WO2011149073A1 - ポリカーボネート樹脂およびそれよりなる透明フィルム - Google Patents
ポリカーボネート樹脂およびそれよりなる透明フィルム Download PDFInfo
- Publication number
- WO2011149073A1 WO2011149073A1 PCT/JP2011/062261 JP2011062261W WO2011149073A1 WO 2011149073 A1 WO2011149073 A1 WO 2011149073A1 JP 2011062261 W JP2011062261 W JP 2011062261W WO 2011149073 A1 WO2011149073 A1 WO 2011149073A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polycarbonate resin
- group
- formula
- dihydroxy compound
- carbon atoms
- Prior art date
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
- C08G64/16—Aliphatic-aromatic or araliphatic polycarbonates
- C08G64/1608—Aliphatic-aromatic or araliphatic polycarbonates saturated
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
- C08G64/02—Aliphatic polycarbonates
- C08G64/0208—Aliphatic polycarbonates saturated
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
- C08G64/16—Aliphatic-aromatic or araliphatic polycarbonates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
- C08G64/20—General preparatory processes
- C08G64/30—General preparatory processes using carbonates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/18—Manufacture of films or sheets
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L69/00—Compositions of polycarbonates; Compositions of derivatives of polycarbonates
-
- G—PHYSICS
- G02—OPTICS
- G02B—OPTICAL ELEMENTS, SYSTEMS OR APPARATUS
- G02B1/00—Optical elements characterised by the material of which they are made; Optical coatings for optical elements
- G02B1/04—Optical elements characterised by the material of which they are made; Optical coatings for optical elements made of organic materials, e.g. plastics
-
- G—PHYSICS
- G02—OPTICS
- G02B—OPTICAL ELEMENTS, SYSTEMS OR APPARATUS
- G02B5/00—Optical elements other than lenses
- G02B5/30—Polarising elements
- G02B5/3083—Birefringent or phase retarding elements
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2369/00—Characterised by the use of polycarbonates; Derivatives of polycarbonates
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Definitions
- the present invention relates to a polycarbonate resin excellent in optical characteristics, hue and thermal stability, and a transparent film comprising the same.
- Polycarbonate resins generally contain bisphenols as monomer components, taking advantage of transparency, heat resistance, mechanical strength, etc., to make electrical and electronic parts, automotive parts, medical parts, building materials, films, sheets, It is widely used as so-called engineering plastics in the fields of bottles, optical recording media, lenses and the like.
- Patent Document 1 describes a polycarbonate resin excellent in optical properties using a bisphenol compound having a fluorene ring in the side chain and 2,2-bis (4-hydroxyphenyl) propane.
- Patent Document 2 describes a polycarbonate resin having a small photoelastic coefficient using a bisphenol compound having a fluorene ring in the side chain and pentacyclodecane dimethanol or isosorbide.
- Patent Document 3 describes a polycarbonate resin using a dihydroxy compound having a fluorene ring in the side chain, tricyclodecane dimethanol and isosorbide.
- Patent Document 4 discloses that a retardation film made of a polycarbonate resin containing a fluorene ring has a low photoelastic coefficient and a reverse wavelength dispersibility that becomes smaller as the retardation becomes shorter. It is disclosed that polycarbonates useful for optical applications such as films can be obtained.
- a polycarbonate resin is produced using a dihydroxy compound having an alcoholic hydroxy group as a raw material, such as the above-mentioned dihydroxy compound having a fluorene ring in the side chain or isosorbide, it is usually a method called a transesterification method or a melting method.
- a transesterification method or a melting method.
- the dihydroxy compound and a carbonic acid diester such as diphenyl carbonate were transesterified at a high temperature in the presence of a basic catalyst, and polymerization was advanced by removing by-product phenol out of the system, thereby obtaining a polycarbonate resin.
- the conventional polycarbonate resin has excellent optical properties in the patent document and the like, since the molecular weight of the resin is very high, when the processing temperature is increased in order to ensure the fluidity of the resin, Appearance defects due to film coloring and the occurrence of burns and silver, etc., on the other hand, when the processing temperature is lowered, there is a problem that the melt viscosity is too high and it becomes difficult to control the thickness of the film. .
- a polycarbonate resin obtained using a dihydroxy compound having an ether bond in the molecule such as isosorbide is inferior in thermal stability to a polycarbonate resin obtained using a monomer having a phenolic hydroxyl group such as bisphenol A. Therefore, coloring may occur during polymerization or molding that is exposed to high temperatures.
- polycarbonate resins made from dihydroxy compounds having a fluorene ring generally have a high melt viscosity, making it difficult to lower the polymerization temperature or lower the processing temperature, and it is also difficult to suppress coloring. there were.
- An object of the present invention is to solve the above-mentioned conventional problems and provide a polycarbonate resin excellent in optical characteristics, opportunity physical properties, hue and thermal stability, and a transparent film comprising the same.
- a polycarbonate resin having a specific ratio of a glass transition temperature, a reduced viscosity, and a phase difference at a specific wavelength has excellent optical properties. It has been found that it has properties, mechanical properties, hue and thermal stability, and has reached the present invention.
- the gist of the present invention resides in the following [1] to [22].
- a ratio (Re450 / Re550) of a retardation (Re450) measured at a wavelength of 450 nm and a retardation (Re550) measured at a wavelength of 550 nm of a film formed by molding the polycarbonate resin is 0.50 or more and 0.00.
- R 1 to R 4 each independently represent a hydrogen atom, a substituted or unsubstituted alkyl group having 1 to 20 carbon atoms, or a substituted or unsubstituted cycloalkyl group having 6 to 20 carbon atoms.
- Group or a substituted or unsubstituted aryl group having 6 to 20 carbon atoms, and the same or different groups are arranged as each of the four substituents on each benzene ring.
- X is a substituted or unsubstituted alkylene group having 2 to 10 carbon atoms, a substituted or unsubstituted cycloalkylene group having 6 to 20 carbon atoms, or a substituted or unsubstituted carbon group having 6 to 20 carbon atoms.
- m and n are each independently an integer of 0 to 5.
- R 1 to R 4 , X, m, and n represent the same group as in the formula (1), and A 1 has a substituent having 1 to 18 carbon atoms.
- the structural unit includes a structural unit derived from a dihydroxy compound having a site represented by the following formula (4) in a part of the structure. ]
- a 1 and A 2 are each independently an aliphatic group which may have a substituent having 1 to 18 carbon atoms, or a substituent having 6 to 18 carbon atoms. Is an aromatic group which may have a group, and A 1 and A 2 may be the same or different.
- Any of [1] to [12], wherein the total content of sodium, potassium and cesium in the polycarbonate resin is 1 ⁇ mol or less per 1 mol of a structural unit derived from a dihydroxy compound constituting the polycarbonate resin.
- the polycarbonate resin as described in any one.
- the total sulfur element content per mol of structural unit derived from the dihydroxy compound constituting the polycarbonate resin in the polycarbonate resin is A [ ⁇ mol], the Group 1 metal element and the second element in the long-period periodic table
- the total content of the compound group represented by the formula (2) relative to the structural unit derived from the dihydroxy compound represented by the formula (1) in the polycarbonate resin is a [ppm]
- the total amount of the long-period periodic table Group 1 metal and Group 2 metal contained in the polycarbonate with respect to 1 mol of the structural unit derived from the dihydroxy compound represented by the formula (1) is b [ ⁇ mol]
- the polycarbonate resin With respect to 1 mol of the structural unit derived from the dihydroxy compound represented by the formula (1) in the polycarbonate resin, at least one selected from the group consisting of a long-period group 2 metal and lithium
- the polycarbonate resin according to any one of [1] to [15], which contains a metal compound in an amount of 0.5 ⁇ mol to 50 ⁇ mol.
- the polycarbonate resin further includes a structural unit derived from at least one compound selected from the group consisting of aliphatic dihydroxy compounds, alicyclic dihydroxy compounds, oxyalkylene glycols, and diols having a cyclic acetal structure.
- the polycarbonate resin according to any one of [1] to [16].
- a dihydroxy compound mixture including a dihydroxy compound represented by the following formula (1), a dihydroxy compound having a portion represented by the following formula (4) in a part of the structure, and the following formula (6):
- the total content of Group 1 metal elements and Group 2 metal elements in the long-period periodic table in the polycarbonate resin is 1 mol of a structural unit derived from a dihydroxy compound constituting the polycarbonate resin.
- the polycarbonate resin characterized by being 20 ⁇ mol or less per hit.
- R 1 to R 4 each independently represent a hydrogen atom, a substituted or unsubstituted alkyl group having 1 to 20 carbon atoms, or a substituted or unsubstituted cycloalkyl group having 6 to 20 carbon atoms.
- Group or a substituted or unsubstituted aryl group having 6 to 20 carbon atoms, and the same or different groups are arranged as each of the four substituents on each benzene ring.
- X is a substituted or unsubstituted alkylene group having 2 to 10 carbon atoms, a substituted or unsubstituted cycloalkylene group having 6 to 20 carbon atoms, or a substituted or unsubstituted carbon group having 6 to 20 carbon atoms.
- m and n are each independently an integer of 0 to 5.
- each of A 1 and A 2 independently represents an aliphatic group which may have a substituent having 1 to 18 carbon atoms, or a substituent having 6 to 18 carbon atoms. It may be an aromatic group, and A 1 and A 2 may be the same or different.
- the ratio (Re450 / Re550) of the phase difference (Re450) measured at a wavelength of 450 nm to the phase difference (Re550) measured at a wavelength of 550 nm is 0.5 or more and 0.93 or less, [21] or [22 ]
- the ratio (Re450 / Re550) of the phase difference (Re450) measured at a wavelength of 450 nm to the phase difference (Re550) measured at a wavelength of 550 nm is 0.5 or more and 0.90 or less, [21] or [22 ]
- the film as described in. [25] The film according to any one of [21] to [24], which has a photoelastic coefficient of 40 ⁇ 10 ⁇ 12 Pa ⁇ 1 or less.
- optical properties not only has excellent optical properties, but also has excellent hue and thermal stability, electric and electronic parts, automotive parts and other injection molding fields, films, sheet fields, bottles, container fields
- lens applications such as camera lenses, viewfinder lenses, CCD and CMOS lenses, retardation films used in liquid crystal and plasma displays, diffusion sheets, polarizing films, and other films, sheets, optical disks, optical materials, optical It becomes possible to provide a polycarbonate resin that can be applied to a wide range of fields such as binder use for immobilizing components, dyes, charge transfer agents and the like.
- FIG. 1 is a drawing showing an LC chart when the total amount of the compound group represented by the formula (2) in the polycarbonate resin of the present invention is analyzed by LC.
- the description of the constituent elements described below is an example (representative example) of an embodiment of the present invention. It is not limited to the contents.
- “weight” is synonymous with “mass”.
- the polycarbonate resin of the present invention includes not only a polymer having a carbonate structure, but also those containing various compounds produced during the production of the polymer, and those obtained by blending various additives with the polymer. It is.
- the polycarbonate resin of the present invention is a polycarbonate resin having a glass transition temperature of 110 ° C. or more and 150 ° C. or less, and a reduced viscosity at 20 ° C. of a methylene chloride solution having a concentration of 0.6 g / dL of 0.30 or more and 0.46 or less,
- the ratio of the retardation (Re450) measured at a wavelength of 450 nm and the retardation (Re550) measured at a wavelength of 550 nm of the film formed by molding the resin is 0.50 or more and 0.93 or less.
- the glass transition temperature of the polycarbonate resin of the present invention is 110 ° C. or higher and 150 ° C. or lower. If the glass transition temperature of the polycarbonate resin is too low, the heat resistance required under the usage environment is insufficient, and the transparency may decrease or the dimensions may change after molding. Further, when used as an optical film, the molecular properties may be relaxed and the optical characteristics may not be maintained, so the temperature is more preferably 115 ° C. or higher, and further preferably 120 ° C. or higher.
- the glass transition temperature of the polycarbonate resin is too high, sufficient mechanical properties cannot be obtained. Furthermore, when the molecular weight is increased in order to improve the mechanical properties, the melt viscosity usually increases. Therefore, it is necessary to increase the temperature during the production of the polycarbonate resin and the temperature during the molding process. It may cause decomposition. Therefore, it is more preferably 148 ° C. or lower, further preferably 146 ° C. or lower.
- the type of the repeating structural unit constituting the polycarbonate resin is adjusted, the polycarbonate resin is used as a copolymer, and the copolymerization ratio of the repeating structural unit is set in the case of the copolymer. It becomes possible by adjusting it.
- the glass transition temperature is measured by the method described later in the examples.
- the polycarbonate resin of the present invention has a reduced viscosity at 20 ° C. of a methylene chloride solution having a concentration of 0.6 g / dL of 0.30 or more and 0.46 or less, but if the reduced viscosity is too low, the mechanical properties decrease, for example, When the toughness is lowered, it becomes difficult to form a thin film such as a film or to stretch the film, so that the thickness is preferably 0.33 or more, more preferably 0.35 or more.
- the reduced viscosity is too high, the fluidity during molding tends to decrease, and there is a tendency to reduce productivity and moldability.
- silver or burnt foreign matter occurs in the molded product during melt processing, resulting in poor appearance. May occur.
- the molecular weight of the resin may be lowered, it is preferably 0.43 or less, more preferably 0.41 or less.
- the ratio of the reduced viscosity of the molded article after melt molding to the reduced viscosity of the polycarbonate resin before molding is preferably 0.95 or more.
- the reduced viscosity is measured by using a Ubbelohde viscometer at a temperature of 20.0 ° C. ⁇ 0.1 ° C., precisely adjusting the polycarbonate concentration to 0.6 g / dL using methylene chloride as a solvent.
- the reduced viscosity of the polycarbonate resin can be adjusted by increasing the molecular weight of the polycarbonate resin or introducing a branched structure into the polycarbonate resin.
- the polycarbonate resin of the present invention is particularly suitable for viewing angle compensation of various displays (liquid crystal display device, organic EL display device, plasma display device, FED field emission display device, SED surface field display device), antireflection of external light, and color compensation. It is useful as an optical compensation film used for, for example, conversion of linearly polarized light into circularly polarized light.
- the ratio (Re450 / Re550) of the retardation (Re450) measured at a wavelength of 450 nm of the film formed by molding the polycarbonate resin to the retardation (Re550) measured at a wavelength of 550 nm is 0.50 or more, 0 .93 or less, more preferably 0.70 or more and 0.91 or less, and particularly preferably 0.85 or more and 0.90 or less.
- the ratio is within this range, ideal phase difference characteristics can be obtained at each wavelength in the visible region.
- a retardation film having such wavelength dependency as a quarter wavelength plate is prepared and bonded to a polarizing plate, whereby a circular polarizing plate or the like can be prepared, and a polarizing plate with less hue wavelength dependency
- a display device can be realized.
- the ratio is out of this range, the wavelength dependence of the hue is increased, optical compensation is not performed at all wavelengths in the visible region, and coloring and contrast due to light passing through the polarizing plate and the display device are lost. Problems such as degradation occur.
- the type of the repeating structural unit constituting the polycarbonate resin is adjusted, the polycarbonate resin is used as a copolymer, or in the case of the copolymer, the repeating structural unit is copolymerized. This is possible by adjusting the ratio.
- the retardation film preferably has an appropriate range of retardation. Since the retardation is determined by the product of the birefringence and the thickness of the film, it is preferable that the birefringence of the film is high in order to make the film thin while having a predetermined retardation.
- the birefringence when the fixed end is stretched 2.0 times under the condition of glass transition temperature + 15 ° C. is preferably 0.001 or more, more preferably 0.0015 or more, particularly 0 0020 or more is preferable.
- the birefringence is less than 0.001, it is necessary to increase the thickness of the film excessively, so the amount of material used increases, and it becomes difficult to control homogeneity in terms of thickness, transparency, and phase difference. . Therefore, when the birefringence is less than 0.001, there is a possibility that the transparent film produced using the polycarbonate resin cannot be adapted to a device that requires precision, thinness, and homogeneity. On the other hand, if the thickness of the film is too thin, handling problems will occur. Therefore, the birefringence is preferably 0.010 or less, more preferably 0.008 or less.
- Birefringence is determined by intrinsic birefringence that depends on the molecular structure of the polycarbonate resin, and processing conditions such as draw ratio and draw temperature.
- the dihydroxy compound constituting the polycarbonate resin preferably has a structure with strong polarization in the main chain direction.
- a flexible structure is incorporated into the polycarbonate resin, and the glass transition temperature and melt viscosity described above are used. It is also important to set the value within an appropriate range.
- the polycarbonate resin of the present invention preferably has a photoelastic coefficient of 40 ⁇ 10 ⁇ 12 Pa ⁇ 1 or less. Furthermore, it is preferably 35 ⁇ 10 ⁇ 12 Pa ⁇ 1 or less, particularly preferably 30 ⁇ 10 ⁇ 12 Pa ⁇ 1 or less. If the photoelastic coefficient is greater than 40 ⁇ 10 ⁇ 12 Pa ⁇ 1 , the transparent film is bonded to a polarizing plate as a retardation film, and when this polarizing plate is further mounted on a display device, due to the stress at the time of bonding.
- melt viscosity In view of the mechanical properties and color tone of the polycarbonate resin and the flowability during melt polymerization and molding, the melt viscosity of the polycarbonate resin needs to be within an appropriate range. If the melt viscosity is too high, the mechanical properties are improved, but a high processing temperature is required, so that it is difficult to suppress coloring and thermal decomposition of the resin.
- the melt viscosity of the polycarbonate resin of the present invention is measured using a capillograph, and is preferably 1500 Pa ⁇ s or more and 3500 Pa ⁇ s or less at a measurement temperature of 240 ° C. and a shear rate of 91.2 sec ⁇ 1 . Furthermore, it is preferably 2000 Pa ⁇ s or more and 3000 Pa ⁇ s or less.
- the polycarbonate resin of the present invention preferably contains a structural unit derived from a dihydroxy compound represented by the following formula (1).
- R 1 to R 4 each independently represent a hydrogen atom, a substituted or unsubstituted alkyl group having 1 to 20 carbon atoms, or a substituted or unsubstituted cycloalkyl group having 6 to 20 carbon atoms.
- Group or a substituted or unsubstituted aryl group having 6 to 20 carbon atoms, and the same or different groups are arranged as each of the four substituents on each benzene ring.
- X is a substituted or unsubstituted alkylene group having 2 to 10 carbon atoms, a substituted or unsubstituted cycloalkylene group having 6 to 20 carbon atoms, or a substituted or unsubstituted carbon group having 6 to 20 carbon atoms.
- m and n are each independently an integer of 0 to 5.
- the dihydroxy compound represented by the formula (1) By including a structural unit derived from the dihydroxy compound represented by the formula (1), it has appropriate optical properties such as retardation, retardation ratio, birefringence and low photoelastic coefficient, and physical properties such as heat resistance and mechanical strength. It becomes easy to make it more preferable.
- the polycarbonate resin of the present invention may be produced by any method, but in the case of producing a dihydroxy compound having an alcoholic hydroxy group as a raw material, melt polycondensation is carried out by transesterification using the dihydroxy compound as a raw material. It is preferable. Therefore, the polycarbonate resin of the present invention is preferably polycondensed by a transesterification reaction using a dihydroxy compound and a carbonic acid diester as raw materials.
- m and n of 1 are preferable because they exhibit excellent mechanical properties and heat resistance. Used for.
- 9,9-bis (4- (2-hydroxyethoxy) phenyl) fluorene is most preferable from the viewpoint of availability and production. These may be used alone or in combination of two or more depending on the required performance of the polycarbonate resin to be obtained.
- the compound represented by the formula (1) may contain a sulfur compound during the production of the compound, and may adversely affect the polymerization process of the polycarbonate. Is preferably 10 ppm or less, more preferably 5 ppm or less. Since the fluorene-based dihydroxy compound has a very high boiling point, it is difficult to purify by distillation. Generally, purification is performed by washing with water, recrystallization, ion exchange resin, activated carbon, or the like. The total amount of sulfur contained can be measured by ion chromatography.
- the polycarbonate of the present invention preferably contains a structural unit derived from a dihydroxy compound other than the dihydroxy compound represented by the formula (1) in order to adjust the desired optical properties.
- the ratio of the dihydroxy compound represented by the formula (1) is preferably 25 mol% or more and 80 mol% or less, more preferably 30 mol% or more and 70 mol%, with respect to the structural units derived from all dihydroxy compounds. It is particularly preferably not more than mol% and not less than 35 mol% and not more than 60 mol%.
- a dihydroxy compound having a site represented by the following formula (3) in a part of the structure (specific dihydroxy) Compound) is preferably used.
- Specific examples include oxyalkylene glycols, dihydroxy compounds having an ether group bonded to an aromatic group, and dihydroxy compounds having a cyclic ether structure.
- Examples of the oxyalkylene glycols include diethylene glycol, triethylene glycol, tetraethylene glycol, polyethylene glycol, and polypropylene glycol.
- dihydroxy compound having an ether group bonded to the aromatic group examples include 2,2-bis (4- (2-hydroxyethoxy) phenyl) propane and 2,2-bis (4- (2-hydroxypropoxy). ) Phenyl) propane, 1,3-bis (2-hydroxyethoxy) benzene, 4,4′-bis (2-hydroxyethoxy) biphenyl, bis (4- (2-hydroxyethoxy) phenyl) sulfone, and the like.
- dihydroxy compound having the cyclic ether structure examples include a dihydroxy compound represented by the following formula (4), a spiro glycol represented by the following formula (6) and the following formula (7), and the like.
- the “cyclic ether structure” of the above “dihydroxy compound having a cyclic ether structure” means a structure having an ether group in the cyclic structure and a structure in which the carbon constituting the cyclic chain is an aliphatic carbon. To do.
- Examples of the dihydroxy compound represented by the above formula (4) include stereosorbent isosorbide (ISB), isomannide, and isoidet, and these may be used alone or in combination of two or more. May be used in combination.
- ISB stereosorbent isosorbide
- isomannide isomannide
- isoidet stereosorbent isosorbide
- the hydroxy compounds represented by the above formulas (4), (6) and (7) are preferred from the viewpoints of availability, handling, reactivity during polymerization, and hue of the resulting polycarbonate.
- a representative dihydroxy compound having a cyclic ether structure is preferable, and a dihydroxy compound having two cyclic ether structures such as a dihydroxy compound represented by the above formula (4) and a spiro glycol represented by the above formula (7) is further included.
- an anhydrous sugar alcohol which is a dihydroxy compound having two sugar-derived cyclic ether structures such as the dihydroxy compound represented by the above formula (4) is particularly preferable.
- dihydroxy compounds it is preferable to use a dihydroxy compound having no aromatic ring structure from the viewpoint of the optical properties of polycarbonate, and among them, it is abundant as a plant-derived resource and is produced from various easily available starches.
- Anhydrosugar alcohols such as dihydroxy compounds represented by the above formula (4) obtained by dehydrating and condensing sorbitol produced are easy to obtain and manufacture, light resistance, optical properties, moldability, heat resistance, carbon neutral From the viewpoint of the above, it is most preferable.
- These specific dihydroxy compounds may be used alone or in combination of two or more according to the required performance of the polycarbonate to be obtained.
- the dihydroxy compound having a bond structure of the above formula (3) may contain a stabilizer such as a reducing agent, an antioxidant, an oxygen scavenger, a light stabilizer, an antacid, a pH stabilizer, and a heat stabilizer. .
- a stabilizer such as a reducing agent, an antioxidant, an oxygen scavenger, a light stabilizer, an antacid, a pH stabilizer, and a heat stabilizer.
- a stabilizer such as a reducing agent, an antioxidant, an oxygen scavenger, a light stabilizer, an antacid, a pH stabilizer, and a heat stabilizer.
- a basic stabilizer since the specific dihydroxy compound of the present invention is easily altered under acidic conditions, it is preferable to include a basic stabilizer.
- Examples of the basic stabilizer include hydroxides, carbonates, phosphates, phosphites, and the like of group 1 or group 2 metals in the long-period periodic table (Nomenclature of Inorganic Chemistry IUPAC Recommendations 2005).
- the content of these basic stabilizers in the dihydroxy compound is not particularly limited, but the dihydroxy compound having the structure represented by the above formula (4) used in the present invention is unstable in an acidic state, and thus the above-mentioned stability. It is preferable to add a stabilizer so that the pH of the aqueous solution of the dihydroxy compound containing the agent is 7 or more. If the amount is too small, there is a possibility that the effect of preventing the alteration of the specific dihydroxy compound may not be obtained. If the amount is too large, the fluorene-based dihydroxy compound or the specific dihydroxy compound may be modified during the polymerization reaction. It is 0.0001% by weight to 1% by weight, preferably 0.001% by weight to 0.1% by weight, based on each dihydroxy compound used in the present invention.
- the specific dihydroxy compound having the structure represented by the formula (4) is easily oxidized by oxygen, moisture is not mixed to prevent decomposition by oxygen during storage or handling during manufacture.
- isosorbide is oxidized, decomposition products such as formic acid are generated.
- polycarbonate is produced using isosorbide containing these decomposition products, it not only causes coloration of the resulting polycarbonate and significantly deteriorates physical properties, but also affects the polymerization reaction, thereby obtaining a high molecular weight polymer. It is not preferable because it may not be possible.
- the polycarbonate of the present invention may contain a structural unit derived from a dihydroxy compound other than the fluorene-based dihydroxy compound and the specific dihydroxy compound (hereinafter may be referred to as “other dihydroxy compound”).
- the dihydroxy compound include a linear aliphatic hydrocarbon dihydroxy compound, a linear branched aliphatic hydrocarbon dihydroxy compound, an alicyclic hydrocarbon dihydroxy compound, and aromatic bisphenols.
- straight-chain aliphatic hydrocarbon dihydroxy compound examples include ethylene glycol, 1,3-propanediol, 1,2-propanediol, 1,4-butanediol, 1,3-butanediol, 1,2 -Butanediol, 1,5-heptanediol, 1,6-hexanediol, 1,10-decanediol, 1,12-dodecanediol and the like.
- dihydroxy compound of the linear branched aliphatic hydrocarbon examples include neopentyl glycol and hexylene glycol.
- Examples of the alicyclic hydrocarbon dihydroxy compound include 1,2-cyclohexanediol, 1,2-cyclohexanedimethanol, 1,3-cyclohexanedimethanol, 1,4-cyclohexanedimethanol, and tricyclodecanedi.
- aromatic bisphenols examples include 2,2-bis (4-hydroxyphenyl) propane, 2,2-bis (4-hydroxy-3,5-dimethylphenyl) propane, and 2,2-bis (4 -Hydroxy-3,5-diethylphenyl) propane, 2,2-bis (4-hydroxy- (3,5-diphenyl) phenyl) propane, 2,2-bis (4-hydroxy-3,5-dibromophenyl) Propane, 2,2-bis (4-hydroxyphenyl) pentane, 2,4'-dihydroxy-diphenylmethane, bis (4-hydroxyphenyl) methane, bis (4-hydroxy-5-nitrophenyl) methane, 1,1- Bis (4-hydroxyphenyl) ethane, 3,3-bis (4-hydroxyphenyl) pentane, 1,1-bis (4-hydroxy) Enyl) cyclohexane, bis (4-hydroxyphenyl) sulfone, 2,4′-dihydroxydiphenylsulfone, bis (4-hydroxyphen
- dihydroxy compounds may be used alone or in combination with the specific dihydroxy compound depending on the required performance of the obtained polycarbonate, and after combining two or more kinds, the fluorene-based dihydroxy compound or the specific dihydroxy compound You may use together.
- a dihydroxy compound having no aromatic ring structure in the molecular structure that is, an aliphatic hydrocarbon dihydroxy compound or an alicyclic hydrocarbon dihydroxy compound is preferable. Also good.
- aliphatic hydrocarbon dihydroxy compounds suitable for the polycarbonate of the present invention include 1,3-propanediol, 1,4-butanediol, 1,5-heptanediol, 1,6-hexanediol and the like.
- a straight-chain aliphatic hydrocarbon dihydroxy compound having 3 to 6 carbon atoms and having hydroxy groups at both ends is preferred.
- 1,2-cyclohexanedimethanol, 1,3-cyclohexanedimethanol, 1,4-cyclohexanedimethanol, and tricyclodecane dimethanol are particularly preferable.
- 2-cyclohexanedimethanol, 1,3-cyclohexanedimethanol, 1,4-cyclohexanedimethanol and other dihydroxy compounds having a cyclohexane structure are particularly preferable.
- the polycarbonate resin of the present invention is subjected to a melt polymerization method, as described later, since it is a reaction under a high temperature and a high vacuum, when a dihydroxy compound having a low boiling point is used for the reaction, the reaction system is left unreacted outside the reaction system. Distilling out makes it difficult to control the copolymer composition of the resulting polycarbonate resin.
- the polycarbonate resin of the present invention can have high physical properties such as optical properties and mechanical properties by controlling the copolymer composition of multiple types of dihydroxy compounds, so the boiling point of the dihydroxy compounds used in the reaction is high, The more difficult the distillation, the easier it is to control the copolymer composition.
- the boiling points at 5 kPa of all dihydroxy compounds constituting the polycarbonate resin are 200 ° C. or higher.
- the polycarbonate of the present invention is preferably obtained by polycondensation by a transesterification reaction using a dihydroxy compound containing the dihydroxy compound represented by the formula (1) and a carbonic acid diester as raw materials.
- Examples of the carbonic acid diester used include those represented by the following formula (5). These carbonic acid diesters may be used alone or in combination of two or more.
- a 1 and A 2 may each independently have an aliphatic group that may have a substituent having 1 to 18 carbon atoms, or a substituent that has 6 to 18 carbon atoms. It is an aromatic group, and A 1 and A 2 may be the same or different.
- Examples of the carbonic acid diester represented by the formula (5) include substituted diphenyl carbonates such as diphenyl carbonate (DPC) and ditolyl carbonate, dimethyl carbonate, diethyl carbonate, and di-t-butyl carbonate. Diphenyl carbonate and substituted diphenyl carbonate are preferable, and diphenyl carbonate is particularly preferable.
- Carbonic acid diesters may contain impurities such as chloride ions, which may hinder the polymerization reaction or worsen the hue of the resulting polycarbonate, and are purified by distillation as necessary. It is preferable to use one.
- the polycarbonate of the present invention is produced by transesterification of a dihydroxy compound containing a fluorene-based dihydroxy compound and a carbonic acid diester represented by the above formula (5) as described above. More specifically, it can be obtained by transesterification and removing by-product monohydroxy compounds and the like out of the system. In this transesterification reaction, polycondensation is carried out in the presence of the transesterification reaction catalyst.
- the transesterification reaction catalyst that can be used in the production of the polycarbonate of the present invention (hereinafter sometimes simply referred to as catalyst or polymerization catalyst). Can greatly affect the reaction rate and the color tone of the polycarbonate obtained by polycondensation.
- the catalyst used is not limited as long as it can satisfy the transparency, hue, heat resistance, thermal stability, and mechanical strength of the produced polycarbonate.
- Examples include basic compounds such as Group 2 (hereinafter simply referred to as “Group 1” and “Group 2”) metal compounds, basic boron compounds, basic phosphorus compounds, basic ammonium compounds, and amine compounds. .
- Group 1 metal compounds and / or Group 2 metal compounds are used.
- Examples of the Group 1 metal compound include sodium hydroxide, potassium hydroxide, lithium hydroxide, cesium hydroxide, sodium hydrogen carbonate, potassium hydrogen carbonate, lithium hydrogen carbonate, cesium hydrogen carbonate, sodium carbonate, potassium carbonate, and carbonic acid. Lithium, cesium carbonate, sodium acetate, potassium acetate, lithium acetate, cesium acetate, sodium stearate, potassium stearate, lithium stearate, cesium stearate, sodium borohydride, potassium borohydride, lithium borohydride, hydrogenated Cesium boron, sodium borohydride, potassium phenyl boronate, lithium phenyl boronide, cesium phenyl borohydride, sodium benzoate, potassium benzoate, lithium benzoate, cesium benzoate, hydrogen phosphate Sodium, 2 potassium hydrogen phosphate, 2 lithium hydrogen phosphate, 2 cesium hydrogen phosphate, 2 sodium phenyl phosphate, 2 potassium phenyl phosphate, 2 lithium
- Examples of the Group 2 metal compound include calcium hydroxide, barium hydroxide, magnesium hydroxide, strontium hydroxide, calcium hydrogen carbonate, barium hydrogen carbonate, magnesium hydrogen carbonate, strontium hydrogen carbonate, calcium carbonate, barium carbonate, carbonic acid.
- a basic compound such as a basic boron compound, a basic phosphorus compound, a basic ammonium compound, and an amine compound can be used in combination with the aforementioned Group 1 metal compound and / or Group 2 metal compound.
- a basic compound such as a basic boron compound, a basic phosphorus compound, a basic ammonium compound, and an amine compound
- Examples of the basic phosphorus compound include triethylphosphine, tri-n-propylphosphine, triisopropylphosphine, tri-n-butylphosphine, triphenylphosphine, tributylphosphine, and quaternary phosphonium salts.
- Examples of the basic ammonium compound include tetramethylammonium hydroxide, tetraethylammonium hydroxide, tetrapropylammonium hydroxide, tetrabutylammonium hydroxide, trimethylethylammonium hydroxide, trimethylbenzylammonium hydroxide, trimethylphenylammonium hydroxide.
- Triethylmethylammonium hydroxide triethylbenzylammonium hydroxide, triethylphenylammonium hydroxide, tributylbenzylammonium hydroxide, tributylphenylammonium hydroxide, tetraphenylammonium hydroxide, benzyltriphenylammonium hydroxide, methyltriphenylammonium Hydroxide, butyl triphenyl ammonium hydroxide, and the like.
- Examples of the amine compound include 4-aminopyridine, 2-aminopyridine, N, N-dimethyl-4-aminopyridine, 4-diethylaminopyridine, 2-hydroxypyridine, 2-methoxypyridine, and 4-methoxypyridine.
- the amount of the polymerization catalyst used is usually 0.1 ⁇ mol to 300 ⁇ mol, preferably 0.5 ⁇ mol to 100 ⁇ mol, per 1 mol of all dihydroxy compounds used in the polymerization.
- the amount of metal is usually 0.1 ⁇ mol or more, preferably 0.3 ⁇ mol or more, particularly preferably 0.5 ⁇ mol or more, per 1 mol of the dihydroxy compound.
- the upper limit is usually 40 ⁇ mol, preferably 30 ⁇ mol, more preferably 20 ⁇ mol.
- the specific dihydroxy compound represented by the formula (1) of the present invention may contain sulfur due to, for example, those derived from the catalyst used in production, and deactivate the polymerization catalyst.
- the polymerization catalyst actually used may need to be used in excess of the above range.
- the ratio of Q to P (Q / P) is 0.1 or more. It is preferable that it is 2 or less.
- the total sulfur element content per mol of all dihydroxy compounds is A [ ⁇ mol]
- the total content of group 1 metal elements and group 2 metal elements in the long-period periodic table is B [ [mu] mol]
- the ratio of B to A (B / A) is preferably 0.1 or more and 2 or less.
- the amount of the catalyst is too small, the polymerization rate is slowed down. Therefore, in order to obtain a polycarbonate having a desired molecular weight, the polymerization temperature must be increased accordingly. For this reason, there is a high possibility that the hue of the resulting polycarbonate will deteriorate, and the unreacted raw material may volatilize during the polymerization, causing the molar ratio of the dihydroxy compound and the carbonic acid diester to collapse and not reaching the desired molecular weight. is there. On the other hand, if the amount of the polymerization catalyst used is too large, undesirable side reactions may occur, which may lead to deterioration of the hue of the resulting polycarbonate and coloring of the resin during molding.
- the metal component contained in the polycarbonate resin may cause the resin to be colored during the polymerization reaction or molding process.
- Dihydroxy compounds and carbonic acid diesters used in the polycarbonate resin of the present invention are sufficiently refined because the metal components can be mixed not only from the aforementioned polymerization catalyst but also from raw materials, reactors, and the environment. It is necessary to avoid contamination of metal components even in the polycarbonate production process.
- the content of the Group 2 metal and lithium in the polycarbonate resin of the present invention is preferably 50 ⁇ mol or less, more preferably 30 ⁇ m or less, and particularly preferably 20 ⁇ mol or less with respect to 1 mol of the fluorene-based dihydroxy compound.
- a minimum polymerization catalyst is added, so the lower limit is 0.1 ⁇ mol, more preferably 0.5 ⁇ mol.
- the Group 1 metals sodium, potassium, and cesium may adversely affect the hue if they are contained in a large amount in the polycarbonate. And these metals may mix not only from the catalyst to be used but from a raw material or a reaction apparatus. Regardless of the source, the total amount of these metal compounds in the polycarbonate is preferably 1 ⁇ mol or less, and preferably 0.5 ⁇ mol or less, relative to 1 mol of the fluorene-based dihydroxy compound as the metal amount.
- the polycarbonate resin of the present invention preferably contains a compound group represented by the following formula (2) in a total amount of 400 ppm or less.
- R 1 to R 4 , X, m, and n represent the same group as in the formula (1), and A 1 has a substituent having 1 to 18 carbon atoms.
- a 1 often has the same structure as A 1 in the carbonic acid diester represented by the formula (5).
- the formula (2 ) Is composed of a plurality of compound groups.
- the total amount of these compound groups is preferably 400 ppm based on the structure derived from the dihydroxy compound represented by the formula (1) in the polycarbonate resin of the present invention.
- the total amount of the compound group represented by the said Formula (2) with respect to the structure originating in the dihydroxy compound represented by the said Formula (1) in polycarbonate resin is represented by a [ppm] and the said Formula (1).
- the ratio of a to b (a / b) is 20 or less.
- it is.
- it is preferably 15 or less, particularly 12 or less. This is because a polycarbonate resin having more excellent color tone and thermal stability can be obtained by setting a / b to 20 or less.
- the amount of metal contained in the polycarbonate resin is measured by ICP-MS. From the content of the dihydroxy compound in the polycarbonate resin measured by liquid chromatography and the total amount of the compound group represented by the formula (2), it is represented by the formula (1) in the polycarbonate resin. The amount of metal with respect to the structure derived from the dihydroxy compound can be calculated.
- the polycarbonate resin of the present invention can be usually obtained by polycondensing a dihydroxy compound and a carbonic acid diester represented by the above formula (5) by an ester exchange reaction in the presence of a catalyst.
- the polycarbonate resin of the present invention includes a dihydroxy compound containing the dihydroxy compound represented by the formula (1), a dihydroxy compound having a site represented by the formula (3) in a part of the structure, and the formula
- the carbonic acid diester represented by (5) is obtained by polycondensation in the presence of a catalyst that is a compound containing at least one metal selected from the group consisting of Group 2 metals and lithium in the long-period periodic table.
- Polycarbonate resin, wherein the total content of Group 1 metal elements and Group 2 metal elements in the long-period periodic table in the polycarbonate resin is 20 ⁇ mol or less per 1 mol of the dihydroxy compound constituting the polycarbonate resin A resin is preferred.
- the raw material dihydroxy compound and carbonic acid diester are uniformly mixed before the transesterification reaction.
- the temperature for uniformly mixing is usually 80 ° C. or higher, preferably 90 ° C. or higher, and the upper limit is usually 250 ° C., preferably 200 ° C., more preferably 150 ° C. Among these, 100 ° C. or higher and 120 ° C. or lower is preferable. If the mixing temperature is too low, the dissolution rate may be slow or the solubility may be insufficient, often causing problems such as solidification. If the mixing temperature is too high, the dihydroxy compound may be thermally deteriorated, and the hue of the resulting polycarbonate resin may be deteriorated.
- the operation of mixing the dihydroxy compound and the carbonic acid diester represented by the formula (5) is an oxygen concentration of 10% by volume or less, more preferably 0.0001% by volume to 10% by volume, and more preferably 0.0001% by volume to 5% by volume. In particular, it is preferably performed in an atmosphere of 0.0001% by volume to 1% by volume from the viewpoint of preventing hue deterioration.
- the carbonic acid diester represented by the above formula (5) is preferably used in a molar ratio of 0.90 to 1.20, more preferably to the dihydroxy compound used in the reaction. Is a molar ratio of 0.95 to 1.10.
- the terminal hydroxyl group of the produced polycarbonate resin is increased, the thermal stability of the polymer is deteriorated, coloring occurs at the time of molding, the rate of transesterification reaction is decreased, and the desired high molecular weight.
- the body may not be obtained.
- the rate of transesterification may be reduced, or it may be difficult to produce a polycarbonate having a desired molecular weight.
- the decrease in the transesterification reaction rate may increase the thermal history during the polymerization reaction and may deteriorate the hue of the resulting polycarbonate resin.
- the amount of residual carbonic acid diester in the obtained polycarbonate resin is increased, which causes odor during molding. Or the amount of deposits on the mold may increase.
- the concentration of the carbonic acid diester remaining in the polycarbonate resin of the present invention is preferably 80 ppm by weight or less, more preferably 70 ppm by weight or less, and particularly preferably 60 ppm by weight or less.
- the polycarbonate resin may contain unreacted carbonic acid diester, and the lower limit of the concentration is usually 1 ppm by weight.
- the method of polycondensing a dihydroxy compound and a carbonic acid diester is usually carried out in multiple stages using a plurality of reactors in the presence of the above-mentioned catalyst.
- the type of reaction may be any of batch type, continuous type, or a combination of batch type and continuous type.
- the initial stage of polymerization it is preferable to obtain a prepolymer at a relatively low temperature and low vacuum, and in the latter stage of polymerization, it is preferable to increase the molecular weight to a predetermined value at a relatively high temperature and high vacuum, but the jacket temperature at each molecular weight stage.
- Appropriate selection of the internal temperature and pressure in the reaction system is important for stable reaction and from the viewpoint of hue. For example, if either the temperature or the pressure is changed too quickly before the polymerization reaction reaches a predetermined value, the unreacted monomer will be distilled, causing the molar ratio of the dihydroxy compound and the carbonic acid diester to change, resulting in a decrease in the polymerization rate. Or a polymer having a predetermined molecular weight or terminal group may not be obtained.
- the temperature of the refrigerant introduced into the reflux condenser can be appropriately selected according to the monomer used. Usually, the temperature of the refrigerant introduced into the reflux condenser is preferably 45 ° C. to 180 ° C. at the inlet of the reflux condenser, more preferably 80 ° C. to 150 ° C., particularly preferably 100 ° C. to 130 ° C. is there.
- the temperature of the refrigerant introduced into the reflux condenser is too high, the amount of reflux is reduced and the effect is reduced.
- the temperature is too low, the distillation efficiency of the monohydroxy compound to be originally distilled tends to be lowered.
- the refrigerant for example, hot water, steam, heat medium oil or the like is used, and steam or heat medium oil is preferable.
- the polycarbonate resin of the present invention is preferably produced by polymerization in a plurality of stages using a plurality of reactors using a catalyst.
- the reason for carrying out the polymerization in a plurality of reactors is that the reaction liquid is used at the initial stage of the polymerization reaction. Since there are many monomers contained therein, it is important to suppress the volatilization of the monomers while maintaining the necessary polymerization rate. Further, in the latter stage of the polymerization reaction, it is important to sufficiently distill off the by-produced monohydroxy compound in order to shift the equilibrium to the polymerization side. Thus, in order to set different polymerization reaction conditions, it is preferable from the viewpoint of production efficiency to use a plurality of polymerization reactors arranged in series.
- the number of reactors used in the method of the present invention may be at least two or more. However, from the viewpoint of production efficiency, three or more are preferable, more preferably 3 to 5, and particularly preferably. Is four.
- a plurality of reaction stages having different conditions may be provided in the reactor, or the temperature and pressure may be continuously changed.
- the polymerization catalyst can be added to the raw material preparation tank, the raw material storage tank, or can be added directly to the polymerization tank. From the viewpoint of supply stability and polymerization control, the polymerization catalyst is supplied to the polymerization tank.
- a catalyst supply line is installed in the middle of the raw material line before being fed, and preferably supplied as an aqueous solution. If the temperature of the polymerization reaction is too low, the productivity is lowered and the thermal history of the product is increased. If it is too high, not only the monomer is volatilized but also decomposition and coloring of the polycarbonate resin may be promoted.
- the first stage reaction is preferably performed at a maximum internal temperature of the polymerization reactor of 130 ° C. to 270 ° C., more preferably 150 ° C. to 240 ° C., and even more preferably 180 ° C. to 230 ° C.
- the monohydroxy compound is distilled out of the reaction system.
- the pressure in the reaction system is gradually reduced from the pressure in the first stage, and the monohydroxy compound that is subsequently generated is removed from the reaction system.
- the pressure is set to 200 Pa or less
- the maximum internal temperature is preferably 200 ° C. to 270 ° C., more preferably 220 ° C. to 260 ° C., usually 0.1 hour to 10 hours, preferably 1 hour to 6 hours. Particularly preferably, it is carried out for 0.5 to 3 hours.
- the maximum internal temperature in all reaction steps is less than 260 ° C., particularly 220 ° C. to 240 ° C.
- the monohydroxy compound produced as a by-product is preferably reused as a raw material for diphenyl carbonate, bisphenol A, etc. after purification as necessary from the viewpoint of effective resource utilization.
- the polycarbonate resin of the present invention is usually cooled and solidified after polycondensation as described above, and pelletized with a rotary cutter or the like.
- the method of pelletization is not limited, but it is extracted from the final polymerization reactor in a molten state, cooled and solidified in the form of a strand, and pelletized, or from the final polymerization reactor in a molten state, uniaxial or biaxial extrusion.
- the resin is supplied to the machine, melt-extruded, cooled and solidified into pellets, or extracted from the final polymerization reactor in a molten state, cooled and solidified in the form of strands, once pelletized, and then uniaxially again
- a method may be mentioned in which resin is supplied to a biaxial extruder, melt-extruded, cooled and solidified, and pelletized.
- the residual monomer under reduced pressure devolatilization and generally known heat stabilizers, neutralizers, UV absorbers, mold release agents, colorants, antistatic agents, lubricants, lubricants, A plasticizer, a compatibilizer, a flame retardant, etc. can be added and kneaded.
- the melt kneading temperature in the extruder depends on the glass transition temperature and molecular weight of the polycarbonate resin, but is usually preferably from 150 ° C to 300 ° C, more preferably from 200 ° C to 270 ° C, still more preferably from 230 ° C to 260 ° C. ° C.
- the melt kneading temperature is usually preferably from 150 ° C to 300 ° C, more preferably from 200 ° C to 270 ° C, still more preferably from 230 ° C to 260 ° C. ° C.
- the polycarbonate resin of the present invention is produced using a substituted diphenyl carbonate such as diphenyl carbonate or ditolyl carbonate as the carbonic acid diester represented by the general formula (5), phenol and substituted phenol are by-produced in the polycarbonate resin.
- phenol and substituted phenol may cause odor during molding and coloring during high temperature exposure.
- the polycarbonate resin contains an aromatic monohydroxy compound having an aromatic ring such as by-product phenol of 1000 ppm by weight or more after a normal batch reaction, but a horizontal reactor or vacuum with excellent devolatilization performance.
- an aromatic monohydroxy compound having an aromatic ring such as by-product phenol of 1000 ppm by weight or more after a normal batch reaction, but a horizontal reactor or vacuum with excellent devolatilization performance.
- a vented extruder it is preferably 700 ppm by weight or less, more preferably 500 ppm by weight or less, and particularly preferably 300 ppm by weight or less.
- the lower limit of the content of the aromatic monohydroxy compound is usually 1 ppm by weight.
- aromatic monohydroxy compounds may naturally have a substituent depending on the raw material to be used, and may have, for example, an alkyl group having 5 or less carbon atoms.
- the filter installation position is preferably on the downstream side of the extruder, and the foreign matter removal size (opening) of the filter is preferably 100 ⁇ m or less as the filtration accuracy for 99% removal. In particular, in the case of disagreeing with the entry of minute foreign matters for film use etc., it is preferably 40 ⁇ m or less, more preferably 20 ⁇ m or less.
- Extrusion of the polycarbonate resin of the present invention is preferably performed in a clean room having a higher degree of cleanliness than class 7, more preferably higher than class 6, as defined in JIS B9920 (2002) in order to prevent foreign matter from being mixed after extrusion. It is preferable.
- the extruded polycarbonate resin is cooled to form chips, it is preferable to use a cooling method such as air cooling or water cooling.
- the air used for air cooling is preferably air from which foreign matters in the air have been removed in advance with a hepa filter or the like to prevent reattachment of foreign matters in the air.
- the opening of the filter to be used is preferably 10 ⁇ m to 0.45 ⁇ m as 99% removal filtration accuracy.
- the polycarbonate resin of the present invention thus produced can be blended with a heat stabilizer in order to prevent a decrease in molecular weight and a deterioration in hue during molding.
- heat stabilizer examples include phosphorous acid, phosphoric acid, phosphonous acid, phosphonic acid, and esters thereof. Specific examples include triphenyl phosphite and tris (nonylphenyl) phosphite.
- heat stabilizers may be used alone or in combination of two or more.
- the blending amount of these heat stabilizers is preferably 0.0001 to 1 part by weight, more preferably 0.0005 to 0.5 part by weight, based on 100 parts by weight of the polycarbonate resin, and 0.001 Part by weight to 0.2 part by weight is more preferable.
- the polycarbonate resin of the present invention can be blended with an antioxidant generally known for the purpose of preventing oxidation.
- antioxidants examples include pentaerythritol tetrakis (3-mercaptopropionate), pentaerythritol tetrakis (3-lauryl thiopropionate), glycerol-3-stearyl thiopropionate, triethylene glycol bis [ 3- (3-tert-butyl-5-methyl-4-hydroxyphenyl) propionate], 1,6-hexanediol-bis [3- (3,5-di-tert-butyl-4-hydroxyphenyl) propionate] Pentaerythritol-tetrakis [3- (3,5-di-tert-butyl-4-hydroxyphenyl) propionate], octadecyl-3- (3,5-di-tert-butyl-4-hydroxyphenyl) propionate, , 3,5-trimethyl-2, , 6-Tris (3,5-di-tert-butyl-4-hydroxybenzyl) benz
- the blending amount of these antioxidants is preferably 0.0001 to 0.5 parts by weight when polycarbonate is 100 parts by weight.
- Polycarbonate resin and the above additives are mixed with a tumbler, super mixer, floater, etc., or at the same time or in any order, single-screw / special single-screw / double-screw extruder, V-type blender, Nauta mixer, It can be manufactured by melt-kneading with a Banbury mixer, a kneading roll or the like.
- the melt kneading temperature of the extruder depends on the glass transition temperature and molecular weight of the polycarbonate resin, it is usually 150 ° C. to 300 ° C., preferably 200 ° C. to 270 ° C.
- the melt-kneading temperature is lower than 150 ° C., the melt viscosity of the polycarbonate resin is high, the load on the extruder is increased, and the productivity is lowered.
- the temperature is higher than 300 ° C., the thermal deterioration of the polycarbonate becomes severe, which causes a decrease in mechanical strength due to a decrease in molecular weight, coloring, and gas generation.
- the above-mentioned heat stabilizer or antioxidant is added to the monomer or prevents coloring during polymerization in order to improve the storage stability of the monomer.
- the above-mentioned heat stabilizer or antioxidant is added to the monomer or prevents coloring during polymerization in order to improve the storage stability of the monomer.
- it can be added during polymerization.
- melt production method melt extrusion method
- the melt extrusion method is preferable from the viewpoint of productivity.
- a method of extruding a resin using a T die and sending it to a cooling roll is preferably used.
- the melting temperature at this time is determined by the molecular weight, Tg, melt flow characteristics, etc. of the polycarbonate, but is preferably in the range of 180 ° C. to 320 ° C., more preferably in the range of 200 ° C. to 300 ° C. If the temperature is too high, problems such as coloring due to thermal degradation, appearance defects due to the generation of foreign matter and silver, and die lines from the T die are likely to occur. If the temperature is too low, the viscosity increases, and polymer orientation and stress strain tend to remain.
- the retardation value of the formed film is preferably 20 nm or less, more preferably 10 nm or less.
- the retardation value of the film is larger than this, it is not preferable because when the film is stretched to obtain a retardation film, the dispersion of the retardation value in the film surface increases.
- a solution casting method can also be used as a film production method.
- the solvent for example, methylene chloride, 1,2-dichloroethane, 1,1,2,2-tetrachloroethane, dioxolane, dioxane and the like are preferably used.
- the amount of residual solvent in the film obtained by the solution casting method is preferably 2% by weight or less, more preferably 1% by weight or less. When the amount of residual solvent is 2% by weight or more, the glass transition temperature of the film is remarkably lowered, which is not preferable from the viewpoint of heat resistance.
- the thickness of the film of the present invention is preferably in the range of 30 ⁇ m to 400 ⁇ m, more preferably in the range of 40 ⁇ m to 300 ⁇ m. In the case where such a film is further stretched to obtain a retardation film, a desired retardation value and thickness of the retardation film may be taken into consideration and appropriately determined within the above range.
- the film thus obtained can be made into a retardation film by being stretched and oriented.
- a known method such as longitudinal uniaxial stretching, lateral uniaxial stretching using a tenter or the like, or simultaneous biaxial stretching or sequential biaxial stretching in combination thereof can be used. Although it may be performed batchwise, it is preferable in terms of productivity to be performed continuously. Further, a continuous retardation film with less variation in retardation within the film surface can be obtained compared to a batch system.
- the stretching temperature is preferably in the range of (Tg-20 ° C) to (Tg + 30 ° C), more preferably in the range of (Tg-10 ° C) to (Tg + 20 ° C) with respect to the glass transition temperature of the polycarbonate.
- the draw ratio is determined by the target retardation value, it is preferably from 1.05 to 4 times, more preferably from 1.1 to 3 times in the vertical and horizontal directions.
- the film formed by molding the polycarbonate resin in the present invention preferably has a birefringence of 0.001 or more, and more preferably 0.0014 or more. If the birefringence is excessively small, when a retardation film is used, in order to develop the same retardation, the film thickness must be increased, which may not be suitable for thin devices.
- the said birefringence is the value which measured the film which carried out fixed uniaxial stretching at the glass transition temperature +15 degreeC extending
- the ratio (Re450 / Re550) of the retardation (Re450) measured at a wavelength of 450 nm to the retardation (Re550) measured at a wavelength of 550 nm is more preferably 0.50 or more and 0.93 or less. Preferably, it is 0.70 or more and 0.90 or less, more preferably 0.85 or more and 0.90 or less.
- a retardation film may be used, and when it is attached to a polarizing plate, image quality may be deteriorated.
- the transparent film of the present invention preferably has a photoelastic coefficient of 40 ⁇ 10 ⁇ 12 Pa ⁇ 1 or less, more preferably 35 ⁇ 10 ⁇ 12 Pa ⁇ 1 or less, particularly 30 ⁇ 10 ⁇ 12 Pa. ⁇ 1 or less is preferable.
- a photoelastic coefficient is excessively large, when a retardation film is used, there is a possibility that image quality is deteriorated such that the periphery of the screen is blurred in white when pasted to a polarizing plate.
- the film of the present invention can be used as a retardation plate for various liquid crystal display devices.
- the retardation value is generally selected in the range from 400 nm to 2000 nm. Moreover, when using the retardation film of this invention as a half-wave plate, the retardation value is selected in the range of 200 nm to 400 nm. When the film of the present invention is used as a quarter-wave plate, the retardation value is selected in the range from 90 nm to 200 nm. A more preferable retardation value as a quarter wavelength plate is from 100 nm to 180 nm.
- the film of the present invention can be used alone, in combination of two or more, or in combination with other films.
- the film of the present invention can be laminated and bonded via a known iodine-based or dye-based polarizing plate and an adhesive.
- a known iodine-based or dye-based polarizing plate and an adhesive When laminating, it is necessary to laminate the polarizing axis of the polarizing plate and the slow axis of the film at a specific angle depending on the application.
- the film of the present invention can be used as a circularly polarizing plate by forming a quarter wave plate and laminating and laminating this with a polarizing plate.
- the polarizing axis of the polarizing plate and the slow axis of the film are laminated while maintaining a relative angle of substantially 45 °.
- the film of the present invention may be laminated using a polarizing protective film constituting a polarizing plate.
- the retardation film of the present invention can be used as a color compensation plate for an STN liquid crystal display device, and can be used as an elliptically polarizing plate by laminating and laminating it with a polarizing plate.
- the polycarbonate resin of the present invention has low birefringence, excellent heat resistance and moldability, and also has hue and transparency, so that it can be used for other optical films, optical disks, optical prisms, pickup lenses, and the like.
- BHEPF 9,9-bis (4- (2-hydroxyethoxy) -phenyl) fluorene (Osaka Gas Chemical Co., Ltd.)
- ISB Isosorbide (Rocket Fleure, trade name: POLYSORB PS)
- CHDM 1,4-cyclohexanedimethanol (manufactured by Shin Nippon Rika Co., Ltd.)
- TCDDM Tricyclodecane dimethanol (Oxea)
- DEG Diethylene glycol (Mitsubishi Chemical Corporation)
- PEG # 1000 Polyethylene glycol number average molecular weight 1000 (manufactured by Sanyo Chemical Co., Ltd.)
- DPC Diphenyl carbonate (Mitsubishi Chemical Corporation)
- the measuring method of the physical property of polycarbonate resin, the evaluation of the characteristic of polycarbonate resin, and the confirmation method of the manufacturing conditions of polycarbonate resin were performed with the following method.
- the glass transition temperature of the polycarbonate resin was measured using a differential scanning calorimeter (DSC6220 manufactured by SII Nanotechnology). About 10 mg of a polycarbonate resin sample was put in an aluminum pan manufactured by the same company and sealed, and the temperature was raised from room temperature to 250 ° C. at a temperature rising rate of 20 ° C./min under a nitrogen stream of 50 mL / min. After maintaining the temperature for 3 minutes, it was cooled to 30 ° C. at a rate of 20 ° C./min. The temperature was maintained at 30 ° C. for 3 minutes, and the temperature was increased again to 200 ° C. at a rate of 20 ° C./min. From the DSC data obtained at the second temperature increase, the extrapolated glass transition start temperature was adopted.
- Quantifying the dihydroxy compound represented by Formula (1) and the component represented by Formula (2), and calculating the content of the component represented by Formula (2) with respect to the dihydroxy compound represented by Formula (1) did.
- a calibration curve was prepared using BHEPF and quantified by the absolute calibration curve method.
- the component represented by the formula (2) was also quantified by a calibration curve using BHEPF, the content of the component is not the absolute value of the content of the compound group represented by the formula (2), but is converted to BHEPF It is a numerical value.
- the LC chart is shown in FIG.
- the structural unit ratio derived from each dihydroxy compound in the polycarbonate resin was obtained by weighing 30 mg of the polycarbonate resin and dissolving it in about 0.7 mL of heavy chloroform. The solution was put into an NMR tube having an inner diameter of 5 mm, and a 1 H NMR spectrum was measured at room temperature using JNM-AL400 (resonance frequency 400 MHz) manufactured by JEOL. The structural unit ratio derived from each dihydroxy compound was determined from the signal intensity ratio based on the structural unit derived from each dihydroxy compound.
- the equipment and conditions used are as follows. ⁇ Equipment: JNM-AL400 manufactured by JEOL Ltd. (resonance frequency 400 MHz) ⁇ Measurement temperature: normal temperature ⁇ Relaxation time: 6 seconds ⁇ Integration count: 128 times
- the hue of the polycarbonate resin was evaluated by measuring the yellow index (YI) value in the reflected light of the pellet in accordance with ASTM D1925.
- YI yellow index
- ASTM D1925 As the apparatus, a spectrocolorimeter CM-5 manufactured by Konica Minolta Co., Ltd. was used, and the measurement conditions were a measurement diameter of 30 mm and SCE.
- a petri dish calibration glass CM-A212 was fitted into the measurement part, and a zero calibration box CM-A124 was placed thereon to perform zero calibration, followed by white calibration using a built-in white calibration plate.
- the measurement of the pellet was performed by putting the pellet to a depth of 30 mm or more in a cylindrical glass container having an inner diameter of 30 mm and a height of 50 mm. The operation of taking out the pellet from the glass container and then performing the measurement again was repeated twice, and the average value of the measurement values of three times in total was used. The smaller the YI value, the better the quality without yellowness.
- Measurement of melt viscosity of polycarbonate resin Measurement was performed with a capillary rheometer (manufactured by Toyo Seiki Co., Ltd.) using a polycarbonate resin sample that had been vacuum-dried at 80 ° C for 5 hours. By heating to the same temperature as the reaction temperature, the melt viscosity was measured at a shear rate of 9.12 to 1824 sec ⁇ 1 , and the value of the melt viscosity at 91.2 sec ⁇ 1 was used. An orifice having a die diameter of ⁇ 1 mm ⁇ 10 mmL was used.
- the emitted laser light is passed through the polarizer, sample, compensator, and analyzer in this order, picked up by a photodetector (photodiode), and passed through a lock-in amplifier with respect to the amplitude and distortion of the waveform of angular frequency ⁇ or 2 ⁇ .
- the phase difference was determined, and the strain optical coefficient O ′ was determined.
- the directions of the polarizer and the analyzer were orthogonal to each other, and each was adjusted so as to form an angle of ⁇ / 4 with respect to the extending direction of the sample.
- the photoelastic coefficient (C) was obtained from the following equation using the storage elastic modulus E ′ and the strain optical coefficient O ′.
- C O '/ E'
- Phase difference of hot press film and wavelength dispersion of phase difference A sample having a width of 6 cm and a length of 6 cm was cut out from the film obtained by the above-described hot press.
- This sample was batch-biaxially stretched (manufactured by Toyo Seiki Co., Ltd.), the stretching temperature was the glass transition temperature of polycarbonate resin + 15 ° C., the stretching speed was 720 mm / min (strain speed 1200% / min), and the stretching ratio was 2. Uniaxial stretching of 0 times was performed. At this time, it extended
- phase difference measuring device manufactured by Oji Scientific Instruments. Dispersibility was measured.
- the ratio (Re450 / Re550) of the phase differences Re450 and Re550 measured at 450 nm and 550 nm was calculated. If the phase difference ratio is greater than 1, the chromatic dispersion is positive, and if it is less than 1, it is negative. It is shown that the smaller the ratio of the respective phase differences is less than 1, the stronger the negative wavelength dispersion.
- phase difference measuring device manufactured by Oji Scientific Instruments. Dispersibility was measured.
- the ratio (Re450 / Re550) of the phase differences Re450 and Re550 measured at 450 nm and 550 nm was calculated. If the phase difference ratio is greater than 1, the chromatic dispersion is positive, and if it is less than 1, it is negative. The smaller the phase difference ratio is less than 1, the stronger the negative wavelength dispersion.
- the phenol vapor produced as a by-product with the polymerization reaction is led to a reflux condenser at 100 ° C., and a monomer component contained in a small amount in the phenol vapor is returned to the polymerization reactor (1), and the phenol vapor not condensed is led to a condenser at 45 ° C. And recovered.
- the oligomerized contents in the polymerization reactor (1) were further provided with a stirring blade and a reflux condenser controlled at 100 ° C. Transferred to the polymerization reactor (2). Subsequently, the temperature increase and pressure reduction in the polymerization reaction apparatus (2) were started, and the internal temperature was 240 ° C. and the pressure was 200 Pa in 50 minutes. Thereafter, the pressure was reduced to 200 Pa or less over 20 minutes, the pressure was restored when the predetermined stirring power was reached, the contents were extracted in the form of strands, and pelletized with a rotary cutter.
- the polycarbonate resin thus obtained had a reduced viscosity of 0.352 dL / g, a glass transition temperature of 145 ° C., and a YI of 26. Thus, a resin having a good color tone was obtained.
- the copolymer composition of the polycarbonate resin was almost as prepared.
- Example 1-1 shows the physical properties and evaluation results of the obtained polycarbonate resin.
- the reduced viscosity was 0.389 dL / g
- the glass transition temperature was 131 ° C.
- a resin having a good color tone was obtained.
- the flexibility of the polycarbonate resin was higher than that of Example 1, and the melt processability was excellent.
- polydiol such as polyethylene glycol
- the procedure was the same as Example 1-1, except that the amount was adjusted to /6.00 ⁇ 10 ⁇ 6 .
- Table 1 shows the physical properties and evaluation results of the obtained polycarbonate resin.
- a polycarbonate resin having a good color tone was obtained and the melt processability was excellent, but the birefringence was slightly small.
- the procedure was the same as Example 1-1, except that the amount was adjusted to /6.00 ⁇ 10 ⁇ 6 .
- Table 1 shows the physical properties and evaluation results of the obtained polycarbonate resin. A polycarbonate resin excellent in color tone, melt processability, and optical properties was obtained.
- Table 1 shows the physical properties and evaluation results of the obtained polycarbonate resin. The reduced viscosity was 0.358 dL / g, and the glass transition temperature was 151 ° C. When film formation was performed, silver was generated in the film, and a film having a good appearance could not be obtained.
- the DPC was purified by distillation and the chloride ion concentration was 10 ppb or less.
- the interior of the polymerization reaction apparatus (1) was sufficiently purged with nitrogen (oxygen concentration: 0.0005 to 0.001 vol%), and then heated with a heating medium, and stirring was started when the internal temperature reached 100 ° C. After 40 minutes from the start of temperature increase, the internal temperature is reached to 220 ° C., and control is performed to maintain this temperature. At the same time, pressure reduction is started, and after reaching 220 ° C., 13.3 kPa (absolute pressure, the same applies hereinafter) in 90 minutes Then, the pressure was maintained for another 30 minutes while maintaining the pressure.
- the phenol vapor produced as a by-product with the polymerization reaction is led to a reflux condenser at 100 ° C., and a monomer component contained in a small amount in the phenol vapor is returned to the polymerization reactor (1), and the phenol vapor not condensed is led to a condenser at 45 ° C. And recovered.
- the oligomerized contents in the polymerization reactor (1) were further provided with a stirring blade and a reflux condenser controlled at 100 ° C. Transferred to the polymerization reactor (2). Subsequently, the temperature increase and pressure reduction in the polymerization reaction apparatus (2) were started, and the internal temperature was 240 ° C. and the pressure was 200 Pa in 50 minutes. Thereafter, the pressure was reduced to 200 Pa or less over 20 minutes, the pressure was restored when the predetermined stirring power was reached, the contents were extracted in the form of strands, and pelletized with a rotary cutter. A resin having a reduced viscosity of 0.368 dL / g and a glass transition temperature of 148 ° C. was obtained.
- Others were carried out in the same manner as in Example 2-1, to obtain a polycarbonate resin.
- Table 2 shows the physical properties and evaluation results of the obtained polycarbonate resin.
- the total content of the compound group represented by the formula (2) with respect to the fluorene compound was 612 ppm and a / b was 24.1, and the color tone and heat resistance of the resin were inferior.
- the film obtained from the polycarbonate resin of the present invention has good wavelength dispersibility and a photoelastic coefficient, and can be suitably used as a quarter wavelength plate.
- the polycarbonate copolymer of the present invention has good hue and transparency and low optical distortion, it is suitable for lens applications such as camera lenses, viewfinder lenses, CCD and CMOS lenses, optical applications such as diffusion sheets and polarizing films. Suitable for use. Furthermore, since the retardation exhibits reverse wavelength dispersion, it can be suitably used for a retardation film used particularly for liquid crystals, plasma displays and the like.
- BHEPF used what contained 4.1 ppm as total sulfur element amount.
- DPC used was purified by distillation to a chloride ion concentration of 10 ppb or less.
- the phenol vapor produced as a by-product with the polymerization reaction is led to a reflux condenser at 100 ° C., and a monomer component contained in a small amount in the phenol vapor is returned to the polymerization reactor (1), and the phenol vapor not condensed is led to a condenser at 45 ° C. And recovered.
- the oligomerized contents in the polymerization reactor (1) were further provided with a stirring blade and a reflux condenser controlled at 100 ° C. Transferred to the polymerization reactor (2). Subsequently, the temperature increase and pressure reduction in the polymerization reaction apparatus (2) were started, and the internal temperature was 240 ° C. and the pressure was 200 Pa in 50 minutes. Thereafter, the pressure was reduced to 133 Pa or less over 20 minutes, the pressure was restored when the predetermined stirring power was reached, the contents were extracted in the form of strands, and pelletized with a rotary cutter.
- the reduced viscosity of the obtained polycarbonate resin was 0.368 dL / g, and the glass transition temperature was 148 ° C. Moreover, when the structural unit ratio derived from each dihydroxy compound was measured, it was found that a resin having a composition almost as prepared was obtained.
- the obtained polycarbonate resin was measured for the metal content, the phenol content, the DPC content, and the YI value of the pellets according to the method described above. Further, the ⁇ YI value was calculated from the measurement of the YI value after the heat aging test. Table 3 shows the physical properties and evaluation results of the obtained polycarbonate resin.
- Table 4 shows the physical properties and evaluation results of the obtained polycarbonate resin. The obtained polycarbonate resin was made into a hot press film by the hot press method described in the above 11), and various optical properties were measured. The results are shown in Table 5.
- Table 4 shows the physical properties and evaluation results of the obtained polycarbonate resin. The obtained polycarbonate resin was made into a hot press film by the hot press method described in the above 11), and various optical properties were measured. The results are shown in Table 5.
- Others were carried out in the same manner as in Example 3-1, to obtain a polycarbonate resin.
- Table 3 shows the physical properties and evaluation results of the obtained polycarbonate resin.
- a polycarbonate resin was obtained in the same manner as in Example 3-1, except that the final polymerization temperature was 260 ° C.
- Table 4 shows the physical properties and evaluation results of the obtained polycarbonate resin. The obtained polycarbonate resin was made into a hot press film by the hot press method described in the above 11), and various optical properties were measured. The results are shown in Table 5.
- the polycarbonate copolymer of the present invention has good hue and transparency and low optical distortion, it is suitable for lens applications such as camera lenses, viewfinder lenses, CCD and CMOS lenses, optical applications such as diffusion sheets and polarizing films. Suitable for use. Furthermore, since the retardation exhibits reverse wavelength dispersion, it is particularly suitable for use in retardation films used for liquid crystals, plasma displays and the like.
- the polycarbonate resin of the present invention is excellent in optical properties, hue, and thermal stability, and is in the field of injection molding such as electrical / electronic parts and automotive parts, film, sheet field, bottle, container field, camera lens, and finder lens.
- Films such as CCD and CMOS lenses, retardation films used for liquid crystal and plasma displays, diffusion sheets, polarizing films, films, sheets, optical disks, optical materials, optical components, dyes, charge transfer agents, etc. It is possible to provide a polycarbonate resin that can be applied to a wide range of fields such as a binder application to be fixed.
Abstract
Description
[1]ガラス転移温度が110℃以上150℃以下、濃度0.6g/dLの塩化メチレン溶液の20℃における還元粘度が0.30以上0.46以下のポリカーボネート樹脂であって、当該樹脂を成形してなるフィルムの波長450nmで測定した位相差(Re450)と波長550nmで測定した位相差(Re550)との比(Re450/Re550)が、0.50以上0.93以下である、ポリカーボネート樹脂。
[2]前記ポリカーボネート樹脂を成形してなるフィルムの波長450nmで測定した位相差(Re450)と波長550nmで測定した位相差(Re550)との比(Re450/Re550)が、0.50以上0.90以下である、[1]に記載のポリカーボネート樹脂。
[3]測定温度240℃、剪断速度91.2sec-1における溶融粘度が、1500Pa・s以上3500Pa・s以下である、[1]または[2]に記載のポリカーボネート樹脂。
[4]光弾性係数が、40×10-12Pa-1以下である、[1]から[3]のいずれか1つに記載のポリカーボネート樹脂。
[5]前記ポリカーボネート樹脂のガラス転移温度より15℃高い温度条件下で、固定端2.0倍延伸したときの複屈折が、0.001以上である、[1]から[4]のいずれか1つに記載のポリカーボネート樹脂。
[6]前記ポリカーボネート樹脂が、下記式(1)で表されるジヒドロキシ化合物に由来する構造単位を含む[1]から[5]のいずれか1つに記載のポリカーボネート樹脂。

[7]前記式(1)で表されるジヒドロキシ化合物に由来する構造単位を含むポリカーボネート樹脂であって、該ジヒドロキシ化合物に対する、下記式(2)で表される化合物群の合計含有量が400ppm以下である、[6]に記載のポリカーボネート樹脂。

[8]前記式(1)で表されるジヒドロキシ化合物以外に、構造の一部に下記式(4)で表される部位を有するジヒドロキシ化合物に由来する構造単位を含む、[1]から[7]のいずれか1つに記載のポリカーボネート樹脂。

[9]前記式(3)で表される部位を有するジヒドロキシ化合物が、環状構造を有し、かつエーテル構造を有する化合物である[8]に記載のポリカーボネート樹脂。
[10]構造の一部に前記式(3)で表される部位を有するジヒドロキシ化合物が、下記式(4)で表されるジヒドロキシ化合物である、[8]または[9]に記載のポリカーボネート樹脂。

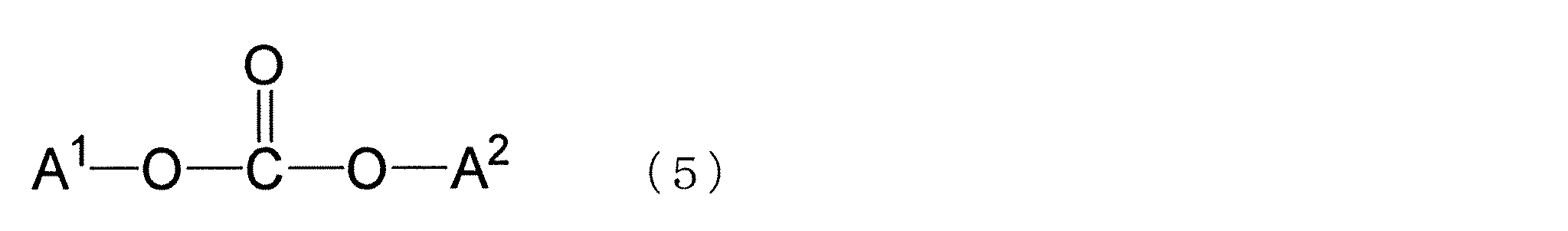
[12]前記ポリカーボネート樹脂が、芳香族モノヒドロキシ化合物を700重量ppm以下含有する、[1]から[11]のいずれか1つに記載のポリカーボネート樹脂。
[13]前記ポリカーボネート樹脂中の、ナトリウム、カリウムおよびセシウムの合計含有量が、前記ポリカーボネート樹脂を構成するジヒドロキシ化合物に由来する構造単位1mol当たり、1μmol以下である、[1]から[12]のいずれか1つに記載のポリカーボネート樹脂。
[14]前記ポリカーボネート樹脂中の、前記ポリカーボネート樹脂を構成するジヒドロキシ化合物に由来する構造単位1mol当たりの全硫黄元素含有量をA[μmol]、長周期型周期表における第1族金属元素および第2族金属元素の合計含有量をB[μmol]とした場合に、Aに対するBの比率(B/A)が2以下である、[1]から[13]のいずれか1つに記載のポリカーボネート樹脂。
[15]前記ポリカーボネート樹脂中の、前記式(1)で表されるジヒドロキシ化合物に由来する構造単位に対する前記式(2)で表される化合物群の合計含有量をa[ppm]、前記ポリカーボネート中の、前記式(1)で表されるジヒドロキシ化合物に由来する構造単位1molに対するポリカーボネート中に含まれる長周期型周期表第1族金属および第2族金属の合計量をb[μmol]とした場合に、bに対するaの比率(a/b)が20以下である、[1]から[14]のいずれか1つに記載のポリカーボネート樹脂。
[16]前記ポリカーボネート樹脂中の前記式(1)で表されるジヒドロキシ化合物に由来する構造単位1molに対して、長周期型周期表第2族金属およびリチウムからなる群から選ばれる少なくとも1種の金属化合物を0.5μmol以上50μmol以下含有する、[1]から[15]のいずれか1つに記載のポリカーボネート樹脂。
[17]前記ポリカーボネート樹脂が、脂肪族ジヒドロキシ化合物、脂環式ジヒドロキシ化合物、オキシアルキレングリコールおよび環状アセタール構造を有するジオールからなる群より選ばれた少なくとも1種の化合物に由来する構成単位を、さらに含むことを特徴とする[1]から[16]のいずれか1つに記載のポリカーボネート樹脂。
[18]前記式(1)で表されるジヒドロキシ化合物を含むジヒドロキシ化合物と、前記式(6)で表される炭酸ジエステルとを、溶融重縮合してなる[1]から[17]に記載のポリカーボネート樹脂。
[19]前記ポリカーボネート樹脂を構成するジヒドロキシ化合物に由来する構造単位の、由来となるすべてのジヒドロキシ化合物が、5kPaにおける沸点が200℃以上のジヒドロキシ化合物である、[1]から[18]のいずれか1つに記載のポリカーボネート樹脂。
[20]下記式(1)で表されるジヒドロキシ化合物と、構造の一部に下記式(4)で表される部位を有するジヒドロキシ化合物とを含むジヒドロキシ化合物混合体と、下記式(6)で表される炭酸ジエステルとを、触媒の存在下、重縮合することにより得られるポリカーボネート樹脂であって、該触媒が長周期型周期表における2族金属およびリチウムからなる群より選ばれた少なくとも1種の金属を含む化合物であり、該ポリカーボネート樹脂中の長周期型周期表における第1族金属元素および第2族金属元素の合計含有量が、該ポリカーボネート樹脂を構成するジヒドロキシ化合物に由来する構造単位1mol当たり、20μmol以下である、ことを特徴とするポリカーボネート樹脂。
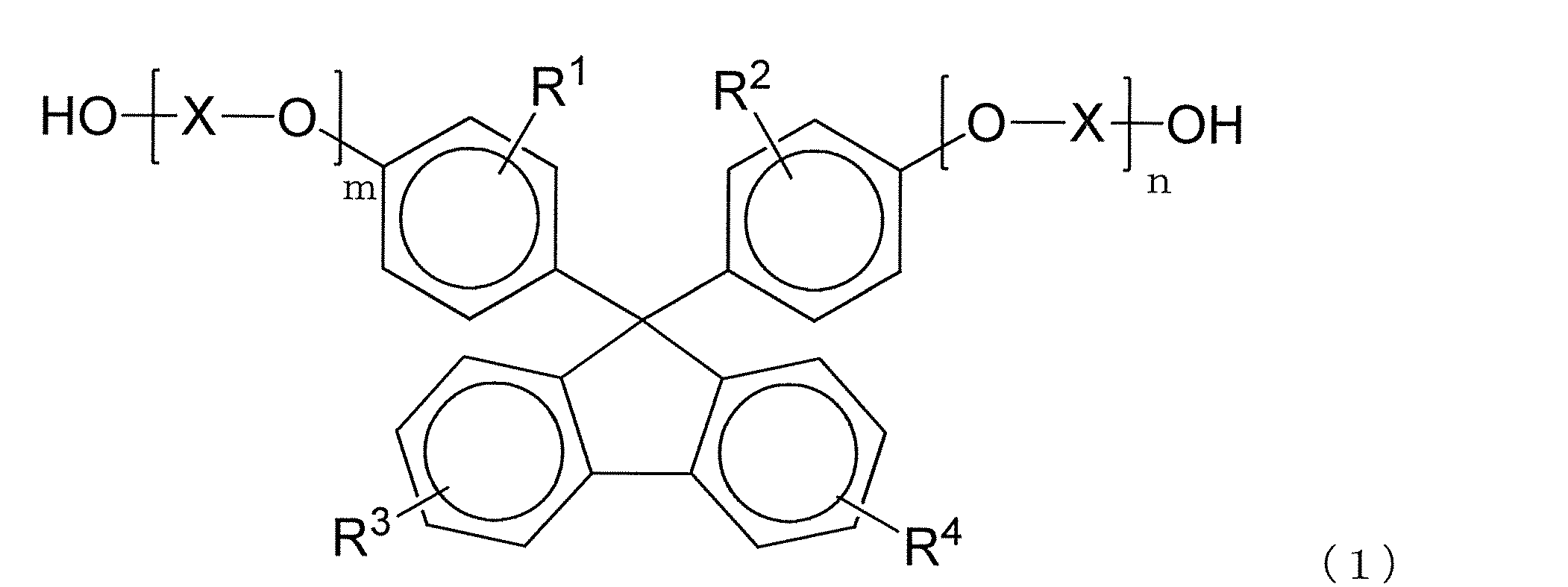


[21][1]から[20]のいずれか1つに記載のポリカーボネート樹脂を製膜してなる、フィルム。
[22]前記透明フィルムが、少なくとも一方向に延伸されてなるものである、[21]に記載のフィルム。
[23]波長450nmで測定した位相差(Re450)と波長550nmで測定した位相差(Re550)の比(Re450/Re550)が、0.5以上0.93以下である、[21]または[22]に記載のフィルム。
[24]波長450nmで測定した位相差(Re450)と波長550nmで測定した位相差(Re550)の比(Re450/Re550)が、0.5以上0.90以下である、[21]または[22]に記載のフィルム。
[25]光弾性係数が40×10-12Pa-1以下である、[21]から[24]のいずれか1つに記載のフィルム。
(ガラス転移温度)
本発明のポリカーボネート樹脂のガラス転移温度は、110℃以上150℃以下である。ポリカーボネート樹脂のガラス転移温度が低すぎると、使用環境下において必要な耐熱性が不足し、成形後に透明性が低下したり寸法変化を起こしたりする可能性がある。また、光学フィルムとして使用した際には、分子鎖の緩和が生じるなどして、光学特性を保てなくなるおそれがあるため、より好ましくは115℃以上であり、更に好ましくは120℃以上である。
本発明のポリカーボネート樹脂は、濃度0.6g/dLの塩化メチレン溶液の20℃における還元粘度が0.30以上0.46以下であるが、還元粘度が低すぎると、機械物性が低下し、例えば靭性などが低下するとフィルムのような厚さの薄いものの成形や、それを延伸加工することが困難となるため、より好ましくは0.33以上、更に好ましくは0.35以上である。
本発明のポリカーボネート樹脂は特に各種ディスプレイ(液晶表示装置、有機EL表示装置、プラズマ表示装置、FED電界放出表示装置、SED表面電界表示装置)の視野角補償用、外光の反射防止用、色補償用、直線偏光の円偏光への変換用などに用いられる光学補償フィルムとして有用である。この用途には、該ポリカーボネート樹脂を成形してなるフィルムの波長450nmで測定した位相差(Re450)の波長550nmで測定した位相差(Re550)に対する比率(Re450/Re550)が0.50以上、0.93以下となることが好ましく、さらには0.70以上、0.91以下であることが好ましく、特には0.85以上、0.90以下であることが好ましい。
上記の位相差フィルムは、更に適切な範囲の位相差を有していることが好ましい。位相差はフィルムの複屈折と厚みの積によって決まるため、所定の位相差を持ちながらフィルムを薄型とするには、フィルムの複屈折が高いことが好ましい。本発明のポリカーボネート樹脂において、ガラス転移温度+15℃の条件下で固定端2.0倍延伸したときの複屈折は、0.001以上であることが好ましく、さらには0.0015以上、特には0.0020以上が好ましい。
本発明のポリカーボネート樹脂は、光弾性係数が40×10-12Pa-1以下であることが好ましい。さらには35×10-12Pa-1以下であることが好ましく、30×10-12Pa-1以下であることが特に好ましい。光弾性係数が40×10-12Pa-1より大きいと、前記透明フィルムを位相差フィルムとして偏光板に貼り合わせ、さらにこの偏光板を表示装置に搭載させたときに、貼り合わせ時の応力により、視認環境やバックライトの熱で位相差フィルムに部分的応力がかかり、不均一な位相差変化が生じ、画面の周囲が白くぼやけるような画像品質の低下が起きる可能性がある。特に大型の表示装置に用いられる場合にはこの問題が顕著に現れる。
ポリカーボネート樹脂の機械物性や色調、あるいは溶融重合時や成形加工時の流動性などの面から、ポリカーボネート樹脂の溶融粘度は適切な範囲に収める必要がある。溶融粘度が高すぎると、機械物性は向上するものの、高い加工温度が必要となるために、樹脂の着色や熱分解を抑制することが難しくなる。
本発明のポリカーボネート樹脂は、下記式(1)で表されるジヒドロキシ化合物に由来する構造単位を含むことが好ましい。
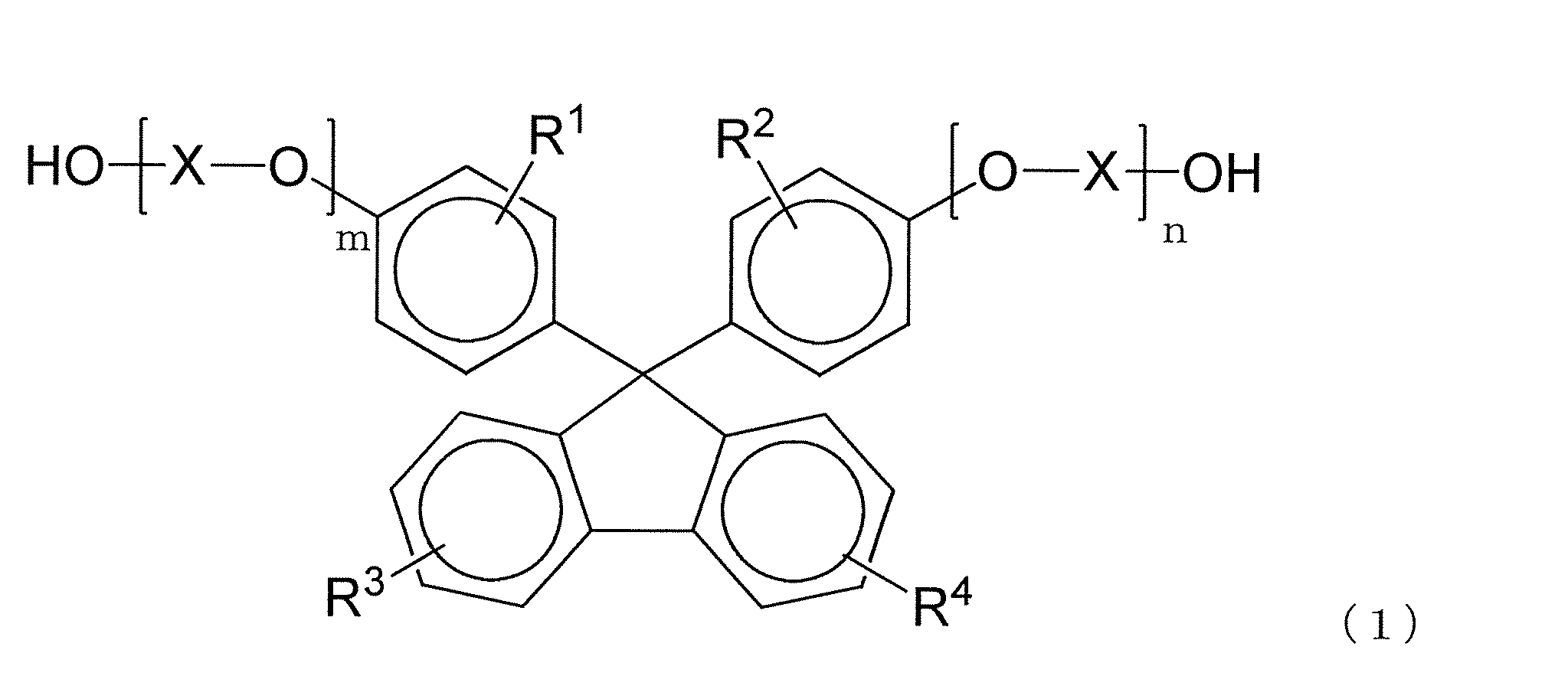
前記式(1)で表されるジヒドロキシ化合物(以下「フルオレン系ジヒドロキシ化合物」と称する場合がある。)において、m、nがともに1のものが優れた機械物性と耐熱性を発現するため、好適に用いられる。


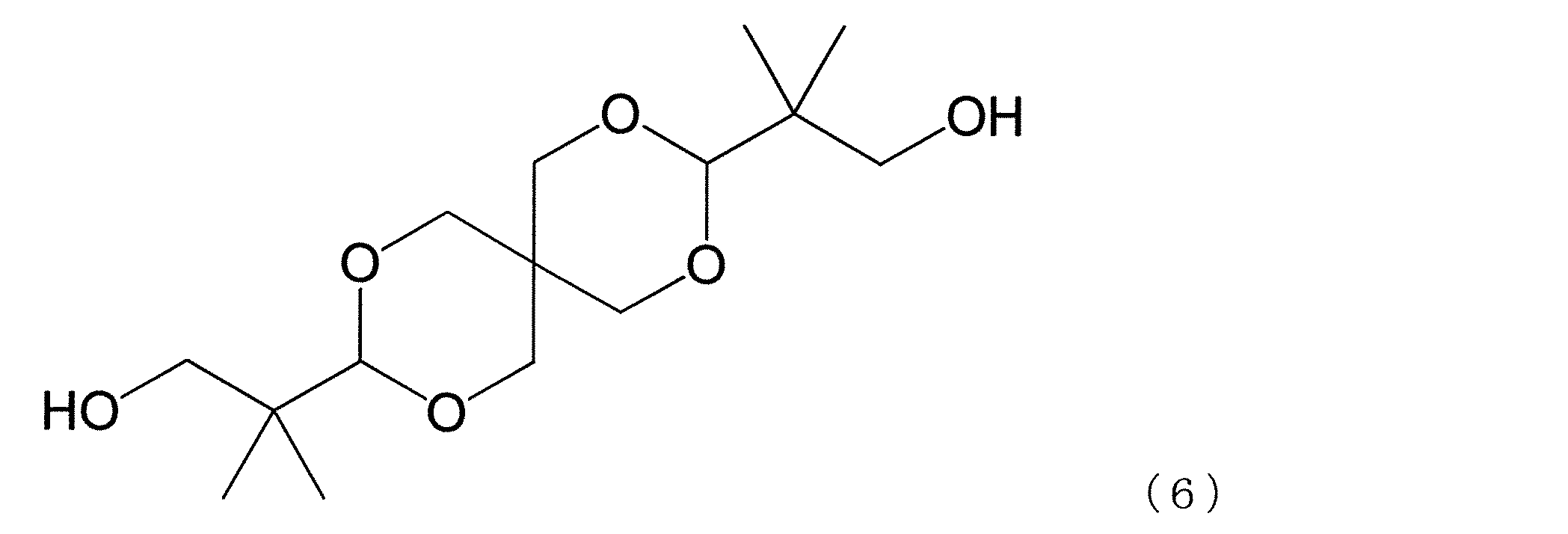
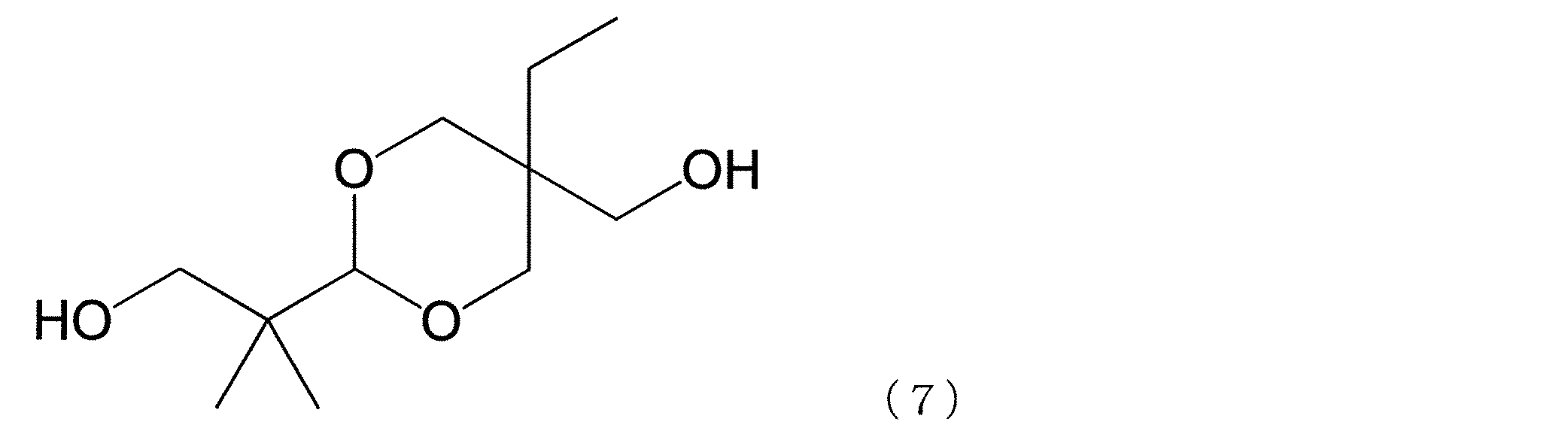
本発明のポリカーボネートは、前記式(1)で表されるジヒドロキシ化合物を含むジヒドロキシ化合物と炭酸ジエステルとを原料として、エステル交換反応により重縮合させて得ることが好ましい。

本発明のポリカーボネートは、上述のようにフルオレン系ジヒドロキシ化合物を含むジヒドロキシ化合物と前記式(5)で表される炭酸ジエステルをエステル交換反応させて製造する。より詳細には、エステル交換させ、副生するモノヒドロキシ化合物等を系外に除去することによって得られる。このエステル交換反応の際には、エステル交換反応触媒存在下で重縮合を行うが、本発明のポリカーボネートの製造時に使用し得るエステル交換反応触媒(以下、単に触媒、重合触媒と言うことがある)は、反応速度や重縮合して得られるポリカーボネートの色調に非常に大きな影響を与え得る。
本発明のポリカーボネート樹脂は、下記式(2)で表される化合物群を合計量で400ppm以下含有するものであることが好ましい。
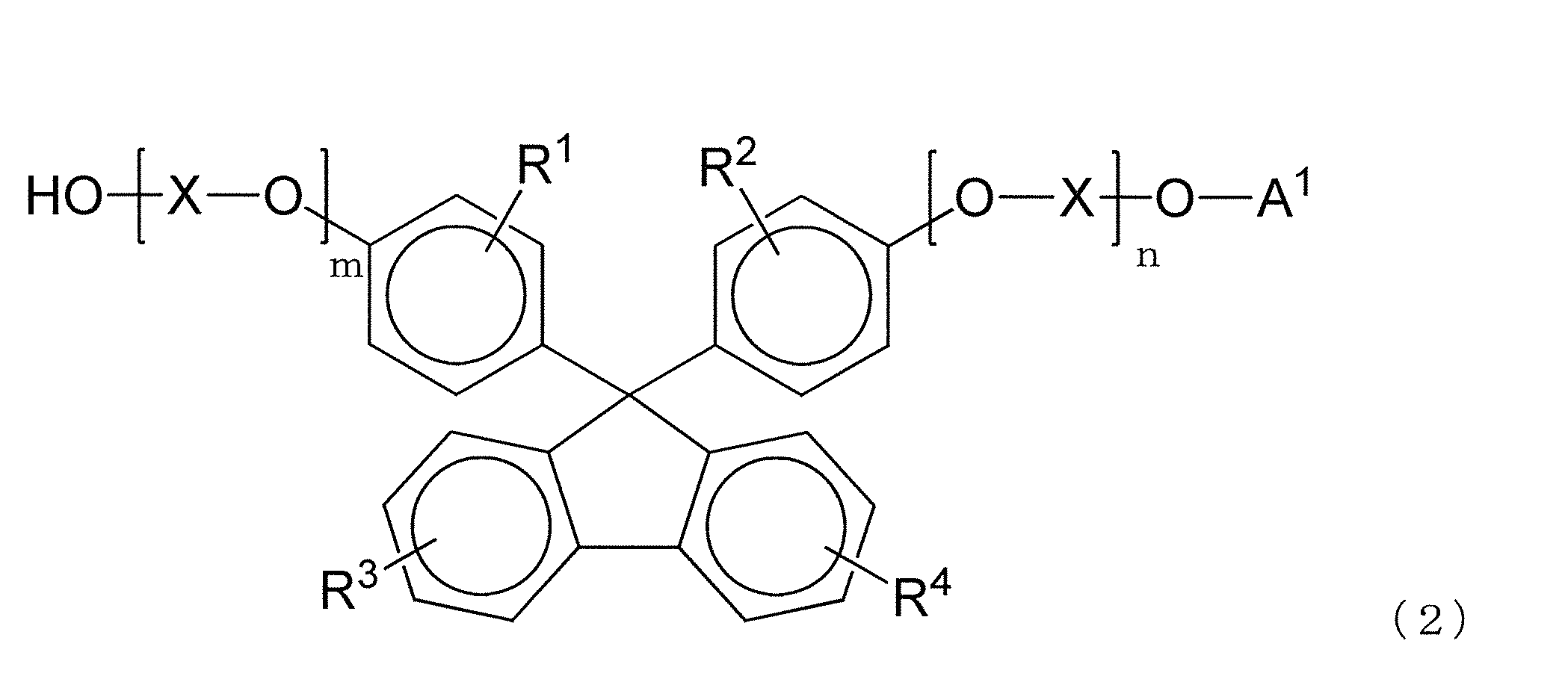
本発明のポリカーボネート樹脂は通常、ジヒドロキシ化合物と前記式(5)で表される炭酸ジエステルとを触媒の存在下、エステル交換反応により重縮合させることによって得ることができる。
本発明に係るフィルムの製造法としては溶融押出法が生産性の点から好ましい。溶融押出法においては、Tダイを用いて樹脂を押し出し、冷却ロールに送る方法が好ましく用いられる。この時の溶融温度はポリカーボネートの分子量、Tg、溶融流動特性などから決められるが、180℃~320℃の範囲であることが好ましく、200℃~300℃の範囲がより好ましい。該温度が高すぎると熱劣化による着色や異物やシルバーの発生による外観不良、Tダイからのダイラインなどの問題が起きやすくなる。該温度が低すぎると粘度が高くなり、ポリマーの配向、応力歪みが残りやすい。
フィルムの製造法としては溶液キャスト法を用いることもできる。溶媒としては、例えば、塩化メチレン、1,2-ジクロロエタン、1,1,2,2-テトラクロロエタン、ジオキソラン、ジオキサンなどが好適に用いられる。溶液キャスト法で得られるフィルム中の残留溶媒量は2重量%以下であることが好ましく、より好ましくは1重量%以下である。2重量%以上と残留溶媒量が多いとフィルムのガラス転移温度の低下が著しくなり耐熱性の点で好ましくない。
本発明のフィルムの厚みとしては、30μm~400μmの範囲が好ましく、より好ましくは40μm~300μmの範囲である。かかるフィルムをさらに延伸して位相差フィルムとする場合には、該位相差フィルムの所望の位相差値、厚みを勘案して上記範囲内で適宜決めればよい。
かくして得られたフィルムは延伸配向されることにより、位相差フィルムとすることができる。延伸方法としては縦一軸延伸、テンター等を用いる横一軸延伸、あるいはそれらを組み合わせた同時二軸延伸、逐次二軸延伸など公知の方法を用いることができる。バッチ式で行ってもよいが、連続で行うことが生産性において好ましい。さらにバッチ式に比べて、連続の方がフィルム面内の位相差のばらつきの少ない位相差フィルムが得られる。
BHEPF:9,9-ビス(4-(2-ヒドロキシエトキシ)-フェニル)フルオレン (大阪ガスケミカル株式会社製)
ISB:イソソルビド (ロケットフルーレ社製、商品名:POLYSORB PS)
CHDM:1,4-シクロヘキサンジメタノール (新日本理化株式会社製)
TCDDM:トリシクロデカンジメタノール(オクセア製)
DEG:ジエチレングリコール (三菱化学株式会社製)
PEG#1000:ポリエチレングリコール数平均分子量1000 (三洋化成株式会社製)
DPC:ジフェニルカーボネート (三菱化学株式会社製)
1)ポリカーボネート樹脂のガラス転移温度
ポリカーボネート樹脂のガラス転移温度は示差走査熱量計(エスアイアイ・ナノテクノロジー社製DSC6220)を用いて測定した。ポリカーボネート樹脂サンプル約10mgを同社製アルミパンに入れて密封し、50mL/分の窒素気流下、昇温速度20℃/分で室温から250℃まで昇温した。3分間温度を保持した後、30℃まで20℃/分の速度で冷却した。30℃で3分保持し、再び200℃まで20℃/分の速度で昇温した。2回目の昇温で得られたDSCデータより、補外ガラス転移開始温度を採用した。
ポリカーボネート樹脂を溶媒として塩化メチレンを用い溶解し、0.6g/dLの濃度のポリカーボネート溶液を調製した。ウベローデ型粘度管(森友理化工業社製)を用いて、温度20.0℃±0.1℃で測定を行い、溶媒の通過時間t0と溶液の通過時間tから次式より相対粘度ηrelを求め、
ηrel=t/t0
相対粘度から次式より比粘度ηspを求めた。
ηsp=(η-η0)/η0=ηrel-1
比粘度を濃度c(g/dL)で割って、還元粘度ηsp/cを求めた。
ポリカーボネート樹脂試料約0.5gを精秤して三角フラスコに入れ、塩化メチレン5mLを加え、攪拌して溶解した後、メタノール45mLと25%水酸化ナトリウム5mLを加えた。溶液を80℃に加熱して、還流下、30分間反応させた。反応後、6N塩酸7mLとTHF10mLを加えて、100mLメスフラスコに溶液を全量移し、純水でメスアップした。調製した溶液を0.2μmディスクフィルターで濾過して、液体クロマトグラフィーの測定を行った。式(1)で表されるジヒドロキシ化合物、および式(2)で表される成分を定量し、式(1)で表されるジヒドロキシ化合物に対する式(2)で表される成分の含有量を計算した。本実施例ではBHEPFを用いて検量線を作成し、絶対検量線法によって定量した。なお、式(2)で表される成分もBHEPFを用いた検量線によって定量したため、該成分の含有量は式(2)で表される化合物群の含有量の絶対値ではなく、BHEPFに換算した数値である。LCチャートを図1に示す。
・装置:(株)島津製作所製
システムコントローラ:CBM-20A
ポンプ:LC-10AD
カラムオーブン:CTO-10ASvp
検出器:SPD-M20A
分析カラム:Cadenza CD-18 Φ4.6mm×250mm
オーブン温度:60℃
・検出波長:300nm
・溶離液:A液:0.1%リン酸水溶液、B液:アセトニトリル、A/B=40/60(vol%)からA/B=0/100(vol%)まで10分間でグラジエント
・流量:1mL/min
・試料注入量:1μL(式(1)で表されるジヒドロキシ化合物の定量の場合)
10μL(式(2)で表される成分の定量の場合)
ポリカーボネート樹脂中の各ジヒドロキシ化合物に由来する構造単位比は、ポリカーボネート樹脂30mgを秤取し、重クロロホルム約0.7mLに溶解し、溶液とし、これを内径5mmのNMR用チューブに入れ、日本電子社製JNM-AL400(共鳴周波数400MHz)を用いて常温で1H NMRスペクトルを測定した。各ジヒドロキシ化合物に由来する構造単位に基づくシグナル強度比より各ジヒドロキシ化合物に由来する構造単位比を求めた。
・装置:日本電子社製JNM-AL400(共鳴周波数400MHz)
・測定温度:常温
・緩和時間:6秒
・積算回数:128回
パーキンエルマー社製マイクロウェーブ分解容器にポリカーボネート樹脂ペレット約0.5gを精秤し、97%硫酸2mLを加え、密閉状態にして230℃で10分間マイクロウェーブ加熱した。室温まで冷却後、68%硝酸1.5mLを加えて、密閉状態にして150℃で10分間マイクロウェーブ加熱した後、再度室温まで冷却を行い、68%硝酸2.5mLを加え、再び密閉状態にして230℃で10分間マイクロウェーブ加熱し、内容物を完全に分解させた。室温まで冷却後、上記で得られた液を純水で希釈し、サーモクエスト社製ICP-MSで定量した。
試料約0.5gを精秤し、塩化メチレン5mLに溶解した後、総量が25mLになるようにアセトンを添加した。溶液を0.2μmディスクフィルターでろ過して、液体クロマトグラフィーにてフェノールとDPCの定量を行った後、含有量を算出した。
・装置:(株)島津製作所製
システムコントローラ:CBM-20A
ポンプ:LC-10AD
カラムオーブン:CTO-10ASvp
検出器:SPD-M20A
分析カラム:Cadenza CD-18 Φ4.6mm×250mm
オーブン温度:40℃
・検出波長:260nm
・溶離液:A液:0.1%リン酸水溶液、B液:アセトニトリル
A/B=40/60(vol%)からA/B=0/100(vol%)まで10分間でグラジエント
・流量:1mL/min
・試料注入量:10μL
・定量法:絶対検量線法
ポリカーボネート樹脂の色相は、ASTM D1925に準拠して、ペレットの反射光におけるイエローインデックス(YI)値を測定して評価した。装置はコニカミノルタ社製分光測色計CM-5を用い、測定条件は測定径30mm、SCEを選択した。シャーレ測定用校正ガラスCM-A212を測定部にはめ込み、その上からゼロ校正ボックスCM-A124をかぶせてゼロ校正を行い、続いて内蔵の白色校正板を用いて白色校正を行った。
内部を空気雰囲気とした120℃のオーブンに樹脂ペレットを入れ、200時間後に取り出し、前記(7)と同じ方法でYI値を測定した。熱老化試験後のYI値と前記(7)のYI値との差をΔYI値として表す。ΔYI値が小さいほど熱安定性が優れることを示す。
ポリカーボネート樹脂試料を白金製ボートに採取し、石英管管状炉(三菱化学株式会社社製AQF-100型)で加熱し、燃焼ガス中の硫黄分を0.03%の過酸化水素水溶液で吸収した。吸収液中のSO42-をイオンクロマトグラフ(Dionex社製ICS-1000型)で測定した。
80℃で5時間、真空乾燥をしたポリカーボネート樹脂試料を用いて、キャピラリーレオメーター(東洋精機(株)製)で測定を行った。反応温度と同じ温度に加熱して、剪断速度9.12~1824sec-1間で溶融粘度を測定し、91.2sec-1における溶融粘度の値を用いた。ダイス径φ1mm×10mmLのオリフィスを使用した。
酸素計(AMI社製:1000RS)を使用し、測定した。
重合反応の留出液3mLを精秤し、アセトニトリルを加えて10mLとした。ガスクロマトグラフィーの測定を行い、留出液中に含有されているジヒドロキシ化合物の含有率を求めた。留出液全量の重量からジヒドロキシ化合物の留出量を計算し、重合反応に用いたジヒドロキシ化合物の仕込み量との比から留出率を求めた。
・装置:アジレント・テクノロジー社製 6850
・カラム:アジレント・テクノロジー社製 DB-1(内径250μm、長さ30m、膜圧0.25μm)
・オーブン温度:50℃ 3分保持 → 昇温10℃/min → 250℃ → 昇温50℃/min → 300℃ 6分保持
・検出器:水素炎イオン化検出器
・注入口温度:250℃
・検出器温度:320℃
・キャリアガス:ヘリウム
・試料注入量:1μL
・定量法:絶対検量線法
He-Neレーザー、偏光子、補償板、検光子、光検出器からなる複屈折測定装置と振動型粘弾性測定装置(レオロジー社製DVE-3)を組み合わせた装置を用いて光弾性係数を測定した。(詳細は、日本レオロジー学会誌Vol.19,p.93~97(1991)を参照。)
C=O’/E’
前述の熱プレスで得られたフィルムからから幅6cm、長さ6cmの試料を切り出した。この試料を、バッチ式二軸延伸装置(東洋精機社製)で、延伸温度をポリカーボネート樹脂のガラス転移温度+15℃、延伸速度を720mm/分(ひずみ速度1200%/min)で、延伸倍率2.0倍の一軸延伸を行った。このとき延伸方向に対して垂直方向は、保持した状態(延伸倍率1.0)で延伸を行った。
80℃で5時間、真空乾燥をしたポリカーボネート樹脂を、単軸押出機(いすず化工機社製、スクリュー径25mm、シリンダー設定温度:220℃)、Tダイ(幅200mm、設定温度:220℃)、チルロール(設定温度:120~130℃)及び巻取機を備えたフィルム製膜装置を用いて、厚み100μmのフィルムを作製した。
前述した方法で製膜した溶融押出フィルムから幅6cm、長さ6cmの試料を切り出した。この試料をバッチ式二軸延伸装置(東洋精機社製)で、延伸温度をポリカーボネート樹脂のガラス転移温度+15℃に設定し、延伸速度720mm/分(ひずみ速度1200%/分)、2.0倍の一軸延伸を行い、透明フィルムを得た。このとき延伸方向に対して垂直方向は、保持した状態(延伸倍率1.0)で延伸を行った。
上記のフィルムを切り出したサンプルを前記位相差測定装置により波長590nmの位相差(Re590)を測定した。前記位相差を前記フィルムの厚み(t)で除し、下記式に従い、複屈折を求めた。
複屈折(Δn)=Re590/t
撹拌翼および100℃に制御された還流冷却器を具備した重合反応装置(1)に、BHEPFとISB、PEG#1000、DPC、および酢酸マグネシウム4水和物を、モル比率でBHEPF/ISB/PEG#1000/DPC/酢酸マグネシウム4水和物=0.445/0.552/0.003/1.015/1.20×10-5となるように仕込んだ。BHEPFは全硫黄元素量として4.1ppm含有するものを使用した。DPCは蒸留精製して塩化物イオン濃度を10ppb以下にしたものを使用した。
BHEPFとISB、PEG#1000、DPC、および酢酸マグネシウム4水和物を、モル比率でBHEPF/ISB/PEG#1000/DPC/酢酸マグネシウム4水和物=0.432/0.556/0.012/1.005/1.20×10-5となるように仕込んだ以外は実施例1-1と同様に行った。得られたポリカーボネート樹脂の物性と評価結果を表1に示す。
BHEPFとISB、TCDDM、DPC、および酢酸カルシウム1水和物を、モル比率でBHEPF/ISB/TCDDM/DPC/酢酸カルシウム1水和物=0.300/0.400/0.300/1.005/6.00×10-6となるように仕込んだ以外は実施例1-1と同様に行った。得られたポリカーボネート樹脂の物性と評価結果を表1に示す。
BHEPFとISB、DEG、DPC、および酢酸カルシウム1水和物を、モル比率でBHEPF/ISB/DEG/DPC/酢酸カルシウム1水和物=0.380/0.435/0.185/1.015/6.00×10-6となるように仕込んだ以外は実施例1-1と同様に行った。得られたポリカーボネート樹脂の物性と評価結果を表1に示す。色調や溶融加工性、および光学特性とも優れたポリカーボネート樹脂が得られた。
重合触媒として炭酸セシウムを用いた。BHEPFとISB、PEG#1000、DPC、および炭酸セシウムを、モル比率でBHEPF/ISB/PEG#1000/DPC/酢酸マグネシウム4水和物=0.445/0.552/0.003/1.005/1.20×10-5となるように仕込んだ以外は実施例1-1と同様に行った。得られたポリカーボネート樹脂の物性と評価結果を表1に示す。得られたポリカーボネート樹脂のYIが37であり、実施例1よりも色調は劣っていた。
BHEPFとISB、DPC、および酢酸マグネシウム4水和物を、モル比率でBHEPF/ISB/DPC/酢酸マグネシウム4水和物=0.400/0.600/1.015/1.20×10-5となるように仕込んだ以外は実施例1-1と同様に行った。得られたポリカーボネート樹脂の物性と評価結果を表1に示す。還元粘度は0.358dL/g、ガラス転移温度は151℃であった。フィルム製膜を行ったところ、フィルムにシルバーが発生し、外観の良好なフィルムが得られなかった。
BHEPFとISB、CHDM、DPC、および酢酸カルシウム1水和物を、モル比率でBHEPF/ISB/CHDM/DPC/酢酸カルシウム1水和物=0.330/0.335/0.335/1.005/6.00×10-6となるように仕込んだ以外は実施例1-1と同様に行った。得られたポリカーボネート樹脂の物性と評価結果を表1に示す。Re450/Re550が0.953であり、波長分散性が不足していた。また、光弾性係数も30×10-12Pa-1であり、若干高い値となった。
表1に示した結果から、適切なジヒドロキシ化合物の組み合わせにより、色調、溶融加工性、および光学特性のバランスに優れたポリカーボネート樹脂が得られることがわかる。
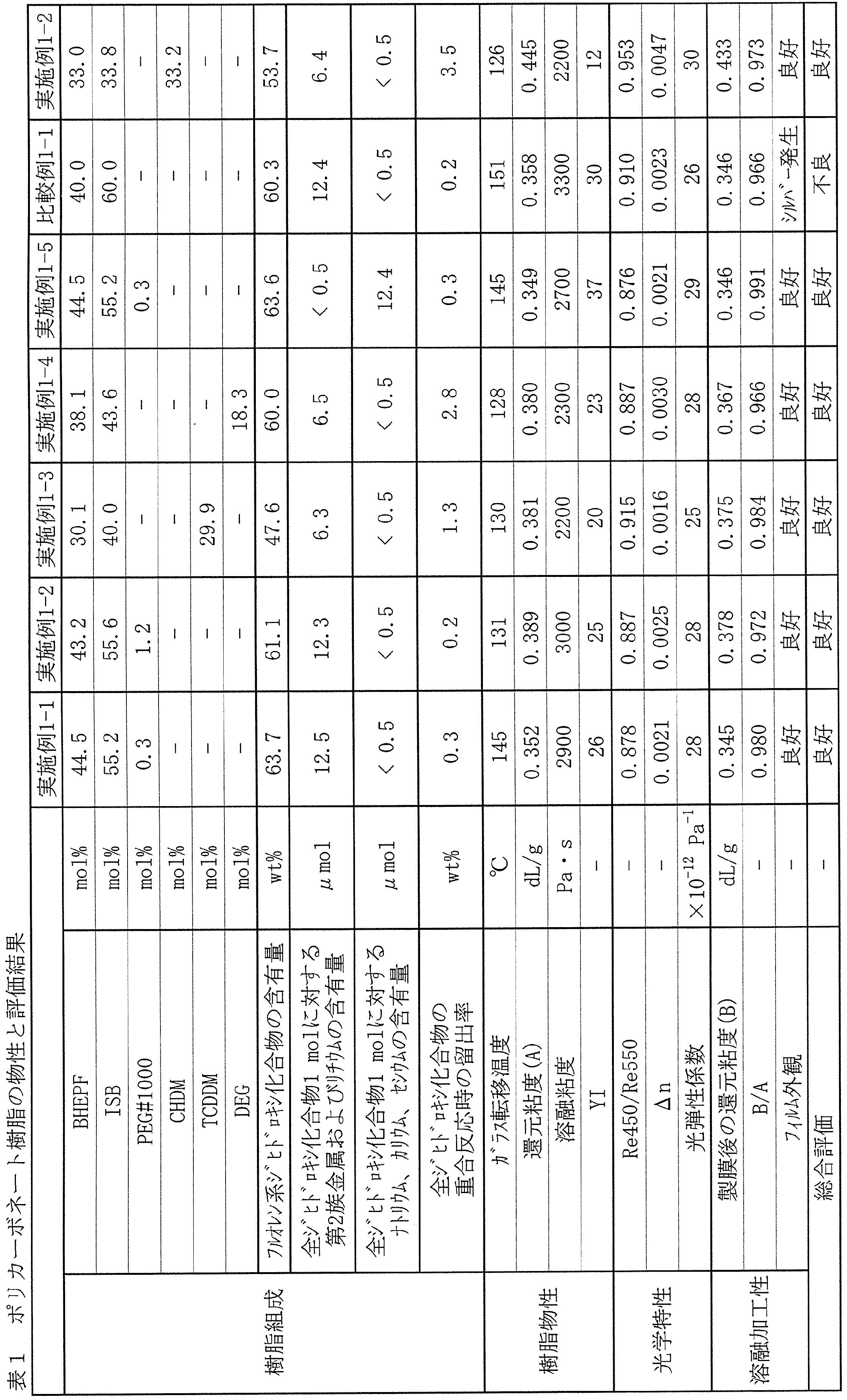
撹拌翼および100℃に制御された還流冷却器を具備した重合反応装置(1)に、BHEPFとISB、DEG、DPC、および酢酸リチウムを、モル比率でBHEPF/ISB/DEG/DPC/酢酸リチウム=0.380/0.435/0.185/1.00/6.00×10-6となるように仕込んだ。BHEPFは全硫黄元素量として4.1ppm含有するものを使用した。
BHEPFとISB、DEG、DPC、および酢酸マグネシウム4水和物を、モル比率でBHEPF/ISB/DEG/DPC/酢酸マグネシウム4水和物=0.380/0.435/0.185/1.00/6.00×10-6となるように仕込んだ。他は実施例2-1と同様に実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂の物性と評価結果を表2に示す。フルオレン系化合物に対する前記式(2)で表される化合物群の合計含有量は155ppm、a/bは9.6であり、実施例2-1と同様に優れた品質のポリカーボネート樹脂が得られた。
BHEPFとISB、DEG、DPC、および酢酸カルシウム1水和物を、モル比率でBHEPF/ISB/DEG/DPC/酢酸カルシウム1水和物=0.380/0.435/0.185/1.00/6.00×10-6となるように仕込んだ。他は実施例2-1と同様に実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂の物性と評価結果を表2に示す。フルオレン系化合物に対する前記式(2)で表される化合物群の合計含有量は146ppm、a/bは9.0であり、実施例2-1と同様に優れた品質のポリカーボネート樹脂が得られた。
BHEPFとISB、DEG、DPC、および酢酸バリウムを、モル比率でBHEPF/ISB/DEG/DPC/酢酸バリウム=0.380/0.435/0.185/1.00/6.00×10-6となるように仕込んだ。他は実施例2-1と同様に実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂の物性と評価結果を表2に示す。フルオレン系化合物に対する前記式(2)で表される化合物群の合計含有量は172ppm、a/bは10.7であり、実施例2-1と同様に優れた品質のポリカーボネート樹脂が得られた。
BHEPFとISB、PEG#1000、DPC、および酢酸カルシウム1水和物を、モル比率でBHEPF/ISB/PEG#1000/DPC/酢酸カルシウム1水和物=0.445/0.552/0.003/1.00/1.20×10-5になるように仕込んだ。他は実施例2-1と同様に実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂の物性と評価結果を表2に示す。フルオレン系化合物に対する前記式(2)で表される化合物群の合計含有量は322ppm、a/bは11.7であり、優れた品質のポリカーボネート樹脂であった。
BHEPFとISB、DPC、および炭酸水素ナトリウムを、モル比率でBHEPF/ISB/DPC/炭酸水素ナトリウム=0.400/0.600/1.00/8.00×10-6となるように仕込んだ。他は実施例2-1と同様に実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂の物性と評価結果を表2に示す。フルオレン系化合物に対する前記式(2)で表される化合物群の合計含有量は612ppm、a/bは24.1であり、樹脂の色調と耐熱性が劣っていた。
BHEPFとISB、CHDM、DPC、および炭酸水素ナトリウムを、モル比率でBHEPF/ISB/CHDM/DPC/炭酸水素ナトリウム=0.330/0.335/0.335/1.00/6.00×10-6になるように仕込んだ。他は実施例2-1と同様に実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂の物性と評価結果を表2に示す。フルオレン系化合物に対する前記式(2)で表される化合物群の合計含有量は490ppm、a/bは26.5であり、樹脂の色調と耐熱性が劣っていた。

撹拌翼および100℃に制御された還流冷却器を具備した重合反応装置(1)に、BHEPFとISB、DEG、DPC、および酢酸リチウムを、モル比率でBHEPF/ISB/DEG/DPC/酢酸リチウム=0.380/0.435/0.185/1.00/6.00×10-6になるように仕込んだ。BHEPFは全硫黄元素量として4.1ppm含有するものを使用した。DPCは蒸留精製して塩化物イオン濃度を10ppb以下にしたものを使用した。
BHEPFとISB、DEG、DPC、および酢酸マグネシウム4水和物を、モル比率でBHEPF/ISB/DEG/DPC/酢酸マグネシウム4水和物=0.380/0.435/0.185/1.00/6.00×10-6になるように仕込んだ。他は実施例1と同様に実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂の物性および評価の結果を表3に示す。
BHEPFとISB、DEG、DPC、および酢酸カルシウム1水和物を、モル比率でBHEPF/ISB/DEG/DPC/酢酸カルシウム1水和物=0.380/0.435/0.185/1.00/6.00×10-6になるように仕込んだ。他は実施例1と同様に実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂の物性および評価の結果を表3に示す。得られたポリカーボネート樹脂は更に前記11)に記載の熱プレス法で熱プレスフィルムとし、各種光学特性を測定した。結果を表5に示す。
BHEPFとISB、DEG、DPC、および酢酸バリウムを、モル比率でBHEPF/ISB/DEG/DPC/酢酸バリウム=0.380/0.435/0.185/1.00/6.00×10-6になるように仕込んだ。他は実施例1と同様に実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂の物性および評価の結果を表3に示す。
BHEPFとISB、DEG、DPC、および酢酸カルシウム1水和物を、モル比率でBHEPF/ISB/DEG/DPC/酢酸カルシウム1水和物=0.380/0.435/0.185/1.00/8.00×10-6になるように仕込んだ。他は実施例1と同様に実施し、ポリカーボネート樹脂を得た。重合速度が非常に速かったため、重合反応装置(2)の反応では圧力が133Pa以下に到達する前に所定の攪拌動力に到達してしまった。得られたポリカーボネート樹脂の物性および評価の結果を表3に示す。
BHEPFとISB、DPC、および酢酸カルシウム1水和物を、モル比率でBHEPF/ISB/DPC/酢酸カルシウム1水和物=0.400/0.600/1.00/8.00×10-6になるように仕込んだ。他は実施例1と同様に実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂の物性および評価の結果を表4に示す。得られたポリカーボネート樹脂は、更に前記11)に記載の熱プレス法で熱プレスフィルムとし、各種光学特性を測定した。結果を表5に示す。
BHEPFとISB、CHDM、DPC、および酢酸カルシウム1水和物を、モル比率でBHEPF/ISB/CHDM/DPC/酢酸カルシウム1水和物=0.330/0.335/0.335/1.00/6.00×10-6になるように仕込んだ。他は実施例1と同様に実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂の物性および評価の結果を表4に示す。得られたポリカーボネート樹脂は、前記11)に記載の熱プレス法で熱プレスフィルムとし、各種光学特性を測定した。結果を表5に示す。
BHEPFとISB、PEG#1000、DPC、および酢酸マグネシウム4水和物を、モル比率でBHEPF/ISB/CHDM/DPC/酢酸マグネシウム4水和物=0.445/0.552/0.003/1.015/1.20×10-5になるように仕込んだ。他は実施例3-1と同様に実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂の物性および評価の結果を表4に示す。得られたポリカーボネート樹脂は、前記11)に記載の熱プレス法で熱プレスフィルムとし、各種光学特性を測定した。結果を表5に示す。
BHEPFとISB、DEG、DPC、および炭酸水素ナトリウムを、モル比率でBHEPF/ISB/DEG/DPC/炭酸水素ナトリウム=0.380/0.435/0.185/1.00/6.00×10-6になるように仕込んだ。他は実施例3-1と同様に実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂の物性および評価の結果を表3に示す。
BHEPFとISB、DEG、DPC、および炭酸セシウムを、モル比率でBHEPF
/ISB/DEG/DPC/炭酸セシウム=0.380/0.435/0.185/1.00/3.00×10-6になるように仕込んだ。他は実施例3-1と同様に実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂の物性および評価の結果を表3に示す。
BHEPFとISB、DPC、および酢酸カルシウム1水和物を、モル比率でBHEPF/ISB/DPC/炭酸水素ナトリウム=0.400/0.600/1.00/8.00×10-6になるように仕込んだ。他は実施例3-1と同様に実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂の物性および評価の結果を表4に示す。
BHEPFは全硫黄元素量として10.1ppmのものを使用した。触媒量が実施例6と同量では十分に反応が進行しなかったため、BHEPFとISB、DPC、および酢酸カルシウム1水和物を、モル比率でBHEPF/ISB/DPC/酢酸カルシウム1水和物=0.400/0.600/1.00/2.00×10-5になるように仕込んだ。他は実施例3-6と同様に実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂の物性および評価の結果を表4に示す。
BHEPFとDPC、および炭酸水素ナトリウムを、モル比率でBHEPF/DPC/炭酸水素ナトリウム=1.00/1.00/3.00×10-5になるように仕込んだ。最終重合温度を260℃にした他は実施例3-1と同様に実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂の物性および評価の結果を表4に示す。得られたポリカーボネート樹脂は、前記11)に記載の熱プレス法で熱プレスフィルムとし、各種光学特性を測定した。結果を表5に示す。
ISBとDPC、および炭酸水素ナトリウムを、モル比率でISB/DPC/炭酸水素ナトリウム=1.00/1.00/1.50×10-6になるように仕込んだ。他は実施例3-1と同様に実施し、ポリカーボネート樹脂を得た。結果を表4に示す。得られたポリカーボネート樹脂は、前記11)に記載の熱プレス法で熱プレスフィルムとし、各種光学特性を測定した。結果を表5に示す。

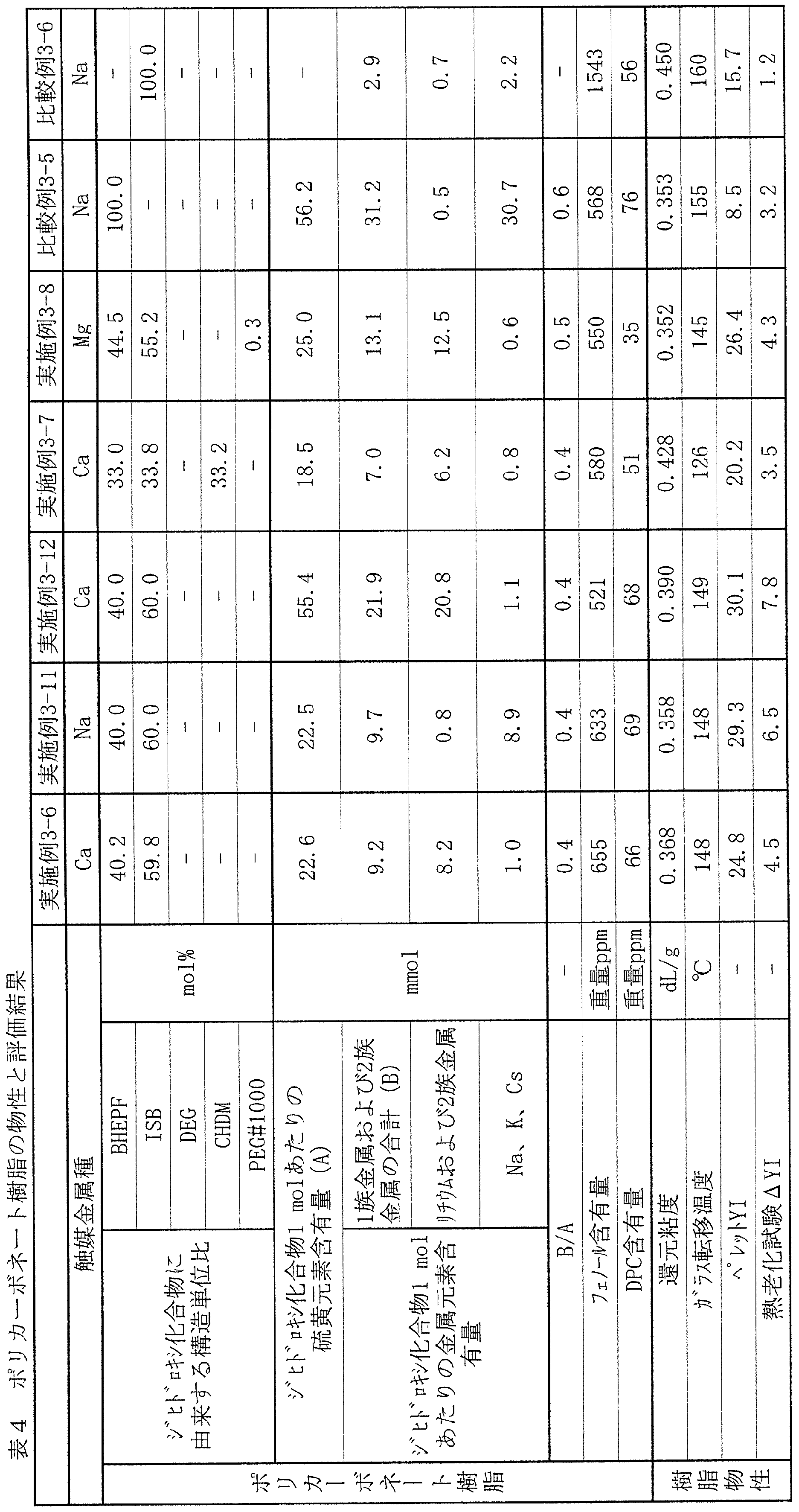
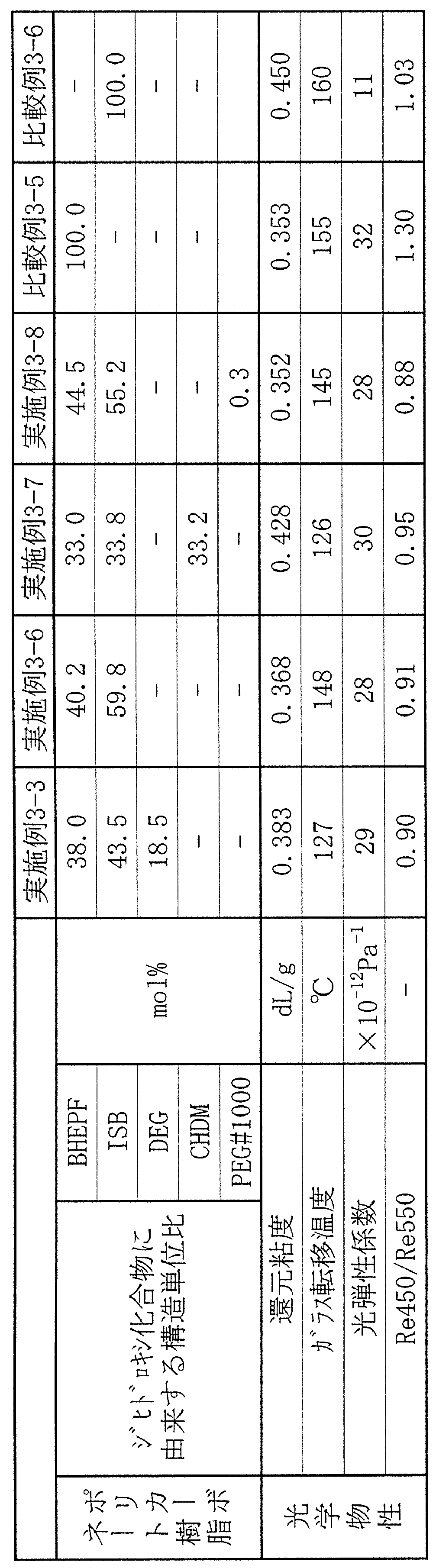
Claims (25)
- ガラス転移温度が110℃以上150℃以下、濃度0.6g/dLの塩化メチレン溶液の20℃における還元粘度が0.30以上0.46以下のポリカーボネート樹脂であって、当該樹脂を成形してなるフィルムの波長450nmで測定した位相差(Re450)と波長550nmで測定した位相差(Re550)との比(Re450/Re550)が、0.50以上0.93以下である、ポリカーボネート樹脂。
- 前記ポリカーボネート樹脂を成形してなるフィルムの波長450nmで測定した位相差(Re450)と波長550nmで測定した位相差(Re550)との比(Re450/Re550)が、0.50以上0.90以下である、請求項1に記載のポリカーボネート樹脂。
- 測定温度240℃、剪断速度91.2sec-1における溶融粘度が、1500Pa・s以上3500Pa・s以下である、請求項1または請求項2に記載のポリカーボネート樹脂。
- 光弾性係数が、40×10-12Pa-1以下である、請求項1から請求項3のいずれか1項に記載のポリカーボネート樹脂。
- 前記ポリカーボネート樹脂のガラス転移温度より15℃高い温度条件下で、固定端2.0倍延伸したときの複屈折が、0.001以上である、請求項1から請求項4のいずれか1項に記載のポリカーボネート樹脂。
- 前記ポリカーボネート樹脂が、下記式(1)で表されるジヒドロキシ化合物に由来する構造単位を含む請求項1から請求項5のいずれか1項に記載のポリカーボネート樹脂。

- 前記式(1)で表されるジヒドロキシ化合物に由来する構造単位を含むポリカーボネート樹脂であって、該ジヒドロキシ化合物に対する、下記式(2)で表される化合物群の合計含有量が400ppm以下である、請求項6に記載のポリカーボネート樹脂。

- 前記式(1)で表されるジヒドロキシ化合物以外に、構造の一部に下記式(4)で表される部位を有するジヒドロキシ化合物に由来する構造単位を含む、請求項1から請求項7のいずれか1項に記載のポリカーボネート樹脂。

- 前記式(3)で表される部位を有するジヒドロキシ化合物が、環状構造を有し、かつエーテル構造を有する化合物である請求項8に記載のポリカーボネート樹脂。
- 構造の一部に前記式(3)で表される部位を有するジヒドロキシ化合物が、下記式(4)で表されるジヒドロキシ化合物である、請求項8または請求項9に記載のポリカーボネート樹脂。

- 前記ポリカーボネート樹脂が、下記式(5)で表される炭酸ジエステルを80重量ppm以下含有する、請求項1から請求項10のいずれか1項に記載のポリカーボネート樹脂。

- 前記ポリカーボネート樹脂が、芳香族モノヒドロキシ化合物を700重量ppm以下含有する、請求項1から請求項11のいずれか1項に記載のポリカーボネート樹脂。
- 前記ポリカーボネート樹脂中の、ナトリウム、カリウムおよびセシウムの合計含有量が、前記ポリカーボネート樹脂を構成するジヒドロキシ化合物に由来する構造単位1mol当たり、1μmol以下である、請求項1から請求項12のいずれか1項に記載のポリカーボネート樹脂。
- 前記ポリカーボネート樹脂中の、前記ポリカーボネート樹脂を構成するジヒドロキシ化合物に由来する構造単位1mol当たりの全硫黄元素含有量をA[μmol]、長周期型周期表における第1族金属元素および第2族金属元素の合計含有量をB[μmol]とした場合に、Aに対するBの比率(B/A)が2以下である、請求項1から請求項13のいずれか1項に記載のポリカーボネート樹脂。
- 前記ポリカーボネート樹脂中の、前記式(1)で表されるジヒドロキシ化合物に由来する構造単位に対する前記式(2)で表される化合物群の合計含有量をa[ppm]、前記ポリカーボネート中の、前記式(1)で表されるジヒドロキシ化合物に由来する構造単位1molに対するポリカーボネート中に含まれる長周期型周期表第1族金属および第2族金属の合計量をb[μmol]とした場合に、bに対するaの比率(a/b)が20以下である、請求項1から請求項14のいずれか1項に記載のポリカーボネート樹脂。
- 前記ポリカーボネート樹脂中の前記式(1)で表されるジヒドロキシ化合物に由来する構造単位1molに対して、長周期型周期表第2族金属およびリチウムからなる群から選ばれる少なくとも1種の金属化合物を0.5μmol以上50μmol以下含有する、請求項1から請求項15のいずれか1項に記載のポリカーボネート樹脂。
- 前記ポリカーボネート樹脂が、脂肪族ジヒドロキシ化合物、脂環式ジヒドロキシ化合物、オキシアルキレングリコールおよび環状アセタール構造を有するジオールからなる群より選ばれた少なくとも1種の化合物に由来する構成単位を、さらに含むことを特徴とする請求項1から請求項16のいずれか1項に記載のポリカーボネート樹脂。
- 前記式(1)で表されるジヒドロキシ化合物を含むジヒドロキシ化合物と、前記式(6)で表される炭酸ジエステルとを、溶融重縮合してなる請求項1から請求項17に記載のポリカーボネート樹脂。
- 前記ポリカーボネート樹脂を構成するジヒドロキシ化合物に由来する構造単位の、由来となるすべてのジヒドロキシ化合物が、5kPaにおける沸点が200℃以上のジヒドロキシ化合物である、請求項1から請求項18のいずれか1項に記載のポリカーボネート樹脂。
- 下記式(1)で表されるジヒドロキシ化合物と、構造の一部に下記式(4)で表される部位を有するジヒドロキシ化合物とを含むジヒドロキシ化合物混合体と、下記式(6)で表される炭酸ジエステルとを、触媒の存在下、重縮合することにより得られるポリカーボネート樹脂であって、該触媒が長周期型周期表における2族金属およびリチウムからなる群より選ばれた少なくとも1種の金属を含む化合物であり、該ポリカーボネート樹脂中の長周期型周期表における第1族金属元素および第2族金属元素の合計含有量が、該ポリカーボネート樹脂を構成するジヒドロキシ化合物に由来する構造単位1mol当たり、20μmol以下である、ことを特徴とするポリカーボネート樹脂。
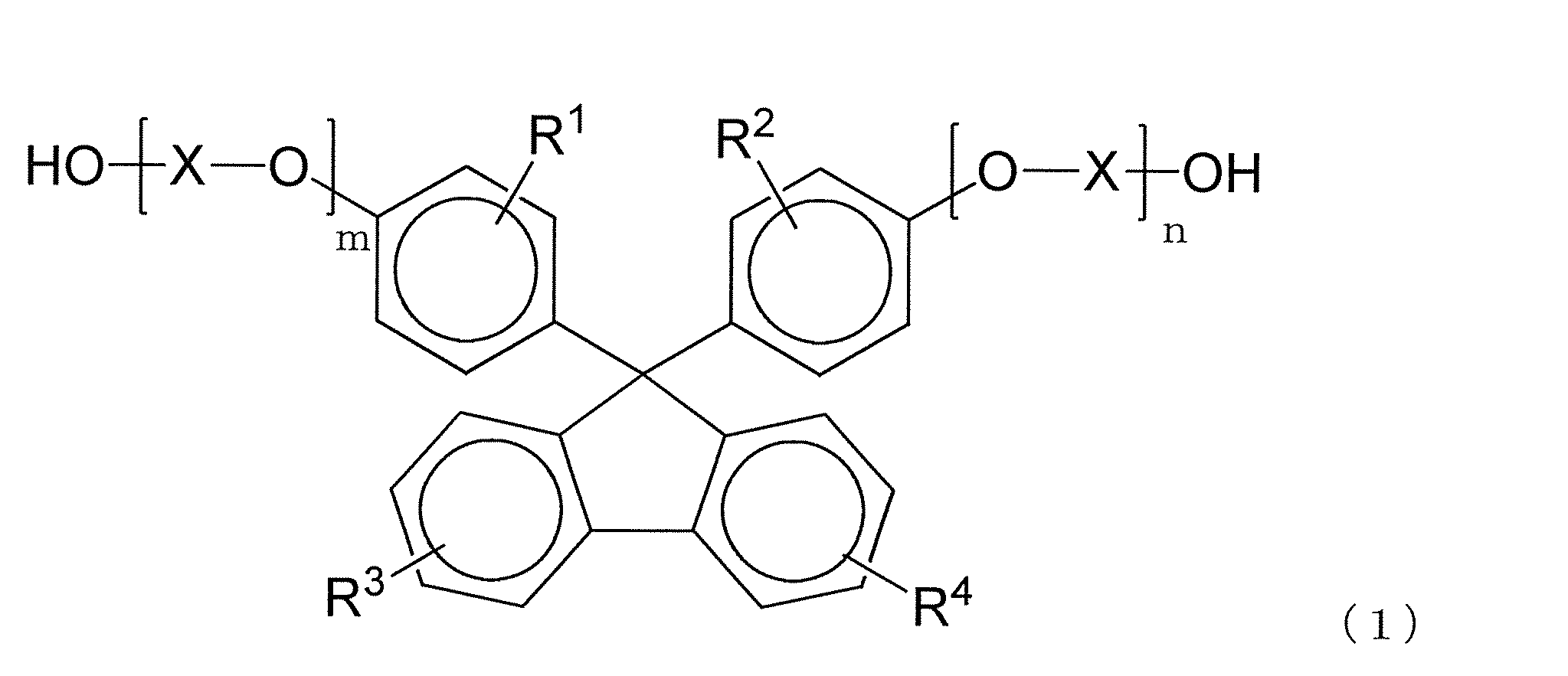

[式(3)で表される部位が-CH2-OHの一部を構成する部位である場合を除く。]
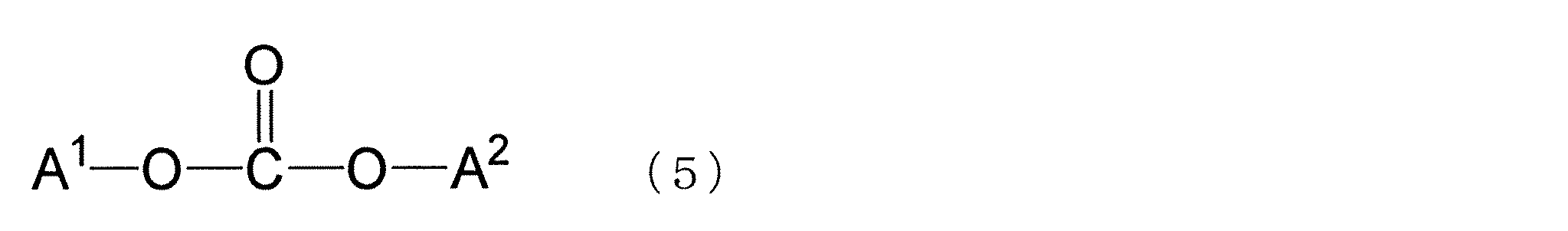
- 請求項1から請求項20のいずれか1項に記載のポリカーボネート樹脂を製膜してなる、フィルム。
- 前記フィルムが、少なくとも一方向に延伸されてなるものである、請求項21に記載のフィルム。
- 波長450nmで測定した位相差(Re450)と波長550nmで測定した位相差(Re550)の比(Re450/Re550)が、0.5以上0.93以下である、請求項21または請求項22に記載のフィルム。
- 波長450nmで測定した位相差(Re450)と波長550nmで測定した位相差(Re550)の比(Re450/Re550)が、0.5以上0.90以下である、請求項21または請求項22に記載のフィルム。
- 光弾性係数が40×10-12Pa-1以下である、請求項21から請求項24のいずれか1項に記載のフィルム。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11786770.5A EP2578616B1 (en) | 2010-05-27 | 2011-05-27 | Transparent film formed from a polycarbonate resin |
| KR1020127029448A KR101801580B1 (ko) | 2010-05-27 | 2011-05-27 | 폴리카보네이트 수지 및 그것으로 이루어지는 투명 필름 |
| CN2011800261434A CN102939318A (zh) | 2010-05-27 | 2011-05-27 | 聚碳酸酯树脂以及由该聚碳酸酯树脂形成的透明膜 |
| US13/686,097 US8487067B2 (en) | 2010-05-27 | 2012-11-27 | Polycarbonate resin and transparent film formed therefrom |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010121982 | 2010-05-27 | ||
| JP2010-121982 | 2010-05-27 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/686,097 Continuation US8487067B2 (en) | 2010-05-27 | 2012-11-27 | Polycarbonate resin and transparent film formed therefrom |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011149073A1 true WO2011149073A1 (ja) | 2011-12-01 |
Family
ID=45004056
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/062261 WO2011149073A1 (ja) | 2010-05-27 | 2011-05-27 | ポリカーボネート樹脂およびそれよりなる透明フィルム |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US8487067B2 (ja) |
| EP (1) | EP2578616B1 (ja) |
| JP (1) | JP5927780B2 (ja) |
| KR (1) | KR101801580B1 (ja) |
| CN (1) | CN102939318A (ja) |
| TW (1) | TWI503349B (ja) |
| WO (1) | WO2011149073A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014061677A1 (ja) | 2012-10-16 | 2014-04-24 | 三菱化学株式会社 | 樹脂組成物、延伸フィルム、円偏光板及び画像表示装置 |
| US20150141577A1 (en) * | 2012-08-01 | 2015-05-21 | Mitsubishi Chemical Corporation | Polycarbonate resin and transparent film comprising the same |
| JP2015199706A (ja) * | 2014-04-04 | 2015-11-12 | 三菱化学株式会社 | オリゴフルオレンジエステル、及びそれを用いた樹脂組成物の製造方法 |
| KR20160126998A (ko) | 2014-02-27 | 2016-11-02 | 미쓰비시 가가꾸 가부시키가이샤 | 중축합계 수지 및 그것으로 이루어지는 광학 필름 |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5448264B2 (ja) * | 2009-11-19 | 2014-03-19 | 三菱化学株式会社 | ポリカーボネート樹脂フィルム並びに透明フィルム |
| CN110577634A (zh) | 2009-11-30 | 2019-12-17 | 三菱化学株式会社 | 聚碳酸酯树脂及聚碳酸酯树脂成型品的制造方法 |
| KR101898307B1 (ko) | 2011-03-30 | 2018-09-12 | 미쯔비시 케미컬 주식회사 | 폴리카보네이트 수지의 제조 방법 |
| CN103476828B (zh) | 2011-03-30 | 2016-11-23 | 三菱化学株式会社 | 聚碳酸酯树脂的制造方法 |
| JP5962148B2 (ja) | 2011-03-31 | 2016-08-03 | 三菱化学株式会社 | ポリカーボネートの製造方法およびポリカーボネートペレット |
| JP5939012B2 (ja) * | 2012-04-19 | 2016-06-22 | 三菱化学株式会社 | ポリカーボネート樹脂の製造方法 |
| JP2014080602A (ja) * | 2012-09-26 | 2014-05-08 | Mitsubishi Chemicals Corp | ポリカーボネート樹脂 |
| JP2014080601A (ja) * | 2012-09-26 | 2014-05-08 | Mitsubishi Chemicals Corp | ポリカーボネート樹脂 |
| JP6264809B2 (ja) * | 2012-09-26 | 2018-01-24 | 三菱ケミカル株式会社 | ポリカーボネート樹脂の製造方法 |
| JP6179318B2 (ja) * | 2012-09-26 | 2017-08-16 | 三菱ケミカル株式会社 | ポリカーボネート樹脂の製造方法 |
| JP2014074106A (ja) * | 2012-10-03 | 2014-04-24 | Mitsubishi Chemicals Corp | ポリカーボネート樹脂の製造方法、ポリカーボネート樹脂ペレットおよび延伸フィルム |
| JP2014198761A (ja) * | 2013-03-29 | 2014-10-23 | 三菱化学株式会社 | ポリカーボネート樹脂成形品 |
| CN107075099B (zh) * | 2014-10-28 | 2020-06-12 | 三菱化学株式会社 | 聚碳酸酯树脂、成型品及光学膜 |
| CN105676318B (zh) | 2014-12-08 | 2018-06-05 | 三星电子株式会社 | 抗反射膜和包括其的有机发光器件 |
| KR101623086B1 (ko) | 2014-12-08 | 2016-05-20 | 삼성전자 주식회사 | 반사방지필름 및 이를 구비한 유기발광장치 |
| KR20160079687A (ko) | 2014-12-26 | 2016-07-06 | 삼성전자주식회사 | 반사방지필름 및 이를 구비한 유기발광장치 |
| KR102304889B1 (ko) | 2015-02-11 | 2021-09-23 | 삼성전자주식회사 | 유기 발광 장치 및 그 제조 방법 |
| CN107533153B (zh) * | 2015-07-13 | 2021-02-19 | 帝人株式会社 | 摄像透镜 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10101786A (ja) | 1996-09-30 | 1998-04-21 | Teijin Ltd | ポリカーボネート共重合体およびその製造方法 |
| JP2004067990A (ja) | 2002-06-12 | 2004-03-04 | Mitsubishi Gas Chem Co Inc | ポリカーボネート共重合体 |
| WO2006041190A1 (ja) | 2004-10-14 | 2006-04-20 | Teijin Limited | 光弾性定数の低いポリカーボネート及びそれからなるフィルム |
| JP2008111047A (ja) | 2006-10-30 | 2008-05-15 | Mitsubishi Gas Chem Co Inc | ポリカーボネート樹脂の製造方法 |
| WO2010064721A1 (ja) | 2008-12-05 | 2010-06-10 | 帝人化成株式会社 | 光学フィルム |
| JP2010134232A (ja) | 2008-12-05 | 2010-06-17 | Teijin Chem Ltd | 光学フィルム |
| WO2010071079A1 (ja) | 2008-12-16 | 2010-06-24 | 帝人化成株式会社 | 光学フィルム |
| JP2010230832A (ja) | 2009-03-26 | 2010-10-14 | Teijin Chem Ltd | 光学フィルム |
| JP2011111614A (ja) * | 2009-11-30 | 2011-06-09 | Mitsubishi Chemicals Corp | ポリカーボネート樹脂 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20140121894A (ko) * | 2007-06-19 | 2014-10-16 | 테이진 카세이 가부시키가이샤 | 광학 필름 |
| EP2738197A1 (en) * | 2007-12-12 | 2014-06-04 | Mitsubishi Chemical Corporation | Molded polycarbonate articles |
| EP2952536B1 (en) | 2007-12-13 | 2021-02-24 | Mitsubishi Chemical Corporation | Polycarbonate and molded article |
| EP2711386B1 (en) * | 2009-11-17 | 2017-05-31 | Mitsubishi Chemical Corporation | Polycarbonate resin and transparent film formed therefrom |
| CN110577634A (zh) | 2009-11-30 | 2019-12-17 | 三菱化学株式会社 | 聚碳酸酯树脂及聚碳酸酯树脂成型品的制造方法 |
-
2011
- 2011-05-27 KR KR1020127029448A patent/KR101801580B1/ko active IP Right Grant
- 2011-05-27 TW TW100118208A patent/TWI503349B/zh active
- 2011-05-27 JP JP2011118991A patent/JP5927780B2/ja active Active
- 2011-05-27 EP EP11786770.5A patent/EP2578616B1/en active Active
- 2011-05-27 CN CN2011800261434A patent/CN102939318A/zh active Pending
- 2011-05-27 WO PCT/JP2011/062261 patent/WO2011149073A1/ja active Application Filing
-
2012
- 2012-11-27 US US13/686,097 patent/US8487067B2/en active Active
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10101786A (ja) | 1996-09-30 | 1998-04-21 | Teijin Ltd | ポリカーボネート共重合体およびその製造方法 |
| JP2004067990A (ja) | 2002-06-12 | 2004-03-04 | Mitsubishi Gas Chem Co Inc | ポリカーボネート共重合体 |
| WO2006041190A1 (ja) | 2004-10-14 | 2006-04-20 | Teijin Limited | 光弾性定数の低いポリカーボネート及びそれからなるフィルム |
| JP2008111047A (ja) | 2006-10-30 | 2008-05-15 | Mitsubishi Gas Chem Co Inc | ポリカーボネート樹脂の製造方法 |
| WO2010064721A1 (ja) | 2008-12-05 | 2010-06-10 | 帝人化成株式会社 | 光学フィルム |
| JP2010134232A (ja) | 2008-12-05 | 2010-06-17 | Teijin Chem Ltd | 光学フィルム |
| WO2010071079A1 (ja) | 2008-12-16 | 2010-06-24 | 帝人化成株式会社 | 光学フィルム |
| JP2010230832A (ja) | 2009-03-26 | 2010-10-14 | Teijin Chem Ltd | 光学フィルム |
| JP2011111614A (ja) * | 2009-11-30 | 2011-06-09 | Mitsubishi Chemicals Corp | ポリカーボネート樹脂 |
Non-Patent Citations (2)
| Title |
|---|
| NIHON REOROGI GAKKAI-SHI, vol. 19, 1991, pages 93 - 97 |
| See also references of EP2578616A4 |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150141577A1 (en) * | 2012-08-01 | 2015-05-21 | Mitsubishi Chemical Corporation | Polycarbonate resin and transparent film comprising the same |
| WO2014061677A1 (ja) | 2012-10-16 | 2014-04-24 | 三菱化学株式会社 | 樹脂組成物、延伸フィルム、円偏光板及び画像表示装置 |
| KR20150073167A (ko) | 2012-10-16 | 2015-06-30 | 미쓰비시 가가꾸 가부시키가이샤 | 수지 조성물, 연신 필름, 원 편광판 및 화상 표시 장치 |
| US9518150B2 (en) | 2012-10-16 | 2016-12-13 | Mitsubishi Chemical Corporation | Resin composition, stretched film, circularly polarizing plate, and image display device |
| EP3385300A1 (en) | 2012-10-16 | 2018-10-10 | Mitsubishi Chemical Corporation | Resin composition, stretched film, circularly polarizing plate, and image display device |
| KR20160126998A (ko) | 2014-02-27 | 2016-11-02 | 미쓰비시 가가꾸 가부시키가이샤 | 중축합계 수지 및 그것으로 이루어지는 광학 필름 |
| KR20210020179A (ko) | 2014-02-27 | 2021-02-23 | 미쯔비시 케미컬 주식회사 | 중축합계 수지 및 그것으로 이루어지는 광학 필름 |
| JP2015199706A (ja) * | 2014-04-04 | 2015-11-12 | 三菱化学株式会社 | オリゴフルオレンジエステル、及びそれを用いた樹脂組成物の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5927780B2 (ja) | 2016-06-01 |
| TW201202297A (en) | 2012-01-16 |
| EP2578616B1 (en) | 2019-12-18 |
| US8487067B2 (en) | 2013-07-16 |
| KR101801580B1 (ko) | 2017-11-27 |
| EP2578616A4 (en) | 2015-09-30 |
| KR20130092398A (ko) | 2013-08-20 |
| US20130085254A1 (en) | 2013-04-04 |
| TWI503349B (zh) | 2015-10-11 |
| EP2578616A1 (en) | 2013-04-10 |
| CN102939318A (zh) | 2013-02-20 |
| JP2012007160A (ja) | 2012-01-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5927780B2 (ja) | ポリカーボネート樹脂及びフィルムの製造方法 | |
| KR101945075B1 (ko) | 위상차 필름, 그리고 이것을 사용한 원편광판 및 화상 표시 장치 | |
| JP5977917B2 (ja) | ポリカーボネート樹脂 | |
| JP2016199760A (ja) | ポリカーボネート樹脂組成物及び成形品 | |
| KR20140069158A (ko) | 위상차 필름, 그리고 이것을 사용한 원편광판 및 화상 표시 장치 | |
| KR20100096157A (ko) | 폴리카보네이트의 제조 방법 및 폴리카보네이트 성형물 | |
| KR20120102637A (ko) | 폴리카보네이트 수지 필름 그리고 투명 필름 및 그 제조 방법 | |
| JP6015071B2 (ja) | ポリカーボネートの製造方法および透明フィルム | |
| WO2011065505A1 (ja) | ポリカーボネート樹脂及びその製造方法 | |
| JP5079150B2 (ja) | 位相差フィルム | |
| JP5936803B2 (ja) | ポリカーボネート樹脂 | |
| JP2011157545A (ja) | ポリカーボネート樹脂組成物及びその成形品 | |
| WO2012153738A1 (ja) | ポリカーボネート樹脂及びそれよりなる透明フィルム | |
| WO2012133856A1 (ja) | ポリカーボネート樹脂の製造方法、ポリカーボネート樹脂ペレットおよび延伸フィルム | |
| JP2017075255A (ja) | 熱可塑性樹脂、及びそれよりなる光学成形体 | |
| JP6597158B2 (ja) | 熱可塑性樹脂、及びそれよりなる光学成形体 | |
| JP5731124B2 (ja) | 光弾性定数が低いポリカーボネート樹脂の製造方法 | |
| JP2011111613A (ja) | ポリカーボネート樹脂 | |
| JP2011079897A (ja) | 光弾性定数が低いポリカーボネート樹脂および光学フィルム | |
| JP2011214015A (ja) | ポリカーボネート樹脂 | |
| JP2011241277A (ja) | 共重合ポリカーボネート | |
| JP5907232B2 (ja) | ポリカーボネート樹脂 | |
| JP2015025139A (ja) | ポリカーボネート樹脂 | |
| JP6801194B2 (ja) | ポリカーボネート樹脂、該ポリカーボネート樹脂の製造方法、該ポリカーボネート樹脂からなる透明フィルムの製造方法、及び位相差フィルム | |
| JP5580017B2 (ja) | 光弾性定数が低いポリカーボネート樹脂および光学フィルム |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180026143.4 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11786770 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20127029448 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011786770 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |