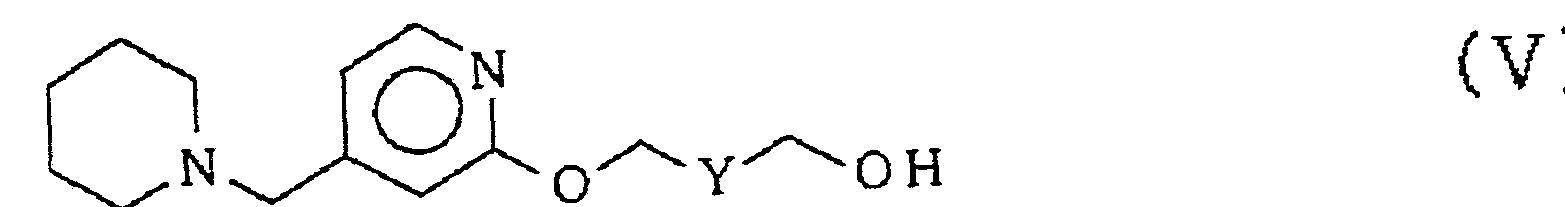明 細 フタルイ ミ ド化合物およびその製造方法
[技術分野]
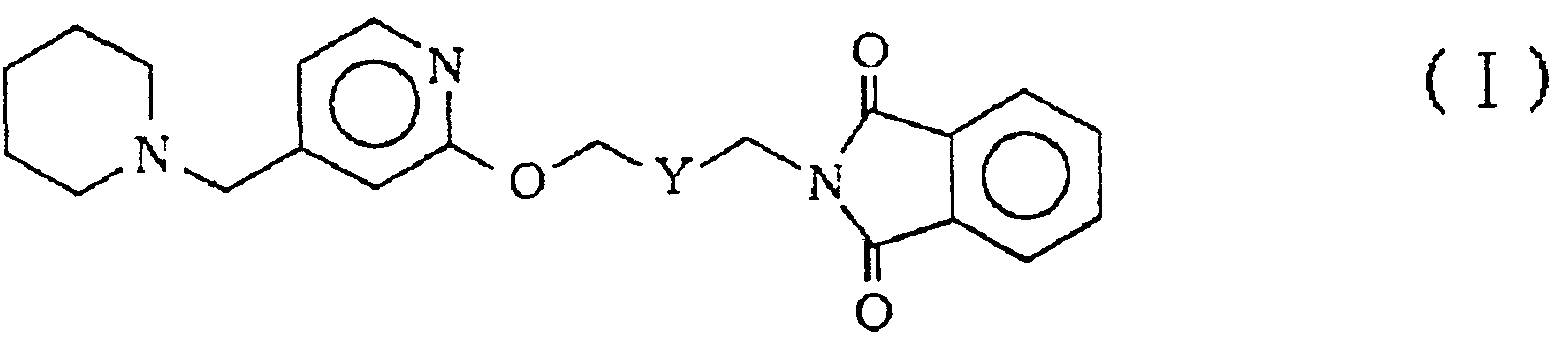
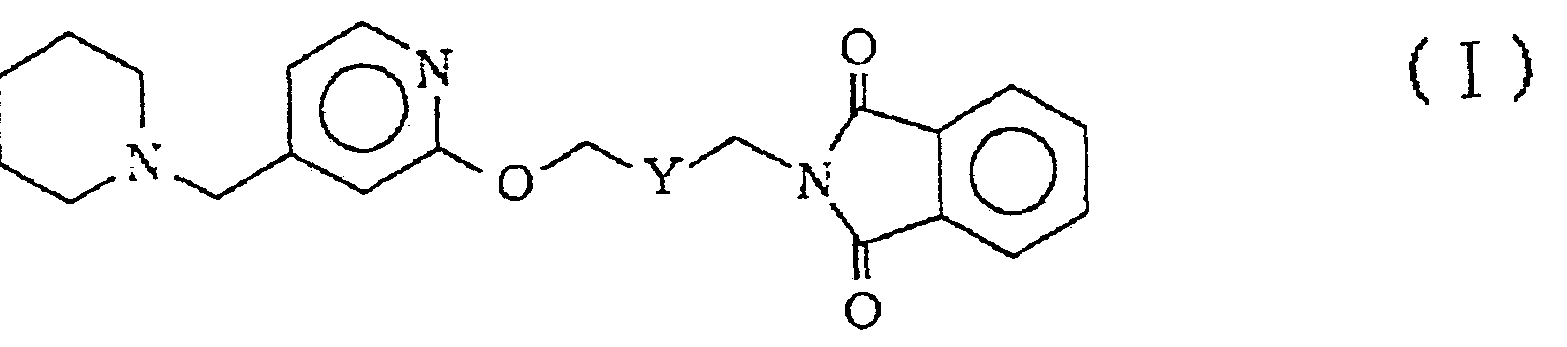
本発明は、 次式 ( I )
(式中、 Υは一 C H2— C H2—または一 C H = C H—である。 ) で示されるフタルイ ミ ド化合物およびその酸付加塩ならびにそれら の製造方法に関するものである。 上式 ( I ) で示されるフタルイ ミ ド化合物およびその酸付加塩は、 医薬品、 特にヒ スタ ミ ン H2受容 体拮抗作用に基づく抗潰瘍剤の製造中間体と して有用な化合物であ る。
[背景技術]
従来、 ヒ スタ ミ ン H2受容体拮抗作用に基づく抗消化性潰瘍剤は、 特開昭 6 1 — 8 5 3 6 5号公報に次式 (W)
(式中、 Ζは一 C Η C Η—である: = )
で示される化合物が開示され、 また、 特開昭 6' 3 — 2 2 5 3 7 1 号 公報に次式 (H)
(式中、 Yは一 C H2— C H 2—または一 C H二 C H—である。 ) で示される化合物が開示されている。 これらの化合物の共通の中間 体として次式 (IX)
(式中、 Υは一 C H2— C H2—または一 C H = C H—である。 ) で示される化合物が用いられている。 この化合物の従来の合成法は 次式 (X)
Η〇Αγ, ヽ ΝΗ2 (X I)
(式中、 Υは一 C H2— C H2—または一 C H C H—である。 ) で示される化合物の両者を反応させることによるものである。 し力、 し この合成法を用いた場合、 原料である上式 (X ) および上式
(X I ) で示される化合物の工業的入手が不可能であり、 製造する と高倚であるという問題がある。 また、 上式 (X ) および上式 ( X
I ) で示される化合物の反応による前記の式 (K) で示される化合 物の製造においては副生成物や分解物が多く、 そのため精製に負荷 がかかるなど前記の中間体 (K) への誘導が極めて煩雑であり、 ェ 業化するには問題が多い方法である。 [発明の開示]
本発明者らは、 かかる問題点に鑑み鋭意検討を行なった結果、 次 式 ( I )
(式中、 Yは一 C H2— C H2—または一 C H = C H—である。 ) で示されるフタ ルイ ミ ド化合物およびその酸付加塩が前記の中間体 (3X) の製造に極めて有用であるこ とを見出し本発明に到達した。 すなわち本発明は、 次式 ( I )
(式中、 Yは一 C H2— C H2—または一 C H C H—である。 ) 示されるフ タ ルイ ミ ド化合物およびその酸付加塩ならびに次式 ( Π )
( Π )
で示される 2—クロ口一 4 ( 1 —ビペ リ ジニルメ チル) ピ リ ジン と次式 (m)
HO八 Y^^OR (m)
(式中、 Yは一 CH2— CH2—または一 CH CH—であり、 Rは テ ト ラ ヒ ドロ ビラ二ル基、 メ トキシメチル基、 ト リ メ チルシ リ ル基 またはべンジル基である。 )
で示されるアルコール誘導体を反応させて次式 (W)
(式中、 Yは一 C H 2— C H2—または一 C H C H—であり、 Rは テ ト ラ ヒ ドロ ビラ二ル基、 メ トキシメチル基、 ト リ メ チルシ リ ル基 またはべンジル基である。 )
で示されるピリ ジルォキシ誘導体と し、 その水酸基の保護基 Rを脱 保護させて次式 (V)
(式中、 Yは一 CH2— C H2—または一 C H-CH—である。 ) で示される ピリ ジルォキシ誘導体と し、 これをハロゲン化またはス ルホニル化して次式 (VI)
(VI)
(式中、 Yは一 C H
2— C H
2—または一 C H = C H—であり、 Xは C I 一、 B r ―、 C H 3 S 0 a - .
33〇
3—または ー(: 1
3— C
6H
4— S 0
3—である。 )
で示されるピリ ジルォキシ誘導体と した後、 フタルイ ミ ドィ匕し、 さ らに必要に応じて酸と反応させることを特徴とする次式 ( I )
で示されるフタルイ ミ ド化合物およびその酸付加塩の製造方法であ る。
本発明において、 上式 ( I ) で示されるフタルイ ミ ド化合物は、 粘性黄色油状物であり、 具体的には N— ( 4 一 ( 4 - ( 1 ーピペ リ ジニルメ チル) ピ リ ジルー 2 —ォキシ) 一 c i s — 2 —ブ亍ニル) フタルイ ミ ド、 N— ( 4 - ( 4 - ( 1 ーピベ リ ジニルメ チル) ピ リ ジルー 2 —ォキシ) - t r a Ti s — 2 —ブテニル) フタルイ ミ ドぉ よび K一 ( 4 — ( 4 - ( 1 —ピベ リ ジニルメ チル) ピ リ ジル一 2 — ォキシ) ブチル) フタルイ ミ ドである。
本発明における上式 ( I ) で示されるフタルイ ミ ド化合物は、 た とえば、 以下に示す工程に従って製造することが可能である。
こ第一工程
+ H O^Y' \〇R (DO
(式中、 Yは一 C H 2— C H 2—または一 C H = C H—であり、 Rは テ ト ラ ヒ ド ビラニル基、 メ ト キシメ チル基、 ト リ ノ チルシ リ ル基 またはべンジル基である。 )
本工程は、 上式 ( II ) で示される 2 —クロ口一 4 一 ( 1 ーピペリ ジニルメチル) ピリ ジンと上式 (ΙΠ ) で示される了ルコール誘導体 を反応させ、 上式 (IV ) で示されるピリ ジルォキシ誘導体を製造す る丄 でめる。
反応は、 2 —ク ロ ロ ー 4— ( 1 ー ピベ リ ジニルメ チル) ピ リ ジン 1 モルに対してアルコール誘導体を 1 モルの割合で用い、 ナ ト リ ウ 厶、 永素化ナ ト リ ウ厶、 力 リ ウム、 氷素化力 リ ウム、 リ チウ ム、 水 素化リ チ ウ ム、 T1 ーブチル リ チウ 厶、 s e c —ブチル リ チウ ム、 1 e r t 一ブチルリチウム、 t e r t 一ブトキシカ リ ウ厶などの存在 下で行ない、 その使用量は基質に対して 1 〜 3当量であるのが望ま し 。 反応は溶媒中で行なうのが望ま しく、 テ ト ラ ヒ ドロフ ラ ン、 ェ一テル、 ジォヰサ ン、 ベンゼン、 ト ルエ ン、 ジメ チルホル厶ア ミ ド どを甩いることができる。 また、 反応温度は、 2 0〜 : ί 2 0 t で行 うのが望ま しい。
[第一工程
(式中、 Υは一 C H2— C H2—または _C H = C H—であり、 Rは テ ト ラ ヒ ド ロ ビラ ニル基、 メ ト キ シメ チル基、 ト リ メ チルシ リ ル基 またはべンジル基である。 )
本工程は、 上式 (IV) で示される ピリ ジルォキシ誘導体の水酸基 の保護基 Rを脱保護させ、 上式 (V) で示される ピリ ジルォキシ誘 導体を製造する工程である。
反応は、 塩酸、 硫酸、 酢酸、 p — ト ルエ ンスルホ ン酸、 p — ト ル エ ンスルホ ン酸ピリ ジン塩などの酸触媒の存在下で行ない、 その使 用量は基質に対して 0. 1〜 3当量であるのが望ま しい。 反応は溶 媒中で行なうのが望ま しく、 メ タ ノ ール、 エタノ ールなどのアルコ ール類、 アセ ト ン一水、 テ ト ラ ヒ ドロ フ ラ ン一氷、 水などを用いる ことができる。 また、 反応温度は、 0〜 8 0 °Cで行なうのが望ま し い。
=第三工程:
(式中、 Yは一 C H2— C H2—または一 C H = C H—であり、 Xは C 1 一、 B r—、 C H a S 03-. 。 35〇 3—または ー(:113— C6H4— S〇3—である。 )
本工程は、 上式 (V) で示される ピリ ジルォヰシ誘導体をハロゲ ン化またはスルホニル化し、 上式 (VI) で示されるピリ ジルォキシ 誘導体を製造する工程である。
反応は、 塩化チォニル、 塩化 P — ト ルェ ンスルホニル、 塩化メタ ンスルホニル、 塩化ト リ フルォ ロ メ タ ンスルホニル、 四塩化炭素一 ト リ フ ヱニルホスフ イ ン、 四臭化炭素一 ト リ フ ヱ ニルホスフ ィ ンな どの存在下で行ない、 その使用量は基質に対して 1〜 3当量である のが望ま しい。 また、 反応には塩基を用いるのが好ま しく、 ト リ エ チルァ ミ ン、 ピ リ ジン、 炭酸力 リ ウ 厶、 炭酸ナ ト リ ウ ムなどを用い ることができ、 その使用量は基質に対して 1〜 3当量であるのが望 ま しい c 反応は溶媒中で行なうのが望ま しく、 ジクロ ロメ タ ン、 ク ロロ ル厶、 ベンゼン、 ト ルエ ン、 酢酸ェチル、 ジメ チルホルム了 ミ ド どを用いることができる。 また、 反応温度は、 0〜; 3 G : 行 うのが望ま しい =
第四工程
(式中、 Yは一 C H2— C H2—または一 C H - C H—であり、 Xは C 1 一、 B r —、 C H 3 S 〇 3—、 C F 3 S 03—または p— C H3— C 6H4— S〇 3—である。 )
本工程は、 上式 (VI) で示される ピ リ ジルォキシ誘導体をフタ ル ィ ミ ドィヒし、 上式 ( I ) で示されるフタルイ ミ ド化合物を製造する _C禾王であ o
反応は、 ピ リ ジルォキ シ誘導体 1 モルに対してフタルイ ミ ド力 リ ゥムを 1 モル用いて行なうが、 相間移動触媒の存在下に反応させる と速やかに反応を完結させる ことができる。 相問移動触媒と しては 硫酸水素テ ト ラ 一 n — ブチル了 ンモニゥ ム、 塩化テ ト ラ 一 11 一プチ ルア ンモニゥ 厶、 吳化テ ト ラ 一 ri —ブチル了 ンモニゥ 厶などを用い る ことができ、 その使用量は基質に対して ϋ . 0 1〜 3当 fiである のが望ま しい。 反応は溶媒中で行 うのが望ま し く 、 ベ ンゼン、 ト ルェ ン、 了セ ト ニ ト リ ル、 酢酸ェチルなどを ¾ る 二 とができる:..
また、 反応温度は、 0 〜 1 0 0 X:で行なうのが望ま しい。
上のようにして上式 ( I ) で示されるフタルイ ミ ド化合物を製 造することができる。
また、 上式 ( I ) で示されるフタルイ ミ ド化合物は、 常法に従つ て酸付加塩とすることができ、 固体と して単離することができる。 酸と しては、 塩酸、 臭化永素酸、 硝酸、 硫酸、 シユウ酸、 コハク酸、 マレイ ン酸、 フマル酸、 酒石酸、 乳酸、 フタル酸などを使用するこ とができる。 フタルイ ミ ド化合物の酸付加塩と しては、 具体的には
X - ( 4 — ( 4 - ( 1 ーピペ リ ジニルメチル) ピ リ ジルー 2 —ォキ シ) 一 c i s— 2 —ブテニル) フタルイ ミ ド ' 2塩酸 ' エタノ ール 塩、 X— ( 4 - ( 4 - ( 1 ーピベ リ ジニルメチル) ピ リ ジルー 2 — ォキシ) 一 c i s - 2 ーブテニル) フタルイ ミ ド · 2硝酸塩、 N— ( 4 一 ( 4 - ( 1 ーピペ リ ジニルメ チル) ピ リ ジルー 2 —ォキシ) - c i s — 2 —ブテニル) フタルイ ミ ド ' マレイ ン酸塩、 N— ( 4 一 ( - ( 1 ーピペ リ ジニルメチル) ピ リ ジルー 2 —ォキシ) ブチ ル) フタルイ ミ ド . フマル酸塩などを例示することができる。 この フタルイ ミ ド化合物の酸付加塩は、 再結晶法により精製することが 可能である。
上式 ( I ) で示されるフタルイ ミ ド化合物およびその酸付加塩は、 フタロイル基の脱保護により容易に前記の式 (K ) で示されるア ミ ン体に変換することができ、 前記の中間体 (K ) の製造に有用 化 合物である。 特にフタルイ ミ ド化合物の酸付加塩は、 再結晶法によ り容易に精製することが可能であることから変換後の前記の式 (DO で示されるアミ ン体の純度を容易に向上させることができ、 また、 それによつてア ミ ン体 (K ) より誘導される前記の式 (W ) や式
( V£ j 等で示される抗清瘍剤の钝度の向上にも寄与することができ、 ¾めて有 な化合物である。
[実施例二
以下、 実施例 より本発明を具体的に説明する
実施例 1
水素化ナ ト リ ウ ム ( 6 0 %純度、 4. 5 6 g ) をテ ト ラ ヒ ドロフ ラ ン ( 1 0 0 m l ) およびジメ チルホルムア ミ ド ( 5 m l ) に懸濁 させ、 氷冷下、 上式 ( Π ) で示される 2 —ク ロ口 一 4 一 ( 1 ーピぺ リ ジニルメ チル) ピリ ジン ( 1 5. 0 g ) および上式 (X ΙΠ ) で示 される 4 — ( 2 —ォキ シテ ト ラ ヒ ド ロ ビラ ニル) 一 c i s — 2 —ブ テ ン一 1 一オール ( 1 2. 3 g ) を滴下した後、 徐々に室温まで温 度を上げ、 さ らに温度を上げて 1 1 時間還流した。 冷却後、 水を加 え、 酢酸ェチルで抽出し、 水、 飽和食塩水で洗浄し、 無水硫酸マグ ネ シゥ ムで乾燥後、 濾過し、 溶媒を留去し、 シ リ カゲルカ ラ ムク ロ マ トグラフィ 一に付して精製し、 上式 (X IV) で示される 2 — ( 4 ― ( 2 —ォキ シテ ト ラ ヒ ド ロ ビラ ニル) 一 c i s — 2 —ブテ ン一 1 —ォキ シ) 一 4 — ( 1 ー ピペ リ ジニルメ チル) ピ リ ジ ン ( 2 2. 5 g、 9 1 . 3 % ) を得た。
実施例 2
i T
z
上式 (XIV) で示される 2 — ( 4 一 ( 2 —ォキシテ ト ラ ヒ ドロ ピ ラニル) 一じ i s— 2 —ブテン一 1 —ォキシ) 一 4 一 ( 1 —ピペ リ ジニルメチル) ピ リ ジン ( 2 2. 5 g ) を酢酸ェチル ( 1 0 0 m l ) に溶解し、 1 N— H C 1 ( 1 0 0 m l ) を加えて室温で 2時間攪拌 した。 反応終了後、 水層を分離し、 酢酸ェチルで洗浄後、 炭酸力 リ ゥムでアル力 リ性と し、 ジク ロ πメ タ ンで抽出し、 飽和食塩水で洗 浄し、 無水硫酸マグネシウムで乾燥後、 濾過し、 溶媒を留去して上 式 (X V) で示される 4 一 ( 4 - ( 1 ーピベ リ ジニルメチル) ピ リ ジルー 2 —ォキシ) 一 c i s — 2 —ブテン一 1 一オール ( 1 5. 5 g、 9 1. 0 %) を得た。
I R ( c m"1, f i l m)
3 3 7 0 , 2 9 3 5 , 1 6 1
1 K - X M R ( δ , C D C )
1. 3 ΰ〜 1 . 了 5 ( 6 Η , m ) ,
H , m ) , 3. 4 0 ( 2 H, s ) ,
4. 3 2 ( 2 H, d , J = 6 H z ) 4. 9 9 ( 2 H, d , J 二 6 H 2 ) , 5 5 7〜 6. 0 9 ( 2 H, m ) , 6. 了 4 ( 1 H, s ) , 6 8 9 ( 1 H, d, J - 5 fl z ) , S . 0
実施例 3
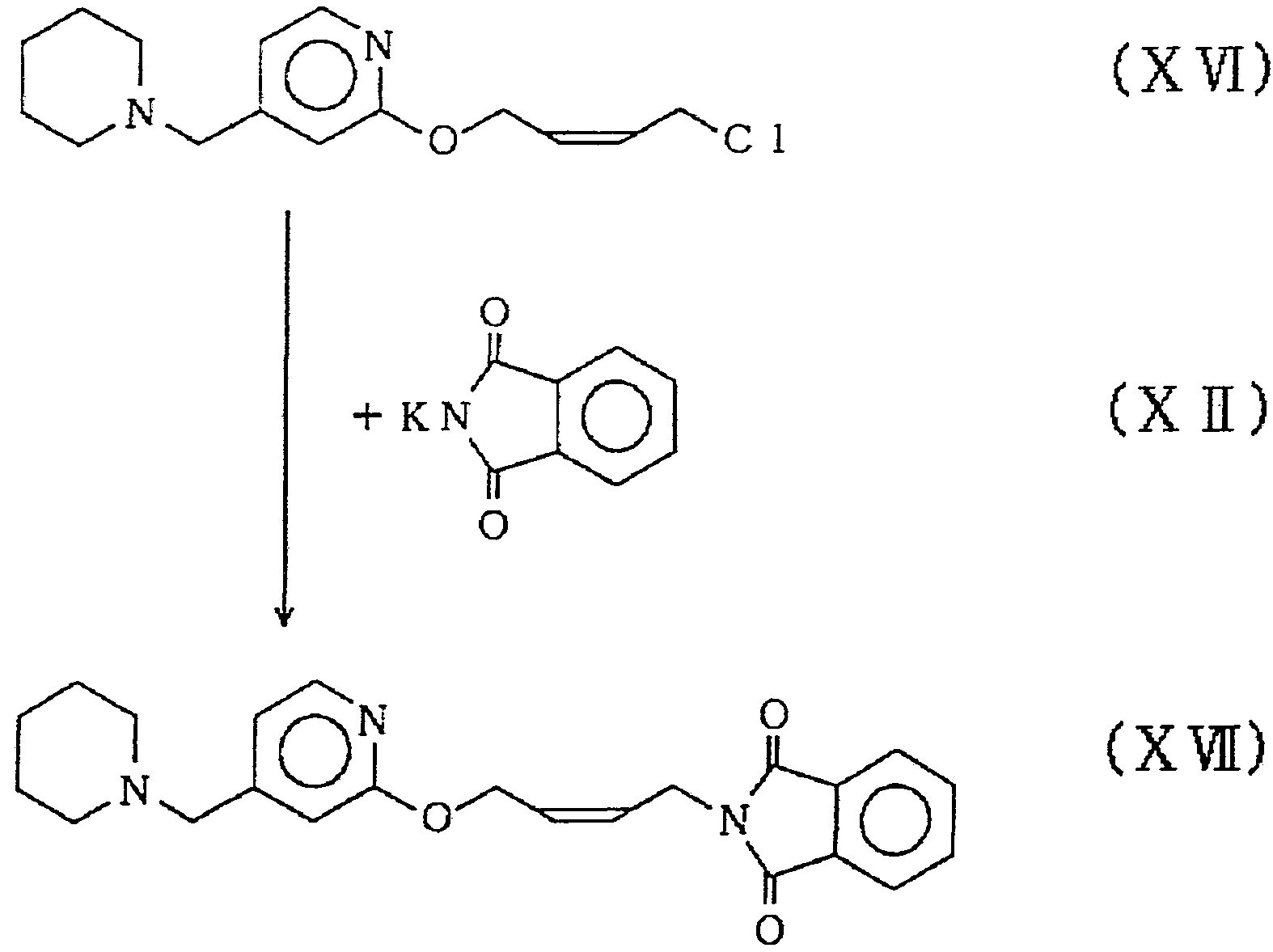
上式 (X V) で示される 4 — ( 4 - ( 1 — ピベ リ ジニルメ チル) ピリ ジルー 2 —ォキシ) 一 c i s — 2 —ブテン一 1 —ォ―ル ( 1 5. 5 g ) をジク ロ ロ メ タ ン ( 2 0 0 m l ) に溶解し、 無氷炭酸力 リ ウ 厶 ( 9. 4 g ) を懸濁させ、 氷冷下、 塩化チォニル ( 1 0. 2 g ) のジク ロロメ タ ン ( 2 0 m l ) 溶液を滴下し、 徐々に室温まで温度 を上げ、 2時間攪拌した。 反応終了後、 5 %炭酸水素ナ ト リ ウム氷 溶液を加え、 有機層を分離し、 1. 5 N— H C 1 で抽出し、 ト ルェ ンで洗浄後、 炭酸力 リ ゥ 厶で了ルカ リ性と し、 ト ルエ ンで抽出し、 飽和食塩水で洗浄し、 無水硫酸マグネ シウ ムで乾燥後、 濾過して上 式 (XVI) で示される 1 一ク ロ口 一 4 一 ( 4 - ( 1 —ピペ リ ジニル メ チル) ピ リ ジル一 2 —ォキ シ) 一 c i s — 2 —ブテ ン ( 1 4. 7 g、 8 8. 6 % ) を含む ト ルェ ン溶液 ( 8 0 m 1 ) を得た。
実施例 4
3
十 (XI)
I,
Π Τ 丁
上式 ( X VI ) で示される 1 —ク ロ 一 4 — ( 4 一 ( 1 ーピペ リ ジ ニルメチル) ピ リ ジルー 2 —ォキシ) 一 c i s _ 2 —ブテン ( 1 4 ,
5
7 g ) を含むトルェ ン溶液 ( 8 G m 1 ) に、 上式 (X Π ) で示され るフタルイ ミ ドカ リ ウム ( 1 3. 2 g ) 、 硫酸水素テ ト ラー π—ブ チルア ンモニゥ厶 ( 1. 4 5 g ) を懸濁させ、 8 0 tで 2時間反応 させた。 冷却後、 反応液を 1 X— N a 〇 Hで洗浄し、 飽和食塩水で 浼浄し、 無水硫酸マグネシウムで乾燥後、 瀘過し、 溶媒を留去して 上式 (XW) で示される N— ( 4 - ( 4 一 ( 1 ーピベ リ ジ二ルメ チ ル) ピ リ ジルー 2 —ォキシ) 一 c i s — 2 —ブテニル) フタルイ ミ ド ( 1 8. Q g、 8 7. 8 %) を得た。
I R ( c m- 1, f i 1 m
2 9 3 5 , 1 7 7 Q , 1 7 1 , 1 6 1 5
1 H - X M R ( o , C D C ")
9
H , m ) , 3. 4 1 ( 2 H , s ) , 4. 4 7 ( 2 H , d , J = 6 H z ) , 5. 1 2 ( 2 H, d , J = 6 H z ) , 5. 5 0 ~ 6. 1 5 ( 2 H, m ) , 6. 7 5 ( 1 H , s ) , 6. 8 9
( 2 H, d , J = 5 H z ) , 7. 6 5〜 7. 9 7 ( 4 H, m) , 8. 0 8 ( 2 H, d , J = 5 H z )
実施例 5
5 2 HC 1 · E t〇H 上式 (X ) で示される N— ( 4 - ( 4 — ( 1 ーピペ リ ジニルメ チル) ピ リ ジルー 2 —ォキ シ) 一 c i s — 2 —ブテニル) フタ リレイ ミ ド ( 3. 2 9 g ) をエタノ ール ( 3 0 m l ) に溶解し、 これに塩 化氷素ガスを通じて氷冷した。 析出した白色結晶を濾別し、 ェタ ノ —ルー n —へキサ ンで洗浄し、 乾燥して上式 (XVIII) で示される N 一 ( 4 一 ( 4 一 ( 1 ー ピベ リ ジニルメ チル) ピ リ ジルー 2 —ォキ シ) - c i s _ 2 —ブテニル) フタ ルイ ミ ド ' 2塩酸 ' エタ ノ ール塩 ( 3. 7 0 g、 8 6. 2 %) を得た。
m . p .
1 5 5〜 1 ? 0 で (分解)
I R ( c m"1, K B r )
3 4 0 8 , 2 9 8 , 2 6 3 2 , 2 5 3 2 , 1 7 6 8 , 1 7 1 4, 1 6 1 6, 1 3 9 4 , 1 2 9
1 Η - X M R ( δ , D S 0 - d 6 )
1 . 0 6 ( 3 Η, t , J = 7 Η ζ ) , 1 . 2 卜 1 . 4 2 ( 1 Η, m ) , 1 . 6 7〜 ; I . 9 4 ( 5 Η , m) , 2. 8 0 〜 2. 1 ( 2 Κ, m) , 3. 2 8 ( 2 Η , d , J = 1 2 Η
3. 4 5 ( 2 H, q , J = 7 H z ) , 4. 2 6 ( 2 H d , J = 6 H z ) , 4. 3 6 ( 2 H, d, J 二 ? H z ) , 5 0 5 ( 2 H, d, J = 6 H z ) , 5. 6 2 ~ . 7 1 ( 1 H m) , o . 8 0 - 5. 8 9 ( 1 H, m) , 7. 1 8 ( 1 H, s ) , 7. 3 2 ( 1 H, d , J = 6 H ) , 7. 8 2〜? . 9 1 ( H, m) , 8. 2 δ ( 1 H, d , J = 6 H z ) , 1 1. 2 0 ( 1 H , b r o a d )
実施例 6
上式 (X W) で示される N— ( 4 - ( 4 一 ( 1 ーピペ リ ジニルメ チル) ピリ ジル一 2 —ォヰシ) 一 c i s — 2 —ブテニル) フタルイ ミ ド ( 2. 2 5 g ) をエタノ ール ( 3 0 m l ) に溶解し、 これに濃 硙酸 ( S 1 %純度、 1. 3 m l ) を加え、 冷蔵庫で 2 日 冷却した, 析出した結晶を濾別し、 乾燥して上式 (XIX) で示される N— ( 4 一 ( - ( 1 ーピペ リ ジニルメチル) ピリ ジルー 2 —ォキシ) 一 c i s— 2 —ブテニル) フタルイ ミ ド · 2硝酸塩 ( 2. ? G g、 9 0. 7 %) を得た。
m . p .
I 1 4〜 1 1 6 °C (分解)
I R ( c m " 1 , K E r )
K, m) , 3. 2 0 ~ 3. 5 5 ( 2 H, m) , 4. 3 6 ( 2 H, s ) , 4. 3 8 ( 2 H, d , J - 6 H z ) , 5. 1 0 ( 2 H, d, J = 6 H z ) , 5. 5 5〜 6. 0 5 ( 2 H, m) , 7. 0 6 ( 1 H, s ) , 7. 1 9 ( 1 H, d , J - 5 I-I z ) , 7. 9 1 ( H, s ) , 8. 3 2 ( 1 H, d , J = 5 H z ) , 9. 5 3 ( H, b s )
実施例 Ί
HO〇C COOH 上式 (X で示される N— ( 4 - ( 4 - ( 1 ーピペ リ ジニルメ チル) ピ リ ジルー 2 —ォキ シ) 一 c i s — 2 —ブテニル) フタ ルイ ミ ド ( 3. 3 3 g ) をエタノ ール ( 3 5 m l ) に溶解し、 これにマ レイ ン酸 ( 1. 0 9 g ) を加え、 2時間氷冷した。 析出した白 9色結 o 1 曰
曰曰を濾別し、 乾燥して上式 (X X) で示される N— ( 4 - ( 4 - 6
( 1 ー ピペ リ ジニルメ チル) ピ リ ジルー 2 —才キ シ) 一 c i s 一 2 ーブテニル) フタ ルイ ミ ド ' マ レイ ン酸塩 ( 4. 0 0 g、 9 2. 6
%) を得た。
ΠΊ . P .
1 4 8〜 1 5 0 C (分解)
I R ( c m-1, K B r )
3 4 6 8 , 2 9 0 , 2 3 6 , 1 7 7 0 , 1 7 1 0
2 0 , 1 5 6 8, 1 4 5 6 , 1 3 9 2 , 1 0 6 6, 9
8 7 4 , 7 1 [-,
1 H一 X M R ( δ , D M S 0 - d B )
1 . 3 6〜 1 . 9 3 ( 6 H, m) 2 . 9 0〜 3 . 2 0 ( 4 H , m) , . 2 3 ( 2 H, s ) 4 . 3 9 ( 2 H, d, J = 6 E z ) , D . 0 9 ( 2 H, d J = 6 H z ) , 5 . δ δ ~ 6 . 0 0 ( 2 H, m ) , 6 . 1 2 ( 2
( 1 H, s ) , Ί . 1 6 ( 1 H, d , J
2 ( H, s ) , 8 . 3 1 ( 1 H, d , J = 5 H z ) 実施例 8
永素化ナ ト リ ウ ム ( 6 0 %純度、 1 3 . 2 9 g ) をテ ト ラ ヒ ドロ フ ラ ン ( 2 5 0 m l ) およぴジメ チルホルム了 ミ ド ( 1 5 τη 1 ) に 態 ¾|させ、 永冷下、 上式 ( Π ) で示される 2 —ク ロ口一 4 一 ( 1 一 ピぺ リ ジニルメ チル) ピ リ ジン ( 5 0 . 0 g ) および上式 ( X X I ) で示される 4 一 ( 2 —ォキ シテ ト ラ ヒ ド ロ ビラ ニル) 一ブタ ン一 i 一; rール ( 4 1 . 4 g ) を滴下した後、 徐々に室温まで温度を上げ、 さらに温度を上げて i 5時間還流した。 冷却後、 水を加え、 酢酸ェ チルで拍 し、 永、 飽¾食塩水で洗浄し、 無水硫酸マグネ シウ ムで 乾燥後、 瀘過し、 溶媒を留去し、 シ リ カゲルカ ラ 厶クロマ トグラ フ ノ ーに して精製し、 上式 (X X H ) で示される 2 — ( 4 一 ( -
ォヰ シテ ト ラ ヒ ドロ ビラニル) 一ブタ ン - 1 ーォキシ) 一 4 一 ( 1 —ピぺ リ ジニルメ チル) ピ リ ジン ( 7 4. 5 g、 9 0. 1 % ) を ί
実施例 δ
5 z
8
上式 ( X X D ) で示される 2 — ( 4 - ( 2 —ォキシテ ト ラ ヒ ドロ ビラニル) —ブタ ン— 1 —ォキシ) 一 4 — ( 1 ー 3 ピペ リ ジニルメ チ ル) ピ リ ジン ( ? 4. 5 g ) を酢酸ェチル ( 4 0 0 m l ) に溶解し
H
1 X - H C 1 ( 3 4 0 m l ) を加えて室温で 1 2時間攪拌した。 反 応終了後、 水層を分離し、 酢酸ェチルで洗浄後、 炭酸力 リ ウ dムでァ ルカ リ性と し、 酢酸ェチルで抽出し、 飽和食塩水で洗浄し、 無水 J C 1 G硫 酸マグネシウムで乾燥後、 濾過し、 溶媒を留去して上式 (xxm) で示さ riる 4 一 ( 4 - ( 1 ーピベ リ ジニルメ チル) ピ リ ジルー 2 — ォキシ) 一ブタ ン一 1 —オール ( 5 0. 4 g、 8 9. 2 % ) を得た
1 K一 X MR ( δ , C D C 1 3)
i . C! 0〜 2 0 4 ( 1 O K, m ) , 2
( 4 H, m ) 2. 7 5 ( 1 H , b r o
( 2 H , s ) 3. 7 1 ( 2 H, t , J
: ( 2 K, t , JJ == 6 H z ) , 6. 7 1 ( 1 1, s )
8 5 ( I II, d , J
5 H 2 )
2 o
i o
3
0
κ
上式 (xxm) で示される 4一 ( 4 - ( 1ーピペ リ ジニルメチル) ピリ ジルー 2 —ォキシ) 一ブタ ン一 1 一オール ( 5 0. 0 g ) をジ ク ロ ロメ タ ン ( 5 0 0 m l ) に溶解し、 ト リ二ェチルァ ミ ン ( 4 7. 8 g ) を加え、 氷冷下、 メ タ ンスルホニルクロ ライ ド ( 3 6. 8 g ) を滴下し、 徐々に室温まで温度を上げ、 1 時間攪拌 ζした。 反応終了 後、 5 %炭酸水素ナ ト リ ゥム水溶液を加え、 有機層を分離し、 無水 硫酸マグネシウムで乾燥後、 瀘過し、 溶媒を留去して上式 (X 82 ο XIV) で示される 1 一メタ ンスルホニルォヰシー 4 一 ( 4 一 ( 1 ーピペ リ ジニルメチル) ピ リ ジルー 2 —ォキシ) 一ブタ ン ( 5 8. 5 g、 9 4. 8 %) を得た。
H - N R ( δ , C D C 1 3)
1. 0 9 ~ 1. 7 5 ( 6 H, τη ) , · 1
Κ, τη ) , 2. 1 卜 2. 5 3 ( 4 H
Η, s ) , 3. 1 ( 2 H , s ) , 4
Κ , m ) , t! . 7 1 ( 1 H , s ) , 6 S 6 ( 1 Η , d, J
— 0 i 2 ) , 8
実旌例 1 1
上式 (xxw) で示される i 一メ タ ンスルホニルォキシ一 4 一
( 4 一 ( 1 —ピベ リ ジニルメ チル) ピ リ ジルー 2 —ォキシ) ーブタ ン ( 5 8. 5 g ) をァセ トニ ト リ ル ( 4 0 0 m l ) に溶解し、 上式
(X Π ) で示されるフタルイ ミ ドカ リ ウ ム ( 5 2. 6 g ) 、 硫酸水 素テ ト ラー n—ブチル了 ンモニゥム ( 3. 9 g ) を懸濁させ、 5時 間還流した。 冷却後、 反応液を濾過し、 溶媒を留去した。 残渣を酢 酸ェチルに溶解し、 1 λ'— N a 〇 Hで洗浄し、 飽和食塩水で洗浄し 無水硫酸マグネ シウ ムで乾燥後、 瀘過し、 溶媒を留去して上式 (X X V ) で示される X— ( 4 一 ( 4 — ( 1 ーピベ リ ジニルメ チル) ピ リ ジルー 2 —ォキ シ) ブチル) フタリレイ ミ ド ( 6 0. 2 g、 8 5. 4 を得た。
m . p .
4 6〜 4 8 。C
ί R ( c m"1, f i l m)
2 9 4 0 , 2 8 5 6 , 2 7 9 6 , 1 7 7 2 , 1 7 1 2, 1 6 1 4 , 1 5 6 0 , 1 4 7 0 , 1 4 2 2 , 1 3 9 8 , 1 3 7 2 1 0 4 4 , 7 2 0
1 H— NM R ( o , C D C ")
1. 2 ト 1. ? 2 ( 6 H, m) , 1. 7 卜 1 . 9 9 ( H, m) , . 2 0〜 2. 4 8 ( 4 H, m) , 3. 3 9 ( 2 H, s ) , 3. 6 0〜 3. 8 8 ( 2 H, m) , 4. 1 4〜 4 4 1 ( 2 H, m) , 6. 6 7 ( 1 H , s ) , 6. 8 3 ( 1 H d , J = 5 H z ) , 7. 5 2〜 7. 9 2 ( 4 H, m) , 8
0 2 ( 1 H, d , J = 5 H z )
実施例 1 2
上式 (X X V) で示される X— ( 4 - ( 4 - ( 1 ー ピペ リ ジニル メ チル) ピ リ ジルー 2 —ォキ シ) ブチル) フタ ルイ ミ ド ( 0. 5 0 g ) とフマル酸 ( 0. 3 ? g ) をエタ ノ ール ( 2 0 m ] ) に熱時溶 解し、 溶液量が約 5 m 1 になるまで減圧下、 濃縮し、 氷冷下に 1 2 時間放置した。 沂 Sした結晶を瀘別し、 乾燥して白色結晶の上式 ( X X VI) で示される X— ( 4 一 ( 4 - ( 1 ーピベ リ ジニルメチル) ピ リ ジルー 2 —ォキ シ) ブチル) フタ ルイ ミ ド · フマル酸塩 ( 0. 5 2 g、 8 0. 3 %) を得た。
IT .
1 0 ト 1 0 6 °C
I R ( c m"1, K B r )
3 4 6 0 , 2 8 , 2 6 0 , 2 5 0 , 1 7 6 8 , 1 7 1 2 , 1 6 5 0 , 1 6 1 2 , 1 5 6 2 , 1 2 8 , 1 3 9 8 , 1 3 6 4 , 1 3 0 6 , 1 1 6 8 , 1 0 5 8 , 9 8 4 , 7 1 , 6 4 6
1 H - - NN MM RR (( δδ ,, D M S 0 - d 6)
1 . 2 0〜 1 9 8 ( 1 0 H, m) , 2. 3 2〜 2. 6 8 ( H, m) 3. 4 2〜 3. 8 0 ( 2 H, s, 2 H , m ) , 4. 0 8 ~ 4 4 0 ( 2 H, m) , 6. 6 2 ( 2 H, s ) ,
6. 7 4 ( 1 H, s ) 6. 9 3 ( 1 H, d, J = 5 H z ) ,
7. 8 4 ( 4 H, s ) 8. 0 5 ( 1 H, d, J = 5 H z ) ,
9. 2 2 ( 2 H, b r o a d )
[発明の効果]
本発明は、 医薬品、 特にヒ スタ ミ ン H 2受容体拮抗作用に基づく 抗潰瘍剤の製造中間体と して有用な化合物である前記の式 ( I ) で 示されるフタ ルイ ミ ド化合物およびその酸付加塩を提供するもので ある。 特にフタ ルイ ミ ド化合物の酸付加塩は、 再結晶法により容易 に精製することが可能であり、 それによつて本発明のフタルイ ミ ド 化合物およびその酸付加塩より誘導される前記の式 (\¾) や式 (\ 〉 等で示される抗潰瘍剤の純度の向上に寄与することができるという 効果を奏する。