WO2014109355A1 - 光電変換素子の製造方法 - Google Patents
光電変換素子の製造方法 Download PDFInfo
- Publication number
- WO2014109355A1 WO2014109355A1 PCT/JP2014/050218 JP2014050218W WO2014109355A1 WO 2014109355 A1 WO2014109355 A1 WO 2014109355A1 JP 2014050218 W JP2014050218 W JP 2014050218W WO 2014109355 A1 WO2014109355 A1 WO 2014109355A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- photoelectric conversion
- layer
- electrode
- short
- Prior art date
Links
- 238000006243 chemical reaction Methods 0.000 title claims abstract description 193
- 238000000034 method Methods 0.000 title claims abstract description 137
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 25
- 239000004065 semiconductor Substances 0.000 claims abstract description 152
- 230000002265 prevention Effects 0.000 claims abstract description 151
- 230000005525 hole transport Effects 0.000 claims abstract description 110
- 239000002243 precursor Substances 0.000 claims abstract description 109
- 229920001940 conductive polymer Polymers 0.000 claims abstract description 95
- 230000001235 sensitizing effect Effects 0.000 claims abstract description 95
- 239000000758 substrate Substances 0.000 claims abstract description 46
- 239000007800 oxidant agent Substances 0.000 claims abstract description 34
- 238000000151 deposition Methods 0.000 claims abstract description 22
- 230000001678 irradiating effect Effects 0.000 claims abstract description 9
- 238000000576 coating method Methods 0.000 claims description 61
- 239000011248 coating agent Substances 0.000 claims description 56
- 239000007788 liquid Substances 0.000 claims description 19
- 230000003746 surface roughness Effects 0.000 claims description 12
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 8
- 230000001590 oxidative effect Effects 0.000 abstract description 17
- 230000000379 polymerizing effect Effects 0.000 abstract description 7
- 239000010410 layer Substances 0.000 description 363
- 239000000975 dye Substances 0.000 description 93
- -1 polyethylene terephthalate Polymers 0.000 description 90
- 239000000463 material Substances 0.000 description 89
- 238000006116 polymerization reaction Methods 0.000 description 69
- 239000000243 solution Substances 0.000 description 57
- 239000010408 film Substances 0.000 description 56
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 41
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 39
- 239000002904 solvent Substances 0.000 description 39
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 38
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 34
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 33
- 239000007789 gas Substances 0.000 description 29
- 238000011282 treatment Methods 0.000 description 25
- 238000010304 firing Methods 0.000 description 23
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 22
- 239000000126 substance Substances 0.000 description 21
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 17
- 150000001875 compounds Chemical class 0.000 description 17
- 125000000217 alkyl group Chemical group 0.000 description 16
- 229920000642 polymer Polymers 0.000 description 16
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 15
- 125000003545 alkoxy group Chemical group 0.000 description 15
- 230000000052 comparative effect Effects 0.000 description 15
- 125000000623 heterocyclic group Chemical group 0.000 description 15
- 229910052751 metal Inorganic materials 0.000 description 15
- 239000002184 metal Substances 0.000 description 15
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 14
- 239000003792 electrolyte Substances 0.000 description 14
- 125000003118 aryl group Chemical group 0.000 description 13
- 239000002245 particle Substances 0.000 description 13
- 239000010936 titanium Substances 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 12
- 230000015572 biosynthetic process Effects 0.000 description 12
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 12
- 239000011261 inert gas Substances 0.000 description 12
- 230000008569 process Effects 0.000 description 12
- 150000003839 salts Chemical class 0.000 description 12
- 239000007921 spray Substances 0.000 description 12
- 238000005507 spraying Methods 0.000 description 12
- 239000000178 monomer Substances 0.000 description 11
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 11
- YRKCREAYFQTBPV-UHFFFAOYSA-N acetylacetone Chemical compound CC(=O)CC(C)=O YRKCREAYFQTBPV-UHFFFAOYSA-N 0.000 description 10
- 230000027756 respiratory electron transport chain Effects 0.000 description 10
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 9
- 125000003342 alkenyl group Chemical group 0.000 description 9
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 9
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 9
- 229920005989 resin Polymers 0.000 description 9
- 239000011347 resin Substances 0.000 description 9
- 230000002441 reversible effect Effects 0.000 description 9
- 125000001424 substituent group Chemical group 0.000 description 9
- 229910052719 titanium Inorganic materials 0.000 description 9
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 9
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 8
- 238000010521 absorption reaction Methods 0.000 description 8
- 239000007864 aqueous solution Substances 0.000 description 8
- 125000000753 cycloalkyl group Chemical group 0.000 description 8
- 239000010419 fine particle Substances 0.000 description 8
- 238000002834 transmittance Methods 0.000 description 8
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 7
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 7
- 239000000654 additive Substances 0.000 description 7
- 238000000231 atomic layer deposition Methods 0.000 description 7
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 7
- 230000000694 effects Effects 0.000 description 7
- 239000011521 glass Substances 0.000 description 7
- 150000002430 hydrocarbons Chemical class 0.000 description 7
- 239000001301 oxygen Substances 0.000 description 7
- 229910052760 oxygen Inorganic materials 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- 238000004381 surface treatment Methods 0.000 description 7
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- 229910006404 SnO 2 Inorganic materials 0.000 description 6
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical group C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 6
- 229910010413 TiO 2 Inorganic materials 0.000 description 6
- 125000000304 alkynyl group Chemical group 0.000 description 6
- 125000006267 biphenyl group Chemical group 0.000 description 6
- 150000001923 cyclic compounds Chemical group 0.000 description 6
- 238000001035 drying Methods 0.000 description 6
- 230000006870 function Effects 0.000 description 6
- 239000012046 mixed solvent Substances 0.000 description 6
- 239000000203 mixture Substances 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 239000003960 organic solvent Substances 0.000 description 6
- 239000002685 polymerization catalyst Substances 0.000 description 6
- 230000001681 protective effect Effects 0.000 description 6
- 238000004528 spin coating Methods 0.000 description 6
- 239000003115 supporting electrolyte Substances 0.000 description 6
- KHMOASUYFVRATF-UHFFFAOYSA-J tin(4+);tetrachloride;pentahydrate Chemical compound O.O.O.O.O.Cl[Sn](Cl)(Cl)Cl KHMOASUYFVRATF-UHFFFAOYSA-J 0.000 description 6
- 229910052724 xenon Inorganic materials 0.000 description 6
- FHNFHKCVQCLJFQ-UHFFFAOYSA-N xenon atom Chemical compound [Xe] FHNFHKCVQCLJFQ-UHFFFAOYSA-N 0.000 description 6
- WZELXJBMMZFDDU-UHFFFAOYSA-N Imidazol-2-one Chemical group O=C1N=CC=N1 WZELXJBMMZFDDU-UHFFFAOYSA-N 0.000 description 5
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 5
- 239000004698 Polyethylene Substances 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 125000002091 cationic group Chemical group 0.000 description 5
- 125000001309 chloro group Chemical group Cl* 0.000 description 5
- 208000028659 discharge Diseases 0.000 description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 5
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 5
- 125000005843 halogen group Chemical group 0.000 description 5
- 125000002883 imidazolyl group Chemical group 0.000 description 5
- 229910052738 indium Inorganic materials 0.000 description 5
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 5
- 238000005259 measurement Methods 0.000 description 5
- 229910044991 metal oxide Inorganic materials 0.000 description 5
- 150000004706 metal oxides Chemical class 0.000 description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 5
- 239000002052 molecular layer Substances 0.000 description 5
- 125000001624 naphthyl group Chemical group 0.000 description 5
- 150000007530 organic bases Chemical class 0.000 description 5
- 150000002978 peroxides Chemical class 0.000 description 5
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 5
- 229920000573 polyethylene Polymers 0.000 description 5
- 125000000168 pyrrolyl group Chemical group 0.000 description 5
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical group [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- 238000001179 sorption measurement Methods 0.000 description 5
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 4
- OVSGBKZKXUMMHS-VGKOASNMSA-L (z)-4-oxopent-2-en-2-olate;propan-2-olate;titanium(4+) Chemical compound [Ti+4].CC(C)[O-].CC(C)[O-].C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O OVSGBKZKXUMMHS-VGKOASNMSA-L 0.000 description 4
- OHZAHWOAMVVGEL-UHFFFAOYSA-N 2,2'-bithiophene Chemical group C1=CSC(C=2SC=CC=2)=C1 OHZAHWOAMVVGEL-UHFFFAOYSA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- 206010070834 Sensitisation Diseases 0.000 description 4
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 4
- 230000002378 acidificating effect Effects 0.000 description 4
- 239000002131 composite material Substances 0.000 description 4
- 125000004093 cyano group Chemical group *C#N 0.000 description 4
- 125000000392 cycloalkenyl group Chemical group 0.000 description 4
- 238000000354 decomposition reaction Methods 0.000 description 4
- 238000007599 discharging Methods 0.000 description 4
- 239000006185 dispersion Substances 0.000 description 4
- 229910052731 fluorine Inorganic materials 0.000 description 4
- 125000001153 fluoro group Chemical group F* 0.000 description 4
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 4
- 229910052737 gold Inorganic materials 0.000 description 4
- 239000010931 gold Substances 0.000 description 4
- 238000005470 impregnation Methods 0.000 description 4
- 125000001041 indolyl group Chemical group 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 239000011259 mixed solution Substances 0.000 description 4
- 229920000139 polyethylene terephthalate Polymers 0.000 description 4
- 239000005020 polyethylene terephthalate Substances 0.000 description 4
- 239000005518 polymer electrolyte Substances 0.000 description 4
- 239000011164 primary particle Substances 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 230000008313 sensitization Effects 0.000 description 4
- 229910052709 silver Inorganic materials 0.000 description 4
- 239000004332 silver Substances 0.000 description 4
- SQGYOTSLMSWVJD-UHFFFAOYSA-N silver(1+) nitrate Chemical compound [Ag+].[O-]N(=O)=O SQGYOTSLMSWVJD-UHFFFAOYSA-N 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- 239000011135 tin Substances 0.000 description 4
- VXUYXOFXAQZZMF-UHFFFAOYSA-N titanium(IV) isopropoxide Chemical compound CC(C)O[Ti](OC(C)C)(OC(C)C)OC(C)C VXUYXOFXAQZZMF-UHFFFAOYSA-N 0.000 description 4
- 238000007740 vapor deposition Methods 0.000 description 4
- 229910052725 zinc Inorganic materials 0.000 description 4
- 239000011701 zinc Substances 0.000 description 4
- 239000011787 zinc oxide Substances 0.000 description 4
- YJTKZCDBKVTVBY-UHFFFAOYSA-N 1,3-Diphenylbenzene Chemical group C1=CC=CC=C1C1=CC=CC(C=2C=CC=CC=2)=C1 YJTKZCDBKVTVBY-UHFFFAOYSA-N 0.000 description 3
- 125000000355 1,3-benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 3
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 101710134784 Agnoprotein Proteins 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- QGJOPFRUJISHPQ-UHFFFAOYSA-N Carbon disulfide Chemical compound S=C=S QGJOPFRUJISHPQ-UHFFFAOYSA-N 0.000 description 3
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 230000000996 additive effect Effects 0.000 description 3
- 125000002947 alkylene group Chemical group 0.000 description 3
- 229910052782 aluminium Inorganic materials 0.000 description 3
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 3
- 125000000732 arylene group Chemical group 0.000 description 3
- 230000004888 barrier function Effects 0.000 description 3
- YHWCPXVTRSHPNY-UHFFFAOYSA-N butan-1-olate;titanium(4+) Chemical compound [Ti+4].CCCC[O-].CCCC[O-].CCCC[O-].CCCC[O-] YHWCPXVTRSHPNY-UHFFFAOYSA-N 0.000 description 3
- 229910052793 cadmium Inorganic materials 0.000 description 3
- BDOSMKKIYDKNTQ-UHFFFAOYSA-N cadmium atom Chemical compound [Cd] BDOSMKKIYDKNTQ-UHFFFAOYSA-N 0.000 description 3
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 238000005229 chemical vapour deposition Methods 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- 238000009833 condensation Methods 0.000 description 3
- 230000005494 condensation Effects 0.000 description 3
- 239000004020 conductor Substances 0.000 description 3
- 239000000470 constituent Substances 0.000 description 3
- 229920001577 copolymer Polymers 0.000 description 3
- 229910052802 copper Inorganic materials 0.000 description 3
- 239000010949 copper Substances 0.000 description 3
- 125000006612 decyloxy group Chemical group 0.000 description 3
- 230000008021 deposition Effects 0.000 description 3
- 239000000539 dimer Substances 0.000 description 3
- 125000006575 electron-withdrawing group Chemical group 0.000 description 3
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 3
- 238000011156 evaluation Methods 0.000 description 3
- JVZRCNQLWOELDU-UHFFFAOYSA-N gamma-Phenylpyridine Natural products C1=CC=CC=C1C1=CC=NC=C1 JVZRCNQLWOELDU-UHFFFAOYSA-N 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 229910052742 iron Inorganic materials 0.000 description 3
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 3
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 3
- 125000002757 morpholinyl group Chemical group 0.000 description 3
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 3
- 125000000962 organic group Chemical group 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 238000010926 purge Methods 0.000 description 3
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 3
- 125000003373 pyrazinyl group Chemical group 0.000 description 3
- JEXVQSWXXUJEMA-UHFFFAOYSA-N pyrazol-3-one Chemical group O=C1C=CN=N1 JEXVQSWXXUJEMA-UHFFFAOYSA-N 0.000 description 3
- 125000002755 pyrazolinyl group Chemical group 0.000 description 3
- 229910052703 rhodium Inorganic materials 0.000 description 3
- 239000010948 rhodium Substances 0.000 description 3
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 238000007650 screen-printing Methods 0.000 description 3
- 238000004544 sputter deposition Methods 0.000 description 3
- 239000010409 thin film Substances 0.000 description 3
- 229910052718 tin Inorganic materials 0.000 description 3
- XOLBLPGZBRYERU-UHFFFAOYSA-N tin dioxide Chemical compound O=[Sn]=O XOLBLPGZBRYERU-UHFFFAOYSA-N 0.000 description 3
- 229910001887 tin oxide Inorganic materials 0.000 description 3
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 3
- UBOXGVDOUJQMTN-UHFFFAOYSA-N 1,1,2-trichloroethane Chemical compound ClCC(Cl)Cl UBOXGVDOUJQMTN-UHFFFAOYSA-N 0.000 description 2
- MEBONNVPKOBPEA-UHFFFAOYSA-N 1,1,2-trimethylcyclohexane Chemical compound CC1CCCCC1(C)C MEBONNVPKOBPEA-UHFFFAOYSA-N 0.000 description 2
- UGUHFDPGDQDVGX-UHFFFAOYSA-N 1,2,3-thiadiazole Chemical group C1=CSN=N1 UGUHFDPGDQDVGX-UHFFFAOYSA-N 0.000 description 2
- RFFLAFLAYFXFSW-UHFFFAOYSA-N 1,2-dichlorobenzene Chemical compound ClC1=CC=CC=C1Cl RFFLAFLAYFXFSW-UHFFFAOYSA-N 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 description 2
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 2
- 125000006039 1-hexenyl group Chemical group 0.000 description 2
- RZFOAVRHEGQZRV-UHFFFAOYSA-N 2,3-diphenylthiophene Chemical group S1C=CC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 RZFOAVRHEGQZRV-UHFFFAOYSA-N 0.000 description 2
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 2
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 2
- 125000006040 2-hexenyl group Chemical group 0.000 description 2
- VQGHOUODWALEFC-UHFFFAOYSA-N 2-phenylpyridine Chemical compound C1=CC=CC=C1C1=CC=CC=N1 VQGHOUODWALEFC-UHFFFAOYSA-N 0.000 description 2
- PJRGDKFLFAYRBV-UHFFFAOYSA-N 2-phenylthiophene Chemical group C1=CSC(C=2C=CC=CC=2)=C1 PJRGDKFLFAYRBV-UHFFFAOYSA-N 0.000 description 2
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 2
- FRIBMENBGGCKPD-UHFFFAOYSA-N 3-(2,3-dimethoxyphenyl)prop-2-enal Chemical compound COC1=CC=CC(C=CC=O)=C1OC FRIBMENBGGCKPD-UHFFFAOYSA-N 0.000 description 2
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 2
- 125000004337 3-ethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000006041 3-hexenyl group Chemical group 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- FBAKMGYRQGFADI-UHFFFAOYSA-K 4-dodecylbenzenesulfonate;iron(3+) Chemical compound [Fe+3].CCCCCCCCCCCCC1=CC=C(S([O-])(=O)=O)C=C1.CCCCCCCCCCCCC1=CC=C(S([O-])(=O)=O)C=C1.CCCCCCCCCCCCC1=CC=C(S([O-])(=O)=O)C=C1 FBAKMGYRQGFADI-UHFFFAOYSA-K 0.000 description 2
- 125000006042 4-hexenyl group Chemical group 0.000 description 2
- YSHMQTRICHYLGF-UHFFFAOYSA-N 4-tert-butylpyridine Chemical compound CC(C)(C)C1=CC=NC=C1 YSHMQTRICHYLGF-UHFFFAOYSA-N 0.000 description 2
- DDFHBQSCUXNBSA-UHFFFAOYSA-N 5-(5-carboxythiophen-2-yl)thiophene-2-carboxylic acid Chemical compound S1C(C(=O)O)=CC=C1C1=CC=C(C(O)=O)S1 DDFHBQSCUXNBSA-UHFFFAOYSA-N 0.000 description 2
- 125000006043 5-hexenyl group Chemical group 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- JBRZTFJDHDCESZ-UHFFFAOYSA-N AsGa Chemical compound [As]#[Ga] JBRZTFJDHDCESZ-UHFFFAOYSA-N 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 229920002284 Cellulose triacetate Polymers 0.000 description 2
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 2
- 229910020366 ClO 4 Inorganic materials 0.000 description 2
- 239000001856 Ethyl cellulose Substances 0.000 description 2
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 241000511976 Hoya Species 0.000 description 2
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 2
- 229910021577 Iron(II) chloride Inorganic materials 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- KFSLWBXXFJQRDL-UHFFFAOYSA-N Peracetic acid Chemical compound CC(=O)OO KFSLWBXXFJQRDL-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 239000004696 Poly ether ether ketone Substances 0.000 description 2
- 239000004743 Polypropylene Substances 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical group C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 2
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- MKYQPGPNVYRMHI-UHFFFAOYSA-N Triphenylethylene Chemical compound C=1C=CC=CC=1C=C(C=1C=CC=CC=1)C1=CC=CC=C1 MKYQPGPNVYRMHI-UHFFFAOYSA-N 0.000 description 2
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 description 2
- NNLVGZFZQQXQNW-ADJNRHBOSA-N [(2r,3r,4s,5r,6s)-4,5-diacetyloxy-3-[(2s,3r,4s,5r,6r)-3,4,5-triacetyloxy-6-(acetyloxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6s)-4,5,6-triacetyloxy-2-(acetyloxymethyl)oxan-3-yl]oxyoxan-2-yl]methyl acetate Chemical compound O([C@@H]1O[C@@H]([C@H]([C@H](OC(C)=O)[C@H]1OC(C)=O)O[C@H]1[C@@H]([C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(C)=O)O1)OC(C)=O)COC(=O)C)[C@@H]1[C@@H](COC(C)=O)O[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O NNLVGZFZQQXQNW-ADJNRHBOSA-N 0.000 description 2
- 125000004054 acenaphthylenyl group Chemical group C1(=CC2=CC=CC3=CC=CC1=C23)* 0.000 description 2
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 125000005577 anthracene group Chemical group 0.000 description 2
- 125000005428 anthryl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C3C(*)=C([H])C([H])=C([H])C3=C([H])C2=C1[H] 0.000 description 2
- 229910052787 antimony Inorganic materials 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 238000000889 atomisation Methods 0.000 description 2
- 125000003828 azulenyl group Chemical group 0.000 description 2
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 2
- 125000004106 butoxy group Chemical group [*]OC([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- UHYPYGJEEGLRJD-UHFFFAOYSA-N cadmium(2+);selenium(2-) Chemical compound [Se-2].[Cd+2] UHYPYGJEEGLRJD-UHFFFAOYSA-N 0.000 description 2
- 238000001354 calcination Methods 0.000 description 2
- 125000004623 carbolinyl group Chemical group 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 150000004770 chalcogenides Chemical class 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- 125000004230 chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 description 2
- 229910052804 chromium Inorganic materials 0.000 description 2
- 239000011651 chromium Substances 0.000 description 2
- MGNZXYYWBUKAII-UHFFFAOYSA-N cyclohexa-1,3-diene Chemical compound C1CC=CC=C1 MGNZXYYWBUKAII-UHFFFAOYSA-N 0.000 description 2
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 2
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000000522 cyclooctenyl group Chemical group C1(=CCCCCCC1)* 0.000 description 2
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 229910001873 dinitrogen Inorganic materials 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 229920001249 ethyl cellulose Polymers 0.000 description 2
- 235000019325 ethyl cellulose Nutrition 0.000 description 2
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 2
- 230000005284 excitation Effects 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 239000011245 gel electrolyte Substances 0.000 description 2
- 229910052736 halogen Inorganic materials 0.000 description 2
- 150000002367 halogens Chemical class 0.000 description 2
- 229910052734 helium Inorganic materials 0.000 description 2
- 239000001307 helium Substances 0.000 description 2
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 2
- 125000003707 hexyloxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- JGJLWPGRMCADHB-UHFFFAOYSA-N hypobromite Chemical compound Br[O-] JGJLWPGRMCADHB-UHFFFAOYSA-N 0.000 description 2
- QWPPOHNGKGFGJK-UHFFFAOYSA-N hypochlorous acid Chemical compound ClO QWPPOHNGKGFGJK-UHFFFAOYSA-N 0.000 description 2
- 238000007654 immersion Methods 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- NMCUIPGRVMDVDB-UHFFFAOYSA-L iron dichloride Chemical compound Cl[Fe]Cl NMCUIPGRVMDVDB-UHFFFAOYSA-L 0.000 description 2
- RBTARNINKXHZNM-UHFFFAOYSA-K iron trichloride Chemical compound Cl[Fe](Cl)Cl RBTARNINKXHZNM-UHFFFAOYSA-K 0.000 description 2
- FYMCOOOLDFPFPN-UHFFFAOYSA-K iron(3+);4-methylbenzenesulfonate Chemical compound [Fe+3].CC1=CC=C(S([O-])(=O)=O)C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 FYMCOOOLDFPFPN-UHFFFAOYSA-K 0.000 description 2
- ACQYKVVPCCAWBZ-UHFFFAOYSA-K iron(3+);methanesulfonate Chemical compound [Fe+3].CS([O-])(=O)=O.CS([O-])(=O)=O.CS([O-])(=O)=O ACQYKVVPCCAWBZ-UHFFFAOYSA-K 0.000 description 2
- VCJMYUPGQJHHFU-UHFFFAOYSA-N iron(3+);trinitrate Chemical compound [Fe+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O VCJMYUPGQJHHFU-UHFFFAOYSA-N 0.000 description 2
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 2
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 2
- 125000001786 isothiazolyl group Chemical group 0.000 description 2
- 125000000842 isoxazolyl group Chemical group 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000011244 liquid electrolyte Substances 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 229910052976 metal sulfide Inorganic materials 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 239000003607 modifier Substances 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 2
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 239000010955 niobium Substances 0.000 description 2
- GUCVJGMIXFAOAE-UHFFFAOYSA-N niobium atom Chemical compound [Nb] GUCVJGMIXFAOAE-UHFFFAOYSA-N 0.000 description 2
- 229910017604 nitric acid Inorganic materials 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- MCCIMQKMMBVWHO-UHFFFAOYSA-N octadecanoic acid;titanium Chemical compound [Ti].CCCCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCC(O)=O MCCIMQKMMBVWHO-UHFFFAOYSA-N 0.000 description 2
- 125000002971 oxazolyl group Chemical group 0.000 description 2
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- 125000004115 pentoxy group Chemical group [*]OC([H])([H])C([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 2
- 230000000737 periodic effect Effects 0.000 description 2
- 125000004934 phenanthridinyl group Chemical group C1(=CC=CC2=NC=C3C=CC=CC3=C12)* 0.000 description 2
- 125000005561 phenanthryl group Chemical group 0.000 description 2
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 2
- 125000005954 phenoxathiinyl group Chemical group 0.000 description 2
- ABLZXFCXXLZCGV-UHFFFAOYSA-N phosphonic acid group Chemical group P(O)(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 2
- 238000001782 photodegradation Methods 0.000 description 2
- 238000007747 plating Methods 0.000 description 2
- 229910052697 platinum Inorganic materials 0.000 description 2
- 229920003207 poly(ethylene-2,6-naphthalate) Polymers 0.000 description 2
- 229920002492 poly(sulfone) Polymers 0.000 description 2
- 229920002037 poly(vinyl butyral) polymer Polymers 0.000 description 2
- 229920000728 polyester Polymers 0.000 description 2
- 229920002530 polyetherether ketone Polymers 0.000 description 2
- 239000011112 polyethylene naphthalate Substances 0.000 description 2
- 229920006254 polymer film Polymers 0.000 description 2
- 229920001155 polypropylene Polymers 0.000 description 2
- 229920000915 polyvinyl chloride Polymers 0.000 description 2
- 229920002717 polyvinylpyridine Polymers 0.000 description 2
- 239000012286 potassium permanganate Substances 0.000 description 2
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 2
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 2
- RUOJZAUFBMNUDX-UHFFFAOYSA-N propylene carbonate Chemical compound CC1COC(=O)O1 RUOJZAUFBMNUDX-UHFFFAOYSA-N 0.000 description 2
- 125000001725 pyrenyl group Chemical group 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 125000004076 pyridyl group Chemical group 0.000 description 2
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 2
- 125000005493 quinolyl group Chemical group 0.000 description 2
- 125000004151 quinonyl group Chemical group 0.000 description 2
- 229910052707 ruthenium Inorganic materials 0.000 description 2
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 238000007086 side reaction Methods 0.000 description 2
- 229910001961 silver nitrate Inorganic materials 0.000 description 2
- 238000003980 solgel method Methods 0.000 description 2
- 239000007784 solid electrolyte Substances 0.000 description 2
- 238000005118 spray pyrolysis Methods 0.000 description 2
- 125000004079 stearyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- 150000003609 titanium compounds Chemical class 0.000 description 2
- 239000004408 titanium dioxide Substances 0.000 description 2
- XJDNKRIXUMDJCW-UHFFFAOYSA-J titanium tetrachloride Chemical compound Cl[Ti](Cl)(Cl)Cl XJDNKRIXUMDJCW-UHFFFAOYSA-J 0.000 description 2
- YONPGGFAJWQGJC-UHFFFAOYSA-K titanium(iii) chloride Chemical compound Cl[Ti](Cl)Cl YONPGGFAJWQGJC-UHFFFAOYSA-K 0.000 description 2
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 2
- 229910052721 tungsten Inorganic materials 0.000 description 2
- 239000010937 tungsten Substances 0.000 description 2
- GPPXJZIENCGNKB-UHFFFAOYSA-N vanadium Chemical compound [V]#[V] GPPXJZIENCGNKB-UHFFFAOYSA-N 0.000 description 2
- 229910052726 zirconium Inorganic materials 0.000 description 2
- HCXVPNKIBYLBIT-UHFFFAOYSA-N (2-methylpropan-2-yl)oxy 3,5,5-trimethylhexaneperoxoate Chemical compound CC(C)(C)CC(C)CC(=O)OOOC(C)(C)C HCXVPNKIBYLBIT-UHFFFAOYSA-N 0.000 description 1
- QEQBMZQFDDDTPN-UHFFFAOYSA-N (2-methylpropan-2-yl)oxy benzenecarboperoxoate Chemical compound CC(C)(C)OOOC(=O)C1=CC=CC=C1 QEQBMZQFDDDTPN-UHFFFAOYSA-N 0.000 description 1
- NLBJAOHLJABDAU-UHFFFAOYSA-N (3-methylbenzoyl) 3-methylbenzenecarboperoxoate Chemical compound CC1=CC=CC(C(=O)OOC(=O)C=2C=C(C)C=CC=2)=C1 NLBJAOHLJABDAU-UHFFFAOYSA-N 0.000 description 1
- RIPYNJLMMFGZSX-UHFFFAOYSA-N (5-benzoylperoxy-2,5-dimethylhexan-2-yl) benzenecarboperoxoate Chemical compound C=1C=CC=CC=1C(=O)OOC(C)(C)CCC(C)(C)OOC(=O)C1=CC=CC=C1 RIPYNJLMMFGZSX-UHFFFAOYSA-N 0.000 description 1
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 1
- BLKRGXCGFRXRNQ-SNAWJCMRSA-N (z)-3-carbonoperoxoyl-4,4-dimethylpent-2-enoic acid Chemical compound OC(=O)/C=C(C(C)(C)C)\C(=O)OO BLKRGXCGFRXRNQ-SNAWJCMRSA-N 0.000 description 1
- NOGBEXBVDOCGDB-NRFIWDAESA-L (z)-4-ethoxy-4-oxobut-2-en-2-olate;propan-2-olate;titanium(4+) Chemical compound [Ti+4].CC(C)[O-].CC(C)[O-].CCOC(=O)\C=C(\C)[O-].CCOC(=O)\C=C(\C)[O-] NOGBEXBVDOCGDB-NRFIWDAESA-L 0.000 description 1
- TYKCBTYOMAUNLH-MTOQALJVSA-J (z)-4-oxopent-2-en-2-olate;titanium(4+) Chemical compound [Ti+4].C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O TYKCBTYOMAUNLH-MTOQALJVSA-J 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N 1,1-Diethoxyethane Chemical compound CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- VBQCFYPTKHCPGI-UHFFFAOYSA-N 1,1-bis(2-methylpentan-2-ylperoxy)cyclohexane Chemical compound CCCC(C)(C)OOC1(OOC(C)(C)CCC)CCCCC1 VBQCFYPTKHCPGI-UHFFFAOYSA-N 0.000 description 1
- HSLFISVKRDQEBY-UHFFFAOYSA-N 1,1-bis(tert-butylperoxy)cyclohexane Chemical compound CC(C)(C)OOC1(OOC(C)(C)C)CCCCC1 HSLFISVKRDQEBY-UHFFFAOYSA-N 0.000 description 1
- CTPYJEXTTINDEM-UHFFFAOYSA-N 1,2-bis(1-tert-butylperoxypropan-2-yl)benzene Chemical compound CC(C)(C)OOCC(C)C1=CC=CC=C1C(C)COOC(C)(C)C CTPYJEXTTINDEM-UHFFFAOYSA-N 0.000 description 1
- 125000005918 1,2-dimethylbutyl group Chemical group 0.000 description 1
- ZXJYAYKVSJJDNO-UHFFFAOYSA-N 1,4-diphenylbuta-1,3-dienylbenzene Chemical compound C=1C=CC=CC=1C=CC=C(C=1C=CC=CC=1)C1=CC=CC=C1 ZXJYAYKVSJJDNO-UHFFFAOYSA-N 0.000 description 1
- XSZYESUNPWGWFQ-UHFFFAOYSA-N 1-(2-hydroperoxypropan-2-yl)-4-methylcyclohexane Chemical compound CC1CCC(C(C)(C)OO)CC1 XSZYESUNPWGWFQ-UHFFFAOYSA-N 0.000 description 1
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 1
- XRPVQXPWEVJKTN-UHFFFAOYSA-N 1-hexyl-4-phenylbenzene Chemical group C1=CC(CCCCCC)=CC=C1C1=CC=CC=C1 XRPVQXPWEVJKTN-UHFFFAOYSA-N 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- HQOVXPHOJANJBR-UHFFFAOYSA-N 2,2-bis(tert-butylperoxy)butane Chemical compound CC(C)(C)OOC(C)(CC)OOC(C)(C)C HQOVXPHOJANJBR-UHFFFAOYSA-N 0.000 description 1
- CRJIYMRJTJWVLU-UHFFFAOYSA-N 2,4,4-trimethylpentan-2-yl 3-(5,5-dimethylhexyl)dioxirane-3-carboxylate Chemical compound CC(C)(C)CCCCC1(C(=O)OC(C)(C)CC(C)(C)C)OO1 CRJIYMRJTJWVLU-UHFFFAOYSA-N 0.000 description 1
- DPGYCJUCJYUHTM-UHFFFAOYSA-N 2,4,4-trimethylpentan-2-yloxy 2-ethylhexaneperoxoate Chemical compound CCCCC(CC)C(=O)OOOC(C)(C)CC(C)(C)C DPGYCJUCJYUHTM-UHFFFAOYSA-N 0.000 description 1
- DMWVYCCGCQPJEA-UHFFFAOYSA-N 2,5-bis(tert-butylperoxy)-2,5-dimethylhexane Chemical compound CC(C)(C)OOC(C)(C)CCC(C)(C)OOC(C)(C)C DMWVYCCGCQPJEA-UHFFFAOYSA-N 0.000 description 1
- DPAPGSNLWBQIGV-UHFFFAOYSA-N 2-(2-phenylethenyl)thiophene Chemical compound C=1C=CSC=1C=CC1=CC=CC=C1 DPAPGSNLWBQIGV-UHFFFAOYSA-N 0.000 description 1
- XMNIXWIUMCBBBL-UHFFFAOYSA-N 2-(2-phenylpropan-2-ylperoxy)propan-2-ylbenzene Chemical compound C=1C=CC=CC=1C(C)(C)OOC(C)(C)C1=CC=CC=C1 XMNIXWIUMCBBBL-UHFFFAOYSA-N 0.000 description 1
- OEPOKWHJYJXUGD-UHFFFAOYSA-N 2-(3-phenylmethoxyphenyl)-1,3-thiazole-4-carbaldehyde Chemical compound O=CC1=CSC(C=2C=C(OCC=3C=CC=CC=3)C=CC=2)=N1 OEPOKWHJYJXUGD-UHFFFAOYSA-N 0.000 description 1
- XXJORRQVWJFCFR-UHFFFAOYSA-N 2-(9h-fluoren-2-yl)thiophene Chemical compound C1=C2CC3=CC=CC=C3C2=CC=C1C1=CC=CS1 XXJORRQVWJFCFR-UHFFFAOYSA-N 0.000 description 1
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 1
- DPAPGSNLWBQIGV-CMDGGOBGSA-N 2-[(e)-2-phenylethenyl]thiophene Chemical compound C=1C=CSC=1/C=C/C1=CC=CC=C1 DPAPGSNLWBQIGV-CMDGGOBGSA-N 0.000 description 1
- XKBHBVFIWWDGQX-UHFFFAOYSA-N 2-bromo-3,3,4,4,5,5,5-heptafluoropent-1-ene Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(Br)=C XKBHBVFIWWDGQX-UHFFFAOYSA-N 0.000 description 1
- GLVYLTSKTCWWJR-UHFFFAOYSA-N 2-carbonoperoxoylbenzoic acid Chemical compound OOC(=O)C1=CC=CC=C1C(O)=O GLVYLTSKTCWWJR-UHFFFAOYSA-N 0.000 description 1
- ZACVGCNKGYYQHA-UHFFFAOYSA-N 2-ethylhexoxycarbonyloxy 2-ethylhexyl carbonate Chemical compound CCCCC(CC)COC(=O)OOC(=O)OCC(CC)CCCC ZACVGCNKGYYQHA-UHFFFAOYSA-N 0.000 description 1
- AIFLGMNWQFPTAJ-UHFFFAOYSA-J 2-hydroxypropanoate;titanium(4+) Chemical compound [Ti+4].CC(O)C([O-])=O.CC(O)C([O-])=O.CC(O)C([O-])=O.CC(O)C([O-])=O AIFLGMNWQFPTAJ-UHFFFAOYSA-J 0.000 description 1
- YAQDPWONDFRAHF-UHFFFAOYSA-N 2-methyl-2-(2-methylpentan-2-ylperoxy)pentane Chemical compound CCCC(C)(C)OOC(C)(C)CCC YAQDPWONDFRAHF-UHFFFAOYSA-N 0.000 description 1
- RTEZVHMDMFEURJ-UHFFFAOYSA-N 2-methylpentan-2-yl 2,2-dimethylpropaneperoxoate Chemical compound CCCC(C)(C)OOC(=O)C(C)(C)C RTEZVHMDMFEURJ-UHFFFAOYSA-N 0.000 description 1
- YMMLZUQDXYPNOG-UHFFFAOYSA-N 2-methylpentan-2-yl 7,7-dimethyloctaneperoxoate Chemical compound CCCC(C)(C)OOC(=O)CCCCCC(C)(C)C YMMLZUQDXYPNOG-UHFFFAOYSA-N 0.000 description 1
- WXDJDZIIPSOZAH-UHFFFAOYSA-N 2-methylpentan-2-yl benzenecarboperoxoate Chemical compound CCCC(C)(C)OOC(=O)C1=CC=CC=C1 WXDJDZIIPSOZAH-UHFFFAOYSA-N 0.000 description 1
- RPBWMJBZQXCSFW-UHFFFAOYSA-N 2-methylpropanoyl 2-methylpropaneperoxoate Chemical compound CC(C)C(=O)OOC(=O)C(C)C RPBWMJBZQXCSFW-UHFFFAOYSA-N 0.000 description 1
- ZYEIXHZLTHTEOI-UHFFFAOYSA-N 2-octyl-5-thiophen-2-ylthiophene Chemical group S1C(CCCCCCCC)=CC=C1C1=CC=CS1 ZYEIXHZLTHTEOI-UHFFFAOYSA-N 0.000 description 1
- LBMHPHUSGIEGHJ-UHFFFAOYSA-N 2-phenyl-1-benzothiophene Chemical compound S1C2=CC=CC=C2C=C1C1=CC=CC=C1 LBMHPHUSGIEGHJ-UHFFFAOYSA-N 0.000 description 1
- OWEUOGSUFUUNQE-UHFFFAOYSA-N 2-phenyldithieno[3,2-a:2',3'-d]thiophene Chemical group S1C=CC(C=2S3)=C1SC=2C=C3C1=CC=CC=C1 OWEUOGSUFUUNQE-UHFFFAOYSA-N 0.000 description 1
- BIISIZOQPWZPPS-UHFFFAOYSA-N 2-tert-butylperoxypropan-2-ylbenzene Chemical compound CC(C)(C)OOC(C)(C)C1=CC=CC=C1 BIISIZOQPWZPPS-UHFFFAOYSA-N 0.000 description 1
- UUIMDJFBHNDZOW-UHFFFAOYSA-N 2-tert-butylpyridine Chemical compound CC(C)(C)C1=CC=CC=N1 UUIMDJFBHNDZOW-UHFFFAOYSA-N 0.000 description 1
- KFGFVPMRLOQXNB-UHFFFAOYSA-N 3,5,5-trimethylhexanoyl 3,5,5-trimethylhexaneperoxoate Chemical compound CC(C)(C)CC(C)CC(=O)OOC(=O)CC(C)CC(C)(C)C KFGFVPMRLOQXNB-UHFFFAOYSA-N 0.000 description 1
- LJGHYPLBDBRCRZ-UHFFFAOYSA-N 3-(3-aminophenyl)sulfonylaniline Chemical compound NC1=CC=CC(S(=O)(=O)C=2C=C(N)C=CC=2)=C1 LJGHYPLBDBRCRZ-UHFFFAOYSA-N 0.000 description 1
- YNJSNEKCXVFDKW-UHFFFAOYSA-N 3-(5-amino-1h-indol-3-yl)-2-azaniumylpropanoate Chemical compound C1=C(N)C=C2C(CC(N)C(O)=O)=CNC2=C1 YNJSNEKCXVFDKW-UHFFFAOYSA-N 0.000 description 1
- JYLNVJYYQQXNEK-UHFFFAOYSA-N 3-amino-2-(4-chlorophenyl)-1-propanesulfonic acid Chemical compound OS(=O)(=O)CC(CN)C1=CC=C(Cl)C=C1 JYLNVJYYQQXNEK-UHFFFAOYSA-N 0.000 description 1
- 125000003542 3-methylbutan-2-yl group Chemical group [H]C([H])([H])C([H])(*)C([H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- ZYVFZUPZEORVHB-UHFFFAOYSA-N 3-phenyl-2-thiophen-2-ylthiophene Chemical compound C1=CSC(C2=C(C=CS2)C=2C=CC=CC=2)=C1 ZYVFZUPZEORVHB-UHFFFAOYSA-N 0.000 description 1
- MKTOIPPVFPJEQO-UHFFFAOYSA-N 4-(3-carboxypropanoylperoxy)-4-oxobutanoic acid Chemical compound OC(=O)CCC(=O)OOC(=O)CCC(O)=O MKTOIPPVFPJEQO-UHFFFAOYSA-N 0.000 description 1
- ZHKCHSNXUCRFSM-UHFFFAOYSA-N 4-[2-[4,4-bis(tert-butylperoxy)cyclohexyl]propan-2-yl]-1,1-bis(tert-butylperoxy)cyclohexane Chemical compound C1CC(OOC(C)(C)C)(OOC(C)(C)C)CCC1C(C)(C)C1CCC(OOC(C)(C)C)(OOC(C)(C)C)CC1 ZHKCHSNXUCRFSM-UHFFFAOYSA-N 0.000 description 1
- KWXICGTUELOLSQ-UHFFFAOYSA-N 4-dodecylbenzenesulfonic acid Chemical compound CCCCCCCCCCCCC1=CC=C(S(O)(=O)=O)C=C1 KWXICGTUELOLSQ-UHFFFAOYSA-N 0.000 description 1
- JQEJJSKHXVZRNM-UHFFFAOYSA-K 4-ethylbenzenesulfonate;iron(3+) Chemical compound [Fe+3].CCC1=CC=C(S([O-])(=O)=O)C=C1.CCC1=CC=C(S([O-])(=O)=O)C=C1.CCC1=CC=C(S([O-])(=O)=O)C=C1 JQEJJSKHXVZRNM-UHFFFAOYSA-K 0.000 description 1
- NFWPZNNZUCPLAX-UHFFFAOYSA-N 4-methoxy-3-methylaniline Chemical compound COC1=CC=C(N)C=C1C NFWPZNNZUCPLAX-UHFFFAOYSA-N 0.000 description 1
- XECCJSBEUDPALF-UHFFFAOYSA-N 5-(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)-2,3-dihydrothieno[3,4-b][1,4]dioxine Chemical compound S1C=C2OCCOC2=C1C1=C2OCCOC2=CS1 XECCJSBEUDPALF-UHFFFAOYSA-N 0.000 description 1
- MARUHZGHZWCEQU-UHFFFAOYSA-N 5-phenyl-2h-tetrazole Chemical compound C1=CC=CC=C1C1=NNN=N1 MARUHZGHZWCEQU-UHFFFAOYSA-N 0.000 description 1
- YPIFGDQKSSMYHQ-UHFFFAOYSA-M 7,7-dimethyloctanoate Chemical compound CC(C)(C)CCCCCC([O-])=O YPIFGDQKSSMYHQ-UHFFFAOYSA-M 0.000 description 1
- LAYHMKGDTFMKGR-UHFFFAOYSA-N 8-ethoxyoctan-1-ol Chemical group CCOCCCCCCCCO LAYHMKGDTFMKGR-UHFFFAOYSA-N 0.000 description 1
- 239000004925 Acrylic resin Substances 0.000 description 1
- 229920000178 Acrylic resin Polymers 0.000 description 1
- FIPWRIJSWJWJAI-UHFFFAOYSA-N Butyl carbitol 6-propylpiperonyl ether Chemical compound C1=C(CCC)C(COCCOCCOCCCC)=CC2=C1OCO2 FIPWRIJSWJWJAI-UHFFFAOYSA-N 0.000 description 1
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 1
- DRTRBNANCRUBEB-UHFFFAOYSA-J C(CCCCCCC)C(C(=O)[O-])(O)CCCCCCCCOCCCCCCCCC(C(=O)[O-])(O)CCCCCCCC.[Ti+4].C(CCCCCCC)C(C(=O)[O-])(O)CCCCCCCCOCCCCCCCCC(C(=O)[O-])(O)CCCCCCCC Chemical compound C(CCCCCCC)C(C(=O)[O-])(O)CCCCCCCCOCCCCCCCCC(C(=O)[O-])(O)CCCCCCCC.[Ti+4].C(CCCCCCC)C(C(=O)[O-])(O)CCCCCCCCOCCCCCCCCC(C(=O)[O-])(O)CCCCCCCC DRTRBNANCRUBEB-UHFFFAOYSA-J 0.000 description 1
- DIICDKVONBLTBM-UHFFFAOYSA-L CC(C)O[Ti++]OC(C)C.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O Chemical compound CC(C)O[Ti++]OC(C)C.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O DIICDKVONBLTBM-UHFFFAOYSA-L 0.000 description 1
- KLQSRTKDOLFPQJ-UHFFFAOYSA-M CCCCO[Ti+](OCCCC)OCCCC.CCCCCCCCCCCCCCCCCC([O-])=O Chemical compound CCCCO[Ti+](OCCCC)OCCCC.CCCCCCCCCCCCCCCCCC([O-])=O KLQSRTKDOLFPQJ-UHFFFAOYSA-M 0.000 description 1
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 1
- ZKQDCIXGCQPQNV-UHFFFAOYSA-N Calcium hypochlorite Chemical compound [Ca+2].Cl[O-].Cl[O-] ZKQDCIXGCQPQNV-UHFFFAOYSA-N 0.000 description 1
- 229910004613 CdTe Inorganic materials 0.000 description 1
- 229910052684 Cerium Inorganic materials 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 1
- 239000004593 Epoxy Substances 0.000 description 1
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- GYHNNYVSQQEPJS-UHFFFAOYSA-N Gallium Chemical compound [Ga] GYHNNYVSQQEPJS-UHFFFAOYSA-N 0.000 description 1
- 229910001218 Gallium arsenide Inorganic materials 0.000 description 1
- 229910003771 Gold(I) chloride Inorganic materials 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 229910021578 Iron(III) chloride Inorganic materials 0.000 description 1
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical group CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 1
- 238000006576 Kolbe electrolysis reaction Methods 0.000 description 1
- YIVJZNGAASQVEM-UHFFFAOYSA-N Lauroyl peroxide Chemical compound CCCCCCCCCCCC(=O)OOC(=O)CCCCCCCCCCC YIVJZNGAASQVEM-UHFFFAOYSA-N 0.000 description 1
- 229910013063 LiBF 4 Inorganic materials 0.000 description 1
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 1
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 1
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229920012266 Poly(ether sulfone) PES Polymers 0.000 description 1
- 239000004952 Polyamide Substances 0.000 description 1
- 239000004695 Polyether sulfone Substances 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 239000005708 Sodium hypochlorite Substances 0.000 description 1
- PJANXHGTPQOBST-VAWYXSNFSA-N Stilbene Natural products C=1C=CC=CC=1/C=C/C1=CC=CC=C1 PJANXHGTPQOBST-VAWYXSNFSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- NRTOMJZYCJJWKI-UHFFFAOYSA-N Titanium nitride Chemical compound [Ti]#N NRTOMJZYCJJWKI-UHFFFAOYSA-N 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- SMEGJBVQLJJKKX-HOTMZDKISA-N [(2R,3S,4S,5R,6R)-5-acetyloxy-3,4,6-trihydroxyoxan-2-yl]methyl acetate Chemical compound CC(=O)OC[C@@H]1[C@H]([C@@H]([C@H]([C@@H](O1)O)OC(=O)C)O)O SMEGJBVQLJJKKX-HOTMZDKISA-N 0.000 description 1
- JUIBLDFFVYKUAC-UHFFFAOYSA-N [5-(2-ethylhexanoylperoxy)-2,5-dimethylhexan-2-yl] 2-ethylhexaneperoxoate Chemical compound CCCCC(CC)C(=O)OOC(C)(C)CCC(C)(C)OOC(=O)C(CC)CCCC JUIBLDFFVYKUAC-UHFFFAOYSA-N 0.000 description 1
- ODMBZECFFGGOSH-UHFFFAOYSA-L [Fe+2].C(C)C1=CC=C(C=C1)S(=O)(=O)[O-].C(C)C1=CC=C(C=C1)S(=O)(=O)[O-] Chemical compound [Fe+2].C(C)C1=CC=C(C=C1)S(=O)(=O)[O-].C(C)C1=CC=C(C=C1)S(=O)(=O)[O-] ODMBZECFFGGOSH-UHFFFAOYSA-L 0.000 description 1
- UMXAKWDYSKLYBU-UHFFFAOYSA-K [Fe+3].[O-]S(=O)(=O)c1cccc2ccccc12.[O-]S(=O)(=O)c1cccc2ccccc12.[O-]S(=O)(=O)c1cccc2ccccc12 Chemical compound [Fe+3].[O-]S(=O)(=O)c1cccc2ccccc12.[O-]S(=O)(=O)c1cccc2ccccc12.[O-]S(=O)(=O)c1cccc2ccccc12 UMXAKWDYSKLYBU-UHFFFAOYSA-K 0.000 description 1
- 239000011354 acetal resin Substances 0.000 description 1
- 229940081735 acetylcellulose Drugs 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 229920006243 acrylic copolymer Polymers 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 238000007754 air knife coating Methods 0.000 description 1
- 239000005456 alcohol based solvent Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 150000001409 amidines Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- ROOXNKNUYICQNP-UHFFFAOYSA-N ammonium persulfate Chemical compound [NH4+].[NH4+].[O-]S(=O)(=O)OOS([O-])(=O)=O ROOXNKNUYICQNP-UHFFFAOYSA-N 0.000 description 1
- 239000012935 ammoniumperoxodisulfate Substances 0.000 description 1
- WATWJIUSRGPENY-UHFFFAOYSA-N antimony atom Chemical compound [Sb] WATWJIUSRGPENY-UHFFFAOYSA-N 0.000 description 1
- 150000004982 aromatic amines Chemical class 0.000 description 1
- 125000004391 aryl sulfonyl group Chemical group 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- XRASGLNHKOPXQL-UHFFFAOYSA-L azane 2-oxidopropanoate titanium(4+) dihydrate Chemical compound N.N.O.O.[Ti+4].CC([O-])C([O-])=O.CC([O-])C([O-])=O XRASGLNHKOPXQL-UHFFFAOYSA-L 0.000 description 1
- ZJRXSAYFZMGQFP-UHFFFAOYSA-N barium peroxide Chemical compound [Ba+2].[O-][O-] ZJRXSAYFZMGQFP-UHFFFAOYSA-N 0.000 description 1
- JRPBQTZRNDNNOP-UHFFFAOYSA-N barium titanate Chemical compound [Ba+2].[Ba+2].[O-][Ti]([O-])([O-])[O-] JRPBQTZRNDNNOP-UHFFFAOYSA-N 0.000 description 1
- 229910002113 barium titanate Inorganic materials 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical group C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 1
- RJNQAWYDOFQOFG-UHFFFAOYSA-N benzoyl 3-methylbenzenecarboperoxoate Chemical compound CC1=CC=CC(C(=O)OOC(=O)C=2C=CC=CC=2)=C1 RJNQAWYDOFQOFG-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- SXDBWCPKPHAZSM-UHFFFAOYSA-N bromic acid Chemical compound OBr(=O)=O SXDBWCPKPHAZSM-UHFFFAOYSA-N 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- QARVLSVVCXYDNA-UHFFFAOYSA-N bromobenzene Chemical compound BrC1=CC=CC=C1 QARVLSVVCXYDNA-UHFFFAOYSA-N 0.000 description 1
- NSGQRLUGQNBHLD-UHFFFAOYSA-N butan-2-yl butan-2-yloxycarbonyloxy carbonate Chemical compound CCC(C)OC(=O)OOC(=O)OC(C)CC NSGQRLUGQNBHLD-UHFFFAOYSA-N 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- BXIQXYOPGBXIEM-UHFFFAOYSA-N butyl 4,4-bis(tert-butylperoxy)pentanoate Chemical compound CCCCOC(=O)CCC(C)(OOC(C)(C)C)OOC(C)(C)C BXIQXYOPGBXIEM-UHFFFAOYSA-N 0.000 description 1
- CXKCTMHTOKXKQT-UHFFFAOYSA-N cadmium oxide Inorganic materials [Cd]=O CXKCTMHTOKXKQT-UHFFFAOYSA-N 0.000 description 1
- AOWKSNWVBZGMTJ-UHFFFAOYSA-N calcium titanate Chemical compound [Ca+2].[O-][Ti]([O-])=O AOWKSNWVBZGMTJ-UHFFFAOYSA-N 0.000 description 1
- NNLOHLDVJGPUFR-UHFFFAOYSA-L calcium;3,4,5,6-tetrahydroxy-2-oxohexanoate Chemical compound [Ca+2].OCC(O)C(O)C(O)C(=O)C([O-])=O.OCC(O)C(O)C(O)C(=O)C([O-])=O NNLOHLDVJGPUFR-UHFFFAOYSA-L 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 239000006229 carbon black Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical class OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- ZMIGMASIKSOYAM-UHFFFAOYSA-N cerium Chemical compound [Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce][Ce] ZMIGMASIKSOYAM-UHFFFAOYSA-N 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- XTEGARKTQYYJKE-UHFFFAOYSA-N chloric acid Chemical compound OCl(=O)=O XTEGARKTQYYJKE-UHFFFAOYSA-N 0.000 description 1
- 229940005991 chloric acid Drugs 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- ZCDOYSPFYFSLEW-UHFFFAOYSA-N chromate(2-) Chemical compound [O-][Cr]([O-])(=O)=O ZCDOYSPFYFSLEW-UHFFFAOYSA-N 0.000 description 1
- KRVSOGSZCMJSLX-UHFFFAOYSA-L chromic acid Substances O[Cr](O)(=O)=O KRVSOGSZCMJSLX-UHFFFAOYSA-L 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 239000008139 complexing agent Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- UIPVMGDJUWUZEI-UHFFFAOYSA-N copper;selanylideneindium Chemical class [Cu].[In]=[Se] UIPVMGDJUWUZEI-UHFFFAOYSA-N 0.000 description 1
- LCUOIYYHNRBAFS-UHFFFAOYSA-N copper;sulfanylideneindium Chemical class [Cu].[In]=S LCUOIYYHNRBAFS-UHFFFAOYSA-N 0.000 description 1
- 238000003851 corona treatment Methods 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- SPTHWAJJMLCAQF-UHFFFAOYSA-M ctk4f8481 Chemical compound [O-]O.CC(C)C1=CC=CC=C1C(C)C SPTHWAJJMLCAQF-UHFFFAOYSA-M 0.000 description 1
- 238000007766 curtain coating Methods 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- IGKQSFOEHVTSER-UHFFFAOYSA-N cyclohexa-2,5-dien-1-ylidenemethylbenzene Chemical compound C1=CCC=CC1=CC1=CC=CC=C1 IGKQSFOEHVTSER-UHFFFAOYSA-N 0.000 description 1
- 150000001934 cyclohexanes Chemical class 0.000 description 1
- 125000006547 cyclononyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000032798 delamination Effects 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- LSXWFXONGKSEMY-UHFFFAOYSA-N di-tert-butyl peroxide Chemical compound CC(C)(C)OOC(C)(C)C LSXWFXONGKSEMY-UHFFFAOYSA-N 0.000 description 1
- 125000005266 diarylamine group Chemical group 0.000 description 1
- SOCTUWSJJQCPFX-UHFFFAOYSA-N dichromate(2-) Chemical compound [O-][Cr](=O)(=O)O[Cr]([O-])(=O)=O SOCTUWSJJQCPFX-UHFFFAOYSA-N 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 125000001664 diethylamino group Chemical group [H]C([H])([H])C([H])([H])N(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- 238000007598 dipping method Methods 0.000 description 1
- VIUKNDFMFRTONS-UHFFFAOYSA-N distrontium;niobium(5+);oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[O-2].[O-2].[O-2].[O-2].[Sr+2].[Sr+2].[Nb+5].[Nb+5] VIUKNDFMFRTONS-UHFFFAOYSA-N 0.000 description 1
- 238000007606 doctor blade method Methods 0.000 description 1
- YRIUSKIDOIARQF-UHFFFAOYSA-N dodecyl benzenesulfonate Chemical compound CCCCCCCCCCCCOS(=O)(=O)C1=CC=CC=C1 YRIUSKIDOIARQF-UHFFFAOYSA-N 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229940071161 dodecylbenzenesulfonate Drugs 0.000 description 1
- 239000002019 doping agent Substances 0.000 description 1
- 230000005684 electric field Effects 0.000 description 1
- 238000005530 etching Methods 0.000 description 1
- 150000002168 ethanoic acid esters Chemical class 0.000 description 1
- 239000004210 ether based solvent Substances 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- XYIBRDXRRQCHLP-UHFFFAOYSA-N ethyl acetoacetate Chemical compound CCOC(=O)CC(C)=O XYIBRDXRRQCHLP-UHFFFAOYSA-N 0.000 description 1
- 229940093858 ethyl acetoacetate Drugs 0.000 description 1
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 238000007765 extrusion coating Methods 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- HKIOYBQGHSTUDB-UHFFFAOYSA-N folpet Chemical group C1=CC=C2C(=O)N(SC(Cl)(Cl)Cl)C(=O)C2=C1 HKIOYBQGHSTUDB-UHFFFAOYSA-N 0.000 description 1
- AWJWCTOOIBYHON-UHFFFAOYSA-N furo[3,4-b]pyrazine-5,7-dione Chemical compound C1=CN=C2C(=O)OC(=O)C2=N1 AWJWCTOOIBYHON-UHFFFAOYSA-N 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 229910052733 gallium Inorganic materials 0.000 description 1
- 229910052732 germanium Inorganic materials 0.000 description 1
- GNPVGFCGXDBREM-UHFFFAOYSA-N germanium atom Chemical compound [Ge] GNPVGFCGXDBREM-UHFFFAOYSA-N 0.000 description 1
- QFWPJPIVLCBXFJ-UHFFFAOYSA-N glymidine Chemical compound N1=CC(OCCOC)=CN=C1NS(=O)(=O)C1=CC=CC=C1 QFWPJPIVLCBXFJ-UHFFFAOYSA-N 0.000 description 1
- FDWREHZXQUYJFJ-UHFFFAOYSA-M gold monochloride Chemical compound [Cl-].[Au+] FDWREHZXQUYJFJ-UHFFFAOYSA-M 0.000 description 1
- 238000007756 gravure coating Methods 0.000 description 1
- 230000005283 ground state Effects 0.000 description 1
- 229910052735 hafnium Inorganic materials 0.000 description 1
- VBJZVLUMGGDVMO-UHFFFAOYSA-N hafnium atom Chemical compound [Hf] VBJZVLUMGGDVMO-UHFFFAOYSA-N 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 125000004836 hexamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- RKKOMEIYHHASIN-UHFFFAOYSA-N hydroperoxyboronic acid Chemical compound OOB(O)O RKKOMEIYHHASIN-UHFFFAOYSA-N 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- GEOVEUCEIQCBKH-UHFFFAOYSA-N hypoiodous acid Chemical compound IO GEOVEUCEIQCBKH-UHFFFAOYSA-N 0.000 description 1
- RHZWSUVWRRXEJF-UHFFFAOYSA-N indium tin Chemical compound [In].[Sn] RHZWSUVWRRXEJF-UHFFFAOYSA-N 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 239000011810 insulating material Substances 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 229910052741 iridium Inorganic materials 0.000 description 1
- GKOZUEZYRPOHIO-UHFFFAOYSA-N iridium atom Chemical compound [Ir] GKOZUEZYRPOHIO-UHFFFAOYSA-N 0.000 description 1
- QXYHCTRKUSRDPG-UHFFFAOYSA-L iron(2+);iron(3+);sulfate Chemical compound [Fe+2].[Fe+3].[O-]S([O-])(=O)=O QXYHCTRKUSRDPG-UHFFFAOYSA-L 0.000 description 1
- GGHPFIYIFKEQCM-UHFFFAOYSA-L iron(2+);naphthalene-1-sulfonate Chemical compound [Fe+2].C1=CC=C2C(S(=O)(=O)[O-])=CC=CC2=C1.C1=CC=C2C(S(=O)(=O)[O-])=CC=CC2=C1 GGHPFIYIFKEQCM-UHFFFAOYSA-L 0.000 description 1
- RUTXIHLAWFEWGM-UHFFFAOYSA-H iron(3+) sulfate Chemical compound [Fe+3].[Fe+3].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O RUTXIHLAWFEWGM-UHFFFAOYSA-H 0.000 description 1
- NPFOYSMITVOQOS-UHFFFAOYSA-K iron(III) citrate Chemical compound [Fe+3].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NPFOYSMITVOQOS-UHFFFAOYSA-K 0.000 description 1
- 229910000360 iron(III) sulfate Inorganic materials 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 229940035429 isobutyl alcohol Drugs 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 239000005453 ketone based solvent Substances 0.000 description 1
- 238000004898 kneading Methods 0.000 description 1
- 229910052743 krypton Inorganic materials 0.000 description 1
- DNNSSWSSYDEUBZ-UHFFFAOYSA-N krypton atom Chemical compound [Kr] DNNSSWSSYDEUBZ-UHFFFAOYSA-N 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- FZLIPJUXYLNCLC-UHFFFAOYSA-N lanthanum atom Chemical compound [La] FZLIPJUXYLNCLC-UHFFFAOYSA-N 0.000 description 1
- MRELNEQAGSRDBK-UHFFFAOYSA-N lanthanum oxide Inorganic materials [O-2].[O-2].[O-2].[La+3].[La+3] MRELNEQAGSRDBK-UHFFFAOYSA-N 0.000 description 1
- 238000013532 laser treatment Methods 0.000 description 1
- 239000011133 lead Substances 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 230000031700 light absorption Effects 0.000 description 1
- 125000002463 lignoceryl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- MHCFAGZWMAWTNR-UHFFFAOYSA-M lithium perchlorate Chemical compound [Li+].[O-]Cl(=O)(=O)=O MHCFAGZWMAWTNR-UHFFFAOYSA-M 0.000 description 1
- 229910001496 lithium tetrafluoroborate Inorganic materials 0.000 description 1
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 150000002736 metal compounds Chemical class 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 229910052750 molybdenum Inorganic materials 0.000 description 1
- 239000011733 molybdenum Substances 0.000 description 1
- 125000002911 monocyclic heterocycle group Chemical group 0.000 description 1
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- WCZAXBXVDLKQGV-UHFFFAOYSA-N n,n-dimethyl-2-(7-oxobenzo[c]fluoren-5-yl)oxyethanamine oxide Chemical compound C12=CC=CC=C2C(OCC[N+](C)([O-])C)=CC2=C1C1=CC=CC=C1C2=O WCZAXBXVDLKQGV-UHFFFAOYSA-N 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- 229910052754 neon Inorganic materials 0.000 description 1
- GKAOGPIIYCISHV-UHFFFAOYSA-N neon atom Chemical compound [Ne] GKAOGPIIYCISHV-UHFFFAOYSA-N 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 229910052758 niobium Inorganic materials 0.000 description 1
- 229910000484 niobium oxide Inorganic materials 0.000 description 1
- 150000004767 nitrides Chemical class 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 125000006611 nonyloxy group Chemical group 0.000 description 1
- 125000005447 octyloxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 239000005416 organic matter Substances 0.000 description 1
- 150000001451 organic peroxides Chemical class 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 125000000369 oxido group Chemical group [*]=O 0.000 description 1
- BPUBBGLMJRNUCC-UHFFFAOYSA-N oxygen(2-);tantalum(5+) Chemical compound [O-2].[O-2].[O-2].[O-2].[O-2].[Ta+5].[Ta+5] BPUBBGLMJRNUCC-UHFFFAOYSA-N 0.000 description 1
- 230000000149 penetrating effect Effects 0.000 description 1
- 125000004817 pentamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- 125000005010 perfluoroalkyl group Chemical group 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 125000005385 peroxodisulfate group Chemical group 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-N peroxydisulfuric acid Chemical compound OS(=O)(=O)OOS(O)(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-N 0.000 description 1
- MPNNOLHYOHFJKL-UHFFFAOYSA-N peroxyphosphoric acid Chemical compound OOP(O)(O)=O MPNNOLHYOHFJKL-UHFFFAOYSA-N 0.000 description 1
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 description 1
- 125000001639 phenylmethylene group Chemical group [H]C(=*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000005328 phosphinyl group Chemical group [PH2](=O)* 0.000 description 1
- 125000001476 phosphono group Chemical group [H]OP(*)(=O)O[H] 0.000 description 1
- 125000005499 phosphonyl group Chemical group 0.000 description 1
- 230000001699 photocatalysis Effects 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- SIOXPEMLGUPBBT-UHFFFAOYSA-N picolinic acid Chemical compound OC(=O)C1=CC=CC=N1 SIOXPEMLGUPBBT-UHFFFAOYSA-N 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 229960005235 piperonyl butoxide Drugs 0.000 description 1
- 238000009832 plasma treatment Methods 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229910052696 pnictogen Inorganic materials 0.000 description 1
- 229920002647 polyamide Polymers 0.000 description 1
- 229920006122 polyamide resin Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 229920006289 polycarbonate film Polymers 0.000 description 1
- 229920001225 polyester resin Polymers 0.000 description 1
- 239000004645 polyester resin Substances 0.000 description 1
- 229920006393 polyether sulfone Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920006290 polyethylene naphthalate film Polymers 0.000 description 1
- 229920001721 polyimide Polymers 0.000 description 1
- 239000009719 polyimide resin Substances 0.000 description 1
- 239000003505 polymerization initiator Substances 0.000 description 1
- 229920005672 polyolefin resin Polymers 0.000 description 1
- 229920006324 polyoxymethylene Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229920002635 polyurethane Polymers 0.000 description 1
- 239000004814 polyurethane Substances 0.000 description 1
- 239000004800 polyvinyl chloride Substances 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- BWJUFXUULUEGMA-UHFFFAOYSA-N propan-2-yl propan-2-yloxycarbonyloxy carbonate Chemical compound CC(C)OC(=O)OOC(=O)OC(C)C BWJUFXUULUEGMA-UHFFFAOYSA-N 0.000 description 1
- YPVDWEHVCUBACK-UHFFFAOYSA-N propoxycarbonyloxy propyl carbonate Chemical compound CCCOC(=O)OOC(=O)OCCC YPVDWEHVCUBACK-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- GGYFMLJDMAMTAB-UHFFFAOYSA-N selanylidenelead Chemical compound [Pb]=[Se] GGYFMLJDMAMTAB-UHFFFAOYSA-N 0.000 description 1
- 239000011669 selenium Substances 0.000 description 1
- 125000003748 selenium group Chemical group *[Se]* 0.000 description 1
- 150000003346 selenoethers Chemical class 0.000 description 1
- 238000004904 shortening Methods 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 description 1
- PFUVRDFDKPNGAV-UHFFFAOYSA-N sodium peroxide Chemical compound [Na+].[Na+].[O-][O-] PFUVRDFDKPNGAV-UHFFFAOYSA-N 0.000 description 1
- PJANXHGTPQOBST-UHFFFAOYSA-N stilbene Chemical compound C=1C=CC=CC=1C=CC1=CC=CC=C1 PJANXHGTPQOBST-UHFFFAOYSA-N 0.000 description 1
- 235000021286 stilbenes Nutrition 0.000 description 1
- 229910052712 strontium Inorganic materials 0.000 description 1
- CIOAGBVUUVVLOB-UHFFFAOYSA-N strontium atom Chemical compound [Sr] CIOAGBVUUVVLOB-UHFFFAOYSA-N 0.000 description 1
- VEALVRVVWBQVSL-UHFFFAOYSA-N strontium titanate Chemical compound [Sr+2].[O-][Ti]([O-])=O VEALVRVVWBQVSL-UHFFFAOYSA-N 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- 150000004763 sulfides Chemical class 0.000 description 1
- 125000000213 sulfino group Chemical group [H]OS(*)=O 0.000 description 1
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 229910052715 tantalum Inorganic materials 0.000 description 1
- GUVRBAGPIYLISA-UHFFFAOYSA-N tantalum atom Chemical compound [Ta] GUVRBAGPIYLISA-UHFFFAOYSA-N 0.000 description 1
- 229910001936 tantalum oxide Inorganic materials 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- PORWMNRCUJJQNO-UHFFFAOYSA-N tellurium atom Chemical group [Te] PORWMNRCUJJQNO-UHFFFAOYSA-N 0.000 description 1
- OPQYOFWUFGEMRZ-UHFFFAOYSA-N tert-butyl 2,2-dimethylpropaneperoxoate Chemical compound CC(C)(C)OOC(=O)C(C)(C)C OPQYOFWUFGEMRZ-UHFFFAOYSA-N 0.000 description 1
- VNJISVYSDHJQFR-UHFFFAOYSA-N tert-butyl 4,4-dimethylpentaneperoxoate Chemical compound CC(C)(C)CCC(=O)OOC(C)(C)C VNJISVYSDHJQFR-UHFFFAOYSA-N 0.000 description 1
- NMOALOSNPWTWRH-UHFFFAOYSA-N tert-butyl 7,7-dimethyloctaneperoxoate Chemical compound CC(C)(C)CCCCCC(=O)OOC(C)(C)C NMOALOSNPWTWRH-UHFFFAOYSA-N 0.000 description 1
- SWAXTRYEYUTSAP-UHFFFAOYSA-N tert-butyl ethaneperoxoate Chemical compound CC(=O)OOC(C)(C)C SWAXTRYEYUTSAP-UHFFFAOYSA-N 0.000 description 1
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- CIHOLLKRGTVIJN-UHFFFAOYSA-N tert‐butyl hydroperoxide Chemical compound CC(C)(C)OO CIHOLLKRGTVIJN-UHFFFAOYSA-N 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- KBLZDCFTQSIIOH-UHFFFAOYSA-M tetrabutylazanium;perchlorate Chemical compound [O-]Cl(=O)(=O)=O.CCCC[N+](CCCC)(CCCC)CCCC KBLZDCFTQSIIOH-UHFFFAOYSA-M 0.000 description 1
- PCCVSPMFGIFTHU-UHFFFAOYSA-N tetracyanoquinodimethane Chemical compound N#CC(C#N)=C1C=CC(=C(C#N)C#N)C=C1 PCCVSPMFGIFTHU-UHFFFAOYSA-N 0.000 description 1
- MNWRORMXBIWXCI-UHFFFAOYSA-N tetrakis(dimethylamido)titanium Chemical compound CN(C)[Ti](N(C)C)(N(C)C)N(C)C MNWRORMXBIWXCI-UHFFFAOYSA-N 0.000 description 1
- 238000009210 therapy by ultrasound Methods 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 239000012780 transparent material Substances 0.000 description 1
- 125000005259 triarylamine group Chemical group 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 239000013638 trimer Substances 0.000 description 1
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 1
- 238000001771 vacuum deposition Methods 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- 229910001935 vanadium oxide Inorganic materials 0.000 description 1
- 229920006163 vinyl copolymer Polymers 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- KAKZBPTYRLMSJV-UHFFFAOYSA-N vinyl-ethylene Natural products C=CC=C KAKZBPTYRLMSJV-UHFFFAOYSA-N 0.000 description 1
- 239000004034 viscosity adjusting agent Substances 0.000 description 1
- 239000011800 void material Substances 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical compound [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D11/00—Inks
- C09D11/30—Inkjet printing inks
- C09D11/32—Inkjet printing inks characterised by colouring agents
- C09D11/322—Pigment inks
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D11/00—Inks
- C09D11/52—Electrically conductive inks
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/10—Organic polymers or oligomers
- H10K85/111—Organic polymers or oligomers comprising aromatic, heteroaromatic, or aryl chains, e.g. polyaniline, polyphenylene or polyphenylene vinylene
- H10K85/113—Heteroaromatic compounds comprising sulfur or selene, e.g. polythiophene
- H10K85/1135—Polyethylene dioxythiophene [PEDOT]; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/10—Definition of the polymer structure
- C08G2261/14—Side-groups
- C08G2261/142—Side-chains containing oxygen
- C08G2261/1424—Side-chains containing oxygen containing ether groups, including alkoxy
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/40—Polymerisation processes
- C08G2261/44—Electrochemical polymerisation, i.e. oxidative or reductive coupling
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/90—Applications
- C08G2261/91—Photovoltaic applications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G61/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G61/12—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule
- C08G61/122—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides
- C08G61/123—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides derived from five-membered heterocyclic compounds
- C08G61/126—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides derived from five-membered heterocyclic compounds with a five-membered ring containing one sulfur atom in the ring
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES, LIGHT-SENSITIVE OR TEMPERATURE-SENSITIVE DEVICES OF THE ELECTROLYTIC TYPE
- H01G9/00—Electrolytic capacitors, rectifiers, detectors, switching devices, light-sensitive or temperature-sensitive devices; Processes of their manufacture
- H01G9/20—Light-sensitive devices
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/631—Amine compounds having at least two aryl rest on at least one amine-nitrogen atom, e.g. triphenylamine
- H10K85/633—Amine compounds having at least two aryl rest on at least one amine-nitrogen atom, e.g. triphenylamine comprising polycyclic condensed aromatic hydrocarbons as substituents on the nitrogen atom
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/631—Amine compounds having at least two aryl rest on at least one amine-nitrogen atom, e.g. triphenylamine
- H10K85/636—Amine compounds having at least two aryl rest on at least one amine-nitrogen atom, e.g. triphenylamine comprising heteroaromatic hydrocarbons as substituents on the nitrogen atom
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/649—Aromatic compounds comprising a hetero atom
- H10K85/655—Aromatic compounds comprising a hetero atom comprising only sulfur as heteroatom
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E10/00—Energy generation through renewable energy sources
- Y02E10/50—Photovoltaic [PV] energy
- Y02E10/549—Organic PV cells
Definitions
- the present invention relates to a method for manufacturing a photoelectric conversion element.
- the photovoltaic effect is a phenomenon in which an electromotive force is generated by irradiating a substance with light.
- a photoelectric conversion element containing the substance By using a photoelectric conversion element containing the substance, light energy can be converted into electric energy.
- the photoelectric conversion element usually has a configuration in which a substrate, a first electrode, a photoelectric conversion layer (a semiconductor layer carrying a sensitizing dye), a hole transport layer, and a second electrode are arranged in this order.
- the sensitizing dye in the photoelectric conversion layer inside the element is excited to emit electrons.
- the excited electrons move to the first electrode, and the electrons move to the second electrode through an external circuit and are supplied to the hole transport layer.
- the oxidized sensitizing dye (by releasing electrons) receives electrons from the hole transport layer and returns to the ground state. By repeating such a cycle, light energy is converted into electrical energy.
- the performance required for the photoelectric conversion element includes excellent photoelectric conversion efficiency and high durability. Attempts have been made so far to improve various elements of the photoelectric conversion element and to improve the performance of the photoelectric conversion element.
- Patent Document 1 focuses on the contact between the hole transport layer constituting the photoelectric conversion element and the counter electrode (second electrode) to improve the photoelectric conversion efficiency.
- Patent Document 1 discloses a dye-sensitized mesoporous titanium dioxide using a combination of an electron conductive material and a polymer electrolyte having a film forming property as a counter electrode constituting material of a solid-type dye-sensitized solar cell.
- a solid dye-sensitized solar cell is disclosed.
- the hole transport polymer solid electrolyte is obtained by immersing porous titanium dioxide carrying a dye in a solution containing a monomer such as pyrrole, and irradiating with light while maintaining a predetermined potential to polymerize the monomer. It is formed by that.
- a solid dye-sensitized solar having a high current density is formed by dropping a kneading liquid containing a predetermined component on the surface of the hole transport layer and expanding the liquid to form a counter electrode. It is described that a battery is obtained.
- Patent Document 2 attempts to improve photoelectric conversion efficiency by providing a short-circuit prevention layer between the first electrode and the photoelectric conversion layer. Specifically, Patent Document 2 discloses a first electrode (first electrode), a second electrode (second electrode) disposed to face the first electrode, and the first electrode.
- a light-receiving layer (heterojunction photoelectric conversion layer in which a p-type organic semiconductor and an n-type inorganic semiconductor are bonded), which is located between the first electrode and the second electrode, and at least a part of which is porous, and the light-receiving layer And a hole transport layer positioned between the second electrode and a rectifying barrier formed at an interface between the light receiving layer and the hole transport layer, the photoelectric conversion element comprising: The photoelectric conversion element characterized by having a short circuit prevention means which prevents or suppresses the short circuit between 1 electrode and the said hole transport layer is described.
- a short circuit prevention means (barrier layer / short circuit prevention layer) is provided between the first electrode and the hole transport layer to prevent a short circuit between the first electrode and the hole transport layer. It is described that a photoelectric conversion element that prevents leakage current and is excellent in photoelectric conversion efficiency can be obtained.
- a hole transport layer is formed using a method called an electrolytic polymerization method.
- the electrolytic polymerization method is a method of obtaining a conductive polymer by polymerizing a conductive polymer precursor by immersing an electrode pair in a solution in which a conductive polymer precursor (monomer or the like) is dissolved and applying a voltage. It is.
- a hole transport layer is formed by electrolytic polymerization on a photoelectric conversion layer containing a sensitizing dye supported on a semiconductor, decomposition (deterioration) of the sensitizing dye due to voltage application. In order to prevent this, it is necessary to perform polymerization under a low voltage.
- Patent Document 2 describes that the barrier layer (short-circuit prevention layer) can be formed by a sol-gel method, a vapor deposition method, a sputtering method, a spray pyrolysis method, a jet mold method, a CVD method, or the like. .
- the short-circuit prevention layer cannot sufficiently cover the first electrode, and a part of the first electrode It has been found that may protrude from the short-circuit prevention layer. As a result, the first electrode and the hole transport layer may be short-circuited, and the photoelectric conversion efficiency may be reduced.
- the photoelectric conversion element still has sufficient photoelectric conversion efficiency and durability even if the photoelectric conversion efficiency and durability are improved by improving the formation method of the hole transport layer and the arrangement of the short-circuit prevention layer. I can't say that.
- an object of the present invention is to provide a photoelectric conversion element that is more excellent in photoelectric conversion efficiency and has higher durability.
- the present inventors contacted the conductive polymer precursor with the photoelectric conversion layer, and irradiated the light to the sensitizing dye in the presence of an oxidant, thereby the conductive polymer precursor. It has been found that the photoelectric conversion efficiency and durability of the photoelectric conversion element are significantly improved by forming a hole transport layer by polymerizing and forming a short-circuit prevention layer by a vertical deposition method. It came to complete.
- a method for manufacturing a photoelectric conversion element reflecting one aspect of the present invention includes a photoelectric conversion element including a substrate, a first electrode, a short-circuit prevention layer, a semiconductor, and a sensitizing dye.
- FIG. 1 is a substrate; 2 is a first electrode; 3 is an anti-shorting layer; 4 is a sensitizing dye; 5 is a semiconductor; 6 is a photoelectric conversion layer; 7 is a hole transport layer; 8 indicates a second electrode; 9 indicates sunlight; and 10 indicates a photoelectric conversion element.
- FIG. 1 is a substrate; 2 is a first electrode; 3 is an anti-shorting layer; 4 is a sensitizing dye; 5 is a semiconductor; 6 is a photoelectric conversion layer; 7 is a hole transport layer; 8 indicates a second electrode; 9 indicates sunlight; and 10 indicates a photoelectric conversion element.
- FIG. 1 is a substrate; 2 is a first electrode; 3 is an anti-shorting layer; 4 is a sensitizing dye; 5 is a semiconductor; 6 is a photoelectric conversion layer; 7 is a hole transport layer; 8 indicates a second electrode; 9 indicates sunlight; and 10 indicates a photoelectric conversion element.
- FIG. 1 is a substrate; 2 is a first electrode
- 2A 1 is a substrate; 2 is a first electrode; 3 is an anti-shorting layer; 4 is a sensitizing dye; 5 is a semiconductor; 6 is a photoelectric conversion layer; 7 is a hole transport layer; 8 represents a second electrode; 10 ′ represents a photoelectric conversion element; and 11 represents a material constituting the hole transport layer.
- 2B 1 is a substrate; 2 is a first electrode; 3 is an anti-shorting layer; 4 is a sensitizing dye; 5 is a semiconductor; 6 is a photoelectric conversion layer; 7 is a hole transport layer; 8 represents a second electrode; 10 represents a photoelectric conversion element; and 11 represents a material constituting the hole transport layer.
- a substrate, a first electrode, a short-circuit prevention layer, a photoelectric conversion layer containing a semiconductor and a sensitizing dye, a hole transport layer containing a conductive polymer, and a second electrode are arranged in this order.
- the manufacturing method of the photoelectric conversion element which has is provided.
- the short-circuit prevention layer is formed by a vertical deposition method (step (1))
- the hole transport layer contacts the conductive polymer precursor with the photoelectric conversion layer, and the oxidant is present in the presence of an oxidizing agent. It is characterized by being formed by irradiating light to a sensitizing dye to polymerize the conductive polymer precursor (step (2)).
- a method for producing a photoelectric conversion element having excellent photoelectric conversion efficiency and high durability can be provided.
- FIG. 1 is a cross-sectional view schematically showing a photoelectric conversion element according to an embodiment of the present invention.
- the photoelectric conversion element 10 has a configuration in which a substrate 1, a first electrode 2, a short-circuit prevention layer 3, a photoelectric conversion layer 6, a hole transport layer 7, and a second electrode 8 are sequentially stacked.
- the photoelectric conversion layer 6 contains the sensitizing dye 4 and the semiconductor 5.
- a short-circuit prevention layer 3 is disposed between the first electrode 2 and the photoelectric conversion layer 6.
- sunlight enters from the direction of the arrow 9 at the bottom of the figure, but the present invention is not limited to this form, and sunlight may enter from the top of the figure.
- the first electrode 2 is formed on the substrate (step of forming the first electrode).
- the short-circuit prevention layer 3 is formed on the first electrode 2 by the vertical deposition method (step / step (1) of forming the short-circuit prevention layer).
- a semiconductor layer made of the semiconductor 5 is formed on the formed short-circuit preventing layer 3, and the photoelectric conversion layer 6 is formed by adsorbing the sensitizing dye 4 on the semiconductor surface (step of forming a photoelectric conversion layer).
- a conductive polymer precursor is brought into contact with the photoelectric conversion layer on the photoelectric conversion layer 6 and the sensitizing dye is irradiated with light in the presence of an oxidizing agent to polymerize the conductive polymer precursor.
- the hole transport layer 7 step of forming the hole transport layer / step (2).
- the hole transport layer 7 penetrates into and exists on the photoelectric conversion layer 6 made of the semiconductor 5 carrying the sensitizing dye 4.
- the photoelectric conversion layer 6 contains the material 11 which comprises a positive hole transport layer in addition to the sensitizing dye 4 and the semiconductor 5.
- the second electrode 8 is formed on the hole transport layer 7 (step of forming the second electrode). A current can be taken out by attaching terminals to the first electrode 2 and the second electrode 8.
- the manufacturing method of a photoelectric conversion element is the process of forming a 1st electrode, the process of forming a short circuit prevention layer (process (1)), the process of forming a photoelectric conversion layer, A step of forming a hole transport layer (step (2)) and a step of forming a second electrode.
- X to Y indicating a range means “X or more and Y or less”.
- ppm means “weight ppm”.
- weight and mass mass and weight
- wt% weight and mass%
- part by weight and “part by mass” are treated as synonyms.
- measurement is performed at room temperature (20 to 25 ° C., further about 23 ° C.) and relative humidity 40 to 50% RH.
- Step of forming first electrode In this step, a substrate is prepared and a first electrode is formed on the substrate.
- the substrate has a function of holding the first electrode.
- the substrate When light is incident from the substrate side, the substrate is preferably a member capable of transmitting this light, that is, a member transparent to the wavelength of light to be subjected to photoelectric conversion.
- the light transmittance is preferably 10% or more, more preferably 50% or more, and particularly preferably 80% to 100%.
- “light transmittance” conforms to “Testing method of total light transmittance of plastic-transparent material” in JIS K 7361-1: 1997 (corresponding to ISO 13468-1: 1996). It shall mean the total light transmittance in the visible light wavelength region measured by the method.
- the material, shape, structure, thickness, hardness and the like can be appropriately selected from known materials, but preferably have high light transmittance as described above.
- a rigid substrate and a flexible substrate can be used as the substrate material.
- a combination of a rigid substrate and a flexible substrate may be used.
- the substrate having rigidity is not particularly limited, and a known substrate can be used. Specifically, a glass plate and an acrylic plate are mentioned. Among these, it is preferable to use a glass plate from the viewpoint of heat resistance.
- the substrate having flexibility is not particularly limited, and a known substrate can be used.
- polyester resin films such as polyethylene terephthalate (PET), polyethylene naphthalate, and modified polyester; polyolefin resin films such as polyethylene (PE), polypropylene (PP), polystyrene, and cyclic olefins; polyvinyl chloride, poly Vinyl resin film such as vinylidene chloride; Polyvinyl acetal resin film such as polyvinyl butyral (PVB); Polyether ether ketone (PEEK) resin film; Polysulfone (PSF) resin film; Polyether sulfone (PES) resin film; Polycarbonate (PC ) Resin film; polyamide resin film; polyimide resin film; acrylic resin film; triacetyl cellulose (TAC) resin film.
- PET polyethylene terephthalate
- PP polypropylene
- PES polystyrene
- cyclic olefins polyvinyl chlor
- a resin film having a transmittance of 80% or more at a wavelength in the visible region (400 to 700 nm) may be used as the substrate.
- the resin film include a biaxially stretched polyethylene terephthalate film, a biaxially stretched polyethylene naphthalate film, a polyethersulfone film, and a polycarbonate film.
- a biaxially stretched polyethylene terephthalate film, a biaxially stretched polyethylene naphthalate It is preferable to use a phthalate film.
- the thickness of the substrate is not particularly limited, but when a rigid substrate is used, it is preferably 0.1 to 100 mm, and more preferably 0.5 to 10 mm. In the case where a flexible substrate is used, the thickness of the substrate is preferably 1 to 1000 ⁇ m, and more preferably 10 to 100 ⁇ m.
- the substrate may be provided with a surface treatment or an easy adhesion layer in order to ensure the wettability and adhesion of the coating solution.
- a conventionally well-known technique can be used about a surface treatment or an easily bonding layer.
- the surface treatment can be performed by a surface activation treatment such as chemical cleaning, corona discharge treatment, flame treatment, ultraviolet treatment, high frequency treatment, glow discharge treatment, active plasma treatment, or laser treatment.
- polyester, polyamide, polyurethane, vinyl copolymer, butadiene copolymer, acrylic copolymer, vinylidene copolymer, epoxy copolymer, and the like can be used as the easy adhesion layer.
- a method for forming the first electrode is not particularly limited, and a known method can be applied.
- the first electrode can be formed by spraying a solution containing the material constituting the first electrode on the substrate and drying it.
- the material constituting the first electrode is not particularly limited, and a known material can be used.
- a composite (dope) material including at least one selected from the group consisting of metals, oxides thereof, and Sn, Sb, F, and Al can be used.
- Examples of the metal include platinum, gold, silver, copper, aluminum, rhodium, and indium.
- Examples of the metal oxide include SnO 2 , CdO, ZnO, and CTO (CdSnO 3 , Cd 2 SnO 4 , CdSnO 4). ), In 2 O 3 , CdIn 2 O 4 and the like.
- examples of the composite (dope) material include In 2 O 3 (ITO) doped with Sn, SnO 2 doped with Sb, SnO 2 (FTO) doped with F, and the like.
- SnO 2 or FTO doped with silver, ITO, or Sb it is preferable to use FTO from the viewpoint of heat resistance.
- a material for forming the first electrode a commercially available material described above may be used, or a precursor of the material may be used.
- FTO fluorine-doped tin oxide
- a first electrode forming material such as tin (IV) chloride pentahydrate is dissolved in an appropriate solvent (for example, an ethanol aqueous solution), mixed with a saturated aqueous solution of ammonium fluoride, and sprayed and dried on the substrate. May be.
- an appropriate solvent for example, an ethanol aqueous solution
- Organic solvents such as alcohol, water, or the mixed solution of alcohol and water, etc. are mentioned.
- the content of the first electrode forming material in the solvent is not particularly limited, but the first electrode forming material is 1 to 20 g, more preferably 3 to 10 g, and particularly preferably 5 to 10 mL in 100 mL of the solvent.
- the amount is preferably such that 8 g is contained. With such an amount, it is possible to easily control the surface roughness (Ra) of the first electrode as described below.
- the amount of the material forming the first electrode applied to the substrate is not particularly limited, but is preferably about 1 to 100 g per 1 m 2 of the substrate.
- the first electrode is not particularly limited, and a coating method, a sputtering method, a chemical vapor deposition (CVD) method, a spray pyrolysis (SPD) method, or the like can be used.
- CVD chemical vapor deposition
- SPD spray pyrolysis
- the first electrode When the first electrode is formed in a slit shape, excimer laser, YAG laser, CO 2 laser, air jet, water jet processing, etching processing, mechanical processing, or the like may be performed. Thereby, the first electrode can be separated into a plurality of regions.
- the pitch of the slits can be appropriately set according to the size of the cell of the photoelectric conversion element.
- the first electrode thus formed has a function for taking out current from the photoelectric conversion element together with the second electrode.
- the first electrode preferably has a light transmittance of 80% or more, more preferably 85% or more, and particularly preferably 90% to 100%.
- the surface roughness (Ra) of the first electrode is preferably 5 to 100 nm, more preferably 10 to 50 nm, and particularly preferably 12 to 30 nm. When the surface roughness of the first electrode is 10 nm or more, it is preferable because dissipation of incident light can be prevented. On the other hand, when the surface roughness of the first electrode is 50 nm or less, it is preferable because a short-circuit prevention layer to be described later can be thinned.
- the surface roughness of the first electrode can be controlled by the type of material constituting the first electrode, the method of forming the first electrode, and the conditions (coating conditions, vapor deposition conditions, etc.).
- the surface roughness (Ra) of the first electrode is measured by a known method. In the present specification, “surface roughness (Ra) of the first electrode” means a value (nm) measured using Surfcom 1400D (manufactured by Tokyo Seimitsu Co., Ltd.).
- the first electrode preferably has a thickness range of about 0.03 to 3 ⁇ m.
- the film thickness of the conductive support is not particularly limited, but is preferably 0.1 to 5 mm.
- the surface resistance value of the conductive support is preferably as low as possible. Specifically, the surface resistance value is preferably 50 ⁇ / ⁇ (square) or less, and more preferably 10 ⁇ / ⁇ or less.
- “conductive support” means a laminate of a substrate and a first electrode formed thereon.
- the short-circuit prevention layer has a function of preventing a short circuit that is a recombination of holes generated by light reception and injected into the hole transport layer and electrons of the first electrode.
- Step (1) is a step of forming a short-circuit prevention layer by a vertical deposition method.
- the vertical deposition method is a method of forming a short-circuit prevention layer by depositing a short-circuit prevention material in a direction perpendicular to the deposition target.
- the vertical deposition method is not particularly limited, and examples thereof include (a) a method using an inkjet method, (b) a method using a spray method, and (c) an atomic layer deposition (ALD) method.
- the method using the inkjet method is to form a coating film by depositing a coating liquid containing a short-circuit prevention material precursor on the first electrode by the inkjet method, and then to prevent short-circuit material precursor This is a method of forming a short-circuit prevention layer by converting the body into a short-circuit prevention material.
- the coating liquid containing a short circuit prevention material precursor contains a short circuit prevention material precursor and a solvent. Furthermore, a known additive may be included.
- the short-circuit prevention material precursor is not particularly limited, but a titanium oxide precursor is preferably used.
- the titanium oxide precursor is preferably one that produces titanium oxide by hydrolysis.
- Specific examples include titanium halides such as titanium trichloride and titanium tetrachloride; methyl orthotitanate, ethyl orthotitanate, Ortho titanate such as isopropyl orthotitanate and butyl orthotitanate; titanium butoxide dimer; titanium alkoxide such as titanium tetraisopropoxide, titanium tetranormal butoxide and titanium butoxide dimer; titanium stearate, diisopropoxy titanium distearate And titanium acylates such as tri-n-butoxy titanium monostearate and polyhydroxy titanium stearate.
- titanium alkoxide and orthotitanate are preferably used.
- titanium oxide precursor examples include ligands such as acetylacetone, aminoethanol, diethanolamine, triethanolamine, ethylenediamine, other amines, pyridinecarboxylic acid, tartaric acid, oxalic acid, lactic acid, glycolic acid, and other hydroxycarboxylic acids. And a complex of titanium oxide precursor may be formed.
- ligands such as acetylacetone, aminoethanol, diethanolamine, triethanolamine, ethylenediamine, other amines, pyridinecarboxylic acid, tartaric acid, oxalic acid, lactic acid, glycolic acid, and other hydroxycarboxylic acids.
- a complex of titanium oxide precursor may be formed.
- the complex includes titanium diisopropoxybis (acetylacetonate), titanium tetraacetylacetonate, titanium dioctyloxybis (octylene glycolate), titanium diisopropoxybis (ethylacetoacetate), titanium diisopropoxybis (Triethanolaminate), titanium lactate ammonium salt, titanium lactate, propanedioxytitanium bis (ethylacetoacetate) and the like.
- Halogen type organic solvents such as chloroform and a dichloromethane
- Aliphatic hydrocarbons such as butane, pentane, hexane
- Aromatic hydrocarbons such as benzene, toluene, xylene, chlorobenzene
- Ethyl acetate acetic acid Esters such as butyl
- Water-insoluble ketones such as methyl isobutyl ketone
- Ethers such as tetrahydrofuran (THF), butyl ether and dioxane
- Aliphatic alcohols such as methanol, ethanol, n-propanol and isopropyl alcohol
- Dimethyl Examples include formamide (DMF); dimethyl sulfoxide (DMSO); carbon disulfide; water and the like. These solvents may be used alone or in combination of two or more.
- the content of the short-circuit preventing material precursor in the coating solution is not particularly limited, but is preferably 0.1 to 65 parts by mass, preferably 0.5 to 13 parts per 100 parts by mass of the solvent in the coating solution. More preferably, it is part by mass.
- the inkjet method is a method of forming a coating film by spraying a coating liquid containing a short-circuit preventing material precursor on the first electrode in units of droplets. By using the inkjet method, the coating is deposited vertically.
- the inkjet head is preferably a piezoelectric element type. Further, the discharge amount is preferably 4 to 42 picoliters per drop, and more preferably 8 to 30 picoliters.
- the number of times of discharging the coating liquid is not particularly limited, and can be appropriately adjusted according to the desired performance of the short-circuit prevention layer.
- the short circuit prevention material precursor in the coating film formed by the inkjet method is converted into a short circuit prevention material. At this time, part or all of the solvent in the coating film volatilizes. Thereby, a short circuit prevention layer is formed.
- the conversion method of the short-circuit preventing material precursor is not particularly limited, and a known method can be used. Of these, firing is preferable.
- the short-circuit prevention material precursor is an organic titanium compound
- the organic titanium compound when firing is performed, the organic titanium compound is converted into a titanium oxide polymer (short-circuit prevention material) by a reaction represented by the following formula.
- Calcination temperature is not particularly limited, but is preferably 200 ° C. or higher, more preferably 250 ° C. or higher, even more preferably 300 ° C. or higher, and particularly preferably 400 ° C. or higher.
- the lower limit of the firing temperature is not particularly limited, but is preferably 700 ° C. or lower, more preferably 650 ° C. or lower, and even more preferably 600 ° C. or lower. That is, the firing temperature is preferably 200 to 700 ° C., more preferably 250 to 650 ° C., further preferably 300 to 600 ° C., and particularly preferably 400 to 600 ° C. It is preferable for the firing temperature to be 200 ° C.
- the firing temperature is 700 ° C. or lower because excessive crystallization of the short-circuit prevention layer can be prevented.
- the firing time is not particularly limited, but is preferably 1 minute to 5 hours, more preferably 5 minutes to 3 hours, further preferably 5 minutes to 120 minutes, and more preferably 5 to 30 minutes. It is particularly preferred.
- a short-circuit prevention layer having suitable crystallinity can be obtained, and a short-circuit prevention layer having low internal resistance can be obtained.
- the degree of denseness and crystallinity of the short-circuit prevention layer can be adjusted by appropriately changing the firing temperature and firing time.
- a process (1) includes the process of apply
- the short-circuit prevention layer preferably contains titanium oxide.
- the short-circuit prevention layer contains titanium oxide means that it is only necessary to have a —Ti—O— bond. That is, it is not limited to the case of containing titanium oxide molecules, but includes the case of containing titanium oxide polymers.
- the short-circuit prevention layer may contain an organic substance such as an unreacted titanium oxide precursor or a titanium oxide polymer having a —Ti—O—R bond.
- the ink jet method can form a coating film by directly spraying the coating liquid on the first electrode, and therefore, the degree of freedom of film formation is very high and a uniform film can be formed.
- the coating liquid can be sprayed on the order of picoliters, a film can be formed with high accuracy, and a thin film can be easily formed.
- it can be performed under atmospheric pressure, the manufacturing cost is low.
- a coating solution is formed by depositing a coating liquid containing a short-circuit prevention material precursor on the first electrode by a spray method, and then a short-circuit prevention material precursor. This is a method of forming a short-circuit prevention layer by converting the body into a short-circuit prevention material.
- a coating solution containing a short-circuit prevention material precursor Since the same coating solution as described in (a) above can be used, the description thereof is omitted here.
- the spray method is a method of forming a coating film by spraying droplets on a first electrode with a coating solution prepared by dissolving a short-circuit prevention material precursor in a solvent. By using the spray method, the coating is deposited vertically.
- the speed of the droplet when spraying is not particularly limited, but is preferably 0.0001 to 10 m / s, and more preferably 0.001 to 1 m / s.
- the amount of spraying the droplets is not particularly limited, but is preferably 3 to 20 mL / min, and more preferably 5 to 15 mL / min.
- the pressure for spraying the droplets is not particularly limited, but is preferably 10 to 0.0001 MPa, and more preferably 1 to 0.001 MPa.
- the spraying time or frequency of the coating liquid is not particularly limited and can be appropriately adjusted according to the desired performance of the short-circuit prevention layer.
- ALD Atomic Layer Deposition
- a short-circuit prevention material precursor gas and an oxidizing gas are alternately introduced onto the first electrode, thereby depositing a short-circuit prevention layer for each layer.
- the gas of the short circuit preventing material precursor is first introduced onto the first electrode to form a gas molecular layer (monoatomic layer).
- an inert gas is introduced to purge (remove) the gas of the short-circuit prevention material precursor.
- the gas molecule layer of the gas of the short-circuit prevention material precursor formed is chemically adsorbed to the first electrode, it is not purged even when an inert gas is introduced.
- an oxidizing gas is introduced to convert the short-circuit prevention material precursor constituting the formed gas molecular layer into a short-circuit prevention material, thereby forming a short-circuit prevention layer.
- an inert gas the oxidizing gas is purged and one cycle of the ALD method is completed. By repeating the cycle, atomic layers are deposited one by one, and a short-circuit prevention layer having a predetermined film thickness can be formed.
- titanium oxide precursors described in said (a) include titanium (IV) tetraisopropoxide (Ti (OiPr) 4 ), tetrakis (dimethylamino) titanium (IV) (Ti (NMe 2 ) 4 ), and the like.
- the oxidizing gas is not particularly limited as long as it can convert the short-circuit prevention material precursor into a short-circuit prevention material.
- ozone (O 3 ) water vapor (H 2 O), hydrogen peroxide (H 2 O 2 ), methanol (CH 3 OH), ethanol (C 2 H 5 OH), and the like can be used.
- the inert gas a rare gas (helium, neon, argon, krypton, xenon), nitrogen gas, or the like can be used.
- a rare gas helium, neon, argon, krypton, xenon
- nitrogen gas or the like.
- the introduction time of the gas for the short circuit preventing material precursor is preferably 0.05 to 10 seconds, more preferably 0.1 to 3 seconds, and further preferably 0.5 to 2 seconds. It is preferable that the gas introduction time of the short-circuit prevention material precursor is 0.05 seconds or longer because a sufficient time for forming the gas molecular layer can be secured. On the other hand, when the gas introduction time of the short circuit preventing material precursor is 10 seconds or less, it is preferable because the time required for one cycle can be reduced.
- the introduction time of the inert gas for purging the gas of the short circuit preventing material precursor is preferably 0.05 to 10 seconds, more preferably 0.5 to 6 seconds. More preferably, it is seconds. It is preferable that the introduction time of the inert gas is 0.05 seconds or more because the gas of the short circuit preventing material precursor can be sufficiently purged. On the other hand, when the introduction time of the inert gas is 10 seconds or less, it is preferable because the time required for one cycle can be reduced and the influence on the formed gas molecular layer is reduced.
- the introduction time of the oxidizing gas is preferably 0.05 to 10 seconds, more preferably 0.1 to 3 seconds, and further preferably 0.5 to 2 seconds.
- An introduction time of the oxidizing gas of 0.05 seconds or more is preferable because a sufficient time for oxidizing the gas molecular layer can be secured.
- the introduction time of the oxidizing gas is 10 seconds or less, the time required for one cycle can be reduced, and side reactions can be prevented.
- the introduction time of the inert gas for purging the oxidizing gas is preferably 0.05 to 10 seconds, more preferably 0.5 to 6 seconds, and 1 to 4 seconds. Is more preferable.
- An inert gas introduction time of 0.05 seconds or longer is preferable because the oxidizing gas can be sufficiently purged.
- an inert gas introduction time of 10 seconds or less is preferable because the time required for one cycle can be reduced and the influence on the formed atomic layer is small.
- the film formation temperature when forming the short-circuit prevention layer is preferably 350 ° C. or less, more preferably 250 ° C. or less, further preferably less than 150 ° C., and less than 120 ° C. It is particularly preferred that The film forming temperature of 350 ° C. or lower is preferable because the heat resistance required for the substrate holding the first electrode is reduced, and the manufacturing cost can be reduced.
- the first electrode having irregularities on the surface may not be sufficiently covered.
- FIG. 2A is a schematic cross-sectional view schematically showing an example of a partial intra-layer configuration of a conventional photoelectric conversion element.
- a conventional photoelectric conversion element 10 ′ includes a substrate 1, a first electrode 2, a short-circuit prevention layer 3, a photoelectric conversion layer 6, a hole transport layer 7, and a second electrode 8 in a part thereof. It has a configuration in which the layers are sequentially stacked.
- the photoelectric conversion layer 6 includes a semiconductor 5 and a sensitizing dye 4 carried on the semiconductor 5.
- the hole transport layer 7 is disposed on the photoelectric conversion layer 6, the material 11 (conductive polymer or the like) constituting the hole transport layer 7 is filled so as to fill the porous gap of the semiconductor 5, It is a constituent element of the photoelectric conversion layer 6 (however, in this specification, the material 11 constituting the hole transport layer may be described as a constituent element of the hole transport layer 7).
- the first electrode 2 is not sufficiently covered with the short-circuit prevention layer 3, the first electrode 2 protrudes from the short-circuit prevention layer, and the material 11 constituting the hole transport layer in the photoelectric conversion layer is the first electrode 2. And direct contact between the short-circuit prevention layer 3 can occur. And when both contact, dark current and reverse electron transfer will increase remarkably, and photoelectric conversion efficiency will fall.
- the dark current does not depend on the intensity of light applied to the photoelectric conversion element. Therefore, the influence is particularly great when power is generated with a small amount of light with weak light and short-circuit current.
- the short-circuit current is 100 mA / cm 2
- the dark current is 500 ⁇ A / cm 2
- the decrease factor is only about 0.5%, but when the short-circuit current is 10 mA / cm 2 .
- the reduction factor is 5%, and the influence becomes larger.
- the reverse electron transfer means electron transfer from the photoelectric conversion layer to the hole transport layer, and is one of the causes of lowering the short circuit current.
- the first electrode can be completely covered by increasing the film thickness of the short-circuit prevention layer.
- a portion where the thickness of the short-circuit prevention layer is excessive with respect to the first electrode may occur.
- the adhesiveness is reduced in the excess portion, so that cracks and bubbles may occur, and the resistance of the short-circuit prevention layer may partially increase, and the photoelectric conversion efficiency may decrease.
- the above-described problem with the coating of the short-circuit prevention layer can be a problem because many commonly used first electrodes have irregularities (surface roughness) on the surface.
- FIG. 2B is a schematic cross-sectional view schematically showing an example of a partial intra-layer configuration of the photoelectric conversion element of the present invention.
- the photoelectric conversion element 10 according to the present invention has the first electrode 2 because the short-circuit prevention layer 3 covers the first electrode 2 so as to follow the uneven structure of the first electrode 2.
- the material 11 constituting the hole transport layer in the photoelectric conversion layer 6 do not come into contact with each other.
- reverse electron transfer and dark current can be reduced, a photoelectric conversion element having high photoelectric conversion efficiency can be obtained.
- Short-circuit prevention layer Although it does not restrict
- metals such as zinc, niobium, tin, titanium, vanadium, indium, tungsten, tantalum, zirconium, molybdenum, manganese, iron, copper, nickel, iridium, rhodium, chromium, ruthenium, or oxides thereof or the like
- Polymer perovskite such as strontium titanate, calcium titanate, barium titanate, magnesium titanate, strontium niobate or a complex oxide or oxide mixture thereof; CdS, CdSe, TiC, Si 3 N 4 , SiC, BN, etc.
- the metal compound of these is mentioned. Among these, it is preferable that it has the electrical conductivity equivalent to the semiconductor material of a photoelectric converting layer, and it is more preferable that it is a titanium oxide or its polymer.
- the titanium oxide may be either anatase-type titanium oxide or rutile-type titanium oxide having a relatively high dielectric constant, or a combination of both.
- the short-circuit prevention layer may contain an organic substance such as an unreacted titanium oxide precursor or a titanium oxide polymer having a —Ti—O—R bond.
- the short-circuit prevention layer is preferably porous together with a semiconductor layer in the photoelectric conversion layer described later.
- the D / C value is preferably 1.1 or more. More preferably, it is more preferably 10 or more.
- the porosity C of the short-circuit prevention layer is preferably 20% by volume or less, more preferably 5% by volume or less, and 2% by volume or less. More preferably it is. That is, the short-circuit prevention layer is preferably a dense layer (dense porous shape).
- the lower limit of the porosity C of the short-circuit preventing layer is preferably as small as possible, it is not particularly required to be specified, but it is usually about 0.05% by volume or more.
- the average thickness (film thickness) of the short-circuit prevention layer is not particularly limited as long as it can exhibit a short-circuit prevention effect. Specifically, the thickness is preferably 10 to 400 nm, more preferably 10 to 200 nm, and particularly preferably 20 to 120 nm. When the average thickness of the short-circuit prevention layer is 10 nm or more, it is preferable because a short circuit between the first electrode and the hole transport layer can be sufficiently prevented. On the other hand, when the thickness of the short-circuit prevention layer is 400 nm or less, the resistance value of the short-circuit prevention layer becomes an appropriate value, which is preferable because high conversion efficiency is obtained. In addition, when the short circuit prevention layer is formed along the unevenness
- the short circuit prevention layer When a p-type semiconductor is used for the hole transport layer and a metal is used for the short circuit prevention layer, the short circuit prevention layer has a work function value smaller than that of the hole transport layer, and has a Schottky contact. It is preferable to use what to do.
- the short-circuit prevention layer In the case of using a metal oxide for the short-circuit prevention layer, the short-circuit prevention layer should be in ohmic contact with the transparent conductive layer and have a conduction band energy level lower than that of the semiconductor layer. It is preferable to use it. By selecting an oxide to be used, it is possible to improve the efficiency of electron transfer from the porous semiconductor layer (photoelectric conversion layer) to the short-circuit prevention layer.
- the photoelectric conversion layer has a function of converting light energy into electric energy using the photovoltaic effect.
- the photoelectric conversion layer contains a semiconductor and a sensitizing dye. More specifically, the photoelectric conversion layer has a configuration in which a sensitizing dye is supported on a semiconductor layer containing a semiconductor.
- the method for forming the photoelectric conversion layer is generally divided into (1) formation of a semiconductor layer on the short-circuit prevention layer and (2) sensitization treatment of the semiconductor.
- a semiconductor dispersion or colloidal solution is applied or sprayed onto the short-circuit prevention layer, and the semiconductor fine particle precursor is applied to the short-circuit prevention layer.
- the semiconductor layer can be formed by a method (sol-gel method) or the like that is applied to, and condensed after hydrolysis with moisture (for example, moisture in the air).
- the semiconductor layer obtained by the above method is preferably fired.
- the semiconductor layer can be formed by bonding the semiconductor onto the short-circuit prevention layer.
- Examples of the sensitizing method (2) include adsorption of a sensitizing dye to a semiconductor layer.
- a sensitizing treatment with a sensitizing dye quickly after baking and before moisture is adsorbed to the semiconductor.
- a coating solution containing a semiconductor, preferably a semiconductor fine powder (semiconductor-containing coating solution) is prepared.
- the semiconductor fine powder preferably has a fine primary particle size.
- the semiconductor-containing coating solution can be prepared by dispersing the semiconductor fine powder in a solvent, and the semiconductor fine powder dispersed in the solvent is dispersed in the form of primary particles.
- the semiconductor material used is not particularly limited, but a simple substance such as silicon or germanium, a group 3 to group 5, or a group 13 to group 15 element of the periodic table (also referred to as an element periodic table).
- Compounds, metal chalcogenides (eg, oxides, sulfides, selenides, etc.), metal nitrides, and the like can be used.
- metal chalcogenides include titanium, tin, zinc, iron, tungsten, zirconium, hafnium, strontium, indium, cerium, yttrium, lanthanum, vanadium, niobium, or tantalum oxide; cadmium, zinc, lead, silver , Antimony or bismuth sulfide; cadmium or lead selenide; cadmium telluride.
- Other semiconductor materials include phosphides such as zinc, gallium, indium, and cadmium; gallium-arsenic or copper-indium selenides; copper-indium sulfides; titanium nitrides, and the like.
- TiO 2 , ZnO, SnO 2 , Fe 2 O 3 , WO 3 , Nb 2 O 5 , CdS, or PbS is preferably used, and TiO 2 or Nb 2 O 5 is more preferably used. It is particularly preferable to use TiO 2 (titanium oxide) because of its excellent adsorbability. These materials may be used alone or in combination of two or more.
- Ti 3 N 4 20% by mass of titanium nitride
- a composite form of zinc oxide / tin oxide a form in which 20% by mass of titanium nitride (Ti 3 N 4 ) is mixed with a titanium oxide semiconductor; Chem. Soc. Chem. Commun. 15 (1999), and a composite form of zinc oxide / tin oxide.
- the mass ratio of the other semiconductor material to the metal oxide or metal sulfide semiconductor may be 30% or less. preferable.
- TiO 2 when used for the semiconductor layer, TiO 2 may be either anatase-type titanium oxide and / or rutile-type titanium oxide having a relatively high dielectric constant.
- the shape of the semiconductor is not particularly limited, and may have any shape such as a filler shape, a spherical shape, a conical shape, a columnar shape, a tubular shape, and a flat plate shape.
- a film-like layer formed by agglomerating these filler-like, particle-like, conical, columnar, tubular, and other semiconductors may be used.
- a semiconductor whose surface is coated with a dye in advance may be used, or the dye may be coated after a semiconductor layer is formed.
- the size of the semiconductor is also not particularly limited.
- the average particle size of the semiconductor is preferably 1 to 5000 nm, and more preferably 2 to 500 nm.
- the “average particle size” of the semiconductor means an average particle size (primary average particle size) of primary particle diameters when 100 or more samples are observed with an electron microscope.
- the surface of the semiconductor may be treated with an organic base.
- the organic base used for the surface treatment is not particularly limited, and examples thereof include diarylamine, triarylamine, pyridine, 4-tert-butylpyridine, polyvinylpyridine, quinoline, piperidine, and amidine. Of these, surface treatment is preferably performed using pyridine, 4-tert-butylpyridine, or polyvinylpyridine.
- the surface treatment method is not particularly limited, and a known method can be used, and the method can be appropriately changed by a person skilled in the art as necessary. For example, as an example of a semiconductor surface treatment method, a method of preparing a solution containing an organic base (organic base solution) and immersing the semiconductor in the organic base solution is exemplified.
- the solvent that can be used for the semiconductor-containing coating solution is not particularly limited as long as it can disperse the semiconductor fine powder, and water, an organic solvent, or a mixed solution of water and an organic solvent can be used.
- the organic solvent include, for example, alcohols such as methanol, ethanol and isopropyl alcohol; ketones such as methyl ethyl ketone, acetone and acetylacetone; hydrocarbons such as hexane and cyclohexane; acetylcellulose, nitrocellulose, acetylbutylcellulose, ethylcellulose, Examples thereof include cellulose derivatives such as methylcellulose.
- a surfactant such as acetic acid and nitric acid
- an acid such as acetic acid and nitric acid
- a viscosity modifier such as a polyhydric alcohol such as polyethylene glycol
- a chelating agent such as acetylacetone
- a known additive may be added to the semiconductor-containing coating solution as necessary.
- the concentration of the semiconductor fine powder in the solvent is preferably 0.1 to 70% by mass, and more preferably 0.1 to 30% by mass.
- the semiconductor layer is formed by applying or spraying the semiconductor-containing coating solution prepared in (1-1) above onto the short-circuit prevention layer and drying.
- the application is not particularly limited, and is performed by a known method such as a doctor blade method, a squeegee method, a spin coating method, or a screen printing method.
- the semiconductor layer obtained by the above application or spraying and drying is composed of an aggregate of semiconductor fine particles, and the particle size of the fine particles corresponds to the primary particle size of the used semiconductor fine powder.
- the semiconductor-containing coating solution may contain two or more semiconductor materials, or may be coated or sprayed using two or more semiconductor materials to form a semiconductor layer having a layered structure.
- the semiconductor layer formed by the above (1-2) is preferably fired in air or in an inert gas.
- the bonding strength between the semiconductor layer formed in (1-2) and the short-circuit prevention layer and the bonding strength between the semiconductor fine particles can be increased, and the mechanical strength can be improved.
- the firing conditions are not particularly limited as long as a semiconductor layer having a desired actual surface area and porosity can be formed.
- Calcination temperature is not particularly limited. From the viewpoint of appropriately preparing the actual surface area of the fired film during the firing treatment and obtaining a fired film having the above porosity, the firing temperature is preferably 900 ° C. or lower, and preferably 200 ° C. to 850 ° C. More preferably, it is 400 ° C. to 850 ° C., even more preferably 450 ° C. to 800 ° C. Further, when the substrate is made of plastic or the like and is inferior in heat resistance, the semiconductor fine particles-short-circuit prevention layer and the semiconductor fine particles may be fixed by pressurization, or only the semiconductor layer may be fired using microwaves. .
- the firing time is not particularly limited, but is preferably 10 seconds to 12 hours, more preferably 1 to 240 minutes, and particularly preferably 10 to 120 minutes.
- the firing atmosphere is not particularly limited, but usually the firing step is performed in air or in an inert gas (eg, argon, helium, nitrogen, etc.) atmosphere.
- an inert gas eg, argon, helium, nitrogen, etc.
- the firing may be performed only once at a single temperature, or may be repeated twice or more by changing the temperature and time.
- the structure of the fired semiconductor layer is not particularly limited, but is preferably a porous structure (a porous structure having voids) from the viewpoint of effectively performing adsorption with a sensitizing dye. Therefore, the porosity (D) of the semiconductor layer is preferably 1 to 90% by volume, more preferably 10 to 80% by volume, and particularly preferably 20 to 70% by volume.
- the porosity of the semiconductor layer means a porosity that is penetrating in the thickness direction of the dielectric, and can be measured using a commercially available device such as a mercury porosimeter (Shimadzu pore sizer 9220 type).
- the photoelectric conversion element is preferably manufactured so that the material constituting the hole transport layer is also present in this void.
- the film thickness of the fired semiconductor layer is not particularly limited, but is preferably 10 nm or more, and more preferably 500 nm to 30 ⁇ m. If it is such a range, it can become a semiconductor layer excellent in characteristics, such as permeability and conversion efficiency.
- the semiconductor layer may be a single layer formed of semiconductor fine particles having the same average particle diameter, or a multilayer film (layered structure) composed of semiconductor layers containing semiconductor fine particles having different average particle diameters and types. Also good.
- the ratio of the actual surface area to the apparent surface area of the obtained semiconductor layer can be controlled by the particle size and specific surface area of the semiconductor fine particles, the firing temperature, and the like.
- the obtained semiconductor layer is subjected to, for example, chemical plating using a titanium tetrachloride aqueous solution or electrochemical plating treatment using a titanium trichloride aqueous solution, so that the surface area of the semiconductor particles and the vicinity of the semiconductor particles are increased. Purity may be controlled to increase the efficiency of electron injection from the dye into the semiconductor particles.
- sensitization treatment with sensitizing dye is performed, for example, by immersing a semiconductor layer in which the sensitizing dye is dissolved in an appropriate solvent and thoroughly dried in the solution for a long time. Is done by doing.
- the sensitizing dye can be adsorbed to the semiconductor by the sensitizing treatment.
- pretreatment such as reduced pressure treatment or heat treatment before immersion to remove bubbles in the film or moisture in the voids.
- the sensitizing dye can be adsorbed inside the semiconductor layer.
- the sensitizing treatment is not limited to the immersion of the semiconductor layer in the sensitizing dye-containing solution, and other known sensitizing treatment methods can be appropriately applied.
- the sensitizing dye that can be used is not particularly limited, but is preferably an arylamine dye, and more preferably a compound represented by the following general formula (1).
- Ar represents a cyclic compound group.
- R 1 represents a hydrogen atom, a halogen atom, a substituted or unsubstituted alkyl group, a substituted or unsubstituted cycloalkyl group, a substituted or unsubstituted alkoxy group, a substituted or unsubstituted alkenyl group, a substituted or unsubstituted cyclohexane.
- An alkenyl group, a substituted or unsubstituted alkynyl group, a substituted or unsubstituted aryl group, an amino group, a cyano group, or a substituted or unsubstituted heterocyclic group is represented.
- R 1 may be linked to another group to form a ring structure.
- a 1 and A 2 are each independently a single bond, a divalent saturated or unsaturated hydrocarbon group, a substituted or unsubstituted alkylene group, a substituted or unsubstituted arylene group, or a substituted or unsubstituted 2 Represents a valent heterocyclic group.
- Z represents an organic group.
- n is an integer of 1 to 3, and when n is 1, the two R 1 may be the same or different from each other.
- p and q are integers from 0 to 6.
- the plurality of A 1 and A 2 may be the same or different from each other.
- the Ar is not particularly limited, and examples thereof include divalent to tetravalent cyclic compound groups.
- Specific examples of the cyclic compound group include, for example, benzene ring, naphthalene ring, anthracene ring, thiophene ring, phenylthiophene ring, diphenylthiophene ring, imidazole ring, oxazole ring, thiazole ring, pyrrole ring, furan ring, benzimidazole.
- aromatic ring such as a ring, a benzoxazole ring, a rhodamine ring, a pyrazolone ring, an imidazolone ring, a pyran ring, a pyridine ring, or a fluorene ring.
- aromatic rings such as a ring, a benzoxazole ring, a rhodamine ring, a pyrazolone ring, an imidazolone ring, a pyran ring, a pyridine ring, or a fluorene ring.
- a plurality of these aromatic rings may be used in combination.
- aromatic rings may have a substituent.
- substituents include a halogen atom (for example, a fluorine atom, a chlorine atom, a bromine atom, etc.), a C1-C24 linear or branched alkyl group ( For example, methyl group, ethyl group, tert-butyl group, isobutyl group, dodecyl group, octadecyl group, 3-ethylpentyl group), hydroxyalkyl group (for example, hydroxymethyl group, hydroxyethyl group), alkoxyalkyl group (for example, Methoxyethyl group, etc.), alkoxy groups having a carbon chain length of 1 to 18 (eg methoxy group, ethoxy group, ethoxy group, propoxy group, isopropoxy group, butoxy group, pentyloxy group, hexyloxy group, etc.), aryl group ( For example, phenyl group, toly
- Ar is preferably a divalent or trivalent aromatic group obtained by removing two or three hydrogen atoms from the above-described aromatic group.
- Preferred examples of Ar include the following chemical formulas (1-A) to (1-G).
- the alkyl group is not particularly limited, and examples thereof include a C1-C24 linear or branched alkyl group. Specifically, methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, isobutyl group, sec-butyl group, tert-butyl group, n-pentyl group, isopentyl group, tert-pentyl group, Neopentyl group, 1,2-dimethylpropyl group, n-hexyl group, isohexyl group, 1,3-dimethylbutyl group, 1-isopropylpropyl group, 1,2-dimethylbutyl group, n-heptyl group, 1,4- Dimethylpentyl group, 3-ethylpentyl group, 2-methyl-1-isopropylpropyl group, 1-ethyl-3-methylbutyl group, n-octyl group, 2-eth
- C1-C6 straight chain alkyl groups such as methyl group, ethyl group, n-propyl group, n-butyl group, n-pentyl group, n-hexyl group, isopropyl group, t-butyl group, etc.
- a C3 to C6 branched chain alkyl group is preferred.
- the cycloalkyl group is not particularly limited, and examples thereof include C3 to C9 cycloalkyl groups. Specific examples include a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, a cycloheptyl group, a cyclooctyl group, and a cyclononyl group. Of these, C3-C6 cycloalkyl groups such as a cyclopentyl group and a cyclohexyl group are preferred.
- the alkoxy group is not particularly limited, and examples thereof include C1-C18 alkoxy groups. Specifically, methoxy group, ethoxy group, propoxy group, isopropoxy group, butoxy group, pentyloxy group, hexyloxy group, 2-ethylhexyloxy group, octyloxy group, nonyloxy group, decyloxy group, undecyloxy group, Examples include dodecyloxy group, tridecyloxy group, tetradecyloxy group, pentadecyloxy group, hexadecyloxy group, heptadecyloxy group, octadecyloxy group and the like. Of these, a C6 to C18 alkoxy group is preferable, and a decyloxy group is more preferable.
- the alkenyl group is not particularly limited, and examples thereof include C2 to C18 linear or branched alkenyl groups. Specifically, vinyl group, allyl group, propenyl group, isopropenyl group, 1-butenyl group, 2-butenyl group, 3-butenyl group, 1-hexenyl group, 2-hexenyl group, 3-hexenyl group, 4- Hexenyl group, 5-hexenyl group and the like can be mentioned. Alkenyl groups other than these may be used.
- the cycloalkenyl group is not particularly limited, and examples thereof include C3-C18 cycloalkenyl groups. Specific examples include a cyclopentenyl group, a cyclohexenyl group, and a cyclooctenyl group.
- the alkynyl group is not particularly limited, and examples thereof include a C2 to C18 linear or branched alkynyl group. Specific examples include an ethynyl group, a 2-propynyl group, and a 2-butynyl group.
- the aryl group is not particularly limited, and examples thereof include C6 to C30 aryl groups. Specific examples include a phenyl group, a naphthyl group, a biphenyl group, a fluorenyl group, an anthryl group, a pyrenyl group, an azulenyl group, an acenaphthylenyl group, a terphenyl group, and a phenanthryl group. Of these, a phenyl group, a biphenyl group, and a naphthyl group are preferable.
- the heterocyclic group is not particularly limited, and examples thereof include a heterocyclic group containing at least one hetero atom selected from a nitrogen atom, an oxygen atom, and a sulfur atom.
- the heterocyclic group is not limited to a monocyclic heterocyclic group, but is a condensed heterocyclic group in which a plurality of heterocyclic rings are condensed, a heterocyclic ring and a hydrocarbon ring (non-aromatic hydrocarbon ring or aromatic hydrocarbon ring), and May be a condensed heterocyclic group obtained by condensation (ortho condensation, ortho-and-peri condensation, etc.).
- the heterocyclic group may be non-aromatic or aromatic.
- either the heterocyclic ring or the hydrocarbon ring may have a bond.
- a pyrrolyl group, an indolyl group, and a carbazolyl group are preferable.
- the hydrocarbon group is a group consisting of a carbon atom and a hydrogen atom, and is not particularly limited, but means a divalent group derived from the above-mentioned cycloalkyl group, alkenyl group, cycloalkenyl group, and alkynyl group.
- the hydrocarbon group may be substituted with a substituent.
- the alkylene group is not particularly limited, and examples thereof include C1-C24 linear or branched alkylene groups. Specific examples include a methylene group, an ethylene group, a propylene group, an isobutylene group, a sec-butylene group, a tert-butylene group, a pentylene group, an iso-pentylene group, and a hexylene group.
- the arylene group is not particularly limited, and examples thereof include C6 to C30 arylene groups. Specific examples include a phenylene group, a biphenyl-diyl group, a terphenyl-diyl group, a naphthalene-diyl group, an anthracene-diyl group, a tetracene-diyl group, a fluorene-diyl group, and a phenanthrene-diyl group.
- substitution in “substituted or unsubstituted” in the general formula (1) means that a hydrogen atom constituting a group described as R 1 , A 1 , or A 2 is substituted with a substituent.
- “Unsubstituted” refers to the case where the hydrogen atom is not substituted with a substituent.
- the substituent is not particularly limited, and may be the alkyl group, cycloalkyl group, alkoxy group, alkenyl group, cycloalkenyl group, alkynyl group, aryl group, and heterocyclic group exemplified above.
- R 1 examples include the following chemical formulas (2-A) to (2-S).
- Y is a hydrogen atom, an alkyl group, or an alkoxy group.
- the wavy line indicates a position where it is connected to another group. By connecting the broken line part with another group, for example, R 1 forms a condensed ring structure together with Ar.
- h is an integer of 1 to 17.
- a 1 and A 2 include the following chemical formulas (3-A) to (3-b).
- Y is a hydrogen atom, an alkyl group, or an alkoxy group.
- h is an integer of 1 to 17.
- Z examples include an organic group having an acidic group and / or a partial structure of Ar having an electron-withdrawing group or an electron-withdrawing ring structure.
- An organic group containing at least one carboxyl group is preferred.
- Z is bonded to Ar, A 1 , or A 2 , preferably Ar in the general formula (1).
- the acidic group is not particularly limited, but is a carboxy group, a sulfo group (—SO 3 H), a sulfino group, a sulfinyl group, a phosphonic acid group (—PO (OH) 2 ), a phosphoryl group, a phosphinyl group, a phosphono group, A thiol group, a hydroxy group, a phosphonyl group, an alkoxysilane group, and a sulfonyl group; and salts thereof.
- the acidic group is preferably a carboxy group, a sulfo group, a phosphonic acid group, or a hydroxy group, and more preferably a carboxyl group.
- the electron-withdrawing group is not particularly limited, but is a cyano group, a nitro group, a fluoro group, a chloro group, a bromo group, an iodo group, a perfluoroalkyl group (for example, a trifluoromethyl group), an alkylsulfonyl group, An arylsulfonyl group, a perfluoroalkylsulfonyl group, a perfluoroarylsulfonyl group, etc. are mentioned.
- a cyano group, a nitro group, a fluoro group, and a chloro group are preferable, and a cyano group and a nitro group are more preferable.
- the electron-withdrawing ring structure is not particularly limited, but rhodamine ring, dirhodamine ring, imidazolone ring, pyrazolone ring, pyrazoline ring, quinone ring, pyran ring, pyrazine ring, pyrimidine ring, imidazole ring, indole ring, benzothiazole Ring, benzimidazole ring, benzoxazole ring, thiadiazole ring and the like.
- a rhodamine ring, a dirhodamine ring, an imidazolone ring, a pyrazoline ring, a quinone ring, and a thiadiazole ring are preferable, and a rhodamine ring, a dirhodamine ring, an imidazolone ring, and a pyrazoline ring are more preferable.
- the Z can effectively inject photoelectrons into a semiconductor (particularly an oxide semiconductor).
- the acidic group and the electron-withdrawing group or the electron-withdrawing ring structure are oxygen atoms (O), sulfur atoms (S), selenium atoms (Se), tellurium atoms (Te), etc. You may combine through an atom.
- the partial structure Z may have a charge, particularly a positive charge. In this case, Cl ⁇ , Br ⁇ , I ⁇ , ClO 4 ⁇ , NO 3 ⁇ , SO 4 2 ⁇ , H 2 PO 4 ⁇ And so on.
- Preferred examples of Z include the following chemical formulas (4-A) to (4-N).
- g is an integer of 1 to 17.
- sensitizing dyes examples include a sensitizing dye having the following structure A-1 is also referred to as sensitizing dye A-1.
- the sensitizing dyes it is preferable to use the dyes of Group A and Group B.
- the sensitizing dyes When performing the sensitization treatment, the sensitizing dyes may be used alone or in combination of two or more.
- the use of the photoelectric conversion element of the present invention is a solar cell to be described later, two or more sensitizing dyes having different absorption wavelengths are used so that the wavelength range of photoelectric conversion can be made as wide as possible to effectively use sunlight. It is preferable to use a mixture.
- the sensitizing treatment method is not particularly limited, and the semiconductor layer may be immersed in a mixed solution of each sensitizing dye, or each sensitizing dye as a separate solution. It is possible to prepare and sequentially immerse the semiconductor layer.
- the solvent used for dissolving the sensitizing dye is not particularly limited as long as it can dissolve the sensitizing dye and does not dissolve the semiconductor or react with the semiconductor. However, in order to prevent moisture and gas dissolved in the solvent from entering the semiconductor film and hindering sensitizing treatment such as adsorption of a sensitizing dye, it is preferable to degas and purify the solvent in advance. .
- Solvents preferably used in dissolving the sensitizing dye include nitrile solvents such as acetonitrile; alcohol solvents such as methanol, ethanol, n-propanol, isopropyl alcohol, and tert-butyl alcohol; ketone solvents such as acetone and methyl ethyl ketone; Examples include ether solvents such as diethyl ether, diisopropyl ether, tetrahydrofuran, and 1,4-dioxane; halogenated hydrocarbon solvents such as methylene chloride and 1,1,2-trichloroethane.
- nitrile solvents such as acetonitrile
- alcohol solvents such as methanol, ethanol, n-propanol, isopropyl alcohol, and tert-butyl alcohol
- ketone solvents such as acetone and methyl ethyl ketone
- Examples include ether solvents such as diethyl ether
- acetonitrile, methanol, ethanol, n-propanol, isopropyl alcohol, tert-butyl alcohol, acetone, methyl ethyl ketone, tetrahydrofuran and methylene chloride, and mixed solvents thereof such as acetonitrile / methanol mixed solvent, acetonitrile / ethanol mixed solvent It is preferable to use a mixed solvent of acetonitrile / tert-butyl alcohol. These solvents may be used alone or in combination of two or more.
- the temperature of the sensitizing treatment is preferably 5 to 100 ° C., and preferably 25 to 80 ° C. More preferred.
- the sensitizing time is preferably 15 minutes to 20 hours, and more preferably 3 to 24 hours.
- the sensitizing treatment is preferably performed at room temperature (25 ° C.) for 2 to 48 hours, particularly 3 to 24 hours.
- the sensitizing treatment time may be appropriately changed depending on the set temperature.
- the sensitizing treatment may be performed under reduced pressure or under vacuum from the viewpoint of shortening the time of the sensitizing treatment and adsorbing to the deep part of the semiconductor layer.
- the total content (total supported amount) of the sensitizing dye per 1 m 2 of the photoelectric conversion layer is not particularly limited, but is preferably 0.01 to 100 mmol / m 2 , and is 0.1 to 50 mmol / m 2 . More preferably, it is more preferably 0.5 to 20 mmol / m 2 .
- the hole transport layer causes the conductive polymer precursor to contact the photoelectric conversion layer, and the sensitizing dye is irradiated with light in the presence of an oxidizing agent to polymerize the conductive polymer precursor. Is formed.
- a method for bringing the conductive polymer precursor into contact with the photoelectric conversion layer is not particularly limited, but when the semiconductor layer is not a porous body, a solution containing the conductive polymer precursor is applied onto the photoelectric conversion layer. The drying method is preferred.
- the photoelectric conversion layer is added to the solution containing the conductive polymer precursor so that the conductive polymer precursor comes into contact with the porous interior carrying the sensitizing dye. Is preferably impregnated and / or applied.
- the solution containing a conductive polymer precursor usually contains a conductive polymer precursor, an oxidizing agent, and a solvent. Other additives may be included as necessary.
- the conductive polymer precursor that can be used is not particularly limited, but is preferably a compound represented by the following general formula (2) or a compound having a repeating unit represented by the following general formula (2). .
- the conductive polymer precursor is preferably a compound having a repeating unit represented by the following general formula (2).
- X is S, NR, or O
- R is a hydrogen atom or a substituted or unsubstituted alkyl group.
- R 2 to R 5 each independently represents a hydrogen atom, a halogen atom, a substituted or unsubstituted alkyl group, a substituted or unsubstituted cycloalkyl group, a substituted or unsubstituted alkoxy group, or a C2 to C30 polyethylene oxide. Or a substituted or unsubstituted C4-C30 cyclic compound-containing group.
- substituted or unsubstituted alkyl group, substituted or unsubstituted cycloalkyl group, or substituted or unsubstituted alkoxy group is the same as in the general formula (1), the description thereof is omitted here.
- the halogen atom is not particularly limited, but includes a fluorine atom, a chlorine atom, a bromine atom or an iodine atom.
- the C2-C30 polyethylene oxido group is not particularly limited, but is preferably a group represented by the formula (a) or the formula (b).
- x is an integer from 1 to 9.
- x is preferably a polyethylene oxy group of 3 to 9, and more preferably x is a polyethylene oxy group of 3.
- the C4-C30 cyclic compound group is not particularly limited, but includes a benzene ring, naphthalene ring, anthracene ring, thiophene group, phenylthiophene group, diphenylthiophene group, imidazole ring, oxazole ring, thiazole ring, pyrrole ring, furan And groups derived from a ring, a benzimidazole ring, a benzoxazole ring, a rhodamine ring, a pyrazolone ring, an imidazolone ring, a pyran ring, a pyridine ring, and a fluorene ring.
- substituted or unsubstituted means that at least one hydrogen atom is substituted with another substituent among an alkyl group, a cycloalkyl group, an alkoxy group, and a cyclic compound-containing group.
- R 2 to R 5 are each independently a hydrogen atom, a halogen atom, a C6 to C24 alkyl group, a C1 to C18 alkoxy group, a phenyl group, a biphenyl group, C1-C8 alkyl group substituted phenyl group, C1-C8 alkyl group substituted biphenyl group, thiophene group, bithiophene group, C1-C8 alkyl group substituted thiophene group, C1-C8 alkyl group And a bithiophene group substituted with a C1-C8 alkoxy group, and a bithiophene group substituted with a C1-C8 alkoxy group.
- R 2 to R 5 are each independently a hydrogen atom, chlorine atom, methyl group, ethyl group, butyl group, isobutyl group, sec-butyl group, tert-butyl group, hexyl group, decyl group.
- Tetradecyl group Tetradecyl group, octadecyl group, tetracosyl group, 1-methyldecyl group, 8-methylnonyl group, cyclopropyl group, cyclooctyl group, methoxy group, decyloxy group, octadecyloxy group, octamethylene glycol ethyl ether group, phenyl group, p- Examples thereof include a hexylphenyl group, a biphenyl group, a 4′-hexylbiphenyl group, a 5-hexyloxy-2-thienyl group, a 5′-octyl-2,2′-bithienyl group, and the like.
- two or more monomers including a dimer or higher multimer or a so-called oligomer.
- the oxidizing agent is not particularly limited as long as the conductive polymer precursor can be polymerized. If the level of the oxidant is higher than that of the excited dye, electrons can be taken away. On the other hand, if the level of the oxidizing agent is too high, the conductive polymer precursor (for example, bis-EDOT) may be directly oxidized and polymerized, making it difficult to form a uniform film near the dye. is there. For this reason, it is preferable to polymerize with an oxidizing agent having an appropriate standard electrode potential.
- the oxidizing agent according to the present invention preferably has a standard electrode potential (E 0 (OX) ) (V) of ⁇ 1.5 to +2.5 V, and is ⁇ 0.5 to +2.0 V. It is more preferable to have a standard electrode potential (E 0 (OX) ) (V) of
- E 0 (OX) standard electrode potential
- the standard electrode potential of the oxidizing agent is equal to or higher than the upper limit, the polymerization can proceed more efficiently.
- the standard electrode potential of the oxidizing agent is lower than the lower limit, the reaction (reaction rate) can be easily controlled, the productivity is excellent, and this is industrially preferable.
- an oxidant having such a standard electrode potential (E 0 (OX) ) (V) can more efficiently consume electrons excited by a dye during light irradiation, and therefore can conduct polymerization of a conductive polymer precursor. It can be further promoted, and a more uniform film can be formed in the vicinity of the pigment.
- standard electrode potential (E 0 (OX) ) (V) means a standard electrode potential (25 ° C.) in an aqueous solution.
- an oxidizing agent used for forming the hole transport layer of the present invention include hydrogen peroxide (+1.763 V), metal salt, peroxide, ozone, oxygen (+1.229 V), and methanol. (+ 0.588V).
- standard electrode potential (E 0 (OX) ) (V) is shown.
- an oxidizing agent is at least 1 sort (s) of hydrogen peroxide, oxygen, ozone, a metal salt, and a peroxide from a viewpoint of stability of material itself.
- the oxidizing agent is a compound that becomes a gas compound or a liquid compound by light irradiation (reduction of itself).
- the oxidizing agent can be hydrogen peroxide, oxygen, ozone.
- the “gaseous compound” means a compound that is gaseous under the conditions of 20 ° C. and 1 atm.
- the “liquid compound” means a compound that is liquid under conditions of 20 ° C. and 1 atm.
- the dye When the dye is irradiated with light in the presence of the oxidizing agent, the electrons excited in the dye are consumed by the oxidizing agent (for example, hydrogen peroxide / hydrogen peroxide solution), and the cationic dye is a monomer. It is considered that the polymerization of the molecular precursor is pulled out and polymerization is started.
- the oxidizing agent for example, hydrogen peroxide / hydrogen peroxide solution
- peroxide examples include permanganic acid or a salt thereof, chromic acid or a salt thereof, peroxo acid or a salt thereof, oxygen acid or a salt thereof, nitric acid, sulfuric acid, and the like.
- sodium peroxide Barium peroxide, potassium permanganate, sodium permanganate, chromate metal salt, dichromate metal salt, peroxodisulfuric acid, ammonium peroxodisulfate, peroxodisulfate metal salt, peroxophosphoric acid, peroxosulfuric acid, peroxoboric acid
- Inorganic peroxides such as sodium, hypochlorous acid, hypobromite, hypoiodous acid, chloric acid, bromic acid, iodic acid, sodium hypochlorite, calcium hypochlorite; cumene hydroperoxide, formic acid, Performic acid, peracetic acid, perbenzoic acid, perphthalic acid, t-butyl hydroperoxide, 1,
- metal salts examples include iron (II) chloride, iron (III) chloride, iron (III) sulfate, iron (III) nitrate, silver nitrate (AgNO 3 ), iron (III) citrate, iron iron (III) sulfate, and the like. Can be mentioned.
- an oxidizing agent having a standard electrode potential (E 0 (OX) ) of ⁇ 0.5 to +2.0 (V) may be used.
- E 0 (OX) a standard electrode potential
- methanol (+0.588 V) may be used.
- Oxygen (+1.229 V) and the like can be used.
- hydrogen peroxide (+1.763 V), potassium permanganate, cumene hydroperoxide, formic acid (+0.034 V), iron (II) chloride ( ⁇ 0.440 V), silver nitrate (AgNO 3 ) (+0 799 V), methanol, oxygen (+1.229 V) and ozone are preferred, hydrogen peroxide, methanol and oxygen (+1.229 V) are more preferred, and hydrogen peroxide is particularly preferred.
- the above oxidizing agents may be used alone or in combination of two or more.
- the content of the oxidizing agent in the solution containing the conductive polymer precursor is preferably 0.01 to 10 parts by weight, and 0.05 to 5 parts by weight with respect to 100 parts by weight of the conductive polymer precursor. It is more preferable that
- solvent The solvent that can be used is not particularly limited as long as it can dissolve the electrolyte and the conductive polymer precursor, but butylene oxide, chloroform, cyclohexanone, acetonitrile, tetrahydrofuran, propylene carbonate, dichloromethane, o-dichlorobenzene, dimethylformamide. Dimethyl sulfoxide, hexamethylphosphoric triamide, dimethoxyethane, acetone, methanol, ethanol, propanol, isobutyl alcohol, tert-butyl alcohol, methylene chloride and the like. Moreover, you may add water and another organic solvent to the said solvent as needed, and may use it as a mixed solvent. The above solvents may be used alone or in combination of two or more.
- the content of the solvent in the solution containing the conductive polymer precursor is preferably 2000 to 5000 parts by mass and more preferably 3000 to 4000 parts by mass with respect to 100 parts by mass of the conductive polymer precursor. preferable.
- additives are not particularly limited, and examples thereof include an electrolyte, a polymerization catalyst, and a polymerization rate adjusting agent.
- Electrolyte examples of the electrolyte include a dispersion of a redox electrolyte and a supporting electrolyte.
- the redox electrolyte is not particularly limited, and examples thereof include I ⁇ / I 3 ⁇ system, Br ⁇ / Br 3 ⁇ system, and quinone / hydroquinone system.
- the dispersion of the redox electrolyte can be obtained by a known method.
- an I ⁇ / I 3 ⁇ system electrolyte can be obtained by mixing iodide ions and iodine.
- the redox electrolyte dispersion is dispersed in a liquid electrolyte when used in a liquid form, a solid polymer electrolyte when dispersed in a solid polymer at room temperature (25 ° C.), and a gel substance. In this case, it is called a gel electrolyte.
- an electrochemically inert solvent is used as the solvent.
- the solvent include acetonitrile, propylene carbonate, and ethylene carbonate.
- the electrolyte described in JP-A No. 2001-160427 is used.
- the electrolyte described in “Surface Science” Vol. 21, No. 5, pages 288 to 293 is used. , Respectively.
- the supporting electrolyte is not particularly limited, but those that can be ionized are used and are not limited to specific ones. However, those that are highly soluble in a solvent and hardly undergo oxidation and reduction are preferably used. Specifically, lithium perchlorate (LiClO 4 ), lithium tetrafluoroborate, tetrabutylammonium perchlorate, Li [(CF 3 SO 2 ) 2 N], (n—C 4 H 9 ) 4 NBF 4 , (NC 4 H 9 ) 4 NPF 4 , p-toluenesulfonate, dodecylbenzenesulfonate, and the like are preferred. Further, polymer electrolytes described in JP-A-2000-106223 (for example, PA-1 to PA-10 in the same publication) may be used as the supporting electrolyte.
- the above electrolytes may be used alone or in combination of two or more.
- the content of the supporting electrolyte in the solution containing the conductive polymer precursor is preferably 30 to 200 parts by mass and preferably 30 to 100 parts by mass with respect to 100 parts by mass of the conductive polymer precursor. More preferred.
- the polymerization catalyst is not particularly limited, but iron (III) tris-p-toluenesulfonate, iron (III) p-dodecylbenzenesulfonate, iron (III) methanesulfonate, iron p-ethylbenzenesulfonate (III), iron (III) naphthalenesulfonate, and hydrates thereof.
- the polymerization rate modifier is not particularly limited, but there is a weak complexing agent for trivalent iron ions in the polymerization catalyst, and there is no particular limitation as long as the polymerization rate is reduced so that a film can be formed. Absent.
- the polymerization catalyst is iron (III) tris-p-toluenesulfonate, iron (III) p-dodecylbenzenesulfonate, iron (III) methanesulfonate, iron (III) p-ethylbenzenesulfonate, iron naphthalenesulfonate In the case of (III) and hydrates thereof, imidazole and the like can be used.
- polymerization catalyst for example, N (PhBr) 3 SbCl 6 , NOPF 6 , SbCl 5 , I 2 , Br 2 , HClO 4 , (nC) 4 H 9 ) 4 ClO 4 , trifluoroacetic acid, 4-dodecylbenzenesulfonic acid, 1-naphthalenesulfonic acid, FeCl 3 , AuCl 3 , NOSbF 6 , AsF 5 , NOBF 4 , LiBF 4 , H 3 [PMo 12 O 40 ]
- Additives such as acceptor doping agents such as 7,7,8,8-tetracyanoquinodimethane (TCNQ), binder resins that do not easily trap holes, and coating improvers such as leveling agents. Also good.
- the said additive may be used individually or may be used in mixture of 2 or more types.
- the impregnation method is not particularly limited, and the photoelectric conversion layer can be impregnated in a solution containing a conductive polymer precursor by a known method. Specifically, an apparatus may be used, or a manufacturer or the like may perform it.
- the impregnation temperature is not particularly limited, but is preferably set in a range where the solvent does not solidify or bump, more preferably -10 ° C to 60 ° C.
- the coating method of the solution containing the conductive polymer precursor is not particularly limited, but dipping, dripping, doctor blade, spin coating, brush coating, spray coating, roll coater, air knife coating, curtain coating, wire bar coating, Various coating methods such as gravure coating, extrusion coating using a hopper described in US Pat. No. 2,681,294, and multilayer simultaneous coating methods described in US Pat. Nos. 2,761,418, 3,508,947, and 2,761791 can be used. Further, the coating may be repeated by repeating such coating operation. The number of coatings in this case is not particularly limited and can be appropriately selected according to the desired thickness of the hole transport layer.
- the coating temperature is not particularly limited, but is preferably set in a range where the solvent does not solidify or bump, more preferably from ⁇ 10 ° C. to 60 ° C.
- the hole transport layer is formed by irradiating the sensitizing dye with light in the presence of an oxidizing agent to polymerize the conductive polymer precursor.
- the polymerization method is also referred to as “photochemical polymerization method”.
- the photochemical polymerization method is a polymerization method of a conductive polymer precursor using excitation of a sensitizing dye in a photoelectric conversion layer. Specifically, (1) when the sensitizing dye is irradiated with light, the sensitizing dye is excited. (2) The electrons of the excited sensitizing dye are consumed by the reduction of an oxidizing agent (for example, hydrogen peroxide), and the sensitizing dye becomes a cationic state. (3) The cationic sensitizing dye extracts electrons from the conductive polymer precursor, and the conductive polymer precursor becomes a cationic state. (4) Polymerization of the conductive polymer precursor is initiated by the conductive polymer precursor in the cationic state serving as a trigger (that is, functioning as a polymerization initiator). The photochemical polymerization reaction is represented by the following reaction formula.
- the photochemical polymerization method does not perform polymerization by applying a voltage unlike conventional electrolytic polymerization.
- the sensitizing dye is not deteriorated by voltage application as compared with electrolytic polymerization.
- a hole transport layer having a uniform composition can be obtained. Therefore, the degree of polymerization in the hole transport layer does not become nonuniform due to a voltage drop due to the conductive polymer deposited by polymerization of the conductive polymer precursor as in electrolytic polymerization.
- it since it is a chemical polymerization method using a cation, it may be possible to form a polymer in a sufficient amount and densely on the surface of the photoelectric conversion layer (semiconductor layer).
- the time required for the polymerization can be long.
- photochemical polymerization polymerization can be performed more rapidly than electrolytic polymerization, and the productivity is excellent.
- the electropolymerization due to the non-uniformity of the degree of polymerization that accompanies the deposition of the conductive polymer, when the photoelectric conversion element having a large area is to be manufactured, the decrease in durability can be more remarkable.
- photochemical polymerization a hole transport layer having a uniform composition in the hole transport layer can be obtained, and thus a photoelectric conversion element having a larger area can be produced more suitably.
- the photochemical polymerization method can be carried out not only in a batch format but also in a continuous format, and is excellent in terms of production.
- the photochemical polymerization method has further advantages. More specifically, so far, of the sensitizing dye
- the sensitizing dye having a carboxyl group (-COOH group) under the conditions of the electrolytic polymerization, CO 2 is desorbed (Kolbe electrolysis) by an applied voltage
- a problem that the dye is decomposed for example, see JP 2011-065751 A.
- the performance of the obtained photoelectric conversion element was low.
- a sensitizing dye having a carboxy group can be preferably used.
- a light source that can be used for light irradiation is not particularly limited, and examples thereof include a xenon lamp, a halogen lamp, and an LED.
- the wavelength of the irradiation light is not particularly limited, but it is preferable to use a light source having a wavelength of 400 nm or more, more preferably using a light source having a wavelength of 400 to 1100 nm (or more than 400 nm to 1100 nm or less), particularly preferably a wavelength exceeding 420 nm to 1100 nm or less. preferable.
- the wavelength is 400 nm or less, decomposition of the sensitizing dye due to the photocatalytic action of titanium oxide generated by light having a wavelength of 400 nm or less can be suppressed, and a long hole transport layer can be formed for a long time. It is preferable because stable characteristics can be obtained when light irradiation is performed.
- the conductive polymer precursor is represented by the general formula (2). Is preferably used because side reactions such as decomposition of the produced conductive polymer by light irradiation can be suppressed.
- the intensity of the irradiation light is not particularly limited, preferably from 10 ⁇ 150mW / cm 2, and more preferably 20 ⁇ 80mW / cm 2.
- the light irradiation time varies depending on the wavelength and intensity of the irradiation light, but is preferably 0.1 to 30 minutes, and more preferably 0.5 to 15 minutes. It is preferable that the light irradiation time be in the above range since the polymerization can proceed sufficiently.
- the light irradiation may be performed from the outside in a state where the photoelectric conversion layer is impregnated with a solution containing a conductive polymer precursor, or may be performed separately after impregnation and / or application on the photoelectric conversion layer.
- a step of washing with the above-mentioned solvent and / or a step of drying under conditions of, for example, 25 to 150 ° C. and 0.2 to 12 hours may be performed as necessary. Good.
- the semiconductor electrode having the hole transport layer is replaced with the above-mentioned solvent, the above-mentioned electrolyte.
- a mixed solution containing at least one selected from the group consisting of the above-mentioned organic salts for example, may be immersed at ⁇ 10 to 70 ° C. for 0.1 to 2 hours. In this case, it is preferable to let it stand for 0.01 to 24 hours by natural drying after being immersed.
- the hole transport layer polymerized by the photochemical polymerization reaction includes a conductive polymer.
- the hole transport layer may include a component that can be contained in a solvent including the conductive polymer precursor.
- the conductive polymer may have a structure obtained by polymerizing the conductive polymer precursor described above.
- the conductive polymer precursor preferably has the above formula (2) and plays a role of polymerizing. Therefore, a multimer in which the above formula (2) is combined alone or a plurality of types of repeating units may be used. Furthermore, it may be a prepolymer (including a dimer or higher multimer or a so-called oligomer) obtained by polymerizing a monomer having the above repeating unit in advance, alone or with a plurality of types of monomers, as necessary.
- the conductive polymer precursor represented by the above general formula (2) when used, it may be a conductive polymer represented by the following general formula (3).
- X and R 2 to R 5 are the same as those in the general formula (2).
- M represents the number of bonds of the monomer.
- m is an integer of 1 or more, preferably an integer of 1 to 10.
- the conductive polymer precursor is preferably M1-1, M1-4, and M1-26, and the conductive polymer precursor is particularly preferably M1-1. That is, the conductive polymer precursor has the following formula:
- the hole transport layer is soaked in tetrahydrofuran (THF) that can dissolve the polymer.
- THF tetrahydrofuran
- the solubility can be determined. Specifically, 60 mg of a compound (conductive polymer) is taken in a 25 mL sample bottle, 10 ml of THF is added, and ultrasonic waves (25 kHz, 150 W, ultrasonic industry COLLECTOR CURRENT 1.5A made by ultrasonic industry 150) When the dissolved compound is 5 mg or less when it is irradiated for 5 minutes, it is judged to be polymerized.
- the polymerization of the conductive polymer can be confirmed by comparing the absorption bands before and after the polymerization.
- polymerization of the conductive polymer can be confirmed by irradiating the obtained polymer with light and confirming new absorption at 600 to 1100 nm.
- the material contained in the hole transport layer preferably has a large band gap so as not to prevent light absorption by the sensitizing dye. Specifically, it preferably has a band cap of 2 eV or more, and more preferably has a band cap of 2.5 eV or more.
- the hole transport layer preferably has a low ionization potential in order to reduce sensitizing dye holes. Although the value of the ionization potential varies depending on the sensitizing dye to be applied, it is usually preferably 4.5 to 5.5 eV, more preferably 4.7 to 5.3 eV.
- the average thickness of the hole transport layer according to the present invention is not easily measured when the semiconductor layer is a porous body, since it penetrates into the porous body and the gaps.
- the short-circuit prevention layer according to this embodiment is formed in a form that follows the first electrode, it is possible to prevent the first electrode from protruding from the short-circuit prevention layer. As a result, the obtained photoelectric conversion element can exhibit high photoelectric conversion efficiency by preventing or reducing the occurrence of reverse electron transfer and dark current.
- the conductive polymer is uniformly formed in the hole transport layer. As a result, the manufactured photoelectric conversion element can have high durability.
- the hole transport layer can contact not only the photoelectric conversion layer but also the short-circuit prevention layer.
- the hole transport layer is also in contact with the short-circuit prevention layer.
- Conductive polymers can exist with high probability.
- contact between the first electrode and the conductive polymer increases reverse electron transfer and dark current, which can reduce photoelectric conversion efficiency. There is a risk of increased sex (FIG. 2A).
- Such a phenomenon can be more problematic than the conventional case of forming a hole transport layer by electrolytic polymerization.
- the hole transport layer is formed by electrolytic polymerization, the conductive polymer is present in the hole transport layer non-uniformly, so even if the first electrode and the hole transport layer are in contact with each other, The possibility of contact with the conductive polymer in the hole transport layer was relatively low (for example, there is a possibility of contact with a monomer or the like that does not exhibit conductivity in the hole transport layer). Therefore, the problem of a decrease in photoelectric conversion efficiency due to the protrusion of the short-circuit prevention layer is a problem that requires more attention when forming the hole transport layer by photochemical polymerization, and it is necessary to carefully adjust the manufacturing conditions.
- the photoelectric conversion efficiency which combined these methods and the improvement effect of durability were expressed to the maximum.
- the reason why the photoelectric conversion element according to the present invention is more excellent in photoelectric conversion efficiency and has higher durability is estimated as follows. That is, in the conventional electric field polymerization, nonuniform film formation was forced due to the partial nonuniformity of the applied voltage generated in the porous minute part. As a result, the conversion efficiency is reduced due to the disconnection of the conduction path due to the use of the photoelectric conversion element for a long period of time.
- a conductive polymer phase can be uniformly formed on the entire porous semiconductor, and a photoelectric conversion element having higher durability can be formed.
- the short-circuit prevention layer by the vertical deposition method, it is estimated that high conversion efficiency can be achieved in order to suppress the increase probability of reverse electron transfer sometimes caused by sample variation in uniform coating formation. .
- the second electrode is not particularly limited and can be formed by a known method. For example, methods such as vapor deposition (including vacuum vapor deposition), sputtering, coating, and screen printing of the material for the second electrode can be applied.
- the second electrode formed in this manner is disposed in contact with the hole transport layer. Even an insulating material can be used if a conductive material layer is provided on the side facing the hole transport layer.
- the second electrode preferably has good contact with the hole transport layer from the viewpoint of reducing the electric resistance of the element.
- the second electrode preferably has a small work function difference from the hole transport layer and is chemically stable.
- the material of the second electrode that can be used is not particularly limited, but a metal thin film such as gold, silver, copper, aluminum, platinum, chromium, rhodium, ruthenium, magnesium, indium, carbon, carbon black, conductive polymer, And organic conductors such as conductive metal oxides (indium-tin composite oxide, tin oxide doped with fluorine, etc.).
- a metal thin film such as gold is preferable.
- the film thickness of the second electrode is not particularly limited, but is preferably 10 to 1000 nm.
- the surface resistance value of the second electrode is not particularly limited and is preferably as low as possible. Specifically, the surface resistance value is preferably 80 ⁇ / ⁇ or less, and more preferably 20 ⁇ / ⁇ or less.
- the photoelectric conversion element manufactured by the method of the present invention can be particularly suitably used for a solar cell.
- the photoelectric conversion element according to the present invention can be used as a dye-sensitized solar cell (cell). That is, the solar cell according to the present invention includes, for example, a plurality of solar cells (photoelectric conversion elements according to the present invention) electrically connected by an interconnector, a pair of protective members sandwiching the solar cells, and a pair of protective members. And a sealing resin filled in gaps between the plurality of solar cells.
- One of the pair of protective members serves as the base of the photoelectric conversion element described above. Both of the pair of protective members may be transparent, or only one of them may be transparent.
- Examples of the structure of the solar cell according to the present invention include a Z-type module and a W-type module.
- the Z-type module is composed of a pair of opposing protective members, one of which is a porous semiconductor layer carrying a plurality of dyes, and the other substrate is formed with a plurality of hole transport layers, which are bonded together.
- the W-type module has a structure in which a laminate of a porous semiconductor layer and a hole transport layer each carrying a dye is formed on each of the protective members, and the cells are bonded so that the cells are staggered.
- the sensitizing dye supported on the semiconductor is excited by absorbing the irradiated light or electromagnetic wave. Electrons generated by excitation move to the semiconductor, and then move to the second electrode via the conductive support and the external load, and are supplied to the hole transporting material of the hole transporting layer.
- the sensitizing dye that has moved the electrons to the semiconductor is an oxidant, but is reduced by the supply of electrons from the second electrode via the polymer of the hole transport layer, thereby returning to the original state.
- the polymer of the hole transport layer is oxidized and returned to a state where it can be reduced again by the electrons supplied from the second electrode. In this way, electrons flow, and a solar cell using the photoelectric conversion element of the present invention can be configured.
- Example 1 Step of forming first electrode A heat-resistant glass plate having a thickness of 1.1 mm was used as the substrate.
- tin (IV) chloride pentahydrate (SnCl 4 .5H 2 O, molecular weight: 350.60) was used.
- tin (IV) chloride pentahydrate was dissolved in a ratio of 10 ml of 30% aqueous ethanol solution to 0.701 g of tin (IV) chloride pentahydrate.
- a coating solution was prepared by adding 0.592 g of a saturated aqueous solution of ammonium fluoride to the solution and subjecting the resulting mixture to ultrasonic treatment for about 20 minutes.
- the surface of the heat-resistant glass plate was chemically washed and dried.
- the heat-resistant glass plate was left in the reactor and heated with a heater.
- the heating temperature of the heater reached 400 ° C.
- the coating liquid was sprayed at a pressure of 0.06 MPa for 25 minutes from a nozzle having a diameter of 0.3 mm with a distance to the glass plate of 600 mm, whereby a thickness of 100 nm was obtained.
- One electrode (FTO layer) was formed.
- Step (1) Step of forming a short-circuit prevention layer TC100 (titanium diisopropoxybis (acetylacetonate), manufactured by Matsumoto Kosho Co., Ltd.) was used as a short-circuit prevention material precursor.
- the short-circuit prevention layer was formed by a method using an ink jet method.
- TC100 titanium diisopropoxybis (acetylacetonate)
- the obtained solution is applied on the first electrode (FTO) formed above by an ink jet method, and then heated at 500 ° C. for 10 minutes to be fired, whereby a short-circuit preventing layer made of a thin layer of titanium oxide. Formed.
- the thickness of the obtained short circuit prevention layer was 100 nm.
- Step of forming photoelectric conversion layer As the semiconductor, titanium oxide (anatase type, primary average particle diameter (microscope observation average) 18 nm) was used. As a sensitizing dye, a compound of A-16 (manufactured by Konica Minolta) represented by the following formula was used.
- a titanium oxide paste (ethyl cellulose dispersed in 10% acetylacetone water) was applied on the short-circuit prevention layer by a screen printing method (application area: 25 mm 2 ).
- the obtained coating film was baked at 200 ° C. for 10 minutes and at 500 ° C. for 15 minutes, and a porous layer of titanium oxide having a thickness of 2.5 ⁇ m and a porosity (D) of 60% by volume (porous) Obtained).
- the sensitizing dye A-16 was dissolved in a mixed solvent of acetonitrile and tert-butyl alcohol (volume ratio 1: 1) so as to have a concentration of 5 ⁇ 10 ⁇ 4 mol / L.
- the FTO glass substrate on which the porous layer of titanium oxide obtained above is formed is immersed at room temperature (25 ° C.) for 3 hours to adsorb the sensitizing dye to the semiconductor layer, thereby supporting the porous dye.
- a semiconductor electrode having a high quality semiconductor layer (photoelectric conversion layer) was obtained.
- the total amount of the sensitizing dye per 1 m 2 of the semiconductor layer was 1 mmol.
- the sensitizing dye used exhibited an absorption band at 350 to 650 nm.
- Step (2) Step of forming a hole transport layer
- a conductive polymer precursor M1-1 represented by the following chemical formula (2,2′-bis [3,4- (ethylenebisoxy) thiophene]) Li [(CF 3 SO 2 ) 2 N], which is a supporting electrolyte, was dissolved in acetonitrile so that the concentrations were 1 ⁇ 10 ⁇ 3 (mol / L) and 0.1 (mol / L), respectively.
- 30% by weight of hydrogen peroxide as an oxidizing agent is added so that hydrogen peroxide is 1 part by mass with respect to 100 parts by mass of the conductive polymer precursor, and the conductive polymer precursor is added.
- a solution containing was prepared.
- a semiconductor electrode (semiconductor electrode / hole transport layer) on which the hole transport layer is formed is formed as Li [(CF 3 SO 2 ) 2 N] was immersed in an acetonitrile solution containing 15 ⁇ 10 ⁇ 3 (mol / L) and tert-butylpyridine at a ratio of 50 ⁇ 10 ⁇ 3 (mol / l) for 10 minutes. , Dried naturally.
- Step of forming second electrode Gold was deposited to 60 nm by a vacuum deposition method to form a second electrode, and a photoelectric conversion element was manufactured.
- Example 2 The photoelectric conversion is performed in the same manner as in Example 1 except that the number of times of discharging the solution containing the short-circuit prevention material precursor in step (1) is changed to 3 and the thickness of the short-circuit prevention layer is 30 nm. A conversion element was manufactured.
- Example 3 The photoelectric conversion is performed in the same manner as in Example 1 except that the number of times of discharging the solution containing the short-circuit prevention material precursor in step (1) is changed to 1 and the thickness of the short-circuit prevention layer is 5 nm. A conversion element was manufactured.
- Example 4 The photoelectric conversion is performed in the same manner as in Example 1 except that the number of times of discharging the solution containing the short-circuit prevention material precursor in step (1) is changed to 40 times and the thickness of the short-circuit prevention layer is set to 400 nm. A conversion element was manufactured.
- Example 5 A photoelectric conversion element was produced in the same manner as in Example 1 except that the firing temperature in step (1) was changed to 200 ° C.
- Example 6 A photoelectric conversion element was produced in the same manner as in Example 1 except that the firing temperature in step (1) was changed to 400 ° C.
- Example 7 A photoelectric conversion element was produced in the same manner as in Example 1 except that the firing temperature in step (1) was changed to 600 ° C.
- Example 8 In the step (1), a photoelectric conversion element was produced in the same manner as in Example 1 except that it was applied on the first electrode (FTO) by a spray method.
- the said spray method was performed by the following method. That is, a two-fluid spray nozzle AM25S (manufactured by Atmax Co.) was used as the spray nozzle, and nitrogen gas having an atomization pressure of 30 kPa was used as the atomization and ejection gas.
- the distance between the nozzle tip and the first electrode was 80 mm.
- An orifice with a diameter of 0.4 mm is provided in the flow path that connects the container containing the solution containing the precursor solution for the short circuit and the nozzle, and the discharge amount of the solution from the nozzle is adjusted by applying a discharge pressure of 1 kPa to the container. did.
- the discharge rate at this time was 11.3 mL / min.
- the solution was applied to the surface of the first electrode by scanning the spray nozzle at a speed of 400 mm / s and a pitch of 15 mm. Subsequently, it heated at 500 degreeC for 10 minute (s), and the short circuit prevention layer which consists of a thin layer of a 100-nm-thick titanium oxide was formed.
- Example 9 In the step of forming the first electrode, the amount of the 30% aqueous solution of ethanol for dissolving the material of the first electrode (tin (IV) chloride pentahydrate) was changed to 8 mL to form the first electrode. Except for this, a photoelectric conversion element was produced in the same manner as in Example 1. In addition, when the surface roughness (Ra) of the formed 1st electrode was measured by the method similar to Example 1, it was 36 nm.
- Example 10 In the step of forming the first electrode, the amount of the 30% aqueous solution of ethanol for dissolving the material of the first electrode (tin (IV) chloride pentahydrate) was changed to 5 mL to form the first electrode. Except for this, a photoelectric conversion element was produced in the same manner as in Example 1. In addition, when the surface roughness (Ra) of the formed 1st electrode was measured by the method similar to Example 1, it was 69 nm.
- the short-circuit prevention layer is formed by a method using a spin coat method
- the hole transport layer is formed by photoelectrolytic polymerization.
- a photoelectric conversion element was produced by the same method.
- TC100 titanium diisopropoxybis (acetylacetonate), manufactured by Matsumoto Kosho Co., Ltd.
- 0.8 mL of acetylacetone 18 mL of ethanol were mixed.
- the obtained solution is dropped by a spin coating method and applied onto the first electrode (FTO), and then heated at 500 ° C. for 10 minutes to perform baking, thereby forming a short-circuit prevention layer composed of a thin layer of titanium oxide. did.
- the thickness of the obtained short circuit prevention layer was 100 nm.
- the positive hole transport layer by photoelectrolytic polymerization was performed as follows. First, the conductive polymer precursor used in Example 1 and the supporting electrolyte Li [(CF 3 SO 2 ) 2 N] were added at concentrations of 1 ⁇ 10 ⁇ 3 (mol / L) and 0.1 ( mol / L) was dissolved in acetonitrile to prepare a solution containing a conductive polymer precursor.
- the semiconductor electrode was immersed in the solution prepared above. Thereafter, the working electrode is the semiconductor electrode, the counter electrode is a platinum wire, the reference electrode is Ag / Ag + (AgNO 3 : 0.01M), the holding voltage is ⁇ 0.16 V, and from the outside of the semiconductor layer (in the semiconductor layer direction), While maintaining the voltage, polymerization was performed by irradiating light from a xenon lamp through a sharp cut filter (manufactured by HOYA: S-L42) that cuts a wavelength of 420 nm or less for 30 minutes. When the absorption bands before and after polymerization were compared after light irradiation, new absorption was observed at 600 to 1100 nm, confirming that the conductive polymer precursor was polymerized.
- the obtained semiconductor electrode having a hole transport layer was washed with acetonitrile and then dried. The obtained hole transport layer was a polymer film insoluble in the solvent.
- Comparative Example 2 A photoelectric conversion element was produced in the same manner as in Example 1 except that the short-circuit prevention layer was formed in the same manner as in Comparative Example 1.
- Comparative Example 3 A photoelectric conversion element was produced in the same manner as in Example 1 except that the hole transport layer was formed in the same manner as in Comparative Example 1.
- Comparative Example 1 in which the short-circuit prevention layer was formed by a method using a spin coating method and the hole transport layer was formed by photoelectrolytic polymerization, the initial photoelectric conversion efficiency was slightly low, and durability was low. The dark current density was a low value.
- Comparative Example 2 in which the method for forming the hole transport layer was changed from photo-electric field polymerization to photochemical polymerization, the durability was significantly improved.
- the dark current density is lower than that of Comparative Example 1. Therefore, the conductive polymer is uniformly present inside the hole transport layer obtained by photochemical polymerization, and the surface where the hole transport layer is in contact with the short-circuit prevention layer is also highly conductive.
- Comparative Example 1 the method of forming the short-circuit prevention layer was changed from the method using the spin coating method to the method using the inkjet method, and the short-circuit prevention layer suitably covered the first electrode. Therefore, it can be seen that the initial photoelectric conversion efficiency has been significantly improved, and the value of dark current density has also been significantly reduced.
- the photoelectric conversion elements of Examples 1 to 10 were formed by forming a short-circuit prevention layer by a vertical deposition method and by positive photopolymerization. It can be seen that by combining the formation of the hole transport layer, it has significantly excellent photoelectric conversion efficiency (initial photoelectric conversion efficiency and dark current density) and high durability.
- the photoelectric conversion element of Example 1 shows superior durability as compared with Comparative Example 2, and more excellent initial photoelectric conversion efficiency and darkness as compared with Comparative Example 3. This shows the effect of preventing current generation.
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Life Sciences & Earth Sciences (AREA)
- Wood Science & Technology (AREA)
- Organic Chemistry (AREA)
- Photovoltaic Devices (AREA)
Abstract
本発明は、光電変換効率に優れ、かつ、高い耐久性を有する光電変換素子の製造方法を提供することを目的とする。本発明は、基板、第一電極、短絡防止層、半導体および増感色素を含有する光電変換層、導電性高分子を含有する正孔輸送層、並びに第二電極をこの順に有する光電変換素子の製造方法であって、垂直堆積法により前記短絡防止層を形成する工程(1)と、導電性高分子前駆体を前記光電変換層と接触させ、酸化剤存在下で前記増感色素に光を照射して前記導電性高分子前駆体を重合することにより前記正孔輸送層を形成する工程(2)と、を含む、製造方法を提供する。
Description
本発明は、光電変換素子の製造方法に関する。
近年、無限で有害物質を発生しない太陽光の利用が精力的に検討されている。このクリーンエネルギー源である太陽光の利用法としては、光起電力効果を利用した太陽電池への適用が挙げられる。光起電力効果とは、物質に光を照射することで起電力を発生させる現象であり、当該物質を含む光電変換素子を用いることにより、光エネルギーを電気エネルギーへと変換することができる。
光電変換素子は、通常、基板、第一電極、光電変換層(増感色素が担持された半導体層)、正孔輸送層、および第二電極がこの順に配置された構成を有する。このような構成を有する光電変換素子に対して、基板の外側から光が照射されると、素子内部の光電変換層の増感色素が励起されて電子を放出する。励起された電子は第一電極に移動し、当該電子は、外部回路を通じて第二電極に移動して、正孔輸送層に供給される。そして、(電子を放出して)酸化された増感色素は、正孔輸送層から電子を受け取り、基底状態に戻る。このようなサイクルを繰り返すことで、光エネルギーが電気エネルギーに変換されるのである。
光電変換素子に求められる性能としては、優れた光電変換効率、高い耐久性等が挙げられる。これまでに、光電変換素子の種々の要素等を改善し、光電変換素子の上記性能向上を図る試みがなされている。
例えば、特許文献1は、光電変換素子を構成する正孔輸送層と対極(第二電極)との接触に着目し、光電変換効率の向上を図っている。具体的には、特許文献1には、固体型色素増感型太陽電池の対極構成材料として電子伝導性材料と成膜性を持つ高分子電解質との組み合わせを用いて、色素増感メゾポーラス二酸化チタン多孔質層(光電変換層)と該層に接して配置された正孔輸送高分子固体電解質(正孔輸送層)を有する太陽電池の対電極(第一電極および第二電極)を形成したことを特徴とする固体色素増感型太陽電池が開示されている。この際、前記正孔輸送高分子固体電解質は、色素を担持した多孔質二酸化チタンをピロール等のモノマーを含む溶液に浸漬し、所定の電位を保持して光照射を行い、前記モノマーを重合することで形成されている。そして、特許文献1によれば、前記正孔輸送層表面に所定の成分を含む混練液を滴下し、該液を拡げて対極を形成することにより、高い電流密度を示す固体色素増感型太陽電池が得られることが記載されている。
また、特許文献2は、第一電極と光電変換層との間に、短絡防止層を設けることで光電変換効率の向上を図っている。具体的には、特許文献2には、第1の電極(第一電極)と、該第1の電極と対向して設置された第2の電極(第二電極)と、前記第1の電極と前記第2の電極との間に位置し、その少なくとも一部が多孔質な受光層(p型有機半導体およびn型無機半導体を接合させたヘテロ接合型の光電変換層)と、該受光層と前記第2の電極との間に位置する正孔輸送層とを有し、前記受光層と前記正孔輸送層との界面に整流障壁が形成されている光電変換素子であって、前記第1の電極と前記正孔輸送層との間での短絡を防止または抑制する短絡防止手段を有することを特徴とする光電変換素子が記載されている。特許文献2によれば、第一電極と正孔輸送層との間に短絡防止手段(バリヤ層・短絡防止層)を設けることによって、第一電極と正孔輸送層との短絡を防止することができ、これによって、漏れ電流を防止し、光電変換効率に優れる光電変換素子が得られることが記載されている。
特許文献1では、電解重合法と呼ばれる方法を利用して正孔輸送層を形成している。当該電解重合法は、導電性高分子前駆体(モノマー等)が溶解した溶液に電極対を浸して電圧を印加することにより、導電性高分子前駆体を重合させて導電性高分子を得る方法である。ここで、特許文献1のように、半導体に担持された増感色素を含む光電変換層上に電解重合により正孔輸送層を形成する場合には、電圧印加による増感色素の分解(劣化)を防止するために、低電圧下で重合を行う必要がある。しかしながら、低電圧下で重合を行うと、モノマーの重合により形成される導電性高分子の析出に伴い電圧降下が生じるため、重合が不十分となり、色素の周りに十分な量の導電性高分子を生成させることが困難となりうる。また、正孔輸送層内の重合の程度が不均一となりうる。その結果、光電変換素子として十分な耐久性が得られないことがある。
また、特許文献2では、バリヤ層(短絡防止層)は、ゾル・ゲル法、蒸着法、スパッタリング法、スプレー熱分解法、ジェットモールド法、CVD法等によって形成することができると記載されている。しかし、これらの形成方法のなかには、例えば、表面に凹凸を有する第一電極を用いた場合等には、短絡防止層が十分に第一電極を被覆することができず、第一電極の一部が短絡防止層から突出する場合があることが判明した。その結果、第一電極と正孔輸送層とが短絡する場合があり、光電変換効率が低下しうる。
このように、光電変換素子は、正孔輸送層の形成方法の改善、短絡防止層の配置等によって光電変換効率および耐久性が改善されるとしても、未だ十分な光電変換効率および耐久性を有しているとはいえない。
そこで、本発明は、より光電変換効率に優れ、かつ、より高い耐久性を有する光電変換素子を提供することを目的とする。
本発明者らは、鋭意研究を行った結果、導電性高分子前駆体を前記光電変換層と接触させ、酸化剤存在下で前記増感色素に光を照射して前記導電性高分子前駆体を重合することにより正孔輸送層を形成し、かつ、短絡防止層を垂直堆積法により形成することにより、光電変換素子の光電変換効率および耐久性が有意に向上することを見出し、本発明を完成させるに至った。
すなわち、上記目的のうち少なくとも一つを実現するために、本発明の一側面を反映した光電変換素子の製造方法は、基板、第一電極、短絡防止層、半導体および増感色素を含有する光電変換層、導電性高分子を含有する正孔輸送層、並びに第二電極をこの順に有する光電変換素子の製造方法であって、垂直堆積法により前記短絡防止層を形成する工程(1)と、導電性高分子前駆体を前記光電変換層と接触させ、酸化剤存在下で前記増感色素に光を照射して前記導電性高分子前駆体を重合することにより前記正孔輸送層を形成する工程(2)と、を含む。
本発明の一形態によれば、基板、第一電極、短絡防止層、半導体および増感色素を含有する光電変換層、導電性高分子を含有する正孔輸送層、並びに第二電極をこの順に有する光電変換素子の製造方法が提供される。この際、前記短絡防止層が、垂直堆積法により形成され(工程(1))、前記正孔輸送層が、導電性高分子前駆体を前記光電変換層と接触させ、酸化剤存在下で前記増感色素に光を照射して前記導電性高分子前駆体を重合することにより形成される(工程(2))点に特徴を有する。本発明により、光電変換効率に優れ、かつ、高い耐久性を有する光電変換素子の製造方法が提供できる。
<光電変換素子の製造方法>
本発明の光電変換素子の製造方法について、図1を参照しながら説明する。
本発明の光電変換素子の製造方法について、図1を参照しながら説明する。
図1は、本発明の一実施形態に係る光電変換素子を模式的に表す断面図である。図1に示すように、光電変換素子10は、基板1、第一電極2、短絡防止層3、光電変換層6、正孔輸送層7、および第二電極8が順次積層されてなる構成を有する。ここで、光電変換層6は、増感色素4および半導体5を含有する。図1に示されるように、第一電極2と光電変換層6との間には、短絡防止層3が配置されている。なお、図1中では、太陽光は、図下方の矢印9の方向から入っているが、本発明は当該形態に限定されず、図上方から太陽光が入射してもよい。
次に、上記光電変換素子の製造方法の好ましい実施形態について説明する。まず、基板上に第一電極2を形成する(第一電極を形成する工程)。その後、第一電極2上に、垂直堆積法により短絡防止層3を形成する(短絡防止層を形成する工程・工程(1))。形成された短絡防止層3上に半導体5からなる半導体層を形成し、その半導体表面に増感色素4を吸着させて光電変換層6を形成する(光電変換層を形成する工程)。その後、光電変換層6上に、導電性高分子前駆体を前記光電変換層と接触させ、酸化剤存在下で前記増感色素に光を照射して前記導電性高分子前駆体を重合することにより正孔輸送層7を形成する(正孔輸送層を形成する工程・工程(2))。この際、正孔輸送層7は、増感色素4を担持した半導体5からなる光電変換層6に侵入し、かつ、その上に存在している。このため、光電変換層6は、増感色素4および半導体5に加えて、正孔輸送層を構成する材料11を含有する。そして、正孔輸送層7の上に第二電極8を形成する(第二電極を形成する工程)。第一電極2および第二電極8に端子を付けることにより電流を取り出すことができる。
すなわち、本発明の一実施形態によれば、光電変換素子の製造方法は、第一電極を形成する工程、短絡防止層を形成する工程(工程(1))、光電変換層を形成する工程、正孔輸送層を形成する工程(工程(2))、および第二電極を形成する工程を含む。
以下、各工程について詳細に説明する。しかしながら、本発明の範囲はこれらの説明に拘束されることなく、以下の例示以外についても、本発明の趣旨を損なわない範囲で適宜変更実施し得る。具体的には、本発明は下記の各実施形態に限定されるものではなく、請求項に示した範囲で種々の変更が可能であり、異なる実施形態にそれぞれ開示された技術的手段を適宜組み合わせて得られる実施形態についても、本発明の技術的範囲に含まれる。
本明細書において、範囲を示す「X~Y」は、「X以上Y以下」であることを意味する。また、特に注釈のない限り、「ppm」は「重量ppm」を意味する。更に、「重量」と「質量」、「重量%」と「質量%」、「重量部」と「質量部」は同義語として扱う。また、物性等の測定に関しては、特に断りのない限り、室温(20~25℃、さらには約23℃)、相対湿度40~50%RHで測定する。
[第一電極を形成する工程]
本工程では、基板を準備し、基板上に第一電極を形成する。
本工程では、基板を準備し、基板上に第一電極を形成する。
(基板)
基板は、第一電極を保持する機能を有する。
基板は、第一電極を保持する機能を有する。
基板側から光が入射する場合、基板はこの光を透過させることが可能な、すなわち、光電変換すべき光の波長に対して透明な部材であることが好ましい。具体的には、光電変換効率の観点から、光透過率が10%以上であることが好ましく、50%以上であることがより好ましく、80%~100%であることが特に好ましい。なお、本明細書において、「光透過率」とは、JIS K 7361-1:1997(ISO 13468-1:1996に対応)の「プラスチック-透明材料の全光線透過率の試験方法」に準拠した方法で測定した可視光波長領域における全光線透過率を意味するものとする。
基板としては、その材料、形状、構造、厚み、硬度等については公知のものの中から適宜選択することができるが、上記のように高い光透過性を有していることが好ましい。
基板の材料としては、剛性を有する基板、および可撓性を有する基板を用いることができる。剛性を有する基板と可撓性を有する基板を組み合わせて用いてもよい。
剛性を有する基板としては、特に制限されず、公知のものを用いることができる。具体的には、ガラス板およびアクリル板が挙げられる。これらのうち、耐熱性の観点からガラス板を用いることが好ましい。
可撓性を有する基板としては、特に制限されず、公知のものを用いることができる。具体的には、ポリエチレンテレフタレート(PET)、ポリエチレンナフタレート、変性ポリエステル等のポリエステル系樹脂フィルム;ポリエチレン(PE)、ポリプロピレン(PP)、ポリスチレン、環状オレフィン等のポリオレフィン類樹脂フィルム;ポリ塩化ビニル、ポリ塩化ビニリデン等のビニル系樹脂フィルム;ポリビニルブチラール(PVB)等のポリビニルアセタール樹脂フィルム;ポリエーテルエーテルケトン(PEEK)樹脂フィルム;ポリスルホン(PSF)樹脂フィルム;ポリエーテルスルホン(PES)樹脂フィルム;ポリカーボネート(PC)樹脂フィルム;ポリアミド樹脂フィルム;ポリイミド樹脂フィルム;アクリル樹脂フィルム;トリアセチルセルロース(TAC)樹脂フィルムが挙げられる。
さらに、太陽光エネルギーを利用することを考慮し、可視領域の波長(400~700nm)における透過率が80%以上である樹脂フィルムを基板として用いてもよい。当該樹脂フィルムとしては、二軸延伸ポリエチレンテレフタレートフィルム、二軸延伸ポリエチレンナフタレートフィルム、ポリエーテルスルホンフィルム、およびポリカーボネートフィルム等が挙げられ、これらのうち、二軸延伸ポリエチレンテレフタレートフィルム、二軸延伸ポリエチレンナフタレートフィルムを用いることが好ましい。
基板の厚さは、特に制限されないが、剛性を有する基板を用いる場合には、0.1~100mmであることが好ましく、0.5~10mmであることがより好ましい。また、可撓性を有する基板を用いる場合には、前記基板の厚さは、1~1000μmであることが好ましく、10~100μmであることがより好ましい。
上記基板には、塗布液の濡れ性や接着性を確保するために、表面処理や易接着層を設けてもよい。表面処理や易接着層については従来公知の技術を使用できる。例えば、化学洗浄、コロナ放電処理、火炎処理、紫外線処理、高周波処理、グロー放電処理、活性プラズマ処理、レーザー処理等の表面活性化処理により表面処理を行うことができる。また、ポリエステル、ポリアミド、ポリウレタン、ビニル系共重合体、ブタジエン系共重合体、アクリル系共重合体、ビニリデン系共重合体、およびエポキシ系共重合体等を易接着層として使用することができる。
(第一電極の形成)
第一電極の形成方法としては、特に制限されず、公知の方法が適用されうる。例えば、第一電極を構成する材料を含む溶液を基板上に噴霧し、乾燥させることによって第一電極を形成することができる。
第一電極の形成方法としては、特に制限されず、公知の方法が適用されうる。例えば、第一電極を構成する材料を含む溶液を基板上に噴霧し、乾燥させることによって第一電極を形成することができる。
前記第一電極を構成する材料としては、特に制限されず、公知の材料が使用できる。例えば、金属およびその酸化物、並びにSn、Sb、FおよびAlからなる群から選択される少なくとも1種を含む複合(ドープ)材料を用いることができる。
前記金属としては、白金、金、銀、銅、アルミニウム、ロジウム、およびインジウム等が挙げられ、金属酸化物としては、SnO2、CdO、ZnO、CTO系(CdSnO3、Cd2SnO4、CdSnO4)、In2O3、およびCdIn2O4等が挙げられる。
また、複合(ドープ)材料としては、SnをドープしたIn2O3(ITO)、SbをドープしたSnO2、FをドープしたSnO2(FTO)等が挙げられる。
上述の材料のうち、銀、ITO、SbをドープしたSnO2、FTOを用いることが好ましく、耐熱性の観点からFTOを用いることがより好ましい。
第1電極を形成する材料としては、市販の前述した材料を用いてもよく、当該材料の前駆体を用いてもよい。例えば、前述の好ましいFTO電極を形成する場合では、市販のフッ素ドープ酸化スズ(FTO)を用いることができる。または、塩化スズ(IV)五水和物などの第一電極形成材料を適宜な溶媒(例えば、エタノール水溶液)に溶かし、フッ化アンモニウムの飽和水溶液と混合させたものを基板上に噴霧・乾燥してもよい。なお、ここでいう適宜な溶媒として、特に制限されず、アルコールなどの有機溶媒、水、またはアルコールと水の混合溶液などが挙げられる。また、この際、第一電極形成材料の溶媒における含有量は、特に制限されないが、第一電極形成材料が、溶媒100mL中に、1~20g、より好ましくは3~10g、特に好ましくは5~8g含まれるような量であることが好ましい。このような量であれば、以下に記載されるような第一電極の表面粗さ(Ra)に容易に制御できる。
ここで、第一電極を形成する材料の基板への塗布量は、特に制限されないが、基板1m2当たり、1~100g程度であることが好ましい。
その他の第一電極の形成方法としては、特に制限されないが、塗布法、スパッタ法、化学気相蒸着(CVD)法、スプレー熱分解(SPD)法等が用いられうる。
なお、第一電極をスリット状に形成する場合には、エキシマレーザー、YAGレーザー、CO2レーザー、エアジェット、ウォータジェットによる加工、エッチング加工、機械的加工等を行ってもよい。これにより、第一電極を複数の領域に分離することができる。スリットのピッチは、光電変換素子のセルのサイズに応じて、適宜設定することができる。
このように形成された第一電極は、第二電極とともに光電変換素子から電流を取り出すための機能を有する。
第一電極は、光電変換効率の観点から、光透過率が80%以上であることが好ましく、85%以上であることがより好ましく、90%~100%であることが特に好ましい。
また、第一電極の表面粗さ(Ra)は、5~100nmであることが好ましく、10~50nmであることがより好ましく、12~30nmであることが特に好ましい。第一電極の表面粗さが10nm以上であると、入射光の散逸を防止できることから好ましい。一方、第一電極の表面粗さが50nm以下であると、後述する短絡防止層を薄膜化できることから好ましい。第一電極の表面粗さは、第一電極を構成する材料の種類、第一電極の形成方法、条件(塗布条件や蒸着条件等)によって制御することができる。第一電極の表面粗さ(Ra)は、公知の方法によって測定される。本明細書では、「第一電極の表面粗さ(Ra)」は、サーフコム1400D(株式会社東京精密製)を用いて測定される値(nm)を意味する。
第一電極は、厚過ぎると光透過性が劣り、一方、薄過ぎると導電性が劣ってしまうことになる。このため、光透過性と導電性の機能を両立させることを考慮すると、第一電極は、0.03~3μm程度の膜厚範囲であることが好ましい。
または、導電性支持体の膜厚としては、特に制限されないが、0.1~5mmであることが好ましい。導電性支持体の表面抵抗値としては、可能な限り低い値であることが好ましい。具体的には、表面抵抗値が50Ω/□(square)以下であることが好ましく、10Ω/□以下であることがより好ましい。なお、本明細書において、「導電性支持体」とは、基板とその上に形成された第一電極との積層体を意味する。
[短絡防止層を形成する工程・工程(1)]
短絡防止層は、受光により発生し、正孔輸送層に注入されたホールと、第一電極の電子との再結合である短絡を防止する機能を有する。
短絡防止層は、受光により発生し、正孔輸送層に注入されたホールと、第一電極の電子との再結合である短絡を防止する機能を有する。
工程(1)は、垂直堆積法により短絡防止層を形成する工程である。
(垂直堆積法)
本形態において、垂直堆積法とは、被堆積体に対して垂直方向に短絡防止材料を堆積させ、短絡防止層を形成する方法である。前記垂直堆積法としては、特に制限されないが、(a)インクジェット法を利用した方法、(b)スプレー法を利用した方法、および(c)原子層堆積(ALD)法が挙げられる。
本形態において、垂直堆積法とは、被堆積体に対して垂直方向に短絡防止材料を堆積させ、短絡防止層を形成する方法である。前記垂直堆積法としては、特に制限されないが、(a)インクジェット法を利用した方法、(b)スプレー法を利用した方法、および(c)原子層堆積(ALD)法が挙げられる。
(a)インクジェット法を利用した方法
インクジェット法を利用した方法は、短絡防止材料前駆体を含む塗布液をインクジェット法により第一電極上に堆積させることで塗膜を形成し、次いで短絡防止材料前駆体を短絡防止材料に変換して短絡防止層を形成する方法である。
インクジェット法を利用した方法は、短絡防止材料前駆体を含む塗布液をインクジェット法により第一電極上に堆積させることで塗膜を形成し、次いで短絡防止材料前駆体を短絡防止材料に変換して短絡防止層を形成する方法である。
短絡防止材料前駆体を含む塗布液
短絡防止材料前駆体を含む塗布液は、短絡防止材料前駆体および溶媒を含む。さらに公知の添加剤を含んでいてもよい。
短絡防止材料前駆体を含む塗布液は、短絡防止材料前駆体および溶媒を含む。さらに公知の添加剤を含んでいてもよい。
前記短絡防止材料前駆体としては、特に制限されないが、酸化チタン前駆体を用いることが好ましい。当該酸化チタン前駆体としては、加水分解により酸化チタンを生じるものであることが好ましく、具体例としては、三塩化チタン、四塩化チタン等のハロゲン化チタン;オルトチタン酸メチル、オルトチタン酸エチル、オルトチタン酸イソプロピル、オルトチタン酸ブチル等のオルトチタン酸エステル;チタンブトキシドダイマー;チタンテトライソプロポキシド、チタンテトラノルマルブトキシド、チタンブトキシドダイマー等のチタンアルコキシド;チタニウムステアレート、ジイソプロポキシチタンジステアレート、トリ-n-ブトキシチタンモノステアレート、ポリヒドロキシチタンステアレート等のチタンアシレートが挙げられる。これらのうち、チタンアルコキシド、オルトチタン酸エステルを用いることが好ましい。
前記酸化チタン前駆体は、例えば、アセチルアセトン、アミノエタノール、ジエタノールアミン、トリエタノールアミン、エチレンジアミン、その他のアミン、ピリジンカルボン酸、酒石酸、シュウ酸、乳酸、グリコール酸、その他のヒドロキシカルボン酸等の配位子と混合し、酸化チタン前駆体の錯体を形成したものであってもよい。当該錯体としては、チタンジイソプロポキシビス(アセチルアセトネート)、チタンテトラアセチルアセトネート、チタンジオクチロキシビス(オクチレングリコレート)、チタンジイソプロポキシビス(エチルアセトアセテート)、チタンジイソプロポキシビス(トリエタノールアミネート)、チタンラクテートアンモニウム塩、チタンラクテート、プロパンジオキシチタンビス(エチルアセトアセテート)等が挙げられる。
前記溶媒としては、特に制限されないが、クロロホルム、ジクロロメタン等のハロゲン系有機溶剤;ブタン、ペンタン、ヘキサン等の脂肪族炭化水素;ベンゼン、トルエン、キシレン、クロロベンゼン等の芳香族炭化水素;酢酸エチル、酢酸ブチル等のエステル類;メチルイソブチルケトン等の非水溶性ケトン類;テトラヒドロフラン(THF)、ブチルエーテル、ジオキサン等のエーテル類;メタノール、エタノール、n-プロパノール、イソプロピルアルコールなどの脂肪族アルコール類;アセトニトリル;ジメチルホルムアミド(DMF);ジメチルスルホキシド(DMSO);二硫化炭素;水等が挙げられる。これらの溶媒は、単独で用いても、2種以上を混合して用いてもよい。
塗布液中の短絡防止材料前駆体の含有量としては、特に制限されないが、塗布液中の溶媒100質量部に対して、0.1~65質量部であることが好ましく、0.5~13質量部であることがより好ましい。
インクジェット法
インクジェット法とは、短絡防止材料前駆体を含む塗布液を、第一電極に液滴単位で吹き付けることで塗膜を形成する方法である。インクジェット法を用いることにより、塗膜は垂直に堆積される。
インクジェット法とは、短絡防止材料前駆体を含む塗布液を、第一電極に液滴単位で吹き付けることで塗膜を形成する方法である。インクジェット法を用いることにより、塗膜は垂直に堆積される。
インクジェット法において、インクジェットヘッドは圧電素子方式であることが好ましい。また、吐出量としては、1滴あたり4~42ピコリットルであることが好ましく、8~30ピコリットルであることがより好ましい。
塗布液の吐出回数は、特に制限されず、所望の短絡防止層の性能等に応じて適宜調節されうる。
変換方法
インクジェット法により形成された塗膜中の短絡防止材料前駆体は、短絡防止材料に変換される。この際、塗膜中の溶媒の一部または全部が揮発する。これにより、短絡防止層が形成される。
インクジェット法により形成された塗膜中の短絡防止材料前駆体は、短絡防止材料に変換される。この際、塗膜中の溶媒の一部または全部が揮発する。これにより、短絡防止層が形成される。
短絡防止材料前駆体の変換方法としては、特に制限されず、公知の方法を使用することができる。なかでも、焼成であることが好ましい。例えば、短絡防止材料前駆体が有機チタン化合物である場合には、焼成を行うと、下記式で表される反応により有機チタン化合物が酸化チタンのポリマー(短絡防止材料)に変換される。

焼成温度としては、特に制限されないが、好ましくは200℃以上、より好ましくは250℃以上、さらにより好ましくは300℃以上、特に好ましくは400℃以上である。また、焼成温度の下限もまた特に制限されないが、好ましくは700℃以下、より好ましくは650℃以下、さらにより好ましくは600℃以下である。すなわち、焼成温度は、200~700℃であることが好ましく、250~650℃であることがより好ましく、300~600℃であることがさらに好ましく、400~600℃であることが特に好ましい。焼成温度が200℃以上であると、上記式において有機物(R)の残存率を低減することができ、より緻密な短絡防止層が形成できることから好ましい。一方、焼成温度が700℃以下であると、短絡防止層の過剰な結晶化を防止できることから好ましい。
焼成時間としては、特に制限されないが、1分~5時間であることが好ましく、5分~3時間であることがより好ましく、5分~120分であることがさらに好ましく、5~30分であることが特に好ましい。
焼成を上記温度および/または時間に設定することにより、好適な結晶性を有する短絡防止層を得ることができ、内部抵抗が低い短絡防止層を得ることができる。また、第一電極と短絡防止層との熱膨張係数の差に起因する第一電極および短絡防止層の界面の隙間の発生を防止することができ、第一電極と短絡防止層とを好適に密着させることができる。
焼成温度および焼成時間を適宜変更することにより、短絡防止層の緻密の度合いや結晶性を調節することができる。
なお、導電性向上の観点から、インクジェット法による塗膜の形成後、直ちに焼成を行うことが好ましい。すなわち、工程(1)が、塗布液をインクジェット法により塗布して塗膜を形成する工程と、得られた塗膜を焼成する工程と、を含むことが好ましい。
短絡防止材料前駆体が酸化チタン前駆体である場合には、短絡防止層が酸化チタンを含むことが好ましい。この際、「短絡防止層が酸化チタンを含む」とは、-Ti-O-結合を有していればよいことを意味する。すなわち、酸化チタンの分子を含む場合に限られず、酸化チタンのポリマーを含む場合を含む。また、前記短絡防止層は、未反応の酸化チタン前駆体等の有機物や-Ti-O-R結合を有する酸化チタンポリマー含んでいてもよい。
インクジェット法は、上述したように、第一電極に塗布液を直接吹き付けることにより塗膜を形成できることから、膜形成の自由度が非常に高く、かつ、均一な膜を形成することができる。また、ピコリットルオーダーで塗布液を噴射できることから、高精度で膜を形成することができ、薄膜化も容易である。さらに、大気圧下で行うことができることから、製造コストも低い。
(b)スプレー法を利用した方法
スプレー法を利用した方法では、短絡防止材料前駆体を含む塗布液をスプレー法により第一電極上に堆積させることで塗膜を形成し、次いで短絡防止材料前駆体を短絡防止材料に変換して短絡防止層を形成する方法である。
スプレー法を利用した方法では、短絡防止材料前駆体を含む塗布液をスプレー法により第一電極上に堆積させることで塗膜を形成し、次いで短絡防止材料前駆体を短絡防止材料に変換して短絡防止層を形成する方法である。
短絡防止材料前駆体を含む塗布液
上記(a)で記載したものと同様のものが用いられうることから、ここでは説明を省略する。
上記(a)で記載したものと同様のものが用いられうることから、ここでは説明を省略する。
スプレー法
スプレー法とは、短絡防止材料前駆体を溶媒に溶解させて調製した塗布液を、第一電極に液滴を噴霧させることで塗膜を形成する方法である。スプレー法を用いることにより、塗膜は垂直に堆積される。
スプレー法とは、短絡防止材料前駆体を溶媒に溶解させて調製した塗布液を、第一電極に液滴を噴霧させることで塗膜を形成する方法である。スプレー法を用いることにより、塗膜は垂直に堆積される。
スプレー法において、噴霧する際の液滴の速度としては、特に制限されないが、0.0001~10m/sであることが好ましく、0.001~1m/sであることがより好ましい。
また、液滴を噴霧する量(吐出量)は、特に制限されないが、3~20mL/分であることが好ましく、5~15mL/分であることがより好ましい。液滴を噴霧する圧力(吐出圧)は、特に制限されないが、10~0.0001MPaであることが好ましく、1~0.001MPaであることがより好ましい。
塗布液の噴霧時間または回数は、特に制限されず、所望の短絡防止層の性能等に応じて適宜調節されうる。
変換方法
上記(a)で記載した方法と同様の方法が適用されうることから、ここでは説明を省略する。
上記(a)で記載した方法と同様の方法が適用されうることから、ここでは説明を省略する。
(c)原子層堆積(ALD)法
ALD法とは、第一電極上に短絡防止材料前駆体のガス、および酸化性ガスを交互に導入することにより、短絡防止層を1層ごとに堆積させる方法である。より詳細には、はじめに短絡防止材料前駆体のガスを第一電極上に導入し、ガス分子層(単原子層)を形成させる。次いで不活性ガスを導入することにより、短絡防止材料前駆体のガスをパージ(除去)する。この際、形成された短絡防止材料前駆体のガスのガス分子層は、第一電極と化学吸着していることから、不活性ガスを導入してもパージされることはない。次に、酸化性ガスを導入して、形成されたガス分子層を構成する短絡防止材料前駆体を短絡防止材料に変換して短絡防止層が形成される。最後に、不活性ガスを導入することにより、酸化性ガスをパージし、ALD法の1サイクルが完了する。上記サイクルを繰り返すことにより、原子層が1層ずつ堆積されて、所定の膜厚を有する短絡防止層を形成することができる。
ALD法とは、第一電極上に短絡防止材料前駆体のガス、および酸化性ガスを交互に導入することにより、短絡防止層を1層ごとに堆積させる方法である。より詳細には、はじめに短絡防止材料前駆体のガスを第一電極上に導入し、ガス分子層(単原子層)を形成させる。次いで不活性ガスを導入することにより、短絡防止材料前駆体のガスをパージ(除去)する。この際、形成された短絡防止材料前駆体のガスのガス分子層は、第一電極と化学吸着していることから、不活性ガスを導入してもパージされることはない。次に、酸化性ガスを導入して、形成されたガス分子層を構成する短絡防止材料前駆体を短絡防止材料に変換して短絡防止層が形成される。最後に、不活性ガスを導入することにより、酸化性ガスをパージし、ALD法の1サイクルが完了する。上記サイクルを繰り返すことにより、原子層が1層ずつ堆積されて、所定の膜厚を有する短絡防止層を形成することができる。
短絡防止材料前駆体としては、特に制限されないが、上記(a)で記載した酸化チタン前駆体のうち、気化するものであることが好ましい。具体的には、チタン(IV)テトライソプロポキシド(Ti(OiPr)4)、テトラキス(ジメチルアミノ)チタン(IV)(Ti(NMe2)4)等が挙げられる。
また、前記酸化性ガスとしては、短絡防止材料前駆体を短絡防止材料に変換できるものであれば特に制限はなく、例えば、オゾン(O3)、水蒸気(H2O)、過酸化水素(H2O2)、メタノール(CH3OH)、およびエタノール(C2H5OH)等が用いられうる。
さらに、前記不活性ガスとしては、希ガス(ヘリウム、ネオン、アルゴン、クリプトン、キセノン)、窒素ガス等が用いられうる。
短絡防止材料前駆体のガスの導入時間は、0.05~10秒であることが好ましく、0.1~3秒であることがより好ましく、0.5~2秒であることがさらに好ましい。短絡防止材料前駆体のガスの導入時間が0.05秒以上であると、ガス分子層を形成できる時間が十分に確保できることから好ましい。一方、短絡防止材料前駆体のガスの導入時間が10秒以下であると、1サイクルに要する時間が低減できることから好ましい。
また、短絡防止材料前駆体のガスをパージするための不活性ガスの導入時間は、0.05~10秒であることが好ましく、0.5~6秒であることがより好ましく、1~4秒であることがさらに好ましい。不活性ガスの導入時間が0.05秒以上であると、短絡防止材料前駆体のガスを十分にパージできることから好ましい。一方、不活性ガスの導入時間が10秒以下であると、1サイクルに要する時間を低減でき、形成されたガス分子層への影響が少なくなることから好ましい。
さらに酸化性ガスの導入時間は、0.05~10秒であることが好ましく、0.1~3秒であることがより好ましく、0.5~2秒であることがさらに好ましい。酸化性ガスの導入時間が0.05秒以上であると、ガス分子層を酸化できる時間が十分に確保できることから好ましい。一方、酸化性ガスの導入時間が10秒以下であると、1サイクルに要する時間が低減でき、副反応が防止されうることから好ましい。
また、酸化性ガスをパージするための不活性ガスの導入時間は、0.05~10秒であることが好ましく、0.5~6秒であることがより好ましく、1~4秒であることがさらに好ましい。不活性ガスの導入時間が0.05秒以上であると、酸化性ガスを十分にパージできることから好ましい。一方、不活性ガスの導入時間が10秒以下であると、1サイクルに要する時間が低減でき、形成された原子層への影響が少ないことから好ましい。
ALD法において、短絡防止層を成膜する際の成膜温度は、350℃以下であることが好ましく、250℃以下であることがより好ましく、150℃未満であることがさらに好ましく、120℃未満であることが特に好ましい。成膜温度が350℃以下であると、第一電極を保持する基板に求められる耐熱性が小さくなり、製造コストを低減できることから好ましい。
上述のように従来の短絡防止層の形成方法のなかには、表面に凹凸を有する第一電極を十分に被覆できない場合がある。
例えば、スピンコート法では、横方向に応力をかけながら製膜するものであることから、第一電極表面の凹部には膜が形成されるものの、凸部には十分に膜が形成されず、結果として第一電極が短絡防止層から突出してしまう場合がある。図2Aは、従来の光電変換素子の一部の層内構成の例を模式的に示す概略断面図である。図2Aに示すように、従来の光電変換素子10’は、その一部に基板1、第一電極2、短絡防止層3、光電変換層6、正孔輸送層7、および第二電極8が順次積層されてなる構成を有する。光電変換層6は、半導体5および前記半導体5に担持された増感色素4を含む。光電変換層6上に正孔輸送層7が配置されるが、前記正孔輸送層7を構成する材料11(導電性高分子等)は、半導体5の多孔の隙間を埋めるように充填され、光電変換層6の構成要素となっている(ただし、本明細書中において、正孔輸送層を構成する材料11を、正孔輸送層7の構成要素と解して説明する場合がある)。短絡防止層3によって十分に第一電極2が被覆されていない場合には、第一電極2が短絡防止層から突出し、光電変換層中の正孔輸送層を構成する材料11が第一電極2と短絡防止層3との間で直接接触が起こりうる。そして、両者が接触することで暗電流や逆電子移動が著しく増加することにより、光電変換効率が低下することとなる。
この際、前記暗電流は、光電変換素子に照射される光の強度に依存しない。そのため、光が弱く、短絡電流が少ない微小光量での発電時には特に影響が大きい。例えば、短絡電流が100mA/cm2である場合、暗電流が500μA/cm2であると、低下要因としては0.5%程度に過ぎないが、短絡電流が10mA/cm2である場合に暗電流が500μA/cm2であると、低下要因としては5%の割合となり、影響がより大きくなる。
一方、前記逆電子移動は、光電変換層から正孔輸送層への電子移動を意味し、短絡電流の低下要因の一つである。
前記問題に対し、短絡防止層の膜厚を増加させることで、完全に第一電極を被覆することもできる。しかしながら、短絡防止層の厚さが第一電極に対して過剰となる部分が生じうる。その結果、当該過剰となった部分において接着性が低減するため、ひび割れや気泡の発生が生じる場合があり、部分的に短絡防止層が高抵抗化し、光電変換効率が低下する可能性がある。
そして、上述の短絡防止層の被覆についての問題は、一般に用いられる第一電極の多くが表面に凹凸(表面粗さ)を有していることから、問題となりうる。
しかしながら、短絡防止層を垂直堆積法により形成することで、上記問題が解決されうる。図2Bは、本発明の光電変換素子の一部の層内構成の例を模式的に示す概略断面図である。図2Bに示すように、本発明に係る光電変換素子10は、短絡防止層3が、第一電極2の凹凸構造に追随する形で第一電極2を被覆しているため、第一電極2と光電変換層6中の正孔輸送層を構成する材料11とが接触しない。その結果、逆電子移動や暗電流を低減できることから、高い光電変換効率を有する光電変換素子を得ることができる。さらに、上記垂直堆積法を用いることにより、短絡防止層を薄膜化することも可能である。
(短絡防止層)
短絡防止層の構成材料(短絡防止材料)としては、特に制限されないが、短絡防止材料前駆体から変換される材料でありうる。具体的には、亜鉛、ニオブ、スズ、チタン、バナジウム、インジウム、タングステン、タンタル、ジルコニウム、モリブデン、マンガン、鉄、銅、ニッケル、イリジウム、ロジウム、クロム、ルテニウム等の金属またはこれらの酸化物もしくはそのポリマー;チタン酸ストロンチウム、チタン酸カルシウム、チタン酸バリウム、チタン酸マグネシウム、ニオブ酸ストロンチウム等のペロブスカイトまたはこれらの複合酸化物もしくは酸化物混合物;CdS、CdSe、TiC、Si3N4、SiC、BN等の金属化合物が挙げられる。これらのうち、光電変換層の半導体材料と同等の電気伝導性を有するものであることが好ましく、酸化チタンまたはそのポリマーであることがより好ましい。前記酸化チタンとしては、アナターゼ型酸化チタンおよび誘電率が比較的高いルチル型の酸化チタンのいずれであってもよく、両者を組み合わせて用いてもよい。
短絡防止層の構成材料(短絡防止材料)としては、特に制限されないが、短絡防止材料前駆体から変換される材料でありうる。具体的には、亜鉛、ニオブ、スズ、チタン、バナジウム、インジウム、タングステン、タンタル、ジルコニウム、モリブデン、マンガン、鉄、銅、ニッケル、イリジウム、ロジウム、クロム、ルテニウム等の金属またはこれらの酸化物もしくはそのポリマー;チタン酸ストロンチウム、チタン酸カルシウム、チタン酸バリウム、チタン酸マグネシウム、ニオブ酸ストロンチウム等のペロブスカイトまたはこれらの複合酸化物もしくは酸化物混合物;CdS、CdSe、TiC、Si3N4、SiC、BN等の金属化合物が挙げられる。これらのうち、光電変換層の半導体材料と同等の電気伝導性を有するものであることが好ましく、酸化チタンまたはそのポリマーであることがより好ましい。前記酸化チタンとしては、アナターゼ型酸化チタンおよび誘電率が比較的高いルチル型の酸化チタンのいずれであってもよく、両者を組み合わせて用いてもよい。
これらの材料は単独で用いても、2種以上を組み合わせて用いてもよい。
なお、上述のように、短絡防止層は、未反応の酸化チタン前駆体等の有機物や-Ti-O-R結合を有する酸化チタンポリマー含んでいてもよい。
短絡防止層は、後述する光電変換層中の半導体層とともに、多孔質であることが好ましい。この場合、短絡防止層の空孔率をC[体積%]とし、半導体層の空孔率をD[体積%]としたとき、D/C値が1.1以上であることが好ましく、5以上であることがより好ましく、10以上であることがさらに好ましい。当該D/C値を上記の値とするために、短絡防止層の空孔率Cは、20体積%以下であることが好ましく、5体積%以下であることがより好ましく、2体積%以下であることがさらに好ましい。すなわち、短絡防止層は、緻密層(緻密な多孔質状)であることが好ましい。これにより、短絡防止層が短絡防止効果を有効に発揮することができる。ここで、短絡防止層の空孔率Cの下限は、可能な限り小さいことが好ましいため、特に規定する必要はないが、通常、0.05体積%以上程度である。
短絡防止層の平均厚さ(膜厚)としては、短絡防止効果を発揮することができる膜厚であれば特に制限はない。具体的には、10~400nmであることが好ましく、10~200nmであることがより好ましく、20~120nmであることが特に好ましい。前記短絡防止層の平均厚さが10nm以上であると、第一電極と正孔輸送層との短絡を十分に防止できることから好ましい。一方、短絡防止層の厚さが400nm以下であると、短絡防止層の抵抗値が適度な値となり高い変換効率が得られることから好ましい。なお、短絡防止層が第一電極の凹凸に沿って形成されている場合には、第一電極表面に対して垂直方向に測定した厚さを短絡防止層の膜厚とする。
正孔輸送層にp型半導体を使用し、短絡防止層に金属を使用する場合には、当該短絡防止層には、正孔輸送層よりも仕事関数の値が小さく、ショットキー型の接触をするものを用いることが好ましい。また、短絡防止層に金属酸化物を用いる場合には、当該短絡防止層には、透明導電層とオーミックに接触し、かつ、伝導帯のエネルギー準位が半導体層よりも低いところにあるものを使用することが好ましい。用いる酸化物を選択することにより、多孔質半導体層(光電変換層)から短絡防止層への電子移動効率を向上させることも可能である。
[光電変換層を形成する工程]
光電変換層は、光起電力効果を利用して光エネルギーを電気エネルギーに変換する機能を有する。本発明において、光電変換層は半導体および増感色素を含む。より詳しくは、当該光電変換層は、半導体を含有する半導体層に増感色素が担持された構成を有する。
光電変換層は、光起電力効果を利用して光エネルギーを電気エネルギーに変換する機能を有する。本発明において、光電変換層は半導体および増感色素を含む。より詳しくは、当該光電変換層は、半導体を含有する半導体層に増感色素が担持された構成を有する。
光電変換層の形成方法は、一般に、(1)短絡防止層上への半導体層の形成、および(2)半導体の増感処理に大別される。
(1)において、半導体の材料が粒子状の場合には、半導体の分散液またはコロイド溶液(半導体含有塗布液)を短絡防止層に塗布あるいは吹き付ける方法、および半導体微粒子の前駆体を短絡防止層上に塗布し、水分(例えば、空気中の水分)によって加水分解後に縮合を行う方法(ゾル-ゲル法)等によって半導体層を形成することができる。上記方法によって得られた半導体層は焼成することが好ましい。また、半導体の材料が膜状であり、短絡防止層上に保持されていない場合には、半導体を短絡防止層上に貼合することによって半導体層を形成することができる。(2)の増感処理方法は、増感色素の半導体層への吸着等が挙げられる。(1)において、半導体層を焼成する場合には、焼成後、半導体に水分が吸着する前に素早く増感色素による増感処理を行うことが好ましい。
以下、本発明に好ましく用いられる光電変換層の作製方法について詳細に説明する。
(1)短絡防止層上への半導体層の形成
(1-1)半導体含有塗布液の調製
まず、半導体、好ましくは半導体の微粉末を含む塗布液(半導体含有塗布液)を調製する。当該半導体微粉末はその一次粒子径が微細であることが好ましい。半導体含有塗布液は、半導体微粉末を溶媒中に分散させることによって調製することができ、溶媒中に分散された半導体微粉末は一次粒子状で分散する。
(1-1)半導体含有塗布液の調製
まず、半導体、好ましくは半導体の微粉末を含む塗布液(半導体含有塗布液)を調製する。当該半導体微粉末はその一次粒子径が微細であることが好ましい。半導体含有塗布液は、半導体微粉末を溶媒中に分散させることによって調製することができ、溶媒中に分散された半導体微粉末は一次粒子状で分散する。
用いられる半導体の材料としては、特に制限されないが、シリコン、ゲルマニウムのような単体、周期表(元素周期表ともいう)の第3族~第5族、第13族~第15族系の元素を有する化合物、金属のカルコゲニド(例えば、酸化物、硫化物、セレン化物等)、金属窒化物等が使用されうる。金属のカルコゲニドの具体例としては、チタン、スズ、亜鉛、鉄、タングステン、ジルコニウム、ハフニウム、ストロンチウム、インジウム、セリウム、イットリウム、ランタン、バナジウム、ニオブ、またはタンタルの酸化物;カドミウム、亜鉛、鉛、銀、アンチモンまたはビスマスの硫化物;カドミウムまたは鉛のセレン化物;カドミウムのテルル化物等が挙げられる。また、その他の半導体の材料としては、亜鉛、ガリウム、インジウム、カドミウム等のリン化物;ガリウム-ヒ素または銅-インジウムのセレン化物;銅-インジウムの硫化物;チタンの窒化物等が挙げられる。より詳細には、TiO2、SnO2、Fe2O3、WO3、ZnO、Nb2O5、CdS、ZnS、PbS、Bi2S3、CdSe、CdTe、GaP、InP、GaAs、CuInS2、CuInSe2、Ti3N4等が挙げられる。これらのうち、TiO2、ZnO、SnO2、Fe2O3、WO3、Nb2O5、CdS、またはPbSを用いることが好ましく、TiO2またはNb2O5を用いることがより好ましく、色素の吸着性が優れていることから、TiO2(酸化チタン)を用いることが特に好ましい。これらの材料は単独で用いても、2種以上を組み合わせて用いてもよい。2種以上を組み合わせた形態としては、例えば、酸化チタン半導体に20質量%の窒化チタン(Ti3N4)を混合する形態、J.Chem.Soc.Chem.Commun.,15(1999)に記載の酸化亜鉛/酸化スズの複合の形態等が挙げられる。なお、金属酸化物または金属硫化物に、その他の半導体材料を組み合わせて使用する場合には、当該その他の半導体材料は、金属酸化物または金属硫化物半導体に対する質量比が30%以下であることが好ましい。
加えて、TiO2を半導体層に使用する場合に、TiO2はアナターゼ型酸化チタンおよび/また誘電率が比較的高いルチル型の酸化チタンのいずれであってもよい。
半導体の形状としては、特に制限されず、フィラー状、球状、円錐状、柱状、管状、平板状等の任意の形状を有しうる。また、半導体層として、これらフィラー状、粒子状、円錐状、柱状、管状等の形状の半導体が凝集して形成された膜状のものを使用してもよい。また、この場合、予め色素が表面に被覆した半導体を使用しても、半導体からなる層を形成した後に色素を被覆してもよい。半導体の大きさもまた、特に制限されず、例えば、半導体が球状である場合には、半導体の平均粒径が1~5000nmであることが好ましく、2~500nmであることがより好ましい。なお、上記半導体の「平均粒径」とは、100個以上のサンプルを電子顕微鏡で観察したときの1次粒子直径の平均粒径(一次平均粒径)を意味する。
上記半導体は、有機塩基を用いて表面処理がされていてもよい。表面処理に用いられる有機塩基としては、特に制限はなく、ジアリールアミン、トリアリールアミン、ピリジン、4-tert-ブチルピリジン、ポリビニルピリジン、キノリン、ピペリジン、アミジン等が挙げられる。これらのうち、ピリジン、4-tert-ブチルピリジン、ポリビニルピリジンを用いて表面処理することが好ましい。表面処理方法は、特に制限されず、公知の方法を用いることができ、当該方法は、当業者が必要に応じて適宜変更することができる。例えば、半導体を表面処理方法の一例として、有機塩基を含む溶液(有機塩基溶液)を準備し、半導体を有機塩基溶液に浸漬する方法が例示される。
半導体含有塗布液に用いられうる溶媒としては、半導体微粉末を分散できるものであれば特に制限されず、水、有機溶媒、水と有機溶媒との混合液が用いられうる。前記有機溶媒の具体例としては、例えば、メタノール、エタノール、イソプロピルアルコール等のアルコール;メチルエチルケトン、アセトン、アセチルアセトン等のケトン;ヘキサン、シクロヘキサン等の炭化水素;アセチルセルロース、ニトロセルロース、アセチルブチルセルロース、エチルセルロース、メチルセルロース等のセルロース誘導体等が挙げられる。塗布液中には、必要に応じて、界面活性剤、酸(酢酸、硝酸など)、粘度調節剤(ポリエチレングリコール等の多価アルコール等)、キレート剤(アセチルアセトンなど)を添加してもよい。
半導体含有塗布液には、必要に応じて公知の添加剤が添加されていてもよい。
溶媒中の半導体微粉末の濃度は0.1~70質量%であることが好ましく、0.1~30質量%であることがより好ましい。
(1-2)半導体含有塗布液の塗布
上記(1-1)によって調製した半導体含有塗布液を、短絡防止層上に塗布または吹き付け、乾燥等を行うことにより、半導体層が形成される。
上記(1-1)によって調製した半導体含有塗布液を、短絡防止層上に塗布または吹き付け、乾燥等を行うことにより、半導体層が形成される。
当該塗布は、特に制限されず、ドクターブレード法、スキージ法、スピンコート法、スクリーン印刷法など公知の方法によって行われる。上記塗布または吹き付け、および乾燥によって得られた半導体層は、半導体微粒子の集合体からなるものであり、その微粒子の粒径は使用した半導体微粉末の一次粒子径に対応する。なお、半導体含有塗布液は2種以上の半導体材料を含むものであってもよいし、2種以上の半導体材料を用いて塗布または吹き付けを行い、層状構造の半導体層を形成してもよい。
(1-3)半導体層の焼成処理
上記(1-2)によって形成された半導体層は、空気中または不活性ガス中で焼成することが好ましい。焼成を行うことにより、(1-2)で形成された半導体層と短絡防止層との結合力および半導体微粒子どうしの結合力を高め、機械的強度が向上しうる。焼成条件は、所望の実表面積や空孔率を有する半導体層を形成することができれば特に制限されない。
上記(1-2)によって形成された半導体層は、空気中または不活性ガス中で焼成することが好ましい。焼成を行うことにより、(1-2)で形成された半導体層と短絡防止層との結合力および半導体微粒子どうしの結合力を高め、機械的強度が向上しうる。焼成条件は、所望の実表面積や空孔率を有する半導体層を形成することができれば特に制限されない。
焼成温度は、特に制限されない。焼成処理時、焼成膜の実表面積を適切に調製し、上記の空隙率を有する焼成膜を得る観点から、焼成温度は、900℃以下であることが好ましく、200℃~850℃であることがより好ましく、400℃~850℃であることがさらにより好ましく、450℃~800℃であることが特に好ましい。また、基板がプラスチック等で耐熱性に劣る場合には、加圧により半導体微粒子-短絡防止層間および半導体微粒子どうしを固着させてもよいし、マイクロ波を用いて半導体層のみを焼成してもよい。
焼成時間も特に制限されないが、10秒~12時間であることが好ましく、1~240分であることがより好ましく、10~120分であることが特に好ましい。
また、焼成雰囲気も特に制限されないが、通常、焼成工程は、大気中または不活性ガス(例えば、アルゴン、ヘリウム、窒素など)雰囲気中で行われる。
なお、上記焼成は、単一の温度で1回のみ行ってもよいし、温度や時間を変化させて2回以上繰り返し行ってもよい。
焼成された半導体層の構造は、特に制限されないが、増感色素との吸着を効果的に行う観点から多孔質構造(空隙を有するポーラスな構造)であることが好ましい。よって、半導体層の空孔率(D)は、1~90体積%であることが好ましく、10~80体積%であることがさらに好ましく、20~70体積%であることが特に好ましい。なお、半導体層の空孔率は、誘電体の厚み方向に貫通性のある空孔率を意味し、水銀ポロシメーター(島津ポアサイザー9220型)等の市販の装置を用いて測定することができる。なお、半導体層が多孔質構造膜である場合には、正孔輸送層を構成する材料がこの空隙にも存在するように光電変換素子を製造することが好ましい。
焼成された半導体層の膜厚は、特に制限されないが、10nm以上であることが好ましく、500nm~30μmであることがさらに好ましい。このような範囲であれば、透過性、変換効率などの特性に優れた半導体層となりうる。なお、半導体層は、平均粒径がほぼ同じ半導体微粒子により形成された単層であっても、あるいは平均粒径や種類の異なる半導体微粒子を含む半導体層からなる多層膜(層状構造)であってもよい。
得られた半導体層の見かけ表面積に対する実表面積の比は、半導体微粒子の粒径および比表面積、並びに焼成温度等により制御することができる。また、得られた半導体層は、焼成後、例えば、四塩化チタン水溶液を用いた化学メッキや三塩化チタン水溶液を用いた電気化学的メッキ処理を行うことにより、半導体粒子の表面積および半導体粒子近傍の純度を制御し、色素から半導体粒子への電子注入効率を高めてもよい。
(2)増感色素による半導体の増感処理
増感色素による半導体の増感処理は、例えば、増感色素を適切な溶媒に溶解し、当該溶液中によく乾燥させた半導体層を長時間浸漬することによって行われる。当該増感処理によって、増感色素が半導体に吸着されうる。この際、半導体層が多孔質構造を有する場合には、浸漬前に減圧処理、加熱処理等の前処理を行い、膜中の気泡や空隙中の水分を除去することが好ましい。当該前処理によって、増感色素が半導体層内部にも吸着されうる。なお、増感処理は、増感色素含有溶液への半導体層の浸漬に限定されず、その他の公知の増感処理方法も適宜適用することができる。
増感色素による半導体の増感処理は、例えば、増感色素を適切な溶媒に溶解し、当該溶液中によく乾燥させた半導体層を長時間浸漬することによって行われる。当該増感処理によって、増感色素が半導体に吸着されうる。この際、半導体層が多孔質構造を有する場合には、浸漬前に減圧処理、加熱処理等の前処理を行い、膜中の気泡や空隙中の水分を除去することが好ましい。当該前処理によって、増感色素が半導体層内部にも吸着されうる。なお、増感処理は、増感色素含有溶液への半導体層の浸漬に限定されず、その他の公知の増感処理方法も適宜適用することができる。
用いられうる増感色素としては、特に制限されないが、アリールアミン系色素であることが好ましく、下記一般式(1)で表される化合物であることがより好ましい。
上記一般式(1)中、Arは環式化合物基を表す。また、R1は水素原子、ハロゲン原子、置換もしくは未置換のアルキル基、置換もしくは未置換のシクロアルキル基、置換もしくは未置換のアルコキシ基、置換もしくは未置換のアルケニル基、置換もしくは未置換のシクロアルケニル基、置換もしくは未置換のアルキニル基、置換もしくは未置換のアリール基、アミノ基、シアノ基、または置換もしくは未置換の複素環基を表す。この際、R1は他の基と連結して環構造を形成されていてもよい。A1およびA2は、それぞれ独立して、単結合、2価の飽和もしくは不飽和の炭化水素基、置換もしくは未置換のアルキレン基、置換もしくは未置換のアリーレン基、または置換もしくは未置換の2価の複素環基を表す。Zは有機基を表す。nは1~3の整数であり、この際、nが1のとき、2つのR1は同じものであっても、互いに異なるものであってもよい。また、nが2以上のとき、複数のA1、A2、Zは同じものであっても、互いに異なるものであってもよい。pおよびqは0~6の整数である。pおよび/またはqが2以上のとき、複数のA1、A2は同じものであっても、互いに異なるものであってもよい。
前記Arとしては、特に制限されないが、2価~4価の環式化合物基が挙げられる。当該環式化合物基の具体例としては、例えば、ベンゼン環、ナフタレン環、アントラセン環、チオフェン環、フェニルチオフェン環、ジフェニルチオフェン環、イミダゾール環、オキサゾール環、チアゾール環、ピロール環、フラン環、ベンズイミダゾール環、ベンズオキサゾール環、ローダミン環、ピラゾロン環、イミダゾロン環、ピラン環、ピリジン環、フルオレン環等の芳香族環から導かれるものである。これらの芳香族環を複数組み合わせて用いてもよい。例えば、ビフェニル基、ターフェニル基、フルオレニル基、ビチオフェン基、4-チエニルフェニル基、ジフェニルスチリル基等、さらには、スチルベン、4-フェニルメチレン-2,5-シクロヘキサジエン、トリフェニルエテン(例えば、1,1,2-トリフェニルエテン)、フェニルピリジン(例えば、4-フェニルピリジン)、スチリルチオフェン(例えば、2-スチリルチオフェン)、2-(9H-フルオレン-2-イル)チオフェン、2-フェニルベンゾ[b]チオフェン、フェニルビチオフェン、(1,1-ジフェニル-4-フェニル)-1,3-ブタジエン、1,4-ジフェニル-1,3-ジブタジエン、4-(フェニルメチレン)-2,5-シクロヘキサジエン、フェニルジチエノチオフェン環由来の基等が用いられうる。
これらの芳香族環は置換基を有していてもよく、置換基としては、ハロゲン原子(例えば、フッ素原子、塩素原子、臭素原子等)、C1~C24の直鎖若しくは分岐状のアルキル基(例えば、メチル基、エチル基、tert-ブチル基、イソブチル基、ドデシル基、オクタデシル基、3-エチルペンチル基)、ヒドロキシアルキル基(例えば、ヒドロキシメチル基、ヒドロキシエチル基)、アルコキシアルキル基(例えば、メトキシエチル基等)、炭素鎖長1~18のアルコキシ基(例えば、メトキシ基、エトキシ基、エトキシ基、プロポキシ基、イソプロポキシ基、ブトキシ基、ペンチルオキシ基、ヘキシルオキシ基等)、アリール基(例えば、フェニル基、トリル基等)、アルケニル基(例えば、ビニル基、アリル基等)、アミノ基(例えば、ジメチルアミノ基、ジエチルアミノ基、ジフェニルアミノ基)、複素環基(例えば、モルホニル基、フラニル基等)等が挙げられる。
これらのうち、Arは、上述した芳香族基から水素原子を2個または3個除いた2価または3価の芳香族基であることが好ましい。
好適なArとして、以下の化学式(1-A)~(1-G)が例示される。

前記アルキル基としては、特に制限されないが、C1~C24の直鎖もしくは分岐鎖アルキル基が挙げられる。具体的には、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、n-ペンチル基、イソペンチル基、tert-ペンチル基、ネオペンチル基、1,2-ジメチルプロピル基、n-ヘキシル基、イソヘキシル基、1,3-ジメチルブチル基、1-イソプロピルプロピル基、1,2-ジメチルブチル基、n-ヘプチル基、1,4-ジメチルペンチル基、3-エチルペンチル基、2-メチル-1-イソプロピルプロピル基、1-エチル-3-メチルブチル基、n-オクチル基、2-エチルヘキシル基、3-メチル-1-イソプロピルブチル基、2-メチル-1-イソプロピル基、1-t-ブチル-2-メチルプロピル基、n-ノニル基、3,5,5-トリメチルヘキシル基、n-デシル基、イソデシル基、n-ウンデシル基、1-メチルデシル基、n-ドデシル基、n-トリデシル基、n-テトラデシル基、n-ペンタデシル基、n-ヘキサデシル基、n-ヘプタデシル基、n-オクタデシル基、n-ノナデシル基、n-エイコシル基、n-ヘンエイコシル基、n-ドコシル基、n-トリコシル基、n-テトラコシル基等が挙げられる。これらのうち、メチル基、エチル基、n-プロピル基、n-ブチル基、n-ペンチル基、n-ヘキシル基等のC1~C6の直鎖鎖アルキル基、およびイソプロピル基、t-ブチル基等のC3~C6の分岐鎖アルキル基であることが好ましい。
前記シクロアルキル基としては、特に制限されないが、C3~C9のシクロアルキル基が挙げられる。具体的には、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、シクロヘプチル基、シクロオクチル基、シクロノニル基等が挙げられる。これらのうち、シクロペンチル基、シクロヘキシル基等のC3~C6のシクロアルキル基等が好ましい。
前記アルコキシ基としては、特に制限されないが、C1~C18のアルコキシ基が挙げられる。具体的には、メトキシ基、エトキシ基、プロポキシ基、イソプロポキシ基、ブトキシ基、ペンチルオキシ基、ヘキシルオキシ基、2-エチルヘキシルオキシ基、オクチルオキシ基、ノニルオキシ基、デシルオキシ基、ウンデシルオキシ基、ドデシルオキシ基、トリデシルオキシ基、テトラデシルオキシ基、ペンタデシルオキシ基、ヘキサデシルオキシ基、ヘプタデシルオキシ基、オクタデシルオキシ基等が挙げられる。これらのうち、C6~C18のアルコキシ基であることが好ましく、デシルオキシ基であることがより好ましい。
前記アルケニル基としては、特に制限されないが、C2~C18の直鎖もしくは分岐鎖アルケニル基が挙げられる。具体的には、ビニル基、アリル基、プロペニル基、イソプロペニル基、1-ブテニル基、2-ブテニル基、3-ブテニル基、1-ヘキセニル基、2-ヘキセニル基、3-ヘキセニル基、4-ヘキセニル基、5-ヘキセニル基等が挙げられる。これら以外のアルケニル基が用いられてもよい。
前記シクロアルケニル基としては、特に制限されないが、C3~C18のシクロアルケニル基が挙げられる。具体的には、シクロペンテニル基、シクロヘキセニル基、シクロオクテニル基等が挙げられる。
前記アルキニル基としては、特に制限されないが、C2~C18の直鎖もしくは分岐鎖アルキニル基が挙げられる。具体的には、エチニル基、2-プロピニル基、2-ブチニル基等が挙げられる。
前記アリール基としては、特に制限されないが、C6~C30のアリール基が挙げられる。具体的には、フェニル基、ナフチル基、ビフェニル基、フルオレニル基、アンスリル基、ピレニル基、アズレニル基、アセナフチレニル基、ターフェニル基、フェナンスリル基などが挙げられる。これらのうち、フェニル基、ビフェニル基、ナフチル基であることが好ましい。
前記複素環基としては、特に制限されないが、窒素原子、酸素原子、および硫黄原子からなる選択された少なくとも1つのヘテロ原子を含む複素環基が挙げられる。前記複素環基は、単環式複素環基に限られず、複数の複素環が縮合した縮合複素環基、複素環と炭化水素環(非芳香族性炭化水素環または芳香族炭化水素環)とが縮合(オルソ縮合、オルソアンドペリ縮合など)した縮合複素環基であってもよい。また、当該複素環基は、非芳香族性または芳香族性のいずれであってもよい。さらに、複素環と炭化水素環とが縮合した縮合複素環基においては、複素環または炭化水素環のいずれかが結合手を有していてもよい。具体的には、ピロリル基、イミダゾリル基、ピリジル基、ピラジニル基、インドリル基、キノリル基、イソキノリル基、キナゾリル基、カルバゾリル基、カルボリニル基、フェナントリジニル基、アクリジニル基、フェナジニル基、イソベンゾフラニル、クロメニル基、チエニル基、チアントレニル基、モルホリニル基、イソチアゾリル基、イソオキサゾリル基、フェノキサチイニル基等が挙げられる。これらのうち、ピロリル基、インドリル基、カルバゾリル基であることが好ましい。
前記炭化水素基とは、炭素原子および水素原子からなる基であり、特に制限されないが、上述のシクロアルキル基、アルケニル基、シクロアルケニル基、およびアルキニル基から導かれる2価の基を意味する。前記炭化水素基は、置換基で置換されていてもよい。
前記アルキレン基としては、特に制限されないが、C1~C24の直鎖もしくは分岐鎖アルキレン基が挙げられる。具体的には、メチレン基、エチレン基、プロピレン基、イソブチレン基、sec-ブチレン基、tert-ブチレン基、ペンチレン基、iso-ペンチレン基、へキシレン基等が挙げられる。
前記アリーレン基としては、特に制限されないが、C6~C30のアリーレン基が挙げられる。具体的には、フェニレン基、ビフェニル-ジイル基、ターフェニル-ジイル基、ナフタレン-ジイル基、アントラセン-ジイル基、テトラセン-ジイル基、フルオレン-ジイル基、フェナントレン-ジイル基等が挙げられる。
なお、一般式(1)における「置換または未置換の」における「置換」とは、R1、A1、およびA2として記載された基を構成する水素原子が置換基に置換されている場合をいう。また、「未置換」とは、前記水素原子が置換基に置換されていない場合をいう。前記置換基としては、特に制限されないが、上記で例示したアルキル基、シクロアルキル基、アルコキシ基、アルケニル基、シクロアルケニル基、アルキニル基、アリール基、および複素環基でありうる。
好適なR1として、以下の化学式(2-A)~(2-S)が例示される。

上記化学式(2-A)および(2-G)において、Yは、水素原子、アルキル基、またはアルコキシ基である。また、上記化学式(2-M)~(2-S)において、波線部は他の基と連結する位置を示す。当該破線部が他の基と連結することにより、例えばR1がArとともに縮合環構造を形成する。さらに、上記化学式(2-S)において、hは1~17の整数である。
また、好適なA1およびA2として、以下の化学式(3-A)~(3-b)が例示される。


上記化学式(3-P)、(3-R)、(3-X)、および(3-Z)において、Yは、水素原子、アルキル基、またはアルコキシ基である。また、上記化学式(3-I)において、hは1~17の整数である。
前記Zとしては、酸性基、および/または電子吸引性基もしくは電子吸引性環構造を有するArのいずれかの部分構造を有する有機基が挙げられる。好ましくは少なくとも1つのカルボキシル基を含む有機基である。なお、前記Zは、一般式(1)において、Ar、A1、またはA2、好ましくはArと結合する。
前記酸性基としては、特に制限されないが、カルボキシ基、スルホ基(-SO3H)、スルフィノ基、スルフィニル基、ホスホン酸基(-PO(OH)2)、ホスホリル基、ホスフィニル基、ホスホノ基、チオール基、ヒドロキシ基、ホスホニル基、アルコキシシラン基、およびスルホニル基;ならびにこれらの塩などが挙げられる。これらのうち、酸性基としては、カルボキシ基、スルホ基、ホスホン酸基、ヒドロキシ基であることが好ましく、カルボキシル基であることがより好ましい。
また、前記電子吸引性基としては、特に制限されないが、シアノ基、ニトロ基、フルオロ基、クロロ基、ブロモ基、ヨード基、パーフルオロアルキル基(例えば、トリフルオロメチル基)、アルキルスルホニル基、アリールスルホニル基、パーフルオロアルキルスルホニル基、パーフルオロアリールスルホニル基等が挙げられる。これらのうち、シアノ基、ニトロ基、フルオロ基、クロロ基であることが好ましく、シアノ基、ニトロ基であることがより好ましい。
さらに、電子吸引性環構造としては、特に制限されないが、ローダミン環、ジローダミン環、イミダゾロン環、ピラゾロン環、ピラゾリン環、キノン環、ピラン環、ピラジン環、ピリミジン環、イミダゾール環、インドール環、ベンゾチアゾール環、ベンゾイミダゾール環、ベンゾオキサゾール環、チアジアゾール環等が挙げられる。これらのうち、ローダミン環、ジローダミン環、イミダゾロン環、ピラゾリン環、キノン環、チアジアゾール環であることが好ましく、ローダミン環、ジローダミン環、イミダゾロン環、ピラゾリン環であることがより好ましい。
当該Zは、光電子を効果的に半導体(特に酸化物半導体)に注入することができる。また、部分構造Zにおいて、酸性基と、電子吸引性基または電子吸引性環構造とは、酸素原子(O)、硫黄原子(S)、セレン原子(Se)、またはテルル原子(Te)等の原子を介して結合してもよい。または、部分構造Zは、電荷、特に正の電荷を帯びていてもよく、この際、Cl-、Br-、I-、ClO4
-、NO3-、SO4
2-、H2PO4
-等の対イオンを有していてもよい。
好適なZとして、以下の化学式(4-A)~(4-N)が例示される。

上記化学式(4-H)において、gは1~17の整数である。
本発明に係る好適な増感色素の例を以下に示す。なお、本明細書では、下記増感色素の番号で増感色素を示す。例えば、下記A-1の構造を有する増感色素は、増感色素A-1とも称する。















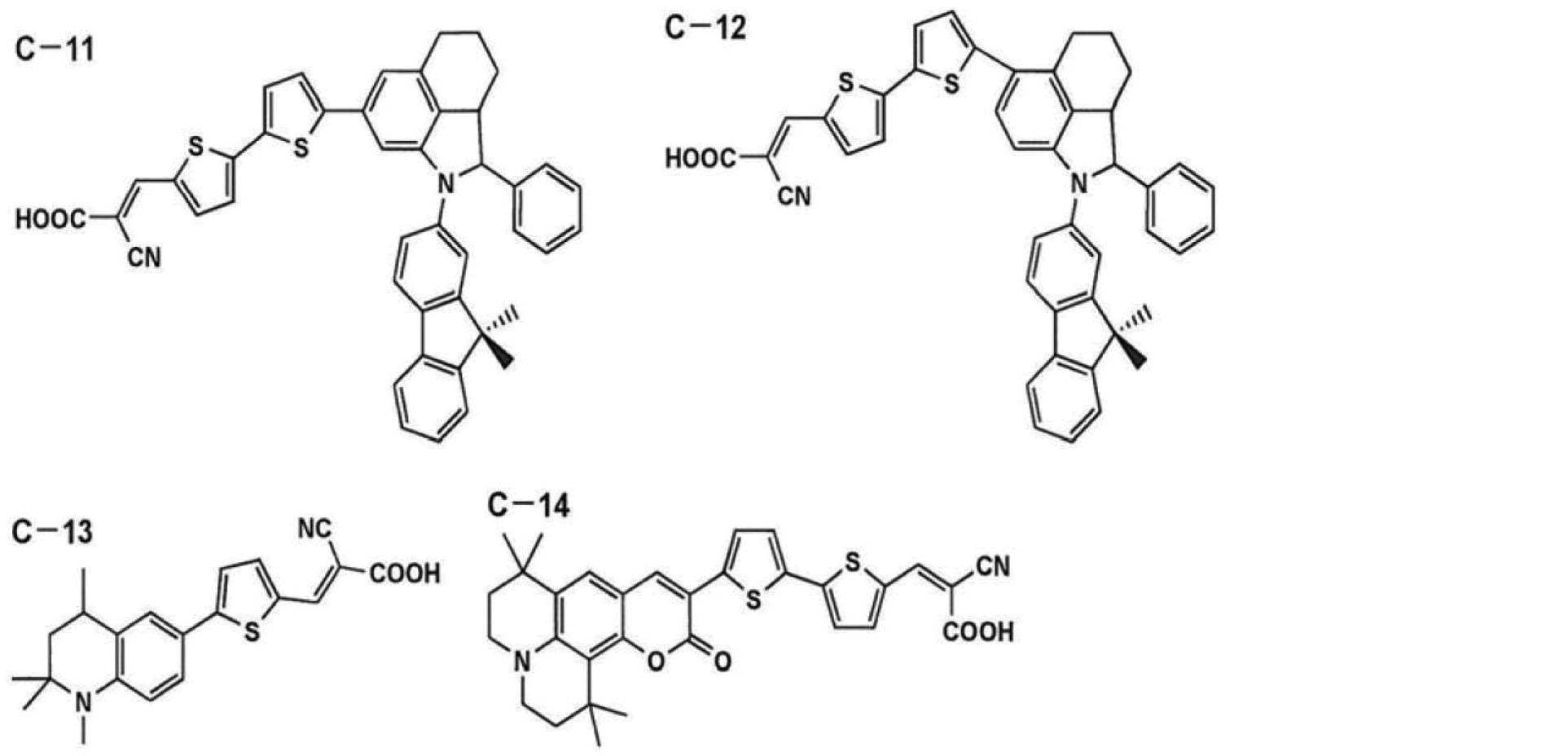

上記増感色素のうち、A群、B群の色素を用いることが好ましい。
また、上記増感色素の他、例えば、米国特許第4,684,537号明細書、同4,927,721号明細書、同5,084,365号明細書、同5,350,644号明細書、同5,463,057号明細書、同5,525,440号明細書、特開平7-249790号公報、特開2000-150007号公報等に記載の化合物を使用してもよい。
増感処理を行う場合、増感色素は単独で用いても、2種以上を混合して用いてもよい。本発明の光電変換素子の用途が後述する太陽電池である場合には、光電変換の波長域をできるだけ広くして太陽光を有効に利用できるように吸収波長の異なる2種以上の増感色素を混合して用いることが好ましい。2種以上の増感色素を用いる場合に、増感処理方法は、特に限定されず、各増感色素の混合溶液に半導体層を浸漬してもよいし、各増感色素を別々の溶液として準備し、順次に半導体層を浸漬してもよい。
増感色素を溶解するのに用いる溶媒は、増感色素を溶解することができ、かつ、半導体を溶解させたり半導体と反応したりすることのないものであれば格別の制限はない。しかしながら、溶媒に溶解している水分および気体が半導体膜に進入して、増感色素の吸着等の増感処理を妨げることを防ぐために、溶媒をあらかじめ脱気および蒸留精製しておくことが好ましい。増感色素の溶解において好ましく用いられる溶媒としては、アセトニトリル等のニトリル系溶媒;メタノール、エタノール、n-プロパノール、イソプロピルアルコール、tert-ブチルアルコール等のアルコール系溶媒;アセトン、メチルエチルケトン等のケトン系溶媒;ジエチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン、1,4-ジオキサン等のエーテル系溶媒;塩化メチレン、1,1,2-トリクロロエタン等のハロゲン化炭化水素溶媒等が挙げられる。これらのうち、アセトニトリル、メタノール、エタノール、n-プロパノール、イソプロピルアルコール、tert-ブチルアルコール、アセトン、メチルエチルケトン、テトラヒドロフランおよび塩化メチレン、並びにこれらの混合溶媒、例えば、アセトニトリル/メタノール混合溶媒、アセトニトリル/エタノール混合溶媒、アセトニトリル/tert-ブチルアルコール混合溶媒を用いることが好ましい。これらの溶媒は、単独で使用しても、2種以上を混合して使用してもよい。
増感処理条件は特に制限はないが、増感色素が半導体層に深く進入して吸着等が充分に進行できるような条件に設定することが好ましい。例えば、溶液中における増感色素の分解および分解物の半導体層への吸着を防止する観点から、増感処理の温度は、5~100℃であることが好ましく、25~80℃であることがより好ましい。また、増感処理の時間は、15分~20時間であることが好ましく、3~24時間であることがより好ましい。特に、室温(25℃)で2~48時間、特に3~24時間、増感処理を行うことが好ましいが、設定する温度によって増感処理の時間は適宜変更してもよい。また、増感処理の時間の短縮および半導体層の深部まで吸着させる観点から、減圧下または真空下で増感処理を行ってもよい。
光電変換層1m2あたりの増感色素の総含有量(総担持量)は、特に制限されないが、0.01~100mmol/m2であることが好ましく、0.1~50mmol/m2であることがより好ましく、0.5~20mmol/m2であることがさらに好ましい。
[正孔輸送層を形成する工程・工程(2)]
本形態において、正孔輸送層は、導電性高分子前駆体を前記光電変換層と接触させ、酸化剤存在下で前記増感色素に光を照射して前記導電性高分子前駆体を重合することにより形成される。
本形態において、正孔輸送層は、導電性高分子前駆体を前記光電変換層と接触させ、酸化剤存在下で前記増感色素に光を照射して前記導電性高分子前駆体を重合することにより形成される。
<前記光電変換層の導電性高分子前駆体への接触>
導電性高分子前駆体を前記光電変換層と接触させる方法としては、特に制限されないが、半導体層が多孔質体でない場合には、導電性高分子前駆体を含む溶液を光電変換層上に塗布、乾燥させる方法であることが好ましい。また、半導体層が多孔質体である場合には、導電性高分子前駆体が増感色素を担持する多孔質内部にまで接触するように、導電性高分子前駆体を含む溶液に光電変換層を含浸および/または塗布することが好ましい。
導電性高分子前駆体を前記光電変換層と接触させる方法としては、特に制限されないが、半導体層が多孔質体でない場合には、導電性高分子前駆体を含む溶液を光電変換層上に塗布、乾燥させる方法であることが好ましい。また、半導体層が多孔質体である場合には、導電性高分子前駆体が増感色素を担持する多孔質内部にまで接触するように、導電性高分子前駆体を含む溶液に光電変換層を含浸および/または塗布することが好ましい。
導電性高分子前駆体を含む溶液は、通常、導電性高分子前駆体、酸化剤、および溶媒を含む。必要に応じてその他の添加剤を含んでもよい。
(導電性高分子前駆体)
用いられうる導電性高分子前駆体としては、特に制限されないが、以下の一般式(2)で表される化合物、以下の一般式(2)に示される繰り返し単位を有する化合物であることが好ましい。これらのうち、導電性高分子前駆体は、以下の一般式(2)に示される繰り返し単位を有する化合物が好ましい。
用いられうる導電性高分子前駆体としては、特に制限されないが、以下の一般式(2)で表される化合物、以下の一般式(2)に示される繰り返し単位を有する化合物であることが好ましい。これらのうち、導電性高分子前駆体は、以下の一般式(2)に示される繰り返し単位を有する化合物が好ましい。

上記一般式(2)中、Xは、S、NR、Oであり、この際、Rは水素原子、または置換もしくは未置換のアルキル基である。R2~R5は、それぞれ独立して、水素原子、ハロゲン原子、置換もしくは未置換のアルキル基、置換もしくは未置換のシクロアルキル基、置換もしくは未置換のアルコキシ基、C2~C30のポリエチレンオシキド基、または置換もしくは未置換のC4~C30の環式化合物含有基である。
上記置換もしくは未置換のアルキル基、置換もしくは未置換のシクロアルキル基、または置換もしくは未置換のアルコキシ基は、上記一般式(1)と同様であることから、ここでは説明を省略する。
上記ハロゲン原子は、特に制限されないが、フッ素原子、塩素原子、臭素原子またはヨウ素原子がある。
前記C2~C30のポリエチレンオシキド基としては、特に制限されないが、式(a)または式(b)で表される基であることが好ましい。
上記式中、xは1~9の整数である。これらのうち、xが3~9のポリエチレンオシキド基であることが好ましく、xが3のポリエチレンオシキド基であることがより好ましい。
前記C4~C30の環式化合物基としては、特に制限されないが、ベンゼン環、ナフタレン環、アントラセン環、チオフェン基、フェニルチオフェン基、ジフェニルチオフェン基、イミダゾール環、オキサゾール環、チアゾール環、ピロール環、フラン環、ベンズイミダゾール環、ベンズオキサゾール環、ローダミン環、ピラゾロン環、イミダゾロン環、ピラン環、ピリジン環、およびフルオレン環等から導かれる基が挙げられる。
また、上記「置換または未置換の」とは、アルキル基、シクロアルキル基、アルコキシ基、環式化合物含有基のうち、少なくとも1個以上の水素原子が他の置換基に置換されていることをいう。上記したアルキル基、上記したアルコキシ基、アルケニル基(例えば、ビニル基、アリル基、プロペニル基、イソプロペニル基、1-ブテニル基、2-ブテニル基、3-ブテニル基、1-ヘキセニル基、2-ヘキセニル基、3-ヘキセニル基、4-ヘキセニル基、5-ヘキセニル基、シクロペンテニル基、シクロヘキセニル基、シクロオクテニル基)、アルキニル基(例えば、エチニル基、2-プロピニル基、2-ブチニル基)、アリール基(例えば、フェニル基、ナフチル基、ビフェニル基、フルオレニル基、アンスリル基、ピレニル基、アズレニル基、アセナフチレニル基、ターフェニル基、フェナンスリル基)、および複素環基(例えば、ピロリル基、イミダゾリル基、ピリジル基、ピラジニル基、インドリル基、キノリル基、イソキノリル基、キナゾリル基、カルバゾリル基、カルボリニル基、フェナントリジニル基、アクリジニル基、フェナジニル基、イソベンゾフラニル、クロメニル基、チエニル基、チアントレニル基、モルホリニル基、イソチアゾリル基、イソオキサゾリル基、フェノキサチイニル基)が炭素数の数を超えない範囲で置換されてもよい。以下同様である。なお、場合によって存在する置換基は、置換する基と同じとなることはない。例えば、Z1~Z4がアルキル基の場合には、さらにアルキル基で置換されることはない。
上記の一般式(2)において、より好ましいR2~R5としては、それぞれ独立して、水素原子、ハロゲン原子、C6~C24のアルキル基、C1~C18のアルコキシ基、フェニル基、ビフェニル基、C1~C8のアルキル基に置換されたフェニル基、C1~C8のアルキル基に置換されたビフェニル基、チオフェン基、ビチオフェン基、C1~C8のアルキル基に置換されたチオフェン基、C1~C8のアルキル基に置換されたビチオフェン基、C1~C8のアルコキシ基に置換されたチオフェン基、C1~C8のアルコキシ基に置換されたビチオフェン基が挙げられる。また、さらに好ましいR2~R5としては、それぞれ独立して、水素原子、塩素原子、メチル基、エチル基、ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、ヘキシル基、デシル基、テトラデシル基、オクタデシル基、テトラコシル基、1-メチルデシル基、8-メチルノニル基、シクロプロピル基、シクロオクチル基、メトキシ基、デシルオキシ基、オクタデシルオキシ基、オクタメチレングリコールエチルエーテル基、フェニル基、p-ヘキシルフェニル基、ビフェニル基、4’-ヘキシルビフェニル基、5-ヘキシルオキシ-2-チエニル基、5’-オクチル-2,2’-ビチエニル基等が挙げられる。
前記導電性高分子前駆体としては、あらかじめ上記一般式(2)で表される化合物を、単独または2種以上のモノマーと共に重合したプレポリマー(二量体以上の多量体やいわゆるオリゴマーを含む)を用いてもよい。ただし、後述する光化学重合において、多孔質体の半導体内部に侵入しやすく、重合した導電性高分子が増感色素を覆う量が多くなる観点から、モノマーを用いることがより好ましい。
(酸化剤)
酸化剤としては、導電性高分子前駆体を重合できるものであれば特に限定されない。酸化剤の準位が励起した色素の順位よりも高ければ電子を奪うことが可能である。一方、酸化剤の準位が高すぎると、導電性高分子前駆体(例えば、bis-EDOT)を直接酸化重合してしまい、色素近傍に均一な膜を形成することが困難になる可能性がある。このため、適度な標準電極電位を有する酸化剤で重合することが好ましい。
酸化剤としては、導電性高分子前駆体を重合できるものであれば特に限定されない。酸化剤の準位が励起した色素の順位よりも高ければ電子を奪うことが可能である。一方、酸化剤の準位が高すぎると、導電性高分子前駆体(例えば、bis-EDOT)を直接酸化重合してしまい、色素近傍に均一な膜を形成することが困難になる可能性がある。このため、適度な標準電極電位を有する酸化剤で重合することが好ましい。
上記点を考慮すると、本発明に係る酸化剤は、-1.5~+2.5Vの標準電極電位(E0
(OX))(V)を有することが好ましく、-0.5~+2.0Vの標準電極電位(E0
(OX))(V)を有することがより好ましい。ここで、酸化剤の標準電極電位が上限以上であれば、重合をより効率的に進行させることができる。また、酸化剤の標準電極電位が下限以下であれば、反応(反応速度)の制御が容易であり、生産性に優れ、産業上好ましい。すなわち、このような標準電極電位(E0
(OX))(V)を有する酸化剤は、光照射時に色素で励起された電子をより効率よく消費できるため、導電性高分子前駆体の重合をより促進でき、また、色素近傍により均一な膜を形成することができる。本明細書において、「標準電極電位(E0
(OX))(V)」は、水溶液中における標準電極電位(25℃)を意味する。
本発明の正孔輸送層を形成させるために用いられる酸化剤としては、具体的には、過酸化水素(+1.763V)、金属塩、過酸化物、オゾン、酸素(+1.229V)、メタノール(+0.588V)などが挙げられる。なお、括弧内は、標準電極電位(E0
(OX))(V)を示す。中でも、材料自体の安定性の観点から、酸化剤が、過酸化水素、酸素、オゾン、金属塩、および過酸化物の少なくとも1種であることが好ましい。
ここで、酸化剤は、光照射(自身が還元されること)により気体化合物または液体化合物となるような化合物であることがより好ましい。このように酸化剤が重合反応後に気体または液体になることによって、重合膜である正孔輸送層中に酸化剤が残らないため、得られる光電変換素子の耐久性をさらに向上できる。上記点を考慮すると、酸化剤は、過酸化水素、酸素、オゾンでありうる。なお、本明細書において、「気体化合物」とは、20℃、1atmの条件下で気体状である化合物を意味する。また、「液体化合物」とは、20℃、1atmの条件下で液体状である化合物を意味する。
酸化剤存在下で色素に光を照射すると、当該色素において励起された電子が酸化剤(例えば、過酸化水素/過酸化水素水など)により消費され、カチオン状態の色素がモノマーである導電性高分子前駆体の電子を引き抜き重合が開始されると考えられる。
上記過酸化物としては、過マンガン酸又はその塩、クロム酸又はその塩、ペルオキソ酸又はその塩、酸素酸又はその塩、硝酸類、硫酸類等が挙げられ、具体的には、過酸化ナトリウム、過酸化バリウム、過マンガン酸カリウム、過マンガン酸ナトリウム、クロム酸金属塩、重クロム酸金属塩、ペルオキソ二硫酸、ペルオキソ二硫酸アンモニウム、ペルオキソ二硫酸金属塩、ペルオキソリン酸、ペルオキソ硫酸、ペルオキソホウ酸ナトリウム、次亜塩素酸、次亜臭素酸、次亜ヨウ素酸、塩素酸、臭素酸、ヨウ素酸、次亜塩素酸ナトリウム、次亜塩素酸カルシウム等の無機過酸化物;クメンヒドロペルオキシド、ギ酸、過ギ酸、過酢酸、過安息香酸、過フタル酸、t-ブチルヒドロペルオキシド、1,1,3,3-テトラメチルブチルヒドロペルオキシド、ジイソプロピルベンゼンヒドロペルオキシド、p-メンタンヒドロペルオキシド、ジ-t-ブチルペルオキシド、t-ブチルクミルペルオキシド、2,5-ジメチル-2,5-ジ(t-ブチルペルオキシ)ヘキサン、ジ-t-ヘキシルペルオキシド、ジクミルペルオキシド、ジ(2-t-ブチルペルオキシイソプロピル)ベンゼン、n-ブチル-4,4-ジ-(t-ブチルペルオキシ)バレレート、t-ブチルペルオキシベンゾエート、2,2-ジ(t-ブチルペルオキシ)ブタン、t-ブチルペルオキシアセテート、2,5-ジ-メチル-2,5-ジ(ベンゾイルペルオキシ)ヘキサン、t-ヘキシルペルオキシベンゾエート、t-ブチルペルオキシ 2-エチルヘキシルモノカルボネート、t-ブチルペルオキシ イソプロピルモノカルボネート、t-ブチルペルオキシラウレート、t-ブチルペルオキシ-3,5,5,-トリメチルヘキサノエート、t-ブチルペルオキシマレイン酸、t-ヘキシルペルオキシイソプロピルモノカルボネート、2,2-ジ(4,4-ジ-(t-ブチルペルオキシ)シクロヘキシル)プロパン、1,1-ジ(t-ブチルペルオキシ)シクロヘキサン、1,1-ジ(t-ヘキシルペルオキシ)シクロヘキサン、ジイソブチリルペルオキシド、クミルペルオキシネオデカノエート、ジ-n-プロピルペルオキシジカルボネート、ジイソプロピルペルオキシジカルボネート、ジ-sec-ブチルペルオキシジカルボネート、1,1,3,3-テトラメチルブチルペルオキシネオデカノエート、ジ(4-tert-ブチルシクロヘキシル)ペルオキシジカルボネート、ジ(2-エチルヘキシル)ペルオキシジカルボネート、t-ヘキシルペルオキシネオデカノエート、t-ブチルペルオキシネオデカノエート、t-ブチルペルオキシネオヘプタノエート、t-ヘキシルペルオキシピバレート、t-ブチルペルオキシピバレート、ジ(3,5,5-トリメチルヘキサノイル)ペルオキシド、ジラウロイルペルオキシド、1,1,3,3-テトラメチルブチルペルオキシ-2-エチルヘキサノエート、二コハク酸ペルオキシド、2,5-ジメチル-2,5-ジ(エチルヘキサノイルペルオキシ)ヘキサン、t-ヘキシルペルオキシ-2-エチルヘキサノエート、ジ(3-メチルベンゾイル)ペルオキシド、ベンゾイル(3-メチルベンゾイル)ペルオキシド、ジベンゾイルペルオキシド、1,1-ジ(t-ブチルペルオキシ)-2-メチルシクロヘキサン、1,1-ジ(t-ヘキシルペルオキシ)-3,3,5-トリメチルシクロヘキサン等の有機過酸化物等が挙げられる。上記過酸化物は、合成してもあるいは市販品を使用してもよい。
上記金属塩としては、塩化鉄(II)、塩化鉄(III)、硫酸鉄(III)、硝酸鉄(III)、硝酸銀(AgNO3)、クエン酸鉄(III)、硫酸アンモニウム鉄(III)等が挙げられる。
上記以外にも、-0.5~+2.0(V)の標準電極電位(E0
(OX))を有する酸化剤を使用してもよく、このような例としては、メタノール(+0.588V)、酸素(+1.229V)などが使用できる。
上記酸化剤のうち、過酸化水素(+1.763V)、過マンガン酸カリウム、クメンヒドロペルオキシド、ギ酸(+0.034V)、塩化鉄(II)(-0.440V)、硝酸銀(AgNO3)(+0.799V)、メタノール、酸素(+1.229V)、オゾンが好ましく、過酸化水素、メタノール、酸素(+1.229V)がより好ましく、過酸化水素が特に好ましい。
上記の酸化剤は、単独で用いても、2種以上を混合して用いてもよい。
導電性高分子前駆体を含む溶液における酸化剤の含有量は、導電性高分子前駆体100質量部に対して、0.01~10質量部であることが好ましく、0.05~5質量部であることがより好ましい。
(溶媒)
用いられうる溶媒としては、電解質および導電性高分子前駆体を溶解できるものであれば特に限定されないが、ブチレンオキシド、クロロホルム、シクロヘキサノン、アセトニトリル、テトラヒドロフラン、プロピレンカーボネイト、ジクロロメタン、o-ジクロロベンゼン、ジメチルホルムアミド、ジメチルスホキシド、ヘキサメチルリン酸トリアミド、ジメトキシエタン、アセトン、メタノール、エタノール、プロパノール、イソブチルアルコール、tert-ブチルアルコール、塩化メチレン等が挙げられる。また、上記溶媒に、必要に応じて水やその他の有機溶剤を加えて混合溶媒として使用してもよい。上記溶媒は、単独で用いても、2種以上を混合して用いてもよい。
用いられうる溶媒としては、電解質および導電性高分子前駆体を溶解できるものであれば特に限定されないが、ブチレンオキシド、クロロホルム、シクロヘキサノン、アセトニトリル、テトラヒドロフラン、プロピレンカーボネイト、ジクロロメタン、o-ジクロロベンゼン、ジメチルホルムアミド、ジメチルスホキシド、ヘキサメチルリン酸トリアミド、ジメトキシエタン、アセトン、メタノール、エタノール、プロパノール、イソブチルアルコール、tert-ブチルアルコール、塩化メチレン等が挙げられる。また、上記溶媒に、必要に応じて水やその他の有機溶剤を加えて混合溶媒として使用してもよい。上記溶媒は、単独で用いても、2種以上を混合して用いてもよい。
導電性高分子前駆体を含む溶液における溶媒の含有量は、導電性高分子前駆体100質量部に対して、2000~5000質量部であることが好ましく、3000~4000質量部であることがより好ましい。
(その他の添加剤)
その他の添加剤としては、特に制限されないが、電解質、重合触媒、重合速度調整剤等が挙げられる。
その他の添加剤としては、特に制限されないが、電解質、重合触媒、重合速度調整剤等が挙げられる。
電解質
前記電解質としては、酸化還元電解質の分散物や支持電解質が挙げられる。
前記電解質としては、酸化還元電解質の分散物や支持電解質が挙げられる。
前記酸化還元電解質としては、特に制限されないが、I-/I3
-系、Br-/Br3
-系、およびキノン/ハイドロキノン系等が挙げられる。上記酸化還元電解質の分散物は、公知の方法によって得ることができる。例えば、I-/I3
-系の電解質は、ヨウ化物イオンとヨウ素とを混合することによって得ることができる。上記酸化還元電解質の分散物は、液状の形態で用いられる場合には液体電解質、室温(25℃)で固体の高分子に分散させた場合には固体高分子電解質、そしてゲル状物質に分散された場合にはゲル電解質と呼ばれる。正孔輸送層として液体電解質が用いられる場合には、その溶媒として電気化学的に不活性なものが用いられる。当該溶媒としては、例えば、アセトニトリル、炭酸プロピレン、およびエチレンカーボネート等が用いられる。固体高分子電解質が用いられる場合としては特開2001-160427号公報記載の電解質が、ゲル電解質が用いられる場合としては「表面科学」21巻、第5号第288~293頁に記載の電解質が、それぞれ参照されうる。
また、前記支持電解質としては、特に制限されないが、イオン電離可能なものが用いられ、特定のものに限定されないが、溶媒に対する溶解性が高く、酸化、還元を受けにくいものが好適に用いられる。具体的には、過塩素酸リチウム(LiClO4)、テトラフルオロホウ酸リチウム、過塩素酸テトラブチルアンモニウム、Li[(CF3SO2)2N]、(n-C4H9)4NBF4、(n-C4H9)4NPF4、p-トルエンスルホン酸塩、ドデシルベンゼンスルホン酸塩などの塩類が好ましく挙げられる。また、特開2000-106223号公報に記載されるポリマー電解質(例えば、同公報中のPA-1~PA-10)を支持電解質として使用してもよい。
上述の電解質は、単独で用いても、2種以上を混合して用いてもよい。
導電性高分子前駆体を含む溶液における支持電解質の含有量は、導電性高分子前駆体100質量部に対して、30~200質量部であることが好ましく、30~100質量部であることがより好ましい。
重合触媒
前記重合触媒としては、特に制限されないが、トリス-p-トルエンスルホン酸鉄(III)、p-ドデシルベンゼンスルホン酸鉄(III)、メタンスルホン酸鉄(III)、p-エチルベンゼンスルホン酸鉄(III)、ナフタレンスルホン酸鉄(III)、およびこれらの水和物等が挙げられる。
前記重合触媒としては、特に制限されないが、トリス-p-トルエンスルホン酸鉄(III)、p-ドデシルベンゼンスルホン酸鉄(III)、メタンスルホン酸鉄(III)、p-エチルベンゼンスルホン酸鉄(III)、ナフタレンスルホン酸鉄(III)、およびこれらの水和物等が挙げられる。
重合速度調整剤
前記重合速度調整剤としては、特に制限されないが、重合触媒における三価鉄イオンに対する弱い錯化剤があり、膜が形成できるように重合速度を低減するものであれば特に制限はない。例えば、重合触媒がトリス-p-トルエンスルホン酸鉄(III)、p-ドデシルベンゼンスルホン酸鉄(III)、メタンスルホン酸鉄(III)、p-エチルベンゼンスルホン酸鉄(III)、ナフタレンスルホン酸鉄(III)、およびこれらの水和物である場合には、イミダゾール等が用いられうる。
前記重合速度調整剤としては、特に制限されないが、重合触媒における三価鉄イオンに対する弱い錯化剤があり、膜が形成できるように重合速度を低減するものであれば特に制限はない。例えば、重合触媒がトリス-p-トルエンスルホン酸鉄(III)、p-ドデシルベンゼンスルホン酸鉄(III)、メタンスルホン酸鉄(III)、p-エチルベンゼンスルホン酸鉄(III)、ナフタレンスルホン酸鉄(III)、およびこれらの水和物である場合には、イミダゾール等が用いられうる。
また、前記電解質、重合触媒、重合速度調整剤等の他、必要に応じて、例えば、N(PhBr)3SbCl6、NOPF6、SbCl5、I2、Br2、HClO4、(n-C4H9)4ClO4、トリフルオロ酢酸、4-ドデシルベンゼンスルホン酸、1-ナフタレンスルホン酸、FeCl3、AuCl3、NOSbF6、AsF5、NOBF4、LiBF4、H3[PMo12O40]、7,7,8,8-テトラシアノキノジメタン(TCNQ)などのアクセプタードーピング剤、ホールをトラップしにくいバインダー樹脂、レベリング剤等の塗布性改良剤等の各種添加剤を添加してもよい。上記添加剤は、単独で使用しても、2種以上を混合して使用してもよい。
(含浸方法)
含浸方法としては、特に制限されず、公知の方法で光電変換層を、導電性高分子前駆体を含む溶液に含浸することができる。具体的には、装置を使用してもよいし、製造者等が自ら行ってもよい。
含浸方法としては、特に制限されず、公知の方法で光電変換層を、導電性高分子前駆体を含む溶液に含浸することができる。具体的には、装置を使用してもよいし、製造者等が自ら行ってもよい。
含浸温度については、特に制限されないが、溶媒が固化・突沸しない範囲に設定することが好ましく、-10℃~60℃であることがより好ましい。
(塗布方法)
導電性高分子前駆体を含む溶液の塗布方法としては、特に制限されないが、ディッピング、滴下、ドクターブレード、スピンコート、刷毛塗り、スプレー塗装、ロールコーター、エアーナイフコート、カーテンコート、ワイヤーバーコート、グラビアコート、米国特許第2681294号記載のホッパーを使用するエクストルージョンコート、および米国特許第2761418号、同3508947号、同2761791号記載の多層同時塗布方法等の各種塗布法を用いることができる。また、このような塗布の操作を繰り返し行って積層するようにしてもよい。この場合の塗布回数は、特に制限されず、所望の正孔輸送層の厚さに応じて適宜選択されうる。
導電性高分子前駆体を含む溶液の塗布方法としては、特に制限されないが、ディッピング、滴下、ドクターブレード、スピンコート、刷毛塗り、スプレー塗装、ロールコーター、エアーナイフコート、カーテンコート、ワイヤーバーコート、グラビアコート、米国特許第2681294号記載のホッパーを使用するエクストルージョンコート、および米国特許第2761418号、同3508947号、同2761791号記載の多層同時塗布方法等の各種塗布法を用いることができる。また、このような塗布の操作を繰り返し行って積層するようにしてもよい。この場合の塗布回数は、特に制限されず、所望の正孔輸送層の厚さに応じて適宜選択されうる。
塗布温度については、特に制限されないが、溶媒が固化・突沸しない範囲に設定することが好ましく、-10℃~60℃であることがより好ましい。
<導電性高分子前駆体の重合>
正孔輸送層は、酸化剤存在下で前記増感色素に光を照射して前記導電性高分子前駆体を重合することにより形成される。以下、前記重合方法を、「光化学重合法」とも称する。
正孔輸送層は、酸化剤存在下で前記増感色素に光を照射して前記導電性高分子前駆体を重合することにより形成される。以下、前記重合方法を、「光化学重合法」とも称する。
前記光化学重合法は、光電変換層中の増感色素の励起を利用した導電性高分子前駆体の重合方法である。具体的には、(1)増感色素に光を照射すると、増感色素が励起する。(2)励起された増感色素の電子は、酸化剤(例えば過酸化水素など)の還元によって消費され、増感色素はカチオン状態となる。(3)カチオン状態の増感色素は導電性高分子前駆体から電子を抜き取り、導電性高分子前駆体がカチオン状態となる。(4)このカチオン状態となった導電性高分子前駆体がトリガーとなる(すなわち、重合開始剤として機能する)ことで導電性高分子前駆体の重合が開始される。上記光化学重合反応は、以下の反応式によって示される。

光化学重合法は、従来の電解重合のように電圧をかけて重合を行うものではない。その結果、電解重合と対比して、電圧印加による増感色素の劣化が生じることがない。また、均一な組成を有する正孔輸送層が得られうる。よって、電解重合のように、導電性高分子前駆体の重合によって析出する導電性高分子による電圧降下により、正孔輸送層内の重合の程度が不均一となることがない。また、カチオンを利用する化学重合法であることから、光電変換層(半導体層)の表面に十分量でかつ緻密に重合体を形成することが可能となりうる。
また、電解重合では、低電圧で重合を行うことから、重合に要する時間は長時間となりうる。しかし、光化学重合においては、電解重合よりも迅速に重合を行うことができ、生産性に優れる。さらに、電解重合では、導電性高分子の析出に伴う重合度の不均一性の問題から、大面積化した光電変換素子を製造しようとする場合には、耐久性の低下がより顕著となりうる。しかしながら、光化学重合においては、正孔輸送層内において均一な組成を有する正孔輸送層が得られうることから、より好適に大面積化した光電変換素子を製造することができる。
光電変換素子の製造面を考慮すると、電解重合は電圧を印加する必要があることから、バッチ形式で行わなければならない。しかし、光化学重合法では、バッチ形式だけでなく、連続形式でも行うことができ、製造面からも優れている。
本発明の一実施形態において、光化学重合法はさらなる利点を有する。具体的には、これまでに、増感色素のうち、カルボキシ基(-COOH基)を有する増感色素については、電解重合の条件下において、印加電圧によりCO2が脱離(コルベ電解)して色素が分解するという問題が報告されている(例えば、特開2011-065751号公報を参照)。その結果、得られる光電変換素子の性能が低いものとなっていた。しかしながら、光化学重合では、電圧を印加することがないため、好適にカルボキシ基を有する増感色素を使用することができる。
光化学重合反応において、光照射に用いられうる光源としては、特に制限されないが、キセノンランプ、ハロゲンランプ、LED等が挙げられる。
照射光の波長としては、特に制限されないが、波長400nm以上の光源を用いることが好ましく、波長400~1100nm(または400nm超1100nm以下)、特に好ましくは波長420nm超1100nm以下の光源を用いることがより好ましい。前記波長が400nm以下であると、400nm以下の波長の光で生じる酸化チタンの光触媒作用による増感色素の分解を抑制することができ、また、厚い正孔輸送層を形成するために長時間の光照射を行う場合に安定した特性が得られうることから好ましい。なお、前記波長が1100nm以下であると、赤外光の照射を抑制することにより、加熱が抑制され層間剥離を抑制でき、また、導電性高分子前駆体として上記一般式(2)で表される化合物を用いる場合には、生成した導電性高分子の光照射による分解等の副反応が抑制されうるから好ましい。
また、照射光の強度としては、特に制限されないが、10~150mW/cm2であることが好ましく、20~80mW/cm2であることがより好ましい。
光照射の時間としては、照射光の波長および強度によっても異なるが、0.1~30分間であることが好ましく、0.5~15分間であることがより好ましい。光照射時間が上記範囲にあると、重合が十分に進行しうることから好ましい。
光照射は、光電変換層を、導電性高分子前駆体を含む溶液に含浸した状態で外部から行ってもよいし、光電変換層上に含浸および/または塗布した後別途に行ってもよい。
光重合を行った後は、必要に応じて、上述の溶媒を用いて洗浄する工程、および/または、例えば、25~150℃、0.2~12時間の条件で乾燥する工程を行ってもよい。
さらに、必要に応じて、導電性高分子のドープ率向上および酸化チタンから正孔輸送層への逆電子移動防止を目的として、正孔輸送層を有する半導体電極を、上述の溶媒、上述の電解質、および上述の有機塩からなる群から選択される少なくとも一種を含む混合液に、例えば、-10~70℃、0.1~2時間浸漬させてもよい。この場合、浸漬させた後、自然乾燥で0.01~24時間静置させることが好ましい。
<正孔輸送層>
光化学重合反応により重合した正孔輸送層は、導電性高分子を含む。また、正孔輸送層は、前記導電性高分子前駆体を含む溶媒に含有されうる成分を含みうる。
光化学重合反応により重合した正孔輸送層は、導電性高分子を含む。また、正孔輸送層は、前記導電性高分子前駆体を含む溶媒に含有されうる成分を含みうる。
(導電性高分子)
前記導電性高分子としては、上述の導電性高分子前駆体が重合してなる構造を有しうる。導電性高分子前駆体は、上記式(2)を有してポリマー化する役割を担うものであることが好ましい。そのため、上記式(2)を単独または複数種類の繰り返し単位が結合した多量体を用いてもよい。さらに、予め上記繰り返し単位を有するモノマーを必要に応じて、単独あるいは複数種類のモノマーと共に重合したプレポリマー(二量体以上の多量体やいわゆるオリゴマーを含む)であってもよい。例えば、上述の一般式(2)で表される導電性高分子前駆体を用いた場合には、以下の一般式(3)で表される導電性高分子でありうる。
前記導電性高分子としては、上述の導電性高分子前駆体が重合してなる構造を有しうる。導電性高分子前駆体は、上記式(2)を有してポリマー化する役割を担うものであることが好ましい。そのため、上記式(2)を単独または複数種類の繰り返し単位が結合した多量体を用いてもよい。さらに、予め上記繰り返し単位を有するモノマーを必要に応じて、単独あるいは複数種類のモノマーと共に重合したプレポリマー(二量体以上の多量体やいわゆるオリゴマーを含む)であってもよい。例えば、上述の一般式(2)で表される導電性高分子前駆体を用いた場合には、以下の一般式(3)で表される導電性高分子でありうる。

上記一般式(3)中、X、R2~R5は、一般式(2)と同一である。また、mは、単量体の結合数を表し、例えばm=2の場合は二量体、m=3の場合は三量体を示す。ここでは、mは、1以上の整数、好ましくは1~10の整数である。
さらに、以下、本発明に係る導電性高分子前駆体の特に好ましい形態を表1に示す。


上記のうち、導電性高分子前駆体がM1-1、M1-4、M1-26であることが好ましく、導電性高分子前駆体がM1-1であることが特に好ましい。すなわち、導電性高分子前駆体は、下記式:

で示されることが好ましい。
導電性高分子の重合度は、その合成方法により得られた重合体から把握することは困難である。しかしながら、重合後に形成された正孔輸送層の溶媒溶解性は大きく低下するため、重合体かどうかの確認については、当該重合体の溶解が可能なテトラヒドロフラン(THF)に正孔輸送層を浸漬させることで、その溶解度により判断できる。具体的には、25mLのサンプル瓶に化合物(導電性高分子)60mgをとり、THF 10mlを添加して、超音波(25kHz、150W 超音波工業(株)COLLECTOR CURRENT1.5A超音波工業製150)を5分間照射したときに、溶解している化合物が5mg以下の場合は重合していると判断する。また、重合前後の吸収帯を対比することで導電性高分子の重合を確認することもできる。または、得られた重合体に、光を照射して、600~1100nmに新たな吸収を確認することによって、導電性高分子の重合を確認することもできる。
正孔輸送層に含まれる材料は、増感色素による光吸収を妨げないように、大きいバンドギャップを持つことが好ましい。具体的には2eV以上のバンドキャップを有することが好ましく、2.5eV以上のバンドキャップを有することがさらに好ましい。また、正孔輸送層は、増感色素ホールを還元させるために低いイオン化ポテンシャルを有することが好ましい。適用する増感色素に応じてイオン化ポテンシャルの値は異なるが、通常、4.5~5.5eVであることが好ましく、4.7~5.3eVであることがより好ましい。
本発明に係る正孔輸送層の平均厚みは、半導体層が多孔質体である場合は、当該多孔質体の内部や隙間にも浸透してため測定が容易ではない。
上述のように、本形態に係る短絡防止層は、第一電極を追随する形で形成されるため、短絡防止層からの第一電極の突出を防止することができる。その結果、得られる光電変換素子は、逆電子移動や暗電流の発生を防止または低減することにより、高い光電変換効率を示しうる。一方、本形態に係る正孔輸送層は、正孔輸送層内で均一に導電性高分子が形成されている。その結果、製造される光電変換素子は高い耐久性を有しうる。
ここで、上記2つの方法を組み合わせて適用することについて検討する。図1、図2A、2Bに示されるように、正孔輸送層は、光電変換層だけでなく、短絡防止層にも接触しうる。そして、上述のように、光化学重合によって得られた正孔輸送層の内部には、導電性高分子が均一に存在していることから、正孔輸送層が短絡防止層と接触する面にも高確率で導電性高分子が存在しうる。その結果、仮に短絡防止層から第一電極が突出している場合には、第一電極と導電性高分子が接触することによって、逆電子移動や暗電流が増加し、光電変換効率が低下する可能性が高まるリスクがある(図2A)。このような現象は、従来の電解重合によって正孔輸送層を形成する場合よりも問題となりうる。ところが、電解重合によって正孔輸送層を形成する場合には、導電性高分子は正孔輸送層内に不均一に存在することから、仮に第一電極と正孔輸送層が接触したとしても、正孔輸送層内の導電性高分子と接触する可能性は相対的に低いものであった(例えば、正孔輸送層内の導電性を示さないモノマー等と接触する可能性がある)。したがって、短絡防止層の突出による光電変換効率の低下の問題は、光化学重合によって正孔輸送層を形成する場合により注意が必要な課題であり、製造条件の調整を慎重にする必要があった。
しかしながら、垂直堆積法により短絡防止層を形成することで、短絡防止層からの第一電極の突出を防止することができるため、上記問題が解決されうる(図2B)。
そして、後述する実施例においても示されるように、これらの方法を組み合わせる光電変換効率および耐久性の向上効果が、最大限発現されたものと推定している。なお、従来の光電変換素子と対比して、本発明に係る光電変換素子がより光電変換効率に優れ、かつ、より高い耐久性を有する理由については以下のように推定している。すなわち、従来の電界重合では、多孔質の微小部分に生じる部分的な印加電圧の不均一性のために、不均一な製膜を強いられていた。その結果、長期間の光電変換素子の使用による導通経路の寸断によって変換効率の低下を生じていた。これに対し、光化学重合法を用いることで、多孔質半導体全体に均一に導電性高分子相を形成することができ、より高い耐久性を有する光電変換素子を形成できる。また、垂直堆積法による短絡防止層の形成により、均一な塗膜形成において、時としてサンプルばらつきにより生じる逆電子移動の増大確率を抑制するため、高い変換効率が達成できたものと推定している。
[第二電極を形成する工程]
第二電極は、特に制限されず、公知の方法により形成することができる。例えば、第二電極の材料を蒸着(真空蒸着を含む)、スパッタリング、塗布、スクリーン印刷等の方法が適用されうる。
第二電極は、特に制限されず、公知の方法により形成することができる。例えば、第二電極の材料を蒸着(真空蒸着を含む)、スパッタリング、塗布、スクリーン印刷等の方法が適用されうる。
(第二電極)
このようにして形成される第二電極は、正孔輸送層と接して配置される。絶縁性の物質でも、正孔輸送層に面している側に導電性物質層が設置されていれば、これも使用することができる。第二電極は、素子の電気抵抗を低減する等の観点から、正孔輸送層との接触が良好であることが好ましい。また、第二電極は、正孔輸送層との仕事関数の差が小さく、化学的に安定であることが好ましい。
このようにして形成される第二電極は、正孔輸送層と接して配置される。絶縁性の物質でも、正孔輸送層に面している側に導電性物質層が設置されていれば、これも使用することができる。第二電極は、素子の電気抵抗を低減する等の観点から、正孔輸送層との接触が良好であることが好ましい。また、第二電極は、正孔輸送層との仕事関数の差が小さく、化学的に安定であることが好ましい。
用いられうる第二電極の材料としては、特に制限されないが、金、銀、銅、アルミニウム、白金、クロム、ロジウム、ルテニウム、マグネシウム、インジウム等の金属薄膜、炭素、カーボンブラック、導電性高分子、導電性の金属酸化物(インジウム-スズ複合酸化物、酸化スズにフッ素をドープしたもの等)等の有機導電体などが挙げられる。好ましくは金などの金属薄膜である。
第二電極の膜厚は、特に制限されないが、10~1000nmであることが好ましい。
第二電極の表面抵抗値は、特に制限されず、可能な限り低い値であることが好ましい。具体的には、表面抵抗値は、80Ω/□以下であることが好ましく、20Ω/□以下であることがより好ましい。
<太陽電池>
本発明の方法によって製造された光電変換素子は、太陽電池に特に好適に使用することができる。
本発明の方法によって製造された光電変換素子は、太陽電池に特に好適に使用することができる。
本発明に係る光電変換素子は、色素増感型の太陽電池(セル)として用いられうる。すなわち、本発明に係る太陽電池は、例えばインターコネクタにより電気的に接続された複数の太陽電池セル(本発明に係る光電変換素子)と、それを挟持する一対の保護部材と、一対の保護部材と複数の太陽電池との間の隙間に充填された封止樹脂とを有する。一対の保護部材のうちの一方は、前述の光電変換素子の基体となる。一対の保護部材は両方が透明であってもよいし、一方のみが透明であってもよい。
本発明に係る太陽電池の構造の例には、Z型モジュール、W型モジュールが含まれる。Z型モジュールは、対向する一対の保護部材のうち、一方の保護部材に複数の色素を担持した多孔質な半導体層を、他方の基体に複数の正孔輸送層を形成し、これらを貼り合わせた構造を有する。W型モジュールは、保護部材のそれぞれに一つおきに色素を担持した多孔質な半導体層および正孔輸送層の積層体を形成し、セルが互い違いとなるように貼り合わせた構造を有する。
本発明に係る太陽電池に、太陽光または太陽光と同等の電磁波を照射すると、半導体に担持された増感色素は照射された光もしくは電磁波を吸収して励起する。励起によって発生した電子は半導体に移動し、次いで導電性支持体および外部負荷を経由して第二電極に移動して、正孔輸送層の正孔輸送性材料に供給される。一方、半導体に電子を移動させた増感色素は酸化体となっているが、第二電極から正孔輸送層の重合体を経由して電子が供給されることにより、還元されて元の状態に戻り、同時に正孔輸送層の重合体は酸化されて、再び第二電極から供給される電子により還元されうる状態に戻る。このようにして電子が流れ、本発明の光電変換素子を用いた太陽電池を構成することができる。
以下、実施例により本発明を説明するが、本発明はこれらに限定されない。
(実施例1)
第一電極を形成する工程
基板として、厚さ1.1mmの耐熱ガラス板を用いた。
第一電極を形成する工程
基板として、厚さ1.1mmの耐熱ガラス板を用いた。
第一電極の材料としては、塩化スズ(IV)五水和物(SnCl4・5H2O、分子量:350.60)を用いた。
はじめに、塩化スズ(IV)五水和物0.701gに対してエタノール30%水溶液10mlの割合で、塩化スズ(IV)五水和物を溶解した。当該溶解液にフッ化アンモニウム0.592gの飽和水溶液を加え、得られた混合液を約20分間超音波処理することにより、塗布液を調製した。
次に、耐熱ガラス板の表面を化学洗浄し、乾燥した。次いで、この耐熱ガラス板を反応器内に静置し、ヒータで加熱した。ヒータの加熱温度が400℃になったところで、上記塗布液を、ガラス板までの距離を600mmとして、口径0.3mmのノズルから圧力0.06MPaで25分間噴霧することで、厚みが100nmの第一電極(FTO層)を形成した。
前記形成された第一電極の表面粗さ(Ra)をサーフコム1400D(株式会社東京精密製)を用いて測定したところ、21nmであった。
工程(1):短絡防止層を形成する工程
短絡防止材料前駆体として、TC100(チタンジイソプロポキシビス(アセチルアセトネート)、株式会社マツモト交商製)を用いた。また、短絡防止層はインクジェット法を利用する方法により形成した。
短絡防止材料前駆体として、TC100(チタンジイソプロポキシビス(アセチルアセトネート)、株式会社マツモト交商製)を用いた。また、短絡防止層はインクジェット法を利用する方法により形成した。
はじめに、5gのTC100(チタンジイソプロポキシビス(アセチルアセトネート))を95gのn-プロパノールに溶解した。
得られた溶液を、上記で形成した第一電極(FTO)上に、インクジェット法により塗布した後、500℃で10分間加熱して焼成を行うことにより、酸化チタンの薄層からなる短絡防止層を形成した。なお、インクジェットヘッドの構成としては圧電素子方式を用い、ヘッドを基板の一片に沿って移動させながら、溶液を吐出する(吐出量=1滴あたり10ピコリットル)ことを10回繰り返した。得られた短絡防止層の厚さは100nmであった。
光電変換層を形成する工程
半導体としては、酸化チタン(アナターゼ型、1次平均粒径(顕微鏡観察平均)18nm)を用いた。また、増感色素としては、下記式で表されるA-16(コニカミノルタ製)の化合物を用いた。
半導体としては、酸化チタン(アナターゼ型、1次平均粒径(顕微鏡観察平均)18nm)を用いた。また、増感色素としては、下記式で表されるA-16(コニカミノルタ製)の化合物を用いた。

はじめに、短絡防止層上に酸化チタンペースト(エチルセルロースを10%アセチルアセトン水に分散)をスクリーン印刷法(塗布面積:25mm2)により塗布した。得られた塗膜を、200℃で10分間、および500℃で15分間焼成して、厚さが2.5μmで空孔率(D)が60体積%の酸化チタンの多孔質層(多孔質の半導体層)を得た。
次に、増感色素A-16を、濃度が5×10-4mol/Lとなるようにアセトニトリルおよびtert-ブチルアルコールの混合溶媒(体積比1:1)に溶解した。得られた溶液に、上記で得た酸化チタンの多孔質層を形成したFTOガラス基板を室温(25℃)で3時間浸漬して半導体層に増感色素を吸着させて、色素を担持する多孔質の半導体層(光電変換層)を有する半導体電極を得た。なお、この際の半導体層1m2当たりの増感色素の総担持量は、1mmolであった。また、用いた増感色素は350~650nmに吸収帯を示した。
工程(2):正孔輸送層を形成する工程
はじめに、下記化学式で表される導電性高分子前駆体M1-1(2,2’-ビス[3,4-(エチレンビスオキシ)チオフェン])および支持電解質であるLi[(CF3SO2)2N]を、それぞれ濃度が1×10-3(mol/L)および0.1(mol/L)となるようにアセトニトリルに溶解した。次に、酸化剤である30重量%の過酸化水素水を、過酸化水素が導電性高分子前駆体100質量部に対して1質量部となるように添加して、導電性高分子前駆体を含む溶液を調製した。
はじめに、下記化学式で表される導電性高分子前駆体M1-1(2,2’-ビス[3,4-(エチレンビスオキシ)チオフェン])および支持電解質であるLi[(CF3SO2)2N]を、それぞれ濃度が1×10-3(mol/L)および0.1(mol/L)となるようにアセトニトリルに溶解した。次に、酸化剤である30重量%の過酸化水素水を、過酸化水素が導電性高分子前駆体100質量部に対して1質量部となるように添加して、導電性高分子前駆体を含む溶液を調製した。

上記で調製した溶液に半導体電極を浸漬させた後、半導体電極の外側から、キセノンランプから420nm以下の波長をカットするシャープカットフィルター(HOYA製:S-L42)を通した光を1分間照射して重合を行った。この際、照射光の強度は、22mW/cm2であった。光照射後、重合前後の吸収帯を対比すると、600~1100nmに新たな吸収が観察され、導電性高分子前駆体が重合していることを確認した。得られた正孔輸送層を有する半導体電極をアセトニトリルで洗浄した後、乾燥させた。なお、得られた正孔輸送層は、溶媒には不溶の重合膜であった。
次いで、導電性高分子のドープ率向上および酸化チタンから正孔輸送層への逆電子移動防止を目的として、正孔輸送層が形成された半導体電極(半導体電極/正孔輸送層)を、Li[(CF3SO2)2N]を15×10-3(mol/L)、tert-ブチルピリジンを50×10-3(モル/l)の割合で含有するアセトニトリル溶液に10分間浸漬させて、自然乾燥させた。
第二電極を形成する工程
真空蒸着法で金を60nm蒸着して、第二電極を形成して、光電変換素子を製造した。
真空蒸着法で金を60nm蒸着して、第二電極を形成して、光電変換素子を製造した。
(実施例2)
工程(1)における短絡防止材料前駆体を含む溶液を吐出する回数を3回に変更して、短絡防止層の厚さを30nmとしたことを除いては、実施例1と同様の方法で光電変換素子を製造した。
工程(1)における短絡防止材料前駆体を含む溶液を吐出する回数を3回に変更して、短絡防止層の厚さを30nmとしたことを除いては、実施例1と同様の方法で光電変換素子を製造した。
(実施例3)
工程(1)における短絡防止材料前駆体を含む溶液を吐出する回数を1回に変更して、短絡防止層の厚さを5nmとしたことを除いては、実施例1と同様の方法で光電変換素子を製造した。
工程(1)における短絡防止材料前駆体を含む溶液を吐出する回数を1回に変更して、短絡防止層の厚さを5nmとしたことを除いては、実施例1と同様の方法で光電変換素子を製造した。
(実施例4)
工程(1)における短絡防止材料前駆体を含む溶液を吐出する回数を40回に変更して、短絡防止層の厚さを400nmとしたことを除いては、実施例1と同様の方法で光電変換素子を製造した。
工程(1)における短絡防止材料前駆体を含む溶液を吐出する回数を40回に変更して、短絡防止層の厚さを400nmとしたことを除いては、実施例1と同様の方法で光電変換素子を製造した。
(実施例5)
工程(1)における焼成温度を200℃に変更したことを除いては、実施例1と同様の方法で光電変換素子を製造した。
工程(1)における焼成温度を200℃に変更したことを除いては、実施例1と同様の方法で光電変換素子を製造した。
(実施例6)
工程(1)における焼成温度を400℃に変更したことを除いては、実施例1と同様の方法で光電変換素子を製造した。
工程(1)における焼成温度を400℃に変更したことを除いては、実施例1と同様の方法で光電変換素子を製造した。
(実施例7)
工程(1)における焼成温度を600℃に変更したことを除いては、実施例1と同様の方法で光電変換素子を製造した。
工程(1)における焼成温度を600℃に変更したことを除いては、実施例1と同様の方法で光電変換素子を製造した。
(実施例8)
工程(1)において、第一電極(FTO)上に、スプレー法により塗布したことを除いては、実施例1と同様の方法で光電変換素子を製造した。
工程(1)において、第一電極(FTO)上に、スプレー法により塗布したことを除いては、実施例1と同様の方法で光電変換素子を製造した。
なお、上記スプレー法は以下の方法により行った。すなわち、スプレーノズルとして、二流体スプレーノズルAM25S(アトマックス社製)を使用し、霧化および噴出用気体としては、霧化圧力30kPaの窒素ガスを使用した。また、ノズル先端と第一電極との距離は80mmとした。短絡防止材料前駆体液を含む溶液が入った容器とノズルとをつなぐ流路に口径0.4mmのオリフィスを設け、容器に1kPaの吐出圧を印加することで、ノズルからの溶液の吐出量を調整した。このときの吐出量は11.3mL/minであった。スプレーノズルを速度400mm/s、ピッチ15mmでスキャンして、第一電極表面に前記溶液を塗布した。次いで、500℃で10分間加熱し、厚さ100nmの酸化チタンの薄層からなる短絡防止層を形成した。
(実施例9)
第一電極を形成する工程において、第一電極の材料(塩化スズ(IV)五水和物)を溶解するためのエタノール30%水溶液の量を8mLに変更して第一電極を形成したことを除いては、実施例1と同様に光電変換素子を製造した。なお、形成された第一電極の表面粗さ(Ra)は、実施例1と同様の方法で測定したところ、36nmであった。
第一電極を形成する工程において、第一電極の材料(塩化スズ(IV)五水和物)を溶解するためのエタノール30%水溶液の量を8mLに変更して第一電極を形成したことを除いては、実施例1と同様に光電変換素子を製造した。なお、形成された第一電極の表面粗さ(Ra)は、実施例1と同様の方法で測定したところ、36nmであった。
(実施例10)
第一電極を形成する工程において、第一電極の材料(塩化スズ(IV)五水和物)を溶解するためのエタノール30%水溶液の量を5mLに変更して第一電極を形成したことを除いては、実施例1と同様に光電変換素子を製造した。なお、形成された第一電極の表面粗さ(Ra)は、実施例1と同様の方法で測定したところ、69nmであった。
第一電極を形成する工程において、第一電極の材料(塩化スズ(IV)五水和物)を溶解するためのエタノール30%水溶液の量を5mLに変更して第一電極を形成したことを除いては、実施例1と同様に光電変換素子を製造した。なお、形成された第一電極の表面粗さ(Ra)は、実施例1と同様の方法で測定したところ、69nmであった。
(比較例1)
工程(1)において、短絡防止層を、スピンコート法を利用した方法により形成し、工程(2)において、正孔輸送層を、光電解重合により形成したことを除いては、実施例1と同様の方法で光電変換素子を製造した。
工程(1)において、短絡防止層を、スピンコート法を利用した方法により形成し、工程(2)において、正孔輸送層を、光電解重合により形成したことを除いては、実施例1と同様の方法で光電変換素子を製造した。
なお、上記スピンコートを利用した短絡防止層の形成は以下のように行った。
はじめに、1.2mLのTC100(チタンジイソプロポキシビス(アセチルアセトネート)、株式会社マツモト交商製)、0.8mLのアセチルアセトン、および18mLのエタノールを混合した。得られた溶液をスピンコート法により滴下して第一電極(FTO)上に塗布した後、500℃で10分間加熱して焼成を行うことにより、酸化チタンの薄層からなる短絡防止層を形成した。得られた短絡防止層の厚さは100nmであった。
また、光電解重合による正孔輸送層の形成は以下のように行った。はじめに、実施例1で使用した導電性高分子前駆体および支持電解質であるLi[(CF3SO2)2N]を、それぞれ濃度が1×10-3(mol/L)および0.1(mol/L)となるようにアセトニトリルに溶解し、導電性高分子前駆体を含む溶液を調製した。
上記で調製した溶液に半導体電極を浸漬させた。その後、作用極を前記半導体電極、対極を白金線、参照電極をAg/Ag+(AgNO3:0.01M)、保持電圧を-0.16Vとし、半導体層の外側(半導体層方向)から、電圧を保持した状態で、キセノンランプから420nm以下の波長をカットするシャープカットフィルター(HOYA製:S-L42)を通した光を30分間照射して重合を行った。光照射後、重合前後の吸収帯を対比すると、600~1100nmに新たな吸収が観察され、導電性高分子前駆体が重合していることを確認した。得られた正孔輸送層を有する半導体電極をアセトニトリルで洗浄した後、乾燥させた。なお、得られた正孔輸送層は、溶媒には不溶の重合膜であった。
(比較例2)
短絡防止層を比較例1と同様の方法で形成したことを除いては、実施例1と同様の方法で光電変換素子を製造した。
短絡防止層を比較例1と同様の方法で形成したことを除いては、実施例1と同様の方法で光電変換素子を製造した。
(比較例3)
正孔輸送層を比較例1と同様の方法で形成したことを除いては、実施例1と同様の方法で光電変換素子を製造した。
正孔輸送層を比較例1と同様の方法で形成したことを除いては、実施例1と同様の方法で光電変換素子を製造した。
上記実施例1~10および比較例1~3で製造した光電変換素子について、下記表2に示す。

[光電変換素子の性能評価]
実施例1~10および比較例1~3で製造した光電変換素子についての性能評価を行った。
実施例1~10および比較例1~3で製造した光電変換素子についての性能評価を行った。
<初期の光電変換効率の測定>
ソーラーシミュレータ(英弘精機製)を用いて、光電変換素子に、キセノンランプからAMフィルター(AM-1.5)を通して強度100mW/cm2の擬似太陽光を照射した。そして、I-Vテスターを用いて、光電変換素子の室温(25℃)での電流-電圧特性を測定し、短絡電流密度(Jsc)、開放電圧(Voc)、および形状因子(F.F.)を測定した。これらの値に基づき、下記式1から光電変換効率η1(%)を算出した。
ソーラーシミュレータ(英弘精機製)を用いて、光電変換素子に、キセノンランプからAMフィルター(AM-1.5)を通して強度100mW/cm2の擬似太陽光を照射した。そして、I-Vテスターを用いて、光電変換素子の室温(25℃)での電流-電圧特性を測定し、短絡電流密度(Jsc)、開放電圧(Voc)、および形状因子(F.F.)を測定した。これらの値に基づき、下記式1から光電変換効率η1(%)を算出した。

<光劣化後の光電変換効率の測定>
開回路状態で、キセノンランプからAMフィルター(AM-1.5)を通して強度100mW/cm2の擬似太陽光を光電変換素子に6時間照射した。次いで、上記「初期の光電変換効率の測定」と同様の方法で、短絡電流密度(Jsc)、開放電圧(Voc)、および形状因子(F.F.)を測定し、光電変換効率η2(%)を算出した。
開回路状態で、キセノンランプからAMフィルター(AM-1.5)を通して強度100mW/cm2の擬似太陽光を光電変換素子に6時間照射した。次いで、上記「初期の光電変換効率の測定」と同様の方法で、短絡電流密度(Jsc)、開放電圧(Voc)、および形状因子(F.F.)を測定し、光電変換効率η2(%)を算出した。
そして、上記で測定した初期の光電変換効率η1に対する光劣化後の光電変換効率η2の比率(光劣化前後の効率比)η2/η1を求めた。
<暗電流密度の測定>
光を完全に遮断した状態で強制的に逆電圧を印加して、漏れ電流性を評価する為、I-Vテスターを用い、素子面積Sの光電変換素子に-0.5Vの電圧を印加した際の電流値Iを測定した。前記電流値Iおよび素子面積Sの値から、下記式2から暗電流密度Jを算出した。
光を完全に遮断した状態で強制的に逆電圧を印加して、漏れ電流性を評価する為、I-Vテスターを用い、素子面積Sの光電変換素子に-0.5Vの電圧を印加した際の電流値Iを測定した。前記電流値Iおよび素子面積Sの値から、下記式2から暗電流密度Jを算出した。
上記性能評価において、得られた結果を下記表3に示す。

表3の結果から、スピンコート法を利用した方法により短絡防止層を形成し、かつ、光電解重合により正孔輸送層を形成した比較例1では、初期の光電変換効率がやや低く、耐久性は低い値であった。また、暗電流密度は低い値であった。比較例1において、正孔輸送層の形成方法を光電界重合から光化学重合に変更した比較例2では、優位に耐久性が向上したことが分かる。一方、暗電流密度については比較例1よりもさらに低い値となっている。このことから、光化学重合によって得られた正孔輸送層の内部には、導電性高分子が均一に存在しており、正孔輸送層が短絡防止層と接触する面にも高確率で導電性高分子が存在することが理解される。さらに、比較例1において、短絡防止層の形成方法を、スピンコート法を利用した方法からインクジェット法を利用した方法に変更した比較例3では、短絡防止層が第一電極を好適に被覆していると考えられることから、優位に初期の光電変換効率が向上し、また、暗電流密度の値も優位に低下したことが分かる。
そして、これら比較例1~3と対比して、実施例1~10の光電変換素子(特に、実施例1の光電変換素子)は、垂直堆積法による短絡防止層の形成、および光化学重合による正孔輸送層の形成を組み合わせることによって、顕著に優れた光電変換効率(初期の光電変換効率および暗電流密度)および高い耐久性を有していることが分かる。より詳細には、実施例1の光電変換素子は、比較例2と対比してより優れた耐久性を示しており、また、比較例3と対比してより優れた初期の光電変換効率および暗電流の発生防止効果を示している。
本出願は、2013年1月11日に出願された日本特許出願番号2013-003976号に基づいており、その開示内容は、参照され、全体として、組み入れられている。
1 基板、
2 第一電極、
3 短絡防止層、
4 増感色素、
5 半導体、
6 光電変換層、
7 正孔輸送層、
8 第二電極、
9 太陽光、
10、10’ 光電変換素子、
11 正孔輸送層を構成する材料。
2 第一電極、
3 短絡防止層、
4 増感色素、
5 半導体、
6 光電変換層、
7 正孔輸送層、
8 第二電極、
9 太陽光、
10、10’ 光電変換素子、
11 正孔輸送層を構成する材料。
Claims (6)
- 基板、第一電極、短絡防止層、半導体および増感色素を含有する光電変換層、導電性高分子を含有する正孔輸送層、並びに第二電極をこの順に有する光電変換素子の製造方法であって、
垂直堆積法により前記短絡防止層を形成する工程(1)と、
導電性高分子前駆体を前記光電変換層と接触させ、酸化剤存在下で前記増感色素に光を照射して前記導電性高分子前駆体を重合することにより前記正孔輸送層を形成する工程(2)と、
を含む、製造方法。 - 前記工程(1)が、塗布液をインクジェット法により塗布して塗膜を形成する工程と、得られた塗膜を焼成する工程と、を含む、請求項1に記載の製造方法。
- 前記焼成が、200~700℃で行われる、請求項2に記載の製造方法。
- 前記短絡防止層の膜厚が10~200nmである、請求項1~3のいずれか1項に記載の製造方法。
- 前記第一電極の表面粗さ(Ra)が、10~50nmである、請求項1~4のいずれか1項に記載の製造方法。
- 前記増感色素が、カルボキシ基を有する、請求項1~5のいずれか1項に記載の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014556433A JPWO2014109355A1 (ja) | 2013-01-11 | 2014-01-09 | 光電変換素子の製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013003976 | 2013-01-11 | ||
| JP2013-003976 | 2013-01-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014109355A1 true WO2014109355A1 (ja) | 2014-07-17 |
Family
ID=51167002
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/050218 WO2014109355A1 (ja) | 2013-01-11 | 2014-01-09 | 光電変換素子の製造方法 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JPWO2014109355A1 (ja) |
| WO (1) | WO2014109355A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114556606A (zh) * | 2019-12-24 | 2022-05-27 | 松下知识产权经营株式会社 | 太阳能电池 |
| EP3660109B1 (en) * | 2017-07-28 | 2023-12-27 | Sumitomo Chemical Company Limited | Ink composition, film, and display |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004063758A (ja) * | 2002-07-29 | 2004-02-26 | Fdk Corp | 色素増感型太陽電池とその製造方法 |
| JP2005222753A (ja) * | 2004-02-04 | 2005-08-18 | Shin Etsu Polymer Co Ltd | 光電変換素子およびその製造方法 |
| WO2007007735A1 (ja) * | 2005-07-12 | 2007-01-18 | Osaka University | 光照射による金属酸化物表面上への有機導電性重合性材料の合成 |
| JP2007042494A (ja) * | 2005-08-04 | 2007-02-15 | Fujikura Ltd | 作用極及びその製造方法、並びに太陽電池及びその製造方法 |
| JP2007134328A (ja) * | 2005-11-11 | 2007-05-31 | Samsung Sdi Co Ltd | 太陽電池及びその製造方法 |
| JP2010177109A (ja) * | 2009-01-30 | 2010-08-12 | Toyota Central R&D Labs Inc | 下地層形成用組成物、下地層の製造方法、光電極の製造方法、太陽電池の製造方法及び太陽電池モジュール |
| JP2010205753A (ja) * | 2009-02-27 | 2010-09-16 | Toyota Central R&D Labs Inc | 色素増感型太陽電池及びその製造方法 |
| JP2011076869A (ja) * | 2009-09-30 | 2011-04-14 | Tdk Corp | 色素増感型太陽電池及びその製造方法、並びに、色素増感型太陽電池用の作用電極の製造方法 |
-
2014
- 2014-01-09 JP JP2014556433A patent/JPWO2014109355A1/ja active Pending
- 2014-01-09 WO PCT/JP2014/050218 patent/WO2014109355A1/ja active Application Filing
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004063758A (ja) * | 2002-07-29 | 2004-02-26 | Fdk Corp | 色素増感型太陽電池とその製造方法 |
| JP2005222753A (ja) * | 2004-02-04 | 2005-08-18 | Shin Etsu Polymer Co Ltd | 光電変換素子およびその製造方法 |
| WO2007007735A1 (ja) * | 2005-07-12 | 2007-01-18 | Osaka University | 光照射による金属酸化物表面上への有機導電性重合性材料の合成 |
| JP2007042494A (ja) * | 2005-08-04 | 2007-02-15 | Fujikura Ltd | 作用極及びその製造方法、並びに太陽電池及びその製造方法 |
| JP2007134328A (ja) * | 2005-11-11 | 2007-05-31 | Samsung Sdi Co Ltd | 太陽電池及びその製造方法 |
| JP2010177109A (ja) * | 2009-01-30 | 2010-08-12 | Toyota Central R&D Labs Inc | 下地層形成用組成物、下地層の製造方法、光電極の製造方法、太陽電池の製造方法及び太陽電池モジュール |
| JP2010205753A (ja) * | 2009-02-27 | 2010-09-16 | Toyota Central R&D Labs Inc | 色素増感型太陽電池及びその製造方法 |
| JP2011076869A (ja) * | 2009-09-30 | 2011-04-14 | Tdk Corp | 色素増感型太陽電池及びその製造方法、並びに、色素増感型太陽電池用の作用電極の製造方法 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3660109B1 (en) * | 2017-07-28 | 2023-12-27 | Sumitomo Chemical Company Limited | Ink composition, film, and display |
| CN114556606A (zh) * | 2019-12-24 | 2022-05-27 | 松下知识产权经营株式会社 | 太阳能电池 |
| US20220285640A1 (en) * | 2019-12-24 | 2022-09-08 | Panasonic Intellectual Property Management Co., Ltd. | Solar cell |
| US11696456B2 (en) * | 2019-12-24 | 2023-07-04 | Panasonic Intellectual Property Management Co., Ltd. | Solar cell |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2014109355A1 (ja) | 2017-01-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6252479B2 (ja) | 光電変換素子およびその製造方法 | |
| WO2015001984A1 (ja) | 光電変換素子モジュールおよびその製造方法 | |
| JP5682189B2 (ja) | 光電変換素子、光電変換素子の製造方法および太陽電池 | |
| JP5621405B2 (ja) | 光電変換素子、光電変換素子の製造方法および太陽電池 | |
| JP5772750B2 (ja) | 光電変換素子および太陽電池 | |
| JP2015012058A (ja) | 光電変換素子およびその製造方法、ならびにそれを用いた太陽電池 | |
| JP2015046298A (ja) | 光電変換素子およびその製造方法、ならびにそれを用いた太陽電池 | |
| JP2012084300A (ja) | 光電変換素子および太陽電池 | |
| JP2013077549A (ja) | 光電変換素子、光電変換素子の製造方法、太陽電池 | |
| JP2015119023A (ja) | 光電変換素子およびその製造方法、ならびにそれを用いた太陽電池 | |
| JP2013149446A (ja) | 光電変換素子およびこれを用いた太陽電池 | |
| JP2014232608A (ja) | 光電変換素子、光電変換素子の製造方法および太陽電池 | |
| WO2014109355A1 (ja) | 光電変換素子の製造方法 | |
| JP5870862B2 (ja) | 光電変換素子、その製造方法およびそれを用いた太陽電池 | |
| JP2016189390A (ja) | 光電変換素子 | |
| JP5673477B2 (ja) | 光電変換素子 | |
| JP5712873B2 (ja) | 光電変換素子およびそれを含む太陽電池 | |
| JP5900175B2 (ja) | 光電変換素子、光電変換素子の製造方法および太陽電池 | |
| JP2013145677A (ja) | 光電変換素子及びこれを含む太陽電池 | |
| JP2014207257A (ja) | 光電変換素子およびその製造方法 | |
| JP5900177B2 (ja) | 色素増感光電変換素子、およびそれを用いた太陽電池 | |
| JP2013186996A (ja) | 光電変換素子及び太陽電池 | |
| JP6160069B2 (ja) | 色素増感型光電変換素子の製造方法 | |
| JP5842774B2 (ja) | 光電変換素子および太陽電池 | |
| JP2015002304A (ja) | 光電変換素子およびその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14737809 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014556433 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14737809 Country of ref document: EP Kind code of ref document: A1 |