JP5252130B2 - ノボラック型フェノール樹脂の製造方法 - Google Patents
ノボラック型フェノール樹脂の製造方法 Download PDFInfo
- Publication number
- JP5252130B2 JP5252130B2 JP2012520422A JP2012520422A JP5252130B2 JP 5252130 B2 JP5252130 B2 JP 5252130B2 JP 2012520422 A JP2012520422 A JP 2012520422A JP 2012520422 A JP2012520422 A JP 2012520422A JP 5252130 B2 JP5252130 B2 JP 5252130B2
- Authority
- JP
- Japan
- Prior art keywords
- reaction
- phenol resin
- phenols
- water
- aldehydes
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 52
- 239000010680 novolac-type phenolic resin Substances 0.000 title description 4
- 238000006243 chemical reaction Methods 0.000 claims abstract description 130
- 239000005011 phenolic resin Substances 0.000 claims abstract description 86
- 150000002989 phenols Chemical class 0.000 claims abstract description 61
- 150000001299 aldehydes Chemical class 0.000 claims abstract description 40
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical compound OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 claims abstract description 34
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Natural products P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 claims abstract description 23
- -1 phosphine compound Chemical group 0.000 claims abstract description 23
- 229910000073 phosphorus hydride Inorganic materials 0.000 claims abstract description 22
- 239000007809 chemical reaction catalyst Substances 0.000 claims abstract description 17
- 229910052799 carbon Inorganic materials 0.000 claims abstract description 5
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 74
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 claims description 44
- 229920003986 novolac Polymers 0.000 claims description 32
- 238000002156 mixing Methods 0.000 claims description 25
- 230000003068 static effect Effects 0.000 claims description 7
- 229920001568 phenolic resin Polymers 0.000 claims description 6
- KXGFMDJXCMQABM-UHFFFAOYSA-N 2-methoxy-6-methylphenol Chemical compound [CH]OC1=CC=CC([CH])=C1O KXGFMDJXCMQABM-UHFFFAOYSA-N 0.000 claims description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 4
- 238000007789 sealing Methods 0.000 claims description 4
- 238000000034 method Methods 0.000 abstract description 20
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 62
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 27
- 239000000203 mixture Substances 0.000 description 21
- 239000008346 aqueous phase Substances 0.000 description 19
- 238000004821 distillation Methods 0.000 description 16
- 239000007864 aqueous solution Substances 0.000 description 15
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 12
- 230000000052 comparative effect Effects 0.000 description 12
- 230000000694 effects Effects 0.000 description 12
- 238000004817 gas chromatography Methods 0.000 description 12
- 229920005989 resin Polymers 0.000 description 12
- 239000011347 resin Substances 0.000 description 12
- 230000008569 process Effects 0.000 description 10
- 238000010992 reflux Methods 0.000 description 10
- 238000005292 vacuum distillation Methods 0.000 description 10
- 238000005406 washing Methods 0.000 description 10
- 239000002253 acid Substances 0.000 description 9
- 239000012074 organic phase Substances 0.000 description 9
- 239000002994 raw material Substances 0.000 description 9
- 238000003756 stirring Methods 0.000 description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 6
- 230000009471 action Effects 0.000 description 6
- 150000003009 phosphonic acids Chemical class 0.000 description 6
- ZTQSAGDEMFDKMZ-UHFFFAOYSA-N Butyraldehyde Chemical compound CCCC=O ZTQSAGDEMFDKMZ-UHFFFAOYSA-N 0.000 description 5
- 241000282320 Panthera leo Species 0.000 description 5
- 239000006087 Silane Coupling Agent Substances 0.000 description 5
- 239000003054 catalyst Substances 0.000 description 5
- 239000003426 co-catalyst Substances 0.000 description 5
- 239000012530 fluid Substances 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 230000003197 catalytic effect Effects 0.000 description 4
- 235000006408 oxalic acid Nutrition 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 4
- HGINCPLSRVDWNT-UHFFFAOYSA-N Acrolein Chemical compound C=CC=O HGINCPLSRVDWNT-UHFFFAOYSA-N 0.000 description 3
- 229930040373 Paraformaldehyde Natural products 0.000 description 3
- YSMRWXYRXBRSND-UHFFFAOYSA-N TOTP Chemical compound CC1=CC=CC=C1OP(=O)(OC=1C(=CC=CC=1)C)OC1=CC=CC=C1C YSMRWXYRXBRSND-UHFFFAOYSA-N 0.000 description 3
- 150000001875 compounds Chemical class 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 238000000354 decomposition reaction Methods 0.000 description 3
- VRIVJOXICYMTAG-IYEMJOQQSA-L iron(ii) gluconate Chemical compound [Fe+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O VRIVJOXICYMTAG-IYEMJOQQSA-L 0.000 description 3
- 238000004811 liquid chromatography Methods 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- NKTOLZVEWDHZMU-UHFFFAOYSA-N 2,5-xylenol Chemical compound CC1=CC=C(C)C(O)=C1 NKTOLZVEWDHZMU-UHFFFAOYSA-N 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- NBBJYMSMWIIQGU-UHFFFAOYSA-N Propionic aldehyde Chemical compound CCC=O NBBJYMSMWIIQGU-UHFFFAOYSA-N 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical compound C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 description 2
- PXKLMJQFEQBVLD-UHFFFAOYSA-N bisphenol F Chemical compound C1=CC(O)=CC=C1CC1=CC=C(O)C=C1 PXKLMJQFEQBVLD-UHFFFAOYSA-N 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 239000002783 friction material Substances 0.000 description 2
- HYBBIBNJHNGZAN-UHFFFAOYSA-N furfural Chemical compound O=CC1=CC=CO1 HYBBIBNJHNGZAN-UHFFFAOYSA-N 0.000 description 2
- LEQAOMBKQFMDFZ-UHFFFAOYSA-N glyoxal Chemical compound O=CC=O LEQAOMBKQFMDFZ-UHFFFAOYSA-N 0.000 description 2
- VKYKSIONXSXAKP-UHFFFAOYSA-N hexamethylenetetramine Chemical compound C1N(C2)CN3CN1CN2C3 VKYKSIONXSXAKP-UHFFFAOYSA-N 0.000 description 2
- JARKCYVAAOWBJS-UHFFFAOYSA-N hexanal Chemical compound CCCCCC=O JARKCYVAAOWBJS-UHFFFAOYSA-N 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 239000012778 molding material Substances 0.000 description 2
- IXQGCWUGDFDQMF-UHFFFAOYSA-N o-Hydroxyethylbenzene Natural products CCC1=CC=CC=C1O IXQGCWUGDFDQMF-UHFFFAOYSA-N 0.000 description 2
- QWVGKYWNOKOFNN-UHFFFAOYSA-N o-cresol Chemical compound CC1=CC=CC=C1O QWVGKYWNOKOFNN-UHFFFAOYSA-N 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 229920002866 paraformaldehyde Polymers 0.000 description 2
- DTUQWGWMVIHBKE-UHFFFAOYSA-N phenylacetaldehyde Chemical compound O=CCC1=CC=CC=C1 DTUQWGWMVIHBKE-UHFFFAOYSA-N 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- GHMLBKRAJCXXBS-UHFFFAOYSA-N resorcinol Chemical compound OC1=CC=CC(O)=C1 GHMLBKRAJCXXBS-UHFFFAOYSA-N 0.000 description 2
- SMQUZDBALVYZAC-UHFFFAOYSA-N salicylaldehyde Chemical compound OC1=CC=CC=C1C=O SMQUZDBALVYZAC-UHFFFAOYSA-N 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- YWWDBCBWQNCYNR-UHFFFAOYSA-N trimethylphosphine Chemical compound CP(C)C YWWDBCBWQNCYNR-UHFFFAOYSA-N 0.000 description 2
- HGBOYTHUEUWSSQ-UHFFFAOYSA-N valeric aldehyde Natural products CCCCC=O HGBOYTHUEUWSSQ-UHFFFAOYSA-N 0.000 description 2
- QQVDJLLNRSOCEL-UHFFFAOYSA-N (2-aminoethyl)phosphonic acid Chemical compound [NH3+]CCP(O)([O-])=O QQVDJLLNRSOCEL-UHFFFAOYSA-N 0.000 description 1
- KYLUAQBYONVMCP-UHFFFAOYSA-N (2-methylphenyl)phosphane Chemical compound CC1=CC=CC=C1P KYLUAQBYONVMCP-UHFFFAOYSA-N 0.000 description 1
- MGRVRXRGTBOSHW-UHFFFAOYSA-N (aminomethyl)phosphonic acid Chemical compound NCP(O)(O)=O MGRVRXRGTBOSHW-UHFFFAOYSA-N 0.000 description 1
- KJCVRFUGPWSIIH-UHFFFAOYSA-N 1-naphthol Chemical compound C1=CC=C2C(O)=CC=CC2=C1 KJCVRFUGPWSIIH-UHFFFAOYSA-N 0.000 description 1
- CJWNFAKWHDOUKL-UHFFFAOYSA-N 2-(2-phenylpropan-2-yl)phenol Chemical compound C=1C=CC=C(O)C=1C(C)(C)C1=CC=CC=C1 CJWNFAKWHDOUKL-UHFFFAOYSA-N 0.000 description 1
- KHTJRKQAETUUQH-UHFFFAOYSA-N 2-(hydroxymethyl)octadecanamide Chemical compound CCCCCCCCCCCCCCCCC(CO)C(N)=O KHTJRKQAETUUQH-UHFFFAOYSA-N 0.000 description 1
- CDAWCLOXVUBKRW-UHFFFAOYSA-N 2-aminophenol Chemical compound NC1=CC=CC=C1O CDAWCLOXVUBKRW-UHFFFAOYSA-N 0.000 description 1
- CRBJBYGJVIBWIY-UHFFFAOYSA-N 2-isopropylphenol Chemical compound CC(C)C1=CC=CC=C1O CRBJBYGJVIBWIY-UHFFFAOYSA-N 0.000 description 1
- IQUPABOKLQSFBK-UHFFFAOYSA-N 2-nitrophenol Chemical compound OC1=CC=CC=C1[N+]([O-])=O IQUPABOKLQSFBK-UHFFFAOYSA-N 0.000 description 1
- SZHQPBJEOCHCKM-UHFFFAOYSA-N 2-phosphonobutane-1,2,4-tricarboxylic acid Chemical compound OC(=O)CCC(P(O)(O)=O)(C(O)=O)CC(O)=O SZHQPBJEOCHCKM-UHFFFAOYSA-N 0.000 description 1
- VPWNQTHUCYMVMZ-UHFFFAOYSA-N 4,4'-sulfonyldiphenol Chemical compound C1=CC(O)=CC=C1S(=O)(=O)C1=CC=C(O)C=C1 VPWNQTHUCYMVMZ-UHFFFAOYSA-N 0.000 description 1
- NTDQQZYCCIDJRK-UHFFFAOYSA-N 4-octylphenol Chemical compound CCCCCCCCC1=CC=C(O)C=C1 NTDQQZYCCIDJRK-UHFFFAOYSA-N 0.000 description 1
- QHPQWRBYOIRBIT-UHFFFAOYSA-N 4-tert-butylphenol Chemical compound CC(C)(C)C1=CC=C(O)C=C1 QHPQWRBYOIRBIT-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 239000004593 Epoxy Substances 0.000 description 1
- IGFHQQFPSIBGKE-UHFFFAOYSA-N Nonylphenol Natural products CCCCCCCCCC1=CC=C(O)C=C1 IGFHQQFPSIBGKE-UHFFFAOYSA-N 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 206010037660 Pyrexia Diseases 0.000 description 1
- IKHGUXGNUITLKF-XPULMUKRSA-N acetaldehyde Chemical compound [14CH]([14CH3])=O IKHGUXGNUITLKF-XPULMUKRSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- YXVFYQXJAXKLAK-UHFFFAOYSA-N biphenyl-4-ol Chemical compound C1=CC(O)=CC=C1C1=CC=CC=C1 YXVFYQXJAXKLAK-UHFFFAOYSA-N 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- MLUCVPSAIODCQM-NSCUHMNNSA-N crotonaldehyde Chemical compound C\C=C\C=O MLUCVPSAIODCQM-NSCUHMNNSA-N 0.000 description 1
- MLUCVPSAIODCQM-UHFFFAOYSA-N crotonaldehyde Natural products CC=CC=O MLUCVPSAIODCQM-UHFFFAOYSA-N 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- HASCQPSFPAKVEK-UHFFFAOYSA-N dimethyl(phenyl)phosphine Chemical compound CP(C)C1=CC=CC=C1 HASCQPSFPAKVEK-UHFFFAOYSA-N 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 239000012156 elution solvent Substances 0.000 description 1
- ZJOLCKGSXLIVAA-UHFFFAOYSA-N ethene;octadecanamide Chemical compound C=C.CCCCCCCCCCCCCCCCCC(N)=O.CCCCCCCCCCCCCCCCCC(N)=O ZJOLCKGSXLIVAA-UHFFFAOYSA-N 0.000 description 1
- 239000008098 formaldehyde solution Substances 0.000 description 1
- 238000001879 gelation Methods 0.000 description 1
- 229940015043 glyoxal Drugs 0.000 description 1
- 230000020169 heat generation Effects 0.000 description 1
- FEEPBTVZSYQUDP-UHFFFAOYSA-N heptatriacontanediamide Chemical compound NC(=O)CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC(N)=O FEEPBTVZSYQUDP-UHFFFAOYSA-N 0.000 description 1
- 239000004312 hexamethylene tetramine Substances 0.000 description 1
- 235000010299 hexamethylene tetramine Nutrition 0.000 description 1
- VLHZUYUOEGBBJB-UHFFFAOYSA-N hydroxy stearic acid Natural products OCCCCCCCCCCCCCCCCCC(O)=O VLHZUYUOEGBBJB-UHFFFAOYSA-N 0.000 description 1
- 238000010813 internal standard method Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- RLSSMJSEOOYNOY-UHFFFAOYSA-N m-cresol Chemical compound CC1=CC=CC(O)=C1 RLSSMJSEOOYNOY-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 229940100630 metacresol Drugs 0.000 description 1
- WSFSSNUMVMOOMR-NJFSPNSNSA-N methanone Chemical compound O=[14CH2] WSFSSNUMVMOOMR-NJFSPNSNSA-N 0.000 description 1
- UJNZOIKQAUQOCN-UHFFFAOYSA-N methyl(diphenyl)phosphane Chemical compound C=1C=CC=CC=1P(C)C1=CC=CC=C1 UJNZOIKQAUQOCN-UHFFFAOYSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000005065 mining Methods 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- NXPPAOGUKPJVDI-UHFFFAOYSA-N naphthalene-1,2-diol Chemical compound C1=CC=CC2=C(O)C(O)=CC=C21 NXPPAOGUKPJVDI-UHFFFAOYSA-N 0.000 description 1
- SNQQPOLDUKLAAF-UHFFFAOYSA-N nonylphenol Chemical compound CCCCCCCCCC1=CC=CC=C1O SNQQPOLDUKLAAF-UHFFFAOYSA-N 0.000 description 1
- LYRFLYHAGKPMFH-UHFFFAOYSA-N octadecanamide Chemical compound CCCCCCCCCCCCCCCCCC(N)=O LYRFLYHAGKPMFH-UHFFFAOYSA-N 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- IWDCLRJOBJJRNH-UHFFFAOYSA-N p-cresol Chemical compound CC1=CC=C(O)C=C1 IWDCLRJOBJJRNH-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 229940100595 phenylacetaldehyde Drugs 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920006324 polyoxymethylene Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 239000004576 sand Substances 0.000 description 1
- 239000003566 sealing material Substances 0.000 description 1
- 239000011257 shell material Substances 0.000 description 1
- FZHAPNGMFPVSLP-UHFFFAOYSA-N silanamine Chemical compound [SiH3]N FZHAPNGMFPVSLP-UHFFFAOYSA-N 0.000 description 1
- 238000001256 steam distillation Methods 0.000 description 1
- ISIJQEHRDSCQIU-UHFFFAOYSA-N tert-butyl 2,7-diazaspiro[4.5]decane-7-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCCC11CNCC1 ISIJQEHRDSCQIU-UHFFFAOYSA-N 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical compound CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 description 1
- HFFLGKNGCAIQMO-UHFFFAOYSA-N trichloroacetaldehyde Chemical compound ClC(Cl)(Cl)C=O HFFLGKNGCAIQMO-UHFFFAOYSA-N 0.000 description 1
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 1
- RXJKFRMDXUJTEX-UHFFFAOYSA-N triethylphosphine Chemical compound CCP(CC)CC RXJKFRMDXUJTEX-UHFFFAOYSA-N 0.000 description 1
- RMZAYIKUYWXQPB-UHFFFAOYSA-N trioctylphosphane Chemical compound CCCCCCCCP(CCCCCCCC)CCCCCCCC RMZAYIKUYWXQPB-UHFFFAOYSA-N 0.000 description 1
- UKRDPEFKFJNXQM-UHFFFAOYSA-N vinylsilane Chemical compound [SiH3]C=C UKRDPEFKFJNXQM-UHFFFAOYSA-N 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 150000003739 xylenols Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G8/00—Condensation polymers of aldehydes or ketones with phenols only
- C08G8/04—Condensation polymers of aldehydes or ketones with phenols only of aldehydes
- C08G8/08—Condensation polymers of aldehydes or ketones with phenols only of aldehydes of formaldehyde, e.g. of formaldehyde formed in situ
- C08G8/10—Condensation polymers of aldehydes or ketones with phenols only of aldehydes of formaldehyde, e.g. of formaldehyde formed in situ with phenol
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Phenolic Resins Or Amino Resins (AREA)
Description
本願は、2010年6月14日に、日本に出願された特願2010−134781号に基づき優先権を主張し、その内容をここに援用する。
このような問題に対して、例えば、フェノール類とアルデヒド類とを、有機ホスホン酸を反応触媒として反応させる、ノボラック型フェノール樹脂の製造方法が開示されている(例えば、特許文献1参照。)。しかしながら、未反応フェノール類の低減、ノボラック型フェノール樹脂の高収率化などについて、更なる向上が望まれている。
[1]フェノール類とアルデヒド類とを反応させるノボラック型フェノール樹脂の製造方法であって、反応触媒として水溶性を有する有機ホスホン酸を用いるとともに、反応助触媒として3級ホスフィン化合物を用い、前記水溶性を有する有機ホスホン酸が、下記一般式(1)で示される構造を有することを特徴とする、ノボラック型フェノール樹脂の製造方法。
R−PO(OH) 2 (1)
(Rは、炭素原子を含み、かつ、−COOH及び/又は−PO(OH) 2 を含む基である)
[2]上記3級ホスフィン化合物を、上記フェノール類に対して100〜5000ppm用いる、上記[1]に記載のノボラック型フェノール樹脂の製造方法。
[3]上記フェノール類とアルデヒド類とを、反応系中の水分量を30重量%以下、反応温度を110〜200℃の条件で反応させる、上記[1]又は[2]に記載のノボラック型フェノール樹脂の製造方法。
[4]上記3級ホスフィン化合物が、トリフェニルホスフィンである、上記[1]ないし[3]のいずれかに記載のノボラック型フェノール樹脂の製造方法。
[5]上記フェノール類とアルデヒド類とを、密閉装置中で反応させる、上記[1]ない
[4]のいずれかに記載のノボラック型フェノール樹脂の製造方法。
[6]上記フェノール類、アルデヒド類、水溶性を有する有機ホスホン酸、及び、3級ホスフィン化合物を一括して仕込み反応させる、上記[1]ないし[5]のいずれかに記載のノボラック型フェノール樹脂の製造方法。
[7]上記フェノール類とアルデヒド類とを、連続式混合装置で反応させる、上記[1]ないし[4]のいずれかに記載のノボラック型フェノール樹脂の製造方法。
[8]上記連続式混合装置として、静止型ミキサーを用いる、上記[7]に記載のノボラック型フェノール樹脂の製造方法。
本発明の製造方法は、フェノール類とアルデヒド類とを反応させるノボラック型フェノール樹脂の製造方法であって、反応触媒として水溶性を有する有機ホスホン酸を用いるとともに、反応助触媒として3級ホスフィン化合物を用いることを特徴とする。
フェノール類とアルデヒド類との反応方法としては特に限定されない。例えば、反応の開始時において、フェノール類とアルデヒド類を全量一括して仕込み、これに反応触媒、及び反応助触媒を添加し反応させてもよい。全てを一括して混合しても良い。あるいは、反応初期の発熱を抑えるため、フェノール類と反応触媒、及び反応助触媒を反応容器に添加してから、アルデヒド類を逐次添加して反応させてもよい。アルデヒド類は2回以上に分けて添加しても良い。
R−PO(OH)2 (1)
(Rは、炭素原子を含み、かつ、−COOH及び又は−PO(OH)2 を含む基である)
この有機ホスホン酸は1以上の−PO(OH)2を含み、式中のRは炭素原子及び−COOH及び−PO(OH)2 の少なくとも1つを含む。前記Rは必要に応じて選択できるが、炭素数が少ない程化合物の水溶性が高くなりより好ましい。
上記一般式(1)で示される有機ホスホン酸の例としては、アミノポリホスホン酸類であるエチレンジアミンテトラキスメチレンホスホン酸、エチレンジアミンビスメチレンホスホン酸、アミノトリスメチレンホスホン酸、β−アミノエチルホスホン酸N,N−ジ酢酸、アミノメチルホスホン酸N,N−ジ酢酸や、1−ヒドロキシエチリデン−1,1’−ジホスホン酸、及び2−ホスホノブタン−1,2,4−トリカルボン酸等がある。
本発明の目的からみて、工業的に大量生産され安価であるアミノトリスメチレンホスホン酸や、1−ヒドロキシエチリデン−1,1’−ジホスホン酸、2−ホスホノブタン−1,2,4−トリカルボン酸が好ましい。
また、本発明の製造方法では、前記触媒等と、シュウ酸、硫酸、塩酸、及びp−トルエンスルホン酸などの通常ノボラック型フェノール樹脂の製造で使用する酸との併用も可能である。これらの酸の併用は特に4核体以上の高分子の領域での反応促進に有効であり、分子量分布を制御する方法として有効な手段と言える。
3級ホスフィン化合物の添加量としては、フェノール類に対して100〜5000ppmであることが好ましく、より好ましくは300〜3500ppm、さらに好ましくは、500〜2000ppmである。添加量が100ppm未満では、3級ホスフィン化合物添加による、未反応フェノール類の含有量が少なく、かつ、分子量分布が狭いノボラック型フェノール樹脂を高収率で得るという効果が低い。5000ppmを超えるとその効果が実質的に変わらない。上記範囲のうちで、必要に応じて3級ホスフィン化合物の添加量を選択できるが、3級ホスフィン化合物の量が少ないと残留する成分が少なくなるという点で有利である。
この反応条件は、未反応フェノール類のみならず、2核体や3核体といった低分子量領域のノボラック型フェノール樹脂が更に選択的に反応するのに有効であり、分子量分布を効果的に狭くすることができる条件である。言い換えれば、未反応フェノール類の反応は、上記反応条件から外れた条件でも、即ち、水分が多く、低温下でも十分に行いうるが、一方で、2核体、3核体等の比較的低分子領域にある樹脂の更なる選択的な反応は、上記反応条件から外れた条件では不十分となることがあり、その結果分子量分布が広くなる傾向がある。
水分には、仕込み時に添加した水分、添加するアルデヒド類に含まれる水分、添加する有機ホスホン酸に含まれる水分、有機ホスホン酸の結晶水等、仕込み原料に由来する水分、及び反応時に発生する縮合水などがある。本発明では、これらの反応系中の水分量が40重量%以下が適当であり、30重量%以下であることが好ましく、より好ましくは1〜20重量%であり、さらに好ましくは1〜15重量%である。反応系中の水分量の計算方法は、仕込み原料中の水分量と反応で生成する縮合水量を反応系中の水分量とし、これを仕込み全量で除した値である。また、水を蒸留して取り除きながら反応させる場合、上記仕込み原料中の水分量及び反応で生成する縮合水量から溜去した水分量を減じた水分量が反応系中の水分量である。この水分量は少ない程、未反応フェノール類の含有量が少なくなり、かつ、分子量分布が狭いノボラック型フェノール樹脂を高収率に得る効果が高くなるので、20重量%以下がより好ましい。しかし、水分量が少なすぎると有機ホスホン酸が高粘度化若しくは固結し、触媒作用が低下するため、結晶水を含む程度の水分量である1重量%以上が好ましい。水分量が30重量%を越えるとそれら効果がほとんど変わらなくなる。
本発明の製造方法に用いる有機ホスホン酸は、非常に水溶性が高い。しかし、有機ホスホン酸はフェノール類には溶解性が小さく、またノボラック型フェノール樹脂には樹脂の分子量増大とともに有機ホスホン酸の溶解性が更に小さくなる性質を有している。このため反応時には、反応触媒である有機ホスホン酸を多量に含んだ水相と、フェノール類、及び形成されたノボラック型フェノール樹脂からなる、反応触媒がほとんど存在しない有機相とに相分離した状態となる。フェノール類及び2核体等の低分子量成分は比較的水相に溶出しやすく、溶出した部分は水相中のアルデヒド類と反応する。しかし、高分子量領域では、すなわち高分子量成分は、水相への溶出がほとんどなく反応が進まない。また、水相で生成したノボラック型フェノール樹脂は速やかに有機相に抽出され、その以上反応は進みにくくなる。
しかしながら、フェノール類、及びノボラック型フェノール樹脂を含有する有機相にも極微量の反応触媒が存在する。このため有機相においても反応が進行し、このことが、高収率を達成しつつ分子量分布を狭くする効果を低下させる。
ここで、反応助触媒として3級ホスフィン化合物を使用することによって、有機相中での有機ホスホン酸の触媒作用を抑えられる。これにより、低分子量領域と高分子量領域の反応速度差が、反応触媒として有機ホスホン酸のみを用いた場合より大きくなるため、結果的に未反応フェノール類の含有量が少なく、かつ、分子量分布が狭いノボラック型フェノール樹脂を高収率に製造する事が可能となる。
本発明の製造方法において、好ましくは上記反応条件とすることにより、分子量分布が狭く、かつ高収率であるノボラック型フェノール樹脂を得ることができる理由は、以下のように考えられる。反応系中の水分が30重量%以下と少なく、反応温度が110℃以上の高温であることにより、以下のような効果を得ることができる。まず、高温であることから、フェノール類だけでなく2核体、3核体等の低分子量領域の成分も水相へ溶出されやすくなり、水相での反応が容易に進む。そして水相中では、水分量が少なく、かつ水相中のイオン濃度が高い状態で維持される。この結果、水相と有機相の界面がよりしっかりと分離するので、有機相側の反応を防止できる。また、有機ホスホン酸は高濃度であると粘度を高めたり固結したりする性質があるが、高温であるため溶融した状態となり触媒機能を失うことが防止できる。これらの作用により、未反応フェノール類の含有量が少なく、かつ、分子量分布が狭いノボラック型フェノール樹脂を高収率に得る効果をより高めることができる。
撹拌装置の一例を挙げると、らせん状のブレードを取付けたスクリュー形状のシャフトを回転させて混合を行うものや、ステーターとタービンとからなり、高速で回転するタービンにより被混合流体を混合するものなどがある。いずれも、撹拌装置を駆動させ、被混合流体を通過させることにより、混合、分散などの作用により被混合流体を均質化させる機能を有する装置である。
上記の方法で行う場合は、好ましくは連続的に1パスで反応を終了させることにより、供給した原材料分の樹脂を連続的に得ることができるので、効率的で好適な形態である。
この他、所定量のフェノール類、アルデヒド類、および水溶性を有する有機ホスホン酸と3級ホスフィン化合物をタンク等に計量して入れ、均一に混合して原材料混合液を調製した後、これを連続式混合装置に供給する方法も挙げられる。この場合、連続式混合装置を通過した原材料混合液は、元のタンクに戻すことにより循環反応を行ってもよいし、別のタンクに送ることもできる。
このような場合でも、連続式混合装置中で反応させていれば、このような問題を回避し、反応を効率よく短時間で進行させることができる。
これは、触媒を含む水相と、フェノール類を含む有機相とは、混じり合わず不均一な状態で反応が進行するため、流速や撹拌速度が速いほど、水相と有機相との接触界面が増加し、反応が効率的に進行するためである。
流速や撹拌速度が遅すぎる場合には、連続式混合装置内部で滞留する部分が発生し、部分的なゲル化が起こる場合がある。
一例を挙げると、連続式混合装置として静止型ミキサーを用い、連続方式にて原材料混合物の流速を5m/分とし、120℃以上で反応させる場合では、静止型ミキサーの長さは10〜15mとなる。
本発明の製造方法により得られるフェノール樹脂をこれらの用途に用いる場合、必要に応じて硬化促進剤、滑剤、及びシランカップリング剤等の改質剤を加えることができる。硬化促進剤としてはサリチル酸、安息香酸、及びマレイン酸等の有機酸やアミン類が挙げられ、アニリン等の塩基性化合物が用いられる滑剤としては、例えば、エチレンビスステアリン酸アマイド、メチレンビスステアリン酸アマイド、オキシステアリン酸アマイド、ステアリン酸アマイド、及びメチロールステアリン酸アマイド等が使用でき、シランカップリング剤としては、例えば、アミノシランカップリング剤、エポキシシランカップリング剤、及びビニルシランカップリング剤等が使用できる。
3Lの三口フラスコ中に1−ヒドロキシエチリデン−1,1’−ジホスホン酸60%水溶液(フェリオックス115、ライオン社製)500部を添加し、常圧蒸留を行い80%の濃度とした。これに、トリフェニルフォスフィン1部、フェノール1000部を添加して100℃に昇温し、37%ホルムアルデヒド水溶液550部を30分間かけて逐次添加し、常圧蒸留を行い、130℃まで昇温させ反応系中の水分量を6%とした。その後、130℃に温度を維持し、水分量を約6%で一定として、常圧蒸留を行いながら37%ホルムアルデヒド水溶液140部を30分間かけて添加した。この間蒸留により失われたフェノール量は仕込んだフェノールに対して0.3%であった。その後、140℃で1時間還流させながら反応を行った。反応時の系中水分は反応初期は6%であり、反応終了時は25%であった。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応フェノール量を測定した。その後、純水500部を添加し、樹脂と分離した水相を除去する水洗工程を3回行った。その後、常圧蒸留を行い130℃まで昇温し、5000Paの減圧度で減圧蒸留を行って150℃まで昇温し、フェノール樹脂A1073部を得た。
実施例1で、トリフェニルフォスフィン添加量を0.1部に変えた以外は実施例1と同様の工程でフェノール樹脂を製造し、フェノール樹脂B1073部を得た。
実施例1で、トリフェニルフォスフィン添加量を5部に変えた以外は実施例1と同様の工程でフェノール樹脂を製造し、フェノール樹脂C1078部を得た。
3Lの三口フラスコ中にフェノール1000部、1−ヒドロキシエチリデン−1,1’−ジホスホン酸(1−1−ヒドロキシエチリデン−1,1’−ジホスホン酸(1水和物)95%以上、キシダ化学社製)300部、トリフェニルフォスフィン1部を添加した。これを140℃に昇温し、92%パラホルムアルデヒド277.5部を30分間かけて逐次添加し、126℃で1時間還流させながら反応させた。この反応時の系中水分は反応初期は2%であり、反応終了時は12%であった。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応フェノール量を測定した。その後、実施例1と同様に、純水500部を添加し、樹脂と分離した水相を除去する水洗工程を3回行った。その後、常圧蒸留を行い130℃まで昇温し、5000Paの減圧度で減圧蒸留を行って150℃まで昇温し、フェノール樹脂D1074部を得た。
3Lの三口フラスコ中にフェノール1000部、1−ヒドロキシエチリデン−1,1’−ジホスホン酸60%水溶液(フェリオックス115、ライオン社製)を200部とトリフェニルフォスフィン1部を添加した。これを、100℃に昇温し、37%ホルムアルデヒド水溶液690部を30分間かけて逐次添加し、100℃で1時間還流させながら反応させた。この反応時の系中水分は反応初期は7%であり、反応終了時は37%であった。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応フェノール量を測定した。その後、実施例1と同様に、純水500部を添加し混合した後、樹脂と分離した水相を除去した。このような水洗工程を3回行った。その後、常圧蒸留を行い130℃まで昇温し、5000Paの減圧度で減圧蒸留を行って150℃まで昇温し、フェノール樹脂E1058部を得た。
3Lの三口フラスコ中にフェノール1000部、アミノトリスメチレンホスホン酸50%水溶液(ディクエスト2000、ソルーシア・ジャパン社製)240部とトリフェニルフォスフィン1部を添加した。これを、100℃に昇温し、37%ホルムアルデヒド水溶液690部を30分間かけて逐次添加し、100℃で1時間還流させながら反応させた。この反応時の系中水分は反応初期は10%であり、反応終了時は38%であった。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応フェノール量を測定した。その後、実施例1と同様に、純水500部を添加し、樹脂と分離した水相を除去する水洗工程を3回行った。その後、常圧蒸留を行い130℃まで昇温し、5000Paの減圧度で減圧蒸留を行って150℃まで昇温し、フェノール樹脂F1054部を得た。
3Lの三口フラスコ中にフェノール1000部、2−ホスホノブタン−1,2,4−トリカルボン酸50%水溶液(PBTC、城北化学社製)240部とトリフェニルフォスフィン1部を添加した。これを100℃に昇温し、37%ホルムアルデヒド水溶液690部を30分間かけて逐次添加し、100℃で1時間還流させながら反応させた。この反応時の系中水分は反応初期は10%であり、反応終了時は38%であった。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応フェノール量を測定した。その後、実施例1と同様に、純水500部を添加し、樹脂と分離した水相を除去する水洗工程を3回行った。その後、常圧蒸留を行い130℃まで昇温し、5000Paの減圧度で減圧蒸留を行って150℃まで昇温し、フェノール樹脂G1049部を得た。
3Lの三口フラスコ中にフェノール1000部、1−ヒドロキシエチリデン−1,1’−ジホスホン酸60%水溶液(フェリオックス115、ライオン社製)1000部、トリフェニルフォスフィン1部を添加した。これを100℃に昇温し、37%ホルムアルデヒド水溶液690部を1時間かけて逐次添加し、100℃で1時間還流させながら反応を行った。この反応時の系中水分は反応初期は20%であり、反応終了時は38%であった。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応フェノール量を測定した。その後、実施例1と同様に、純水500部を添加し、樹脂と分離した水相を除去する水洗工程を3回行った。その後、常圧蒸留を行い130℃まで昇温し、5000Paの減圧度で減圧蒸留を行って150℃まで昇温し、フェノール樹脂H1066部を得た。
攪拌装置及び温度計を備えた10Lの密閉装置(オートクレーブ)にフェノール3000部、1−ヒドロキシエチリデン−1,1’−ジホスホン酸60%水溶液(フェリオックス115、ライオン社製)3000部、37%ホルムアルデヒド水溶液1811部(モル比F/P=0.7)、トリフェニルフォスフィン3部を添加して密閉状態で加熱した。内温が80℃に上昇した時点で急激な発熱反応が起こり、内温が150℃までおよそ3分間で上昇した。同時に圧力も0.45MPaまで上昇した。内温が130℃まで低下した時点で圧力抜きコックを徐々に開き、冷却管経由で内圧を0MPaに戻した。内温を80℃まで低下させ、反応物を取り出した。この反応時の系中水分は反応初期は30%であり、反応終了時は34%であった。反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応フェノール量を測定した。その後、実施例1と同様に、純水500部を添加し、樹脂と分離した水相を除去する水洗工程を3回行った。その後、常圧蒸留を行い130℃まで昇温し、5000Paの減圧度で減圧蒸留を行って150℃まで昇温し、フェノール樹脂I3242部を得た。
攪拌装置および温度計を備えた10Lの密閉容器(装置1)の下部に、蒸気で温調できる10m長さの静止型ミキサー(エレメント形状:長方形の板を180度捻ったスパイラル形状をつなげたもの)を開閉式のコックを介し接続した。静止型ミキサーの出口は、攪拌装置、冷却管、温度計および冷却装置を備えた10Lの密閉容器(装置2)の上部に、開閉式のコックを介し接続した。
装置1にフェノール1000部、37%ホルマリン溶液604部(モル比F/P=0.7)、1−ヒドロキシエチリデン−1,1’−ジホスホン酸60%水溶液(フェリオックス115、ライオン社製)1000部、トリフェニルフォスフィン1部を添加し、60℃にて密閉状態で充分に混合した。静止型ミキサーは160℃で保温した。装置1内を0.1MPaまでエアーで加圧し、装置1の下部コックを全開にした。この際、装置1内の圧力が0.1MPaを保つようにエアー圧を調整した。上記混合物が静止型ミキサーを通過するのに所要する時間が2分間となるように装置2上部のコックの開度を調整した。装置2は、攪拌しながら冷却した。静止型ミキサー内の内温(装置1から1mの位置に温度計を設置)は133℃まで上昇した。装置1内部が空になり、装置2へ全量移送終了後、エアーを停止し、装置1、2のコックを閉じた。この時点で装置2の内温は、64℃であった。この反応時の系中水分は反応初期は30%であり、反応終了時は34%であった。その後、水500部を加え、内温80〜90℃で15分間攪拌した。内温を60℃まで冷却し、10分間静置した。反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応フェノール量を測定した。その後、実施例1と同様に、純水500部を添加し、樹脂と分離した水相を除去する水洗工程を3回行った。その後、常圧蒸留を行い130℃まで昇温し、5000Paの減圧度で減圧蒸留を行って150℃まで昇温し、フェノール樹脂J1072部を得た。
実施例1でトリフェニルフォスフィン添加を行わなかった以外は実施例1と同様の工程でフェノール樹脂を製造し、フェノール樹脂K1070部を得た。
実施例4でトリフェニルフォスフィン添加を行わなかった以外は実施例4と同様の工程でフェノール樹脂を製造し、フェノール樹脂L1071部を得た。
実施例5でトリフェニルフォスフィン添加を行わなかった以外は実施例5と同様の工程でフェノール樹脂を製造し、フェノール樹脂M1056部を得た。
実施例6でトリフェニルフォスフィン添加を行わなかった以外は実施例6と同様の工程でフェノール樹脂を製造し、フェノール樹脂N1052部を得た。
実施例7でトリフェニルフォスフィン添加を行わなかった以外は実施例7と同様の工程でフェノール樹脂を製造し、フェノール樹脂O1047部を得た。
実施例8でトリフェニルフォスフィン添加を行わなかった以外は実施例8と同様の工程でフェノール樹脂を製造し、フェノール樹脂P1065部を得た。
実施例9でトリフェニルフォスフィン添加を行わなかった以外は実施例9と同様の工程でフェノール樹脂を製造し、フェノール樹脂Q3225部を得た。
実施例10でトリフェニルフォスフィン添加を行わなかった以外は実施例10と同様の工程でフェノール樹脂を製造し、フェノール樹脂R1062部を得た。
3Lの三口フラスコ中にフェノール1000部、シュウ酸10部を添加し、100℃に昇温し、37%ホルムアルデヒド水溶液690部を30分間かけて逐次添加し、100℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応フェノール量を測定した。その後、常圧蒸留を行い130℃まで昇温し、5000Paの減圧下で減圧蒸留を行って190℃まで昇温し、フェノール樹脂S957部を得た。
3Lの三口フラスコ中にフェノール1000部、シュウ酸10部を添加し、100℃に昇温し、37%ホルムアルデヒド水溶液690部を30分間かけて逐次添加し、100℃で1時間還流させながら反応させた。反応終了後、反応組成物をサンプリングしガスクロマトグラフィーを用いて未反応フェノール量を測定した。その後、実施例1と同様に、純水500部を添加し、樹脂と分離した水相を除去する水洗工程を3回行った。その後、常圧蒸留を行い130℃まで昇温し、5000Paの減圧下で減圧蒸留を行って150℃まで昇温し、フェノール樹脂T972部を得た。
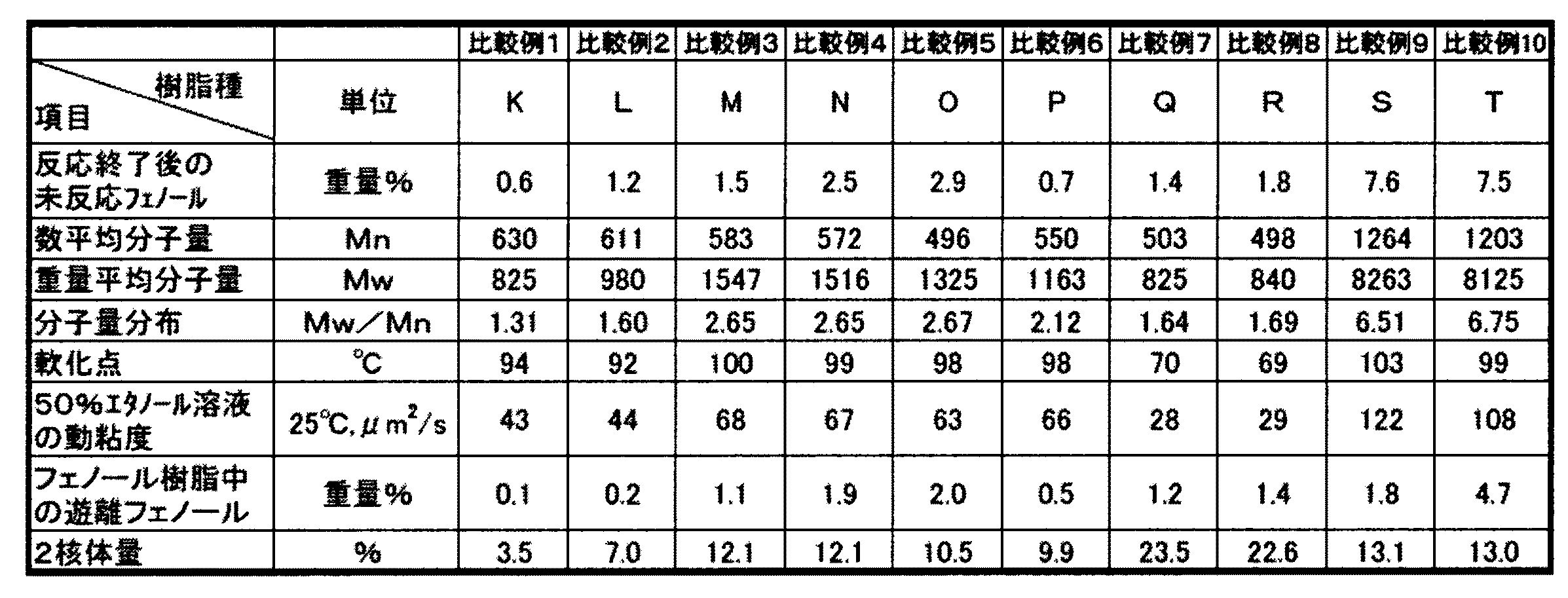
1.反応終了後の未反応フェノール量及びフェノール樹脂中の遊離フェノール量:ガスクロマトグラフィーで測定した。
・ガスクロマトグラフィー:JIS K0114に準拠し、2,5−キシレノールを内部標準として内部標準法で測定した。
2.軟化点:JIS K2207に準拠して測定した。
3.50%エタノール溶液の動粘度:50重量%のエタノール溶液を25℃でキャノンフェンスケを用いて測定した。
4.数平均分子量、重量平均分子量、2核体量:液体クロマトグラフィー
液体クロマトグラフィー:東ソー製GPCカラム(G1000HXL:1本、G2000HXL:2本、G3000HXL:1本)を用い、流量1.0ml/分、溶出溶媒テトラヒドロフラン、カラム温度40℃の分析条件で示差屈折計を検出器として用いてGPC測定し、分子量は標準ポリスチレンにより換算した。また、2核体量は、液体クロマトグラフィーで測定したチャートの面積比から求めた。
Claims (8)
- フェノール類とアルデヒド類とを反応させるノボラック型フェノール樹脂の製造方法であって、反応触媒として水溶性を有する有機ホスホン酸を用いるとともに、反応助触媒として3級ホスフィン化合物を用い、
前記水溶性を有する有機ホスホン酸が、下記一般式(1)で示される構造を有することを特徴とする、ノボラック型フェノール樹脂の製造方法。
R−PO(OH) 2 (1)
(Rは、炭素原子を含み、かつ、−COOH及び/又は−PO(OH) 2 を含む基である) - 前記3級ホスフィン化合物を、前記フェノール類に対して100〜5000ppm用いる、請求項1に記載のノボラック型フェノール樹脂の製造方法。
- 前記フェノール類とアルデヒド類とを、反応系中の水分量を30重量%以下、反応温度を110〜200℃の条件で反応させる、請求項1又は2に記載のノボラック型フェノール樹脂の製造方法。
- 前記3級ホスフィン化合物が、トリフェニルホスフィンである、請求項1ないし3いずれかに記載のノボラック型フェノール樹脂の製造方法。
- 前記フェノール類とアルデヒド類とを、密閉装置中で反応させる、請求項1ないし4のいずれかに記載のノボラック型フェノール樹脂の製造方法。
- 前記フェノール類、アルデヒド類、水溶性を有する有機ホスホン酸、及び、3級ホスフィン化合物を一括して仕込み反応させる、請求項1ないし5のいずれかに記載のノボラック型フェノール樹脂の製造方法。
- 前記フェノール類とアルデヒド類とを、連続式混合装置で反応させる、請求項1ないし4のいずれかに記載のノボラック型フェノール樹脂の製造方法。
- 前記連続式混合装置として、静止型ミキサーを用いる、請求項7に記載のノボラック型フェノール樹脂の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012520422A JP5252130B2 (ja) | 2010-06-14 | 2011-06-10 | ノボラック型フェノール樹脂の製造方法 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010134781 | 2010-06-14 | ||
| JP2010134781 | 2010-06-14 | ||
| PCT/JP2011/063362 WO2011158751A1 (ja) | 2010-06-14 | 2011-06-10 | ノボラック型フェノール樹脂の製造方法 |
| JP2012520422A JP5252130B2 (ja) | 2010-06-14 | 2011-06-10 | ノボラック型フェノール樹脂の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP5252130B2 true JP5252130B2 (ja) | 2013-07-31 |
| JPWO2011158751A1 JPWO2011158751A1 (ja) | 2013-08-19 |
Family
ID=45348151
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012520422A Expired - Fee Related JP5252130B2 (ja) | 2010-06-14 | 2011-06-10 | ノボラック型フェノール樹脂の製造方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US8822627B2 (ja) |
| JP (1) | JP5252130B2 (ja) |
| CN (1) | CN102933630B (ja) |
| WO (1) | WO2011158751A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20160053161A1 (en) * | 2013-04-16 | 2016-02-25 | Sumitomo Bakelite Company Limited | Resin composition, injection material and packing method |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH04226181A (ja) * | 1990-06-08 | 1992-08-14 | Dainippon Ink & Chem Inc | 熱硬化性塗装用組成物および塗料 |
| JP2001329034A (ja) * | 2000-05-22 | 2001-11-27 | Sumitomo Bakelite Co Ltd | フェノール樹脂の製造方法 |
| JP2002105157A (ja) * | 2000-09-28 | 2002-04-10 | Sumitomo Bakelite Co Ltd | フェノール樹脂の製造方法 |
| JP2002128849A (ja) * | 2000-10-26 | 2002-05-09 | Sumitomo Bakelite Co Ltd | フェノール樹脂の製造方法 |
| JP2002194041A (ja) * | 1999-12-16 | 2002-07-10 | Sumitomo Bakelite Co Ltd | フェノール樹脂の製造方法 |
| JP2004238618A (ja) * | 2003-01-16 | 2004-08-26 | Sumitomo Bakelite Co Ltd | ノボラック型アルキルフェノール樹脂の製造方法 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4016348A (en) * | 1972-02-22 | 1977-04-05 | Adams George F | Reactor process and apparatus for continuous polymerization |
| US6326453B2 (en) * | 1999-12-16 | 2001-12-04 | Sumitomo Durez Company, Ltd. | Process for producing phenol resin |
| JP2004339256A (ja) * | 2003-05-13 | 2004-12-02 | Asahi Organic Chem Ind Co Ltd | ノボラック型フェノール樹脂の製造方法 |
| EP2409979B1 (en) * | 2009-03-18 | 2019-06-26 | DIC Corporation | Process for production of phosphorus-atom-containing phenol, novel phosphorus-atom-containing phenol, curable resin composition, cured product thereof, printed circuit board, and semiconductor sealing material |
-
2011
- 2011-06-10 JP JP2012520422A patent/JP5252130B2/ja not_active Expired - Fee Related
- 2011-06-10 US US13/699,085 patent/US8822627B2/en not_active Expired - Fee Related
- 2011-06-10 CN CN201180028110.3A patent/CN102933630B/zh not_active Expired - Fee Related
- 2011-06-10 WO PCT/JP2011/063362 patent/WO2011158751A1/ja active Application Filing
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH04226181A (ja) * | 1990-06-08 | 1992-08-14 | Dainippon Ink & Chem Inc | 熱硬化性塗装用組成物および塗料 |
| JP2002194041A (ja) * | 1999-12-16 | 2002-07-10 | Sumitomo Bakelite Co Ltd | フェノール樹脂の製造方法 |
| JP2001329034A (ja) * | 2000-05-22 | 2001-11-27 | Sumitomo Bakelite Co Ltd | フェノール樹脂の製造方法 |
| JP2002105157A (ja) * | 2000-09-28 | 2002-04-10 | Sumitomo Bakelite Co Ltd | フェノール樹脂の製造方法 |
| JP2002128849A (ja) * | 2000-10-26 | 2002-05-09 | Sumitomo Bakelite Co Ltd | フェノール樹脂の製造方法 |
| JP2004238618A (ja) * | 2003-01-16 | 2004-08-26 | Sumitomo Bakelite Co Ltd | ノボラック型アルキルフェノール樹脂の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2011158751A1 (ja) | 2011-12-22 |
| CN102933630B (zh) | 2014-08-20 |
| JPWO2011158751A1 (ja) | 2013-08-19 |
| US20130066036A1 (en) | 2013-03-14 |
| CN102933630A (zh) | 2013-02-13 |
| US8822627B2 (en) | 2014-09-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4998271B2 (ja) | フェノール樹脂及び樹脂組成物 | |
| CN103304778A (zh) | 酚树脂和热固性树脂组合物 | |
| JP5252130B2 (ja) | ノボラック型フェノール樹脂の製造方法 | |
| CN103597001B (zh) | 尿素改性酚醛清漆型酚醛树脂的制造方法和由该方法得到的尿素改性酚醛清漆型酚醛树脂及用其得到的树脂覆膜砂 | |
| CN102282187B (zh) | 酚醛清漆树脂和热固性树脂组合物 | |
| JP3651843B2 (ja) | フェノール樹脂の製造方法 | |
| JP2013177524A (ja) | バイオマス誘導体、バイオマス誘導体組成物及びバイオマス誘導体硬化物 | |
| JP7180815B2 (ja) | フェノール樹脂組成物およびその製造方法 | |
| JP2005075939A (ja) | ノボラック型フェノール樹脂の製造方法 | |
| JP2003212945A (ja) | ノボラック型フェノール樹脂の製造方法 | |
| JP2005154480A (ja) | ノボラック型フェノール樹脂の製造方法 | |
| JP2006016489A (ja) | レゾール型フェノール樹脂及びその製造方法 | |
| JP4206909B2 (ja) | トリアジン変性ノボラック型フェノール樹脂の製造方法 | |
| JP2005095933A (ja) | シェルモールド用ノボラック型フェノール樹脂 | |
| JP2005179382A (ja) | ノボラック型フェノール樹脂の製造方法 | |
| KR100561898B1 (ko) | 고-분자량 고-오르토 노볼락형 페놀수지 | |
| JPH10195158A (ja) | ノボラック型フェノール樹脂の製造方法 | |
| JP3874338B2 (ja) | ノボラック型フェノール樹脂の製造方法 | |
| JP2002167417A (ja) | 芳香族炭化水素変性フェノール樹脂の製造方法 | |
| JPH0867728A (ja) | ノボラック型フェノール樹脂の製造方法 | |
| JP2002179751A (ja) | 芳香族炭化水素フェノール樹脂の製造方法 | |
| JP2006265427A (ja) | アルキルエーテル化フェノール樹脂とその製造方法 | |
| JP2006056960A (ja) | レゾール型フェノール樹脂乳濁液及びその製造方法 | |
| JP2004091726A (ja) | 摩擦材用フェノール樹脂組成物およびその製造方法 | |
| JP2003292555A (ja) | 固形レゾール型フェノール樹脂の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20130319 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20130401 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 Ref document number: 5252130 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20160426 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |