WO2007023824A1 - 3-アラルキルオキシピロリジン誘導体の製造法 - Google Patents
3-アラルキルオキシピロリジン誘導体の製造法 Download PDFInfo
- Publication number
- WO2007023824A1 WO2007023824A1 PCT/JP2006/316435 JP2006316435W WO2007023824A1 WO 2007023824 A1 WO2007023824 A1 WO 2007023824A1 JP 2006316435 W JP2006316435 W JP 2006316435W WO 2007023824 A1 WO2007023824 A1 WO 2007023824A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- aralkyloxypyrrolidine
- derivative
- acid
- benzyloxypyrrolidine
- solvent
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/12—Oxygen or sulfur atoms
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention provides the following general formula (2);
- R represents an optionally substituted aralkyl group having 7 to 15 carbon atoms
- HX represents a mineral acid, sulfonic acid, carboxylic acid, or amino acid
- (R) -3-benzyloxypyrrolidine or a salt thereof with an acid is known as a useful compound as an intermediate for production of pharmaceutical products.
- the 3 aralkyloxypyrrolidine derivative (2) has an inadequate affinity with various solvents, particularly non-polar solvents such as hydrocarbon solvents.
- the 3-aralkyloxypyrrolidine derivative (2) is oily and difficult to crystallize suitably.
- the substance for example, physical properties such as moisture absorption characteristics are hardly known, and no problems or solutions for handling on an industrial scale have been disclosed.
- N-protected aralkyloxypyrrolidine (1) which is a raw material of the present invention
- Patent Document 1 JP-A-1-311059
- Patent Document 2 WO9604274
- Non-patent literature l Synlett 2004, No. 10, 1763-1764
- Non-Patent Document 2 J. Med. Chem., 1999, 42, 677-690
- the present invention provides a 3-alkyloxypyrrolidine derivative (2) such as high-purity 3-benzyloxypyrrolidine, which is simple, efficient, extremely high productivity, and industrial scale. It aims at providing the method of manufacturing by. Another object of the present invention is to provide an appropriate method for handling the 3-aralkyloxypyrrolidine derivative (2) on an industrial scale. It is another object of the present invention to provide a method for easily and safely producing N-protected 13-alkyloxypyrrolidine (1) as a raw material on an industrial scale.
- the aqueous layer is treated with a base, and then the 3-aralkyloxy
- the chemical purity is improved by a series of operations in which the free amine of the pyrrolidine derivative (2) is extracted with an organic solvent, and the free amine is treated with an acid to convert it to the 3-aralkyloxypyrrolidine derivative (2). It was found that it can be improved.
- N-protected 1-hydroxypyrrolidine (5) is subjected to O-aralkylation by the action of a halogenaralkyl in the presence of a base, a metal halide and Z or a phase transfer catalyst.
- a halogenaralkyl in the presence of a base, a metal halide and Z or a phase transfer catalyst.
- the present invention is characterized in that the 3-aralkyloxypyrrolidine derivative represented by the formula (2) is subjected to a crystallization process using a solvent containing a polar organic solvent, and is obtained as a crystal. And a method for producing a 3-aralkyloxypyrrolidine derivative (2).
- the present invention provides a two-phase mixture of water and an organic solvent containing a 3-aralkyloxypyrrolidine derivative represented by the above formula (2).
- the aqueous layer is treated with a base
- the free amine is treated with an acid to convert it to a 3-aralkyloxypyrrolidine derivative (2).
- the present invention relates to a method for producing a 3-aralkyloxypyrrolidine derivative (2) characterized by improving chemical purity by a series of operations.
- the present invention provides an N-protected 1-3-hydroxypyrrolidine represented by the above formula (5) as a base, and a halogenaralkyl in the presence of a metal halide and Z or a phase transfer catalyst.
- the present invention relates to a method for producing N-protected-3-aralkyloxypyrrolidine represented by the above formula (1), wherein the O-aralkyl is allowed to act.
- HX represents a mineral acid, a sulfonic acid, a carboxylic acid or an amino acid
- HX represents a mineral acid, a sulfonic acid, a carboxylic acid or an amino acid
- the present invention provides a general formula (3)
- R ' is an optionally substituted aralkyl group having 7 to 15 carbon atoms
- HY is a halogenated halogen
- a 3-aralkyloxypyrrolidine halogen represented by The present invention relates to a method for handling a halogenated hydrobromide of 3-aralkyloxypyrrolidine, characterized in that a crystal of hydrohalide is handled in an environment with an absolute humidity of 12 gZm 3 or less.
- the 3-aralkyloxypyrrolidine derivative (2) can be produced easily and efficiently on a commercial scale with extremely high productivity.
- the crystals of 3-aralkyloxypyrrolidine halogen hydrohydrogen salt (3) can be handled stably on an industrial scale.
- the raw material N-protected 13-aralkyloxypyrrolidine (1) can be produced efficiently and safely on an industrial scale.
- the present invention has the following process power.
- 3-hydroxypyrrolidine is represented by the general formula (5);
- P means a protecting group for an amino group.
- This amino-protecting group is a group that protects the amino group, and generally usable groups are PROTECTIVE GROUPS IN ORGANIC SYNTHESIS 2nd. Ed., John's It is described in JOHN WILEY & SONS Publishing (1991).
- a preferred protecting group is a urethane-type protecting group (also referred to as a strong rubamate-type protecting group).
- a methoxycarbo group is preferred, which is a lower alkoxycarbo group (wherein the alkyl group has 1 to 6, preferably 1 to 4 carbon atoms). More preferred is a tert-butoxycarbonyl group, more preferably a tert-butoxycarbonyl group, or a tert-butoxycarbonyl group.
- This reaction can be carried out using a known method described in the above-mentioned technical book.
- the method is not particularly limited, for example, by treating 3-hydroxypyrrolidine with ditert-butoxydicanolate, a powerful rubamate-type protecting group can be introduced into the amino group.
- the 3-hydroxypyrrolidine to be used may be an optically active substance or a non-optically active substance.
- N-protected 1-hydroxypyrrolidine (5) is O-aralkylated to give a general formula (1);
- O-Aralkylation refers to conversion of a hydroxy group to an aralkyloxy group.
- the group R bonded to the hydroxyl group at the 3-position is an aralkyl group having 7 to 15 carbon atoms which may have a substituent.
- the substituent include a hydroxyl group, a halogen atom, an amino group, a nitro group, and an alkoxy group.
- Examples of the aralkyl group having 7 to 15 carbon atoms which may have a substituent include, for example, benzyl group, p-chlorobenzoyl group, p-hydroxybenzyl group, p-fluorobenzyl group, m, m-difluoro group. Examples include a robenzyl group, a phenyl group, a naphthylmethyl group, and the like.
- N-protected-3-hydroxypyrrolidine (5) used in this step may be synthesized by the above method, or may be synthesized separately by a known method.
- N-Protected 3-Aralkyloxypyrrolidine (1) may be an optically active substance or a non-active substance. It may be an optically active substance.
- the reaction in this step is preferably carried out in the presence of a base and a metal halide and Z or a phase transfer catalyst.
- the base may be an inorganic base or an organic base, but is preferably an inorganic base, specifically, although not particularly limited, such as sodium hydroxide and potassium hydroxide hydroxide.
- examples thereof include alkali metal hydroxides; alkali metal carbonates such as sodium carbonate and potassium carbonate; alkali metal hydrogen carbonates such as sodium hydrogen carbonate and the like. Among these, the above-mentioned alkali metal hydroxides are preferable.
- the amount of the base used is not particularly limited, but is usually 1 to 30 times mol, preferably 1 to 20 times mol, more preferably 2 to 10 times the amount of N-protected 1-3 hydroxypyrrolidine (5). Double mol, particularly preferably 2 to 5 mol.
- the metal halide is not particularly limited, but alkali metal halides such as lithium bromide, sodium bromide, potassium bromide, lithium iodide, sodium iodide and potassium iodide; magnesium bromide Alkaline earth metal halides such as calcium bromide, magnesium iodide and calcium iodide; aluminum halides such as aluminum bromide and aluminum iodide, and the like. Preferred are alkali metal halides. Yes, more preferably potassium iodide.
- alkali metal halide is not particularly limited, with respect to general N- protected-3-hydroxypyrrolidine (5), from 0.01 to 5 mol 0/0 preferably instrument more preferably , 0.05 mol force is also a 1 mol 0/0.
- the phase transfer catalyst is not particularly limited, and examples thereof include 12 crown-4, 15 crown-1, 18 crown-1, 24 crown-1, 8, dibenzo-18 crown-1, 6, dibenzo24 crown-8. , Dicyclohexylux 18 crown-6, dicyclohexylenole 24 —crown ethers such as crown 1-8; cryptands such as cryptand [2, 2], cryptand [2, 2, 1], and talybutand [2, 2, 2] Class: Trioctylmethylammo-chloride (trade name: ALIQUAT 336), trioctylmethylammonium bromide, methyltrialkyl (carbon number 8 to 10) ammochloride (trade name: Adogen 464), tetraptylammomuchloride , Tetraptylammomubromide, tetraptylammomuumide, etc. 4th grade ammonium salt.
- the amount of the phase transfer catalyst used is not particularly limited, but generally 0.01 to 5 mol 0/0 is more preferable, preferably 0. 0 to 0 with respect to N-protected 1-hydroxy pyrrolidine (5). 05 mol force et al. 1 mole 0/0.
- the reaction in this step is performed by a) base and metal halide, b) base and phase transfer catalyst, c) base, metal halide and phase transfer catalyst.
- the ability to carry out the reaction in the presence of A is preferably the embodiment of a) or c). It is further preferred to carry out the reaction in the presence of a base represented by a) and a metal halide.
- the reaction solvent is not particularly limited as long as it is an essentially inert solvent.
- a hydrocarbon solvent it is preferable to use an ether solvent, a nitrogen-containing solvent, or a sulfur-containing solvent.
- the hydrocarbon solvent is not particularly limited, but aromatic hydrocarbons such as benzene, toluene, xylene, salt-benzene, salt-toluene, cyclohexane, methylcyclohexane, heptane, etc. Aliphatic hydrocarbons can be mentioned, but aromatic hydrocarbons are preferable, and toluene is more preferable.
- the ether solvent is not particularly limited. Specifically, it is a power capable of mentioning tetrahydrofuran, 1,4 dioxane, dimethoxyethane, ethylene glycol dimethyl ether, etc. Preferably, 1, 4 Dioxane.
- nitrogen-containing solvent examples include acetonitrile, dimethylformamide, dimethylacetamide, jetylacetamide, dimethylbutyramide, N-methyl-2-pyrrolidone, hexamethyl phosphate triamide, and the like.
- acetonitrile is preferred.
- the sulfur-containing solvent is not particularly limited, and examples thereof include dimethyl sulfoxide.
- the amount of the organic solvent used is not particularly limited, but for N-protected-3hydroxypyrrolidine (5), the lower limit is usually 10 wt%, preferably 50 wt%, more preferably 100 wt%. is there.
- the upper limit is not particularly limited, but is usually 10000 wt%, preferably in consideration of economy. 5000 wt%, more preferably 2000 wt%.
- reaction solvent used in the O-aralkyl group reaction of the present invention may be used alone or in combination of two or more.
- water may coexist if necessary.
- the reaction in this step can be carried out using an aralkyl halide.
- the aralkyl group of the halogenated aralkyl is the same as the aralkyl group described as R in the formula (1).
- the halogen of the halogen aralkyl is not particularly limited, but chlorine, bromine or iodine is preferred, chlorine or iodine is more preferred, and chlorine is particularly preferred.
- benzyl halide is preferable, and benzyl chloride is particularly preferable.
- the lower limit is not particularly limited as long as it is equal to or higher than the freezing point of the reaction solvent, but generally 0 ° C or higher is more preferable, 20 ° C or higher is preferable, and 40 ° C or higher.
- the upper limit is not particularly limited as long as it is not higher than the boiling point of the reaction solvent, but is preferably 120 ° C or lower, more preferably 100 ° C or lower, and further preferably 80 ° C or lower.
- the reaction time is not particularly limited, but it is generally preferably 1 to 100 hours, more preferably 5 to 80 hours, and further preferably 10 to 50 hours.
- a general post-treatment may be performed.
- finish of reaction can be liquid-separated and the organic layer containing a product can be acquired.
- the obtained organic layer can be washed with water as necessary, and the reaction solvent can be removed by heating under reduced pressure.
- N-protected-3-aralkyloxypyrrolidine (1) is obtained.
- the product thus obtained can be further purified by a general technique such as crystallization purification, fractional distillation, column chromatography or the like, if necessary, to increase the purity.
- the 3-aralkyloxypyrrolidine derivative (2) is an acid salt of 3-aralkyloxypyrrolidine.
- HX in the formula (2) include mineral acids, sulfonic acids, carboxylic acids, and amino acids. These are not limited to monovalent acids, and may be divalent or trivalent.
- the mineral acid is not particularly limited, and examples thereof include hydrogen halides such as hydrogen chloride and hydrogen bromide, sulfuric acid, and phosphoric acid.
- the sulfonic acid is not particularly limited.
- methanesulfonic acid methanesulfonic acid, ethanesulfonic acid, propanesulfonic acid, butanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, camphorsulfonic acid, 1-phenolethanesulfonic acid, etc.
- the carboxylic acid is not particularly limited, and examples thereof include formic acid, acetic acid, trifluoroacetic acid, non-optically active carboxylic acid of benzoic acid, and optically active rubonic acid such as tartaric acid.
- amino acids include, but are not limited to, optically active amino acids such as alanine, parin, ferrolanine, and aspartic acid.
- the salts with the resulting acid have good crystallinity, such as hydrogen chloride, hydrogen bromide, p-toluenesulfonic acid, and benzoic acid, among which hydrogen chloride, bromide Hydrogen Particularly preferred is salt hydrogen.
- the 3-aralkyloxypyrrolidine derivative (2) to be crystallized is an optically active substance and its optical purity needs to be increased, it is preferable to use an optically active acid as the acid to be used.
- optically active tartaric acid and optically active aspartic acid are preferred, although optically active carboxylic acids and optically active amino acids are preferred.
- N-protected 13-aralkyloxypyrrolidine derivative (1) may be synthesized by the above method or may be synthesized by a known method.
- N-protected 1-aralkyloxypyrrolidine derivative (1) contains N-protected 1-hydroxypyrrolidine (5) and / or aralkyl alcohol as impurities!
- the aralkyl alcohol is a general formula;
- Aralkyl alcohol represented by Examples of R include those described above, and the aralkyl alcohol (6) is preferably an aralkyl alcohol having 7 or 8 carbon atoms, more preferably benzyl alcohol.
- the N-protected-3-aralkyloxypyrrolidine derivative (1) may be used as an optically active substance or a non-optically active substance.
- oxypyrrolidine (1) is an optically active form
- the resulting 3-aralkyloxypyrrolidine derivative (2) is also an optically active form
- N-protected-3 aralkyloxypyrrolidine (1 ) In the 3-position configuration of (3R), the 3-position configuration of the 3-aralkyloxypyrrolidine derivative (2) is (3R), and when the former is (3S), the latter is (3S ).
- an appropriate method can be selected as appropriate depending on the type of P representing the N-protecting group.
- An example of the deprotection method is the method described in PROTECTIVE GROUPS IN ORGANIC SYNTHESIS 2nd. Ed-.
- the above-mentioned deprotection is performed, or N-protected-3-aralkyloxypyrrolidine derivative (1) is described later.
- An operation of forming an acid salt may be performed, and deprotection and acid salt formation may be performed simultaneously.
- Examples of the acid used include the acids mentioned above for HX.
- the amount of acid used should be about the theoretical amount or more, but since it is not economical even if used in large amounts, it is usually 1 for N-protected 1-alkyloxypyrrolidine. -10 mol times, preferably 1-3 mol times, more preferably 1-2 mol times.
- the acid addition rate is not particularly limited, but in order to avoid the progress of deprotection of the 3-position hydroxyl group, the total amount of acid used should be added over 1Z6 hours or more. Is preferred. More preferably, it is 1 hour or longer, further preferably 3 hours or longer, particularly preferably 6 hours or longer.
- the upper limit of the covering time is not particularly limited, but is preferably 24 hours or less, preferably 12 hours or less.
- This reaction is usually carried out in a solvent.
- the solvent is not particularly limited, and examples thereof include organic solvents such as alcohol solvents, ether solvents, ester solvents, and aromatic hydrocarbon solvents.
- the alcohol solvent is not particularly limited !, but examples include methanol, ethanol, n propanol, isopropanol, n-butanol, sec butanol, isobutanol, and tert-butanol. Of these, isopropanol is preferred.
- the ether solvent is not particularly limited, and examples thereof include tetrahydrofuran, 1,4 dioxane, 1,3 dioxolane, 1,2 dimethoxyethane, diethylene glycol dimethyl ether, methyl tert butyl ether and the like. Of these, tetrahydrofuran is preferred.
- the ester solvent is not particularly limited but is preferably an ester having 2 to 8 carbon atoms, more preferably 4 to 6 carbon atoms. Examples include pill, isopropyl acetate, n-butyl acetate, tert butyl acetate, and the like, and ethyl acetate is preferred.
- the aromatic hydrocarbon solvent is not particularly limited, but is preferably an aromatic hydrocarbon having 6 to 12 carbon atoms, more preferably 6 to 10 carbon atoms, and further preferably 6 to 8 carbon atoms. Specific examples include benzene, toluene, xylene and the like. in Are also preferably aromatic hydrocarbons having 7 or 8 carbon atoms, specifically, toluene and xylene.
- Toluene is most preferably used.
- isopropanol is preferred from the viewpoints of high reactivity and acid stability, among alcohol solvents.
- the reaction solvents may be used alone or in combination of two or more. Further, from the viewpoint of further increasing the reactivity, water may coexist in the organic solvent.
- the amount of water to be used is not particularly limited, but is usually 0.1 to 50 wt%, preferably 0.5 to 30 wt%, more preferably 1 to 20 wt% based on the total weight of the reaction solution.
- the reaction temperature is not particularly limited, but is usually 20 to 100 ° C, preferably 0 to 80 ° C, more preferably 20 to 50 ° C.
- the reaction time is not particularly limited, but usually 1
- This method comprises a two-phase mixture of water and an organic solvent containing a 3-alkyloxypyrrolidine derivative (2),
- the aqueous layer is treated with a base
- the 3-aralkyloxypyrrolidine derivative (2) may be synthesized by the above-described method, or may be synthesized in a separately known method and used in this method.
- the 3 aralkyloxypyrrolidine derivative (2) synthesized by the above method it is not particularly limited, but it is also possible to use the reaction end solution as it is.
- the 3-aralkyloxypyrrolidine derivative (2) used in the present method may be a racemate or an optically active (R)-or (S) -3-aralkyloxypyrrolidine. Derivatives may also be used. Usefulness as a pharmaceutical intermediate The optically active 3-aralkyloxypyrrolidine derivative is preferred.
- Impurities contained in the 3-aralkyloxypyrrolidine derivative (2) are removed. It can be effectively removed. Impurities include 3-hydroxypyrrolidine and Z or aralkyl alcohol (6).
- the aralkyl alcohol (6) is the same as described above.
- the purity of the 3-aralkyloxypyrrolidine derivative (2) used in the present method is not particularly limited, but usually 70% or more is preferred, more preferably 80% or more, and 85% or more. Especially preferred.
- the two-phase mixture of water and an organic solvent containing the 3-aralkyloxypyrrolidine derivative (2) is, for example, water and an organic solvent in the 3-aralkyloxypyrrolidine derivative (2).
- a mixture that includes The organic solvent to be added is not particularly limited, but a solvent having no compatibility with water is preferable. Examples thereof include toluene, ethyl acetate, methyl tert-butyl ether, and methylene chloride. Of these, toluene and ethyl acetate are preferred, and toluene is particularly preferred.
- water and Z or an organic solvent may be added as appropriate.
- the 3-aralkyloxypyrrolidine derivative (2) may be dissolved in the two-phase mixture, or a part thereof may be precipitated.
- the aqueous solution of the 3-aralkyloxypyrrolidine derivative (2) transferred to the aqueous layer may be further washed with an organic solvent.
- the organic solvent to be washed is not particularly limited, but an organic solvent added after completion of the reaction is preferably used.
- the 3-aralkyloxypyrrolidine derivative (2) that has migrated to the aqueous layer can be converted to the free amine of the 3-aralkyloxypyrrolidine derivative (2) by salting with a base.
- a base an inorganic base and Z or an organic base can be used, and the inorganic base is not particularly limited, but examples thereof include alkali metal hydroxides such as lithium hydroxide, sodium hydroxide, potassium hydroxide and the like.
- the organic base is not particularly limited, and examples thereof include methylamine, ethylamine, and propylamine.
- Primary amines such as amine and butylamine; secondary amines such as dimethylamine, jetylamine, dipropylamine, diisopropylamine and dibutylamine; tertiary amines such as trimethylamine, triethylamine and tripropylamine, preferably triethylamine. It is a third grade amine such as Min.
- the free amine of the 3-aralkyloxypyrrolidine derivative (2) is extracted into an organic solvent, and further washed with water as necessary to improve the chemical purity of the 3-aralkyloxypyrrolidine derivative (2 ) Can be obtained as a free amine.
- the organic solvent to be used is not particularly limited, but solvents that are not compatible with water are preferred, and examples thereof include toluene, ethyl acetate, methyl tert-butyl ether, and methylene chloride. Of these, toluene and ethyl acetate are preferred, and toluene is particularly preferred.
- the free amine of the 3-aralkyloxypyrrolidine derivative (2) thus obtained can be treated with an acid to synthesize the 3-aralkyloxypyrrolidine derivative (2).
- Examples of the acid used include the acids described above as HX. Of these, the salt with the resulting acid has good crystallinity, particularly preferred is hydrogen chloride, hydrogen bromide, p-toluenesulfonic acid, benzoic acid is preferred, and hydrogen bromide is more preferred. Is salty hydrogen.
- the 3-aralkyloxypyrrolidine derivative (2) used in this step may be one prepared from N-protected-3-hydroxypyrrolidine (5) by the method described above, or may be publicly known. It may be synthesized by a method. In addition, the chemical purity of the 3-aralkyloxypyrrolidine derivative (2) may be increased by the method described above! ,.
- the 3-aralkyloxypyrrolidine derivative (2) used in the present method may be a racemate or an optically active (R)-or (S) -3-aralkyloxy derivative. It may be a pyrrolidine derivative. Usefulness as a pharmaceutical intermediate The optically active 3-aralkyloxypyrrolidine derivative is preferred.
- the purity of the 3-aralkyloxypyrrolidine derivative (2) used in this step is particularly limited. Although 70% or more is preferred, 80% or more is preferred, and 85% or more is particularly preferred.
- Examples of impurities contained in the 3-aralkyloxypyrrolidine derivative (2) used in the present method include 3-hydroxypyrrolidine and Z or N-protected mono-hydroxypyrrolidine deprotected.
- Aralkyl alcohol (6) Aralkyl alcohol (6).
- the amount of 3-hydroxypyrrolidine contained in the 3-aralkyloxypyrrolidine derivative (2) is 10 wt% or less, more preferably 5 wt% or less, more preferably 5 wt% or less based on the 3-aralkyloxypyrrolidine derivative (2). Preferably, it is 3 wt% or less, particularly preferably 2 wt% or less.
- the amount of aralkyl alcohol (6) is preferably 4 wt% or less, more preferably 3 wt% or less, and even more preferably 2 wt% or less.
- the crystallization method is not particularly limited.
- a reaction crystallization method, a cooling crystallization method, a concentrated crystallization method, a crystallization method using solvent substitution, a crystallization method by mixing a poor solvent In addition, commonly used crystallization methods such as Z or salting out methods can be carried out alone or in appropriate combination.
- N-protected 1-aralkyloxypyrrolidine (1) or 3-aralkyloxypyrrolidine can also be used to form an acid salt by using a polar organic solvent.
- a reaction crystallization method for obtaining crystals of the xylpyrrolidine derivative (2) can also be used as appropriate. In this crystallization, seed crystals can be added as necessary.
- This crystallization method can be applied to a solution of 3-aralkyloxypyrrolidine, an oily concentrate, or the like, and the 3-aralkyloxypyrrolidine obtained by this method can be used. It may be carried out to dissolve and further increase the purity of the crystal.
- the crystallization method is usually carried out in a polar solvent or a mixed solvent of a polar solvent and a nonpolar solvent.
- the mixing ratio is not particularly limited, but it is sufficient if the ratio of polar solvent in the mixed solvent is 5% or more.
- the polar solvent is not particularly limited, and examples thereof include water, ester solvents, ether solvents, halogenated hydrocarbon solvents, nitrogen-containing solvents, alcohol solvents, ketone solvents, and the like. be able to.
- the ester solvent is not particularly limited, but preferably has 2 to 8 carbon atoms. Or an ester having 4 to 6 carbon atoms. Specific examples include ethyl acetate, n-propyl acetate, isopropyl acetate, n-butyl acetate, tert-butyl acetate, and the like. Is preferred.
- the ether solvent is not particularly limited, and examples thereof include tetrahydrofuran, 1,3-dioxolane, 1,2-dimethoxyethane, diethylene glycol dimethyl ether, methyl tert-butyl ether, and the like. Of these, methyl tert-butyl ether is preferred.
- the halogenated hydrocarbon solvent is not particularly limited, and examples thereof include methylene chloride, black benzene, dichlorobenzene, 1,2-dichloroethane, and the like. Of these, chlorobenzene or dichloroethane is preferred.
- the nitrogen-containing solvent is not particularly limited, but is acetonitrile, dimethylformamide.
- the alcohol solvent is not particularly limited, and examples thereof include methanol, ethanol, n-propanol, isopropanol, n-butanol, sec-butanol, isobutanol, and tert-butanol. Of these, isopropanol is preferred.
- the ketone solvent is not particularly limited, and examples thereof include acetone and methyl ethyl ketone. Of these, acetone is preferred.
- the polar solvent is particularly preferably an ester solvent or an alcohol solvent, preferably an ester solvent, a halogenated hydrocarbon solvent, a nitrogen-containing solvent, an alcohol solvent, or a ketone solvent.
- the amount of the polar solvent used is not particularly limited, but the lower limit is usually 0.01 times weight, preferably 0.05 times weight, with respect to the 3-aralkyloxypyrrolidine derivative (2). More preferably, it is 0.1 times weight, more preferably 0.5 times weight, particularly preferably 1 time weight, and the upper limit is 100 times weight, preferably 50 times weight, more preferably 30 times weight. .
- the nonpolar solvent is not particularly limited, and examples thereof include hydrocarbon solvents such as aliphatic hydrocarbon solvents and aromatic hydrocarbon solvents.
- the aliphatic hydrocarbon solvent is not particularly limited, and examples thereof include aliphatic hydrocarbons having 5 to 8 carbon atoms such as pentane, hexane, heptane, and methylcyclohexane. Of these, aliphatic hydrocarbons having 6 or 7 carbon atoms, specifically, hexane, heptane, methylcyclohexane and the like are preferable.
- the aromatic hydrocarbon solvent is not particularly limited, but is preferably an aromatic hydrocarbon having 6 to 8 carbon atoms, specifically, for example, benzene, toluene, xylene and the like. Of these, aromatic hydrocarbons having 7 or 8 carbon atoms, preferably toluene, xylene and the like are preferable.
- a nonpolar solvent when used, it may be mixed with a polar solvent in advance, but if necessary, after a crystal is precipitated by a reaction crystallization method, a cooling crystallization method, a concentrated crystallization method, or the like. May be added as appropriate. Preferably, it is a method of adding after the crystals are precipitated.
- the crystallization temperature is not particularly limited, but is usually 100 ° C or lower, preferably 80 ° C or lower, more preferably 60 ° C or lower, further preferably 40 ° C or lower, particularly preferably 30 ° C. It is as follows.
- the lower limit is usually ⁇ 20 ° C., preferably ⁇ 10 ° C., more preferably 0 ° C.
- the 3-aralkyloxypyrrolidine derivative (2) thus obtained can be collected by using a general solid-liquid separation method such as centrifugation, pressure separation, and vacuum filtration. it can .
- the obtained crystal can be obtained as a dried crystal, for example, by drying under reduced pressure (vacuum drying) as necessary.
- R is benzyl, represented by the general formula (4);
- the 3-benzyloxypyrrolidine derivative crystal represented by the formula is a novel crystal isolated for the first time in the present invention, and is very useful as a pharmaceutical intermediate because of its ease of handling.
- HX is the same as described above.
- the crystal of the 3-benzyloxypyrrolidine derivative (4) of the present invention may be a racemate or an optically active substance.
- the usefulness as a pharmaceutical intermediate is also preferably an optically active substance.
- the crystal (2) is a high-purity crystal. That is, the chemical purity is 95% or higher, preferably 97% or higher, more preferably 98% or higher, especially 99% or higher. Further, in the case of an optically active substance, the optical purity is 98% ee or higher, preferably 99% ee or higher, more preferably 99.5% ee or higher, in addition to the above chemical purity. Further, the content of 3-hydroxypyrrolidine contained in the 3-aralkyloxypyrrolidine derivative (2) obtained by this crystallization step is usually 2% or less, preferably 1% or less, more preferably 0.5%. In the following, it is particularly preferably 0.3% or less. Further, the impurities contained in the 3-aralkyloxypyrrolidine derivative (2) are usually 1% or less, preferably 0.5% or less, more preferably 0.3% or less, and particularly preferably 0.1%. It is as follows.
- R 'in the formula (3) is a aralkyl group having 7 to 15 carbon atoms which may have a substituent, or a hydrogen atom.
- the aralkyl group having 7 to 15 carbon atoms which may have a substituent is the same as the group described as R in the formula (1).
- HY in the formula (3) is hydrogen chloride, hydrogen bromide, hydrogen iodide, hydrogen fluoride, or a mixture thereof.
- Preferred is salt hydrogen or hydrogen bromide, more preferably salt hydrogen.
- the 3-haloalkyloxypyrrolidine hydrohalide (3) may be a racemate or an optically active substance, but is preferably an optically active substance.
- the halogenated hydrohydrogen salt of 3-aralkyloxypyrrolidine (3) can be produced, for example, by the method described for the 3-aralkyloxypyrrolidine derivative (2).
- the handling method of the present invention is particularly effective for dry crystals of the 3-haloalkyloxypyrrolidine hydrohalide (3).
- the dry crystal means a crystal that absorbs moisture (increases the water content) with a time when the initial water content is less than the equilibrium water content in the environment, and is preferably a halogenated 3-alkyloxypyrrolidine.
- This is a crystal obtained by subjecting a crystal of hydrosulfate (3) to a drying treatment such as drying under reduced pressure.
- the water content of the above-mentioned 3-aralkyloxypyrrolidine halogen hydrohydrogen salt (3) depends on the type of handling and operation and the time required, but for example, the water content is 3% by weight or less, preferably 2% by weight or less. More preferably, it is 1% by weight or less, further preferably 0.5% by weight or less, and particularly preferably 0.3% by weight or less.
- the present invention provides 3-haloalkyloxypyrrolidine hydrohalide (3) crystals having an absolute humidity of 12 g / m 3 or less, preferably 10 g / It is characterized by being handled in an environment of m 3 or less, particularly preferably 8 gZm 3 or less.
- moisture absorption is suitably suppressed, and it is possible to prevent dehalogenation of the 3-aralkyloxypyrrolidine halide hydrate (3).
- the above-mentioned humidity environment is provided by dehumidification with environmental power or introduction of a dehumidified gas (preferably, dry inert gas such as dry nitrogen) into the environment.
- a dehumidified gas preferably, dry inert gas such as dry nitrogen
- an environment with an absolute humidity of 12 gZm 3 or less means, for example, in a sealed box.
- the absolute humidity in the box is 12 gZm 3 or less
- the crystal around 3-haloalkyloxypyrrolidine halogen hydrohydrogen salt (3) The range for measuring the absolute humidity is not particularly limited.
- the absolute humidity may be measured in the same room of 1 to 30 m.
- the absolute humidity may be measured directly using a commercially available absolute hygrometer, but another method is to measure the temperature and relative humidity in the above environment, and the saturated water vapor concentration (gZm at that temperature). 3 ) Calculated from the product of relative humidity (%).
- the method of dehumidification is not particularly limited, and can be achieved by freezing moisture, using a dehumidifier or a desiccant (such as silica gel).
- a dehumidifier or a desiccant such as silica gel.
- a particularly preferred method there can be mentioned a method of handling an environment in which a crystal of 3-hydroalkyloxy lysine halogen hydrohydrogen salt (3) is treated at a low temperature. That is, since the saturated water vapor concentration in the atmosphere essentially decreases in a low-temperature environment, the low-humidity environment can be easily provided even in areas with high humidity, for example, Japan. From this, it is possible to favorably suppress the moisture absorption of the crystal of the hydrohalated salt of 3-aralkyloxypyrrolidine (3) and prevent its deliquescence.
- the low temperature is usually 25 ° C or lower, preferably 20 ° C or lower, more preferably 10 ° C or lower, especially 5 ° C or lower.
- the 3-aralkyloxypyrrolidine is stable, even on a large scale (industrial scale). It is possible to handle the crystal of Rogen dehydrogenate (3) while maintaining high quality.
- 3-haloxylpyrrolidine hydrohalide salt (3) useful as an intermediate for pharmaceuticals and the like can be produced on a large scale (industrial scale). Even if it is, it can handle suitably.
- the method is not particularly limited as long as the absolute humidity environment is given. Further, the practice of the present invention can be carried out under normal pressure, under pressure, or under reduced pressure, and can usually be suitably carried out under normal pressure.
- the moisture absorption of the 3-haloalkyloxypyrrolidine hydrohalide salt (3) is suppressed, so that even when contacted with a metal material, the solid Incorporation of metal components into the inside is reduced, and corrosion of metallic materials is also reduced. Meanwhile, phase In an environment with high humidity, moisture absorption proceeds to such an extent that it has an adverse effect.
- the metal component is significantly mixed into the solid and the metal material is significantly corroded.
- metal materials include stainless steel, hastelloy, and iron.
- SUS304, SUS304L, SUS316, SUS316L, SUS309S, SUS310S, SUS321 or SUS347 which are austenitic stainless steels, especially SUS304 or SUS316.
- Examples of the metal component that can be mixed into the solid include transition metals such as iron, chromium, nickel, and salts thereof contained in stainless steel.
- transition metals such as iron, chromium, nickel, and salts thereof contained in stainless steel.
- the most likely metal components are iron, chromium, nickel or their salts and mixtures, especially iron or its salts.
- the crystal package of 3-haloalkyloxypyrrolidine hydrohalide (3) is stored in, for example, a single or double aluminum laminate bag, polyethylene bag, etc.
- the outer bag can be preferably an aluminum laminated bag and the inner bag can be a polyethylene bag.
- a desiccant (silica gel or the like) can be enclosed if necessary or preferably. When the desiccant is enclosed, it may be enclosed in the inner bag, or it may be packed inside the outer bag and outside the inner bag.
- the above-mentioned bag in which the crystal is packed can be placed in an exterior such as a steel drum, a fiber drum, or a cardboard as necessary or preferably.
- the present invention can exert its effect to the maximum on a large scale (industrial scale).
- the scale is not particularly limited, but is usually 1 kg or more, preferably 10 kg or more, more preferably 100 kg or more, and especially 1000 kg as a 3-haloalkyloxypyrrolidine hydrohalide (3) crystal. That's it.
- the ambient environment is preferable to an absolute humidity of 12 gZm 3 or less.
- the moisture content is preferably controlled to 10 g / m 3 or less, particularly preferably 8 g / m 3 or less to suppress moisture absorption up to the equilibrium moisture content (that is, its water content is 3% by weight or less, preferably 2 % Or less, more preferably 1% by weight or less, even more preferably 0.5% by weight or less, particularly preferably 0.3% by weight or less), and the above-mentioned problem can be solved satisfactorily.
- the crystals of the halogenated hydrogen salt of triaralkyloxypyrrolidine (3) obtained according to the present invention are white to light brown and highly pure. That is, the chemical purity is preferably 95% or more, preferably 96% or more, more preferably 97% or more, still more preferably 98% or more, and particularly 99% or more. Further, in the case of an optically active substance, in addition to the above chemical purity, the optical purity is 98% ee or higher, preferably 99% ee or higher, more preferably 99.5% ee or higher, and particularly preferably 99.8%. % ee or higher.
- 3 hydroxypyrrolidine hydrochloride is derivatized to tert butoxycarbo-lamino 3-hydroxypyrrolidine and quantified.
- N-tert-butoxycarbo-lamino 3-hydroxypyrrolidine (Boc protector of 3-hydroxypyrrolidine hydrochloride) 10. 4 min, N-tert-butoxy-carbo-lamino mono 3-benzyloxypyrrolidine (3 —Boc protected form of benzyloxypyrrolidine hydrochloride) 24. 1 min
- 3-aralkyloxypyrrolidine is derivatized into tert-butoxycarbo-amino-3 monobenzyloxypyrrolidine and the optical purity is measured.
- 3-Benzyl pyrrolidine hydrochloride containing the enantiomer is dissolved in isopropanol, neutralized with 30% aqueous sodium hydroxide solution, and then sonicated with di-tert-butyl dicarbonate. . The resulting mixture is injected into the HPLC and analyzed.
- Retention time (R) —N—tert-butoxycarbo-lamino—3-benzyloxypyrrolidine (Boc protector of (R) —3 benzyloxypyrrolidine hydrochloride) 13. 4 minutes, (S) — N-tert Butoxycarbonylamino-3-benzyloxypyrrolidine (Boc protector of (S) -3-benzyloxypyrrolidine hydrochloride) 12. 1 min.
- the obtained organic layer was concentrated to obtain 244 g of N tert butoxycarbol- (R) -3-benzyloxypyrrolidine (yield 92%).
- the residual amount of N tert butoxycarbol- (R) -3 hydroxypyrrolidine with respect to N-tert butoxycarbolulu (R) -3-benzyloxypyrrolidine was 1. lwt%.
- N-tert-butoxycarboru (R) -3-hydroxypyrrolidine was dissolved in 296 g of toluene, and 6.4 g of tetra-n-butylammonium bromide was added. More 264 g of 30% aqueous sodium hydroxide solution was added and heated to 72 ° C., and then 65 g of benzyl chloride was added dropwise. N-tert-Butoxycarboro (R) -3-benzyloxypyrrolidine remaining N-tert-Butoxycarboro (R) -3-hydroxypyrrolidine reached 1.3 wt% The reaction temperature was cooled to 25 ° C. to stop the reaction. The obtained organic layer was washed with water and concentrated to obtain 83 g of N-tert-butoxycarbol- (R) -3-benzyloxypyrrolidine (yield 97%).
- N-tert-butoxycarbolu (R) -3-benzyloxypyrrolidine lO lg was dissolved in 149 g of isopropanol, heated to 40 ° C, and then 138 g of isopropanolic hydrochloric acid was added dropwise over 6 hours. did. After completion of the reaction, the mixture was cooled to 23 ° C and concentrated to distill off the solvent. Further, 1400 g of ethyl acetate was added and concentrated to obtain about 300 g of concentrate. The mixture was heated to 40 ° C, and isopropanol was added until the (R) -3-benzyloxypyrrolidine hydrochloride, which had separated as an oil, was completely dissolved.
- (R) -3-hydroxypyrrolidine hydrochloride was reduced from 0.92 wt% to 0.42 wt% by this crystallization operation.
- Benzyl alcohol was also reduced from 1.9 wt% to 0.01% by crystallization.
- This crude product was dispersed in 4.Og of isopropanol and heated to 40 ° C., and 506 mg of distilled water was added to dissolve it completely. The solution was gradually cooled to 22 ° C, and the precipitated crystals were filtered under reduced pressure.
- Example 23 (R) -3-Benzyloxypyrrolidine mono (D) -aspartate Concentrated 0.25 g (purity: 86.6%) containing (R) -3-benzyloxypyrrolidine separately obtained, (R) —3-hydroxypyrrolidine hydrochloride (content 0.12 wt%) and benzyl alcohol ( Content 1. Owt%) was mixed as an impurity, optical purity 97.0% ee) was dissolved in 10 ml of distilled water, and (D) -aspartic acid 325 mg was added. After stirring for a while, isopropanol was added for azeotropic dehydration, followed by concentration and drying under reduced pressure to obtain white crystals.
- (R) 3-Benzyloxypyrrolidine hydrochloride (purity 99.2%, water content 0.25 wt%) in a container, constant humidity (relative humidity 22%, 33%, 80% (25 ° C) ) Was left open in the container marked with).
- the water content of (R) -3-benzyloxypyrrolidine hydrochloride after standing was measured with a Karl Fischer moisture meter.
- Absolute humidity 8g / m 3 Saturated magnesium chloride aqueous solution (relative humidity 33%, 25 ° C)
- Absolute humidity 18gZm 3 Saturated sodium chloride aqueous solution (relative humidity 80%, 25 ° C) [0171] [Table 3]
- N-tert-butoxycarbol- (R) -3-hydroxypyrrolidine was dissolved in 748 mg of toluene, and 16 mg of tetra-n-butylammonium bromide and 9 mg of sodium iodide were added. . Further, 666 mg of 30% aqueous sodium hydroxide solution was added and heated to 50 ° C., and then 164 mg of benzyl chloride was added dropwise.
- N-tert-Butoxycarbol-(R) -3-Benzyloxypyrrolidine is compared to 6.4 wt% of the remaining N-tert-butoxycarbolulu (R) -3-hydroxypyrrolidine. The reaction temperature was cooled to 25 ° C to stop the reaction.
- N-tert-butoxycarbolu (R) -3-hydroxypyrrolidine (20 g) in a solution of approximately 60% toluene is charged with dimethyl sulfoxide (50 g), and potassium iodide (5.32 g) is added thereto. 85% solid potassium hydroxide (14. lg) was added. After this slurry solution was heated to 70 ° C., benzyl chloride (17.58 g) was added dropwise over 5 hours. Subsequently, when the mixture was stirred at 0 ° C for 16 hours, the conversion rate (*) reached 98%.
- Examples 27 to 28 The same reaction operation was carried out except that the solvent of Example 26 and toluene and dimethyl sulfoxide were replaced with the single solvent shown below and the reaction temperature and reaction time were changed. The results are shown in Table 4.
Abstract
本発明は、医薬品等の製造上重要な3-アラルキルオキシピロリジン誘導体の製造法を提供する。本発明においては、N-保護-3-ヒドロキシピロリジンを塩基、並びに、金属ハロゲン化物及び/または相間移動触媒の存在下にハロゲン化アラルキルを作用させてN-保護-3-アラルキルオキシピロリジンに変換した後、N-保護基を脱保護させて3-アラルキルオキシピロリジン誘導体に変換させ、次いで、極性溶剤を含有する溶剤中で処理することにより、該3-アラルキルオキシピロリジン誘導体を結晶として取得する。本発明によれば、高純度の3-アラルキルオキシピロリジン誘導体を簡便かつ効率的に工業的規模で製造することができる。
Description
明 細 書
3 -ァラルキルォキシピロリジン誘導体の製造法
技術分野
[0001] 本発明は、下記一般式 (2);
[0002] [化 9]

[0003] (式中、 Rは置換基を有していても良い炭素数 7〜 15のァラルキル基を表し、 HXは 鉱酸、スルホン酸、カルボン酸、アミノ酸を表す。)で表される 3—ァラルキルォキシピ 口リジン誘導体の製造法に関する。特に、 (R)—3—ベンジルォキシピロリジン、又は その酸との塩は、医薬品などの製造中間体として有用な化合物として知られている。 背景技術
[0004] 3—ァラルキルォキシピロリジン誘導体(2)の製造法としては、従来、以下のような 方法が用いられている。
(i)無水ジォキサン中の 4M塩化水素を N— Boc— 3—ベンジルォキシピロリジンに 0 °Cで加え、混合物を濃縮後、トルエンに溶解し、攪拌し、シロップ状の 3—ベンジルォ キシピロリジンを得る(例えば、特許文献 1参照)。
(ii) 4一べンジルォキシピロリジンー2—ォンを1^ 111で還元し、 3—べンジルォキシ
4
ピロリジンを得る (例えば、非特許文献 1参照)。
(iii) (R)— N— Boc— 3—ベンジルォキシピロリジンを 90%蟻酸水溶液中で 0°C〜 室温下に攪拌し、溶剤を減圧下に留去し、得られた残渣を K CO水溶液で中和する
2 3
。水溶液を n—ブタノールで抽出後、溶剤を減圧下に除去し、シリカゲルカラムクロマ トグラフィ一で処理し、 (R)—3—ベンジルォキシピロリジンを得る(例えば、特許文献 2、非特許文献 2参照)。
[0005] しかし、(i)の方法で記載されているように、 3—べンジルォキシピロリジン塩酸塩等
の 3 ァラルキルォキシピロリジン誘導体 (2)は、各種溶剤、特に炭化水素系溶剤等 の非極性溶剤との親和性が適切でない為に、これらの溶剤を主溶剤とする溶剤中で は、該 3 ァラルキルォキシピロリジン誘導体(2)は油状ィ匕し、好適に結晶化させるの が難しい。また、当該物質としては、例えば、吸湿特性など物性に関しても殆ど知ら れておらず、工業的規模で取り扱う上での問題点、その解決策については何ら開示 されていなかった。
[0006] また、(ii)につ 、ては、反応時に水素が発生する危険性を有し、且つ反応後に副 生するアルミニウムを処理する際に煩雑な処理が必要となる LiAlHを反応試剤とし
4
て使用しているため、商業規模で生産するに際しては、大きな課題があった。また、こ の方法で 4 ベンジルォキシピロリジン 2 オンを還元した場合、得られる 3 ヒドロ キシピロリジンはラセミ体である為、 目的物が光学活性 3—ァラルキルォキシピロリジ ンの場合は、適切な方法ではなかった。
[0007] また、 3 ァラルキルォキシピロリジンの精製法に関しては、(iii)の方法でカラムクロ マトグラフィ一による精製法が開示されているが、好ましくない溶剤の多量使用、工程 の煩雑さ、それに伴う時間の消費、製造装置の数や容量の増大、収量の低下等、商 業的規模で生産を行う上で大きな問題があった。
[0008] このように、 3 べンジルォキシピロリジンをはじめとする 3 ァラルキルォキシピロリ ジン誘導体 (2)に関しては、その結晶化方法、工業的に効率的な精製法、並びにそ の取り扱 、方法の 、ずれも開示されて 、な 、ことから、該化合物を工業的に簡便に 結晶化し、且つ高純度の該化合物を取得する精製法、並びに工業的規模での適切 な取り扱い方法の開発が求められていた。
[0009] さらに、本発明の原料である N—保護一 3 ァラルキルォキシピロリジン(1)
[0010] [化 10]
(1)
 [0011] (式中、 Pはァミノ基の保護基を表し、 Rは前記に同じ。)の製法に関しては、 N—保護 3—ヒドロキシピロリジン(5);
[0011] (式中、 Pはァミノ基の保護基を表し、 Rは前記に同じ。)の製法に関しては、 N—保護 3—ヒドロキシピロリジン(5);
[0012] [化 11]

[0013] (式中、 Pは、ァミノ基の保護基を表す。)から変換する方法、例えば、有機溶剤中、 N aH存在下にベンジルブ口ミドを作用させる方法 (例えば、非特許文献 2)が知られて いる。し力しながら、この方法では、水分との接触による水素発生、発火の危険性が ある NaHを使用していること、強い催涙性を有することから実質的に工業的規模では 入手困難なベンジルブ口ミドが用いられていること、等の点力も N 保護一 3—ァラル キルォキシピロリジンを工業的に製造する方法としては、必ずしも適切な方法ではな かった。
特許文献 1 :特開平 1—311059
特許文献 2: WO9604274
非特許文献 l : Synlett 2004, No.10, 1763-1764
非特許文献 2 : J. Med. Chem., 1999, 42, 677-690
発明の開示
発明が解決しょうとする課題
[0014] 本発明は、上記課題を鑑み、高純度の 3 べンジルォキシピロリジン等の 3 ァラ ルキルォキシピロリジン誘導体(2)を、簡便かつ効率的、極めて高い生産性で、工業 的規模で製造する方法を提供することを目的とするものである。また、 3—ァラルキル ォキシピロリジン誘導体 (2)の工業的規模での適切な取り扱い方法を提供することを も目的とする。さらに、原料である N—保護一 3 ァラルキルォキシピロリジン(1)を簡 便かつ安全に工業的規模で製造する方法を提供することをも目的とする。
課題を解決するための手段
[0015] 本発明者らは前記課題を解決すべく鋭意検討した結果、 3—ァラルキルォキシピロ リジン誘導体 (2)を、極性有機溶剤を含有する溶剤を晶析溶剤として使用すること〖こ より、 3—ァラルキルォキシピロリジン誘導体(2)を結晶化させることができ、これにより 高純度の 3—ァラルキルォキシピロリジン誘導体(2)を製造できることを見出した。
[0016] また、 3—ァラルキルォキシピロリジン誘導体(2)を含有する水と有機溶剤の 2相系 混合物から有機層を除去した後、水層を塩基で処理し、次いで、 3—ァラルキルォキ シピロリジン誘導体(2)の遊離のアミンを有機溶剤で抽出し、該遊離のアミンを酸で 処理して 3—ァラルキルォキシピロリジン誘導体(2)に変換する、一連の操作により化 学純度を向上させることができることを見出した。
[0017] また、 N—保護一 3—ヒドロキシピロリジン(5)を塩基、並びに、金属ハロゲン化物及 び Zまたは相関移動触媒の存在下に、ハロゲンィ匕ァラルキルを作用させて O—ァラ ルキルイ匕することにより、安全かつ簡便に N—保護一 3—ァラルキルォキシピロリジン を製造できることを見出した。
[0018] さらに、 3—ァラルキルォキシピロリジンのハロゲン化水素酸塩(3)の結晶に関し、 その平衡含水率 (又は、吸湿挙動)は、絶対湿度約 12gZm3を境に大きく変化するこ とから取り扱う際の周囲の環境を絶対湿度約 12gZm3以下に制御することにより、 3 ーァラルキルォキシピロリジンのハロゲンィ匕水素酸塩(3)の結晶の吸湿を好適に抑 制して、潮解等の問題を解決しうることを見出した。
[0019] 以上の一連の知見に基づき、本発明を完成するに至った。すなわち本発明は、前 記式(2)で表される 3—ァラルキルォキシピロリジン誘導体を、極性有機溶剤を含有 する溶剤を用いた晶析工程に付し、結晶として取得することを特徴とする 3—ァラル キルォキシピロリジン誘導体(2)の製造法に関する。
[0020] また、本発明は、前記式(2)で表される 3—ァラルキルォキシピロリジン誘導体を含 有する水と有機溶剤の 2相系混合物から、
A)有機層を除去した後、水層を塩基で処理し、
B) 3—ァラルキルォキシピロリジン誘導体(2)の遊離のアミンを有機溶剤で抽出し、
C)該遊離のアミンを酸で処理して 3—ァラルキルォキシピロリジン誘導体(2)に変換 する、
一連の操作により化学純度を向上させることを特徴とする 3—ァラルキルォキシピロリ ジン誘導体 (2)の製造法に関する。
[0021] また、本発明は、前記式(5)で表される N—保護一 3—ヒドロキシピロリジンを塩基、 並びに、金属ハロゲンィ匕物及び Zまたは相関移動触媒の存在下にハロゲンィ匕ァラル キルを作用させて O—ァラルキルィ匕することを特徴とする前記式(1)で表される N— 保護— 3—ァラルキルォキシピロリジンの製造法に関する。
[0022] また、本発明は、下記一般式 (4);
[0023] [化 12]

[0024] (式中、 HXは鉱酸、スルホン酸、カルボン酸またはアミノ酸を表す)で表される 3—べ ンジルォキシピロリジン誘導体の結晶に関する。
[0025] また、本発明は、一般式 (3);
[0026] [化 13]

[0027] (式中、 R'は置換基を有していても良い炭素数 7〜 15のァラルキル基、 HYは、ハロ ゲン化水素)で表される 3—ァラルキルォキシピロリジンのハロゲン化水素酸塩の結 晶を、絶対湿度 12gZm3以下の環境下で取り扱うことを特徴とする 3—ァラルキルォ キシピロリジンのハロゲンィ匕水素酸塩の取り扱い方法に関する。
発明の効果
[0028] 本発明によれば、 3—ァラルキルォキシピロリジン誘導体(2)を簡便且つ効率的に 、極めて高い生産性で、商業規模で製造することができる。また、 3—ァラルキルォキ シピロリジンのハロゲンィ匕水素酸塩 (3)の結晶を工業的規模で安定的に取り扱うこと ができる。さらに、原料である N—保護一 3—ァラルキルォキシピロリジン(1)を工業 的規模で効率的かつ安全に製造することができる。
発明を実施するための最良の形態
[0029] 本発明は下記の工程力 なる。
[0030] [化 14]

3 - Hydroxypyrrondme 、
[0031] 以下、本発明の各工程について詳述する。
まず、 3—ヒドロキシピロリジンを一般式(5);
[0032] [化 15]

[0033] で表される N—保護一 3—ヒドロキシピロリジンに変換する工程について説明する。
[0034] 前記式(5)にお 、て、 Pはァミノ基の保護基を意味する。このアミノ基の保護基は、 アミノ基を保護する基であり、一般に使用しうる基は、プロテクテイヴ'グループス 'イン •オーガニックシンセシス第 2版(PROTECTIVE GROUPS IN ORGANIC SYNTHESIS 2nd. Ed.)、ジョン'ゥイリ一'アンド'サンズ (JOHN WILEY&SONS)出版(1991年)に 記載されている。上記式(1)において保護基として好ましいものは、ウレタン型保護 基 (力ルバメート型保護基ともいう)である。なかでも、低級アルコキシカルボ-ル基( アルキル基の炭素数が 1〜6、好ましくは 1〜4であるもの)が好ましぐメトキシカルボ
-ル基、エトキシカルボ-ル基、または tert—ブトキシカルボ-ル基がより好ましぐ te rt—ブトキシカルボニル基が特に好まし 、。
[0035] 本反応は、前記専門書に記載の公知の方法を用いて行うことができる。特に方法 は限定されな ヽが、例えば、 3—ヒドロキシピロリジンをジ tert—ブトキシジカノレボネー トで処理することにより、ァミノ基に力ルバメート型保護基を導入することができる。
[0036] 用いる 3—ヒドロキシピロリジンは光学活性体であってもよいし、非光学活性体であ つて 良い。
[0037] 次に、 N—保護一 3—ヒドロキシピロリジン(5)を O—ァラルキルィ匕して、一般式(1); [0038] [化 16]

[0039] で表される N—保護一 3—ァラルキルォキシピロリジンへ変換する工程について説明 する。
[0040] O—ァラルキル化とはヒドロキシ基をァラルキルォキシ基に変換することを指す。前 記式(1)において、 3位の水酸基に結合する基 Rは、置換基を有していても良い炭素 数 7〜 15のァラルキル基である。置換基としては、水酸基、ハロゲン原子、アミノ基、 ニトロ基、アルコキシ基などがあげられる。置換基を有していても良い炭素数 7〜 15 のァラルキル基としては、例えば、ベンジル基、 p—クロ口べンジル基、 p—ヒドロキシ ベンジル基、 p—フルォロベンジル基、 m, m—ジフルォロベンジル基、フエ-ルェチ ル基、ナフチルメチル基等が挙げられる。好ましくは置換基を有していてもよい炭素 数 7または 8のァラルキル基であり、さらに好ましくは置換基を有していてもよいべンジ ル基であり、特に好ましくはべンジル基である。
[0041] 本工程に用いる N—保護— 3—ヒドロキシピロリジン(5)は、前記の方法で合成した ものでも良 、し、公知な方法で別途合成したものでも良 、。
[0042] N—保護— 3—ァラルキルォキシピロリジン(1)は光学活性体であってもよいし、非
光学活性体であっても良 、。
[0043] 本工程の反応は、塩基、並びに、金属ハロゲン化物及び Zまたは相関移動触媒の 存在下に行うのが好ましい。
[0044] 上記塩基としては、無機塩基、有機塩基を問わず使用しうるが、好ましくは無機塩 基であり、具体的には、特に限定されないが、水酸化ナトリウム、水酸ィ匕カリウム等の アルカリ金属水酸化物;炭酸ナトリウム、炭酸カリウム等アルカリ金属炭酸塩;炭酸水 素ナトリウム等のアルカリ金属炭酸水素塩等を挙げることができ、なかでも上記アル力 リ金属水酸化物が好ましい。
[0045] 塩基の使用量としては、特に制限されないが、普通 N—保護一 3 ヒドロキシピロリ ジン(5)に対して 1〜30倍モル、好ましくは 1〜20倍モル、より好ましくは 2〜10倍モ ル、特に好ましくは 2〜5倍モルである。
[0046] 上記金属ハロゲン化物としては、特に限定されないが、臭化リチウム、臭化ナトリウ ム、臭化カリウム、ヨウ化リチウム、ヨウ化ナトリウム、ヨウ化カリウム等のアルカリ金属ハ ロゲン化物;臭化マグネシウム、臭化カルシウム、ヨウ化マグネシウム、ヨウ化カルシゥ ム等のアルカリ土類金属ハロゲン化物;臭化アルミニウム、ヨウ化アルミニウム等のハ ロゲン化アルミニウム等を挙げることができ、好ましくは、アルカリ金属ハロゲン化物で あり、さらに好ましくは、ヨウ化カリウムである。
[0047] ハロゲン化アルカリ金属の使用量としては、特に制限されないが、一般に N—保護 —3—ヒドロキシピロリジン(5)に対して、 0. 01から 5モル0 /0が好ましぐより好ましくは 、 0. 05モル力も 1モル0 /0である。
[0048] 相関移動触媒としては、特に制限されないが、例えば、 12 クラウン— 4、 15 クラ ゥン一 5、 18 クラウン一 6、 24 クラウン一 8、ジベンゾ一 18 クラウン一 6、ジベン ゾ 24 クラウンー8、ジシクロへキシルー 18 クラウンー6、ジシクロへキシノレ 24 —クラウン一 8等のクラウンエーテル類;クリプタンド [2, 2]、クリプタンド [2, 2, 1]、タリ ブタンド [2, 2, 2]等のクリプタンド類;トリオクチルメチルアンモ -ゥムクロリド [商品名: ALIQUAT 336]、トリオクチルメチルアンモ-ゥムブロミド、メチルトリアルキル(炭 素数 8から 10)アンモ-ゥムクロリド [商品名: Adogen 464]、テトラプチルアンモ-ゥ ムクロリド、テトラプチルアンモ-ゥムブロミド、テトラプチルアンモ -ゥムョーダイド等の
4級アンモニゥム塩が挙げられる。上記相関移動触媒の内、一般に 4級アンモニゥム 塩が好ましぐなかでもテトラブチルアンモ-ゥムブロミドが好適に用いられる。
[0049] 相関移動触媒の使用量は、特に制限されないが、一般に N—保護一 3 ヒドロキシ ピロリジン(5)に対して、 0. 01から 5モル0 /0が好ましぐより好ましくは、 0. 05モル力 ら 1モル0 /0である。
[0050] 本工程の反応は、 a)塩基及び金属ハロゲンィ匕物、 b)塩基及び相関移動触媒、 c) 塩基、金属ハロゲン化物及び相関移動触媒で表される a)〜c) V、ずれかの存在下、 反応を実施することができる力 a)または c)の態様が好ましぐ a)で表される塩基及 び金属ハロゲンィ匕物の存在下で反応させることがさらに好ましい。
[0051] 反応溶剤としては、本質的に不活性な溶剤であれば特に限定されな 、が、例えば 、 N— 3—ヒドロキシピロリジン(5)の残存を低減させる為には、炭化水素系溶剤、ェ 一テル系溶剤、含窒素系溶剤、又は含硫黄系溶剤を用いるのが好ましい。
[0052] 上記炭化水素系溶剤としては、特に限定されないが、ベンゼン、トルエン、キシレン 、塩ィ匕ベンゼン、塩ィ匕トルエン等の芳香族炭化水素類、シクロへキサン、メチルシクロ へキサン、ヘプタン等の脂肪族炭化水素類を挙げることができるが、好ましくは、芳香 族炭化水素類であり、さらに好ましくはトルエンである。
[0053] 上記エーテル系溶剤としては、特に限定されないが、具体的には、テトラヒドロフラ ン、 1, 4 ジォキサン、ジメトキシェタン、エチレングリコールジメチルエーテル等を挙 げることができる力 好ましくは、 1, 4 ジォキサンである。
[0054] 上記含窒素系溶剤としては、具体的には、ァセトニトリル、ジメチルホルムアミド、ジ メチルァセトアミド、ジェチルァセトアミド、ジメチルブチルアミド、 N—メチルー 2—ピロ リドン、へキサメチル燐酸トリアミド等の含窒素系化合物を挙げることができるが、これ らに限定されるものではない。好ましくは、ァセトニトリルである。
[0055] 上記含硫黄系溶剤としては、特に限定されな 、が、例えば、ジメチルスルホキシド 等を挙げることができる。
[0056] 上記の有機溶剤の使用量は、特に制限されないが、 N—保護— 3ヒドロキシピロリジ ン(5)に対して、下限は普通 10wt%、好ましくは 50wt%、より好ましくは 100wt%で ある。上限は特に制限されないが、経済性を考慮して、普通 10000wt%、好ましくは
5000wt%、より好ましくは 2000wt%である。
[0057] 本発明の O—ァラルキルィ匕反応において用いられる反応溶剤は、上記溶剤を単独 で用いても良いし、 2種以上を併用しても良い。又、必要に応じて水を共存させても 良い。
[0058] 本工程の反応は、ハロゲン化ァラルキルを用いて行うことができる。ハロゲン化ァラ ルキルのァラルキル基としては、前記式(1)における Rとして説明したァラルキル基と 同様である。ハロゲンィ匕ァラルキルのハロゲンとしては、特に制限されないが、塩素、 臭素又はヨウ素が好ましぐ塩素またはヨウ素がさらに好ましぐとりわけ好ましくは塩 素である。ハロゲン化ァラルキルとしては、ハロゲン化ベンジルが好ましく塩化べンジ ルが特に好ましい。
[0059] 反応温度に関しては、下限は、反応溶媒の凝固点以上であれば、特に限定され ないが、一般に 0°C以上が好ましぐより好ましくは、 20°C以上であり、さらに好ましく は 40°C以上である。上限は、反応溶媒の沸点以下であれば、特に限定されないが、 好ましくは 120°C以下であり、より好ましくは 100°C以下であり、さらに好ましくは 80°C 以下である。
[0060] 反応時間に関しては、特に制限されないが、一般には 1〜100時間が好ましぐより 好ましくは 5〜80時間、さらに好ましくは 10〜50時間である。
[0061] 上記反応においては、反応終了時に N—保護一 3—ヒドロキシピロリジン(5)が残 存した場合、次工程の N -保護基の脱保護工程で上記 N—保護— 3—ヒドロキシピ 口リジン(5)が脱保護され、そのまま一般式(2)で表される 3—ァラルキルォキシピロリ ジン誘導体に不純物として混入する問題が生じる。従い、本反応における反応終了 時の N—保護一 3—ヒドロキシピロリジン(5)の残存量は少な!/、方が好ましぐその残 存量は、普通、生成物である N—保護— 3—ァラルキルォキシピロリジン(1)に対して 、 10wt%以下、より好ましくは 5wt%以下、さらにこのましくは 3wt%以下、特に好ま しくは 2wt%以下である。本反応は、反応中の N—保護一 3—ヒドロキシピロリジン(5 )の残存量が N—保護— 3—ァラルキルォキシピロリジン(1)に対して上記値になるま で反応を継続させることが好まし 、。
[0062] この反応液力 生成物を取得する為には、一般的な後処理を行えばよい。例えば、
反応終了後の液を分液し、生成物を含む有機層を取得することができる。得られた 有機層は、必要に応じて水洗してもよぐついで、減圧加熱等の操作により反応溶剤
、および抽出溶剤を留去すると、 N—保護— 3—ァラルキルォキシピロリジン(1)が得 られる。
[0063] このようにして得られる生成物は、さらに必要に応じて晶析精製、分別蒸留、カラム クロマトグラフィー等の一般的な手法により精製を加え、純度を高めることができる。
[0064] 次に、 N—保護— 3—ァラルキルォキシピロリジン(1)から一般式(2);
[0065] [化 17]

[0066] で表される 3—ァラルキルォキシピロリジン誘導体(2)へ変換する工程について説明 する。
[0067] 3—ァラルキルォキシピロリジン誘導体(2)は 3—ァラルキルォキシピロリジンの酸塩 である。前記式(2)における HXは、例えば、鉱酸、スルホン酸、カルボン酸、又はァ ミノ酸が挙げられる。これらは、 1価の酸に限定されず、 2価でも 3価でもよい。鉱酸と しては、特に制限されないが、例えば、塩化水素や臭化水素などのハロゲン化水素、 硫酸、燐酸等が挙げられる。スルホン酸としては、特に制限されないが、例えば、メタ ンスルホン酸、エタンスルホン酸、プロパンスルホン酸、ブタンスルホン酸、ベンゼンス ルホン酸、 p—トルエンスルホン酸、カンファースルホン酸、 1—フエ-ルェタンスルホ ン酸等が挙げられる。カルボン酸としては、特に制限されないが、例えば、蟻酸、酢 酸、トリフルォロ酢酸、安息香酸の非光学活性カルボン酸、酒石酸等の光学活性力 ルボン酸等が挙げられる。アミノ酸としては、特に制限されないが、ァラニン、パリン、 フエ-ルァラニン、ァスパラギン酸等の光学活性アミノ酸を挙げることができる。
[0068] これらの酸のうち、得られる酸との塩が良好な結晶性を有する塩ィ匕水素、臭化水素 、 p—トルエンスルホン酸、安息香酸が好ましいが、なかでも塩化水素、臭化水素が
好ましぐ特に好ましくは、塩ィ匕水素である。また、晶析に供する 3—ァラルキルォキ シピロリジン誘導体 (2)が光学活性体であり、且つその光学純度を高める必要がある 場合は、用いる酸としては光学活性な酸を用いるのが好ましぐ光学活性な酸として は、光学活性カルボン酸や光学活性アミノ酸を使用するのが好ましぐなかでも光学 活性酒石酸や光学活性ァスパラギン酸が好まし 、。
[0069] N—保護一 3—ァラルキルォキシピロリジン誘導体(1)は前記の方法で合成したも のでも良いし、別途公知な方法で合成したものでも良い。 N—保護一 3—ァラルキル ォキシピロリジン誘導体(1)は不純物として N—保護一 3—ヒドロキシピロリジン(5)及 び/又はァラルキルアルコールを含有して!/、ても良! 、。ァラルキルアルコールとして は一般式;
ROH (6)
で表されるァラルキルアルコールがあげられる。 Rとしては前述のものがあげられ、ァ ラルキルアルコール(6)としては、好ましくは炭素数 7または 8のァラルキルアルコー ルであり、より好ましくはべンジルアルコールである。
[0070] 上記 N—保護— 3—ァラルキルォキシピロリジン誘導体(1)は、光学活性体でも非 光学活性体の 、ずれを用いても良 、が、 N—保護一 3—ァラルキルォキシピロリジン (1)が光学活性体の場合は、得られる 3—ァラルキルォキシピロリジン誘導体(2)も光 学活性体であり、 N—保護— 3—ァラルキルォキシピロリジン(1)の 3位の立体配置が (3R)の場合、 3—ァラルキルォキシピロリジン誘導体(2)の 3位の立体配置は(3R)、 前者が(3S)の場合は、後者は(3S)となる。
[0071] 本工程の反応は、 N—保護基を示す Pの種類に応じて、適宜適切な方法を選ぶこ とができる。脱保護の方法としては、例えば、プロテクティヴ'グループス'イン'オーガ ニックシンセシス第 2版(PROTECTIVE GROUPS IN ORGANIC SYNTHESIS 2nd. Ed -)記載の方法があげられる。例えば、 P力 酸で脱保護される tert—ブトキシカルボ- ルの場合は、上記脱保護を行うか、或いは、 N—保護— 3—ァラルキルォキシピロリ ジン誘導体(1)に、後述する酸塩を形成する操作を実施し、脱保護及び酸塩の形成 を同時に行っても良い。
[0072] 次に、 3—ァラルキルォキシピロリジンまたは、酸で脱保護される保護基を有する N
—保護一 3 ァラルキルォキシピロリジン誘導体(1)力 酸との塩である 3 ァラルキ ルォキシピロリジン誘導体(2)を形成する方法につ!ヽて説明する。
[0073] 使用する酸としては上記 HXであげた酸があげられる。酸の使用量としては、およそ 理論量以上であればよいが、多量に使用しても経済的ではないだけであるので、普 通 N—保護一 3 ァラルキルォキシピロリジンに対して、 1〜10モル倍量、好ましくは 1〜3モル倍量、より好ましくは、 1〜2モル倍量である。
[0074] 酸の添加速度としては、特に制限されないが、 3—位の水酸基の脱保護が進行す るのを回避するためには、酸の使用量の全量を 1Z6時間以上かけて添加するのが 好ましい。より好ましくは 1時間以上であり、さらに好ましくは 3時間以上、特に好ましく は 6時間以上である。添カ卩時間の上限は特に制限されないが、好ましくは 24時間以 下、好ましくは 12時間以下である。
[0075] 本反応は、普通溶剤中で実施される。溶剤としては、特に制限されないが、例えば 、アルコール系溶剤、エーテル系溶剤、エステル系溶剤又は芳香族炭化水素系溶 剤等の有機溶剤が挙げられる。
[0076] アルコール系溶剤としては、特に制限されな!、が、例えば、メタノール、エタノール、 n プロパノール、イソプロパノール、 n—ブタノール、 sec ブタノール、イソブタノー ル、 tert—ブタノール等を挙げることができる。なかでもイソプロパノールが好ましい。
[0077] エーテル系溶剤としては、特に制限されないが、例えば、テトラヒドロフラン、 1, 4 ジォキサン、 1, 3 ジォキソラン、 1, 2 ジメトキシェタン、ジエチレングリコールジメ チルエーテル、メチル tert ブチルエーテル等が挙げられる。なかでもテトラヒドロフ ランが好ましい。
[0078] エステル系溶剤としては、特に制限されないが、好ましくは炭素数 2〜8、より好まし くは炭素数 4〜6のエステルであり、具体的には、例えば、酢酸ェチル、酢酸 n プロ ピル、酢酸イソプロピル、酢酸 n—ブチル、酢酸 tert ブチル等が挙げられ、なかで も酢酸ェチルが好ましい。
[0079] 芳香族炭化水素系溶剤としては、特に制限されないが、好ましくは炭素数 6〜12、 より好ましくは炭素数 6〜10、さらに好ましくは炭素数 6〜8の芳香族炭化水素であり 、具体的には、例えば、ベンゼン、トルエン、キシレン等を挙げることができる。なかで
も炭素数 7又は 8の芳香族炭化水素、具体的には、トルエン、キシレンが特に好ましく
、トルエンが最も好適に用いられる。
[0080] 上記溶剤の中でも、反応性の高さ、酸に対する安定性の観点から、アルコール系 溶剤が好ましぐなかでもイソプロパノールが好まし!/、。
[0081] なお、言うまでもなく上記反応溶剤は単独で用いても良いが、 2種以上併用すること もできる。また、反応性をさらに高める観点から、上記有機溶剤に水を共存させてもよ い。用いる水の量は特に制限されないが、反応液の総重量に対して、普通 0. 1〜50 wt%、好ましくは 0. 5〜30wt%、より好ましくは l〜20wt%である。
[0082] 反応温度は、特に制限されないが、普通— 20〜100°C、好ましくは 0〜80°C、より 好ましくは、 20〜50°Cである。また、反応時間としては、特に制限されないが、普通 1
〜100時間、好ましくは 1〜48時間、より好ましくは 1〜24時間である。
[0083] 次に、 3 ァラルキルォキシピロリジン誘導体(2)の化学純度を向上させる方法に ついて説明する。本方法は、 3 ァラルキルォキシピロリジン誘導体(2)を含有する 水と有機溶剤の 2相系混合物から、
A)有機層を除去した後、水層を塩基で処理し、
B) 3 ァラルキルォキシピロリジン誘導体(2)の遊離のアミンを有機溶剤で抽出し、
C)該遊離のアミンを酸で処理して 3 ァラルキルォキシピロリジン誘導体(2)に変換 する、
一連の操作により化学純度を向上させることを特徴とする。
[0084] 3 ァラルキルォキシピロリジン誘導体(2)は、前記の方法で合成したものでも良!ヽ し、別途公知な方法で合成したものでも本方法に使用することができる。前記の方法 で合成した 3 ァラルキルォキシピロリジン誘導体 (2)を使用する場合、特に制限さ れな 、が、反応終了液をそのまま用いたものでも良 、。
[0085] 本方法に用いる 3 ァラルキルォキシピロリジン誘導体(2)は、ラセミ体であっても 良いし、光学活性体の (R)—又は(S)—3—ァラルキルォキシピロリジン誘導体あつ ても良い。医薬中間体としての有用性力 光学活性体 3—ァラルキルォキシピロリジ ン誘導体が好ましい。
[0086] 本方法において、 3 ァラルキルォキシピロリジン誘導体(2)に含まれる不純物を
効果的に除去することができる。不純物としては、 3—ヒドロキシピロリジン及び Z又は ァラルキルアルコール(6)があげられる。ァラルキルアルコール(6)は前記に同じで ある。
[0087] 本方法に用いる 3—ァラルキルォキシピロリジン誘導体(2)の純度は特に制限され ないが、普通、 70%以上が好ましぐより好ましくは 80%以上であり、 85%以上が特 に好ましい。
[0088] 3—ァラルキルォキシピロリジン誘導体(2)を含有する水と有機溶剤の 2相系混合 物とは、例えば、 3—ァラルキルォキシピロリジン誘導体(2)に水及び有機溶剤を添 カロした混合物があげられる。添加する有機溶剤としては、特に制限されないが、水と の相溶性を有さない溶剤が好ましぐ例えば、トルエン、酢酸ェチル、メチル tert—ブ チルエーテル、塩化メチレン等を挙げることができる。なかでも、トルエン、酢酸ェチ ルが好ましぐ特にトルエンが好適に用いられる。前記の方法で合成された 3—ァラ ルキルォキシピロリジン誘導体(2)の反応終了液を用いる場合は、水及び Zまたは 有機溶剤を適宜添加すればょ ヽ。
[0089] 2相系混合物中の水と有機溶剤の量に特に制限はない。また、 3—ァラルキルォキ シピロリジン誘導体(2)は 2相系混合物に溶解していてもよいし、一部が析出したまま でも良い。
[0090] ー且、水層に移行した 3—ァラルキルォキシピロリジン誘導体(2)の水溶液は、さら に有機溶剤で洗浄しても良い。洗浄する有機溶剤としては、特に制限されないが、好 ましくは前記反応終了後に添加した有機溶剤が用いられる。
[0091] 水層に移行した 3—ァラルキルォキシピロリジン誘導体(2)は、塩基で解塩すること により、 3—ァラルキルォキシピロリジン誘導体(2)の遊離のァミンに変換することがで きる。塩基としては、無機塩基および Zまたは有機塩基を用いることができ、無機塩 基としては、特に制限されないが、例えば、水酸化リチウム、水酸化ナトリウム、水酸 化カリウム等のアルカリ金属水酸ィ匕物;炭酸ナトリウム、炭酸カリウム等のアルカリ金属 炭酸塩、炭酸水素ナトリウム等のアルカリ金属炭酸水素塩等を挙げることができ、好 ましくはアルカリ金属水酸ィ匕物であり、特に好ましくは水酸ィ匕ナトリウムである。有機 塩基としては、特に制限されないが、例えば、メチルァミン、ェチルァミン、プロピルァ
ミン、ブチルァミン等の 1級ァミン;ジメチルァミン、ジェチルァミン、ジプロピルァミン、 ジイソプロピルァミン、ジブチルァミン等の 2級ァミン;トリメチルァミン、トリェチルァミン 、トリプロピルアミン等の 3級ァミンを挙げることができ、好ましくはトリエチルァミン等の 3級ァミンである。
[0092] この 3—ァラルキルォキシピロリジン誘導体(2)の遊離のアミンは、有機溶剤に抽出 し、さらに必要に応じて水洗することにより、化学純度が向上した 3—ァラルキルォキ シピロリジン誘導体(2)の遊離のァミンとして取得することができる。用いる有機溶剤 としては、特に制限されないが、水との相溶性を有さない溶剤が好ましぐ例えば、ト ルェン、酢酸ェチル、メチル tert—ブチルエーテル、塩化メチレン等を挙げることが できる。なかでも、トルエン、酢酸ェチルが好ましぐ特にトルエンが好適に用いられる 。 3—ァラルキルォキシピロリジン誘導体の遊離のァミンが溶解する量であれば、使 用量に特に制限はな 、。このようにして得た 3—ァラルキルォキシピロリジン誘導体( 2)の遊離のアミンを酸で処理し、 3—ァラルキルォキシピロリジン誘導体(2)を合成す ることがでさる。
[0093] 用いる酸としては、前記 HXとして説明した酸が挙げられる。なかでも、得られる酸と の塩が良好な結晶性を有する塩ィヒ水素、臭化水素、 p—トルエンスルホン酸、安息香 酸が好ましぐ塩化水素、臭化水素がより好ましぐ特に好ましくは、塩ィ匕水素である。
[0094] 次に 3—ァラルキルォキシピロリジン誘導体(2)を晶析工程に付し、結晶として取得 する方法について説明する。
[0095] 本工程に用いる 3—ァラルキルォキシピロリジン誘導体(2)は、前述の方法で N— 保護— 3—ヒドロキシピロリジン(5)から製造したものであっても良いし、別途公知な方 法で合成したものであっても良い。また、前述の方法で 3—ァラルキルォキシピロリジ ン誘導体(2)の化学純度を高めたものであっても良!、。
[0096] 本方法に用いる 3—ァラルキルォキシピロリジン誘導体(2)は、ラセミ体であっても 良いし、光学活性体の (R)—又は(S)—3—ァラルキルォキシピロリジン誘導体であ つても良い。医薬中間体としての有用性力 光学活性体 3—ァラルキルォキシピロリ ジン誘導体が好ましい。
[0097] 本工程に用いる 3—ァラルキルォキシピロリジン誘導体(2)の純度は特に制限され
ないが、 70%以上が好ましぐより好ましくは 80%以上であり、 85%以上が特に好ま しい。
[0098] 本方法に用いる 3—ァラルキルォキシピロリジン誘導体(2)に含まれる不純物として は、例えば、 N—保護一 3—ヒドロキシピロリジンが脱保護された 3—ヒドロキシピロリジ ン及び Z又はァラルキルアルコール(6)があげられる。 3—ァラルキルォキシピロリジ ン誘導体(2)に含まれる 3—ヒドロキシピロリジンの量としては、 3—ァラルキルォキシ ピロリジン誘導体(2)に対して、 10wt%以下、より好ましくは 5wt%以下、さらに好ま しくは 3wt%以下、特に好ましくは 2wt%以下である。ァラルキルアルコール(6)の量 としては、好ましくは 4wt%以下、より好ましくは 3wt%以下、さらに好ましくは 2wt% 以下である。
[0099] 晶析方法としては、特に制限されないが、例えば、反応晶析法、冷却晶析法、濃縮 晶析法、溶剤置換を用いる晶析法、貧溶剤を混合することによる晶析法、及び Z又 は塩析法等の一般に用いられる晶析法を、単独又は適宜組み合わせて実施すること ができる。また、 N—保護一 3—ァラルキルォキシピロリジン(1)、または、 3—ァラル キルォキシピロリジン力も酸塩を形成する際に、極性有機溶剤を用いることで、 3—ァ ラルキルォキシピロリジン誘導体(2)の結晶を得る反応晶析法も適宜使用することが できる。なお、本晶析では必要に応じて種晶を添加することもできる。
[0100] 本晶析方法は、 3—ァラルキルォキシピロリジンの溶液や油状の濃縮物などに適用 することもできるし、ー且本方法にて取得した 3—ァラルキルォキシピロリジンを溶解 して、さらに結晶の純度を高めるために実施してもよい。
[0101] 上記結晶化方法は、通常、極性溶剤、または、極性溶剤と非極性溶剤の混合溶剤 中で実施される。極性溶剤と非極性溶剤の混合溶剤中で実施する場合、その混合 割合に特に制限はないが、混合溶剤中、極性溶剤の割合が 5%以上であればよぐ
10%以上がより好ましぐ 30%以上がさらに好ましぐ 50%以上が特に好ましい。
[0102] 極性溶剤としては、特に制限されな!ヽが、例えば、水、エステル系溶剤、エーテル 系溶剤、ハロゲン化炭化水素系溶剤、含窒素系溶剤、アルコール系溶剤、ケトン系 溶剤等を挙げることができる。
[0103] エステル系溶剤としては、特に制限されないが、好ましくは炭素数 2〜8、より好まし
くは炭素数 4〜6のエステルであり、具体的には、例えば、酢酸ェチル、酢酸 n—プロ ピル、酢酸イソプロピル、酢酸 n—ブチル、酢酸 tert—ブチル等が挙げられ、なかで も酢酸ェチルが好ましい。
[0104] エーテル系溶剤としては、特に制限されないが、例えば、テトラヒドロフラン、 1, 3— ジォキソラン、 1, 2—ジメトキシェタン、ジエチレングリコールジメチルエーテル、メチ ル tert -ブチルエーテル等が挙げられる。なかでもメチル tert -ブチルエーテルが 好ましい。
[0105] ハロゲン化炭化水素系溶剤としては、特に制限されないが、例えば、塩化メチレン、 クロ口ベンゼン、ジクロロベンゼン、 1, 2—ジクロロェタン等が挙げられる。なかでも、 クロ口ベンゼン、又はジクロロェタンが好ましい。
[0106] 含窒素系溶剤としては、特に制限されないが、ァセトニトリル、ジメチルホルムアミド
、ジメチルァセトアミド、ジェチルァセトアミド、ジメチルブチルアミド、 N—メチルー 2— ピロリドン等が挙げられる。なかでもァセトニトリルが好まし 、。
[0107] アルコール系溶剤としては、特に制限されないが、例えば、メタノール、エタノール、 n—プロパノール、イソプロパノール、 n—ブタノール、 sec—ブタノール、イソブタノー ル、 tert—ブタノール等を挙げることができる。なかでもイソプロパノールが好ましい。
[0108] ケトン系溶剤としては、特に制限されないが、例えば、アセトン、メチルェチルケトン 等が挙げられる。なかでもアセトンが好ましい。
[0109] 極性溶剤としてはエステル系溶剤、ハロゲン化炭化水素溶剤、含窒素系溶剤、ァ ルコール系溶剤、ケトン系溶剤が好ましぐエステル系溶剤またはアルコール系溶剤 が特に好ましい。
[0110] これらの溶剤は単独で用いてもよぐ 2種以上を併用しても良い。
[0111] 極性溶剤の使用量としては、特に制限されないが、 3—ァラルキルォキシピロリジン 誘導体(2)に対して、下限は普通、 0. 01倍重量、好ましくは 0. 05倍重量、より好ま しくは 0. 1倍重量、さらに好ましくは 0. 5倍重量、特に好ましくは 1倍重量であり、上 限は、 100倍重量、好ましくは 50倍重量、より好ましくは 30倍重量である。
[0112] 非極性溶剤としては、特に制限されないが、脂肪族炭化水素系溶剤、芳香族炭化 水素系溶剤等の炭化水素系溶剤を挙げることができる。
[0113] 脂肪族炭化水素系溶剤としては、特に制限されないが、炭素数 5〜8の脂肪族炭 化水素、例えば、ペンタン、へキサン、ヘプタン、メチルシクロへキサン等を挙げること ができる。なかでも炭素数 6又は 7の脂肪族炭化水素、具体的には、へキサン、ヘプ タン、メチルシクロへキサン等が好ましい。
[0114] 芳香族炭化水素系溶剤としては、特に制限されないが、好ましくは炭素数 6〜8の 芳香族炭化水素、具体的には、例えば、ベンゼン、トルエン、キシレン等を挙げること ができる。なかでも炭素数 7又は 8の芳香族炭化水素、好ましくは、トルエン、キシレン 等が好ましい。
[0115] 非極性溶剤を使用する場合、あらかじめ極性溶剤と混合して用いてもよいが、必要 に応じて、反応晶析法、冷却晶析法や濃縮晶析法などで結晶が析出した後で適宜 添加しても良い。好ましくは、結晶が析出した後で添加する方法である。
[0116] 晶析温度は、特に制限されないが、普通 100°C以下、好ましくは 80°C以下、より好 ましくは 60°C以下、さらに好ましくは 40°C以下、とりわけ好ましくは 30°C以下である。 下限は、普通— 20°C、好ましくは— 10°C、より好ましくは 0°Cである。
[0117] 晶析に際しては、単位容積当たりの攪拌所要動力が 0. lkWZm3以上、好ましくは 0. 3kWZm3以上、より好ましくは 0. 5kWZm3以上の強攪拌下で析出させるのが好 ましい。
[0118] このようにして得られる 3—ァラルキルォキシピロリジン誘導体(2)は、遠心分離、加 圧分離、減圧濾過等の一般的な固液分離方法を用いて結晶を採取することができる 。得られた結晶は、必要に応じて、例えば、減圧乾燥 (真空乾燥)することにより乾燥 結晶として取得することができる。
[0119] 本工程により得られた 3—ァラルキルォキシピロリジン誘導体(2)において、 Rがべ ンジルである一般式 (4) ;
[0120] [化 18]

[0121] であらわされる 3—べンジルォキシピロリジン誘導体の結晶は、本発明において、初 めて単離された新規な結晶であり、取り扱いの容易性から、医薬中間体として非常に 有用である。 3—べンジルォキシピロリジン誘導体 (4)において HXは前記に同じであ る。本発明の 3—ベンジルォキシピロリジン誘導体 (4)の結晶は、ラセミ体であっても よいし、光学活性体であっても良い。医薬中間体としての有用性力も光学活性体で あることが好ましい。
[0122] 尚、言うまでも無ぐ本発明により取得される 3—ァラルキルォキシピロリジン誘導体
(2)の結晶は、高純度の結晶である。すなわち、化学純度が 95%以上、好ましくは 9 7%以上、より好ましくは 98%以上、とりわけ 99%以上である。さらに、光学活性体の 場合は、上記の化学純度にカ卩え、光学純度が 98%ee以上、好ましくは 99%ee以上 、より好ましくは 99. 5%ee以上である。また、本結晶化工程により得られる 3—ァラル キルォキシピロリジン誘導体(2)に含まれる 3—ヒドロキシピロリジンの含有量は、普通 2%以下、好ましくは 1%以下、より好ましくは 0. 5%以下、特に好ましくは 0. 3%以 下である。さらに、 3—ァラルキルォキシピロリジン誘導体(2)に含まれる不純物は、 普通 1%以下、好ましくは 0. 5%以下、より好ましくは、 0. 3%以下、とりわけ好ましく は 0. 1%以下である。
[0123] 次に、 3—ァラルキルォキシピロリジン誘導体(2)において、一般式(3);
[0124] [化 19]

[0126] 前記式(3)の R'は、置換基を有していても良い炭素数 7〜 15のァラルキル基、又 は水素原子である。置換基を有していても良い炭素数 7〜 15のァラルキル基は、前 記式(1)の Rとして説明した基と同様である。また、前記式(3)における HYは、塩ィ匕 水素、臭化水素、ヨウ化水素、フッ化水素、或いはこれらの混合物である。好ましくは 、塩ィ匕水素又は臭化水素であり、より好ましくは、塩ィ匕水素である。
[0127] 上記 3—ァラルキルォキシピロリジンのハロゲン化水素酸塩(3)は、ラセミ体であつ てもよく、光学活性体であってもよいが、好ましくは光学活性体である。
[0128] 上記 3—ァラルキルォキシピロリジンのハロゲンィ匕水素酸塩(3)は、例えば、前記 3 ーァラルキルォキシピロリジン誘導体(2)で述べた方法により製造することができる。
[0129] 本発明の取り扱い方法は、 3—ァラルキルォキシピロリジンのハロゲン化水素酸塩( 3)の乾燥結晶に対して特に効果的である。ここで乾燥結晶とは、当初の水分含量が 当該環境における平衡含水量より少なぐ時間と共に吸湿する (水分含量が増加す る)結晶を表し、好ましくは、 3—ァラルキルォキシピロリジンのハロゲンィ匕水素酸塩(3 )の結晶を減圧乾燥等により乾燥処理を施した結晶である。上記 3—ァラルキルォキ シピロリジンのハロゲンィ匕水素酸塩 (3)の水分含量としては、取り扱 、操作の種類や 所要時間にもよるが、例えば、水分含量 3重量%以下、好ましくは 2重量%以下、より 好ましくは 1重量%以下、さらに好ましくは 0. 5重量%以下、特に好ましくは 0. 3重量 %以下である。
[0130] 本発明は、上述の問題を解決するために、 3—ァラルキルォキシピロリジンのハロゲ ン化水素酸塩(3)の結晶を、絶対湿度 12g/m3以下、好ましくは 10g/m3以下、特 に好ましくは 8gZm3以下の環境下で取り扱うことを特徴とする。上記の湿度条件取り 扱うことで、吸湿が好適に抑制され、 3—ァラルキルォキシピロリジンのハロゲン化水 素酸塩 (3)が潮解するのを防ぐことができる。
[0131] 本発明において、上記の湿度の環境は、環境力もの除湿、或いは、除湿された気 体 (好ましくは、乾燥不活性ガス、例えば乾燥窒素等)の環境への導入等により与え られる。例えば、絶対湿度 12gZm3以下の環境とは、例えば密閉されたボックス内で
取り扱う場合にはボックス内の絶対湿度が 12gZm3以下であればよぐ屋内で取り扱 う場合には、 3—ァラルキルォキシピロリジンのハロゲンィ匕水素酸塩(3)の結晶の周 囲の絶対湿度を測定すればよぐ範囲は特に制限されないが、例えば、 l〜30mの 同屋内で絶対湿度を測定すればよい。絶対湿度の測定方法は、市販の絶対湿度計 を用いて直接測定してもよいが、別の方法としては、上記環境における温度と相対湿 度を各々測定し、その温度における飽和水蒸気濃度 (gZm3)と相対湿度(%)の積 より求めることちでさる。
[0132] 除湿の方法は、特に制限されないが、湿気の氷結、除湿機や乾燥剤 (シリカゲル等 )の使用により達成される。また、特に好ましい方法としては、 3—ァラルキルォキシピ 口リジンのハロゲンィ匕水素酸塩 (3)の結晶を取り扱う環境を低温下で扱う方法を挙げ ることができる。すなわち、低温環境下では、本質的に大気中の飽和水蒸気濃度が 低下することから、湿度の高い地域、例えば、日本国内においてさえ、容易に前記低 湿度の環境を提供することができる。これ〖こより、 3—ァラルキルォキシピロリジンのハ ロゲン化水素酸塩 (3)の結晶の吸湿を好適に抑制し、その潮解を防ぐことができる。 尚、前記低温下とは、普通 25°C以下、好ましくは 20°C以下、より好ましくは 10°C以下 、とりわけ 5°C以下である。
[0133] 従って、本発明によれば、前記絶対湿度の環境下、とりわけ低温環境下では、大規 模スケール(工業的規模)においても、安定的に、 3—ァラルキルォキシピロリジンの ノ、ロゲンィ匕水素酸塩(3)の結晶を、高品質を維持して取り扱うことができる。
[0134] 以上述べたように、本発明によれば、医薬品等の中間体として有用な 3—ァラルキ ルォキシピロリジンのハロゲン化水素酸塩 (3)を、大規模スケール (工業的規模)に おいても、好適に取り扱うことができる。
[0135] 尚、言うまでもなぐ前記絶対湿度の環境が与えられれば、その方法は、特に問わ ない。又、本発明の実施は、常圧下、加圧下、減圧下におこなうことができ、通常、常 圧下に好適に行うことができる。
[0136] 本取り扱い方法によれば、 3—ァラルキルォキシピロリジンのハロゲン化水素酸塩( 3)の結晶の吸湿が抑制されるため、金属製材料と接触させた場合でさえも、固体中 への金属成分の混入が軽減されると共に金属製材料の腐食も軽減される。一方、相
対湿度の高い環境下では、悪影響を及ぼす程度まで吸湿が進行する。更には固体 中へ金属成分が著しく混入すると共に金属製材料が著しく腐食する。尚、金属製材 料とはステンレススチール、ハステロイ、鉄などが挙げられる。例えばオーステナイト 系ステンレススチールである SUS304、 SUS304L, SUS316、 SUS316L, SUS3 09S、 SUS310S、 SUS321或いは SUS347であり、とりわけ SUS304或いは SUS 316である。
[0137] また、固体中へ混入しうる金属成分はステンレススチールに含まれる鉄、クロム、二 ッケル等の遷移金属、或いはそれらの塩を挙げることができる。最も混入しうる金属成 分は鉄、クロム、ニッケル、或いはこれらの塩やこれらの混合物であり、とりわけ鉄或 いはその塩である。
[0138] 尚、本発明において、 3—ァラルキルォキシピロリジンのハロゲン化水素酸塩(3)の 結晶の梱包'保管は、たとえば、 1重又は 2重以上のアルミラミネート袋やポリエチレン 袋等の袋中で、好ましくは外袋をアルミラミネート袋、内袋をポリエチレン袋として行う ことができる。更に、必要に応じ、或いは、好ましくは、乾燥剤(シリカゲル等)を同封 できる。乾燥剤を同封する場合は、内袋に同梱してもよいし、外袋の内側、かつ、内 袋の外側に梱包してもよい。結晶を梱包した上記の袋を、必要に応じ、或いは、好ま しくは、鋼製のドラム、或いは、ファイバードラム、或いは、ダンボール等の外装中に 人れることができる。
[0139] 本発明は、大規模スケール (工業的規模)で、その効果を最大限に発揮しうる。上 記スケールとしては、特に限定されないが、 3—ァラルキルォキシピロリジンのハロゲ ン化水素酸塩(3)の結晶として、普通 lkg以上、好ましくは 10kg以上、より好ましくは 100kg以上、とりわけ 1000kg以上である。
[0140] 本発明によれば、 3—ァラルキルォキシピロリジンのハロゲン化水素酸塩(3)の結 晶の取り扱いが長期間に及ぶ場合でも、周囲の環境を絶対湿度 12gZm3以下、好 ましくは 10g/m3以下、特に好ましくは 8g/m3以下に制御することによって、その平 衡含水率までの吸湿に抑えて (すなわち、その水分含量を 3重量%以下、好ましくは 2重量%以下、より好ましくは 1重量%以下、さらに好ましくは 0. 5重量%以下、特に 好ましくは 0. 3重量%以下に制御して)、上述の問題を見事に解決しうる。
[0141] 尚、言うまでも無ぐ本発明により取得される 3 ァラルキルォキシピロリジンのハロ ゲンィ匕水素酸塩(3)の結晶は、白色〜淡褐色で、高純度である。すなわち、化学純 度が 95%以上、好ましくは 96%以上、より好ましくは 97%以上、さらに好ましくは 98 %以上、とりわけ 99%以上が好ましい。さらに、光学活性体の場合は、上記の化学純 度に加え、光学純度が 98%ee以上、好ましくは 99%ee以上、より好ましくは 99. 5% ee以上であり、特に好ましくは 99. 8%ee以上である。
実施例
[0142] 以下、実施例を示して本発明を詳細に説明する。これらの実施例は無論本発明を 何ら限定するものではな 、。
[0143] なお、実施例に記載している 3 ヒドロキシピロリジン誘導体の含量、不純物含量、 並びに光学純度は、以下の HPLC法により分析した。
[含量、不純物含量分析法]
カラム:コスモシル 5C18ARII (250X4. 6mm;ナカライ製)、
移動相: KH POバッファー(pH3) Zァセトニトリル =80Z20 (vZv)、
2 4
流速: 0. 5mlZmin(0〜20分)、 1. 5mlZmin(20〜30分)、
検出: UV 210nm、
カラム温度: 40°C
保持時間: 3 べンジルォキシピロリジン塩酸塩 2. 4分、ベンジルアルコール 3. 4 分
[0144] [3 ヒドロキシピロリジン含量の分析法]
本方法では、 3 ヒドロキシピロリジン塩酸塩を tert ブトキシカルボ-ルァミノ 3 —ヒドロキシピロリジンに誘導ィ匕し、定量する。
(3—ヒドロキシピロリジン塩酸塩の誘導ィ匕方法)
3 ヒドロキシピロリジン塩酸塩を含有する 3—ベンジルォキシピロリジン塩酸塩をィ ソプロパノールに溶解し、 30%水酸ィ匕ナトリウム水溶液で中和した後、ジ一 tert ブ チルジカルボナートをカ卩え、超音波処理する。得られた混合物を HPLCに注入し、分 析する。
[0145] (分析条件)
カラム: AQ303 (250X4. 6mm, 5 πι;ΥΜ。製)、
移動相 Α: 0. 5%ΚΗ ΡΟ水溶液、
2 4
移動相 Β :ァセトニトリル、
流速:丄 . OmlZ mm、
検出: UV 210nm、
カラム温度: 40°C
グラジェント条件:
時間(分) 八液(%) 8液(%)
0 80 20
10 40 60
30 40 60
31 80 20
40 80 20
保持時間: N— tert ブトキシカルボ-ルァミノ 3—ヒドロキシピロリジン(3—ヒドロ キシピロリジン塩酸塩の Boc保護体) 10. 4分、 N— tert ブトキシ—カルボ-ルァ ミノ一 3—ベンジルォキシピロリジン(3—ベンジルォキシピロリジン塩酸塩の Boc保護 体) 24. 1分
[0146] [光学純度分析法]
本方法では、 3 ァラルキルォキシピロリジンを tert ブトキシカルボ-ルァミノ 3 一べンジルォキシピロリジンに誘導ィ匕し、光学純度を測定する。
(3—ァラルキルォキシピロリジン塩酸塩の誘導ィ匕方法)
鏡像異性体を含有する 3—ベンジルォキシピロリジン塩酸塩をイソプロパノールに 溶解し、 30%水酸ィ匕ナトリウム水溶液で中和した後、ジ— tert—ブチルジカルボナ ートを加え、超音波処理する。得られた混合物を HPLCに注入し、分析する。
[0147] (分析条件)
カラム: CHIRALPAK AD— H (4. 6mmX250mm;ダイセル製)、
移動相:へキサン ZlPA= 98/2 (v/v)、
検出: UV 210nm、
カラム温度: 13°C
保持時間:(R)—N— tert—ブトキシカルボ-ルァミノ— 3—ベンジルォキシピロリジ ン((R)— 3 ベンジルォキシピロリジン塩酸塩の Boc保護体) 13. 4分、(S)— N— tert ブトキシカルボニルアミノー 3—べンジルォキシピロリジン((S)—3—べンジル ォキシピロリジン塩酸塩の Boc保護体) 12. 1分。
[0148] (実施例 1) N— tert ブトキシカルボ二ルー(R)—3 ヒドロキシピロリジン
(R)— 3 ヒドロキシピロリジン塩酸塩 124gを蒸留水 187gで溶解し、炭酸カリウム 水溶液 217g (炭酸カリウム 72g含有)をカ卩え中和した。この溶液に二炭酸ジ― tert - ブチルの THF溶液 431g (二炭酸ジ— tert—ブチル 214g含有)を滴下した。しばらく 撹拌した後、トルエン 565gで抽出を行い、濃縮して N— tert ブトキシカルボ-ルー (R) - 3 ヒドロキシピロリジン 180gを取得した(収率 96%)。
— NMR(CDCl ): δ (ppm) l. 48 (s, 9H) , 1. 86— 2. 04 (m, 3H) , 3. 26— 3
3
. 56 (m, 4H) , 4. 45 (m, 1H)。
[0149] (実施例 2) N— tert ブトキシカルボ二ルー(R)— 3 べンジルォキシピロリジン
N— tert—ブトキシカルボ二ルー(R)—3 ヒドロキシピロリジン 180gをトルエン 72 5gに溶解し、臭化テトラ— n—プチルアンモ -ゥム 31gと触媒量のヨウ化カリウム 16g を添カ卩した。更に 10%水酸ィ匕ナトリウム水溶液 1924gをカ卩え、 50°Cまで加温した後 、ベンジルクロライド 158gを滴下した。反応終了後、 25°Cまで冷却し、有機層を水洗 した。得られた有機層を濃縮し、 N tert ブトキシカルボ-ル—(R)—3—ベンジ ルォキシピロリジン 244gを取得した(収率 92%)。 N— tert ブトキシカルボ-ルー( R)—3—ベンジルォキシピロリジンに対する N tert ブトキシカルボ-ル—(R) - 3 ヒドロキシピロリジン残存量は 1. lwt%であった。
NMR(CDCl ): δ (ppm) l. 46 (s, 9H) , 1. 87— 2. 11 (m, 2H) , 3. 42— 3
3
. 50 (m, 4H) , 4. 13 (m, 1H) , 4. 53 (s, 2H) , 7. 26— 7. 39 (m, 5H)。
[0150] (実施例 3) N— tert ブトキシカルボ二ルー(R)— 3 べンジルォキシピロリジン
別途取得した N— tert—ブトキシカルボ-ルー(R)—3—ヒドロキシピロリジン 74gを トルエン 296g〖こ溶解し、臭化テトラ一 n—ブチルアンモ -ゥム 6. 4gを添加した。更に
30%水酸ィ匕ナトリウム水溶液 264gをカ卩え、 72°Cまで加温した後、ベンジルクロライド 65gを滴下した。 N—tert—ブトキシカルボ-ルー(R)—3—ベンジルォキシピロリジ ンに対し、残存する N— tert—ブトキシカルボ-ルー(R)—3—ヒドロキシピロリジンが 1. 3wt%になった段階で、反応温度を 25°Cまで冷却し、反応を停止した。得られた 有機層を水洗、濃縮し、 N— tert—ブトキシカルボ-ル—(R)—3—ベンジルォキシ ピロリジン 83gを取得した(収率 97%)。
— NMR (CDCl ): δ (ppm) l . 46 (s, 9H) , 1. 87— 2. 11 (m, 2H) , 3. 42— 3
3
. 50 (m, 4H) , 4. 13 (m, 1H) , 4. 53 (s, 2H) , 7. 26— 7. 39 (m, 5H)。
[0151] (実施例 4) (R)—3—ベンジルォキシピロリジン塩酸塩
別途取得した N— tert—ブトキシカルボ-ルー(R)—3—ベンジルォキシピロリジン lO lgをイソプロパノール 149gに溶解し、 40°Cまでカ卩温した後、イソプロパノール性 塩酸 138gを 6時間かけて滴下した。反応終了後、 23°Cまで冷却し、濃縮して溶剤を 留去した。さらに酢酸ェチル 1400gを加えて濃縮し、約 300gの濃縮物を得た。 40°C に加温し、油状物として分離している (R)—3—ベンジルォキシピロリジン塩酸塩が 全溶するまでイソプロパノールを添加した。徐々に冷却しつつ、適宜種晶を添加する と (R)—3—ベンジルォキシピロリジン塩酸塩が結晶として析出したので、更にへキサ ン 195g添カ卩した。この時の晶析溶剤比は、イソプロパノール/酢酸ェチル /へキサ ン = 1Z71. 5/71. 5であった。析出した結晶を減圧濾過し、得られた湿結晶を酢 酸ェチル 389gで洗浄、次いで湿結晶を減圧乾燥して、(R)—3—ベンジルォキシピ 口リジン塩酸塩の乾燥晶 64gを取得した (収率 82%、純度 99. 8%)。尚、(R)— 3— ヒドロキシピロリジン塩酸塩は、この晶析操作により、 0. 92wt%から 0. 42wt%まで 低減した。また、ベンジルアルコールも晶析操作により、 1. 9wt%から 0. 01 %ま で低減した。
NMR (D O) : δ (ppm) 2. 12— 2. 30 (m, 2H) , 3. 34— 3. 54 (m, 4H) , 4.
2
48 (m, 1H) , 4. 60 (s, 2H) , 7. 40— 7. 48 (m, 5H)。融点: 70. 4〜71. 5。C。
[0152] (実施例 5) (R)—3—ベンジルォキシピロリジン塩酸塩
実施例 2にて取得した N—tert—ブトキシカルボ-ル—(R)—3—ベンジルォキシ ピロリジン 242gをイソプロパノール 360gに溶解し、 40°Cまで加温した後、イソプロパ
ノール性塩酸 254gを 6時間かけて滴下した。反応終了後、 25°Cまで冷却し、濃縮し て溶剤を留去した。さらに酢酸ェチル 3600gを加えて濃縮し、約 900gの濃縮物を得 た。 40°Cに加温し、油状物として分離している(R)—3 ベンジルォキシピロリジン塩 酸塩が全溶するまでイソプロパノールを添加した。徐々に冷却しつつ、適宜種晶を添 加すると (R)—3—ベンジルォキシピロリジン塩酸塩が結晶として析出したので、更に へキサン 93gを添カ卩した。この時の晶析溶剤比は、イソプロパノール Z酢酸ェチル Z へキサン =1Z25Z6. 3であった。析出した結晶を減圧濾過し、得られた湿結晶を 酢酸ェチル 558gで洗浄、次いで湿結晶を減圧乾燥して、(R)— 3 べンジルォキシ ピロリジン塩酸塩の乾燥晶 146gを取得した (収率 78%、純度 99. 2%) 0尚、(R)— 3 ヒドロキシピロリジン塩酸塩は、この晶析操作により、 1. 2wt%力 0. 3wt%まで 低減した。また、ベンジルアルコールも晶析操作により、 2. 9wt%から 0. 05 %ま で低減した。
— NMR(D O) : δ (ppm) 2. 12— 2. 30 (m, 2H) , 3. 34— 3. 54 (m, 4H) , 4.
2
48 (m, 1H) , 4. 60 (s, 2H) , 7. 40— 7. 48 (m, 5H)。
[0153] (比較例 1) (R)—3 ベンジルォキシピロリジン塩酸塩
別途取得した (R)— 3 べンジルォキシピロリジン塩酸塩 6. 6gを含有する 1, 4 ジォキサン溶液 10gを 40°Cに加温した後、トルエンを 170g添カ卩したが、(R)— 3— ベンジルォキシピロリジン塩酸塩は油状ィ匕した。
[0154] (実施例 6〜8)
別途取得した (R)—3—ベンジルォキシピロリジン塩酸塩の粗製物(純度 98. 4%) を用い、上記実施例 4、 5以外の晶析条件についても検討を行った。操作及び結果( 表 1)を示す。
[0155] [実験操作]
(R)— 3 ベンジルォキシピロリジン塩酸塩の粗製物 1. Og ( (R)— 3 ヒドロキシピ 口リジン塩酸塩(含量 0. 13wt%)及びべンジルアルコール(含量 1. Owt%)が不純 物として混入)を各種溶剤 4. Og中に分散し、 40°Cに加温した後、全溶するまでイソ プロパノールを添カ卩した。 24°Cまで徐々に冷却し、必要に応じて種晶を添カ卩して結 晶を析出させた。析出した結晶を減圧濾過し、得られた湿結晶を酢酸ェチル 10mlで
洗浄した。次いで湿結晶を減圧乾燥し、(R)— 3—ベンジルォキシピロリジン塩酸塩 を取得した。
[0156] [表 1]

[0157] (実施例 9〜14)
別途取得した (R)—3—ベンジルォキシピロリジン塩酸塩の粗製物(純度 98.4%) を用い、上記実施例 4〜8以外の晶析条件についても検討を行った。操作及び結果
(表 2)を示す。
[0158] [実験操作]
(R)— 3 ベンジルォキシピロリジン塩酸塩の粗製物 1. Og ( (R)— 3 ヒドロキシピ 口リジン塩酸塩(含量 0.13wt%)及びべンジルアルコール(含量 1.0wt%)が不純 物として混入)に、 40°Cで全溶する量の各種溶剤を添加した。 22〜24°Cまで (ァセト ンの場合は 3°Cまで)徐々に冷却し、必要に応じて種晶を添加して結晶を析出させた 。析出した結晶を減圧濾過し、得られた湿結晶を減圧乾燥して、 (R) 3—べンジル ォキシピロリジン塩酸塩の乾燥晶を取得した。
[0159] [表 2] 実 溶剤 溶剤添加 収率 純度 不純物含量 (w t %)
施 量 (g) (%) (%) (R) 3—ヒ ドロ ぺンンノレ 例 キシピロリジン塩 ァノレコーノレ 酸塩
9 テトラヒ ド 4. 0 20 99. 9 0. 0 1以下 0. 0 1以下 口フラン
1 0 クロ αベン 4 - 0 72 99. 8 0. 0 1以下 0. 0 1以下 ゼン
1 1 ジク Π Ρェ 0. 44 5 1 99. 9 0. 0 1以下 0. 0 1以下 タン
1 2 ァセトュト 0. 1 3 56 1 00. 0 0. 0 1以下 0. 0 1以下 リル
1 3 イソプロパ 0. 22 45 1 00. 0 0. 0 1以下 0. 0 1以 ノール
1 4 アセ トン 0. 1 7 70 99. 9 0. 0 1以下 0. 0 1以下
[0160] (実施例 15) (R)—3—ベンジルォキシピロリジン
別途取得した (R)— 3—ベンジルォキシピロリジン塩酸塩 39gに酢酸ェチル 156gと 水 100gを添加してしばらく攪拌後、有機層を分離した。得られた水層に酢酸ェチル 156gを添加し、氷冷後、 30%水酸ィ匕ナトリウム水溶液 24gを滴下した。しばらく撹拌 後、水層を分離した後、次に、有機層を水 20gで洗浄した。得られた有機層を濃縮、 減圧乾燥を行い、(R)— 3—ベンジルォキシピロリジンの濃縮物 33g (純度 86. 6%) を取得した。尚、(R)— 3—ベンジルォキシピロリジンに対する(R)— 3—ヒドロキシピ 口リジンの量は、この転溶操作により、 0. 84wt%力 0. 12wt%まで低減した。
— NMR(CDCl ): δ (ppm) l. 85— 1. 92 (m, 3H) , 2. 80— 2. 85 (m, 2H) ,
3
3. 06- 3. 13 (m, 2H) , 4. 10 (m, 1H) , 4. 48 (s, 2H) , 7. 24— 7. 34 (m, 5H)
[0161] (実施例 16) (R)—3—ベンジルォキシピロリジン阜化水素酸塩
実施例 15で取得した (R)— 3—ベンジルォキシピロリジンを含む濃縮物 1. Og (純 度 86. 6%、(R)— 3—ヒドロキシピロリジン塩酸塩(含量 0. 12wt%)とべンジルアル コール (含量 1. 9wt%)が不純物として混入)をイソプロパノール 9mlに溶解し、氷冷 下で 48%臭化水素酸 0. 82gを添加した。しばらく撹拌した後、濃縮、減圧乾燥を行 い、濃縮物を得た。この濃縮物を酢酸ェチル 4. Og中に分散し、 40°Cに加温して全 溶させ、 22°Cまで徐々に冷却した。析出した結晶を減圧濾過し、得られた湿結晶を 減圧乾燥して、(R)— 3—ベンジルォキシピロリジン臭化水素酸塩の乾燥晶 0. 74g を取得した (収率 59%、純度 100. 0%)。尚、結晶中の (R)— 3—ヒドロキシピロリジ ン塩酸塩及びべンジルアルコールは共に 0. 01 wt %以下であつた。
NMR(D O) : δ (ppm) 2. 10— 2. 32 (m, 2H) , 3. 34— 3. 54 (m, 4H) , 4.
2
49 (m, 1H) , 4. 61 (s, 2H) , 7. 41— 7. 48 (m, 5H)。
[0162] (実施例 17) (R)—3—ベンジルォキシピロリジン r>—トルエンスルホン酸塩
実施例 15で取得した (R)— 3—ベンジルォキシピロリジンを含む濃縮物 1. 0g (純 度 86. 6%、(R)— 3—ヒドロキシピロリジン塩酸塩(含量 0. 12wt%)とべンジルアル コール (含量 1. 9wt%)が不純物として混入)をイソプロパノール 9mlに溶解し、氷冷 下で P—トルエンスルホン酸一水和物 0. 93gを添加した。しばらく撹拌した後、濃縮、
減圧乾燥を行い、白色結晶を得た。この粗製物を酢酸ェチル 4. OOg中に分散させ、 40°Cに加温し、イソプロパノール 621mgを添カ卩して全溶させた。 22°Cまで徐々に冷 却し、析出した結晶を減圧濾過した。得られた湿結晶を酢酸ェチル 10mlで洗浄し、 次 ヽで湿結晶を減圧乾燥して、(R)— 3—ベンジルォキシピロリジン p—トルエンスル ホン酸塩の乾燥晶 1. 15gを取得した (収率 68%、純度 100. 0%)。尚、結晶中の (R ) ー3—ヒドロキシピロリジン塩酸塩及びべンジルアルコールは共に 0. 01wt%以下 であった。
— NMR(D O) : δ (ppm) 2. 06— 2. 30 (m, 2H) , 2. 39 (s, 3H) , 3. 32— 3.
2
51 (m, 4H) , 4. 46 (m, 1H) , 4. 57 (s, 2H) , 7. 36 (d, J = 7. 8Hz, 2H) , 7. 40 - 7. 47 (m, 5H) , 7. 69 (d, J = 8. 3Hz, 2H)。
[0163] (実施例 18) (R)—3—ベンジルォキシピロリジン硫酸塩
実施例 15で取得した (R)— 3—ベンジルォキシピロリジンを含む濃縮物 1. 0g (純 度 86. 6%)、(R)— 3—ヒドロキシピロリジン(含量 0. 12wt%)とべンジルアルコール (含量 1. 9wt%)が不純物として混入)をイソプロパノール 9mlに溶解し、氷冷下で硫 酸 240mgを添加した。しばらく撹拌した後、濃縮、減圧乾燥を行い、濃縮物を得た。 この濃縮物を酢酸ェチル 4. 0g中に分散させ、 40°Cに加温し、イソプロパノール 209 mgを添カ卩して全溶させた。 22°Cまで徐々に冷却した後、へキサン 207mgを添カロし、 更に種晶を添加し、析出した結晶を減圧濾過した。得られた湿結晶を減圧乾燥して 、 (R)—3—ベンジルォキシピロリジン硫酸塩の乾燥晶 75mgを取得した (収率 7%、 純度 99. 9%)。尚、結晶中の(R)— 3—ヒドロキシピロリジン塩酸塩及びベンジルァ ノレコーノレは共〖こ 0. 01wt%以下であった。
NMR(D O) : δ (ppm) 2. 10— 2. 32 (m, 2H) , 3. 34— 3. 54 (m, 4H) , 4.
2
49 (m, 1H) , 4. 61 (s, 2H) , 7. 39— 7. 48 (m, 5H)。
[0164] (実施例 19) (R)—3—ベンジルォキシピロリジン安息香酸塩
実施例 15で取得した (R)— 3—ベンジルォキシピロリジンを含む濃縮物 1. 0g ( (純 度 86. 6%)、(R)— 3—ヒドロキシピロリジン塩酸塩 (含量 0. 12wt%)とベンジルァ ルコール (含量 1. 9wt%)が不純物として混入)をイソプロパノール 9mlに溶解し、安 息香酸 0. 60gを添加した。しばらく撹拌した後、濃縮、減圧乾燥を行い、白色結晶を
得た。この粗製物を酢酸ェチル 4. Og中に分散させ、 40°Cに加温し、イソプロノ V— ル 1. 19gを添加して全溶させた。 22°Cまで徐々に冷却し、析出した結晶を減圧濾過 した。得られた湿結晶を酢酸ェチル 10mlで洗浄し、次いで湿結晶を減圧乾燥して、 (R)—3—ベンジルォキシピロリジン安息香酸塩の乾燥晶 0. 69gを取得した(収率 4 7%、純度 100. 0%)。尚、結晶中の (R)— 3 ヒドロキシピロリジン塩酸塩及びベン ジルアルコールは共に 0. 0 lwt%以下であつた。
— NMR(D O) : δ (ppm) 2. 08— 2. 32 (m, 2H) , 3. 33— 3. 52 (m, 4H) , 4.
2
47 (m, 1H) , 4. 58 (s, 2H) , 7. 41— 7. 55 (m, 8H) , 7. 87 (d, J = 7. 8Hz, 2H
) o
[0165] (実施例 20) (R)—3—ベンジルォキシピロリジン—(L)—酒石酸塩
別途取得した (R)—3 ベンジルォキシピロリジンを含む濃縮物 0. 25g (純度 86. 6%)、(R)— 3 ヒドロキシピロリジン塩酸塩(含量 0. 12wt%)とべンジルアルコール (含量 1. 0wt%)が不純物として混入、光学純度 97. 0%ee)をイソプロパノール 40 mlに溶解し、 (L)—酒石酸 0. 18gを添加した。しばらく撹拌した後、濃縮、減圧乾燥 を行い、白色結晶を得た。この粗製物をイソプロパノール 4. Og中に分散させ、 40°C に加温し、蒸留水 400mgを添加して全溶させた。 22°Cまで徐々に冷却し、析出した 結晶を減圧濾過した。得られた湿結晶を酢酸ェチル 10mlで洗浄し、次いで湿結晶 を減圧乾燥して、(R)—3—べンジルォキシピロリジン一(L) 酒石酸塩の乾燥晶 0. 16gを取得した (収率 52%、純度 100. 0%、光学純度 98. 2%ee) 0尚、結晶中の( R)—3 ヒドロキシピロリジン塩酸塩及びべンジルアルコールは共に 0. 01wt%以下 であった。
NMR(D O) : δ (ppm) 2. 08— 2. 32 (m, 2H) , 3. 32— 3. 54 (m, 4H) , 4.
2
31 (d, J = 3. 2Hz, 1H) , 4. 49 (m, 1H) , 4. 61 (s, 2H) , 7. 40— 7. 48 (m, 5H
) o
[0166] (実施例 21) (R)—3—ベンジルォキシピロリジン—(D)—酒石酸塩
別途取得した (R)—3 ベンジルォキシピロリジンを含む濃縮物 0. 25g (純度 86. 6%)、(R)— 3 ヒドロキシピロリジン塩酸塩(含量 0. 12wt%)とべンジルアルコール (含量 1. 0wt%)が不純物として混入、光学純度 97. 0%ee)をイソプロパノール 40
mlに溶解し、 (D)—酒石酸 183mgを添加した。しばらく撹拌した後、濃縮、減圧乾 燥を行い、白色結晶を得た。この粗製物をイソプロパノール 4. Og中に分散させ、 40 °Cに加温し、蒸留水 400mgを添加して全溶させた。 22°Cまで徐々に冷却し、析出し た結晶を減圧濾過した。得られた湿結晶を酢酸ェチル 10mlで洗浄し、次いで湿結 晶を減圧乾燥して、 (R)—3—ベンジルォキシピロリジン—(D)—酒石酸塩の乾燥晶 93mgを取得した (収率 30%、純度 100. 0%、光学純度 97. 5%ee) 0尚、結晶中の (R)—3—ヒドロキシピロリジン及びべンジルアルコールは共に 0. 01wt%以下であ つた o
— NMR(D O) : δ (ppm) 2. 08— 2. 32 (m, 2H) , 3. 34— 3. 54 (m, 4H) , 4.
2
32 (d, J = 3. 2Hz, 1H) , 4. 49 (m, 1H) , 4. 61 (s, 2H) , 7. 40— 7. 48 (m, 5H
) o
[0167] (実施例 22) (R)—3—ベンジルォキシピロリジン一(L)—ァスパラギン酸塩
別途取得した (R)—3—ベンジルォキシピロリジンを含む濃縮物 0. 25g (純度 86. 6%)、 (R)— 3—ヒドロキシピロリジン塩酸塩(含量 0. 12wt%)とべンジルアルコール (含量 1. 0wt%)が不純物として混入、光学純度 97. 0%ee)を蒸留水 10mlに溶解 し、(L)—ァスパラギン酸 0. 33gを添加した。しばらく撹拌した後、イソプロノ V—ルを 添加して共沸脱水し、濃縮、減圧乾燥を行い、白色結晶を得た。この粗製物をイソプ ロパノール 4. Og中に分散させ、 40°Cに加温し、蒸留水 506mgを添カ卩して全溶させ た。 22°Cまで徐々に冷却し、析出した結晶を減圧濾過した。得られた湿結晶を酢酸 ェチル 10mlで洗浄し、次いで湿結晶を減圧乾燥して、(R)—3—ベンジルォキシピ 口リジン一(L)ーァスパラギン酸塩の乾燥晶 0. 37gを取得した (収率 49%、純度 100 . 0%、光学純度 99. 4%ee) 0尚、結晶中の (R)—3—ヒドロキシピロリジン塩酸塩及 びべンジルアルコールは共に 0. 01 %以下であった。
NMR(D O) : δ (ppm) 2. 08— 2. 34 (m, 2H) , 2. 67 (dd, J = 8. 9Hz, 17.
2
4Hz, 1H) , 2. 82 (dd, J = 3. 7Hz, 17. 4Hz, 1H) , 3. 34— 3. 54 (m, 4H) , 3. 89 (dd, J = 3. 7Hz, 8. 9Hz, 1H) , 4. 49 (m, 1H) , 4. 61 (s, 2H) , 7. 40— 7. 4 8 (m, 5H)。
[0168] (実施例 23) (R)—3—ベンジルォキシピロリジン一(D)—ァスパラギン酸塩
別途取得した (R)—3—ベンジルォキシピロリジンを含む濃縮物 0. 25g (純度 86. 6%)、 (R)— 3—ヒドロキシピロリジン塩酸塩(含量 0. 12wt%)とべンジルアルコール (含量 1. Owt%)が不純物として混入、光学純度 97. 0%ee)を蒸留水 10mlに溶解 し、(D)—ァスパラギン酸 325mgを添加した。しばらく撹拌した後、イソプロパノール を添加して共沸脱水し、濃縮、減圧乾燥を行い、白色結晶を得た。この粗製物をイソ プロパノール 4. 00g中〖こ分散させ、 40°Cに加温し、蒸留水 466mgを添カ卩して全溶 させた。 22°Cまで徐々に冷却し、析出した結晶を減圧濾過した。得られた湿結晶を 酢酸ェチル 10mlで洗浄し、次いで湿結晶を減圧乾燥して、(R)—3—ベンジルォキ シピロリジン—(D)—ァスパラギン酸塩の乾燥晶 0. 17gを取得した (収率 22%、純度 98. 7%、光学純度 99. 8%ee)。尚、結晶中の(R)—3—ヒドロキシピロリジン塩酸塩 及びべンジルアルコールは共に 0. 01 %以下であった。
— NMR(D O) : δ (ppm) 2. 08— 2. 34 (m, 2H) , 2. 67 (dd, J = 8. 9Hz, 17.
2
4Hz, 1H) , 2. 82 (dd, J = 3. 7Hz, 17. 4Hz, 1H) , 3. 34— 3. 54 (m, 4H) , 3. 89 (dd, J = 3. 7Hz, 8. 9Hz, 1H) , 4. 49 (m, 1H) , 4. 61 (s, 2H) , 7. 40— 7. 4 8 (m, 5H)。
[0169] (実施例 24)
別途取得した (R)— 3—ベンジルォキシピロリジン塩酸塩を用いて吸湿測定を行つ た。操作及び結果 (表 3)を示す。相対湿度はいす 製作所製温湿計を用いて測定し た。
[0170] [実験操作]
(R)— 3—べンジルォキシピロリジン塩酸塩(純度 99. 2%,水分量 0. 25wt%)を 容器に入れ、湿度一定 (相対湿度 22%、 33%、 80% (25°C) )になっている容器内 に開放状態で静置した。静置後の (R)—3—ベンジルォキシピロリジン塩酸塩の水分 量をカールフィッシャー水分計で測定した。
尚、各湿度の環境は、 25°C下に各溶液をデシケータ底部に置くことにより作った。 絶対湿度 5gZm3:飽和酢酸カリウム水溶液 (相対湿度 22%、 25°C)
絶対湿度 8g/m3:飽和塩化マグネシウム水溶液 (相対湿度 33%、 25°C) 絶対湿度 18gZm3:飽和塩化ナトリウム水溶液 (相対湿度 80%、 25°C)
[0171] [表 3]
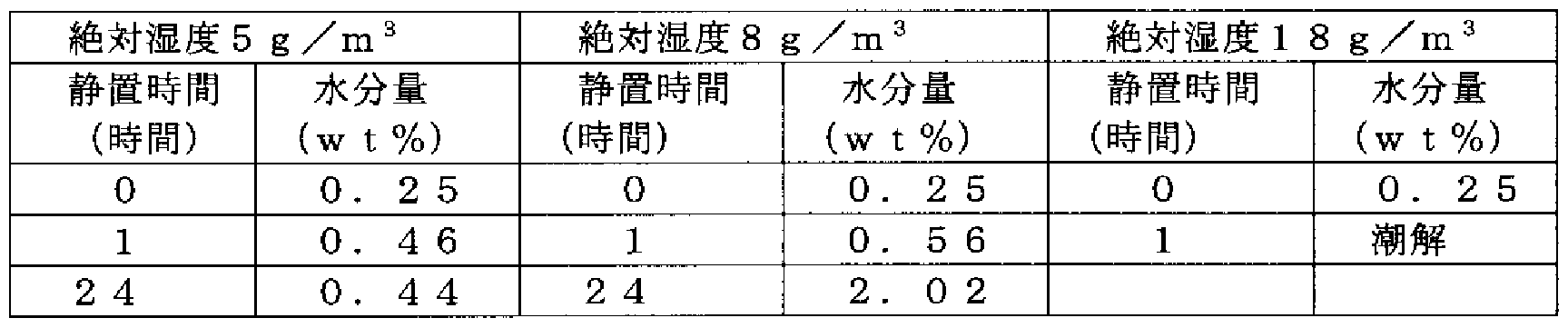
[0172] (実施例 25) N—tert—ブトキシカルボ二ルー(R)— 3—べンジルォキシピロリジン
別途調製した N— tert—ブトキシカルボ-ル—(R)—3—ヒドロキシピロリジン 187 mgをトルエン 748mgに溶解し、臭化テトラ— n—ブチルアンモ -ゥム 16mgとヨウ化 ナトリウム 9mgを添カ卩した。更に 30%水酸化ナトリウム水溶液 666mgをカ卩え、 50°Cま で加温した後、ベンジルクロライド 164mgを滴下した。 N—tert—ブトキシカルボ-ル - (R)—3—ベンジルォキシピロリジンに対し、残存する N— tert—ブトキシカルボ- ルー(R)—3—ヒドロキシピロリジンが 6. 4wt%になった段階で、反応温度を 25°Cま で冷却し、反応を停止した。
[0173] (実施例 26) N—tert—ブトキシカルボ二ルー(R)— 3—べンジルォキシピロリジン
別途取得した N—tert—ブトキシカルボ-ルー(R)—3—ヒドロキシピロリジン(20g )のトルエン約 60%溶液にジメチルスルホキシド(50g)をカ卩え、そこにヨウ化カリウム( 5. 32g)と 85%水酸ィ匕カリウムの固体(14. lg)を添加した。このスラリー溶液を 70°C まで加温した後、ベンジルクロライド(17. 58g)を 5時間かけて滴下した。引き続き、 0 °Cで 16時間攪拌したところ、変換率(* )は 98%に到達した。反応終了後、 20°Cま で冷却し、トルエン、及び 30%チォ硫酸ナトリウム水溶液をカ卩え、水層を除去した後 、有機層を水洗した。得られた有機層を減圧濃縮することにより、 N— tert—ブトキシ カルボニル一 (R)—3—ベンジルォキシピロリジンのトルエン溶液を取得した(収率: 98%) o
( * )変換率(。/。) = (N— tert—ブトキシカルボ-ル—(R)—3—ベンジロキシピロリジ ンの生成量(g) ) Z[ (N— tert—ブトキシカルボ-ルー(R)—3—ベンジロキシピロリ ジンの生成量(g) ) + (N— tert—ブトキシカルボ-ル—(R)—3—ヒドロキシピロリジ ンの残存量 (g) )]
[0174] (実施例 27〜28)
実施例 26の溶媒であるトルエンとジメチルスルホキシドを下記で表される単一溶媒 に置き換えて、反応温度及び反応時間を変更した以外は同様な反応操作を実施し た。結果を表 4に示す。
[表 4]

Claims
[化 1]

(式中、 Rは置換基を有していても良い炭素数 7〜 15のァラルキル基を表し、 HXは 鉱酸、スルホン酸、カルボン酸またはアミノ酸を表す)で表される 3—ァラルキルォキ シピロリジン誘導体を、極性有機溶剤を含有する溶剤を用いた晶析工程に付し、結 晶として取得することを特徴とする 3—ァラルキルォキシピロリジン誘導体(2)の製造 法。
[2] 極性溶剤がエステル系溶剤及び Z又はアルコール系溶剤である請求項 1に記載の
[3] 種晶を添加して結晶を析出させる請求項 1または 2に記載の製造法。
[4] 下記一般式 (2) ;
[化 2]

(式中、 Rは置換基を有していても良い炭素数 7〜 15のァラルキル基を表し、 HXは 鉱酸、スルホン酸、カルボン酸またはアミノ酸を表す)で表される 3—ァラルキルォキ シピロリジン誘導体を含有する水と有機溶剤の 2相系混合物から、
A)有機層を除去した後、水層を塩基で処理し、
B) 3—ァラルキルォキシピロリジン誘導体(2)の遊離のアミンを有機溶剤で抽出し、
C)該遊離のアミンを酸で処理して 3—ァラルキルォキシピロリジン誘導体(2)に変換
する、
一連の操作により化学純度を向上させることを特徴とする 3—ァラルキルォキシピロリ ジン誘導体 (2)の製造法。
[5] 請求項 4記載の方法により得られた 3—ァラルキルォキシピロリジン誘導体(2)を使用 する請求項 1〜3のいずれかに記載の製造法。
[6] 3—ァラルキルォキシピロリジン誘導体(2)が一般式(1);
[化 3]

(式中、 Rは置換基を有していても良い炭素数 7〜 15のァラルキル基を表す。 Pは、 ァミノ基の保護基を表す。)

の N—保護基を脱離させることにより得られたものである請求項 1〜5のいずれかに 記載の製造法。
N—保護— 3—ァラルキルォキシピロリジン(1)は、一般式(5);
[化 4]

(式中、 Pは前記に同じ。)で表される N—保護一 3—ヒドロキシピロリジンの水酸基を O—ァラルキルィ匕することにより得られたものである請求項 6に記載の製造法。
[8] 3—ァラルキルォキシピロリジン誘導体(2)が光学活性体である請求項 1〜7のいず れかに記載の製造法。
[9] 一般式 (5) ;

(式中、 Pは、ァミノ基の保護基を表す。)で表される N—保護— 3—ヒドロキシピロリジ ンを塩基、並びに、金属ハロゲンィ匕物及び Zまたは相関移動触媒存在下に、ハロゲ ン化ァラルキルを作用させて O—ァラルキルィ匕することを特徴とする一般式(1) ; [化 6]

(式中、 Rは置換基を有していても良い炭素数 7〜 15のァラルキル基を表す。 Pは、 前記に同じ。)で表される N—保護— 3—ァラルキルォキシピロリジンの製造法。
[10] 塩基及び金属ハロゲン化物存在下、ハロゲン化ァラルキルを作用させて O—ァラル キル化することを特徴とする請求項 9記載の製造法。
[11] N—保護— 3—ァラルキルォキシピロリジン(1)は請求項 9または 10に記載の製造法 で得られたものである請求項 6に記載の製造法。
[12] 前記式(1)及び(5)で表されるピロリジン誘導体が光学活性体である請求項 6〜11 の!、ずれかに記載の製造法。
[13] 下記一般式 (4) ;
[化 7]

(式中、 HXは鉱酸、スルホン酸、カルボン酸またはアミノ酸を表す)で表される 3—べ ンジルォキシピロリジン誘導体の結晶。
[14] 3 ベンジルォキシピロリジン誘導体 (4)が光学活性 3 ベンジルォキシピロリジン誘 導体である請求項 13記載の結晶。
[15] 一般式 (3) ;
[化 8]

(式中、 R'は置換基を有していても良い炭素数 7〜 15のァラルキル基、 HYは、ハロ ゲン化水素)で表される 3—ァラルキルォキシピロリジンのハロゲン化水素酸塩の結 晶を、絶対湿度 12g/m3以下の環境下で取り扱うことを特徴とする 3 ァラルキルォ キシピロリジンのハロゲン化水素酸塩の取り扱い方法。
[16] 3 ァラルキルォキシピロリジンのハロゲンィ匕水素酸塩(3)の結晶を取り扱うに際し、 周囲の環境を絶対湿度 12gZm3以下に制御することを特徴とする請求項 15記載の 取り扱い方法。
[17] 3 ァラルキルォキシピロリジンのハロゲン化水素酸塩(3)の結晶を 25°C以下の環 境で取り扱うことを特徴とする請求項 15または 16に記載の取り扱い方法。
[18] 3 ァラルキルォキシピロリジンのハロゲンィ匕水素酸塩(3)の結晶を取り扱うに際し、 周辺の環境を 25°C以下に制御することを特徴とする請求項 15〜17のいずれかに記
載の取り扱い方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06796649A EP1947083B1 (en) | 2005-08-23 | 2006-08-22 | Process for production of aralkyloxypyrrolidine derivative |
| JP2007532139A JP4167297B2 (ja) | 2005-08-23 | 2006-08-22 | 3−アラルキルオキシピロリジン誘導体の製造法 |
| US11/990,766 US7973176B2 (en) | 2005-08-23 | 2006-08-22 | Process for production of aralkyloxypyrrolidine derivative |
| DE602006018182T DE602006018182D1 (de) | 2005-08-23 | 2006-08-22 | Verfahren zur herstellung von aralkyloxypyrrolidinderivaten |
| US13/115,385 US8247578B2 (en) | 2005-08-23 | 2011-05-25 | Process for production of aralkyloxypyrrolidine derivative |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005-241742 | 2005-08-23 | ||
| JP2005241742 | 2005-08-23 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/990,766 A-371-Of-International US7973176B2 (en) | 2005-08-23 | 2006-08-22 | Process for production of aralkyloxypyrrolidine derivative |
| US13/115,385 Division US8247578B2 (en) | 2005-08-23 | 2011-05-25 | Process for production of aralkyloxypyrrolidine derivative |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007023824A1 true WO2007023824A1 (ja) | 2007-03-01 |
Family
ID=37771567
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2006/316435 WO2007023824A1 (ja) | 2005-08-23 | 2006-08-22 | 3-アラルキルオキシピロリジン誘導体の製造法 |
Country Status (5)
| Country | Link |
|---|---|
| US (2) | US7973176B2 (ja) |
| EP (1) | EP1947083B1 (ja) |
| JP (2) | JP4167297B2 (ja) |
| DE (1) | DE602006018182D1 (ja) |
| WO (1) | WO2007023824A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009122997A1 (ja) * | 2008-04-02 | 2009-10-08 | 株式会社カネカ | (s)-3-(1-シアノ-1,1-ジフェニルメチル)-ピロリジンの製造法 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102633704A (zh) * | 2012-03-28 | 2012-08-15 | 扬州天和药业有限公司 | R-4-苄氧吡咯烷的一种制备方法 |
| CN104610122A (zh) * | 2015-02-06 | 2015-05-13 | 中国药科大学 | 反-2-[(3r)-3-苄氧基-1-吡咯烷基]-1-环己醇的制备方法 |
| CA3079834C (en) * | 2016-07-13 | 2022-07-26 | Revelant IP Holdings LLC | Band-pass filter |
| WO2018022873A1 (en) | 2016-07-27 | 2018-02-01 | Revelant | Device and methods for increasing the solubility of crystals in water |
| CN108203517B (zh) * | 2017-06-26 | 2021-03-30 | 哈尔滨工业大学(威海) | 一种芳香聚合物材料表面接枝阳离子抑菌涂层的制备方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01311059A (ja) * | 1988-04-19 | 1989-12-15 | Bayer Ag | 1,3―二置換されたピロリジン |
| JPH10503768A (ja) * | 1994-08-02 | 1998-04-07 | メルク シヤープ エンド ドーム リミテツド | アゼチジン、ピロリジンおよびピペリジン誘導体 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS60178837A (ja) * | 1984-02-24 | 1985-09-12 | Yamanouchi Pharmaceut Co Ltd | カテコ−ル誘導体 |
| US4618627A (en) | 1983-05-13 | 1986-10-21 | Yamanouchi Pharmaceutical Co., Ltd. | Catechol derivatives and pharmaceutical compositions thereof for inhibiting anaphylaxis (SRS-A) |
| JPS61243081A (ja) * | 1985-04-19 | 1986-10-29 | Dainippon Pharmaceut Co Ltd | ピリドンカルボン酸誘導体、そのエステルおよびその塩 |
| AU578793B2 (en) | 1985-02-15 | 1988-11-03 | Dainippon Pharmaceutical Co. Ltd. | Novel 1,8-naphthyridine derivatives and processes for preparation thereof |
| AR006793A1 (es) | 1996-04-26 | 1999-09-29 | Ishihara Sangyo Kaisha | Compuestos pirazol o sus sales y herbicidas conteniendo los mismos |
| JPH10109976A (ja) * | 1996-04-26 | 1998-04-28 | Ishihara Sangyo Kaisha Ltd | ピラゾール系化合物、それらの製造方法及びそれらを含有する除草剤 |
| JP4481659B2 (ja) | 2002-03-29 | 2010-06-16 | 株式会社カネカ | 1−ベンジル−3−アミノピロリジンの光学純度向上方法及びそれに用いられる塩 |
| NZ543921A (en) * | 2003-05-02 | 2008-11-28 | Cardiome Pharma Corp | Aminocyclohexyl ether compounds and uses thereof |
-
2006
- 2006-08-22 US US11/990,766 patent/US7973176B2/en not_active Expired - Fee Related
- 2006-08-22 WO PCT/JP2006/316435 patent/WO2007023824A1/ja active Application Filing
- 2006-08-22 EP EP06796649A patent/EP1947083B1/en active Active
- 2006-08-22 DE DE602006018182T patent/DE602006018182D1/de active Active
- 2006-08-22 JP JP2007532139A patent/JP4167297B2/ja active Active
-
2008
- 2008-03-10 JP JP2008060163A patent/JP2008169225A/ja active Pending
-
2011
- 2011-05-25 US US13/115,385 patent/US8247578B2/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01311059A (ja) * | 1988-04-19 | 1989-12-15 | Bayer Ag | 1,3―二置換されたピロリジン |
| JPH10503768A (ja) * | 1994-08-02 | 1998-04-07 | メルク シヤープ エンド ドーム リミテツド | アゼチジン、ピロリジンおよびピペリジン誘導体 |
Non-Patent Citations (8)
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009122997A1 (ja) * | 2008-04-02 | 2009-10-08 | 株式会社カネカ | (s)-3-(1-シアノ-1,1-ジフェニルメチル)-ピロリジンの製造法 |
| JP5450387B2 (ja) * | 2008-04-02 | 2014-03-26 | 株式会社カネカ | (s)−3−(1−シアノ−1,1−ジフェニルメチル)−ピロリジンの製造法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1947083A1 (en) | 2008-07-23 |
| EP1947083A4 (en) | 2009-05-27 |
| JP4167297B2 (ja) | 2008-10-15 |
| US20080306283A1 (en) | 2008-12-11 |
| JP2008169225A (ja) | 2008-07-24 |
| DE602006018182D1 (de) | 2010-12-23 |
| EP1947083B1 (en) | 2010-11-10 |
| US7973176B2 (en) | 2011-07-05 |
| US8247578B2 (en) | 2012-08-21 |
| US20110224444A1 (en) | 2011-09-15 |
| JPWO2007023824A1 (ja) | 2009-03-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007023824A1 (ja) | 3-アラルキルオキシピロリジン誘導体の製造法 | |
| KR100706567B1 (ko) | N-카바메이트-보호된 β-아미노알콜의 제조방법 및N-카바메이트 보호된 β-아미노에폭사이드의 제조방법 | |
| JP4940790B2 (ja) | 脱ヒドロキシフッ素化剤 | |
| EA020088B1 (ru) | Способ получения производных бисфуранового спирта, соединения, соль, фармацевтическая композиция на ее основе, способ лечения или профилактики ретровирусных инфекций с ее помощью | |
| CA2919317A1 (en) | Synthesis of biphenylalaninol via novel intermediates | |
| WO2015145467A1 (en) | An improved process for preparing vildagliptin | |
| JP4149668B2 (ja) | β−ハロゲノ−α−アミノカルボン酸並びにフェニルシステイン誘導体及びその中間体の製造方法 | |
| US7528264B2 (en) | Hydride reduction process for preparing quinolone intermediates | |
| JP2008169225A5 (ja) | ||
| CA2627502C (en) | Process for production of benzyloxypyrrolidine derivative, and process for production of hydrochloride salt powder of optically active benzyloxypyrrolidine derivative | |
| WO2010071129A1 (ja) | ヒドロキシル基置換生成物の製造方法 | |
| WO2009047797A2 (en) | Process for the preparation of perhydroisoindole derivative | |
| JP5260062B2 (ja) | β−アミノ−α−ヒドロキシ酸アミド誘導体の製造法 | |
| KR100915551B1 (ko) | 3-히드록시 피롤리딘 및 이의 유도체의 효율적 제조방법 | |
| JP5023443B2 (ja) | 4−フルオロプロリン誘導体の製造方法 | |
| WO2000044706A1 (fr) | Procede relatif a l'elaboration d'alpha-aminocetones | |
| JP2004238322A (ja) | (r)−3−アミノペンタンニトリルメタンスルホン酸塩の製造方法 | |
| JP5704763B2 (ja) | トランス−4−アミノシクロペンタ−2−エン−1−カルボン酸誘導体の製造 | |
| AU776101B2 (en) | Production method of 2-cyclohexyl-2-hydroxy-2-phenylacetic acid intermediate therefor and production method thereof | |
| WO2001055074A1 (fr) | Processus de production d'acide carboxylique optiquement actif substitue en position 2 | |
| EP1300391A1 (en) | Process for producing 3-amino-2-hydroxypropionic acid derivatives | |
| WO2005000810A1 (ja) | 含窒素複素環化合物の製造方法 | |
| JP4421802B2 (ja) | クロロ炭酸エステルの製造方法 | |
| JP5981719B2 (ja) | 光学活性チアゾリルアラニン誘導体およびその塩の製造方法 | |
| Coti | Investigation on the mechanism of action of pseudoephedrine as a chiral auxiliary: synthesis and use of a novel pseudoephedrine based chiral auxiliary |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2007532139 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11990766 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006796649 Country of ref document: EP |