JP5884005B2 - 新規なセファロタキサン誘導体の製造に用いられるカルボン酸誘導体 - Google Patents
新規なセファロタキサン誘導体の製造に用いられるカルボン酸誘導体 Download PDFInfo
- Publication number
- JP5884005B2 JP5884005B2 JP2009063260A JP2009063260A JP5884005B2 JP 5884005 B2 JP5884005 B2 JP 5884005B2 JP 2009063260 A JP2009063260 A JP 2009063260A JP 2009063260 A JP2009063260 A JP 2009063260A JP 5884005 B2 JP5884005 B2 JP 5884005B2
- Authority
- JP
- Japan
- Prior art keywords
- acid
- structural formula
- mmol
- compound
- mixture
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 *C1(C(OC(C2c3c4)C(*)=CC2(CCC2)N2CCc3cc2c4OCO2)=O)OC(c2ccccc2)=NC1c1ccccc1 Chemical compound *C1(C(OC(C2c3c4)C(*)=CC2(CCC2)N2CCc3cc2c4OCO2)=O)OC(c2ccccc2)=NC1c1ccccc1 0.000 description 21
- GRHKYQAEFWQFIQ-UHFFFAOYSA-N CC(/[O]=C(\C12)/C(OC)=CC1(CCC1)N1CCc1c2cc2O[IH]Oc2c1)=O Chemical compound CC(/[O]=C(\C12)/C(OC)=CC1(CCC1)N1CCc1c2cc2O[IH]Oc2c1)=O GRHKYQAEFWQFIQ-UHFFFAOYSA-N 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/14—Ortho-condensed systems
- C07D491/147—Ortho-condensed systems the condensed system containing one ring with oxygen as ring hetero atom and two rings with nitrogen as ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
- A61P33/06—Antimalarials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C57/00—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms
- C07C57/02—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms with only carbon-to-carbon double bonds as unsaturation
- C07C57/13—Dicarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/40—Unsaturated compounds
- C07C59/42—Unsaturated compounds containing hydroxy or O-metal groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/40—Unsaturated compounds
- C07C59/42—Unsaturated compounds containing hydroxy or O-metal groups
- C07C59/48—Unsaturated compounds containing hydroxy or O-metal groups containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/66—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety
- C07C69/73—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of unsaturated acids
- C07C69/732—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of unsaturated acids of unsaturated hydroxy carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D203/00—Heterocyclic compounds containing three-membered rings with one nitrogen atom as the only ring hetero atom
- C07D203/04—Heterocyclic compounds containing three-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D203/06—Heterocyclic compounds containing three-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D203/08—Heterocyclic compounds containing three-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/08—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D263/16—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D303/00—Compounds containing three-membered rings having one oxygen atom as the only ring hetero atom
- C07D303/02—Compounds containing oxirane rings
- C07D303/48—Compounds containing oxirane rings with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D305/00—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms
- C07D305/02—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D305/04—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D305/08—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/04—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D307/10—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D307/16—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/08—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/10—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings
- C07D317/32—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D453/00—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids
- C07D453/02—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids containing not further condensed quinuclidine ring systems
- C07D453/04—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids containing not further condensed quinuclidine ring systems having a quinolyl-4, a substituted quinolyl-4 or a alkylenedioxy-quinolyl-4 radical linked through only one carbon atom, attached in position 2, e.g. quinine
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Tropical Medicine & Parasitology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Cephalosporin Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Epoxy Compounds (AREA)
- Pyrane Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Description
pは1〜2であり(2個の単位は同一であるかまたは異なり、単結合または酸素原子によって結合することができる)、様々な酸素化置換基(脂肪族または芳香族エーテル、遊離またはエステル化アルコール、置換または遊離エノールおよび/またはフェノール、橋かけエーテル、および更に一般的には、この種の化合物について天然状態で普通に見られる任意の置換基)を含むことができる]
の基本骨格を有する化合物またはその塩を表す。
ことを考慮すれば、
多環骨格である完全構造の不活性な生合成前駆体からなり、それらに親水性および疎水性置換基の同様な組合せを含む側鎖がグラフトしており、
タキサン(広義のバカチン)およびハリングトニン(セファロタキシン)の多環部は植物の再生可能な部分が比較的多量に含まれているが、活性分子(ハリングトニンおよびタキサン)は1/10〜100しか含まれておらず、
イヌガヤ(Cephalotaxus)は珍しい木であり、イチイ(Taxus)よりも珍しく、後者に比較して遙かに偏在しない
という2個以上の点において、タキサンと類似している。
最初に)亜鉛の存在下でブロモ酢酸メチルとセファロタキシンに予め結合した側鎖の(真のまたは潜在的な)カルボニルとのリフォーマツキー反応によって、または
二番目に)有機リチウム試薬を先行して形成することによる、
第二の側鎖−CH2CO2Meの結合を伴う。
立体選択性の欠如、
収束性に乏しい、
平凡な収率、
稀少で高価な基剤での鎖の官能化および構築、
キラルのホモ−ハリングトニンは今日まで得られていない
という欠点を有する。
ので、ラセミセファロタキシンからでも鏡像異性体的に純粋なハリングトニンが得られるという利点を有する。
セファロタキシンのヒンダード遊離アルコールあるいは相当する金属アルコキシドを、骨格および官能化のいずれについても完全に予備形成した適当に置換した第三カルボキシルオキサシクロアルカン酸の形態の鎖を用いてエステル化して、半合成によるアンヒドロ−ホモ−ハリングトン酸を調製し、
ことからなっている。
水素、
線状または分岐状および/または環状の飽和、不飽和または芳香族の炭化水素を基剤とした基、特にアルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、アリール、ヘテロシクロアルキルであって、上記の炭化水素を基剤とした基は(複数の)ヘテロ原子を含みまたは含まず、R5およびR8は互いに結合して環を形成することができ、
上記基の1個を有する酸素化エーテルである。
nは0〜8である。
の環状無水物についての場合であり、
これは相当する二酸から容易に調製して、メタノールのエステル化によって再度3kを得ることができ、あるいは上記のように型2のセファロタキシンのアルコール官能基に結合させることができるが、上記よりも収率はよくなく、次に、第一酸官能基をプロトン酸またはルイス酸の存在下にてメタノールを用いて常法により、あるいは三フッ化ホウ素エーテレート/メタノール複合体またはジアゾメタンを用いてメチル化する。
(a)酸触媒での、遊離酸のアルコールによるエステル化、
(b)無水物またはハロゲン化物を介するアシル移動によるエステル化、
(c)活性化エステルを用いるアシル移動によるエステル化、
(d)スカンジウムトリフレートを用いるエステル化、
(e)三フッ化ホウ素エーテレートを用いるエステル化、
(f)チオエステル法によるエステル化
の6種類の具体的様式に従って行うことができる。
を用いることができ、
これは、下記の混合無水物の調製のための一般的操作条件下などで相当する二酸を無水酢酸で処理することによって極めて簡単に調製することができる。
式4cにおいて、n、R5、R6、R8およびCTX−は上記と同じ意味を有する)
として知られている4c型(R5=CH2CO2Me)の形成した2種類のジアステレオ異性体は、いわゆる正常相では、例えば固定相としての天然のままのシリカゲルおよび移動相としての有機溶媒の混合物で、または好ましくは逆相では、例えば有機シリル、シアノアルキル、フェニルアルキル、好ましくはオクタデシルアルキルシラン鎖のような無極性基をグラフトした不活性シリカ、および移動相としての水性溶媒の混合物で、合成用クロマトグラフィーによって分離することができる。
n、R5、R6、R8、R9、XおよびCTX−は、上記で定義した通りである。
2型のセファロタキシンは、文献記載の方法に従って合成によりまたは抽出によって調製することができる。後者の場合には、いずれの方法も植物出発材料を酸水溶液と直接接触させる方法を用いていないので、本発明においてこれを説明することが有利であることを見出した。新鮮なまたは乾燥した植物出発材料を希無機酸または弱有機酸を用いて酸性化水性−有機混合物と24時間接触させ、pHを1〜4、好ましくは3とした。無機酸は、例えば硫酸または塩酸であり、有機酸はクエン酸、乳酸または酒石酸などとすることができ、有機溶媒は、例えば低級アルカノール、ケトン、テトラヒドロフラン、または当業者が抽出に用いる任意の他の水混和性溶媒とすることができる。水含量は、20〜80%であり、好ましくは50%である。得られる溶液を直接クロマトグラフィー処理を行い、または文献記載の方法とは対照的に、葉緑素および/または植物脂肪を含まないので、塩基性にして向流抽出を行うことができる。低級エステル、または好ましくは低級ハロゲン化炭化水素、更に具体的にはジクロロメタンのような水不混和性の有機溶媒を用いる向流抽出により、白色粉末の形態で単離された総アルカロイドの混合物を得た。イヌガヤアルカロイドの精製には幾つかの方法があるが、いずれも、特に逆相では、特にセファロタキシン、および更に具体的には式2aのセファロタキシンの精製には適合しない。
に従って3f型の置換エチレン性第三(−ヒドロキシ酸の環化によって行うことができる。
を有する3k型の適当に置換されたカルボン酸シクロエーテルは、相当するジエステル3j(但し、R5=CH2CO2Rである)の全ケン化の後、中間体の二酸3r(但し、R5=CH2CO2Hである)の温和な選択的メチル化によって調製することができる。
を有する3t型の誘導体(但し、R=GP)の製造法が得られ、これを上記の方法を用いてセファロタキシンとカップリングして、本発明の主題を形成する第三酸のカップリングの際にエステル交換反応が起こらないようにすることができる。
3i型のそれらの前駆体
3s型の二酸(但し、R5=CH2CO2Hである)
得ることができる。
に従って調製することができる。
は、オクタデシルシラン型のグラフト逆相を固定相として、また適当に調整したメタノール/水混合物を移動相として用いる合成用逆相クロマトグラフィーによって、他の関連不純物と同時にそのエピマーから分離することができる。
Ω−CO−O−CTX
[上記式中、
Ω(「オメガ」)は鎖の末端残基の代表的基であり、−CO−はセファロタキサンに結合したエステル基のカルボニルであり、
基Ω−CO−は、
下記式
nは0〜8であり、
Zは酸素、窒素または硫黄ヘテロ原子であり、
R5、R6およびR8は、独立して
水素、
飽和、不飽和または芳香族の線状または分岐状および/または環状の炭化水素基、特にアルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、アリール、ヘテロシクロアルキルであって、上記基は(複数の)ヘテロ原子を含みまたは含まず、R6およびR8は1つの環に含まれていてもよく、
上記基の1個を有する酸素エーテルである)の置換ヘテロシクロアルカン、または
下記式
mは1〜8であり、
R5、R6およびR8は上記で定義した通りである)の線状アルケン、または
下記式
n、R5、R6およびR8は上記で定義した通りであり、
ZおよびQ2は、独立して酸素、窒素または硫黄ヘテロ原子であり、
Q1は炭素、ケイ素またはリン原子であり、
R9およびR10は、独立して水素、アルコキシ、(複数の)ヘテロ原子を含むまたは含まない、飽和、不飽和または芳香族性の線状または分岐状および/または環状の炭化水素基、特にアルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、アリール、ヘテロシクロアルキルであり、
R9および/またはR10は、ゼロであるかまたは一緒になってヘテロ原子を作成しおよび/または多重結合を作成することができ、但し、Q1、R9およびR11はゼロであって、それらを有する炭素の2個の原子の間に多重結合を作成することができ、
R11は水素、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニルまたはアルキルカルボニルである)
に相当し、
−O−CTXは、下記式
pは1または2である)、
のセファロタキシン残基 その塩であり、
上記の2種類の基−Ωおよび−CTXは、エステル結合−CO−O−によって結合されている]
の側鎖を有するセファロタキサンおよび/またはその塩の製造法であって、
一般式Ω−CO−Aを有する酸の活性化形態またはその塩であって、Ω−COが下記式
Ω−COが下記式
Ω−COが下記式
を有し、
Aが、
下記式
の環状無水物であり、
この反応はこのようにして形成された第一カルボキシルのメチル化によって完成されているものを、
式H−O−CTX(式中、CTXは上記で定義した通りである)のヒドロキシル基を有するセファロタキサンまたはその塩、または
式M−O−CTX(式中、CTXは上記で定義した通りであり、Mは金属である)の金属アルコキシド、または
式Y−O−CTX(式中、−O−CTXは上記で定義した通りであり、Yは、Y−と−O−CTXとの間で開裂することによって酸素原子に陰電荷を与え、またはY−O−と−CTXとの間で開裂することによってカルボカチオンを与える脱離基である)のそのヒドロキシル基の活性化形態
と結合させ、1または数個の反応添加剤が存在することができ、上記側鎖を有するセファロタキサンおよび/またはその塩を形成する、方法に関する。
のセファロタキシン、またはその塩である。
式MeOCOO−のメトキシホルミルオキシ、
式CF3COO−のトリフルオロアセチルオキシ、
式RSO3−のアルキルスルホンオキシ、
式(RO)2PO−のホスホキシ、
式ROP(Cl)O−のハロホスホキシ、
式R3SiO−のトリアルキルシリルオキシ、
(式中、Rはアルキルである)
式
式
から選択される基である。
に相当するセファロタキシンアルコキシドは、式H−O−CTXのセファロタキシンを、金属自身、アミド、金属水素化物、またはアルキル−金属と接触させることによって得ることができる。
nは0〜8であり、
Zは酸素、窒素または硫黄ヘテロ原子であり、
R5、R6およびR8は、独立して
水素、
飽和、不飽和または芳香族の線状または分岐状および/または環状の炭化水素基、特にアルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、アリール、ヘテロシクロアルキルであって、上記基は(複数の)ヘテロ原子を含みまたは含まず、
上記基の1個を有する酸素エーテルであり、
CTXは、上記で定義した通りである)に相当する側鎖を有するセファロタキサンであって、
1°)n=2または3、同時にR6=R8=メチルおよびR5=OMeまたはヒドロキシル、
2°)n=2、同時にR6=R8=メチルおよびR5=OMeまたはヒドロキシル、
3°)R8がメチルでありかつR5が基−CH2CO2CH3であるとき、n=3、同時にR6がヒドロキシル
である化合物を除く、セファロタキシン、および/またはその塩、
m、R5、R6、R8、およびCTXは上記で定義した通りである)
に相当し、
m=2、R5=CH2CO2CH3、R6=R8=メチルであり、かつCTXが上記で定義した通りである化合物を除く、側鎖を有するセファロタキサン、および/またはその塩、
n、Z、Q1、Q2、R5、R6、R8、R9、R10、R11、およびCTXは上記で定義した通りである)
に相当する、側鎖を有するセファロタキサン、および/またはその塩、
のような新規化合物の調製でもある。
上記鎖を、水性または非水性媒質中で薬剤および/またはプロトン性または非プロトン性の親電子性基Eで開環し、下記式
の中間体化合物を提供し、
上記鎖を、場合によっては活性化および/または開環添加剤の存在下で、加水分解または慎重な加溶媒分解によって開環し、
Z′は、
ハロゲンであるか、または水素または請求項1で定義したような基R11を有するヘテロ原子であるか、または
水素、炭化水素基であって、(複数の)ヘテロ原子を有しまたは有さない、飽和、不飽和または芳香族性の線状、分岐状および/または環状の上記基、特にアルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、アリール、またはヘテロシクロアルキル
である)の開環した側鎖を有するセファロタキサンを提供する。
上記鎖をハロゲン化溶媒、好ましくはジクロロメタン中で臭化水素酸を酢酸に溶解したもので処理した後、インシテューでの加水分解によって開環し、中間体を単離することなく、下記式
Ω−CO−OH
(式中、基Ωは上記で定義した通りである)
に相当し、
式(+)−Ω−CO−OHおよび(−)−Ω−CO−OHの化合物であって、例えば(+)−Ω−CO−OHが右旋性鏡像異性体を表し、(−)−Ω−CO−OHが左旋性鏡像異性体を表すものを含むラセミ混合物と同等な上式を、
a)上記ラセミ混合物、または上記で定義した通りである式
Ω−CO−A
の活性化形態の1つであって、
上記のラセミ混合物または上記の活性化形態は、それぞれ
式(Ω−CO−O)−に相当するアニオン、または
式(Ω−CO)+に相当するカチオン
を生成するものを、キラル体(chiral entity)の純粋な鏡像異性形態と接触させ、Δ*(星印付きデルタ)の記号で表される上記の「分割剤」は、
共有結合による安定な組合せ、または
水素結合によるまたは疎水性相互作用による容易に逆転可能な不安定な組合せ、または
静電的相互作用による中間的不安定性の組合せ
を形成する能力を有し、
Ω−CO−O−Δ*およびΩ−CO−Δ*のジアステレオマー混合物を提供し、
下記式
に相当する基であるか、または
に相当する基であるか、または
に相当する基である。
Ω−CO−O−[NH−Δ*]+
Ω−CO−O−[NH2−Δ*]+
Ω−CO−O−[NH3−Δ*]+
(式中、ΩおよびΔ*は上記で定義した通りである)のいずれかに相当するキラルアミンと接触させることによって調製された塩によって表すことができる。
に関する。
に相当する第三ヘテロシクロアルカンカルボン酸であり、上記酸は、究極的には環化添加剤および/または脱水剤の存在下で非プロトン性またはプロトン性溶媒中で処理することによって得られ、上記処理は、究極的には形成された水を物理的に担持することによって支持されるとき、または
に相当する開環した第三エチレン酸であるとき、または
に相当する開環した第三エチレン酸であるとき、
R12を後で、ケン化により、または水素化分解により、または更に一般的には酸の保護基を除去するための当該技術分野で通常の方法によって除去する。
1°)n=0、およびR5は基−CH2CO2Hまたは−CH2CO2CH3ではない、
2°)n=0、およびR5は基−CH2CO2Hまたは−CH2CO2CH3であり、
R6=R8=メチルまたは基−CH2CO2Hまたは−CH2CO2CH3、
3°)n=2、同時にR6=R8=メチルおよびR5=OMeまたはヒドロキシル、
4°)n=2、同時にR6=R8=メチルおよびR5=基−CH2CO2Hまたは−CH2CO2CH3、またはメチル、
5°)n=3、同時にR6がヒドロキシル、およびR8=メチルおよびR5=基−CH2CO2CH3、
6°)n=3、同時にR6=R8=メチルおよびR5=OHまたはメチルまたはエチル
である化合物を除く、
第三ヘテロシクロアルカンカルボン酸であって、その塩およびその純粋な鏡像異性体のそれぞれを包含し、またはラセミ混合物または可変組成物でのもの、
第三オキサシクロアルカンカルボン酸であって、その塩およびその純粋な鏡像異性体のそれぞれを包含し、またはラセミ混合物または可変組成物でのものであり、 上記で定義した例外項目1〜6に相当する化合物を除くもの、
第三ヘテロシクロアルカンカルボン酸であって、その塩およびその純粋な鏡像異性体のそれぞれを包含し、またはラセミ混合物または可変組成物でのものであり、 Zが酸素原子でありかつ上記で定義した例外項目1〜6に相当する化合物を除くもの、
第三オキサシクロアルカンカルボン酸ヘミエステルであって、その塩およびその純粋な鏡像異性体のそれぞれを包含し、またはラセミ混合物または可変組成物でのものであり、
上記で定義した例外項目1〜6に相当する化合物を除くもの、
第三オキサシクロアルカンカルボン酸ヘミエステルであって、その塩およびその純粋な鏡像異性体のそれぞれを包含し、またはラセミ混合物または可変組成物でのものであり、
上記で定義した例外項目1〜6に相当する化合物を除くもの、
上記で定義した例外項目1〜6に相当する化合物を除くもの、
に相当する、第三アルケンカルボン酸であって、その塩およびその純粋な鏡像異性体のそれぞれを包含し、またはラセミ混合物または可変組成物でのもの、
に相当する、第三アルケンカルボン酸であって、その塩およびその純粋な鏡像異性体のそれぞれを含み、またはラセミ混合物または可変組成物でのもの、
に相当する、第三アルケンカルボン酸であって、その塩およびその純粋な鏡像異性体のそれぞれを含み、またはラセミ混合物または可変組成物でのもの、
に相当する第三アルケンカルボン酸であって、その塩およびその純粋な鏡像異性体のそれぞれを包含し、またはラセミ混合物または可変組成物でのもの、
式MeOCOO−のメトキシホルミルオキシ、
式CF3COO−のトリフルオロアセチルオキシ、
式RSO3−のアルキルスルホンオキシ、
式(RO)2PO−のホスホキシ、
式ROP(Cl)O−のハロホスホキシ、
式R3SiO−のトリアルキルシリルオキシ、
(式中、Rはアルキルである)
式
式
2,4,6−トリクロロベンゾイルオキシ
から選択される基である)の酸の混合無水物、
に相当する、環状無水物、
の製造でもある。
A)固定相:
アルキル−またはフェニル−またはアルキルフェニル−またはフェニルアルキル−シラン、好ましくはn−オクタデシルシラン、
B)移動相:
水−テトラヒドロフラン、水−メタノール、水−アセトニトリル、または水の代わりのpH2〜6.5の緩衝液、または同等な選択性を有する総ての他の移動相
の一つを用いて精製した。
ハリングトイド:この包括的用語は、非天然のハリングトニンであって、側鎖が少なくとも3個の炭素原子を有するエステル基であるものを表す。
ハリングトイドの側鎖:この包括的用語は、ヒドロキシル基の1個と、少なくとも3個の炭素原子を有するカルボン酸との間に形成したエステルであって、通常はカルボニル基に対して第三アルコールはα位にあり、疎水性置換体はω位にあるものを表す。
ビスホモ:2個の余分な炭素。
ノル:1個の炭素が少ない側鎖。
イソ:末端付近の炭素の位置にヒドロキシル基を有するメチレンを有する側鎖。
デオキシ:末端付近の炭素のヒドロキシル基が水素に置換されている。
アンヒドロ:2個の第三ヒドロキシル基が水の分子を失って、相当する飽和の酸素化ヘテロ環を生じる。
ネオ:R6が2−ヒドロキシイソプロピルの位置におけるフェニル基。
NMR:核磁気共鳴。
エチル=2−メトキシカルボニルメチル−2−ヒドロキシ−6−メチルヘプト−5−エノエートまたはエチル=6−デスオキシ−5,6−デヒドロホモハリングトネートの調製
5−ブロモ−2−メチル−ペント−2−エン(15.6g,95.6ミリモル)を、無水テトラヒドロフラン(75ml)にマグネシウム(2.32g,95.5ミリモル)(追加のヨウ素結晶で活性化)を混合攪拌したものに滴加した。反応の開始と同時に、反応混合物が激しく過熱し、還流した。マグネシウムのほとんどが反応してしまうまで還流を維持し、反応混合物を無水テトラヒドロフラン(150ml)で希釈した。シュウ酸ジメチル(10.8ml,80ミリモル)を無水テトラヒドロフラン(75ml)中で混合攪拌したものに、−78℃で生成するグリニャール試薬を20分間かけて加えた。攪拌を−78℃±5℃で30分間持続した後、温度を−10℃まで1.5時間かけて上げた。混合物を15%塩化アンモニウム溶液(300ml)で反応停止し、蒸発乾固した。水層をエーテル(2×300ml)で抽出した。有機層を濃縮物と合わせて、塩水(300ml)で洗浄し、硫酸マグネシウム上で乾燥し、蒸発乾固した。粗生成物を球入り蒸留装置(bulb-to-bulb distillation apparatus)で精製した後、無色油状生成物(10.3g,70%)を得た。中間体であるα−ケトエステルは、下記の特性を示した。
1HNMR 400MHz(CDCl3)(δppm,JHz):
5.08(1H,t,J=7.2,HC=),4.32(2H,q,J=7.1,OCH2);2.86(2H,t,J=7.2,CH2CO);2.32(2H,q,J=7.2,CH2−C=);1.68(3H,s,CH3);1.62(3H,s,CH3);1.37(3H,t,J=7.1,OCH2CH3)。
無水酢酸メチル(0.6ml,7.5ミリモル)を、1Mリチウム=ビス−(トリメチルシリルアミド)/テトラヒドロフランの攪拌市販溶液(7.5ml,7.5ミリモル)に−78℃で1分間かけて加え、これを−78±5℃で20分間反応させた。上記で調製したエチル=2−オキソ−6−メチルヘプト−5−エノエート(480mg,2.6ミリモル)を無水テトラヒドロフラン(10ml)に−78℃で混合攪拌したものに、リチウムエノレートを5分間かけて加え、生成する混合物を−78±5℃で30分間攪拌した。CCMで観察した後、冷凍浴を外し、混合物を15%塩化アンモニウム溶液(10ml)で反応停止した。分離した有機層を15%塩化アンモニウム溶液(10ml)で洗浄し、蒸発乾固した。水層をエーテル(2×10ml)で抽出した。有機層を濃縮物と合わせ、塩水(10ml)で洗浄し、硫酸マグネシウム上で乾燥して、蒸発乾固した。生成する粗生成物(1.13g)をカラムクロマトグラフィー(シクロヘキサン/酢酸エチル(95:5),シリカ(15〜40μm)38g)によって精製し、無色油状生成物(482mg,72%)を得た。このようにして得た生成物は、下記の特性を示した。
IR(ATR)(cm−1):2790;2916;1725;1068。
1HNMR 400MHz(CDCl3)(δppm,JHz):
5.05(1H,t,J=7.1,HC=),4.27(2H,q,J=7.1,OCH2);3.70(1H,s,OH);3.68(3H,s,OCH3);2.92および2.70(2H,2d,JAB=16.1,CH 2CO2);2.12(1H,m);1.88(1H,m);1.72(2H,m);1.67(3H,s,CH3);1.58(3H,s,CH3);1.31(3H,t,J=7.1,OCH2CH3)。
エチル=2−メトキシカルボニルメチル−2−ヒドロキシ−6−ジメチル−2−テトラヒドロフランカルボキシレートまたはまたはエチル=アンヒドロホモハリングトネートの調製
p−トルエンスルホン酸(2.06g,10.8ミリモル)を、例1で得たエチレン性エステル(2.8g,10.8ミリモル)をトルエン(30ml)に溶解攪拌したものに加え、生成混合物を65℃で5時間攪拌した。室温で冷却した後、混合物を飽和炭酸水素ナトリウム溶液で加水分解した。水層をエーテル(3×50ml)で抽出し、有機層を合わせて、塩水(100ml)で洗浄し、硫酸マグネシウム上で乾燥し、蒸発乾固した。生成する粗生成物(2.8g)をカラムクロマトグラフィー(シクロヘキサン/エーテル(95:5)、シリカ(15〜40μm110g)によって精製し、無色油状生成物(1.94g,69%)を得た。このようにして得た生成物は、下記の特性を示した。
1HNMR 400MHz(CDCl3)(δppm,JHz):
4.21(2H,m,OCH 2 CH3);3.64(3H,s,OCH3);2.85および2.60(2H,2d,JAB=14.0,CH 2 CO2);2.30(1H,dt,J=13.3および3.7);1.87(1H,qt,J=13.8および3.6);1.62(1H,m);1.51(2H,m);1.43(1H,m);1.31(3H,t,J=7.1,OCH2 CH 3 );1.22(3H,s,CH3);1.13(3H,s,CH3)。
2°)方法B
例1で得たエチレン性エステル(50mg,0.19ミリモル)をメタノール(30ml)に溶解攪拌したものに、1Nえんさん(0.5ml)を加え、生成混合物を65℃で15時間攪拌した。ジクロロメタンで希釈した後、有機層を硫酸マグネシウム上で乾燥し、蒸発乾固した。生成する粗生成物(32mg)をカラムクロマトグラフィー(ジクロロメタン、次いでジクロロメタン/メタノール(9:1),シリカ(15〜40μm)2.2g)によって精製し、予想した中間体ジオール(20mg,37%)を得た。このようにして得た生成物は、下記の特性を示した。
1HNMR 400MHz(CDCl3)(δppm,JHz):
4.28(2H,q,J=7.2,OCH2);3.75(1H,s,OH),3.68(3H,s,OCH3);2.93および2.69(2H,2d,JAB=16.2,CH 2 CO2);1.70(2H,m);1.53(1H,m);1.44(1H,m);1.30(3H,t,J=7.1,OCH2CH3);1.20(3H,s,CH3);1.19(3H,s,CH3)。
例1で得たエチレン性エステル(400mg,1.55ミリモル)をギ酸(4ml)と水(4ml)との混合物に溶解したものを、50℃で15時間攪拌した。ギ酸を真空留去した後、残渣を5%炭酸水素ナトリウム溶液で処理した。水層をジクロロメタン3回抽出した後、合わせた有機層を硫酸マグネシウム上で乾燥し、蒸発乾固した。生成する粗生成物(375mg)をカラムクロマトグラフィー(ジクロロメタン、次いでジクロロメタン/メタノール(98:2)、シリカ(15〜40μm)16g)によって精製し、無色油状生成物(235mg,55%)を得た。このようにして得た生成物は、方法Aで得たものと同じ特性を示した。このようにして得たジオールを上記の例2の方法Bと同様にして塩化亜鉛で環化し、方法Aで得たものと同じ特性を示す環状ジエステルを得た。
2−カルボキシメチル−2−ヒドロキシ−6−メチルヘプト−5−エン酸またはO−デメチル−6−デソキシホモハリングトン酸の調製
IR(ATR)(cm−1):3500;3019;2966;2931;1716;1691;1656;1219;1199;1111。
1HNMR 400MHz(CDCl3)(δppm,JHz):
5.06(1H,t,J=6.9,HC=),3.04および2.78(2H,2d,JAB=17.1,CH 2CO2);2.25−1.20(4H,m,2×CH2);1.67(3H,s,CH3);1.60(3H,s,CH3)。
2−カルボキシメチル−6,6−ジメチル−2−テトラヒドロピランカルボン酸またはO−デメチルアンヒドロホモハリングトン酸の調製
水酸化カリウム(4.2g,75ミリモル)を水(45ml)と混合したものを、例2で得た環状ジエステル(1.94g,7.5ミリモル)をエタノール(75ml)に溶解攪拌したものに加え、生成混合物を還流温度5時間攪拌した。室温で冷却し、エタノールを真空留去した後、残渣を水(10ml)で処理し、生成する水層をエーテル(2×50ml)で抽出した。2Nえんさん(35ml)で酸性にした後、水層を塩化ナトリウムで飽和した後、エーテル(3×50ml)で抽出した。合わせた有機層を塩水(2×100ml)で洗浄し、硫酸マグネシウム上で乾燥し、蒸発乾固して、淡黄色油状生成物(1.66g,98%)を得た。このようにして得た粗生成物は、下記の特性を示した。
IR(ATR)(cm−1):2974;2941;1709;1215。
1HNMR 400MHz(CDCl3)(δppm,JHz):
3.01および2.95(2H,2d,JAB=16.1,CH 2CO2);1.89(1H,m);1.75(2H,m,CH2);1.58(3H,m);1.31(6H,s,2×CH3)。
例3で得たエチレン性二酸(50mg,23ミリモル)を無水トルエン(500μl)に溶解攪拌したものに、塩化亜鉛(6mg,0.04ミリモル)を加え、生成混合物を80℃で15時間攪拌した。室温で冷却した後、混合物を10%塩酸で加水分解し、生成する水層を酢酸エチルで3回抽出した。合わせた有機層を硫酸マグネシウム上で乾燥して、蒸発乾固し、淡黄色固形生成物(38mg,76%)を得た。このようにして得た粗生成物は、方法Aで得たものと同じ特性を示した。
例3で得たエチレン性二酸(50mg,0.23ミリモル)をギ酸(500μl)と水(500μl)との混合物に溶解したものを、60℃で3時間攪拌した。室温で冷却し、ギ酸を真空留去した後、残渣を酢酸エチルで処理した。生成する有機層を10%塩酸で洗浄し、水層を酢酸エチルで3回抽出した。合わせた有機層を硫酸マグネシウム上で乾燥し、蒸発乾固して、淡黄色固形生成物(50mg,100%)を得た。このようにして得た粗生成物は、方法Aで得たものと同じ特性を示した。
2−メトキシカルボニルメチル−2−ヒドロキシ−6−メチルヘプト−5−エン酸または6−デソキシ−5,6−デヒドロホモハリングトン酸の調製
IR(ATR)(cm−1):3483;2954;1731;1197;1173。
1HNMR 400MHz(CDCl3)(δppm,JHz):
5.06(1H,m,HC=),4.12(2H,brs,CO2H+OH);3.73(3H,s,OCH3);2.99および2.74(2H,2d,JAB=16.7,CH 2CO2);2.16(1H,m);1.98(1H,m);1.85〜1.60(4H,m);1.67(3H,s,CH3);1.60(3H,s,CH3)。
2−メトキシカルボニルメチル−6,6−ジメチル−2−テトラヒドロピランカルボン酸またはアンヒドロホモハリングトン酸の調製
例4で得た環状二酸(1.6mg,7.4ミリモル)と、三フッ化ホウ素−メタノール複合体をメタノールに溶解した市販溶液(15.5ml,BF312%w/w)との混合物を、18±5℃で15時間攪拌した。反応混合物を飽和炭酸水素ナトリウム溶液(50ml)に注意しながら加えた後、生成する水層をエーテル(2×50ml)で洗浄し(下記の添付調製を参照)、2N塩酸(15ml)で酸性にし(pH1)、エーテル(3×75ml)で抽出した。合わせた有機層を硫酸マグネシウム上で乾燥し、蒸発乾固して、黄色油状生成物(1.17g,69%)を得た。このようにして得た粗生成物は、下記の特性を示した。
IR(ATR)(cm−1):2974;2951;1740;1718;1437。
1HNMR 400MHz(CDCl3)(δppm,JHz):
3.70(3H,s,OCH3);3.03および2.98(2H,2d,JAB=16.1,CH 2CO2);1.82(1H,m);1.74(3H,m);1.62(1H,m);1.48(1H,m);1.31(3H,s,CH3);1.26(3H,s,CH3)。
a)ジエステルの調製
1HNMR 400MHz(CDCl3)(δppm,JHz):
3.75(3H,s,OCH3);3.65(3H,s,OCH3);2.85および2.61(2H,2d,JAB=14.1,CH 2CO2);1.85(1H,m);1.62(1H,m);1.50(2H,m);1.43(1H,m);1.21(3H,s,CH3);1.11(3H,s,CH3)。
IR(ATR)(cm−1):3421;2960;2929;1744;1705;1209
1HNMR 400MHz(CDCl3)(δppm,JHz):
3.76(3H,s,OCH3);2.76および2.67(2H,2d,JAB=15.3,CH 2CO2);2.36(1H,m,JAB=13.7,J3−4=3.5,J3−5=1.2,H−3eq);1.85(1H,m,JAB〜Jax−ax=14.0,Jax−eq=3.7,H−4ax);1.67(1H,m,JAB=14.1,J4−3,5=3.9,H−4eq);1.59(1H,m,JAB=13.4,J5−4=3.6,J5−3=1.0,H−5eq);1.49(1H,m,JAB〜Jax−ax=13.2,Jax−eq=4.0,H−3ax);1.42(1H,m,JAB〜Jax−ax=13.2,Jax−eq=4.5,H−5ax);1.33(3H,s,CH3);1.16(3H,s,CH3)。
例5で得たエチレン性ヘミエステル(4.6g,20ミリモル)をトルエン(125ml)に溶解攪拌したものに、p−トルエンスルホン酸(3.8g,20ミリモル)を加え、生成する混合物を65℃で5時間攪拌した。室温で冷却した後、混合物を飽和炭酸水素ナトリウム溶液(100ml)で加水分解した。水層をエーテル(2×100ml)で洗浄し、有機層を廃棄した(反応の生成するジエステルを除去するため)。1N塩酸(35ml)で酸性(pH1)にした後、水層を塩化ナトリウムで飽和した後、エーテル(3×100ml)で抽出した。合わせた有機層を塩水(100ml)で洗浄し、硫酸マグネシウム上で乾燥して、蒸発乾固した。
2−カルボキシメチル−2−ヒドロキシ−6−メチルヘプト−5−エン酸の環状無水物の調製
IR(ATR)(cm−1):2976;1732;1188;1170。
1HNMR 400MHz(CDCl3)(δppm,JHz):
3.02(2H,s,CH 2CO2);1.98(2H,m,CH2);1.8−1.5(4H,m,CH2);1.31(3H,s,CH3);1.22(3H,s,CH3)。
(−)−セファロタキシルピバレートの調製
ピバリン酸(100mg,0.98ミリモル)を無水トルエン(2ml)と混合攪拌したものに、室温でトリエチルアミン(水酸化カリウム上で乾燥)(138μl,0.98ミリモル)および2,4,6−トリクロロベンゾイルクロリド(153μl,0.98ミリモル)を加えた。(赤外で出発酸の消失を制御しながら)18±5℃で1.5時間攪拌した後、4−ジメチルアミノピリジン(139mg,1.14ミリモル)を加え、反応混合物を5分間反応させ、セファロタキシン(103mg,0.33ミリモル)を加えた。18±5℃で15時間攪拌した後、反応混合物を濾紙で濾過し、エーテル(5ml)で希釈した。生成する有機層を、水(5ml)、飽和炭酸水素ナトリウム溶液(5ml)および再度水(5ml)で連続的に洗浄した後、硫酸マグネシウム上で乾燥し、真空留去した。生成する粗生成物をカラムクロマトグラフィー(ジクロロメタン/メタノール(98:2),シリカ(15〜40μm))によって精製し、固形生成物(130mg,93%)を得た。このようにして得た生成物は、下記の特性を示した。
1HNMR 400MHz(CDCl3)(δppm,JHz):
6.60(1H,s,H−17*);6.58(1H,s,H−14*);5.84および5.83(2H,2d,JAB=1.5,OCH2O);5.83(1H,d,H−3);5.02(1H,s,H−1);3.77(1H,d,J4−3=9.6,H−4);3.69(3H,s,OCH3);3.21(1H,m,JAB=14.0,J=12.5,7.8,H−11b);3.09(1H,m,H−8a);2.94(1H,td,J=11.5,7.1,H−10a);2.57(2H,m,H−8b+H−10b);2.35(1H,dd,JAB=14.5,J=6.9,H−11a);2.03(1H,td,JAB=12.1,J=9.7,H−6A);1.89(1H,m,JAB=12.1,J=7.9,4.0,H−6B);1.75(2H,m,CH2−7);0.83(9H,s,C(CH3)3)。
不活性雰囲気中に保持されたピバリン酸(50mg,0.49ミリモル)と無水トルエン(2ml)とを混合攪拌したものに、1,3−ジクロロヘキシルカルボジイミド(130mg,0.63ミリモル)を加えた。室温で10分間攪拌した後、セファロタキシン(50mg,0.16ミリモル)およびピロリジノピリジン(24mg,0.16ミリモル)を加えた。(CCM、溶離液ジクロロメタン/メタノール;9:1で反応を制御しながら)18±5℃で2時間攪拌し、50℃で15時間攪拌した後、反応混合物を磨りガラスフィルターで濾過し、ケーキをトルエン(5ml)で洗浄し、濾液を真空留去した。生成する粗生成物(130mg)をカラムクロマトグラフィー(ジクロロメタン/メタノール(9:1),シリカ(15〜40μm)3g)によって精製し、白色固形生成物(36mg,57%)を得た。このようにして得た粗生成物は、混合無水物により上記で得たものと同じ特性を示した。
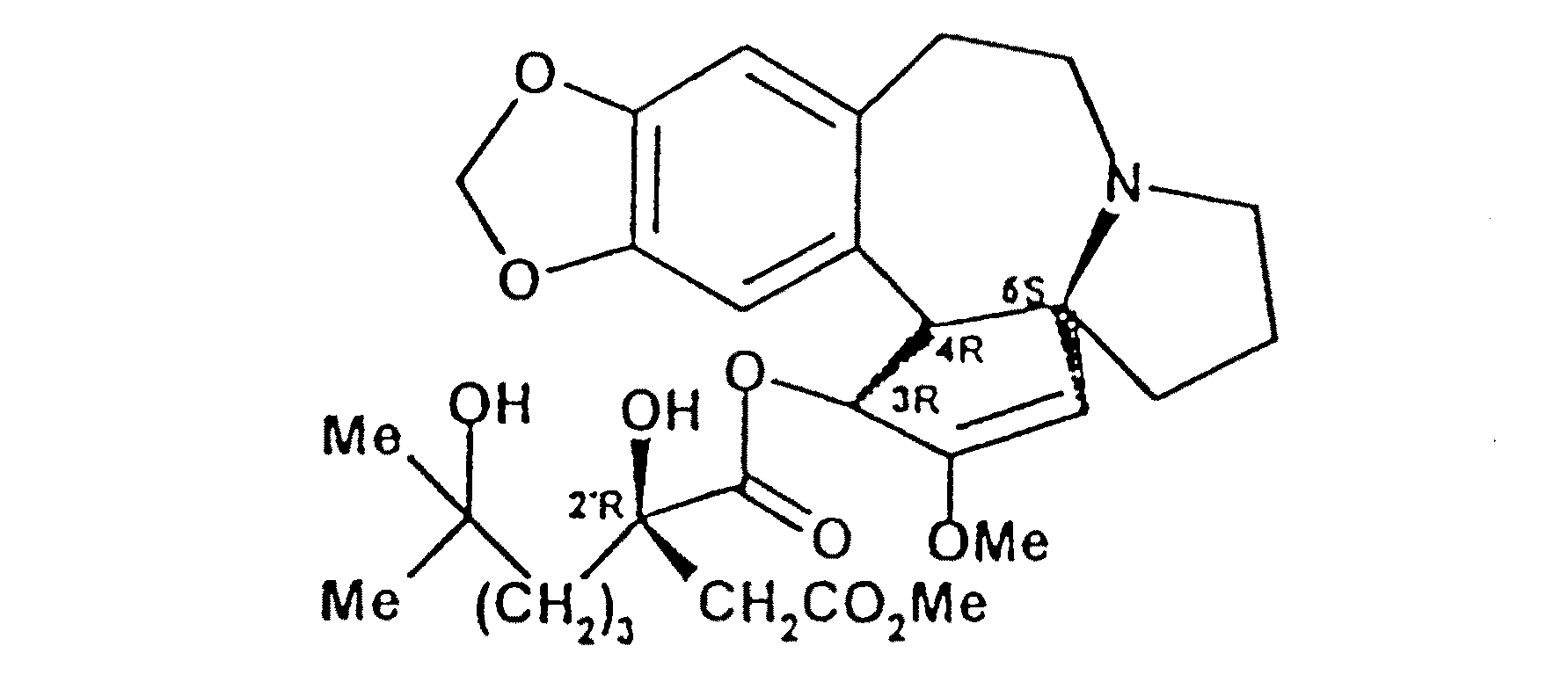
例7で得た環状アンハイドライドから(−)−セファロタキシル=2−メトキシカルボニルメチル−6,6−ジメチル−2−テトラヒドロピランカルボキシレートまたはアンヒドロホモハリングトニンおよびメチル=2−セファロタキシル−カルボニルメチル−6,6−ジメチル−2−テトラヒドロピランカルボキシレートの調製
1HNMR 400MHz(CDCl3)(δppm,JHz):
6.61(1H,s,H−17*);6.57(1H,s,H−14*);5.91(J3−4=9.8)および5.84(2H,2d,H−3);5.84および5.79(2d,JAB=1.4,OCH2O);5.84および5.82(2d,JAB=1.4,OCH2O);5.04および5.01(1H,2s,H−1);3.79および3.78(1H,2d,J4−3=9.6,H−4);3.70および3.65(3H,2s,OCH3);3.59(3H,s,OCH3);3.15(1H,m,H−11β);3.09(1H,m,H−8α);2.94(1H,m,H−10α);2.58(2H,m,H−8β+H−10β);2.37(1H,m,H−11α);2.16および1.81(2d,JAB=14.4,CH 2CO2);2.13および1.66(2d,JAB=14.3,CH2CO2);2.02(1H,m,H−6A);1.88(1H,m,H−6B);1.75(2H,m,CH2−7);1.8〜1.2(6H,m,3×CH2);1.11および1.02(2s,2×CH3);1.10および1.04(2s,2×CH3)。
例6の方法Cで得たヘミエステル(100mg,0.43ミリモル)を室温で不活性雰囲気中に保持した無水トルエン(1ml)と混合攪拌したものに、1,3−ジシクロヘキシルカルボジイミド(120mg,0.58ミリモル)を加えた。5分間攪拌した後、セファロタキシン(45mg,0.15ミリモル)およびピロリジノピリジン(21mg,0.14ミリモル)を加えた。(CCM、溶離液ジクロロメタン/メタノール;9:1で反応を制御しながら)35℃で45分間、次いで8℃で15時間攪拌した後、反応混合物を濾過し、ケーキをトルエン(5ml)で洗浄し、濾液を真空留去した。生成する粗生成物をカラムクロマトグラフィー(ジクロロメタン/メタノール(98:2),シリカ(15〜40μm)4g)によって精製し、予想生成物(23mg,30%,2種類のジアステレオ異性体)を得た。このようにして得た生成物は、下記の特性を示した。
1HNMR 400MHz(CDCl3)(δppm,JHz):
6.61および6.58(1H,2s,H−17*);6.57および6.53(1H,2s,H−14*);5.89および5.86(2d,JAB=1.5,OCH2O);5.76(1H,d,J3−4=9.4,H−3);5.02(1H,2s,H−1);3.73および3.72(1H,2d,J4−3=9.4,H−4);3.70および3.68(3H,2s,OCH3);3.69および3.65(3H,2s,OCH3);3.15(1H,m,H−11α);3.07(1H,m,H−8α);2.90(1H,m,H−10α);2.74および1.95(2d,JAB=15.3,CH 2CO2);2.56(2H,m,H−8β+H−10β);2.33(1H,m,H−11α);2.28および2.23(2d,JAB=15.4,CH 2CO2);2.16(m,H−3′eq);1.97(1H,m,H−6A);1.9〜1.1(5H,m,CH2);1.86(1H,m,H−6B);1.73(2H,m,CH2−7);1.14(3H,s,CH3);1.03(3H,s,CH3)。
例6で得たテトラヒドロピランカルボン酸から(−)−セファロタキシル=2−メトキシカルボニルメチル−6,6−ジメチル−2−テトラヒドロピランカルボキシレートまたはアンヒドロホモハリングトニンの調製
例9の式
1°)混合無水物による方法
例6で得たヘミエステル(50mg,0.22ミリモル)を無水トルエン(1ml)と室温で混合攪拌したものに、トリエチルアミン(水酸化カリウム上で乾燥)(29.4μl,0.22ミリモル)および2,4,6−トリクロロベンゾイルクロリド(32.7μl,0.22ミリモル)を加えた。(赤外で出発酸の消失を制御しながら)25℃で20時間攪拌した後、4−ジメチルアミノピリジン(29mg,0.24ミリモル)を加え、反応混合物を5分間反応させ、セファロタキシン(16.5mg,0.05ミリモル)を加えた。24℃で24時間攪拌した後、反応混合物を濾紙で濾過し、エーテル(5ml)で希釈した。生成する有機層を、水(5ml)、飽和炭酸水素ナトリウム溶液(5ml)および再度水(5ml)で連続的に洗浄した後、硫酸マグネシウム上で乾燥し、真空留去した。生成する粗生成物をカラムクロマトグラフィー(ジクロロメタン/メタノール(98:2),シリカ(15〜40μm))によって精製し、予想生成物(16mg,56%,2種類のジアステレオ異性体)を得た。このようにして得た生成物は、例9で得たものと同じ特性を示した。
例6で得たヘミエステル(100mg,0.43ミリモル)を室温で不活性雰囲気中に保持した無水トルエン(1ml)と混合攪拌したものに、1,3−ジクロロヘキシルカルボジイミド(180mg,0.87ミリモル)を加えた。10分間攪拌した後、セファロタキシン(165mg,0.52ミリモル)およびピロリジノピリジン(77mg,0.52ミリモル)を加えた。18±5℃で18時間攪拌した後、エーテルを加え、反応混合物を磨りガラスフィルターで濾過し、ケーキをエーテルで洗浄した。生成する有機層を10%炭酸水素ナトリウム溶液、水で連続して洗浄した後、硫酸マグネシウム上で乾燥し、真空留去した。生成する粗生成物をカラムクロマトグラフィー(ジクロロメタン/メタノール(98:2),シリカ(15〜40μm)9g)によって精製し、固形生成物(110mg,48%)を得た。このようにして得た粗生成物は、例9で得たものと同じ特性を示した。
例6で得たエチレン酸から(−)−セファロタキシル=(2′RS)−2−メトキシカルボニルメチル−6,6−ジメチル−2−テトラヒドロピランカルボキシレートまたはアンヒドロホモハリングトニンの調製
例9の式
1°)方法A:混合無水物による環化カップリング
例5で得たエチレン性エステル(50mg,0.22ミリモル)を無水トルエン(1ml)と室温で混合攪拌したものに、トリエチルアミン(水酸化カリウム上で乾燥)(29.4μl,0.22ミリモル)および2,4,6−トリクロロベンゾイルクロリド(34μl,0.22ミリモル)を加えた。(赤外で出発酸の消失を制御しながら)30分間攪拌した後、4−ジメチルアミノピリジン(30mg,0.25ミリモル)を加え、反応混合物を5分間反応させ、セファロタキシン(31mg,0.1ミリモル)を加えた。18±5℃で65時間攪拌した後、反応混合物を濾紙で濾過し、エーテル(5ml)で希釈した。生成する有機層を、水(5ml)、飽和炭酸水素ナトリウム溶液(5ml)および再度水(5ml)で連続的に洗浄した後、硫酸マグネシウム上で乾燥し、真空留去した。生成する粗生成物をカラムクロマトグラフィー(ジクロロメタン/メタノール(98:2),シリカ(15〜40μm))によって精製し、予想生成物(46mg,96%,2種類のジアステレオ異性体40/60)を得た。このようにして得た生成物は、例9で得たものと同じ特性を示した。
例5で得たエチレン酸(50mg,0.22ミリモル)を室温で不活性雰囲気中に保持した無水トルエン(2ml)と混合攪拌したものに、1,3−ジクロロヘキシルカルボジイミド(270mg,1.31ミリモル)を加えた。5分間攪拌した後、セファロタキシン(70mg,0.22ミリモル)およびピロリジノピリジン(32mg,0.22ミリモル)を加えた。(CCM、溶離液ジクロロメタン/メタノール;9:1で反応を制御しながら)18±5℃で65時間攪拌した後、反応混合物を磨りガラスフィルターで濾過し、ケーキをトルエン(5ml)で洗浄し、濾液を真空留去した。生成する粗生成物をカラムクロマトグラフィー(ジクロロメタン/メタノール(98:2),シリカ(15〜40μm)9g)によって精製し、固形生成物(40mg,35%)を得た。このようにして得た粗生成物は、例9で得たものと同じ特性を示した。
イヌガヤ種の全アルカロイド抽出物からの精製(−)−セファロタキシンの調製
30リットルタンクで、イヌガヤ種(10kg)の圧潰した葉(新鮮なものまたは乾燥物)をメタノール(20リットル)と混合し、65時間スチーム処理した後、浸出した(50リットル)。溶液を濾過し、容積が5リットルになるまで真空濃縮した。濃縮溶液を、6%酒石酸水溶液で酸性にした。次いで、水−アルコール溶液をジクロロメタン(5×5リットル)で洗浄し、脂肪性物質と色素を除去した。水溶液をアンモニア水溶液(2.5%)でpH9まで塩基性にした後、ジクロロメタン(5×5リットル)で抽出した。減圧濃縮した後、粗製のアルカロイド抽出物が白色結晶性固形生成物(24.5g)として回収された。セファロタキシン含量は、71%であった(HPLC)。
上記の粗製アルカロイドを、移動相(トリエチルアミン(1.55/1000)/脱イオン水およびオルトリン酸に溶解し、pH3に調整した。次に、溶液を、アキシャル・コンプレッションおよび高圧ポンプを備えた合成用高性能液体クロマトグラフィー装置(固定相:n−オクタデシルシラン、15μm、多孔度100、1kg)に注入した。溶出は、0.2リットル/分の流速で行った。画分の含量は、UV検出器およびTLCによって観察した。保持画分を最後にHPLCでチェックした後、合わせて、2.5%アンモニア水溶液でアルカリ性にして、ジクロロメタン(4×400ml)で抽出した。減圧濃縮の後、樹脂状生成物が得られ、これをメタノールで粉砕したところ、(−)−セファロタキシン(18g)を白色結晶性固形生成物(HPLC純度=99.8%)として得た。このようにして得た生成物は、下記の特性を示した。
[αD 20]:−174.1(c=0.20;CHCl3)。
1HNMR 400MHz(CDCl3)(δppm,JHz):
6.68(1H,s,H−17*);6.65(1H,s,H−14*);5.91および5.90(2H,2d,JAB=1.5,OCH2O);4.93(1H,s,H−1);4.77(1H,dd,J3−4=9.4,J3−OH=3.4,H−3);3.73(3H,s,OCH3);3.68(1H,d,J4−3=9.4,H−4);3.35(1H,m,JAB=14.3,J=12.2および7.9,H−11β);3.08(1H,m,J=9.1および4.9,H−8α);2.92(1H,td,J=11.6および7.1,H−10α);2.59(2H,H−8α+H−10α);2.35(1H,dd,JAB=14.4,J=6.9,H−11α);2.02(1H,td,JAB=12.1,J=9.7,H−6A);1.87(1H,m,JAB=12.1,J=7.9および4.4,H−6B);1.74(2H,m,CH2−7);1.62(1H,d,J3−OH=3.5,3−OH)。
(−)−セファロタキシンのリチウムアルコレートの調製(3−O−アセチル誘導体のようにトラッピング)
1°)ブチルリチウム法
ブチルリチウムをヘキサンに溶解した市販溶液(0.44ml,1.6Mヘキサン溶液,0.70ミリモル)を、(−)−セファロタキシン(200mg,0.63ミリモル)を無水テトラヒドロフラン(6.8ml)に混合攪拌したものに加えた。反応混合物を−60℃で20分間、次いで−48℃で30分間保持し、無水酢酸(90μl,.095ミリモル)を8分間かけて加え、攪拌を−48℃で20分間、次いで0℃で1時間保持した。混合物を飽和塩化アンモニウム溶液(5ml)で反応停止した後、酢酸エチル(3×8ml)で抽出した。合わせた有機層を塩水(15ml)で洗浄し、硫酸マグネシウム上で乾燥し、蒸発乾固した。生成する粗生成物をカラムクロマトグラフィー(ジクロロメタン/メタノール(98:2),シリカ(15〜40μm)6g)によって精製し、白色固形生成物(60mg,26%)を得た。このようにして得た生成物は、下記の特性を示した。
1HNMR 400MHz(CDCl3)(δppm,JHz):
6.60(1H,s,H−17*);6.57(1H,s,H−14*);5.89および5.86(2H,2d,JAB=1.4,OCH2O);5.80(1H,d,J3−4=9.3,H−3);5.05(1H,s,H−1);3.77(1H,d,J4−3=9.4,H−4);3.72(3H,s,OCH3);3.23(1H,m,JAB=14.3,J=12.3および7.9,H−11β);3.08(1H,m,H−8α);2.92(1H,td,J=11.5および7.1,H−10α);2.57(2H,m,H−8β+H−10β);2.36(1H,dd,JAB=14.4,J=7.0,H−11α);2.02(1H,td,JAB=12.1,J=9.7,H−6A);1.88(1H,m,JAB=12.1,J=8.0および4.0,H−6B);1.74(2H,m,CH2−7);1.57(3H,s,OAc)。
1Mリチウムビス−(トリメチルシリル)アミドをテトラヒドロフラン(0.95ml,0.95ミリモル)に溶解した市販溶液を、(−)−セファロタキシン(200mg,0.63ミリモル)を無水テトラヒドロフランに−40℃で溶解攪拌したものに加えた。5分間攪拌した後、無水酢酸(90μl,0.95ミリモル)を加え、反応混合物を1°)に記載した方法と同様に処理した。このようにして得た生成物は、上記のブチルリチウム法で得たのと同じ特性を示した。
2Mリチウムジイソプロピルアミドをテトラヒドロフラン(0.35ml,0.70ミリモル)に溶解した市販溶液を、(−)−セファロタキシン(200mg,0.63ミリモル)を無水テトラヒドロフラン(6.8ml)に−60℃で溶解攪拌したものに20分間かけて加えた。−60℃で20分間、−48℃で30分間攪拌した後、無水酢酸(90μl,0.95ミリモル)を加えた。溶液を−48℃20分間、次いで0℃で1時間攪拌し、反応混合物を上記の1°)の方法と同様にこのようにして得た生成物は、上記のブチルリチウム法で得たのと同じ特性を示した。
水素化ナトリウム(1.5g)を新たに蒸留したジメチルホルムアミド(3ml)と混合攪拌したものに、−60℃でセファロタキシン(200mg,0.63ミリモル)をジメチルホルムアミド(3ml)および無水酢酸(90μl,0.95ミリモル)に溶解したものに加えた。周囲温度で24時間攪拌した後、反応混合物を0℃で水(3ml)で処理し、エーテル(3×5ml)で抽出した。合わせた有機層を硫酸マグネシウム上で乾燥し、真空留去した。このようにして得た生成物は、上記のブチルリチウム法で得たのと同じ特性を示した。
セファロタキシンのリチウムアルコレートを用いる(−)−セファロタキシル=2−メトキシカルボニルメチル−6,6−ジメチル−2−テトラヒドロピランカルボキシレートまたはアンヒドロホモハリングトニンの調製
例9の式
例13に従って調製した(−)−セファロタキシンのリチウムアルコレート(158mg,0.5ミリモル)を無水テトラヒドロフランに溶解攪拌したものに、例10で得た混合無水物(0.75ミリモル)を−50℃で10分間かけて加えた。−50℃で30分間、次いで0℃で2時間攪拌した後、反応混合物を飽和塩化アンモニウム溶液(5ml)で反応停止し、酢酸エチル(3×10ml)で抽出した。合わせた有機層を塩水(15ml)で洗浄し、硫酸マグネシウム上で乾燥し、真空留去した。生成する粗生成物をカラムクロマトグラフィー(ジクロロメタン/メタノール(98:2),シリカ(15〜40μm)7g)によって精製し、白色固形生成物(48mg)を得た。このようにして得た生成物は、例9で得たものと同じ特性を示した。
例6で得たテトラヒドロピランカルボン酸から(−)−キニジル=2−メトキシカルボニルメチル−6,6−ジメチル−2−テトラヒドロピランカルボキシレートのジアステレオ異性体混合物の調製
例6で得た酸(458mg,1.99ミリモル)を無水トルエン(8ml)と室温で混合攪拌したものに、トリエチルアミン(水酸化カリウム上で乾燥)(270μl,1.92ミリモル)および2,4,6−トリクロロベンゾイルクロリド(300μl,1.91ミリモル)を加えた。(赤外で出発酸の消失を制御しながら)3時間攪拌した後、4−ジメチルアミノピリジン(352mg,2.88ミリモル)を加え、反応混合物を5分間反応させ、キニン(936mg,2.88ミリモル)を加えた。18±5℃で65時間攪拌した後、反応混合物を濾紙上で濾過し、エーテル(15ml)で希釈した。生成する有機層を、水(5ml)、飽和炭酸水素ナトリウム溶液(15ml)、再度水(15ml)で連続して洗浄した後、硫酸マグネシウム上で乾燥し、真空留去した。生成する粗生成物をカラムクロマトグラフィー(ジクロロメタン/メタノール(99:1),シリカ(15〜40μm)32g)によって精製し、予想生成物(930mg,84%,2種類のジアステレオ異性体50/50)を得た。このようにして得た生成物は、下記の特性を示した。
1HNMR 400MHz(CDCl3)(δppm,JHz):
8.73(1H,m,H−2qn);8.0および7.98(1H,2d,J=9.2,H−8qn);7.63および7.50(1H,2brs);7.45(brs)および7.39(d,J=4.5)(1H,H−3qn);7.36(1H,dd,J=9.1および3.97(3H,2s,OCH3);3.54および3.33(3H,2brs,OCH3);3.2〜1.0(m,7×CH2+3CH);2.92および2.67(2d,JAB=14.9,CH 2CO2);2.87(d,JAB=14.8,CH 2CO2);1.17および0.99(2s,2×CH3);1.03および0.42(2brs,2×CH3)。
例6で得たテトラヒドロカルボン酸(200mg,0.87ミリモル)を無水トルエン(4ml)と混合攪拌して室温で不活性雰囲気に保持したものに、1,3−ジクロロヘキシルカルボジイミド(239mg,1.16ミリモル)を加えた。5分間攪拌した後、キニン(94mg,0.29ミリモル)およびピロリジノピリジン(43mg,0.29ミリモル)を加えた。(CCM、溶離液ジクロロメタン/メタノール;9:1で反応を制御しながら)18±5℃で65時間攪拌した後、反応混合物を磨りガラスフィルター上で濾過し、ケーキをトルエン(5ml)で洗浄し、濾液を真空留去した。生成する粗生成物をカラムクロマトグラフィー(ジクロロメタン/メタノール(9:1),シリカ(15〜40μm))によって精製し、予想生成物(96mg,60%,2種類のジアステレオ異性体50/50)を得た。このようにして得た生成物は、上記で得たのと同じ特性を示した。
例6で得たテトラヒドロピラン−カルボン酸から(−)−メンチル=2−メトキシカルボニルメチル−6,6−ジメチル−2−テトラヒドロ−ピランカルボキシレートの調製
1HNMR 400MHz(CDCl3)(δppm,JHz):
4.68(1H,m,H−1men);3.64(3H,s,OCH3);2.84および2.64(2d,JAB=14.6,CH 2CO2);2.83および2.63(2d,JAB=14.3,CH 2CO2);2.29(1H,m,H−3eq);2.1〜0.8(m,CHおよびCH2);1.21(3H,2s,CH3);1.17および1.16(3H,2s,CH3);0.9および0.88(6H,2d,J=6.4,2×CH3men);0.74および0.72(3H,2d,J=6.8,CH3men)。
例6で得たテトラヒドロピランカルボン酸から(−)−メチルマンデレート=2−メトキシカルボニルメチル−6,6−ジメチル−2−テトラヒドロピランカルボキシレートの調製
1HNMR 400MHz(CDCl3)(δppm,JHz):
7.47(2H,m,Ph);7.38(3H,m,Ph);5.96(1H,s,CH);3.73および3.72(3H,2s,OCH3);3.54(3H,2s,OCH3);2.88および2.72(2d,JAB=14.4,CH 2CO2);2.85および2.65(2d,JAB=14.2,CH 2CO2);2.35(1H,m,H−3eq);2.0〜1.15(5H,m,CH2);1.23および1.22(3H,2s,CH3);1.19および1.07(3H,2s,CH3)。
例15で得たジアステレオ異性体混合物から(−)−キニル=(2′R)−アンヒドロホモハリングトネートおよび(−)−キニル=(2′S)−アンヒドロホモハリングトネートの分離
(−)−キニル=(2′R)−アンヒドロホモハリングトネートと(−)−キニル=(2′S)−アンヒドロホモハリングトネートとのジアステレオ異性体混合物(5g)に、合成用HPLCを施した。上記混合物を、緩衝液(トリエチルアミン(1.55/1000)/脱イオン水およびオルトリン酸に溶解し、pH3に調整した。次に、溶液を、アキシャル・コンプレッションおよび高圧ポンプを備えた合成用高性能液体クロマトグラフィー装置(固定相:n−オクタデシルシラン、15μm、多孔度100、1kg;移動相:緩衝液/アセトニトリル70/30)に注入した。溶出は、0.2リットル/分の流速で行った。画分の含量は、UV検出器およびTLCによって観察した。保持画分を最後にHPLCでチェックした後、合わせて、2.5%アンモニア水溶液でアルカリ性にして、ジクロロメタン(4×400ml)で抽出した。減圧濃縮の後、2種類の分離した異性体が、(−)−キニル=(2′R)−アンヒドロホモハリングトネート(2g)と(−)−キニル=(2′S)−アンヒドロホモハリングトネート(2.2g)に相当する白色結晶性固形生成物として得られた。このようにして得た生成物は、下記の特性を示した。
1HNMR 400MHz(CDCl3)(δppm,JHz):
8.73(1H,d,J=4.4,H−2qn);8.0(1H,d,J=9.2,H−8qn);7.50(1H,brs);7.39(1H,d,J=4.5,H−3qn);7.36(1H,dd,H−7qn);6.39(1H,brs);5.88(1H,m,=CHqn);5.03(2H,m,=CH2qn);3.97(3H,s,OCH3);3.31(3H,brs,OCH3);3.5〜1.2(m,7×CH2+3CH);2.86および2.64(2H,2d,JAB=15.0,CH 2CO2);1.17(3H,s,CH3);0.99(3H,s,CH3)。
1HNMR 400MHz(CDCl3)(δppm,JHz):
8.74(1H,d,J=4.4,H−2qn);7.99(1H,d,J=9.2,H−8qn);7.65(1H,brs,H−3qn);7.44(1H,brs,H−5qn);7.36(1H,dd,J=9.2および2.7,H−7qn);6.55(1H,brs);5.89(1H,m,=CHqn);5.05(2H,m,=CH2qn);3.99(3H,s,OCH3);3.54(3H,s,OCH3);3.1〜1.0(m,7×CH2+3CH);2.91および2.67(2H,2d,JAB=15.0,CH 2CO2);1.03(3H,brs,CH3);0.44(3H,brs,CH3)。
例18で得た(−)−キニル=(2′R)−アンヒドロホモハリングトネートから(2R)−アンヒドロホモハリングトン酸の調製
(−)−キニル=(2′R)−アンヒドロホモハリングトネート(100mg,0.19ミリモル)を酢酸エチル(11ml)に溶解攪拌したものに、10%パラジウム/炭(40mg)を加えた。生成する混合物を室温で水素圧下(50psi)20時間攪拌し、CCM制御の後、反応混合物を濾過して、生成する有機層を飽和炭酸水素ナトリウム溶液で処理した。水層を酢酸エチルで洗浄し、1N塩酸で酸性にした後、酢酸エチルで抽出した。合わせた有機層を塩水で洗浄し、硫酸マグネシウム上で乾燥し、蒸発乾固して、黄色固形生成物(20mg,50%)を得た。このようにして得た生成物は、下記の特性を示した。
[α]D 20:−23(c=0.38;CHCl3)。
IR(NaClフィルム)(cm−1):2974;2951;1740;1718;1437。
1HNMR 400MHz(CDCl3)(δppm,JHz):このようにして得た生成物の1HNMRスペクトルは、例6−1で記載したものと同じであった。
水酸化カリウム(396mg,7.1ミリモル)を水(8ml)と混合したものを、キニル=(2′R)−アンヒドロホモハリングトネート(396g,0.72ミリモル)をエタノール(15ml)に溶解攪拌したものに加え、生成する混合物を還流温度で24時間攪拌した。室温で冷却して、エタノールを真空留去した後、残渣を水(10ml)で処理し、生成する水層をエーテル(4×15ml)で抽出した。2N塩酸で酸性(pH1)にし、塩化ナトリウムで飽和した後、水層を酢酸エチル(3×15ml)で抽出した。合わせた有機層を硫酸マグネシウム上で乾燥し、蒸発乾固して、黄色固形生成物(110mg,72%)を得た。このようにして得た中間体二酸は、下記の特性を示した。
[α]D 20:−14(c=0.54;CHCl3)。
IR(NaClフィルム)(cm−1):2975;2941;1716;1217。
1HNMR 400MHz(CDCl3)(δppm,JHz):このようにして得た生成物の1HNMRスペクトルは、例4で記載したものと同じであった。
例18から得た(−)−キニル=(2′R)−アンヒドロホモハリングトネートから(2S)−アンヒドロホモハリングトン酸の調製
キニル=(2′S)−アンヒドロホモハリングトネート(100mg,0.19ミリモル)を酢酸エチル(11ml)に溶解攪拌したものに、10%パラジウム/炭(40mg)を加えた。生成する混合物を室温で水素圧下(50psi)で攪拌し、CCM制御の後、反応混合物を濾過して、生成する有機層を飽和炭酸水素ナトリウム溶液で処理した。水層を酢酸エチルで洗浄し、1N塩酸で酸性にした後、酢酸エチルで抽出した。合わせた有機層を塩水で洗浄し、硫酸マグネシウム上で乾燥し、蒸発乾固して、黄色固形生成物(23mg,53%)を得た。このようにして得た生成物は、下記の特性を示した。
[α]D 20:+30(c=0.36;CHCl3)。
IR(NaClフィルム)(cm−1):2975;2951;1740;1718;1439。
1HNMR 400MHz(CDCl3)(δppm,JHz):このようにして得た生成物の1HNMRスペクトルは、例6−1で記載したものと同じであった。
水酸化カリウム(430mg,7.7ミリモル)を水(9ml)と混合したものを、キニル=(2′S)−アンヒドロホモハリングトネート(447g,0.81ミリモル)をエタノール(16ml)に溶解攪拌したものに加え、生成する混合物を還流温度で24時間攪拌した。室温で冷却して、エタノールを真空留去した後、残渣を水(10ml)で処理し、生成する水層をエーテル(4×15ml)で抽出した。2N塩酸で酸性(pH1)にし、塩化ナトリウムで飽和した後、水層を酢酸エチル(3×15ml)で抽出した。合わせた有機層を硫酸マグネシウム上で乾燥し、蒸発乾固して、黄色固形生成物(140mg,80%)を得た。このようにして得た環状二酸は、下記の特性を示した。
[α]D 20:+8(c=0.19;CHCl3)。
IR(NaClフィルム)(cm−1):2975;2945;1717。
1HNMR 400MHz(CDCl3)(δppm,JHz):このようにして得た生成物の1HNMRスペクトルは、例4で記載したものと同じであった。
(2R)−(+)−アンヒドロホモハリングトン酸を用いるセファロタキシンのエステル化によるアンヒドロホモハリングトニンの調製
1HNMR 400MHz(CDCl3)(δppm,JHz):
6.61(1H,s,H−17*);6.58(1H,s,H−14*);5.92(1H,d,J3−4=9.6,H−3);5.87および5.79(2H,2s,OCH2O);5.04(1H,brs,H−1);3.80(1H,d,J4−3=9.6,H−4);3.70(3H,s,OCH3);3.59(3H,s,OCH3);3.12(2H,m,H−11β+H−8α);2.95(1H,m,H−10α);2.60(2H,m,H−8β+H−10β);2.38(1H,m,H−11α);2.13および1.66(2H,2d,JAB=14.3,CH 2CO2);2.02(1H,m,H−6A);1.90(1H,m,H−6B);1.76(2H,m,CH2−7);1.8〜1.2(6H,m,3×CH2);1.10(3H,s,CH3);1.04(3H,s,CH3)。
(2S)−(+)−アンヒドロホモハリングトン酸を用いるセファロタキシンのエステル化によるアンヒドロエピホモハリングトニンの調製
1HNMR 400MHz(CDCl3)(δppm,JHz):
6.61(1H,s,H−17*);6.57(1H,s,H−14*);5.84(3H,m,H−3)+OCH2O);5.04(1H,s,H−1);3.78(1H,d,J4−3=9.7,H−4);3.65(3H,s,OCH3);3.59(3H,s,OCH3);3.23(1H,m,H−11β);3.09(1H,m,H−8α);2.93(1H,m,H−10α);2.58(2H,m,H−8β+H−10β);2.39(1H,dd,JAB=14.4,J=7.0,H−11α);2.16および1.83(2H,2d,JAB=14.5,CH 2CO2);2.06(1H,m,H−6A);1.88(1H,m,H−6B);1.74(2H,m,CH2−7);1.5〜1.2(6H,m,3×CH2);1.11(3H,s,CH3);1.02(3H,s,CH3)。
例21で得たアンヒドロホモハリングトニンから6′−ブロモ−6′−デソキシ−ホモハリングトニンの調製
IR(ATR)(cm−1):2957;1744;1653;1487;1223。
1HNMR 400MHz(CDCl3)(δppm,JHz):
6.63(1H,s,H−17*);6.54(1H,s,H−14*);5.99(1H,d,J3−4=9.8,H−3);5.87(2H,m,OCH2O);5.05(1H,s,H−1);3.78(1H,d,J4−3=9.8,H−4);3.69(3H,s,OCH3);3.58(3H,s,OCH3);3.54(1H,s,2′−OH);3.10(2H,m,H−11β+H−8α);2.94(1H,m,H−10α);2.60(2H,m,H−8β+H−10β);2.39(1H,dd,JAB=14.0,J=6.8,H−11α);2.26および1.89(2H,2d,JAB=16.5,CH 2CO2);2.03(1H,m,H−6A);1.91(1H,m,H−6B);1.75(2H,m,CH2−7);1.74(3H,s,CH3);1.72(3H,s,CH3);1.6〜1.2(6H,m,3×CH2)。
例22で得たアンヒドロエピホモハリングトニンから6′−ブロモ−6′−デソキシ−エピホモハリングトニンの調製
IR(ATR)(cm−1):2957;1744;1653;1487;1223。
1HNMR 400MHz(CDCl3)(δppm,JHz):
6.64(1H,s,H−17*);6.59(1H,s,H−14*);5.97および5.87(2H,2d,JAB=1.1,OCH2O);5.95(1H,d,J3−4=9.7,H−3);5.04(1H,s,H−1);3.78(1H,d,J4−3=9.7,H−4);3.67(3H,s,OCH3);3.66(3H,s,OCH3);3.49(1H,s,2′−OH);3.10(2H,m,H−11β+H−8α);2.93(1H,m,H−10α);2.62および2.54(2H,2d,JAB=16.5,CH 2CO2);2.60(2H,m,H−8β+H−10β);2.40(1H,m,H−11α);2.03(1H,m,H−6A);1.89(1H,m,H−6B);1.74(2H,m,CH2−7);1.72(3H,s,CH3);1.70(3H,s,CH3);1.6〜0.7(6H,m,3×CH2)。
例23で得た6′−ブロモ−6′−デソキシ−ホモハリングトニンからホモハリングトニンの調製
5%炭酸水素ナトリウム溶液(3ml)を、例23で得た生成物(60mg,0.099ミリモル)をアセトン(1.5ml)に溶解攪拌したものに加えた。室温で2時間攪拌した後、反応混合物を真空留去し、残渣水層をジクロロメタンで3回抽出した。合わせた有機層を硫酸マグネシウム上で乾燥し、蒸発乾固した。生成する粗生成物(55mg)をカラムクロマトグラフィー(ジクロロメタン、次いでジクロロメタン/メタノール(99:1、次に95:5),シリカ(15〜40μm)2.75g)によって精製し、ホモハリングトニン(29mg,47%)を得た。このようにして得た生成物は、下記の特性を示した。
[α]D 20:−110(c=0.24;CHCl3)。
IR(NaClフィルム)(cm−1):3468;2961;1745;1656;1487;1224;1033。
1HNMR 400MHz(CDCl3)(δppm,JHz):
6.63(1H,s,H−17*);6.55(1H,s,H−14*);6.01(1H,d,J3−4=9.8,H−3);5.87(2H,m,OCH2O);5.05(1H,s,H−1);3.78(1H,d,J4−3=9.8,H−4);3.68(3H,s,OCH3);3.58(3H,s,OCH3);3.54(1H,s,2′−OH);3.10(2H,m,H−11β+H−8α);2.95(1H,m,H−10α);2.59(2H,m,H−8β+H−10β);2.38(1H,dd,JAB=14.0,J=6.7,H−11α);2.27および1.90(2H,2d,JAB=16.5,CH 2CO2);2.02(1H,m,H−6A);1.90(1H,m,H−6B);1.76(2H,m,CH2−7);1.5〜1.15(6H,m,3×CH2)1.30(1H,s,6′−OH);1.19(6H,2s,2×CH3)。
飽和炭酸カルシウム溶液(3ml)を、例23で得た生成物(60mg,0.099ミリモル)をアセトン(3ml)に溶解攪拌したものに加えた。室温で2時間攪拌した後、方法Aで得た生成物の入手をCCMによって記載した。
飽和炭酸バリウム溶液(9ml)を、例23で得た生成物(60mg,0.099ミリモル)をアセトン(3ml)に溶解攪拌したものに加えた。室温で2時間攪拌した後、方法Aで得た生成物の入手をCCMによって記載した。
例23で得た生成物(60mg,0.099ミリモル)をアセトン/水混合物(3/2,2.15ml)に溶解攪拌したものに、硝酸銀(25mg,0.149ミリモル)加えた。室温で2時間攪拌した後、方法Aで得た生成物の入手をCCMによって記載した。
エピホモハリングトニンの調製
a)方法A:
5%炭酸水素ナトリウム溶液(3ml)を、例23で得た生成物(60mg,0.099ミリモル)をアセトン(1.75ml)に溶解攪拌したものに加えた。室温で2時間攪拌した後、反応混合物を真空留去し、残渣水層をジクロロメタンで3回抽出した。合わせた有機層を硫酸マグネシウム上で乾燥し、蒸発乾固した。生成する粗生成物(60mg)をカラムクロマトグラフィー(ジクロロメタン、次いでジクロロメタン/メタノール(99:1、次に97:3),シリカ(15〜40μm)3g)によって精製し、エピホモハリングトニン(29mg,47%)を得た。このようにして得た生成物は、下記の特性を示した。
[α]D 20:−92(c=0.29;CHCl3)。
IR(NaClフィルム)(cm−1):3514;2961;1744;1655;1488;1223;1035。
1HNMR 400MHz(CDCl3)(δppm,JHz):
6.65(1H,s,H−17*);6.60(1H,s,H−14*);5.95(1H,d,H−3);5.95および5.86(2H,2d,OCH2O);5.05(1H,s,H−1);3.78(1H,d,J4−3=9.7,H−4);3.68(3H,s,OCH3);3.66(3H,s,OCH3);3.52(1H,brs,2′−OH);3.13(2H,m,H−11β+H−8α);2.97(1H,m,H−10α);2.63(2H,m,H−8β+H−10β);2.61および2.52(2H,2d,JAB=16.5,CH 2CO2,2.40(1H,dd,JAB=13.8,J=6.3,H−11α);2.04(1H,m,H−6A);1.94(1H,m,H−6B);1.78(2H,m,CH2−7);1.45〜0.7(6H,m,3×CH2);1.16(1H,s,CH3);1.15(3H,s,CH3)。
飽和炭酸カルシウム溶液(3ml)を、例24で得た生成物(60mg,0.099ミリモル)をアセトン(3ml)に溶解攪拌したものに加えた。室温で2時間攪拌した後、方法Aで得た生成物の入手をCCMによって記載した。
飽和炭酸バリウム溶液(9ml)を、例24で得た生成物(60mg,0.099ミリモル)をアセトン(3ml)に溶解攪拌したものに加えた。室温で2時間攪拌した後、方法Aで得た生成物の入手をCCMによって記載した。
例24で得た生成物(60mg,0.099ミリモル)をアセトン/水混合物(3/2,2.15ml)に溶解攪拌したものに、硝酸銀(25mg,0.149ミリモル)加えた。室温で2時間攪拌した後、方法Aで得た生成物の入手をCCMによって記載した。
例22で得たアンヒドロエピホモハリングトニン(58mg,0.109ミリモル)を無水ジクロロメタン(0.3ml)に溶解攪拌したものに、臭化水素酸を酢酸に溶解した市販溶液(0.195ml,0.98ミリモル,30%(w/w)HBr)を−10℃で加えた。−10℃で3時間攪拌した後、水(2.8ml)を加えた後、温度を20℃に上昇させた。20℃で3時間攪拌した後、炭酸ナトリウム溶液(0.76M;6ml)を加えて、pH8とした。生成する水層を塩化ナトリウムで飽和した後、ジクロロメタン(3×10ml)で抽出し、合わせた有機層を硫酸マグネシウム上で乾燥し、蒸発乾固して、エピホモハリングトニン(45mg粗生成物,75%)を得た。このようにして得た粗生成物は、方法Aで得たのと同じ特性を示した。
合成用高性能液体クロマトグラフィーによる例25で得た粗製の半合成ホモハリングトニンから医薬用としてのホモハリングトニンの調製
粗製のホモハリングトニン(35g)を、緩衝液(トリエチルアミン(1.55/1000)/脱イオン水およびオルトリン酸に溶解し、pH3に調整した。次に、溶液を、アキシャル・コンプレッションおよび高圧ポンプを備えた合成用高性能液体クロマトグラフィー装置(固定相:n−オクタデシルシラン、15μm、多孔度100、1kg;移動相:緩衝液/テトラヒドロフラン85/15)に注入した。溶出は、0.2リットル/分の流速で行った。画分の含量は、UV検出器およびTLCによって観察した。保持画分を最後にHPLCでチェックした後、合わせて、2.5%アンモニア水溶液でアルカリ性にして、ジクロロメタン(4×400ml)で抽出した。減圧濃縮の後、ホモハリングトニンが淡黄色樹脂状生成物として得られ、これを8/2水−メタノール混合物で粉砕すると、純粋なホモハリングトニンが白色結晶性固形生成物(融点=127℃)として得られ、HPLC純度は99.8%を上回った。
緩衝液/テトラヒドロフランの代わりに、移動相として緩衝液/メタノール(68/32)を用いることを除き、方法Aと同じ精製手続きを行った。
緩衝液/テトラヒドロフランの代わりに、移動相として緩衝液/アセトニトリル(85/15)を用いることを除き、方法Aと同じ精製手続きを行った。
半精製天然セファロタキシンから医薬用としてのホモハリングトニンの調製
部分的にラセミ化した天然セファロタキシンから例25に従って調製し、例27の方法Aに従ってクロマトグラフィーおよび結晶化によって精製した粗製のホモハリングトニンは、非天然の鏡像異性体であるエピ−ホモハリングトニン含量が0.05%未満であるホモハリングトニンを生成した。
セファロタキシンとフェニルグリシド酸とのエステル化による2′−デ−(メトキシカルボニルメチル)−(2−O−3′)−デヒドロネオハリングトニンまたはセファロタキシルフェニルグリシデートの調製
1HNMR 400MHz(CDCl3)(δppm,JHz):
7.27(3H,m,Ph);7.18(2H,m,Ph);6.63(1H,s,H−17*);6.40(1H,s,H−14*);5.96および5.85(2H,2d,JAB=1.5,OCH2O);5.73(1H,d,J3−4=9.4,H−3);5.01(1H,2s,H−1);4.01(1H,d,J3′−2′=4.6,H−3′);3.65(3H,s,OCH3);3.62(1H,d,J4−3=9.3,H−4);3.40(1H,d,J2′−3′=4.5,H−2′);3.27(1H,m,JAB=14.3,J=12.1および7.8,H−11β);3.05(1H,m,H−8α);2.91(1H,td,J=11.7および7.4,H−10α);2.57(2H,m,H−8β+H−10β);2.43(1H,dd,JAB=14.5,J=7.0,H−11α);1.93(1H,m,H−6A);1.84(1H,m,H−6B);1.68(2H,m,CH2−7)。
例29で得たセファロタキシルフェニルグリシデートの水素化分解による2′−デ−(メトキシカルボニルメチル)−ネオハリングトニンの調製
IR(KBr錠剤)(cm−1):3436;2937;1747;1655;1487;1224および1035。
1HNMR 400MHz(CDCl3)(δppm,JHz):
7.25(3H,m,m,p−Ph);7.0(2H,m,o−Ph);6.65(1H,s,H−17*);6.63(1H,s,H−14*);5.98(1H,d,J3−4=9.3,H−3);5.85(2H,2d,JAB=1.2,OCH2O);5.09(1H,s,H−1);4.17(1H,m,H−2′);3.85(1H,d,J4−3=9.6,H−4);3.71(3H,s,OCH3);3.20(1H,m,H−11β);3.10(1H,m,H−8α);2.95(1H,m,H−10α);2.60(2H,m,H−8β+H−10β);2.39(2H,m,H−11α+H−3′A);2.04(1H,m,H−6A);2.0(1H,dd,JAB=14.3,J3′B−2′=9.5,H−3′B);1.91(1H,m,H−6B);1.77(2H,m,CH2−7)。
例29で得たセファロタキシルフェニルグリシデートから2′R−デ−(メトキシカルボニルメチル)−3′S−アジド−ネオハリングトニンの調製
IR(ATR)(cm−1):3488;2935;2105;1748;1654;1486;1223;1034。
1HNMR 400MHz(CDCl3)(δppm,JHz):
7.38(3H,m,m,p−Ph);7.29(2H,m,o−Ph);6.74(1H,s,H−17*);6.67(1H,s,H−14*);6.08(1H,d,J3−4=9.8,H−3);5.90(2H,2d,JAB=1.4,OCH2O);5.08(1H,s,H−1);4.07(1H,大きなd,H−2′);3.85(1H,d,J4−3=9.7,H−4);3.78(1H,brs,H−3′);3.69(3H,s,OCH3);3.23(1H,m,H−11β);3.11(1H,m,H−8α);2.98(1H,m,H−10α);2.90(1H,d,J2′−OH=8.2,2′−OH);2.63(2H,m,H−8β+H−10β);2.47(1H,dd,JAB=14.2,J=6.9,H−11α);2.05(1H,m,H−6A);1.92(1H,m,H−6B);1.78(2H,m,CH2−7)。
例31で得たアジドの水素化分解による2′R−デ−(メトキシカルボニルメチル)−3′S−アミノ−ネオハリングトニンの調製
IR(ATR)(cm−1):3299;2935;1740;1654;1486;1222;および1034。
1HNMR 400MHz(CDCl3)(δppm,JHz):
7.27(5H,m,Ph);6.69(1H,s,H−17*);6.67(1H,s,H−14*);6.0(1H,d,J3−4=9.7,H−3);5.85(2H,m,OCH2O);5.09(1H,brs,H−1);4.06(1H,d,J=1.2,H−2′);3.86(1H,d,J4−3=9.5,H−4);3.72(3H,s,OCH3);3.38(1H,brs);3.25(1H,m,H−11β);3.14(1H,m,H−8α);2.99(1H,m,H−10α);2.64(2H,m,H−8β+H−10β);2.49(1H,m,H−11α);2.05(1H,m,H−6A);1.94(1H,m,H−6B);1.79(2H,m,CH2−7)。
セファロタキシンのエステル化による2′−デ−(メトキシカルボニルメチル)−3′−ヒドロキシ−ネオハリングトニンのアセトニドの調製
1HNMR 400MHz(CDCl3)(δppm,JHz):
7.27(5H,Ph);6.63,6.62,6.60および6.57(1H,4s,H−14*);6.51,6.49,6.42および6.41(1H,s,H−17*);5.93(J3−4=9.6),5.89,5.43(J3−4=9.5)および5.31(J3−4=9.3)(1H,4d,H−3);5.89(s),5.87+5.84(2d,JAB=1.5),5.85+5.80(2d)および5.84+5.77(2d,JAB=1.5)(2H,OCH2O);5.23(J5′−4′=7.3),5.20(J5′−4′=7.4),4.58(J5′−4′=8.0)および4.49(J5′−4′=6.2)(1H,4d,H−5′);5.07,5.03および4.83(1H,3s,H−1);4.32(J4′−5′=7.4),4.21(J4′−5′=6.2)4.18(J4′−5′=7.4)および3.75(1H,4d,H−4′);3.86(J4−3=9.6),3.76および3.60(J4−3=9.5)(1H,4d,H−4);3.76,3.75,3.70および3.43(3H,4s,OCH2);3.3〜1.6(10H,m);1.66+1.41,1.65+1.37,1.51+1.44および1.47+1.22(6H,8s,2×CH3)。
セファロタキシンのエステル化によるセファロタキシル=N−ベンジル−3−フェニル−アジリジン−1−カルボキシレートの調製
1HNMR 400MHz(CDCl3)(δppm,JHz):
7.24(10H,m,2×Ph);6.63(1H,s,H−17*);6.60(1H,s,H−14*);5.85および5.80(2H,2d,JAB=1.4,OCH2O);5.64(1H,d,J3−4=9.3,H−3);4.97(1H,s,H−1);3.92および3.20(2H,2d,JAB=13.7,CH 2Ph);3.71(1H,d,J4−3=9.4,H−4);3.56(3H,s,OCH3);3.25(1H,m,H−11β);3.07(1H,m,H−8α);2.93(1H,m,H−10α);2.86(1H,d,J3′−2′=6.8,H−3′);2.57(2H,m,H−8β+H−10β);2.38(1H,dd,JAB=14.4,J=7.0,H−11α);2.07(1H,d,J2′−3′=6.8,H−2′);1.96(1H,m,H−6A);1.82(1H,m,H−6B);1.70(2H,m,CH2−7)。
セファロタキシンのエステル化によるN,O−アンヒドロ−2′−デ−(メトキシカルボニルメチル)−3′−ベンズアミドネオ−ハリングトニンまたはセファロタキシル=N,O−アンヒドロ−N−ベンゾイル−フェニルイソセリネートの調製
1HNMR 400MHz(CDCl3)(δppm,JHz):
8.0(2H,d,J=7.3,o−PhC=N);7.52(1H,t,J=7.4,p−PhC=N);7.44(2H,t,J=7.5,m−PhC=N);7.32(2H,t,J=7.2,m−Ph);7.26(1H,m,p−Ph);7.15(2H,d,J=7.1,o−Ph);6.58(1H,s,H−17*);6.51(1H,s,H−14*);5.98(1H,d,J3−4=9.5,H−3);5.85および5.76(2H,2d,JAB=1.3,OCH2O);5.08(1H,s,H−1);4.67(1H,d,J4′−5′=5.6,H−4′);4.52(1H,d,J5′−4′=5.6,H−5′);3.85(1H,d,J4−3=9.6,H−4);3.70(3H,s,OCH3);3.17(1H,m,H−11β);3.08(1H,m,H−8α);2.93(1H,m,H−10α);2.59(2H,m,H−8β+H−10β);2.31(1H,dd,JAB=14.2,J=6.8,H−11α);2.04(1H,m,H−6A);1.91(1H,m,H−6B);1.75(2H,m,CH2−7)。
セファロタキシンのエステル化によるN,O−メトキシメチレン−2′−デ−(メトキシカルボニルメチル)−3′−ベンズ−アミドネオハリングトニンまたはセファロタキシル=N,O−メトキシメチレン−N−ベンゾイル−フェニルイソセリネートの調製
1HNMR 400MHz(CDCl3)(δppm,JHz):
7.66(2H,brs,o−BzN);7.41(4H,m,BzN+Ph);7.32(2H,m,Ph);7.26(2H,m,Ph);6.56(1H,s,H−17*);6.54(1H,s,H−14*);5.89(1H,d,J3−4=9.5,H−3);5.83および5.80(2H,2m,OCH2O);5.76(1H,brs,H−2′);5.10(1H,s,H−1);4.85(1H,brs,H−4′);4.42(1H,brs,H−5′);3.84(1H,d,J4−3=9.5,H−4);3.72(3H,s,OCH3);3.28(3H,brs,2′−OCH3);3.19(1H,m,H−11β);3.09(1H,m,H−8α);2.93(1H,m,H−10α);2.60(2H,m,H−8β+H−10β);2.37(1H,dd,JAB=14.4,J=6.6,H−11α);2.03(1H,m,H−6A);1.90(1H,m,JAB=12.2,J=7.8および4.4,H−6B);1.76(2H,m,CH2−7)。
2′−デ−(メトキシカルボニルメチル)−3′−ベンズアミド−ネオハリングトニンまたはセファロタキシル=N−ベンゾイル−フェニルイソセリネートの調製
例35で得たセファロタキシル=(4S,5R)−2,4−ジフェニル−4,5−ジヒドロオキサゾール−5−カルボキシレート(300mg,0.53ミリモル)をメタノール/テトラヒドロフランの混合物50/50(10ml)に溶解攪拌したものに、1N塩酸を室温で加えた。(CCMで反応を制御しながら)18±5℃で3時間攪拌した後、飽和炭酸水素ナトリウム溶液(19ml)とメタノール/テトラヒドロフランの混合物50/50(50ml)を加えた。(CCMで反応を制御しながら)18±5℃で20時間攪拌した後、反応混合物を酢酸エチルと水とで処理した。生成する水層を酢酸エチルで抽出し、合わせた有機層を硫酸マグネシウム上で乾燥し、蒸発乾固した。生成する粗生成物(170mg)をカラムクロマトグラフィー(ジクロロメタン/メタノール(98:2),シリカ(15〜40μm)8g)によって精製し、白色固形生成物(180mg,58%;HPLC純度92.3%)を得た。このようにして得た予想生成物は、下記の特性を示した。
[α]D 20:−119.2(c=0.141;CHCl3)。
1HNMR 400MHz(CDCl3)(δppm,JHz):
7.75(2H,d,J=7.3,o−BzN);7.51(1H,t,J=7.3,p−BzN);7.13(2H,t,J=7.3,m−BzN;Ph);7.27(5H,m,Ph);6.88(1H,d,J3′−NH=7.9,3′NH);6.59(1H,s,H−17*);6.57(1H,s,H−14*);5.93(1H,d,J3−4=9.7,H−3);5.78および5.69(2H,2d,JAB=1.5,OCH2O);5.06(1H,s,H−1);4.98(1H,dd,J3′−NH=7.9,H−3′);4.22(1H,brs,H−2′);3.81(1H,d,J4−3=9.6,H−4);3.58(3H,s,OCH3);3.19(1H,m,J=12.8,7.9,H−11b);3.07(1H,m,H−8a);2.93(1H,m,H−10a);2.72(1H,brs,2′−OH);2.58(2H,m,H−8b+H−10b);2.43(1H,dd,JAB=14.2,J=7.0,H−11a);2.01(1H,m,H−6A);1.88(1H,m,JAB=12.0,J=7.8,3.8,H−6B);1.75(2H,m,CH2−7)。
例32で得た生成物(60mg,0.125ミリモル)を酢酸エチル(850μl)に溶解攪拌したものに、飽和炭酸水素ナトリウム溶液(850μl)および塩化ベンゾイル(19μl,0.163ミリモル)を加えた。反応の経過中に、白色沈澱が生成した。室温で1時間攪拌した後、反応混合物を酢酸エチルで希釈し、有機層を飽和炭酸水素ナトリウム溶液で洗浄した。生成する水層を酢酸エチルで3回抽出し、合わせた有機層を硫酸マグネシウム上で乾燥し、蒸発乾固した。生成する粗生成物(65mg)をカラムクロマトグラフィー(ジクロロメタン/メタノール(98:2),シリカ(15〜40μm)2.5g)によって精製し、白色固形生成物(41mg,56%)を得た。このようにして得た予想生成物は、上記の方法で得たのと同じ特性を示した。
例32で得た生成物のアミド化によるN−第三ブトキシカルバモイル−2′−デ−(メトキシカルボニルメチル)−3′−アミノ−ネオハリングトニンまたはセファロタキシル=N−第三ブトキシカルバモイル−フェニルイソセリネートの調製
1HNMR 400MHz(CDCl3)(δppm,JHz):
7.27(3H,m,m,p−Ph);6.94(2H,d,J=6.6,o−Ph);6.71(1H,s,H−17*);6.66(1H,s,H−14*);6.01(1H,d,J3−4=9.7,H−3);5.90(2H,s,OCH2O);5.06(1H,s,H−1);5.05(1H,m,NH);4.56(1H,m,H−3′);4.15(1H,m,H−2′);3.81(1H,d,J4−3=9.7,H−4);3.69(3H,s,OCH3);3.19(1H,m,H−11β);3.10(1H,m,H−8α);2.93(1H,m,H−10α);2.61(2H,m,H−8β+H−10β);2.51(1H,m,H−11α);2.05(1H,m,H−6A);1.89(1H,m,H−6B);1.77(2H,m,CH2−7);1.44(9H,s,OC(CH3)3)。
第三ブチル=2−メトキシカルバモイルメチル−2−ヒドロキシ−6−メチルヘプト−5−エノエートの調製
5−ブロモ−2−メチル−ペント−2−エン(1.34g,8.2ミリモル)を、無水テトラヒドロフラン(8ml)にマグネシウム(240g,10ミリモル)(追加のヨウ素結晶で活性化)を混合攪拌したものに滴加した。反応の開始と同時に、反応混合物が激しく過熱し、還流した。マグネシウムのほとんどが反応してしまうまで還流を維持し、反応混合物を無水テトラヒドロフラン(16ml)で希釈した。第三ブチルエチルオキザレート(1.4g,8ミリモル)を無水テトラヒドロフラン(8ml)中で混合攪拌したものに、生成するグリニャール試薬を−78℃で20分間かけて加えた。温度を−15℃まで2時間かけて上げ、混合物を1N塩酸で反応停止した。分離した有機層を塩水で3回洗浄し、硫酸マグネシウム上で乾燥し、蒸発乾固した。生成する粗生成物(2g)をカラムクロマトグラフィー(シクロヘキサン/酢酸エチル(98:2),シリカ(15〜40μm)80g)によって精製し、油状生成物(660mg,39%)を得た。中間体であるα−ケトエステルは、下記の特性を示した。
5.08(1H,m,H−5),2.80(2H,t,J=7.3,CH2−3);2.28(2H,m,CH2−4);1.68(3H,s,CH3);1.62(3H,s,CH3);1.54(9H,s,O−第三ブチル)。
無水酢酸メチル(0.7ml,8.75ミリモル)を、1Mリチウム=ビス−(トリメチルシリルアミド)/テトラヒドロフランの攪拌溶液(9ml,9ミリモル)に−78℃で1分間かけて加え、これを−78±5℃で20分間反応させた。上記で調製した第三ブチル=2−オキソ−6−メチルヘプト−5−エノエート(640mg,3ミリモル)を無水テトラヒドロフラン(10ml)に混合攪拌したものに、リチウムエノレートを−78℃で5分間かけて加え、生成する混合物を−78±5℃で30分間攪拌した。CCMで観察した後、冷凍浴を外し、混合物を15%塩化アンモニウム溶液(10ml)で反応停止した。分離した有機層を15%塩化アンモニウム溶液(10ml)で洗浄し、蒸発乾固した。水層をエーテル(2×10ml)で抽出した。有機層を濃縮物と合わせ、塩水(10ml)で洗浄し、硫酸マグネシウム上で乾燥して、蒸発乾固した。生成する粗生成物(1.3g)をカラムクロマトグラフィー(シクロヘキサン/酢酸エチル(95:5),シリカ(15〜40μm)60g)によって精製し、油状生成物(222mg,26%)を得た。このようにして得た生成物は、下記の特性を示した。
1HNMR 400MHz(CDCl3)(δppm,JHz):
5.07(1H,m,HC=),3.67(3H,s,OCH3);3.66(1H,s,OH);2.86および2.67(2H,2d,JAB=15.8,CH 2CO2);2.13(1H,m,CH2);1.85(1H,m,CH2);1.67(3H,s,CH3)および(2H,m,CH2);1.59(3H,s,CH3);1.51(9H,s,第三−BuO)。
エチル=2−N−[(4′S)−イソプロピル−2′−オキサゾリジノ]カルボニルメチル−2−ヒドロキシ−6−メチルヘプト−5−エノエートの調製
1HNMR 400MHz(CDCl3)(δppm,JHz):
5.06(1H,m,H−3″),4.41(1H,m,H−4″);4.25(4H,m,CH2−5″およびOCH 2CH3);3.72(1H,s,OH);3.52および3.41(2H,2d,JAB=17.9,CH2−3);2.36(1H,m,H−6″);2.16(1H,m,CH2′);1.92(1H,m,CH2′);1.75(2H,m,CH2′);1.67(3H,s,CH3′);1.59(3H,s,CH3′);1.30(3H,t,J=7.1,OCH2 CH 3);0.89(3H,d,J=7.0,CH3″);0.87(3H,d,J=6.9,CH3″)。
1HNMR 400MHz(CDCl3)(δppm,JHz):
5.06(1H,m,H−3″),4.40(1H,m,H−4″);4.23(4H,m,CH2−5″およびOCH 2CH3);3.68(1H,s,OH);3.46(2H,s,CH2−3);2.33(1H,m,H−6″);2.16(1H,m,CH2′);1.91(1H,m,CH2′);1.75(2H,m,CH2′);1.67(3H,s,CH3′);1.59(3H,s,CH3′);1.28(3H,t,J=7.1,OCH2 CH 3);0.90(3H,d,J=7.0,CH3″);0.87(3H,d,J=6.9,CH3″)。
エチル=2−(1′R,2′S,5′R)−メントキシカルボニルメチル−2−ヒドロキシ−6−メチルヘプト−5−エノエートの調製
1HNMR 400MHz(CDCl3)(δppm,JHz):
5.03(1H,t,J=7.0,H−3′),4.69(1H,td,J=10.9および4.3,H−1Men);4.24(2H,q,J=7.0,OCH 2CH3);3.77(1H,s,2−OH);2.91および2.67(2H,2d,JAB=16.4,CH2CO2);2.13(1H,m,H−6eqMen);1.85(2H,m,CH′およびH−7Men);1.75〜1.6(4H,m,CH2′およびH−3eq,4eqMen);1.67(3H,s,CH3′);1.58(3H,s,CH3′);1.45(1H,m,H−5Men);1.35(1H,m,H−2axMen);1.30(3H,t,J=6.9,OCH2 CH 3);1.03(1H,m,H−3axMen);0.93(1H,m,H−6axMen);0.89(6H,d,J=6.8,2×CH3(Men));0.87(1H,m,H−4axMen);0.73(3H,d,J=6.9,CH3(Men))。
1HNMR 400MHz(CDCl3)(δppm,JHz):
5.05(1H,t,J=7.0,H−3′),4.67(1H,td,J=10.7および4.7,H−1Men);4.25(2H,m,OCH 2CH3);3.74(1H,s,2−OH);2.92および2.65(2H,2d,JAB=15.9,CH 2CO2);2.12(1H,m,CH′);1.97(1H,m,H−6eqMen);1.86(2H,m,CH′およびH−7Men);1.75〜1.6(4H,m,CH2′およびH−3eq,4eqMen);1.67(3H,s,CH3′);1.58(3H,s,CH3′);1.48(1H,m,H−5Men);1.36(1H,m,H−2axMen);1.31(3H,t,J=7.0,OCH2 CH 3);1.15〜0.8(3H,m,H−3ax,6ax,4axMen);0.89(6H,d,J=6.9,2×CH3(Men));0.76(3H,d,J=7.0,CH3(Men))。
2−[(R)−1′,2′,2′−トリフェニル−エタン−2′−オール]エトキシカルボニルメチル−2−ヒドロキシ−6−メチルヘプト−5−エノエートの調製
7.66(2H,d,J=7.5,o−Ph);7.43(2H,t,J=7.7,m−Ph);7.35〜7.0(11H,m,Ph);6.72(1H,s,H−1″);4.95(1H,m,H−3′);4.41(2H,m,OCH 2CH3);3.42(1H,s,2−OH);2.90および2.67(2H,2d,JAB=16.5,CH2−3);2.53(1H,s,2″−OH);1.98(1H,m,CH2);1.8〜1.5(3H,m,CH2);1.63(3H,s,CH3);1.52(3H,s,CH3);1.38(3H,t,J=7.1,OCH2 CH 3)。
1HNMR 400MHz(CDCl3)(δppm,JHz):
7.52(2H,d,J=7.5,o−Ph);7.36(2H,t,J=7.6,m−Ph);7.27(1H,t,J=7.3,p−Ph);7.2〜7.0(10H,m,Ph);6.59(1H,s,H−1″);4.98(1H,m,H−3′);3.90および3.34(2H,2m,OCH 2CH3);3.56(1H,s,2−OH);3.22(1H,s,2″−OH);2.88および2.69(2H,2d,JAB=16.7,CH2−3);2.06および1.79(2H,2m,CH2);1.7〜1.5(2H,m,CH2);1.64(3H,s,CH3);1.54(3H,s,CH3);0.99(3H,t,J=7.1,OCH2 CH 3)。
第三ブチル=2−[(R)−1′,2′,2′−トリフェニル−エタン−2′−オール]エトキシカルボニルメチル−2−ヒドロキシ−6−メチルヘプト−5−エノエートの調製
1HNMR 400MHz(CDCl3)(δppm,JHz):
7.53(2H,d,J=7.4,o−Ph);7.36(2H,t,J=7.6,m−Ph);7.28(1H,t,J=7.3,p−Ph);7.2〜7.0(10H,m,Ph);6.66(1H,s,H−1″);5.00(1H,m,H−3′);3.50(1H,s,2−OH);2.94(1H,s,2″−OH);2.76および2.61(2H,2d,JAB=16.3,CH 2−CO2);2.06および1.78(2H,2m,CH2);1.65(3H,s,CH3);1.55(3H,s,CH3)および2H,m,CH2);1.23(9H,s,第三−BuO)。
2−カルボキシメチル−6,6−ジメチル−2−テトラヒドロピランカルボン酸またはO−デメチルアンヒドロホモハリングトン酸の調製
例3で得たエチレン性二酸(1.5g,6.94ミリモル)をギ酸(2.6ml)に溶解したものを、60℃で16時間攪拌した。周囲温度に戻した後、ギ酸を真空留去し、生成する粗生成物を40℃で20時間真空乾燥した(1.5g,100%)。
例39で得た生成物から2−メトキシカルボニルメチル−6,6−ジメチル−2−テトラヒドロピランカルボン酸またはアンヒドロホモハリングトン酸の調製
イヌガヤ種の全アルカロイド抽出物から精製した(−)−セファロタキシンの調製
(上記と同じ条件で抽出した)セファロタキシンの2バッチの1HNMRは、光学活性なNMRシフト試薬であるユーロピウム(III)=トリス[3−(ヘプタフルオロプロピルヒドロキシメチレン)−(+)−カンファレート(1当量)を用いると、下記の結果を示した。
(+)−セファロタキシン11±5%の存在。
[α]22=−134.0°(c=0.214;CHCl3);計算比25±5%。
バッチB:僅かにラセミ化(1%)
[α]22=−173.3°(c=0.208;CHCl3)。
粗製のクロマトグラフィー処理を行ったセファロタキシン(20g)を、55℃で乾燥メタノール(100ml)に溶解した。ロータリーエバポレーターで冷却することにより、結晶化が起こり、濾過の後にこのようにして得た生成物のHPLC純度は99.9%であった。
[α]20 D=−130°(C1,CHD3)10%のラセミ化に相当。このようにして得られた結晶化生成物(20g)を、再度熱メタノール(100ml)に溶解した。
濾過の後、母液を室温で徐々に蒸発させ、ラセミセファロタキシンからのみナル双晶状針状結晶の形態の結晶[α]20 D=0.5°(C1,CHCl3)を得た。
濾過の後、第二の母液から、最初の結晶化で得たのと同一の(−)−セファロタキシンからなる柱状晶を得た。
濾過の後、第三の母液から(±)−セファロタキシンからなる双晶状針状結晶(アーチン)も得た。
サイクルは3回繰返した。合わせた柱状結晶を1回再結晶し、鏡像異性体的に純粋な(−)−セファロタキシンを得て、一方、合わせた双晶状針状結晶を同様の方法で処理すると、100%ラセミ性のセファロタキシンを得る。
部分的にラセミ化した天然セファロタキシンの試料を、例19で得た純粋な(2R)−ホモハリングトン酸を用いて、順序が例1、2、3、4、5、6、15、19および21に記載されている工程に挿入した。
このようにして得たアンヒドロ−ホモハリングトニンのジアステレオマー混合物のHPLC分析では、供給源のラセミ混合物での(+)−セファロタキシン含量に相当する有意なエナンシオ−エピ−ホモハリングトニン比(11%±3%)を示した(ホモハリングトン酸の2種類の鏡像異性体は、純粋な鏡像異性セファロタキシンに相当し、化学量論的に反応することが示されている)。
アンヒドロ−ホモハリングトニンからのホモハリングトニンの調製
臭化水素酸を酢酸に溶解した市販溶液(17.4ml,86.6ミリモル,30%(w/w)HBr)を、例21で得たアンヒドロホモハリングトニン(50.8g,9.63ミリモル)を−10℃で無水ジクロロメタン(25.6ml)に溶解攪拌したものに加えた。−10℃で3時間攪拌した後、水(240ml)を加えると、反応混合物は粘稠になった。温度を室温まで上昇させ、2.5時間攪拌した後、0.76M炭酸ナトリウム(406ml)を加えてpH8とした。生成する水層を塩化ナトリウムで飽和した後、ジクロロメタン(3×230ml)で抽出し、合わせた有機層を硫酸マグネシウム上で乾燥し、蒸発乾固して、フォーム状生成物を得た。下記の逆相クロマトグラフィーの後、ホモハリングトニン4.03g(77%)を得た。このようにして得た生成物は、例25で得たのと同じ特性を示した。
例21で得たアンヒドロホモハリングトニン(214mg,0.406ミリモル)を無水ジクロロメタン(1.1ml)に溶解攪拌したものに、−10℃で臭化水素酸を酢酸に溶解した市販溶液(0.728ml,3.6ミリモル,30%(w/w)HBr)を加えた。−10℃で3時間攪拌した後、水(13ml)を加え、温度を20℃に上昇させた。20℃で3時間攪拌した後、炭酸ナトリウム溶液(0.76M;31.5ml)を加えてpH8とした。生成する水層を塩化ナトリウムで飽和した後、ジクロロメタン(3×20ml)で抽出し、合わせた有機層を硫酸マグネシウム上で乾燥し、蒸発乾固した。生成する粗生成物を下記の逆相クロマトグラフィーによって精製し、ホモハリングトニン(166mg,75%)を得た。このようにして得た生成物は、例25で得たものと同じ特性を示した。
(1)下記式
Ω−CO−O−CTX
[上記式中、
Ω(「オメガ」)は鎖の末端残基の代表的基であり、−CO−はセファロタキサンに結合したエステル基のカルボニルであり、
基Ω−CO−は、
下記式
nは0〜8であり、
Zは酸素、窒素または硫黄ヘテロ原子であり、
R5、R6およびR8は、独立して
水素、
飽和、不飽和または芳香族の線状または分岐状および/または環状の炭化水素基、特にアルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、アリール、ヘテロシクロアルキルであって、上記基は(複数の)ヘテロ原子を含みまたは含まず、R6およびR8は1つの環に含まれていてもよく、
上記基の1個を有する酸素エーテルである)の置換ヘテロシクロアルカン、または
下記式
mは1〜8であり、
R5、R6およびR8は上記で定義した通りである)の線状アルケン、または
下記式
n、R5、R6およびR8は上記で定義した通りであり、
ZおよびQ2は、独立して酸素、窒素または硫黄ヘテロ原子であり、
Q1は炭素、ケイ素またはリン原子であり、
R9およびR10は、独立して水素、アルコキシ、(複数の)ヘテロ原子を含むまたは含まない、飽和、不飽和または芳香族性の線状または分岐状および/または環状の炭化水素基、特にアルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、アリール、ヘテロシクロアルキルであり、
R9および/またはR10は、ゼロであるかまたは一緒になってヘテロ原子を作成しおよび/または多重結合を作成することができ、但し、Q1、R9およびR11はゼロであって、それらを有する炭素の2個の原子の間に多重結合を作成することができ、
R11は水素、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニルまたはアルキルカルボニルである)
に相当し、
−O−CTXは、下記式
pは1または2である)、
のセファロタキシン残基およびその塩であり、
上記の2種類の基−Ωおよび−CTXは、エステル結合−CO−O−によって結合されている]
の側鎖を有するセファロタキサンおよび/またはその塩の製造法であって、
一般式Ω−CO−OHを有するカルボン酸またはその塩、または
一般式Ω−CO−Aを有する酸の活性化形態またはその塩であって、Ω−COが下記式
Ω−COが下記式
Ω−COが下記式
を有し、
Aが、
下記式
の環状無水物であるものを、
式H−O−CTX(式中、CTXは上記で定義した通りである)のヒドロキシル基を有するセファロタキサンまたはその塩、または
式M−O−CTX(式中、CTXは上記で定義した通りであり、Mは金属である)の金属アルコキシド、または
式Y−O−CTX(式中、−O−CTXは上記で定義した通りであり、Yは、Y−と−O−CTXとの間で開裂することによって酸素原子に陰電荷を与え、またはY−O−と−CTXとの間で開裂することによってカルボカチオンを与える脱離基である)のそのヒドロキシル基の活性化形態
と反応させ、1または数個の反応添加剤が存在し、上記側鎖を有するセファロタキサンおよび/またはその塩を形成することができる方法であって、この反応はこのようにして形成された第一カルボキシルのメチル化によって完成される方法。
のセファロタキシン、またはその塩である、(1)に記載の方法。
式MeOCOO−のメトキシホルミルオキシ、
式CF3COO−のトリフルオロアセチルオキシ、
式RSO3−のアルキルスルホンオキシ、
式(RO)2PO−のホスホキシ、
式ROP(Cl)O−のハロホスホキシ、
式R3SiO−のトリアルキルシリルオキシ、
式
式
から選択される基である、(1)〜(8)のいずれか1項に記載の方法。
M−O−CTX
(式中、MおよびCTXは、(1)で定義した通りである)
に相当し、
下記式
H−O−CTX
(式中、CTXは、(1)で定義した通りである)
のセファロタキシンを、金属自身、アミド、金属水素化物、またはアルキル−金属と接触させることによって得られる、(1)〜(19)のいずれか1項に記載の方法。
nは0〜8であり、
Zは酸素、窒素または硫黄ヘテロ原子であり、
R5、R6およびR8は、独立して
水素、
飽和、不飽和または芳香族の線状または分岐状および/または環状の炭化水素基、特にアルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、アリール、ヘテロシクロアルキルであって、上記基は(複数の)ヘテロ原子を含みまたは含まず、
上記基の1個を有する酸素エーテルであり、
CTXは、(1)〜(3)のいずれか1項で定義した通りである)に相当する側鎖を有するセファロタキサンであって、
Zが酸素原子であり、かつ
1°)n=2または3、同時にR6=R8=メチルおよびR5=OMeまたはヒドロキシル、
2°)n=2、同時にR6=R8=メチルおよびR5=OMeまたはヒドロキシル、
3°)n=3、同時にR8がメチルでありかつR5が基−CH2CO2CH3であるとき、R6がヒドロキシル
である化合物を除く、セファロタキシン、および/またはその塩。
m、R5、R6およびR8は(1)で定義した通りであり、CTXは(1)〜(3)のいずれか1項で定義した通りである)
に相当し、
m=2、R5=CH2CO2CH3、R6=R8=メチルであり、かつCTXが(3)で定義した通りである化合物を除く、側鎖を有するセファロタキサン、および/またはその塩。
n、Z、Q1、Q2、R5、R6、R8、R9、R10およびR11は(1)で定義した通りであり、CTXは(1)〜(3)のいずれか1項で定義した通りである)
に相当する、側鎖を有するセファロタキサン、および/またはその塩。
上記鎖を、水性または非水性媒質中で薬剤および/またはプロトン性または非プロトン性の親電子性基Eで開環し、下記式
の中間体化合物を提供し、
上記中間体化合物を、媒質に計画的に添加されまたは含まれている可能性がある薬剤または親核性基Z′で攻撃することができ、および
側鎖を有するセファロタキサンおよび/またはその塩の環状側鎖が、下記式
上記鎖を、場合によっては活性化および/または開環添加剤の存在下で、加水分解または慎重な加溶媒分解によって開環し、
下記式
Z′は、
ハロゲンであるか、または水素または(1)で定義したような基R11を有するヘテロ原子であるか、または
水素、炭化水素基であって、(複数の)ヘテロ原子を有しまたは有さない、飽和、不飽和または芳香族性の線状、分岐状および/または環状の上記基、特にアルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、アリール、またはヘテロシクロアルキル
である)の開環した側鎖を有するセファロタキサンを提供する、(1)〜(21)のいずれか1項に記載の方法。
Ω−CO−OH
(式中、基Ωは(1)で定義した通りである)
に相当し、
式(+)−Ω−CO−OHおよび(−)−Ω−CO−OHの化合物であって、例えば(+)−Ω−CO−OHが右旋性鏡像異性体を表し、(−)−Ω−CO−OHが左旋性鏡像異性体を表すものを含むラセミ混合物と同等な上式を、
a)上記ラセミ混合物、または(1)で定義した通りである式
Ω−CO−A
の活性化形態の1つであって、
上記のラセミ混合物または上記の活性化形態は、それぞれ
式(Ω−CO−O)−に相当するアニオン、または
式(Ω−CO)+に相当するカチオン
を生成するものを、キラル体(chiral entity)の純粋な鏡像異性形態と接触させ、Δ*(星印付きデルタ)の記号で表される上記の「分割剤」は、
共有結合による安定な組合せ、または
水素結合によるまたは疎水性相互作用による容易に逆転可能な不安定な組合せ、または
静電的相互作用による中程度の不安定性の組合せ
を形成する能力を有し、
Ω−CO−O−Δ*およびΩ−CO−Δ*のジアステレオマー混合物を提供し、
b)2個のジアステレオマーまたは2個の複合体化合物の混合物、または更に一般的には次いで得られた物理的および/または化学的に異なる2種類の新規なものを物理的に分離し、
c)一般式Ω*−CO−OH
(式中、Ω*(「星付きオメガ」)は下記式(+)−Ω−CO−OHおよび(−)−Ω−CO−OH(上記で定義した通りである)に相当する一方または他方の純粋な鏡像異性形態における同一キラル基の包括的記号を表す)
の鏡像異性体の1個ずつを再生し、最終的に分離することによって得る、(1)〜(21)および(34)のいずれか1項に記載の方法。
に相当する基である、(36)に記載の方法。
に相当する基である、(36)に記載の方法。
に相当する基である、(36)に記載の方法。
Ω−CO−O−[NH−Δ*]+
Ω−CO−O−[NH2−Δ*]+
Ω−CO−O−[NH3−Δ*]+
(式中、ΩおよびΔ*は(36)で定義した通りである)のいずれかに相当するキラルアミンと接触させることによって調製された塩によって表される、(36)〜(39)のいずれか1項に記載の方法。
下記式
に相当する第三ヘテロシクロアルカンカルボン酸であり、上記酸は、究極的には環化添加剤および/または脱水剤の存在下で非プロトン性またはプロトン性溶媒中で処理することによって得られ、上記処理は、究極的には形成された水を物理的に担持することによって支持され、または
下記式
に相当する開環した第三エチレン酸、または
下記式
に相当する開環した第三エチレン酸であり、
次いで、R12を後で、ケン化により、または水素化分解により、または更に一般的には酸の保護基を除去するための当該技術分野で通常の方法によって除去する、(1)〜(21)および(34)〜(49)のいずれか1項に記載の方法。
Zが酸素原子であり、かつ
1°)n=0、およびR5は基−CH2CO2Hまたは−CH2CO2CH3基であり、R6およびR8は同時に水素原子とはならない、
2°)n=2、同時にR6=R8=メチルであり、R5=OMeまたはヒドロキシル基、
3°)n=2、同時にR6=R8=メチル、およびR5=基−CH2CO2Hまたは−CH2CO2CH3、
4°)n=3、同時にR6はヒドロキシル基、R8はメチル基およびR5は基−CH2CO2CH3、
5°)n=2または3でありかつR5=基CH3であるときには、R6およびR8は互いに独立してHまたはMe、
6°)n=3およびR6=R8=Hであるとき、R5はフェニル、エチル、またはプロピル基、
7°)n=3およびR6=R8=Hであるとき、R5は
−CH(CH3)=CHCH3、−CH2CH(CH3)=CH2、−CH2CH=CH2、または3−シクロヘキセニル基、
8°)n=3、R6=R8=HおよびR5=エチル
である化合物を除く、
第三ヘテロシクロアルカンカルボン酸であって、その塩およびその純粋な鏡像異性体のそれぞれを包含し、またはラセミ混合物または可変組成物での第三ヘテロシクロアルカンカルボン酸。
第三オキサシクロアルカンカルボン酸であって、その塩およびその純粋な鏡像異性体のそれぞれを包含し、またはラセミ混合物または可変組成物での第三オキサシクロアルカンカルボン酸。
第三ヘテロシクロアルカンカルボン酸であって、その塩およびその純粋な鏡像異性体のそれぞれを包含し、またはラセミ混合物または可変組成物での第三オキサシクロアルカンカルボン酸。
第三オキサシクロアルカンカルボン酸ヘミエステルであって、その塩およびその純粋な鏡像異性体のそれぞれを包含し、またはラセミ混合物または可変組成物での第三オキサシクロアルカンカルボン酸ヘミエステル。
第三オキサシクロアルカンカルボン酸ヘミエステルであって、その塩およびその純粋な鏡像異性体のそれぞれを包含し、またはラセミ混合物または可変組成物での第三オキサシクロアルカンカルボン酸ヘミエステル。
に相当する、第三アルケンカルボン酸であって、その塩およびその純粋な鏡像異性体のそれぞれを包含し、またはラセミ混合物または可変組成物での第三アルケンカルボン酸。
に相当する、第三アルケンカルボン酸であって、その塩およびその純粋な鏡像異性体のそれぞれを含み、またはラセミ混合物または可変組成物での、第三アルケンカルボン酸。
に相当する、第三アルケンカルボン酸であって、その塩およびその純粋な鏡像異性体のそれぞれを含み、またはラセミ混合物または可変組成物での、第三アルケンカルボン酸。
に相当する第三アルケンカルボン酸であって、その塩およびその純粋な鏡像異性体のそれぞれを包含し、またはラセミ混合物または可変組成物での第三アルケンカルボン酸。
に相当する、環状無水物。
a)不適切な鏡像異性体純度の合成ホモハリングトン酸を導入する半合成法であって、生成した不純物が、下記式
b)植物での生合成法であって、不適切な鏡像異性体純度を有するセファロタキシンを導入し、またはセファロタキシン残基の部分ラセミ化による人工生成物の形態であって、生成した不純物が、非キラル系と厳密に同一のクロマトグラフィー特性を示し、絶対位置が上記のもの(鏡像異性体)と反対であり、下記式
から生成する2−エピ−ホモハリングトニンと命名された望ましくない関連不純物を除去し、
具体的には、下記のクロマトグラフィー系:
A)固定相:
アルキル−またはフェニル−またはアルキルフェニル−またはフェニルアルキル−シラン、好ましくはn−オクタデシルシラン、
B)移動相:
水−テトラヒドロフラン、水−メタノール、水−アセトニトリル、または水の代わりのpH2〜6.5の緩衝液、または同等な選択性を有する総ての他の移動相の一つを用いる、段階を含んでなる、(1)〜(21)および(34)〜(49)のいずれか1項に記載の方法。
Claims (25)
- 下記の構造式(1)または(2)から選択される化合物、もしくはそれらの混合物。
- 下記の構造式(3)または(4)から選択される化合物、もしくはそれらの混合物。
- 下記の構造式(5)または(6)から選択される化合物、もしくはそれらの混合物。
- 下記の構造式(7)または(8)から選択される化合物、もしくはそれらの混合物。
- 下記の構造式の化合物
R 5 は、水素原子、−CH 2 COOH、−CH 2 COOCH 3 、−CH 3 、−OH、−OCH 3 または−CH 2 CH 3 であり、
かつ、
R6およびR8は、独立して水素原子かフェニルであり(ただし、R 6 およびR 8 は同時に水素原子とはならない)、さらにR 5 が−CH 2 COOHまたは−CH 2 COOCH 3 の場合は、R 6 およびR 8 は独立して−CH 3 であってもよい)、
その塩、その純粋な鏡像異性体のそれぞれの化合物、それらのラセミ混合物、または該構造式に包含される各化合物の量比が可変である組成物。 - R6およびR8は、独立して、アルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、またはアリールである、請求項5に記載の化合物。
- 下記の構造式
- 下記の構造式の化合物
- 下記の構造式
- 下記の構造式
- 下記の構造式
- 下記の構造式
- 下記の構造式
- 下記の構造式
- 下記の構造式
- 下記の構造式
- 下記の構造式
の化合物、その塩、その純粋な鏡像異性体のそれぞれの化合物、それらのラセミ混合物、または該構造式に包含される各化合物の量比が可変である組成物。 - 下記の構造式
の化合物、その塩、その純粋な鏡像異性体のそれぞれの化合物、それらのラセミ混合物、または該構造式に包含される各化合物の量比が可変である組成物。 - 下記の構造式
- 下記の構造式
- 下記の構造式
の化合物、その塩、その純粋な鏡像異性体のそれぞれの化合物、それらのラセミ混合物、または該構造式に包含される各化合物の量比が可変である組成物。 - m=1である、請求項21に記載の化合物。
- 下記式
- 一般式
- 一般式
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9803492A FR2776292B1 (fr) | 1998-03-20 | 1998-03-20 | Cephalotaxanes porteurs de chaine laterale et leur procede de synthese |
| FR98/03492 | 1998-03-20 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2000537877A Division JP4942871B2 (ja) | 1998-03-20 | 1999-03-17 | 新規なセファロタキサン誘導体およびそれらの製造法 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015056022A Division JP6087381B2 (ja) | 1998-03-20 | 2015-03-19 | 新規なセファロタキサン誘導体の製造に用いられるカルボン酸誘導体 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2009132735A JP2009132735A (ja) | 2009-06-18 |
| JP5884005B2 true JP5884005B2 (ja) | 2016-03-15 |
Family
ID=9524328
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2000537877A Expired - Fee Related JP4942871B2 (ja) | 1998-03-20 | 1999-03-17 | 新規なセファロタキサン誘導体およびそれらの製造法 |
| JP2009063260A Expired - Fee Related JP5884005B2 (ja) | 1998-03-20 | 2009-03-16 | 新規なセファロタキサン誘導体の製造に用いられるカルボン酸誘導体 |
| JP2015056022A Expired - Lifetime JP6087381B2 (ja) | 1998-03-20 | 2015-03-19 | 新規なセファロタキサン誘導体の製造に用いられるカルボン酸誘導体 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2000537877A Expired - Fee Related JP4942871B2 (ja) | 1998-03-20 | 1999-03-17 | 新規なセファロタキサン誘導体およびそれらの製造法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015056022A Expired - Lifetime JP6087381B2 (ja) | 1998-03-20 | 2015-03-19 | 新規なセファロタキサン誘導体の製造に用いられるカルボン酸誘導体 |
Country Status (19)
| Country | Link |
|---|---|
| US (5) | US6831180B1 (ja) |
| EP (1) | EP1064285B1 (ja) |
| JP (3) | JP4942871B2 (ja) |
| KR (1) | KR20010042085A (ja) |
| CN (1) | CN1220692C (ja) |
| AT (1) | ATE289312T1 (ja) |
| AU (1) | AU3270699A (ja) |
| CA (1) | CA2324895A1 (ja) |
| DE (1) | DE69923768T2 (ja) |
| DK (1) | DK1064285T3 (ja) |
| ES (1) | ES2237144T3 (ja) |
| FR (1) | FR2776292B1 (ja) |
| HU (1) | HUP0101375A3 (ja) |
| IL (1) | IL138563A0 (ja) |
| PL (1) | PL343032A1 (ja) |
| PT (1) | PT1064285E (ja) |
| TR (1) | TR200003613T2 (ja) |
| WO (1) | WO1999048894A1 (ja) |
| ZA (1) | ZA200005018B (ja) |
Families Citing this family (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2776292B1 (fr) | 1998-03-20 | 2004-09-10 | Oncopharm | Cephalotaxanes porteurs de chaine laterale et leur procede de synthese |
| AU7940500A (en) * | 2000-10-17 | 2002-04-29 | Oncopharm Corp | New cephalotaxanes, their method of preparation and their use in treatment of cancers, leukemias, parasites including thus resistant to usual chemotherapeutic agents and as reversal agents |
| US7285546B2 (en) | 2000-10-17 | 2007-10-23 | Stragen Pharma S.A. | Cephalotaxanes, their method of preparation and their use in treatment of cancers, leukemias, parasites including those resistant to usual chemotherapeutic agents and as reversal agents |
| JP2004523586A (ja) * | 2001-03-21 | 2004-08-05 | オンコファーム コーポレーション | 高純度で結晶性形状のハリントニン、および経口投与法を使用してのガンの治療にとくに有益な医薬品組成物への配合に使用することを可能にする、天然、合成または半合成の供給源から得られる粗アルカロイドの精製によるハリントニンの調製法 |
| JP2006501199A (ja) | 2002-07-17 | 2006-01-12 | ケムジェネックス・ファーマシューティカルズ・リミテッド | ホモハリングトニンを含むセファロタキシンの製剤および投与方法 |
| CN100396286C (zh) * | 2002-12-30 | 2008-06-25 | 北京大学第一医院 | 高三尖杉酯碱和三尖杉酯碱在制备抑制血管生成药物中的应用 |
| ITMI20051352A1 (it) * | 2005-07-15 | 2007-01-16 | Erregierre Spa | Processo di preparazi0ne di acido 2-metossicarbonilmetil-6,6-dimetil-2-tetraidropiran carbossilico |
| CN101677951A (zh) * | 2007-04-13 | 2010-03-24 | 化学基因制药公司 | 三尖杉碱口服剂型 |
| EP2260041A4 (en) * | 2008-03-03 | 2012-04-25 | Sloan Kettering Inst Cancer | CEPHALOTAXUS ESTERS, SYNTHESIS METHODS AND USES THEREOF |
| US20120022250A1 (en) * | 2009-03-11 | 2012-01-26 | Jean-Pierre Robin | Process for preparing cephalotaxine esters |
| US20100240887A1 (en) * | 2009-03-23 | 2010-09-23 | Yaguang Liu | New methods of producing HHT |
| WO2013023622A1 (zh) * | 2011-08-18 | 2013-02-21 | 杭州本生药业有限公司 | 高三尖杉酯碱的酰化衍生物、及其制备方法和应用 |
| CN103635476B (zh) * | 2011-08-18 | 2016-03-09 | 杭州本生药业有限公司 | 高三尖杉酯碱的酰化衍生物、及其制备方法和应用 |
| CN103687859B (zh) * | 2011-08-18 | 2016-08-17 | 杭州本生药业有限公司 | 高三尖杉酯碱的胺化衍生物、及其制备方法和应用 |
| JP6068472B2 (ja) * | 2011-08-18 | 2017-01-25 | ハンジョウ ベンシェン ファーマシューティカル シーオー., エルティーディー.Hangzhou Bensheng Pharmaceutical Co., Ltd. | アミノ化ホモハリントニン誘導体、その調製方法及び使用 |
| CN102675327B (zh) * | 2012-03-01 | 2014-12-10 | 南开大学 | 三尖杉酯碱类似物及制备方法和应用 |
| CN102633806A (zh) * | 2012-04-05 | 2012-08-15 | 贵州博丰生物科技产业开发有限公司 | 一种三尖杉酯碱的提取分离方法 |
| TN2016000269A1 (en) * | 2013-12-31 | 2017-10-06 | Thierry Bataille | Water soluble crystallin salts of certain harringtonines unambiguously protonated on their alkaloid nitrogen and their use as chemotherapeutic drugs. |
| WO2015128463A1 (en) * | 2014-02-28 | 2015-09-03 | Jean-Pierre Robin | Water soluble salts of harringtonine and their pharmaceutical applications |
| WO2015162217A2 (en) * | 2014-04-23 | 2015-10-29 | Jean-Pierre Robin | Crystalline salts of cephalotaxine and their use for its purification including enantiomers |
| WO2016182850A1 (en) * | 2015-05-08 | 2016-11-17 | Albany Molecular Research, Inc. | Methods and intermediates for the preparation of omacetaxine and cephalotaxine derivatives thereof |
| CN106866690B (zh) * | 2015-12-10 | 2019-10-11 | 南开大学 | 三尖杉酯类生物碱、其制备方法和用途 |
| EP3275875A1 (en) | 2016-07-28 | 2018-01-31 | INDENA S.p.A. | Dioxolanone intermediate useful in the synthesis of homoharringtonine |
| CN110577506B (zh) * | 2018-06-07 | 2023-04-07 | 中国科学院上海有机化学研究所 | (﹣)-三尖杉碱的酯类衍生物的合成方法及其中间体 |
| EP3763715A1 (en) | 2019-07-11 | 2021-01-13 | Robin, Jean-Pierre | Harringtonines salts, in particular retinoates, their process of preparation and their uses in the treatment of leukemias, cancers, autoimmune, skin, alzheimer's and inflammatory bowel diseases and viral infections, combined with myelopoiesis stimulating agents |
| CN114544849B (zh) * | 2022-02-22 | 2024-07-19 | 苏州正济医药研究有限公司 | 一种1h-1,2,4-三氮唑-1-甲脒单盐酸盐及杂质的检测方法 |
Family Cites Families (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2889340A (en) * | 1956-07-02 | 1959-06-02 | Trubek Lab | Unsaturated dialkyl substituted 2, 3-epoxyacid esters |
| US2889339A (en) * | 1956-07-02 | 1959-06-02 | Trubek Lab | Dialkyl substituted 2, 3-epoxyacid esters |
| DE1142595B (de) * | 1961-05-05 | 1963-01-24 | Bayer Ag | Verfahren zur Herstellung von 4-(2,6,6-Trimethyl-cyclohexen-(1)-yl)-2-methylbuten-(3)-al-(1) |
| US3225069A (en) * | 1963-04-18 | 1965-12-21 | Atlas Chem Ind | Process of producing esters of hydroxymethyletrahydrofuroic acid |
| US3870727A (en) * | 1970-04-09 | 1975-03-11 | Us Agriculture | Production of harringtonine and isoharringtonine |
| US3725437A (en) * | 1970-09-16 | 1973-04-03 | Sankyo Chem Ind Ltd | Process for the preparation of {60 -hydroxy-{62 -phenypropionic acid derivatives and alkalimetal-or ammonium salts thereof |
| JPS5227147B1 (ja) * | 1971-06-16 | 1977-07-19 | ||
| US3793454A (en) | 1971-11-12 | 1974-02-19 | Us Agriculture | Harringtonine and isopharringtonine for treating l1210 or p388 leukemic tumors in mice |
| GB1521904A (en) * | 1974-07-27 | 1978-08-16 | Nisshin Flour Milling Co | 3-methyl-3-(substituted aryl)-pyruvic acids and a process of preparing them and their lower alkyl esters |
| US3959312A (en) | 1974-12-20 | 1976-05-25 | The United States Of America As Represented By The Secretary Of Agriculture | Synthesis of antitumor alkaloid deoxyharringtonine and its precursor 3'-0-(5-methyl-2-oxohexanoyl)-cephalotaxine |
| JPS5262237A (en) * | 1975-11-19 | 1977-05-23 | Ono Pharmaceut Co Ltd | Process for preparation of alpha-substituted phenylalkane carboxylic a cid |
| US4046889A (en) * | 1976-02-13 | 1977-09-06 | E. R. Squibb & Sons, Inc. | Azetidine-2-carboxylic acid derivatives |
| US4178286A (en) * | 1976-10-28 | 1979-12-11 | Research Corporation | β-Lactams, their production, intermediates thereto and derivatives thereof |
| US4152214A (en) * | 1977-10-07 | 1979-05-01 | The United States Of America As Represented By The Secretary Of Agriculture | Production of homodeoxyharringtonine and other cephalotaxine esters by tissue culture |
| US4152333A (en) | 1978-02-22 | 1979-05-01 | The United States Of America As Represented By The Secretary Of Agriculture | Synthetic cephalotaxine esters having antileukemic activity |
| US4203996A (en) * | 1978-02-22 | 1980-05-20 | The United States Of America As Represented By The Secretary Of Agriculture | Synthetic cephalotaxine esters having antileukemic P388 activity |
| US4252728A (en) * | 1978-04-17 | 1981-02-24 | Firmenich Sa | Norbornane and norbornene derivatives |
| US4132720A (en) * | 1978-04-20 | 1979-01-02 | Mcneil Laboratories, Inc. | Alkenylglycidic acid derivatives |
| DE2833986A1 (de) * | 1978-08-03 | 1980-02-21 | Boehringer Mannheim Gmbh | Immunstimulierende n-substituierte aziridin-2-carbonsaeurederivate, verfahren zu ihrer herstellung sowie arzneimittel, die diese substanzen enthalten |
| WO1982000822A1 (en) * | 1980-08-29 | 1982-03-18 | Eistetter K | Substituted oxiranecarboxylic acids,their preparation and their use as medicaments |
| JPS5764695A (en) * | 1980-10-09 | 1982-04-19 | Kyowa Hakko Kogyo Co Ltd | Novel cephalotaxine derivative |
| JPS57102194A (en) * | 1980-12-17 | 1982-06-25 | Kyowa Hakko Kogyo Co Ltd | Preparation of cephalotaxine and ester thereof |
| JPS5832880A (ja) | 1981-08-24 | 1983-02-25 | Isukura Sangyo Kk | ハリントニンの部分合成方法 |
| USH271H (en) | 1984-08-06 | 1987-05-05 | The United States Of America As Represented By The Secretary Of The Army | Treatment of malaria with esters of cephalotaxine |
| JPS6153635A (ja) * | 1984-08-24 | 1986-03-17 | Fuji Photo Film Co Ltd | 熱現像感光材料 |
| US4808629A (en) | 1985-05-28 | 1989-02-28 | Yaguang Liu | Safe antileukemia drug, SAL |
| US4675318A (en) | 1985-05-28 | 1987-06-23 | Yaguang Liu | Safe antileukemia drug, SAL |
| US4783454A (en) | 1985-05-28 | 1988-11-08 | Yaguang Liu | Process for producing harringtonine and homoharringtonine |
| DE3639879A1 (de) * | 1986-11-21 | 1988-06-01 | Hoechst Ag | Verfahren zur herstellung von mono-, bi- und tricyclischen aminosaeuren, zwischenprodukte dieses verfahrens sowie ein verfahren zu deren herstellung |
| US4870208A (en) * | 1988-06-03 | 1989-09-26 | Monsanto Company | Asymmetric hydrogenolysis of epoxides |
| FR2633620B1 (fr) * | 1988-07-01 | 1992-02-21 | Elf Aquitaine | Oxydation d'epoxy-alcools en epoxy-acides carboxyliques |
| FR2649980A1 (ja) * | 1989-07-18 | 1991-01-25 | Sanofi Sa | |
| FR2658513B1 (fr) * | 1990-02-21 | 1994-02-04 | Rhone Poulenc Sante | Procede de preparation de l'acide cis-beta-phenylglycidique-(2r,3r). |
| MY106399A (en) * | 1990-07-24 | 1995-05-30 | Pfizer | Cephalosporins and homologeus, preparation and pharmaceutical composition |
| US5442105A (en) * | 1991-06-21 | 1995-08-15 | Takasago International Corporation | Process for producing cyclohexylbutyric acid derivative |
| DE4122218A1 (de) * | 1991-07-04 | 1993-01-07 | Consortium Elektrochem Ind | Verfahren zur herstellung optisch aktiver carbonsaeuren |
| US5484926A (en) * | 1993-10-07 | 1996-01-16 | Agouron Pharmaceuticals, Inc. | HIV protease inhibitors |
| US5677470A (en) * | 1994-06-28 | 1997-10-14 | Tanabe Seiyaku Co., Ltd. | Baccatin derivatives and processes for preparing the same |
| US6107291A (en) * | 1997-12-19 | 2000-08-22 | Amgen Inc. | Azepine or larger medium ring derivatives and methods of use |
| FR2776292B1 (fr) * | 1998-03-20 | 2004-09-10 | Oncopharm | Cephalotaxanes porteurs de chaine laterale et leur procede de synthese |
| FR2818970A1 (fr) | 2000-12-29 | 2002-07-05 | Oncopharm | Procede de preparation enantioselective d'adduits de phenylethylimino-cycloalcanes alpha substitues et de 2-acetoxyacrylonitrile, intermediaires dans la synthese enantoselective de la chaine laterale des harringtonines |
| US20020128258A1 (en) * | 2001-03-09 | 2002-09-12 | Jean-Pierre Robin | Therapeutical method involving subcutaneous administration of drugs containing cephalotaxine derivatives |
| JP2006501199A (ja) | 2002-07-17 | 2006-01-12 | ケムジェネックス・ファーマシューティカルズ・リミテッド | ホモハリングトニンを含むセファロタキシンの製剤および投与方法 |
-
1998
- 1998-03-20 FR FR9803492A patent/FR2776292B1/fr not_active Expired - Lifetime
-
1999
- 1999-03-16 US US09/270,006 patent/US6831180B1/en not_active Expired - Lifetime
- 1999-03-17 WO PCT/IB1999/000491 patent/WO1999048894A1/en active IP Right Grant
- 1999-03-17 CN CNB998060445A patent/CN1220692C/zh not_active Expired - Lifetime
- 1999-03-17 PL PL99343032A patent/PL343032A1/xx unknown
- 1999-03-17 HU HU0101375A patent/HUP0101375A3/hu unknown
- 1999-03-17 TR TR2000/03613T patent/TR200003613T2/xx unknown
- 1999-03-17 PT PT99942587T patent/PT1064285E/pt unknown
- 1999-03-17 JP JP2000537877A patent/JP4942871B2/ja not_active Expired - Fee Related
- 1999-03-17 IL IL13856399A patent/IL138563A0/xx unknown
- 1999-03-17 KR KR1020007010440A patent/KR20010042085A/ko not_active Application Discontinuation
- 1999-03-17 DK DK99942587T patent/DK1064285T3/da active
- 1999-03-17 AU AU32706/99A patent/AU3270699A/en not_active Abandoned
- 1999-03-17 EP EP99942587A patent/EP1064285B1/en not_active Expired - Lifetime
- 1999-03-17 AT AT99942587T patent/ATE289312T1/de active
- 1999-03-17 CA CA002324895A patent/CA2324895A1/en not_active Abandoned
- 1999-03-17 ES ES99942587T patent/ES2237144T3/es not_active Expired - Lifetime
- 1999-03-17 DE DE69923768T patent/DE69923768T2/de not_active Expired - Lifetime
-
2000
- 2000-09-20 ZA ZA200005018A patent/ZA200005018B/en unknown
-
2001
- 2001-03-27 US US09/817,176 patent/US6613900B2/en not_active Expired - Lifetime
-
2004
- 2004-06-25 US US10/877,067 patent/US7169774B2/en not_active Expired - Lifetime
-
2006
- 2006-05-25 US US11/440,648 patent/US7842687B2/en not_active Ceased
-
2009
- 2009-03-16 JP JP2009063260A patent/JP5884005B2/ja not_active Expired - Fee Related
-
2012
- 2012-10-26 US US13/661,677 patent/USRE45128E1/en not_active Expired - Fee Related
-
2015
- 2015-03-19 JP JP2015056022A patent/JP6087381B2/ja not_active Expired - Lifetime
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6087381B2 (ja) | 新規なセファロタキサン誘導体の製造に用いられるカルボン酸誘導体 | |
| EP0559019B1 (en) | 14-beta-hydroxy-10-deacytyl-baccatine III and its derivatives as antitumour agents | |
| Nicolaou et al. | Synthesis of zaragozic acid A/squalestatin S1 | |
| DE602004007145T2 (de) | Regioselektive synthese von cci-779 | |
| SI9520078A (en) | Semi-synthetic taxanes with anti-tumoural activity | |
| JP2009512662A (ja) | 中間体、並びにかかる中間体を用いたエクチナサイジン583、597などのエクチナサイジン類の調製方法 | |
| RU2168513C2 (ru) | Производные таксана, способ их получения и фармацевтическая композиция | |
| DE60106057T2 (de) | Verfahren zur herstellung von baccatin iii-derivativen | |
| US5698712A (en) | Baccatine III derivatives | |
| WO2002022618A1 (fr) | Preparation de la camptothecine et de ses derives | |
| Riehs et al. | EPC synthesis of (+)-heptelidic acid | |
| KR20080056633A (ko) | 탁산유도체의 제조방법 및 이에 사용되는 중간체 | |
| CZ20003451A3 (cs) | Nové cefalotaxanové deriváty a způsob jejich přípravy | |
| DE19813821A1 (de) | Verfahren zur Herstellung von C1-C6-Bausteinen zur Totalsynthese von Epothilon und Epothilon-Derivaten | |
| Riehs et al. | EPC-Synthese von (+)-heptelidsäure | |
| EP2493885B1 (fr) | Molecules polyketides comme agents anticancereux | |
| JPS6143354B2 (ja) | ||
| Baars | Towards a Total Synthesis of Peloruside A and the Preparation of Selected Inositol Derivatives | |
| Yue | Towards the total synthesis of Azaspiracid-3 | |
| JPS63316777A (ja) | ボルネン誘導体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20090316 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A711 Effective date: 20091222 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20091222 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120508 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20120807 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20120810 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20120910 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20120913 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20121005 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130730 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20131030 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140318 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20140618 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20140623 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140718 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140919 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20141219 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20141225 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20150219 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20150319 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20150403 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20150422 |
|
| R155 | Notification before disposition of declining of application |
Free format text: JAPANESE INTERMEDIATE CODE: R155 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20150602 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A711 Effective date: 20160114 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20160114 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5884005 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |