルコキシ基を示す。Zは、単結合、炭素数1~8のアルキレン基、炭素数2~8のアルキリデン基、炭素数5~15のシクロアルキレン基、炭素数5~15のシクロアルキリデン基、フルオレンジイル基、炭素数7~15のアリールアルキレン基、炭素数7~15のアリールアルキリデン基、-S-、-SO-、-SO2-、-O-又は-CO-を示す。a及びbは、それぞれ独立に0~4の整数を示す。
二価フェノールとしては、ポリカーボネートの製造に使用される二価フェノールが用いられる。前記二価フェノールとしては、下記一般式(1)で表される二価フェノールを用いることが好ましい。
上記一般式(1)中、R11及びR12は、それぞれ独立にハロゲン原子、炭素数1~6のアルキル基又は炭素数1~6のアルコキシ基を示す。Zは、単結合、炭素数1~8のアルキレン基、炭素数2~8のアルキリデン基、炭素数5~15のシクロアルキレン基、炭素数5~15のシクロアルキリデン基、フルオレンジイル基、炭素数7~15のアリールアルキレン基、炭素数7~15のアリールアルキリデン基、-S-、-SO-、-SO2-、-O-又は-CO-を示す。a及びbは、それぞれ独立に0~4の整数を示す。
一般式(1)で表される二価フェノールとしては、特に限定されないが、2,2-ビス(4-ヒドロキシフェニル)プロパン〔通称:ビスフェノールA〕が好適である。
ビスフェノールA以外の二価フェノールとしては、例えば、ビス(4-ヒドロキシフェニル)メタン、1,1-ビス(4-ヒドロキシフェニル)エタン、2,2-ビス(4-ヒドロキシフェニル)ブタン、2,2-ビス(4-ヒドロキシフェニル)オクタン、ビス(4-ヒドロキシフェニル)フェニルメタン、ビス(4-ヒドロキシフェニル)ジフェニルメタン、2,2-ビス(4-ヒドロキシ-3-メチルフェニル)プロパン、ビス(4-ヒドロキシフェニル)ナフチルメタン、1,1-ビス(4-ヒドロキシ-t-ブチルフェニル)プロパン、2,2-ビス(4-ヒドロキシ-3-ブロモフェニル)プロパン、2,2-ビス(4-ヒドロキシ-3,5-ジメチルフェニル)プロパン、2,2-ビス(4-ヒドロキシ-3-クロロフェニル)プロパン、2,2-ビス(4-ヒドロキシ-3,5-ジクロロフェニル)プロパン、2,2-ビス(4-ヒドロキシ-3,5-ジブロモフェニル)プロパン等のビス(ヒドロキシアリール)アルカン類、1,1-ビス(4-ヒドロキシフェニル)シクロペンタン、1,1-ビス(4-ヒドロキシフェニル)シクロヘキサン、1,1-ビス(4-ヒドロキシフェニル)-3,5,5-トリメチルシクロヘキサン、2,2-ビス(4-ヒドロキシフェニル)ノルボルナン、1,1-ビス(4-ヒドロキシフェニル)シクロドデカン等のビス(ヒドロキシアリール)シクロアルカン類、4,4’-ジヒドロキシジフェニルエーテル、4,4’-ジヒドロキシ-3,3’-ジメチルフェニルエーテル等のジヒドロキシアリールエーテル類、4,4’-ジヒドロキシジフェニルスルフィド、4,4’-ジヒドロキシ-3,3’-ジメチルジフェニルスルフィド等のジヒドロキシジアリールスルフィド類、4,4’-ジヒドロキシジフェニルスルホキシド、4,4’-ジヒドロキシ-3,3’-ジメチルジフェニルスルホキシド等のジヒドロキシジアリールスルホキシド類、4,4’-ジヒドロキシジフェニルスルホン、4,4’-ジヒドロキシ-3,3’-ジメチルジフェニルスルホン等のジヒドロキシジアリールスルホン類、4,4’-ジヒドロキシジフェニル等のジヒドロキシジフェニル類、9,9-ビス(4-ヒドロキシフェニル)フルオレン、9,9-ビス(4-ヒドロキシ-3-メチルフェニル)フルオレン等のジヒドロキシジアリールフルオレン類、1,3-ビス(4-ヒドロキシフェニル)アダマンタン、2,2-ビス(4-ヒドロキシフェニル)アダマンタン、1,3-ビス(4-ヒドロキシフェニル)-5,7-ジメチルアダマンタン等のジヒドロキシジアリールアダマンタン類、4,4’-[1,3-フェニレンビス(1-メチルエチリデン)]ビスフェノール、10,10-ビス(4-ヒドロキシフェニル)-9-アントロン、1,5-ビス(4-ヒドロキシフェニルチオ)-2,3-ジオキサペンタン等が挙げられる。
これらの二価フェノールは、単独で又は二種以上を混合して用いてもよい。
ビスフェノールA以外の二価フェノールとしては、例えば、ビス(4-ヒドロキシフェニル)メタン、1,1-ビス(4-ヒドロキシフェニル)エタン、2,2-ビス(4-ヒドロキシフェニル)ブタン、2,2-ビス(4-ヒドロキシフェニル)オクタン、ビス(4-ヒドロキシフェニル)フェニルメタン、ビス(4-ヒドロキシフェニル)ジフェニルメタン、2,2-ビス(4-ヒドロキシ-3-メチルフェニル)プロパン、ビス(4-ヒドロキシフェニル)ナフチルメタン、1,1-ビス(4-ヒドロキシ-t-ブチルフェニル)プロパン、2,2-ビス(4-ヒドロキシ-3-ブロモフェニル)プロパン、2,2-ビス(4-ヒドロキシ-3,5-ジメチルフェニル)プロパン、2,2-ビス(4-ヒドロキシ-3-クロロフェニル)プロパン、2,2-ビス(4-ヒドロキシ-3,5-ジクロロフェニル)プロパン、2,2-ビス(4-ヒドロキシ-3,5-ジブロモフェニル)プロパン等のビス(ヒドロキシアリール)アルカン類、1,1-ビス(4-ヒドロキシフェニル)シクロペンタン、1,1-ビス(4-ヒドロキシフェニル)シクロヘキサン、1,1-ビス(4-ヒドロキシフェニル)-3,5,5-トリメチルシクロヘキサン、2,2-ビス(4-ヒドロキシフェニル)ノルボルナン、1,1-ビス(4-ヒドロキシフェニル)シクロドデカン等のビス(ヒドロキシアリール)シクロアルカン類、4,4’-ジヒドロキシジフェニルエーテル、4,4’-ジヒドロキシ-3,3’-ジメチルフェニルエーテル等のジヒドロキシアリールエーテル類、4,4’-ジヒドロキシジフェニルスルフィド、4,4’-ジヒドロキシ-3,3’-ジメチルジフェニルスルフィド等のジヒドロキシジアリールスルフィド類、4,4’-ジヒドロキシジフェニルスルホキシド、4,4’-ジヒドロキシ-3,3’-ジメチルジフェニルスルホキシド等のジヒドロキシジアリールスルホキシド類、4,4’-ジヒドロキシジフェニルスルホン、4,4’-ジヒドロキシ-3,3’-ジメチルジフェニルスルホン等のジヒドロキシジアリールスルホン類、4,4’-ジヒドロキシジフェニル等のジヒドロキシジフェニル類、9,9-ビス(4-ヒドロキシフェニル)フルオレン、9,9-ビス(4-ヒドロキシ-3-メチルフェニル)フルオレン等のジヒドロキシジアリールフルオレン類、1,3-ビス(4-ヒドロキシフェニル)アダマンタン、2,2-ビス(4-ヒドロキシフェニル)アダマンタン、1,3-ビス(4-ヒドロキシフェニル)-5,7-ジメチルアダマンタン等のジヒドロキシジアリールアダマンタン類、4,4’-[1,3-フェニレンビス(1-メチルエチリデン)]ビスフェノール、10,10-ビス(4-ヒドロキシフェニル)-9-アントロン、1,5-ビス(4-ヒドロキシフェニルチオ)-2,3-ジオキサペンタン等が挙げられる。
これらの二価フェノールは、単独で又は二種以上を混合して用いてもよい。
前記二価フェノールは、アルカリ水溶液として使用されるが、この際に使用されるアルカリとしては、水酸化アルカリ、特に強塩基性の水酸化物、例えば、水酸化ナトリウム、水酸化カリウムを挙げることができる。アルカリ水溶液のアルカリ濃度としては、通常、1~15質量%のものが好ましく用いられる。また、アルカリ水溶液中の二価フェノールの含有量は、通常0.5~20質量%の範囲で選ばれる。
<ホスゲン>
工程(a)で使用されるホスゲンは、通常、塩素および一酸化炭素を、塩素1モルに対し一酸化炭素1.01~1.3モルの割合で触媒として活性炭を使用して反応させて得られる化合物である。使用するホスゲン中には、ホスゲンガスとして使用する場合、未反応の一酸化炭素を1~30容量%程度含んだホスゲンガスを使用することができる。また、液化状態のホスゲンも使用することができる。
工程(a)で使用されるホスゲンは、通常、塩素および一酸化炭素を、塩素1モルに対し一酸化炭素1.01~1.3モルの割合で触媒として活性炭を使用して反応させて得られる化合物である。使用するホスゲン中には、ホスゲンガスとして使用する場合、未反応の一酸化炭素を1~30容量%程度含んだホスゲンガスを使用することができる。また、液化状態のホスゲンも使用することができる。
<分岐剤>
工程(a)で使用される分岐剤は、特に限定されるものではなく、公知の分岐剤を用いることができる。公知の分岐剤の中でも、下記一般式(I)で表わされる化合物を用いることにより、溶融時にドローダウンを生じ難い分岐ポリカーボネートを得ることができる。
工程(a)で使用される分岐剤は、特に限定されるものではなく、公知の分岐剤を用いることができる。公知の分岐剤の中でも、下記一般式(I)で表わされる化合物を用いることにより、溶融時にドローダウンを生じ難い分岐ポリカーボネートを得ることができる。

[式中、Rは水素原子又は炭素数1~5のアルキル基であり、R1~R6は、それぞれ水素原子又は炭素数1~5のアルキル基あるいはハロゲン原子を示す。]
上記一般式(I)中、Rが炭素数1~5のアルキル基としては、例えば、メチル基、エチル基、n-プロピル基、n-ブチル基、n-ペンチル基などである。また、R1~R6が炭素数1~5のアルキル基としては、例えば、メチル基、エチル基、n-プロピル基、n-ブチル基、n-ペンチル基などであり、ハロゲン原子である場合は、塩素原子、臭素原子、フッ素原子などである。一般式(I)の分岐剤は、具体的には、1,1,1-トリス(4-ヒドロキシフェニル)-メタン;1,1,1-トリス(4-ヒドロキシフェニル)-エタン;1,1,1-トリス(4-ヒドロキシフェニル)-プロパン;1,1,1-トリス(2-メチル-4-ヒドロキシフェニル)-メタン;1,1,1-トリス(2-メチル-4-ヒドロキシフェニル)-エタン;1,1,1-トリス(3-メチル-4-ヒドロキシフェニル)-メタン;1,1,1-トリス(3-メチル-4-ヒドロキシフェニル)-エタン;1,1,1-トリス(3,5-ジメチル-4-ヒドロキシフェニル)-メタン;1,1,1-トリス(3,5-ジメチル-4-ヒドロキシフェニル)-エタン;1,1,1-トリス(3-クロロ-4-ヒドロキシフェニル)-メタン;1,1,1-トリス(3-クロロ-4-ヒドロキシフェニル)エタン;1,1,1-トリス(3,5-ジクロロ-4-ヒドロキシフェニル)-メタン;1,1,1-トリス(3,5-ジクロロ-4-ヒドロキシフェニル)-エタン;1,1,1-トリス(3-ブロモ-4-ヒドロキシフェニル)-メタン;1,1,1-トリス(3-ブロモ-4-ヒドロキシフェニル)-エタン;1,1,1-トリス(3,5-ジブロモ-4-ヒドロキシフェニル)-メタン;1,1,1-トリス(3,5-ジブロモ-4-ヒドロキシフェニル)-エタンなどである。上記一般式(I)の分岐剤の中でも、1,1,1-トリス(4-ヒドロキシフェニル)-エタン[以下、THPEと称することもある。]を用いることが、分岐ポリカーボネートの分岐性の観点から、特に好ましい。
<有機溶媒>
工程(a)で使用される有機溶媒としては、ポリカーボネートオリゴマーおよび分岐ポリカーボネートを溶解する溶媒が挙げられる。具体的にはジクロロメタン(塩化メチレン)、ジクロロエタン、トリクロロエタン、テトラクロロエタン、ペンタクロロエタン、ヘキサクロロエタン、ジクロロエチレン、クロロベンゼン、ジクロロベンゼンなどのハロゲン化炭化水素溶媒が挙げられ、特にジクロロメタン(塩化メチレン)が好ましい。
工程(a)で使用される有機溶媒としては、ポリカーボネートオリゴマーおよび分岐ポリカーボネートを溶解する溶媒が挙げられる。具体的にはジクロロメタン(塩化メチレン)、ジクロロエタン、トリクロロエタン、テトラクロロエタン、ペンタクロロエタン、ヘキサクロロエタン、ジクロロエチレン、クロロベンゼン、ジクロロベンゼンなどのハロゲン化炭化水素溶媒が挙げられ、特にジクロロメタン(塩化メチレン)が好ましい。
<反応器>
工程(a)では、二価フェノールのアルカリ水溶液及び分岐剤とホスゲンとが激しく反応して、発熱を伴うため、副反応を抑制するために反応生成物を0~50℃に冷却することが望ましい。従って、工程(a)に使用する反応器としては、反応生成物を冷却するための冷却設備を備えた反応器を用いることが好ましい。また、二価フェノールのアルカリ水溶液、ホスゲン、分岐剤及び有機溶媒を連続的に反応器に導入した際に、反応器内で反応液が乱流状態となるような状態でホスゲン化反応させることが好ましい。このような反応器としては、混合反応器を用いることが望ましく、混合反応器としては、静止型混合器、即ちスタティックミキサーであることが好ましい。静止型混合器は、流体を分割、転換、反転させる作用を有するエレメントを内部に有した管状の反応器であることが好ましく、エレメントは一般的に、長方形の板を180度ねじった形状を有する。反応器内に導入された反応混合物は、ひとつのエレメントを通過するごとに2分割される。また、反応混合物流体又は反応生成物流体は、エレメント内のねじれ面に沿って管中央部から壁部へ、管壁部から中央部へと並び替えられる。また、流体は、1エレメントごとに回転方向が替わり、急激な慣性力の反転を受け乱流撹拌される。
上記に記載の管状の静止型混合器を反応器として用いた場合、反応器内では、液体中の気泡が微細化され、接触界面が大きくなり、これにより反応効率が飛躍的に高まる。
工程(a)では、二価フェノールのアルカリ水溶液及び分岐剤とホスゲンとが激しく反応して、発熱を伴うため、副反応を抑制するために反応生成物を0~50℃に冷却することが望ましい。従って、工程(a)に使用する反応器としては、反応生成物を冷却するための冷却設備を備えた反応器を用いることが好ましい。また、二価フェノールのアルカリ水溶液、ホスゲン、分岐剤及び有機溶媒を連続的に反応器に導入した際に、反応器内で反応液が乱流状態となるような状態でホスゲン化反応させることが好ましい。このような反応器としては、混合反応器を用いることが望ましく、混合反応器としては、静止型混合器、即ちスタティックミキサーであることが好ましい。静止型混合器は、流体を分割、転換、反転させる作用を有するエレメントを内部に有した管状の反応器であることが好ましく、エレメントは一般的に、長方形の板を180度ねじった形状を有する。反応器内に導入された反応混合物は、ひとつのエレメントを通過するごとに2分割される。また、反応混合物流体又は反応生成物流体は、エレメント内のねじれ面に沿って管中央部から壁部へ、管壁部から中央部へと並び替えられる。また、流体は、1エレメントごとに回転方向が替わり、急激な慣性力の反転を受け乱流撹拌される。
上記に記載の管状の静止型混合器を反応器として用いた場合、反応器内では、液体中の気泡が微細化され、接触界面が大きくなり、これにより反応効率が飛躍的に高まる。
<反応器への各原料の導入比率>
工程(a)に使用する反応器には、二価フェノールのアルカリ水溶液、ホスゲン、分岐剤及び有機溶媒を導入し、混合してホスゲン化反応させるが、有機溶媒の使用量は、有機溶媒相と水相の容量比が5/1~1/7、好ましくは2/1~1/4となるように選定するのが望ましい。また、ホスゲンの使用量は、二価フェノール1モルに対して、通常1.05~1.5モル、好ましくは、1.1~1.3モルとなるように、ホスゲンを過剰に使用することが好ましい。また、二価フェノールと分岐剤との使用モル比率は、通常、二価フェノール:分岐剤が、99:1~90:10の範囲内のモル比率で、好ましくは98:2~92:8の範囲内のモル比率で使用することが好ましい。分岐剤は、使用する分岐剤により相違するが、前記一般式(I)で表わされる分岐剤は、アルカリ水溶液に溶解させることができるので、アルカリ水溶液に溶解させて導入することが望ましい。また、アルカリ水溶液に溶解させることが困難な分岐剤は、塩化メチレン等の有機溶媒に溶解させて導入することが望ましい。
ホスゲン化反応器内では、二価フェノール及び分岐剤の末端基がホスゲンによりクロロフォーメート化される反応やホスゲンがアルカリにより分解される反応により、発熱し反応生成物の温度が高くなるので、反応生成物の温度が0~80℃、好ましくは5~70℃となるように冷却することが好ましい。前述した管状の静止型混合器を反応器として用いた場合、二価フェノールのアルカリ水溶液及び分岐剤とホスゲンとの合流点から発熱反応するので、この合流点においても冷却しておくことが好ましい。管状の静止型混合器の反応器内を反応生成物が反応器出口に流れるに従い、ホスゲンは消費されていき、激しい反応熱も発生しなくなる。この工程(a)での反応の主目的は、二価フェノール及び分岐剤の末端基をホスゲンによりクロロフォーメート化することであり、オリゴマー化反応はほとんど進行していない。
工程(a)に使用する反応器には、二価フェノールのアルカリ水溶液、ホスゲン、分岐剤及び有機溶媒を導入し、混合してホスゲン化反応させるが、有機溶媒の使用量は、有機溶媒相と水相の容量比が5/1~1/7、好ましくは2/1~1/4となるように選定するのが望ましい。また、ホスゲンの使用量は、二価フェノール1モルに対して、通常1.05~1.5モル、好ましくは、1.1~1.3モルとなるように、ホスゲンを過剰に使用することが好ましい。また、二価フェノールと分岐剤との使用モル比率は、通常、二価フェノール:分岐剤が、99:1~90:10の範囲内のモル比率で、好ましくは98:2~92:8の範囲内のモル比率で使用することが好ましい。分岐剤は、使用する分岐剤により相違するが、前記一般式(I)で表わされる分岐剤は、アルカリ水溶液に溶解させることができるので、アルカリ水溶液に溶解させて導入することが望ましい。また、アルカリ水溶液に溶解させることが困難な分岐剤は、塩化メチレン等の有機溶媒に溶解させて導入することが望ましい。
ホスゲン化反応器内では、二価フェノール及び分岐剤の末端基がホスゲンによりクロロフォーメート化される反応やホスゲンがアルカリにより分解される反応により、発熱し反応生成物の温度が高くなるので、反応生成物の温度が0~80℃、好ましくは5~70℃となるように冷却することが好ましい。前述した管状の静止型混合器を反応器として用いた場合、二価フェノールのアルカリ水溶液及び分岐剤とホスゲンとの合流点から発熱反応するので、この合流点においても冷却しておくことが好ましい。管状の静止型混合器の反応器内を反応生成物が反応器出口に流れるに従い、ホスゲンは消費されていき、激しい反応熱も発生しなくなる。この工程(a)での反応の主目的は、二価フェノール及び分岐剤の末端基をホスゲンによりクロロフォーメート化することであり、オリゴマー化反応はほとんど進行していない。
[工程(b)]
工程(b)は、前記工程(a)から得られる該反応液に、二価フェノールのアルカリ水溶液及び重合触媒を添加し、ポリカーボネートオリゴマーを含む反応液を得る工程である。この工程(b)に使用される原料、反応条件について説明する。
工程(b)は、前記工程(a)から得られる該反応液に、二価フェノールのアルカリ水溶液及び重合触媒を添加し、ポリカーボネートオリゴマーを含む反応液を得る工程である。この工程(b)に使用される原料、反応条件について説明する。
前述したとおり、工程(a)では、オリゴマー化反応はほとんど進行していないため、工程(b)でオリゴマー化反応させることで分子量を上げて、ポリカーボネートオリゴマーを製造する。この工程(b)では、工程(a)から得られる反応液に、二価フェノールのアルカリ水溶液及び重合触媒を添加し、オリゴマー化反応を行うが、使用される二価フェノールのアルカリ水溶液としては、工程(a)で説明した二価フェノールのアルカリ水溶液が使用される。
<重合触媒>
工程(b)で使用する重合触媒としては、ポリカーボネート樹脂の界面重合時に使用される公知の触媒を用いることができる。触媒としては、相間移動触媒、例えば三級アミン又はその塩、四級アンモニウム塩、四級ホスホニウム塩などを好ましく用いることができる。三級アミンとしては、例えばトリエチルアミン、トリブチルアミン、N,N-ジメチルシクロヘキシルアミン、ピリジン、ジメチルアニリンなどが挙げられ、また三級アミン塩としては、例えばこれらの三級アミンの塩酸塩、臭素酸塩などが挙げられる。四級アンモニウム塩としては、例えばトリメチルベンジルアンモニウムクロリド、トリエチルベンジルアンモニウムクロリド、トリブチルベンジルアンモニウムクロリド、トリオクチルメチルアンモニウムクロリド、テトラブチルアンモニウムクロリド、テトラブチルアンモニウムブロミドなどが、四級ホスホニウム塩としては、例えばテトラブチルホスホニウムクロリド、テトラブチルホスホニウムブロミドなどが挙げられる。これらの触媒は、それぞれ単独で用いてもよく、二種以上を組み合わせて用いてもよい。前記触媒の中では、三級アミンが好ましく、特にトリエチルアミンが好適である。これらの触媒は、液体状態のものであればそのまま、または有機溶媒や水に溶解させて導入することができる。また固体状態のものは、有機溶媒や水に溶解させて導入することができる。
工程(b)で使用する重合触媒としては、ポリカーボネート樹脂の界面重合時に使用される公知の触媒を用いることができる。触媒としては、相間移動触媒、例えば三級アミン又はその塩、四級アンモニウム塩、四級ホスホニウム塩などを好ましく用いることができる。三級アミンとしては、例えばトリエチルアミン、トリブチルアミン、N,N-ジメチルシクロヘキシルアミン、ピリジン、ジメチルアニリンなどが挙げられ、また三級アミン塩としては、例えばこれらの三級アミンの塩酸塩、臭素酸塩などが挙げられる。四級アンモニウム塩としては、例えばトリメチルベンジルアンモニウムクロリド、トリエチルベンジルアンモニウムクロリド、トリブチルベンジルアンモニウムクロリド、トリオクチルメチルアンモニウムクロリド、テトラブチルアンモニウムクロリド、テトラブチルアンモニウムブロミドなどが、四級ホスホニウム塩としては、例えばテトラブチルホスホニウムクロリド、テトラブチルホスホニウムブロミドなどが挙げられる。これらの触媒は、それぞれ単独で用いてもよく、二種以上を組み合わせて用いてもよい。前記触媒の中では、三級アミンが好ましく、特にトリエチルアミンが好適である。これらの触媒は、液体状態のものであればそのまま、または有機溶媒や水に溶解させて導入することができる。また固体状態のものは、有機溶媒や水に溶解させて導入することができる。
工程(b)に用いられる反応器としては、一般的に攪拌槽が用いられる。攪拌槽としては、攪拌機を有する槽型の攪拌槽であれば特に限定されない。
このオリゴマー化反応を進めるための反応器には、工程(a)から得られた反応液が導入される。工程(a)から得られた反応液中には、未反応の二価フェノール及び残留するアルカリ成分の残存量は少なく、オリゴマー化反応を進めるためには、二価フェノール及びアルカリ成分を追加して反応させることが必要である。
このオリゴマー化反応を進めるための反応器には、工程(a)から得られた反応液が導入される。工程(a)から得られた反応液中には、未反応の二価フェノール及び残留するアルカリ成分の残存量は少なく、オリゴマー化反応を進めるためには、二価フェノール及びアルカリ成分を追加して反応させることが必要である。
工程(b)のオリゴマー化反応は、用いられる反応器内において、工程(a)から得られた反応液中に含まれる二価フェノール及び分岐剤の末端基がホスゲンによりクロロフォーメート化された化合物と二価フェノールとが、アルカリの存在下で反応することにより進行する。本発明のポリカーボネートの製造方法では、前記二価フェノールは、工程(a)で使用される予め調製しておいた二価フェノールのアルカリ水溶液を反応器に導入したり、さらにこれに加えて、予め調製されたアルカリ水溶液を反応器に導入する等して、オリゴマー化反応を進めることができる。
また、別の方法として、重縮合工程後に得られた分岐ポリカーボネートを含む反応液を有機溶媒相と水相に分離したうちの水相[後述する工程(e)で得られた水相]をリサイクルして、工程(b)の反応器に導入して、オリゴマー化反応を進めることができる。工程(e)で得られた水相中には、未反応の二価フェノール及びアルカリが含まれており、該水相をリサイクル使用することにより、二価フェノール及びアルカリを有効活用することができる。なお、重縮合工程後の水相には、重縮合工程時にポリカーボネートオリゴマーのクロロフォーメート基が重合に使用されず水酸化ナトリウムと分解反応することで発生する炭酸ナトリウムが含まれることがある。
工程(b)に添加される二価フェノール濃度は、通常0.05~0.15モル/リットル、好ましくは0.06~0.12モル/リットル、より好ましくは0.06~0.08モル/リットルとして添加することが望ましく、工程(b)に添加されるアルカリ濃度は、通常0.03~0.25モル/リットル、好ましくは0.05~0.22モル/リットル、より好ましくは0.10~0.22モル/リットルとして添加することが望ましい。この工程(b)の反応液中の有機溶媒の使用量は、通常、有機相と水相との容量比が、好ましくは5/1~1/7、より好ましくは2/1~1/4となるように選択される。
工程(b)は、ポリカーボネートオリゴマーを含む反応液を得る工程であるが、好ましくは、重量平均分子量の上限値は5000であり、下限値は、通常、約500程度である。工程(b)では、ポリカーボネートオリゴマーの重量平均分子量を5000以下とするために、末端停止剤を添加することが好ましい。末端停止剤の添加で、工程(b)におけるポリカーボネートオリゴマーの重量平均分子量を5000以下に調整することが容易になる。末端停止剤としては、特に限定されるものではなく、ポリカーボネートの製造に使用されるものを用いることができる。具体的には、末端停止剤に用いられる化合物としては、例えば、フェノール,p-クレゾール,p-tert-ブチルフェノール,p-tert-オクチルフェノール,p-クミルフェノール,3-ペンタデシルフェノール,ブロモフェノール,トリブロモフェノール,ノニルフェノールなどの一価フェノールを挙げることができる。これらの中で、経済性、入手の容易さなどの点から、p-tert-ブチルフェノール、p-クミルフェノールおよびフェノールから選ばれる少なくとも1種が好ましい。これらの末端停止剤は、塩化メチレン等の有機溶媒に溶解させて、工程(a)から得られる反応液に添加して、工程(b)に導入してもよいし、工程(b)に使用される反応器に直接添加して導入してもよい。
工程(b)での反応器内の温度は、通常、5~50℃、好ましくは5~40℃の範囲内の温度に保ち反応させる。攪拌条件は、比較的緩やかな、層流となる条件で攪拌される。反応器内における反応液の滞留時間は、目的とするポリカーボネートオリゴマーの分子量、工程(a)から得られた反応液の性状等によって相違するが、一般的には、15~60分間である。
本発明の分岐ポリカーボネートの製造方法では、工程(b)に添加される重合触媒の添加量をxモル/hrとし、前記工程(b)から得られる反応液に含まれるポリカーボネートオリゴマーのクロロフォーメート基量をyモル/hrとするときに、(x/y)が0.0035以上になることを要す。(x/y)が0.0035未満であると、前記工程(b)から得られる反応液を、工程(c)でポリカーボネートオリゴマーを含む有機溶媒相と水相に分離させる際に、中間相が多く発生して反応液の分離性が悪化し、分岐ポリカーボネートの生産効率が低下するので好ましくない。(x/y)は、好ましくは、0.0038以上、より好ましくは、0.0042以上である。(x/y)の上限は、中間相発生量の低減効果が変化しなくなる観点から通常、0.023以下である。(x/y)を上記の値となるようにするためには、通常、工程(b)から抜き出される反応液に含まれるポリカーボネートオリゴマーのクロロフォーメート基量に応じて、工程(b)に添加される重合触媒の添加量を調整すればよい。
なお、工程(b)から得られる反応液に含まれるポリカーボネートオリゴマーのクロロフォーメート基量の測定は、工程(b)から得られるポリカーボネートオリゴマーを含む反応液をサンプリングし、静置分離や遠心分離等により、ポリカーボネートオリゴマーを含む有機溶媒相と水相に分離して、得られた有機溶媒相中に含まれるポリカーボネートオリゴマーのクロロフォーメート基濃度を求め、そのクロロフォーメート基濃度と工程(b)から得られる反応液から分離したポリカーボネートオリゴマーを含む有機溶媒相の量(流量)から単位時間当たりのモル量を求めることができる。また、工程(b)から得られる反応液を工程(c)で分離して、分離したポリカーボネートオリゴマーを含む有機溶媒相の抜き出し量(流量)とそのクロロフォーメート基濃度からも単位時間当たりのモル量を求めることもできる。
[工程(c)]
工程(c)は、前記工程(b)で得られたポリカーボネートオリゴマーを含む反応液を、ポリカーボネートオリゴマーを含む有機溶媒相と水相に分離する工程である。工程(c)に用いられる機器としては、静置分離槽が好ましく使用される。工程(b)で得られたポリカーボネートオリゴマーを含む反応液は、静置分離槽内に導入され、比重差により、ポリカーボネートオリゴマーを含む有機溶媒相と水相に分離される。下層のポリカーボネートオリゴマーを含む有機溶媒相は、静置分離槽の下側から連続的にあるいは断続的に抜き出される。上層の水相は、連続的にあるいは断続的に抜き出され、静置分離槽内の各相のレベルは、一定のレベル範囲内となるように保たれる。工程(c)では、ポリカーボネートオリゴマーを含む有機溶媒相が、静置分離槽の下側から連続的にあるいは断続的に抜き出されるが、前述したとおり、この有機溶媒相に含まれるポリカーボネートオリゴマーのクロロフォーメート基濃度を求め、かつ、その抜き出し量とから、前記(x/y)の値を求めることができる。
工程(c)は、前記工程(b)で得られたポリカーボネートオリゴマーを含む反応液を、ポリカーボネートオリゴマーを含む有機溶媒相と水相に分離する工程である。工程(c)に用いられる機器としては、静置分離槽が好ましく使用される。工程(b)で得られたポリカーボネートオリゴマーを含む反応液は、静置分離槽内に導入され、比重差により、ポリカーボネートオリゴマーを含む有機溶媒相と水相に分離される。下層のポリカーボネートオリゴマーを含む有機溶媒相は、静置分離槽の下側から連続的にあるいは断続的に抜き出される。上層の水相は、連続的にあるいは断続的に抜き出され、静置分離槽内の各相のレベルは、一定のレベル範囲内となるように保たれる。工程(c)では、ポリカーボネートオリゴマーを含む有機溶媒相が、静置分離槽の下側から連続的にあるいは断続的に抜き出されるが、前述したとおり、この有機溶媒相に含まれるポリカーボネートオリゴマーのクロロフォーメート基濃度を求め、かつ、その抜き出し量とから、前記(x/y)の値を求めることができる。
[工程(d)]
工程(d)は、工程(c)で分離されたポリカーボネートオリゴマーを含む有機溶媒相と二価フェノールのアルカリ水溶液を反応させて、分岐ポリカーボネートを含む反応液を得る工程である。この工程(d)では、アルカリ水溶液及び有機溶媒の存在下に、ポリカーボネートオリゴマーと二価フェノールとを重縮合反応させ、分岐ポリカーボネートの分子量が目的の分子量範囲に調整される。得られる分岐ポリカーボネートの分子量は、粘度平均分子量で、通常、10,000~50,000程度の範囲内となるまで重縮合反応を行う。
具体的には、工程(c)で分離されたポリカーボネートオリゴマーを含む有機溶媒相と、所望により用いられる末端停止剤と、所望により用いられる触媒と、有機溶媒と、アルカリ水溶液と、二価フェノールのアルカリ水溶液とを混合し、通常0~50℃、好ましくは20~40℃の範囲の温度において界面重縮合させる。
工程(d)は、工程(c)で分離されたポリカーボネートオリゴマーを含む有機溶媒相と二価フェノールのアルカリ水溶液を反応させて、分岐ポリカーボネートを含む反応液を得る工程である。この工程(d)では、アルカリ水溶液及び有機溶媒の存在下に、ポリカーボネートオリゴマーと二価フェノールとを重縮合反応させ、分岐ポリカーボネートの分子量が目的の分子量範囲に調整される。得られる分岐ポリカーボネートの分子量は、粘度平均分子量で、通常、10,000~50,000程度の範囲内となるまで重縮合反応を行う。
具体的には、工程(c)で分離されたポリカーボネートオリゴマーを含む有機溶媒相と、所望により用いられる末端停止剤と、所望により用いられる触媒と、有機溶媒と、アルカリ水溶液と、二価フェノールのアルカリ水溶液とを混合し、通常0~50℃、好ましくは20~40℃の範囲の温度において界面重縮合させる。
工程(d)で使用するアルカリ水溶液のアルカリ、有機溶媒、末端停止剤及び触媒としては、前記工程(a)又は(b)で説明したものと同じものを挙げることができる。また、この工程(d)において、界面重縮合における有機溶媒の使用量は、通常、有機相と水相との容量比が、好ましくは7/1~1/1、より好ましくは5/1~2/1となるように選択される。
なお、工程(d)で使用される反応器は、反応器の能力次第では1基の反応器のみで反応を完結することができるが、必要に応じてさらに、それに後続する2基目の反応器、更には3基目の反応器等の複数の反応器を構築して、使用することができる。これらの反応器としては、撹拌槽,多段塔型撹拌槽,無撹拌槽,スタティックミキサー,ラインミキサー,オリフィスミキサー,配管などを用いることができる。これらの反応器は、任意に組み合わせて、複数の反応器として用いてもよい。
なお、工程(d)で使用される反応器は、反応器の能力次第では1基の反応器のみで反応を完結することができるが、必要に応じてさらに、それに後続する2基目の反応器、更には3基目の反応器等の複数の反応器を構築して、使用することができる。これらの反応器としては、撹拌槽,多段塔型撹拌槽,無撹拌槽,スタティックミキサー,ラインミキサー,オリフィスミキサー,配管などを用いることができる。これらの反応器は、任意に組み合わせて、複数の反応器として用いてもよい。
上記で説明したとおり、工程(a)~工程(d)により、分岐ポリカーボネートを含む反応液が得られる。本発明の分岐ポリカーボネートの製造方法は、さらに下記工程(e)を有し、下記工程(e)からの未反応の二価フェノールを含む水相の少なくとも一部を工程(b)に添加される二価フェノールのアルカリ水溶液として使用することが好ましい。以下、工程(e)について説明する。
[工程(e)]
工程(e)は、前記工程(d)で得られた分岐ポリカーボネートを含む反応液を、分岐ポリカーボネートを含む有機溶媒相と未反応の二価フェノールを含む水相に分離する工程である。この工程(e)で、分岐ポリカーボネートを含む有機溶媒相と未反応の二価フェノールを含む水相に分離するために使用される機器としては、静置分離槽や遠心分離機を挙げることができる。この工程(e)で分離された分岐ポリカーボネートを含む有機溶媒相は、アルカリ洗浄、酸洗浄及び純水洗浄を順に行い精製された分岐ポリカーボネートを含む有機溶媒相を得る。精製されたポリカーボネートを含む有機溶媒相は、必要に応じて濃縮され、精製されたポリカーボネートを含む有機溶媒溶液とし、ニーダー処理したり、温水造粒等を行い、分岐ポリカーボネート粉体を得ることができる。得られた分岐ポリカーボネート粉体中には、有機溶媒が残留しているので、加熱処理等の乾燥処理を行うことにより、有機溶媒を除去した分岐ポリカーボネート粉体を得ることができる。得られた分岐ポリカーボネート粉体は、ペレタイザー等を使用してペレット化して、各種の成形体とすることができる。
工程(e)で分離された水相中には、未反応の二価フェノール及びアルカリを含んでおり、原材料を有効的に利用する観点から、工程(b)にその全量もしくは一部をリサイクルさせることが好ましい。
工程(e)は、前記工程(d)で得られた分岐ポリカーボネートを含む反応液を、分岐ポリカーボネートを含む有機溶媒相と未反応の二価フェノールを含む水相に分離する工程である。この工程(e)で、分岐ポリカーボネートを含む有機溶媒相と未反応の二価フェノールを含む水相に分離するために使用される機器としては、静置分離槽や遠心分離機を挙げることができる。この工程(e)で分離された分岐ポリカーボネートを含む有機溶媒相は、アルカリ洗浄、酸洗浄及び純水洗浄を順に行い精製された分岐ポリカーボネートを含む有機溶媒相を得る。精製されたポリカーボネートを含む有機溶媒相は、必要に応じて濃縮され、精製されたポリカーボネートを含む有機溶媒溶液とし、ニーダー処理したり、温水造粒等を行い、分岐ポリカーボネート粉体を得ることができる。得られた分岐ポリカーボネート粉体中には、有機溶媒が残留しているので、加熱処理等の乾燥処理を行うことにより、有機溶媒を除去した分岐ポリカーボネート粉体を得ることができる。得られた分岐ポリカーボネート粉体は、ペレタイザー等を使用してペレット化して、各種の成形体とすることができる。
工程(e)で分離された水相中には、未反応の二価フェノール及びアルカリを含んでおり、原材料を有効的に利用する観点から、工程(b)にその全量もしくは一部をリサイクルさせることが好ましい。
以下に実施例を挙げ、本発明をさらに詳しく説明する。なお、本発明はこれらの例によって限定されるものではない。なお、実施例及び比較例中の測定評価は以下に示す方法で行った。
<重量平均分子量(Mw)の測定>
重量平均分子量(Mw)は、展開溶媒としてTHF(テトラヒドロフラン)を用い、GPC〔カラム:TOSOH TSK-GEL MULTIPORE HXL-M(2本)+Shodex KF801(1本)、温度40℃、流速1.0ml/分、検出器:RI〕にて、標準ポリスチレン換算分子量(重量平均分子量:Mw)として測定した。
重量平均分子量(Mw)は、展開溶媒としてTHF(テトラヒドロフラン)を用い、GPC〔カラム:TOSOH TSK-GEL MULTIPORE HXL-M(2本)+Shodex KF801(1本)、温度40℃、流速1.0ml/分、検出器:RI〕にて、標準ポリスチレン換算分子量(重量平均分子量:Mw)として測定した。
<クロロフォーメート基濃度(CF値)の測定>
塩素イオン濃度基準で、JIS K8203を参考とし、酸化・還元滴定、硝酸銀滴定を用いて測定した。
塩素イオン濃度基準で、JIS K8203を参考とし、酸化・還元滴定、硝酸銀滴定を用いて測定した。
<オリゴマー化反応液の静置分離槽における評価>
オリゴマー化反応液の静置分離槽における分離性については、60分静置後の有機溶媒相中の水分濃度と中間相の厚みを測定した。それぞれの数値が大きいほど分離性が悪いことを示す。また、水相中の固形分含有率は、水相に塩化メチレンを入れて混合した後、油水分離して得られた塩化メチレン相を蒸発乾固し、残渣分の重量を測定し溶液中の質量分率とすることにより求めた。固形分含有率が大きいほど中間相が水相側に流出していることを示す。
オリゴマー化反応液の静置分離槽における分離性については、60分静置後の有機溶媒相中の水分濃度と中間相の厚みを測定した。それぞれの数値が大きいほど分離性が悪いことを示す。また、水相中の固形分含有率は、水相に塩化メチレンを入れて混合した後、油水分離して得られた塩化メチレン相を蒸発乾固し、残渣分の重量を測定し溶液中の質量分率とすることにより求めた。固形分含有率が大きいほど中間相が水相側に流出していることを示す。
<粘度平均分子量(Mv)の測定>
ポリカーボネートの粘度平均分子量(Mv)は、ウベローデ型粘度計を用いて、20℃における塩化メチレン溶液の粘度を測定し、これより極限粘度[η]を求め、次式にて算出するものである。
[η]=1.23×10-5Mv0.83
ポリカーボネートの粘度平均分子量(Mv)は、ウベローデ型粘度計を用いて、20℃における塩化メチレン溶液の粘度を測定し、これより極限粘度[η]を求め、次式にて算出するものである。
[η]=1.23×10-5Mv0.83
実施例1
<ポリカーボネートオリゴマーの製造>
ポリカーボネートオリゴマーの製造は、図1に示す流れに沿って製造した。
まず、6.0質量%の水酸化ナトリウム水溶液を調製し、さらにこれにビスフェノールA(BPAと略する。)を溶解して、13.5質量%(固形物換算)のBPA水酸化ナトリウム水溶液を調製した。次に、p-tert-ブチルフェノール(PTBP)を塩化メチレンに溶解し、24質量%の溶液を調製した。
<ポリカーボネートオリゴマーの製造>
ポリカーボネートオリゴマーの製造は、図1に示す流れに沿って製造した。
まず、6.0質量%の水酸化ナトリウム水溶液を調製し、さらにこれにビスフェノールA(BPAと略する。)を溶解して、13.5質量%(固形物換算)のBPA水酸化ナトリウム水溶液を調製した。次に、p-tert-ブチルフェノール(PTBP)を塩化メチレンに溶解し、24質量%の溶液を調製した。
次いで、内径6mm、長さ26mの管型反応器に、上記BPA水酸化ナトリウム水溶液を36リットル/hrの流量で、塩化メチレンを15.4リットル/hrの流量で、さらに分岐剤として1,1,1-トリス(4-ヒドロキシフェニル)-エタン(THPEと略する。)を5.1質量%の水酸化ナトリウムで溶解して調製した11質量%(固形物換算)のTHPE水酸化ナトリウム水溶液を0.7リットル/hrの流量で連続的に供給した。二価フェノール(BPA):分岐剤(THPE)のモル比は98.9:1.1である。これと同時にホスゲンを3.1kg/hrの流量で連続的に上記管型反応器に吹き込んで、ホスゲン化反応を行い、PTBP溶液を310ミリリットル/hrの流量で供給し、ホスゲン化反応生成物を含有する反応液を得た。この時、管型反応器の出口における反応生成物の温度は30℃となるように管型反応器を冷却した。なお、ホスゲンは、一酸化炭素(CO)と塩素(Cl2)から別途合成したものを用いた。
次いで、内容積100リットルの攪拌機を有するオリゴマー化反応器[工程(b)]に、上記反応液と、触媒としてあらかじめ調製したトリエチルアミン(TEAと略する。)の3質量%水溶液を210ミリリットル/hr(0.062モル/hr)を、連続的に供給すると共に、後述する重縮合反応[工程(d)]から得られた反応液を水相とポリカーボネートを含む有機溶媒相とに分離する工程[工程(e)]から得られたリサイクル水相を15.7リットル/hrをオリゴマー化反応器に導入して、オリゴマー化反応を行った。なお、オリゴマー化反応器に導入する前の水相『[工程(e)]からのリサイクル水相と濃度調整のために追加導入する水相(純水)の合流後の水相』中のBPA濃度は、0.07モル/リットルであり、水酸化ナトリウム濃度は、0.13モル/リットルであり、炭酸ナトリウム濃度は、0.08モル/リットルであった。このオリゴマー化反応器内を350rpmで回転させ、層流状態でオリゴマー化反応を行った。オリゴマー化反応器の底部から抜き出した反応液を、移送配管[SUS製、配管径12.7mm(1/2インチ)]を通じて、横型静置分離槽(内径350mm、内容積100リットル)に連続的に供給し、水相と有機溶媒相との分離[工程(c)]を行った。ポリカーボネートオリゴマーを含有する反応液は、横型静置分離槽で水相及び有機溶媒相に分離した。横型静置分離槽から有機溶媒相を20リットル/hrの流量で連続的に抜出し、抜き出した有機溶媒相中のクロロフォーメート基濃度は0.72モル/リットルであった。また、有機溶媒相中のポリカーボネートオリゴマーの重量平均分子量は、3,100であった。上記連続運転を24時間実施した後に、横型静置分離槽内の水相及び有機溶媒相を観察したところ、水相及び有機溶媒相との間には中間相がわずかに発生したが、その中間相の厚みは、約9.0mm(3.2リットルに相当)であり、連続して静置分離を行っても、中間相の厚みは増大せず、良好に水相と有機溶媒相とに分離することができた。なお、分離後の有機溶媒相中の水分量は2000質量ppmであり、分離後の水相中の固形分量は、10質量ppm未満であった。横型静置分離槽から連続的に抜出された有機溶媒相中に含まれるポリカーボネートオリゴマーのクロロフォーメート基量(y)は、20×0.72=14.4モル/hrである。また、重合触媒として用いたTEAのオリゴマー化反応器への添加量(x)は、0.062モル/hrであり、(x/y)=0.0043であった。
<分岐ポリカーボネートの製造>
上記の横型静置分離槽から分離されたポリカーボネートオリゴマーを含む有機溶媒相(PCOと略することがある。)を使用して図1に示す流れで、工程(d)で重縮合反応を行った。前記ポリカーボネートオリゴマーを含む有機溶媒相(PCO)を20リットル/hr、BPAの水酸化ナトリウム溶液(ポリカーボネートオリゴマーの製造に使用したもの)を9.8L/hr、触媒として濃度3質量%のTEA水溶液を0.10リットル/hr、末端停止剤としてPTBP溶液を0.34リットル/hr、濃度20質量%の水酸化ナトリウム水溶液を1.3リットル/hr、溶媒として塩化メチレンを13.5リットル/hrの流量で工程(d)の重縮合反応器に導入し重縮合反応を行った。重縮合反応器に使用した反応器は、ラインミキサー及び塔型反応器の2基の反応器を用いた。塔型反応器の上部からオーバーフローして出てきた反応混合物を静置分離させ、水相と有機溶媒相に分離した[工程(e)]。分離された水相の全量を工程(b)のオリゴマー化反応器に導入しリサイクルした。また、得られた有機溶媒相は、pHを13.5に調製した水酸化ナトリウム水溶液、pHを1.5に調製した塩酸水溶液、および純水をそれぞれ使用して順次洗浄して、清澄な分岐ポリカーボネートの塩化メチレン溶液を得た。
得られた分岐ポリカーボネートの塩化メチレン溶液からニーダーで塩化メチレンを蒸発除去し、分岐ポリカーボネート粉末を得た。さらに、残留する塩化メチレンを加熱乾燥して100ppm以下まで除去し、白色の分岐ポリカーボネート粉末を得た。この粉末について,粘度平均分子量(Mv)を測定したところ、23,000であった。
上記の横型静置分離槽から分離されたポリカーボネートオリゴマーを含む有機溶媒相(PCOと略することがある。)を使用して図1に示す流れで、工程(d)で重縮合反応を行った。前記ポリカーボネートオリゴマーを含む有機溶媒相(PCO)を20リットル/hr、BPAの水酸化ナトリウム溶液(ポリカーボネートオリゴマーの製造に使用したもの)を9.8L/hr、触媒として濃度3質量%のTEA水溶液を0.10リットル/hr、末端停止剤としてPTBP溶液を0.34リットル/hr、濃度20質量%の水酸化ナトリウム水溶液を1.3リットル/hr、溶媒として塩化メチレンを13.5リットル/hrの流量で工程(d)の重縮合反応器に導入し重縮合反応を行った。重縮合反応器に使用した反応器は、ラインミキサー及び塔型反応器の2基の反応器を用いた。塔型反応器の上部からオーバーフローして出てきた反応混合物を静置分離させ、水相と有機溶媒相に分離した[工程(e)]。分離された水相の全量を工程(b)のオリゴマー化反応器に導入しリサイクルした。また、得られた有機溶媒相は、pHを13.5に調製した水酸化ナトリウム水溶液、pHを1.5に調製した塩酸水溶液、および純水をそれぞれ使用して順次洗浄して、清澄な分岐ポリカーボネートの塩化メチレン溶液を得た。
得られた分岐ポリカーボネートの塩化メチレン溶液からニーダーで塩化メチレンを蒸発除去し、分岐ポリカーボネート粉末を得た。さらに、残留する塩化メチレンを加熱乾燥して100ppm以下まで除去し、白色の分岐ポリカーボネート粉末を得た。この粉末について,粘度平均分子量(Mv)を測定したところ、23,000であった。
実施例2
実施例1において、オリゴマー化反応器[工程(b)]に添加したTEAの3質量%水溶液を310ミリリットル/hr(0.092モル/hr)に変更し、(x/y)=0.0064とした以外は、実施例1と同様に実施した。その際の水相及び有機溶媒相との間には中間相がわずかに発生したが、その中間相の厚みは、約4mm(1.4リットルに相当)であり、連続して静置分離を行っても、中間相の厚みは増大せず、良好に水相と有機溶媒相とに分離することができた。分岐ポリカーボネートの粘度平均分子量(Mv)を測定したところ、23,000であった。なお、有機溶媒相中のポリカーボネートオリゴマーの重量平均分子量は3,100、分離後の有機溶媒相中の水分量は2000質量ppmであり、分離後の水相中の固形分量は10質量ppm未満であった。
実施例1において、オリゴマー化反応器[工程(b)]に添加したTEAの3質量%水溶液を310ミリリットル/hr(0.092モル/hr)に変更し、(x/y)=0.0064とした以外は、実施例1と同様に実施した。その際の水相及び有機溶媒相との間には中間相がわずかに発生したが、その中間相の厚みは、約4mm(1.4リットルに相当)であり、連続して静置分離を行っても、中間相の厚みは増大せず、良好に水相と有機溶媒相とに分離することができた。分岐ポリカーボネートの粘度平均分子量(Mv)を測定したところ、23,000であった。なお、有機溶媒相中のポリカーボネートオリゴマーの重量平均分子量は3,100、分離後の有機溶媒相中の水分量は2000質量ppmであり、分離後の水相中の固形分量は10質量ppm未満であった。
比較例1
実施例1において、オリゴマー化反応器[工程(b)]に、添加したTEAの3質量%水溶液を110ミリリットル/hr(0.033モル/hr)に変更し、(x/y)=0.0023とした以外は、実施例1と同様に実施した。その際の水相及び有機溶媒相との間には中間相が多量に発生し、その中間相の厚みは、約42mm(14.8リットルに相当)に達した。このように中間相が多量に発生することにより、連続して静置分離を行うことが不可能となり、連続して分岐ポリカーボネートを製造することが困難となった。なお、有機溶媒相中のポリカーボネートオリゴマーの重量平均分子量は3,100、分離後の有機溶媒相中の水分量は2000質量ppmであり、分離後の水相中の固形分量は10質量ppm未満であった。
実施例1において、オリゴマー化反応器[工程(b)]に、添加したTEAの3質量%水溶液を110ミリリットル/hr(0.033モル/hr)に変更し、(x/y)=0.0023とした以外は、実施例1と同様に実施した。その際の水相及び有機溶媒相との間には中間相が多量に発生し、その中間相の厚みは、約42mm(14.8リットルに相当)に達した。このように中間相が多量に発生することにより、連続して静置分離を行うことが不可能となり、連続して分岐ポリカーボネートを製造することが困難となった。なお、有機溶媒相中のポリカーボネートオリゴマーの重量平均分子量は3,100、分離後の有機溶媒相中の水分量は2000質量ppmであり、分離後の水相中の固形分量は10質量ppm未満であった。
参考例1
実施例1において、ホスゲン化反応時に、THPEの水酸化ナトリウム水溶液を供給せずに、ホスゲン化反応を行った。得られたホスゲン化反応生成物を含有する反応液をオリゴマー化反応器[工程(b)]に、添加したTEAの3質量%水溶液を110ミリリットル/hr(0.033モル/hr)に変更し、(x/y)=0.0023とした以外は、実施例1と同様に実施した。その際の水相及び有機溶媒相との間には中間相がわずかに発生したが、その中間相の厚みは、約4mm(1.4リットルに相当)であり、連続して静置分離を行っても、中間相の厚みは増大せず、良好に水相と有機溶媒相とに分離することができた。得られた非分岐ポリカーボネートの粘度平均分子量(Mv)を測定したところ、23,000であった。
実施例1において、ホスゲン化反応時に、THPEの水酸化ナトリウム水溶液を供給せずに、ホスゲン化反応を行った。得られたホスゲン化反応生成物を含有する反応液をオリゴマー化反応器[工程(b)]に、添加したTEAの3質量%水溶液を110ミリリットル/hr(0.033モル/hr)に変更し、(x/y)=0.0023とした以外は、実施例1と同様に実施した。その際の水相及び有機溶媒相との間には中間相がわずかに発生したが、その中間相の厚みは、約4mm(1.4リットルに相当)であり、連続して静置分離を行っても、中間相の厚みは増大せず、良好に水相と有機溶媒相とに分離することができた。得られた非分岐ポリカーボネートの粘度平均分子量(Mv)を測定したところ、23,000であった。
本発明の実施例1、2及び比較例1から、分岐ポリカーボネートを製造するに際して、ポリカーボネートオリゴマーを含む反応液を得る工程において、添加される重合触媒量と抜き出される反応液に含まれるポリカーボネートオリゴマーのクロロフォーメート基量との比率が0.0035以上である実施例1、2は、中間相の発生が少なく、連続して静置分離を行うことができることが示された。一方、その比率が0.0035未満である比較例1では、中間相が多量に発生することにより、連続して静置分離を行うことが不可能となることが示された。また、分岐剤を使用しない参考例1では、重合触媒量が比較例1と同じであっても、実施例1及び2と同様に、中間相の発生が少なく、連続して静置分離を行うことができることが示されている。
本発明の分岐ポリカーボネートの製造方法では、ポリカーボネートオリゴマーを含む反応液の分離工程において、反応液の分離性が良好であり、分岐ポリカーボネートの生産効率を上げることができる。
Claims (11)
- 二価フェノールのアルカリ水溶液、ホスゲン及び分岐剤を有機溶媒の存在下でホスゲン化反応させて、反応液を得る工程(a)と、
前記工程(a)から得られる該反応液に、二価フェノールのアルカリ水溶液及び重合触媒を添加し、ポリカーボネートオリゴマーを含む反応液を得る工程(b)と、
前記工程(b)で得られたポリカーボネートオリゴマーを含む反応液を、ポリカーボネートオリゴマーを含む有機溶媒相と水相に分離する工程(c)と、
前記工程(c)で分離されたポリカーボネートオリゴマーを含む有機溶媒相と二価フェノールのアルカリ水溶液を反応させて、分岐ポリカーボネートを含む反応液を得る工程(d)を有する分岐ポリカーボネートの製造方法であって、
前記工程(b)に添加される重合触媒の添加量をxモル/hrとし、前記工程(b)から得られる反応液に含まれるポリカーボネートオリゴマーのクロロフォーメート基量をyモル/hrとするときに、(x/y)が0.0035以上である、分岐ポリカーボネートの製造方法。 - 前記ポリカーボネートオリゴマーの重量平均分子量が5000以下である、請求項1に記載の分岐ポリカーボネートの製造方法。
- 分岐剤が下記一般式(I)で表わされる化合物である、請求項1又は2に記載の分岐ポリカーボネートの製造方法。

[式中、Rは水素原子又は炭素数1~5のアルキル基であり、R1~R6は、それぞれ水素原子又は炭素数1~5のアルキル基あるいはハロゲン原子を示す。] - 前記一般式(I)で表わされる化合物が、1,1,1-トリス(4-ヒドロキシフェニル)エタンである、請求項3に記載の分岐ポリカーボネートの製造方法。
- 前記二価フェノールが、下記一般式(1)で表わされる化合物である、請求項1~4のいずれかに記載の分岐ポリカーボネートの製造方法。

[式中、R11及びR12は、それぞれ独立にハロゲン原子、炭素数1~6のアルキル基又は炭素数1~6のアルコキシ基を示す。Zは、単結合、炭素数1~8のアルキレン基、炭素数2~8のアルキリデン基、炭素数5~15のシクロアルキレン基、炭素数5~15のシクロアルキリデン基、フルオレンジイル基、炭素数7~15のアリールアルキレン基、炭素数7~15のアリールアルキリデン基、-S-、-SO-、-SO2-、-O-又は-CO-を示す。a及びbは、それぞれ独立に0~4の整数を示す。] - 前記一般式(1)で表わされる化合物が、2,2-ビス(4-ヒドロキシフェニル)プロパンである、請求項5に記載の分岐ポリカーボネートの製造方法。
- 前記工程(a)から得られる該反応液に、さらに末端停止剤を添加する、請求項1~6のいずれかに記載の分岐ポリカーボネートの製造方法。
- 前記工程(d)で得られた分岐ポリカーボネートを含む反応液を、分岐ポリカーボネートを含む有機溶媒相と未反応の二価フェノールを含む水相に分離する工程(e)を有する、請求項1~7のいずれかに記載の分岐ポリカーボネートの製造方法。
- 前記工程(b)に添加される二価フェノールのアルカリ水溶液として、前記工程(e)で分離された未反応の二価フェノールを含む水相の少なくとも一部を使用する、請求項8に記載の分岐ポリカーボネートの製造方法。
- 前記工程(b)から抜き出される反応液に含まれるポリカーボネートオリゴマーのクロロフォーメート基量を、前記工程(c)で分離されたポリカーボネートオリゴマーを含む有機溶媒相の単位時間当たりのモル量とする、請求項1~9のいずれかに記載の分岐ポリカーボネートの製造方法。
- 前記重合触媒がトリエチルアミンである、請求項1~10のいずれかに記載の分岐ポリカーボネートの製造方法。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US15/523,918 US10030101B2 (en) | 2014-11-07 | 2015-11-06 | Branched polycarbonate production method |
| CN201580059760.2A CN107075102B (zh) | 2014-11-07 | 2015-11-06 | 支化聚碳酸酯的制造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014-227409 | 2014-11-07 | ||
| JP2014227409A JP6357408B2 (ja) | 2014-11-07 | 2014-11-07 | 分岐ポリカーボネートの製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016072491A1 true WO2016072491A1 (ja) | 2016-05-12 |
Family
ID=55909214
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/081309 WO2016072491A1 (ja) | 2014-11-07 | 2015-11-06 | 分岐ポリカーボネートの製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US10030101B2 (ja) |
| JP (1) | JP6357408B2 (ja) |
| CN (1) | CN107075102B (ja) |
| TW (1) | TWI675863B (ja) |
| WO (1) | WO2016072491A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6976716B2 (ja) * | 2017-05-23 | 2021-12-08 | 本州化学工業株式会社 | 芳香族ポリカーボネートオリゴマー固形体 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59191717A (ja) * | 1983-04-13 | 1984-10-30 | Mitsubishi Gas Chem Co Inc | 分岐化ポリカ−ボネ−トの製造法 |
| JPH07102055A (ja) * | 1993-10-05 | 1995-04-18 | Idemitsu Petrochem Co Ltd | 分岐状ポリカーボネートおよびその製造方法 |
| JPH07103235B2 (ja) * | 1989-12-13 | 1995-11-08 | 出光石油化学株式会社 | 分岐ポリカーボネート及びその製造方法 |
| JP2005126478A (ja) * | 2003-10-21 | 2005-05-19 | Teijin Chem Ltd | 分岐状ポリカーボネート樹脂及びその製造方法 |
| JP2005239876A (ja) * | 2004-02-26 | 2005-09-08 | Idemitsu Kosan Co Ltd | ポリカーボネートの製造方法 |
| WO2011043484A1 (ja) * | 2009-10-07 | 2011-04-14 | 帝人化成株式会社 | 分岐状ポリカーボネート樹脂およびその製造方法 |
| JP2015229766A (ja) * | 2014-06-06 | 2015-12-21 | 出光興産株式会社 | ポリカーボネートの製造方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4415722A (en) * | 1982-03-19 | 1983-11-15 | General Electric Company | Branched aromatic polycarbonate from aliphatic polyol |
| US5104964A (en) * | 1989-08-03 | 1992-04-14 | Idemitsu Petrochemical Co., Ltd. | Branched polycarbonate having branching parameter and degree of branching |
| JP2628562B2 (ja) * | 1989-08-03 | 1997-07-09 | 出光石油化学株式会社 | 分岐状ポリカーボネート及びその製造方法 |
| JP3995945B2 (ja) * | 2002-02-04 | 2007-10-24 | 出光興産株式会社 | 直鎖状ポリカーボネートの製造方法 |
| CN101525431B (zh) * | 2008-01-30 | 2011-06-29 | 赵云 | 管道式高剪切分散乳化机连续界面光气法合成聚碳酸酯 |
| CN101735443B (zh) * | 2009-11-30 | 2011-07-06 | 中国蓝星(集团)股份有限公司 | 聚碳酸酯的制备方法 |
| JP5775347B2 (ja) * | 2011-03-31 | 2015-09-09 | 出光興産株式会社 | ポリカーボネートオリゴマー連続製造の制御方法 |
| CN102516519B (zh) * | 2011-11-18 | 2014-05-21 | 万华化学(宁波)有限公司 | 一种制备聚碳酸酯的方法 |
-
2014
- 2014-11-07 JP JP2014227409A patent/JP6357408B2/ja active Active
-
2015
- 2015-11-06 TW TW104136723A patent/TWI675863B/zh active
- 2015-11-06 CN CN201580059760.2A patent/CN107075102B/zh active Active
- 2015-11-06 WO PCT/JP2015/081309 patent/WO2016072491A1/ja active Application Filing
- 2015-11-06 US US15/523,918 patent/US10030101B2/en active Active
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59191717A (ja) * | 1983-04-13 | 1984-10-30 | Mitsubishi Gas Chem Co Inc | 分岐化ポリカ−ボネ−トの製造法 |
| JPH07103235B2 (ja) * | 1989-12-13 | 1995-11-08 | 出光石油化学株式会社 | 分岐ポリカーボネート及びその製造方法 |
| JPH07102055A (ja) * | 1993-10-05 | 1995-04-18 | Idemitsu Petrochem Co Ltd | 分岐状ポリカーボネートおよびその製造方法 |
| JP2005126478A (ja) * | 2003-10-21 | 2005-05-19 | Teijin Chem Ltd | 分岐状ポリカーボネート樹脂及びその製造方法 |
| JP2005239876A (ja) * | 2004-02-26 | 2005-09-08 | Idemitsu Kosan Co Ltd | ポリカーボネートの製造方法 |
| WO2011043484A1 (ja) * | 2009-10-07 | 2011-04-14 | 帝人化成株式会社 | 分岐状ポリカーボネート樹脂およびその製造方法 |
| JP2015229766A (ja) * | 2014-06-06 | 2015-12-21 | 出光興産株式会社 | ポリカーボネートの製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20170313817A1 (en) | 2017-11-02 |
| CN107075102B (zh) | 2019-06-11 |
| TWI675863B (zh) | 2019-11-01 |
| CN107075102A (zh) | 2017-08-18 |
| TW201619236A (zh) | 2016-06-01 |
| JP6357408B2 (ja) | 2018-07-11 |
| US10030101B2 (en) | 2018-07-24 |
| JP2016089102A (ja) | 2016-05-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6711502B2 (ja) | ポリカーボネート−ポリオルガノシロキサン共重合体の製造方法 | |
| JP2014080462A (ja) | ポリカーボネート−ポリオルガノシロキサン共重合体の連続的な製造方法 | |
| WO2015186773A1 (ja) | ポリカーボネートの製造方法 | |
| WO2016088814A1 (ja) | ポリカーボネート-ポリオルガノシロキサン共重合体の製造方法 | |
| US10738154B2 (en) | Method for producing polycarbonate-polyorganosiloxane copolymer | |
| JP6357408B2 (ja) | 分岐ポリカーボネートの製造方法 | |
| WO2016104532A1 (ja) | ポリカーボネートの製造方法 | |
| WO2015147198A1 (ja) | 3-ペンタデシルフェノールの塩化メチレン溶液、その製造方法、及び該溶液を用いるポリカーボネート樹脂の製造方法 | |
| JP6657543B2 (ja) | ポリカーボネートの製造方法 | |
| WO2015159958A1 (ja) | ポリカーボネート樹脂の製造方法 | |
| WO2016080382A1 (ja) | ポリカーボネート-ポリオルガノシロキサン共重合体の製造方法 | |
| JP6035182B2 (ja) | ポリカーボネート共重合体及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15857401 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15523918 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 15857401 Country of ref document: EP Kind code of ref document: A1 |