WO2010071073A1 - 硬化性組成物及びその硬化物 - Google Patents
硬化性組成物及びその硬化物 Download PDFInfo
- Publication number
- WO2010071073A1 WO2010071073A1 PCT/JP2009/070684 JP2009070684W WO2010071073A1 WO 2010071073 A1 WO2010071073 A1 WO 2010071073A1 JP 2009070684 W JP2009070684 W JP 2009070684W WO 2010071073 A1 WO2010071073 A1 WO 2010071073A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- curable composition
- fine particles
- mass
- black
- meth
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/44—Polymerisation in the presence of compounding ingredients, e.g. plasticisers, dyestuffs, fillers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/08—Anhydrides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/10—Esters
- C08F220/12—Esters of monohydric alcohols or phenols
- C08F220/16—Esters of monohydric alcohols or phenols of phenols or of alcohols containing two or more carbon atoms
- C08F220/18—Esters of monohydric alcohols or phenols of phenols or of alcohols containing two or more carbon atoms with acrylic or methacrylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/10—Esters
- C08F220/12—Esters of monohydric alcohols or phenols
- C08F220/16—Esters of monohydric alcohols or phenols of phenols or of alcohols containing two or more carbon atoms
- C08F220/18—Esters of monohydric alcohols or phenols of phenols or of alcohols containing two or more carbon atoms with acrylic or methacrylic acids
- C08F220/1811—C10or C11-(Meth)acrylate, e.g. isodecyl (meth)acrylate, isobornyl (meth)acrylate or 2-naphthyl (meth)acrylate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F222/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a carboxyl radical and containing at least one other carboxyl radical in the molecule; Salts, anhydrides, esters, amides, imides, or nitriles thereof
- C08F222/10—Esters
- C08F222/1006—Esters of polyhydric alcohols or polyhydric phenols
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F222/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a carboxyl radical and containing at least one other carboxyl radical in the molecule; Salts, anhydrides, esters, amides, imides, or nitriles thereof
- C08F222/10—Esters
- C08F222/1006—Esters of polyhydric alcohols or polyhydric phenols
- C08F222/103—Esters of polyhydric alcohols or polyhydric phenols of trialcohols, e.g. trimethylolpropane tri(meth)acrylate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F222/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a carboxyl radical and containing at least one other carboxyl radical in the molecule; Salts, anhydrides, esters, amides, imides, or nitriles thereof
- C08F222/10—Esters
- C08F222/1006—Esters of polyhydric alcohols or polyhydric phenols
- C08F222/106—Esters of polycondensation macromers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F292/00—Macromolecular compounds obtained by polymerising monomers on to inorganic materials
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/02—Elements
- C08K3/04—Carbon
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/02—Elements
- C08K3/04—Carbon
- C08K3/041—Carbon nanotubes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/34—Silicon-containing compounds
- C08K3/36—Silica
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K9/00—Use of pretreated ingredients
- C08K9/04—Ingredients treated with organic substances
- C08K9/06—Ingredients treated with organic substances with silicon-containing compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L33/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides or nitriles thereof; Compositions of derivatives of such polymers
- C08L33/04—Homopolymers or copolymers of esters
- C08L33/06—Homopolymers or copolymers of esters of esters containing only carbon, hydrogen and oxygen, which oxygen atoms are present only as part of the carboxyl radical
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2666/00—Composition of polymers characterized by a further compound in the blend, being organic macromolecular compounds, natural resins, waxes or and bituminous materials, non-macromolecular organic substances, inorganic substances or characterized by their function in the composition
- C08L2666/02—Organic macromolecular compounds, natural resins, waxes or and bituminous materials
- C08L2666/04—Macromolecular compounds according to groups C08L7/00 - C08L49/00, or C08L55/00 - C08L57/00; Derivatives thereof
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/29—Coated or structually defined flake, particle, cell, strand, strand portion, rod, filament, macroscopic fiber or mass thereof
- Y10T428/2982—Particulate matter [e.g., sphere, flake, etc.]
- Y10T428/2991—Coated
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/29—Coated or structually defined flake, particle, cell, strand, strand portion, rod, filament, macroscopic fiber or mass thereof
- Y10T428/2982—Particulate matter [e.g., sphere, flake, etc.]
- Y10T428/2991—Coated
- Y10T428/2993—Silicic or refractory material containing [e.g., tungsten oxide, glass, cement, etc.]
- Y10T428/2995—Silane, siloxane or silicone coating
Definitions
- the present invention is a curable composition having a low viscosity and excellent handling properties, and excellent in light-shielding properties, heat resistance and molding processability obtained by curing the curable composition, and further in electrical conductivity if necessary. Also relates to an excellent cured product.
- the cured product is useful as a light shielding member for various optical devices such as cameras, video cameras, copying machines, and developing machines.
- Patent Document 1 JP-A-2008-138068
- Patent Document 2 JP-A-2008-138068
- Patent Document 3 JP-A-2008-138068
- Patent Document 2 discloses a method of obtaining a light-shielding film by applying a thermosetting resin composition containing carbon black on both surfaces of a synthetic resin film.
- the light-shielding films listed above cannot be manufactured by a manufacturing process including a solder reflow process because the glass transition temperature (Tg) of the member is low and there is no solder reflow resistance, and productivity cannot be improved. It was.
- the light shielding material is required to have good moldability, and when used for a precision instrument member, it is desired to have conductivity in order to reduce troubles caused by static electricity as necessary.
- the present invention provides a curable composition that can provide a light-shielding member for various optical devices such as a camera, a video camera, a copying machine, and a developing machine, and that can solve the problems associated with the prior art. For the purpose.
- an object of the present invention is to provide a curable composition that can provide a light-shielding property, heat resistance, molding processability, and if necessary, a cured product excellent in conductivity and excellent in handling properties. .
- a curable composition containing an acrylate, a mono (meth) acrylate having an ethylenically unsaturated group and an alicyclic structure, a polymerization initiator, and black inorganic fine particles has low viscosity and good handling properties.
- a light shielding member of various optical devices such as a camera, a video camera, a copying machine, and a developing machine.
- the present inventors have found that a cured product having excellent conductivity and, if necessary, excellent conductivity can be obtained.
- the gist of the present invention is as follows. [1] (a) silica fine particles; (B) a (meth) acrylate having two or more ethylenically unsaturated groups and having no ring structure; (C) a (meth) acrylate having an ethylenically unsaturated group and an alicyclic structure; (D) a polymerization initiator; (E) black inorganic fine particles; Including The silica fine particles (a) are surface-treated with a silane compound (f) represented by the following general formula (1) and a silane compound (g) represented by the following general formula (2). Curable composition:
- R 1 represents a hydrogen atom or a methyl group
- R 2 represents an alkyl group having 1 to 3 carbon atoms or a phenyl group
- R 3 represents a hydrogen atom or a hydrocarbon group having 1 to 10 carbon atoms.
- Q is an integer from 1 to 6, and r is an integer from 0 to 2.
- R 4 represents an alkyl group having 1 to 3 carbon atoms or an optionally substituted phenyl group
- R 5 represents a hydrogen atom or a hydrocarbon group having 1 to 10 carbon atoms.
- S is an integer from 0 to 6
- t is an integer from 0 to 2.
- the silica fine particles (a) Silica fine particles before the surface treatment, 5 to 25 parts by mass of the silane compound (f) with respect to 100 parts by mass of the silica fine particles,
- the glass transition temperature of the (meth) acrylate (b) homopolymer and the glass transition temperature of the (meth) acrylate (c) homopolymer are both 150 ° C. or higher.
- a curable composition that can provide a light-shielding property, heat resistance, molding processability, and, if necessary, a cured product excellent in conductivity, and excellent in handling properties.
- a cured product of the curable composition is also provided.
- the curable composition of the present invention includes (a) silica fine particles, (b) (meth) acrylate having two or more ethylenically unsaturated groups and having no ring structure, and (c) one ethylenic group.
- (meth) acrylate means methacrylate and / or acrylate.
- silica fine particles (a) used in the present invention those having an average particle diameter of 1 to 100 nm can be suitably used.
- the average particle size is less than 1 nm, the viscosity of the prepared curable composition increases, the content of the silica fine particles (a) in the curable composition is limited, and in the curable composition Dispersibility deteriorates, and a cured product obtained by curing a curable composition (hereinafter, also simply referred to as a cured product) tends not to have sufficient heat resistance.
- the average particle diameter exceeds 100 nm, appearance performance and mechanical properties may be deteriorated.
- the average particle diameter of the silica fine particles (a) is more preferably 1 to 50 nm, further preferably 5 to 50 nm, and most preferably 5 to 40 nm from the viewpoint of adjusting the viscosity of the curable composition to a suitable value.
- the average particle size of the silica fine particles (a) was determined by observing the silica fine particles with a high-resolution transmission electron microscope (Hitachi Corp., H-9000 type), and arbitrarily selecting 100 silica particles from the observed fine particle image. A particle image can be selected and obtained as a number average particle size by a known image data statistical processing technique.
- silica fine particles having different average particle diameters may be mixed and used in order to increase the filling amount of the silica fine particles (a) into the cured product.
- silica fine particles (a) porous silica sol, or a composite metal oxide of aluminum, magnesium, zinc or the like and silicon may be used.
- the content of the silica fine particles (a) in the curable composition is preferably 20 to 80% by mass, from the viewpoint of the balance between the heat resistance and environmental resistance of the cured product and the viscosity of the curable composition. More preferably, it is 40 to 60% by mass. If the content is 20 to 80% by mass, the fluidity of the curable composition and the dispersibility of the silica fine particles (a) in the curable composition are good. A cured product having strength, heat resistance and environmental resistance can be easily produced.
- the silica fine particles (a) used in the present invention are surface-treated with a silane compound (f) and a silane compound (g). That is, the silica fine particles (a) are obtained by surface-treating the silica fine particles before the surface treatment with the silane compound (f) and the silane compound (g). Each of these silane compounds will be described below. The surface treatment method for the silica fine particles (a) will be described later.
- the silane compound (f) is represented by the following general formula (1).
- R 1 represents a hydrogen atom or a methyl group
- R 2 represents an alkyl group having 1 to 3 carbon atoms or a phenyl group
- R 3 represents a hydrogen atom or a hydrocarbon group having 1 to 10 carbon atoms.
- Q is an integer from 1 to 6
- r is an integer from 0 to 2.
- R 3 is a methyl group, preferable q is 3, and preferable r is 0.
- R 2 is present, preferred R 2 is a methyl group.
- the silane compound (f) improves the dispersion stability of the silica fine particles (a) in the curable composition by reducing the viscosity of the curable composition and reacting with the (meth) acrylate (b) described later. Therefore, it is used for reducing shrinkage during curing of the curable composition and imparting moldability to the cured product. That is, when silica fine particles that are not surface-treated with the silane compound (f) are used, the viscosity of the curable composition increases, the shrinkage during curing increases, the cured product becomes brittle, and the cured product becomes Since cracks are generated, it is not preferable.
- silane compound (f) examples include ⁇ -acryloxypropyldimethylmethoxysilane, ⁇ -acryloxypropylmethyldimethoxysilane, ⁇ -acryloxypropyldiethylmethoxysilane, ⁇ -acryloxypropylethyldimethoxysilane, and ⁇ -acryloxy.
- ⁇ -acryloxypropyldimethylmethoxysilane ⁇ -acrylic Roxypropylmethyldimethoxysilane, ⁇ -methacryloxypropyldimethylmethoxysilane, ⁇ -methacryloxypropylmethyldimethoxysilane, ⁇ -acryloxypropyltrimethoxysilane, and ⁇ -methacryloxypropyltrimethoxysilane are preferable, and ⁇ - Acryloxypropyltrimethoxysilane. Moreover, these can be used in combination of 2 or more types.
- silane compound (f) can be produced by a known method and is commercially available.
- the amount of the silane compound (f) used for the surface treatment of the silica fine particles is usually 5 to 25 parts by mass, preferably 10 to 20 parts by mass, more preferably 12 to 100 parts by mass of the silica fine particles before the surface treatment. ⁇ 18 parts by mass.
- the amount of the silane compound (f) used is less than 5 parts by mass, the viscosity of the curable composition is increased, and the dispersibility of the silica fine particles (a) in the curable composition is deteriorated to cause gelation. there is a possibility.
- it exceeds 20 mass parts aggregation of a silica fine particle (a) may be caused.
- the mass of the silica fine particles refers to the mass of only the silica fine particles themselves dispersed in the organic solvent.
- the silane compound (f) has an acrylic group, that is, R 1 is a hydrogen atom. It is preferable to use a silane compound represented by the formula (1), and when the curable composition contains a large amount of methacrylate (methacrylate (b) and methacrylate (c) described later), the silane compound (f) It is preferable to use a silane compound represented by the general formula (1) containing a methacryl group, that is, R 1 is a methyl group. In such a case, a curing reaction tends to occur when the curable composition of the present invention is cured.
- silane compound (g) used in the present invention is represented by the following general formula (2).
- R 4 represents an alkyl group having 1 to 3 carbon atoms or an optionally substituted phenyl group
- R 5 represents a hydrogen atom or a hydrocarbon group having 1 to 10 carbon atoms.
- S is an integer from 0 to 6
- t is an integer from 0 to 2.
- a substituent may be bonded to the phenyl group as long as the effects of the present invention are not impaired.
- Examples of the substituent in the group formed by bonding a substituent to such a phenyl group include a methyl group, an ethyl group, a methoxy group, a hydroxyl group, a carboxyl group, a fluoro group, a phenyl group, and a naphthyl group.
- preferred R 5 is a methyl group, preferred s is 0 or 1, and preferred t is 0.
- preferred R 4 is a methyl group.
- the silica fine particles react with the silane compound (g), hydrophobicity is imparted to the surface of the silica fine particles, the dispersibility of the silica fine particles in the organic solvent is improved, and the (meth) acrylate (c) described later With good compatibility, the viscosity of the curable composition can be reduced, the storage stability of the curable composition can be improved, and at the same time, the water absorption can be lowered.
- silane compound (g) examples include phenyldimethylmethoxysilane, phenylmethyldimethoxysilane, phenyldiethylmethoxysilane, phenylethyldimethoxysilane, phenyltrimethoxysilane, phenyldimethylethoxysilane, phenylmethyldiethoxysilane, and phenyldiethylethoxysilane.
- phenyldimethylmethoxysilane, phenylmethyldimethoxysilane, phenyldiethylmethoxysilane are used from the viewpoint of improving the environmental resistance including the reduction of the viscosity of the curable composition, the improvement of storage stability, and the reduction of water absorption.
- Phenylethyldimethoxysilane, and phenyltrimethoxysilane are preferable, and phenyltrimethoxysilane is more preferable.
- These silane compounds can be used in combination of two or more.
- silane compound (g) can be produced by a known method and is commercially available.
- the amount of the silane compound (g) used in the surface treatment of the silica fine particles is usually 5 to 25 parts by mass, preferably 10 to 20 parts by mass, more preferably 12 to 100 parts by mass of the silica fine particles before the surface treatment. ⁇ 18 parts by mass.
- the usage-amount of a silane compound (g) is less than 5 mass parts, the viscosity of a curable composition may become high, gelling may be produced, or the heat resistance of hardened
- it exceeds 20 mass parts aggregation of a silica fine particle (a) may be caused.
- the mass of the silica fine particles refers to the mass of only the silica fine particles themselves dispersed in the organic solvent.
- the total amount of the silane compound (f) and the silane compound (g) used exceeds 50 parts by mass with respect to 100 parts by mass of the silica fine particles, the amount of the treating agent is too large. Aggregation and gelation may occur due to reaction between particles.
- Examples of the (meth) acrylate (b) having two or more ethylenically unsaturated groups and having no ring structure used in the present invention include trimethylolpropane tri (meth) acrylate and pentaerythritol tri (meth).
- ring structure means a benzene ring, a heterocyclic ring, or a cycloalkyl ring.
- those having three ethylenically unsaturated groups are preferable from the viewpoint of heat resistance of the cured product, and those having a glass transition temperature of a homopolymer of 150 ° C. or higher are preferable.
- trimethylolpropane tri (meth) acrylate having a glass transition temperature of a homopolymer of 200 ° C. or higher and relatively low curing shrinkage among polyfunctional (meth) acrylates is most preferable.
- the glass transition temperature of the homopolymer is measured by the following method. 100 parts by weight of (meth) acrylate (b) and 1 part by weight of diphenyl- (2,4,6-trimethylbenzoyl) phosphine oxide (trade name Speedcure TPO-L; manufactured by Nippon Sebel Hegner) as a photopolymerization initiator
- the solution is applied onto a glass substrate (50 mm ⁇ 50 mm) so that the thickness of the cured film is 100 ⁇ m, and exposed at 3 J / cm 2 using an exposure apparatus incorporating an ultrahigh pressure mercury lamp. Cure the coating.
- the cured film cut into a strip of 5 mm ⁇ 30 mm was used as a test piece, and in DMS6100 (manufactured by Seiko Denshi Kogyo Co., Ltd.) in a tensile mode, a temperature range of 20 ° C. to 300 ° C., and a temperature increase rate of 2 ° C./min.
- the peak temperature of the tan ⁇ value measured at a frequency of 1 Hz is defined as the glass transition temperature.
- the amount of the (meth) acrylate (b) used in the present invention is preferably 20 to 500 parts by mass with respect to 100 parts by mass of the silica fine particles before the surface treatment, and the viscosity and curability of the curable composition. From the viewpoint of dispersion stability of the silica fine particles (a) in the composition and heat resistance of the cured product, the amount is more preferably 30 to 300 parts by mass, and still more preferably 50 to 200 parts by mass. If the blending amount is less than 20 parts by mass, the viscosity of the curable composition is increased, and gelation may occur. When the blending amount exceeds 500 parts by mass, shrinkage at the time of curing of the curable composition is increased, and the cured product may be warped or cracked.
- the (meth) acrylate (c) having an ethylenically unsaturated group and an alicyclic structure used in the present invention imparts heat resistance and environmental resistance to the cured product, and reduces shrinkage during curing. Used to do.
- (meth) acrylates having one ethylenically unsaturated group and having an alicyclic structure are preferably used.
- Examples of such (meth) acrylates include cyclohexyl (meth) acrylate, 4-butylcyclohexyl (meth) acrylate, dicyclopentanyl (meth) acrylate, dicyclopentenyl (meth) acrylate, dicyclopentadienyl (meth) ) Acrylate, bornyl (meth) acrylate, isobornyl (meth) acrylate, tricyclodecanyl (meth) acrylate, tricyclodecane dimethanol diacrylate, adamantyl (meth) acrylate, etc .; benzyl (meth) ) Acrylate, tetrahydrofurfuryl (meth) acrylate, and the like.
- urethane (meth) acrylate having an alicyclic structure can also be mentioned. These may be used alone or in combination of two or more.
- (meth) acrylates having a glass transition temperature of a homopolymer of 150 ° C. or higher are preferable from the viewpoint of heat resistance of the cured product.
- the method for measuring the glass transition temperature of the homopolymer is the same as described above.
- dicyclopentanyl (meth) acrylate and adamantyl (meth) acrylate are preferable from the viewpoint of heat resistance and environmental resistance of the cured product, and the glass transition temperature of the homopolymer is high.
- adamantyl (meth) acrylate is most preferred.
- the alicyclic structure is a structure in which an aromatic ring structure is excluded from a structure in which carbon atoms are bonded cyclically.
- the amount of the (meth) acrylate (c) used in the present invention is preferably 5 to 400 parts by mass with respect to 100 parts by mass of the silica fine particles before the surface treatment, and the viscosity and curability of the curable composition. From the viewpoint of the dispersion stability of the silica fine particles (a) in the composition and the heat resistance of the cured product, the amount is more preferably 10 to 200 parts by weight, still more preferably 20 to 100 parts by weight. When the blending amount is less than 5 parts by mass, the viscosity of the curable composition is increased, and gelation may occur. If the blending amount exceeds 400 parts by mass, cracks may occur in the cured product, and the heat resistance and environmental resistance of the cured product may decrease.
- Examples of the polymerization initiator (d) used in the present invention include a photopolymerization initiator that generates radicals and a thermal polymerization initiator. These may be used alone or in combination.
- photopolymerization initiator examples include benzophenone, benzoin methyl ether, benzoin propyl ether, diethoxyacetophenone, 1-hydroxy-phenylphenyl ketone, 2,6-dimethylbenzoyldiphenylphosphine oxide, 2,4,6-trimethylbenzoyldiphenylphosphine. Oxides and diphenyl- (2,4,6-trimethylbenzoyl) phosphine oxide. Two or more of these photopolymerization initiators may be used in combination.
- the content of the photopolymerization initiator in the curable composition may be an amount that allows the curable composition to be appropriately cured, and is 0.01 to 10% by mass with respect to the entire curable composition. More preferably, it is 0.02 to 5% by mass, and further preferably 0.1 to 2% by mass. If the addition amount of the photopolymerization initiator is too large, the storage stability of the curable composition is lowered, colored, or the crosslinking at the time of crosslinking to obtain a cured product rapidly proceeds, such as cracks during curing. In addition to problems, there is a risk of increasing the outgas component during high temperature processing and contaminating the equipment. On the other hand, when there is too little addition amount of a photoinitiator, there exists a possibility that hardening of a curable composition may become inadequate.
- thermal polymerization initiator examples include benzoyl peroxide, diisopropyl peroxycarbonate, t-butylperoxy (2-ethylhexanoate), 1,1-di (t-hexylperoxy) cyclohexane, 1,1-di ( t-butylperoxy) cyclohexane, 2,2-di (4,4-di- (t-butylperoxy) cyclohexyl) propane, t-hexylperoxysopropyl monocarbonate, t-butylperoxymaleic acid, t-butylperoxy 3,5,5-trimethylhexanoate, t-butylperoxylaurate, t-butylperoxysopropyl monocarbonate, t-butylperoxy-2-ethylhexyl monocarbonate, t-hexylperoxybenzoate, 2,5-dimethyl- 2,5-di Benzoy
- the content of the thermal polymerization initiator in the curable composition is preferably 0.01 to 5% by mass and more preferably 0.1 to 2% by mass with respect to the entire curable composition. . If the amount of the thermal polymerization initiator added is too large, the storage stability of the curable composition will be reduced, colored, or cross-linking will proceed rapidly when obtaining a cured product by crosslinking, such as cracks during curing. In addition to problems, there is a risk of increasing the outgas component during high temperature processing and contaminating the equipment. On the other hand, when there is too little addition amount of a thermal-polymerization initiator, there exists a possibility that hardening of a curable composition may become inadequate.
- the total amount of these polymerization initiators used is preferably 0.01 to 10% by mass relative to the entire curable composition.
- the content is preferably 0.02 to 5% by mass.
- Black inorganic fine particles (e) As the black inorganic fine particles (e) used in the present invention, acetylene black, lamp black, furnace black, ketjen black, thermal black, etc., which are collectively referred to as carbon black, graphite (graphite), carbon nanotube, activated carbon, perylene Carbon particles such as black, titanium black, copper oxide, chromium oxide, iron oxide (including mars black, ferrite, magnetite, etc.), metal oxides such as manganese oxide, cobalt oxide, titanium nitride, titanium oxynitride, disulfide Complex oxide black pigments formed from molybdenum or alloys of these metals, azo black pigments such as aniline black, organic black pigments such as cyanine black, ivory black, peach black, anthraquinone organic black pigments Etc. It is. Further, a black pigment obtained by mixing organic pigments of three colors of red, green and blue can be used as the black inorganic fine particles (e).
- carbon black and titanium black are preferable, and carbon black is particularly preferable in terms of light shielding properties and image characteristics.
- a mixture of carbon black and titanium black can also be used.
- the carbon black commercially available ones can be used, and the number average particle diameter is preferably 5 to 200 nm, more preferably 10 to 100 nm in consideration of dispersibility and resolution. When the number average particle size is less than 5 nm, uniform dispersion becomes difficult, and when the number average particle size exceeds 200 nm, the resolution tends to decrease.
- carbon nanotubes are preferable from the viewpoint of conductivity.
- Commercially available carbon nanotubes can be used, and the average diameter is preferably 0.5 to 300 nm, more preferably 1 to 200 nm.
- the average length is preferably 100 nm to 50 ⁇ m, more preferably 200 nm to 20 ⁇ m.
- the average particle diameter of black inorganic fine particles (e) having a relatively small aspect ratio such as carbon black and titanium black is observed with a high-resolution transmission electron microscope (Hitachi Ltd. H-9000 type). It is possible to arbitrarily select 100 black inorganic fine particles from the observed fine particle image and obtain the number average particle diameter by a known image data statistical processing technique.
- the average diameter and average length are measured as follows. That is, carbon nanotubes are dispersed and fixed on a sample table of an SEM (scanning electron microscope) with a double-sided tape, etc., observed with a FESEM (field emission scanning electron microscope), and arbitrarily tens of Several hundred carbon nanotubes can be selected and averaged by a known statistical processing method to determine the average diameter.
- the length of the carbon nanotubes is arbitrarily determined from the observed image by dispersing a small amount of carbon nanotubes in a solvent such as ethanol, pouring a small amount of the dispersion on an aluminum foil and drying it. An average length can be obtained by selecting several tens to several hundreds of carbon nanotubes and averaging them by a known data statistical processing technique.
- carbon black Asahi Carbon Corporation Asahi # 120, Asahi # 90, Asahi # 78, Asahi # 80, Asahi # 80L, Asahi # 75, Asahi # 73, Asahi # 70, SANBLACK900, SANBLACK300, SANBLACK200, SANBLACK 905, SANBLACK 305, SANBLACK 215, SANBLACK X15, Special Black 550 manufactured by Degussa, Special Black 350, Special Black 250, Special Black 100, 57 manufactured by Mitsubishi Chemical Co., Ltd.
- carbon nanotubes include VGCF (registered trademark), VGCF (registered trademark) -H, VGCF (registered trademark) -S, VGCF (registered trademark) -X, manufactured by Showa Denko KK, and Meijo Nanocarbon Company. Meijo Arc APJ, Meijo Arc FH, Meijo Arc SO and the like.
- the black inorganic fine particles (e) those having hydrophobicity on the surface are desirable from the viewpoint of improving the light shielding property by improving the dispersibility.
- the surface is treated with one or more hydrophobicity imparting agents selected from the group consisting of silicone resins, alkoxysilanes, silane coupling agents, higher fatty acid salts, etc. to impart hydrophobicity. Examples thereof include a method of coating the surface with an agent.
- hydrophobicity-imparting agent examples include higher fatty acids such as higher fatty acid glyceryl, higher fatty acid, higher fatty acid polyvalent metal salt, higher fatty acid sulfate polyvalent metal salt, higher alcohol and derivatives thereof, perfluorination or partial Organic fluorine compounds such as fluorinated higher fatty acids and higher alcohols, silicone resins, silane coupling agents, alkoxysilanes, chlorosilanes, and organic silicon compounds such as silazanes can be used. In view of practical effects, silicone resins and silane coupling agents are preferably used, and silicone resins are more preferable.
- silicone resin for example, dimethylpolysiloxane, methylhydrogenpolysiloxane, and methylphenylpolysiloxane are preferable, and dimethylpolysiloxane is more preferable.
- silane coupling agents include ⁇ -acryloxypropyldimethylmethoxysilane, ⁇ -acryloxypropylmethyldimethoxysilane, ⁇ -methacryloxypropyldimethylmethoxysilane, ⁇ -methacryloxypropylmethyldimethoxysilane, ⁇ -acryloxypropyltri Methoxysilane, ⁇ -methacryloxypropyltrimethoxysilane, phenyldimethylmethoxysilane, phenylmethyldimethoxysilane, phenyldiethylmethoxysilane, phenylethyldimethoxysilane, and phenyltrimethoxysilane are preferred. Among them, ⁇ -acryloxypropyltrimethoxysilane, More preferred is phenyltrimethoxysilane.
- the surface treatment method using a silicone resin or a silane coupling agent is not limited, and for example, either a wet method or a dry method is possible.
- the wet method is preferable in that a uniform surface treatment can be performed in which the surface of the particles is completely wetted with the hydrophobicity-imparting agent.
- the addition amount of the hydrophobicity-imparting agent may be an amount that can cover a part or all of the surface of the black inorganic fine particles from which the hydrophobicity-imparting agent is a raw material.
- the amount of the hydrophobicity-imparting agent to be added is not generally determined, but if excessive, the amount deposited on the surface other than the surface of the black inorganic fine particles is not economical.
- the addition amount of the hydrophobicity imparting agent is usually 0.5 to 20% by mass, preferably 0.5 to 10% by mass, more preferably 1 to 6% by mass with respect to the black inorganic fine particles. If the addition amount is less than 0.5% by mass, the hydrophobicity is low and the dispersibility is not sufficient, and if it exceeds 20% by mass, the resolution may be lowered.
- the content of the black inorganic fine particles (e) in the curable composition may be an amount that can ensure a sufficient light-shielding property and can maintain the fluidity of the curable composition appropriately.
- the entire curable composition The content is preferably 0.1 to 20% by mass, more preferably 0.5 to 15% by mass, and still more preferably 1.0 to 12% by mass. If the added amount of the black inorganic fine particles (e) is too large, the dispersion in the curable composition may be hindered, and the handling property may be deteriorated due to aggregation or a significant increase in viscosity. If the added amount of e) is too small, there is a possibility that the light-shielding property or conductivity cannot be sufficiently exhibited.
- the curable composition of the present invention is a leveling agent, an antioxidant, an ultraviolet absorber, a solvent, a pigment, as long as it does not impair the properties such as the viscosity of the composition and the heat resistance of the cured product. It may contain other fillers such as inorganic fillers, reactive diluents, dispersants and other modifiers.
- leveling agents include polyether-modified dimethylpolysiloxane copolymer, polyester-modified dimethylpolysiloxane copolymer, polyether-modified methylalkylpolysiloxane copolymer, aralkyl-modified methylalkylpolysiloxane copolymer, and polyether-modified. Examples thereof include methylalkylpolysiloxane copolymer.
- filler or pigment examples include calcium carbonate, talc, mica, clay, Aerosil (registered trademark), barium sulfate, aluminum hydroxide, zinc stearate, zinc white, bengara, azo pigment, and the like.
- solder 3000 for example, Solsperse 3000, Solsperse 5000, Solsperse 9000, Solsperse 11200, Solsperse 12000, Solsperse 13240, Solsperse 13650, Solsperse 13940, Solsperse 16000, Solsperse 17000, Solsperse 18000, Solsperse 20000, Solsperse manufactured by Nippon Lubrizol Co., Ltd.
- the amount of the dispersant added is usually 5 wt% to 100 wt%, preferably 10 wt% to 80 wt%, more preferably 20 wt% to 60 wt%, in terms of solid content with respect to the black inorganic fine particles. If the addition amount is less than 5 wt%, the dispersibility is not sufficient, and if it is 100 wt% or more, the physical properties of the cured product may be seriously affected.
- the curable composition of the present invention containing such various components was measured using a B-type viscometer DV-III URTRA (manufactured by BROOKFIELD) under spindle conditions of 25 ° C.
- the viscosity using 41 is usually 100 (rotation speed 4 rpm) to 20000 (rotation speed 0.4 rpm) mPa ⁇ s.
- it is usually 500 (rotation speed 4 rpm) to 400000 (rotation speed 0.03 rpm) mPa ⁇ s.
- the curable composition of the present invention has an extremely low viscosity even when it does not contain a solvent, and has good handling properties. This is due to the high dispersion stability due to the surface treatment of the silica fine particles (a) and the black inorganic fine particles (e) described above.
- the curable composition of the present invention includes, for example, a step of obtaining silica fine particles (a) by surface-treating silica fine particles dispersed in an organic solvent with silane compounds (f) and (g) (step 1), silica fine particles (a ) (Meth) acrylates (b) and (c) are added and mixed uniformly (step 2), silica fine particles (a) and (meth) acrylates (b) and (c) obtained in step 2
- the step of distilling off and removing the organic solvent and water from the homogeneous mixed solution (step 3), the composition obtained by distilling off and removing the solvent in step 3, the polymerization initiator (d), and black inorganic fine particles It can manufacture by performing sequentially the process (process 4) of adding (e), uniformly mixing, and disperse
- step 1 silica fine particles are surface-treated with silane compounds (f) and (g).
- the surface treatment is necessary for adding an organic solvent dispersion of silica fine particles into a reactor, adding and stirring the silane compounds (f) and (g) while stirring, and further hydrolyzing the silane compound. While adding water and a catalyst and stirring, the silane compound is hydrolyzed and subjected to condensation polymerization on the surface of the silica fine particles.
- the silica fine particles (a) are preferably used in a state dispersed in an organic solvent.
- an organic solvent dispersion of silica fine particles for the surface treatment because the silica fine particles (a) are obtained in a state dispersed in an organic solvent.
- Such an organic solvent dispersion of silica fine particles can be produced by a conventionally known method, and is commercially available, for example, under the trade name Snowtech IPA-ST (manufactured by Nissan Chemical Co., Ltd.).
- the disappearance due to hydrolysis of the silane compound can be confirmed by gas chromatography.
- a non-polar column DB-1 manufactured by J & W
- gas chromatography Alignment Co., Ltd. Model 6850
- temperature 50 to 300 ° C. heating rate 10 ° C./min
- He He
- the residual amount of the silane compound can be measured by an internal standard method with a flow rate of 1.2 cc / min and a flame ionization detector, disappearance due to hydrolysis of the silane compound can be confirmed.
- the amount of the silane compound (f) used in the surface treatment of the silica fine particles is usually 5 to 25 parts by mass, preferably 10 to 20 parts by mass with respect to 100 parts by mass of the silica fine particles before the surface treatment. Part, more preferably 12 to 18 parts by weight.
- the amount of the silane compound (g) used is usually 5 to 25 parts by mass, preferably 10 to 20 parts by mass, more preferably 12 to 18 parts by mass with respect to 100 parts by mass of the silica fine particles.
- the lower limit of the amount of water required for carrying out the hydrolysis reaction is 1 times the total number of moles of alkoxy groups and hydroxy groups bonded to the silane compounds (f) and (g), and the upper limit is 10 times. . If the amount of water is too small, the hydrolysis rate may become extremely slow, resulting in lack of economic efficiency, or the surface treatment may not proceed sufficiently. Conversely, if the amount of water is excessively large, the silica fine particles (a) may form a gel.
- a catalyst for the hydrolysis reaction When performing the hydrolysis reaction, a catalyst for the hydrolysis reaction is usually used.
- a catalyst include, for example, inorganic acids such as hydrochloric acid, acetic acid, sulfuric acid, phosphoric acid; Organic acids such as formic acid, propionic acid, oxalic acid, p-toluenesulfonic acid, benzoic acid, phthalic acid, maleic acid; Alkaline catalysts such as potassium hydroxide, sodium hydroxide, calcium hydroxide, ammonia; Organometallics; Metal alkoxides; Organotin compounds such as dibutyltin dilaurate, dibutyltin dioctylate, dibutyltin diacetate; Aluminum tris (acetylacetonate), titanium tetrakis (acetylacetonate), titanium bis (butoxy) bis (acetylacetonate), titanium bis (isopropoxy) bis (acetylacetonate),
- hydrochloric acid, acetic acid, maleic acid, and boron compounds are preferable from the viewpoints of solubility in water and sufficient hydrolysis rate.
- These catalysts can be used alone or in combination of two or more.
- Step 1 when the hydrolysis reaction of the silane compounds (f) and (g) is performed, a water-insoluble catalyst may be used, but a water-soluble catalyst is preferably used.
- a water-soluble catalyst for hydrolysis reaction it is preferable to dissolve the water-soluble catalyst in an appropriate amount of water and add it to the reaction system because the catalyst can be uniformly dispersed.
- the addition amount of the catalyst used for the hydrolysis reaction is not particularly limited, but is usually 0.1 to 10 parts by mass, preferably 0.5 to 5 parts by mass with respect to 100 parts by mass of silica fine particles.
- the mass of the silica fine particles refers to the mass of only the silica fine particles themselves dispersed in the organic solvent.
- the reaction temperature of the hydrolysis reaction is not particularly limited, but is usually in the range of 10 to 80 ° C, preferably in the range of 20 to 50 ° C. If the reaction temperature is excessively low, the hydrolysis rate may be extremely slow, resulting in lack of economic efficiency, and the surface treatment may not proceed sufficiently. When the reaction temperature is excessively high, the gelation reaction tends to occur.
- the reaction time for performing the hydrolysis reaction is not particularly limited, but is usually in the range of 10 minutes to 48 hours, preferably 30 minutes to 24 hours.
- the surface treatment with the silane compound (f) and the silane compound (g) in Step 1 may be performed sequentially, but it is preferable to perform the surface treatment at the same time in terms of simplification and efficiency of the reaction process.
- the method of mixing the silica fine particles (a) and the (meth) acrylates (b) and (c) is not particularly limited.
- silica fine particles (a) silica fine particles dispersed in an organic solvent are preferably used from the viewpoint of dispersibility in the curable composition.
- organic solvent it is preferable to use an organic solvent that can dissolve organic components (such as (meth) acrylate (b) and (meth) acrylate (c) described later) contained in the curable composition.
- organic solvent examples include alcohols, ketones, esters, and glycol ethers.
- Alcohol solvents such as butyl alcohol and n-propyl alcohol, and ketone organic solvents such as methyl ethyl ketone and methyl isobutyl ketone are preferred.
- isopropyl alcohol is particularly preferable.
- silica fine particles (a) dispersed in isopropyl alcohol are used, the viscosity of the curable composition after desolvation is lower than when other solvents are used, and the curable composition having a low viscosity is stabilized. Can be produced.
- the temperature is preferably maintained at 20 to 100 ° C., and more preferably 30 to 70 ° C., and further preferably 30 to 50 ° C. in terms of the balance between aggregation gelation prevention and the solvent removal speed. If the temperature is raised too much, the fluidity of the curable composition may be extremely lowered, or the curable composition may be gelled.
- the degree of vacuum at the time of depressurization is usually 10 to 4000 kPa, more preferably 10 to 1000 kPa, and most preferably 10 to 500 kPa in order to balance the solvent removal speed and the prevention of aggregation gelation. If the value of the degree of vacuum is too large, the desolvation speed becomes extremely slow and the economy is lacking.
- the composition after desolvation contains substantially no solvent.
- substantially means that when a cured product is actually obtained using the curable composition of the present invention, it is not necessary to go through a step of removing the solvent again.
- the remaining amount of the organic solvent and water in the curable composition is preferably 1% by mass or less, preferably 0.5% by mass or less, and more preferably 0.1% by mass or less.
- a polymerization inhibitor may be added to 100 parts by mass of the composition after desolvation before desolvation.
- the polymerization inhibitor is used to prevent components contained in the composition from undergoing a polymerization reaction during the solvent removal process or during storage of the composition and the curable composition after the solvent removal.
- the polymerization inhibitor include hydroquinone, hydroquinone monomethyl ether, benzoquinone, pt-butylcatechol, 2,6-di-tert-butyl-4-methylphenol, and the like. These may be used alone or in combination of two or more.
- Step 3 can be carried out by transferring the homogeneous mixed liquid of silica fine particles (a) and (meth) acrylates (b) and (c) that have undergone Step 2 to a dedicated apparatus. If carried out in the reactor which has been carried out, it can also be carried out in the reactor subsequent to step 2.
- step 4 the method of adding the polymerization initiator (d) and the black inorganic fine particles (e) to the composition obtained by removing the solvent in step 3 and uniformly mixing and dispersing is not particularly limited. Mixing with a mixer such as a mixer, ball mill, three rolls, etc., and adding and mixing the polymerization initiator (d) with continuous stirring in the reactor in which Steps 1 to 3 were performed, black inorganic fine particles After adding (e) separately, a method of mixing at room temperature with a mixer such as a mixer, ball mill, three rolls, bead mill, etc., and black inorganic fine particles (e And a polymerization initiator (d) is added and mixed after mixing with a mixer such as a mixer, ball mill, three rolls, or bead mill at room temperature.
- a mixer such as a mixer, ball mill, three rolls, or bead mill at room temperature.
- the curable composition obtained by adding, mixing, and dispersing such a polymerization initiator (d) and black inorganic fine particles (e) may be filtered as necessary. This filtration is performed for the purpose of removing foreign substances such as dust in the curable composition.
- the filtration method is not particularly limited, but a method of pressure filtration using a membrane type or cartridge type filter having a pressure filtration pore size of 1.0 ⁇ m is preferable.
- the curable composition of the present invention can be produced through the above steps.
- the silica fine particles (a) which is a constituent component, are treated with a specific silane compound, so that the viscosity is low and the handling property is good even if it does not contain a solvent.
- the curable composition of the present invention When the curable composition of the present invention is cured, it becomes a cured product that can be used as a light-shielding member for various optical devices such as cameras, video cameras, copying machines, and developing machines. Moreover, it can apply also to the curable black resin composition for inkjet used for color filter manufacture by the inkjet method by using it as it is.
- a cured product is obtained by curing the curable composition of the present invention.
- the curing method include a method of crosslinking the ethylenically unsaturated groups of (meth) acrylates (b) and (c) by irradiation with active energy rays, a method of thermally polymerizing the ethylenically unsaturated groups by applying heat, and the like. Yes, these can be used in combination.
- a photopolymerization initiator is contained in the curable composition in Step 4 described above.
- a thermal polymerization initiator is contained in the curable composition.
- both a photoinitiator and a thermal-polymerization initiator are contained.
- the cured product of the present invention is applied, for example, to the surface of an electronic device member made by combining the curable composition of the present invention with a glass plate, a plastic plate, a metal plate, a silicon wafer, or a combination of these materials. It can be obtained by irradiation or heating with active energy rays. Moreover, you may perform both irradiation of an active energy ray, and a heating for hardening.
- Examples of the method of applying the curable composition include application by a bar coater, applicator, die coater, spin coater, spray coater, curtain coater, roll coater, etc., application by screen printing, and application by dipping.
- the amount of application of the curable composition of the present invention on the substrate is not particularly limited, and can be appropriately adjusted according to the purpose.
- the amount of the coating film obtained after the curing treatment by active energy ray irradiation and / or heating is preferably 10 ⁇ m to 5 mm, and more preferably 20 ⁇ m to 3 mm.
- the active energy rays used for curing are preferably electron beams and light in the ultraviolet to infrared wavelength range.
- the light source for example, an ultra-high pressure mercury light source and a metal halide light source can be used for ultraviolet rays, a metal halide light source and a halogen light source can be used for visible rays, and a halogen light source can be used for infrared rays. Can be used.
- curing may be further advanced by heat treatment (annealing treatment).
- the heating temperature at that time is preferably in the range of 80 to 200 ° C.
- the heating time is preferably in the range of 10 minutes to 60 minutes.
- the heating temperature is preferably in the range of 80 to 200 ° C, more preferably in the range of 100 to 150 ° C.
- the heating temperature is lower than 80 ° C., it is necessary to lengthen the heating time, and there is a tendency that it is not economical. Therefore, it tends to lack economic efficiency.
- the curing may be further advanced by heat treatment (annealing treatment) as necessary.
- the heating temperature at that time is preferably in the range of 150 to 200 ° C.
- the heating time is preferably in the range of 5 minutes to 60 minutes.
- the cured product of the present invention is excellent in light shielding properties, heat resistance and molding processability, and therefore can be suitably used as a light shielding member for various optical devices such as a camera, a video camera, a copying machine, and a developing machine, for example, a light shielding film. .
- the cured product of the present invention is obtained by curing a curable composition containing (meth) acrylates (b) and (c) having a high glass transition temperature of the homopolymer, it is excellent in heat resistance. .
- the 5% weight reduction temperature when heated in a nitrogen atmosphere is usually 280 ° C. or higher, preferably 300 ° C. or higher, more preferably 320 ° C. or higher.
- the 5% weight loss temperature when heated is lower than 280 ° C., for example, when this cured product is used for an active matrix display element substrate, problems such as warpage and deflection in the manufacturing process, and generation of cracks may occur.
- the cured product of the present invention has excellent light shielding properties.
- the light shielding property can be evaluated using the optical density (OD value). Since the optical density depends on the thickness of the cured film and the amount of fine particles contained, it is necessary to adjust the film thickness or the amount of black inorganic fine particles according to the target optical density.
- the cured product obtained from the curable composition of the present invention can be used as a light shielding member for various optical devices such as a camera, a video camera, a copying machine, and a developing machine.
- the optical density required for use in these applications is 1.0 or more, preferably 2.0 or more, and more preferably 4.0 or more. If the optical density is less than 1.0, the light shielding property may be insufficient.
- Example 1 [Preparation of curable composition]
- 100 parts by mass of isopropyl alcohol-dispersed colloidal silica (silica content 30% by mass, average particle size 10 to 20 nm, trade name Snowtech IPA-ST; manufactured by Nissan Chemical Co., Ltd.) is placed.
- the silica fine particles were surface-treated by stirring for 24 hours to obtain a dispersion of silica fine particles (a).
- silica fine particle (a) dispersion 22.5 parts by mass of trimethylolpropane triacrylate (trade name: Biscoat # 295; manufactured by Osaka Organic Chemical Co., Ltd., Tg> 250 ° C. of homopolymer) was added to 100 parts by mass of the silica fine particle (a) dispersion.
- 22.5 parts by mass of adamantyl methacrylate (trade name ADMA; manufactured by Osaka Organic Chemical Co., Ltd., homopolymer Tg 180 ° C.) was added and mixed uniformly.
- the silica dispersion resin composition was obtained by heating under reduced pressure at 40 degreeC and 100 kPa, stirring, and removing a volatile matter. The amount of volatile matter removed was 72.4 parts by mass.
- carbon black whose surface is treated with dimethylpolysiloxane as black inorganic fine particles with respect to 95 parts by mass of the silica-dispersed resin composition (dimethylpolysiloxane-treated denka black granular product, trade name SI06-5; Daito) 5 parts by mass (manufactured by Kasei Kogyo Co., Ltd., number average particle size: 30 to 50 nm) was added and kneaded using a planetary mixer to completely disperse the carbon black, whereby a light-shielding resin composition 1 was obtained.
- the above surface treatment with dimethylpolysiloxane of carbon black was performed by the following method. That is, while adding 100 g of carbon black in a 1 L Henschel mixer and flowing, 100 g of isopropyl alcohol dissolved with 5 g of dimethylpolysiloxane ⁇ Shin-Etsu Chemical Co., Ltd. KF96-100CS) was added dropwise and mixed, and then vacuum dried at 80 ° C. The isopropyl alcohol was removed. Furthermore, after heating at 180 degreeC for 4 hours, it air-cooled to room temperature and prepared surface treatment carbon black.
- 100 parts by mass of the light-shielding resin composition 1 includes 4 parts by mass of diphenyl- (2,4,6-trimethylbenzoyl) phosphine oxide (trade name Speedcure TPO-L; manufactured by Nippon Sebel Hegner) as a photopolymerization initiator. And 1 part by mass of t-butylperoxy (2-ethylhexanoate) (trade name perbutyl O; manufactured by NOF Corporation) as a thermal polymerization initiator were mixed with the curable composition 1 (BR -1) was obtained.
- diphenyl- (2,4,6-trimethylbenzoyl) phosphine oxide trade name Speedcure TPO-L; manufactured by Nippon Sebel Hegner
- t-butylperoxy (2-ethylhexanoate) trade name perbutyl O; manufactured by NOF Corporation
- the viscosity of the resulting curable composition 1 was 300,000 mPa ⁇ s.
- the viscosity was measured using a B-type viscometer DV-III ULTRA (manufactured by BROOKFIELD), spindle no. 41, measured at a rotation speed of 0.05 rpm and 25 ° C.
- Table 1 below shows the composition of the components used in the preparation of the curable composition 1 described above.
- A It can be processed (peeled) without causing cracks or cracks.
- the total light transmittance (%) of the obtained cured film was measured using a color / turbidity simultaneous measuring machine (COH400 manufactured by Nippon Denshoku Co., Ltd.), and the OD value was calculated from the obtained value, and its light shielding property. Were evaluated according to the following indices.
- Example 2 [Preparation of curable composition] A curable composition 2 (BR-2) was prepared by performing the same operation as in Example 1 except that the addition amount of trimethylolpropane triacrylate was changed to 50 parts by mass and the addition amount of adamantyl methacrylate was changed to 10 parts by mass. Got.
- composition of the components used for the preparation of the above curable composition 2 is shown in Table 1 below.
- Example 2 The same performance evaluation as in Example 1 was performed using this cured film. The results are shown in Table 2.
- Example 3 [Preparation of curable composition] A curable composition was obtained by performing the same operation as in Example 1 except that adamantyl methacrylate was changed to dicyclopentanyl methacrylate (trade name FA-513M; manufactured by Hitachi Chemical Co., Ltd., homopolymer Tg 175 ° C.). Product 3 (BR-3) was obtained.
- composition of the components used for the preparation of the above curable composition 3 is shown in Table 1 below.
- Example 2 The same performance evaluation as in Example 1 was performed using this cured film. The results are shown in Table 2.
- Example 4 [Preparation of curable composition]
- the curable composition 4 (BR-4) was obtained by performing the same operation as in Example 1 except that the composition of the light-shielding resin composition was changed to 98 parts by mass of the silica-dispersed resin composition and 2 parts by mass of the black inorganic fine particles. )
- composition of the components used for the preparation of the above curable composition 4 is shown in Table 1 below.
- Example 2 The same performance evaluation as in Example 1 was performed using this cured film. The results are shown in Table 2.
- Example 5 [Preparation of curable composition] A curable composition 5 (BR-5) was obtained in the same manner as in Example 1 except that the photopolymerization initiator was not used.
- composition of the components used for the preparation of the above curable composition 5 is shown in Table 1 below.
- Example 6 [Preparation of curable composition] A curable composition 6 (BR-6) was obtained in the same manner as in Example 2 except that no photopolymerization initiator was used.
- composition of the components used for the preparation of the above curable composition 6 is shown in Table 1 below.
- Example 7 [Preparation of curable composition] A curable composition 7 (BR-7) was obtained in the same manner as in Example 3 except that no photopolymerization initiator was used.
- composition of the components used for the preparation of the above curable composition 7 is shown in Table 1 below.
- Example 8 [Preparation of curable composition] A curable composition 8 (BR-8) was obtained in the same manner as in Example 4 except that no photopolymerization initiator was used.
- composition of the components used for the preparation of the curable composition 8 is shown in Table 1 below.
- Example 9 [Preparation of curable composition] A curable composition 9 (BR-9) was obtained in the same manner as in Example 5 except that the composition amount was changed to the values shown in Table 1. The composition of the components used for the preparation of the curable composition 9 is shown in Table 1 below.
- a cured film was obtained by heat-curing in the same manner as in Example 5 except that the heating temperature and time were changed to 130 ° C. and 16 minutes, respectively.
- Example 10 [Preparation of curable composition] A curable composition 10 (BR-10) was obtained by performing the same operation as in Example 5 except that the composition amount was changed to the values shown in Table 1. The composition of the components used for the preparation of the curable composition 10 is shown in Table 1 below.
- Example 11 [Preparation of curable composition] A curable composition 11 (BR-11) was obtained by performing the same operation as in Example 9, except that the composition amount was changed to the values shown in Table 1. The composition of the components used for the preparation of the curable composition 11 is shown in Table 1 below.
- t-butyl peroxy (2-ethylhexanoate) (trade name Perbutyl O; manufactured by NOF Corporation) is mixed as a thermal polymerization initiator with 100 parts by mass of the light-shielding resin composition 15.
- a paste-like curable composition 15 (BR-15) was obtained.
- the composition of the components used for the preparation of the curable composition 15 is shown in Table 3 below. [Preparation of cured film] The curable composition 15 was heated and cured at the same heating temperature and heating time as in Example 9 to obtain a cured film.
- the measuring method of volume resistivity is as follows.
- ⁇ Volume resistivity> The above cured film was prepared on a glass substrate, and a measurement point was prepared on the cured film using a silver paste (trade name: Dotite D-550, manufactured by Fujikura Kasei Kogyo Co., Ltd.). Next, the resistance value between two points was measured using an ohmmeter, and the volume resistivity was determined by converting the volume between the measurement points.
- Example 13 [Preparation of curable composition] Carbon nanotubes (trade name VGCF (registered trademark); manufactured by Showa Denko KK) were changed to carbon nanotubes (trade name VGCF (registered trademark) -H; manufactured by Showa Denko KK, average diameter 150 nm, average length 6 ⁇ m). Were the same as in Example 12 to obtain a curable composition 16 (BR-16).
- composition of the components used for the preparation of the above curable composition 16 is shown in Table 3 below.
- Example 16 [Preparation of curable composition] Except that the carbon black of Example 9 was changed to a carbon nanotube (trade name VGCF (registered trademark) -H; manufactured by Showa Denko KK, average diameter 150 nm, average length 6 ⁇ m), the composition was the same as that of Example 9, The same operation as in Example 12 was performed to obtain curable composition 19 (BR-19). The composition of the components used for the preparation of the above curable composition 19 is shown in Table 3 below.
- curable composition 23 (BR-23) was obtained in the same manner as in Example 16 except that trimethylolpropane trimethacrylate and adamantyl methacrylate were replaced with only trimethylolpropane trimethacrylate.
- the composition of the components used for the preparation of the curable composition 23 is shown in Table 3 below.
- the curable composition of the present invention containing silica fine particles surface-treated with two specific types of silane compounds, two specific types of (meth) acrylates, a polymerization initiator, and black inorganic fine particles is handled with low viscosity. Good properties.
- the curable composition by curing the curable composition, it can be suitably used as a light shielding member for various optical devices such as a camera, a video camera, a copying machine, and a developing machine.
- An excellent cured product can be obtained.
- carbon nanotubes are used as the black inorganic fine particles, the conductivity is further improved.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Polymers & Plastics (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Materials Engineering (AREA)
- Engineering & Computer Science (AREA)
- Inorganic Chemistry (AREA)
- Nanotechnology (AREA)
- Polymerisation Methods In General (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Pigments, Carbon Blacks, Or Wood Stains (AREA)
- Macromonomer-Based Addition Polymer (AREA)
- Optical Elements Other Than Lenses (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
Abstract
Description
[1](a)シリカ微粒子と、
(b)2つ以上のエチレン性不飽和基を有し且つ環構造を有しない(メタ)アクリレートと、
(c)エチレン性不飽和基を有し且つ脂環式構造を有する(メタ)アクリレートと、
(d)重合開始剤と、
(e)黒色無機微粒子と、
を含み、
前記シリカ微粒子(a)が、下記一般式(1)で表されるシラン化合物(f)及び下記一般式(2)で表されるシラン化合物(g)で表面処理されていることを特徴とする硬化性組成物:
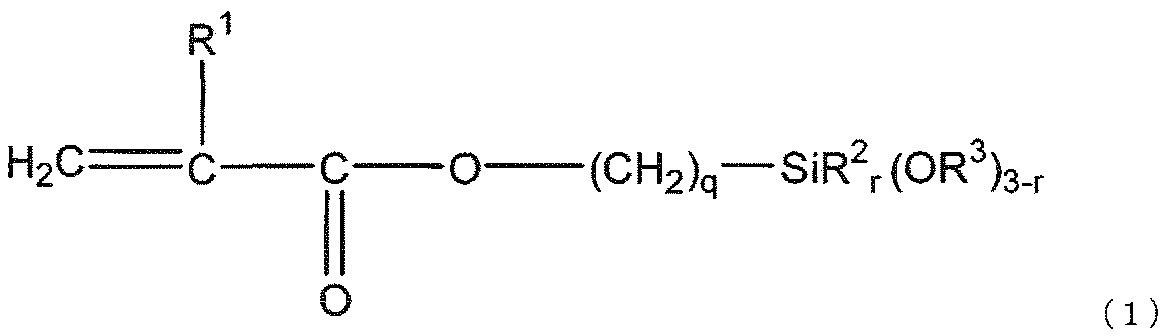
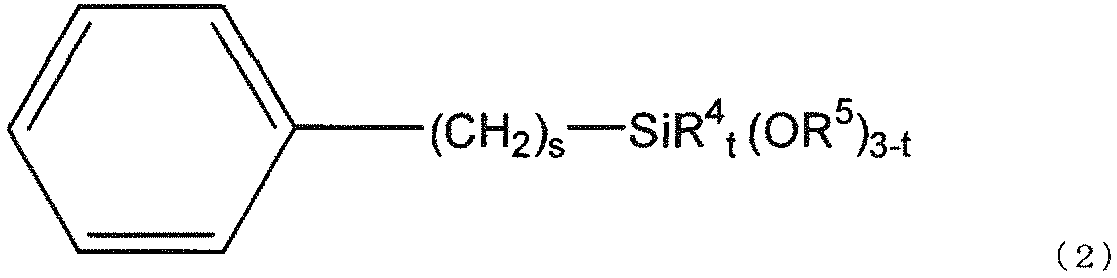
[2]前記(メタ)アクリレート(b)が、3つのエチレン性不飽和基を有し且つ環構造を有しない(メタ)アクリレートであることを特徴とする[1]に記載の硬化性組成物。
[3]前記シリカ微粒子(a)が、
表面処理される前のシリカ微粒子を、該シリカ微粒子100質量部に対して5~25質量部の前記シラン化合物(f)と、
前記シリカ微粒子100質量部に対して5~25質量部の前記シラン化合物(g)とで表面処理して得られることを特徴とする[1]又は[2]に記載の硬化性組成物。
[4]前記(メタ)アクリレート(b)の単独重合体のガラス転移温度及び前記(メタ)アクリレート(c)の単独重合体のガラス転移温度がともに150℃以上であることを特徴とする[1]~[3]のいずれかに記載の硬化性組成物。
[5]黒色無機微粒子(e)が、カーボンブラック、チタンブラック及びカーボンナノチューブからなる群から選ばれる少なくとも1種であることを特徴とする[1]~[4]のいずれかに記載の硬化性組成物。
[6]黒色無機微粒子(e)が、カーボンブラック及び/又はチタンブラックであることを特徴とする[1]~[4]のいずれかに記載の硬化性組成物。
[7]カーボンブラック及び/又はチタンブラックの数平均粒子径が5~200nmであることを特徴とする[6]に記載の硬化性組成物。
[8]前記黒色無機微粒子(e)が、シリコーン樹脂によって表面処理を施されたカーボンブラックであることを特徴とする[1]~[4]のいずれかに記載の硬化性組成物。
[9]シリコーン樹脂によって表面処理を施されたカーボンブラックの数平均粒子径が5~200nmであることを特徴とする[8]に記載の硬化性組成物。
[10]黒色無機微粒子(e)が、カーボンナノチューブであることを特徴とする[1]~[4]のいずれかに記載の硬化性組成物。
[11]カーボンナノチューブの平均直径が0.5~200nmであり、平均長さが100nm~50μmであることを特徴とする[10]に記載の硬化性組成物。
[12]粘度が30~2000mPa・sであることを特徴とする[1]~[11]のいずれかに記載の硬化性組成物。
[13][1]~[12]のいずれかに記載の硬化性組成物を硬化させてなる硬化物。
[14][13]に記載の硬化物からなる遮光膜。
本発明の硬化性組成物は、(a)シリカ微粒子と、(b)2つ以上のエチレン性不飽和基を有し且つ環構造を有しない(メタ)アクリレートと、(c)1つのエチレン性不飽和基を有し且つ脂環式構造を有する(メタ)アクリレートと、(d)重合開始剤と、(e)黒色無機微粒子とを含み、前記シリカ微粒子(a)が、特定のシラン化合物で表面処理されていることを特徴している。以下これら各構成要素について説明する。尚、ここで(メタ)アクリレートとは、メタクリレート及び/又はアクリレートを意味する。
本発明で用いられるシリカ微粒子(a)としては、平均粒子径が1~100nmのものを好適に用いることができる。平均粒子径が1nm未満であると、作製した硬化性組成物の粘度が増大し、シリカ微粒子(a)の硬化性組成物中での含有量が制限されるとともに、硬化性組成物中での分散性が悪化し、硬化性組成物を硬化させて得られる硬化物(以下単に硬化物とも言う)が十分な耐熱性を得ることができない傾向がある。また、平均粒子径が100nmを越えると、外観性能や機械特性が低下する場合がある。
前記シラン化合物(f)は、下記一般式(1)で表される。
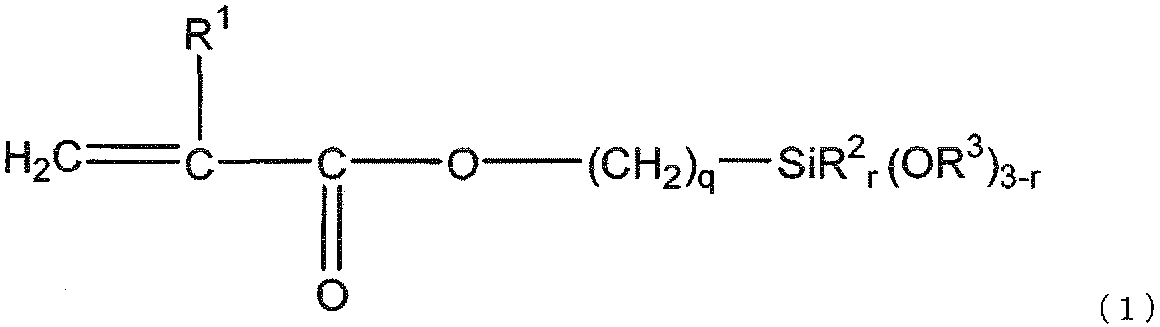
本発明で用いられるシラン化合物(g)は、下記一般式(2)で表される。
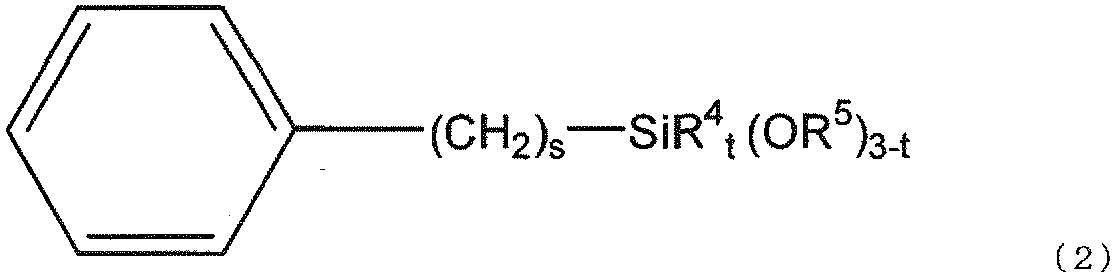
本発明で用いられる2つ以上のエチレン性不飽和基を有し且つ環構造を有しない(メタ)アクリレート(b)としては、例えば、トリメチロールプロパントリ(メタ)アクリレート、ペンタエリスリトールトリ(メタ)アクリレート、ペンタエリスリトールテトラ(メタ)アクリレート、ジペンタエリスリトールテトラ(メタ)アクリレート、ジペンタエリスリトールペンタ(メタ)アクリレート、ジペンタエリスリトールヘキサ(メタ)アクリレート、トリメチロールプロパントリオキシエチル(メタ)アクリレート等の多官能(メタ)アクリレートが挙げられる。また上記の他、環構造を有しない多官能ウレタン(メタ)アクリレートも挙げることができる。これらは、1種単独で用いても、2種以上を併用してもよい。ここで、「環構造」とは、ベンゼン環、複素環、シクロアルキル環をいう。
本発明で用いられるエチレン性不飽和基を有し且つ脂環式構造を有する(メタ)アクリレート(c)は、硬化物に耐熱性、耐環境性を付与するため、及び硬化時の収縮を低減するために用いられる。なかでも1つのエチレン性不飽和基を有し且つ脂環式構造を有する(メタ)アクリレートが好適に用いられる。そのような(メタ)アクリレートとして、たとえば、シクロヘキシル(メタ)アクリレート、4-ブチルシクロヘキシル(メタ)アクリレート、ジシクロペンタニル(メタ)アクリレート、ジシクロペンテニル(メタ)アクリレート、ジシクロペンタジエニル(メタ)アクリレート、ボルニル(メタ)アクリレート、イソボルニル(メタ)アクリレート、トリシクロデカニル(メタ)アクリレート、トリシクロデカンジメタノールジアクリレート、アダマンチル(メタ)アクリレートなどのシクロアルキル(メタ)アクリレート類;ベンジル(メタ)アクリレート、テトラヒドロフルフリル(メタ)アクリレート等が挙げられる。また上記の他、脂環式構造を有するウレタン(メタ)アクリレートも挙げることができる。これらは、1種単独で用いても、2種以上を併用してもよい。
本発明で用いられる重合開始剤(d)としては、ラジカルを発生する光重合開始剤及び熱重合開始剤が挙げられる。これらは、それぞれ単独に用いても良く、併用してもよい。
本発明で用いられる黒色無機微粒子(e)としては、カーボンブラックとして総称される、アセチレンブラック、ランプブラック、ファーネスブラック、ケッチェンブラック、サーマルブラック等や、グラファイト(黒鉛)、カーボンナノチューブ、活性炭、ペリレンブラック、などの炭素系粒子、チタンブラック、酸化銅、酸化クロム、酸化鉄(マルスブラック、フェライト、マグネタイトなど含む)、酸化マンガン、酸化コバルトなどの金属酸化物、窒化チタン、酸窒化チタン、二硫化モリブデンまたこれらに含まれる金属の合金から形成される複合酸化物系黒色色素、アニリンブラックのようなアゾ系黒色顔料、シアニンブラックのような有機黒色顔料、アイボリーブラック、ピーチブラック、アントラキノン系有機黒色色素などが挙げられる。また、赤色、緑色、青色の三色の有機顔料を混合して得られる黒色系顔料を黒色無機微粒子(e)として用いることもできる。
本発明の硬化性組成物は、例えば、有機溶媒に分散したシリカ微粒子をシラン化合物(f)及び(g)で表面処理してシリカ微粒子(a)を得る工程(工程1)、シリカ微粒子(a)に(メタ)アクリレート(b)及び(c)を添加し、均一混合する工程(工程2)、工程2で得られた、シリカ微粒子(a)と(メタ)アクリレート(b)及び(c)との均一混合液から有機溶媒及び水を留去・脱溶媒する工程(工程3)、工程3で留去・脱溶媒して得られた組成物に重合開始剤(d)と、黒色無機微粒子(e)を添加、均一混合、分散して硬化性組成物とする工程(工程4)を順次行うことにより製造することができる。以下各工程について説明する。
工程1では、シリカ微粒子をシラン化合物(f)及び(g)で表面処理する。表面処理は、シリカ微粒子の有機溶媒分散液を反応器に入れ、攪拌しながら、シラン化合物(f)及び(g)を添加、攪拌混合し、さらに該シラン化合物の加水分解を行うのに必要な水と触媒を添加、攪拌しながら、該シラン化合物を加水分解し、シリカ微粒子表面にて縮重合させることにより行う。
蟻酸、プロピオン酸、シュウ酸、パラトルエンスルホン酸、安息香酸、フタル酸、マレイン酸等の有機酸;
水酸化カリウム、水酸化ナトリウム、水酸化カルシウム、アンモニア等のアルカリ触媒;
有機金属;
金属アルコキシド;
ジブチルスズジラウレート、ジブチルスズジオクチレート、ジブチルスズジアセテート等の有機スズ化合物;
アルミニウムトリス(アセチルアセトネート)、チタニウムテトラキス(アセチルアセトネート)、チタニウムビス(ブトキシ)ビス(アセチルアセトネート)、チタニウムビス(イソプロポキシ)ビス(アセチルアセトネート)、ジルコニウムビス(ブトキシ)ビス(アセチルアセトネート)、ジルコニウムビス(イソプロポキシ)ビス(アセチルアセトネート)等の金属キレート化合物;
ホウ素ブトキシド、ホウ酸等のホウ素化合物;
等が挙げられる。
工程2において、シリカ微粒子(a)と(メタ)アクリレート(b)及び(c)とを混合する方法は、特に制限は無いが、たとえば、室温又は加熱条件下でミキサー、ボールミル、3本ロールなどの混合機により混合する方法や、工程1を行った反応器の中で連続的に攪拌しながら(メタ)アクリレート(b)及び(c)を添加、混合する方法が挙げられる。
工程3において、シリカ微粒子(a)と(メタ)アクリレート(b)及び(c)との均一混合液から有機溶媒及び水を留去・脱溶媒(以下これらをまとめて脱溶媒という)するには、減圧状態で加熱することが好ましい。
工程4において、工程3で脱溶媒して得られた組成物に重合開始剤(d)、黒色無機微粒子(e)を添加、均一混合、分散する方法は、特に制限はないが、たとえば、室温でミキサー、ボールミル、3本ロールなどの混合機により混合する方法、工程1~3を行った反応器の中で連続的に攪拌しながら重合開始剤(d)を添加、混合し、黒色無機微粒子(e)を別途添加したのちに、室温でミキサー、ボールミル、3本ロール、ビーズミルなどの混合機により混合する方法、及び工程1~3を行うことで得られた組成物に黒色無機微粒子(e)を添加し室温でミキサー、ボールミル、3本ロール、ビーズミルなどの混合機により混合したのち、重合開始剤(d)を添加、混合する方法等が挙げられる。
本発明の硬化性組成物を硬化させることにより、硬化物が得られる。硬化の方法としては、活性エネルギー線の照射により(メタ)アクリレート(b)及び(c)のエチレン性不飽和基を架橋させる方法、熱をかけてエチレン性不飽和基を熱重合させる方法等があり、これらを併用することもできる。
本発明の硬化物は、遮光性、耐熱性及び成形加工性に優れることから、カメラ、ビデオカメラ、複写機、現像機等の各種光学機器の遮光部材、たとえば遮光膜として好適に用いることができる。
[硬化性組成物の調製]
セパラブルフラスコに、イソプロピルアルコール分散型コロイダルシリカ(シリカ含量30質量%、平均粒子径10~20nm、商品名スノーテックIPA-ST;日産化学(株)製)100質量部を入れ、該セパラブルフラスコにγ-メタクリロキシプロピルトリメトキシシラン4.5質量部とフェニルトリメトキシシラン4.5質量部を加え、攪拌混合し、さらに0.1825質量%のHCl溶液2.9質量部を加え、20℃で24hr撹拌することにより、シリカ微粒子の表面処理を行い、シリカ微粒子(a)の分散液を得た。
<活性エネルギー線硬化、加熱硬化>
硬化性組成物1を基板上に塗布し、200μmのスペーサーを挟んで、その上部と下部をガラス板で挟み、超高圧水銀ランプを組み込んだ露光装置で3J/cm2露光し塗膜を硬化させた。その後、更に120℃で9分間加熱処理を行うことで、塗膜を完全に硬化させた。
<成形加工性>
上記の硬化膜をガラス基板から剥がす際に、硬化膜に割れ又はクラックが生じることなく、加工できる度合いを下記の指標で評価した。
得られた硬化膜の全光線透過率(%)を色彩・濁度同時測定機(日本電飾社製 COH400)を用いて測定し、また得られた値よりOD値を算出し、その遮光性を下記の指標で評価した。
B: 0.01%<全光線透過率≦1%(4>OD値≧2)
C: 1%<全光線透過率≦10%(2>OD値≧1)
D: 全光線透過率>10%(OD値<1)
<耐熱形状安定性>
得られた硬化膜について、270℃で1分加熱したあと、1分間室温で放冷する工程を3回行った後の硬化物の形状安定性をそれぞれ下記の指標で評価した。
得られた硬化膜について、TG-DTA(セイコー電子工業社製)を用いて、窒素雰囲気下、温度範囲20℃~500℃、昇温速度10℃/minで処理した際の、5%重量減少温度を求めた。その5%重量減少温度の値が高いほど、耐熱性が良好な硬化膜である。
[硬化性組成物の調製]
トリメチロールプロパントリアクリレートの添加量を50質量部、アダマンチルメタクリレートの添加量を10質量部に変更した点以外は実施例1と同様の操作を行うことで、硬化性組成物2(BR-2)を得た。
実施例1と同様に、硬化性組成物2を活性エネルギー線硬化及び加熱硬化させることにより硬化膜を得た。
[硬化性組成物の調製]
アダマンチルメタクリレートをジシクロペンタニルメタクリレート(商品名FA-513M;日立化成(株)製、単独重合体のTg175℃)に変更した点以外は実施例1と同様の操作を行うことで、硬化性組成物3(BR-3)を得た。
実施例1と同様に、硬化性組成物3を活性エネルギー線硬化及び加熱硬化させることにより硬化膜を得た。
[硬化性組成物の調製]
遮光樹脂組成物の組成を、シリカ分散樹脂組成物98質量部、黒色無機微粒子2質量部に変更した点以外は実施例1と同様の操作を行うことで、硬化性組成物4(BR-4)を得た。
実施例1と同様に、硬化性組成物4を活性エネルギー線硬化及び加熱硬化させることにより硬化膜を得た。
[硬化性組成物の調製]
光重合開始剤を用いない点以外は実施例1と同様の操作を行うことで、硬化性組成物5(BR-5)を得た。
<加熱硬化>
硬化性組成物5を基板上に塗布し、200μmのスペーサーを挟んで、その上部と下部をガラス板で挟み、120℃で9分間加熱処理を行うことで、塗膜を完全に硬化させた。
[硬化性組成物の調製]
光重合開始剤を用いない点以外は実施例2と同様の操作を行うことで、硬化性組成物6(BR-6)を得た。
実施例5と同様に、硬化性組成物6を加熱硬化させることにより硬化膜を得た。
[硬化性組成物の調製]
光重合開始剤を用いない点以外は実施例3と同様の操作を行うことで、硬化性組成物7(BR-7)を得た。
実施例5と同様に、硬化性組成物7を加熱硬化させることにより硬化膜を得た。
[硬化性組成物の調製]
光重合開始剤を用いない点以外は実施例4と同様の操作を行うことで、硬化性組成物8(BR-8)を得た。
実施例5と同様に、硬化性組成物8を加熱硬化させることにより硬化膜を得た。
(実施例9)
[硬化性組成物の調製]
組成量を表1に記載の値に変更した点以外は実施例5と同様の操作を行うことで、硬化性組成物9(BR-9)を得た。硬化性組成物9の調製に用いた成分の組成を下記表1に示す。
加熱温度及び時間を、それぞれ130℃、16分間に変更した点以外は実施例5と同様に加熱硬化させることにより硬化膜を得た。
(実施例10)
[硬化性組成物の調製]
組成量を表1に記載の値に変更した点以外は実施例5と同様の操作を行うことで、硬化性組成物10(BR-10)を得た。硬化性組成物10の調製に用いた成分の組成を下記表1に示す。
実施例5と同様に、硬化性組成物10を加熱硬化させることにより硬化膜を得た。
(実施例11)
[硬化性組成物の調製]
組成量を表1に記載の値に変更した点以外は実施例9と同様の操作を行うことで、硬化性組成物11(BR-11)を得た。硬化性組成物11の調製に用いた成分の組成を下記表1に示す。
実施例5と同様に、硬化性組成物11を加熱硬化させることにより硬化膜を得た。
(比較例1)
[硬化性組成物の調製]
トリメチロールプロパントリアクリレート及びアダマンチルメタクリレートを、アダマンチルメタクリレートのみに置き換えた点以外は実施例9と同様の操作を行うことで、硬化性組成物12(BR-12)を得た。硬化性組成物12の調製に用いた成分の組成を下記表1に示す。
調製中に組成物がゲル状となり、硬化膜を調製することができなかった。
(比較例2)
[硬化性組成物の調製]
トリメチロールプロパントリメタクリレート及びアダマンチルメタクリレートを、トリメチロールプロパントリメタクリレートのみに置き換えた点以外は、実施例9と同様の操作を行うことで、硬化性組成物13(BR-13)を得た。硬化性組成物13の調製に用いた成分の組成を下記表1に示す。
実施例9と同様の加熱温度及び加熱時間にて、硬化性組成物11を加熱硬化させることにより硬化膜を得た。
(比較例3)
[硬化性組成物の調製]
シリカ微粒子を使用せず、表1に示す組成を用いた以外は実施例9と同様の操作を行うことで、硬化性組成物14(BR-14)を得た。硬化性組成物14の調製に用いた成分の組成を下記表1に示す。
実施例9と同様の加熱温度及び加熱時間にて加熱硬化を試みたが、評価可能な硬化膜を得ることはできなかった。
(実施例12)
[導電性硬化性組成物の調製]
セパラブルフラスコに、イソプロピルアルコール分散型コロイダルシリカ(シリカ含量30質量%、平均粒子径10~20nm、商品名スノーテックIPA-ST;日産化学(株)製)125.3質量部を入れ、該セパラブルフラスコにγ-メタクリロキシプロピルトリメトキシシラン6.8質量部とフェニルトリメトキシシラン4.5質量部を加え、攪拌混合し、さらに0.1825質量%のHCl溶液3.6質量部を加え、20℃で24hr撹拌することにより、シリカ微粒子の表面処理を行い、シリカ微粒子(a)の分散液を得た。
[硬化膜の調製]
実施例9と同様の加熱温度及び加熱時間にて、硬化性組成物15を加熱硬化させることにより硬化膜を得た。
ガラス基板上に上記の硬化膜を作成し、硬化膜上に銀ペースト(商品名:ドータイトD-550、藤倉化成工業社製)を用いて測定点を作成した。次に、抵抗計を用いて2点間の抵抗値を測定し、測定点間の体積で換算することで体積抵抗率を求めた。
(実施例13)
[硬化性組成物の調製]
カーボンナノチューブ(商品名VGCF(登録商標);昭和電工社製)を、カーボンナノチューブ(商品名VGCF(登録商標)-H;昭和電工社製、平均直径150nm、平均長さ6μm)に変更した点以外は実施例12と同様の操作を行うことで、硬化性組成物16(BR-16)を得た。
実施例12と同様に、硬化性組成物16を加熱硬化させることにより硬化膜を得た。
(実施例14)
[硬化性組成物の調製]
カーボンナノチューブ(商品名VGCF(登録商標);昭和電工社製)を、カーボンナノチューブ(商品名VGCF(登録商標)-S;昭和電工社製、平均直径80nm、平均長さ10μm)に変更した点以外は実施例12と同様の操作を行うことで、硬化性組成物17(BR-17)を得た。以上の硬化性組成物17の調製に用いた成分の組成を下記表3に示す。
実施例12と同様に、硬化性組成物17を加熱硬化させることにより硬化膜を得た。
(実施例15)
[硬化性組成物の調製]
カーボンナノチューブ(商品名VGCF(登録商標);昭和電工社製)を、カーボンナノチューブ(商品名:Meijo-Arc AP-J、名城ナノカーボン社製、平均直径1.4nm、平均長さ5μm)に変更した点以外は実施例12と同様の操作を行うことで、硬化性組成物18(BR-18)を得た。以上の硬化性組成物18の調製に用いた成分の組成を下記表3に示す。
実施例12と同様に、硬化性組成物18を加熱硬化させることにより硬化膜を得た。
(実施例16)
[硬化性組成物の調製]
実施例9のカーボンブラックをカーボンナノチューブ(商品名VGCF(登録商標)-H;昭和電工社製、平均直径150nm、平均長さ6μm)に変更した点以外は、実施例9と同等の組成とし、実施例12と同様の操作を行うことで、硬化性組成物19(BR-19)を得た。以上の硬化性組成物19の調製に用いた成分の組成を下記表3に示す。
実施例12と同様に、硬化性組成物19を加熱硬化させることにより硬化膜を得た。
(実施例17)
[硬化性組成物の調製]
実施例16のトリメチロールプロパントリメタクリレートをジペンタエリスリトールヘキサアクリレートに変更した点以外は、実施例16と同等の組成とし、実施例12と同様の操作を行うことで、硬化性組成物20(BR-20)を得た。以上の硬化性組成物20の調製に用いた成分の組成を下記表3に示す。
実施例12と同様に、硬化性組成物20を加熱硬化させることにより硬化膜を得た。
(実施例18)
[硬化性組成物の調製]
実施例16のトリメチロールプロパントリメタクリレートをペンタエリスリトールトリアクリレートに変更した点以外は、実施例16と同等の組成とし、実施例12と同様の操作を行うことで、硬化性組成物21(BR-21)を得た。以上の硬化性組成物21の調製に用いた成分の組成を下記表3に示す。
実施例12と同様に、硬化性組成物21を加熱硬化させることにより硬化膜を得た。
(比較例4)
[硬化性組成物の調製]
トリメチロールプロパントリメタクリレート及びアダマンチルメタクリレートを、アダマンチルメタクリレートのみに置き換えた点以外は実施例16と同様の操作を行ったが、合成中に組成物がゲル状となってしまい、目標の硬化性組成物22(BR-22)は得られなかった。以上の硬化性組成物22の調製に用いた成分の組成を下記表3に示す。
(比較例5)
[硬化性組成物の調製]
トリメチロールプロパントリメタクリレート及びアダマンチルメタクリレートを、トリメチロールプロパントリメタクリレートのみに置き換えた点以外は、実施例16と同様の操作を行うことで、硬化性組成物23(BR-23)を得た。以上の硬化性組成物23の調製に用いた成分の組成を下記表3に示す。
実施例16と同様に、硬化性組成物23を加熱硬化させることにより硬化膜を得た。
(比較例6)
[硬化性組成物の調製]
シリカ微粒子を使用せず、表3に示す組成を用いた以外は実施例16と同様の操作を行うことで、硬化性組成物24(BR-24)を得た。以上の硬化性組成物24の調製に用いた成分の組成を下記表3に示す。
実施例16と同様に、硬化性組成物24の加熱硬化を試みたが、組成物は全く硬化せず、硬化膜を得ることはできなかった。
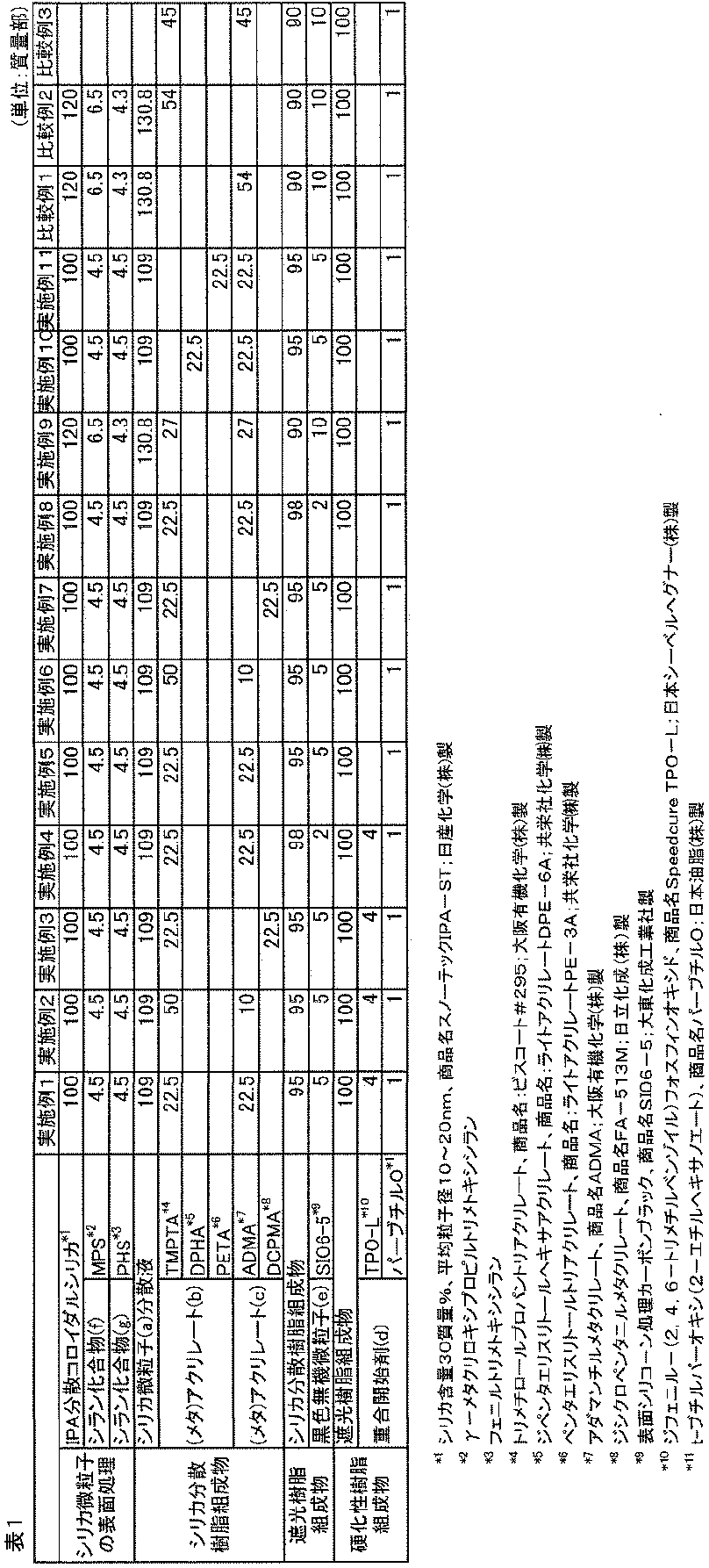
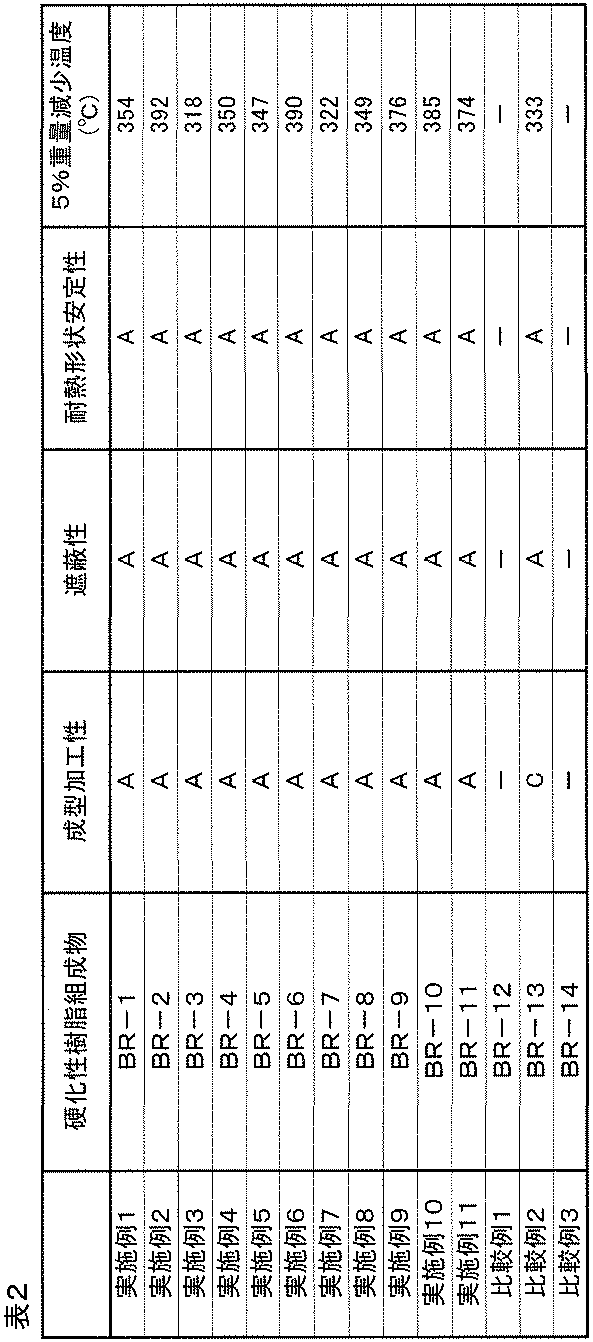
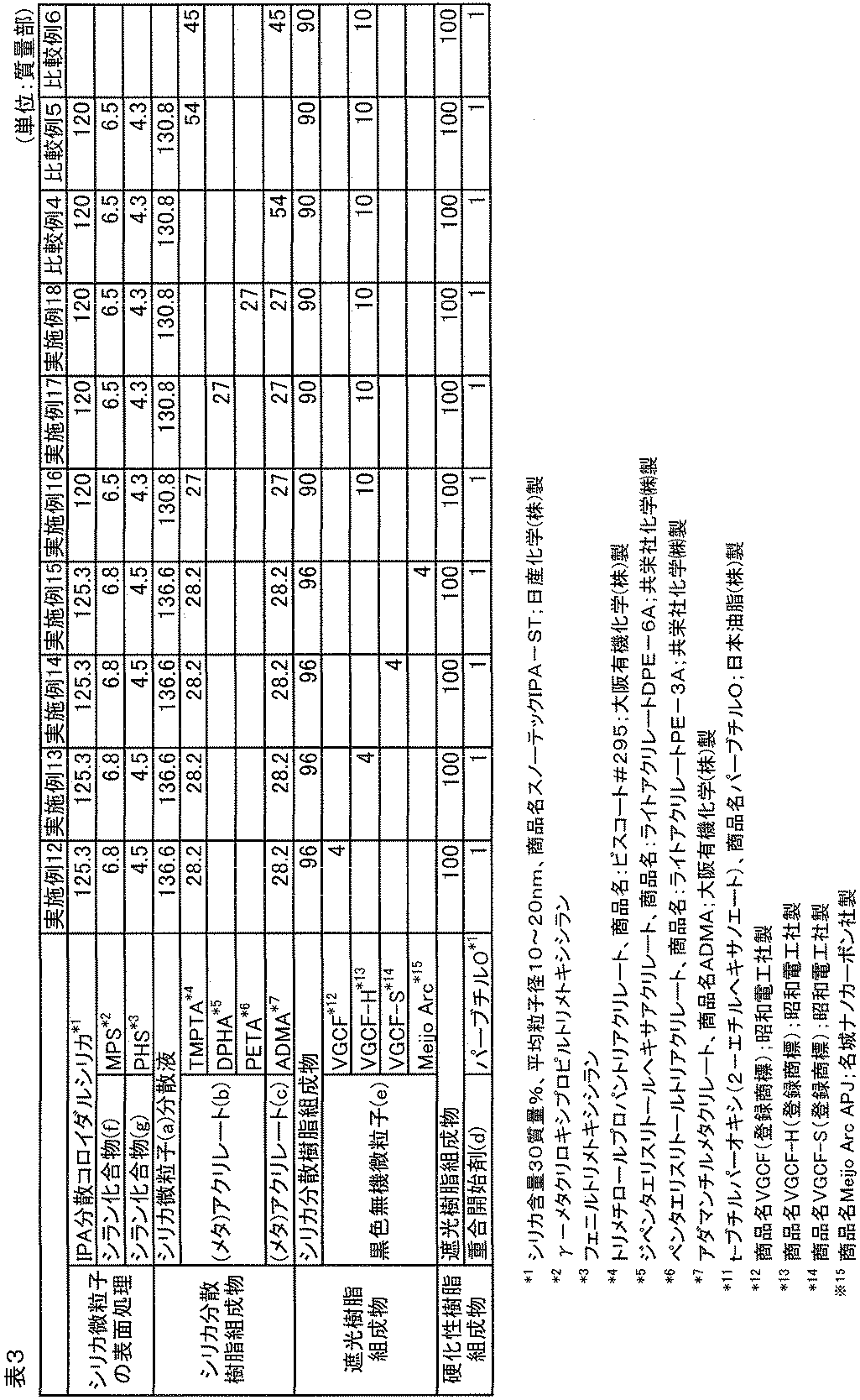

Claims (14)
- (a)シリカ微粒子と、
(b)2つ以上のエチレン性不飽和基を有し且つ環構造を有しない(メタ)アクリレートと、
(c)エチレン性不飽和基を有し且つ脂環式構造を有する(メタ)アクリレートと、
(d)重合開始剤と、
(e)黒色無機微粒子と、
を含み、
前記シリカ微粒子(a)が、下記一般式(1)で表されるシラン化合物(f)及び下記一般式(2)で表されるシラン化合物(g)で表面処理されていることを特徴とする硬化性組成物:

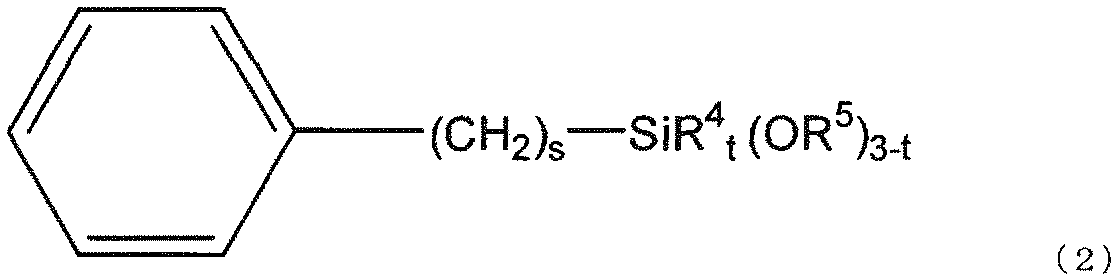
- 前記(メタ)アクリレート(b)が、3つのエチレン性不飽和基を有し且つ環構造を有しない(メタ)アクリレートであることを特徴とする請求項1に記載の硬化性組成物。
- 前記シリカ微粒子(a)が、
表面処理される前のシリカ微粒子を、該シリカ微粒子100質量部に対して5~25質量部の前記シラン化合物(f)と、
前記シリカ微粒子100質量部に対して5~25質量部の前記シラン化合物(g)と
で表面処理して得られることを特徴とする請求項1又は2に記載の硬化性組成物。 - 前記(メタ)アクリレート(b)の単独重合体のガラス転移温度及び前記(メタ)アクリレート(c)の単独重合体のガラス転移温度がともに150℃以上であることを特徴とする請求項1~3のいずれかに記載の硬化性組成物。
- 黒色無機微粒子(e)が、カーボンブラック、チタンブラック及びカーボンナノチューブからなる群から選ばれる少なくとも1種であることを特徴とする請求項1~4のいずれかに記載の硬化性組成物。
- 黒色無機微粒子(e)が、カーボンブラック及び/又はチタンブラックであることを特徴とする請求項1~4のいずれかに記載の硬化性組成物。
- カーボンブラック及び/又はチタンブラックの数平均粒子径が5~200nmであることを特徴とする請求項6に記載の硬化性組成物。
- 前記黒色無機微粒子(e)が、シリコーン樹脂によって表面処理を施されたカーボンブラックであることを特徴とする請求項1~4のいずれかに記載の硬化性組成物。
- シリコーン樹脂によって表面処理を施されたカーボンブラックの数平均粒子径が5~200nmであることを特徴とする請求項8に記載の硬化性組成物。
- 黒色無機微粒子(e)が、カーボンナノチューブであることを特徴とする請求項1~4のいずれかに記載の硬化性組成物。
- カーボンナノチューブの平均直径が0.5~200nmであり、平均長さが100nm~50μmであることを特徴とする請求項10に記載の硬化性組成物。
- 粘度が30~2000mPa・sであることを特徴とする請求項1~11のいずれかに記載の硬化性組成物。
- 請求項1~12のいずれかに記載の硬化性組成物を硬化させてなる硬化物。
- 請求項13に記載の硬化物からなる遮光膜。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2009801502996A CN102245645B (zh) | 2008-12-16 | 2009-12-10 | 固化性组合物和其固化物 |
| US13/139,847 US8349934B2 (en) | 2008-12-16 | 2009-12-10 | Hardening composition and hardened product thereof |
| SG2011043957A SG172185A1 (en) | 2008-12-16 | 2009-12-10 | Hardening composition and hardened product of same |
| KR1020117016460A KR101203301B1 (ko) | 2008-12-16 | 2009-12-10 | 경화성 조성물 및 그의 경화물 |
| EP09833376.8A EP2380916B1 (en) | 2008-12-16 | 2009-12-10 | Hardening composition and hardened product of same |
| JP2010542947A JP5689320B2 (ja) | 2008-12-16 | 2009-12-10 | 硬化性組成物及びその硬化物 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008319433 | 2008-12-16 | ||
| JP2008-319433 | 2008-12-16 | ||
| JP2009-139043 | 2009-06-10 | ||
| JP2009139043 | 2009-06-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010071073A1 true WO2010071073A1 (ja) | 2010-06-24 |
Family
ID=42268743
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2009/070684 WO2010071073A1 (ja) | 2008-12-16 | 2009-12-10 | 硬化性組成物及びその硬化物 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US8349934B2 (ja) |
| EP (1) | EP2380916B1 (ja) |
| JP (1) | JP5689320B2 (ja) |
| KR (1) | KR101203301B1 (ja) |
| CN (1) | CN102245645B (ja) |
| SG (1) | SG172185A1 (ja) |
| TW (1) | TWI500686B (ja) |
| WO (1) | WO2010071073A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010241985A (ja) * | 2009-04-07 | 2010-10-28 | Olympus Corp | 有機−無機ハイブリッド樹脂組成物、それを用いた光学素子、及び該組成物の製造方法 |
| JP2013014690A (ja) * | 2011-07-04 | 2013-01-24 | Tokuyama Dental Corp | 重合性組成物の製造方法 |
| WO2014077111A1 (ja) * | 2012-11-16 | 2014-05-22 | 富士フイルム株式会社 | 非アルカリ現像型着色組成物、非アルカリ現像型着色転写材料、着色パターン、静電容量型入力装置および画像表示装置 |
| WO2015019941A1 (ja) * | 2013-08-09 | 2015-02-12 | 昭和電工株式会社 | 半導体ナノ粒子含有硬化性組成物、硬化物、光学材料および電子材料 |
| JP2017032798A (ja) * | 2015-07-31 | 2017-02-09 | ソニーセミコンダクタソリューションズ株式会社 | レンズ付き基板、積層レンズ構造体、カメラモジュール、並びに、製造装置および方法 |
| JP2019011447A (ja) * | 2017-06-30 | 2019-01-24 | 出光興産株式会社 | 熱硬化性材料、及び当該熱硬化性材料の成形方法 |
| JP2019157062A (ja) * | 2018-03-16 | 2019-09-19 | 株式会社リコー | 活性エネルギー線硬化型組成物、活性エネルギー線硬化型インクジェットインクおよびインクジェット記録装置 |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5466640B2 (ja) * | 2008-07-03 | 2014-04-09 | 昭和電工株式会社 | 硬化性組成物及びその硬化物 |
| WO2010064534A1 (ja) * | 2008-12-01 | 2010-06-10 | 昭和電工株式会社 | 造形方法 |
| JP5643623B2 (ja) * | 2010-12-02 | 2014-12-17 | デクセリアルズ株式会社 | 異方性導電材料及びその製造方法 |
| WO2013118713A1 (ja) * | 2012-02-10 | 2013-08-15 | 昭和電工株式会社 | 硬化性組成物およびその用途 |
| JP5885585B2 (ja) * | 2012-05-21 | 2016-03-15 | 昭和電工株式会社 | 硬化性組成物およびその硬化物 |
| US10040954B2 (en) | 2015-05-28 | 2018-08-07 | E Ink California, Llc | Electrophoretic medium comprising a mixture of charge control agents |
| US11098206B2 (en) | 2015-10-06 | 2021-08-24 | E Ink Corporation | Electrophoretic media including charge control agents comprising quartenary amines and unsaturated polymeric tails |
| JP6893721B2 (ja) * | 2016-10-03 | 2021-06-23 | モメンティブ・パフォーマンス・マテリアルズ・ジャパン合同会社 | 紫外線硬化型シリコーン樹脂組成物、及びそれを用いた物品 |
| CN109280309A (zh) * | 2017-07-21 | 2019-01-29 | 住友化学株式会社 | 固化性组合物、成型体和其制造方法 |
| KR102354305B1 (ko) * | 2019-06-20 | 2022-01-21 | 주식회사 포스코 | 열전도-전기절연성 도료 조성물 및 이를 포함하는 태양전지용 외장재 강판 |
| RU2731635C9 (ru) * | 2019-11-05 | 2020-11-12 | МСД Текнолоджис С.а.р.л. | Электропроводящая резиновая композиция для сплошных шин и не оставляющая следов сплошная шина |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH049802A (ja) | 1990-04-27 | 1992-01-14 | Kimoto & Co Ltd | 光学機器用遮光部材 |
| JPH07258582A (ja) * | 1994-02-02 | 1995-10-09 | Mitsubishi Rayon Co Ltd | 被覆用組成物、それを用いた表面被覆成形物、及びその製造方法 |
| JPH1090512A (ja) * | 1996-09-13 | 1998-04-10 | Dainippon Printing Co Ltd | 非導電性遮光層用組成物、非導電性遮光層及びカラーフイルター |
| JP2008138068A (ja) | 2006-12-01 | 2008-06-19 | Toray Ind Inc | 遮光性ポリエステルフィルム |
| JP2008241767A (ja) * | 2007-03-26 | 2008-10-09 | Somar Corp | 遮光フィルム |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5502087A (en) * | 1993-06-23 | 1996-03-26 | Dentsply Research & Development Corp. | Dental composition, prosthesis, and method for making dental prosthesis |
| CA2159331C (en) | 1994-02-02 | 2008-05-06 | Hiroyuki Watanabe | Coating composition and surface-coated molding produced therewith |
| JP2000226488A (ja) * | 1999-02-04 | 2000-08-15 | Mitsubishi Rayon Co Ltd | 硬化性組成物およびそれからなる歯科用修復材料 |
| CN100396737C (zh) | 2003-06-06 | 2008-06-25 | 浙江弘晟材料科技股份有限公司 | 乳胶漆及其制备方法 |
| JP2005003772A (ja) | 2003-06-10 | 2005-01-06 | Olympus Corp | 光学材料用組成物および光学素子 |
| US20060160917A1 (en) * | 2004-12-21 | 2006-07-20 | Seiko Epson Corporation | Ink composition |
| US20060251901A1 (en) * | 2005-05-09 | 2006-11-09 | Armstrong Sean E | Curable composition and substrates possessing protective layer obtained therefrom |
| JP4896502B2 (ja) * | 2005-11-22 | 2012-03-14 | 富士フイルム株式会社 | インク組成物、インクジェット記録方法、平版印刷版の製造方法、及び平版印刷版 |
| JPWO2008015999A1 (ja) | 2006-08-04 | 2009-12-24 | コニカミノルタオプト株式会社 | 複合材料及び光学素子 |
| WO2009142237A1 (ja) * | 2008-05-23 | 2009-11-26 | 昭和電工株式会社 | 反応性(メタ)アクリレートポリマーを含有した硬化性組成物およびその硬化物 |
-
2009
- 2009-12-10 WO PCT/JP2009/070684 patent/WO2010071073A1/ja active Application Filing
- 2009-12-10 EP EP09833376.8A patent/EP2380916B1/en not_active Not-in-force
- 2009-12-10 JP JP2010542947A patent/JP5689320B2/ja active Active
- 2009-12-10 US US13/139,847 patent/US8349934B2/en not_active Expired - Fee Related
- 2009-12-10 KR KR1020117016460A patent/KR101203301B1/ko not_active IP Right Cessation
- 2009-12-10 CN CN2009801502996A patent/CN102245645B/zh not_active Expired - Fee Related
- 2009-12-10 SG SG2011043957A patent/SG172185A1/en unknown
- 2009-12-15 TW TW098142889A patent/TWI500686B/zh not_active IP Right Cessation
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH049802A (ja) | 1990-04-27 | 1992-01-14 | Kimoto & Co Ltd | 光学機器用遮光部材 |
| JPH07258582A (ja) * | 1994-02-02 | 1995-10-09 | Mitsubishi Rayon Co Ltd | 被覆用組成物、それを用いた表面被覆成形物、及びその製造方法 |
| JPH1090512A (ja) * | 1996-09-13 | 1998-04-10 | Dainippon Printing Co Ltd | 非導電性遮光層用組成物、非導電性遮光層及びカラーフイルター |
| JP2008138068A (ja) | 2006-12-01 | 2008-06-19 | Toray Ind Inc | 遮光性ポリエステルフィルム |
| JP2008241767A (ja) * | 2007-03-26 | 2008-10-09 | Somar Corp | 遮光フィルム |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2380916A4 * |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010241985A (ja) * | 2009-04-07 | 2010-10-28 | Olympus Corp | 有機−無機ハイブリッド樹脂組成物、それを用いた光学素子、及び該組成物の製造方法 |
| JP2013014690A (ja) * | 2011-07-04 | 2013-01-24 | Tokuyama Dental Corp | 重合性組成物の製造方法 |
| WO2014077111A1 (ja) * | 2012-11-16 | 2014-05-22 | 富士フイルム株式会社 | 非アルカリ現像型着色組成物、非アルカリ現像型着色転写材料、着色パターン、静電容量型入力装置および画像表示装置 |
| JP2014102289A (ja) * | 2012-11-16 | 2014-06-05 | Fujifilm Corp | 非アルカリ現像型着色組成物、非アルカリ現像型着色転写材料および着色パターン |
| WO2015019941A1 (ja) * | 2013-08-09 | 2015-02-12 | 昭和電工株式会社 | 半導体ナノ粒子含有硬化性組成物、硬化物、光学材料および電子材料 |
| JPWO2015019941A1 (ja) * | 2013-08-09 | 2017-03-02 | 昭和電工株式会社 | 半導体ナノ粒子含有硬化性組成物、硬化物、光学材料および電子材料 |
| JP2017032798A (ja) * | 2015-07-31 | 2017-02-09 | ソニーセミコンダクタソリューションズ株式会社 | レンズ付き基板、積層レンズ構造体、カメラモジュール、並びに、製造装置および方法 |
| US11194135B2 (en) | 2015-07-31 | 2021-12-07 | Sony Semiconductor Solutions Corporation | Lens attached substrate, layered lens structure, camera module, manufacturing apparatus, and manufacturing method |
| JP2019011447A (ja) * | 2017-06-30 | 2019-01-24 | 出光興産株式会社 | 熱硬化性材料、及び当該熱硬化性材料の成形方法 |
| US11926683B2 (en) | 2017-06-30 | 2024-03-12 | Idemitsu Kosan Co., Ltd. | Curable material and method for molding said thermally curable material |
| JP2019157062A (ja) * | 2018-03-16 | 2019-09-19 | 株式会社リコー | 活性エネルギー線硬化型組成物、活性エネルギー線硬化型インクジェットインクおよびインクジェット記録装置 |
| JP7010089B2 (ja) | 2018-03-16 | 2022-01-26 | 株式会社リコー | 活性エネルギー線硬化型組成物、活性エネルギー線硬化型インクジェットインクおよびインクジェット記録装置 |
Also Published As
| Publication number | Publication date |
|---|---|
| SG172185A1 (en) | 2011-07-28 |
| EP2380916B1 (en) | 2013-11-13 |
| CN102245645B (zh) | 2013-06-19 |
| JP5689320B2 (ja) | 2015-03-25 |
| TWI500686B (zh) | 2015-09-21 |
| KR20110104048A (ko) | 2011-09-21 |
| KR101203301B1 (ko) | 2012-11-21 |
| EP2380916A4 (en) | 2012-11-21 |
| TW201035224A (en) | 2010-10-01 |
| EP2380916A1 (en) | 2011-10-26 |
| US8349934B2 (en) | 2013-01-08 |
| US20110263779A1 (en) | 2011-10-27 |
| JPWO2010071073A1 (ja) | 2012-05-31 |
| CN102245645A (zh) | 2011-11-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5689320B2 (ja) | 硬化性組成物及びその硬化物 | |
| EP2298822B1 (en) | Hardening composition and resultant hardened material | |
| WO2015019941A1 (ja) | 半導体ナノ粒子含有硬化性組成物、硬化物、光学材料および電子材料 | |
| WO2012053543A1 (ja) | 光硬化性ナノインプリント用組成物、該組成物を用いたパターンの形成方法及び該組成物の硬化体を有するナノインプリント用レプリカ金型 | |
| KR101268700B1 (ko) | 액상 경화성 조성물, 경화막 및 대전 방지용 적층체 | |
| JP5804987B2 (ja) | 光硬化性ナノインプリント用組成物、該組成物を用いたパターンの形成方法、及び該組成物の硬化体を有するナノインプリント用レプリカ金型 | |
| WO2012114986A1 (ja) | 硬化性組成物及びその硬化物 | |
| JP4963813B2 (ja) | 帯電防止性ハードコート樹脂組成物とその用途 | |
| JP2011201930A (ja) | ハードコート用組成物およびそれを塗工した基材 | |
| JP7021710B2 (ja) | インプリント用光硬化性組成物 | |
| JP2003216060A (ja) | 表示素子用プラスチック基板 | |
| JP5405861B2 (ja) | 透明導電膜及びその製造方法 | |
| JP5151113B2 (ja) | 硬化性組成物、硬化膜及び帯電防止用積層体 | |
| JP6547749B2 (ja) | 酸化ジルコニウム、酸化ジルコニウム分散液、酸化ジルコニウム含有組成物、塗膜、および表示装置 | |
| JP4958142B2 (ja) | 透明導電膜形成用組成物、透明導電膜及びディスプレイ | |
| WO2012133586A1 (ja) | 低屈折率膜形成用組成物、低屈折率膜、プラスチック基材、及び表示装置 | |
| CN113867100B (zh) | 一种光刻胶组合物及其制备方法和用途 | |
| JP2007019002A (ja) | 透明導電膜形成用組成物、透明導電膜及びディスプレイ | |
| CN115877655A (zh) | 导电性树脂组合物、及形成有发热电阻体的电路基板的制造方法 | |
| KR101238578B1 (ko) | 카본블랙 복합체, 이를 포함하는 블랙 매트릭스 조성물, 및칼라필터 | |
| JP2012247607A (ja) | 反射防止用積層体およびその製造方法、ならびに硬化性組成物 | |
| JP2007231112A (ja) | 液状硬化性組成物、硬化膜及び帯電防止用積層体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980150299.6 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09833376 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2010542947 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13139847 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009833376 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20117016460 Country of ref document: KR Kind code of ref document: A |